Область техники, к которой относится изобретение
Настоящее изобретение имеет отношение к новым производным пиперазинил пиримидина, обладающим антагонизмом к рецептору хемокина 4 человека (далее называемый hCCR4), способу получения данных соединений, фармацевтических композиций, содержащих эти соединения и применению данных соединений для изготовления лекарственного средства, предназначенного для лечения и/или предотвращения болезней или нарушений, опосредованных hCCR4.
Уровень техники
CCR4 (хемокиновый рецептор 4) впервые был открыт в 1995 Christine A. Power et al. (Christine A.P. et al., J. Biol. Chem., 1995, 270 (8):19495-19500). Этот рецептор является одним из членов семейства хемокиновых рецепторов (CCR) и представляет собой сопряженный с G-белком рецептор, состоящий из семи трансмембранных доменов. Он имеет два специфических лиганда природного происхождения: MDC (хемокин, получаемый из макрофагов) и TARC (хемокин, регулируемый тимусом и активацией) (Sadatoshi Maeda et al., Veterinary Immunology и Immunopathology 2002 (90):145-154). Недавно открытый хемокин-подобный фактор 1 (CKLF1) также является одним из его лигандов (Han W.L. et al., Biochem. J., 2001, 357 (Pt1): 127-135).
CCR4 может экспрессироваться в лейкоцитах периферической крови, клетках тимуса, базофилах, моноцитах, макрофагах, тромбоцитах, IL-активированных NK-клетках, селезенке и мозге, и может играть важную роль в развитии различных болезней. Например, в случае аллергического дерматита (AD) человека, в мононуклеарных клетках периферической крови (РВМС) наблюдается повышенная экспрессия CCR4, экспрессируемого CD4+Т-клетками, при этом уровень TARC в сыворотке также соответственно увеличивается. Это показывает, что хемотаксический ответ CCR4, экспрессированного в клетках, индуцируется TARC, и, в том случае, когда у человека возникает аллергический дерматит, Th2 клетки выборочно мигрируют в поврежденную кожу. Лекарственные средства, пригодные для лечения аллергического дерматита включают антигистаминные препараты, бронхолитические средства, однако, они могут только улучшить симптомы, не оказывая воздействия на развитие болезни. В дополнение к этому, некоторое воздействие на аллергический дерматит также оказывают кортикостероиды, однако, в этом случае имеется потенциальная угроза безопасности. Существуют исследования, показывающие, что противодействие MDC или TARC может уменьшать накопление Т-клеток в местах воспаления, и антагонисты CCR4 могут быть очень эффективными при лечении аллергического дерматита.
Увеличение экспрессия CCR4 наблюдается при ревматоидном артрите, системной красной волчанке, рассеянном склерозе и т.д. Не исключено, что MDC- и TARC-активирование тромбоцитов происходит посредством CCR4, означая, что CCR4 может играть важную роль в активации тромбоцитов и связанной с этим тромботической болезни. Кроме того, CCR4 может опосредованно объединяться с HIV-1, и в то же время он также является ко-рецептором HIV-2.
Кроме того, CCR4 тесно связан с такими легочными болезнями, как хроническая обструктивная пневмония, хронический бронхит и астма. CCR4 может ограниченно экспрессироваться в клетках, вовлеченных в астматическую реакцию, и считается хорошей мишенью для лечения астмы. В настоящее время, антагонисты рецептора хемокина, пригодные для лечения астмы, которые входят в клиническую фазу I в основном включают антагонисты рецепторов CXCR2, CXCR4, CCR1 и CCR5, но не антагонист рецептора CCR4. Поэтому разработка антагонистов рецептора CCR4 имеет хорошую перспективу.
Сущность изобретения
Цель настоящего изобретения заключается в поиске и разработке маломолекулярных соединений, в качестве антагонистов CCR4 рецептора, пригодных для лечения астмы, аллергического дерматита, болезней, связанных с CCR4, факторов риска или патологических состояний.
Настоящие изобретатели обнаружили, что соединения формулы I являются эффективными для оказания противодействия рецептору CCR4.
Соответственно, в первом аспекте настоящего изобретения предоставляется соединение формулы I,
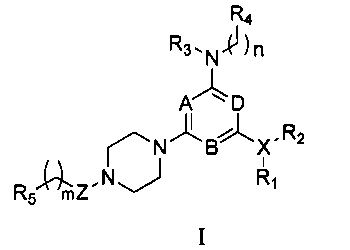
в которой:
любые два из A, B и D являются N, а оставшийся является СН, предпочтительно, A и B являются N, a D является СН;
Z выбирают из группы, состоящей из -СН2-, -С(О)- и -S(O)2-;
X представляет собой галоген или N, при условии, что когда X является галогеном, R1 и R2 в формуле I отсутствуют;
R1 и R2 каждый независимо выбирают из группы, состоящей из Н, линейного или разветвленного алкила, имеющего от 1 до 6 атомов углерода, С1-С6 линейного или разветвленного гетероалкила, содержащего от 1 до 2 гетероатомов, независимо выбранных из группы, состоящей из О, N и S; или
R1 и R2, вместе с N, с которым они связаны, образуют 5-8-членный гетероциклоалкил, содержащий от 1 до 2 гетероатомов, независимо выбранных из группы, состоящей из О, N и S; или
R1 отсутствует, D и R2, вместе с N и другими атомами, с которыми они связаны, образуют гетероарил, имеющий от 5 до 10 атомов;
R3 и R4 каждый независимо выбирают из группы, состоящей из Н, арила, гетероарила, конденсированного арила или конденсированного гетероарила, содержащего от 5 до 10 атомов; при этом указанный арил, гетероарил, конденсированный арил или конденсированный гетероарил необязательно и независимо является моно-, ди- или полизамещенным заместителем, выбранным из группы, состоящей из галогена, цианогруппы, трифторметила, гидроксигруппы и нитрогруппы;
R5 выбирают из группы, состоящей из линейного или разветвленного алкила, имеющего от 1 до 6 атомов углерода, С1-С6 линейного или разветвленного гетероалкила, содержащего 1-2 гетероатома, независимо выбранных из группы, состоящей из О, N и S, циклоалкильной группы, содержащей от 4 до 8, предпочтительно от 5 до 8, более предпочтительно 5 или 6 атомов углерода, 5-8-членного, предпочтительно 5- или 6-членного гетероциклоалкила, содержащего 1-2 гетероатома, независимо выбранных из группы, состоящей из О, N и S;
m и n каждый независимо представляют собой 0, 1 или 2;
или его стереоизомер, или фармацевтически приемлемая соль, или сольват.
В одном варианте осуществления первого аспекта настоящего изобретения предоставляется соединение формулы I согласно настоящему изобретению или его стереоизомер, или фармацевтически приемлемая соль или сольват, в котором A и B являются N, a D является СН.
В одном варианте осуществления первого аспекта настоящего изобретения предоставляется соединение формулы I согласно настоящему изобретению или его стереоизомер, или фармацевтически приемлемая соль или сольват, в котором Z представляет собой -C(O)-.
В одном варианте осуществления первого аспекта настоящего изобретения предоставляется соединение формулы I согласно настоящему изобретению или его стереоизомер, или фармацевтически приемлемая соль или сольват, в котором X является N.
В одном варианте осуществления первого аспекта настоящего изобретения предоставляется соединение формулы I согласно настоящему изобретению или его стереоизомер, или фармацевтически приемлемая соль или сольват, в котором R3 является Н.
В одном варианте осуществления первого аспекта настоящего изобретения предоставляется соединение формулы I согласно настоящему изобретению или его стереоизомер, или фармацевтически приемлемая соль или сольват, в котором n является 1.
В одном варианте осуществления первого аспекта настоящего изобретения предоставляется соединение формулы I согласно настоящему изобретению или его стереоизомер, или фармацевтически приемлемая соль или сольват, в котором R4 представляет собой фенил необязательно и независимо моно-, ди- или полизамещенный заместителем, выбранным из группы, состоящей из галогена, цианогруппы, трифторметила, гидроксигруппы и нитрогруппы.
В одном варианте осуществления первого аспекта настоящего изобретения предоставляется соединение формулы I согласно настоящему изобретению или его стереоизомер, или фармацевтически приемлемая соль или сольват, в котором m является 0.
В одном варианте осуществления первого аспекта настоящего изобретения предоставляется соединение формулы I согласно настоящему изобретению или его стереоизомер, или фармацевтически приемлемая соль или сольват, в котором R5 выбирают из группы, состоящей из линейного или разветвленного алкила, имеющего от 1 до 6 атомов углерода, С1-С6 линейного или разветвленного гетероалкила, содержащего 1-2 гетероатома, независимо выбранных из группы, состоящей из O, N и S, циклоалкильной группы, содержащей 5 или 6 атомов углерода, 5- или 6-членного гетероциклоалкила, содержащего 1-2 гетероатома, независимо выбранных из группы, состоящей из О, N и S.
В одном варианте осуществления первого аспекта настоящего изобретения предоставляется соединение формулы I согласно настоящему изобретению, при этом арил выбирают из группы, состоящей из фенила, нафтила, антрила, фенантрила, инденила, фторенила и аценафтиленила, предпочтительно выбирают из группы, состоящей из фенила и нафтила, более предпочтительно выбирают из фенила.
В одном варианте осуществления первого аспекта настоящего изобретения предоставляется соединение формулы I согласно настоящему изобретению, в котором гетероарил выбирают из группы, состоящей из пиридила, пирролила, фурила, тиенила, пиразолила, имидазолила, тиазолила, оксазолила, изоксазолила, индолила, бензофуранила, карбазолила, пиридазинила, пиразинила, хинолила, изохинолила, пиринила, фенотиазинила и феноксазинила, предпочтительно выбирают из группы, состоящей из пиридила, пирролила и пиразолила, более предпочтительно выбирают из группы, состоящей из пиридила и пирролила, и наиболее предпочтительно выбирают из пиридила.
В одном варианте осуществления первого аспекта настоящего изобретения предоставляется соединение формулы I согласно изобретению или его фармацевтически приемлемая соль или сольват, при этом соединение выбирают из группы, состоящей из:
1-{4-{4-[(2,4-дихлорбензил)амино]-6-[(2,2-диметоксиэтил)метиламино]пиримидин-2-ил}пиперазин-1-ил}пропанона;
1-{4-{4-[(2,4-дихлорбензил)амино]-6-[(2,2-диметоксиэтил)метиламино]пиримидин-2-ил}пиперазин-1-ил}-2-метилпропанона;
1-{4-{4-[(2,4-дихлорбензил)амино]-6-[(2,2-диметоксиэтил)метиламино]пиримидин-2-ил}пиперазин-1-ил}-3-метилтиопропанона;
1-{4-{4-[бис(2-метоксиэтил)амино]-6-(2,4-дихлорбензиламино)-пиримидин-2-ил}пиперазин-1-ил}пропанона;
1-{4-{4-[бис(2-метоксиэтил)амино]-6-(2,4-дихлорбензиламино)-пиримидин-2-ил}пиперазин-1-ил}-2-метилпропанона;
1-{4-{4-[бис(2-метоксиэтил)амино]-6-(2,4-дихлорбензиламино)-пиримидин-2-ил}пиперазин-1-ил}-3-метилтиопропанона;
{4-{4-[бис(2-метоксиэтил)амино]-6-(2,4-дихлорбензиламино)-пиримидин-2-ил}пиперазин-1-ил}циклогексилметанона;
(R)-{4-{4-хлор-6-[(2,4-дихлорбензил)амино]пиримидин-2-ил}пиперазин-1-ил}(пиперидин-2-ил)метанона;
(R)-{4-{4-хлор-6-[(2,4-дихлорбензил)амино]пиримидин-2-ил}пиперазин-1-ил}(тиоморфолин-3-ил)метанона;
1-{4-{4-[(2,4-дихлорбензил)амино]пиридо[2,3-d]пиримидин-2-ил}пиперазин-1-ил}пропанона;
1-{4-{4-[(2,4-дихлорбензил)амино]пиридо[2,3-d]пиримидин-2-ил}пиперазин-1-ил}-2-метилпропанона;
циклогексил {4-{4-[(2,4-дихлорбензил)амино]пиридо[2,3-d]пиримидин-2-ил}пиперазин-1-ил}метанона;
4-[(2,4-дихлорбензил)амино]-2-(4-пропилпиперазин-1-ил)пиридо[2,3-d]пиримидин; и
(R)-{4-{4-[(2,4-дихлорбензил)амино]пиридо[2,3-d]пиримидин-2-ил}пиперазин-1-ил}(тиоморфолин-3-ил)метанона.
Во втором аспекте настоящего изобретения обеспечивается применение соединения формулы I, его рацемата или стереоизомера или фармацевтически приемлемой соли или сольвата согласно первому аспекту настоящего изобретения для лечения или предотвращения болезней или нарушений, связанных с CCR4.
