Изобретение относится к радиохимической технологии и может быть использовано для получения порошка диоксида урана, идущего на изготовление керамических таблеток уранового и смешанного уран-плутониевого оксидного ядерного топлива для сборки тепловыделяющих элементов ядерных реакторов атомных электростанций, в том числе реакторных установок на быстрых нейтронах.
Известен способ получения диоксида урана [RU 2158971 C1, МПК G21C 3/62, опубл. 10.11.2000], по которому проводят гидролиз гексафторида урана, экстракцию урана из азотнокислого раствора 30% раствором трибутилфосфата в органическом разбавителе, его реэкстракцию в водный кислый раствор, осаждения полиураната аммония гидроксидом аммония при рН 6,6-8,0 с последующей фильтрацией, сушкой-прокалкой при 450-600°С и восстановлением в токе водорода при 680-720°С.
По другому способу [RU 2296106, МПК C01G 43/025, опубл. 27.03.2007] диоксид урана получают обработкой 25%-ным раствором аммиака, предварительно приготовленного водного раствора уранилнитрата с содержанием урана 50-100 г/дм3 с поддержанием значения рН не менее 6,6 при осаждении полиураната аммония. Далее следуют промежуточные стадии (фильтрация, сушка) с получением порошка полиураната аммония, его термическим разложением при 500°С и последующим восстановлением при 670-730°С, что приводит к получению диоксида урана.
Известен способ получения диоксида урана [Заявка ФРГ, N 2693977, МПК С01С 43/02, 1978], пригодного для изготовления таблетированного ядерного топлива, методом осаждения пероксида урана. Для получения пероксида урана через раствор уранилнитрата (~100 г/л по урану) пропускают смесь аммиака и воздуха для нейтрализации избыточной кислотности до рН среды ~2, затем в раствор добавляют до ~20 масс. % водного раствора перекиси водорода и молярного соотношение уран : перекись водорода = 1 : 1,5-3. Пероксид урана после отделения от маточного раствора прокаливают при температуре 500-800°С с последующим восстановлением до диоксида урана при 550-750°С.
Известен способ получения диоксида урана [RU 2415084, МПК C01G 43/025, опубл. 27.03.2011], согласно которому при добавлении к азотнокислому раствору уранилнитрата восстановителя - хлорида гидроксиламина и аммиачной воды до рН 7 получают, в зависимости от температурных условий, малорастворимые соединения: моногидрат диаквадигидроксиламинат уранила [UO2(Н2О)2(NH2O)2]·Н2О или безводный дигидроксиламинат уранила UO2(NH2O)2, термическое разложение которых приводит к образованию UO2 в инертной атмосфере в температурном интервале 200-400°С. Восстановителем U(VI) до U(IV) при термическом разложении указанного соединения служит координированный с ионом уранила лиганд гидроксиламина.
Во всех перечисленных способах исходным веществом является раствор уранилнитрата, из которого, добавляя соответствующие реагенты и используя многоступенчатых процессы, получают диоксид урана. По своей сути все описанные выше способы переработки раствора уранилнитрата являются способами многостадийной термической реагентной денитрации, осуществляемой в растворах и приводящей к образованию маточных растворов, подлежащих дальнейшей утилизации тем или иным способом. Это является их главным недостатком.
Известен способ [RU 2404925 С2, МПК C01G 43/01, опубл. 27.11.2010], при котором оксиды урана получают нагреванием смеси раствора уранилнитрата и аминоуксусной кислоты (глицин) при температуре 180-220°С в автоклавном режиме. Глицин берут в количестве 90-140% от стехиометрии. При этом в зависимости от соотношения уранилнитрат : аминокислота, могут образовываться UO3, U3O8 или UO2.
Недостатком и этого метода также является использование для термической денитрации водного раствора уранилнитрата с вытекающими отсюда последствиями по переработке остающегося маточного раствора, являющимся жидким радиоактивным отходом.
Наиболее близким к предлагаемому способу и выбранным в качестве прототипа является способ [US 4364859 G21F 9/08, опубл. 21.12.82], при котором оксиды урана получают денитрацией раствора нитрата уранила до 500 г/л по урану в две стадии: выпаривание раствора под действием СВЧ-излучения с последующей денитрацией полученного продукта в другом устройстве в атмосфере водорода с получением оксидов. Недостаток метода - необходимость использования двух стадий при получении диоксида урана, первая из которых связана с упариванием раствора.
Задачей, на решение которой направлено предлагаемое изобретение, является разработка технологичного способа получения порошков оксидов урана в одну стадию, что сокращает время проведения процесса.
Техническим результатом является упрощение способа получения оксидов урана за счет использования твердого уранилнитрата в виде соли гексагидрата нитрата уранила (ГНУ), в процессе микроволновой термической денитрации при взаимодействии с гидразингидратом с исключением образования водных растворов-отходов при проведении процесса, уменьшение времени проведения процесса.
