Настоящее изобретение относится к катализаторам, включающим один или несколько переходных металлов, диспергированных на пористом носителе.
Катализаторы, включающие металлы и соединения металлов, диспергированных на материале пористого носителя, были хорошо известны и использовались в химической промышленности в течение многих лет для различных целей, включая гидрирование и дегидрирование химического сырья. US 4490480 описывает такие катализаторы, используемые для различных реакций гидрирования, которые состоят из 5-40% масс. никеля на переходном оксиде алюминия, в частности на носителе из гамма-оксида алюминия. Эти катализаторы имеют площадь поверхности активного никеля между 80 и 300, предпочтительно 100-250 м2/г никеля, и кристаллиты никеля имеют средний диаметр от 1 до 5, предпочтительно от 1,5 до 3 нм. Кристаллиты никеля диспергированы на по меньшей мере 95% в порах оксида алюминия. Патент описывает способ изготовления катализаторов путем нагрева водной суспензии или смеси переходного оксида алюминия в растворенном комплексе никель-оксид алюминия в течение некоторого времени до температуры 60-100°С, предпочтительно 75-95°С, которое вызывает осаждение гидроксида никеля. Суспензию катализатора отделяют и, если требуется, промывают, после этого сушат и прокаливают до оксида никеля и, если будет необходимо, восстанавливают. Альтернативно гранулы оксида алюминия или экструдаты пропитывают концентрированным раствором аммиачного комплекса никеля, после чего никель осаждают повышением температуры.
US 4920089 предлагает катализатор никель на переходном оксиде алюминия, содержащий 5-40% масс. никеля с площадью поверхности активного никеля между 80 и 300 м2/г Ni, с переходным оксидом алюминия, который соответствует определенной картине дифракции рентгеновских лучей. Предпочтительно общая площадь поверхности катализатора по БЭТ находится между 50 и 200 м2/г катализатора и практически не содержит пор с радиусом ниже 2,0 нм. Катализаторы готовили пропиткой формованных частиц тета-оксида алюминия аммиачным раствором никеля, имеющим особенно высокую величину рН, а именно между 9 и 11, с последующим выпариванием пропитанных частиц оксида алюминия до сухости, прокаливанием и восстановлением.
Способы, описанные в вышеупомянутых патентах предшествующего уровня техники, позволяют образовать высокодисперсные и активные металлические катализаторы, которые были весьма удачными. Авторы, однако, обнаружили, что невозможно приготовить катализаторы в форме гранул или в фасонных формах, подходящих для типа реакций с неподвижным слоем, которые имеют содержание металла выше чем примерно 33% масс. и которые имеют высокую дисперсию металла в сочетании с высокой прочностью на раздавливание. Целью изобретения является предложить такой катализатор.
Согласно данному изобретению катализатор представляет собой дисперсный катализатор в форме частиц, имеющих минимальный размер по меньшей части 0,8 мм, включающий переходный металл или его соединение, диспергированные на материале пористого носителя, отличающийся тем, что частицы указанного катализатора включают по меньшей мере 35% масс. суммарного переходного металла, и площадь поверхности переходного металла в указанном катализаторе составляет по меньшей мере 110 м2/г переходного металла, и насыпной вес после уплотнения постукиванием слоя частиц катализатора составляет по меньшей мере 0,7 г/мл.
Согласно второму аспекту изобретения предложен способ изготовления катализатора, включающего переходный металл или его соединение, диспергированные на материале пористого носителя, где указанный катализатор содержит по меньшей мере 35% масс. суммарного переходного металла, включающий стадии:
a) обеспечения раствором аммиачного комплекса указанного переходного металла;
b) пропитки указанным раствором аммиачного комплекса материала пористого носителя в форме частиц, имеющих минимальный размер по меньшей мере 0,8 мм, где общий объем пор больше 1,0 мл/г;
c) сушки пропитанных частиц носителя, полученных на стадии (b);
d) повторения стадий (b) и (с) по меньшей мере еще четыре раза до тех пор, пока количество переходного металла в частице не достигнет требуемого уровня, и включения стадии
e) прокаливания высушенных пропитанных частиц со стадии (с) при температуре и длительности, достаточных для превращения по меньшей мере большей части соединений переходного металла, пропитавших носитель, в оксиды переходного металла;
f) необязательно восстановления по меньшей мере 50% оставшегося соединения переходного металла и оксида переходного металла в элементарный металл.
Переходный металл предпочтительно выбирают из кобальта, никеля или меди, и он может включать более чем один переходный металл.
Суммарное содержание переходного металла в конкретном катализаторе составляет по меньшей мере 35% масс. Суммарное содержание переходного металла означает массу металла независимо от того, присутствует ли он в виде элементарного восстановленного металла или в виде соединения металла, выраженное в процентах от общей массы катализатора. Количество металла может быть измерено рентгеновской флюоресценцией (XRF) или эмиссионной спектроскопией индуктивно связанной плазмы (ICP-AES). Для целей определения суммарного содержания металла в катализаторе, описанном и заявленном здесь, применяли ICP-AES, используя внутренний стандарт сульфат меди, калиброванной с использованием стандартных растворов сульфата никеля. Оба метода хорошо известны для использования при определении содержания металла в таких материалах, как катализаторы, и специалист должен иметь такие методы доступными для него.