В третьем аспекте настоящего изобретения предоставляется способ получения соединения формулы I согласно первому аспекту настоящего изобретения, данный способ включает стадии:
1) реагирования 2,4,6-трихлорпиримидина с монозащищенным пиперазином в присутствии связывающего кислоту вещества, с получением соединения формулы 1,
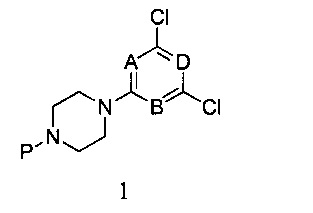
в которой A, B и D являются, как определено выше в формуле I, а P представляет собой азот-защитную группу;
2) реагирования соединения формулы 1 с R3- и -(CH2)n-R4-замещенным амином в присутствии связывающего кислоту вещества, с получением соединения формулы 2,
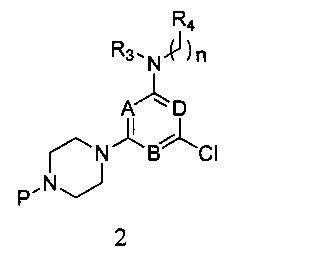
в которой A, B, D, R3, R4 и n являются, как определено выше в формуле I, а P представляет собой азот-защитную группу;
3) реагирования соединения формулы 2 с R1- и R2-замещенным амином в присутствии связывающего кислоту вещества, с получением соединения формулы 3,
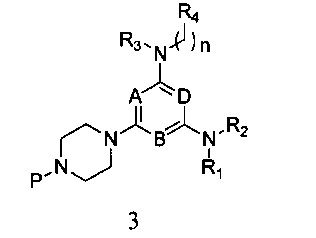
в которой A, B, D, R1, R2, R3, R4 и n являются, как определено выше в формуле I, а P представляет собой азот-защитную группу;
4) удаления защитной группы P из соединения формулы 3, и затем вступления его в реакцию с R5-замещенной карбоновой кислотой, ацилгалоидом, сульфонилхлоридом или галоидзамещенным углеводородом с получением соединения формулы 4,
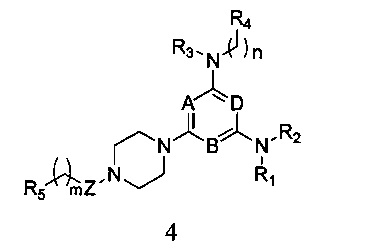
в которой A, B, D, R1, R2, R3, R4, R5, n и m являются, как определено выше в формуле I;
или
5) когда X является галогеном, соединение формулы I получают согласно следующему пути реакции: удаление защитной группы P из соединения формулы 2, и реагирования его с R5-замещенной карбоновой кислотой, ацилгалоидом, сульфонилхлоридом или галоидзамещенным углеводородом с получением соединения формулы 5,
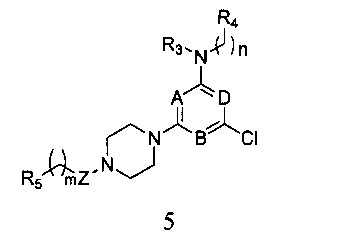
в которой A, B, D, Z, R3, R4, R5, n и m являются, как определено выше в формуле I;
или
6) когда R1 отсутствует, D и R2, вместе с N и другими атомами, с которыми они связаны, образуют гетероарил, имеющий от 5 до 10 атомов, соединение формулы I получают в соответствии со следующей схемой реакции:
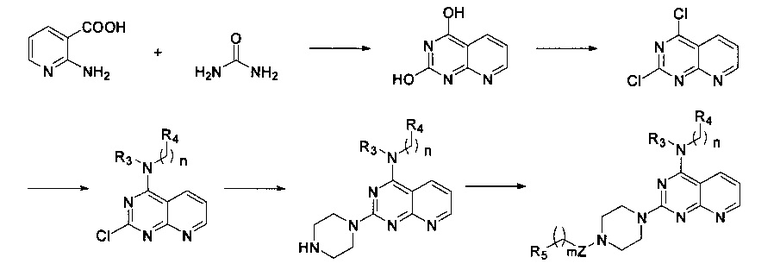
в которой Z, R3, R4, R5 и n являются, как определено выше в формуле I.
В четвертом аспекте настоящего изобретения предоставляется фармацевтическая композиция, содержащая соединение формулы I, его рацемат или стереоизомер или фармацевтически приемлемую соль или сольват согласно первому аспекту изобретения и, по меньшей мере, один фармацевтически приемлемый носитель, разбавитель или эксципиент.
В пятом аспекте настоящего изобретения обеспечивается применение соединения формулы I, его рацемата или стереоизомера или фармацевтически приемлемой соли или сольвата согласно первому аспекту изобретения для изготовления лекарственного средства, предназначенного для лечения или предотвращения болезней или нарушений, связанных с CCR4.
В шестом аспекте настоящего изобретения предоставляется способ лечения или предотвращения болезней или нарушений, связанных с CCR4, причем данный способ включает введение терапевтически или профилактически эффективного количества соединения формулы I, его рацемата или стереоизомера или фармацевтически приемлемой соли или сольвата согласно первому аспекту изобретения нуждающемуся в этом субъекту.
Болезни или нарушения, связанные с CCR4, согласно настоящему изобретению включают, но не ограничиваются этим, аутоиммунные болезни, аллергическое воспаление, тромботические болезни, атопический дерматит, аллергический ринит, астму, аллергический дерматит, инфекционный неспецифический артрит, ревматоидный артрит и волчанку. В частности, болезнью или нарушением, связанным с CCR4 согласно настоящему изобретению, является аллергический ринит, астма, аллергический дерматит, инфекционный неспецифический артрит или ревматоидный артрит.
Осуществление изобретения
Использованный в описании термин “алкил” относится к насыщенной линейной или разветвленной одновалентной углеводородной группе, имеющей 1-20 атомов углерода (т.е., С1-20алкил). В некоторых вариантах осуществления алкил имеет от 1 до 10 атомов углерода (т.е., С1-10алкил), предпочтительно от 1 до 6 атомов углерода (т.е., С1-6алкил), от 1 до 4 атомов углерода (т.е., С1-4алкил) или от 1 до 3 атомов углерода (т.е., С1-3алкил). Примеры “алкила” включают, но не ограничиваются этим, метил, этил, н-пропил, изопропил, н-бутил, трет-бутил, н-пентил, неопентил, н-гексил, 2-метилпентил, 2,2-диметилбутил, 3,3-диметилбутил и тому подобное.
Использованный в описании термин “циклоалкил” относится к циклической углеводородной группе, имеющей 3-12 атомов углерода, предпочтительно 3-8 атомов углерода, более предпочтительно 5-6 атомов углерода и имеющей единственное кольцо или несколько конденсированных колец или мостиковую кольцевую систему. В качестве примеров, такой циклоалкил может включать единичные кольцевые структуры, такие как циклопропил, циклобутил, циклопентил, циклооктил, 1-метил-цикл опропил, 2-метил-циклопентил, 2-метил-циклооктил и тому подобное, и полициклические структуры, такие как адамантил и тому подобное.
Использованный в описании термин “гетероциклоалкил” относится к циклоалкилу, как определено выше, у которого один или более атомов углерода являются независимо замещенными гетероатомами, выбранными из группы, состоящей из N, O и S. Примеры гетероциклоалкила включают, но не ограничиваются этим, пирролидинил, морфолинил, пиперазинил, пиперидинил и тому подобное.
Использованный в описании термин “арил” относится к ароматической углеводородной группе, имеющей 5-18 атомов углерода и имеющей единственное кольцо или несколько конденсированных колец. Арил предпочтительно имеет 5-10, 5-8 или 5-6 или 6 атомов углерода. Примеры “арила” включают, но не ограничиваются этим, фенил, нафтил, антрил, фенантрил, инденил, фторенил и аценафтиленил, который необязательно может быть моно- или полизамещенным.
Использованный в описании термин “гетероарил” относится к гетероароматической кольцевой группе, имеющей 5-18, предпочтительно 5-14, более предпочтительно 5-10 членов, включая моноциклические гетероароматические кольца и полициклические гетероароматические кольца, при этом моноциклическое гетероароматическое кольцо является конденсированным с одним или более другими ароматическими кольцами. Гетероарил имеет один или более гетероатомов в кольце, независимо выбранных из группы, состоящей из N, O и S. Использованный в описании термин “гетероарил” также включает группы, в которых ароматическое кольцо является конденсированным с одним или более неароматическими кольцами (карбоциклическими кольцами и гетероциклическими кольцами), причем соединяющая группа или место соединения располагается на ароматическом кольце. Примеры “гетероарила” включают, но не ограничиваются этим, пиридил, пирролил, фурил, тиенил, пиразолил, имидазолил, тиазолил, оксазолил, изоксазолил, индолил, бензофуранил, бензимидазолил, карбазолил, пиридазинил, пиримидинил, пиразинил, хинолил, изохинолил, пиринил, фенотиазинил, феноксазолил и тому подобное, который необязательно может быть моно- или полизамещенным.
Использованный в описании термин “конденсированный арил” имеет свое общее значение, хорошо известное в данной области техники, и составляет основную часть в соединении формулы I, и как правило, включает, но не ограничивается этим, перечисленные в описании примеры конденсированного арила.
Использованный в описании термин “конденсированный гетероарил” имеет свое общее значение, хорошо известное в данной области техники, и составляет основную часть в соединении формулы I, и, как правило, включает, но не ограничивается этим, перечисленные в описании примеры конденсированного гетероарила.
Использованный в описании термин “галоген” относится к фтору, хлору, брому или йоду. Предпочтительно, галогруппа является фтором или хлором, более предпочтительно хлором. Использованный в описании термин “галоген” также может включать его изотопную форму.
При использовании в описании группы, представленные следующими терминами, имеют их общие значения, хорошо известные в данной области техники: нитрил, трифторметил, трифторметокси, гидроксил, нитро, карбоксиалкил, алкоксикарбонил, алкоксикарбонилалкил, ацил, карбоксамидоалкил, алкил, циклоалкил, алкилтио, алкилсульфинил, алкилсульфонил, сульфамоил, амидино, циано, амино, амидо, алкиламино, диалкиламино, алкиламиноалкил.
Использованная в описании фраза “С1-С6 линейный или разветвленный гетероалкил, содержащий 1-2 гетероатома, независимо выбранных из группы, состоящей из O, N и S” имеет свое общее значение, хорошо известное в данной области техники, и помимо этого, в данном описании она может, в частности, относиться к линейному или разветвленному алкилу, у которого атом углерода заменяется O, N или S.
Использованная в описании фраза “5-8-членный гетероциклоалкил, содержащий 1-2 гетероатома, независимо выбранных из группы, состоящей из O, N и S” имеет свое общее значение, хорошо известное в данной области техники, и помимо этого, в данном описании она может, в частности, относиться к гетероциклоалкилу, у которого атом углерода заменяется O, N или S.
Использованные в описании термины “рацемат” и “энантиомеры” имеют свои общепринятые значения, хорошо известные в данной области техники.
Использованные в описании термины “защитная группа” и “блокирующая группа” могут использоваться взаимозаменяемым образом и относятся к реагенту, который используется, чтобы временно блокировать одну или более желательных функциональных групп у соединения, имеющего несколько реакционных центров. В некоторых вариантах осуществления защитная группа имеет одну или более или предпочтительно все из следующих характеристик: а) она селективно добавляется с хорошим выходом к функциональной группе с целью получения защищенного субстрата; b) защищенный субстрат является устойчивым в отношении реакции(ий), происходящей в одном или более других реакционных центрах; и c) она селективно удаляется с хорошим выходом с помощью реагента, который не действует на восстановленную незащищенную функциональную группу. Специалистам ясно, что реагент не должен действовать на другие реакционноспособные группы в соединении. В других случаях реагент также может вступать в реакцию с другими реакционноспособными группами в соединении. Примеры защитных групп подробно описаны в Greene, T.W., Wuts, P.G., “Protective Groups in Organic Synthesis”, 3rd Edition, John Wiley & Sons, New York: 1999 (и других изданиях этой книги). Полное содержание этих печатных материалов включается в описание путем отсылки. Использованный в описании термин “азот-защитная группа” относится к реагенту, который используется, чтобы временно блокировать один или более желательных азот-реакционноспособных центров на полифункциональном соединении. Предпочтительные азот-защитные группы имеют типичные характеристики упомянутых выше защитных групп, а некоторые типичные азот-защитные группы также подробно описаны в Greene, T.W., Wuts, P.G.ʹs “Protective Groups in Organic Synthesis”, 3rd Edition, John Wiley & Sons, New York: 1999, Chapter 7. Полное содержание этого печатного материала включается в описание путем отсылки.
Использованный в описании термин “фармацевтически приемлемый” в основном имеет отношение к форме, являющейся пригодной с фармацевтической или медицинской точки зрения, или если она не является напрямую пригодной с фармацевтической или медицинской точки зрения, она может быть пригодна в качестве промежуточного продукта фармацевтического или медицинского продукта, а затем может быть удалена с помощью подходящего метода перед окончательным применением в фармацевтике или медицине. Например, фармацевтически приемлемая соль включает не только фармацевтически приемлемые соли, которые могут использоваться в клиническом аспекте, но также те, которые не могут напрямую использоваться клинически, но могут использоваться при получении соединения настоящего изобретения, а затем удаляться в последующей процедуре.
Использованный в описании термин “фармацевтически приемлемый носитель, разбавитель или эксципиент” означает фармацевтические вспомогательные средства, обычно используемые при производстве препаратов, например, перечисленные в Luo Mingsheng et al., “Encyclopedia of Pharmaceutical Adjuvants”, Science и Technology Press, Sichuan, 1995.
Использованный в описании термин “изомер” включает все возможные изомеры (например, энантиомеры, диастереомеры, геометрические изомеры, конформационные изомеры, эпимеры и ротамеры) соединения формулы I настоящего изобретения. Например, в настоящем изобретении рассматриваются соответствующие R и S конфигурации асимметричных центров, (Z) и (Е) изомеры с двойной связью и (Z) и (Е) конформационные изомеры.
Соединение настоящего изобретения может существовать в форме несольвата и сольвата, включая гидратную форму, например, полугидрат. В большинстве случаев для цели настоящего изобретения сольватная форма с фармацевтически приемлемым растворителем, таким как вода и этанол, соответствует несольватной форме.