Технический результат достигается в способе получения оксидов урана под действием микроволнового излучения путем нагревания уранилнитрата, причем используют твердый уранилнитрат, предварительно обработанный гидразингидратом, причем процесс проводят при температуре 600-1000°С в течение 10-30 минут.
Процесс осуществляют в восстановительной атмосфере смеси водорода с аргоном при содержании до 10 объемных % водорода с получением диоксида урана или процесс осуществляют в атмосфере воздуха с получением октооксидатриурана.
Способ осуществляется следующим способом.
Порошок оксида урана получают при микроволновом воздействии на твердый уранилнитрат, находящийся в кварцевом сосуде, через горловину которого проходит трубка для подачи в объем сосуда требуемой атмосферы. На поверхность порошка уранилнитрата вносится гидразингидрат при мольном соотношении ГНУ к N2H4, равном 1:0,05-0,15. Гидразингидрат является инициатором процесса поглощения СВЧ-излучения, и разогрев всей массы уранилнитрата происходит лавинообразно от точки внесения гидразингидрата. Процесс проводят в диапазоне 600-1000°С в течение 10-30 мин.
В этих условиях при подаче микроволнового излучения гидразингидрат выступает в роли инициатора процесса поглощения СВЧ-излучения и разогрев всей массы уранилнитрата происходит лавинообразно от точки внесения гидразингидрата. При этом уранилнитрат подвергается процессу денитрации с образованием оксидов урана.
При подаче в колбу восстановительной атмосферы смеси водорода с аргоном в процессе денитрации уранилнитрата сразу образуется диоксид урана.
Если процесс проводится при контакте с атмосферой воздуха, образуется триуранооктооксид.
Сущность заявляемого изобретения поясняется следующими таблицами и прилагаемыми иллюстрациями.
В табл. 1. приведены данные гравиметрических определений исследуемых образцов и расчетные величины потери веса при микроволновом термическом разложении ГНУ (1000°С) в восстановительной атмосфере.
В табл. 2 даны результаты радиометрического определения содержания урана в образцах оксидов, полученных при микроволновой денитрации уранилнитрата при температуре 1000°С в восстановительной атмосфере.
Для определения содержания валентных форм урана в полученных оксидах проводили их растворение в смеси 4 моль/л HCl с 0,05 моль/л HF.
После чего записывали спектр поглощения полученного раствора. Содержание U(IV) в растворе определяли при длине волны 650 нм. Для определения U(VI) в образце этот же раствор контактировали с амальгамой цинка, при этом ионы уранила восстанавливаются до U(IV). После этого повторно записывали спектр раствора и по разнице оптической плотности при длине волны 650 нм двух спектров определяли содержание U(VI) в анализируемом образце.
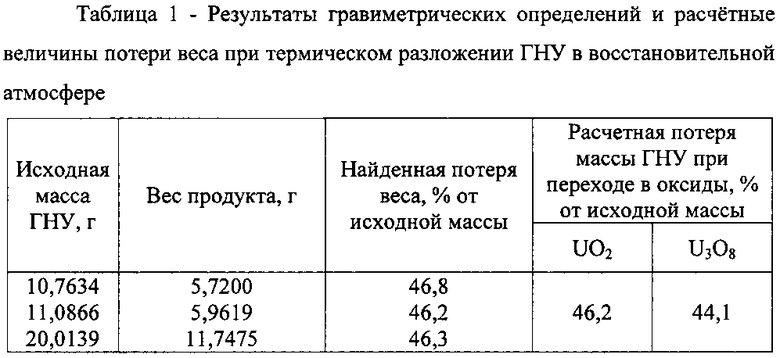
На фиг. 1 показаны спектры 1 и 2 раствора оксида, полученного при термическом разложении соли гексагидрата нитрата уранила (ГНУ) в восстановительной атмосфере до (1) и после контакта этого же раствора (2) с амальгамой цинка.
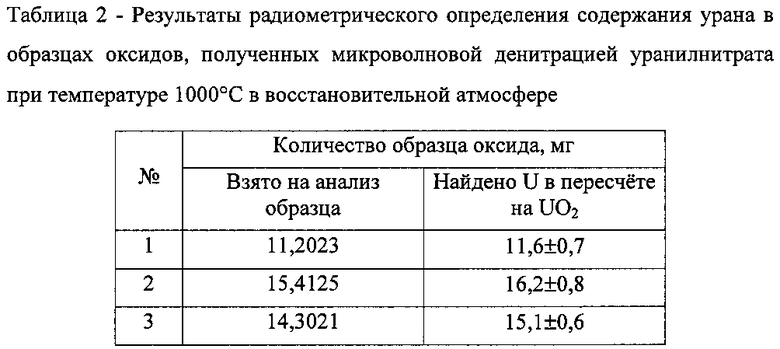
На фиг. 2 - спектры 1 и 2 раствора оксида, полученного при термическом разложении ГНУ в атмосфере воздуха до (1) и после контакта раствора (2) с амальгамой цинка.