Насыпной вес катализатора после уплотнения постукиванием составляет по меньшей мере 0,7 г/мл, когда он измерен, и использованием следующего метода. 100 г (приблизительно) образца частиц катализатора точно взвешивают в стандартном 250 мл мерном цилиндре, помещают на платформу автоматического анализатора насыпного веса после уплотнения постукиванием и постукивают 2000 раз перед измерением объема образца. Насыпной вес рассчитывают затем как точный вес катализатора в граммах, деленный на объем образца после уплотнения постукиванием. Насыпные веса, приведенные в этом документе, измерены с использованием прибора Quantachrome Dual Autotap™, доступного от Quantachrome Instruments. Предпочтительно насыпной вес катализатора после уплотнения постукиванием составляет по меньшей мере 0,75 г/мл.
Хотя катализаторы, содержащие 35% никеля и имеющие относительно высокую площадь поверхности металлического никеля, известны в форме порошков и мелких частиц, авторы изобретения считают, что такие катализаторы в форме частиц, имеющих минимальный размер по меньшей мере 0,8 мм и насыпной вес после уплотнения постукиванием по меньшей мере 0,7, являются новыми. Частицы, имеющие минимальный размер по меньшей мере 0,8 мм, включают фигурные конфигурации катализатора, такие как сферы, цилиндры, лопастные цилиндры (т.е. фигуры, имеющие поперечный разрез в форме многолопастного круга, такие как трехлопастные, четырехлопастные или пятилопастные), колеса, кольца, седла и другие известные формы. Некоторые формы могут включать каналы, выточки или отверстия. Минимальным размером может быть диаметр или длина, как целесообразно. Предпочтительно минимальный размер составляет по меньшей мере 1 мм. Обычно частицы катализатора измеряют и устанавливают средний минимальный размер. Величины минимального размера, использованные в этом документе, рассчитаны как средний минимальный размер 40 частиц катализатора.
Частицы могут быть сформированы гранулированием, таблетированием, экструзией или другими известными способами. Экструдированные частицы являются предпочтительными. В предпочтительном осуществлении изобретения частицы катализатора включают экструдированные цилиндры или лопастные цилиндры. Такие частицы подходят для использования в реакторах неподвижного слоя, где исходное соединение или смесь соединений вынуждают двигаться над слоем и/или через слой частиц катализатора. Для использования в слое катализатора необходимо, чтобы частицы катализатора были достаточно прочны для того, чтобы оставаться неповрежденными внутри слоя. Предпочтительно частицы катализатора имеют прочность на раздавливание по меньшей мере 10 Н/мм, более предпочтительно по меньшей мере 12 Н/мм, выраженную как средняя прочность на раздавливание 25 частиц. Прочность на раздавливание 25 частиц измеряли, используя полутонную машину для испытаний на раздавливание Engineering System CT5. Среднюю прочность на раздавливание на единицу длины рассчитывали затем из среднего усилия, деленного на среднюю длину частиц. Частицы нагревали до 500°С в никелевом тигле в течение одного часа перед испытанием и затем держали при 100°С, чтобы избежать абсорбции влаги.
Материалом пористого носителя предпочтительно является переходный оксид алюминия. Переходный оксид алюминия может включать дельта-, эта-, гамма- и/или тета-оксид алюминия. Одно предпочтительное осуществление использует переходный оксид алюминия, включающий главным образом гамма-фазу оксида алюминия. Пористый материал находится обычно в виде фасонных, например экструдированных, частиц, как описано выше для катализатора. Материал пористого носителя предпочтительно имеет суммарный объем пор, измеренный ртутной порозиметрией, по меньшей мере 1,0 мл/г. Материал пористого носителя предпочтительно имеет общую площадь поверхности, измеренную методами БЭТ, по меньшей мере 250 м2/г. Особенно предпочтительным материалом пористого носителя является носитель из переходного оксида алюминия, имеющий объем пор по меньшей мере 1,0 мл/г и бимодальное распределение размеров пор. Авторы изобретения обнаружили, что, используя носитель катализатора этого типа, в материал носителя можно ввести большее количество переходного металла, чем в носитель, имеющий одномодальное распределение размера пор, сохраняя в то же время прочность на раздавливание, достаточную для катализатора, который должен быть использован в неподвижном слое. Предпочтительный носитель катализатора имеет распределение размеров пор, измеренное ртутной порозиметрией, в котором по меньшей мере 20% (предпочтительно по меньшей мере 25%) от общего объема пор содержится в порах, имеющих диаметр от 100 нм до 700 нм, а по меньшей мере 30%, более предпочтительно по меньшей мере 40% от общего объема пор содержится в порах, имеющих диаметр от 5 нм до 20 нм.
