ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
Настоящая заявка испрашивает приоритет предварительной заявки на патент США № 60/791701, поданной 13 апреля 2006 года.
Настоящее изобретение направлено на фармацевтические композиции, включающие либо человеческий глюкагон-подобный пептид-1, либо эксендин-4 и/или аналоги и производные hGLP-1 или эксендина-4, а также на способы применения таких фармацевтических композиций для лечения некоторых заболеваний и/или состояний у людей.
Природный или синтетический человеческий GLP-1 и его производные являются метаболически нестабильными, имеющими период полужизни в плазме in vivo лишь от одной до двух минут. При их введении в организм они также быстро разрушаются in vivo. Метаболическая неустойчивость ограничивает терапевтическое применение GLP-1. Следовательно, существует необходимость в особых фармацевтических композициях, обеспечивающих длительный профиль высвобождения.
Цель настоящего изобретения заключается в разработке и получении состава, способного сохранять биологическую активность на протяжении продолжительного периода времени благодаря образованию депо препарата в месте инъекции непосредственно после введения.
Кроме того, PK профиль, получаемый благодаря этому депо, должен быть по возможности максимально плоским, с учетом узкого терапевтического окна пептида.
Настоящее изобретение охватывает фармацевтические композиции, которые обеспечивают высвобождение за период от одного дня до более чем одной недели.
Фармацевтические композиции по настоящему изобретению могли бы представлять собой прозрачные растворы, водные суспензии или водные смеси суспензий и растворов, или же являться полутвердыми веществами.
Амид глюкагон-подобного пептида-1 (7-36) (GLP-1(7-36)-NH2) синтезируется в кишечных L-клетках путем ткань-специфичного посттрансляционного процессинга предшественника глюкагона препроглюкагона (Varndell, J.M., et al., J.Histochem Cytochem, 1985:33:1080-6) и выделяется в кровь в качестве реакции на поступление пищи. Концентрация GLP-1 повышается от уровня, соответствующего голодному состоянию, равного примерно 15 пмоль/л до максимального уровня после приема пищи, равного 40 пмоль/л. Было показано, что при одном и том же увеличении концентрации глюкозы в плазме, увеличение содержания инсулина в плазме приблизительно в три раза выше, если глюкозу вводят перорально, по сравнению с внутривенным введением (Kreymann, B., et al., Lancet 1987:2, 1300-4). Это связанное с пищей усиление выделения инсулина, известное как инкретиновый эффект, в первую очередь является гуморальным, и, как полагают в настоящее время, GLP-1 представляет собой наиболее мощный физиологический инкретин в организме людей. Помимо инсулинотропного эффекта GLP-1 подавляет секрецию глюкагона, замедляет опорожнение желудка (Wettergren A., et al., Dig Dis Sci 1993:38:665-73) и может улучшать периферическое удаление глюкозы (D'Alessio, D.A. et al., J.Clin.Invest 1994:93:2293-6).
В 1994 г. было выдвинуто предположение о терапевтическом потенциале GLP-1, после наблюдения, что однократное подкожное (s/c) введение GLP-1 могло полностью нормализовать уровни глюкозы после приема пищи у пациентов с не инсулин-зависимым сахарным диабетом (NIDDM) (Gutniak, M.K., et al., Diabetes Care 1994:17:1039-44). Считалось, что этот эффект опосредуется как увеличением выделения инсулина, так и уменьшением секреции глюкагона. Кроме того, было показано, что внутривенная инфузия GLP-1 замедляет опорожнение желудка после приема пищи у пациентов с NIDDM (Williams, B., et al., J.Clin Endo Metab 1996:81:327-32). В отличие от сульфонилмочевин, инсулинотропное действие GLP-1 зависит от концентрации глюкозы в плазме (Holz, G.G. 4th, et al., Nature 1993:361:362-5). Так, например, уменьшение выделения инсулина, опосредованное GLP-1, при низких концентрациях глюкозы в плазме, защищает от тяжелой гипогликемии. Описанная совокупность свойств дает GLP-1 особенные значительные терапевтические преимущества по сравнению с другими средствами, применяемыми в настоящее время для лечения NIDDM.
Многочисленные исследования показали, что если вводить GLP-1 здоровым субъектам, он оказывает мощное воздействие на уровни глюкозы, а также на концентрации инсулина и глюкагона (Orskov, C, Diabetologia 35:701-711, 1992; Holst, J.J., et al., Potential of GLP-1 in diabetes management in Glucagon III, Handbook of Experimental Pharmacology, Lefevbre PJ, Ed., Berlin, Springer Verlag, 1996, p. 311-326), эффекты, которые находятся в зависимости от глюкозы (Kreymann, B., et al., Lancet ii: 1300-1304, 1987; Weir, G.C., et al., Diabetes 38:338-342, 1989). Кроме того, GLP-1 также эффективен у пациентов с диабетом (Gutniak, M., N.Engl J Med 226:1316-1322, 1992; Nathan, D.M., et al., Diabetes Care 15:270-276, 1992), нормализуя уровни глюкозы в крови у субъектов с диабетом типа 2 (Nauck, M.A., et al., Diabetologia 36:741-744, 1993) и улучшая контроль за содержанием глюкозы у пациентов с диабетом типа 1 (Creutzfeldt, W.O., et al., Diabetes Care 19:580-586, 1996), что увеличивает возможность его применения в качестве терапевтического средства.
Однако GLP-1 является метаболически неустойчивым, имея период полужизни в плазме (t1/2), равный лишь 1-2 минутам in vivo. При введении экзогенного GLP-1, он также претерпевает быстрое разрушение (Deacon, C.F., et al., Diabetes 44:1126-1131, 1995). Эта метаболическая неустойчивость ограничивает терапевтический потенциал природного GLP-1.
Был предпринят ряд попыток улучшения терапевтического потенциала GLP-1 и его аналогов за счет усовершенствования состава лекарственных препаратов. Например, в международной патентной публикации № WO 01/57084 описан способ получения кристаллов аналогов GLP-1, которые, как утверждается, применимы для получения фармацевтических композиций, например, пригодных для инъекций препаратов, включающих кристаллы и фармацевтически приемлемый носитель. Гетерогенные микрокристаллические кластеры GLP-1(7-37)-OH выращивали из солевых растворов и после отмачивания кристаллов испытывали обработкой цинком и/или м-крезолом (Kim and Haren, Pharma.Res.Vol. 12 No. 11 (1995)). Неочищенные кристаллические суспензии GLP(7-36)-NH2, содержащие игольчатые кристаллы и аморфный осадок, получали из фосфатных растворов, содержащих цинк или протамин (Pridal, et al., International Journal of Pharmaceutics Vol. 136, pp. 53-59 (1996)). В европейской патентной публикации № EP 0619322A2 описано получение микрокристаллических форм GLP-1(7-37)-OH путем смешивания растворов белка в буфере с pH 7-8,5 с определенными комбинациями солей и полиэтиленгликолей (ПЭГ) низкой молекулярной массы. В патенте США № 6566490 описано высевание микрокристаллов, в т.ч. GLP-1, которые, как утверждается, содействуют получению очищенных пептидных продуктов. В патенте США 6555521 (US '521) раскрыты кристаллы GLP-1, имеющие форму тетрагонального плоского бруска или пластинчатую форму, которые, как утверждается, имеют высокую степень чистоты и проявляют продолжительную активность in vivo. В US '521 сообщается, что такие кристаллы являются относительно однородными и остаются в суспензии в течение более продолжительного периода времени, чем кристаллические кластеры и аморфные кристаллические суспензии известного уровня техники, которые, как заявлено, быстро осаждаются, агрегируются или слеживаются, засоряют иглы шприцев и, как правило, приводят к непредсказуемой дозировке.
Биоразрушаемый трехблочный сополимер поли[(dl-лактид-гликолид)-b-этиленгликоль-b-(лактид-гликолид)] был предложен для применения в составе с регулируемым высвобождением GLP-1. Однако, как и в случае других полимерных систем, производство трехблочного сополимера включает сложные методики и образование нестабильных частиц.
Аналогично биоразрушаемые полимеры, например поли(молочная кислота-гликолевая кислота) (PLGA), также предлагались для применения в составах для длительной доставки пептидов. Однако применение таких биоразрушаемых полимеров не приветствуется в технике, поскольку эти полимеры в основном имеют плохую растворимость в воде и требуют не смешивающихся с водой органических растворителей, например хлористого метилена и/или жестких условий получения при производстве. Считается, что упомянутые органические растворители и/или жесткие условия получения увеличивают опасность возникновения конформационных изменений в действующем пептиде или белке, приводя к уменьшению структурной целостности и нарушению биологической активности (Choi et al., Pharm.Research, Vol.21, No.5, (2004)). Аналогичные недостатки обнаружены у полоксамеров (Id).
