Настоящая заявка заявляет приоритет предварительной заявки на патент США №60/603479, опубликованной 20 августа 2004.
Настоящее изобретение относится к никотиновым антагонистам, в частности к антагонистам и неполным антагонистам, которые обладают большей потенциальной антагонистической активностью в отношении высвобождения допамина, чем α4β2-рецептор, фармацевтическим композициям, включающим эти соединения, и применению этих соединений для лечения зависимости, включая зависимость от курения, зависимость от наркотических веществ и других лекарственных средств, и ожирения, которое возникает после прекращения приема лекарственных средств.
Зависимость от курения представляет собой сложное явление, которое, как полагают, включает усиление когнитивных способностей, психологическое обусловливание, адаптацию к стрессу, подкрепляющие свойства и облегчение от устранения негативных симптомов. Следовательно, терапевтическое лечение зависимости от курения представляет собой очень сложную задачу.
Никотин, содержащийся в табаке, может быть отчасти ответственным за трудности, с которыми сталкиваются некоторые индивидуумы в процессе преодоления зависимости от курения. Были разработаны многочисленные способы помощи в прекращении курения, включая уменьшение потребления с течением времени и предоставление альтернативных средств подачи никотина, включая жвачки и кожные пластыри.
Нейронные никотиновые ацетилхолиновые рецепторы (nAChR) широко распространены во всей центральной и периферийной нервной системе, включая некоторые участки головного мозга. Два наиболее известных подтипа nAChR ЦНС представляют собой α4β2 и α7. Однако преобладание конкретного подтипа никотиновых рецепторов в головном мозге не обязательно отражает его функциональную важность. Например, полагают, что подтипы, содержащие α3β2-рецепторы, несмотря на их малую распространенность в головном мозге, являются, по меньшей мере, отчасти ответственными за посредничество в высвобождении допамина, основываясь на исследованиях, в которых антагонисты этих рецепторов (т.e. бунгаротоксин и α-коноксин) частично ингибируют высвобождение допамина (Dworsin et al., J. Pharm. Ex. Ther., 10(10): 1561-1581 (2000)). Соответственно предполагается, что в полосатом теле существует множество подтипов рецепторов, включенных в индуцируемое никотином высвобождение допамина. Никотиновые антагонисты, действующие против одного или нескольких из этих рецепторов, хорошо известны в данной области техники и описаны, например, у Dwoskin et al., J. Pharm. Ex. Ther. 298(2): 395 (2001).
Один из фармацевтических подходов к стимулированию прекращения курения включает блокирование никотинового сигнала от табака другим агентом, таким как бупроприон. В низких микромолярных концентрациях бупроприон неконкурентно ингибирует α3β2, α4β2 и α7 nAChR и в настоящее время реализуется на рынке как средство, способствующее прекращению курения. Другие неконкурентные никотиновые антагонисты также рассматривались в качестве подхода к прекращению курения. Одной из теорий является то, что никотиновые антагонисты блокируют сигнал подкрепления от никотина, связанный с зависимостью от курения. Мекамиламин, антагонист и α4β2-, и α7-рецепторов, является примером никотинового антагониста, который использовали отдельно и в комбинации с терапией замены никотина, чтобы способствовать прекращению курения.
Несмотря на известные способы лечения зависимости от курения, остается интерес к новым способам и фармацевтическим композициям для лечения зависимости от курения.
Также трудно преодолеть зависимость от других соединений, включая опиаты, кокаин и другие запрещенные лекарственные средства. Для преодоления зависимости от указанных запрещенных лекарственных средств предлагали использовать мекамиламин и другие никотиновые соединения (см., например, Reid, Neuropsychopharmacology, 20(3): 297-307 (1999); Campiani et al., J. Med Chem., 46: 3822-39 (2003) (обсуждение роли D3/D2-рецепторов допамина), Chi and de Wit H, Alcoholism: Clinical and Experimental Research, 27: 780-786 (2003); Pilla et al., Nature, 400: 371-5 (1999) (обсуждение роли неполного агониста D3 рецептора допамина); Reid et al., Neuropsychopharmacology, 20: 297-307 (1999); Slemmer et al., J. Pharmacol. Exp.Ther. 295: 321-327 (2000); Vorel et al., J. Neurosci., 22: 9595-603 (2002) (обсуждение того, как антагонизм к D3-рецепторам допамина подавляет поиск кокаина и усиленное кокаином “поощрение” мозга у крыс), и Zachariou et al., Neuropsychopharmacology, 24: 576-589 (2001), содержание каждого из которых включено в настоящее описание во всей своей полноте в качестве ссылки).
Увеличение веса часто связано с прекращением приема лекарственного средства (см., например, Dwoskin et al., “Recent developments in neuronal nicotinic acetylcholine receptor antagonists”, Exp.Opin. Ther. Patents 10: 1561-1581 (2000)). Было бы желательным разработать способы и композиции, которые препятствуют такому увеличению веса.
Предполагается, что высвобождение допамина связано с психологической “компенсацией”, связанной с потреблением указанных веществ, к которым возникает зависимость. Для лечения зависимостей было предложено использовать модуляцию высвобождения допамина. Модуляция α4β2-рецептора представляет собой один из способов модуляции высвобождения допамина и может быть, по меньшей мере, частью механизма, благодаря которому мекамиламин является эффективным при лечении лекарственной зависимости. Однако в некоторых случаях может быть желательным модулировать высвобождение допамина без антагонизации активности
α4β2-рецепторов. Таким образом, вызывает интерес пригодность множества лигандов, которые с высокой аффинностью и селективностью связываются с рецепторами, отличными от α4β2, и которые модулируют высвобождение допамина.
Кроме того, ограничение некоторых никотиновых соединений заключается в том, что они связаны с различными нежелательными побочными эффектами, например стимуляцией мышечных и ганглионарных рецепторов. Было бы желательным иметь соединения, композиции и способы для предупреждения и/или лечения лекарственной зависимости, побуждения к прекращению курения и которые препятствуют ожирению, связанному с преодолением зависимости, в которых соединения фармакологически проявляют полезный эффект (например, ингибирование секреции допамина), без значимых ассоциированных побочных эффектов.
Настоящее изобретение предоставляет такие соединения, композиции и способы.
Раскрыты соединения, фармацевтические композиции и способы лечения никотиновой зависимости, лекарственной зависимости и/или ожирения, связанного с прекращением приема лекарственного средства и/или никотина. Соединения функционируют, уменьшая высвобождение допамина, по существу не затрагивая α4β2-рецепторы. Уменьшение высвобождения допамина приводит к снижению психологической “компенсации”, связанной с введением никотина или запрещенных лекарственных средств, и таким образом помогает преодолеть зависимость.
Соединения представляют собой N-арилдиазаспироциклические соединения, мостиковые аналоги N-гетероарилдиазаспироциклических соединений или пролекарства или метаболиты этих соединений. Арильная группа может представлять собой пяти- или шестичленное гетероциклическое кольцо (гетероарил). Примеры N-арилдиазаспироциклических соединений включают 7-(3-пиридил)-1,7-диазаспиро[4.4]нонан и 1-(3-пиридил)-1,7-диазаспиро[4.4]нонан. Примеры мостиковых аналогов N-гетероарилдиазаспироциклических соединений включают 1'-(3-пиридил)спиро[1-азабицикло[2.2.1]гептан-2,3'-пирролидин].
Соединения и композиции могут применяться для лечения и/или предупреждения большого разнообразия состояний или нарушений, в частности таких нарушений, которые характеризуются дисфункцией никотиновой холинэргической нейротрансмиссии, включая нарушения, затрагивающие нейромодуляцию высвобождения нейротрансмиттеров, например высвобождения допамина. Нарушения ЦНС, которые характеризуются изменениями в нормальном высвобождении нейротрансмиттеров, представляют собой другой пример нарушений, которые можно вылечить и/или предупредить. Соединения и композиции также могут быть использованы для облегчения боли. Способы включают введение субъекту эффективного количества N-арилдиазаспироциклического соединения, мостикового аналога N-гетероарилдиазаспироциклического соединения или пролекарства или его метаболита для облегчения конкретного нарушения.
Фармацевтические композиции включают эффективное количество соединений, раскрытых в настоящем описании. При использовании эффективного количества соединения могут вызывать у субъекта снижение высвобождения допамина, не проявляя свойств повышенной чувствительности.
Фармацевтические композиции обеспечивают терапевтически благоприятное действие на индивидуумов, которые страдают от таких нарушений и которые имеют клинические проявления таких нарушений. Предполагается, что терапевтические композиции безопасны и эффективны для лечения указанных нарушений.
Вышеприведенные и другие аспекты настоящего изобретения подробно объяснены в нижеприведенном подробном описании и примерах.
Подробное описание изобретения
Раскрыты соединения, фармацевтические композиции, включающие эти соединения, и способы их изготовления и применения.
Нижеследующие определения полезны для понимания границ и пределов настоящего изобретения, как раскрыто в настоящем описании.
Как используется в настоящем описании, "алкил" относится к алкильным радикалам с прямой или разветвленной цепью, включающим C1-C8, предпочтительно C1-C5, например метил, этил или изопропил; "замещенный алкил" относится к алкильным радикалам, дополнительно несущим одну или несколько замещающих групп, таких как гидрокси, алкокси, арилокси, меркапто, арил, гетероцикло, галоген, амино, карбоксил, карбамил, циано и т.п.; "алкенил" относится к углеводородным радикалам с прямой или разветвленной цепью, включающей C1-C8, предпочтительно C1-C5, и имеющих, по меньшей мере, одну двойную связь углерод-углерод; "замещенный алкенил" относится к алкенильным радикалам, дополнительно несущим одну или несколько замещающих групп, как определено выше; "циклоалкил" относится к радикалам, содержащим насыщенные или ненасыщенные, неароматические, циклические кольца, содержащие три-восемь атомов углерода, предпочтительно три-шесть атомов углерода; "замещенный циклоалкил" относится к циклоалкильным радикалам, дополнительно несущим одну или несколько замещающих групп, как определено выше; "арил" относится к ароматическим радикалам, имеющим шесть-десять атомов углерода; "замещенный арил" относится к арильным радикалам, дополнительно несущим одну или несколько замещающих групп, как определено выше; "алкиларил" относится к алкилзамещенным арильным радикалам; "замещенный алкиларил" относится к алкиларильным радикалам, дополнительно несущим одну или несколько замещающих групп, как определено выше; “арилалкил” относится к арилзамещенным алкильным радикалам; "замещенный арилалкил" относится к арилалкильным радикалам, дополнительно несущим одну или несколько замещающих групп, как определено выше; "гетероциклил" относится к насыщенным или ненасыщенным циклическим радикалам, содержащим один или несколько гетероатомов (например, O, N, S) в виде части кольцевой структуры и имеющим два-семь атомов углерода в кольце; "замещенный гетероциклил" относится к гетероциклильным радикалам, дополнительно несущим одну или несколько замещающих групп, как определено выше.
Как используется в настоящем описании, "агонист" представляет собой вещество, которое стимулирует своего связывающего партнера, обычно рецептор. Стимуляция определена в контексте конкретного исследования или может быть очевидной из литературы, обсуждаемой в настоящем описании, где дается сравнение с фактором или веществом, которое принимается в качестве "агониста" или "антагониста" конкретного связывающего партнера, по существу, в аналогичных обстоятельствах, как очевидно специалистам в данной области техники. Стимуляция может быть определена в отношении усиления конкретного эффекта или функции, которая индуцируется взаимодействием агониста или неполного агониста со связывающим партнером и может включать аллостерические эффекты.
Как используется в настоящем описании, "антагонист" представляет собой вещество, которое ингибирует своего связывающего партнера, обычно рецептор. Ингибирование определено в контексте конкретного исследования или может быть очевидным из литературы, обсуждаемой в настоящем описании, где дается сравнение с фактором или веществом, которое принимается в качестве "агониста" или "антагониста" конкретного связывающего партнера, по существу, в аналогичных обстоятельствах, как очевидно специалистам в данной области техники. Ингибирование может быть определено в отношении уменьшения конкретного эффекта или функции, которая индуцируется взаимодействием антагониста со связывающим партнером и может включать аллостерические эффекты.
Как используется в настоящем описании, "неполный агонист" представляет собой вещество, обеспечивающее уровень стимуляции своего связывающего партнера, который является промежуточным между уровнем стимуляции полного или абсолютного антагониста и агониста, определенного при помощи любого общепринятого стандарта для агонистической активности.
Как используется в настоящем описании, "неполный антагонист" представляет собой вещество, обеспечивающее уровень ингибирования своего связывающего партнера, который является промежуточным между уровнем ингибирования полного или абсолютного антагониста и неактивного лиганда.
Очевидно, что стимуляция, и в данном случае ингибирование, определена, по существу, для любого вещества или категории веществ, предназначенных для определения в качестве агонистов, антагонистов или неполных агонистов. Как используется в настоящем описании, "внутренняя активность" или "эффективность" относится к некоторым показателям биологической эффективности комплекса связывающих партнеров. Относительно рецепторной фармакологии контекст, в котором следует определять внутреннюю активность или эффективность, будет зависеть от контекста комплекса связывающих партнеров (например, рецептор/лиганд) и анализа активности, релевантной для конкретного биологического результата. Например, в некоторых случаях внутренняя активность может изменяться в зависимости от вовлеченной конкретной второй сигнальной системы. См. Hoyer D. и Boddeke, H., Trends Pharmacol. Set. 14(7): 270-5 (1993). Где такие контекстуально специфические оценки являются релевантными и каким образом они могут быть релевантными в контексте настоящего изобретения, очевидно специалистам в данной области техники.
Как используются в настоящем изобретении, нейротрансмиттеры, высвобождение которых опосредуется соединениями, раскрытыми в настоящем описании, включают без ограничений ацетилхолин, допамин, норэпинефрин, серотонин и глутамин, и соединения, раскрытые в настоящем описании, функционируют в качестве агонистов или неполных агонистов для одного или нескольких nAChR центральной нервной системы (ЦНС).
I. Соединения
Соединения представляют собой N-арилдиазаспироциклические соединения, мостиковые аналоги N-гетероарилдиазаспироциклических соединений, пролекарства или метаболиты этих соединений и их фармацевтически приемлемые соли.
Соединения могут связывать и модулировать никотиновые ацетилхолиновые рецепторы в головном мозге пациента, в кортексе, гипокампе, таламусе, базальных ганглиях и спинном мозге. При таком связывании соединения проявляют фармакологию никотина и, в частности, могут антагонизировать высвобождение допамина в эффективных концентрациях, которые по существу не антагонизируют
α4β2-рецептор.
Константы связывания рецепторов предоставляют показатель способности соединения связываться с половиной релевантных рецепторных участков некоторых клеток головного мозга пациента. См., например, Cheng et al., Biochem. Pharmacol. 22: 3099 (1973). Константы связывания рецепторов соединений, раскрытых в настоящем описании, для одного или нескольких рецепторов, отличных от α4β2-рецептора, которые опосредуют высвобождение допамина, обычно превышают примерно 0,1 нМ, часто превышают примерно 1 нМ и наиболее часто превышают примерно 10 нМ и бывают меньше примерно 100 мкМ, часто - меньше примерно 10 мкМ и наиболее часто - меньше примерно 5 мкМ. Предпочтительные соединения обычно имеют константы связывания рецепторов меньше чем примерно 2,5 мкМ, иногда меньше чем примерно 1 мкМ и могут быть меньше чем примерно 100 нМ.
Предпочтительно, соединения могут преодолевать гематоэнцефалический барьер и таким образом проникать в центральную нервную систему пациента. Значения log P предоставляют показатель способности соединения проходить сквозь пористую перегородку, такую как биологическая мембрана, включая гематоэнцефалический барьер. См., например, Hansch et al., J. Med. Chem. 11: 1 (1968). Обычные значения log P для соединений, раскрытых в настоящем описании, в общем случае превышают примерно -0,5, часто превышают примерно 0 и наиболее часто превышают примерно 0,5 и обычно бывают ниже примерно 3, часто - ниже примерно 2 и наиболее часто - ниже примерно 1.
В одном из вариантов осуществления соединения имеют структуру, представленную ниже формулой 1
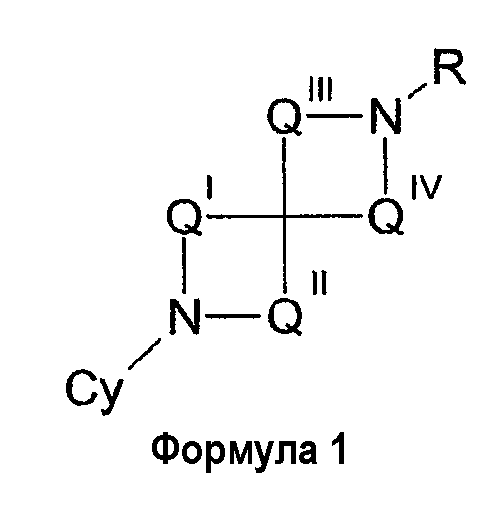
В формуле QI представляет собой (CZ2)u, QII представляет собой (CZ2)v, QIII представляет собой (CZ2)w и QIV представляет собой (CZ2)x, где u, v, w и x отдельно равны 0, 1, 2, 3 или 4, предпочтительно 0, 1, 2 или 3. R представляет собой водород, низший алкил, ацил, алкоксикарбонил или арилоксикарбонил, предпочтительно водород или низший алкил. Когда значение u равно 0, значение v должно быть больше 0, и в случае формулы 1, когда значение w равно 0, значение x должно быть больше 0. Кроме того, значения u, v, w и x выбирают таким образом, чтобы диазаспироциклическое кольцо содержало 7, 8, 9, 10 или 11 членов, предпочтительно 8, 9 или 10 членов.
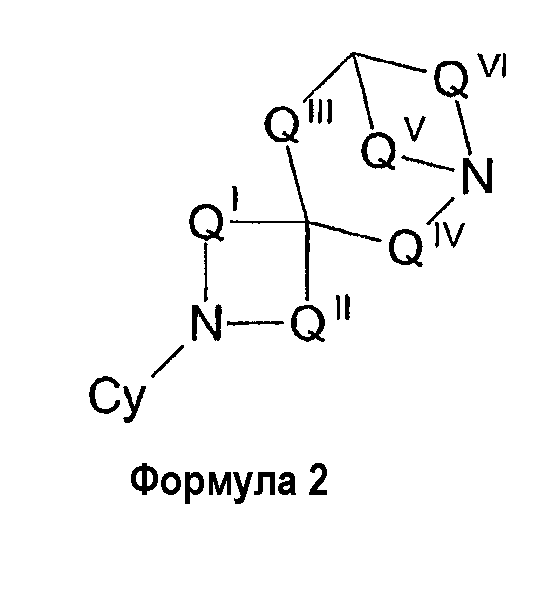
В другом варианте осуществления соединения представлены формулой 2, приведенной выше. В формуле 2 QI представляет собой (CZ2)u, QII представляет собой (CZ2)v, QIII представляет собой (CZ2)w, QIV представляет собой (CZ2)x, Qv представляет собой (CZ2)y и QVI представляет собой (CZ2)z, где u, v, w, x, y и z отдельно равны 0, 1, 2, 3 или 4, предпочтительно - 0, 1 или 2. Значения u, v, w, x, y и z выбирают таким образом, чтобы мостиковое диазаспироциклическое кольцо имело 8, 9, 10, 11, 12 или 13 членов, предпочтительно 9, 10, 11 или 12 членов. В случае формулы 2 значения w и x одновременно могут быть равны 0. Кроме того, R представляет собой водород, низший алкил, ацил, алкоксикарбонил или арилалкоксикарбонил, предпочтительно водород или низший алкил.
Каждый отдельный Z представляет либо водород либо подходящий вид заместителя, который не является водородом (например, алкил, замещенный алкил, циклоалкил, замещенный циклоалкил, гетероциклил, замещенный гетероциклил, арил, замещенный арил, алкиларил, замещенный алкиларил, арилалкил или замещенный арилалкил; но предпочтительно низший алкил или арил).
В любой из формул Cy представляет подходящее пяти- или шестичленное гетероароматическое кольцо. В одном из вариантов осуществления Cy представляет собой шестичленное кольцо формулы
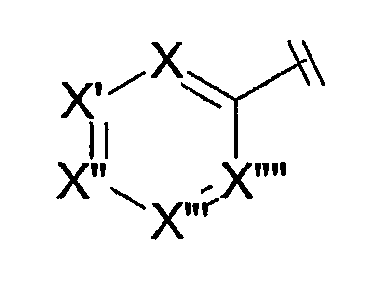
Каждый из X, X', X'', X''' и X'''' отдельно представляют собой азот, азот, связанный с кислородом (например, функциональную группу N-оксид или N-О), или углерод, связанный с заместителем. Не более чем три из X, X', X'', X''' и X'''' представляют собой азот или азот, связанный с кислородом, и предпочтительным является, чтобы только один или два из X, X', X'', X''' и X'''' представляли собой азот или азот, связанный с кислородом. Кроме того, наиболее предпочтительно, чтобы не более чем один из X, X', X'', X''' и X'''' представлял собой азот, связанный с кислородом; и в случае, если один из таких видов представляет собой азот, связанный с кислородом, то предпочтительным является, чтобы таким видом был X'''. Наиболее предпочтительно, чтобы X''' представлял собой азот. В некоторых предпочтительных случаях как X', так и X''' представляют собой азот. Обычно X, X'' и X'''' представляют собой углерод, связанный с заместителем, и обычным является, чтобы вид заместителя при X, X'' и X'''' представлял собой водород. В некоторых других предпочтительных соединениях, в которых X''' представляет собой углерод, связанный с заместителем, например водородом, X и X'', оба, представляют собой азот. В некоторых других предпочтительных соединениях, где X' представляет собой углерод, связанный с заместителем, например водородом, X и X''', оба представляют собой азот.
