ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к соединениям, которые являются обратными агонистами/антагонистами рецептора грелина, полезными для лечения расстройств сна. Изобретение также относится к фармацевтическим композициям, содержащим такие соединения.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Бессонницей, самым распространенным расстройством сна, страдает приблизительно 50-70 миллионов взрослых американцев. Она характеризуется трудностью засыпания, частым пробуждением ночью, пробуждением слишком рано и неспособностью опять заснуть или пробуждением, не чувствуя себя отдохнувшим.
Раньше для лечения бессонницы обычно использовали депрессанты центральной нервной системы (ЦНС), такие как барбитураты. Эти соединения обычно имеют длительный срок полувыведения и имеют общеизвестный спектр побочных эффектов, включая летаргию, спутанное сознание, депрессию и похмельный синдром на следующий день. Кроме того, постоянное применение ассоциируется с высоким потенциалом развития пристрастия, включающего как физическую, так и психологическую зависимость. Способы лечения отошли от барбитуратов, и им на смену пришли другие депрессанты ЦНС бензодиазепинового класса седативных снотворных агентов. Этот класс соединений оказывает успокоительное действие, которое приводит к подобному сну состоянию у пациентов и животных, причем предел их безопасности выше, чем у предшествующих снотворных. Однако многие бензодиазепины обладают побочными эффектами, которые ограничивают их применимость для некоторой части пациентов. Эти проблемы включают синергию с другими депрессантами ЦНС (особенно алкоголем), развитию толерантности после повторных приемов, возобновление бессонницы после прекращения приема, синдром похмелья на следующий день и нарушение психомоторной деятельности и памяти. В современных способах лечения бессонницы используются небензодиазепиновые соединения. Ambien (золпидем), Sonata (залеплон) являются примерами разрешенных для применения лекарственных продуктов. В данной области существует потребность в безопасных и терапевтически эффективных небензодиазепиновых агентах для лечения расстройств сна.
Синдром ночной еды (NES) обычно ассоциируется с недостаточным потреблением пищи в первой половине дня, вечерней гиперфагией и затем бессонницей, а также пробуждениями (смотри, например, Vander Wal, Jillon S., Clinical Psychology Review (2012) 32(1), 49-59). Несмотря на то, что знания о NES расширилось, он все еще не изучен и, что более важно, продолжающиеся исследования подходов к лечению остаются безуспешными.
Настоящее изобретение относится к применению обратных агонистов или антагонистов рецептора грелина для лечения расстройств сна.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к способам лечения расстройств сна у пациентов, включающим введение пациенту, нуждающемуся в таком лечении, терапевтически эффективного количества обратного агониста или антагониста рецептора грелина.
В другом воплощении настоящее изобретение относится к способам лечения расстройств сна у пациентов, включающим введение пациенту, нуждающемуся в таком лечении, фармацевтической композиции, содержащей терапевтически эффективное количество обратного агониста или антагониста рецептора грелина и по меньшей мере один фармацевтически приемлемый носитель, разбавитель или эксципиент.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
Фиг. 1 представляет собой графическую иллюстрацию эффектов соединения Примера 1 по снижению локомоторной активности у самцов крыс Wistar Han через 35-65 минут после введения дозы через пероральный зонд.
Фиг. 2 представляет собой графическую иллюстрацию эффектов сонливости соединения Примера 1 у людей.
Фиг. 3 является примером индивидуальной картины активности животного после введения один раз в сутки (QD) соединения Примера 1 или носителя.
Фиг. 4 является примером индивидуальной картины приема пищи животного после введения один раз в сутки (QD) соединения Примера 1 или носителя.
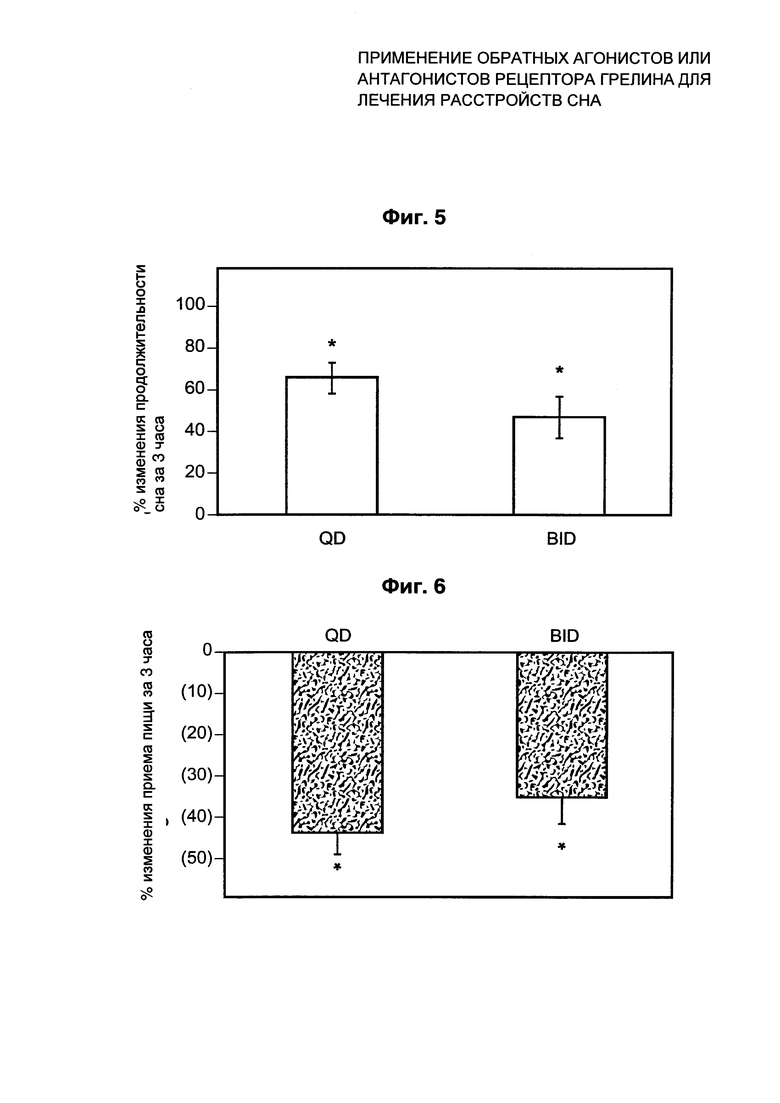
Фиг. 5 иллюстрирует изменение продолжительности сна в пределах 3 часов после вечернего введения соединения Примера 1.
Фиг. 6 иллюстрирует изменение приема пищи в пределах 3 часов после вечернего введения соединения Примера 1.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В другом воплощении настоящее изобретение относится к способам лечения первичной бессонницы у пациентов, включающим введение пациенту, нуждающемуся в таком лечении, терапевтически эффективного количества обратного агониста или антагониста рецептора грелина.
В другом воплощении настоящее изобретение относится к способам лечения чрезмерной сонливости в дневное время у пациентов, включающим введение пациенту, нуждающемуся в таком лечении, терапевтически эффективного количества обратного агониста или антагониста рецептора грелина. Способы по настоящему изобретению включают лечение чрезмерной сонливости в дневное время у пациентов с диагнозом синдром Прадера-Вилли.
В другом воплощении настоящее изобретение относится к способам лечения NES у пациентов, включающим введение пациенту, нуждающемуся в таком лечении, терапевтически эффективного количества обратного агониста или антагониста рецептора грелина.
В другом воплощении настоящее изобретение относится к способам лечения первичной бессонницы у пациентов, включающим введение пациенту, нуждающемуся в таком лечении, фармацевтической композиции, содержащей терапевтически эффективное количество обратного агониста или антагониста рецептора грелина и по меньшей мере один фармацевтически приемлемый носитель, разбавитель или эксципиент.
В другом воплощении настоящее изобретение относится к способам лечения чрезмерной сонливости в дневное время у пациентов, включающим введение пациенту, нуждающемуся в таком лечении, фармацевтической композиции, содержащей терапевтически эффективное количество обратного агониста или антагониста рецептора грелина и по меньшей мере один фармацевтически приемлемый носитель, разбавитель или эксципиент. Способы по настоящему изобретению включают лечение чрезмерной сонливости в дневное время у пациентов с диагнозом синдром Прадера-Вилли.
В другом воплощении настоящее изобретение относится к способам лечения NES у пациентов, включающим введение пациенту, нуждающемуся в таком лечении, фармацевтической композиции, содержащей терапевтически эффективное количество обратного агониста или антагониста рецептора грелина и по меньшей мере один фармацевтически приемлемый носитель, разбавитель или эксципиент.
В другом воплощении настоящее изобретение относится к способу лечения расстройств сна у пациентов, включающему введение пациенту, нуждающемуся в таком лечении, терапевтически эффективного количества (R)-2-(2-метилимидазо[2,1-b]тиазол-6-ил)-1-(2-(5-(6-метилпиримидин-4-ил)-2,3-дигидро-1Н-инден-1-ил)-2,7-диазаспиро[3.5]нонан-7-ил)этанона или его фармацевтически приемлемой соли.
В другом воплощении настоящее изобретение относится к способу лечения первичной бессонницы у пациентов, включающему введение пациенту, нуждающемуся в таком лечении, терапевтически эффективного количества (R)-2-(2-метилимидазо[2,1-b]тиазол-6-ил)-1-(2-(5-(6-метилпиримидин-4-ил)-2,3-дигидро-1Н-инден-1-ил)-2,7-диазаспиро[3.5]нонан-7-ил)этанона или его фармацевтически приемлемой соли.
В другом воплощении настоящее изобретение относится к способу лечения чрезмерной сонливости в дневное время у пациентов, включающему введение пациенту, нуждающемуся в таком лечении, терапевтически эффективного количества (R)-2-(2-метилимидазо[2,1-b]тиазол-6-ил)-1-(2-(5-(6-метилпиримидин-4-ил)-2,3-дигидро-1Н-инден-1-ил)-2,7-диазаспиро[3.5]нонан-7-ил)этанона или его фармацевтически приемлемой соли. Способы по настоящему изобретению включают лечение чрезмерной сонливости в дневное время у пациентов с диагнозом синдром Прадера-Вилли.
В другом воплощении настоящее изобретение относится к способам лечения NES у пациентов, включающим введение пациенту, нуждающемуся в таком лечении, терапевтически эффективного количества (R)-2-(2-метилимидазо[2,1-b]тиазол-6-ил)-1-(2-(5-(6-метилпиримидин-4-ил)-2,3-дигидро-1Н-инден-1-ил)-2,7-диазаспиро[3.5]нонан-7-ил)этанона или его фармацевтически приемлемой соли.
В другом воплощении настоящее изобретение относится к способам лечения расстройств сна у пациентов, включающим введение пациенту, нуждающемуся в таком лечении, фармацевтической композиции, содержащей терапевтически эффективное количество (R)-2-(2-метилимидазо[2,1-b]тиазол-6-ил)-1-(2-(5-(6-метилпиримидин-4-ил)-2,3-дигидро-1Н-инден-1-ил)-2,7-диазаспиро[3.5]нонан-7-ил)этанона или его фармацевтически приемлемой соли и по меньшей мере один фармацевтически приемлемый носитель, разбавитель или эксципиент.
В другом воплощении настоящее изобретение относится к способам лечения первичной бессонницы у пациентов, включающим введение пациенту, нуждающемуся в таком лечении, фармацевтической композиции, содержащей терапевтически эффективное количество (R)-2-(2-метилимидазо[2,1-b]тиазол-6-ил)-1-(2-(5-(6-метилпиримидин-4-ил)-2,3-дигидро-1Н-инден-1-ил)-2,7-диазаспиро[3.5]нонан-7-ил)этанона или его фармацевтически приемлемой соли и по меньшей мере один фармацевтически приемлемый носитель, разбавитель или эксципиент.
В другом воплощении настоящее изобретение относится к способам лечения чрезмерной сонливости в дневное время у пациентов, включающим введение пациенту, нуждающемуся в таком лечении, фармацевтической композиции, содержащей терапевтически эффективное количество (R)-2-(2-метилимидазо[2,1-b]тиазол-6-ил)-1-(2-(5-(6-метилпиримидин-4-ил)-2,3-дигидро-1Н-инден-1-ил)-2,7-диазаспиро[3.5]нонан-7-ил)этанона или его фармацевтически приемлемой соли и по меньшей мере один фармацевтически приемлемый носитель, разбавитель или эксципиент. Способы по настоящему изобретению включают лечение чрезмерной сонливости в дневное время у пациентов с диагнозом синдром Прадера-Вилли.
