Область техники, к которой относится изобретение
Настоящее изобретение относится к способу получения подвергнутого обработке наполнителя, подвергнутому обработке наполнителю, полученному данным способом, и композициям и изделиям, включающим такой подвергнутый обработке наполнитель.
Уровень техники
На современном уровне техники известно использование систем наполнителей диоксид кремния/силан для уменьшения сопротивления качению и улучшения силы сцепления с мокрой дорогой для покрышек легковых автомобилей. Уменьшение сопротивления качению и поэтому меньшее потребление топлива имеют значение также и для покрышек грузовых автомобилей.
Одновременное улучшение сопротивления качению, износа и силы сцепления, известное как расширение «магического треугольника», требует новых подходов к разработке каучукового композита. Осажденный диоксид кремния сыграл основную роль в появлении невулканизованной покрышки, которая отличается большим улучшением сопротивления качению в сопоставлении с тем, что имело место для технологий прошлого. Непосредственное вшивание диоксида кремния в высокосшитую полимерную матрицу при одновременном сведении к минимуму взаимодействий между частицами диоксида кремния, как представляется, имеет огромное значение для желательных динамических механических свойств. В натуральном каучуке белки, присутствующие вследствие его биосинтеза, могут преимущественно адсорбироваться на поверхности диоксида кремния, создавая помехи для реакции сочетания «по месту». Кроме того, как также было показано, увеличенные температуры выгрузки, которые могут улучшить эффективность реакции сочетания, приводят к деструкции натурального каучука.
Агентство по охране окружающей среды (АООС) США в рамках партнерского взаимодействия с Министерством транспорта (МТ) США недавно объявило о первых в истории стандартах по экономии топлива и выбросам парниковых газов для грузовых автомобилей средней и большой грузоподъемности. Преимущества от наполнения составов протекторов покрышек пассажирских автомобилей, полученных из стирол-бутадиенового каучука (SBR) и/или бутадиенового каучука (BR), аморфным осажденным диоксидом кремния были известны уже с 1976 года. К сожалению, технология диоксид кремния/силан не нашла для себя широкого применения в покрышках грузовых автомобилей.
Покрышки грузовых автомобилей обычно изготавливают из натурального каучука, который представляет собой биосинтетический латекс, производимый каучуконосным деревом Hevia Brasiliensis. Твердый натуральный каучук, получаемый в результате коагулирования, обычно содержит некоторые некаучуковые компоненты. Данные компоненты могут состоять из белков, липидов, Сахаров, золы и других примесей. Как предполагалось, традиционные способы переработки каучука, в которых агент реакции сочетания и наполнитель добавляют в смеситель независимо, в результате приводят к преимущественному адсорбированию белков на поверхности диоксида кремния, что, тем самым, своим результатом имеет неполноту реакции сочетания для силановых фрагментов. Белки, как давно было известно, адсорбируются на диоксиде кремния в результате действия сильных межмолекулярных взаимодействий. Кроме того, согласно теории неполнота реакции сочетания для функциональности диоксида кремния по отношению к концевым группам полимерных цепей в натуральном каучуке имеет место вследствие отсутствия в натуральном каучуке необходимого количества высокореакционноспособных боковых 1,2-винильных групп (которые присутствуют с переменными количествами в синтетических полимерах, использующихся в покрышках легковых автомобилей).
Кроме того, повышенные температуры перемешивания (в некоторых случаях доходящие вплоть до 150°C) для обеспечения оптимальной реакции сочетания «по месту» между силаном и диоксидом кремния, как прежде было показано, приводят к деструкции натурального каучука.
В соответствии с этим, для устранения вышеупомянутых недостатков желательны дополнительные усовершенствования технологии диоксид кремния/силан.
Раскрытие изобретения
Настоящее изобретение относится к способу получения подвергнутого обработке наполнителя, включающему:
(a) обработку суспензии, включающей неподвергнутый обработке наполнитель, где неподвергнутый обработке наполнитель прежде не высушивали, при использовании композиции для обработки, содержащей органосилан, описывающийся общей формулой (I)
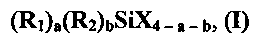
где в общей формуле (I) R1 независимо представляет собой органофункциональный углеводород, содержащий от 1 до 36 атомов углерода, где функциональная группа органофункционального углеводорода представляет собой винил, аллил, гексенил, эпокси, глицидокси, (мет)акрилокси, сульфид, изоцианато, полисульфид, меркапто или галоген; каждый R2 независимо представляет собой углеводородную группу, содержащую от 1 до 36 атомов углерода, или водород, каждый X независимо представляет собой галоген или алкокси, содержащий от 1 до 36 атомов углерода; а составляет 0, 1, 2 или 3; b составляет 0, 1 или 2; (a+b) составляет 1, 2 или 3; при том условии, что в случае, когда b=1, (a+b) составит 2 или 3, для получения суспензии подвергнутого обработке наполнителя и
(b) высушивание упомянутой суспензии подвергнутого обработке наполнителя для получения подвергнутого обработке наполнителя.
Настоящее изобретение также предлагает способ получения подвергнутого обработке наполнителя, включающий:
(a) объединение силиката щелочного металла и кислоты для получения суспензии, включающей неподвергнутый обработке наполнитель, где упомянутый неподвергнутый обработке наполнитель прежде не высушивали;
(b) обработку упомянутой суспензии при использовании композиции для обработки, содержащей описанный выше органосилан, описьшающийся общей формулой (I), и
(c) высушивание упомянутой суспензии подвергнутого обработке наполнителя для получения подвергнутого обработке наполнителя.
Настоящее изобретение также относится к подвергнутому обработке наполнителю, полученному по данному способу, а также композициям для составления каучуковых смесей, включающим подвергнутый обработке наполнитель, и протектору покрышки, включающему подвергнутый обработке наполнитель настоящего изобретения.
Осуществление изобретения
Как упоминалось прежде, настоящее изобретение предлагает способ получения подвергнутого обработке наполнителя. Способ включает (a) обработку суспензии, включающей неподвергнутый обработке наполнитель, где упомянутый неподвергнутый обработке наполнитель прежде не высушивали, и высушивание суспензии подвергнутого обработке наполнителя для получения подвергнутого обработке наполнителя. Суспензию неподвергнутого обработке наполнителя (a) подвергают обработке при использовании композиции для обработки, содержащей органосилан, описьшающийся общей формулой (I)
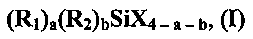
где в общей формуле (I) R1 независимо представляет собой органофункциональный углеводород, содержащий от 1 до 36 атомов углерода, где функциональная группа органофункционального углеводорода представляет собой винил, аллил, гексенил, эпокси, глицидокси, (мет)акрилокси, сульфид, изоцианато, полисульфид, меркапто или, что является предпочтительным, галоген; каждый R2 независимо представляет собой углеводородную группу, содержащую от 1 до 36 атомов углерода, или водород, каждый X независимо представляет собой галоген или, что является предпочтительным, алкокси, содержащий от 1 до 36 атомов углерода; а составляет 0, 1, 2 или 3; b составляет 0, 1 или 2; (a+b) составляет 1, 2 или 3; при том условии, что в случае, когда b=1, (a+b) составит 2 или 3. В одном конкретном варианте осуществления настоящего изобретения композиция для обработки содержит органосилан, включающий галогенфункциональный органосилан, где X представляет собой алкокси; а составляет 1; b составляет 0; и функциональная группа органофункционального углеводорода R1 представляет собой галоген.
В общем случае в способе настоящего изобретения силикат щелочного металла объединяют с водным раствором кислоты для получения суспензии неподвергнутого обработке наполнителя; после этого суспензию неподвергнутого обработке наполнителя подвергают обработке при использовании композиции для обработки, содержащей описанный выше органосилан, описывающийся формулой (I), и необязательно материал для обработки, описанный в настоящем документе ниже, для получения суспензии подвергнутого обработке наполнителя; и после этого суспензия подвергнутого обработке наполнителя может быть высушена при использовании обычных методик высушивания, известных на современном уровне техники, для получения подвергнутого обработке наполнителя настоящего изобретения.
В соответствии с использованием в настоящем документе, в описании изобретения и формуле изобретения, при обращении к наполнителю (то есть подвергнутому обработке и/или неподвергнутому обработке) термин «прежде не высушивали» обозначает наполнитель, который перед реализацией способа обработки не высушивали до уровня содержания влаги, меньшего, чем 20 массовых процентов. Для целей настоящего изобретения неподвергнутый обработке наполнитель не включает наполнитель, который прежде высушивали до уровня содержания влаги, меньшего, чем 20 массовых процентов, а после этого повторно гидратировали.
В соответствии с использованием в настоящем документе и формуле изобретения термин «наполнитель» обозначает неорганический оксид, который может быть использован в полимерной композиции, по существу, для улучшения по меньшей мере одного свойства упомянутого полимера. В соответствии с использованием в настоящем документе и формуле изобретения термин «суспензия» обозначает смесь, включающую по меньшей мере наполнитель и воду.
Неподвергнутые обработке наполнители, подходящие для использования в способе настоящего изобретения, могут включать широкий спектр материалов, известных специалисту в соответствующей области техники. Неограничивающие примеры могут включать неорганические оксиды, такие как неорганические дисперсные и аморфные твердые материалы, которые включают на обнаженной поверхности либо кислород (хемисорбированный или ковалентно связанный), либо гидроксил (связанный или свободный), такие как нижеследующие, но не ограничиваясь только этим: оксиды металлов в периодах 2, 3, 4, 5 и 6 из групп Ib, IIb, IIIa, IVa, IVb (за исключением углерода), Va, VIa, VIIa и VIII Периодической таблицы элементов из публикации Advanced Inorganic Chemistry: Ф Comprehensive Text by F. Albert Cotton et al., Fourth Edition, John Wiley and Sons, 1980. Неограничивающие примеры подходящих для использования неорганических оксидов могут включать нижеследующее, но не ограничиваются только этим: силикаты алюминия, диоксид кремния, такой как силикагель, коллоидальный диоксид кремния, осажденный диоксид кремния и их смеси.
