Изобретение относится к области органической химии и касается способа синтеза 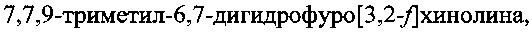 который может быть использован в качестве основы для синтеза различных производных триметилзамещенных фуродигидрохинолинов (ФДГХ), представляющих практический интерес в качестве потенциальных сенсибилизаторов для применения в PUVA-терапии кожных заболеваний, а также могут быть использованы как антиоксиданты и антиозонанты для стабилизации каучуков и резин, ракетных топлив и смазочных масел, а также как аналитические реагенты и ловушки для радикалов.
который может быть использован в качестве основы для синтеза различных производных триметилзамещенных фуродигидрохинолинов (ФДГХ), представляющих практический интерес в качестве потенциальных сенсибилизаторов для применения в PUVA-терапии кожных заболеваний, а также могут быть использованы как антиоксиданты и антиозонанты для стабилизации каучуков и резин, ракетных топлив и смазочных масел, а также как аналитические реагенты и ловушки для радикалов.
Анализ информации, касающейся фотосенсибилизаторов, используемых в PUVA-терапии, показывает, что для лечения тяжелых кожных патологий в настоящее время применяют, главным образом, псоралены - линейные фурокумарины [US 4129576, опубл. 12.12.1978; US 4130568, опубл. 19.12.1978; US 4960408, опубл. 02.10.1990; US 4279922, опубл. 21.07.1981], которые в эффективных терапевтических дозах проявляют целый ряд нежелательных побочных действий. К ним относится развитие тяжелой эритемы кожи, а также общая генотоксичность, приводящая к возникновению точечных мутаций и хромосомных аберраций и, как следствие, - к повышению риска развития рака кожи при облучении УФ-светом. Поиск соединений, сохраняющих полезные свойства псораленов, но лишенных фото- и генотоксичности, привел к незамещенным по первому положению соединениям ряда фурохинолона, которые, как было показано, в 20-40 раз менее опасны для пациентов по показателям фото- и генотоксичности, чем псоралены [Marzano et al., 2002, Bioorganic and Medicinal Chem., 10, 2835-2844]. Однако для этих соединений характерна высокая гидрофобность, препятствующая доставке соединения в очаги кератоза, что не позволило внедрить их медицинскую в практику.
Исследования, проведенные авторами, показывают, что к соединениям, наиболее близким по строению к фурохинолонам и обладающим комплексом свойств, позволяющих рассматривать их в качестве потенциальных перспективных фотосенсибилизаторов для PUVA-терапии кожных заболеваний, можно отнести замещенные фуродигидрохинолины (ФДГХ), представляющие собой триметилзамещенные в хинолиновое кольцо, частично гидрированные, возможно содержащие заместители в бензольном кольце, хинолины с аннелированным фурановым кольцом. Эти соединения так же, как псоралены и фурохинолоны, имеют высокий выход триплетного состояния при фотовозбуждении, но, в отличие от последних, не образуют сшивок и диаддуктов с молекулами ДНК, выгодно отличаются по основным фотохимическим параметрам от фурокумаринов и могут образовывать водорастворимые солянокислые соли. Кроме того, замещенные фуродигидрохинолины могут выступать в качестве ловушек свободных радикалов и, обладая антиоксидантной активностью, способствовать уменьшению эритемы при УФ-облучении.
Таким образом, разработка способа химического синтеза этих соединений является важной практической задачей, на решение которой направлено настоящее изобретение.
Известен способ синтеза производного дигидрохинолина 6-этокси-2,2,4-триметил-1,2-дигидрохинолина (этоксихина), согласно которому формирование азотсодержащего гетероцикла хинолина осуществляют конденсацией ацетона с анизидином при температуре 175-180°C в присутствии йода [WO 2011013159, опубл. 03.02.2011].
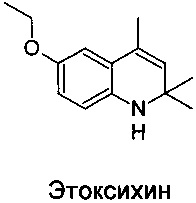
Недостаток этого способа состоит в том, что циклизация требует очень жестких условий проведения реакции и при наличии в молекуле исходного n-метоксианилина лабильных групп выход целевого продукта может резко уменьшиться.
В качестве прототипа заявляемого изобретения взят способ синтеза 7,7,9-триметил-6,7-дигидрофуро[3,2-f]хинолина (ФДГХ-1), описанный в работе В.А. Кузьмин, Л.И. Мазалецкая, Т.Д. Некипелова, Е.Н. Ходот. Известия Академии наук. Серия химическая, 2008, №11, стр. 2356-2360. В соответствии с известным способом формирование азотсодержащего триметилзамещенного кольца дигидрохинолина осуществляют путем конденсации солянокислой соли 5-аминобензофурана с ацетоном в присутствии кислоты Льюиса - трифлата скандия (скандий III трифторметансульфонат) при комнатной температуре. При этом 5-аминобензофуран получают восстановлением 5-нитробензофурана гидразингидратом в присутствии никель-алюминиевого сплава, а 5-нитробензофуран получают из n-нитросалицилового альдегида, как описано в П.И. Абраменко, В.Г. Жиряков, Т.К. Пономарева. Химия гетероциклических соединений, 1975, с. 1603, путем трехстадийного синтеза.
Описанный метод имеет следующие недостатки и ограничения:
1. Труднодоступность и дороговизна 5-нитросалицилового альдегида, который получают нитрованием салицилового альдегида дымящейся азотной кислотой (d=1,5). При этом получают смесь изомеров 5-нитро- и 3-нитросалицилового альдегида, для разделения и очистки которых через их натриевые соли требуются большие затраты труда и времени.
2. Получение нитробензофурана, не замещенного во 2 и 3 положении, проходит через энергозатратную стадию декарбоксилирования при высокой температуре.
3. Использование на последней стадии в качестве кислоты Льюиса труднодоступного дорогостоящего препарата трифлата скандия удорожает процесс и затрудняет его масштабирование.
Следует отметить, что способ по прототипу позволяет получить только 7,7,9-триметил-6,7-дигидрофуро[3,2-f]хинолин и не может быть использован в качестве основы для получения других, перспективных для применения в качестве сенсибилизаторов при PUVA-терапии соединений этого класса, что значительно снижает его практическую ценность.
Задачей настоящего изобретения является создание способа синтеза 7,7,9-триметил-6,7-дигидрофуро[3,2-f]хинолина, лишенного недостатков и ограничений способа по прототипу и позволяющего при меньших трудозатратах и с высоким выходом получить целевой продукт при использовании доступных недорогих исходных соединений и реагентов.
