Настоящее изобретение относится к новому классу ингибиторов киназ, включая их фармацевтически приемлемые соли, пролекарства и метаболиты, которые полезны для модулирования активности протеинкиназ для модулирования клеточных активностей, таких как передача сигналов, пролиферация и секреция цитокинов. Более конкретно в изобретении предложены соединения, которые ингибируют, регулируют и/или модулируют активность киназ, в частности активность mTOR, и метаболические пути передачи сигналов, относящиеся к вышеупомянутым клеточным активностям. Кроме того, настоящее изобретение относится к фармацевтическим композициям, содержащим указанные соединения, например, для лечения таких заболеваний, как иммунологические, воспалительные, аутоиммунные, аллергические расстройства, или пролиферативных заболеваний, таких как рак.
Киназы катализируют фосфорилирование белков, липидов, сахаров, нуклеозидов и других клеточных метаболитов и играют ключевые роли во всех аспектах физиологии эукариотических клеток. В частности, протеинкиназы и липидкиназы принимают участие в событиях передачи сигналов, которые контролируют активацию, рост, дифференциацию и выживание клеток в ответ на внеклеточные медиаторы или стимулы, такие как факторы роста, цитокины или хемокины. В общем протеинкиназы классифицируют на две группы, а именно те, которые преимущественно фосфорилируют остатки тирозина, и те, которые преимущественно фосфорилируют остатки серина и/или треонина.
Неадекватно высокая активность протеинкиназ вовлечена в многочисленные заболевания, включая рак, метаболические заболевания и аутоиммунные/воспалительные расстройства. Это может быть вызвано либо прямо, либо косвенно нарушением механизмов контроля из-за мутации, сверхэкспрессии или неадекватной активации фермента. Во всех этих случаях ожидается, что селективное ингибирование киназы будет иметь благоприятный эффект.
mTOR ("мишень рапамицина у млекопитающих", также известная как FRAP или RAFT1) стала новым средоточием для попыток создания новых лекарственных средств (Tsang et al., 2007, Drug Discovery Today 12, 112-124). Было установлено, что белок mTOR является лекарственной мишенью для иммуносуппрессивного эффекта рапамицина, лекарственного средства, которое используется для предотвращения отторжения трансплантатов. Рапамицин работает через механизм «приобретения функции» (gain-of-function) посредством связывания с внутриклеточным белком "FK-506-связывающий белок 12 кДа" (FKBP12) с образованием комплекса лекарственное средство-рецептор, который затем связывается с mTOR и ингибирует ее. Таким образом, рапамицин индуцирует образование трехкомпонентного комплекса, состоящего из рапамицина и двух белков, FKBP12 и mTOR.
Белок mTOR представляет собой крупную киназу 289 кДа, которая присутствует у всех исследованных к настоящему времени эукариотических организмов (Schmelzle and Hall, 2000, Cell 103, 253-262). Последовательность карбокси-концевого домена "фосфатидилинозит-3-киназа (PI3K)-родственной киназы" (PIKK) является высококонсервативной между видами и демонстрирует серии- и треонин-киназную активность и отсутствие обнаружимой липидкиназной активности. Интактный домен PIKK необходим для всех известных функций mTOR. FKBP12-рапамицин-связывающий (FRB) домен локализован вблизи PIKK домена и образует гидрофобный карман, который связывается с рапамицином, связанным с FKBP12. FRB домен, по-видимому, не ингибирует ферментативную активность киназного домена непосредственно. Одним объяснением является то, что FKBP12-рапамицин предотвращает взаимодействие mTOR с ее субстратами благодаря стерическим затруднениям. N-конец mTOR состоит из приблизительно 20 тандемных повторов из 37-43 аминокислот, именуемых HEAT повторами. HEAT повторы взаимодействуют с белковыми партнерами связывания, такими как Raptor.
mTOR может образовывать по меньшей мере два различных белковых комплекса, mTORC1 и mTORC2. В mTORC1 белковом комплексе mTOR взаимодействует с белками Raptor и mLST8/GβL и регулирует клеточный рост посредством фосфорилирования эффекторов, таких как p70S6K и 4Е-ВР1, способствуя трансляции мРНК (матричная РНК) и белковому синтезу. Комплекс mTORC1 ответственен за считывание пищевых сигналов (например доступность аминокислот) в сочетании с передачей сигналов инсулина. Активность mTOR в mTORC1 может ингибироваться рапамицином.
Второй белковый комплекс, mTORC2, состоит из белков mTOR, Rictor, mLST8/GβL и Sin1 и вовлечен с организацию актина. mTORC2 изначально описан как нечувствительный к рапамицину. Последняя публикация продемонстрировала, что рапамицин воздействует на функционирование mTORC2 после прологированной обработки через непрямой механизм посредством нарушения сборки mTORC2 белкового комплекса (Sarbassov et al., 2006, Molecular Cell 22, 159-168).
Биологическая функция mTOR заключается в том, что она является центральным регулятором различных внеклеточных и внутриклеточных сигналов, включая факторы роста, питательные вещества, энергию и стресс. Индуцированная фактором роста и гормоном (например инсулином) активация mTOR опосредуется PI3 киназами, Akt и белковым комплексом туберозного склероза (TSC). Например, mTOR действует в качестве центрального регулятора клеточной пролиферации, ангиогенеза и клеточного метаболизма (Tsang et al., 2007, Drug Discovery Today 12, 112-124). В дополнение к своим иммуносуппрессивным эффектам рапамицин (Sirolimus) является мощным ингибитором пролиферации гладкомышечных клеток сосудов и одобрен FDA (Управление по санитарному надзору за качеством пищевых продуктов и медикаментов) в качестве лекарственного средства против рестеноза, используемого в коронарных стентах. Кроме того, установлено, что рапамицин проявляет противоопухолевую активность в некоторых моделях in vitro и животных моделях (Faivre et al., 2006. Nat. Rev. Drug. Discov. 5(8):671-688).
Из-за терапевтического потенциала рапамицина некоторые фармацевтические компании начали создание аналогов рапамицина для улучшения фармакокинетических свойств этой молекулы (Tsang et al., 2007, Drug Discovery Today 12, 112-124). Например, CCI-779 (темсиролимус, temsirolimus) представляет собой более водорастворимое сложно-эфирное производное рапамицина для внутривенных и пероральных композиций. CCI-779 обладает противоопухолевой активностью как сам по себе, так и в комбинации с цитотоксическими агентами в клеточных линиях. RAD-001 (эверолимус, everolimus) представляет собой производное в виде гидроксиэтилового эфира рапамицина, которое было создано для перорального введения. АР23573 (деферолимус, deferolimus) разработан для перорального или внутривенного введения.
В общем, производные рапамицина действуют через тот же самый молекулярный механизм, индукцию трехкомпонентного комплекса рапамицин-FKBP12-mTOR. Вероятно, что эта функция mTOR может быть в той же степени или даже более эффективно ингибироваться ингибиторами киназной функции. Например, это могло бы быть осуществлено посредстовм обнаружения соединений, которые взаимодействуют с АТР-связывающим карманом mTOR киназного домена. Например, Torin1 является мощным и селективным АТР-конкурентным ингибитором mTOR, который непосредственно связывается с обоими mTOR комплексами и нарушает клеточный рост и пролиферацию более эффективно, чем рапамицин (Thoreen et al., 2009. J Biol. Chem. 284(12):8023-32; Feldman et al., 2009. PLOSBiology 7(2):e38).
Заболевания и расстройства, ассоциированные с mTOR, также описаны, например, в WO-A 2008/116129, WO-A 2008/115974, WO-A 2008/023159, WO-A 2009/007748, WO-A 2009/007749, WO-A 2009/007750, WO-A 2009/007751, WO-A 2011/011716.
В литературе имеются сообщения о некоторых ингибиторах mTOR, которые могут быть полезны в медицинской сфере, например в качестве противораковых агентов (Faivre et al., 2006. Nat. Rev. Drug. Discov. 5(8):671-688). В WO-A 2008/116129 описаны имидазолопиримидиновые аналоги в качестве смешанных ингибиторов mTOR и PI3K киназ. Пиразолопиримидиновые аналоги описаны в качестве смешанных ингибиторов mTOR и PI3K киназ в WO-A 2008/115974. Дополнительные пиримидиновые производные в качестве соединений, активных в отношении mTOR киназы и/или фермента PI3K, описаны в WO-A 2008/023159, WO-A 2009/007748, WO-A 2009/007749, WO-A 2009/007750, WO-A 2009/007751, WO-A 2010/103094, WO-A 2010/120994 и WO-А 2010/120998.
Триазиновые соединения в качестве ингибиторов PI3K киназы и MTOR описаны в WO 2009/143313 А1, WO 2009/143317 А1 и WO 2010/096619 А1.
Кроме того, ингибиторы mTOR описаны в международных заявках на патент с номерами РСТ/ЕР2012/055953 и РСТ/ЕР2012/068590, а также в WO2011/107585 А1.
Незамещенные конформационно-ограниченные циклические сульфоны были раскрыты как мощные и селективные ингибиторы киназы mTOR в Bioorganic and Medicinal Chemistry Letters, 2012, 22 (15), 5114-5117.
Ожидается, что селективный ингибитор mTOR, который ингибирует mTOR с большей эффективностью, чем другие киназы, может обладать благоприятными терапевтическими свойствами, поскольку ингибирование других киназ может вызывать нежелательные побочные эффекты (Richard et al., 2011. Current Opinion Drug Discovery and Development 13(4):428-440). Может быть важной особенно селективность в отношении членов семейства фосфатидилинозит-3-киназ (PI3K) (например ΡΙ3Κα, ΡΙ3Κβ, ΡΙ3Κγ и ΡΙ3Κδ) и PI3K-родственных киназ (например, DMA-PK, ATM и ATR).
Даже несмотря на то, что ингибиторы mTOR известны из уровня техники, существует потребность в предложении дополнительных ингибиторов mTOR, обладающих, по меньшей мере частично, более эффективными фармацевтически релевантными свойствами, такими как активность, селективность и ADME свойства.
Соответственно, в настоящем изобретении предложены соединения формулы (I)
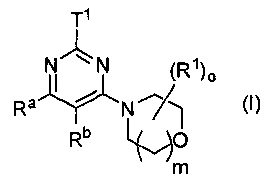
или их фармацевтически приемлемая соль, где
m равно 1 или 2;
о равно 1, 2, 3 или 4;
каждый R1 независимо выбран из группы, состоящей из Н; галогена; CN; C(O)OR2; OR2a; оксо (=O); C(O)R2; C(O)N(R2R2a); S(O)2N(R2R2a); S(O)N(R2R2a); S(O)2R2; S(O)R2; N(R2)S(O)2N(R2aR2b); N(R2)S(O)N(R2aR2b); SR2; N(R2R2a); NO2; OC(O)R2; N(R2)C(O)R2a; N(R2)S(O)2R2a; N(R2)S(O)R2a; N(R2)C(O)N(R2aR2b); N(R2)C(O)OR2a; OC(O)N(R2R2a) и С1-6алкила, где С1-6алкил возможно замещен одним или более R3, которые являются одинаковыми или разными;
возможно два R1 объединены с образованием, вместе с кольцом, к которому они присоединены, 8-11-членного гетеробицикла;
R2, R2a, R2b независимо выбраны из группы, состоящей из Н; C1-6алкила, где C1-6алкил возможно замещен одним или более атомами галогена, которые являются одинаковыми или разными;
R3 представляет собой галоген; CN; C(O)OR4; OR4; C(O)R4; C(O)N(R4R4a); S(O)2N(R4R4a); S(O)N(R4R4a); S(O)2R4; S(O)R4; N(R4)S(O)2N(R4aR4b); N(R4)S(O)N(R4aR4b); SR4; N(R4R4a); NO2; OC(O)R4; N(R4)C(O)R4a; N(R4)S(O)2R4a; N(R4)S(O)R4a; N(R4)C(O)N(R4aR4b); N(R4)C(O)OR4a или OC(O)N(R4R4a);
R4, R4a, R4b независимо выбраны из группы, состоящей из H и С1-6алкила, где C1-6алкил возможно замещен одним или более атомами галогена, которые являются одинаковыми или разными;
Т1 представляет собой фенил или 5-6-членный ароматический гетероцикл, где Т1 замещен группой N(R5a)C(O)N(R5bR5) или N(R5a)C(O)OR5 и возможно дополнительно замещен одним или более R6, которые являются одинаковыми или разными;
R6 представляет собой галоген; CN; C(O)OR7; OR7; C(O)R7; C(O)N(R7R7a); S(O)2N(R7R7a); S(O)N(R7R7a); S(O)2R7; S(O)R7; N(R7)S(O)2N(R7aR7b); N(R7)S(O)N(R7aR7b); SR7; N(R7R7a); NO2; OC(O)R7; N(R7)C(O)R7a; N(R7)S(O)2R7a; N(R7)S(O)R7a; N(R7)C(O)N(R7aR7b); N(R7)C(O)OR7a; OC(O)N(R7R7a) или С1-6алкил, где C1-6алкил возможно замещен одним или более атомами галогена, которые являются одинаковыми или разными;
R5a, R5b, R7, R7a, R7b независимо выбраны из группы, состоящей из Н; С1-6алкила, где C1-6алкил возможно замещен одним или более атомами галогена, которые являются одинаковыми или разными;
R5 представляет собой H; Т2 и С1-6алкил, где С1-6алкил возможно замещен одним или более R8, которые являются одинаковыми или разными;
R8 представляет собой галоген; CN; C(O)OR9; OR9; C(O)R9; C(O)N(R9R9a); S(O)2N(R9R9a); S(O)N(R9R9a); S(O)2R9; S(O)R9; N(R9)S(O)2N(R9aR9b); N(R9)S(O)N(R9aR9b); SR9; N(R9R9a); NO2; OC(O)R9; N(R9)C(O)R9a; N(R9)S(O)2R9a; N(R9)S(O)R9a; N(R9)C(O)N(R9aR9b); N(R9)C(O)OR9a; OC(O)N(R9R9a) или Τ2;
R9, R9a, R9b независимо выбраны из группы, состоящей из H и С1-6алкила, где C1-6алкил возможно замещен одним или более атомами галогена, которые являются одинаковыми или разными;
возможно R5, R5b объединены с образованием, вместе с атомом азота, к которому они присоединены, содержащего по меньшей мере атом азота в качестве кольцевого гетероатома 4-7-членного гетероциклильного кольца или 8-11-членного гетеробициклильного кольца, где эти 4-7-членное гетероциклильное кольцо и 8-11-членное гетеробициклильное кольца возможно замещены одним или более R10, которые являются одинаковыми или разными;
Т2 представляет собой С3-7циклоалкил; 4-7-членный гетероциклил; 4-7-членный гетероарил; 8-11-членный гетеробициклил; фенил; нафтил; инденил или инданил, где Т2 возможно замещен одним или более R10, которые являются одинаковыми или разными;
R10 представляет собой галоген; CN; C(O)OR11; OR11; оксо (=O), где кольцо является по меньшей мере частично насыщенным; C(O)R11; C(O)N(R11R11a); S(O)2N(R11R11a); S(O)N(R11R11a); S(O)2R11; S(O)R11; N(R11)S(O)2N(R11aR11b); N(R11)S(O)N(R11aR11b); SR11; N(R11R11a); NO2; OC(O)R11; N(R11)C(O)R11a; N(R11)S(O)2R11a; N(R11)S(O)R11a; N(R11)C(O)N(R11aR11b); N(R11)C(O)OR11a; OC(O)N(R11R11a) или С1-6алкил, где С1-6алкил возможно замещен одним или более R12, которые являются одинаковыми или разными;
R11, R11a, R11b независимо выбраны из группы, состоящей из Н; C1-6алкила, где C1-6алкил возможно замещен одним или более атомами галогена, которые являются одинаковыми или разными;
R12 представляет собой галоген; CN; C(O)OR13; OR13; C(O)R13; C(O)N(R13R13a); S(O)2N(R13R13a); S(O)N(R13R13a); S(O)2R13; S(O)R13; N(R13)S(O)2N(R13aR13b); N(R13)S(O)N(R13aR13b); SR13; N(R13R13a); NO2; OC(O)R13; N(R13)C(O)R13a; N(R13)S(O)2R13a; N(R13)S(O)R13a; N(R13)C(O)N(R13aR13b); N(R13)C(O)OR13a или OC(O)N(R13R13a);
R13, R13a, R13b независимо выбраны из группы, состоящей из H и C1-6алкила, где C1-6алкил возможно замещен одним или более атомами галогена, которые являются одинаковыми или разными;
Ra, Rb объединены с образованием -(CR14R14a)p-S(O)r-(CR14bR14c)q-;
r равно 0; 1 или 2;
p, q равны 0; 1; 2 или 3, при условии, что p+q равно 2; 3 или 4;
R14, R14a, R14b, R14c независимо выбраны из группы, состоящей из Н; галогена; CN; C(O)OR15; OR15; C(O)R15; C(O)N(R15R15a); S(O)2N(R15R15a); S(O)N(R15R15a); S(O)2R15; S(O)R15; N(R15)S(O)2N(R15aR15b); N(R15)S(O)N(R15aR15b); SR15; N(R15R15a); NO2; OC(O)R15; N(R15)C(O)R15a; N(R15)S(O)2R15a; N(R15)S(O)R15a; N(R15)C(O)N(R15aR15b); N(R15)C(O)OR15a; OC(ON(R15R15a) или С1-6алкила, где С1-6алкил возможно замещен одним или более R16, которые являются одинаковыми или разными;
возможно одна из пар R14, R14a и R14b, R14c или обе пары образует(ют) оксо-группу (=O);
возможно члены одной из пар, выбранных из группы, состоящей из R14, R14a; R14, R14b; двух соседних R14 в случае, когда p>1; и двух соседних R14b в случае, когда q>1, объединены с образованием, вместе с кольцом, к которому они присоединены, 6-11-членного гетеробицикла;
R15, R15a, R15b независимо выбраны из группы, состоящей из Н; C1-6алкила, где С1-6алкил возможно замещен одним или более атомами галогена, которые являются одинаковыми или разными;
R16 представляет собой галоген; CN; C(O)OR17; OR17; C(O)R17; C(O)N(R17R17a); S(O)2N(R17R17a); S(O)N(R17R17a); S(O)2R17; S(O)R17; N(R17)S(O)2N(R17aR17b); N(R17)S(O)N(R17aR17b); SR17; N(R17R17a); NO2; OC(O)R17; N(R17)C(O)R17a; N(R17)S(O)2R17a; N(R17)S(OR17a; N(R17)C(O)N(R17aR17b); N(R17)C(O)OR17a или OC(O)N(R17R17a);
R17, R17a, R17b независимо выбраны из группы, состоящей из Н; C1-6алкила, где С1-6алкил возможно замещен одним или более атомами галогена, которые являются одинаковыми или разными.
В случае, когда переменная или заместитель могут быть выбраны из группы разных вариантов, и такие переменная или заместитель присутствуют в количестве более одного, соответствующие варианты могут быть одинаковыми или разными.
В пределах настоящего изобретения используемые термины имеют следующие значения.
Термин "возможно замещенный" означает незамещенный или замещенный. Как правило, но без ограничения указанным, "одним или более заместителями" означает одним, двумя или тремя, предпочтительно одним или двумя и более предпочтительно одним заместителем. В общем эти заместители могут быть одинаковыми или разными.
"Алкил" означает прямую или разветвленную углеродную цепь. Каждый водород при алкильном углероде может быть заменен заместителем, как далее раскрыто в данном описании.
"С1-4алкил" означает алкильную цепь, имеющую 1-4 атома углерода, например, если он присутствует в конце молекулы: метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил трет-бутил, или, например, -CH2-, -СН2-СН2-, -СН(СН3)-, -С(СН2)-, -CH2-CH2-CH2-, -СН(С2Н5)-, -С(СН3)2-, когда две группировки молекулы соединены такой алкильной группой. Каждый водород при углероде в С1-4алкиле может быть заменен заместителем, как далее раскрыто в данном описании.
"С1-6алкил" означает алкильную цепь, имеющую 1-6 атомов углерода, например, если он присутствует в конце молекулы: С1-4алкил, метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил; трет-бутил, н-пентил, н-гексил, или, например, -CH2-, -СН2-СН2-, -СН(СН3)-, -СН2-СН2-СН2-, -СН(С2Н5)-, -С(СН3)2-, когда две группировки молекулы соединены такой алкильной группой. Каждый водород при углероде в С1-6алкиле может быть заменен заместителем, как далее раскрыто в данном описании.
"С3-7циклоалкил" или "С3-7циклоалкильное кольцо" означает циклическую алкильную цепь, имеющую 3-7 атомов углерода, например циклопропил, циклобутил, циклопентил, циклогексил, циклогексенил, циклогептил. Каждый водород при циклоалкильном углероде может быть заменен заместителем, как далее раскрыто в данном описании.
"Галоген" означает фтор, хлор, бром или йод. В общем предпочтительно, чтобы галоген представлял собой фтор или хлор.
"4-7-членный гетероциклил" или "4-7-членный гетероцикл" означает кольцо с 4, 5, 6 или 7 кольцевыми атомами, которое может содержать вплоть до максимального числа двойных связей (ароматическое или неароматическое кольцо, которое является полностью, частично насыщенным или ненасыщенным), где по меньшей мере один кольцевой атом и вплоть до 4 кольцевых атомов заменены гетероатомом, выбранным из группы, состоящей из серы (включая -S(O)-, -S(O)2-), кислорода и азота (включая =N(O)-), и где это кольцо соединено с остальной частью молекулы через атом углерода или азота. Примерами 4-7-членных гетероциклов являются азетидин, оксетан, тиетан, фуран, тиофен, пиррол, пирролин, имидазол, имидазолин, пиразол, пиразолин, оксазол, оксазолин, изоксазол, изоксазолин, тиазол, тиазолин, изотиазол, изотиазолин, тиадиазол, тиадиазолин, тетрагидрофуран, тетрагидротиофен, пирролидин, имидазолидин, пиразолидин, оксазолидин, изоксазолидин, тиазолидин, изотиазолидин, тиадиазолидин, сульфолан, пиран, дигидропиран, тетрагидропиран, имидазолидин, пиридин, пиридазин, пиразин, пиримидин, пиперазин, пиперидин, морфолин, тетразол, триазол, триазолидин, тетразолидин, диазепан, азепин или гомопиперазин. Термин "5-6-членный гетероциклил" или "5-6-членный гетероцикл" определен соответственно.
Термин "6-11-членный гетеробициклил" или "6-11-членный гетеробицикл" означает гетероциклическую систему из двух колец с 6-11 кольцевыми атомами, где по меньшей мере один кольцевой атом является общим для обоих колец, и которая может содержать вплоть до максимального числа двойных связей (ароматическое или неароматическое кольцо, которое является полностью, частично насыщенным или ненасыщенным), где по меньшей мере один кольцевой атом и вплоть до 6 кольцевых атомов (предпочтительно вплоть до 5, более предпочтительно вплоть до 4, более предпочтительно вплоть до 3 кольцевых атомов) заменены гетероатомом, выбранным из группы, состоящей из серы (включая -S(O)-, -S(O)2-), кислорода и азота (включая =N(O)-), и где это кольцо соединено с остальной частью молекулы через атом углерода или азота. Примерами 6-11-членного гетеробицикла являются индол, индолин, бензофуран, бензотиофен, бензоксазол, бензизоксазол, бензотиазол, бензизотиазол, бензимидазол, бензимидазолин, хинолин, хиназолин, дигидрохиназолин, хинолин, дигидрохинолин, тетрагидрохинолин, декагидрохинолин, изохинолин, декагидроизохинолин, тетрагидроизохинолин, дигидроизохинолин, бензазепин, пурин или птеридин. Термин "6-11-членный гетеробицикл" также включает спиро-структуры из двух колец, такие как 1,4-диокса-8-азаспиро[4.5]декан, или мостиковые гетероциклы, такие как 8-аза-бицикло[3.2.1]октан. Термин "8-11-членный гетеробициклил" или "8-11-членный гетеробицикл" определен соответственно.
Термин "5-6-членный ароматический гетероциклил" или "5-6-членный ароматический гетероцикл" означает гетероцикл, полученный из циклопентадиенила или бензола, где по меньшей мере один атом углерода заменен гетероатомом, выбранным из группы, состоящей из серы (включая -S(O)-, -S(O)2-), кислорода и азота (включая =N(O)-). Примерами таких гетероциклов являются фуран, тиофен, пиррол, имидазол, пиразол, оксазол, изоксазол, тиазол, изотиазол, тиадиазол, пираний, пиридин, пиридазин, пиримидин, триазол, тетразол.
Предпочтительными соединениями формулы (I) являются те соединения, в которых один или более из содержащихся в них остатков имеют значения, приведенные ниже, со всеми комбинациями предпочтительных определений заместителей, являющимися объектом настоящего изобретения. Что касается всех предпочтительных соединений формулы (I), то настоящее изобретение также включает все таутомерные и стереоизомерные формы и их смеси во всех соотношениях, и их фармацевтически приемлемые соли.
В предпочтительных воплощениях настоящего изобретения упомянутые ниже заместители независимо имеют следующее значение. Таким образом, один или более из этих заместителей могут иметь предпочтительные или более предпочтительные значения, приведенные ниже.
Предпочтительно в формуле (I) Ra и Rb выбраны с получением одной из формул (Ia)-(Ij):
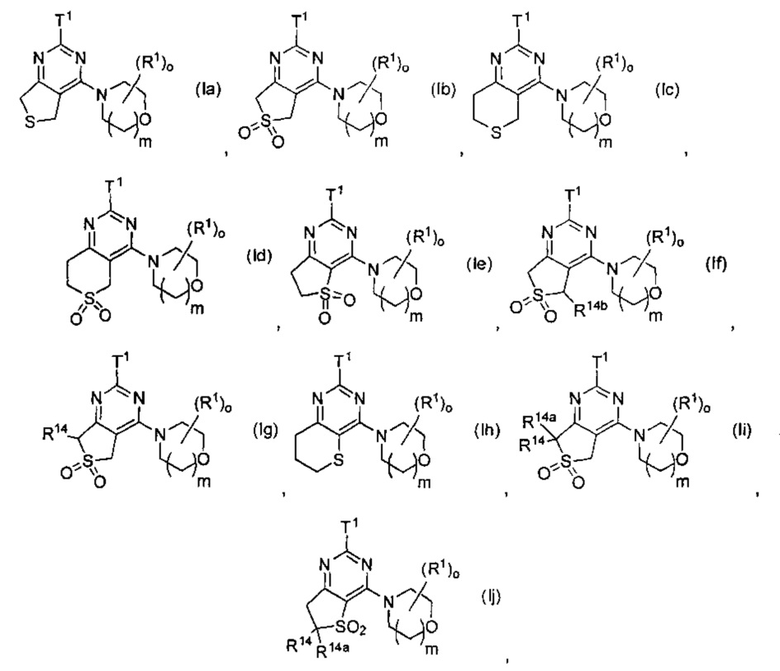
где Τ1, R1, ο, m, R14, R14a, R14b имеют значения, как указано выше. Более предпочтительными являются (Ib), (If) и (1i), особенно (Ii).
Предпочтительно в формуле (I) Ra и Rb выбраны с получением одной из формул (Ik)-(Ip):
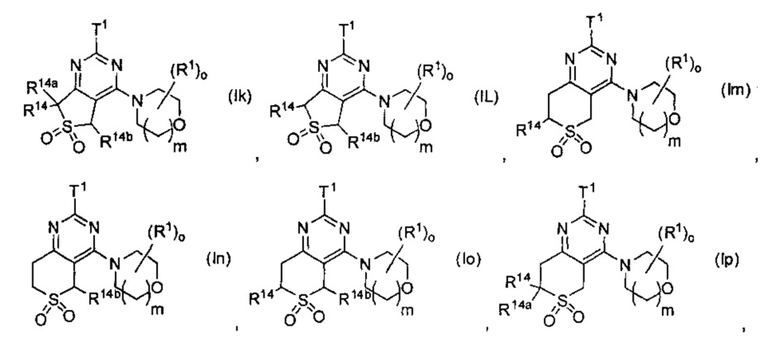
где Τ1, R1, о, m, R14, R14a, R14b имеют значения, как указано выше. Наиболее предпочтительной является (Ik).
Предпочтительно Т1 представляет собой фенил, где Т1 замещен группой N(R5a)C(O)N(R5bR5) или N(R5a)C(O)OR5 и возможно дополнительно замещен одним или более R6, которые являются одинаковыми или разными.
Предпочтительно Т1 замещен группой N(R5a)C(O)N(R5bR5) и возможно дополнительно замещен одним или более R6, которые являются одинаковыми или разными.
Предпочтительно Т1 не является дополнительно замещенным одним или более R6.
Предпочтительно в формуле (I) Т1 определен так, чтобы получалась формула (Ik)
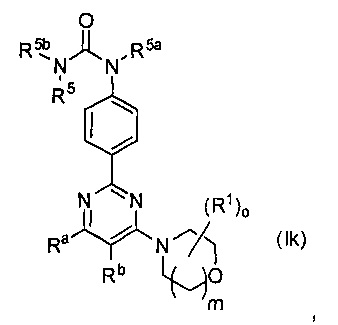
где о, m, R1, Ra, Rb, R5, R5a, R5b имеют значения, как определено выше.
Предпочтительно R5a, R5b представляют собой Н.
Предпочтительно R5 представляет собой Т2, где Т2 является незамещенным или замещен одним или более R10, которые являются одинаковыми или разными, и где Т2 представляет собой фенил; пиридил; циклопропил; циклобутил; циклопентил; циклогексил; оксетанил или тетрагидрофуранил. Более предпочтительно Т2 представляет собой циклопропил. Более предпочтительно Т2 является незамещенным.
Предпочтительно R5 представляет собой незамещенный C1-6алкил.
Предпочтительно R5 представляет собой C1-6алкил, замещенный одним или более R8, которые являются одинаковыми или разными и выбраны из группы, состоящей из F; OR9 и N(R9R9a).
Предпочтительно r равен 0 или 2. Предпочтительно r равно 1 или 2. Еще более предпочтительно r равно 2.
Предпочтительно p, q равны 1; 2 или 3. Таким образом, предпочтительно, чтобы ни p, ни q не равнялись 0.
Предпочтительно p+q равно 2 или 3.
Более предпочтительно p и q оба равны 1.
Еще более предпочтительно p и q оба равны 1, и r равно 2.
Предпочтительно максимально два из R14, R14a, R14b, R14c отличны от H. Соответственно, в одном воплощении ни один из R14, R14a, R14b, R14c не является отличным от Н; в другом воплощении один из R14, R14a, R14b, R14c отличен от H; и в третьем воплощении два из R14, R14a, R14b, R14c отличны от H. Предпочтительно по меньшей мере один из R14, R14a, R14b и R14c отличен от Н.
Предпочтительно три из R14, R14a, R14b, R14c отличны от H.
Предпочтительно R14, R14a, R14b, R14c независимо выбраны из группы, состоящей из H; F; этила и метила. Более предпочтительно R14, R14a, R14b, R14c независимо выбраны из группы, состоящей из H и метила. В одном воплощении R14 представляет собой метил, R14a представляет собой водород, R14b представляет собой водород, и R14c представляет собой водород. В другом воплощении R14 представляет собой метил, R14a представляет собой метил, R14b представляет собой водород, и R14c представляет собой водород. В другом воплощении R14 представляет собой метил, R14a представляет собой метил, R14b представляет собой метил, и R14c представляет собой водород. В другом воплощении R14 представляет собой метил, R14a представляет собой F, R14b представляет собой водород, и R14c представляет собой водород. В другом воплощении R14 представляет собой метил, R14a представляет собой F, R14b представляет собой метил, и R14c представляет собой водород. В другом воплощении R14 представляет собой метил, R14a представляет собой метил, R14b представляет собой F, и R14c представляет собой водород. В другом воплощении R14 представляет собой F, R14a представляет собой F, R14b представляет собой водород, и R14c представляет собой водород.
Предпочтительно m равно 1.
Предпочтительно о равно 1 или 2.
Предпочтительно каждый R1 независимо выбран из группы, состоящей из Н; галогена; CN; оксо (=O) или С1-6алкила, где С1-6алкил возможно замещен одним или более R3, которые являются одинаковыми или разными.
Предпочтительно R1 представляет собой незамещенный С1-6алкил (более предпочтительно метил или этил, еще более предпочтительно метил) или С1-6алкил, замещенный одним R3.
Предпочтительно два R1 объединены с образованием, вместе с кольцом, к которому они присоединены, 8-окса-3-азабицикло[3.2.1]октан-3-ильного или 3-окса-8-азабицикло[3.2.1]октан-8-ильного кольца.
Один подкласс соединений согласно настоящему изобретению представлен соединениями формулы (Iq):
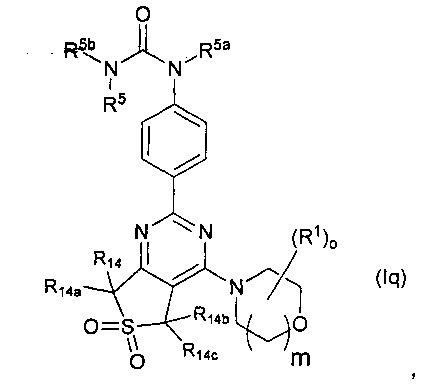
где R5, R5a, R5b, R14, R14a, R14b, R14c, R1, o, m являются такими, как определено в данном описании выше.
В одном воплощении R5b и R5a представляют собой Н, и R5 представляет собой С1-6алкил или С3-7циклоалкил, где С1-6алкил возможно замещен одним или более R8, которые являются одинаковыми или разными, и С3-7циклоалкил возможно замещен одним или более R10, которые являются одинаковыми или разными.
В другом воплощении R5b и R5a представляют собой Н, и R5 представляет собой С1-6алкил (например, метил, этил, пропил, или изопропил), который возможно замещен одним или более атомами галогена (например. фтора) или R8 (например, OR9), которые являются одинаковыми или разными, или R5 представляет собой С3-7циклоалкил (например циклопропил), который возможно замещен одним или более атомами галогена (например фтора) или R10 (например, OR11), которые являются одинаковыми или разными.
В другом воплощении R5b и R5a представляют собой Н, и R5 представляет собой C1-6алкил (например, метил, этил, пропил, или изопропил), который возможно замещен одним или более атомами галогена (например, фтора) или R8 (например, OR9), которые являются одинаковыми или разными, или R5 представляет собой С3-7циклоалкил (например, циклопропил), который возможно замещен одним или более атомами галогена (например, фтора) или R10 (например, OR11), которые являются одинаковыми или разными, и где R8 и R10 представляют собой Н.
В другом воплощении R5b и R5a представляют собой Н, и R5 представляет собой C1-6алкил (например, метил, этил или пропил) и замещен одним или более фторами или гидрокси.
Конкретные примеры R5 включают циклопропил, метил, этил, фторэтил, гидроксиэтил, дифторэтил, изопропил, фторпропил, пиридинил и оксетанил.
В одном воплощении R1 представляет собой С1-6алкил (например, метил, или этил), и о равно 1.
В другом воплощении (R1)o присоединен в положении 3.
В одном воплощении R14, R14a, R14b и R14c выбраны из С1-6алкила или Н. В одном воплощении R14, R14a, R14b и R14c выбраны из С1-6алкила; F или Н.
В одном воплощении три из R14, R14a, R14b и R14c отличны от Н.
В другом воплощении три из R14, R14a, R14b и R14c выбраны из С1-6алкила (например, метила, этила или пропила). В другом воплощении три из R14, R14a, R14b и R14c выбраны из С1-6алкила (например, метила, этила или пропила) или F.
В другом воплощении три из R14, R14a, R14b и R14c выбраны из метила. В другом воплощении три из R14, R14a, R14b и R14c выбраны из метила или F.
В другом воплощении R14, R14a, R14b представляют собой метил, и R14c представляет собой Н.
Соединения формулы (I), в которых некоторые или все из вышеупомянутых групп имеют предпочтительные значения, также являются объектом настоящего изобретения.
Другие предпочтительные соединения по настоящему изобретению выбраны из группы, состоящей из
1-циклопропил-3-(4-(4-морфолино-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
1-циклопропил-3-(4-(4-((2S,6R)-2,6-диметилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
(S)-1-циклопропил-3-(4-(4-(3-метилморфолино)-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
(S)-1-циклопропил-3-(4-(4-(3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
(S)-1-этил-3-(4-(4-(3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
(S)-1-метил-3-(4-(4-(3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
(S)-1-(2-гидроксиэтил)-3-(4-(4-(3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
(S)-1-(2-фторэтил)-3-(4-(4-(3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
1-(4-(4-(8-окса-3-азабицикло[3.2.1]октан-3-ил)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)-3-этилмочевины;
1-(4-(4-(8-окса-3-азабицикло[3.2.1]октан-3-ил)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)-3-(2-гидроксиэтил)мочевины;
(R)-1-циклопропил-3-(4-(4-(3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
1-(4-(4-(8-окса-3-азабицикло[3.2.1]октан-3-ил)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)-3-циклопропилмочевины;
1-(4-(4-(8-окса-3-азабицикло[3.2.1]октан-3-ил)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)-3-метилмочевины;
1-(4-(4-(8-окса-3-азабицикло[3.2.1]октан-3-ил)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)-3-(2-фторэтил)мочевины;
(S)-1-циклопропил-3-(4-(4-(3-метилморфолино)-6,6-диоксидо-7,8-дигидро-5Н-тиопирано[4,3-d]пиримидин-2-ил)фенил)мочевины;
(S)-1-циклопропил-3-(4-(4-(3-метилморфолино)-7,8-дигидро-5Н-тиопирано[4,3-d]пиримидин-2-ил)фенил)мочевины;
(S)-1-(2,2-дифторэтил)-3-(4-(4-(3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
1-(4-(4-(8-окса-3-азабицикло[3.2.1]октан-3-ил)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)-3-(2,2-дифторэтил)мочевины;
(S)-1-изопропил-3-(4-(4-(3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
1-(4-(4-(3-окса-8-азабицикло[3.2.1]октан-8-ил)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)-3-циклопропилмочевины;
1-циклопропил-3-(4-(4-(3-этилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
(S)-1-(3-фторпропил)-3-(4-(4-(3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
1-(4-(4-(8-окса-3-азабицикло[3.2.1]октан-3-ил)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)-3-(3-фторпропил)мочевины;
1-циклопропил-3-(4-(7-метил-4-((S)-3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
(S)-1-циклопропил-3-(4-(4-(3-метилморфолино)-5,5-диоксидо-6,7-дигидротиено[3,2-d]пиримидин-2-ил)фенил)мочевины;
(S)-1-циклопропил-3-(4-(4-(3-метилморфолино)-7,8-дигидро-6Н-тиопирано[3,2-d]пиримидин-2-ил)фенил)мочевины;
1-циклопропил-3-(4-(5-метил-4-((S)-3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
(S)-1-циклопропил-3-(4-(7,7-диметил-4-(3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
1-циклопропил-3-(4-(5-метил-4-морфолино-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
1-циклопропил-3-(4-(4-(3-этилморфолино)-5-метил-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
1-(4-(4-(3-окса-8-азабицикло[3.2.1]октан-8-ил)-5-метил-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)-3-циклопропилмочевины;
1-(4-(4-(8-окса-3-азабицикло[3.2.1]октан-3-ил)-5-метил-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)-3-циклопропилмочевины;
(S)-1-(4-(4-(3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4 d]пиримидин-2-ил)фенил)-3-(пиридин-4-ил)мочевины;
(S)-1-(4-(4-(3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)-3-(пиридин-3-ил)мочевины;
1-циклопропил-3-(4-((R)-5-метил-4-((S)-3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
1-циклопропил-3-(4-((S)-5-метил-4-((S)-3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины и
(S)-1-циклопропил-3-(4-(6,6-диметил-4-(3-метилморфолино)-5,5-диоксидо-6,7-дигидротиено[3,2-d]пиримидин-2-ил)фенил)мочевины.
Другие предпочтительные соединения по настоящему изобретению выбраны из группы, состоящей из
1-этил-3-(4-(5-метил-4-((S)-3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
1-этил-3-(4-((R)-5-метил-4-((S)-3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
1-этил-3-(4-((S)-5-метил-4-((S)-3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
1-этил-3-(4-(4-((S-3-этилморфолино)-7-метил-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
1-этил-3-(4-(5,7,7-триметил-4-((S)-3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
1-циклопропил-3-(4-(7-фтор-7-метил-4-((S)-3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
1-циклопропил-3-(4-(5,7-диметил-4-((S)-3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
1-(4-(5-метил-4-((S)-3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)-3-пропилмочевины;
1-(циклопропилметил)-3-(4-(5-метил-4-((S)-3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
1-циклопропил-3-(4-(4-((S)-3-этилморфолино)-5-метил-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
1-циклопропил-3-(4-(5-метил-4-((R)-3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
1-циклопропил-3-(4-(5-этил-4-((S)-3-метилморфолино)-6,6-диоксидо-5,7 дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
1-этил-3-(2-фтор-4-(5-метил-4-((S)-3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
(S)-1-циклопропил-3-(4-(4-(3-этилморфолино)-7,7-диметил-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
1-этил-3-(4-((R)-4-((S)-3-этилморфолино)-5-метил-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
1-этил-3-(4-((S)-4-((S)-3-этилморфолино)-5-метил-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
(R)-1-циклопропил-3-(4-(7,7-диметил-4-(3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
(R)-1-(4-(7,7-диметил-4-(3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)-3-этилмочевины;
(S)-1-этил-3-(4-(4-(3-этилморфолино)-7,7-диметил-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
(S)-1-(4-(7,7-диметил-4-(3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)-3-метилмочевины;
1-метил-3-(4-(5-метил-4-((S)-3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
1-(4-(4-(3-окса-8-азабицикло[3.2.1]октан-8-ил)-7,7-диметил-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)-3-циклопропилмочевины;
1-(4-(4-(3-окса-8-азабицикло[3.2.1]октан-8-ил)-7,7-диметил-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)-3-этилмочевины;
(S)-1-(4-(7,7-диметил-4-(3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)-3-(2-фторэтил)мочевины;
(S)-1-(4-(7,7-диметил-4-(3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)-3-этилмочевины;
1-циклопропил-3-(4-((S)-5-метил-4-((R)-3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
(S)-1-(4-(4-(3-этилморфолино)-7,7-диметил-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)-2-фторфенил)-3-метилмочевины;
(S)-1-(4-(7,7-диметил-4-(3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)-2-фторфенил)-3-метилмочевины;
(S)-1-(4-(7,7-диметил-4-(3-метилморфолино)-6,6-диоксидо-5,7 дигидротиено[3,4-d]пиримидин-2-ил)-3-фторфенил)-3-метилмочевины;
1-этил-3-(4-(5-этил-4-((S)-3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
(S)-1-(4-(7,7-диметил-4-(3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)-3-(оксетан-3-ил)мочевины;
(S)-1-(4-(7,7-диметил-4-(3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)-3-фторфенил)-3-этилмочевины;
(S)-1-циклопропил-3-(4-(7,7-диметил-4-(3-метилморфолино)-6,6-диоксидо-7,8-дигидро-5Н-тиопирано[4,3-d]пиримидин-2-ил)фенил)мочевины;
1-циклопропил-3-(4-(7-метил-4-((S)-3-метилморфолино)-6,6-диоксидо-7,8-дигидро-5Н-тиопирано[4,3-d]пиримидин-2-ил)фенил)мочевины;
1-циклопропил-3-(4-(5,7-диметил-4-((S)-3-метилморфолино)-6,6-диоксидо-7,8-дигидро-5Н-тиопирано[4,3-d]пиримидин-2-ил)фенил)мочевины;
(S)-1-(4-(7,7-диметил-4-(3-метилморфолино)-6,6-диоксидо-7,8-дигидро-5Н-тиопирано[4,3-d]пиримидин-2-ил)фенил)-3-этилмочевины;
1-циклопропил-3-(4-(5-метил-4-((S)-3-метилморфолино)-6,6-диоксидо-7,8-дигидро-5Н-тиопирано[4,3-d]пиримидин-2-ил)фенил)мочевины;
1-(2-фторэтил)-3-(4-(5,7,7-триметил-4-((S)-3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
1-(4-(7,7-диметил-4-морфолино-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)-3-этилмочевины;
1-метил-3-(4-(5,7,7-триметил-4-((S)-3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
1-(3-фтор-4-(5,7,7-триметил-4-((S)-3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)-3-метилмочевины;
1-(4-(4-((S)-3-этилморфолино)-5,7,7-триметил-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)-3-фторфенил)-3-метилмочевины;
1-(4-(4-((S)-3-этилморфолино)-5,7,7-триметил-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)-3-метилмочевины;
1-(2-фторэтил)-3-(4-(5,7,7-триметил-4-морфолино-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины и
1-циклопропил-3-(4-((R)-5,7,7-триметил-4-((S)-3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины.
Там, где может иметь место таутомеризм, такой как, например, кетоенольный таутомеризм, соединений общей формулы (I), индивидуальные формы, такие как, например, кето- и енольная форма, охватываются по-раздельности и совместно в виде смесей в любом соотношении. То же самое относится и к стереоизомерам, таким как, например, энантиомеры, цис/транс-изомеры, конформеры и тому подобное.
В частности, соединения формулы (I), где морфолиновое кольцо замещено одним R1 (отличным от Н) в положении 3 и/или разными заместителями R14/R14a, R14b/R14c, охватываются настоящим изобретением в виде их изомеров, энантиомеров, диастереомеров или их смесей в отношении соответствующего(их) хирального(ых) углеродного(ых) центра(ов).
Если необходимо, изомеры могут быть разделены способами, хорошо известными в уровне техники, например, посредством жидкостной хроматографии. То же самое применимо к энантиомерам посредством использования, например, хиральных стационарных фаз. Аналогичным образом диастереомеры могут быть разделены посредством обычной жидкостной хроматографии или путем использования хиральных стационарных фаз. Кроме того, энантиомеры могут быть выделены путем превращения их в диастереомеры, например посредством сочетания с энантиомерно чистым вспомогательным соединением с последующим разделением полученных диастереомеров и отщепления вспомогательного остатка. Альтернативно любой энантиомер или диастереомер соединения формулы (I) может быть получен в результате стереоселективного синтеза с использованием оптически чистых исходных материалов.
Соединения формулы (I) могут существовать в кристаллической или аморфной форме. Кроме того, некоторые кристаллические формы соединений формулы (I) могут существовать как полиморфы, которые включены в объем защиты согласно настоящему изобретению. Полиморфные формы соединений формулы (I) могут быть охарактеризованы и дифференцированы с помощью ряда обычных аналитических методик, включая, без ограничения ими, картины дифракции рентгеновских лучей на порошке (XRPD), инфракрасные (IR) спектры, рамановские спектры, дифференциальную сканирующую калориметрию (DSC), термогравиметрический анализ (TGA) и ядерный магнитный резонанс твердого состояния (тсЯМР).
В случае, когда соединения согласно формуле (I) содержат одну или более кислотных или основных групп, изобретение также включает их соответствующие фармацевтически или токсикологически приемлемые соли, в частности их фармацевтически используемые соли. Таким образом, согласно изобретению могут быть использованы соединения формулы (I), которые содержат кислотные группы, например, в виде солей щелочных металлов, солей щелочно-земельных металлов или солей аммония. Более конкретные примеры таких солей включают соли натрия, соли калия, соли кальция, соли магния или соли с аммонием или органическими аминами, такими как, например, этиламин, этаноламин, триэтаноламин или аминокислоты. Соединения формулы (I), которые содержат одну или более основных групп, то есть групп, которые могут быть протонированы, могут быть представлены и использованы по изобретению в форме их солей присоединения с неорганическими или органическими кислотами. Примеры приемлемых кислот включают хлористый водород, бромистый водород, фосфорную кислоту, серную кислоту, азотную кислоту, метансульфоновую кислоту, п-толуолсульфоновую кислоту, нафталиндисульфоновые кислоты, оксалиновую кислоту, уксусную кислоту, винную кислоту, молочную кислоту, салициловую кислоту, бензойную кислоту, муравьиную кислоту, пропионовую кислоту, пивалоильную кислоту, диэтилуксусную кислоту, малоновую кислоту, янтарную кислоту, пимелиновую кислоту, фумаровую кислоту, малеиновую кислоту, яблочную кислоту, сульфаминовую кислоту, фенилпропионовую кислоту, глюконовую кислоту, аскорбиновую кислоту, изоникотиновую кислоту, лимонную кислоту, адипиновая кислоту и другие кислоты, известные специалистам в данной области техники. Если соединения формулы (I) одновременно содержат в молекуле кислотные и основные группы, изобретение также включает, в дополнение к упомянутым солевым формам, внутренние соли или бетаины (цвиттерионы). Соответствующие соли согласно формуле (I) могут быть получены традиционными методами, которые известны специалистам в данной области техники, такими как, например, посредством приведения их в контакт с органическими или неорганическими кислотой или основанием в растворителе или диспергирующем агенте, или посредством ионообмена или катионообмена в другими солями. Настоящее изобретение также включает все соли соединений формулы (I), которые благодаря их низкой физиологической совместимости непосредственно не пригодны для применения в фармацевтических средствах, но которые могут быть использованы, например, как промежуточные соединения для химических взаимодействий или для получения фармацевтически приемлемых солей.
В данном изобретении термин "фармацевтически приемлемый" означает, что соответствующее соединение, носитель или молекула являются приемлемыми для введения людям. Предпочтительно этот термин означает «одобренный регулирующим ведомством, таким как ЕМЕА (Европейское агенство лекарственных средств, Европа) и/или FDA (Управление по санитарному надзору за качеством пищевых продуктов и медикаментов, США) и/или любое другое национальное регулирующее ведомство, для применения у животных, предпочтительно у людей».
Настоящее изобретение дополнительно включает все сольваты соединений по изобретению.
При необходимости эффекты заявленных соединений в отношении активности mTOR могут, например, быть протестированы с помощью временно экспрессируемой меченной эпитопом mTOR в клеточной линии млекопитающих, такой как HEK293, которая образует иммунопреципитат с моноклональным антителом, направленным против метки-эпитопа (Knight et al. 2004, Bioorganic and Medicinal Chemistry 12, 4749-4759). В другом анализе используется белок mTOR, выделенный из клеточных или тканевых лизатов с помощью обычных методов очистки белков. В этом анализе в качестве субстрата используют GST-слитый белок Р70 S6 киназы. Фосфорилирование Р70 S6 обнаруживают с помощью первичного фосфо-специфичного антитела (направленного против фосфорилированного треонина 389) и связанного с ферментом вторичного антитела в анализе ELISA (твердофазный иммуноферментный анализ, US-A 2004/0191836).
Согласно настоящему изобретению, выражение "mTOR" или "киназа mTOR" означает белок mTOR (Tsang et al., 2007, Drug Discovery Today 12, 112-124). Ген, кодирующий mTOR, локализован в локусе 1р36.2 на карте человеческих хромосом, и он широко экспрессируется в человеческих тканях.
Как продемонстрировано в примерах, соединения по изобретению тестировали в отношении их селективности в отношении mTOR относительно других киназ. Как показано, протестированные соединения связывают mTOR более селективно, чем киназы РI3Kдельта или DNA-PK (смотри таблицы 9 и 10 ниже). Соответственно, соединения по настоящему изобретению считают полезными для предупреждения или лечения заболеваний и расстройств, ассоциированных с mTOR, например иммунологических, воспалительных, аутоиммунных или аллергических расстройств, или пролиферативных заболеваний, отторжения трансплантатов, болезни «трансплантат-против-хозяина», сердечно-сосудистых заболеваний, метаболических заболеваний или нейродегенеративных заболеваний.
Таким образом, в настоящем изобретении предложены фармацевтические композиции, содержащие соединение формулы (I) или его фармацевтически приемлемую соль в качестве активного ингредиента вместе с фармацевтически приемлемым носителем, возможно в комбинации с одной или более другими фармацевтическими композициями.
"Фармацевтическая композиция" означает один или более активных ингредиентов и один или более инертных ингредиентов, которые составляют носитель, а также любой продукт, который является результатом, прямо или косвенно, комбинирования, комплексообразования или агрегирования любых двух или более ингредиентов, или диссоциации одного или более ингредиентов, или других типов реакций или взаимодействий одного или более ингредиентов. Соответственно, фармацевтическая композиция по настоящему изобретению охватывает любую композицию, полученную посредством смешивания соединения по настоящему изобретению и фармацевтически приемлемого носителя.
Термин "носитель" относится к разбавителю, адъюванту, эксципиенту или наполнителю, с которым вводят терапевтическое средство. Такие фармацевтические носители могут представлять собой стерильные жидкости, такие как вода и масла, включая вазелиновое масло, масла животного, растительного или синтетического происхождения, включая, без ограничения ими, арахисовое масло, соевое масло, минеральное масло, кунжутное масло и тому подобное. Вода является предпочтительным носителем, когда фармацевтическую композицию вводят перорально. Физиологический раствор и водная декстроза являются предпочтительными носителями, когда фармацевтическую композицию вводят внутривенно. Физиологические растворы и водные растворы декстрозы и глицерина предпочтительно используют в качестве жидких носителей для растворов для инъекций. Подходящие фармацевтические эксципиенты включают крахмал, глюкозу, лактозу, сахарозу, желатин, солод, рис, муку, мел, силикагель, стеарат натрия, глицерина моностеарат, тальк, хлорид натрия, высушенное снятое молоко, глицерин, пропиленгликоль, воду, этанол и тому подобное. При необходимости композиция может также содержать небольшие количества увлажняющих или эмульгирующих агентов или забуферивающих pH агентов. Эти композиции могут быть представлены в форме растворов, суспензий, эмульсий, таблеток, пилюль, капсул, порошков, препаратов замедленного высвобождения и тому подобного. Композиция может быть приготовлена в форме суппозитория с традиционными связующими и носителями, такими как триглицериды. Пероральная композиция может включать стандартные носители фармацевтического качества, такие как маннит, лактоза, крахмал, стеарат магния, сахаринат натрия, целлюлоза, карбонат магния и так далее. Примеры подходящих фармацевтических носителей описаны в "Remington's Pharmaceutical Sciences", E.W. Martin. Такие композиции будут содержать терапевтически эффективное количество терапевтического средства, предпочтительно в очищенной форме, вместе с подходящим количеством носителя так, чтобы получить форму для соответствующего введения пациенту. Композиция должна соответствовать пути введения.
Фармацевтическая композиция по настоящему изобретению может содержать в качестве активных ингредиентов один или более дополнительных соединений, таких как одно или более соединений формулы (I), не являющихся первым соединением в композиции или mTOR ингибиторами. Дополнительные биоактивные соединения могут представлять собой стероиды, антагонисты лейкотриенов, циклоспорин или рапамицин.
Соединения по настоящему изобретению или его фармацевтически приемлемая(ые) соль(и) и другой(ие) фармацевтически активный(е) агент(ы) может(гут) быть введен(ы) совместно или раздельно и, при их раздельном введении, это может происходить по-раздельности или последовательно в любом порядке. При объединении в одной и той же композиции следует понимать, что два соединения должны быть стабильными и совместимыми друг с другом и с другими компонентами композиции. При их раздельном приготовлении они могут быть представлены в любой традиционной композиции, обычно таким образом, как известно для таких соединений в уровне техники.
Настоящее изобретение предполагает, что соединение формулы (I) или его фармацевтически приемлемую соль или фармацевтическую композицию, содержащую соединение формулы (I), вводят в комбинации с другим лекарственным средством или фармацевтически активным агентом, и/или что фармацевтическая композиция по изобретению дополнительно содержит такое лекарственное средство или фармацевтически активный агент.
В этом контексте термин "лекарственное средство или фармацевтически активный агент" включает лекарственное средство или фармацевтический агент, которые будут вызывать биологический или медицинский ответ в ткани, системе, животном или человеке, предполагаемый, например, исследователем или клиницистом.
"Объединенные", или "в комбинации", или "комбинация" следует понимать как функциональное совместное введение, где некоторые или все соединения могут быть введены раздельно, в разных композициях, разными путями введения (например, подкожно, внутривенно или перорально) и в разные моменты времени введения. Индивидуальные соединения таких комбинаций могут быть введены либо последовательно в раздельных фармацевтических композициях, а также одновременно в объединенных фармацевтических композициях.
Например, терапия ревматоидного артрита предусматривает комбинирование с другими химиотерапевтическими или антительными агентами. Подходящие примеры фармацевтически активных агентов, которые могут быть использованы в комбинации с соединениями по настоящему изобретению и их солями для терапии ревматоидного артрита, включают следующее: иммуносупрессанты, такие как амтолметин гуацил, мизорибин и римексолон; анти-TNFα (фактор некроза опухолей) агенты, такие как этанерцепт, инфликсимаб, адалимумаб, анакинра, абатацепт, ритуксимаб; ингибиторы тирозинкиназ, такие как лефлуномид; антагонисты калликреина, такие как субреум; агонисты интерлейкина 11, такие как опрелвекин; агонисты интерферона бета 1; агонисты гиалуроновой кислоты, такие как NRD-101 (Aventis); антагонисты рецепторов интерлейкина 1, такие как анакинра; антагонисты CD8, такие как гидрохлорид амиприлозы; антагонисты белка-предшественника бета-амилоида, такие как реумакон; ингибиторы матриксных металлопротеиназ, такие как ципемастат, и другие базовые противоревматические препараты, модифицирующие течение болезни (DMARD), такие как метотрексат, сулфасалазин, циклоспорин А, гидроксихороквин, ауранофин, ауротиоглюкоза, тиомалат натрия и золота и пенициламин.
В частности, определенное в данном описании лечение может применяться как монотерапия или может включать, в дополнение к соединениям по изобретению, обычную хирургию, или радиотерапию, или химиотерапию. Соответственно, соединения по изобретению могут также быть использованы в комбинации с существующими терапевтическими агентами для лечения пролиферативных заболеваний, таких как рак. Приемлемые агенты для использования в комбинации включают следующее:
(1) антипролиферативные/антинеопластические лекарственные средства и их комбинации, как применяется в медицинской онкологии, такие как алкилирующие агенты (например, цис-платин, карбоплатин, циклофосфамид, азотистый иприт, мелфалан, хлорамбуцил, бусульфан и нитрозомочевины); антиметаболиты (например, антифолаты, такие как фторпиримидины, например 5-фторурацил и тегафур, ралтитрексед, метотрексат, цитозин арабинозид, гидроксимочевина и гемцитабин); противоопухолевые антибиотики (например, антрациклины, такие как адриамицин, блеомицин, доксорубицин, дауномицин, эпирубицин, идарубицин, митомицин-С, дактиномицин и митрамицин); антимитотические агенты (например, алкалоиды барвинка, такие как винкристин, винбластин, виндезин и винорелбин, и таксоиды, такие как паклитаксел и таксотере); и ингибиторы топоизомераз (например, зпиподофиллотоксины, такие как этопозид и тенипозид, амсакрин, топотекан и камптотецины);
(2) цитостатические агенты, такие как антиэстрогены (например, тамоксифен, торемифен, ралоксифен, дролоксифен и йодоксифен), суппрессирующие регуляторы эстрогеновых рецепторов (например, фулвестрант), антиандрогены (например, бикалутамид, флутамид, нилутамид и ципротерона ацетат), антагонисты LHRH (рилизинг-фактор лютеинизирующего гормона) или агонисты LHRH (например, гозерелин, лейпрорелин и бузерелин), прогестогены (например, мегестрола ацетат), ингибиторы ароматазы (например, анастрозол, летрозол, воразол и эксеместан) и ингибиторы 5α-редуктазы, такие как финастерид;
(3) противоинвазивные агенты (например, ингибиторы семейства c-Src киназ, такие как 4-(6-хлор-2,3-метилендиоксианилино)-7-[2-(4-метилпиперазин-1-ил)этокси]-5-тетрагидропиран-4-илокси-хиназолин (AZD0530) и N-(2-хлор-6-метилфенил)-2-{6-[4-(2-гидроксиэтил)пиперазин-1-ил]-2-метилпиримидин-4-иламино}тиазол-5-карбоксамид (дасатиниб, BMS-354825), и ингибиторы металлопротеиназ, такие как маримастат, и ингибиторы функционирования рецептора урокиназного активатора плазминогена;
(4) ингибиторы функционирования факторов роста, например, такие ингибиторы включают антитела к факторам роста и антитела к рецепторам факторов роста (например, анти-erB2 антитело трастузумаб [Herceptin™] и анти-erbBI антитело цетуксимаб [С225]); такие ингибиторы также включают, например, ингибиторы тирозинкиназ, например, ингибиторы семейства эпидермальных факторов роста (например ингибиторы тирозинкиназ семейства EGFR (рецепторы эпидермальных факторов роста), такие как N-(3-хлор-4-фторфенил)-7-метокси-6-(3-морфолинопропокси)хиназолин-4-амин (гефитиниб, ZD 1839), N-(3-этинилфенил)-6,7-бис(2-метоксиэтокси)хиназолин-4-амин (эрлотиниб, OSI-774) и 6-акриламидо-N-(3-хлор-4-фторфенил)-7-(3-морфолинопропокси)-хиназолин-4-амин (CI 1033), и ингибиторы erB2 тирозинкиназы, такие как лапатиниб), ингибиторы семейства факторов роста гепатоцитов, ингибиторы семейства факторов роста тромбоцитов, такие как иматиниб, ингибиторы серин/треонинкиназ (например ингибиторы передачи сигнала Ras/Raf, такие как ингибиторы фарнезилтрансферазы, например сорафениб (BAY 43-9006)) и ингибиторы клеточной передачи сигнала посредством МЕК и/или Akt киназ;
(5) антиангиогенные агенты, такие как агенты, которые ингибируют эффекты васкулярного эндотелиального фактора роста, например антитело против васкулярного эндотелиального фактора роста бевацизумаб (Avastin™), и ингибиторы VEGF рецепторных тирозинкиназ, такие как 4-(4-бром-2-фторанилино)-6-метокси-7-(1-метилпиперидин-4-илметокси)хиназолин (ZD6474; Пример 2 в WO 01/32651), 4-(4-фтор-2-метилиндол-5-илокси)-6-метокси-7-(3-пирролидин-1-илпропокси)хиназолин (AZD2171; Пример 240 в WO 00/47212), ваталаниб (РТК787; WO 98/35985) и SUI 1248 (сунитиниб; WO 01/60814), и соединения, которые действуют по другим механизмам (например, линомид, ингибиторы функционирования интегрина αvβ3 и ангиостатина);
(6) агенты, повреждающие сосуды, такие как комбретастатин A4 и соединения, раскрытые в международной заявке на патент WO 99/02166;
(7) антисмысловые терапии, например терапии, направленные на перечисленные выше мишени, такие как ISIS 2503, анти-ras антисмысловой агент;
(8) подходы генной терапии, включая подходы с заменой аберрантных генов, таких как аберрантный р53 или аберрантный BRCAI или BRCA2, GDEPT (ген-направленная ферментная пролекарственная терапия) подходы, такие как подходы с использованием цитозиндезаминазы, тимидинкиназы или бактериального нитроредуктазного фермента, и подходы для повышения переносимости пациентом химиотерапии или радиотерапии, такие как генная терапия множественной лекарственной устойчивости; и
(9) иммунотерапевтические подходы, включая ex-vivo и in-vivo подходы для увеличения иммуногенности опухолевых клеток пациента, такие как трансфекция цитокинами, такими как интерлейкин 2, интерлейкин 4 или гранулоцитарно-макрофагальный колониестимулирующий фактор, подходы по снижению Т-клеточной анэргии, подходы с использованием трансфицированных иммунных клеток, таких как цитокин-трансфицированные дендритные клетки, подходы с использованием цитокин-транисфицированных опухолевых клеточных линий и подходы с использованием анти-идиотидиотипических антител.
Другие комбинированные типы лечения описаны в WO-A 2009/008992, которая включена в данное описание посредством ссылки.
Соответственно, индивидуальные соединения таких комбинаций могут быть введены либо последовательно в отдельных фармацевтических композициях, либо одновременно в объединенных фармацевтических композициях.
Фармацевтические композиции по настоящему изобретению включают композиции, пригодные для перорального, ректального, местного, парентерального введения (включая подкожное, внутримышечное и внутривенное), введения в глаз (офтальмическое), легочного (ингаляция через нос или рот) или назального введения, хотя выбор наиболее приемлемого пути в любом конкретном случае будет зависеть от природы и тяжести подлежащих лечению состояний и от природы активного ингредиента. Они могут быть удобным образом представлены в стандартной лекарственной форме и приготовлены любым из способов, хорошо известных в области фармацевтики.
Для практического применения соединения формулы (I) могут быть объединены в качестве активного ингредиента в гомогенную смесь с фармацевтическим носителем в соответствии с традиционными технологиями приготовления фармацевтических препаратов. Носитель может принимать большое разнообразие форм в зависимости от формы препарата, подлежащего введению, например перорального или парентерального (включая внутривенное). При изготовлении композиций для пероральной лекарственной формы могут быть использованы любые фармацевтические среды, такие как вода, гликоли, масла, спирты, корригенты, консерванты, красители и тому подобное в случае жидких пероральных препаратов, таких как, например, суспензии, эликсиры и растворы; или такие носители, как крахмалы, сахара, микрокристаллическая целлюлоза, разбавители, гранулирующие агенты, смазывающие агенты, связывающие вещества, разрыхлители и тому подобное в случае твердых пероральных препаратов, таких как порошки, твердые и мягкие капсулы и таблетки, причем твердые пероральные препараты являются более предпочтительными, чем жидкие препараты.
В силу легкости их введения, таблетки и капсулы представляют собой наиболее удобную пероральную стандартную лекарственную форму, в которой обычно используют твердые фармацевтические носители. При необходимости на таблетки может быть нанесено покрытие с помощью стандартных водных и неводных технологий. Такие композиции и препараты должны содержать по меньшей мере 0,1 процент активного соединения. Процентное содержание активного соединения в этих композициях может, конечно, меняться и может удобным образом находиться между примерно 2 процентами и примерно 60 процентами от массы стандартной формы. Количество активного соединения в таких терапевтически полезных композициях является таким, чтобы получать эффективную дозировку. Активные соединения также могут вводиться интраназально, например, в виде жидких капель или спрея.
Таблетки, пилюли, капсулы и тому подобное также могут содержать связующий агент в виде трагаканта, аравийской камеди, кукурузного крахмала или желатина; эксципиенты, такие как дикальция фосфат; разрыхлитель, такой как кукурузный крахмал, картофельный крахмал, альгиновая кислота; смазывающее вещество, такое как стеарат магния; и подсластитель, такой как сахароза, лактоза или сахарин. Когда стандартная лекарственная форма представляет собой капсулу, она может содержать, в дополнение к вышеперечисленным материалам, жидкий носитель, такой как жирное масло.
Различные другие материалы могут присутствовать в качестве покрытий или для модифицирования физической формы лекарственной формы. Например, таблетки могут быть покрыты шеллаком, сахаром или ими обоими. Сироп или эликсир могут содержать, в дополнение к активному ингредиенту, сахарозу в качестве подсластителя, метил- и пропилпарабены в качестве консервантов, красители и корригенты, такие как вишневый или апельсиновый ароматизатор.
Соединения формулы (I) также могут быть введены парентерально. Растворы или суспензии этих активных соединений могут быть получены в воде, предпочтительно при смешивании с поверхностно-активным веществом, таким как гидроксипропил-целлюлоза. Дисперсии также могут быть приготовлены в глицерине, жидких полиэтиленгликолях и их смесях в маслах. При обычных условиях хранения и применения эти препараты содержат консерванты для предотвращения роста микроорганизмов.
Фармацевтические формы, подходящие для инъекционного применения, включают стерильные водные растворы или дисперсии и стерильные порошки для немедленного приготовления стерильных растворов или дисперсий для инъекций. Во всех случаях форма должна быть стерильной и должна быть жидкой до такой степени, чтобы она могла быть легко введена посредством шприца. Она должна быть стабильной при условиях изготовления и хранения и должна содержать консерванты для предотвращения заражения микроорганизмами, такими как бактерии и грибы. Носитель может представлять собой растворитель или дисперсионную среду, содержащую, например, воду, этанол, полиол (например, глицерин, пропиленгликоль и жидкий полиэтиленгликоль), их подходящие смеси и растительные масла.
Для обеспечения млекопитающего, в частности человека, эффективной дозой соединения по настоящему изобретению может быть использован любой приемлемый путь введения, например пероральный, ректальный, местный, парентеральный, введение в глаз, в легкое, в нос и тому подобное. Лекарственные формы включают таблетки, пастилки, дисперсии, суспензии, растворы, капсулы, крема, мази, аэрозоли и тому подобное. Предпочтительно соединения формулы (I) вводят перорально.
Эффективные дозировки используемого активного ингредиента могут варьировать в зависимости от конкретного используемого соединения, способа введения, подлежащего лечению состояния и тяжести подлежащего лечению состояния. Такие дозировки могут быть легко установлены специалистом в данной области.
Терапевтически эффективное количество соединения по настоящему изобретению будет обычно зависеть от ряда факторов, включая, например, возраст и массу животного, конкретное требующее лечения состояние и его тяжесть, природу композиции и путь введения. Однако эффективное количество соединения формулы (I) для лечения воспалительного заболевания, например ревматоидного артрита (RA), будет в общем находиться в диапазоне от 0,1 до 100 мг/кг массы тела реципиента (млекопитающее) в сутки и более традиционно в диапазоне от 1 до 10 мг/кг массы тела в сутки. Таким образом, для взрослого млекопитающего массой 70 кг действительное количество в сутки будет составлять от 70 до 700 мг и это количество может быть введено в однократной дозе в сутки или более традиционно в виде ряда (например, двух, трех, четырех, пяти или шести) субдоз в сутки так, что общая суточная доза будет такой же. Эффективное количество их фармацевтически приемлемой соли, пролекарства или метаболита может быть определено как доля эффективного количества соединения формулы (I) самого по себе. Предполагается, что аналогичные дозировки будут приемлемы для лечения других упомянутых выше состояний.
Как он используется в данном описании, термин "эффективное количество" означает количество лекарственного средства или фармацевтического агента, которое будет вызывать биологический или медицинский ответ в ткани, системе, у животного или человека, которое определяется, например, исследователем или клиницистом.
Кроме того, термин "терапевтически эффективное количество" означает любое количество, которое, в сравнении с соответствующим субъектом, который не получил такого количества, имеет результатом улучшенное лечение, излечение, предупреждение или облегчение заболевания, расстройства или побочного эффекта, либо снижение степени развития заболевания или расстройства. Термин также включает в свой объем количества, эффективные для усиления нормального физиологического функционирования.
Другой аспект настоящего изобретения составляет соединение по настоящему изобретению или его фармацевтически приемлемая соль для применения в качестве лекарственного средства.
Другой аспект настоящего изобретения составляет соединение по настоящему изобретению или его фармацевтически приемлемая соль для применения в способе лечения или предупреждения заболевания или расстройства, ассоциированного с mTOR.
В контексте настоящего изобретения заболевание или расстройство, ассоциированное с mTOR, определяют как заболевание или расстройство, в которое вовлечена mTOR.
Связь между киназой mTOR и иммунологическими заболеваниями демонстрируется тем фактом, что FDA одобрило в 1997 году ингибитор mTOR рапамицин (Sirolimus®) в качестве лекарственного средства для предупреждения отторжения трансплантатов почки (Tsang et al., 2007, Drug Discovery Today 12, 112-124). Рапамицин блокирует интерлейкин 2 (IL-2)-опосредованную Т-клеточную пролиферацию и активацию. Поэтому ингибиторы mTOR могут быть полезны для лечения других иммунологических, воспалительных, аутоиммунных и аллергических заболеваний, в которых играют роль Τ клетки, например ревматоидного артрита (RA), воспалительного заболевания кишечника (IBD; болезнь Крона (Crohns's) и неспецифический язвенный колит), псориаза, системной красной волчанки (SLE) и рассеянного склероза (MS).
Кроме того, FDA одобрило в 2003 году рапамицин в качестве противорестенозного лекарственного средства, используемого в коронарно-артериальных стентах, поскольку рапамицин является мощным ингибитором пролиферации гладкомышечных клеток сосудов (Tsang et al., 2007, Drug Discovery Today 12, 112-124). Таким образом, ингибиторы mTOR могут быть полезны для лечения других заболеваний, в которых играет роль избыточная клеточная пролиферация.
Аналоги рапамицина (рапалоги) темсиролимус и эверолимус одобрены для применения при развившейся почечно-клеточной карциноме, демонстрируя полезность ингибирования mTOR метаболического пути при лечении рака (Richard et al., 2011. Current Opinion Drug Discovery and Development 13(4):428-440).
Хотя соединения по настоящему изобретению действуют как ингибиторы киназы mTOR и не обладают тем же типом действия, что и рапамицин, можно ожидать, что этот класс ингибиторов mTOR будет полезен по тем же показаниям, что и рапамицин, и по дополнительным показаниям, как описано ниже.
В предпочтительном воплощении заболевание или расстройство, ассоциированное с mTOR, представляет собой иммунологическое, воспалительное, аутоиммунное или аллергическое расстройство или заболевание, или отторжение трансплантата, или заболевание «трансплантат-против-хозяина».
Соответственно, другой аспект настоящего изобретения составляет соединение или его фармацевтически приемлемая соль по настоящему изобретению для применения в способе лечения или предупреждения иммунологического, воспалительного, аутоиммунного или аллергического расстройства или заболевания, или отторжения трансплантата, или заболевания «трансплантат-против-хозяина».
Согласно настоящему изобретению, аутоиммунное заболевание представляет собой заболевание, которое по меньшей мере частично провоцируется иммунной реакцией организма против своих собственных компонентов, например белков, липидов или ДНК.
В предпочтительном воплощении аутоиммунное заболевание выбрано из группы, состоящей из ревматоидного артрита (RA), воспалительного заболевания кишечника (IBD; болезнь Крона (Crohns's) и неспецифический язвенный колит), псориаза, системной красной волчанки (SLE) и рассеянного склероза (MS).
Ревматоидный артрит (RA) представляет собой хроническое прогрессирующее изнуряющее воспалительное заболевание, которое поражает приблизительно 1% населения в мире. RA представляет собой симметричный многосуставный артрит, который сначала поражает малые суставы рук и ног. В дополнение к воспалению в синовиальной оболочке, выстилке суставов, агрессивная поверхность ткани, так называемый поверхностный диффузный сосудистый кератит, вторгается в локальные суставные структуры и разрушает их (Firestein 2003, Nature 423:356-361).
Воспалительное заболевание кишечника (IBD) характеризуется хроническим рецидивирующим воспалением кишечника. IBD подразделяется на фенотипы болезни Крона и неспецифического язвенного колита. Болезнь Крона охватывает наиболее часто концевую часть подвздошной кишки и толстую кишку, является трансмуральной и прерывистой. При язвенном колите, напротив, воспаление является непрерывным и ограничено слоями слизистой оболочки прямой и толстой кишки. В приблизительно 10% случаев, ограниченных прямой и толстой кишкой, определенная классификация заболевания как болезни Крона или язвенного колита не может быть осуществлена и их называют 'неопределенным колитом'. Оба заболевания включают внекишечное воспаление кожи, глаз или суставов. Нейтрофил-индуцированные поражения могут быть предотвращены посредством применения ингибиторов миграции нейтрофилов (Asakura et al., 2007, World J Gastroenterol. 13(15):2145-9).
Псориаз - это хронический воспалительный дерматоз, который поражает приблизительно 2% населения. Он характеризуется красными, чешуйчатыми бляшками на коже, которые обычно обнаруживаются на волосистой части головы, локтях и коленях, и могут быть ассоциированы с тяжелым артритом. Поражения вызываются ненормальной пролиферацией кератиноцитов и инфильтрацией воспалительных клеток в дермис и эпидермис (Schön et al., 2005, New Engl. J. Med. 352:1899-1912).
Системная красная волчанка (SLE) представляет собой хроническое воспалительное заболевание, вызываемое опосредованной Т-клетками В-клеточной активацией, что приводит к гломерулонефриту и почечной недостаточности. SLE у людей характеризуется на ранних стадиях экспансией долгоживущих аутореактивных CD4+ клеток памяти (D'Cruz et al., 2007, Lancet 369(9561):587-596).
Рассеянный склероз (MS) представляет собой воспалительное и демиелинизирующее заболевание. Его считали аутоиммунным расстройством, опосредованным Τ хелперными CD4+ клетками типа 1, но недавние исследования выявили участие других иммунных клеток (Hemmer et al., 2002, Nat. Rev. Neuroscience 3, 291-301).
Заболевание «трансплантат-против-хозяина» (GVDH) является основным осложнением при трансплантации аллогенного костного мозга. GVDH вызывается донорными Τ клетками, которые распознают и реагируют на отличия реципиента в комплексной системе гистосовместимости, приводя к значительной заболеваемости и смертности.
Отторжение трансплантатов (отторжение аллотрансплантатов) включает, без ограничения ими, острое и хроническое отторжение аллотрансплантата после, например, трансплантатции почки, сердца, печени, легкого, костного мозга, кожи и роговицы. Известно, что Τ клетки играют центральную роль в специфическом иммунном ответе при оттржении аллотрансплантата.
В другом предпочтительном воплощении заболевание или расстройство, ассоциированное с mTOR, представляет собой пролиферативное заболевание, в частности рак.
Заболевания и расстройства, ассоциированные главным образом с mTOR, представляют собой пролиферативные расстройства или заболевания, в частности рак.
Таким образом, другой аспект настоящего изобретения составляют соединение или его фармацевтически приемлемая соль по настоящему изобретению для применения в способе лечения или предупреждения пролиферативного заболевания, в частности рака.
Рак включает группу заболеваний, характризующихся неконтролируемым ростом и распространением ненормальных клеток. Все типы рака в общем включают определенную ненормальность в контроле клеточного роста, деления и выживания, ведущую к злокачественному росту клеток. Ключевыми факторами, вносящими вклад в указанный злокачественный рост клеток, являются независимость от ростовых сигналов, нечувствительность к антиростовым сигналам, усиление апоптоза, неограниченный репликативный потенциал, продленный ангиогенез, тканевая инвазия и метастазирование и нестабильность генома (Hanahan and Weinberg, 2000. The Hallmarks of Cancer. Cell 100, 57-70).
Как правило, рак классифицируют как гематологический рак (например лейкемии и лимфомы) и солидный рак, такой как саркомы и карциномы (например рак головного мозга, молочной железы, легкого, прямой кишки, желудка, печени, поджелудочной железы, простаты, яичников).
Ожидают, что особенно чувствительным к лечению ингибиторами mTOR будет рак, при котором активирован путь передачи сигнала PI3K/Akt, например, из-за дезактивации опухолевого супрессора PTEN или активирующих мутаций в PIK3A, гене, кодирующем каталитическую субъединицу р110α (p110alpha) фосфоинозитид-3 киназы (Garcia-Echeverria and Sellers, 2008, Oncogene 27, 5511-5526). Примеры рака с высокой долей PTEN мутаций и/или активации PI3K/Akt являются эндометриальная карцинома, глиобластома, рак головы и шеи, рак прямой кишки, рак поджелудочной железы, рак желудка, гепатокарцинома, рак яичника, рак щитовидной железы, почечно-клеточный рак, рак молочной железы, рак простаты и стромальные опухоли желудочно-кишечного тракта (GIST). Наиболее обнадеживающие результаты для ингибиторов mTOR были получены на почечно-клеточной карциноме (RCC), лимфоме мантийных клеток и эндометриальном раке (Faivre et al., 2006. Nat. Rev. Drug. Discov. 5(8):671-688). Кроме того, ингибиторы mTOR могут быть полезны для лечения лейкемий, включая острую миелоидную лейкемию (AML) и хроническую миелоидную лейкемию (CML), множественную миелому и лимфомы.
Кроме того, лечению ингибиторами mTOR может быть подвергнут рак, несущий активирующие mTOR мутации, например одиночные аминокислотные замены, которые придают конституитивную активацию mTOR, такие как S2215Y или R2505P (Sato et al., 2010, Oncogene 29(18):2746-2752).
mTOR играет важную роль в ангиогенезе, образовании новых кровеносных сосудов с доставкой кислорода и питательных веществ к растущим и делящимся клеткам. В этом контексте mTOR контролирует продуцирование белков HIF1-α и HIFI-β, которые представляют собой субъединицы индуцируемого гипоксией фактора (HIF), фактора транскрипции, который контролирует экспрессию генов, продукты которых играют роль в ангиогенезе, клеточной проилиферации, подвижности и выживании. Двумя важными белками, индуцируемыми HIF, являются факторы роста эндотелия сосудов (VEGFs) и ангиопоэтин-2. В недавнем времени появились сообщения о том, что низкомолекулярный ингибитор mTOR может подавлять опухолевый рост, опухолевый ангиогенез, проницаемость сосудов (Xue et al., 2008. Cancer Research 68(22): 9551-9557).
В дополнение к опухолегензу имеются свидетельства о том, что mTOR играет роль в синдромах гамартомы. Недавние исследования показали, что опухолевые супрессорные белки, такие как TSC1, TSC2, PTEN и LKB1, плотно контролируют передачу сигнала mTOR. Потеря этих опухолевых супрессорных белков приводит к ряду гамартомных состояний как результату усиленной передачи сигнала mTOR (Rosner et al., 2008. Mutation Research 659(3):284-292). Синдромы с установленной молекулярной связью с нарушением регулирования mTOR включают синдром Пейтса-Егерца (Peutz-Jeghers (PJS)), болезнь Коудена (Cowden), синдром Баньяна-Райли-Рувалкаба (Bannayan-Riley-Ruvalcaba (BRRS)), синдром Протеуса (Proteus), болезнь Лермитт-Дуклос (Lhermitte-Duclos) и туберозный склероз (TSC). У пациентов с этими синдромами характерным образом развиваются доброкачественные гамартоматозные опухоли в разнообразных органах. Другими опухолевыми супрессорными белками, оказывающими воздействие на активность mTOR, являются VHL (болезнь Гиппеля-Ландау), NF1 (нейрофиброматоз) и PKD (поликистозное заболевание почек), чья потеря может запускать болезнь Гиппеля-Ландау (Hippel-Lindau), нейрофиброматоз типа 1 и поликистозную болезнь почек, соответственно.
Пролиферативные заболевания или расстройства включают группу заболеваний, характеризующихся усиленным размножением клеток. Одним из примеров является рестеноз, вызываемый избыточным ростом гладкомышечных клеток сосудов (VSM) после коронарной ангиопластики стентами. Для нарушения этого явления были разработаны стенты, элюирующие лекарственное средство, для ингибирования роста VSM клеток. Покрытые рапамицином стенты эффективно снижают рестеноз и были одобрены FDA (Serruys et al., 2006. N. Engl. J. Med. 354(5):483-95).
В еще одном предпочтительном воплощении заболевание или расстройство, ассоциированное с mTOR, представляет собой сердечнососудистое заболевание, метаболическое заболевание или нейродегенеративное заболевание.
Таким образом, другой аспект настоящего изобретения составляет соединение или его фармацевтически приемлемая соль по настоящему изобретению для применения в способе лечения или предупреждения сердечно-сосудистого заболевания, метаболического заболевания или нейроденеративного заболевания.
Недавние исследования выявили роль mTOR в сердечно-сосудистых заболеваниях, например, повышенная активность mTOR киназы ассоциирована с гипертрофией сердца (расширение сердца), которое является основным фактором риска для развития сердечной недостаточности. На клеточном уровне гипертрофия сердца характеризуется увеличение размеров клеток и усиленным белковым синтезом. Хотя имеются различные гипертрофические стимулы, такие как нейрогормоны и пептидные факторы роста, и некоторые протеинкиназные каскады вовлечены в сердечную гипертрофию, очевидно, что все формы гипертрофических стимулов активируют общий белковый трансляционный механизм mTOR-зависимым образом. Примечательно, что ингибирование mTOR рапамицином предупреждает гипертрофию сердца в многочисленных моделях трансгенных мышей. Кроме того, индуцированная стрессом сердечная гипертрофия является у мышей mTOR-зависимой. Эти результаты свидетельствуют о том, что mTOR является ключевой для аномального сердечного сверхроста, и что ингибиторы mTOR могут быть полезными для лечения гипертрофии сердца у людей (Tsang et al., 2007, Drug Discovery Today 12, 112-124).
Метаболические заболевания, которые могут лечиться ингибиторами mTOR, включают диабет типа 1, диабет типа 2 и ожирение (Tsang et al., 2007, Drug Discovery Today 12, 112-124). Диабет типа 1 вызывается потерей продуцирования инсулина из-за деструкции панкреатических β-клеток. Клинические исследования с использованием иммуносуппрессивного режима, который включает рапамицин, для предупреждения отторжения трансплантантов островков выявили значительную эффективность у пациентов с диабетом типа 1. Диабет типа 2 возникает, когда секреция из панкреатических β-клеток не может компенсировать периферическую резистентность к инсулину (или нечуствительность к инсулину) в скелетных мышцах, печени и жировых клетках. Последние данные свидетельствуют о том, что длительная активация mTOR передачи сигнала является ключевым событием, которое делает субстрат инсулиновых рецепторов (IRS) нечувствительным к инсулину. Более того, было продемонстрировано, что рапамицин восстанавливает чувствительность IRS к инсулину (Shah et al., 2004. Curr. Biol. 14(18):1650-1656). Таким образом, ингибиторы mTOR являются потенциально полезными в регулировании диабета типа 2. Ожирение представляет собой меаболическое заболевание с монотонно возрастающим риском для здоровья во всем мире. Последние открытия подтверждают, что mTOR играет определенную роль в липидном метаболизме. При адипогенезе экспрессия mTOR драматически возрастает от едва обнаружимых уровней в преадипоцитах до высокой экспрессии в полностью дифференцированных адипоцитах, и рапамицин ингибирует дифференциацию адипоцитов (Yen et al., 1995. Proc. Natl. Acad. Sci. USA. 92(24): 11086-90).
Последние сообщения свидетельствуют о том, что ингибиторы mTOR могут быть полезными для лечения нейродегенеративных заболеваний, таких как болезнь Гентингтона (Huntingtons's), Альцгеймера (Alzheimer's) и Паркинсона (Parkinson's). Болезнь Гентингтона представляет собой нейроденеративное расстройство, вызываемое мутантной формой белка гентингтина с аномально длинными глутаминовыми повторами на амино-конце. Мутантный белок агрегируется в нейрональных клетках и может вызывать повреждение нервных клеток и токсичность. Рапамицин ослабляет аккумулирование гентингтина и клеточную гибель и защищает против нейродегенерации в животных моделях болезни Гентингтона (Ravikumar et al., 2004. Nat Genet. 36(6):585-95). Кроме того, рапамицин индуцирует аутофаговый ответ, который, как было высказано предположение, играет роль в клиренсе гентингтиновых агрегатов.
Внутриклеточные белковые агрегаты также имеют место при других нейродегенеративных заболеваниях, например болезни Альцгеймера. Тау белок часто обнаруживают в головном мозге пациентов с болезнью Альцгеймера и он, как полагают, вносит вклад в образование нейрофибриллярных клубков (например, при таутопатиях, таких как фронто-темпоральная деменция). В мушиной модели рапамицин снижает концентрацию тау белка и снижает токсичность, вызванную тау-аккумуляцией (Berger et al., 2006. Hum Mol Genet. 2006 Feb 1; 15(3):433-42). Поэтому ингибиторы mTOR могут быть полезными в предупреждении аккумулирования токсического тау белка у пациентов с болезнью Альцгеймера.
Болезнь Паркинсона (PD) представляет собой нейродегенеративное заболевание, ассоциированное с аккумулированием и агрегацией неправильно упакованных белков. Предупреждение агрегации и дезагрегация неправильно упакованных белков может обеспечить терапевтическую пользу путем замедления или предупреждения прогрессирования PD. Убиквитин-протеасомная система (UPS) является важным механизмом деградации, воздействующим на агрегированные белки. Сообщают, что рапамицин обеспечивает нейропротекцию против гибели нейрональных допаминэргических клеток, индуцированной ингибитором протеасом лактацистином. Предполагают, что эффект рапамицина частично опосредован усилением аутофагии посредством усиленной деградации неправильно упакованных белков (Pan et al., 2008. Neurobiol. Dis. 32(1): 16-25). Поэтому соединения, которые могут усиливать аутофагию, могут представлять собой многообещающую стратегию для лечения PD пациентов.
В дополнительном предпочтительном воплощении заболевание или расстройство, ассоциированное с mTOR, представляет собой заболевание, ассоциированное с аутофагией.
Таким образом, другой аспект настоящего изобретения составляет соединение или его фармацевтически приемлемая соль по настоящему изобретению для применения в способе лечения или предупрежедения заболевания, ассоциированного с аутофагией.
Аутофагия представляет собой лизосома-зависимый процесс, посредством которого белки или поврежденные органеллы подвергаются деградации в клетке (Mizushima et al., 2008. Nature 451 (7182): 1069-75). В ходе этого процесса аутофагосома с двойной мембраной захватывает компонент клетки, который должен быть подвергнут деградации. Затем аутофагосома сливается с лизосомой, которая, например, подвергает деградации белки, что приводит к рециклингу аминокислот. Аутофагия первично вовлечена в деградацию долгоживующих белков, белковых агрегатов и клеточных органелл и других клеточных компонентов. В дополнение к ее физиологической функции аутофагия могла бы предполагаться для лечения ряда заболеваний, вызываемых агрегатами неправильно упакованных белков, например нейродегенеративных заболеваний, таких как болезнь Гентингтона, Альцгеймера или Паркинсона. Другие ассоциированные с аутофагией заболевания раскрыты в WO-A 2009/049242, включенной в данное описание посредством ссылки.
Термин «соединение, индуцирующее аутофагию» относится к соединению, которое индуцирует аутофагию в клетке. Термин «заболевание, ассоциированное с аутофагией» относится к заболеванию, которое можно лечить посредством индуцирования аутофагии. Недавно было показано, что АТР-конкурентный ингибитор mTOR киназы может индуцировать аутофагию (Thoreen et al., 2009. J. Biol. Chem. 284(12):8023-32). Интересно, что ATP (аденозинтрифосфат)-конкурентные ингибиторы mTOR киназы, по-видимому, индуцируют аутофагию в клетках млекопитающих более эффективно, чем рапамицин. Взятые в совокупности, соединения по настоящему изобретению могут быть полезными для индуцирования аутофагии в клетках и для лечения заболеваний, ассоциированных с аутофагией.
В дополнительном предпочтительном воплощении заболевание или расстройство представляет собой вирусную инфекцию.
Таким образом, другой аспект настоящего изобретения составляет соединение или его фармацевтически приемлемая соль по настоящему изобретению для применения в способе лечения или предупреждения вирусной инфекции.
Все вирусы нуждаются в клеточных рибосомах для трансляции их мРНК. Например, было показано, что инфекция цитомегаловирусом человека (HCMV) активирует метаболический путь передачи сигнала mTORC1. Обработка инфицированных клеток Torin1, ингибитором mTOR, который нацелен на каталитический сайт mTOR киназы, блокирует продуцирование вирусного потомства. Кроме того, показано, что Torin1 ингибирует репликацию репрезентативных членов семейств альфа-, бета- и гамма-герпесвирусов, демонстрируя потенциал ингибиторов киназы mTOR как противовирусных агентов широкого спектра действия (Moorman and Shenk, 2010. J. Virol. 84(10):5260-9). Другие вирусные инфекции, которые можно лечить или предупреждать ингибиторами mTOR, описаны в WO-A 2011/011716, включенной в данное описание посредством ссылки.
Еще один аспект настоящего изобретения составляет применение соединения по настоящему изобретению или его фармацевтически приемлемой соли для изготовления лекарственного средства для лечения или профилактики заболеваний и расстройств, ассоциированных с mTOR.
Еще один аспект настоящего изобретения составляет применение соединения по настоящему изобретению или его фармацевтически приемлемой соли для изготовления лекарственного средства для лечения или предупреждения иммунологического, воспалительного, аутоиммунного или аллергического расстройства или заболевания, или отторжения трансплантата, или болезни «трансплантат-против-хозяина».
Еще один аспект настоящего изобретения составляет применение соединения по настоящему изобретению или его фармацевтически приемлемой соли для изготовления лекарственного средства для лечения или предупреждения пролиферативного заболевания, в особенности рака.
Еще один аспект настоящего изобретения составляет применение соединения по настоящему изобретению или его фармацевтически приемлемой соли для изготовления лекарственного средства для лечения или предупреждения сердечно-сосудистого заболевания, метаболического заболевания или нейродегенеративного заболевания.
Еще один аспект настоящего изобретения составляет применение соединения по настоящему изобретению или его фармацевтически приемлемой соли для изготовления лекарственного средства для лечения или предупреждения заболевания, ассоциированного с аутофагией.
Еще один аспект настоящего изобретения составляет применение соединения по настоящему изобретению или его фармацевтически приемлемой соли для изготовления лекарственного средства для лечения или предупреждения вирусной инфекции.
В контексте этих применений по изобретению заболевания и расстройства, ассоциированные mTOR, являются такими, как определено выше.
Еще один аспект настоящего изобретения составляет способ лечения, контролирования, замедления или предупреждения у нуждающего в этом пациента - млекопитающего одного или более состояний, выбранных из группы, состоящей из заболеваний и расстройств, ассоциированных с mTOR, который включает введение указанному пациенту терапевтически эффективного количества соединения по настоящему изобретению или его фармацевтически приемлемой соли.
Еще один аспект настоящего изобретения составляет способ лечения, контролирования, замедления или предупреждения у нуждающего в этом пациента - млекопитающего одного или более состояний, выбранных из группы, состоящей из иммунологического, воспалительного, аутоиммунного или аллергического расстройства или заболевания, или отторжения рансплантата, или болезни «трансплантат-против-хозяина», который включает введение указанному пациенту терапевтически эффективного количества соединения по настоящему изобретению или его фармацевтически приемлемой соли.
Еще один аспект настоящего изобретения составляет способ лечения, контролирования, замедления, или предупреждения у нуждающего в этом пациента - млекопитающего пролиферативного заболевания, в частности рака, который включает введение указанному пациенту терапевтически эффективного количества соединения по настоящему изобретению или его фармацевтически приемлемой соли.
Еще один аспект настоящего изобретения составляет способ лечения, контролирования, замедления или предупреждения у нуждающего в этом пациента - млекопитающего одного или более состояний, выбранных из группы, состоящей из сердечно-сосудистого заболевания, метаболического заболевания или нейродегенеративного заболевания, который включает введение указанному пациенту терапевтически эффективного количества соединения по настоящему изобретению или его фармацевтически приемлемой соли.
Еще один аспект настоящего изобретения составляет способ лечения, контролирования, замедления или предупреждения у нуждающего в этом пациента - млекопитающего ассоциированного с аутофагией заболевания, который включает введение указанному пациенту терапевтически эффективного количества соединения по настоящему изобретению или его фармацевтически приемлемой соли.
Еще один аспект настоящего изобретения составляет способ лечения, контролирования, замедления или предупреждения у нуждающего в этом пациента - млекопитающего вирусной инфекции, который включает введение указанному пациенту терапевтически эффективного количества соединения по настоящему изобретению или его фармацевтически приемлемой соли.
В контексте этих способов по изобретению заболевания и расстройства, ассоциированные с mTOR, являются такими, как определено выше.
Как он используется в данном описании, термин "лечить" или "лечение" относится ко всем процессам, где может иметь место замедление, прерывание, приостановка или остановка прогрессирования заболевания, но не обязательно указывает на общее элиминирование всех симптомов.
Все обсуждаемые выше воплощения в отношении фармацевтической композиции по изобретению также применимы к вышеуказанным первому или второму медицинским применениям или способам по изобретению.
Варианты путей получения соединений по настоящему изобретению описаны ниже. Для специалиста-практика в области техники понятно, как комбинировать или корректировать такие пути, особенно в комбинации с введением активирующих или защитных химических групп.
Общий путь относится к способу получения соединения по настоящему изобретению, включающему стадию
- взаимодействия соединения формулы (II)
где Ra, Rb, R1, o, m имеют значения, как указано выше, с соединением формулы Т1-Х, где Т1 имеет значение, как указано выше, и X представляет собой подходящую группу для реакции Сузуки, такую как 4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил, с получением соединения формулы (I).
В качестве примеров приведены следующие Схемы. Для специалиста-практика в данной области понятно, как соответствующим образом варьровать реакционные условия, особенно для введения стадий активации функциональных групп и/или для защиты функциональных групп.
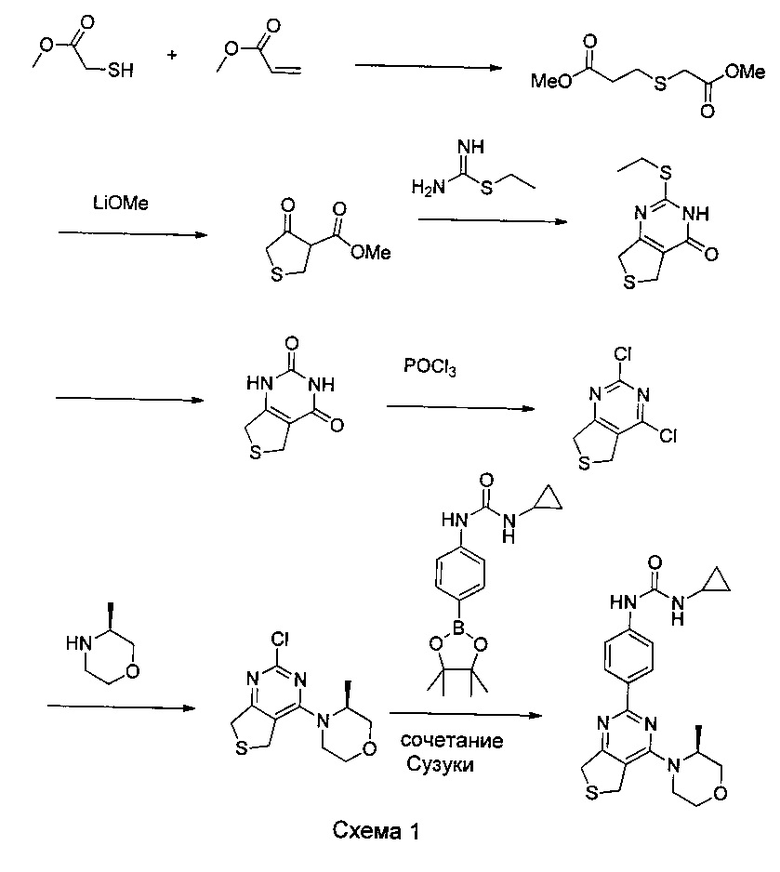
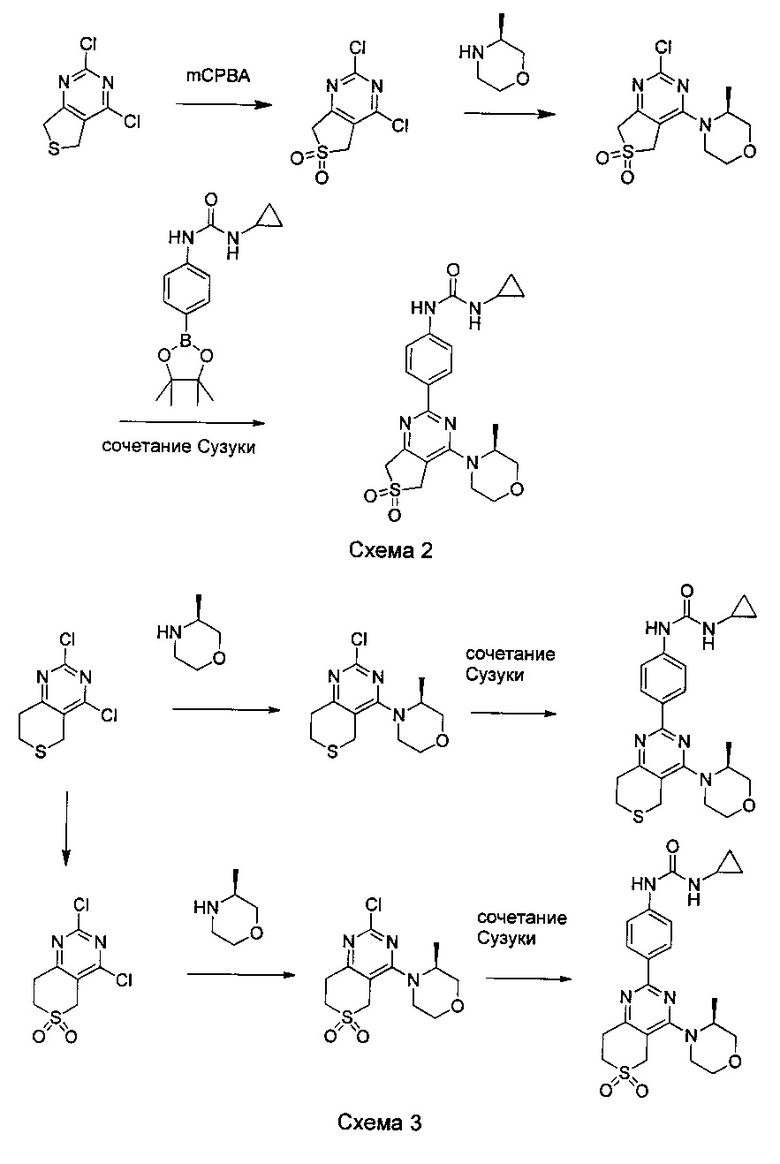
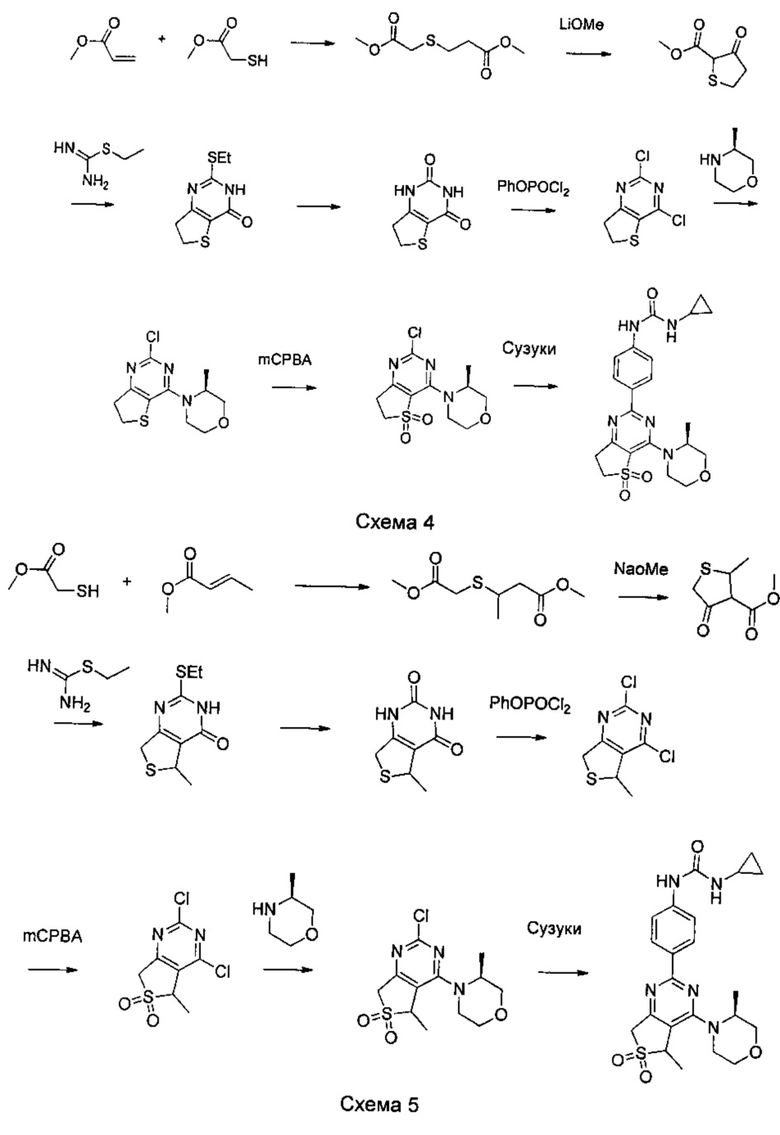
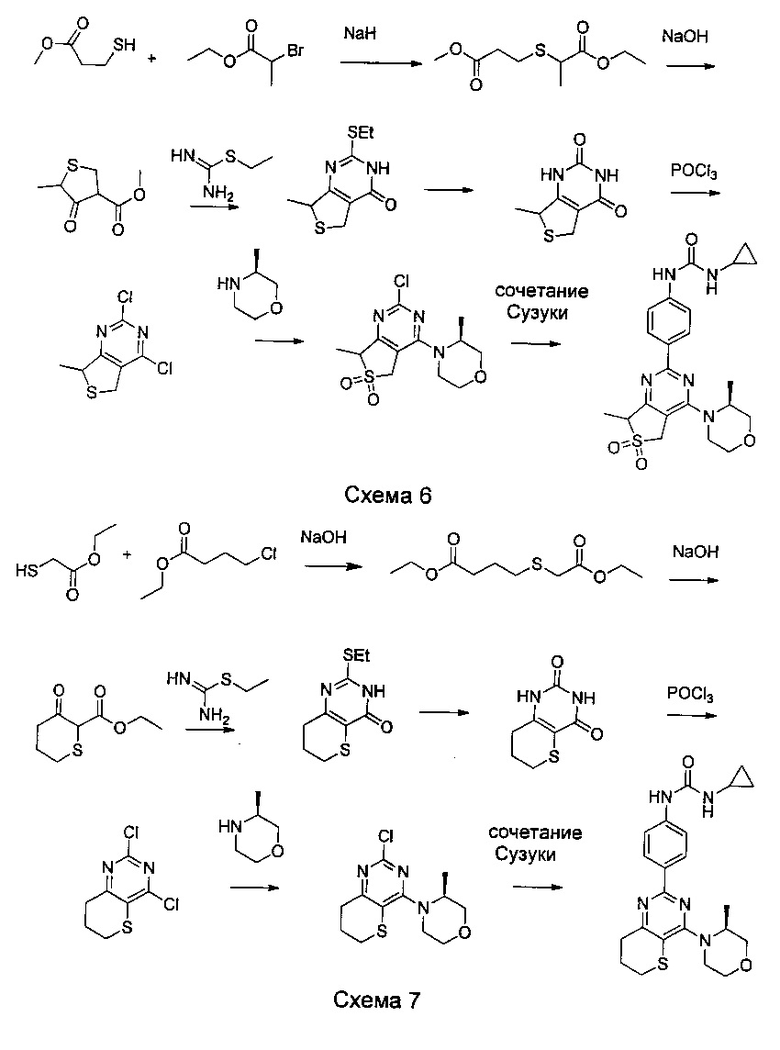
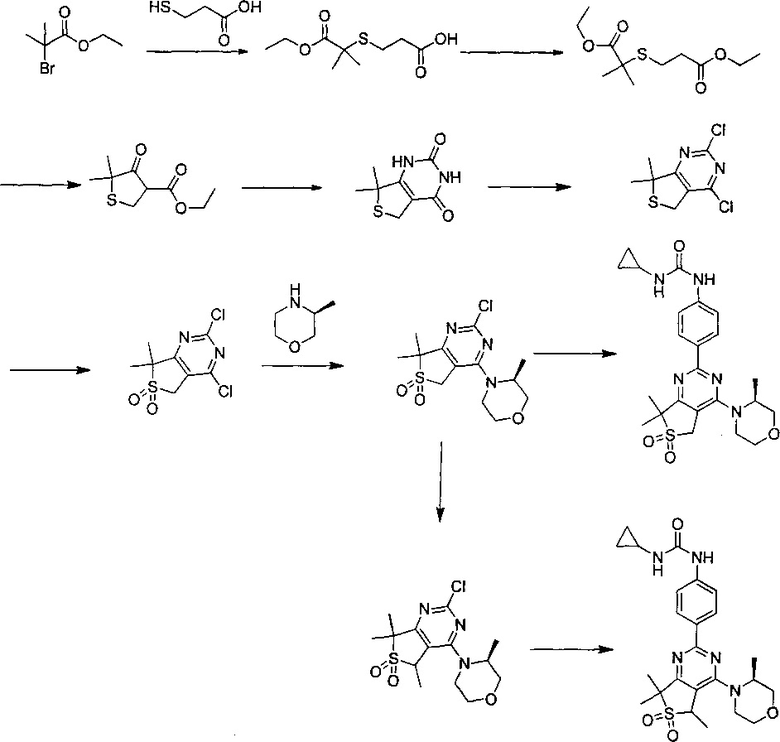
Примеры
Материалы и методы
ЯМР
1Н ЯМР спектры снимали при 400 МГц, используя спектрометр Bruker AVANCE 400 МГц.
Оборудование и условия для ЖХ-МС
ЖХ-МС метод А
Анализ проводили на приборе серии Agilent Technologies 1200 с бинарным насосом.
Колонка: Waters xTerra, 3,5 мкм, 2,1×50 мм
Растворители: А = вода + 0,07% муравьиная кислота
В = метанол
Скорость потока: 0,4 мл/мин
Температура: 25°С
Длина волны: PDA детектирование в диапазоне 200-400 нм
Масс-спектральные условия: G6110A, Quadrupole LC. Масс-спектральные данные собирали в позитивной моде, сканируя в отношении масс между 50 и 900 а.е.м. (атомная единица массы).
ЖХ/МС метод В
Анализ проводили на системе Waters - ZQ, используя следующие условия.
Колонка: Phenomenex Gemini NX С18 30×3 мм, 3 мкм
Растворители:
А = вода + 0,1% муравьиная кислота
В = (95% ацетонитрил: 5% вода) + 0,1% муравьиная кислота
Скорость потока: 1,5 мл/мин
Температура: комнатная температура
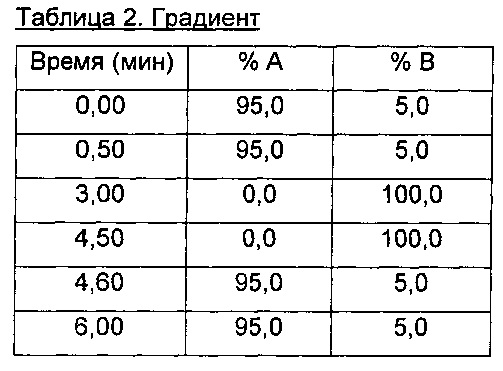
Длина волны: PDA детектирование в диапазоне 200-400 нм
Масс-спектральные условия: масс-спектральные данные собирали в позитивной и негативной моде, сканируя в отношении масс между 150 и 700 а.е.м., используя напряжение на конусе 20 В.
ЖХ-МС метод С
Анализ проводили на приборе серии Agilent Technologies 1200 с бинарным насосом.
Колонка: Venusil ХВР-С18 2,1×50 мм, 5 мкм
Растворители: А = вода + 0,04% TFA
В = ацетонитрил + 0,02% TFA
Скорость потока: 0,6 мл/мин
Температура: 40°C
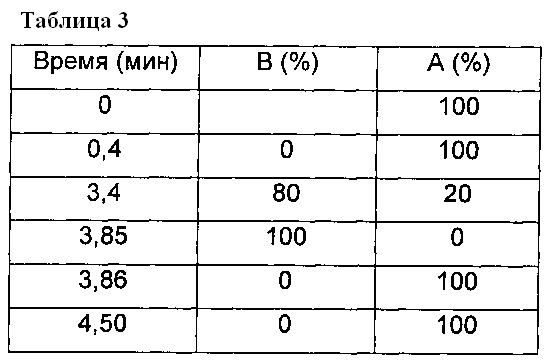
Длина волны: PDA детектирование в диапазоне 200-400 нм
Масс-спектральные условия: G6110A, Quadrupole LC. Масс-спектальные данные собирали в позитивной моде, сканируя в отношении масс между 100 и 1000 а.е.м.
ЖХ-МС метод D
Анализ проводили на приборе серии Agilent Technologies 1200 с бинарным насосом.
Колонка: Venusil ХВР-С18 2,1x50 мм, 5 мкм
Растворители: А = вода+0,04% TFA
В = ацетонитрил + 0,02% TFA
Скорость потока: 0,8 мл/мин
Температура: 40°С