Композиции GLP-1, описанные в приведенных выше ссылках, не особенно хорошо подходят для получения фармацевтических составов GLP's, поскольку они имеют тенденцию захватывать примеси и/или в других случаях затруднено их воспроизводимое производство и введение. Кроме того, известно, что аналоги GLP в повышенных концентрациях вызывают тошноту, из-за чего существует необходимость обеспечения продолжительного действия лекарственного препарата при пониженных начальных концентрациях в плазме. Следовательно, существует потребность в составах GLP-1 которые производятся более легко и надежно, которые более легко и воспроизводимо вводятся пациенту и которые обеспечивают невысокие начальные концентрации в плазме для снижения или устранения нежелательных побочных эффектов.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение можно суммировать в приведенных ниже параграфах (1)-(28), а также в приведенной ниже формуле изобретения. Соответственно:
(I) в первом аспекте настоящее изобретение направлено на фармацевтическую композицию, включающую прозрачный раствор (a) как минимум одного пептидного соединения, имеющего растворимость в воде более 1 мг/мл при комнатной температуре и нейтральном значении pH, которое выбрано из группы, состоящей из hGLP-1(7-36)-NH2, а также его аналогов и производных, hGLP-1(7-37)-OH, а также его аналогов и производных, эксендина-4, а также его аналогов и производных
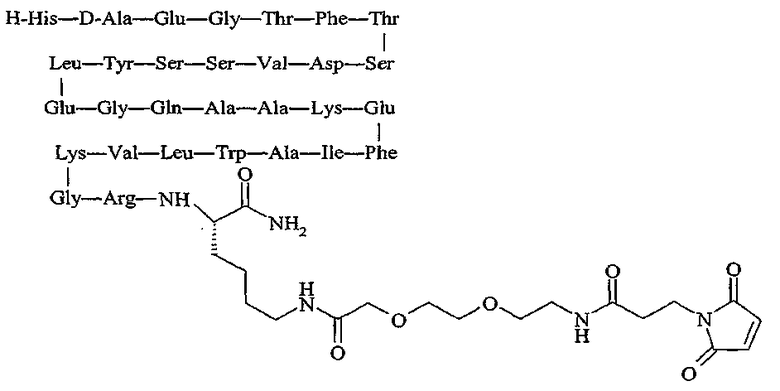 ,
,
а также его аналогов и производных,
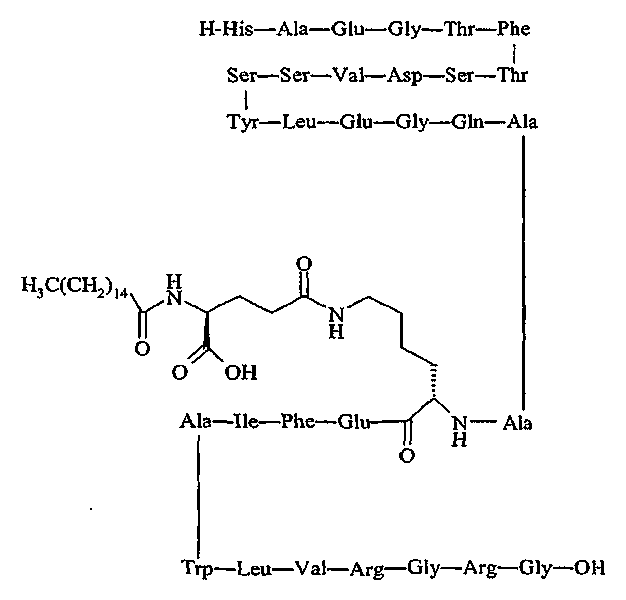 ,
,
а также его аналогов и производных, и H-His-Gly-Glu-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Leu-Ser-Lys-Gln-Met-Glu-Glu-Glu-Ala-Val-Arg-Leu-Phe-Ile-Glu-Trp-Leu-Lys-Asn-Gly-Gly-Pro-Ser-Ser-Gly-Ala-Pro-Pro-Ser-Lys-Lys-Lys-Lys-Lys-Lys-NH2, а также его аналогов и производных;
(b) ион двухвалентного металла; и
(c) растворитель,
при условии, что не менее 95% указанного пептида растворено указанным растворителем.
1. Композиция по параграфу (I), где указанный ион двухвалентного металла представляет собой цинк.
2. В одном из вариантов осуществления изобретение относится к композиции по параграфам (I) и (1), где указанный растворитель представляет собой воду.
3. Композиция по параграфу (I), включающая неводную среду.
4. Композиция по любому из параграфов (I)-(3), где указанное пептидное соединение присутствует в концентрации примерно 0,00001-500 мг/мл, предпочтительно примерно 0,0001-10 мг/мл.
5. Композиция по параграфу (1), где указанный цинк присутствует в концентрации от 0,0005 мг/мл до 50 мг/мл.
6. Композиция по любому из параграфов (I)-(5) дополнительно включающая консервант.
7. Композиция по параграфу (6), где указанный консервант выбран из группы, состоящей из м-крезола, фенола, бензилового спирта и метилпарабена.
8. Композиция по параграфу (7), где указанный консервант присутствует в концентрации от 0,01 мг/мл до 50 мг/мл.
9. Композиция по любому из параграфов (I)-(8), дополнительно включающая изотоническое средство.
10. Композиция по параграфам (I)-(9), где указанное изотоническое средство присутствует в концентрации от 0,01 мг/мл до 50 мг/мл.
11. Композиция по любому из параграфов (I)-(10), дополнительно включающая стабилизатор.
12. Композиция по параграфу (11), где указанный стабилизатор выбран из группы, состоящей из имидазола, аргинина и гистидина.
13. Композиция по любому из параграфов (1)-(12), дополнительно включающая ПАВ.
14. Композиция по любому из параграфов (1)-(13), дополнительно включающая хелатирующее средство.
15. Композиция по любому из параграфов (1)-(14), дополнительно включающая буфер.
16. Композиция по параграфу (15), где указанный буфер выбран из группы, состоящей из Tris, ацетата аммония, ацетата натрия, глицина, аспарагиновой кислоты и Bis-Tris.
17. Композиция по любому из параграфов (1)-(16), дополнительно включающая основной полипептид.
18. Композиция по параграфу (17), где указанный основной полипептид выбран из группы, состоящей из полилизина, полиаргинина, полиорнитина, протамина, путресцина, спермина, спермидина и гистона.
19. Композиция по любому из параграфов (1)-(18), дополнительно включающая спирт или моно-, или дисахариды.
20. Композиция по параграфу (19), где указанный спирт или моно- или дисахарид выбран из группы, состоящей из метанола, этанола, пропанола, глицерина, трегалозы, маннита, глюкозы, эритрозы, рибозы, галактозы, фруктозы, мальтозы, сахарозы и лактозы.
21. Композиция по любому из параграфов (1)-(20), дополнительно включающая сульфат аммония.
22. Фармацевтическая композиция, включающая эффективные количества соединений по параграфам (1)-(21) или их фармацевтически приемлемых солей, а также фармацевтически приемлемый носитель или разбавитель.
23. Способ достижения агонистического эффекта от рецептора GLP-1 у субъекта при наличии такой необходимости, который включает введение указанному субъекту эффективного количества соединения по параграфу (1) или параграфу (22) или их фармацевтически приемлемых солей.
24. Способ лечения заболевания, выбранного из группы, состоящей из диабета типа I, диабета типа II, ожирения, глюкагоном, секреторных расстройств дыхательных путей, метаболических расстройств, артрита, остеопороза, заболеваний центральной нервной системы, рестеноза и нейродегенеративных заболеваний у субъекта при наличии такой необходимости, который включает введение указанному субъекту эффективного количества композиции по параграфу (1) или ее фармацевтически приемлемой соли.
25. В еще одном аспекте настоящее изобретение относится к способу достижения агонистического эффекта от рецептора GLP-1 у субъекта при наличии такой необходимости, который включает введение упомянутому субъекту состава по настоящему изобретению, включающего эффективное количество определенного выше соединения по параграфу (I) или его фармацевтически приемлемой соли.
26. В еще одном аспекте настоящее изобретение относится к способу лечения заболевания, выбранного из группы, состоящей из диабета типа I, диабета типа II, ожирения, глюкагоном, секреторных расстройств дыхательных путей, метаболических расстройств, артрита, остеопороза, заболеваний центральной нервной системы, рестеноза, нейродегенеративных заболеваний, почечной недостаточности, застойной сердечной недостаточности, нефротического синдрома, цирроза, отека легких, гипертонии, а также расстройств, при которых желательно сокращение поступления пищи, у субъекта при наличии такой необходимости, который включает введение указанному субъекту состава по настоящему изобретению, включающего эффективное количество определенного выше соединения по параграфу (I) или его фармацевтически приемлемой соли.
27. Предпочтительным способом по параграфу (26) является способ, в котором подвергаемое лечению заболевание представляет собой диабет типа I или диабет типа II.