В другом воплощении настоящее изобретение относится к способам лечения NES у пациентов включающим введение пациенту, нуждающемуся в таком лечении, фармацевтической композиции, содержащей терапевтически эффективное количество (R)-2-(2-метилимидазо[2,1-b]тиазол-6-ил)-1-(2-(5-(6-метилпиримидин-4-ил)-2,3-дигидро-1Н-инден-1-ил)-2,7-диазаспиро[3.5]нонан-7-ил)этанона или его фармацевтически приемлемой соли и по меньшей мере один фармацевтически приемлемый носитель, разбавитель или эксципиент.
В другом воплощении настоящего изобретения также предложены:
применение обратного агониста или антагониста рецептора грелина, как описано в данном документе, для изготовления лекарственного средства для лечения расстройства сна, в частности первичной бессонницы, чрезмерной сонливости в дневное время или NES;
обратный агонист или антагонист рецептора грелина, как описано в данном документе, для применения в качестве лекарственного средства;
обратный агонист или антагонист рецептора грелина, как описано в данном документе, для применения в лечении расстройства сна, в частности первичной бессонницы, чрезмерной сонливости в дневное время или NES;
фармацевтическая композиция, содержащая обратный агонист или антагонист рецептора грелина, как описано в данном документе, и фармацевтически приемлемый эксципиент; и
фармацевтическая композиция для лечения расстройства сна, в частности первичной бессонницы, чрезмерной сонливости в дневное время или NES, содержащая обратный агонист или антагонист рецептора грелина, как описано в данном документе;
в частности где обратный агонист или антагонист рецептора грелина может представлять собой (R)-2-(2-метилимидазо[2,1-b]тиазол-6-ил)-1-(2-(5-(6-метилпиримидин-4-ил)-2,3-дигидро-1Н-инден-1-ил)-2,7-диазаспиро[3.5]нонан-7-ил)этанон или его фармацевтически приемлемую соль.
(R)-2-(2-метилимидазо[2,1-b]тиазол-6-ил)-1-(2-(5-(6-метилпиримидин-4-ил)-2,3-дигидро-1Н-инден-1-ил)-2,7-диазаспиро[3.5]нонан-7-ил)этанон или его фармацевтически приемлемая соль может упоминаться в данном документе просто как соединение Примера 1.
В другом воплощении настоящего изобретения также предложен любой способ, рассматриваемый в данном документе, при котором обратный агонист или антагонист рецептора грелина, в частности (R)-2-(2-метилимидазо[2,1-b]тиазол-6-ил)-1-(2-(5-(6-метилпиримидин-4-ил)-2,3-дигидро-1Н-инден-1-ил)-2,7-диазаспиро[3.5]нонан-7-ил)этанон или его фармацевтически приемлемую соль, вводят в комбинации с другим фармакологически активным агентом.
В другом воплощении обратный агонист или антагонист рецептора грелина, в частности соединение Примера 1, можно использовать в комбинации с другими фармакологически активными агентами, также упоминаемыми как соединения, известные в данной области, которые вводят либо отдельно, либо в той же фармацевтической композиции, и они включают, без ограничения, сенсибилизаторы инсулина, в том числе (i) антагонисты PPAR гамма (активируемый пролифератором пероксисом рецептор гамма), такие как глитазоны (например циглитазон, дарглитазон, энглитазон, исаглитазон (МСС-555), пиоглитазон, росиглитазон, троглитазон, туларик, BRL49653, CLX-0921, 5-BTZD), GW-0207, LG-100641 и LY-300512 и т.п.); (iii) бигуаниды, такие как метформин и фенформин; (b) инсулин или миметики инсулина, такие как биота, LP-100, новарапид, инсулин детемир, инсулин лиспро, инсулин гларгин, суспензия цинк-инсулина (ленте и ультраленте); Lys-Pro инсулин, GLP-1 (73-7) (инсулинтропин); и GLP-1 (7-36)-NH2); (с) сульфонилмочевины, такие как ацетогексамид, хлорпропамид, диабенез, глибенкламид, глипизид, глибурид, глимепирид, гликлазид, глипентид, гликвидон, глисоламид, толазамид и толбутамид; (d) ингибиторы альфа-глюкозидазы, такие как акарбоза, адипозин, камиглибоза, эмиглитат, миглитол, воглибоза, прадимицин-Q, сальбостатин, CKD-711, MDL-25,637, MDL-73,945 и MOR 14 и т.п.; (д) агенты, снижающие уровни холестерина, такие как (i) ингибиторы HMG-CoA-редуктазы (гидроксиметилглутарил-коэнзим А-редуктаза) (аторвастатин, итавастатин, флувастатин, ловастатин, правастатин, ривастатин, розувастатин, симвастатин и другие статины), (ii) абсорберы/секвестранты желчных кислот, такие как холестирамин, колестипол, диалкиламиноалкильные производные поперечно-сшитого декстрана, Colestid.RTM., LoCholest.RTM. и т.п.; (ii) никотиниловый спирт, никотиновую кислоту или ее соль; (iii) агонисты рецептора альфа пролифератора-активатора, такие как производные фенофибриновой кислоты (гемфиброзил, клофибрат, фенофибрат и бензафибрат); (iv) ингибиторы всасывания холестерина, такие как сложные эфиры станола, бета-ситостерол, стероловые гликозиды, такие как тиквезид, и азетидиноны, такие как эзетимиб, и т.п., и ингибиторы ацил-СоА:холестерол-ацилтрансферазы (АСАТ), такие как авасимиб и мелинамид; (v) антиоксиданты, такие как пробукол, (vi) витамин Е; и (vii) тиреомиметики; (f) агонисты PPAR альфа, такие как беклофибрат, бензафибрат, ципрофибрат, клофибрат, этофибрат, фенофибрат и гемфиброзил и другие производные фибриновой кислоты, такие как Atromid.RTM., Lopid.RTM. и Tricor.RTM. и т.п., и агонисты PPAR альфа, которые описаны в WO 97/36579, Glaxo; (g) агонисты PPAR дельта; (h) агонисты PPAR альфа/дельта, такие как мураглитазар и соединения, раскрытые в патенте США №6414002; и (i) агенты против ожирения, такие как (1) секретагоги гормона роста, агонисты/антагонисты рецепторов секретагогов гормона роста, такие как NN703, гексарелин, МК-0677, SM-130686, СР-424,391, L-692,429 и L-163,255; (2) ингибиторы протеинтирозинфосфатазы-1В (РТР-1В); (3) лиганды каннабиноидных рецепторов, такие как антагонисты или обратные агонисты каннабиноидного рецептора CB1, такие как римонабант (Sanofi Synthelabo), АМТ-251 и SR-14778 и SR 141716А (Sanofi Synthelabo), SLV-319 (Solvay), BAY 65-2520 (Bayer); (4) серотонинергические агенты против ожирения, такие как фенфлурамид, дексфенфлурамин, фентермин и сибутрамин; (5) агонисты бета-3-адренорецептора, такие как AD9677/TAK677 (Dainippon/Takeda), CL-316,243, SB 418790, BRL-37344, L-796568, BMS-196085, BRL-35135A, CGP12177A, BTA-243, трекадрин, Zeneca D7114, SR 59119A; (6) ингибиторы панкреатической липазы, такие как орлистат (Xenical.RTM.), Triton WR1339, RHC80267, липстатин, тетрагидролипстатин, теасапонин, диэтилумбеллиферилфосфат; (7) антагонисты нейропептида Y1, такие как BIBP3226, J-115814, BIBO 3304, LY-357897, СР-671906, GI-264879A; (8) антагонисты нейропептида Y5, такие как GW-569180A, GW-594884A, GW-587081X, GW-548118X, FR226928, FR 240662, FR252384, 1229U91, GI-264879A, CGP71683A, LY-377897, PD-160170, SR-120562А, SR-120819A и JCF-104; (9) антагонисты рецепторов меланин-концентрирующего гормона (МСН); (10) антагонисты рецептора меланин-концентрирующего гормона 1 (MCH1R), такие как Т-226296 (Takeda); (11) агонисты/антагонисты рецептора меланин-концентрирующего гормона 2 (MCH2R); (12) антагонисты рецепторов орексина, такие как SB-334867-A и те, которые раскрыты в патентных публикациях, указанных в данном описании; (13) ингибиторы обратного захвата серотонина, такие как флуксетин, пароксетин и сертралин; (14) агонисты меланокортина, такие как Melanotan II; (15) другие агонисты Mc4r (рецептор меланокртина 4), такие как CHIR86036 (Chiron), МЕ-10142 и МЕ-10145 (Melacure), CHIR86036 (Chiron); РТ-141 и РТ-14 (Palatin); (16) агонисты 5НТ-2; (17) агонисты 5НТ2С (рецептор серотонина 2С), такие как BVT933, DPCA37215, WAY161503, R-1065; (18) антагонисты галанина; (19) агонисты ССК; (20) агонисты ССК-А (холецистокинин-А), такие как AR-R 15849, GI 181771, JMV-180, А-71378, А-71623 и SR14613; (22) агонисты кортикотропи-высвобождающего гормона; (23) модуляторы гистаминового рецептора-3 (Н3); (24) антагонисты/обратные агонисты гистаминового рецептора-3 (Н3), такие как гиоперамид, 3-(1Н-имидазол-4-ил)пропил-N-(4-пентенил)карбамат, клобенпропит, йодофенпропит, имопроксифан, GT2394 (Gliatech) и O-[3-(1Н-имидазол-4-ил)пропанол]-карбаматы; (25) ингибиторы бета-гидрокси стероидной дегидрогеназы-1 (бета-HSD-1); 26) ингибиторы PDE (фосфодиэстеразы), такие как теофиллин, пентоксифиллин, запринаст, силденафил, аминон, милринон, цилостамид, ролипрам и циломиласт; (27) ингибиторы фосфодиэстеразы-3В (PDE3B); (28) ингибиторы транспорта NE (норепинефрина), такие как GW 320659, деспирамин, талсупрам и номифензин; (29) обратный агонист или антагонист второго или третьего рецептора грелина; (30) лептин, в том числе рекомбинантный лептин человека (PEG-OB, Hoffman La Roche) и рекомбинантный метионильный лептин человека (Amgen); (31) производные лептина; (32) агонисты BRS3 (рецептора бомбезина подтипа 3), такие как [D-Phe6,beta-Ala11,Phe13,Nle14]Bn(6-14) и [D-Phe6,Phe13]Bn(6-13)пропиламид и те соединения, которые раскрыты в Pept. Sci. 2002 August; 8(8): 461-75); (33) CNTF (цилиарные нейротрофические факторы), такие как GI-181771 (GlaxoSmithKline), SR146131 (Sanofi Synthelabo), бутабиндид, PD170,292 и PD 149164 (Pfizer); (34) производные CNTF, такие как аксокин (Regeneron); (35) ингибиторы обратного захвата моноаминов, такие как сибутрамин; (36) активаторы UCP-1 (разобщающий белок-1), 2 или 3, такие как фитановая кислота, 4-[(Е)-2-(5,6,7,8-тетрагидро-5,5,8,8-тетраметил-2-нафталенил)-1-пропенил]бензойная кислота (TTNPB), ретиноевая кислота; (37) агонисты тиреоидного гормона бета, такие как КВ-2611 (KaroBioBMS); (38) ингибиторы FAS (синтаза жирных кислот), такие как Cerulenin и С75; (39) ингибиторы DGAT1 (диацилглицеролацилтрансфераза 1); (40) ингибиторы DGAT2 (диацилглицеролацилтрансфераза 2); (41) ингибиторы АСС2 (ацетил-СоА-карбоксилаза-2); (42) глюкортикоидные антагонисты; (43) ацил-эстрогены, такие как олеоил-эстрон, раскрытый в del Mar-Grasa, М. et al., Obesity Research, 9:202-9 (2001); (44) ингибиторы дипептидилпептидазы IV (DP-IV), такие как тиазолидид изолейцина, пирролидид валина, NVP-DPP728, LAF237, МК-431, Р93/01, TSL 225, ТМС-2А/2 В/2С, FE 999011, Р9310/К364, VIP 0177, SDZ 274-444; (46) ингибиторы переносчика дикарбоксилатов; (47) ингибиторы переносчика глюкозы; (48) ингибиторы переносчика фосфатов; (49) метформин (Glucophage.RTM.); и (50) топирамат (Topimax.RTM.); и (50) пептид YY, PYY 3-36, аналоги, производные и фрагменты пептида YY, такие как BIM-43073D, BIM-43004С (Olitvak, D.A. et al., Dig. Dis. Sci. 44(3):643-48 (1999)); (51) агонисты рецепторов нейропептида Y2 (NPY2), такие как NPY3-36, N-ацетил-[Leu(28,31)] NPY 24-36, TASP-V и цикло-(28/32)-Ac-[Lys28-Glu32]-(25-36)-pNPY; (52) агонисты нейропептида Y4 (NPY4), такие как панкреатический пептид (РР), и другие агонисты Y4, такие как 1229U91; (54) ингибиторы циклооксигеназы-2, такие как эторикоксиб, целекоксиб, вальдекоксиб, парекоксиб, лумиракоксиб, BMS347070, тиракоксиб или JTE522, АВТ963, CS502 и GW406381 и их фармацевтически приемлемые соли; (55) антагонисты нейропепетида Y1 (NPY1), такие как BIBP3226, J-115814, BIBO 3304, LY-357897, СР-671906, GI-264879А; (56) антагонисты опиоидов, такие как налмефен (Revex.RTM.), 3-метоксиналтрексон, налоксон, налтрексон; (57) ингибитор 11-6eTa-HSD-1 (11-бета-гидроксистероид-дегидрогеназа типа 1), такой как BVT 3498, BVT 2733; (58) минорекс; (59) амфехлорал; (60) амфетамин; (61) бензфетамин; (62) хлорфентермин; (63) клобензорекс; (64) клофорекс; (65) кломинорекс; (66) клотермин; (67) циклекседрин; (68) дектроамфетамин; (69) дифеметоксидин, (70) N-этиламфетамин; (71) фенбутразат; (72) фенисорекс; (73) фенпропорекс; (74) флудорекс; (75) флуминорекс; (76) фурфурилметиламфетамин; (77) левафетамин; (78) левофацетоперан; (79) мефенорекс; (80) метамфепрамон; (81) метамфетамин; (82) норпсевдоэфедрин; (83) пенторекс; (84) фендиметразин; (85) фенметразин; (86) пицилорекс; (87) фитофарм 57; и (88) зонисамид.
