ОБЛАСТЬ ТЕХНИКИ
Изобретение относится к новым замещенным производным фенокси-этил-амина, полезным в качестве модуляторов дофаминергической и опосредованной N-метил-D-аспартатным (NMDA) рецептором глутаматергической нейротрансмиссии в коре головного мозга и базальных ганглиях. В других аспектах изобретение относится к применению этих соединений в способе лечения и к фармацевтическим композициям, содержащим соединения по изобретению.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Дофамин является нейротрансмиттером в головном мозге. С момента открытия в 1950-х годах функция дофамина в головном мозге интенсивно изучалась. К настоящему времени установлено, что дофамин важен в нескольких аспектах функционирования головного мозга, включая двигательную, когнитивную, сенсорную, эмоциональную и автономную функции (например регуляция аппетита, температуры тела, сна). Поэтому модулирование дофаминергической функции может быть полезным в лечении целого ряда расстройств, нарушающих функции головного мозга. Фактически, лекарственные средства, которые прямо или опосредованно воздействуют на центральные дофаминовые рецепторы, обычно используют в лечении неврологических и психиатрических расстройств, например болезни Гентингтона, болезни Паркинсона и шизофрении.
Антипсихотические лекарственные средства (или нейролептики) представляют собой класс соединений с разнообразными воздействиями на разные рецепторные системы. Однако в целом они обладают способностью блокировать рецепторы дофамина D2 в базальных ганглиях (т.е. в полосатом теле), и их применяют для лечения психоза (включая бред или галлюцинации, а также нарушение мышления), в частности при шизофрении и биполярном расстройстве.
Кора головного мозга охватывает несколько основных областей, которые вовлечены в высшие функции, такие как мышление, чувства, память и планирование. Биогенные амины, такие как дофамин, важны для кортикальной функции млекопитающих. Восходящие дофаминовые пути иннервируют кору головного мозга. Первичные или вторичные дисфункции в активности этих путей приводят к нарушению регуляции активности дофамина в этих участках головного мозга и впоследствии к проявлениям психиатрических и неврологических симптомов. Рецепторы дофамина D1 и N-метил-D-аспартата (NMDA) в префронтальной коре головного мозга играют решающую роль в синаптической пластичности, механизмах памяти и когнитивной способности.
Болезнь Гентингтона (HD) является редким нейродегенеративным расстройством центральной нервной системы, которое характеризуется прогрессивным ухудшением двигательной и когнитивной функции, а также поведенческими и психиатрическими нарушениями. Общепризнано, что при болезни Гентингтона также нарушены некоторые аспекты дофаминергических функций. Невропатологические изменения при болезни Гентингтона включают заметную потерю и атрофию клеток в полосатом теле, а также во многих других областях головного мозга, таких как кора головного мозга, черная субстанция, гипоталамус, мозжечок и таламус.
При HD изменяется глутаматная и дофаминовая (DA) трансмиссия, что по всей вероятности индуцирует дисбаланс в активности прямых и опосредованных путей и вносит вклад в двигательные, когнитивные и психиатрические симптомы HD (т.е. передачу между корой головного мозга и полосатым телом, Capeda et al; ASN Neuro 2010, 2(2), е00033). Поэтому соединения, которые могут усиливать кортикальную дофаминовую и NMDA трансмиссию и вызывать антагонизм избыточной субкортикальной дофаминовой трансмиссии, могут уравновешивать аберрантное функционирование кортико-стриато-таламической сети, контролирующей двигательные функции (Alexander et al; Ann. Rev. Neurosci. 1986, 9, 357-381).
В JP 2006-193494 (Dainippon Ink and Chemicals, Inc) описаны некоторые четвертичные аммониевые соединения, полезные в качестве терапевтического агента для лечения заболеваний сердца.
В WO 2009/133107 (NSAB, Filial at NeuroSearch Sweden AB, Sverige) описаны некоторые производные 1-(2,3-дигидро-1,4-бензодиоксин-2-ил)метанамина, в WO 2009/133109 (NSAB, Filial af NeuroSearch Sweden AB, Sverige) описаны некоторые производные 1-(2,3-дигидро-1,4-бензодиоксин-2-ил)метанамина, и в WO 2009/133110 (NSAB, Filial af NeuroSearch Sweden AB, Sverige) описаны некоторые производные 1-(4H-1,3-бензодиоксин-2-ил)метанамина, полезные в качестве модуляторов дофаминовой нейротрансмиссии и, более конкретно, в качестве дофаминергических стабилизаторов. Однако о производных фенокси-этил-амина по настоящему изобретению ранее не сообщалось.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Задача настоящего изобретения заключается в предоставлении новых фармацевтически активных соединений, в частности полезных в лечении расстройств центральной нервной системы. Задача также заключается в предоставлении соединений для модулирования дофаминергической и глутаматергической систем в головном мозге млекопитающих, включая головной мозг человека.
В первом аспекте изобретения предложено производное фенокси-этил-амина формулы 1
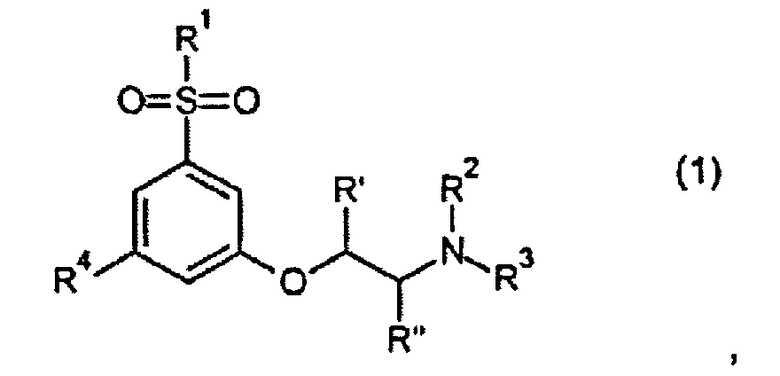
его стереоизомер или смесь его стереоизомеров, или его N-оксид, или его полностью или частично дейтерированный аналог, или его фармацевтически приемлемая соль, где R1, R2, R3, R4, R′ и R′′ такие, как определено ниже.
Во втором аспекте изобретения предложена фармацевтическая композиция, содержащая терапевтически эффективное количество производного фенокси-этил-амина по изобретению, его стереоизомера или смеси его стереоизомеров, или его N-оксида, или его фармацевтически приемлемой соли вместе с по меньшей мере одним фармацевтически приемлемым носителем, эксципиентом или разбавителем.
В следующем аспекте изобретения предложено применение производного фенокси-этил-амина по изобретению, его стереоизомера или смеси его стереоизомеров, или его N-оксида, или его фармацевтически приемлемой соли для изготовления фармацевтической композиции для лечения, предупреждения или облегчения заболевания или расстройства или состояния у млекопитающего, включая человека, причем заболевания, или расстройства, или состояния, которое реагирует на модулирование дофаминергической функции в центральной нервной системе.
В еще одном аспекте изобретение относится к способу лечения, предупреждения или облегчения заболевания или расстройства или состояния организма живого животного, включая человека, причем заболевания или расстройства или состояния, которое реагирует на модулирование дофаминергической функции в центральной нервной системе, включающему стадию введения в такой организм живого животного, нуждающегося в этом, терапевтически эффективного количества производного фенокси-этил-амина по изобретению, его стереоизомера или смеси его стереоизомеров, или его N-оксида, или его фармацевтически приемлемой соли.
Другие аспекты изобретения станут понятны специалисту в данной области из следующего ниже подробного описания и примеров.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Производные феноксиэтиламина
В первом аспекте настоящего изобретения предложены производные фенокси-этил-амина формулы 1
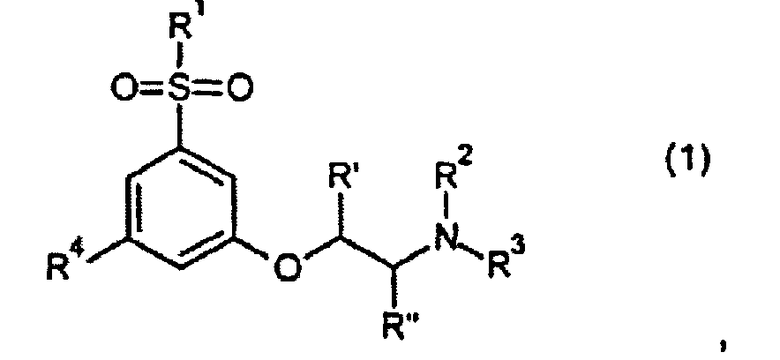
их стереоизомеры или смеси их стереоизомеров, или их N-оксиды, или их полностью или частично дейтерированные аналоги, или их фармацевтически приемлемые соли, где
R1 представляет собой CH3 или CF3;
R2 выбран из группы, состоящей из C1-C4-алкила, аллила, CH2CH2OCH3, C(CH3)2CH2CH3, группы CH2-циклопропил, циклобутила, циклопентила, CH2CH2CH2F, CH2CH2CHF2, CH2CH2F, 3,3,3-трифторпропила и 4,4,4-трифторбутила; и
R3 выбран из группы, состоящей из H, CH3 и CH2CH3; или
R2 и R3 вместе образуют CH2(CH2)CH2 или CH2(CH2)3CH2;
R4 представляет собой F или Cl; и
R′ и R′′ независимо представляют собой водород или метил.
В предпочтительном воплощении производное фенокси-этил-амина по изобретению представляет собой соединение формулы 1a
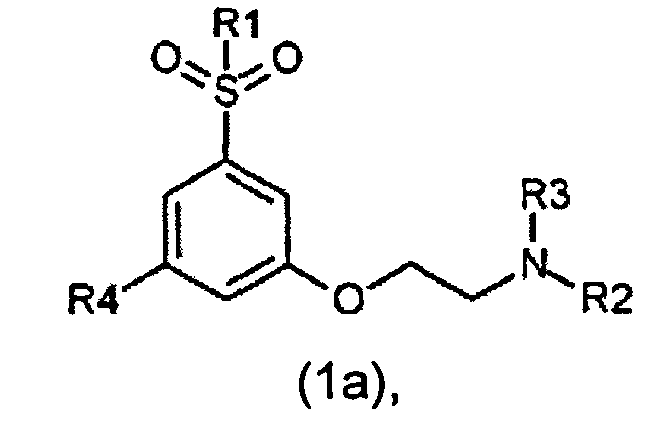
его стереоизомер или смесь его стереоизомеров, или его N-оксид, или его фармацевтически приемлемую соль, где
R1 выбран из группы, состоящей из CH3 или CF3;
R2 выбран из группы, состоящей из C1-C4-алкила, аллила, CH2CH2OCH3, С(CH3)2CH2CH3, группы CH2-циклопропил, CH2CH2CH2F, CH2CH2CHF2, CH2CH2F, 3,3,3-трифторпропила и 4,4,4-трифторбутила;
R3 выбран из группы, состоящей из H, CH3 и CH2CH3; и
R4 выбран из группы, состоящей из F и Cl.
В другом предпочтительном воплощении производное фенокси-этил-амина по изобретению представляет собой соединение формулы 1a, его стереоизомер или смесь его стереоизомеров, или его N-оксид, или его фармацевтически приемлемую соль, где
R1 выбран из группы, состоящей из CH3 или CF3;
R2 выбран из группы, состоящей из C1-C4-алкила, аллила, CH2CH2OCH3, C(CH3)2CH2CH3, группы CH2-циклопропил, CH2CH2CH2F, CH2CH2CHF2, CH2CH2F, 3,3,3-трифторпропила и 4,4,4-трифторбутила;
R3 выбран из группы, состоящей из H, CH3 и CH2CH3;
или R2 и R3 вместе образуют CH2(CH2)3CH2; и
R4 выбран из группы, состоящей из F и Cl.
В третьем предпочтительном воплощении производное фенокси-этил-амина по изобретению представляет собой соединение формулы 1 или 1a, его стереоизомер или смесь его стереоизомеров, или его N-оксид, или его фармацевтически приемлемую соль, где R1 представляет собой CH3 или CF3.
В более предпочтительном воплощении R1 представляет собой CH3.
В еще одном более предпочтительном воплощении R1 представляет собой CF3.
В четвертом предпочтительном воплощении производное фенокси-этил-амина по изобретению представляет собой соединение формулы 1 или 1a, его стереоизомер или смесь его стереоизомеров, или его N-оксид, или его фармацевтически приемлемую соль, где R2 выбран из группы, состоящей из C1-C4-алкила, аллила, CH2CH2OCH3, С(CH3)2CH2CH3, группы CH2-циклопропил, циклобутила, циклопентила, CH2CH2CH2F, CH2CH2CHF2, CH2CH2F, 3,3,3-трифторпропила и 4,4,4-трифторбутила.
В более предпочтительном воплощении R2 выбран из группы, состоящей из C1-C4-алкила, группы CH2-циклопропил, циклобутила и циклопентила.
В другом более предпочтительном воплощении R2 представляет собой C1-C4-алкил.
В третьем более предпочтительном воплощении R2 представляет собой CH2-циклопропил.
В четвертом более предпочтительном воплощении R2 представляет собой циклобутил.
В пятом более предпочтительном воплощении R2 представляет собой циклопентил.
В пятом предпочтительном воплощении производное фенокси-этил-амина по изобретению представляет собой соединение формулы 1 или 1a, его стереоизомер или смесь его стереоизомеров, или его N-оксид, или его фармацевтически приемлемую соль, где R3 выбран из группы, состоящей из Н и CH3.
