Область изобретения
Настоящее изобретение относится к новым модуляторам нейротрансмиссии допамина и, более конкретно, к новым дизамещенным фенил-пиперидинам и их применению.
Предшествующий уровень техники
Допамин является нейротрансмиттером в мозге. После этого открытия, сделанного в 1950-х годах, функцию допамина в мозге активно исследовали. В настоящее время окончательно установлено, что допамин жизненно важен в нескольких аспектах функции мозга, включая моторную, когнитивную, сенсорную, эмоциональную и автономную функции (например, регуляция аппетита, температура тела, сон). Таким образом, модулирование допаминергической функции полезно в лечении множества разных расстройств, поражающих функции мозга. В действительности лекарственные средства, которые прямо или опосредованно действуют на центральные рецепторы допамина, широко применяются в лечении неврологических и психиатрических расстройств, например болезни Паркинсона и шизофрении. Однако доступные в настоящее время допаминергические фармацевтические препараты могут оказывать тяжелые побочные эффекты. Например, известно, что антагонисты допамина вызывают как моторные (эктрапирамидные побочные эффекты, EPS), так и ментальные побочные эффекты (например ангедонию, дисфорию и ухудшение когнитивной способности), и известно, что допаминергические агонисты вызывают дискинезии и психозы (Goodman and Gilman's the Pharmacological Basis of Therapeutics, 9th ed./McGraw-Hill, USA. Chapter 18, pp.407-416, Chapter 22, pp.509-512, pp.515-516).
Подход, выбранный многими исследователями для повышения эффективности и снижения побочных эффектов допаминергических фармацевтических препаратов, состоит в разработке новых лигандов рецепторов допамина, проявляющих селективность в отношении конкретных подтипов рецепторов допамина или региональную селективность. Еще один класс соединений, действующих через допаминовые системы мозга, составляют допаминергические стабилизаторы, которые, как было показано, полезны в лечении как неврологических, так и психиатрических расстройств (А.Ekesbo, PhD Thesis, Uppsala University, Sweden: Functional consequences of dopaminergic degeneration; clinical and experimental studies using a novel stabilizer of dopaminergic systems: Ekesbo et al., (-)-OSU6162 inhibits levodopa-induced dyskinesias in a monkey model of Parkinson's disease, Neuroreport, 8, 2567, 1997; Tedroff et al. Long-lasting improvement in motor function following (-)-OSU6162 in a patient with Huntington's disease. Neurology, 22:53:1605-6, 1999; Gefvert O. et al., (-)-OSU6162 induces a rapid onset of antipsychotic effect after a single dose. A double-blind placebo-controlled pilot study. Scandinavian Society for Psychopharmacology, 41st Annual Meeting, Copenhagen Denmark Nordic Journal of Psychiatry 54/2 93-94, April 2000: Carlsson et al., Annu. Rev. Pharmacol. Toxicol., 41, 237, 2001; Carlsson et al. Current Medicinal Chemistry, 11, 267, 2004).
Другим допаминергическим соединением, о котором сообщалось как о стабилизаторе допамин-серотониновой системы, а также как о частичном агонисте DA D2-рецепторов, является недавно выпущенное антипсихотическое соединение арипипразол (Burris et al., Pharm. Exp.Ther, vol. 302, 381, 2002). Соединения, являющиеся допаминергическими стабилизаторами, описаны также в WO 01/46145; WO 01/46146; Pettersson et al. The development of ACR16. A new class of dopaminergic stabilizers. Society for Neuroscience 32nd Annual Meeting, Abstract 2002, Vol.28 part 1 1028, Orlando USA, 2002, и Nyberg et al., Efficacy and tolerability of the new dopamine stabiliser ACR16 a randomised placebo-controlled add-on study in patients with schizophrenia, 12th BIENNIAL WINTER WORKSHOP ON SCHIZOPHRENIA, 7-13 February 2004, Davos, Switzerland.
Типичные фармакологические эффекты, характерные для допаминергических стабилизаторов, описанных в WO 01/46145, WO 01/46146 and Pettersson et al. 2002, можно суммировать как 1) усиление обратного захвата допамина в терминальных областях восходящих допаминергических проекций мозга млекопитающего, 2) отсутствие или только слабые поведенческие эффекты у крыс, не подвергавшихся лечению иным способом, и 3) ингибирование у крыс поведенческих эффектов, индуцированных психостимуляторами или психотомиметическими соединениями. В настоящем изобретении это называется профилем допаминергического стабилизатора.
Известно, что некоторые фармацевтически активные соединения, которые применяются в лечении неврологических и психиатрических расстройств (особенно антипсихотические соединения и соединения-антидепрессанты), могут оказывать нежелательные эффекты на те сердечные калиевые каналы, которые вовлечены в электрическую реполяризацию клеток сердца и которые обычно называются hERG-каналами (потенциал-зависимый калиевый канал, кодируемый геном ether-a-go-go человека) или 1 кг (быстро активируемый калиевый ток задержанного выпрямления). Лекарственные средства, которые блокируют эти каналы, могут вызывать желудочковую аритмию (двунаправленную желудочковую тахикардию, TdP), приводящую к внезапной смерти у здоровых во всех других отношениях субъектов. Признаком того, что лекарственное средство, возможно, оказывает нежелательные эффекты на реполяризацию сердца, является удлинение интервала QT на электрокардиограмме, которое считают суррогатным маркером риска TdP. Многие лекарственные средства были изъяты из продажи из-за неприемлемых побочных эффектов, относящихся к сердечной аритмии (J. Cardiovasc. Electrophysiol. 15, 475, 2004.; Eur. J. Pharm., 450, 37, 2002.; Cardiovascular Research, 58, 32, 2003).
Данное изобретение относится к области лечения млекопитающих, страдающих расстройствами ЦНС, у которых симптомы могут быть обусловлены допаминергическими функциями, где лечение включает введение указанному млекопитающему определенного количества соединения нового типа с профилем допаминергического стабилизатора. Кроме того, указанные соединения проявляют низкую аффинность к сердечным калиевым каналам, снижая риск серьезных кардиотонических побочных эффектов.
Описание предшествующего уровня техники
Ранее сообщалось о соединениях класса замещенных 4-(фенил)-N-алкил-пиперидинов. Среди этих соединений некоторые неактивны в ЦНС, некоторые проявляют серотонинергические или смешанные серотонинергические/допаминергические фармакологические свойства, а некоторые являются полными или неполными агонистами или антагонистами допаминовых рецепторов с высокой аффинностью к допаминовым рецепторам.
Известен целый ряд производных 4-фенил-пиперидина. В ЕР 0369887 раскрыты замещенные 4-(мета-трифторметилфенил)-1,2,3,6-тетрагидропиридины для лечения тревоги. В WO 00/03713 раскрыт способ лечения шизофрении и других дисфункций допаминовой системы с использованием замещенных 1-метил-4-фенил-1,2,3,6-тетрагидропиридинов.
Glennon et al. (патент США 6057371) заявляют способ лечения связанного с сигма-рецепторами расстройства ЦНС, включающий введение ариламинов, в том числе арилпиперидинов, которые либо не замещены, либо моно-замещены по арильному кольцу. Соединения проявляют высокую аффинность связывания с сигма-рецепторами. В WO 91/095954 утверждается, что термин «высокая аффинность» означает, что соединение демонстрирует IC50 менее 100 нМ в анализе против 3Н-DTG, описанном в Weber et al. Proc. Natl. Acad. Sci. (USA) 83: 8784-8788. В частности, в WO 91/095954 раскрыты композиции, относящиеся к «открытию, что определенные производные фенилалкил-амина, аминотетралина, пиперазина, пиперидина и родственных соединений проявляют высокую степень связывания с сигма-рецептором и неожиданно низкую степень связывания с РСР (фенциклидиновым) и DA (допаминовым) рецепторами» (смотри с.11, строки 33-36).
И в WO 91/095954, и в WO 93/00313 указано, что соединения должны иметь высокую аффинность связывания с сигма-рецептором, но не сообщается, что соединения фамакологически активны в отсутствие аффинности к сигма-рецепторам. Кроме того, клинические испытания, направленные на исследование свойств лигандов сигма-рецепторов у пациентов с шизофренией, не свидетельствуют ни об антипсихотической активности, ни об активности при любом другом расстройстве ЦНС. Два из наиболее всесторонне исследованных селективных антагонистов сигма-рецепторов, BW234U (Римказол) и BMY14802, не прошли клинические испытания у пациентов с шизофренией (Borison et al., 1991, Psychopharmacol Bull 27(2): 103-106; Gewirtz et al., 1994, Neuropsychopharmacology 10:37-40).
В WO 97/23216 раскрыты аналоги 4-замещенного пиперидина формулы:
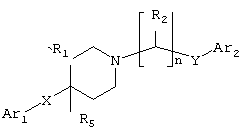
в которой R5 может быть выбран из ОН, а Ar1 может быть замещенным. Такие соединения применяют для лечения травм ЦНС, психоза и нейродегенеративных расстройств посредством, среди прочего, селективной блокады подтипов рецепторов NMDA (N-метил-D-аспарагиновая кислота).
В US 4485109 раскрыты соединения формулы:
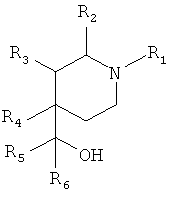
которые применяются в качестве психотерапевтических агентов, в частности в качестве антидепрессантов.