Предлагаемый способ синтеза 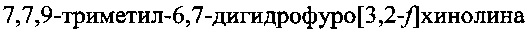 включает конденсацию 5-аминобензофурана с ацетоном в присутствии кислоты и отличается тем, что в качестве кислоты используют n-толуолсульфокислоту (p-TsOH), а конденсацию проводят в присутствии перхлората магния при температуре от 70-125°C в автоклаве, при этом 5-аминобензофуран получают путем осуществления следующей последовательности стадий, включающей:
включает конденсацию 5-аминобензофурана с ацетоном в присутствии кислоты и отличается тем, что в качестве кислоты используют n-толуолсульфокислоту (p-TsOH), а конденсацию проводят в присутствии перхлората магния при температуре от 70-125°C в автоклаве, при этом 5-аминобензофуран получают путем осуществления следующей последовательности стадий, включающей:
а - ацилирование n-анизидина уксусным ангидридом с получением N-(4-метоксифенил)ацетамида;
b - взаимодействие N-(4-метоксифенил)ацетамида, полученного на стадии а, с хлорангидридом хлоруксусной кислоты с получением N-[3-(хлорацетил)-4-гидроксифенил]ацетамида (реакция Фриделя-Крафтса);
с - замыкание дигидрофуранового цикла путем обработки продукта, полученного на стадии b, ацетатом натрия и последующего восстановления интермедиата без его выделения из реакционной смеси боргидридом натрия с получением N-(3-гидрокси-2,3-дигидро-1-бензофуран-5-ил)ацетамида (циклизация с последующим восстановлением кето-группы);
d - дегидратацию продукта, полученного на стадии с, путем его обработки n-толуолсульфокислотой и перхлоратом магния с получением N-1-бензофуран-5-ил ацетамида;
е - кислый гидролиз продукта, полученного на стадии d, с получением 5-аминобензофурана.
Технический результат изобретения состоит в упрощении и удешевлении процесса получения целевого продукта и повышении его суммарного выхода по сравнению с прототипом. Упрощение и удешевление процесса получения целевого продукта 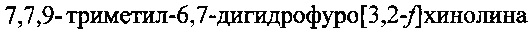 обеспечено уменьшением трудоемкости при одинаковом количестве стадий синтеза по сравнению с прототипом, использованием более доступных и дешевых реагентов и исходных соединений, отсутствием высокотемпературной стадии декарбоксилирования и более высоким суммарным выходом, который по совокупности шести стадий более чем втрое превышает выход по прототипу: 18,8% - по изобретению, 5,8% - по прототипу.
обеспечено уменьшением трудоемкости при одинаковом количестве стадий синтеза по сравнению с прототипом, использованием более доступных и дешевых реагентов и исходных соединений, отсутствием высокотемпературной стадии декарбоксилирования и более высоким суммарным выходом, который по совокупности шести стадий более чем втрое превышает выход по прототипу: 18,8% - по изобретению, 5,8% - по прототипу.
Предлагаемая последовательность реакций в сочетании с выбранными условиями их осуществления, обеспечивающие в совокупности достижение технического результата - получение целевого продукта с максимальным выходом и минимальными затратами труда, времени и финансовых средств, ранее не описаны и не являются очевидными для специалиста.
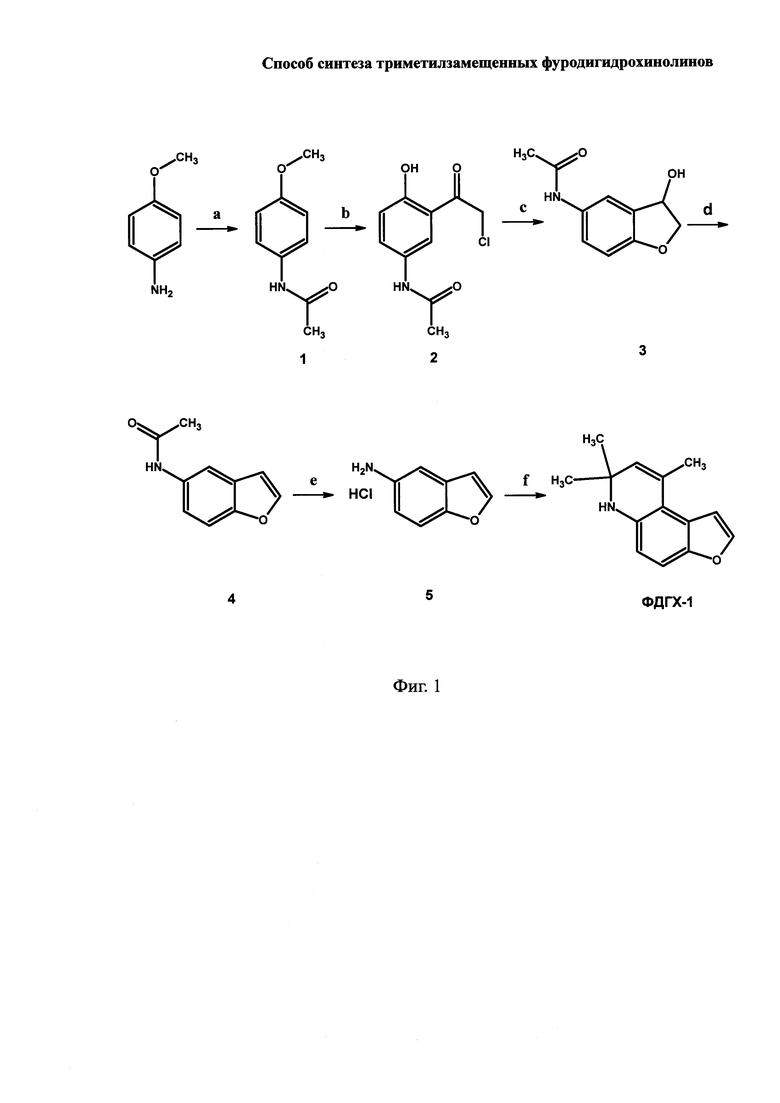
На Фиг. 1 показана предлагаемая схема синтеза целевого продукта 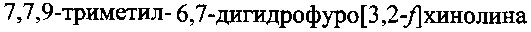 (ФДГХ-1). Использованные реагенты и выходы продуктов на каждой стадии приведены в Примере 1.
(ФДГХ-1). Использованные реагенты и выходы продуктов на каждой стадии приведены в Примере 1.
