Описание
Область техники, к которой относится настоящее изобретение
Настоящее изобретение относится к гетероциклическим соединениям или их фармацевтически приемлемым солям, подходящим для регулирования или ингибирования активности Janus-киназы (JAK), в частности, тирозинкиназы 2 (TYK2). Настоящее изобретение также относится к способам получения указанных гетероциклических соединений. Настоящее изобретение дополнительно относится к способам лечения и/или предупреждения заболеваний, опосредованных указанной киназой, в частности, аутоиммунных заболеваний, воспалительных заболеваний или злокачественных опухолей.
Предшествующий уровень техники настоящего изобретения
JAK представляет собой нерецепторную тирозинпротеинкиназу, сформированную четырьмя представителями семейства, а именно: JAK1, JAK2, JAK3 и TYK2. JAK имеет в своей структуре 7 гомологичных доменов (JAK-гомологичный домен, JH), среди которых JH1 представляет собой киназный домен, JH2 представляет собой псевдокиназный домен (который регулирует киназную активность JH1), и JH6 и JH7 представляют собой рецептор-связывающие домены. Если поверхностно-клеточная область цитокинового рецептора связана с цитокином, то ее внутриклеточная область, к которой присоединены JAK. являются фосфорилированной, что формирует сайт докинга для белков-трансдукторов сигнала и активаторов транскрипции (STAT). Белки STAT дополнительно фосфорилируются активированными JAK с формированием димера, который входит в ядро, регулирует экспрессию и транскрипцию связанных генов, и обеспечивает возможность передачи сигналов от клеточной мембраны к ядру (Lionard et. al, Ann. Rev. Immunol. 1998, 16, 293-322). Поэтому, JAK передает цитокин-опосредованные сигналы посредством пути JAK-STAT и играет важную роль во многих клеточных функциях, таких как цитокин-зависимая регуляция пролиферации клеток, дифференцировки, апоптоза и иммунного ответа, и представляет собой известную мишень для лечения воспалительных заболеваний, аутоиммунных заболеваний и злокачественных опухолей (Alicea-Velazquez et. al, Curr. Drug Targets 2011, 12, 546-55). Некоторые фармацевтические средства, которые регулируют JAK, были допущены к продаже или были представлены для получения разрешения, включая ингибитор JAK1/JAK2 руксолитиниб и ингибитор JAK2 федратиниб для лечения миелофиброза, пан-ингибитор JAK тофацитиниб, ингибитор JAK1/JAK2 барицитиниб, пан-ингибитор JAK пефицитиниб и ингибитор JAK1 упадацитиниб для лечения ревматоидного артрита, и т.д.
Исследования с нокаутом генов продемонстрировали, что JAK и STAT играют высокоспецифичную роль в регуляции различных иммунных ответов. Фермент JAK может принимать участие в процессах передачи сигналов, индуцированных множеством цитокинов, и цитокиновый путь передачи сигналов также может активировать множество ферментов JAK, но сам по себе цитокин обладает определенной селективностью активации STAT. Например, интерлейкин-4 (IL-4) активирует STAT6, тогда как IL-12 специфически активирует STAT4. JAK1, JAK2 и TYK2 широко представлены в различных тканях и клетках. JAK1 тесно связана с активацией воспалительных факторов, таких как IL-6 и интерферон (IFN), поэтому считается, что селективный ингибитор JAK1 характеризуется потенциальным терапевтическим эффектом в отношении аутоиммунных заболеваний, таких как ревматоидный артрит (RA) и псориаз. JAK2 независимо опосредует цитокины. такие как эритропоэтин (ЕРО) и тромбопоэтин (ТРО) (Won et. al, ВМС Bioinformatics 2009. 10, S53), и тесно связана с пролиферацией и дифференцировкой клеток крови. JAK3 присутствует только в костном мозге и лимфатической системе, и опосредует передачу сигналов IL-2, IL-4, IL-7, IL-9, IL-15 и IL-21. Указанные цитокины играют важную роль в индукции пролиферации и дифференцировке Т-клеток, в активации В-клеток с продукцией антител, в активации макрофагов, в усилении активности естественных клеток-киллеров (NK клетки), и в индукции других цитокинов, таких как IFN. Поэтому считается, что селективный ингибитор JAK3 будет играть важную роль при трансплантации органов и при лечении аутоиммунных заболеваний и воспалительной пневмонии.
Путь JAK/STAT может быть чрезмерно активирован аутокринными и паракринными цитокинами, а также за счет некоторых мутаций, и это связано с целым рядом злокачественных опухолей, таких как злокачественная опухоль молочной железы, злокачественная опухоль печени, злокачественная опухоль предстательной железы, злокачественная опухоль толстого кишечника, злокачественная опухоль легких, злокачественная опухоль поджелудочной железы, злокачественная опухоль мочевого пузыря и диффузная В-крупноклеточная лимфома (Tan et. al, Curr. Drug Targets 2014, 15, 1341-53; Lam et. al, Blood 2008, 111, 3701-13). Мутация JAK2 JAK2/V617F возникает в псевдокиназном домене JH2, вызывая изменение конформации JAK2, что приводит к продолжительной активации киназного домена JH1, независимой от внеклеточных цитокиновых сигналов, что, в свою очередь, вызывает гиперплазию клеток и злокачественную опухоль крови, и тесно связана с истинной полицитемией (PV), эссенциальной тромбоцитемией и миелофиброзом (MF) (O'Shea et. al, Ann. Rev. Med. 2015, 66, 311-28). Ингибитор JAK2 руксолитиниб может быть использован для лечения таких заболеваний крови, но его эффективность не связана с наличием мутации JAK2/V617F, что указывает на то, что противоопухолевая активность основана не только на ингибировании передачи сигналов, обусловленном мутацией JAK2/V617F, а также может быть связана с регуляцией JAK1-STAT.
TYK2 вовлечена в передачу сигналов воспалительных цитокинов, таких как интерфероны (IFN), IL-12 и IL-23, и играет ключевую роль во врожденном иммунитете и приобретенном иммунитете. Нокаутные по TYK2 мыши имеют нормальное количество красных клеток крови и способны выживать, но мыши с дефицитом JAK3 характеризуются тяжелым иммунодефицитом, а нокаутные по JAK1 или JAK2 мыши могут погибать в эмбриональном периоде. При этом, заболевания, обусловленные нарушением функции JAK1/2, не были обнаружены у людей, косвенно указывая на важность физиологических функций JAK1/2. Один пациент с нулевой мутацией гена TYK2 имел синдром гипериммуноглобулинемии Е, но в семи других случаях нулевой мутации гомозитозы TYK2 синдром гипериммуноглобулинемии Е отсутствовал, хотя вследствие сниженного ответа на IL-12 и INF-α/β возрастала чувствительность к микобактериальным или вирусным инфекциям. Поэтому, ингибирование TYK2 не вызывает острой токсичности. Отсутствие экспрессии TYK2 проявляется в виде ослабленной передача сигналов множества провоспалительных цитокинов и тяжелого нарушения баланса дифференцировки Т-хелперов. Кроме того, связанные с генетикой исследования свидетельствуют в пользу предположения о том, что TYK2 является характерным геном предрасположенности к аутоиммунным заболеваниям. Важность регулируемых TYK2 путей была дополнительно подтверждена эффективностью терапии антителами при лечении заболеваний в клинической практике, как например, устекинумаба с направленным воздействием на IL-12/IL-23 для лечения псориаза и анифролумаба с направленным воздействием на рецептор интерферона I типа для лечения системной красной волчанки (SLE). Поэтому, TYK2 привлекает большое внимание в качестве мишени для лекарств при аутоиммунных заболеваниях. Например, ингибитор TYK2 может быть потенциально использован для лечения псориаза, SLE и воспалительного заболевания кишечника (IBD), и т.д.
TYK2 также ассоциирована с некоторыми злокачественными опухолями. Например, аномальная выживаемость клеток острого лимфобластного лейкоза (T-ALL) связана с активацией TYK2. Эксперименты с нокаутом генов продемонстрировали, что линий клеток 88% T-ALL и 63% клеток T-ALL, полученных от пациентов, зависимы от TYK2, то есть TYK2 является онкогеном T-ALL (Sanda et. al, Cancer Disc. 2013, 3, 564-77). Селективный ингибитор TYK2 NDI-031301 может индуцировать апоптоз для ингибирования роста линий клеток T-ALL человека и обладает требуемой безопасностью и эффективностью в мышиных моделях с T-ALL опухолевыми клетками KOPT-K1 (Akahane et. al, British J. Haematol. 2017, 177, 271-82), демонстрируя перспективу селективного ингибитора TYK2 для лечения T-ALL.
В дополнение ко многим ингибиторам, преимущественно нацеленным на JAK1/2/3, специфический ингибитор TYK2 BMS-986165 и двойной ингибитор JAK1/TYK2 PF-06700841 также были выведены на позднюю стадию клинических испытаний. Поскольку JAK регулирует различные иммунные ответы в JAK-STAT, различные селективные ингибиторы JAK демонстрируют разные токсические и побочные эффекты при клиническом применении и имеют различные клинические применения. Руксолитиниб используется для лечения фиброза костного мозга, демонстрирует хорошую безопасность и не имеет токсических и побочных эффектов в отношении нецелевых органов. Тофацитиниб ингибирует активность JAK2 в дополнении к JAK1, воздействует на продукцию клеток крови и лимфоцитов, и демонстрирует определенный токсический побочный эффект в виде анемии, ограничивая тем самым его клиническую эффективность при RA. В связи с перспективой ингибиторов JAK при лечении воспалительных заболеваний, аутоиммунных заболеваний и злокачественных опухолей, разработка селективных ингибиторов JAK привлекает большое внимание фармацевтической индустрии. Однако, по причине высокого сходства последовательностей в активных сайтах семейства киназ JAK, селективный ингибитор JAK разработать сложно. Несмотря на то, что было раскрыто несколько патентных заявок на селективные ингибиторы TYK2, включая WO 2010142752, WO 2012062704, WO 2013180265, WO 2015032423, WO 2015131080 и WO 2017040757, существует постоянная потребность в разработке новых соединений, которые обладают лучшей нацеленностью на мишень, большей эффективностью и более высокой селективностью в отношении TYK2 или TYK2/JAK1.
Подробное описание настоящего изобретения
Определения
Если не указано иное, т следующие термины, использованные в настоящей заявке, характеризуются следующими значениями.
«Сх-у» относится к численному диапазону атомов углерода, где х и у представляют собой целые числа, например, С3-8 циклоалкил обозначает циклоалкил, содержащий от 3 до 8 атомов углерода. Также следует понимать, что «С3-8» дополнительно включает в себя любые поддиапазоны, такие как С3-7, С3-6, С4-7, С4-6 и С5-6.
«Алкил» относится к насыщенному неразветвленному или разветвленному гидрокарбильному заместителю, содержащему от 1 до 20 атомов углерода, например, от 1 до 8 атомов углерода, от 1 до 6 атомов углерода или от 1 до 4 атомов углерода. Неограничивающие примеры алкила включают в себя без ограничения метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, втор-бутил, н-пентил, 1,1-диметилпропил, 1,2-диметилпропил, 2,2-диметилпропил, 1-этилпропил, 2-метилбутил, 3-метилбутил, н-гексил, 1-этил-2-метилпропил, 1,1,2-триметилпропил, 1,1-диметилбутил. 1,2-диметилбутил, 2,2-диметилбутил, 1,3-диметилбутил, 2-этилбутил.
«Алкилен» относится к насыщенному неразветвленному или разветвленному гидрокарбильному двухвалентному заместителю, содержащему от 1 до 20 атомов углерода, например, от 1 до 6 атомов углерода или от 1 до 4 атомов углерода. Неограничивающие примеры алкилена включают в себя без ограничения -СН2-, -СН(СН3)-, -СН2СН2-, -СН2СН2СН2-, -(СН3)С(СН3)-, -СН2СН2СН2СН2- и -СН2СН(СН3)СН2-.
«Циклоалкил» относится к насыщенному циклическому гидрокарбильному заместителю, содержащему от 3 до 14 кольцевых атомов углерода. Циклоалкил может представлять собой моноуглеродный кольцевой заместитель, обычно содержащий от 3 до 8, от 3 до 7 или от 3 до 6 атомов углерода. Неограничивающие примеры моноциклического циклоалкила включают в себя без ограничения циклопропил, циклобутил, циклопентил, циклогексил и циклогептил. Циклоалкил также может представлять собой заместитель с двумя или тремя моноуглеродными кольцами, которые конденсированы вместе, такой как декагидронафтил.
«Гетероциклил или гетероцикл» относится к насыщенной или частично ненасыщенной моноциклической или полициклической группе, содержащей от 3 до 20 кольцевых атомов, например, от 3 до 14, от 3 до 12, от 3 до 10, от 3 до 8, от 3 до 6 или от 5 до 6 кольцевых атомов, в которой один или более кольцевых атомов выбраны из N, О и S(O)m (где m представляет собой целое число от 0 до 2), но не содержат -О-О-, -O-S- или -S-S- в кольцевой структуре, и оставшиеся представляют собой атомы углерода. Предпочтительно, она может содержать от 3 до 12 кольцевых атомов, от 3 до 10 кольцевых атомов, от 4 до 7 кольцевых атомов и от 4 до 6 кольцевых атомов, где от 1 до 4 являются гетероатомами, от 1 до 3 являются гетероатомами или от 1 до 2 являются гетероатомами. Неограничивающие примеры моноциклического гетероциклила включают в себя без ограничения пирролидинил, оксетанил, пиперидил, пиперазинил, тетерагидрофуранил, тетрагидропиранил, тетрагидротиопиранил, морфолинил, тиоморфолинил, гомопиперазинил и азетидинил. Полициклический гетероциклил включает в себя конденсированный, связанный мостиковой связью или спиро полициклический гетероцикл, такой как октагидроциклопента[с]пиррол, октагидропиррол[1,2-а]пиразин, 3,8-диазабицикло[3.2.1]октан, 5-азаспиро[2.4]гептан и 2-окса-7-азаспиро[3.5]нонан.
«Арил или арильное кольцо» относится к ароматической моноциклической или конденсированной полициклической группе, содержащей то 6 до 14 атомов углерода, предпочтительно 6-10-членной, такой как фенил или нафтил, наиболее предпочтительно фенил. Арильное кольцо может быть конденсировано с гетероарильным, гетероциклильным или циклоалкильным кольцом, и неограничивающие примеры включают в себя без ограничения:
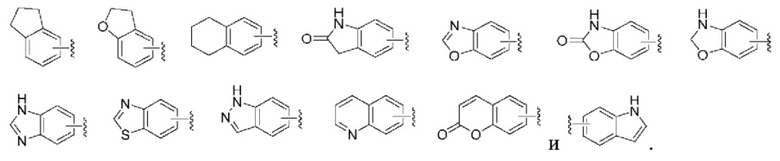
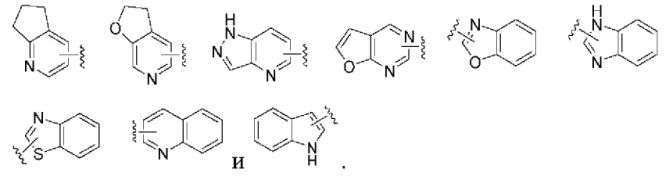
«Гетероарил или гетероарильное кольцо» относится к гетероароматической системе, содержащей от 5 до 14 кольцевых атомов, из которых от 1 до 4 кольцевых атомов выбраны из гетероатомов, включающих О, S и N. Гетероарил предпочтительно является 5-10-членным, и более предпочтительно 5- или 6-членным, таким как фурил, тиенил, пиридил, пирролил, пиримидил, пиразинил, пиразолил, имидазолил, тетразолил, оксазолил, изоксазолил, тиазолил, изотиазолил, хинолинил, изохинолинил, индолил и изоиндолил. Гетероарильное кольцо может быть конденсировано с гетероарильным, гетероциклильным или циклоалкильным кольцом, и неограничивающие примеры включают в себя без ограничения:
«Галоген» относится к F, Cl, Br или I.
«Циано» относится к -CN.
«Оксо» относится к =O.
«Карбонил» относится к группе -С(=O)-.
«Сульфонил» относится к группе -S(O)2-.
«Сульфинил» относится к группе -S(O)-.
«Необязательное замещение или необязательно замещенный» относится к тому, что один или более атомов водорода в группе, предпочтительно 5, более предпочтительно от 1 до 3 атомов водорода, независимо замещены соответствующим числом заместителей. Само собой разумеется, что заместители расположены исключительно в химических положениях, в которых они могут быть расположены, и специалисты в данной области техники смогут определить возможные или невозможные замещения без особого труда (посредством эксперимента или теоретически). Например, амино или гидроксильные группы со свободным атомом водорода могут быть нестабильны, будучи связанными с атомами углерода ненасыщенных связей (таких как олефиновые). Заместители включают в себя без ограничения галоген, циано, нитро, оксо, -SF5, С1-4 алкил, С3-7 циклоалкил, и т.д.
«Изомеры» относится к соединениям, которые характеризуются той же самой молекулярной формулой, но природа или последовательность их атомных связей или пространственное расположение различаются. Изомеры с различным расположением их атомов в пространстве называют «стереоизомерами». Стереоизомеры включают в себя оптические изомеры, геометрические изомеры и конформационные изомеры.
Соединения согласно настоящему изобретению могут существовать в форме оптических изомеров. Оптические изомеры включают в себя энантиомеры и диастереоизомеры. Энантиомер представляет собой один из двух стереоизомеров, которые являются зеркальными отображениями друг друга, не налагаемыми друг на друга. Рацемическая смесь или рацемат представляет собой смесь, которая содержит равные количества лево- и правовращающих энантиомеров хиральной молекулы. Диастереоизомеры представляют собой стереоизомеры, которые не являются зеркальными отображениями друг друга и не налагаются друг на друга. Способы получения и разделения оптических изомеров известны из уровня техники. Если соединение представляет собой отдельный изомер, и его абсолютная конфигурация определена, то его называют «R» или «S» изомером в зависимости от конфигурации заместителей вокруг хирального атома углерода; если его абсолютная конфигурация не определена, то его называют (+) или (-) изомером в соответствии с его измеренным значением оптического вращения.
Соединения согласно настоящему изобретению могут также иметь геометрические изомеры, являющиеся результатом распределения заместителей вокруг углерод-углеродных двойных связей, углерод-азотных двойных связей, циклоалкильных или гетероциклильных групп. Заместители вокруг углерод-углеродной двойной связи или углерод-азотной связи обозначают как находящиеся в Z или Е конфигурации, и заместители вокруг циклоалкила или гетероцикла обозначают как находящиеся в цис или транс конфигурации.
Соединения согласно настоящему изобретению также могут демонстрировать таутомерию, такую как кето-енольная таутомерия.
Настоящее изобретение включает в себя любые таутомерные или стереоизомерные формы или их смеси и не ограничивается какими-либо таутомерными или стереоизомерными формами, использованными в номенклатуре соединений или в химических структурных формулах.
«Изотопы» включают в себя все изотопы атомов, встречающихся в соединениях согласно настоящему изобретению. Изотопы включают в себя атомы с одним и тем же атомным числом, но различными массами. Примеры изотопов, подходящих для включения в состав соединений согласно настоящему изобретению, включают в себя изотопы водорода, углерода, азота, кислорода, фосфора, фтора и хлора, например без ограничения 2Н (D), 3Н, 13С, 14С, 15N, 18O, 17O, 31Р, 32Р, 35S, 18F и 36Cl. Меченые изотопами соединения согласно настоящему изобретению как правило могут быть получены традиционными методиками, известными специалистам в данной области техники, или способами, сходными с описанными в вариантах осуществления, с использованием соответствующих меченых изотопами реагентов вместо немеченых изотопами реагентов. Такие соединения имеют различные потенциальные применения, например, в качестве стандартов и реагентов при определении биологических видов активности. В случае стабильных изотопов, таких как дейтерий 2Н (D), 13С и 15N, такие соединения характеризуются потенциалом благоприятного изменения биологических, фармакологических или фармакокинетических свойств. Дейтерий 2Н (D) является предпочтительным изотопом согласно настоящему изобретению. Например, атомы водорода в -СН3 могут быть замещены D на -CD3. Например, один или более атомов водорода циклопропила могут быть замещены D.
Соединения согласно настоящему изобретению могут вводиться в форме пролекарств. «Пролекарства» относятся к производным, которые преобразуются в биологически активные соединения в физиологических условиях in vivo, например, посредством окисления, восстановления или гидролиза (каждый из которых происходит с участием или без участия ферментов). Примеры пролекарства представляют собой соединение согласно настоящему изобретению, в котором аминогруппа ацилирована, алкилирована или фосфорилирована, например эйкозаниламино, аланиламино и пивалоилоксиметиламино; гидроксил ацилирован, алкилирован, фосфорилирован или преобразован в боран, например ацетокси, пальмитоилокси, пивалоилокси, сукцинилокси, фумароилокси и аланилокси; карбонил эстерифицирован или амидирован; и тиол формирует дисульфидную связь с молекулой-носителем, которая селективно доставляет лекарство к мишени и/или в цитозоль клеток, такой как пептид. Пролекарства могут быть получены из соединений согласно настоящему изобретению в соответствии с хорошо известными способами.
«Фармацевтически приемлемые соли» относятся к солям, полученным из соединений согласно настоящему изобретению с фармацевтически приемлемыми основаниями или кислотами, включая неорганические щелочи или кислоты и органические основания или кислоты, при условии, что соединения содержат одну или более кислых или основных групп. Поэтому, соединения согласно настоящему изобретению, которые содержат кислые группы, могут существовать в форме солей, например, в виде солей щелочных металлов, солей щелочно-земельных металлов или солей аммония. Например, такие соли включают в себя соли натрия, соли калия, соли кальция, соли магния или аммония, или соли органического амина, такие как соли этиламина, этаноламина, триэтаноламина или аминокислот. Соединения согласно настоящему изобретению, которые содержат основные группы, могут существовать в форме солей неорганических или органических кислот. Примеры подходящих кислот включают в себя соляную кислоту, бромистоводородную кислоту, фосфорную кислоту, серную кислоту, азотную кислоту, метансульфоновую кислоту, пара-толуолсульфоновую кислоту, нафталиндисульфоновую кислоту, щавелевую кислоту, уксусную кислоту, винную кислоту, молочную кислоту, салициловую кислоту, бензойную кислоту, муравьиную кислоту, пропановую кислоту, пивалевую кислоту, малоновую кислоту, янтарную кислоту, пимелиновую кислоту, фумаровую кислоту, малеиновую кислоту, яблочную кислоту, сульфамовую кислоту, фенилпропионовую кислоту, глюконовую кислоту, аскорбиновую кислоту, изоникотиновую кислоту, лимонную кислоту, адипиновую кислоту и другие кислоты, известные специалистам в данной области техники. Если соединения согласно настоящему изобретению содержат в молекуле и кислую, и основную группы, то настоящее изобретение дополнительно включает в себя внутренние соли в дополнение у упомянутым выше солевым формам. Каждая соль может быть получены традиционными способами, известными специалистам в данной области техники, например, путем смешивания соединения согласно настоящему изобретению с органической или неорганической кислотой или основанием в растворителе или диспергирующем агенте, или путем анионного обмена или катионного обмена с другой солью.
Термин «фармацевтическая композиция» относится к композиции, содержащей одно или более соединений, описанных в настоящем документе, или их фармацевтически приемлемые соли, пролекарства, стабильные изотопные производные и изомеры, и другие компоненты, такие как фармацевтически приемлемые носители и вспомогательные вещества.
Если в настоящей заявке упоминается термин «соединения», то в него включены все формы соединения, такие как его фармацевтически приемлемые соли, пролекарства, стабильные изотопные производные и изомеры, а также их смеси.
«Аутоиммунные заболевания или воспалительные заболевания» включают в себя без ограничения артрит, тиреоидит Хашимото, аутоиммунную гемолитическую анемию, аутоиммунный атрофический гастрит при пернициозной анемии, аутоиммунный энцефаломиелит, аутоиммунный орхит, болезнь Гудпасчера, аутоиммунную тромбоцитопению, симпатическую офтальмию, миастению гравис, болезнь Грейвса, первичный билиарный цирроз, гепатит, первичный склерозирующий холангит, хронический инвазивный гепатит, неалкогольный стеатоз печени, неалкогольный стеатогепатит, язвенный колит, мембранозную гломерулопатию, системную красную волчанку, ревматоидный артрит, псориатический артрит, синдром Шегрена, синдром Рейтера, полимиозит, дерматомиозит, заболевание интерферона I типа, включая синдром Айкарди-Гутьереса и другой системный склероз с повышенной экспрессией интерферона I типа, менделевская болезнь, узелковый полиартериит, рассеянный склероз, рецидивирующий рассеянный склероз, первичный прогрессирующий рассеянный склероз, вторичный прогрессирующий рассеянный склероз и буллезный пемфигоид; дополнительно, аутоиммунные заболевания, связанные с О-клетками (биологические жидкости организма) или Т-клетками, включая синдром Когана, анкилозирующий спондилит, гранулематоз Вегенера, аутоиммунную алопецию, сахарный диабет I типа или ювенильный сахарный диабет, и тиреоидит.
В настоящем документе термин «энтерит» включает в себя без ограничения болезнь Крона, язвенный колит, воспалительное заболевание кишечника, целиакию, проктит, эозинофильный гастроэнтерит и мастоцитоз.
«Злокачественные опухоли/опухоли» включают в себя без ограничения злокачественную опухоль кишечника/желудочно-кишечного тракта, злокачественную опухоль толстого кишечника, злокачественную опухоль печени, злокачественную опухоль кожи (включая опухоль тучных клеток и сквамозно-клеточную карциному), злокачественную опухоль молочной железы, злокачественную опухоль яичника, злокачественную опухоль предстательной железы, лимфому, лейкоз (включая острый миелоидный лейкоз и хронический миелогенный лейкоз), злокачественную опухоль почки, злокачественную опухоль легкого, злокачественную опухоль мышц, злокачественную опухоль кости, злокачественную опухоль мочевого пузыря, злокачественную опухоль головного мозга, меланому (включая внутриротовую и метастатическую меланому), саркому Капоши (миелому, включая множественную миелому), миелопролиферативные заболевания, пролиферативную диабетическую ретинопатию и связанные с сосудистой гиперплазией нарушения/опухоли.
«Заболевания кожи» включают в себя без ограничения атопический дерматит, экзему, псориаз, склеродермию, прурит и другие симптомы зуда, витилиго и выпадение волос.
«Диабет» включает в себя без ограничения сахарный диабет I типа и осложнения диабета.
«Заболевания глаз» включают в себя без ограничения кератоконъюнктивит, увеит (включая увеит, ассоциированный с болезнью Бехчета, и увеит, вызванный контактными линзами), кератит, герпетический кератит, кератоконус, эпителиальную дистрофию роговицы, лейкопению роговицы, передний увеит, язву Мурена, склерит, заболевание глаз Грейвса, синдром Фогта-Коянаги-Харада, сухой кератоконъюнктивит (сухой глаз), волдыри, иридоциклит при саркоидозе, эндокринную офтальмопатию, симпатическую офтальмию, аллергический конъюнктивит и неоваскуляризацию глаза.
«Нейродегенеративные заболевания» включают в себя без ограничения болезнь моторных нейронов, болезнь Альцгеймера, болезнь Паркинсона, боковой амиотрофический склероз, болезнь Хантингтона, церебральную ишемию; нейродегенеративное заболевание, вызванное травмой, повреждением, нейротоксичностью глутамата или гипоксией; ишемическое/реперфузионное повреждение при инсульте, ишемии миокарда, почечной ишемии, сердечном приступе, гипертрофии миокарда, атеросклерозе и артериосклерозе, органной гипоксии или агрегации тромбоцитов.
«Анафилаксия» включает в себя без ограничения аллергический дерматит у млекопитающих (включая анафилактические заболевания лошадей, такие как гиперчувствительность к укусам), экзему в летний период, itchy horseshoes, спазм, воспалительные заболевания дыхательных путей, повторную обструкцию дыхательных путей, гиперреактивность дыхательных путей и хроническую обструктивную болезнь легких.
«Астма и другие обструктивные болезни дыхательных путей» включают в себя без ограничения хроническую или усиленную астму, отсроченную астму, бронхит, бронхиальную астму, аллергическую астму, эндогенную астму, экзогенную астму и пылевую астму.
«Отторжение трансплантата» включает в себя без ограничения отторжение трансплантата островковых клеток, отторжение трансплантата костного мозга, болезнь «трансплантат против хозяина», отторжение трансплантата органа или клеток (таких как костный мозг, хрящ, роговица, сердце, межпозвоночный диск, островковые клетки, почка, конечность, печень, легкое, мышца, миобласт, нерв, поджелудочная железа, кожа, тонкий кишечник или трахея) и ксенотрансплантацию.
«Терапевтически эффективное количество» относится к количеству соединения согласно настоящему изобретению, которое может эффективно ингибировать функцию JAK, в частности TYK2, и/или лечить или предупреждать заболевание, опосредованное этой киназой.
«Пациенты» относятся к млекопитающим, предпочтительно к людям.
Настоящее изобретение относится к соединениям, применимым в качестве ингибиторов JAK, в частности TYK2. Соединения представлены формулой (I) или их пролекарствами, стабильными изотопными производными, фармацевтически приемлемыми солями и изомерами.
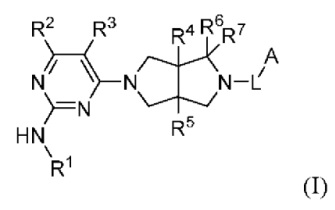
где:
R1 представляет собой арил или гетероарил, где один или более атомов водорода арила и гетероарила необязательно замещены D, галогеном, циано, -ORb, -NRbRc, -COORb, -C(O)Rb, -NRbC(O)Rc, -C(O)NRbRc, -S(O)2Rb, -S(O)2NRbRc, -S(O)(NRb)Rc, -P(O)(CH3)2, C1-6 алкилом, С3-6 циклоалкилом, 3-8-членным гетероциклилом или 5-6-членным гетероарилом;
R2 представляет собой Н, D или -NHRa;
R3 представляет собой Н, D, галоген, циано, С1-6 алкил, С3-6 циклоалкил или OC1-6 алкил, где один или более атомов водорода алкила и циклоалкила необязательно замещены D или F;
R4 и R5 независимо выбраны из Н, D, галогена, C1-6 алкила или ОС1-6 алкила, где один или более атомов водорода алкила необязательно замещены D или F;
R6 и R7 независимо выбраны из Н, D, циано или C1-6 алкила, где один или более атомов водорода алкила необязательно замещен D или F, или R6 и R7 объединены в виде оксо;
L представляет собой связь, С1-6 алкилен, -С(О)-, -С(O)O-, -C(O)N(Ra)-, -S(O)2- или -S(O)2N(Ra)-;
А представляет собой Н, С1-6 алкил, С3-6 циклоалкил, 3-8-членный гетероциклил, арил или гетероарил, где один или более атомов водорода алкила, циклоалкила, гетероциклила, арила и гетероарила необязательно замещены D, галогеном, циано, -ORd, -NRdRe, C1-6 алкилом, С3-6 циклоалкилом или 3-8-членным гетероциклилом;
Ra представляет собой Н, С1-6 алкил или С3-6 циклоалкил, где один или более атомов водорода алкила и циклоалкила необязательно замещены D или F;
Rb и Rc независимо выбраны из Н, С1-6 алкила, С3-6 циклоалкила или 3-8-членного гетероциклила, содержащего N и/или О, где один или более атомов водорода алкила, циклоалкила и гетероциклила необязательно дополнительно замещены D, галогеном, CN, -ОН, -NH2, C1-6 алкилом и -OC1-6 алкилом, и
Rd и Re независимо выбраны из Н, С1-6 алкила, С3-6 циклоалкила или 3-8-членного гетероциклила, где один или более атомов водорода алкила, циклоалкила и гетероциклила необязательно дополнительно замещены D или F.
Согласно одному варианту осуществления, R1 представляет собой фенил.
Согласно одному варианту осуществления, R1 представляет собой 5-6-членный гетероарил, например, R1 представляет собой пиридил, пиримидил или пиразолил.
Согласно одному варианту осуществления, один или более атомов водорода фенила и гетероарила R1 необязательно замещены галогеном. -COORb, -C(O)Rb, -C(O)NRbRc, C1-6 алкилом, С3-6 циклоалкилом или 4-6-членным гетероциклилом, содержащим N и/или О, предпочтительно C1-6 алкилом, 4-6-членным гетероциклилом, -C(O)Rb или -C(O)NRbRc, где один или более атомов водорода алкила, циклоалкила и гетероциклила необязательно дополнительно замещены D, галогеном, CN, -ОН, -NH2, C1-6 алкилом и -ОС1-6 алкилом. предпочтительно D, F, CN, -ОН или С1-6 алкилом.
Согласно одному варианту осуществления, Rb и Rc независимо выбраны из Н, C1-6 алкила, С3-6 циклоалкила или 4-6-членного гетероциклила содержащего N и/или О, где один или более атомов водорода алкила, циклоалкила и гетероциклила необязательно дополнительно замещены С1-6 алкилом.
Согласно одному предпочтительному варианту осуществления, R2 представляет собой Н.
Согласно одному предпочтительному варианту осуществления, R3 представляет собой Н, галоген, циано, C1-6 алкил или OC1-6 алкил.
Согласно одному предпочтительному варианту осуществления, R4 и R5 независимо представляют собой Н или C1-6 алкил.
Согласно одному предпочтительному варианту осуществления, R6 и R7 представляют собой Н, или R6 и R7 объединены в виде оксо.
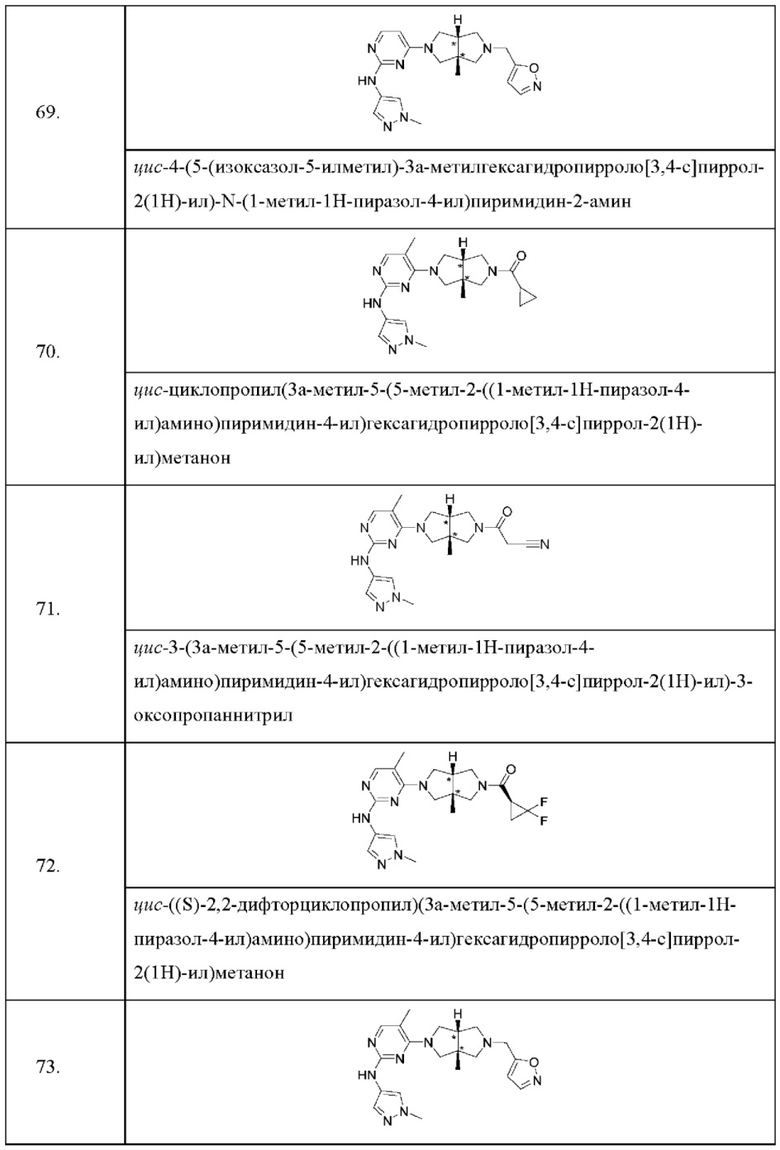
Согласно одному предпочтительному варианту осуществления, L представляет собой связь, C1-6 алкилен, -С(О)-, -С(O)O-, -C(O)NH- или -S(O)2-.
Согласно одному варианту осуществления, А представляет собой C1-6 алкил или С3-6 циклоалкил, где один или более атомов водорода алкила и циклоалкила необязательно замещены галогеном, циано, -ОН, -ОС1-2 алкилом или С1-2 алкилом.
Согласно одному варианту осуществления, А представляет собой пиридил, пиримидил, 5-членный гетероарил или 4-6-членный гетероциклил, где один или более атомов водорода гетероциклила и гетероарила необязательно замещены галогеном, циано, -ОН, -ОС1-2 алкилом или С1-2 алкилом, предпочтительно С1-2 алкилом.
Согласно некоторым вариантам осуществления, соединения, представленные формулой (I), характеризуются следующей формулой (II):
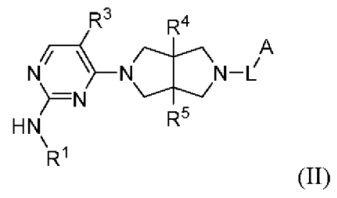
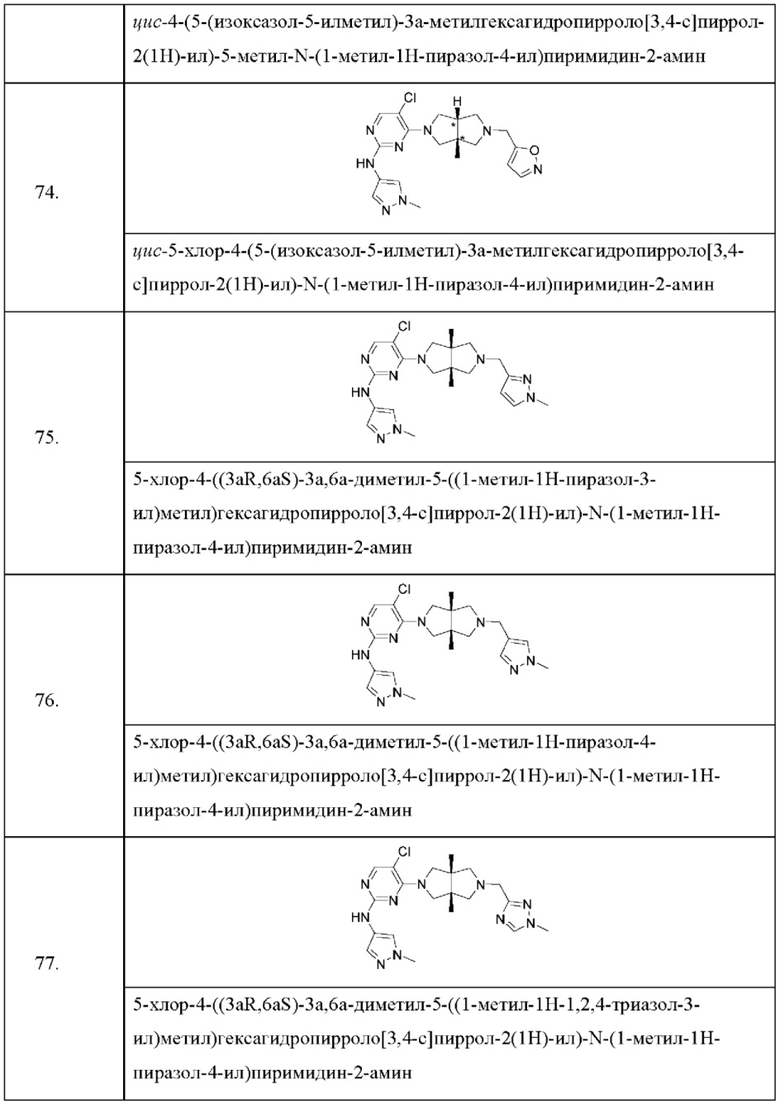
где:
R1 представляет собой фенил, пиридил или пиразолил, где один или более атомов водорода фенила, пиридила и пиразолила необязательно замещены галогеном, С1-6 алкилом, 4-6-членным гетероциклилом, содержащим N и/или О (например, морфолином, пиперазином, пиперидином, пирролидином и оксетаном), -C(O)Rb или -C(O)NRbRc, и один или более атомов водорода алкила и гетероциклила необязательно дополнительно замещены D, F, CN, -ОН или C1-6 алкилом;
Rb и Rc независимо выбраны из Н, C1-6 алкила, С3-6 циклоалкила или 4-6-членного гетероциклила, содержащего N и/или О (например, морфолина, пиперазина, пиперидина, пирролидина и оксетана), где один или более атомов водорода алкила, циклоалкила и гетероциклила необязательно дополнительно замещены С1-2 алкилом;
R3 представляет собой Н, галоген, циано, C1-6 алкил или OC1-6 алкил;
R4 и R5 независимо представляют собой Н или C1-6 алкил;
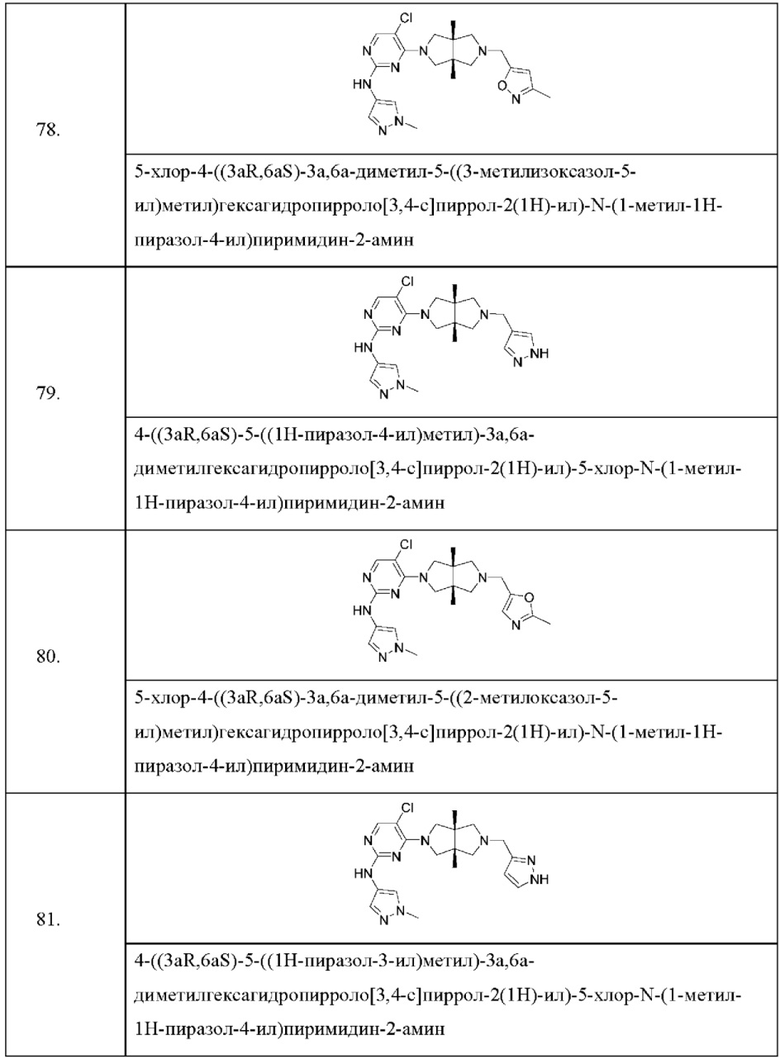
L представляет собой связь, C1-6 алкилен, -С(О)-, -С(O)O-, -C(O)NH- или -S(O)2-, и
А представляет собой С1-6 алкил, С3-6 циклоалкил, 4-6-членный гетероциклил, содержащий N и/или О (например, морфолин, пиперазин, пиперидин, пирролидин и оксетан), пиридил, пиримидил или 5-членный гетероарил (например, изоксазол и пиразол), где один или более атомов водорода алкила и циклоалкила необязательно замещены галогеном, циано, -ОН, -ОС1-2 алкилом или С1-2 алкилом, и один или более атомов водорода гетероциклила и гетероарила необязательно замещены С1-2 алкилом.