Согласно настоящему изобретению соединение формулы I и его фармацевтически приемлемая соль или сольват может быть получено с помощью следующего типичного иллюстративного метода, данный метод включает стадии:
1) При комнатной температуре раствор монозащищенного пиперазина и связывающего кислоту агента, такого как N,N-диизопропилэтиламин в дихлорметане, добавляют по каплям к раствору 2,4,6-трихлорпиримидина в дихлорметане, и перемешивают в течение двух часов. После этого органические соли отмывают водой и насыщенным рассолом, соответственно. Продукт реакции подвергается разделению и очистке методом колоночной хроматографии с силикагелем при использовании элюента петролейный эфир/этилацетат с получением соединения формулы 1,
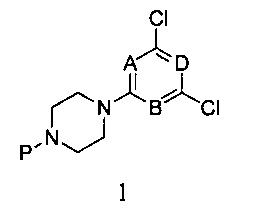
в которой A, B и D являются, как определено выше в формуле I, при этом P является азот-защитной группой;
2) Соединение формулы 1, R3- и -(СН2)n-R4-замещенный амин и связывающий кислоту агент, такой как N,N-диизопропилэтиламин, растворяют в NMP, нагревают до 90°C и перемешивают в течение ночи. После этого продукт реакции в 5 раз разбавляют этилацетатом, 6 раз промывают водой, 3 раза промывают насыщенным раствором и затем подвергают разделению и очистке методом колоночной хроматографии с силикагелем при использовании элюента петролейный эфир/этилацетат с получением соединения формулы 2,
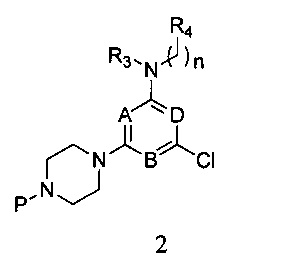
в которой A, B, D, R3, R4 и n являются, как определено выше в формуле I, а P является азот-защитной группой;
3) Соединение формулы 2 смешивают с 15-кратным (количеством) R1- и R2-замещенного амина, при этом амин действует и как реактант и как связывающий кислоту агент, и нагревают с обратным холодильником в течение 24 часов. После этого продукт реакции в 5 раз разбавляют этилацетатом, 6 раз промывают водой, 3 раза промывают насыщенным раствором и затем подвергают разделению и очистке методом колоночной хроматографии с силикагелем при использовании элюента петролейный эфир/этилацетат с получением соединения формулы 3,
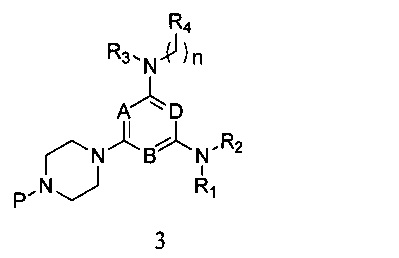
в которой A, B, D, R1, R2, R3, R4 и n являются, как определено выше в формуле I, а P является азот-защитной группой;
4) Смешивают равные объемы этанола и 10% водного раствора гидроксида натрия, в этой смеси растворяют соединение формулы 3, а затем перемешивают при нагревании с обратным холодильником в течение 24 часов. После удаления защитной группы P полученный продукт вступает в реакцию с R5-замещенной карбоновой кислотой, ацилгалоидом, сульфонилхлоридом или галоидзамещенным углеводородом с получением соединения формулы 4,
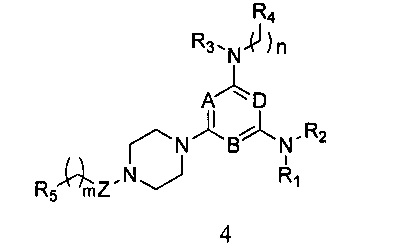
в которой A, B, D, Z, R1, R2, R3, R4, R5, n и m являются, как определено выше в формуле I;
5) Когда X является галогеном, соединение формулы I получают в соответствии со следующим путем реакции: смешивают равные объемы этанола и 10% водного раствора гидроксида натрия, в этой смеси растворяют соединение формулы 2, а затем перемешивают при нагревании с обратным холодильником в течение 24 часов. После удаления защитной группы P полученный продукт вступает в реакцию с R5-замещенной карбоновой кислотой, ацилгалоидом, сульфонилхлоридом или галоидзамещенным углеводородом с получением соединения формулы 5,
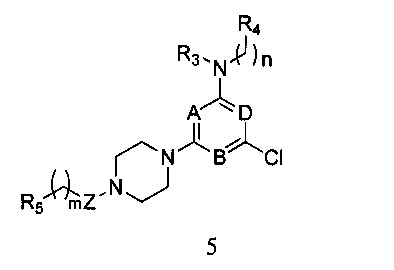
в которой A, B, D, Z, R3, R4, R5, n и m являются, как определено выше в формуле I;
6) Когда R1 отсутствует, D и R2 вместе с N и другими атомами, с которыми они связаны, образуют гетероарил, имеющий от 5 до 10 атомов, соединение формулы I получают в соответствии со следующим путем реакции: 2-амино-никотиновую кислоту и мочевину тонко измельчают, равномерно перемешивают, нагревают до 210°C, выдерживают при этой температуре в течение 15 минут, а затем охлаждают для перекристаллизации; полученный продукт подвергают реакции хлорирования путем нагревания с обратным холодильником в хлорангидриде фосфорной кислоты для осуществления реакции нуклеофильного замещения с R3- и -(СН2)n-R4-замещенным амином, а затем реакции нуклеофильного замещения с пиперазином, и, наконец, вступает в реакцию с R5-замещенной карбоновой кислотой, ацилгалоидом, сульфонилхлоридом или галоидзамещенным углеводородом с получением соединения формулы I,
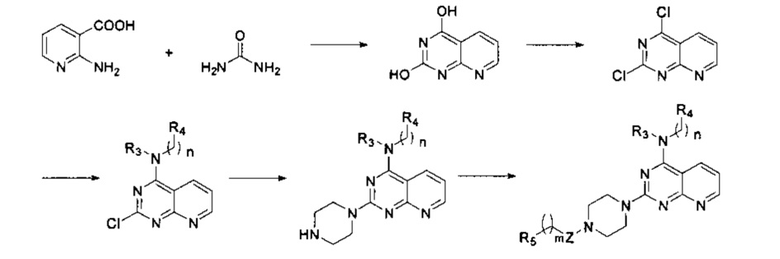
в которой Z, R3, R4, R5, n и m являются, как определено выше в формуле I.
Специалисту в данной области техники должно быть понятно, что соединение формулы I настоящего изобретения также может использоваться в форме фармацевтически приемлемой соли или сольвата. Фармацевтически приемлемая соль соединения формулы I включает обычные соли, образованные фармацевтически приемлемой неорганической кислотой или органической кислотой или неорганическим основанием или органическим основанием, и соли присоединения кислоты четвертичного аммония. Более конкретные примеры подходящих кислых солей включают соли хлористоводородной кислоты, бромистоводородной кислоты, серной кислоты, фосфорной кислоты, азотной кислоты, перхлорной кислоты, фумаровой кислоты, уксусной кислоты, пропионовой кислоты, янтарной кислоты, гликолиевой кислоты, муравьиной кислоты, молочной кислоты, малеиновой кислоты, винной кислоты, лимонной кислоты, памовой кислоты, малоновой кислоты, гидроксималеиновой кислоты, фенилуксусной кислоты, глутаминовой кислоты, бензойной кислоты, салициловой кислоты, фумаровой кислоты, толуолсульфоновой кислоты, метансульфоновой кислоты, нафталин-2-сульфоновой кислоты, бензолсульфоновой кислоты, гидроксинафтойной кислоты, йодистоводородной кислоты, яблочной кислоты, стеариновой кислоты, танниновой кислоты и тому подобного. Что касается других кислот, таких как щавелевая кислота, несмотря на то, что они не являются фармацевтически приемлемыми сами по себе, их можно использовать для получения соли, пригодной в качестве промежуточного продукта, чтобы таким образом получить соединение настоящего изобретения и его фармацевтически приемлемую соль. Более конкретные примеры подходящих основных солей включают соли натрия, лития, калия, магния, алюминия, кальция, цинка, N,Nʹ-дибензилэтилендиамина, хлорпрокаина, холина, диэтаноламина, этилендиамина, N-метилглюкамина и прокаина.
Настоящее изобретение также включает пролекарство соединения формулы I, при этом пролекарство после введения может подвергаться химическому превращению в метаболическом процессе, чтобы превратиться в активное лекарственное средство. В общем, такое пролекарство является функциональным производным соединения настоящего изобретения, которое легко превращается in vivo в необходимое соединение формулы I. Например, общепринятые методы отбора и получения подходящих производных пролекарств описаны в “Design Of Prodrugs”, H Bund Saard, Elsevier, 1985. Полное содержание этого печатного материала включается в описание путем отсылки.
Настоящее изобретение также включает активный метаболит соединения формулы I.
Другой аспект настоящего изобретения имеет отношение к фармацевтической композиции, содержащей соединение формулы I, рацемат или стереоизомер или его фармацевтически приемлемую соль или сольват и, по меньшей мере, один фармацевтически приемлемый носитель, который может использоваться при лечении in vivo и обладает биосовместимостью. Фармацевтическая композиция может быть приготовлена в различных формах, соответствующих разным способам введения. Соединения, упомянутые в настоящем изобретении, также могут быть получены в виде различных фармацевтически приемлемых солей.
Фармацевтическая композиция настоящего изобретения содержит эффективную дозу соединения формулы I, рацемата или стереоизомера или его фармацевтически приемлемой соли или сольвата, такого как гидрат, согласно настоящему изобретению и один или более пригодных фармацевтически приемлемых носителей. Фармацевтически приемлемый носитель в данном описании включает, но не ограничивается этим, ионообменник, окись алюминия, стеарат алюминия, лецитин, сывороточный белок, такой как сывороточный альбумин человека, буферное вещество, такое как фосфат, глицерин, сорбиновая кислота, сорбат калия, неполные глицеридные смеси насыщенных растительных жирных кислот, вода, соль и электролит, такой как протаминсульфат, двузамещенный фосфорнокислый натрий, вторичный кислый фосфат калия, хлористый натрий, соль цинка, коллоидная двуокись кремния, трисиликат магния, поливинилпирролидон, материалы на основе целлюлозы, полиэтиленгликоль, натрий карбоксиметилцеллюлоза, полиакрилаты, пчелиный воск, ланолин.
Фармацевтическая композиция, содержащая соединение настоящего изобретения, может быть введена любым из следующих способов: перорально, ингаляцией путем распыления, ректально, назально, буккально, местно, парентерально, например, подкожно, внутривенно, внутримышечно, внутрибрюшинно, подоболочечно, внутрь желудочка, с помощью внутригрудинной или внутричерепной инъекции или инфузии, или при помощи резервуара эксплантата. Из числа этих способов предпочтительным способом введения является пероральный, внутрибрюшинный и внутривенный.
Для перорального введения соединение настоящего изобретения может быть изготовлено в любой форме, подходящей для перорального приема, включая, но не ограничиваясь этим, таблетки, капсулы, водные растворы или суспензии. Используемый в таблетках носитель, как правило, включает лактозу и кукурузный крахмал, и дополнительно, может добавляться смазывающее вещество, такое как стеарат магния. Используемый в капсулах разбавитель, как правило, включает лактозу и сухой кукурузный крахмал. Общепринятой является водная суспензия, в которой активный ингредиент смешивается с подходящим эмульгирующим веществом и суспендирущим веществом. При необходимости, в перечисленные выше пероральные препараты может добавляться определенный подсластитель, ароматизатор или краситель.
При местном применении, особенно в случае лечения пораженной поверхности или органа, когда местное нанесение легко достигается, таком как окулярное, дермальное или при нервном расстройстве нижнего отдела кишечника, соединение настоящего изобретения может быть изготовлено в формах для местного применения в соответствии с разными пораженными поверхностями или органами, как описано ниже.
В случае местного окулярного введения соединение настоящего изобретения может быть изготовлено в форме микронизированной суспензии или раствора, в котором используемый носитель представляет собой изотонический стерильный солевой раствор с определенным значением pH, в который может добавляться или не добавляться консервирующее вещество, такое как бензилхлорид алкоксид. С целью окулярного введения соединение может быть приготовлено в форме мази, например, вазелиновой мази.
В случае местного применения на коже соединение настоящего изобретения может быть изготовлено в форме мази, лосьона или крема, в котором активный ингредиент суспендирован или растворен в одном или более носителях. Носитель, который может использоваться в препаратах в виде мази, включает, но не ограничивается этим, минеральное масло, жидкий вазелин, белый вазелин, пропиленгликоль, полиоксиэтилен, полиоксипропилен, эмульгированный воск и воду; носитель, который может использоваться в лосьоне или креме, включает, но не ограничивается этим, минеральное масло, сорбитмоностеарат, Твин 60, воск цетиловых эфиров, гексадецен ариловый спирт, 2-октилдодеканол, бензиловый спирт и воду.
Соединение настоящего изобретения также может применяться в виде стерильных инъекционных форм, включая стерильную инъекционную водную или масляную суспензию или стерильный инъекционный раствор. Причем, используемый носитель и растворитель включают воду, раствор Рингера и изотонический раствор хлорида натрия. Кроме того, стерильное нелетучее масло также может использоваться в качестве растворителя или суспендирующей среды, например, моно- или диглицериды.