В других вариантах осуществления Cy представляет собой пятичленное гетероароматическое кольцо, такое как пиррол, фуран, тиофен, изоксазол, изотиазол, оксазол, тиазол, пиразол, 1,2,4-оксадиазол, 1,3,4-оксадиазол и 1,2,4-триазол. Другие примеры таких колец описаны в патенте США №6022868 Olesen et al., содержание которого включено в настоящее описание во всей своей полноте в качестве ссылки. Один из способов изображения Cy является следующим:
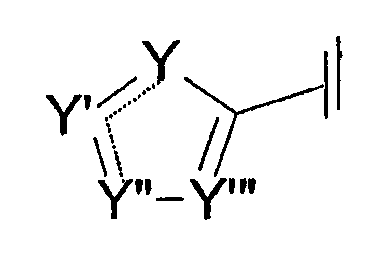
где Y и Y'' отдельно представляют собой азот, азот, связанный с заместителем, кислород, серу или углерод, связанный с заместителем, и Y' и Y''' представляют собой азот или углерод, связанный с заместителем. Пунктирные линии указывают, что связи (между Y и Y' и между Y' и Y'') могут быть либо одинарными, либо двойными связями. Однако, если связь между Y и Y' представляет собой одинарную связь, то связь между Y' и Y'' должна быть двойной, и наоборот. В случае, при котором Y или Y'' представляет собой кислород или серу, только один из Y и Y'' представляет собой либо кислород, либо серу. По меньшей мере, один из Y, Y', Y'' и Y''' должен быть кислородом, серой, азотом или азотом, связанным с заместителем. Предпочтительным является, чтобы не более чем три из Y, Y', Y'' и Y''' представляли собой кислород, серу, азот или азот, связанный с заместителем. Более предпочтительным является, чтобы, по меньшей мере, один, но не более чем три из Y, Y', Y'' и Y''' представляли собой азот.
Заместители, связанные с любым из X, X', X'', X''', X'''', Y, Y', Y'' и Y''' (когда любой представляет собой углерод, связанный с заместителем, или азот, связанный с заместителем), обычно имеют значение σm между примерно -0,3 и примерно 0,75, часто между примерно -0,25 и примерно 0,6; и каждое значение σm отдельно может быть равно 0 или не равно нулю; как определено согласно Hansch et al., Chem. Rev. 91: 165 (1991).
Примеры подходящих видов заместителей, связанных с любым из X, X', X'', X''', X'''', Y, Y', Y'' и Y''' (когда любой представляет собой углерод, связанный с заместителем, или азот, связанный с заместителем), включают водород, алкил, замещенный алкил, алкенил, замещенный алкенил, гетероциклил, замещенный гетероциклил, циклоалкил, замещенный циклоалкил, арил, замещенный арил, алкиларил, замещенный алкиларил, арилалкил, замещенный арилалкил, галоген (например, F, Cl, Br или I), -OR', -NR'R'', -CF3, -CN, -NO2, -C2R', -SR', -N3, -C(=O)NR'R'', -NR'C(=O)R'', -C(=O)R', -C(=O)OR', -OC(=O)R', -O(CR'R'')rC(=O)R', -O(CR'R'')rNR''C(=O)R', -O(CR'R'')rNR''SO2R', -OC(=O)NR'R'', -NR'C(=O)OR'', -SO2R', -SO2NR'R'' и -NR'SO2R'', где R' и R'' отдельно представляют собой водород, низший алкил (например, алкил с линейной или разветвленной цепью, включающей С1-C8, предпочтительно С1-C5, такие как метил, этил или изопропил), циклоалкил, гетероциклил, арил или арилалкил (такой как бензил), а r равно целому числу от 1 до 6. R' и R'' могут быть объединены для формирования циклической функциональной группы. Термин “замещенный”, как он применяется для алкила, арила, циклоалкила и т.п., относится к заместителям, описанным выше, начиная с галогена и заканчивая -NR'SO2R''.
Примеры, подходящих Cy групп включают 3-пиридил (незамещенный или замещенный в 5 и/или 6 позиции(позициях) любым из вышеупомянутых заместителей), 5-пиримидинил (незамещенный или замещенный во 2-й позиции любым из вышеупомянутых заместителей), 4- и 5-изоксазолил, 4- и 5-изотиазолил, 5-оксазолил, 5-тиазолил, 5-(1,2,4-оксадиазолил), 2-(1,3,4-оксадиазолил) или 3-(1,2,4-триазолил).
Характерные арильные группы включают фенил, нафтил, фуранил, тиенил, пиридинил, пиримидинил, пиразинил, пиридазинил, хинолинил и индолил. Другие характерные ароматические кольцевые системы представляют собой набор, приведенный у Gibson et al., J., Med. Chem. 39: 4065 (1996). Любой из видов, содержащих такую ароматическую группу, может быть замещен, по меньшей мере, одной группой из вышеуказанных заместителей, которые связаны с x' и т.п. Типичные представители, по существу, включают алкил, арил, галоген, гидрокси, алкокси, арилокси или аминозаместитель.
Смежные заместители при X, X', X'', X''', X'''', Y, Y', Y'' и Y''' (если имеется заместитель) могут объединяться для формирования одного или нескольких насыщенных или ненасыщенных, замещенных или незамещенных карбоциклических или гетероциклических колец, содержащих без ограничения либо ацетильную, кетальную, аминную, кетонную, лактонную, лактамовую, карбаматную или карбомидную функциональные группы.
Соединения могут встречаться в стереоизомерных формах, включающих как отдельные энантиомеры, так и рацемические смеси таких соединений, а также смеси с различной степенью избытка энантиомеров.
Соединения могут находиться в виде свободного основания или в виде соли (например, в виде фармацевтически приемлемых солей). Примеры подходящих фармацевтически приемлемых солей включают неорганические кислотные аддитивные соли, такие как сульфат, фосфат и нитрат; органические кислотные аддитивные соли, такие как ацетат, галактарат, пропионат, сукцинат, лактат, гликолят, малат, тартрат, цитрат, малеат, фумарат, метансульфонат, п-толуолсульфонат и аскорбат; соли с кислыми аминокислотами, такими как аспартат и глутамат; соли щелочных металлов, таких как натрий и калий; соли щелочноземельных металлов, таких как магний и кальций; соли аммония; соли органических оснований, таких как триметиламин, триэтиламин, пиридин, пиколин, дициклогексиламин и N,N'-дибензилэтилендиамин; и соли с основными аминокислотами, такими как лизин и аргинин. В некоторых случаях соли могут представлять собой гидраты или спиртовые сольваты. Стехиометрия соли может изменяться в зависимости от природы компонентов. Характерные соли представлены, как описано в патентах США №5597919, Dull et al., 5616716 Dull et al. и 5663356 Ruecroft et al., раскрытие которых приведено в настоящем описании во всей своей полноте в качестве ссылки.
Характерные соединения включают следующие:
7-(3-пиридил)-1,7-диазаспиро[4.4]нонан
7-(5-пиримидинил)-1,7-диазаспиро[4.4]нонан
7-(5-изоксазолил)-1,7-диазаспиро[4.4]нонан
7-(5-изотиазолил)-1,7-диазаспиро[4.4]нонан
7-(5-(1,2,4-оксадиазол)ил)-1,7-диазаспиро[4.4]нонан
7-(2-(1,3,4-оксадиазол)ил)-1,7-диазаспиро[4.4]нонан
7-(2-пиразинил)-1,7-диазаспиро[4.4]нонан
7-(3-пиридазинил)-1,7-диазаспиро[4.4]нонан
7-(5-метокси-3-пиридил)-1,7-диазаспиро[4.4]нонан
7-(5-циклопентилокси-3-пиридил)-1,7-диазаспиро[4.4]нонан
7-(5-фенокси-3-пиридил)-1,7-диазаспиро[4.4]нонан
7-(5-(4-гидроксифенокси)-3-пиридил)-1,7-диазаспиро[4.4]нонан
7-(5-этинил-3-пиридил)-1,7-диазаспиро[4.4]нонан
7-(6-хлор-3-пиридил)-1,7-диазаспиро[4.4]нонан
7-(6-метокси-3-пиридазинил)-1,7-диазаспиро[4.4]нонан
1-(3-пиридил)-1,7-диазаспиро[4.4]нонан
1-(5-пиримидинил)-1,7-диазаспиро[4.4]нонан
1-(5-изоксазолил)-1,7-диазаспиро[4.4]нонан
1-(5-изотиазолил)-1,7-диазаспиро[4.4]нонан
1-(5-(1,2,4-оксадиазол)ил)-1,7-диазаспиро[4.4]нонан
1-(2-(1,3,4-оксадиазол)ил)-1,7-диазаспиро[4.4]нонан
1-(2-пиразинил)-1,7-диазаспиро[4.4]нонан
1-(3-пиридазинил)-1,7-диазаспиро[4.4]нонан
1-метил-7-(3-пиридил)-1,7-диазаспиро[4.4]нонан
1-метил-7-(5-пиримидинил)-1,7-диазаспиро[4.4]нонан
1-метил-7-(5-изоксазолил)-1,7-диазаспиро[4.4]нонан
1-метил-7-(5-изотиазолил)-1,7-диазаспиро[4.4]нонан
1-метил-7-(5-(1,2,4-оксадиазол)ил)-1,7-диазаспиро[4.4]нонан
1-метил-7-(2-(1,3,4-оксадиазол)ил)-1,7-диазаспиро[4.4]нонан
1-метил-7-(2-пиразинил)-1,7-диазаспиро[4.4]нонан
1-метил-7-(3-пиридазинил)-1,7-диазаспиро[4.4]нонан
1-метил-7-(5-метокси-3-пиридил)-1,7-диазаспиро[4.4]нонан
1-метил-7-(5-циклопентилокси-3-пиридил)-1,7-диазаспиро[4.4]нонан
1-метил-7-(5-фенокси-3-пиридил)-1,7-диазаспиро[4.4]нонан
1-метил-7-(5-(4-гидроксифенокси)-3-пиридил)-1,7-диазаспиро[4.4]нонан
1-метил-7-(5-этинил-3-пиридил)-1,7-диазаспиро[4.4]нонан
1-метил-7-(6-хлор-3-пиридил)-1,7-диазаспиро[4.4]нонан
1-метил-7-(6-метокси-3-пиридазинил)-1,7-диазаспиро[4.4]нонан
7-метил-1-(3-пиридил)-1,7-диазаспиро[4.4]нонан
7-метил-1-(5-пиримидинил)-1,7-диазаспиро[4.4]нонан
7-метил-1-(5-изоксазолил)-1,7-диазаспиро[4.4]нонан
7-метил-1-(5-изотиазолил)-1,7-диазаспиро[4.4]нонан
7-метил-1-(5-(l,2,4-оксадиазол)ил)-1,7-диазаспиро[4.4]нонан
7-метил-1-(2-(1,3,4-оксадиазол)ил)-1,7-диазаспиро[4.4]нонан
7-метил-1-(2-пиразинил)-1,7-диазаспиро[4.4]нонан
7-метил-1-(3-пиридазинил)-1,7-диазаспиро[4.4]нонан
2-(3-пиридил)-2,7-диазаспиро[4.4]нонан
2-(5-пиримидинил)-2,7-диазаспиро[4.4]нонан
2-(5-изоксазолил)-2,7-диазаспиро[4.4]нонан
2-(5-изотиазолил)-2,7-диазаспиро[4.4]нонан
2-(5-(1,2,4-оксадиазол)ил)-2,7-диазаспиро[4.4]нонан
2-(2-(1,3,4-оксадиазол)ил)-2,7-диазаспиро[4.4]нонан
2-(2-пиразинил)-2,7-диазаспиро[4.4]нонан
2-(3-пиридазинил)-2,7-диазаспиро[4.4]нонан
2-(5-метокси-3-пиридил)-2,7-диазаспиро[4.4]нонан
2-(5-циклопентилокси-3-пиридил)-2,7-диазаспиро[4.4]нонан
2-(5-фенокси-3-пиридил)-2,7-диазаспиро[4.4]нонан
2-(5-(4-гидроксифенокси)-3-пиридил)-2,7-диазаспиро[4.4]нонан
2-(5-этинил-3-пиридил)-2,7-диазаспиро[4.4]нонан
2-(6-хлор-3-пиридил)-2,7-диазаспиро[4.4]нонан
2-(6-метокси-3-пиридазинил)-2,7-диазаспиро[4.4]нонан
2-метил-7-(3-пиридил)-2,7-диазаспиро[4.4]нонан
2-метил-7-(5-метокси-3-пиридил)-2,7-диазаспиро[4.4]нонан
2-метил-7-(5-фенокси-3-пиридил)-2,7-диазаспиро[4.4]нонан
6-(3-пиридил)-1,6-диазаспиро[3.4]октан
1-метил-6-(3-пиридил)-1,6-диазаспиро[3.4]октан
2-(3-пиридил)-2,5-диазаспиро[3.4]октан
5-метил-2-(3-пиридил)-2,5-диазаспиро[3.4]октан
6-(3-пиридил)-1,6-диазаспиро[3.5]нонан
1-метил-6-(3-пиридил)-1,6-диазаспиро[3.5]нонан
2-(3-пиридил)-2,5-диазаспиро[3.5]нонан
5-метил-2-(3-пиридил)-2,5-диазаспиро[3.5]нонан
2-(3-пиридил)-2,6-диазаспиро[4.5]декан
6-метил-2-(3-пиридил)-2,6-диазаспиро[4.5]декан
7-(3-пиридил)-1,7-диазаспиро[4.5]декан
1-метил-7-(3-пиридил)-1,7-диазаспиро[4.5]декан
8-(3-пиридил)-1,8-диазаспиро[5.5]ундекан
1-метил-8-(3-пиридил)-1,8-диазаспиро[5.5]ундекан
Другие характерные соединения настоящего соединения включают следующие:
1'-(3-пиридил)спиро[1-азабицикло[2.2.1]гептан-2,3'-пирролидин]
1'-(5-этокси-3-пиридил)спиро[1-азабицикло[2.2.1]гептан-2,3'-пирролидин]
1'-(5-циклопентилокси-3-пиридил)спиро[1-азабицикло[2,2,1]гептан-2,3'-пирролидин]
1'-(5-фенокси-3-пиридил)спиро[1-азабицикло[2.2.1]гептан-2,3'-пирролидин]
1'-(5-(4-гидроксифенокси)-3-пиридил)спиро[1-азабицикло[2.2.1]гептан-2,3'-пирролидин]
1'-(5-пиримидинил)спиро[1-азабицикло[2.2.1]гептан-2,3'-пирролидин]
1'-(5-изоксазолил)спиро[1-азабицикло[2.2.1]гептан-2,3'-пирролидин]
1'-(5-изотиазолил)спиро[1-азабицикло[2.2.1]гептан-2,3'-пирролидин]
1'-(5-(1,2,4-оксадиазол)ил)спиро[1-азабицикло[2.2.1]гептан-2,3'-пирролидин]
1'-(2-(1,3,4-оксадиазол)ил)спиро[1-азабицикло[2.2.1]гептан-2,3'-пирролидин]
1'-(2-пиразинил)спиро[1-азабицикло[2.2.1]гептан-2,3'-пирролидин]
1'-(3-пиридазинил)спиро[1-азабицикло[2.2.1]гептан-2,3'-пирролидин]
1'-(5-этинил-3-пиридил)спиро[1-азабицикло[2.2.1]гептан-2,3'-пирролидин]
1'-(6-хлор-3-пиридил)спиро[1-азабицикло[2.2.1]гептан-2,3'-пирролидин]
1'-(6-метокси-3-пиридазинил)спиро[1-азабицикло[2.2.1]гептан-2,3'-пирролидин]
1'-(3-пиридил)спиро[1-азабицикло[2.2.2]октан-2,3'-пирролидин]
1'-(5-метокси-3-пиридил)спиро[1-азабицикло[2.2.2]октан-2,3'-пирролидин]
1'-(5-циклопентилокси-3-пиридил)спиро[1-азабицикло[2.2.2]октан-2,3'-пирролидин]
1'-(5-фенокси-3-пиридил)спиро[1-азабицикло[2.2.2]октан-2,3'-пирролидин]
1'-(5-(4-гидроксифенокси)-3-пиридил)спиро[1-азабицикло[2.2.2]октан-2,3'-пирролидин]
1'-(5-этинил-3-пиридил)спиро[1-азабицикло[2.2.2]октан-2,3'-пирролидин]
1'-(6-хлор-3-пиридил)спиро[1-азабицикло[2.2.2]октан-2,3'-пирролидин]
1'-(5-пиримидинил)спиро[1-азабицикло[2.2.2]октан-2,3'-пирролидин]
1'-(2-пиразинил)спиро[1-азабицикло[2.2.2]октан-2,3'-пирролидин]
1'-(3-пиридазинил)спиро[1-азабицикло[2.2.2]октан-2,3'-пирролидин]
1'-(6-метокси-3-пиридазинил)спиро[1-азабицикло[2.2.2]октан-2,3'-пирролидин]
1'-(5-изоксазолил)спиро[1-азабицикло[2.2.2]октан-2,3'-пирролидин]
1'-(5-изотиазолил)спиро[1-азабицикло[2.2.2]октан-2,3'-пирролидин]
1'-(5-(1,2,4-оксадиазол)ил)спиро[1-азабицикло[2.2.2]октан-2,3'-пирролидин]
1'-(2-(1,3,4-оксадиазол)ил)спиро[1-азабицикло[2.2.2]октан-2,3'-пирролидин]
1'-(3-пиридил)-2'H-спиро[1-азабицикло[2.2.1]гептан-7,3'-пирролидин]
1'-(5-метокси-3-пиридил)-2'H-спиро[1-азабицикло[2.2.1]гептан-7,3'-пирролидин]
1'-(5-циклопентилокси-3-пиридил)-2'H-спиро[1-азабицикло[2.2.1]гептан-7,3'-пирролидин]
1'-(5-фенокси-3-пиридил)-2'H-спиро[1-азабицикло[2.2.1]гептан-7,3'-пирролидин]
1'-(5-(4-гидроксифенокси)-3-пиридил)-2'H-спиро[1-азабицикло[2.2.1]гептан-7,3'-пирролидин]
1'-(6-хлор-3-пиридил)-2'H-спиро[1-азабицикло[2.2.1]гептан-7,3'-пирролидин]
1'-(5-пиримидинил)-2'H-спиро[1-азабицикло[2.2.1]гептан-7,3'-пирролидин]
1'-(2-пиразинил)-2'H-спиро[1-азабицикло[2.2.1]гептан-7,3'-пирролидин]
1'-(3-пиридазинил)-2'H-спиро[1-азабицикло[2.2.1]гептан-7,3'-пирролидин]
1'-(6-метокси-3-пиридазинил)-2'H-спиро[1-азабицикло[2.2.1]гептан-7,3'-пирролидин]
1'-(5-изоксазолил)-2'H-спиро[1-азабицикло[2.2.1]гептан-7,3'-пирролидин]
1'-(5-изотиазолил)-2'H-спиро[1-азабицикло[2.2.1]гептан-7,3'-пирролидин]
1'-(5-(1,2,4-оксадиазол)ил)-2'H-спиро[1-азабицикло[2.2.1]гептан-7,3'-пирролидин]
1'-(2-(1,3,4-оксадиазол)ил)-2'H-спиро[1-азабицикло[2.2.1]гептан-7,3'-пирролидин]
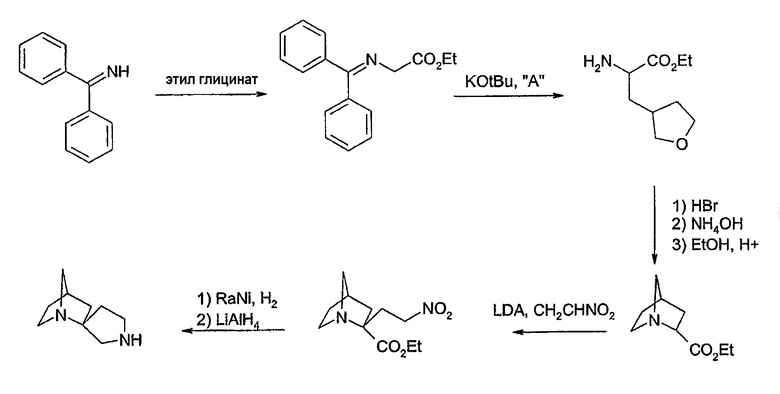
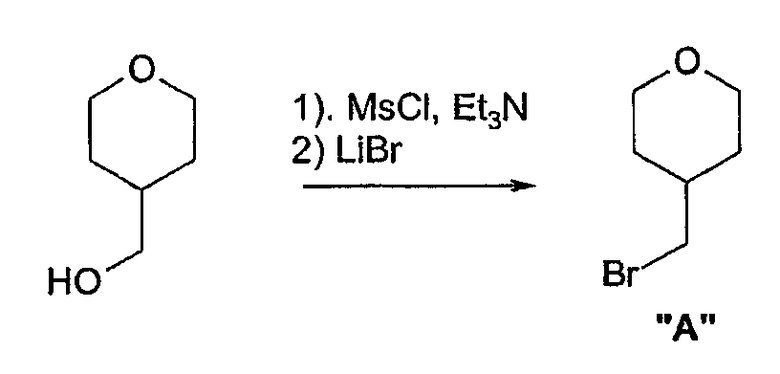
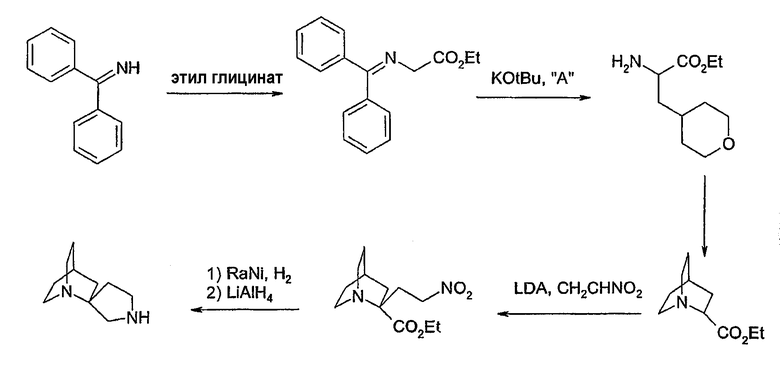
II. Способы получения соединений
Схема 1
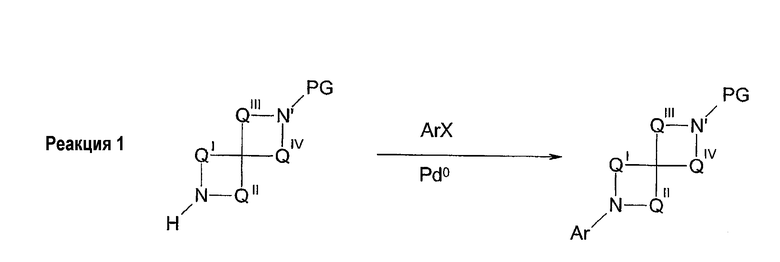
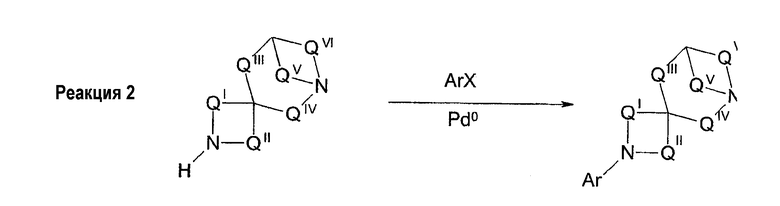
Соединения формул 1 и 2 могут быть получены обычным способом, включающим арилирование одной из аминогрупп, необязательно защищенной диазаспироалканом (схема 1). Арилирование при N подходящим арилом или предпочтительно гетероарилом, галогеном или трифлатом может быть выполнено согласно способам, известным специалистам в данной области техники, например, используя катализ металлами (например, соединений меди или платины). Предпочтительный обычный способ настоящего изобретения использует технологии Buchwald или Hartwig (Buchwald et al., J. Org. Chem., 61: 7240 (1996); Hartwig et al., J. Org. Chem., 64: 5575 (1999); см. также Old et al., J. Am. Chem. Soc. 120: 9722 (1998)), в которой амин обрабатывают палладиевым (0) катализатором, лигандом-фосфином и основанием. Таким образом, выполняют реакцию 1-бензил-1,7-диазаспиро[4.4]нонана с 3-бромпиридином в присутствии трис(дибензилиденацетон)дипалладия (0), 2,2'-бис(дифенилфосфино)-1,1'-бинафтила и трет-бутоксида натрия в толуоле для получения 1-бензил-7-(3-пиридил)диазаспиро[4.4]нонана. Удаление бензильной группы гидрированием, более 10% палладия-на-угле, дает 7-(3-пиридил)диазаспиро[4.4]нонан. Специалистам в данной области техники очевидно, что в качестве альтернативы могут использоваться различные стратегии защитных групп для получения продуктов, несущих арильную группу на азоте N', в отличие от N (реакция 1, схема 1). Особенно полезная комбинация защитных групп в настоящем изобретении представляет собой бензил и карбамат, в особенности трет-бутилкарбамат. Таким образом, 1-бензил-1,7-диазаспиро[4.4]нонан превращается в 1-бензил-7-(трет-бутоксикарбонил)-1,7-диазаспиро[4.4]нонан обработкой ди-трет-бутилкарбонатом. Последующее гидрирование и катализируемое палладием арилирование с 3-бромпиридином дает 7-(трет-бутоксикарбонил)-1-(3-пиридил)диазаспиро[4.4]нонан. Удаление трет-бутоксикарбонильной группы гидрохлорной кислотой обеспечивает 1-(3-пиридил)диазаспиро[4.4]нонан. И, наконец, во многих случаях, где N и N' пространственно отличаются, и всякий раз, когда N является третичным (как в реакции 2, схема 1), селективное арилирование N может происходить без первого защищенного N'. Таким образом, реакция 1,7-диазаспиро[4.4]нонана с 3-бромпиридином в катализируемых палладием условиях, описанных ранее, дает практически исключительно 7-(3-пиридил)диазаспиро[4.4]нонан.