В более предпочтительном воплощении R3 представляет собой Н.
В другом более предпочтительном воплощении R3 представляет собой CH3.
В шестом предпочтительном воплощении производное фенокси-этил-амина по изобретению представляет собой соединение формулы 1 или 1a, его стереоизомер или смесь его стереоизомеров, или его N-оксид, или его фармацевтически приемлемую соль, где R2 и R3 вместе образуют CH2(CH2)CH2 или CH2(CH2)3CH2 .
В более предпочтительном воплощении R2 и R3 вместе образуют CH2(CH2)CH2.
В другом более предпочтительном воплощении R2 и R3 вместе образуют CH2(CH2)3CH2.
В седьмом предпочтительном воплощении производное фенокси-этил-амина по изобретению представляет собой соединение формулы 1 или 1a, его стереоизомер или смесь его стереоизомеров, или его N-оксид, или его фармацевтически приемлемую соль, где R4 представляет собой F или Cl.
В более предпочтительном воплощении R4 представляет собой F.
В другом более предпочтительном воплощении R4 представляет собой Cl.
В восьмом предпочтительном воплощении производное фенокси-этил-амина по изобретению представляет собой соединение формулы 1, его стереоизомер или смесь его стереоизомеров, или его N-оксид, или его фармацевтически приемлемую соль, где R′ и R′′ независимо представляют собой водород или метил.
В более предпочтительном воплощении один из R′ и R′′ представляет собой водород, а другой из R′ и R′′ представляет собой метил.
В другом более предпочтительном воплощении R′ представляет собой водород, и R′′ представляет собой метил.
В третьем более предпочтительном воплощении R′ представляет собой метил, и R′′ представляет собой водород.
В четвертом более предпочтительном воплощении R′ и R′′ оба представляют собой водород.
В пятом более предпочтительном воплощении R′ и R′′ оба представляют собой метил.
В наиболее предпочтительном воплощении производное фенокси-этил-амина по изобретению представляет собой
N-[2-(3-фтор-5-метилсульфонил-фенокси)этил]пропан-1-амин;
N-[2-(3-фтор-5-метилсульфонил-фенокси)этил]бутан-1-амин;
N-этил-2-(3-фтор-5-метилсульфонил-фенокси)этанамин;
N-[2-(3-фтор-5-метилсульфонил-фенокси)этил]-N-метил-пропан-1-амин;
N-[2-(3-хлор-5-метилсульфонил-фенокси)этил]пропан-1-амин;
N-[2-(3-фтор-5-метилсульфонил-фенокси)этил]бутан-2-амин;
N-[2-(3-фтор-5-метилсульфонил-фенокси)этил]циклопентамин;
N-[2-(3-фтор-5-метилсульфонил-фенокси)этил]-2-метил-бутан-2-амин;
N-[2-(3-фтор-5-метилсульфонил-фенокси)этил]циклобутамин;
N-[2-(3-фтор-5-метилсульфонил-фенокси)этил]пропан-2-амин;
1-[2-(3-фтор-5-метилсульфонил-фенокси)этил]пиперидин;
N,N-диэтил-2-(3-фтор-5-метилсульфонил-фенокси)этанамин;
N-1,1-ди-дейтерий-пропил-[2-(3-фтор-5-метансульфонил-фенокси)-этил]-амин;
N-(циклопропилметил)-N-{2-[3-фтор-5-(метилсульфонил)-фенокси]этил}амин;
N-{2-[3-фтор-5-(метилсульфонил)фенокси]этил}-N-изобутиламин;
1-[2-(3-фтор-5-метилсульфонил-фенокси)этил]азетидин;
1-(3-фтор-5-метилсульфонил-фенокси)-N-пропил-пропан-2-амин; или
2-(3-фтор-5-метилсульфонил-фенокси)-N-пропил-пропан-1-амин;
его стереоизомер или смесь его стереоизомеров, или его N-оксид, или его фармацевтически приемлемую соль.
Любая комбинация двух или более воплощений, описанных выше, входит в объем настоящего изобретения.
Определение терминов
В контексте данного изобретения C1-C4-алкил означает прямую цепь или разветвленную цепь из одного-четырех атомов углерода, включая, без ограничения, метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил.
Термин "аллил" относится к группе -CH2-CH=CH2.
Термин "лечение" здесь означает проведение лечения пациента и оказание помощи пациенту в целях борьбы с заболеванием, расстройством или состоянием. Этот термин охватывает замедление прогрессирования заболевания, расстройства или состояния, облегчение или ослабление симптомов и осложнений и/или излечение или устранение заболевания, расстройства или состояния. Пациентом, которого лечат, предпочтительно является млекопитающее, в частности человек.
Термины "заболевание", "состояние" и "расстройство" здесь используются взаимозаменяемым образом для определения состояния пациента, которое не является нормальным физиологическим состоянием человека.
Термин "лекарственное средство" здесь означает фармацевтическую композицию, пригодную для введения фармацевтически активного соединения пациенту.
Термин "фармацевтически приемлемый" здесь означает пригодный для нормальных фармацевтический применений, т.е. не вызывающий никаких вредных явлений у пациентов и т.д.
Термин "эффективное количество" здесь означает дозировку, которая достаточна для того, чтобы лечение пациента было эффективным по сравнению с отсутствием лечения.
Термин "терапевтически эффективное количество" соединения здесь означает количество, достаточное для излечения, облегчения или частичной остановки клинических проявлений данного заболевания и его осложнений. Количество, достаточное для осуществления этого, называется "терапевтически эффективным количеством". Эффективные количества для каждой цели будут зависеть от тяжести заболевания или повреждения, а также от массы тела и общего состояния субъекта. Понятно, что определение соответствующей дозировки может быть осуществлено путем рутинного экспериментирования, путем построения матрицы значений и разных точек тестирования в матрице, и это все находится в компетенции квалифицированного врача или ветеринара.
Фармацевтически приемлемые соли
Соединение по изобретению может быть предоставлено в любой форме, подходящей для назначенного введения. Подходящие формы включают фармацевтически (т.е. физиологические) приемлемые соли соединения по изобретению.
Такие фармацевтически приемлемые соли и общая методология их получения известны в данной области. Дополнительные подробности можно найти в Р Stahl et al, Handbook of Pharmaceutical Salts: Properties, Selection and Use; Wiley-VCH, 2002.
Химическое соединение по изобретению может быть предоставлено в растворимой форме или нерастворимой форме вместе с фармацевтически приемлемым растворителем, таким как вода, этанол и т.п. Растворимые формы также включают гидратированные формы, такие как моногидрат, дигидрат, гемигидрат, тригидрат, тетрагидрат и т.п. Как правило, растворимые формы считаются эквивалентными нерастворимым формам для целей данного изобретения.
Пространственные изомеры
Специалистам в данной области будет понятно, что соединения по настоящему изобретению могут существовать в разных стереоизомерных формах, включая энантиомеры, диастереоизомеры или цис-транс-изомеры.
Изобретение охватывает все такие изомеры и любые их смеси, включая рацемические смеси.
Рацемические формы могут быть разделены на оптические антиподы известными способами и методами. Одним из способов разделения энантиомерных соединений (включая энантиомерные промежуточные соединения) является, в случае если соединение представляет собой хиральную кислоту, использование оптически активного амина и высвобождение диастереомерной разделенной соли путем обработки кислотой. Другой способ разделения рацематов на оптические антиподы основан на хроматографии на оптически активной фазе. Рацемические соединения по настоящему изобретению могут быть разделены на их оптические антиподы, например, фракционной кристаллизацией D- или L-солей (тартратов, манделатов или камфорсульфонатов, например).
Химические соединения по настоящему изобретению могут быть также разделены путем образования диастереомерных амидов в результате реакции химических соединений по настоящему изобретению с оптически активной карбоновой кислотой, такой как (+) или (-) фенилаланин, (+) или (-) фенилглицин, (+) или (-) камфановая кислота, или путем образования диастереомерных карбаматов в результате реакции химического соединения по настоящему изобретению с оптически активным хлорформиатом или т.п.
Дополнительные методы разделения оптических изомеров известны в данной области. Такие методы включают методы, описанные в Jaques J, Collet А, & Wilen S, "Enantiomers, Racemates, and Resolutions", John Wiley and Sons, New York (1981).
Оптически активные соединения могут быть также получены из оптически активных исходных веществ.
N-оксиды
В контексте данного изобретения N-оксид означает оксидное производное третичного амина, включая атом азота ароматического N-гетероциклического соединения, неароматических N-гетероциклических соединений, триалкиламина и триалкениламина.
N-оксиды соединений по изобретению могут быть получены в результате окисления соответствующего азотсодержащего основания с использованием обычного окислителя, такого как перекись водорода, в присутствии кислоты, такой как уксусная кислота, при повышенной температуре или в результате реакции с надкислотой, такой как надуксусная кислота, в подходящем растворителе, например в дихлорметане, этилацетате или метилацетате, или в хлороформе или дихлорметане, с 3-хлорпероксибензойной кислотой.
Меченые соединения
Соединения по изобретению можно использовать в их меченой или немеченой форме. В контексте данного изобретения меченое соединение имеет один или более атомов, замещенных атомом, имеющим атомную массу или массовое число, отличающееся от атомной массы или массового числа, обычно встречающихся в природе. Мечение облегчает количественное детектирование указанного соединения.
Меченые соединения по изобретению могут быть полезны в качестве диагностических средств, радиоактивных индикаторов или контрольных агентов в различных диагностических методах и для визуализации рецепторов in vivo.
Меченый изомер по изобретению предпочтительно содержит по меньшей мере один радионуклид в качестве метки. Позитрон-испускающие радионуклиды все являются кандидатами для использования. В контексте данного изобретения радионуклид предпочтительно выбран из 2H (дейтерия), 3H (трития), 11C, 13C, 1C, 131I, 125I, 123I и 18F.
Физический метод детектирования меченого изомера по настоящему изобретению может быть выбран из позитронно-эмиссионной томографии (PET), однофотонной компьютерной томографии (SPECT), магнитно-резонансной томографии (MRS), магнитно-резонансной визуализации (MRI) и компьютерной аксиальной рентгеновской томографии (CAT) или их комбинаций.
Дейтерированные аналоги
Соединения по изобретению могут быть предоставлены в форме их дейтерированных аналогов. Дейтерий образует связи с углеродом с низкой частотой колебаний и поэтому более сильные, чем связи C-H. Следовательно, варианты лекарственных средств с "тяжелым водородом" (дейтерием) могут быть более устойчивыми к разложению и могут дольше присутствовать в живом организме.
Способы получения
Химические соединения по изобретению могут быть получены обычными способами химического синтеза, например способами, описанными в рабочих примерах. Исходные вещества для способов, описанных в настоящей заявке, известны или легко могут быть получены стандартными способами из коммерчески доступных химических реагентов.
Кроме того, одно соединение по изобретению может быть превращено в другое соединение по изобретению стандартными способами.
Конечные продукты описанных здесь реакций могут быть выделены стандартными методами, например экстракцией, кристаллизацией, дистилляцией, хроматографией и т.д.
Для специалистов в данной области очевидно, что для получения соединений по изобретению альтернативным и, в некоторых случаях, более удобным способом, отдельные стадии способа, упомянутого выше, можно выполнять в другом порядке, и/или отдельные реакции можно осуществлять на другой стадии общего пути синтеза (т.е. можно осуществлять химические превращения других промежуточных соединений в те промежуточные соединения, которые упомянуты здесь выше в связи с конкретной реакцией).
Биологическая активность
Соединения по настоящему изобретению обладают способностью модулировать дофаминергическую и опосредованную рецепторами N-метил-D-аспартата (NMDA) глутаматергическую нейротрансмиссию в коре головного мозга и базальных ганглиях, и как сами эти соединения, так и содержащие их фармацевтические композиции полезны в лечении многочисленных расстройств центральной нервной системы, включая как психиатрические, так и неврологические расстройства. В частности, соединения и содержащие их фармацевтические композиции можно применять в лечении расстройств ЦНС, при которых функции дофаминергической и глутаматергической системы нарушены вследствие прямого или опосредованного воздействия.
Соединения и композиции по изобретению можно применять для улучшения состояния при всех формах психоза, включая шизофрению и шизофреноформные и биполярные расстройства, а также психотические расстройства, вызванные лекарственными средствами, ятрогенные психозы и галлюцинации, и неятрогенные психозы и галлюцинации также можно лечить.
В конкретном воплощении заболевание, расстройство или состояние согласно изобретению представляет собой форму психоза, в частности шизофрению, шизофреноформное расстройство, биполярное расстройство или психотическое расстройство, вызванное лекарственным средством.
Тревожные расстройства и расстройства настроения, депрессию и обсессивно-компульсивное заболевание также можно лечить соединениями и композициями по изобретению.
Соединения, оказывающие модулирующее воздействие на дофаминергическую и глутаматергическую системы, можно также применять для улучшения двигательных и когнитивных функций и для лечения эмоциональных нарушений, связанных со старением, нейродегенеративных расстройств (например деменции и возрастного нарушения когнитивной функции) и расстройств развития (таких как расстройства аутистического спектра, ADHD (синдром дефицита внимания и гиперактивности), центрального паралича, синдрома Жиля де ля Туретта), а также после повреждения головного мозга. Такие повреждения головного мозга могут быть вызваны травматическими, воспалительными, инфекционными, неопластическими, сосудистыми, гипоксическими или метаболическими причинами или токсическими реакциями на экзогенные химические вещества, где экзогенные химические вещества выбраны из группы, состоящей из веществ, вызывающих пристрастие, фармацевтических соединений и токсинов окружающей среды.
