Данная заявка испрашивает приоритет согласно предварительной заявке США на патент №61/487,640, поданной 18 мая 2011 г., и предварительной заявки США на патент №61/543,716, поданной 05 октября 2011 г., которые включены в данную заявку во всей полноте посредством отсылок.
Область техники, к которой относится изобретение
Увеличивающаяся частота сахарного диабета является глобальной проблемой колоссальных масштабов, основным виновником заболеваемости и смертности пациентов и основным экономическим бременем. Ожирение представляет собой важный фактором риска заболеваемости диабетом типа 2, и примерно у 90% пациентов с диабетом типа 2 наблюдается избыточный вес или ожирение. Ожирение является быстро увеличивающейся в масштабах общемировой проблемой, и в настоящее время избыточный вес наблюдается более чем у 65% взрослых жителей США (Hedley, A.A., et аl. (2004) JAMA 291: 2847-2850). Существует необходимость в разработке безопасных и эффективных лекарственных средств против ожирения и сахарного диабета.
Раскрытие изобретения
В настоящей заявке раскрываются композиции и способы лечения или предупреждения нарушений, связанных с инсулинорезистентностью, включая, но без ограничения, ожирение, метаболический синдром, диабет типа 2, гипертонию, атеросклероз и т.п.Согласно некоторым вариантам изобретения способ включает профилактическое и/или терапевтическое лечение с применением пептидов и/или белков. Некоторые недостатки лекарственных препаратов на основе пептидов и/или белков ограничивают их применение в медицине (Nestor, J.J., Jr. (2007) Comprehensive Medicinal Chemistry II 2: 573-601) - кратковременное действие, низкая биодоступность и недостаточная селективность к подтипам рецепторов. Кроме того, пептиды и/или белки неустойчивы в лекарственных препаратах, часто подвергаются агрегации.
В настоящей заявке описаны некоторые пептиды и/или белки, модифицированные за счет ковалентного связывания (например, GLP-1, глюкагон, родственные аналоги или т.п.), что способствует более продолжительному действию и/или повышенной биодоступности при введении модифицированных пептидов и/или белков. Такие пептиды и/или белки, модифицированные за счет ковалентного связывания, применимы для предупреждения и/или лечения состояний, связанных с ожирением, метаболическим синдромом, резистентностью к инсулину (инсулинорезистентностью), диабетом типа 2, гипертонией, атеросклерозом и т.п.
Согласно некоторым вариантам изобретения модифицированные за счет ковалентного связывания пептиды и/или белки, раскрываемые в настоящей заявке, связаны с поверхностно-активными гликозидами. В одном аспекте модифицированные за счет ковалентного связывания пептиды и/или белки связываются с поверхностно-активным гликозидом, причем пептид и/или белок связывается с поверхностно-активным гликозидом в поверхностно-активном веществе (ПАВ), а гликозид затем связывается с гидрофобной группой. Также согласно некоторым вариантам настоящего изобретения предусматриваются реагенты и интермедиаты для синтеза модифицированных пептидов и/или белков (например, модифицированный GLP-1, глюкагон, аналоги глюкагона или GLP-1 или т.п.), получаемые с помощью включения поверхностно-активных веществ (ПАВ).
Согласно некоторым вариантам изобретения предусматриваются пептидные продукты, содержащие поверхностно-активное вещество (ПАВ) X, связанное с пептидом ковалентной связью, причем пептид содержит линкерную аминокислоту U и по меньшей мере одну другую аминокислоту:
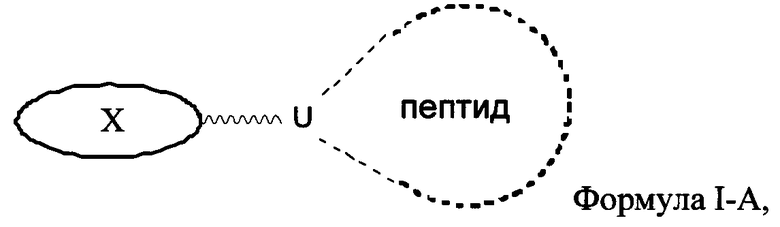
где ПАВ Х обозначает группу Формулы I:
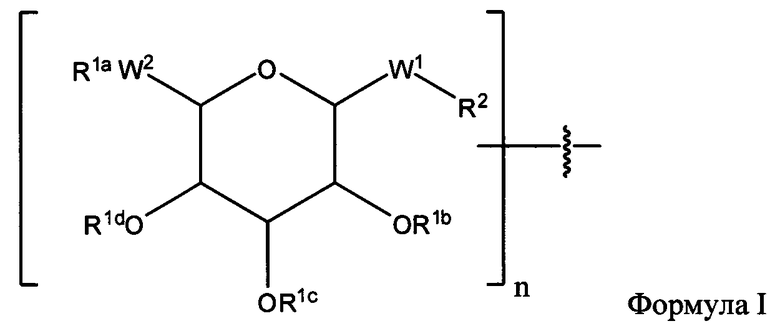
где:
R1a, независимо, в каждом случае обозначает связь, Н, замещенную или незамещенную C1-С30 алкильную группу, замещенную или незамещенную алкоксиарильную группу или замещенную или незамещенную аралкильную группу;
R1b, R1c и R1d каждый независимо, в каждом случае обозначает связь, Н, замещенную или незамещенную C1-С30 алкильную группу, замещенную или незамещенную алкоксиарильную группу или замещенную или незамещенную аралкильную группу;
W1, независимо, в каждом случае обозначает -СН2-, -CH2-O-, -(C=O), -(C=O)-O-, -(C=O)-NH-, -(C=S)-, -(C=S)-NH- или -CH2-S-;
W2 обозначает -O-, -СН2- или -S-;
R2 независимо, в каждом случае обозначает связь, Н, замещенную или незамещенную C1-С30 алкильную группу, замещенную или незамещенную алкоксиарильную группу или замещенную или незамещенную аралкильную группу, -NH2, -SH, С2-С4-алкен, С2-С4-алкин, -NH(C=O)-CH2-Br, -(CH2)m-малеимид или -N3;
n обозначает 1, 2 и 3; и
m обозначает 1-10;
пептид выбран из соединения Формулы II:
где:
Z обозначает ОН, или -NH-R3, где R3 обозначает Н или С1-С12 замещенный или незамещенный алкил, или цепь PEG (ПЭГ) протяженностью менее 10 Да;
aa1 обозначает His, N-Ac-His, pGlu-His или N-R3-His;
аа2 обозначает Ser, Ala, Gly, Aib, Ac4c или Ас5с;
аа3 обозначает Gln или Cit;
aa4 обозначает Gly или D-Ala;
aa5 обозначает Thr или Ser;
аа6 обозначает Phe, Trp, F2Phe, Me2Phe или Nal2;
aa7 обозначает Thr или Ser;
aa8 обозначает Ser или Asp;
aa9 обозначает Asp обозначает Glu;
аа10 обозначает Tyr, Leu, Met, Nal2, Bip или Bip2EtMeO;
аа11 обозначает Ser, Asn или U;
aa12 обозначает Lys, Glu, Ser, Arg или U;
aa13 отсутствует или обозначает Tyr, Gln, Cit или U;
aa14 отсутствует или обозначает Leu, Met, Nle или U;
aa15 отсутствует или обозначает Asp, Glu или U;
aa16 отсутствует или обозначает Ser, Gly, Glu, Aib, Ac5c, Lys, Arg или U;
aa17 отсутствует или обозначает Arg, hArg, Gln, Glu, Cit, Aib, Ac4c, Ac5c или U;
aa18 отсутствует или обозначает Arg, hArg, Ala, Aib, Ac4c, Ac5c или U;
aa19 отсутствует или обозначает Ala, Val, Aib, Ac4c, Ac5c или U;
aa20 отсутствует или обозначает Gln, Lys, Arg, Cit, Glu, Aib, Ac4c, Ac5c или U;
аа21 отсутствует или обозначает Asp, Glu, Leu, Aib, Ac4c Ac5c или U;
aa22 отсутствует или обозначает Phe, Trp, Nal2, Aib, Ac4c, Ac5c или U
aa23 отсутствует или обозначает Val, He, Aib, Ac4c, Ac5c или U;
aa24 отсутствует или обозначает Gln, Ala, Glu, Cit или U;
aa25 отсутствует или обозначает Trp, Nal2 или U;
аа26 отсутствует или обозначает Leu или U;
aа27 отсутствует или обозначает Met, Val, Nle, Lys или U;
аа28 отсутствует или обозначает Asn, Lys или U;
аа29 отсутствует или обозначает Thr, Gly, Aib, Ac4c, Ac5c или U;
аа30 отсутствует или обозначает Lys, Aib, Ac4c, Ac5c или U;
аа31 отсутствует или обозначает Arg, Aib, Ac4c, Ac5c или U;
аа32 отсутствует или обозначает Asn, Aib, Ac4c, Ac5c или U;
аа33 отсутствует или обозначает Arg, Aib, Ac4c, Ac5c или U;
аа34 отсутствует или обозначает Asn, Aib, Ac4c, Ac5c или U;
аа35 отсутствует или обозначает Asn, Aib, Ac4c, Ac5c или U;
аа36 отсутствует или обозначает Ilе, Aib, Ac4c, Ас5С или U;
аа36 отсутствует или обозначает Ala, Aib, Ac4c, Ас5С или U;
аа37 отсутствует или обозначает U;
U обозначает природную или не встречающуюся в природе аминокислоту, содержащую функциональную группу, применяемую для ковалентного связывания с ПАВ X;
где любые две из aa1-аа37, необязательно, циклизуются по своим боковым цепям с образованием лактамной связи; и
при условии, что одна или по меньшей мере одна из аминокислот аа11-аа37 представляет собой линкерную аминокислоту U, связанную с Х ковалентной связью.
Согласно некоторым вариантам изобретения n обозначает 1. Согласно некоторым вариантам изобретения n обозначает 2, а первый гликозид связан со вторым гликозидом за счет связи между W2 первого гликозида и любой из групп OR1b, OR1c или OR1d второго гликозида. Согласно некоторым вариантам изобретения n обозначает 3, первый гликозид связан со вторым гликозидом за счет связи между W2 первого гликозида и любой из групп OR1b, OR1c или OR1d второго гликозида, а второй гликозид связан с третьим гликозидом за счет связи между W2 второго гликозида и любой из групп OR1b, OR1c или OR1d третьего гликозида.
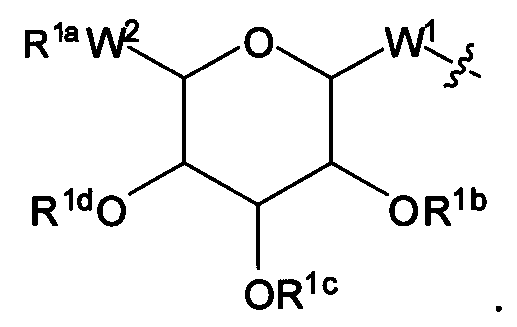
Согласно одному варианту изобретения соединения Формулы I-A представляют собой соединения, в которых Х имеет структуру:
где:
R1a обозначает Н, защитную группу, замещенную или незамещенную C1-С30 алкильную группу или фрагмент, содержащий стероидное ядро;
R1b, R1c и R1d, каждый, независимо, в каждом случае, обозначает Н, защитную группу, замещенную или незамещенную C1-С30 алкильную группу;
W1, независимо, в каждом случае, обозначает -СН2-, -СН2-O-, -(C=O), -(C=O)-O-, -(C=O)-NH-, -(C=S)-, -(C=S)-NH-, или -CH2-S-;
W2 обозначает -O-, -S-;
R2 обозначает связь, С2-С4-алкен, С2-С4-алкин или -(СН2)m-малеимид; и
m обозначает 1-10.
Согласно другому варианту изобретения соединения Формулы I-A представляют собой соединения, в которых Х имеет структуру:
Соответственно, согласно описанному выше варианту изобретения R2 обозначает связь.
Например, согласно типичному варианту структуры X, описанному выше, W1 обозначает -C(=O)NH-, R2 обозначает связь между W1 и аминокислотным остатком U в пептиде (например, в боковой цепи остатка лизина, присутствующего в пептиде).
Согласно другому варианту изобретения соединения Формулы I-A представляют собой соединения, в которых Х имеет структуру:
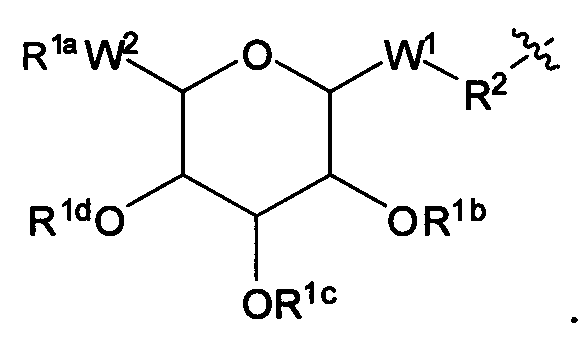
Например, согласно типичному варианту структуры X, описанному выше, W1 обозначает -СН2-, R2 обозначает связанную с алкилом малеимидную функциональную группу в Х и группа R связана с соответствующей группой аминокислотного остатка U в пептиде (Например, тиольная группа в цистеиновом остатке пептида образует тиоэфир с малеимидом в X).
Согласно еще одному варианту изобретения соединения Формулы I-A представляют собой соединения, в которых Х имеет структуру:
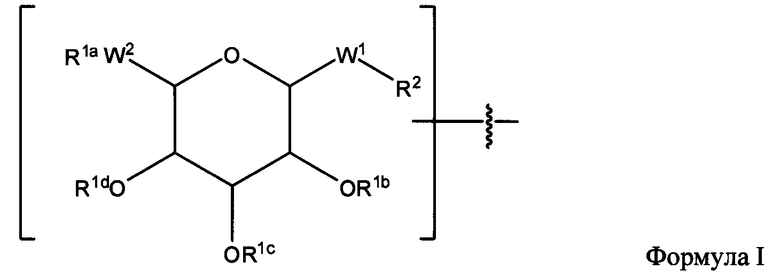
где:
R1a обозначает Н, защитную группу, замещенную или незамещенную C1-С30 алкильную группу или фрагмент, содержащий стероидное ядро;
R1b, R1c и R1d, каждый, независимо, в каждом случае, обозначает Н, защитную группу, замещенную или незамещенную C1-С30 алкильную группу;
W1 обозначает -(C=O)-NH-;
W2 обозначает -O-;
R2 обозначает связь.
Согласно другому варианту изобретения соединения Формулы I-A представляют собой соединения, в которых Х имеет структуру:
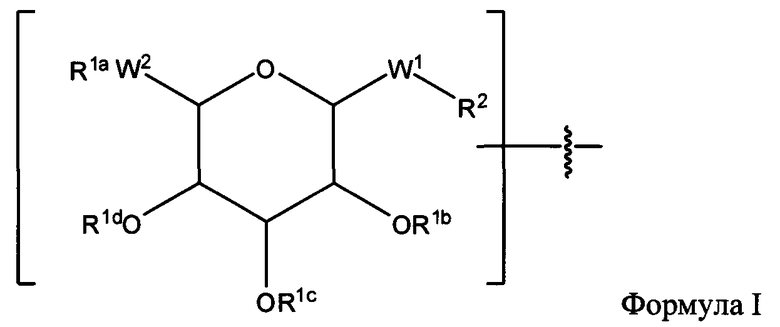
где:
R1a обозначает замещенную или незамещенную C1-С30 алкильную группу;
R1b, R1c и R1d обозначают Н;
W1 обозначает -(C=O)-NH-;
W2 обозначает -O-; и
R2 обозначает связь.
Согласно некоторым вариантам изобретения, описанным выше и ниже в данной заявке, R1a обозначает замещенную или незамещенную C1-С30 алкильную группу.
Согласно некоторым вариантам изобретения, описанным выше и ниже в данной заявке, R1a обозначает замещенную или незамещенную С6-С20 алкильную группу.
Также в настоящем описании представлены альтернативные варианты изобретения, где Х в Формуле I-A имеет структуру:
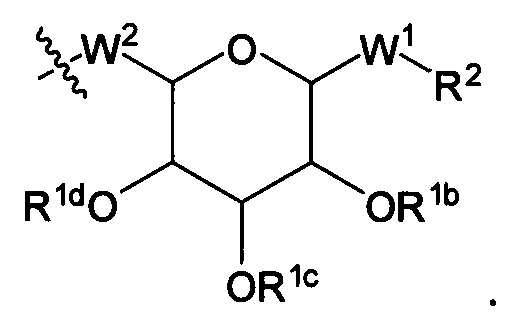
Например, в типичном варианте описанной выше структуры Х W1 обозначает -S-, R2 обозначает C1-С30 алкильную группу, W2 обозначает S, R1a обозначает связь между W2 и соответствующим фрагментом аминокислотного остатка U в пептиде (например, тиольная группа в цистеиновом остатке пептида образует тиоэфир с X).
В другом типичном варианте описанной выше структуры Х W1 обозначает -O-, R2 обозначает C1-С30 алкильную группу, W обозначает О, R1a обозначает связь между W2 и соответствующим фрагментом аминокислотного остатка U в пептиде (например, гидроксильная группа в сериновом или треониновом остатке пептида образует простой эфир с X).
Согласно некоторым вариантам изобретения U используется для ковалентного связывания с Х и представляет собой двухосновную природную или не встречающуюся в природе аминокислоту, природную или не встречающуюся в природе аминокислоту, содержащую тиольную группу, не встречающуюся в природе аминокислоту, содержащую группу -N3, не встречающуюся в природе аминокислоту, содержащую ацетиленовую группу, или не встречающуюся в природе аминокислоту, содержащую группу -NH-C(=O)-СН2-Br или группу -(СН2)m-малеимид, где m обозначает 1-10.
Согласно некоторым вариантам изобретения в пептидном продукте поверхностно-активное вещество представляет собой ПАВ класса 1-алкилгликозидов. Согласно некоторым вариантам пептидного продукта ПАВ связано с пептидом амидной связью.
Согласно некоторым вариантам изобретения в пептидном продукте ПАВ Х представляет собой 1-эйкозил бета-D-глюкуроновую кислоту, 1-октадецил бета-D-глюкуроновую кислоту, 1-гексадецил бета-D-глюкуроновую кислоту, 1-тетрадецил бета-D-глюкуроновую кислоту, 1-додедецил бета-D-глюкуроновую кислоту, 1-децил бета-D-глюкуроновую кислоту, 1-октил бета-D-глюкуроновую кислоту, 1-эйкозил бета-D-диглюкуроновую кислоту, 1-октадецил бета-D-диглюкуроновую кислоту, 1-гексадецил бета-D-диглюкуроновую кислоту, 1-тетрадецил бета-D-диглюкуроновую кислоту, 1 додецил бета-D-диглюкуроновую кислоту, 1-децил-бета-D-диглюкуроновую кислоту, 1-октил-бета-D-диглюкуроновую кислоту, или функционализованную 1-эйкозил бета-D-глюкозу, 1-октадецил бета-D-глюкозу, 1-гексадецил бета-D-глюкозу, 1-тетрадецил бета-D-глюкозу, 1-додецил бета-D-глюкозу, 1-децил бета-D-глюкозу, 1- октил бета-D-глюкозу, 1-эйкозил бета D-мальтозид, 1-октадецил бета-D-мальтозид, 1-гексадецил бета-D-мальтозид, 1-додецил бета-D-мальтозид, 1-децил бета-D-мальтозид, 1-октил бета-D-мальтозид и т.п., и пептидный продукт получают за счет образования связи между вышеуказанными группами и группой на пептиде (например, между -СООН группой в вышеуказанных группах и аминогруппой пептида).
Согласно некоторым вариантам изобретения в пептидном продукте U обозначает концевую аминокислоту пептида. Согласно некоторым вариантам изобретения в пептидном продукте U обозначает неконцевую аминокислоту пептида. Согласно некоторым вариантам изобретения в пептидном продукте U обозначает природную D- или L-аминокислоту. Согласно некоторым вариантам изобретения в пептидном продукте U обозначает не встречающуюся в природе аминокислоту. Согласно некоторым вариантам изобретения в пептидном продукте U выбран из Lys, Cys, Orn, или неприродной аминокислоты, содержащей функциональную группу, используемую для ковалентного связывания с ПАВ X.
Согласно некоторым вариантам изобретения в пептидном продукте функциональная группа, используемая для ковалентного связывания пептида с ПАВ X, представляет собой -NH2, -SH, -ОН, -N3, галогенацетильную, -(СН2)m-малеимидную (где m обозначает 1-10) или ацетиленовую группу.
Согласно некоторым вариантам изобретения функциональные группы боковых цепей двух различных аминокислотных остатков связываются с образованием циклического лактама. Например, согласно некоторым вариантам изобретения боковая цепь Lys образует циклический пактам с боковой цепью Glu. Согласно некоторым вариантам изобретения такие лактамные структуры обращаются и образуются из Glu и Lys. Известно, что в некоторых случаях такие лактамные связи стабилизируют альфа-спиральные участки структуры в пептидах (Condon, S.M., et al. (2002) Bioorg Med Chem 10: 731-736; Murage, E.N., et al (2008) Bioorg Med Chem 16: 10106-12); Murage, E.N., et al. (2010) J Med Chem 53: 6412-20). Согласно некоторым вариантам изобретения цистеиновые остатки могут связываться с образованием дисульфидных мостиков, при этом достигается сходное по форме конформационное ограничение и содействие образованию спиральных структур (Li, Y., et al. (2011) Peptides 32: 1400-1407). Согласно некоторым вариантам изобретения функциональные группы боковых цепей двух различных аминокислотных остатков связываются с образованием гетероцикла за счет "клик-реакции" между азидной группой в боковой цепи и алкином, при этом достигается сходное по форме конформационное ограничение и образуются стабилизированные спиральные конформации (Le Chevalier Isaad A., et al. (2009) J Peptide Sci 15: 451-4).
Согласно некоторым вариантам изобретения пептидный продукт, содержащий связанный ковалентной связью алкилгликозид, представляет собой модифицированный глюкагон или его аналог. Согласно некоторым из таких вариантов пептидный продукт представляет собой связанную ковалентной связью 1-O-алкил β-D-глюкуроновую кислоту, а пептид является аналогом глюкагона.
Согласно некоторым вариантам изобретения пептидный продукт, содержащий связанный ковалентной связью алкилгликозид, представляет собой модифицированный за счет ковалентного связывания GLP-1 или его аналог. Согласно некоторым из таких вариантов пептидный продукт представляет собой аналог GLP-1.
Согласно некоторым вариантам изобретения пептидный продукт Формулы I-A имеет структуру Формулы III-A
аа1-аа2-аа3-аа4-аа5-аа6-аа7-аа8-аа9-аа10-aa11-aa12-aa13-aa14-aa15-aa16-aa17-aa18-aa19-aa20-aa21-aa22-aa23-aa24-aa25-aa26-aa27-aa28-aa29-Z Формула III-A (SEQ. ID. NO. 2)
в которой:
Z обозначает ОН или -NH-R3, где R3 обозначает Н, или С1-С12 замещенный или незамещенный алкил, или цепь PEG (ПЭГ) протяженностью менее 10 Да;
aa1 обозначает His, N-Ac-His, pGlu-His или N-R3-His;
аа2 обозначает Ser, Ala, Gly, Aib, Ac4c или Ас5с;
аа3 обозначает Gln или Cit;
аа4 обозначает Gly или D-Ala;
аа5 обозначает Thr или Ser;
аа6 обозначает Phe, Trp, F2Phe, Me2Phe или Nal2;
аа7 обозначает Thr или Ser;
aa8 обозначает Ser или Asp;
аа9 обозначает Asp или Glu;
аа10 обозначает Tyr, Leu, Met, Nal2, Bip или Bip2EtMeO;
аа11 обозначает Ser, Asn или U;
aa12 обозначает Lys, Glu, Ser, Arg или U(X);
aa13 отсутствует или обозначает Tyr, Gln, Cit или U(X);
aa14 отсутствует или обозначает Leu, Met, Nle или U(X);
aa15 отсутствует или обозначает Asp, Glu или U(X);
aa16 отсутствует или обозначает Ser, Gly, Glu, Aib, Ac5c, Lys, Arg, or U(X);
aa17 отсутствует или обозначает Arg, hArg, Gln, Glu, Cit, Aib, Ac4c, Ac5c или U(X);
aa18 отсутствует или обозначает Arg, hArg, Ala, Aib, Ac4c, Ac5c или U(X);
aa19 отсутствует или обозначает Ala, Val, Aib, Ac4c, Ac5c или U(X);
aa20 отсутствует или обозначает Gln, Lys, Arg, Cit, Glu, Aib, Ac4c, Ac5c или U(X);
аа21 отсутствует или обозначает Asp, Glu, Leu, Aib, Ac4c, Ac5c или U(X);
аа22 отсутствует или обозначает Phe, Trp, Nal2, Aib, Ac4c, Ac5c или U(X);
аа23 отсутствует или обозначает Val, Не, Aib, Ac4c, Ac5c или U(X);
аа24 отсутствует или обозначает Gln, Ala, Glu, Cit или U(X);
аа25 отсутствует или обозначает Trp, Nal2 или U(X);
аа26 отсутствует или обозначает Leu или U(X);
аа27 отсутствует или обозначает Met, Val, Nle, Lys или U(X);
аа28 отсутствует или обозначает Asn, Lys или U(X);
аа29 отсутствует или обозначает Thr, Gly, Aib, Ac4c, Ac5c или U(X);
где любые две из aa1-aa29, необязательно, циклизуются за счет своих боковых цепей с образованием лактамной связи; и
при условии, что одна или по меньшей мере одна из аминокислот aa16, aa17, aa18, aa19, аа20, аа21, aa22, аа23, аа24, aa25, аа26, аа27, aa28 or aa29 представляет собой природную или не встречающуюся в природе (неприродную) аминокислоту U, ковалентно связанную с X.
Согласно некоторым вариантам изобретения пептидный продукт Формулы I-A имеет структуру Формулы III-B:
His1-aa2-aa3-Gly4-Thr5-aa6-Thr7-Ser8-Asp9-aa10-aa11-aa12-aa13-aa14-aa15-aa16-aa17-aa18-aa19-aa20-aa21-aa22-aa23-Z Формула III-B (SEQ. ID. NO. 3)
В которой:
Z обозначает ОН или -NH-R3, где R3 обозначает Н или замещенный или незамещенный С1-С12 алкил; или цепь PEG (ПЭГ) протяженностью менее 10 Да;
аа2 обозначает Ser, Ala, Gly, Aib, Ac4c или Ас5с;
аа3 обозначает Gln или Cit;
аа6 обозначает Phe, Trp, F2Phe, Me2Phe, MePhe или Nal2;
аа10 обозначает Tyr, Leu, Met, Nal2, Bip или Bip2EtMeO;
аа11 обозначает Ser, Asn или U(X);
aa12 обозначает Lys, Glu, Ser или U(X);
aa13 отсутствует или обозначает Tyr, Gln, Cit или U(X);
aa14 отсутствует или обозначает Leu, Met, Nle или U(X);
aa15 отсутствует или обозначает Asp, Glu или U(X);
aa16 отсутствует или обозначает Ser, Gly, Glu, Aib, Ac4c, Ас5с, Lys, R или U(X);
aa17 отсутствует или обозначает Arg, hArg, Gln, Glu, Cit, Aib, Ac4c, Ас5с или U(X);
aa18 отсутствует или обозначает Arg, hArg, Ala, Aib, Ac4c, Ас5с или U(X);
aa19 отсутствует или обозначает Ala, Val, Aib, Ac4c, Ас5с или U(X);
aa20 отсутствует или обозначает Gln, Lys, Arg, Cit, Glu, Aib, Ac4c, Ас5с или U(X);
aa21 отсутствует или обозначает Asp, Glu, Leu, Aib, Ac4c, Ас5с или U(X);
aa22 отсутствует или обозначает Phe, Aib, Ac4c, Ас5с или U(X)
аа23 отсутствует или обозначает Val, Ile, Aib, Ac4c, Ас5с или U(X);
где любые две из aa1-aa23, необязательно, циклизуются за счет своих боковых цепей с образованием лактамной связи; и
при условии, что одна, или по меньшей мере одна из аминокислот aa16, aa17, aa18, aa19, аа20, aa21, aa22, аа23 или аа24 представляет собой природную или не встречающуюся в природе (неприродную) аминокислоту U, ковалентно связанную с X.
Согласно некоторым вариантам изобретения в соединениях Формулы I-A, III-A, III-В или Формулы V U обозначает любую линкерную аминокислоту по настоящему описанию.
Согласно некоторым вариантам изобретения в соединениях Формулы I-A, III-A, III-В или Формулы V аа12 обозначает лизин. Согласно некоторым вариантам изобретения в соединениях Формулы I-A, III-A, III-В или Формулы V аа14 обозначает лейцин.
Согласно некоторым вариантам изобретения в соединениях Формулы I-A, III-A, III-В или Формулы V aa18 обозначает лизиновый остаток, связанный с X.
Согласно некоторым вариантам изобретения в соединениях Формулы I-A, III-A, III-В или Формулы V аа17 обозначает остаток гомоаргинина (hArg).
Согласно некоторым вариантам изобретения в соединениях Формулы I-A, III-A, III-В или Формулы V aa17 обозначает глициновый остаток.
Согласно некоторым вариантам изобретения в соединениях Формулы I-A, III-A, III-В или Формулы V аа2 обозначает остаток Aib или Ас4с.
Согласно некоторым вариантам изобретения в соединениях Формулы I-A, III-A, III-В или Формулы V пептид содержит один или более остатков Aib.
Согласно некоторым вариантам изобретения в соединениях Формулы I-A, III-A, III-В или Формулы V пептид содержит один или более остатков Aib на С-конце.
Согласно некоторым вариантам изобретения пептидный продукт Формулы I-A, III-А, III-В или Формулы V имеет строение:
His1-aa2-Gln3-Gly4-Thr5-Phe6-Thr7-Ser8-Asp9-Tyr10-Ser11-Lys12-Tyr13-Leu14-Asp15-Aib16-aa17-Lys(N-омега-1′-алкил бета-D-глюкуронил)18-аа19-NH2; (SEQ. ID. NO. 318)
где
aa2 обозначает Aib или Ас4с;
aa17 обозначает Arg, hArg или Gln;
aa19 обозначает Aib, Ас4с или Ас5с; и
алкил обозначает C8-С20 линейную алкильную цепь.
Согласно некоторым вариантам изобретения пептидный продукт Формулы I-A, III-А, III-В или Формулы V имеет строение:
His1-aa2-Gln3-Gly4-Thr5-Phe6-Thr7-Ser8-Asp9-Tyr10-Ser11-Lys12-Tyr13-Leu14-Asp15-Aib16-aa17-Lys(N-омега-1′-алкил бета-D-глюкуронил)18-aa19-aa20-NP2; (SEQ. ID. NO.319)
где
аа2 обозначает Aib или Ас4с,
aa17 обозначает Arg, hArg или Gln,
aa19 и аа20, независимо, обозначают Aib, Ас4с или Ас5с; и
алкил обозначает C8-С20 линейную алкильную цепь.
Согласно некоторым вариантам изобретения пептидный продукт Формулы I-A, III-А, III-В или Формулы V имеет строение:
His1-aa2-Gln3-Gly4-Thr5-Phe6-Thr7-Ser8-Asp9-Tyr10-Ser11-Lys12-Tyr13-Leu14-Asp15-aa16-aa17-Lys(N-омега-1′-алкил бета-D-глюкуронил)18-аа19-NH2; (SEQ. ID. NO. 320)
где
aa2 обозначает Aib или Ас4с;
aa16 обозначает Aib или Ас4с;
aa17 обозначает Arg, hArg или Gln;
aa19 обозначает Aib, Ас4с или Ас5с; и
алкил обозначает C8 - C20 линейную алкильную цепь.
Согласно некоторым вариантам изобретения в соединениях Формулы I-A, III-A, III-В или Формулы V aa16 и аа20 циклизуются с образованием лактамной связи.
Согласно некоторым вариантам изобретения пептидный продукт Формулы I-A, III-А, III-В или Формулы V имеет строение:
His1-aa2-Gln3-Gly4-Thr5-Phe6-Thr7-Ser8-Asp9-Tyr10-Ser11-Lys12-Tyr13-Leu14-Asp15-aa16-aa17 Ala18-Ala19-aa20-Glu21-Phe22-Ile23-Lys(N-омега-1′-алкил бета-D-глюкуронил)24-Trp25-Leu26-aa27-Asn28-Thr29-NH2; (SEQ. ID. NO. 321)
где
aa2 обозначает Aib или Ас4с;
aa16 и аа20, каждый независимо, обозначают либо Lys, либо Glu и циклизуются за счет своих боковых цепей с образованием лактамной связи;
aa17 обозначает Arg, hArg или Gln;
аа27 обозначает Met или Nle; и
алкил обозначает C8-C20 линейную алкильную цепь.
Согласно некоторым вариантам изобретения пептидный продукт Формулы I-A, III-А, III-В или Формулы V имеет строение:
His1-aa2-Gln3-Gly4-Thr5-Phe6-Thr7-Ser8-Asp9-Tyr10-Ser11-Lys12-Tyr13-Leu14-Asp15-циклический(Glu16-Gln17-Ala18-Ala19-Lys20)-Glu21-Phe22-Ile23-Lys(N-омега-1′-алкил бета-D-глюкуронил)24-Trp25-Leu26-Met27-Asn28-aa29-NH2; (SEQ. ID. NO. 322)
где aa2 обозначает Aib или Ас4с, aa29 обозначает Thr, Aib, Ас4с или Ас5с, а 1′-алкильная группа выбрана из додецила, тетрадецила, гексадецила или октадецила;
и боковые цепи аминокислот в положении 16 и 20 циклизуются с образованием лактамного цикла в боковой цепи.
Согласно некоторым вариантам изобретения в соединениях Формулы I-A, III-A, III-В или Формулы V aa12 и aa16 циклизуются с образованием лактамной связи.
Согласно некоторым вариантам изобретения пептидный продукт Формулы I-A, III-А, III-В или Формулы V имеет строение:
His1-aa2-Gln3-Gly4-Thr5-Phe6-Thr7-Ser8-Asp9-Tyr10-Ser11-aa12-Tyr13-Leu14-Asp15-aa16-aa17-Lys(N-омега-1′-алкил бета-D-глюкуронил)18-аа19-аа20-NH2; (SEQ. ID. NO. 323)
где
аа2 обозначает Aib или Ас4с;
аа12 и aa16, каждый независимо, обозначают либо Lys, либо Glu и циклизуются за счет своих боковых цепей с образованием лактамной связи;
aa17 обозначает Arg, hArg;
aa19 и аа20, независимо, обозначают либо Aib, Ас4с, либо Ас5с; и
алкил обозначает C8-C20 линейную алкильную цепь.
Согласно некоторым вариантам изобретения пептидный продукт Формулы I-A, III-А, III-В или Формулы V имеет строение:
His1-Ac4c2-Gln3-Gly4-Thr5-Phe6-Thr7-Ser8-Asp9-Tyr10-Ser11-цикло(Glu12-Tyr13-Leu14-Asp15-Lys16)-aa17-Lys(N-омега-1′-алкил бета-D-глюкуронил)18-Aib19-Aib20-NP2; (SEQ. ID. NO. 324),
где
aa12 и aa16 циклизуются за счет своих боковых цепей с образованием лактамной связи;
aa17 обозначает Arg или hArg; и
алкил обозначает С12, С14, C16 или C18 линейную алкильную цепь.
Согласно некоторым вариантам изобретения пептидный продукт Формулы I-A, III-А, III-В или Формулы V имеет строение:
His1-aa2-Gln3-Gly4-Thr5-Phe6-Thr7-Ser8-Asp9-Tyr10-Ser11-aa12-Tyr13-Leu14-Asp15-aa16-aa17-Lys(N-омега-1′-алкил бета-D-глюкуронил)18-аа19-аа20-NH2; (SEQ. ID. NO. 325)
где
aa12 и aa16, каждый независимо, обозначает либо Lys, либо Glu
и aa12 и aa16 циклизуются за счет своих боковых цепей с образованиемлактамной связи;
aa17 обозначает Arg или hArg; aa19 и aa20, независимо, обозначает либо Aib, Ас4с, либо Ас5с; и
1′-алкильная группа выбрана из додецила, тетрадецила, гексадецила или октадецила.
Согласно некоторым вариантам изобретения пептидный продукт Формулы I-A, III-А, III-В или Формулы V имеет строение:
His1-aa2-Gln3-Gly4-Thr5-aa6-Thr7-Ser8-Asp9-Tyr10-Ser11-Lys12-Tyr13-Leu14-Asp15-Ser16-Aib17-Lys(N-омега-1′-додецил бета-D-глюкуронил)18-aa19-NH2; (SEQ. ID. NO. 326)
где аа2 обозначает Aib или Ас4с, аа6 обозначает Me2Phe, MePhe или Phe; и aa19 обозначает Aib, Ас4с или Ас5с.
Согласно некоторым вариантам изобретения пептидный продукт Формулы I-A, III-А, III-В или Формулы V имеет строение:
His1-aa2-Gln3-Gly4-Thr5-aa6-Thr7-Ser8-Asp9-Tyr10-Ser11-Lys12-Tyr13-Leu14-Asp15-Ser16-aa17-Lys(N-омега-1′-додецил бета-D-глюкуронил)18-aa19-aa20-NH2; (SEQ. ID. NO. 327),
где аа2 обозначает Aib или Ас4с, аа6 обозначает Me2Phe, MePhe или Phe; аа17 обозначает Arg или hArg и aa19 или аа20 обозначает Aib, Ас4с или Ас5с.
Согласно некоторым вариантам изобретения пептидный продукт Формулы I-A, III-А, III-В или Формулы V имеет строение:
His1-Aib2-Gln3-Gly4-Thr5-Phe6-Thr7-Ser8-Asp9-Tyr10-Ser11-Lys12-Tyr13- Leu14-Asp15-цикло(Glu16-Arg17-Ala18-Ala19-Lys20)-Lys(N-омега-1′-алкил бета-D-глюкуронил)21-Phe22-aa23-NH2; (SEQ. ID. NO. 328)
где аа23 обозначает Aib, Ас4с или Ас5с, а 1′-алкильная группа выбрана из додецила, тетрадецила, гексадецила или октадецила.