(II) Во втором аспекте настоящее изобретение направлено на фармацевтическую композицию, в форме прозрачного раствора или водной смеси, суспензии, или на полутвердую фармацевтическую композицию, включающую (a) по крайней мере одно пептидное соединение, имеющее растворимость в воде более 1 мг/мл при комнатной температуре и имеющее pH от 3,0 до 8,0 и предпочтительно pH от 4,0 до 6,0, которое выбрано из группы, состоящей из hGLP-1(7-36)-NH2, а также его аналогов и производных, hGLP-1(7-37)-OH, а также его аналогов и производных, эксендина-4, а также его аналогов и производных
 ,
,
а также его аналогов и производных,
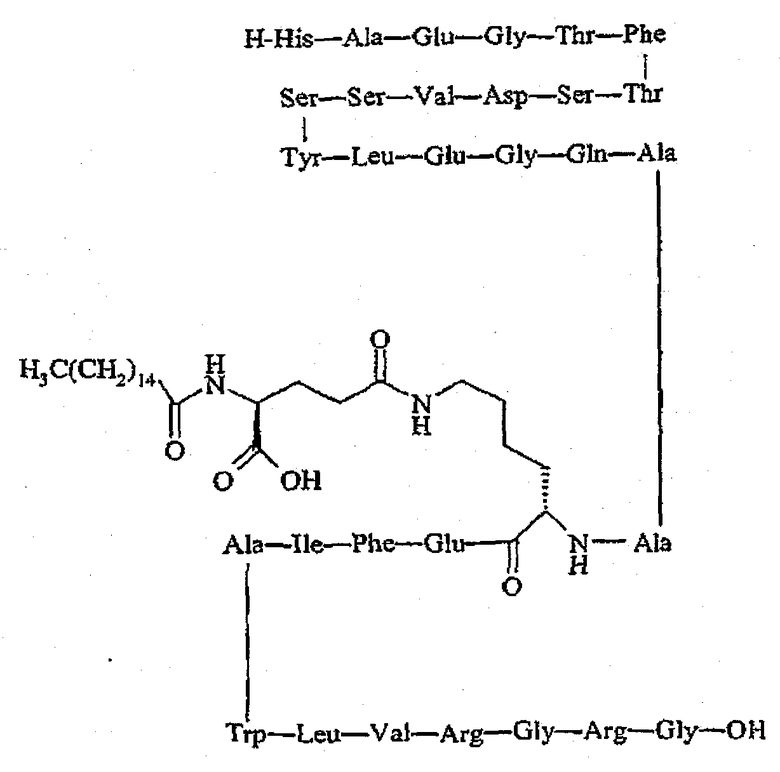
а также его аналогов и производных, и H-His-Gly-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Leu-Ser-Lys-Gln-Met-Glu-Glu-Glu-Ala-Val-Arg-Leu-Phe-Ile-Glu-Trp-Leu-Lys-Asn-Gly-Gly-Pro-Ser-Ser-Gly-Ala-Pro-Pro-Ser-Lys-Lys-Lys-Lys-Lys-Lys-NH2, а также его аналогов и производных;
(b) иона двухвалентного металла; и
(с) растворитель,
при условии, что менее 95% указанного пептидного соединения растворено указанным растворителем.
Указанные ниже номера 1-27 по второму аспекту настоящего изобретения являются номерами, относящимися к параграфу (II).
1. Композиция по параграфу (II), где указанный ион двухвалентного металла представляет собой цинк.
2. В одном из вариантов осуществления изобретение относится к композиции по параграфам (II) и (1), где указанный растворитель представляет собой воду.
3. Композиция по параграфу (II), включающая неводную среду.
4. Композиция по любому из параграфов (II)-(3), где указанное пептидное соединение присутствует в концентрации примерно 0,00001-500 мг/мл, или 0,00001-500 мг/г, предпочтительно примерно 50-350 мг/мл или 50-350 мг/г.
5. Композиция по параграфу (1), где указанный цинк присутствует в концентрации от 0,0005 мг/мл до 50 мг/мл.
6. Композиция по любому из параграфов (II)-(5), дополнительно включающая консервант.
7. Композиция по параграфу (6), где указанный консервант выбран из группы, состоящей из м-крезола, фенола, бензилового спирта и метилпарабена.
8. Композиция по параграфу (7), где указанный консервант присутствует в концентрации от 0,01 мг/мл до 50 мг/мл.
9. Композиция по любому из параграфов (II)-(8), дополнительно включающая изотоническое средство.
10. Композиция по параграфам (II)-(9), где указанное изотоническое средство присутствует в концентрации от 0,01 мг/мл до 50 мг/мл.
11. Композиция по любому из параграфов (II)-(10), дополнительно включающая стабилизатор.
12. Композиция по параграфу (11), где указанный стабилизатор выбран из группы, состоящей из имидазола, аргинина и гистидина.
13. Композиция по любому из параграфов (II)-(12), дополнительно включающая ПАВ.
14. Композиция по любому из параграфов (II)-(13), дополнительно включающая хелатирующее средство.
15. Композиция по любому из параграфов (II)-(14), дополнительно включающая буфер.
16. Композиция по параграфу (15), где указанный буфер выбран из группы, состоящей из Tris, ацетата аммония, ацетата натрия, глицина, аспарагиновой кислоты и Bis-Tris.
17. Композиция по любому из параграфов (II)-(16), дополнительно включающая основной полипептид.
18. Композиция по параграфу (17), где указанный основной полипептид выбран из группы, состоящей из полилизина, полиаргинина, полиорнитина, протамина, путресцина, спермина, спермидина и гистона.
19. Композиция по любому из параграфов (II)-(18), дополнительно включающая спирт или моно-, или дисахариды.
20. Композиция по параграфу (19), где указанный спирт или моно-, или дисахарид выбран из группы, состоящей из метанола, этанола, пропанола, глицерина, трегалозы, маннита, глюкозы, эритрозы, рибозы, галактозы, фруктозы, мальтозы, сахарозы и лактозы.
21. Композиция по любому из параграфов (II)-(20), дополнительно включающая сульфат аммония.
22. Фармацевтическая композиция, включающая эффективные количества соединений по параграфам (II)-(21) или их фармацевтически приемлемых солей, а также фармацевтически приемлемый носитель или разбавитель.
23. Способ достижения агонистического эффекта от рецептора GLP-1 у субъекта при наличии такой необходимости, который включает введение указанному субъекту эффективного количества соединения по параграфу (II) или параграфу (22) или его фармацевтически приемлемой соли.
24. Способ лечения заболевания, выбранного из группы, состоящей из диабета типа I, диабета типа II, ожирения, глюкагоном, секреторных расстройств дыхательных путей, метаболических расстройств, артрита, остеопороза, заболеваний центральной нервной системы, рестеноза и нейродегенеративных заболеваний у субъекта при наличии такой необходимости, который включает введение указанному субъекту эффективного количества композиции по параграфу (II) или ее фармацевтически приемлемой соли.
25. В еще одном аспекте настоящее изобретение относится к способу достижения агонистического эффекта от рецептора GLP-1 у субъекта при наличии такой необходимости, который включает введение упомянутому субъекту состава по настоящему изобретению, включающего эффективное количество определенного выше соединения по параграфу (29) или его фармацевтически приемлемой соли.
26. В еще одном аспекте настоящее изобретение относится к способу лечения заболевания, выбранного из группы, состоящей из диабета типа I, диабета типа II, ожирения, глюкагоном, секреторных расстройств дыхательных путей, метаболических расстройств, артрита, остеопороза, заболеваний центральной нервной системы, рестеноза, нейродегенеративных заболеваний, почечной недостаточности, застойной сердечной недостаточности, нефротического синдрома, цирроза, отека легких, гипертонии, а также расстройств, при которых желательно сокращение поступления пищи, у субъекта при наличии такой необходимости, который включает введение указанному субъекту состава по настоящему изобретению, включающего эффективное количество определенного выше соединения по параграфу (II) или его фармацевтически приемлемой соли.
27. Предпочтительным способом по параграфу (26) является способ, в котором подвергаемое лечению заболевание представляет собой диабет типа I или диабет типа II.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
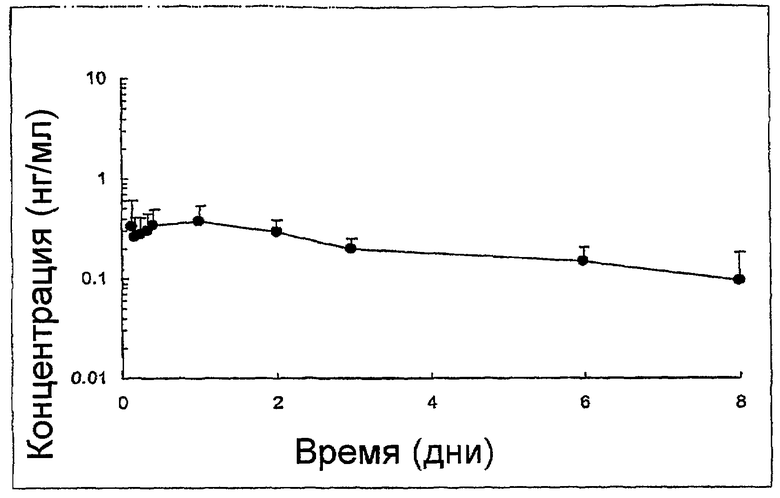
На чертеже показан профиль концентрации пептида в плазме, полученный после одного подкожного введения собаке водной композиции 100 мг/г hGLP-1(7-36)-NH2 с Zn при D=15 мг пептида.