Длина волны: PDA детектирование в диапазоне 200-400 нм
Масс-спектральные условия: масс-спектральные данные собирали в позитивной и негативной моде, сканируя в отношении масс между 150 и 700 а.е.м., используя напряжение на конусе 20 В.
ЖХ-МС метод Ε
Анализ проводили на приборе серии Agilent Technologies 1200 с бинарным насосом.
Колонка: Waters Acquity UPLC ВЕН С18, 30×2,1 мм, 1,7 мкм
Растворители: А = вода + 0,1% муравьиная кислота
В = ацетонитрил + 0,1% муравьиная кислота
Скорость потока: 0,5 мл/мин
Температура: 40°C
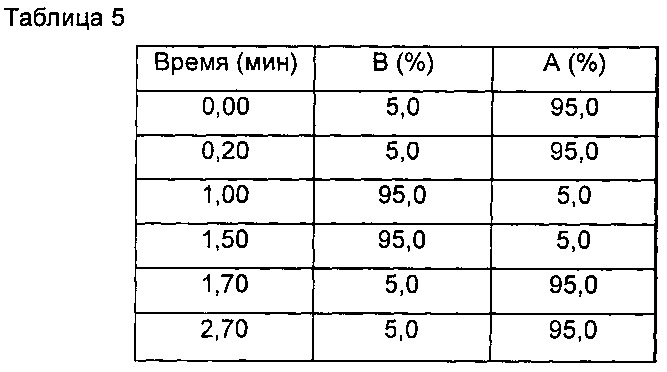
Длина волны: PDA детектирование в диапазоне 210-400 нм
Масс-спектральные условия: масс-спектральные данные собирали в позитивной и негативной моде, сканируя в отношении масс между 150 и 1000 а.е.м., используя напряжение на конусе 25 В.
ЖХ-МС метод F
Анализ проводили на приборе серии Agilent Technologies 1200 с бинарным наосом.
Колонка: Waters Acquity UPLC ВЕН С18, 50×2,1 мм, 1,7 мкм
Растворители: А = вода + 0,1% муравьиная кислота
В = ацетонитрил + 0,1% муравьиная кислота
Скорость потока: 0,5 мл/мин
Температура: 40°C
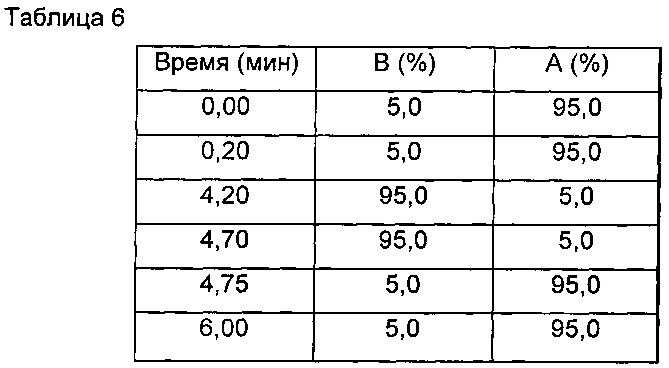
Длина волны: PDA детектирование в диапазоне 210-400 нм
Масс-спектральные условия: масс-спектральные данные собирали в позитивной и негативной моде, сканируя в отношении масс между 150 и 1000 а.е.м., используя напряжение на конусе 25 В.
ЖХ-МС метод G:
Анализ проводили на бинарном насосе Shimadzu Technologies LC-20AD.
Колонка: Halogen-C18 2,1×30 мм, 2,7 мкм
Растворители: А = 4 л вода (0,04% TFA)
В = 4 л ацетонитрил (0,02% TFA)
Скорость потока: 1,0 мл/мин
Температура: 40°C
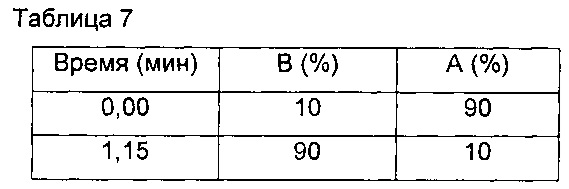
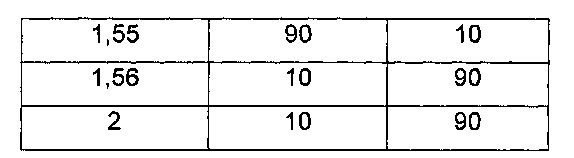
Длина волны: PDA детектирование в диапазоне 200-400 нм
Масс-спектральные условия: LCMC-2010EV, Quadrupole LC. Масс-спектральные данные собирали в позитивной моде, сканируя в отношении масс между 100 и 1000 а.е.м.