Специалистам в данной области техники очевидно, что на практике можно осуществить введение заместителей в гетероарильное кольцо, введенное в диазаспироалкан. Такие заместители могут обеспечивать полезные свойства или могут быть полезными сами по себе и могут давать возможность для дальнейшей синтетической обработки. Подходящий защищенный гетероарильный диазаспироалкан может быть обработан с целью получения полезных соединений, имеющих заместители на гетероарильном кольце. Например, 1-бензил-7-(5-бромо-3-пиридил)-1,7-диазаспиро[4.4]нонан может быть получен при помощи реакции 3,5-дибромпиридина с 1-бензил-1,7-диазаспиро[4.4]нонаном согласно вышеописанной процедуре. Для удобства 1-бензил-7-(5-бром-3-пиридил)-1,7-диазаспиро[4.4]нонан в соответствующем 5-аминозамещенном соединении можно получить при помощи обычного способа Zwart et al., Recueil Trav. Chim. Pays-Bas 74: 1062 (1995), в котором бромсоединение нагревают с водным аммиаком в присутствии медного катализатора. 5-алкиламинозамещенные соединения могут быть получены аналогичным способом. 5-этинилзамещенные соединения могут быть получены из 5-бромсоединений путем взаимодействия, катализируемого палладием, с 2-метил-3-бутин-2-олом с последующим удалением ацетона, которое катализируется основанием (гидридом натрия), согласно обычному способу, описанному у Cosford et al., J. Med. Chem. 39: 3235 (1996). 5-этинильные аналоги могут быть превращены в соответствующие 5-этенильные и затем в соответствующие 5-этиловые аналоги при помощи реакций гидрирования с избытком катализатора. 5-азидозамещенные аналоги могут быть получены из 5-бромсоединения реакцией с азидом лития в N,N-диметилформамиде. 5-алкилтиозамещенные аналоги могут быть получены из 5-бромсоединения реакцией с подходящим алкилмеркаптидом натрия (алкантиолат натрия), используя способ, хорошо известный в области органического синтеза.
Несколько других аналогов, несущих заместители в 5-й позиции пиридинового кольца, могут быть синтезированы из соответствующих аминосоединений и, наоборот, через промежуточное соединение соли 5-диазония. Примеры других 5-замещенных аналогов, которые могут быть получены из промежуточного соединения соли 5-диазония, включают без ограничений 5-гидрокси, 5-алкокси, 5-фтор, 5-хлор, 5-йод, 5-циано и 5-меркапто. Эти соединения могут быть синтезированы обычными способами, приведенными у Zwart et al., см. выше. Например, 1-бензил-7-(5-гидрокси-3-пиридил)-1,7-диазаспиро[4.4]нонан может быть получен реакцией соответствующей промежуточной соли 5-диазония с водой. Аналогично, 1-бензил-7-(5-алкокси-3-пиридил)-1,7-диазаспиро[4.4]нонан может быть синтезирован реакцией соли диазония со спиртами. Подходящие соли 5-диазония могут быть использованы для синтеза циано или галогенпроизводных соединений, как известно специалистам в данной области техники. 5-меркаптозаместители могут быть получены способом, описанным у Hoffman et al., J. Med. Chem. 36: 953 (1993). 5-меркаптан, полученный таким образом, можно, в свою очередь, превратить в 5-алкилтиозаместитель реакцией с гидридом натрия и подходящим алкилбромидом. Затем можно провести процесс окисления для получения сульфона. 5-ациламидные аналоги вышеупомянутых соединений могут быть получены реакцией соответствующих 5-аминосоединений с подходящим кислым ангидридом или кислым хлоридом способами, хорошо известными в области органического синтеза.
5-гидроксизамещенные аналоги вышеуказанных соединений могут быть использованы для получения соответствующих 5-алканоилоксизамещенных соединений реакцией с подходящей кислотой, кислым хлоридом или кислым гидридом. Аналогично, 5-гидроксисоединения являются предшественниками как для 5-арилокси, так и для 5-гетероарилокси через нуклеофильное ароматическое замещение в ароматических кольцах с дефицитом электрона (например, 4-фторбензонитрил и 2,4-дихлорпиримидин). Такая химия хорошо известна специалистам в области органического синтеза. Любое производное может быть получено из 5-гидроксисоединений алкилированием алкилгалогенами и соответствующим основанием или реакцией Митсунобу (Mitsunobu), в которой обычно используется триалкил- или триарилфосфин и диэтилазодикарбоксилат. См. Hughes, Org. React. (N.Y.) 42: 335 (1992) и Hughes, Org. Prep.Proced. Int. 28: 127 (1996) в обычных условиях Митсунобу (Mitsunobu).
5-цианозамещенные аналоги вышеуказанных соединений могут быть гидролизованы для получения соответствующих 5-карбоксамидозамещенных соединений. Дальнейший гидролиз приводит к образованию соответствующих аналогов замещенной 5-карбоксильной кислоты. Восстановление 5-цианозамещенных аналогов алюмогидридом лития дает соответствующие 5-аминометильные аналоги. 5-ацилзамещенные аналоги могут быть получены из соответствующих аналогов замещенной 5-карбоксильной кислоты реакцией с подходящим алкиллитием, используя способы, известные специалистам в области органического синтеза.
Аналоги замещенной 5-карбоксильной кислоты вышеуказанных соединений могут быть превращены в соответствующие эфиры реакцией с подходящим спиртом и кислым катализатором. Соединения с эфирной группой в 5-пиридильной позиции могут быть восстановлены боргидридом натрия или алюмогидридом лития для получения соответствующих 5-гидроксиметилзамещенных аналогов. В свою очередь, эти аналоги могут быть преобразованы в соединения, несущие эфирные фрагменты в 5-пиридильной позиции, при помощи реакции с гидридом натрия и подходящим алкилгалогеном, используя способы превращений. В качестве альтернативы, можно провести реакцию 5-гидроксиметилзамещенных аналогов с тозилхлоридом для получения соответствующих 5-тозилоксиметиловых аналогов. Аналоги замещенной 5-карбоксильной кислоты также могут быть превращены в 5-алкиламиноациловые аналоги последовательной обработкой тионилхлоридом и подходящим алкиламином. Хорошо известно, что некоторые из этих амидов действительно подвержены нуклеофильному ацильному замещению при получении кетонов. Таким образом, так называемые амиды Вейнреба (Weinreb) (N-метокси-N-метиламиды) реагируют с ариллитиевыми реагентами, давая соответствующие диарилкетоны. Например, см. Selnick et al., Tet. Lett. 34: 2043 (1993).
5-тозилоксиметилзамещенные аналоги вышеуказанных соединений могут быть превращены в соответствующие 5-метилзамещенные соединения восстановлением алюмогидридом лития. 5-тозилоксиметилзамещенные аналоги вышеуказанных соединений также могут быть использованы для получения 5-алкилзамещенных соединений реакцией с алкиллитиевым реагентом. 5-гидроксизамещенные аналоги вышеуказанных соединений могут быть использованы для получения 5-N-алкил- или 5-N-арилкарбамоилоксизамещенных соединений реакцией с N-алкил- или N-арилизоцианатами. 5-аминозамещенные аналоги вышеуказанных соединений могут быть использованы для получения 5-алкоксикарбоксамидозамещенных соединений и производных 5-карбамида реакцией с алкиловыми сложными эфирами хлормуравьиной кислоты и N-алкил- или N-арилизоционатами соответственно, используя способы, известные специалистам в области органического синтеза.
Может быть разработана химия, аналогичная таковой, раскрытой выше в настоящем описании, для получения 5-замещенных пиридиновых аналогов диазаспиросоединений для синтеза аналогов, несущих заместители во 2, 4 и 6 позициях пиридинового кольца. Например, несколько 2-, 4- и 6-аминопиридилдиазаспироалканов может быть превращено в соответствующие промежуточные соединения соли диазония, которые могут быть трансформированы во множество соединений с заместителями во 2, 4 и 6 позициях пиридинового кольца, как описано выше для 5-замещенных аналогов. Требуемые 2-, 4- и 6-аминопиридилдиазаспироалканы можно получить при помощи реакции Чичибабина, реакцией незамещенных пиридилдиазаспироалканов (например, описанного выше 1-бензил-7-(3-пиридил)-1,7-диазаспиро[4.4]нонана) с амидом натрия. Аналогичные реакции описаны в Chemistry of Heterocyclic Compounds, vol.14, part 3, pp.3-5 (Intescience Publishes, 1962) и Lahti et al., J. Med. Chem. 42: 2227 (1999).
После получения описанной функциональной группы гетероарильного кольца в подходящих условиях из диазабицикла может быть удалена необязательная защитная группа. Таким образом, например, гидрогенолиз 1-бензил-7-(5-алкокси-3-пиридил)-1,7-диазаспиро[4.4]нонана будет давать 7-(5-алкокси-3-пиридил)-1,7-диазаспиро[4.4]нонан. Специалистам в области органической химии очевидна необходимость объединения защитных групп при помощи химии, необходимой для создания конкретных функциональных групп. В некоторых случаях для сохранения конкретной функциональности может потребоваться замена одной защитной группы другой.
В альтернативном подходе синтеза пиридинзамещенных пиридилдиазаспироалканов 3,5-дибромпиридин может быть превращен в соответствующие 5-алкокси-3-бром- и 5-арилокси-3-бромпиридины под воздействием алкоксидов натрия или арилоксидов натрия при помощи процедур, которые описаны у Comins et al., J. Org. Chem. 55: 69 (1990) и Hertog et al., Recueil Trav. Chim. Pays-Bas 74: 1171 (1955). Это проиллюстрировано получением 7-(5-(4-метоксифенокси)-3-пиридил)-1,7-диазаспиро[4.4]нонана. Реакция 3,5-дибромпиридина с 4-метоксифеноксидом натрия в N,N-диметилформамиде дает 3-бром-5-(4-метоксифенокси)пиридин. Взаимодействие 3-бром-5-(4-метоксифенокси)пиридина с 1-бензил-7-(3-пиридил)-1,7-диазаспиро[4.4]нонаном в присутствии трет-бутоксида натрия и каталитического количества трис(дибензилиденацетон)дипалладия (0) и 2,2'-бис(дифенилфосфино)-1,1'-бинафтила в толуоле с последующим гидрогенолизом бензильной защитной группы будет давать 7-(5-(4-метоксифенокси)-3-пиридил)-1,7-диазаспиро[4.4]нонан.
Вышеописанной реакции взаимодействия, катализируемой палладием, подвергают другие арилгалогены. Таким образом, получают 7-(5-пиримидинил)-1,7-диазаспиро[4.4]нонан из 5-бромпиримидина и необязательно 1,7-диазаспиро[4.4]нонан, защищенный в 1-й позиции, с последующим удалением защитной группы, при необходимости. Этот способ особенно применим в таком случае, как 3-бромпиридин, 3,5-дибромпиридин и 5-бромпиридин, где ароматическое кольцо не активно относительно ароматического нуклеофильного замещения.
В некоторых случаях взаимодействие гетероароматического кольца с диазациклом может проходить без использования палладиевого катализатора. Примеры как пяти-, так и шестичленных гетероароматических соединений кольцевой структуры, которые активируются в направлении нуклеофильного ароматического замещения, известны специалистам в области органического синтеза. Например, 7-(6-хлор-3-пиридазинил)-1,7-диазаспиро[4.4]нонан может быть синтезирован из 3,6-дихлорпиридазина и 1,7-диазаспиро[4.4]нонана. Аналогично, 2,6-дихлорпиразин и 2-бромтиазол будут реагировать с 1,7-диазаспиро[4.4]нонаном, давая 7-(6-хлор-2-пиразинил)-1,7-диазаспиро[4.4]нонан и 7-(2-тиазоил)-1,7-диазаспиро[4.4]нонан соответственно.
Реакции взаимодействия, описанные в настоящей заявке, независимо от того, катализируются ли они палладием или нет, представляют собой высокопроизводительный способ синтеза. Такие библиотеки соединений настоящего изобретения могут быть получены путем взаимодействия в формате 96-луночного планшета, например, различных галогенаренов с различными диазаспиросоединениями.
Специфические диазаспиросистемы кольцевой структуры
Промежуточные соединения необязательно защищенных диазаспироалканов, используемых для получения соединений формулы I и II, могут быть образованы множеством различных способов. Известны некоторые из таких промежуточных соединений диазаспироалканов, которые могут быть получены при помощи способов, известных в данной области техники. Однако синтез промежуточных соединений с использованием химии палладия является новым для данной области техники, а фармацевтическая активность промежуточных соединений в данной области техники не является подходящей.
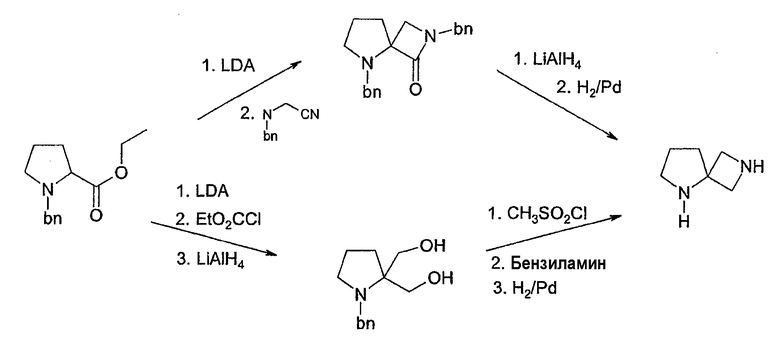
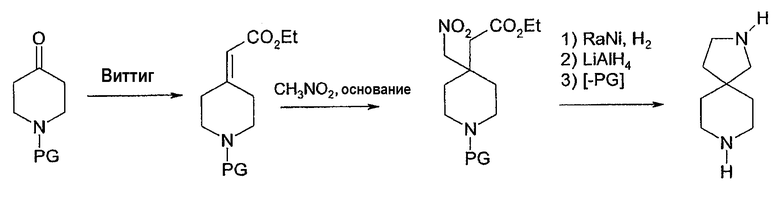
Соединения формулы 1, в которых u=v=1, w=0 и x=3, имеют 2,5-диазаспиро[3,4]октановое ядро, которое может быть получено, как описано в схеме 2.
Алкилирование N-бензил-L-пролинэтилового эфира (Aldrich Chemical), используя сильное основание, такое как диизопропиламид лития (LDA), и цианометилбензиламин, эквивалентный аминометилу, дает бета-лактам согласно процедуре, опубликованной у Overman, J. Am. Chem. Soc. 107: 1698 (1985) и Tet. Lett. 25:1635 (1985). Затем его можно восстановить алюмогидридом лития для получения 2,5-дибензилового производного от 2,5-диазаспиро[3,4]октана. Удаление защитных бензильных групп при помощи либо гидрирования, либо окислительного расщепления, например, нитратом церия-аммония даст 2,5-диазаспиро[3.4]октан. В качестве альтернативы, может быть использована химия, аналогичная химии, описанной в ЕР заявке на патент 90117078.7 (ЕР регистрационный номер 0417631) для получения геминального бис(гидроксиметил)производного и последующего превращения его в желательный 2,5-диазаспиро[3.4]октан (схема 2). Предполагается, что дальнейшее арилирование, катализируемое палладием, как описано выше, будет проходить селективно для менее пространственно заблокированного азота азетидинила, давая 2-арил-2,5-диазаспиро[3.4]октаны. Изомерные 5-арил-2,5-диазаспиро[3.4]октаны могут быть получены путем сначала защиты азетидинильного азота (например, карбаматом) и затем путем арилирования с последующим удалением защитной группы.
Схема 2
Соединения формулы 1, где u=2, v=1, w=0 и x=3, имеют систему 1,7-диазаспиро[3.4]нонана, которая может быть получена согласно многочисленным способам, некоторые из которых показаны ниже в схеме 3. В одном из вариантов осуществления (способ А) в подходящим образом защищенном сложном эфире пролина, например N-бензил-L-пролинэтиловом эфире, может быть удалена защитная группа при помощи диизопропиламида лития и может быть выполнена реакция присоединения по Михаэлю к нитроэтилену. Это дает метил-2-(2-нитроэтил)-1-бензилпирролидин-2-карбоксилат. Последующее восстановление нитрогруппы никелем Ренея с последующей лактамизацией способом, известным специалистам в данной области техники (например, нагреванием в подходящем растворителе в присутствии или без кислого или основного катализатора), дает 1-бензил-1,7-диазаспиро[4.4]нонан-6-он.
В качестве альтернативы 1,7-диазаспиро[4.4]нонан-6-он может быть получен согласно одному из некоторых других способов, опубликованных в литературе. Такие описанные способы указывают, что подходящим образом защищенный эфир пролина может быть лишен защиты при помощи диизопропиламида лития и может вступить в реакцию с алкилирующим агентом, таким как хлорацетонитрил, затем подвержен восстановлению нитрила и циклизации (способ В, схема 3), как опубликовано у Culbertson et al., J. Med. Chem. 33: 2270 (1990).
Другие публикации указывают, что подходящим образом защищенный эфир пролина может быть лишен защиты при помощи диизопропиламида лития и может вступить в реакцию с алкилирующим агентом, таким как аллилбромид (способ С, схема 3). Затем может быть проведено окислительное расщепление полученного олефина до альдегида, как описано у Genin et al., J. Org. Chem. 58: 2334 (1993); Hinds et al., J. Med. Chem. 34: 1777 (1991); Kim et al., J. Org. Chem. 61: 3138 (1996); EP 0360390 и патент США №5733912. После чего альдегид может быть подвергнут восстановительному аминированию солью аммония или первичным алифатическим или ароматическим амином согласно способу, известному специалистам в данной области техники. В качестве альтернативы, альдегид может быть восстановлен соответствующим спиртом, и затем спирт может быть трансформирован в амин путем превращения в уходящую группу с последующей заменой подходящим амином. Это также может быть достигнуто путем замены уходящей группы ионом азида и последующим восстановлением первичного амина способом, хорошо известным специалистам в данной области техники. Спирт может быть превращен в амин в условиях Митсунобу, как описано выше. Алкил 2-аминоэтилпирролидин-2-карбоксилат, полученный согласно одному из вышеописанных способов, может быть циклизован в спиролактам способами, известными специалистам в данной области техники, такими как нагревание в подходящем растворителе в присутствии или без кислого или основного катализатора.