Согласно некоторым вариантам изобретения пептидный продукт Формулы I-A, III-А, III-В или Формулы V имеет строение:
His1-aa2-Gln3-Gly4-Thr5-aa6-Thr7-Ser8-Asp9-Tyr10-Ser11-aa12-Tyr13-Leu14-Asp15-aa16-aa17-aa18-Ala19-aa20-Lys(N-омега-1′-алкил бета-D-глюкуронил)21-Phe22-аа23-NH2; (SEQ. ID. NO. 329)
где
аа2 обозначает Aib или Ас4с:
аа6 обозначает Me2Phe, MePhe или Phe;
aa12 и aa16, каждый независимо, обозначает либо Lys, либо Glu;
a aa16 и аа20 циклизуются за счет своих боковых цепей с образованием лактамной связи;
aa17 обозначает Arg, hArg или Gln;
aa18 обозначает Aib или Аlа;
аа23 обозначает Aib, Ас4с или Ас5с, а 1′-алкильная группа выбрана из додецила, тетрадецила, гексадецила или октадецила.
Согласно некоторым вариантам изобретения пептидный продукт Формулы I-A, III-А, III-В или Формулы V имеет строение:
His1-aa2-Gln3-Gly4-Thr5-aa6-Thr7-Ser8-Asp9-Tyr10-Ser11-аа12-Tyr13-Leu14-Asp15-aa16-aa17-Lys(N-омега-1′-алкил бета-D-глюкуронил)18-aa19-aa20-NH2; (SEQ. ID. NO. 330)
где
аа2 обозначает Aib или Ас4с:
аа6 обозначает Phe;
aa12 и aa16, каждый независимо, обозначает либо Lys, либо Glu; и aa12 и aa16 циклизуются с помощью своих боковых цепей с образованием лактамной связи;
aa17 обозначает Arg или hArg;
aa19 обозначает Aib, Ас4с или Ас5с;
аа20 обозначает Aib, Ас4с или Ас5с, а 1′-алкильная группа выбрана из додецила, тетрадецила, гексадецила или октадецила.
Согласно некоторым вариантам изобретения в любом соединении Формулы I-A, Формулы III-A, Формулы III-B или Формулы V Х содержит додецильную алкильную цепь.
Согласно некоторым вариантам изобретения пептидный продукт представляет собой биологически активный продукт, который связывается с GLP1R и/или с GLCR.
Согласно конкретному варианту изобретения пептидные продукты Формулы I-A, III-A, III-B или Формулы V по описанию выше и ниже имеют нижеприведенную структуру:
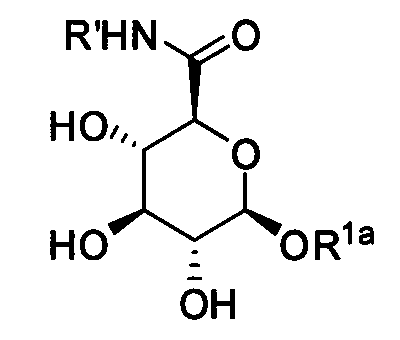
где R1a обозначает C1-C20 алкильную цепь по описанию в Таблице 1 на Фигуре 1, R′ обозначает пептид по описанию в Таблице 1 на Фигуре 1 и в Таблице 2 на Фигуре 2, W2 в Формуле I-A обозначает -O-, a W1 в Формуле I-A обозначает -(C=O)NH- и является частью амидной связи с пептидом R′. Согласно некоторым из таких вариантов изобретения R1a обозначает С6-С20 алкильную цепь. Согласно некоторым из таких вариантов изобретения R1a обозначает C8-C20 алкильную цепь. Согласно некоторым из таких вариантов изобретения R1a обозначает С12-С20 алкильную цепь. Согласно некоторым из таких вариантов изобретения R1a обозначает C12-C16 алкильную цепь.
Согласно описанным выше изобретениям аминогруппа аминокислоты и/или пептида R′ (например, аминогруппа аминокислотного остатка, такого как лизин, или лизинового остатка в пептиде R′) используется для образования ковалентной связи с соединением строения:
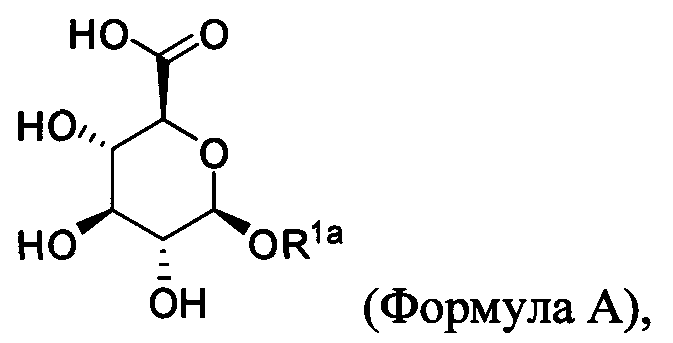
где R1a обозначает C1-C20 алкильную цепь по описанию выше и в Таблице 1 на Фигуре 1 и в Таблице 2 на Фигуре 2.
В таких случаях аминокислотный остаток, содержащий аминогруппу (например, лизин в пептиде R′), который используется для образования ковалентной связи с описанным выше соединением А, представляет собой линкерную аминокислоту U, которая связывается с ПАВ X, имеющим структуру, представленную Формулой А. Соответственно, в качестве примера, Lys(C12) в Таблице 1 на Фигуре 1 или в Таблице 2 на Фигуре 2 имеет следующую структуру:
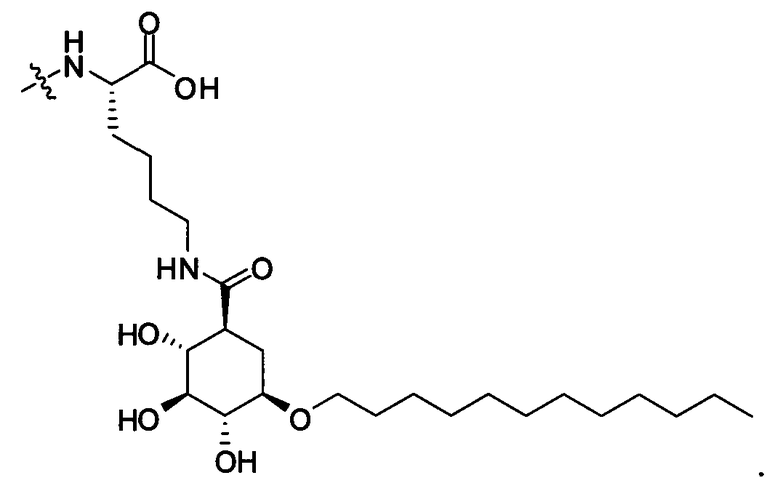
Также в объеме вариантов изобретения, представленных в настоящей заявке, рассматриваются пептидные продукты Формулы I-A, полученные при использовании ПАВ на основе мальтоуроновой кислоты путем связывания по любой или по обеим карбоксильным группам. Так, в качестве примера, пептиды в Таблице 1 на Фигуре 1 или в Таблице 2 на Фигуре 2 содержат лизиновый (аминокислотный) линкер, связанный с ПАВ Х на основе мальтоуроновой кислоты и имеющий структуру:
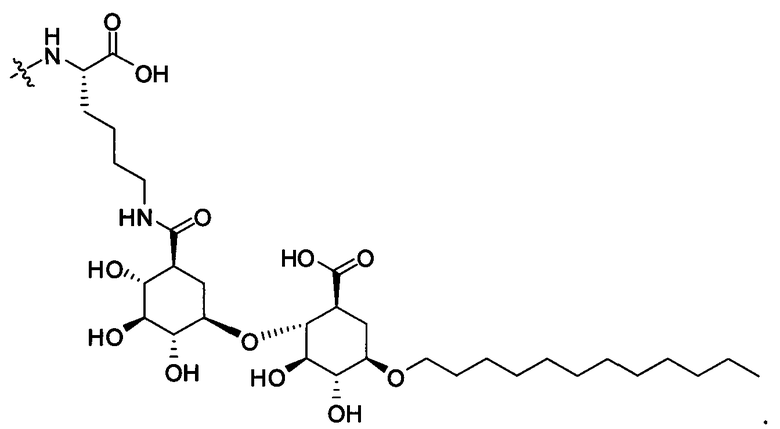
В одном варианте изобретения соединения Формулы I-A получают, связывая лизин с группой X, с последующим связыванием дополнительных аминокислотных остатков, и/или пептиды связываются с соединением лизин-Х с образованием соединения Формулы I-A. Очевидно, что согласно другому варианту изобретения для связывания с ПАВ Х и для связывания с другой(ими) аминокислотой/пептидами с образованием соединений Формулы I-A также могут применяться другие природные и неприродные аминокислоты по настоящему описанию. Согласно другому варианту изобретения соединения Формулы I-A получают путем связывания полноразмерного или неполноразмерного пептида с группой Х при необходимости с последующим связыванием дополнительных аминокислотных остатков, и/или пептиды связываются с образованием соединений Формулы I-A.
Согласно конкретному варианту изобретения предусматривается соединение, выбранное из соединений из Таблицы 1 на Фигуре 1 или Таблицы 2 на Фигуре 2.
Настоящее изобретение предусматривает также фармацевтические композиции, содержащие терапевтически эффективное количество пептидного продукта по описанию выше, или его приемлемой соли, и по меньшей мере один фармацевтически приемлемый носитель или эксципиент.
В фармацевтических композициях в соответствии с некоторыми вариантами изобретения носитель представляет собой носитель на водной основе. Согласно некоторым вариантам изобретения носитель в фармацевтических композициях представляет собой неводный носитель. Согласно некоторым вариантам изобретения неводный носитель в фармацевтических композициях представляет собой растворитель, подобный гидрофторалкану, который может содержать субмикронную безводную α-лактозу или другие эксципиенты.
В объем вариантов настоящего изобретения входит реакция аминокислоты и/или пептида, содержащего линкерную аминокислоту U, несущую нуклеофил, и группы X, содержащей уходящую группу или функциональную группу, которую можно активировать таким образом, чтобы она стала содержать уходящую группу, например, карбоксильную группу или любую другую реакционноспособную группу, тем самым способствуя ковалентному связыванию аминокислоты и/или пептида с ПАВ Х за счет линкерной аминокислоты U с образованием пептидного продукта Формулы I-A.
Также в объем вариантов по настоящему изобретению входит реакция аминокислоты и/или пептида, содержащего линкерную аминокислоту U, несущую уходящую группу или функциональную группу, которую можно активировать таким образом, чтобы она стала содержать уходящую группу, например, карбоксильную группу или любую другую реакционноспособную группу, и группы X, содержащей нуклеофильную группу, тем самым способствуя ковалентному связыванию аминокислоты и/или пептида с ПАВ Х за счет линкерной аминокислоты U с образованием пептидного продукта Формулы I-A.
Согласно одному варианту изобретения соединения Формулы I-A получают реакцией линкерной аминокислоты U с Х с последующим присоединением других (дополнительных) остатков к U с образованием пептидного продукта Формулы I-A. Согласно альтернативному варианту изобретения соединения Формулы I-A получают реакцией соответствующего пептида, содержащего линкерную аминокислоту U, с Х с последующим необязательным присоединением дополнительных остатков к U с образованием пептидного продукта Формулы I-A.
Помимо этого, в настоящей заявке предусматриваются способы синтеза пептидных продуктов, описанных выше, включающие последовательные стадии:
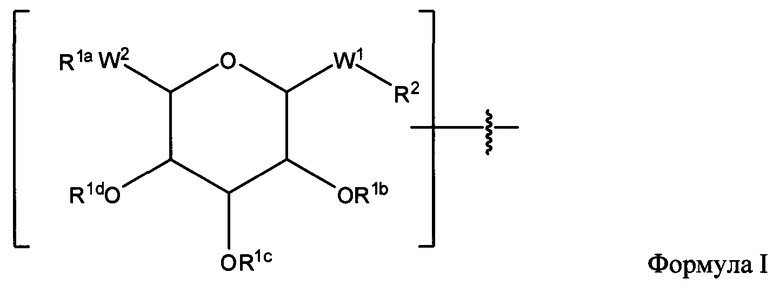
(а) Взаимодействие пептида с интермедиатом, а именно, с соединением Формулы IV:
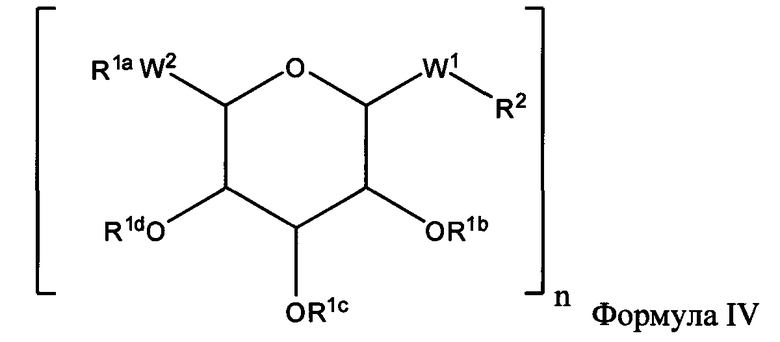
где:
R1a, независимо, каждый раз обозначает связь, Н, уходящую группу, защитную группу, природную или неприродную аминокислоту, замещенную или незамещенную C1-С30 алкильную группу, замещенную или незамещенную алкоксиарильную группу или замещенную или незамещенную аралкильную группу;
R1b, R1c и R1d, каждый независимо, каждый раз обозначают связь, Н, уходящую группу, защитную группу, природную или неприродную аминокислоту с обратимо снимаемой защитной группой, замещенную или незамещенную C1-С30 алкильную группу, замещенную или незамещенную алкоксиарильную группу или замещенную или незамещенную аралкильную группу;
W1 обозначает -СН2-, -СН2-О-, -(С=O), -(C=O)-O-, -(C=O)-NH-, -(C=S)-, -(C=S)-NH- или -CH2-S-;
W2 обозначает -O-, -CH2- or -S-;
R2, независимо, каждый раз обозначает связь, Н, уходящую группу, защитную группу, природную или неприродную аминокислоту с обратимо снимаемой защитной группой, замещенную или незамещенную C1-С30 алкильную группу, замещенную или незамещенную алкоксиарильную группу или замещенную или незамещенную аралкильную группу, -NH2, -SH, С2-С4-алкен, С2-С4-алкин, -NH(C=O)-CH2-Br, -(CH2)m-малеимид или -N3;
n обозначает 1, 2 или 3;
m обозначает 1-10;
и
(б) необязательно, депротекция связанного пептида, полученного на стадии (а).
Согласно некоторым вариантам в способах по изобретению каждая природная или неприродная аминокислота, независимо, каждый раз обозначает линкерную аминокислоту с обратимо снимаемой защитной группой. Согласно некоторым вариантам в способах по изобретению каждая природная или неприродная аминокислота, независимо, каждый раз обозначает лизин с обратимо снимаемой защитной группой или свободный лизин.
Согласно некоторым вариантам в способах по изобретению пептид обозначает пептид Формулы II по описанию выше.
Согласно некоторым вариантам в способах по изобретению
n обозначает 1;
W1 обозначает -(C=O)-;
R1a обозначает замещенную или незамещенную C1-С30 алкильную группу, замещенную или незамещенную 1-алкоксиарильную группу или замещенную или незамещенную 1-аралкильную группу,
R2 обозначает лизин с обратимо снимаемой защитной группой, имеющий D- или L-конфигурацию.
Согласно некоторым вариантам в способах по изобретению
n обозначает 1;
W1 обозначает -(C=O)-;
R1a обозначает замещенную или незамещенную C8-С30 алкильную группу, замещенную или незамещенную 1-алкоксиарильную группу или замещенную или незамещенную 1-аралкильную группу,
R2 обозначает лизин с обратимо снимаемой защитной группой, имеющий D- или L-конфигурацию.
Согласно некоторым вариантам в способах по изобретению R1a обозначает октильную, децильную, додецильную, тетрадецильную или гексадецильную группу.
Согласно некоторым вариантам в способах по изобретению
n обозначает 1;
W1 обозначает -(C=O)-NH- или -(C=O)-O-;
R2 обозначает замещенную или незамещенную C1-С30 алкильную гидрофобную группу, замещенную или незамещенную 1-алкоксиарильную группу или замещенную или незамещенную 1-аралкильную группу,
R1a обозначает серин или треонин с обратимо снимаемой защитной группой, имеющий D- или L-конфигурацию.
Согласно некоторым вариантам в способах по изобретению R2 обозначает октильную, децильную, додецильную, тетрадецильную или гексадецильную группу.
Согласно некоторым вариантам в способах по изобретению
n обозначает 1;
m обозначает 1-6;
W1 обозначает -CH2-;
R1a обозначает замещенную или незамещенную C1-С30 алкильную гидрофобную группу, замещенную или незамещенную 1-алкоксиарильную группу или замещенную или незамещенную 1-аралкильную группу,
R2 обозначает -N3, NFb, -С2-алкин, -(СН2)m-малеимид, NH-(C=O)-CH2-Br или NH-(C=O)-CH2-I.
Согласно некоторым вариантам изобретения в Формуле IV
n обозначает 1;
W1 обозначает -(C=O)-O-;
R2 обозначает Н,
R1a обозначает замещенную или незамещенную C1-С30 алкильную гидрофобную группу.
Согласно некоторым вариантам в способах по изобретению W1 обозначает -(СH2)О. Согласно некоторым вариантам в способах по изобретению n обозначает 1. Согласно некоторым вариантам в способах по изобретению n обозначает 2, а первый гликозид связан со вторым гликозидом за счет связи между W2 первого гликозида и любой из групп OR1b, OR1c или OR1d второго гликозида.
Согласно некоторым вариантам в способах по изобретению n обозначает 3, первый гликозид связан со вторым гликозидом за счет связи между W2 первого гликозида и любой из групп OR1b, OR1c или OR1d второго гликозида, а второй гликозид связан с третьим гликозидом за счет связи между W2 второго гликозида и любой из групп OR1b, OR1c или OR1d третьего гликозида.
Согласно некоторым вариантам в способах по изобретению соединение Формулы IV представляет собой N-ε-(1′-алкил глюкуронил)-лизин с обратимо снимаемой защитной группой, имеющий D- или L-конфигурацию, где R1a обозначает замещенную или незамещенную C1-C20 алкильную цепь, замещенную или незамещенную 1-алкоксиарильную группу или замещенную или незамещенную 1-аралкильную группу.
Согласно некоторым вариантам в способах по изобретению соединение Формулы IV представляет собой N-ε-(1′-додецил β-D-глюкуронил)-лизин с обратимо снимаемой защитной группой, имеющий D- или L-конфигурацию.
Согласно некоторым вариантам в способах по изобретению депротекция включает обработку слабой кислотой и/или слабым основанием. Согласно некоторым вариантам в способах по изобретению депротекция включает применение сильных кислот.
Согласно некоторым вариантам способы по изобретению дополнительно включают стадии хроматографии, обессоливания интермедиатов обращенно-фазовой, высокоэффективной жидкостной хроматографией или ионообменной хроматографией интермедиатов.
Настоящее изобретение предусматривает фармацевтическую композицию, содержащую терапевтически эффективное количество пептидного продукта, описанного выше и ниже, или его приемлемую соль и по меньшей мере один фармацевтически приемлемый носитель или эксципиент.
Настоящее изобретение предусматривает способ лечения состояния, ассоциированного инсулинорезистентностью, включающий введение любого соединения по настоящей заявке нуждающемуся в этом субъекту.
Настоящее изобретение предусматривает лечение диабета, диабетической ретинопатии, диабетической нейропатии, диабетической нефропатии, заживления ран, лечения резистентности к инсулину, гипергликемии, гиперинсулинемии, метаболического синдрома, диабетических осложнений, повышенного уровня свободных жирных кислот или глицерина в крови, гиперлипидемии, ожирения, гипертриглицеридемии, атеросклероза, острого сердечно-сосудистого синдрома, инфаркта, ишемии-реперфузии или гипертонии, включающие введение терапевтически эффективного количества описанного выше и ниже пептидного продукта нуждающемуся в этом субъекту.
В настоящем изобретении предусматриваются способы увеличения массы тела в меньшей степени или стимулирования уменьшения массы тела, включающие введение нуждающемуся в этом субъекту терапевтически эффективного количества пептидного продукта, описанного в данной заявке.
В настоящем изобретении предусматриваются способы лечения у млекопитающих состояний, характеризующихся связанными с ожирением резистентностью к инсулину или метаболическим синдромом, включающие введение нуждающемуся в этом субъекту стимулирующего снижение веса или повышающего чувствительность к инсулину количества пептидного продукта, описанного в данной заявке.
Согласно некоторым вариантам изобретения состояние, подлежащее лечению, представляет собой метаболический синдром (Синдром X). Согласно некоторым вариантам изобретения состояние, подлежащее лечению, представляет собой диабет. Согласно некоторым вариантам изобретения состояние, подлежащее лечению, представляет собой гиперлипидемию. Согласно некоторым вариантам изобретения состояние, подлежащее лечению, представляет собой гипертонию. Согласно некоторым вариантам изобретения состояние, подлежащее лечению, представляет собой сосудистое заболевание, включающее атеросклероз, или системное воспаление, характеризующееся повышенным уровнем С-реактивного белка.
Согласно некоторым вариантам в способах по изобретению эффективное количество пептидного продукта для введения составляет от около 0.1 мкг/кг/день до около 100.0 мкг/кг/день, или от 0.01 мкг/кг/день до около 1 мг/кг/день или от 0.1 мкг/кг/день до около 50 мг/кг/день. Согласно некоторым вариантам изобретения способом введения пептидного продукта является вдувание в нос.
Подразумевается, однако, что конкретный уровень дозы и конкретная частота введения дозы для любого конкретного субъекта, нуждающегося в лечении, может меняться и зависит от различных факторов, включая активность конкретного применяемого соединения, метаболическую устойчивость и продолжительность действия этого соединения, возраст, массу тела, общее состояния здоровья, пол, диету, способ и время введения, скорость выведения (экскреции), комбинацию лекарственных средств, тяжесть конкретного состояния и лечение, которое проходит хозяин.
Настоящее изобретение предусматривает способы лечения метаболического синдрома, или составляющих его факторов, включающие введение нуждающемуся в этом субъекту терапевтически эффективного количества пептидного продукта, описанного выше. Согласно некоторым вариантам изобретения метаболический синдром прогрессировал с развитием диабета.
Также настоящее изобретение предусматривает модифицированный за счет ковалентного связывания GLCR и/или GLP1R связывающий пептид или его аналог, содержащий гидрофильную группу по настоящей заявке; и гидрофобную группу, ковалентно связанную с гидрофильной группой. Согласно конкретным вариантам изобретения модифицированный за счет ковалентного связывания пептидный и/или белковый продукт содержит гидрофильную группу, которая представляет собой сахарид, и гидрофобную группу, которая представляет собой C1-C20 алкильную цепь или аралкильную цепь.
Согласно одному варианту изобретение предусматривает способ химической модификации молекулы с помощью ковалентного связывания с ПАВ для усиления или обеспечения биологического действия композиции или молекулы, например, рецепторного связывания или ферментативной активности. Согласно некоторым вариантам изобретения молекула представляет собой пептид. Помимо этого, способ может дополнительно включать модификацию, содержащую ковалентное связывание молекулы в композиции с полимером, таким как полиэтиленгликоль.
Согласно другому варианту изобретения предусматривается способ снижения или исключения иммуногенности пептидного и/или белкового лекарства с помощью ковалентного связывания пептидной цепи по меньшей мере с одним алкилгликозидом, причем алкил содержит от 1 до 30 углеродных атомов.
Также предусматривается способ лечения состояний, ассоциированных с инсулинорезистентностью, включая, но без ограничения, ожирение, метаболический синдром, типа 2 диабет, гипертонию, атеросклероз и т.п., включающий введение лекарственной композиции, содержащей пептид, ковалентно связанный по меньшей мере с одним алкилгликозидом и доставляемый позвоночному, причем алкил имеет от 1 до 30 углеродных атомов, от 1 до 20 углеродных атомов или в интервале от 6 до 16 углеродных атомов, или от 6 до 18 углеродных атомов, и при этом ковалентное связывание алкилгликозида с пептидом повышает устойчивость, биодоступность и/или продолжительность действия лекарства.
Кроме того, в настоящем изобретении предусматривается применение пептидного продукта по данному описанию (например, пептидного продукта Формулы I-A, Формулы III-A, Формулы III-B или Формулы V) для получения лекарственного средства для лечения любого состояния по данному описанию.
Краткое описание фигур
Фигура 1 В Таблице 1 на Фигуре 1 показаны соединения, полученные способами по настоящему описанию. В описании представлены последовательности SEQ. ID. No. 1-3 и SEQ. ID. No. 318-343. Помимо этого, в Таблице 1 на Фигуре 1 представлены последовательности соединений от EU-А300 до EU-A425, имеющие порядковые номера SEQ. ID. NO. 4-129, соответственно, как показано в Таблице 1 на Фигуре 1. Соединения в Таблице 1 на Фигуре 1 и их соответствующие SEQ. ID. NOs, показанные в Таблице 1 на Фигуре 1, тем самым вводятся в настоящую заявку.
Фигура 2 В Таблице 2 на Фигуре 2 показаны соединения, полученные способами по настоящему описанию. В описании представлены последовательности SEQ. ID. Nos. l-3 и SEQ. ID. Nos. 318-343. Помимо этого, в Таблице 2 на Фигуре 2 представлены последовательности соединений от EU-A426 до EU-599, имеющие порядковые номера SEQ. ID. NOs. 130-317, соответственно, как показано в Таблице 2 на Фигуре 2. Соединения в Таблице 2 на Фигуре 2 и их соответствующие SEQ. ID. NOs, показанные в Таблице 2 на Фигуре 2, тем самым вводятся в настоящую заявку..
Фигура 3 На Фигуре 3 представлена полученная методом РСА (рентгеноструктурного анализа) кристаллическая структура (Runge, S., et al. (2008) J Biol Chem 283: 11340- 7) сайта связывания внеклеточного домена GLP-1 рецептора и иллюстрируются важнейшие элементы гидрофобного связывания рецептора и лиганда-4 (Val19*, Phe22*, Trp25*, Leu26*), которые в пептидах по изобретению имитируются и заменяются гидрофобным 1′-алкильным участком ПАВ (поверхностно-активного вещества).
Осуществление изобретения
В настоящей заявке описываются модифицированные с помощью ковалентного связывания пептиды и/или белки, обладающие более эффективными фармацевтическими свойствами. Также в настоящей заявке предусматриваются способы применения ковалентно модифицированных пептидов и/или белков для лечения расстройств, связанных с ожирением и метаболическим синдромом.
Согласно некоторым вариантам изобретения модифицированные пептиды и/или белки содержат пептид и/или белок, связанный ковалентной связью с гидрофильной группой, "головкой" (например, полиолом (например, сахаридом)); гидрофильная группа ковалентно связана с гидрофобной группой, "хвостом", тем самым образуя ПАВ. Согласно некоторым вариантам изобретения применение связанных с гидрофобной группой гликозидных ПАВ-фрагментов (например, алкилгликозидов) для ковалентной модификации пептидов или белков (например, родственных глюкагону или GLP-1 и т.п.) пролонгирует действие пептидов и/или белков по множеству механизмов, включая образование депо лекарственного средства в месте введения и связывание с гидрофобными белками-носителями. Согласно некоторым вариантам изобретения включение в пептидную и/или белковую структуру пространственного затруднения может предупредить сближение протеаз с пептидным и/или белковым продуктом и тем самым предотвратить протеолиз. Согласно некоторым вариантам изобретения модификация пептидов и/или белков с помощью ПАВ (например, ковалентное связывание ПАВ класса алкилгликозидов) по данному описанию повышает перенос через слизистый барьер. Соответственно, модификации пептидов и/или белков по настоящему описанию предоставляют желательные преимущества, включая, но без ограничения, защиту от протеолиза и замедленное движение от места введения, тем самым обеспечивая пролонгированные фармакокинетические характеристики (например, пролонгирование полупериода циркуляции tin) и повышенную биодоступность при чресслизистом (трансмукозальном) введении.
Согласно некоторым вариантам изобретения взаимодействие более эффективных пептидов и/или белков со своими рецепторами модифицируется благоприятным образом с помощью процессирования (усечения) последовательности, введения ограничения степеней свободы и/или включения пространственного затруднения. В настоящей заявке представлены новые алкилгликозидные реагенты, которые способствуют включению в модифицированные пептиды и/или белки как жесткости, так и пространственного затруднения. Согласно некоторым вариантам изобретения пространственное затруднение придает модифицированным пептидам и/или белкам по настоящей заявке рецепторную селективность. Согласно некоторым вариантам изобретения пространственное затруднение обеспечивает защиту от протеолиза.
Пептиды и белки подвергаются множеству физических и химических изменений, которые могут повлиять на активность и безопасность. Среди этих изменений агрегация, которая включает димеризацию, тримеризацию и образование агрегатов более высокого порядка, таких как амилоиды. Агрегация является главной проблемой, лежащей в основе многочисленных потенциально вредных эффектов лекарственных препаратов на основе пептидов и/или белков, включая потерю активности, измененную фармакокинетику, пониженную устойчивость или меньший срок годности (время хранения) продукта и индукцию нежелательной иммуногенности. На биодоступность и фармакокинетику самоассоциированного пептида может влиять размер агрегата и легкость нарушения нековалентных внутримолекулярных взаимодействий в месте подкожного введения (Maji, S.K., et al. (2008) PLoS Biol 6: el7). В некоторых случаях пептиды могут объединяться в подкожные депо, которые диссоциируют с t1/2 30 или более дней. Такое медленное растворение может привести к благоприятным эффектам, так как доставка в течение одного месяца при использовании одной sc (подкожной) инъекции обеспечивает такую низкую концентрацию в крови, что пептид оказывается неактивным in vivo. Так, в некоторых случаях гидрофобная агрегация препятствует биодоступности и эффективности пептида (Clodfelter, D.K., et al. (1998) Pharm Res 15: 254-262). Модифицированные пептидные продукты по настоящему описанию являются связанными с ПАВ и, необязательно, разработаны таким образом, чтобы, при необходимости, либо помешать агрегации, либо содействовать повышенной агрегации.
Обычно природные олигосахариды, которые связаны с белками ковалентной связью, не обладают свойствами ПАВ. Согласно некоторым вариантам изобретения пептидные и/или белковые продукты по настоящему описанию содержат связанный ковалентной связью сахарид и дополнительную гидрофобную группу, которая придает модифицированным пептидам свойства ПАВ, что позволяет регулировать биодоступность, иммуногенность и/или фармакокинетические свойства пептидов, модифицированных с помощью ПАВ.
Белки и пептиды, модифицированные с помощью олигосахаридов, описаны, например, в Jensen, K.J. and Brask, J. (2005) Biopolymers 80: 747-761, путем введения сахаридных или олигосахаридных структур с применением ферментативных (Gijsen, H.J., et al. (1996) Chem Rev 96: 443-474; Sears, P. and Wong, C.H. (1998) Cell Mol Life Sci 54: 223-252; Guo, Z. and Shao, N. (2005) Med Res Rev 25: 655-678) или химических методов (Urge, L., et al. (1992) Biochem Biophys Res Commun 184: 1125-1132; Salvador, L.A., et al. (1995) Tetrahedron 51: 5643-5656; Kihiberg, J., et al. (1997) Methods Enzymol 289: 221-245; Gregoriadis, G., et al. (2000) Cell Mol Life Sci 57: 1964-1969; Chakraborty, Т.К., et al. (2005) Glycoconj J 22: 83-93; Liu, M., et al. (2005) Carbohydr Res 340: 2111-2122; Payne, R.J., et al. (2007) J Am Chem Soc 129: 13527-13536; Pedersen, S.L., et al. (2010) Chembiochem 11: 366-374). Пептиды, а также белки были модифицированы с помощью гликозилирования (Filira, F., et al. (2003) Org Biomol Chem 1: 3059-3063); (Negri, L., et al. (1999) J Med Chem 42: 400-404); (Negri, L., et al. (1998) Br J Pharmacol 124: 1516-1522); Rocchi, R., et al. (1987) Int J Pept Protein Res 29: 250-261; Filira, F., et al. (1990) Int J Biol Macromol 12: 41-49; Gobbo, M., et al. (1992) Int J Pept Protein Res 40: 54-61; Urge, L., et al. (1992) Biochem Biophys Res Commun 184: 1125-1132; Djedaini-Pilard, F., et al. (1993) Tetrahedron Lett 34: 2457-2460; Drouillat, В., et al. (1997) Bioorg Med Chem Lett 7: 2247-2250; Lohof, E., et al. (2000) Angew Chem Int Ed Engi 39: 2761-2764; Gruner, S.A., et al. (2001) Org Lett 3: 3723-3725; Pean, С., et al. (2001) Biochim Biophys Acta 1541: 150-160; Filira, F., et al. (2003) Org Biomol Chem 1: 3059-3063; Grotenbreg, G.M., et al. (2004) J Org Chem 69: 7851-7859; Biondi, L., et al. (2007) J Pept Sci 13: 179-189; Koda, Y., et al. (2008) Bioorg Med Chem 16: 6286-6296; Yamamoto, Т., et al. (2009) J Med Chem 52: 5164-5175).
Однако, в вышеуказанных статьях не описывается дополнительная гидрофобная группа, связанная с олигосахаридом, соединенным с пептидом. Поэтому в настоящей заявке предусматриваются модифицированные пептиды и/или белки, включающие гидрофобную группу, связанную с сахаридом и/или олигосахаридом, который ковалентно связан с пептидом и/или белком, что позволяет регулировать биодоступность, иммуногенность и фармакокинетические свойства. Соответственно, в настоящей заявке также предусматриваются ПАВ реагенты, содержащие олигосахарид и гидрофобную группу, которые способствуют ковалентной модификации пептидов и/или белков, например, таких как глюкагон и/или GLP-1 и/или их аналоги.
В настоящей заявке предусматривается применение ПАВ на основе сахаридов в ковалентном связывании с пептидом для улучшения свойств пептидов и/или белков. Согласно некоторым вариантам изобретения модификация пептидов и/или белков с помощью ПАВ (например, ковалентное связывание ПАВ класса алкилгликозидов) по настоящему описанию повышает перенос через слизистый барьер. Согласно некоторым вариантам изобретения ковалентное связывание ПАВ с пептидным и/или белковым продуктом уменьшает или предупреждает агрегацию пептида и/или белка. Согласно некоторым вариантам изобретения ковалентно модифицированные пептиды и/или белки представляют собой глюкагон или GLP-1 пептиды, или их аналоги, которые модифицируются с целью улучшить их фармацевтические и медицинские свойства путем ковалентной модификации с помощью фрагментов алкилгликозид-ПАВ. В молекулах этих модифицированных с помощью ПАВ аналогов наблюдаются повышенные пространственные затруднения, которые препятствуют протеолизу, замедляют всасывание и клиренс из организма.
В некоторых случаях действие ПАВ является благотворным в том, что касается физических свойств или характеристик фармацевтических препаратов, но раздражающим для кожи и/или других тканей и, в частности, раздражающим для слизистых оболочек, таких как слизистые оболочки носа, полости рта, глаза, влагалища, прямой кишки, защечной и подъязычной областей. Кроме того, в некоторых случаях ПАВ денатурируют белки, аннулируя при этом их биологическую функцию. В силу того, что ПАВ оказывают вышеуказанное действие на критическую концентрацию мицеллообразования (CMC, ККМ), желательными являются ПАВ с низкой CMC с тем, чтобы их можно было эффективно использовать в фармацевтических препаратах в низких концентрациях или в малых количествах. Поэтому, согласно некоторым вариантам изобретения, ПАВ (например, алкилгликозиды), пригодные для модификации пептидов по настоящей заявке, имеют значение CMC в чистой воде или в водных растворах менее, примерно, 1 мМ. Только в качестве примера, ниже представлены некоторые значения CMC для алкилгликозидов в воде: октил мальтозид 19.5 мМ; децил мальтозид 1.8 мМ; додецил-β-D-мальтозид 0.17 мМ; тридецил мальтозид 0.03 мМ; тетрадецил мальтозид 0.01 мМ; сахарозы додеканоат 0.3 мМ. Следует иметь в виду, что подходящий ПАВ агент может иметь более высокое или более низкое значение CMC в зависимости от модифицируемого пептида и/или белка. Термин "критическая концентрация мицеллообразования" или "CMC" в настоящем описании обозначает концентрацию амфифильного компонента (алкилгликозида) в растворе, при которой начинается образование мицелл (сферических мицелл, цилиндрических мицелл, ламеллярных структур и т.д.) в растворе. Согласно некоторым вариантам изобретения алкилгликозиды додецил, тридецил и тетрадецил мальтозид или глюкозид, а также сахарозы додеканоат, тридеканоат и тетрадеканоат имеют пониженные значения CMC и применимы для модификации пептидов по настоящему описанию.
Инсулинорезистентность
Риски, связанные с продолжительной гипергликемией, включают повышенный риск микрососудистых осложнений, сенсорной нейропатии, инфаркта миокарда, удара, смертности, вызванной поражением макросудов и общей смертности. Диабет типа 2 также связан причинно-следственной связью с ожирением, другой глобальной эпидемией. В 2007 году по меньшей мере 232 миллиарда долларов было потрачено во всем мире на лечение и предупреждение диабета, причем три четверти этой суммы было потрачено в промышленно развитых странах на лечение длительных осложнений и на общую медицинскую помощь, такую как попытки предупредить микро- и макрососудистые осложнения. По оценкам в 2007 году непрямые затраты экономики Соединенных Штатов на диабет (инвалидность, снижение производительности труда и преждевременную смерть вследствие диабета) составляли 58 миллиарда долларов.
Ожирение приводит к инсулинорезистентности, пониженной способности клеток организма реагировать на стимуляцию инсулином вследствие меньшего числа рецепторов инсулина и более слабого взаимодействия этих рецепторов с важными системами внутриклеточной сигнализации. Помимо этого, состояние ожирения приводит к "метаболическому синдрому", совокупности заболеваний (инсулинорезистентности, гипертонии, атеросклерозу и т.д.) с очень большими медицинскими последствиями. Если диагноз "инсулинорезистентность" поставлен достаточно рано, клинические проявления сахарного диабета типа 2 можно предупредить или задержать, изменив образ жизни с целью уменьшить калорийность потребляемой пищи и содержание жира в организме и с помощью лекарственного лечения нормализовать гликемический контроль. Несмотря на инструкции по лечению, рекомендующие раннее резкое изменение образа жизни, многим пациентам не удается выполнить план по гликемическому контролю. На неудачу успешного сдерживания развития диабета типа 2 влияют многие факторы, включая психосоциальные и экономические причины и недостатки профилей эффективности, пригодности и переносимости имеющихся антидиабетических лекарственных препаратов. Пептидные и/или белковые продукты по настоящей заявке разработаны с целью преодолеть эти недостатки.