Пример 1:
(S)-1-циклопропил-3-(4-(4-(3-метилморфолино)-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевина
Стадия (1)
Метилакрилат (99,2 мл, 1,1 ммоль) медленно добавляли к раствору метилтиогликолята (91 мл, 1,0 ммоль) и пиперидина (2,0 мл, 0,02 моль), поддерживания температуру реакционной смеси равной 50°C. Реакционную смесь перемешивали в течение 2 ч, затем избыток метилакрилата и пиперидина отгоняли под высоким вакуумом с получением целевого продукта, метил-3-((метоксикарбонил)метилтио)пропаноата (185 г, 96%).
ЖХ-МС (метод А): (ЭР (электрораспыление)+) 193, RT=4,29 мин.
Стадия (2)
Металлический литий (2,12 г, 0,30 моль) в толуоле (250 мл) при к.т.обрабатывали метанолом (80 мл). После того, как весь литий растворился, в течение 0,5 ч при 70°C добавляли метил-3-((метоксикарбонил)метилтио)-пропаноат. Температуру реакционной смеси затем поднимали (метанол отгонялся) до конечной температуры 110°C и эту температуру поддерживали в течение 18 ч. Полученная смесь содержала целевой продукт и метил-3-оксо-тетрагидротиофен-2-карбоксилат в виде смеси 1:1. Реакционную смесь охлаждали и твердое вещество собирали фильтрованием с получением обогащенного твердого вещества, содержавшего литиевую соль целевого продукта. Твердое вещество подкисляли с помощью 1 н. раствора HCl и экстрагировали DCM. Органическую фазу концентрировали и остаток очищали колоночной хроматографией, элюируя EtoAc в петролейном эфире (от 2% до 10%) для удаления всего остаточного метил-3-оксо-тетрагидротиофен-2-карбоксилата. Метил-тетрагидро-4-оксотиофен-3-карбоксилат (14,7 г, 33%) получали как вторую элюируемую фракцию из колонки.
ЖХ-МС (метод А), (ЭР+) 161, RT=4,92 мин
Стадия (3)
К раствору бромида S-этил-изотиоурония (3,0 г, 16,2 ммоль) в воде (60 мл) по каплям добавляли карбонат натрия (1,71 г, 16,2 ммоль) и метил-тетрагидро-4-оксотиофен-3-карбоксилат (2,51 г, 16,2 ммоль). Смесь перемешивали в течение ночи при к.т. в темноте. Твердое вещество, выпавшее в осадок в полученной смеси, собирали фильтрованием, промывали водой, диэтиловым эфиром, метанолом и ацетоном, затем твердое вещество сушили под вакуумом с получением 2-(этилтио)тиено[3,4-d]пиримидин-4-(3Н,5Н,7Н)-она в виде белого твердого вещества (2,88 г, 83%). Неочищенный продукт использовали непосредственно на следующей стадии без дополнительной очистки.
ЖХ-МС (метод А), (ЭР+) 215, RT=4,99 мин.
Стадия (4)
К суспензии 2-(этилтио)тиено[3,4-d]пиримидин-4-(3Н,5Н,7Н)-она (2,88 г, 13,45 ммоль) в воде (20 мл) добавляли 2,0 мл конц. HCl и 4,0 мл АсОН. Реакционную смесь кипятили с обратным холодильником в течение ночи, затем охлаждали и твердое вещество собирали фильтрованием, промывали водой и метанолом, выпаривали и сушили с получением белого твердого вещества, тиено[3,4-d]пиримидин-2,4-(1H,3H,5H,7H)-диона (1,80 г, 80%). Неочищенный продукт использовали непосредственно на следующей стадии без дополнительной очистки.
ЖХ-МС (метод А), (ЭР+) 171, RT=1,66 мин
Стадия (5)
Тиено[3,4-d]пиримидин-2,4-(1Н,3Н,5Н,7Н)-дион (1,6 г, 9,41 ммоль) суспендировали в 4,0 мл фенилфосфонилдихлорида и суспензию нагревали в течение ночи при 135°С, затем в течение 1 часа при 165°С. Полученную реакционную смесь охлаждали и вливали в смесь лед-вода. Водный слой экстрагировали этилацетатом. Органические слои объединяли, промывали насыщенным раствором NaHCO3 и рассолом, сушили над безводным Na2SO4 и выпаривали. Неочищенный продукт затем очищали колоночной хроматографией на силикагеле (элюент: РЕ (петролейный эфир)/ЕА (этилацетат) от 20/1 до 10/1) с получением 2,4-дихлор-5,7-дигидротиено[3,4-d]пиримидина в виде светло-желтого твердого вещества (1,6 г, 83%).
ЖХ-МС (метод А), (ЭР+)207/209, RT=4,96 мин.
Стадия (6)
К раствору 2,4-дихлор-5,7-дигидротиено[3,4-d]пиримидина (300 мг, 1,45 ммоль) и Et3N (294 мг, 2,9 ммоль) в DMF (5,0 мл) при 0°С по каплям добавляли 3-(S)-метилморфолин (161 мг,1,59 ммоль). После завершения добавления смесь перемешивали в течение ночи при к.т. Растворитель удаляли под вакуумом с получением остатка, который распределяли между водой и этилацетатом. Органический слой промывали рассолом, сушили и концентрировали с получением неочищенного продукта, который очищали посредством препаративной ТСХ (тонкослойная хроматография, элюент: петролейный эфир/этилацетат, 2/1) с получением 2-хлор-5,7-дигидро-4-((S)-3-метилморфолино)тиено[3,4-d]-пиримидина в виде светло-желтого твердого вещества (283 мг, 72%).
ЖХ-МС (метод А), (ЭР+) 272, RT=5,00 мин.
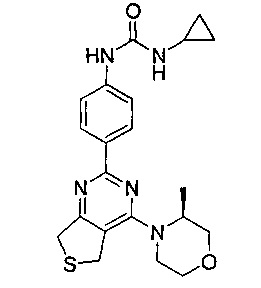
Стадия (7)
К раствору 2-хлор-5,7-дигидро-4-((S)-3-метилморфолино)тиено[3,4-d]-пиримидина (100 мг, 0,37 ммоль) в смеси DME/H2O (4:1, 10 мл) добавляли 1-циклопропил-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)-мочевину (124 мг, 0,41 ммоль) и Na2CO3 (118 мг, 1,11 ммоль) с последующим добавлением PdCl2(dppf) (1,1'-бис(дифенилфосфин)ферроцен]дипалладий(II), комплекс с дихлорметаном, 15 мг, 0,02 ммоль). Полученную смесь нагревали до 70°C и перемешивали в течение ночи под азотом. Растворитель удаляли при пониженном давлении с получением остатка, который распределяли между этилацетатом и водой. Органический слой промывали рассолом, сушили (Na2SO4) и концентрировали с получением неочищенного продукта, который очищали посредством препаративной ТСХ (DCM/MeOH, 20/1) с получением целевого продукта в виде желтого твердого вещества (20 мг, 13%).
1Н ЯМР (d4-Метанол) δ 8.24 (d, 2Н), 7.51 (d, 2Н), 4.69 (s, 1Н), 4.60-4.58 (m, 1Н), 4.37-4.35 (m, 1Н), 4.24-4.17 (m, 3Н), 4.03-3.99 (m, 1Н), 3.80 (s, 2Н), 3.70-3.65 (m, 1Н), 3.55-3.48 (m, 1Н), 2.64-2.60 (m, 1Н), 1.40 (d, 3Н), 0.79-0.77 (m, 2Н), 0.55-0.54 (m, 2Н).
ЖХ-МС (метод А), (ЭР+) 412, RT=4,90 мин.
Пример 2:
(S)-1-циклопропил-3-(4-(4-(3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевина
Стадия (1)
К раствору 2,4-дихлор-5,7-дигидротиено[3,4-d]пиримидина (400 мг, 1,93 ммоль) (Пример 1, Стадия (5)) в CH2Cl2 (5 мл) порциями добавляли m-СРВА (мета-хлорпероксибензойная кислота, 830 мг, 4,83 ммоль). Реакционную смесь перемешивали при к.т.в течение ночи. Полученную смесь промывали насыщенным NaHSO3 и насыщенным Na2CO3, экстрагировали CH2Cl2, сушили над безводным Na2SO4 и выпаривали. Остаток затем очищали колоночной хроматографией на силикагеле (элюент: петролейный эфир/этилацетат, 2/1) с получением 6,6-диоксида 2,4-дихлор-5,7-дигидротиено[3,4-d]пиримидина в виде белого твердого вещества (260 мг, 57%).
ЖХ-МС (метод А), (ЭР+) 239/241, RT=4,04 мин.
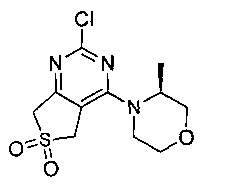
Стадия (2)
К раствору 6,6-диоксида 2,4-дихлор-5,7-дигидротиено[3,4-d]пиримидина (120 мг, 0,5 ммоль) и Et3N (101 мг, 1,0 ммоль) в DMF (5,0 мл) при 0°C по каплям добавляли смесь (S)-3-метилморфолина (51 мг, 0,5 ммоль). После завершения добавления смесь перемешивали в течение ночи при к.т. Растворитель удаляли под вакуумом с получением остатка, который распределяли между водой и этилацетатом. Органический слой промывали рассолом, сушили (Na2SO4) и концентрировали с получением продукта, который очищали посредством препаративной ТСХ (элюент: петролейный эфир/этилацетат, 2/1) с получением 6,6-диоксида 2-хлор-5,7-дигидро-4-((5)-3-метилморфолино)-тиено[3,4-d]пиримидина в виде светло-желтого твердого вещества (100 мг, 66%).
ЖХ-МС (метод А), (ЭР+) 304, RT=2,50 мин.