Лактам, полученный любым из вышеуказанных способов (способы А, В или С), может быть обработан подходящим восстанавливающим агентом, таким как алюмогидрид лития, для получения защищенного 1,7-диазаспиро[4.4]нонана, в этом примере 1-бензил-1,7-диазаспиро[4.4]нонана. Защитная группа может быть удалена способом, известным специалистам в данной области техники, для получения 1,7-диазаспиро[4.4]нонана. Арилирование любого азота может быть выполнено способом, раскрытым в настоящем описании.
Схема 3
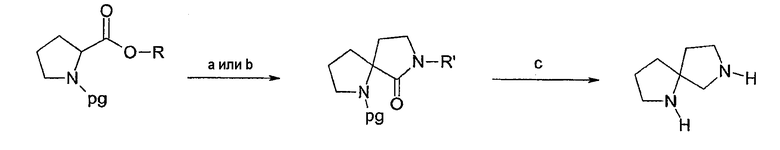
а) (i) LDA; (ii) нитроэтилен (способ А) или ClCH2CN (способ В); (iii) RaNi; (iv) PhCH3, нагревание
b) (i) LDA; (ii) аллилбромид; (iii) О3 или OsO4, NaIO4; (iv) NH4Cl, NaBH(OAc)3 (способ С)
с) (i) LiAlH4 или BH3; (ii) [-PG]
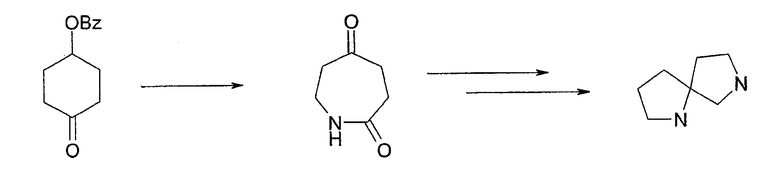
В качестве альтернативы, ядро 1,7-диазаспиро[4.4]нонана также может быть получено согласно схеме 4. Превращение 1,4-диоксаспиро[4.5]декан-8-она в 4-бензоилоксициклогексанон действительно может быть достигнуто специалистами в данной области техники. Последующая трансформация 4-бензоилоксициклогексанона в 1,7-диазосиро[4.4]нонан (при помощи промежуточного образования 4-оксокапролактама, как показано) может быть выполнена, как описано у Majer et al., Coll. Czech. Chem. Comm. 47: 950 (1982).
Схема 4
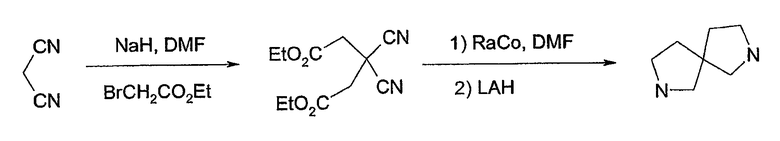
Соединения формулы 1, в которых u=2, v=1, w=1 и х=2, имеют симметричную систему 2,7-диазаспиро[4.4]нонана, могут быть получены согласно схеме 5. Этот способ описан у Overman et al., J. Org. Chem. 46: 2757 (1981) и Culbertson et al., J. Med. Chem. 33:2270 (1990).
Схема 5
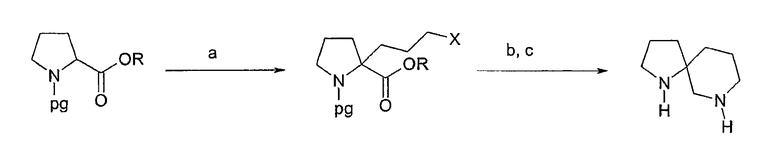
Соединения формулы 1, в которых u=3, v=1, w=0 и х=3, имеют систему 1,7-диазаспиро[4.5]декана, могут быть получены согласно схеме 6. Как описано у Kim et al., J. Org. Chem. 61: 3138 (1996), в патенте ЕР 360390 и патенте США №5733912, соответствующим образом защищенный эфир пролина (например, N-бензил-L-пролинэтиловый эфир) может быть лишен защиты при помощи диизопропиламида лития и может вступить в реакцию с алкилирующим агентом, таким как аллилбромид. В патенте США №5733912 также описано, что может быть выполнено гидроборирование/окисление аллильной боковой цепи для получения 2-(3-гидроксипропил)группы. Специалистам в данной области техники очевидно, что гидроксильная группа может быть затем превращена в аминогруппу несколькими способами, например окислением с последующим восстановительным аминированием. В качестве альтернативы, соответствующим образом защищенный эфир пролина может быть лишен защиты при помощи диизопропиламида лития и может вступить в реакцию с алкилирующим агентом, таким как дийодопропан. Затем может быть выполнено превращение первичного йодида в амин согласно известным способам, например обработкой аммонием в присутствии медного катализатора. Полученный аминоэфир может быть циклизован для получения защищенного 1,7-диазаспиро[4.5]декана-6-она с использованием любого количества известных процедур, например нагреванием в подходящем растворителе в присутствии или без кислого или основного катализатора, как описано выше. В качестве альтернативы, указанный 1,7-диазаспиро[4.5]декана-6-он может быть получен, как описано у Loefas et al., J. Het. Chem. 21: 583 (1984), где используется сужение кольца 2,10-диазабицикло[4.4.0]дек-1-ена.
1,7-диазаспиро[4.5]декана-6-он, полученный любым из вышеуказанных способов, затем может быть обработан восстанавливающим агентом, таким как алюмогидрид лития, с последующим удалением защитной группы для получения желательного 1,7-диазоспиро[4.5]декана. После этого может быть выполнено арилирование по любому азоту при помощи способа, раскрытого в настоящем описании.
Схема 6
а) Х=ОН: (i) LDA, аллилбромид; (ii) BH3, H2O
X=I: LDA, 1,3-дийодопропан
b) X=OH: (i) PCC или Swern; (ii) NH4Cl, NaBH(OAc)3; (iii) нагревание (+катализатор?) х=I: (i) NH3, CuI; (ii) нагревание (+катализатор?)
с) (i) BH3 или LiAlH4; (ii) [-PG]
Соединения формулы 1, в которых u=2, v=1, w=0 и х=4, имеют ядро 2,6-диазаспиро[4.5]декана, могут быть получены согласно способу, описанному у Ciblat, et al., Tet. Lett. 42: 4815 (2001). Таким образом, коммерчески доступный 1-бензил-3-пирролидинон может вступать в реакцию с 2-метил-2-(2-аминоэтил)-1,3-диоксоланом (Islam and Raphael, J. Chem. Soc. 3151 (1955)) во внутримолекулярной реакции Манниха (Mannich). Продукт, этиленкеталь 2-бензил-2,10-диазаспиро[4.5]декан-7-она, может быть затем гидролизован в кетон при помощи водного раствора гидрохлорной кислоты. После чего может быть выполнено деоксигенирование кетона стандартным способом, таким как превращение в соответствующий 1,3-дитиан с последующей обработкой никелем Ренея. 2-бензил-2,6-диазаспиро[4.5]декан, полученный таким образом, может быть непосредственно арилирован по азоту в 6-й позиции или превращен в 6-(трет-бутоксикарбонил)-2,6-диазаспиро[4.5]декан обработкой ди-трет-бутилдикарбонатом с последующим гидрированием. Далее, последнее производное может быть арилировано по азоту во 2-й позиции. Аналогичная химия может быть использована для превращения других азациклических кетонов в соответствующие спиродиазасоединения. Таким образом, реакция любого из различных N-защищенных 3-азетидинонов (синтез которых описан у Lall, et al., J. Org. Chem. 67: 1536 (2002) и Marchand, et al., Heterocycles 49: 149 (1998)) с 2-метил-2-(2-аминоэтил)-1,3-диоксоланом с последующим деоксигенированием (как описано выше) даст соответствующий защищенный 2,5-диазаспиро[3.5]нонан (формула 1, в которой u=1, v=1, w=0 и х=4).
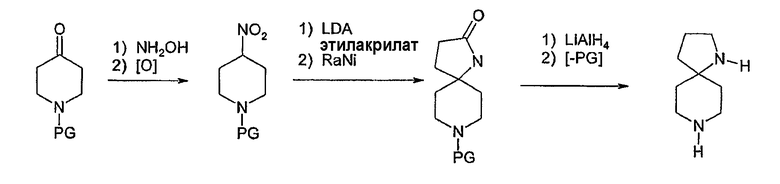
Соединения формулы 1, у которых u=v=2, w=0 и х=3, имеют ядро 1,8-диазаспиро[4.5]декана, могут быть получены согласно схеме 7. Как написано у Wittekind et al., J. Het. Chem. 9: 11 (1972), защищенный 4-пиперидон может быть превращен в 4-нитропиперидин. Последующая реакция присоединения по Михаэлю этилакрилата, например, с последующим восстановлением нитрогруппы никелем Ренея дает 1,8-диазоспиро[4.5]декан-2-он. Этот лактам может быть восстановлен соответствующим восстанавливающим агентом, таким как алюмогидрид лития, с последующим удалением защитной группы для получения необязательно замещенного 1,8-диазаспиро[4.5]декана. Арилирование по любому азоту может быть выполнено при помощи способов, раскрытых в настоящем описании.
Схема 7
Соединения формулы 1, в которых u=2, v=1 и w=х=2, имеют ядро 2,8-диазаспиро[4.5]декана, могут быть получены согласно схеме 8. Согласно различным описаниям (Helv. Chim. Acta 60: 1650 (1977); Smith et al., J. Med. Chem. 19: 3772 (1995); Elliott et al., Biorg. Med. Chem. Lett. 8: 1851 (1998)), защищенный 4-пиперидон может быть превращен в 4-пиперидинилиденовый эфир уксусной кислоты путем олефинирования по Виттигу (Wittig). Последующая реакция присоединения по Михаэлю аниона нитрометана с последующим восстановлением нитрогруппы и произвольной циклизацией при помощи никеля Ренея дает защищенный 2,8-диазоспиро[4.5]декан-3-он. Обработка защищенного 2,8-диазоспиро[4.5]декан-3-она восстанавливающим агентом, таким как алюмогидрид лития, и последующее удаление защитной группы дает 2,8-диазаспиро[4.5]декан. Арилирование может быть выполнено по любому азоту способом, раскрытым в настоящем описании.
Схема 8
Соединения формулы 1, в которых u=2, v=1, w=4 и х=0, имеют ядро 1,8-диазаспиро[5.5]декана и могут быть получены согласно процедурам, используемым для аналогов 1,7-диазаспиро[4.4]нонанов путем замещения эфиром пипеколината вместо эфира пролина. В качестве альтернативы, может быть использована процедура, описанная у Zhu et al., J. Org. Chem. 58: 6451 (1993).
Соединения формулы 1, в которых u=3, v=1, w=1 и х=3, имеют симметричное ядро 2,8-диазаспиро[5.5]декана и могут быть получены согласно процедурам, опубликованным у Helv. Chim. Acta 36: 1815 (1953), J. Org. Chem. 28: 336 (1963) или предпочтительно согласно Culbertson et al., J. Med. Chem. 33: 2270 (1990).
Соединения формулы 1, в которых u=v=2 и w=х=2, имеют симметричное ядро 3,9-диазаспиро[5.5]ундекана и могут быть получены согласно процедурам, описанным у Rice et al., J. Het. Chem. 1: 125 (1964), патенте США 3282947 или J. Med. Chem. 8: 62 (1965).
Отдельные энантиомерные соединения настоящего изобретения могут быть образованы различными способами. Один из способов, хорошо известных специалистам в области органического синтеза, включает разрешение при помощи диастереомерных солей. Соединения настоящего изобретения содержат основные атомы азота и реагируют с кислотами с образованием кристаллических солей. Различные кислоты, карбоксильная и сульфоновая, являются коммерчески доступными в энантиомерно чистой форме. Примеры включают винную, дибензоил- и ди-п-толуоилвинную и камфорсульфоновую кислоты. В реакции любой из этих или других отдельных энантиомерных кислот с рацемическим аминным основанием получаются диастереомерные соли. Фракционная кристаллизация солей и последующее восстановление основания дает в результате разрешение их энантиомеров.
Другое средство разделения включает превращение смеси энантиомеров в диастереомерные амиды или карбаматы при помощи хиральной кислоты или хлорформиата. Таким образом, при взаимодействии рацемического 7-(3-пиридил)-1,7-диазаспиро[4.4]нонана с N-(трет-бутоксикарбонил)-S-пролином, используя дифенилхлорфосфат, и удаляемой (при помощи трифторуксусной кислоты) защитной группой получаемые диастереомерные амиды пролина 7-(3-пиридил)-1,7-диазаспиро[4.4]нонан могут быть разделены при помощи жидкой хроматографии. Затем разделенные амиды трансформируются в (+) и (-) 7-(3-пиридил)-1,7-диазаспиро[4.4]нонан деградацией по Эдману (Edman).
Селективный синтез отдельных энантиомеров также может быть выполнен способами, известными специалистам в данной области техники. Такие способы будут отличаться химией, используемой для образования вариантов диазоспирокольца. Например, для синтеза, при котором для образования диазаспиросистемы (такой, как описано для системы 1,7-диазаспиро[4.4]нонана) используется алкилирование производного пролина, алкилирование пролина может быть выполнено стереоспецифическим способом. Таким образом, такие способы, как описано у Beck et al., Org. Synth. 72: 62 (1993) или Wang and Germanas, Synlett: 33 (1999) (и ссылки, приведенные в настоящем описании), могут быть использованы для управления стереохимией этапа алкилирования. Если в качестве начального вещества для такого процесса используется энантиомерно чистый эфир пролина (коммерчески доступный от Aldrich), то продукт алкилирования также будет представлять собой отдельный энантиомер. В таких процессах алкилирования может быть использовано множество электрофилов, включая аллилгалогены, которые были использованы при сборке спиросистем в отношении соединений настоящего изобретения Genin and Johnson, J. Amer. Chem. Soc. 114: 8778 (1992).
Мостиковые спирокольцевые системы
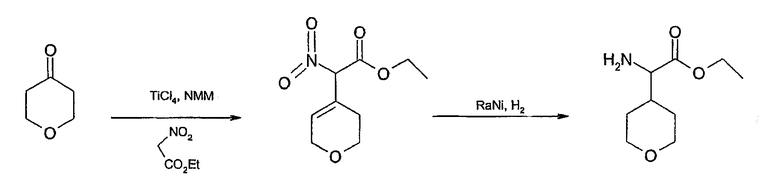
Соединения формулы 2, в которых u=1, v=2, w=0, x=0, y=2 и z=2, имеют ядро спиро[1-азабицикло[2.2.1]гептан-7,3'-пирролидин] и могут быть получены согласно схеме 9. Анион этилнитроацетата, образованный в присутствии основания амина, может быть конденсирован с тетрагидропиран-4-оном процедурой, описанной у Fornicola et al., J. Org. Chem. 63: 3528 (1998). Одновременное восстановление нитрогруппы и олефина в условиях каталитического гидрирования дает 2-(4-оксанил)глициновый эфир. Это соединение может быть обработано гидробромной кислотой для получения дибромида, который циклизуется в присутствии основания в азабицикло[2.2.1]гептан-7-карбоксильную кислоту. Обработка кислоты этанолом и серной кислотой дает этил азабицикло[2.2.1]гептан-7-карбоксилат. Это соединение затем подвергают депротонированию диизопропиламидом лития и реакции присоединения по Михаэлю с нитроэтиленом для получения этил аза-7-(2-нитроэтил)бицикло[2.2.1]гептан-7-карбоксилата. Восстановление нитрогруппы никелем Ренея с последующей самопроизвольной циклизацией дает спиролактам. Обработка лактама алюмогидридом лития дает спиро[1-азабицикло[2.2.1]гептан-7,3'-пирролидин], который затем арилируют по азоту пирролидина для получения соединений настоящего изобретения.
Схема 9
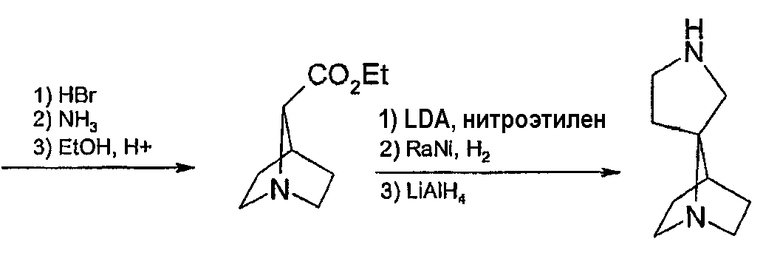
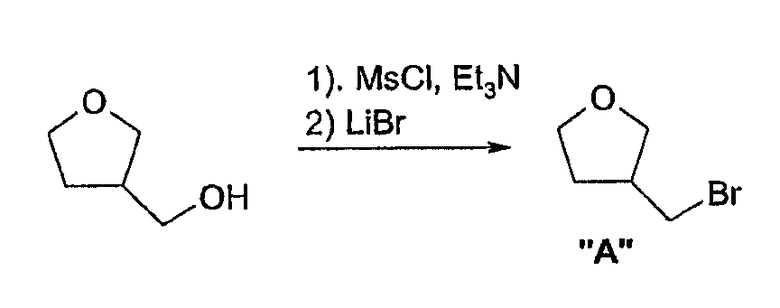
Соединения формулы 2, в которых u=1, v=2, w=1, x=0, y=1 и z=2, имеют спиро[1-азабицикло[2.2.1]гептан-2,3'-пирролидин]кольцевую систему и могут быть получены согласно схеме 10. Превращение тетрагидрофуран-3-илметанола (Aldrich) в 3-(бромметил)тетрагидрофуран может быть достигнуто при помощи мезилирования и последующей обработкой бромидом лития. Реакция этил глицината с имином бензофенона дает этил 3-аза-4,4-дифенил-бут-3-еноат, который служит как для защиты амина, так и для активации углерода метилена по пути алкилирования. Алкилирование этого имина может быть выполнено согласно способу Hansen, J. Org. Chem. 63: 775 (1998) депротонированием трет-бутоксидом калия и реакцией с 3-(бромметил)тетрагидрофураном. Удаление защиты в кислых условиях дает желательный 2-амино-3-(тетрагидрофуран-3-ил)пропионовый эфир. Раскрытие кольца тетрагидрофурана может быть достигнуто путем обработки гидробромной кислотой для получения дибромамино-кислого промежуточного соединения, которое при нагревании в присутствии основания циклизуется в 1-азабицикло[2.2.1]гептан-2-карбоксильную кислоту. Эта кислота затем может быть превращена в этиловый эфир при помощи этанола и серной кислоты. Затем может быть выполнено алкилирование депротонированием диизопропилмамидом лития и проведена реакция с нитроэтиленом. Последующее восстановление нитрогруппы никелем Ренея с последующей лактамизацией способами, известными специалистам в данной области техники, дает спиро[1-азабицикло[2.2.1]гептан-2,3'-пирролидин]-2'-он. Обработка лактама алюмогидридом лития дает желательный спиро[1-азабицикло[2.2.1]гептан-2,3'-пирролидин], который затем арилируют по азоту пирролидина для получения соединений настоящего изобретения.
Схема 10
Соединения формулы 2, в которых u=1, v=2, w=1, x=0, y=2 и z=2, имеют ядро спиро[1-азабицикло[2.2.2]октан-2,3'-пирролидина] и могут быть получены способом, аналогичным способам для соответствующего спиро[1-азабицикло[2.2.1]гептан-2,3'-пирролидина], как показано на схеме 11. Этилхинуклидин-2-карбоксилат может быть получен из (4-бромметил)тетрагидропирана при помощи вышеописанных процедур для этил 1-азабицикло[2.2.1]гептан-2-карбоксилата. Требуемый 4-(бромметил)тетрагидропиран может быть получен согласно процедурам, найденным у Burger, et al., J. Am. Chem. Soc. 72: 5512 (1950), Thomas, et al., J. Pharm. Pharmacol. 15: 167 (1963) и J. Am. Chem. Soc. 115: 8401 (1993). Затем этилхинуклидин-2-карбоксилат может быть депротонирован диизопропиламидом лития и использован в реакции с нитроэтиленом. Последующая обработка никелем Ренея даст непосредственно спиролактам, спиро[азабицикло[2.2.2]октан-2,3'-пирролидин]-2'-он, путем восстановления нитрогруппы с последующей самопроизвольной циклизацией. Затем такой лактам может быть восстановлен алюмогидридом лития для получения желательного спиро[1-азабицикло[2.2.2]октан-2,3'-пирролидин], который затем арилируют по азоту пирролидина.
Схема 11
Альтернативные способы синтеза
Соединения могут быть получены другими отличающимися способами. Могут быть использованы описанные альтернативные варианты протокола реакций взаимодействий, катализируемых палладием. Например, специалистам в области органического синтеза очевидно, что одно или несколько колец, содержащих азот, могут быть образованы любым из многочисленных обычных способов синтеза амина. Таким образом, ариламин может вступить в реакцию с защищенным циклическим производным амина (см. схему 12), который содержит два реактивных электрофила, образуя N-арилдиазаспиросоединение. В такой химии участвует множество электрофилов (например, галогены и сульфонаты путем нуклеофильной замены, альдегиды через восстановительное аминирование, эфиры и другие производные кислот через замещение ацила с последующим восстановлением).
Схема 12
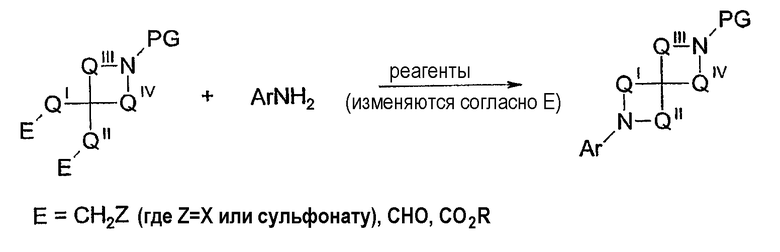
Требуемые бис-электрофилы могут быть синтезированы многочисленными различными способами. Схемы 2, 3 и 6, все, содержат такие промежуточные соединения (в реакции с бензиламином или аммонием). Pedersen et al., J. Org. Chem. 58: 6966 (1993) и Berkowits, et al., J. Org. Chem. 60: 1233 (1965) описывают алкилирование дианионов N-ацил α-аминоэфиров. Такие способы алкилирования также могут быть использованы для синтеза N-арилдиазаспиросоединений. Таким образом, дианион коммерчески доступного (Acros) этил 2-пирролидон-5-карбоксилата может быть алкилирован этилбромацетатом для образования этил 5-(карбоэтоксиметил)-2-пирролидон-5-карбоксилата. Второе спирокольцо может быть образовано при помощи реакции этил 5-(карбоэтоксиметил)-2-пирролидон-5-карбоксилата с ариламином. Полученный 2-арил-2,6-диазаспиро[4.4]нонан-1,3,7-трион может быть восстановлен дибораном, давая 7-арил-1,7-диазаспиро[4.4]нонан. Порядок этапов синтеза может меняться в зависимости от природы арильной группы. Аналогично, могут потребоваться дополнительные этапы введения защитной группы/удаления защитной группы.