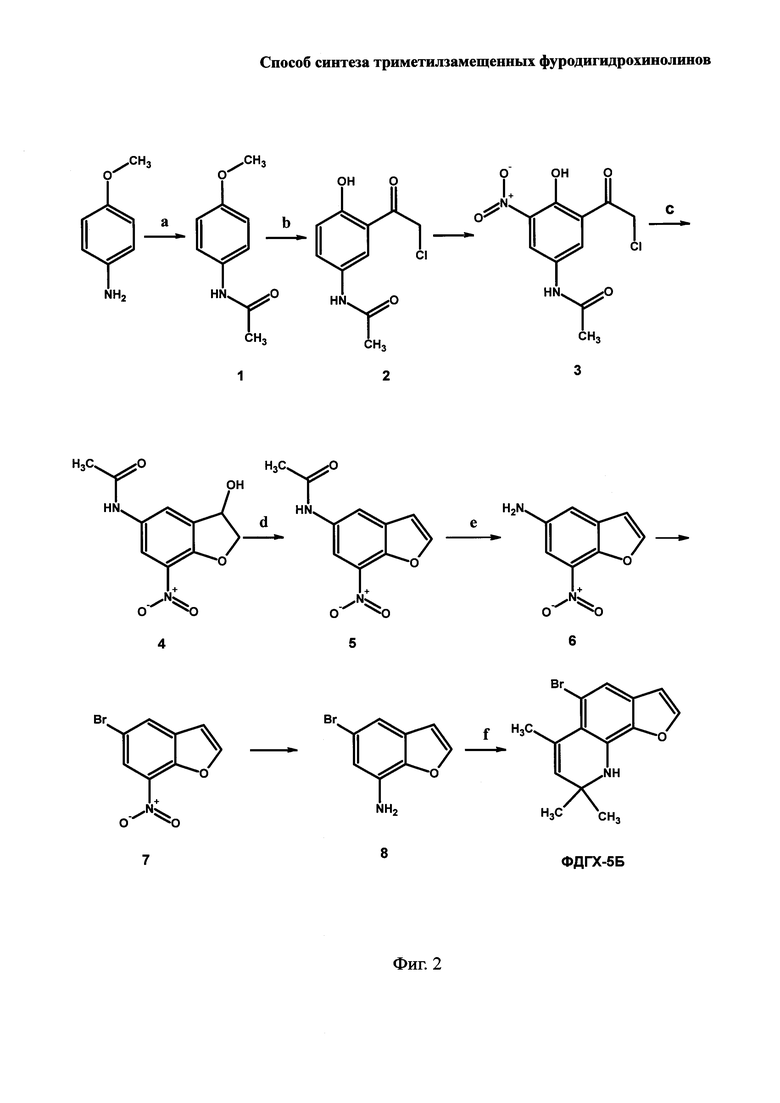
На Фиг. 2 показана схема синтеза 5-бром-6,8,8-триметил-8,9-дигидрофуро[3,2-h]хинолина (ФДГХ-5Б). Использованные реагенты и выходы продуктов на каждой стадии приведены в Примере 2.
Как и в способе по прототипу, триметилзамещенные по хинолиновому кольцу фуродигидрохинолины получают реакцией Скраупа из не замещенного по второму и третьему положениям 5-аминобензфурана и ацетона в присутствии кислотного катализатора. Отличия от прототипа заключаются в следующем: во-первых, в качестве катализатора вместо дорогостоящего и труднодоступного трифлата скандия используют недорогую n-толуолсульфокислоту, а повышение выхода достигают за счет того, что конденсацию проводят в присутствии перхлората магния при температуре от 70-125°C в автоклаве, во-вторых, исходный для этой стадии 5-аминобензфуран получают новым высокоэффективным способом из доступного сырья с применением энергосберегающего способа, исключающего стадию декарбоксилирования при высокой температуре.
Исходным соединением для получения целевого продукта, а также других возможных производных ФДГХ, которые, как будет показано ниже, могут быть синтезированы на основе заявляемого способа, является p-анизидин (p-метоксианилин) - доступный недорогой продукт крупнотоннажного синтеза. Его ацетилирование на стадии а приводит к получению N-(4-метоксифенил)ацетамида (соединение 1 на Фиг. 1) с выходом, превышающим 90%. На стадии b по реакции Фриделя-Крафтса получают N-[3-(хлорацетилl)-4-гидроксифенил]ацетамид (соединение 2 на Фиг. I), при этом вместо пожароопасного сероуглерода, используемого в известной методике [Kunckell F. (1901) Darstellung von oxyamido- und oxyamidochlor-ketonen. Berichte der deutschen chemischen Gesellschaft, 34, 124-129], нами использован дихлорэтан. Это позволяет упростить процесс и получить выход N-[3-(хлорацетилl)-4-гидроксифенил]ацетамида, превышающий 90%. На стадии с, которая ранее не была описана и осуществлена впервые, проводят циклизацию соединения 2 с образованием гидрофуранона и последующим восстановлением его до гидрофуранола путем добавления ацетата натрия в метаноле с последующим восстановлением получающегося промежуточного оксосоединения боргидридом натрия. Стадию осуществляют без выделения промежуточного N-(3-оксо-2,3-дигидро-1-бензофуран-5-ил)ацетамида, поскольку его выделение приводит к резкому снижению выхода продукта циклизации N-(3-гидрокси-2,3-дигидро-1-бензофуран-5-ил)ацетамида (соединение 3 на Фиг. I). Далее на стадии d с количественным выходом осуществляют дегидратацию соединения 3 под действием p-толуолсульфокислоты в присутствии ангидрона (безводный перхлорат магния) с получением N-1-бензофуран-5-ил ацетамида (соединение 4 на Фиг. I), который на следующей стадии е гидролизуют соляной кислотой с получением 5-аминобензофурана (соединение 5 на Фиг. 1) с выходом, превышающим 90%.
Таким образом, итоговый выход последовательности реакций, приводящих, в соответствии с заявляемым изобретением, к 5-аминобензофурану - непосредственному предшественнику целевого продукта 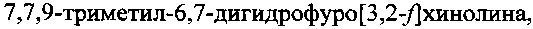 составляет 28,5% на пяти стадиях, что в 3,5 раза выше, чем на пяти стадиях по прототипу (8%). К тому же отсутствуют: стадия нитрования, на которой образуются трудноразделяемые 3- и 5-нитросалициловые альдегиды, и высокотемпературная стадия декарбоксилирования.
составляет 28,5% на пяти стадиях, что в 3,5 раза выше, чем на пяти стадиях по прототипу (8%). К тому же отсутствуют: стадия нитрования, на которой образуются трудноразделяемые 3- и 5-нитросалициловые альдегиды, и высокотемпературная стадия декарбоксилирования.
На завершающем этапе синтеза (стадия f) в качестве кислоты используют легкодоступную недорогую n-толуолсульфокислоту, а конденсацию проводят в присутствии перхлората магния при температуре от 70-125°C в автоклаве. Используемый по прототипу трифлат скандия, хотя и обеспечивает высокий выход целевого продукта (73%), однако дорог и труднодоступен, что ограничивает возможность его применения при масштабировании процесса. Необходимость использования более жестких условий в случае применения n-толуолсульфокислоты компенсируется доступностью и дешевизной используемых реагентов, при этом выход целевого продукта приближается к 70%.
Возможность практического реализации изобретения проиллюстрирована ниже конкретным примером его осуществления.