Использованный в данном документе термин "фармацевтически приемлемая соль" означает те соли, которые в рамках обоснованного медицинского суждения являются подходящими для использования в контакте с тканями пациентов и низших животных без чрезмерной токсичности, раздражения, аллергической реакции и т.п. и соразмерными с приемлемым соотношением польза/риск. Фармацевтически приемлемые соли общеизвестны в данной области. Например, S. М. Berge et al. подробно описывают фармацевтически приемлемые соли в Berge et al., J. Pharmaceutal Sciences, 1977, 66: 1-19. Соли могут быть получены in situ в процессе конечного выделения и очистки соединения Примера 1 по настоящему изобретению или отдельно в результате взаимодействия соединения Примера 1 в форме свободного основания с органической или неорганической кислотой. Репрезентативные соли присоединения кислоты включают, без ограничения, ацетат, адипат, альгинат, цитрат, аспартат, бензоат, бензолсульфонат, бикарбонат, бисульфат, бутират, камфорат, камфорсульфонат, цитрат, диглюконат, глицерофосфат, гемисульфат, гептаноат, гексаноат, фумарат, гидрохлорид, гидробромид, гидройодид, 2-гидроксиэтансульфонат (изетионат), лактат, малеат, метансульфонат, никотинат, 2-нафталинсульфонат, оксалат, памоат, пектинат, персульфат, 3-фенилпропионат, фосфат, пикрат, пивалат, пропионат, сукцинат, сульфат, тартрат, тиоцианат и пара-толуолсульфонат.
Использованный в данном документе термин "рецептор грелина" означает сопряженный с G-белком рецептор, известный как рецептор секретагога гормона роста (GHSR1a или GHS-1aR).
Использованный в данном документе термин "расстройства сна" охватывает приблизительно 70 синдромов, характеризующихся нарушением количества сна, качества сна или распределения сна во времени, или поведенческих или физиологических состояний, связанных со сном, у пациента. Репрезентативные примеры синдромов расстройства сна включают, без ограничения, бессонницу, первичную бессонницу, апноэ во сне, нарколепсию, синдром беспокойных ног, нарушение циркадного ритма сна, нарушение поведения в фазе сна REM (быстрый сон), сомнабулизм (хождение во сне), бруксизм во сне (скрежетание зубами), гиперсомнию, синдром "взрывающейся головы", сонный паралич и чрезмерную сонливость в дневное время (EDS).
EDS является одним из типичных признаков у пациентов с синдромом Прадера-Вилли (PWS). Те, кто ухаживает за пациентами, описывают детей с PWS как вялых и очень часто дремлющих в дневное время. При многочисленном тестировании периода ожидания сна EDS характеризуется пониженным периодом ожидания начала сна как в ночное время, так и в дневное время. Родительские сообщения и вопросники свидетельствуют о наличии EDS у 90-100% взрослых с PWS. Пациенты с PWS по их собственным оценкам также имеют более высокие уровни EDS по сравнению с другими группами с интеллектуальными нарушениями и контрольными группами без PWS.
"NES характеризуется по типу характера приема пищи, нарушений сна, клинического курса и семейной агрегации. В сравнении со здоровыми контрольными группами, лица с NES расходуют значительно больше калорий после вечерней еды, чаще просыпаются ночью и, по-видимому, больше едят, просыпаясь, чем контрольные группы" (Vander Wal, указанный выше источник информации, р. 50, упоминание внутренней Таблицы и ссылки опущены).
Использованный в данном документе термин "пациент" означает человек.
Согласно настоящему изобретению предложены также фармацевтические композиции, содержащие обратный агонист или антагонист рецептора грелина, в частности соединение Примера 1, приготовленный вместе с одним или более нетоксичными фармацевтически приемлемыми носителями. Фармацевтические композиции могут быть приготовлены специально для перорального введения в твердой или жидкой форме, для парентеральных инъекций или для ректального введения.
Использованный в данном документе термин "фармацевтически приемлемый носитель" означает нетоксичный инертный твердый, полутвердый или жидкий наполнитель, разбавитель, инкапсулирующий материал или технологическое вспомогательное вещество любого типа. Некоторыми примерами веществ, которые могут служить фармацевтически приемлемыми носителями, являются сахара, такие как лактоза, глюкоза и сахароза; крахмалы, такие как кукурузный крахмал и картофельный крахмал; целлюлоза и ее производные, такие как натрий-карбоксиметилцеллюлоза, этилцеллюлоза и ацетат целлюлозы; порошкообразный трагакант; солод; желатин; тальк; эксципиенты, такие как масло какао и суппозиторные воски; масла, такие как арахисовое масло, хлопковое масло, подсолнечное масло, кунжутное масло, оливковое масло, кукурузное масло и соевое масло; гликоли; такие как пропиленгликоль; сложные эфиры, такие как этилолеат и этиллаурат; агар; буферные агенты, такие как гидроксид магния и гидроксид алюминия; альгиновая кислота; апирогенная вода; изотонический физиологический раствор; раствор Рингера; этиловый спирт и фосфатные буферные растворы, а также другие нетоксичные совместимые смазывающие вещества, такие как лаурилсульфат натрия и стеарат магния; окрашивающие агенты, высвобождающие агенты, покрывающие агенты, подсластители, корригенты и отдушки, консерванты и антиоксиданты также могут присутствовать в композиции по усмотрению изготовителя. Согласно настоящему изобретению предложены фармацевтические композиции, содержащие обратный агонист или антагонист рецептора грелина, в частности соединение Примера 1, приготовленные вместе с одним или более нетоксичными фармацевтически приемлемыми носителями. Фармацевтические композиции могут быть приготовлены для перорального введения в твердой или жидкой форме, для парентеральных инъекций или для ректального введения.
Фармацевтические композиции по данному изобретению можно вводить пациентам перорально, парентерально, интраперитонеально, местно (в виде порошков, мазей или капель), трансбуккально или в виде перорального или назального спрея. Использованный в данном документе термин "парентерально" относится к способу введения, включающему внутривенную, внутримышечную, интраперитонеальную, интрастернальную, подкожную, интраартрикулярную инъекцию и инфузию.
Фармацевтические композиции по данному изобретению для парентеральных инъекций включают фармацевтически приемлемые стерильные водные или неводные растворы, дисперсии, суспензии или эмульсии и стерильные порошки для разведения в стерильных растворах или дисперсиях для инъекций. Примеры подходящих водных и неводных носителей, разбавителей, растворителей или наполнителей включают воду, этанол, полиолы (пропиленгликоль, полиэтиленгликоль, глицерин и т.п.), их подходящие смеси, растительные масла (такие как оливковое масло) и инъекционные органические сложные эфиры, такие как этилолеат. Надлежащую текучесть можно поддерживать, например, за счет использования покрытия, такого как лецитин, за счет поддерживания требуемого размера частиц в случае дисперсий и за счет использования поверхностно-активных веществ.
Эти композиции могут содержать также вспомогательные вещества, такие как консерванты, увлажняющие агенты, эмульгаторы и диспергирующие агенты. Предотвращение воздействия микроорганизмов может быть обеспечено различными антибактериальными и противогрибковыми агентами, например парабенами, хлорбутанолом, фенолом, сорбиновой кислотой и т.п. Может также потребоваться включение в состав изотонических агентов, например сахаров, хлорида натрия и т.п. Пролонгированное всасывание инъекционной фармацевтической формы может быть осуществлено за счет использования агентов, задерживающих всасывание, например моностеарата алюминия и желатина.
В некоторых случаях, для того чтобы пролонгировать эффект лекарственного средства, часто бывает желательным замедление всасывания лекарственного средства при подкожной или внутримышечной инъекции. Это осуществимо за счет использования жидкой суспензии кристаллического или аморфного вещества с плохой растворимостью в воде. Скорость всасывания лекарственного средства в таком случае зависит от скорости его растворения, которая, в свою очередь, может зависеть от размера кристаллов и кристаллической формы. Альтернативно, замедленное всасывание парентерально вводимой лекарственной формы осуществляют путем растворения или суспендирования лекарственного средства в масляном носителе.
Суспензии, в дополнение к обратному агонисту или антагонисту рецептора грелина, в частности соединению Примера 1, могут содержать суспендирующие агенты, такие как, например, этоксилированные изостеариловые спирты, сложные эфиры полиоксиэтиленсорбита и сорбитана, микрокристаллическая целлюлоза, метагидроксид алюминия, бентонит, агар-агар, трагакант и их смеси.
Если желательно и с целью более эффективного распределения, обратный агонист или антагонист рецептора грелина, в частности соединение Примера 1 по настоящему изобретению, может быть включен в системы медленного высвобождения или системы, обеспечивающие направленную доставку к мишени, такие как полимерные матрицы, липосомы и микросферы. Они могут быть подвергнуты стерилизации, например посредством фильтрации через удерживающий бактерии фильтр или за счет включения в состав стерилизующих агентов в форме стерильных твердых композиций, которые могут быть растворены в стерильной воде или другой стерильной инъекционной среде непосредственно перед использованием.
Обратный агонист или антагонист рецептора грелина, в частности соединение Примера 1, может также находиться в микроинкапсулированной форме, если это целесообразно, с одним или более фармацевтически приемлемыми носителями, которые указаны выше. Твердые лекарственные формы таблеток, драже, капсул, пилюль и гранул могут быть изготовлены с покрытиями или оболочками, такими как энтеросолюбильные покрытия, покрытия, контролирующие высвобождение, и другие покрытия, известные в области приготовления фармацевтических препаратов. В таких твердых лекарственных формах обратный агонист или антагонист рецептора грелина, в частности соединение Примера 1, может быть смешан с по меньшей мере одним инертным разбавителем, таким как сахароза, лактоза или крахмал. Такие лекарственные формы могут также содержать, как это принято на практике, дополнительные вещества, иные, чем инертные разбавители, например смазывающие вещества для таблетирования и другие вспомогательные вещества для таблетирования, такие как стеарат магния и микрокристаллическая целлюлоза. В случае капсул, таблеток и пилюль лекарственные формы могут также содержать буферные агенты. Они возможно могут содержать матирующие агенты и могут также представлять собой такую композицию, которая обеспечивает высвобождение только активного(ых) ингредиента(ов) или, предпочтительно, в определенной части кишечного тракта замедленным образом. Примеры заливочных композиций, которые могут быть использованы, включают полимерные вещества и воски.
Инъецируемые депо-формы изготавливают путем формирования микроинкасулированных матриц лекарственного средства в биоразлагаемых полимерах, таких как полиактид-полигликолид. В зависимости от соотношения лекарственного средства к полимеру и природы конкретного используемого полимера, скорость высвобождения лекарственного средства можно контролировать. Примеры других биоразлагаемых полимеров включают поли(ортоэфиры) и поли(ангидриды). Инъецируемые депо-формы также изготавливают путем захватывания лекарственного средства в липосомы или микроэмульсии, которые совместимы с тканями организма.
Инъекционные композиции могут быть подвергнуты стерилизации, например фильтрованием через удерживающий бактерии фильтр или за счет включения стерилизующих агентов в форме стерильных твердых композиций, которые могут быть растворены или диспергированы в стерильной воде или другой стерильной среде для инъекций непосредственно перед использованием.