Согласно одному предпочтительному варианту осуществления, R1 представляет собой пиразолил.
Согласно одному предпочтительному варианту осуществления, R3 представляет собой Н, галоген или С1-6 алкил.
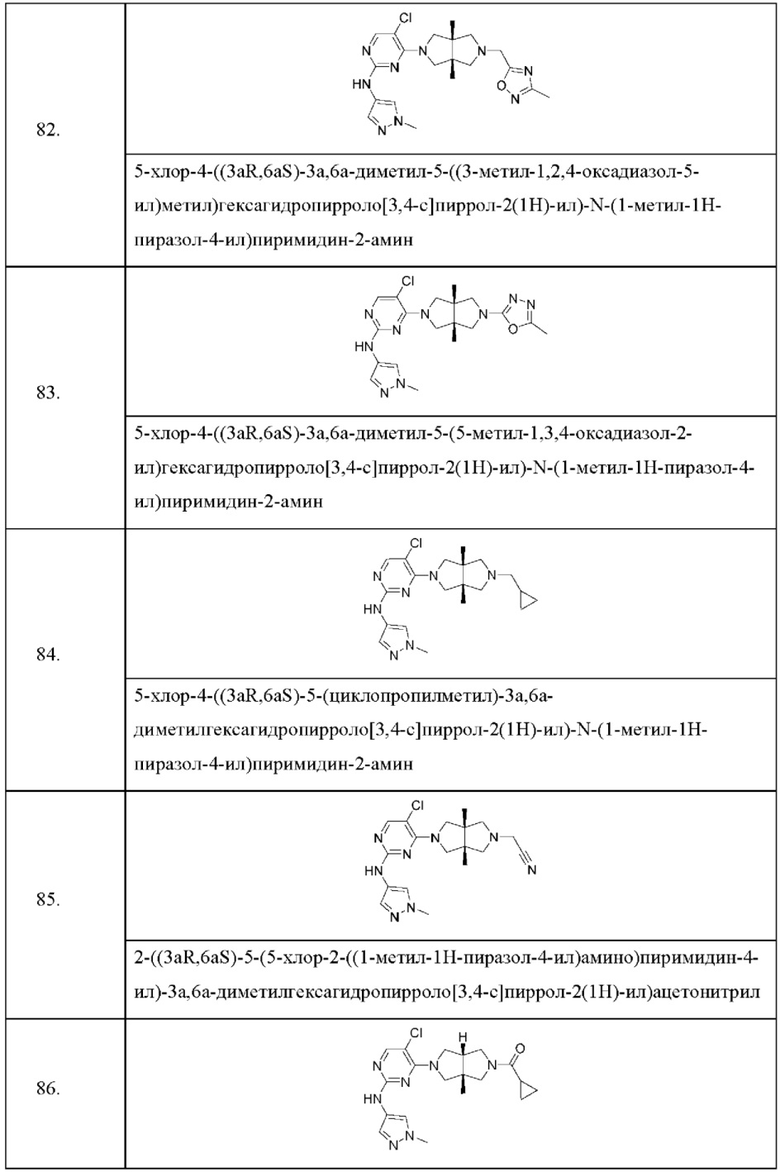
Согласно одному предпочтительному варианту осуществления, R4 представляет собой Н или метил.
Согласно одному предпочтительному варианту осуществления, R5 представляет собой Н или метил.
Согласно одному предпочтительному варианту осуществления, L представляет собой -С(О)-.
Согласно одному предпочтительному варианту осуществления, А представляет собой С1-6 алкил или С3-6 циклоалкил, где один или более атомов водорода алкила и циклоалкила необязательно замещены галогеном, циано, -ОН или -ОС1-2 алкилом.
Согласно некоторым вариантам осуществления, соединения, представленные формулой (II), характеризуются следующей формулой (III):
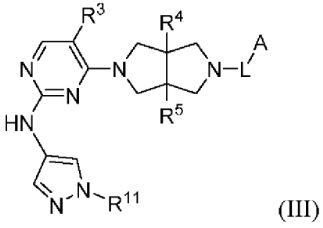
где:
R3 представляет собой Н, галоген, циано, С1-6 алкил или OC1-6 алкил;
R4 и R5 независимо представляют собой Н или С1-6 алкил;
L представляет собой связь, С1-6 алкилен, -С(О)-, -С(O)O-, -C(O)NH- или -S(O)2-;
А представляет собой С1-6 алкил, С3-6 циклоалкил, 4-6-членный гетероциклил, содержащий N и/или О (например, морфолин, пиперазин, пиперидин, пирролидин и оксетан), пиридил, пиримидил или 5-членный гетероарил (например, изоксазол и пиразол), где один или более атомов водорода алкила и циклоалкила необязательно замещены D, галогеном, циано, -ОН, -ОС1-2 алкилом или С1-2 алкилом, и один или более атомов водорода гетероциклила и гетероарила необязательно замещены С1-2 алкилом, и
R11 представляет собой Н, C1-6 алкил или 4-6-членный гетероциклил, содержащий N и/или О (например, морфолин, пиперазин, пиперидин, пирролидин и оксетан), где один или более атомов водорода алкила и гетероциклила необязательно замещены D, F, CN, -ОН или С1-6 алкилом.
Предпочтительный А в соединении формулы III представляет собой С1-6 алкил или С3-6 циклоалкил, где один или более атомов водорода алкила и циклоалкила необязательно замещены галогеном, циано, -ОН или -ОС1-2 алкилом.
Предпочтительный R11 представляет собой С1-6 алкил (например, метил), где один или более атомов водорода алкила необязательно замещены D, F, CN, -ОН или C1-6 алкилом.
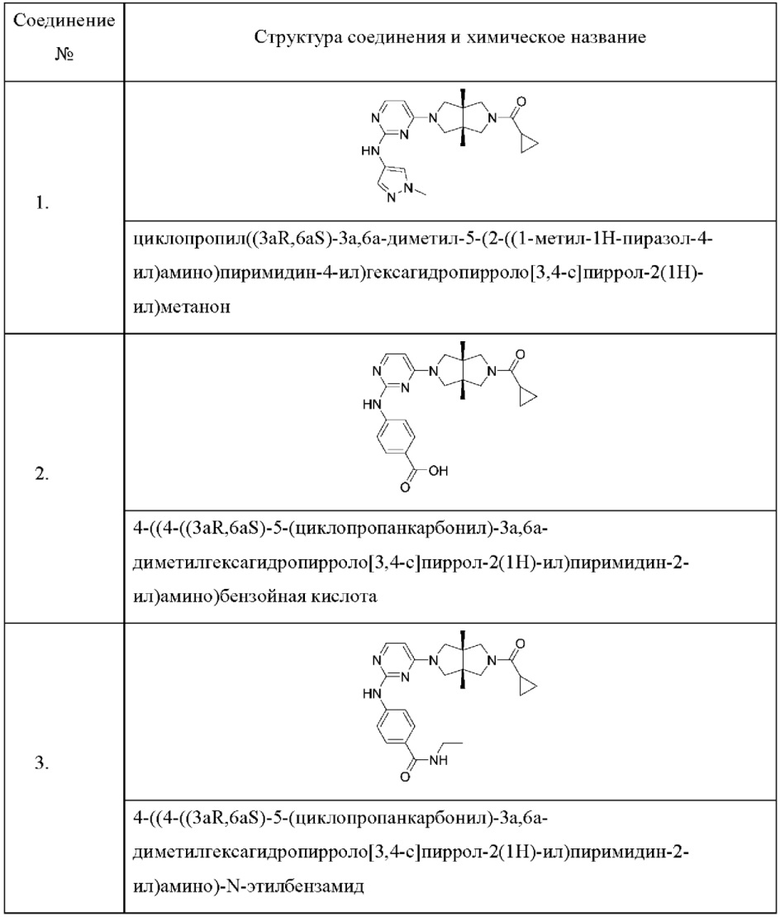
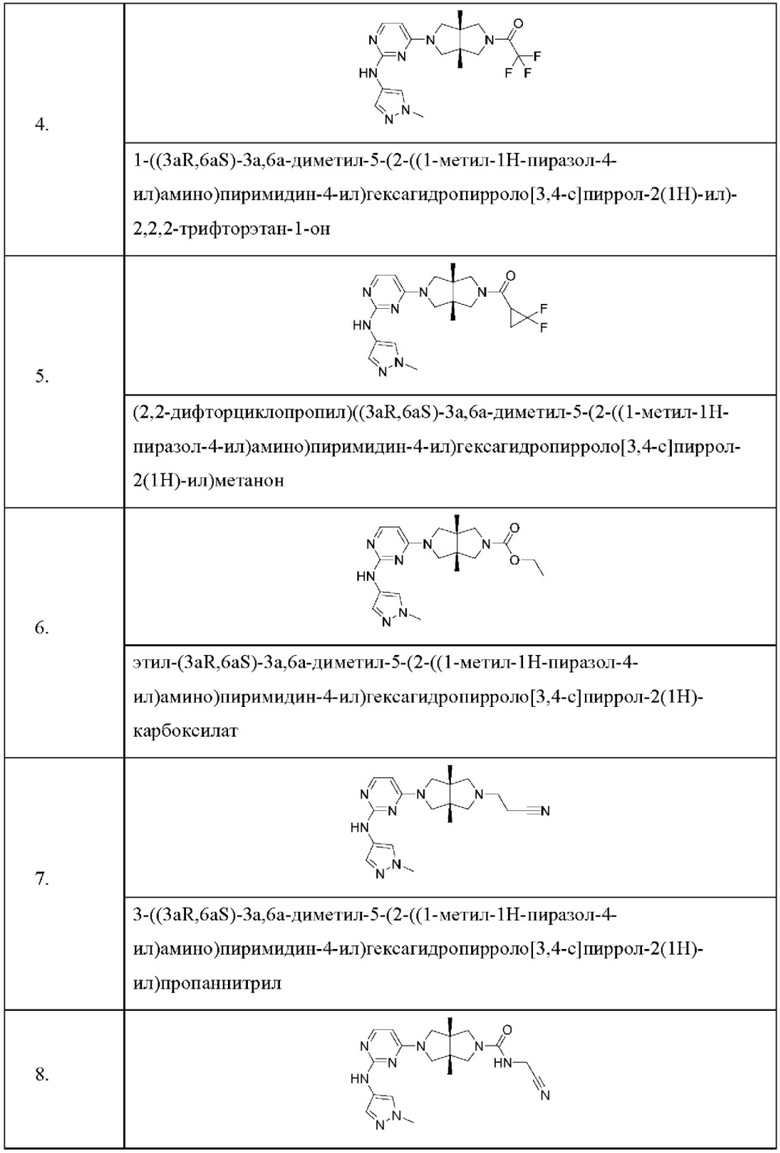
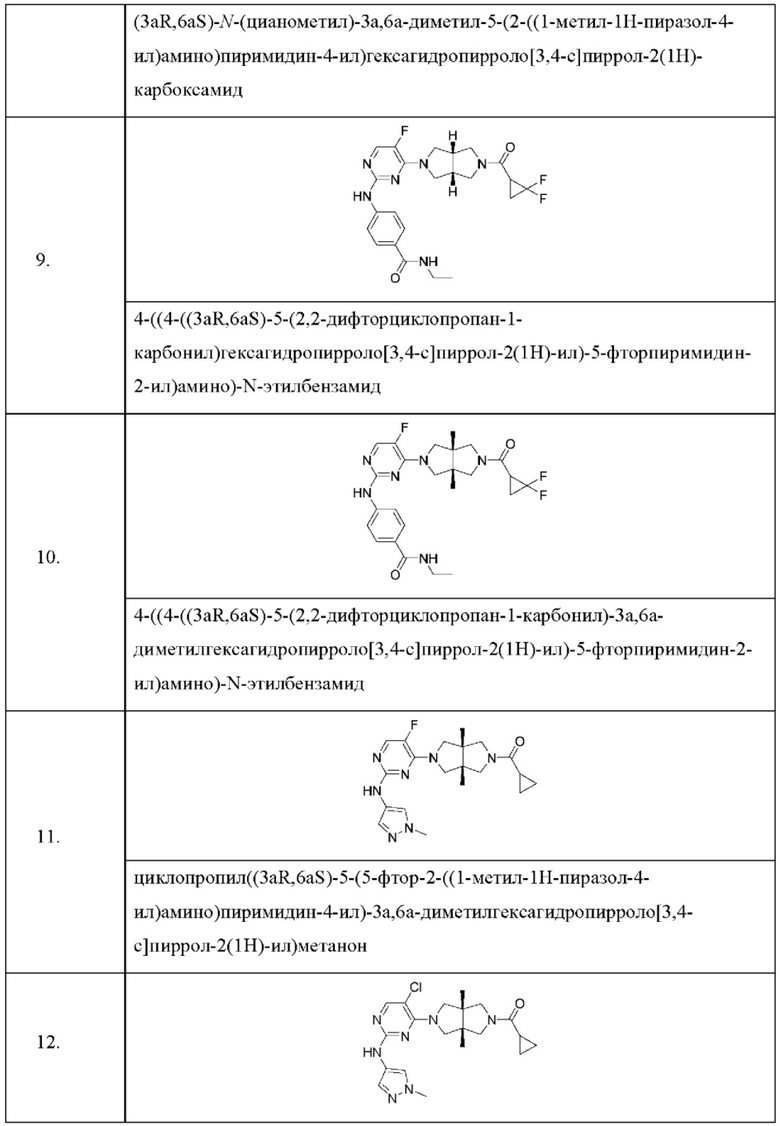
Настоящее изобретение дополнительно относится к следующим соединениям 1-100 или их фармацевтически приемлемым солям, пролекарствам, стабильным изотопным производным, изомерам и их смесям.
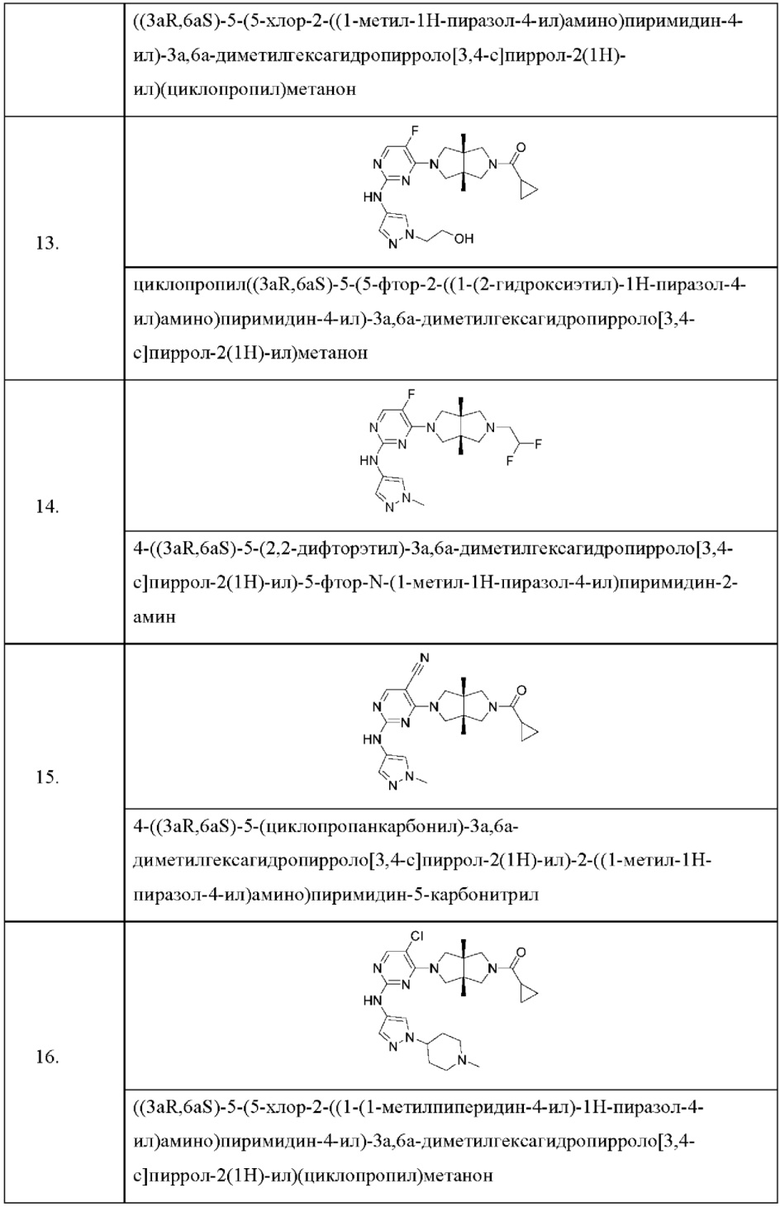
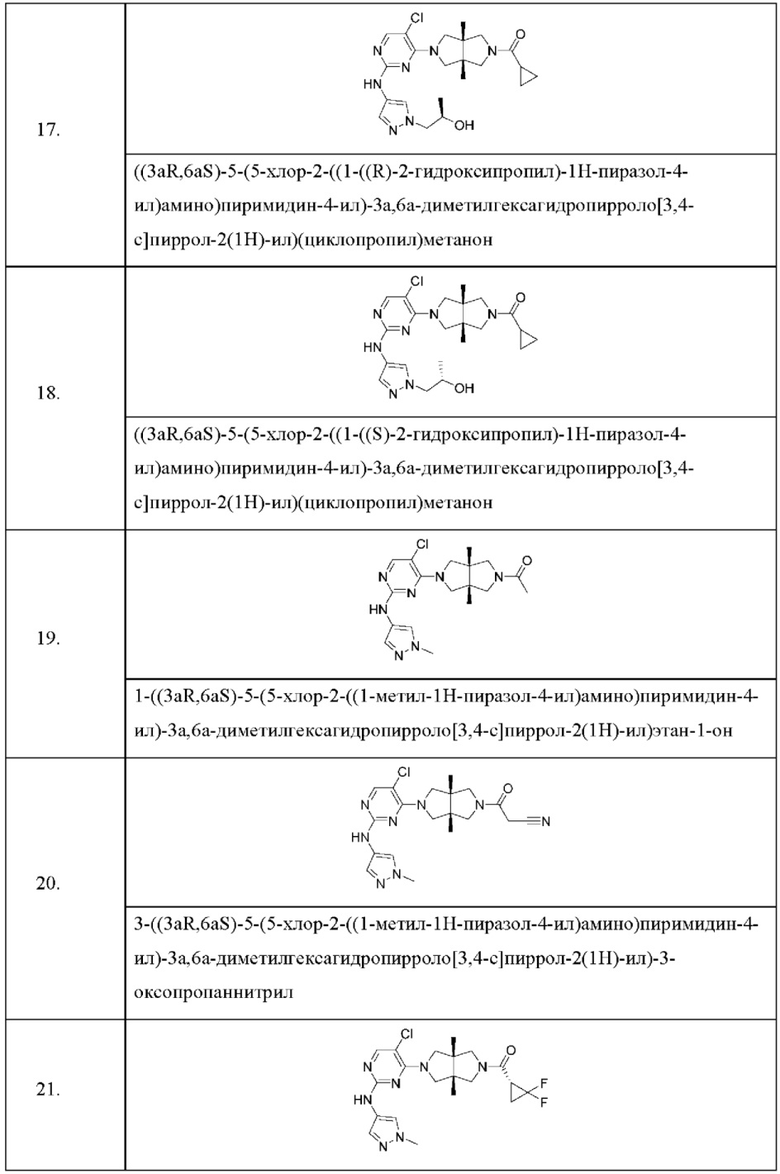
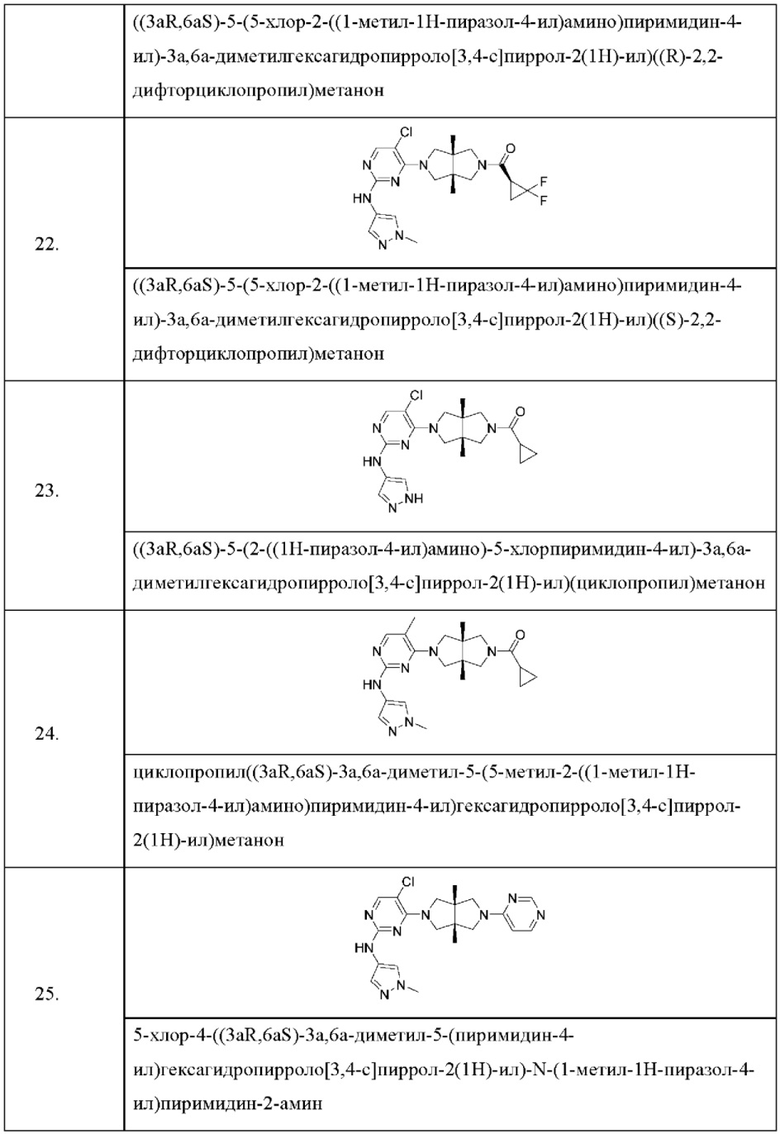
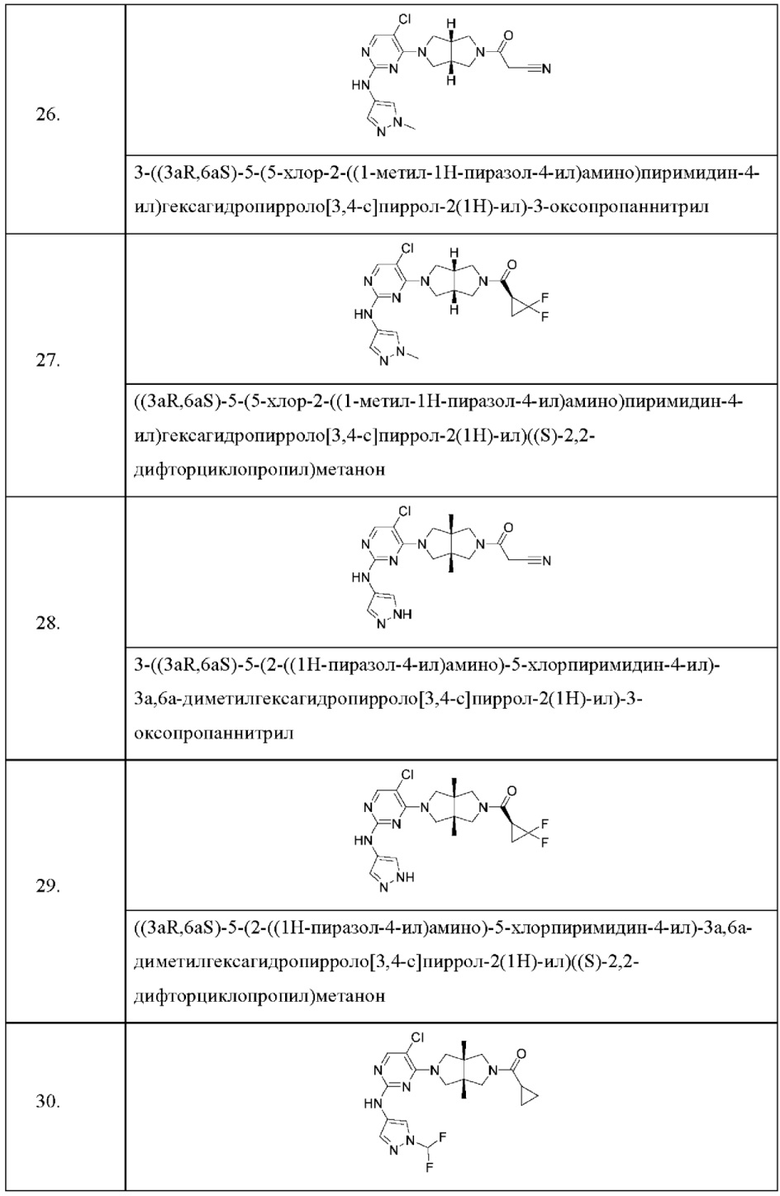
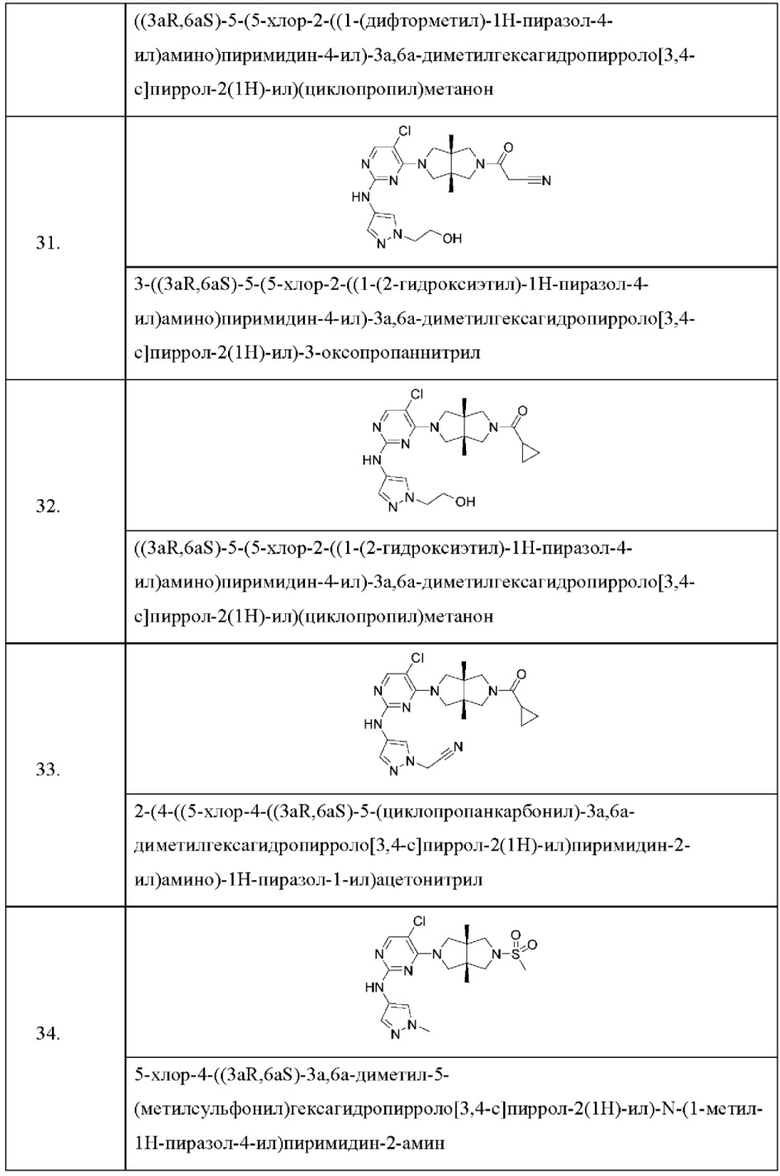
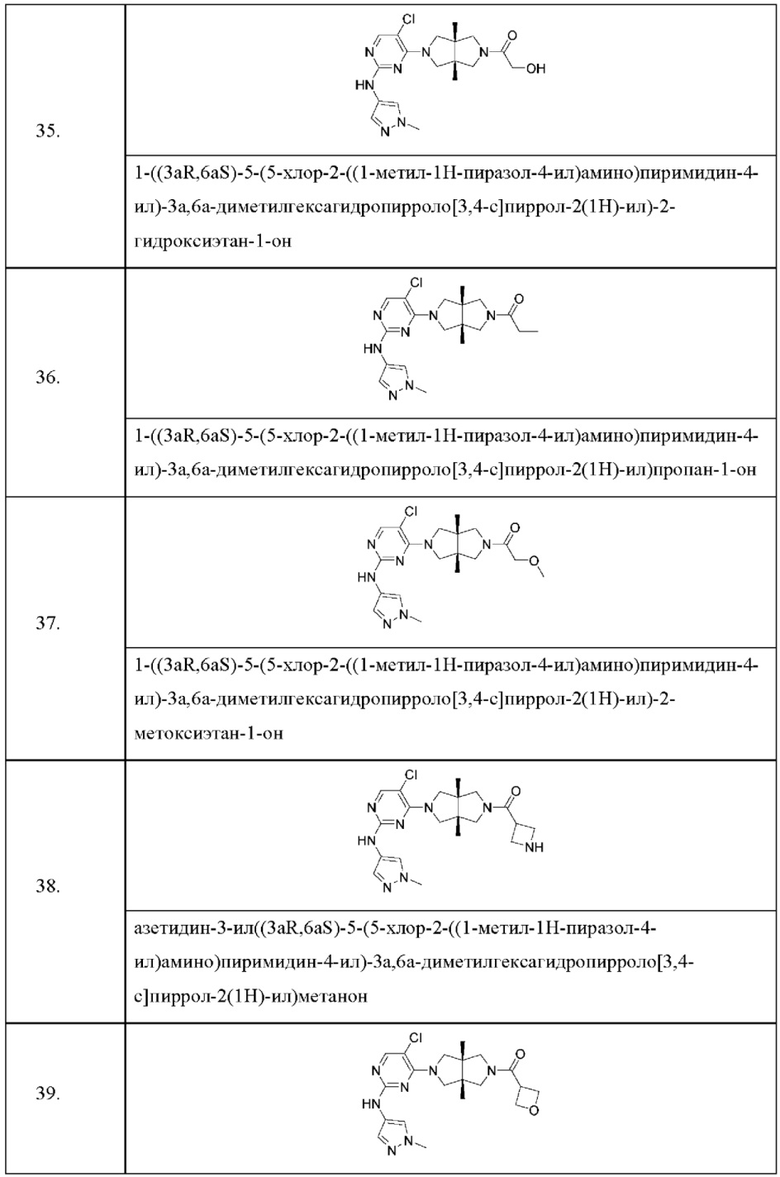
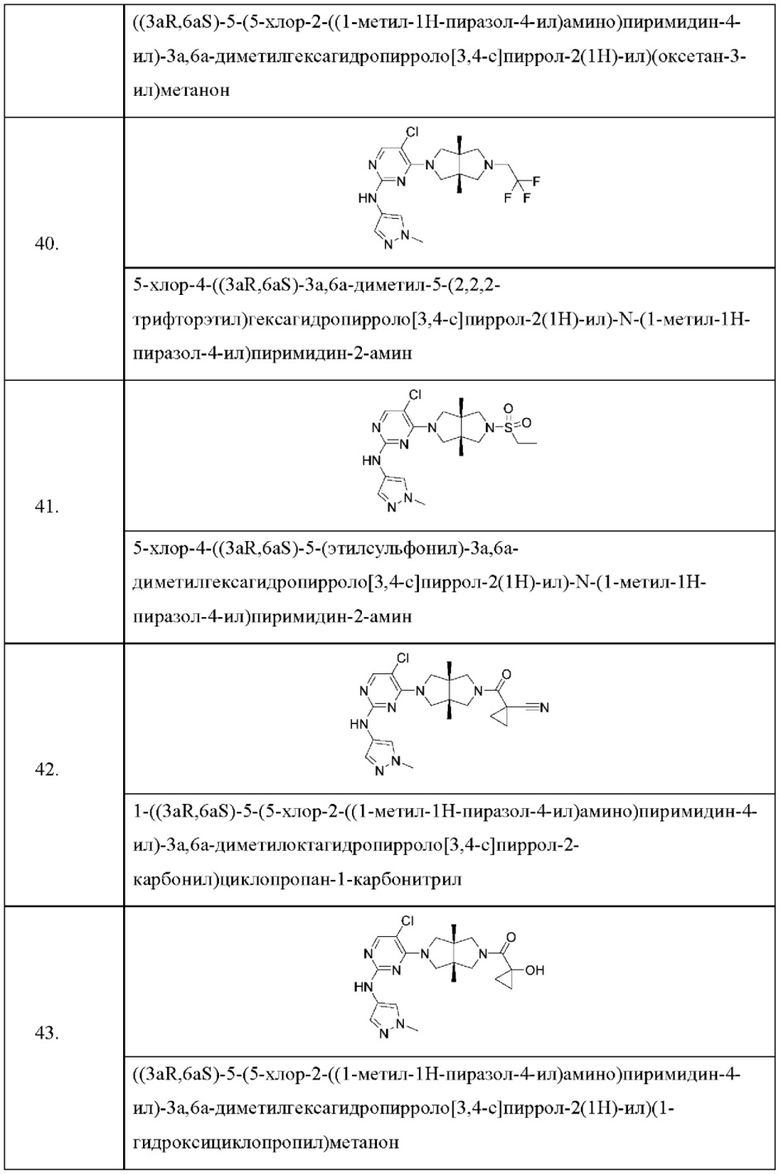
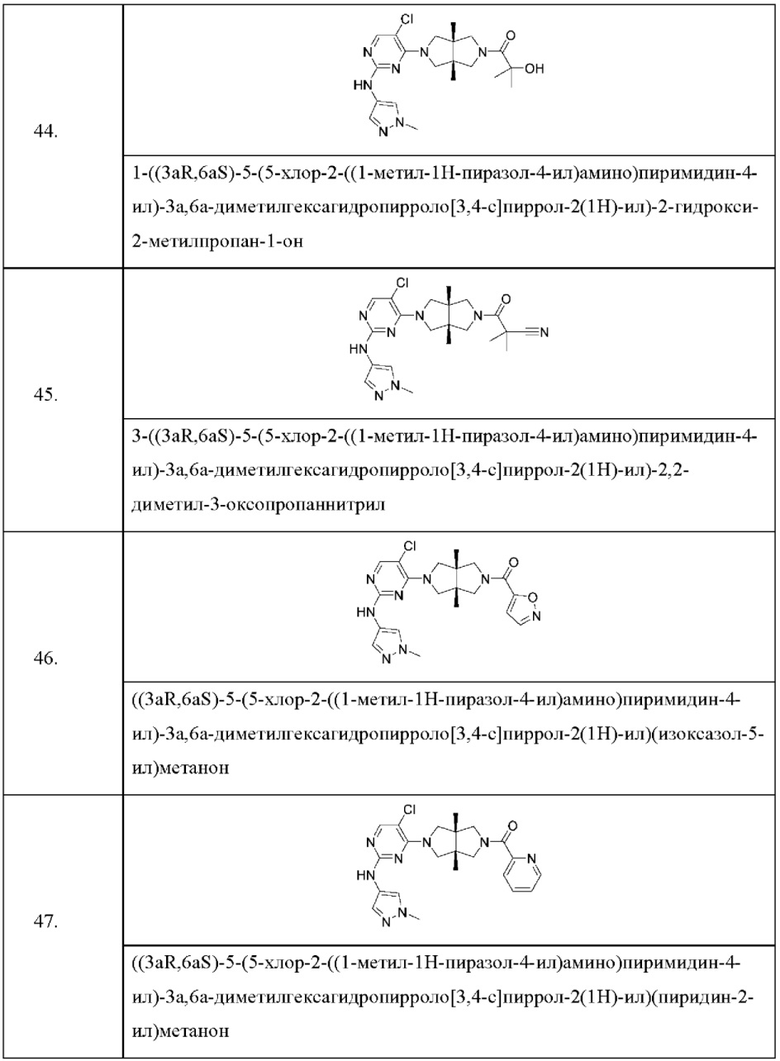
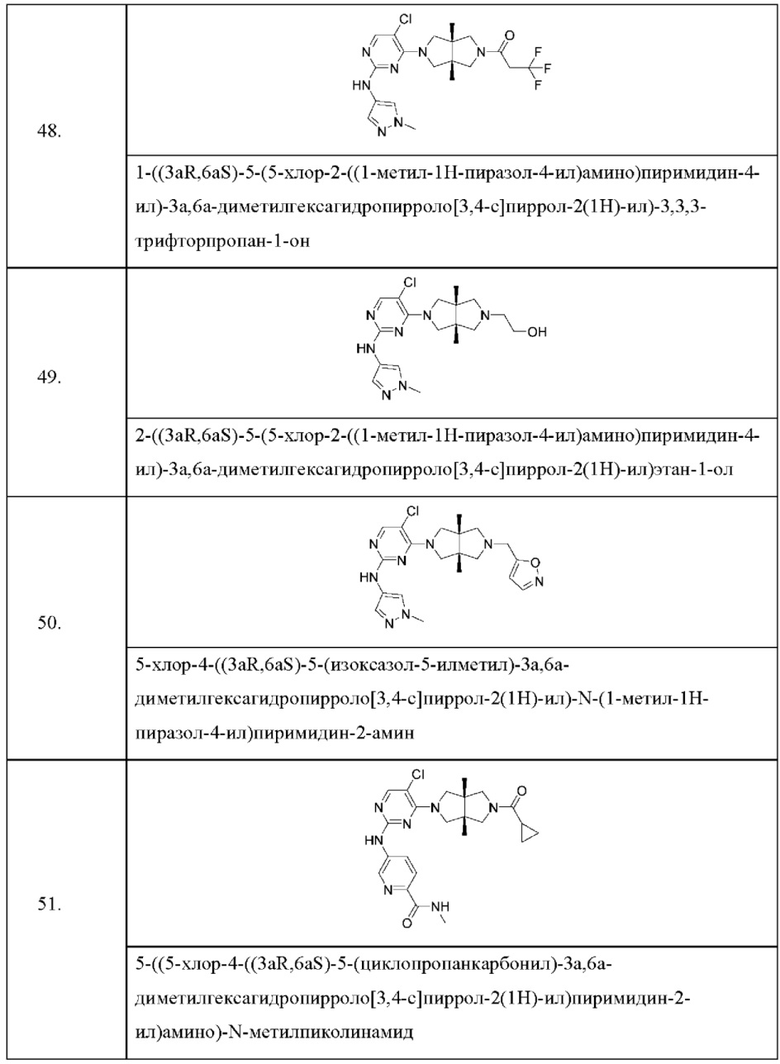
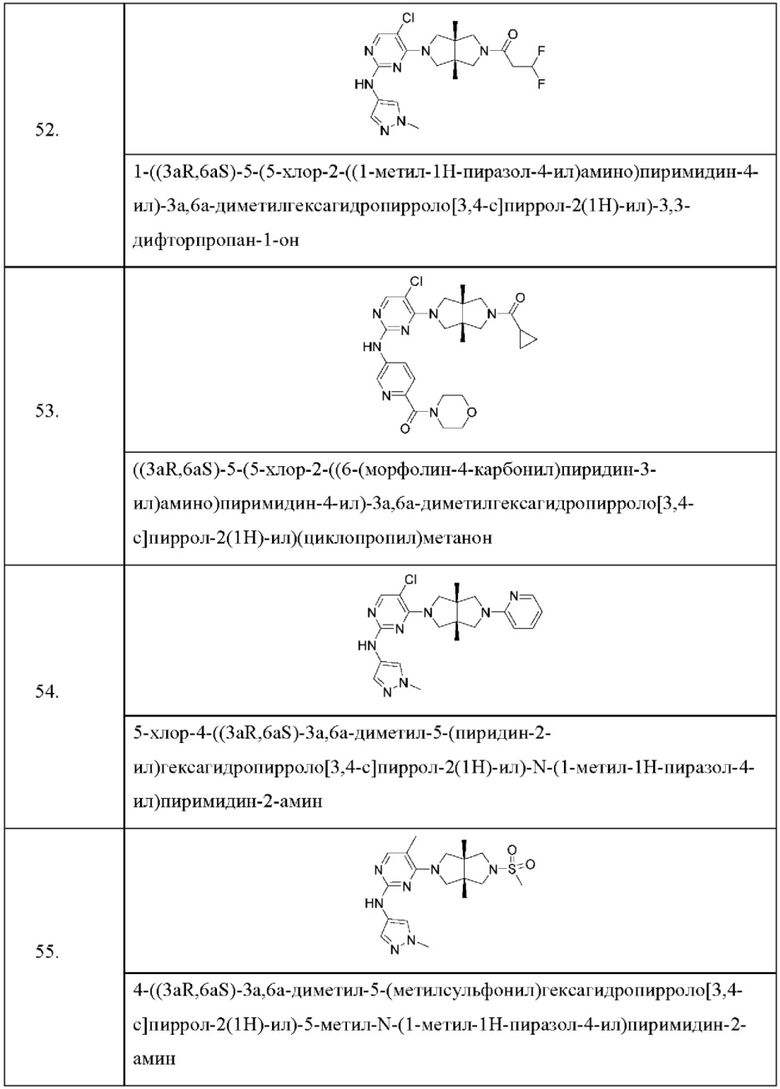
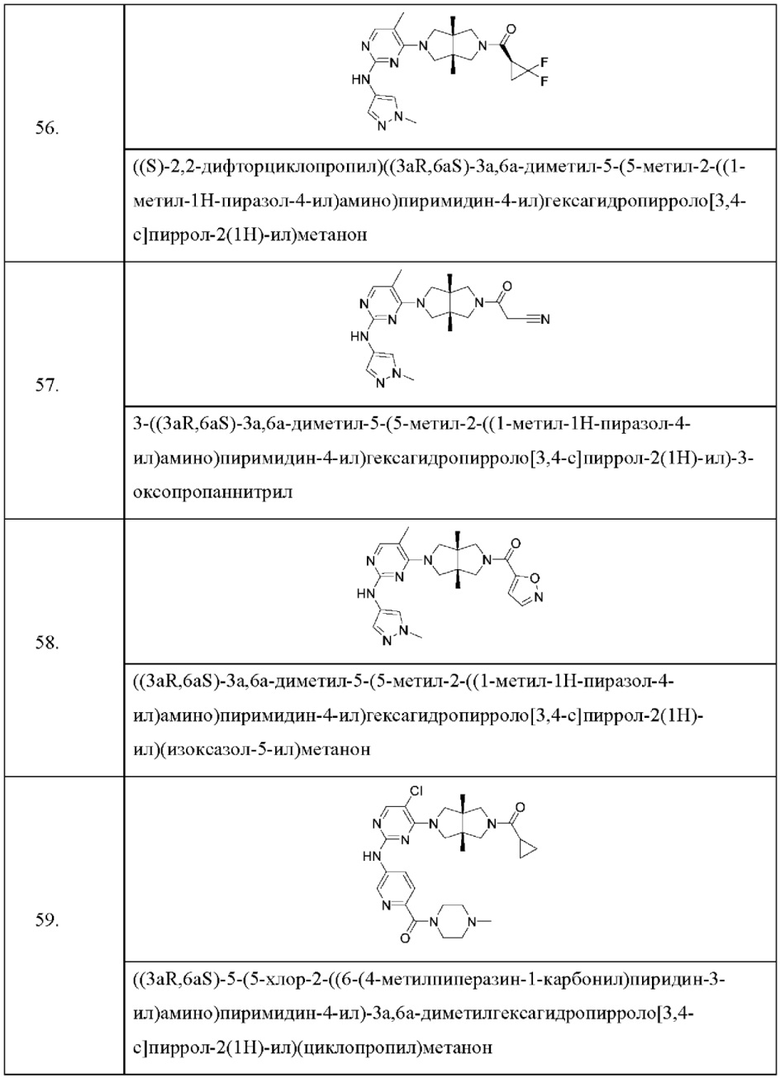
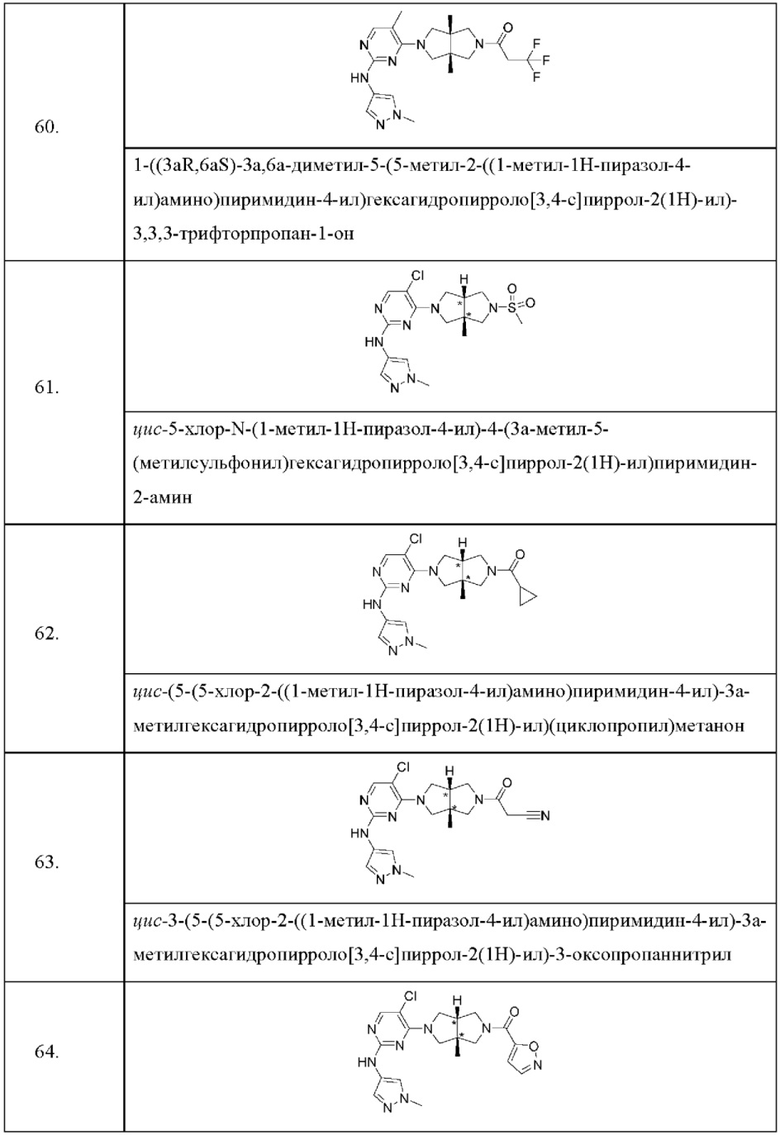

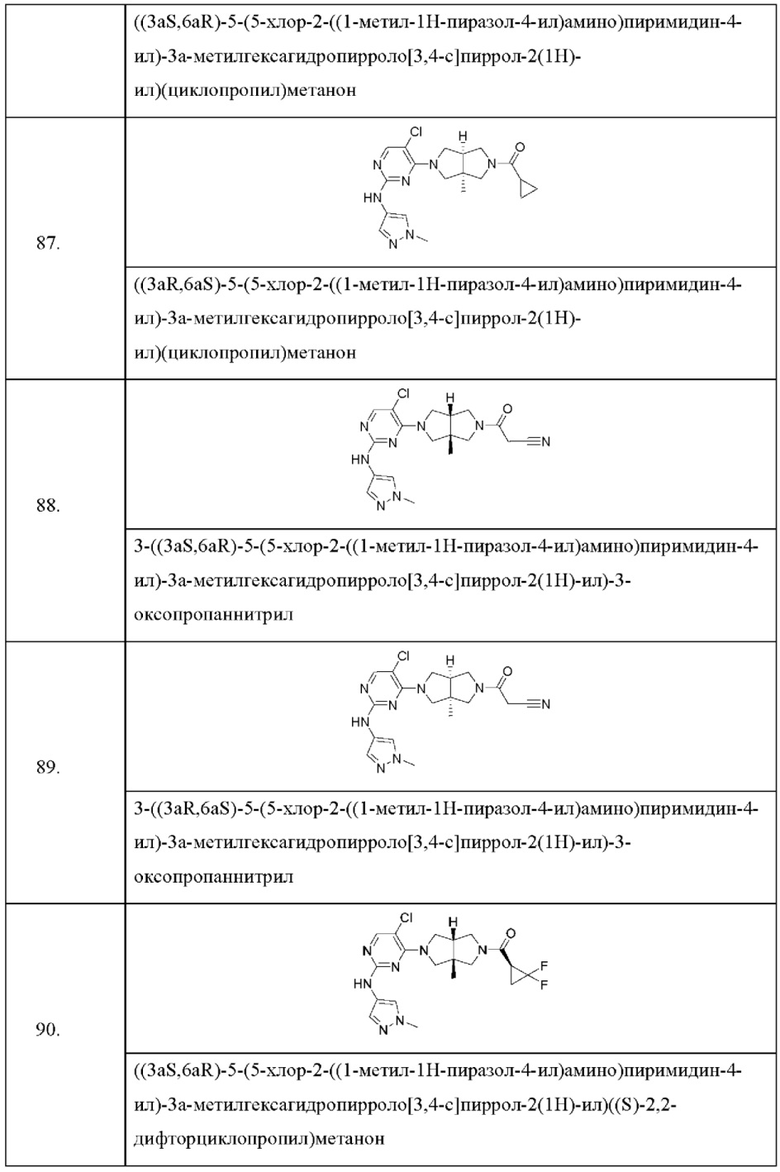
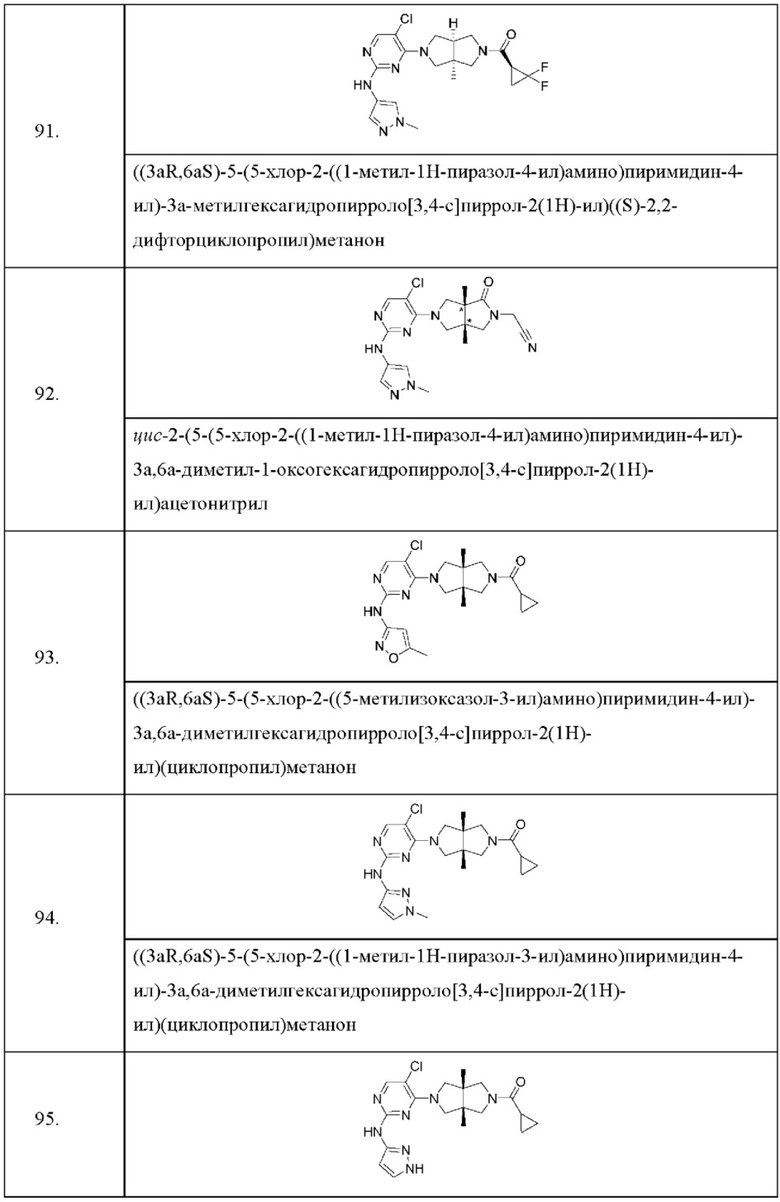
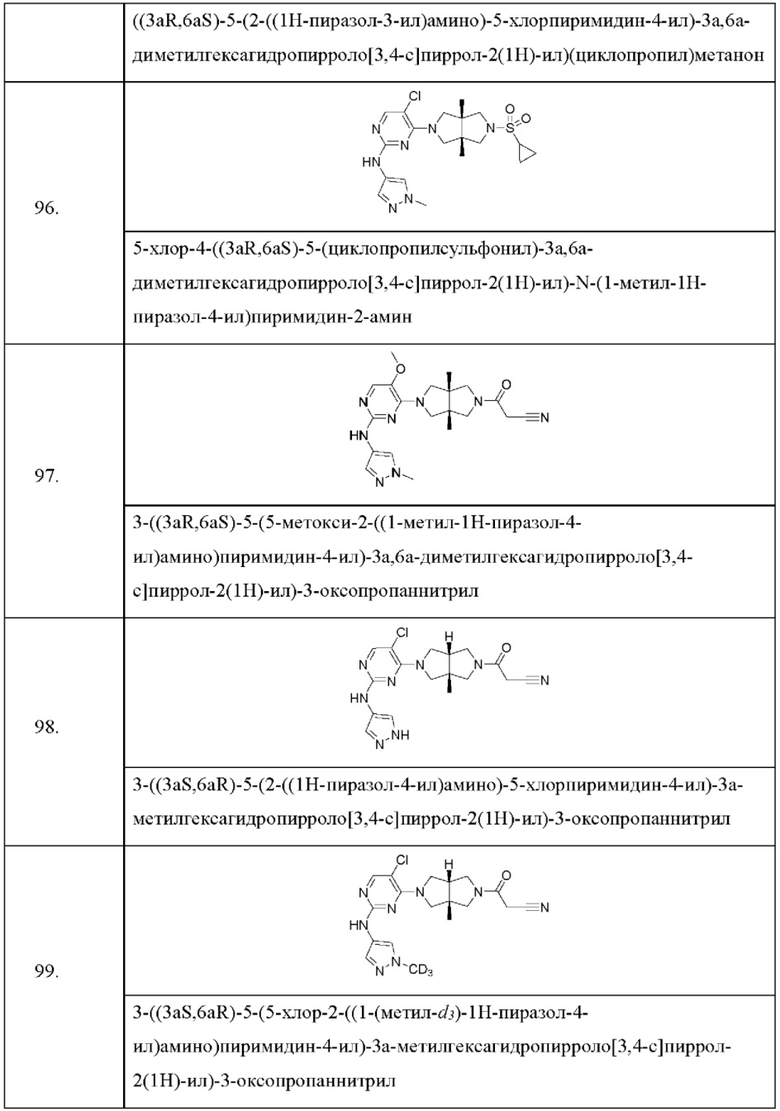
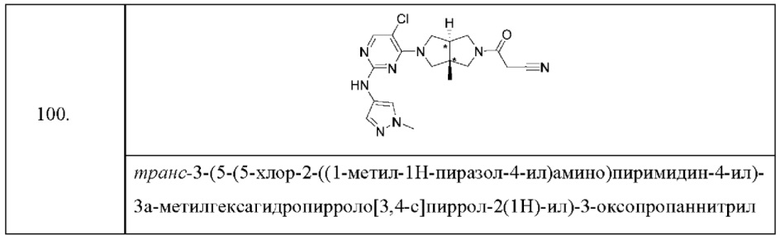
Соединения 61, 62, 63, 64, 65, 68, 69, 70, 71, 72, 73, 74, 92 и 100 являются рацематами или парой диастереоизомеров. Соединения 86, 87, 88, 89, 90, 91, 98 и 99 являются отдельными оптическими изомерами. Соединения 86/87 и соединения 88/89 представляют собой пару энантиомеров, соответственно. Соединения 90 и 91 являются парой диастереоизомеров.