Стадия (3)
К раствору 6,6-диоксида 2-хлор-5,7-дигидро-4-((S)-3-метилморфолино)-тиено[3,4-d]пиримидина (96 мг, 0,32 ммоль) в смеси DME/H2O (4:1, 10 мл) добавляли 1-циклопропил-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-фенил)мочевину (106 мг, 0,35 ммоль) и Na2CO3 (102 мг, 0,96 ммоль), а затем PdCl2(dppf) (15 мг, 0,02 ммоль). Полученную смесь нагревали до 70°C и перемешивали в течение ночи под азотом. Растворитель удаляли под вакуумом с получением остатка, который распределяли между этилацетатом и водой. Органический слой отделяли, сушили (Na2SO4) и растворитель удаляли с получением неочищенного продукта, который очищали посредством препаративной ТСХ (DCM/MeOH, 20/1) с получением целевого продукта в виде желтого твердого вещества (15 мг, 11%).
1Н ЯМР (d6-DMSO) δ 8.66 (s, 1Н) 8.20 (d, 2Н), 7.53 (d, 2Н), 6.52 (s, 1H), 4.75-4.44 (m, 5H), 3.98-3.93 (m, 2H), 3.66 (s, 2H), 3.54-3.40 (m, 2H), 2.50 (m, 1H, скрыт под DMSO), 1.29 (d, 3Н), 0.66-0.64 (m, 2Н), 0.43-0.39 (m, 2Н).
ЖХ-МС (метод А), (ЭР+) 444, RT=4,64 мин.
Пример 3:
(S)-1-циклопропил-3-(4-(4-(3-метилморфолино)-7,8-дигидро-5Н-тиопирано[4,3-d]пиримидин-2-ил)фенил)мочевина
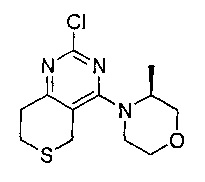
Стадия (1)
К раствору 2,4-дихлор-7,8-дигидро-5Н-тиопирано[4,3-d]пиримидина (200 мг, 0,9 ммоль) и Et3N (182 мг, 1,8 ммоль) в DMF (3 мл) при 0°C добавляли 3-(S)-метилморфолин (100 мг, 0,99 ммоль). Полученную смесь перемешивали при к.т. в течение ночи. Растворитель удаляли под вакуумом с получением остатка, который распределяли между водой и этилацетатом. Органический слой промывали рассолом, сушили и концентрировали с получением продукта, который очищали посредством препаративной ТСХ (петролейный эфир/этилацетат, 2/1) с получением (S)-2-хлор-4-(3-метилморфолино)-7,8-дигидро-5Н-тиопирано[4,3-d]пиримидина (150 мг, 58%).
ЖХ-МС (метод А), (ЭР+) 286, RT=4,75 мин.
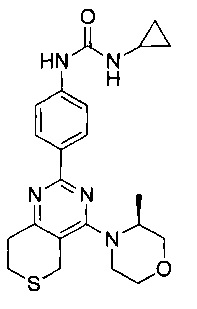
Стадия (2)
К раствору (S)-2-хлор-4-(3-метилморфолино)-7,8-дигидро-5Н-тиопирано-[4,3-d]пиримидина (100 мг, 0,35 ммоль) в смеси DME/H2O (4:1, 10 мл) добавляли 1-циклопропил-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-фенил)мочевину (116 мг, 0,38 ммоль) и Na2CO3 (111 мг, 1,05 ммоль), а затем PdCl2(dppf) (15 мг, 0,02 ммоль). Полученную смесь перемешивали в течение ночи при 70°C под азотом. Растворитель удаляли под вакуумом с получением остатка, который распределяли между этилацетатом и водой. Органический слой отделяли, сушили над Na2SO4, фильтровали и выпаривали с получением неочищенного продукта, который очищали посредством препаративной ТСХ (DCM/MeOH, 20/1) с получением целевого продукта в виде желтого твердого вещества (40 мг, 27%).
1Н ЯМР (CDCl3) 8.37 (d, 2Н), 7.53 (d, 2Н), 7.12 (s, 1Н), 5.09 (s, 1Н), 3.93-3.75 (m, 4Н), 3.67-3.58 (m, 3Н), 3.52-3.40 (m, 1Н),3.35-3.32 (m, 1Н), 3.20 (t, 2Н),3.03 (t, 2Н), 2.66-2.63 (m, 1Н), 1.30-1.27 (m, 3Н), 0.89-0.87 (m, 2Н), 0.72-0.68 (m, 2Н).
ЖХ-МС (метод А), (ЭР+) 426, RT=4,50 мин.
Пример 4:
(S)-1-циклопропил-3-(4-(4-(3-метилморфолино)-6,6-диоксидо-7,8-дигидро-5Н-тиопирано[4,3-d]пиримидин-2-ил)фенил)мочевина
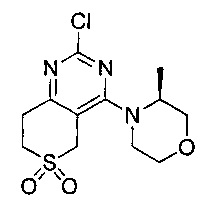
Стадия (1)
Используя способ, аналогичный использованному в примере 2, стадия (2), начиная с 6,6-диоксида 2,4-дихлор-7,8-дигидро-5Н-тиопирано[4,3-d]пиримидина, синтезировали 6,6 диоксид (S)-2-хлор-4-(3-метилморфолино)-7,8-дигидро-5Н-тиопирано[4,3-d]пиримидина.
ЖХ-МС (метод А), (ЭР+) 318/320, RT=4,43 мин.
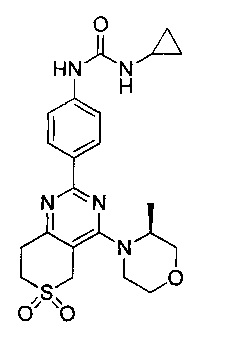
Стадия (2)
К раствору 6,6 диоксида (S)-2-хлор-4-(3-метилморфолино)-7,8-дигидро-5Н-тиопирано[4,3-d]пиримидина (160 мг, 0,5 ммоль) в смеси DME/Н2О (4:1, 10 мл) добавляли 1-циклопропил-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)мочевину (167 мг, 0,55 ммоль) и Na2CO3 (159 мг, 1,5 ммоль), а затем PdCl2(dppf) (20 мг, 0,025 ммоль). Полученную смесь нагревали до 70°C и перемешивали в течение ночи под азотом. Растворитель удаляли под вакуумом с получением остатка, который распределяли между этилацетатом и водой. Органический слой отделяли, сушили над Na2SO4, фильтровали и растворитель удаляли с получением неочищенного продукта, который очищали посредством препаративной ТСХ (DCM/MeOH, 20/1) с получением целевого продукта в виде желтого твердого вещества (36 мг, 16%).
1Н ЯМР (CDCl3) 8.61 (s, 1Н), 8.22 (d,, 2Н), 7.53 (d, 2Н), 6.48 (s, 1Н), 4.41-4.37 (m, 1Н), 4.27-4.23 (m, 1Н), 3.87-3.27 (m, 10Н), 2.60-2.54 (m, 1Н), 1.20 (d, 3Н), 0.65-0.63 (m, 2Н), 0.43-0.40 (m, 2Н).
ЖХ-МС (метод А), (ЭР+) 458, RT=4,52 мин.
Пример 5:
(5)-1-циклопропил-3-(4-(4-(3-метилморфолино)-5,5-диоксидо-6,7-дигидро-тиено[3,2-d]пиримидин-2-ил)фенил)мочевина
Стадия (1)
К раствору метилтиогликолята (91,0 мл, 1,0 моль) и пиперидина (2 мл, 4,05 ммоль) по каплям добавляли метилакрилат (99,2 мл, 1,1 моль). Реакционную смесь перемешивали при 50°C в течение 2 ч. Избыток метилакрилата и пиперидина затем удаляли посредством отгонки с получением метил-3-(2-метокси-2-оксоэтилтио)-пропаноата в виде бесцветного масла (185 г, 96%). Неочищенный продукт использовали непосредственно на следующей стадии без дополнительной очистки.
ЖХ-МС (метод А), (ЭР+)193, RT=4,9 мин.