В ЕР 1177792 среди прочих раскрыты соединения, имеющие структуру:
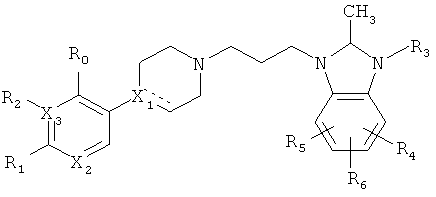 ,
,
обладающие допаминергической активностью, в частности активностью лигандов D4-рецепторов, и полезны для лечения расстройств, характеризующихся поиском новизны.
В WO 98/51668 раскрыты производные замещенного пиперидина формулы:
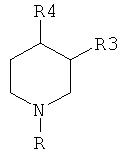 ,
,
которые обладают свойствами ингибиторов обратного захвата моноаминного нейротрансмиттера, то есть допамина, серотонина, норадреналина. Указано, что эти соединения полезны в лечении паркинсонизма, депрессии, псевдодеменции, ожирения, нарколепсии, наркомании и/или злоупотребления лекарственными средствами, синдрома дефицита внимания с гиперактивностью, сенильной деменции или нарушений памяти.
Известно также, что соединения формул II (WO 01/46145) и III (WO 01/46146) обладают свойствами допаминергических стабилизаторов.
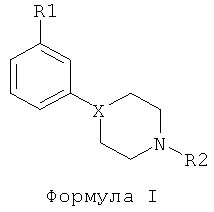
В формуле I:
Х представляет собой, среди прочего, СН; R1 выбран из группы, состоящей из OSO2CF3, OSO2CH3, SOR3, SO2R3, COR3, CN, NO2, CONHR3, CF3 (при условии, что Х представляет собой СН или С), F, Cl, Br, I (где R3 определен ниже);
R2 выбран из группы, состоящей из С1-С4алкила, аллила, CH2SCH3, CH2CH2OCH3, CH2CH2CH2F, CH2CF3, 3,3,3-трифторпропила, 4,4,4-трифторбутила или -(CH2)-R4 (где R4 определен ниже);
R3 выбран из группы, состоящей из С1-С3алкила, CF3 или N(R2)2;
R4 выбран из группы, состоящей из С3-С6циклоалкила, 2-тетрагидрофурана, 3-тетрагидрофурана.
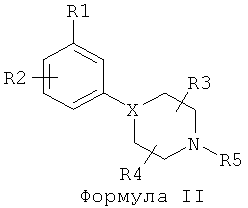
В формуле II:
Х представляет собой, среди прочего, СН; R1 выбран из группы, состоящей из OSO2CF3, OSO2CH3, SOR7, SO2R7, COR7, CN, NO2, CONHR3, CF3, F, Cl, Br, I (где R3 определен ниже), 3-тиофена, 2-тиофена, 3-фурана, 2-фурана;
R2 выбран из группы, состоящей из F, Cl, Br, I, CN, CF3, СН3, ОСН3, ОН, NH2;
R3 и R4 независимо представляют собой Н или С1-C4алкил;
R5 выбран из группы, состоящей из С1-С4алкила, аллила, CH2SCH3, CH2CH2OCH3, CH2CH2CH2F, CH2CF3, 3,3,3-трифторпропила, 4,4,4-трифторбутила или -(CH2)-R6;
R6 выбран из группы, состоящей из С3-С6циклоалкила, 2-тетрагидрофурана, 3-тетрагидрофурана;
R7 выбран из группы, состоящей из C1-С3алкила, CF3 или N(R4)2.
Однако ни в WO 01/46145 (Формула I), ни в WO 01/46146 (Формула II) не приведены фармакологические данные для 3,5-дизамещения в фенольном кольце, раскрытого в настоящем изобретении. Приведенная ниже структура известна из примера синтеза, приведенного в WO 01/46146 (Пример 44. 4-[3-фтор-5-(трифторметил)фенил-]1-пропилпиперидин):
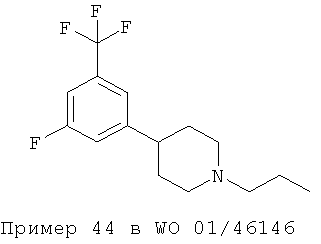
Кроме того, ни в WO 01/46145 (Формула I), ни в WO 01/46146 (Формула II) нет никаких указаний для получения эффективных допаминергических стабилизаторов.
Сохраняется потребность в новых фармацевтически активных соединениях, в частности полезных в лечении расстройств центральной нервной системы, обладающих повышенной эффективностью в качестве допаминергических стабилизаторов. Желательно также, чтобы любое такое фармацевтически активное соединение оказывало меньше побочных эффектов, в частности, относящихся к сердечной аритмии.
Краткое описание сущности изобретения
Задача настоящего изобретения заключается в том, чтобы предоставить новые фармацевтически активные соединения, в частности, полезные в лечении расстройств центральной нервной системы, обладающие повышенной эффективностью в качестве допаминергических стабилизаторов (смотри Таблицу 1, колонка 1) с низкой склонностью к блокированию hERG-канала (смотри Таблицу 1, колонка 2). Эти соединения имеют определенные преимущества, относящиеся к уменьшению побочных эффектов, в частности сердечных побочных эффектов.
Согласно настоящему изобретению 3,5-дизамещение неожиданно повышает силу действия и эффективность по сравнению с альтернативными картинами замещения (например, 3,4-дизамещение, где в положении 4 находится галоген) или монозамещением (в положении 3). Кроме того, соединения по настоящему изобретению проявляют пониженную аффинность к hERG-каналу по сравнению с соединениями предшествующего уровня техники.
Вещества по настоящему изобретению были испытаны в биологических тестах на крысах, и было обнаружено, что они действуют преимущественно на допаминергические системы в мозге. Они воздействуют на биохимические показатели в мозге с характерными признаками антагонистов допамина. Однако вещества по изобретению не оказывают ингибирующего воздействия на спонтанную двигательную активность в широком диапазоне доз. Кроме того, вещества по изобретению могут вызывать легкую поведенческую активацию, особенно когда исходная двигательная активность незначительна. Однако вещества по настоящему изобретению ингибируют поведенческую активацию, вызванную психостимуляторами и психотомиметиками.
Вещества по настоящему изобретению проявляют низкую эффективность ингибирования hERG-канала, которую определяли по значениям IC50 в анализе Rapid ICE (подробности изложены в экспериментальном разделе), что указывает на низкий риск удлинения интервала QT и аритмии у человека.
Подробное описание изобретения
Настоящее изобретение относится к новым пиперидинам в форме свободного основания или их фармацевтически приемлемых солей, к фармацевтическим композициям, содержащим указанные соединения, и к применению указанных соединений в изготовлении фармацевтических препаратов, являющихся нейротрансмиттерами допамина, и в терапии.
Более точно, настоящее изобретение относится к соединениям пиперидина формулы 1;
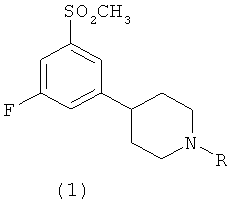 ,
,
где R выбран из группы, состоящей из C1-С3алкилов и аллила, и их фармацевтически приемлемым солям.
В конкретных воплощениях R выбран из группы, состоящей из н-пропила и этила.
Еще один аспект изобретения относится к способу лечения расстройств центральной нервной системы путем введения терапевтически активного количества соединения формулы 1 или его фармацевтически приемлемой соли млекопитающему, включая человека, страдающему расстройством центральной нервной системы. Дополнительно, настоящее изобретение относится к способу лечения любых расстройств, перечисленных в данном описании, путем введения терапевтически активного количества соединения формулы 1 или его фармацевтически приемлемой соли млекопитающему, включая человека, страдающему указанным расстройством.
Наличие двух заместителей на арильном кольце таких соединений - одного в положении 3 (мета 1), а другого в положении 5 (мета 2) - повышает их эффективность модулирования нейротрансмиссии допамина. Беспрецедентное увеличение эффективности этих 3,5-дизамещенных соединений по сравнению с монозамещенными или 3,4-дизамещенными соединениями иллюстрируется в Таблице 1.
Обнаружено также, что 3,5-дизамещение в соединениях по настоящему изобретению снижает побочные эффекты, относящиеся к сердечной аритмии, которые определяют по воздействию этих соединений на hERG калиевый канал (Rapid Ice). Беспрецедентное снижение побочных эффектов таких замещенных соединений иллюстрируется в Таблице 1.
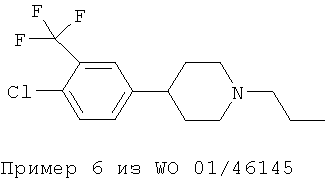
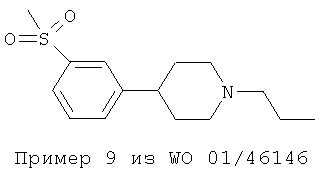
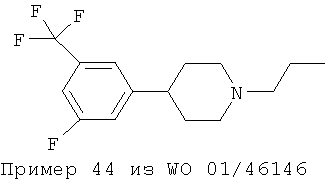
Важным наблюдением является то, что присутствие заместителей F и SO2CH3 в положениях мета 1 и мета 2 в фенильном кольце повышает эффективность и силу действия допаминергического стабилизатора по сравнению с моно- или 3,4-дизамещением (например Пример 6 из WO 01/46145 и Пример 9 из WO 01/46146), а также снижает аффинность к hERG-каналу. Такой результат не мог быть предсказуемым как общее правило.
Задача настоящего изобретения заключается в том, чтобы предоставить новые соединения для терапевтического применения и, более точно, соединения для модулирования допаминергических систем в мозге млекопитающих, включая мозг человека. Предпочтительно такие соединения оказывают меньше побочных эффектов, относящихся к ингибированию калиевых каналов сердца.