Соединения и фармацевтические композиции по изобретению можно также применять при поведенческих расстройствах, которые обычно впервые диагностируются в младенческом, детском или подростковом возрасте, а также при расстройствах контроля побуждений.
Их можно также применять для лечения расстройств, связанных со злоупотреблением веществами, а также расстройств, характеризующихся неправильным употреблением пищи. Они также полезны для лечения состояния, выбранного из группы, состоящей из расстройств сна, сексуальных расстройств, расстройств приема пищи, ожирений и головных болей и других болей при состояниях, характеризующихся повышением мышечного тонуса.
Неврологические показания включают применение соединений и содержащих их фармацевтических композиций для улучшения ментальной и двигательной функции при болезни Паркинсона и при родственных паркинсоновских синдромах, дискинезиях (включая дискинезии, индуцированные L-DOPA (L-3,4-дигидроксифенилаланином), и поздние дискинезии) и дистониях. Их можно также применять для уменьшения тиков и тремора различного происхождения.
Их можно также применять в лечении болезни Гентингтона и других двигательных расстройств, а также двигательных расстройств, вызванных лекарственными средствами.
Синдром беспокойных ног и родственные расстройства, а также нарколепсию также можно лечить соединениями по изобретению.
Соединения и содержащие их фармацевтические композиции по настоящему изобретению можно применять для лечения болезни Альцгеймера или связанных с ней деменций.
В еще одном воплощении заболевание, расстройство или состояние согласно изобретению выбрано из группы, состоящей из шизофрении, дискинезии, индуцированных L-DOPA, и болезни Гентингтона.
Фармацевтические композиции
В другом аспекте изобретения предложены новые фармацевтические композиции, содержащие терапевтически эффективное количество соединения по изобретению.
Хотя соединение по изобретению для применения в терапии можно вводить в форме необработанного соединения, предпочтительно вводить активный ингредиент, возможно в форме физиологически приемлемой соли, в фармацевтической композиции вместе с одним или более адъювантами, эксципиентами, носителями, буферными агентами, разбавителями и/или другими обычными фармацевтическими вспомогательными веществами.
В предпочтительном воплощении изобретения предложены фармацевтические композиции, содержащие соединение по изобретению, или его фармацевтически приемлемую соль или производное, вместе с одним или более фармацевтически приемлемыми носителями и, возможно, другими терапевтическими и/или профилактическими ингредиентами, известными и используемыми в данной области. Носитель(и) должен(должны) быть "приемлемым(и)" в смысле совместимости с другими ингредиентами препарата и не наносящим(и) вреда реципиенту.
Фармацевтическую композицию по изобретению можно вводить любым обычным путем, который подходит для желательной терапии. Предпочтительные пути введения включают пероральное введение, в частности в форме таблетки, капсулы, драже, порошка или в жидкой форме, и парентеральное введение, в частности кожную, подкожную, внутримышечную или внутривенную инъекцию. Фармацевтическая композиция по изобретению может быть изготовлена специалистом с использованием стандартных способов и общепринятых методов, подходящих для целевого препарата. При желании, могут быть использованы композиции, изготовленные с возможностью длительного высвобождения активного ингредиента.
Фармацевтические композиции по изобретению могут представлять собой композиции, подходящие для перорального, ректального, бронхиального, назального, легочного, местного (включая буккальное и сублингвальное), трансдермального, вагинального или парентерального (включая кожную, подкожную, внутримышечную, интраперитонеальную, внутривенную, внутриартериальную, интрацеребральную, внутриглазную инъекцию или инфузию) введения, или композиции в форме, подходящей для введения ингаляцией или инсуффляцией, включая аэрозольное введение порошков и жидкости, или посредством систем длительного высвобождения. Примерами подходящих систем длительного высвобождения являются полупроницаемые матрицы из твердых гидрофобных полимеров, содержащие соединение по изобретению, которые могут быть изготовлены в форме сформованных изделий, например пленок или микрокапсул.
Химическое соединение по изобретению, вместе с традиционным вспомогательным веществом, носителем или разбавителем, может быть приготовлено в виде фармацевтических композиций и их стандартных лекарственных формах. Такие формы включают твердые формы, в частности, таблетки, заполненные капсулы, порошок и гранулы, и жидкие формы, в частности водные или неводные растворы, суспензии, эмульсии, эликсиры и капсулы, заполненные ими, все для перорального применения, суппозитории для ректального введения и стерильные инъекционные растворы для парентерального применения. Такие фармацевтические композиции и их стандартные лекарственные формы могут содержать традиционные ингредиенты в общепринятых пропорциях, с дополнительными активными соединениями или активными началами или без них, и такие стандартные лекарственные формы могут содержать любое подходящее эффективное количество активного ингредиента, соизмеримое с назначенным диапазоном используемых суточных дозировок.
Химическое соединение по настоящему изобретению можно вводить в разнообразных пероральных и парентеральных лекарственных формах. Для специалистов в данной области очевидно, что указанные ниже лекарственные формы могут содержать в качестве активного компонента либо химическое соединение по изобретению, либо фармацевтически приемлемую соль химического соединения по изобретению.
Для приготовления фармацевтических композиций из химического соединения по настоящему изобретению фармацевтически приемлемые носители могут быть либо твердыми, либо жидкими. Препараты в твердой форме включают порошки, таблетки, пилюли, капсулы, облатки, суппозитории и диспергируемые гранулы. Твердый носитель может представлять собой одно или более веществ, которые могут также действовать как разбавители, корригенты, солюбилизаторы, смазывающие вещества, суспендирующие агенты, связывающие вещества, консерванты, агенты, разрыхляющие таблетку, или инкапсулирующее вещество.
В порошках носитель является тонкодисперсным твердым веществом, которое находится в смеси с тонкодисперсным активным компонентом.
В таблетках активный компонент смешан в подходящих пропорциях с носителем, обладающим необходимой связывающей способностью, и спрессован в желаемую форму желаемого размера.
Порошки и таблетки предпочтительно содержат от пяти или десяти до примерно семидесяти процентов активного соединения. Подходящими носителями являются карбонат магния, стеарат магния, тальк, сахар, лактоза, пектин, декстрин, целлюлоза, крахмал, желатин, трагакант, метилцеллюлоза, натрийкарбоксиметилцеллюлоза, низкоплавкий воск, масло какао и т.п. Термин "приготовление" охватывает технологию приготовления активного соединения с инкапсулирующим веществом в качестве носителя, образующего капсулу, в которой активный компонент, с носителями или без них, окружен носителем, который находится, таким образом, в соединении с ним. Аналогично, охвачены облатки и леденцы. Таблетки, капсулы, пилюли, облатки и леденцы можно применять как твердые формы, подходящие для перорального введения.
Для приготовления суппозиториев низкоплавкий воск, такой как смесь глицеридов жирных кислот и масла какао, сначала расплавляют, и в нем до гомогенного состояния диспергируют активные компоненты путем перемешивания. Расплавленную гомогенную смесь затем вливают в удобные калиброванные формы, оставляют охлаждаться и одновременно затвердевать.
Композиции, подходящие для вагинального введения, могут быть представлены в виде пессариев, тампонов, кремов, гелей, паст, пенок или спреев, содержащих в дополнение к активному ингредиенту такие носители, которые известны в данной области как подходящие.
Жидкие препараты включают растворы, суспензии и эмульсии, например водные или водно-пропиленгликолевые растворы. Например, жидкие препараты для парентеральной инъекции могут быть приготовлены в виде растворов в водно-полиэтиленгликолевом растворе.
Химическое соединение по настоящему изобретению может быть приготовлено, таким образом, для парентерального введения (например инъекцией, например болюсной инъекцией или непрерывной инфузией), и может быть представлено в стандартной лекарственной форме в ампулах, предварительно заполненных шприцах, контейнерах для инфузии малого объема или многодозовых контейнерах с добавлением консерванта. Композиции могут также принимать такие формы, как суспензии, растворы или эмульсии в масляных или водных разбавителях, и могут содержать технологические агенты, такие как суспендирующие, стабилизирующие и/или диспергирующие агенты.
Альтернативно, активный ингредиент может быть приготовлен в порошковой форме, которую получат путем асептического выделения стерильного твердого вещества или лиофилизации из раствора, для разведения перед использованием подходящим разбавителем, например стерильной апирогенной водой.
Водные растворы, подходящие для перорального применения, могут быть приготовлены путем растворения активного компонента в воде и добавления подходящих красителей, корригентов, стабилизирующих и загущающих агентов, когда это целесообразно.
Водные суспензии, подходящие для перорального применения, могут быть приготовлены путем диспергирования тонкодисперсного активного компонента в воде с вязким веществом, таким как природные или синтетические камеди, смолы, метилцеллюлоза, натрийкарбоксиметилцеллюлоза или другие известные суспендирующие агенты.
Охвачены также препараты в твердой форме, предназначенные для быстрого превращения перед использованием в препараты в жидкой форме для перорального введения. Такие жидкие формы включают растворы, суспензии и эмульсии. В добавление к активному компоненту такие препараты могут содержать красители, корригенты, стабилизаторы, буферные агенты, искусственные и природные подсластители, диспергирующие агенты, загустители, солюбилизирующие агенты и т.п.
Для местного введения в эпидермис химическое соединение по изобретению может быть приготовлено в виде мазей, кремов или лосьонов, или в виде трансдермального пластыря. Мази и кремы могут быть приготовлены, например, на водной или масляной основе с добавлением подходящих загустителей и/или гелеобразующих агентов. Лосьоны могут быть приготовлены на водной или масляной основе и обычно будут содержать один или более эмульгаторов, стабилизаторов, диспергирующих агентов, суспендирующих агентов, загустителей или окрашивающих агентов.
Композиции, подходящие для местного введения в ротовую полость, включают леденцы, содержащие активный агент в корригированной основе, обычно сахарозе и аравийской камеди или трагаканте; пастилки, содержащие активный ингредиент в инертной основе, такой как желатин и глицерин или сахароза и аравийская камедь; и полоскания для рта, содержащие активный ингредиент в подходящем жидком носителе.
Растворы или суспензии наносят прямо в носовую полость обычными средствами, например капельницей, пипеткой или спреем. Композиции могут быть предоставлены в форме однократной дозы или многократных доз.
Введение в дыхательные пути можно осуществлять посредством аэрозольной композиции, в которой активный ингредиент находится в упаковке под давлением с подходящим пропеллентом, таким как хлорфторуглерод (CFC), например дихлордифторметан, трихлорфторметан или дихлортетрафторэтан, диоксид углерода или другой подходящий газ. Аэрозоль обычно может также содержать поверхностно-активное вещество, такое как лецитин. Дозу лекарственного средства можно контролировать посредством дозирующего клапана.
Альтернативно, активные ингредиенты могут быть предоставлены в форме сухого порошка, например порошковой смеси соединения в подходящей порошковой основе, такой как лактоза, крахмал, производные крахмала, такие как гидроксипропилметилцеллюлоза и поливинилпирролидон (PVP). Обычно порошковый носитель будет образовывать гель в носовой полости. Порошковая композиция может присутствовать в стандартной лекарственной форме, например в капсулах или картриджах, например желатиновых, или в блистерных упаковках, из которых порошок можно вводить посредством ингалятора.
В композициях, предназначенных для введения в дыхательные пути, включая интраназальные композиции, соединение обычно будет иметь небольшой размер частиц, например порядка 5 микрон или менее. Такого размера частиц можно достичь способами, известными в данной области, например микронизацией.
При желании, можно применять композиции, приготовленные с возможностью длительного высвобождения активного ингредиента.
Фармацевтические препараты предпочтительно находятся в стандартных лекарственных формах. В такой форме препарат разделен на стандартные дозы, содержащие подходящие количества активного компонента. Стандартная лекарственная форма может быть упакована, причем упаковка может содержать дискретные количества препарата, например упакованные таблетки, капсулы и порошки во флаконах или ампулах. Кроме того, стандартная лекарственная форма может представлять собой саму капсулу, таблетку, облатку или леденец, или она может представлять собой соответствующее их количество в упакованной форме.
Таблетки или капсулы для перорального введения и жидкости для внутривенного введения и непрерывной инфузии являются предпочтительными композициями.
В одном из воплощений, когда фармацевтическая композиция по изобретению предназначена для лечения пациентов со склонностью к злоупотреблению и симптомами отмены, возникающими в результате пристрастия к никотину, предусмотрены такие препараты, как жевательные резинки, пластыри, спреи, ингаляторы, аэрозоли и т.д.
Дополнительные подробности по методам приготовления и введения можно найти в самом последнем издании Remington′s Pharmaceutical Sciences (Maack Publishing Co., Easton, PA).
Терапевтически эффективная доза относится к количеству активного ингредиента, которое ослабляет симптомы или облегчает состояние. Терапевтическая эффективность и токсичность, например ED50 и LD50, могут быть определены по стандартным фармакологическим методикам в культурах клеток или экспериментальных животных. Соотношение доз, оказывающих терапевтический эффект и токсический эффект, называется терапевтическим индексом и выражается соотношением LD50/ED50. Предпочтительными являются фармацевтические композиции, показывающие большие терапевтические индексы.