Неорганическим оксидом могут быть диоксид кремния, например осажденный диоксид кремния, коллоидальный диоксид кремния и их смеси. Диоксид кремния характеризуется средним размером первичных частиц, меньшим, чем 0,1 микрона или большим, чем 0,001 микрона или находящимся в диапазоне от 0,01 до 0,05 микрона или от 0,015 до 0,02 микрона, согласно измерению при использовании электронного микроскопа. Кроме того, диоксид кремния может характеризоваться площадью поверхности в диапазоне от 25 до 1000 квадратных метров в расчете на один грамм или от 75 до 250 квадратных метров в расчете на один грамм или от 100 до 200 квадратных метров в расчете на один грамм согласно определению по методу Брунауэра, Эмметта и Теллера (БЭТ) в соответствии с документом ASTM D1993-91.
Как упоминалось прежде, суспензию неподвергнутого обработке наполнителя подвергают обработке при использовании композиции для обработки, содержащей подробно описанный выше органосилан, описывающийся общей формулой (I). В особенности подходящим для использования органосиланом, обладающим такой структурой, является галогенфункциональный органосилан, где X представляет собой алкокси, а составляет 1, b составляет 0, а функциональная группа органофункционального углеводорода R1 представляет собой галоген. Например, композиция для обработки может содержать галогенфункциональный органосилан, выбираемый из (4-хлорметилфенил)триметоксисилана, (4-хлорметилфенил)триэтоксисилана, [2-(4-хлорметилфенил)этил]триметоксисилана, [2-(4-хлорметилфенил)этил]триэтоксисилана, (3-хлорпропенил)триметоксисилана, (3-хлорпропенил)триэтоксисилана, (3-хлорпропил)триэтоксисилана, (3-хлорпропил)триметоксисилана, триметокси(2-п-толилэтил)силана и/или триэтокси(2-п-толилэтил)силана.
В способе настоящего изобретения могут быть использованы и смеси любых из вышеупомянутых органосиланов.
Органосилан, описывающийся общей формулой (I), может присутствовать в суспензии в количестве в диапазоне от 0,25 до 30,0 массового процента, таком как от 1 до 15 массовых процентов или от 5 до 10 массовых процентов, в расчете на совокупную массу реагента SiO2, который был осажден.
В дополнение к вышеупомянутому органосилану композиция для обработки может, кроме того, содержать материал для обработки (то есть материал, не подвергающийся реакции сочетания), выбираемый из анионных, неионных и амфотерных поверхностно-активных веществ и их смесей, где материал для обработки присутствует в количестве в диапазоне от более чем 1% вплоть до и с включением 25% (мас.) в расчете на совокупную массу реагента SiO2, который был осажден. Например, материал для обработки может быть выбран из солей жирных кислот, алкилсаркозинатов, солей алкилсаркозинатов и их смесей. Конкретные неограничивающие примеры таких материалов для обработки могут быть найдены в публикации U.S. 7569107, во фрагменте от столбца 5, строки 9 до столбца 7, строки 21, процитированные части которой посредством ссылки включаются в настоящий документ. В одном конкретном варианте осуществления настоящего изобретения материал для обработки содержит один или несколько анионных поверхностно-активных веществ, выбираемых из группы, состоящей из стеарата натрия, стеарата аммония, кокоата аммония, лаурината натрия, кокоилсаркозината натрия, лауроилсаркозината натрия, натриевого мыла на основе твердого животного жира, натриевого мыла на основе кокосового масла, миристоилсаркозината натрия и стеароилсаркозиновой кислоты.
Материал для обработки обычно присутствует в количестве в диапазоне от более чем 1% вплоть до и с включением 25% (мас), например, от 2,0 до 20,0% или от 4 до 15%) или от 5 до 12% (мас.) в расчете на совокупную массу реагента SiO2, который был осажден.
Композиция для обработки, подходящая для использования в способе настоящего изобретения, также может, кроме того, содержать серосодержащий органосилан, отличный от вышеупомянутого органосилана, использующегося при обработке суспензии стадии (a). Неограничивающие примеры таких материалов могут включать нижеследующее, но не ограничиваются только этим: органосиланы, описывающиеся общей формулой:
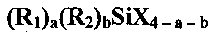
где каждый R1 независимо может представлять собой органофункциональный углеводородный радикал, содержащий от 1 до 12 атомов углерода, где органофункциональная группа представляет собой сульфид, полисульфид или меркапто; каждый R2 независимо может представлять собой углеводородную группу, содержащую от 1 до 18 атомов углерода, или водород, каждый X независимо может представлять собой галоген или радикал алкокси, содержащий от 1 до 12 атомов углерода, а может составлять 0, 1, 2 или 3, b может составлять 0, 1 или 2, и a+b может составлять 1, 2 или 3, при том условии, что в случае, когда b=1, a+b составит 2 или 3. Группы R1 и R2 могут быть выбраны таким образом, чтобы они могли бы вступать в реакцию с полимерной композицией, в которой может быть использован подвергнутый обработке наполнитель. В дополнение к этому, серосодержащий органосилан может включать бис(алкоксисилилалкил)полисульфиды, описывающиеся следующей далее структурной формулой
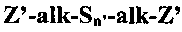 ,
,
в которой «alk» представляет собой двухвалентный углеводородный радикал, содержащий от 1 до 18 атомов углерода; n′ представляет собой целое число в диапазоне от 2 до 12; и Z′ представляет собой:
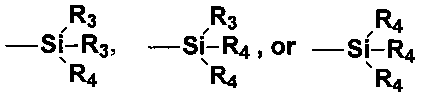
где R3 представляет собой алкильную группу, содержащую от 1 до 4 атомов углерода, или фенил, а R4 представляет собой группу алкокси, содержащую от 1 до 8 атомов углерода, группу циклоалкокси, содержащую от 5 до 8 атомов углерода, или с прямой или разветвленной цепью группу алкилмеркапто, содержащую от 1 до 8 атомов углерода. Группы R3 и R4 могут быть идентичными или различными. Кроме того, двухвалентная группа alk может быть с прямой или разветвленной цепью, насыщенной или ненасыщенной алифатической углеводородной группой или циклической углеводородной группой. Неограничивающие примеры бис(алкоксисилилалкил)полисульфидов могут включать бис(2-триалкоксисилилэтил)полисульфиды, в которых группа триалкокси может представлять собой триметокси, триэтокси, три(метилэтокси), трипропокси, трибутокси и тому подобное вплоть до триоктилокси, а полисульфид может представлять собой либо ди-, либо три-, либо тетра-, либо пента-, либо гексасульфид, либо их смеси. Кроме того, неограничивающие примеры могут включать соответствующие бис(3-триалкоксисилилпропил)-, бис(3-триалкоксисилилизобутил)-, бис(4-триалкоксисилилбутил)- и тому подобное вплоть до бис(6-триалкоксисилилгексил)полисульфидов. Кроме того, неограничивающие примеры бис(алкоксисилилалкил)полисульфидов описываются в патенте США №3873489, в столбце 6, в строках 5-55 и в патенте США №5580919, в столбце 11, в строках 11-41. Кроме того, неограничивающие примеры таких соединений могут включать: 3,3′-бис(триметоксисилилпропил) дисульфид, 3,3′-бис(триэтоксисилилпропил)тетрасульфид, 3,3′-бис(триметоксисилилпропил)тетрасульфид, 2,2′-бис(триэтоксисилилэтил)тетрасульфид, 3,3′-бис(триметоксисилилпропил)трисульфид, 3,3′-бис(триэтоксисилилпропил)трисульфид, 3,3′-бис(трибутоксисилилпропил)дисульфид, 3,3′-бис(триметоксисилилпропил)гексасульфид и 3,3′-бис(триоктоксисилилпропил)тетрасульфид и их смеси.
Серосодержащий органосилан также может представлять собой меркаптометаллоорганическое соединение, описывающееся следующей далее структурной формулой
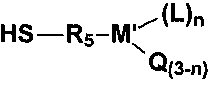
где M′ представляет собой кремний, L представляет собой галоген или -OR6, Q представляет собой водород, C1-C12 алкил или галогензамещенный C1-C12 алкил, R5 представляет собой C1-C12 алкилен, R6 представляет собой C1-C12 алкил или алкоксиалкил, содержащий от 2 до 12 атомов углерода, при этом упомянутые галоген или (галогеновые) группы представляют собой хлор, бром, йод или фтор, а n составляет 1, 2 или 3. В одном неограничивающем варианте осуществления могут быть использованы меркаптометаллоорганические реагенты, содержащие две группы меркапто.
Неограничивающие примеры подходящего меркаптометаллоорганического соединения (соединений) могут включать нижеследующее, но не ограничиваются только этим: меркаптометилтриметоксисилан, меркаптоэтилтриметоксисилан, меркаптопропилтриметоксисилан, меркаптометилтриэтоксисилан, меркаптоэтилтрипропоксисилан, меркаптопропилтриэтоксисилан, (меркаптометил)диметилэтоксисилан, (меркаптометил)метилдиэтоксисилан, 3-меркаптопропилметилдиметоксисилан и их смеси.