Кроме того, следует отметить, что дозировка и способ применения соединения настоящего изобретения зависит от многих факторов, включая возраст пациента, вес, пол, состояние здоровья, пищевой статус, активность соединения, время приема, уровень метаболизма, тяжесть болезни и субъективное мнение врача. Предпочтительная дозировка составляет от 0,001 до 100 мг/кг веса тела/день, более предпочтительно между 0,01 и 50 мг/кг веса тела/день, еще более предпочтительно между 0,1 и 25 мг/кг веса тела/день и наиболее предпочтительно между 1 и 10 мг/кг веса тела/день. При необходимости эффективная ежедневная доза может быть разделена на несколько доз для осуществления введения. Поэтому, однократная доза композиции может содержать такую дозу или ее дробные дозы, чтобы получить суточную дозу. Частота, с которой вводится соединение формулы I, определяется согласно опыту практикующего врача и различными факторами, включая возраст пациента, вес, пол, состояние здоровья, тип и тяжесть заболевания и т.п., например, соединение может быть введено 1, 2, 3, 4, 5 или более раз в день или один раз в два дня, один раз каждые три дня, один раз в неделю, один раз в две недели и т.п.
Что касается патентов, патентных заявок, публикаций и т.п., упомянутых в настоящем изобретении, их полное содержание, как одна часть настоящего изобретения, включается в описание путем отсылки.
Варианты осуществления настоящего изобретения
Далее настоящее изобретение дополнительно описывается с использованием специфических промежуточных продуктов и примеров. Однако, следует понимать, что эти промежуточные продукты и примеры используются только для более подробного и специального описания настоящего изобретения и не должны истолковываться как ограничивающие настоящее изобретение каким-либо образом.
В настоящем изобретении использованные материалы и методы тестирования предоставляются в общем и/или частичном описании. Хотя многие материалы и методы, использованные для достижения цели настоящего изобретения, хорошо известны в данной области техники, они, тем не менее, описаны по возможности подробно в настоящем изобретении. Для разъяснения специалисту, в дальнейшем тексте, если не указано иначе, материалы и методы, использованные в настоящем изобретении, являются хорошо известными в данной области техники.
Температура плавления соединения измеряется прибором для определения точки плавления YRT-3, температура является нескорректированной. Спектр 1Н-ЯМР измеряется с помощью ЯМР-спектрометра Bruker ARX 400. Для FAB масс-спектрометрии используется магнитный спектрометр с высокой разрешающей способностью Zabspect.
Получение промежуточных соединений
Промежуточный продукт 1
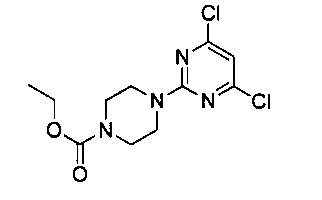
В 1000 мл двугорлую кругло донную колбу, снабженную термометром и воронкой постоянного давления, добавили дихлорметан (350 мл) и 2,4,6-трихлорпиримидин (20 г, 0.109 моль); воронку постоянного давления наполняли раствором 1-этоксикарбонил пиперазина (19 г, 0.120 моль) и N,N-диизопропилэтиламина (15.5 г, 0.120 моль) в дихлорметане (150 мл) и медленно добавляли по каплям, поддерживая температуру ниже 30°C. После завершения добавления по каплям систему перемешивали в течение 2 часов. После завершения реакции продукт реакции 3 раза промыли водой 200 мл, 3 раза промыли 200 мл насыщенного рассола, концентрировали и разделили на колонке с силикагелем (элюент: петролейный эфир/этилацетат) с получением 8,0 г белого твердого вещества, выход 24.05%.
1H-ЯМР (400 MHz, CDCl3) δ ppm: 6.42 (1Н, s), 4.18 (2H, m), 3.65-3.52 (8H, brm), 1.29 (3H, t, J=7.0 Hz, J=7.28 Hz); EI-MS (m/z): 305.1 [M+H]+.
Промежуточный продукт 2
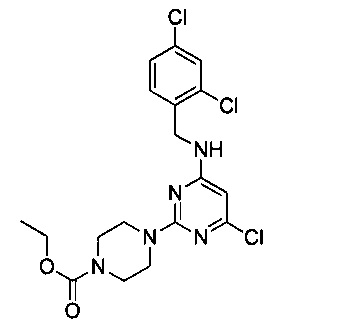
В 250 мл двугорлую круглодонную колбу, снабженную термометром и обратным холодильником, добавили промежуточный продукт 1 (8.0 г, 0.026 моль), 2,4-дихлорбензиламин (4.75 г, 0.027 моль), N,N-диизопропилэтиламин (6.72 г, 0.052 моль) и NMP (80 мл), смешали до однородности, нагрели до 90°C, и перемешивали в течение 12 часов. После этого продукт реакции разбавили в 5 раз этилацетатом, 6 раз промыли водой, 3 раза промыли насыщенным рассолом, концентрировали и разделили на колонке с силикагелем (элюент: петролейный эфир/этилацетат) с получением 10,8 г белого твердого вещества, выход 92.6%.
1H-ЯМР (400 MHz, CDCl3) δ ppm: 7.39 (1H, d, J=1.68 Hz), 7.34 (1H, d, J=8.4 Hz), 7.21 (1H, d, J=8.6 Hz), 4.94 (1H, s), 4.55 (2H, d, J=6.16 Hz), 4.18 (2H, m), 3.71-3.55 (8H, brm), 1.30 (3H, t, J=7.0 Hz, J=7.28 Hz); EI-MS (m/z): 444.2 [M+H]+.
Промежуточный продукт 3
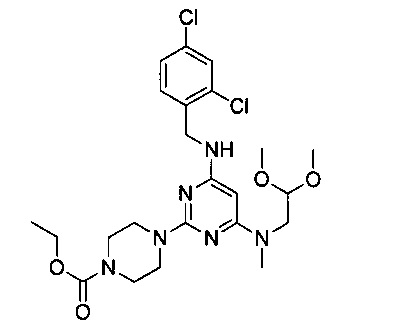
В 100 мл двугорлую круглодонную колбу, снабженную термометром и обратным холодильником, добавили промежуточный продукт 2 (5.0 г, 0.011 моль) и 2-метиламиноацетальдегид диметил ацеталь (19.64 г, 0.165 моль), смешали до однородности, нагревали с обратным холодильником при 140°C и перемешивали в течение 24 часов. После этого продукт реакции разбавили в 5 раз этилацетатом, 3 раза промыли водой, 3 раза промыли насыщенным рассолом, концентрировали и разделили на колонке с силикагелем (элюент: петролейный эфир/этилацетат) с получением 4.22 г белого твердого продукта, выход 71.2%.
1Н-ЯМР (400 MHz, CDCl3) δ ppm: 7.38 (2Н, m), 7.19 (1Н, d, J=8.4 Hz), 4.85 (1H, m), 4.82 (1H, s), 4.53 (2H, d, J=6.16 Hz), 4.49 (1H, m), 4.13 (2H, m), 3.72 (4H, m), 3.58 (2H, d, J=5.2 Hz), 3.37 (6H, s), 2.95 (3H, s), 2.9 (4H, m), 1.32 (3H, t, J=7.0 Hz, J=7.28 Hz); EI-MS (m/z): 527.2 [M+H]+.
Промежуточный продукт 4
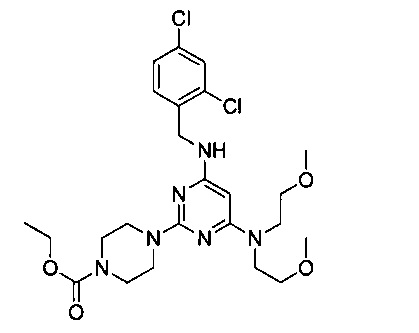
В 100 мл двугорлую круглодонную колбу, снабженную термометром и обратным холодильником, добавили промежуточный продукт 2 (5.0 г, 0.011 моль) и диметоксиэтиламин (21.98 г, 0.165 моль), смешали до однородности, нагревали с обратным холодильником при 168°C и перемешивали в течение 24 часов. После этого продукт реакции разбавили в 5 раз этилацетатом, 3 раза промыли водой, 3 раза промыли насыщенным рассолом, концентрировали и разделили на колонке с силикагелем (элюент: петролейный эфир/этилацетат) с получением 3.63 г белого твердого продукта, выход 74.6%.
1Н-ЯМР (400 MHz, CDCl3) δ ppm: 7.37 (2Н, m), 7.18 (1Н, d, J=8.4 Hz), 4.84 (2H, m), 4.51 (2H, d, J=6.4 Hz), 4.12 (2H, m), 3.68-3.47 (12H, brm), 3.3 (6H, s), 2.88 (4H, m), 1.31 (3H, t, J=7.0 Hz, J=7.28 Hz); EI-MS (m/z): 541.2 [M+H]+.
Промежуточный продукт 5
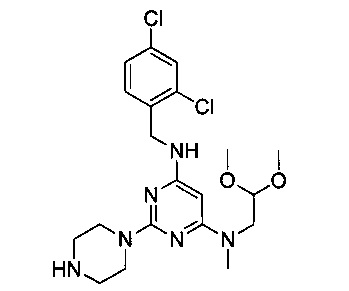
В 500 мл двугорлую круглодонную колбу, снабженную термометром и обратным холодильником, добавили промежуточный продукт 3 (4.22 г, 0.009 моль), этанол (90 мл) и 10% водный раствор гидроксида натрия (90 мл), смешали до однородности, нагревали с обратным холодильником при 90°C и перемешивали в течение 24 часов. После этого продукт реакции был подвергнут вакуумной перегонке для удаления этанола, затем его 3 раза экстрагировали этилацетатом. Экстракты объединили, 3 раза промыли насыщенным рассолом, концентрировали и разделили на колонке с силикагелем (элюент: петролейный эфир/этил ацетат/метано л) с получением 3.61 г маслянистого продукта, выход 88.1%.
1Н-ЯМР (400 MHz, CDCl3) δ ppm: 7.38 (2Н, m), 7.19 (1H, d, J=8.4 Hz), 4.85 (1H, m), 4.82 (1H, s), 4.53 (2H, d, J=6.16 Hz), 4.49 (1H, m), 3.72 (4H, m), 3.58 (2H, d, J=5.2 Hz), 3.37 (6H, s), 2.95 (3H, s), 2.9 (4H, m); EI-MS (m/z): 455.2 [M+H]+.
Промежуточный продукт 6
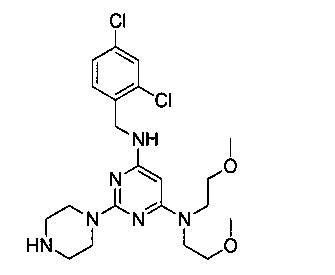
В 500 мл двугорлую круглодонную колбу, снабженную термометром и обратным холодильником, добавили промежуточный продукт 4 (5.41 г, 0.010 моль), этанол (100 мл) и а 10% водный раствор гидроксида натрия (100 мл), смешали до однородности, нагревали с обратным холодильником при 90°C и перемешивали в течение 24 часов. После этого продукт реакции был подвергнут вакуумной перегонке для удаления этанола, затем его 3 раза экстрагировали этилацетатом. Экстракты объединили, 3 раза промыли насыщенным рассолом, концентрировали и разделили на колонке с силикагелем (элюент: петролейный эфир/этилацетат/метанол) с получением 4,46 г маслянистого продукта, выход 95,0%.
1H-ЯМР (400 MHz, CDCl3) δ ppm: 7.37 (2H, m), 7.18 (1H, d, J=8.4 Hz), 4.84 (2H, m), 4.51 (2H, d, J=6.4 Hz), 3.68-3.47 (12H, brm), 3.3 (6H, s), 2.88 (4H, m); EI-MS (m/z): 469.4 [M+H]+.
Промежуточный продукт 7
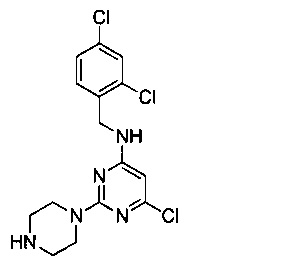
В 500 мл двугорлую круглодонную колбу, снабженную термометром и обратным холодильником, добавили промежуточный продукт 2 (4.45 г, 0.010 моль), этанол (100 мл) и а 10% водный раствор гидроксида натрия (100 мл), смешали до однородности, нагрели с обратным холодильником при 90°C и перемешивали в течение 24 часов. После этого продукт реакции был подвергнут вакуумной перегонке для удаления этанола, затем его 3 раза экстрагировали этилацетатом. Экстракты объединили, 3 раза промыли насыщенным рассолом, концентрировали и разделили на колонке с силикагелем (элюент: петролейный эфир/этилацетат/метанол) с получением 3.54 г белого пенящегося твердого продукта, выход 95,0%.
1H-ЯМР (400 MHz, CDCl3) δ ppm: 7.42 (1H, d, J=1.12 Hz), 7.27 (2H, m), 5.75 (1H, s), 5.19 (1H, s), 4.58 (2H, s), 3.73 (4H, m), 3.01 (4H, m); EI-MS (m/z): 372.1 [M+H]+.
Промежуточный продукт 8
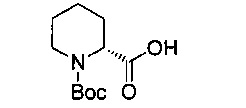
В 1000 мл круглодонную колбу добавили 2R-пиперидин-карбоновую кислоту (8.0 г, 0.062 моль), ди-трет-бутил дикарбонат (14.8 г, 0.068 моль), бикарбонат натрия (26.04 г, 0.310 моль) и метанол (400 мл) и перемешивали при комнатной температуре в течение 24 часов. После этого продукт реакции был подвергнут вакуумной перегонке для удаления метанола, затем его развели водой, 3 раза промыли этиловым эфиром, довели до значения рН=2 насыщенным гидросульфатом калия и 3 раза экстрагировали дихлорметаном. Экстракты объединили, 3 раза промыли насыщенным рассолом, концентрировали и разделили на колонке с силикагелем (элюент: петролейный эфир/этилацетат/уксусная кислота) с получением 12,48 г белого продукта, выход 87,8%.