В настоящем описании все сокращения аминокислот (например, Ala) применяются для обозначения структуры общей формулы -NH-CR1R2-CO-, где R1 и R2 являются боковыми цепями аминокислоты (например, в случае Ala R1=CH3 и R2=H). Amp, 1-Nal, 2-Nal, Nle, Cha, 3-Pal, 4-Pal и Aib являются сокращениями для обозначения следующих α-аминокислот: 4-аминофенилаланина, β-(1-нафтил)аланина, β-(2-нафтил)аланина, норлейцина, циклогексилаланина, β-(3-пиридил)аланина, β-(4-пиридил)аланина и α-аминомасляной кислоты соответственно. Обозначения других аминокислот являются следующими: Ura означает урокановую кислоту; Pta означает (4-пиридилтио)уксусную кислоту; Paa означает транс-3-(3-пиридил)акриловую кислоту; Tma-His означает N,N-тетраметиламидиногистидин; N-Me-Ala означает N-метилаланин; N-Me-Gly означает N-метилглицин; N-Me-Glu означает N-метилглутаминовую кислоту; Tle означает трет-бутилглицин; Abu означает α-аминомасляную кислоту; Tba означает трет-бутилаланин; Orn означает орнитин; Aib означает α-аминоизомасляную кислоту; β-ala означает β-аланин; Gaba означает γ-аминомасляную кислоту; Ava означает 5-аминовалериановую кислоту; Ado означает 12-аминододекановую кислоту; Aic означает 2-аминоиндан-2-карбоновую кислоту; Aun означает 11-аминоундекановую кислоту; и Aec означает 4-(2-аминоэтил)-1-карбоксиметилпиперазин, представленный следующей структурной формулой:
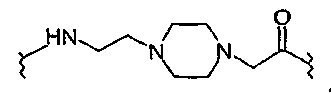
Под обозначением Acc подразумевается аминокислота, выбранная из группы, состоящей из 1-амино-1-циклопропанкарбоновой кислоты (A3c); 1-амино-1-циклобутаннкарбоновой кислоты (A4c); 1-амино-1-циклопентанкарбоновой кислоты (A5c); 1-амино-1-циклогексанкарбоновой кислоты (A6c); 1-амино-1-циклогептанкарбоновой кислоты (A7c); 1-амино-1-циклооктанкарбоновой кислоты (A8c); и 1-амино-1-циклононанкарбоновой кислоты (A9c). В упомянутых выше формулах гидроксиалкил, гидроксифенилалкил и гидроксинафтилалкил могут содержать 1-4 гидроксизаместителя. COX5 означает -C(=O)X5. Примеры -C(=O)X5 включают, не ограничиваясь указанным, ацетил и фенилпропионил.
Полные наименования соединений, обозначаемых в настоящей заявке другими сокращениями, являются следующими: Boc означает т-бутоксикарбонил, HF означает фтористый водород, Fm означает формил, Xan означает ксантил, Bzl означает бензил, Tos означает тозил, DNP означает 2,4-динитрофенил, DMF(ДМФА) означает диметилформамид, DCM означает дихлорметан, HBTU означает гексафторфосфат 2-(1H-бензотриазол-1-ил)-1,1,3,3-тетраметилурония, DIEA означает диизопропилэтиламин, HOAc означает уксусную кислоту, TFA (ТФУ) означает трифторуксусную кислоту, 2CIZ означает 2-хлорбензилоксикарбонил, 2BrZ означает 2-бромбензилоксикарбонил, OcHex означает O-циклогексил, Fmoc означает 9-флуоренилметоксикарбонил, HOBt означает N-гидроксибензотриазол; полимер PAM означает полимер 4-гидроксиметилфенилацетамидометила; Tris означает трис(гидроксиметил)аминометан; и Bis-Tris означает бис(2-гидроксиэтил)амино-трис(гидроксиметил)метан (т.е. 2-бис(2-гидроксиэтил)амино-2-(гидроксиметил)-1,3-пропандиол). Термин «гало» или «галоген» охватывает фтор, хлор, бром и йод.
Термины «углеводородный фрагмент (C1-C12)», «углеводородный фрагмент (C1-C30)» и т.п. охватывают алкильные, алкенильные и алкинильные группы с разветвленной или линейной цепью, включающие указанное число атомов углерода, при условии, что в случае алкенила и алкинила они включают минимум два атома углерода.
Пептид по настоящему изобретению обозначается в данной заявке также и в другом формате, например (A5c8)hGLP-1(7-36)NH2, где замещенные аминокислоты из природной последовательности располагаются между первой парой скобок (например, A5c8 вместо Ala8 в hGLP-1). Сокращение GLP-1 означает глюкагон-подобный пептид-1; hGLP-1 означает человеческий глюкагон-подобный пептид-1. Цифры в скобках относятся к номерам аминокислотных остатков, присутствующих в пептиде (например, обозначение hGLP-1(7-36) соответствует аминокислотным остаткам 7-36 пептидной последовательности человеческого GLP-1). Последовательность hGLP-1(7-37) приведена в Mojsov, S., Int.J.Peptide Protein Res., 40, 1992, pp. 333-342. Обозначение «NH2» в hGlP-1(7-36)NH2 показывает, что C-конец пептида амидирован. hGLP-1(7-36) означает, что C-конец представляет собой свободную кислоту. В hGLP-1(7-38) остатки в положениях 37 и 38 представляют собой Gly и Arg соответственно, если не указано другое. Последовательность эксендина-4 приведена в J.W.Neidigh, et al. Biochemistry, 2001, 40, pp 13188-13200.
Под «прозрачным раствором» подразумевается раствор, включающий растворитель и одно или несколько растворенных веществ, в котором 95%±5%, предпочтительно 99% растворенных веществ полностью растворены, так что раствор является относительно прозрачным. Прозрачный раствор может содержать следовые количества не растворившихся видимых растворенных веществ и/или другие неактивные частицы, в зависимости от чистоты примененного растворителя, однако такие частицы присутствуют в количестве, которое недостаточно для возникновения раствора мутного или непрозрачного вида. Термин «прозрачный раствор» не применим к суспензии, которая представляет собой гетерогенную смесь, состоящую из непрерывной и дискретной фазы, в то время как раствор является гомогенной однофазной смесью двух или нескольких веществ.
Под водной смесью, имеющей вид суспензии или полутвердого вещества, подразумевается состав, включающий растворитель и одно или несколько растворенных веществ, где растворенные вещества могут быть растворены только частично, так что состав не является прозрачной композицией, которая могла бы представлять собой жидкость, прозрачный раствор или более вязкую смесь, в зависимости от концентрации растворенного вещества, но все еще пригоден для инъекций с применением тонких игл.
Применяемые в настоящем изобретении пептиды преимущественно могут иметь форму фармацевтически приемлемых солей. Примеры таких солей включают, не ограничиваясь перечисленными, соли, полученные из органических кислот (например, уксусной, молочной, малеиновой, лимонной, яблочной, аскорбиновой, янтарной, бензойной, метансульфоновой, толуолсульфоновой или памовой кислот, а также трифторуксусной кислоты (ТФУ)), неорганических кислот (например, хлористоводородной кислоты, серной кислоты или фосфорной кислоты) и полимерных кислот (например, дубильной кислоты, карбоксиметилцеллюлозы, полимолочной кислоты, полигликолевой кислоты или сополимеров молочной и гликолевой кислот).
Типовой способ получения солей пептидов по настоящему изобретению хорошо известен в технике, и эти соли можно получить по стандартным методикам солевого обмена.
Как хорошо известно специалистам в данной области техники, уже известные и потенциальные способы применения GLP-1 являются многочисленными и разнообразными (см., Todd J.F., et al., Clinical Science, 1998, 95, pp.325-329; и Todd, J.F. et al., European Journal of Clinical Investigation, 1997, 27, pp. 533-536).
Так, например, введение природного GLP-1 (т.е. hGLP-1(7-36)-NH2 и hGLP-1(7-37)-OH), экседина-4, PC-DAC®, Liraglutide® и/или AVE-0010/ZP-10 по настоящему изобретению с целью достижения агонистического эффекта может значительно улучшить лечение различных ослабляющих заболеваний и состояний, которые, как известно, могут подвергаться лечению действием GLP-1, а именно диабета типа I, диабета типа II, ожирения, глюкагоном, секреторных расстройств дыхательных путей, метаболических расстройств, артрита, остеопороза, заболеваний центральной нервной системы, рестеноза, нейродегенеративных заболеваний, почечной недостаточности, застойной сердечной недостаточности, нефротического синдрома, цирроза, отека легких, гипертонии, а также расстройств, при которых желательно сокращение поступления пищи.