Препараты для инъекций, например стерильные водные или масляные суспензии могут быть приготовлены известными в данной области способами с использованием подходящих диспергирующих или увлажняющих агентов и суспендирующих агентов. Стерильный препарат для инъекций может также представлять собой инъекционный стерильный раствор, суспензию или эмульсию в нетоксичном, парентерально приемлемом разбавителе или растворителе, например раствор в 1,3-бутандиоле. Среди доступных носителей и растворителей, которые могут быть использованы, находятся вода, раствор Рингера в соответствии с Фармакопеей США и изотонический раствор хлорида натрия. Кроме того, в качестве растворителя или суспендирующей среды обычно используют стерильные нелетучие масла. Для этой цели можно использовать любое мягкое нелетучее масло, в том числе синтетические моно- или диглицериды. Кроме того, в приготовлении композиций для инъекций используют жирные кислоты, такие как олеиновая кислота.
Твердые лекарственные формы для перорального введения включают капсулы, таблетки, пилюли, порошки и гранулы. В таких твердых лекарственных формах обратный агонист или антагонист рецептора грелина, в частности соединение Примера 1, смешан с по меньшей мере одним инертным фармацевтически приемлемым носителем, таким как цитрат натрия или фосфат кальция, и/или а) наполнителями или веществами, увеличивающими объем, такими как крахмалы, лактоза, сахароза, глюкоза, маннит и салициловая кислота; b) связывающими веществами, такими как карбоксиметилцеллюлоза, альгинаты, желатин, поливинилпирролидон, сахароза и аравийская камедь; с) увлажнителями, такими как глицерин; d) разрыхлителями, такими как агар-агар, карбонат кальция, картофельный или тапиоковый крахмал, альгиновая кислота, некоторые силикаты и карбонат натрия; е) агентами, замедляющими растворение, такими как парафин; f) ускорителями всасывания, такими как четвертичные аммониевые соединения; g) смачивающими агентами, такими как цетиловый спирт и моностеарат глицерина; h) абсорбентами, такими как каолин и бентонитовая глина; и i) смазывающими веществами, такими как тальк, стеарат кальция, стеарат магния, твердые полиэтиленгликоли, лаурилсульфат натрия и их смеси. В случае капсул, таблеток и пилюль лекарственная форма может также содержать буферные агенты.
Твердые композиции подобного типа могут быть использованы также в качестве наполнителей в мягких и твердых желатиновых капсулах с использованием лактозы или молочного сахара, а также высокомолекулярных полиэтиленгликолей и т.п.
Твердые лекарственные формы таблеток, драже, капсул, пилюль и гранул могут быть изготовлены с покрытиями и оболочками, такими как энтеросолюбильные покрытия и другие покрытия, известные в области приготовления фармацевтических препаратов. Они возможно могут содержать матирующие агенты и могут также представлять собой такую композицию, которая обеспечивает высвобождение только активного(ых) ингредиента(ов) или, предпочтительно, в определенной части кишечного тракта задержанным образом. Примеры заливочных композиций, которые могут быть использованы, включают полимерные вещества и воски.
Жидкие лекарственные формы для перорального введения включают фармацевтически приемлемые эмульсии, микроэмульсии, растворы, суспензии, сиропы и эликсиры. В дополнение к обратному агонисту или антагонисту рецептора грелина, в частности соединению Примера 1, жидкие лекарственные формы могут содержать инертные разбавители, обычно используемые в данной области, такие как, например, вода или другие растворители, солюбилизирующие агенты и эмульгаторы, такие как этиловый спирт, изопропиловый спирт, этилкарбонат, этилацетат, бензиловый спирт, бензилбензоат, пропиленгликоль, 1,3-бутиленгликоль, диметилформамид, масла (в частности, хлопковое, арахисовое, кукурузное, масло проростков пшеницы, оливковое, касторовое и кунжутное масло), глицерин, тетрагидрофурфуриловый спирт, полиэтиленгликоли и сложные эфиры жирных кислот с сорбитаном и их смеси.
Помимо инертных разбавителей пероральные композиции могут также содержать вспомогательные вещества, такие как смачивающие агенты, эмульгаторы и суспендирующие агенты, подсластители, корригенты и отдушки.
Фактические уровни дозировок обратного агониста или антагониста рецептора грелина, в частности соединения Примера 1, в фармацевтических композициях по данному изобретению можно варьировать, чтобы получить количество обратного агониста или антагониста рецептора грелина, в частности соединения Примера 1, которое является эффективным для достижения желаемого терапевтического эффекта для конкретного пациента, композиций и способа введения. Выбранный уровень дозировки будет зависеть от активности конкретного обратного агониста или антагониста рецептора грелина, пути введения, тяжести состояния, которое лечат, и состояния и предшествующей истории болезни пациента, которого лечат.
Суммарная суточная доза обратного агониста или антагониста рецептора грелина, в частности соединения Примера 1 по данному изобретению, вводимая пациенту, составляет от примерно 0,003 до примерно 10 мг/кг/сутки. В целях перорального введения более предпочтительные дозы могут находиться в диапазоне от примерно 0,01 до примерно 5 мг/кг/сутки. Предпочтительным является диапазон доз от 25 до 300 мг в сутки. Наиболее предпочтительным является диапазон от 75 до 200 мг в сутки. При желании, эффективная суточная доза может быть разделена на множество доз в целях введения, например, от двух до четырех доз в сутки.
Все патенты, патентные заявки и литературные источники информации, процитированные в данном описании изобретения, во всей их полноте включены в него посредством ссылки.
(R)-2-(2-Метилимидазо[2,1-b]тиазол-6-ил)-1-(2-(5-(6-метилпиримидин-4-ил)-2,3-дигидро-1Н-инден-1-ил)-2,7-диазаспиро[3.5]нонан-7-ил)этанон может быть получен с использованием синтетической методологии, раскрытой в данном документе или в US 2011/0230461.
Пример 1
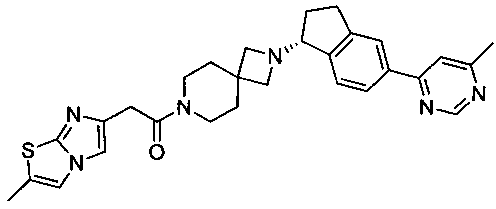
(R)-2-(2-Метилимидазо[2,1-b]тиазол-6-ил)-1-(2-(5-(6-метилпиримидин-4-ил)-2,3-дигидро-1Н-инден-1-ил)-2,7-диазаспиро[3.5]нонан-7-ил)этанон
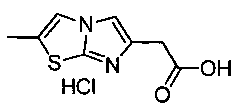
Гидрохлорид 2-(2-метилимидазо[2,1-b]тиазол-6-ил)уксусной кислоты
Раствор брома (436 г, 2,73 моль) в уксусной кислоте (750 мл) добавляли в раствор этил-3-оксобутаноата (355 г, 2,73 моль) в уксусной кислоте (1000 мл). Эту смесь перемешивали при комнатной температуре в течение 72 часов и концентрировали при пониженном давлении при 45°С для удаления уксусной кислоты. Остаток распределяли между метиленхлоридом (400 мл) и водой (250 мл). Органический слой промывали насыщенным раствором бикарбоната натрия (2×300 мл), водой (300 мл), рассолом (125 мл) и сушили над безводным сульфатом магния. Раствор фильтровали и концентрировали с получением этил-4-бром-3-оксобутаноата в виде желтого масла (421 г).
В раствор 2-амино-5-метилтиазола (150 г, 1,31 моль) в ацетоне (1500 мл) медленно добавляли этил-4-бром-3-оксобутаноат (345 г, 1,65 моль). Поддерживали температуру реакционной смеси 22-40°С. Смесь превращалась в густую пасту, и для облегчения перемешивания добавляли ацетон (300 мл). После перемешивания при комнатной температуре в течение ночи смесь фильтровали, и остаток на фильтре промывали ацетоном с получением белого твердого вещества. Это твердое вещество промывали гексанами и сушили в вакуумном шкафу при 40°С в течение 4 часов с получением гидробромида этилового эфира 4-(2-амино-5-метил-тиазол)-3-оксомасляной кислоты (272 г).
В гидробромид этилового эфира 4-(2-амино-5-метил-тиазол)-3-оксомасляной кислоты (272 г, 0,84 моль) добавляли безводный этанол (675 мл), и густую смесь нагревали при 90°С в течение 2 часов. За это время твердое вещество переходило в раствор. Реакционную смесь концентрировали с получением коричневого полутвердого вещества, которое растирали с этанолом с получением белого рыхлого твердого вещества, которое собирали фильтрованием. Твердое вещество промывали Et2O и сушили в вакууме при 40°С в течение 4 часов с получением гидробромида этил-(2-метилимидазо[2,1-b][1,3]тиазол-6-ил)ацетата (226 г).
Гидробромид этил-(2-метилимидазо[2,1-b][1,3]тиазол-6-ил)ацетата (226 г, 0,74 моль) растворяли в воде (350 мл), и раствор доводили до рН 7 добавлением карбоната калия (51,0 г, 0,37 моль). Водный раствор экстрагировали метиленхлоридом (300 мл), и органическую фазу промывали рассолом (150 мл), сушили над безводным сульфатом магния, фильтровали и концентрировали с получением этил-(2-метилимидазо[2,1-b][1,3]тиазол-6-ил)ацетата в виде коричневого масла (151,3 г).
Этил-(2-метилимидазо[2,1-b][1,3]тиазол-6-ил)ацетат (151,3 г, 0,67 моль) растворяли в 10%-ном водном растворе HCl (435 мл), и смесь нагревали при температуре дефлегмации в течение 2 часов. Реакционную смесь охлаждали до комнатной температуры и концентрировали в вакууме с получением желтого масла. Добавляли этанол (100 мл) и диэтиловый эфир (200 мл), и полученный белый осадок собирали фильтрованием и сушили в вакуумном шкафу в течение ночи с получением 144,3 г (93%) указанного в заголовке соединения. МС (ЭРИ+) (масс-спектрометрия с электрораспылительной ионизацией с регистрацией положительных ионов) 197,1 (М+Н)+. 1Н ЯМР (CD3OD) δ 2.48 (s, 3Н), 3.88 (s, 2Н), 7.81 (s, 1Н), 7.85 (s, 1Н).
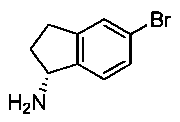
(R)-5-Бром-2,3-дигидро-1Н-инден-1-амин
В 5-горлую круглодонную колбу вместимостью 22 л загружали 5-бром-1-инданон (1,0 кг, 4,72 моль), безводный THF (8 л) и (R)-метил-CBS-оксаазаборолидин (730 мл, 0,73 моль), и эту смесь охлаждали до -10°С в атмосфере N2. По каплям добавляли боран-метилсульфид (10,0 М, 650 мл, 6,5 моль) в течение 1 часа, поддерживая температуру ниже -5°С. Смесь перемешивали при температуре от -10°С до 0°С в течение 3 часов и гасили водой (4 л) с такой скоростью, чтобы реакционная температура поддерживалась ниже 5°С. Смесь экстрагировали EtOAc (3×3 л). Объединенные органические экстракты промывали рассолом (2 л), сушили над MgSO4, фильтровали и концентрировали с получением желтого твердого вещества. Этот неочищенный продукт пропускали через короткую силикагелевую колонку (упакованную 3 л силикагеля с 1% Et3N в гексанах) и элюировали смесью EtOAc/гексаны (1/3). Фильтрат концентрировали, и остаток суспендировали в 10% EtOAc в гексанах, фильтровали и сушили с получением 585 г (S)-5-бром-индан-1-ола в виде не совсем белого твердого вещества. Маточные жидкости снова концентрировали, суспендировали в 10% EtOAc в гексанах и фильтровали с получением еще 200 г (S)-5-бром-индан-1-ола в виде желтого твердого вещества. Объединенные партии (785 г, 78%) переносили на следующую стадию без дополнительной очистки.
Раствор (S)-5-бром-индан-1-ола (288 г, 1,35 моль) в толуоле (2 л) охлаждали в ледяной бане в атмосфере N2 и обрабатывали дифенилфосфорилазидом (DPPA, 400 мл, 1,85 моль) одной порцией, затем раствором 1,8-диазабицикло[5,4,0]ундец-7-ена (300 мл, 2,01 моль) в толуоле (600 мл). Реакционную температуру поддерживали от 3 до 10°С во время добавления в течение 3 часов, и смесь нагревали до 15°С в течение 3 часов (ТСХ (тонкослойная хроматография) показала отсутствие исходного вещества). Смесь разбавляли EtOAc (1 л) и промывали водой (3×2 л). Органический слой сушили над MgSO4, фильтровали и концентрировали с получением 516 г темного масла. Этот неочищенный продукт очищали на силикагелевой колонке (упакованной 1% Et3N в гексанах, гексановый элюент) с получением (R)-1-азидо-5-бром-индана (291 г, 90%) в виде масла, которое напрямую использовали на следующей стадии.