Соединения согласно настоящему изобретению эффективно ингибируют активность JAK, в частности TYK2, предпочтительно характеризуясь значением IC50 от 10 до 100 нМ, и более предпочтительно характеризуясь значением IC50 менее 10 нМ. Соединения согласно настоящему изобретению обладают значительным эффектом ингибирования индуцированной IL-12 секреции IFN-γ в клетках NK92, предпочтительно характеризуясь значением IC50 менее 1000 нМ.
Настоящее изобретение дополнительно относится к фармацевтическим композициям, содержащим соединения формулы (I) или их фармацевтически приемлемые соли, пролекарства, стабильные производные изотопов или изомеры и фармацевтически приемлемые носители или вспомогательные вещества. Фармацевтические композиции применимы для лечения или предупреждения заболеваний, опосредованных JAK, в частности TYK2, включая без ограничения аутоиммунные заболевания, воспалительные заболевания, включая кишечные заболевания, злокачественные опухоли, заболевания кожи, диабет, заболевания глаз, нейродегенеративные заболевания, анафилаксию, астму, обструктивные заболевания дыхательных путей и отторжение трансплантата.
Настоящее изобретение дополнительно относится к способу лечения или предупреждения заболеваний, опосредованных JAK, в частности TYK2. Указанный способ включает в себя введение нуждающемуся в этом пациенту терапевтически эффективного количества соединений, представленных формулой (I), или их фармацевтически приемлемых солей, пролекарств, стабильных производных изотопов и изомеров. Заболевания включают в себя без ограничения аутоиммунные заболевания, воспалительные заболевания, включая кишечные заболевания, злокачественные опухоли, заболевания кожи, диабет, заболевания глаз, нейродегенеративные заболевания, анафилаксию, астму и другие обструктивные заболевания дыхательных путей, такие как COPD, и отторжение трансплантата. Соединения согласно настоящему изобретению особенно применимы для лечения псориаза, псориатического артрита, язвенного колита, болезни Крона, SLE, волчаночного нефрита, витилиго, очаговой алопеции, дерматита, астмы, атопической экземы.
В соответствии с настоящим изобретением, фармацевтические средства могут находиться в любой лекарственной форме, включая без ограничения таблетки, капсулы, раствор, лиофилизированный препарат и инъекционный препарат.
Лекарственную форму согласно настоящему изобретению можно вводить в виде стандартной лекарственной формы, содержащей предопределенное количество активного ингредиента. Такая стандартная форма может содержать от 0,5 мг до 1 г, предпочтительно от 1 мг до 700 мг, более предпочтительно от 5 мг до 300 мг, соединения согласно настоящему изобретению в зависимости от подлежащего лечению заболевания, способа введения, а также возраста, веса и состояния пациентов. Кроме того, лекарственная форма может быть приготовлена с использованием способов, хорошо известных в области фармации, например, путем включения активного ингредиента в состав с одним или более вспомогательными веществами или с одним или более адъювантами.
Лекарственная форма согласно настоящему изобретению подходит для введения любым подходящим способом, например, путем перорального (включая буккальное или сублингвальное), ректального, назального, местного (включая буккальное, сублингвальное или чрескожное), вагинального или парентерального (включая подкожное, внутримышечное, внутривенное или внутрикожное) введения.
Настоящее изобретение дополнительно относится к способу получения соединений. Получение соединений согласно настоящему изобретению, представленных формулой (I), может проводиться следующими иллюстративными способами и вариантами осуществления, но указанные способы и варианты осуществления никоим образом не должны рассматриваться как ограничения объема настоящего изобретения. В качестве альтернативы, соединения согласно настоящему изобретению могут быть синтезированы методиками синтеза, известными специалистам в данной области техники, или всесторонне используя способы, известные из уровня техники, и способы, описанные в настоящем изобретении. Продукты, полученные на каждой реакционной стадии, выделяют методиками разделения, известными из уровня техники, включая без ограничения разделение путем экстрагирования, фильтрования, дистилляции, кристаллизации и хроматографии. Исходные вещества и химические реагенты, использованные для синтеза, могут быть традиционным способом получены, основываясь на литературе (могут быт быть найдены в SciFinder), или приобретены.
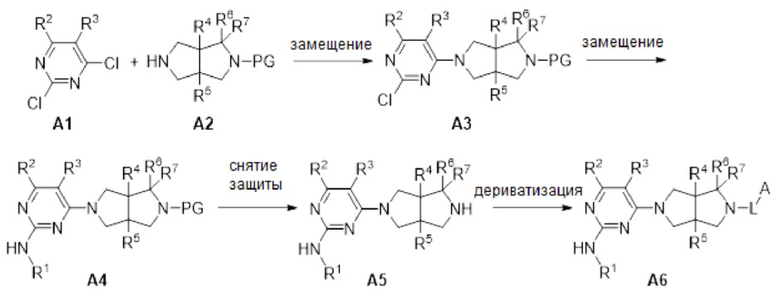
Гетероциклические соединения, представленные формулой (I) согласно настоящему изобретению, могут быть синтезированы в соответствии с путем, представленным ниже: 1) реакция замещения между исходными веществами А1 и А2, катализируемая органическим основанием, с получением промежуточного соединения A3; 2) катализируемая кислотой реакция замещения или сочетание по Бухвальду-Хартвигу соединения A3 с первичным амином (R1-NH2) с получением соединения А4; 3) снятие защиты с соединения А4 в кислоте (например, PG представляет собой Boc) или в условиях гидрирования (например, PG представляет собой Bn) с получением соединения А5; 4) дериватизация соединения А5 с получением целевых соединений, например, путем амидирования хлор ангидридом или ангидридом, амидного сочетания с кислотой, сульфонирования сульфонилхлоридом, формирования мочевины с амином, сочетания по Бухвальду-Хартвигу с (гетеро)арилгалогенидом, и т.д.
Примеры
Структуру соединений определяли методом ядерного магнитного резонанса (ЯМР) или масс-спектрометрии (MS). Для регистрации ЯМР использовали ЯМР-спектрометр Bruker ASCEND-400, растворитель для регистрации представлял собой дейтерированный диметилсульфоксид (DMSO-d6), дейтерированный хлороформ (CDCl3) или дейтерированный метанол (CD3OD), внутренний стандарт представлял собой тетраметилсилан (TMS), и химический сдвиг приводили в миллионных долях (м. д.).
Для регистрации MS использовали масс-спектрометр Agilent SQD (ESI) (Agilent 6120).
Для регистрации HPLC использовали жидкостной хроматограф высокого давления Agilent 1260 DAD (колонка: Poroshell 120 ЕС-С18, 50×3,0 мм, 2,7 мкм) или жидкостной хроматограф высокого давления Waters Arc (колонка: Sunfire С18, 150×4,6 мм, 5 мкм).
Для тонкослойной хроматографии (TLC) использовали пластины с силикагелем GF254 производства Qingdao Haiyang Chemical Co., Ltd. С толщиной 0,15-0,2 мм, и для разделения/очистки продуктов методом тонкослойной хроматографии использовали пластины с силикагелем толщиной 0,4-0,5 мм.
Для колоночной хроматографии как правило использовали силикагеле 200-300 меш производства Qingdao Haiyang Chemical Co., Ltd.
Известные исходные вещества для настоящего изобретения синтезировали в соответствии со способами, известными из уровня техники, или приобретали у ABCR GmbH&Co. KG, Acros Organics, Aldrich Chemical Company, Accela ChemBio Inc., Beijing Ouhe Technology Co., Ltd., и т.д.
Если в вариантах осуществления не указано иное, то взаимодействия проводили в атмосфере аргона или азота с использованием баллона объемом около 1 л.
Гидрирование проводили в атмосфере водорода с использованием баллона объемом около 1 л, который был подсоединен к реакционному сосуду после трехкратного вакуумирования и заполнения водородом в повторяющемся режиме.
Для проведения реакции с обработкой микроволнами использовали микроволновой реактор СЕМ Discover-SP.
Если в вариантах осуществления не указано иное, то взаимодействия проводили при комнатной температуре.
Реакцию отслеживали с использованием Agilent LCMS (1260/6120) или методом тонкослойной хроматографии. Системы элюирующих растворителей для колоночной хроматографии и TLC включали в себя а) дихлорметан/метанол, b) петролейный эфир/этилацетат, или другие указанные системы. Соотношение растворителей корректировали в соответствии с полярностью соединения и при необходимости дополнительно корректировали путем добавления небольшого количества TEA и кислого или щелочного реагента. Очистку соединения проводили в порядке альтернативы с использованием MS-управляемой автоматизированной препаративной системы Waters (сокращенно преп-HPLC) с MS-детектором (SQD2), проводя элюирование со скоростью потока 20 мл/мин в соответствующем градиенте ацетонитрил/вода (содержащем 0,1% TFA или муравьиной кислоты) или ацетонитрил/вода (содержащем 0,05% аммиака) (XBridge-С18, 19×150 мм, 5 мкм). После очистки методом преп-HPLC, некоторые примеры получали в виде гидрохлоридов путем добавления 1н HCl к собранным фракциям с последующей сушкой в условиях пониженного давления.
Пример 1. Циклопропил((3aR,6aS)-3а,6а-диметил-5-(2-((1-метил-1H-пиразол-4-ил)амино)пиримидин-4-ил)гексагидропирроло[3,4-с]пиррол-2(1H)-ил)метанон
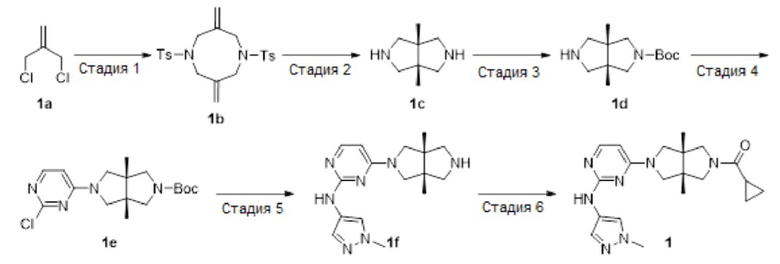
Стадия 1. 3,7-Диметилен-1,5-дитозил-1,5-диазокан (1b)
К смеси 4-метилбензолсульфонамида (17,12 г, 100 ммоль) и безводного карбоната калия (27,6 г, 200 ммоль) в MeCN (200 мл) в течение 10 мин медленно добавляли раствор 3-хлор-2-хлорметил-1-пропилена 1а (12,5 г, 100 ммоль) в MeCN (20 мл). Затем, смесь нагревали до температуры возгонки и перемешивали в течение 18 ч. После охлаждения до комнатной температуры, к смеси добавляли воду (250 мл), и перемешивали в течение 30 мин. Полученную смесь фильтровали, осадок собирали и очищали методом колоночной хроматографии на силикагеле (петролейный эфир/дихлорметан = 100/0 - 0/100) с получением указанного в заголовке соединения 1b (10,8 г, 48%).
MS m/z (ESI): 447 [M+1]
1H-ЯМР (400 МГц, CDCl3) δ 7,70 - 7,64 (м, 4Н), 7,31 (д, J=8,0 Гц, 4Н), 5,19 (с, 4Н), 3,82 (с, 8H), 2,43 (с, 6Н).
Стадия 2. (3ar,6ar)-3а,6а-диметилоктагидропирроло[3,4-с]пиррол (1с)
К раствору соединения 1b (10,8 г, 24,2 ммоль) в THF (400 мл) при 0°С порциями добавляли LAH (9,2 г, 242 ммоль). После перемешивания в течение 4 суток, смесь охлаждали до 0°С, и по каплям добавляли к ней 20% водный раствор хлорида натрия (18 мл). Полученную смесь постепенно нагревали до комнатной температуры и перемешивали в течение 1 ч. Смесь фильтровали через слой Celite, и промывали осадок на фильтре THF (3×200 мл). Объединенный фильтрат концентрировали досуха в условиях вакуума с получением указанного в заголовке соединения 1с (2,25 г, 66%). Неочищенный продукт использовали непосредственно на следующей стадии без дополнительной очистки.
MS m/z (ESI): 141 [M+1]
1H-ЯМР (400 МГц, CDCl3) δ 2,90 (д, J=11,0 Гц, 4Н), 2,69 (д, J=11,0 Гц, 4Н), 2,39 (ушир. с, 2Н), 0,98 (с, 6Н).
Стадия 3. трет-Бутил-(3aR,6aS)-3а,6а-диметилгексагидропирроло[3,4-с]пиррол-2(1Н)-карбоксилат (1d)
К раствору соединения 1с (2,25 г, 16 ммоль) в CH2Cl2 (100 мл) при 0°С добавляли TEA (4,8 г, 48 ммоль) и раствор ди-трет-бутилдикарбоната (3,5 г, 16 ммоль) в CH2Cl2 (100 мл). Смесь постепенно нагревали до комнатной температуры и перемешивали в течение 1 ч. Реакционную смесь гасили добавлением воды (100 мл) и экстрагировали CH2Cl2 (2×100 мл). Объединенную органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали досуха. Остаток очищали методом колоночной хроматографии на силикагеле (дихлорметан/метанол = 100/1 - 10/1) с получением указанного в заголовке соединения 1d (1,37 г, 36%).
MS m/z (ESI): 241 [M+l]
1H-ЯМР (400 МГц, CDCl3) δ 3,47 (с, 2Н), 3,20 (д, J=10,2 Гц, 2Н), 3,09 (д, J=11,4 Гц, 2Н), 2,90 (д, J=11,4 Гц, 2Н), 1,45 (с, 9Н), 1,06 (с, 6Н).
Стадия 4. трет-Бутил-(3aR,6aS)-5-(2-хлорпиримидин-4-ил)-3а,6а-диметилгексагидропирроло[3,4-с]пиррол-2(1Н)-карбоксилат (1е)
К раствору соединения 1d (1,37 г, 5,7 ммоль) в MeCN (50 мл) добавляли 2,4-дихлорпиримидин (850 мг, 5,7 ммоль) и DIEA (2,21 г, 17,1 ммоль). Смесь нагревали до температуры возгонки и перемешивали в течение 16 ч. После охлаждения до комнатной температуры, смесь концентрировали досуха в условиях вакуума, и разбавляли остаток водой (50 мл). Затем, смесь экстрагировали этилацетатом (3×100 мл). Объединенную органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали досуха. Остаток очищали методом колоночной хроматографии на силикагеле (петролейный эфир/этилацетат = 5/1 - 1/1) с получением указанного в заголовке соединения 1е (1,47 г, 73%).
MS m/z (ESI): 353 [M+1]
1H-ЯМР (400 МГц, CDCl3) δ 8,03 (д, J=6,0 Гц, 1H), 6,17 (д, J=5,8 Гц, 1H), 3,77 (д, J=12,3 Гц, 1H), 3,57 (т, J=10,9 Гц, 1H), 3,53 - 3,39 (м, 3Н), 3,35 (д, J=11,4 Гц, 1H), 3,32 -3,24 (м, 2Н), 1,45 (с, 9Н), 1,15 (д, J=6,5 Гц, 6Н).
Стадия 5. 4-((3aR,6aS)-3а,6а-диметилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)-N-(1-метил-1Н-пиразол-4-ил)пиримидин-2-амина гидрохлорид (1f)
В реакционный сосуд для микроволновой обработки емкостью 30 мл добавляли соединение 1е (353 мг, 1 ммоль), 1-метил-1Н-пиразол-4-амин (102 мг, 1,05 ммоль), pTsOH (4 мг, 0,02 ммоль) и изопропанол (25 мл). Сосуд герметизировали и нагревали в микроволновом реакторе до 100°С в течение 1 ч. После охлаждения до комнатной температуры, к смеси добавляли раствор HCl (33% в этаноле, 2 мл), а затем нагревали в микроволновом реакторе при 80°С в течение 30 мин. Синтез повторяли еще для трех партий, следуя той же методике, что и описанная выше. Четыре партии объединяли и фильтровали. Осадок на фильтре собирали с получением указанного в заголовке соединения 1f (1 г, 54%) в виде гидрохлорида.
MS m/z (ESI): 314 [M+1]
Стадия 6. Циклопропил((3aR,6aS)-3а,6а-диметил-5-(2-((1-метил-1H-пиразол-4-ил)амино)пиримидин-4-ил)гексагидропирроло[3,4-с]пиррол-2(1H)-ил)метанон (1)
К раствору соединения 1f (46 мг, 0,1 ммоль) и TEA (50 мг, 0,5 ммоль) в CH2Cl2 (5 мл) при 0°С добавляли циклопропанкарбонилхлорид (11 мг, 0,1 ммоль). Смесь перемешивали при 0°С в течение 20 мин, затем гасили добавлением воды (20 мл) и экстрагировали CH2Cl2 (2×20 мл). Объединенную органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали досуха в условиях вакуума. Остаток очищали методом преп-HPLC с получением указанного в заголовке соединения 1 (23 мг, твердое вещество, 61%).
MS m/z (ESI): 382 [M+1]
1H-ЯМР (400 МГц, CD3OD) δ 7,85 (д, J=6,3 Гц, 2Н), 7,60 (с, 1Н), 5,92 (д, J=6,0 Гц, 1Н), 3,90 (д, J=10,0 Гц, 2Н), 3,89 (с, 3Н), 3,76 (д, J=10,8 Гц, 2Н), 3,61 (д, J=12,4 Гц, 2Н), 3,48 (д, 1=12,3 Гц, 2Н), 1,84 - 1,78 (м, 1Н), 1,26 (с, 3Н), 1,23 (с, 3Н), 0,96 - 0,82 (м, 4Н).
Пример 2. 4-((4-((3aR,6aS)-5-(циклопропанкарбонил)-3а,6а-диметилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)пиримидин-2-ил)амино)бензойная кислота
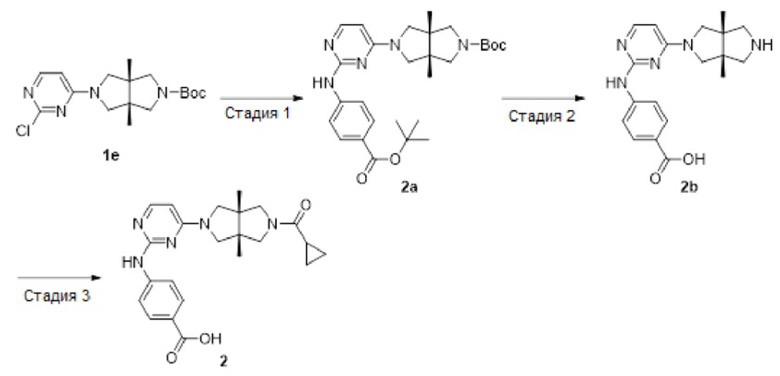
Стадия 1. трет-Бутил-(3aR,6aS)-5-(2-((4-(трет-бутоксикарбонил)фенил)амино)пиримидин-4-ил)-3а,6а-диметилгексагидропирроло[3,4-с]пиррол-2(1Н)-карбоксилат (2а)
В реакционный сосуд для микроволновой обработки емкостью 10 мл добавляли соединение 1е (100 мг, 0,283 ммоль), трет-бутил-4-аминобензоат (55 мг, 0,283 ммоль), pTsOH (5 мг, 0,0283 ммоль) и изопропанол (4 мл). Сосуд перемешивали в микроволновом реакторе при 100°С в течение 1 ч. После охлаждения до комнатной температуры, смесь концентрировали досуха в условиях вакуума, и очищали остаток методом колоночной хроматографии на силикагеле (петролейный эфир/этилацетат = 2/1 - 1/2) с получением указанного в заголовке соединения 2а (102 мг, 71%).
MS m/z (ESI): 510 [М+1]
Стадия 2. 4-((4-((3aR,6aS)-3а,6а-диметилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)пиримидин-2-ил)амино)бензойная кислота (2b)
К раствору соединения 2а (30 мг, 0,06 ммоль) в этаноле (2 мл) добавляли раствор HCl (33% в этаноле, 2 мл). Смесь перемешивали при комнатной температуре в течение 2 ч, а затем концентрировали досуха в условиях вакуума с получением указанного в заголовке соединения 2b (36 мг). Неочищенный продукт использовали непосредственно на следующей стадии без дополнительной очистки.
MS m/z (ESI): 354 [M+1]
Стадия 3. 4-((4-((3aR,6aS)-5-(циклопропанкарбонил)-3а,6а-диметилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)пиримидин-2-ил)амино)бензойная кислота (2)
К раствору соединения 2b (36 мг, неочищенное) и TEA (30 мг, 0,3 ммоль) в CH2Cl2 (5 мл) добавляли циклопропанкарбонилхлорид (6 мг, 0,06 ммоль). Смесь перемешивали в течение 10 мин, а затем гасили добавлением воды (0,5 мл). Смесь концентрировали досуха, и растворяли остаток в THF (2 мл). К полученной смеси добавляли 20% водный раствор хлорида натрия (2 мл), и перемешивали при комнатной температуре в течение 2 ч. Смесь корректировали до рН=3 ~ 4 добавлением 1н HCl и экстрагировали этилацетатом (3×20 мл). Объединенную органическую фазу сушили над безводным сульфатом натрия, фильтровали, концентрировали досуха в условиях вакуума и очищали методом преп-HPLC с получением указанного в заголовке соединения 2 (20 мг, твердое вещество, 79%).
MS m/z (ESI): 422 [M+1]
1H-ЯМР (400 МГц, CD3OD) δ 7,96 (дд, J=10,3, 7,5 Гц, 3Н), 7,81 (д, J=8,7 Гц, 2Н), 6,07 (д, J=6,1 Гц, 1H), 3,92 (д, J=10,9 Гц, 2Н), 3,76 (д, J=10,8 Гц, 2Н), 3,62 (д, J=12,4 Гц, 2Н), 3,49 (д, J=12,4 Гц, 2Н), 1,84 - 1,77 (м, 1H), 1,27 (с, 3Н), 1,24 (с, 3Н), 0,95 - 0,84 (м, 4Н).
Пример 3. 4-((4-((3aR,6aS)-5-(циклопропанкарбонил)-3а,6а-диметилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)пиримидин-2-ил)амино)-N-этилбензамид
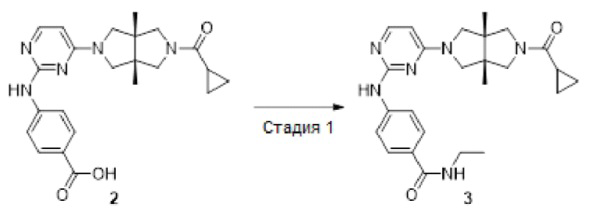
К раствору соединения 2 (56 мг, 0,133 ммоль) в CH2Cl2 (10 мл) добавляли DIEA (59 мг, 0,153 ммоль) и HATU (58 мг, 0,153 ммоль). Смесь перемешивали при комнатной температуре в течение 5 мин, а затем добавляли 3 капли водного раствора этиламина (65%-70 масс. %). Смесь перемешивали при комнатной температуре в течение 30 мин, концентрировали и очищали методом преп-HPLC с получением указанного в заголовке соединения 3 (3,3 мг, твердое вещество, 6%).
MS m/z (ESI): 449 [M+1]
1H-ЯМР (400 МГц, CD3OD) δ 7,94 (д, J=6,1 Гц, 1Н),7,84 - 7,77 (м, 4Н), 6,04 (д, J=6,1 Гц, 1Н), 3,90 (д, J=11,1 Гц,2Н),3,76 (д, J=10,9 Гц, 2Н), 3,62 (д, 1=12,4 Гц, 2Н), 3,48 (д, J=12,5 Гц, 2Н), 3,43 (кв, J=14,5, 7,3 Гц, 2Н), 1,85 - 1,77 (м, 1Н), 1,28 - 1,19 (м, 9Н), 0,95 - 0,83 (м, 4Н).
Пример 4. 1-((3aR,6aS)-3а,6а-диметил-5-(2-((1-метил-1H-пиразол-4-ил)амино)пиримидин-4-ил)гексагидропирроло[3,4-с]пиррол-2(1Н)-ил)-2,2,2-трифторэтан-1-она гидрохлорид
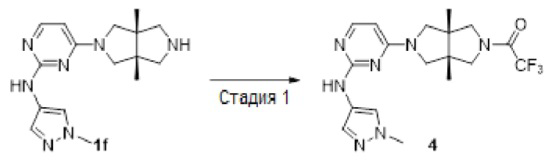
К смеси соединения 1f (69 мг, 0,15 ммоль) и DIEA (116 мг, 0,9 ммоль) в CH2Cl2 (2 мл) добавляли раствор трифторуксусного ангидрида (32 мг, 0,15 ммоль) в CH2Cl2 (2 мл). Смесь перемешивали в течение 20 мин, концентрировали и очищали методом преп-HPLC с получением указанного в заголовке соединения 4 (36,4 мг, твердое вещество, 54%) в виде гидрохлорида.
MS m/z (ESI): 410 [M+1]
1H-ЯМР (400 МГц, CD3OD) δ 7,89 (с, 1Н), 7,74 (с, 1H), 7,65 (с, 1Н), 6,31 (дд, J=7,4, 5,0 Гц, 1Н), 4,00 - 3,73 (м, 9Н), 3,70 - 3,61 (м, 2Н), 1,27 (с, 6Н).
Пример 6 получали в соответствии с методикой, описанной для Примера 4, за исключением того, что этилхлороформиат использовали вместо трифторуксусного ангидрида.

Пример 5. (2,2-Дифторциклопропил)((3aR,6aS)-3а,6а-диметил-5-(2-((1-метил-1Н-пиразол-4-ил)амино)пиримидин-4-ил)гексагидропирроло[3,4-с]пиррол-2(1Н)-ил)метанона гидрохлорид
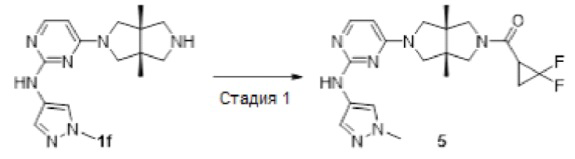
К смеси соединения 1f (69 мг, 0,15 ммоль) в CH2Cl2 (5 мл) добавляли DIEA (116 мг, 0,9 ммоль), HATU (57 мг, 0,15 ммоль) и 2,2-дифторциклопропан-1-карбоновую кислоту (19 мг, 0,15 ммоль). Смесь перемешивали при комнатной температуре в течение 20 мин, а затем концентрировали и очищали методом преп-HPLC с получением указанного в заголовке соединения 5 (32,8 мг, твердое вещество, 52%) в виде гидрохлорида.
MS m/z (ESI): 418 [M+1]
1H-ЯМР (400 МГц, CD3OD) δ 7,85 (т, J=4,0 Гц, 2Н), 7,59 (с, 1Н), 5,92 (д, J=6,1 Гц, 1Н), 3,90 (д, J=10,9 Гц, 4Н), 3,78 (кв, J=11,0 Гц, 2Н), 3,66 (д, J=11,9 Гц, 3Н), 3,52 (д, J=12,5 Гц, 2Н), 2,89 - 2,79 (м, 1Н), 2,09 - 2,01 (м, 1Н), 1,85 - 1,74 (м, 1H), 1,28 -1,19 (м, 6Н).
Пример 7. 3-((3aR,6aS)-3а,6а-диметил-5-(2-((1-метил-1H-пиразол-4-ил)амино)пиримидин-4-ил)гексагидропирроло[3,4-с]пиррол-2(1Н)-ил)пропаннитрил
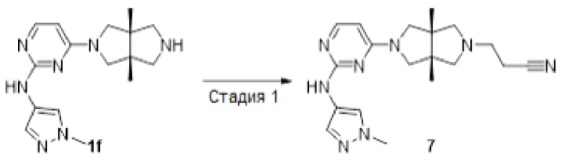
К смеси соединения 1f (69 мг, 0,15 ммоль) в MeCN (5 мл) добавляли 3-бромпропаннитрил (20 мг, 0,15 ммоль) и DIEA (116 мг, 0,9 ммоль). Смесь перемешивали в течение 20 ч, а затем нагревали до 90°С в течение 24 ч (методом LCMS обнаруживали лишь следовые количества образовавшегося целевого продукта). Реакционную смесь переносили в герметизированную пробирку емкостью 15 мл и нагревали до 120°С в течение 72 ч. После охлаждения до комнатной температуры, реакционную смесь концентрировали досуха в условиях вакуума и очищали методом преп-HPLC с получением указанного в заголовке соединения 7 (3,2 мг, твердое вещество, 4%).
MS m/z (ESI): 367 [M+1]
1H-ЯМР (400 МГц, CD3OD) δ 7,81 (д, J=5,6 Гц, 2Н),7,61 (с, 1Н), 5,92 (д, J=6,1 Гц, 1Н), 3,88 (с, 3Н), 3,77 (дд, J=19,3, 12,2 Гц, 2Н), 3,48 - 3,39 (м, 2Н), 2,97 (д, J=9,3 Гц, 2Н), 2,79 (т, J=6,8 Гц, 2Н), 2,61 (дд, J=12,8, 8,0 Гц, 4Н), 1,20 (с, 6Н).
Пример 8. (3aR,6aS)-N-(цианометил)-3а,6а-диметил-5-(2-((1-метил-1Н-пиразол-4-ил)амино)пиримидин-4-ил)гексагидропирроло[3,4-с]пиррол-2(1Н)-карбоксамид
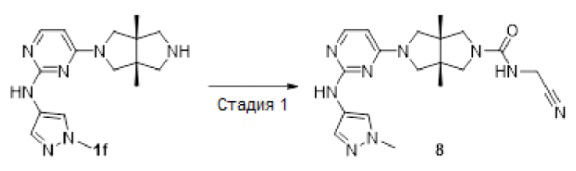
К раствору 2-аминоацетонитрила (14 мг, 0,24 ммоль) в DMF (3 мл) добавляли N,N'-карбонилдиимидазол (49 мг, 0,3 ммоль). Полученную смесь нагревали до 65°С и перемешивали в течение 2 ч, а затем добавляли соединение 1f (69 мг, 0,15 ммоль). Смесь перемешивали при 65°С дополнительно в течение 2 ч и концентрировали в условиях вакуума. Остаток диспергировали в насыщенном водном растворе гидрокарбоната натрия (20 мл) и экстрагировали этилацетатом (3×20 мл). Объединенную органическую фазу сушили над безводным сульфатом натрия, фильтровали, концентрировали и очищали методом преп-HPLC с получением указанного в заголовке соединения 8 (19,2 мг, твердое вещество, 32%).
MS m/z (ESI): 396 [M+1]
1H-ЯМР (400 МГц, CD3OD) δ 7,81 (д, J=8,0 Гц, 2Н), 7,57 (с, 1Н), 5,88 (д, J=6,0 Гц, 1Н), 4,09 (с, 2Н), 3,85 (с, 3Н), 3,76 (с, 1H), 3,59 (с, 1H), 3,51 (д, J=9,9 Гц, 3Н), 3,37 (д, J=10,3 Гц, 3Н), 1,19 (с, 6Н).
Пример 9. 4-((4-((3aR,6aS)-5-(2,2-дифторциклопропан-1-карбонил)гексагидропирроло[3,4-с]пиррол-2(1Н)-ил)-5-фторпиримидин-2-ил)амино)-N-этилбензамид

Стадия 1. N-этил-4-нитробензамид (9b)
К раствору соединения 4-нитробензойной кислоты (5 г, 30 ммоль) в CH2Cl2 (100 мл) по каплям добавляли оксалилхлорид (7,6 г, 60 ммоль), а затем DME (0,1 мл). Смесь перемешивали при в течение 30 мин, а затем концентрировали досуха в условиях вакуума. Остаток растворяли в THF (50 мл) и по каплям добавляли к водному раствору этиламина (60-70 масс. %, 10 мл). Смесь перемешивали при комнатной температуре в течение 30 мин, вливали в воду (200 мл) и экстрагировали этилацетатом (2×100 мл). Объединенную органическую фазу сушили над безводным сульфатом натрия, фильтровали, концентрировали и очищали методом колоночной хроматографии на силикагеле (петролейный эфир/этилацетат = 1/1 - 0/100) с получением указанного в заголовке соединения 9b (5,13 г, 88%).