Соответственно в объем настоящего изобретения входят описанные в заявке фармацевтические композиции, включающие в качестве действующего ингредиента хотя бы одно из соединений по параграфу (I).
Дозировка действующего ингредиента в составах по настоящему изобретению может меняться; однако необходимо, чтобы количество действующего ингредиента было таким, чтобы достигалась подходящая дозировка. Выбранная дозировка зависит от желаемого терапевтического эффекта, пути введения и от продолжительности лечения, и обычно определяется лечащим врачом. Как правило, эффективная дозировка действующих веществ по настоящему изобретению находится в пределах от 1×10-7 до 200 мг/кг/день, предпочтительно от 1×10-4 до 100 мг/кг/день, причем это количество можно вводить в виде одной дозы или разделять на несколько доз.
Составы по настоящему изобретению предпочтительно вводят парентерально, например внутримышечно, интраперитонеально, внутривенно, подкожно и т.п.
Лекарственные формы по настоящему изобретению, предназначенные для парентерального введения, включают стерильные водные и неводные растворы, суспензии, гели или эмульсии, при условии, что достигнут желаемый профиль высвобождения in vivo. Примерами неводных растворителей или носителей являются пропиленгликоль, полиэтиленгликоль, растительные масла, например оливковое масло и кукурузное масло, желатин и пригодные для инъекций органические сложные эфиры, такие как этилолеат. Указанные лекарственные формы могут также содержать вспомогательные вещества, такие как консервирующие, смачивающие эмульгирующие и диспергирующие средства. Их можно стерилизовать, например, пропусканием через фильтры, задерживающие бактерии, путем включения в композиции стерилизующих средств, путем облучения композиций или путем нагревания композиций. Кроме того, лекарственные формы могут выпускаться в форме стерильных твердых композиций, которые могут быть растворены в стерильной воде или некоторых других стерильных средах, пригодных для инъекций, непосредственно перед применением.
Если не указано иное, все технические и научные термины, использованные в настоящем описании, имеют те же значения, которые обычно понимают под ними рядовые специалисты в той области техники, к которой относится изобретение. Также все публикации, заявки на патенты, патенты и другие источники, упомянутые в настоящем описании, включены в него с помощью ссылок.
ПОДРОБНОЕ ОПИСАНИЕ
Синтез пептидов
Пептиды, применимые для практической реализации настоящего изобретения, могут быть и были получены стандартным твердофазным пептидным синтезом. См., например, Stewart J.M. et al., Solid Phase Synthesis (Pierce Chemical Co., ed. 1984). Заместители могут быть присоединены к свободной аминогруппе остатка Lys или другим аминокислотным остаткам с помощью стандартных методик, известных в технике. Например, ацильная группа может быть присоединена путем сочетания свободной кислоты со свободной аминогруппой остатка при смешивании частично защищенного пептида, прикрепленного к смоле, с 3-мольными эквивалентами свободной кислоты и 3-мольными эквивалентами карбодиимида в хлористом метилене в течение одного часа.
Пептид hGLP-1(7-36)-NH2 синтезировали на синтезаторе пептидов Applied Biosystems (Foster City, CA) модели 430A, который был модифицирован для ускоренного твердофазного синтеза пептидов на основе группы Boc. См. Schnolzer et al., Int.J.Peptide Protein Res., 90:180 (1992). Применяли смолу на основе 4-метилбензгидриламина (MBHA) (Peninsula, Belmont, CA). Применяли Boc-аминокислоты (Bachem, CA, Torrance, CA; Nova Biochem., LaJolla, CA) со следующей защитой боковых цепей: Boc-Ala-OH, Boc-Arg(Tos)-OH, Boc-Asp(OcHex)-OH, Boc-Tyr(2BrZ)-OH, Boc-His(DNP)-OH, Boc-Val-OH, Boc-Leu-OH, Boc-Gly-OH, Boc-Gln-OH, Boc-Ile-OH, Boc-Lys(2CIZ)-OH, Boc-Thr(Bzl)-OH, Boc-Ser(Bzl)-OH, Boc-Phe-OH, Boc-Glu(OcHex)-OH и Boc-Trp(Fm)-OH. Группы Boc удаляли обработкой 100% ТФУ 2×1 мин. Boc-аминокислоты подвергали предварительной активации действием HBTU и DIEA в ДМФА и затем вводили в реакцию сочетания без предварительной нейтрализации соли пептида на смоле и ТФУ. Время сочетания составляло 5 минут.
По завершении сборки пептидной цепи смолу обрабатывали раствором 20% меркаптоэтанола/10% DIEA в ДМФА 2×30 мин. Затем N-концевую группу Boc удаляли обработкой 100% ТФУ 2×2 мин. После нейтрализации пептида на смоле 10% DIEA в ДМФА (1×1 мин) удаляли формильную группу на боковой цепи Trp обработкой раствором, содержавшим 15% этаноламина/15% воды/70% ДМФА 2×30 мин. Пептид на смоле промывали ДМФА и DCM и высушивали при пониженном давлении. Заключительное отщепление выполняли путем перемешивания пептида на смоле в HF, содержавшем анизол и дитиотрейтол, при 0°C в течение 75 мин. HF удаляли током азота. Остаток промывали эфиром и экстрагировали 4н HOAc.
Пептидную смесь в водном экстракте очищали с помощью препаративной жидкостной хроматографии высокого давления (ВЭЖХ) на обращенной фазе, применяя колонку VYDAC®C18 с обращенной фазой (Nest Group, Southborough, MA). Колонку элюировали линейным градиентом (20%-50% раствора B в течение 105 минут) при скорости потока 10 мл/мин (раствор A=вода, содержавшая 0,1% ТФУ; раствор B ацетонитрил, содержавший 0,1% ТФУ). Фракции собирали и проверяли с помощью аналитической ВЭЖХ. Фракции, содержавшие чистый продукт, объединяли и лиофилизировали до сухого состояния. Чистоту полученного пептида проверяли с помощью системы аналитической ВЭЖХ. Анализ на масс-спектрометре с электрораспылением (MS(ES))S применяли для проверки молекулярной массы конечного продукта.
ТФУ соли пептидов по настоящему изобретению получали в результате очистки пептидов с применением препаративной ВЭЖХ при элюировании ТФУ-содержащими буферными растворами. ТФУ соли могли быть превращены в другие соли, например, ацетаты, растворением пептида в небольшом количестве 0,25 н. водного раствора уксусной кислоты. Полученный раствор вводили в полупрепаративную ВЭЖХ колонку (Zorbax, 300 SB, C-8). Осуществляли элюирование колонки (1) 0,1 н. водным раствором ацетата аммония в течение 0,5 ч, (2) 0,25 н. водным раствором уксусной кислоты в течение 0,54 ч и (3) линейным градиентом (от 20% до 100% раствора B в течение 30 мин) при скорости потока 4 мл/мин (раствор A представлял собой 0,25 н. водный раствор уксусной кислоты; раствор B представлял собой 0,25 н. уксусную кислоту в смеси ацетонитрил/вода, 80:20). Фракции, содержавшие пептиды, собирали и лиофилизовали до сухого состояния.
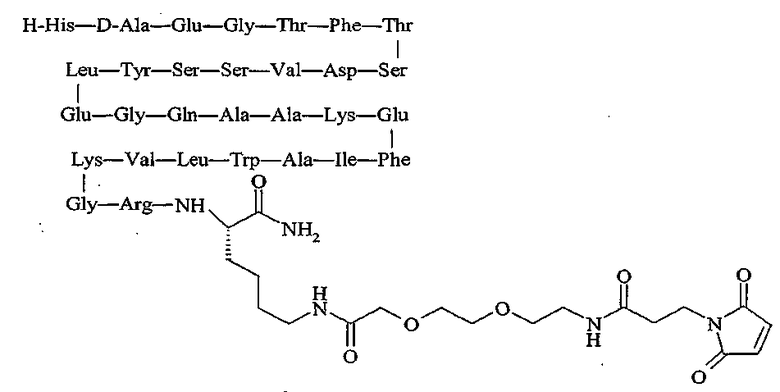
продается под торговой маркой PC-DAC® и является собственностью Conjuchem, Montreal, Quebec, Canada.