Раствор (R)-1-азидо-5-бром-индана (154 г, 0,645 моль) растворяли в метаноле (2,4 л) и добавляли SnCl2·2H2O (265 г, 1,18 моль). Смесь перемешивали при комнатной температуре в течение ночи (ТСХ показала отсутствие исходного вещества) и концентрировали досуха. Полученный остаток обрабатывали 2 н. водным раствором NaOH (2,5 л) и EtOAc (1,5 л). Смесь перемешивали в течение 1 часа и фильтровали через Celite® с помощью EtOAc (3×250 мл). Органический раствор отделяли, и водный слой экстрагировали EtOAc (3×2 л). Объединенные органические экстракты промывали 1 н. HCl (2×2 л), затем водой (2 л). рН объединенных водных слоев доводили до значения 11 холодным насыщенным раствором NaOH, и смесь экстрагировали EtOAc (3×2 л). Объединенные органические экстракты сушили над MgSO4, фильтровали и концентрировали с получением (87,5 г, 64,0%) (R)-5-бром-2,3-дигидро-1Н-инден-1-амина в виде темно-желтого масла, которое затвердевало после охлаждения. МС (ЭРИ+) 213,9 (М+Н)+. 1Н ЯМР (CDCl3) δ 1.70-1.75 (m, 1Н), 2.40-2.45 (m, 1Н), 2.77-2.82 (m, 1Н), 2.93-2.97 (m, 1Н), 4.28-4.33 (m, 1Н), 7.18-7.23 (m, 1Н), 7.36-7.41 (m, 2Н).
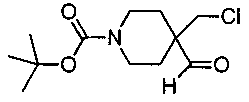
трет-Бутил-4-(хлорметил)-4-формилпиперидин-1-карбоксилат
В раствор диизопропиламина (22,6 мл, 159 ммоль) в безводном THF (140 мл) в высушенной в термостате круглодонной колбе по каплям добавляли n-BuLi (65,4 мл, 163 ммоль, 2,50 М в гексанах) при 0°С. Раствор перемешивали в течение 45 минут и по каплям добавляли 1-трет-бутил-4-метил-пиперидин-1,4-дикарбоксилат (20 г, 80 ммоль) в THF (60 мл) при 0°С, и смесь перемешивали при 0°С в течение 1 часа. По каплям добавляли хлорйодметан (17,9 мл, 239 ммоль), и смесь перемешивали в течение 1 ч. Смесь гасили 250 мл насыщенного водного раствора NaHCO3, затем экстрагировали этилацетатом (3×250 мл). Объединенные органические слои промывали (рассолом, 250 мл), сушили (Na2SO4) и концентрировали при пониженном давлении с получением желтого масла, которое очищали хроматографией на силикагеле с использованием системы очистки Combiflash ISCO (Teledyne Isco Inc., Lincoln, NE) с получением 1-трет-бутил-4-метил-4-(хлорметил)пиперидин-1,4-дикарбоксилата (12 г, 52%). 1Н ЯМР (CDCl3) δ 1.43 (s, 9Н), 2.10-2.17 (m, 4Н), 2.97 (br s, 2H), 3.56 (s, 2H), 3.74 (s, 3Н), 3.83 (br s, 2H).
Раствор 1-трет-бутил-4-метил-4-(хлорметил)пиперидин-1,4-дикарбоксилата (11,7 г, 40,2 ммоль) в безводном THF (100 мл) охлаждали до 0°С. Медленно (в течение 15-20 мин) добавляли алюмогидрид лития (1 н. в THF, 44,3 мл, 44,3 ммоль), и раствор перемешивали при 0°С в течение 25 минут. Смесь гасили добавлением воды (1,8 мл) по каплям с большой осторожностью. По каплям добавляли водный 1 н. NaOH (1,8 мл), и смесь перемешивали в течение 5 минут. Охлаждающую баню удаляли, твердое вещество отфильтровывали, и остаток на фильтре промывали Et2O (2×100 мл). Фильтрат промывали водой (2×100 мл), рассолом (100 мл), сушили (Na2SO4) и концентрировали при пониженном давлении с получением трет-бутил-4-(хлорметил)-4-(гидроксиметил)пиперидин-1-карбоксилата в виде твердого вещества (9,96 г, 93,8%). 1Н ЯМР (CDCl3) δ 1.43 (s, 9Н), 1.48-1.55 (m, 4Н), 3.36-3.41 (m, 4Н), 3.57 (s, 2Н), 3.59 (br s, 2Н).
В раствор оксалилхлорида (5,1 мл, 57 ммоль) в дихлорметане (100 мл) в высушенной в термостате круглодонной колбе при -78°С добавляли диметилсульфоксид (8,2 мл, 114 ммоль) в дихлорметане (17 мл). Смесь перемешивали в течение 2 минут и добавляли трет-бутил-4-(хлорметил)-4-(гидроксиметил)пиперидин-1-карбоксилат (13,7 г, 52 ммоль) в дихлорметане (50 мл) в течение 10 минут. Раствор перемешивали в течение 15 минут при -78°С и добавляли триэтиламин (36 мл, 260 ммоль). Смесь перемешивали при -78°С в течение 15 минут и нагревали до комнатной температуры. Смесь перемешивали в течение 15 минут при комнатной температуре и гасили насыщенным водным раствором NaHCO3 (200 мл). Водный раствор промывали Et2O (2×300 мл). Объединенные органические слои промывали рассолом, сушили (Na2SO4) и концентрировали при пониженном давлении с получением указанного в заголовке соединения в виде желтого масла, которое затвердевало после стояния в атмосфере азота при комнатной температуре (13,7 г, 99%). 1Н ЯМР (CDCl3) δ 1.43 (s, 9Н), 1.48-1.60 (m, 2Н), 2.00-2.07 (m, 2Н), 3.07 (t, 2Н), 3.57 (s, 2Н), 3.69-3.79 (m, 2Н), 9.55 (s, 1Н).
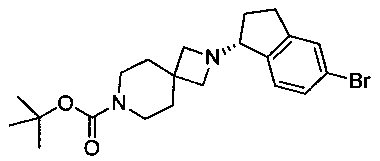
(R)-трет-Бутил-2-(5-бром-2,3-дигидро-1Н-инден-1-ил)-2,7-диазаспиро[3.5]нонан-7-карбоксилат
В раствор (R)-5-бром-2,3-дигидро-1Н-инден-1-амина (1835 г, 8,66 моль) в безводном метаноле (24 л) добавляли трет-бутил-4-(хлорметил)-4-формилпиперидин-1-карбоксилат (2310 г, 8,83 моль). Эту смесь перемешивали при 50°С в течение 16 ч и охлаждали до комнатной температуры. Добавляли цианоборгидрид натрия (1000 г, 15,9 моль) в THF (15 л) посредством шприцевого насоса в течение 2 часов. Смесь перемешивали при 60°С в течение 24 часов в атмосфере азота с отводом в отбеливающую ванну. Реакционную смесь охлаждали до 20°С и переносили через канюлю в сосуд, содержащий 24 л 2,5 М гидроксида натрия и 30 л DCM. Слои разделяли, и водный слой экстрагировали DCM (2×5 л). Водный слой подвергали обработке с целью разложения остаточного цианоборгидрида натрия. Объединенные органические слои сушили (MgSO4) и концентрировали при пониженном давлении. Неочищенное вещество суспендировали в МТВЕ (7 л) путем перемешивания при 40°С в течение 1 ч и при комнатной температуре в течение 1 ч. Твердое вещество отфильтровывали, промывали МТВЕ (2×500 мл) и сушили в вакуумном шкафу при 50°С с получением указанного в заголовке продукта в виде белых кристаллов (3657 г, 90%). МС (ЭРИ+) 422,3 (М+Н)+. 1Н ЯМР (CDCl3) δ 1.44 (s, 9Н), 1.67 (dd, 4Н), 1.84-1.93 (m, 1Н), 2.07-2.16 (m, 1Н), 2.72-2.81 (m, 1Н), 2.95-3.15 (m, 5Н), 3.31 (dd, 4Н), 3.85 (br s, 1Н), 7.12 (d, 1Н), 7.28 (br s, 1Н), 7.35 (br s, 1H). 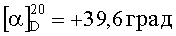 (с=1,06 мг/мл, МеОН).
(с=1,06 мг/мл, МеОН).
(R)-трет-Бутил-2-(5-(6-метилпиримидин-4-ил)-2,3-дигидро-1Н-инден-1-ил)-2,7-диазаспиро[3.5]нонан-7-карбоксилат
В колбу вместимостью 50 мл, содержащую (R)-трет-бутил-2-(5-бром-2,3-дигидро-1Н-инден-1-ил)-2,7-диазаспиро[3.5]нонан-7-карбоксилат (4,0 г, 9,49 ммоль), добавляли хлорид бис(трифенилфосфин)палладия(II) (0,17 г, 0,24 ммоль), ацетат калия (3,73 г, 37,97 ммоль), бис(пинаколато)дибор (2,65 г, 10,44 ммоль), затем дегазировали вакуумированием, после чего заполняли азотом, повторяя 5 раз. Деоксигенированный (поток азота в течение 30 минут перед добавлением) толуол (40 мл) добавляли в смесь, и реакционную смесь нагревали при 100°С в течение 1,5 часов. Ход реакции контролировали методом ВЭЖХ (высокоэффективная жидкостная хроматография). После образования промежуточного сложного эфира бороновой кислоты реакционную смесь охлаждали до 40°С, и в нее загружали дегазированный 4 М раствор гидроксида натрия (11,87 мл, 47,46 ммоль), затем добавляли 4-хлор-6-метилпиримидин (1,53 г, 11,87 ммоль). Полученную смесь затем нагревали до 90°С в течение 5 часов в атмосфере азота. Реакционную смесь охлаждали до комнатной температуры, и в нее загружали воду (25 мл). После перемешивания в течение 20 минут смесь фильтровали для удаления черного твердого вещества. Органический слой экстрагировали в водный раствор, содержащий 1,5 экв. HCl (40 мл). Органический слой удаляли, и полученный раствор обрабатывали силикагелем (4 г) ISOLUTE® Ultra Pure Si-Thiol в течение 1,5 часов и фильтровали. рН водного раствора доводили до значения 7,8 добавлением 4 н. NaOH и экстрагировали толуолом (40 мл). Толуольный слой концентрировали до приблизительно 15 мл в вакууме при 45°С, медленно добавляли гептан (75 мл), и смесь перемешивали при 20°С в течение 1 часа. Продукт фильтровали и сушили в вакууме при 45°С в течение 8 часов с получением указанного в заголовке соединения в виде белого твердого вещества (3,56 г, 86%). МС (ЭРИ+) 435,5 (М+Н)+. 1Н ЯМР (CDCl3) δ 1.46 (s, 9Н), 1.70-1.74 (m, 4Н), 1.90-2.01 (m, 1Н), 2.13-2.26 (m, 1Н), 2.59 (s, 3Н), 2.84-2.93 (m, 1Н), 3.04-3.21 (m, 5Н), 3.30-3.38 (m, 4Н), 3.95-4.02 (m, 1Н), 7.40 (d, 1Н), 7.56 (s, 1Н), 7.87 (d, 1Н), 7.95 (s, 1Н), 9.12 (s, 1Н).
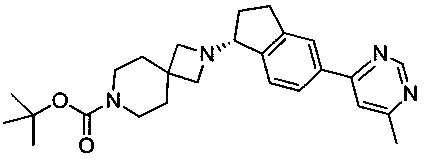
Дигидрохлорид 2-[(E)-5-(6-метил-пиримидин-4-ил)-индан-1-ил]-2,7-диазаспиро[3.5]нонана
(R)-трет-Бутил-2-(5-(6-метилпиримидин-4-ил)-2,3-дигидро-1Н-инден-1-ил)-2,7-диазаспиро[3.5]нонан-7-карбоксилат (72,6 г, 167 ммоль) суспендировали в метаноле (363 мл) и добавляли 4 М HCl в 1,4-диоксане (251 мл). После перемешивания в течение 2 часов суспензию концентрировали досуха. Неочищенное вещество ресуспендировали в МеОН (500 мл) и концентрировали (3×). Полученное твердое вещество затем сушили в вакууме при 45°С с получением указанного в заголовке соединения (74,1 г, 99,9%). МС (ЭРИ+) 335,2 (М+Н)+. 1Н ЯМР (CD3OD) δ 2.16-2.23 (m, 5Н), 2.59 (br s, 1 Н), 2.78-2.80 (m, 3H), 3.12 (brs, 1Н), 3.19-3.24 (m, 4Н), 3.37-3.49 (m, 1Н), 4.14-4.23 (m, 3Н), 4.49 (br s, 1Н), 5.11 (br s, 1Н), 7.84 (d, 1Н), 8.30-8.34 (m, 2Н), 8.46 (s, 1Н), 9.36 (s, 1Н).