1Н-ЯМР (400 MHz, DMSO) δ ppm: 12.71 (1Н, s), 4.61 (1H, d, J=28.8 Hz), 3.82 (1H, d, J=12 Hz), 2.93 (1H, m), 2.06 (1H, s), 1.62 (3H, m), 1.39 (11H, m); EI-MS (m/z): 229.1 [M]+.
Промежуточный продукт 9
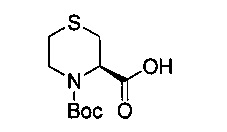
В 1000 мл круглодонную колбу добавили 2R-тиоморфолин карбоновую кислоту, гидрохлорид (10.0 г, 0.054 моль), ди-трет-бутил дикарбонат (13.0 г, 0.060 моль), бикарбонат натрия (45.0 г, 0.536 моль) и метанол (500 мл), и перемешивали при комнатной температуре в течение 24 часов. После этого продукт реакции был подвергнут вакуумной перегонке для удаления метанола, затем его развели водой, 3 раза промыли этиловым эфиром, довели до значения рН=2 насыщенным гидросульфатом калия и 3 раза экстрагировали дихлорметаном. Экстракты объединили, 3 раза промыли насыщенным рассолом, концентрировали и разделили на колонке с силикагелем (элюент: петролейный эфир/этилацетат/уксусная кислота) с получением 8,63 г белого продукта, выход 64,6%.
1Н-ЯМР (400 MHz, CDCl3) δ ppm: 5.33 (1H, d, J=90.4 Hz), 4.39 (1H, brm), 3.30 (2H, brm), 2.95 (1H, m), 2.71 (1H, m), 2.53 (1H, m), 1.53 (9H, s); EI-MS (m/z): 247.1 [M]+.
Промежуточный продукт 10
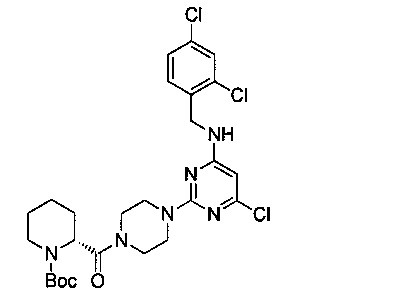
В 25 мл круглодонную колбу добавили промежуточный продукт 7 (540 мг, 1.45 ммоль), промежуточный продукт 8 (332 мг, 1.45 ммоль), EDCI (418 мг, 2.18 ммоль), HOBt (294 мг, 2.18 ммоль), DIEA (374 мг, 2.9 ммоль) и тетрагидрофуран (9 мл) и перемешивали при комнатной температуре в течение ночи. После этого, продукт реакции был подвергнут вакуумной перегонке для удаления растворителя, затем его разбавили водой и 3 раза экстрагировали этилацетатом. Экстракты объединили, 3 раза промыли насыщенным рассолом, концентрировали и разделили на колонке с силикагелем (элюент: петролейный эфир/этилацетат) с получением 650 мг белого пенящегося твердого продукта, выход 76,8%.
1Н-ЯМР (400 MHz, DMSO) δ ppm: 9.13 (1Н, s), 8.63 (1H, s), 8.08 (1H, s), 7.64 (1H, d, J=1.52 Hz), 7.27 (2H, m), 5.97 (1H, s), 4.56 (2H, d, J=4.48 Hz), 4.39 (1H, d, J=9.24 Hz), 3.76-3.22 (9H, brm), 2.87 (1H, s), 1.99 (1H, m), 1.74 (4H, m), 1.45 (10H, m); EI-MS (m/z): 583.2 [M+H]+.
Промежуточный продукт 11
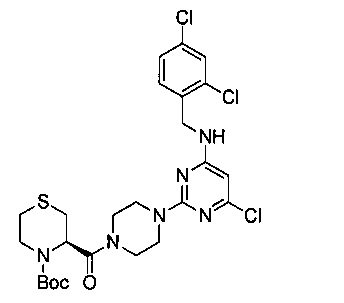
В 25 мл круглодонную колбу добавили промежуточный продукт 7 (540 мг, 1.45 ммоль), промежуточный продукт 9 (359 мг, 1.45 ммоль), EDCI (418 мг, 2.18 ммоль), HOBt (294 мг, 2.18 ммоль), DIEA (374 мг, 2.9 ммоль) и тетрагидрофуран (9 мл) и перемешивали при комнатной температуре в течение ночи. После этого, продукт реакции был подвергнут вакуумной перегонке для удаления растворителя, затем его разбавили водой и 3 раза экстрагировали этилацетатом. Экстракты объединили, 3 раза промыли насыщенным рассолом, концентрировали и разделили на колонке с силикагелем (элюент: петролейный эфир/этилацетат) с получением 688 мг белого пенящегося твердого продукта, выход 78.8%.
1Н-ЯМР (400 MHz, CDCl3) δ ppm: 7.42 (1Н, d, J=1.12 Hz), 7.27 (2H, m), 5.75 (1H, s), 5.19 (1H, s), 4.58 (2H, s), 3.95-3.44 (10H, brm), 3.12 (1H, m), 2.82 (2H, m), 2.43 (2H, m), 1.92 (9H, s); EI-MS (m/z): 601.1 [M+H]+.
Промежуточный продукт 12 ОН
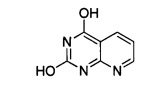
2-Аминоникотиновую кислоту (5 г, 0.036 моль) и мочевину (9 г, 0.150 моль) поместили в ступку, тонко измельчили и равномерно перемешали. Полученную смесь вылили в фарфоровую чашку для выпаривания, нагревали в колбонагревателе при постоянном перемешивании, и плавили, когда температура увеличивалась до 185°C. Затем дополнительно нагревали до 210°C, и держали при этой температуре в течение 15 минут. Затем, отключили подачу энергии. Продукт реакции охладили, растворили в 100 мл водного раствора NaOH 2 моль/л, затем нагревали до 80°C до полного растворения, а затем добавили по каплям уксусную кислоту до нейтрального значения с получением 3,2 г белого кристаллического продукта, выход 54.2%.
1H-ЯМР (400 MHz, DMSO) δ ppm: 11.69 (1Н, s), 11.48 (1H, s), 8.61 (1H, m), 8.27 (1H, m), 7.26 (1H, m).
Промежуточный продукт 13
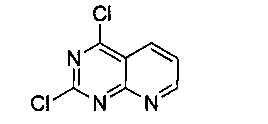
В 500 мл двугорлую круглодонную колбу, снабженную термометром и обратным холодильником, добавили промежуточный продукт 12 (10.0 г, 0.061 моль) и хлорокись фосфора (200 мл), смешали до однородности, нагревали с обратным холодильником при 105°C и перемешивали в течение 24 часов. После этого продукт реакции был подвергнут вакуумной перегонке для удаления оксихлорида фосфора. Оставшееся имеющее консистенцию сиропа вещество вылили на 200 г дробленого льда, и немедленно 3 раза экстрагировали хлороформом, используя каждый раз 150 мл. Экстракты объединили, 3 раза промыли насыщенным рассолом, концентрировали и разделили на колонке с силикагелем (элюент: петролейный эфир/этилацетат) с получением 10,36 г белого твердого продукта, выход 84.9%.
1H-ЯМР (400 MHz, CDCl3) δ ppm: 9.34 (1H, m), 8.66 (1H, m), 7.76 (1H, m); EI-MS (m/z): 199.0 [M]+.
Промежуточный продукт 14
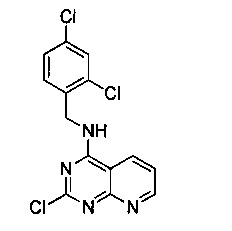
В 250 мл двугорлую круглодонную колбу, снабженную термометром и воронкой постоянного давления, добавили промежуточный продукт 13 (10.36 г, 0.052 моль), N,N-диизопропилэтиламин (7.35 г, 0.057 моль) и 1,2-дихлорэтан (80 мл); в воронку постоянного давления пометили раствор 2,4-дихлорбензиламина (10.03 г, 0.057 моль) в 1,2-дихлорэтане (10 мл) и медленно добавляли по каплям при -10°C. После завершения добавления по каплям систему перемешивали в течение ночи. Осажденное твердое вещество профильтровали, и фильтрат очистили на колонке с силикагелем (элюент: петролейный эфир/этилацетат) с получением 16,35 г белого твердого продукта, выход 92,6%.
1Н-ЯМР (400 MHz, DMSO) δ ppm: 9.57 (1Н, m), 9.03 (1H, m), 8.79 (1H, m), 7.69 (1H, m), 7.64 (1H, m), 7.45 (2H, m), 4.79 (2H, d, J=5.2 Hz); EI-MS (m/z): 339.2 [M+H]+.
Промежуточный продукт 15
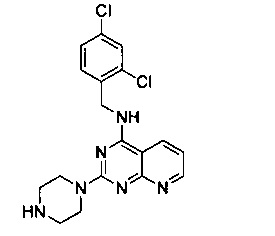
В 2000 мл двугорлую круглодонную колбу, снабженную термометром и обратным холодильником, добавили промежуточный продукт 14 (16.35 г, 0.048 моль), пиперазин (8.27 г, 0.096 моль) и этанол (1200 мл), нагрели до 60°C и перемешивали в течение 15 часов. После концентрирования продукт реакции разбавили 300 мл дихлорметана, 3 раза промыли насыщенным бульоном и очистили на колонке с силикагелем (элюент: этилацетат/метанол/аммоний) с получением 13,86 г белого твердого продукта, выход 74,2%.
1H-ЯМР (400 MHz, DMSO) δ ppm: 8.85 (1H, m), 8.66 (1H, m), 8.46 (1H, m), 7.63 (1H, m), 7.38 (2H, m), 7.10 (1H, m), 4.73 (2H, d, J=5.2 Hz), 3.63 (4H, s), 2.61 (4H, s); EI-MS (m/z): 389.2 [M+H]+.
Промежуточный продукт 16
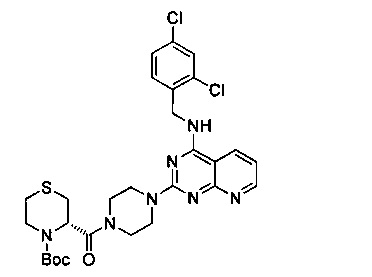
В 100 мл круглодонную колбу добавили промежуточный продукт 15 (1.28 г, 3.28 ммоль), промежуточный продукт 9 (0.81 г, 3.28 ммоль), EDCI (0.94 г, 4.92 ммоль), HOBt (0.66 г, 4.92 ммоль), DIEA (0.85 г, 6.56 ммоль) и тетрагидрофуран (40 мл), и перемешивали при комнатной температуре в течение ночи. После этого, продукт реакции был подвергнут вакуумной перегонке для удаления растворителя, затем его разбавили водой и 3 раза экстрагировали этилацетатом. Экстракты объединили, 3 раза промыли насыщенным рассолом, концентрировали и разделили на колонке с силикагелем (элюент: этилацетат/метанол/аммоний) с получением 1,63 г белого пенящегося твердого продукта, выход 80,5%.
1H-ЯМР (400 MHz, CDCl3) δ ppm: 8.77 (1Н, m), 8.02 (1H, m), 7.43 (1H, m), 7.31 (1H, m), 7.20 (1H, m), 7.05 (1H, m), 6.56 (1H, m), 4.85 (2H, d, J=5.2 Hz), 4.02-3.44 (10H, brm), 3.13 (1H, m), 2.83 (2H, m), 2.44 (2H, m), 2.02 (9H, s); EI-MS (m/z): 618.2 [M+H]+.
Примеры
Пример 1
1-{4-{4-[(2,4-дихлорбензил)амино]-6-[(2,2-диметоксиэтил)метиламино]пиримидин-2-ил}пиперазин-1-ил}пропанон (Соединение 1)
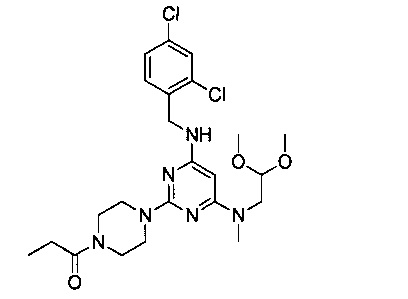
Промежуточный продукт 5 (300 мг, 0.66 ммоль), пропионовую кислоту (54 мг, 0.73 ммоль), EDCI (190 мг, 0.99 ммоль), HOBt (134 мг, 0.99 ммоль), DIEA (170 мг, 1.32 ммоль) растворили в тетрагидрофуране (9 мл) и перемешивали при комнатной температуре в течение ночи. После этого, продукт реакции был подвергнут вакуумной перегонке для удаления растворителя, затем его разбавили водой и 3 раза экстрагировали этилацетатом. Экстракты объединили, 3 раза промыли насыщенным рассолом, концентрировали и разделили на колонке с силикагелем (элюент: петролейный эфир/этилацетат/аммоний), и перекристаллизовали с этилацетатом, н-гексаном с получением белого твердого продукта.