Эффект инкретина
Термин "эффект инкретина" применяется для описания явления, при котором глюкозная нагрузка, доставляемая перорально, вызывает заметно более высокую секрецию инсулина, чем та же самая глюкозная нагрузка, вводимая внутривенно. Этот эффект опосредуется по меньшей мере двумя гормонами семейства инкретинов, секретируемыми L-клетками кишечника. Глюкозозависимый инсулинотропный полипептид (GIP) и глюкагоноподобный пептид 1 (GLP-1) были идентифицированы как инкретины, и предполагается, что у здоровых субъектов до 70% секреторного выброса инсулина в ответ на прием пищи может быть обусловлено эффектом инкретина.
Обычно пептиды инкретины секретируются, при необходимости, в ответ на прием пищи и имеют короткий период полужизни в плазме вследствие расщепления ферментом дипептидил пептидазой IV (DPP-4). У людей с диабетом типа 2 восприимчивость к GLP-1 ослаблена, но инсулин-секреторный ответ может быть восстановлен с помощью фармакологических доз человеческого GLP-1 (Kieffer, T.J., et al. (1995) Endocrinology 136: 3585-3596). Кроме того, GLP-1 стимулирует восстановление и сохранение бета-клеток (Aaboe, К., et al. (2008) Diabetes Obes Metab 10: 994-1003). GLP-1 оказывает дополнительное благотворное действие, например, на сердечную функцию: например, он улучшает функцию левого желудочка (Sokos, G.G., et al. (2006) J Card Fail 12: 694-699) у людей. GLP-1 также замедляет опорожнение желудка у людей и снижает аппетит (Toft-Nlelsen, M.B., et al. (1999) Diabetes Care 22: 1137-1143).
Лечение больных диабетом метаболически устойчивыми аналогами GLP-1 продолжительного действия, описанное, например, в Drab, S.R. (2010) Pharmacotherapy 30: 609-624, страдает недостатками, связанными с удобством применения и риском развития панкреатита и тиреоидной карциномы. Аналоги GLP-1 обусловливают глюкозозависимую стимуляцию секреции инсулина и пониженный риск гипогликемии. Кроме того, в то время как ряд современных лекарственных средств против диабета вызывает увеличение веса, как описано ниже, аналоги GLP-1 вызывают насыщение и слабое увеличение веса. Поэтому, согласно некоторым вариантам, в настоящем изобретении предусматриваются аналоги GLP-1 пролонгированного действия, вводимые в низких дозах, что снижает побочные эффекты, вызываемые современными лекарственными средствами.
Известно, что ряд пептидных гормонов желудочно-кишечного тракта (ЖКТ) модулируют аппетит (Sanger, G.J. and Lee, К. (2008) Nat Rev Drug Discov 7: 241-254). Некоторые пептиды образуются в процессе тканеспецифического процессирования под действием ферментов (прогормон конвертазы; PC) генного продукта пропреглюкагона: например, глюкагон, GLP-1, глюкагоноподобный пептид-2 (GLP-2), глицентин и оксинтомодулин (ОХМ) (Drucker, D.J. (2005) Nat Clin Pract Endocrinol Metab 1: 22-31; Sinclair, E.M. and Drucker, D.J. (2005) Physiology (Bethesda) 20: 357- 365). GLP-1, GLP-2, глицентин и ОХМ совместно секретируются L-клетками ЖКТ под действием пищи. Или же препроглюкагон процессируется (РС2), продуцируя глюкагон в альфа-клетках панкреатических островков. Структура ОХМ представляет собой по существу глюкагон с С-концевым расширением из 8 остатков.
Помимо стимуляции биосинтеза инсулина и глюкозозависимой секреции инсулина, GLP-1 и его устойчивые миметики (например, баета (Byetta)) вызывают также умеренное снижение веса в исследованиях на животных моделях (Mack, С.М., et al. (2006) hit J Obes (Lond) 30: 1332-1340) и у больных диабетом типа 2 (DeFronzo, R.A., et al. (2005) Diabetes Care 28: 1092-1100; Buse, J.B., et al. (2010) Diabetes Care 33: 1255-1261). Инфузия глюкагона снижает потребление пищи у человека (Geary, N., et al. (1992) Am J Physiol 262: R975- 980), в то время как продолжительное действие глюкагона на жировую ткань также стимулирует липолиз (Heckemeyer, С.М., et al. (1983) Endocrinology 113: 270-276) и потерю в весе (Salter, J.M., et al. (1960) Metabolism 9: 753-768; Chan, E.K., et al. (1984) Exp Mol Pathol 40: 320-327). Глюкагон оказывает действие на энергетический метаболизм в широком диапазоне (Heppner, K.M., et al. (2010) Physiol Behav)). Глюкагон, или его аналоги, можно применять для диагностики временного паралича кишечника. Таким образом, по меньшей мере два из продуктов PC процессинга белка препроглюкагона связаны с насыщением и метаболическими эффектами.
У грызунов повторное (многократное) интраперитонеальное введение ОХМ, третьего продукта препроглюкагона, ассоциировано с понижением содержания белой жировой ткани и снижением веса по сравнению с контрольными животными (Dakin, C.L., et al. (2004) Endocrinology 145: 2687-2695). ОХМ снижал потребление пищи на 19.3% в процессе введения с помощью внутривенной инфузии людям с нормальным весом, и этот эффект сохранялся в течение более 12 час. после инфузии (Cohen, М.А., et al. (2003) J Clin Endocrinol Metab 88: 4696-4701). Лечение добровольцев в течение 4 недель привело к устойчивому эффекту насыщения и потере в весе, отражающем снижение содержания жировой ткани (Wynne, К., et al. (2005) Diabetes 54: 2390-2395).
По своей структуре ОХМ является гомологом GLP-1 и глюкагона и активирует как рецептор глюкагона (GCGR), так и рецептор GLP-1 (GLP1R), но с активностью, в 10-100 раз меньшей, чем одноименные лиганды. Кроме того, изучение взаимодействия ОХМ с GLP1R наводит на мысль, что он может оказывать действие на рекрутинг бета-аррестина, отличное от действия GLP-1 (Jorgensen, R., et al. (2007) J Pharmacol Exp Ther 322: 148-154), таким образом действуя как "смещенный" ("пристрастный") лиганд. Поиск уникального рецептора для ОХМ проводили многие годы, но так и не выявили его, и принято считать, что он действует за счет использования GLP1R и GCGR путей. Поэтому в настоящем изобретении предусматриваются методы ПАВ-модификации пептидов ЖКТ, которые содействуют стимуляции насыщения, потере в весе, ослаблению инсулинорезистентности и/или замедлению прогрессирования предиабета в диабет.
GLP-1
Ввиду сложного поведения и взаимодействия продуктов белка препроглюкагона в процессе действия на насыщение и метаболизм, описанные выше, сотрудники многих групп изучали зависимость активностей от структуры GLP-1 и глюкагона. Было показано, что на всем протяжении последовательности остатки допускают замену. Например, замена на Аlа вполне приемлема в N-концевой области GLP-1, в особенности, в остатках 2, 3, 5, 8, 11 и 12 (Adelhorst, К., et al. (1994) J Biol Chem 269: 6275-6278).
Было показано, что химерные аналоги, способные связываться с GLP1R и GLCR, можно получить прививкой С- концевых остатков GLP-1 на N-конце глюкагона (Hjorth, S.A., et al. (1994) J Biol Chem 269: 30121-30124). Остаток в положении 3 (кислый Glu в GLP1 или нейтральный Gln в глюкагоне или ОХМ) снижает аффинность глюкагона (Runge, S., et al. (2003) J Biol Chem 278: 28005-28010) или ОХМ (Pocai, A., et al. (2009) Diabetes 58: 2258-2266) к G1P1R. Было изучено влияние обработки стабилизированными аналогами GLP-1 или глюкагона или ОХМ с Gln в положении 3 на метаболический профиль животных (Day, J.W., et al. (2009) Nat Chem Biol 5: 749-757; Druce, M.R., et al. (2009) Endocrinology 150: 1712-1722; Pocai, A., et al. (2009) Diabetes 58: 2258-2266). Были созданы такие аналоги, оказывающие агонистическое действие как на GLP1R, так и на GCGR (Day, J.W., et al. Заявка на патент США 2010/0190701 A1).
Химерные аналоги должны оказывать нужное действие на исходные гормоны, действуя на их рецепторы и, следовательно, аналогичное действию ОХМ, который, по-видимому, действует как на GLP-1R, так и на GLCR: т.е. вызывает глюкозозависимую секрецию инсулина и насыщение в сочетании с липолизом и повышенным сжиганием жира благодаря глюкагону. Было показано, что аналоги вызывают нужные эффекты снижения веса и повышенного сжигания жира. Такой профиль кажется привлекательным при лечении ожирения, но при лечении ожирения большой проблемой является соблюдение предписанного режима терапии. Хотя известные в настоящее время полноразмерные аналоги глюкагона и ОХМ, соответственно, с аффинностью как к GLP1R, так и к GLCR могут привести к потере в весе, высокая биодоступность, фармацевтические свойства и удобная доставка пациентам для этих аналогов не оптимизированы, а это необходимо для оптимальных схем лекарственной терапии. Поэтому в настоящей заявке предусматриваются аналоги пептидов желудочно-кишечного тракта (например, GLP, ОХМ, глюкагона и т.п.), которые способствуют высокой биодоступности и/или продолжительным эффектам для достижения лучшего терапевтического результата при лечении состояний, таких как ожирение и/или диабет и/или метаболический синдром.
Другие факторы оптимизированного лечения метаболического синдрома и диабета с помощью ОХМ- подобных молекул относятся к длительности лечения и количеству действующего глюкагона. Например, постоянное лечение аналогами, которые активируют рецепторы GLP-1 и глюкагона (ОХМ фармалогический профиль), может привести с быстрой потере большой массы жировой ткани (Day, J.W., et al. (2009) Nat Chem Biol 5: 749-757), но оно также может быть причиной потери сухой мышечной массы (Kosinski, J.R., et al. (2012) Obesity (Silver Spring): dot: 10.1038/oby.2012.67), что не годится для лекарственных препаратов этого класса. Например, в научной статье Kosinski, J.R., et al., натуральный гормон Oxm вводится непрерывно в течение 14 дней с помощью мининасоса (минипомпы) Alzet, это приводит к снижению жировой массы на 30%, но также вызывает снижение сухой (тощей, мышечной) массы на 7%.
Известно, что глюкагон стимулирует гликогенолиз, липолиз и повышенное сжигание жира, но оказывает также катаболическое действие на мышцы. Успешное лечение с применением агента, который сочетает действие GLP-1 и глюкагона (ОХМ профиль), необходимо для того, чтобы вызвать оптимальное насыщение и усиленную глюкозозависимую секрецию инсулина под действием GLP-1 аналога в сочетании с количественно разумной активностью глюкагона (сжигание жира). Помимо этого, применение такого агента в интермиттирующем режиме обеспечивает нужный клинический профиль умеренной, непрерывной потери веса за счет потери жировой массы при минимальной потере тощей (сухой) массы. В настоящей заявке предусматриваются молекулы с нужной комбинацией GLP-1 и ОХМ действием, а также с регулируемым фармакокинетическим/фармакодинамическим профилем, способствующим оптимальному применению в терапии (например, для лечения метаболического синдрома, диабета, ожирения и т.п.).
Согласно одному варианту изобретения соединения Формулы I-A, III-A, III-B и Формулы V созданы таким образом, чтобы обеспечить либо глюкагоноподобную, либо GLP-1-подобную активность. Согласно другому варианту изобретения соединения Формулы I-A, III-A, III-B и Формулы V обеспечивают регулируемую активность. Например, в одном случает пептидные продукты по настоящей заявке (например, соединения в Таблице 1 на Фигуре 1 и в Таблице 2 на Фигуре 2) имеют значение ЕС50 менее, примерно, 500 нМ, предпочтительно, менее, примерно, 50 нМ, более предпочтительно, менее, примерно, 20 нМ по рецепторам как к глюкагону, так и к GLP-1. В другом примере пептидные продукты по настоящей заявке (например, соединения в Таблице 1 на Фигуре 1 и в Таблице 2 на Фигуре 2) являются более активными (например, значение ЕС50 менее 10 нМ, предпочтительно, менее 5 нМ, более предпочтительно, около 1 нМ) к GLP-1 рецептору и менее активными к рецептору глюкагона (например, ЕС50 менее 50 нМ, предпочтительно, менее 20 нМ, более предпочтительно, около 5 нМ) к рецептору глюкагона. Такая регулируемость биологической активности до некоторой степени способствует сохранению действия глюкагона в разумных пределах, тем самым содействуя сжиганию жира при одновременном сохранении положительного воздействия усиленной глюкозозависимой секреции инсулина. По своей структуре ОХМ является гомологом GLP-1 и глюкагона и активирует как рецептор глюкагона (GCGR), так и рецептор GLP-1 (GLP1R). Соответственно, согласно некоторым вариантам изобретения соединения Формулы I-A, Формулы III-A, Формулы III-B и Формулы V обеспечивают регулируемую ОХМ-подобную биологическую активность. Согласно некоторым конкретным вариантам изобретения пептидные продукты по настоящей заявке содержат пептид, имеющий аминокислотные остатки 1-17 из GLP-1 и/или его аналогов (например, аналогов, содержащих модифицированные неприродные замены, описанные в настоящей заявке, лактамные (циклические) связи, описанные в настоящей заявке, ПАВ-модификации, описанные в настоящей заявке, или их комбинацию). Согласно некоторым другим вариантам изобретения пептидные продукты по настоящему описанию содержат пептид, имеющий аминокислотные остатки 1-16 из GLP-1 и/или его аналогов (например, аналогов, содержащих модифицированные неприродные замены, описанные в настоящей заявке, лактамные (циклические) связи, описанные в настоящей заявке, ПАВ-модификации, описанные в настоящей заявке, или их комбинацию). Согласно другим вариантам изобретения пептидные продукты по настоящему описанию содержат пептид, имеющий аминокислотные остатки 1-18 из GLP-1 и/или его аналогов (например, аналогов, содержащих модифицированные неприродные замены, описанные в настоящей заявке, лактамные (циклические) связи, описанные в настоящей заявке, ПАВ-модификации, описанные в настоящей заявке, или их комбинацию). Помимо этого, пептидные продукты по настоящему описанию содержат один или более остатков (например, Aib, Ac4C), которые обеспечивают стабилизацию спиральных структур синтезированных соединений Формулы I-A, Формулы III-A, Формулы III-B, Формулы V и соединений в Таблице 1 на Фигуре 1 и в Таблице 2 на Фигуре 2.
Принято считать, что лиганды подсемейства глюкагона связываются со своими рецепторами как двухдоменные молекулы (по двум доменам), что обычно для ряда рецепторов класса В (класс секретина, рецепторы, сопряженные с G белком-(GPCR)). По общему мнению, в случае GLP-1 этими доменами являются N-концевая область от остатка 1, примерно, до остатка 16, которая связывается с верхними участками трансмембранных спиралей (околомембранная область), и С-концевая спираль протяженностью от остатка 17 до остатка 31, которая связывается с большим внеклеточным N-концевым удлиняющим участком (ECD) рецептора. Связывание этих лигандов основано на том факте, что усеченные (процессированные) по N-концу аналоги этих пептидных лигандов все еще могут сохранять значительную аффинность связывания и селективность именно к изолированной ECD области рецептора. Поэтому было высказано предположение, что N-концевая область отвечает за активацию рецептора, тогда как С-концевая область отвечает за связывание. В последнее время было показано, что короткие N-концевые аналоги GLP-1 могут являться как сильными связывающими агентами, так и активаторами рецепторов (Mapelli, С., et al. (2009) J Med Chem 52: 7788-7799; Haque, T.S, et al. (2010) Peptides 31: 950- 955; Haque, T.S., et al. (2010) Peptides 31: 1353-1360).
Помимо этого исследование методом РСА кристаллической структуры (Runge, S., et al. (2008) J Biol Chem 283: 11340-7) N-концевой области GLP1R с усеченными (процессированными) аналогами миметика GLP-1, эксендина-4 (баета, Byetta), связанного в этой области, показывает, что важнейшая лиганд-связывающая область в ECD обладает высокой гидрофобностью (Фигура 3). Последовательность эксендина-4 за пределами Glul5 как амфифильная спираль взаимодействует с этой в высокой степени гидрофобной областью (Val19*, Phe22*, Trp25*, Leu26*). Согласно одному варианту изобретения процессированные N-концевые фрагменты GLP-1 или глюкагона модифицируются так, чтобы связываться с GLCR, и ковалентно связываются с ПАВ. Гидрофобный 1′-алкильный участок ПАВ имитирует и заменяет С- концевую область нативного гормона/лиганда и повышает активность, эффективность и продолжительность действия пептида. Кроме того, такие аналоги, вследствие их малого размера, имеют основные преимущества, заключающиеся в том, что это уменьшает их сложность, стоимость синтеза и чувствительность к протеолизу. Помимо этого, пептиды меньшего размера легче всасываются через слизистую оболочку носа или кишечного барьера.
Гипогликемия представляет собой патологическое состояние с низким содержанием сахара, которое может быть опасным для жизни и тем чаще рассматривается как результат более агрессивного лечения повышенного содержания сахара в крови, чем больше пациентов подвергается интенсивному лечению инсулином. Гипогликемия наблюдается, когда уровни глюкозы в крови падают слишком низко для того, чтобы обеспечить достаточной энергией мозг и мышцы для активной работы организма. Глюкагон может применяться для лечения этого состояния и делает это, стимулируя печень расщеплять гликоген, вырабатывая глюкозу, и повышать уровни глюкозы до нормального значения. Для достижения этого искомого воздействия на уровни глюкозы в крови можно применять аналоги глюкагона, которые сохраняют способность активировать GLCR.
Аналоги GLP-1, которые активируют GLP1R, стимулируют продуцирование и, в присутствии повышенных уровней глюкозы в крови, высвобождают инсулин из поджелудочной железы. Это дает возможность эффективно регулировать и нормализовать уровни глюкозы в крови, что наблюдается при лечении современными продуктами, такими как эксенатид (Byetta®). Помимо этого, такие продукты, по-видимому, вызывают пониженный аппетит и замедляют движение пищи по желудочно-кишечному тракту. Следовательно, они эффективны при лечении диабета за счет использования многих механизмов. Аналоги, сочетающие эффекты глюкагона и GLP-1, которые активируют как GLCR, так и GLP1R, могут дать преимущество при лечении диабета за счет согласованного действия, способствующего подавлению аппетита, глюкозозависимому высвобождению инсулина, содействию защите от гипогликемии и ускорению сжигания жира.
Предполагается, что такие методы лечения гипергликемии, включая диабет, сахарный диабет типа I, сахарный диабет типа II или гестационный диабет, как инсулинозависимый, так и инсулиннезависимый, будут применимы для уменьшения осложнений диабета, включая нефропатию, ретинопатию и заболевание сосудов. Применение при сердечно-сосудистом заболевании охватывает как микроваскулярное, так и макроваскулярное заболевание (Davidson, M.H., (2011) Am J Cardiol 108[suppl]:33B-41B; Gejl, M., et al. (2012) J Clin Endocrinol Metab 97:doi:10.1210/jc. 2011-3456), и включает лечение инфаркта миокарда. Полагают, что такие методы снижения аппетита или стимулирования потери веса будут применяться для снижения массы тела, предупреждения увеличения массы тела или лечения ожирения, вызванного различными причинами, включая лекарственное ожирение, и для уменьшения осложнений, обусловленных ожирением, включая заболевание сосудов (ишемическую болезнь сердца, удар, болезнь периферических сосудов, ишемически-реперфузионные повреждения и т.д.), гипертонию, начальную стадию диабета типа II, гиперлипидемию и болезни скелетно-мышечной системы.
В данном контексте термин "аналоги глюкагона или GLP-1" включает все их фармацевтически приемлемые соли и сложные эфиры.
Пептиды и их аналоги
В одном аспекте изобретения пептиды, ковалентно модифицированные и пригодные для применения в способах по настоящему описанию, представляют собой процессированные аналоги глюкагона и/или родственного гормона GLP-1, включая, но без ограничения:
Глюкагон:
His1-Ser2-Gln3-Gly4-Thr5 Phe6-Thr7-Ser8-Asp9-Tyr10-Ser11-Lys12-Tyr13-Leu14-Asp15-Ser16-Arg17-Arg18-Ala19-Gln20-Asp21-Phe22-Val23-Gln24-Trp25-Leu26-Met27-Asn28-Thr29 (SEQ. ID. NO. 331)
Оксинтомодулин:
His1-Ser2-Gln3-Gly4-Thr5 Phe6-Thr7-Ser8-Asp9-Tyr10-Ser11-Lys12-Tyr13-Leu14-Asp15-Ser16-Arg17-Arg18-Ala19-Gln20-Asp21-Phe22-Val23-Gln24-Trp25-Leu26-Met27-Asn28-Thr29-Lys30-Arg31-Asn32-Arg33-Asn34-Asn35-Ile36-Ala37 (SEQ. ID. NO. 332)
GLP-1 (используется нумерация в последовательности глюкагона):
His1-Ala2-Glu3-Gly4-Thr5 Phe6-Thr7-Ser8-Asp9-Val10-Ser11-Ser12-Tyr13-Leu14-Glu15-Gly16-Gln17-Ala18-Ala19-Lys20-Glu21-Phe22-Ile23-Ala24-Trp25-Leu26-Val27-Lys28-Gly29-Arg30 (SEQ. ID. NO. 333)
Согласно некоторым вариантам изобретения пептидный продукт по настоящему описанию имеет структуру Формулы V:
где:
U обозначает связывающую аминокислоту;
Х обозначает остаток, связанный через ПАВ с боковой цепью U;
Z обозначает ОН или -NH-R3, где R3 обозначает Н или С1-C12-замещенный или незамещенный алкил;
aa1 обозначает His, N-Ac-His, pGlu-His или N-R3-His;
аа2 обозначает Ser, Ala, Gly, Aib, Ac4c или Ас5с;
аа3 обозначает Gln или Cit;
aa4 обозначает Gly или D-Ala;
aa5 обозначает Thr или Ser;
аа6 обозначает Phe, Trp, F2Phe, Me2Phe или Nal(2);
аа7 обозначает Thr или Ser;
aa8 обозначает Ser или Asp;
аа9 обозначает Asp или Glu;
аа10 обозначает Tyr, Leu, Met, Nal(2), Bip или Bip2EtMeO;
аа11 обозначает Ser, Asn или U(X);
aa12 обозначает Lys, Glu, Ser, Arg или U(X);
aa13 отсутствует или обозначает Tyr, Gln, Cit или U(X);
aa14 отсутствует или обозначает Leu, Met, Nle или U(X);
aa15 отсутствует или обозначает Asp, Glu или U(X);
aa16 отсутствует или обозначает Ser, Gly, Glu, Aib, Ас5с, Lys, Arg или U(X);
aa17 отсутствует или обозначает Arg, hArg, Gln, Glu, Cit, Aib, Ac4c, Ас5с или U(X);
aa18 отсутствует или обозначает Arg, hArg, Ala, Aib, Ac4c, Ас5с или U(X);
aa19 отсутствует или обозначает Ala, Val, Aib, Ac4c, Ас5с или U(X);
aa20 отсутствует или обозначает Gln, Lys, Arg, Cit, Glu, Aib, Ac4c, Ас5с или U(X);
aa21 отсутствует или обозначает Asp, Glu, Leu, Aib, Ac4c, Ас5с отсутствует или обозначает U(X);
аа22 отсутствует или обозначает Phe, Trp, Nal(2), Aib, Ас4с, Ас5с или U(X);
aa23 отсутствует или обозначает Val, He, Aib, Ас4с, Ас5с или U(X);
аа24 отсутствует или обозначает Gln, Ala, Glu, Cit или U(X);
aa25 отсутствует или обозначает Trp, Nal(2) или U(X);
аа26 отсутствует или обозначает Leu, U(X);
аа27 отсутствует или обозначает Met, Val, Nle, Lys или U(X);
аа28 отсутствует или обозначает Asn, Lys или U(X);
аа29 отсутствует или обозначает Thr, Gly, Aib, Ас4с, Ас5с или U(X);
аа30 отсутствует или обозначает Lys, Aib, Ас4с, Ас5с или U(X);
аа31 отсутствует или обозначает Arg, Aib, Ас4с, Ас5с или U(X);
аа32 отсутствует или обозначает Asn, Aib, Ас4с, Ас5с или U(X);
аа33 отсутствует или обозначает Arg, Aib, Ас5с или U(X);
аа34 отсутствует или обозначает Asn, Aib, Ас4с, Ас5с или U(X);
аа35 отсутствует или обозначает Asn, Aib, Ас4с, Ас5с или U(X);
аа36 отсутствует или обозначает Не, Aib, Ас4с, Ас5С или U(X);
аа36 отсутствует или обозначает Ala, Aib, Ас4с, Ас5С или U(X);
aа37 отсутствует или обозначает U(X);
при условии, что одна или по меньшей мере одна из аминокислот аа11-аа37 обозначает U(X).
В конкретных вариантах изобретения связывающая аминокислота U представляет собой диаминокислоту, подобную Lys или От, Х обозначает модифицированное ПАВ класса 1-алкилгликозидов, связанное с U, a Z обозначает ОН или -NH-R2, где R3 обозначает Н или C1-C12; или цепь ПЭГ протяженностью менее 10 Да.
Согласно некоторым вариантам изобретения пептидный продукт по настоящему описанию имеет структуру Формулы III-B:
где:
Z обозначает ОН или -NH-R3, где R3 обозначает Н или замещенный или незамещенный C1-C12 алкил; или цепь ПЭГ протяженностью менее 10 Da;
аа2 обозначает Ser, Ala, Gly, Aib, Ас4с или Ас5с;
аа3 обозначает Gln или Cit;
аа6 обозначает Phe, Trp, F2Phe, Me2Phe, MePhe или Nal2;
аа10 обозначает Tyr, Leu, Met, Nal2, Bip или Bip2EtMeO;
аа11 обозначает Ser, Asn или U;
aa12 обозначает Lys, Glu, Ser или U(X);
aa13 отсутствует или обозначает Tyr, Gln, Cit или U(X);
aa14 отсутствует или обозначает Leu, Met, Nle или U(X);
aa15 отсутствует или обозначает Asp, Glu или U(X);
aa16 отсутствует или обозначает Ser, Gly, Glu, Aib, Ac4c, Ac5c, Lys, R или U(X);
aa17 отсутствует или обозначает Arg, hArg, Gln, Glu, Cit, Aib, Ac4c, Ac5c или U(X);
aa18 отсутствует или обозначает Arg, hArg, Ala, Aib, Ac4c, Ac5c или U(X);
aa19 отсутствует или обозначает Ala, Val, Aib, Ac4c, Ac5c или U(X);
аа20 отсутствует или обозначает Gln, Lys, Arg, Cit, Glu, Aib, Ac4c, Ac5c или U(X);
aa21 отсутствует или обозначает Asp, Glu, Leu, Aib, Ac4c, Ac5c или U(X);
aa22 отсутствует или обозначает Phe, Aib, Ac4c, Ac5c или U(X);
aa23 отсутствует или обозначает Val, He, Aib, Ac4c, Ac5c или U(X);
где любые две из aa1-aa23, необязательно, циклизуются по своим боковым цепям с образованием лактамной связи; и
при условии, что одна, или по меньшей мере одна, аминокислота из aa16, aa17, aa18, aa19, аа20, aa21, aa22, аа23 или аа24 обозначает природную или неприродную аминокислоту U, ковалентно связанную с X.
Согласно некоторым конкретным вариантам изобретения в соединениях Формулы III-А, Формулы III-B и Формулы V Х имеет строение:
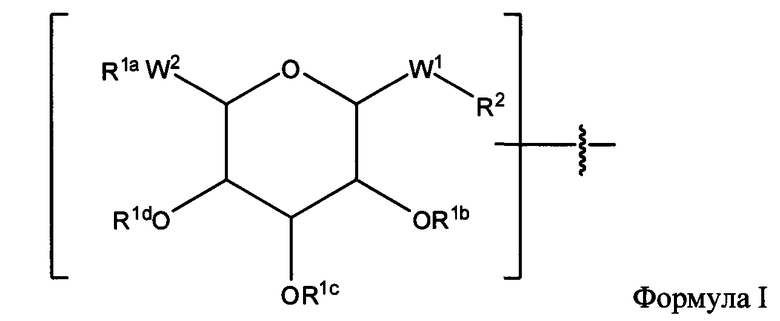
где:
R1a обозначает замещенную или незамещенную C1-С30 алкильную группу;
R1b, R1c и R1d обозначают Н;
W1 обозначает -(C=O)-NH-;
W2 обозначает -O-; и
R2 обозначает связь.
Согласно некоторым вариантам изобретения, описанным выше, R1a обозначает C1-C20 алкильную группу, C8-C20 алкильную группу, C12-C18 алкильную группу или C14-C18 алкильную группу.
Согласно некоторым вариантам в соединениях Формулы III-B U обозначает линкерную аминокислоту по настоящему описанию. В Таблице 1 на Фигуре 1 и в Таблице 2 на Фигуре 2 показаны некоторые примеры пептидов, которые ковалентно связаны с ПАВ по настоящему описанию.
В объеме вариантов настоящего изобретения рассматриваются пептидные продукты Формулы I-A, Формулы III-A, Формулы III-B или Формулы V, в которых пептидный продукт содержит одну, или более чем одну, ПАВ-группу (например, группу X, имеющую структуру, представленную Формулой I). Согласно одному варианту изобретения пептидный продукт Формулы I-A, Формулы III-A, Формулы III-B или Формулы V содержит одну поверхностно-активную (ПАВ) группу. Согласно другому варианту изобретения пептидный продукт Формулы I-A, Формулы III-A, Формулы III-B или Формулы V содержит две поверхностно-активные (ПАВ) группы. Согласно другому варианту изобретения пептидный продукт Формулы I-A, Формулы III-A, Формулы III-B или Формулы V содержит три поверхностно-активные (ПАВ) группы.
В настоящем описании обнаруживается (показана) важность определенных участков последовательности SEQ. ID. NO. 331 для лечения патологических состояний, ассоциированных с инсулинорезистентностью и/или сердечно-сосудистыми заболеваниями. Соответственно, в настоящем описании предусматривается способ лечения диабета у нуждающегося в этом субъекта, включающий введение нуждающемуся в этом субъекту терапевтически эффективного количества аналога глюкагона, содержащего аминокислотные остатки aa1-aa17 последовательности SEQ. ID. NO. 331.
Согласно другому варианту изобретения в настоящей заявке предусматривается способ лечения диабета у нуждающегося в этом субъекта, включающий введение нуждающемуся в этом субъекту терапевтически эффективного количества аналога глюкагона, содержащего аминокислотные остатки aa1-aa18 последовательности SEQ. ID. NO. 331.
Согласно другому варианту изобретения в настоящем описании предусматривается способ лечения диабета у нуждающегося в этом субъекта, включающий введение нуждающемуся в этом субъекту терапевтически эффективного количества аналога глюкагона, содержащего аминокислотные остатки aa1-aa19 последовательности SEQ. ID. NO. 331.
Согласно другому варианту изобретения в настоящем описании предусматривается способ лечения диабета у нуждающегося в этом субъекта, включающий введение нуждающемуся в этом субъекту терапевтически эффективного количества аналога глюкагона, содержащего аминокислотные остатки aa1-аа20 последовательности SEQ. ID. NO. 331.
Согласно дополнительному варианту изобретения введение указанного глюкагона по описанию выше вызывает потерю веса.
В настоящем описании обнаруживается (показана) важность определенных участков последовательности SEQ. ID. NO. 1 для лечения патологических состояний, ассоциированных с инсулинорезистентностью и/или сердечно-сосудистыми заболеваниями. Соответственно, в настоящем описании предусматривается способ лечения диабета у нуждающегося в этом субъекта, включающий введение нуждающемуся в этом субъекту терапевтически эффективного количества аналога глюкагона, содержащего аминокислотные остатки aa1-аа17 последовательности SEQ. ID. NO. 1.
Согласно другому варианту изобретения в настоящей заявке предусматривается способ лечения диабета у нуждающегося в этом субъекта, включающий введение нуждающемуся в этом субъекту терапевтически эффективного количества аналога глюкагона, содержащего аминокислотные остатки aa1-aa18 последовательности SEQ. ID. NO. 1.
Согласно другому варианту изобретения в настоящей заявке предусматривается способ лечения диабета у нуждающегося в этом субъекта, включающий введение нуждающемуся в этом субъекту терапевтически эффективного количества аналога глюкагона, содержащего аминокислотные остатки aa1-aa19 последовательности SEQ. ID. NO. 1.
Согласно другому варианту изобретения в настоящей заявке предусматривается способ лечения диабета у нуждающегося в этом субъекта, включающий введение нуждающемуся в этом субъекту терапевтически эффективного количества аналога глюкагона, содержащего аминокислотные остатки aa1-aa20 последовательности SEQ. ID. NO. 1.
Согласно дополнительному варианту изобретения введение указанного глюкагона по описанию выше вызывает потерю веса.
Согласно любому из описанных выше вариантов изобретения указанный аналог глюкагона модифицирован с помощью поверхностно-активного вещества (ПАВ) Х Формулы I:
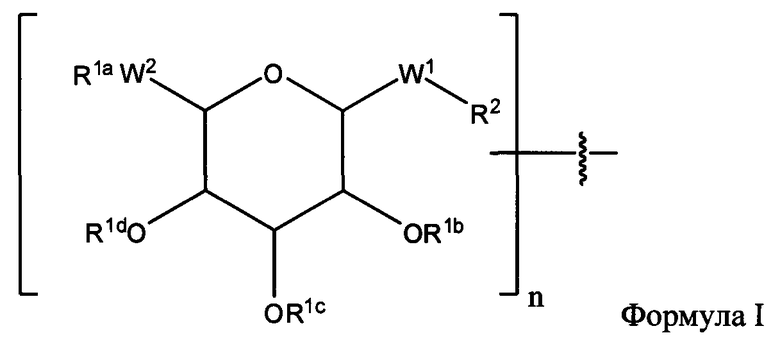
где:
R1a, независимо, в каждом случае обозначает связь, Н, замещенную или незамещенную C1-С30 алкильную группу, замещенную или незамещенную алкоксиарильную группу, замещенную или незамещенную аралкильную группу или фрагмент, содержащий стероидное ядро;
R1b, R1c и R1d каждый независимо, в каждом случае обозначает связь, Н, замещенную или незамещенную C1-С30 алкильную группу, замещенную или незамещенную алкоксиарильную группу или замещенную или незамещенную аралкильную группу;
W1, независимо, в каждом случае обозначает -СН2-, -СН2-O-, -(C=O), -(C=O)-O-, -(C=O)-NH-, -(C=S)-, -(C=S)-NH- или -CH2-S-;
W2 обозначает -O-, -CH2- или -S-;
R2, независимо, в каждом случае обозначает связь с U, Н, замещенную или незамещенную C1-С30 алкильную группу, замещенную или незамещенную алкоксиарильную группу или замещенную или незамещенную аралкильную группу, -NH2, -SH, С2-С4-алкен, С2-С4-алкин, -NH(C=O)-CH2-Br, -(СH2)m-малеимид или -N3;
n обозначает 1, 2 и 3; и
m обозначает 1-10.
Согласно конкретному варианту изобретения указанный аналог глюкагона модифицирован с помощью ПАВ, X, имеющего структуру:
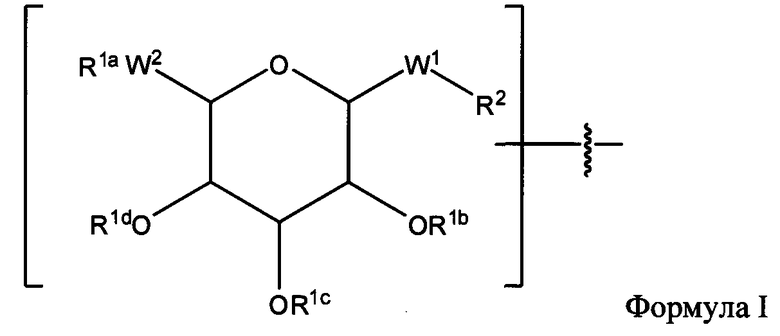
где:
R1a обозначает замещенную или незамещенную C1-С30 алкильную группу;
R1b, R1c и R1d обозначают Н;
W1 обозначает -(C=O)-NH-;
W2 обозначает -O-; и
R2 обозначает связь.
Согласно некоторым описанным выше вариантам изобретения R1a обозначает C1-C20 алкильную группу, C8-C20 алкильную группу, C12-C18 алкильную группу или C14-C18 алкильную группу.
В данном контексте термин "диабет" включает как диабет типа 1, так и диабет типа 2. В соответствии с этим согласно некоторым вариантам изобретения способы, описанные в настоящей заявке, включают введение любого соединения по настоящему изобретению, включая введение соединений Формулы II, III-A, III-B и/или Формулы V и/или соединений, представленных в Таблице 1 на Фигуре 1 и в Таблице 2 на Фигуре 2, субъекту, страдающему диабетом типа 1. Согласно некоторым другим вариантам изобретения способы, описанные в настоящей заявке, включают введение любого соединения по настоящему изобретению, включая введение соединений Формулы II, III-А, III-B и/или Формулы V и/или соединений, представленных в Таблице 1 на Фигуре 1 и в Таблице 2 на Фигуре 2, субъекту, страдающему диабетом типа 2.
Также в настоящей заявке предусматривается способ лечения сердечнососудистого заболевания у нуждающегося в этом субъекта, включающий введение нуждающемуся в этом субъекту терапевтически эффективного количества аналога глюкагона, содержащего аминокислотные остатки aa1-аа17 последовательности SEQ. ID. NO. 331.
Также в настоящей заявке предусматривается способ лечения сердечнососудистого заболевания у нуждающегося в этом субъекта, включающий введение нуждающемуся в этом субъекту терапевтически эффективного количества аналога глюкагона, содержащего аминокислотные остатки aa1-aa18 последовательности SEQ. ID. NO. 331.
Также в настоящей заявке предусматривается способ лечения сердечнососудистого заболевания у нуждающегося в этом субъекта, включающий введение нуждающемуся в этом субъекту терапевтически эффективного количества аналога глюкагона, содержащего аминокислотные остатки aa1-aa19 последовательности SEQ. ID. NO. 331.
Также в настоящей заявке предусматривается способ лечения сердечнососудистого заболевания у нуждающегося в этом субъекта, включающий введение нуждающемуся в этом субъекту терапевтически эффективного количества аналога глюкагона, содержащего аминокислотные остатки aa1-aa20 последовательности SEQ. ID. NO. 331.