Вводимая доза, разумеется, должна быть тщательно выверена в зависимости от возраста, массы тела и состояния индивидуума, которого лечат, а также от пути введения, лекарственной формы и режима и желаемого результата, и точную дозировку, разумеется, должен определять практикующий врач.
Реальная дозировка зависит от природы и тяжести заболевания, которое лечат, точного способа введения и формы введения, и находится на усмотрении лечащего врача, и ее можно варьировать путем титрования в соответствии с конкретными условиями данного изобретения для продуцирования желаемого терапевтического эффекта. Однако на данный момент подходящими для терапевтического лечения являются фармацевтические композиции, содержащие от примерно 0,1 до примерно 500 мг активного ингредиента на индивидуальную дозу, предпочтительно от примерно 1 до примерно 100 мг, наиболее предпочтительно от примерно 1 до примерно 10 мг.
Активный ингредиент можно вводить одной или несколькими дозами в сутки. Удовлетворительный результат в некоторых случаях может быть получен при такой низкой дозировке, как 0,1 мкг/кг в.в. (внутривенно) и 1 мкг/кг п.о. (перорально). На данный момент считается, что верхний предел диапазона дозировок составляет примерно 10 мг/кг в.в. и 100 мг/кг п.о. Предпочтительными диапазонами являются от примерно 0,1 мкг/кг до примерно 10 мг/кг/сутки в.в. и от примерно 1 мкг/кг до примерно 100 мг/кг/сутки п.о.
Способы лечения
Соединения по настоящему изобретению являются модуляторами дофаминергической и опосредованной рецепторами N-метил-D-аспартата (NMDA) глутаматергической нейротрансмиссии в коре головного мозга и базальных ганглиях и, следовательно, полезны для лечения целого ряда болезней с вовлечением модулирования дофаминергической и глутаматергической функции.
В еще одном аспекте изобретения предложен способ лечения, предупреждения или облегчения заболевания или расстройства или состояния организма живого животного, включая человека, причем заболевания или расстройства или состояния, которое реагирует на модулирование дофаминергической функции в центральной нервной системе, включающий введение в такой организм живого животного, включая человека, нуждающегося в этом, эффективного количества соединения по изобретению, его стереоизомера, или смеси его стереоизомеров, или его фармацевтически приемлемой соли.
Показания, предусмотренные согласно изобретению, представляют собой показания, изложенные выше.
На данный момент предполагается, что подходящие диапазоны дозировок составляют от 0,1 до 1000 миллиграммов в сутки, 10-500 миллиграммов в сутки и особенно 30-100 миллиграммов в сутки в зависимости, как обычно, от способа введения, формы, в которой вводят, показания, на которое направлено введение, субъекта и массы тела субъекта, а также предпочтения и опыта ответственного лечащего врача или ветеринара.
ПРИМЕРЫ
Изобретение дополнительно иллюстрируется приведенными ниже примерами и Схемой 1, которые никоим образом не ограничивают объем изобретения.
Схема 1
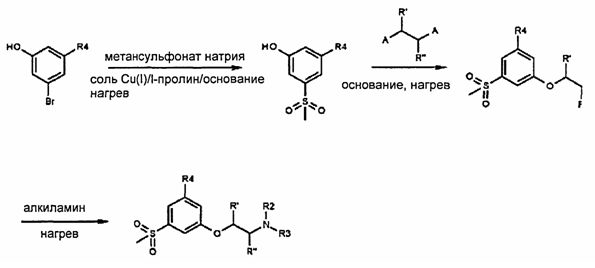
Заместители на Схеме 1 следующие: А представляет собой уходящую группу, a R2, R3, R4, R′ и R′′ такие, как определено выше.
Пример 1
N-[2-(3-ФТОР-5-МЕТИЛСУЛЬФОНИЛ-ФЕНОКСИ)ЭТИЛ]ПРОПАН-1-АМИН
Смесь 1-(2-бромэтокси)-3-фтор-5-метилсульфонил-бензола (2,65 г, 8,9 ммоль) и пропан-1-амина (5,24 мл, 63,7 ммоль) в этаноле (32 мл) разделяли на 3 аликвоты, каждую из которых нагревали под воздействием микроволнового излучения при 120°С в течение 30 мин. Реакционные смеси охлаждали до комнатной температуры, фильтровали и собирали вместе, затем летучие вещества выпаривали. После очистки колоночной хроматографией (этилацетат/метанол, от 1:0 до 1:1) получили указанное в заголовке соединение (2,14 г, 87%). Амин превращали в гидрохлоридную соль и подвергали перекристаллизации из смеси метанол/диэтиловый эфир: Т.пл. 191°C. МС (масс-спектрометрия) m/z (относительная интенсивность, 70 эВ) 275 0 (M+, 2), 246 (32), 73 (5), 72 (bp), 56 (11).
Пример 2
N-[2-(3-ФТОР-5-МЕТИЛСУЛЬФОНИЛ-ФЕНОКСИ)ЭТИЛ]БУТАН-1-АМИН
Получение осуществляли согласно Примеру 1, но одной порцией: 1-(2-бромэтокси)-3-фтор-5-метилсульфонил-бензол (0,3 г, 1,01 ммоль) и бутан-1-амин (0,61 мл, 0,5907 ммоль) в этаноле (4 мл). Выход: 290 мг (99%). Амин превращали в гидрохлоридную соль и подвергали перекристаллизации из смеси этанол/диэтиловый эфир: Т.пл. 204°C. МС m/z (относительная интенсивность, 70 эВ) 289 (M+, 1), 246 (35), 86 (bp), 56 (12), 87 (12).
Пример 3
N-ЭТИЛ-2-(3-ФТОР-5-МЕТИЛСУЛЬФОНИЛ-ФЕНОКСИ)ЭТАНАМИН
Получение осуществляли согласно Примеру 1, но одной порцией: 1-(2-бромэтокси)-3-фтор-5-метилсульфонил-бензол (0,321 г, 1,08 ммоль) и этанамин (4,3 мл, 8,6 ммоль 2 М в метаноле) в этаноле (6 мл). Выход: 255 мг (90%). Амин превращали в гидрохлоридную соль и подвергали перекристаллизации из смеси этанол/диэтиловый эфир: Т.пл. 200,8-201,1°C. МС m/z (относительная интенсивность, 70 эВ) 261 (M+, 3), 94 (6), 59 (12), 58 (bp), 56 (8).
Пример 4
N-[2-(3-ФТОР-5-МЕТИЛСУЛЬФОНИЛ-ФЕНОКСИ)ЭТИЛ]-N-МЕТИЛ-ПРОПАН-1-АМИН
Смесь из Примера 1 (0,54 г, 1,95 ммоль) в муравьиной кислоте (5,75 мл) и формальдегиде (40%-ный раствор, 5,1 мл) нагревали при 85°C в течение 5 ч. Раствору давали возможность достичь температуры окружающей среды, добавляли воду (5 мл) и диэтиловый эфир, фазы разделяли, и водную фазу подщелачивали добавлением водного раствора гидроксида натрия (5 М). Водную фазу экстрагировали дважды этилацетатом, объединенные органические фазы сушили (Na2SO4) и упаривали под давлением с получением неочищенного продукта, который затем очищали флэш-хроматографией (EtOAc:МеОН 100:0, затем постепенно изменяли соотношение до 0:100). Амин превращали в гидрохлоридную соль и подвергали перекристаллизации из смеси метанол/диэтиловый эфир: Т.пл. 128-130°C. МС m/z (относительная интенсивность, 70 эВ). 289 (M+, 1), 260 (26), 87 (6), 86 (bp), 58 (6).
Пример 5
N-[2-(3-ФТОР-5-МЕТИЛСУЛЬФОНИЛ-ФЕНОКСИ)ЭТИЛ]БУТАН-2-АМИН
Получение осуществляли согласно Примеру 1, но одной порцией: 1-(2-бромэтокси)-3-фтор-5-метилсульфонил-бензол (0,5 г, 1,68 ммоль) и втор-бутиламин 1,37 мл, 13,46 ммоль) в этаноле (5 мл). После очистки колоночной хроматографией (этилацетат/метанол, от 100:0 до 85:15) получили указанное в заголовке соединение (342 мг, 70%). Амин превращали в гидрохлоридную соль и подвергали перекристаллизации из смеси метанол/диэтиловый эфир: Т.пл. 166,5°C. МС m/z (относительная интенсивность, 70 эВ) 289 (M+, 1), 261 (25), 260 (bp), 86 (49), 70 (12).
Пример 6
N2-(3-ФТОР-5-МЕТИЛСУЛЬФОНИЛ-ФЕНОКСИ)ЭТИЛ]ЦИКЛОПЕНТАН-АМИН
Получение осуществляли согласно Примеру 1, но одной порцией: 1-(2-бромэтокси)-3-фтор-5-метилсульфонил-бензола (0,5 г, 1,68 ммоль) и циклопентиламин (1,34 мл, 13,46 ммоль) в этаноле (5 мл). После очистки колоночной хроматографией (этилацетат/метанол, от 100:0 до 85:15) получили указанное в заголовке соединение (443 мг, 87,4%). Амин превращали в гидрохлоридную соль и подвергали перекристаллизации из смеси метанол/диэтиловый эфир: Т.пл. 207,5°C. МС m/z (относительная интенсивность, 70 эВ) 301 (M+, 2), 272 (7), 99 (24), 98 (bp), 70 (7).
Пример 7
N-[2-(3-ФТОР-5-МЕТИЛСУЛЬФОНИЛ-ФЕНОКСИ)ЭТИЛ]-2-МЕТИЛ-БУТАН-2-АМИН
Получение осуществляли согласно Примеру 1, но одной порцией: 1-(2-бромэтокси)-3-фтор-5-метилсульфонил-бензол (0,5 г, 1,68 ммоль) и трет-амиламин (1,61 мл, 13,46 ммоль) в этаноле (5 мл). После очистки колоночной хроматографией (этилацетат/метанол, от 100:0 до 85:15) получили указанное в заголовке соединение (389 мг, 76,2%). Амин превращали в гидрохлоридную соль и подвергали перекристаллизации из смеси метанол/диэтиловый эфир: Т.пл. 190,2°C. МС m/z (относительная интенсивность, 70 эВ) 303 (M+, 0), 288 (21), 276 (10), 275 (41), 274 (bp), 84 (10).
Пример 8
N-[2-(3-ФТОР-5-МЕТИЛСУЛЬФОНИЛ-ФЕНОКСИ)ЭТИЛ]ЦИКЛОБУТАН-АМИН
Получение осуществляли согласно Примеру 1, но одной порцией: 1-(2-бромэтокси)-3-фтор-5-метилсульфонил-бензол (0,5 г, 1,68 ммоль) и циклобутиламин (1,17 мл, 13,46 ммоль) в этаноле (4 мл). После очистки колоночной хроматографией (этилацетат/метанол, от 100:0 до 85:15) получили указанное в заголовке соединение (400 мг, 82,7%). Амин превращали в гидрохлоридную соль и подвергали перекристаллизации из смеси метанол/диэтиловый эфир: Т.пл. 202°C. МС m/z (относительная интенсивность, 70 эВ) 287 (M+, 0), 260 (33), 259 (91), 216 (bp), 215 (23), 56 (73).
Пример 9
N-[2-(3-ФТОР-5-МЕТИЛСУЛЬФОНИЛ-ФЕНОКСИ)ЭТИЛ]ПРОПАН-2-АМИН
Получение осуществляли согласно Примеру 1, но одной порцией: 1-(2-бромэтокси)-3-фтор-5-метилсульфонил-бензол (0,5 г, 1,68 ммоль) и изопропиламин (1,15 мл, 13,46 ммоль) в этаноле (4 мл). После очистки колоночной хроматографией (этилацетат/метанол, от 100:0 до 85:15) получили указанное в заголовке соединение (421 мг, 90,9%). Амин превращали в гидрохлоридную соль и подвергали перекристаллизации из смеси метанол/диэтиловый эфир: Т.пл. 173°C. МС m/z (относительная интенсивность, 70 эВ) 275 (M+, 4), 261 (16), 260 (41), 73 (32), 72 (bp).
Пример 10
1-[2-(3-ФТОР-5-МЕТИЛСУЛЬФОНИЛ-ФЕНОКСИ)ЭТИЛ]ПИПЕРИДИН
Получение осуществляли согласно Примеру 1, но одной порцией: 1-(2-бромэтокси)-3-фтор-5-метилсульфонил-бензол (0,5 г, 1,68 ммоль) и пиперидин (1,33 мл, 13,46 ммоль) в этаноле (5 мл). После очистки колоночной хроматографией (этилацетат/метанол, от 100:0 до 85:15) получили указанное в заголовке соединение (480 мг, 94,7%). Амин превращали в гидрохлоридную соль и подвергали перекристаллизации из смеси метанол/диэтиловый эфир: Т.пл. 194,6°C. МС m/z (относительная интенсивность, 70 эВ) 301 (M+, 1), 99 (8), 98 (bp), 96 (4), 55 (5).