В одном конкретном варианте осуществления настоящего изобретения серосодержащий органосилан может представлять собой меркаптометаллоорганическое соединение, такое как меркаптосилан, отличный от органосилана, использующегося в композиции для обработки стадии (a), например, меркаптопропилтриметоксисилан и/или меркаптометилтриэтоксисилан.
Кроме того, предполагается то, что сераорганический органосилан, отличный от вышеупомянутого органосилана, использующегося на стадии (a) способа настоящего изобретения, может представлять собой меркаптометаллоорганическое соединение, в котором группа меркапто является блокированной, то есть, атом водорода меркапто замещен другой группой. Блокированные меркаптометаллоорганические соединения могут содержать ненасыщенный гетероатом или атом углерода, связанный непосредственно с атомом серы через одинарную связь. Неограничивающие примеры конкретных блокирующих групп могут включать тиокарбоксилатный сложный эфир, дитиокарбаматный сложный эфир, тиосульфонатный сложный эфир, тиосульфатный сложный эфир, тиофосфатный сложный эфир, тиофосфонатный сложный эфир, тиофосфинатный сложный эфир и тому подобное. В одном неограничивающем варианте осуществления, где в качестве материала, подвергающегося реакции сочетания, используют блокированное меркпатометаллоорганическое соединение, к смеси полимерных соединений может быть добавлен деблокирующий агент для деблокирования блокированного меркаптометаллоорганического соединения. В одном неограничивающем варианте осуществления, в котором в смеси присутствуют вода и/или спирт, для инициирования и промотирования потери блокирующей группы в результате гидролиза или алкоголиза в целях высвобождения соответствующих меркаптометаллоорганических соединений может быть использован катализатор, например, третичные амины, кислоты Льюиса или тиолы. Неограничивающие примеры блокированных меркаптосиланов могут включать нижеследующее, но не ограничиваются только этим: 2-триэтоксисилил-1-этилтиоацетат, 3-триметоксисилил-1-пропилтиооктаноат, бис(3-триэтоксисилил-1-пропил)метилдитиофосфонат, 3-триэтоксисилил-1-пропилдиметилтиофосфинат, 3-триэтоксисилил-1-пропилметилтиосульфат, 3-триэтоксисилил-1-пропилтолуолтиосульфонат и их смеси.
Количество данных необязательных серосодержащих органосиланов может варьироваться в широких пределах и может зависеть от конкретного выбранного материала. Например, количество данных необязательных серосодержащих органосиланов может быть большим, чем 0,1% в расчете на массу неподвергнутого обработке наполнителя, таким как величина в диапазоне от 0,5 до 25% в расчете на массу неподвергнутого обработке наполнителя или от 1 до 20% или от 2 до 15%.
В одном дополнительном варианте осуществления настоящего изобретения композиция для обработки также может, кроме того, содержать галогенфункциональный органосилан, который включает мономерное, димерное, олигомерное и/или полимерное соединение, содержащее галогеновую функциональность и алкандиоксисилильную функциональность, произведенную из (i) полигидроксилсодержащих соединений, в которых группа алкандиокси ковалентно связывается с одним атомом Si через связи Si-O с образованием кольца, и/или (ii) группы алкандиокси ковалентно связываются по меньшей мере с двумя атомами Si через связи Si-O с образованием димера, олигомера или полимера, в которых соседние силильные звенья связываются друг с другом через мостиковые структуры алканалкокси. Такие галогенфункциональные органосиланы подробно описываются в опубликованной патентной заявке Соединенных Штатов №2011/0003922 A1, опубликованной 6 января 2011 года, в абзацах от [0020] до [0057], процитированные части которой посредством ссылки включаются в настоящий документ.
Кроме того, композиция для обработки, подходящая для использования в способе настоящего изобретения, также может включать «органосиланы, не подвергающиеся реакции сочетания». В соответствии с использованием в настоящем документе термин «органосилан, не подвергающийся реакции сочетания» обозначает органосилан, который используется для улучшения совместимости подвергнутого обработке наполнителя настоящего изобретения с полимерной композицией, в которой, в конечном счете, используют подвергнутый обработке наполнитель. То есть органосилан, не подвергающийся реакции сочетания, может оказывать воздействие на свободную энергию поверхности частиц подвергнутого обработке наполнителя, приводя к получению частиц подвергнутого обработке наполнителя, имеющих энергию поверхности, подобную той, что и у полимерной композиции. Это облегчает включение подвергнутого обработке наполнителя в полимерную композицию и может использоваться для улучшения (то есть уменьшения) вязкости смеси для композиции. Необходимо отметить то, что силаны, не подвергающиеся реакции сочетания, предположительно не вступают в реакцию сочетания с каучуковой матрицей сверх того, что имеет место для ван-дер-ваальсовых взаимодействий. Некоторые неограничивающие примеры таких силанов, не подвергающихся реакции сочетания, включают октадецилтриэтоксисилан, октадецилтрихлорсилан, октадецилтриметоксисилан, пропилтриэтоксисилан, пропилтриметоксисилан, пропилтрихлорсилан, н-октилтриметоксисилан, н-октилтриэтоксисилан, н-октилтрихлорсилан, н-гексилтриметоксисилан, н-гексилтриэтоксисилан и/или н-гексилтрихлорсилан.
Необходимо понять то, что для целей настоящего изобретения любые из вышеупомянутых органосиланов, включая органосилан, обладающий описанной выше структурой (I), включают их неполные гидролизаты.
Подвергнутый обработке наполнитель настоящего изобретения может быть получен при использовании любых из широкого спектра способов, известных специалистам в соответствующей области техники. Например, в случае подвергнутого обработке наполнителя в виде подвергнутого обработке диоксида кремния подвергнутый обработке наполнитель будут получать в результате объединения водного раствора растворимого силиката металла с раствором кислоты для получения суспензии диоксида кремния; суспензия диоксида кремния необязательно может быть подвергнута старению; к необязательно подвергнутой старению суспензии диоксида кремния могут быть добавлены кислота или основание для регулирования значения pH суспензии; суспензия диоксида кремния может быть отфильтрована, необязательно промыта, а после этого высушена при использовании обычных методик, известных специалистам в соответствующей области техники. Композиция для обработки может быть добавлена на любой стадии в описанном выше способе перед высушиванием.
Дополнительное подробное описание способа получения подвергнутого обработке диоксида кремния может быть найдено ниже в настоящем документе в примерах.
Подходящие для использования силикаты металлов могут включать широкий спектр материалов, известных на современном уровне техники. Неограничивающие примеры могут включать нижеследующее, но не ограничиваются только этим: оксид алюминия, силикат лития, натрия, калия и их смеси. Кроме того, силикат металла может быть представлен следующей далее структурной формулой M2O(SiO2)x, где M может представлять собой оксид алюминия, литий, натрий или калий, а x может представлять собой целое число в диапазоне от 0,1 до 4.
Подходящие для использования кислоты могут быть выбраны из широкого спектра кислот, известных на современном уровне техники. Неограничивающие примеры могут включать нижеследующее, но не ограничиваются только этим: минеральные кислоты, органические кислоты, диоксид углерода и их смеси. Предпочтительной является серная кислота.
Как упоминалось выше, подвергнутые обработке наполнители, которые получают по способу настоящего изобретения, являются в особенности хорошо подходящими для использования при включении в органические полимерные композиции, например, любую органическую полимерную композицию, в которую в выгодном случае может быть включен подвергнутый обработке наполнитель. Материалы подвергнутых обработке наполнителей, полученные по способу настоящего изобретения, являются в особенности хорошо подходящими для использования в композициях для составления каучуковых смесей и, в частности, в каучуковых композициях, использующихся при изготовлении покрышек и компонентов покрышек, таких как протекторы покрышек.
Такие полимеры описываются в публикации Kirk Othmer Encyclopedia of Chemical Technology, Fourth Edition, 1996, Volume 19, pp. 881-904, описание которой посредством ссылки включается в настоящий документ. Подвергнутый обработке наполнитель настоящего изобретения может быть примешан к полимеру или его полимеризуемым компонентам тогда, когда физическая форма полимера или полимеризуемых компонентов соответствует любой жидкости или позволяющей составлять смеси форме, такой как раствор, суспензия, латекс, дисперсия и тому подобное. Полимерные композиции, включающие подвергнутый обработке наполнитель настоящего изобретения, могут быть пропущены через вальцы, перемешаны, сформованы и отверждены по любому способу, известному на современном уровне техники, для изготовления полимерного изделия. Подходящие для использования полимеры могут включать нижеследующее, но не ограничиваются только этим: термопластические и термоотверждающиеся смолы, каучуковые составы и другие полимеры, обладающие эластомерными свойствами.
Вышеупомянутые полимеры могут включать, например, алкидные смолы, алкидные смолы, модифицированные маслом, ненасыщенные сложные полиэфиры, натуральные масла (например, льняное, тунговое, соевое), эпоксиды, найлоны, термопластический сложный полиэфир (например, полиэтилентерефталат, полибутилентерефталат), поликарбонаты, то есть термопластические и термоотверждающиеся, полиэтилены, полибутилены, полистиролы, полипропилены, этилен-пропиленовые со- и терполимеры, акриловые полимеры (гомополимер и сополимеры акриловой кислоты, акрилатов, метакрилатов, акриламидов, их солей, гидрогалогенидов и тому подобного), фенольные смолы, полиоксиметилен (гомополимеры и сополимеры), полиуретаны, полисульфоны, полисульфидные каучуки, нитроцеллюлозы, винилбутираты, виниловые полимеры (полимеры, включающие винилхлорид и/или винилацетат), этилцеллюлозу, ацетаты и бутираты целлюлозы, вискозное волокно, шеллак, воска, этиленовые сополимеры (например, сополимеры этилена-винилацетата, сополимеры этилена-акриловой кислоты, этиленакрилатные сополимеры), органические каучуки (как синтетические, так и природные каучуки) и тому подробное.