1H-ЯМР (400 MHz, CDCl3) δ ppm: 7.39 (1Н, d, J=1.96 Hz), 7.35 (1H, d, J=7.84 Hz), 7.20 (1H, d, J=8.4 Hz), 4.85 (1H, s), 4.53 (2H, d, J=6.2 Hz), 4.50 (1H, t, J=5.04 Hz, J=4.76 Hz), 3.71-3.47 (10H, brm), 3.37 (6H, s), 2.96 (3H, s), 2.41 (1H, m), 1.19 (3H, m); EI-MS (m/z): 511.3 [M+H]+.
Пример 2
1-{4-{4-[(2,4-дихлорбензил)амино]-6-[(2,2-диметоксиэтил)метиламино]пиримидин-2-ил}пиперазин-1-ил}-2-метилпропанон (Соединение 2)
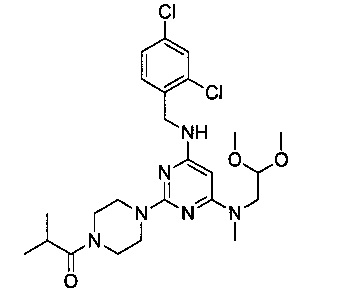
В качестве исходных материалов использовали промежуточный продукт 5 и изомасляную кислоту для прохождения стадий в соответствии с примером 1, чтобы получить белый твердый продукт.
1H-ЯМР (400 MHz, CDCl3) δ ppm: 7.38 (1Н, d, J=2.00 Hz), 7.35 (1H, d, J=8.12 Hz), 7.20 (1H, dd, J=2.24 Hz, J=2.36 Hz), 4.85 (1H, s), 4.53 (2H, d, J=6.44 Hz), 4.50 (1H, t, J=5.36 Hz, J=5.32 Hz), 3.75-3.50 (10H, brm), 3.37 (6H, s), 2.96 (3H, s), 2.84 (1H, m), 1.16 (6H, d, J=6.72 Hz); EI-MS (m/z): 525.3 [M+H]+.
Пример 3
1-{4-{4-[(2,4-дихлорбензил)амино]-6-[(2,2-диметоксиэтил)метиламино]пиримидин-2-ил}пиперазин-1-ил}-3-метилтиопропанон (Соединение 3)
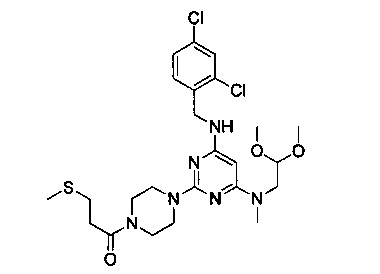
В качестве исходных материалов использовали промежуточный продукт 5 и 3-метилтиопропионовую кислоту для прохождения стадий в соответствии с примером 1, чтобы получить белый твердый продукт.
1H-ЯМР (400 MHz, CDCl3) δ ppm: 7.39 (1H, d, J=1.96 Hz), 7.35 (1H, d, J=8.12 Hz), 7.20 (1H, dd, J=1.96 Hz, J=2.24 Hz), 4.85 (1H, s), 4.53 (2H, d, J=6-16 Hz), 4.48 (1H, t), 3.76-3.46 (10H, brm), 3.37 (6H, s), 2.96 (3H, s), 2.86 (2H, t, J=7.00 Hz, J=7.84 Hz), 2.68 (2H, t, J=8.12 Hz, J=7.04 Hz), 2.16 (3H, s); EI-MS (m/z): 557.2 [M+H]+.
Пример 4
1-{4-{4-[бис(2-метоксиэтил)амино]-6-(2,4-дихлорбензиламино)-пиримидин-2-ил}пиперазин-1-ил}пропанон (Соединение 4)
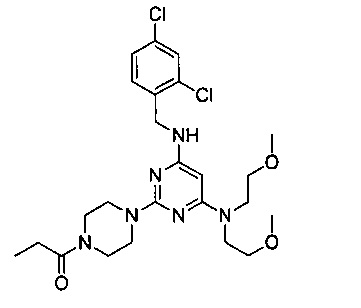
В качестве исходных материалов использовали промежуточный продукт 6 и пропионовую кислоту для прохождения стадий в соответствии с примером 1, чтобы получить белый твердый продукт.
1H-ЯМР (400 MHz, CDCl3) δ ppm: 7.38 (1Н, d, J=1.96 Hz), 7.34 (1H, d, J=8.4 Hz), 7.20 (1H, dd, J=1.96 Hz, J=2.24 Hz), 4.88 (1H, s), 4.52 (2H, d, J=6.44 Hz), 3.73-3.59 (10H, brm), 3.49-3.44 (6H, brm), 3.30 (6H, s), 2.39 (2H, q, J=7.56 Hz, J=7.32 Hz, J=7.56 Hz), 1.19 (3H, t, J=7.32 Hz, J=7.56 Hz); EI-MS (m/z): 525.0 [M+H]+.
Пример 5
1-{4-{4-[бис(2-метоксиэтил)амино]-6-(2,4-дихлорбензиламино)-пиримидин-2-ил}пиперазин-1-ил}-2-метилпропанон (Соединение 5)
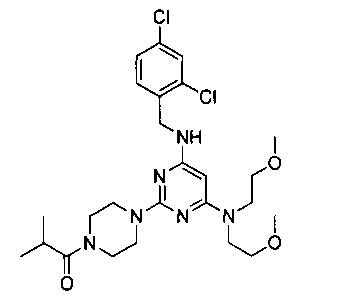
В качестве исходных материалов использовали промежуточный продукт 6 и изомасляную кислоту для прохождения стадий в соответствии с примером 1, чтобы получить белый твердый продукт.
1Н-ЯМР (400 MHz, CDCl3) δ ppm: 7.38 (1Н, d, J=1.96 Hz), 7.34 (1H, d, J=8.4 Hz), 7.20 (1H, dd, J=1.96 Hz, J=2.24 Hz), 4.88 (1H, s), 4.52 (2H, d, J=6.2 Hz), 3.72-3.46 (16H, brm), 3.30 (6H, s), 2.84 (1H, m), 1.16 (6H, d, J=6.76 Hz); EI-MS (m/z): 539.1 [M+H]+.
Пример 6
1-{4-{4-[бис(2-метоксиэтил)амино]-6-(2,4-дихлорбензиламино)-пиримидин-2-ил}пиперазин-1-ил}-3-метилтиопропанон (Соединение 6)
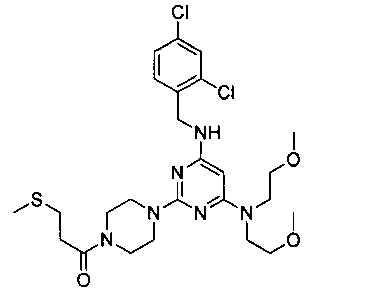
В качестве исходных материалов использовали промежуточный продукт 6 и 3-метилтиопропионовую кислоту для прохождения стадий в соответствии с примером 1, чтобы получить белый твердый продукт.
1H-ЯМР (400 MHz, CDCl3) δ ppm: 7.38 (1Н, d, J=1.96 Hz), 7.34 (1H, d, J=8.4 Hz), 7.20 (1H, dd, J=1.68 Hz, J=1.96 Hz), 4.88 (1H, s), 4.51 (2H, d, J=6.2 Hz), 3.74-3.47 (16H, brm), 3.30 (6H, s), 2.86 (2H, t, J=7.28 Hz, J=7.84 Hz), 2.67 (2H, t, J=7.84 Hz, J=6.96 Hz), 2.15 (3H, s); EI-MS (m/z): 571.2 [M+H]+.
Пример 7
{4-{4-[бис(2-метоксиэтил)амино]-6-(2,4-дихлорбензиламино)-пиримидин-2-ил}пиперазин-1-ил}циклогексилметанон (Соединение 7)
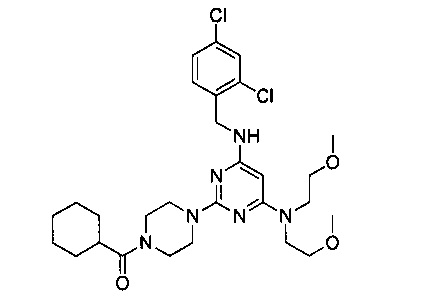
В качестве исходных материалов использовали промежуточный продукт 6 и циклогексил-карбновую кислоту для прохождения стадий в соответствии с примером 1, чтобы получить белый твердый продукт.
1H-ЯМР (400 MHz, CDCl3) δ ppm: 7.38 (1H, d, J=1.96 Hz), 7.34 (1H, d, J=8.12 Hz), 7.20 (1H, dd, J=1.96 Hz, J=1.96 Hz), 4.88 (1H, s), 4.83 (1H, s), 4.52 (2H, d, J=6.16 Hz), 3.75-3.46(16H, brm), 3.30 (6H, s), 2.50 (1H, m), 1, 82 (5H, m), 1.55 (2H, m), 1.27 (3H, m); EI-MS (m/z): 579.1 [M+H]+.
Пример 8
(R)-{4-{4-хлор-6-[(2,4-дихлорбензил)амино]пиримидин-2-ил}пиперазин-1-ил}(пиперидин-2-ил)метанон (Соединение 8)
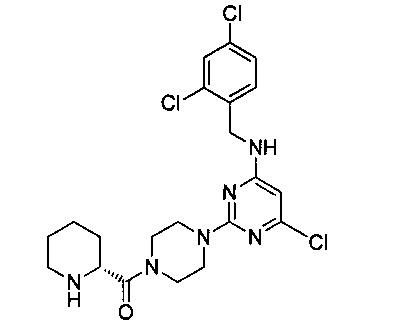
Промежуточный продукт 10 (300 мг, 0.514 ммоль) растворили в 4 мл дихлорметана, добавили 1 мл трифторуксусной кислоты и перемешивали при комнатной температуре в течение 2 часов. Продукт реакции концентрировали, к нему добавили 10 мл воды, затем 3 раза экстрагировали дихлорметаном. Экстракты объединили, 2 раза промыли насыщенным рассолом, концентрировали и очистили на колонке с силикагелем (элюент: этилацетат/метанол/аммоний), с получением белого твердого продукта.
1H-ЯМР (400 MHz, DMSO) δ ppm: 9.13 (1H, s), 8.63 (1H, s), 8.08 (1H, s), 7.64 (1H, d, J=1.52 Hz), 7.27 (2H, m), 5.97 (1H, s), 4.56 (2H, d, J=4.48 Hz), 4.39 (1H, d, J=9.24 Hz), 3.76-3.22 (9H, brm), 2.87 (1H, s), 1.99 (1H, m), 1.74 (4H, m), 1.48 (1H, m); EI-MS (m/z): 483.2 [M+H]+.
Пример 9
(R)-{4-{4-хлор-6-[(2,4-дихлорбензил)амино]пиримидин-2-ил}пиперазин-1-ил}(тиоморфолин-3-ил)метанон (Соединение 9)
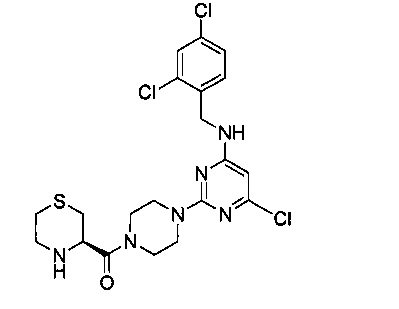
В качестве исходного материала использовали промежуточный продукт 11 для прохождения стадий в соответствии с примером 8, чтобы получить белый твердый продукт.
1Н-ЯМР (400 MHz, CDCl3) δ ppm: 7.42 (1Н, d, J=1.12 Hz), 7.27 (2H, m), 5.75 (1H, s), 5.19 (1H, s), 4.58 (2H, s), 3.95-3.44 (10H, brm), 3.12 (1H, m), 2.82 (2H, m), 2.43 (2H, m); EI-MS (m/z): 501.1 [M+H]+.
Пример 10
1-{4-{4-[(2,4-дихлорбензил)амино]пиридо[2,3-(1]пиримидин-2-ил}пиперазин-1-у1}пропанон (Соединение 10)
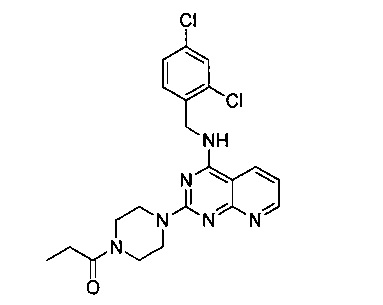
В качестве исходных материалов использовали промежуточный продукт 15 и пропионовую кислоту для прохождения стадий в соответствии с примером 1, чтобы получить белый твердый продукт.
1H-ЯМР (400 MHz, CDCl3) δ ppm: 8.76 (1Н, m), 8.07 (1H, d, J=7.28 Hz), 7.42 (1H, d, J=2.24 Hz), 7.35 (1H, d, J=8.4 Hz), 7.2 (1H, m), 7.05 (1H, m), 6.61 (1H, s), 4.86 (2H, d, J=5.92 Hz), 3.97 (4H, d, J=19.32 Hz), 3.66 (2H, m), 3.50 (2H, m), 2.43 (2H, q), 1.20 (3H, t); EI-MS (m/z): 444.9 [M+H]+.
Пример 11
1-{4-{4-[(2,4-дихлорбензил)амино]пиридо[2,3-d]пиримидин-2-ил}пиперазин-1-у1}-2-метилпропанон (Соединение 11)
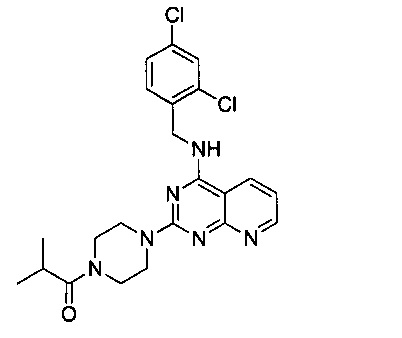
В качестве исходных материалов использовали промежуточный продукт 15 и изомасляную кислоту для прохождения стадий в соответствии с примером 1, чтобы получить белый твердый продукт.