Задача изобретения также заключается в том, чтобы предоставить соединения, обладающие терапевтическими эффектами после перорального введения.
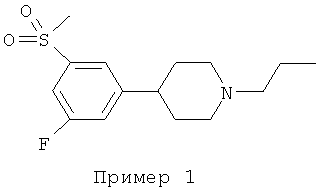
Предпочтительными замещенными структурами являются:
1-этил-4-[3-фтор-5-(метилсульфонил)фенил]пиперидин,
4-[3-фтор-5-(метилсульфонил)фенил]-1-пропилпиперидин,
1-аллил-4-[3-фтор-5-(метилсульфонил)фенил]пиперидин.
Соединения и композиции по настоящему изобретению обладают допамин-модулирующими свойствами и полезны в лечении многих расстройств центральной нервной системы, включая как психиатрические, так и неврологические расстройства. В частности, соединения и содержащие их фармацевтические композиции можно применять в лечении расстройств ЦНС, при которых функция допаминергической системы нарушена по прямым или косвенным причинам.
Соединения и композиции по настоящему изобретению можно применять для улучшения состояния при всех формах психоза, включая шизофрению и шизофреноформные расстройства, а также психотические расстройства, вызванные лекарственными средствами, и биполярное расстройство. Их можно применять также в лечении состояния, выбранного из группы, состоящей из ятрогенных и неятрогенных психозов и галлюцинозов.
Расстройства настроения и тревожные расстройства, включая депрессию и обсессивно-компульсивное расстройство, также можно лечить соединениями и композициями по изобретению.
Соединения, обладающие модулирующими эффектами в отношении допаминергических систем, можно применять также для улучшения когнитивных функций и для лечения эмоциональных нарушений, связанных со старением, нейродегенеративных расстройств (например, деменции и возрастного ухудшения когнитивной функции) и расстройств развития (таких как расстройства типа аутизма, ADHD, корковый паралич, синдром Жиля де ля Туретта), а также после повреждения мозга. Такое повреждение мозга может быть вызвано травматическими, воспалительными, инфекционными, неопластическими, сосудистыми, гипоксическими или метаболическими факторами или токсическими реакциями на экзогенные химические вещества, где экзогенные химические вещества выбраны из группы, состоящей из веществ, которыми злоупотребляют, фармацевтических соединений и токсинов окружающей среды. Соединения и содержащие их фармакологические композиции полезны для лечения состояния, выбранного из группы, состоящей из расстройств сна, сексуальных расстройств, расстройств приема пищи, ожирения, а также головных болей и других болей при состояниях, характеризующихся повышенным мышечным тонусом. Их также можно применять в лечении болезни Альцгеймера или родственных расстройств с деменцией.
Соединения и композиции по изобретению можно применять также при поведенческих расстройствах, обычно впервые диагностированных в младенческом, детском или пубертатном возрасте, а также при расстройствах контроля мотивации.
Их можно применять также для лечения расстройств, связанных со злоупотреблением веществами, а также расстройств, характеризующихся злоупотреблением пищи.
Неврологические показания включают применение соединений и содержащих их композиций для улучшения ментальной и моторной функции при болезни Паркинсона, дискинезиях (включая дискинезии, вызванные L-DOPA) и родственных паркинсонических синдромах. Их также можно применять для улучшения состояния при тиках и треморе различного происхождения. Более того, их можно применять для снятия боли при состояниях, характеризующихся повышенным мышечным тонусом.
Их можно применять также в лечении болезни Гентингтона и других двигательных расстройств, а также двигательных расстройств, вызванных лекарственными средствами. Синдром беспокойных ног и родственные расстройства, а также нарколепсию тоже можно лечить соединениями по изобретению.
Настоящее изобретение также относится к применению соединения формулы 1, указанной выше, где R выбран из группы, состоящей из С1-С3алкилов и аллила, или его фармацевтически приемлемой соли в изготовлении фармацевтически активных препаратов для лечения расстройства центральной нервной системы. Расстройство центральной нервной системы может быть одним или более чем одним расстройством, описанным выше. В конкретном воплощении этого применения R выбран из группы, состоящей из н-пропила и этила.
Было показано, что соединения по настоящему изобретению проявляют свойства допаминергического стабилизатора с улучшенной эффективностью (Таблица 1). Они оказывают эффекты на биохимические показатели в мозге с характерными признаками антагонистов допамина, например, продуцируя увеличение концентрации допаминных метаболитов. У крыс соединение Примера 1 повышает концентрацию 3,4-дигидроксифенилуксусной кислоты (DOPAC) в полосатом теле до 318% по сравнению с контролем при 100 мкмоль/кг п.к. (подкожно). Соединение Примера 2 повышает концентрацию DOPAC до 292% при 100 мкмоль/кг п.к.
Соединения по данному изобретению не оказывают влияния на спонтанную двигательную активность в широком диапазоне доз (1-100 мкмоль/кг п.к.).
В некоторых случаях, в частности когда исходная активность низкая, они могут вызывать незначительную поведенческую активацию. Поведенческая активация ограничивается, не достигая полного повышения активности, вызванного прямыми или непрямыми допаминергическими агонистами. С другой стороны, предпочтительные вещества снижают повышение активности, вызванное прямыми или непрямыми допаминергическими агонистами, то есть d-амфетамином и представителями того же класса соединений (Таблица 1).
Таким образом, соединения по данному изобретению проявляют свойства допаминергического стабилизатора с повышенной или сохраненной эффективностью (Таблица 1) по сравнению с соединениями формулы I и II. Кроме того, за счет конкретного характера замещения снизилась эффективность ингибирования HERG-канала.
Учитывая вовлечение допамина в большое количество функций ЦНС и клинические недостатки доступных в настоящее время фармацевтических препаратов, действующих на допаминергические системы, новый класс допаминергических модуляторов, представленных в этом изобретении, может превзойти известные в настоящее время допаминергические соединения в лечении некоторых расстройств, связанных с дисфункциями ЦНС, по показателям эффективности, а также по сниженным побочным эффектам.
Было показано, что соединения по настоящему изобретению проявляют высокую метаболическую стабильность в микросомах печени крыс, измеренную как обновление за 15 минут (Пример 1-5%, Пример 2-0%), и высокую пероральную биодоступность у крыс, подтвержденную Примером 2 (приблизительно 85%).
Таким образом, эти соединения подходят для изготовления фармацевтических препаратов для перорального введения. В предшествующем уровне техники отсутствует информация по получению соединений с таким воздействием на поведение и допаминергические системы в мозге.
Фармакология
Имеется доказательство того, что при психиатрических и неврологических заболеваниях нарушается допаминергическая нейротрансмиссия в ЦНС. Во многих случаях, например при шизофрении, болезни Паркинсона, болезни Гентингтона, биполярном расстройстве и при деменции, фармакотерапии, основанные на антагонизме или агонизме рецепторов допамина, полезны, но не являются оптимальными.
В последние годы было предпринято много попыток поиска новых и селективных в отношении подтипов рецепторов допамина (D1, D2, D3, D4, D5) соединений с целью повышения эффективности и снижения побочных эффектов.
Настоящее изобретение предлагает другой принцип для новых терапевтических средств, основанный на взаимодействиях с допаминергической системой. Согласно изобретению предложены соединения, оказывающие, в качестве их главной особенности, стабилизирующее воздействие на допаминергическую систему в мозге.
Описание животных моделей, использованных в изобретении
Соединения по изобретению оказывают эффекты на нейрохимию мозга, подобные эффектам антагонистов D2-рецепторов допамина (то есть дозозависимо повышают концентрацию метаболита допамина DOPAC в кортикальной, стриарной и лимбической областях мозга). Соединения по изобретению не оказывают или оказывают только ограниченное ингибирующее воздействие на спонтанную двигательную активность. При определенных условиях они могут вызывать поведенческую активацию. Поведенческая активация ограничивается, не достигая полного увеличения активности, вызываемого прямыми или непрямыми агонистами рецепторов допамина. Однако предпочтительные вещества снижают повышение активности, вызванное непрямым допаминергическим агонистом d-амфетамином. Повышение активности после лечения d-амфетамином представляет собой стандартную модель гипердопаминергии (Таблица 1). В этой модели допаминергическая нейротрансмиссия повышается в результате системного введения d-амфетамина в дозе, которая достаточно высока для того, чтобы вызвать значительное повышение двигательной активности. Способность соединения противодействовать этой гиперактивности отражает антидопаминергические свойства, которые составляют часть профиля допаминергического стабилизатора. Кроме того, антагонизм гиперактивности, вызванной d-амфетамином, широко используется в качестве стандартного анализа антипсихотической активности (смотри Psychopharmacology, 4th Generation of progress, Chapter 68, p.793-795).
Другая животная модель антипсихотической активности основана на введении глутаматного антагониста МK-801. Глутаматные антагонисты (то есть антагонисты NMDA) могут вызывать психозы у человека (смотри Psychopharmacology, 4th Generation of progress, Chapter 101, p.1205 and 1207) и вызывать поведенческие аберрации у животных. Так, способность лекарственного средства воздействовать на шизофрению и психотические состояния может быть измерена с использованием поведенческих моделей, основанных на экспериментально вызванных гипоглутаматергических состояниях. В этом исследовании, чтобы вызвать гипоглутаматергическое состояние, при котором крысы демонстрируют атипичное гиперактивное поведение, использовали антагонист NMDA МK-801 (0,7 мг/кг в.б. (внутрибрюшинно)). Соединения по настоящему изобретнию дозозависимо реверсируют поведенческую аберрацию, вызванную МK-801 (смотри Таблицу 2).