В некоторых случаях, согласно описанным выше вариантам изобретения, указанный аналог глюкагона вводят, когда сердечно-сосудистое заболевание ассоциировано с ишемическим событием.
Также в настоящей заявке предусматривается способ лечения сердечнососудистого заболевания у нуждающегося в этом субъекта, включающий введение нуждающемуся в этом субъекту терапевтически эффективного количества аналога глюкагона, содержащего аминокислотные остатки aa1-aa17 последовательности SEQ. ID. NO. 1.
Также в настоящей заявке предусматривается способ лечения сердечнососудистого заболевания у нуждающегося в этом субъекта, включающий введение нуждающемуся в этом субъекту терапевтически эффективного количества аналога глюкагона, содержащего аминокислотные остатки aa1-aa18 последовательности SEQ. ID. NO. 1.
Также в настоящей заявке предусматривается способ лечения сердечнососудистого заболевания у нуждающегося в этом субъекта, включающий введение нуждающемуся в этом субъекту терапевтически эффективного количества аналога глюкагона, содержащего аминокислотные остатки aa1-aa19 последовательности SEQ. ID. NO. 1.
Также в настоящей заявке предусматривается способ лечения сердечнососудистого заболевания у нуждающегося в этом субъекта, включающий введение нуждающемуся в этом субъекту терапевтически эффективного количества аналога глюкагона, содержащего аминокислотные остатки aa1-аа20 последовательности SEQ. ID. NO. 1.
В некоторых случаях, согласно описанным выше вариантам изобретения, указанный аналог глюкагона вводят, когда сердечно-сосудистое заболевание ассоциировано с ишемическим событием.
Согласно любому из описанных выше вариантов изобретения указанный аналог глюкагона модифицирован с помощью ПАВ, X, имеющего структуру Формулы I:
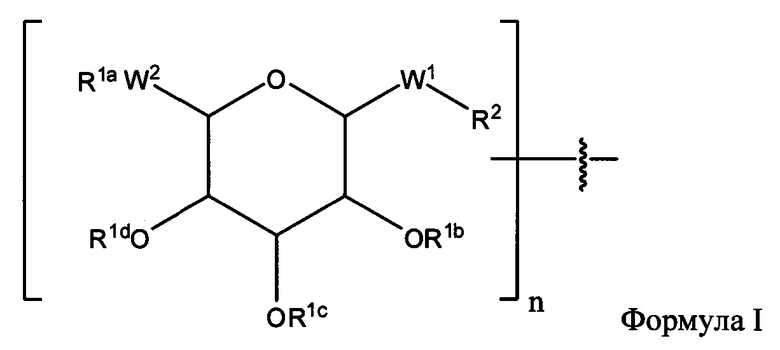
где:
R1a, независимо, в каждом случае обозначает связь, Н, замещенную или незамещенную C1-С30 алкильную группу, замещенную или незамещенную алкоксиарильную группу, замещенную или незамещенную аралкильную группу или фрагмент, содержащий стероидное ядро;
R1b, R1c и R1d каждый независимо, в каждом случае обозначает связь, Н, замещенную или незамещенную C1-С30 алкильную группу, замещенную или незамещенную алкоксиарильную группу или замещенную или незамещенную аралкильную группу;
W1, независимо, в каждом случае обозначает -CH2-, -CH2-O-, -(C=O), -(C=O)-O-, -(C=O)-NH-, -(C=S)-, -(C=S)-NH- или -CH2-S-;
W2 обозначает -O-, -СН2- или -S-;
R2, независимо, в каждом случае обозначает связь с U, Н, замещенную или незамещенную C1-С30 алкильную группу, замещенную или незамещенную алкоксиарильную группу или замещенную или незамещенную аралкильную группу, -NH2, -SH, С2-С4-алкен, С2-С4-алкин, -NH(C=O)-CH2-Br, -(CH2)m -малеимид или -N3;
n обозначает 1, 2 и 3; и
m обозначает 1-10;
Согласно конкретному варианту изобретения указанный аналог глюкагона модифицирован с помощью ПАВ, X, имеющего структуру:
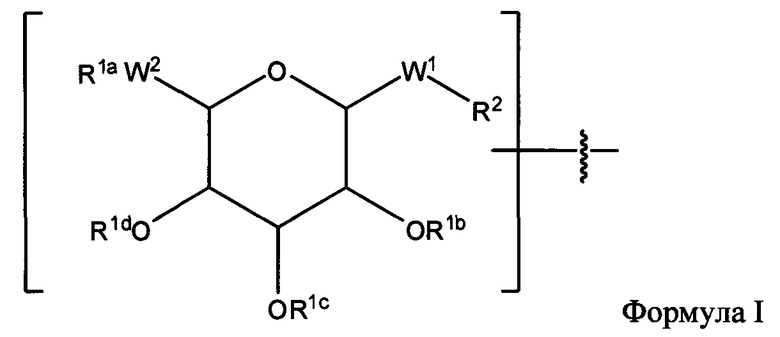
где:
R1a обозначает замещенную или незамещенную C1-С30 алкильную группу;
R1b, R1c и R1d обозначают Н;
W1 обозначает -(C=O)-NH-;
W2 обозначает -O-; и
R2 обозначает связь.
Согласно некоторым описанным выше вариантам изобретения R1a обозначает C1-С20 алкильную группу, C8-C20 алкильную группу, C12-C18 алкильную группу или C14-C18 алкильную группу.
В пептиды (например, глюкагон или GLP-1) можно вводить модификации на амино- или карбоксильном конце (Nestor, J.J., Jr. (2009) Current Medicinal Chemistry 16: 4399-4418). Например, можно осуществлять процессирование или ацилирование пептидов на N-конце, получая пептиды-аналоги, проявляющие низкую эффективность, частичную агонистическую или антагонистическую активность, наблюдаемую для некоторых пептидов (статьи Gourlet, P., et al. (1998) Eur J Pharmacol 354: 105-111, Gozes, I. and Furman, S. (2003) Curr Pharm Des 9: 483-494, содержание которых включено в настоящее изобретение посредством отсылки). Например, делеция первых 6 остатков bPTH дает антагонистические аналоги (Mahaffey, J.E., et al. (1979) J Biol Chem 254: 6496-6498; Goldman, M.E., et al. (1988) Endocrinology 123: 2597-2599), а аналогичный процесс в случае пептидов по настоящему изобретению дает высокоактивные антагонистические аналоги. Другие модификации по N-концу пептидов, такие как делеции или включение D-аминокислот, таких как D-Phe, также могут дать высокоактивные агонисты длительного действия при замене модификациями по настоящему описанию, такими как алкилгликозиды с длинной цепью. Такие агонисты и антагонисты также имеют промышленную применимость и входят в объем рассматриваемых вариантов настоящего изобретения.
Также в объеме настоящего изобретения рассматриваются поверхностно-активные вещества (ПАВ), связанные ковалентной связью с пептидными аналогами, где нативный пептид модифицирован посредством ацетилирования, ацилирования, пегилирования, ADP-рибозилирования, амидирования, связывания ковалентной связью с липидом или производным липида, связывания ковалентной связью с фосфотидилинозитом, сшивания, циклизации, образования дисульфидных связей, деметилирования, образования ковалентной поперечной связи (сшивки), образования цистеина, образования пироглутамата, формилирования, гамма-карбоксилирования, гликозилирования, образования GPI якоря, гидроксилирования, иодирования, метилирования, миристоилирования, окисления, протеолитического процессирования, фосфорилирования, пренилирования, рацемизации, гликозилирования, связывания с липидом, сульфирования, гамма-карбоксилирования остатков глутаминовой кислоты, гидроксилирования и ADP-рибозилирования, селеноилирования, опосредуемого транспортной (т) РНК присоединения аминокислот к белкам, такого как аргинилирование и убиквитинирование. См., например, Nestor, J.J., Jr. (2007) Comprehensive Medicinal Chemistry II 2: 573-601, Nestor, J.J., Jr. (2009) Current Medicinal Chemistry 16: 4399-4418, Creighton, Т.Е. (1993, Wold, F. (1983) Posttranslational Covalent Modification of Proteins 1-12, Seifter, S. and Englard, S. (1990) Methods Enzymol 182: 626-646, Rattan, S.I., et al. (1992) Ann N Y Acad Sci 663: 48-62). Также в объеме настоящего изобретения рассматриваются пептиды разветвленные или циклические с разветвлением или без разветвления. Циклические, разветвленные и циклические разветвленные пептиды образуются в результате посттрансляционных природных процессов, а также получаются соответствующими синтетическими способами. Согласно некоторым вариантам изобретения любой пептидный продукт по настоящему изобретению содержит пептидный аналог по описанию выше, который далее связан ковалентной связью с алкилгликозидным ПАВ-фрагментом.
Также в объеме настоящего изобретения рассматриваются пептидные цепи, замещенные в соответствующем положении, с помощью замены аналогами по настоящему изобретению, полученными путем ацилирования линкерной аминокислоты, например, в е- положении остатка Lys, жирными аминокислотами, такими как октановая, декановая, додекановая, тетрадекановая, гексадекановая, октадекановая, 3-фенилпропановая кислота и т.п., с насыщенными или ненасыщенными алкильными цепями (Zhang, L. and Bulaj, G. (2012) Curr Med Chem 19: 1602-1618). Неограничивающие типичные примеры таких аналогов представлены ниже:
His1-Aib2-Gln3-Gly4-Thr5-Phe6-Thr7-Ser8-Asp9-Tyr10-Ser11-Lys12-Tyr13-Leu14-Asp15-Ser16-Arg17-Lys(N-эпсилон-додеканоил)18-Aib19-NH2, (SEQ. ID. NO. 335)
His1-Aib2-Gln3-Gly4-Thr5-Phe6-Thr7-Ser8-Asp9-Tyr10-Ser11-Lys12-Tyr13-Leu14-Asp15-Ser16-Arg17-Lys(N-эпсилон-тетрадеканоил)18-Ас4с19-NH2, (SEQ. ID. NO. 336)
His1-Aib2-Gln3-Gly4-Thr5-Phe6-Thr7-Ser8-Asp9-Tyr10-Ser11-Lys12-Tyr13-Leu14-Asp15-Ser16-Arg17-Lys(N-эпсилон-гексадеканоил)18-Aib19-NH2, (SEQ. ID. NO. 337)
His1-Aib2-Gln3-Gly4-Thr5-Phe6-Thr7-Ser8-Asp9-Tyr10-Ser11-Lys12-Tyr13-Leu14-Asp15-Aib16-Arg17-Lys(N-эпсилон-додеканоил)18-NH2, (SEQ. ID. NO. 338)
His1-Aib2-Gln3-Gly4-Thr5-Phe6-Thr7-Ser8-Asp9-Tyr10-Ser11-Lys12-Tyr13-Leu14-Asp15-Aib16-Arg17-Lys(N-эпсилон-тетрадеканоил)18-NH2, (SEQ. ID. NO. 339)
His1-Aib2-Gln3-Gly4-Thr5-Phe6-Thr7-Ser8-Asp9-Tyr10-Ser11-Lys12-Tyr13-Leu14-Asp15-Aib16-Arg17-Lys(N-эпсилон-гексадеканоил)18-NH2, (SEQ. ID. NO. 340)
His1-Aib2-Gln3-Gly4-Thr5-Phe6-Thr7-Ser8-Asp9-Tyr10-Ser11-Lys12-Tyr13-Leu14-Asp15-Ser16-Arg17-Lys(N-эпсилон-(гамма-глутамил)-N-альфа-тетрадеканоил))18-Aib19-NH2, (SEQ. ID. NO. 341) и т.п.
Согласно другим вариантам изобретения пептидная цепь, необязательно, замещена в соответствующем положении путем взаимодействия линкерной аминокислоты, например, сульфгидрильной группы Cys, со спейсером и гидрофобным фрагментом, таким как стероидное ядро, например, молекулой холестерина. Согласно некоторым таким вариантам изобретения модифицированный пептид дополнительно содержит одну или более цепей ПЭГ. Ниже представлены неограничивающие примеры таких молекул:
His1-Aib2-Gln3-Gly4-Thr5-Phe6-Thr7-Ser8-Asp9-Tyr10-Ser11-Lys12-Tyr13-Leu14-Asp15-Aib16-Arg17-Cys(S-(3-(PEG4-аминоэтилацетамид-холестирин)))18-Aib19-NH2, (SEQ. ID. NO. 342)
His1-Aib2-Gln3-Gly4-Thr5-Phe6-Thr7-Ser8-Asp9-Tyr10-Ser11-цикло(Glu12-Tyr13-Leu14-Asp15-Lys16)-Arg17-Cys(S-(3-(PEG4-аминоэтилацетамид-холестерин)))18-NH2. (SEQ. ID. NO. 343)
Помимо двадцати стандартных аминокислот имеется огромное количество "нестандартных аминокислот" или не встречающихся в природе (неприродных) аминокислот, которые известны в уровне техники и могут быть включены в описанные выше соединения по настоящему изобретению. Другие нестандартные аминокислоты модифицируются реакционноспособными боковыми цепями с целью конъюгации (Gauthier, M.A. and Klok, H.A. (2008) Chem Commun (Camb) 2591-2611; de Graaf, A.J., et al. (2009) Bioconjug Chem 20: 1281-1295). Согласно одному подходу образующаяся пара тРНК/тРНК-синтетаза кодируется в экспрессионной плазмиде амбер-кодоном (амбер-супрессор) (Deiters, A, et al. (2004). Bio-org. Med. Chem. Lett. 14, 5743-5). Например, в пептиды вводился п-азидофенилаланин, а затем он реагировал с функционализированным ПАВ или полимером, содержащим ацетиленовую связь, в присутствии восстановителя и ионов меди, способствующих реакции, известной как "реакция [3+2] циклоприсоединения Хьюсгена (Huisgen)." Аналогичная цепь реакций с применением реагентов по настоящему описанию, содержащих алкилгликозид, ацетилен- или PEG (ПЭГ)-модифицированный гликозид, дает пегилированные пептиды или пептиды, модифицированные алкилгликозидами. В случае пептидов с числом остатков менее 50 для введения указанных реакционноспособных аминокислотных остатков в нужное положение цепи применяют стандартный твердофазный синтез. Такие ПАВ-модифицированные пептиды и/или белки предлагают иной спектр фармакологических и медицинских свойств, нежели пептиды, модифицированные только лишь путем введения одного ПЭГ.
Специалист в данной области техники понимает, что возможны многочисленные изменения пептидных аналогов при условии, что аминокислотная последовательность содержит включенный ПАВ-фрагмент и обладает заданными свойствами ПАВ-модифицированных пептидных продуктов по настоящему изобретению.
Некоторые определения
В данном описании термины в единственном числе относятся также к множественному числу. В Формуле изобретения при употреблении в сочетании со словом "содержащий" термины в единственном числе относятся к единственному и множественному числу. Слово "другой" обозначает по меньшей мере "второй" или более.
В настоящем описании одно- и трехбуквенные сокращения различных аминокислот даются как указано в статьях Pure Appl. Chem. 31, 639-645 (1972) и 40, 277-290 (1974) и соответствуют требованиям 37 CFR § 1.822 (55 FR 18245, May 1, 1990). Если нет обозначения D- или DL, сокращения обозначают L-аминокислоты. Некоторые аминокислоты, как природные, так и неприродные, являются ахиральными, например, глицин, Сα-диэтилглицин (Deg), α-аминоизомасляная кислота (Aib), 1-аминоциклобутан-1-карбоновая кислота (Ас4с), 1-аминоциклопентан-1-карбоновая кислота (Ас5с), 1-аминоциклогексан-1-карбоновая кислота (Ас6с). Аналоги глутамина включают цитруллин (Cit). Во всех пептидных последовательностях слева указывается аминокислота со свободной аминогруппой (N-концевая аминокислота), а справа аминокислота со свободной карбоксильной группой (С-концевая аминокислота).
Определение "алкильная" группа относится к алифатической углеводородной группе. Ссылка на алкильную группу включает "насыщенный алкил" ("насыщенную алкильную группу)" и/или "ненасыщенный алкил" ("ненасыщенную алкильную группу"). Алкильная группа, либо насыщенная, либо ненасыщенная, включает разветвленные, линейные или циклические группы. "Замещенная" алкильная группа замещена одной или более дополнительных групп. Согласно некоторым вариантам изобретения одна или более дополнительных групп индивидуально и независимо выбраны из амида, сложного эфира, алкила, циклоалкила, гетероалкила, арила, гетероарила, гетероалициклической группы, гидрокси, алкокси, арилокси, алкилтио, арилтио, алкилсульфоксида, арилсульфоксида, сложного эфира, алкилсульфона, арилсульфона, циано, галогена, алкоила, алкоилоксо, изоцианатной, тиоцианатной, изотиоцианатной группы, нитро, галогеналкила, галогеналкокси, фторалкила, амино, алкиламино, диалкиламино, амидо, оксо, гидрофобного природного продукта, такого как стероид, аралкильной цепи (включая алкоксиарил), алкильной цепи, содержащей ацильную группу и т.п. Согласно некоторым вариантам изобретения алкильная группа связана с Nα-положением остатка (например, Tyr или Dmt) в пептиде. Этот класс известен как N-алкил(пептиды) и содержит линейные или разветвленные алкильные группы C1-С10 или арилзамещенную арильную группу, такую как бензил, фенилэтил и т.п. Согласно некоторым вариантам изобретения алкильный фрагмент представляет собой 1-алкильную группу, гликозидной связью соединенную с сахаридным фрагментом (обычно с положением 1, например, глюкозы). Такая 1-алкильная группа представляет собой C1-С30 алкильную группу.
"Арильная" группа относится к ароматическому циклу, в котором каждый из атомов, образующих цикл, представляет собой углеродный атом. Арильные циклы по настоящему описанию включают циклы, содержащие пять, шесть, семь, восемь, девять или более девяти углеродных атомов. Арильные группы, необязательно, имеют заместители, выбранные из галогена, алкила, ацила, алкокси, алкилтио, сульфонила, диалкиламино, эфиров карбоновых кислот, пиано и т.п. Примеры арильных групп включают, но без ограничения, фенил и нафталенил.
Термин "ацил" относится к C1-C20 ацильной цепи. Эта цепь может представлять собой линейную алифатическую цепь, разветвленную алифатическую цепь, цепь, содержащую циклический алкил, гидрофобный природный продукт, такой как стероид, аралкильную цепь или алкильную цепь, содержащую ацильную группу.
Термин "стероидное ядро" относится к ядру стероида, представляющего собой структуру из четырех конденсированных циклов, обозначенных А, В, С и D, показанную ниже:
 . Примеры молекул, содержащих стероидное ядро, включают, но без ограничения, холестерин и т.п.
. Примеры молекул, содержащих стероидное ядро, включают, но без ограничения, холестерин и т.п.
"Терапевтическая композиция" по настоящему описанию может содержать водный или органический носитель или эксципиент и может быть приготовлена, например, с добавлением обычных нетоксических фармацевтически приемлемых носителей, пригодных для получения таблеток, гранул, капсул, лиофилизатов, суппозиториев, растворов, эмульсий, суспензий или других форм, подходящих для применения. Носители, помимо раскрываемых выше, могут включать альгинат, коллаген, глюкозу, лактозу, маннозу, гуммиарабик (аравийскую камедь), желатин, маннит, крахмальный клейстер, трисиликат магния, тальк, кукурузный крахмал, кератин, коллоидный диоксид кремния, картофельный крахмал, мочевину, триглицериды с цепью средней длины, декстраны и другие носители, пригодные для получения препаратов в твердой, полутвердой или жидкой форме. Помимо этого, могут применяться вспомогательные стабилизаторы, загустители или красители, например, сухой стабилизатор, такой как triulose (кетотриоза, дигидроксиацетон).
Выражение "фармацевтически приемлемый носитель", или "терапевтически эффективный носитель", обозначает неводный (твердый), например, спиртовой или масляный носитель, или их смесь, и может содержать ПАВ, смягчающее вещество, смазывающее (скользящее) вещество, стабилизатор, краситель, ароматизатор, консервант, кислоту или основание для корректировки рН, растворитель, эмульгатор, гелеобразующий агент, увлажняющий агент, смачивающий агент, агент для замедленного высвобождения, увлажняющий компонент или другой компонент, обычно включаемый в конкретную форму фармацевтической композиции. Фармацевтически приемлемые носители хорошо известны в уровне техники и включают, например, водные растворы, такие как вода или физиологический (буферный солевой) раствор или другие растворители или носители, такие как гликоли, глицерин и масла, такие как оливковое масло или органические сложные эфиры для инъекций. Фармацевтически приемлемый носитель может содержать физиологически приемлемые соединения, которые, например, стабилизируют или повышают всасывание специфического ингибитора, например, углеводы, такие как глюкоза, сахароза или декстраны, антиоксиданты, такие как аскорбиновая кислота или глутатион, хелатирующие агенты, низкомолекулярные белки или другие стабилизаторы или эксципиенты.
Выражение "возвращающее восприимчивость к инсулину" ("инсулин-ресенсибилизирующее") количество пептидного продукта обозначает количество, которое усиливает реакцию организма на эндогенный или экзогенный (вводимый) инсулин, как правило, снижая массу тела, у нуждающегося в этом субъекта, по показаниям, например, перорального глюкозотолерантного теста или эугликемического гиперинсулинемического "клэмп"-теста.
Фармацевтические композиции могут также содержать другие фармацевтически приемлемые добавки, необходимые для аппроксимации физиологических состояний, такие "вещества" включают, но без ограничения, агенты, корректирующие рН, и буферизующие агенты, агенты, корректирующие тоничность, и т.п., например, ацетат натрия, лактат натрия, хлорид натрия, хлорид калия, хлорид кальция и т.п. Кроме того, суспензия пептида, или его варианта, может включать липид-протективные агенты, которые защищают липиды от повреждения свободными радикалами и переокисления при хранении. Применимы липофильные гасители свободных радикалов, такие как альфа-токоферол, и водорастворимые хелатирующие вещества, специфические к железу, такие как ферри-оксамин.
Термин "поверхностно-активное вещество (ПАВ)" по настоящему описанию обозначает поверхностно-активный агент, который модифицирует поверхностное натяжение воды (на границе раздела фаз). Как правило, в молекуле ПАВ имеется одна липофильная и одна гидрофильная группа. В широком смысле группа включает мыла, детергенты, эмульгаторы, диспергирующие и смачивающие агенты и некоторые группы антисептиков. Более конкретно, ПАВ включают стеарилтриэтаноламин, лаурилсульфат натрия, таурохолат натрия, лауриламинопропионовую кислоту, лецитин, бензалкония хлорид, бензетония хлорид и глицерин моностеарат; и гидрофильные полимеры, такие как поливиниловый спирт, полиэтиленгликоль (ПЭГ, PEG), натрий-карбоксиметилцеллюлозу, метилцеллюлозу, гидроксиметилцеллюлозу, гидроксиэтилцеллюлозу и гидроксипропилцеллюлозу или алкилгликозиды. Согласно некоторым вариантам изобретения ПАВ представляет собой неионное ПАВ (например, ПАВ-алкилглюкозид). Согласно некоторым вариантам изобретения ПАВ представляет собой ионное ПАВ.
Термин "алкилглюкозид" по настоящему описанию относится к любому сахару, соединенному связью с любым гидрофобным алкилом, известным в уровне техники. Гидрофобный алкил может быть выбран любого нужного размера в зависимости от требуемой гидрофобности и гидрофильности сахаридного фрагмента. В одном аспекте изобретения алкильная цепь содержит от 1 до 30 углеродных атомов; или от 6 до 16 углеродных атомов.
В данном контексте термин "сахарид" включает моносахариды, олигосахариды или полисахариды в линейной или в циклической форме. Олигосахариды представляют собой сахариды, содержащие два или более остатков моносахаридов. Некоторые примеры множества возможных сахаридов, пригодных для применения в функционализированной форме, включают глюкозу, галактозу, мальтозу, мальтотриозу, мальтотетраозу, сахарозу, трегалозу и т.п.
"Сложные эфиры сахарозы" в данном контексте означают сложные эфиры сахарозы и жирных кислот. Сложные эфиры сахарозы могут иметь множество форм из-за наличия восьми гидроксильных групп в сахарозе, пригодных для реакции, и множества групп жирных кислот, от ацетата до более объемных жирных кислот, которые могут реагировать с сахарозой. Такая гибкость означает, что на основе применяемой жирной кислоты можно создать множество продуктов и функциональных групп. Сложные эфиры сахарозы применяются в пищевых продуктах и не в пищевых продуктах, в особенности в качестве ПАВ и эмульгаторов при все возрастающем их применении в лекарственных препаратах, косметических средствах, детергентах и пищевых добавках. Они разлагаются микроорганизмами, являются нетоксическими и мягко действуют на кожу.
"Подходящий", "соответствующий" алкилгликозид в данном контексте означает нетоксический и неионный алкилгликозид. В некоторых случаях подходящий алкилгликозид снижает иммуногенность или агрегацию и повышает биодоступность соединения при введении совместно с соединением в глаз, нос, слезно-носовой канал, подъязычно, трансбуккально или в виде инъекции, такой как подкожная, внутримышечная или внутривенная инъекция.
"Линкерная аминокислота" обозначает любую природную или неприродную аминокислоту, которая содержит реакционноспособную функциональную группу (de Graaf, A.J., et al. (2009) Bioconjug Chem 20: 1281-1295), применяемую для ковалентного связывания с функционализированным ПАВ. Например, согласно некоторым вариантам изобретения линкерная аминокислота представляет собой Lys или Orn, имеющие реакционноспособную функциональную группу -NH2; или Cys, имеющий реакционноспособную функциональную группу -SH; или Asp или Glu, имеющие реакционноспособную функциональную группу -С(=O)-ОН. Например, согласно некоторым другим вариантам изобретения линкерная аминокислота представляет собой любую аминокислоту, имеющую реакционноспособную функциональную группу, такую как -ОН, -N3, галогенацетильную или ацетиленовую группу, которая используется для образования ковалентной связи с соответствующим образом функционализированным ПАВ.
Термин "функционализированное ПАВ" в данном контексте означает ПАВ, содержащее реакционноспособную группу, применимую для ковалентного связывания с линкерной аминокислотой. Например, согласно некоторым вариантам изобретения функционализированное ПАВ содержит карбоксильную группу (например, в положении моносахарида) в качестве реакционноспособной группы, пригодной для ковалентного связывания с линкерной аминокислотой. Например, согласно некоторым вариантам изобретения функционализированное ПАВ содержит группу -NH2, группу -N3, ацетиленовую группу, галогенацетиленовую группу, группу -O-NH2 или -(СН2-)m-малеимидную группу в положении 6 моносахарида (как показано на Схеме 6), что способствует ковалентному связыванию с соответствующей линкерной аминокислотой. Согласно некоторым вариантам изобретения в данном контексте функционализированное ПАВ представляет собой соединение Формулы II. Необязательно, согласно конкретным вариантам изобретения функционализированное ПАВ содержит связанную ковалентной связью линкерную аминокислоту; затем, путем последовательного присоединения одной или более аминокислот к линкерной аминокислоте образуется пептид, модифицированный с помощью ПАВ.
Термин "пептид" в данном контексте обозначает любой пептид, содержащий две или более аминокислот. Термин "пептид" включает полипептиды, короткие пептиды (например, пептиды, содержащие 2-14 аминокислот), средние (средней длины) пептиды (15-50 аминокислот) или длинные (с длинной цепью) пептиды (например, белки). Термины пептид, полипептид, пептид средней длины и белок в данном контексте могут применяться как синонимы. В данном контексте термин "пептид" истолковывается как полимер, состоящий из аминокислотных остатков, родственных природных структурных вариантов и их синтетических неприродных аналогов, связанных пептидными связями, родственные природные структурные варианты и их синтетические неприродные аналоги. Синтетические пептиды можно синтезировать, например, на автоматическом синтезаторе пептидов.
Пептиды могут содержать аминокислоты, отличные от аминокислот, кодируемых 20 генами. "Пептид(ы)" включае(ю)т пептид(ы), модифицированный(е) либо с помощью естественных процессов, таких как процессирование и другие посттрансляционные модификации, а также химическими методами модификации. Такие модификации хорошо описаны в основных учебных пособиях и в более подробных монографиях и хорошо известны специалистам в данной области техники. Следует понимать, что согласно некоторым вариантам изобретения модификация такого же типа присутствует в данном пептиде в такой же самой или в меняющейся степени в нескольких сайтах. Также согласно некоторым вариантам изобретения данный пептид содержит модификации более чем одного типа. Модификации встречаются в пептиде в любом месте, включая пептидный остов, боковые цепи аминокислот и амино- или карбоксильные концы..
Термин пептид включает пептиды или белки, которые содержат природные и неприродные аминокислоты или аналоги природных аминокислот.В данной заявке "аналоги" пептидов и/или белков содержат неприродные аминокислоты на основе природных аминокислот, такие как аналоги тирозина, которые включают пара-замещенные тирозины, орто-замещенные тирозины и мета-замещенные тирозины, в которых заместитель в тирозине содержит ацетильную группу, бензоильную группу, аминогруппу, гидразин, гидроксиламин, тиольную группу, карбоксигруппу, метальную группу, изопропильную группу, С2-С20 линейный или разветвленный углеводород, насыщенный или ненасыщенный углеводород, O-метильную группу, простую полиэфирную группу, галоген, нитрогруппу и т.п. Примеры аналогов Tyr включают 2,4-диметилтирозин (Dmt), 2,4-диэтилтирозин, О-4-аллилтирозин, 4-пропилтирозин, Сα-метилтирозин и т.п. Примеры аналогов лизина включают орнитин (Orn), гомолизин, Сα-метиллизин (CMeLys) и т.п. Примеры аналогов фенилаланина включают, но без ограничения, мета-замещенные фенилаланины, в которых заместитель содержит метоксигруппу, C1-C20 алкильную группу, например, метальную группу, аллильную группу, ацетильную группу и т.п. Конкретные примеры включают, но без ограничения, 2,4,6-триметил-L-фенилаланин (Tmt), О-метилтирозин, 3-(2-нафтил)аланин (Nal(2)), 3-(1-нафтил)аланин (Nal(l)), 3-метилфенилаланин, 1,2,3,4-тетрагидроизохинолин-3-карбоновую кислоту (Tic), фторированные фенилаланины, изопропилфенилаланин, по-азидофенилаланин, п-ацилфенилаланин, п-бензоилфенилаланин, п-иодфенилаланин, п-бромфенилаланин, п-аминофенилаланин и изопропилфенилаланин, и т.п. другие нестандартные или неприродные аминокислоты, применяемые в создании пептидных аналогов, включают, но без ограничения, С-альфа-дизамещенные аминокислоты, такие как Aib, Сα-диэтилглицин (Deg), аминоциклопентан-1-карбоновую кислоту (Ас5с) и т.п. Такие аминокислоты часто позволяют получать конформационно-жесткие структуры (с заторможенной конформацией), часто смещенные в сторону спиральной структуры (Kaul, R. and Balaram, P. (1999) Bioorg Med Chem 7: 105-117). Другими примерами таких неприродных аминокислот, применимых в дизайне аналогов, являются гомоаргинин (Har) и т.п. Замена восстановленных амидных связей в некоторых случаях обеспечивает повышенную устойчивость к ферментативному расщеплению или изменяет связывание с рецептором. Например, с помощью включения дипептидного звена Tic-Phe с восстановленной амидной связью между остатками (обозначенного как Тic-ψ[СH2-NH]-ψ-Phe) уменьшает ферментативное расщепление. Соответственно, также в объеме вариантов настоящего изобретения рассматриваются ПАВ, связанные с пептидами ковалентной связью, которые содержат описанные выше модифицированные аминокислоты и/или пептидные аналоги. Некоторые неприродные аминокислоты представлены ниже.
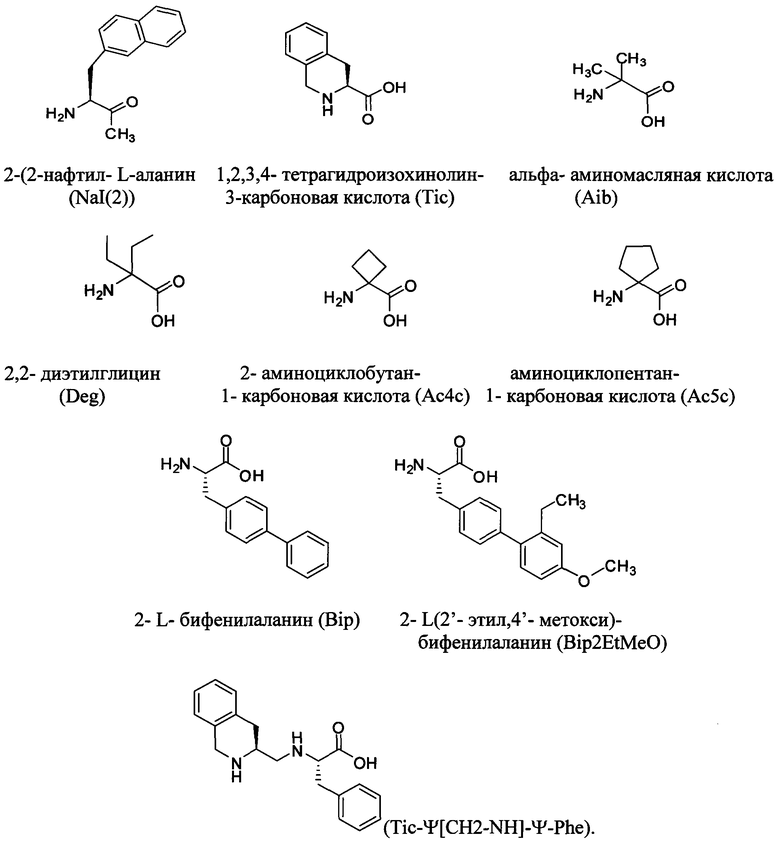
Термин "вариант" в данном контексте означает пептид, который отличается от эталонного (основного, исходного) пептида, но сохраняет основные свойства. Аминокислотная последовательность типичного варианта пептида отличается от аминокислотной последовательности другого, эталонного, основного пептида. Как правило, различия ограничены таким образом, чтобы последовательности эталонного пептида и варианта от одного конца до другого были очень похожи, а во многих областях идентичны. Аминокислотные последовательности вариантного и эталонного пептида могут различаться одной или более замен, добавлений, делеций в любой комбинации. Замещенный или встроенный (инсерция) аминокислотный остаток может или не может представлять собой остаток, кодированный генетическим кодом. Неприродные варианты пептидов могут быть получены методами мутагенеза, прямым синтезом и другими подходящими методами рекомбинантной ДНК.
Методы
Согласно некоторым вариантам настоящего изобретения предусматриваются методы (способы) предупреждения и/или лечения состояний, ассоциированных со снижением чувствительности к инсулину, включающие введение терапевтически эффективного количества модифицированного с помощью ПАВ пептидного и/или белкового продукта по настоящему изобретению (например, пептидного продукта Формулы I-A, III-A, III-B или Формулы V) нуждающемуся в этом субъекту. Согласно некоторым вариантам изобретения патологические состояния, характеризующиеся снижением чувствительности к инсулину, включают, но без ограничения, метаболический синдром, инсулинорезистентность, связанную с ожирением, гипертонию, системное воспалительное заболевание, ассоциированное с высоким содержанием С-реактивного белка, диабет и т.п.
Также в настоящей заявке предусматриваются способы лечения инсулинорезистентности, включающие введение нуждающимся в этом субъектам терапевтически эффективного количества пептидного и/или белкового продукта, модифицированного с помощью ПАВ (например, пептидного продукта Формулы I-A, III-А, III-B или Формулы V). Согласно некоторым вариантам изобретения инсулинорезистентность обусловлена метаболическим синдромом (Синдромом X) и/или диабетом.
Помимо этого, в настоящем изобретении предусматриваются способы стимуляции восстановления чувствительности (ресенсибилизации) организма к инсулину, включающие введение нуждающимся в этом субъектам терапевтически эффективного количества пептидного и/или белкового продукта, модифицированного с помощью ПАВ (например, пептидного продукта Формулы I-A, III-A, III-B или Формулы V).
Согласно другим вариантам настоящего изобретения предусматриваются способы повышения чувствительности к инсулину посредством потери веса, включающие введение нуждающимся в этом субъектам терапевтически эффективного количества пептидного и/или белкового продукта, модифицированного с помощью ПАВ (например, пептидного продукта Формулы I-A, III-А, III-B или Формулы V и представленного в Таблице 1 на Фигуре 1 и в Таблице 2 на Фигуре 2).
Также в настоящем изобретении предусматриваются способы лечения диабета или предиабета, включающие введение нуждающемуся в этом субъекту терапевтически эффективного количества пептидного продукта по настоящему описанию и представленного в Таблице 1 на Фигуре 1 и в Таблице 2 на Фигуре 2.
В настоящем изобретении предусматриваются способы лечения или замедления прогрессирования или начала заболеваний, выбранных из диабета, диабетической ретинопатии, диабетической нейропатии, диабетической нефропатии, инсулинорезистентности, гипергликемии, гиперинсулинемии, метаболического синдрома, диабетических осложнений, повышенного содержания в крови свободных жирных кислот или глицерина, гиперлипидемии, ожирения, гипертриглицеридемии, атеросклероза, острого сердечно-сосудистого синдрома, инфаркта, метаболического синдрома, ишемии-реперфузии или гипертонии, включающие введение терапевтически эффективного количества пептидного продукта по настоящему описанию и в Таблице 1 на Фигуре 1 и в Таблице 2 на Фигуре 2, нуждающемуся в этом субъекту. В дополнительном варианте изобретения предусматриваются способы лечения замедленного заживления ран, включающие введение терапевтически эффективного количества пептидного продукта, представленного в настоящем описании и в Таблице 1 на Фигуре 1 и в Таблице 2 на Фигуре 2, нуждающемуся в этом субъекту.
Согласно одному варианту изобретения указанным заболеванием, подлежащим лечению, является диабет. Согласно одному варианту изобретения указанным заболеванием, подлежащим лечению, является инсулинорезистентность. Согласно одному варианту изобретения указанным заболеванием, подлежащим лечению, является метаболический синдром. Согласно одному варианту изобретения указанное эффективное количество указанного пептида составляет, примерно, от 0.1 мкг/кг/день, примерно, до 100.0 мкг/кг/день.
Согласно одному варианту изобретения способ введения является парентеральным. Согласно одному варианту изобретения способ введения является пероральным. Согласно одному варианту изобретения способ введения является подкожным. Согласно одному варианту изобретения способ введения представляет собой назальную инсуффляцию (вдувание).