Количество подвергнутого обработке наполнителя, который может быть использован в полимерной композиции, может варьироваться в широких пределах в зависимости от полимерной композиции и желательных свойств изделия, изготавливаемого из полимерной композиции. Например, количество подвергнутого обработке наполнителя, присутствующего в полимерной композиции, может находиться в диапазоне от 5 вплоть до 70% (мас.) в расчете на совокупную массу полимерной композиции.
В одном неограничивающем варианте осуществления полимерная композиция может содержать органический каучук. Неограничивающие примеры таких каучуков могут включать нижеследующее, но не ограничиваются только этим: натуральный каучук; каучуки, которые получают в результате гомополимеризации бутадиена и его гомологов и производных, такие как: цис-1,4-полиизопрен; 3,4-полиизопрен; цис-1,4-полибутадиен; транс-1,4-полибутадиен; 1,2-полибутадиен; и каучуки, которые получают в результате сополимеризации бутадиена и его гомологов и производных совместно с одним или несколькими сополимеризуемыми мономерами, содержащими этиленовую ненасыщенность, такими как стирол и его производные, винилпиридин и его производные, акрилонитрил, изобутилен и алкилзамещенные акрилаты, такие как метилметакрилат. Кроме того, неограничивающие примеры могут включать стирол-бутадиеновый сополимерный каучук, образованный при различных уровнях процентного содержания стирола и бутадиена и использующий различные изомеры бутадиена в соответствии с пожеланиями (ниже в настоящем документе «SBR»); терполимеры стирольных, изопреновых и бутадиеновых полимеров и их различных изомеров; композиции сополимерного и терполимерного каучуков на акрилонитрильной основе; и композиции каучуков на изобутиленовой основе; или их смесь в соответствии с описанием, например, в патентах Соединенных Штатов №№4530959; 4616065; 4748199; 4866131; 4894420; 4925894; 5082901; и 5162409.
Неограничивающие примеры подходящих для использования органических полимеров могут включать сополимеры этилена с другими высшими альфа-олефинами, такими как пропилен, бутен-1 и пентен-1, и диеновым мономером. Органические полимеры могут быть блочными, статистическими или последовательными и могут быть получены по способам, известным на современном уровне техники, таким как нижеследующее, но не ограничивающимся только этим: способы эмульсионной (например, e-SBR) или растворной (например, s-SBR) полимеризации. Кроме того, неограничивающие примеры полимеров, предназначенных для использования в настоящем изобретении, могут включать полимеры, которые являются частично или полностью функционализованными, включая подвергнутые реакции сочетания или звездообразно-разветвленные полимеры. Дополнительные неограничивающие примеры функционализованных органических каучуков могут включать полихлоропрен, хлорбутил- и бромбутилкаучук, а также бромированный сополимерный каучук изобутилена-пара-метилстирола. В одном неограничивающем варианте осуществления органический каучук может представлять собой полибутадиен, полимер 5-SBR и их смеси.
Полимерная композиция может представлять собой отверждаемый каучук. Термин «отверждаемый каучук» предназначается для включения натурального каучука и его различных исходных и регенерированных форм, а также различных синтетических каучуков. В альтернативных неограничивающих вариантах осуществления отверждаемый каучук может включать комбинации из каучука SBR и бутадиенового каучука (BR), каучуки SBR, BR и натуральный каучук и любые другие комбинации из материалов, прежде описанных в качестве органических каучуков. При описании данного изобретения термины «каучук», «эластомер» и «эластомерный каучук» могут быть использованы взаимозаменяющим образом, если только не будет указано другого. Термины «каучуковая композиция», «составленная каучуковая смесь» и «каучуковый состав» используются взаимозаменяющим образом для обозначения каучука, который был смешан или перемешан с различными ингредиентами и материалами, и такие термины хорошо известны специалистам в области смешивания каучуков или составления каучуковых смесей.
Каучуковые композиции, содержащие подвергнутый обработке наполнитель, полученный по способу настоящего изобретения, могут быть использованы при изготовлении мириада каучуковых изделий, например, покрышки по меньшей мере один компонент которой, например, протектор, содержит отвержденную каучуковую композицию, а также других каучуковых изделий, таких как обувные подошвы, шланги, уплотнения, кабельные оболочки, прокладки, ремни и тому подобное. Каучуковые композиции, включающие подвергнутый обработке наполнитель, полученный по способу настоящего изобретения, являются в особенности выгодными для использования при изготовлении протекторов покрышек, характеризующихся низким сопротивлением качению и высоким сопротивлением износу, в особенности в случае протекторов покрышек на основе натурального каучука. Кроме того, согласно наблюдениям для таких композиций натурального каучука, включающих подвергнутый обработке наполнитель, полученный по способу настоящего изобретения, могут быть достигнуты меньшие температуры отверждения.
Настоящее изобретение более конкретно описывается в следующих далее примерах, которые предназначаются только для исполнения функции иллюстрации, поскольку специалистам в соответствующей области техники будут очевидными и многочисленные модификации и вариации настоящего изобретения. Если только не будет указано другого, то все части и все уровни процентного содержания будут массовыми.
Примеры
В части 1 описывается аналитическое испытание, проведенное для определения физических свойств образцов диоксида кремния. В части 2 описывается получение примера 1 и сравнительных примеров 1 и 2 и их характеристические свойства, приведенные в таблице 1. В части 3 описывается получение образцов каучуков и сравнительного примера 3 согласно представлению в таблицах 2 и 3. В части 4 описываются испытание и физические свойства образцов каучуков согласно представлению в таблицах 4 и 5.
Часть 1 - Аналитическое испытание
В следующем далее методе для определения площади поверхности используют раствор бромида цетилтриметиламмония (БЦТА) для анализирования площади внешней удельной поверхности подвергнутого обработке наполнителя, соответствующего данному изобретению. Анализ проводили при использовании автотитратора Metrohm 751 Titrino, снабженного бюреткой на 50 миллилитров Metrohm Interchangeable «Snap-In» и устройством Brinkmann Probe Colorimeter Model PC 910, снабженным фильтром в области 550 нм. В дополнение к этому прибор Mettler Toledo HB43 использовали для определения потери влаги наполнителем, а устройство Fisher Scientific CentrificTM Centrifuge Model 225 - для разделения наполнителя и остаточного раствора реагента БЦТА. Избыток реагента БЦТА определяли в результате проведения автоматического титрования при использовании раствора поверхностно-активного вещества Aerosol® ОТ, согласно сообщениям являющегося сульфосукцинатом натрия, вплоть до достижения максимальной мутности, что детектировали при использовании зондового колориметра. За точку максимальной мутности принимали точку, соответствующую милливольтовому показанию 150. Знание количества реагента БЦТА, адсорбированного для заданной массы наполнителя, и пространства, занимаемого молекулой реагента БЦТА, сделало возможным вычисление площади внешней удельной поверхности подвергнутого обработке наполнителя, что приводили при выражении в квадратных метрах в расчете на один грамм в пересчете на сухую массу в таблице 2.
Растворы, требуемые для испытания и получения, включали буфер при pH 9,6, раствор реагента БЦТА, поверхностно-активное вещество Aerosol ОТ и гидроксид натрия при концентрации 1 н. Буферный раствор при pH 9,6 получали в результате растворения 3,101 г ортоборной кислоты (99%; Fisher Scientific, Inc., технический сорт, кристаллическая фаза) в однолитровой мерной колбе, содержащей 500 миллилитров деионизированной воды и 3,708 г твердого вещества хлорида калия (Fisher Scientific, Inc., технический сорт, кристаллическая фаза). При использовании бюретки добавляли 36,85 миллилитра раствора гидроксида натрия при концентрации 1 н. Раствор перемешивали, разбавляли до заданного объема. Раствор реагента БЦТА получали при использовании 11,0 г порошкообразного реагента БЦТА (Fisher Scientific, Inc., технический сорт) на весовой кювете. Порошкообразный реагент БЦТА переводили в 2-литровый химический стакан, промывая весовую кювету при использовании деионизированной воды. В 2-литровый химический стакан добавляли приблизительно 700 миллилитров раствора буфера при pH 9,6 и 1000 миллилитров дистиллированной или деионизированной воды и производили перемешивание при использовании якоря магнитной мешалки. На химическом стакане размещали большое предметное стекло и химический стакан перемешивали при комнатной температуре вплоть до полного растворения реагента БЦТА. Раствор переводили в 2-литровую мерную колбу, промывая химический стакан и якорь магнитной мешалки при использовании деионизированной воды. Пузырькам позволяли рассеяться и проводили разбавление до заданного объема при использовании деионизированной воды. Добавляли большой якорь магнитной мешалки и проводили перемешивание на магнитной мешалке в течение приблизительно 10 часов. Раствор (Aerosol ОТ® surfactant Fisher Scientific Inc., 100% твердого вещества) получали при использовании 3,46 г на весовой кювете. Реагент Aerosol® ОТ вымывали в 2-литровый химический стакан, который содержал приблизительно 1500 миллилитров деионизированной воды и большой якорь магнитной мешалки. Раствор реагента Aerosol® ОТ растворяли и вымывали в 2-литровую мерную колбу. Раствор разбавляли до метки объема 2 литра в мерной колбе. Раствору реагента Aerosol® ОТ давали возможность стареть в течение, как минимум, 12 дней перед использованием и использовали до истечения срока годности в 2 месяца от даты получения.