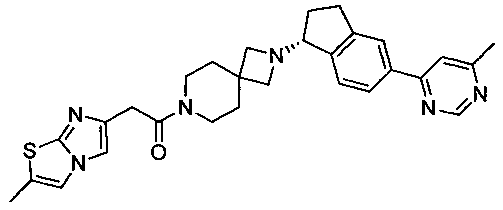
(R)-2-(2-Метилимидазо[2,1-b]тиазол-6-ил)-1-(2-(5-(6-метилпиримидин-4-ил)-2,3-дигидро-1Н-инден-1-ил)-2,7-диазаспиро[3.5]нонан-7-ил)этанон
В суспензию дигидрохлорида 2-[(R)-5-(6-метил-пиримидин-4-ил)-индан-1-ил]-2,7-диаза-спиро[3.5]нонана (540 мг, 1,22 ммоль) в 10 мл дихлорметана добавляли триэтиламин (492 мг, 4,90 ммоль). Как только смесь стала гомогенной, ее добавляли в раствор гидрохлорида 2-(2-метилимидазо[2,1-b]тиазол-6-ил)уксусной кислоты (251 мг, 1,28 ммоль) в 3 мл дихлорметана. Смесь перемешивали в течение 5 минут и добавляли HBTU (гексафторфосфат O-бензотриазол-1-ил-N,N,N′,N′-тетраметилурония) (462 мг, 1,22 ммоль) в 2 мл DMF (диметилформамид). Реакционную смесь перемешивали при комнатной температуре в течение 1,5 часов. Реакционную смесь гасили добавлением 10 мл NaHCO3 и разбавляли 50 мл дихлорметана. Органический слой промывали насыщенным соляным раствором, сушили над Na2SO4, фильтровали, и фильтрат концентрировали. Остаток растворяли в 5 мл CH3CN, и раствор нагревали до 100°С в течение 1 часа при перемешивании. Смесь охлаждали до комнатной температуры, и полученное твердое вещество подвергали вакуумной фильтрации с получением целевого продукта в виде не совсем белого порошка (428 мг, 69%). МС (ЭРИ+) 513,5 (М+Н)+. 1Н ЯМР (CD3OD) δ 1.70-1.74 (m, 4Н), 1.88-1.96 (m, 1Н), 2.27-2.34 (m, 2Н), 2.40 (s, 3Н), 2.58 (s, 3Н), 2.86-2.97 (m, 1Н), 3.11-3.15 (m, 1Н), 3.31-3.34 (m, 3Н), 3.52-3.55 (m, 4Н), 3.78 (s, 2Н), 4.05-4.09 (m, 1Н), 7.33 (s, 2Н), 7.55 (d, 1Н), 7.78-7.79 (m, 1Н), 7.94-8.03 (m, 2Н), 8.93 (s, 1Н). 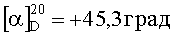 (с=2,5 мг/мл, МеОН).
(с=2,5 мг/мл, МеОН).
АНАЛИЗЫ IN VITRO
АНАЛИЗЫ СВЯЗЫВАНИЯ РАДИОЛИГАНДА
Для измерения способности тестируемого соединения связываться с рецептором грелина и, следовательно, возможности модулировать активность грелина выполняли анализы на вытеснение. Формат SPA (сцинтилляционный проксимальный анализ) использовали для высокопроизводительного скрининга тестируемых соединений, и формат связывания на фильтре служил для более полной характеристики связывания. В обоих форматах аффинность тестируемого соединения выражали в виде значения Ki, определяемого как концентрация соединения, необходимая для снижения связывания [125I]грелина на 50% для конкретной партии мембран при заданной концентрации радиолиганда.
SPA анализ связывания грелина человека
SPA анализ связывания грелина выполняли в конечном объеме 90 мкл, содержащем 250 нг GHSR1a человека (тетрациклин-индуцибельная клеточная линия НЕК293, экспрессирующая рецептор секретагога гормона роста 1а человека; приготовленная в виде мембран), связанных с 0,5 мг SPA гранул (покрытых агглютинином из проростков пшеницы, GE Healthcare, RPNQ0060), и 50 пМ [125I]-грелина (Perkin Elmer Life Sciences, NEX-388) плюс различные концентрации тестируемого соединения или носителя.
Кратко, анализы проводили при комнатной температуре в 384-луночных планшетах (Matrix, 4322), содержащих 2 мкл тестируемого соединения в DMSO (диметилсульфоксид) (или DMSO в качестве носителя). Анализы инициировали добавлением 28 мкл аналитического буфера (50 мМ HEPES, 10 мМ MgCl2, 0,2% BSA (бычий сывороточный альбумин), не содержащие EDTA (этилендиаминтетрауксусная кислота) ингибиторы протеаз - 1 таблетка/50 мл буфера, рН 7,4), 30 мкл 8,3 мкг/мл hGHSR1a мембран и 30 мкл 150 рМ [125I]-грелина, оба в аналитическом буфере.
Смесь инкубировали в течение 8 часов, чтобы дать возможность связыванию достичь равновесия, и количество комплекса рецептор-лиганд определяли жидкостным сцинтилляционным методом подсчета импульсов, используя 1450 Microbeta Trilux (Wallac).
Анализ связывания грелина человека на фильтре
Анализы связывания грелина выполняли в конечном объеме 100 мкл, содержащем 100 нг GHSR1a человека (тетрациклин-индуцибельная клеточная линия НЕК293, экспрессирующая рецептор секретагога гормона роста человека 1а; приготовленная в виде мембран) и 50 пМ [125I]-грелина (Perkin Elmer Life Sciences, NEX-388) плюс различные концентрации тестируемого соединения или носителя.
Кратко, анализы проводили при комнатной температуре в 96-луночных планшетах (Costar, 3357), содержащих 2 мкл тестируемого соединения в DMSO (или DMSO в качестве носителя). Анализы инициировали добавлением 23 мкл аналитического буфера (50 мМ HEPES, 10 мМ MgCl2, 0,2% BSA, не содержащие EDTA таблетки ингибитора протеаз - 1 таблетка/50 мл буфера, рН 7,4), 25 мкл 4 мкг/мл hGHSR1a мембран и 50 мкл 100 рМ [125I]грелина, оба в аналитическом буфере.
Смесь инкубировали в течение 90 минут при комнатной температуре, после чего переносили в обработанный 0,3% PEI (полиэтиленимином) 96-луночный стекловолокнистный фильтр-планшет (Perkin Elmer, 6005174). Смесь сушили вакуум-отсосом и сразу промывали 3 раза по 200 мкл охлажденным во льду 50 мМ Tris рН 7,5. Планшеты оставляли высушиваться в течение ночи при комнатной температуре, и в каждую лунку добавляли 30 мкл сцинтиллятора Supermix (Perkin Elmer, 1200-439). Количество комплекса рецептор-лиганд определяли жидкостным сцинтилляционным методом подсчета импульсов, используя 1450 Microbeta Trilux (Wallac).
Анализы связывания радиолигандов в формате фильтрации для GHSR1a собаки (NM_001099945.1), обезьяны (ХМ_001084886.1), мыши (NM_177330) и крысы (NM_032075) (все экспрессируются в уникальных тетрациклин-индуцибельных клеточных линиях НЕК293) выполняли идентичным образом, как описано для GHSR1a человека, за исключением того, что конечное количество используемых мембран было следующим: 2 мкг GHSR собаки, 250 нг GHSR обезьяны, 200 нг GHSR мыши или 125 нг GHSR крысы.
Функциональный анализ грелина человека
Для измерения способности соединения модулировать активность GHSR1a человека (агонист, антагонист, частичный агонист, обратный агонист) выполняли анализ GTP-связывания DELFIA (флуоресцентный иммуноанализ с лантанидной меткой, флуоресценция которой усилена за счет диссоциации) (Perkin Elmer, AD0260 и AD0261). Этот анализ контролирует лиганд-зависимый обмен GDP (гуанозиндифосфат) на GTP (гуанозинтрифосфат). Активация GPCR (сопряженного с G-белком рецептора) приводит к увеличению флуоресценции, так как связанный с рецептором GDP замещается европий-меченым GTP. Связывание антагониста предотвращает GDP-GTP обмен, тогда как связывание обратного агониста смещает рецептор к GDP-связанному (неактивному) состоянию, причем и то, и другое приводит к снижению флуоресценции.
Функциональные анализы грелина выполняли в конечном объеме 39,5 мкл, содержащем 720 нг GHSR1a человека (тетрациклин-индуцибельная клеточная линия НЕК293, экспрессирующая рецептор секретагога гормона роста 1а человека, приготовленная в виде мембран), 9 нМ GTP-европий и различные концентрации тестируемого соединения или носителя. Для тестирования антагонистического действия на рецептор мембраны инкубировали в присутствии агониста грелина (Anaspec, 24158) в концентрации ЕС80 плюс тестируемое соединение или носитель.
Кратко, тестируемое соединение готовили при комнатной температуре в 384-луночных планшетах (Matrix, 4340). Тестируемое соединение сначала разводили в DMSO, затем добавляли в количестве 15 мкл к 10 мкл исходного буфера (50 мМ HEPES рН 7,4, 3,7 мМ MgCl2, 250 мкМ EGTA (этиленгликольтетрауксусная кислота), 125 нМ GDP) без грелинового пептида и с 9 нМ грелинового пептида. Образцы затем переносили в количестве 6 мкл в 384-луночные фильтр-планшеты (Pall, 5071), содержащие 30 мкл 24 мкг/мл hGHSR1a мембран и 0,35 мг/мл сапонина (Perkin Elmer, AD0261) в исходном буфере.
Смесь инкубировали в течение 24 минут при комнатной температуре при осторожном встряхивании, после чего добавляли 3,5 мкл 100 нМ GTP-европия в 50 мМ HEPES, рН 7,4. Образцы защищали от света и инкубировали в течение 90 минут дополнительно при комнатной температуре при осторожном встряхивании. Реакционные смеси сушили вакуум-отсосом, промывали три раза 75 мкл охлажденным во льду 1× GTP промывочным раствором (Perkin Elmer, AD0261) и сразу считывали на планшет-ридере Envision 2101 Multilabel Reader (Perkin Elmer), используя фильтр возбуждения длиной волны 320 нм и фильтр эмиссии длиной волны 615 нм.
Анализ диспергированных островковых клеток человека
День 1: Островковые клетки человека получают в мешке для внутривенных (в/в) инъекций. Островковые клетки декантируют путем присоединения соединителя к в/в мешку, и жидкость декантируют в конические пробирки на 50 мл. Мешок смывают 20 мл среды и собирают пул. Клетки центрифугируют в течение 1 минут при 1000 оборотах в минуту (об/мин). Клетки затем инкубируют в течение ночи при 37°С, 5% CO2 (чашки 10 см2 для суспензии, 10 мл среды/планшет).
День 2: Островковые клетки переносят в коническую пробирку на 50 мл, добавляют рабочий буфер Хэнкса без кальция и смешивают, затем смесь центрифугируют в течение 1 минуты при 1000 об/мин. Островковые клетки затем промывают рабочим буфером Хэнкса без кальция, смешивают и затем центрифугируют при 1000 об/мин в течение 1 минуты. Затем все, кроме 15 мл буфера, удаляют пипеткой. Затем добавляют 30 мкл 500 мМ EDTA [1 мМ] и инкубируют в течение 8 минут при комнатной температуре. Туда же затем добавляют 75 мкл смеси 0,25% трипсина-EDTA и 15 мкл 2 мг/мл ДНКазы I [2 мкг/мл]. Смесь инкубируют в течение 10 минут при 30°С при встряхивании при 60 об/мин. Сгусток разбивают посредством пипетирования 1 мл пипеткой (50 раз). Добавляют 50 мл культуральной среды и пропускают через нейлоновую мембрану 63 мкМ. Смесь центрифугируют при 1000 об/мин в течение 1 минуты, затем среду удаляют пипеткой. Остаток после центрифугирования ресуспендируют, и клетки снова промывают приблизительно 25 мл культуральной среды и центрифугируют при 1000 об/мин в течение 1 минуты. Надосадочную жидкость удаляют, затем осадок ресуспендируют в приблизительно 5 мл культуральной среды, и клетки подсчитывают. В планшеты с V-образным дном засевают 5000 клеток на лунку (200 мкл на лунку). Планшеты центрифугируют при 1000 об/мин в течение 5 минут и помещают в инкубатор клеточных культур. 600000 клеток удаляют для кальциевой визуализации.