Пример 1. Получение гидрохлорида 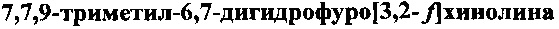 (ФДГХ-1)
(ФДГХ-1)
a. Получение N-(4-метоксифенил)ацетамида (соединение 1 на Фиг. 1)
К перемешиваемой суспензии n-анизидина (120,4 г, 0,98 моль) в метиленхлориде (400 мл) в течение 1 часа прибавляют уксусный ангидрид (95 мл, 0,98 моль) так, чтобы температура в реакционной смеси не поднималась выше 25-30°C. Для этого используют баню с холодной водой. После прибавления всего ангидрида смесь перемешивают 1,5 часа при 25°C. Затем гексан (1200 мл) прибавляют за час и сметанообразную массу выдерживают еще 1 час. Продукт отфильтровывают и сушат в вакууме при 40°C до постоянного веса. Получают 150 г вещества (выход 92,8%). 1H NMR (300 MHz, CDCl3) δ: 7.39 (m, 2Н), 7.28 (bs, NH), 6.86 (m, 2H), 3.79 (s, 3H), 2.16 (s, 3H).
b. Получение N-[3-(хлорацетилl)-4-гидроксифенил]ацетамида (соединение 2 на Фиг. 1)
К суспензии безводного хлорида алюминия (103,17 г, 0,77 моль) в 270 мл дихлорэтана при 0°C прибавляют хлорангидрид хлоруксусной кислоты (31,8 мл). После часа перемешивания порциями при комнатной температуре прибавляют 4-метоксиацетанилид (65,95 г, 0,4 моль) (охлаждение обеспечивается инкубацией колбы с реакционной смесью в бане лед-вода). После окончания прибавления 4-метоксиацетанилида реакционную смесь помещают в масляную баню, медленно поднимают температуру до 70°C и перемешивают при этой температуре в течение 1 часа. После остывания реакционную массу осторожно разбавляют ледяной водой (~1200 мл) и перемешивают до выпадения светло-желтого осадка, который отфильтровывают, промывают водой, петролейным эфиром (200 мл) и снова водой. Сушат на воздухе. Получают 83 г продукта (выход 91,3%), который без очистки используют на следующей стадии. 1H NMR (300 MHz, DMSO-D6) δ, ppm: 2.01 (s, 3H), 5.03 (s, 2Н), 6.95 (d J=9.0 Hz 1H), 7.69 (dd J=9.0, 3.0 Hz 1H), 7.93 (d J=3.0 Hz 1H), 9.88 (s, 1H), 10.81 (s, 1H).
c. Получение N-(3-гидрокси-2,3-дигидро-1-бензофуран-5-ил)ацетамида (соединение 3 на Фиг. 1)
Растворяют N-[3-(хлорацетил)-4-гидроксифенил]ацетамида (3,8 г, 16,7 ммоль) и ацетат натрия (1,7 г, 20 ммоль) в 100 мл метанола. Смесь кипятят 4 часа. После охлаждения до комнатной температуры к реакционной смеси осторожно прибавляют NaBH4 (0,98 г, 26 ммоль). Реакционную смесь перемешивают 2 часа. Растворитель отгоняют при пониженном давлении, остаток обрабатывают 100 мл воды и экстрагируют этилацетатом (4 раза по 50 мл). Органический слой сушат над прокаленным сульфатом магния. Растворитель отгоняют при пониженном давлении. Получают 1,26 г продукта (выход 39,1%), который без очистки используют на следующей стадии. 1H NMR (300 MHz, DMSO-D6) δ, ppm: 2.0 (s, 3 Н), 4.2 (dd, J=9.9, 3.3 Hz, 1 H), 4.5 (m, 1 H), 5.2 (s, 1 H), 5.6 (d, J=5.9 Hz, 1 H), 6.7 (d, J=8.8 Hz, 1 H), 7.3 (d, J=8.8 Hz, 1 H), 7.7 (s, 1 H), 9.8 (s, 1 H).
d. Получение N-1-бензофуран-5-ил ацетамида (соединение 4 на Фиг. 1)
К суспензии N-(3-гидрокси-2,3-дигидро-1-бензофуран-5-ил)ацетамида (24,34 г, 0,126 моль) в смеси 100 мл сухого ТГФ и 400 мл сухого метиленхлорида прибавляют 1,9 г p-TsOH и 8 г перхлората магния. Смесь кипятят с перемешиванием 26 часов. Растворитель отгоняют при пониженном давлении, остаток обрабатывают водой. Выпавший осадок отфильтровывают и промывают несколько раз водой. Сушат на воздухе. Получают 20,86 г продукта (выход 94,5%), который без очистки используют на следующей стадии. 1H NMR (500 MHz, DMSO-D6) δ, ppm: 2,05 (s, 3H); 6,93 (d, J=1,3 Hz, 1H); 7,37 (dd, J=8,8; 1,7 Hz, 1H); 7,50 (d, J=8,8 1H); 7,94 (d, J=1,6 Hz, 1H); 8,00 (d, J=1,6 Hz, 1H); 9,94 (s, 1H).
e. Получение солянокислого 1-бензофуран-5-амина (соединение 5 на Фиг. 1)
N-1-бензофуран-5-ил ацетамид (17,52 г, 0,1 моль) помещают в колбу, содержащую 60 мл 15% HCl. Смесь нагревают при 80°C в течение 40 минут. Раствор становится гомогенным. После остывания до комнатной температуры реакционную смесь концентрируют при пониженном давлении и нейтрализуют водным аммиаком. Выпавший осадок переносят в этилацетат (200 мл). Экстракт промывают насыщенным раствором соли, сушат над прокаленным сульфатом магния. Растворитель отгоняют при пониженном давлении, остаток растворяют в 70 мл метил-трет-бутилового эфира и фильтруют. Фильтрат насыщают сухим HCl. Выпавший осадок отфильтровывают, промывают МТБЭ и диэтиловым эфиром, сушат под вакуумом при 40°C. Получают 15,4 г продукта (выход 90,8%). Температура плавления 241-243°C (лит. 244-246°C, Kakimoto et. al., Chem. Zentralbl., 1954, v. 125, p. 12071).
f. Получение  (ФДГХ-1)
(ФДГХ-1)
Растворяют 1,69 г (10 ммоль) солянокислого 5-аминобензофурана в 4 мл воды и осторожно вносят сухой бикарбонат натрия массой 1,8 г (20 ммоль). После прекращения выделения газа экстрагируют эфиром (3 раза по 5 мл), органическую фазу объединяют и сушат над прокаленным MgSO4. После упаривания растворителя при пониженном давлении к остатку приливают 8 мл сухого ацетона, смесь помещают в автоклав и прибавляют p-TsOH (0,08 г) и Mg(ClO4)2 (1,7 г). Смесь нагревают при 120-125°C 6 часов. По окончании реакции реакционную смесь упаривают при пониженном давлении досуха, прибавляют 3 мл хлороформа и отфильтровывают через слой силикагеля. Очистку производят методом флеш-хроматографии (этилацетат:гексан - 1:3). Выход продукта составляет 1,42 г (66%) 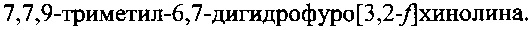 Т пл. 58°C из бензола. Реакцию можно проводить при более низкой температуре, увеличив соответственно время нагревания. Так, при 70°C реакция закачивается через 14 часов.
Т пл. 58°C из бензола. Реакцию можно проводить при более низкой температуре, увеличив соответственно время нагревания. Так, при 70°C реакция закачивается через 14 часов.
Результаты элементного анализа: C (78,79%), H (7,12%), N (6,59%). Вычислено: C (78,84%), H (7,09%), N (6,57%).