MS m/z (ESI): 195 [M+1]
1H-ЯМР (400 МГц, CDCl3) δ 8,33 - 8,23 (м, 2Н), 7,98 -7,85 (м, 2Н), 6,16 (с, 1H), 3,54 (кв.д, J=7,3, 5,7 Гц, 2Н), 1,29 (т, J=7,3 Гц, 3Н).
Стадия 2. 4-амино-N-этилбензамид (9с)
К смеси воды (40 мл), уксусной кислоты (4 мл), хлорида аммония (14,13 г, 264,2 ммоль), этанола (100 мл) и соединения 9b (5,13 г, 26,42 ммоль) порциями добавляли цинковый порошок (8,64 г, 132 ммоль). Смесь перемешивали в течение 1 ч, и порциями снова добавляли цинковый порошок (8,64 г, 132 ммоль). Полученную смесь перемешивали при 60°С в течение 1 ч. После охлаждения до комнатной температуры, смесь фильтровали через слой Celite. Фильтрат концентрировали досуха в условиях вакуума, остаток диспергировали в насыщенном водном растворе гидрокарбоната натрия (50 мл) и экстрагировали этилацетатом (4×50 мл). Объединенную органическую фазу сушили над безводным сульфатом натрия, фильтровали, концентрировали и очищали методом колоночной хроматографии на силикагеле (петролейный эфир/этилацетат = 1/1 - 0/100) с получением указанного в заголовке соединения 9с (3,25 г, 75%).
MS m/z (ESI): 165 [M+1]
1H-ЯМР (400 МГц, DMSO-d6) δ 7,94 (т, J=5,2 Гц, 1Н), 7,55 (дд, J=9,0, 2,2 Гц, 2Н), 6,52 (дд, J=9,0, 2,2 Гц, 2Н), 5,54 (с, 2Н), 3,26 - 3,16 (м, 2Н), 1,08 (т, J=7,2 Гц, 3Н).
Стадия 3. трет-Бутил-(3aR,6aS)-5-(2-хлор-5-фторпиримидин-4-ил)гексагидропирроло[3,4-с]пиррол-2(1Н)-карбоксилат (9е)
К раствору соединения 9d (900 мг, 4,24 ммоль) в MeCN (50 мл) добавляли 2,4-дихлор-5-фторпиримидин (708 мг, 4,24 ммоль) и DIEA (1,64 г, 12,72 ммоль). Смесь перемешивали в течение 2 ч, а затем концентрировали досуха в условиях вакуума. Остаток очищали методом колоночной хроматографии на силикагеле (петролейный эфир/этилацетат = 1/1) с получением указанного в заголовке соединения 9е (1,13 г, 78%).
MS m/z (ESI): 343 [M+1]
Стадия 4. трет-Бутил-(3aR,6aS)-5-(2-((4-(этилкарбамоил)фенил)амино)-5-фторпиримидин-4-ил)гексагидропирроло[3,4-с]пиррол-2(1Н)-карбоксилат (9f)
В сосуд для микроволновой обработки емкостью 10 мл, содержащий соединение 9е (68 мг, 0,2 ммоль), 4-амино-N-этилбензамид (33 мг, 0,2 ммоль), KOtBu (48 мг, 0,5 ммоль), и 1,4-диоксан (3 мл) в атмосфере азота добавляли RuPhos-Pd-G2 (7 мг, 0,01 ммоль). Затем, сосуд нагревали в микроволновом реакторе при 120°С в течение 1 ч. После охлаждения до комнатной температуры, реакционную смесь вливали в воду (20 мл) и экстрагировали этилацетатом (3×20 мл). Объединенную органическую фазу сушили над безводным сульфатом натрия, фильтровали, концентрировали и очищали методом колоночной хроматографии на силикагеле (дихлорметан/метанол = 10/1 - 5/1) с получением указанного в заголовке продукта 9f (56 мг, 63%).
MS m/z (ESI): 471 [M+1]
Стадия 5. N-этил-4-((5-фтор-4-((3aR,6aS)-гексагидропирроло[3,4-с]пиррол-2(1Н)-ил)пиримидин-2-ил)амино)бензамида гидрохлорид (9g)
К раствору соединения 9f (56 мг, 0,112 ммоль) в этаноле (2 мл) добавляли раствор HCl (33% в этаноле, 2 мл). Смесь перемешивали в течение 14 ч, а затем концентрировали досуха в условиях вакуума с получением указанного в заголовке соединения 9g (57 мг). Неочищенный продукт использовали непосредственно на следующей стадии без дополнительной очистки.
MS m/z (ESI): 371 [M+1]
Стадия 6. 4-((4-((3aR,6aS)-5-(2,2-дифторциклопропан-1-кар6онил)гексагидропирроло[3,4-с]пиррол-2(1Н)-ил)-5-фторпиримидин-2-ил)амино)-N-этилбензамид (9)
К смеси соединения 9g (57 мг, 0,112 ммоль) в CH2Cl2 (5 мл) добавляли DIEA (91 мг, 0,696 ммоль), 2,2-дифторциклопропан-1-карбоновую кислоту (15 мг, 0,12 ммоль) и HATU (46 мг, 0,12 ммоль). Смесь перемешивали в течение 1 ч и концентрировали досуха в условиях вакуума. Остаток очищали методом преп-HPLC с получением указанного в заголовке соединения 9 (21 мг, твердое вещество, 40%).
MS m/z (ESI): 475 [M+1]
1H-ЯМР (400 МГц, CD3OD) δ 7,84 (д, J=5,8 Гц, 1H), 7,77 - 7,70 (м, 4Н), 4,09 - 3,90 (м, 3Н), 3,82 - 3,56 (м, 4Н), 3,52 - 3,43 (м, 1Н), 3,40 (кв, J=7,2 Гц, 2Н), 3,22 - 3,05 (м, 2Н), 2,88 -2,77 (м, 1H), 2,08 - 1,97 (м, 1Н), 1,83 - 1,70 (м, 1Н), 1,22 (т, J=7,2 Гц, 3Н).
Пример 10 получали в соответствии с методикой, описанной для Примера 9, за исключением того, что соединение 1d использовали вместо соединения 9d.
Пример 11. Циклопропил((3aR,6aS)-5-(5-фтор-2-((1-метил-1Н-пиразол-4-ил)амино)пиримидин-4-ил)-3а,6а-диметилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)метанон
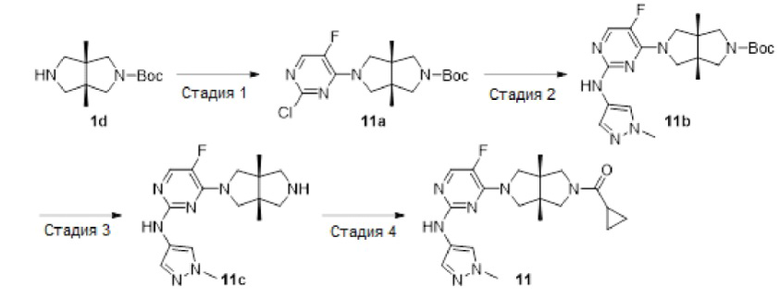
Стадия 1. трет-Бутил-(3aR,6aS)-5-(2-хлор-5-фторпиримидин-4-ил)-3а,6а-диметилгексагидропирроло[3,4-с]пиррол-2(1Н)-кар6оксилат (11а)
К раствору 2,4-дихлор-5-фторпиримидина (680 мг, 4 ммоль) и соединения 1d (1,96 г, неочищенный продукт, чистота около 50%) в MeCN (40 мл) добавляли TEA (1,55 г, 12 ммоль). Смесь нагревали до 90°С и перемешивали в течение 18 ч. После охлаждения до комнатной температуры, к смеси добавляли насыщенный водный раствор хлорида аммония (100 мл) и экстрагировали этилацетатом (3×50 мл). Объединенную органическую фазу промывали насыщенным солевым раствором (100 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали досуха в условиях вакуума. Остаток очищали методом колоночной хроматографии на силикагеле (петролейный эфир/этилацетат = 100/0 - 7/3) с получением указанного в заголовке соединения 11а (245 мг, 17%).
MS m/z (ESI): 371 [M+l]
1H-ЯМР (400 МГц, CDCl3) δ 7,91 (д, J=4,9 Гц, 1H), 3,83 (с, 2Н), 3,67 (с, 2Н), 3,46 (с, 2Н), 3,33 (с, 2Н), 1,46 (с, 9Н), 1,15 (с, 6Н).
Стадия 2. трет-Бутил-(3aR,6aS)-5-(5-фтор-2-((1-метил-1Н-пиразол-4-ил)амино)-пиримидин-4-ил)-3а,6а-диметилгексагидропирроло[3,4-с]пиррол-2(1Н)-карбоксилат (11b)
В сосуд для микроволновой обработки емкостью 30 мл добавляли соединение 11а (245 мг, 0,66 ммоль), 1-метил-1H-пиразол-4-амин (64 мг, 0,66 ммоль), NaOtBu (159 мг, 1,65 ммоль), 1,4-диоксан (8 мл) и RuPhos-Pd-G2 (24 мг, 0,033 ммоль), и нагревали реакционную смесь до 120°С в микроволновом реакторе в течение 1 ч в атмосфере азота. После охлаждения до комнатной температуры, реакционную смесь фильтровали через слой Celite. Фильтрат концентрировали досуха в условиях вакуума, и очищали остаток методом колоночной хроматографии на силикагеле (дихлорметан/этилацетат = 100/0 - 0/100) с получением указанного в заголовке соединения 11b (147 мг, 52%).
MS m/z (ESI): 432 [M+1]
Стадия 3. 4-((3aR,6aS)-3а,6а-диметилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)-5-фтор-N-(1-метил-1Н-пиразол-4-ил)пиримидин-2-амина гидрохлорид (11с)
К раствору соединения 11b (147 мг, 0,34 ммоль) в CH2Cl2 (10 мл) добавляли TFA (2 мл). Смесь перемешивали в течение 18 ч, а затем концентрировали досуха в условиях вакуума. Остаток растворяли в CH2Cl2 (10 мл), и добавляли к нему раствор HCl в этаноле (9 М, 1 мл). Полученную смесь концентрировали досуха с получением указанного в заголовке соединения 11с в виде гидрохлорида (160 мг, 98%).
MS m/z (ESI): 332 [M+1]
Стадия 4. Циклопропил((3aR,6aS)-5-(5-фтор-2-((1-метил-1Н-пиразол-4-ил)амино)-пиримидин-4-ил)-3а,6а-диметилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)метанон (11)
К смеси соединения 11 с (160 мг, 0,34 ммоль) и TEA (243 мг, 2,4 моль) в CH2Cl2 (10 мл) при 0°С добавляли циклопропанкарбонилхлорид (42 мг, 0,4 ммоль). Смесь перемешивали при 0°С в течение 30 мин, разбавляли CH2Cl2 (20 мл) и промывали водой (20 мл). Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали досуха. Остаток очищали методом преп-HPLC с получением указанного в заголовке соединения 11 (12,5 мг, твердое вещество, 9%).
MS m/z (ESI): 400 [M+1]
1H-ЯМР (400 МГц, CDCl3) δ 7,76 (д, J=5,7 Гц, 1H), 7,61 (с, 1H), 7,53 (с, 1H), 6,80 (с, 1Н), 3,87 (с, 3Н), 3,81 (дд, J=20,0, 10,7 Гц, 3Н), 3,64 (т, J=11,8 Гц, 4Н), 3,48 (д, J=12,4 Гц, 1Н), 1,60 - 1,52 (м, 1Н), 1,20 (с, 3Н), 1,16 (с, 3Н), 1,05 - 0,96 (м, 2Н), 0,83 - 0,73 (м, 2Н).
Пример 13 синтезировали в соответствии с методикой, описанной для Примера 11, за исключением того, что на стадии 3 2-(4-амино-1Н-пиразол-1-ил)этан-1-ол использовали вместо 1-метил-1H-пиразол-4-амина.
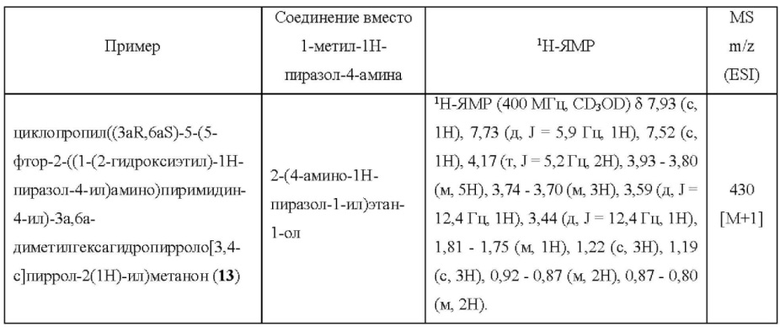
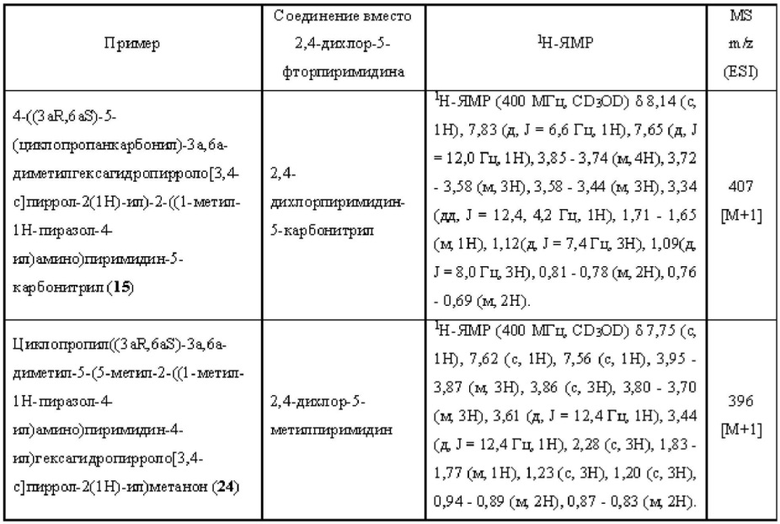
Пример 15 и Пример 24 синтезировали в соответствии с методикой, описанной для Примера 11, за исключением того, что на стадии 1 различные соединения использовали вместо 2,4-дихлор-5-фторпиримидина.
Пример 12. ((3aR,6aS)-5-(5-хлор-2-((1-метил-1H-пиразол-4-ил)амино)пиримидин-4-ил)-3а,6а-диметилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)(циклопропил)метанон (12)
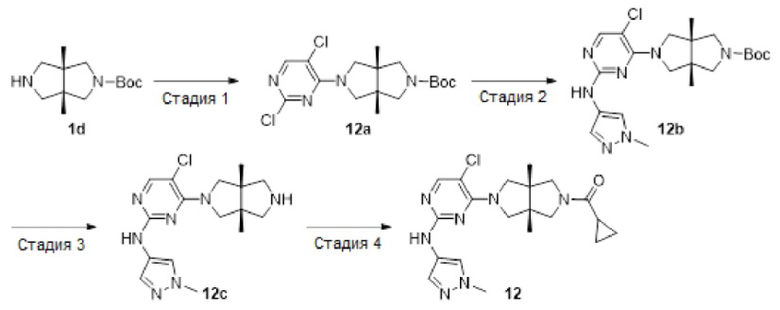
Стадия 1. трет-Бутил-(3aR,6aS)-5-(2,5-диклорпиримидин-4-ил)-3а,6а-диметилгексагидропирроло[3,4-с]пиррол-2(1H)-карбоксилат (12а)
К раствору 2,4,5-трихлорпиримидина (183 мг, 1 ммоль) и соединения 1d (770 мг, неочищенный продукт, около 1 ммоль) в MeCN (40 мл) добавляли DIEA (388 мг, 3 ммоль). Смесь нагревали до 90°С и перемешивали в течение 24 ч. После охлаждения до комнатной температуры, к реакционной смеси добавляли насыщенный водный раствор хлорида аммония (50 мл), и экстрагировали этилацетатом (3×20 мл). Объединенную органическую фазу промывали насыщенным солевым раствором (50 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали досуха в условиях вакуума. Остаток очищали методом колоночной хроматографии на силикагеле (петролейный эфир/этилацетат = 100/0 - 7/3) с получением указанного в заголовке соединения 12а (90 мг.23%).
MS m/z (ESI): 387 [M+1]
Стадия 2. трет-Бутил-(3aR,6aS)-5-(5-хлор-2-((1-метил-1Н-пиразол-4-ил)амино)-пиримидин-4-ил)-3а,6а-диметилгексагидропирроло[3,4-с]пиррол-2(1Н)-карбоксилат (12b)
В сосуд для микроволновой обработки емкостью 30 мл добавляли соединение 12а (90 мг, 0,23 ммоль), 1-метил-1H-пиразол-4-амин (23 мг, 0,23 ммоль), 1,4-диоксан (5 мл), NaOtBu (49 мг, 0,506 ммоль) и RuPhos-Pd-G2 (9 мг, 0,0115 ммоль). Реакционную смесь нагревали до 110°С в микроволновом реакторе в течение 1 ч в атмосфере азота. После охлаждения до комнатной температуры, смесь фильтровали через слой Celite. Фильтрат концентрировали досуха в условиях вакуума, и очищали остаток методом колоночной хроматографии на силикагеле (дихлорметан/этилацетат = 100/0 - 0/100) с получением указанного в заголовке соединения 12b (61 мг, 59%).
MS m/z (ESI): 448 [M+1]
Стадия 3. 5-Хлор-4-((3aR,6aS)-3а,6а-диметилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)-N-(1-метил-1Н-пиразол-4-ил)пиримидин-2-амина гидрохлорид (12с)
К раствору соединения 12b (61 мг, 0,136 ммоль) в CH2Cl2 (5 мл) добавляли TFA (2 мл). Смесь перемешивали в течение 20 ч, а затем концентрировали досуха в условиях вакуума. Остаток растворяли в смеси метанола (2 мл) и воды (10 мл), и добавляли раствор HCl (9 М в этаноле, 0,2 мл). Смесь снова концентрировали досуха с получением указанного в заголовке соединения 12с (67 мг, 100%) в виде гидрохлорида.
MS m/z (ESI): 348 [M+1]
Стадия 4. ((3aR,6aS)-5-(5-хлор-2-((1-метил-1Н-пиразол-4-ил)амино)пиримидин-4-ил)-3а,6а-диметилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)(циклопропил)метанон (12)
К смеси соединения 12с (67 мг, 0,136 ммоль) и TEA (55 мг, 0,544 моль) в CH2Cl2 (20 мл) при 0°С добавляли циклопропанкарбонилхлорид (14 мг, 0,136 ммоль). Смесь перемешивали при 0°С в течение 30 мин, а затем промывали насыщенным раствором хлорида аммония (20 мл). Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали досуха в условиях вакуума. Остаток очищали методом преп-HPLC с получением указанного в заголовке соединения 12 (1,9 мг, твердое вещество, 3%).
MS m/z (ESI): 416 [M+1]
1H-ЯМР (400 МГц, CD3OD) δ 7,83 (с, 1H), 7,75 (с, 1H), 7,53 (с, 1H), 3,97 (дд, J=15,1, 11,8 Гц, 2Н), 3,90 - 3,78 (м, 6Н), 3,71 (д, J=10,8 Гц, 1H), 3,59 (д, J=12,4 Гц, 1H), 3,42 (д, J=12,4 Гц, 1H), 1,78 (дд, J=8,7, 3,9 Гц, 1Н), 1,21 (с, 3Н), 1,18 (с, 3Н), 0,92 - 0,87 (м, 2Н), 0,86 - 0,82 (м, 2Н).
Примеры 19, 34, 37, 41 и 96 синтезировали в соответствии с методикой, описанной для Примера 12, за исключением того, что на стадии 4 различные соединения использовали вместо циклопропанкарбонилхлорида.


Примеры 23, 30 и 32 синтезировали в соответствии с методикой, описанной для Примера 12, за исключением того, что на стадии 2 различные соединения использовали вместо 1-метил-1H-пиразол-4-амина.
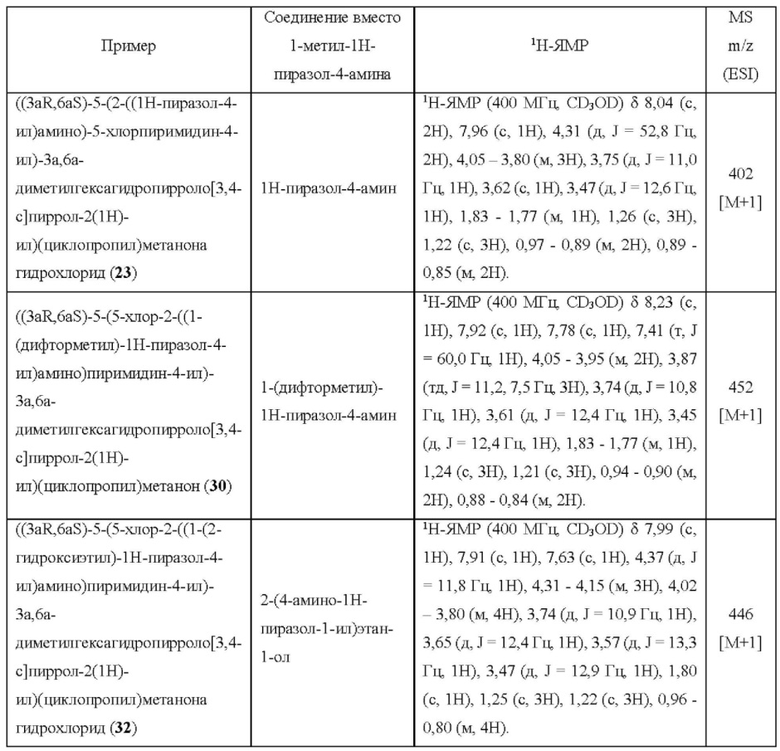
Пример 14. 4-((3aR,6aS)-5-(2,2-дифторэтил)-3а,6а-диметилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)-5-фтор-N-(1-метил-1Н-пиразол-4-ил)пиримидин-2-амин
К раствору соединения 11с (135 мг, 0,255 ммоль) в DMF (2 мл) добавляли DIEA (132 мг, 1,02 ммоль) и 1,1-дифтор-2-йодэтан (49 мг, 0,255 ммоль), и перемешивали смесь при 70°С в течение 24 ч. После охлаждения до комнатной температуры, смесь переносили в герметизированную пробирку емкостью 15 мл, а затем добавляли DIEA (132 мг, 1,02 ммоль) и 1,1-дифтор-2-йодэтан (98 мг, 0,51 ммоль). Смесь перемешивали в герметизированной пробирке при 80°С в течение 72 ч. После охлаждения до комнатной температуры, смесь очищали методом преп-HPLC с получением указанного в заголовке соединения 14 (16,9 мг, твердое вещество, 17%).
MS m/z (ESI): 396 [M+1]
1H-ЯМР (400 МГц, CD3OD) δ 7,77 (с, 1Н), 7,74 (д, J=6,2 Гц, 1H), 7,55 (с, 1Н), 5,89 (тт, J=56,1, 4,3 Гц, 1H), 3,96 (дд, J=11,7, 1,9 Гц, 2Н), 3,87 (с, 3Н), 3,58 (дд, J=11,6, 1,2 Гц, 2Н), 2,99 (д, J=9,5 Гц, 2Н), 2,89 (тд, J=15,3, 4,3 Гц, 2Н), 2,66 (д, J=9,5 Гц, 2Н), 1,17 (с, 6Н).
Пример 16. ((3aR,6aS)-5-(5-Хлор-2-((1-(1-метилпиперидин-4-ил)-1Н-пиразол-4-ил)амино)пиримидин-4-ил)-3а,6а-диметилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)(циклопропил)метанон
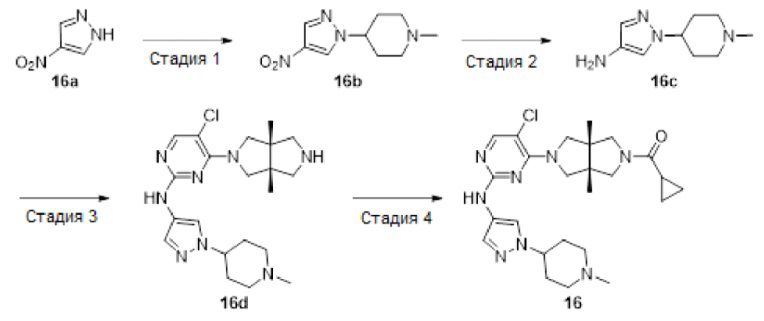
Стадия 1. 1-Метил-4-(4-нитро-1H-пиразол-1-ил)пиперидин (16b)
К смеси 4-нитро-1Н-пиразола 16а (1,13 г, 10 ммоль), 1-метилпиперидин-4-ола (1,15 г, 10 ммоль) и трифенилфосфина (3,15 г, 12 ммоль) в THF (25 мл) при 0°С в атмосфере азота по каплям добавляли диизопропилазодикарбоксилат (2,63 г, 13 ммоль). После перемешивания в течение 4 ч, смесь разбавляли водой (100 мл) и корректировали до рН=1 добавлением 6н HCl. Полученную смесь промывали этилацетатом (2×50 мл). К водному слою добавляли твердый LiOH для корректировки до рН=10, а затем экстрагировали этилацетатом (3×50 мл). Объединенную органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали досуха в условиях вакуума. Остаток очищали методом колоночной хроматографии на силикагеле (дихлорметан/метанол = 100/0 - 94/6) с получением указанного в заголовке соединения 16b (800 мг, 38%).
MS m/z (ESI): 211 [М+1]
1H-ЯМР (400 МГц, CDCl3) δ 8,17 (с, 1H), 8,08 (с, 1Н), 4,19 - 4,11 (м, 1Н), 3,04 - 2,96 (м, 2Н), 2,35 (с, 3Н), 2,24 - 2,12 (м, 4Н), 2,10 - 2,00 (м, 2Н).
Стадия 2. 1-(1-Метилпиперидин-4-ил)-1Н-пиразол-4-амин (16с)
К раствору соединения 16b (800 мг, 3,8 ммоль) в МеОН (10 мл) добавляли 10% Pd/C (400 мг). Смесь перемешивали в атмосфере водорода в течение 2 ч, а затем фильтровали. Фильтрат концентрировали досуха в условиях вакуума с получением указанного в заголовке соединения 16с (688 мг, 100%). Неочищенный продукт использовали непосредственно на следующей стадии без дополнительной очистки.
MS m/z (ESI): 181 [М+1]
Стадия 3. 5-Хлор-4-((3aR,6aS)-3а,6а-диметилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)-N-(1-(1-метилпиперидин-4-ил)-1Н-пиразол-4-ил)пиримидин-2-амин (16d)
В сосуд для микроволновой обработки емкостью 10 мл добавляли соединение 12а (39 мг, 0,1 ммоль), соединение 16с (18 мг, 0,1 ммоль), pTsOH (38 мг, 0,2 ммоль) и изопропанол (2 мл). Смесь перемешивали в микроволновом реакторе при 100°С в течение 1 ч. После охлаждения до комнатной температуры, смесь концентрировали досуха в условиях вакуума с получением указанного в заголовке соединения 16d (60 мг). Неочищенный продукт использовали непосредственно на следующей стадии без дополнительной очистки.
MS m/z (ESI): 431 [М+1]
Стадия 4. ((3aR,6aS)-5-(5-хлор-2-((1-(1-метилпиперидин-4-ил)-1Н-пиразол-4-ил)амино)пиримидин-4-ил)-3а,6а-диметилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)(циклопропил)метанон (16)
К раствору соединения 16d (60 мг) и TEA (51 мг, 0,5 ммоль) в CH2Cl2 (5 мл) добавляли раствор циклопропанкарбонилхлорид (11 мг, 0,1 ммоль) в CH2Cl2 (1 мл). Смесь перемешивали в течение 30 мин, а затем концентрировали досуха в условиях вакуума. Остаток очищали методом преп-HPLC с получением указанного в заголовке соединения 16 (22,3 мг, твердое вещество, 45% после двух стадий).
MS m/z (ESI): 499 [M+1]
1H-ЯМР (400 МГц, CD3OD) δ 7,92 (с, 1Н),7,85 (с, 1Н),7,56 (с, 1H), 4,19-4,11 (м, 1H), 4,19-4,11 (дд, J=18,7, 11,6 Гц, 2Н), 3,89 - 3,83 (м, 3Н), 3,73 (д, J=10,8 Гц, 1Н), 3,62 (д, J=12,4 Гц, 1Н), 3,44 (д, J=12,4 Гц, 1Н), 3,03 (д, J=12,5 Гц, 2Н), 2,37 (с, 3Н), 2,29 (т, J=11,7 Гц, 2Н), 2,16 (д, J=11,6 Гц, 2Н), 2,07 (дд, J=16,7, 8,0 Гц, 2Н), 1,84 - 1,76 (м, 1Н), 1,24 (с, 3Н), 1,21 (с, 3Н), 0,94 - 0,89 (м, 2Н), 0,88 - 0,82 (м, 2Н).
Пример 17. ((3aR,6aS)-5-(5-Хлор-2-((1-((R)-2-гидроксипропил)-1Н-пиразол-4-ил)амино)пиримидин-4-ил)-3а,6а-диметилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)(циклопропил)метанон
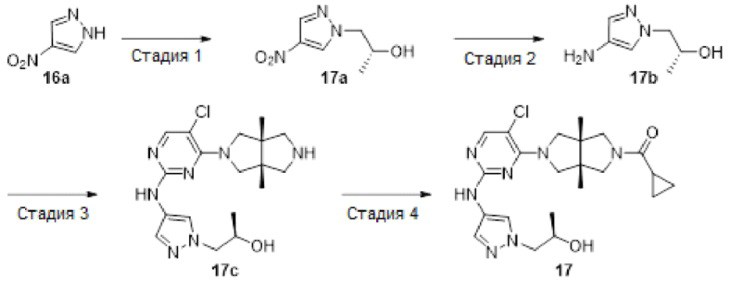
Стадия 1. (R)-1-(4-нитро-1Н-пиразол-1-ил)пропан-2-ол (17а)
К раствору 4-нитро-1Н-пиразола 16а (2,26 г, 20 ммоль) и (R)-2-метилоксирана (3,48 г, 60 ммоль) в DMF (15 мл) добавляли карбонат цезия (13 г, 40 ммоль). Смесь перемешивали в герметизированной пробирке при 100°С в течение 4 ч. После охлаждения до комнатной температуры, смесь вливали в воду (100 мл) и экстрагировали этилацетатом (3×100 мл). Объединенную органическую фазу промывали водой (3×100 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали досуха в условиях вакуума. Остаток очищали методом колоночной хроматографии на силикагеле (петролейный эфир/этилацетат = 100/0 - 7/3), а затем очищали методом преп-HPLC с получением указанного в заголовке соединения 17а (1,2 г, 35%).
MS m/z (ESI): 172 [M+1]
1H-ЯМР (400 МГц, DMSO-d6) δ 8,77 (с, 1Н),8,26(с, 1Н), 5,03 (д, J=4,8 Гц, 1Н),4,17 - 4,09 (м, 1H), 4,06 - 3,97 (м, 2Н), 1,08 (д, J=6,0 Гц, 3Н).
Стадия 2. (R)-1-(4-амино-1H-пиразол-1-ил)пропан-2-ол (17b)
К раствору соединения 17а (1,2 г, 7 ммоль) в MeOH (20 мл) добавляли 10% Pd/С (120 мг). Смесь перемешивали в атмосфере водорода в течение 16 ч, а затем фильтровали. Фильтрат концентрировали досуха, и очищали остаток методом колоночной хроматографии на силикагеле (дихлорметан/метанол = 100/0 - 19/1) с получением указанного в заголовке соединения 17b (848 мг, 86%).
MS m/z (ESI): 142 [M+1]
Стадия 3. (R)-1-(4-((5-хлор-4-((3aR,6aS)-3a,6a-диметилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)пиримидин-2-ил)амино)-1Н-пиразол-1-ил)пропан-2-ол (17с)
В сосуд для микроволновой обработки емкостью 10 мл добавляли соединение 12а (39 мг, 0,1 ммоль), соединение 17b (15 мг, 0,1 ммоль), pTsOH (38 мг, 0,2 ммоль) и изопропанол (2 мл). Сосуд перемешивали в микроволновом реакторе при 100°С в течение 1 ч. После охлаждения до комнатной температуры, смесь концентрировали досуха с получением указанного в заголовке соединения 17 с (60 мг). Неочищенный продукт использовали непосредственно на следующей стадии без дополнительной очистки.
MS m/z (ESI): 392 [M+1]
Стадия 4. ((3aR,6aS)-5-(5-хлор-2-((1-((R)-2-гидроксипропил)-1Н-пиразол-4-ил)амино)пиримидин-4-ил)-3а,6а-диметилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)(циклопропил)метанон (17)
К раствору соединения 17с (60 мг, неочищенное) и TEA (51 мг, 0,5 ммоль) в CH2Cl2 (5 мл) добавляли раствор циклопропанкарбонилхлорида (11 мг, 0,1 ммоль) в CH2Cl2 (1 мл). Смесь перемешивали в течение 30 мин, а затем концентрировали досуха в условиях вакуума. Остаток очищали методом преп-HPLC с получением указанного в заголовке соединения 17 (20,1 мг, твердое вещество, 44% после двух стадий).
MS m/z (ESI): 460 [M+1]
1H-ЯМР (400 МГц, CD3OD) δ 7,92 (с, 1H), 7,84 (с, 1H), 7,54 (с, 1H), 4,13 - 3,97 (м, 5Н), 3,91 - 3,83 (м, 3Н), 3,72 (д, J=10,8 Гц, 1H), 3,61 (дд, J=12,4, 3,7 Гц, 1H), 3,44 (дд, J=12,4, 1,7 Гц, 1H), 1,83 - 1,77 (м, 1H), 1,23 (с, 3Н), 1,20 (с, 3Н), 1,17 (д, J=6,1 Гц, 3Н), 0,94 - 0,89 (м, 2Н), 0,88 - 0,82 (м, 2Н).
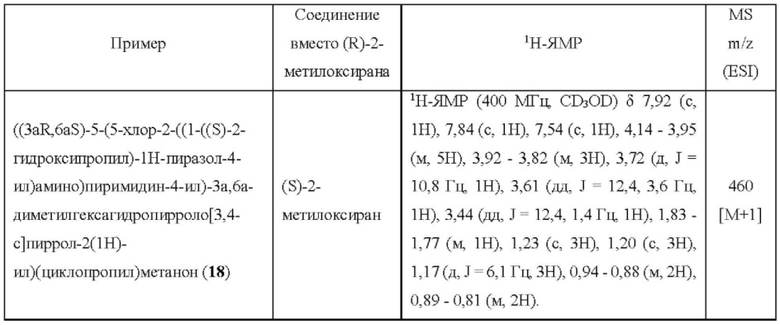
Пример 18 синтезировали в соответствии с методикой, описанной для Примера 12, за исключением того, что на стадии 1 (S)-2-метилоксиран использовали вместо (R)-2-метилоксирана.
Пример 20. 3-((3aR,6aS)-5-(5-Хлор-2-((1-метил-1Н-пиразол-4-ил)амино)пиримидин-4-ил)-3а,6а-диметилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)-3-оксопропаннитрил
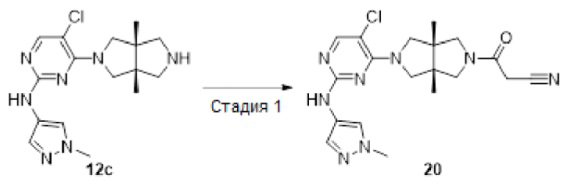
Смесь соединения 12с (35 мг, 0,1 ммоль), 2-цианоуксусной кислоты (9 мг, 0,1 ммоль), DIEA (39 мг, 0,3 ммоль) и HATU (46 мг, 0,12 ммоль) в CH2Cl2 (5 мл) перемешивали в течение 30 мин. Смесь гасили добавлением воды (20 мл) и экстрагировали CH2Cl2 (2×100 мл). Объединенную органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали досуха в условиях вакуума. Остаток очищали методом преп-HPLC с получениемуказанного взаголовке соединения 20 (10,5 мг, твердое вещество, 25%).
MS m/z (ESI): 415 [M+1]
1H-ЯМР (400 МГц, CD3OD) δ 7,84 (с, 1Н), 7,77 (с, 1H), 7,55 (с, 1H), 3,99 (т, J=10,8 Гц, 2Н), 3,86 (с, 3Н), 3,84 (д, J=11,8 Гц, 2Н), 3,66 (дд, J=18,8, 11,6 Гц, 2Н), 3,50 (дд, J=19,0, 11,6 Гц, 2Н), 1,21 (с, 3Н), 1,20 (с, 3Н).
Примеры 21, 22, 35, 36, 39, 42, 43, 44, 45, 46, 47, 48 и 52 синтезировали в соответствии с методикой, описанной для Примера 20, за исключением того, что на стадии 1 различные соединения использовали вместо 2-цианоуксусной кислоты.
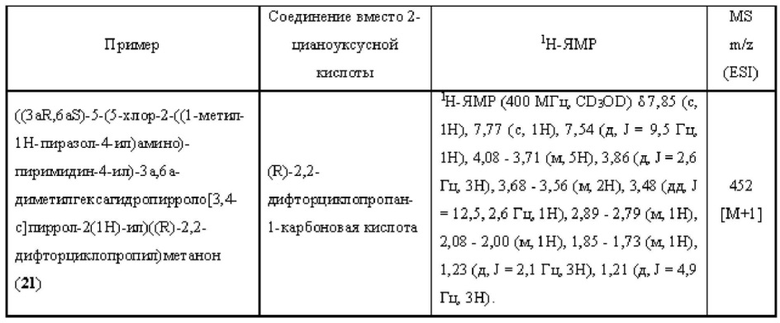

Пример 25. 5-Хлор-4-((3aR,6aS)-3а,6а-диметил-5-(пиримидин-4-ил)-гексагидропирроло[3,4-с]пиррол-2(1Н)-ил)-N-(1-метил-1Н-пиразол-4-ил) пиримидин-2-амин
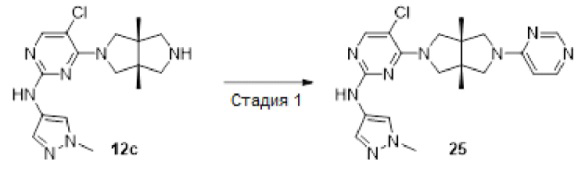
Смесь соединения 12с (100 мг, 0,26 ммоль), 4-хлорпиримидина гидрохлорида (59 мг, 0,39 ммоль) и DIEA (336 мг, 2,6 ммоль) в MeCN (4 мл) нагревали до 80°С и перемешивали в течение 8 ч. После охлаждения до комнатной температуры, смесь концентрировали досуха в условиях вакуума, и очищали остаток методом преп-HPLC с получением указанного в заголовке соединения 25 (65,1 мг, твердое вещество, 58%).
MS m/z (ESI): 426 [M+1]
1H-ЯМР (400 МГц, DMSO-d6) δ 9,06 (с, 1Н), 8,46 (с, 1H), 8,14 (д, J=6,1, 1Н), 7,89 (с, 1Н), 7,69 (с, 1H), 7,43 (с, 1Н), 6,46 (дд, J=6,1, 1,0, 1Н), 3,88 (с, 2Н), 3,76 (с, 3Н), 3,74 (с, 2Н), 3,67 (с, 2Н), 3,52 (с, 2Н), 1,15 (с, 6Н).
Пример 26. 3-((3aR,6aS)-5-(5-Хлор-2-((1-метил-1Н-пиразол-4-ил)амино)пиримидин-4-ил)гексагидропирроло[3,4-с]пиррол-2(1Н)-ил)-3-оксопропаннитрил
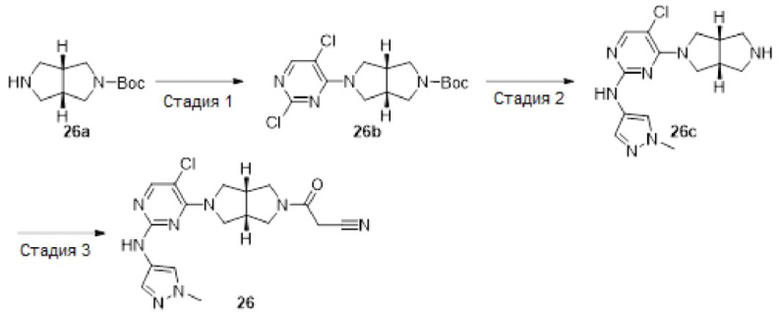
Стадия 1. трет-Бутил-(3aR,6aS)-5-(2,5-дихл op пиримидин-4-ил)гексагидропирроло[3,4-с]пиррол-2(1Н)-карбоксилат (26b)
К раствору 2,4,5-трихлорпиримидина (182 mg, 1 ммоль) в MeCN (10 мл) добавляли соединение 26а (212 мг, 1 ммоль) и карбонат калия (207 мг, 1,5 ммоль). Смесь перемешивали при 80°С в течение 3 ч, а затем концентрировали досуха в условиях вакуума. Остаток очищали методом колоночной хроматографии на силикагеле (петролейный эфир/этилацетат = 100/0 - 3/1) с получением указанного в заголовке соединения 26b (300 мг, 84%).
MS m/z (ESI): 359 [M+1]
Стадия 2. 5-Хлор-4-((3aR,6aS)-гексагидропирроло[3,4-с]пиррол-2(1H)-ил)-N-(1-метил-1H-пиразол-4-ил)пиримидин-2-амин(26с)
К смеси соединения 26b (300 мг, 0,84 ммоль) и 1-метил-1H-пиразол-4-амина гидрохлорида (81,3 мг, 0,84 ммоль) в изопропаноле (50 мл) добавляли pTsOH (318 мг, 1,68 ммоль). Смесь нагревали до 100°С в микроволновом реакторе в течение 2 ч. После охлаждения до комнатной температуры, осадок собирали путем фильтрования с получением указанного в заголовке соединения 26с (200 мг, 75%).
MS m/z (ESI): 320 [M+1]
Стадия 3. 3-((3aR,6aS)-5-(5-Хлор-2-((1-метил-1H-пиразол-4-ил)амино)пиримидин-4-ил)гексагидропирроло[3,4-с]пиррол-2(1Н)-ил)-3-оксопропаннитрил (26)
К смеси соединения 26 с (100 мг, 0,313 ммоль), 2-цианоуксусной кислоты (26,6 мг, 0,313 ммоль) и TEA (47,5 мг, 0,47 ммоль) в DMF (5 мл) добавляли HATU (178,6 мг, 0,47 ммоль). Смесь перемешивали в течение 1 ч, а затем концентрировали досуха в условиях вакуума. Остаток очищали методом преп-HPLC с получением указанного в заголовке соединения 26 (61,9 мг, твердое вещество, 50%).
MS m/z (ESI): 387 [M+1]
1H-ЯМР (400 МГц, CD3OD) δ 7,85 (с, 1H), 7,78 (с, 1Н), 7,54 (с, 1H), 4,19 - 4,11 (м, 2Н), 3,93 - 3,72 (м, 9Н), 3,52 - 3,45 (м, 2Н), 3,18 - 3,12 (м, 1Н), 3,09 - 3,03 (м, 1Н).
Пример 27 синтезировали в соответствии с методикой, описанной для Примера 26, за исключением того, что на стадии 3 (S)-2,2-дифторциклопропан-1-карбоновую кислоту использовали вместо 2-цианоуксусной кислоты.