Для использования в подходе, показанном на схеме 12, большое количество ариламинов является доступным. Кроме того, для аминопиридинов и аминопиримидинов коммерчески доступным (Aldrich) является 3-аминоизоксазол. Это обеспечивает средство для синтеза N-изоксазолилдиазаспиросоединений. Изомерный 4-аминоизоксазол может быть синтезирован восстановлением соответствующего нитросоединения способом, описанным у Reiter, J. Org. Chem. 52: 2714 (1987). Примеры других аминопроизводных 5-членных ароматических колец включают 3-аминоизотиазол, полученный согласно Holland, et al., J. Chem. Soc., 7277 (1965), и 4-аминоизотиазол, полученный согласно Avalos, et al., An. Quim 72: 922 (1976). Таким образом, может быть получено множество N-арилдиазаспиросоединений настоящего изобретения, в которых арильная группа представляет собой пятичленный гетероцикл.
III. Фармацевтические композиции
Соединения, раскрытые в настоящем описании, могут быть включены в состав фармацевтических композиций и использованы с целью прекращения курения, лечения лекарственной зависимости или лечения или предупреждения ожирения, связанного с прекращением приема лекарственных средств. Фармацевтические композиции, раскрытые в настоящем описании, включают одно или несколько соединений формул 1 или 2 и/или их фармацевтически приемлемые соли. Необязательно активные соединения могут быть использованы в виде рацемических смесей или в виде чистых энантиомеров.
Способ, которым вводят соединения, может меняться. Композиции предпочтительно вводят перорально (например, в жидком виде в растворителе, таком как вода или жидкость, отличная от воды, или в твердом носителе). Предпочтительные композиции для перорального введения включают пилюли, таблетки, капсулы, таблетки в виде капсул, сиропы и растворы, включая твердые желатиновые капсулы и капсулы с замедленным высвобождением. Композиции могут быть составлены в лекарственные формы в виде разовой дозы или в виде многочисленных или субъединичных доз. Предпочтительные композиции являются жидкими или полутвердыми. Могут быть использованы композиции, включающие жидкий фармацевтически инертный носитель, такой как вода или другие фармацевтически совместимые жидкости или полутвердые вещества. Использование таких жидкостей или полутвердых веществ хорошо известно специалистам в данной области техники.
Композиции также могут быть введены при помощи инъекции, т.е. внутривенно, внутримышечно, подкожно, интраперитонеально, интраартериально, интратекально и интрацеребровентрикулярно. Внутривенное введение представляет собой предпочтительный способ инъекции. Подходящие носители для инъекции хорошо известны специалистам в данной области техники и включают 5% раствор декстрозы, физиологический раствор и забуференный фосфатом физиологический раствор. Соединения также могут быть введены в виде инфузии или инъекции (например, в виде суспензии или в виде эмульсии в фармакологически приемлемой жидкости или смеси жидкостей).
Лекарственные формы также могут быть введены другими способами, например трансдермально (например, при помощи трансдермального пластыря, используя технологию, которая является коммерчески доступной от Novartis and Alza Corporation). Лекарственные формы, используемые для трансдермального введения, хорошо известны специалистам в данной области техники. Соединения также могут быть введены при помощи ингаляции (например, в виде аэрозолей либо назально, либо путем использования изделий для введения типа, приведенного в патенте США №4922901, Brooks et al., включенного в настоящее описание во всей своей полноте в качестве ссылки); локально (например, в виде примочек) или ректально. Хотя существует возможность для введения композиций в виде химического вещества в большом количестве, предпочтительным является предоставление каждого соединения в виде фармацевтически приемлемой композиции или лекарственной формы для действенного и эффективного введения.
Иллюстративные способы введения таких соединений очевидны специалистам в данной области техники. Применимость этих лекарственных форм может зависеть от конкретной используемой композиции и конкретного пациента, получающего лечение. Указанные лекарственные формы могут содержать жидкий носитель, который может быть масляным, водным, в виде эмульсии или может содержать некоторые растворители, подходящие для режима введения.
Композиции могут вводиться периодически или с постепенно увеличивающейся, непрерывной, постоянной или управляемой скоростью теплокровному животному (например, млекопитающему, такому как мышь, крыса, кошка, кролик, собака, свинья, корова или обезьяна), но преимущественно вводятся человеку. Кроме того, время суток и количество введений фармацевтической лекарственной формы могут меняться.
Предпочтительно, при введении активные ингредиенты взаимодействуют с рецепторными участками в теле пациента, которые управляют высвобождением допамина. Соединения могут представлять собой антагонисты как для α4β2 подтипа, так и для тех NNR подтипов, которые влияют на высвобождение допамина, при условии, что эффективная концентрация, необходимая для эффективного управляемого высвобождения допамина, представляет собой, по меньшей мере, величину меньшего порядка, чем ту, которая необходима для значимого влияния на α4β2 рецептор. В одном из вариантов осуществления соединения представляют собой неполные антагонисты, и неполный антагонизм дает возможность соединениям давать предпочтительный профиль побочных эффектов относительно полных антагонистов.
Способность этих соединений к неполному ингибированию высвобождения допамина особенно значима, поскольку она указывает, что соединения могут быть полезными для нарушения системы компенсации допамина, и таким образом для лечения нарушений, которые ею опосредованы. Такие нарушения включают наркотическую зависимость, применение табака и увеличение веса, которое происходит при прекращении приема лекарственных средств.
Таким образом, соединения, раскрытые в настоящем описании, представляют собой полезные альтернативные варианты для лечения зависимостей, связанных с неправильным применением лекарственных средств, включая спирт, амфетамины, барбитураты, бензодиазепины, кофеин, каннабиноиды, кокаин, галлюциногены, опиаты, фенциклидин и табак, и для лечения нарушений, связанных с питанием, таких как ожирение, которое возникает после прекращения приема лекарственных средств, несмотря на уменьшение побочных эффектов, связанных с применением психотропных стимуляторов (тревожность, бессонницу, привыкание и т.д.).
Соединения также преимущественно воздействуют на функционирование ЦНС, оптимизируя воздействие на такие релевантные подтипы рецепторов, которые влияют на высвобождение допамина, при этом минимизируется влияние на подтипы рецепторов мышечного типа.
Предпочтительно, композиции вводят таким образом, чтобы были задействованы или происходило взаимодействие активных ингредиентов с областями продуцирования допамина. Соединения, раскрытые в настоящем описании, даже в небольших концентрациях являются достаточно эффективными для воздействия на продуцирование и/или секрецию допамина, и достаточно действенными (например, они ингибируют продуцирование и/или секрецию допамина достаточно эффективно).
В некоторых обстоятельствах соединения, раскрытые в настоящем описании, могут использоваться в виде части фармацевтической композиции с другими соединениями, предназначенными для предупреждения или лечения лекарственной зависимости, никотиновой зависимости и/или ожирения. Кроме того, наряду с эффективными количествами соединений, раскрытых в настоящем описании, в фармацевтические композиции могут быть включены различные другие компоненты в качестве добавок или вспомогательных веществ. Иллюстративные фармацевтически приемлемые компоненты или вспомогательные вещества, которые используются в релевантных обстоятельствах, включают антидепрессанты, антиоксиданты, агенты, удаляющие свободные радикалы, пептиды, факторы роста, антибиотики, бактериостатические агенты, иммуносупрессанты, антикоагулянты, буферные агенты, противовоспалительные агенты, жаропонижающие вещества, связующие вещества с замедленным высвобождением, анестетики, стероиды, витамины, минералы и кортикостероиды. Такие соединения могут обеспечивать дополнительные преимущества, влияя на терапевтическое действие фармацевтических композиций или предотвращая любые потенциальные побочные эффекты, которые могут возникать в результате введения фармацевтической композиции.
Подходящая доза соединения представляет собой такое количество, которое является эффективным для предупреждения появления симптомов нарушения или для лечения некоторых симптомов нарушения, которым страдает пациент. “Эффективное количество” или “эффективная доза” означает количество, достаточное для получения желательных фармакологических или терапевтических эффектов, таким образом, приводя к эффективному предупреждению или лечению нарушения.
Эффективное количество соединения представляет собой количество, эффективное для преодоления гемоэнцефалического барьера пациента для связывания с релевантными рецепторными участками в головном мозге пациента и для активации релевантных подтипов никотиновых рецепторов (например, для антагонизирования или неполного антагонизирования продуцирования и/или секреции допамина, таким образом, приводя к эффективному предупреждению или лечению нарушения). Предупреждение нарушения проявляется в задержке проявления симптомов нарушения. Лечение нарушения проявляется в уменьшении симптомов, связанных с нарушением или уменьшением вероятности рецидивов симптомов нарушения. Предпочтительно, эффективное количество является достаточным для получения желательного результата, но недостаточно для того, чтобы вызывать заметные побочные эффекты.
Эффективная доза может меняться в зависимости от таких факторов, как состояние пациента, тяжесть симптомов или тяжесть нарушения и способ, которым вводится фармацевтическая композиция. Для человека эффективная доза обычных соединений в общем случае требует введения соединения в количестве, достаточном для уменьшения высвобождения допамина, но такое количество должно быть недостаточным для оказания любого влияния на скелетные мышцы и ганглии. Конечно, эффективная доза соединений будет меняться от пациента к пациенту, но обычно она содержит такое начальное количество, при котором происходит желательный терапевтический эффект (т.е. при котором продуцирование и/или секреция допамина существенно уменьшается), но ниже того количества, при котором наблюдаются мышечные эффекты.
Соединения при использовании эффективного количества согласно способу, раскрытому в настоящем описании, выбирают для некоторых релевантных никотиновых рецепторов, при этом они по существу не активируют рецепторы, связанные с нежелательными побочными эффектами, в концентрациях, по меньшей мере, больших, чем концентрации, требуемые для подавления высвобождения допамина или других нейромедиаторов. Это означает, что конкретная доза соединения, эффективная для предупреждения и/или лечения лекарственной зависимости, никотиновой зависимости и/или ожирения (главным образом, но без необходимости, ожирения, связанного с прекращением приема лекарственного средства или поступления в организм никотина), по существу является эффективной для активации некоторых никотиновых рецепторов ганглионарного типа в концентрации, превышающей в 5 раз, предпочтительно в 100 раз и более предпочтительно в 1000 раз концентрацию, необходимую для подавления продуцирования и/или высвобождения допамина. Такая селективность некоторых соединений, раскрытых в настоящем описании, в отношении таких рецепторов ганглионарного типа, ответственная за сердечно-сосудистые осложнения, проявляется в отсутствие способности у таких соединений активировать никотиновую функцию надпочечной хромаффинной ткани в концентрациях, превышающих концентрации, необходимые для подавления продуцирования и/или высвобождения допамина.
Для человека эффективная доза обычных соединений главным образом требует введения соединения в количестве, по меньшей мере, примерно 1, часто, по меньшей мере, 10, и наиболее часто, по меньшей мере, примерно 25 мкг/24 ч/пациент. Эффективная доза обычно представляет собой дозу, не превышающую примерно 500, часто не превышающую 400 и наиболее часто не превышающую 300 мкг/24 ч/пациент. Кроме того, введение эффективной дозы в плазму пациента выполняют при такой концентрации соединения, которая обычно не превышает 500 нг/мл и часто не превышает 100 нг/мл.
Соединения, раскрытые в настоящем описании, при использовании эффективного количества согласно способам, раскрытым в настоящем описании, в некоторой степени могут обеспечивать предупреждение развития нарушений ЦНС, уменьшение симптомов нарушений ЦНС и уменьшение до некоторой степени вероятности рецидивов нарушений ЦНС. Эффективное количество таких соединений обычно находится ниже пороговой концентрации, необходимой для появления любых видимых побочных эффектов, например таких эффектов, которые имеют отношение к скелетным мышцам. Соединения могут быть введены в терапевтическое окно, во время которого лечат некоторые нарушения ЦНС и во время которого не возникают некоторые побочные эффекты. В идеале, эффективная доза соединений, раскрытых в настоящем описании, является эффективной для получения желательного влияния на ЦНС, но является неэффективной (т.е. недостаточно высокого уровня) для получения нежелательных побочных эффектов. Предпочтительно, соединения вводят в дозированной форме, эффективной для лечения нарушений ЦНС, но в количестве, меньшем чем 1/5 и часто меньшем чем 1/10 количества, требуемого для появления побочных эффектов в любой значительной степени.
Наиболее предпочтительно эффективные дозы представляют собой достаточно низкие концентрации, при которых могут происходить максимально наблюдаемые эффекты с минимальными побочными эффектами. Концентрации, определенные как количество соединения на объем релевантной ткани, обычно обеспечивают показатель степени, при которой такое соединение действует на продуцирование цитокинов. Обычно эффективная доза таких соединений в основном требует введения соединения в количестве, меньшем чем 5 мг/кг веса тела пациента. Часто соединения настоящего изобретения вводят в количестве, меньшем чем примерно 1 мг/кг веса тела пациента и обычно меньшем чем примерно 100 мг/кг веса тела пациента, но часто от примерно 10 мг до менее чем 100 мг/кг веса тела пациента. Для соединений, которые не оказывают влияния на никотиновые рецепторы мышечного типа в низких концентрациях, эффективная доза составляет менее 5 мг/кг веса тела пациента; и часто такие соединения вводят в количестве от 50 мкг до менее чем 5 мг/кг веса тела пациента. Вышеуказанные эффективные дозы обычно представляют такое количество, которое вводят в виде разовой дозы или в виде одной или нескольких доз, вводимых в течение 24 часов.
Для человека эффективная доза обычных соединений главным образом требует введения соединения в количестве, по меньшей мере, примерно 1, часто, по меньшей мере, примерно 10, и наиболее часто, по меньшей мере, примерно 25 мг/24 ч/пациент. Для человека эффективные дозы обычных соединений требуют введения соединения в количестве, которое обычно не превышает примерно 500, часто не превышает примерно 400 и наиболее часто не превышает примерно 300 мкг/24 ч/пациент. Кроме того, соединения преимущественно вводят в эффективной дозе, такой, при которой концентрация соединения в плазме пациента обычно не превышает 500 пг/мл, часто не превышает 300 пг/мл и наиболее часто не превышает 100 пг/мл. При применении соединений таким образом соединения зависят от применяемой дозы, и по существу ингибируют продуцирование и/или секрецию цитокинов при использовании низких концентраций, но не проявляют таких ингибирующих эффектов при более высоких концентрациях. Соединения оказывают ингибиторные эффекты на продуцирование и/или секрецию допамина при использовании в количествах, не превышающих количества, которые необходимы для активации в любой значимой степени подтипов никотиновых рецепторов, связанных с побочными эффектами.
IV. Способы использования соединений
и/или фармацевтических композиций
Соединения могут быть использованы для лечения лекарственной зависимости, никотиновой зависимости и/или ожирения, такого как ожирение, связанное с прекращением приема лекарственных средств. Соединения также могут быть использованы в качестве вспомогательной терапии в комбинации с существующими методами терапии для управления вышеупомянутыми типами заболеваний и нарушений. В таких ситуациях предпочтительным является введение активных ингредиентов способом, который оптимизирует влияние на продуцирование и/или секрецию допамина, при этом минимизируя влияние на подтипы рецепторов, такие как подтипы рецепторов, которые связаны с мышцами и ганглиями. Это может происходить благодаря целевой доставке лекарственного средства и/или путем подбора дозированной формы, при которой можно получить желательные эффекты без превышения пороговой дозированной формы, при которой достигаются значимые побочные эффекты.
В большинстве случаев соединения могут связываться, антагонизируя или не полностью антагонизируя один или несколько никотиновых рецепторов головного мозга пациента, которые отличны от α4β2-рецепторов и которые модулируют высвобождение допамина, в концентрациях, при которых в значительной степени не затрагиваются α4β2-рецепторы. По существу такие соединения имеют способность проявлять никотиновую фармакологию и, в частности, действовать в качестве антагонистов допамина. Константы связывания рецепторов обычных соединений, полезных для осуществления настоящего изобретения, обычно превышают примерно 0,1 нМ, часто превышают примерно 1 нМ и наиболее часто превышают примерно 10 нМ. Константы связывания рецепторов таких обычных соединений в общем случае меньше примерно 1 мкМ, часто меньше примерно 100 нМ и наиболее часто меньше примерно 50 нМ. Константы связывания рецепторов предоставляют показатель способности соединения связываться с половиной релевантных рецепторных участков некоторых клеток головного мозга пациента. См. Cheng, et al., Biochem. Pharmacol. 22: 3099 (1973).
Соединения при их использовании в эффективных количествах выбирают для некоторых релевантных никотиновых рецепторов, но эти соединения в значительной степени не активируют рецепторы, связанные с нежелательными побочными эффектами. Это означает, что конкретная доза соединения, которая эффективно подавляет продуцирование и/или высвобождение допамина, по существу не эффективна для того, чтобы активировать некоторые никотиновые рецепторы ганглионарного типа. Такая селективность соединений настоящего изобретения по отношению к тем рецепторам, которые ответственны за сердечно-сосудистые осложнения, доказывается отсутствием способности у таких соединений активировать никотиновую функцию надпочечной хромаффинной ткани.
Соединения проявляют слабую способность вызывать ионный поток изотопного рубидия через никотиновые рецепторы в клеточных препаратах, экспрессирующих никотиновые ацетилхолиновые рецепторы мышечного типа. Таким образом, соединения показывают константы активации рецепторов или ЕС50 значения (т.е. которые предоставляют показатель концентрации соединения, необходимой для активации половины релевантных рецепторных участков скелетных мышц пациента), которые являются чрезвычайно высокими (т.е. выше примерно 100 мкМ). В общем случае, обычные предпочтительные соединения, полезные для осуществления настоящего изобретения, активируют ионный поток изотопного рубидия величиной, меньшей чем 10 процентов, часто меньшей чем 5 процентов от максимально обеспечиваемого S(-)никотином.
Следовательно, соединения являются эффективными для подавления продуцирования и/или высвобождения допамина и могут быть использованы для лечения лекарственной зависимости, никотиновой зависимости и/или ожирения в эффективных концентрациях, которые недостаточны для того, чтобы вызывать любые соответствующие побочные эффекты, как показано при помощи уменьшенного воздействия на препаратах, исходя из предположения, что они отражают влияние на сердечно-сосудистую систему или влияние на скелетные мышцы. По существу, введение соединения обеспечивает терапевтическое окно, во время которого лечение лекарственной зависимости, никотиновой зависимости и/или ожирения является эффективным и устранены побочные эффекты. То есть эффективная доза соединения настоящего изобретения является достаточной для обеспечения желательного антагонистического влияния на продуцирование и/или секрецию допамина, но не является достаточной (т.е. не на достаточно высоком уровне) для обеспечения нежелательных побочных эффектов. Предпочтительно, соединения приводят к лечению лекарственной зависимости, никотиновой зависимости и/или ожирения при введении менее 1/3, часто менее 1/5 и чаще всего менее 1/10 такого эффективного количества, при котором в значительной степени возникают побочные эффекты.
Следующие примеры предоставлены с целью иллюстрации настоящего изобретения, и их не следует рассматривать в качестве ограничения. В этих примерах все части и проценты даны по весу, кроме обозначенных иным образом. Выходы реакций даны в молярных процентах. В нижеследующих примерах используются некоторые коммерчески доступные исходные вещества. 3-бромпиридин, 3,5-дибромпиридин, 5-бромникотиновая кислота, 5-бромпиримидин и 4-пентен-2-ол были приобретены у Aldrich Chemical Company или Lancaster Synthesis Inc. 2-амино-5-бром-3-метилпиридин был приобретен у Maybridge Chemical Company Ltd. (R)-(+)-пропиленоксид был получен от Fluka Chemical Company и (S)-(-)-пропиленоксид был получен от Aldrich Chemical Company. Колоночную хроматографию проводили с использованием либо Merck силикагеля 60 (70-230 меш), либо оксида алюминия (активированный, нейтральный, Brockmann I, стандартный размер, примерно 150 меш). Реакции под высоким давлением проводили в толстостенных стеклянных пробирках для высокого давления (емкостью 185 мл) с Ace-Thread, и поршневым клапаном, доступным от Ace Glass Inc. Реакционные смеси обычно нагревали, используя баню с высокотемпературным силиконовым маслом, а температура дана относительно температуры масляной бани. В нижеприведенных примерах используется следующая аббревиатура: CHCl3 для хлороформа, CH2Cl2 для дихлорметана, CH3OH для метанола, DMF для N,N-диметилформамида, EtOAc для этилацетата, THF для тетрагидрофурана, Et3N для триэтиламина.
V. Анализ
Анализ связывания
Способность соединения связываться с релевантными рецепторными участками определяли согласно технологиям, описанным в патенте США №5597919, Dull et al. Константы ингибирования (значения Ki) вычисляли, исходя из значений IC50, используя способ Cheng et al., Biochem. Pharmacol. 22: 3099 (1973). Для подтипа α4β2 значение Ki для каждого из примеров настоящего описания было меньше, чем 1 мкМ, что указывало на сильное связывание этих соединений настоящего изобретения с рецептором.
Определение значения log P
Значения log P, которые использовались для анализа относительной способности соединений преодолевать гематоэнцефалический барьер (Hansch, et al., J. Med. Chem. 11: 1 (1968)), вычисляли при помощи пакета программ Cerius2, версии 3,5 от Molecular Stimulations, Inc.