1H NMR (300 MHz, DMSO-D6) δ, ppm: 7.52 (s, 1H); 7,17 (d, J=8.54 Hz, 1H); 6,91 (s, 1H); 6,49 (d, J=8,54 Hz, 1H); 5.35 (s, 1H); 3.64 (br s, 1H); 2.27 (s, 3H); 1.28 (s, 6H).
ЯМР (δ, м.д.) 13C: 149,76 (2); 144,67 (10); 139,33 (5); 129,68 (8); 129,10 (9); 123,94 (4); 114,01 (3); 111,95 (13); 110,81 (12); 106,91 (11); 51,34 (7); 29,71 (14,15); 21,85 (16).
Результаты масс-спектроскопического анализа: m/z 213 [М]+; 198 [М-CH3]+; 183 [М-2CH3]+; 168 [М-3CH3]+; 154 [C10H4NO]+.
Спектр поглощения, (этанол), λмакс, нм (ε, М-1см-1): 362 (3160), 260 (14140), 213 (22400).
Суммарный выход по совокупности шести описанных выше стадий более чем втрое превышает выход по прототипу и составляет 18,8% - по изобретению, 5,8% - по прототипу.
В работе В.А. Кузьмин, Л.И. Мазалецкая, Т.Д. Некипелова, Е.Н. Ходот. Известия Академии наук. Серия химическая, 2008, №11, стр. 2356-2360 было показано, что ФДГХ-1 является эффективным антиоксидантом. Спектр поглощения ФДГХ-1 имеет полосу поглощения в области 400-330 нм, которая обычно используется в PUVA-терапии, с максимумом поглощения при 340-360 нм. ФДГХ-1 имеет высокий выход триплетных состояний при фотовозбуждении этой полосы поглощения. Спектры триплет-триплетного поглощения имеют максимумы поглощения в области 550-750 нм. Таким образом, это соединение имеет перспективу применения в качестве сенсибилизатора для применения в PUVA-терапии, а также может быть использовано в качестве антиоксиданта и антиозонанта для стабилизации каучуков, резин, и других продуктов.
Важно отметить, что заявляемый способ может быть положен в основу получения других, перспективных для применения в PUVA-терапии и других областях техники, производных ФДГХ с различным взаимным расположением O- и N-содержащих гетероциклических колец и содержащих в бензольном кольце дополнительные заместители. При этом, кроме стадий а-е, необходимых для получения аминобензофурана, являющегося предшественником целевого продукта, в схему синтеза включают дополнительные стадии, обеспечивающие введение дополнительного заместителя в бензольное кольцо. В частности, при использовании способа для синтеза 5-бром-6,8,8-триметил-8,9-дигидрофуро[3,2-h]хинолина (ФДГХ-5Б) или других производных, содержащих заместители в бензольном кольце, стадии с предшествует реакция нитрования продукта, полученного на стадии b, с получением 5-нитрофенилзамещенного производного (соединение 3 на Фиг. 2), которое затем вводят, в соответствии с заявляемым способом, в реакцию циклизация с последующим восстановлением кето-группы (стадия с) с получением ацетамидного производного нитродигидробензофурана (соединение 4 на Фиг. 2). Далее, в соответствии с заявляемым способом, на стадии d под действием n-толуолсульфокислоты в присутствии ангидрона с количественным выходом осуществляют дегидратацию продукта, полученного на стадии с, с получением ацетамидного производного нитробензофурана (соединение 5 на Фиг. 2), которое на следующей стадии е гидролизуют соляной кислотой с получением 7-нитро-1-бензофуран-5-амина (соединение 6 на Фиг. 2). Это соединение служит ключевым для получения аминобензофуранов с дополнительными заместителями в бензольном кольце, которые далее на стадии f согласно изобретению конденсируют с ацетоном в присутствии n-толуолсульфокислоты и перхлората магния при кипячении в автоклаве. Таким образом, на основе заявляемого способа могут быть получены триметилзамещенные фуродигидрохинолины, содержащие в бензольном кольце различные заместители (например, бром-, нитро-, ацетамидо- и др.).
В качестве примера возможность получения бром-замещенного производного проиллюстрирована ниже синтезом 5-бром-6,8,8-триметил-8,9-дигидрофуро[3,2-h]хинолина (ФДГХ-5Б).
Схема синтеза ФДГХ-5Б приведена на Фиг. 2 (стадии, дополнительные по отношению к заявляемому способу, на Фиг. 2 и в описании примера не имеют буквенных обозначений).
Пример 2. Получение 5-бром-6,8,8-триметил-8,9-дигидрофуро[3,2-h]хинолина (ФДГХ-5Б)
a. Получение N-(4-метоксифенил)ацетамида (соединение 1 на Фиг. 2)
К перемешиваемой суспензии n-анизидина (120,4 г, 0,98 моль) в метиленхлориде (400 мл) в течение 1 часа прибавляют уксусный ангидрид (95 мл, 0,98 моль) так, чтобы температура в реакционной смеси не поднималась выше 25-30°C. Для этого используют баню с холодной водой. После прибавления всего ангидрида смесь перемешивают в течение 1,5 часов при 25°C. Затем прибавляют гексан (1200 мл) за 1 час и сметанообразную массу выдерживают еще в течение 1 часа. Продукт отфильтровывают на стеклянном фильтре и сушат в вакууме при 40°C до постоянного веса. Выход продукта составляет 150 г (92,8%), который без очистки используют на следующей стадии.
Характеристики ЯМР-спектра N-(4-метоксифенил)ацетамида: 1H NMR (300 MHz, CDCl3) δ: 7,39 (m, 2Н), 7,28 (bs, NH), 6,86 (m, 2H), 3,79 (s, 3H), 2,16 (s, 3H).
b. Получение N-[3-(хлорацетил-l)-4-гидроксифенил]ацетамида (соединение 2 на Фиг. 2)
К суспензии безводного хлорида алюминия (103,2 г=0,77 моль) в 270 мл дихлорэтана при 0°C прибавляют хлорангидрид хлоруксусной кислоты (31,8 мл). После часа перемешивания порциями при комнатной температуре прибавляют 4-метоксиацетанилид (65,95 г, 0,4 моль) (охлаждение в бане лед-вода). После окончания прибавления 4-метоксиацетанилида реакционную смесь помещают в масляную баню, медленно повышают температуру до 70°C и перемешивают при данной температуре в течение 1 часа. После остывания реакционную смесь осторожно разбавляют ледяной водой (~1200 мл) и перемешивают до выпадения светло-желтого осадка, который отфильтровывают, промывают водой, петролейным эфиром (200 мл) и снова водой. Продукт высушивают на воздухе. Выход продукта составляет 83 г (~91,3%), который без очистки используется на следующей стадии.
Характеристики ЯМР-спектра N-[3-(хлорацетил-l)-4-гидроксифенил]ацетамида: 1H NMR (300 MHz, DMSO-D6) δ, ppm: 2,01 (s, 3H), 5,03 (s, 2Н), 6,95 (d J=9,0 Hz 1H), 7,69 (dd J=9,0, 3,0 Hz 1H), 7,93 (d J=3,0 Hz 1H), 9,88 (s, 1H), 10,81 (s, 1H).