Известно, что допаминергические системы мозга интенсивно взаимодействуют с другими медиаторными системами (смотри Psychopharmacology, 4th Generation of progress, Chapter 101, pages 1208-1209). Такие взаимодействия могут объяснить мощное воздействие допаминергических стабилизаторов на поведенческие аберрации, вызванные глутаматным антагонистом МK-801, несмотря на то что эти аберрации первоначально не основаны на или не вызваны изменениями в допаминергической трансмиссии.
Терапевтическое применение допаминергических стабилизаторов
Согласно изобретению предложены соединения, обладающие, в качестве их главного признака, стабилизирующим воздействием на допаминергическую систему в мозге. Эти соединения полезны для лечения расстройств ЦНС, на симптомы которых могут влиять допаминергические функции. Подтверждение этого утверждения можно найти в следующих источниках информации:
- Подтверждение для шизофрении и психоза можно найти в Psychopharmacology, 4th Generation of progress, Chapter 26, p.295-301;
- Болезнь Паркинсона (Psychopharmacology, 4th Generation of progress, Chapter 26, p.295, Chapter 1479-1482);
- Тревожные расстройства (Psychopharmacology, 4th Generation of progress, Chapter 21, p.227 and 237, Chapter 111, p.1317-1318 and 1320);
- Расстройства настроения (Psychopharmacology, 4th Generation of progress, Chapter 80, p.921-928) и
- Злоупотребление веществами (Psychopharmacology, 4th Generation of progress, Chapter 25, p.283 and 292, Chapter 66, p.759-760, Chapter 147, p.1725 (смотри также Nisell et al., "Systemic Nicotine-Induced Dopamine Release in the Rat Nucleus Accumbens is Regulated by Nicotinic receptors in the Ventral Tegmental Area; Synapse (1994) 16: 36-44), Chapter 149, p.1745-1747 and 1751-1752). Лекарственные средства, злоупотребляемые людьми, преимущественно повышают синаптические концентрации допамина в мезолимбической системе свободно двигающихся крыс (Di Chiara et al. Proc Natl Acad Sci USA 85, 5274, 1988. Drug addiction as a disorder of associative learning. Role of nucleus accumbens shell/extended amygdala dopamine Ann N.Y. Acad Sci 877, 461, 1999).
Как показано в этих источниках информации, указанные состояния признаны в известном уровне техники как заболевания, которые имеют отношение к допаминергической нейротрансмиссии.
Кроме того, общепризнано, что фармакологическое взаимодействие с допаминергической нейротрансмиссией полезно в лечении ряда расстройств ЦНС, которые обычно не считаются прямо вызванными нарушениями допаминергической нейротрансмиссии. Например, симптомы болезни Гентингтона и других двигательных расстройств можно лечить допаминергическими агентами за счет вовлечения допамина в моторные функции (смотри Psychopharmacology, 4th Generation of progress, Chapter 26, p.295-301). Известно также, что когнитивные расстройства (смотри Psychopharmacology, 4th Generation of progress, Chapter 25, p.292, Chapter 120, p.1417 and 1420, Chapter 123, p.1447 and 1452 and 1455-1457), аутизм (смотри Psychopharmacology, 4th Generation of progress, Chapter 142, p.1653 and 1661), синдром дефицита внимания с гиперактивностью (смотри Psychopharmacology, 4th Generation of progress, Chapter 141, p.1643 and 1649-1650), сексуальные расстройства (смотри Psychopharmacology, 4th Generation of progress, Chapters 65, p.743-746 and Chapter 22, p.245 and 254) и расстройства приема пищи (смотри Psychopharmacology, 4th Generation of progress, Chapters 137, p.1600, Chapter 138, p.1609-1610 and 1612) можно лечить агентами, усиливающими допаминергическую трансмиссию. Таким образом, вышеизложенные ссылки являются подтверждением тому, что соединения по изобретению можно применять для лечения таких заболеваний.
В данной области признано, что ингибирование hERG-канала может иметь тяжелые побочные эффекты со стороны сердца, в том числе летальную аритмию (J.Cardiovasc. Electrophysiol. 15, 475, 2004; Eur. J. Pharm., 450, 37, 2002; Cardiovascular Research, 58, 32, 2003). Таким образом, при разработке новых лекарственных средств, действующих на ЦНС, искомыми являются соединения с минимальной аффинностью к hERG-каналу и, следовательно, с широкими пределами безопасности.
Способы получения
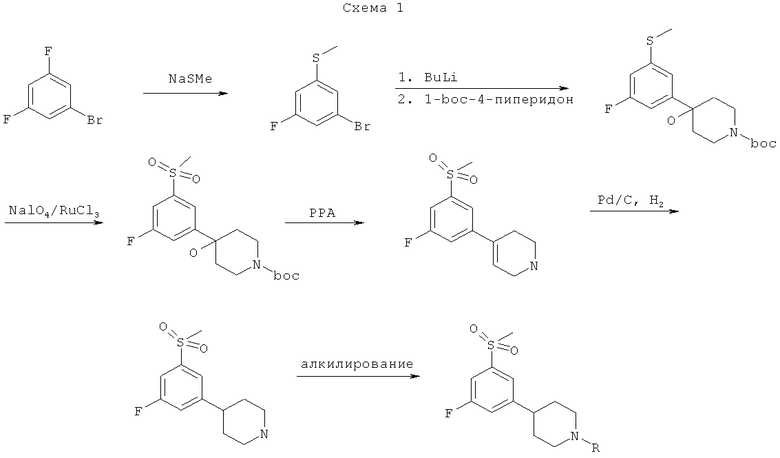
Соединения по изобретению могут быть получены, как в общем показано на Схеме 1. Однако изобретение не ограничивается этими способами. Соединения могут быть также получены способами, описанными для структурно родственных соединений в предшествующем уровне техники. Реакции можно проводить по стандартным методикам1,2 или как описано в рабочих примерах. Исходные вещества для процессов, описанных в настоящей заявке, известны или без труда могут быть получены общепринятыми способами из коммерчески доступных химических веществ.
Специалистам в данной области очевидно, что для получения соединений по изобретению альтернативным или в некоторых случаях более удобным способом отдельные стадии способа, упомянутые выше, можно выполнять в другом порядке и/или отдельные реакции можно осуществлять на другой стадии общего пути синтеза (т.е. химические превращения можно осуществлять с промежуточными соединениями, которые отличаются от промежуточных соединений, которые здесь выше ассоциированы с конкретными реакциями).
Ссылки:
1. Comprehensive Organic Transformations: A Guide to Functional Group Preparations
Richard C. Larock, 22 October, 1999 Wiley-VCH
ISBN: 0471190314
2. March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 5th Edition.
Michael B. Smith, Jerry March, January 15, 2001 Wiley-lnterscience
ISBN: 0471585890
Используемый здесь термин "C1-С3алкил" относится к алкилу, содержащему 1-3 атома углерода, в любой изомерной форме. Различные углеродные группировки определены следующим образом: алкил относится к алифатическому углеводородному радикалу и включает неразветвленные формы, такие как метил, этил, н-пропил. Термин «аллил» относится к группе -CH2-CH=CH2.
Используемый здесь термин «пациент» относится к индивидууму, нуждающемуся в лечении согласно изобретению.
Используемый здесь термин «лечение» относится как к лечению с целью излечения или облегчения симптомов заболевания или состояния, так и к лечению с целью предупреждения развития заболевания или состояния. Можно проводить либо экстренное, либо длительное лечение.
Для образования нетоксичных фармацевтически приемлемых солей присоединения кислоты соединений по изобретению могут быть использованы как органические, так и неорганические кислоты. Подходящие соли присоединения кислоты соединений по настоящему изобретению включают фармацевтически приемлемые соли, такие как толуолсульфонат, метансульфонат, фумарат, гидрохлорид, гидробромид, гидройодид, нитрат, ацетат, лактат, цитрат, кислый цитрат, тартрат, битартрат, алифатический, алициклический, ароматический или гетероциклический карбоксилат, сукцинат, малеат, фумарат, глюконат, гликолят, сахарат, аскорбат, ацетат, пропионат, бензоат, пируват, памоат [то есть 1,1'-метилен-бис-(2-гидрокси-3-нафтоат)], фосфат, кислый фосфат, сульфат или бисульфат. Эти соли получают способами, известными в уровне техники. Понятно также, что соединения по настоящему изобретению могут существовать как в сольватированной, так и в несольватированной форме, например в гидратированной форме.
Фармацевтическая композиция, содержащая соединение по изобретению, также может содержать вещества, используемые для облегчения изготовления фармацевтического препарата или введения препаратов. Такие вещества общеизвестны специалистам в данной области и могут представлять собой, например, фармацевтически приемлемые адъюванты, носители и консерванты.
В клинической практике соединения, применяемые согласно настоящему изобретению, будут вводить, как правило, перорально, ректально, интраназально или инъекцией в форме фармацевтических препаратов, содержащих активный ингредиент либо в виде свободного основания, либо в виде фармацевтически приемлемой нетоксичной соли присоединения кислоты, такой как гидрохлорид, лактат, ацетат, сульфамат, совместно с фармацевтически приемлемым носителем. Носитель может представлять собой твердое, полутвердое или жидкое вещество. Обычно активное вещество будет составлять от 0,1 до 99 мас.% от массы препарата, более конкретно от 0,5 до 20 мас.% для препаратов, предназначенных для инъекций, и от 0,2 до 50 мас.% для препаратов, подходящих для перорального введения.