Упоминавшийся пептид:
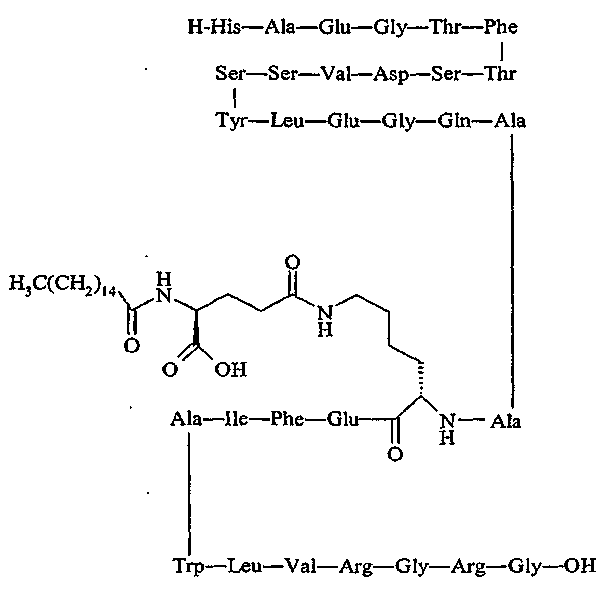
продается под названием Liraglutide® и является собственностью Novo Nordisk, Bagsværd, Denmark.
Упоминавшийся пептид H-His-Gly-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Leu-Ser-Lys-Gln-Met-Glu-Glu-Glu-Ala-Val-Arg-Leu-Phe-Ile-Glu-Trp-Leu-Lys-Asn-Gly-Gly-Pro-Ser-Ser-Gly-Ala-Pro-Pro-Ser-Lys-Lys-Lys-Lys-Lys-Lys-NH2 именуется в известном уровне техники как «AVE-0010/ZP-10» и является совместной собственностью Sanofi-Aventis, Paris, France и Zealand Pharma, Glostrup, Denmark.
МЕТОДИКИ ЭКСПЕРИМЕНТОВ
A. Определение сродства к рецептору GLP-1
Соединения, применимые для практической реализации настоящего изобретения, можно протестировать на способность связываться с рецептором GLP-1 с использованием следующей методики.
Клеточная культура
Клетки инсулиномы крысы RIN 5F (ATCC #CRL-2058, American Type Culture Collection, Manassas, VA), экспрессирующие рецептор GLP-1, культивировали в среде Игла, модифицированной Дульбекко (DMEM), содержавшей 10% фетальной телячьей сыворотки, и хранили при 37°C в увлажненной атмосфере, содержавшей 5% CO2/95% воздуха.
Связывание радиоактивного лиганда
Для исследования связывания радиоактивного лиганда получали мембраны путем гомогенизации клеток RIN в 20 мл ледяного 50 мМ Tris-HCl с помощью прибора Brinkman Polytron (Westbury, NY) (скорость 6, 15 с). Гомогенаты дважды промывали, используя центрифугирование (39000 g/10 мин), и полученный в результате осадок ресуспендировали в 50 мМ Tris-HCl, содержавшем 2,5 мМ MgCl2, 0,1 мг/мл бацитрацина (Sigma Chemical, St.Louis,MO) и 0,1% BSA. Для проведения анализа, аликвоты (0,4 мл) инкубировали с 0,05 пМ (125I)GLP-1(7-36) (~2200 Ки/моль, New England Nuclear, Boston, MA) в присутствии и в отсутствие 0,05 мл не меченных конкурирующих тестируемых пептидов. После инкубирования в течение 100 минут (25°C) связанный (125I)GLP-1(7-36) отделяли от свободного путем быстрого фильтрования через фильтры GF/C (Brandel, Gaithersburg, MD), которые предварительно замачивали в 0,5% полиэтиленимине. Затем фильтры три раза промывали 5 мл аликвотами ледяного 50 мМ Tris-HCl, и связанную радиоактивность, оставшуюся на фильтрах, подсчитывали с помощью гамма-спектрометра (Wallac LKB, Gaithersburg, MD). Специфичное связывание определяли как общее связывание (125I)GLP-1(7-36) минус количество, которое было связано в присутствии 1000 нМ GLP1(7-36) (Bachem, Torrence, CA).
B. Определение зависимости растворимости от pH
Соединения, предназначенные для применения по настоящему изобретению, преимущественно относительно хорошо растворимы в водных растворах при определенных значениях pH, и относительно мало растворимы в водных растворах в присутствии ионов двухвалентных металлов, например цинка. Соединения, предназначенные для применения по настоящему изобретению, обладают растворимостью в воде, превышающей 1 мг/мл при нейтральных значениях pH и комнатной температуре.
Определение растворимости соединения в воде при pH 7
Соединения, которые преимущественно могут использоваться при практической реализации настоящего изобретения, могут быть исследованы с целью определения их растворимости в воде либо при комнатной температуре, либо приблизительно при 37°C с применением следующей методики.
Для определения растворимости при комнатной температуре 2 мг hGLP-1(7-36)-NH2 взвешивали, помещали в стеклянный пузырек и затем туда добавляли 200 мкл аликвоту деионизированной воды. Эту операцию проводили в помещении, где поддерживалась температура примерно 25°C. Измеренное значение pH полученного раствора составляло приблизительно 5. Образец пептида моментально растворялся и наблюдался прозрачный раствор. Нейтрального значения pH (pH 7) добивались путем обработки образца раствора небольшим количеством 0,1 н. NaOH. Наблюдали, что нейтральный раствор оказался прозрачным, и это указывало на то, что растворимость hGLP-1(7-36)-NH2 превышает 10 мг/мл при комнатной температуре и нейтральном pH.
Для определения растворимости при 37°C 2 мг hGLP-1(7-36)-NH2 взвешивали, помещали в стеклянный пузырек и затем туда добавляли 200 мкл аликвоту деионизированной воды. Эту операцию проводили в помещении, где поддерживалась температура примерно 37°C. Измеренное значение pH полученного раствора составляло приблизительно 5. Образец пептида моментально растворялся и наблюдался прозрачный раствор. Нейтральное значение pH (pH 7) получали путем обработки образца раствора небольшим количеством 0,1 н. NaOH. Наблюдали, что нейтральный раствор оказался прозрачным, и это указывало на то, что растворимость hGLP-1(7-36)-NH2 превышает 10 мг/мл при 37°C.
C. Определение растворимости соединений в воде в зависимости от концентрации цинка
Соединения, которые могут преимущественно применяться при практической реализации настоящего изобретения, могут быть исследованы с целью определения их растворимости в воде при pH 7 при различных концентрациях цинка с применением следующей методики.
Исходный раствор цинка получали растворением ZnCl2 в деионизированной воде до достижения концентрации 100 мг/мл и доведением pH до 2,7 добавлением HCl. Рабочие растворы, имеющие различные концентрации цинка («тестовые растворы Zn»), получали, используя соответствующие разбавления исходного раствора.
1 мг образец тестируемого соединения растворяли в 250 мкл каждого из тестовых растворов цинка, получая раствор с концентрацией тестируемого соединения 4 мг/мл. Затем повышали значение pH раствора, используя 0,2 н. NaOH, до образования белого осадка. Раствор с осадком центрифугировали и анализировали маточный раствор с применением ВЭЖХ. Измеряли площадь пика тестируемого соединения в спектре поглощения УФ, и определяли концентрацию тестируемого соединения в маточном растворе путем сравнения с калибровочной кривой.
D. Исследования in vivo
Композиции по настоящему изобретению могли быть и были исследованы с целью определения их способности стимулировать и улучшать эффект in vivo с использованием следующих методик.
Методика эксперимента - 24 часа
В день, предшествовавший эксперименту, взрослым самцам крыс Sprague-Dawley (Taconic, Germantown, NY) массой приблизительно 300-350 г имплантировали катетер в правую атриальную яремную вену с применением анестезии хлоргидратом. Затем крысы голодали в течение 18 часов, после чего им проводили инъекцию соответствующей тестируемой композиции или контрольную инъекцию носителя в момент времени 0. Крыс продолжали поддерживать в голодном состоянии на протяжении всего эксперимента.
В нулевой момент времени крысам подкожно (sc) вводили тестируемые соединения при pH 4,0 или при pH 7,0 в виде прозрачных растворов. В обоих случаях объем инъекции был весьма незначительным (4-6 мкл) и доза GLP-1 соединения, вводимого субъекту, составляла 75 мкг/кг. В соответствующий момент времени после подкожных инъекций, через внутривенный (iv) катетер отбирали образец крови 500 мкл, и крысы получали iv стимулирующую дозу глюкозы с целью выявления наличия усиленной секреции инсулина. Время введения стимулирующей дозы глюкозы было равно 0,25, 1, 6, 12 и 24 часов после инъекции соединений. После отбора первых образцов крови проводили iv инъекцию глюкозы (1 г/кг) и вливали 500 мкл гепаринизированного физиологического раствора (10 Ед/мл). Затем отбирали 500-мкл образцы крови через 2,5, 5, 10 и 20 минут после инъекции глюкозы. После отбора каждого из упомянутых образцов немедленно проводили iv введение через катетер 500 мкл гепаринизированного физиологического раствора (10 Ед/мл). Образцы крови центрифугировали, от каждого образца отделяли плазму, и образцы хранили при -20°C до проведения анализа на содержание инсулина. Количество инсулина в каждом из образцов определяли с применением набора для определения инсулина у крыс с помощью ферментного иммуносорбентного анализа (ELISA) (American Laboratory Products Co., Windham, NH).