День 3: Анализ диспергированных островковых клеток
Культуральную среду заменяют 100 мкл инкубационного буфера, содержащего 3 мМ глюкозы. Планшеты центрифугируют в течение 5 минут при 1000 об/мин до повторного осаждения островковых клеток. Планшеты инкубируют на водяной бане с температурой 37°С, непрерывно газируемой смесью 95%O2/5%CO2 в течение 45 минут. Преинкубационный буфер заменяют 50 мкл инкубационного буфера, содержащего различные тестируемые соединения с соответствующей концентрацией глюкозы (n=4 для каждого образца). Планшеты центрифугируют в течение 5 минут при 1000 об/мин для повторного осаждения клеток. Планшеты возвращают в водяную баню, непрерывно газируемую 95%O2/5%CO2 в течение 60 минут. 40 мкл переносят в другой планшет и анализируют на инсулин с использованием ELISA-анализа на инсулин человека (ALPCO Patient Insulin ELISA; Cat. No. 80-INSHU-E10, доступный от ALPCO, Salem, New Hampshire, USA).
Фармакологические данные, представленные в Таблице 1, были получены для соединения Примера 1. Значения IC50 (5,02 нМ) и Ki (4,37 нМ) были получены в результате SPA анализа грелина человека. В графе, обозначенной "n", указано сколько раз анализировали тестируемое соединение. Функциональность тестируемого соединения была определена как обратный агонист с использованием функционального анализа грелина человека.
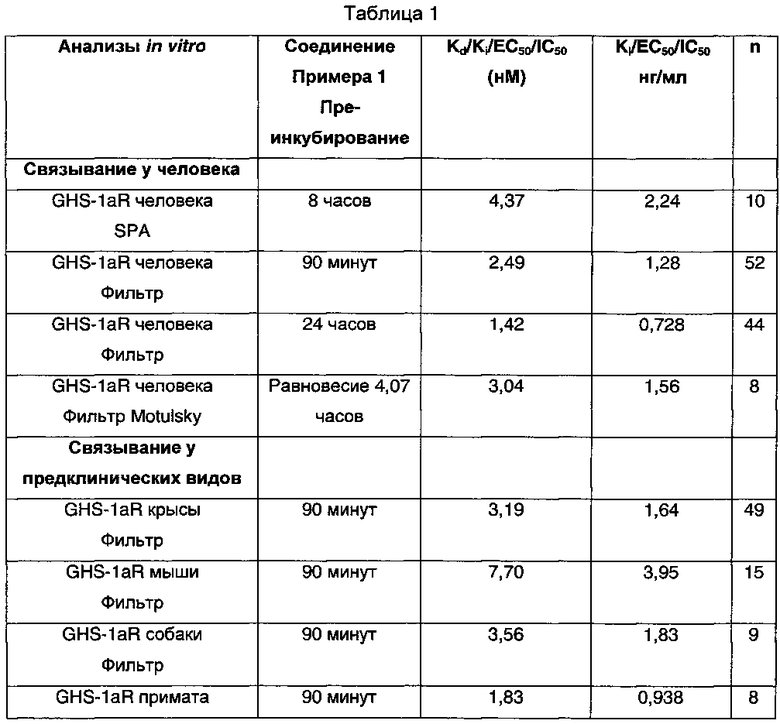
Соединение Примера 1 является мощным (по результатам анализа связывания грелина человека на фильтре Kd=3 нМ; подобно всем видам), селективным (IC50>1 мкМ по сравнению с многочисленной группой рецепторов, переносчиков, ионных каналов и ферментов) и умеренным, действующим по принципу "включено-выключено" обратным агонистом, конкурентным антагонистом рецептора секретагога гормона роста (GHS-1aR).
АНАЛИЗЫ IN VIVO
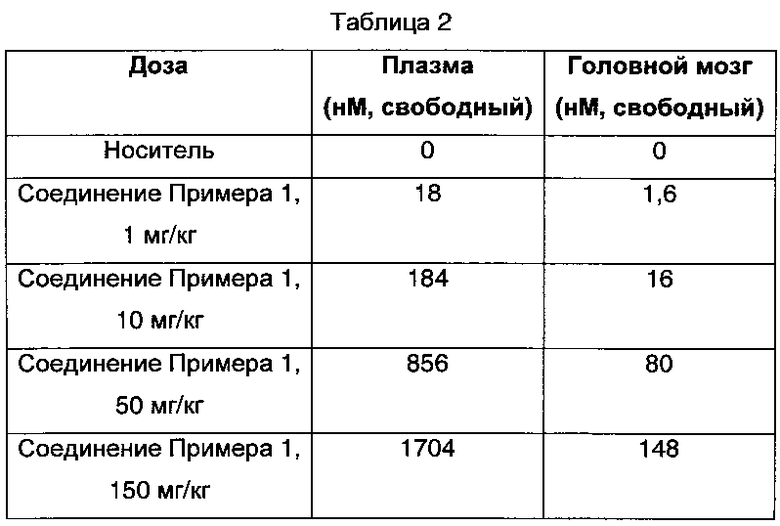
Самцам крыс Wistar Han вводили однократную дозу (1, 10, 50 или 150 мг/кг) (R)-2-(2-метилимидазо[2,1-b]тиазол-6-ил)-1-(2-(5-(6-метилпиримидин-4-ил)-2,3-дигидро-1Н-инден-1-ил)-2,7-диазаспиро[3.5]нонан-7-ил)этанона (соединение Примера 1) через пероральный зонд для оценки воздействий соединения Примера 1 на локомоторную активность за 21-часовой период времени (Фиг. 1). Животных помещали в локомоторную установку за одни час до введения дозы. Лампы выключали в 16 часов и включали в 4 часа. Животным вводили дозу в 16 часов. Мониторинг активности после введения дозы проводили в течение 20 часов. Плазму, мозг и CSF (спинномозговую жидкость) брали у сателлитной группы животных в момент времени Tmax (через 30 минут после введения дозы).
Через 35-65 минут после введения дозы никакого биологически или статистически значимого эффекта не наблюдалось в дозе 1 мг/кг при сравнении с животными, которым вводили носитель. В дозе 10 мг/кг соединение Примера 1 снижало подъем на задние лапы на 56% и общую локомоторную активность на 60% через 35-65 минут после введения дозы. В дозе 50 мг/кг соединение Примера 1 снижало подъем на задние лапы на 43% и общую локомоторную активность на 48%. Соединение Примера 1 в дозе 150 мг/кг вызывало снижение общей активности, которое было статистически значимым по сравнению с контрольными крысами, которым вводили носитель (Р<0,5), через 35-65 минут после введения дозы. Соединение Примера 1 не вызывало статистически или биологически значимого снижения общей активности через 20 часов после введения дозы. Соединение Примера 1 не вызывало гиперактивность ни в одной из доз.
Уровни соединения Примера 1 в плазме и головном мозге, которые были определены через 30 минут после введения дозы, указаны в Таблице 2.
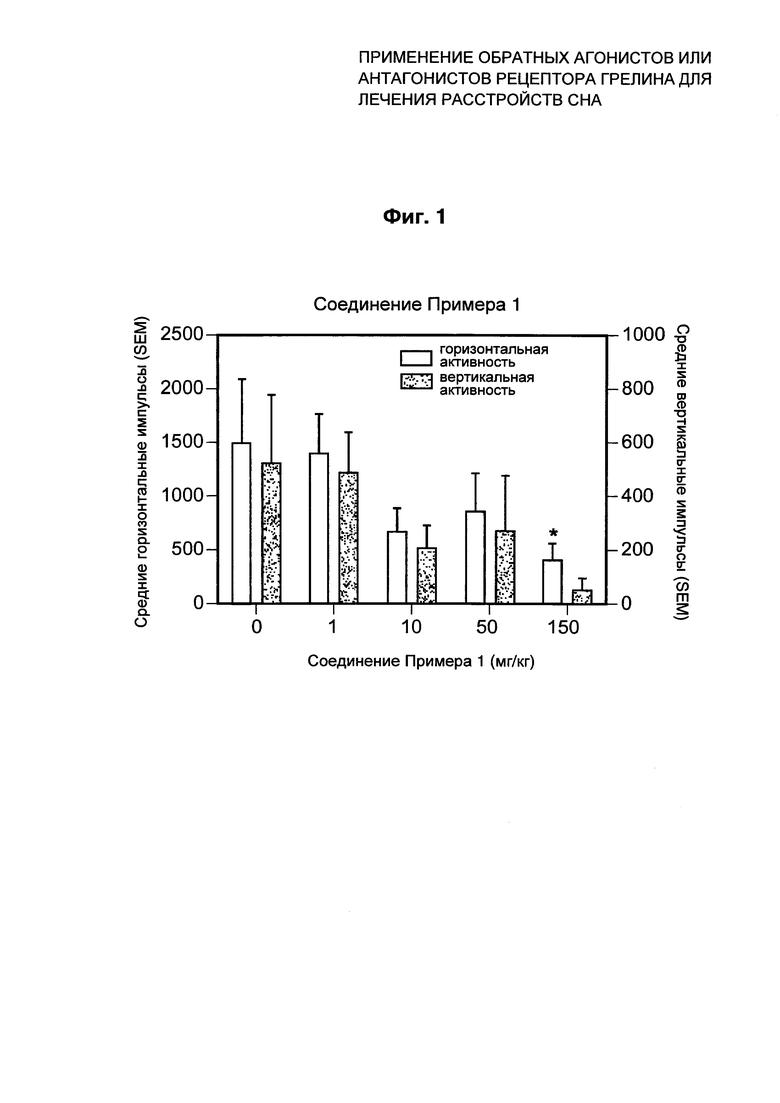
В трех клинических исследованиях (Фиг. 2), в которых определяли соотношение доза-эффект у людей с сонливостью, при наивысших дозах 100 мг BID (два раза в сутки) в течение 14 суток и однократных дозах 300 мг от 75 до 100% субъектов сообщили о сонливости или о связанном со сном эффекте. Пациентам вводили соединение Примера 1 в виде приготовленной для немедленного введения суспензии в исследованиях В3301001, В3301002, и IR в исследовании В3301007 и в виде приготовленной для немедленного введения осмотической капсулы (EPOC) в исследовании В3301007. Соединение Примера 1 было безопасным и обычно хорошо переносилось людьми в дозе вплоть до 100 мг BID в течение 14 суток, что приравнивается к оценочной 80%-ной системной занятости рецептора в течение 20 ч и 70%-ной центрально в течение приблизительно 3 часов. Зависящее от дозы увеличение числа сердечных сокращений (примерно на 10 ударов в минуту), а также ослабление как грелин-индуцированной секреции гормона роста, так и послеобеденного уровня глюкозы наблюдалось после введения критической дозы. Все эти три эффекта полностью исчезали к 14-му дню введения дозы два раза в сутки.
В исследовании на приматах эффект соединения Примера 1 в отношении продолжительности сна и приема пищи через три часа после вечернего введения дозы соединения Примера 1 исследовали при введении дозы либо один раз в сутки (QD), либо два раза в сутки (BID). Использовали восемь здоровых взрослых (в возрасте от 5,8 до 9,7 лет) самцов макак-резус (Масаса mulatta родом из Индии).
Исследования на животных проводили параллельно. Согласно перекрестной схеме четверо животных получали соединение Примера 1 (или носитель в качестве контроля) QD во время первой фазы исследования и BID во время второй фазы исследования. Для остальных четырех животных использовали схему введения доз в обратном порядке. Когда соединение Примера 1 вводили QD, эту однократную дозу вводили вечером. Каждый период введения доз длился 28-30 следующих друг за другом суток. Введение носителя длилось 10-14 суток, непосредственно предшествующих фазе, когда вводили соединение Примера 1. Животных также держали в их отсеках для регистрации сна/околосуточного ритма, чтобы они освоились. Дозу вводили в виде жидкого раствора, подаваемого автоматически с использованием контролируемого таймером насоса.
На протяжении всего периода исследования животных держали в отсеках для регистрации сна/околосуточного ритма с контролируемым светом и ослабленным звуком, в постоянных условиях тусклого света, чтобы определить внутренние ритмы сна/активности и самостоятельного приема пищи.
Сбор данных:
Цикл сон/активность и продолжительность сна документировали с использованием анализа методом непрерывной интерактивной актиграфической визуализации, как описано в Masuda et al., J Biol Rhythms. 2010 Oct; 25(5): 361-71, Intrinsic activity rhythms in Macaca mulatta: their entrainment to light и melatonin. Программно-реализованный алгоритм для анализа актиграфических данных сна относительно бодрствования у макак-резус разъясняется в Zhdanova et al., J Biol Rhythms. 2011 Apr; 26(2): 149-59. Aging of intrinsic circadian rhythms и sleep in a diurnal nonhuman primate, Macaca mulatta. Оценки начала и продолжительности сна основаны на сравнениях между полисомнографическим и актиграфическим сном у этого вида.