Для получения фармацевтических препаратов, содержащих соединение по изобретению в стандартных лекарственных формах для перорального применения, выбранное соединение может быть смешано с твердым эксципиентом, например лактозой, сахарозой, сорбитом, маннитом, крахмалами, такими как картофельный крахмал, кукурузный крахмал или амилопектин, производными целлюлозы, связывающим веществом, таким как желатин или поливинилпирролидон, и смазывающим веществом, таким как стеарат магния, стеарат кальция, полиэтиленгликоль, воски, парафин и т.п., и затем спрессовано в таблетки. Если требуются таблетки, покрытые оболочкой, то ядра, изготовленные, как описано выше, могут быть покрыты концентрированным раствором сахара, который может содержать, например, аравийскую камедь, желатин, тальк, диоксид титана и т.п. Альтернативно, таблетка может быть покрыта полимером, известным специалисту в данной области, растворенным в быстро улетучивающемся органическом растворителе или в смеси органических растворителей. В эти покрытия могут быть добавлены красители, чтобы без труда различать таблетки, содержащие разные активные вещества или разные дозы активного соединения.
Для изготовления мягких желатиновых капсул активное вещество может быть смешано, например, с растительным маслом или полиэтиленгликолем. Твердые желатиновые капсулы могут содержать гранулы активного вещества с использованием либо упомянутых эксципиентов для таблеток, например лактозы, сахарозы, сорбита, маннита, крахмала (например картофельного крахмала, кукурузного крахмала или амилопектина), производных целлюлозы, либо желатина. Твердые желатиновые капсулы могут быть также заполнены жидким или полутвердым лекарственным средством. Ниже приведены примеры композиций таблеток или капсул, подходящих для перорального введения:
Стандартные лекарственные формы для ректального применения могут представлять собой растворы или суспензии или могут быть изготовлены в форме суппозиториев, содержащих активное вещество в смеси с нейтральной жировой основой, или в форме ректальных желатиновых капсул, содержащих активное вещество в смеси с растительным маслом или вазелиновым маслом. Жидкие препараты для перорального применения могут быть в форме сиропов или суспензий, например в форме растворов, содержащих от приблизительно 0,2% до приблизительно 20 мас.% активного вещества, охарактеризованного в данном описании, и остальное сахар и смесь этанола, воды, глицерина и пропиленгликоля. Такие жидкие препараты, возможно, могут содержать красители, ароматизаторы, сахарин и карбоксиметилцеллюлозу в качестве загустителя или другие эксципиенты, известные специалисту в данной области.
Растворы для парентерального введения инъекцией могут быть изготовлены в виде водного раствора водорастворимой фармацевтически приемлемой соли активного вещества, предпочтительно в концентрации от 0,5% до приблизительно 10 мас.%. Эти растворы могут содержать также стабилизаторы и/или буферные агенты и удобно могут быть предоставлены в стандартных ампулах. Применение и введение пациенту, которого лечат в клинике, будет очевидно специалисту в данной области.
Для интраназального введения или введения ингаляцией соединения по настоящему изобретению можно доставлять в форме раствора, сухого порошка или суспензии. Введение можно осуществлять с посредством распылителя, который сжимает или сдавливает пациент, или посредством создания аэрозольного спрея из контейнера под давлением, или посредством небулайзера с использованием подходящего пропеллента, например дихлордифторметана, трихлорфторметана, дихлортетрафторэтана, диоксида углерода или другого подходящего газа. Соединения по изобретению также можно вводить посредством сухого порошкового ингалятора либо в виде тонкоизмельченного порошка в комбинации с веществом-носителем (например, сахаридом), либо в виде микросфер. Ингалятор, пульверизатор или аэрозольный распылитель может быть рассчитан на однократную дозу или многократные дозы. Дозирование можно контролировать посредством клапана, который доставляет отмеренное количество активного соединения.
Соединения по изобретению также можно вводить в виде препарата с контролируемым высвобождением. Соединения высвобождаются с требуемой скоростью для поддержания постоянной фармакологической активности в течение желаемого периода времени. Такие стандартные лекарственные формы обеспечивают доставку лекарственного средства в организм в течение заданного периода времени и, следовательно, поддерживают уровни лекарственного средства в пределах терапевтического диапазона в течение более длительных периодов времени, чем традиционные препараты с неконтролируемым высвобождением. Соединения также могут быть приготовлены в виде препаратов с контролируемым высвобождением, которые обеспечивают направленную доставку активного соединения к мишени. Например, высвобождение соединения может быть ограничено конкретной областью пищеварительной системы через чувствительность препарата к рН. Такие препараты общеизвестны специалистам в данной области.
В зависимости от расстройства, которое лечат, и пациента, которого лечат, и пути введения композиции можно вводить в различных дозах. Дозирование также будет зависеть от отношения эффективности к абсорбционной способности и от частоты и пути введения. Такие дозы можно водить однократно, два раза или три раза или более раз в сутки. Соединения по данному изобретению можно вводить субъекту в дозах в пределах от 0,01 мг до 500 мг на кг массы тела в сутки, хотя вариации будут обязательно иметь место в зависимости от массы, пола и состояния субъекта, которого лечат, болезненного состояния, которое лечат, и выбранного конкретного пути введения. Однако для лечения заболеваний у человека наиболее желательно использовать уровень дозы, который находится в пределах от 0,1 мг до 10 мг на кг массы тела в сутки, однократной или разделенной дозировкой. Альтернативно, уровень дозы является таким, чтобы получить концентрацию соединения в сыворотке от 0,1 нМ до 10 мкМ.
Подразумевается, что любая химическая формула или название, приведенные в данном описании, охватывает все стерео и оптические изомеры и рацематы и их смеси в любом соотношении. Различные изомеры могут быть получены стандартными способами, общеизвестными специалистам в данной области, например хроматографией или фракционной кристаллизацией. Например, цис/транс смеси могут быть разделены на индивидуальные стереоизомеры стереоселективным синтезом. Энантиомеры или диастереомеры могут быть выделены разделением их смесей, например фракционной кристаллизацией, перерастворением или высокоэффективной жидкостной хроматографией (ВЭЖХ). Альтернативно, разделение может быть осуществлено путем дериватизации хиральным реагентом. Стереоизомеры могут быть получены стереоселективным синтезом из стереохимически чистых исходных веществ в условиях, которые не вызывают потерю стереохимической целостности. Все стереоизомеры входят в объем изобретения.
Соединения по настоящему изобретению могут быть выделены с любой степенью чистоты стандартными способами, и очистка может быть достигнута с использованием традиционных методов, известных специалисту в данной области, таких как дистилляция, перекристаллизация и хроматография.
Изобретение дополнительно иллюстрируется приведенными ниже примерами, которые никоим образом не предназначены для ограничения объема изобретения.
Пример 1:
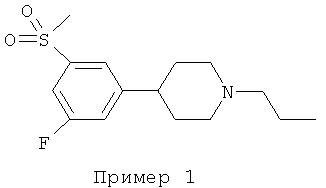
4-[3-ФТОР-5-(МЕТИЛСУЛЬФОНИЛ)ФЕНИЛ]-1-ПРОПИЛПИПЕРИДИН
К раствору 4-[3-фтор-5-(метилсульфонил)фенил]-1-пропилпиперидина (0,5 г, 1,94 ммоль) в ацетонитриле (5 мл) добавляли карбонат калия (0,53 г, 3,83 ммоль) и йодпропан (0,189 мл, 1,94 ммоль) и эту смесь нагревали с использованием микроволнового излучения в течение 20 мин при температуре 150°С. Смесь охлаждали до температуры окружающей среды и добавляли воду (50 мл). Водный остаток экстрагировали этилацетатом (3×50 мл) и объединенные органические фазы сушили, концентрировали и очищали колоночной флэш-хроматографией (этилацетат/метанол, 1:1) с получением указанного в заголовке соединения (0,27 г, 46%). Амин превращали в гидрохлоридную соль и подвергали перекристаллизации из смеси этанол/диэтиловый эфир: т.пл. 187-189°С. MS (масс-спектрометрия) m/z (относительная интенсивность, 70 эВ) 299 (М+, 3), 271 (15), 270 (bp), 147 (5) 133 (5).
Пример 2:
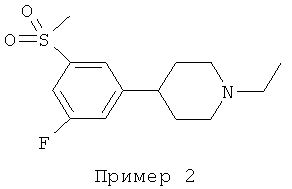
1-ЭТИЛ-4-[3-ФТОР-5-(МЕТИЛСУЛЬФОНИЛ)ФЕНИЛ]ПИПЕРИДИН
Получение по Примеру 1: 4-[3-фтор-5-(метилсульфонил)фенил]-пиперидин (0,4 г, 1,55 ммоль), ацетонитрил (5 мл), карбонат калия (0,42 г, 3,0 ммоль), 1-йодэтан (0,147 мл, 1,5 ммоль). Выход: 0,28 г (63%). Амин превращали в гидрохлоридную соль и подвергали перекристаллизации из смеси этанол/диэтиловый эфир: т.пл. 176-178°С. MS m/z (относительная интенсивность, 70 эВ) 285 (М+, 15), 284 (16), 271 (16), 270 (bp), 84 (15).
Синтез промежуточных соединений, использованных в приведенных выше Примерах, описан в Подготовительных примерах ниже.