Результаты
На протяжении всех 24 часов эксперимента наблюдалась длительная активность, приводившая к повышению уровня инсулина, в качестве реакции на инъекцию глюкозы.
Методика эксперимента - увеличение продолжительности
Общая методика совпадала с описанной выше. В данном эксперименте в нулевой момент времени подкожно (sc) вводили либо тестируемое соединение, либо носитель в качестве контроля. Моменты времени для введения стимулирующих доз глюкозы были 1, 6, 12, 24, 48 и 72 часа после первой инъекции. Инъекции глюкозы через внутривенный катетер и последующий отбор образцов крови проводили как и в ранее описанном эксперименте. Поскольку период голодания был расширен, в момент каждой инъекции осуществляли контрольные эксперименты, в которых вводили носитель и только глюкозу.
Результаты
На протяжении как минимум 48 часов после подкожной инъекции тестируемой композиции наблюдалась длительная активность, приводившая к повышению уровня инсулина, в качестве реакции на инъекцию глюкозы. Кроме того, как и в ранее описанном эксперименте, не наблюдалось высокого первоначального увеличения уровня инсулина в качестве реакции на введение глюкозы.
E. Тесты in vivo
Композиции по настоящему изобретению могут быть и были протестированы с целью определения их способности содействовать продолжительному высвобождению действующего соединения in vivo с применением тестов E.1-E.4., описанных ниже.
Композиции, которые использовались в описанных ниже исследованиях получали согласно следующей общей методике.
Исходный раствор, содержавший 100 мг/мл ZnCl2, получали растворением хлорида цинка (Merck, Mollet del Valles, Barcelona, Spain) в стерильной воде для инъекций (Braun, Rubi, Spain) и значение pH раствора доводили до 2,7, используя HCl. Растворы, содержавшие цинк в различных концентрациях, например 0,1 мг/мл, 0,5 мг/мл, 2 мг/мл и т.д., получали разбавлением исходного раствора. Растворы с более низкой концентрацией цинка, например 10 мкг/мл, 20 мкг/мл, 30 мкг/мл, получали аналогичным способом путем разбавления исходного раствора с содержанием ZnCl2 1 мг/мл. Взвешивали необходимое количество соединения, которое предполагалось подвергнуть исследованию, и растворяли его в соответствующем объеме каждого из полученных растворов соединения цинка, получая прозрачные растворы, имеющие желаемую концентрацию соединения, например 4 мг/мл. Затем полученные растворы подвергали микрофильтрованию и, при необходимости, до введения сохраняли в сосудах, защищающих от действия света.
Концентрацию исследуемого соединения в плазме участвующих в тесте субъектов можно определить рядом известных в технике способов. В одном из традиционных способов, концентрацию соединения определяют с помощью радиоиммуноанализа, применяя выделенные из кролика антитела против тестируемого соединения и осуществляя конкуренцию с известным количеством тестируемого соединения, меченного радиоактивным изотопом йода, например 125I.
E.1. Фармакокинетическое исследование 1
Влияние цинка на биодоступность биологически активного соединения, вводимого субъекту с применением композиции по настоящему изобретению, может быть и было определено следующим образом.
Следуя описанным выше методикам, получали четыре водные композиции, содержавшие 4 мг/мл тестируемых соединений при pH 2,7 и 0,0, 0,1, 0,5 и 2,0 мг/мл ZnCl2 соответственно. Каждую из этих четырех композиций подкожно вводили 16 крысам Sprague-Dawley (Charles River Laboratories, Wilmington, Mass., USA). Средний возраст крыс составлял примерно 8-9 недель, и средняя масса составляла примерно 260-430 г. Пища и вода давалась крысам без ограничения.
E.2. Фармакокинетическое исследование 2
Влияние объема инъекции на биодоступность биологически активного соединения, вводимого субъекту с применением композиции по настоящему изобретению, может быть и была определена следующим образом.
Следуя описанным выше методикам, получали три водные композиции, так, чтобы они содержали 3000, 300 и 75 мкг/мл соответственно при pH 2,7 и концентрация цинка составляла 0,5 мг/мл. Каждую из трех композиций подкожно вводили 16 крысам Sprague-Dawley (Charles River Laboratories, Wilmington, Mass., USA). Средний возраст крыс составлял примерно 8-10 недель, и средняя масса составляла примерно 330-460 г. Перед началом исследования крысам не давали пищу в течение ночи. Объем инъекции выбирали таким образом, чтобы обеспечить для каждой крысы дозировку тестируемого соединения 75 мкг/кг (0,025 мл/кг, 0,25 мл/кг и 1 мл/кг соответственно).
E.3. Фармакокинетическое исследование 3
Влияние цинка на биодоступность биологически активного соединения, вводимого субъекту с применением композиции по настоящему изобретению, может быть и была определена следующим образом.
Следуя описанным выше методикам, получали три водные композиции, так чтобы они содержали 4 мг/мл тестируемых соединений при pH 2,7 и 10, 20 и 30 мкг/мл цинка соответственно. Каждую из этих трех композиций подкожно вводили 16 самцам белых крыс Sprague-Dawley (St.Feliu de Codines, Barcelona, ES). Перед началом исследования крысам не давали пищу в течение ночи.
E.4. Фармакокинетическое исследование 4
Влияние цинка и концентрации биологически активного соединения на биодоступность биологически активного соединения, вводимого субъекту с применением композиции по настоящему изобретению, может быть и была определена следующим образом.
Следуя описанной выше методике, получали две композиции на водной основе. Первый раствор включал 1,45 мг/мл тестируемых соединений и 30 мкг/мл цинка, второй раствор включал 1,45 мг/мл соединения, но не включал цинк. Оба раствора имели значение pH=2,7. Каждый из растворов вводили подкожно самцам собак породы Бигль (Isoquimen, Barcelona, Spain), возраст которых находился в диапазоне примерно 54-65 месяцев, и вес находился в пределах 16-21 кг. Собакам не давали пищи в течение ночи до начала исследования. Дополнительно проводили внутривенное введение второго раствора, содержавшего только действующее соединение.
E.5. Примеры фармакокинетических исследований
В этом разделе описано получение и введение композиции, включающей водный состав 100 мг/г природного человеческого глюкагон-подобного пептида-1, т.е. hGLP-1(7-36)-NH2 и цинк (в составе ZnCl2) в мольном соотношении [пептид:Zn]=1,5:1.
Тестируемое вещество представляло собой природный hGLP-1(7-36)-NH2, который был предоставлен (Polypeptide, USA).
E.5.1. Методика получения
Пептидное соединение PThe взвешивали и смешивали с отвешенным количеством раствора ZnCl2, содержавшим 1,474 мг Zn/мл, получая конечную концентрацию пептида 100 мг/г и конечное мольное соотношение [пептид:Zn]=1,5:1.
Шприцы с иглой 29G (0,33 мм) заполняли композицией в количестве, необходимом для введения дозы пептида 15 мг. После приготовления образцы анализировали и композицию вводили самцам собак породы Бигль.
Были получены следующие результаты анализа:
Содержание пептида: 10,31±0,03% масс/масс
Введенная доза: 15,71±0,18 мг
Чистота по данным ВЭЖХ: 98,5% Ar
4,5
Значение мольного соотношения компонентов композиции составляло [пептид:Zn]=1,44:1.
E.5.2. Фармакокинетическое исследование, биоанализ и результаты
Цель данного исследования заключается в получении фармакокинетического профиля природного hGLP-1(7-36)-NH2 в сыворотке после одного подкожного введения самцам собак породы Бигль состава, содержащего 100 мг/г ацетата GLP-1(7-36)-NH2 и ZnCl2 в мольном соотношении [пептид:Zn]=1,5:1, при общей вычисленной дозе 15 мг чистого пептида.
Композицию вводили в день приготовления в расчетной дозе 15 мг чистого пептида (приблизительно 150 мкл) самцам собаки породы Бигль.
В исследовании использовали в общей сложности 6 самцов собаки породы Бигль в возрасте от 33 до 84 месяцев и массой тела от 12 до 25 кг. Их содержали на свободном доступе к стандартному сухому корму и питьевой воде, которые периодически проверяли.
Животные не получали пищи на 6 ч больше обычного (примерно 18 ч голодание перед введением препарата), чтобы избежать возможного взаимодействия с пищей.
Было отобрано шесть особей животных для получения полного фармакокинетического профиля.