Получение N-[3-(хлорацетил)-4-гидрокси-5-нитрофенил-l]ацетамида (соединение 3 на Фиг. 2)
К смеси N-[3-(хлорацетил)-4-гидроксифенил]ацетамида (соединение 2 на Фиг. 2) (14,5 г=63,7 ммоль) в 145 мл ледяной уксусной кислоты за 2 часа прибавляют раствор азотной кислоты (6,1 мл, d=1,42 в 17 мл ледяной уксусной кислоты) при температуре 15-20°C. Образовавшийся осадок отфильтровывают, промывают водой до нейтральной реакции, сушат на воздухе. Выход продукта составляет 12,6 г (72,5%), который без очистки может быть использован на следующей стадии.
Характеристики ЯМР-спектра N-[3-(хлорацетил)-4-гидрокси-5-нитрофенил-l]ацетамида: 1H NMR (300 MHz, DMSO-D6) δ, ppm: 2,06 (s, 3H), 5,08 (s, 2Н), 8,15 (d, J=2,98 Hz, 1H), 8,58 (d, J=2,99 Hz, 1H), 10,30 (s, 1H), 11,38 (br s).
c. Получение N-(3-гидрокси-7-нитро-2,3-дигидро-1-бензофуран-5-ил)ацетамида (соединение 4 на Фиг. 2)
К раствору N-[(3-(хлорацетил)-4гидрокси-5-нитрофенил]ацетамида (2,7 г, 11,3 ммоль) в 150 мл метанола прибавляют ацетат натрия (0,93 г, 11,3 ммоль). Реакционная масса приобретает темно-красную окраску. Смесь кипятят 2,5 часа, охлаждают до комнатной температуры и отфильтровывают. К фильтрату осторожно прибавляют NaBH4 (0,75 г, 19,8 ммоль), поддерживая температуру ≤25°C. В процессе реакции наблюдается сильное вспенивание. Смесь перемешивают в течение 2 часов, после чего растворитель отгоняют и к остатку прибавляют 100 мл воды. Полученный темно-красный, гомогенный раствор экстрагируют этилацетатом (5×50 мл). Органический слой сушат сульфатом магния. Растворитель отгоняют при пониженном давлении. Выход продукта составляет 0,76 г (31,6% от теоретического). Продукт без промежуточной очистки используют на следующей стадии.
Характеристики ЯМР-спектра N-(3-гидрокси-7-нитро-2,3-дигидро-1-бензофуран-5-ил)ацетамида: 1H NMR (300 MHz, DMSO-D6) δ, ppm: 2,1 (s, 3H), 4,5 (dd, J=11,0, 2,9 Hz, 1H), 4,7 (dd, J=10,3, 6,6 Hz, 1H), 5,3 (dd, J=6,6, 2,9 Hz, 1H), 8,0 (s, 1H), 8,3 (s, 1H), 10,2 (s, 1H).
d. Получение N-(7-нитро-1-бензофуран-5-ил)ацетамида (соединение 5 на Фиг. 2)
К суспензии N-(3-гидрокси-7-нитро-2,3-дигидро-1-бензофуран-5-ил)ацетамида (соединение 4 на Фиг. 2) (30 г, 0,126 моль) в смеси 100 мл сухого ТГФ и 400 мл сухого метиленхлорида прибавляют 1,9 г p-TsOH и 8 г перхлората магния. Смесь кипятят с перемешиванием в течение 18 часов. Растворитель отгоняют при пониженном давлении, остаток обрабатывают водой. Выпавший осадок отфильтровывают и промывают несколько раз водой и сушат на воздухе. Выход продукта составляет 25 г (90%), который без очистки используют на следующей стадии.
Характеристики ЯМР-спектра N-(7-нитро-1-бензофуран-5-ил-1)ацетамида: 1H NMR (300 MHz, DMSO-D6) δ, ppm: 2,1 (s, 3 H), 7,2 (s, 1 H), 8,2 (s, 1 H), 8,3 (s, 1 H), 8,4 (s, 1 H), 10,4 (s, 1 H).
е. Получение 7-нитро-1-бензофуран-5-амина (соединение 6 на Фиг. 2)
N-(7-нитро-1-бензофуран-5-ил)ацетамид (соединение 5 на Фиг. 2) (38,2 г=0,173 моль) нагревают с перемешиванием при 80°C в смеси 250 мл 1,5N HCl и 1200 мл метанола до израсходования ацетамида (контроль ТСХ в системе этилацетата, ~28 часов). После окончания реакции растворитель отгоняют при пониженном давлении, остаток нагревают с 200 мл спирта, смесь охлаждают до -12°C, выпавший осадок отфильтровывают и промывают холодным спиртом, сушат на воздухе и в вакууме. Выход продукта составляет 36,9 г (99%), который без очистки используют на следующей стадии.
Характеристики ЯМР-спектра 7-нитро-1-бензофуран-5-амина: 1H NMR (300 MHz, DMSO-D6) δ, ppm: 7,27 (s, 1H), 8,04 (s, 1H), 8,12 (s, 1Н), 8,34 (s, 1H), 8,80 (br s).
Получение 5-бром-7-нитро-1-бензофурана (соединение 7 на Фиг. 2)
К раствору 7-нитро-1-бензофуран-5-амина (17,8 г=0,1 моль) в концентрированной HCl (30 мл) и H2O (30 мл), охлажденному до 0-5°C, медленно прибавляют охлажденный раствор NaNO2 (6,9 г, 0,1 моль) в H2O (15 мл). Полученный раствор фильтруют, прибавляют к нему охлажденный раствор NH4BF4 (13,7 г=0,13 моль) в H2O (50 мл). Примерно через 30 минут перемешивания при 0°C выпадает осадок, который отфильтровывают, промывают холодным 5% раствором NH4BF4, холодным MeOH и диэтиловым эфиром. Полученную соль диазония сушат на воздухе и хранят при 0°C до использования. Выход составляет 24 г (87%).
К перемешиваемой суспензии KBr (7,14 г=60 ммоль) в сухом MeCN прибавляют дибензо-18-краун-6 (1,08 г=3 ммоль=10% моль), CuBr (0,43 г=3 ммоль,=10% моль) и CuBr2 (0,67 г=3 ммоль=10% моль). После перемешивания в течение двух минут прибавляют 1,10-фенантролин (0,54 г=3 ммоль=10% моль). К полученной смеси прикапывают раствор соли диазония в безводном MeCN (100 мл). Смесь перемешивают 20 минут при комнатной температуре, после чего разбавляют 250 мл МТБЭ, профильтровывают через слой силикагеля, чтобы удалить неорганические соли. Органический растворитель удаляют сушкой под вакуумом. Соединение используют на следующей стадии без дополнительной очистки. Выход: 23,7 г (98% от теоретического).