Помимо этого, настоящее изобретение предусматривает способ менее значительного увеличения веса тела или стимулирования потери веса, включающий введение нуждающемуся в этом субъекту терапевтически эффективного количества пептидного продукта, представленного в настоящем описании и в Таблице 1 на Фигуре 1 и в Таблице 2 на Фигуре 2. Согласно некоторым вариантам изобретения увеличение веса тела ассоциировано с метаболическим синдромом.
Настоящее изобретение предусматривает способ лечения гипогликемии, включающий введение нуждающемуся в этом субъекту терапевтически эффективного количества пептидного продукта, представленного в настоящем описании и в Таблице 1 на Фигуре 1 и в Таблице 2 на Фигуре 2.
Настоящее изобретение предусматривает также способ лечения диабета, включающий введение нуждающемуся в этом субъекту терапевтически эффективного количества пептидного продукта, представленного в настоящем описании и в Таблице 1 на Фигуре 1 и в Таблице 2 на Фигуре 2.
Также настоящее изобретение предусматривает способ лечения диабета, включающий введение нуждающемуся в этом субъекту терапевтически эффективного количества пептидного продукта, представленного в настоящем описании и в Таблице 1 на Фигуре 1 и в Таблице 2 на Фигуре 2, и по меньшей мере одного дополнительного терапевтического агента; причем указанный терапевтический агент выбран из антидиабетического агента, агента против ожирения, агента, вызывающего чувство насыщения, противовоспалительного агента, гипотензивного агента, антиатеросклеротического агента и агента, понижающего содержание липидов.
В некоторых способах согласно описанным выше вариантам изобретения пептид и/или белок, ковалентно связанный с ПАВ, представляет собой глюкагон или GLP-1 пептид, или его аналог. Согласно некоторым вариантам изобретения пептид и/или белок, модифицированный с помощью ПАВ (например, пептидный продукт Формулы I-A, III-A, III-B или Формулы V), вводится профилактически и замедляет возникновение любого состояния, ассоциированного с инсулинорезистентностью, включая, но без ограничения, метаболический синдром, гипертонию, диабет, диабет типа 2, гестационный диабет, гиперлипидемию, атеросклероз, системное воспаление и т.п. Согласно некоторым вариантам изобретения пептид и/или белок, модифицированный с помощью ПАВ (например, пептидный продукт Формулы I-A, III-A, III-B или Формулы V), вводится с терапевтической целью и замедляет прогрессирование любого состояния, ассоциированного с метаболическим синдромом, гипертонией, диабетом, диабетом типа 2, гестационным диабетом, гиперлипидемией, атеросклерозом, системным воспалением и т.п. Согласно некоторым вариантам изобретения пептид и/или белок, модифицированный с помощью ПАВ (например, пептидный продукт Формулы I-A, III-A, III-B или Формулы V), вводится профилактически и/или с терапевтической целью и замедляет прогрессирование инсулинорезистентности до диабета. Согласно некоторым вариантам изобретения пептид и/или белок, модифицированный с помощью ПАВ (например, пептидный продукт Формулы I-A, III-A, III-B или Формулы V), вводится профилактически и/или с терапевтической целью и снижает или прекращает дальнейшее развитие инсулинорезистентности, тем самым стабилизируя заболевание, (halts further loss of insulin resistance?).
Согласно некоторым вариантам изобретения ПАВ-модифицированный пептид и/или белок (например, пептидный продукт Формулы I-A, III-A, III-B или Формулы V) вводят парентерально. Согласно некоторым вариантам изобретения ПАВ-модифицированный пептид и/или белок (например, пептидный продукт Формулы I-A, III-А, III-B или Формулы V) вводят подкожно. Согласно некоторым вариантам изобретения ПАВ-модифицированный пептид и/или белок (например, пептидный продукт Формулы I-А, III-A, III-B или Формулы V) вводят с помощью назальной инсуффляции.
Согласно некоторым вариантам изобретения в описанных выше методах действие ПАВ-модифицированного пептида и/или белка (например, пептидного продукта Формулы I-A, III-А, III-B или Формулы V) является более продолжительным по сравнению с действием фармацевтических препаратов, содержащих современные лекарственные средства (например, эксенатид, метформин и т.п.).
Комбинированная терапия
Согласно некоторым вариантам изобретения в описанных выше методах ПАВ-модифицированный пептид и/или белок (например, пептидный продукт Формулы I-A, III-А, III-B или Формулы V) вводят в сочетании с другими лекарственными средствами для лечения метаболического синдрома, выбранными из группы, содержащей антидиабетический агент, гипотензивный агент, агент против атеросклероза и агент, снижающий содержание липидов. Например, эффективные антидиабетические агенты, применимые для введения в комбинации с ПАВ-модифицированным пептидным и/или белковым продуктом по настоящему изобретению, включают бигуанид, сульфонилмочевину, ингибитор глюкозидазы, агонист PPAR γ, двойной агонист PPAR α/γ, ингибитор аР2, ингибитор DPP4, сенсибилизатор инсулина, аналог GLP-1, инсулин и меглитинид. Дополнительные примеры включают метформин, глибурид, глимепирид, глипирид, глипизид, хлорпропамид, гликлазид, акарбозу, миглитол, пиоглитазон, троглитазон, розиглитазон, мураглитазар, инсулин, G1-262570, изаглитазон, JTT-501, NN-2344, L895 645, YM-440, R-119702, A19677, репаглинид, натеглинид, KAD 1129, AR-HO 39242, GW-4015 44, KRP217, AC2993, LY3 I 5902, NVP-DPP-728A и саксаглиптин.
Согласно некоторым вариантам изобретения в описанных выше методах ПАВ-модифицированный пептид и/или белок (например, пептидный продукт Формулы I-A, III-А, III-B или Формулы V) вводят в сочетании с другими лекарственными средствами для лечения метаболического синдрома, выбранными из группы эффективных агентов против ожирения. Например, эффективные агенты против ожирения, применимые для введения с пептидными продуктами по настоящему изобретению, включают бета 3 адренергический агонист, ингибитор липазы, ингибитор обратного захвата серотонина (и дофамина), бета рецептор тиреоидного гормона, антагонист СВ-1, агонист рецепторов NPY-Y2 и NPY-Y4 и анорексигенный агент. Конкретные представители этих классов включают орлистат, AfL-962, А19671, L750355, СР331648, сибутрамин, топирамат, аксокин, дексамфетамин, фентермин, фенилпропаноламин, римонабат (SRI 4I7164) и мазиндол.
Согласно некоторым вариантам изобретения в описанных выше методах ПАВ-модифицированный пептид и/или белок (например, пептидный продукт Формулы I-A, III-А, III-B или Формулы V) вводят в сочетании с другими лекарственными средствами для лечения метаболического синдрома, выбранными из группы агентов, эффективно снижающих содержание липидов. Например, эффективные агенты для снижения содержания липидов, применимые для введения с пептидными продуктами по настоящему изобретению, включают агенты, выбранные из группы, состоящей из ингибитора МТР, белка-переносчика сложных эфиров холестерина, ингибитора HMG СоА редуктазы, ингибитора сквален-синтетазы (сквален-синтазы), производного фибриновой кислоты, позитивного регулятора LDL рецепторной активности, ингибитора липоксигеназы и ингибитора АСАТ. Конкретные примеры представителей этих классов включают правастатин, ловастатин, симвастатин, аторвастатин, церивастатин, флувастатин, нисвастатин, висастатин (visastatin), фенофибрат, гемфиброзил, клофибрат, авасимиб, TS-962, MD-700, СР-52941 4, and LY295 427.
Согласно некоторым вариантам изобретения в описанных выше методах ПАВ-модифицированный пептид и/или белок (например, пептидный продукт Формулы I-A, III-А, III-B или Формулы V) вводят в сочетании с пептидными гормонами и их аналогами, заведомо вызывающими эффект насыщения на животных моделях и у человека. В объеме вариантов настоящего изобретения рассматривается комбинация пептидных продуктов по настоящему описанию и агентов, вызывающих продолжительное чувство насыщения, для лечения ожирения. Примеры таких пептидных агентов, вызывающих чувство насыщения, включают GLP-1, панкреатический полипептид (РР), холецистокинин (ССК), пептид YY (PYY), амилин, кальцитонин, ОХМ, нейропептид Y (NPY) и их аналоги (Bloom, S.R., et al. (2008) Mol Interv 8: 82-98; Field, B.C., et al. (2009) Br J Clin Pharmacol 68: 830-843).
Также в объеме вариантов настоящего изобретения рассматриваются методы лечения ожирения, включающие введение пептидных продуктов по настоящему изобретению в комбинации с пептидными гормонами, включая, но без ограничения, аналоги и антагонисты лептина, грелина и CART (кокаин- и амфетамин-регулируемого транскрипта).
Дополнительные пептидные продукты в организме ассоциированы с жировыми клетками или с состоянием ожирения (адипокины) и заведомо проявляют провоспалительный эффект (Gonzalez-Periz, A. and Claria, J. (2010) ScientificWorld Journal 10: 832-856). Такие агенты проявляют дополнительное благотворное действие в случае применения в сочетании с пептидными продуктами по настоящему изобретению. Примеры агентов, которые оказывают благотворное действие в сочетании с пептидными продуктами по настоящему изобретению, включают аналоги и антагонисты адипонектина, хемерина, висфатина, несфатина, оментина, резистина, ТМРальфа, IL-6 и обестатина.
Интермедиаты
Согласно одному варианту настоящее изобретение предусматривает интермедиаты и/или реагенты, содержащие молекулу ПАВ и реакционноспособную функциональную группу, способные образовывать связь с помощью реакционноспособной функциональной группы природной или неприродной аминокислоты. Эти интермедиаты и/или реагенты позволяют повышать биодоступность и фармацевтические, фармакокинетические и/или фармакодинамические характеристики пептидов и/или белков, применяемых при заболеваниях человека и животных. Ковалентное связывание таких интермедиатов и/или реагентов за счет функциональной группы в боковой цепи аминокислоты, например, эпсилон-аминогруппы Lys, сульфгидрильной группы Cys, или на амино- или карбоксильном конце пептида- и/или белка-мишени позволяет синтезировать пептидные продукты по настоящему изобретению Согласно конкретным вариантам изобретения неионные ПАВ-молекулы представляют собой моно- или дисахариды с О- алкильным заместителем в гликозиде, причем указанная гликозидная связь имеет как альфа-, так и бета-конфигурацию. Согласно конкретным вариантам изобретения О-алкильные цепи представляют собой C1-C20 или C6-C16 алкильные цепи.
Согласно другому варианту настоящее изобретение предусматривает интермедиаты и/или реагенты, содержащие молекулу неионного ПАВ с определенной алкилгликозидной связью, которая имитирует О-алкилгликозидные связи, и реакционноспособную функциональную группу, способную образовывать связь с реакционноспособной функциональной группой в природной или неприродной аминокислоте. Такие интермедиаты и/или реагенты содержат S-связанные алкильные цепи или N-связанные алкильные цепи и проявляют химическую и/или ферментативную устойчивость, измененную по сравнению с продуктами, связанными с O-алкилгликозидами.
Согласно некоторым вариантам интермедиат и/или реагент, предусматриваемый в настоящем изобретении, представляет собой соединение, в котором гидрофильной группой является модифицированная глюкоза, галактоза, мальтоза, глюкуроновая кислота, диглюкуроновая кислота и т.п. Согласно некоторым вариантам изобретения гидрофильная группа представляет собой глюкозу, мальтозу, глюкуроновую кислоту или диглюкуроновую кислоту, а гидрофобная группа представляет собой C1-С30 алкильную цепь или аралкильную цепь. В некоторых вариантах изобретения гликозидная связь с гидрофобной группой имеет альфа-конфигурацию, а в некоторых вариантах эта связь имеет бета конфигурацию при аномерном центре сахарида.
Согласно некоторым вариантам изобретения гидрофильная группа обозначает глюкозу, мальтозу, глюкуроновую кислоту или диглюкуроновую кислоту, а гидрофобная группа обозначает C1-C20 алкильную или аралкильную цепь.
Согласно некоторым вариантам изобретения интермедиат и/или реагент по настоящему изобретению содержит ПАВ, включающее реакционноспособную функциональную группу, которая представляет собой карбоксильную группу, аминогруппу, азидную, альдегидную, малеимидную, сульфгидрильную группу, гидроксиламиногруппу, алкин и т.п.
Согласно некоторым вариантам изобретения интермедиат и/или реагент представляет собой 0-связанный алкилгликозид, за счет одной из гидроксильных функций модифицированный с образованием функциональной карбоксильной или аминогруппы. Согласно некоторым вариантам изобретения реагент представляет собой 1-O-алкил глюкуроновую кислоту в альфа или бета-конфигурации, а алкильная цепь имеет протяженность от C1 до С20. Согласно некоторым таким вариантам изобретения алкильная цепь имеет протяженность от С6 до C16.
Согласно некоторым вариантам изобретения реагент содержит 1-О-алкил глюкуроновую кислоту, а алкильная цепь имеет протяженность от C1 до С20. Согласно некоторым таким вариантам изобретения алкильная цепь имеет протяженность от С6 до C16.
Согласно некоторым вариантам изобретения реагент представляет собой S-связанный алкилгликозид в альфа или в бета-конфигурации, за счет одной из гидроксильных функций модифицированный с образованием функциональной карбоксильной или аминогруппы.
Согласно некоторым вариантам изобретения реагент представляет собой N-связанный алкилгликозид в альфа или в бета-конфигурации, за счет одной из гидроксильных функций модифицированный с образованием функциональной карбоксильной или аминогруппы.
Согласно еще одному варианту в настоящем изобретении предусматриваются пептидные и/или белковые продукты, содержащие ковалентно связанный алкилгликозид, обладающий свойствами, приемлемыми для применения при заболеваниях человека и животных. На Схеме 1 приводятся типичные неионные ПАВ, которые можно модифицировать, получая реагенты и/или интермедиаты, применимые для синтеза ПАВ-модифицированных пептидных продуктов по настоящему изобретению.
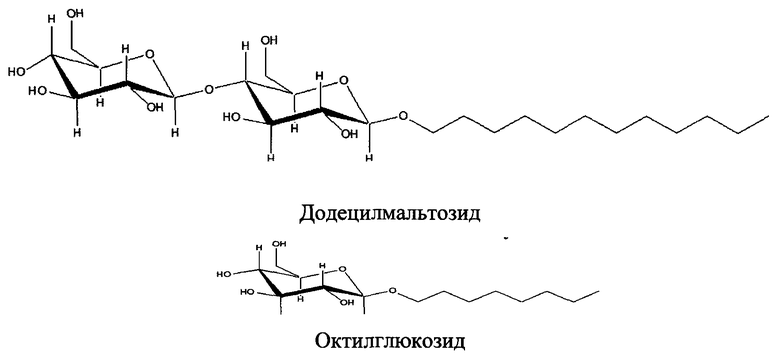
Пример 1. Примеры имеющихся в продаже неионных ПАВ класса алкилгликозидов.
Согласно некоторым вариантам изобретения модифицированные за счет ковалентного связывания пептиды и/или белки по настоящему изобретению включают в структуру пептида ПАВ-фрагмент. Согласно конкретному варианту изобретения модифицированные за счет ковалентного связывания пептиды и/или белки по настоящему изобретению включают неионный ПАВ-агент из класса алкил-, алкоксиарил- или аралкилгликозидов. Алкилгликозиды представляют собой важные товары и широко применяются в пищевой промышленности, в сфере услуг и в индустрии чистоты. Поэтому их производство в промышленных масштабах было предметом всестороннего изучения. Имеются очень дешевые ферментативные и химические методы их получения (Park, D.W., et al. (2000) Biotechnology Letters 22: 951-956). Эти алкилгликозиды можно модифировать далее, чтобы получить интермедиаты для синтеза модифицированных за счет ковалентной связи пептидов и/или белков по настоящему изобретению. Так, известно, что 1-додецил бета-D-глюкозид, в отсутствие защитных групп и в присутствии в качестве катализатора платиновой черни в атмосфере кислорода, окисляется, предпочтительно, по 6-положению с образованием соответствующего аналога глюкуроновой кислоты с высоким выходом (van Bekkum, H. (1990) Carbohydrates as Organic Raw Materials 289-310). Имеются другие химиоселективные методы окисления первичного спирта в положении 6 алкилглюкозидов. Например, путем окисления первичной гидроксильной группы в присутствии каталитических количеств 2,2,6,6-тетраметил-1-пиперидиноксила (TEMPO) и стехиометрических количеств органического окислителя [бис(ацетокси)иод]бензола (BAIB) (De Mico, A., et al. (1997) J Org Chem 1997: 6974-6977) получали нуклеозид-5′-карбоновые кислоты с прекрасным выходом (Ерр, J.B. and Widlanski, T.S. (1999) J Org Chem 64: 293-295). Этот процесс окисления является хемоселективным по отношению к первичной гидроксильной группе, даже если другие, вторичные, гидроксильные группы не имеют защитных групп (Codee, J.D., et al. (2005) J Am Chem Soc 127: 3767-3773). Аналогичным образом 1-додецил β-D-глюкопиранозид, 1-тетрадецил β-D-глюкопиранозид, 1-гексадецил β-D-глюкопиранозид, 1-октадецил β-D-глюкопиранозид и 1-эйкозил β-D-глюкопиранозид были окислены в соответствующие уроновые кислоты (1-додецил β-D-глюкуроновую кислоту, 1-тетрадецил β-D-глюкуроновую кислоту, 1-гексадецил β-D-глюкуроновую кислоту, 1-октадецил β-D-глюкуроновую кислоту, 1-эйкозил β-D-глюкуроновую кислоту) в присутствии TEMPO при использовании KBr и стехиометрических количеств гипохлорита натрия в качестве окислителя (Milkereit, G., et al. (2004) Chem Phys Lipids 127: 47-63) в воде. Методика мягкого окисления с применением (диацетоксииод)бензола (DAIB aka (он же) BAIB) дается в разделе Примеры. Некоторые из этих промежуточных глюкуроновых кислот имеются в продаже (например, октил b-D-глюкуроновая кислота; Carbosynth, МО 07928) и, как указано, большой ряд этих соединений получают обычными методами (Schamann, М. and Schafer, H.J. (2003) Eur J Org Chem 351-358; Van den Bos, L.J., et al. (2007) Eur J Org Chem 3963-3976) или, по запросу, из коммерческих источников. На Схеме 2, в качестве примера, некоторые функционализированные ПАВ-интермедиаты, применяемые для получения интермедиатов и/или реагентов по настоящему изобретению, содержащие в качестве реакционноспособной функциональной группы -СООН группу.
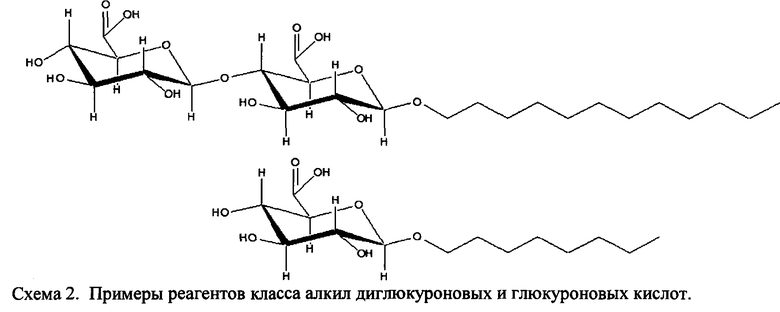
Аналогично, аралкил гликозиды (включая алкоксиарил) могут составлять основу для родственных неионных ПАВ-реагентов. Например, 4-алкоксифенил β-D-глюкопиранозиды легко получаются реакцией 4-алкоксифенолов с пента-О-ацетил β-D-глюкозой в присутствии эфирата трехфтористого бора. Последующее деацетилирование с применением триметиламина в смеси метанол/вода и селективное окисление по описанию выше и в примерах дает реагенты -алкоксиарил глюкуроновые кислоты, пригодные для получения реагентов и пептидов по настоящему описанию ((Smits, E., et al. (1996) J Chem Soc, Perkin Trails I 2873-2877; Smits, E., et al. (1997) Liquid Crystals 23: 481-488).
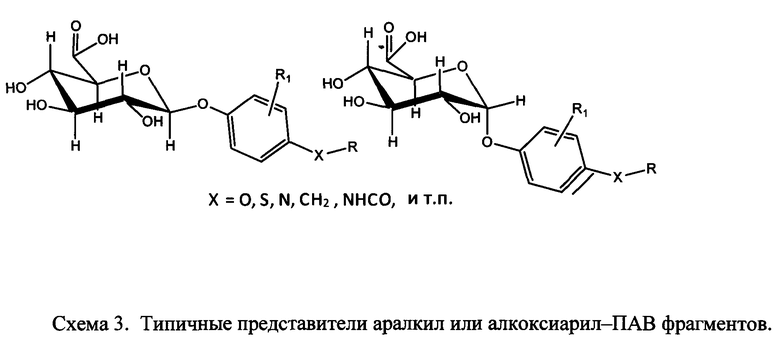
Интермедиаты класса глюкуроновых кислот легко активируется обычными конденсирующими агентами со связыванием с боковой цепью аминокислоты, например аминокислоты Lys. Так, Fmoc-Lys-O-TMS (триметилсилил = ТМ8) может реагировать с октил бета-D-глюкуроновой кислотой в присутствии конденсирующего агента, а О-TMS защитную группу можно затем гидролизовать обработкой водой с образованием Fmoc-Lys(1-октил бета-D-глюкуронамид), как показано на Схеме 4. Этот реагент можно применять для включения в твердофазный синтез пептидов в соответствии со стандартным протоколом конденсации, когда требуется включить ПАВ-фрагмент рядом с N-концевой областью молекулы. Вторичные гидроксильные группы можно оставить без защиты из-за очень высокой реакционной способности функциональной аминогруппы Lys или их можно защитить перацетилированием. Если в качестве защитной группы используется ацетильная группа, снятие защитных ацетильных групп с высоким выходом можно проводить, обрабатывая либо с помощью MeOH/NaOMe, либо с помощью MeOH/EtsN. На Схеме 4 показано получение реагентов по настоящему изобретению.
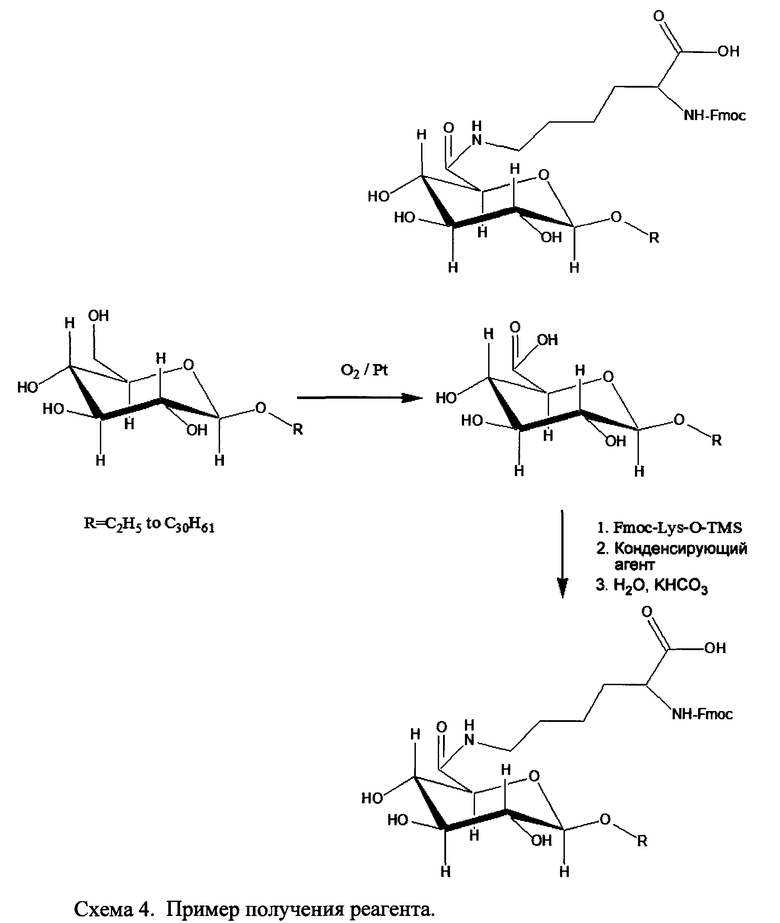
Согласно некоторым вариантам изобретения реагенты и/или интермедиаты для получения биологически активных пептидных продуктов по настоящему изобретению включают семейство модифицированных с помощью ПАВ линкерных аминокислот для встраивания в синтетические пептидные продукты. Так, согласно одному варианту изобретения пептидные продукты по настоящему изобретению синтезируют последовательно (линейно), причем функционализированный ПАВ-агент присоединяют к имеющей обратимую защиту линкерной аминокислоте за счет функциональной группы в боковой цепи линкерной аминокислоты (например, аминогруппы лизинового остатка), получая патентованный реагент (показанный на Схеме 4.), который можно вводить в растущую пептидную цепь, а затем оставшуюся пептидную цепь синтезируют, связывая следующие аминокислоты с цистеиновым остатком. Защитные группы, пригодные для синтеза модифицированных пептидов и/или белков по настоящему изобретению, описаны, например, в книге Т. W. Green, P. G. M. Wuts, Protective Groups in Organic Synthesis, Wiley-Interscience, New York, 1999, 503-507, 736-739, содержание которой вводится в настоящее изобретение посредством отсылки.
Согласно другому варианту изобретения пептидные продукты по настоящему изобретению синтезируются ковалентным связыванием функционализированного ПАВ с полноразмерным пептидом за счет соответствующей функциональной группы в линкерной аминокислоте в пептидной цепи.
Или же функционализированный ПАВ-агент можно присоединять к боковой цепи линкерной аминокислоты, депротекция которой осуществлялась в ходе твердофазного синтеза пептида. Например, алкил глюкуронильную группу можно связывать непосредственно с боковой цепью линкерной аминокислоты (например, с лишенной защиты боковой цепью Lys) в процессе твердофазного синтеза пептида. Например, использование Fmoc-Lys(Alloc)-OH в качестве субъединицы обеспечивает ортогональную защиту, которую можно удалять в то время как пептид еще находится на смоле. Таким образом, депротекция боковой цепи Lys с использованием Pd/тиобарбитала или другого способа снятия Alloc защитной группы позволяет экспонировать аминогруппу с целью связывания с ацил-защищенной или незащищенной единичной 1- октил бета-D-глюкуроновой кислотой. Затем заключительная депротекция с помощью расщепляющего коктейля с низким % CF3СО2Н (TFA) дает нужный продукт. Хотя гликозидная связь неустойчива по отношению к сильной кислоте, эксперименты, проведенные нами и другими исследователями, показывают, что она относительно устойчива к расщеплению при низкой концентрации % TFA. Или же, для того, чтобы обеспечить повышенную защиту гликозидной связи, можно использовать ацильную защитную группу (например, ацетильную. Ас; бензоильную, Bz) или триалкилсилильную защитную группу для защиты ОН функциональных групп сахаридов. Последующая депротекция под действием основания (NH2NH2/MeOH; NH2/MeOH, NaOMe/MeOH) дает нужные незащищенные продукты. На Схеме 4 показаны реагенты по настоящему изобретению. На Схеме 5 представлен неограничивающий пример пептидного интермедиата по настоящему описанию. Хотя данный пример показывает пептид с ПАВ-связью на N-конце пептида, методы по настоящему изобретению применимы для синтеза пептидных интермедиатов со связью с ПАВ в срединной области, в С-концевой области или в любом положении внутри пептида.
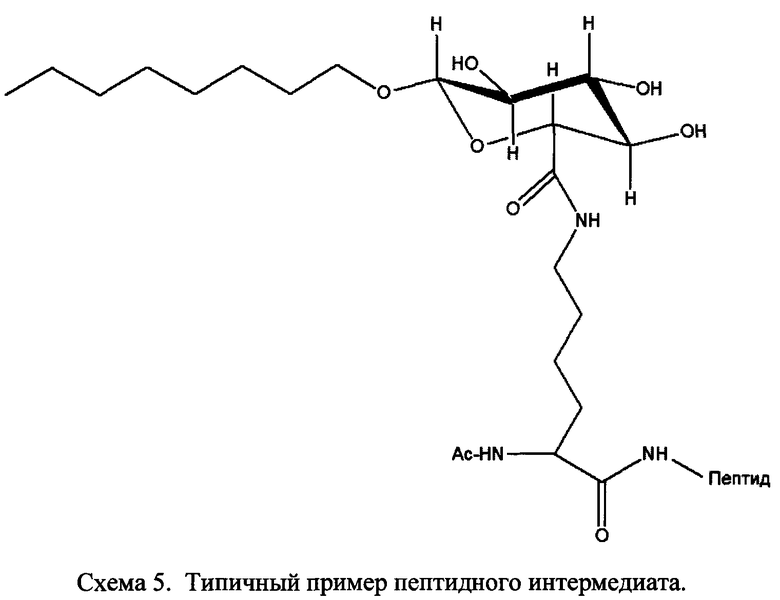
Другие реагенты получают модификацией функциональной группы в положении 6 с образованием различных связей с функциональными группами в боковой цепи аминокислот, как показано ниже, на Схеме 6. Так, можно использовать замену на аминогруппу для связывания с боковыми цепями Asp или Glu. Замену на азидогруппу или алкин или можно применять для связывания с неприродными аминокислотами, содержащими комплементарный акцептор для 3+2 циклоприсоединения по реакции Huisgen (Gauthier, M.A. and Klok, H.A. (2008) Chem Commun (Camb) 2591-2611). Аминоокси- или альдегидные функциональные группы можно использовать для связывания с альдегидной (т.е. оксимная связь) или амино функциональными группами (т.е. восстановительное алкилирование), соответственно. Малеимидная или -NH-(C=O)-СН2-Br функциональная группа может селективно связываться с группой SH в Cys или с другой SH функциональной группой. Такая стратегия связывания является эффективной в сочетании с реагентами по настоящему изобретению. Взаимное превращение функциональных групп широко применяется в органическом синтезе, и имеются подробные списки множества способов модификации каждой функциональной группы (Larock, R.C. (1999)) "Comprehensive Organic Transformations", VCH Publishers, New York.
Так например, первичную гидроксильную группу в положении 6 октил 1-β-D-гюкозида превращают в азид активацией и заменой на азидный анион в соответствии с реакциями, используемыми в химии углеводов (например, тозилированием с последующей реакцией с NaN3). Соответствующий азид восстанавливают до аминогруппы тиолуксусной кислотой в пиридине (Elofsson, M., et al. (1997) Tetrahedron 53: 369-390) или похожими методами получения аминогрупп (Slangier, P., et al. (1994) Liquid Crystals 17: 589-595). Реакции получения ацетиленовых, аминоокси- и альдегидных групп лучше всего проводить в виде триацетокси производных, получаемых из продажного глюкозида обработкой с помощью Ac2O с последующим гидролизом первичного амина в мягких условиях. Это 6-гидроксисоединение можно селективно окислить до альдегида или активировать в виде тозилата или трифлата и заместить с помощью NH2OH или ацетиленида натрия. Малеимидную связь можно получать, как показано, через углеродную связь или, предпочтительно, через О или амидную связь, опять же заменой активированной гидроксильной группы или конденсацией производного глюкуроновой кислоты со связанным с аминогруппой малеимидным реагентом, что хорошо известно в уровне техники. Другие взаимопревращения функциональных групп хорошо известны специалистам в области медицинской химии и входят в объем настоящего изобретения.
Также в рамках методов синтеза по настоящему изобретению рассматриваются ПАВЮ в молекулах которых сахарид и гидрофобная цепь ковалентно связаны через альфа-гликозидную связь. Синтетические способы получения преимущественно α-связанных гликозидов хорошо известны в уровне техники и обычно исходят из перацетильного производного сахара и используют кислый катализатор (например, SnCl4, BF3 или НСl) для осуществления α-гликозилирования (Cudic, M. and Burstein, G.D. (2008) Methods Mol Biol 494: 187-208; Vill, V., et al. (2000) Chem Phys Lipids 104: 75-91, вводимые в настоящее изобретение посредством отсылок). Аналогичные синтетические методы существуют для дисахаридных гликозидов (von Minden, H.M., et al. (2000) Chem Phys Lipids 106: 157-179, вводится в настоящее изобретение посредством отсылки). Затем осуществляются указанные выше взаимные превращения функциональных групп, в результате образуется 6-карбоновая кислота и т.д., применяемая(-ые) для получения соответствующих реагентов с α-связью.
На Схеме 6 приводятся некоторые соединения и реагенты, применимые для синтеза ковалентно модифицированных пептидов и/или белков по настоящему изобретению. Используют стандартную номенклатуру с однобуквенными сокращениями.
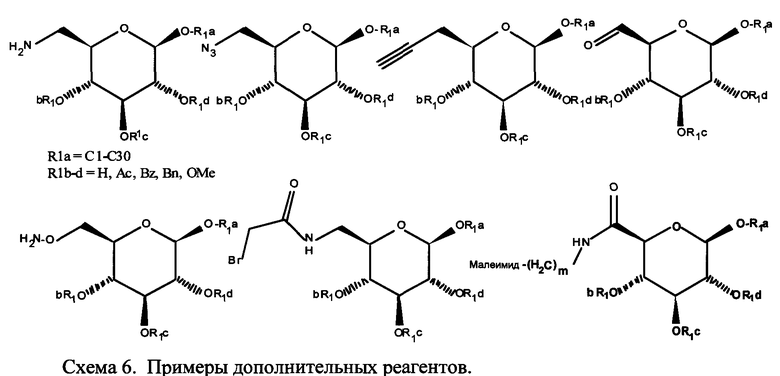
Многие алкилгликозиды можно синтезировать известными методами, описанными, например, в Rosevear, P., et al. (1980) Biochemistry 19: 4108-4115, Li, Y.T., et al. (1991) J Biol Chem 266: 10723-10726) или Koeltzow and Urfer, J. Am. Oil Chem. Soc., 61:1651-1655 (1984), патенте США No. 3,219,656 и патенте США No. 3,839,318, или ферментативными методами, описанными, например, в Li, Y.T., et al. (1991) J Biol Chem 266: 10723-10726, Gopalan, V., et al. (1992) J Biol Chem 267: 9629-9638). Образование O-алкильных связей с природными аминокислотами, такими как Ser, можно наблюдать при использовании Fmoc-Ser-OH и перацетилглюкозы, при этом получают Na-Fmoc-4-O-(2,3,4,6-тетра-O-ацетил-β-D-глюкопиранозил)-L-серин. В этом соединении селективно снимают защитную группу у первичного углеродного атома (в положении 6) и его селективно окисляют с применением TEMPO/BAIB, как описано выше, получают соответствующую 6-карбоксильную функциональную группу, которая может конденсироваться с липофильными аминами с образованием нового класса неионных ПАВ-реагентов (Схема 7).
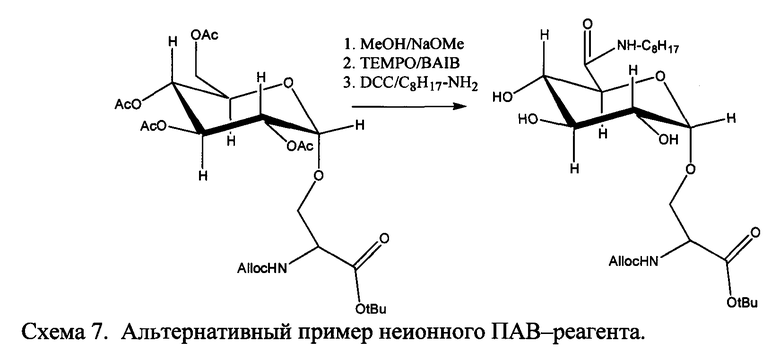
Связь между гидрофобной алкильной группой и гидрофильным сахаридом может включать, наряду с другими возможными, гликозидную, тиогликозидную, амидную (Carbohydrates as Organic Raw Materials, F. W. Lichtenthaler ed., VCH Publishers, New York, 1991), уреидо (Austrian Pat. 386, 414 (1988); Chem. Abstr. 110:137536p (1989); см. Gruber, H. and Greber, G., "Reactive Sucrose Derivatives" in Carbohydrates as Organic Raw Materials, pp. 95-116) или сложноэфирную связь (Sugar Esters: Preparation and Application, J. C. Colbert ed., (Noyes Data Corp., New Jersey), (1974)).
Примеры, из которых можно выбрать алкилгликозиды, применимые для модификации в реагенты или для получения продуктов по настоящему изобретению, включают: алкилгликозиды, такие как октил-, нонил-, децил-, ундецил-, додецил-, тридецил-, тетрадецил, пентацецил-, гексадецил-, гептадецил- и октадецил-D-мальтозид, -глюкозид или -сахарозу (т.е. сложный эфир сахарозы) (синтезированные согласно Koeltzow and Urfer; Anatrace Inc., Maumee, Ohio; Calbiochem, San Diego, Calif.; Fluka Chemie, Switzerland); алкилтиомальтозиды, такие как гептил-, октил-, додецил-, тридецил и тетрадецил-β-D-тиомальтозид (синтезированный по Defaye, J. and Pederson, С., "Hydrogen Fluoride, Solvent and Reagent for Carbohydrate Conversion Technology" в Carbohydrates as Organic Raw Materials, 247-265 (F. W. Lichtenthaler, ed.) VCH Publishers, New York (1991); Ferenci, Т., J. Bacteriol, 144:7-11 (1980)); алкилтиогликозиды, такие как 1-додецил-или 1-октилтио α- или β-D-глюкопиранозид (Anatrace, hie., Maumee, Ohio; см. Saito, S. and Tsuchiya, T. Chem. Pharm. Bull. 33:503-508 (1985)); алкилтио сахарозы (синтезированные, например, по Binder, Т. Р. and Robyt, J. F., Carbohydr. Res. 140:9-20 (1985)); алкил мальтотриозиды (синтезированные по Koeltzow and Urfer); длинноцепные алифатические амиды простых аминоалкиловых эфиров сахарозы (полученные в соответствии с австрийским патентом 382, 381 (1987); Chem. Abstr., 108:114719 (1988) и Gruber and Greber pp. 95-116); производные палатинозы и изомальтамина, связанные амидной связью с алкильной цепью (синтезированные по Kunz, M., "Sucrose-based Hydrophilic Building Blocks as Intermediates for the Synthesis of Surfactants and Polymers" в Carbohydrates as Organic Raw Materials, 127-153); производные изомальтамина, связанные через мочевинную группу с алкильной цепью (синтезированные по методике в Kunz);
длинноцепные алифатические уреиды простых аминоалкиловых эфиров сахарозы (синтезированные по Gruber and Greber, pp.95-116); и длинноцепные алифатические амиды простых аминоалкиловых эфиров сахарозы (полученные в соответствии с австрийским патентом 382, 381 (1987); Chem. Abstr., 108:114719 (1988) и Gruber and Greber pp. 95-116).