Подготовительный пример 1:
1-БРОМ-3-ФТОР-5-(МЕТИЛТИО)БЕНЗОЛ
К раствору 1-бром-3,5-дифторбензола (5,0 г, 25,9 ммоль) в диметилформамиде (40 мл) добавляли тиометилат натрия (1,81 г, 25,9 ммоль) и эту смесь нагревали до 150°С в течение 10 мин. Реакционную смесь доводили до температуры окружающей среды, гасили насыщенным водным раствором хлорида аммония (100 мл) и экстрагировали этилацетатом (3×100 мл). Объединенные органические фазы сушили и концентрировали в вакууме с получением чистого указанного в заголовке соединения (1,84 г). MS m/z (относительная интенсивность, 70 эВ) 222 (М+, 100), 220 (М+, 100), 189 (49), 187 (50), 126 (75).
Подготовительный пример 2:
ГРЕТ-БУТИЛ-4-[3-ФТОР-5-(МЕТИЛТИО)ФЕНИЛ]-4-ГИДРОКСИ-ПИПЕРИДИН-1-КАРБОКСИЛАТ
К раствору 1-бром-3-фтор-5-(метилтио)бензол (3,7 г, 16,7 ммоль) в сухом диэтиловом эфире (100 мл) в атмосфере азота при -78°С по каплям добавляли н-бутиллитий (2,5 М в гексане, 6,7 мл, 16,7 ммоль). Смесь перемешивали в течение 30 мин при -78°С, а затем температуру доводили до -20°С в течение 2 мин и снова охлаждали до -78°С. В полученную смесь при температуре -78°С по каплям добавляли раствор 4-Вос-1-пиперидона (3,3 г, 16,7 ммоль) в сухом диэтиловом эфире (50 мл). Смесь перемешивали при температуре -78°С в течение 10 мин, а затем температуру доводили до температуры окружающей среды. Реакционную смесь гасили насыщенным водным раствором хлорида аммония (100 мл) и экстрагировали этилацетатом (3×100 мл). Объединенные органические фазы сушили, концентрировали и очищали колоночной флэш-хроматографией (изооктан/этилацетат 2:1) с получением указанного в заголовке соединения (3,76 г). MS m/z (относительная интенсивность, 70 эВ) 341 (М+, 7), 285 (11), 241 (11), 196 (4), 57 (bp).
Подготовительный пример 3:
ТРЕТ-БУТИЛ-4-[3-ФТОР-5-(МЕТИЛСУЛЬФОНИЛ)ФЕНИЛ]-4-ГИДРОКСИ-ПИПЕРИДИН-1-КАРБОКСИЛАТ
К раствору трет-бутил-4-[3-фтор-5-(метилтио)фенил]-4-гидроксипиперидин-1-карбоксилата (3,66 г, 10,6 ммоль) в тетрахлориде углерода (13 мл), ацетонитриле (13 мл) и воде (26 мл) добавляли перйодат натрия (6,8 г, 31,8 ммоль) и трихлорид рутения (3 мг, 0,05 мол.%) и эту смесь перемешивали в течение 20 мин при температуре окружающей среды. Добавляли воду и продукт экстрагировали этилацетатом (3×100 мл). Объединенные органические фазы сушили и концентрировали в вакууме с получением чистого указанного в заголовке соединения (3,3 г). MS m/z (относительная интенсивность, 70 эВ) 373 (М+, 0), 273 (25), 255 (74), 133 (28), 56 (bp).
Подготовительный пример 4:
4-[3-ФТОР-5-(МЕТИЛСУЛЬФОНИЛ)ФЕНИЛ]-1,2,3,6-ТЕТРАГИДРО-ПИРИДИН
Смесь трет-бутил-4-[3-фтор-5-(метилсульфонил)фенил]-4-гидрокси-пиперидин-1-карбоксилата (3,3 г, 8,8 ммоль) и полифосфорной кислоты (20 мл) нагревали при 120°С в течение 3 часов. Смесь выливали на лед и подщелачивали 5М гидроксидом натрия. Смесь экстрагировали этилацетатом (3×100 мл) и объединенные органические фазы сушили (MgSO4), концентрировали и очищали колоночной флэш-хроматографией (метанол/этилацетат 1:1) с получением указанного в заголовке соединения (2,02 г). MS m/z (относительная интенсивность, 70 эВ) 255 (М+, bp), 254 (50), 251 (87), 172 (87), 146 (53).
Подготовительный пример 5:
4-[3-ФТОР-5-(МЕТИЛСУЛЬФОНИЛ)ФЕНИЛ]ПИПЕРИДИН
Смесь 4-[3-фтор-5-(метилсульфонил)фенил]-1,2,3,6-тетрагидропиридин (2,02 г, 7,9 ммоль), палладия на углероде (0,56 г) и муравьиной кислоты (1,9 мл) в изопропиловом спирте (60 мл) гидрировали при давлении водорода 50 фунт/кв.дюйм (344,75 кПа) в течение 24 часов. Реакционную смесь фильтровали через слой целита и фильтрат концентрировали и упаривали до сухости с получением 1,66 г неочищенного продукта. MS m/z (относительная интенсивность, 70 эВ) 257 (М+, bp), 256 (80), 133 (21), 69 (25) 56 (99).
Приведенные ниже тесты использовали для оценки соединений по изобретению.
Тест in vivo: Поведение
Поведенческую активность измеряли с использованием восьми мониторов активности Digiscan (RXYZM (16) ТАО, Omnitech Electronics, Columbus, OH, USA), соединенных с анализатором Omnitech Digiscan и компьютером Apple Macintosh, оснащенным панелью управления с цифровым интерфейсом (NB DIO-24, National Instruments, USA). Каждый монитор активности состоял из квадратной металлической рамки (ширина × длина 40×40 см), оснащенной фотолучевыми датчиками. Во время измерений поведенческой активности крыса находилась в прозрачной акриловой клетке (ширина × длина × высота 40×40×30 см), которая, в свою очередь, находилась в мониторе активности. Каждый монитор активности был оснащен тремя рядами инфракрасных фотолучевых датчиков, причем каждый ряд состоял из 16 датчиков. Два ряда были размещены вдоль передней и боковой сторонам пола клетки под углом 90°, а третий ряд был размещен на 10 см выше уровня пола для измерения вертикальной активности. Фотолучевые датчики были расположены на расстоянии 2,5 см друг от друга. Каждый монитор активности был установлен в идентичном звуко- и светоизолирующем боксе со слабым электрическим освещением и вентилятором.
Компьютерное программное обеспечение было написано с использованием объектно-ориентированного программирования (LabVIEW®, National instruments, Austin, TX, USA).
Поведенческие данные с каждого монитора активности, отражающие положение (горизонтальный центр тяжести и вертикальная активность) животного в каждый момент времени, регистрировали с частотой отсчетов 25 Гц и собирали с использованием специально написанного приложения LABView™. Данные каждой сессии регистрации сохраняли и анализировали относительно пройденного расстояния. Каждая сессия регистрации поведения длилась 60 минут, начиная приблизительно с 4 минуты после инъекции тестируемого соединения. Аналогичные процедуры регистрации поведения применяли для крыс, не получивших лекарственное средство, и крыс, предварительно получивших лекарственное средство. Крысам, предварительно получившим d-амфетамин, вводили дозу 1,5 мг/кг и.п. (интраперитонеально) за 10 минут до сессии регистрации в мониторе активности. Крысам, предварительно получившим МK-801, вводили дозу 0,7 мг/кг и.п. за 90 минут до сессии регистрации в мониторе активности. Результаты представлены в виде отношения отсчеты/60 минут или отсчеты/30 минут в произвольных единицах протяженности. Статистические сравнения проводили с использованием t-теста Стьюдента против контрольной группы. У животных, предварительно получивших МK-801 или амфетамин, статистические сравнения выполняли против контролей с МK801 или d-амфетамином соответственно.
Значения ED50 для снижения индуцированной амфетамином гиперлокомоции рассчитывают путем выравнивания кривой. Для большинства соединений оценка основана на 16 животных, предварительно получивших амфетамин в пределах доз 0, 11, 33 и 100 мкмоль/кг п.к. в одном единичном эксперименте с дополнительными дозами в отдельных экспериментах. Расчеты основаны на расстоянии за последние 45 минут одного часа измерения. Расстояния нормализуют к контролю с амфетамином и выравнивают методом наименьших квадратов с функцией «Конец-(Конец-Контроль)/(1+(доза/ED50)Угловой коэффициент)». Четыре параметра выравнивают с ограничениями: ED50>0, 0,5<Угловой коэффициент<3, Конец>0% контроля. Для оценки доверительных интервалов для параметров выравнивание повторяют 100 раз с использованием случайной равномерно распределенной массы в квадрате (от 0 до 1) для каждого значения измерения. Представленные ED50-диапазоны охватывают 95% этих значений.
Тест in vivo: Нейрохимия
После сессий регистрации поведенческой активности крыс обезглавливали и быстро извлекали их мозг и помещали его в чашку Петри, охлаждаемую льдом. Лимбический отдел переднего мозга, полосатое тело, лобную часть коры головного мозга и остальные части полушарий каждой крысы препарировали и замораживали. Каждую часть мозга последовательно анализировали на содержание моноаминов и их метаболитов.
Количество моноаминных веществ-трансмиттеров (NA (норадреналин), DA (допамин), 5-НТ (серотонин)), а также их аминных (NM (норметанэфрин), 3-МТ (3-метокситирамин)) и кислотных (DOPAC (3,4-дигидроксифенилуксусная кислота)), 5-HIAA (5-гидроксииндолуксусная кислота), HVA (гомованильная кислота)) метаболитов определяют в гомогенатах ткани мозга путем HPLC-выделения и электрохимического детектирования.