Характеристики ЯМР-спектра 5-бром-7-нитро-1-бензофурана: 1H NMR (400 MHz, DMSO-d6) δ, ppm: 7.13 (d, J=2.3 Hz, 1 H), 8.20 (d,. J=2.0 Hz, 1 H), 8.28 (d, J=2.3 Hz, 1 H), 8.32 (d, J=2.0 Hz, 1 H).
Получение 5-бром-1-бензофуран-7-амина (соединение 8 на Фиг. 2)
Смесь 5-бром-7-нитро-1-бензофурана (10) (3,8 г, 15,8 ммоль), порошок железа (4,6 г), метанола (40 мл), 1,4-диоксана (40 мл) и раствора хлористого аммония (4,6 г в 20 мл воды) кипятят 16 часов. Теплый раствор профильтровывают через слой силикагеля, который промывают горячим метанолом. Фильтрат упаривают и остаток перекристаллизовывают из 50% этанола (200 мл). Выпавшие кристаллы отфильтровывают и сушат в вакууме. Выход 2,1 г (62% от теоретического).
Характеристики ЯМР-спектра 5-бром-1-бензофуран-7-амина: 1H NMR (400 MHz, DMSO-d6) δ, ppm: 5.63 (s, 2 H), 6.68 (d, J=2.0 Hz, 1 H), 6.81 (d, J=2.3 Hz, 1 H), 6.96 (d, J=2.0 Hz, 1 H), 7.92 (d, J=2.3 Hz, 1 H).
f. Получение 5-бром-6,8,8-триметил-8,9-дигидрофуро[3,2-h]хинолина (ФДГХ-5Б соединение на Фиг. 2)
В автоклав помещают 5-бром-1-бензофуран-7-амина (2,12 г, 10 ммоль) в 15 мл сухого ацетона p-TsOH (0,08 г), Mg(ClO4)2 (1,7 г). Смесь нагревают в автоклаве при 120°C в течение 24 часов. Контролируют полноту протекания реакции методом ТСХ (силикагель Sorbfil ПТСХ-АФ-В-УФ, этилацетат, Rf=0,53). После расходования исходного амина растворитель отгоняют при пониженном давлении. Продукт очищают методом колоночной хроматографии (силикагель, этилацетат). Выход 1,72 г (59% от теоретического).
Характеристики ЯМР-спектра 5-бром-6,8,8-триметил-8,9-дигидрофуро[3,2-h]хинолина: 1H NMR (300 MHz, DMSO-D6) δ, ppm: 1.25 (s, 6Н), 1.98 (s, 3H), 5.24 (br s, 1H), 6.24 (s, 1H), 6.62 (s, 1H), 6.85 (d, J=2.4 Hz, 1H), 7.92 (d, J=2.6, 1H).
13C NMR (400 MHz, DMSO-D6) δ=21.54 (CH3), 29.46 (CH3), 31.96 (CH3), 69.93 (C), 101.55 (CH), 110.66 (C), 124.37 (C), 124.67 (CH), 124.87 (C), 130.37 (C), 132.33 (C), 136.80 (C), 138.91 (C), 141.58 (CH).
Масс-спектр [M+H] 293 (ESI).
Соединение 5-бром-6,8,8-триметил-8,9-дигидрофуро[3,2-h]хинолин ФДГХ-Б хорошо растворимо в спиртах, ароматических углеводородах, ацетонитриле, ДМСО, менее растворимо в алканах. Растворимость в воде составляет 2,1×10-5 моль/л.
Как и в случае ФДГХ-1, спектр поглощения ФДГХ-5Б имеет полосу поглощения в области 400-330 нм, которая обычно используется в PUVA-терапии, с максимумом поглощения при 340-360 нм. ФДГХ-5Б имеет высокий выход триплетных состояний при фотовозбуждении этой полосы поглощения, что делает его потенциально эффективным средством для PUVA-терапии псориаза. Спектры триплет-триплетного поглощения имеют максимумы поглощения в области 550-750 нм.
Методы анализа и приборы
Структуру и чистоту полученных промежуточных и конечных продуктов контролируют:
- спектроскопией ядерного магнитного резонанса на приборе АМ-300 (Bruker, Германия, 300 МГц) и DRX-500 (Bruker, Германия, 500 МГц);
- тонкослойной хроматографией на силикагеле на пластинках Kieselgel 60 F254 (Merck, Германия) и sorbfil ПТСХ-АФ-В-УФ, в системе этилацетат:гексан (1:1), если специально не оговорено иное.
Масс-спектры получают на установке Finigan MAT INCOS 50 (70 эВ) с прямым вводом и масс-спектрометре высокого разрешения MicrOTOF II (Bruker Daltonics, Германия) (ESI).
Спектры поглощения фурогидрохинолинов в УФ и видимой области измеряют на спектрофотометре Shimadzu UV-3101 PC (Япония) в кварцевых кюветах с длиной оптического пути 1 см. Растворы ФДГХ с концентрациями от 10-4 моль/л до 10-5 моль/л в этаноле готовят разбавлением маточного раствора с концентрацией ~2×10-3 моль/л (5 мг вещества в 10 мл этанола).
Спектры триплет-триплетного поглощения ФДГХ регистрируют на установке импульсного фотолиза на диапазоне. Возбуждение проводят с использованием третьей гармоники Nd-YAG лазера (λвозб=353 нм). Промежуточное поглощение регистрируют с помощью системы, состоящей из импульсной Xe лампы, расположенной под прямым углом к возбуждающему лазерному свету, в интервале 300-800 нм. Используют кювету с длиной оптического пути 1 см, сигнал записывают с помощью осциллографа Tektronix TDS 5052. Для удаления кислорода продувают кювету с исследуемым раствором аргона особой чистоты в течение 30 мин, как описано в работе Некипелова и соавт., Химия выс. энергий, 2012, т. 46, №3, 211.
Величина ΔФ ФДГХ-5Б при λ=553 нм составляет не менее 0,08 ОЕ по сравнению с 0,01 OE для ФДГХ-1, что подтверждает высокую фотохимическую активность ФДГХ-5Б.