Пример 11
N,N-ДИЭТИЛ-2-(3-ФТОР-5-МЕТИЛСУЛЬФОНИЛ-ФЕНОКСИ)ЭТАНАМИН
Получение осуществляли согласно Примеру 1, но одной порцией: 1-(2-бромэтокси)-3-фтор-5-метилсульфонил-бензол (0,5 г, 1,68 ммоль) и диэтиламин (1,39 мл, 13,46 ммоль) в этаноле (4 мл). После очистки колоночной хроматографией (этилацетат/метанол, от 1:0 до 1:1) получили указанное в заголовке соединение (300 мг, 61,6%). Амин превращали в гидрохлоридную соль и подвергали перекристаллизации из смеси метанол/диэтиловый эфир: Т.пл. 172,3°C. МС m/z (относительная интенсивность, 70 эВ) 289 (M+, 1), 274 (6), 87 (6), 86 (bp), 58 (5).
Пример 12
N-[2-(3-ХЛОР-5-МЕТИЛСУЛЬФОНИЛ-ФЕНОКСИ)ЭТИЛ]ПРОПАН-1-АМИН
Получение осуществляли согласно Примеру 1, но одной порцией: 1-(2-бромэтокси)-3-хлор-5-метилсульфонил-бензол (0,5 г, 1,59 ммоль) и пропан-1-амин (1,04 мл, 12,76 ммоль) в этаноле (5 мл). Выход: 368 мг (79,1%). Амин превращали в гидрохлоридную соль и подвергали перекристаллизации из смеси этанол/диэтиловый эфир: Т.пл. 195-197°C. МС m/z (относительная интенсивность, 70 эВ) 291 (M+, 1), 264 (8), 262 (22), 73 (9), 72 (bp).
Пример 13
N-{2-[3-ФТОР-5-(МЕТИЛСУЛЬФОНИЛ)ФЕНОКСИ]ЭТИЛ}-N-ПРОПИЛАМИН D2
Смесь 2-[3-фтор-5-(метилсульфонил)фенокси]этанамина (0,3 г, 1,26 ммоль), пропил-4-метилбензолсульфоната D2 (1,04 мл, 12,76 ммоль) и карбоната калия (0,35 г, 2,52 ммоль) в ацетонитриле (10 мл) нагревали под воздействием микроволнового излучения при 120°C в течение 45 мин. Реакционные смеси охлаждали до комнатной температуры, фильтровали и собирали вместе, затем летучие вещества выпаривали. После очистки колоночной хроматографией (этилацетат/метанол, от 1:0 до 1:1) получили указанное в заголовке соединение (0,15 г, 44%). Амин превращали в гидрохлоридную соль и подвергали перекристаллизации из смеси метанол/диэтиловый эфир. МС m/z (относительная интенсивность, 70 эВ) 277 (M+, 2), 248 (26), 138 (3), 94 (3), 74 (bp).
Пример 14
N-(ЦИКЛОПРОПИЛМЕТИЛ)-N-{2-[3-ФТОР-5-(МЕТИЛСУЛЬФОНИЛ-ФЕНОКСИ]ЭТИЛ}АМИН
Получение осуществляли согласно Примеру 1, но одной порцией: 1-(2-бромэтокси)-3-фтор-5-метилсульфонил-бензол (0,05 г, 0,16 ммоль) и аминометилциклопропан (0,11 мл, 1,3 ммоль) в этаноле (5 мл). МС m/z (относительная интенсивность, 70 эВ) 287 (M+, 2), 246 (6), 138 (2), 84 (bp), 56 (14).
Пример 15
N-(2-[3-ФТОР-5-(МЕТИЛСУЛЬФОНИЛ)ФЕНОКСИ]ЭТИЛ}-N-ИЗОБУТИЛ-АМИН
Получение осуществляли согласно Примеру 1, но одной порцией: 1-(2-бромэтокси)-3-хлор-5-метилсульфонил-бензол (0,05 г, 0,16 ммоль) и изобутиламин (0,2 мл, 1,3 ммоль) в этаноле (5 мл). МС m/z (относительная интенсивность, 70 эВ) 289 (M+, 1), 246 (bp), 138 (2), 86 (70), 56 (17).
Пример 16
1-[2-(3-ФТОР-5-МЕТИЛСУЛЬФОНИЛ-ФЕНОКСИ)ЭТИЛ]АЗЕТИДИН
1-(2-Бромэтокси)-3-фтор-5-метилсульфонил-бензол (0,5 г, 1,6 ммоль), азетидин (0,23 мл, 3,2 ммоль) и карбонат калия (0,58 г, 4,2 ммоль) растворяли в ацетонитриле (10 мл). Эту смесь нагревали в герметичном контейнере при 120°C в течение 2 ч. Реакционную смесь охлаждали до комнатной температуры, добавляли CH2Cl2 (100 мл), твердое вещество отфильтровывали, и летучие вещества выпаривали. Амин превращали в соль с фумаровой кислотой и подвергали перекристаллизации из смеси метанол/диэтиловый эфир. МС m/z (относительная интенсивность, 70 эВ) 273 (M+, 1), 94 (5), 82 (3), 71 (5), 70 (bp).
Пример 17
1-(3-ФТОР-5-МЕТИЛСУЛЬФОНИЛ-ФЕНОКСИ)-N-ПРОПИЛ-ПРОПАН-2-АМИН
Триацетоксиборгидрид натрия (1,16 г, 5,51 ммоль) добавляли в перемешиваемую смесь 1-(3-фтор-5-метилсульфонил-фенокси)пропан-2-она (0,905 г, 3,67 ммоль), пропиламина (0,24 г, 4,04 ммоль), уксусной кислоты (0,5 г, 8,33 ммоль) и молекулярных сит (2 г, 4Å, 4-8 меш) в сухом 1,2-дихлорэтане (20 мл). Эту смесь перемешивали при температуре окружающей среды в течение 24 ч, суспензию фильтровали и добавляли карбонат натрия (100 мл, 10%-ный водный раствор). Водную фазу экстрагировали дихлорметаном (3×50 мл). Объединенную органическую фазу сушили над - Na2SO4 и упаривали. Выход после очистки флэш-хроматографией (этилацетат: метанол от 1:0 до 4:1) 0,3 г, 28%. МС m/z (относительная интенсивность, 70 эВ) 289 (M+, 1), 260 (9), 152 (4), 87 (6), 86 (bp).
Пример 18
2-(3-ФТОР-5-МЕТИЛСУЛЬФОНИЛ-ФЕНОКСИ)-N-ПРОПИЛ-ПРОПАН-1-АМИН
Толуолсульфонилхлорид (0,60 г, 3,1 ммоль) добавляли в перемешиваемую смесь 4-диметиламинопиридина (0,41 г, 3,4 ммоль), триэтиламина (0,53 г, 5,26 ммоль) и смеси 1-(3-фтор-5-метилсульфонил-фенокси)пропан-2-ола и 2-(3-фтор-5-метилсульфонил-фенокси)пропан-1-ола (0,65 г, 2,63 ммоль) в сухом дихлорметане (20 мл). Полученную смесь перемешивали при температуре окружающей среды в течение 5 ч, добавляли HCl (100 мл, 5%-ный водный раствор), водную фазу экстрагировали дихлорметаном (2×50 мл), объединенную органическую фазу промывали рассолом (50 мл) и карбонатом натрия (50 мл, 5%-ный водный раствор).
Полученное масло растворяли в этаноле (5 мл) и метаноле (5 мл) и добавляли пропиламин (3,24 мл, 39,51 ммоль). Полученную смесь нагревали до температуры дефлегмации в течение 15 ч. Неочищенную смесь концентрировали под вакуумом и очищали флэш-хроматографией (этилацетат:метанол от 1:0 до 5:1). Получили 0,52 г (в виде смеси 2-(3-фтор-5-метилсульфонил-фенокси)-N-пропил-пропан-1-амина и 1-(3-фтор-5-метилсульфонил-фенокси)-N-пропил-пропан-2-амина), 68%. МС m/z (относительная интенсивность, 70 эВ) 289 (M+, 1), 260 (3), 73 (5), 72 (bp), 70 (9).
Подготовительные примеры
Подготовительный пример 1
3-ФТОР-5-МЕТИЛСУЛЬФОНИЛ-ФЕНОЛ
Азот барботировали через раствор 3-бром-5-фторфенола (10 г, 51,31 ммоль) в сухом диметилсульфоксиде (70 мл) в течение 10 минут, после чего добавляли метансульфинат натрия (8,27 г, 76,96 ммоль), йодид меди(I) (5,86 г, 30,79 ммоль), L-пролин (7,09 г, 61,57 ммоль) и карбонат калия (4,25 г, 30,79 ммоль). Поток азота пропускали в течение еще 10 минут, после чего смесь нагревали при 100°C в течение 24 ч. Добавляли этилацетат (100 мл) и воду (100 мл) и фазы разделяли. Водную фазу экстрагировали этилацетатом (2×100 мл), объединенную органическую фазу промывали водным раствором хлорида лития (100 мл, 5%) и водным раствором соляной кислоты (100 мл, 5%), сушили (Na2SO4) и упаривали. После очистки колоночной хроматографией (этилацетат/изооктан, от 0:1 до 1:1) получили указанное в заголовке соединение (7,17 г, 73%). МС m/z (относительная интенсивность, 70 эВ) 190 (M+, 81), 175 (27), 128 (50), 111 (bp), 83 (58).
Подготовительный пример 2
1-(2-БРОМЭТОКСИ)-3-ФТОР-5-МЕТИЛСУЛЬФОНИЛ-БЕНЗОЛ
Смесь 3-фтор-5-метилсульфонил-фенола (4,2 г, 22,08 ммоль), 1,2-дибромэтана (24 мл, 27,7 ммоль) и карбоната калия (6,1 г, 44,2 ммоль) в ацетонитриле (36 мл) разделяли на 6 аликвот, каждую из которых нагревали под воздействием микроволнового излучения при 120°C в течение 30 мин. Реакционные смеси охлаждали до комнатной температуры, фильтровали и собирали вместе. Летучие вещества выпаривали, добавляли водный раствор карбоната натрия (100 мл, 10%) и смесь экстрагировали этилацетатом (2×100 мл). Объединенную органическую фазу промывали рассолом (75 мл), сушили (Na2SO4) и упаривали. После очистки колоночной хроматографией (этилацетат/изооктан, от 0:1 до 1:0) получили указанное в заголовке соединение (4,77 г, 73%). МС m/z (относительная интенсивность, 70 эВ) 298 (M+, 18), 296 (M+, 18), 109 (bp), 107 (99), 82 (15).
Подготовительный пример 3
трет-БУТИЛ-2-[3-ФТОР-5-(МЕТИЛСУЛЬФОНИЛ)ФЕНОКСИ]ЭТИЛ-КАРБАМАТ
Смесь трифенилфосфина (1,9 г, 7,3 ммоль) в сухом THF (20 мл) продували N2 (г) и по каплям добавляли DEAD (3,1 мл, 6,9 ммоль), затем добавляли 1,2-дибромэтан, затем 3-фтор-5-метилсульфонил-фенол (1,1 г, 6,1 ммоль) и затем boc-глицинол (1,0 мл, 6,1 ммоль) порциями. Эту смесь перемешивали при 70°C в течение 20 ч, охлаждали до комнатной температуры и добавляли воду и EtOAc. Водную фазу экстрагировали этилацетатом (2×100 мл). Объединенную органическую фазу промывали водным раствором NaOH (3 М, 50 мл) и рассолом (75 мл), сушили (Na2SO4) и упаривали. После очистки колоночной хроматографией (этилацетат/изооктан, от 0:1 до 1:0) получили указанное в заголовке соединение (1,4 г, 70%). МС m/z (относительная интенсивность, 70 эВ) 298 (M+, 18), 296 (M+, 18), 109 (bp), 107 (99), 82 (15).
Подготовительный пример 4
2-[3-ФТОР-5-(МЕТИЛСУЛЬФОНИЛ)ФЕНОКСИ]ЭТАНАМИН
В смесь трет-бутил-2-[3-фтор-5-(метилсульфонил)фенокси]этил-карбамата (1,4 г, 4,2 ммоль) в EtOH (18 мл) добавляли HCl (1,25 М в EtOH, 6 мл). Эту смесь перемешивали при температуре окружающей среды в течение 20 ч. Водную фазу подщелачивали добавлением водного раствора NaOH (1 M, 50 мл) и экстрагировали этилацетатом (2×100 мл). Объединенную органическую фазу промывали рассолом (75 мл), сушили (Na2SO4) и упаривали с получением указанного в заголовке соединения (0,77 г, 77%). МС m/z (относительная интенсивность, 70 эВ) 298 (M+, 18), 296 (M+, 18), 109 (bp), 107 (99), 82 (15).
Подготовительный пример 5
3-ХЛОР-5-МЕТИЛСУЛЬФОНИЛ-ФЕНОЛ
Получение осуществляли согласно Подготовительному примеру 1: 3-бром-5-хлорфенол (4,0 г, 19,3 ммоль), метансульфинат натрия (3,1 г, 28,9 ммоль), йодид меди(I) (2,2 г, 11,5 ммоль), L-пролин (2,7 г, 23,1 ммоль), карбонат калия (1,6 г, 11,6 ммоль), сухой диметилсульфоксид (70 мл). Выход: 3,7 г (92%). МС m/z (относительная интенсивность, 70 эВ) 206 (M+, 88), 191 (36), 144 (58), 127 (bp), 99 (76).