Примеры реализации способов
Пример 1
Порошок соли гексагидрата нитрата уранила (ГНУ) 12 г помещали в реакционный кварцевый сосуд. Поверхность смачивали 0,1 мл гидразингидрата (мольное соотношение ГНУ к N2H4 составляет 1:0,1) и помещали в МВ-печь «SAMSUNG, модель MW83UR» с частотой излучения 2450 МГц и мощностью 800 Вт. Через тефлоновую трубку, проходящую через горловину колбы, во внутренний объем печи подавали смесь водорода с аргоном, содержащую до 10 объемных % Н2 для создания восстановительной атмосферы. Процесс микроволновой денитрации начинали кратковременной ~2-минутной подачей излучения при мощности 800 Вт, после чего переходили на 10-минутный режим излучения при мощности 300 Вт, при котором температура образующегося продукта достигала 1000°С. В этом режиме магнетрон включался на 12 сек, затем отключался на 18 сек, в течение которых продукт остывал. В общей сложности процесс длился 10 мин.
Образовавшийся в денитрационном процессе продукт анализировали методами гравиметрии, спектрофотометрии и радиометрии. Полученные данные приведены в табл. 1 и 2 и на фиг. 1. Как видно из таблицы 1, найденная потеря веса всех обработанных образцов соответствует теоритическому значению перехода UO2(NO3)2·6H2O в UO2. Общее содержание урана в полученных образцах также соответствует химической форме UO2. Спектр раствора (фиг. 1 кривая А) незначительно изменился после обработки раствора с амальгамой цинка. Все выполненные аналитические измерения однозначно свидетельствуют о получении диоксида урана в процессе денитрационного термолиза в восстановительной атмосфере.
Пример 2
Способ осуществляли как в примере 1 в том же микроволновом режиме термической денитрации образца, однако процесс проводили в условиях доступа атмосферы воздуха в реакционный объем колбы. Из разницы оптических плотностей раствора (кривая А фиг. 2) полученного образца и его контакта с амальгамой цинка (кривая Б фиг. 2) определено, что содержание U(VI) близко к теоретическому содержанию U(VI) в оксиде U3O8.
Пример 3
Порошок соли гексагидрата нитрата уранила (ГНУ) 67 г помещали в реакционный кварцевый сосуд. Поверхность смачивали 0,25 мл гидразингидрата (мольное соотношение ГНУ к N2H4 составляет 1:0,05) и помещали в МВ-печь. Через тефлоновую трубку, проходящую через горловину колбы, во внутренний объем печи подавали смесь водорода с аргоном, содержащую до 10 объемных % Н2 для создания восстановительной атмосферы. Процесс микроволновой денитрации проводили при мощности 300 Вт при котором температура образующегося продукта достигала 600°С. В ходе процесса денитрации через каждые 10 минут проводили остановку и перемешивание полученного продукта. Общее время СВЧ-обработки составило 30 минут. По результатам анализа полученный продукт состоял на 99% из диоксида урана.
Пример 4.
Способ осуществляли как в примере 3, но время СВЧ-обработки составляло 15 минут, перемешивание не осуществлялось. По результатам анализа полученный порошок содержал 85% UO2.
При проведении процесса при температуре менее 600°С происходит неполное разложение уранилнитрата и восстановление до диоксида урана. Уменьшение температуры менее 10 минут приводит к замедлению скорости восстановления.
Увеличение времени денитрации выше 30 минут и температуры более 1000°С нецелесообразно в связи с экономически необоснованными затратами энергии и расхода восстановительной атмосферы водорода с аргоном.