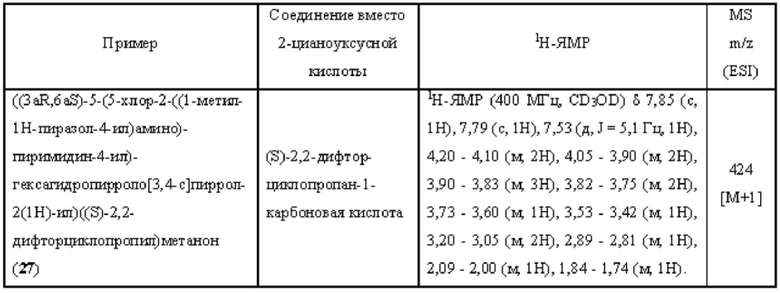
Пример 28. 3-((3aR,6aS)-5-(2-((1H-пиразол-4-ил)амино)-5-хлорпиримидин-4-ил)-3а,6а-диметилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)-3-оксопропаннитрил
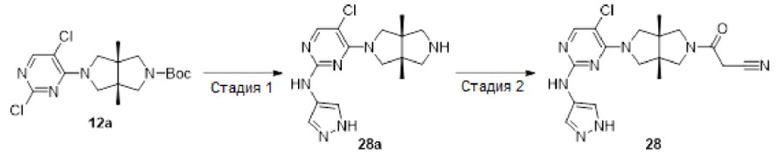
Стадия 1. 5-хлор-4-((3aR,6aS)-3а,6а-диметилгексагидропирроло[3,4-с]пиррол-2(1H)-ил)-N-(1H-пиразол-4-ил)пиримидин-2-амин (28а)
В сосуд для микроволновой обработки емкостью 30 мл добавляли соединение 12а (161 мг, 0,42 ммоль), Ш-пиразол-4-амин (35 мг, 0,42 ммоль), pTsOH (160 мг, 0,84 ммоль) и изопропанол (10 мл). Смесь нагревали до 100°С в микроволновом реакторе в течение 1 ч. Смесь охлаждали до комнатной температуры и концентрировали досуха в условиях вакуума с получением указанного в заголовке соединения 28а (260 мг). Неочищенный продукт использовали непосредственно на следующей стадии без очистки.
MS m/z (ESI): 334 [M+1]
Стадия 2. 3-((3aR,6aS)-5-(2-((1Н-Пиразол-4-ил)амино)-5-хлорпиримид ин-4-ил)-3а,6а-диметилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)-3-оксопропаннитрил (28)
К смеси соединения 28а (130 мг, неочищенное вещество, 0,21 ммоль), 2-цианоуксусной кислоты (18 мг, 0,21 ммоль) и DIEA (82 мг, 0,63 ммоль) в DMF (5 мл) добавляли HATU (80 мг, 0,21 ммоль). Смесь перемешивали в течение 1 ч, а затем очищали методом преп-HPLC с получением указанного в заголовке соединения 28 (7,6 мг, твердое вещество, 9%).
MS m/z (ESI): 401 [M+1]
1H-ЯМР (400 МГц, CD3OD) δ 7,85 (с, 2Н), 7,65 (с, 1Н), 4,02 (д, J=11,8 Гц, 1H), 3,98 (д, J=11,8 Гц, 1Н), 3,85 (дд, J=11,6, 2,8 Гц, 2Н), 3,66 (т, J=12,1 Гц, 2Н), 3,53 (д, J=10,7 Гц, 1Н), 3,47 (д, J=12,5 Гц, 1Н), 1,21 (с, 3Н), 1,20 (с, 3Н).
Пример 29 синтезировали в соответствии с методикой, описанной для Примера 28, за исключением того, что на стадии 2 (S)-2,2-дифторциклопропан-1-карбоновую кислоту использовали вместо 2-цианоуксусной кислоты.

Пример 31. 3-((3aR,6aS)-5-(5-Хлор-2-((1-(2-гидроксиэтил)-1Н-пиразол-4-ил)амино)пиримидин-4-ил)-3а,6а-диметилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)-3-оксопропаннитрил
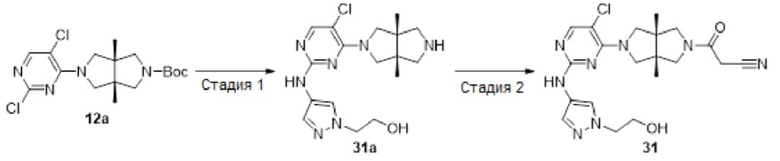
Стадия 1. 2-(4-((5-Хлор-4-((3aR,6aS)-3а,6а-диметилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)пиримидин-2-ил)амино)-1Н-пиразол-1-ил)этан-1-ол (31а)
В сосуд для микроволновой обработки емкостью 30 мл добавляли соединение 12а (330 мг, 0,852 ммоль), 2-(4-амино-1Н-пиразол-1-ил)этан-1-ол (108 мг, 0,852 ммоль), pTsOH (324 мг, 1,704 ммоль) и изопропанол (10 мл). Смесь нагревали до 100°С в микроволновом реакторе в течение 1 ч. После охлаждения до комнатной температуры, смесь концентрировали досуха в условиях вакуума, и очищали остаток методом преп-HPLC с получением указанного в заголовке соединения 31а (210 мг, 65%).
MS m/z (ESI): 378 [M+1]
Стадия 2. 3-((3aR,6aS)-5-(5-Хлор-2-((1-(2-гидроксиэтил)-1Н-пиразол-4-ил)амино)пиримидин-4-ил)-3а,6а-диметилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)-3-оксопропаннитрил (31)
К смеси соединения 31а (100 мг, 0,26 ммоль), 2-цианоуксусной кислоты (23 мг, 0,26 ммоль) и DIEA (101 мг, 0,87 ммоль) в CH2Cl2 (10 мл) добавляли HATU (101 мг, 0,26 ммоль). Смесь перемешивали в течение 1 ч, а затем концентрировали досуха в условиях вакуума. Остаток очищали методом преп-HPLC с получением указанного в заголовке соединения 31 (18,6 мг, твердое вещество, 16%).
MS m/z (ESI): 445 [M+1]
1H-ЯМР (400 МГц, CD3OD) δ 7,96 (с, 1Н),7,84 (с, 1H), 7,53 (с, 1Н),4,20 (т, J=5,2 Гц, 2Н), 4,02 (д, J=5,8 Гц, 1H), 3,99 (д, J=5,7 Гц, 1Н), 3,92 - 3,81 (м, 4Н), 3,68 (д, J=10,7 Гц, 1Н), 3,63 (д, J=12,5 Гц, 1H), 3,52 (д, J=10,7 Гц, 1Н), 3,48 (д, J=12,5 Гц, 1Н), 1,21 (с, 3Н), 1,20 (с, 3Н).
Пример 33. 2-(4-((5-Хлор-4-((3aR,6aS)-5-(циклопропанкарбонил)-3а,6а-диметилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)пиримидин-2-ил)амино)-1Н-пиразол-1-ил)ацетонитрил
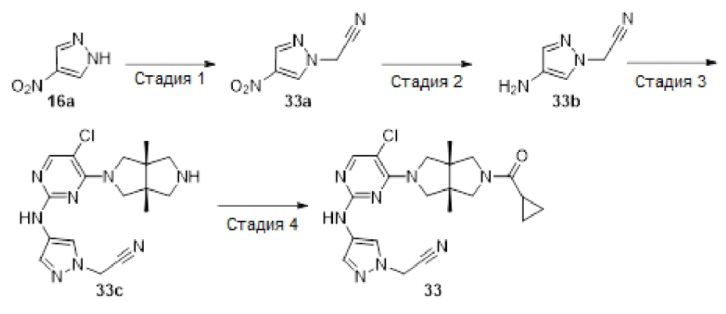
Стадия 1. 2-(4-Нитро-1Н-пиразол-1-ил)ацетонитрил (33а)
К раствору 4-нитро-1Н-пиразола 16а (1,13 г, 10 ммоль) в DMF (12 мл) добавляли карбонат цезия (9,75 г, 30 ммоль). После охлаждения до 0°С, к смеси по каплям добавляли 2-бромацетонитрил (2,4 г, 20 ммоль). Смесь перемешивали в течение 1 ч, разбавляли водой (150 мл) и экстрагировали этилацетатом (3×100 мл). Объединенную органическую фазу промывали водой (3×100 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали досуха в условиях вакуума. Остаток очищали методом колоночной хроматографии на силикагеле (петролейный эфир/этилацетат = 100/0 - 1/1) с получением указанного в заголовке соединения 33а (960 мг, 63%).
MS m/z (ESI): 153 [M+1]
1H-ЯМР (400 МГц, CDCl3) δ 8,36 (с, 1H), 8,16 (с, 1H), 5,15 (с, 2Н).
Стадия 2. 2-(4-Амино-1Н-пиразол-1-ил)ацетонитрил (33b)
Смесь соединения 33а (940 мг, 6,18 ммоль), хлорида аммония (6,6 г, 123,6 ммоль), этанола (40 мл) и воды (10 мл) нагревали до 60°С, а затем порциями добавляли цинковый порошок (4 г, 61,8 ммоль). Смесь перемешивали при 60°С в течение 10 мин, охлаждали до комнатной температуры и фильтровали. Фильтрат разбавляли водой (80 мл) и экстрагировали этилацетатом (3×100 мл). Объединенную органическую фазу промывали насыщенным солевым раствором (50 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали досуха в условиях вакуума, и очищали остаток методом колоночной хроматографии на силикагеле (дихлорметан/метанол = 100/0 - 19/1) с получением указанного в заголовке соединения 33b (130 мг, 17%).
MS m/z (ESI): 123 [M+1]
Стадия 3. 2-(4-((5-Хлор-4-((3aR,6aS)-3a,6a-диметилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)пиримидин-2-ил)амино)-1Н-пиразол-1-ил)ацетонитрил (33с)
В колбу для микроволновой обработки емкостью 10 мл добавляли соединение 12а (155 мг, 0,4 ммоль), соединение 33b (49 мг, 0,4 ммоль), pTsOH (16 мг, 0,08 ммоль) и изопропанол (4 мл). Смесь перемешивали в микроволновом реакторе при 100°С в течение 1 ч. После охлаждения до комнатной температуры, смесь концентрировали досуха в условиях вакуума с получением указанного в заголовке соединения 33с. Неочищенный продукт использовали непосредственно на следующей стадии без очистки.
MS m/z (ESI): 373 [M+1]
Стадия 4. 2-(4-((5-Хлор-4-((3aR,6aS)-5-(циклопропанкарбонил)-3a,6a-диметилгексагидропирроло[3,4-с]пиррол-2(1H)-ил)пиримидин-2-ил)амино)-1Н-пиразол-1-ил)ацетонитрил (33)
К смеси соединения 33 с (неочищенное вещество, 0,4 ммоль) в CH2Cl2 (20 мл) добавляли TEA (121 мг, 1,2 ммоль) и раствор циклопропанкарбонилхлорида (42 мг, 0,4 ммоль) в CH2Cl2 (1 мл). Смесь перемешивали в течение 1 ч, а затем концентрировали досуха в условиях вакуума. Остаток очищали методом преп-HPLC с получением указанного в заголовке соединения 2 33 (81 мг, твердое вещество, 46% после двух стадий).
MS m/z (ESI): 441 [M+1]
1H-ЯМР (400 МГц, CD3OD) δ 8,04 (с, 1H), 7,87 (с, 1Н), 7,63 (с, 1Н), 5,29 (с, 2Н), 4,06 - 3,96 (м, 2Н), 3,95 - 3,82 (м, 3Н), 3,72 (д, J=10,8 Гц, 1H), 3,62 (д, J=12,4 Гц, 1H), 3,45 (д, J=12,4 Гц, 1H), 1,83 - 1,77 (м, 1H), 1,24 (с, 3Н), 1,20 (с, 3Н), 0,94 - 0,88 (м, 2Н), 0,88 - 0,82 (м, 2Н).
Пример 38. Азетидин-3-ил((3aR,6aS)-5-(5-хлор-2-((1-метил-1Н-пиразол-4-ил)-амино)пиримидин-4-ил)-3а,6а-диметилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)метанон
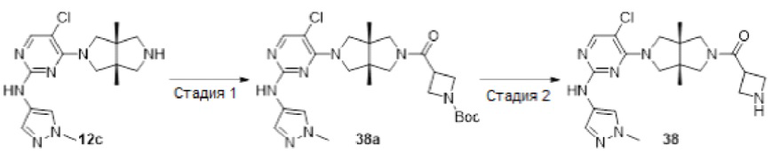
Стадия 1. трет-Бутил-3-((3aR,6aS)-5-(5-хлор-2-((1-метил-1Н-пиразол-4-ил)амино)-пиримидин-4-ил)-3а,6а-диметилоктагидропирроло[3,4-с]пиррол-2-кар6онил)азетидин-1-карбоксилат (38а)
К смеси соединения 12с (35 мг, 0,1 ммоль), 1-(трет-бутоксикарбонил)азетидин-3-карбоновой кислоты (21 мг, 0,1 ммоль) и DIEA (39 мг, 0,3 ммоль) в CH2Cl2 (5 мл) добавляли HATU (38 мг, 0,1 ммоль). После перемешивания при комнатной температуре в течение 30 мин, смесь разбавляли насыщенным раствором хлорида аммония (20 мл) и экстрагировали CH2Cl2 (3×20 мл). Объединенную органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали досуха в условиях вакуума с получением указанного в заголовке соединения 38а. Этот неочищенный продукт использовали непосредственно на следующей стадии без очистки.
MS m/z (ESI): 531 [M+1]
Стадия 2. Азетидин-3-ил((3aR,6aS)-5-(5-хлор-2-((1-метил-1H-пиразол-4-ил)амино)-пиримидин-4-ил)-3а,6а-диметилгексагидропирроло[3,4-с]пиррол-2(1H)-ил)метанон (38)
К раствору соединения 38а (неочищенное вещество, около 0,1 ммоль) в CH2Cl2 (2 мл) добавляли TEA (2 мл). Смесь перемешивали в течение 30 мин, а затем концентрировали досуха в условиях вакуума. Остаток очищали методом преп-HPLC с получением указанного в заголовке соединения 38 (19,9 мг, твердое вещество, 46% после двух стадий.
MS m/z (ESI): 431 [M+1]
1Н-ЯМР(400 МГц, CD3OD) δ 7,85 (с, 1H), 7,76 (с, 1Н),7,55 (с, 1Н), 4,05 - 3,93 (м, 4Н), 3,90 - 3,78 (м, 8Н), 3,63 (д, J=12,5 Гц, 1Н), 3,57 (д, J=10,8 Гц, 1Н), 3,46 (д, J=12,5 Гц, 1Н), 3,41 [д, J=10,8 Гц, 1Н), 1,19 (с, 6Н).
Пример 40. 5-Хлор-4-((3aR,6aS)-3а,6а-диметил-5-(2,2,2-трифторэтил)-гексагидропирроло[3,4-с]пиррол-2(1H)-ил)-N-(1-метил-1Н-пиразол-4-ил)пиримидин-2-амин
К смеси соединения 12 с (50 мг, 0,14 ммоль) в 1,4-диоксане (4 мл) добавляли DIEA (54 мг, 0,42 ммоль) и 2,2,2-трифторэтилтрифторметансульфонат (65 мг, 0,28 ммоль). Смесь перемешивали в герметизированной пробирке при 100°C в течение 2 ч. После охлаждения до комнатной температуры, смесь концентрировали досуха в условиях вакуума, и очищали остаток методом преп-HPLC с получением указанного в заголовке соединения 40 (7,3 мг, твердое вещество, 12%).
MS m/z (ESI): 430 [M+1]
1H-ЯМР (400 МГц, CD3OD) δ 7,84 (с, 1H), 7,77 (с, 1Н), 7,56 (с, 1Н), 4,06 (д, J=11,5 Гц, 2Н), 3,86 (с, 3Н), 3,69 (д, J=11,5 Гц, 2Н), 3,22 (кв, J=9,8 Гц, 2Н), 3,04 (д, J=9,3 Гц, 2Н), 2,79 (д, J=9,3 Гц, 2Н), 1,16 (с, 6Н).
Пример 49. 2-((3aR,6aS)-5-(5-Хлор-2-((1-метил-1Н-пиразол-4-ил)амино)пиримидин-4-ил)-3а,6а-диметилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)этан-1-ол
Смесь соединения 12с (35 мг, 0,1 ммоль), 2-бромэтанола (15 мг, 0,12 ммоль) и карбоната калия (42 мг, 0,3 ммоль) в MeCN (5 мл) нагревали до температуры возгонки в течение 20 ч при перемешивании. После охлаждения до комнатной температуры, смесь концентрировали досуха в условиях вакуума. Остаток растворяли в DMSO (5 мл), а затем добавляли 2-бромэтанол (0,2 мл) и карбонат цезия (98 мг, 0,3 ммоль). Смесь нагревали до 100°С в течение 2 ч. После охлаждения до комнатной температуры, смесь фильтровали, и очищали фильтрат методом преп-HPLC с получением указанного в заголовке соединения 49 (16,7 мг, твердое вещество, 43%).
MS m/z (ESI): 392 [M+1]
1H-ЯМР (400 МГц, CD3OD) δ 7,84 (с, 1H), 7,77 (с, 1Н), 7,56 (с, 1Н), 4,12 (д, J=11,5 Гц, 2Н), 3,87 (с, 3Н), 3,65 (дд, J=8,7, 5,1 Гц, 4Н), 2,93 (д, J=9,6 Гц, 2Н), 2,70-2,61 (м, 4Н), 1,17 (с, 6Н).
Пример 50. 5-Хлор-4-((3aR,6aS)-5-(изоксазол-5-илметил)-3а,6а-диметилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)-N-(1-метил-1H-пиразол-4-ил)пиримидин-2-амин
Стадия 1. Из оксазол-5-ил-метилметансульфонат (50b)
К раствору из оксазол-5-илметанола 50а (76 мг, 0,77 ммоль) в CH2Cl2 (10 мл) при 0°С добавляли TEA (390 мг, 3,85 ммоль) и метилсульфонилхлорид (93 мг, 0,81 ммоль). После перемешивания при комнатной температуре в течение 1 ч, полученный раствор использовали непосредственно на следующей стадии.
MS m/z (ESI): 178 [M+1]
Стадия 2. 5-Хлор-4-((3aR,6aS)-5-(изоксазол-5-илметил)-3а,6а-диметилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)-N-(1-метил-1H-пиразол-4-ил)пиримидин-2-амин (50)
К смеси соединения 12с (70 мг, 0,2 ммоль) в CH2Cl2 (10 мл) по каплям добавляли раствор, полученный на стадии 1. Смесь перемешивали в течение 2 ч и концентрировали досуха в условиях вакуума. Остаток растворяли в DMSO (2 мл), и добавляли карбонат цезия (195 мг, 0,6 ммоль). Смесь нагревали до 50°С, а затем немедленно охлаждали до комнатной температуры. Затем, смесь фильтровали, и очищали фильтрат методом преп-HPLC с получением указанного в заголовке соединения 50 (7,7 мг, твердое вещество, 9%).
MS m/z (ESI): 429 [M+1]
1H-ЯМР (400 МГц, CD3OD) δ 8,31 (д, J=1,4 Гц, 1Н),7,84 (с, 1Н), 7,76 (с, 1Н),7,56 (с, 1Н), 6,32 (д, J=1,1 Гц, 1H), 4,08 (д, J=11,5 Гц, 2Н), 3,87 (с, 2Н), 3,87 (с, 3Н), 3,62 (д, J=11,4 Гц, 2Н), 2,94 (д, J=9,4 Гц, 2Н), 2,66 (д, J=9,4 Гц, 2Н), 1,15 (с, 6Н).
Пример 51. 5-((5-Хлор-4-((3aR,6aS)-5-(циклопропанкарбонил)-3а,6а-диметилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)пиримидин-2-ил)амино)-N-метилпиколинамид

Стадия 1. (3aR,6aS)-5-Бензил-3а,6а-диметилтетрагидро-1Н-фуро[3,4-с]пиррол-1,3(3аН)-дион (51b)
К раствору 3,4-диметилфуран-2,5-диона 51а (6 г, 47,6 ммоль) в CH2Cl2 (120 мл) добавляли N-бензил-1-метокси-N-((триметилсилил)метил)метанамин (14,8 г, 61,9 ммоль). После охлаждения до 0°С, к смеси в атмосфере азота по каплям добавляли раствор TFA (543 мг, 4,76 ммоль) в CH2Cl2 (30 мл). Полученную смесь перемешивали в течение 3 ч, а затем концентрировали досуха в условиях вакуума с получением указанного в заголовке соединения 51b (17 г). Неочищенный продукт использовали непосредственно на следующей стадии без дополнительной очистки.
MS m/z (ESI): 260 [M+1]
1H-ЯМР (400 МГц, CDCl3) δ 7,45 - 7,18 (м, 5Н), 3,53 (с, 2Н), 3,46 (д, J=10,2 Гц, 2Н), 2,18 (д, J=10,1 Гц, 2Н), 1,28 (с, 6Н).
Стадия 2. (3aR,6aS)-5-Бензил-3а,6а-диметилтетрагидропирроло[3,4-с]пиррол-1,3(2Н,3аН)-дион (51с)
К раствору соединения 51b (17 г, неочищенный продукт, 47,6 ммоль) в THF (60 мл) добавляли гидроксид аммония (60 мл). Смесь нагревали в герметизированной пробирке при 100°С в течение 4 ч. После охлаждения до комнатной температуры, смесь концентрировали досуха в условиях вакуума. Остаток разбавляли водой (150 мл) и экстрагировали CH2Cl2 (3×150 мл). Объединенную органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали досуха в условиях вакуума. Остаток очищали методом колоночной хроматографии на силикагеле (петролейный эфир/этилацетат = 100/0 - 1/4) с получением указанного в заголовке соединения 51с (7 г, 57% после двух стадий).
MS m/z (ESI): 259 [M+1]
1H-ЯМР (400 МГц, DMSO-d6) δ 11,24 (с, 1H), 7,35 - 7,12 (м, 5Н), 3,48 (с, 2Н), 3,12 (д, J=9,7 Гц, 2Н), 2,03 (д, J=9,6 Гц, 2Н), 1,09 (с, 6Н).
Стадия 3. (3aR,6aS)-2-Бензил-3а,6а-диметилоктагидропирроло[3,4-с]пиррол (51d)
К раствору соединения 51с (2,67 г, 10,34 ммоль) в THF (30 мл) порциями добавляли LAH (1,18 г, 31 ммоль). Смесь нагревали до температуры возгонки в течение 2 ч при перемешивании. После охлаждения до 0°С, к смеси добавляли воду (8 мл), 20% водный раствор гидроксида натрия (16 мл) и воду (8 мл). Смесь перемешивали в течение 10 мин и фильтровали. Фильтрат концентрировали досуха в условиях вакуума с получением указанного в заголовке соединения 51d (2,44 г, 100%).
MS m/z(ESI): 231 [М+1]
1H-ЯМР (400 МГц, DMSO-d6) δ 7,34 - 7,17 (м, 5Н), 3,45 (с, 2Н), 2,61 (д, J=8,8 Гц, 2Н), 2,46 (д, J=8,7 Гц, 2Н), 2,24 (дд, J=10,6, 8,9 Гц, 4Н), 0,97 (с, 6Н).
Стадия 4. ((3aR,6aS)-5-Бензил-3а,6а-диметилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)(циклопропил)метанон (51е)
К раствору соединения 51d (2,44 г, 10,34 ммоль) в CH2Cl2 (40 мл) добавляли TEA (3,14 г, 31 ммоль) и циклопропанкарбонилхлорид (1,08 г, 10,34 ммоль). Смесь перемешивали в течение 1 ч, а затем концентрировали досуха в условиях вакуума. Остаток очищали методом колоночной хроматографии на силикагеле (петролейный эфир/этилацетат = 100/0 - 1/4) с получением указанного в заголовке соединения 51е (1,59 г, 52%).
MS m/z (ESI): 299 [M+1]
1H-ЯМР (400 МГц, CDCl3) δ 7,31 - 7,27 (м, 4Н), 7,25 - 7,20 (м, 1H), 3,81 (д, J=10,4 Гц, 1H), 3,75 (д, J=12,1 Гц, 1H), 3,59 (кв, J=13,3 Гц, 2Н), 3,46 (д, J=10,4 Гц, 1Н), 3,34 (д, J=12,1 Гц, 1H), 2,81 (д, J=9,3 Гц, 1H), 2,77 (д, J=9,2 Гц, 1H), 2,37 (д, J=9,1 Гц, 2Н), 1,59 (тт, J=8,0, 4,7 Гц, 1H), 1,09 (с, 3Н), 1,05 (с, 3Н), 1,03 - 0,95 (м, 2Н), 0,79 - 0,68 (м, 2Н).
Стадия 5. Циклопропил((3aR,6aS)-3а,6а-диметилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)метанон (51f)
К раствору соединения 51е (1,59 г, 5,38 ммоль) в этаноле (30 мл) добавляли 10% Pd/C (320 мг). Смесь перемешивали в атмосфере водорода в течение 18 ч, а затем фильтровали. Фильтрат концентрировали досуха в условиях вакуума с получением указанного в заголовке соединения 51f (1,11 г, 100%).
MS m/z (ESI): 209 [M+1]
Стадия 6. Циклопропил((3aR,6aS)-5-(2,5-дихлорпиримидин-4-ил)-3а,6а-диметилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)метанон (51g)
К раствору соединения 51f (1,11 г, 5,33 ммоль) в MeCN (20 мл) добавляли DIEA (2,07 г, 16 ммоль) и 2,4,5-трихлорпиримидин (977 мг, 5,33 ммоль). Смесь перемешивали в течение 1 ч, а затем концентрировали досуха в условиях вакуума. Остаток очищали методом колоночной хроматографии на силикагеле (петролейный эфир/этилацетат = 100/0 - 3/7) с получением указанного в заголовке соединения 51g (1,59 г, 86%).
MS m/z (ESI): 355 [M+1]
1H-ЯМР (400 МГц, CDCl3) δ 8,02 (с, 1H), 3,96 (с, 2Н), 3,87 - 3,72 (м, 3Н), 3,62 (д, J=7,0 Гц, 2Н), 3,48 (д, J=12,3 Гц, 1Н), 1,60 - 1,51 (м, 1H), 1,19 (с, 3Н), 1,16 (с, 3Н), 1,03 - 1,00 (м, 2Н), 0,82 - 0,75 (м, 2Н).
Стадия 7. Метил-5-((5-хлор-4-((3aR,6aS)-5-(циклопропанкарбонил)-3а,6а-диметилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)пиримидин-2-ил)амино)пиколинат (51h)
Смесь соединения 51g (355 мг, 4,74 ммоль), метил-5-аминопиколината (167 мг, 1,1 ммоль), карбоната цезия (975 мг, 3 ммоль), трис(дибензилиденацетон)дипалладия (92 мг, 0,1 ммоль) и 9,9-диметил-4,5-бис(дифенилфосфино)ксантен (116 мг, 0,2 ммоль) в 1,4-диоксане (15 мл) нагревали в микроволновом реакторе до 100°С в течение 1 ч в атмосфере азота. После охлаждения до комнатной температуры, смесь фильтровали, и концентрировали фильтрат досуха в условиях вакуума. Остаток очищали методом колоночной хроматографии на силикагеле (дихлорметан/метанол = 100/0 - 9/1) с получением указанного в заголовке соединения 51h (88 мг, 19%).
MS m/z (ESI): 471 [M+1]
Стадия 8. 5-((5-Хлор-4-((3aR,6aS)-5-(циклопропанкарбонил)-3а,6а-диметилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)пиримидин-2-ил)амино)пиколиновая кислота (51i)
К смеси соединения 51h (88 мг, 0,19 ммоль) в THF (2 мл) и МеОН (2 мл) добавляли 1н NaOH (4 мл). Смесь перемешивали в течение 2 ч, а затем концентрировали досуха в условиях вакуума. К остатку добавляли воду (10 мл) и промывали этилацетатом (20 мл). Водный слой корректировали до рН=3 добавлением 1н HCl и экстрагировали этилацетатом (3×20 мл). Объединенную органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали досуха в условиях вакуума с получением указанного в заголовке соединения 51i (30 мг, 35%).
MS m/z (ESI): 457 [M+1]
Стадия 9. ,5-((5-Хлор-4-((3aR,6aS)-5-(циклопропанкарбонил)-3а,6а-диметилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)пиримидин-2-ил)амино)-N-метилпиколинамид (51)
К раствору соединения 51i (30 мг, 0,066 ммоль) в CH2Cl2 (10 мл) последовательно добавляли DIEA (74 мг, 0,57 ммоль), HATU (72 мг, 0,19 ммоль) и раствор метиламина (2 М в THF, 0,05 мл, 1 ммоль). Полученную смесь перемешивали в течение 1 ч, а затем концентрировали досуха в условиях вакуума. Остаток очищали методом преп-HPLC с получением указанного в заголовке соединения 51 (8,2 мг, твердое вещество, 27%).
MS m/z (ESI): 470 [M+1]
1H-ЯМР (400 МГц, CD3OD) δ 8,77 (д, J=2,3 Гц, 1H), 8,17 (дд, J=8,6, 2,6 Гц, 1H), 7,88 (д, J=10,1 Гц, 2Н), 3,91 (т, J=12,2 Гц, 2Н), 3,82 - 3,73 (м, 3Н), 3,62 (д, J=10,8 Гц, 1H), 3,50 (д, J=12,5 Гц, 1H), 3,34 (д, J=12,4 Гц, 1Н), 2,85 (с, 3Н), 1,72 - 1,66 (м, 1H), 1,13 (с, 3Н), 1,09 (с, 3Н), 0,82 - 0,77 (м, 2Н), 0,77 - 0,71 (м, 2Н).
Пример 53. ((3aR,6aS)-5-(5-Хлор-2-((6-(морфолин-4-карбонил)пиридин-3-ил)амино)пиримидин-4-ил)-3а,6а-диметилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)(циклопропил)метанон
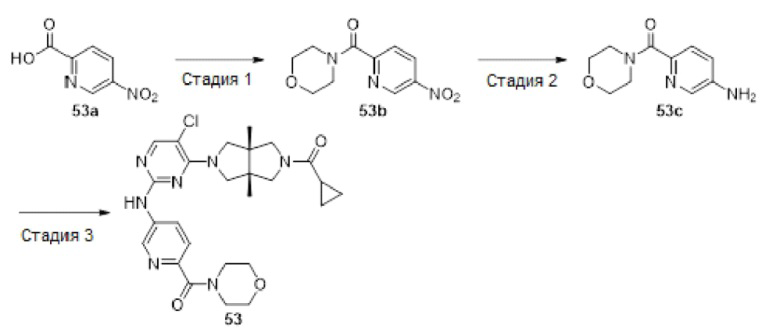
Стадия 1. Морфолино(5-нитропиридин-2-ил)метанон (53b)
К раствору 5-нитропиколиновой кислоты 53а (505 мг, 3 ммоль) в CH2Cl2 (30 мл) добавляли морфолин (262 мг, 3 ммоль), DIEA (1,16 г, 9 ммоль) и HATU (1,14 г, 3 ммоль). Смесь перемешивали в течение 1 ч, а затем концентрировали досуха в условиях вакуума. Остаток очищали методом колоночной хроматографии на силикагеле (дихлор метан/этил ацетат = 100/0 - 1/4) с получением указанного в заголовке соединения 53b (880 мг, с примесью DIEA и тетраметилмочевины).
MS m/z (ESI): 238 [M+1]
Стадия 2. (5-Аминопиридин-2-ил)(морфолино)метанон (53с)
К раствору соединения 53b (880 мг, около 3 ммоль) в МеОН (20 мл) добавляли 10% Pd/С (200 мг). Смесь перемешивали в атмосфере водорода в течение 2 ч, а затем фильтровали. Фильтрат концентрировали досуха в условиях вакуума с получением указанного в заголовке соединения 53 с (740 мг, с примесью DIEA и тетраметилмочевины).
MS m/z (ESI): 208 [M+1]
Стадия 3. ((3aR,6aS)-5-(5-Хлор-2-((6-(морфолин-4-карбонил)пиридин-3-ил)амино)пиримидин-4-ил)-3а,6а-диметилгексагидропирроло[3,4-с]пиррол-2(1H)-ил)(циклопропил)метанон (53)
К смеси соединения 51g (71 мг, 0,2 ммоль) и соединения 53с (42 мг, 0,2 ммоль) в 1,4-диоксане (2 мл) добавляли NaOEt (30 мг, 0,44 ммоль) и RuPhos-Pd-G2 (8 мг, 0,01 ммоль). Смесь нагревали до 100°С в микроволновом реакторе в атмосф ере аз отав течение 1 ч. После охлаждения до комнатной температуры, смесь фильтровали, и концентрировали фильтрат досуха в условиях вакуума. Остаток очищали методом колоночной хроматографии на силикагеле (дихлорметан/метанол = 100/0 - 9/1), а затем преп-HPLC, с получением указанного в заголовке соединения 53 (11,4 мг, твердое вещество, 11%).
MS m/z (ESI): 526 [M+1]
1H-ЯМР (400 МГц, CD3OD) δ 8,92 (д, J=2,4 Гц, 1H), 8,29 (дд, J=8,6, 2,6 Гц, 1Н), 7,99 (с, 1Н),7,62 (д, J=8,6 Гц, 1Н),4,02 (т, J=11,0 Гц, 2Н), 3,95 - 3,58 (м, 13Н), 3,46 (д, J=12,4 Гц, 1H), 1,81 (ддд, J=12,8, 7,9,4,7 Гц, 1Н), 1,24 (с, 3Н), 1,21 (с, 3Н), 0,92 (дт, J=5,4, 3,8 Гц, 2Н), 0,89 - 0,81 (м, 2Н).
Пример 59 синтезировали в соответствии с методикой, описанной для Примера 53, за исключением того, что на стадии 1 1-мети л пиперазин использовали вместо морфолина.
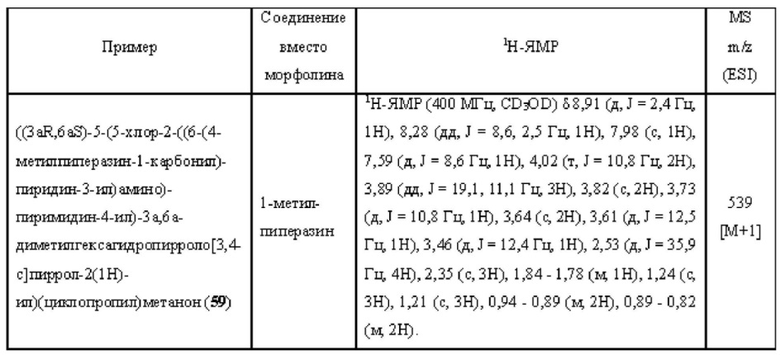
Пример 54. 5-Хлор-4-((3aR,6aS)-3а,6а-диметил-5-(пиридин-2-ил)гексагидропирроло[3,4-с]пиррол-2(1Н)-ил)-N-(1-метил-1H-пиразол-4-ил) пиримидин-2-амин
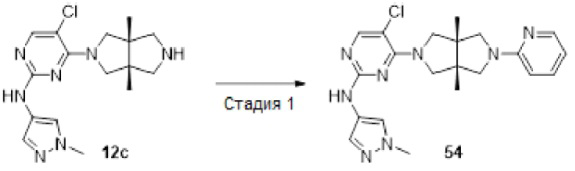
Смесь соединения 12с (100 мг, 0,26 ммоль), 2-6ромпиридина (62 мг, 0,39 ммоль), DBU (4 мл) нагревали до 140°С и перемешивали в течение 16 ч. После охлаждения до комнатной температуры, к смеси добавляли воду (4 мл), и экстрагировали этилацетатом (3×5 мл). Объединенную органическую фазу промывали водой (10 мл) и концентрировали досуха в условиях вакуума. Остаток очищали методом преп-HPLC с получением указанного в заголовке соединения 54 (36,4 мг, твердое вещество, 33%).
MS m/z (ESI): 425 [M+1]
1H-ЯМР (400 МГц, DMSO-d6) δ 9,05 (с, 1H), 8,04 (д, J=3,7 Гц, 1Н), 7,89 (с, 1Н), 7,69 (с, 1H), 7,47 (с, 1Н), 7,43 (с, 1Н), 6,54 (д, J=1,6 Гц, 1Н), 6,40 (д, J=8,4 Гц, 1Н), 3,89 (д, J=11,4 Гц, 2Н), 3,75 (с, 3Н), 3,75 - 3,71 (м, 2Н), 3,56 (д, J=10,9 Гц, 2Н), 3,38 (д, J=10,8 Гц, 2Н), 1,15 (с, 6Н).
Пример 55. 4-((3aR,6aS)-3а,6а-Диметил-5-(метилсульфонил)гексагидропирроло[3,4-с]пиррол-2(1H)-ил)-5-метил-N-(1-метил-1Н-пиразол-4-ил)пиримидин-2-амин
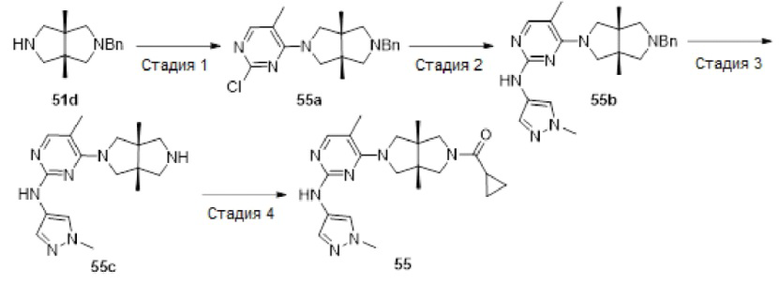
Стадия 1. (3aR,6aS)-2-Бензил-5-(2-хлор-5-метилпиримидин-4-ил)-3а,6а-диметилоктагидропирроло[3,4-с]пиррол (55а)
К раствору соединения 51d (2,02 г, 8,8 ммоль) в MeCN (20 мл) добавляли 2,4-дихлор-5-метилпиримидин (1,63 г, 10 ммоль) и DIEA (3,88 г, 30 ммоль). Смесь перемешивали в течение 2 ч, а затем концентрировали досуха в условиях вакуума. Остаток очищали методом колоночной хроматографии на силикагеле (петролейный эфир/этилацетат = 100/0 - 45/55) с получением указанного в заголовке соединения 55а (1,45 г, 46%).
MS m/z (ESI): 357 [M+1]
Стадия 2. 4-((3aR,6aS)-5-Бензил-3а,6а-диметилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)-5-метил-N-(1-метил-1Н-пиразол-4-ил)пиримидин-2-амин (55b)
К смеси соединения 55а (1,45 г, 4,1 ммоль) и 1-метил-1H-пиразол-4-амина гидрохлорида (4,1 г, 1 ммоль) в изопропаноле (40 мл) добавляли pTsOH (78 мг, 0,41 ммоль). Смесь перемешивали при 100°С в течение 18 ч. После охлаждения до комнатной температуры, к смеси добавляли МеОН (20 мл), а затем TEA (0,5 мл). Смесь перемешивали дополнительно в течение 5 мин и концентрировали досуха в условиях вакуума. Остаток очищали методом колоночной хроматографии на силикагеле (дихлорметан/метанол = 19/1) с получением указанного в заголовке соединения 55b (540 мг, 32%).
MS m/z (ESI): 418 [M+1]
1H-ЯМР (400 МГц, CDCl3) δ 7,68 (с, 2Н), 7,51 (с, 1H), 7,30 (дд, J=6,6, 3,5 Гц, 4Н), 7,25 - 7,20 (м, 1Н), 6,86 (с, 1H), 3,85 (д, J=10,8 Гц, 2Н), 3,85 (с, 3Н), 3,60 (с, 2Н), 3,48 (д, J=10,8 Гц, 2Н), 2,80 (д, J=9,3 Гц, 2Н), 2,41 (д, J=9,3 Гц, 2Н), 2,20 (с, 3Н), 1,10 (с, 6Н).
Стадия 3. 4-((3aR,6aS)-3а,6а-Диметилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)-5-метил-N-(1-метил-1H-пиразол-4-ил)пиримидин-2-амин (55с)
К раствору соединения 55b (540 мг, 1,29 ммоль) в МеОН (30 мл) добавляли 10% Pd/C (300 мг). Смесь перемешивали в атмосфере водорода при 36°С в течение 16 ч, а затем фильтровали. Фильтрат концентрировали досуха в условиях вакуума с получением указанного в заголовке соединения 55с (340 мг, 81%).
MS m/z (ESI): 328 [M+1]
Стадия 4. 4-((3aR,6aS)-3а,6а-Диметил-5-(метилсульфонил)гексагидропирроло[3,4-с]пиррол-2(1Н)-ил)-5-метил-N-(1-метил-1Н-пиразол-4-ил)пиримидин-2-амин (55)
К раствору соединения 55с (33 мг, 0,1 ммоль) в CH2Cl2 (10 мл) добавляли TEA (30 мг, 0,3 ммоль), а затем раствор MsCl (12 мг, 0,1 ммоль) в CH2Cl2 (1 мл). Смесь перемешивали в течение 1 ч, а затем концентрировали досуха в условиях вакуума. Остаток очищали методом преп-HPLC с получением указанного в заголовке соединения 55 (18,7 мг, твердое вещество, 46%).
MS m/z (ESI): 406 [M+1]
1H-ЯМР (400 МГц, CD3OD) δ 7,64 (с, 1H), 7,50 (с, 1Н), 7,44 (с, 1Н), 3,82 (д, J=11,2 Гц, 2Н), 3,74 (с, 3Н), 3,60 (д, J=11,2 Гц, 2Н), 3,40 (д, J=10,1 Гц, 2Н), 3,23 (д, J=10,1 Гц, 2Н), 2,83 (с, 3Н), 2,15 (с, 3Н), 1,09 (с, 6Н).
Пример 56. ((S)-2,2-дифторциклопропил)((3aR,6aS)-3а,6а-диметил-5-(5-метил-2-((1-метил-1H-пиразол-4-ил)амино)пиримидин-4-ил)гексагидропирроло[3,4-с]пиррол-2(1Н)-ил)метанон
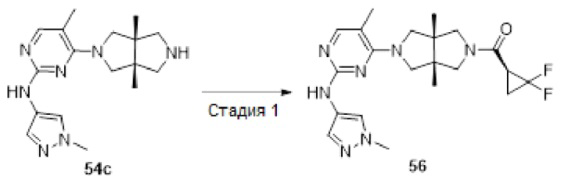
К раствору соединения 54с (33 мг, 0,1 ммоль) в CH2Cl2 (10 мл) добавляли (S)-2,2-дифторциклопропан-1-карбоновую кислоту (13 мг, 0,1 ммоль) и DIEA (39 мг, 0,3 ммоль), а затем HATU (38 мг, 0,1 ммоль). Смесь перемешивали в течение 1 ч, а затем концентрировали досуха в условиях вакуума. Остаток очищали методом преп-HPLC с получением указанного в заголовке соединения 56 (21,3 мг, твердое вещество, 49%).
MS m/z (ESI): 432 [M+1]
1H-ЯМР (400 МГц, CD3OD) δ 7,76 (д, J=3,1 Гц, 1H), 7,62 (с, 1Н),7,54 (д, J=11,0 Гц, 1Н), 3,98 - 3,88 (м, 2Н), 3,86 (д, J=2,7 Гц, 3Н), 3,82 - 3,71 (м, 3Н), 3,68 - 3,56 (м, 2Н), 3,48 (дд, J=12,6, 2,7 Гц, 1Н), 2,89 - 2,79 (м, 1H), 2,28 (д, J=5,7 Гц, 3Н), 2,08 - 2,00 (м, 1H), 1,85 - 1,73 (м, 1H), 1,24 (д, J=3,4 Гц, 2Н), 1,21 (д, J=3,9 Гц, 3Н).
Примеры 57, 58 и 60 синтезировали в соответствии с методикой, описанной для Примера 56, за исключением того, что на стадии 1 различные соединения использовали вместо (S)-2,2-дифторциклопропан-1-карбоновой кислоты.