Определение высвобождения допамина
Высвобождение допамина измеряли при помощи технологий, описанных в патенте США №5597919, Dull et al. Высвобождение выражали в виде процентов от высвобождения, полученного при концентрации (S)-(-)-никотина, оказывающей максимальное воздействие. Опубликованные ЕС50 значения выражены в нМ, и значения Emax представляют количество, которое высвобождается относительно
(S)-(-)-никотина, в процентном отношении.
Антагонизм в отношении высвобождения допамина также может быть оценен при помощи анализа, описанного у Gradyet al., “Characterization of nicotinic receptor mediated [3H]dopamine release from synaptosomes prepared from mouse striatum”, J. Neurochem. 59: 848-856 (1992) и Soliakov and Wonnacott, “Voltage-sensitive Ca2+ channels involved in nicotinic receptor-mediated [3H]dopamine release from rat striatal synaptosomes”, J. Neurochem. 67: 163-170 (1996).
Определение высвобождения ионов рубидия
Высвобождение рубидия измеряли, используя технологии, описанные у Bencherif et al., JPET 279: 1413-1421 (1996). Опубликованные значения ЕС50 выражены в нМ, а значения Emax представляют количество ионов рубидия, которое высвобождается, относительно 300 мкМ иона тетраметиламмония, в процентном отношении.
Определение взаимодействия с мышечными рецепторами
Определение взаимодействия соединений с мышечными рецепторами осуществляли согласно технологиям, описанным в патенте США №5597919, Dull et al. Максимальную активацию отдельных соединений (Emax) определяли в виде процента от максимальной активации, индуцируемой (S)-(-)-никотином. Опубликованные значения Emax представляют количество, которое высвобождается относительно
(S)-(-)-никотина, в процентном отношении.
Определение взаимодействия с ганглионарными рецепторами
Определение взаимодействия соединений с ганглионарными рецепторами осуществляли согласно технологиям, описанным в патенте США №5597919, Dull et al. Максимальную активацию отдельных соединений (Emax) определяли в виде процента от максимальной активации, индуцируемой (S)-(-)-никотином. Опубликованные значения Emax представляют количество, которое высвобождается относительно
(S)-(-)-никотина, в процентном отношении.
Селективность
Селективность соединений для данного рецептора можно оценить путем сравнения связывания соединений с различными рецепторами, используя известные технологии.
VI. Примеры синтеза
Нижеследующие примеры синтеза приведены с целью иллюстрации настоящего изобретения и не должны рассматриваться в качестве ограничения его объема. В этих примерах все части и проценты указаны по весу, кроме обозначенных иным способом. Выходы реакций даны в процентах.
Пример 1
Образец 1 представляет собой дигидрохлорид 7-(3-пиридил)-1,7-диазаспиро[4.4]нонана, который получен согласно следующим способам.
Нитроэтилен
Нитроэтилен получали согласно процедуре, описанной у Ranganathan, et al., J. Org. Chem. 45: 1185 (1980).
Этил 2-(2-нитроэтил)-1-бензилпирролидин-2-карбоксилат
В атмосфере азота раствор диизопропиламина (4,34 г, 6,01 мл, 42,9 ммол) в сухом THF (50 мл) охлаждали на ледяной бане, в это время при помощи шприца добавляли н-бутиллитий (17,1 мл 2,5 M в гексане, 42,8 ммол). Ледяную баню убирали и раствор диизопропиламида лития сначала нагревали до температуры окружающей среды, а затем переносили при помощи канюли в перемешиваемый раствор этил (S)-N-бензилпирролидин-2-карбоксилата (10,0 г, 42,9 ммол) (Fluka) в сухом THF (50 мл), находящийся при температуре -78°C в атмосфере азота. Процесс добавления занимал 10 мин. После перемешивания в течение 30 мин при -78°C раствор енолятов обрабатывали (при помощи канюли) раствором нитроэтилена (3,13 г, 42,9 ммол) в сухом THF (20 мл). Затем смесь перемешивали в течение 1 ч при температуре -78°C. После этого добавляли насыщенный водный раствор хлорида аммония (при -78°C) и смесь нагревали до температуры окружающей среды и экстрагировали этилацетатом (4×30 мл). Экстракты сушили (K2CO3) и концентрировали ротационным испарением. Остаток очищали хроматографией на Merck 60 (70-230 меш) силикагелевой колонке смесью 9:1 (об./об.) гексан/этилацетат. Концентрации выделенных фракций дали 10,0 г (76,3%) вязкого масла желто-коричневого цвета.
6-Бензил-2,6-диазаспиро[4.4]нонан-1-он
Добавляли никель Ренея (~2 г) в раствор этил 2-(2-нитроэтил)-1-бензилпирролидин-2-карбоксилата (6,00 г, 19,6 ммол) в абсолютном этаноле (200 мл) в бутыль для гидрирования. Смесь встряхивали в течение 12 ч в атмосфере водорода (50 psi) в Parr устройстве для гидрирования, фильтровали через слой целита и концентрировали роторным испарением. ХМС (хромато-масс-спектрометрический, GCMS) анализ указывал на то, что продукт гидрирования представлял собой смесь первичного амина и лактама, который получается в результате циклизации амина в эфир. Смесь растворяли в толуоле (150 мл). Добавляли каталитическое количество п-толуолсульфоновой кислоты (~30 мг) и смесь нагревали противотоком в атмосфере азота в течение 24 ч. При испарении толуола остаток (в данном случае исключительно лактам, при помощи ХМС) кристаллизовали до получения 4,20 г (93,1%) твердого вещества желто-коричневого цвета (т.пл. 152-153°C).
1-Бензил-1,7-диазаспиро[4.4]нонан
Алюмогидрид лития (1,98 г, 52,2 ммол) добавляли по частям в атмосфере аргона к охлажденному на ледяной бане раствору 6-бензил-2,6-диазаспиро[4.4]нонан-1-она (4,00 г, 17,4 ммол) в сухом THF (100 мл). Дополнительную воронку заменяли обратным холодильником и смесь нагревали при кипячении с обратным холодильником в течение 24 ч. Смесь охлаждали до 0°C и обрабатывали по каплям (осторожно: экзотермическая реакция) 10 M водным раствором гидроксида натрия до тех пор, пока не перестал образовываться водород, и не появились гранулированные соли алюмината. Смесь перемешивали в течение 1 ч при 0°C и фильтровали через слой целита. Фильтрат сушили (K2CO3) и концентрировали, получая 3,60 г (95,7%) вязкой бесцветной жидкости.
1-Бензил-7-(3-пиридил)-1,7-диазаспиро[4.4]нонан
Смесь 1-бензил-1,7-диазаспиро[4.4]нонана (2,00 г, 9,26 ммол), 3-бромпиридина (1,38 г, 8,73 ммол), трет-бутоксида калия (2,50 г, 22,3 ммол), трис(дибензилиденацетон)-дипалладия (0) (0,318 г, 0,347 ммол), 2,2'-бис(дифенилфосфино)-1,1'-бинафтила (0,324 г, 0,520 ммол) и сухого толуола (50 мл) помещали в пробирку для высокого давления в атмосфере аргона. Смесь перемешивали и нагревали при 90°C (температура бани) в течение 24 ч и охлаждали. Добавляли воду (20 мл) и смесь экстрагировали этилацетатом (6×25 мл). Экстракты сушили (K2CO3) и концентрировали. Колоночная хроматография остатка на Merck силикагеле 60 (70-230 меш) смесью 6:4 (об./об.) хлороформ/ацетон дала 1,80 г (66,2%) светло-коричневого масла после концентрации выделенных фракций.
7-(3-Пиридил)-1,7-диазаспиро[4.4]нонан дигидрохлорид
Водный раствор соляной кислоты (0,5 мл 12 M) и 10% палладия-на-угле (0,100 г) добавляли в раствор 1-бензил-7-(3-пиридил)-1,7-диазаспиро[4.4]нонана (1,0 г, 3,41 ммол) в метаноле (30 мл). Смесь встряхивали в атмосфере водорода (50 psi) в Parr устройстве для гидрирования в течение 24 ч и фильтровали через слой целита. Фильтрат концентрировали ротационным испарением и выполняли колоночную хроматографию на Merck силикагеле 60 (70-230 меш). Элюция смесью 0,01:1:9 (об./об.) водный раствор аммония/метанол/хлороформ и концентрация отобранных фракций дали 0,650 г (93,8%) вязкого коричневого масла. Часть (300 мг, 1,48 мМ) этого вещества обрабатывали водным раствором соляной кислоты (2 мл). Воду азеотропно удаляли повторной обработкой небольшими объемами этанола (~5 мл) и ротационным испарением. Полученное твердое вещество повторно кристаллизовали из горячего изопропанола для получения 360 мг (88,2%) мелких кристаллов желто-коричневого цвета.
Пример 2
Образец 2 представляет собой дигидрохлорид 1-(3-пиридил)-1,7-диазаспиро[4.4]нонана, который получали согласно нижеописанным способам.
трет-Бутил 6-бензил-2,6-диазаспиро[4.4]нонан-2-карбоксилат
Ди-трет-бутилдикарбонат (1,45 г, 6,64 ммол) добавляли в раствор 1-бензил-1,7-диазаспиро[4.4]нонана (1,30 г, 6,01 ммол) и триэтиламина (1 мл) в дихлорметане (25 мл) и смесь перемешивали в течение ночи при комнатной температуре. Смесь выливали в перемешиваемый водный раствор бикарбоната натрия (10 мл) и экстрагировали хлороформом (4×25 мл). Экстракты сушили (K2CO3) и концентрировали ротационным испарением. Остаток элюировали колоночной хроматографией на Merck силикагеле 60 (70-230 меш) с получением 1,85 г (97,4%) вязкого бесцветного масла после концентрации выделенных фракций.
трет-Бутил 2,6-диазаспиро[4.4]нонан-2-карбоксилат
Раствор трет-бутил 6-бензил-2,6-диазаспиро[4.4]нонан-2-карбоксилата (1,70 г, 5,37 ммол) в метаноле (30 мл) смешивали с 10% палладия-на-угле (50 мг). Смесь встряхивали в атмосфере водорода (50 psi) в Parr устройстве для гидрирования в течение 8 ч и фильтровали через слой целита. Фильтрат концентрировали ротационным испарением и выполняли обработку в высоком вакууме, получая 1,26 г вязкого светло-коричневого масла (>100%), которое имело достаточную чистоту для использования в следующей реакции.
трет-Бутил 6-(3-пиридил)-2,6-диазаспиро[4.4]нонан-2-карбоксилат
Смесь трет-бутил 2,6-диазаспиро[4.4]нонан-2-карбоксилата (1,00 г, ~4,4 ммол), 3-бромпиридина (0,736 г, 4,66 ммол), трет-бутоксида калия (1,22 г, 10,9 ммол), трис(дибензилиденацетон)дипалладия (0) (0,155 г, 0,169 ммол), 2,2'-бис(дифенилфосфино)-1,1'-бинафтила (0,158 г, 0,254 ммол) и сухого толуола (25 мл) помещали в пробирку для высокого давления в атмосфере аргона. Смесь перемешивали и нагревали при 180°C (температура бани) в течение 8 ч и охлаждали. Тонкослойный анализ указывал, что имело место незначительное превращение. Вторую загрузку, количественно равную первой, всех реагентов, кроме трет-бутил 2,6-диазаспиро[4.4]нонан-2-карбоксилата, добавляли в пробирку для высокого давления и пробирку возвращали в баню на следующие 8 ч. Оказалось, что снова произошла слабая реакция, поэтому добавляли третью загрузку реагентов и продолжали нагревать (при 180°C) в течение еще 8 ч. Добавляли воду (20 мл) и смесь экстрагировали этилацетатом (6×25 мл). Экстракты сушили (K2CO3) и концентрировали. Колоночная хроматография остатка на Merck силикагеле 60 (70-230 меш) смесью 6:4 (об./об.) хлороформ/ацетон дала 150 мг (~11%) светло-коричневого масла после концентрации выделенных фракций.
Дигидрохлорид 1-(3-пиридил)-1,7-диазаспиро[4.4]нонана
Раствор трет-бутил 6-(3-пиридил)-2,6-диазаспиро[4.4]нонан-2-карбоксилата (100 мг, 0,330 ммол) в дихлорметане (5 мл) быстро перемешивали с 1 мл 12M соляной кислоты при комнатной температуре в течение 1 ч, в течение этого времени двухфазная смесь становилась однофазной. Дихлорметан испаряли и остаток растворяли в воде (3 мл) и делали его сильно основным (pH 9), используя карбонат калия. Смесь перемешивали с хлоридом натрия и экстрагировали хлороформом (4×10 мл). Экстракты сушили (K2CO3) и концентрировали сначала ротационным испарением, а затем обработкой в высоком вакууме. Вязкое коричневое масло, которое получилось, имело 98% чистоту благодаря ХСМ и весило 50 мг (73%). Образец указанного свободного основания (40 мг, 0,20 ммол) растворяли в 10 каплях 12 M соляной кислоты. Воду азеотропно удаляли повторной обработкой небольшими объемами этанола (~5 мл) и ротационным испарением. Полученное твердое вещество повторно кристаллизовали из горячего изопропанола для получения 40 мг (72%) мелких кристаллов желто-коричневого цвета (т.пл. 170-175°C).
Пример 3
Образец 3 представляет собой 1-метил-7-(3-пиридил)-1,7-диазаспиро[4.4]нонан, который получали согласно следующим способам.
1-Метил-7-(3-пиридил)-1,7-диазаспиро[4.4]нонан
7-(3-пиридил)-1,7-диазаспиро[4.4]нонан (30 мг, 0,15 ммол) растворяли в 98% муравьиной кислоте (0,5 мл) и формальдегиде (1 мл, 28% водный раствор). Реакционную смесь нагревали при кипячении с обратным холодильником в течение 8 ч. Реакционную смесь охлаждали до комнатной температуры, доводили pH до 9-10 насыщенным водным раствором бикарбоната натрия и экстрагировали хлороформом (4×3 мл). Объединенные экстракты хлороформа сушили (K2CO3), фильтровали и концентрировали ротационным испарением до получения 30 мг желаемого соединения (93,6%) в виде светло-коричневой жидкости.
Пример 4
Образец 4 представляет собой 1-метил-7-(5-этокси-3-пиридил)-1,7-диазаспиро[4.4]нонан, который получали согласно следующим способам.
5-Бром-3-этоксипиридин
В атмосфере азота добавляли натрий (4,60 г, 200 ммол) в абсолютный этанол (100 мл) при 0-5°C и перемешивали смесь при комнатной температуре в течение 18 ч. В полученный раствор добавляли 3,5-дибромпиридина (31,5 г, 133 ммол), затем DMF (100 мл). Смесь нагревали при 70°C в течение 48 ч. Коричневую смесь охлаждали, выливали в воду (600 мл) и экстрагировали эфиром (3×500 мл). Объединенные экстракты сушили (Na2SO4), фильтровали и концентрировали ротационным испарением. Очистка дистилляцией под вакуумом дала 22,85 г (85,0%) масла, точка кипения 89-90°C при 2,8 мм Hg (точка кипения согласно литературным источникам - 111°C при 5 мм Hg, см. K.Clarke, et al., J. Chem. Soc. 1885 (1960)).
1-Бензил-7-(5-этокси-3-пиридил)-1,7-диазаспиро[4.4]нонан
1-Бензил-1,7-диазаспиро[4.4]нонан (500,0 мг, 2,4 ммол) растворяли в сухом толуоле (15 мл) в 50-мл круглодонной колбе, снабженной магнитной мешалкой. Через раствор пропускали азот потоком пузырей с низкой скоростью. В перемешиваемый раствор добавляли 3-бром-5-этоксипиридин (513,8 мг, 2,55 ммол), трет-бутоксид калия (1039,0 мг, 9,26 ммол), rac-2,2'-бис(дифенилфосфино)-1,1'-бинафтил (86,4 мг, 0,14 ммол) и трис(дибензилиденацетон)дипалладий (0) (63,6 мг, 0,06 ммол), продолжая очищать азотом. Поток азота прекратили, бутыль закупорили и нагревали при 90°C в течение 8 ч. Реакционную смесь охлаждали и растворитель удаляли ротационным испарением. Полученный остаток суспендировали в насыщенном водном растворе бикарбоната натрия (10 мл) и экстрагировали хлороформом (4×25 мл). Объединенные органические экстракты сушили (Na2SO4), фильтровали и концентрировали ротационным испарением до густой темной массы. Очистка колоночной хроматографией с использованием смеси метанол/хлороформ (2:98, об./об.) в качестве элюента дала 0,54 г желаемого соединения в виде светло-коричневой вязкой жидкости (69%).
7-(5-Этокси-3-пиридил)-1,7-диазаспиро[4.4]нонан
В раствор 1-бензил-7-(5-этокси-3-пиридил)-1,7-диазаспиро[4.4]нонана (540 мг, 1,6 ммол) в этаноле (25 мл) в бутыль для высокого давления добавляли концентрированную HCl (1 мл) и Pearlman катализатор (Pd(OH)2, 20% на углероде, 50 мг). Раствор встряхивали под давлением 50 psi в атмосфере водорода в течение 8 ч. Катализатор удаляли при помощи целита и фильтровальную лепешку промывали этанолом (20 мл). Растворитель удаляли ротационным испарением и pH остатка доводили до 8-9 насыщенным водным раствором бикарбоната натрия. Добавляли твердый хлорид натрия (2 г) и смесь экстрагировали хлороформом (4×20 мл). Объединенные экстракты хлороформа сушили (Na2SO4), фильтровали и концентрировали ротационным испарением для получения 360,7 мг желаемого соединения в виде светло-коричневой вязкой жидкости (91,1%).
1-Метил-7-(5-этокси-3-пиридил)-1,7-диазаспиро[4.4]нонан
В перемешиваемый раствор 7-(5-этокси-3-пиридил)-1,7-диазаспиро[4.4]нонана (360,4 мг, 1,4 ммол) в 37% водном растворе формальдегида (4 мл) добавляли 98% муравьиную кислоту (2 мл) в атмосфере азота. Реакционную смесь нагревали при кипячении с обратным холодильником в течение 8 ч. Реакционную смесь охлаждали до комнатной температуры, затем доводили pH до 8-9 насыщенным водным раствором бикарбоната натрия и экстрагировали хлороформом (4×15 мл). Объединенные экстракты хлороформа сушили (Na2SO4), фильтровали и концентрировали ротационным испарением до получения вязкой коричневой жидкости. Ее дистиллировали, используя аппарат Кугельрора (Kugelrohr) (2 мм, 180°C) до получения очень светлого сиропа кремового цвета (340 мг, 89,3%).
Пример 5
Образец 5 представляет собой 1-метил-7-(5-фенокси-3-пиридил)-1,7-диазаспиро[4.4]нонан, который получали согласно нижеприведенным способам.
3-Бром-5-феноксипиридин
Гидрид натрия (1,35 г 80% в минеральном масле, 45,0 ммол) добавляли в насыщенный раствор фенола (4,26 г, 45,3 ммол) в DMF (30 мл) при 0°C в атмосфере азота. Смесь перемешивали при комнатной температуре в течение 3 ч, обрабатывали 3,5-дибромпиридином (4,0 г, 16,9 ммол) и нагревали при 100°C в течение 48 ч. Реакционную смесь охлаждали до комнатной температуры, выливали в смесь воды (100 мл) и 5M гидроксида натрия (10 мл) и экстрагировали эфиром (3×60 мл). Объединенные экстракты эфира сушили (Na2SO4), фильтровали и концентрировали ротационным испарением до светло-желтого полутвердого вещества (4,9 г). Очистку выполняли колоночной хроматографией на силикагеле (200 г) смесью гексан/этилацетат/хлороформ (8:1:1, об./об.) в качестве элюента до получения 2,86 г (выход 68%) бесцветного масла.
1-Бензил-7-(5-фенокси-3-пиридил)-1,7-диазаспиро[4.4]нонан
1-Бензил-1,7-диазаспиро[4.4]нонан (500,0 мг, 2,4 ммол) растворяли в сухом толуоле (15 мл) в 50-мл круглодонной колбе, оборудованной магнитной мешалкой. Через раствор пропускали азот потоком пузырей с низкой скоростью. В перемешиваемый раствор добавляли 3-бром-5-феноксипиридин (636,8 мг, 2,55 ммол), трет-бутоксид калия (1039,0 мг, 9,26 ммол), rac-2,2'-бис(дифенилфосфино)-1,1'-бинафтил (86,4 мг, 0,14 ммол) и трис(дибензилиденацетон)дипалладий (0) (63,6 мг, 0,06 ммол), продолжая очищать азотом. Поток азота прекращали, и колбу плотно закрывали и нагревали при 90°C в течение 8 ч. Реакционную смесь охлаждали и растворитель удаляли ротационным испарением. Полученный остаток суспендировали в насыщенном водном растворе бикарбоната натрия (10 мл) и экстрагировали хлороформом (4×25 мл). Объединенные органические экстракты сушили (Na2SO4), фильтровали, концентрировали ротационным испарением до густой темной массы. Ее чистили колоночной хроматографией, используя в качестве элюента смесь метанол/хлороформ (2:98, об./об.), до получения 0,70 г желаемого соединения в виде светло-коричневой вязкой жидкости (78,6%).
7-(5-Фенокси-3-пиридил)-1,7-диазаспиро[4.4]нонан
В раствор 1-бензил-7-(5-фенокси-3-пиридил)-1,7-диазаспиро[4.4]нонана (690 мг, 1,79 ммол) в этаноле (25 мл) в сосуде для высокого давления добавляли концентрированную HCl (1 мл) и Pearlman катализатор (Pd(OH)2, 20%, на углероде, 50 мг). Раствор встряхивали под давлением 50 psi в атмосфере водорода в течение 8 ч. Катализатор удаляли фильтрованием через слой целита и фильтровальную лепешку промывали этанолом (20 мл). Растворитель удаляли ротационным испарением и pH остатка доводили до 8-9 насыщенным водным раствором бикарбоната натрия. Добавляли твердый хлорид натрия (2 г) и раствор экстрагировали хлороформом (4×20 мл). Объединенные экстракты хлороформа сушили (Na2SO4), фильтровали и концентрировали ротационным испарением до получения 490 мг желаемого соединения в виде светло-коричневой вязкой жидкости (92,7%).