1Н-ЯМР (400 MHz, CDCl3) δ ppm: 8.77 (1Н, m), 8.02 (1H, d, J=7.56 Hz), 7.42 (1H, d, J=1.96 Hz), 7.35 (1H, d, J=8.16 Hz), 7.21 (1H, m), 7.05 (1H, m), 6.49 (1H, s), 4.86 (2H, d, J=5.6 Hz), 3.98 (4H, d, J=21.56 Hz), 3.66 (2H, m), 3.54 (2H, m), 2.86 (1H, m), 1.17 (6H, d, J=6.72 Hz); EI-MS (m/z): 459.2 [M+H]+.
Пример 12
Циклогексил {4-{4-[(2,4-дихлорбензил)амино]пиридо[2,3-ил]пиримидин-2-ил}пиперазин-1-ил}метанон (Соединение 12)
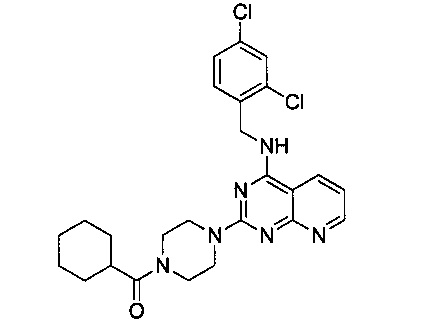
В качестве исходных материалов использовали промежуточный продукт 15 и циклогексил-карбоновую кислоту для прохождения стадий в соответствии с примером 1, чтобы получить белый твердый продукт.
1Н-ЯМР (400 MHz, CDCl3) δ ppm: 8.76 (1Н, m), 8.05 (1H, d, J=7.00 Hz), 7.41 (1H, d, J=1.96 Hz), 7.35 (1H, d, J=8.12 Hz), 7.21 (1H, m), 7.04 (1H, m), 6.56 (1H, s), 4.86 (2H, d, J=5.88 Hz), 3.96 (4H, d, J=20.76 Hz), 3.65 (2H, s), 3.52 (2H, s), 2.51 (1H, m), 1.83 (5H, m), 1.52 (2H, m), 1.28 (3H, m); EI-MS (m/z): 499.1 [M+H]+.
Пример 13
4-[(2,4-дихлорбензил)амино]-2-(4-пропилпиперазин-1-ил)пиридо[2,3-d]пиримидин (Соединение 13)
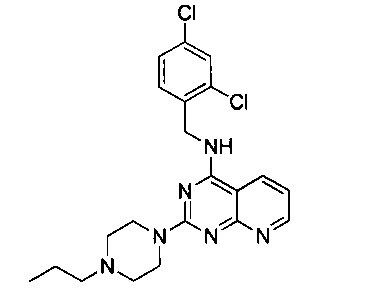
Промежуточный продукт 15 (400 мг, 1.03 ммоль), 1-бромпропан (139 мг, 1.13 ммоль) и DIEA (146 мг, 1.13 ммоль) растворили в NMP (10 мл) и перемешивали при комнатной температуре в течение ночи. После этого, продукт реакции разбавили в 5 раз этилацетатом, 6 раз промыли водой, 3 раза промыли насыщенным рассолом, концентрировали, разделили на колонке с силикагелем (элюент: этилацетат/метанол/аммоний) и перекристаллизовали с этилацетатом с получением белого твердого продукта.
1Н-ЯМР (400 MHz, CDCl3) δ ppm: 8.74 (1Н, m), 7.90 (1H, d, J=6.72 Hz), 7.41 (1H, d, J=2.24 Hz), 7.36 (1H, d, J=8.12 Hz), 7.21 (1H, m), 6.99 (1H, m), 6.18 (1H, s), 4.86 (2H, d, J=5.6 Hz), 3.97 (4H, s), 2.48 (4H, s), 2.36 (2H, t, J=7.56 Hz, J=7.84 Hz), 1.57 (2H, m), 0.95 (3H, t, J=7.28 Hz, J=7.56 Hz); EI-MS (m/z): 431.2 [M+H]+.
Пример 14
(R)-{4-{4-[(2,4-дихлорбензил)амино]пиридо[2,3-d]пиримидин-2-ил}пиперазин-1-ил}(тиоморфолин-3-ил)метанон (Соединение 14)
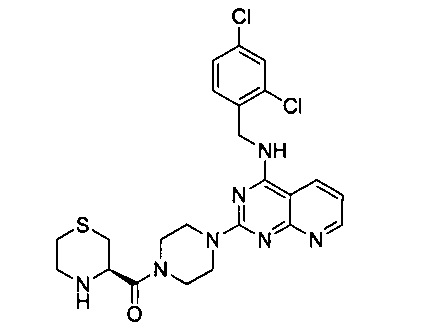
В качестве исходного материала использовали промежуточный продукт 16 для прохождения стадий в соответствии с примером 8, чтобы получить белый твердый продукт.
1H-ЯМР (400 MHz, CDCl3) δ ppm: 8.79 (1Н, m), 7.93 (1H, dd, J=1.96 Hz, J=1.68 Hz), 7.44 (1H, d, J=1.96 Hz), 7.34 (1H, d, J=8.4 Hz), 7.22 (1H, m), 7.05 (1H, m), 6.26 (1H, t), 4.86 (2H, d, J=5.88 Hz), 4.09-3.44 (10H, brm), 3.14 (1H, m), 2.82 (2H, m), 2.44 (2H, m); EI-MS (m/z): 518.3 [M+H]+.
Антагонистическая активность соединений настоящего изобретения в отношении CCR4 может быть проверена с помощью следующего метода.
Пример 15. Оценка CCR4-антагонистической активности соединений настоящего изобретения.
Способность соединений 1-14 настоящего изобретения ингибировать опосредованный MDC (Peprotech) хемотаксический ответ HEK293 клеток исследовали с использованием камеры Бойдена (Neuro Probe, Inc.).
1. Создание плазмиды, экспрессирующей рецептор:
Фрагменты кДНК, включая открытую рамку считывания человеческого рецептора хемокина, были получены следующим способом: был клонирован CCR4 рецептор из кДНК библиотеки K562 клеток с помощью цепной полимеразной реакции (ПЦР). Были созданы праймеры согласно последовательности Gen-Bank™ учетный номер: CCR4 (NM_005508.2). кДНК фрагменты открытой рамки считывания рецептора были соответственно введены в pcDI (вектор, преобразованный в камере: эукариотический вектор экспрессии, полученный путем замещения Bg1II KpnΙ фрагмента pcDNA3 (Invitrogen Corporation) плазмиды фрагментом Bg1II KpnI плазмиды pCI (Promega Corporation)) вектор экспрессии, для того, чтобы они эффективно экспрессировались в HEK293 клетках. Секвенирование ДНК показало, что кодирующая последовательность была правильной и соответствовала последовательности Gen-Bank™.
2. Культура клеток:
Клетки HEK293 культивировали в среде RPMI 1640 (Life Technologies, Inc.), содержащей 10% инактивированной нагреванием эмбриональной бычьей сыворотки, 100 U/мл пенициллина, 100 мкг/мл стрептомицина. Каждые 4×106 HEK293 клеток/400 мкл транзиентно трансфицировали с помощью электропорации 15 мкг плазмид, экспрессирующих хемокиновый рецептор, при условиях 120 V в течение 20 мс, для этого использовали генератор электрических импульсов (Electro Square porator ЕСМ 830, ВТХ, San Diego, Калифорния). Анализ хемотаксиса проводили через 36-48 ч.
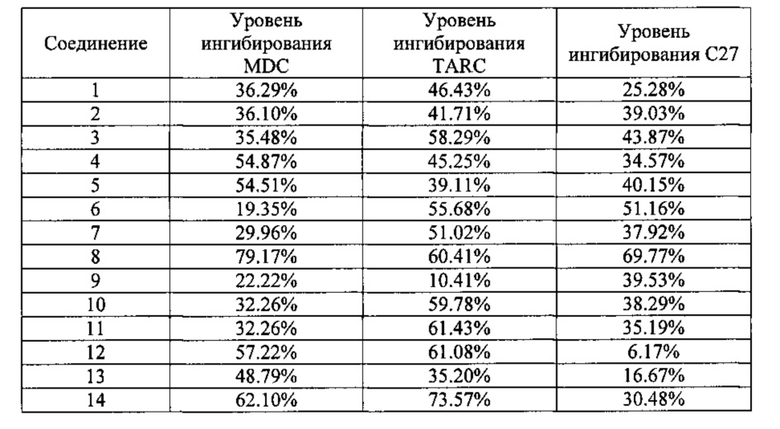
3. Ингибирующее действие на опосредованный MDC/TARC/C27 хемотаксический ответ HEK293 клеток
Хемокины MDC/TARC/C27 разбавили до 10 нг/мл средой RPMI1640 (Life Technologies, Inc.), 0,1% BSA (Sigma) и добавили в нижние лунки в 48-луночную камеру для хемотаксиса, 27,5 мкл в каждую лунку. Эукариотические экспрессирующие плазмиды pCDI-CCR4 с помощью электричества переместили в HEK293 клетки и культивировали обычным порядком в течение 36 часов. Клетки обрабатывали, ресуспендировали RPMI1640, 10% FBS (Life Technologies, Inc.) и проводили ротационную инкубацию при 37°C в течение 6,5 часов. Клетки дважды промыли RPMI1640 и суспендировали в RPMI1640, 0.1% BSA, при окончательной концентрации 1×106/мл. Провели ротационную инкубацию клеток и соединения-кандидата, растворенного в DMSO (Sigma), при комнатной температуре в течение получаса, окончательная концентрация соединения-кандидата составляла 1 мкМ, а окончательная концентрация DMSO составляла 0.1%, а затем добавили к верхним лункам камеры для хемотаксиса, 55 мкл в каждую лунку. Два слоя были разделены полиуглеродной мембраной с размером пор 10 мкМ (Neuro Probe, Inc.). Реакцию хемотаксиса проводили в атмосфере 5% СО2 при 37°C в течение 5 часов. В эксперименте было три группы контроля, при этом первая группа была положительным контролем, в котором трансфицированные клетки были непосредственно добавлены в верхние лунки без инкубации с соединением-кандидатом, тогда как в нижние лунки были добавлены хемокины MDC/TARC/C27. Вторая группа была отрицательным контролем, в котором трансфицированные клетки были непосредственно добавлены в верхние лунки без инкубации с соединением-кандидатом, тогда как в нижние лунки были добавлены RPMI1640, 0.1% BSA. Третья группа была контролем соединения-кандидата с растворителем DMSO, в котором трансфицированные клетки и DMSO инкубировали при комнатной температуре, окончательная концентрация DMSO составляла 0.1%, тогда как в нижние лунки были добавлены хемокины MDC/TARC/C27. После завершения реакции хемотаксиса мембрану удаляли, фиксировали и окрашивали, для подсчета клеток при большом увеличении 400Х случайным образом были выбраны пять зон, которые затем суммировали. Отношение суммы количества клеток в пяти зонах при большом увеличении в каждой экспериментальной группе к сумме количества клеток в пяти зонах при большом увеличении в отрицательной контрольной группе считается хемотаксическим индексом (CI). Уровень ингибирования хемотаксиса вычисляли, как указано далее:
100% × (1 - хемотаксический индекс клеток, инкубированных с соединением: хемотаксический индекс клеток, инкубированных с DMSO)
Соединения 1-14 настоящего изобретения в состоянии преимущественно ингибировать опосредованный MDC/TARC/C27 хемотаксический ответ НЕК293 клеток.
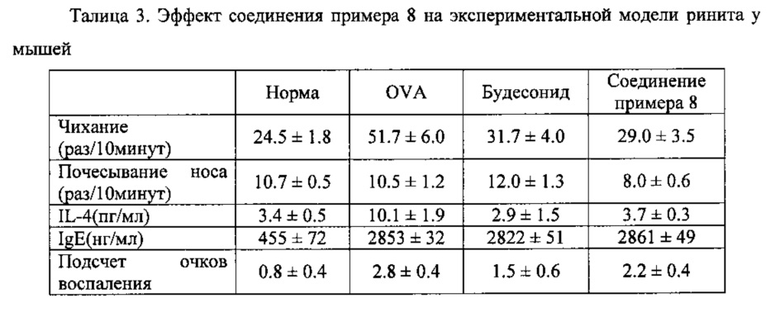
Пример 16. Оценка активности соединений настоящего изобретения на экспериментальной мышиной модели ринита
Экспериментальная модель ринита у мышей Метод:
Самок мышей BALB/c (6-8 недель) сенсибилизировали куриным овальбумином (OVA, Sigma-Aldrich, St Louis, МО, США). Соединение настоящего изобретения (например, соединение Примера 8) наносили на слизистую носа в дозе 1 мкг/кг. В качестве положительного контроля использовали глюкокортикоидное гормональное лекарственное средство будесонид, клинический препарат первой линии, в дозе 1,28 мг/кг. При этом были обеспечены нормальная группа, в которой мышей не сенсибилизировали и не лечили, и группа OVA в которой мышей сенсибилизировали и не лечили. Определяли следующие пять показателей: количество чиханий, количество случаев почесывания носа в течение 10 минут после воздействия; уровень IL-4 в жидкости бронхоальвеолярного лаважа; уровень IgE в сыворотке; подсчет очков в отношении воспаления легочной ткани. Подсчет очков в отношении воспаления легочной ткани проводили в соответствии с односторонним слепым методом: воспаление подразделяли на четыре уровня от легкого до тяжелого, используя счет от 0 до 3:0 - нет обнаружимого воспаления; 1 - видны редкие воспалительные клетки; 2 - бронх или кровеносный сосуд окружен эозинофилами, но меньше, чем в пять клеточных слоев; 3 - бронх или кровеносный сосуд окружен большим количеством эозинофилов в 5 или более клеточных слоев.