Аммиачный комплекс переходного металла для использования при приготовлении катализаторов согласно способу по изобретению может быть изготовлен растворением соединения переходного металла или количества переходного металла в металлической форме в растворе соединения аммония, такого как карбонат аммония, в гидроксиде аммония. Аммиачно-карбонатные комплексы переходного металла являются предпочтительными, но могут быть использованы другие анионы, такие как сульфаты, ацетаты или формиаты.
Аммиачным комплексом кобальта наиболее предпочтительно является аммиачно-карбонатный комплекс кобальта, который образуется in situ в водном растворе при растворении основного карбоната кобальта в растворе карбоната аммония в водном гидроксиде аммония, чтобы получить продукт с желаемым содержанием кобальта. Альтернативно могут быть использованы другие соли кобальта, включая органические соли, такие как ацетат кобальта или формиат кобальта. Аммиачно-карбонатные растворы никеля и меди могут быть приготовлены подобным образом.
Аммиачно-карбонатный комплекс кобальта является продуктом растворения основного карбоната кобальта предпочтительно эмпирической формулы Co(OH)2-2x(CO3)x в растворе карбоната аммония в водном гидроксиде аммония, чтобы получить продукт с желаемым содержанием кобальта. Аммиачно-карбонатный раствор кобальта может быть изготовлен растворением основного карбоната кобальта в водном растворе карбоната аммония или карбамата аммония, содержащем дополнительный гидроксид аммония. Относительные количества должны быть такими, чтобы рН раствора было в интервале от 7,5 до 12, предпочтительно от 9 до 12. Раствор предпочтительно содержит от 0,1 до 2,5 молей комплекса кобальта на литр. Когда концентрация кобальта возрастает, обычно доля карбонатных ионов по отношению к гидроксидным ионам в исходном основном карбонате кобальта должна быть увеличена. Дополнительный раствор гидроксида аммония может быть добавлен, если требуется повысить вязкость раствора. В качестве альтернативы аммиачно-карбонатный раствор кобальта может быть изготовлен растворением металлического кобальта, предпочтительно в форме порошка, в водном аммиаке с рН 11-12 в присутствии кислорода или воздуха или с добавлением карбоната аммония или с добавлением газообразного СО2. Раствору аммиачного комплекса кобальта может быть предоставлена возможность окисляться до того, как им будут пропитывать носитель катализатора, или путем старения в контакте с богатым кислородом газом, или путем химического или электрохимического окисления, для того, чтобы комплекс Co(II) был превращен, по меньшей мере частично, в комплекс Co(III).
Когда переходным металлом является никель, катализаторы могут быть приготовлены путем пропитки пористого носителя должным количеством водного раствора аммиачного комплекса никеля. Аммиачно-карбонатный комплекс никеля может быть изготовлен растворением основного карбоната никеля в растворе карбоната аммония в водном гидроксиде аммония, чтобы получить продукт с желаемым содержанием никеля. Раствор аммиачного комплекса никеля предпочтительно имеет рН в интервале от 9 до 10,5.
Аммиачно-карбонатный раствор меди может быть приготовлен растворением основного карбоната меди в водном растворе карбоната аммония, содержащем дополнительно гидроксид аммония. Относительные количества должны быть такими, чтобы рН раствора был в интервале 7-12, предпочтительно 8-11. Раствор предпочтительно содержит 2-5, предпочтительно 2-4 моля комплекса меди на литр. Когда концентрация меди возрастает, обычно доля карбонатных ионов по отношению к гидроксидным ионам в исходном основном карбонате меди должна быть увеличена.
Предпочтительно суммарное число стадий пропитки (b) составляет по меньшей мере пять, более предпочтительно по меньшей мере шесть, и пропитанный катализатор, полученный на каждой стадии (b), затем сушат на стадии (с). В предпочтительном способе стадии (b) и (с) проводят три раза, затем проводят стадию (е), и затем стадии (b) и (с) проводят по меньшей мере еще один раз (более предпочтительно по меньшей мере еще два раза, особенно предпочтительно еще три раза), и затем проводят во второй раз стадию прокаливания (е). Предпочтительно практически все соединения переходного металла на стадии (е) превращаются в оксиды переходного металла.
Предпочтительно прокаленный катализатор с конечной стадии прокаливания (е) восстанавливают водородсодержащим газом. Предпочтительно восстановленный катализатор затем пассивируют, используя кислородсодержащий газ, используя обычные методы пассивации, такие, чтобы температура катализатора не превышала 100°С, когда он подвергается воздействию воздуха. Должная пассивация катализатора позволяет хранить его и транспортировать на воздухе без воспламенения, вызванного пирофорной природой тонко диспергированных частиц металла, которая хорошо известна.