Таким образом, заявляемый способ приводит к быстрому образованию целевого продукта без каких-либо промежуточных стадий и без возникновения растворов-отходов, за исключением образования газовой фазы с парами воды, аммиака и окислов азота.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ПОРОШКА ДИОКСИДА УРАНА | 2013 |
|
RU2542317C2 |
| СПОСОБ ПОЛУЧЕНИЯ НИТРАТА УРАНИЛА | 2013 |
|
RU2563480C2 |
| СПОСОБ ПЕРЕРАБОТКИ ОТРАБОТАВШЕГО ЯДЕРНОГО ТОПЛИВА | 2014 |
|
RU2560119C1 |
| СПОСОБ ПОЛУЧЕНИЯ ТАБЛЕТИРОВАННОГО ТОПЛИВА НА ОСНОВЕ ПОРОШКА ДИОКСИДА УРАНА | 2004 |
|
RU2296106C2 |
| СПОСОБ КОНТРОЛЯ ОКСИДОВ УРАНА UO И UO НА ПРИМЕСИ | 2015 |
|
RU2605456C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПОРОШКА ДИОКСИДА УРАНА | 2009 |
|
RU2415084C1 |
| СПОСОБ ИЗГОТОВЛЕНИЯ ТАБЛЕТОК ЯДЕРНОГО ОКСИДНОГО ТОПЛИВА | 2010 |
|
RU2428757C1 |
| СПОСОБ ИЗГОТОВЛЕНИЯ КЕРАМИЧЕСКИХ ТОПЛИВНЫХ ТАБЛЕТОК ДЛЯ ТЕПЛОВЫДЕЛЯЮЩИХ ЭЛЕМЕНТОВ ЯДЕРНОГО РЕАКТОРА | 2010 |
|
RU2421834C1 |
| СПОСОБ ПОЛУЧЕНИЯ ОКСИДА УРАНА ИЗ РАСТВОРА УРАНИЛНИТРАТА И УСТРОЙСТВО ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ | 2015 |
|
RU2601765C1 |
| СПОСОБ ИЗГОТОВЛЕНИЯ ТАБЛЕТИРОВАННОГО ТОПЛИВА ИЗ ДИОКСИДА УРАНА | 2001 |
|
RU2209476C2 |
Изобретение относится к радиохимической технологии и может быть использовано для получения порошка диоксида урана, идущего на изготовление керамических таблеток уранового оксидного ядерного топлива. Способ получения оксидов урана под действием микроволнового излучения осуществляют путем нагревания уранилнитрата. При этом используют твердый уранилнитрат, предварительно обработанный гидразингидратом. Процесс проводят при температуре 600-1000°С в течение 10-30 минут. Изобретение позволяет упростить способ получения оксидов урана за счет использования твердого уранилнитрата в процессе микроволновой термической денитрации при взаимодействии с гидразингидратом с исключением образования водных растворов-отходов при проведении процесса, уменьшить время проведения процесса. 2 з.п. ф-лы, 2 ил., 2 табл., 4 пр.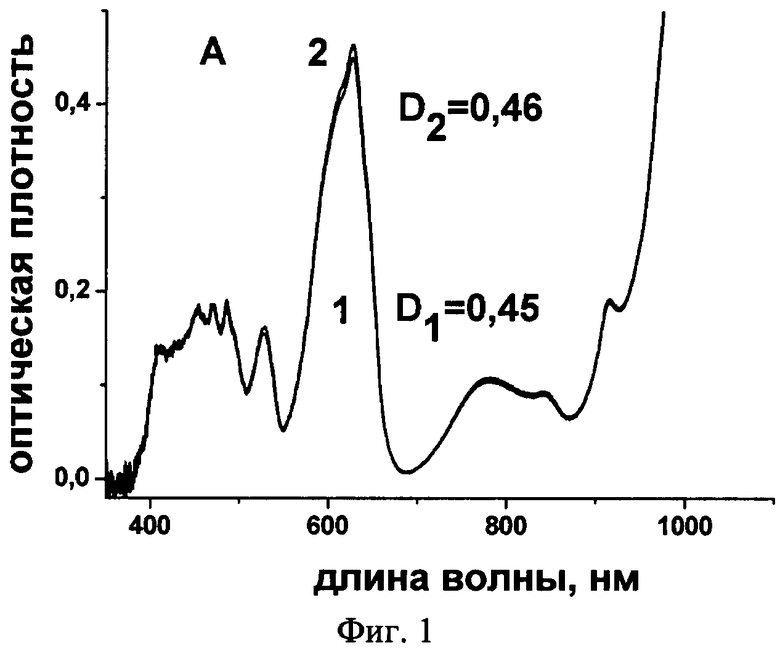
1. Способ получения оксидов урана под действием микроволнового излучения путем нагревания уранилнитрата, отличающийся тем, что используют твердый уранилнитрат, предварительно обработанный гидразингидратом, а процесс проводят при температуре 600-1000°С в течение 10-30 минут.
2. Способ по п. 1, отличающийся тем, что процесс осуществляют в восстановительной атмосфере смеси водорода с аргоном, содержащей до 10 объемных % водорода, с получением диоксида урана.
3. Способ по п. 1, отличающийся тем, что процесс осуществляют в атмосфере воздуха с получением октооксидатриурана.
| US 4364859 A, 21.12.1982;RU 2542317 C2, 20.02.2015 | |||
| СПОСОБ ПОЛУЧЕНИЯ ТРИОКСИДА УРАНА ПУТЕМ ПРЯМОГО ТЕРМИЧЕСКОГО ДЕНИТРИРОВАНИЯ УРАНИЛНИТРАТА | 1996 |
|
RU2106308C1 |
| US 4035468 A, 12.07.1977 | |||
| JP 2004131348 A, 30.04.2004. | |||