Стадия (2)
Металлический литий (2,70 г, 390,15 ммоль) добавляли к 500 мл метанола в ледяной бане. Затем к раствору при к.т. по каплям добавляли метил-3-(2-метокси-2-оксоэтилтио)пропаноат (50,0 г, 260,10 ммоль). Реакционную смесь перемешивали в течение ночи. Полученную реакционную смесь упаривали для удаления растворителя. Смесь нейтрализовали до pH=7-8 с помощью 3 н. HCl и экстрагировали дихлорметаном. Органические слои объединяли, промывали рассолом, сушили над безводным Na2SO4, фильтровали и выпаривали. Полученный неочищенный остаток содержал целевое соединение в виде единственного выделяемого продукта. В результате очистки посредством колоночной хроматографии на силикагеле (элюент: РЕ/ЕА от 20/1 до 5/1) получали метил-3-оксо-тетрагидротиофен-2-карбоксилат в виде бесцветного масла (18,0 г, 43%).
1Н ЯМР (CDCl3) 4.04 (s, 1Н), 3.78 (s, 3Н), 3.34 (m,1Н), 3.06 (m,1Н), 2.83 (m, 1Н), 2.69 (m, 1Н).
ЖХ-МС (метод А), (ЭР+) 183, RT=4,15 мин.
Стадия (3)
К раствору бромида S-этил-изотиоурония (6,06 г, 32,7 ммоль) в воде (60 мл) по каплям добавляли карбонат натрия (3,64 г, 34,33 ммоль) и метил-3-оксо-тетрагидротиофен-2-карбоксилат (5,00 г, 31,20 ммоль). Смесь перемешивали в темноте в течение 3 суток. Твердое вещество, выпадавшее в осадок в полученной смеси, собирали фильтрованием, промывали водой, эфиром и этилацетатом и затем сушили под вакуумом с получением 2-(этилтио)-6,7-дигидротиено[3,2-d]пиримидин-4(3Н)-она в виде белого твердого вещества (2,8 г, 42,4%). Неочищенный продукт использовали непосредственно на следующей стадии без дополнительной очистки.
ЖХ-МС (метод А), (ЭР+)215, RT=4,51 мин.
Стадия (4)
К суспензии 2-(этилтио)-6,7-дигидротиено[3,2-d]пиримидин-4(3Н)-она (2,80 г, 13,06 ммоль) в воде (2,0 мл) добавляли 2,0 мл конц. HCl и 4,0 мл АсОН. Смесь нагревали при 110°C в течение ночи. Реакционную смесь охлаждали и твердое вещество, которое образовывалось, собирали фильтрованием, промывали водой и метанолом и сушили с получением белого твердого вещества, 6,7-дигидротиено[3,2-d]пиримидин-2,4(1Н,3Н)-диона (1,80 г, 82%). Неочищенный продукт использовали непосредственно на следующей стадии без дополнительной очистки.
ЖХ-МС (метод А), (ЭР+) 171, RT=4,21 мин.
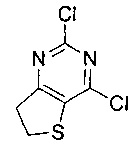
Стадия (5)
6,7-Дигидротиено[3,2-d]пиримидин-2,4(1Н,3Н)-дион (1,8 г, 10,57 ммоль) суспендировали в 4,0 мл фенилфосфонилдихлорида. Суспензию нагревали до 130-140°C и перемешивали в течение 6 ч. Полученную реакционную смесь охлаждали и вливали в 10,0 мл смеси лед-вода. Смесь экстрагировали этилацетатом, органические слои объединяли, промывали насыщенным МаНСО3 и рассолом, сушили над безводным Na2O4, фильтровали и выпаривали. Неочищенный продукт затем очищали колоночной хроматографией на силикагеле (элюент: РЕ/ЕА, от 20:1 до 10/1) с получением 2,4-дихлор-6,7-дигидротиено[3,2-d]пиримидина в виде не совсем белого твердого вещества (1,0 г, 46%).
ЖХ-МС (метод А), (ЭР+) 207/209, RT=4,68 мин.
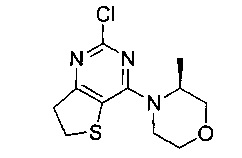
Стадия (6)
К раствору 2,4-дихлор-6,7-дигидротиено[3,2-d]пиримидина (200,0 мг, 0,96 ммоль) и Et3N (195,5 мг, 1,93 ммоль) в DMF (2,0 мл) при 0°C по каплям добавляли (S)-3-метилморфолин (97,7 мг). Смесь перемешивали в течение ночи при к.т., затем распределяли между H2O и этилацетатом. Водный слой экстрагировали этилацетатом и объединенные органические слои промывали рассолом, сушили над безводным Na2SO4, фильтровали, выпаривали и затем неочищенный продукт очищали колоночной хроматографией на силикагеле (элюент: РЕ/ЕА, от 20:1 до 2/1) с получением (S)-2-хлор-4-(3-метилморфолино)-6,7-дигидротиено[3,2-d]пиримидина в виде светло-желтого твердого вещества (120 мг, 46%).
ЖХ-МС (метод А), (ЭР+) 272/274, RT=4,88 мин.
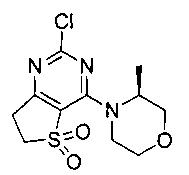
Стадия (7)
(S)-2-Хлор-4-(3-метилморфолино)-6,7-дигидротиено[3,2-d]пиримидин (120 мг, 0,44 ммоль) растворяли в 4,0 мл дихлорметана. К раствору порциями добавляли m-СРВА (190,4 мг, 1,10 ммоль). Реакционную смесь перемешивали при к.т. в течение 5 ч. Полученную смесь разбавляли дихлорметаном, промывали насыщенным NaHSO3 и насыщенным Na2CO3. Органический слой сушили над безводным Na2SO4, фильтровали и выпаривали. Остаток затем очищали посредством преп. ТСХ (элюент: петролейный эфир/этилацетат, 2/1) с получением 5,5-диоксида 2-хлор-4-[(3S)-3-метилморфолин-4-ил]-6,7-дигидротиено[3,2-d]пиримидина в виде белого твердого вещества, (50,0 мг, 37%).
ЖХ-МС (метод А), (ЭР+) 304/306, RT=3,44 мин.
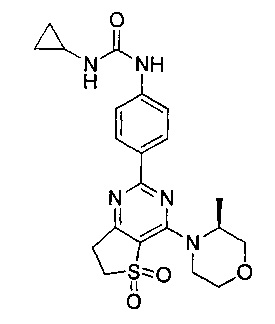
Стадия (8)
К раствору 5,5-диоксида 2-хлор-4-[(3S)-3-метилморфолин-4-ил]-6,7-дигидротиено[3,2-d]пиримидина (50 мг, 0,16 ммоль) в 2,0 мл смеси DME/Н2О (4/1, об./об.) добавляли 1-циклопропил-3-(4-(4,4,5,5-тетраметил-1,3,2-диокса-боролан-2-ил)фенил)мочевину (59,7 мг, 0,20 ммоль) и Na2CO3 (52,3 мг, 0,49 ммоль). Смесь дважды дегазировали и затем добавляли каталитическое количество Pd(dppf)Cl2 (10 мг, 0,013 ммоль). Смесь нагревали до 80°C и перемешивали в течение 3 ч под азотом. Полученную смесь затем охлаждали и разбавляли EtOc и H2O, водный слой экстрагировали этилацетатом и объединенные органические слои объединяли, промывали рассолом, сушили над безводным Na2SO4, фильтровали и затем выпаривали с получением неочищенного продукта, который очищали колоночной хроматографией на силикагеле (элюент: CH2Cl2/этилацетат от 30/1 до приблизительно 1/1) с получением 1-циклопропил-3-[4-[4-[(3S)-3-метилморфолин-4-ил]-5,5-диоксо-6,7-дигидротиено[3,2-d] пиримидин-2-ил]фенил]мочевины в виде белого твердого вещества (30,0 мг, 41%).
1Н ЯМР (d6-DMSO): 8.68 (s, 1Н), 8.24 (d, 2Н), 7.54 (d,, 2Н), 6.51 (s, 1Н), 4.90 (s, 1Н), 4.39 (s, 1Н), 4.01-4.02 (d, 1Н), 3.80(d, 1Н), 3.69-3.66 (m, 3Н), 3.53-3.51 (d, 2Н), 2.56-2.51 (m, 1Н), 1.35 (d, 3Н), 1.24 (s, 1Н), 0.66-0.64 (m, 2Н), 0.42 (m, 2Н).
ЖХ-МС (метод А), (ЭР+) 444, RT=3,32 мин.
Пример 6:
1-Циклопропил-3-(4-(5-метил-4-((S)-3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевина
Стадия (1)
К смеси метилтиогликолята (21,2 г, 199,7 ммоль) и пиперидина (0,5 мл, 5,06 ммоль) при 0°C по каплям добавляли метилкротонат (25,0 г, 249,70 ммоль). Спустя 10 минут двумя порциями добавляли дополнительное количество пиперидина (0,3 мл, 3,04 ммоль). Смесь затем перемешивали при к.т. в течение ночи. Избыток метилкротоната и пиперидина удаляли посредством отгонки с получением в остатке метил-3-(2-метокси-2-оксоэтилтио)бутаноата в виде светло-желтого масла (40 г, 97%). Неочищенный продукт использовали непосредственно на следующей стадии без дополнительной очистки.
ЖХ-МС (метод А), (ЭР+) 207, RT=4,78 мин.
Стадия (2)
К суспензии метоксида натрия (5,89 г, 109,2 ммоль) в толуоле (150 мл) по каплям добавляли метил-3-(2-метокси-2-оксоэтилтио)бутаноат (150 г, 72,8 ммоль). Полученную смесь нагревали до образования флегмы и перемешивали в течение ночи. Полученную реакционную смесь охлаждали, затем вливали в смесь 10,0 мл уксусной кислоты в 150,0 г молотого льда. Раствор подщелачивали (pH=8-9) посредством добавления насыщенного Na2CO3 и полученную смесь экстрагировали этилацетатом. Объединенные органические слои затем объединяли и промывали рассолом, сушили над безводным Na2SO4, фильтровали, выпаривали и остаток очищали колоночной хроматографией (элюент: петролейный эфир/этилацетат, от 20/1 до 5/1) с получением метил-2-метил-4-оксо-тетрагидротиофен-3-карбоксилата в виде бесцветного масла (4,0 г, 32%).
ЖХ-МС (метод А), (ЭР+) 175, RT=4,40 мин.
Стадия (3)
К раствору бромида 3-этилизотиоурония (5,09 г, 22,7 ммоль) в воде (35,0 мл) порциями добавляли карбонат натрия (3,65 г, 34,4 ммоль) и метил-2-метил-4-оксо-тетрагидротиофен-3-карбоксилат (4,00 г, 22,9 ммоль). Полученную смесь перемешивали в темноте в течение 3 суток и твердое вещество, которое выпадало в осадок из реакционной смеси, собирали фильтрованием, промывали водой, эфиром и этилацетатом, затем сушили под вакуумом с получением 2-(этилтио)-5-метилтиено[3,4-d]пиримидин-4(3Н,5Н,7Н)-она в виде белого твердого вещества (4,0 г, 76%). Неочищенный продукт использовали непосредственно на следующей стадии без дополнительной очистки.
ЖХ-МС (метод А), (ЭР+) 229, РТ=4,90 мин.

Стадия (4)
2-(Этилтио)-5-метилтиено[3,4-d]пиримидин-4(3Н,5Н,7Н)-он (4,0 г, 17,5 ммоль) суспендировали в 4,0 мл конц. HCl и 6,0 мл ледяной уксусной кислоты и суспензию кипятили с обратным холодильником в течение ночи. Смесь охлаждали и выпавшее в осадок твердое вещество собирали фильтрованием, промывали водой и сушили с получением 5-метилтиено[3,4-d]пиримидин-2,4(1Н,3Н,5Н,7Н)-диона в виде белого твердого вещества (3,20 г,100%).
1Н ЯМР (d6-DMSO): 11.19 (s, 1Н), 11.04 (s, 1Н), 4.36-4.34 (m, 1Н), 4.07-4.02 (dd, J=16, 4,4 Гц, 1Н), 3.87-3.83 (dd, J=16, 1,2 Гц, 1Н), 1.48-1.48 (d, J=6,4 Гц, 3Н).
ЖХ-МС (метод А), (ЭР+) 185, РТ=4,11 мин.
Стадия (5)
5-Метилтиено[3,4-d]пиримидин-2)4(1Н,3Н,5Н,7Н)-дион (3,2 г, 17,2 ммоль) суспендировали в фенилфосфонилдихлориде (8,0 мл), затем суспензию нагревали до 130-140°C и перемешивали в течение 6 ч. Полученную реакционную смесь охлаждали и вливали в 30 мл смеси лед-вода. Смесь экстрагировали этилацетатом и объединенные органические слои промывали насыщенным NaHCO3, рассолом, сушили над безводным Na2SO4, фильтровали и выпаривали. Неочищенный продукт затем очищали колоночной хроматографией на силикагеле (элюент: петролейный эфир/этилацетат от 20/1 до 10/1) с получением 2,4-дихлор-5-метил-5,7-дигидротиено[3,4-d]пиримидина в виде светло-желтого твердого вещества (2,0 г, 53%).
1Н ЯМР (CDCl3): 4.66-4.64 (m, 1Н), 4.47-4.42 (dd, J=2,8, 2,4 Гц, 1Н), 4.19-4.14 (m, 1Н), 1.71-1.69 (d, J=6,8 Гц, 3Н).
ЖХ-МС (метод А), (ЭР+) 221/223, RT=4,94 мин.
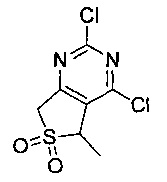
Стадия (6)
2,4-Дихлор-5-метил-5,7-дигидротиено[3,4-d]пиримидин (1,0 г, 4,5 ммоль) растворяли в дихлорметане (10 мл). Порциями добавляли m-СРВА (2,3 г, 13,6 ммоль) и реакционную смесь перемешивали при к.т. в течение ночи. Реакционную смесь разбавляли дихлорметаном и промывали насыщенным NaHSO3 и насыщенным Na2CO3. Органический слой сушили с помощью безводного Na2SO4, фильтровали и выпаривали с получением неочищенного остатка, который очищали колоночной хроматографией на силикагеле (элюент: петролейный эфир/этилацетат, 2/1). Продукт, 6,6-диоксид 2,4-дихлор-5-метил-5,7-дигидротиено[3,4-d]пиримидина, получали в виде белого твердого вещества (0,60 г, 53%).
ЖХ-МС (метод А), (ЭР+) 253/255, RT=4,36 мин.
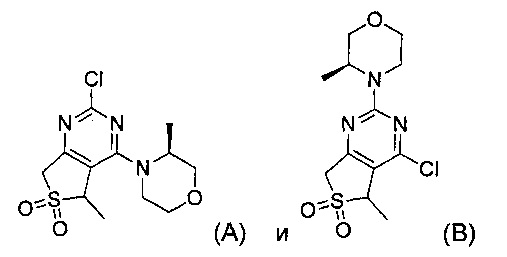
Стадия (7)
К раствору 6,6-диоксида 2,4-дихлор-5-метил-5,7-дигидротиено[3,4-d]-пиримидина (150,0 мг, 0,5926 ммоль) и Et3N (119,9 мг, 1,2 ммоль) в DMF (2,0 мл) по каплям добавляли (S)-3-метилморфолин (59,9 мг, 0,6 ммоль). Реакционную смесь перемешивали в течение ночи, затем гасили посредством добавления Н2О и полученную смесь экстрагировали этил ацетатом. Органические слои объединяли, промывали рассолом, сушили над безводным Na2SO4, фильтровали и выпаривали. Неочищенный продукт очищали колоночной хроматографией на силикагеле (элюент: петролейный эфир/этилацетат от 20:1 до 2/1) с получением 6,6-диоксида 4-хлор-5-метил-2-[(3S)-3-метилморфолин-4-ил]-5,7-дигидротиено[3,4-d]пиримидина (В) в виде светло-желтого твердого вещества (50 мг, 13,5%). Дальнейшее элюирование затем давало 6,6-диоксид 2-хлор-5-метил-4-[(3S)-3-метилморфолин-4-ил]-5,7-дигидротиено-[3,4-d]-пиримидина (А) в виде не совсем белого твердого вещества (100 мг, 27%).
А: 1Н ЯМР (CDCl3): 4.35-4.29 (m, 2Н), 4.26-4.21 (m, 3Н), 4.06-4.00 (m, 1Н), 3.89-3.86 (m, 1Н), 3.80 (s, 2Н), 3.73-3.72 (m, 1Н), 3.62-3.59 (m, 1Н), 3.57-3.51 (m, 2Н), 1.37-1.36 (d, J=7,8 Гц, 3Н).
ЖХ-МС (метод А), (ЭР+) 318/320, RT=4,51 мин.
В: 1Н ЯМР (CDCl3): 4.69-4.67 (m, 1Н), 4.35-4.24 (m, 3Н), 4.20-4.15 (m, 1Н), 4.02-3.98 (m, 1Н), 3.80-3.77 (m, 1Н), 3.69-3.65 (m, 1Н), 3.55-3.49 (m, 1Н), 3.34-3.27 (m, 1Н), 1.68-1.66 (d, J=7,2 Гц, ЗН), 1.34-1.66 (d, J=6,8 Гц, 3Н).
ЖХ-МС (метод А), (ЭР+) 318/320, RT=4,73 мин.
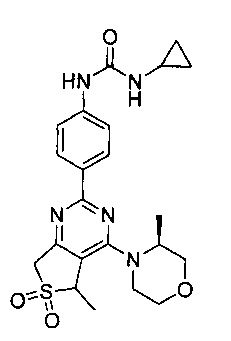
Стадия (8)
К раствору 6,6-диоксида 2-хлор-5-метил-4-[(3S)-3-метилморфолин-4-ил]-5,7-дигидротиено[3,4-d]пиримидина (100 мг, 0,3147 ммоль) в смеси DME/H2O (2 мл, 4/1, об./об.) добавляли 1-циклопропил-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)мочевину (114,1 мг, 0,37 ммоль) и карбонат натрия (100,1 мг, 0,94 ммоль). Реакционную смесь дважды дегазировали и к суспензии добавляли каталитическое количество Pd(dppf)Cl2 (10 мг, 0,013 ммоль). Смесь нагревали при 80°C в течение 3 ч в атмосфере азота. Полученную смесь охлаждали и добавляли H2O. Смесь экстрагировали этилацетатом и объединенные органические слои промывали рассолом, сушили над безводным Na2SO4, фильтровали и затем выпаривали. Остаток очищали посредством колоночной хроматографии на силикагеле (элюент: CH2Cl2/этилацетат от 20/1 до приблизительно 5/1) и препаративной ВЭЖХ с получением продукта в виде белого твердого вещества (30,0 мг, 21%).
1Н ЯМР (CDCl3): 8.32-8.24 (m, 2Н), 7.52 (d, 2Н), 7.12 (s, 1Н), 5.05 (s, 1Н), 4.42-4.20(m, 4Н), 4.07-3.98 (m, 1Н), 3.88-3.77 (m, 3Н), 3.75-3.49 (m, 2Н), 2.67-2.62 (m, 1Н), 1.65-1.62 (m, 3Н), 0.90-0.84 (m, 2Н), 0.72-0.67 (m 2Н).
ЖХ-МС (метод А), (ЭР+) 458, RT=3,47 мин.
Пример 7:
1-циклопропил-3-(4-(7-метил-4-((S)-3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевина
Стадия (1)
К смеси метил-3-меркаптопропаноата (34,3 г, 0,29 моль) и этил-2-бром-пропаноата (54,3 г, 0,30 моль) в THF (200 мл) медленно добавляли NaH (60%-ная дисперсия в минеральном масле, 12,76 г, 0,32 моль) и реакционную смесь перемешивали при к.т. в течение ночи. Растворитель удаляли при пониженном давлении и добавляли воду. Смесь экстрагировали DCM и органическую фазу сушили над Na2SO4 и концентрировали с получением неочищенного продукта, метил-3-(2-этокси-1-метил-2-оксо-этил)сульфанилпропаноата (65 г, 100%), который использовали непосредственно на следующей стадии.
ЖХ-МС (метод А), (ЭР+) 221, RT=4,63 мин.
Стадия (2)
К раствору метил-3-(2-этокси-1-метил-2-оксо-этил)сульфанилпропаноата (2,2 г, 10 ммоль) в метаноле (15 мл) при 0°C добавляли метоксид натрия (886 мг, 16,4 ммоль). Реакционную смесь нагревали до к.т. и перемешивали в течение 3 ч. Смесь подкисляли 1 н. HCl до pH 2 и экстрагировали DCM. Органическую фазу промывали водой и рассолом, сушили (Na2SO4), концентрировали и очищали колоночной хроматографией, элюируя ЕЮАс в петролейном эфире (3%-10%), с получением метил-тетрагидро-5-метил-4-оксотиофен-3-карбоксилата в виде бесцветного масла (0,85 г, 50%-ный выход).
ЖХ-МС (метод А), (ЭР+) 175, RT=4,30 мин.
Стадия (3)
К раствору бромида S-этил-изотиоурония (4,89 г, 26,4 ммоль) в воде (60 мл) по каплям добавляли карбонат натрия (2,80 г, 26,4 ммоль) и метил-тетрагидро-5-метил-4-оксотиофен-3-карбоксилат (4,60 г, 26,4 ммоль). Смесь перемешивали в течение ночи в темноте при к.т. Твердое вещество, которое выпадало в осадок в полученной смеси, собирали фильтрованием, промывали водой, диэтиловым эфиром, метанолом и ацетоном, затем твердое вещество сушили под вакуумом с получением 2-(этилтио)-7-метилтиено[3,4-d]пиримидин-4(3Н,5Н,7Н)-она в виде белого твердого вещества (3,96 г, 66%).
ЖХ-МС (метод А), (ЭР+) 229, RT=3,10 мин.
Стадия (4)
К суспензии 2-(этилтио)-7-метилтиено[3,4-d]пиримидин-4(3Н,5Н,7Н)-она (3,96 г, 17,3 ммоль) в воде (20 мл) добавляли 2,0 мл конц. HCl и 4,0 мл АсОН. Смесь нагревали до образования флегмы в течение ночи, затем охлаждали и выпадавшее в осадок твердое вещество собирали фильтрованием, промывали водой и метанолом, выпаривали и сушили с получением 7-метилтиено[3,4-d]пиримидин-2,4(1Н,3Н,5Н,7Н)-диона в виде белого твердого вещества (2,52 г, 80%). Неочищенный продукт использовали непосредственно на следующей стадии без дополнительной очистки.
ЖХ-МС (метод А), (ЭР+) 185, RT=2,58 мин.
Стадия (5)
7-Метилтиено[3,4-d]пиримидин-2,4(1Н,3Н,5Н,7Н)-дион (2,52 г, 13,7 ммоль) суспендировали в POCl3 (20 мл) и кипятили с обратным холодильником в течение ночи. Растворитель удаляли при пониженном давлении и остаток вливали в 10,0 мл смеси лед-вода. Смесь экстрагировали этилацетатом (3×10,0 мл) и объединенные органические слои промывали насыщенным NaHCO3 и рассолом, сушили над безводным Na2SO4, фильтровали и концентрировали. Неочищенный продукт затем очищали посредством колоночной хроматографии на силикагеле (элюирование: РЕ/ЕА от 20:1 до приблизительно 10/1) с получением 2,4-дихлор-5,7-дигидро-7-метилтиено[3,4-d]пиримидина в виде не совсем белого твердого вещества (670 мг, 23%).
ЖХ-МС (метод А), (ЭР+) 221/223, RT=3,32 мин.
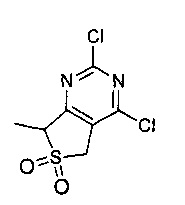
Стадия (6)
К раствору 2,4-дихлор-5,7-дигидро-7-метилтиено[3,4-d]пиримидина (221 мг, 1,0 ммоль) в DCM (10 мл) при 0°C порциями добавляли m-СРВА (432 мг, 2,5 ммоль). Реакционную смесь перемешивали при к.т. в течение 2 ч, затем гасили посредством добавления насыщенного раствора Na2CO3. Смесь экстрагировали DCM и объединенные органические слои промывали водой и рассолом, сушили и концентрировали с получением неочищенного продукта, 6,6-диоксида 2,4-дихлор-7-метил-5,7-дигидротиено[3,4-d]пиримидина, в виде белого твердого вещества (230 мг, 92%-ный выход). Этот материал использовали непосредственно на следующей стадии без дополнительной очистки.
ЖХ-МС (метод А), (ЭР+) 275 (М+Na), RT=2,73 мин.
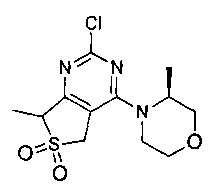
Стадия (7)
К раствору 6,6-диоксида 2,4-дихлор-7-метил-5,7-дигидротиено[3,4-d]пиримидина (160 мг, 0,63 ммоль) и триэтиламина (64 мг, 0,63 ммоль) в DMF (3 мл) при 0°C добавляли 3-(S)-метилморфолин (57 мг, 0,57 ммоль). Полученную смесь перемешивали при к.т. в течение 2 ч. Растворитель удаляли под вакуумом и полученный остаток распределяли между водой и этилацетатом. Органический слой сушили (Na2SO4) и концентрировали с получением продукта, который очищали посредством препаративной ТСХ (петролейный эфир/этилацетат, 2/1) с получением 6,6-диоксида 2-хлор-7-метил-4-[(3S)-3-метилморфолин-4-ил]-5,7-дигидротиено[3,4-d]пиримидина (72 мг, 36%-ный выход).
ЖХ-МС (метод А), (ЭР+) 318/320, RT=2,95 мин.
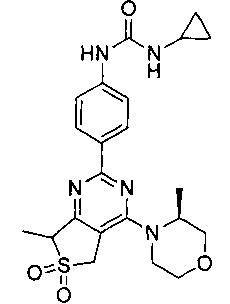
Стадия (8)
К раствору 6,6-диоксида 2-хлор-7-метил-4-[(3S)-3-метилморфолин-4-ил]-5,7-дигидротиено[3,4-d]пиримидина (32 мг, 0,10 ммоль) в смеси DME/H2O (4:1, 10 мл) добавляли 1-циклопропил-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)мочевину (45 мг, 0,15 ммоль) и Na2CO3 (32 мг, 0,30 ммоль), а затем PdCl2(dppf) (10 мг, 0,013 ммоль). Полученную смесь перемешивали под азотом в течение ночи при 80°C. Растворитель удаляли под вакуумом с получением остатка, который распределяли между этилацетатом и водой. Органический слой отделяли, сушили (Na2SO4), фильтровали и выпаривали с получением неочищенного продукта, который очищали посредством препаративной ТСХ (DCM/MeOH, 10/1) с получением целевого продукта в виде белого твердого вещества (10 мг, 22%).
1Н ЯМР (d6-DMSO) 9.30 (s, 1Н), 8.20 (d, 2Н), 7.52 (d, 2Н), 7.00 (s, 1Н), 4.75-4.35 (m, 4Н), 4.02-3.85 (m, 2Н), 3.73-3.42 (m, 4Н), 2.57-2.49 (m, 1Н частично под DMSO) 1.54 (d, 3Н), 1.24 (d, 3Н), 0.65-0.59 (m, 2Н), 0.41-0.35 (m, 2Н).
ЖХ-МС (метод А), (ЭР+) 458, RT=3,06 мин.
Пример 8:
(S)-1-циклопропил-3-(4-(4-(3-метилморфолино)-7,8-дигидро-6Н-тиопирано[3,2-d]пиримидин-2-ил)фенил)мочевина

Стадия (1)
Металлический натрий (4,3 г, 0,18 моль) осторожно небольшими порциями добавляли к этанолу (150 мл). После полного растворения натрия смесь охлаждали до 0°C и добавляли этил-2-сульфанилацетат (21,9 г, 0,18 моль). Затем медленно добавляли этил-4-хлорбутаноат (27,4 г, 0,18 моль) и полученную смесь перемешивали в течение ночи при к.т. Твердое вещество отфильтровывали и затем фильтрат концентрировали при пониженном давлении. Полученное масло распределяли между водой и этилацетатом. Органический слой собирали, сушили над безводным Na2SO4 и концентрировали под вакуумом с получением целевого этил-4-((этоксикарбонил)метилтио)бутаноата (40,2 г, 95%) в виде бесцветного масла. Неочищенный продукт использовали непосредственно на следующей стадии без дополнительной очистки.
ЖХ-МС (метод А), (ЭР+) 235, RT=4,74 мин.
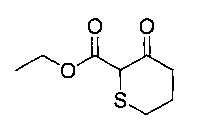
Стадия (2)
Металлический натрий (4,6 г, 0,2 моль) осторожно небольшими порциями добавляли к этанолу (150 мл). После полного растворения натрия смесь охлаждали до 0°C и добавляли этил-4-((этоксикарбонил)метилтио)бутаноат (40,2 г, 0,17 моль) в толуоле (80 мл). Смесь перемешивали при к.т.в течение 3 часов и затем нагревали до 50°C в течение еще одного часа. Смесь оставляли при к.т. на 24 часа. Растворитель затем удаляли при пониженном давлении и остаток распределяли между этилацетатом и водой. Органический слой промывали рассолом, сушили над безводным Na2SO4 и выпаривали. Неочищенный продукт затем очищали колоночной хроматографией на силикагеле (элюент: петролейный эфир/этилацетат, 30/1) с получением смеси этил-тетрагидро-3-оксо-2Н-тиопиран-2-карбоксилата и метил-3-оксотетрагидро-2Н-тиопиран-4-карбоксилата в соотношении 10:1 (4,8 г, 15%). Этот продукт использовали на следующей стадии без дополнительной очистки.
ЖХ-МС (метод А), (ЭР+) 189, RT=4,39 мин.
Стадия (3)
К раствору бромида S-этил-изотиоурония (4,4 г, 24 ммоль) в воде (50 мл) по каплям добавляли карбонат натрия (2,5 г, 24 ммоль) и этил-тетрагидро-3-оксо-2Н-тиопиран-2-карбоксилат (4,5 г, 24 ммоль). Смесь перемешивали в течение ночи в темноте при к.т. Твердое вещество, которое выпадало в осадок из реакционной смеси, собирали фильтрованием, промывали водой, диэтиловым эфиром, метанолом и ацетоном. Твердое вещество сушили под вакуумом с получением 2-(этилтио)-7,8-дигидро-3Н-тиопирано[3,2-d]пиримидин-4(6Н)-она в виде белого твердого вещества (4,5 г, 82%). Неочищенный продукт использовали непосредственно на следующей стадии без дополнительной очистки.
ЖХ-МС (метод А), (ЭР+) 229, RT=4,60 мин.
Стадия (4)
К суспензии 2-(этилтио)-7,8-дигидро-3Н-тиопирано[3,2-d]пиримидин-4(6Н)-она (4,5 г, 19,7 ммоль) в воде (20 мл) добавляли 3,0 мл конц. HCl и 6,0 мл АсОН. Смесь нагревали до образования флегмы и перемешивали в течение ночи. Реакционную смесь охлаждали и образовавшееся твердое вещество собирали фильтрованием, промывали водой и метанолом, выпаривали и сушили с получением 7,8-дигидро-1Н-тиопирано[3,2-d]пиримидин-2,4(3Н,6Н)-диона в виде белого твердого вещества (2,6 г, 72%). Неочищенный продукт использовали непосредственно на следующей стадии без дополнительной очистки.
ЖХ-МС (метод А), (ЭР+)185, RT=2,17 мин.
Стадия (5)
К 7,8-дигидро-1Н-тиопирано[3,2-d]пиримидин-2,4(3Н,6Н)-диону (2,6 г, 14,13 ммоль) добавляли 6,0 мл фосфорилтрихлорида и суспензию нагревали в течение ночи при 135°C и затем еще в течение одного часа при 165°C. Полученную реакционную смесь охлаждали и вливали в смесь лед-вода. Водный слой экстрагировали этилацетатом и объединенные органические слои промывали насыщенным NaHCO3, рассолом, сушили над безводным Na2SO4 и выпаривали. Неочищенный продукт затем очищали колоночной хроматографией на силикагеле (элюент: петролейный эфир/этилацетат, 20/1) с получением 2,4-дихлор-7,8-дигидро-6Н-тиопирано[3,2-d]пиримидина в виде светло-желтого твердого вещества (2,7 г, 87%). Региоизомерный продукт, происходящий из метил-3-оксотетрагидро-2Н-тиопиран-4-карбоксилата (Пример 8, Стадия (2)), не обнаруживался и считался утерянным в процессе очистки.
ЖХ-МС (метод А), (ЭР+) 221, RT=2,88 мин.
Стадия (6)
К раствору 2,4-дихлор-7,8-дигидро-6Н-тиопирано[3,2-d]пиримидина (500 мг, 2,26 ммоль) и Et3N (457 мг, 4,52 ммоль) в DMF (5,0 мл) при 0°C по каплям добавляли 3-(S)-метилморфолин (252 мг, 2,49 ммоль). Реакционную смесь перемешивали в течение ночи при к.т., затем растворитель удаляли под вакуумом с получением остатка, который распределяли между водой и этилацетатом. Органический слой отделяли, сушили и концентрировали с получением неочищенного продукта, который очищали колоночной хроматографией на силикагеле (элюент: петролейный эфир/этилацетат, 10/1) с получением 2-хлор-7,8-дигидро-4-((S)-3-метилморфолино)-6Н-тиопирано[3,2-d]пиримидина (380 мг, 87%) в виде светло-желтого твердого вещества.
ЖХ-МС (метод А), (ЭР+) 286, RT=3,69 мин.
Стадия (7)
К раствору 2-хлор-7,8-дигидро-4-((S)-3-метилморфолино)-6Н-тиопирано-[3,2-d]пиримидина (150 мг, 0,53 ммоль) в смеси DME/H2O (4:1, 10 мл) добавляли 1-циклопропил-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-фенил)мочевину (175 мг, 0,58 ммоль) и Na2CO3 (170 мг, 1,60 ммоль), а затем PdCl2(dppf) (15 мг, 0,02 ммоль). Полученную смесь перемешивали в течение ночи под азотом при 70°C. Растворитель удаляли под вакуумом с получением остатка, который распределяли между этилацетатом и водой. Органический слой отделяли и сушили над безводным Na2SO4. Растворитель удаляли с получением неочищенного материала, который очищали посредством препаративной ТСХ (DCM/MeOH, 20/1) с получением целевого продукта в виде белого твердого вещества (180 мг, 80%).
1Н ЯМР (CDCl3) δ 8.34 (d, 2Н), 7.52 (d, 2Н), 7.10 (s, 1Н), 5.08 (s, 1Н), 4.20-4.14 (m, 1Н), 3.92-3.84 (m, 3Н), 3.65-3.58 (m, 3Н), 3.04-2.98 (m, 4Н), 2.66-2.63 (m, 1Н), 2.30-2.24 (qu, 2Н), 1.30 (d, 3Н), 0.88-0.86 (m, 2Н), 0.71-0.68 (m, 2Н).
ЖХ-МС (метод А), (ЭР+) 426, RT=3,29 мин.
Примеры 9 и 10:
1-Циклопропил-3-(4-((R)-5-метил-4-((S)-3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевина и 1-циклопропил-3-(4-((S)-5-метил-4-((S)-3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]-пиримидин-2-ил)фенил)мочевина
Соединение из примера 6(1-циклопропил-3-(4-(5-метил-4-((S)-3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)-фенил)мочевина) представляет собой смесь диастереоизомеров. Индивидуальные диастереоизомеры выделяли при следующих условиях сверхкритической жидкостной хроматографии (SFC).
Прибор: MG II для препаративной SFC
Колонка: ChiralPak AS-Η, 250×30 мм I.D. (внутренний диаметр)
Подвижная фаза: А - для CO2 и В - для этанола
Градиент: В 40%
Скорость потока: 50 мл/мин
Обратное давление: 100 бар
Температура колонки: 38°C
Длина волны: 220 нм
Время цикла: 14 мин
Приготовление образца: соединение растворяли в этаноле до концентрации приблизительно 5 мг/мл
Инъекция: 4,7 мл на инъекцию.
Обработка: после разделения фракции высушивали с помощью роторного испарителя при температуре бани 40°C с получением целевых диастереоизомеров.
Из 275 мг смеси диастереомеров:
соединение из примера 9 - первый элюируемый диастереомер (92 мг)
1Н ЯМР (DMSO) δ 8.65 (s, 1Н), 8.18 (d, 2Н), 7.52 (d, 2Н), 6.53 (s, 1Н), 4.77 (q, 1Н), 4.71 (d, 1Н), 4.61-4.51 (m, 1Н), 4.40 (d, 1Н), 3.99-3.83 (m, 2Н), 3.81-3.65 (m, 2H), 3.57 (ddd, 7.9 Гц, 1H), 3.46-3.38 (m, 1H), 2.56 (m, 1H), 1.50 (d, 3H), 1.35 (d, 3H), 0.65 (ddd, 2H), 0.40 (ddd, 2H);
соединение из пример 10 - второй элюируемый диастереомер (145 мг)
1Н ЯМР (DMSO) δ 8.70 (s, 1Н), 8.24 (d, 2Н), 7.57 (d, 2Н), 6.58 (d, 1Н), 4.77 (q, 1Н), 4.73 (d, 1H), 4.54 (d, 1H), 4.50-4.41 (m, 1H), 4.08-3.93 (m, 2H), 3.85-3.73 (m, 2H), 3.67-3.55 (m, 1H), 3.55-3.43 (m, 1H), 2.64-2.58 (m, 1H), 1.54 (d, 3H), 1.27 (d, 3H), 0.70 (ddd, 2H), 0.47 (ddd, 2H).
Следующие соединения синтезировали посредством процедур, аналогичных описанным выше.
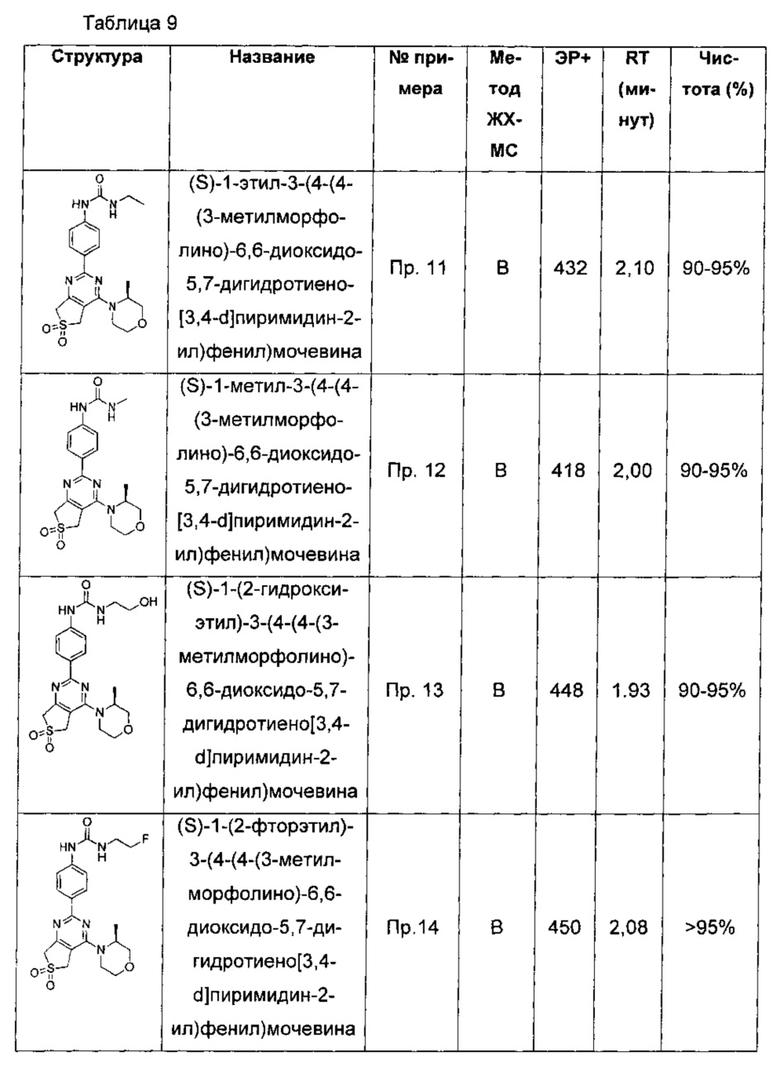
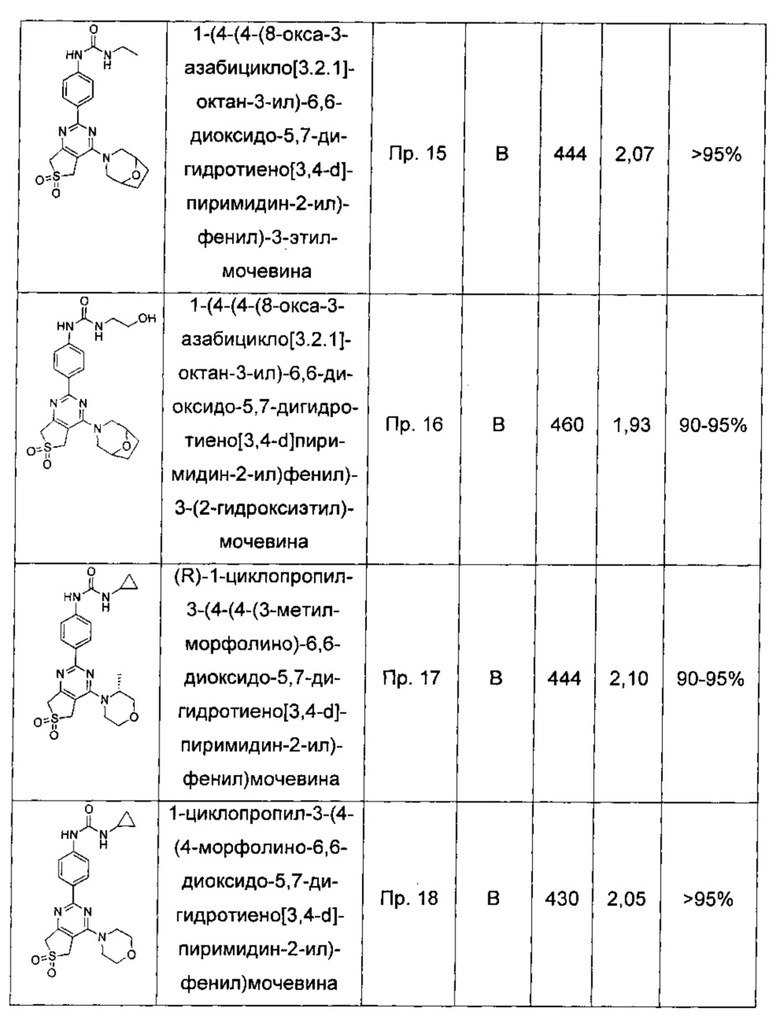