Частицы высушенного пропитанного носителя прокаливают согласно стадии (е) способа предпочтительно после по меньшей мере каждых трех стадий пропитки и сушки. Предпочтительно стадию прокаливания проводят, когда количество переходного металла в частице посредством достаточно успешных стадий пропитки достигло требуемого уровня.
Площадь поверхности переходного металла катализатора составляет по меньшей мере 110 м2/г переходного металла. Площадь поверхности переходного металла измеряют после восстановления металла до его элементарного состояния. Когда переходным металлом является никель, поверхность металлического никеля составляет по меньшей мере 110 м2/г суммарного никеля, присутствующего в катализаторе, при измерении хемосорбцией водорода после того, как катализатор был восстановлен в токе водорода при заранее определенной температуре в течение одного часа. Стадия восстановления является частью процедуры определения площади поверхности и делается для того, чтобы восстановить соединения никеля до элементарного никеля для измерения хемосорбции водорода. Когда никель в катализаторе находится в значительной степени или полностью в форме соединений оксида никеля, заранее заданная температура восстановления составляет 430°С. Когда никель в катализаторе находится в основном или в значительной степени в элементарной форме, т.е. когда катализатор был предварительно восстановлен и пассивирован, то заранее заданная температура восстановления составляет 240°С. 0,7-0,8 г образца точно взвешивали и переносили в кювету для образца аппарата хемосорбции. Устанавливали поток водорода через кювету для образца 250 см3/мин. Затем температуру поднимали со скоростью повышения 3°С×мин-1 до выбранной температуры восстановления и поддерживали постоянной в течение одного часа. После восстановления поток Н2 останавливали, и кювету для образца дегазировали при 450°С под вакуумом в течение шести часов и затем давали ей остыть до 50°С, поддерживая в это время вакуум. Хемосорбция Н2 проводится в интервале давления между 100 и 760 мм рт.ст. (между 1,133 кПа и 101,08 кПа). Образцу давали придти в равновесие при каждом давлении в течение 60 секунд. Объем водорода, хемосорбировавшегося при каждом давлении, наносили на график как функцию давления. Выбирали наиболее линейную часть изотермы и экстраполировали назад до пересечения с нулевым давлением, чтобы определить емкость монослоя. Количеством монослоя является его емкость, деленная на массу образца в граммах.
Удельную площадь поверхности никеля определяли из следующего уравнения:
SNi=(Nm×NA×S)/D,
где:
SNi - удельная площадь поверхности никеля, м2/г;
Nm - количество монослоя, моль × г-1;
NA - число Авогадро, 6×1023 моль-1;
S - стехиометрия абсорбции Н2, которая была принята равной 2;
D - поверхностная плотность атомов никеля, 1,54×1019 атомов на м2.
Предпочтительно площадь поверхности никеля составляет по меньшей мере 120 м2/г.
Площадь поверхности кобальта определяли хемосорбцией Н2. Этот способ использовали, когда измерение площади поверхности кобальта упоминается в данном описании для катализаторов по изобретению, содержащих кобальт в качестве переходного металла (если не указано иное). Приблизительно от 0,2 до 0,5 г образца материала вначале дегазировали и сушили нагреванием до 140°С со скоростью 10°С/мин в потоке гелия и поддержанием при 140°С в течение 60 минут. Дегазированный и высушенный образец затем восстанавливали, нагревая от 140°С до 425°С со скоростью 3°С/мин в токе 50 мл/мин водорода и затем поддерживая поток водорода при 425°С в течение 6 часов. После этого образец нагревали под вакуумом до 450°С со скоростью 10°С/мин и выдерживали при этих условиях в течение 2 часов. Затем образец охлаждали до 150°С и выдерживали под вакуумом дополнительно 60 минут. Затем проводили анализ на хемосорбцию при 150°С, используя чистый газообразный водород. Программу автоматического анализа использовали для измерения полной изотермы в интервале давления водорода от 100 мм рт.ст. до 760 мм рт. ст. Анализ проводили дважды, вначале измеряя "суммарное" поглощение водорода (т.е. включающее хемосорбированный водород и физически адсорбированный водород), и немедленно после первого анализа образец помещали под вакуум (<5 мм рт.ст.) на 30 минут. Затем анализ повторяли, чтобы измерить поглощение физической адсорбцией. К данным по "суммарному" поглощению применяли линейную регрессию с экстраполяцией назад до нулевого давления, чтобы рассчитать объем (V) хемосорбированного газа.
Площади поверхности кобальта во всех случаях рассчитывали, используя следующее уравнение:
Площадь поверхности кобальта=(6,023×1023×V×SF×A)/22414,
где:
V - поглощение H2 в мл/г;
SF - стехиометрический фактор (для хемосорбции Н2 на Со предполагается равным 2);
A - площадь, занимаемая одним атомом кобальта (предполагается равной 0,0662 нм2).
Это уравнение описано в Operators Manual for Micrometerics ASAP 2010 Chemi System V 2.01, Appendix C, Part № 201-42808-01, October 1996.