1-Метил-7-(5-фенокси-3-пиридил)-1,7-диазаспиро[4.4]нонан
В перемешиваемый раствор 7-(5-фенокси-3-пиридил)-1,7-диазаспиро[4.4]нонана (420 мг, 1,42 ммол) в 37% водном растворе формальдегида (5 мл) добавляли 98% муравьиную кислоту (3 мл) в атмосфере азота. Реакционную смесь нагревали при кипячении с обратным холодильником в течение 8 ч. Реакционную смесь охлаждали до комнатной температуры, затем доводили pH до 8-9 насыщенным водным раствором бикарбоната натрия и экстрагировали хлороформом (4×15 мл). Объединенные экстракты хлороформа сушили (Na2SO4), фильтровали и концентрировали ротационным испарением до получения вязкой жидкости насыщенного коричневого цвета. Ее дистиллировали при помощи аппарата Кугельрора (2 мм, 180°C) до получения сиропа бледно-кремового цвета (400 мг, 90,9%).
Дихлорид 1-метил-7-(5-фенокси-3-пиридил)-1,7-диазаспиро[4.4]нонана
1-метил-7-(5-фенокси-3-пиридил)-1,7-диазаспиро[4.4]нонан (200 мг, 0,65 ммол) добавляли в концентрированную HCl (1 мл) и обрабатывали ультразвуком в течение 5 мин. Избыток кислоты и воды удаляли повторным азеотропным испарением небольшими частями этанола. Получали бледно-желтое твердое вещество. Твердое вещество растворяли в минимальном количестве абсолютного этанола (~1 мл) и затем по каплям добавляли эфир до тех пор, пока раствор не становился мутным. Охлаждение в холодильнике в течение ночи дало кристаллы кремового цвета, которые фильтровали, промывали эфиром и сушили в вакуумной печи до получения 210 мг (85,4%) чистой соли дигидрохлорида, т.пл. 180-191°C.
Пример 6
Образец 6 представляет собой 1'-(3-пиридил)спиро[1-азабицикло[2.2.1]гептан-2,3'-пирролидин]дигидрохлорид, который получали согласно нижеприведенным способам.
(3-Оксоланил)метила метансульфонат
В перемешиваемый раствор (3-оксоланил)метан-1-ола (25 г, 245 ммол) и триэтиламина (34,37 мл, 245 ммол) в сухом дихорметане (250 мл) при 0°C в атмосфере N2 добавляли по каплям метансульфонилхлорид (18,94 мл, 245 ммол). Реакционную смесь перемешивали всю ночь после того, как нагрели до комнатной температуры, затем добавляли насыщенный раствор NaHCO3 (100 мл) и смесь перемешивали в течение следующих 30 мин. Двухфазную смесь разделяли и удаляли органический слой. Водный слой экстрагировали дихлорметаном (3×25 мл) и объединенные экстракты дихлорметана сушили (Na2SO4), фильтровали и концентрировали до получения 42,16 г (3-оксоланил)метилметансульфоната (99%) в виде светло-коричневой жидкости.
3-(Бромметил)оксолан
В перемешиваемый раствор (3-оксоланил)метилметансульфоната (42,16 г, 239,5 ммол) в сухом ацетоне (600 мл) добавляли бромид лития (101,7 г, 1198 ммол). Реакционную смесь нагревали при кипячении с обратным холодильником в течение 3 ч, затем охлаждали и растворитель удаляли ротационным испарением. Остаток растворяли в воде (200 мл) и экстрагировали дихлорметаном (2×100 мл). Объединенные экстракты сушили (Na2SO4), фильтровали и концентрировали ротационным испарением до получения светло-коричневой жидкости. Ее дистиллировали при 70°C и давлении 1 мм для получения 33,00 г (86,77%) 3-(бромметил)оксолана в виде бесцветной жидкости.
Метил 3-аза-4,4-дифенил-бут-3-еноат
В перемешиваемый раствор глицин метилового эфира гидрохлорида (17,49 г, 139 ммол) в сухом дихлорметане (150 мл) в атмосфере N2 при комнатной температуре добавляли дифениламин (25,00 г, 137 ммол) одной частью. Реакционную смесь перемешивали в течение 24 ч, в течение которого осаждался хлорид аммония. Добавляли воду (20 мл) и слои разделяли. Органический слой промывали насыщенным раствором Na2CO3 (2×20 мл) и физиологическим раствором (20 мл). Органический слой сушили (Na2SO4), фильтровали и концентрировали ротационным испарением до получения ~35 г густого светло-коричневого сиропа (99% чистоты) с выходом ~100%. Он был использован в следующей реакции без дополнительной очистки.
Метил 3-(3-оксоланил)-2-аминопропаноат
В перемешиваемый раствор метил 3-аза-4,4-дифенилбут-3-еноата (23,00 г, 90 ммол) в атмосфере N2 в сухом DMF (25 мл) и толуоле (25 мл) добавляли трет-бутоксид калия (10,20 г, 90 ммол) одной порцией. Реакционную смесь перемешивали в течение 15 мин; она изменила цвет с желтого на темный красновато-коричневый. Затем при помощи канюли добавляли раствор 3-(бромметил)оксолана (15 г, 90 мМ) в DMF (20 мл) и сухом толуоле (20 мл) в течение 30 мин. Реакционную смесь перемешивали в течение следующих 16 ч при температуре окружающей среды. Затем в реакционную добавляли 1 н. HCl (100 мл), которую перемешивали в течение следующих 30 мин. Смесь экстрагировали этилацетатом (3×50 мл). Доводили рН водного слоя до 8-9 при помощи K2CO3, затем раствор переводили в насыщенное состояние, используя твердый NaCl, и экстрагировали этилацетатом (4×50 мл). Объединенные слои этилацетата сушили (K2CO3), фильтровали и концентрировали до получения метил 3-(3-оксоланил)-2-аминопропаноата (10 г, 59,37%) в виде коричневой жидкости.
Этил 1-азабицикло[2.2.1]гептан-2-карбоксилат
Метил 3-(3-оксоланил)-2-аминопропаноат (6,00 г, 3,46 ммол) помещали в герметичную пробирку для высокого давления, затем добавляли 48% водный HBr (20 мл) и раствор насыщали газообразным HBr. Пробирку тщательно герметизировали и нагревали при 110-120°C в течение 8 ч. Затем реакционную смесь охлаждали и содержимое переносили в 250-мл круглодонную колбу, содержащую 20 мл воды. Избыток кислоты удаляли ротационным испарением для получения полутвердой коричневой массы. Затем добавляли 30% водный раствор гидрохлорида аммония (150 мл) при 0°C и смесь осторожно нагревали при кипячении с обратным холодильником в течение 4 ч. Растворитель удаляли ротационным испарением для получения твердого вещества коричневого цвета, которое затем растворяли в абсолютном этаноле (50 мл). Добавляли концентрированную H2SO4 (10 мл) и раствор кипятили с обратным холодильником в течение 8 ч. Содержимое охлаждали на ледяной бане и затем доводили pH до 8-9 концентрированным раствором NaHCO3 и экстрагировали хлороформом (4×40 мл). Объединенные экстракты хлороформа сушили (K2CO3), фильтровали и концентрировали для получения коричнево-черной жидкости, которую дистиллировали, используя аппарат Кугельрора (1 мм, 140°C) для получения бесцветной жидкости (4 г, 68,25%) в виде смеси экзо- и эндоизомеров этил 1-азабицикло[2.2.1]гептан-2-карбоксилата.
Этил 1-аза-2-(нитроэтил)бицикло[2.2.1]гептан-2-карбоксилат
Диизопропиламид лития (LDA) получали при 0°C из диизопропиламина (2,078 г, 20,53 ммол) и н-бутиллития (8,21 мл, 20,53 ммол) в сухом THF (20 мл) в атмосфере N2. В перемешиваемый раствор экзо- и эндоизомеров этил 1-азабицикло[2.2.1]гептан-2-карбоксилата (2,67 г, 15,79 ммол) в сухом THF (35 мл) при -78°C в атмосфере N2 при помощи канюли добавляли раствор LDA в течение 15 мин. Реакционную смесь перемешивали в течение следующих 40 минут. Затем в течение 15 мин в реакционную смесь при помощи канюли по каплям добавляли раствор нитроэтилена (1,45 г, 20,53 мМ) в сухом THF (20 мл). После перемешивания в течение 2 ч при -78°C реакцию гасили добавлением насыщенного раствора хлорида аммония (20 мл). Реакционную смесь экстрагировали этилацетатом (5×25 мл), сушили (Na2SO4), фильтровали и концентрировали ротационным испарением до получения 3,82 г желательного продукта (86% чистоты) в виде светло-коричневой жидкости, которую использовали в следующей реакции без дополнительной очистки.
2'Η-Спиро[азабицикло[2.2.1]гептан-2,3'-пирролидин]-2'-он
Этил 1-аза-2-(нитроэтил)бицикло[2.2.1]гептан-2-карбоксилат (3,82 г, 86% чистоты, 15,78 ммол) растворяли в этаноле (50 мл) в сосуде для гидрогенолиза. Добавляли каталитическое количество никеля Ренея и смесь подвергали гидрогенолизу под давлением 50 psi в Parr устройстве в течение 16 ч. Катализатор удаляли фильтрованием через слой целита и промывали этанолом (20 мл), добавляли каталитическое количество (5 мг) п-толуолсульфоновой кислоты и реакционную смесь кипятили с обратным холодильником в течение 12 ч. Растворитель удаляли ротационным испарением для получения твердого вещества светло-коричневого цвета. Это вещество растворяли в концентрированном растворе NaHCO3 (10 мл), переводили в насыщенное состояние, используя NaCl и экстрагировали хлороформом (4×40 мл). Объединенные экстракты хлороформа сушили (K2CO3), фильтровали и концентрировали ротационным испарением для получения твердого вещества светло-коричневого цвета. Его очищали колоночной хроматографией, используя в качестве элюента смесь MeOH:CHCl3:NH4OH (9:1:0,01, об./об.), для получения 1,96 г (75%) 2'H-спиро[азабицикло[2.2.1]гептан-2,3'-пирролидин]-2'-она в виде твердого вещества кремового цвета (точка плавления 98°C).
Спиро[1-азабицикло[2.2.1]гептан-2,3'-пирролидин]
В раствор 2'Η-спиро[азабицикло[2.2.1]гептан-2,3'-пирролидин]-2'-она (1,00 г, 6,02 ммол) в сухом THF (20 мл) при 0°C в атмосфере N2 добавляли алюмогидрид лития (647 мг, 17,7 ммол) и смесь кипятили с обратным холодильником в течение 24 ч. Реакционную смесь охлаждали на ледяной бане и затем добавляли простой эфир (20 мл). Избыток гидрида гасили добавлением по каплям 5M раствора NaOH. Полученные твердые соли алюмината удаляли фильтрованием через слой целита. Фильтрат сушили (Na2SO4), фильтровали и концентрировали ротационным испарением для получения 800 мг спиро[1-азабицикло[2.2.1]гептан-2,3'-пирролидин] в виде бесцветной жидкости (87,43%).
1'-(3-Пиридил)спиро[1-азабицикло[2.2.1]гептан-2,3'-пирролидин]дигидрохлорид
Смесь спиро[1-азабицикло[2.2.1]гептан-2,3'-пирролидин] (300 мг, 1,98 ммол), 3-бромпиридина (344 мг, 2,18 ммол), трис(дибензилиденацетон)дипалладия (0) (54,57 мг, 0,0654 ммол), rac-2,2'-бис(дифенилфосфино)-1,1'-бинафтила (74,22 мг, 0,131 ммол) и трет-бутоксида калия (668,8 мг, 5,96 ммол) в сухом толуоле (20 мл) нагревали в плотно закрытой пробирке, продуваемой аргоном при 90°C в течение 8 ч. Реакционную смесь охлаждали до 0°C и содержимое переносили в 100 мл круглодонную колбу. Растворитель удаляли ротационным испарением и остаток растворяли в насыщенном растворе NaHCO3 (10 мл) и экстрагировали хлороформом (4×15 мл). Объединенные экстракты хлороформа сушили (K2CO3), фильтровали и концентрировали ротационным испарением до получения темноокрашенного сиропа. Его очищали колоночной хроматографией, используя в качестве элюента раствор MeOH:CHCl3:NH4OH (8:2:0,01, об./об.), для получения 350 мг (79,0%) 1'-(3-пиридил)спиро[1-азабицикло[2.2.1]гептан-2,3'-пирролидин] в виде светло-коричневого сиропа. Часть свободного основания (200 мг) превращали в соль гидрохлорида, которую кристаллизовали из изопропанола и этанола для получения 200 мг (76%) твердого вещества светло-коричневого цвета (т.пл. 232-236°C).
Пример 7
Образец 7 представляет собой 1'-(5-этокси-3-пиридил)спиро[1-азабицикло[2.2.1]гептан-2,3'-пирролидин], который получали согласно нижеприведенным способам.
1'-(5-этокси-3-пиридил)спиро[1-азабицикло[2.2.1]гептан-2,3'-пирролидин]
Смесь спиро[1-азабицикло[2.2.1]гептан-2,3'-пирролидин] (50 мг, 0,3 ммол), трис(дибензилиденацетон)дипалладия (0) (9 мг, 0,009 ммол), rac-2,2'-бис(дифенилфосфино)-1,1'-бинафтила (12 мг, 0,018 ммол), трет-бутоксида калия (147 мг, 1,2 ммол) и 5-бром-3-этоксипиридина (73 мг, 0,36 ммол) в сухом толуоле (5 мл) помещали в герметичную пробирку в атмосфере аргона и нагревали при 160°C в течение 17 ч. Реакционную смесь охлаждали до 0°C и содержимое переносили в 100-мл круглодонную колбу. Растворитель удаляли ротационным испарением и остаток растворяли в насыщенном растворе NaHCO3 (10 мл) и экстрагировали хлороформом (4×15 мл). Объединенные экстракты хлороформа сушили (K2CO3), фильтровали и концентрировали до получения темноокрашенного сиропа. Его очищали колоночной хроматографией, используя в качестве элюента смесь MeOH:CHCl3:NH4OH (8:2:0,01, об./об.), для получения 28 мг (27%) 1'-(5-этокси-3-пиридил)спиро[1-азабицикло[2.2.1]гептан-2,3'-пирролидина] в виде вязкого коричневого масла.
Пример 8
Образец 8 представляет собой 1'-(5-фенокси-3-пиридил)спиро[1-азабицикло[2.2.1]гептан-2,3'-пирролидин], который получали согласно нижеприведенным способам.
1'-(5-Фенокси-3-пиридил)спиро[1-азабицикло[2.2.1]гептан-2,3'-пирролидин]
Смесь спиро[1-азабицикло[2.2.1]гептан-2,3'-пирролидин] (50 мг, 0,3 ммол), трис(дибензилиденацетон)дипалладия (0) (9 мг, 0,009 ммол), rac-2,2'-бис(дифенилфосфино)-1,1'-бинафтила (12 мг, 0,018 ммол), трет-бутоксид калия (147 мг, 1,3 ммол) и 5-бром-3-феноксипиридина (90 мг, 0,36 ммол) в сухом толуоле (5 мл) нагревали в герметичной пробирке в атмосфере аргона при 160°C в течение 17 ч. Реакционную смесь охлаждали до 0°C и содержание переносили в 100-мл круглодонную колбу. Растворитель удаляли ротационным испарением и остаток растворяли в насыщенном растворе NaHCO3 (10 мл) и экстрагировали хлороформом (4×15 мл). Объединенные экстракты хлороформа сушили (K2CO3), фильтровали и концентрировали ротационным испарением до получения темноокрашенного сиропа. Его очищали колоночной хроматографией, используя в качестве элюента смесь MeOH:CHCl3:NH4OH (8:2:0,01, об./об.), для получения 55,8 мг 1'-(5-фенокси-3-пиридил)спиро[1-азабицикло[2.2.1]гептан-2,3'-пирролидин] (52%) в виде вязкого желто-коричневого масла.
Пример 9
Образец 9 представляет собой 1'-(5-пиримидинил)спиро[1-азабицикло[2.2.1]гептан-2,3'-пирролидин], который получали согласно нижеприведенным способам.
1'-(5-Пиримидинил)спиро[1-азабицикло[2.2.1]гептан-2,3'-пирролидин]
Смесь спиро[1-азабицикло[2.2.1]гептан-2,3'-пирролидин] (100 мг, 0,06 ммол), трис(дибензилиденацетон)дипалладия (0) (18 мг, 0,0018 ммол), rac-2,2'-бис(дифенилфосфино)-1,1'-бинафтила (24 мг, 0,0036 ммол), трет-бутоксид калия (300 мг, 2,6 ммол) и 5-бромпиридина (114 мг, 0,7 ммол) в сухом толуоле (10 мл) помещали в герметичную колбу в атмосфере аргона и нагревали при 125°C в течение 17 ч. Реакционную смесь охлаждали до 0°C и содержимое переносили в 100-мл круглодонную колбу. Растворитель удаляли ротационным испарением, и остаток растворяли в насыщенном растворе NaHCO3 (10 мл) и экстрагировали хлороформом (4×15 мл). Объединенные экстракты хлороформа сушили (K2CO3), фильтровали и концентрировали ротационным испарением для получения темноокрашенного сиропа. Его очищали колоночной хроматографией, используя в качестве элюента смесь MeOH:CHCl3:NH4OH (8:2:0,01, об./об.), для получения 49,0 мг 1'-(5-пиримидинил)спиро[1-азабицикло[2.2.1]гептан-2,3'-пирролидина] (32%) в виде вязкого коричневого масла.
Пример 10
Образец 10 представляет собой 1'-(3-пиридил)спиро[1-азабицикло[2.2.2]октан-2,3'-пирролидин], который получали согласно нижеприведенным способам.
Этил хинуклидин-2-карбоксилат
Этил хинуклидин-2-карбоксилат для этого синтеза получали согласно способу, описанному у Ricciardi and Doukas (Heterocycles 24: 971 (1986)). Также получали этил хинуклидин-2-карбоксилат при помощи химии, аналогичной химии, используемой для синтеза этил 1-азабицикло[2.2.1]гептан-2-карбоксилата, но вместо 3-(бромметил)оксолана используя 4-(бромметил)оксан.
Этил 2-(2-нитроэтил)хинуклидин-2-карбоксилат
Диизопропиламид лития получали при 0°C из изопропиламина лития (193,53 мг, 1,91 ммол) и н-бутиллития (0,764 мл, 1,91 ммол) в атмосфере N2. Его добавляли при помощи канюли в перемешиваемый раствор этил хинуклидина-2-карбоксилата (320 мг, 1,74 ммол) в сухом THF (10 мл) при -78°C. Спустя 1 ч в реакционную смесь добавляли по каплям раствор нитроэтилена (140,41 мг, 1,91 мМ) в THF (5 мл). После перемешивания в течение 2 ч при -78°C реакцию гасили добавлением насыщенного раствора хлорида аммония (20 мл). Проводили экстракцию этилацетатом (5×25 мл), сушили (Na2SO4), фильтровали и концентрировали ротационным испарением для получения 325 мг (70% чистоты) этил 2-(2-нитроэтил)хинуклидин-2-карбоксилата в виде светло-коричневой жидкости, которую использовали на следующем этапе без дополнительной очистки.
2'Η-Спиро[азабицикло[2.2.2]октан-2,3'-пирролидин]-2'-он
Раствор этил 2-(2-нитроэтил)хинуклидин-2-карбоксилата (320 мг) в этаноле (10 мл) подвергали гидрогенолизу под давлением 50 psi в Parr устройстве в течение 16 ч, используя никель Ренея в качестве катализатора. Катализатор удаляли фильтрованием через слой целита и промывали этанолом (20 мл). Добавляли каталитическое количество (5 мг) п-толуолсульфоновой кислоты и реакционную смесь кипятили с обратным холодильником в течение 12 ч. Растворитель удаляли ротационным испарением для получения твердого вещества светло-коричневого цвета. Это вещество растворяли в концентрированном растворе NaHCO3 (10 мл), полученный раствор переводили в насыщенное состояние NaCl и экстрагировали хлороформом (4×40 мл). Объединенные экстракты хлороформа сушили (K2CO3), фильтровали и концентрировали ротационным испарением для получения твердого вещества светло-коричневого цвета. Его очищали хроматографией, используя в качестве элюента смесь MeOH:CHCl3:NH4OH (8:2:0,01, об./об.), для получения 120 мг (38,2%) желаемого соединения в виде твердого вещества светло-кремового цвета (т.пл. 103-105°C).
Спиро[1-азабицикло[2.2.2]октан-2,3'-пирролидин]
В раствор 2'Η-спиро[азабицикло[2.2.2]октан-2,3'-пирролидин]-2'-она (100 мг, 0,55 ммол) в сухом THF (10 мл) при 0°C в атмосфере N2 добавляли алюмогидрид лития (74 мг, 1,94 ммол), и смесь кипятили с обратным холодильником в течение 24 ч. Реакционную смесь охлаждали на ледяной бане и затем добавляли эфир (20 мл). Избыток гидрида гасили добавлением по каплям 5 M раствора NaOH. Полученные твердые соли алюмината удаляли фильтрацией через слой целита. Фильтрат сушили (Na2SO4), фильтровали и концентрировали ротационным испарением для получения 83 мг спиро[1-азабицикло[2.2.2]октан-2,3'-пирролид] в виде бесцветной жидкости (90%).
1'-(3-Пиридил)спиро[1-азабицикло[2.2.2]октан-2,3'-пирролидин]
Перемешиваемый раствор спиро[1-азабицикло[2.2.2]октан-2,3'-пирролидин] (80 мг, 0,48 ммол), трис(дибензилиденацетон)дипалладия (0) (26,47 мг, 0,024 ммол), rac-2,2'-бис(дифенилфосфино)-1,1'-бинафтила (30 мг, 0,048 ммол) и трет-бутоксид калия (215 мг, 1,92 ммол) в сухом толуоле (15 мл) помещали в герметичную пробирку в атмосфере аргона и нагревали при 90°C в течение 16 ч. Реакционную смесь охлаждали до 0°C и содержимое переносили в 100-мл круглодонную колбу. Растворитель удаляли ротационным испарением и остаток растворяли в насыщенном растворе NaHCO3 (10 мл) и экстрагировали хлороформом (4×15 мл). Объединенные экстракты хлороформа сушили (K2CO3), фильтровали и концентрировали ротационным испарением до получения темноокрашенного сиропа. Его очищали колоночной хроматографией, используя в качестве элюента смесь MeOH:CHCl3:NH4OH (8:2:0,01, об./об.), для получения 102 мг (85,7%) 1'-(3-пиридил)спиро[1-азабицикло[2.2.2]октан-2,3'-пирролидин] в виде светло-коричневого сиропа.