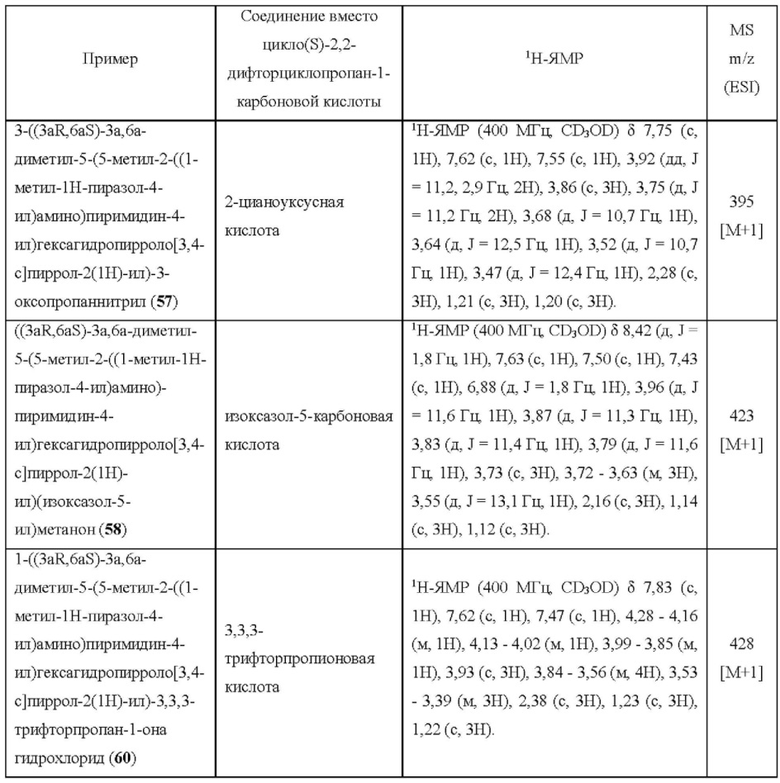
Пример 61. цис-5-хлор-N-(1-метил-1Н-пиразол-4-ил)-4-(3а-метил-5-(метилсульфонил)гексагидропирроло[3,4-с]пиррол-2(1Н)-ил)пиримидин-2-амин
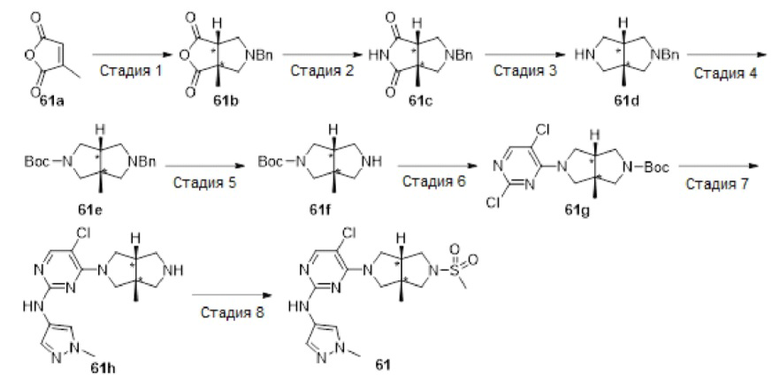
Стадия 1. цис-5-Бензил-3а-метилтетрагидро-1H-фуро[3,4-с]пиррол-1,3(3аН)-дион (61b)
К раствору 3-метилфуран-2,5-диона (5,33 г, 47,6 ммоль) и N-бензил-1-метокси-N-((триметилсилил)метил)метанамина (14,8 г, 61,9 ммоль) в CH2Cl2 (170 мл) добавляли трифторуксусную кислоту (543 мг, 4,76 ммоль). Смесь перемешивали в течение 3 ч, а затем концентрировали досуха в условиях вакуума с получением указанного в заголовке соединения 61b (11,62 г). Неочищенный продукт использовали непосредственно на следующей стадии без дополнительной очистки.
MS m/z (ESI): 246 [M+1]
Стадия 2. цис-5-Бензил-3а-метилтетрагидропирроло[3,4-с]пиррол-1,3(2Н,3аН)-дион (61с)
К раствору соединения 61b (11,66 г, около 47,6 ммоль) в THF (60 мл) добавляли гидроксид аммония (60 мл). Смесь нагревали в герметизированной пробирке при 100°С в течение 5 ч. После охлаждения до комнатной температуры, смесь концентрировали досуха в условиях вакуума, и растворяли остаток в CH2Cl2 (200 мл). Полученную смесь промывали водой (20 мл) и концентрировали досуха в условиях вакуума. Остаток очищали методом колоночной хроматографии на силикагеле (петролейный эфир/этилацетат = 100/1 - 1/1) с получением указанного в заголовке соединения 61с (8 г, 68%).
MS m/z (ESI): 245 [M+1]
Стадия 3. цис-2-Бензил-3а-метилоктагидропирроло[3,4-с]пиррол (61d)
К раствору соединения 61с (8 г, 32. 8 ммоль) в THF (100 мл) добавляли LAH (3,73 г, 98,36 ммоль). Смесь нагревали до 70°С и перемешивали в течение 3 ч. После охлаждения до 0°С, к смеси последовательно добавляли воду (10 мл), 20% раствор NaOH (20 мл) и воду (10 мл). Смесь перемешивали в течение 10 мин, а затем фильтровали. Фильтрат концентрировали досуха в условиях вакуума с получением указанного в заголовке соединения (8 г). Неочищенный продукт использовали непосредственно на следующей стадии без дополнительной очистки.
MS m/z(ESI): 217 [М+1]
Стадия 4. трет-Бутил-цис-5-бензил-3а-метилгексагидропирроло[3,4-с]пиррол-2(1Н)-карбоксилат (61е)
К раствору соединения 61d (8 г, неочищенное) и TEA (7,48 г, 74,07 ммоль) в CH2Cl2 (100 мл) добавляли ди-трет-бутилбикарбонат (8,07 г, 37,03 ммоль). Смесь перемешивали в течение 3 ч и концентрировали досуха в условиях вакуума. Остаток очищали методом колоночной хроматографии на силикагеле (петролейный эфир/этилацетат = 100/1 - 7/3) с получением указанного в заголовке соединения 61е (10 г, 96% после двух стадий).
MS m/z (ESI): 317 [М+1]
Стадия 5. трет-Бутил-цис-3а-метилгексагидропирроло[3,4-с]пиррол-2(1Н)-карбоксилат (61f)
К раствору соединения 61е (10 г, 31,64 ммоль) в МеОН (100 мл) добавляли 10% Pd/C (1 г). Смесь перемешивали в атмосфере водорода в течение 18 ч, а затем фильтровали. Фильтрат концентрировали досуха в условиях вакуума с получением указанного в заголовке соединения 61f (7 г, 97%).
MS m/z (ESI): 227 [M+1]
Стадия 6. трет-Бутил-цис-5-(2,5-дихлорпиримидин-4-ил)-3а-метилгексагидропирроло[3,4-с]пиррол-2(1Н)-карбоксилат (61g)
К раствору 2,4,5-трихлорпиримидина (546 мг, 3 ммоль) в MeCN (10 мл) добавляли соединение 61f (678 мг, 3 ммоль) и карбонат калия (828 мг, 6 ммоль). Смесь нагревали до 80°С и перемешивали в течение 3 ч. После охлаждения до комнатной температуры, смесь концентрировали досуха в условиях вакуума, и очищали остаток методом колоночной хроматографии на силикагеле (петролейный эфир/этилацетат = 100/0 - 9/1) с получением указанного в заголовке соединения 61g (850 мг, 76%).
MS m/z (ESI): 373 [M+1]
Стадия 7. цис-5-Хлор-N-(1-метил-1Н-пиразол-4-ил)-4-(3а-метилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)пиримидин-2-амина гидрохлорид (61h)
К смеси соединения 61 g (850 мг, 2,28 ммоль) и 1-метил-1H-пиразол-4-амина гидрохлорида (221 мг, 2,28 ммоль) в изопропаноле (10 мл) добавляли pTsOH (868 мг, 4,57 ммоль). Смесь нагревали в микроволновом реакторе при 100°С в течение 2 ч. После охлаждения до комнатной температуры, осадок собирали путем фильтрования с получением указанного в заголовке соединения 61h (200 мг, 75%) в виде гидрохлорида.
MS m/z (ESI): 334 [M+1]
Стадия 8. цис-5-Хлор-N-(1-метил-1Н-пиразол-4-ил)-4-(3а-метил-5-(метилсульфонил)гексагидропирроло[3,4-с]пиррол-2(1H)-ил)пиримидин-2-амин (61)
К смеси соединения 61h (33,3 мг, 0,1 ммоль) и TEA (20,2 мг, 0,2 ммоль) в CH2Cl2 (5 мл) добавляли MsCl (11,4 мг, 0,1 ммоль). Смесь перемешивали в течение 1 ч и концентрировали досуха в условиях вакуума. Остаток очищали методом преп-HPLC с получением указанного в заголовке соединения 61 (15,4 мг, твердое вещество, 37%).
MS m/z (ESI): 412 [M+1]
1H-ЯМР (400МГц, CD3OD) δ 7,74 (с, 1H),7,66 (с, 1Н),7,43 (с, 1H), 4,06 - 4,01 (м, 1H), 3,88 (д, J=11,5 Гц, 1H), 3,76 - 3,64 (м, 6Н), 3,61 - 3,56 (м, 1Н), 3,35 (д, J=9,9 Гц, 2Н), 2,83 (с, 3Н), 2,56 - 2,51 (м, 1Н), 1,22 (с, 3Н).
Пример 62 синтезировали в соответствии с методикой, описанной для Примера 61, за исключением того, что на стадии 8 циклопропанкарбонилхлорид использовали вместо MsCl.
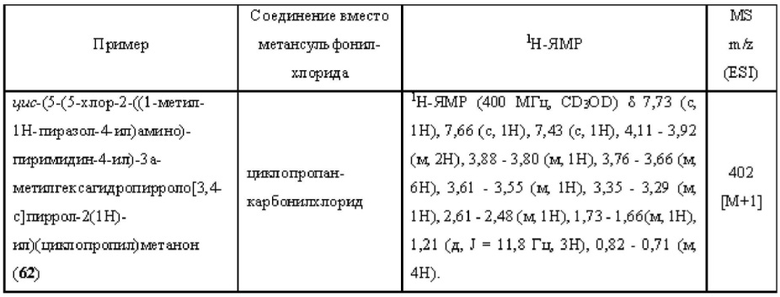
Пример 63. цис-3-(5-(5-хлор-2-((1-метил-1Н-пиразол-4-ил)амино)пиримидин-4-ил)-3а-метилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)-3-оксопропаннитрил
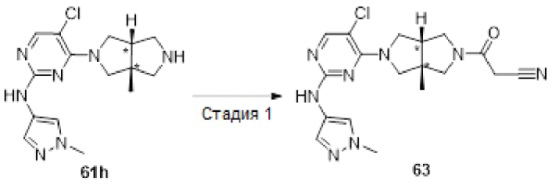
К раствору соединения 61h (66,6 мг, 0,2 ммоль), HATU (152 мг, 0,4 ммоль) и TEA (40,4 мг, 0,4 ммоль) в CH2Cl2 (5 мл) добавляли 2-цианоуксусную кислоту (17 мг, 0,2 ммоль). Смесь перемешивали в течение 2 ч, а затем концентрировали досуха в условиях вакуума. Остаток очищали методом преп-HPLC с получением указанного в заголовке соединения 63 (12,4 мг, твердое вещество, 16%).
MS m/z (ESI): 401 [M+1]
1H-ЯМР (400 МГц, CD3OD) δ 7,85 (с, 1H), 7,78 (с, 1H), 7,54 (с, 1H), 4,20 - 4,15 (м, 1H), 4,00 - 3,92 (м, 1H), 3,88 - 3,79 (м, 6Н), 3,66 - 3,61 (м, 1H), 3,52 - 3,45 (м, 2Н), 2,71 - 2,62 (м, 1Н), 1,32 (д, J=4,0 Гц, 3Н).
Примеры 64, 65 и 68 синтезировали в соответствии с методикой, описанной для Примера 63, за исключением того, что на стадии 1 различные соединения использовали вместо 2-цианоуксусной кислоты.
Пример 66. 5-Хлор-4-((3aR,6aS)-5-(изоксазол-5-ил)-3а,6а-диметилгексагидропирроло [3,4-с]пиррол-2(1Н)-ил)-N-(1-метил-1 Н-пиразол-4-ил)пиримидин-2-амин
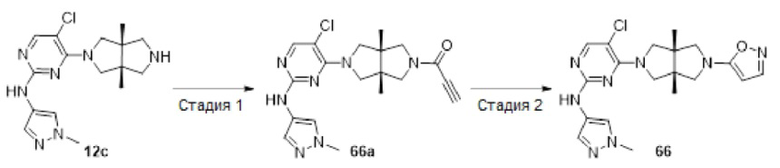
Стадия 1. 01-((3aR,6aS)-5-(5-Хлор-2-((1-метил-1Н-пиразол-4-ил)амино)пиримидин-4-ил)-3а,6а-диметилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)проп-2-ин-1-он (66а)
К смеси соединения 12с (139 мг, 0,4 ммоль) в CH2Cl2 (20 мл) добавляли пропиоловую кислоту (28 мг, 0,4 ммоль), DIEA (155 мг, 1,2 ммоль) и HATU (152 мг, 0,4 ммоль). Смесь перемешивали в течение 1 ч, а затем концентрировали досуха в условиях вакуума. Остаток очищали методом колоночной хроматографии на силикагеле (дихлорметан/метанол = 100/0 - 19/1) с получением указанного в заголовке соединения 66а (90 мг, 56%).
MS m/z (ESI): 400 [M+1]
Стадия 2. 5-Хлор-4-((3aR,6aS)-5-(изоксазол-5-ил)-3а,6a-диметилгексагидропирроло[3,4-с]пиррол-2(1H)-ил)-N-(1-метил-1H-пиразол-4-ил)пиримидин-2-амин (66)
К раствору соединения 66а (90 мг, 0,225 ммоль) в THF (2 мл) добавляли воду (2 мл), хлорид аммония (60 мг, 1,125 ммоль) и азид натрия (73 мг, 1,125 ммоль). Смесь перемешивали в течение 18 ч, а затем нагревали до 70°С и перемешивали дополнительно в течение 2 ч. После охлаждения до комнатной температуры, смесь разбавляли водой (20 мл) и экстрагировали этилацетатом (2×20 мл). Объединенную органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали досуха в условиях вакуума. Остаток очищали методом преп-HPLC с получением указанного в заголовке соединения 66 (12,1 мг, твердое вещество, 13%).
MS m/z (ESI): 415 [M+1]
1H-ЯМР (400 МГц, CD3OD) δ 8,06 (д, J=2,1 Гц, 1Н),7,85 (с, 1Н), 7,76 (с, 1Н),7,54 (с, 1Н), 5,00 (д, J=2,1 Гц, 1Н), 4,01 (д, J=11,6 Гц, 2Н), 3,86 (д, J=11,5 Гц, 2Н), 3,85 (с, 3Н), 3,62 (д, J=10,3 Гц, 2Н), 3,47 (д, J=10,3 Гц, 2Н), 1,24 (с, 6Н).
Пример 69. цис-4-(5-(Изоксазол-5-илметил)-3а-метилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)-N-(1-метил-1Н-пиразол-4-ил)пиримидин-2-амин
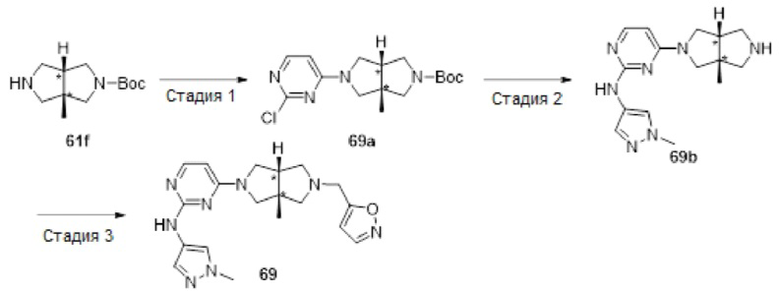
Стадия 1. трет-Бутил-цис-5-(2-хлорпиримидин-4-ил)-3а-метилгексагидропирроло[3,4-с]пиррол-2(1Н)-карбоксилат (69а)
К раствору 2,4-дих лор пиримидина (444 мг, 3 ммоль) в MeCN (10 мл) добавляли соединение 61f (678 мг, 3 ммоль) и карбонат калия (621 мг, 4,5 ммоль). Смесь перемешивали при 80°С в течение 3 ч, а затем охлаждали до комнатной температуры. Смесь фильтровали, и концентрировали фильтрат досуха в условиях вакуума с получением указанного в заголовке соединения 69а (700 мг, 69%).
MS m/z (ESI): 339 [M+1]
Стадия 2. цис-N-(1-метил-1Н-пиразол-4-ил)-4-(3а-метилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)пиримидин-2-амин (69b)
К смеси соединения 69а (700 мг, 2,07 ммоль) и 1-метил-1H-пиразол-4-амина гидрохлорида (200 мг, 2,07 ммоль) в изопропаноле (15 мл) добавляли pTsOH (787 мг, 4,14 ммоль). Смесь перемешивали при 100°С в течение 18 ч. После охлаждения до комнатной температуры, смесь концентрировали досуха в условиях вакуума. Остаток очищали методом колоночной хроматографии на силикагеле (CH2Cl2/МеОН = 100/1 - 4/1) с получением указанного в заголовке соединения 69b (300 мг, 48%).
MS m/z (ESI): 300 [M+1]
Стадия 3. цис-4-(5-(Изоксазол-5-илметил)-3а-метилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)-Н-(1-метил-1Н-пиразол-4-ил)пиримидин-2-амин (69)
Соединение 69 синтезировали в соответствии со стадией 2 Примера 50 за исключением того, что соединение 69b использовали вместо соединение 12с.
MS m/z (ESI): 381 [M+1]
1H-ЯМР (400 МГц, CD3OD) δ 8,33 (д, J=1,8 Гц, 1H), 7,83 - 7,80 (м, 2Н), 7,58 (с, 1H), 6,35 (д, J=1,8 Гц, 1H), 5,92 (д, J=6,1 Гц, 1H), 3,87 (с, 5Н), 3,82 (с, 2Н), 3,63 (с, 1H), 3,52 (с, 1Н), 2,98 (дд, J=9,4, 7,8 Гц, 1H), 2,84 (д, J=9,2 Гц, 1H), 2,69 (дд, J=9,5, 4,6 Гц, 1H), 2,58 (д, J=9,3 Гц, 1H), 2,55 - 2,49 (м, 1Н), 1,32 (с, 3Н).
Примеры 73 и 74 синтезировали в соответствии со стадией 2 Примера 50 за исключением того, что различные химические соединения использовали вместо соединения 12с.
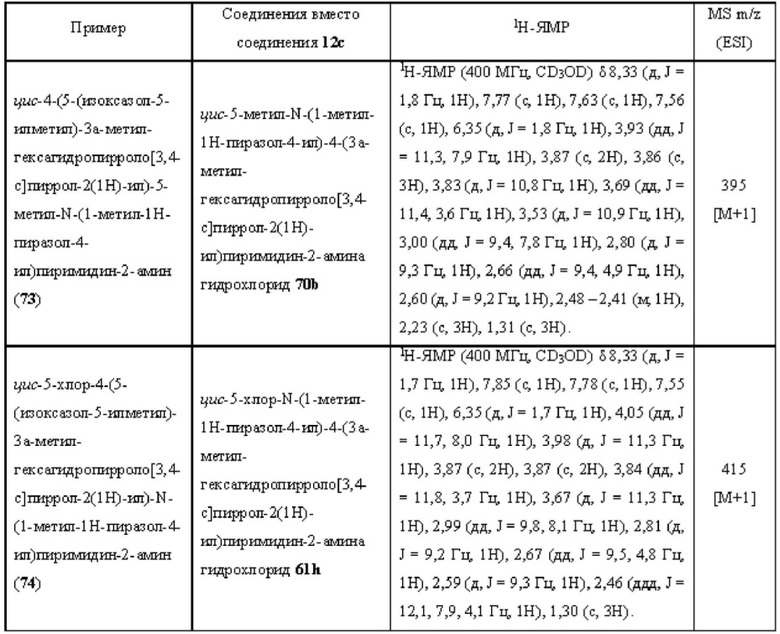
Пример 70. цис-Циклопропил(3а-метил-5-(5-метил-2-((1-метил-1Н-пиразол-4-ил)амино)пиримидин-4-ил)гексагидропирроло[3,4-с]пиррол-2(1H)-ил)метанон
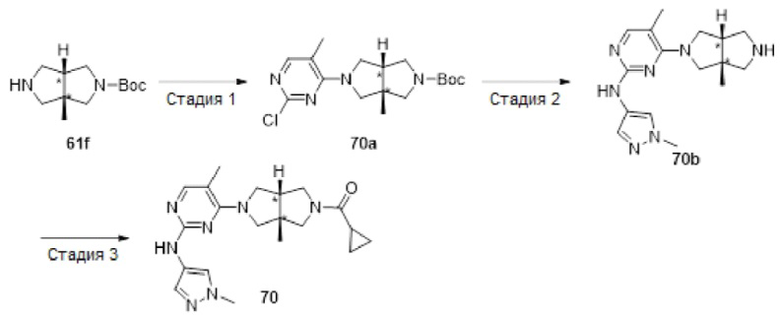
Стадия 1. трет-Бутил-цис-5-(2-хлор-5-метилпиримидин-4-ил)-3а-метилгексагидропирроло[3,4-с]пиррол-2(1Н)-кар6оксилат (70а)
К раствору соединения 2,4-дихлор-5-метил пиримидин (324 мг, 2 ммоль) в MeCN (10 мл) добавляли соединение 61f (452 мг, 2 ммоль) и карбонат калия (552 мг, 4 ммоль). Смесь перемешивали при 80°С втечение 3 ч, азатем охлаждали до комнатной температуры. Смесь концентрировали досуха в условиях вакуума, и очищали остаток методом колоночной хроматографии на силикагеле (петролейный эфир/этилацетат = 100/0 - 7/3) с получением указанного в заголовке соединения 70а (600 мг, 85%).
MS m/z (ESI): 353 [M+1]
Стадия 2. цис-5-Метил-N-(1-метил-1H-пиразол-4-ил)-4-(3а-метилгексагидропирроло[3,4-с]пиррол-2(1H)-ил)пиримидин-2-амина гидрохлорид (70b)
К смеси соединения 70а (600 мг, 1,7 ммоль) и 1-метил-1H-пиразол-4-амина гидрохлорида (165 мг, 1,7 ммоль) в изопропаноле (15 мл) добавляли pTsOH (646 мг, 3,4 ммоль). Смесь нагревали при 100°С в микроволновом реакторе в течение 1 ч. После охлаждения до комнатной температуры, смесь фильтровали, и концентрировали фильтрат досуха в условиях вакуума. Остаток очищали методом колоночной хроматографии на силикагеле (CH2Cl2/МеОН=100/1 - 67/33) с получением указанного в заголовке соединения 70b в виде гидрохлорида (400 мг, 75%).
MS m/z (ESI): 314 [M+1]
Стадия 3. цис-Циклопропил(3а-метил-5-(5-метил-2-((1-метил-1Н-пиразол-4-ил)амино)пиримидин-4-ил)гексагидропирроло[3,4-с]пиррол-2(1Н)-ил)метанон (70)
К смеси соединения 70b (62,6 мг, 0,2 ммоль) в CH2Cl2 (5 мл) добавляли TEA (30,3 мг, 0,3 ммоль) и циклопропанкарбонилхлорид (20,8 мг, 0,2 ммоль). Смесь перемешивали при комнатной температуре в течение 1 ч, а затем концентрировали досуха в условиях вакуума. Остаток очищали методом преп-HPLC с получением указанного в заголовке соединения 70 (7,3 мг, твердое вещество, 10%).
MS m/z (ESI): 382 [M+1]
1H-ЯМР (400МГц, CD3OD) δ 7,65 (с, 1Н),7,49 (с, 1Н),7,43 (с, 1H), 4,05 - 3,92 (м, 2Н), 3,81 - 3,71 (м, 4Н), 3,69 - 3,48 (м, 4Н), 3,36 - 3,29 (м, 1Н), 2,60 - 2,47 (м, 1H), 2,17 (с, 3Н), 1,74 - 1,66 (м, 1H), 1,22 (д, J=12,1 Гц, 3Н), 0,84 - 0,66 (м, 4Н).
Пример 71. цис-3-(3а-метил-5-(5-метил-2-((1-метил-1Н-пиразол-4-ил)амино)-пиримидин-4-ил)гексагидропирроло[3,4-с]пиррол-2(1Н)-ил)-3-оксопропаннитрил
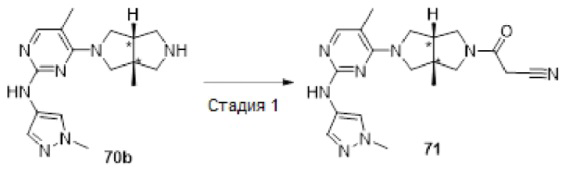
К смеси соединения 70b (62,6 мг, 0,2 ммоль) в CH2Cl2 (5 мл) добавляли 2-цианоуксусную кислоту (17 мг, 0,2 ммоль), TEA (40,4 мг, 0,4 ммоль) и HATU (114 мг, 0,3 ммоль). Смесь перемешивали в течение 2 ч, а затем концентрировали досуха в условиях вакуума. Остаток очищали методом преп-HPLC с получением указанного в заголовке соединения 71 (9,4 мг, твердое вещество, 10%).
MS m/z (ESI): 382 [M+1]
1H-ЯМР (400МГц, CD3OD) δ 7,64 (с, 1H),7,50 (с, 1Н),7,43 (с, 1H), 4,01 - 3,93 (м, 1H), 3,83 -3,56 (м, 8Н), 3,54 - 3,49(м, 1H), 3,43 -3,27 (м, 3Н), 2,59 - 2,49 (м, 1Н), 2,15 (д, J=12,8 Гц, 3Н), 1,22 (дд, J=10,7, 3,4 Гц, 3Н).
Пример 72 синтезировали в соответствии с методикой, описанной для Примера 7, за исключением того, что на стадии 1 (S)-2,2-дифторциклопропан-1-карбоновую кислоту использовали вместо 2-цианоуксусной кислоты.

Пример 75. 5-Хлор-4-((3aR,6aS)-3а,6а-диметил-5-((1-метил-1H-пиразол-3-ил)метил)гексагидропирроло[3,4-с]пиррол-2(1Н)-ил)-N-(1-метил-1H-пиразол-4-ил)пиримидин-2-амина гидрохлорид
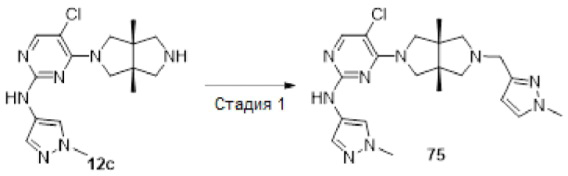
К смеси соединения 12с (35 мг, 0,1 ммоль) в DMF (4 мл) добавляли карбонат цезия (98 мг, 0,3 ммоль) и 33-(хлорметил)-1-метил-1H-пиразол (14 мг, 0,11 ммоль). Смесь перемешивали в течение 20 ч, азатем фильтровали. Фильтрат очищали непосредственно методом преп-HPLC с получением указанного в заголовке соединения 75 в виде гидрохлорида (3,4 мг, твердое вещество, 8%).
MS m/z (ESI): 442 [M+1]
1H-ЯМР (400 МГц, CD3OD) δ 7,94 (т, J=31,0 Гц, 2H), 7,74 - 7,56 (м, 2Н), 6,56 (д, J=5,3 Гц, 1H), 4,45 (д, J=6,5 Гц, 2Н), 4,15 - 3,85 (м, 6Н), 3,81 (д, J=12,8 Гц, 2Н), 3,71 (д, J=11,8 Гц, 2Н), 3,60 (д, J=11,9 Гц, 2Н), 3,54 - 3,48 (м, 2Н), 1,28 (с, 3Н), 1,22 (с, 3Н).
Примеры 76, 77, 78, 79, 80, 81 и 82 синтезировали в соответствии с методикой, описанной для Примера 75, за исключением того, что на стадии 1 различные соединения использовали вместо 1,3-(хлорметил)-1-метил-1H-пиразола.
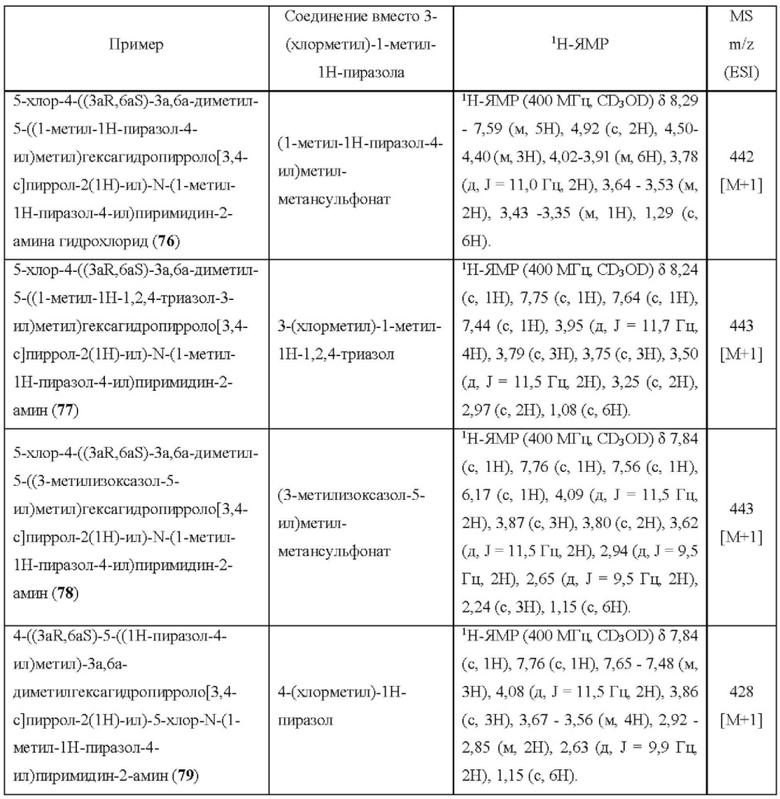

Пример 83. 5-Хлор-4-((3aR,6aS)-3а,6а-диметил-5-(5-метил-1,3,4-оксадиазол-2-ил)гексагидропирроло[3,4-с]пиррол-2(1H)-ил)-N-(1-метил-1H-пиразол-4-ил)пиримидин-2-амин
К смеси соединения 12с (80 мг, 0,208 ммоль) в DMF (3 мл) добавляли 5-метил-1,3,4-оксадиазол-2(3Н)-он (21 мг, 0,208 ммоль) и DIEA (161 мг, 1,25 ммоль). Смесь перемешивали при в течение 5 мин, а затем добавляли (бензотриазол-1-илокси)трис(диметиламино)фосфония гексафторфосфат (101 мг, 0,229 ммоль). Полученную смесь нагревали до 50°С и перемешивали в течение 12 ч. После охлаждения до комнатной температуры, к смеси добавляли воду (5 мл) и экстрагировали этилацетатом (3×5 мл). Объединенную органическую фазу концентрировали досуха в условиях вакуума, и очищали остаток методом преп-HPLC с получением указанного в заголовке соединения 83 (8,5 мг, твердое вещество, 10%).
MS m/z (ESI): 430 [M+1]
1H-ЯМР (400 МГц, DMSO-d6) δ 9,06 (с, 1H), 7,89 (с, 1H), 7,70 (с, 1H), 7,44 (с, 1H), 3,92 (д, J=11,6 Гц, 2Н), 3,77 (с, 3Н), 3,73 (д, J=11,3 Гц, 2Н), 3,57 (д, J=10,4 Гц, 2Н), 3,40 (д, 1=10,4 Гц, 2Н), 2,31 (с, 3Н), 1,14 (с, 6Н).
Пример 84. 5-Хлор-4-((3aR,6aS)-5-(циклопропилметил)-3а,6а-диметилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)-N-(1-метил-1H-пиразол-4-ил)пиримидин-2-амин
Стадия 1. (3aR,6aS)-2-бензил-5-(2,5-дихлорпиримидин-4-ил)-3а,6а-диметилоктагидропирроло[3,4-с]пиррол (84а)
К раствору соединения 51d (400 мг, 1,74 ммоль) в MeCN (10 мл) добавляли карбонат калия (720 мг, 5,22 ммоль) и 2,4,5-трихлорпиримидин (410 мг, 2,26 ммоль). Смесь перемешивали в течение 2 ч, а затем концентрировали досуха в условиях вакуума. Остаток очищали методом колоночной хроматографии на силикагеле (петролейный эфир/этилацетат = 100/0 - 3/2) с получением указанного в заголовке соединения 84а (200 мг, 31%).
MS m/z (ESI): 377 [M+1]
Стадия 2. (3aR,6aS)-2-(2,5-дихлорпиримидин-4-ил)-3а,6а-диметилоктагидропирроло[3,4-с]пиррол (84b)
К раствору соединения 84а (0,2 г, 0,53 ммоль) в CH2Cl2 (10 мл) добавляли 1-хлорэтилхлороф ормиат (226 мг, 1,59 ммоль). Смесь нагревали до температуры возгонки в течение 2 ч. После охлаждения до комнатной температуры, смесь концентрировали досуха в условиях вакуума, а затем добавляли МеОН (10 мл). Смесь нагревали до температуры возгонки дополнительно в течение 2 ч, а затем охлаждали до комнатной температуры. Смесь концентрировали досуха в условиях вакуума, и очищали остаток методом колоночной хроматографии на силикагеле (CH2Cl2/MeOH=9/1) с получением указанного в заголовке соединения 84b (140 мг, 92%).
MS m/z (ESI): 287 [M+1]
Стадия 3. (3aR,6aS)-2-(циклопропилметил)-5-(2,5-дихлорпиримидин-4-ил)-3а,6а-диметилоктагидропирроло[3,4-с]пиррол (84с)
Смесь (бромметил)циклопропана (56,3 мг, 0,42 ммоль), соединения 84b (60 мг, 0,21 ммоль), карбоната калия (87 мг, 0,63 ммоль) в MeCN (4 мл) нагревали до 50°С и перемешивали в течение 16 ч. После охлаждения до комнатной температуры, смесь концентрировали досуха в условиях вакуума, и очищали остаток методом колоночной хроматографии на силикагеле (петролейный эфир/этилацетат = 100/0 - 2/3) с получением указанного в заголовке соединения 84с (50 мг, 70%).
MS m/z (ESI): 341 [M+1]
Стадия 4. 5-Хлор-4-((3aR,6aS)-5-(циклопропилметил)-3а,6а-диметилгексагидропирроло[3,4-с]пиррол-2(1H)-ил)-N-(1-метил-1H-пиразол-4-ил)пиримидин-2-амин (84)
Смесь соединения 84с (50 мг, 0,147 ммоль), 1-метил-1Н-пиразол-4-амина (21 мг, 0,122 ммоль) и pTsOH (55 мг, 0,284 ммоль) в изопропаноле (2 мл) нагревали до 100°С и перемешивали в течение 4 ч. После охлаждения до комнатной температуры, смесь концентрировали досуха в условиях вакуума, и очищали остаток методом преп-HPLC с получением указанного в заголовке соединения 84 (16,4 мг, твердое вещество, 28%).
MS m/z (ESI): 402 [M+1]
1H-ЯМР (400 МГц, DMSO-d6) δ 9,06 (с, 1H), 7,89 (с, 1H), 7,70 (с, 1H), 7,45 (с, 1H), 3,98 (д, J=10,9 Гц, 2Н), 3,77 (с, 3Н), 3,56 (д, J=11,2 Гц, 2Н), 2,82 (с, 2Н), 2,32 (д, J=63,4 Гц, 4Н), 1,07 (с, 6Н), 0,79 (с, 1H), 0,41 (д, J=6,1 Гц, 2Н), 0,05 (с, 2Н).
Пример 85. 2-((3aR,6aS)-5-(5-Хлор-2-((1-метил-1Н-пиразол-4-ил)амино)пиримидин-4-ил)-3а,6а-диметилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)ацетонитрил
Смесь соединения 12с (42 мг, 0,115 ммоль), 2-бромацетонитрила (30 мг, 0,23 ммоль), TEA (50 мг, 0,46 ммоль) в THF (1 мл) перемешивали в течение 2 ч, а затем концентрировали досуха в условиях вакуума. Остаток очищали методом преп-HPLC с получением указанного в заголовке соединения 85 (16 мг, твердое вещество, 34%).
MS m/z (ESI): 402 [M+1]
1H-ЯМР (400 МГц, DMSO-d6) δ 9,07 (с, 1H), 7,89 (с, 1H), 7,71 (с, 1H), 7,44 (с, 1H), 4,00 (д, J=11,3 Гц, 2Н), 3,80 (с, 2Н), 3,77 (с, 3Н), 3,56 (д, J=11,3 Гц, 2Н), 2,85 (д, J=9,2 Гц, 2Н), 2,55 (д, J=9,3 Гц, 2Н), 1,09 (с, 6Н).
Пример 86. ((3aS,6aR)-5-(5-Хлор-2-((1-метил-1Н-пиразол-4-ил)амино)пиримидин-4-ил)-3а-метилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)(циклопропил)метанон
Стадия 1. (S)-1-Фенил-N-((триметилсилил)метил)этан-1-амин (86b)
Смесь (хлорметил)триметилсилана (20,24 г, 165 ммоль), соединения 86а (20 г, 165 ммоль) и TEA (20 г, 165 ммоль) нагревали до температуры возгонки в течение 16 ч. После охлаждения до комнатной температуры, к смеси добавляли н-гептан (500 мл), перемешивали и фильтровали. Фильтрат концентрировали досуха в условиях вакуума, и очищали остаток методом колоночной хроматографии на силикагеле (петролейный эфир/этилацетат = 1/20) с получением указанного в заголовке соединения 86b (26 г, 76%).
MS m/z (ESI): 208 [M+1]
Стадия 2. (S)-N-(метоксиметил)-1-фенил-N-((триметилсилил)метил)этан-1-амин (86с)
К 37% водному раствору формальдегида (6,6 г, 81,49 ммоль) при 0°С добавляли соединение 86b (13,0 г, 62,68 ммоль). Смесь перемеш ивали при 0°С в течение 0,5 ч, а затем добавляли МеОН (3,4 г, 106,56 ммоль) и карбонат калия (8,7 г, 62,68 ммоль). Смесь перемешивали при 0°С дополнительно в течение 2 ч, добавляли воду (50 мл) и экстрагировали этилацетатом (3×50 мл). Объединенную органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали досуха в условиях вакуума. Остаток очищали путем перегонки с получением указанного в заголовке соединения 86с (6,816 г, 43%).
1H-ЯМР (400 МГц, DMSO-d6) δ 7,35 - 7,30 (м, 4Н), 7,23 (с, 1H), 4,18 (д, J=9,3 Гц, 1Н), 3,99 (д, J=9,3 Гц, 1Н), 3,90 (д, J=6,8 Гц, 1Н), 3,14 (с, 3Н), 2,06 (кв, J=14,4 Гц, 2Н), 1,33 (д, J=6,8 Гц, 3Н), 0,00 (с,9Н).
Стадия 3. цис-3а-метил-5-((R)-1-фенилэтил)тетрагидро-1Н-фуро[3,4-с]пиррол-1,3(3аН)-дион (86d)
К раствору соединения 86с (6,62 г, 26,34 ммоль) в CH2Cl2 (25 мл) при 0°С добавляли раствор 3-метилфуран-2,5-диона (2,95 г, 26,34 ммоль) в CH2Cl2 (10 мл), а затем по каплям добавляли раствор TFA (0,3 г, 2,63 ммоль) в CH2Cl2 (5 мл). Полученную смесь перемешивали в течение 6 ч, а затем концентрировали досуха в условиях вакуума с получением указанного в заголовке соединения 86d (6,83 г). Неочищенный продукт использовали непосредственно на следующей стадии без дополнительной очистки.
MS m/z (ESI): 260 [M+1]
Стадия 4. цис-3а-Метил-5-((R)-1-фенилэтил)тетрагидропирроло[3,4-с]пиррол-1,3(2Н,3аН)-дион (86е)
К раствору соединения 86d (6,83 г, 26,34 ммоль) в THF (30 мл) добавляли гидроксид аммония (30 мл). Полученную смесь нагревали до 100°С в герметизированной пробирке и перемешивали в течение 3 ч. После охлаждения до комнатной температуры, смесь концентрировали досуха в условиях вакуума. Остаток очищали методом колоночной хроматографии на силикагеле (петролейный эфир/этилацетат = 1/2) с получением указанного в заголовке соединения 86е (4,30 г, 63%).
MS m/z (ESI): 259 [M+1]
Стадия 5. цис-3а-Метил-2-((R)-1-фенилэтил)октагидропирроло[3,4-с]пиррол (86f)
К раствору соединения 86е (4,3 г, 16,65 ммоль) в THF (80 мл) при 0°С порциями добавляли LAH (1,90 г, 49,95 ммоль). Смесь нагревали до 70°С и перемешивали в течение 2 ч. После охлаждения до 0°С, смесь гасили добавлением насыщенного раствора сульфата натрия (20 мл) и фильтровали. Фильтрат концентрировали досуха в условиях вакуума с получением указанного в заголовке соединения 86f (3,62 г, 94%).
MS m/z(ESI): 231 [М+1]
Стадия 6. трет-Бутил-(3aS,6aR)-3а-метил-5-((R)-1-фенилэтил)-гексагидропирроло[3,4-с]пиррол-2(1Н)-карбоксилат (86g) и трет-бутил-(3aR,6aS)-3a-метил-5-((R)-1-фенилэтил)гексагидропирроло[3,4-с]пиррол-2(1Н)-карбоксилат (86h)
К раствору соединения 86f (3,62 г, 15,73 ммоль) в CH2Cl2 (40 мл) при 0°С добавляли TEA (3,18 г, 31,46 ммоль) и раствор ди-трет-бутилпирокарбоната (4,12 г, 18,88 ммоль) в CH2Cl2 (10 мл). Смесь перемешивали в течение 1 ч, добавляли воду (20 мл) и экстрагировали CH2Cl2 (3×50 мл). Объединенную органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали досуха в условиях вакуума. Остаток очищали методом колоночной хроматографии на силикагеле (петролейный эфир/этилацетат = 20/1) с получением указанных в заголовке соединения 86g (1,89 г) и соединения 86h (1,65 г), а также смеси соединения 86g и соединения 86h (1,04 г). Общий выход соединения 86g и соединения 86h составил 88%.
трет-Бутил-(3aS,6aR)-За-метил-5-((R)-1-фенилэтил)гексагидропирроло[3,4-с]пиррол-2(1Н)-карбоксилат 86g:
MS m/z (ESI): 331 [M+1]
1H-ЯМР (400 МГц, DMSO-d6) δ 7,29 (д, J=4,5 Гц, 4H), 7,21 (д, J=4,2 Гц, 1H), 3,57 -3,48 (м,1Н), 3,25 (д, J=10,8 Гц, 1H), 3,22 - 3,14 (м, 2Н), 3,06 (д, J=10,8 Гц, 1H), 2,54 (дд, J=5,5, 3,8 Гц, 2Н), 2,35 (д, J=9,1 Гц, 1Н), 2,18 (с, 1Н), 2,12 (д, J=9,1 Гц, 1H), 1,40 (с, 9Н), 1,24 (д, J=6,5 Гц, 3Н), 1,08 (с, 3Н).
трет-Бутил-(3aR,6aS)-3а-метил-5-((R)-1-фенилэтил)гексагидропирроло[3,4-с]пиррол-2(1Н)-карбоксилат 86h:
MS m/z (ESI): 331 [M+1]
1H-ЯМР (400 МГц, DMSO-d6) δ 7,29 (д, J=4,4 Гц, 4Н), 7,21 (д, J=4,3 Гц, 1H), 3,49 -3,41 (м, 1H), 3,34 (д, J=10,9 Гц, 1H), 3,20 (д, J=6,6 Гц, 1Н), 3,10 (дд, J=11,3, 3,5 Гц, 1H), 3,06 (д, J=11,0 Гц, 1Н), 2,65 (с, 1H), 2,57 (д, J=8,9 Гц, 1H), 2,18 (д, J=3,5 Гц, 1H), 2,16 (с, 2Н), 1,39 (с, 9Н), 1,24 (д, J=6,5 Гц, 3Н), 1,11 (с, 3Н).
Стадия 7. трет-Бутил-(3aS,6aR)-3а-метилгексагидропирроло[3,4-с]пиррол-2(1Н)-карбоксилат (86i)
К раствору соединения 86g (1,89 г, 5,72 ммоль) в МеОН (50 мл) добавляли 10% Pd/C (0,19 г). Смесь перемешивали в атмосфере водорода в течение 12 ч, а затем концентрировали досуха в условиях вакуума с получением указанного в заголовке соединения 86i (1,30 г, 100%).
MS m/z (ESI): 227 [M+1]
Стадия 8. трет-Бутил-(3aS,6aR)-5-(2,5-дихлорпиримидин-4-ил)-3а-метилгексагидропирроло[3,4-с]пиррол-2(1Н)-карбоксилат (86j)
К раствору соединения 86i (1,3 г, 5,73 ммоль) в MeCN (40 мл) добавляли DIEA (1,48 г, 11,46 ммоль) и 2,4,5-трихлорпиримидин (1,05 г, 5,73 ммоль). Смесь перемешивали в течение 2 ч и концентрировали досуха в условиях вакуума. Остаток очищали методом колоночной хроматографии на силикагеле (петролейный эфир/этилацетат = 7/3) с получением указанного в заголовке соединения 86j (1,83 г, 86%).