Площадь поверхности меди удобно определять методом разложения закиси азота, как описано, например, Evans et al. в "Applied Catalysis", 7 (1983), pp. 76-83; особенно подходящая методика описана в ЕР 0202824. Метод основан на разложении молекулы закиси азота на поверхности меди, которое сопровождается высвобождением одного атома азота. В нижеследующем уравнении нижний индекс S указывает на атомы поверхности
N2O(газ)+2Cus→N2(газ)+(Cu-O-Cu)s
Восстановление образцов проводили до определения площади поверхности меди, нагревая образец со скоростью 200 K/ч в токе водорода, разбавленного аргоном (67% Н2/33% Ar по объему), до температуры 393 K (120°С), выдерживая образец при этой температуре в течение 30 мин, затем повышая температуру со скоростью 100 K/ч до желаемой температуры восстановления и поддерживая при этой желаемой температуре в течение 1 ч. После восстановления образец охлаждали до 90°С, при каковой температуре осуществляется разложение закиси азота, используя смесь закиси азота и аргона (1% N2O/99% Ar по объему). Принято, что стехиометрия адсорбции Cus/Oабс была равна 2, и площадь, занятая атомом меди, составляет 5,18 Å2 (т.е. при плотности упаковки 73% 1,46×1019 поверхности атомов меди на м2).
Катализаторы по изобретению особенно полезны при реакциях гидрирования, т.е. для гидрирования способных гидрироваться органических соединений, включающих ненасыщенность в олефиновых или ароматических соединениях и функциональные группы, включающие карбонильные соединения, нитрогруппы, нитрилы и гидрогенолизы сложных эфиров. Катализаторы могут быть особенно полезными, например, при деароматизации растворителей.
Сравнительный пример 1
250 г карбоната аммония растворяли в 1 л 33%-ного водного раствора аммиака перемешиванием в течение 3 часов. Затем в аммиачно-карбонатный раствор добавляли 350 г основного карбоната никеля порциями по 50 г, перемешивая в течение 30 минут после каждого добавления. Полученный раствор гексаммиаката никеля хранили, пока не потребуется.
Используемым носителем являлся экструдированный носитель катализатора из переходного оксида алюминия, (тета/дельта) в форме трехлопастников, имевших номинальный диаметр 1,2 мм, среднюю длину 2,9 мм, и имевший объем пор 0,67 мл/г, одномодальное распределение размера пор и площадь поверхности по БЭТ 110 м2/г. 100 г носителя помещали в химический стакан и добавляли достаточное количество раствора гексаммиаката никеля, чтобы покрыть гранулы носителя и поддерживать их покрытыми в течение времени пропитки 2 минуты. Избыток раствора затем отфильтровывали под давлением воды, и влажные гранулы сушили при 150°С во вращающейся прокалочной печи в течение 30 минут под потоком воздуха. Комплекс гексаммиаката никеля разлагался во время этой стадии сушки с выделением аммиака для получения "сырого" гидроксикарбоната никеля, диспергированного в порах носителя. Пропитку и сушку повторяли еще дважды, и затем продукт прокаливали на воздухе при 280°С в течение 45 минут, чтобы превратить гидроксикарбонат никеля в оксид никеля. Катализатор затем восстанавливали в потоке водорода при нагреве до 450°С, чтобы достичь конечной степени восстановления по меньшей мере 90%. После восстановления катализатор пассивировали в смеси азот/кислород контролируемого состава до стабильного на воздухе. Содержание никеля составляло 21%, и удельная площадь поверхности металлического Ni составляла 140 м2/г. Насыпной вес после уплотнения постукиванием составлял 0,73.
Пример 2. Приготовление катализатора согласно изобретению
Раствор гексамминкарбоната никеля готовили, как описано в сравнительном примере 1, использованным носителем были экструдированные трехлопастники из переходного оксида алюминия диаметром 1,2 мм, имевшие площадь поверхности по БЭТ 265 м2/г. Объем пор составлял 1,1 мл/г, и распределение размеров пор было бимодальным.
100 г носителя пропитывали раствором гексаммиаката никеля трехкратными пропиткой и сушкой с последующим прокаливанием, как описано в сравнительном примере 1. Полученный катализатор затем пропитывали еще три раза, причем каждая пропитка сопровождалась стадией сушки, как и до этого. После последней стадии сушки продукт прокаливали, восстанавливали и пассивировали. Было найдено, что содержание никеля составляло 39%, и удельная площадь поверхности металлического никеля составляла 120 м2/г. Насыпной вес после уплотнения постукиванием составлял 0,86. Прочность на раздавливание составляла 22 Н/мм.
Сравнительный пример 3
Катализатор готовили, как описано в сравнительном примере 1, за исключением того, что после стадии прокаливания прокаленный материал дополнительно пропитывали и сушили три раза. Материал затем прокаливали еще один раз и затем восстанавливали и пассивировали, следуя той же методике, которая описана в сравнительном примере 1. Суммарное содержание никеля составляло 30,2% и удельная площадь поверхности металлического Ni составляла 121 м2/г. Насыпной вес после уплотнения постукиванием составлял 0,89.