Подготовительный пример 6
1-(2-БРОМЭТОКСИ)-3-ХЛОР-5-МЕТИЛСУЛЬФОНИЛ-БЕНЗОЛ
Получение осуществляли согласно Подготовительному примеру 2: 3-хлор-5-метилсульфонил-фенол (2,8 г, 13,5 ммоль), 1,2-дибромэтан (12 мл, 135 ммоль), карбонат калия (3,7 г, 27,1 ммоль), ацетонитрил (15 мл). Выход: 2,1 г, 50%). МС m/z (относительная интенсивность, 70 эВ 314 (M+, 35), 312 (M+, 25), 206 (7), 126 (9), 109 (98), 107 (bp).
Подготовительный пример 7
2-(3-ФТОР-5-МЕТИЛСУЛЬФОНИЛ-ФЕНОКСИ)ПРОПАН-1-ОЛ
Смесь 1-бром-2-пропанола (70%) и 2-бром-1-пропанола (30%) (5,81 г, 41,8 ммоль) добавляли в раствор 3-фтор-5-метилсульфонил-фенола (1,59 г, 8,36 ммоль) и карбоната калия (2,37 г, 16,71 ммоль) в сухом диметилформамиде (10 мл). Эту смесь нагревали до 120°C в течение 20 часов, смесь оставляли охлаждаться до температуры окружающей среды и добавляли воду (100 мл). Водную фазу экстрагировали этилацетатом (3×100 мл), объединенную органическую фазу промывали LiCl (5%-ный водный раствор, 4×50 мл), рассолом (50 мл) и сушили над Na2SO4 и упаривали. После очистки флэш-хроматографией (изооктан:этилацетат от 1:0 до 3:2) получили 0,65 г (смесь 1-(3-фтор-5-метилсульфонил-фенокси)пропан-2-ола и 2-(3-фтор-5-метилсульфонил-фенокси)пропан-1-ола), 31%. МС m/z (относительная интенсивность, 70 эВ) 248 (M+, 14), 204 (54), 203 (bp), 191 (37), 190 (44).
Подготовительный пример 8
1-(3-ФТОР-5-МЕТИЛСУЛЬФОНИЛ-ФЕНОКСИ)ПРОПАН-2-ОН
1-Хлорацетон (95%, 2,66 г, 27,34 ммоль) добавляли в перемешиваемый раствор 3-фтор-5-метилсульфонил-фенола (80%, 1,3 г, 5,46 ммоль) и карбоната калия (2,26 г, 16,40 ммоль) в сухом диметилформамиде (10 мл). Эту смесь нагревали до 120°C в течение 20 минут, смесь оставляли охлаждаться до температуры окружающей среды и добавляли воду (100 мл). Водную фазу экстрагировали этилацетатом (3×100 мл), объединенную органическую фазу промывали LiCl (5%-ный водный раствор, 4×50 мл), рассолом (50 мл) и сушили над Na2SO4 и упаривали. После очистки флэш-хроматографией (изооктан:этилацетат от 1:0 до 3:2) выход составил 0,905 г, 67%. МС m/z (относительная интенсивность, 70 эВ) 246 (M+, 79), 204 (55), 203 (bp), 141 (30), 94 (67).
Биологическая активность
3,4-Дигидроксифенил-уксусная кислота (DOPAC) в полосатом теле
Повышенный оборот дофамина в терминальных участках восходящих дофаминергических проекций головного мозга млекопитающего может быть проиллюстрирован результатами измерения изменений биохимических показателей в головном мозге с характерными признаками антагонистов дофамина, например вызывающих увеличение концентрации метаболитов дофамина, таких как 3,4-дигидроксифенил-уксусная кислота (DOPAC), в полосатом теле.
Результаты теста представлены в Таблице 1.
Влияние на спонтанную локомоцию
Известно, что антагонисты дофаминовых рецепторов предшествующего уровня техники вызывают сильное снижение локомоторной активности (каталепсию). Воздействие соединений по изобретению на спонтанную локомоцию показано в Таблице 2.
Гиперлокомоция, индуцированная амфетамином
Увеличение активности после введения d-амфетамина является стандартной моделью гипердофаминергии. В этой модели дофаминергическая нейротрансмиссия увеличивается при системном введении d-амфетамина в дозе, которая является достаточно высокой, чтобы вызывать большое увеличение локомоторной активности. Способность соединения оказывать антагонистическое действие на эту гиперактивность отражает анти-дофаминергические свойства. К тому же, антагонизм гиперактивности, индуцированной d-амфетамином, широко используется в качестве стандартного анализа антипсихотической активности (см. Psychopharmacology 4th Generation of progress Chapter 68, p 793-795).
Воздействие соединений по изобретению на увеличение активности, индуцированной прямыми или непрямыми дофаминергическими агонистами, т.е. d-амфетамином и представителями того же рода, представлено в Таблице 3.
Снижение гиперлокомоции, индуцированной МК-801
Другая животная модель антипсихотической активности основана на введении глутаматного антагониста МК-801. Глутаматные антагонисты (т.е. NMDA-антагонисты) могут вызывать психозы у человека (см. Psychopharmacoiogy, 4th Generation of progress Chapter 101, p.1205 и 1207) и поведенческие отклонения у животных. Так, способность лекарственного средства воздействовать на шизофрению и психотические состояния может быть измерена с использованием поведенческих моделей на основе экспериментально вызываемых гипоглутаматергических состояний. В этом исследовании NMDA-антагонист МК-801 (0,7 мг/кг интраперитонеально) использовали для создания гипоглутаматергического состояния, при котором крысы проявляют ненормальное, гиперактивное поведение.
Результаты тестов для соединений по настоящему изобретению приведены в Таблице 4.
Увеличение экспрессии гена Arc
Известно, что дофаминергические системы головного мозга интенсивно взаимодействуют с другими трансмиттерными системами (см. Psychopharmacoiogy, 4th Generation of progress. Chapter 101, pages 1208-1209).
Для исследования потенциального воздействия соединений по настоящему изобретению на кортикальную и стриатальную, связанную с NMDA-рецептором синаптическую сигнализацию, индуцирование мРНК Arc оценивали сразу после введения соединения по настоящему изобретению. Arc (Arc/Arg3.1 - activity-regulated cytoskeleton-associated protein/activity regulated gene 3.1 (активность-регулируемый цитоскелет-ассоциированный белок/активность-регулируемый ген 3.1)); (Link Wet al.; Proc Natl Acad Sci USA 1995 92 5734-5738; Lyford GL et al; Neuron 1995 14 433-445))), представляет собой немедленно-ранний ген (IEG), индуцируемый синаптической активностью, и его экспрессия и локализация в синаптических сайтах запускается специфически активацией NMDA-рецепторов и в значительной степени связана с нейропластичностью (Steward О, Worley PF; Neuron 2001 30 227-240; Takashi Kawashima et al.; PNAS 2009 106 (1) 316-321; Clive R. Bramham et al.; Exp. Brain Res. 2010 200 125-140).
Результаты для соединения по изобретению представлены в Таблице 5.
Методы тестирования
Описанные ниже тесты были использованы для оценки соединений по изобретению.
Тест in vivo: Поведение
Поведенческую активность измеряли с использованием восьми мониторов для регистрации активности Digiscan (RXYZM (16) ТАО, Omnitech Electronics, Columbus, ОН, США), соединенных с анализатором Omnitech Digiscan и компьютером Apple Macintosh с цифровым интерфейсом (NB DIO-24, National Instruments, CIF). Каждый монитор для регистрации активности состоит из квадратной металлической рамки (Ш×Д = 40 см × 40 см), оснащенной фотодатчиками. Во время измерений поведенческой активности крыса находится в прозрачной акриловой коробке (Ш×Д×В=40×40×30 см), которая в свою очередь помещена в монитор для регистрации активности. Каждый монитор для регистрации активности оснащен тремя рядами инфракрасных фотодатчиков, и каждый ряд состоит из 16 датчиков. Два ряда размещены вдоль передней и боковой стороны на уровне пола клетки под углом 90°, а третий ряд размещен на высоте 10 см от пола для измерения вертикальной активности. Фотодатчики расположены на расстоянии 2,5 см друг от друга. Каждый монитор для регистрации активности установлен в идентичном заглушающем шум и ослабляющем свет боксе с лампой слабого освещения и вентилятором.
Программное обеспечение написано с использованием объектно-ориентированного программирования (LabVIEW®, National instruments, Austin, TX, США).
Поведенческие данные из каждого монитора для регистрации активности, отображающие положение (горизонтальный центр тяжести и вертикальная активность) животного в каждый момент времени, регистрируют с частотой замеров 25 Гц и собирают с использованием специально написанного приложения LABView™. Данные каждого сеанса записи сохраняют и анализируют в отношении пройденного расстояния. Каждый сеанс записи поведения длится 60 мин, начиная приблизительно через 4 минуты после инъекции тестируемого соединения. Аналогичные процедуры записи поведения применяют для крыс, ранее не подвергавшихся воздействию лекарственных средств, и крыс, ранее получавших лекарственное средство. Крысам, которым предварительно вводят d-амфетамин, вводят дозу 1,5 мг/кг интраперитонеально за 10 минут до сеанса записи в мониторе для регистрации активности. Крысам, которым предварительно вводят МК-801, вводят дозу 0,7 мг/кг интраперитонеально за 90 минут до сеанса записи в мониторе активности. Результаты представлены как число отсчетов / 60 минут или число отсчетов / 30 минут в произвольных единицах длины. Статистические сравнения проводят с использованием t-критерия Стьюдента относительно контрольной группы. У животных, которым предварительно вводили МК-801 или амфетамин, статистические сравнения проводят относительно контролей с МК801 или d-амфетамином соответственно.
Значение ED50 для снижения гиперлокомоции, индуцированной амфетамином, вычисляют путем подгонки кривой. Оценка базируется на 16 животных, которым предварительно вводили амфетамин, в диапазоне доз 0, 11, 33 и 100 пмоль/кг подкожно в единственном эксперименте. Вычисления базируются на пройденном расстоянии в течение последних 45 минут одного часа измерений. Расстояния нормализуют к контролю с амфетамином и подгоняют методом наименьших квадратов к функции "End-(End-Контроль)/(1+(доза/ED50)slope)". Четыре параметра (Контроль, End, ED50 и Slope) подгоняют с ограничениями: ED50>0, 0,5<Slope<3, End=0% от контроля. Ограничение фиксированным End делают, чтобы сконцентрировать внимание скорее на силе действия, чем на эффективности. Чтобы оценить доверительные уровни для параметров, подгонку повторяют 100 раз со случайной равномерно распределенной квадратичной массой (от 0 до 1) для каждого значения измерения. Представленные диапазоны ED50 охватывают 95% этих значений.
Значение ED50 для снижения МК-801-индуцированной гиперлокомоции вычисляют путем подгонки кривой. Оценка базируется на 16 животных, которым предварительно вводили МК-801, в диапазоне доз 0, 11, 33 и 100 пмоль/кг подкожно в единственном эксперименте. Вычисления основаны на пройденном расстоянии в течение последних 15 минут одного часа измерений. Расстояния нормализуют к контролю с МК-801 и подгоняют методом наименьших квадратов к функции "End-(End-Контроль)/(1+(доза/ED50)slope)". Четыре параметра (Control, End, ED50 и Slope) подгоняют с ограничениями: ED50>0, 0,5<Slope<3, End=0% от контроля. Ограничение фиксированным End делают, чтобы сконцентрировать внимание скорее на силе действия, чем на эффективности. Чтобы оценить доверительные уровни для параметров, подгонку повторяют 100 раз со случайной равномерно распределенной квадратичной массой (от 0 до 1) для каждого значения измерения. Представленные диапазоны ED50 охватывают 95% этих значений.
Тест in vivo: Нейрохимия
После сеансов записи поведенческой активности крыс обезглавливают, их головной мозг извлекают и помещают на охлаждаемую во льду чашку Петри. Лимбический передний мозг, полосатое тело, фронтальную кору головного мозга и остальные полусферические части иссекают из каждой крысы и замораживают. Каждую часть головного мозга затем анализируют на содержание в ней моноаминов и их метаболитов.
Трансмиттеры моноамины (NA (норадреналин), DA (дофамин), 5-НТ (серотонин)), а также их аминные метаболиты (NM (норметанефрин), 3-МТ (3-метокситирамин)) и кислотные метаболиты (DOPAC (3,4-дигидроксифенилуксусная кислота), 5-HIAA (5-гидроксииндолуксусная кислота), HVA (гомованилиновая кислота)) количественно определяют в гомогенатах ткани головного мозга методом ВЭЖХ-разделений и электрохимического детектирования.