Пример 11
Образец 11 представляет собой 1'-(3-пиридил)-2'H-спиро[1-азабицикло[2.2.1]гептан-7,3'-пирролидин], который получали согласно нижеприведенным способам.
Этил 2-(2H,3H,5H-4-оксинил)-2-нитроацетат
2 M раствор тетрахлорида титана в THF получали медленным добавлением тетрахлорида титана (7,59 г, 40 ммол) в сухом THF (20 мл) при 0°C в атмосфере азота. Затем в перемешиваемый раствор добавляли этилнитроацетат (2,66 г, 20 ммол), и смесь перемешивали в течение 5 мин. После этого одной частью добавляли татрагидро-4-H-пиран-4-он (2,00 г, 20 ммол). Затем 1,0 M раствор N-метилморфолина в THF (8,09 г, 80 ммол) добавляли по каплям в течение 2 ч при 0°C. После чего смесь оставляли для нагревания до комнатной температуры и перемешивали в течение 18 ч. Затем выливали в воду (20 мл) и экстрагировали этилацетатом (5×40 мл). Объединенные экстракты сушили в течение Na2SO4, фильтровали и концентрировали ротационным испарением. Густой коричневый сироп очищали колоночной хроматографией, используя в качестве элюента смесь этилацетат:гексан (1:9, об./об.), для получения 3,00 г чистого соединения в виде светло-коричневого сиропа (70%).
Этил 2-(4-оксанил)-2-аминоацетат
Никель Ренея (~2 г) добавляли в раствор этил 2-(2H,3H,5H-4-оксинил)-2-нитроацетата (2,50 г, 11,62 ммол) в этаноле (50 мл) и концентрированной HCl (1 мл). Смесь подвергали гидрогенолизу под давлением 50 psi в Parr устройстве в течение 18 ч. Катализатор осторожно удаляли фильтрованием через слой целита. Растворитель удаляли ротационным испарением. Доводили pH остатка до 8-9 насыщенным водным раствором NaHCO3, затем полученный раствор переводили в насыщенное состояние при помощи NaCl и экстрагировали хлороформом (4×25 мл). Объединенные экстракты сушили в течение K2CO3, фильтровали и концентрировали для получения 2,40 г (~100%) желаемого соединения в виде желто-коричневой жидкости.
Гидрохлорид 1-азабицикло[2.2.1]гептан-7-карбоксильной кислоты
Этил 2-(оксанил)-2-аминоацетат (1,50 г, 8,02 ммол) растворяли в 48% HBr (10 мл) в пробирке для высокого давления и насыщали газообразным HBr. Пробирку осторожно плотно закрывали и нагревали в течение 12 ч при 120-130°C. Реакционную смесь охлаждали до комнатной температуры, переносили в 250-мл круглодонную колбу и удаляли кислоту ротационным испарением. Темноокрашенный осадок растворяли в 30% растворе аммиака (50 мл). Эту смесь перемешивали в течение 5 ч при комнатной температуре до тех пор, пока не завершилась циклизация желаемой кислоты. Раствор аммиака удаляли ротационным испарением для получения твердого вещества светло-коричневого цвета, которое повторно растворяли в 5 мл воды и очищали на ионно-обменной смоле, используя в качестве элюента воду и аммиак (30% водный). Аммиачные фракции, содержащие желательную кислоту, объединяли и концентрировали для получения чистой кислоты, которую превращали в соль HCl и кристаллизовали из изопропанола и диэтилового эфира для получения 1,21 г (85%) твердого вещества кремового цвета (т.пл. 232°С, превращаясь в коричневое, плавится при 253-254°C).
Этил 1-азабицикло[2.2.1]гептан-7-карбоксилат
Раствор гидрохлорида 1-азабицикло[2.2.1]гептан-7-карбоксильной кислоты (1,20 г, 6,76 ммол) в абсолютном этаноле (10 мл) и концентрированной серной кислоты (2 мл) кипятили с обратным холодильником в течение 8 ч. Реакционную смесь охлаждали и доводили pH до 8-9 насыщенным водным раствором NaHCO3. Раствор переводили в насыщенное состояние, используя твердый NaCl, и экстрагировали хлороформом (4×20 мл). Объединенные экстракты сушили при помощи Na2SO4, фильтровали и концентрировали ротационным испарением, получая светло-коричневую жидкость. Ее очищали дистилляцией Кугельрора при 120°C и давлении 2,5 мм для получения 1,00 г (90%) в виде бесцветной жидкости.
Этил 1-аза-7-(2-нитроэтил)бицикло[2.2.1]гептан-7-карбоксилат
Диизопропиламид лития получали добавлением н-бутиллития (1,70 мл, 6,26 ммол) в диизопропиламин (431,1 мг, 6,26 ммол) в сухом THF (5 мл) при 0°С в атмосфере N2. Реакционную смесь перемешивали при комнатной температуре в течение 15 мин и затем при помощи канюли переносили в перемешиваемый раствор этил 1-азабицикло[2.2.1]гептан-7-карбоксилата (600 мг, 3,55 мМ) в THF (20 мл) при -78°C в атмосфере N2. Реакционную смесь перемешивали в течение 30 мин при -78°C, затем при помощи канюли добавляли раствор нитроэтилена (285,3 мг, 3,91 мМ) в THF (10 мл) и реакционную смесь перемешивали в течение следующих 2 ч при -78°C. После этого реакцию гасили насыщенным раствором NH4Cl (10 мл). Реакционную смесь оставляли нагреваться до комнатной температуры и затем экстрагировали этилацетатом (4×20 мл). Объединенные фракции сушили (K2CO3), фильтровали и концентрировали, получая 650 мг светло-коричневой жидкости. Ее очищали колоночной хроматографией, используя смесь этилацетат:дихлорметан (8:2, об./об.), получая 600 мг (85%) желто-коричневой жидкости.
2'Η-спиро[1-азабицикло[2.2.1]гептан-7,3'-пирролидин]-2'-он
Этил 1-аза-7-(2-нитроэтил)бицикло[2.2.1]гептан-7-карбоксилат (550 мг, 2,27 ммол) растворяли в этаноле (25 мл) и подвергали гидрогенолизу под давлением 50 psi в течение 18 ч, используя в качестве катализатора никель Ренея. Катализатор удаляли фильтрованием через слой целита. Растворитель удаляли ротационным испарением. Полученный остаток растворяли в толуоле (50 мл) и добавляли каталитическое количество п-толуолсульфоновой кислоты (10 мг). Раствор кипятили с обратным холодильником в течение 12 ч и затем удаляли растворитель ротационным испарением. Остаток добавляли в насыщенный раствор NaHCO3 (10 мл) и экстрагировали хлороформом (5×15 мл). Объединенные экстракты хлороформа сушили (K2CO3), фильтровали и концентрировали. Остаток очищали колоночной хроматографией, используя в качестве элюента смесь CHCl3:MeOH:NH4OH (9:1:0,01, об./об.), получая 320 мг (85%) чистого соединения в виде густого сиропа кремового цвета.
2'Η-спиро[1-азабицикло[2.2.1]гептан-7,3'-пирролидин]
В перемешиваемый раствор 2'H-спиро[1-азабицикло[2.2.1]гептан-7,3'-пирролидин]-2'-она (300 мг, 1,80 ммол) в сухом THF (30 мл) при 0° в атмосфере N2 добавляли LiAlH4 (274,33 мг, 7,22 ммол). Ледяную баню удаляли и реакционную смесь кипятили с обратным холодильником в течение 24 ч. Реакционную смесь охлаждали до 0°C, добавляли диэтилэфир (20 мл) и по каплям добавляли 5M NaOH при постоянном помешивании до тех пор, пока весь непрореагировавший LiAlH4 не выпал в осадок в виде твердого вещества. Реакционную смесь фильтровали через слой целита и фильтрат сушили (K2CO3), фильтровали и концентрировали, получая 250 мг (70%) бесцветного сиропа.
1'-(3-Пиридил)-2'H-спиро[1-азабицикло[2.2.1]гептан-7,3'-пирролидин]
2'Η-спиро[1-азабицикло[2.2.1]гептан-7,3'-пирролидин] (100 мг, 0,66 ммол), трис(дибензилиденацетон)дипалладий (0) (30 мг, 0,020 ммол), rac-2,2'-бис(дифенилфосфино)-1,1'-нафтил (45 мг, 0,040 ммол), трет-бутоксид калия (369 мг, 3,3 ммол), 3-бромпиридин (114 мг, 0,72 ммол) и сухой толуол (10 мл) помещали в пробирку для высокого давления, которую прочищали аргоном. Пробирку осторожно плотно закрывали и нагревали в течение 8 ч при 90°C. Реакционную смесь охлаждали, переносили в круглодонную колбу, и растворитель удаляли ротационным испарением. Остаток выливали в насыщенный раствор NaHCO3 (5 мл) и экстрагировали хлороформом (4×15 мл). Объединенные экстракты хлороформа сушили над K2CO3, фильтровали и концентрировали ротационным испарением. Остаток очищали колоночной хроматографией, используя в качестве элюента смесь CHCl3:MeOH:NH4OH (8:2:0,01, об./об.) до получения 130 мг (86,7%) светло-коричневого сиропа. Под воздействием света и воздуха продукт становится темно-коричневым.
Примеры 12 и 13
Образцы 12 и 13 представляют собой соответственно (+) и (-)7-(3-пиридил)-1,7-диазаспиро[4.4]нонан, которые получали согласно нижеприведенным способам.
Диастереомерные S-пролинамиды 7-(3-пиридил)-1,7-диазаспиро[4.4]нонана
В суспензию для перемешивания N-(трет-бутоксикарбонил)-S-пролина (4,67 г, 21,7 ммол) в дихлорметане (100 мл) в атмосфере азота добавляли в указанном порядке триэтиламин (6,0 мл, 43 ммол) и дифенилхлорфосфат (4,0 мл, 19 ммол). После перемешивания в течение 1,5 ч при температуре окружающей среды реакционную смесь обрабатывали раствором 7-(3-пиридил)-1,7-диазаспиро[4.4]нонана (4,40 г, 21,6 ммол) в дихлорметане (10 мл). Смесь перемешивали в течение 3 дней при температуре окружающей среды. Затем добавляли раствор гидроксида натрия (30 мл, 5 M). После перемешивания в течение еще одного часа смесь выливали в делительную воронку с хлороформом (30 мл) и водой (30 мл). Смесь энергично встряхивали и слои отделяли. Органический слой и 30 мл хлороформного экстракта из водного слоя объединяли, сушили (MgSO4) и концентрировали ротационным испарением. Остаток (7,2 г) растворяли в дихлорметане (100 мл) и объединяли с трифторуксусной кислотой (50 мл). Смесь перемешивали при температуре окружающей среды в течение 1 ч. Летучие компоненты испаряли сначала ротационным испарением, затем при помощи вакуумного насоса. Остаток очищали препаративной ВЭЖХ, используя в качестве элюента 10% ацетонитрил, 0,1% трифторуксусную кислоту в воде. Отобранные фракции объединяли и концентрировали, получая 3,13 г (выход 79%) диастереомера, который элюировал за 11,4 мин, и 2,90 г (выход 74%) диастереомера, который элюировал за 13,2 мин, оба в виде белых пенообразных веществ (предположительно соли монотрифторуксусной кислоты).
(+) и (-)7-(3-Пиридил)-1,7-диазаспиро[4.4]нонан
Каждый из двух диастереомерных S-пролинамидов растворяли в дихлорметане (50 мл) и триэтиламине (2-3 мл) и затем объединяли с фенилизотиоционатом (1,73 г, 12,8 ммол для более быстро элюирующего диастереомера и 1,57 г, 11,6 ммол для более поздно элюирующего диастереомера). Две реакционные смеси перемешивали при температуре окружающей среды в течение 16 ч, время, при котором тонкослойная хроматография показала полное завершение реакции. Смеси концентрировали ротационным испарением и каждый остаток помещали в дихлорметан (10 мл) и обрабатывали трифторуксусной кислотой (10 мл). Эти реакции проводили при 50°C в течение 16 ч и смеси концентрировали до сухого состояния. Колоночная хроматография на силикагеле с использованием смеси 80:20:2 хлороформ/метанол/аммиак дала 620 мг (производное от более быстро элюирующего диастереомера, выход 40,5%) и 720 мг (производное от медленнее элюирующего диастереомера, выход 50,7%), в виде светло-коричневого масла. Хиральный анализ ВЭЖХ выполняли на колонке Chiralcel OD®, используя смесь 7:3 гексан/этанол. Изомер, производный от более быстро элюирующего диастереомера, имел большее время отстаивания на хиральной колонке (10,9 мин); изомер, производный от медленнее элюирующего диастереомера, показал время отстаивания 8,7 мин на хиральной колонке. Образцы были энантиомерно чистыми в пределах детектирования (-2%).
Пример 14
Исследование in vitro фармакологии 7-(3-пиридил)-1,7-диазаспиро[4.4]нонана показало, что он является антагонистом как подтипа α4β2 (IC50=193 мкМ; Imax=50%), так и подтипов NNR, влияющих на высвобождение допамина (IC50=901 нМ; Imax=67%). Способность этого соединения к неполному ингибированию высвобождения допамина является особенно значимой, поскольку указывает, что это соединение (и другие в классе N-арилспиродиазаалканов) может быть полезным для нарушения системы компенсации допамина и таким образом для лечения нарушений, которые ею опосредованы. Такие нарушения включают наркотическую зависимость и токсикоманию, потребление табака и увеличение веса, которое происходит при прекращении приема лекарственного средства.
Данные in vivo о том, что N-арилспиродиазаалканы могут быть полезны таким образом, были получены в результате 14-дневного безопасного профилактического фармакологического исследования, в котором 7-(3-пиридил)-1,7-диазаспиро[4.4]нонан уменьшил увеличение веса у крыс, не проявляя свойств, приводящих к сенсибилизации.
Основываясь на этих данных, очевидно, что соединения класса N-арилспиродиазаалканов, раскрытых в настоящем описании, представляют собой полезную альтернативу для лечения зависимостей от лекарственных средств, допускающих злоупотребление, включая спирт, амфетамины, барбитураты, бензодиазепины, кофеин, каннабиноиды, кокаин, галлюциногены, опиаты, фенциклидин и табак, и для лечения нарушений, связанных с питанием, таких как ожирение, которое происходит после прекращения приема лекарственного средства, при этом уменьшая побочные эффекты, связанные с использованием психомоторных стимуляторов (тревожность, сонливость, привыкание и т.д.).
Имея таким образом раскрытую сущность настоящего изобретения, следует отметить, что возможны многие модификации, замены и варианты настоящего изобретения. Должно быть ясно, что настоящее изобретение может быть реализовано иначе, чем, в частности, описано. Такие модификации, замены и варианты должны находиться в объеме настоящего описания.
| название | год | авторы | номер документа |
|---|---|---|---|
| Спироконденсированные пирролидиновые производные в качестве ингибиторов деубиквитилирующих ферментов (DUB) | 2017 |
|
RU2730552C2 |
| ОКСАДИАЗОЛЫ В КАЧЕСТВЕ АГОНИСТОВ МУСКАРИНОВОГО РЕЦЕПТОРА M1 И/ИЛИ M4 | 2019 |
|
RU2817018C2 |
| ПРОИЗВОДНЫЕ 2,3-ДИГИДРО-1Н-ИНДЕН-1-ИЛ-2,7-ДИАЗАСПИРО[3.5]НОНАНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ АНТАГОНИСТОВ ИЛИ ОБРАТНЫХ АГОНИСТОВ ГРЕЛИНОВОГО РЕЦЕПТОРА | 2011 |
|
RU2524341C2 |
| ИНГИБИТОРЫ ВЗАИМОДЕЙСТВИЯ МЕНИН-MLL | 2017 |
|
RU2799820C2 |
| СПОСОБ ПРОГНОЗИРОВАНИЯ ТЕРАПЕВТИЧЕСКОГО ЭФФЕКТА ИНГИБИТОРА LSD1 НА ОСНОВЕ ЭКСПРЕССИИ INSM1 | 2018 |
|
RU2789449C2 |
| НОВЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ | 2019 |
|
RU2809257C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ ПРОСТОГО ЭФИРА ИЗОКСАЗОЛИЛА В КАЧЕСТВЕ ПАМ ГАМК A АЛЬФА5 | 2019 |
|
RU2800160C2 |
| 2-ОКСА-5-АЗАБИЦИКЛО[2.2.1]ГЕПТАН-3-ИЛЬНЫЕ ПРОИЗВОДНЫЕ | 2015 |
|
RU2697651C2 |
| ПИРИДО[3,4-D]ПИРИМИДИНОВОЕ ПРОИЗВОДНОЕ И ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМАЯ СОЛЬ | 2017 |
|
RU2796400C2 |
| НОВОЕ СОЕДИНЕНИЕ БИФЕНИЛА ИЛИ ЕГО СОЛЬ | 2018 |
|
RU2765152C2 |
Изобретение относится к способу лечения одной или более лекарственной зависимости, никотиновой зависимости и/или ожирения, включающему введение эффективного количества соединения, достаточного для уменьшения продуцирования и/или секреции допамина, причем соединение имеет следующую формулу:
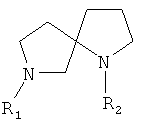
или его фармацевтически приемлемых солей, где R1 представляет собой водород или метил, когда R2 представляет собой Су; R2 представляет собой водород или метил, когда R1 представляет собой Су; и где Су представляет собой группу
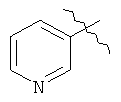
которая необязательно замещена -R, -OR, -NR2 или галогеном, где каждый R независимо является необязательно замещенным низшим алкилом, арилом или гетероарилом. Изобретение также относится к способам лечения одной или более лекарственной зависимости, никотиновой зависимости или ожирения. Технический результат - получение новых способов лечения зависимости. 5 н.п. ф-лы.
1. Способ лечения одной или более лекарственной зависимости, никотиновой зависимости и/или ожирения, включающий введение эффективного количества соединения, достаточного для уменьшения продуцирования и/или секреции допамина, причем соединение имеет следующую формулу:
или его фармацевтически приемлемых солей,
где R1 представляет собой водород или метил, когда R2 представляет собой Су;
R2 представляет собой водород или метил, когда R1 представляет собой Су;
и где Су представляет собой группу
, которая необязательно замещена -R, -OR,
-NR2 или галогеном, где каждый R независимо является необязательно замещенным низшим алкилом, арилом или гетероарилом.
2. Способ лечения одной или более лекарственной зависимости, никотиновой зависимости или ожирения, включающий введение эффективного количества соединения, достаточного для уменьшения продуцирования и/или секреции допамина, причем соединение имеет следующую формулу:
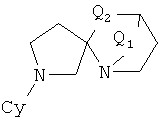
или его фармацевтически приемлемых солей,
где Q1 представляет собой -СН2- или -CH2CH2-;
Q2 представляет собой простую связь или -CH2-;
при условии, что когда Q2 является простой связью, Q1 является -СН2-;
Су представляет собой группу
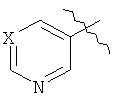 , которая необязательно замещена -R, -OR,
, которая необязательно замещена -R, -OR,
-NR2 или галогеном, где каждый R независимо является необязательно замещенным низшим алкилом, арилом или гетероарилом; и
Х представляет собой -СН или N.
3. Способ лечения одной или более лекарственной зависимости, никотиновой зависимости и/или ожирения, включающий введение эффективного количества соединения, достаточного для уменьшения продуцирования и/или секреции допамина, выбранного из группы, состоящей из
7-(3-пиридил)-1,7-диазаспиро[4.4]нонана;
7-(5-этокси-3-пиридил)-1,7-диазаспиро[4.4]нонана;
7-(5-фенокси-3-пиридил)-1,7-диазаспиро[4.4]нонана;
1-(3-пиридил)-1,7-диазаспиро[4.4]нонана;
1-метил-7-(3-пиридил)-1,7-диазаспиро[4.4]нонана;
1-метил-7-(5-этокси-3-пиридил)-1,7-диазаспиро[4.4]нонана;
1-метил-7-(5-фенокси-3-пиридил)-1,7-диазаспиро[4.4]нонана;
или их фармацевтически приемлемых солей.
4. Способ лечения одной или более лекарственной зависимости, никотиновой зависимости и/или ожирения, включающий введение эффективного количества соединения, достаточного для уменьшения продуцирования и/или секреции допамина, выбранного из группы, состоящей из
1'-(3-пиридил)спиро[1-азабицикло[2.2.1]гептан-2,3'-пирролидин];
1'-(5-этокси-3-пиридил)спиро[1-азабицикло[2.2.1]гептан-2,3'-пирролидин];
1'-(5-фенокси-3-пиридил)спиро[1-азабицикло[2.2.1]гептан-2,3'-пирролидин];
1'-(5-пиримидинил)спиро[1-азабицикло[2.2.1]гептан-2,3'-пирролидин];
1'-(3-пиридил)спиро[1-азабицикло[2.2.2]октан-2,3'-пирролидин];
1'-(3-пиридил)-2'Н-спиро[1-азабицикло[2.2.1]гептан-7,3'-пирролидин];
или их фармацевтически приемлемых солей.
5. Способ лечения одной или более лекарственной зависимости, никотиновой зависимости и/или ожирения, включающий введение эффективного количества 7-(3-пиридил)-1,7-диазаспиро[4.4]нонана или его фармацевтически приемлемой соли.
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| EP 0970957 A, 12.01.2000 | |||
| Экономайзер | 0 |
|
SU94A1 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| RU 94045948 A1, 10.09.1996. | |||