Некоторые предпочтительные гликозиды, которые можно далее модифицировать, чтобы включить реакционноспособную функциональную группу для связывания с пептидом, включают сахариды мальтозу, сахарозу, глюкозу и галактозу, связанные гликозидной или сложноэфирной связью с алкилыюй цепью из 6, 8, 10, 12, 14 или 16 углеродных атомов, например, гексил-, октил-, децил-, додецил-, тетрадецил- и гексадецил- мальтозид, сахароза, глюкозид и галактозид. В организме эти гликозиды расщепляются до нетоксического спирта или жирной кислоты и олигосахарида или сахарида. Вышеприведенные примеры иллюстрируют типы алкилгликозидов, применяемых в способах по настоящему изобретению, однако не предполагается, что список является исчерпывающим.
Как правило, эти ПАВ (например, алкилгликозиды), необязательно, синтезированы или выбраны с целью модифицировать биологические свойства пептида, например, модулировать биодоступность, период полужизни, рецепторную селективность, токсичность, биораспределение (поиск тканей-мишеней), растворимость, устойчивость, например, термическую, гидролитическую, окислительную, устойчивость к ферментативному расщеплению и т.п., легкость очистки и процессирования, структурные свойства, спектроскопические свойства, химические и/или фотохимические свойства, каталитическую активность, редокс-потенциал, способность реагировать с другими молекулами, например, за счет ковалентного или нековалентного взаимодействия, и т.п.
ПАВ
Термин "ПАВ" представляет собой сокращение выражения "поверхностно-активное вещество (агент)". В фармацевтике ПАВ применяются в жидких фармацевтических препаратах, в которых они служат множеству целей, действуя как эмульгаторы, стабилизаторы, солюбилизаторы и увлажняющие агенты (увлажнители). Эмульгаторы стабилизируют водные растворы липофильных или частично липофильных веществ. Солюбилизаторы повышают растворимость компонентов фармацевтических композиций, увеличивая концентрацию, которой можно достичь. Увлажняющий агент представляет собой химическую добавку, которая снижает поверхностное натяжение жидкости, заставляя ее быстро распределяться на поверхности, на которую она наносится, тем самым вызывая тем самым равномерное "увлажнение" поверхности жидкостями. Увлажнители дают возможность жидким препаратом достичь тесного контакта со слизистой оболочкой или другими областями поверхности, с которыми фармацевтическая композиция контактирует. Таким образом, ПАВ могут являться полезными добавками для стабилизации препарата пептидных продуктов по настоящему изобретению, а также для модификации свойств самого пептида.
Согласно конкретным вариантам изобретения алкилгликозиды, которые можно получать синтетическими методами, например, алкилгликозиды додецил, тридецил и тетрадецил мальтозид или глюкозид или глюкозид, а также сахарозы додеканоат, тридеканоат и тетрадеканоат, применимы для ковалентного связывания с пептидами по настоящему описанию. Аналогично, соответствующие алкилтиогликозиды представляют собой устойчивые, получаемые доступными методами синтеза ПАВ, приемлемые для разработки препаратов.
Широкий ряд физических и поверхностно-активных свойств можно получить путем соответствующей модификации гидрофобных или гидрофильных областей ПАВ (например, алкилгликозида). Например, сравнительное исследование активности додецил мальтозида (DM) по отношению к (липидному) бислою с активностью додецил глюкозида (DG) показало, что активность DM более чем в три раза выше, чем активность DG, несмотря на одинаковую длину гидрофобного хвоста (Lopez, О., et al. (2002) Colloid Polym Sci 280: 352-357). В данном конкретном примере на характеристики ПАВ влияет идентичность полярной области (дисахарида относительно моносахарида). В случае ПАВ, связанного с пептидом, например, в пептидных продуктах по настоящему изобретению пептидная область также может вносить свой вклад в гидрофобный или гидрофильный характер целой молекулы. Регулируя таким образом физические и поверхностно-активные свойства, можно получать конкретные физические и фармацевтические свойства, соответствующие отдельным целевым пептидам.
Модификация с помощью ПЭГ (PEG)
Согласно некоторым вариантам изобретения модифицированные с помощью ПАВ пептидные продукты далее модифицируют с целью включить один или более фрагментов ПЭГ (PEG) (Veronese, F.M. and Mero, A. (2008) BioDrugs 22: 315-329). В некоторых случаях включение больших цепей ПЭГ (PEG) предупреждает клубочковую фильтрацию пептида в почках с образованием там гипотонической мочи (Nestor, J.J., Jr. (2009) Current Medicinal Chemistry 16: 4399-4418, Caliceti, P. and Veronese, F.M. (2003) Adv Drug Deliv Rev 55: 1261-1277). Согласно некоторым вариантам изобретения необязательная PEG гидрофильная цепь позволяет уравновешивать растворимость и физические свойства пептидов или белков, которым была предоставлена гидрофобность путем введения более длинного алкилгликозидного фрагмента.
Пегилирование белка может оказывать также потенциально отрицательное воздействие. Так, пегилирование может вызывать значительную потерю биологической активности у некоторых белков, и это может относиться к лигандам для специфических классов рецепторов. В таких случаях может быть эффективным обратимое пегилирование (Peleg-Shulman, Т., et al. (2004) J Med Chem 47: 4897-4904, Greenwald, R.B., et al. (2003) Adv Drug Deliv Rev 55: 217-250, Roberts, M.J. and Harris, J.M. (1998) J Pharm Sci 87: 1440-1445).
Помимо этого, повышенная молекулярная масса может препятствовать проникновению, через биологические барьеры, отличные от гломерулярного барьера. Например, было высказано предположение, что высокомолекулярные формы продуктов пегилирования могут препятствовать проникновению в некоторые ткани и тем самым снижать терапевтическую эффективность. Помимо этого, высокая молекулярная масса может препятствовать всасыванию. Помимо этого, высокая молекулярная масса может предупредить всасывание через слизистые оболочки (при назальной, трансбуккальной (защечной), вагинальной, пероральной, ректальной, легочной доставке). Однако, замедленное всасывание (накопление) может быть в высшей степени предпочтительным для введения устойчивых молекул в легкое, значительно пролонгируя продолжительность действия. Пептидные и/или белковые продукты по настоящему изобретению обладают повышенной биодоступностью при введении через слизистую оболочку, и это позволяет применять более длинноцепные ПЭГ-модификации в сочетании с ПАВ-модификацией, достигая при этом с коммерческой точки зрения значимой биодоступности после интраназального введения или любого другого пути введения через слизистую оболочку.
Согласно некоторым вариантам изобретения длинноцепные ПЭГ-полимеры и короткоцепные ПЭГ-полимеры применимы для модификации белков и пептидов по настоящему изобретению. Введение лекарственных средств против диабета с помощью ингаляции является новым подходом к доставке лекарства, а проницаемость барьера легкого высока (например, эксубера). Для такого применения, с отсроченным (замедленным) проникновением через легочный барьер, предпочтительными формами пегилирования являются более низкомолекулярные полимеры в интервале от С10 до C400 (примерно, от 250 до 10000 Да). Таким образом, в то время как основной способ пролонгирования с помощью ПЭГ позволяет достичь "эффективной молекулярной массы" выше порогового значения для клубочковой фильтрации (более 68 кДа), применение более коротких цепей может быть способом пролонгирования пребывания в легком для лечения легочных заболеваний и других респираторных заболеваний. Этих ПЭГ цепей размером от около 500 до 3000 Да достаточно для замедления поступления в периферический кровоток, но недостаточно для того, чтобы значительно пролонгировать время циркуляции. Согласно некоторым вариантам изобретения пегилирование применяют для того, чтобы придать повышенную локальную эффективность легочной ткани с пониженной способностью к системным побочным эффектам ковалентно модифицированных пептидов и/или белков по данному изобретению. Согласно некоторым таким вариантам изобретения ПЭГ цепи в интервале от около 750 до около 1500 Да в общем называют "PEG1K."
Помимо этого, можно применять другие полимеры в сочетании с соединениями по настоящему изобретению с целью оптимизировать их физические свойства. Например, конъюгаты поли(2-этил 2-оксазолина) имеют переменную гидрофобность и достаточный размер для повышения длительности действия (Mero, A., et al. (2008) J Control Release 125: 87-95). Связывание такого полимера с сахаридами дает класс ПАВ, пригодный для применения при модификации пептидов и/или белков по настоящему изобретению.
Цепи полиэтиленгиколя функционализируют для содействия их конъюгации с реакционноспособными группами в пептидной и/или белковой цепи. Типичные функциональные группы содействуют реакции с амино, карбоксильными или сульфгидрильными группами в пептиде за счет соответствующих карбоксильных, амино и малеимидогрупп (и т.п.) в цепи полиэтиленгликоля. Согласно одному варианту изобретения ПЭГ содержит цепь С10-С3000. Согласно другому варианту изобретения ПЭГ имеет молекулярную массу выше 40000 Дальтон. Согласно еще одному варианту изобретения ПЭГ имеет молекулярную массу выше 10000 Дальтон. ПЭГ в качестве реагента для модификации белков хорошо известен в уровне техники и его применение описано, например, в патентах США No. 4,640,835; 4,496,689; 4,301,144; 4,670,417; 4,791,192; и 4,179,337.
Нетрадиционный тип цепи ПЭГ модифицируется таким образом, чтобы иметь амфифильную природу. То-есть, он имеет как гидрофильную структуру ПЭГ, но он модифицирован таким образом, что содержит гидрофобные области, такие как эфиры жирных кислот и другие гидрофобные компоненты. См., например. Miller, M.A., et al. (2006) Bioconjug Chem 17: 267-274; Ekwuribe, et al. патент США 6,309,633; Ekwuribe, et al. патент CIIIA US 6,815,530; Ekwuribe, et al. патент США 6,835,802. Хотя эти амфифильные ПЭГ-конъюгаты с белками первоначально были разработаны для повышения биодоступности при пероральном введении, они были относительно неэффективны в этой роли. Однако применение таких амфифильных ПЭГ-конъюгатов с амфипатическими пептидами сообщает значительно более продолжительное время удержания в легком для того, чтобы продлить полезную биологическую активность этих лекарственных препаратов. Предпочтительные цепи ПЭГ имеют молекулярную массу в интервале от 500 до 3000 Да. Подробное описание методов синтеза этих конъюгатов дано в представленных выше ссылках, полное содержание которых вводится таким образом в данное изобретение.
ПЭГ сам по себе не содержит функциональной группы, которую можно связать с целевой молекулой, такой как пептид. Следовательно, для связывания ПЭГ сам ПЭГ следует сначала функционализировать, затем использовать связывающую функцию для связывания молекулы ПЭГ с целевой молекулой, такой как пептид (Greenwald, R.B., et al. (2003) Adv Drug Deliv Rev 55: 217-250, Veronese, F.M. and Pasut, G. (2005) Drug Discov Today 10: 1451-1458, Roberts, M.J., et al. (2002) Adv Drug Deliv Rev 54: 459-476). Согласно одному варианту изобретения сайт-специфическое пегилирование можно осуществить путем Cys замены в молекуле пептида. Целевой пептид можно синтезировать твердофазным синтезом, методами рекомбинантной ДНК или другим методом по настоящему описанию.
Так, согласно некоторым вариантам изобретения пептидный продукт по настоящему изобретению содержит Lys или другой реактивный остаток, модифицированный алкилгликозидом и специфическим пегилированием по меньшей мере по одному Cys остатку, Lys остатку или по другому реактивному аминокислотному остатку в другом месте молекулы.
Согласно другому варианту изобретения Lys или другой остаток с нуклеофильной боковой цепью можно использовать для включения ПЭГ-остатка. Это можно осуществить, используя связывание через амидную или карбаматную группу с ПЭГ-цепью, содержащей карбоксильную или карбонатную группу. См., например, описание в статье Veronese, F.M. and Pasut, G. (2005) Drug Discov Today 10: 1451-1458. Альтернативный подход состоит в модификации функциональной аминогруппы боковой цепи Lys за счет связывания остатка, содержащего группу SH, такого как меркаптоацетил, меркаптопропионил (СО-СH2-CH2-CH2-SH) и т.п. Или же цепь ПЭГ можно включать на С-конце в виде амида в процессе синтеза. В других методах связывания цепей ПЭГ используют реакцию с боковыми цепями аминокислотных остатков His и Trp. Известны другие аналогичные методы модификации пептидной цепи, позволяющие связывать цепь ПЭГ, они вводятся в настоящее изобретение посредством отсылки (Roberts, M.J., et al. (2002) Adv Drug Deliv Rev 54: 459-476).
Препараты
Согласно одному варианту изобретения ковалентно модифицированные пептиды или белки по настоящему изобретению предусматриваются в виде состава, который, при введении его субъекту, дополнительно уменьшает, предупреждает или сглаживает ассоциацию или агрегацию пептидов и/или белков в композиции, уменьшает самоассоциацию или самоагрегацию пептидов и/или белков, или уменьшает ассоциацию или агрегацию с другими пептидами или белками.
Самоассоциация при высокой концентрации белка в терапевтических препаратах является проблемой. Например, самоассоциация повышает вязкость водного раствора концентрированного моноклонального антитела. Концентрированные препараты инсулина в результате самоагрегации инактивируются. Эти взаимодействия белков, вызывающие самоассоциацию, в частности при высокой концентрации белка, снижают, модулируют или аннулируют биологическую активность многих терапевтических средств (Clodfelter, D.K., et al. (1998) Pharm Res 15: 254-262). Препараты, содержащие терапевтические белки в высоких концентрациях для доставки в виде инъекции или другим способом, могут быть физически неустойчивыми или могут становиться нерастворимыми в результате этих белковых взаимодействий.
Значительную проблему при приготовлении пептидных и белковых препаратов представляет разработка технологичных и устойчивых лекарственных форм. Свойства физической устойчивости, важные для процессирования и обработки, часто недостаточно охарактеризованы и их трудно предсказать. Встречается множество явлений физической неустойчивости (нестабильности), таких как ассоциация, агрегация, кристаллизация и осаждение (преципитация), определяемые взаимодействием и растворимостью белков. Это вызывает значительные проблемы при приготовлении, проблемы устойчивости, проблемы при анализе и доставке. Разработка препаратов пептидных и белковых лекарственных средств, требующих введения высоких доз (порядка мг/кг), необходима во многих клинических случаях. Например, при подкожной (SC) инъекции допустимо вводить примерный объем <1.5 мл. Для введения адекватной дозы в этом случае могут потребоваться концентрации белка >100 мг/мл. Аналогичные концентрации наблюдаются при приготовлении высококонцентрированного лиофилизированного препарата моноклональных антител. Как правило, при более высоких концентрациях белка можно делать инъекции меньших объемов, что очень важно для комфорта и удобства пациента и приверженности его лечению (комплаентности). ПАВ-модифицированные соединения по настоящему изобретению разрабатывают (синтезируют) с целью минимизировать такие события агрегации, чему может способствовать применение малых количеств ПАВ по настоящему описанию.
Поскольку инъекция для многих людей является неудобным, некомфортным способом введения, проводился поиск других способов введения пептидных лекарственных средств. Некоторые пептидные и белковые лекарственные средства можно вводить, например, интраназально, трансбуккально, перорально, вагинально, с помощью ингаляции или другим способом введения через слизистую оболочку. Примерами являются нафарелин (Синарел, Synarel®) и кальцитонин, которые вводят в виде продажных препаратов-назальных спреев. Ковалентно модифицированные пептиды и/или белки по настоящему изобретению созданы с целью упростить такое введение через слизистую, и такие препараты можно далее упростить за счет применения малых количеств ПАВ по настоящему изобретению.
Типичные свойства (критерии) препарата включают выбор раствора с оптимальным значением рН, буфера и стабилизирующих эксципиентов. Кроме того, восстановление (реконституция) лиофилизированного осадка (брикета) является важным для лиофилизированных или порошковых препаратов. Еще одной, и значительной, проблемой является изменение вязкости белкового препарата при самоассоциации. Изменение вязкости может значительно изменить свойства доставки, например, при доставке в виде спрея (аэрозоля) для интраназальной доставки, доставки в легкие и в ротовую полость. Кроме того, повышенная вязкость может затруднить или сделать невозможной доставку в виде инъекции с помощью шприца или капельницы.
Сообщалось о множестве попыток стабилизировать и поддерживать целостность и физиологическую активность пептидов. Некоторые попытки сделали возможной стабилизацию против термической денатурации и агрегации, в частности, для систем с инсулиновым насосом. Полимерные ПАВ описаны в Thurow, H. and Geisen, К. (1984) Diabetologia 27: 212-218; Chawla, A.S., et al. (1985) Diabetes 34: 420-424). Полагают, что стабилизация инсулина с помощью этих соединений имеет пространственную (стерическую) природу. Среди других применяемых систем сахариды (Arakawa, Т. and Timasheff, S.N. (1982) Biochemistry 21: 6536-6544), осмолиты, такие как аминокислоты (Arakawa, Т. and Timasheff, S.N. (1985) Biophys J 47: 411-414), и вещества, разрушающие структуру воды, такие как мочевина (Sato, S., et al. (1983) J Pharm Sci 72: 228-232). Действие этих веществ проявляется в модуляции внутримолекулярного гидрофобного взаимодействия белка или пептида.
Различные пептиды или белки описаны в настоящей заявке и могут быть модифицированы с помощью любых ковалентно связанных ПАВ-реагентов по настоящему описанию. Предпочтительно, модификации пептидов по настоящему описанию включают ковалентное связывание ПАВ-реагента, который содержит как гидрофильные (например, сахаридные), так и гидрофобные (например, алкильную цепь) группы, тем самым содействуя стабилизации пептида в физиологических условиях. Согласно некоторым вариантам изобретения ковалентное связывание молекулы, содержащей как гидрофильную группу, так и гидрофобную группу (например, гликозидное ПАВ), с пептидом и/или белком по настоящей заявке отменяет необходимость в модификации аминокислотной последовательности пептида и/или белка для повышения устойчивости (стабильности) (например, уменьшения агрегации).
Согласно некоторым вариантам изобретения препараты содержат по меньшей мере одно лекарство, включающее пептид, модифицированный реагентом, образованным с помощью ПАВ по настоящему изобретению, и, кроме того, в препарате оно может быть ассоциировано с ПАВ, причем ПАВ, помимо этого, состоит, например, из сахарида, алкилгликозида или другого эксципиента, и препарат можно вводить в виде, выбранном из группы, состоящей из капли, спрея, аэрозоля, лиофилизата, продукта, высушенного распылительной сушкой, в виде инъекций и в виде препарата с пролонгированным высвобождением. Спрей и аэрозоль можно доставлять с применением подходящего дозирующего устройства, диспенсера и вводить интраназально, трансбуккально, в виде ингаляции или другим способом введения через слизистую. Лиофилизат может содержать другие соединения, такие как маннит, сахариды, субмикронные количества безводной α-лактозы, желатин, биосовместимые гели или полимеры. Препараты в форме с пролонгированным высвобождением могут представлять собой глазной вкладыш, разрушаемые микрочастицы, гидролизуемые полимеры, набухающие мукоадгезивные частицы, чувствительные к рН микрочастицы, наночастицы/латексные системы, ионообменные смолы и другие полимерные гели и имплантаты (Ocusert, Alza Corp., California; Joshi, A., S. Ping and K. J. Himmelstein, патентная заявка WO 91/19481). Также достигается биодоступность при пероральном введении.
Модификации пептидов и белков по настоящему изобретению уменьшают, а в некоторых случаях могут исключить, необходимость в органических растворителях. Для предупреждения агрегации применяли трегалозу, лактозу и маннит и другие сахариды. Агрегация гуманизированного моноклонального антитела против IgE была минимизирована с помощью препарата с трегалозой в молярном соотношении выше или равном от 300:1 до 500:1 (эксципиент: белок). Однако порошки обладали избыточным межмолекулярным сцеплением и не подходили для введения в виде аэрозоля, или у них наблюдалось нежелательное гликозилирование при хранении (Andya, J.D., et al. (1999) Pharm Res 16: 350-358). Каждое из раскрываемых вспомогательных веществ имеет ограничения в качестве добавки к терапевтическим веществам, включая метаболизм ксенобиотиков, раздражение или токсичность или высокую стоимость. Для применения с ковалентно модифицированными пептидами и/или белками по настоящему изобретению рассматриваются эксципиенты, которые являются эффективными, нераздражающими и нетоксическими, не требуют метаболизма ксенобиотиков, так как они состоят из природных Сахаров, жирных кислот или длинноцепных спиртов, и которые также можно использовать для минимизации агрегации в водных растворах или при восстановлении водой высушенных препаратов пептидов и/или белков in situ при физиологическом восстановлении водными физиологическими жидкостями, такими как плазма или слюна.
Другие компоненты препарата могут включать буферные растворы и физиологические соли, нетоксические ингибиторы протеаз, такие как апротинин, соевый ингибитор трипсина, альфа-1-антитрипсин и, среди прочего, инактивирующие протеазу моноклональные антитела. Буферы могут включать органические вещества (соли), такие как ацетат, цитрат, глюконат, фумарат, малат, полилизин, полиглутамат, хитозан, сульфат декстрана и т.п., или нерганические вещества, такие как фосфат и сульфат. Такие препараты могут, кроме того, содержать малые концентрации бактериостатических агентов, таких как бензиловый спирт и т.п.
Препараты, подходящие для интраназального введения, также представляют собой растворы или суспензии модифицированных пептидных и/или белковых продуктов по настоящему изобретению в приемлемых летучих растворителях, таких как гидрофторалканы. Такие препараты применимы для введения с помощью дозирующих ингаляторов (MDI) и их преимущество заключается в отсутствии передвижения от места введения, малое раздражение и отсутствие необходимости в стерилизации. Такие препараты могут также содержать приемлемые эксципиенты или наполнители, такие как субмикронные частицы безводной α-лактозы.
Согласно еще одному варианту изобретения ковалентно модифицированные пептиды и/или белки по настоящему описанию имеют повышенное время хранения. В данном контексте выражение "время хранения" в широком смысле означает, насколько долго продукт может храниться, не становясь непригодным для применения или потребления. "Время хранения" композиции по настоящему изобретению может также указывать на период времени, в течение которого происходит допустимая потеря качества композиции. Время хранения композиции в данном контексте отличается от даты истечения срока годности; "время хранения" относится к качеству композиции по настоящему изобретению, тогда как "срок годности" больше относится к требованиям к производству и испытаниям композиции. Например, композиция, "срок годности" которой прошел, может все еще сохранять безопасность и эффективность, но оптимальное качество более не гарантируется производителем.
Введение доз
Ковалентно модифицированные пептиды и/или белки по настоящему изобретению можно вводить в любом количестве, способном оказать благотворное терапевтическое действие в ряде болезненных состояний. Согласно некоторым вариантам изобретения ковалентно модифицированные пептиды и/или белки по настоящему изобретению применимы для лечения воспаления. Согласно некоторым вариантам изобретения соединения по настоящему изобретению оказывают благотворное действие на модуляцию послеоперационной или хронической боли. Согласно одному варианту изобретения пептиды вводят пациенту в концентрациях выше или ниже концентраций других видов лекарственного средства, которые модулируют боль. Согласно еще одному варианту изобретения пептиды вводят с другими соединениями, чтобы вызвать синергический терапевтический эффект.
Типичные схемы лечения включают пероральное, чресслизистое введение, парентеральное (включая подкожную, интраперитонеальную, внутримышечную и внутривенную инъекцию) введение, ректальное, трансбуккальное (включая подъязычное), трансдермальное введение, ингаляцию, введение в глаз и чресслизистое (включая интраназальное) введение. Привлекательный широко применяемый способ доставки пептидов предусматривает подкожную инъекцию инъецируемого препарата с контролируемым высвобождением. Согласно некоторым вариантам изобретения ковалентно модифицированные пептиды и/или белки по настоящему изобретению применимы для подкожного, интраназального введения и введения в виде ингаляций. Кроме того, в зависимости от заболевания, подлежащего лечению, эти терапевтические композиции вводятся системно или местно. Методы приготовления и применения можно найти в последнем издании "Remington′s Pharmaceutical Sciences" (Mack Publishing Co, Easton Pa.).
На выбор точной дозы и композиции и наиболее подходящей схемы доставки влияют, среди прочего, фармакологические свойства выбранного пептида, характер и тяжесть подлежащего лечению заболевания и физическое и умственное состояние реципиента. Кроме того, способ введения определяет различные количества всасываемого материала. Наблюдаемая биодоступность при введении пептидов различными способами очень меняется, от менее 1% до почти 100%. Как правило, биодоступность при введении с помощью внутривенной, интраперитонеальной или подкожной инъекции составляет 50% или менее.
Обычно ковалентно модифицированные пептиды и/или белки по настоящему изобретению, или их соли, в виде подкожной инъекции вводят в количествах примерно от 0.1 до 1000 мкг/кг веса тела в день, или примерно от 0.1 до 100 мкг/кг веса тела в день. Для женщины весом 50 кг при введении в виде подкожной инъекции суточная доза активного ингредиента составляет примерно от 5 до 5000 мкг, или примерно от 5 до 5000 мкг подкожной инъекцией. В зависимости от способа введения, активности соединения, фармакокинетического профиля и установленной биодоступности требуются различные дозы. При введении в виде ингаляции суточная доза составляет от 1000 примерно до 20000 мкг, дважды в день. Для других млекопитающих, таких как лошади, собаки и крупный рогатый скот, могут потребоваться более высокие дозы. Эту дозу можно доставлять в виде обычной фармацевтической композиции с помощью однократного введения, многократного введения или введения с контролируемым высвобождением, для достижения наиболее эффективных результатов.
Фармацевтически приемлемые соли сохраняют нужную биологическую активность исходного пептида без токсических побочных эффектов. Примерами таких солей являются (а) кислотно-аддитивные соли, образованные из неорганических кислот, например, соляной кислоты, бромистоводородной кислоты, серной кислоты, фосфорной кислоты, азотной кислоты и т.п.; и соли, образованные из органических кислот, например, таких как уксусная кислота, трифторуксусная кислота, винная кислота, янтарная кислота, малеиновая кислота, фумаровая кислота, глюконовая кислота, лимонная кислота, яблочная кислота, бензойная кислота, дубильная кислота, памовая кислота, альгиновая кислота, полиглутаминовая кислота, нафталинсульфокислоты, нафталиндисульфокислоты, полигалактуроновая кислота и т.п.; (б) основно-аддитивные соли или комплексы, образуемые с катионами поливалентных металлов, такими как цинк, кальций, висмут, барий, магний, алюминий, медь, кобальт, никель, кадмий и т.п.; или с органическим катионом, образуемым из N,N′-дибензилэтилендиамина или этилендиамина; или (в) комбинации (а) и (б), например, соль таннат цинка и т.п.
Также в некоторых вариантах изобретения рассматриваются фармацевтические композиции, содержащие в качестве активного ингредиента ковалентно модифицированные пептиды и/или белки по настоящему изобретению, или их фармацевтически приемлемые соли, в смеси с фармацевтически приемлемым нетоксическим носителем. Как указывается выше, такие композиции можно приготовить для парентерального (подкожного, внутримышечного или внутривенного) введения, в частности, в виде жидких растворов или суспензий; для перорального или трансбуккального введения, в частности, в виде таблеток или капсул; для интраназального введения, в частности, в виде порошков, капель в нос, растворов в летучих растворителях или аэрозолей; для ингаляции, в частности, в виде жидких растворов или сухих порошков с эксципиентами в широком смысле; и для ректального или трансдермального введения.
Композиции удобно вводить в виде разовой (однократной) дозы, и их можно приготовить любым из методов, хорошо известных в фармацевтике, например, описанных в книге Remington′s Pharmaceutical Sciences, 17th ed.. Mack Publishing Company, Easton, Pa., (1985), которая включена в настоящее изобретение посредством отсылки. Препараты для парентерального введения могут содержать в качестве эксципиентов стерильную воду или физиологический солевой раствор, алкиленгликоли, такие как пропиленгликоль, полиалкиленгликоли, такие как полиэтиленгликоль, сахариды, масла растительного происхождения, гидрированные нафталины, наночастицы альбумина бычьей сыворотки (применяемого в препарате Abraxane™, American Pharmaceutical Partners, Inc. Schaumburg IL), и т.п.Для перорального введения действие препарата можно усилить, добавляя соли желчных кислот или ацилкарнитины. Препараты для назального введения могут представлять собой твердые вещества или растворы в летучих растворителях, таких как гидрофторуглероды, и могут содержать эксципиенты для стабилизации, например, сахариды, ПАВ, субмикронные частицы безводной а- лактозы или декстрана, или могут представлять собой водные или масляные растворы для применения в виде капель в нос или дозированного спрея. Типичные эксципиенты для трансбуккального применения включают сахара, стеарат кальция, стеарат магния, прежелатинизированный крахмал и т.п.
В препаратах для назального введения всасывание через слизистую оболочку носа можно дополнительно увеличить с помощью ПАВ, например, таких как гликохолевая кислота, холевая кислота, таурохолевая кислота, этохолевая кислота, дезоксихолевая кислота, хенодезоксихолевая кислота, дегидрохолевая кислота, гликодезоксихолевая кислота, циклодекстрины и т.п., в количественном диапазоне примерно от 0.1 до 15 весовых процентов, примерно от 0.5 до 4 весовых процентов или в количестве примерно 2 весовых процента. Другим классом энхансеров всасывания, по сообщениям, проявляющих повышенную активность с пониженным раздражающим действием, является класс алкилмальтозидов, таких как тетрадецилмальтозид (Arnold, J.J., et al. (2004) J Pharm Sci 93: 2205-2213, Ahsan, F., et al. (2001) Pharm Res 18: 1742-1746 и приведенные в этих источниках ссылки, все из которых включены в настоящее изобретение посредством отсылки).
Среди препаратов для доставки с помощью ингаляции ряд препаратов дает преимущества. Адсорбция активного пептида на легко диспергируемых твердых веществах, таких как дикетопиперазины (например, частицы Technosphere; (Pfutzner, A. and Forst, T. (2005) Expert Opin Drug Deliv 2: 1097-1106) или подобные структуры дают препарат, который вызывает быстрое первичное всасывание терапевтического агента). Лиофилизированные порошки, в особенности гладкие частицы, содержащие активный пептид и эксципиент, применимы для доставки в легкое с хорошей биодоступностью, например, см. Exubera® (эксубера, инсулин для инъекций от фирмы Pfizer and Aventis Pharmaceuticals Inc.). Другие системы для доставки пептидов с помощью ингаляции описаны в Mandal, Т.К., Am. J. Health Syst. Pharm. 62: 1359-64 (2005).
Доставка ковалентно модифицированных пептидов и/или белков по настоящему изобретению субъекту в течение продолжительного периода времени, например, в течение от одной недели до одного года, можно осуществлять с помощью однократного введения системы с контролируемым высвобождением, содержащей достаточно активный ингредиент, в течение заданного периода высвобождения. Для этой цели можно применять различные системы с контролируемым высвобождением, такие как монолитные микрокапсулы или микрокапсулы типа хранилища, депо-имплантаты, полимерные гидрогели, осмотические насосы, везикулы, мицеллы, липосомы, трансдермальные пластыри, ионофоретические устройства и альтернативные лекарственные формы для инъекций. Также были разработаны эксципиенты с контролируемым высвобождением для введения дважды в неделю или раз в неделю, например, защищенную систему с привитым сополимером (Castillo, G.M., et al. (2012) Pharm Res 29: 306-18) можно применять для гидрофобных пептидов или пептидов, модифицированных с помощью гидрофобных агентов, таких как пептиды по настоящему изобретению. Локализация в месте, куда необходимо доставить активный ингредиент, является дополнительным признаком некоторых устройств с контролируемым высвобождением, которое может оказаться полезным при лечении некоторых расстройств.
Одна из форм препарата с контролируемым высвобождением содержит пептид или его соль, диспергированные или инкапсулированные в медленно разрушающемся нетоксическом неантигенном полимере, таком как сополимер молочной/гликолевой кислот, описанный в пионерской работе Kent, Lewis, Sanders, and Tice, патент США No. 4,675,189, которая включена в настоящее изобретение посредством отсылки. Соединения, или их соли, можно также готовить в виде гранул в холестериновой или другой липидной матрице, или в виде имплантатов из силиконовых (кремнийорганических) эластомеров. Другие депо-имплантаты или препараты для инъекций с пролонгированным освобождением будут очевидны для специалиста в данной области техники. См., например, Sustained and Controlled Release Drug Delivery Systems, J. R. Robinson ed., Marcel Dekker, Inc., New York, 1978, и R. W. Baker, Controlled Release of Biologically Active Agents, John Wiley & Sons, New York, 1987.
Другая форма препарата с контролируемым высвобождением включает раствор биоразрушаемого полимера, такого как сополимер молочной/гликолевой кислот или блок сополимеры молочной кислоты и ПЭГ, биологически приемлемый растворитель, который инъецируют подкожно или внутримышечно, получая депо-препарат. Объединение пептидов по настоящему изобретению с таким полимерным препаратом применимо для получения препаратов с очень продолжительным периодом действия.
В данном контексте выражение "терапевтически эффективное количество" является синонимом выражения "эффективное количество" и в связи с этим определяется такими соображениями, которые известны в уровне техники. Это количество должно быть эффективным для достижения заданного обусловленного лекарством эффекта у проходящих лечение субъектов, страдающих данным заболеванием. Терапевтически эффективное количество также включает, но без ограничения, соответствующие показатели, выбранные специалистами в данной области техники, например, повышенный коэффициент выживаемости, более быстрое восстановление, или уменьшение интенсивности, или прекращение симптомов, или другие приемлемые биомаркеры или суррогатные маркеры.
Однако следует принять во внимание, что конкретный уровень дозы и частота введения доз для каждого конкретного субъекта, нуждающегося в лечении, могут меняться и зависят от различных факторов, включая активность конкретного применяемого соединения, метаболическую стабильность и продолжительность действия этого соединения, возраст, массу тела, общее состояние здоровья, пол, диету, способ и время введения, скорость выведения, лекарственную комбинацию, тяжесть конкретного состояния и лечение, которое проходит хозяин.
Метод(ы) приготовления доз лекарств включае(ю)т все аспекты композиций по настоящему изобретению, в том числе, но без ограничения, композиции, которые снижают или исключают иммуногенность пептидных и/или белковых лекарств, не вызывают раздражения, обладают антибактериальной или противогрибковой активностью, повышенной стабильностью или биодоступностью лекарственного вещества, снижают колебания биодоступности этого лекарства, позволяют избежать клиренса при первом прохождении через печень и уменьшить или исключить любые побочные эффекты. В данном контексте термин "иммуногенность" означает способность конкретного вещества или конкретной композиции или конкретного агента вызывать иммунологическую реакцию. Иммуногенность ковалентно модифицированных пептидов и/или белков по настоящему описанию подтверждена методами, известными в уровне техники.
Все публикации и патентные заявки, упоминаемые в настоящем описании, включены в настоящее изобретение посредством отсылки в той степени, как если бы указывалось, что каждая независимая публикация или патентная заявка конкретно и индивидуально включается посредством отсылки.
Ковалентно модифицированные пептиды и/или белки по настоящему изобретению и реагенты для их синтеза более конкретно описаны в нижеприведенных примерах, которые предлагаются только для иллюстрации, так как их многочисленные модификации и варианты будут очевидны для специалистов в данной области техники.
ПРИМЕРЫ
Пример 1: Реагенты-N-α-Fmoc, N-ε-(1-октил β-D-глюкуронид-6-ил)-L-лизин
В высушенную в сушильном шкафу колбу Эрленмейера на 250 мл поместили 1-октил β-D-глюкуроновую кислоту (Carbosynth Ltd., 3.06 г, 10 ммолей), 50 мл безводного DMF безводный 1- гидроксибензотриазол (1.62 г, 12 ммолей). При перемешивании добавляли охлажденный (4°С) раствор N,N′- дициклогексилкарбодиимида (2.48 г, 12 ммолей) в 50 мл DMF и реакцию проводили 5 минут. Обильный белый осадок N,N′-дициклогексилмочевины отфильтровывали на воронке с пористым стеклянным фильтром и фильтрат добавляли к раствору N-α-Fmoc-L-лизина (3.68 г, 10 ммолей) в 25 мл безводного DMF. Реакцию проводили в течение 25 мин при нагревании до комнатной температуры или до тех пор, пока цвет нингидрина не станет совсем бледным. Реакционную смесь фильтровали, отжимали досуха и кристаллизовали из смеси MeOH/Et2O, растворяя в МеОН и медленно добавляя Et2O до точки помутнения с последующим охлаждением. Дальнейшую очистку можно проводить хроматографией на силикагеле в градиенте растворителя от EtOAc до EtOAc/EtOH/AcOH.
Аналогичным образом, но замещая с помощью N-α-Boc-L-лизина, получали N-α-Boc,N-ε-(1-октил β-D-глюкуронид-6-yl)-L-лизин, применимый для N-концевого включения и расщепления по свободному N-концу. Аналогичным способом, но замещая с помощью N-α-Ac-L-лизина, получали N-α-Ac,N-ε-(1-октил β-D-глюкуронид-6-ил)-L-лизин, применимый для включения на N-конце пептида блокированного N-конца. Аналогичным способом, но замещая соответствующим количеством N-α-Fmoc-L-орнитина, получали N-α-Fmoc,N-δ-(1-октил β-D-глюкуронид-6-ил)-L-орнитин. Аналогичным способом, но замещая на другие N-монозамещенные диаминокислоты, получали соответствующие реагенты. Или же, применение во время конденсации защитной группы для неустойчивого эфира Me3Si и без предварительной активации 1-октил β-D-глюкуроновой кислоты предоставлял простой путь к получению реагентов. Неустойчивый Me3Si эфир получают по реакции Fmoc-Lys-OH с эквимолекулярным количеством N,O-бис(триметилсилил)ацетамида в хлористом метилене (CH2Cl2). Органический слой содержал нужный реагент в виде раствора в СН2Сl2, готового для описанной выше конденсации с 1-алкил глюкуронидом. Отфильтрованную реакционную смесь промывали водным раствором NaHSO4 для гидролиза Me3Si эфира, сушили MgSO4 и растворитель отгоняли.