Аналитические методы основаны на двух хроматографических выделениях, предназначенных для аминов и кислот. Две хроматографические системы имеют общий автоинжектор с 10-портовым клапаном и две сэмплерные петли для одновременного впрыска на две системы. Обе системы оснащены обращенно-фазовой колонкой (Luna C18(2), dp 3 мкм, 50×2 мм в.д. (внутренний диаметр), Phenomenex), и электрохимическое детектирование осуществляют при двух потенциалах на стеклоуглеродных электродах (MF-1000, Bioanalytical Systems, Inc.). Через Т-образное соединение элюат проходит в детекторную камеру или в емкость для отходов. Это осуществляется двумя электромагнитными клапанами, которые блокируют либо емкость для отходов, либо детекторную камеру. Лучшие условия детектирования достигаются, если не давать хроматографическому фронту доходить до детектора. Водная подвижная фаза (0,4 мл/мин) для системы для кислот содержит 14 мМ лимонной кислоты, 10 мМ цитрата натрия, 15% МеОН (об./об.) и 0,1 мМ EDTA (этилендиаминтетрауксусная кислота). Потенциалы детектирования относительно стандарта Ag/AgCl составляют 0,45 и 0,60 В. Водная подвижная фаза с ионной парой (0,5 мл/мин) для аминной системы содержит 5 мМ лимонной кислоты, 10 мМ цитрата натрия, 9% (об./об.) МеОН, 10,5% (об./об.) MeCN, 0,45 мМ декансульфоновой кислоты и 0,1 мМ EDTA. Потенциалы детектирования относительно стандарта Ag/AgCl составляют 0,45 и 0,65 В.
Тест in vivo: Пероральная биодоступность
Эксперименты проводят через 24 часа после имплантации артериальных и венозных катетеров. Тестируемое соединение вводят перорально в дозе 12,5 мкмоль/кг или внутривенно в дозе 5 мкмоль/кг, используя венозные катетеры, n=3 на группу. Затем отбирают образцы артериальной крови в течение восьми часов через 0, 3, 9, 27, 60, 120, 180, 240, 300 и 360 минут после введения тестируемого соединения. Пероральную биодоступность рассчитывали как отношение AUC (площадь под кривой), полученной после перорального введения, к AUC, полученной после внутривенного введения, для каждой крысы. Параметр AUC рассчитывали следующим образом:
AUC: площадь под кривой зависимости концентрации в плазме от момента времени ноль до последней измеренной концентрации (Споследняя), рассчитанной логарифмическим/линейным методом трапеций.
Уровни тестируемого соединения измеряют посредством жидкостной хроматографии/масс-спектрометрии (LC-MS) (Hewlett-Packard 1100MSD Series). Модуль включает в себя четвертичную систему нагнетания, вакуумный дегазатор, термостатированный автосэмплер, термостатированный колоночный отсек, детектор на диодной матрице и камеру впрыска API-ES. Обработку данных осуществляли с использованием системы HP ChemStation rev. A.06.03. Параметры настройки: режим MSD (масс-селективный детектор): мониторинг выбранных ионов (SIM); полярность MSD; положительная температура газа: 350°С; осушающий газ: 13,0 л/мин; газ в небулайзере: 50 фунт/кв.дюйм (344,75 кПа); напряжение на капилляре: 5000 В; параметр Fragmentor (напряжение на выходе из капилляра): 70 В.
Аналитическая колонка: Zorbax eclipse XDB-C8 (4.6×150 мм, 5 мкм) при 20°С. Подвижной фазой была уксусная кислота (0,03%) (растворитель А) и ацетонитрил (растворитель Б). Скорость потока подвижной фазы составляла 0,8 мл/мин. Элюирование начинали при 12% растворителя Б изократически в течение 4,5 минуты, затем линейно увеличивали до 60% в течение 4,5 минут.
Методика экстракции: образцы плазмы (0,25-0,5 мл) разбавляли водой до 1 мл и добавляли 60 пмоль (100 мкмоль) внутреннего стандарта (-)-OSU6241. рН доводили до 11 добавлением 25 мкл насыщенного водного раствора карбоната натрия. После смешивания образцы экстрагировали 4 мл дихлорметана при встряхивании в течение 20 минут. После центрифугирования органический слой переносили в пробирку меньшей емкости и упаривали до сухости в потоке азота. Остаток затем растворяли в 120 мкл подвижной фазы (уксусная кислота (0,03%): ацетонитрил, 95:5) для LC-MS анализа (впрыскивали 10 мкл). Селективный ион (МН+) регистрировали для соединения каждого Примера и МН+ 296 для (-)-OSU6241 ((3-[3-(этилсульфонил)фенил]-1-пропилпиперидин).
Стандартную кривую в диапазоне 1-500 пмоль получили добавлением соответствующих количеств тестируемого соединения в пустые образцы плазмы.
Тест in vitro: Метаболическая стабильность в микросомах печени крысы
Микросомы печени крысы выделяли, как описано в Förlin (1980) Effects of Clophen A50, 3-methylcholantrene, pregnenolone-16aq-carbonitrile and Phenobarbital on the hepatic microsomal cytochrome P-450-dependent monooxygenaser system in rainbow trout, salmo garirdneri, of different age and sex. Tox Appl Pharm. 54(3) 420-430, с незначительными модификациями, например: перед гомогенизацией добавляли 3 мл/г печени 0,1М Na/K×PO4 буфера с 0,15М KCl, рН 7,4 (буфер 1), гомогенат центрифугировали в течение 20 минут вместо 15 минут, супернатант подвергли ульрацентрифугированию при 100000 g вместо 105000 g, и осадок после ультрацентрифугирования ресуспендировали в 1 мл/г печени 20% об./об. раствора 87% глицерина в буфере 1.
1 мкл 0,2 или 1 мМ тестируемого вещества разводили в воде и 10 мкл 20 мг/мл микросом печени крысы смешивали с 149 мкл 37°С буфера 1, и реакцию инициировали добавлением 40 мкл 4,1 мг/мл NADPH. После 0 или 15 минут инкубирования при 37°С в нагревательном блоке (LAB-LINE, MULTI-BLOK Heater или lab4you, TS-100 Thermo shaker при 700 об/мин) реакцию останавливали добавлением 100 мкл чистого ацетонитрила. Белковый преципитат затем удаляли отбрасыванием осадка после центрифугирования при 10000 g в течение 10 минут (Heraeus, Biofuge fresco) при 4°С. Тестируемое соединение анализировали с использованием HPLC-MS (Hewlett-Packard 1100MSD Series) с колонкой Zorbax SB-C18 (2,1×150 мм, 5 мкм) с использованием 0,03% муравьиной кислоты и ацетонитрила в качестве подвижной фазы (градиент) или Zorbax Eclipse XDB-C18 (3×75 мм, 3,5 мкм) с использованием 0,03% уксусной кислоты и ацетонитрила в качестве подвижной фазы (градиент). Обращение за 15 минут рассчитывали как долю тестируемого соединения, элиминированную через 15 минут, выраженную в процентах от уровней в минуту 0, то есть 100×[концентрация тестируемого соединения в минуту 0 - концентрация в минуту 15]/концентрация в минуту 0.
Получение микросом печени осуществили, как описано в Forlin (1980), Protocols for incubation with liver microsomes are provided in Crespi & Stresser (2000), and Renwick et al. (2001).
Crespi С L, and DM Stressser (2000). Fluorometric screening for metabolism based drug-drug interactions. J. Pharm. Tox. Meth. 44. 325-331.
Förlin L. (1980) Effects of Clophen A50, 3-methylcholantrene, pregnenolone-16aq-carbonitrile and Phenobarbital on the hepatic microsomal cytochrome P-450-dependent monooxygenaser system in rainbow trout, salmo gairdneri, of different age and sex. Tox Appl Pharm. 54(3) 420-430.
Renwick, AB et al. (2001). Metabolism of 2,5-bis(trifluoromethyl)-7-benzyloxy-4-trifluoromethylcoumarin by human hepatic CYP isoforms: evidence for selectivity towards CYP3A4. Xenobiotica 31(4): 187-204.
Аффинность к hERG
Оценка аффинности к hERG посредством Rapid ICE™ была осуществлена Quintiles Limited, Research Avenue South, Heriot-Watt University Research Park, Riccarton Edinburgh, Scotland. Rapid ICE™ (быстрая электрофизиология ионных каналов) представляет собой автоматизированный пэтч-клэмп анализ с использованием системы PatchXpress 7000A (Ахоn Instruments). Rapid ICE™ оценивает эффект тестируемых веществ на хвостовой ток hERG, регистрируемый на клетках НЕK293, стабильно трансфицированных кДНК hERG. Было показано, что соединения, которые ингибируют hERG ток, пролонгируют сердечный потенциал действия и, следовательно, интервал QT у человека.
HERG.T.HEK (клетки НЕK293, стабильно трансфицированные кДНК hERG) были получены из Университета Висконсина. Эти клетки хранят в криогенном хранилище в Quintiles, а также поддерживают в культуре. Клетки постоянно поддерживают в и осуществляют пассажи с использованием минимально необходимой среды, дополненной 10% фетальной бычей сыворотки, 1% заменимых аминокислот, 1% пирувата натрия и 0,4 мг/мл генетицина. Для использования в Rapid ICE™ исследованиях 4 мл клеток помещают в фальконовские пробирки при плотности 2,5×10 5 /мл. Фальконовские пробирки хранят в увлажненном, заполненном газом (5% CO2) инкубаторе при 37°С и клетки используют в пределах 2,5 часов хранения. Непосредственно перед проведением эксперимента эти клетки центрифугируют при 1000 об/мин в течение 1 минуты, супернатант декантируют и клетки ресуспендируют в 150 мкл промывного раствора в эппендорфовской пробирке на 1,5 мл.