| название | год | авторы | номер документа |
|---|---|---|---|
| Применение N-(6,8,8-триметил-8,9-дигидрофуро[3,2-h]хинолин-5-ил)ацетамида в качестве средства для фототерапии псориаза и псориатического артрита | 2018 |
|
RU2686692C1 |
| ПРОИЗВОДНОЕ 1', 2', 3'-ТРИМЕТОКСИБЕНЗО[5', 6:5, 4]-1H-6, 7-ДИГИДРОЦИКЛОГЕПТА[3, 2-f]БЕНЗОФУРАНА И ЕГО ПРИМЕНЕНИЕ | 2015 |
|
RU2593998C1 |
| ПРОИЗВОДНОЕ N-(1S)-1',2',3'-ТРИМЕТОКСИ-6,7-ДИГИДРО-1Н-БЕНЗО[5',6':5,4]ЦИКЛОГЕПТА-[3,2-F]БЕНЗОФУРАН-1-ИЛ)АЦЕТАМИДА И ЕГО ПРИМЕНЕНИЕ | 2013 |
|
RU2538982C1 |
| ПРОИЗВОДНЫЕ НЕЙРАМИНОВОЙ КИСЛОТЫ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЛЕЧЕНИЯ И ПРОФИЛАКТИКИ ГРИППА | 1997 |
|
RU2169145C2 |
| 1,3-Диметоксипропаны, содержащие 6,6-диметилбицикло[3.1.1]гептановый фрагмент природного происхождения и н-алкильный заместитель, способ их получения и титан-магниевый катализатор полимеризации пропилена, содержащий эти соединения в своем составе | 2024 |
|
RU2839765C1 |
| ДИ(3-СУЛЬФОФЕНИЛФОСФИНИЛ)ПРОИЗВОДНЫЕ 2,2'-БИПИРИДИЛА, 1,10-ФЕНАНТРОЛИНА И ПИРИДИНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2015 |
|
RU2620265C1 |
| ПРОИЗВОДНЫЕ ИЗОХИНОЛИНОНА, ПОЛЕЗНЫЕ ДЛЯ ЛЕЧЕНИЯ РАКА | 2015 |
|
RU2690853C2 |
| НОВЫЕ ПИРИДАЗОНЫ И ТРИАЗИНОНЫ ДЛЯ ЛЕЧЕНИЯ И ПРОФИЛАКТИКИ ЗАРАЖЕНИЯ ВИРУСОМ ГЕПАТИТА B | 2015 |
|
RU2664329C1 |
| ПРОИЗВОДНЫЕ ЦИКЛИЧЕСКОГО ЦИАНОЕНОНА КАК МОДУЛЯТОРЫ KEAP1 | 2021 |
|
RU2822828C1 |
| СПОСОБ ПОЛУЧЕНИЯ (±)-(12R*,13AR*,13BS*)-2,3,5,6,12,13,13A,13B-ОКТАГИДРО-1H-ИНДОЛО[3,2,1-DE]ПИРИДО[3,2,1-IJ]НАФТИРИДИН-12-ОЛА | 2023 |
|
RU2815974C1 |
Изобретение относится к способу синтеза 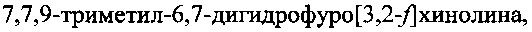 включающему конденсацию 5-аминобензофурана с ацетоном в присутствии n-толуолсульфокислоты и перхлората магния при температуре от 70-125°C в автоклаве, при этом 5-аминобензофуран получают путем осуществления последовательности стадий, включающей: а - ацилирование n-анизидина уксусным ангидридом с получением N-(4-метоксифенил)ацетамида; b - взаимодействие N-(4-метоксифенил)ацетамида, полученного на стадии а, с хлорангидридом хлоруксусной кислоты с получением N-[3-(хлорацетил)-4-гидроксифенил]ацетамида; с - замыкание дигидрофуранового цикла путем обработки продукта, полученного на стадии b, ацетатом натрия и последующего восстановления интермедиата без его выделения из реакционной смеси боргидридом натрия с получением N-(3-гидрокси-2,3-дигидро-1-бензофуран-5-ил)ацетамида; d - дегидратацию продукта, полученного на стадии с, путем его обработки n-толуолсульфокислотой и перхлоратом магния с получением N-1-бензофуран-5-ил ацетамида; е - кислый гидролиз продукта, полученного на стадии d, с получением 5-аминобензофурана. Способ может быть использован в качестве основы для синтеза различных производных триметилзамещенных по хинолиновому кольцу фуродигидрохинолинов (ФДГХ), представляющих практический интерес в качестве потенциальных сенсибилизаторов для применения в PUVA-терапии кожных заболеваний, а также они могут быть использованы в различных областях техники как антиоксиданты и антиозонанты. Технический результат состоит в упрощении и удешевлении процесса получения целевого продукта и повышении его суммарного выхода по сравнению с прототипом. 2 ил., 2 пр.
включающему конденсацию 5-аминобензофурана с ацетоном в присутствии n-толуолсульфокислоты и перхлората магния при температуре от 70-125°C в автоклаве, при этом 5-аминобензофуран получают путем осуществления последовательности стадий, включающей: а - ацилирование n-анизидина уксусным ангидридом с получением N-(4-метоксифенил)ацетамида; b - взаимодействие N-(4-метоксифенил)ацетамида, полученного на стадии а, с хлорангидридом хлоруксусной кислоты с получением N-[3-(хлорацетил)-4-гидроксифенил]ацетамида; с - замыкание дигидрофуранового цикла путем обработки продукта, полученного на стадии b, ацетатом натрия и последующего восстановления интермедиата без его выделения из реакционной смеси боргидридом натрия с получением N-(3-гидрокси-2,3-дигидро-1-бензофуран-5-ил)ацетамида; d - дегидратацию продукта, полученного на стадии с, путем его обработки n-толуолсульфокислотой и перхлоратом магния с получением N-1-бензофуран-5-ил ацетамида; е - кислый гидролиз продукта, полученного на стадии d, с получением 5-аминобензофурана. Способ может быть использован в качестве основы для синтеза различных производных триметилзамещенных по хинолиновому кольцу фуродигидрохинолинов (ФДГХ), представляющих практический интерес в качестве потенциальных сенсибилизаторов для применения в PUVA-терапии кожных заболеваний, а также они могут быть использованы в различных областях техники как антиоксиданты и антиозонанты. Технический результат состоит в упрощении и удешевлении процесса получения целевого продукта и повышении его суммарного выхода по сравнению с прототипом. 2 ил., 2 пр.
Способ синтеза 7,7,9-триметил-6,7-дигидрофуро[3,2-f]хинолина, включающий конденсацию 5-аминобензофурана с ацетоном в присутствии кислоты, отличающийся тем, что в качестве кислоты используют n-толуолсульфокислоту, а конденсацию проводят в присутствии перхлората магния при температуре 70-125°C в автоклаве, при этом 5-аминобензофуран получают путем осуществления следующей последовательности стадий, включающей:
а - ацилирование n-анизидина уксусным ангидридом с получением N-(4-метоксифенил)ацетамида;
b - взаимодействие N-(4-метоксифенил)ацетамида, полученного на стадии а, с хлорангидридом хлоруксусной кислоты с получением N-[3-(хлорацетил)-4-гидроксифенил]ацетамида;
с - замыкание дигидрофуранового цикла путем обработки продукта, полученного на стадии b, ацетатом натрия и последующего восстановления интермедиата без его выделения из реакционной смеси боргидридом натрия с получением N-(3-гидрокси-2,3-дигидро-1-бензофуран-5-ил)ацетамида;
d - дегидратацию продукта, полученного на стадии с, путем его обработки n-толуолсульфокислотой и перхлоратом магния с получением N-бензофуран-5-ил ацетамида;
е - кислый гидролиз продукта, полученного на стадии d, с получением 5-аминобензофурана.
| В.А.Кузьмин и др., Синтез и антиоксидантные свойства 7,7,9-триметил-6,7-дигидрофуро[3,2-f]хинолина, Изв | |||
| АН, серия химическая,2008 | |||
| WO 2011013159 A1 03.02.2011 | |||
| CN 103666452 A 26.03.2014. |