Аналитический метод основан на двух хроматографических разделениях, предназначенных для аминов или кислот. Две хроматографические системы имеют общий автоинжектор с 10-ходовым клапаном и двумя пробоотборными петлями для одновременного впрыскивания на две системы. Обе системы оснащены обращено-фазовой колонкой (Luna C18(2), средний размер частиц 3 мкм, 50×2 мм внутренний диаметр, Phenomenex), и электрохимическое детектирование осуществляют при двух потенциалах на стеклоуглеродных электродах (MF-1000, Bioanalytical Systems, Inc.). Элюирующийся из колонки поток пропускают через Т-соединение в ячейку для детектирования или в выход для сброса. Эту процедуру осуществляют с помощью двух соленоидных клапанов, которые блокируют либо выход, либо детектор. Предотвращая попадание хроматографического фронта в детектор, добиваются лучших условий детектирования. Водная подвижная фаза (0,4 мл/мин) для кислотной системы содержит лимонную кислоту, 14 мМ; цитрат натрия, 10 мМ; МеОН, 15% (об./об.); и EDTA, 0,1 мМ. Потенциалы детектирования относительно стандарта Ag/AgCl составляют 0,45 и 0,60 В. Образующая ионные пары водная подвижная фаза (0,5 мл/мин) для аминной системы содержит лимонную кислоту, 5 мМ; цитрат натрия, 10 мМ; МеОН, 9% (об./об.); MeCN, 10,5% (об./об.); декансульфоновую кислоту, 0,45 мМ; и EDTA, 0,1 мМ. Потенциалы детектирования относительно стандарта Ag/AgCl составляют 0,45 и 0,65 В.
Значение ED50 для увеличения уровня DOPAC в полосатом теле вычисляют путем подгонки кривой. Оценка базируется на 40 животных в диапазоне доз 0, 3, 7, 11, 33 и 100 пмоль/кг подкожно в двух совместных экспериментах. Уровни DOPAC нормализуют к контролю и подгоняют методом наименьших квадратов к функции "End-(End-Контроль)/(1+(доза/ED50)slope)". Четыре параметра (Контроль, End, ED50 и Slope) подгоняют с ограничениями: ED50>0, 0,5<Slope<3, End=0% от контроля. Чтобы оценить доверительные уровни для параметров, подгонку повторяют 100 раз со случайной равномерно распределенной квадратичной массой (от 0 до 1) для каждого значения измерения. Представленные диапазоны ED50 охватывают 95% этих значений.
Тест in vivo: Пероральная биодоступность
Эксперименты выполняют через 24 часа после имплантации артериальных и венозных катетеров. Тестируемое соединение вводят перорально в дозе 12,5 пмоль/кг или внутривенно в дозе 5 пмоль/кг, используя венозные катетеры, n=3 в группе. Образцы артериальной крови затем берут в течение шести часов через 0, 3, 9, 27, 60, 120, 180, 240, 300 и 360 минут после введения тестируемого соединения. Пероральную биодоступность вычисляют как соотношение AUC (площадь под кривой), полученной после перорального введения, относительно AUC, полученной после внутривенного введения, для каждой крысы. Параметр AUC вычисляют следующим образом:
AUC: площадь под кривой зависимости концентрации в плазме от времени с момента времени ноль до последней измеренной концентрации (Clast), вычисленная логарифмическим/линейным трапецеидальным методом.
Уровни тестируемого соединения измеряют методом жидкостной хроматографии/масс-спектрометрии (ЖХ/МС) (Hewlett-Packard 1100MSD Series). Модуль ЖХ/МС включает в себя насосную систему четверного действия, вакуумный дегазатор, термостатируемый автосамплер, термостатируемый колоночный отсек, детектор на диодной матрице и камеру АД-ЭРИ (электрораспылительная ионизация при атмосферном давлении). Обработку данных выполняют с использованием системы HP ChemStation rev.A.06.03. Настройки прибора: MSD режим: мониторинг выбранных ионов (SIM); MSD полярность: положительная; температура газа: 350°C; осушающий газ: 13,0 л/мин; распыляющий газ: избыточное давление 50 фунт/кв.дюйм (345 кПа); напряжение на капилляре: 5000 В; напряжение на фрагментаторе: 70 В.
Аналитическая колонка: Zorbax eclipse XDB-C8 (4,6×150 мм, 5 мкм) при 20°C. Мобильная фаза представляет собой уксусную кислоту (0,03%) (растворитель A) и ацетонитрил (растворитель B). Скорость потока подвижной фазы составляет 0,8 мл/мин. Элюирование начинают при 12% растворителя B изократически в течение 4,5 мин с последующим линейным увеличением до 60% за 4,5 мин.
Процедура экстракции: образцы плазмы (0,25-0,5 мл) разводят водой до 1 мл и добавляют 60 пмоль (100 мкл) внутреннего стандарта (-)-OSU6241. pH доводят до значения 11 добавлением 25 мкл насыщенного раствора Na2CO3. После смешивания образцы экстрагируют 4 мл дихлорметана при встряхивании в течение 20 мин. Органический слой после центрифугирования переносят в меньшую пробирку и упаривают досуха в потоке азота. Остаток затем растворяют в 120 мкл подвижной фазы (уксусная кислота (0,03%): ацетонитрил, 95:5) для ЖХ/МС анализа (впрыскивают 10 мкл). Селективный ион (MH+) регистрируют для каждого примера, и MH+ 296 для (-)-OSU6241 ((3-[3-(этилсульфонил)фенил]-1-пропилпиперидин).
Стандартную кривую в диапазоне 1-500 пмоль получают путем добавления соответствующих количеств тестируемого соединения к пустым образцам плазмы крови.
Тест in vitro: Метаболическая стабильность в микросомах печени крысы
Микросомы печени крысы изолируют, как описано в Förlin L: Тох Appl Pharm. 54 (3) 420-430, 1980, с незначительными модификациями, а именно: перед гомогенизацией добавляют, в количестве 3 мл/г печени, буфер 0,1 М Na/K*PO4 с 0,15 М KCl, pH 7,4 (буфер 1), гомогенат центрифугируют в течение 20 минут вместо 15, надосадочную жидкость подвергают ультрацентрифугированию при 100000 g вместо 105000 g, и остаток после ультрацентрифугирования ресуспендируют в 1 мл/г печени 20% (об./об.) 87% глицерина в буфере 1.
1 мкл 0,2 или 1 мМ тестируемого вещества, разведенного в воде, и 10 мкл 15 мг/мл микросом печени крысы смешивают с 149 мкл буфера 1 при 37°C, и реакцию начинают добавлением 40 мкл 4,1 мг/мл NADPH. После инкубирования в течение 0 или 15 минут при 37°C в нагревательном блоке (LAB-LINE, MULTI-BLOK нагреватель или lab4you, TS-100 термошейкер при 700 об/мин) реакцию останавливают добавлением 100 мкл чистого ацетонитрила. Выпавший в осадок белок затем удаляют режекцией остатка после центрифугирования при 10000 g в течение 10 минут (Heraeus, Biofuge fresco) при 4°C. Тестируемое соединение анализируют методом ВЭЖХ/МС (Hewlett-Packard 1100MSD Series) на колонке Zorbax SB-C18 (2,1×150 мм, 5 мкм), используя 0,03% муравьиную кислоту и ацетонитрил в качестве подвижной фазы (градиент), или на колонке Zorbax Eclipse XDB-C18 (3×75 мм, 3,5 мкм), используя 0,03% уксусную кислоту и ацетонитрил в качестве подвижной фазы (градиент). 15-минутный оборот вычисляют как долю тестируемого соединения, элиминированную через 15 минут, выраженную в процентах от 0-минутных уровней, т.е. 100 × [концентрация тестируемого соединения в 0 минут - концентрация через 15 мин] / концентрация в 0 минут.
Получение микросом печени осуществляют, как описано в Förlin L: Тох Appl Pharm. 54, (3) 420-430, 1980. Протоколы для инкубирования с микросомами печени приведены в Crespi С L, DM Stressser J. Pharm. Тох. Meth. 44 325-331, 2000, и Renwick AB et al.; Xenobiotica 2001 31 (4) 187-204.
Микродиализ
Во всех экспериментах используют самцов крыс Sprague-Dawley с массой тела 220-320 г. Перед экспериментом животных размещают группами по пять животных в каждой клетке и обеспечивают им свободный доступ к воде и корму. После прибытия животные находятся в клетках по меньшей мере в течение одной недели до хирургического вмешательства и использования в экспериментах. Каждую крысу используют для микродиализа только один раз.
Авторы изобретения используют модифицированную версию Waters et al. (Waters et al.; J. Neural. Transm. Gen. Sect. 1994 98 (1) 39-55) I-образного зонда, который описан в Santiago M, Westerink BHC; Naunyn-Schm iedeberg s Arch. Pharmacol. 1990 342 407-414. Используемая для диализа мембрана представляет собой сополимер полиакрилонитрила и металилсульфоната натрия AN69 (HOSPAL; внешний диаметр/внутренний диаметр 310/220 мкм: Gambro, Lund, Швеция). В дорсальном полосатом теле авторы изобретения используют зонды с экспонированной длиной 3 мм мембраны для диализа, а в префронтальной коре головного мозга соответствующая длина составляет 2,5 мм. Крыс оперируют под ингаляционной изофлурановой анестезией, когда они были зафиксированы в стереотаксическом приборе Корфа. Координаты рассчитывают относительно брегмы; дорсального полосатого тела АР +1, ML ±2,6, DV -6,3; префронтальной коры головного мозга, АР +3,2, 8° ML ±1,2, DV -4,0, согласно Paxinos G, Watson C: The Rat Brain in Stereotaxic Coordinates; New York, Academic Press 1986. Зонд для диализа позиционируют в трепанационном отверстии при стереотаксическом наведении и фиксируют фосфатиновым зубным цементом.
Крыс размещают в клетках индивидуально за 48 ч до экспериментов с диализом, что дает им возможность восстановиться после хирургического вмешательства и сводит к минимуму риск взаимодействия лекарственного вещества с анестетиком во время последующих экспериментов. В течение этого периода времени крысы имеют свободный доступ к корму и воде. В день эксперимента крыс подсоединяют к микроперфузионному насосу через шарнирное соединение и снова помещают в клетку, в пределах которой они могут свободно передвигаться. Перфузионная среда представляет собой раствор Рингера, содержащий в ммоль/л: NaCl, 140; CaCl2, 1,2; KCl, 3,0; MgCl2, 1,0; и аскорбиновую кислоту, 0,04 (Moghaddam В, Bunney BS; J. Neurochem. 1989 53 652-654). Насос работает на скорости перфузии 2 мкл/мин, и образцы по 40 мкл собирают каждые 20 мин.
Каждый образец анализируют на двух системах ВЭЖХ. На автоинжекторе (СМА 200) с 10-ходовым клапаном (Valco C10WE), подсоединяющем две пробоотборные петли последовательно (4 мкл и 20 мкл), каждый образец диализата головного мозга впрыскивают в обе петли одновременно. При впрыскивании образец 20 мкл вводится в систему переключения колонок (обращено-фазовая в комбинации с обращено-фазовой ион-парной) для определения дофамина (DA), норадреналина (NA), норметанэфрина (NM), 3-метокситирамина (3-МТ) и серотонина (5-гидрокситрптамин, 5-НТ), а образец 4 мкл вводится в обращено-фазовую колонку для хроматографии кислотных метаболитов моноаминов, а именно 3,4-ди-гидроксифенилуксусной кислоты (DOPAC), гомованилиновой кислоты (HVA) и 5-гидроксииндолуксусной кислоты (5-HIAA). Токи, генерируемые двумя ЕС детекторами, преобразуют в цифровые данные и оценивают с использованием программного продукта Chromeleon (Dionex) на персональном компьютере. Время сменяемости образца составляет 4,5 мин, и два параллельных эксперимента обычно анализируются на данной системе одновременно. После эксперимента крыс отсоединяют от перфузионного насоса и обезглавливают. Их мозг быстро извлекают и фиксируют в растворе Neo-fix (Kebo-lab, Швеция) для последующего обследования локализации зонда. Методики, применяемые в этих экспериментах, одобрены Комитетом по этике проведения экспериментов с животными в Гетеборге, Швеция.
ПЦР
Тотальную РНК получают гуанидинизоцианатным методом, который изложен в Chomczynski Р & Sacchi N; Anal. Biochem., 1987, 162, 156-159. Осажденную РНК растворяют в воде MQ и хранят при -80°C. Концентрацию образца определяли на спектрофотометре NanoDrop ND-1000. Индекс качественной характеристики и индекс целостности рРНК измеряли с использованием Experion (Bio-Rad) на случайных образцах.
Обратную транскрипцию осуществляли с использованием набора Superscript III (Invitrogen). 1 мкг тотальной РНК подвергали обратной транскрипции с использованием 5 мкл 2×RT Reaction Mix (смесь реагентов для обратной транскрипции), 1 мкл RT Enzyme Mix (смесь ферментов для обратной транскрипции), объем доводили до 10 мкл воды, обработанной DEPC (диэтилпилокарбонатом). Добавляли 1 ед. E.coli RNase H. кДНК разводили в 40 раз и хранили при -20°C.
Для ПЦР измерений в реальном времени 0,7 мкл кДНК амплифицировали в 25 мкл смеси реагентов, содержащей 1 × ПЦР-буфер, 0,2 мМ dNTP, 3,7 мМ MgCl2, 0,15 мМ SYBR зеленого, 0,4 мкМ каждого праймера и 1 ед. JumpStart Taq ДНК-полимеразы. ПЦР в реальном времени измеряли на CFX96 (Biorad), используя следующую процедуру для всех генов: предварительное инкубирование в течение 60 с при 95°C, затем 40 циклов денатурации при 95°C в течение 10 с, отжиг при 56°C в течение 10 с и элонгация при 72°C в течение 10 с.