MS m/z (ESI): 373 [M+1]
Стадия 9. 5-Хлор-N-(1-метил-1Н-пиразол-4-ил)-4-((3aR,6aS)-3а-метилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)пиримидин-2-амин (86k)
К раствору соединения 86j (1,83 г, 4,91 ммоль) в изопропаноле (30 мл) добавляли 1-метил-1Н-пиразол-4-амина гидрохлорид (656 мг, 4,91 ммоль) и pTsOH (93 мг, 0,49 ммоль). Смесь нагревали до 100°С и перемешивали в течение 12 ч. После охлаждения до комнатной температуры, реакционную смесь фильтровали, и собирали осадок на фильтре с получением указанного в заголовке соединения 86k (1,28 г, 82%) в виде гидрохлорида.
MS m/z (ESI): 334 [M+1]
Стадия 10. ((3aS,6aR)-5-(5-Хлор-2-((1-метил-1Н-пиразол-4-ил)амино)пиримидин-4-ил)-3а-метилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)(циклопропил)метанон (86)
К раствору соединения 86k (100 мг, 0,27 ммоль) в CH2Cl2 (5 мл) добавляли циклопропанкарбоновую кислоту (23 мг, 0,27 ммоль) и DIEA (175 мг, 1,35 ммоль). Смесь перемешивали в течение 10 мин, а затем добавляли HATU (205 мг, 0,54 ммоль). Смесь перемешивали дополнительно в течение 2 ч, добавляли воду (5 мл) и экстрагировали этилацетатом (3×5 мл). Объединенную органическую фазу концентрировали досуха в условиях вакуума, и очищали остаток методом преп-HPLC с получением указанного в заголовке соединения 86 (64,3 мг, твердое вещество, 59%).
MS m/z (ESI): 402 [M+1]
Удельное вращение  =-9,2°×дм2/кг
=-9,2°×дм2/кг
1H-ЯМР (400 МГц, DMSO-d6) δ 9,06 (с, 1H), 7,89 (с, 1H), 7,71 (с, 1H), 7,44 (с, 1H), 4,04 (с, 1H), 3,95 (дд, J=10,7, 7,7 Гц, 1H), 3,88 - 3,80 (м, 1H), 3,78 (с, 3Н), 3,75 - 3,69 (м, 1H), 3,65 (дд, J=13,6, 6,6 Гц, 1Н), 3,58 (дд, J=12,7, 8,0 Гц, 1Н), 3,44 (д, J=12,2 Гц, 1H), 3,28 (с, 1Н), 2,69 - 2,60 (м, 1H), 1,73 (дд, J=13,0, 6,2 Гц, 1H), 1,23 (д, J=12,9 Гц, 3Н), 0,77 - 0,66 (м, 4Н).
Примеры 87, 88, 89, 90 и 91 синтезировали в соответствии с методикой, описанной для Примера 86, за исключением того, что на стадии 7 различные соединения использовали вместо 86g, и на стадии 10 различные соединения использовали вместо циклопропанкарбоновой кислоты.
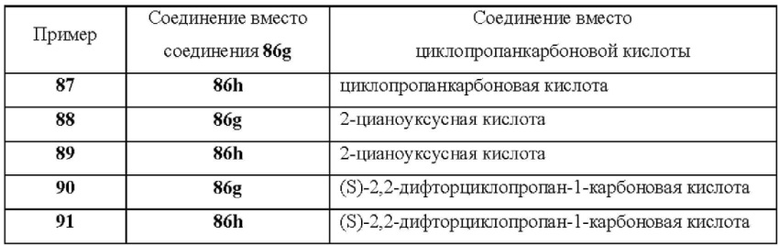
Характеристические данные Примеров 87, 88, 89, 90 и 91 представлены ниже.
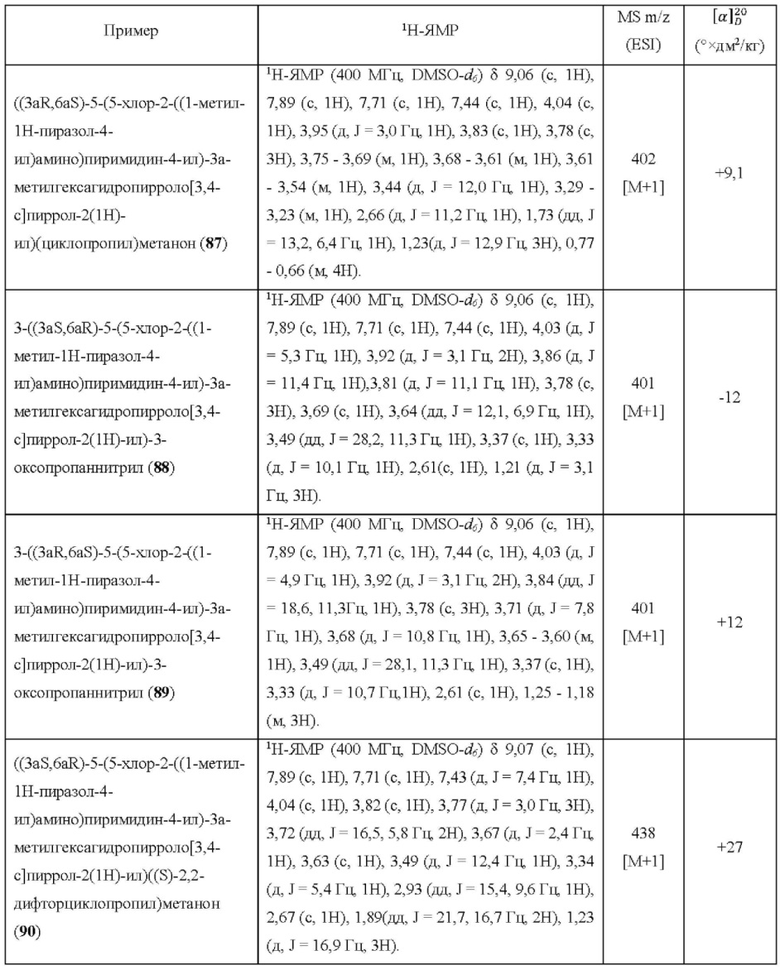

Пример 92. цис-2-(5-(5-Хлор-2-((1-метил-1Н-пиразол-4-ил)амино)пиримидин-4-ил)-3а,6а-диметил-1-оксогексагидропирроло[3,4-с]пиррол-2(1Н)-ил)ацетонитрил

Стадия 1. 2-((3aR,6aS)-5-Бензил-3а,6а-диметил-1,3-диоксогексагидропирроло[3,4-с]пиррол-2(1Н)-ил)ацетонитрил (92а)
К смеси соединения 51b (1,5 г, 5,4 ммоль) и 2-аминоацетонитрила (500 мг, 5,4 ммоль) в 1,2-дихлорэтане (6 мл) добавляли TEA (1,6 г, 16,2 ммоль). Полученный раствор нагревали до 60°С и перемешивали в течение 12 ч. После охлаждения до комнатной температуры, смесь концентрировали досуха в условиях вакуума, и очищали остаток методом преп-HPLC с получением указанного в заголовке соединения 92а (1 г, 62%).
MS m/z (ESI): 298 [M+1]
Стадия 2. цис-2-(5-Бензил-1-гидрокси-3a,6а-диметил-3-оксогексагидропирроло[3,4-с]пиррол-2(1Н)-ил)ацетамид (92b)
К раствору соединения 92а (800 мг, 2,7 ммоль) в EtOH (10 мл) при 0°С добавляли боргидрид натрия (260 мг, 6,7 ммоль). Смесь нагревали до температуры возгонки в течение 12 ч. После охлаждения до комнатной температуры, остаток концентрировали досуха в условиях вакуума. Остаток разбавляли CH2Cl2 (10 мл) и фильтровали. Фильтрат концентрировали досуха в условиях вакуума с получением указанного в заголовке соединения 92b (900 мг). Неочищенный продукт использовали непосредственно на следующей стадии без дополнительной очистки.
MS m/z (ESI): 318 [М+1]
Стадия 3. цис-2-(5-Бензил-3а,6а-диметил-1-оксогексагидропирроло[3,4-с]пиррол-2(1Н)-ил)ацетамид (92с)
К раствору соединения 92b (900 мг, неочищенный продукт) в TFA (8 мл) добавляли триэтилсилан (4 мл). Раствор нагревали до 120°С в герметизированной пробирке и перемешивали в течение 48 ч. После охлаждения до комнатной температуры, раствор концентрировали досуха в условиях вакуума. Остаток очищали методом преп-HPLC с получением указанного в заголовке соединения 92с (130 мг, 16% после двух стадий).
MS m/z (ESI): 302 [M+1]
Стадия 4. цис-2-(3а,6а-Диметил-1-оксогексагидропирроло[3,4-с]пиррол-2(1Н)-ил)ацетамид (92d)
К раствору соединения 92 с (130 мг, 0,42 ммоль) в МеОН (5 мл) добавляли 10% Pd/C (50 мг). Смесь перемешивали в атмосфере водорода в течение 3 ч, а затем фильтровали. Фильтрат концентрировали досуха в условиях вакуума с получением указанного в заголовке соединения 92d (90 мг, 100%).
MS m/z(ESI): 212 [М+1]
Стадия 5. цис-2-(5-(2,5-Дихлорпиримидин-4-ил)-3а,6а-диметил-1-оксогексагидропирроло[3,4-с]пиррол-2(1Н)-ил)ацетамид (92е)
Смесь соединения 92d (90 мг, 0,42 ммоль), 2,4,5-трихлорпиримидина (91 мг, 0,5 ммоль) и DIEA (0,3 мл) в MeCN (2 мл) нагревали до 50°С и перемешивали в течение 12 ч. После охлаждения до комнатной температуры, смесь концентрировали досуха в условиях вакуума, и очищали остаток методом преп-HPLC с получением указанного в заголовке соединения 92е (150 мг, 99%).
MS m/z (ESI): 358 [M+1]
Стадия 6. цис-2-(5-(5-Хлор-2-((1-метил-1H-пиразол-4-ил)амино)пиримидин-4-ил)-3а,6а-диметил-1-оксогексагидропирроло[3,4-с]пиррол-2(1Н)-ил)ацетамид (92f)
Смесь соединения 92е (100 мг, 0,26 ммоль), 1-метил-1H-пиразол-4-амина гидрохлорида (40 мг, 0,29 ммоль) и pTsOH (6 мг, 0,026 ммоль) в изопропаноле (4 мл) нагревали до 120°С и перемешивали в течение 1 ч. После охлаждения до комнатной температуры, смесь концентрировали досуха в условиях вакуума с получением указанного в заголовке соединения 92f (150 мг). Неочищенный продукт использовали непосредственно на следующей стадии без дополнительной очистки.
MS m/z (ESI): 419 [M+1]
Стадия 7. цис-2-(5-(5-Хлор-2-((1-метил-1Н-пиразол-4-ил)амино)пиримидин-4-ил)-3а,6а-диметил-1-оксогексагидропирроло[3,4-с]пиррол-2(1Н)-ил)ацетонитрил (92)
Смесь соединения 92f (150 мг, неочищенный продукт) в оксиде треххлористого фосфора (2 мл) нагревали до 100°С в течение 12 ч при перемешивании. После охлаждения до комнатной температуры, смесь концентрировали досуха в условиях вакуума и растворяли в MeCN (5 мл), к которому медленно добавляли TEA до рН=7. Смесь концентрировали досуха в условиях вакуума, и очищали остаток методом преп-HPLC с получением указанного в заголовке соединения 92 (15 мг, твердое вещество, 14% после двух стадий).
MS m/z (ESI): 401 [M+1]
1H-ЯМР (400 МГц, DMSO-d6) δ 9,11 (ушир. с, 1H), 7,91 (с, 1Н), 7,70 (с, 1Н),7,44 (с, 1Н), 4,44 (с, 2Н), 4,15 (д, J=11,4 Гц, 1Н), 3,81 (д, 1=11,8 Гц, 1Н), 3,78 (с, 3Н), 3,70 (д, 1=11,8 Гц, 1Н), 3,65 (д, J=12,1 Гц, 1H), 3,43 (д, J=10,0 Гц, 1H), 3,34 (д, J=4,8 Гц, 1H), 1,18 (с, 3Н), 1,09 (с, 3Н).
Пример 93. ((3aR,6aS)-5-(5-хлор-2-((5-метилизоксазол-3-ил)амино)пиримидин-4-ил)-3а,6а-диметилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)(циклопропил)метанон
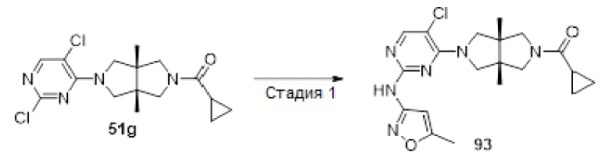
К раствору соединения 51g (0,25 г, 0,71 ммоль) и 5-метилизоксазол-3-амина (83 мг, 0,85 ммоль) в изопропаноле (6 мл) добавляли pTsOH (270 мг, 1,42 ммоль). Смесь перемешивали при 100°С в течение 18 ч, а затем охлаждали до комнатной температуры. Смесь концентрировали досуха в условиях вакуума, и очищали остаток методом преп-HPLC с получением указанного в заголовке соединения 93 (51,3 мг, твердое вещество, 17%).
MS m/z (ESI): 417 [M+1]
1H-ЯМР (400 МГц, DMSO-d6) δ 9,91 (с, 1Н), 7,97 (с, 1H), 6,65 (с, 1Н), 3,92 - 3,66 (м, 5Н), 3,59 (д, J=10,7 Гц, 1Н), 3,47 (д, J=12,3 Гц, 1Н), 3,29 (д, J=12,5 Гц, 1H), 2,36 (с, 3Н), 1,76 - 1,64 (м, 1H), 1,10 (д, J=12,9 Гц, 6Н), 0,77 - 0,65 (м, 4Н).
Примеры 94 и 95 синтезировали в соответствии с методикой, описанной для Примера 93, за исключением того, что на стадии 1 различные соединения использовали вместо 5-метилизоксазол-3-амина.
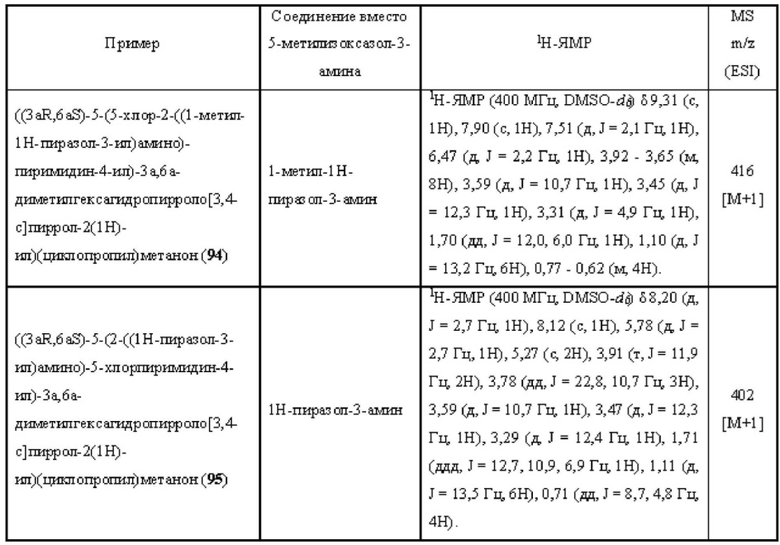
Пример 97. 3-((3aR,6aS)-5-(5-Метокси-2-((1-метил-1H-пиразол-4-ил)амино)пиримидин-4-ил)-3а,6а-диметилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)-3-оксопропаннитрил
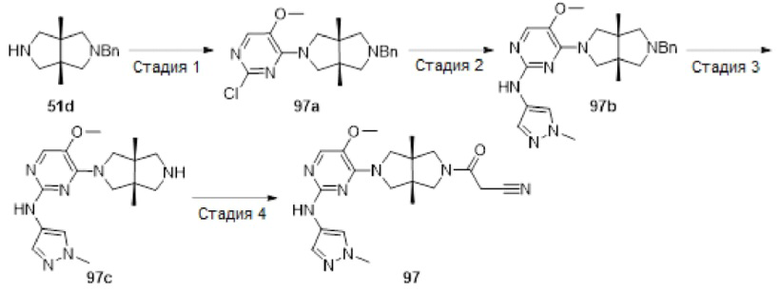
Стадия 1. (3aR,6aS)-2-Бензил-5-(2-хлор-5-метоксипиримидин-4-ил)-3а,6а-диметилоктагидропирроло[3,4-с]пиррол (97а)
К раствору соединения 51d (0,26 г, 1,13 ммоль) в MeCN (10 мл) добавляли карбонат калия (0,47 г, 3,39 ммоль) и 2,4-дихлор-5-метоксипиримидин (0,263 мг, 1,47 ммоль). Смесь перемешивали в течение 2 ч, а затем концентрировали досуха в условиях вакуума. Остаток очищали методом колоночной хроматографии на силикагеле (петролейный эфир/этилацетат = 100/0 - 0/100) с получением указанного в заголовке соединения 97а (70 мг, 20%).
MS m/z (ESI): 373 [M+1]
Стадия 2. 4-((3aR,6aS)-5-Бензил-3а,6а-диметилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)-5-метокси-N-(1-метил-1H-пиразол-4-ил)пиримидин-2-амин (97b)
Смесь соединения 97а (70 мг, 0,188 ммоль), 1-метил-1H-пиразол-4-амина (27,3 мг, 0,182 ммоль) и pTsOH (71,4 мг, 0,376 ммоль) в н-бутаноле (5 мл) перемешивали при 120°С в течение 8 ч. После охлаждения до комнатной температуры, смесь концентрировали досуха в условиях вакуума, и очищали остаток методом колоночной хроматографии на силикагеле (CH2Cl2/МеОН=9/1) с получением указанного в заголовке соединения 97b (70 мг, 86%).
MS m/z (ESI): 434 [M+1]
Стадия 3. 4-((3aR,6aS)-3а,6а-Диметилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)-5-метокси-N-(1-метил-1Н-пиразол-4-ил)пиримидин-2-амин (97с)
К раствору соединения 97b (70 мг, 0,16 ммоль) в МеОН (5 мл) добавляли 10% Pd/С (34 мг, 0,32 ммоль). Смесь перемешивали в атмосфере водорода в течение 3 ч, а затем фильтровали. Фильтрат концентрировали досуха в условиях вакуума с получением указанного в заголовке соединения 97с (50 мг, 91%).
MS m/z (ESI): 344 [M+1]
Стадия 4. 3-((3aR,6aS)-5-(5-Метокси-2-((1-метил-1H-пиразол-4-ил)амино)-пиримидин-4-ил)-3а,6а-диметилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)-3-оксопропаннитрил (97)
К смеси соединения 97с (50 мг, 0,146 ммоль), 2-цианоуксусной кислоты (25 мг, 0,292 ммоль), DIEA (38 мг, 0,292 ммоль) в DMF (1 мл) добавляли HATU (67 мг, 0,175 ммоль). Смесь перемешивали в течение 1 ч, а затем очищали методом преп-HPLC с получением указанного в заголовке соединения 97 (31,9 мг, твердое вещество, 53%).
MS m/z (ESI): 411[М+1]
1H-ЯМР (400 МГц, DMSO-d6) δ 8,56 (с, 1H), 7,70 (с, 1H), 7,67 (с, 1H), 7,41 (с, 1H), 3,91 (д, J=1,1 Гц, 2Н), 3,82 - 3,71 (м, 5Н), 3,66 (с, 3Н), 3,63 - 3,52 (м, 3Н), 3,47 (д, J=12,2 Гц, 1Н), 3,39 (д, J=10,5 Гц, 1Н), 3,31 (д, J=5,3 Гц, 1H), 1,07 (т, J=7,5 Гц, 6Н).
Пример 98. 3-((3aS,6aR)-5-(2-((1H-пиразол-4-ил)амино)-5-хлорпиримидин-4-ил)-3а-метилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)-3-оксопропаннитрил
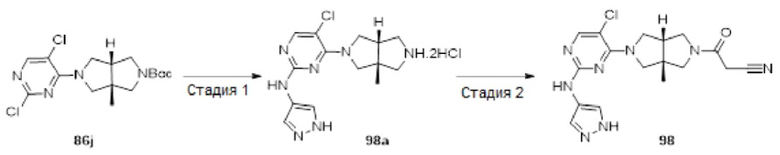
Стадия 1. 5-Хлор-4-((3aR,6aS)-3а-метилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)-N-(1Н-пиразол-4-ил)пиримидин-2-амина дигидрохлорид (98а)
К раствору соединения 86j (4,00 г, 10,72 ммоль) в EtOH (50 мл) добавляли 1H-пиразол-4-амин (1,16 г, 13,94 ммоль) и pTsOH моногидрат (0,21 г, 1,07 ммоль). Смесь нагревали до 70°С и перемешивали в течение 16 ч. После охлаждения до комнатной температуры, смесь концентрировали досуха в условиях вакуума, и добавляли раствор HCl (10н в этаноле, 50 мл). Смесь нагревали до 50°С и перемешивали дополнительно в течение 1 ч. После охлаждения до комнатной температуры, смесь концентрировали досуха в условиях вакуума, и добавляли к ней смесь метанола и 2-метокси-2-метилпропана (60 мл, 1/1 об./об.). Полученную смесь перемешивали в течение 30 мин, а затем фильтровали. Осадок на фильтре собирали и сушили с получением указанного в заголовке соединения 98а (3,4 г, 81%) в виде гидрохлорида.
MS m/z (ESI): 320 [M+1]
Стадия 2. 3-((3aS,6aR)-5-(2-((1Н-пиразол-4-ил)амино)-5-хлорпиримидин-4-ил)-3а-метилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)-3-оксопропаннитрила гемиформиат (98)
К раствору соединения 98а (3,30 г, 8,40 ммоль) в метаноле (40 мл) добавляли этилцианоацетат (1,90 г, 16,80 ммоль) и 1,8-диазабицикло[5.4.0]ундец-7-ен (5,10 г, 33,60 ммоль). Смесь перемешивали в течение 5 ч и концентрировали досуха в условиях вакуума. Остаток очищали методом преп-HPLC с получением указанного в заголовке соединения 98 (1,04 г, твердое вещество, 30%) в виде формиата.
MS m/z (ESI): 387 [M+1]
Удельное вращение 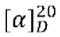 =-4,6°×дм2/кг
=-4,6°×дм2/кг
1H-ЯМР (400 МГц, DMSO-d6) δ 12,50 (с, 1,5Н), 9,07 (с, 1Н), 8,13 (с, 0,5Н), 7,89 (с, 1Н), 7,65 (с, 2Н), 4,09 - 3,99 (м, 1H), 3,92 (д, J=2,7 Гц, 2Н), 3,84 (дд, J=26,5, 11,3 Гц, 1H), 3,76 - 3,72 (м, 1Н), 3,69 (д, J=11,1 Гц, 1H), 3,64 (дд, J=12,5, 7,3 Гц, 1Н), 3,49 (дд, J=22,9, 11,3 Гц, 1H), 3,38 (дд, J=15,7, 7,9 Гц, 1H), 3,29 (д, J=12,3 Гц, 1H), 2,57 (д, J=33,9 Гц, 1H), 1,21 (д, J=2,7 Гц, 3Н).
Пример 99. 3-((3aS,6aR)-5-(5-Хлор-2-((1-(метил-d3)-1Н-пиразол-4-ил)амино)-пиримидин-4-ил)-3а-метилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)-3-оксопропаннитрил
Стадия 1. 1-(Метил-d3)-4-нитро-1Н-пиразол (99b)
К раствору 4-нитро-1H-пиразола 99а (2,5 г, 22,11 ммоль) в MeCN (20 мл) добавляли йодметан-d3 (3,85 г, 26,53 ммоль) и карбонат калия (4,58 г, 33,17 ммоль). Смесь нагревали до 70°С и перемешивали в течение 12 ч. После охлаждения до комнатной температуры, смесь концентрировали досуха в условиях вакуума. К остатку добавляли воду (50 мл) и экстрагировали CH2Cl2 (3×50 мл). Объединенную органическую фазу сушили над безводным сульф атом натрия, фильтровали и концентрировали досуха в условиях вакуума. Остаток очищали методом колоночной хроматографии на силикагеле (петролейный эфир/этилацетат = 4/1) с получением указанного в заголовке соединения 99b (2,50 г, 87%).
MS m/z (ESI): 131 [M+1]
Стадия 2. 1-(Метил-d3)-1Н-пиразол-4-амин (99с)
К раствору соединения 99b (2,50 г, 19,2 ммоль) в МеОН (30 мл) добавляли 10% Pd/С (0,5 г). Смесь перемешивали в атмосфере водорода в течение 16 ч, а затем фильтровали. Фильтрат концентрировали досуха в условиях вакуума с получением указанного в заголовке соединения 99с (1,92 г, 100%).
MS m/z (ESI): 101 [M+1]
Стадия 3. 5-Хлор-N-(1-(метил-d3)-1H-пиразол-4-ил)-4-((3aR,6aS)-3а-метилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)пиримидин-2-амина дигидрохлорид (99d)
К раствору соединения 86j (3,00 г, 8,04 ммоль) в EtOH (50 мл) добавляли соединение 99 с (1,05 г, 10,45 ммоль) и pTsOH моногидрат (0,15 г, 0,80 ммоль). Смесь нагревали до 70°С и перемешивали в течение 8 ч. После охлаждения до комнатной температуры, к смеси добавляли этанольный раствор HCl (10н, 50 мл). Смесь снова нагревали до 50°С и перемешивали в течение 1 ч. После охлаждения до комнатной температуры, смесь концентрировали досуха в условиях вакуума. К остатку добавляли смесь метанола и 2-метокси-2-метилпропана (60 мл, 1/1 об./об.). После перемешивания в течение 30 мин, смесь фильтровали. Осадок на фильтре собирали и сушили с получением указанного в заголовке соединения 99d (3,3 г, 100%) в виде гидрохлорида.
MS m/z (ESI): 337 [M+1]
Стадия 4. 3-((3aS,6aR)-5-(5-Хлор-2-((1-(метил-d3)-1H-пиразол-4-ил)амино)-пиримидин-4-ил)-3а-метилгексагидропирроло[3,4-с]пиррол-2(1H)-ил)-3-оксопропаннитрил (99)
К смеси соединения 99d (3,30 г, 8,05 ммоль) в MeCN (35 мл) добавляли этилцианоацетат (1,82 г, 16,10 ммоль) и DBU (4,90 г, 32,2 ммоль). Смесь нагревали до 30°С и перемешивали в течение 4 ч. Смесь концентрировали досуха в условиях вакуума, а затем добавляли EtOH (20 мл). После перемешивания в течение 1 ч, смесь фильтровали, и собирали осадок на фильтре с получением указанного в заголовке соединения 99 (2,60 г, твердое вещество, 80%).
MS m/z (ESI): 404 [M+1]
Удельное вращение 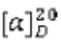 =-7,2°×дм2/кг
=-7,2°×дм2/кг
1H-ЯМР (400 МГц, DMSO-d6) δ 9,07 (с, 1H), 7,89 (с, 1H), 7,71 (с, 1H), 7,44 (с, 1H), 4,03 (дд, J=11,2, 5,9 Гц, 1H), 3,93 (д, J=3,0 Гц, 2Н), 3,84 (дд, J=18,6, 11,5 Гц, 1H), 3,73 (д, J=10,2 Гц, 1H), 3,69 (с, 1H), 3,64 (дд, J=12,1,7,1 Гц, 1Н), 3,49 (дд, J=27,8, 11,2 Гц, 1Н), 3,42 - 3,33 (м, 1H), 3,33 - 3,24 (м, 1Н), 2,57 (д, J=31,9 Гц, 1Н), 1,20 (т, J=8,1 Гц, 3Н).
Пример 100. транс-3-(5-(5-Хлор-2-((1-метил-1H-пиразол-4-ил)амино)пиримидин-4-ил)-3а-метилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)-3-оксопропаннитрил
Стадия 1. Диметил-транс-1-бензил-3-метилпирролидин-3,4-дикарбоксилат (100b)
К смеси диметил-2-метилфумарата 100а (9,63 г, 60,89 ммоль) и TFA (0,69 г, 6,09 ммоль) в CH2Cl2 (10 мл) при 0°С медленно добавляли раствор N-бензил-1-метокси-N-((триметилсилил)метил)метанамина (17,35 г, 73,07 ммоль) в CH2Cl2 (200 мл). После добавления, полученный желтый раствор постепенно нагревали до комнатной температуры в течение 16 ч, а затем добавляли насыщенный водный раствор бикарбоната натрия (200 мл) и CH2Cl2 (200 мл). Смесь перемешивали в течение 15 мин. Разделенную органическую фазу промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали досуха в условиях вакуума с получением остатка, который очищали методом колоночной хроматографии на силикагеле (CH2Cl2/МеОН=100/3) с получением указанного в заголовке соединения 100b (17,74 г, 100%).
MS m/z (ESI): 292 [M+1]
Стадия 2. 1-(трет-Бутил)-3,4-диметил-транс-3-метилпирролидин-1,3,4-трикарбоксилат(100с)
К смеси соединения 100b (17,74 г, 60,79 ммоль) и ди-трет-бутилпирокарбоната (14,62 г, 66,67 ммоль) в МеОН (200 мл) добавляли 10% Pd/C (1,8 г). Полученную смесь нагревали до 50°С в атмосфере водорода и перемешивали в течение 20 ч. После охлаждения до комнатной температуры, смесь фильтровали, и концентрировали фильтрат досуха в условиях вакуума. Остаток очищали методом колоночной хроматографии на силикагеле (петролейный эфир/этилацетат = 1/1) с получением указанного в заголовке соединения 100с (18,00 г, 98%).
MS m/z (ESI): 202 [M+1-100]
Стадия 3. трет-Бутил-транс-3,4-бис(гидроксиметил)-3-метилпирролидин-1-карбоксилат (100d)
К раствору соединения 100 с (18,00 г, 22,75 ммоль) в THF (200 мл) при 0°С добавляли раствор боргидрида лития (2 M в THF, 65 мл, 131,41 ммоль). После перемешивания в течение 30 мин, смесь постепенно нагревали до комнатной температуры и перемешивали дополнительно в течение 16 ч. Смесь снова охлаждали до 0°С, и медленно добавляли 1н HCl до рН=1. Полученный раствор экстрагировали CH2Cl2 (400 мл). Органическую фазу промывали водой и солевым раствором в указанной последовательности, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали досуха в условиях вакуума, и очищали остаток методом колоночной хроматографии на силикагеле (петролейный эфир/этилацетат = 2/1) с получением указанного в заголовке соединения 100d (14,00 г, 96%).
MS m/z (ESI): 190 [M+1-56]
Стадия 4. трет-Бутил-транс-3-метил-3,4-бис(((метилсульфонил)окси)метил)-пирролидин-1-карбоксилат (100е)
К раствору соединения 100d (14,00 г, 57,07 ммоль) и DIEA (44,00 г, 342,41 ммоль) в CH2Cl2 (200 мл) при 0°С по каплям добавляли MsCl (20,00 г, 171,21 ммоль). Смесь перемешивали в течение 1 ч, а затем добавляли насыщенный водный раствор хлорида аммония (100 мл) и CH2Cl2 (200 мл). Смесь перемешивали в течение 10 мин и оставляли отстаиваться. Разделенный органический слой промывали насыщенным солевым раствором и насыщенным раствором бикарбоната натрия в указанной последовательности, сушили над безводным сульфатом натрия, фильтровали и концентрировали досуха в условиях вакуума. Остаток очищали методом колоночной хроматографии на силикагеле (петролейный эфир/этилацетат = 10/1) с получением указанного в заголовке соединения 100е (8,00 г, 35%).
MS m/z (ESI): 346 [M+1-56]
Стадия 5. трет-Бутил-транс-5-бензил-3а-метилгексагидропирроло[3,4-с]пиррол-2(1Н)-карбоксилат (100f)
Смесь соединения 100е (8,00 г, 19,93 ммоль), TEA (12,10 г, 119,55 ммоль) и бензиламина (8,54 г, 79,72 ммоль) в толуоле (100 мл) нагревали до температуры возгонки в течение 16 ч. После охлаждения до комнатной температуры, к реакционной смеси добавляли 1н NaOH (100 мл) и насыщенный солевой раствор. Смесь перемешивали в течение 10 мин, и сушили разделенную органическую фазу над безводным сульфатом натрия. После фильтрования, фильтрат концентрировали досуха в условиях вакуума, и очищали остаток методом колоночной хроматографии на силикагеле (петролейный эфир/этилацетат = 5/1) с получением указанного в заголовке соединения 100f (3,00 г, 48%).
MS m/z(ESI): 317 [М+1]
Стадия 6. трет-Бутил-транс-3а-метилгексагидропирроло[3,4-с]пиррол-2(1Н)-карбоксилат(100g)
К раствору соединения 100f (3,00 г, 9,48 ммоль) в МеОН (100 мл) добавляли 20% Pd/C (0,6 г). Смесь нагревали до 50°С в атмосфере водорода и перемешивали в течение 12 ч. После охлаждения до комнатной температуры, смесь фильтровали и концентрировали фильтрат досуха в условиях вакуума с получением указанного в заголовке соединения 100g (2,15 г, 100%).
MS m/z (ESI): 227 [M+1]
Стадия 7. трет-Бутил-транс-5-(2,5-дихлорпиримидин-4-ил)-3а-метилгексагидропирроло[3,4-с]пиррол-2(1Н)-карбоксилат (100h)
К раствору соединения 100g (2,15 г, 9,50 ммоль) в этилацетате (40 мл) при 0°С добавляли DIEA (1,47 г, 11,40 ммоль) и 2,4,5-трихлорпиримидин (1,74 г, 9,50 ммоль). Смесь перемешивали в течение 1 ч, а затем концентрировали досуха в условиях вакуума. Остаток очищали методом колоночной хроматографии на силикагеле (петролейный эфир/этилацетат = 10/3) с получением указанного в заголовке соединения 100h (3,30 г, 93%).
MS m/z (ESI): 373 [M+1]
Стадия 8. транс-5-Хлор-N-(1-метил-1Н-пиразол-4-ил)-4-(3а-метилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)пиримидин-2-амина дигидрохлорид (100i)
К раствору соединения 100h (3,30 г, 8,84 ммоль) в этаноле (40 мл) добавляли 1-метил-1Н-пиразол-4-амина гидрохлорид (1,54 г, 11,49 ммоль) и pTsOH моногидрат (0,17 г, 0,88 ммоль). Смесь нагревали до 70°С и перемешивали в течение 16 ч. После охлаждения до комнатной температуры, смесь концентрировали досуха, и добавляли к ней смесь 2-метокси-2-метилпропана и метанола (40 мл, 1/1 об./об.). После перемешивания в течение 30 мин, смесь фильтровали, и собирали осадок на фильтре с получением указанного в заголовке соединения 100i (3,15 г, 88%) в виде гидрохлорида.
MS m/z (ESI): 334 [M+1]
Стадия 9. транс-3-(5-(5-Хлор-2-((1-метил-1H-пиразол-4-ил)амино)пиримидин-4-ил)-3а-метилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)-3-оксопропаннитрил (100)
Смесь соединения 100i (2,00 г, 4,92 ммоль) в ацетонитриле (40 мл) добавляли этил-2-цианоацетат (1,11 г, 9,84 ммоль) и DBU (3,74 г, 24,60 ммоль). Смесь нагревали до 30°С и перемешивали в течение 2 ч. Смесь концентрировали досуха, добавляли этанол (30 мл), и перемешивали при комнатной температуре в течение 1 ч. Смесь фильтровали, и собирали осадок на фильтре с получением указанного в заголовке соединения 100 (1,15 г, твердое вещество, 58%).
MS m/z (ESI): 401 [M+1]
1H-ЯМР (400 МГц, DMSO-d6) δ 9,10 (с, 1H), 7,90 (с, 1H), 7,70 (с, 1H), 7,46 (с, 1H), 4,00 - 3,90 (м, 3Н), 3,78 (с, 3Н), 3,69 - 3,47 (м, 4Н), 3,42 - 3,33 (м, 1H), 3,28 (дд, J=11,9, 9,3 Гц, 1H), 3,09 (дд, J=20,0, 10,8 Гц, 1H), 2,40 (дд, J=24,7, 17,6 Гц, 1Н), 0,94 (д, J=8,8 Гц, 3Н).
Пример 101. Биологические эксперименты
Анализ ингибирования JAK2
Эффект соединений согласно настоящему изобретению на ферментативную активность JAK2 оценивали путем определения уровня фосфорилирования субстрата в киназной реакции с использованием аналитического набора для детектирования активности HTRF киназы (Cisbio, кат. №62TK0PEC).
Экспериментальный способ описан ниже в общих чертах:
Реакционный буфер содержал следующие компоненты: буфер для ферментативной реакции (1×), 5 мМ MgCl2, 1 мМ DTT и 0,01% Brij35 из набора. Раствор JAK2 киназы содержал рекомбинантный белок JAK2 человека (Carna Biosciences, 08-045), разбавленный реакционным буфером до 0,15 нг/мкл. Реакционный раствор субстрата содержал 2,5 мкМ АТР и 0,25 мкМ биотинилированного субстрата тирозинкиназы из набора в реакционном буфере. Детектирующий раствор содержал 0,1 нг/мкл меченых Eu3+ кейдж антител (Cisbio, 61T66KLB) и 12,5 нМ меченого стрептавидином XL665 (Cisbio, 610SAXLB) в реакционном буфере.
Тестируемое соединение растворяли в DMSO до 10 мкМ с последующим серийным 4-кратным разбавлением DMSO до минимальной концентрации в 0,061 нМ. Каждую концентрацию дополнительно 40-кратно разбавляли реакционным буфером.
В 384-луночный аналитический планшет (Corning, 3674) добавляли 4 мкл раствора соединения и 2 мкл раствора JAK2 киназы. Смесь перемешивали и инкубировали при комнатной температуре в течение 15 минут, а затем добавляли 4 мкл раствора реакционного субстрата. После дополнительного инкубирования при комнатной температуре в течение 30 минут, в реакционную смесь добавляли равный 10 мкл объем детектирующего раствора, и оставляли отстаиваться при комнатной температуре в течение 30 минут. Затем планшетный ридер Envision (Perkin Elmer) использовали для измерения развития реакции при 620 нм и 665 нм. Соотношение значений оптической плотности при 665 нм и 620 нм положительно коррелировало со степенью фосфорилирования субстрата, обнаруживая тем самым активность JAK2 киназы. В эксперименте, группу без белка JAK2 киназы рассматривали как группу 100% ингибирования, а группу с белком JAK2 киназы, но без тестируемого соединения, рассматривали как группу 0% ингибирования. Выраженное в процентах ингибирование активности JAK2 киназы тестируемым соединением рассчитывали с использованием следующей формулы:
Выраженное в процентах ингибирование = 100-100 × (степень соединение - степень 100% ингибирование) / (степень 0% ингибирование - степень 100% ингибирование)
Значение IC50 тестируемого соединения рассчитывали по 8 концентрационным точкам с использованием программного обеспечения XLfit (ID Business Solutions Ltd., UK) с использованием следующей формулы:
Y = Минимальное значение + (Максимальное значение - Минимальное значение) / (1+10^((log IC50 - X) × Коэффициент наклона)),
где Y представлял собой выраженное в процентах ингибирование, X представлял собой логарифм концентрации тестируемого соединения, «Минимальное значение» представляло собой значение минимума плато S-образной кривой, «Максимальное значение» представляло собой значение максимума плато S-образной кривой, и «Коэффициент наклона» представлял собой коэффициент наклона кривой.
Метод анализа ингибирования TYK2
Эффект соединений согласно настоящему изобретению на ферментативную активность TYK2 оценивали путем определения уровня фосфорилирования субстрата в киназной реакции с использованием аналитического набора для детектирования активности HTRF киназы (Cisbio, кат. №62TK0PEC).
Экспериментальный способ описан ниже в общих чертах:
Реакционный буфер содержал следующие компоненты: буфер для ферментативной реакции (1×), 5 мМ MgCl2, 1 мМ DTT и 0,01% Brij35 из набора. Раствор TYK2 киназы содержал рекомбинантный белок TYK2 человека (Carna Biosciences, 08-147), разбавленный реакционным буфером до 0,25 нг/мкл. Реакционный раствор субстрата содержал 11,25 мкМ АТР и 0,5 мкМ биотинилированного субстрата тирозинкиназы из набора в реакционном буфере. Детектирующий раствор содержал 0,1 нг/мкл меченых Eu3+ кейдж антител (Cisbio, 61T66KLB) и 12,5 нМ меченого стрептавидином XL665 (Cisbio, 610SAXLB) в реакционном буфере.
Тестируемое соединение растворяли в DMSO до 10 мкМ с последующим серийным 4-кратным разбавлением DMSO до минимальной концентрации в 0,061 нМ. Каждую концентрацию дополнительно 40-кратно разбавляли реакционным буфером.
В 384-луночный аналитический планшет (Corning, 3674) добавляли 4 мкл раствора соединения и 2 мкл раствора TYK2 киназы. Смесь перемешивали и инкубировали при комнатной температуре в течение 15 минут, а затем добавляли 4 мкл раствора реакционного субстрата. После дополнительного инкубирования при комнатной температуре в течение 40 минут, в реакционную смесь добавляли равный 10 мкл объем детектирующего раствора, и оставляли отстаиваться при комнатной температуре в течение 30 минут. Затем планшетный ридер Envision (Perkin Elmer) использовали для измерения развития реакции при 620 нм и 665 нм. Соотношение значений оптической плотности при 665 нм и 620 нм положительно коррелировало со степенью фосфорилирования субстрата, обнаруживая тем самым активность TYK2 киназы. В эксперименте, группу без белка TYK2 киназы рассматривали как группу 100% ингибирования, а группу с белком TYK2 киназы, но без тестируемого соединения, рассматривали как группу 0% ингибирования. Выраженное в процентах ингибирование активности TYK2 киназы тестируемым соединением рассчитывали с использованием следующей формулы:
Выраженное в процентах ингибирование = 100 - 100 × (степень соединение - степень 100% ингибирование) / (степень 0% ингибирование - степень 100% ингибирование)
Значение IC50 тестируемого соединения рассчитывали по 8 концентрационным точкам с использованием программного обеспечения XLfit (ID Business Solutions Ltd., UK) с использованием следующей формулы:
Y = Минимальное значение + (Максимальное значение - Минимальное значение) / (1+10^((log IC50 - X) × Коэффициент наклона)),
где Y представлял собой выраженное в процентах ингибирование, X представлял собой логарифм концентрации тестируемого соединения, «Минимальное значение» представляло собой значение минимума плато S-образной кривой, «Максимальное значение» представляло собой значение максимума плато S-образной кривой, и «Коэффициент наклона» представлял собой коэффициент наклона кривой.