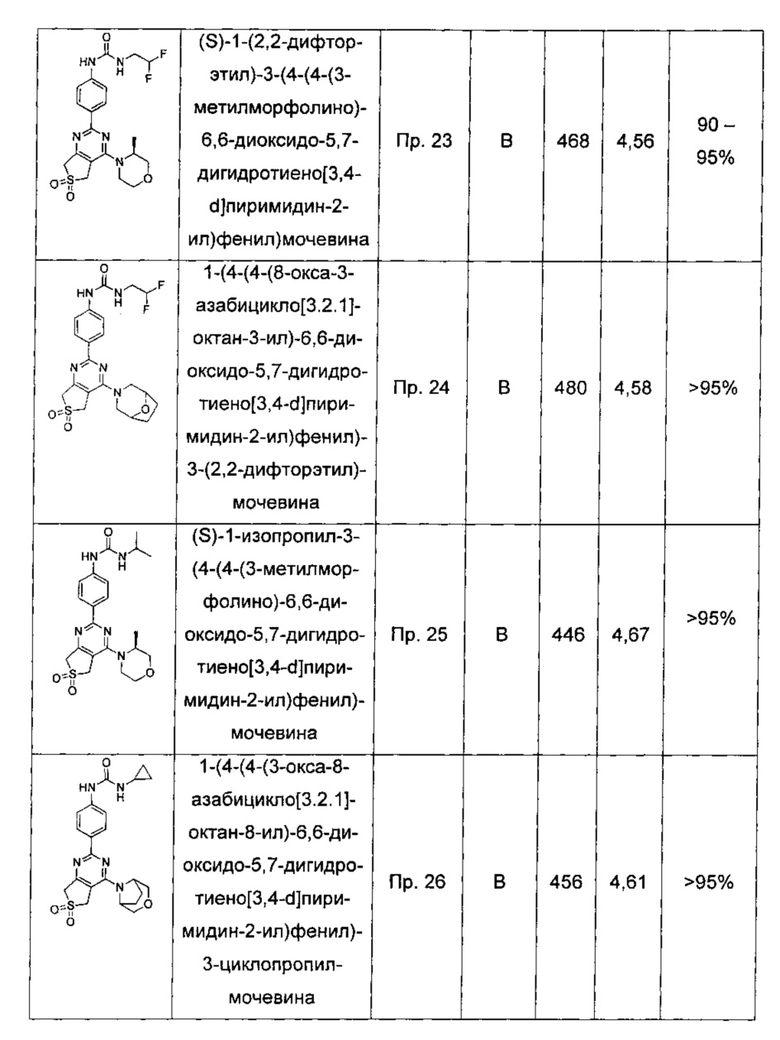
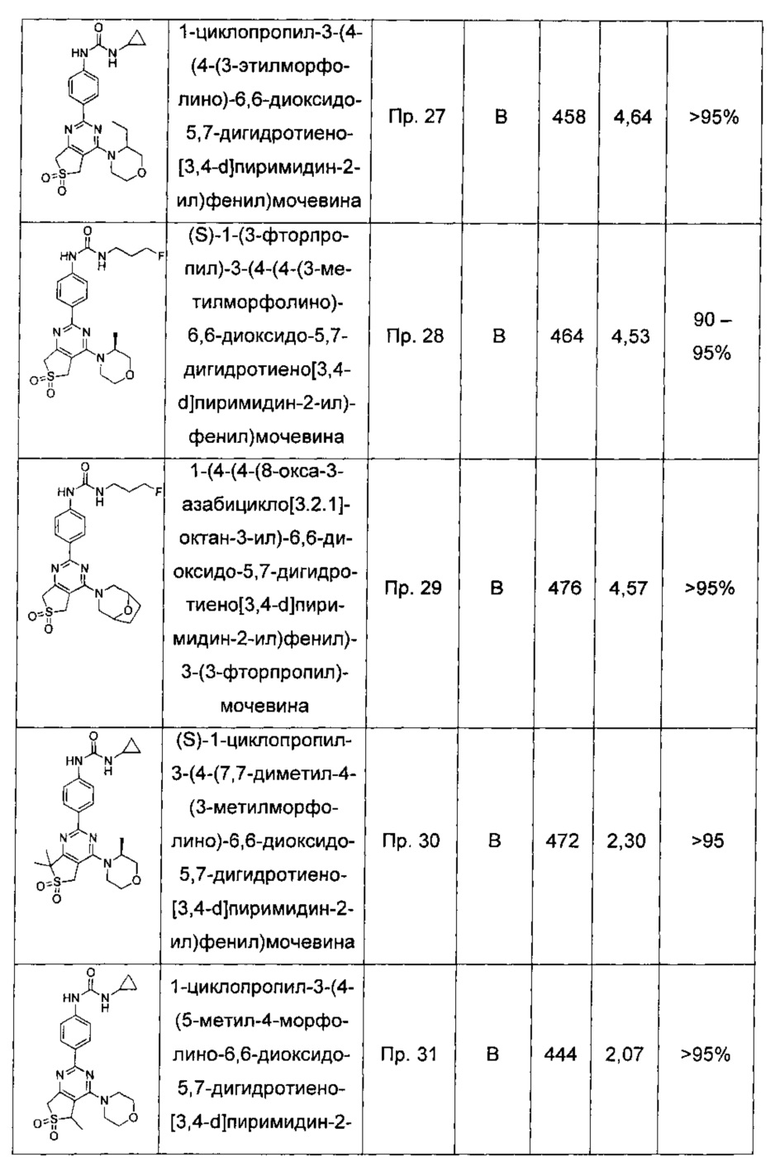

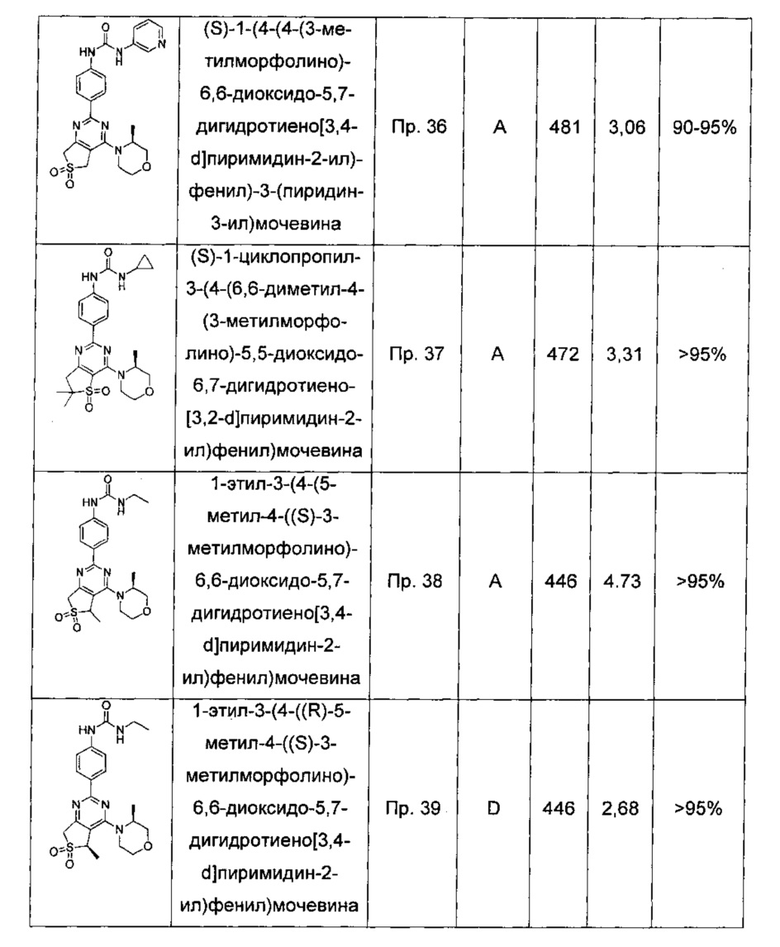
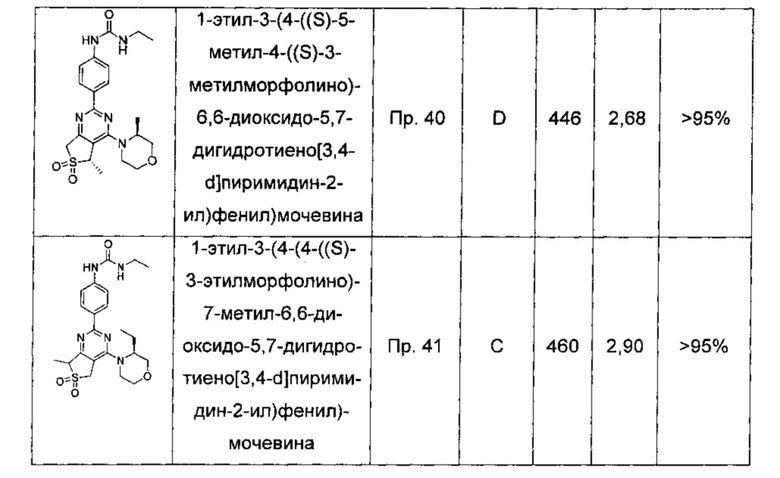
Пример 42: 1-Этил-3-(4-(5,7,7-триметил-4-((S)-3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевина
Стадия (1)
К раствору EtONa (6,5 г, 95,4 ммоль) в EtOH (185 мл) добавляли соединение 1 (18,50 г, 95,4 ммоль) и этиловый эфир 3-меркапто-пропионовой кислоты (1,28 г, 95,4 ммоль). Смесь перемешивали при комнатной температуре в течение ночи. После завершения реакции смесь фильтровали и осадок на фильтре промывали этилацетатом, затем фильтрат концентрировали в вакууме. Концентрат разбавляли этилацетатом и три раза промывали водой. Органическую фазу сушили над MgSO4 и концентрировали в вакууме с получением соединения 2 (2,1 г) в виде жидкости, которую использовали непосредственно на следующей стадии.
Стадия (2)
К раствору EtONa (5,76 г, 84,60 ммоль) в THF (105 мл) по каплям добавляли соединение 2 (10,50 г, 42,30 ммоль), затем смесь нагревали до 60°C и перемешивали в течение в течение ночи. После завершения реакции смесь вливали в насыщ. раствор NH4Cl и экстрагировали этилацетатом. Органическую фазу сушили над MgSO4, затем фильтровали и концентрировали в вакууме с получением неочищенного соединения 3 (7,5 г), которое использовали непосредственно на следующей стадии.
Стадия (3)
Смесь соединения 3 (2,50 г, 12,30 ммоль) и мочевины (2,25 г, 37,50 ммоль) нагревали до 160° в открытой колбе (воду и EtOH, образовашиеся в реакционной смеси, упаривали при высокой температуре), затем коричневое масло дополнительно перемешивали при той же температуре в течение 3 часов. После завершения реакции к го рячей ре акционной смеси добавляли воду (12 мл), осадок отфильтровывали, а осадок на фильтре промывали водой, остаток сушили в вакууме с получением неочищенного соединения 4 (1,6 г) в виде желтого твердого вещества.
Стадия (4)
К раствору соединения 4 (2,0 г, 10 ммоль) в PhPOCl2 (6,0 г, 31 ммоль) нагревали до 160°C, затем смесь перемешивали в течение 3 часов. После завершения реакции смесь охлаждали до комнатной температуры и затем вливали в ледяную воду, водную фазу экстрагировали этилацетатом. Органическую фазу сушили над MgSO4 и концентрировали в вакууме. Остаток очищали посредством хроматографии на силикагеле (петролейный эфир:этилацетат, 50:1) с получением дихлор-ядра (1,35 г, выход 57%) в виде белого твердого вещества. 1H ЯМР (CDCl3, 400 МГц) δ 1.710 (s, 6Н), 4.107 (s, 2Н); ЖХ-МС (ИЭР- (ионизация электрораспылением)): m/z 235 (М+Н)+.
Стадия (5)
К раствору дихлор-ядра (6,0 г, 23,8 ммоль) в DCM (60 мл) в ледяной бане порциями добавляли m-СРВА (12,26 г, 71,30 ммоль) и перемешивали при комнатной температуре в течение ночи. После завершения реакции добавляли раствор DCM (40 мл) и водн. NH4Cl (100 мл). Твердое вещество удаляли фильтрованием и органический слой промывали водн. NaHCO3 и рассолом, сушили над Na2SO4 и концентрировали с получением соединения 6 (6 г, выход 91,6%) в виде белого твердого вещества. 1Н ЯМР (CDCl3, 400 МГц) δ 1.70 (s, 6Н), 4.34 (s, 2Н); ЖХ-МС (ИЭР): m/z 267 (М+Н)+.
Стадия (6)
К раствору соединения 6 (1,4 г, 5,2 ммоль) и TEA (1,1 г, 10,5 ммоль) в DMF (14 мл) по каплям добавляли (S)-3-метилморфолин (0,52 г, 5,20 ммоль) и перемешивали при комнатной температуре в течение ночи. После завершения реакции добавляли EtOAc (20 мл) и H2O (50 мл). Органический слой промывали рассолом, сушили над Na2SO4 и концентрировали с получением неочищенного продукта, который очищали колоночной хроматографией на силикагеле (элюент: петролейный эфир/этилацетат от 5/1 до 2/1) с получением 6,6-диоксида (S)-2-хлор-7,7-диметил-4-(3-метилморфолино)-5,7-дигидротиено-[3,4-d]пиримидина (0,8 г, выход 46%) в виде белого твердого вещества. 1Н ЯМР (CDCl3, 400 МГц) δ 1.39 (d, 3Н), 1.60 (s, 6Н), 3.46-3.56 (m, 2Н), 3.70-3.77 (m, 2Н), 3.96-3.98 (m, 2Н), 4.16-4.26 (m, 3Н); ЖХ-МС (ИЭР-): m/z 332 (М+Н)+.
Стадия (7)
LDA (диизопропиламид лития, 2,5 мл, 5 ммоль) при -78°C добавляли по каплям к раствору 6,6-диоксида (S)-2-хлор-7,7-диметил-4-(3-метилморфолино)-5,7-дигидротиено[3,4-d]пиримидина (Стадия (6), 1,5 г, 4,5 ммоль) в THF (30 мл). Смесь перемешивали при этой температуре в течение 0,5 ч, к вышеуказанной смеси по каплям добавляли Mel (0,7 г, 5 ммоль) в THF (5 мл). Полученную смесь перемешивали при комнатной температуре в течение 2 ч. Затем смесь вливали в насыщ. NH4Cl, экстрагировали EtOAc (3×30 мл). Органический слой промывали рассолом, сушили над Na2SO4 и концентрировали в вакууме. Остаток очищали колоночной хроматографией с получением 6,6-диоксида 2-хлор-5,7,7-триметил-4-((S)-3-метилморфолино)-5,7-дигидротиено[3,4-d]пиримидина (1,2 г, выход 77%) в виде желтого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) 1.38-4.31 (m, 1Н), 4.29-4.21 (m, 1Н), 3.99-3.96 (m, 1Н), 3.78-3.43 (m, 5Н), 1.65-1.25 (m, 12Н).
Стадия (3)
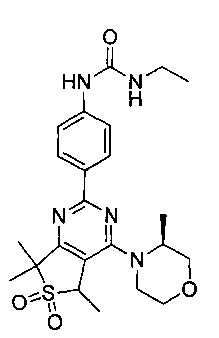
Указанное в заголовке соединение синтезировали согласно процедуре в Примере 5 (стадия (8)), используя 6,6-диоксид 2-хлор-5,7,7-триметил-4-((S)-3-метилморфолино)-5,7-дигидротиено[3,4-d]пиримидина и 1-этил-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)мочевину. ЖХ-МС (метод D), (ЭР+) 474, RT=3,00 мин.
Пример 43: 1-циклопропил-3-(4-(7-фтор-7-метил-4-((S)-3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)-фенил)мочевина
Стадия (1)
К раствору 6,6-диоксида 2,4-дихлор-7-метил-5,7-дигидротиено[3,4-d]-пиримидина (Пример 7, стадия (6)) (160 мг, 0,63 ммоль) и триэтиламина (64 мг, 0,63 ммоль) в DMF (3 мл) при 0°C добавляли 3-(S)-метилморфолин (57 мг, 0,57 ммоль). Полученную смесь перемешивали при к.т. в течение 2 ч. Растворитель удаляли под вакуумом и полученный остаток распределяли между водой и этилацетатом. Органический слой сушили (Na2SO4) и концентрировали с получением продукта, который очищали посредством препаративной ТСХ (петролейный эфир/этилацетат, 2/1) с получением 6,6-диоксида 2-хлор-7-метил-4-[(3S)-3-метилморфолин-4-ил]-5,7-дигидротиено[3,4-d]пиримидина (72 мг, 36%-ный выход). ЖХ-МС (метод А), (ЭР+) 318/320, RT=2,95 мин.
Стадия (2)
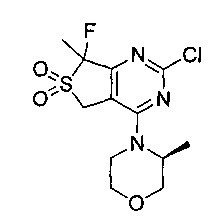
Раствор 6,6-диоксида 2-хлор-7-метил-4-[(3S)-3-метилморфолин-4-ил]-5,7-дигидротиено[3,4-d]пиримидина (0,15 г, 0,47 ммоль) в абсолютном THF (10 мл) охлаждали до -70°C, по каплям добавляли LDA (2 М, 0,26 мл) и перемешивали в течение 30 мин. Медленно добавляли NFSI (N-фторбензолсульфонимид, 0,15 г, 0,47 ммоль) в THF (2 мл). Затем смесь оставляли постепенно нагреваться до комнатной температуры и перемешивали в течение 1 часа. После завершения реакции смесь вливали в раствор NH4Cl, экстрагировали ЕА, органический слой промывали рассолом, сушили над Na2SO4, выпаривали и очищали посредством препаративной ТСХ с получением 6,6-диоксида 2-хлор-7-фтор-7-метил-4-((S)-3-метилморфолино)-5,7-дигидротиено[3,4-d]пиримидина (0,10 г, выход 63%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) 4.44-4.38 (m, 1Н), 4.19-3.99 (m, 3Н), 3.78-3.40 (m, 5Н), 2.03 (d, 3Н), 1.57-1.35 (m, 3Н).
Стадия (3)
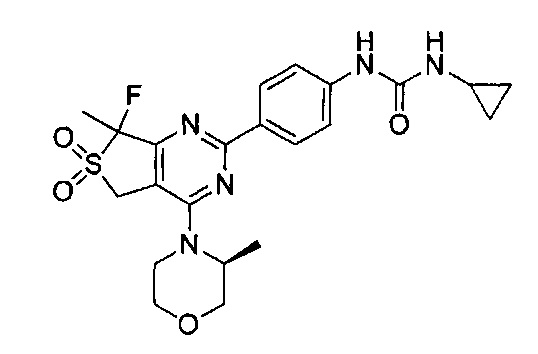
Указанное в заголовке соединение синтезировали согласно процедуре в Примере 5 (стадия (8)), используя 6,6-диоксид 2-хлор-7-фтор-7-метил-4-((S)-3-метилморфолино)-5,7-дигидротиено[3,4-d]пиримидина и 1-циклопропил-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)мочевину. ЖХ-МС (метод D), (ЭР+) 476, RT=2,90 мин.
Пример 44: 1-Циклопропил-3-(4-(5,7-диметил-4-((S)-3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевина
Стадия (1)

Раствор 2-хлор-7-метил-4-[(3S)-3-метилморфолин-4-ил]-5,7-дигидро-тиено[3,4-d]пиримидина (Пример 6, стадия (7)) (0,5 г, 1,58 ммоль) в THF (10 мл) охлаждали до -70°C, по каплям добавляли LDA (2 М, 0,86 мл) и перемешивали в течение 30 мин. Медленно добавляли Mel (0,24 г, 1,73 ммоль) в THF (2 мл). Смесь оставляли постепенно нагреваться до комнатной температуры и перемешивали в течение 2 ч. После завершения реакции реакционную смесь вливали в раствор NH4Cl, экстрагировали EtOAc, органический слой промывали рассолом, сушили над Na2SO4, выпаривали и очищали с помощью колонки с получением 6,6-диоксида 2-хлор-5,7-диметил-4-((S)-3-метилморфолино)-5,7-дигидротиено[3,4-d]пиримидина (0,2 г, выход 38%) в виде слегка желтоватого твердого вещества. ЖХ-МС (метод G), (ЭР+) 332, RT=1,27 мин.
Стадия (2)
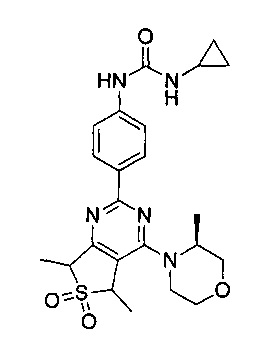
Указанное в заголовке соединение синтезировали согласно процедуре в Примере 5 (стадия (8)), используя 6,6-диоксид 2-хлор-5,7-диметил-4-((S)-3-метилморфолино)-5,7-дигидротиено[3,4-d]пиримидина и 1-циклопропил-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)мочевину. ЖХ-МС (метод D), (ЭР+) 472, RT=2,90 мин.
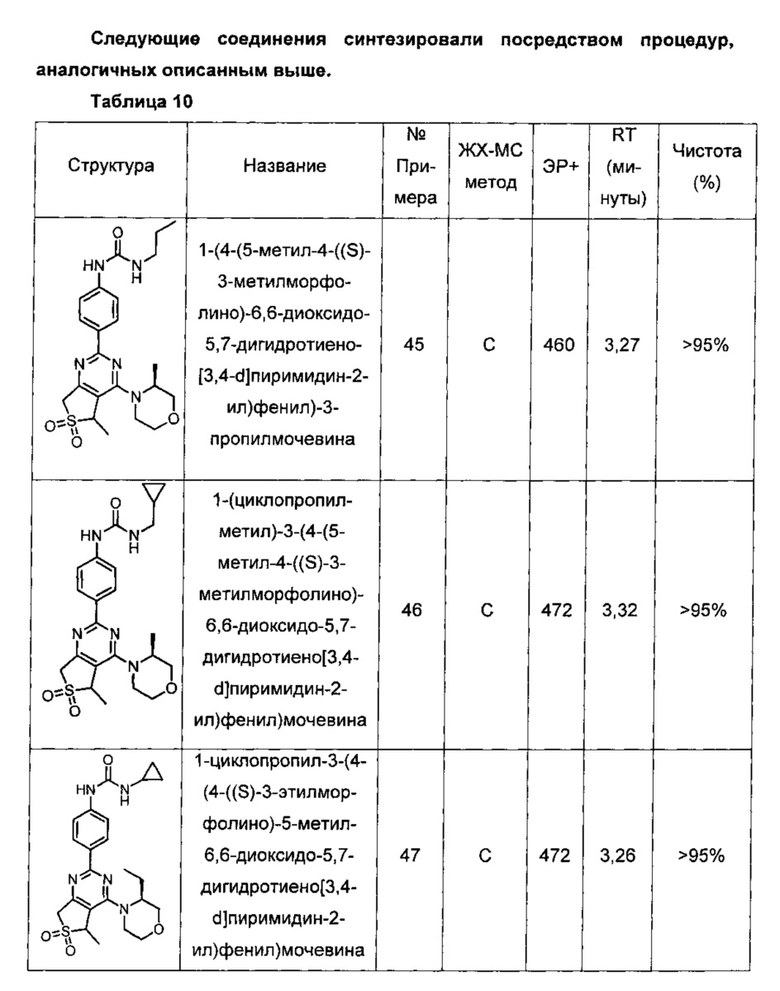
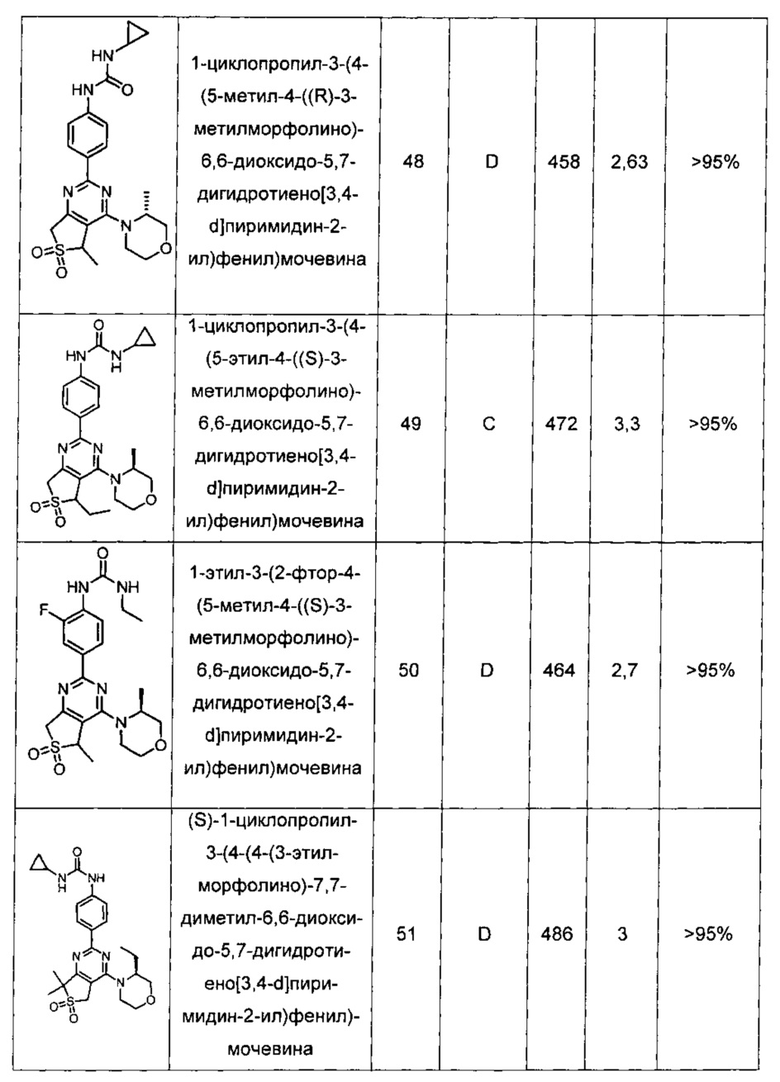
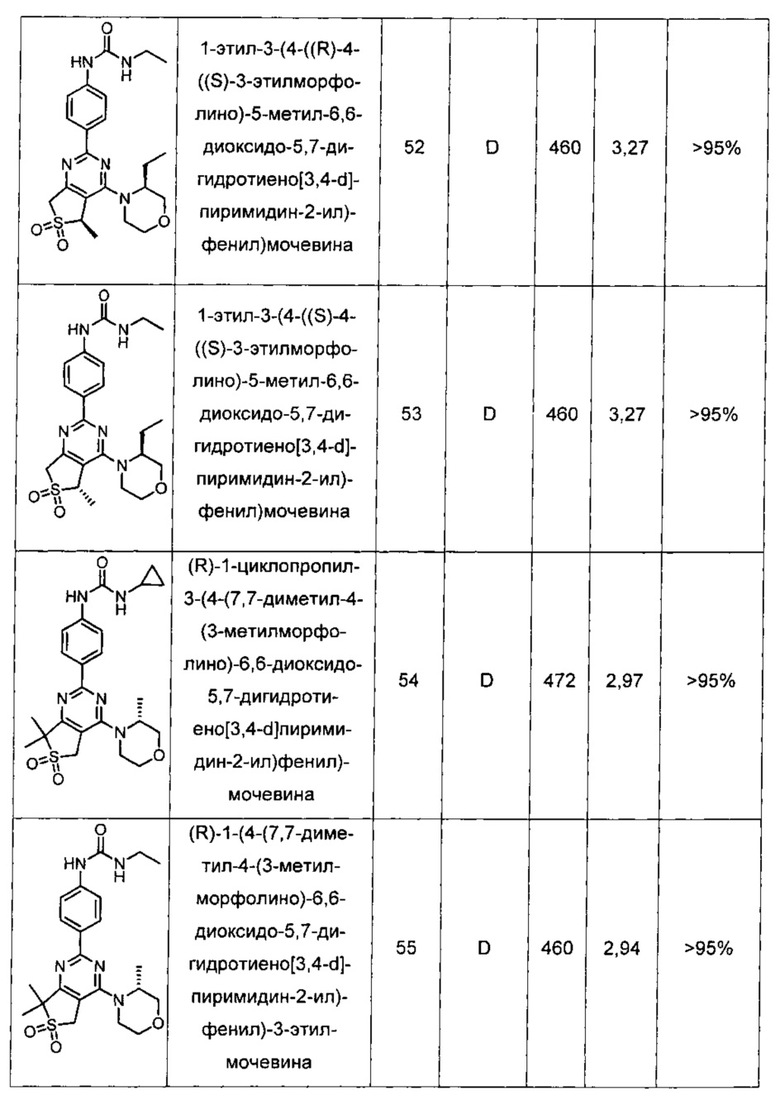
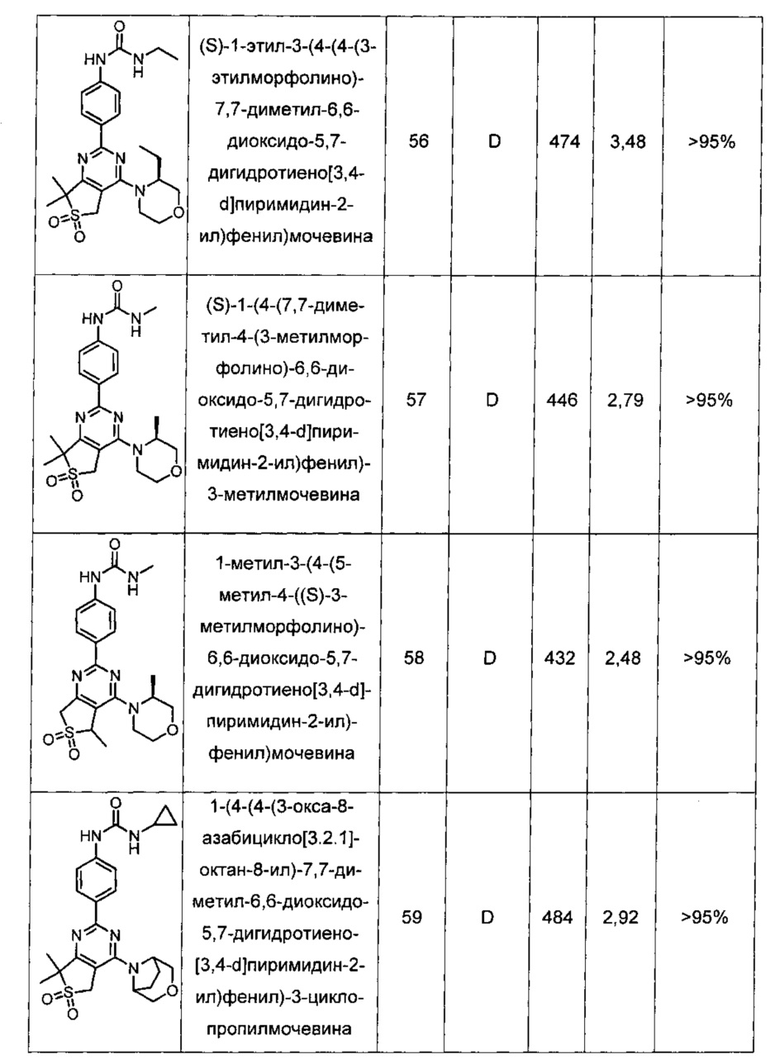
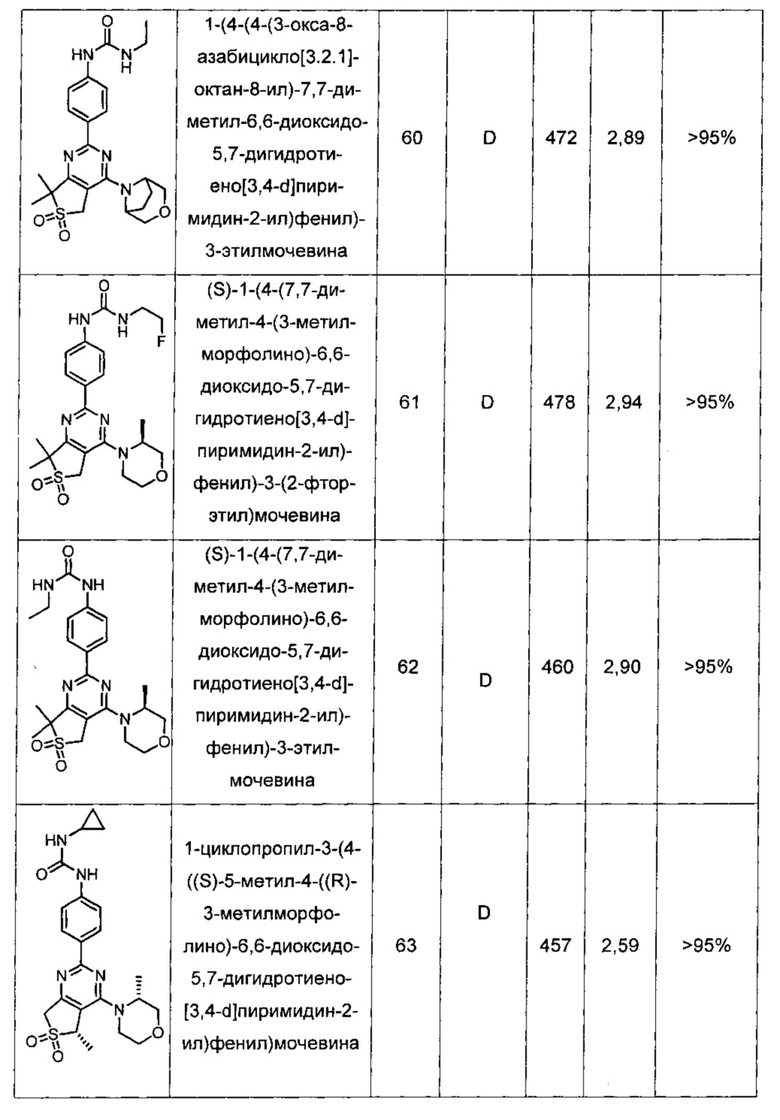
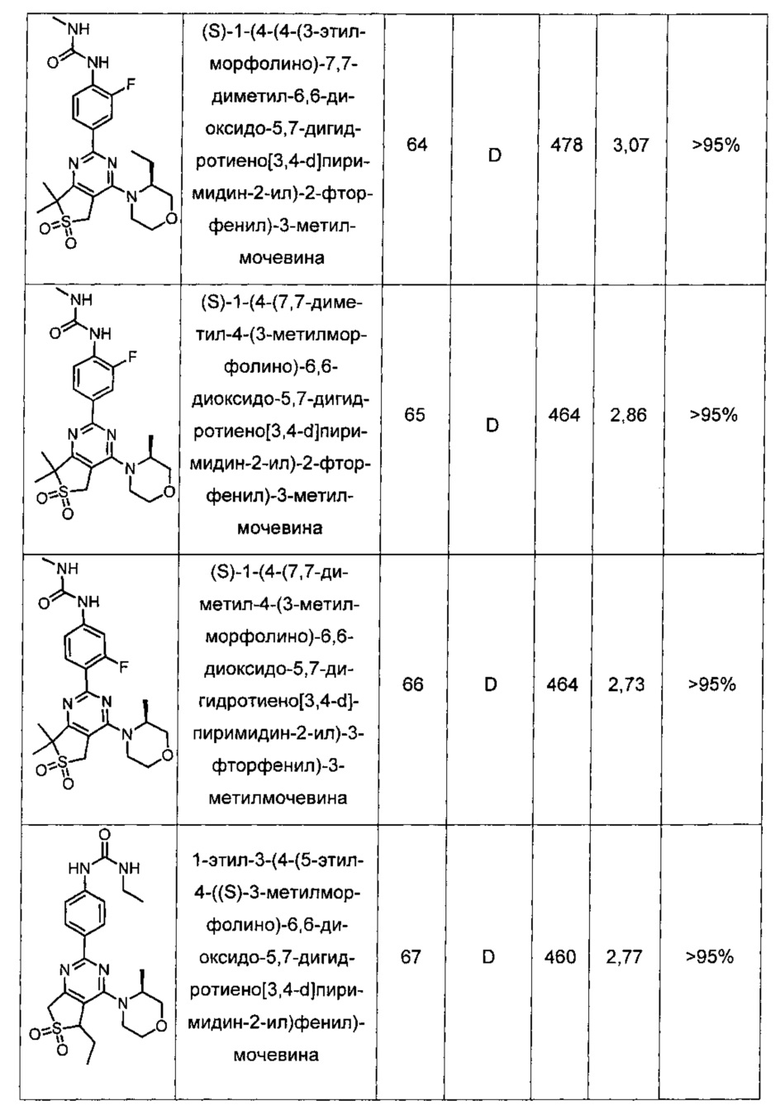