Аналогичным способом, но с применением перацетил или пербензоил 1-октил β-D-глюкуроновой кислоты, получали Ас- или Bz-защищенные формы реагентов (например, 2,3,4-триацетил 1-октил β-D-глюкуроновой кислоты и т.п., образующей обработкой Ac2O). Такие реагенты обладают повышенной устойчивостью при отщеплении кислотой от полимерной подложки и применяются, когда обнаруживается неустойчивость при отщеплении, см. Kihiberg, J., et al. (1997) Methods Enzymol 289: 221-245 и приведенные в этой статье отсылки. Конечную депротекцию таких продуктов проводили переэтерификацией в присутствии катализатора после отщепления, с применением MeOH/NH3, MeOH/NaOMe, MeOH/NH2NH2, как описано выше.
Пример 2: Синтетические аналоги пептидов
Как правило, методы пептидного синтеза включают последовательное добавление защищенных аминокислот к растущей пептидной цепи. Обычно защищают либо амино-, либо карбоксильную группу первой аминокислоты и любую реакционноспособную группу в боковой цепи. Эту защищенную аминокислоту затем либо связывают с инертной твердой подложкой (носителем), либо применяют в растворе, а следующую аминокислоту в последовательности, также соответствующим образом защищенную, добавляют в условиях, способствующих образованию амидной связи. После того, как все нужные аминокислоты соединяются в соответствующей последовательности, защитные группы и любая твердая подложка удаляются и получают сырой пептид. Обессоливание и очистку пептида проводят хроматографическим методом.
Предпочтительным методом получения аналогов физиологически активных процессированных пептидов, содержащих менее примерно пятидесяти аминокислот, является тверд офазный пептидный синтез. В соответствии с этим методом α-амино (Nα) функции и любые реакционноспособные боковые цепи защищают группами, чувствительными к кислоте или основанию. Защитная группа должна быть устойчива в условиях образования пептидной связи, но при этом она должна легко удаляться, не затрагивая имеющуюся пептидную цепь. Соответствующие защитные группы для защиты α-аминогрупп включают, но без ограничения трет-бутилоксикарбинольную (Воc), бензилоксикарбонильную (Cbz), о-хлорбензилоксикарбонильную, бифенилизопропилоксикарбонильную, трет-амилоксикарбонильную (Amoc), изоборнилоксикарбонильную, α,α-димегил-3,5-диметоксибензоил-оксикарбонильную, о-нитрофенилсульфенильную, 2-циано-трет-бутоксикарбонильную, 9-фторфенил-метоксикарбонильную (Fmoc) группы и т.п., предпочтительно. Вое или, более предпочтительно, Fmoc. Соответствующие защитные группы для защиты боковых цепей включают, но без ограничения: ацетильную, бензильную (Bzl), бензилоксиметильную (Bom), Воc, трет-бутильную, о-бромбензоилоксикарбонильную, трет-бутилдиметилсилильную, 2-хлорбензильную (Cl-z), 2,6-дихлорбензильную, циклогексильную, циклопентильную, изопропильную, пивалильную, тетрагидрофуран-2-ильную, тозильную (Tos), 2,2,4,6,7-пентаметилдигидробензофуран-5-сульфонильную (Pbf), триметилсилильную и тритильную. Предпочтительной Nα-защитной группой для синтеза соединений является Fmoc группа. Предпочтительными защитными группами для боковых цепей являются O-трет-бутильная группа для Glu, Tyr, Thr, Asp и Ser; Boc группа для боковых цепей Lys и Trp; Pbf группа для Arg; Trt группа для Asn, Gln и His. Для селективной модификации остатка Lys предпочтительной является ортогональная защита с помощью защитной группы, которая не снимается реагентами, отщепляющими защитные группы на основе Fmoc или трет-бутильной группы. Предпочтительные примеры для модификации боковой цепи Lys включают, но без ограничения, защитные группы, которые удаляются под действием гидразина, но не пиперидина; например, 1-(4,4-диметил-2,6-диоксициклогекс-1-илиден)-3-метилбутил (ivDde) или 1-(4,4-ди-2,6-диоксициклогекс-1-илиден)этил (Dde) и аллилоксикарбонил (Alloc). Схема с защитной группой Fmoc-Lys(ivDde) или Fmoc-Lys(Dde) является предпочтительной в тех случаях, когда предпочтительным является образование лактама в боковой цепи (Houston, M.E., Jr., et аl. (1995) J Pept Sci 1: 274-282; Murage, E.N., et al. (2010) J Med Chem), так как в этом случае можно включать Fmoc-Glu(O-Allyl) и Fmoc-Lys(Alloc) и применять для временной защиты, затем снимать с целью образования лактама, в то время как Lys(Dde) защитная группа остается и удаляется позднее в реакции с функционализированным ПАВ.
Схема с защитной группой Fmoc-Lys(ivDde) или Fmoc-Lys(Dde) является предпочтительной в тех случаях, когда желательно образование лактама в боковой цепи (Houston, M.E., Jr., et al. (1995) J Pept Sci 1: 274-282; Murage, E.N., et al. (2010) J Med Chem), так как в этом случае Fmoc-Glu(O-Allyl) и Fmoc-Lys(Alloc) можно включать и использовать для обеспечения временной защиты, затем снимать (депротекционировать) с целью образования лактама, в то время как Lys(Dde) защитная группа остается и удаляется позднее в реакции с функционализированным ПАВ.
При проведении твердофазного синтеза С-концевую аминокислоту сначала связывают с подходящей полимерной подложкой. Подходящими полимерными подложками являются такие материалы, которые инертны по отношению к реагентам и условиям реакций постадийной конденсации и депротекции, а также нерастворимы в применяемых средах. Примеры имеющихся в продаже полимеров (смол) включают смолы на основе сополимеров стирола/дивинилбензола, модифицированные реакционноспособной группой, например, хлорметилированный сополимер стирола с дивинилбензолом, гидроксиметилированный сополимер стирола с дивинилбензолом и т.п. Для получения пептидных кислот предпочтительным является бензилированная, гидроксиметилированная фенилацетамидометил (РАМ)-смола. Если С-концевая группа соединения представляет собой амидную группу, предпочтительной смолой является п-метилбензгидриламино-сополимер(стирол-дивинилбензол)-смола, смола на основе 2,4-диметоксибензгидриламино ("Амид Ринка") и т.п. Особенно предпочтительной подложкой для синтеза пептидов большего размера являются имеющиеся в продаже смолы, содержащие ПЭГ последовательности, привитые на другие полимерные матрицы, такие как смолы Амид Ринка-PEG (ПЭГ) и PAL-PEG-PS (Applied Biosystems) или аналогичные смолы, созданные для получения пептид амидов с применением Fmoc протокола. Так, в некоторых случаях желательно иметь амидную связь с ПЭГ-цепью. В этих случаях удобно связывать N-Fmoc-амино-ПЭГ(РЕО)-карбоновую кислоту со смолой, образующей амидную связь, представленную выше (например смола Ринка (Амид Ринка) и т.п.). Первая аминокислота в цепи может конденсироваться как N-Fmoc-аминокислота с (функциональной) аминогруппой цепи ПЭГ. Конечная депротекция дает заданный продукт Пептид-NH-PEG-СО-NH2.
Связывание с РАМ- смолой можно осуществлять по реакции Nα-защищенной аминокислоты, например, Воc-аминокислоты, в виде ее аммониевой, цезиевой, триэтиламмониевой соли, солей 1,5- диазабицикло-[5.4.0]ундец-5-ена, тетраметиламмония или аналогичной соли в этаноле, ацетонитриле, N,N-диметилформамиде (DMF) и т.п., предпочтительно, цезиевой соли в DMF, со смолой при повышенной температуре, например, при температуре примерно от 40° до 60°С, предпочтительно, примерно при 50°С, в течение примерно от 12 до 72 часов, предпочтительно, в течение примерно 48 часов. В конечном счете получали пептидный продукт в виде кислоты после кислотного расщепления или амида после аминолиза.
Nα-Boc-аминокислоту можно связывать с бензгидриламин-смолой по реакции конденсации, опосредуемой, например, N,N′-диизопропилкарбодиимидом (DIC)/1-гидроксибензотриазолом (HOBt), в течение примерно от 2 до 24 часов, предпочтительно, в течение 2 часов при температуре примерно от 10° до 50°С, предпочтительно, при температуре 25°С в таком растворителе как СН2Сl2 или DMF, предпочтительно, СН2Сl2.
В соответствии с протоколами на основе Вое успешную конденсацию защищенных аминокислот можно проводить методами, хорошо известными в уровне техники, как правило, на автоматическом синтезаторе пептидов. После нейтрализации триэтиламином, N,N-диизопропилэтиламином (DIEA), N-метилморфолином (NMM), коллидином и аналогичным основанием каждую защищенную аминогруппу вводили примерно в 1.5-2.5-кратном молярном избытке и конденсацию проводили в инертном неводном полярном растворителе, таком как СН2Сl2, DMF, N-метилпирролидон (NMP), N,N-диметилацетамид (DMA) или их смеси, предпочтительно, в хлористом метилене при комнатной температуре. Согласно протоколам на основе Fmoc для депротекции не используют никакой кислоты, но только основание, предпочтительно, DIEA или NMM, которые обычно вводят в состав смеси для конденсации. Конденсацию обычно проводили в DMF, NMP, DMA или в смеси растворителей, предпочтительно, в DMF. Типичными конденсирующими агентами являются N,N′-дициклогексилкарбодиимид (DCC), N,N′-диизопропилкарбодиимид (DIC) или другие карбодиимиды, либо самостоятельно, либо в присутствии HOBt, О-ацилмочевин, бензотриазол-1-ил-окситрис(пирролидино)фосфония гексафторфосфата (РуВор), N-гидроксисукцинимида, других N-гидроксиимидов или оксимов. Или же можно применять защищенные активные сложные эфиры аминокислот (например, п-нитрофенил, пентафторфенил и т.п.) или симметричные ангидриды. Предпочтительными конденсирующими агентами являются соединения класса аммоний(аминий)/уроний (альтернативные номенклатуры, применяемые поставщиками), такие как 2-(1Н-бензотриазол-1-ил)-1,1,3,3-тетраметиламмония гексафторфосфат (HBTU), O-(7-азабензотриазол-1-ил)-1,1,3,3-тетраметилурония гексафторфосфат (HATU), 2-(6-хлор-1Н-бензотриазол-1-ил)-1,1,3,3-тетраметиламмония гексафторфосфат (HCTU) и т.п.
Предпочтительный метод связывания с Fmoc-PAL-PEG-PS смолой можно осуществлять депротекцией полимерного линкера с помощью 20% раствора пиперидина в DMF с последующей реакцией N-α-Fmoc-защищенной аминокислоты, примерно при 5-кратном молярном избытке N-α-Fmoc-аминокислоты, с применением HBTU: диизопропилэтиламин (DIEA) (1:2) в DMF в микроволновом синтезаторе пептидов, цикл конденсации 5 мин, максимальная температура 75°.
В соответствии с этим протоколом на основе Fmoc в микроволновом синтезаторе пептидов N-α-Fmoc-защитные группы аминокислот удаляли 20% раствором пиперидина в DMF, содержащим 0.1 М 1-гидроксибензотриазола (HOBt), по протоколу двойной защиты в течение 30 секунд, а затем в течение 3 минут при максимальной температуре 75°С. В раствор для депротекции добавляли HOBt, чтобы уменьшить образование аспартимида. Для добавления (конденсации) следующей аминокислоты применяли пятикратный молярный избыток, используя HBTU:DIEA (1:2), цикл двойного присоединения (конденсации): 5 мин, максимальная температура 75°.
По окончании твердофазного синтеза полностью защищенный пептид снимали со смолы. Если связь с полимерной подложкой относится к типу бензилового эфира, отщепление можно проводить аминолизом с помощью алкиламина или фторалкиламина для пептидов с алкиламидным С- концом, или аммонолизом, например, аммиаком/в метаноле или аммиаком/ в этаноле для пептидов с незамещенным амидом на С-конце, при температуре примерно от -10° до 50°С, предпочтительно, примерно при 25°С, в течение примерно от 12 до 24 часов, предпочтительно, около 18 часов. Пептиды с гидроксигруппой на С-конце можно отщеплять с помощью HF или используя другую схему депротекции сильной кислотой, или омылением. Или же пептид можно удалять со смолы переэтерификацией, например, метанолом, с последующим аминолизом или омылением. Защищенный пептид можно очищать ВЭЖХ на силикагеле или обращенно-фазовой ВЭЖХ.
Защитные группы в боковой цепи можно удалять из пептидов, обрабатывая продукт аминолиза, например, безводным жидким фтористьм водородом в присутствии анизола или другого "утилизатора" карбониевых ионов, обработкой комплексом фтористый водород/пиридин, обработкой трис(трифторацетил)бораном и трифторуксусной кислотой, восстановлением водородом в присутствии палладия на угле или поливинилпирролидоне, или восстановлением натрием в жидком аммиаке, предпочтительно, жидким фтористым водородом и анизолом при температуре примерно от -10° до +10°С, предпочтительно, при 0°С, в течение примерно от 15 минут до 2 часов, предпочтительно, около 1.5 часов.
Для пептидов на смолах типа бензгидриламина стадию отщепления от смолы и стадию депротекции можно объединять в одну стадию, используя жидкий фтористый водород и анизол, как описано выше, или, предпочтительно, используя для отщепления более мягкие "коктейли". Например, в случае смолы PAL-PEG-PS предпочтительным методом является метод с использованием протокола двойной депротекции в микроволновом синтезаторе пептидов с помощью одного из мягких "коктейлей" для отщепления, известных в уровне техники, таком как TFA/вода/ триизопропилсилан/3,6-диокса-1,8-октандитиол (DODT) (92.5/2.5/2.5/2.5), в течение 18 мин при 38°С каждый раз. Отщепление материалов, содержащих алкилгликозид, показало устойчивость гликозидной связи при использовании протокола при соотношении TFA/вода в интервале от 9/1 до 19/1. Типичным коктейлем является смесь состава 94% TFA: 2% EDT; 2% Н2О; 2% TIS. Как правило, полностью депротекционированный продукт осаждали и промывали холодным (от -70° до 4°С) Et2O, растворяли в деионизированной воде и лиофилизировали.
Раствор пептида можно обессоливать (например, на анионообменной смоле BioRad AG-3®), и пептид очищали, проводя ряд стадий хроматографической очистки, используя любой из нижеприведенных методов хроматографии или все эти методы: ионообменную хроматографию на слабоосновной смоле в виде ацетата; гидрофобную адсорбционную хроматографию на недериватизированном сополимере стирол-дивинилбензол, например, Amberlite® XAD; адсорбционную хроматографию на силикагеле; ионообменную хроматографию на карбоксиметилцеллюлозе; распределительную хроматографию, например, на Sephadex® G-25; противоточную хроматографию; сверхкритическую жидкостную хроматографию; или ВЭЖХ, в особенности обращенно-фазовую ВЭЖХ на колонке с октил- или октадецилсилил силикагелем (ODS).
Также в настоящем изобретении предусматриваются способы получения ковалентно модифицированных пептидов и/или белков по настоящему изобретению и их фармацевтически приемлемых солей, эти способы включают последовательную конденсацию защищенных аминокислот на подходящей полимерной подложке, удаление защитных групп и полимерной подложки и очистку продукта и получение аналогов физиологически активных процессированных гомологов и аналогов ковалентно модифицированных пептидов и/или белков по настоящему изобретению. Согласно некоторым вариантам изобретения ковалентно модифицированные пептиды и/или белки по настоящему изобретению включают описанные выше алкилгликозидные модификации. Другой аспект изобретения относится к способам получения ковалентно модифицированных пептидов и/или белков по настоящему изобретению и их фармацевтически приемлемых солей, эти способы включают твердофазный синтез в микроволновом синтезаторе пептидов или стандартный пептидный синтез с последовательной конденсацией защищенных аминокислот на полимерном ной подложке, снятие защитных групп и удаление полимерной подложки и очистку продукта, получение аналогов физиологически активных пептидов по определению выше.
Пример 3. Общий метод окисления в уроновые кислоты
К раствору 1-додецил β-D-глюкопиранозида (Carbosynth) [2.0 г, 5.74 ммоля] в 20 мл ацетонитрила и 20 мл DI (деионизированной) воды прибавляли (диацетоксииод)бензол (Fluka) [4.4 г, 13.7 ммоля] и TEMPO (SigmaAldrich) [0.180 г, 1.15 ммоля]. Полученную смесь перемешивали при комнатной температуре в течение 20 час. К реакционной смеси прибавляли воду и лиофилизировали досуха, получили 1.52 г (выход сырого продукта 73.1%) сырой 1-додецил β-D-глюкуроновой кислоты в виде порошка белого цвета, которую без дополнительной очистки использовали непосредственно для твердофазного синтеза. Этот продукт был ранее получен альтернативным методом с применением NaOCl в качестве окислителя, как указано в описании, и также был применен для более длинных алкильных групп. Аналогичным способом получали нужные алкилсахарид уроновые кислоты, применяемые для получения продуктов и реагентов по настоящему изобретению.
Аналогичным способом, но применяя соответствующие 1-тетрадецил, 1-гексадецил и 1-октадецил β-D-глюкопиранозиды (полученные от фирмы Anatrace. Maumee. ОН), получали заданные 1-алкил сахарид уроновые кислоты, которые применяли для получения продуктов и реагентов по настоящему изобретению.
Пример 4: Получение аналога EU-A387.
Препарат Fmoc-His-Aib-Gln-Gly-Thr-Phe-Thr-Ser-Asp-Bip-Ser-Lys-Tyr-Leu-Glu-Ser-Lys(Alloc)-Амид (смола) Ринка получали последовательным добавлением N-альфа-Fmoc-защищенных аминокислот, как описано в Примере 1, и депротекционировали по положению Lys-N-эпсилон, инкубируя с Pd(PPh3)4 (0.5 экв.) и DMBA (20 экв.) в DMF/ СН2Сl2 (1:1) в течение ночи в темноте при комнатной температуре. После промывки смесью DMF/CH2Cl2 боковую цепь Lys ацилировали с помощью 1′- додецил β-D-глюкуроновой кислоты в DMF/CH2Cl2 с применением DIC/HOBt. Окончание конденсации проверяли с помощью нингидрина и продукт тщательно промывали CH2Cl2.
Продукт на смоле подвергали окончательной депротекции и отщепляли от смолы, обрабатывая коктейлем для отщепления (94% TFA: 2% EDT; 2% Н2О; 2% TIS) в течение 240 минут при комнатной температуре. К смеси прибавляли Et2O для осаждения продукта и тщательно промывали Et2О, после сушки в вакууме получали сырой титульный пептидный продукт.
Продукт разделили на две партии и очистку каждой партии проводили обращенно-фазовой ВЭЖХ (С18). Сырой продукт загружали на колонку ВЭЖХ 4.1×25 см со скоростью потока 15 мл/мин (15% органического модулятора; ацетатный буфер) и элюировали в градиенте 15-45% буфера В в течение 60 мин при 50°С. Фракцию продукта лиофилизировали, получали титульный пептидный продукт с чистотой 98.03% по данным аналитической ВЭЖХ (18.6 мин; 30-60% СН3CN в 0.1% ТРА)/масс-спектрометрии (М+1 пик = 2382.14).
Соответствующий 1-метальный и 1-октильный аналоги титульного соединения получали аналогичным методом, но используя реагенты 1′-метил β-D-глюкуроновую кислоту и 1′-октил β-D-глюкуроновую кислоту (Carbosynth). Соответствующие аналоги: 1-децил, 1-додецил, 1-тетрадецил, 1-гексадецил, 1-октадецил и 1-эйкозил получали применяя соответствующие глюкуроновые кислоты, полученные как указано выше. Или же, 1-алкил глюкуронил или другие ацилированные аналоги можно получать, сначала очищая депротекционированный или частично депротекционированный пептид с последующим ацилированием реагентом заданной уроновой кислоты.
Анализ проводили методом ВЭЖХ/масс- спектрометрии положительных ионов, используя элюент в градиенте, результаты приводятся в Таблице, см. ниже.
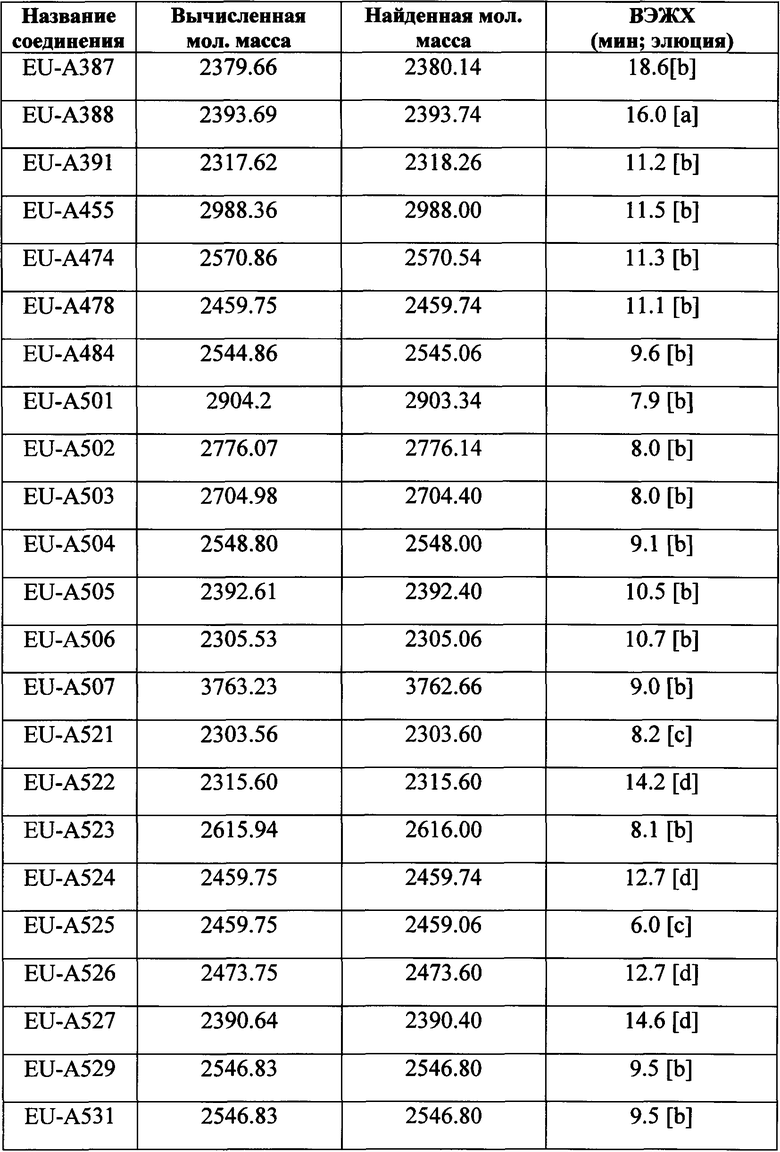
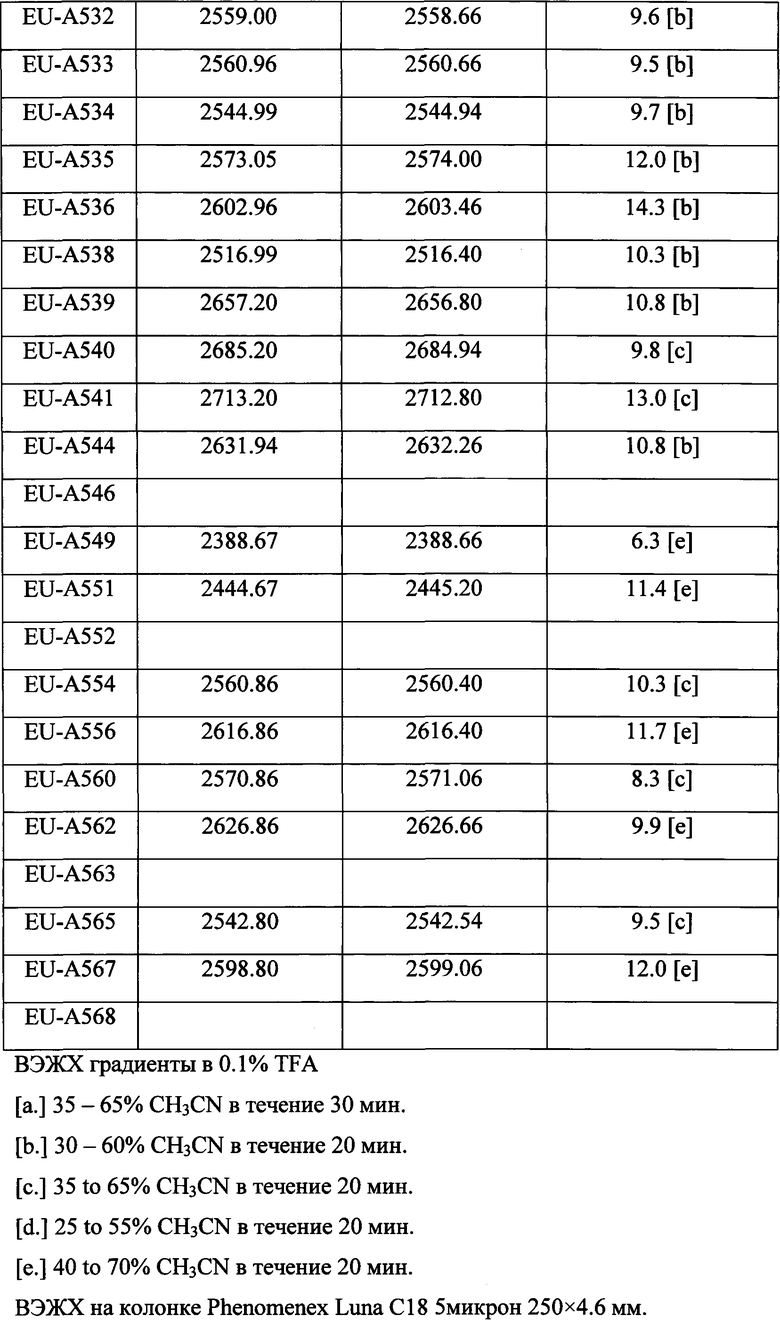
Пример 5: Тестирование соединений в анализах клеток
Соединения точно взвешивали в количестве около 1 мг и проверяли в стандартных анализах клеток (компания Cerep SA). Регистрировали количество сАМР (пАМФ), образующейся в клетках, обработанных тестируемыми соединениями, либо в качестве агонистов, либо в качестве антагонистов. Проводимый анализ включал стимуляцию уровней сАМР в анализах на глюкагон и GLP-1. Анализы описаны Chicchi, G.G., et al. (1997) J Biol Chem 272: 7765- 7769 и Runge, S., et al. (2003) Br J Pharmacol 138: 787-794.
Для соединения EU-A391 GLCR клеточный ответ не менялся, a GLP1R клеточный ответ резко возрастал при ЕС50 420 нМ
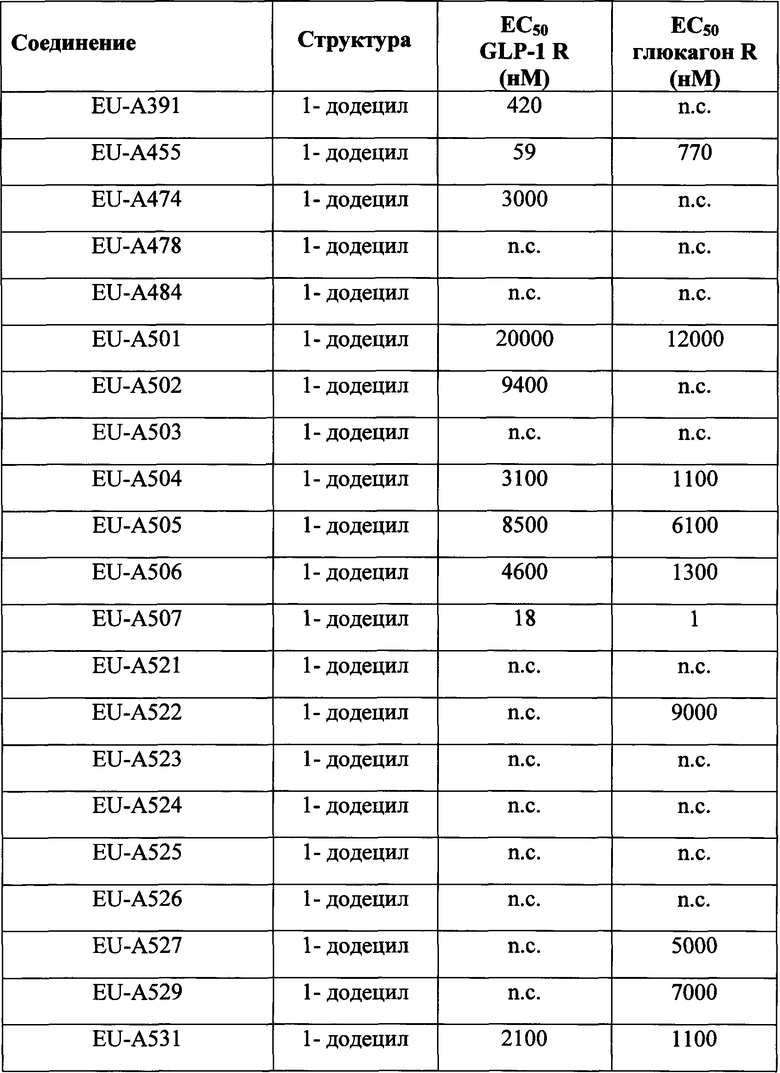
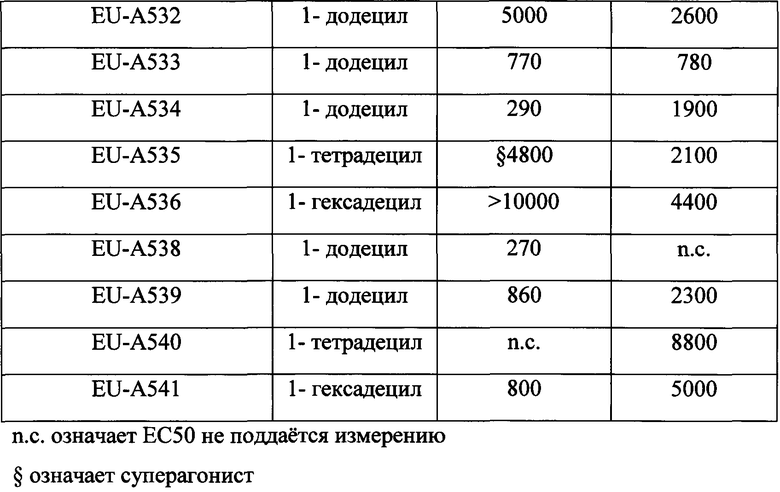
Пример 6: Анализ соединений in vivo
Шестьдесят (60) самцов мышей C57BL/6J, страдающих ожирением, вызванным диетой, получали из лабораторий JAX в возрасте 14 недель. Уши мышей метили выщипом для идентификации и помещали в индивидуально и принудительно вентилируемых клетках из поликарбоната с воздушными фильтрами НЕРА при плотности одна мышь в клетке. Помещение для животных (виварий) освещали исключительно с помощью искусственного флуоресцентного освещения с контролируемым 12-часовым световым циклом. Диапазоны нормальной (комнатной) температуры и относительной влажности в помещениях для животных составляли 22±4°С и 50±15%, соответственно. Отфильтрованную водопроводную воду, подкисленную до рН 2.8-3.1, и корм с высоким содержанием жира (60 kcal %) давали ad libitum (неограниченно).
После акклиматизации в течение 2 недель выбрали 40 мышей с учетом нужной массы тела и мышей произвольно делили на группы (n=10), как указано ниже. Группа 1. Получала носитель; Группа 2. Малая доза тестируемого соединения; Группа 3. Средняя доза тестируемого соединения; Группа 4. Высокая доза тестируемого соединения. Мышам вводили дозу SC (подкожно) ежедневно в течение 28 дней. Массу тела и наблюдения за животными в клетке записывали ежедневно. Потребляемую пищу и воду регистрировали еженедельно. Проводили измерения методом ЯМР для определения состава общей жировой и нежировой (тощей) ткани в 1 день (до введения препарата) и на 26 день. В0, 14 и 27 дни мышам не давали корм в течение ночи для проведения перорального теста на толерантность к глюкозе. На следующий день из надреза на хвосте отбирали первую пробу крови (t=0). Затем мышам вводили болюсную инъекцию 1.0 г/кг глюкозы. Образцы крови из надреза на хвосте отбирали через 5, 30, 60 и 120 мин после введения глюкозы и сразу же определяли содержание глюкозы в плазме с помощью глюкометра.
Умерщвление и взятие образцов ткани. Мышей умерщвляли на 29 день. Конечную кровь перерабатывали в сыворотку/плазму и аликвоты отдавали на анализ на глюкозу, инсулин и липидный профиль. Жировые ткани собирали, взвешивали и замораживали для анализа. Оптимальный профиль соединения показывает пониженное колебание уровня глюкозы в OGTT, пониженную базальную секрецию инсулина наряду с усиленной глюкозозависимой секрецией инсулина, пониженным увеличением массы тела, пониженной массой жировой ткани, но минимальным действием на тощую (нежировую) массу.
Пример 7: Применение соединений
Ковалентно модифицированные пептиды и/или белки по настоящему изобретению применимы для предупреждения и лечения различных заболеваний, связанных с ожирением, метаболического синдрома, сердечно-сосудистого заболевания и диабета. Пептиды, модифицированные меченным соответствующим образом ПАВ, можно применять в качестве диагностических зондов.
Типичные схемы доставки включают пероральное, парентеральное (включая подкожную, внутримышечную и внутривенную инъекцию), ректальное, трансбуккальное (включая подъязычное), трансдермальное введение, ингаляцию, глазное и интраназальное введение. Привлекательный и широко применяемый метод доставки пептидов предусматривает подкожную инъекцию препарата с контролируемым высвобождением. Другие способы введения для применения ковалентно модифицированных пептидов и/или белков по настоящему изобретению включают подкожное, интраназальное введение и ингаляции.
Пример 8. Фармацевтическое применение для лечения инсулинорезистентности.
Больного человека с симптомами инсулинового или метаболического синдрома лечили соединением EU-A596, которое вводили интраназально (200 мкл), с помощью стандартного атомизатора, применяемого в уровне техники, в виде раствора фармацевтического агента в физиологическом растворе, содержащем от 0.5 до 10 мг/мл фармацевтического агента и стандартные эксципиенты, такие как бензиловый спирт. Лечение повторяли при необходимости смягчить симптомы, такие как ожирение, повышенный уровень глюкозы в крови и т.п.Аналогичным образом раствор EU-A596 и выбранные эксципиенты в летучем растворителе, таком как гидрофторалкан, вводили интраназально с помощью дозирующего ингалятора (MDI) при необходимости понизить инсулинорезистентность. Эффект лечения определяли, применяя стандартные тесты, включая определения уровней глюкозы в крови, индекс массы тела (ИМТ) и/или массу тела и/или определение соотношения окружностей талии и бедер.
Аналогичным образом тестируют введение скорректированного количества трансбуккальным, интравагинальным методом, ингаляцией, подкожным, внутривенным, внутриглазным или пероральным методами для определения уровня стимуляции GLP1R и/или GLCR на клетках в организме и для определения терапевтического эффекта.