Перед получением образца для определения площади поверхности значение pH раствора реагента БЦТА проверяли и регулировали, доводя до pH 9,6±0,1 при использовании раствора гидроксида натрия при концентрации 1 н. Для вычислений при испытаниях получали и анализировали холостой образец. Пипеткой отбирали 5 миллилитров раствора БЦТА и в 150-миллилитровый химический стакан добавляли 55 миллилитров деионизированной воды и проводили анализ при использовании автотитратора Metrohm 751 TITRINO®. Автотитратор запрограммировали на определение для холостого образца и образцов при следующих далее параметрах: плотность точек измерения = 2, дрейф сигнала = 20, время равновесия = 20 секунд, объем начала = 0 мл, объем завершения = 35 мл и фиксированная конечная точка = 150 мВ. Кончик бюретки и зонд колориметра размещали непосредственно под поверхностью раствора, располагая таким образом, чтобы кончик и длина траектории измерительного фотозонда были бы полностью погружены. Как кончик, так и фотозонд были, по существу, эквидистантны от дна химического стакана и не соприкасались друг с другом. При минимальном перемешивании (установка 1 на перемешивающем устройстве Metrohm 728) колориметр выставляли на 100% T перед каждым определением для холостого образца и других образцов и при использовании раствора реагента Aerosol®OT начинали титрование. Конечную точку регистрировали в виде объема (мл) титрирующего раствора при 150 мВ.
Для получения образца для испытания приблизительно 0,30 грамма порошкообразного наполнителя отвешивали в 50-миллилитровый контейнер с якорем магнитной мешалки. Раствор реагента БЦТА, отрегулированный по значению pH, (30 миллилитров) пипеткой отбирали в контейнер для образцов с 0,30 грамма порошкообразного наполнителя. После этого наполнитель и раствор реагента БЦТА перемешивали при использовании мешалки в течение 35 минут. По завершении перемешивания наполнитель и раствор реагента БЦТА центрифугировали в течение 20 минут для разделения наполнителя и избыточного раствора реагента БЦТА. При завершении центрифугирования пипеткой отбирали в чистый контейнер раствор реагента БЦТА за вычетом отделенного твердого вещества, что называют «центрифугатом». Для анализа образца 50 миллилитров деионизированной воды размещали в 150-миллилитровом химическом стакане с якорем магнитной мешалки. 10 миллилитров образца центрифугата пипеткой отбирали для анализа в тот же самый химический стакан. Образец анализировали при использовании тех же самых технических приемов и порядка действий, что и описанные в настоящем документе.
Для определения уровня содержания влаги приблизительно 0,2 грамма диоксида кремния отвешивали на устройстве Mettler Toledo HB43 при одновременном определении числа реагента БЦТА. Анализатор влаги запрограммировали на 105°C при 5 критериях отсечки при высушивании. Потерю влаги регистрировали с точностью до ближайшего значения +0,1%.
Площадь внешней поверхности рассчитывали при использовании следующего далее уравнения
Площадь поверхности, определенная при использовании реагента БЦТА (в пересчете на сухую массу) [м2/г]=(2V0-V)×(4774)/(V0W)×(100-Vol),
где Vo = объем в мл для реагента Aerosol OT®, использующегося при титровании холостого образца,
V = объем в мл для реагента Aerosol OT®, использующегося при титровании образца,
W = масса образца в граммах,
Vol = % потери влаги (Vol представляет собой «летучие вещества»).
Значения площади поверхности по методу БЭТ, приведенные в примерах данной заявки, определяли согласно методу Брюнера-Эммета-Теллера (БЭТ) в соответствии с документом ASTM D 1993-91. Определение массового процента углерода для подвергнутых обработке диоксидов кремния проводили на элементном анализаторе Flash 2000. Данную систему устанавливали на отслеживание только углерода. Типичные параметры включали: установку для печи сжигания на 1060°C, установку температуры для печи по методу ГХ на 60°C, установку расхода газообразного носителя на 140 мл/мин и время нагнетания кислорода 15 секунд. Для заданного прогона обычно осуществляли прогон калибровочных стандартов, образцов, контрольных образцов и поверочных стандартов. Размер образцов составлял 1-3 мг, и их герметизировали в жестяных капсулах перед анализом. В случае попадания контрольного образца или известного стандартного контрольного образца не в пределы ±0,5% по отношению к известному принятому значению или в случае несоответствия друг другу образцов, пропущенных через два параллельных определения, ±0,6% (по отношению друг к другу)) весь прогон для образца анализировали повторно.
Массовый процент метанола в диоксиде кремния определяли при использовании газового хроматографа Hewlitt Packard 7890 с автодозатором. Для анализа использовали устройство Rxi-624Sil MS, 30 м × 0,32 мм с колонкой при толщине пленки 1,8 мкм. Отверстие для ввода пробы устанавливали на 200°C и использовали объем вводимой пробы 2,0 мкл. Детектором являлся пламенно-ионизационный детектор. Калибровочный стандарт получали в результате разбавления метанола в ацетонитриле. Для получения образцов диоксида кремния для анализа 1 грамм диоксида кремния диспергировали в 10-20 мл ацетонитрила в зависимости от уровня содержания метанола. В случае непредполагаемого большого количества метанола использовали меньше разбавляющего растворителя. Смесь центрифугировали при 19000 об/мин в течение 30 мин. Анализировали только жидкую часть в результате непосредственного ее нагнетания в хроматограф.
Титрование Na2O:
1. Отбирают пипеткой 20 мл образца, подвергаемого испытанию.
2. Выгружают содержимое пипетки в химический стакан, снабженный якорем магнитной мешалки.
3. Разбавляют образец в химическом стакане при использовании ориентировочно 100 мл деионизированной воды.
4. Размещают химический стакан на плите магнитной мешалки и проводят умеренное перемешивание образца.
5. Добавляют приблизительно 10 капель индикатора метилоранж-ксиленцианол. Окраска раствора в химическом стакане должна быть зеленой.
6. Титруют при использовании HCl при концентрации 0,645 н. из бюретки на 50 мл. Конец титрования будет обозначен при переходе окраски раствора в пурпурную.
7. Считывают величину миллилитров добавленной кислоты HCl при концентрации 0,645 н. Данное значение представляет собой количество граммов в расчете на один литр для Na2O в образце.
Титрование кислотного числа:
1. Отбирают пипеткой 50 мл содержимого реактора.
2. Выгружают содержимое пипетки в химический стакан, снабженный якорем магнитной мешалки.
3. Разбавляют образец в химическом стакане при использовании ориентировочно 100 мл деионизированной воды.
4. Размещают химический стакан на плите магнитной мешалки и проводят умеренное перемешивание образца.
5. Добавляют приблизительно 6 капель индикатора фенолфталеина. Окраска раствора в химическом стакане должна быть розовой.
6. Титруют при использовании HCl при концентрации 0,645 н. из бюретки на 50 мл. Конец титрования будет обозначен при переходе окраски раствора в бесцветную.
7. Считывают величину миллилитров добавленной кислоты HCl при концентрации 0,645 н.
8. Кислотное число = (мл HCl при концентрации 0,645 н.)*(64,5)/50.
Часть 2 - Получение примера 1 и сравнительных примеров 1 и 2
Используемое оборудование для осаждения
Реактор представлял собой круглодонный резервуар из нержавеющей стали на 150 литров. Резервуар имел две перегородки на 5 см, расположенные вертикально на противоположных сторонах внутреннего пространства резервуара для дополнительного перемешивания. Производили нагревание при использовании парового змеевика, расположенного на 46,4 см ниже верха резервуара. Резервуар имел два перемешивающих устройства. Основное перемешивание производили при использовании лопасти типа EKATO® MIG, а для добавления кислоты использовали вторичное высокоскоростное перемешивающее устройство при вращении лопасти типа лопасти Коулса при 1750 об/мин. Вторичное высокоскоростное перемешивающее устройство действовало только при добавлении в резервуар кислоты.
Использующиеся материалы исходного сырья:
Силикат натрия - 80 г/л Na2O при соотношении SiO2/Na2O 3,2
Серная кислота - 96%, 36 н.
Пример 1
В реакторный резервуар на 150 л добавляли воду (74,1 л) и производили нагревание до 87°C в результате непрямого обогрева при использовании парового змеевика. Силикат натрия (2,5 л) добавляли при расходе 553 мл/мин для достижения концентрации Na2O 2,6 г/л и кислотного числа 7,8. Концентрацию Na2O и кислотное число подтверждали в результате титрования смеси силикат натрия/вода при использовании описанных выше метода титрования Na2O и метода титрования кислотного числа. Температуру регулировали по мере надобности, доводя до 87°C в результате непрямого обогрева при использовании парового змеевика, и инициировали стадию осаждения. Реактор на 150 л перемешивали при использовании основного перемешивающего устройства резервуара.
Основное перемешивающее устройство оставляли включенным и начинали стадию одновременных добавления и осаждения. В течение периода в 90 мин одновременно добавляли 50,0 литра силиката натрия и 3,7 л серной кислоты. Силикат натрия добавляли через открытую трубку поблизости от дна резервуара при расходе 553 мл/мин, а серную кислоту добавляли непосредственно над лопастями вторичного высокоскоростного перемешивающего устройства. Расход при добавлении кислоты усредняли до 41,0 мл/мин в течение 90-минутной стадии одновременного добавления.