Последовательности праймеров были следующие:
Гипоксантинфосфорибозилтрансфераза (HPRT) (инвентарный номер AF001282)
Смысловая: 5′-GGCCAGACTTGTTGGATTTG-3′
Антисмысловая: 5′-CCGCTGTCTTTTAGGCTTTG-3′
Циклофилин А (инвентарный номер М19533)
Смысловая: 5′-GTCTCTTTTCGCCGCTTGCT-3′
Антисмысловая: 5′-TCTGCTGTCTTTGGAACTTTGTCTG-3′
Активность-регулируемый ген (Arc) (инвентарный номер U19866)
Смысловая: 5′-GTCCCAGATCCAGAACCACA-3′
Антисмысловая: 5′-CCTCCTCAGCGTCCACATAC-3′
Количества исходной ДНК определяли путем построения стандартной кривой для каждого гена, используя 6 последовательных 4-кратных разведении очищенных продуктов ПЦР. Корректные продукты ПЦР были подтверждены электрофорезом на агарозном геле (2%). Продукты ПЦР очищали с использованием набора для ПЦР очистки Qiagen (Valencia, CA, США). Все гены секвенировали в MWG, Германия, и рутинно посредством анализа кривой плавления.
Количества гена Arc нормализовали с использованием среднего геометрического из количеств двух оцениваемых генов "домашнего хозяйства" (HPRT и циклофилина А).
Значение ED50 для увеличения Arc в полосатом теле вычисляли путем подгонки кривой. Оценивание базировалось на 20 животных в диапазоне доз 0, 11, 33 и 100 пмоль/кг подкожно в единственном эксперименте. Уровни Arc нормализовали к контролю и подгоняли методом наименьших квадратов к функции "End - (End - Контроль) / (1+(доза/ED50)slope)". Четыре параметра (Контроль, End, ED50 и Slope) подгоняли с ограничениями: ED50>0, 0,5<Slope<3, 300<End<600% от контроля.
Для оценки доверительных уровней для параметров подгонку повторяли 100 раз со случайной равномерно распределенной квадратичной массой (от 0 до 1) для каждого измеренного значения. Представленные диапазоны ED50 охватывают 95% этих значений.
Значение ED50 для увеличения уровня Arc в (префронтальной) коре головного мозга оценивается как, безусловно, ниже самой низкой дозы.
Изобретение относится к производному фенокси-этил-амина формулы 1:
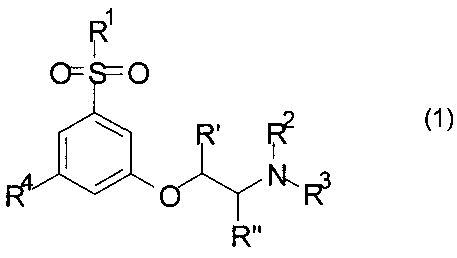
его стереоизомеру или смеси его стереоизомеров, или его дейтерированному аналогу, или его фармацевтически приемлемой соли. Значения радикалов следующие: R1 представляет собой СН3 или CF3; R2 выбран из группы, состоящей из С1-С4-алкила, аллила, С(СН3)2СН2СН3, группы СН2-циклопропил, циклобутила, циклопентила, CH2CH2CH2F, CH2CH2CHF2, CH2CH2F, 3,3,3-трифторпропила и 4,4,4-трифторбутила; R3 выбран из группы, состоящей из Н, СН3 и СН2СН3; или R2 и R3 вместе образуют СН2(СН2)СН2 или СН2(СН2)3СН2; R4 представляет собой F или Cl; R′ и R′′ независимо представляют собой водород или метил. Также предложены фармацевтическая композиция, применение для изготовления лекарственного средства и способ лечения. Производные фенокси-этил-амина применяются в качестве модуляторов дофаминергической и глутаматергической систем в головном мозге млекопитающих и полезны в лечении расстройств центральной нервной системы. 4 н. и 15 з.п. ф-лы, 5 табл., 26 пр.
1. Производное фенокси-этил-амина формулы 1:
его стереоизомер или смесь его стереоизомеров, или его дейтерированный аналог, или его фармацевтически приемлемая соль, где
R1 представляет собой СН3 или CF3;
R2 выбран из группы, состоящей из С1-С4-алкила, аллила, С(СН3)2СН2СН3, группы СН2-циклопропил, циклобутила, циклопентила, CH2CH2CH2F, CH2CH2CHF2, CH2CH2F, 3,3,3-трифторпропила и 4,4,4-трифторбутила; и
R3 выбран из группы, состоящей из Н, СН3 и СН2СН3; или
R2 и R3 вместе образуют СН2(СН2)СН2 или СН2(СН2)3СН2;
R4 представляет собой F или Cl; и
R′ и R′′ независимо представляют собой водород или метил.
2. Производное фенокси-этил-амина по п.1, его стереоизомер или смесь его стереоизомеров, или его дейтерированный аналог, или его фармацевтически приемлемая соль, где R1 представляет собой СН3.
3. Производное фенокси-этил-амина по п.1, его стереоизомер или смесь его стереоизомеров, или его дейтерированный аналог, или его фармацевтически приемлемая соль, где R2 выбран из группы, состоящей из С1-С4-алкила, С(СН3)2СН2СН3, группы СН2-циклопропил, циклобутила, циклопентила.
4. Производное фенокси-этил-амина по п.1, его стереоизомер или смесь его стереоизомеров, или его дейтерированный аналог, или его фармацевтически приемлемая соль, где R3 выбран из группы, состоящей из Н и СН3.
5. Производное фенокси-этил-амина по п.1, его стереоизомер или смесь его стереоизомеров, или его дейтерированный аналог, или его фармацевтически приемлемая соль, где R2 и R3 вместе образуют СН2(СН2)СН2 или СН2(СН2)3СН2.
6. Производное фенокси-этил-амина по п.1, его стереоизомер или смесь его стереоизомеров, или его дейтерированный аналог, или его фармацевтически приемлемая соль, где R4 представляет собой F.
7. Производное фенокси-этил-амина по п.1, или его дейтерированный аналог, или его фармацевтически приемлемая соль, где R' и R" независимо представляют собой водород.
8. Производное фенокси-этил-амина по п.1, представляющее собой
N-[2-(3-фтор-5-метилсульфонил-фенокси)этил]пропан-1-амин;
N-[2-(3-фтор-5-метилсульфонил-фенокси)этил]бутан-1-амин;
N-этил-2-(3-фтор-5-метилсульфонил-фенокси)этанамин;
N-[2-(3-фтор-5-метилсульфонил-фенокси)этил]-N-метил-пропан-1-амин;
N-[2-(3-хлор-5-метилсульфонил-фенокси)этил]пропан-1-амин;
N-[2-(3-фтор-5-метилсульфонил-фенокси)этил]бутан-2-амин;
N-[2-(3-фтор-5-метилсульфонил-фенокси)этил]циклопентамин;
N-[2-(3-фтор-5-метилсульфонил-фенокси)этил]-2-метил-бутан-2-амин;
N-[2-(3-фтор-5-метилсульфонил-фенокси)этил]циклобутамин;
N-[2-(3-фтор-5-метилсульфонил-фенокси)этил]пропан-2-амин;
1-[2-(3-фтор-5-метилсульфонил-фенокси)этил]пиперидин;
N,N-диэтил-2-(3-фтор-5-метилсульфонил-фенокси)этанамин;
N-1,1-ди-дейтерий-пропил-[2-(3-фтор-5-метансульфонил-фенокси)-этил]-амин;
N-(циклопропилметил)-N-{2-[3-фтор-5-(метилсульфонил)-фенокси]этил}амин;
N-{2-[3-фтор-5-(метилсульфонил)фенокси]этил}-N-изобутиламин;
1-[2-(3-фтор-5-метилсульфонил-фенокси)этил]азетидин;
1-(3-фтор-5-метилсульфонил-фенокси)-N-пропил-пропан-2-амин; или
2-(3-фтор-5-метилсульфонил-фенокси)-N-пропил-пропан-1-амин;
его стереоизомер или смесь его стереоизомеров, или его фармацевтически приемлемая соль.
9. Производное фенокси-этил-амина по п.1, представляющее собой N-[2-(3-фтор-5-метилсульфонил-фенокси)этил]пропан-1-амин,
или его фармацевтически приемлемая соль.
10. Производное фенокси-этил-амина по п.1, представляющее собой N-[2-(3-фтор-5-метилсульфонил-фенокси)этил]бутан-1-амин,
или его фармацевтически приемлемая соль.
11. Производное фенокси-этил-амина по п.1, представляющее собой N-этил-2-(3-фтор-5-метилсульфонил-фенокси)этанамин,
или его фармацевтически приемлемая соль.
12. Производное фенокси-этил-амина по любому из пп.1-11, его стереоизомер или смесь его стереоизомеров, или его дейтерированный аналог, или его фармацевтически приемлемая соль для применения в качестве лекарственного средства для лечения, предупреждения или облегчения заболевания или расстройства или состояния у млекопитающего, включая человека, причем заболевания, или расстройства, или состояния, которое реагирует на модулирование дофаминергической функции в центральной нервной системе.
13. Фармацевтическая композиция для лечения, предупреждения или облегчения заболевания, или расстройства, или состояния у млекопитающего, которое реагирует на модулирование дофаминергической функции в центральной нервной системе, содержащая терапевтически эффективное количество производного фенокси-этил-амина по любому из пп. 1-11, его стереоизомера или смеси его стереоизомеров, или его дейтерированного аналога, или его фармацевтически приемлемой соли вместе с по меньшей мере одним фармацевтически приемлемым носителем или разбавителем.
14. Применение производного фенокси-этил-амина по любому из пп.1-11, его стереоизомера или смеси его стереоизомеров, или его дейтерированного аналога, или его фармацевтически приемлемой соли для изготовления лекарственного средства для лечения, предупреждения или облегчения заболевания, или расстройства, или состояния у млекопитающего, включая человека, причем заболевания, или расстройства, или состояния, которое реагирует на модулирование дофаминергической функции в центральной нервной системе.
15. Применение по п.14, где заболевание, расстройство или состояние выбрано из группы психоза, шизофрении, шизофренического расстройства, биполярного расстройства, психотического расстройства, психотического расстройства, вызванного лекарственным средством, расстройства настроения, тревожного расстройства, депрессии, обсессивно-компульсивного заболевания, деменции, возрастного нарушения когнитивной функции, расстройств аутистического спектра, ADHD (синдром дефицита внимания и гиперактивности), центрального паралича, синдрома Жиля де ля Туретта, повреждения головного мозга, расстройства сна, сексуального расстройства, расстройства приема пищи, ожирения, головной боли, болей при состояниях, характеризующихся повышением мышечного тонуса, болезни Паркинсона, паркинсоновского синдрома, дискинезий, дискинезий, индуцированных L-DOPA (L-3,4-дигидроксифенилаланин), поздних дискинезий, дистонии, тиков и тремора, деменции, болезни Гентингтона, двигательного расстройства, вызванного лекарственными средствами, синдрома беспокойных ног, нарколепсии, болезни Альцгеймера и расстройства, связанного с болезнью Альцгеймера.
16. Применение по п.14, где заболевание, расстройство или состояние выбрано из группы психоза, шизофрении, шизофренического расстройства, биполярного расстройства, тревожного расстройства, депрессии, обсессивно-компульсивного заболевания, деменции, возрастного нарушения когнитивной функции, расстройств аутистического спектра, ADHD (синдром дефицита внимания и гиперактивности), синдрома Жиля де ля Туретта, расстройства приема пищи, болезни Паркинсона, паркинсоновского синдрома, дискинезий, индуцированных L-DOPA (L-3,4-дигидроксифенилаланин), поздних дискинезий, дистонии, болезни Гентингтона, болезни Альцгеймера.
17. Применение по п.14, где заболевание, расстройство или состояние выбрано из группы шизофрении, дискинезий, индуцированных L-DOPA, и болезни Гентингтона.
18. Применение по п.14, где заболевание, расстройство или состояние представляет собой дискинезии, индуцированные L-DOPA.
19. Способ лечения, предупреждения или облегчения заболевания, или расстройства, или состояния организма живого животного, включая человека, причем заболевания, или расстройства, или состояния, которое реагирует на модулирование дофаминергической функции в центральной нервной системе, включающий стадию введения в такой организм живого животного, нуждающегося в этом, терапевтически эффективного количества производного фенокси-этил-амина по любому из пп.1-11, его стереоизомера или смеси его стереоизомеров, или его дейтерированного аналога, или его фармацевтически приемлемой соли.
| WO 2007063789 A, 07.06.2007 | |||
| ПРОПОРЦИОНАЛЬНЫЙ ВОДОДЕЛИТЕЛЬ ДЛЯ ОРОСИТЕЛЬНЫХ СИСТЕМ | 0 |
|
SU363782A1 |
| WO 2002000602 A1, 03.01.2002 | |||
| RAUBO P | |||
| et al., Aminoalkyl phenyl sulfones - a novel series of 5-HT7 receptor ligands, Bioorganic & Medicinal Chemistry Letters, 2006, v | |||
| Устройство для электрической сигнализации | 1918 |
|
SU16A1 |
| Кипятильник для воды | 1921 |
|
SU5A1 |
| Украшение для пуговицы, запонки или броши | 1923 |
|
SU1255A1 |
| WO 2007072041 A1, 28.06.2007 | |||
| СПОСОБ ПОЛУЧЕНИЯ АМИНОФЕНОКСИПРОИЗВОДНЫХ | 1990 |
|
RU2032669C1 |