ПОСЛЕДОВАТЕЛЬНОСТИ
Описание включает последовательности SEQ. ID. No. 1-3 и SEQ. ID. No. 318-343. Кроме того, в Таблице 1 на Фигуре 1 приводятся номера последовательностей SEQ. ID для соединений с EU-A300 до EU-A425, имеющих SEQ. ID. NO. 4-129, соответственно, как показано в Таблице 1 на Фигуре 1. Тем самым соединения в Таблице 1 на Фигуре 1 и их соответствующие SEQ. ID. NOs, показанные в Таблице 1 на Фигуре 1, вводятся в подаваемую заявку. Кроме того, в Таблице 2 на Фигуре 2 представлены SEQ. ID номера для соединений с EU-A426 до EU-599, имеющих SEQ. ID. NOs. 130-317, соответственно, показанные в Таблице 2 на Фигуре 2. Тем самым соединения в Таблице 2 на Фигуре 2, и их соответствующие SEQ. ID. NOs, показанные в Таблице 2 на Фигуре 2, вводятся в подаваемую заявку.
| название | год | авторы | номер документа |
|---|---|---|---|
| УЛУЧШЕННЫЕ ПЕПТИДНЫЕ ЛЕКАРСТВЕННЫЕ СРЕДСТВА ДЛЯ ЛЕЧЕНИЯ NASH И ДРУГИХ РАССТРОЙСТВ | 2019 |
|
RU2790209C2 |
| АНАЛОГИ ГЛЮКАГОНА, ОБЛАДАЮЩИЕ ПОВЫШЕННОЙ РАСТВОРИМОСТЬЮ И СТАБИЛЬНОСТЬЮ В БУФЕРАХ С ФИЗИОЛОГИЧЕСКИМИ ЗНАЧЕНИЯМИ Ph | 2009 |
|
RU2560254C2 |
| КОНЪЮГАТЫ ИНСУЛИН-ИНКРЕТИН | 2014 |
|
RU2678134C2 |
| ПЕРОРАЛЬНЫЕ АГОНИСТЫ РЕЦЕПТОРОВ GLP | 2021 |
|
RU2836443C1 |
| ОСНОВАННЫЕ НА АМИДАХ ПРОЛЕКАРСТВА ПЕПТИДОВ ГЛЮКАГОНОВОГО НАДСЕМЕЙСТВА | 2009 |
|
RU2550696C2 |
| ПЕПТИДЫ ГЛЮКАГОНОВОГО СУПЕРСЕМЕЙСТВА, ОБЛАДАЮЩИЕ АКТИВНОСТЬЮ ОТНОСИТЕЛЬНО ЯДЕРНЫХ ГОРМОНАЛЬНЫХ РЕЦЕПТОРОВ | 2011 |
|
RU2604067C2 |
| ДВОЙНЫЕ АГОНИСТЫ GLP1/GIP ИЛИ ТРОЙНЫЕ АГОНИСТЫ GLP1/GIP/ГЛЮКАГОНА | 2013 |
|
RU2652783C2 |
| НОВЫЕ АНАЛОГИ ГЛЮКАГОН-ПОДОБНОГО ПЕПТИДА, КОМПОЗИЦИЯ И СПОСОБ ПРИМЕНЕНИЯ | 2010 |
|
RU2557301C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ hGLP-1, ЭКСЕНДИНА-4 И ИХ АНАЛОГОВ | 2007 |
|
RU2419452C2 |
| СОЕДИНЕНИЯ-АГОНИСТЫ GIP И СПОСОБЫ | 2015 |
|
RU2716985C2 |

























































































































































































































































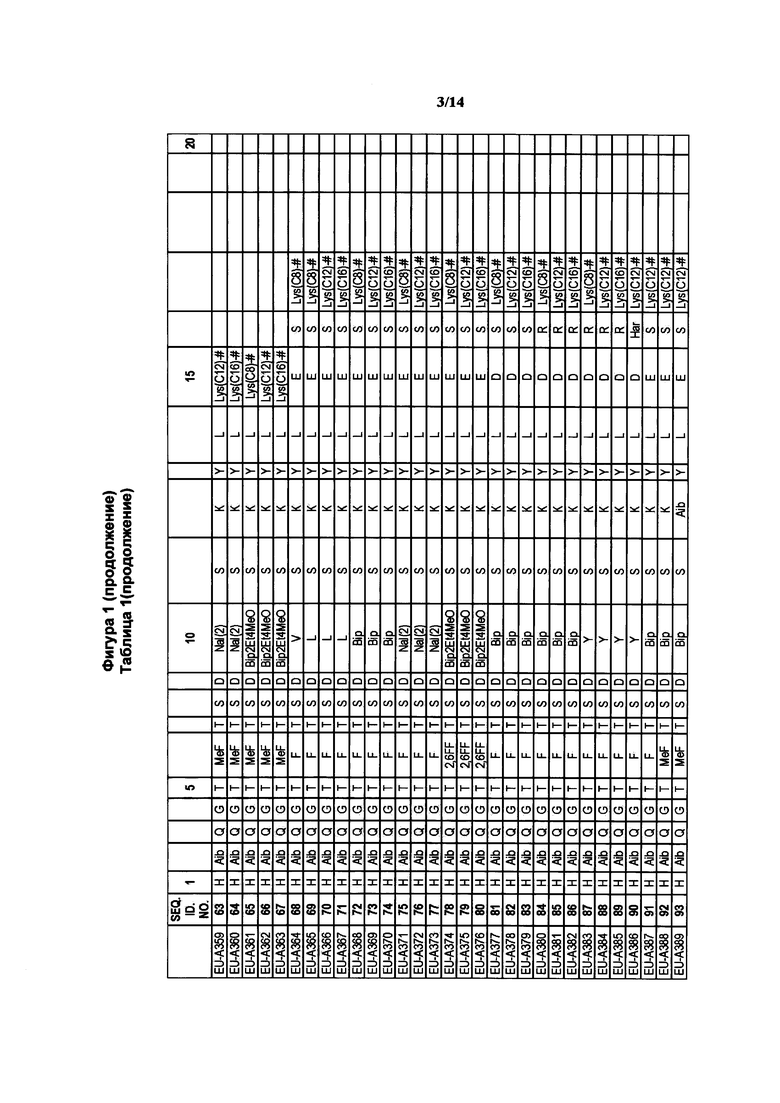
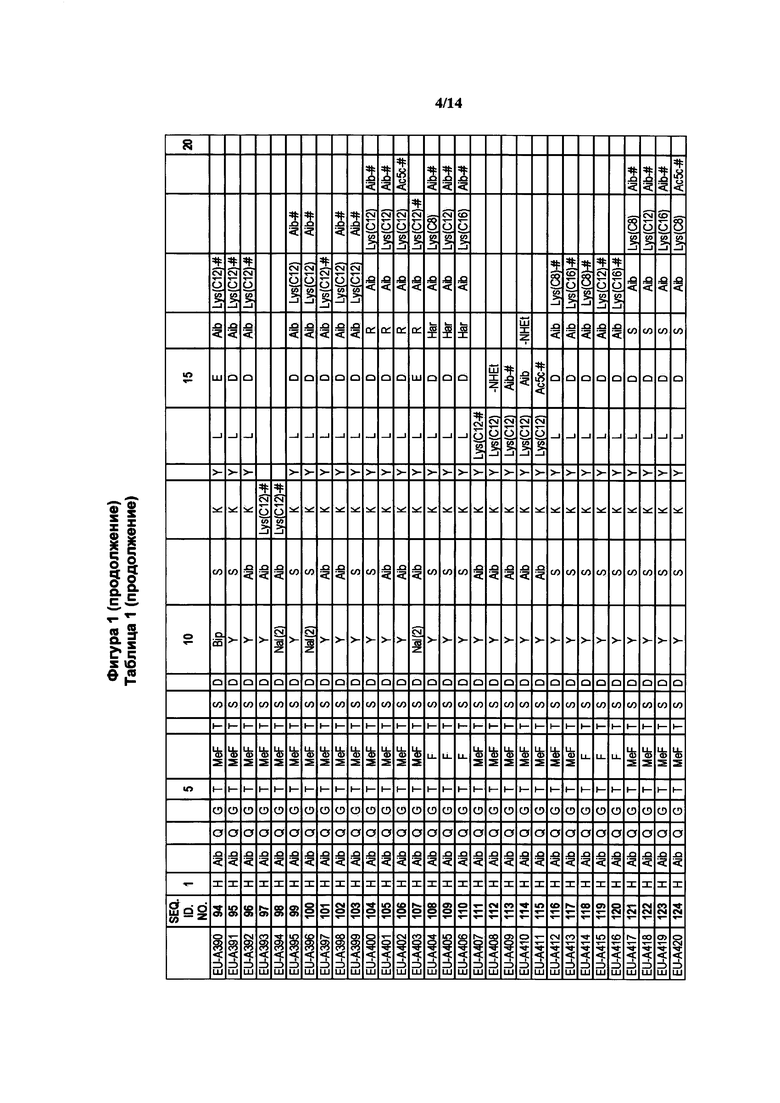

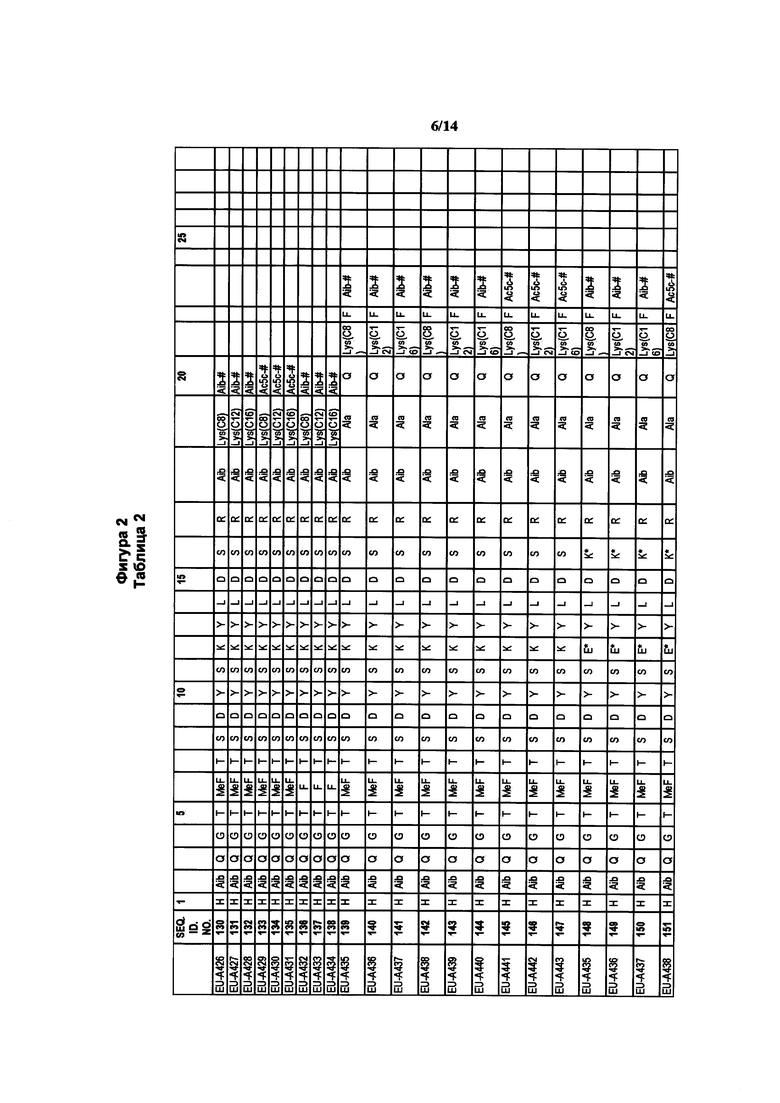
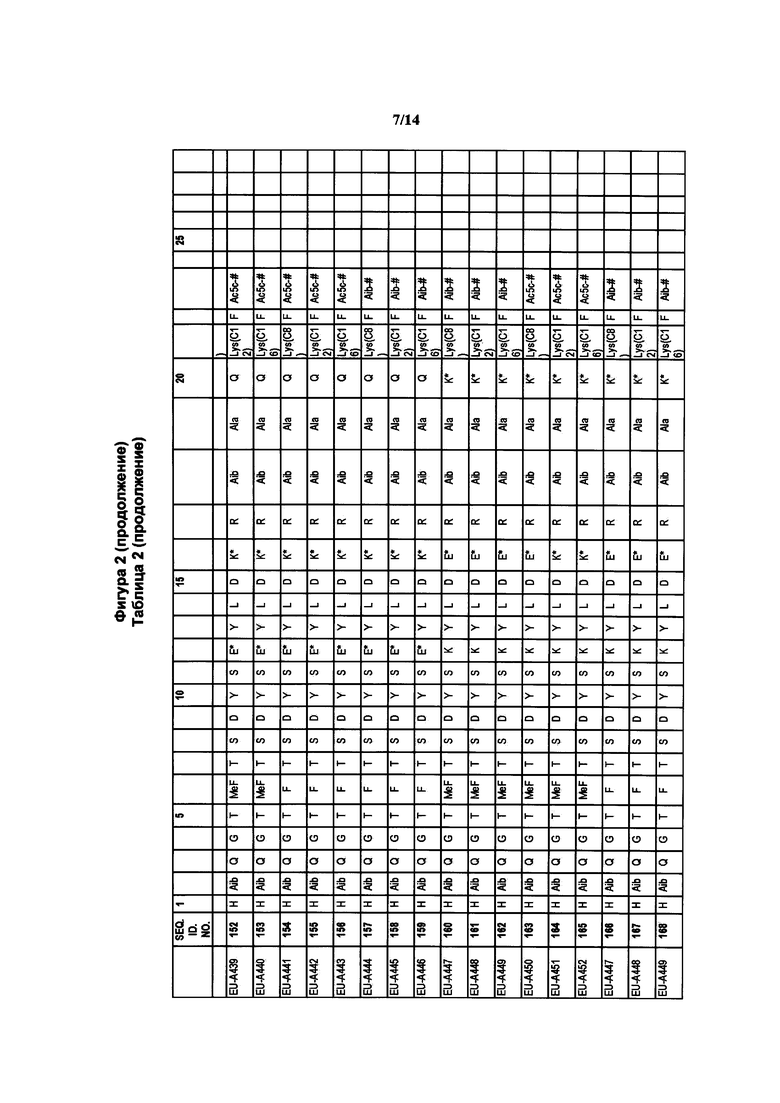
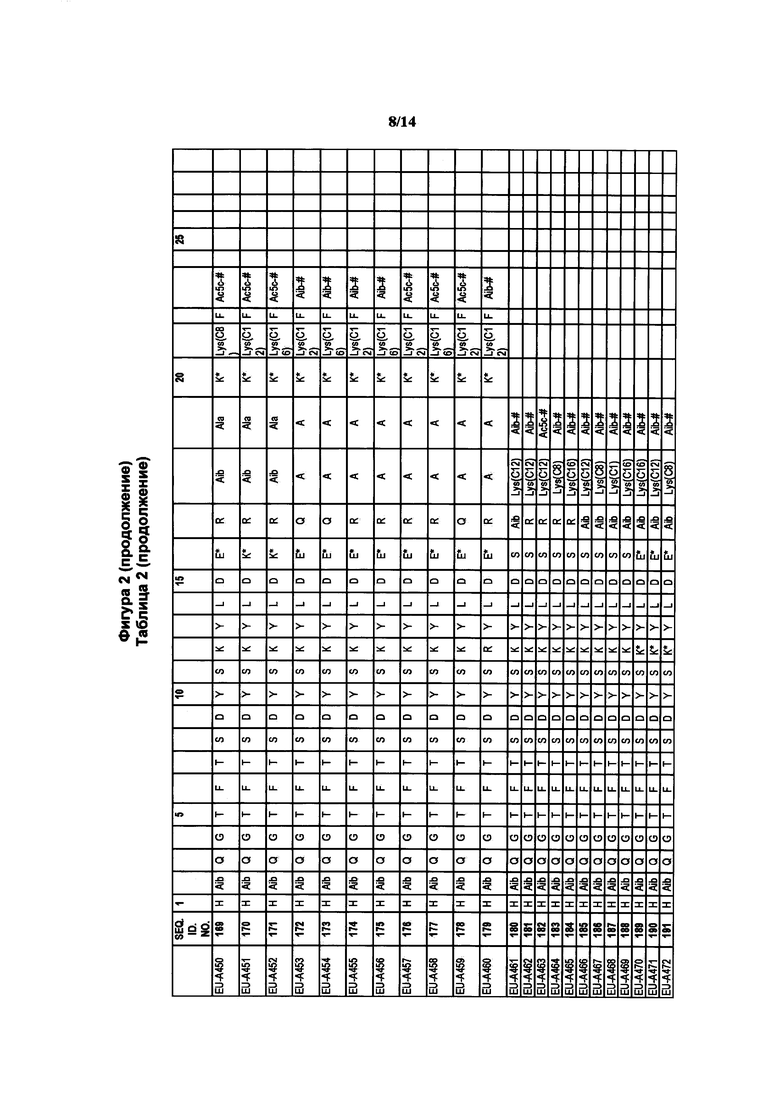

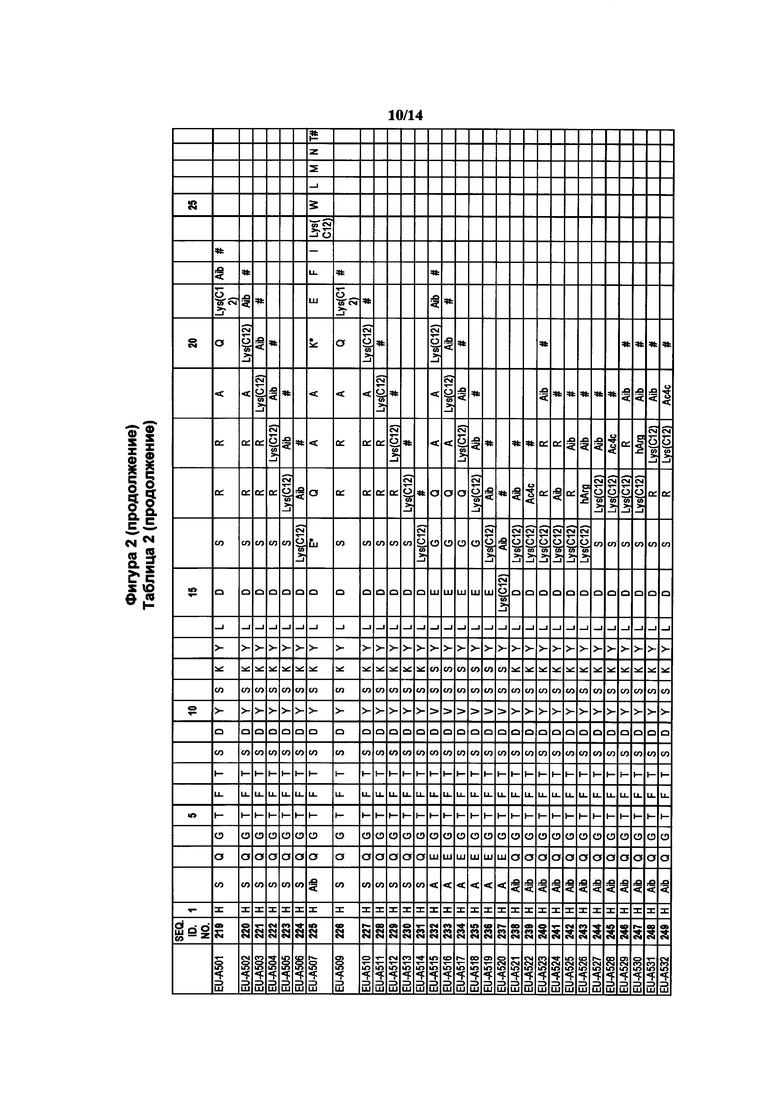
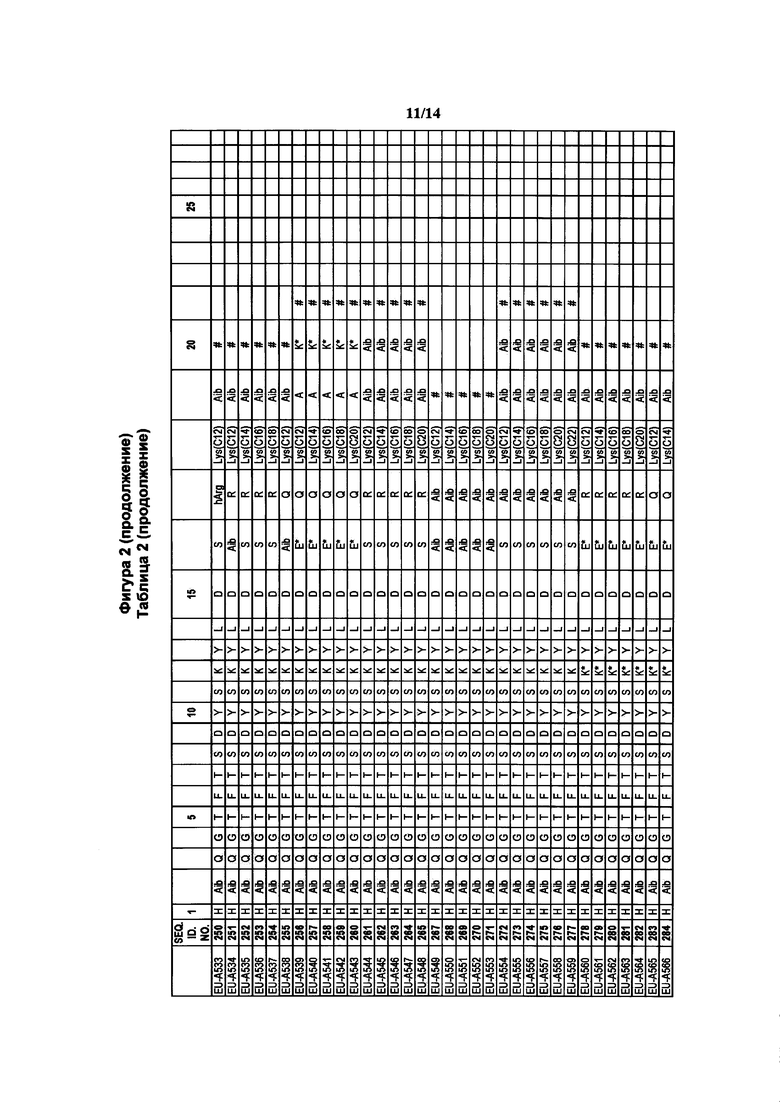
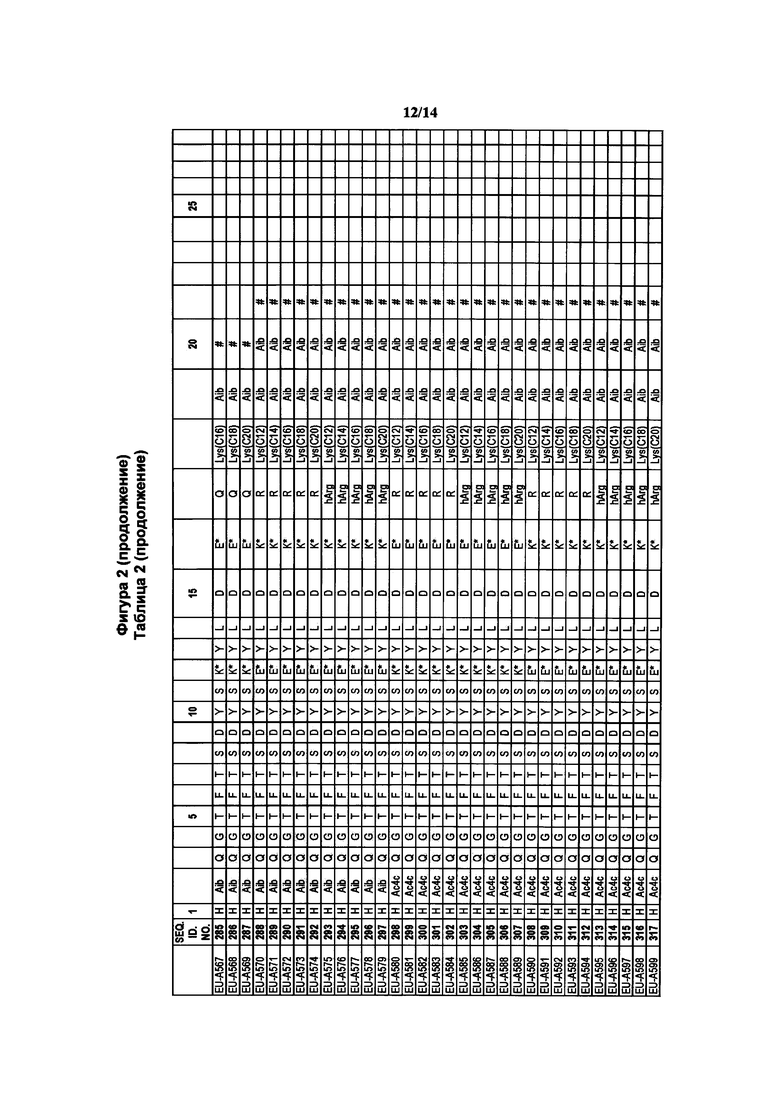

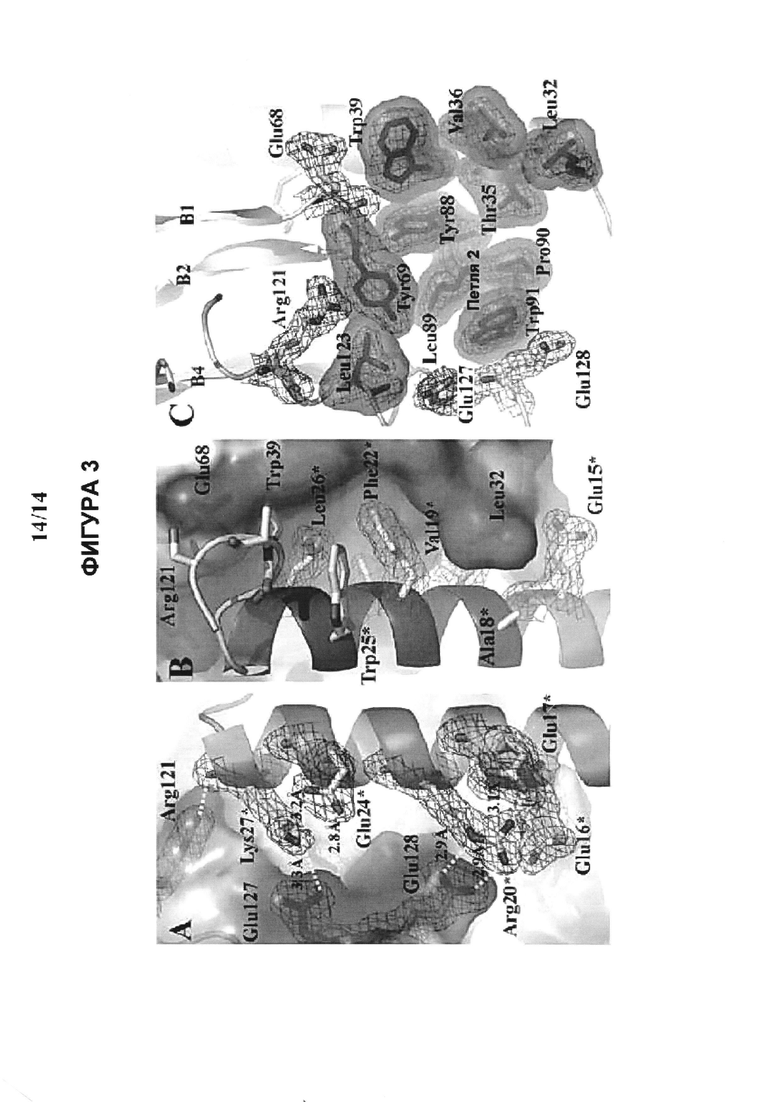
Изобретение относится к пептидам (глюкагону и/или GLP-1 и их аналогам), ковалентно связанным с поверхностно-активными гликозидами, предназначенным для лечения или предупреждения нарушений, связанных с инсулинорезистентностью. Модификация пептидов способствует более продолжительному действию и/или повышенной биодоступности пептидов. 3 н. и 14 з.п. ф-лы, 1 табл., 3 ил., 8 пр.
1. Пептидный продукт, содержащий поверхностно-активное вещество X, ковалентно связанное с пептидом, причем пептид содержит линкерную аминокислоту U и по меньшей мере одну другую аминокислоту:
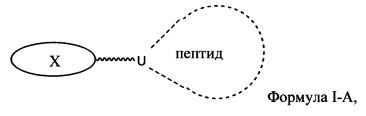
где поверхностно-активное вещество X обозначает группу Формулы I:
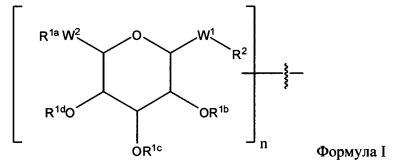
где:
R1a обозначает C1-С30 алкильную группу;
R1b, R1c и R1d, каждый обозначает Н;
W1 обозначает -(C=O)-NH-;
W2 обозначает -О-;
R2 обозначает связь с U; и
n обозначает 1;
пептид выбран из соединения Формулы II:
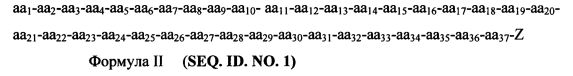
где:
Z обозначает ОН или -NH-R3, где R3 обозначает Н или С1-С12 замещенный или незамещенный алкил, или цепь PEG (ПЭГ) протяженностью менее 10 Да;
aa1 обозначает His, N-Ac-His, pGlu-His или N-R3-His;
aa2 обозначает Ser, Ala, Gly, Aib, Ac4c или Ac5c;
аа3 обозначает Gln или Cit;
аа4 обозначает Gly или D-Ala;
aa5 обозначает Thr или Ser;
aa6 обозначает Phe, Trp, F2Phe, Me2Phe или Nal2;
аа7 обозначает Thr или Ser;
аа8 обозначает Ser или Asp;
аа9 обозначает Asp или Glu;
аа10 обозначает Tyr, Leu, Met, Nal2, Bip или Bip2EtMeO;
aa11 обозначает Ser, Asn или U;
aa12 обозначает Lys, Glu, Ser, Arg или U;
аа13 отсутствует или обозначает Tyr, Gln, Cit или U;
аа14 отсутствует или обозначает Leu, Met, Nle или U;
aa15 отсутствует или обозначает Asp, Glu или U;
aa16 отсутствует или обозначает Ser, Gly, Glu, Aib, Ac5c, Lys, Arg или U;
аа17 отсутствует или обозначает Arg, hArg, Gln, Glu, Cit, Aib, Ac4c, Ac5c или U;
aa18 отсутствует или обозначает Arg, hArg, Ala, Aib, Ac4c, Ac5c или U;
aa19 отсутствует или обозначает Ala, Val, Aib, Ac4c, Ac5c или U;
аа20 отсутствует или обозначает Gln, Lys, Arg, Cit, Glu, Aib, Ac4c, Ac5c или U;
aa21 отсутствует или обозначает Asp, Glu, Leu, Aib, Ac4c Ac5c или U;
aa22 отсутствует или обозначает Phe, Trp, Nal2, Aib, Ac4c, Ac5c или U
аа23 отсутствует или обозначает Val, Ile, Aib, Ас4с, Ас5с или U;
аа24 отсутствует или обозначает Gln, Ala, Glu, Cit или U;
aa25 отсутствует или обозначает Trp, Nal2 или U;
аа26 отсутствует или обозначает Leu или U;
аа27 отсутствует или обозначает Met, Val, Nle, Lys или U;
аа28 отсутствует или обозначает Asn, Lys или U;
аа29 отсутствует или обозначает Thr, Gly, Aib, Ac4c, Ac5c или U;
аа30 отсутствует или обозначает Lys, Aib, Ac4c, Ac5c или U;
аа31 отсутствует или обозначает Arg, Aib, Ac4c, Ac5c или U;
аа32 отсутствует или обозначает Asn, Aib, Ac4c, Ac5c или U;
аа33 отсутствует или обозначает Arg, Aib, Ac4c, Ac5c или U;
аа34 отсутствует или обозначает Asn, Aib, Ac4c, Ac5c или U;
аа35 отсутствует или обозначает Asn, Aib, Ac4c, Ac5c или U;
аа36 отсутствует или обозначает Ile, Ala, Aib, Ас4с, Ас5С или U;
аа37 отсутствует или обозначает U; и
U обозначает природную или неприродную аминокислоту, содержащую функциональную группу, применяемую для ковалентного связывания с поверхностно-активным веществом X;
где любые две из aa1-аа37, необязательно, циклизуются по своим боковым цепям с образованием лактамной связи; и
при условии, что одна или по меньшей мере одна из аа11-аа37 представляет собой линкерную аминокислоту U, связанную с X ковалентной связью.
2. Пептидный продукт по п. 1, в котором R1a обозначает С6-С20 алкильную группу.
3. Пептидный продукт по п. 1, в котором X представляет собой 1-эйкозил бета-D-глюкуроновую кислоту, 1-октадецил бета-D-глюкуроновую кислоту, 1-гексадецил бета-D-глюкуроновую кислоту, 1-тетрадецил бета-D-глюкуроновую кислоту, 1-додедецил бета-D-глюкуроновую кислоту, 1-децил бета-D-глюкуроновую кислоту, 1-октил бета-D-глюкуроновую кислоту, или функционализованную 1-эйкозил бета-D-глюкозу, 1-октадецил бета-D-глюкозу, 1-гексадецил бета-D-глюкозу, 1-тетрадецил бета-D-глюкозу, 1-додецил бета-D-глюкозу, 1-децил бета-D-глюкозу, 1-октил бета-D-глюкозу, или соответствующий 1-алкилгликозид в альфа-конфигурации, или соответствующую функционализированную 1-алкилгалактозу в альфа- или бета-конфигурации.
4. Пептидный продукт по п. 1, в котором U выбран из Lys, Orn, гемолизина и Сα-метиллизина (CMeLys).
5. Пептидный продукт по п. 1 имеет структуру Формулы III-А:
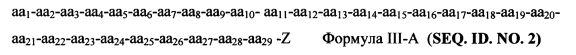
в которой:
Z обозначает ОН или -NH-R3, где R3 обозначает Н или С1-С12 замещенный или незамещенный алкил, или цепь PEG (ПЭГ) протяженностью менее 10 Да;
aa1 обозначает His, N-Ac-His, pGlu-His или N-R3-His;
aa2 обозначает Ser, Ala, Gly, Aib, Ac4c или Ac5c;
аа3 обозначает Gln или Cit;
аа4 обозначает Gly или D-Ala;
aa5 обозначает Thr или Ser;
aa6 обозначает Phe, Trp, F2Phe, Me2Phe или Nal2;
aa7 обозначает Thr или Ser;
аа8 обозначает Ser или Asp;
аа9 обозначает Asp или Glu;
аа10 обозначает Tyr, Leu, Met, Nal2, Bip или Bip2EtMeO;
aa11 обозначает Ser, Asn или U;
аа12 обозначает Lys, Glu, Ser, Arg или U(X);
аа13 отсутствует или обозначает Tyr, Gln, Cit или U(X);
аа14 отсутствует или обозначает Leu, Met, Nle или U(X);
aa15 отсутствует или обозначает Asp, Glu или U(X);
aa16 отсутствует или обозначает Ser, Gly, Glu, Aib, Ac5c, Lys, Arg или U(X);
aa17 отсутствует или обозначает Arg, hArg, Gln, Glu, Cit, Aib, Ac4c, Ac5c или U(X);
aa18 отсутствует или обозначает Arg, hArg, Ala, Aib, Ac4c, Ac5c или U(X);
aa19 отсутствует или обозначает Ala, Val, Aib, Ac4c, Ac5c или U(X);
aa20 отсутствует или обозначает Gln, Lys, Arg, Cit, Glu, Aib, Ac4c, Ac5c или U(X);
aa21 отсутствует или обозначает Asp, Glu, Leu, Aib, Ac4c, Ac5c или U(X);
аа22 отсутствует или обозначает Phe, Trp, Nal2, Aib, Ac4c, Ac5c или U(X);
аа23 отсутствует или обозначает Val, Ile, Aib, Ас4с, Ас5с или U(X);
аа24 отсутствует или обозначает Gln, Ala, Glu, Cit или U(X);
аа25 отсутствует или обозначает Trp, Nal2 или U(X);
аа26 отсутствует или обозначает Leu или U(X);
аа27 отсутствует или обозначает Met, Val, Nle, Lys или U(X);
аа28 отсутствует или обозначает Asn, Lys или U(X); и
аа29 отсутствует или обозначает Thr, Gly, Aib, Ac4c, Ac5c или U(X);
где любые две из aa1-aa29, необязательно, циклизуются за счет своих боковых цепей с образованием лактамной связи; и
при условии, что одна, или по меньшей мере одна из от aa16 до аа29 представляет собой природную или неприродную аминокислоту U, ковалентно связанную с X.
6. Пептидный продукт по п. 1, который имеет структуру Формулы III-В:

в которой:
Z обозначает ОН или -NH-R3, где R3 обозначает Н, С1-С12 алкил; или цепь PEG (ПЭГ) протяженностью менее 10 Да;
аа2 обозначает Ser, Ala, Gly, Aib, Ac4c или Ac5c;
аа3 обозначает Gln или Cit;
аа6 обозначает Phe, Trp, F2Phe, Me2Phe, MePhe или Nal2;
аа10 обозначает Tyr, Leu, Met, Nal2, Bip или Bip2EtMeO;
аа11 обозначает Ser, Asn или U(X);
aa12 обозначает Lys, Glu, Ser или U(X);
аа13 отсутствует или обозначает Tyr, Gln, Cit или U(X);
аа14 отсутствует или обозначает Leu, Met, Nle или U(X);
aa15 отсутствует или обозначает Asp, Glu или U(X);
aa16 отсутствует или обозначает Ser, Gly, Glu, Aib, Ac4c, Ac5c, Lys, Arg или U(X);
аа17 отсутствует или обозначает Arg, hArg, Gln, Glu, Cit, Aib, Ac4c, Ac5c или U(X);
aa18 отсутствует или обозначает Arg, hArg, Ala, Aib, Ac4c, Ac5c или U(X);
aa19 отсутствует или обозначает Ala, Val, Aib, Ac4c, Ac5c или U(X);
аа20 отсутствует или обозначает Gln, Lys, Arg, Cit, Glu, Aib, Ac4c, Ac5c или U(X);
aa21 отсутствует или обозначает Asp, Glu, Leu, Aib, Ac4c, Ac5c или U(X);
aa22 отсутствует или обозначает Phe, Aib, Ac4c, Ac5c или U(X) и
аа23 отсутствует или обозначает Val, Ile, Aib, Ас4с, Ас5с или U(X);
где любые две из aa1-аа23, необязательно, циклизуются за счет своих боковых цепей с образованием лактамной связи; и
при условии, что одна или по меньшей мере одна из аминокислот от aa16 до аа23 или представляет собой природную или неприродную аминокислоту U, ковалентно связанную с X.
7. Пептидный продукт по п. 1, в котором aa18 обозначает остаток лизина, связанный с X.
8. Пептидный продукт по п. 1, в котором аа17 обозначает остаток гомоаргинина (hArg) или остаток глицина.
9. Пептидный продукт по п. 1, в котором аа2 обозначает остаток Aib или Ас4с.
10. Пептидный продукт по п. 1, где пептид содержит один или более остатков Aib.
11. Пептидный продукт по п. 1, где пептид содержит один или более остатков Aib на С-конце.
12. Пептидный продукт по п. 1, в котором aa16 и аа20 циклизуются с образованием лактамной связи.
13. Пептидный продукт по п. 1, имеющий структуру:

где
aa2 обозначает Aib или Ас4с;
aa16 и аа20, каждый независимо, обозначают либо Lys, либо Glu и циклизуются за счет своих боковых цепей с образованием лактамной связи;
аа17 обозначает Arg, hArg или Gln;
аа27 обозначает Met или Nle; и
алкил обозначает С8-С20 линейную алкильную цепь.
14. Пептидный продукт по п. 1, в котором aa12 и aa16 циклизуются с образованием лактамной связи.
15. Пептидный продукт по п. 1, в котором X содержит додецил алкильную цепь.
16. Способ лечения диабета или сердечно-сосудистого заболевания у нуждающегося в этом субъекта, включающий введение нуждающемуся в этом субъекту терапевтически эффективного количества пептидного продукта по п. 1, причем пептидный продукт включает аналог глюкагона, содержащий аминокислотные остатки aa1-aa17 или aa1-aa18 или aa1-aa19 или aa1-аа20 последовательности SEQ. ID. NO. 331.
17. Способ лечения диабета или сердечно-сосудистого заболевания у нуждающегося в этом субъекта, включающий введение нуждающемуся в этом субъекту терапевтически эффективного количества пептидного продукта по п. 1, причем пептидный продукт включает аналог глюкагона, содержащий аминокислотные остатки aa1-аа17 или aa1-aa18 или aa1-aa19 или аа1-аа20 последовательности SEQ. ID. NO. 1.
| ГЛИЦЕРИН-СВЯЗАННЫЕ ПЭГИЛИРОВАННЫЕ САХАРА И ГЛИКОПЕПТИДЫ | 2007 |
|
RU2460543C2 |
| WO 2000078302 A1, 28.12.2000 | |||
| WO 2006121860 A2, 16.11.2006 | |||
| WO 2009155258 A2, 23.12.2009. | |||