По окончании стадии одновременного добавления инициировали стадию 100-минутного старения. Значение pH раствора регулировали, доводя до 8,5 при использовании серной кислоты. Вторичное высокоскоростное перемешивающее устройство выключали. Под свободной поверхностью добавляли 1,4 л п-(хлорметил)фенилтриметоксисилана, 75% (Gelest), через открытую трубку, расположенную приблизительно на половине высоты вниз по резервуару, при расходе 460 мл/мин. После завершения добавления силана интенсивность перемешивания для основного перемешивающего устройства увеличивали до 162 об/мин. Медленно в течение 15-20 минут в реакторе разбрызгивали 735,4 г стеарата натрия. Реакционная смесь завершала старение при перемешивании в течение оставшихся 100 минут. Температуру выдерживали на уровне 87°C.
После завершения стадии старения добавляли серную кислоту для достижения конечного значения pH партии 4,5. Смесь перекачивали в фильтр-пресс и промывали вплоть до получения при измерении проводимости промывных вод, меньшей, чем 1000 микросименс. Получающийся в результате осадок на фильтре повторно суспендировали при использовании воды для получения перекачиваемой суспензии и подвергали распылительному высушиванию при использовании распылительной сушилки Niro (Utility Model 5 с центробежным распылителем Type FU-11, Niro Inc.). Подвергнутый распылительному высушиванию порошок гранулировали при использовании роликового пресса Alexanderwerk WP120X40 Roller Compactor в следующих далее условиях: скорость вращения червяка=55 об/мин, скорость вращения валка 4,5 об/мин, скорость вращения ротора дробилки = 55 об/мин, гидравлическое давление = 25 бар и размер ячеек сита = 7 меш.
Сравнительный пример 1
В V-образный смеситель Patterson Kelley на 8 кварт (7,57 л), снабженный планкой интенсификатора, добавляли осажденный диоксид кремния, характеризующийся площадью поверхности по методу БЭТ, составляющей приблизительно 175 м2/г, и площадью поверхности, определенной при использовании реагента БЦТА, составляющей приблизительно 153 м2/г. Смеситель запускали и действие планки интенсификатора инициировали при непосредственном добавлении 193 г п-(хлорметил)фенилтриметоксисилана, 75% (Gelest), в интенсификатор через трубку добавления и их подаче самотеком из пластиковой воронки. Планке интенсификатора давали возможность продолжать действовать в течение приблизительно двух минут после добавления всего количества силана. V-образный смеситель продолжал перемешивание в течение приблизительно 45 мин после добавления всего количества силана. Материал вываливали в пластиковый пакет и использовали в течение пяти дней. Для данного примера стеарат натрия не использовали вследствие нерастворимости материала и быстрого затвердевания, прохождение чего наблюдали внутри планки интенсификатора.
Подвергнутый обработке порошок гранулировали при использовании роликового пресса Alexanderwerk WP120X40 Roller Compactor в следующих далее условиях: скорость вращения червяка = 55 об/мин, скорость вращения валка 4,5 об/мин, скорость вращения ротора дробилки = 55 об/мин, гидравлическое давление = 25 бар и размер ячеек сита = 7 меш.
Сравнительный пример 2
В реакторный резервуар на 150 л добавляли воду (74,9 л) и производили нагревание до 70°C в результате непрямого обогрева при использовании парового змеевика. Силикат натрия (2,5 л) добавляли при расходе 559 мл/мин для достижения концентрации Na2O 2,6 г/л и кислотного числа 7,8. Концентрацию Na2O и кислотное число подтверждали в результате титрования смеси силикат натрия/вода при использовании описанных выше метода титрования Na2O и метода титрования кислотного числа. Температуру регулировали по мере надобности, доводя до 70°C в результате непрямого обогрева при использовании парового змеевика, и инициировали стадию осаждения. Реактор на 150 л перемешивали при использовании основного перемешивающего устройства резервуара.
Основное перемешивающее устройство оставляли включенным и начинали стадию одновременных добавления и осаждения. В течение периода в 90 мин одновременно добавляли силикат натрия (50,3 л) и 3,2 л серной кислоты. Силикат натрия добавляли через открытую трубку поблизости от дна резервуара при расходе 559 мл/мин, а серную кислоту добавляли непосредственно над лопастями вторичного высокоскоростного перемешивающего устройства. Расход при добавлении кислоты усредняли до 36,0 мл/мин в течение 90-минутной стадии одновременного добавления.
По окончании стадии одновременного добавления инициировали стадию 100-минутного старения. Значение pH раствора регулировали, доводя до 8,5 при использовании серной кислоты. Вторичное высокоскоростное перемешивающее устройство выключали. Под свободной поверхностью добавляли 0,79 л гамма-меркаптопропилтриэтоксисилана через открытую трубку, расположенную приблизительно на половине высоты вниз по резервуару при расходе 39 мл/мин. После завершения добавления силана интенсивность перемешивания для основного перемешивающего устройства увеличивали до 162 об/мин. Медленно в течение 15-20 мин в реакторе разбрызгивали стеарат натрия (1244 г). Реакционная смесь завершала старение при перемешивании в течение оставшихся 100 минут. Температуру выдерживали на уровне 70°C. После завершения стадии старения добавляли серную кислоту для достижения конечного значения pH партии 4,8.
Смесь перекачивали в фильтр-пресс и промывали вплоть до получения при измерении проводимости промывных вод, меньшей, чем 1000 микросименс. Получающийся в результате осадок на фильтре повторно суспендировали при использовании воды для получения перекачиваемой суспензии и подвергали распылительному высушиванию при использовании распылительной сушилки Niro (Utility Model 5 с центробежным распылителем Type FU-11, Niro Inc.). Подвергнутый распылительному высушиванию порошок гранулировали при использовании роликового пресса Alexanderwerk WP120X40 Roller Compactor в следующих далее условиях: скорость вращения червяка = 55 об/мин, скорость вращения валка 4,5 об/мин, скорость вращения ротора дробилки = 55 об/мин, гидравлическое давление = 25 бар и размер ячеек сита = 7 меш. Физические свойства синтезированных диоксидов кремния обобщенно представлены в таблице 1.

Часть 3 - Получение образцов каучука и сравнительного примера 3
Для получения образцов для испытания из составленных рецептур каучуковых композиций, включающих диоксид кремния из примеров и сравнительных примеров (СП), использовали стандартный протокол составления смесей. Сравнительный пример 3 получали в результате добавления в смеситель гранулированного осажденного диоксида кремния, характеризующегося площадью поверхности по методу БЭТ, составляющей приблизительно 157 м2/г, и площадью поверхности, определенной при использовании реагента БЦТА, составляющей приблизительно 158 м2/г, а также п-(хлорметил)фенилтриметоксисилана при дозировках и временах, предписанных в таблицах 2 и 3.
Следующие далее ингредиенты в количествах массовых частей в расчете на сто частей каучука (чек) добавляли в описанном порядке в полиэтиленовый пакет, удерживаемый в прямостоячем положении в пластиковой чаше на 500 миллилитров (мл).

предварительной реакции)
(1) Ароматическое углеводородное технологическое масло Vivatec 500, коммерчески получаемое в компании Н&К Group.
(2) Подвергнутый поверхностной обработке оксид цинка KADOX®, коммерчески получаемый в компании Zinc Corporation of America.
(3) SANTOFLEX® 13 (6-PPD), согласно сообщению представляющий собой N-(1,3-диметилбутил)-N′-фенил-п-фенилендиамин и 4-аминодифениламин от компании Flexsys.
(4) Стеариновая кислота марки для переработки каучука, коммерчески получаемая в компании C.Р. Hall.
(5) Технический углерод наномарки от компании Sid Richardson Carbon and Energy Company.
(6) STANGARD® TMQ, распределяемый компанией Harwick Standard.
(7) SUNPROOF® Improved от компании Chemtura.
Для перемешивания различных ингредиентов использовали закрытый резиносмеситель Kobelco на 1,89 литра (л) (Model «BR00»). Непосредственно перед добавлением ингредиентов партии в смеситель через смеситель для его очищения от любых остатков от предшествующих прогонов и увеличения температуры до приблизительно 93°C (200°F) пропускали 800 граммов (г) натурального каучука марки CV-60. После удаления каучука смеситель охлаждали до приблизительно 65°C (150°F) перед добавлением ингредиентов для получения образца каучука для испытания.
Каучуковую композицию получали при использовании испытываемого наполнителя, следующих далее других перечисленных ингредиентов и методики, описанной ниже в настоящем документе.
Первый проход инициировали в результате добавления в смеситель каучука и проведения перемешивания при 30 об/мин. Скорость вращения ротора выдерживали на уровне 30 об/мин и добавляли 3,0 чек технического углерода. По истечении одной минуты добавляли половину испытываемого наполнителя при добавлении остального количества по истечении еще одной минуты. Совместно со второй частью испытываемого наполнителя добавляли реагент Vivatex 500. При трех минутах затвор поднимали и желоб прочищали, то есть крышку на загрузочном желобе поднимали и любой материал, который находили в желобе, вычищали обратно в смеситель. Скорость смесителя увеличивали до 70 об/мин. Содержимое в смесителе перемешивали в течение еще двух минут для достижения максимальной температуры в диапазоне от 145 до 150°C (от 293 до 302°F) и для завершения первого прохода в смесителе. По истечении 4 минут в зависимости от типа образца скорость вращения ротора смесителя может быть увеличена или уменьшена для достижения температуры в вышеупомянутом диапазоне в течение указанного периода перемешивания. Материал из смесителя удаляли.
После завершения первого прохода удаленный материал взвешивали и раскатывали в лист в двухвалковых вальцах для каучука Farrel на 12 дюймов (304,8 мм) при уровне 2,032±0,127 мм (0,080 дюйма ±0,005 дюйма). Получающуюся в результате смесь, раскатанную на вальцах, использовали для второго прохода в смесителе.