Каждый отсек для регистрации околосуточного ритма был оснащен индивидуальным монитором с сенсорным экраном, соединенным с автоматическим дозатором гранул (ENV-203-1000, Med Associates Inc., St. Albans, VT). Это обеспечивало возможность круглосуточного самостоятельного приема 1 г пищевых гранул (полностью укомплектованные питательными веществами точные гранулы без пыли, Bio-Serv, Frenchtown, NJ) при нажатии на движущийся на экране целевой объект. Время каждого дозирования гранул документировали.
Анализ данных проводили с использованием линейной смешанной модели (SPSS) при уровне значимости р<0,05. Данные представляли в виде средних значений для группы (стандартная погрешность средних значений (SEM)) для каждого условия лечения: введение QD или BID носителя или соединения Примера 1.
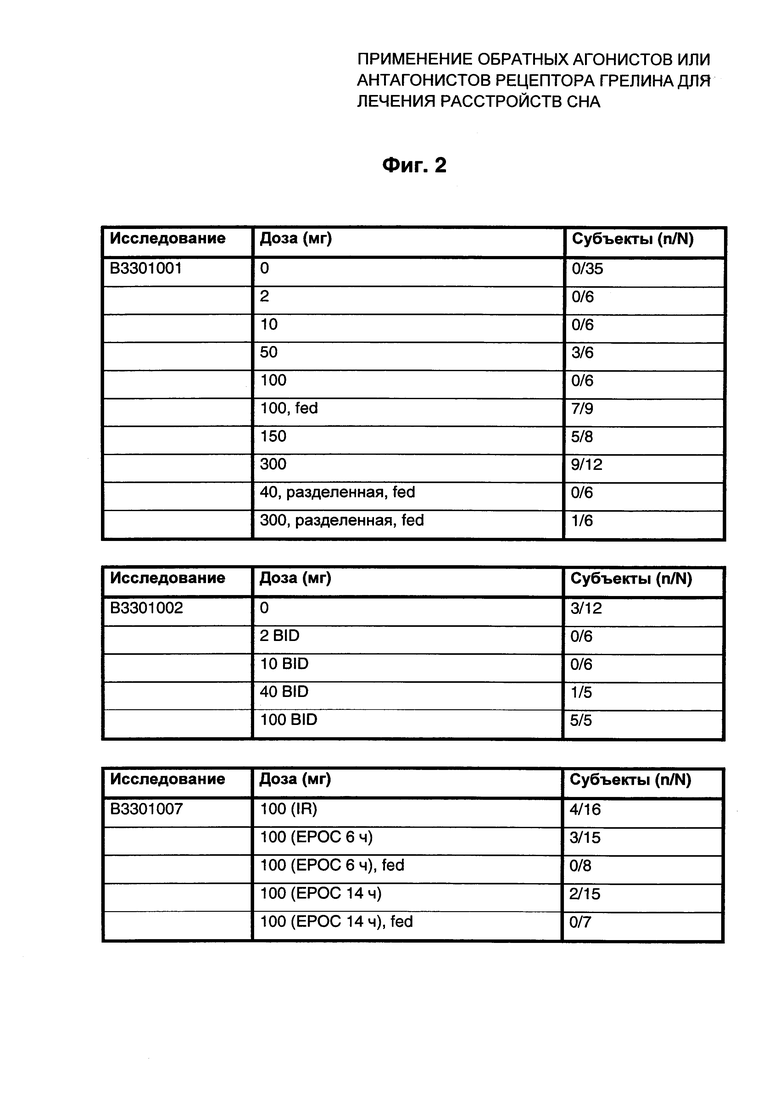
На Фиг. 3 и Фиг. 4 представлены данные в виде диаграммы активности индивидуального животного (Фиг. 3) или диаграммы приема пищи (Фиг. 4) при вечернем приеме QD носителя или соединения Примера 1. Взяты актиграфические записи от животных во время цикла свет-темнота (LD) 12:12 часов с последующими условиями постоянного тусклого света (стрелка вдоль правой стороны графика). Этот индивидуум имел период внутреннего околосуточного ритма дольше 24 часов, проиллюстрированный полной картиной активности с задержкой по времени (постепенный сдвиг вправо). Носитель или соединение Примера 1 вводили в одно и то же время (17:00 часов). В начале периода введения носителя (пунктирная линия вдоль правой стороны диаграммы) это соответствовало 2 часам до ожидаемого начала сна (начало субъективной ночи). Ко времени начала введения соединения Примера 1 (сплошная линия вдоль правой стороны диаграммы) это время соответствовало примерно 3,5 часам до ожидаемого начала сна.
Введение соединения Примера 1 значительно снижало время ожидания сна, способствуя началу сна, и этот эффект продолжался на протяжении всего периода введений. Этот эффект также приводил к общему сдвигу картины активности влево (опережение по фазе по времени пробуждения), причем более раннее время начала сна приводило к сходному более раннему времени пробуждения субъективным утром.
Введение соединения Примера 1 значительно снижало прием пищи вскоре после введения, и этот эффект продолжался на протяжении всего периода введений. Этот эффект также приводил к общему сдвигу картины приема пищи влево (опережение по фазе по времени утренней еды). Картина приема пищи у этого животного соответствовала картине активности (сравните Фиг. 3 и Фиг. 4).
На Фиг. 5 показано изменение продолжительности сна через 3 часа после вечернего введения соединения Примера 1 по сравнению с носителем (N=8 в группе (QD по сравнению с BID)). Для обеих групп значительное увеличение продолжительности сна документировали через 3 часа после введения соединения Примера 1 по сравнению с введением только носителя. Данные выражены в виде процента изменения продолжительности сна в случае, когда животные получают носитель, по сравнению с другими животными в группе, получающей соединение Примера 1 (планки погрешностей, показывающие SEM; р<0,01 относительно контроля).
На Фиг. 6 показано изменение вечернего приема пищи через 3 часа после вечернего введения соединения Примера 1 или носителя. На Фиг. 6 показано изменение для животных, получающих соединение Примера 1 QD или BID (N=8 в группе (QD по сравнению с BID)), для которых значительное снижение приема пищи документировали для обеих групп, по сравнению с введением только носителя, через 3 часа после введения соединения Примера 1. Данные выражены в виде процента приема пищи через 3 часа после введения носителя или соединения Примера 1 (планки погрешностей, показывающие SEM; р<0,01 относительно носителя).
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ 2,3-ДИГИДРО-1Н-ИНДЕН-1-ИЛ-2,7-ДИАЗАСПИРО[3.5]НОНАНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ АНТАГОНИСТОВ ИЛИ ОБРАТНЫХ АГОНИСТОВ ГРЕЛИНОВОГО РЕЦЕПТОРА | 2011 |
|
RU2524341C2 |
| АРИЛПИРИМИДИНОВЫЕ ПРОИЗВОДНЫЕ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1997 |
|
RU2189976C2 |
| ЗАМЕЩЕННЫЕ 2-АЦИЛ-2-АМИНОТИАЗОЛЫ | 2005 |
|
RU2371438C2 |
| ПРИМЕНЕНИЕ N-АРИЛДИАЗАСПИРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ ДЛЯ ЛЕЧЕНИЯ ЗАВИСИМОСТЕЙ | 2005 |
|
RU2387647C9 |
| СОЕДИНЕНИЯ, КОТОРЫЕ УСИЛИВАЮТ РЕЦЕПТОР ГЛУТАМАТА, И ИХ ПРИМЕНЕНИЕ В МЕДИЦИНЕ | 2005 |
|
RU2403242C2 |
| ИНГИБИТОРЫ ВЗАИМОДЕЙСТВИЯ МЕНИН-MLL | 2017 |
|
RU2799820C2 |
| СУЛЬФОНАМИДНЫЕ СОЕДИНЕНИЯ ИЛИ ИХ СОЛИ | 2009 |
|
RU2481329C2 |
| ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ ТЕТРАГИДРОПИРОЛЛО[3,2-c]ПИРИДИН-4-ОНОВЫХ ПРОИЗВОДНЫХ ДЛЯ ЛЕЧЕНИЯ ОЖИРЕНИЯ, ПСИХИАТРИЧЕСКИХ И НЕВРОЛОГИЧЕСКИХ РАССТРОЙСТВ | 2006 |
|
RU2415856C2 |
| ПРОИЗВОДНЫЕ 3-БЕНЗОФУРАНИЛИНДОЛ-2-ОНА, ЗАМЕЩЕННЫЕ В ПОЛОЖЕНИИ 3, ИХ ПОЛУЧЕНИЕ И ИХ ПРИМЕНЕНИЕ В ТЕРАПИИ | 2010 |
|
RU2542991C2 |
| СЕЛЕКТИВНЫЕ К ПОДТИПУ РЕЦЕПТОРА АЗАБИЦИКЛОАЛКАНОВЫЕ ПРОИЗВОДНЫЕ | 2008 |
|
RU2417984C1 |

Группа изобретений относится к медицине, а именно к неврологии, и может быть использована для лечения первичного расстройства сна. Для этого пациенту вводят терапевтически эффективное количество (R)-2-(2-метилимидазо[2,1-b]тиазол-6-ил)-1-(2-(5-(6-метилпиримидин-4-ил)-2,3-дигидро-1Н-инден-1-ил)-2,7-диазаспиро[3.5]нонан-7-ил)этанона или его фармацевтически приемлемой соли, либо фармацевтическую композицию, содержащую терапевтически эффективное количество (R)-2-(2-метилимидазо[2,1-b]тиазол-6-ил)-1-(2-(5-(6-метилпиримидин-4-ил)-2,3-дигидро-1Н-инден-1-ил)-2,7-диазаспиро[3.5]нонан-7-ил)этанона или его фармацевтически приемлемой соли и по меньшей мере один фармацевтически приемлемый носитель, разбавитель или эксципиент. Группа изобретений обеспечивает более раннее время начала сна и значительное увеличение его продолжительности, а также снижение аппетита. 2 н. и 2 з.п. ф-лы, 6 ил., 2 табл.
1. Способ лечения расстройства сна у пациента, включающий стадию введения пациенту, нуждающемуся в таком лечении, терапевтически эффективного количества (R)-2-(2-метилимидазо[2,1-b]тиазол-6-ил)-1-(2-(5-(6-метилпиримидин-4-ил)-2,3-дигидро-1Н-инден-1-ил)-2,7-диазаспиро[3.5]нонан-7-ил)этанона или его фармацевтически приемлемой соли, где указанное расстройство сна представляет собой первичную бессонницу.
2. Способ лечения расстройства сна у пациента, включающий стадию введения пациенту, нуждающемуся в таком лечении, фармацевтической композиции, содержащей терапевтически эффективное количество (R)-2-(2-метилимидазо[2,1-b]тиазол-6-ил)-1-(2-(5-(6-метилпиримидин-4-ил)-2,3-дигидро-1Н-инден-1-ил)-2,7-диазаспиро[3.5]нонан-7-ил)этанона или его фармацевтически приемлемой соли и по меньшей мере один фармацевтически приемлемый носитель, разбавитель или эксципиент, где указанное расстройство сна представляет собой первичную бессонницу.
3. Способ по п. 1, где (R)-2-(2-метилимидазо[2,1-b]тиазол-6-ил)-1-(2-(5-(6-метилпиримидин-4-ил)-2,3-дигидро-1Н-инден-1-ил)-2,7-диазаспиро[3.5]нонан-7-ил)этанон или его фармацевтически приемлемую соль вводят в комбинации с другим фармакологически активным агентом.
4. Способ по п. 2, где (R)-2-(2-метилимидазо[2,1-b]тиазол-6-ил)-1-(2-(5-(6-метилпиримидин-4-ил)-2,3-дигидро-1Н-инден-1-ил)-2,7-диазаспиро[3.5]нонан-7-ил)этанон или его фармацевтически приемлемую соль вводят в комбинации с другим фармакологически активным агентом.
| WO 2008008286 A2, 17.01.2008;US 20110230461 A1, 22.09.2011 | |||
| СОЕДИНЕНИЯ ДЛЯ НОРМАЛИЗАЦИИ ЦИКЛА СНА/БОДРСТВОВАНИЯ | 2003 |
|
RU2366428C2 |
| КОВРОВ Г.В | |||
| и др | |||
| Инсомния и ее лечение//Качество жизни | |||
| Медицина, 2004, No4(7), c.54-57 | |||
| GOLDSTONE A.P | |||
| Prader-Willi syndrome: advances in genetics, pathophysiology and treatment// TRENDS IN ENDOCRINOLOGY&METABOLISM, Vol.15, No1, 01.01.2004, p.12-20. | |||