Систему PatchXpress заправляют соотвествующими внеклеточным (ванна) и внутриклеточным (пипетка) растворами до проведения исследования. В систему загружают 16-луночный герметичный чип (Sealchip 16, Aviva Biosciences Corp) и перед приготовлением клеток в виде суспензии в промывного раствором заправляют. Клеточный эппендорф помещают в назначанную позицию и начинают процедуру тритурации и диспергирования клеток в каждую лунку (записывая камеру) герметичного чипа. Система PatchXpress придерживается основных принципов традиционного цельноклеточного пэтч-клэмпинга: между пэтч-электродом и индивидуальной клеткой формируется изоляция высокого сопротивления, мембрана через рабочий конец электрода разрывается и оценивается цельноклеточная пэтч-клэмп конфигурация. Если качество клеток признают плохим, эксперимент можно остановить в этой точке и при необходимости повторить на другом герметичном чипе.
Сразу после достижения стабильного пэтча начинается регистрация методом фиксации потенциала с исходно фиксированной при -80 мВ клеткой. Стандартный профиль напряжения следующий: шаг с -80 мВ до -50 мВ в течение 200 мс, +20 мВ в течение 4,8 с, шаг до -50 мВ в течение 5 с, затем шаг до фиксации потенциала -80 мВ. Шаг от -80 мВ до тестовой команды (+20 мВ) приводит к выходящему току (то есть ток течет из клетки), а шаг от тестовой команды (+20 мВ) до -50 мВ приводит к хвостовому току (хвостовой ток отражает дезактивацию тока со временем). Получают значения хвостового тока. Каждое значение отражает средний ток, зарегистрированный от 4 последовательных импульсов напряжения. Для каждой клетки эффекты тестируемого вещества определяют вычислением отстаточного тока (% контроль), сравниваемого с предварительной обработкой носителем.
Значение IC50 (мкМ) или другого маркера эффективности оценивают исходя из зависимости концентрация-ответ.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ ДИЗАМЕЩЕННЫЕ ФЕНИЛПИПЕРИДИНЫ/ПИПЕРАЗИНЫ В КАЧЕСТВЕ МОДУЛЯТОРОВ ДОПАМИНОВОЙ НЕЙРОТРАНСМИССИИ | 2005 |
|
RU2366654C2 |
| НОВЫЕ МОДУЛЯТОРЫ КОРТИКАЛЬНОЙ ДОФАМИНЕРГИЧЕСКОЙ И ОПОСРЕДОВАННОЙ NMDA-РЕЦЕПТОРОМ ГЛУТАМАТЕРГИЧЕСКОЙ НЕЙРОТРАНСМИССИИ | 2012 |
|
RU2593500C2 |
| ЛЕЧЕНИЕ ЗАВИСИМОСТИ И РАССТРОЙСТВ ПОБУЖДЕНИЙ С ПРИМЕНЕНИЕМ ИНГИБИТОРОВ ФДЭ7 | 2013 |
|
RU2665134C2 |
| ЗАМЕЩЕННЫЕ 2-ФЕНИЛ-1-(3,4-ДИГИДРОКСИ-5-НИТРОФЕНИЛ)-1-ЭТАНОНЫ, СПОСОБ ЛЕЧЕНИЯ НЕКОТОРЫХ НАРУШЕНИЙ ЦЕНТРАЛЬНОЙ И ПЕРИФЕРИЧЕСКОЙ НЕРВНОЙ СИСТЕМЫ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ УКАЗАННЫЕ ВЕЩЕСТВА | 1999 |
|
RU2232748C2 |
| ЗАМЕЩЕННЫЕ 4-(ФЕНИЛ-N-АЛКИЛ)ПИПЕРИДИНЫ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБЫ ЛЕЧЕНИЯ | 2000 |
|
RU2262504C2 |
| НОВЫЕ МОДУЛЯТОРЫ ДОФАМИНОВОЙ НЕЙРОТРАНСМИССИИ | 2000 |
|
RU2386623C2 |
| 3-ЗАМЕЩЕННЫЕ 4-(ФЕНИЛ-N-АЛКИЛ)-ПИПЕРИДИНЫ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБЫ ЛЕЧЕНИЯ | 2000 |
|
RU2265013C2 |
| ПРОИЗВОДНЫЕ α-АМИНОАМИДА, ПОЛЕЗНЫЕ ПРИ ЛЕЧЕНИИ СИНДРОМА УСТАЛЫХ НОГ И ВЫЗЫВАЮЩИХ ПРИВЫКАНИЕ РАССТРОЙСТВ | 2005 |
|
RU2403030C2 |
| ИСПОЛЬЗОВАНИЕ ИНГИБИТОРОВ PDE7 ДЛЯ ЛЕЧЕНИЯ НАРУШЕНИЙ ДВИЖЕНИЙ | 2010 |
|
RU2600869C2 |
| ПРИМЕНЕНИЕ ПРОИЗВОДНЫХ N-(ИНДОЛКАРБОНИЛ)ПИПЕРАЗИНА | 2002 |
|
RU2317083C2 |
Изобретение относится к новым соединениям формулы 1:
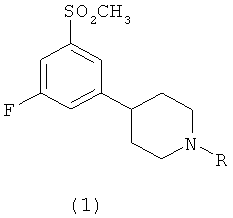 ,
,
где R выбран из группы, состоящей из C1-С3алкилов, и к их фармацевтически приемлемым солям.
Изобретение также относится к фармацевтической композиции.
Технический результат - получение новых биологически активных соединений, которые обладают активностью модулятора нейротрансмиссии допамина. 2 н. и 14 з.п. ф-лы, 2 табл.
1. Соединение формулы 1
,
где R выбран из группы, состоящей из C1-С3алкилов, и его фармацевтически приемлемые соли.
2. Соединение по п.1, где R выбран из группы, состоящей из н-пропила и этила.
3. Соединение по любому из пп.1, 2, выбранное из группы, включающей
1-этил-4-[3-фтор-5-(метилсульфонил)фенил]пиперидин,
4-[3-фтор-5-(метилсульфонил)фенил]-1 -пропилпиперидин
и их фармацевтически приемлемые соли.
4. Соединение по п.1, которое представляет собой
1-этил-4-[3-фтор-5-(метилсульфонил)фенил]пиперидин
или его фармацевтически приемлемую соль.
5. Соединение по п.1, которое представляет собой соль гидрохлорид 1-этил-4-[3-фтор-5-(метилсульфонил)фенил]пиперидина.
6. Фармацевтическая композиция, обладающая активностью модулятора нейротрансмиссии допамина, содержащая соединение по любому из пп.1-5 и один или более фармацевтически приемлемых носителей или разбавителей.
7. Фармацевтическая композиция по п.6 для лечения расстройства центральной нервной системы.
8. Фармацевтическая композиция по п.6 для лечения двигательных расстройств, выбранных из группы, состоящей из болезни Паркинсона, паркинсонизма, дискинезий (включая дискинезии, индуцированные L-DOPA), дистоний, тиков, тремора и болезни Гентингтона.
9. Фармацевтическая композиция по п.6 для лечения состояния, выбранного из группы, состоящей из ятрогенных и неятрогенных психозов и галлюцинозов.
10. Фармацевтическая композиция по п.6 для лечения состояния, выбранного из группы, состоящей из шизофрении и шизофреноформных расстройств и биполярного расстройства.
11. Фармацевтическая композиция по п.6 для лечения состояния, выбранного из группы, состоящей из расстройства настроения и тревожного расстройства, депрессии и обсессивно-компульсивного расстройства.
12. Фармацевтическая композиция по п.6 для лечения расстройств развития нервной системы, выбранных из группы, состоящей из расстройств типа аутизма, ADHD (синдром дефицита внимания с гиперактивностью), коркового паралича, синдрома Жиля де ля Туретта, и нейродегенеративных расстройств, выбранных из группы, состоящей из деменции и возрастного ухудшения когнитивных функций.
13. Фармацевтическая композиция по п.6 для лечения состояния, выбранного из группы, состоящей из расстройств сна, сексуальных расстройств, расстройств приема пищи, ожирения и головных болей или других болей при состояниях, характеризующихся повышенным мышечным тонусом.
14. Фармацевтическая композиция по п.6 для улучшения двигательных функций, когнитивных функций и родственных эмоциональных расстройств, и после повреждения мозга, вызванного травматическими, воспалительными, инфекционными, неопластическими, сосудистыми, гипоксическими или метаболическими причинами, или повреждения мозга, вызванного токсическими реакциями на экзогенные химические вещества, где экзогенные химические вещества выбраны из группы, состоящей из веществ, которыми злоупотребляют, фармацевтических соединений, токсинов окружающей среды.
15. Фармацевтическая композиция по п.6 для лечения расстройства, связанного со злоупотреблением веществом.
16. Фармацевтическая композиция по п.6 для лечения болезни Альцгеймера или родственных расстройств с деменцией.
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| ЗАМЕЩЕННЫЕ БЕНЗИЛАМИНОПИПЕРИДИНЫ И ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, СПОСОБ ЛЕЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1996 |
|
RU2152930C2 |