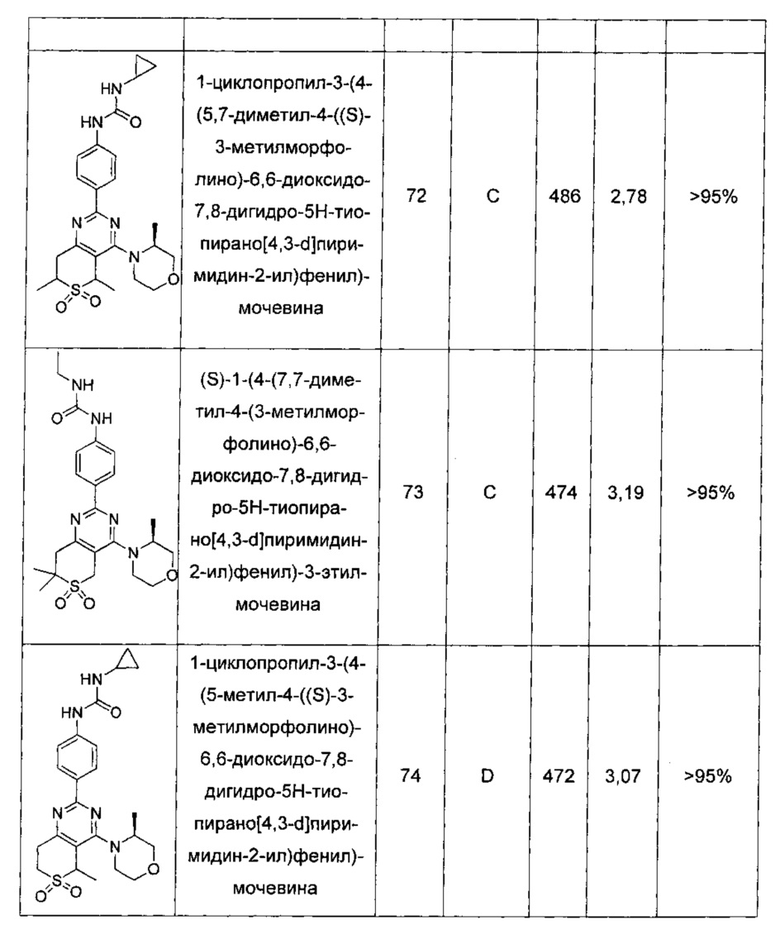
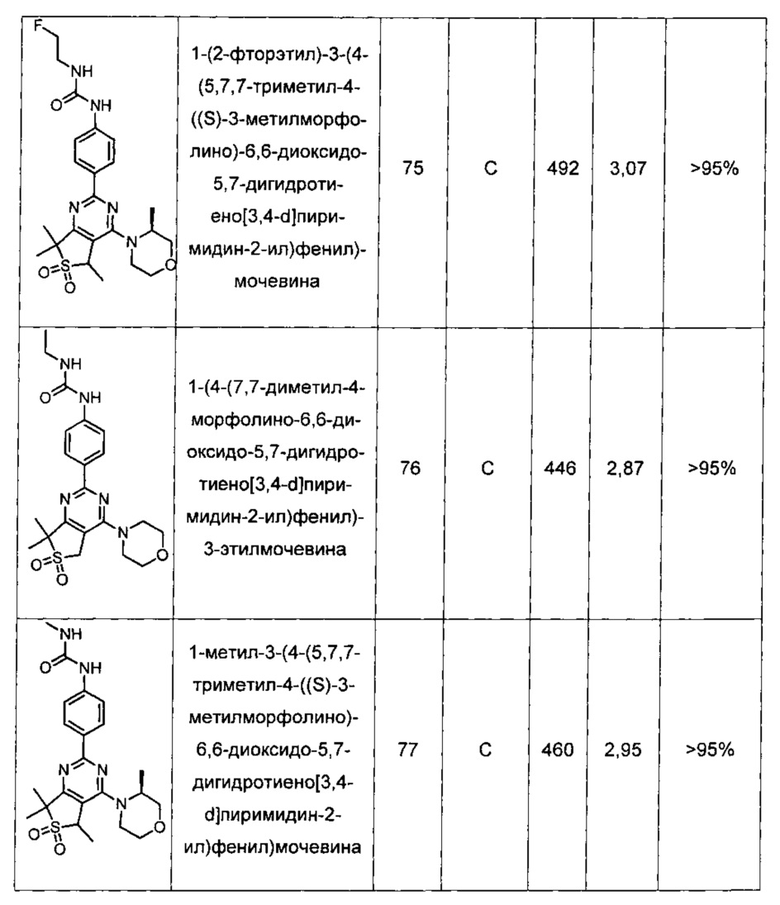
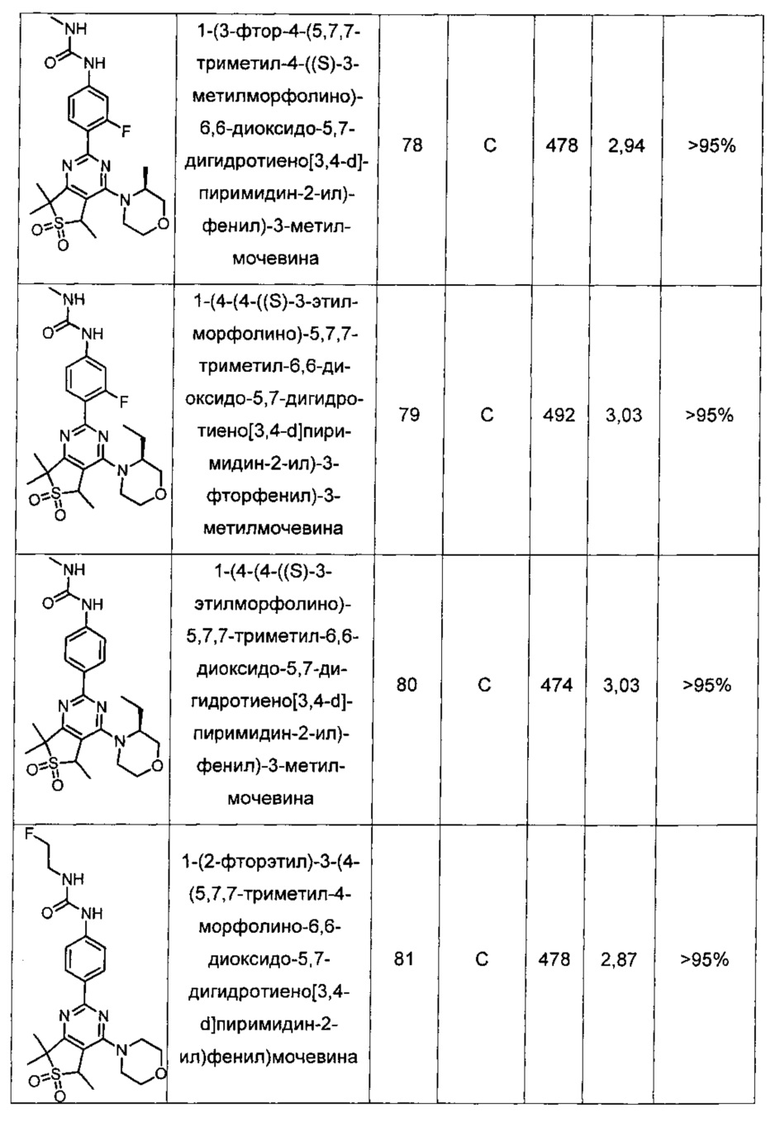

Описание анализа в Kinobeads
Определение эффекта соединения по изобретению в отношении mTOR
Соединения по настоящему изобретению, как они раскрыты в данном описании, тестировали в mTOR Kinobeads анализе, как описано ниже. Кратко, тестируемые соединения (в разных концентрациях) и аффинную матрицу (смесь 1:1 шариков с иммобилизованным фенилтиазольным лигандом 1 и шариков с иммобилизованным фенилморфолин-хроменовым лигандом; WO 2009/098021) добавляли к аликвотам клеточного лизата и оставляли связываться с белками в образце лизата. По прошествии времени инкубирования шарики с захваченными белками отделяли от лизата. Связанные белки затем элюировали, детектировали присутствие mTOR, PI3K (фосфатидилинозит-3-киназа) альфа (PI3Ka), PI3K бета (PI3Kb), PI3K гамма (PI3Kg), PI3K дельта (PI3Kd) и ДНК-зависимой протеинкиназы (DNA-PK) и оценивали их количественно, используя специфическое антитело в процедуре дотблоттинга и системе инфракрасной детекции Odyssey. Строили кривые доза-ответ для индивидуальных киназ и рассчитывали величины IC50. Kinobeads анализы для PI3 киназ (WO-A 2008/015013) и для получения профилей селективности киназ (WO 2009/098021) были описаны ранее.
Промывка аффинной матрицы
Аффинную матрицу (шарики с иммобилизованным фенилморфолин-хроменовым лигандом) промывали три раза 15 мл 1×DP буфера, содержавшего 0,2% NP40 (IGEPAL® СА-630, Sigma, #13021), и затем ресуспендировали в 5,5 мл 1×DP буфера, содержавшего 0,2% NP40 (10%-ная суспензия шариков).
5×DP буфер: 250 мМ трис-HCl (трис(гидроксиметил)аминометан гидрохлорид) pH 7,4, 25% глицерина, 7,5 мМ MgCl2, 750 мМ NaCl, 5 мМ Na3VO4, 5х-лизирующий буфер фильтруют через фильтр 0,22 мкм и хранят в аликвотах при -80°C. 5×DP буфер разбавляют до 1×DP буфера, содержащего 1 мМ DTT (дитиотреитол) и 25 мМ NaF.
Приготовление тестируемых соединений
Исходные растворы тестируемых соединений готовили в DMSO. В 96-луночном планшете готовили 30 мкл раствора разбавленных тестируемых соединений в концентрации 5 мМ в DMSO. Начиная с этого раствора, готовили серийные разведения 1:3 (9 стадий). В качестве контрольных экспериментов (в отсутствие тестируемого соединения) использовали буфер, содержащий 2% DMSO. Соединение PI-103 служило в качестве положительного контроля (Calbiochem каталожный номер 528100).
Клеточная культура и приготовление клеточных лизатов
Клетки Jurkat (АТСС каталожный номер TIB-152 Jurkat, клон Е6-1) выращивали в 1-литровых вращающихся колбах (Integra Biosciences, #182101) в суспензии в среде RPMI 1640 (Invitrogen, #21875-034), обогащенной 10% фетальной телячьей сыворотки (Invitrogen), при плотности между 0,15×106 и 1,2×106 клеток/мл. Клетки собирали центрифугированием, промывали один раз буфером 1×PBS (забуференный фосфатом физиологический раствор, Invitrogen, #14190-094) и клеточные осадки замораживали в жидком азоте и затем хранили при -80°C.
Клетки Jurkat гомогенизировали в гомогенизаторе Potter S в следующем лизирующем буфере: 50 мМ трис-HCl, 0,8% NP40, 5% глицерина, 150 мМ NaCl, 1,5 мМ MgCl2, 25 мМ NaF, 1 мМ ванадата натрия, 1 мМ DTT, pH 7,5. На 25 мл буфера добавляли одну полную таблетку, не содержащую EDTA (этилендиаминтетрауксусная кислота) (коктейль ингибиторов протеаз, Roche Diagnostics, 1873580). Материал 10 раз подвергали гомогенизации в гомогенизаторе Даунса, используя механизированный POTTER S, переносили в цетрифужные пробирки falcon на 50 мл, инкубировали в течение 30 минут на льду и центрифугировали в течение 10 мин при 20000 g при 4°C (10000 об./мин в Sorvall SLA600, предварительное охлаждение). Супернатант переносили в ультрацентрифужную (UZ)-поликарбонатную пробирку (Beckmann, 355654) и центрифугировали в течение 1 часа при 100000 g при 4°C (33500 об./мин в Τi50.2, предварительное охлаждение). Супернатант снова переносили в чистую центрифужную пробирку falcon на 50 мл, определяли концентрацию белка анализом по Брэдфорду (Bradford, BioRad) и готовили образцы, содержащие 50 мг белка на аликвоту. Образцы немедленно использовали для экспериментов или замораживали в жидком азоте и хранили замороженными при -80°C.
Разведение клеточного лизата
Лизат клеток Jurkat (приблизительно 50 мг белка на планшет) оттаивали в водяной бане при комнатной температуре и затем хранили на льду. К оттаявшему клеточному лизату добавляли 1×DP 0,8% NP40 буфер, содержащий ингибиторы протеаз (1 таблетка на 25 мл буфера; коктейль ингибиторов протеаз, не содержащий EDTA; Roche Diagnostics 1873580) для того, чтобы достичь финальной концентрации белка 5 мг/мл общего белка. Разбавленный клеточный лизат хранили на льду.
Инкубирование лизата с тестируемым соединением и аффинной матрицей
В 96-луночный фильтровальный планшет (Multiscreen HTS, BV Filter Plates, Millipore #MSBVN1250) добавляли на лунку: 50 мкл аффинной матрицы (10%-ная суспензия шариков), 3 мкл раствора соединения и 100 мкл разбавленного клеточного лизата. Планшеты плотно закрывали и инкубировали в течение трех часов в холодной комнате на Thermomixer со встряхиванием (750 об./мин). Затем планшет промывали три раза 230 мкл промывочного буфера (1×DP 0,4% NP40). Фильтровальный планшет помещали на поверхность планшета для сбора (Greiner bio-one, РР (полипропилен) - микропланшет 96-луночный с V-образным дном лунок, 65120) и шарики затем элюировали 20 мкл буфера для образцов (100 мМ трис, pH 7,4, 4% SDS (додецилсульфат натрия), 0,00025% бромфеноловый голубой, 20% глицерин, 50 мМ DTT). Элюат быстро замораживали при -80°C и хранили при -20°C.
Обнаружение и количественная оценка элюированных киназ
Киназы в элюатах обнаруживали и оценивали количественно посредством нанесения на нитроцеллюлозные мембраны и использования первого антитела, направленного против представляющей интерес киназы, и меченного флуоресцентной меткой второго антитела (анти-мышиные или анти-кроличьи антитела IRDye™ от Rockland). Систему инфракрасной визуализации Odyssey от LI-COR Biosciences (Lincoln, Nebraska, USA) использовали в соответствии с инструкциями, предоставленными изготовителем (Schutz-Geschwendener et al., 2004. Quantitative, two-color Western blot d etection with infrared fluorescence. Опубликовано в мае 2004 г. LI-COR Biosciences, www.licor.com).
После нанесения элюатов нитроцеллюлозную мембрану (BioTrace NT; PALL, #BTNT30R) сначала блокировали посредством инкубирования с Odyssey блокирующим буфером (LICOR, 927-40000) в течение одного часа при комнатной температуре. Блокированные мембраны затем инкубировали в течение 16 часов при 25°C (или при 4°C) с первым антителом, разбавленным в Odyssey блокирующем буфере (LICOR #927-40000). После этого мембрану промывали дважды в течение 10 минут PBS буфером, содержащим 0,1% Твин 20, при комнатной температуре. Затем мембрану инкубировали в течение 60 минут при комнатной температуре с детектирующим антителом (IRDye™ меченое антитело от Rockland), разведенным в Odyssey блокирующем буфере (LICOR #927-40000). После этого мембрану промывали дважды каждый раз в течение 10 минут 1×PBS буфером, содержащим 0,1% Твин 20, при комнатной температуре. Затем мембрану один раз промывали PBS буфером для удаления остаточного Tween 20. Мембрану хранили в PBS буфере при 4°C и затем сканировали с помощью прибора Odyssey. Сигналы флуореценции регистрировали и анализировали согласно инструкциям изготовителя.
Источники и разведения антител
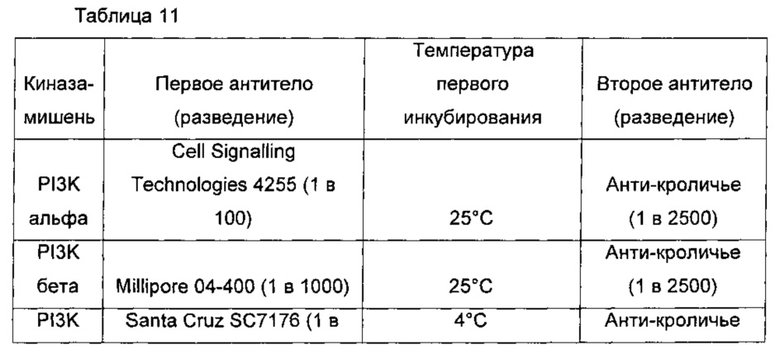
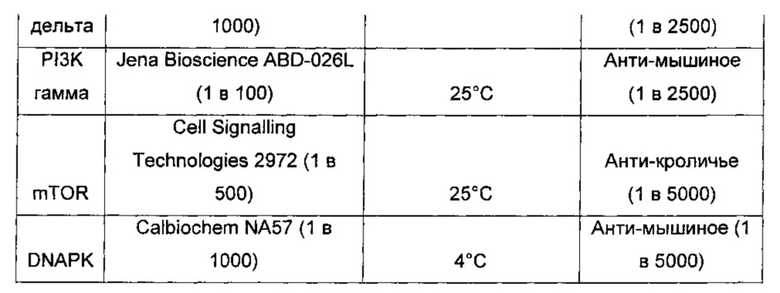
Результаты с Kinobeads
Таблица 12: Показатели ингибирования (IC50 в мкМ), как определено в анализе kinobeads™ (уровень активности: А<0,1 мкМ≤В≤1 мкМ≤С≤10 мкМ<D)

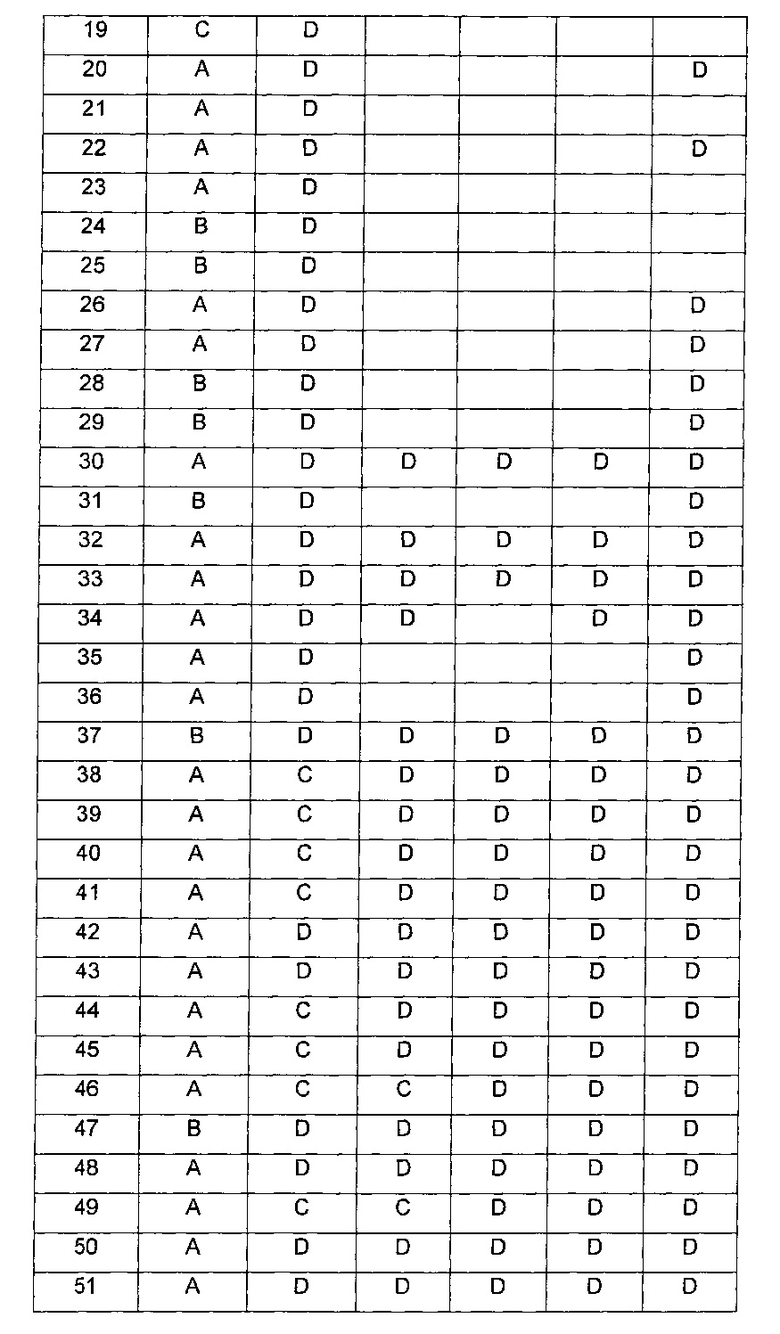
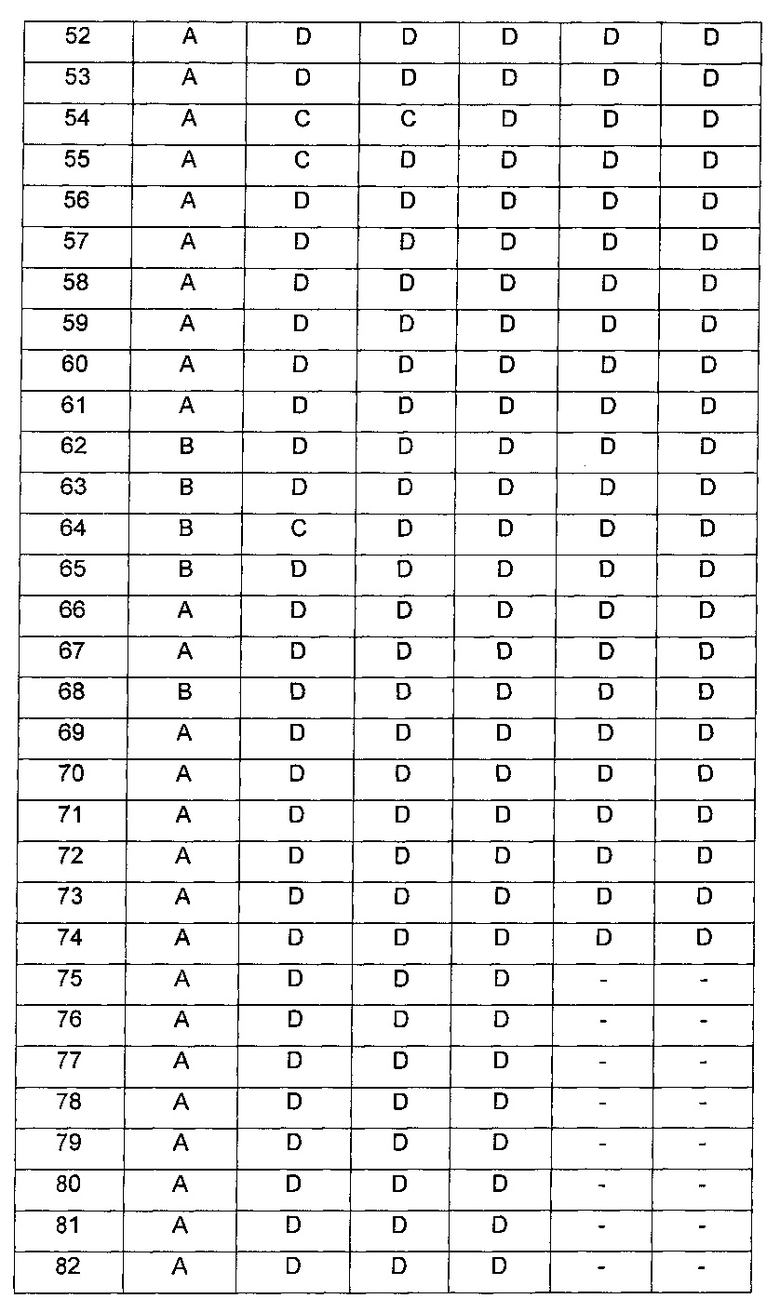
In vitro фосфо-56 и фосфо-Akt клеточный анализ
Активация передачи сигнала mTOR имеет результатом фосфорилирование некоторых находящихся в нисходящем направлении мишеней. В клетках mTOR существует в двух разных белковых комплексах. mTOR комплекс-1 (mTORC1) фосфорилирует и активирует S6 киназу 1 (S6K1) и S6 киназу 2 (S6K2) (также известную как p70S6K), которые затем фосфорилируют S6 рибосомальный белок (S6RP) (также известный как RPS6)3. S6RP фосфорилируется по серину 235, серину 236, серину 240 и серину 244 как pS6K1, так и pS6K2. mTOR комплекс-2 (mTORC2) фософрилирует AKT по серину 473, что активирует путь передачи сигнала AKT.
Анализ измеряет ингибирование тестируемыми соединениями S6RP серин-240/244 фосфорилирования и ингибирование Akt серин-473 фосфорилирования в клетках, полученных из почки эмбриона человека HEK293T/17 (АТСС CRL-11268).
Клеточную линию HEK293T/17 поддерживают в среде DMEM (Invitrogen каталожный номер 41965-039), обогащенной 10% FCS (фетальная телячья сыворотка), при 37°C в увлажненном инкубаторе при 5% CO2.
Клетки высевают в 96-луночные планшеты в концентрации 40000 клеток/лунку (pS6RP S240/244 анализ) или 80000 клеток/лунку (pAkt S473 анализ) в 90 мкл ростовой среды (DMEM (модифицированная по способу Дульбекко среда Игла), 2% FCS). Планшеты инкубируют в течение 1 часа в увлажненном инкубаторе для того, чтобы дать клеткам возможность закрепиться. Клетки обрабатывают либо 8 концентрациями тестируемого соединения, либо DMSO в качестве контролей (финальная концентрация DMSO составляет 0,1%) и инкубируют при 37°C в течение 2 часов. Затем добавляют 20 мкл 5× концентрированного лизирующего буфера (750 мМ NaCl, 100 мМ трис pH 7,4, 5 мМ ADTA, 5 мМ EGTA (этиленгликоль-бис(2-аминоэтиловый эфир)-N,N,N,N-тетрауксусной кислоты), 5% Triton Х-100), планшеты плотно закрывают и инкубируют в течение 15 минут при 4°C с осторожным перемешиванием. После завершения лизиса клеток 25 мкл клеточного лизата переносят в планшет MesoScale, покрытый антителом к pS6RP Ser240/244 (MesoScale Discovery K150DGD-3) или антителом к pAkt Ser 473 (MesoScale Discovery K151DGD-3). Планшеты перед инкубированием блокировали 150 мкл MesoScale Discovery блокирующего раствора-A в течение 1 часа при комнатной температуре, а затем промывали 150 мкл 1× Трис промывочного буфера на лунку. После переноса клеточного лизата в MSD планшет белок pS6RP (или pAkt) захватывается на покрытом антителе посредством инкубирования при комнатной температуре в течение 1 часа с осторожным перемешиванием. После завершения стадии захвата планшет промывают три раза 150 мкл 1× Трис промывочного буфера на лунку. Затем добавляют 25 мкл детектирующего антитела, конъюгированного с Sulfo-Tag, и инкубируют в течение 1 часа при комнатной температуре с осторожным перемешиванием. Затем раствор антитела удаляют и планшет промывают 3 раза 150 мкл 1× Трис промывочного буфера на лунку и добавляют 150 мкл буфера для регистрации. Планшеты анализируют на планшетном ридере MSD 2400 (MesoScale Discovery). Анализ данных осуществляют, используя нелинейную регрессию для сигмоидальной зависимости доза-ответ с переменным наклоном.
Результаты клеточного анализа
Таблица 13: показатели ингибирования (IC50 в мкМ) (уровень активности: А<0,1 мкМ≤В≤1 мкМ≤С≤10 мкМ<D)
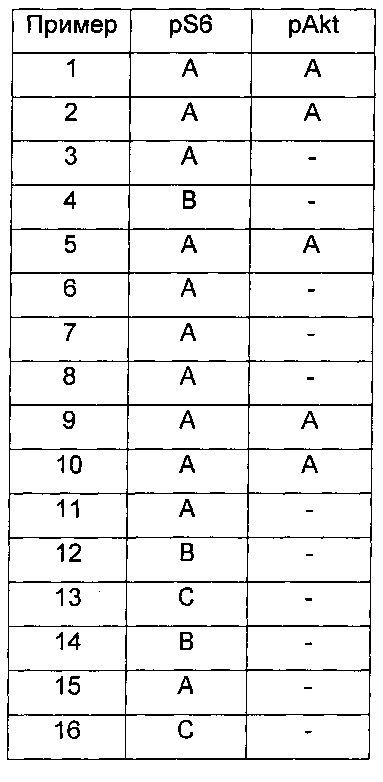