Второй проход инициировали в результате добавления смеси первого прохода в смеситель, функционирующий при 60 об/мин. По истечении одной минуты в смеситель добавляли предварительно отвешенные оксид цинка, стеариновую кислоту, противоозоностаритель SANTOFLEX® 13, антидеградант STANGARD® TMQ и восковой противоозоностаритель SUNPROOF® Improved. По истечении еще одной минуты затвор поднимали и желоб прочищали. Скорость перемешивания уменьшали до 30 об/мин. Второй проход завершали в результате перемешивания смеси в течение еще 3,0 минуты при одновременном выдерживании температуры на уровне или менее величины в диапазоне от 135°C (257°F) до 140°C (284°F).
Двухвалковые вальцы для каучука Farrel на 12 дюймов (304,8 мм) нагревали до приблизительно 60°C (140°F). Смесь со второго прохода части В подавали в действующие вальцы при установке зазора 2,032±0,127 мм (0,080 дюйма ±0,005 дюйма). В вальцы добавляли и перемешивали друг с другом серу RM, реагенты ЦВС и ДФГ. Совокупное время на вальцах составляло приблизительно пять минут при 5 боковых фрезах и 5 концевых валках. Получающийся в результате лист размещали на плоской поверхности вплоть до достижения температурой листа комнатной температуры. Обычно лист охлаждался в течение приблизительно 30 минут. Листовой материал, отобранный из вальцев, размещали на плоской чистой поверхности. При использовании трафарета из раскатанной в лист смеси вырезали прямоугольный образец 203,2 × 152,4 мм (8 дюймов × 6 дюймов). Образец кондиционировали, то есть хранили между чистыми полиэтиленовыми листами и выдерживали в течение от 15 до 18 ч при температуре 23±2°C и относительной влажности 50±5%.
После кондиционирования образец размещали в стальной пресс-форме для прямого прессования стандартной формовочной машины 203,2 × 152,4 × 2,286 мм (8 дюймов × 6 дюймов × 0,09 дюйма), имеющей полированную поверхность. Образец отверждали в 4-позиционном прессе для прямого прессования с электрическим обогревом 61 сантиметр × 61 сантиметр (24 дюйма × 24 дюйма), 890 килоньютон (100 тонн) для T90, то есть времени, которое требуется для прохождения отверждения на 90 процентов в соответствии с документом ASTM D-2084, плюс 5 мин при 150°C (302°F) под давлением 13,79 мегапаскаль (2000 фунтов в расчете на один квадратный дюйм). Обычно отверждение завершали в течение приблизительно 10 мин. Получающийся в результате лист отвержденного каучука удаляли из пресс-формы и выдерживали в течение от 15 до 18 ч при температуре 23±2°C (73,4±3,6°F) и относительной влажности 50±5% перед проведением следующего далее испытания.
Часть 4 - Испытание для отвержденных каучуковых продуктов
Испытание проводили в соответствии с документом ASTM D 412-98a - Test Method A. Образцы для испытания в виде двойной лопатки получали при использовании штампа Die С.Использовали устройство Instron model 4204 с прибором автоматизированного контактного удлинения для измерения относительного удлинения. Скорость траверсы, как было установлено, была равна 508 мм/мин. Все вычисления проводили при использовании программного обеспечения Series IX Automated Materials Testing software, поставляемого производителем.
Подвергнутый вальцеванию и раскатанный в лист каучук из части 3 использовали для определения вязкости по Муни, динамических данных Rheometrics и значений TS2 и TS50. Вязкость по Муни (ML 1+4) определяли при использовании вискозиметра Муни (MV 2000) с большим ротором в соответствии с документом ASTM D 1646-00 part А. Соответствующие данные MDR 2000 (TS2, ТС50 и ТС90) определяли при использовании вискозиметра с пуансоном (MDR 2000) в соответствии с документом ASTM D 5289-95 (2001).
Динамические данные Rheometrics (развертку по температуре и деформации) определяли в условиях параллельных пластин при использовании устройства Rheometrics Dynamic Spectrometer 2 (RDS 2). Образец составленных смесей эластомеров, который отверждали между двумя параллельными пластинами, подвергали воздействию колебательного изменения деформации для оценки вязкоупругих свойств, таких как модуль упругости (С), модуль вязкости (G″) и коэффициент затухания (тангенс дельты = G″/G′). Развертки по температуре проводили в диапазоне от - 45°C до 75°C при 1 Гц и деформации 2%. Развертки по деформации проводили в диапазоне деформаций 0,1-20% при 1 Гц и 30°C.
Образец для динамических данных Rheometrics получали из части подвергнутого вальцеванию и раскатанного в лист переработанного каучука, который подвергали повторному вальцеванию до толщины 0,450 дюйма (11,4 мм). Из листа вырезали блок 2 дюйма на 2 дюйма (50,8 мм на 50,8 мм). После этого из блока при использовании пуансона на 11 мм и машины для вырубания изделий вырезали два цилиндрических образца диаметром в 11 мм. Вырубленные образцы каучуков обрезали до 0,86±0,01 грамма. Образцы размещали в полости с диаметром 11 мм в пресс-форме для прямого прессования между параллельными пластинами, которые представляли собой подвергнутые машинной обработке алюминиевые цилиндры с приподнятой цилиндрической платформой. Параллельные пластины имели совокупную толщину 0,188 дюйма (4,8 мм) и диаметр 0,985 дюйма (25,0 мм). Часть в виде приподнятой цилиндрической платформы параллельных пластин имела толщину 0,125 дюйма (3,2 мм) и диаметр 0,793 дюйма (20,1 мм). Пластины прежде очищали при использовании ацетона и грунтовали при использовании реагента Chemlok 205. Образцы отверждали при 150°C для значения T90 плюс 10 минут под давлением в 15 тонн.
Для испытания напряжение/деформация использовали тонкие листы. Испытание напряжение/деформация проводили в соответствии с документом ASTM D 412-98a - Test Method A. Образцы для испытания в виде двойной лопатки получали при использовании штампа Die С. Использовали устройство Instron model 4204 с прибором автоматизированного контактного удлинения для измерения относительного удлинения. Скорость траверсы была равной 508 мм/мин. Все вычисления проводили при использовании программного обеспечения Series IX Automated Materials Testing software, поставляемого производителем.
Для испытания упругости по отскоку Zwick, истирания DIN и величины белой площади для устройства dispergrader использовали круглые образцы высотой в 12 мм и диаметром в 29 мм. Упругость по отскоку Zwick определяли при использовании устройства Zwick 5109 Rebound Resilience Tester в соответствии с документом ASTM D 1054-91 (2000). Индекс (стойкость к истиранию) DIN определяли согласно методу B в соответствии с документом ASTM D 5963-97A (2001).
% белой площади для устройства dispergrader определяли при использовании прибора DisperGrader 1000 NT+(100X). Оптический прибор, управляемый при помощи компьютера, фиксировал изображения топографии поверхности свежесрезанного образца отвержденного каучука. Недиспергированные неподвергнутые обработке или подвергнутые обработке или подвергающиеся реакции сочетания частицы наполнителя на топографии проявлялись в виде «вздутий» или «накладок». Программное обеспечение для анализа изображений производило измерение размера каждой характеристики в поле обзора 40 микронов × 35 микронов при увеличении 100X. Диаметры и количества частиц группировали в различные диапазоны размеров и рассчитывали % площади. Программное обеспечение делало возможным сопоставление дисперсии подвергнутого обработке или подвергающегося реакции сочетания или неподвергнутого обработке наполнителя с внутренними библиотеками эталонных фотографий. Таблица 4 подробно представляет количество технического углерода в контрольном образце на основе технического углерода и образцах, количества загрузки наполнителя на основе диоксида кремния и количество силана, добавленного в сравнительный пример 3 (СП-3). Тип использующегося технического углерода представлял собой марку N220. Вариант СП-3 представляет собой сравнительный пример с «выбросом», в котором диоксид кремния представлял собой реагент Hi-Sil™ HDS, и к смеси для получения каучука добавляли п-(хлорметил)фенилтриметоксисилан, 75% (Gelest). В таблице 5 продемонстрировано суммарное представление свойств, ассоциированных с каучуковыми продуктами, наполненными при использовании диоксида кремния.