Испытание активности катализатора
Образец испытывали в виде целых частиц в реакторе неподвижного слоя с опускающимся потоком (внутренний диаметр 22,22 мм). Катализатор испытывали в форме целых частиц. Частицы катализатора (60 см3) смешивали с 24 г карбида кремния для образования слоя катализатора, и реактор также включал слой карбида кремния на каждом конце слоя катализатора. Катализатор активировали медленным нагревом при 1°/мин в токе водорода при 50 л/ч (давление 1,02 ати) до 120°С, поддерживая температуру постоянной в течение одного часа и затем поднимая температуру со скоростью 2°/мин до 230°С, затем поддерживая температуру в течение одного часа. Затем катализатор охлаждали до комнатной температуры, поддерживая поток водорода. Испытание проводили под давлением водорода 204 атм при температуре слоя 200°С и объемной часовой скоростью жидкого продукта 4,5 ч-1. Используемым сырьем был Varsol™ 120 от Exxon, который является лигроиновым растворителем, допированным бензотиофеном, чтобы довести содержание S до 24 ч/млн. Сырье содержало приблизительно 30% ароматических углеводородов. Использованное отношение Н2/сырье составляло 89. Испытание проводили до тех пор, пока через катализатор не пропускали 6 кг сырья. Образцы жидкого продукта отбирали через интервалы на протяжении времени реакции и анализировали многоканальной УФ-спектроскопией на содержание ароматических соединений. Конверсию (%) для каждого образца получали из следующего уравнения:
Конверсия (%)=[(AIN-AOUT)/AIN]×100,
где:
AIN - содержание ароматических соединений на входе;
AOUT - содержание ароматических соединений на выходе.
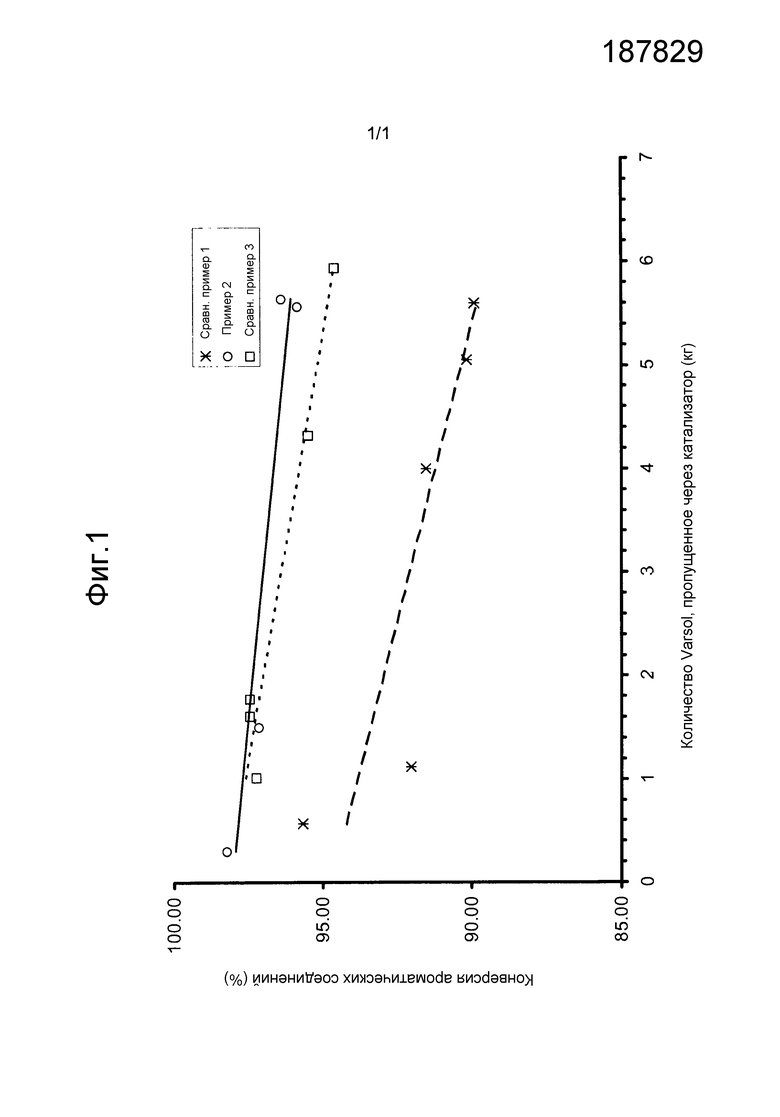
График зависимости конверсии от количества переработанного сырья был построен с использованием линейной регрессии, чтобы получить наиболее гладкую линию. Фиг.1 показывает графики результатов использования катализаторов, приготовленных в сравнительном примере 1, примере 2 и сравнительном примере 3. График показывает, что катализатор примера 2 на протяжении всего испытания имеет более высокую активность, чем катализатор сравнительного примера 1, и сохраняет высокую активность в течение более длительного периода, чем катализатор сравнительного примера 3. Катализатор сравнительного примера 3 был изготовлен с использованием того же способа, что и катализатор примера 2, но количество никеля в готовом катализаторе было значительно ниже. Без желания быть связанным теорией авторы считают, что более высокий объем пор носителя, использованного для изготовления катализатора примера 2, позволяет большему количеству никеля быть абсорбированным в пористой структуре носителя катализатора.