In vitro анализ с цельной кровью человека
В этом анализе измеряют высвобождение IFNy в цельной крови после обработки αCD3/αCD28 и IL-2. Цельная кровь содержит циркулирующие Τ лимфоциты; αCD3/αCD28 стимуляция имитирует антиген-рецепторную передачу сигнала, результатом которой являются эпигенетические изменения в локусе гена IFNG, так что при добавлении IL-2 продуцируется IFNγ.
Цельную кровь человека с Na-гепарином в качестве антикоагулянта получают от Clinical Trials Laboratory Services. Кровь разбавляют в 1,4 раза средой RPMI 1640 (Lonza, BE12-167F) и по 175 мкл/лунку распределяют в 96-луночные планшеты низкого испарентия (Falcon, BD Labware, 353072). Кровь обрабатывают 10 концентрациями тестируемых соединений или одним только DMSO для контролей (финальная концентрация DMSO составляет 0,2%) и инкубируют при 37°C в течение 1 часа. Затем кровь стимулируют 1 мкг/мл αCD3 (R&D Systems, МАВ100), 1 мкг/мл αCD 28 (BD Pharmingen, 555725) и IL-2 в концентрации 10 нг/мл (Peprotech, 200-02). Затем кровь оставляют инкубироваться при 37°C в течение 18 часов. Затем планшеты центрифугируют при 250 g в течение 5 минут для осаждения клеток крови и 25 мкл плазмы переносят в MesoScale планшет, покрытый антителом к IFNy (MesoScale Discovery K151AEC-2), содержащий 25 мкл/лунку блокирующего раствора-2 (MesoScale Discovery Blocking Solution-2). Планшеты блокировали посредством инкубирования с 25 мкл/лунку блокирующего раствора-2 (MesoScale Discovery Blocking Solution-2) в течение 30 минут при комнатной температуре. После переноса сыворотки в MSD планшет белок IFNγ захватывается на покрытом антителе посредством инкубирования при комнатной температуре в течение 2 часов с осторожным перемешиванием. После стадии захвата планшет промывают три раза 150 мкл/лунку 1× PBS-Tween промывочным буфером и добавляют 25 мкл/лунку антитела для детекции, конъюгированного с Sulfo-Tag, и инкубируют в течение 2 часов при комнатной температуре с осторожным перемешиванием. Затем раствор антитела удаляют и планшет промывают 3 раза 150 мкл/лунку буфера 1× PBS-Tween и добавляют 150 мкл/лунку буфера MesoScale Discovery Read. Планшеты анализируют на планшетном ридере MSD 2400 (MesoScale Discovery). Анализ данных осуществляют, используя нелинейную регрессию для сигмоидальной кривой доза-ответ с переменным наклоном.
Результаты анализа цельной крови
Таблица 14: показатели ингибирования (pIC50)
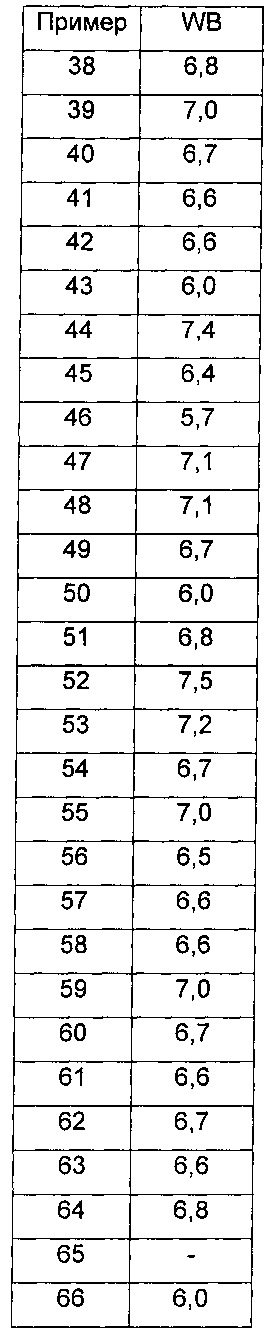
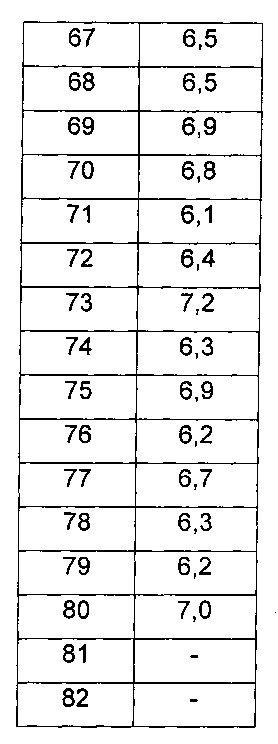
Сасо-2 проницаемость
Двунаправленные Сасо-2 анализы для оценки проницаемости соединения в желудочно-кишечном тракте осуществляют, как описано ниже.
Клетки Сасо-2 получали из Европейской коллекции клеточных культур (ЕСАСС, кат. 86010202) и использовали после 21 суток культивирования клеток в 24-луночных Transwell планшетах (Fisher TKT-545-020В). 2×105 клеток/лунку засевали в ростовой среде, состоящей из DMEM + GlutaMAXI + 1% NEAA (незаменимые аминокислоты) + 10% FBS (FetalClone II) + 1% Pen/Strep (пенициллин/стрептомицин). Среду меняли каждые 2-3 суток.
Тестируемые референсные соединения (пропранолол и родамин 123 или винбластин, все получены от Sigma) готовили в сбалансированном солевом растворе Хэнкса (Hanks'), содержащем 25 мМ HEPES (4-(2-оксиэтил)1-пиперазинэтансульфоновая кислота, pH 7,4) и добавляли либо в апикальную (125 мкл), либо базолатеральную (600 мкл) камеру сборного планшета Transwell в концентрации 10 мкМ с финальной концентрацией DMSO 0,25%.
50 мкМ Lucifer желтого (Sigma) добавляли к донорному буферу во всех лунках для оценки целостности клеточных слоев посредством мониторинга проникновения Lucifer желтого. Поскольку Lucifer желтый (LY) не может свободно проникать через липофильные барьеры, высокая степень транспорта LY указывает на низкую целостность клеточного слоя. После 1 часа инкубирования при 37°C при встряхивании на орбитальном шейкере при 150 об./мин отбирали аликвоты по 70 мкл как из апикальной (А), так и базальной (В) камеры и добавляли к 100 мкл раствора 50:50 ацетонитрил : вода, содержащего аналитический внутренний стандарт (0,5 мкМ карбамазепин), в 96-луночном планшете.
Lucifer желтый измеряли с помощью Spectramax Gemini XS (Ex 426 нм и Em 538 нм) в чистом 96-луночном планшете, содержащем 150 мкл жидкости с базолатеральной и апикальной стороны.
Концентрации соединения в образцах измеряли с помощью высокоэффективной жидкостной хроматографии/масс-спектроскопии (ЖХ-МС/МС).
Величины кажущейся проницаемости (Рарр) рассчитывали из соотношения:
Рарр = [соединение]акцептор финальный × Vакцептор/([соединение]донор исходный)Тинк⋅ × Vдонор/площадь поверхности×60×10-6 см/с
где V = объем камеры
Тинк. = время инкубирования
Площадь поверхности = 0,33 см2
Скорость извлечения как показатель активного извлечения с апикальной клеточной поверхности клеток Сасо-2 рассчитывали, используя соотношение Рарр В>А/ Рарр А>В.
Результаты анализа Сасо-2
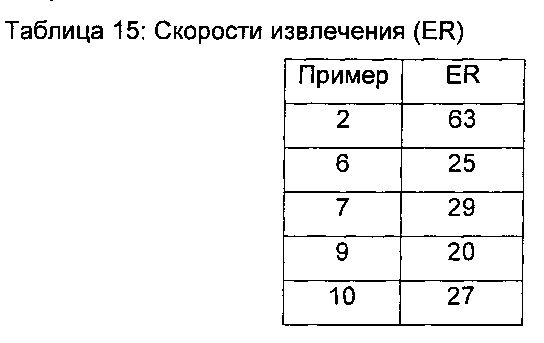

Фармакркинетические исследования на грызунах
Животные
Крыс Sprague-Dawley (самцы, возраст 7-9 недель) получали из SLAC Laboratory Animal Со Ltd (China). Крыс акклиматизировали в течение примерно 3 суток до обработки и содержали с циклом 12 ч свет/темнота. Температуру поддерживали между 18 и 26°C при относительной влажности между 30 и 70%. Пища и вода были в свободном доступе. Животным хирургическим путем имплантировали либо катетер в сонную артерию (для забора образцов), либо как в яремную вену (для дозирования), так и сонную артерию (для забора образцов), используя пропиленовую трубку и растворы гепарина (50 м.е. (международная единица)/мл)/глюкозы (50%) в качестве раствора для повышения вязкости (lumenlock). Животных оставляли восстанавливаться в течение по меньшей мере 3 суток после хирургического вмешательства.
Фармакокинетические исследования
Соединения готовили в 5% DMSO (об./об.), 95% (10% клептоза НРВ (масс/об.)) в физиологическом растворе для внутривенного введения и в 0,5% НРМС (гидроксипропилметилцеллюлоза), 2% масс/об. Полоксамер 188 (Pluronic F68) в воде для перорального введения. Тестируемые соединения вводили перорально однократно через пищеводный зонд при 10 мг/кг в дозовом объеме 2 мг/мл и внутривенно в виде болюса через шейную вену при 1 мг/кг в дозовом объеме 0,5 мг/мл. Каждая группа состояла из 3 крыс. Образцы крови отбирали из шейной вены с K-EDTA в качестве антикоагулянта в следующие временные точки: 0,08, 0,25, 0,5, 1, 2, 4 и 6 часов (внутривенный путь), и 0,25, 0,5, 1, 2, 4, 6 и 8 часов (пероральный путь). Образцы цельной крови центрифугировали при 7000 об./мин при 4°C в пределах 30 минут сбора для 10 мин и полученные образцы плазмы хранили при -20°C до анализа.
Количественное определение уровней соединения в плазме крови
Концентрации каждого тестируемого соединения определяли посредством метода ЖХ-МС/МС с внутренними стандартами.
Определение фармакокинетических параметров
Фармакокинетические параметры рассчитывали, используя программное обеспечение WinNonlin® (Pharsight®, Mountain View, CA)
Фармакокинетика у крысы: например, соединения 2, 6 и 30 давали уровни в плазме крови (Cmax), равные 0,16 мкМ, 0,27 мкМ и 0,32 мкМ, и показатели пероральной биодоступности 7%, 26% и 69%, соответственно.
| название | год | авторы | номер документа |
|---|---|---|---|
| N-9-ЗАМЕЩЕННЫЕ ПУРИНОВЫЕ СОЕДИНЕНИЯ, КОМПОЗИЦИИ И СПОСОБЫ ПРИМЕНЕНИЯ | 2010 |
|
RU2607635C2 |
| N-7 ЗАМЕЩЕННЫЕ ПУРИНЫ И ПИРАЗОЛОПИРИМИДИНЫ, ИХ КОМПОЗИЦИИ И СПОСОБЫ ПРИМЕНЕНИЯ | 2010 |
|
RU2515541C2 |
| МОРФОЛИНО-ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ МОЧЕВИНЫ ИЛИ КАРБАМАТА В КАЧЕСТВЕ ИНГИБИТОРОВ MTOR | 2012 |
|
RU2616619C2 |
| ПИРИМИДИНОВЫЕ СОЕДИНЕНИЯ, КОМПОЗИЦИИ И СПОСОБЫ ПРИМЕНЕНИЯ | 2009 |
|
RU2473549C2 |
| ПРОИЗВОДНЫЕ МОРФОЛИНОПИРИМИДИНА, ПОЛЕЗНЫЕ ДЛЯ ЛЕЧЕНИЯ ПРОЛИФЕРАТИВНЫХ НАРУШЕНИЙ | 2007 |
|
RU2440349C2 |
| БИЦИКЛИЧЕСКИЕ ГЕТЕРОЦИКЛИЛЬНЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЯ | 2020 |
|
RU2811612C2 |
| Новые соединения пиридопиримидинона для модулирования каталитической активности гистонлизиндеметилаз (KDMS) | 2015 |
|
RU2684396C2 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛАМИНОПИРИМИДИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ ОБОГАЩЕННОЙ ЛЕЙЦИНОВЫМИ ПОВТОРАМИ КИНАЗЫ 2 (LRRK2) ДЛЯ ПРИМЕНЕНИЯ ПРИ ЛЕЧЕНИИ БОЛЕЗНИ ПАРКИНСОНА | 2013 |
|
RU2637948C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ ДЛЯ ОПОСРЕДОВАНИЯ АКТИВНОСТИ ТИРОЗИНКИНАЗЫ 2 | 2020 |
|
RU2826012C2 |
| ПИРИДОПИРИМИДИНОНЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ РЕЦЕПТОРА N-МЕТИЛ-D-АСПАРТАТА | 2016 |
|
RU2717665C2 |
Изобретение относится к соединениям формулы (I) и их фармацевтически приемлемым солям

В формуле (I) m равно 1; о равно 1 или 2; каждый R1 независимо выбран из группы, состоящей из Н и незамещенного С1-6алкила; возможно два R1 объединены с образованием, вместе с кольцом, к которому они присоединены, 8-окса-3-азабицикло[3.2.1]октан-3-ильного или 3-окса-8-азабицикло[3.2.1]октан-8-ильного кольца; Т1 представляет собой фенил, где Т1 замещен группой N(R5a)C(O)N(R5bR5) и Т1 возможно дополнительно замещен одним или более R6, которые являются одинаковыми или разными; R6 представляет собой галоген; каждый из R5a, R5b представляет собой Н; R5 представляет собой Н; Т2; и С1-6алкил, где С1-6алкил возможно замещен одним или более R8, которые являются одинаковыми или разными; R8 представляет собой галоген или OR9; R9 представляет собой Н; Т2 является незамещенным и выбран из группы, состоящей из фенила, пиридила, циклопропила, циклобутила, циклопентила, циклогексила, оксетанила или тетрагидрофуранила; Ra и Rb выбраны с получением одной из формул (Ik)-(Ip)
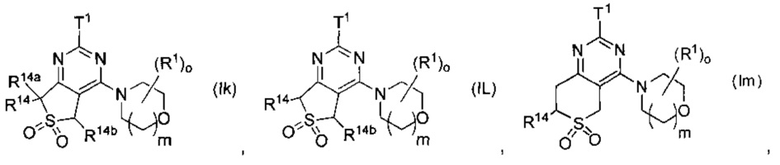
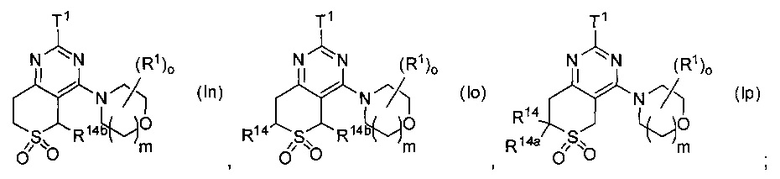
или Ra, Rb, T1 определены так, чтобы получалась формула (Iq)
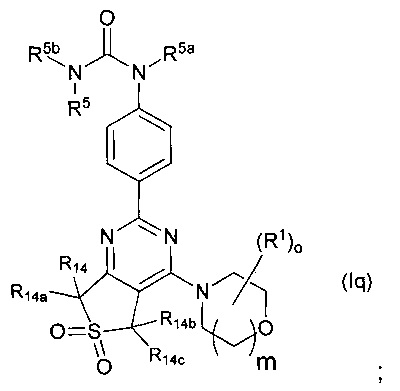
R14, R14a, R14b, R14c независимо выбраны из группы, состоящей из Н; галогена или незамещенного С1-6алкила. Соединения формулы (I) обладают ингибирующей активностью в отношении mTOR. Изобретение также относится к фармацевтической композиции, включающей соединения формулы (I), к применению соединений для изготовления лекарственного средства и к способу лечения. Технический результат: получены новые соединения формулы (I), обладающие свойствами ингибитора mTOR, которые могут применяться для лечения или профилактики связанных с mTOR заболеваний и расстройств. 4 н. и 12 з.п. ф-лы, 15 табл., 82 пр.
1. Соединение формулы (I)
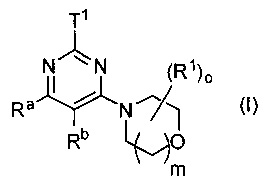
или его фармацевтически приемлемая соль,
где m равно 1;
о равно 1 или 2;
каждый R1 независимо выбран из группы, состоящей из Н и незамещенного С1-6алкила;
возможно два R1 объединены с образованием, вместе с кольцом, к которому они присоединены, 8-окса-3-азабицикло[3.2.1]октан-3-ильного или 3-окса-8-азабицикло[3.2.1]октан-8-ильного кольца;
Т1 представляет собой фенил, где Т1 замещен группой N(R5a)C(O)N(R5bR5) и Т1 возможно дополнительно замещен одним или более R6, которые являются одинаковыми или разными;
R6 представляет собой галоген;
каждый из R5a, R5b представляет собой Н;
R5 представляет собой Н; Т2; и С1-6алкил, где С1-6алкил возможно замещен одним или более R8, которые являются одинаковыми или разными;
R8 представляет собой галоген или OR9;
R9 представляет собой Н;
Т2 является незамещенным и выбран из группы, состоящей из фенила, пиридила, циклопропила, циклобутила, циклопентила, циклогексила, оксетанила или тетрагидрофуранила;
Ra и Rb выбраны с получением одной из формул (Ik)-(Ip)
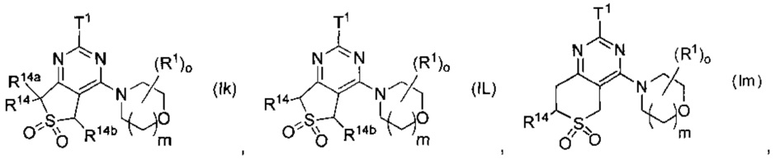
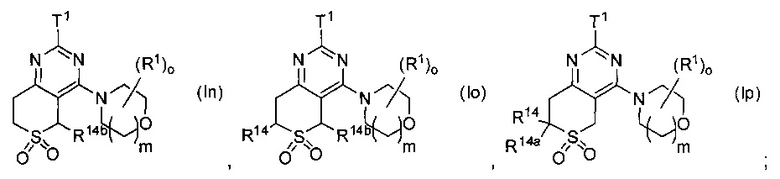
или Ra, Rb, T1 определены так, чтобы получалась формула (Iq)
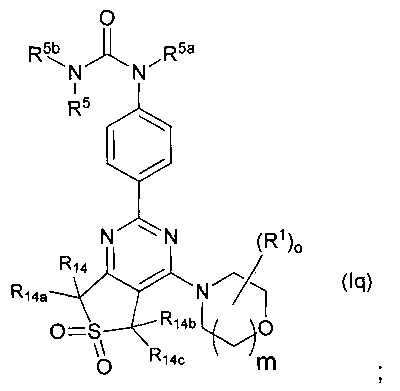
R14, R14a, R14b, R14c независимо выбраны из группы, состоящей из Н, галогена или незамещенного С1-6алкила.
2. Соединение по п. 1, где в формуле (I) Ra и Rb выбраны с получением одной из формул (Ib), (Id), (If), (Ig) или (Ii)
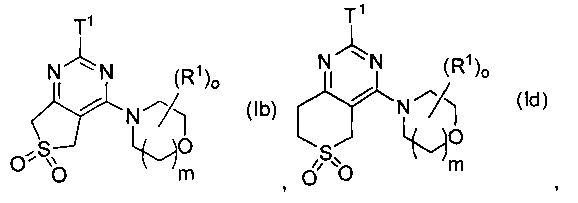
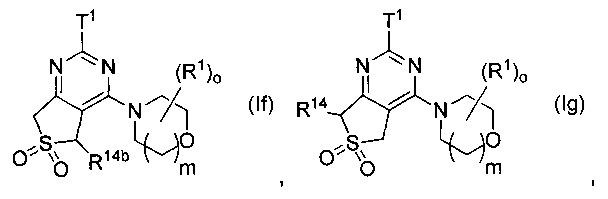
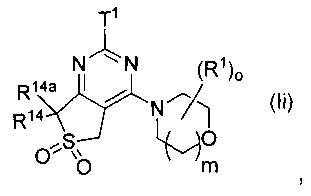
где Т1, R1, о, m, R14, R14a, R14b имеют значения, как указано в п. 1.
3. Соединение по п. 1, где в формуле (I) Т1 определен так, чтобы получалась формула (Iq)
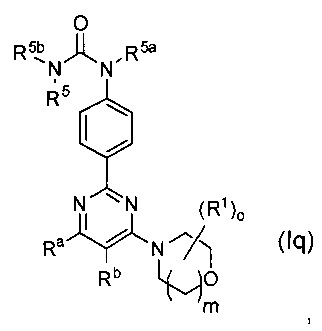
где о, m, R1, Ra, Rb, R5, R5a, R5b имеют значения, как указано в любом из пп. 1 и 2.
4. Соединение по п. 1, где R5 представляет собой С1-6алкил, возможно замещенный одним или более R8, которые являются одинаковыми или разными и выбраны из группы, состоящей из F и OR9.
5. Соединение по п. 1, где R14, R14a, R14b и R14c независимо выбраны из группы, состоящей из Н; F; этила и метила.
6. Соединение по п. 1, где R1 представляет собой незамещенный С1-6алкил.
7. Соединение по п. 1, где два R1 объединены с образованием, вместе с кольцом, к которому они присоединены, 8-окса-3-азабицикло[3.2.1]октан-3-ильного или 3-окса-8-азабицикло[3.2.1]октан-8-ильного кольца.
8. Соединение по п. 1, где R5b и R5a представляют собой Н, и R5 представляет собой С1-6алкил, где С1-6алкил возможно замещен одним или более R8, которые являются одинаковыми или разными, или R5 незамещен и выбран из группы, состоящей из циклопропила, циклобутила, циклопентила и циклогексила.
9. Соединение по п. 1, где R5 представляет собой циклопропил, метил, этил, фторэтил, гидроксиэтил, дифторэтил, изопропил, фторпропил, пиридинил и оксетанил.
10. Соединение по п. 1, где (R1)o присоединен по положению 3.
11. Соединение по п. 1 или его фармацевтически приемлемая соль, где соединение выбрано из группы, состоящей из
1-циклопропил-3-(4-(4-морфолино-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
1-циклопропил-3-(4-(4-((2S,6R)-2,6-диметилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
(S)-1-циклопропил-3-(4-(4-(3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
(S)-1-этил-3-(4-(4-(3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
(S)-1-метил-3-(4-(4-(3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
(S)-1-(2-гидроксиэтил)-3-(4-(4-(3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
(S)-1-(2-фторэтил)-3-(4-(4-(3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
1-(4-(4-(8-окса-3-азабицикло[3.2.1]октан-3-ил)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)-3-этилмочевины;
1-(4-(4-(8-окса-3-азабицикло[3.2.1]октан-3-ил)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)-3-(2-гидроксиэтил)мочевины;
(R)-1-циклопропил-3-(4-(4-(3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
1-(4-(4-(8-окса-3-азабицикло[3.2.1]октан-3-ил)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)-3-циклопропилмочевины;
1-(4-(4-(8-окса-3-азабицикло[3.2.1]октан-3-ил)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)-3-метилмочевины;
1-(4-(4-(8-окса-3-азабицикло[3.2.1]октан-3-ил)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)-3-(2-фторэтил)мочевины;
(S)-1-циклопропил-3-(4-(4-(3-метилморфолино)-6,6-диоксидо-7,8-дигидро-5Н-тиопирано[4,3-d]пиримидин-2-ил)фенил)мочевины;
(S)-1-(2,2-дифторэтил)-3-(4-(4-(3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
1-(4-(4-(8-окса-3-азабицикло[3.2.1]октан-3-ил)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)-3-(2,2-дифторэтил)мочевины;
(S)-1-изопропил-3-(4-(4-(3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
1-(4-(4-(3-окса-8-азабицикло[3.2.1]октан-8-ил)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)-3-циклопропилмочевины;
1-циклопропил-3-(4-(4-(3-этилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
(S)-1-(3-фторпропил)-3-(4-(4-(3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
1-(4-(4-(8-окса-3-азабицикло[3.2.1]октан-3-ил)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)-3-(3-фторпропил)мочевины;
1-циклопропил-3-(4-(7-метил-4-((S)-3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
1-циклопропил-3-(4-(5-метил-4-((S)-3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
(S)-1-циклопропил-3-(4-(7,7-диметил-4-(3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
1-циклопропил-3-(4-(5-метил-4-морфолино-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
1-циклопропил-3-(4-(4-(3-этилморфолино)-5-метил-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
1-(4-(4-(3-окса-8-азабицикло[3.2.1]октан-8-ил)-5-метил-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)-3-циклопропилмочевины;
1-(4-(4-(8-окса-3-азабицикло[3.2.1]октан-3-ил)-5-метил-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)-3-циклопропилмочевины;
(S)-1-(4-(4-(3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)-3-(пиридин-4-ил)мочевины;
(S)-1-(4-(4-(3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)-3-(пиридин-3-ил)мочевины;
1-циклопропил-3-(4-((R)-5-метил-4-((S)-3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
1-циклопропил-3-(4-((S)-5-метил-4-((S)-3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
1-этил-3-(4-(5-метил-4-((S)-3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
1-этил-3-(4-((R)-5-метил-4-((S)-3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
1-этил-3-(4-((S)-5-метил-4-((S)-3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
1-этил-3-(4-(4-((S)-3-этилморфолино)-7-метил-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
1-этил-3-(4-(5,7,7-триметил-4-((S)-3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
1-циклопропил-3-(4-(7-фтор-7-метил-4-((S)-3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
1-циклопропил-3-(4-(5,7-диметил-4-((S)-3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
1-(4-(5-метил-4-((S)-3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)-3-пропилмочевины;
1-(циклопропилметил)-3-(4-(5-метил-4-((S)-3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
1-циклопропил-3-(4-(4-((S)-3-этилморфолино)-5-метил-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
1-циклопропил-3-(4-(5-метил-4-((R)-3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
1-циклопропил-3-(4-(5-этил-4-((S)-3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
1-этил-3-(2-фтор-4-(5-метил-4-((S)-3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
(S)-1-циклопропил-3-(4-(4-(3-этилморфолино)-7,7-диметил-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
1-этил-3-(4-((R)-4-((S)-3-этилморфолино)-5-метил-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
1-этил-3-(4-((S)-4-((S)-3-этилморфолино)-5-метил-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
(R)-1-циклопропил-3-(4-(7,7-диметил-4-(3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
(R)-1-(4-(7,7-диметил-4-(3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)-3-этилмочевины;
(S)-1-этил-3-(4-(4-(3-этилморфолино)-7,7-диметил-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
(S)-1-(4-(7,7-диметил-4-(3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)-3-метилмочевины;
1-метил-3-(4-(5-метил-4-((S)-3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
1-(4-(4-(3-окса-8-азабицикло[3.2.1]октан-8-ил)-7,7-диметил-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)-3-циклопропилмочевины;
1-(4-(4-(3-окса-8-азабицикло[3.2.1]октан-8-ил)-7,7-диметил-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)-3-этилмочевины;
(S)-1-(4-(7,7-диметил-4-(3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)-3-(2-фторэтил)мочевины;
(S)-1-(4-(7,7-диметил-4-(3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)-3-этилмочевины;
1-циклопропил-3-(4-((S)-5-метил-4-((R)-3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
(S)-1-(4-(4-(3-этилморфолино)-7,7-диметил-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)-2-фторфенил)-3-метилмочевины;
(S)-1-(4-(7,7-диметил-4-(3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)-2-фторфенил)-3-метилмочевины;
(S)-1-(4-(7,7-диметил-4-(3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)-3-фторфенил)-3-метилмочевины;
1-этил-3-(4-(5-этил-4-((S)-3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
(S)-1-(4-(7,7-диметил-4-(3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)-3-(оксетан-3-ил)мочевины;
(S)-1-(4-(7,7-диметил-4-(3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)-3-фторфенил)-3-этилмочевины;
(S)-1-циклопропил-3-(4-(7,7-диметил-4-(3-метилморфолино)-6,6-диоксидо-7,8-дигидро-5Н-тиопирано[4,3-d]пиримидин-2-ил)фенил)мочевины;
1-циклопропил-3-(4-(7-метил-4-((S)-3-метилморфолино)-6,6-диоксидо-7,8-дигидро-5Н-тиопирано[4,3-d]пиримидин-2-ил)фенил)мочевины;
1-циклопропил-3-(4-(5,7-диметил-4-((S)-3-метилморфолино)-6,6-диоксидо-7,8-дигидро-5Н-тиопирано[4,3-d]пиримидин-2-ил)фенил)мочевины;
(S)-1-(4-(7,7-диметил-4-(3-метилморфолино)-6,6-диоксидо-7,8-дигидро-5Н-тиопирано[4,3-d]пиримидин-2-ил)фенил)-3-этилмочевины;
1-циклопропил-3-(4-(5-метил-4-((S)-3-метилморфолино)-6,6-диоксидо-7,8-дигидро-5Н-тиопирано[4,3-d]пиримидин-2-ил)фенил)мочевины;
1-(2-фторэтил)-3-(4-(5,7,7-триметил-4-((S)-3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
1-(4-(7,7-диметил-4-морфолино-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)-3-этилмочевины;
1-метил-3-(4-(5,7,7-триметил-4-((S)-3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины;
1-(3-фтор-4-(5,7,7-триметил-4-((S)-3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)-3-метилмочевины;
1-(4-(4-((S)-3-этилморфолино)-5,7,7-триметил-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)-3-фторфенил)-3-метилмочевины;
1-(4-(4-((S)-3-этилморфолино)-5,7,7-триметил-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)-3-метилмочевины;
1-(2-фторэтил)-3-(4-(5,7,7-триметил-4-морфолино-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины и
1-циклопропил-3-(4-((R)-5,7,7-триметил-4-((S)-3-метилморфолино)-6,6-диоксидо-5,7-дигидротиено[3,4-d]пиримидин-2-ил)фенил)мочевины.
12. Фармацевтическая композиция, обладающая ингибирующей активностью в отношении mTOR, содержащая соединение или его фармацевтически приемлемую соль по любому из пп. 1-11 вместе с фармацевтически приемлемым носителем.
13. Соединение или его фармацевтически приемлемая соль по любому из пп. 1-11 для применения в качестве лекарственного средства, обладающего ингибирующей активностью в отношении mTOR.
14. Соединение или его фармацевтически приемлемая соль по любому из пп. 1-11 для применения в способе лечения или предупреждения заболевания или расстройства, ассоциированного с mTOR, выбранного из иммунологического, воспалительного или аутоиммунного заболевания, отторжения трансплантата, болезни «трансплантат-против-хозяина», пролиферативного заболевания (в особенности рака), сердечно-сосудистого, метаболического или нейродегенеративного заболевания, заболеваний, ассоциированных с аутофагией, или вирусной инфекции.
15. Применение соединения по любому из пп. 1-11 или его фармацевтически приемлемой соли для изготовления лекарственного средства для использования в способе лечения или предупреждения заболевания или расстройства, ассоциированного с mTOR, выбранного из иммунологического, воспалительного или аутоиммунного заболевания, отторжения трансплантата, болезни «трансплантат-против-хозяина», пролиферативного заболевания (в особенности рака), сердечно-сосудистого, метаболического или нейродегенеративного заболевания, заболеваний, ассоциированных с аутофагией, или вирусной инфекции.
16. Способ лечения, контролирования, замедления или предупреждения у нуждающегося в этом пациента - млекопитающего одного или более состояний, выбранных из иммунологического, воспалительного или аутоиммунного заболевания, отторжения трансплантата, болезни «трансплантат-против-хозяина», пролиферативного заболевания (в особенности рака), сердечно-сосудистого, метаболического или нейродегенеративного заболевания, заболеваний, ассоциированных с аутофагией, или вирусной инфекции, включающий введение указанному пациенту терапевтически эффективного количества соединения по любому из пп. 1-11 или его фармацевтически приемлемой соли.
| J | |||
| C | |||
| Verheijen et al | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| WO 2010103094 А1, 16.09.2010 | |||
| RU 2009125897 A, 05.12.2007 | |||
| WO 2010052569 A2, 14.05.2010. | |||