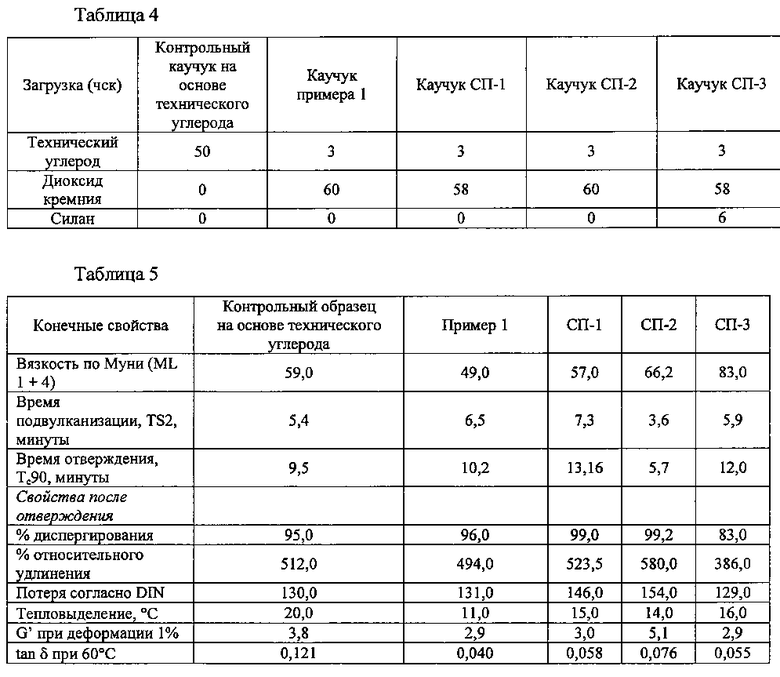
Пример 1 продемонстрировал меньшую вязкость при перемешивании, то есть вязкость по Муни (ML 1+4), в сопоставлении с контрольным примером и всеми сравнительными примерами. Время отверждения из примера 1, на которое указывало значение Tc90, было сопоставимым с тем, что имело место для контрольного образца на основе технического углерода, и меньшим того, что имело место для вариантов СП-1 и СП-3. Вариант СП-2, который включал меркаптосилан, предположительно обеспечивал получение более краткого времени отверждения. Процент диспергирования, на который указывал анализ при использовании устройства dispergrader, продемонстрировал то, что пример 1 обнаруживал сопоставимые характеристики диспергирования по отношению к контрольному образцу на основе технического углерода. Пример 1 также продемонстрировал большие плотности сшивания (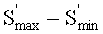
Несмотря на описание настоящего изобретения при обращении к конкретным подробностям его определенных вариантов осуществления не предполагается то, что такие подробности должны рассматриваться в качестве ограничений объема изобретения за исключением тех случаев, в которых они включены в формулу изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ОБРАБОТАННЫЕ НАПОЛНИТЕЛИ, КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ, И ИЗДЕЛИЯ, ИЗГОТОВЛЕННЫЕ ИЗ НИХ | 2014 |
|
RU2658402C2 |
| ОБРАБОТАННЫЕ НАПОЛНИТЕЛИ, КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ, И ИЗДЕЛИЯ, ИЗГОТОВЛЕННЫЕ ИЗ НИХ | 2014 |
|
RU2640075C2 |
| ОБРАБОТАННЫЕ НАПОЛНИТЕЛИ, КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ, И ИЗДЕЛИЯ, ИЗГОТОВЛЕННЫЕ ИЗ НИХ | 2014 |
|
RU2644863C2 |
| ОБРАБОТАННЫЕ НАПОЛНИТЕЛИ, КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ, И ИЗДЕЛИЯ, ИЗГОТОВЛЕННЫЕ ИЗ НИХ | 2014 |
|
RU2642795C2 |
| СПОСОБ ПОЛУЧЕНИЯ КОМПАУНДОВ ДЛЯ ШИН И ШИНЫ, ВКЛЮЧАЮЩИЕ ИХ | 2019 |
|
RU2786563C1 |
| ПОКРЫШКА, ИЗГОТОВЛЕННАЯ С ИСПОЛЬЗОВАНИЕМ КАУЧУКОВОЙ КОМПОЗИЦИИ, СОДЕРЖАЩЕЙ МОДИФИЦИРОВАННЫЙ ПОЛИМЕР | 2009 |
|
RU2475368C2 |
| ФОРМОВОЧНАЯ КОМПОЗИЦИЯ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2008 |
|
RU2584949C2 |
| КАУЧУКОВАЯ КОМПОЗИЦИЯ, СШИТАЯ КАУЧУКОВАЯ КОМПОЗИЦИЯ И ШИНА | 2012 |
|
RU2573869C2 |
| СПОСОБ ПОЛУЧЕНИЯ ВУЛКАНИЗОВАННОЙ КАУЧУКОВОЙ КОМПОЗИЦИИ | 2011 |
|
RU2575657C2 |
| СМЕСИ НА ОСНОВЕ БУТИЛОВОГО КАУЧУКА, СОДЕРЖАЩИЕ ТРЕХКОМПОНЕНТНУЮ СМЕШАННУЮ СИСТЕМУ МОДИФИКАТОРОВ | 2008 |
|
RU2485148C9 |
Изобретение относится к химической промышленности и может быть использовано при получении наполнителей диоксид кремния/силан для покрышек легковых автомобилей. Суспензию необработанного наполнителя обрабатывают композицией для обработки, содержащей органосилан, после чего суспензию высушивают. Органосилан выбирают из (4-хлорметилфенил)триметоксисилана, (4-хлорметилфенил)триэтоксисилана, [2-(4-хлорметилфенил)этил]триметоксисилана, [2-(4-хлорметилфенил)этил]триэтоксисилана, (3-хлорпропенил)триметоксисилана, (3-хлорпропенил)триэтоксисилана, (3-хлорпропил)триэтоксисилана, (3-хлорпропил)триметоксисилана, триметокси(2-п-толилэтил)силана и/или триэтокси-(2-п-толилэтил)силана. Изобретение позволяет уменьшить сопротивление качению. 6 н. и 10 з.п. ф-лы, 5 табл., 1 пр.
1. Способ получения подвергнутого обработке наполнителя, включающий:
(c) обработку суспензии, включающей неподвергнутый обработке наполнитель, где неподвергнутый обработке наполнитель прежде не высушивали, при использовании композиции для обработки, содержащей органосилан, выбираемый из (4-хлорметилфенил)триметоксисилана, (4-хлорметилфенил)триэтоксисилана, [2-(4-хлорметилфенил)этил]триметоксисилана, [2-(4-хлорметилфенил)этил]триэтоксисилана, (3-хлорпропенил)триметоксисилана, (3-хлорпропенил)триэтоксисилана, (3-хлорпропил)триэтоксисилана, (3-хлорпропил)триметоксисилана, триметокси(2-п-толилэтил)силана и/или триэтокси-(2-п-толилэтил)силана, для получения суспензии подвергнутого обработке наполнителя и
(d) высушивание упомянутой суспензии подвергнутого обработке наполнителя для получения подвергнутого обработке наполнителя.
2. Способ по п. 1, где композиция для обработки, кроме того, содержит материал для обработки, выбираемый из анионных, неионных и амфотерных поверхностно-активных веществ и их смесей, где материал для обработки присутствует в количестве в диапазоне от более чем 1 до 25 мас.% в расчете на неподвергнутый обработке наполнитель.
3. Способ по п. 1, где композиция для обработки, кроме того, содержит меркаптосилан, отличный от органосилана.
4. Способ по п. 3, где меркаптосилан включает меркаптопропилтриметоксисилан.
5. Способ по п. 3, где композиция для обработки, кроме того, содержит материал для обработки, выбираемый из анионных, неионных и амфотерных поверхностно-активных веществ и их смесей, где материал для обработки присутствует в количестве в диапазоне от более чем 1 до 25 мас.% в расчете на неподвергнутый обработке наполнитель.
6. Способ по п. 1, где упомянутый неподвергнутый обработке наполнитель выбирают из силиката алюминия, силикагеля, коллоидального диоксида кремния, осажденного диоксида кремния и их смесей.
7. Способ по п. 5, где упомянутый материал для обработки выбирают из солей жирных кислот, алкилсаркозинатов, солей алкилсаркозинатов и их смесей.
8. Способ по п. 1, где наполнитель включает осажденный диоксид кремния.
9. Способ получения подвергнутого обработке наполнителя, включающий:
(а) объединение силиката щелочного металла и кислоты для получения суспензии, включающей неподвергнутый обработке наполнитель, где упомянутый неподвергнутый обработке наполнитель прежде не высушивали;
(b) обработку упомянутой суспензии при использовании композиции для обработки, содержащей органосилан, выбираемый из (4-хлорметилфенил)триметоксисилана, (4-хлорметилфенил)триэтоксисилана, [2-(4-хлорметилфенил)этил]триметоксисилана, [2-(4-хлорметилфенил)этил]триэтоксисилана, (3-хлорпропенил)триметоксисилана, (3-хлорпропенил)триэтоксисилана, (3-хлорпропил)триэтоксисилана, (3-хлорпропил)триметоксисилана, триметокси(2-п-толилэтил)силана и/или триэтокси-(2-п-толилэтил)силана, для получения суспензии подвергнутого обработке наполнителя и
(c) высушивание упомянутой суспензии подвергнутого обработке наполнителя для получения подвергнутого обработке наполнителя.
10. Способ по п. 9, где упомянутый силикат щелочного металла включает силикат алюминия, силикат лития, силикат натрия и/или силикат калия.
11. Способ по п. 9, где композиция для обработки, кроме того, содержит материал для обработки, выбираемый из анионных, неионных и амфотерных поверхностно-активных веществ и их смесей, где материал для обработки присутствует в количестве в диапазоне от более чем 1 до 25 мас.% в расчете на неподвергнутый обработке наполнитель.
12. Материал подвергнутого обработке наполнителя, полученный способом по п. 1 для использования в каучуковой композиции.
13. Каучуковая композиция, включающая подвергнутый обработке наполнитель, полученный способом по п. 1.
14. Каучуковая композиция по п. 13, содержащая натуральный каучук.
15. Каучуковое изделие, включающее подвергнутый обработке наполнитель, полученный способом по п. 1.
16. Протектор покрышки, включающий подвергнутый обработке наполнитель, полученный способом по п. 1.
| EP 1048696 A2, 02.11.2000 | |||
| WO 2006065578 A2, 22.06.2006 | |||
| ГИДРОФОБНЫЙ ДИОКСИД КРЕМНИЯ И ЕГО ПРИМЕНЕНИЕ В СИЛИКОНОВОМ КАУЧУКЕ | 2005 |
|
RU2358908C2 |
| US 20090111923 A1, 30.04.2009. | |||