| название | год | авторы | номер документа |
|---|---|---|---|
| ГРАНУЛИРОВАННЫЙ КАТАЛИЗАТОР ГИДРИРОВАНИЯ НИКЕЛЬ/ПЕРЕХОДНЫЙ ОКСИД АЛЮМИНИЯ (ВАРИАНТЫ), ПРЕДШЕСТВЕННИК КАТАЛИЗАТОРА, КОНЦЕНТРАТ И СПОСОБ ПОЛУЧЕНИЯ КАТАЛИЗАТОРА | 2000 |
|
RU2235587C2 |
| СПОСОБ ПОЛУЧЕНИЯ КАТАЛИЗАТОРА ДЛЯ ОБЕССЕРИВАНИЯ ПОТОКОВ УГЛЕВОДОРОДОВ | 2006 |
|
RU2361668C2 |
| КАТАЛИЗАТОР, СПОСОБ ПОЛУЧЕНИЯ НОСИТЕЛЯ, СПОСОБ ПОЛУЧЕНИЯ КАТАЛИЗАТОРА И ПРОЦЕСС ГИДРООБЕССЕРИВАНИЯ ДИЗЕЛЬНЫХ ФРАКЦИЙ | 2006 |
|
RU2311959C1 |
| СПОСОБ ПРИГОТОВЛЕНИЯ КАТАЛИЗАТОРА | 2015 |
|
RU2710375C2 |
| КАТАЛИЗАТОР ГИДРОДЕСУЛЬФУРИЗАЦИИ ДЛЯ ДИЗЕЛЬНОГО ТОПЛИВА И СПОСОБ ГИДРООЧИСТКИ ДИЗЕЛЬНОГО ТОПЛИВА | 2014 |
|
RU2640583C2 |
| КАТАЛИЗАТОР ГИДРИРОВАНИЯ И СПОСОБ ЕГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ | 2021 |
|
RU2836692C1 |
| КАТАЛИЗАТОРЫ С ВЫСОКИМ СОДЕРЖАНИЕМ КОБАЛЬТА И ВЫСОКОЙ ПЛОЩАДЬЮ ПОВЕРХНОСТИ КОБАЛЬТА, ИХ ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ | 2005 |
|
RU2367521C2 |
| КАТАЛИЗАТОРЫ ГИДРОКОНВЕРСИИ И СПОСОБЫ ИХ ИЗГОТОВЛЕНИЯ И ПРИМЕНЕНИЯ | 2004 |
|
RU2342995C2 |
| СПОСОБ ПОЛУЧЕНИЯ КАТАЛИЗАТОРА ГИДРООБРАБОТКИ | 2006 |
|
RU2415708C2 |
| КОМПОЗИЦИЯ КАТАЛИЗАТОРА ГИДРОКРЕКИНГА | 2005 |
|
RU2387480C2 |
Изобретение относится к катализатору, способу его получения и к способам гидрирования органического соединения в присутствии указанного катализатора. Предложен дисперсный катализатор для гидрирования и дегидрирования в форме частиц, имеющих минимальный размер по меньшей части 0,8 мм, включающий переходный металл или его соединение, диспергированный на материале пористого носителя. Частицы указанного катализатора включают по меньшей мере 35 мас.% суммарного переходного металла, и площадь поверхности переходного металла в указанном катализаторе составляет по меньшей мере 110 м2/г переходного металла, и насыпной вес после уплотнения постукиванием слоя частиц катализатора составляет по меньшей мере 0,7 г/мл, в котором материал пористого носителя имеет бимодальное распределение пор по размерам, и материал пористого носителя имеет объем пор по меньшей мере 1,0 мл/г. Способ изготовления такого катализатора включает несколько стадий пропитки пористого носителя аммиачным раствором металла с последующей сушкой, прокаливанием и восстановлением высушенного материала. Катализатор используют в реакции гидрирования. 4 н. и 12 з.п. ф-лы, 1 ил., 3 пр.
1. Дисперсный катализатор для гидрирования и дегидрирования в форме частиц, имеющих минимальный размер по меньшей части 0,8 мм, включающий переходный металл или его соединение, диспергированные на материале пористого носителя, отличающийся тем, что частицы указанного катализатора включают по меньшей мере 35 мас.% суммарного переходного металла, и площадь поверхности переходного металла в указанном катализаторе составляет по меньшей мере 110 м2/г переходного металла, и насыпной вес после уплотнения постукиванием слоя частиц катализатора составляет по меньшей мере 0,7 г/мл, в котором материал пористого носителя имеет бимодальное распределение пор по размерам, и материал пористого носителя имеет объем пор по меньшей мере 1,0 мл/г.