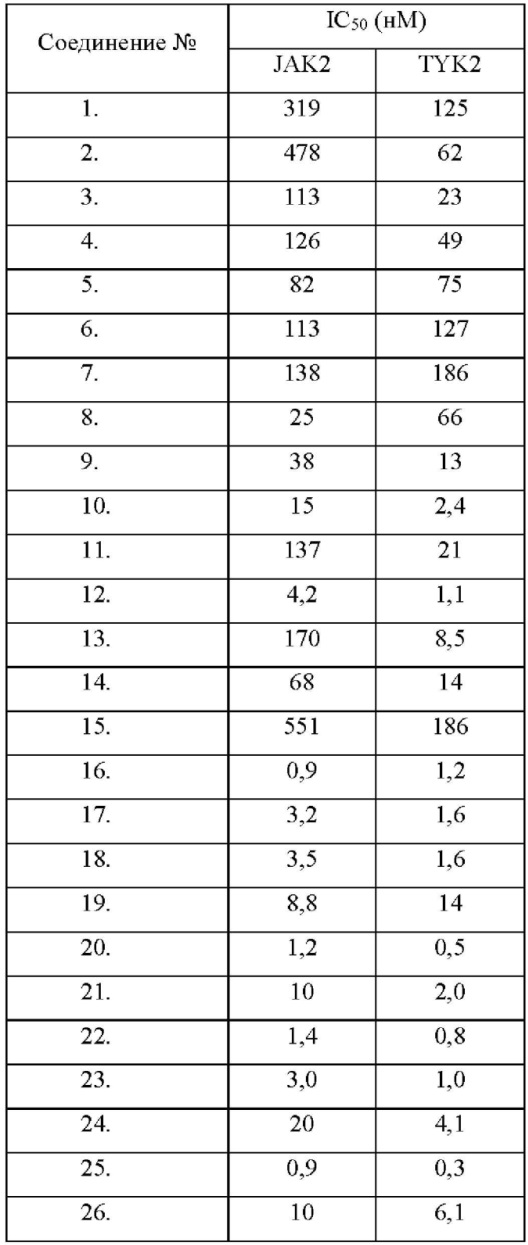
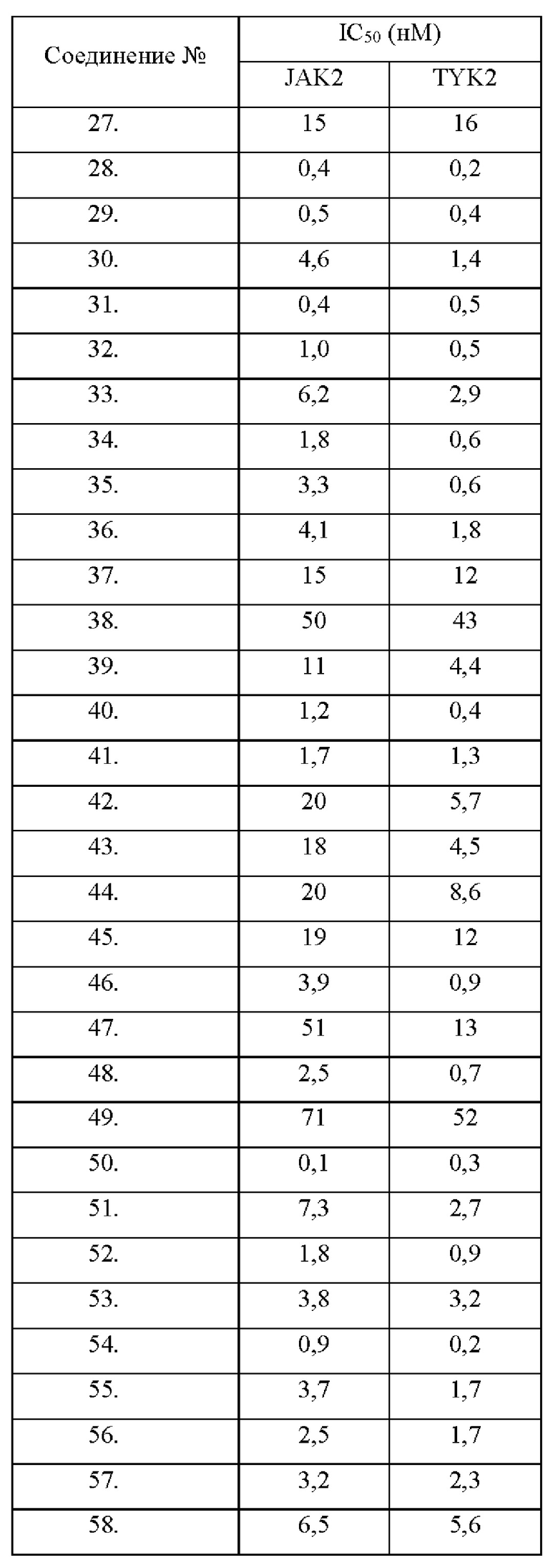
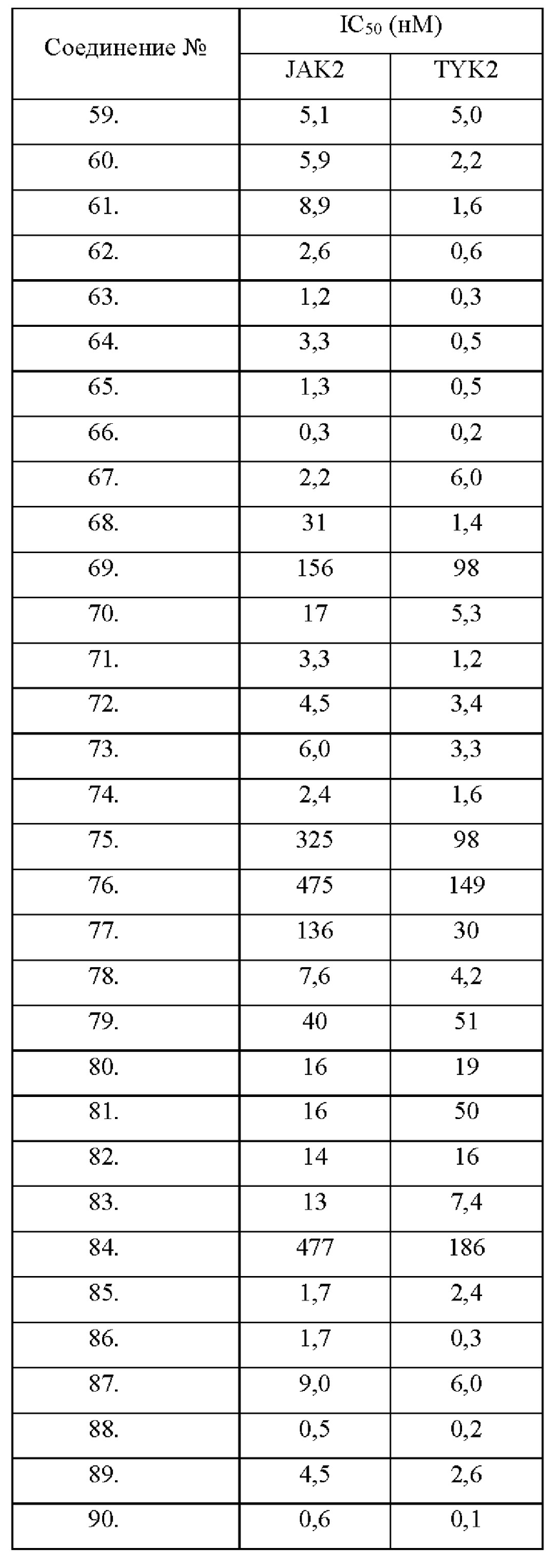
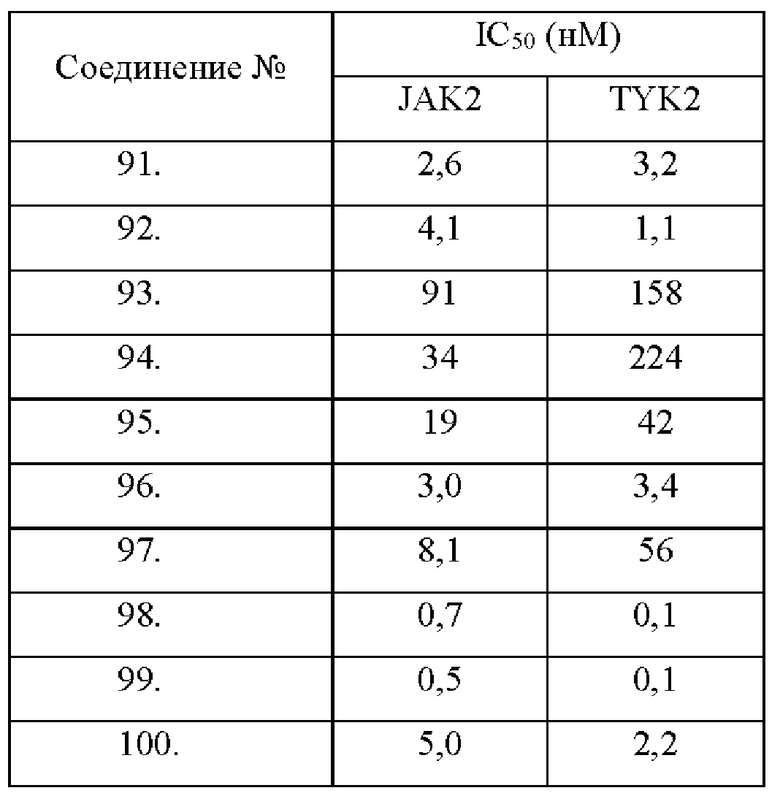
Соединения согласно настоящему изобретению обладают значительным эффектом ингибирования активности TYK2, предпочтительно со значением IC50 от 10 до 100 нМ, и более предпочтительно со значением IC50 менее 10 нМ.
Ингибирование секреции IFN-γ в клетках NK92, индуцированной IL-12
Эффект соединений согласно настоящему изобретению на секрецию IFN-γ в клетках NK92, индуцированную IL-12, оценивали методом твердофазного иммуноферментного анализа (ELISA).
Рецептор IL-12 экспрессируется главным образом в активированных Т-клетках, NK клетках (NK92 представляет собой линию NK клеток), DC клетках и В-клетках. Будучи связанным с IL-12, он активирует путь передачи сигнала JAK2/TYK2 в NK клетках и Т-лимфоцитах, индуцируя тем самым секрецию IFN-γ.
Экспериментальный способ описан ниже в общих чертах:
Тестируемое соединение растворяли в DMSO до 2,5 мМ с последующим серийным 4-кратным разбавлением DMSO до минимальной концентрации в 0,31 нМ. Каждую концентрацию дополнительно 50-кратно разбавляли средой МЕМα без FBS (Gibco, 12561-056).
Клетки NK92 (Nanjing Cobioer, СВР60980) культивировали в полной среде МЕМα, содержащей 12,5% FBS (Ausbian, VS500T), 12,5% лошадиной сыворотки (Gibco, 16050-122), 0,02 мМ фолиевой кислоты (Sigma, F8758), 0,2 мМ инозитола (Sigma, 17850), 0,55 мМ β-меркаптоэтанола (Gibco, 21985-023), 200 ед./мл IL-2 (R&D Systems, 202-1L) и 100 ед./мл пенициллина (Thermofisher, 15140122). При покрытии 80-90% поверхности культурального контейнера, клетки диспергировали и помещали в 96-луночный планшет (Thermofisher, 167425) в количестве 100000 клеток на лунку (80 мкл полной среды МЕМа без IL-2). Затем, 96-луночный планшет инкубировали в течение ночи в инкубаторе при 37°С/5% СО2.
После инкубирования в течение ночи, в каждую лунку добавляли 10 мкл тестируемого соединения и 10 мкл 50 нг/мл IL-12 (R&D Systems, 219-1L), аккуратно перемешивали, и инкубировали 96-луночный планшет в инкубаторе при 37°С/5% СО2 дополнительно в течение 24 часов. Планшет центрифугировали при 800 об./мин в течение 10 минут при комнатной температуре, и переносили 50 мкл супернатанта из каждой лунки в другой 96-луночный планшет (Sigma, CLS3695), покрытый анти-IFN-γ антителами. Величину секреции IFN-γ определяли, следуя инструкции из ELISA-набора для IFN-γ человека DuoSet (R&D Systems, DY285B). В эксперименте группа, в которой IL-12 и тестируемое соединение было заменены средой МЕМа, представляла собой контрольную группу без стимуляции (100% ингибирования), и группа с обработкой IL-12 и 0,2% DMSO представляла собой группу стимуляции (0% ингибирования). Выраженное в процентах ингибирование секреции IFN-γ в клетках NK-92, индуцированной IL-12, тестируемым соединением рассчитывали с использованием следующей формулы:
Выраженное в процентах ингибирование = 100 - 100 × (сигнал соединение - сигнал без стимуляции) / (сигнал стимуляция - сигнал без стимуляции)
Значение IC50 тестируемого соединения рассчитывали по 8 концентрационным точкам с использованием программного обеспечения XLfit (ID Business Solutions Ltd., UK) с использованием следующей формулы:
Y = Минимальное значение + (Максимальное значение - Минимальное значение) / (1+10^((log IC50 - X) × Коэффициент наклона)),
где Y представлял собой выраженное в процентах ингибирование, X представлял собой логарифм концентрации тестируемого соединения, «Минимальное значение» представляло собой значение минимума плато S-образной кривой, «Максимальное значение» представляло собой значение максимума плато S-образной кривой, и «Коэффициент наклона» представлял собой коэффициент наклона кривой.
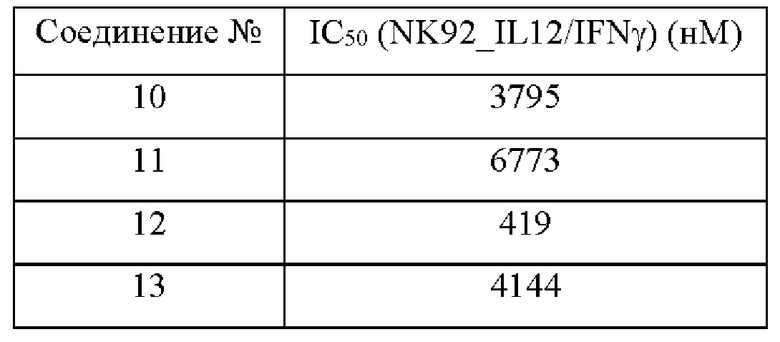

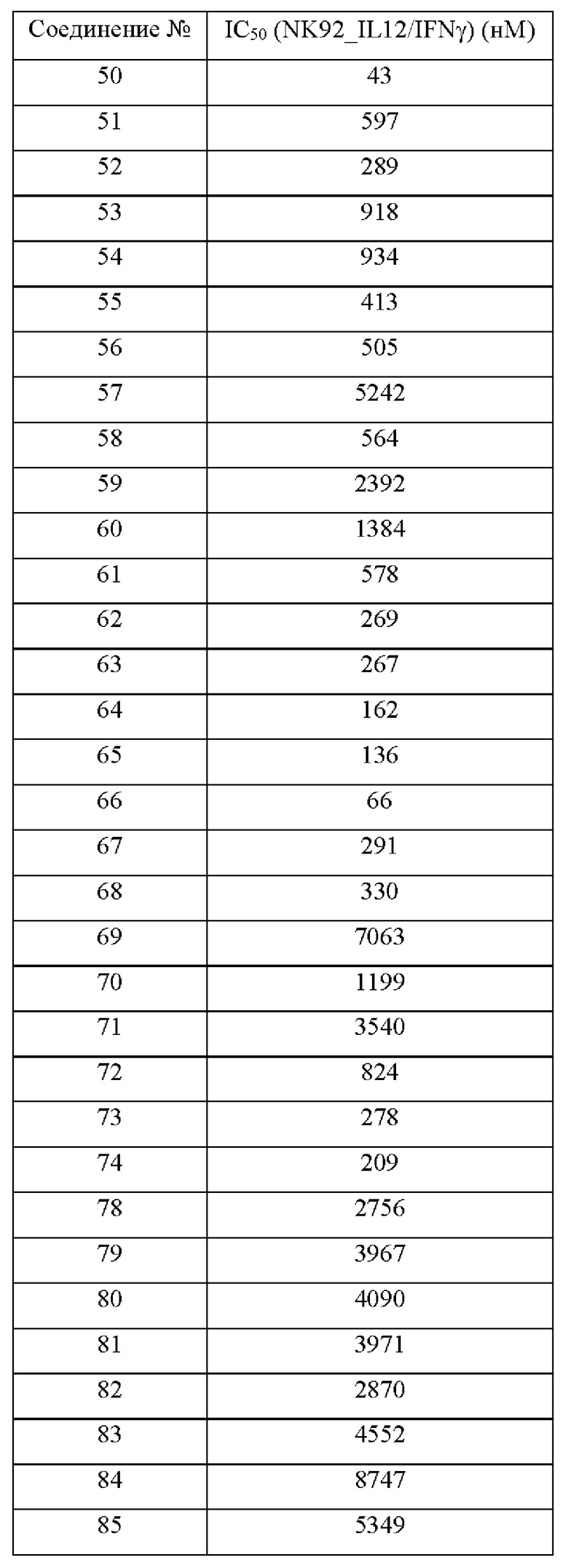
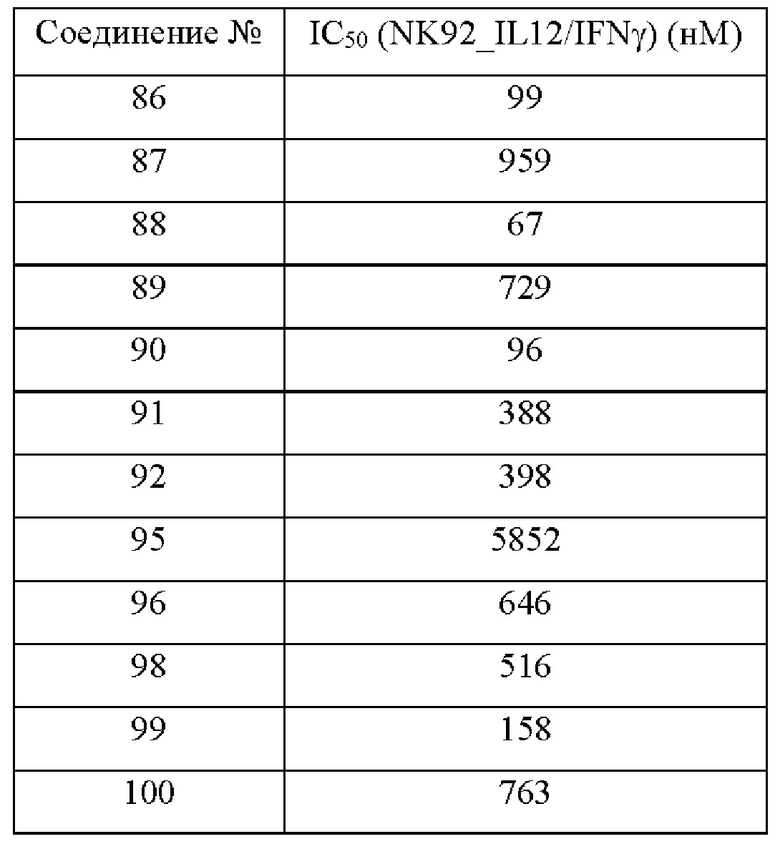
Соединения согласно настоящему изобретению обладают значительным эффектом ингибирования секреции IFN-γ в клетках NK92, индуцированной IL-12, и значение IC50 предпочтительно составляет менее 1000 нМ.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ПИРРОЛОПИРИМИДИНА, ПОЛЕЗНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ JAK-КИНАЗЫ | 2012 |
|
RU2618673C2 |
| ИНГИБИТОРЫ MAGL НА ОСНОВЕ ПИРАЗОЛА | 2018 |
|
RU2789157C2 |
| Соединения 1-циано-пирролидинов в качестве ингибиторов USP30 | 2016 |
|
RU2717238C2 |
| ЗАМЕЩЕННЫЕ ПУРИНОВЫЕ И 7-ДЕАЗАПУРИНОВЫЕ СОЕДИНЕНИЯ | 2011 |
|
RU2606514C2 |
| ПРОИЗВОДНЫЕ ОКТАГИДРОПИРРОЛО[3,4-b]ПИРРОЛА (ВАРИАНТЫ), СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ОКТАГИДРОПИРРОЛО[3,4-b]ПИРРОЛА (ВАРИАНТЫ), ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЯ ИЛИ НАРУШЕНИЯ СОСТОЯНИЯ ОРГАНИЗМА, ЧУВСТВИТЕЛЬНОГО К МОДУЛЯЦИИ АКТИВНОСТИ РЕЦЕПТОРА ГИСТАМИНА-3 | 2007 |
|
RU2492172C2 |
| КЛАСС БИФУНКЦИОНАЛЬНЫХ ХИМЕРНЫХ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ ДЛЯ НАПРАВЛЕННОГО РАЗРУШЕНИЯ АНДРОГЕННЫХ РЕЦЕПТОРОВ И ИХ ПРИМЕНЕНИЕ | 2020 |
|
RU2825000C2 |
| ГИДРОКСИПУРИНОВЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2016 |
|
RU2685417C1 |
| Новые соединения пиридопиримидинона для модулирования каталитической активности гистонлизиндеметилаз (KDMS) | 2015 |
|
RU2684396C2 |
| РЕГУЛЯТОРЫ-ПРОИЗВОДНЫЕ СТЕРОИДОВ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2019 |
|
RU2797408C2 |
| ЗАМЕЩЕННЫЕ НИКОТИНИМИДНЫЕ ИНГИБИТОРЫ ВТК, ИХ ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ В ТЕРАПИИ РАКОВЫХ, ВОСПАЛИТЕЛЬНЫХ И АУТОИММУННЫХ ЗАБОЛЕВАНИЙ | 2014 |
|
RU2677884C2 |
Настоящее изобретение относится к гетероциклическим соединениям или их фармацевтически приемлемым солям, подходящим для регулирования или ингибирования активности Janus-киназы (JAK), в частности тирозинкиназы 2 (TYK2), а именно к соединению формулы (I) или его фармацевтически приемлемой соли, стабильному изотопу, стереоизомеру, в котором R1 представляет собой пиразолил, где один водород пиразолила необязательно замещен -COORb, -C(O)Rb, -C(O)NRbRc, C1-6алкилом, CD3 или пиперидинилом; R2 представляет собой Н; R3 представляет собой Н, галоген, циано, С1-6 алкил, или ОС1-6 алкил; R4 и R5 независимо выбраны из Н, или С1-6 алкила; R6 и R7 независимо выбраны из Н, или R6 и R7 объединены в виде оксо; L представляет собой связь, С1-6алкилен, -С(О)-, -С(O)O-, -C(O)N(Ra)- или -S(O)2-; А представляет собой Н, С1-6алкил, С3-6циклоалкил, 4-членный гетероциклил, в котором один кольцевой атом представляет собой N или О, или 5-6-членный гетероарил, в котором от 1 до 3 кольцевых атомов представляют собой гетероатомы N или О, где один или более атомов водорода алкила, циклоалкила и гетероарила необязательно замещены галогеном, циано или -ORd; Ra представляет собой Н; Rb и Rc независимо выбраны из Н или С1-6алкила, и Rd независимо выбран из Н, C1-6алкила или морфолинила. Изобретение также относится к конкретным соединениям, а также к фармацевтической композиции на основе указанных соединений. Технический результат заключается в получении альтернативных соединений для лечения TYK2-опосредованных заболеваний. 3 н. и 8 з.п. ф-лы, 101 пр.
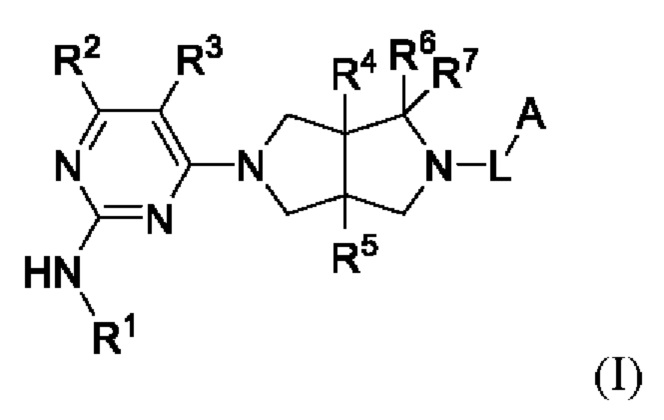
1. Соединение формулы (I) или его фармацевтически приемлемая соль, стабильный изотоп, стереоизомер:
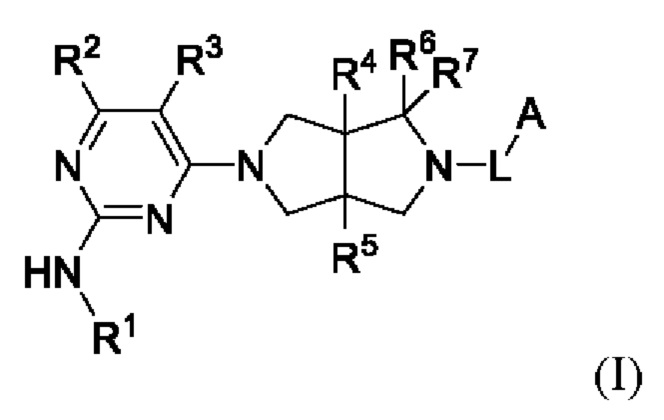
где:
R1 представляет собой пиразолил, где один водород пиразолила необязательно замещен -COORb, -C(O)Rb, -C(O)NRbRc, C1-6алкилом, CD3 или пиперидинилом;
R2 представляет собой Н;
R3 представляет собой Н, галоген, циано, С1-6 алкил, или ОС1-6 алкил;
R4 и R5 независимо выбраны из Н, или С1-6 алкила;
R6 и R7 независимо выбраны из Н, или R6 и R7 объединены в виде оксо;
L представляет собой связь, С1-6алкилен, -С(О)-, -С(O)O-, -C(O)N(Ra)- или -S(O)2-;
А представляет собой Н, С1-6алкил, С3-6циклоалкил, 4-членный гетероциклил, в котором один кольцевой атом представляет собой N или О, или 5-6-членный гетероарил, в котором от 1 до 3 кольцевых атомов представляют собой гетероатомы N или О, где один или более атомов водорода алкила, циклоалкила и гетероарила необязательно замещены галогеном, циано или -ORd;
Ra представляет собой Н;
Rb и Rc независимо выбраны из Н или С1-6алкила, и
Rd независимо выбран из Н, C1-6алкила или морфолинила.
2. Соединение по п. 1, которое представляет собой соединение формулы (II) или его фармацевтически приемлемую кислую соль, стабильный изотоп, стереоизомер:
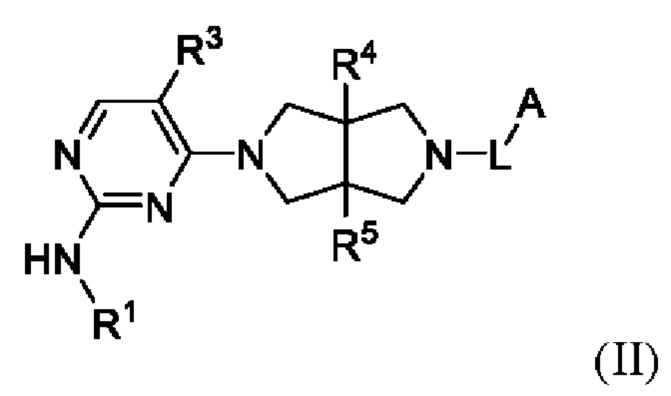
где:
L представляет собой связь, С1-6алкилен, -С(О)-, -С(O)O-, -C(O)NH- или -S(O)2-.
3. Соединение по п. 1 или 2, где L представляет собой -С(О)-.
4. Соединение по п. 1 или 2, где А представляет собой С1-6алкил или С3-6циклоалкил, где один или более атомов водорода алкила и циклоалкила необязательно замещены галогеном, циано или -OC1-2алкилом.
5. Соединение по п. 2, которое представляет собой соединение формулы (III) или его фармацевтически приемлемую кислую соль, стабильный изотоп, стереоизомер:
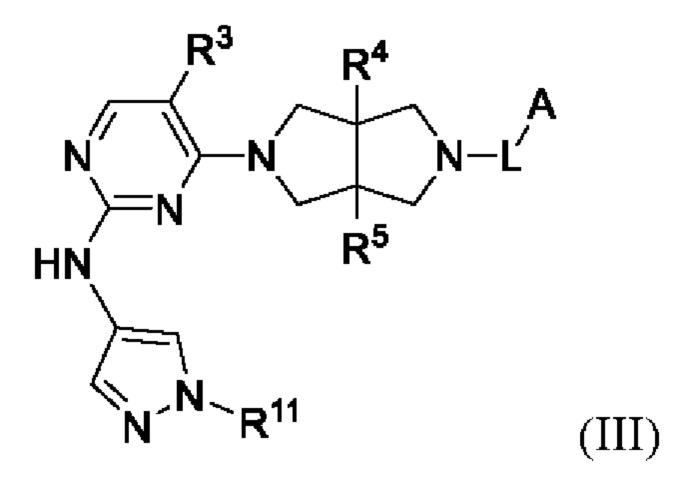
где:
L представляет собой связь, С1-6алкилен, -С(О)-, -С(O)O-, -C(O)NH- или -S(O)2-;
R11 представляет собой Н, COORb, -C(O)Rb, -C(O)NRbRc, С1-6алкил или пиперидинил.
6. Соединение по п. 5, где А представляет собой С1-6алкил или С3-6циклоалкил, где один или более атомов водорода алкила и циклоалкила необязательно замещены галогеном, циано или -OC1-2алкилом.
7. Соединение по п. 5, где R11 представляет собой С1-6алкил, где один или более атомов водорода алкила необязательно замещены D.
8. Соединение, имеющее следующую структуру, или его фармацевтически приемлемая соль, стабильный изотоп, стереоизомер:
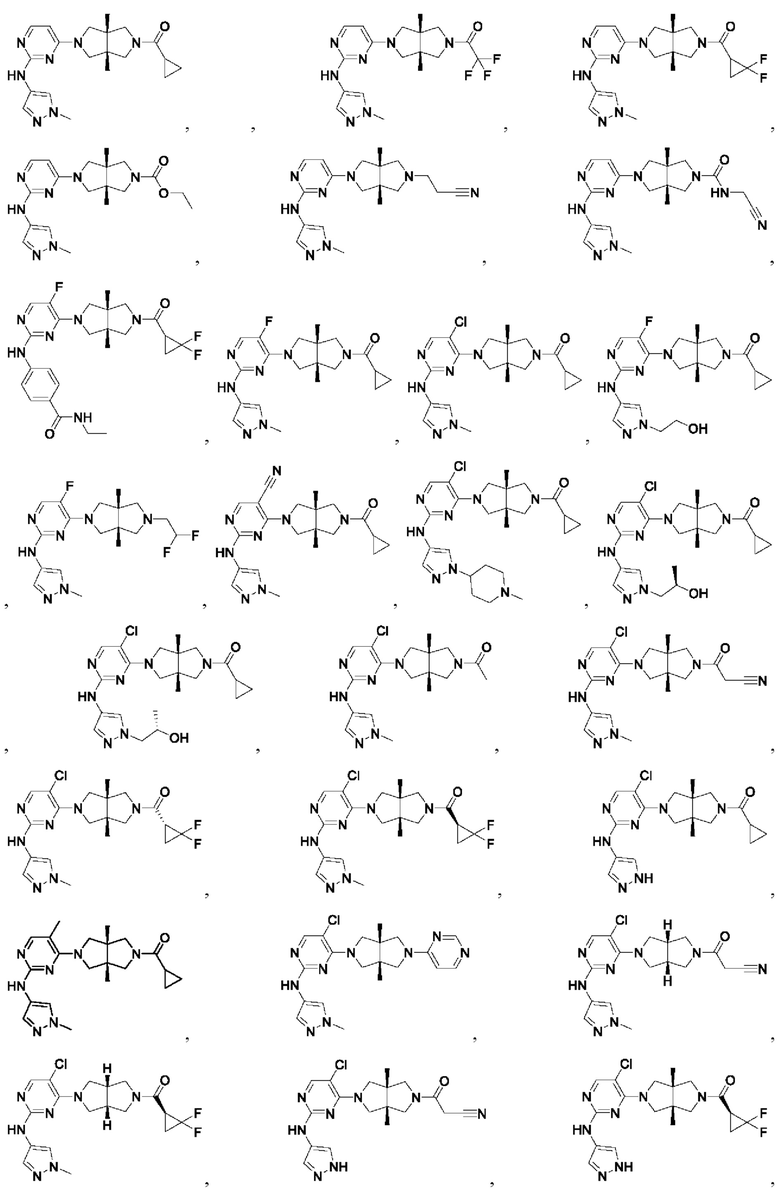
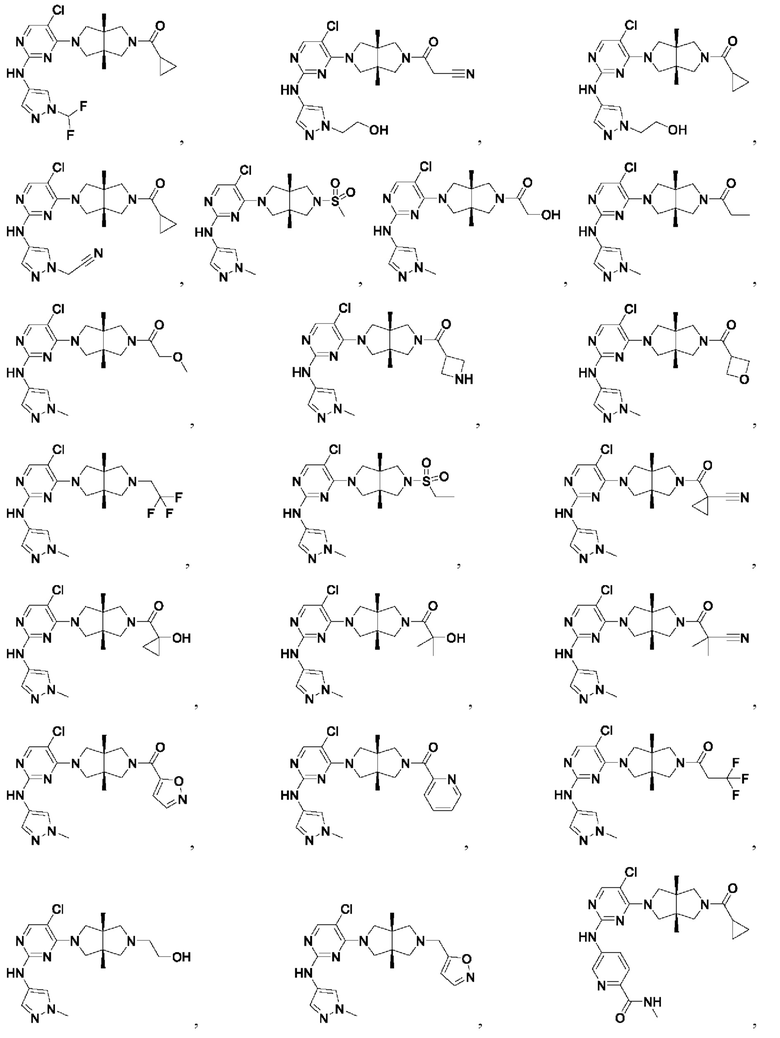
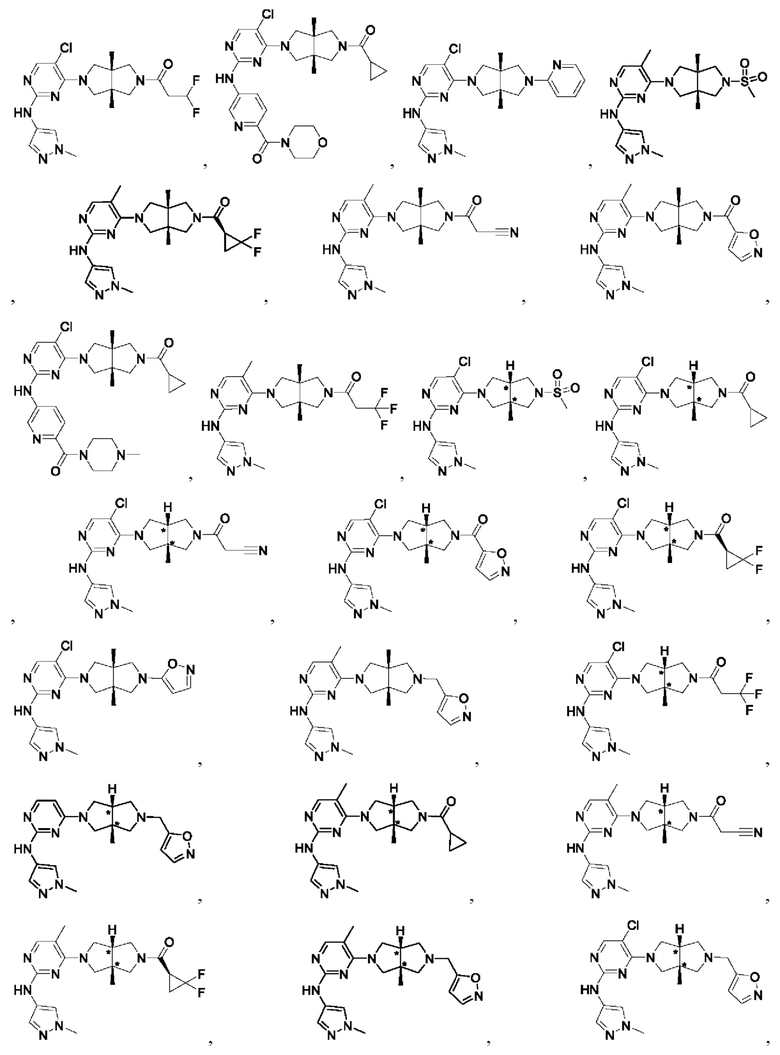
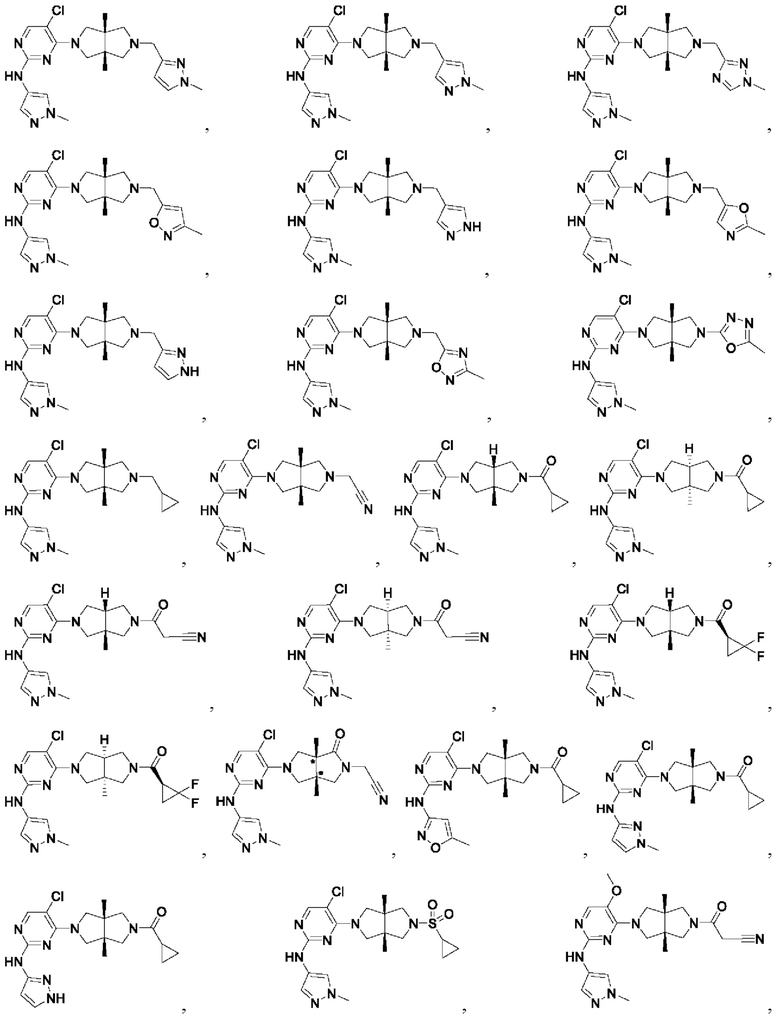
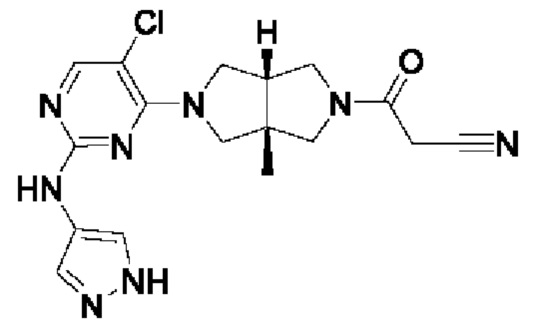 или
или 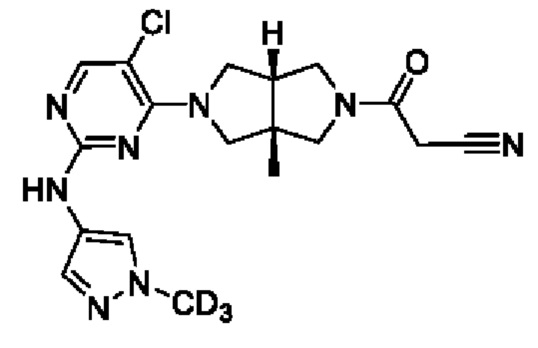 (стереохимия атомов углерода, отмеченных *, не определена).
(стереохимия атомов углерода, отмеченных *, не определена).
9. Соединение по п. 8, которое представляет собой:
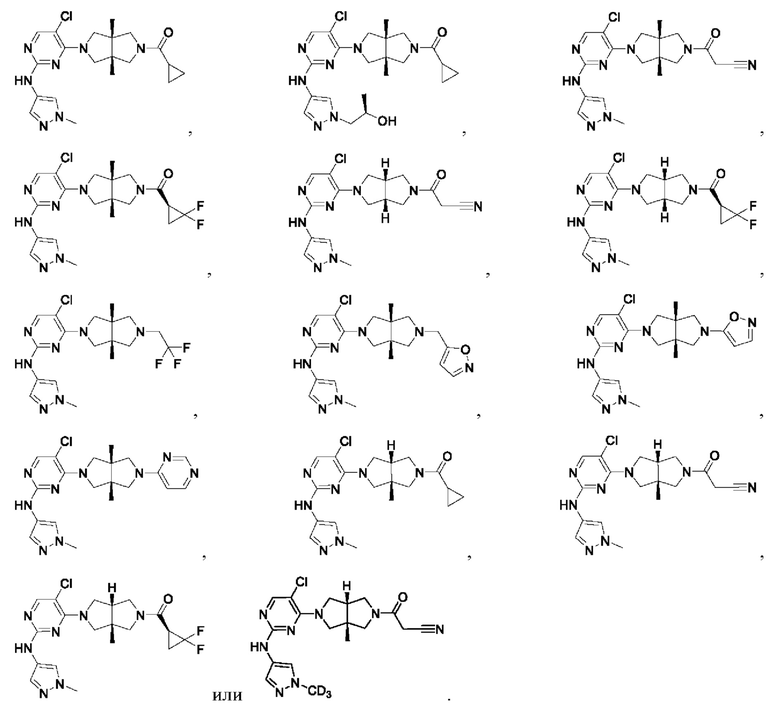
10. Соединение по п. 8, которое представляет собой:
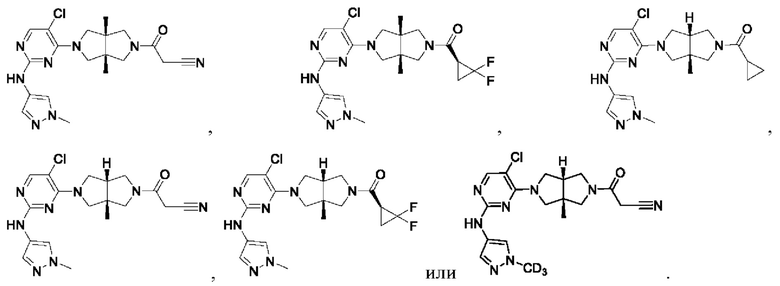
11. Фармацевтическая композиция для предупреждения или лечения заболевания, опосредованного тирозинкиназой 2 (TYK2), содержащая терапевтически эффективное количество соединения по любому из пп. 1-10 и его фармацевтически приемлемый носитель или вспомогательное вещество.
| WO 2012062704 A1, 18.05.2012 | |||
| WO 2017007987 A1, 12.01.2017 | |||
| CN 105622638 B, 02.10.2018 | |||
| WO 2012145581 A1, 26.10.2012 | |||
| WO 2019060365 A1, 28.03.2019 | |||
| КАТАЛИЗАТОР КРЕКИНГА И СПОСОБ ДЛЯ ЕГО ПРИГОТОВЛЕНИЯ | 2021 |
|
RU2832734C1 |
| Асинхронный двигатель однофазного тока | 1928 |
|
SU22802A1 |
| Устройство для сушки и пропитки электрических кабелей с бумажной изоляцией | 1929 |
|
SU22766A1 |