Результаты:
Результаты представлены в таблице ниже. На примере количества чиханий, экспериментальная группа с соединением примера 8 и экспериментальная группа с будесонидом показали количество, которое было значительно ниже, чем в OVA-сенсибилизированной группе, и немного выше, чем в нормальной контрольной группе, показывая, что соединение примера 8 и будесонид могут значительно уменьшать количество чиханий у мыши, причем их эффекты были почти одинаковыми. На примере количества случаев почесывания носа, экспериментальная группа с соединением примера 8 показала количество меньше, чем количество в других группах, что означает, что соединение настоящего изобретения может уменьшить количество случаев почесывания носа на модели ринита у мышей. В отношении уровня IL-4 в жидкости бронхоальвеолярного лаважа, экспериментальная группа с соединением примера 8 и экспериментальная группа с будесонидом показали уровень, который был значительно ниже, чем уровень в OVA-сенсибилизированной группе, и близкий к нормальной контрольной группе, показывая, что соединение настоящего изобретения и будесонид могут значительно уменьшать уровень IL-4 в легких мышей, причем их эффекты были почти одинаковыми. На примере уровня IgE в сыворотке, OVA-сенсибилизированная группа, экспериментальная группа с будесонидом и экспериментальная группа с соединением примера показали почти одинаковые уровни, и все они были выше, чем уровни в нормальной контрольной группе, показывая, что ни соединение настоящего изобретения, ни будесонид не оказывали влияния на уменьшение уровня IgE у мышей с моделью ринита. На примере подсчета очков по поводу воспаления легочной ткани было обнаружено, что экспериментальная группа с соединением примера 8 и экспериментальная группа с будесонидом показали счет очков ниже, чем у OVA-сенсибилизированной группы, и выше, чем в нормальной контрольной группе, что означает, что соединение настоящего изобретения и будесонид могут уменьшать легочное воспаление у мышей с ринитом, и их эффекты были близки друг другу.
Таким образом, на модели ринита у мышей показано, что соединение настоящего изобретения в низкой дозе (1 мкг/кг) может обеспечивать терапевтический эффект, который может быть достигнут при применении одного будесонида только в высокой дозе (1,28 мг/кг).
| название | год | авторы | номер документа |
|---|---|---|---|
| БЕНЗОПРОИЗВОДНЫЕ С ШЕСТИЧЛЕННЫМ КОЛЬЦОМ В КАЧЕСТВЕ ИНГИБИТОРА DPP-4 И ИХ ПРИМЕНЕНИЕ | 2015 |
|
RU2702644C2 |
| КОНДЕНСИРОВАННЫЕ ПЯТИЧЛЕННЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ ПИРИДИНОВЫЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПРОИЗВОДСТВА И ПРИМЕНЕНИЕ | 2014 |
|
RU2668074C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2015 |
|
RU2667498C2 |
| СОЕДИНЕНИЯ ДЛЯ ЛЕЧЕНИЯ РАКА | 2011 |
|
RU2609018C2 |
| СОЕДИНЕНИЯ ТИЕНИЛ[3, 2-d]ПИРИМИДИН-4-ОН, СПОСОБ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ И ИХ ПРИМЕНЕНИЕ | 2012 |
|
RU2624021C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ ТРИАЗОЛОПИРАЗИНА И ИХ ПРИМЕНЕНИЕ | 2013 |
|
RU2643361C2 |
| СОЕДИНЕНИЯ БЕНЗАЗЕПИНА ДИКАРБОКСАМИДА | 2016 |
|
RU2712248C2 |
| ПРОИЗВОДНЫЕ ПИРИМИДИНА ПРОТИВ ВИРУСА ГРИППА | 2017 |
|
RU2727772C1 |
| СОЕДИНЕНИЯ ДЛЯ ЛЕЧЕНИЯ РАКА | 2011 |
|
RU2762111C1 |
| ДИАРИЛГИДАНТОИНЫ | 2011 |
|
RU2638833C2 |
Изобретение относится к производным пиперазинил пиримидина формулы I, обладающим антагонизмом к CCR4, способу их получения, фармацевтической композиции на их основе, их применению для изготовления лекарственного средства, а также к способу лечения. В общей формуле I любые два из А, В и D являются N, а оставшийся является СН; Z выбирают из группы, состоящей из -СН2- и -С(О)-; X представляет собой галоген или N, при условии, что когда X является галогеном, R1 и R2 в формуле I отсутствуют; R1 и R2 каждый независимо выбирают из группы, состоящей из линейного или разветвленного алкила, имеющего от 1 до 6 атомов углерода, С1-С6 линейного или разветвленного гетероалкила, содержащего от 1 до 2 гетероатомов, независимо выбранных из группы, состоящей из О; R3 и R4 каждый независимо выбирают из группы, состоящей из Н, фенила и нафтила; при этом указанный фенил или нафтил необязательно и независимо является моно- или дизамещенным заместителем, выбранным из группы, состоящей из галогена; R5 выбирают из группы, состоящей из линейного или разветвленного алкила, имеющего от 1 до 6 атомов углерода, С1-С6 линейного или разветвленного гетероалкила, содержащего от 1 до 2 гетероатомов, независимо выбранных из группы, состоящей из S, циклоалкильной группы, содержащей от 4 до 8 атомов углерода, 5-8-членного гетероциклоалкила, содержащего от 1 до 2 гетероатомов, независимо выбранных из группы, состоящей из N и S; m и n каждый независимо представляют собой 0 или 1. 5 н. и 2 з.п. ф-лы, 2 табл., 16 пр.
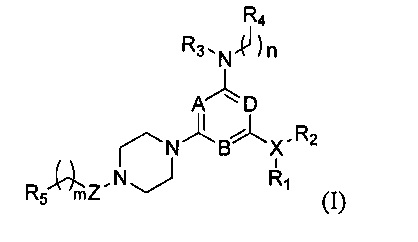
1. Соединение формулы I
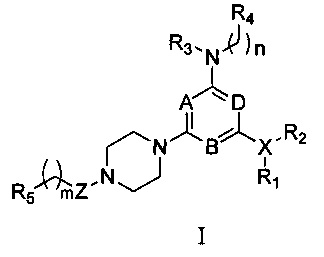
в которой
любые два из А, В и D являются N, а оставшийся является СН;
Z выбирают из группы, состоящей из -СН2- и -С(О)-;
X представляет собой галоген или N, при условии, что когда X является галогеном, R1 и R2 в формуле I отсутствуют;
R1 и R2 каждый независимо выбирают из группы, состоящей из линейного или разветвленного алкила, имеющего от 1 до 6 атомов углерода, С1-С6 линейного или разветвленного гетероалкила, содержащего от 1 до 2 гетероатомов, независимо выбранных из группы, состоящей из О;
R3 и R4 каждый независимо выбирают из группы, состоящей из Н, фенила и нафтила; при этом указанный фенил или нафтил необязательно и независимо является моно- или дизамещенным заместителем, выбранным из группы, состоящей из галогена;
R5 выбирают из группы, состоящей из линейного или разветвленного алкила, имеющего от 1 до 6 атомов углерода, С1-С6 линейного или разветвленного гетероалкила, содержащего от 1 до 2 гетероатомов, независимо выбранных из группы, состоящей из S, циклоалкильной группы, содержащей от 4 до 8 атомов углерода, 5-8-членного гетероциклоалкила, содержащего от 1 до 2 гетероатомов, независимо выбранных из группы, состоящей из N и S;
m и n каждый независимо представляют собой 0 или 1;
или его фармацевтически приемлемая соль.
2. Соединение формулы I или его фармацевтически приемлемая соль по п. 1, при этом соединение выбирают из группы, состоящей из:
1-{4-{4-[(2,4-дихлорбензил)амино]-6-[(2,2-диметоксиэтил)метиламино]пиримидин-2-ил}пиперазин-1-ил}пропанона;
1-{4-{4-[(2,4-дихлорбензил)амино]-6-[(2,2-диметоксиэтил)метиламино]пиримидин-2-ил}пиперазин-1-ил}-2-метилпропанона;
1-{4-{4-[(2,4-дихлорбензил)амино]-6-[(2,2-диметоксиэтил)метиламино]пиримидин-2-ил}пиперазин-1-ил}-3-метилтиопропанона;
1-{4-{4-[бис(2-метоксиэтил)амино]-6-(2,4-дихлорбензиламино)-пиримидин-2-ил}пиперазин-1-ил}пропанона;
1-{4-{4-[бис(2-метоксиэтил)амино]-6-(2,4-дихлорбензиламино)-пиримидин-2-ил}пиперазин-1-ил}-2-метилпропанона;
1-{4-{4-[бис(2-метоксиэтил)амино]-6-(2,4-дихлорбензиламино)-пиримидин-2-ил}пиперазин-1-ил}-3-метилтиопропанона;
{4-{4-[бис(2-метоксиэтил)амино]-6-(2,4-дихлорбензиламино)-пиримидин-2-ил}пиперазин-1-ил}циклогексилметанона;
(R)-{4-{4-хлор-6-[(2,4-дихлорбензил)амино]пиримидин-2-ил}пиперазин-1-ил}(пиперидин-2-ил)метанона; и
(R)-{4-{4-хлор-6-[(2,4-дихлорбензил)амино]пиримидин-2-ил}пиперазин-1-ил}(тиоморфолин-3-ил)метанона.
3. Способ получения соединения формулы I или его фармацевтически приемлемой соли по любому из пп. 1, 2, включающий стадии:
1) реагирования 2,4,6-трихлорпиримидина с монозащищенным пиперазином в присутствии связывающего кислоту вещества с получением соединения формулы 1
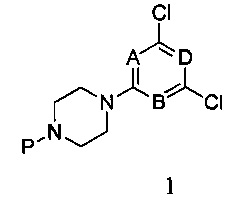
в которой А, В и D являются, как определено выше в формуле I, а Р представляет собой азот-защитную группу;
2) реагирования соединения формулы 1 с R3- и -(СН2)n-R4-замещенным амином в присутствии связывающего кислоту вещества с получением соединения формулы 2
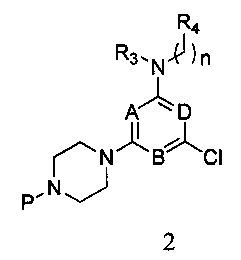
в которой А, В, D, R3, R4 и n являются, как определено выше в формуле I, а Р представляет собой азот-защитную группу;
3) реагирования соединения формулы 2 с R1- и R2-замещенным амином в присутствии связывающего кислоту вещества с получением соединения формулы 3
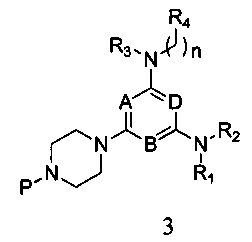
в которой А, В, D, R1, R2, R3, R4 и n являются, как определено выше в формуле I, а Р представляет собой азот-защитную группу;
4) удаления защитной группы Р из соединения формулы 3 и затем вступления его в реакцию с R5-замещенной карбоновой кислотой, ацилгалоидом, сульфонилхлоридом или галоидзамещенным углеводородом с получением соединения формулы 4
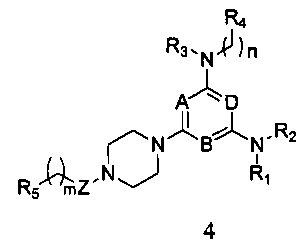
в которой А, В, D, R1, R2, R3, R4, R5, n и m являются, как определено выше в формуле I; или
5) когда X является галогеном, соединение формулы I получают согласно следующему пути реакции: удаление защитной группы Р из соединения формулы 2 и реагирование его с R5-замещенной карбоновой кислотой, ацилгалоидом, сульфонилхлоридом или галоидзамещенным углеводородом с получением соединения формулы 5
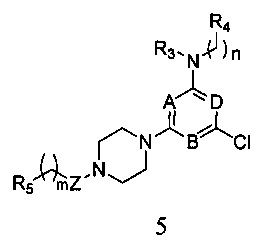
в которой А, В, D, Z, R3, R4, R5, n и m являются, как определено выше в формуле I.
4. Фармацевтическая композиция, обладающая активностью антагониста CCR4 рецептора, содержащая соединение формулы I или его фармацевтически приемлемую соль по любому из пп. 1, 2 и по меньшей мере один фармацевтически приемлемый носитель, разбавитель или эксципиент.
5. Применение соединения формулы I или его фармацевтически приемлемой соли по любому из пп. 1, 2 для изготовления лекарственного средства для лечения или предотвращения болезни или нарушения, связанного с CCR4.
6. Способ лечения и предотвращения болезни или нарушения, связанного с CCR4, где данный способ включает введение терапевтически или профилактически эффективного количества соединения формулы I или его фармацевтически приемлемой соли по любому из пп. 1, 2 нуждающемуся в этом субъекту.
7. Соединение формулы I или его фармацевтически приемлемая соль по любому из пп. 1, 2 для лечения или предотвращения болезни или нарушения, связанного с CCR4.
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| EP 1970373 A1, 17.09.2008 | |||
| EA 200800047 A1, 29.08.2008. | |||