2. Катализатор по п. 1, в котором пористый носитель включает переходный оксид алюминия.
3. Катализатор по п. 1, в котором материал пористого носителя имеет распределение пор по размерам, измеренное ртутной порометрией, в котором по меньшей мере 20% суммарного объема пор содержится в порах, имеющих диаметр от 100 нм до 700 нм, и по меньшей мере 30% суммарного объема пор содержится в порах, имеющих диаметр от 5 нм до 20 нм.
4. Катализатор по любому из пп. 1-3, в котором пористый носитель находится в форме экструдированных цилиндров или лопастных цилиндров.
5. Катализатор по любому из пп. 1-3, в котором переходный металл выбран из кобальта, никеля или меди и который может содержать более чем один переходный металл.
6. Катализатор по п. 4, в котором переходный металл выбран из кобальта, никеля или меди и который может содержать более чем один переходный металл.
7. Катализатор по п. 5, в котором переходный металл включает никель.
8. Катализатор по п. 6, в котором переходный металл включает никель.
9. Катализатор по п. 7 или 8, в котором площадь поверхности металла указанного катализатора составляет по меньшей мере 120 м2 на грамм переходного металла.
10. Способ получения катализатора для гидрирования и дегидрирования, включающего переходный металл или его соединение, диспергированные на материале пористого носителя, где указанный катализатор содержит по меньшей мере 35 мас.% суммарного переходного металла, включающий стадии:
(a) обеспечения раствором аммиачного комплекса указанного переходного металла;
(b) пропитки указанным раствором аммиачного комплекса материала пористого носителя в форме частиц, имеющих минимальный размер по меньшей мере 0,8 мм, где общий объем пор больше 1,0 мл/г, причем материал пористого носителя имеет бимодальное распределение пор по размерам;
(c) сушки пропитанных частиц носителя, полученных на стадии (b);
(d) повторения стадий (b) и (с) по меньшей мере еще четыре раза до тех пор, пока количество переходного металла в частице не достигнет требуемого уровня, и включения стадии
(e) прокаливания высушенных пропитанных частиц со стадии (с) при температуре и длительности, достаточных для превращения по меньшей мере большей части соединений переходного металла, пропитавших носитель, в оксиды переходного металла;
(f) необязательно восстановления по меньшей мере 50% оставшегося соединения переходного металла и оксида переходного металла в элементарный металл.
11. Способ по п. 10, в котором указанный раствор аммиачного комплекса является аммиачно-карбонатным раствором никеля.
12. Способ по п. 10 или 11, в котором стадии (b) и (с) проводят три раза, затем проводят стадию (е), и затем проводят стадии (b) и (с) еще три раза, и затем еще раз проводят стадию (е).
13. Способ по п. 10 или 11, в котором прокаленный катализатор с конечной стадии (е) восстанавливают в водородсодержащем газе и затем пассивируют, используя кислородсодержащий газ, так, чтобы температура катализатора не превышала 100°С, когда он подвергается воздействию воздуха.
14. Способ по п. 13, в котором прокаленный катализатор с конечной стадии (е) восстанавливают в водородсодержащем газе и затем пассивируют, используя кислородсодержащий газ, так, чтобы температура катализатора не превышала 100°С, когда он подвергается воздействию воздуха.
15. Способ осуществления гидрирования способного к гидрированию органического соединения путем контактирования указанного способного к гидрированию органического соединения с водородсодержащим газом, отличающийся тем, что контактирование осуществляют в присутствии дисперсного катализатора по любому из пп. 1-9.
16. Способ осуществления гидрирования способного к гидрированию органического соединения путем контактирования указанного способного к гидрированию органического соединения с водородсодержащим газом, отличающийся тем, что контактирование осуществляют в присутствии дисперсного катализатора, изготовленного согласно способу по любому из пп. 10-14.
| US 4920089 А1, 24.04.1990 | |||
| US 4490480 A1, 25.12.1984 | |||
| Металлический водоудерживающий щит висячей системы | 1922 |
|
SU1999A1 |
| Приспособление к токарному, револьверному и т.п. станкам для нанесения делений на линейках или прямоугольных пластинах | 1926 |
|
SU5420A1 |
| ГРАНУЛИРОВАННЫЙ КАТАЛИЗАТОР ГИДРИРОВАНИЯ НИКЕЛЬ/ПЕРЕХОДНЫЙ ОКСИД АЛЮМИНИЯ (ВАРИАНТЫ), ПРЕДШЕСТВЕННИК КАТАЛИЗАТОРА, КОНЦЕНТРАТ И СПОСОБ ПОЛУЧЕНИЯ КАТАЛИЗАТОРА | 2000 |
|
RU2235587C2 |
| US 6043187 A1, 28.03.2000. | |||