Состав вводили животным индивидуально подкожно в лопаточную область. Область введения дезинфицировали спиртовым раствором (Diolina®, Braun-Dexon). Расчетный уровень дозировки hGLP-1(7-36)-NH2 составлял 15 мг (приблизительно 150 мкл состава на одну собаку) в предварительно заполненных индивидуальных 0,3-миллилитровых шприцах Terumo Myjector с иглами 12х0,33 мм Unimed.
Через вены верхней части тела отбирали образцы крови примерно 2,0 мл, до инъекции (время 0) и в несколько моментов времени после введения композиции на протяжении 35 дней.
После этого кровь помещали в предварительно охлажденные 4-миллилитровые полиэтиленовые пробирки, содержавшие 15% водный раствор EDTA-K3 (12 мкл на мл крови) в качестве антикоагулянта, добавляли консерванты Trasylol® (50 KIU или 5 мкл на мл крови) и ингибитор DPP-IV (10 мкл на мл крови). Образцы крови хранили в бане с холодной водой до центрифугирования (1600 g в течение 20 мин при 4°C на центрифуге Sigma K4-15). В заключении плазму декантировали в полипропиленовые криопробирки и быстро переносили в морозильник с температурой -80°C, где хранили до проведения анализа.
Концентрацию GLP-1(7-36)-NH2 в образцах плазмы определяли после твердофазной экстракции 0,3 мл плазмы собаки с последующей твердофазной экстракцией в сочетании с ЖХ-МС/МС (API4000), применяя аналог GLP-1 в качестве внутреннего стандарта. Этот способ применяли для измерения концентраций GLP-1(7-36)-NH2 в плазме собаки в диапазоне концентраций от 0,25 нг/мл до 25 нг/мл.
Профиль пептида в плазме, полученный после однократного подкожного введения собакам раскрытой в примере композиции при уровне дозировки D=15 мг пептида (906,1 мкг/кг), показан на чертеже.
E.6. Дополнительное фармакокинетическое исследование A
Композицию, раскрытую в E.5.1, хранили при 5°C в течение как минимум 1 недели и испытывали, как описано в предыдущем примере (E.5.2).
E.7. Дополнительное фармакокинетическое исследование B
Композицию, раскрытую в E.5.1, подвергали тестированию при дозировке пептида, превышающей 15 мг.
E.8. Дополнительное фармакокинетическое исследование C
Композицию, подобную полученной в E.5.1, подвергали тестированию при концентрации пептида менее 100 мг/г.
E.9. Дополнительное фармакокинетическое исследование D
Композицию, подобную полученной в E.5.1, подвергали тестированию при молярном соотношении пептид/Zn, превышающем 1,5:1.
E.10. Дополнительное фармакокинетическое исследование E
Композицию, подобную полученной в E.5.1, подвергали тестированию при молярном соотношении пептид/Zn, превышающем 1,5:1, и концентрации пептида менее 100 мг/г.
| название | год | авторы | номер документа |
|---|---|---|---|
| АНАЛОГИ GLP-1 | 1999 |
|
RU2288232C9 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ GLP-1 | 2007 |
|
RU2445972C2 |
| ПРОЛЕКАРСТВА, СОДЕРЖАЩИЕ КОНЬЮГАТ ГИАЛУРОНОВОЙ КИСЛОТЫ, ЛИНКЕРА И ДВОЙНОГО АГОНИСТА GLP-1/ГЛЮКАГОНА | 2016 |
|
RU2719482C2 |
| СПОСОБ ВВЕДЕНИЯ МОЛЕКУЛ GLP-1 | 2003 |
|
RU2332229C2 |
| GLP-1-МОЛЕКУЛЯРНЫЙ КОМПЛЕКС, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ПОЛУЧЕНИЯ КОМПЛЕКСА | 1995 |
|
RU2147588C1 |
| СОСТАВЫ АНАЛОГОВ ГЛЮКАГОНОПОДОБНОГО ПЕПТИДА-2 (GLP-2) | 2019 |
|
RU2833459C2 |
| СХЕМЫ ДОЗИРОВАНИЯ ДЛЯ ВВЕДЕНИЯ АНАЛОГОВ ГЛЮКАГОНОПОДОБНОГО ПЕПТИДА 2 (ГПП-2) | 2018 |
|
RU2785662C2 |
| ДВОЙНЫЕ АГОНИСТЫ GLP1/GIP ИЛИ ТРОЙНЫЕ АГОНИСТЫ GLP1/GIP/ГЛЮКАГОНА | 2013 |
|
RU2652783C2 |
| ПЕПТИДНЫЕ АГОНИСТЫ GLP-2 | 2010 |
|
RU2551977C2 |
| АНАЛОГ ЭКСЕНАТИДА | 2020 |
|
RU2823623C1 |
Изобретение направлено на фармацевтическую композицию в форме прозрачного раствора или водной смеси, суспензии или полутвердой композиции, включающей как минимум одно пептидное соединение, имеющее растворимость в воде более 1 мг/мл при комнатной температуре и имеющим значение pH от 4,0 до 6,0, выбранное из группы, состоящей из hGLP-1(7-36)-NH2, а также его аналогов и производных, hGLP-1(7-37)-ОН, а также его аналогов и производных и/или эксендина-4, а также его аналогов и производных, цинк и растворитель, где менее 95% указанного пептидного соединения растворено растворителем. Изобретение обеспечивает продолжительное действие препарата при пониженных начальных концентрациях в плазме. 2 н. и 18 з.п. ф-лы, 1 ил.
1. Фармацевтическая композиция, включающая прозрачный раствор или водную смесь, суспензию или полутвердую фармацевтическую композицию с (а) по меньшей мере одним пептидным соединением, имеющим растворимость в воде более 1 мг/мл при комнатной температуре и имеющим значение pH от 4,0 до 6,0, которое выбрано из группы, состоящей из hGLP-1(7-36)-NH2, а также его аналогов и производных, hGLP-1(7-37)-OH, а также его аналогов и производных, эксендина-4, а также его аналогов и производных
 ,
,
а также его аналогов и производных,
 ,
,
а также его аналогов и производных, и H-His-Gly-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Leu-Ser-Lys-Gln-Met-Glu-Glu-Glu-Ala-Val-Arg-Leu-Phe-Ile-Glu-Trp-Leu-Lys-Asn-Gly-Gly-Pro-Ser-Ser-Gly-Ala-Pro-Pro-Ser-Lys-Lys-Lys-Lys-Lys-Lys-NH2, а также его аналогов и производных;
(b) ионом двухвалентного металла; и
(c) растворителем,
при условии, что менее 95% указанного пептида растворено указанным растворителем.
2. Композиция по п.1, где указанный ион двухвалентного металла представляет собой цинк.
3. Композиция по п.1, где указанный растворитель представляет собой воду.
4. Композиция по п.3, где указанное пептидное соединение присутствует в концентрации примерно 0,00001-500 мг/мл, предпочтительно примерно 0,0001-10 мг/мл.
5. Композиция по п.2, где указанный цинк присутствует в концентрации от 0,0005 до 50 мг/мл.
6. Композиция по п.1, дополнительно включающая консервант.
7. Композиция по п.6, где указанный консервант выбран из группы, состоящей из м-крезола, фенола, бензилового спирта и метилпарабена.
8. Композиция по п.6 или 7, где указанный консервант присутствует в концентрации от 0,01 до 50 мг/мл.
9. Композиция по п.1, дополнительно включающая изотоническое средство.
10. Композиция по п.9, где указанное изотоническое средство присутствует в концентрации от 0,01 до 50 мг/мл.
11. Композиция по п.1, дополнительно включающая стабилизатор.
12. Композиция по п.11, где указанный стабилизатор выбран из группы, состоящей из имидазола, аргинина и гистидина
13. Композиция по п.1, дополнительно включающая ПАВ.
14. Композиция по п.1, дополнительно включающая хелатирующее средство.
15. Композиция по п.1, дополнительно включающая буфер.
16. Композиция по п.15, где указанный буфер выбран из группы, состоящей из Tris, ацетата аммония, ацетата натрия, глицина, аспарагиновой кислоты и Bis-Tris.
17. Композиция по п.1, дополнительно включающая спирт или моно- или ди-сахариды.
18. Композиция по п.17, где указанный спирт или моно- или дисахарид выбран из группы, состоящей из метанола, этанола, пропанола, глицерина, трегалозы, маннита, глюкозы, эритрозы, рибозы, галактозы, фруктозы, мальтозы, сахарозы и лактозы.
19. Композиция по п.1, дополнительно включающая сульфат аммония.
20. Фармацевтическая композиция, включающая эффективное количество композиции по п.1 и фармацевтически приемлемый носитель или разбавитель.
| Устройство для закрепления изделий | 1977 |
|
SU619322A1 |
| АГОНИСТЫ РЕЦЕПТОРОВ НЕЙРОМЕДИНА В И СОМАТОСТАТИНА | 2000 |
|
RU2263680C2 |
| Choi S et al | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Pharm | |||
| Res | |||
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| Edwards CM et al | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |

