Изобретение относится к производным нейраминовой кислоты или их фармацевтически приемлемым солям, обладающим превосходной способностью in vivo ингибировать сиалидазу; композициям, содержащим их в качестве активных ингредиентов, для лечения или профилактики заражений вирусом гриппа; их применению для производства лекарственного средства для лечения или профилактики заражений вирусом гриппа, способу лечения или профилактики заражений вирусом гриппа путем введения их фармацевтически эффективных количеств теплокровным животным, или способу их получения.
Грипп представляет собой заболевание, которое вызывается вирусной инфекцией. В одном из процессов, благодаря которым распространяется этот вирус, участвуют субвирусы, которые развились на поверхности клетки и отделяются от клетки. Такие субвирусы соединяются с сиаловой кислотой на поверхности клетки, причем связующим звеном служит гемагглютинин поверхности субвируса. Cубвирусы отсоединяются от клетки в результате действия на поверхности субвирусной частицы сиалидазы, которая разрушает сиаловую кислоту, приводя, тем самым, к вторичному заражению окружающих клеток. Таким образом, ингибирование сиалидазы должно сделать возможным ингибирование отделения субвирусных частиц от поверхности клетки, предотвращая тем самым вторичное инфицирование. Соответственно, полагают, что вещество, которое обладает способностью ингибировать сиалидазу, является эффективным для лечения или предотвращения (но предпочтительно - для лечения) заболевания гриппом.
Известные соединения, обладающие ингибиторной активностью в отношении сиалидазы и основой которых является сиаловая кислота (нейраминовая кислота), описаны в WO 91/16320 (PCT заявка Японии (Kokai) N Hei 5-507068). Среди них Соединение A (GG-167), которое испытывается в качестве лекарства для лечения гриппа: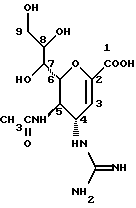
Авторы изобретения провели серьезное изыскание способа синтеза соединения, обладающего лучшим терапевтическим эффектом против гриппа, по сравнению с эффектом от Соединения А (GG-167), описанного в WO 91/16320 (PCT заявка Японии (Kokai) N Hei 5-507068), и исследование его фармакологической активности. В результате они обнаружили, что ацильные производные гидроксильной группы в 7-ом и 8-ом положениях и/или 9-ом положении и эфирные производные карбоксильной группы в 1-ом положении Соединения А показывают превосходную способность in vivo ингибировать репликацию вируса и ингибиторную активность в отношении сиалидазы, аналогичные Соединению А, в то же время проявляя лучший противоинфекционный лечебный эффект, чем Соединение А, при введении мышам, инфицированным вирусом гриппа. Таким образом, являясь полезными в качестве противогриппозных лекарственных средств, эти соединения представляют собой предмет настоящего изобретения.
Настоящее изобретение предлагает производное нейраминовой кислоты или его фармацевтически приемлемые соли, обладающие превосходной способностью in vivo ингибировать сиалидазу, композиции для лечения или профилактики заражений вирусом гриппа, содержащие их в качестве активных ингредиентов, их применение для производства лекарственного средства для лечения или профилактики заражений вирусом гриппа, способ лечения или профилактики заражений вирусом гриппа путем введения их фармакологически эффективных количеств теплокровным животным или способ их получения.
Hейраминовая кислота настоящего изобретения описывается формулой I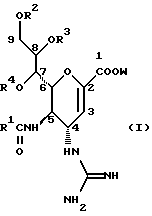
[в которой R1 представляет собой алкильную группу, имеющую от 1 до 4 атомов углерода, которая может быть замещена атомом галогена; R2, R3 и R4 - одинаковы или различны и каждый представляет собой атом водорода или алифатическую ацильную группу, имеющую от 3 до 25 атомов углерода, a W представляет собой атом водорода или сложноэфирный остаток, при условии, что случай, когда R1 - метильная группа, а каждый из R2, R3, R4 и W- атом водорода, исключается].
В вышеприведенной общей формуле (I):
"Алкильная группа, имеющая от 1 до 4 атомов углерода", "алкильной группы, имеющей от 1 до 4 атомов углерода, которая может быть замещена атомом галогена", R1 включает, например, метил, этил, н-пропил, изопропил, н-бутил, изобутил, вторбутил и трет-бутильную группы, предпочтительно метильную группу.
"Атом галогена" "алкильной группы, имеющей от 1 до 4 атомов углерода, которая может быть замещена атомом галогена" R1, включает, например, атом фтора, хлора или брома, предпочтительно атом фтора.
"Алкильная группа, имеющая от 1 до 4 атомов углерода, замещенная атомом галогена" "алкильной группы, имеющей от 1 до 4 атомов углерода, которая может быть замещена атомом галогена" R2, включает, например, монофторметильную, дифторметильную, трифторметильную, 1-фторэтильную, 2-фторэтильную, 1- фторпропильную, 2-фторпропильную, 3-фторпропильную, 4- фторбутильную, монохлорметильную, дихлорметильную, трихлорметильную, 1-хлорэтильную, 2-хлорэтильную, 1-хлорпропильную, 2- хлорпропильную, 3-хлорпропильную, 4-хлорбутильную, монобромметильную, 1-бромэтильную, 2-бромэтильную, 1-бромпропильную, 2-бромпропильную, 3-бромпропильную, 4-бромбутильную и фторхлорметильную группы, предпочтительно метильную группу, замещенную атомом фтора, более предпочтительны монофторметильная и дифторметильная группы.
Поэтому "алкильная группа, имеющая от 1 до 4 атомов углерода, которая может быть замещена атомом галогена" R1, в целом, включает предпочтительно метильную группу, которая может быть замещена атомом фтора, более предпочтительны метильная, монофторметильная и дифторметильная группы, наиболее предпочтительна метильная группа.
"Алифатическая ацильная группа, имеющая от 3 до 25 атомов углерода", R2, R3 и R4 включает, например, алкилкарбонильную группу, такую как пропионил, бутирил, изобутирил, пентаноил, пивалоил, валерил, изовалерил, октаноил, нонилкарбонил, децилкарбонил, 3-метилнонилкарбонил, 8-метилнонилкарбонил, 3-этилоктилкарбонил, 3,7-диметилоктилкарбонил, ундецилкарбонил, додецилкарбонил, тридецилкарбонил, тетрадецилкарбонил, пентадецилкарбонил, гексадецилкарбонил, 1-метилпентадецилкарбонил, 14-метилпентадецилкарбонил, 13,13-диметилтетрадецилкарбонил, гептадецилкарбонил, 15-метилгексадецилкарбонил, октадецилкарбонил, 1-метилгептадецилкарбонил, нонадецилкарбонил, эйкозилкарбонил, трикозилкарбонил и тетракозилкарбонил, предпочтительно алифатическую ацильную группу, имеющую от 6 до 25 атомов углерода, более предпочтительно алифатическую ацильную группу, имеющую от 6 до 20 атомов углерода, особенно предпочтительна гексаноильная, октаноильная, деканоильная, додеканоильная, миристоильная, пальмитоильная или стеароильная группа.
R2, R3, R4 в целом каждый представляет атом водорода или алифатическую ацильную группу с 6-25 атомами углерода, более предпочтительно атом водорода или алифатическую группу с 6-20 атомами углерода, особенно предпочтительно атом водорода и гексаноильную, октаноильную, деканоильную, додеканоильную, миристоильную, пальмитоильную или стеароильную группу.
Как комбинации R2, R3 и R4 возможны:
(а) комбинация, в которой R2 - алифатическая ацильная группа, имеющая от 3 до 25 атомов углерода (предпочтительно, алифатическая ацильная группа, имеющая от 6 до 25 атомов углерода, более предпочтительно, алифатическая ацильная группа, имеющая от 6 до 20 атомов углерода, особенно предпочтительны гексаноильная, октаноильная, деканоильная, додеканоильная, миристоильная, пальмитоильная или стеароильная группа), а каждый из R3 и R4 - атом водорода;
(b) комбинация, в которой R3 - алифатическая ацильная группа, имеющая от 3 до 25 атомов углерода (предпочтительно, алифатическая ацильная группа, имеющая от 6 до 25 атомов углерода, более предпочтительно, алифатическая ацильная группа, имеющая от 6 до 20 атомов углерода, особенно предпочтительны гексаноильная, октаноильная, деканоильная, додеканоильная, миристоильная, пальмитоильная или стеароильная группа), а каждый из R2 и R4 - атом водорода;
(с) комбинация, в которой R4 - алифатическая ацильная группа, имеющая от 3 до 25 атомов углерода (предпочтительно, алифатическая ацильная группа, имеющая от 6 до 25 атомов углерода, более предпочтительно алифатическая ацильная группа, имеющая от 6 до 20 атомов углерода, особенно предпочтительны гексаноильная, октаноильная, деканоильная, додеканоильная, миристоильная, пальмитоильная или стеароильная группа), а каждый из R2 и R3 - атом водорода;
(d) комбинация, в которой каждый из R2 и R3 - алифатическая ацильная группа, имеющая от 3 до 25 атомов углерода (предпочтительно алифатическая ацильная группа, имеющая от 6 до 25 атомов углерода, более предпочтительно алифатическая ацильная группа, имеющая от 6 до 20 атомов углерода, особенно предпочтительны гексаноильная, октаноильная, деканоильная, додеканоильная, миристоильная, пальмитоильная или стеароильная группа), a R4 - атом водорода;
(е) комбинация, в которой каждый из R2 и R4 алифатическая ацильная группа, имеющая от 3 до 25 атомов углерода (предпочтительно алифатическая ацильная группа, имеющая от 6 до 25 атомов углерода, более предпочтительно алифатическая ацильная группа, имеющая от 6 до 20 атомов углерода, особенно предпочтительны гексаноильная, октаноильная, деканоильная, додеканоильная, миристоильная, пальмитоильная или стеароильная группа), a R3 - атом водорода;
(f) комбинация, в которой каждый из R3 и R4- алифатическая ацильная группа, имеющая от 3 до 25 атомов углерода (предпочтительно, алифатическая ацильная группа, имеющая от 6 до 25 атомов углерода, более предпочтительно алифатическая ацильная группа, имеющая от 6 до 20 атомов углерода, особенно предпочтительны гексаноильная, октаноильная, деканоильная, додеканоильная, миристоильная, пальмитоильная или стеароильная группа), a R2 - атом водорода;
(q) комбинация, в которой каждый из R2, R3 и R4 - алифатическая ацильная группа, имеющая от 3 до 25 атомов углерода (предпочтительно алифатическая ацильная группа, имеющая от 6 до 25 атомов углерода, более предпочтительно алифатическая ацильная группа, имеющая от 6 до 20 атомов углерода, особенно предпочтительны гексаноильная, октаноильная, деканоильная, додеканоильная, миристоильная, пальмитоильная и стеароильная группа); и
(h) комбинация, когда каждый из R2, R3 и R4 - атом водорода.
Из этих комбинаций предпочтительными являются комбинации (а) и (h).
"Сложноэфирный остаток" W включает, к примеру, "алкильную группу", такую как метил, этил, н-пропил, изопропил, н- бутил, изобутил, втор-бутил, трет-бутил, н-пентил, изопентил, 2- метилбутил, неопентил, 1-этилпропил, н-гексил, изогексил, 4- метилпентил, 3-метилпентил, 2-метилпентил, 1-метилпентил, 3,3- диметилбутил, 2,2-диметилбутил, 1,1-диметилбутил, 1,2- диметилбутил, 1,3-диметилбутил, 2,3-диметилбутил, 2-этилбутил, 1-метилгексил, 2-метилгексил, 3-метилгексил, 4-метилгексил, 5- метилгексил, 1-пропилбутил, 4,4-диметилпентил, октил, 1- метилгептил, 2-метилгептил, 3-метилгептил, 4-метилгептил, 5- метилгептил, 6-метилгептил, 1-пропилпентил, 2-этилгексил, 5,5- диметилгексил, нонил, 3-метилоктил, 4-метилоктил, 5-метилоктил, 6- метилоктил, 1-пропилгексил, 2-этилгептил, 6,6-диметилгептил, децил, 1-метилнонил, 3-метилнонил, 8-метилнонил, 3-этилоктил, 3,7- диметилоктил, 7,7-диметилоктил, ундецил, 4,8-диметилнонил, додецил, тридецил, тетрадецил, пентадецил, 3,7,11-триметилдодецил, гексадецил, 4,8,12-триметилтридецил, 1-метилпентадецил, 14- метилпентадецил, 13,13-диметилтетрадецил, гептадецил, 15- метилгексадецил, октадецил, 1-метилгептадецил, нонадецил, эйкозил, 3,7,11,15-тетраметилгексадецил, хенейкозил, докозил, трикозил и тетракозил; "алкенильную группу", такую как этенил, 1-пропенил, 2-пропенил, 1-метил-2-пропенил, 1-метил-1-пропенил, 2-метил-1-пропенил, 2-метил-2-пропенил, 2-этил-2-пропенил, 1-бутенил, 2-бутенил, 1-метил-2-бутенил, 1-метил-1-бутенил, 3-метил-2-бутенил, 1-этил-2-бутенил, 3-бутенил, 1-метил-3-бутенил, 2-метил-3-бутенил, 1- этил-3-бутенил, 1-пентенил, 2-пентенил, 1-метил-2-пентенил, 2- метил-2-пентенил, 3-пентенил, 1-метил-3-пентенил, 2-метил- 3-пентенил, 4-пентенил, 1-метил-4-пентенил, 2-метил-4-пентенил, 1-гексенил, 2-гексенил, 3-гексенил, 4-гексенил и 5-гек- сенил; "алкинильную группу", такую как этинил, 2-пропинил, 1- метил-2-пропинил, 2-метил-2-пропинил, 2-этил-2-пропинил, 2- бутинил, 1-метил-2-бутинил, 2-метил-2-бутинил, 1-этил-2-бутинил, 3-бутинил, 1-метил-3-бутинил, 2-метил-3-бутинил, 1-этил-3-бутинил, 2-пентинил, 1-метил-2-пентинил, 2-метил-2-пентинил, 3-пентинил, 1- метил-3-пентинил, 2-мeтил-3-пeнтинил, 4-пeнтинил, 1-метил-4- пентинил, 2-метил-4-пентинил, 2-гексинил, 3-гексинил, 4-гексинил и 5-гексинил; "содержащую галоген низшую алкильную группу", такую как трифторметил, трихлорметил, дифторметил, дихлорметил, дибромметил, фторметил, 2,2,2-трифторэтил, 2,2,2-трихлорэтил, 2- бромэтил, 2-хлорэтил, 2-фторэтил, 2-йодэтил, 3-хлорпропил, 4- фторбутил, 6-йодгексил и 2,2-дибромэтил; "гидрокси низшую алкильную группу", такую как 2-гидроксиэтил, 2,3-дигидроксипропил, 3-гидроксипропил, 3,4-дигидроксибутил и 4-гидроксибутил; "содержащую алифатический ацил низшую алкильную группу", такую как ацетилметил; "низшую алкильную группу, замещенную 1-3 арилами", такую как бензил, фенэтил, 3-фенилпропил, α-нафтилметил, β-нафтилметил, дифенилметил, трифенилметил, 6- фенилгексил, α-нафтилдифенилметил и 9-антрилметил; "аралкильную группу, в которой арильное кольцо содержит в качестве заместителей низший алкил, низший алкокси, нитро, галоген, циано или алкоксикарбонил", такую как 4-метилбензил, 2,4,6- триметилбензил, 3,4,5-триметилбензил, 4-метоксибензил, 4- метоксифенилдифенилметил, 2-нитробензил, 4-нитробензил, 4- хлорбензил, 4-бромбензил, 4-цианобензил, 4-цианобензилдифенилметил, бис(2-нитрофенил)метил, пиперонил и 4-метоксикарбонилбензил; "три(алкил и/или фенил) силил группу", такую как триметилсилил, триэтилсилил, изопропилдиметилсилил, трет-бутилдиметилсилил, метилдиизопропилсилил, метилдитретбутилсилил, триизопропилсилил, метилдифенилсилил, изопропилдифенилсилил, бутилдифенилсилил и фенилдиизопропилсилил; "защищающую группу, способную к отщеплению биологическим способом, например в результате гидролиза в живом организме", то есть эфир, который образует свободную кислоту или ее соль путем гидролиза в организме человека, например "низший алкокси - низшая алкильная группа", такая как метоксиметил, 1-этоксиэтил, 1-метил-1-метоксиэтил, 1-(изопропокси) этил, 2-метоксиэтил, 2-этоксиэтил, 1,1-диметил-1-метоксиметил, этоксиметил, н-пропоксиметил, изопропоксиметил, н-бутоксиметил и трет-бутоксиметил; "низший алкоксилированный низший алкокси - низшая алкильная группа", такая как 2-метоксиэтоксиметил; "арил-окси - низшая алкильная группа", такая как феноксиметил; "галогенированный низший алкокси - низшая алкильная группа", такая как 2,2,2-трихлорэтоксиметил и бис(2-хлорэтокси)метил; "низшая алкильная группа, замещенная низшим алкоксикарбонилом", например метоксикарбонилметил; "низшая алкильная группа, замещенная циано", например цианометил и 2-цианоэтил; "низшая алкилтиометильная группа", например метилтиометил и этилтиометил; "арилтиометильная группа", например фенилтиометил и нафтилтиометил; "низшая алкильная группа, содержащая в качестве заместителя низший алкилсульфонил и которая может быть замещена галогеном", такая как 2-метансульфонилэтил и 2- трифторметансульфонилэтил; "арилсульфонил - низшая алкильная группа", например 2-бензолсульфонилэтил и 2-толуолсульфонилэтил; "замещенная алифатическим ацилокси низшая алкильная группа", такая как формилоксиметил, ацетоксиметил, пропионилоксиметил, бутирилоксиметил, пивалоилоксиметил, валерилоксиметил, изовалерилоксиметил, гексаноилоксиметил, 1-формилоксиметил, 1- ацетоксиэтил, 1-пропионилоксиэтил, 1-бутирилоксиэтил, 1- пивалоилоксиэтил, 1-валерилоксиэтил, 1-изовалерилоксиэтил, 1- гексаноилоксиэтил, 2-формилоксиэтил, 2-ацетоксиэтил, 2- пропионилоксиэтил, 2-бутирилоксиэтил, 2-пивалоилоксиэтил, 2- валерилоксиэтил, 2-изовалерилоксиэтил, 2-гексаноилоксиэтил, 1- формилоксипропил, 1-ацетоксипропил, 1-пропионилоксипропил, 1- бутирилоксипропил, 1-пивалоилоксипропил, 1-валерилоксипропил, 1-изовалерилоксипропил, 1-гексаноилоксипропил, 1-ацетоксибутил, 1-пропионилоксибутил, 1- бутирилоксибутил, 1-пивалоилоксибутил, 1-ацетоксипентил, 1- пропионилоксипентил, 1-бутирилоксипентил, 1-пивалоилоксипентил и 1-пивалоилоксигексил; "циклоалкил-карбонилокси-низшая алкильная группа", такая как циклопентаноилоксиметил, циклогексаноилоксиметил, 1-циклопентаноилоксиэтил, 1-циклогексаноилоксиэтил, 1-циклопентаноилоксипропил, 1-циклогексаноилоксипропил, 1-циклопентаноилоксибутил и 1-циклогексаноилоксибутил; "низшая алкильная группа, содержащая в качестве заместителя ароматический ацилокси", например бензоилоксиметил; "(алкоксикарбонилокси) алкильная группа", такая как метоксикарбонилоксиметил, этоксикарбонилоксиметил, пропоксикарбонилоксиметил, изопропоксикарбонилоксиметил, бутоксикарбонилоксиметил, изобутоксикарбонилоксиметил, пентилоксикарбонилоксиметил, гексилоксикарбонилоксиметил, циклогексилоксикарбонилоксиметил, циклогексилоксикарбонилокси-(циклогексил)метил, 1-(метоксикарбонилокси) этил, 1-(этоксикарбонилокси) этил, 1-пропоксикарбонилоксиэтил, 1-(изопропоксикарбонилокси) этил, 1-бутоксикарбонилоксиэтил, 1-изобутоксикарбонилоксиэтил, 1-(трет-бутоксикарбонилокси) этил, 1-пентилоксикарбонилоксиэтил, 1-гексилоксикарбонилоксиэтил, 1-циклопентилоксикарбонилоксиэтил, 1-циклопентилоксикарбонилоксипропил, 1-циклогексилоксикарбонилоксипропил, 1-циклопентилоксикарбонилоксибутил, 1-циклогексилоксикарбонилоксибутил, 1- (циклогексилоксикарбонилокси)этил, 1-(этоксикарбонилокси) пропил, 2-метоксикарбонилоксиэтил, 2-этоксикарбонилоксиэтил, 2-пропоксикарбонилоксиэтил, 2-изопропоксикарбонилоксиэтил, 2-бутоксикарбонилоксиэтил, 2-изобутоксикарбонилоксиэтил, 2-пeнтилoкcикapбoнилoкcиэтил, 2-гексилоксикарбонилоксиэтил, 1-метоксикарбонилоксипропил, 1-эгоксикарбонилоксипропил, 1-пропоксикарбонилоксипропил, 1-изопропоксикарбонилоксипропил, 1-бутoкcикapбoнилoкcипpoпил, 1-изобутоксикарбонилоксипропил, 1-пентилоксикарбонилоксипропил, 1-гексилоксикарбонилоксипропил, 1-метоксикарбонилоксибутил, 1- этоксикарбонилоксибутил, 1-пропоксикарбонилоксибутил, 1- изопропоксикарбонилоксибутил, 1-бутоксикарбонилоксибутил, 1- изобутоксикарбонилоксибутил, 1-метоксикарбонилоксипентил, 1- этоксикарбонилоксипентил, 1-метoкcикapбoнилoкcигeкcил и 1- этоксикарбонилоксигексил; "оксодиоксоленилметильная группа", такая, как (5-фенил-2-оксо-1,3-диоксолен-4-ил)метил, [5-(4- метилфенил)-2-оксо-1,3-диоксолен-4-ил] метил, [5-(4-метоксифенил) -2-оксо-1,3-диоксолен-4-ил] метил, [5-(4-фторфенил)-2- оксо-1,3-диоксолен-4-ил]метил, [5-(4-хлорфенил)-2-оксо-1,3- диоксолен-4-ил]метил, (2-оксо-1,3-диоксолен-4-ил] метил, (5-метил- 2-оксо-1,3-диоксолен-4-ил]метил, (5-этил-2-оксо-1,3-диоксолен-4- ил) метил, (5-пропил-2-оксо-1, 3-диоксолен-4-ил)-метил, (5- изопропил-2-оксо-1,3-диоксолен-4-ил)метил, (5-бутил-2-оксо-1,3- диоксолен-4-ил)метил; "фталидильная группа", такая как фталидил, диметилфталидил и диметоксифталидил; "арильная группа", такая как фенил и инданил; и "карбоксиалкильная группа", например карбоксиметил, предпочтительно "алкильную группа", более предпочтительно, алкильную группу, имеющую от 1 до 18 атомов углерода.
В случае если по крайней мере один из R2, R3 и R4 - алифатическая ацильная группа с 3-25 атомами углерода, "эфирный остаток" W - предпочтительно алкильная группа с 1-18 атомами углерода. В этом случае W, в целом, является атомом водорода или алкильной группой с 1-18 атомами углерода, более предпочтительно атом водорода.
В случае если каждый из R2, R3, R4 - атом водорода, "эфирный остаток" W - предпочтительно алкильная группа, имеющая от 1 до 18 атомов углерода, более предпочтительно от 6 до 18 атомов углерода. В этом случае W, в целом, представляет собой предпочтительно остаток эфира, более предпочтительно - алкильную группу с 6-18 атомами углерода.
"Фармакологически приемлемая соль" включает соли щелочных металлов, такие как соли натрия, калия и лития, соли щелочноземельных металлов, такие как соли кальция и магния, соли металлов, например соли алюминия, железа, цинка, меди, никеля и кобальта; соли неорганического амина, такие как аммониевые соли, соли органических аминов, такие как соли третоктиламина, дибензиламина, морфолина, глюкозамина, фенилглициналкилового эфира, этилендиамина, N-метилглюкамина, гуанидина, диэтиламина, триэтиламина, дициклогексиламина, N, N'-дибензилэтилендиамина, хлорпрокаина, прюкаина, диэтаноламина, N-бензилфенэтиламина, пиперазина, тетраметиламмония и трис (гидроксиметил) аминометана; галогенгидратные соли, например фторгидраты, хлоргидраты, бромгидраты и йодгидраты, соли неорганических кислот, такие как нитраты, перхлораты, сульфаты и фосфаты; соли органических кислот, включающие низшие алкансульфонаты, такие как метансульфонаты, трифторметансульфонаты и этансульфонаты, арилсульфонаты, такие как бензолсульфонаты и п-толуолсульфонаты, ацетаты, трифторацетаты, малаты, фумараты, сукцинаты, цитраты, тартраты, оксалаты и малеаты; и соли аминокислот, например соли глицина, лизина, аргинина, орнитина, глутаматы и аспартаты, предпочтительно соли щелочных металлов, таких как натриевые, калиевые и литиевые соли и соли органических кислот, такие как ацетаты и трифторацетаты, и соли неорганических кислот, такие как хлоргидраты и сульфаты.
Из всех соединений настоящего изобретения предпочтительные включают следующие:
(1) Соединения, в которых R1 - метильная группа, которая может быть замещена атомом фтора;
(2) Соединения, в которых R1 - метил, монофторметил или дифторметильная группа;
(3) Соединения, в которых R1 - метильная группа;
(4) Соединения, в которых R2 - атом водорода или алифатическая ацильная группа, имеющая от 6 до 25 атомов углерода;
(5) Соединения, в которых R2 - атом водорода или алифатическая ацильная группа, имеющая от 6 до 20 атомов углерода;
(6) Соединения, в которых R2 - атом водорода или гексаноильная, октаноильная, деканоильная, додеканоильная, миристоильная, пальмитоильная или стеароильная группа;
(7) Соединения, в которых R3 - атом водорода или алифатическая ацильная группа, имеющая от 6 до 25 атомов углерода;
18) Соединения, в которых R3 - атом водорода или алифатическая ацильная группа, имеющая от 6 до 20 атомов углерода;
19) Соединения, в которых R3 - атом водорода или гексаноильная, октаноильная, деканоильная, додеканоильная, миристоильная, пальмитоильная или стеароильная группа;
(10) Соединения, в которых R4 - атом водорода или алифатическая ацильная группа, имеющая от 6 до 25 атомов углерода;
(11) Соединения, в которых R4 - атом водорода или алифатическая ацильная группа, имеющая от 6 до 20 атомов углерода;
(12) Соединения, в которых R4 - атом водорода или гексаноильная, октаноильная деканоильная, додеканоильная, миристоильная, пальмитоильная или стеароильная группа;
(13) Соединения, в которых R2 - алифатическая ацильная группа, имеющая от 3 до 25 атомов углерода, и каждый из R3 и R4 - атом водорода;
(14) Соединения, в которых R2- алифатическая ацильная группа, имеющая от 6 до 25 атомов углерода, и каждый из R3 и R4 - атом водорода;
(15) Соединения, в которых R2 - алифатическая ацильная группа, имеющая от 6 до 20 атомов углерода, и каждый из R3 и R4 - атом водорода;
(16) Соединения, в которых R2 - гексаноильная, октаноильная, деканоильная, додеканоильная, миристоильная, пальмитоильная или стеароильная группа, и каждый из R3 и R4 - атом водорода;
(17) Соединения, в которых W - атом водорода или алкильная группа с 1-10 атомами углерода;
(18) Соединения, в которых W - атом водорода;
(19) Соединения, в которых W - остаток эфира;
(20) Соединения, в которых W - алкильная группа с 6-18 атомами углерода.
Далее, соединения, полученные комбинированием значений заместителей R1, R2, R3, R4 и W, отобранных среди значений для вышеприведенных соединений с (1) по (20), являются более предпочтительными и включают, например, следующие соединения.
(21) Соединения, в которых каждый из R2, R3 и R4 - атом водорода, а W - остаток эфира;
(22) Соединения, в которых каждый из R2, R3 и R4 - атом водорода, а W - алкильная группа с 6-18 атомами углерода;
(23) Соединения, в которых R1 - метильная группа, которая может быть замещена атомом фтора, R2 - алифатическая ацильная группа, имеющая от 3 до 25 атомов углерода, каждый из R3 и R4 - атом водорода, a W - атом водорода или остаток эфира;
(24) Соединения, в которых R1 - метильная группа, R2 - алифатическая ацильная группа, имеющая от 6 до 25 атомов углерода, каждый из R3 и R4 - атом водорода, a W - атом водорода или алкильная группа с 1-18 атомами углерода,
(25) Соединения, в которых R1 - метильная группа, R2 - алифатическая ацильная группа, имеющая от 6 до 20 атомов углерода, а каждый из R3, R4 и W - атом водорода.
(26) Соединения, в которых R1 - метильная группа, которая может быть замещена атомом фтора, каждый из R2, R3 и R4 - атом водорода, а W - остаток эфира, и
(27) Соединения, в которых R1 - метильная группа, каждый из R2, R3 и R4 - атом водорода, а W - алкильная группа с 6-18 атомами углерода.
Далее в Таблице 1 представлены возможные соединения настоящего изобретения.
Из приведенных в табл. 1 соединений предпочтительны 36, 37, 38, 38a, 39, 39a, 40, 40a, 41, 41a, 42, 42a, 43, 43a, 44, 44a, 45, 45a, 45b, 45c, 87, 87a, 87b, 87c, 87d, 87e, 87f, 87g, 88, 88a, 89, 90,91, 92, 92a, 93, 94, 94a, 94b, 214, 215, 216, 217, 218, 219, 220, 221, 222, 223, 265, 266, 267, 268, 269, 270, 271, 272, 393, 394, 395, 396, 397, 398, 399, 400, 401, 402, 444, 445, 446, 447, 448, 449, 450 и 451.
Более предпочтительными являются соединения 36, 37, 38, 38a, 39, 39a, 40, 40a, 41, 41a, 42, 42a, 43, 43a, 44, 44a, 45, 45a, 45b, 45c, 87, 87a, 87b, 87c, 87d, 87e, 87f, 87g, 88, 88a, 89, 90, 91, 92, 92a, 93, 94, 94a, 94b, 219, 220, 221, 222, 269, 270, 271, 272, 398, 399, 400, 401, 448, 449, 450 и 451.
Следующие соединения являются самыми предпочтительными:
5-ацетамидо-2,3,4,5- тетрадеокси-4-гуанидино-9-O-гексаноил-D-глицеро-D-галакто-нон- 2-энопиранозойная кислота (примерное соединение N 36)
5-ацетамидо-2,3,4,5-тетрадеокси-4-гуанидино-9-O-октаноил-D- глицеро-D-галакто-нон-2-энопиранозойная кислота (примерное соединение N 38)
5-ацетамидо-2,3,4,5-тетрадеокси-4-гуанидино-9-O- деканоил-D-глицеро-D-галакто-нон-2-энопиранозойная кислота (примерное соединение N 39)
5-ацетамидо-2,3,4,5-тетрадеокси-4- гуанидино-9-O-додеканоил-D-глицеро-D-галакто-нон-2- энопиранозойная кислота (примерное соединение N 40)
5-ацетамидо-2,3,4,5-тетрадеокси-4- гуанидино-9-O-миристоил-D-глицеро-D-галакто-нон- энопиранозойная кислота (примерное соединение N 41)
5-ацетамидо-2,3,4,5-тетрадеокси-4-гуанидино-9-O-пальмитоил- D-глицеро-D-галакто-нон-2-энопиранозойная кислота (примерное соединение N 42)
5-ацетамидо-2,3,4,5-тетрадеокси-4-гуанидино-9-O- стеароил-D-глицеро-D-галакто-нон-2-энопиранозойная кислота (примерное соединение N 43)
гексил 5-ацетамидо-2, 3, 4, 5- тетрадеокси-4-гуанидино-D-глицеро-D-галакто-нон-2-энопиранозоат (примерное соединение N 87)
миристил 5-ацетамидо-2, 3, 4, 5- тетрадеокси-4-гуанидино-глицеро-D-галакто-нон-2-энопиранозоат (примерное соединение N 88)
цетил 5-ацетамидо-2, 3, 4, 5- тетрадеокси-4-гуанидино-D-глицеро-D-галакто-нон-2-энопиранозоат (примерное соединение N 89)
стеарил 5-ацетамидо-2, 3, 4, 5- тетрадеокси-4-гуанидино-D-глицеро-D-галакто-нон-2-энопиранозоат (примерное соединение N 91)
Далее будет описан способ получения соединения (1) настоящего изобретения.
Соединение (1) настоящего изобретения может быть получено в соответствии со способом, описанным в Схемах A, B или C. Далее, соединение (1) может быть также получено по Схеме J, описанной позже.
Исходное соединение (2), используемое в Схемах А и В, может быть получено способами, которые отражены в Схемах D, E, F или G, показанных ниже. Далее, исходное соединение (5), используемое в Схеме C, может быть получено в соответствии со способом, описанным в Схеме H, показанной ниже.
Значения R1, R2, R2a, R2b, R3, R3a, R4, R4a, R6, R7, R8, W, Wa, Me, Ac и Boc, используемых на стадиях Схем от А до J, показаны ниже.
То есть, R1, R2, R3, R4 и W имеют те же самые значения, что определены выше.
R2a имеет то же значение, что и указанное выше для R2, или представляет собой защитную группу гидроксила (предпочтительно трет-бутилдиметилсилильную группу или изопропилиденовую группу, взятую вместе с защитной группой гидроксила R3a).
R2b представляет собой защитную группу гидроксила (предпочтительно трет-бутилдиметилсилильную группу).
R3a имеет то же значение, что указанное выше для R3, или представляет собой защитную группу гидроксила (предпочтительно трет-бутилдиметилсилильную группу или изопропилиденовую группу, взятую вместе с защитной группой гидроксила R2a).
R4a имеет то же значение, что и указанное выше для R4, или представляет собой защитную группу гидроксила (предпочтительно трет-бутилдиметилсилильную группу).
R6, R7 и R8 могут быть одинаковыми и различными и каждый может представлять собой алифатическую ацильную группу, имеющую от 3 до 25 атомов углерода.
Wa имеет такое же значение, как и W, или представляет собой защитную группу карбоксильной группы (предпочтительно метил, этил, бензил, аллил, метоксиметил, метилтиометил, 2-(триметилсилил)этоксиметокси или дифенилметильную группу, более предпочтительны метил, бензил или дифенилметильная группа).
Ас представляет собой ацетильную группу,
Boc представляет собой трет-бутоксикарбонильную группу, и
Me представляет собой метильную группу.
В последующем каждая Схема (Схемы приведены в конце описания) будет расписана в деталях.
Схема A - это способ получения соединения (1) настоящего изобретения снятием защитной группы соединения (3), полученного взаимодействием исходного соединения (2), которое легко получают по способу, описанному ниже, с N, N'-ди-трет- бутоксикарбонилтиомочевиной.
(Стадия А-1)
На данной стадии получают соединение (3) взаимодействием соединения (2) с N,N'-ди- трет-бутоксикарбонилтиомочевиной в инертном растворителе в присутствии основания и хлорида ртути.
Природа применяемого растворителя не является особенно критической, поскольку она не оказывает влияния на реакцию, и это может быть ароматический углеводород, такой как бензол, толуол и ксилол, эфиры, такие как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран, диоксан, диметоксиэтан и диметиловый эфир диэтиленгликоля, и амиды, такие как N,N-диметилацетамид и диметилформамид, предпочтительно амиды (в частности, N,N-диметилацетамид и диметилформамид).
Применяемое основание включает здесь, предпочтительно, органические основания, такие как триэтиламин и диметиламинопиридин.
Температура реакции обычно составляет от -10 до 50oC, предпочтительно 10 - 30oC.
Продолжительность реакции варьирует в зависимости от используемых материалов, основания, температуры реакции и т.д. и составляет обычно 1 - 24 ч, предпочтительно 5 - 10 ч.
После реакции целевое соединение получают, например, фильтрованием реакционной смеси при пониженном давлении для удаления нерастворимых примесей, добавлением в нее несмешивающегося с водой органического растворителя, например этилацетата, промыванием смеси водой с отделением органической фазы, содержащей целевое соединение, сушкой этого слоя над безводным сульфатом магния и выпариванием растворителя.
В случае необходимости, целевое соединение может быть в последующем очищено перекристаллизацией или с помощью различных видов хроматографии.
(Стадия А-2)
На данной стадии получают соединение (1) настоящего изобретения взаимодействием соединения (3) с реагентом для снятия защитной трет-бутоксикарбонильной группы в инертном растворителе.
Природа применяемого растворителя не является особенно важной, поскольку она не оказывает влияния на реакцию, и это могут быть, предпочтительно, спирты, такие как метанол и этанол, вода и их смесь.
Реагент для снятия защиты предпочтительно является кислотой, но не особенно важно какой, поскольку она используется как кислотный катализатор в обычной реакции и включает кислоты Bronsted'a, такие как неорганические кислоты, т.е. соляная кислота, бромистая кислота, серная кислота, перхлорная кислота и фосфорная кислота, и органические кислоты, т.е. уксусная кислота, муравьиная кислота, щавелевая кислота, метансульфокислота, пара-толуолсульфокислота, трифторуксусная кислота и трифторметансульфокислота; кислоты Льюиса, такие как хлорид цинка, тетрахлорид олова, трихлорид бора, трифторид бора и трибромид бора; и кислые ионообменные смолы, предпочтительно органических кислот (в частности, уксусной кислоты и трифторуксусной кислоты).
Температура реакции обычно составляет 10 - 50oC , предпочтительно 10 - 30oC.
Продолжительность реакции варьирует в зависимости от исходных материалов, основания, температуры реакции и т.д. и составляет обычно от 15 мин до 10 ч предпочтительно 1 - 5 ч.
После реакции целевое соединение получают, например, нейтрализацией реакционной смеси, отгонкой растворителя при пониженном давлении с последующей очисткой остатка на хроматографической колонке с силикагелем.
Далее, в случае когда R2a, R3a или R4a является защитной группой гидроксильной группы или Wa - защитная группа карбоксильной группы, они далее удаляются с получением соединения (1) настоящего изобретения.
Способ снятия защитной группы варьирует в зависимости от типа защитной группы и может выполняться в соответствии с обычными общепринятыми методиками, например, методами, описанными в Protective Groups in Organic Synthesis, Second Edition (1991, Green et al.).
В случае если защитной группой гидроксила является триалкилсилильная группа, такая как трет-бутилдиметилсилильная группа, уксусная кислота предпочтительно используется в смеси с водой и тетрагидрофураном или используется фторид тетрабутиламмония в тетрагидрофуране.
В случае если защитной группой гидроксила является изопропилиденовая группа, используют описанный ниже метод стадии Е-2 или Е-4.
В случае если защитной группой карбоксила являются дифенилметильная группа, осуществляют каталитическое восстановление, используемая кислота является, например, уксусной или трифторуксусной кислотой, или используют комплекс три-фторборан-диэтиловый сложный эфир.
В случае если защитной группой карбоксила является бензильная группа, проводят каталитическое восстановление, а в случае, если защитной группой является метильная группа, проводят гидролиз.
Схема В - это процесс получения соединения (1) настоящего изобретения взаимодействием исходного соединения (2), которое легко можно получить в соответствии со способом, описанным ниже, с цианирующим агентом, взаимодействием полученного соединения с аммиаком и последующим, если необходимо, удалением защитной группы.
(Стадия B-1)
На данной стадии получают соединение (4) взаимодействием соединения (2) с цианирующим агентом в инертном растворителе. Применяемый растворитель не является особенно важным, поскольку он не оказывает влияния на реакцию и включает спирты, например метанол, этанол, н-пропанол, изопропанол, н- бутанол, изобутанол, трет-бутанол, изоамиловый спирт, диэтиленгликоль, глицерин, октанол, циклогексанол и метилцеллозольв; амиды, например формамид, N,N-диметилформамид, N, N-диметилацетамид, N-метил-2-пирролидон, N-метилпирролидинон и гексаметилфосфорный триамид; и сульфоксиды, такие как диметилсульфоксид и сульфолан, предпочтительны спирты (особенно метанол).
Приемлемый цианирующий агент, предпочтительно, включает бромциан, и в качестве основания одновременно используют ацетат натрия.
Температура реакции обычно составляет от -10 до 50oC, предпочтительно 10 - 40oC.
Продолжительность реакции варьирует в зависимости от используемого материала, основания, температуры реакции и т.д. и составляет обычно от 15 мин до 10 ч, предпочтительно 1 - 5 ч.
После реакции целевое соединение может быть получено, например, выпариванием растворителя и последующей очисткой остатка перекристаллизацией и колоночной хроматографией на силикагеле.
(Стадия В-2)
На данной стадии получают соединение (1) настоящего изобретения взаимодействием соединения (4) с аммиаком в инертном растворителе.
Растворитель, который может здесь использоваться, не является особенно критическим, поскольку он не влияет на реакцию и включает, предпочтительно, спирты (особенно метанол).
Температура реакции составляет обычно от -10 до 50oC, предпочтительно 10 - 40oC.
Продолжительность реакции варьирует в зависимости от используемого материала, основания, температуры реакции и т.д. и обычно составляет от 15 мин до 10 ч, предпочтительно 1 - 5 ч.
После реакции целевое соединение может быть получено, например, отгонкой растворителя с последующей очисткой остатка перекристаллизацией или колоночной хроматографией на силикагеле.
Между прочим, в случае когда R2a, R3a или R4a - защитная группа гидроксила или Wa - защитная группа карбоксила, соединение настоящего изобретения получают удалением защитной группы способом, аналогичным Cхеме А.
Схема C - способ получения соединения (1) настоящего изобретения полным или частичным ацилированием гидроксильных групп исходного соединения (5), которое легко получить в соответствии с методом, описанным ниже, и дальнейшим снятием защитной группы с полученного соединения.
(Стадия C-1)
На данной стадии получают соединение (6) введением желаемой ацильной группы в соединение (5) в инертном растворителе.
Метод ацилирования включает следующие методы с 1 по 3.
(Метод 1)
Метод 1 призван обеспечить взаимодействие соединения общей формулы: RCO-Hal или соединения общей формулы: RCO-О-COR [где
R представляет собой алкильную группу, Hal представляет собой группу, которая должна быть удалена, а группа, которая должна быть удалена, не является особенно критической, поскольку она является группой, которую необходимо удалить как нуклеофильный остаток, и включает, предпочтительно, атом галогена, например хлор, бром, йод; низшую алкоксикарбонил-оксигруппу, например метоксикарбонилокси и этоксикарбонилокси; галогенированную алкилкарбонилоксигруппу, например хлорацетилокси, дихлорацетилокси, трихлорацетилокси и трифторацетилокси; низшую алкансульфонилоксигруппу, например метансульфонилокси и этансульфонилокси; галоген-низшую-алкансульфонилгруппу, такую как трифторметансульфонилокси и пентафторэтансульфонилокси; и арилсульфонилокси-группу, такую как бензолсульфонилокси, п-толуолсульфонилокси и п-нитробензолсульфонилокси, более предпочтительны> атом галогена, галоген-низшая-алкансульфонилокси-группа и арилсульфонилокси-группа]
с соединением (5) в растворителе в присутствии или в отсутствие основания.
Приемлемый для применения растворитель не является особенно критическим, поскольку он не ингибирует реакцию и растворяет исходные вещества до определенной степени, он включает, предпочтительно, алифатические углеводороды, такие как гексан и гептан; ароматические углеводороды, такие как бензол, толуол и ксилол; галогенированные углеводороды, такие как метиленхлорид, хлороформ, четыреххлористый углерод, дихлорэтан, хлорбензол и дихлорбензол; эфиры, например этилформиат, этилацетат, пропилацетат, бутилацетат и диэтилкарбонат; эфиры, такие как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран, диоксан, диметоксиэтан и диметиловый эфир диэтиленгликоля; нитрилы, такие как ацетонитрил и изо-бутиронитрил; и амиды, такие как формамид, N, N-диметилформ-амид, N, N-диметилацетамид, N-метил- 2-пирролидон, N-метил-пирролидинон и триамид гексаметилфосфорной кислоты.
Основание, которое может применяться, не является особенно критическим, поскольку оно используется как основание в обычной реакции и включает, предпочтительно, органические основания, такие как N-метилморфолин, триэтиламин, трибутиламин, диизопропилэтиламин, дициклогексиламин, N-метилпиперидин, пиридин, 4-пирролидинопиридин, пиколин, 4-(N,N-(диметиламино) пиридин, 2, 6-ди(трет-бутил)-4-метилпиридин, хинолин, N,N-диметиланилин и N,N-диэтиланилин.
Между прочим, 4-(N,N-диметиламино) пиридин и 4-пирролидинопиридин могут использоваться в каталитическом количестве при сочетании с другими, основаниями и, в последующем, четвертичная аммониевая соль, например хлорид бензилтриэтил-аммония и хлорид тетрабутиламмония, а также Крауновские эфиры, например дибензо- 18-краун-6, могут также быть добавлены туда же для эффективного осуществления реакции.
Реакция обычно проводится при от -20oC до температуры дефлегмации используемого растворителя, предпочтительно от 0oC до температуры дефлегмации используемого растворителя.
Продолжительность реакции зависит, главным образом, от температуры реакции, используемого основания и типа растворителя и обычно составляет от 10 мин до 3 дней, предпочтительно 1 - 6 ч.
(Метод 2)
Метод 2 призван обеспечить взаимодействие соединения общей формулы: RCOOH [где R имеет то же значение, что и определенное выше] с соединением (5) в растворителе в присутствии или в отсутствие "этерифицирующего агента" и каталитического количества основания.
"Этерифицирующий агент", который пригоден для применения, включает конденсирующий агент; галогенированные формиаты, такие как метилхлорформиат и этилхлорформиат; диэфиры цианофосфорной кислоты, например диэтилцианофосфат. Такие "конденсирующие агенты" включают N-гидроксипроизводные, например N-гидроксисукцинимид, 1-гидроксибензотриазол и N-гидрокси-5-норборнен-2,3-дикарбоксиимид; дисульфидные соединения, такие как 2,2'-дипиридилдисульфид; соединения янтарной кислоты, например N,N'-дисукцинимидилкарбонат; соединения хлорида фосфиновой кислоты, например хлорид N,N'-бис(2-оксо-3-оксазолидинил)фосфиновой кислоты; производные оксалатов, такие как N,N'-дисукцинимидилоксалат (DSO), N,N'-дифтaлимид-оксалат (DPO), N,N'-бис(норборненилсукцинимидил)оксалат (BNO), 1,1'-бис(бензотриазолил)оксалат (ВВТО), 1,1'-бис(6-хлорбензотриазолил) оксалат (ВCТО) и 1,1'-бис(6-трифторметилбензотриазолил) оксалат (ВТВО); триарилфосфины, такие как триарилфосфины, например трифенилфосфин и ди-низшие алкилтриарилфосфины азодикарбоновой кислоты, такие как диэтилазодикарбоксилат- трифенилфосфин; N-низшие алкил-5-арилизоизоксазолиум-3'- сульфонаты, такие как N-этил-5-фенилизоксазолиум-3'-сульфонат; производные карбодиимида, такие как N',N'-ди-циклоалкилкарбодиимиды, например N',N'-дициклогексилкарбодиимид (DCC) и 1-этил-3-(3-диметиламинопропил)карбодиимид (EDAPC); дигетероарилдиселениды, такие как ди-2-пиридилдиселенид; арилсульфонилтриазолиды, такие как п-нитробензолсуль фонилтриазолид; галоиды 2-гало-1-низший-алкилпиридиния, такие как йодид 2-хлор-1-метилпиридиния; диарилфосфорил-азиды, такие как дифенилфосфорилазид (DPPA); и производные имидазола, такие как 1,1'-оксазолилдиимидазол и N, N'-кapбoнилдиимидазол, предпочтительно диарилфосфорилазиды.
Растворитель, пригодный к применению, не является особенно критическим, поскольку он не ингибирует реакцию, а растворяет исходное вещество до определенной степени и включает, предпочтительно, алифатические углеводороды, такие как гексан или гептан; ароматические углеводороды, такие как бензол, толуол и ксилол; галогенированные углеводороды, такие как метиленхлорид, хлороформ, четыреххлористый углерод, дихлорэтан, хлорбензол и дихлорбензол; эфиры, такие как этилформиат, этилацетат, пропилацетат, бутилацетат и диэтил- карбонат; эфиры, такие как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран, диоксан, диметоксиэтан и диметиловый эфир диэтиленгликоля; нитрилы, такие как ацетонитрил или изобутиронитрил; амиды, например, формамид, N,N-диметилформамид, N,N-диметилацетамид, N-метил-2-пирролидон, N-метилпирролидинон и триамид гексаметилфосфорной кислоты.
Поскольку необходимо использовать основание, могут использоваться основания, подобные тем, которые описаны в вышеприведенном Методе 1.
Реакцию проводят при -20 до 80oC, предпочтительно от 0oC до комнатной температуры.
Продолжительность реакции варьирует в зависимости, в основном, от температуры реакции, исходного соединения, реакционного реагента или типа используемого растворителя, и составляет обычно от 10 минут до 3 дней, предпочтительно от 30 минут до 1 дня.
(Метод 3)
Метод 3 должен обеспечивать взаимодействие соединения общей формулы: RCOOH [где R имеет то же значение, которое определено выше] с соединением (5) в растворителе в присутствии диалкильного эфира галогенированной фосфорной кислоты, например диэтилхлорфосфата, и основания.
Растворитель, пригодный к применению, не является особенно критическим, поскольку он не ингибирует реакцию и растворяет исходный материал до определенной степени, и включает, предпочтительно, алифатические углеводороды, такие как гексан и гептан; ароматические углеводороды, такие как бензол, толуол и ксилол; галогенированные углеводороды, такие как метиленхлорид, хлороформ, четыреххлористый углерод, дихлорэтан, хлорбензол и дихлорбензол; эфиры, такие как этилформиат, этилацетат, пропилацетат, бутилацетат и диэтил-карбонат; эфиры, такие как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран, диоксан, диметоксиэтан и диметиловый эфир диэтиленгликоля; нитрилы, например ацетонитрил и изо-бутиронитрил; и амиды, такие как формамид, N, N-диметилформамид, N, N-диметилацетамид, N-метил-2-пирролидон, N- метилпирролидинон и триамид гексаметилфосфорной кислоты.
Поскольку используется основание, могут быть использованы основания, подобные тем, что описаны в вышеприведенном Методе 1.
Реакцию проводят при температуре от 0oC до температуры кипения растворителя с обратным холодильником, предпочтительно от комнатной температуры до 50oC.
Продолжительность реакции варьирует, в основном, в зависимости от температуры реакции, исходного материала, реагента реакции и типа используемого растворителя, и обычно составляет от 10 мин до 3 дней, предпочтительно от 30 мин до 1 дня.
В вышеохарактеризованных Методе 1, Методе 2 и Методе 3 соединение (6), от одной до трех ацильных групп которого вводят в соединение (5), может быть получено соответствующим регулированием эквивалентного количества используемого ацилирующего агента относительно соединения (5).
По завершении реакции целевое соединение (6) настоящего изобретения отделяют от реакционной смеси общепринятым способом.
Например, целевое соединение может быть получено соответственно нейтрализацией реакционной смеси, удалением нерастворимых примесей, если они есть, фильтрацией, добавлением туда несмешивающегося с водой органического растворителя, такого как этилацетат, промывкой его водой, отделением органического слоя, содержащего целевое соединение, сушкой этого слоя над сульфатом магния и выпариванием растворителя.
Полученное таким образом соединение можно выделить и очистить, если необходимо, соответствующей комбинацией общеизвестных методов, например перекристаллизацией, переосаждением или методом, обычно используемым для выделения и очистки органических соединений, например адсорбционной колоночной хроматографией с использованием носителей, таких как силикагель, алюминий, Флорисил системы Mg-силикагель; методом с использованием синтезированного адсорбирующего агента, таким как разделительная колоночная хроматография с использованием носителей, например Cефадекса LH-20 (производство Pharmacia Co. Ltd.), Amberlite XAD- 11 (производство Rhom and Haas Co., Ltd.) и Диаион HP-20 (Mitsubishi Chemical Corp. ) или обычная колоночная хроматография с обращенной фазой с использованием силикагеля или алкилированного силикагеля (предпочтительна ВЭЖХ) и элюированием соответствующими элюирующими растворами.
(Стадия C-2)
Данная стадия предназначена для получения соединения (1) настоящего изобретения снятием трет- бутоксикарбонильной группы в соединении (6) в инертном растворителе.
Настоящая стадия может выполняться аналогично процедурам Стадии А-2.
Далее, если Wa - защитная группа карбоксильной группы, соединение (1) может быть получено дальнейшим снятием их подобно процедурам Стадии А-2.
Схема D предназначена для получения соединения (2а), которое является одним из исходных соединений в Схемах А и В, с использованием исходного соединения (7), легко получаемого по способу, описанному ниже.
(Стадия D-l)
Данная стадия предназначена для получения соединения (8) взаимодействием соединения (7) с использованием основания в инертном растворителе.
Растворитель, пригодный для использования, не является особенно критическим, поскольку он не ингибирует реакцию, а растворяет исходный материал до определенной степени и включает, предпочтительно, алифатические углеводороды, такие как гексан, гептан, лигроин и петролейный эфир; ароматические углеводороды, такие как бензол, толуол и ксилол; галогенированные углеводороды, такие как метиленхлорид, хлороформ, четыреххлористый углерод, дихлорэтан, хлорбензол и дихлорбензол; эфиры, например диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран, диоксан, диметоксиэтан и диметиловый эфир диэтиленгликоля; и метанол; предпочтительно галогенированные углеводороды и метанол.
Пригодное для использования основание не является особенно критическим, поскольку оно не влияет на другие функциональные группы (например, метиловый эфир) и включает, предпочтительно, метоксиды щелочных металлов, например метоксид натрия или метоксид калия.
Температура реакции обычно составляет от -10 до 50oC, предпочтительно 10 - 30oC.
Продолжительность реакции варьирует в зависимости от используемых веществ, основания, температуры реакции и т.д. и составляет обычно от 15 мин до 10 ч, предпочтительно 1 - 5 ч.
После реакции целевое соединение можно получить, например, нейтрализацией реакционной смеси соляной кислотой/раствором диоксана, выпариванием растворителя при пониженном давлении с последующей очисткой остатка колоночной хроматографией на силикателе.
(Стадия D-2)
Данная стадия предназначена для получения соединения (9) взаимодействием соединения (8) с реагентом для введения изопропилиденовой группы в инертном растворителе.
Растворитель, пригодный для использования, не является особенно критическим, поскольку он не ингибирует реакцию, а растворяет исходные вещества до определенной степени и включают алифатические углеводороды, такие как гексан, гептан, лигроин и петролейный эфир; ароматические углеводороды, такие как бензол, толуол и ксилол; эфиры, такие как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран, диоксан, диметоксиэтан и диметиловый эфир диэтиленгликоля; и кетоны, например ацетон, метилэтилкетон, метилизобутилкетон, изофорон и циклогексанон, предпочтительно кетоны (особенно ацетон).
Пригодный для использования реагент включает, предпочтительно, 2,2- диметоксипропан и кислоты, например п-толуол-сульфокислоту, которые используются в качестве катализаторов.
Температура реакции составляет обычно от -10 до 50oC, предпочтительно 10 - 30oC.
Продолжительность реакции варьирует в зависимости от используемого материала, основания, температуры реакции и т.д. и обычно составляет от 15 мин до 10 ч, предпочтительно 1 - 5 ч.
После реакции целевое соединение может быть получено, например, добавлением к реакционной смеси несмешивающегося с водой растворителя, такого как этилацетат, и водного раствора кислого углекислого натрия, экстрагированием целевого соединения этилацетатом и выпариванием растворителя. Целевое соединение может быть в дальнейшем очищено, если необходимо, перекристаллизацией или различного рода хроматографией.
(Стадия D-3)
Данная стадия предназначена, если необходимо, 1) для замещения метильной группы метилкарбоксилатного фрагмента на другой эфирный остаток, 2) для гидролиза метилкарбоксилатного фрагмента или 3) для введения защитной группы карбоксильной группы или эфирного остатка после гидролиза 2).
(Эфирный обмен)
Данная стадия предназначена для получения соединения (10) взаимодействием соединения (9) со спиртом, который может дать желаемую эфирную группу, в инертном растворителе в присутствии основания.
Растворитель, пригодный для использования, не является особенно критическим, поскольку он не ингибирует реакцию, и включает, например, алифатические углеводороды, такие как гексан, гептан, лигроин и петролейный эфир; ароматические углеводороды, такие как бензол, толуол и ксилол; галогенированные углеводороды, такие как метиленхлорид, хлороформ, четыреххлористый углерод, дихлорэтан, хлорбензол и дихлорбензол; эфиры, такие как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран, диоксан, диметоксиэтан и диметиловый эфир диэтиленгликоля; и спирты, такие как метанол, этанол, н-пропанол, изопропанол, н-бутанол, изобутанол, трет-бутанол, изоамиловый спирт, диэтиленгликоль, глицерин, октанол, циклогексанол и метилцеллозольв. Предпочтительно, спирт, образующий желаемую эфирную группу, может использоваться как растворитель.
Основание, которое может использоваться, включает, предпочтительно, органические основания, такие как пиридин, триэтиламин, диэтиламин и 4-N, N-диметиламинопиридин.
После реакции желаемое соединение может быть получено, к примеру, нейтрализацией реакционной смеси кислотой, добавлением к смеси несмешивающегося с водой растворителя, такого как этилацетат, экстрагированием целевого соединения этилацетатом и т.д., промывкой его водой и выпариванием растворителя. Целевое соединение может быть в дальнейшем очищено, если необходимо, перекристаллизацией или различного типа хроматографией.
(Дифенилметилирование)
Настоящая стадия предназначена для получения соединения (10) взаимодействием соединения (9) с дифенилдиазометаном в инертном растворителе в присутствии кислоты Льюиса.
Пригодный для применения растворитель не является особенно критическим, поскольку он не ингибирует реакцию и растворяет исходное вещество до определенной степени и включает, предпочтительно, алифатические углеводороды, такие как гексан, гептан, лигроин и петролейный эфир; ароматические углеводороды, такие как бензол, толуол и ксилол; галогенированные углеводороды, такие как метиленхлорид, хлороформ, четыреххлористый углерод, дихлорэтан и хлорбензол; эфиры, такие как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран, диоксан, диметоксиэтан и диметиловый эфир этиленгликоля; и спирты, например метанол, этанол, н-пропанол, изопропанол, н-бутанол, изобутанол, трет-бутанол, изоамиловый спирт, диэтиленгликоль, глицерин, октанол, циклогексанол и метилцеллозольв, более предпочтительны спирты (особенно метанол), галогенированные углеводороды (дихлорметан); и их смеси.
Приемлемая для использования кислота Льюиса включает предпочтительно комплекс эфира и трифторида бора.
Температура реакции обычно от 0 до 50oC, предпочтительна комнатная температура.
Время реакции варьирует в зависимости от исходного материала, кислоты Льюиса и температуры реакции, и обычно составляет от 10 мин до 5 ч, предпочтительно 1 - 3 ч.
После реакции растворитель отгоняют и для получения целевого соединения осуществляют очистку перекристаллизацией или хроматографией.
(Гидролиз)
Данная стадия предназначена для получения соединения (10), в котором Wa - атом водорода, гидролизом соединения (9) в инертном растворителе в присутствии основания.
Пригодный для применения растворитель не является особенно критическим, поскольку он не ингибирует реакцию и растворяет исходное вещество до определенной степени и включает ароматические углеводороды, такие как бензол и толуол; эфиры, такие как диэтиловый эфир и тетрагидрофуран; галогенированные углеводороды, такие как дихлорметан и хлороформ; кетоны, такие как ацетон, метилэтилкетон; воду; смеси этих органических растворителей и воды.
Пригодное основание включает гидроксиды щелочных металлов, такие как гидроксид лития, гидроксид натрия и гидроксид калия; и кислые карбонаты щелочных металлов, такие как кислый карбонат натрия и кислый карбонат калия.
Температура реакции обычно составляет от -10 до 50oC, предпочтительно 10 - 30oC.
Продолжительность реакции варьирует в зависимости от используемого исходного материала, основания, температуры реакции и т.д. и обычно составляет от 15 мин до 10 ч, предпочтительно 1 - 5 ч.
После реакции целевое соединение может быть получено, к примеру, охлаждением реакционной смеси, мягким подкислением ее разбавленной соляной кислотой, добавлением к реакционной смеси несмешивающегося с водой растворителя, такого как этилацетат, экстрагированием целевого соединения этилацетатом и выпариванием растворителя. Целевое соединение может быть в дальнейшем очищено перекристаллизацией или различного типа хроматографией.
(Введение защитной группы карбоксильной группы или остатка эфира)
Данная стадия предназначена для получения соединения (10) введением защитной группы или эфирного остатка в карбоксильную группу в положении 1 соединения (9), в котором Wa - атом водорода.
Введение защитной группы или эфирного остатка варьирует в зависимости от типа эфирного остатка или защитной группы и может осуществляться в соответствии с методами, обычно используемыми в области химического органического синтеза, например методами, описанными в Protective Groups in Organic Synthesis, Second Edition (1991, Green et al.).
(Стадия D-4)
Данная стадия предназначена для получения соединения (2a) из соединения (10) с помощью восстанавливающего агента в инертном растворителе.
Пригодный для применения растворитель не является особенно критическим, поскольку он не ингибирует реакцию, а растворяет до некоторой степени исходные вещества и включает, предпочтительно, алифатические углеводороды, такие как гексан, гептан, лигроин и петролейный эфир; ароматические углеводороды, такие как бензол, толуол и ксилол, эфиры, такие как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран, диоксан, диметоксиэтан и диметиловый эфир диэтиленгликоля; спирты, такие как метанол, этанол, н-пропанол, изо-пропанол, н-бутанол, изобутанол, трет- бутанол, изоамиловый спирт, диэтиленгликоль, глицерин, октанол, циклогексанол и метилцеллозольв; кетоны, такие как ацетон, метилэтилкетон, метилизобутилкетон, изофорон и циклогексанон; нитрилы, такие как ацетонитрил и изобутиронитрил; амиды, такие как формамид, N, N-диметилформамид, N, N-диметилацетамид, N-метил-2- пирролидон, N-метилпирролидинон и триамид гексаметилфосфорной кислоты; сульфоксиды, такие как диметилсульфоксид и сульфолан; алифатические карбоновые кислоты, такие как уксусная кислота; и смеси этих органических растворителей и воды, предпочтительно спирты (особенно метанол), эфиры, такие как тетрагидрофуран и диоксан, алифатические карбоновые кислоты, например уксусная кислота, и смеси этих органических растворителей и воды.
В качестве восстановителя может быть использован катализатор, такой как палладий-на-угле, платина и никель Ренея в присутствии газообразного водорода и катализатор Линдлара (Pd-BaSO4 или Pd-CaCO3 и в сочетании используются хинолин или ацетат свинца), использование которого особенно предпочтительно.
Температура реакции составляет обычно от -10 C до 50oC, предпочтительно 10 - 30oC.)
Продолжительность реакции варьирует в зависимости от используемого исходного сырья, основания, температуры реакции и т.д. и составляет обычно от 15 мин до 10 ч, предпочтительно 1 - 5 ч.
После реакции целевое соединение может быть получено, например, фильтрованием реакционной смеси при пониженном давлении для удаления катализатора и выпариванием растворителя при пониженном давлении. Целевое соединение может быть в дальнейшем очищено, если это необходимо, перекристаллизацией или различными видами хроматографии.
Схема E - это процесс получения соединения (2b) или (2c), которое является одним из исходных веществ в Схемах А и В, с использованием исходного соединения (10), которое легко получить в соответствии с вышеописанным способом.
(Стадия Е-1)
Данная стадия предназначена для получения соединения (12) введением желаемой ацильной группы в соединение (10) в инертном растворителе.
Далее, данная стадия может выполняться аналогично процедурам вышеописанной стадии C-1.
(Стадия Е-2)
Данная стадия предназначена для получения соединения (13) обработкой соединения (12) реагентом для удаления изопропилиденовой группы в инертном растворителе.
Пригодный к применению растворитель не является особенно критическим, поскольку он не ингибирует реакцию, а растворяет до некоторой степени исходные вещества, он включает, предпочтительно, галогенированные углеводороды, такие как метиленхлорид и хлороформ.
В качестве реагента для удаления указанной группы предпочтительна кислота, и не особенно существенно, какая кислота, т.к. она используется как кислотный катализатор в обычной реакции и включает кислоты Bronsted'a, такие как неорганические кислоты, например соляная кислота, бромистая кислота, серная кислота, хлорная кислота и фосфорная кислота, и органические кислоты, например, муравьиная кислота, щавелевая кислота, метансульфокислота, п-толуолсульфокислота, трифторуксусная кислота и трифторметансульфокислота; и кислоты Льюиса, например хлорид цинка, тетрахлорид олова, трихлорид бора, трифторид бора и трибромид бора, а также кислотные ионообменные смолы, предпочтительны органические кислоты (особенно трифторуксусная кислота).
Температура реакции обычно составляет от -10 до 50oC, предпочтительно 10 - 30oC.
Продолжительность реакции варьирует в зависимости от используемого исходного материала, основания, температуры реакции и т.д. и составляет обычно от 15 мин до 10 ч., предпочтительно 1 - 5 ч.
После окончания реакции целевое соединение получают, например, путем нейтрализации реакционной смеси, выпаривания растворителя при пониженном давлении и последующей очистки остатка хроматографией на силикагеле.
Далее, на данной стадии ацильная группа (R6) в положении 7 перемещается в положение 9.
(Стадия Е-3)
Данная стадия предназначена для приготовления исходного вещества (2b) из соединения (13) с помощью восстанавливающего агента в инертном растворителе.
Данная стадия выполняется аналогично процедурам вышеописанной Стадии D-4.
(Стадия E-4)
Данная стадия предназначена для приготовления соединения (14) настоящего изобретения обработкой соединения (12) реагентом для снятия изопропилиденовой группы в инертном растворителе в присутствии катализатора.
Пригодный к применению растворитель не является особенно критическим, поскольку он не ингибирует реакцию, а растворяет до некоторой степени исходные материалы, он включает, предпочтительно, смесь уксусной кислоты (если она одновременно используется и как кислотный катализатор) и воды.
Температура реакции обычно составляет 10 - 70oC, предпочтительно 30 - 60oC.
Продолжительность реакции варьирует в зависимости от используемого исходного вещества, основания, температуры реакции и т.д. и составляет обычно от 15 мин до 24 ч, предпочтительно 10 - 20 ч.
По окончании реакции целевое соединение может быть получено, например, выпариванием растворителя при пониженном давлении, добавлением в реакционную смесь несмешивающегося с водой растворителя, например этилацетата, и водного раствора кислого углекислого натрия, экстрагированием целевого соединения этилацетатом и выпариванием растворителя. Целевое соединение может быть в дальнейшем очищено, если это необходимо, перекристаллизацией или различными типами хроматографии.
(Стадия E-5)
Данная стадия предназначена для получения желаемого исходного вещества (2c) из соединения (14) с помощью восстанавливающего агента в инертном растворителе.
Данная стадия выполняется аналогично процедурам вышеописанной Стадии D-4.
Схема F - это способ получения соединения (2d) или (2c), которое является одним из исходных соединений в Схемах А и B, с использованием исходного соединения (14), которое легко получить в соответствии с вышеописанным способом.
(Стадия F-1)
Данная стадия предназначена для получения соединения (15) взаимодействием соединения (14) с реагентом, защищающим гидроксильную группу, в инертном растворителе.
Защитная группа не является особенно критической и включает, предпочтительно, трет-бутилдиметилсилильную группу и трет-бутилдифенилсилильную группу.
Cилилирование можно осуществлять известными способами. Например, силилирование может выполняться взаимодействием с трет-бутилдиметилсилилгалоидом (особенно хлоридом) в диметилформамиде в присутствии основания, такого как триэтиламин и 4- (N,N-диметиламино) пиридин.
Температура реакции составляет обычно 10 - 50oC, предпочтительно 10 - 40oC.
Продолжительность реакции варьирует в зависимости от используемого исходного вещества, основания, температуры реакции и т.д. и составляет обычно от 15 мин до 24 ч., предпочтительно 10 - 20 ч.
По окончании реакции целевое соединение может быть получено путем добавления в реакционную смесь несмешивающегося с водой растворителя, такого как этилацетат, и водного раствора кислого углекислого натрия, экстрагирования целевого соединения этилацетатом и выпаривания растворителя. Целевое соединение может быть в дальнейшем очищено, если это необходимо, перекристаллизацией или различными типами хроматографии.
(Стадия F-2)
Данная стадия предназначена для получения соединения (16) введением желаемой ацильной группы в соединение (15) в инертном растворителе. Данная стадия может выполняться аналогично процедурам Стадии C-1.
(Стадия F-3)
Данная стадия предназначена для получения соединения (17) взаимодействием соединения (16) с реагентом для удаления защитной группы (предпочтительно трет-бутилдиметилсилильной группы или трет-бутилдифенилсилильной группы) гидроксильной группы в инертном растворителе.
Пригодный растворитель включает, предпочтительно, спирты, такие как метанол и этанол, воду и их смеси.
Обычно в качестве реагентов для такого снятия используют кислоты и выбор кислоты не является особенно критическим, поскольку она используется как кислотный катализатор в обычных реакциях и включает кислоты Bronsted'a, например неорганические кислоты, например соляную кислоту, бромистую кислоту, серную кислоту, хлорную кислоту и фосфорную кислоту, а также органические кислоты, например уксусную кислоту, муравьиную кислоту, щавелевую кислоту, метансульфокислоту, п-толуолсульфокислоту, трифторуксусную кислоту и трифторметансульфокислоту, и кислоты Льюиса, такие как хлорид цинка, тетрахлорид олова, трихлорид бора, трифторид бора и трибромид бора, а также кислотные ионообменные смолы; предпочтительны органические кислоты (особенно уксусная кислота и трифторуксусная кислота).
При желании могут также использоваться реагенты, которые дают фторид-ион, например фторид тетрабутиламмония.
Температура реакции обычно составляет от -10 до 50oC, предпочтительно 10 -30oC.
Продолжительность реакции варьирует в зависимости от используемого исходного материала, основания, температуры реакции и т.д. и обычно составляет от 15 мин до 10 ч, предпочтительно 1 - 5 ч.
По окончании реакции целевое соединение может быть получено, например, нейтрализацией реакционной смеси, выпариванием растворителя при пониженном давлении с последующей очисткой остатка хроматографией на силикагеле.
На данной стадии ацильная группа (R7) в положении 8 переносится в положение 9.
(Стадия F-4)
Данная стадия предназначена для получения соединения (18) путем 1) защиты гидроксильной группы в 8-ом положении соединения (17) или, при желании, 2) введения желаемой ацильной группы.
(Введение ацильной группы)
Данная стадия может выполняться аналогично процедурам Стадии C-1.
(Введение защитной группы)
Как защитная группа предпочтительна трет-бутилдиметил-силильная группа, и введение защитной группы проводят при использовании трифлата трет-бутилдиметилсилила и лутидина в метиленхлориде.
(Стадия F-5)
Данная стадия предназначена для получения целевого исходного соединения (2d) из соединения (18) с использованием восстанавливающего агента в инертном растворителе.
Данная стадия может выполняться аналогично процедурам Стадии D-4.
(Стадия F-6)
Данная стадия предназначена для получения желаемого исходного соединения (2e) из соединения (16) с использованием восстанавливающего агента в инертном растворителе.
Данная стадия может выполняться аналогично процедурам Стадии D-4.
Схема G - процесс получения соединения (2f) или (2q), которое является одним из исходных соединений в Схемах А и В, с использованием вышеуказанного соединения (13) или (14), описанных выше.
(Стадия G-1)
Данная стадия предназначена для получения соединения (19) введением желаемой ацильной группы в соединение (13) в инертном растворителе.
Данная стадия выполняется аналогично процедурам Стадии C-1.
(Стадия G-2)
Данная стадия предназначена для получения желаемого исходного соединения (2f) из соединения (19) с использованием восстанавливающего агента в инертном растворителе. Данная стадия выполняется аналогично процедурам Стадии D-4.
(Стадия G-3)
Данная стадия предназначена для получения соединения (20) введением желаемой ацильной группы в соединение (14) в инертном растворителе.
Данная стадия выполняется аналогично процедурам Стадии C-1.
(Стадия G-4)
Данная стадия предназначена для получения желаемого исходного вещества (2q) из соединения (20) с использованием восстанавливающего агента в инертном растворителе.
Данная стадия выполняется аналогично процедурам Стадии D-4.
Процесс H - процесс получения соединения (5), которое является исходным соединением в процессе C, с использованием хорошо известного соединения (26), описанного в Carbohydrate Research, 83, 163-169 (1980) или WO 95/32955.
(Стадия H-1)
Данная стадия предназначена для получения соединения (27) взаимодействием известного соединения (26) с азидирующим агентом в инертном растворителе.
Пригодный к применению растворитель не является особенно критическим, поскольку он не ингибирует реакцию и растворяет в некоторой степени исходное вещество, он включает, предпочтительно, ароматические углеводороды, такие как бензол, толуол и ксилол; галогенированные углеводороды, такие как метиленхлорид и хлороформ; эфиры, такие как диэтиловый эфир, тетрагидрофуран, диоксан и диметоксиэтан; и нитрилы, такие как ацетонитрил.
Реагент, пригодный к применению, не является особенно критическим, поскольку он обычно используется для азидирования и включает, предпочтительно, азидные производные диарилфосфорной кислоты, например азид дифенилфосфорной кислоты; триалкилсилилазиды, такие как триметилсилилазид и триэтилсилилазид, и азиды солей щелочных металлов, такие как азид натрия и азид калия, предпочтительно азид натрия.
Температура реакции обычно составляет от -10 до 50oC предпочтительно 10 - 30oC.
Продолжительность реакции варьирует в зависимости от используемого исходного вещества, основания, температуры реакции и т.д. и она обычно составляет от 15 мин до 10 ч., предпочтительно 1 - 5 ч.
По завершении реакции целевое соединение получают, например, нейтрализацией реакционной смеси раствором соляной кислоты в диоксане и очисткой остатка, полученного выпариванием растворителя при пониженном давлении, хроматографией на силикагеле.
(Стадия H-2)
Данная стадия предназначена для получения соединения (28) взаимодействием соединения (27) с т-бутоксикарбонилирущим агентом в инертном растворителе.
Т-бутоксикарбонилирование можно осуществлять путем взаимодействия с ди-трет-бутилкарбонатом и 2-(трет- бутоксикарбонилоксиимино)-2-фенилацетонитрилом в инертном растворителе (например, ароматические углеводороды, такие как бензол и толуол; галогенированные углеводороды, такие как метилен-хлорид и хлороформ; эфиры, такие как диэтиловый эфир, тетрагидрофуран и диоксан; и амиды, например диметилформамид, в присутствии основания (например, 4-(N,N-диметиламино) пиридина).
По окончании реакции целевое соединение может быть получено, например, нейтрализацией реакционной смеси, выпариванием растворителя при пониженном давлении, добавлением несмешивающегося с водой растворителя, такого как этилацетат, экстрагированием целевого соединения этилацетатом и выпариванием растворителя. Целевое соединение может быть в дальнейшем очищено, если необходимо, перекристаллизацией или различными видами хроматографии.
(Стадия H-3)
Данная стадия предназначена для получения соединения (29) взаимодействием соединения (28) с основанием в инертном растворителе.
Данная стадия может осуществляться аналогично процедурам Стадии D-1.
(Стадия H-4)
Данная стадия предназначена для ацетилирования соединения (29) в инертном растворителе. Ацетилирование осуществляют в соответствии с обычной методикой защиты гидроксильной группы. Например, ацетилирование может выполняться 1) взаимодействием с ацетоангидридом в пиридине или 2) взаимодействием с ацетилгалогенидом (в частности, хлоридом) в метиленхлориде в присутствии основного катализатора (например, триэтиламина и 4-N,N-диметиламинопиридина).
По окончании реакции целевое соединение получают отгонкой растворителя при пониженном давлении, добавлением к остатку несмешивающегося с водой растворителя, такого как этилацетат, и водного раствора кислого углекислого натрия, экстрагированием целевого соединения этилацетатом и отгонкой растворителя. Целевое соединение может быть далее очищено, если это требуется, перекристаллизацией или различными видами хроматографии.
(Стадия H-5)
Данная стадия предназначена для получения соединения (30) обработкой соединения, полученного на Стадии H-4, реагентом, который удаляет трет-бутоксикарбонильную группу, в инертном растворителе.
Удаление трет-бутоксикарбонильной группы выполняется в соответствии с обычными методами.
Растворитель, пригодный к применению, не является особенно критическим, поскольку он не ингибирует реакцию и растворяет до некоторой степени исходное вещество, он включает, предпочтительно, алифатические углеводороды, такие как гексан, гептан, лигроин и петролейный эфир; ароматические углеводороды, такие как бензол, толуол и ксилол; галогенированные углеводороды, такие как метиленхлорид, хлороформ, четыреххлористый углерод, дихлорэтан, хлорбензол, дихлорбензол; эфиры, такие как этилформиат, этилацетат, пропилацетат, бутилацетат и диэтилкарбонат; эфиры, такие как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран, диоксан, диметоксиэтан и диметиловый эфир диэтиленгликоля; амиды, такие как формамид, N,N-диметилформамид, N,N-диметилацетамид, N-метил-2-пирролидон, N-метилпирролидинон и триамид гексаметилфосфорной кислоты; и сульфоксиды, такие как диметилсульфоксид и сульфолан, предпочтительны галогенированные углеводороды (особенно метиленхлорид).
Реагент, пригодный к применению, не является особенно критическим, поскольку это - его обычное применение и, предпочтительно, это - соляная кислота.
Температура реакции обычно составляет от -10 до 50oC, предпочтительно 10 - 30oC.
Продолжительность реакции варьирует в зависимости от используемого исходного вещества, основания, температуры реакции и т.д. и составляет обычно от 15 мин до 24 ч, предпочтительно 1 - 10 ч.
После реакции целевое соединение получают, например, отгонкой растворителя при пониженном давлении, добавлением к реакционной смеси несмешивающегося с водой растворителя,
например этилацетата, и водного раствора кислого углекислого натрия, экстрагированием целевого соединения этилацетатом и отгонкой растворителя. Целевое соединение может быть в дальнейшем очищено, если требуется, перекристаллизацией или различными типами хроматографии.
(Стадия H-6)
Данная стадия предназначена для получения соединения (7) введением желаемой ацильной группы в соединение (30) в инертном растворителе.
Данная стадия может осуществляться аналогично процедурам Стадии C-1.
(Стадия H-7)
Данная стадия предназначена для получения соединения (32) из соединения (7) с помощью восстанавливающего агента в инертном растворителе.
Данная стадия может выполняться аналогично процедурам Стадии D-4.
(Стадия H-8)
Данная стадия предназначена для получения соединения (33) взаимодействием соединения (32) с N,N'-ди-трет-бутоксикарбонилтиомочевиной в инертном растворителе в присутствии основания и хлорида ртути.
Данная стадия может выполняться аналогично процедурам Стадии А-1.
(Стадия H-9)
Данная стадия предназначена для получения соединения (34) взаимодействием соединения (33) с основанием в инертном растворителе.
Данная стадия может выполняться аналогично процедурам Стадии D-1.
(Стадия H-10) Эфирный взаимообмен, гидролиз, защита или этерификация.
Данная стадия предназначена для 1) замещения метильной группы метилкарбоксилатной части на остаток другого эфира, 2) гидролиза метилкарбоксилатного фрагмента или 3) введения защитной группы гидроксильной группы или эфирного остатка после гидролиза в 2), по желанию.
Данная стадия может выполняться аналогично процедурам Стадии D-3.
Соединение (1) настоящего изобретения может быть также получено другими способами, отличающимися от описанных выше. В частности, возможно получение соединения (1) изменением последовательности стадий Схем A-H, в зависимости от ситуаций. Например, соединение (1) настоящего изобретения может быть получено по Схеме J с использованием соединения (10), полученного как промежуточное в Схеме D.
(Стадия J-1)
Данная стадия выполняется в случае необходимости и предназначена для получения соединения (35) введением желаемой ацильной группы в соединение (10) в инертном растворителе.
Данная стадия может выполняться аналогично процедурам Стадии C-1, описанной выше.
(Стадия J-2)
Данная стадия предназначена для получения соединения (36) путем взаимодействия соединения (35) с восстанавливающим агентом в инертном растворителе.
Данная стадия может выполняться аналогично процедурам Стадии D-4, описанной выше.
(Стадия J-3)
Данная стадия предназначена для получения соединения (37) взаимодействием соединения (36) с N,N'- ди-трет-бутоксикарбонилтиомочевиной в инертном растворителе в присутствии основания и хлорида ртути.
Данная стадия может выполняться аналогично процедурам Стадии А-1, описанной выше.
(Стадия J-4)
Данная стадия выполняется в случае необходимости и предназначена для получения соединения (38) удалением защитной группы карбоксильной группы соединения (37).
Метод снятия защитной группы варьирует в зависимости от типа защитной группы и может осуществляться в соответствии с обычно применяемыми способами, например методами, описанными в Protective Groups in Organic Synthesis, Second Edition (1991, Green et al.).
В случае если защитной группой является дифенилметильная группа, осуществляют каталитическое восстановление, используются такие кислоты, как уксусная и трифторуксусная, или используется комплекс трифторида бора и диэтилового эфира.
В случае когда защитной группой карбоксильной группы является бензильная группа, проводят каталитическое восстановление, а в случае когда защитной группой является алкильная группа, например метильная или этильная, проводят гидролиз.
(Стадия J-5)
Данная стадия предназначена для получения соединения (1j) настоящего изобретения взаимодействием соединения (38) с реагентом, удаляющим трет-бутоксикарбонильную группу и изопропилиденовую группу, в инертном растворителе.
Данная стадия может быть выполнена аналогично процедурам Стадии Е-2.
Полученное таким образом соединение нейраминовой кислоты (1) или ее соль, в случае необходимости, могут быть переведены в другие фармакологически приемлемые соли.
Производное нейраминовой кислоты (1) настоящего изобретения подвергается гидролизу гидролазой, присутствующей в живом организме, и показывает отличную способность ингибировать репликацию вируса и ингибиторную активность в отношении сиалидазы. Кроме того, при введении соединения нейраминовой кислоты (1) мышам, инфицированным вирусом гриппа, данное соединение проявляет противоинфекционные лечебные эффекты, превосходящие эффект Соединения A (GG-167), описанного в WO 91/16320 (японская заявка PCT (Kokai) N Hei 5-507068). Поэтому, производное нейраминовой кислоты (1) настоящего изобретения является полезным в качестве терапевтического средства или средства для профилактики (предпочтительно, терапевтического средства) вирусных заражений (предпочтительно, заражения вирусом гриппа).
Назначение нейраминовой кислоты настоящего изобретения предусматривает, например, оральное введение или интраназальное введение растворов, таких как жидкие формы, водные сорастворители и т.д., аэрозоли, порошки и т.д. Из этих препаративных форм растворы, такие как жидкие формы и водные сорастворители, готовятся хорошо известными способами с использованием дистиллированной воды, фармакологически приемлемых органических растворителей (например этанол, пропилен- гликоль, PEG 400 и т.д.); и стабилизаторов (пара-оксибензоаты, такие как метилпарабен и пропилпарабен; спирты, такие как хлорбутанол, бензиловый спирт и фенилэтиловый спирт; бензалкониум хлорид; фенолы, такие как фенол и крезол; тимерозал; дегидроуксусная кислота и т.д.). Аэрозоли готовятся хорошо известными методами с использованием пропеллентов, таких как различные виды фреонов и газообразный азот, и поверхностно- активных агентов, таких, как лецитин. Порошки готовят хорошо известными методами с использованием инертных наполнителей (например, органических наполнителей, таких как caxapa, например лактоза, сахароза, глюкоза, маннитол и сорбитол; крахмалы, например кукурузный крахмал, картофельный крахмал, α-крахмал, декстрин и карбоксиметилкрахмал; целлюлозы, например кристаллическая целлюлоза, с низкой степенью замещения гидроксипропилцеллюлоза, гидроксипропилметилцеллюлоза, карбоксиметилцеллюлоза, Ca-карбоксиметилцеллюлоза и внутренне сшитая Na-карбоксиметилцеллюлоза; аравийская камедь; декстран; и пуллулан; и неорганические носители, такие как силикаты, например светлый кремниевый ангидрид, синтезированные силикат алюминия, Mg-Al-метасиликат; фосфаты, например кальцийфосфат; карбонаты, например карбонат кальция; и сульфаты, например сульфат кальция), смазывающих веществ (например, стеариновая кислота, соли металлов стеариновой кислоты, такие как стеарат кальция и стеарат магния; тальк; коллоидный кремний; воски, такие как пчелиный воск и спермацет; борная кислота; адипиновая кислота; сульфаты, например сульфат натрия; гликоль; фумаровая кислота; бензоат натрия; DL-лейцин, натриевые соли жирных кислот; лаурилсульфаты, такие как лаурилсульфат натрия и лаурилсульфат магния; кремниевые кислоты, такие как кремниевый ангидрид и гидрат кремниевой кислоты; и вышеуказанные виды крахмала), стабилизаторов (пара-оксибензоаты, такие как метилпарабен и пропилпарабен; спирты, такие как хлорбутанол, бензиловый спирт и фенилэтиловый спирт; бензалкониум- хлорид; фенолы, такие как фенол и крезол; тимерозал; дегидроуксусная кислота; и сорбиновая кислота), улучшителей (например, подсластители, уксусы, ароматизаторы и т.п. обычно используемые средства), разбавители и т.п.
Хотя дозы активных ингредиентов будут меняться в зависимости от тяжести заболевания, возраста больного, методов введения и т.д., например в случае растворов, желательно назначать активный ингредиент в количестве 0,1 мг (предпочтительно 1 мг) как нижний предел и 1000 мг (предпочтительно 500 мг) как верхний предел; в случае сухих порошков желательно назначать активный ингредиент в количестве 0,1 мг (предпочтительно 1 мг) как нижний предел и 1000 мг (предпочтительно 500 мг) как верхний предел; и в случае аэрозолей желательно назначать активный ингредиент в количестве 0,1 мг (предпочтительно 1 мг) как нижний предел и 1000 мг (предпочтительно 500 мг) как верхний предел, один или несколько раз в день при вышеуказанных видах назначения, в зависимости от состояния тяжести заболевания.
Изобретение иллюстрируется рабочими примерами, препаративными примерами и испытательными примерами, которые не ограничивают настоящего изобретения.
В описании данных спектров ЯМР примеров приняты следующие обозначения:
d - дублет; dd - двойной дублет; ddd - тройной дублет;
m - мультиплет; s - синглет; bs - широкий синглет;
dt - дублет-триплет; t - триплет; tt - двойной триплет;
q - квинтет; Hz - герц; TMS - триметилсилан;
NMR - ядерный магнитный резонанс.
ПРИМЕР 1. Cоль миристил 5-ацетамидо-2,3,4,5-тетрадеокси-4- гуанидино-D-глицеро-D-галакто-нон-2-энопиранозоата и трифторуксусной кислоты (E8) (образцовое соединение N 88)
i) Метил 5-ацетамидо-4-азидо-2,3,4,5-тетрадеокси- 8,9-O- изопропилиден-D-глицеро-D-галакто-нон-2-энопиранозоат (E3)
1,3 г (2,8 ммоль) метил 5-ацетамидо-7,8,9-три-O-ацетил-4- азидо-2,3,4,5-тетрадеокси-D-глицеро-D-галакто-нон-2-энопиранозоната (E1) (который был синтезирован согласно способам, описанным в Mark von ltzstein et ai., Carbohydr. Res. , 244, 181-185 (1993) и Carbohydr.Res., 259, 293-299 (1993)) были растворены в 26 мл (20-кратный объем) при комнатной температуре и в тех же условиях в систему добавили каталитическое количество метоксида натрия с последующим перемешиванием смеси в течение 1 ч. После того как удостоверились в окончании реакции, в систему добавили Дауэкс-50х8 (H+) для нейтрализации смеси. Реакционную смесь отфильтровали и отфильтрованный продукт промыли метанолом. Фильтрат и промывочную воду отогнали при пониженном давлении и остаток очищали хроматографией на колонке с силикагелем (Кизельгель 60, 100 г, метиленхлорид: метанол = 5:1) с получением 620 мг (выход: 66%) соединения (E2) в виде твердого продукта бледно-желтого цвета.
Величина Rf:0,3 (метиленхлорид:метанол = 5:1).
580 мг (1,8 ммоль) полученного соединения (E2) были растворены в 29 мл (50-кратный объем) ацетона при комнатной температуре и затем в систему при охлаждении льдом были добавлены 0,7 мл (5,2 ммоль) 2,2- диметоксипропана и 42 мг (0,18 ммоль) DL-10-камфарсульфокислоты с последующим перемешиванием смеси при комнатной температуре в течение 1 часа. После подтверждения завершения реакции в систему влили триэтиламин для нейтрализации смеси. Реакционную смесь перегнали при пониженном давлении, а остаток очищали хроматографией на колонке с силикагелем (Кизельгель 60, 60 г, бензол: ацетонитрил = 1: 1) с получением 540 мг (выход: 84%) указанного соединения (E3) в виде белого твердого продукта.
Величина Rf: 0,76 (бензол:ацетонитрил = 1:1)
1H-NMR (270 MHz, CDCl3, TMS): δ (ppm) 5.97(1H, d, J=2.4 Hz), 5.63(1H, d, J=7.1 Hz), 4.36(1H, ddd, J=7.9, 6.4, 4.8 Hz), 4.30-4.00(6H, m), 3.81(3H, s), 3.57(1H, dd, J=7.9, 5.3 Hz), 2.12(3H, s), 1.40(3H, s), 1.36(3H, s);
[α]
ii) 5-Ацетамидо-4-азидо-2, 3, 4, 5-тетрадеокси-8, 9-O- изoпpoпилидeн-D-глицepo-D-гaлaкто-нoн-2-энoпиpaнoзойная кислота (E4).
460 мг (1,2 ммоль) соединения (E3) растворили в 24 мл (30- кратный объем) метанола при комнатной температуре и впоследствии в систему при комнатной температуре добавили 2,9 мл (1,4 ммоль) 1М водного раствора гидроксида натрия с последующим перемешиванием смеси при комнатной температуре в течение 1 ч. После подтверждения завершения реакции к системе добавили Дауэкс-50W для нейтрализации смеси. Реакционную смесь фильтровали, отфильтрованный продукт промывали водой. Фильтрат и промывные воды собирали и перегоняли при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем (Кизельгель 60, 60 г, этилацетат:2-пропанол:вода = 2:2:1) с получением 0,44 г (выход: 100%) указанного соединения (E4) в виде твердого белого продукта.
Величина Rf: 0,25 (этилацетат:2-пропанол:вода = 5:2:1)
1H-NMR (270 MHz, CD3OD, TMS): δ (ppm) 5.72(1H, d, J=2.1 Hz), 4.37(1H, dt, J=8.8, 5.5 Hz), 4.27(1H, dd, J=6.9, 1.4 Hz), 4.20-4.04(3H, m), 4.00(1H, dd, J= 8.8, 5.0 Hz), 3.54(1H, d, J= 8.8 Hz), 2.03(3H, s), 1.37(3H, s), 1.32(3H, s);
[α]
iii) Миристил 5-ацетамидо-4-азидо-2,3,4,5-тетрадеокси-8,9-O- изопропилиден-D-глицеро-D-галакто-нон-2-энопиранозоат (E5)
460 мг (1,2 ммоль) соединения (E4), 530 мг (2,5 ммоль) миристилового спирта и 510 мг (1,9 ммоль) тетрафторбората 2- бромо-1-этилпиридиния растворили в 22 мл (50-кратный объем) формамида, затем в систему влили 0,9 мл (3,8 ммоль) три-н- бутиламина и смесь перемешивали на масляной бане при 50oC в течение 6 ч. Реакционная смесь разделялась этилацетатом и насыщенным водным раствором NaCl, водный слой экстрагировали этилацетатом. Органический слой высушивали над сульфатом магния и фильтровали, после чего отгоняли растворитель при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем (Кизельгель 60, 100 г, бензол: этил-ацетат = 1:1) с получением 0,19 г (выход: 28%) указанного соединения (E5) в виде твердого белого продукта.
Величина Rf: 0,4 (бензол:этилацетат = 1:1)
1H-NMR (270 MHz, CDCl3, TMS): δ (ppm) 5.95(1H, d, J=2.4 Hz), 5.60(1H, d, J= 7.0 Hz), 4.36(1H, dt, J=7.7, 5.7 Hz), 4.30-4.00(8H, m), 3.58(1H, d, J=7.7 Hz), 2.12(3H, s), 1.69 (2H, квинтет, J=6.3 Hz), 1.40(3H, s), 1.35(3H, s), 1.26(22H, bs), 0.88(3H, t, J=6.3 Hz);
[α]
iv) Миристил 5-ацетамидо-4-амино-2,3,4,5-тетрадеокси- 8,9-O-изопропилиден-D-глицеро-D-галакто-нон-2-энопиранозоат (E6)
134 мг (0,24 ммоль) соединения (E5) растворяли в 6,7 мл (50- кратный объем) этанола при комнатной температуре. Затем к системе добавили при комнатной температуре 47 мг (0,35-кратный объем) катализатора Линдлара, с последующим перемешиванием смеси при комнатной температуре под атмосферой водорода в 1 атм в течение 2 ч. После подтверждения окончания реакции реакционную смесь фильтровали и отфильтрованный продукт промывали этанолом. Фильтрат и промывной раствор объединяли и перегоняли при пониженном давлении. Полученный таким образом остаток очищали хроматографией на колонке с силикагелем (Кизельгель 60, 60 г, этилацетат:2- пропанол:вода = 5:2;1) с получением 0,11 г (выход: 88%) указанного соединения (E6) в виде белого твердого продукта.
Величина Rf: 0,28 (этилацетат:2-пропанол:вода = 8:2:1).
1H-NMR (270 MHz, CD3OD, TMS): δ (ppm) 5.93(1H, d, J=2.6 Hz), 4.31(1H, квинтет, J= 6.9 Hz), 4.23-3.93(5H, m), 3.88(1H, t, J=9.0 Hz), 3.67-3.50(2H, m), 2.05(3H, s), 1.68(2H, квинтет, J=8.1 Hz), 1.37(3H, 3), 1.33(3H, s), 1.27(22H, bs), 0.88(3H, t, J=8.1 Hz).
v) Миристил 5-ацетамидо-4-N-N'-бис-трет-бутоксикарбонил) гуанидино-2, 3, 4, 5-тетрадеокси-8,9-O-изопропилиден-D-глицеро- D-галакто-нон-2-энопиранозоат (E7)
110 мг (0,21 ммоль) соединения (E6), 72 мг (0,26 ммоль) N,N'-бис-тpeт-бутоксикарбонилтиомочевины и 80 мг (0,57 ммоль) триэтиламина растворяли в 22 мл (50-кратный объем) диметил- формамида при комнатной температуре. Далее в систему при охлаждении льдом добавили 70,5 мг (0,26 ммоль) хлорида ртути, после чего смесь перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь разбавили этилацетатом и смесь отфильтровали с использованием Целита, отфильтрованный продукт промывали этилацетатом. Полученный таким образом фильтрат и промывной раствор объединили, к смеси добавили этилацетат и насыщенный водный раствор NaCI с последующим разделением. Органический слой высушивали над сульфатом магния и отфильтровывали с последующей отгонкой растворителя при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем (Кизельгель 60, 100 г, гексан: этилацетат = 3: 2) с получением 120 мг (выход: 71%) указанного соединения (E7) в виде белого твердого продукта.
Величина Rf: 0,65 (гексан:этилацетат = 3:2)
1H-NMR (270 MHz, CDCl3, TMS): δ (ppm) 11.4(1H, s), 8.63 (1H, d, J=7.8 Hz), 7.99(1H, d, J=4.8 Hz), 5.78(1H, d, J=2.4 Hz), 5.32(1H, d, J=4.4 Hz), 5.14(1H, tt, J=7.8, 1.6 Hz), 4.40(1H, dt, J=10.6, 4.8 Hz), 4.33-3.90(6H, m), 3.52(1H, dd, J= 8.7, 3.9 Hz), 2.02(3H, s), 1.64(2H, m), 1.52(9H, s), 1.49 (9H, s), 1.43(3H, s), 1.36(3H, з), 1.26(22H, bs), 0.88(3H, t, J=6.7 Hz);,
[α]
vi) Соль миристил 5-aцeтамидо-2,3,4,5-тeтpaдeoкcи-4- гуанидино-D-глицеро-D-галакто-нон-2-энопиранозоата и трифторуксусной кислоты (E8)
84 мг (0,11 ммоль) соединения (E7) растворили в 47 мл (50- кратный объем) метиленхлорида при комнатной температуре и затем при комнатной температуре в систему добавили 0,85 мл (10-кратный объем) трифторуксусной кислоты с последующим перемешиванием смеси в течение 22 ч. После подтверждения завершения реакции реакционную смесь концентрировали при пониженном давлении, остаток очищали хроматографией на колонку с силикагелем (Кизельгель 60, 15 г, этилацетат: 2-пропанол: вода = 8:2:1) с получением 82 мг (выход: 88%) указанного соединения (E8) в виде твердого белого продукта.
Величина Rf: 0,6 (этилацетат:2-пропанол:вода = 8:2:1);
1H-NMR (270 MHz, CD3OD): δ (ppm) 5.83(1H, d, J=2.7 Hz), 4.44(1H, dd, J= 9.0, 2.7 Hz), 4.38(1H, dd, J=9.0, <1 Hz), 4.18(2H, t, J=6.2 Hz), 4.17(1H, t, J=9.0 Hz), 3.90-3.74(2H, m), 3.68(1H, dd, J=12.0, 4.5 Hz), 3.65(1H, d, J=9.0 Hz), 1.99(3H, s), 1.67(2H, квинтет, J=6.2 Hz), 1.26(24H, bs), 0.87(3H, t, J= 6.2 Hz);
FAB-MS (положит.): 529 (M+H)+;
HR-MS: Вычис. для C26H49N4O7, 529.3597; Найдено 529.3605 (М+H)+.
ПРИМЕР 2. Соль гексил 5-ацетамидо-2,3,4,5-тетрадеокси-4- гуанидино-D-глицеро-D-галакто-нон-2-энопиранозоата и трифторуксусной кислоты (E12) (образцовое соединение N 87)
i) Гексил 5-aцeтамидo-4-aзидo-2,3,4,5-тетрадеокси-8,9- O-изопропилиден-D-глитаро-D-галакто-нон-2-энопиранозоат (E9)
940 мг (2,6 ммоль) соединения (E4), 0,66 мл (5,3 ммоль) 1- гексанола и 1,1 г (4,0 ммоль) тетрафторбората 2-бром-1- этилпиридиния растворили в 47 мл (50-кратный объем) диметил- формамида при комнатной температуре. Далее, в систему влили 1,9 мл (8,0 ммоль) три-н-бутиламина и смесь перемешивали на масляной бане при 50oC в течение 6 ч. В реакционную смесь добавили этилацетат и насыщенный водный раствор NaCl. Полученный таким образом органический слой был высушен над сульфатом магния и отфильтрован с последующим концентрированием при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем (Кизельгель 60, 150 т, бензол: ацетонитрил = 2:1) с получением 560 мг (выход: 44%) указанного соединения (E9) в виде твердого продукта бледно- коричневого цвета.
Величина Rf: 0,43 (бензол: ацетонитрил = 2:1);
1H-NMR (270 MHz, CDCl3, TMS): δ (ppm) 6.53(1H, d, J=6.9 Hz), 5.92(1H, d, J=2.5 Hz), 4.48(1H, bs), 4.41-4.29(2H, m), 4.24-4.00(7H, m), 3.63(1H, bd, J= 7.3 Hz), 1.70(2H, квинтет, J=6.5 Hz), 1.40(3H, s), 1.35(9H, bs), 1.30(3H, s), 0.89(3H, t, J=6.5Hz);
[α]
ii) Гексил 5-ацетамидо-4-(N,N'-бис-трет-бутоксикарбонил) гуанидино-2, 3, 4, 5-тетрадеокси-8,9-O-изопропилиден-O-глицеро-D-галакто-нон-2-энопиранозоат (E11)
533 мг (1,1 ммоль) соединения (E9) растворили в 25 мл (50- кратный объем) этанола при комнатной температуре и затем в систему при той же температуре добавили 190 мг (0,35-кратный объем) катализатора Линдлара с последующим перемешиванием смеси при комнатной температуре и давлении водорода в 1 атм в течение 2,5 ч. После подтверждения завершения реакции реакционную смесь отфильтровали. Отфильтрованный продукт промыли этанолом, фильтрат и промывной раствор объединили с последующим концентрированием при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем (Кизельгель 60, 50 г, этилацетат:2-пропанол:вода = 5:2:1) с получением 303 мг (выход: 60%) указанного соединения (E10) в виде твердого пенистого продукта бледно-коричневого цвета.
Величина Rf:0,44 (этилацетат:2-пропанол:вода = 5:2:1)
290 мг (0,63 ммоль) полученного соединения (E10), 210 мг (0,75 ммоль) N, N'-бис-трет-бутоксикарбонилтиомочевины и 0,22 мл (1,6 ммоль) триэтиламина растворили в 15 мл (50-кратный объем) диметилформамида при комнатной температуре. Затем в систему при охлаждении льдом добавили 220 мг (0,8 ммоль) хлорида ртути и смесь перемешивали при комнатной температуре в течение 2 ч. Реакционную смесь разбавили этилацетатом и отфильтровали с использованием Целита, а отфильтрованный продукт промыли этилацетатом. Полученный таким образом фильтрат и промывной раствор объединили, для разделения смеси в нее добавили этилацетат и насыщенный водный раствор NaCl с последующей экстракцией водного слоя этилацетатом. Органический слой высушили над сульфатом магния и отфильтровали с последующим концентрированием при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем (Кизельгель 60, 75 г, гексан: этилацетат = 1:1) с получением 273 мг (выход: 66%) целевого соединения (E11) в виде твердого белого продукта.
Величина Rf:0,38 (бензол:этилацетат = 5:1);
1H-NMR (270 MHz, CDCl3, TMS): δ (ppm) 11.4(1H, s), 8.64 (1H, d, J=7.7 Hz), 7.99(1H, d, J=4.7 Hz), 5.78(1H, d, J=2.4 Hz), 5.33(1H, d, J=4.4 Hz), 5.14(1H, tt, J=7.9, <1 Hz), 4.40 (1H, dt, J=7.8, 5.7 Hz), 4.23-3.90(7H, m), 3.51(1H, dd, J= 8.2, 4.5 Hz), 2.01 (3H, s), 1.68(2H, квинтет, J=6.4 Hz), 1.51 (9H, s), 1.49(9H, s), 1.43(3H, s), 1.36(3H, s), 1.35-1.20 (6H, m), 0.89(3H, t, J=6.4 Hz);
[α]
iii) Соль гексил 5-ацетамидо-2,3,4,5-тетрадеокси-4- гуaнидинo-D-глицepо)-D-гaлaктo-нoн-2-энoпиpaнoзoaтa и трихлоруксусной кислоты (E12)
158 мг (0,24 ммоль) соединения (E11) растворяли в 8,0 мл (50-кратный объем) метиленхлорида при комнатной температуре и затем к системе при той же температуре добавили 1,6 мл (10-кратный объем) трифторуксусной кислоты с последующим перемешиванием смеси при комнатной температуре в течение 18 часов. После подтверждения завершения реакции реакционную смесь концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем (Кизельгель 60, 15 г, этилацетат:2-пропанол:вода = 5:2:1) с получением 155 мг (выход: 100%) указанного соединения (E12) в виде твердого продукта белого цвета.
Величина Rf: 0,8 (этилацетат:2-пропанол:вода = 5:2:1);
1H-NMR (270 MHz, CD3OD): δ (ppm) 5.94 (1H, d, J=2.4 Hz), 4.46(1H, dd, J= 9.3, 2.4 Hz), 4.38(1H, dd, J=10.4, 1.3 Hz), 4.20(1H, t, J=9.3 Hz), 4.20(2H, t, J= 6.2 Hz), 3.91-3.76(2H, m), 3.66 (1H, bd, J=9.3 Hz), 3.61(1H, dd, J= 12.5, 6.3 Hz), 1.97(3H, s), 1.63(2H, квинтет, J=6.3 Hz), 1.40-1.10(6H, m), 0.80(3H, t, J=6.3 Hz);
[α]
ПРИМЕР 3. Соль 5-ацетамидо-2,3,4,5-тетрадеокси-4-гуанидино-9-O-миристил-D-глицеро-D-галакто-нон- -2-энопиранозойной кислоты и трифторуксусной кислоты (E17) (образцовое соединение N 41)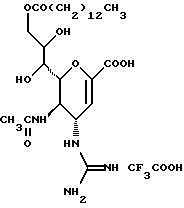
i) Метил 5-ацетамидо-4-азидо-2,3,4,5-тетрадеокси-8,9-O изопропилиден-7-O-миристоил-D-глицеро-D-галакто-нон-2- энопиранозоат (E13)
61 мг (0,16 ммоль) соединения (E3) растворили в 3,0 мл (50- кратный объем) метиленхлорида при комнатной температуре и затем в систему при охлаждении льдом добавили 54 мл (0,2 ммоль) миристоилхлорида и 24,1 мг (0,2 ммоль) 4-диметиламинопиридина с последующим перемешиванием смеси при комнатной температуре в течение 30 мин. Затем в реакционную смесь при комнатной температуре влили 27 мл (0,2 ммоль) триэтиламина с последующим перемешиванием смеси в течение 6 часов. После подтверждения завершения реакции в систему был влит метанол и смесь перемешивали в течение 30 минут. Затем в реакционную смесь добавили этилацетат и водный насыщенный раствор NaCl для разделения смеси. Полученный органический слой высушили над сульфатом магния и отфильтровали с последующим концентрированием при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем (Кизельгель 60, 15 г, бензол: этилацетат = 1:1) с получением 70 г (выход: 74%) целевого соединения (E13) в виде бесцветного прозрачного сиропа.
Величина Rf: 0,42 бензол:этилацетат = 2:1);
1H-NMR (270 MHz, CDCl3, TMS): δ (ppm) 5.95(1H, d, J=2.7 Hz), 5.88(1H, d, J= 7.9 Hz), 5.35(1H, dd, J= 6.0, 1.8 Hz), 4.80(1H, dd, J=9.1, 2.7 Hz), 4.71(1H, dd, J=10.5, 1.8 Hz), 4.39(1H, q, J=6.0 Hz), 4.14(1H, dd, J=8.8, 6.0 Hz), 4.71(1H, dd, J=10.5, 1.8 Hz), 4.39(1H, q, J=6.0 Hz), 4.14(1H, dd, J= 8.8, 6.0 Hz), 3.945(1H, dd, J=8.8, 6.0 Hz), 3.81(3H, s), 3.45(1H, ddd, J= 10.5, 9.1, 7.9 Hz), 2.41(1H, t, J=7.5 Hz), 2.39(1H, t, J=7.5 Hz), 2.02(3H, s), 1.63(2H, квинтет, J= 7.5 Hz), 1.37(3H, s), 1.35(3H, s), 1.26(20H, s), 0.88(3H, t, J= 7.5 Hz).
ii) Метил 5-ацетамидо-4-амино-2, 3,4, 5-тетрадеокси-8,9-O- изопропилиден-7-O-миристоил-D-глицеро-D-галакто-нон-2- энопиранозоат (E14)
70 мг (0,12 ммоль) соединения (E13) растворили в 3,5 мл (50- кратный объем) этанола при комнатной температуре и затем в систему при комнатной температуре добавили 25 мг (0,35- кратный объем) катализатора Линдлара с последующим перемешиванием смеси при комнатной температуре под давлением водорода в 1 атм в течение 1,5 ч. После подтверждения завершения реакции реакционную смесь фильтруют. Отфильтрованный продукт промывали этанолом, фильтрат и промывной раствор объединяли с последующим концентрированием при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем (Кизельгель 60, 15 г, этилацетат:2-пропанол:вода = 5:2:1) с получением 57 мг (выход: 84%) указанного соединения (E14) в виде белого твердого продукта.
Величина Rf: 0,49 (метиленхлорид:метанол = 10:1);
1H-NMR (270 MHz, CD3OD, TMS): δ (ppm) 5.94(1H, d, J=2.4 Hs), 5.42(1H, dd, J= 4.7, 1.8 Hz), 4.39(1H, dt, J=7.1, 6.0 Hz), 4.18(1H, dd, J=9.5, 1.6 Hz), 4.14(1H, dd, J=8.7, 6.4 Hz), 3.93(1H, dd, J=8.7, 6.4 Hz), 3.87(1H, t, J= 9.5 Hz), 3.78(3H s), 3.44(1H, dd, J=9.5, 2.4 Hz), 2.35(2H, q, J=7.3 Hz), 1.94(3H, 3), 1.60(2H, квинтет, J= 7.3 Hz), 1.32(3H, s), 1.31(3H, s), 1.27(20H, s), 0.88(3H, t, J=7.3 Hz).
iii) Метил 5-ацетамидо-4-(N, N'-бис-трет- бутоксикарбонил) гуанидино-2,3,4,5-тетрадеокси-8,9-O- изопропилиден-7-O-миристоил-D-глицеро-D-галакто-нон-2-энопиранозоат (E15)
57 мг (0,1 ммоль) соединения (E14), 34 мг (0,12 ммоль) N,N'-бис-трет-бутоксикарбонилтиомочевины и 35 мл (0,25 ммоль) триэтиламина растворили в 28 мл (50-кратный объем) диметилформамида при комнатной температуре. Затем в систему при охлаждении льдом добавили 35 мг (0,13 ммоль) хлорида ртути и смесь перемешивали при комнатной температуре в течение 2 часов. К реакционной смеси добавили этилацетат, чтобы разбавить ее, с последующей фильтрацией через Целит. Отфильтрованный продукт промыли этилацетатом. Полученный фильтрат и промывной раствор объединили и к смеси, чтобы ее разделить, добавили этилацетат и водный насыщенный раствор NaCl. Органический слой высушили над сульфатом магния и отфильтровали с последующим концентрированием при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем (Кизельгель 60, 15 г, гексан: этилацетат = 2:1) с получением 75 мг (выход: 100%) указанного соединения в виде бесцветного прозрачного сиропа.
Величина Rf: 0,33 (гексан: этилацетат = 2:1);
1H-NMR (270 MHz, CDCl3, TMS): δ (ppm) 11.4(1H, s), 8.46 (1H, d, J=8.7 Hz), 6.06(1H, d, J=8.7 Hz), 5.88(1H, d, J=2.4 Hz), 5.37(1H, dd, J=6.4, 1.5 Hz), 5.15(1H, dt, J=8.7, 2.4 4.38(1H, q, J=6.4 Hz), 4.28(1H, dd, J=8.7, 1.5 Hz), 4.23(1H, t, J=8.7 Hz), 4.10(1H, dd, J=9.6, 6.4 Hz), 3.95(1H, dd, J=9.6, 6.4 Hz), 3.80(3H, s), 2.453(1H, dt, J=16.0, 7.5 Hz), 2.33(1H, dt, J=16.0, 7.5 Hz), 1.87(3H, s), 1.61(2H, квинтет, J=7.5 Hz), 1.49(9H, s), 1.48(9H, s), 1.38(3H, s), 1.35(3H, s), 1.25(20H, bs), 0.88(3H, t, J=7.5Hz).
iv) 5-Ацетамидо-4-(N, N'-бис-трет-бутоксикарбонил)гуанидино- 2,3,4,5-тетрадеокси-8,9-O-изопропилиден-7-O-миристоил-D- глицеро-D-галакто-нон-2-энопиранозойная кислота (E16).
51 мг (0,07 ммоль) соединения (E15) растворили в 2 мл (40-кратный объем) метанола и в систему при комнатной температурю добавили 0,5 мл (10-кратный объем) воды и 3,3 мг (0,078 ммоль) моногидратного гидроксида лития с последующим перемешиванием смеси при комнатной температуре в течение 8 часов. После подтверждения завершения реакции к системе добавили Дауэкс-50W для нейтрализации смеси. Реакционную смесь отфильтровали и отфильтрованный продукт промыли метанолом. Фильтрат и промывочный раствор объединили и сконцентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем (Кизельгель 60, 60 г, этилацетат:2-пропанол:вода = 10: 2: 1) с получением 33 мг (выход: 66%) указанного целевого соединения (E16) в виде белого твердого продукта.
Величина Rf: 0,45 (метиленхлорид:метанол = 10:1);
1H-NMR (270 MHz, CDCl3, TMS): δ (ppm) 11.4(1H, s), 8.48 (1H, d, J=8.0 Hz), 6.31(1H, слабый s), 3.90(1H, bs), 5.30 (1H, bs), 5.10(1H, bs), 4.60-3.30(7H, m), 2.48(1H, dt, J= 13.5, 6.5 Hz), 2.32(1H, dt, J=13.5, 6.5 Hz), 1.88(3H, s), 1.60(2H, квинтет, J=6.5 Hz), 1.48(18H, s), 1.39(3H, s), 1.37 (3H, s), 1.25(20H, bs), 0.88(3H, t, J=6.5 Hz).
v) Соль 5-ацетамидо-2,3,4,5-тетрадеокси-4-гуанидино-9-O- миристоил-D-глицеро-D-галакто-нон-2-энопиранозойной кислоты и трифторуксусной кислоты (E17).
33 мг (0,046 ммоль) соединения (E16) растворили в 1,6 мл (50-кратный объем) метиленхлорида при комнатной температуре и затем к системе при комнатной температуре добавили 60 мл (10- кратный объем) трифторуксусной кислоты с последующим перемешиванием смеси при комнатной температуре в течение 22 часов. После подтверждения завершения реакции реакционную смесь концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем (Кизельгель 60, 5 г, 2-пропанол:вода = 5:1) с получением 30 мг (выход: 84%) указанного соединения (E17) в виде твердого продукта бледно-желтого цвета.
Величина Rf: 0,4 (2-пропанол:вода = 5:1);
1H-NMR (270 MHz, CD3OD): δ (ppm) 5.63(1H, d, J=2.3 Hz), 4.49(1H, dd, J= 9.3, 2.3 Hz), 4.39(1H, d, J=10.6 Hz), 4.25-4.00(4H, m), 3.77(1H, d, J=9.3 Hz), 2.36(2H, t, J=7.4 Hz), 1.92(3H, s), 1.70-1.50(2H, m), 1.30(20H, bs), 0.90(3H, t, J=7.4 Hz);
FAB-MS (положит.): 543 (M+H)+;
HR-MS Вычис. для C26H47O8N4, 543.3378, Найдено 543.3412 (M+H)+;
[α]
ПРИМЕР 4. Соль 5-ацетамидо-2,3,4,5-тетрадеокси-4-гуанидино- 9-O-гексаноил-D-глицеро-D-галакто-нон-2-энопиранозойной кислоты и трифторуксусной кислоты (E22) (образцовое соединение N 36)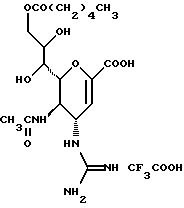
i) Метил 5-ацетамидо-4-азидо-2,3,4,5-тетрадеокси-8,9-O- изoпpoпилидeн-7-O-гeкcaнoил-D-глицеpo-D-гaлaктo-нoн-2- энопиранозоат (E18)
270 мг (0,73 ммоль) соединения (E3) растворяли в 13,5 мл (50-кратный объем) метиленхлорида и затем в систему при охлаждении льдом добавили 0,14 мл (1,0 ммоль) гексаноилхлорида и 110 мг (0,9 ммоль) 4-диметиламинопиридина с последующим перемешиванием смеси при комнатной температуре в течение 30 мин. Далее, 0,13 мл (0,9 ммоль) триэтиламина влили в реакционную смесь при комнатной температуре с последующим перемешиванием смеси в течение 19 ч. После подтверждения завершения реакции в систему влили метанол с последующим перемешиванием в течение 30 мин. Затем к реакционной смеси для ее разделения добавили этилацетат и водный насыщенный раствор NaCl. Органический слой высушили над сульфатом магния и отфильтровали с последующим концентрированием при пониженном давлении. Полученный таким образом остаток очищали хроматографией на колонке с силикагелем (Кизельгель 60, 75 г, бензол:этилацетат = 1: 1) до получения 220 мг (выход: 74%) указанного соединения (E18) в виде бесцветного прозрачного сиропа.
Величина Rf: 0,45 (бензол:этилацетат = 1:1);
1H-NMR (270 MHz, CDCl3, TMS): δ (ppm) 6.11(1H, d, J=8.0 Hz), 5.95(1H, d, J= 2.6 Hz), 5.38(1H, d, J= 5.3, 2.0 Hz), 4.69 (1H, dd, J=9.2, 2.6 Hz), 4.65(1H, dd, J= 10.6, 2.0 Hz), 4.38 (1H, q, J=5.5 Hz), 4.14(1H, dd, J=9.5, 5.5 Hz), 3.95(1H, dd, J=9.5, 5.4 Hz), 3.81(3H, s), 3.59(1H, ddd, J=10.6, 9.2, 8.0 Hz), 2.44(1H, dt, J=15.9, 7.4 Hz), 2.36(1H, dt, J=15.9,7.4 Hz), 2.01(3H, s), 1.64(2H, квинтет, J=7.4 Hz), 1.37(3H, s), 1.35(3H, s), 1.35- 1.30(4H, m), 0.90(3H, t, J=7.4 Hz);
[α]
ii) Метил 5-ацетамидо-4-(N, N'-бис-трет-бутоксикарбонил)- гуанидино-2,3,4,5-тетрадеокси-8,9-O-изопропилиден-7-O- гексаноил-D-глицеро-D-галакто-нон-2-энопиранозоат (E20)
190 мг (0,41 ммоль) соединения (E18) растворили в 9,3 мл (50-кратный объем) этанола при комнатной температуре и затем к системе при комнатной температуре добавили 70 мг (0,38-кратный объем) катализатора Линдлара с последующим перемешиванием смеси при комнатной температуре под давлением водорода 1 атм в течение 2,5 ч. После подтверждения завершения реакции реакционную смесь отфильтровали. Отфильтрованный продукт промыли этанолом, фильтрат и промывочный раствор объединили с последующим концентрированием при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем (Кизельгель 60, 60 г, этилацетет:2-пропанол:вода = 5:2:1) с получением 140 мг (выход: 80%) соединения (E19) в виде твердого пенящегося продукта светло-желтого цвета.
Величина Rf: 0,4 (этилацецатат:2-пропанол = 5:2:1)
140 мг (0,3 ммоль) соединения (E19), 120 мг (0,43 ммоль) N,N'-бис-трет-бутоксикарбонилтиомочевины и 0,12 мл (0,86 ммоль) триэтиламина растворили в 7 мл (50-кратный объем) диметилформамида при комнатной температуре. Затем к системе добавили при охлаждении льдом 110 мг (0,41 ммоль) хлорида ртути с последующим перемешиванием смеси при комнатной температуре в течение 1,5 ч. К реакционной смеси добавили этилацетат, чтобы разбавить ее, и смесь отфильтровали с использованием Целита. Отфильтрованный продукт промывали этилацетатом. Полученный таким образом фильтрат и промывочный раствор объединили и к смеси добавили этилацетат и водный насыщенный раствор NaCl, чтобы разделить ее. Органический слой высушили над сульфатом магния и отфильтровали с последующим концентрированием при пониженном давлении. Остаток очищали на колонке с силикагелем (Кизельгель 60, 50 г, бензол: этилацетат = 3:1) с получением 220 мг (выход: 100%) указанного соединения (E20) в виде белой твердой пены.
Величина Rf: 0,5 (бензол:этилацетат = 3:1);
1H-NMR (270 MHz, CDCl3, TMS): δ (ppm) 11.4(1H, s), 8.46 (1H, d, J=8.7 Hz), 5.97(1H, d, J=8.7 Hz), 5.88(1H, d, J=2.3 Hz), 5.37 (1H, dd, J=6.3, 1.6 Hz), 5.15(1H, dt, J=8.7, 2.3 Hz), 4.37 (1H, q, J=6.4 Hz), 4.2 9(1H, dd, J= 8.7, 1.5 Hz), 4.24(1H, t, J=8.7 Hz), 4.11(1H, dd, J=8.8, 6.4 Hz), 3.96 (1H, dd, J=8.8, 6.4 Hz), 3.81(3H, s), 2.46(1H, dt, J=15.9, 7.4 Hz), 2.33(1H, dt, J= 15.9, 7.4 Hz), 1.88(3H, з), 1.70-1.50(2H, m), 1.49;9H,з), 1.4S(9H, s), 1.38(3H, s), 1.37(3H, з), 1.40-1.25(4H, m), 089 (3H, t, J=7.4 Hz);
[α]
iii) 5-Ацетамидо-4-(N, N'-бис-трет-бутоксикарбонил) гуанидино-2,3,4,5-тeтpадeoкcи-8,9-O-изопропилиден-7-O- гексаноил-D-глицеро-D-галакто-нон-2-энопиранозойная кислота (E21)
180 мг (0,26 ммоль) соединения (E20) растворили в смеси 10,8 мл (60-кратный объем) метанола и 1,8 мл (10-кратный объем) воды и к системе при комнатной температуре добавили 12 мг (0,29 ммоль) моногидратного гидроксида лития с последующим перемешиванием смеси при комнатной температуре в течение 17 ч. После подтверждения завершения реакции в систему для ее нейтрализации был добавлен Дауэкс-50W. Реакционную смесь отфильтровали, отфильтрованный продукт промыли метанолом. Фильтрат и промывочный раствор объединили и сконцентрировали при пониженном давления. Полученный остаток очищали хроматографией на колонке с силикагелем (Кизельгель 60, 50 г, метиленхлорид:метанол = 10: 1) с получением 0,13 г (выход: 75%) указанного целевого соединения (E21) в виде белого пенистого продукта.
Величина Rf: 0,24 (метиленхлорид: метанол = 10:1);
1H-NMR(270 MHz, CDCl3, TMS): δ (ppm) 11.4(1H, bs), 8.52 (1H, bs), 5.95(2H, bs), 5.32(1H, bs), 5.15(1H, bs), 4.60-4.10(3H, m), 3.95(1H, bs), 2.70-2.20(2H, m), 1.89(3H, s). 1.75-1.00(28H, m), 0.85>(3H, m).
iv) Соль 5-ацетамидо-2,3,4,5-тетрадеокси-4-гуанидино-9-O- гексаноил-D-глицеро-D-галакто-нон-2-энопиранозойной кислоты и трифторуксусной кислоты (E22).
95 мг (0,15 моль) соединения (E21) растворили в 4,8 мл (50-кратный объем) метиленхлорида при комнатной температуре и затем к системе при той же температуре добавили 0,95 мл (10- кратный объем) трифторуксусной кислоты, с последующим перемешиванием смеси при комнатной температуре в течение 4,5 ч. После подтверждения завершения реакции реакционную смесь концентрируют при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем (Кизельгель 60, 20 г, этилацетат:2-пропанол:вода = 4:2:1) с получением 54 мг (выход: 57%) указанного соединения в виде твердого белого продукта.
Величина Rа: 0,3 (этилацетат:2-пропанол:вода = 4:2:1);
1H-NMR (270 MHz, CD3OD): δ (ppm) 5.53(1H, d, J=2.2 Hz), 4.50-4.00(6H, m), 3.59(1H, d, J=9.2 Hz), 2.35(2H, t, J=7.5 Hz), 2.01(3H, s), 1.70-1.50(2H, m), 1.40-1.20(4H, m), 0.91 (3H, t, J=7.5 Hz);
IR (KBr) (см-1) 3333, 1665, 1617, 1401, 1385, 1176;
FAB-MS (положит.) 431 (M+H)+;
HR-MS Вычис. для C18H31O8 431.2165; Найдено 431.2125 (М+H)+
[α]
ПРИМЕР 5. Соль 5-ацетамило-2,3,4,5-тетрадеокси-4-гуанидино-9-O- октаноил-D-глицеро-D-галакто-нон-2-энопиранозойной кислоты и трифторуксусной кислоты (E27) (образцовое соединение N 38)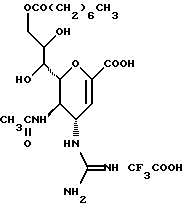
1) Метил 5-aцeтaмидо-4-aзидo-2,3,4,5-тетрадеокси-8,9-O- изопропилиден-7-O-миристил-D-глицеро-D-галакто-нон-2-энопиранозоат (E23)
185 мг (0,5 ммоль) соединения (E3) растворили в 5,0 мл метиленхлорида при комнатной температуре и затем в систему при охлаждении льдом добавили 132 мкл (0,75 ммоль) октаноилхлорида и 91 мл (0,75 ммоль) 4-диметиламинопиридина с
последующим перемешиванием смеси при комнатной температуре в течение 30 мин. Далее, 104 мкл (0,75 ммоль) триэтиламина влили в реакционную смесь при комнатной температуре с последующим перемешиванием смеси в течение 6 ч. После подтверждения завершения реакции в систему влили метанол с последующим перемешиванием в течение 30 мин. Затем к реакционной смеси для ее разделения добавили этилацетат и водный насыщенный раствор NaCl. Органический слой высушили над сульфатом магния и отфильтровали с последующим концентрированием при пониженном давлении. Полученный таким образом остаток очищали хроматографией на колонке с силикагелем (Кизельгель 60, метиленхлорид:метанол = 50: 1) до получения 150 мг (выход: 64%) указанного соединения (E23) в виде бесцветного прозрачного сиропа.
Величина Rf: 0,50 (метиленхлорид:метанол = 20:1);
1H-NMR (270 MHz, CDCl3, TMS): δ (ppm) 5.95(1H, d, J=2.7 Hz), 5.88(1H, d, J= 7.9 Hz), 5.35(1H, dd, J= 6.0, 1.8 Hz), 4.80(1H, dd, J=9.1, 2.7 Hz), 4.71(1H, dd, J=10.5, 1.8 Hz), 4.39(1H, q, J=6.0 Hz), 4.14(1H, dd, J=8.8, 6.0 Hz), 3.945 (1H, dd, J=8.8, 6.0 Hz), 3.81(3H, s), 3.45(1H, ddd, J=10.5, 9.1, 7.9 Hz), 2.41(1H, t, J=7.5 Hz), 2.39(1H, t, J=7.5 Hz), 2.02(3H, s), 1.63(2H, квинтет, J= 7.5 Hz), 1.37(3H, s), 1.35 (3H, s), 1.26(8H, s), 0.88(3H, t, J= 7.5 Hz).
ii) Метил 5-ацетамидо-4-амино-2,3,4,5-тетрадеокси-8,9-O- изопропилиден-7-O-октаноил-D-глицеро-D-галакто-нон-2- энопиранозоат (E24)
130 мг (0,28 ммоль) соединения (E23) растворили в 6,0 мл этанола при комнатной температуре и затем к системе при комнатной температуре добавили 46 мг (0,35-кратный объем) катализатора Линдлара с последующим перемешиванием смеси при комнатной температуре под давлением водорода 1 атм в течение 1,5 ч. После подтверждения завершения реакции реакционную смесь отфильтровали. Отфильтрованный продукт промыли этанолом, фильтрат и промывочный раствор объединили с последующим концентрированием при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем (Кизельгель 60, 15 г, этилацетат: 2-пропанол:вода = 5:2:1) с получением 95 мг (выход: 77%) соединения (E24) в виде твердого белого продукта.
Величина Rf: 0,35 (гексан:этилацетат = 2:1);
1H-NMR (270 MHz, CD3OD, TMS): δ (ppm) 5.94(1H, d, J=2.4 Hz), 5.42(1H, dd, J= 4.7, 1.8 Hz), 4.39(1H, dt, J=7.1, 6.0 Hz), 4.18(1H, dd, J=9.5, 1.6 Hz), 4.14(1H, dd, J=8.7, 6.4 Hz), 3.93(1H, dd, J=8.7, 6.4 Hz), 3.87(1H, t, J= 9.5 Hz), 3.78(3H, s), 3.44(1H, dd, J=9.5, 2.4 Hz), 2.35(2H, q, J=7.3 Hz), 1.94(3H, s), 1.60(2H, квинтет, J=7.3 Hz), 1.32(3H, s), 1.31(3H, s), 1.27(8H, s), 0.88(3H, t, J=7.3 Hz).
iii) Метил 5-ацетамидо-4-(N, N'-бис-трет-бутоксикарбонил) гуанидино-2,3,4,5-тетрадеокси-8,9-O-изопропилиден-7-O- октаноил-D-глицеро-D-галакто-нон-2-энопиранозоат (E25)
90 мг (0,2 ммоль) соединения (E24), 83 мг (0,30 ммоль) N,N'-бис-трет-бутоксикарбонилтиомочевины и 84 мкл (0,25 ммоль) триэтиламина растворили в 5 мл диметилформамида при комнатной температуре. Затем к системе добавили при охлаждении льдом 82 мг (0,30 ммоль) хлорида ртути с последующим перемешиванием смеси при комнатной температуре в течение 2 ч. К реакционной смеси добавили этилацетат, чтобы разбавить ее, и смесь отфильтровали с использованием Целита. Отфильтрованный продукт промывали этилацетатом. Полученный таким образом фильтрат и промывочный раствор объединили и к смеси добавили этилацетат и водный насыщенный раствор NaCl, чтобы разделить ее. Органический слой высушили над сульфатом магния и отфильтровали с последующим концентрированием при пониженном давлении. Остаток очищали на колонке с силикагелем (Кизельгель 60, 15 г, гексан: этилацетат = 2:1) с получением 120 мг (выход: 88%) указанного соединения (E25) в виде бесцветного прозрачного сиропа.
Величина Rf: 0,30 (гексан: этилацетат = 2:1);
1H-NMR (270 MHz, CDCl3, TMS): δ (ppm) 11.4(1H, s), 8.46(1H, d, J=8.7 Hz), 6.06(1H, d, J=8.7 Hz), 5.88(1H, d, J= 2.4 Hz), 5.37(1H, dd, J=6.4, 1.5 Hz), 5.15(1H, dt, J=8.7, 2.4 Hz), 4.38(1H, q, J=6.4 Hz), 4.28(1H, dd, J=8.7, 1.5 Hz), 4.23(1H, t, J=8.7 Hz), 4.10(1H, dd, J=9.6, 6.4 Hz), 3.95(1H, dd, J= 9.6, 6.4 Hz), 3.80(3H, s), 2.453(1H, dt, J=16.0, 7.5 Hz), 2.33(1H, dt, J= 16.0, 7.5 Hz), 1.87(3H, s), 1.61(2H, квинтет, J=7.5 Hz), 1.49(9H, s), 1.48(9H, s), 1.38(3H, s), 1.35(3H, s), 1.25(8H, bs), 0.88(3H, t, J=7.5Hz).
iv) 5-Aцeтaмидo-4-(N, N'-бис-трет-бутоксикарбонил)гуанидино- 2,3,4,5-тетрадеокси-8,9-O-изопропилиден-7-O-oктaнoил-D- глицepo-D-гaлaктo-нoн-2-энoпиpaнoзoйнaя кислота (E26)
100 мг (0,15 ммоль) соединения (E25) растворили в смеси 4 мл (40-кратный объем) метанола и 1 мл (10-кратный объем) воды и к системе при комнатной температуре добавили 7,0 мг (0,165 ммоль) моногидратного гидроксида лития с последующим перемешиванием смеси при комнатной температуре в течение 8 часов. После подтверждения завершения реакции в систему для ее нейтрализации был добавлен Дауэкс-50W. Реакционную смесь отфильтровали, отфильтрованный продукт промыли метанолом. Фильтрат и промывочный раствор объединили и сконцентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем (Кизельгель 60, этилацетат:2-пропанол:вода = 10:2:1) с получением 56 мг (выход: 56%) указанного целевого соединения (E26) в виде белого твердого продукта.
Величина Rf: 0,35 (метиленхлорид:метанол = 10:1);
1H-NMR (270 MHz, CDCl3, TMS): δ (ppm) 11.4(1H, s), 8.48 (1H, d, J=8.0 Hz), 6.31(1H, слаб. s), 5.90(1H, bs), 5.30(1H, bs), 5.10(1H, bs), 4.60-3.30(7H, m), 2.48(1H, dt, J=13.5, 6.5 Hz), 2.32(1H, dt, J=13.5, 6.5 Hz), 1.88(3H, s), 1.60(2H, квинтет, J= 6.5 Hz), 1.48(18H, s), 1.39(3H, s), 1.37(3H, s), 1.25(8H, bs), 0.88(3H, t, J=6.5 Hz).
v) Соль 5-ацетамидо-2,3,4,5-тетрадеокси-4-гуанидино-9-O- октаноил-D-глицеро-D-галакто-нон-2-энопиранозойной кислоты и трифторуксусной кислоты (E27)
50 мг (0,075 ммоль) соединения (E26) растворили в 3 мл (50- кратный объем) метиленхлорида при комнатной температуре и затем к системе при той же температуре добавили 1 мл (10-кратный объем) трифторуксусной кислоты с последующим перемешиванием смеси при комнатной температуре в течение 22 часов. После подтверждения завершения реакции реакционную смесь концентрируют при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем (Кизельгель 60, 5 г, 2-пропанол:вода = 5:1) с получением 38 мг (выход: 88%) указанного соединения (E27) твердого светло-желтого продукта.
Величина Rf: 0,3 (2-пропанол:вода = 5:1);
1H-NMR (270 MHz, CD3OD): δ (ppm) 5.55 (1H, bs), 4.40-4.10 (7H, m), 3.65(1H, d, J= 9.0 Hz), 2.36(2H, t, J=7.0 Hz), 2.00 (3H, s), 1.70-1.50(2H, m), 1.30(8H, bs), 0.90(3H, t, J=7.0 Hz);
FAB-MS (положит.): 459 (M+H)+;
[α]
ПРИМЕР 6. Соль 5-ацетамидо-2,3,4,5-тетрадеокси-4-гуанидино- 9-O-деканоил-D-глицеро-D-галакто-нон-2-энопиранозойной кислоты и трифторуксусной кислоты (E32) (образцовое соединение N 39)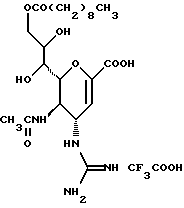
i) Метил 5-ацетамидо-4-азидо-2,3,4,5-тетрадеокси-8,9-O- изопропилиден-7-O-деканоил-D-глицеро-D-галакто-нон-2- энопиранозоат (E28)
214 мг (0,58 ммоль) соединения (E3) растворили в 6,0 мл метиленхлорида при комнатной температуре и затем в систему при охлаждении льдом добавили 152 мкл (0,87 ммоль) деканоилхлорида и 106 мг (0,87 ммоль) 4-диметиламинопиридина с последующим перемешиванием смеси при комнатной температуре в течение 30 мин. Далее, 106 мкл (0,87 ммоль) триэтиламина влили в реакционную смесь при комнатной температуре с последующим перемешиванием смеси в течение 6 ч. После подтверждения завершения реакции в систему влили метанол с последующим перемешиванием в течение 30 мин. Затем к реакционной смеси для ее разделения добавили этилацетат и водный насыщенный раствор NaCl. Органический слой высушили над сульфатом магния и отфильтровали с последующим концентрированием при пониженном давлении. Полученный таким образом остаток очищали хроматографией на колонке с силикагелем (Кизельгель 60, метиленхлорид: метанол = 50:1) до получения 180 мг (выход: 63%) указанного соединения (E28) в виде бесцветного прозрачного сиропа.
Величина Rf: 0,56 (метиленхлорид:метанол = 20:1);
1H-NMR (270 MHz, CDCl3, TMS): δ (ppm) 5.95(1H, d, J=2.7 Hz), 5.88(1H, d, J= 7.9 Hz), 5.35(1H, dd, J= 6.0, 1.8 Hz), 4.80(1H, dd, J=9.1, 2.7 Hz), 4.71(1H, dd, J=10.5, 1.8 Hz), 4.39(1H, q, J=6.0 Hz), 4.14(1H, dd, J=8.8, 6.0 Hz), 3.945 (1H, dd, J=8.8, 6.0 Hz), 3.81(3H, s), 3.45(1H, ddd, J=10.5, 9.1, 7.9 Hz), 2.41(1H, t, J=7.5 Hz), 2.39(1H, t, J=7.5 Hz), 2.02(3H, s), 1.63(2H, квинтет, J=7.5 Hz), 1.37(3H, s), 1.35 (3H, s), 1.26(12H, s), 0.88(3H, t, J= 7.5 Hz).
ii) Метил 5-ацетамидо-4-амино-2,3,4,5-тетрадеокси-8,9-O- изопропилиден-7-O-деканоил-D-глицеро-D-галакто-нон-2- энопиранозоат (E29)
170 мг (0,32 ммоль) соединения (E28) растворили в 10 мл этанола при комнатной температуре и затем к системе при комнатной температуре добавили 60 мг (0,35-кратный объем) катализатора Линдлара с последующим перемешиванием смеси при комнатной температуре под давлением водорода 1 атм в течение 1,5 ч. После подтверждения завершения реакции реакционную смесь отфильтровали. Отфильтрованный продукт промыли этанолом, фильтрат и промывочный раствор объединили с последующим концентрированием при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем (Кизельгель 60, 15 г, этилацетат:2-пропанол:вода = 5:2:1) с получением 120 мг (выход: 80%) соединения (E29) в виде твердого белого продукта.
Величина Rf: 0,40 (н-гексан:этилацетат 2:1);
1H-NMR (270 MHz, CD3OD, TMS): δ (ppm) 5.94(1H, d, J=2.4 Hz), 5.42(1H, dd, J= 4.7, 1.8 Hz), 4.39(1H, dt, J=7.1, 6.0 Hz), 4.18(1H, dd, J=9.5, 1.6 Hz), 4.14(1H, dd, J=8.7, 6.4 Hz), 3.93(1H, dd, J=8.7, 6.4 Hz), 3.87(1H, t, J= 9.5 Hz), 3.78(3H, s), 3.44(1H, dd, J=9.5, 2.4 Hz), 2.35(2H, q, J=7.3 Hz), 1.94(3H, s), 1.60(2H, квинтет, J= 7.3 Hz), 1.32(3H, s), 1.31(3H, s), 1.27(12H, s), 0.88(3H, t, J=7.3 Hz).
iii) Метил 5-ацетамидо-4-(N, N'-бис-трет-бутоксикарбонил) гуанидино-2,3,4,5-тетрадеокси-8,9-O-изопропилиден-7-O- деканоил-D-глицеро-D-галакто-нон-2-энопиранозоат (E30)
100 мг (0,21 ммоль) соединения (E29), 87 мг (0,31 ммоль) N,N'-бис-трет-бутоксикарбонилтиомочевины и 87 мкл (0,63 ммоль) триэтиламина растворили в 5 мл диметилформамида при комнатной температуре. Затем к системе добавили при охлаждении льдом 84 мг (0,31 ммоль) хлорида ртути с последующим перемешиванием смеси при комнатной температуре в течение 2 ч. К реакционной смеси добавили этилацетат, чтобы разбавить ее, и смесь отфильтровали с использованием Целита. Отфильтрованный продукт промывали этилацетатом. Полученный таким образом фильтрат и промывочный раствор объединили и к смеси добавили этилацетат и водный насыщенный раствор NaC1, чтобы разделить ее. Органический слой высушили над сульфатом магния и отфильтровали с последующим концентрированием при пониженном давлении. Остаток очищали на колонке с силикагелем (Кизельгель 60, гексан: этилацетат = 2:1) с получением 120 мг (выход: 87%) указанного соединения (E30) в виде бесцветного прозрачного сиропа.
Величина Rf: 0,30 (гексан:этилацетат = 2:1);
1H-NMR (270 MHz, CDCl3 TMS): δ (ppm) 11.4(1H, s), 8.46 (1H, d, J=8.7 Hz), 6.06(1H, d, J=8.7 Hz), 5.88(1H, d, J=2.4 Hz), 5.37(1H, dd, J=6.4, 1.5 Hz), 5.15(1H, dt, J=8.7, 2.4 Hz), 4.38 (1H, q, J=6.4 Hz), 4.28 (1H, dd, J= 8.7, 1.5 Hz), 4.23(1H, t, J=8.7 Hz), 4.10(1H, dd, J=9.6, 6.4 Hz), 3.95(1H, dd, J=9.6, 6.4 Hz), 3.80(3H, s), 2.453(1H, dt, J=16.0, 7.5 Hz), 2.33(1H, dt, J= 16.0, 7.5 Hz), 1.87(3H, s), 1.61(2H, квинтет, J=7.5 Hz), 1.49(9H, s), 1.48(9H, s), 1.38(3H, s), 1.35(3H, s), 1.25(12H, bs), 0.88(3H, t, J=7.5 Hz).
iv) 5-ацетамидо-4-(N, N'-бис-трет-бутоксикарбонил)гуанидино- 2,3,4,5-тетрадеокси-8,9-O-изопропилиден-7-O-деканоил-D- глицеро-D-галакто-нон-2-энопиранозойная кислота (E31)
100 мг (0,14 ммоль) соединения (E30) растворили в смеси 4 мл (40-кратный объем) метанола и 1 мл (10-кратный объем) воды и к системе при комнатной температуре добавили 6,5 мг (0,154 ммоль) моногидратного гидроксида лития с последующим перемешиванием смеси при комнатной температуре в течение 8 ч. После подтверждения завершения реакции в систему для ее нейтрализации был добавлен Дауэкс-50W. Реакционную смесь отфильтровали, отфильтрованный продукт промыли метанолом. Фильтрат и промывочный раствор объединили и сконцентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем (Кизельгель 60, этилацетат:2-пропанол:вода = 10:2:1) с получением 53 мг (выход: 55%) указанного целевого соединения (E31) в виде белого твердого продукта.
Величина Rf: 0,38 (метиленхлорид:метанол = 10:1);
1H-NMR (270 MHz, CDCl3, TMS): δ (ppm) 11.4(1H, s), 8.48 (1H, d, J=8.0 Hz), 6.31(1H, слаб. s), 5.90(1H, bs), 5.30(1H, bs), 5.10(1H, bs), 4.60-3.30(7H, m), 2.48(1H, dt, J=13.5, 6.5 Hz), 2.32(1H, dt, J=13.5, 6.5 Hz), 1.88(3H, s), 1.60(2H, квинтет, J= 6.5 Hz), 1.48(18H, s), 1.39(3H, s), 1.37(3H, s), 1.25(12H, bs), 0.88(3H, t, J=6.5 Hz).
v) Соль 5-ацетамидо-2,3,4,5-тетрадеокси-4-гуанидино-9-O- деканоил-D-глицеро-D-галакто-нон-2-энопиранозойной кислоты и трифторуксусной кислоты (E32)
40 мг (0,057 ммоль) соединения (E31) растворили в 3 мл (50- кратный объем) метиленхлорида при комнатной температуре и затем к системе при той же температуре добавили 1 мл (10- кратный объем) трифторуксусной кислоты с последующим перемешиванием смеси при комнатной температуре в течение 22 ч. После подтверждения завершения реакции реакционную смесь концентрируют при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем (Кизельгель 60, 5 г, 2- пропанол:вода = 5:1) с получением 30 мг (выход: 87%) указанного соединения (E32) в виде твердого светло-желтого продукта.
Величина Rf: 0,3 (2-пропанол:вода = 5:1);
1H-NMR (270 MHz, CD3OD): δ (ppm) 5.55(1H, bs), 4.40-4.10 (7H, m), 3.65(1H, d, J= 9.0 Hz), 2.36(2H, t, J=7.0 Hz), 2.00 (3H, s), 1.70-1.50(2H, m), 1.30(12H, bs), 0.90(3H, t, J=7.0 Hz);
FAB-MS (положит.): 487 (M+H)+;
[α]
ПРИМЕР 7. Соль 5-ацетамидо-2,3,4,5-тетрадеокси-4-гуанидино- 9-O-пальмитоил-D-глицеро-D-галакто-нон-2- энопиранозойной кислоты и трифторуксусной кислоты (E37) (образцовое соединение N 42)
i) Метил 5-ацетамидо-4-азидо-2,3,4,5-тетрадеокси-8,9-O- изопропилиден-7-O-пальмитоил-D-глицеро-D-галакто-нон-2- энопиранозоат (E28)
1,35 г (3,66 ммоль) соединения (E3) растворили в 10 мл метиленхлорида при комнатной температуре и затем в систему при охлаждении льдом добавили 1,88 мл (6,22 ммоль) пальмитоилхлорида и 759 мг (4,33 ммоль) 4-диметиламинопиридина с последующим перемешиванием смеси при комнатной температуре в течение 30 минут. Затем в реакционную смесь при комнатной температуре влили 104 мкл (6,21 ммоль) триэтиламина с последующим перемешиванием смеси в течение 15 ч. После подтверждения завершения реакции в систему был влит метанол и смесь перемешивали в течение 30 мин. Затем в реакционную смесь добавили этилацетат и водный насыщенный раствор NaCl для разделения смеси. Полученный органический слой высушили над сульфатом магния и отфильтровали с последующим концентрированием при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем (Кизельгель 60, н-гексан: этилацетат = 2:1) с получением 1,42 г (выход: 64%) целевого соединения (E33) в виде бесцветной пены.
Величина Rf: 0,53 (н-гексан:этилацетат = 1:1);
1H-NMR (270 MHz, CDCl3, TMS): δ (ppm) 5.95(1H, d, J=2.7 Hz), 5.88(1H, d, J= 7.9 Hz), 5.35(1H, dd, J= 6.0, 1.8 Hz), 4.80(1H, dd, J=9.1, 2.7 Hz), 4.71(1H, dd, J=10.5, 1.8 Hz), 4.39(1H, q, J=6.0 Hz), 4.14(1H, dd, J=8.8, 6.0 Hz), 3.945 (1H, dd, J=8.8, 6.0 Hz), 3.81(3H, s), 3.45(1H, ddd, J=10.5, 9.1, 7.9 Hz), 2.41(1H, t, J=7.5 Hz), 2.39(1H, t, J=7.5 Hz), 2.02(3H, s), 1.63(2H, квинтет, J=7.5 Hz), 1.37(3H, s), 1.35 (3H, s), 1.26(24H, s), 0.88(3H, t, J= 7.5 Hz).
ii) Метил 5-ацетамидо-4-амино-2,3,4,5-тетрадеокси-8,9-O- изопропилиден-7-O-пальмитоил-D-глицеро-D-галакто-нон-2- энопиранозоат (E34)
1,42 г (2,34 ммоль) соединения (E33) растворили в 15 мл этанола при комнатной температуре и затем в систему при комнатной температуре добавили 477 мг (0,34-кратный объем) катализатора Линдлара с последующим перемешиванием смеси при комнатной температуре под давлением водорода 1 атм в течение 2 часов. После подтверждения завершения реакции реакционную смесь фильтруют. Отфильтрованный продукт промывали этанолом, фильтрат и промывочный раствор объединяли с последующим концентрированием при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем (Кизельгель 60, 15 г, этилацетат:-метанол = 10:1) с получением 1,16 г (выход: 85%) указанного соединения (E34) в виде бесцветной пены.
Величина Rf: 0,18 (этилацетат:метанол = 5:1);
1H-NMR (270 MHz, CD3OD, TMS): δ (ppm) 5.94(1H, d, J=2.4 Hz), 5.42(1H, dd, J= 4.7, 1.8 Hz), 4.39(1H, dt, J=7.1, 6.0 Hz), 4.18(1H, dd, J=9.5, 1.6 Hz), 4.14(1H, dd, J=8.7, 6.4 Hz), 3.93 (1H, dd, J=8.7, 6.4 Hz), 3.87 (1H, t, J= 9.5 Hz), 3.78(3H, 3), 3.44(1H, dd, J=9.5, 2.4 Hz), 2.35(2H, q, J=7.3 Hz), 1.94(3H, s), 1.60(2H, квинтет, J= 7.3 Hz), 1.32(3H, s), 1.31(3H, s), 1.27(24H, s), 0.88(3H, t, J=7.3 Hz).
iii) Метил 5-ацетамидо-4-(N, N'-бис-трет-бутоксикарбонил) гуанидино-2,3,4,5-тетрадеокси-8,9-O-изопропилидeн-7-O- пaльмитoил-D-глицepo-D-галaктo-нoн-2-энoпиранозоат (E35)
1,16 г (2,0 ммоль) соединения (E34), 828 мг (2,99 ммоль) N,N'-бис-трет-бутоксикарбонилтиомочевины и 0,42 мл (3,03 ммоль) триэтиламина растворили в 12 мл диметилформамида при комнатной температуре. Затем в систему при охлаждении льдом добавили 813 мг (2,99 ммоль) хлорида ртути и смесь перемешивали при комнатной температуре в течение 2 ч. К реакционной смеси добавили этилацетат, чтобы разбавить ее, с последующей фильтрацией через Целит. Отфильтрованный продукт промыли этилацетатом. Полученный фильтрат и промывной раствор объединили и к смеси, чтоб ее разделить, добавили этилацетат и водный насыщенный раствор NaCl. Органический слой высушили над сульфатом магния и отфильтровали с последующим концентрированием при пониженном давлении. Остаток очищали на колонке с силикагелем (Кизельгель 60, 15 г, гексан: этилацетат = 2:1) с получением 1,0 г (выход: 61%) указанного соединения (E35) в виде бесцветной пены.
Величина Rf: 0,52 (гексан: этилацетат = 2:1);
1H-NMR (270 MHz, CDCl3, TMS): δ (ppm) 11.4(1H, s), 8.46 (1H, d, J=8.7 Hz), 6.06(1H, d, J=8.7 Hz), 5.88(1H, d, J=2.4 Hz), 5.37(1H, dd, J=6.4, 1.5 Hz), 5.15(1H, dt, J=8.7, 2.4 Hz), 4.38(1H, q, J=6.4 Hz), 4.28(1H, dd, J=8.7, 1.5 Hz), 4.23(1H, t, J=8.7 Hz), 4.10(1H, dd, J=9.6, 6.4 Hz), 3.95(1H, dd, J= 9.6, 6.4 Hz), 3.80(3H, s), 2.453(1H, dt, J=16.0, 7.5 Hz), 2.33(1H, dt, J= 16.0, 7.5 Hz), 1.87(3H, s), 1.61(2H, квинтет, J=7.5 Hz), 1.49(9H, s), 1.48(9H, s), 1.38(3H, s), 1.35(3H, s), 1.25(24H, bs), 0.88(3H, t, J=7.5 Hz).
iv) 5-Ацетамидо-4-(N, N'-бис-трет-бутоксикарбонил)гуанидино- 2,3,4,5-тетрадеокси-8,9-O-изопропилиден-7-O-пальмитоил- D-глицеро-D-галакто-нон-2-энопиранозойная кислота (E36)
61 мг (0,07 ммоль) соединения (E35) растворили в 1,2 мл (40- кратный объем) метанола и в систему при комнатной температуре добавили 0,15 мл (10-кратный объем) воды и 3,4 мг (0,08 ммоль) моногидратного гидроксида лития с последующим перемешиванием смеси при комнатной температуре в течение 3 часов. После подтверждения завершения реакции к системе добавили Дауэкс-50W для нейтрализации смеси. Реакционную смесь отфильтровали и отфильтрованный продукт промыли метанолом. Фильтрат и промывочный раствор объединили и сконцентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем (Кизельгель 60, этилацетат:метанол = 10:1) с получением 33 мг (выход: 55%) указанного целевого соединения (E36) в виде бесцветной пены.
Величина Rf: 0,61 (этилацетат: метанол = 5:1);
1H-NMR (270 MHz, CDCl3, TMS): δ (ppm) 11.4(1H, s), 8.48 (1H, d, J=8.0 Hz), 6.31(1H, слаб. s), 5.90(1H, bs), 5.30(1H, bs), 5.10(1H, bs), 4.60- 3.30(7H, m), 2.48(1H, dt, J=13.5, 6.5 Hz), 2.32(1H, dt, J=13.5, 6.5 Hz), 1.88(3H, s), 1.60(2H, квинтет, J= 6.5 Hz), 1.48(24H, s), 1.39(3H, s), 1.37(3H, s), 1.25(8H, bs), 0.88(3H, t, J=6.5 Hz).
v) Соль 5-ацетамидо-2,3,4,5-тетрадеокси-4-гуанидино-9-O- пaльмитoил-D-глицepo-D-гaлaктo-нoн-2-энoпиpaнoзoйной кислоты и трифторуксусной кислоты (E37)
289 мг (0,36 ммоль) соединения (E36) растворили в 3 мл (50- кратный объем) метиленхлорида при комнатной температуре и затем к системе при комнатной температуре добавили 1 мл (10-кратный объем) трифторуксусной кислоты с последующим перемешиванием смеси при комнатной температуре в течение 22 ч. После подтверждения завершения реакции реакционную смесь концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем (Кизельгель 60, 5 г, этилацетат:2-пропанол:вода = 5:2:1) с получением 206 мг (выход: 84%) указанного соединения (E37) - бесцветная пена.
Величина Rf: 0,44 (2-пропанол:вода =5:1);
1H-NMR (270 MHz, CD3OD): δ (ppm) 5.55 (1H, bs), 4.40-4.10 (7H, m), 3.65(1H, d, J= 9.0 Hz), 2.36(2H, t, J=7.0 Hz), 2.00 (3H, s), 1.70-1.50(2H, m), 1.30(24H, bs), 0.90(3H, t, J=7.0 Hz);
FAB-MS (положит.): 571 (М+H)+;
[α]
ПРИМЕР 8. Соль 5-ацетамидо-2,3,4,5-тетрадеокси-4-гуанидино- 9-O-додеканоил-D-глицеро-D-галакто-нон-2-энопиранозойной кислоты и трифторуксусной кислоты (E42) (образцовое соединение N 40)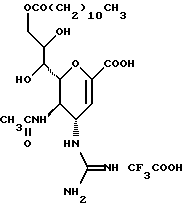
i) Метил 5-ацетамидо-4-азидо-2,3,4,5-тетрадеокси-8,9-O- изопропилиден-7-O-додеканоил-D-глицеро-D-галакто-нон- 2- энопиранозоат (E38)
1,07 г (2,89 ммоль) соединения (E3) растворили в 11 мл метиленхлорида при комнатной температуре и затем в систему при охлаждении льдом добавили 1,03 мл (4,33 ммоль) додеканоилхлорида и 529 мг (4,33 ммоль) 4-диметиламинопиридина с последующим перемешиванием смеси при комнатной температуре в течение 30 мин. Затем в реакционную смесь при комнатной температуре влили 0,6 мл (4,33 ммоль) триэтиламина с последующим перемешиванием смеси в течение 2,5 ч. После подтверждения завершения реакции в систему был влит метанол и смесь перемешивали в течение 30 мин. Затем в реакционную смесь добавили этилацетат и водный насыщенный раствор NaCl для разделения смеси. Органический слой высушили над сульфатом магния и отфильтровали с последующим концентрированием при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем (Кизельгель 60, н-гексан:этилацетат = 2:1) до получения 1,07 мг (выход: 67%) целевого соединения (E38) в виде бесцветной пены.
Величина Rf: 0,44 (н-гексан:этилацетат = 1:1);
1H-NMR (270 MHz, CDCl3, TMS): δ(ppm) 5.95(1H, d, J=2.7 Hz), 5.88(1H, d, J= 7.9 Hz), 5.35(1H, dd, J= 6.0, 1.8 Hz), 4.80(1H, dd, J=9.1, 2.7 Hz), 4.71(1H, dd, J=10.5, 1.8 Hz), 4.39(1H, q, J=6.0 Hz), 4.14(1H, dd, J=8.8, 6.0 Hz), 3.945 (1H, dd, J=8.8, 6.0 Hz), 3.81(3H, s), 3.45(1H, ddd, J=10.5, 9.1, 7.9 Hz), 2.41(1H, t, J=7.5 Hz), 2.39(1H, t, J=7.5 Hz), 2.02(3H, s), 1.63(2H, квинтет, J=7.5 Hz), 1.37(3H, s), 1.35 (3H, s), 1.26(16H, s), 0.88(3H, t, J= 7.5 Hz).
ii) Метил 5-ацетамидо-4-амино-2,3,4,5-тетрадеокси-8,9-O- изопропилиден-7-O-додеканоил-D-глицеро-D-галакто-нон-2- энопиранозоат (E39)
1,06 г (1,92 ммоль) соединения (E38) растворили в 10 мл этанола при комнатной температуре и затем в систему при комнатной температуре добавили 353 мг (0,33-кратный объем) катализатора Линдлара с последующим перемешиванием смеси при комнатной температуре под давлением водорода 1 атм в течение 1,5 ч. После подтверждения завершения реакции реакционную смесь фильтруют. Отфильтрованный продукт промыли этанолом, фильтрат и промывочный раствор объединяли с последующим концентрированием при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем (Кизельгель 60, 15 г, этилацетат: метанол = 5:1) с получением 871 мг (выход: 86%) указанного соединения (E39) в виде бесцветной пены.
Величина Rf: 0,36 (этилацетат:метанол = 5:1);
1H-NMR (270 MHz, CD3OD, TMS): δ (ppm) 5.94(1H, d, J=2.4 Hz), 5.42(1H, dd, J= 4.7, 1.8 Hz), 4.39(1H, dt, J=7.1, 6.0 Hz), 4.18(1H, dd, J=9.5, 1.6 Hz), 4.14(1H, dd, J=8.7, 6.4 Hz), 3.93(1H, dd, J=8.7, 6.4 Hz), 3.87(1H, t, J= 9.5 Hz), 3.78(3H, s), 3.44(1H, dd, J=9.5, 2.4 Hz), 2.35(2H, q, J=7.3 Hz), 1.94(3H, s), 1.60(2H, квинтет, J=7.3 Hz), 1.32(3H,s), 1.31(3H, s), 1.27(16H, s), 0.88(3H, t, J=7.3 Hz).
iii) Метил 5-ацетамидо-4-(N, N'-бис-трет-бутоксикарбонил) гуанидино-2,3,4,5-тетрадеокси-8,9-O-изопропилиден-7-O- додеканоил-D-глицеро-D-галакто-нон-2-энопиранозоат (E40)
868 мг (1,65 ммоль) соединения (E39), 940 мг (3,4 ммоль) N,N'-бис-трет-бутоксикарбонилтиомочевины и 0,95 мл (6,90 ммоль) триэтиламина растворили в 10 мл диметилформамида при комнатной температуре. Затем в систему при охлаждении льдом добавили 926 мг (3,4 ммоль) хлорида ртути и смесь перемешивали при комнатной температуре в течение 2 ч. К реакционной смеси добавили этилацетат, чтобы разбавить ее, с последующей фильтрацией через Целит. Отфильтрованный продукт промыли этилацетатом. Полученный фильтрат и промывной раствор объединили и к смеси, чтобы ее разделить, добавили этилацетат и водный насыщенный раствор NaC1. Органический слой высушили над сульфатом магния и отфильтровали с последующим концентрированием при пониженном давлении. Остаток очищали на колонке с силикагелем (Кизельгель 60, 15 г, гексан: этилацетат = 2:1) с получением 1,3 г (выход: 100%) указанного соединения (E40) в виде бесцветной пены.
Величина Rf: 0,47 (гексан: этилацетат = 2:1);
1H-NMR (270 MHz, CDCl3, TMS): δ (ppm) 11.4(1H, s), 8.46 (1H, d, J=8.7 Hz), 6.06(1H, d, J=8.7 Hz), 5.88(1H, d, J=2.4 Hz), 5.37(1H, dd, J=6.4, 1.5 Hz), 5.15(1H, dt, J=8.7, 2.4 Hz), 4.38 (1H, q, J=6.4 Hz), 4.28 (1H, dd, J= 8.7, 1.5 Hz), 4.23(1H, t, J=8.7 Hz), 4.10(1H, dd, J=9.6, 6.4 Hz), 3.95(1H, dd, - J= 9.6, 6.4 Hz), 3.80(3H, s), 2.453(1H, dt, J=16.0, 7.5 Hz), 2.33(1H, dt, J=16.0, 7.5 Hz), 1.87(3H, s), 1.61(2H, квинтет, J=7.5 Hz), 1.49(9H, s), 1.48(9H, s), 1.38(3H, s), 1.35(3H, s), 1.25(16H, bs), 0.88(3H, t, J=7.5 Hz).
iv) 5-Ацетамидо-4-(N, N'-бис-трет-бутоксикарбонил)гуанидино- 2,3,4,5-тетрадеокси-8,9-O-изопропилиден-7-O-додеканоил- D-глицеро-D-галакто-нон-2-энопиранозойная кислота (E41)
100 мг (0,15 ммоль) соединения (E40) растворили в 4 мл (40- кратный объем) метанола и в систему при комнатной температуре добавили 1 мл (10-кратный объем) воды и 7,0 мг (0,165 ммоль) моногидратного гидроксида лития с последующим перемешиванием смеси при комнатной температуре в течение 8 часов. После подтверждения завершения реакции к системе добавили Дауэкс-50W для нейтрализации смеси. Реакционную смесь отфильтровали и отфильтрованный продукт промыли метанолом. Фильтрат и промывочный раствор объединили и сконцентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем (Кизельгель 60, этилацетат:2-пропанол:вода = 10:2:1) с получением 56 мг (выход: 56%) указанного целевого соединения (E41) в виде белого твердого продукта.
Величина Rf: 0,35 (метиленхлорид:метанол = 10:1);
1H-NMR (270 MHz, CDCl3, TMS): δ (ppm) 11.4(1H, s), 8.48 (1H, d, J=8.0 Hz), 6.31(1H, слаб. s), 5.90(1H, bs), 5.30(1H, bs), 5.10(1H, bs), 4.60-3.30(7H, m), 2.48(1H, dt, J=13.5, 6.5 Hz), 2.32(1H, dt, J=13.5, 6.5 Hz), 1.88(3H, s), 1.60(2H, квинтет, J= 6.5 Hz), 1.48(18H, s), 1.39(3H, s), 1.37(3H, s), 1.25(8H, bs), 0.88(3H, t, J=6.5 Hz).
v) Соль 5-ацетамидо-2,3,4,5-тетрадеокси-4-гуанидино-9-O- додеканоил-D-глицеро-D-галакто-нон-2-энопиранозойной кислоты и трифторуксусной кислоты (E42)
50 мг (0,075 ммоль) соединения (E41) растворили в 3 мл (50- кратный объем) метиленхлорида при комнатной температуре и затем к системе при комнатной температуре добавили 1 мл (10-кратный объем) трифторуксусной кислоты с последующим перемешиванием смеси при комнатной температуре в течение 22 ч. После подтверждения завершения реакции реакционную смесь концентрируют при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем (Кизельгель 60, 5 г, 2-пропанол:вода = 5:1) с получением 38 мг (выход: 88%) указанного соединения (E42) в виде твердого продукта светло-желтого цвета.
Величина Rf: 0,3 (2-пропанол:вода = 5:1);
1H-NMR (270 MHz, CD3OD): δ (ppm) 5.55(1H, bs), 4.40-4.10 (7H, m), 3.65(1H, d, J= 9.0 Hz), 2.36(2H, t, J=7.0 Hz), 2.00 (3H, s), 1.70-1.50(2H, m), 1.30(8H, bs), 0.90(3H, t, J=7.0 Hz);
FAB-MS (положит.): 515 (M+H)+;
[α]
ПРИМЕР 9. Соль 5-ацетамидо-2,3,4,5-тетрадеокси-4-гуанидино-9-O- октадеканоил-D-глицеро-D-галакто-нон-2-энопиранозойной кислоты и трифторуксусной кислоты (E47) (образцовое соединение N 43)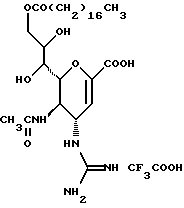
i) Метил 5-ацетамидо-4-азидо-2,3,4,5-тетрадеокси-8,9-O- изопропилиден-7-O-октадеканоил-D-глицеро-D-галакто-нон-2- энопиранозоат (E43)
1,10 г (2,98 ммоль) соединения (E3) растворили в 11 мл метиленхлорида при комнатной температуре и затем в систему при охлаждении льдом добавили 1,70 мл (5,03 ммоль) стеароилхлорида и 620 мг (5,07 ммоль) 4-диметиламинопиридина с последующим перемешиванием смеси при комнатной температуре в течение 30 минут. Затем в реакционного смесь при комнатной температуре влили 0,70 мл (5,05 ммоль) триэтиламина с последующим перемешиванием смеси в течение 15 ч. После подтверждения завершения реакции в систему был влит метанол и смесь перемешивали в течение 30 мин. Затем в реакционную смесь добавили этилацетат и водный насыщенный раствор NaCl для разделения смеси. Полученный органический слой высушили над сульфатом магния и отфильтровали с последующим концентрированием при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем (Кизельгель 60, н-гексан: этилацетат = 1:1) с получением 1,21 г (выход: 64%) целевого соединения (Е43) в виде бесцветной пены.
Величина Rf: 0,51 (н-гексан:этилацетат = 1:1);
1 H-NMR (270 MHz, CDCl3, TMS): δ (ppm) 5.95(1H, d, J=2.7 Hz), 5.88(1H, d, J= 7.9 Hz), 5.35(1H, dd, J= 6.0, 1.8 Hz), 4.80(1H, dd, J=9.1, 2.7 Hz), 4.71(1H, dd, J=10.5, 1.8 Hz), 4.39(1H, q, J=6.0 Hz), 4.14(1H, dd, J=8.8, 6.0 Hz), 3.945 (1H, dd, J=8.8, 6.0 Hz), 3.81(3H, s), 3.45(1H, ddd, J=10.5, 9.1, 7.9 Hz), 2.41(1H, t, J=7.5 Hz), 2.39(1H, t, J=7.5 Hz), 2.02(3H, s), 1.63(2H, квинтет, J=7.5 Hz), 1.37(3H, s), 1.35 (3H, s), 1.26(28H, s), 0.88(3H, t, J= 7.5 Hz).
ii) Метил 5-ацетамидо-4-амино-2,3,4,5-тетрадеокси-8,9-O- изопропилиден-7-O-октадеканоил-D-глицеро-D-галакто-нон-2- энопиранозоат (E44)
1,20 г (1,88 ммоль) соединения (E43) растворили в 12 мл этанола при комнатной температуре и затем в систему при комнатной температуре добавили 399 мг (0,33-кратный объем) катализатора Линдлара с последующим перемешиванием смеси при комнатной температуре под давлением водорода 1 атм в течение 3 ч. После подтверждения завершения реакции реакционную смесь фильтруют. Отфильтрованный продукт промывали этанолом, фильтрат и промывочный раствор объединяли с последующим концентрированием при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем (Кизельгель 60, 15 г, этилацетат: метанол = 10:1) с получением 924 мг (выход: 80%) соединения (E44) в виде бесцветной пены.
Величина Rf: 0,37 (этилацетат: метанол = 4:1);
1H-NMR (270 MHz, CD3OD, TMS): δ (ppm) 5.94(1H, d, J=2.4 Hz), 5.42(1H, dd, J= 4.7, 1.8 Hz), 4.39(1H, dt, J=7.1, 6.0 Hz), 4.18(1H, dd, J=9.5, 1.6 Hz), 4.14(1H, dd, J=8.7, 6.4 Hz), 3.93(1H, dd, J=8.7, 6.4 Hz), 3.87(1H, t, J= 9.5 Hz), 3.78(3H, s), 3.44(1H, dd, J=9.5, 2.4 Hz), 2.35(2H, q, J=7.3 Hz), 1.94(3H, s), 1.60(2H, квинтет, J= 7.3 Hz), 1.32(3H, s), 1.31(3H, s), 1.27(28H, s), 0.88(3H, t, J=7.3 Hz).
iii) Метил 5-ацетамидо-4-(N, N'-бис-трет-бутоксикарбонил) гуанидино-2,3,4,5-тетрадеокси-8,9-O-изопропилиден-7-O- октадеканоил-D-глицеро-D-галакто-нон-2-энопиранозоат (E45)
916 мг (1,50 ммоль) соединения (E44), 539 мг (1,95 ммоль) N,N'-бис-трет-бутоксикарбонилтиомочевины и 0,27 мл (1,95 ммоль) триэтиламина растворили в 12 мл диметилформамида при комнатной температуре. Затем в систему при охлаждении льдом добавили 529 мг (1,95 ммоль) хлорида ртути и смесь перемешивали при комнатной температуре в течение 4 ч. К реакционной смеси добавили этилацетат, чтобы разбавить ее, с последующей фильтрацией через Целит. Отфильтрованный продукт промыли этилацетатом. Полученный фильтрат и промывной раствор объединили и к смеси, чтобы ее разделить, добавили этил-ацетат и водный насыщенный раствор NaC1. Органический слой высушили над сульфатом магния и отфильтровали с последующим концентрированием при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем (Кизельгель 60, 15 г, гексан: этилацетат = 2:1) с получением 750 мг (выход: 59%) указанного соединения (E45) в виде бесцветной пены.
Величина Rf: 0,29 (гексан: этилацетат = 2:1);
1H-NMR (270 MHz, CDCl3, TMS): δ (ppm) 11.4(1H, s), 8.46 (1H, d, J=8.7 Hz), 6.06(1H, d, J=8.7 Hz), 5.88(1H, d, J=2.4 Hz), 5.37(1H, dd, J=6.4, 1.5 Hz), 5.15(1H, dt, J=8.7, 2.4 Hz), 4.38(1H, q, J=6.4 Hz), 4.28(1H, dd, J=8.7, 1.5 Hz), 4.23(1H, t, J=8.7 Hz), 4.10(1H, dd, J=9.6, 6.4 Hz), 3.95(1H, dd, J= 9.6, 6.4 Hz), 3.80(3H, s), 2.453(1H, dt, J=16.0, 7.5 Hz), 2.33(1H, dt, J= 16.0, 7.5 Hz), 1.87(3H, s), 1.61(2H, квинтет, J=7.5 Hz), 1.49(9H, s), 1.48(9H, s), 1.38(3H, s), 1.35(3H, s), 1.25(28H, bs), 0.88(3H, t, J=7.5 Hz).
iv) 5-Ацетамидо-4-(N, N'-бис-трет- бутоксикарбонил)гуанидино-2,3,4,5-тетрадеокси-8,9-O- изопропилиден-7-O-октадеканоил-D-глицеро-D-галакто-нон-2- энопиранозойная кислота (E46)
741 мг (0,87 ммоль) соединения (E45) растворили в смеси 15 мл метанола и 1,5 мл воды, и в систему при комнатной температуре добавили 38 мг (0,91 ммоль) моногидратного гидроксида лития с последующим перемешиванием смеси при комнатной температуре в течение 6 ч. После подтверждения завершения реакции к системе добавили Дауэкс-50W для нейтрализации смеси. Реакционную смесь отфильтровали и отфильтрованный продукт промыли метанолом. Фильтрат и промывочный раствор объединили и сконцентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем (Кизельгель 60, 60 г, этилацетат:метанол = 10:1) с получением 430 мг (выход: 59%) указанного целевого соединения (E16) в виде бесцветной пены.
Величина Rf: 0,40 (этилацетат:метанол = 5:1);
1H-NMR (270 MHz, CDCl3, TMS): δ (ppm) 11.4(1H, s), 8.48 (1H, d, J=8.0 Hz), 6.31(1H, слаб. s), 5.90(1H, bs), 5.30(1H, bs), 5.10(1H, bs), 4.60-3.30(7H, m), 2.48(1H, dt, J=13.5, 6.5 Hz), 2.32(1H, dt, J=13.5, 6.5 Hz), 1.88(3H, s), 1.60(2H, квинтет, J= 6.5 Hz), 1.48(24H, s), 1.39(3H, s), 1.37(3H, s), 1.25(28H, bs), 0.88(3H, t, J=6.5 Hz).
v) Соль 5-ацетамидо-2,3,4,5-тетрадеокси-4-гуанидино-9- O-октадеканоил-D-глицеро-D-галакто-нон-2-энопиранозойной кислоты и трифторуксусной кислоты (E47)
422 мг (0,50 ммоль) соединения (E46) растворили в 3 мл метиленхлорида при комнатной температуре и затем к системе при комнатной температуре добавили 1 мл трифторуксусной кислоты с последующим перемешиванием смеси при комнатной температуре в течение 22 ч. После подтверждения завершения реакции реакционную смесь концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем (Кизельгель 60, 5 г, этилацетат:2-пропанол:вода = 5:2:1) с получением 300 мг (выход: 84%) указанного соединения (E47) в виде бесцветной пены.
Величина Rf: 0,44 (2-пропанол:вода = 5:1);
1H-NMR (270 MHz, CD3OD): δ (ppm) 5.55(1H, bs), 4.40-4.10 (7H, m), 3.65(1H, d, J= 9.0 Hz), 2.36(2H, t, J=7.0 Hz), 2.00 (3H, s), 1.70-1.50(2H, m), 1.30(28H, bs), 0.90(3H, t, J=7.0 Hz);
FAB-MS (положит.): 599 (M+H)+;
[α]
ПРИМЕР 10. Соль цетил 5-ацетамидо-2,3,4,5-тетрадеокси-4- гуанидино-D-глицеро-D-галакто-нон-2-энопиранозойной кислоты и трифторуксусной кислоты (E48) (образцовое соединение N 89)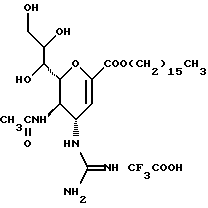
Процедуры выполняются аналогично тем, что описаны в Примере 1 с использованием цетилового спирта вместо миристилового спирта для получения вышеуказанного соединения.
1H-NMR (270 MHz, CD3OD, TMS): δ (ppm) 5.83(1H, d, J=2.7 Hz), 4.44(1H, dd, J= 9.0, 2.7 Hz), 4.38(1H, dd, J=9.0, <1 Hz), 4.18(2H, t, J=6.2 Hz), 4.17(1H, t, J= 9.0 Hz), 3.90-3.74 (2H, m), 3.68(1H, dd, J=12.0, 4.5 Hz), 3.65(1H, d, J=9.0 Hz), 1.99(3H, s), 1.67(2H, квинтет, J=6.2 Hz), 1.26(28H, bs), 0.87(3H, t, J=6.2 Hz).
ПРИМЕР 11. Соль стеарил 5-ацетамидо-2,3,4,5-тетрадеокси-4- гуанидино-D-глицеро-D-галакто-нон-2-энопиранозойной кислоты и трифторуксусной кислоты (E49) (образцовое соединение N 91)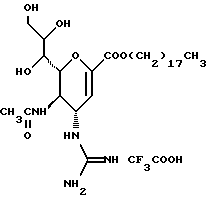
Процедуры выполняются аналогично тем, что описаны в Примере 1 с использованием стеарилового спирта вместо миристилового спирта для получения вышеуказанного соединения.
1H-NMR (270 MHz, CD3OD): δ (ppm) 5.83(1H, d, J=2.7 Hz), 4.44(1H, dd, J= 9.0, 2.7 Hz), 4.38(1H, dd, J=9.0, <1 Hz), 4.18(2H, t, J=6.2 Hz), 4.17(1H, t, J= 9.0 Hz), 3.90-3.74 (2H, m), 3.68(1H, dd, J=12.0, 4.5 Hz), 3.65(1H, d, J= 9.0 Hz), 1.99(3H, s), 1.67(2H, квинтет, J=6.2 Hz), 1.26(32H, bs), 0.87(3H, t, J=6.2 Hz).
Пример 1 получения препарата.
Водный раствор был приготовлен таким образом, чтобы соединение Примера 4 составляло 10% (вес/вес), бензалконий-хлорид 0,04% (вес/вес), фенилэтиловый спирт 0,40% (вес/вес) и очищенная вода 89,56% (вес/вес).
Пример 2 получения препарата.
Водный раствор с сорастворителем был приготовлен таким образом, чтобы соединение Примера 4 составляло 10% (вес/вес), бензалконийхлорид 0,04% (вес/вес), полиэтиленгликоль 400 10% (вес/вес), пропиленгликоль 30% (вес/вес) и очищенная вода 39,96% (вес/вес).
Пример 3 получения препарата.
Сухой порошок готовили таким образом, чтобы соединение Примера 4 было 40% (вес/вес) и лактозы было 60% (вес/вес)
Пример 4 получения препарата.
Аэрозольное средство было приготовлено таким образом, что соединение Примера 4 составило в нем 10% (вес/вес), лецитин 0,5% (вес/вес), Фреон 11 34,5% и Фреон 12 55%.
Пример испытания 1. Активность ингибирования сиалидазы вируса гриппа
Трахеотомическую трубку вводили в трахею мышам (BALB/C, самки, возраст 5-6 недель, масса тела 20 г) при анестезии с последующей инъекцией 0,5 мл забуференного фосфатом физиологического раствора. Осуществляли промывание легких, собирая жидкость аспирацией, повторяя эту процедуру трижды. 1 мкм испытуемого соединения (проба), 10 мкг легочной промывной жидкости в зависимости от количества белка и забуференный фосфатом физраствор перемешивали и инкубировали при 37oC в течение 1-3 дней при конечном объеме 100 мкл. После отбора пробы 10 мкл этой реакционной смеси проводили реакцию с
сиалидазой в 32,5 мМ 2-(N-морфолино)этансульфонатном буфере (pH 6,5), содержащем 40 мМ хлорида кальция, используя вирус гриппа A/PR/8/34 (эквивалентно 5•105 бляшкообразующих единиц) в качестве сиалидазного фермента и 0,1 мМ 4-метилум- беллиферил-N-ацетил -α- D-нейрамината аммония в качестве субстрата. Интенсивность флуоресценции 4-метилумбеллиферона, образующегося в реакционной смеси, измеряли при возбуждении длины волны 360 нм и длине волны для определений в 460 нм. C другой стороны, концентрация в сравнении с кривой ингибирования исследовалась проведением аналогичных реакций с использованием проб различных концентраций Соединения A (GG-167). Количество вещества, обладающего способностью ингибировать сиалидазу, образовавшегося в легочных промывочных жидкостях от испытуемого соединения, определяли количественно, так же как и количество для Соединения А с использованием вышеуказанной кривой ингибирования.
Несмотря на то, что соединение настоящего изобретения не показывало способности непосредственно ингибировать сиалидазу вируса гриппа при обработке биологическими фракциями, содержащими гидролазу (например, промывки легкого мыши), соединения настоящего изобретения продемонстрировали активность подавления сиалидазы вируса гриппа, подобную активности Соединения А.
Пример испытания 2. Эксперимент 1 по лечению заражения у мышей
Готовили раствор, содержащий 500 pfu (бляшкообразующих единиц) адаптированного к мышам штамма A/PR/8/34 вируса гриппа в 50 мкл фосфатного буфера, содержащего 0,42% БСА, который затем использовали для заражения мышей (BALB/C, самки, возраст 5-6 недель, 20 г) закапыванием в нос для заражения. Соединения настоящего изобретения готовили в дозе 0,6 мкмоль/кг/50 мкл суспендированием в физрастворе и вводили животным закапыванием в нос при 3 параллельных за 4 ч до заражения, 4 ч спустя и через 17 ч после вирусного заражения. Опыт проводили на группах 7 или 8 животных и результаты показали число выживших мышей по сравнению с общим количеством испытуемых мышей на 6, 8 и 10 день после заражения. Соединение A (GG-167) использовали для сравнения.
Хотя все животные из группы, получившие дозу Соединения А, погибли на 10-ый день после заражения, 3 или 1 животное в группах, получивших соединения Примера 1 или Примера 2, остались в живых. Эти результаты показывают, что соединения Примеров 1 или 2 данного изобретения обладают большим терапевтическим эффектом против заражения гриппом, по сравнению с аналогичным эффектом Соединения А.
Пример испытания 3. Эксперимент 2 по лечению заражения у мышей
Готовили раствор, содержащий 500 pfu (бляшкообразующих единиц) адаптированного к мышам штамма A/PR/8/34 вируса гриппа в 50 мкл фосфатного буфера, содержащего 0,42% БСА, который затем использовали для заражения мышей (BALB/C, самки, возраст 5-6 недель, 20 г) закапыванием в нос для заражения. Соединения настоящего изобретения готовили в дозе 0,9 ммоль/кг/50 мкл суспендированием в физрастворе и вводили животным закапыванием в нос при 3 параллельных за 4 ч до заражения, 4 ч спустя и через 17 ч спустя вирусного заражения. Опыт проводили на группах 4 - 12 животных и результаты показали число выживших мышей, по сравнению с общим количеством испытуемых мышей на 8 и 10 день после заражения. Соединение A (GG- 167) использовали для сравнения.
Хотя все животные из группы, получившие дозу Соединения А, погибли на 10-ый день после заражения, от 2 до 5 животных в группах, получивших соединения Примеров 1, 10 или 11 остались в живых. Эти результаты показывают, что соединения Примеров 1, 10 или 11 данного изобретения обладают терапевтическим эффектом против заражения гриппом, по сравнению с аналогичным эффектом Соединения А.
Пример испытания 4. Эксперимент 3 по лечению заражения у мышей
Готовили раствор, содержащий 500 pfu (бляшкообразующих единиц) адаптированного к мышам штамма A/PR/8/34 вируса гриппа в 50 мкл фосфатного буфера, содержащего 0,42% БСА, который затем использовали для заражения мышей (BALB/C, самки, возраст 5-6 недель, 20 г) закапыванием в нос для заражения. Соединения настоящего изобретения готовили в дозе 0,3 ммоль/кг/50 мкл суспендированием в физрастворе и вводили животным закапыванием в нос при 3 параллельных за 4 ч до заражения, 4 ч спустя и через 17 ч спустя вирусного заражения. Опыт проводили на группах 10 или 11 животных и результаты показали число выживших мышей по сравнению с общим количеством испытуемых мышей на 6 и 8 день после заражения. Соединение A (GG-167) использовали для сравнения.
Хотя из группы животных, получивших Соединение 4, только одно осталось живым на 8-ой день после заражения, в группах, получавших какое-либо соединение, соответствующее Примерам 3-9, остались живыми от 2 до 10 животных. Эти результаты показывают, что соединения Примеров 3-9 настоящего изобретения обладают терапевтическим эффектом против заражения гриппом, превосходящим эффект Соединения А.
Соединение нейраминовой кислоты подвергается гидролизу гидролазой, присутствующей в живом организме, и проявляет превосходную способность ингибировать репликацию вируса и способность подавлять действие сиалидазы. Кроме того, при введении соединения нейраминовой кислоты (1) настоящего изобретения мышам, инфицированным вирусом гриппа, соединение показывает лучший противоинфекционный терапевтический эффект, по сравнению с таковым Соединения A (GG-167), описанным в WO 91/16320 (японская заявка PCT (Kokai) N Hei 5- 507068. Таким образом, соединение нейраминовой кислоты (1) настоящего изобретения является полезным в качестве лекарственного средства или средства для профилактики (предпочтительно, терапевтическое средство) вирусных инфекций (предпочтительно, вирусных инфекций гриппа).
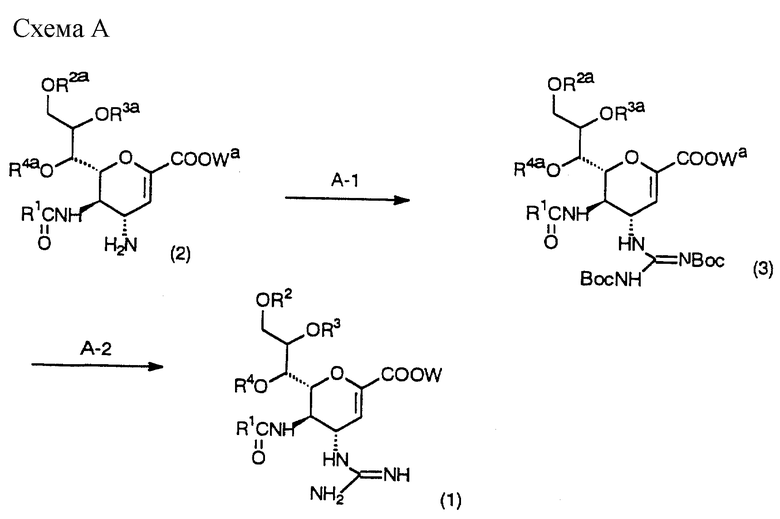
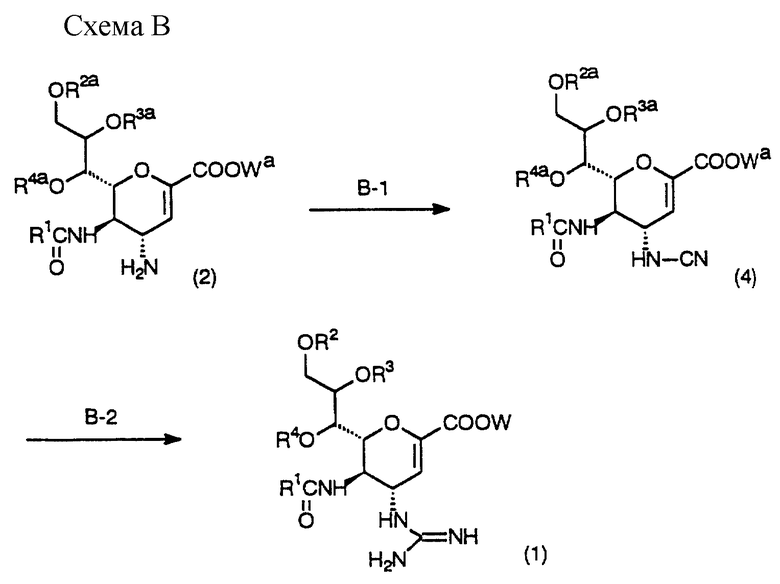

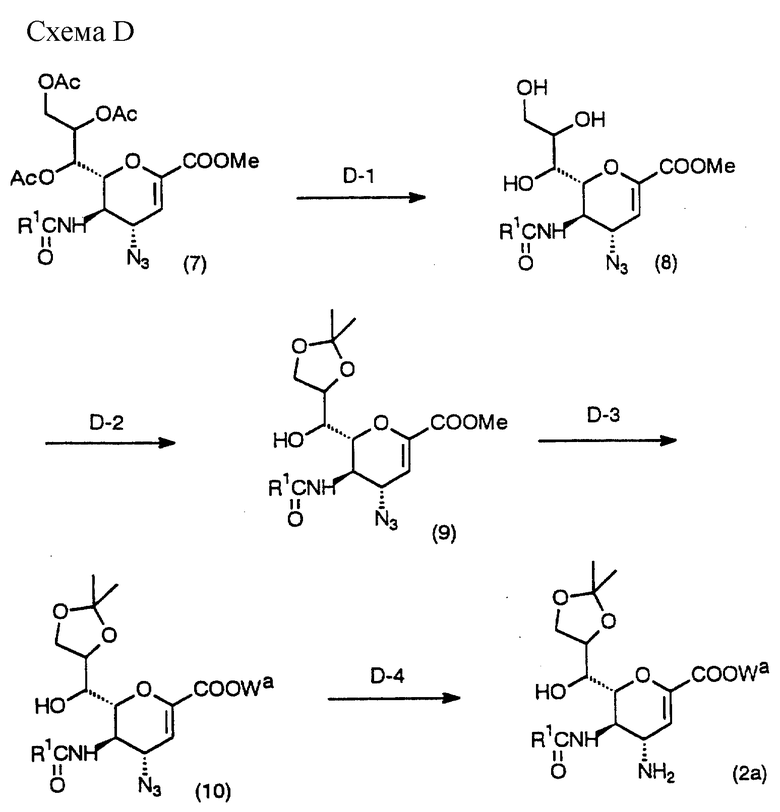
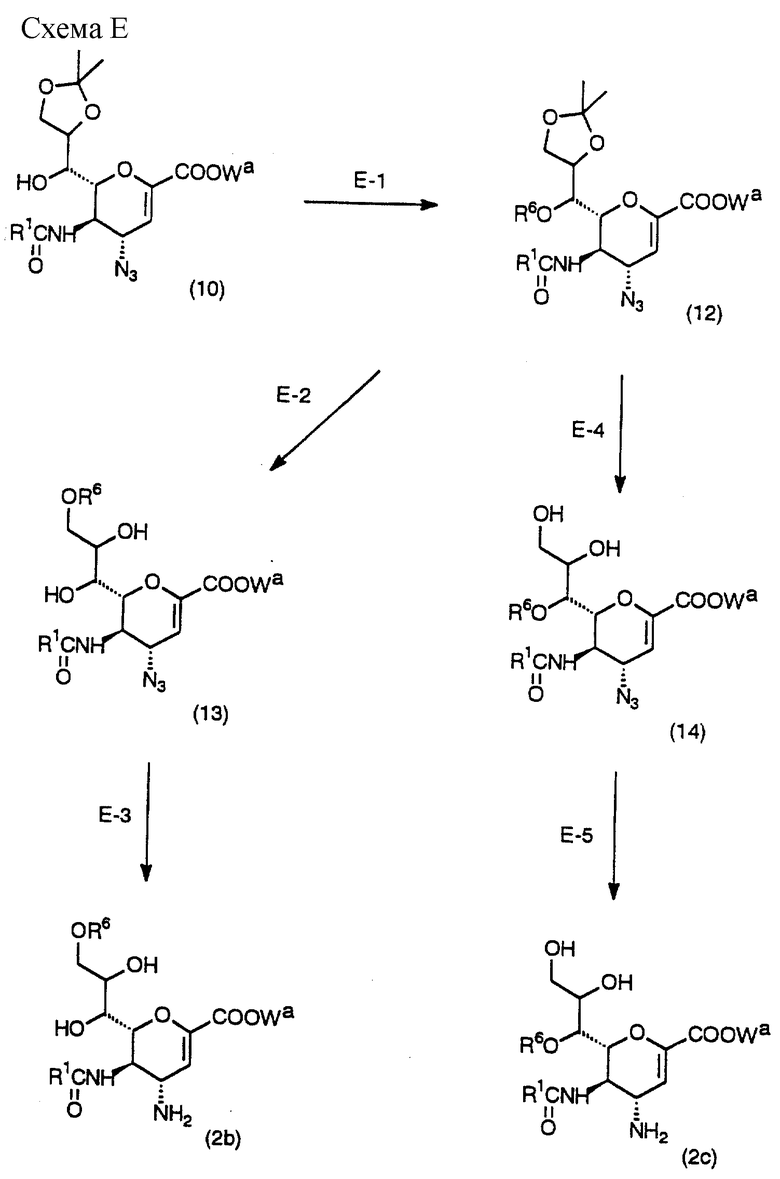

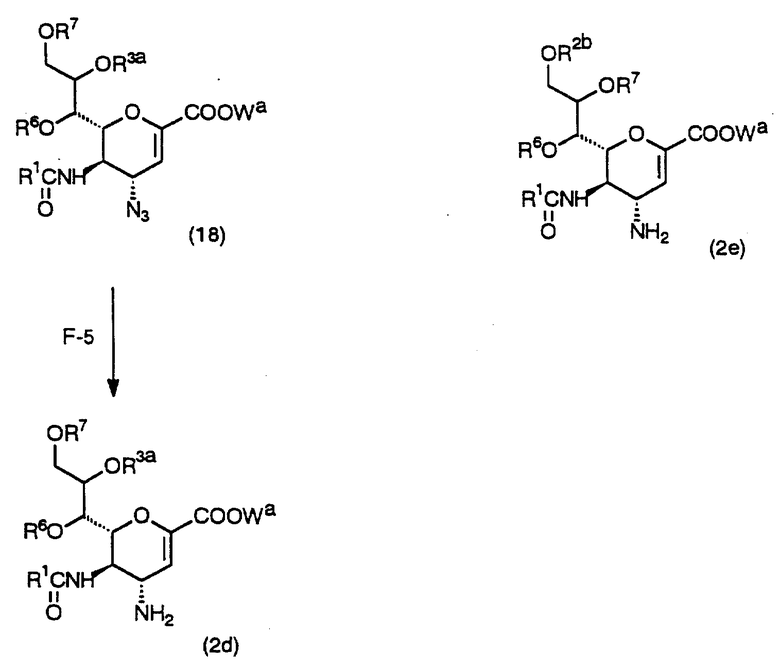
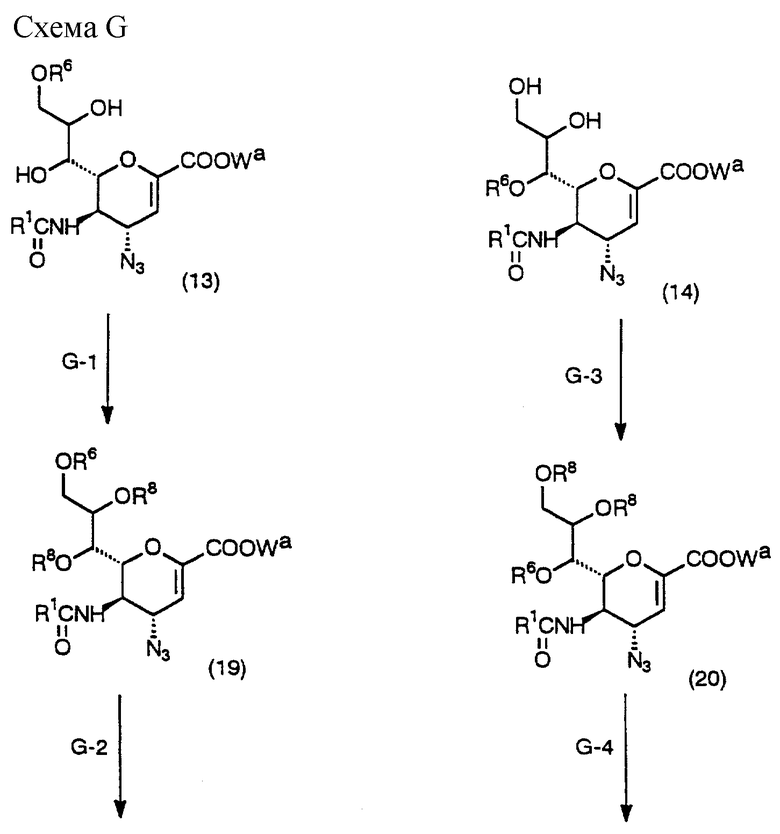
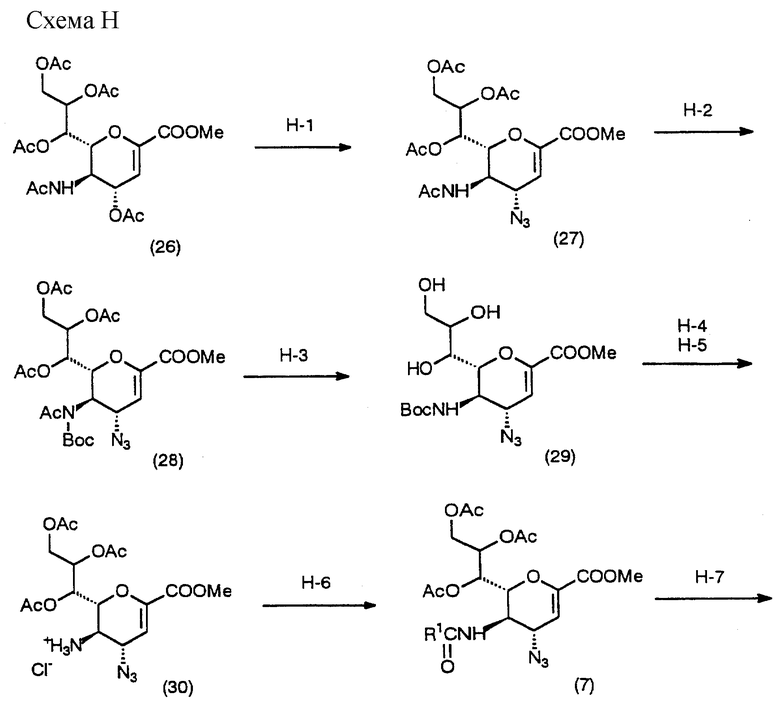
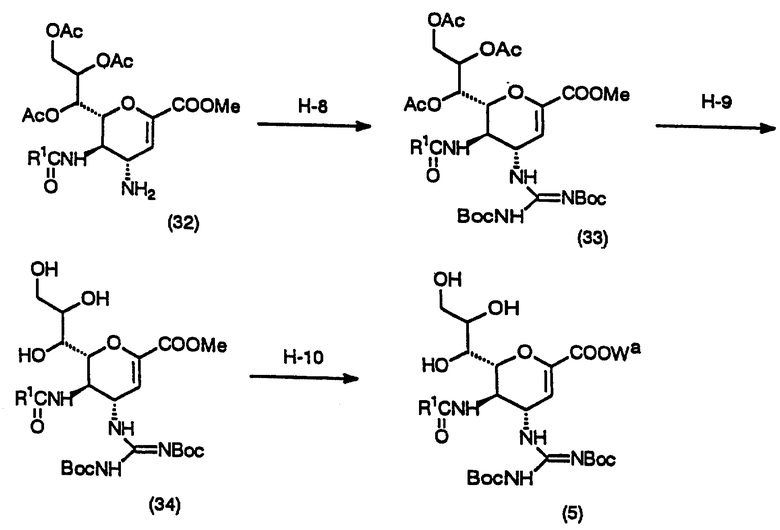
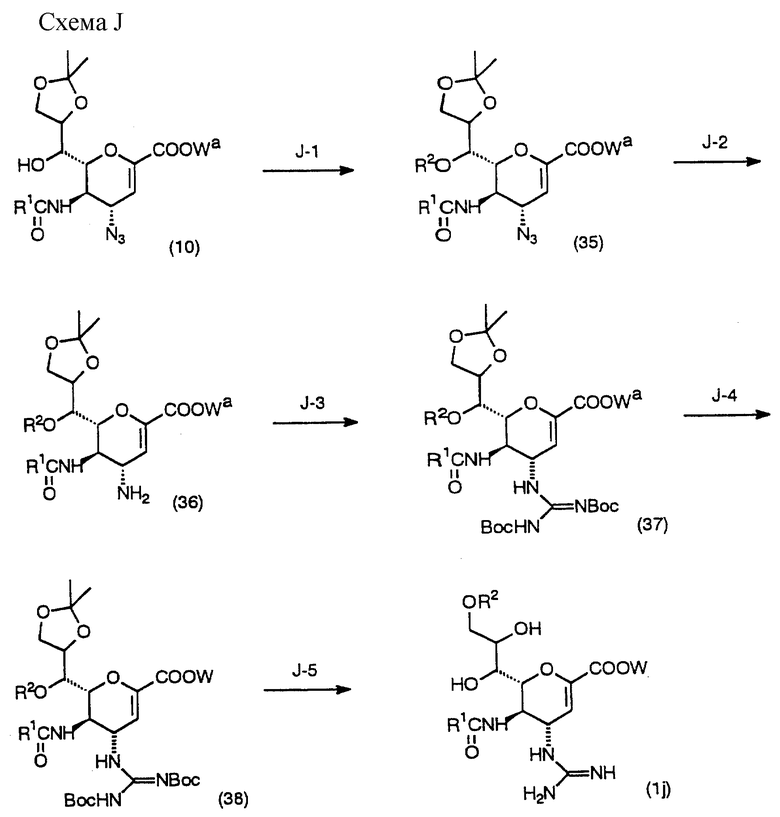
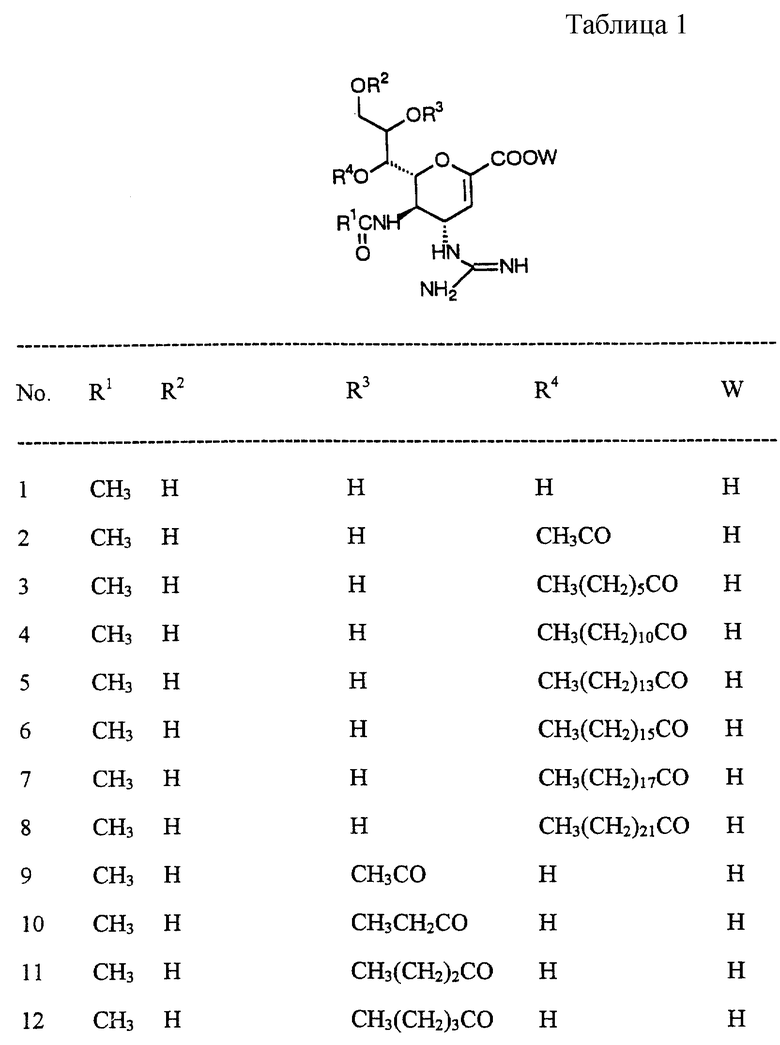
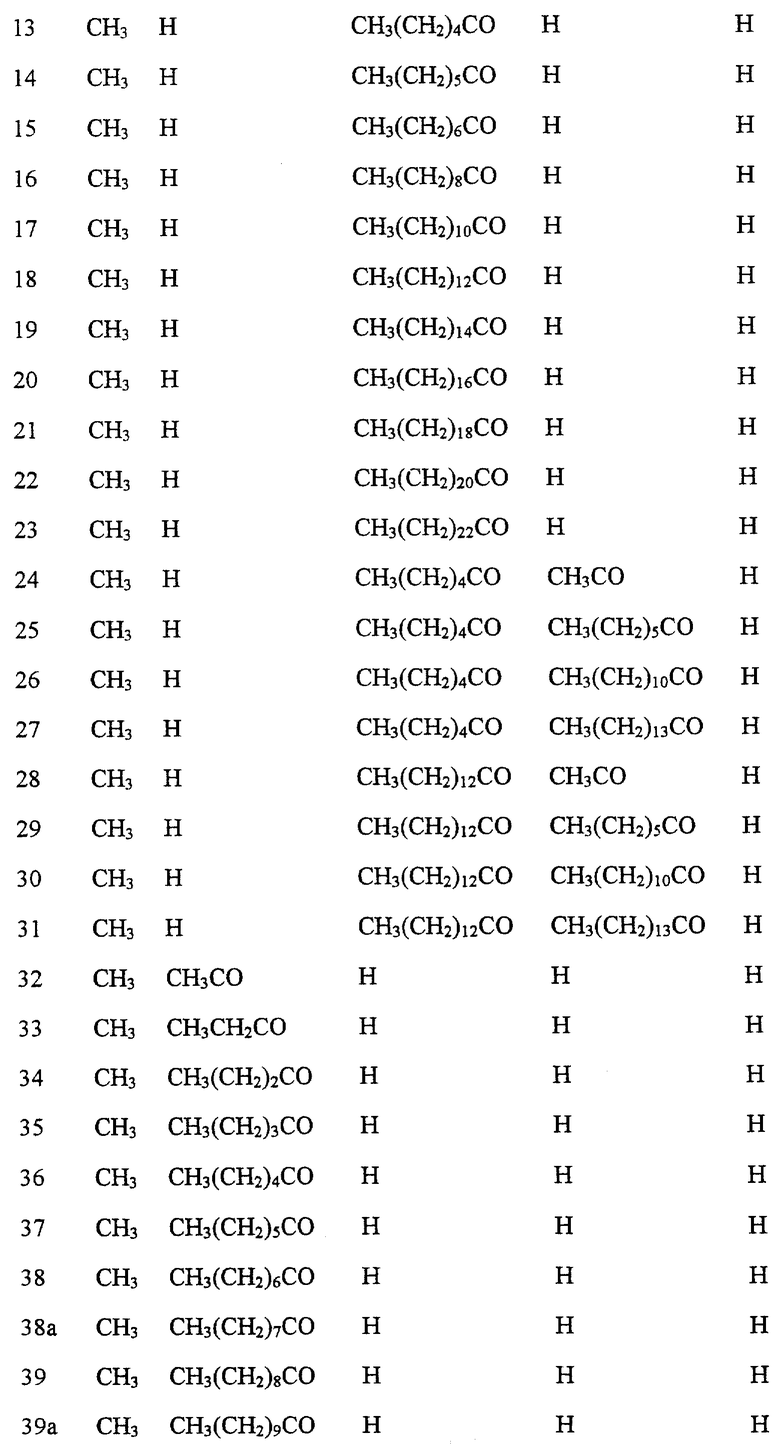
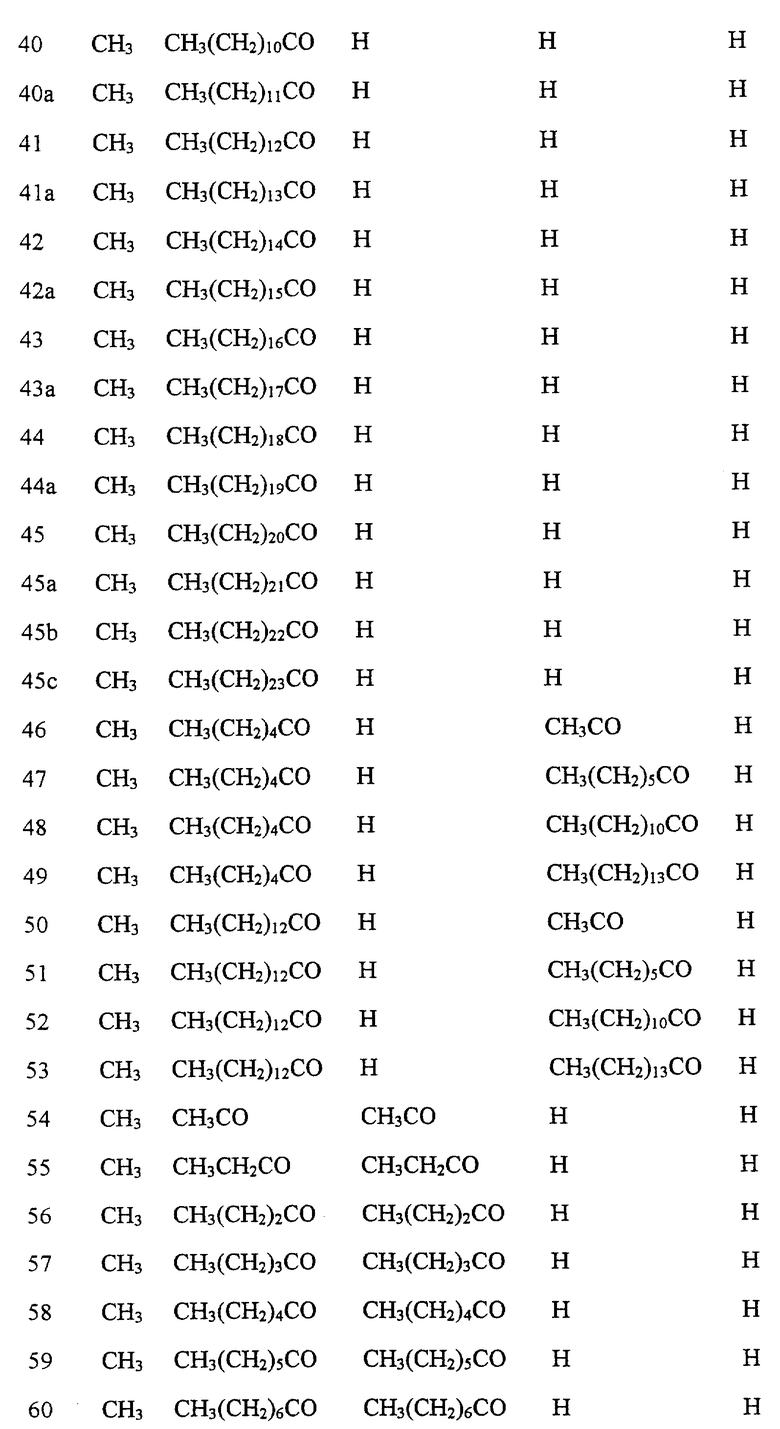
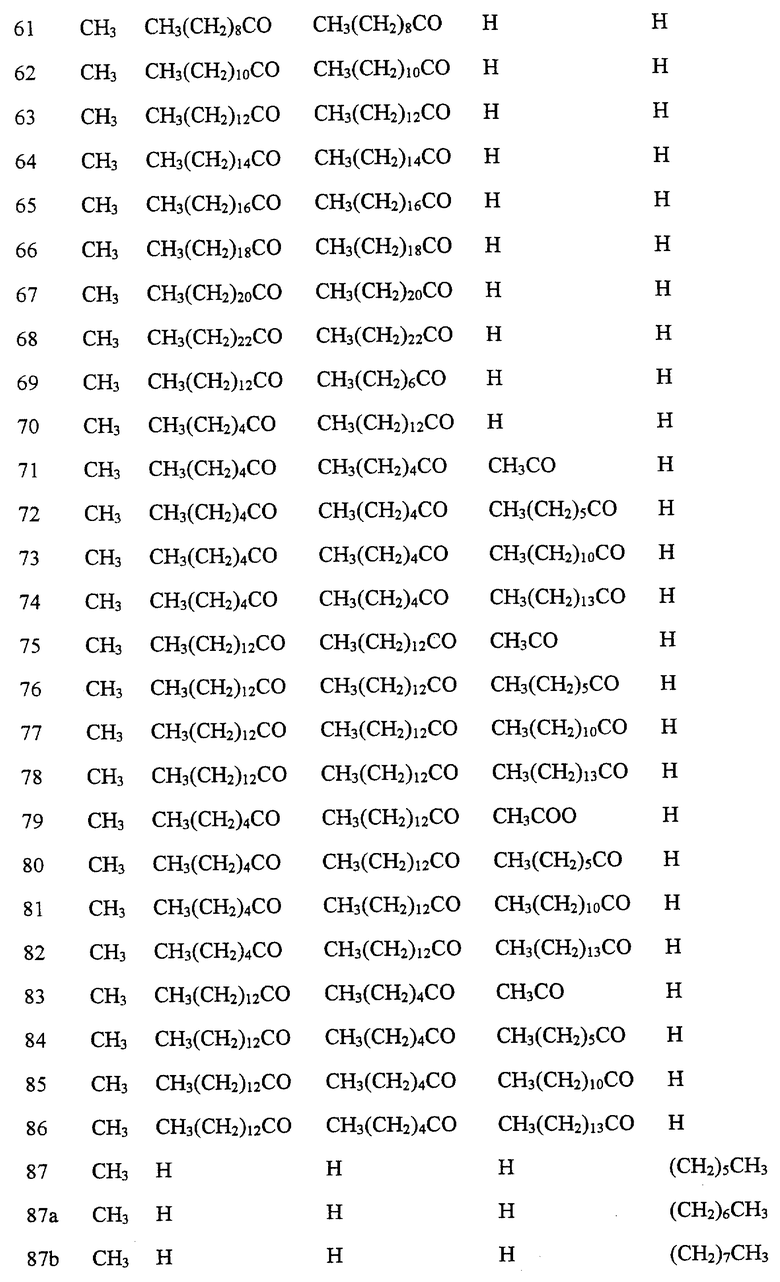
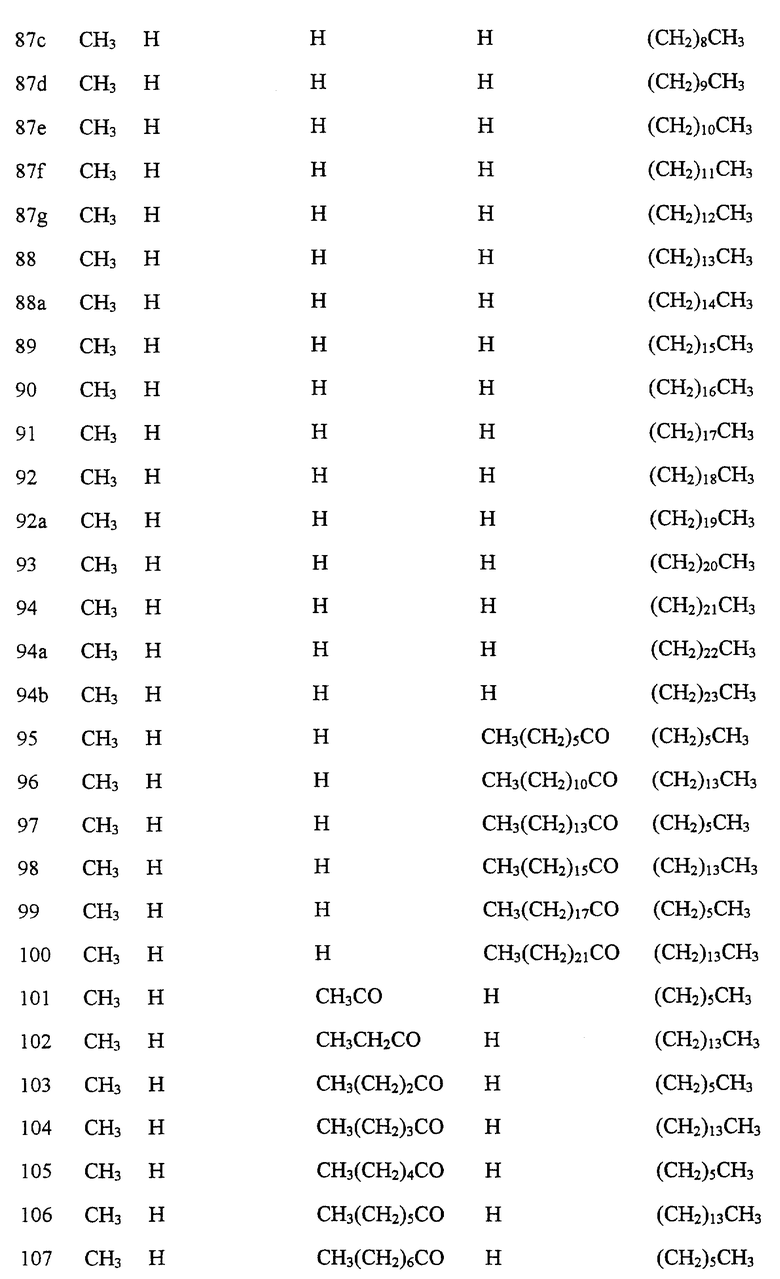
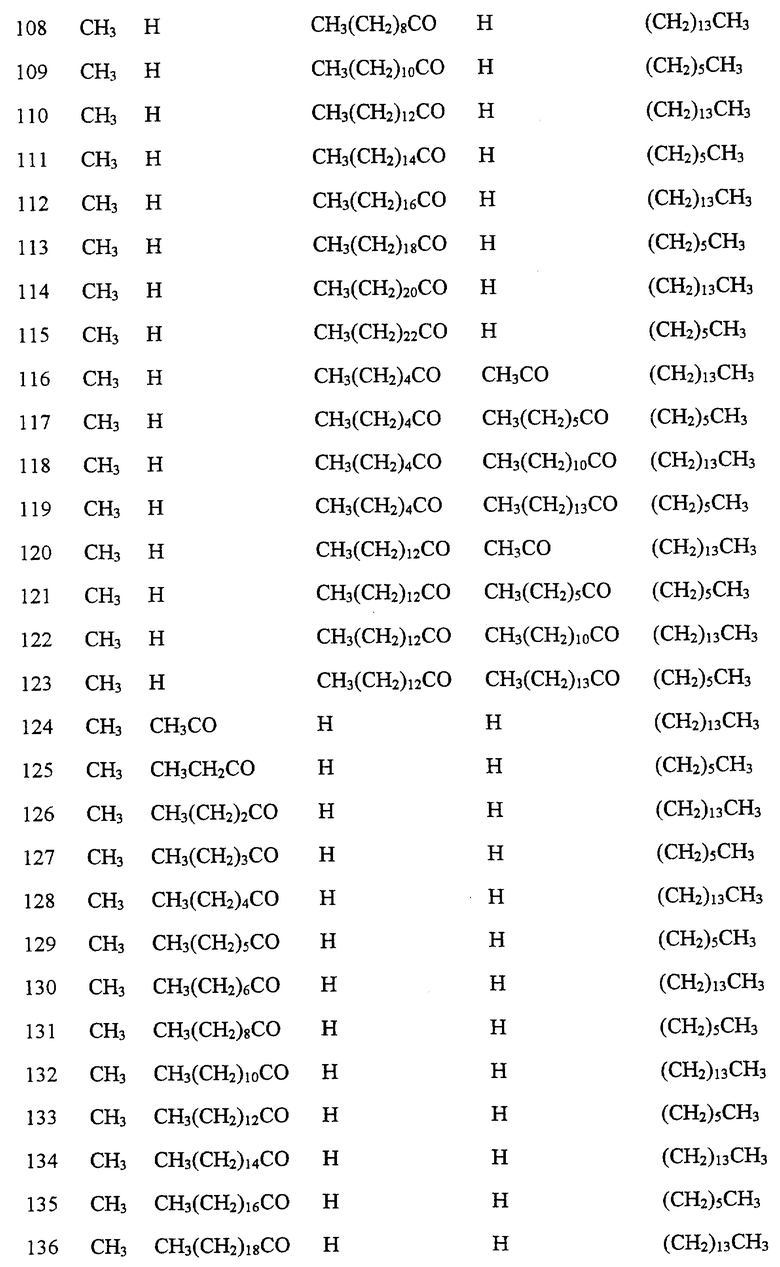
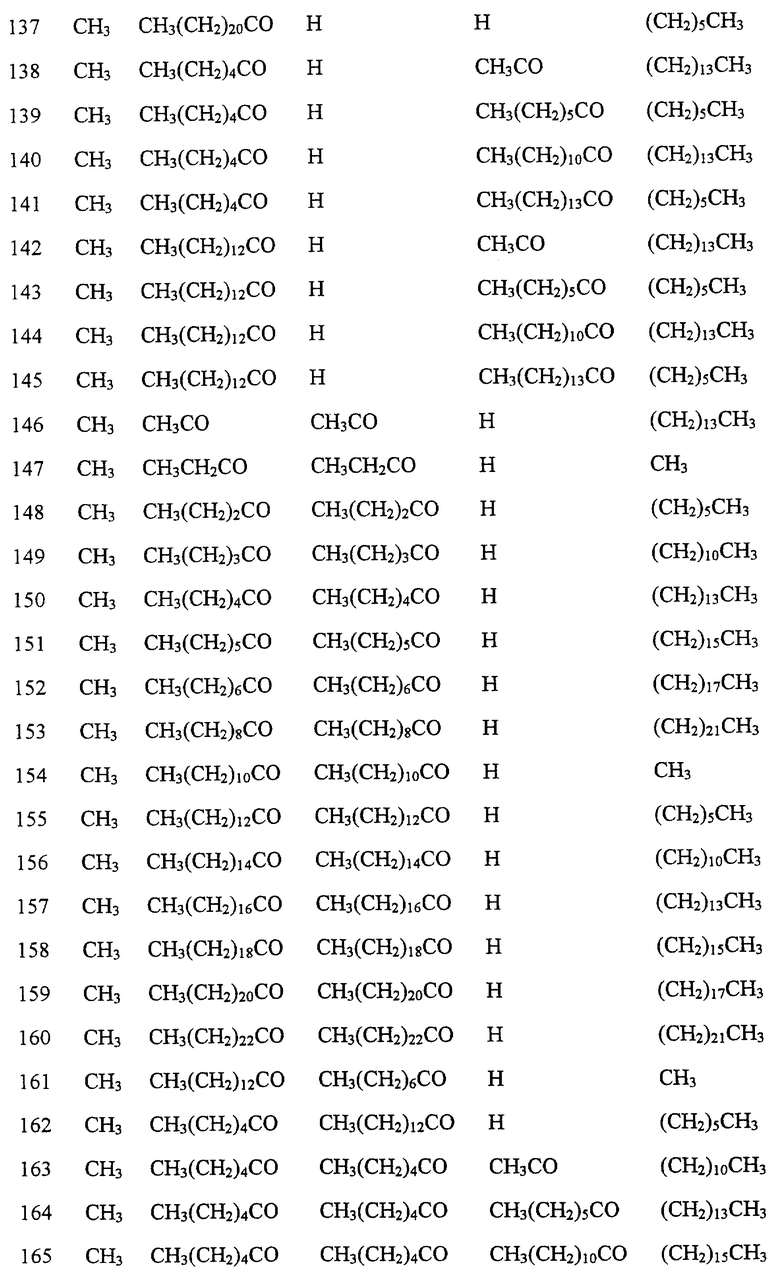
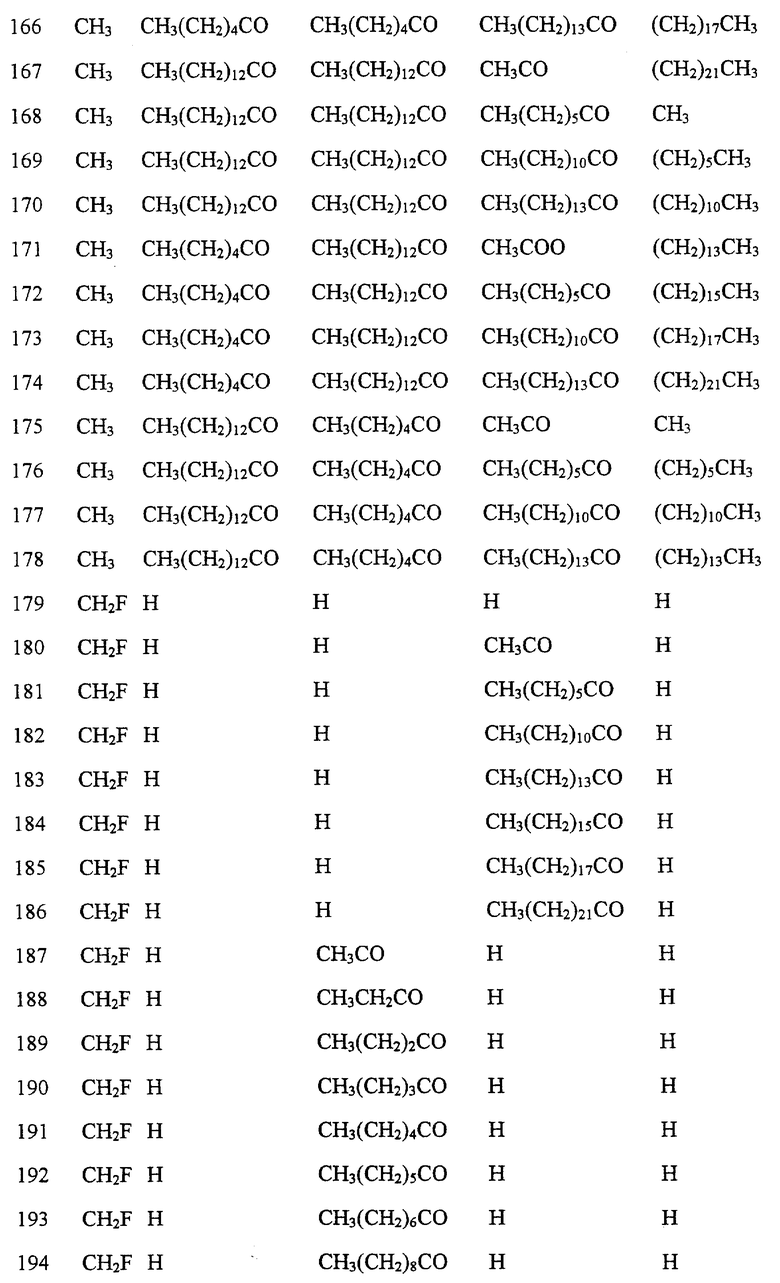
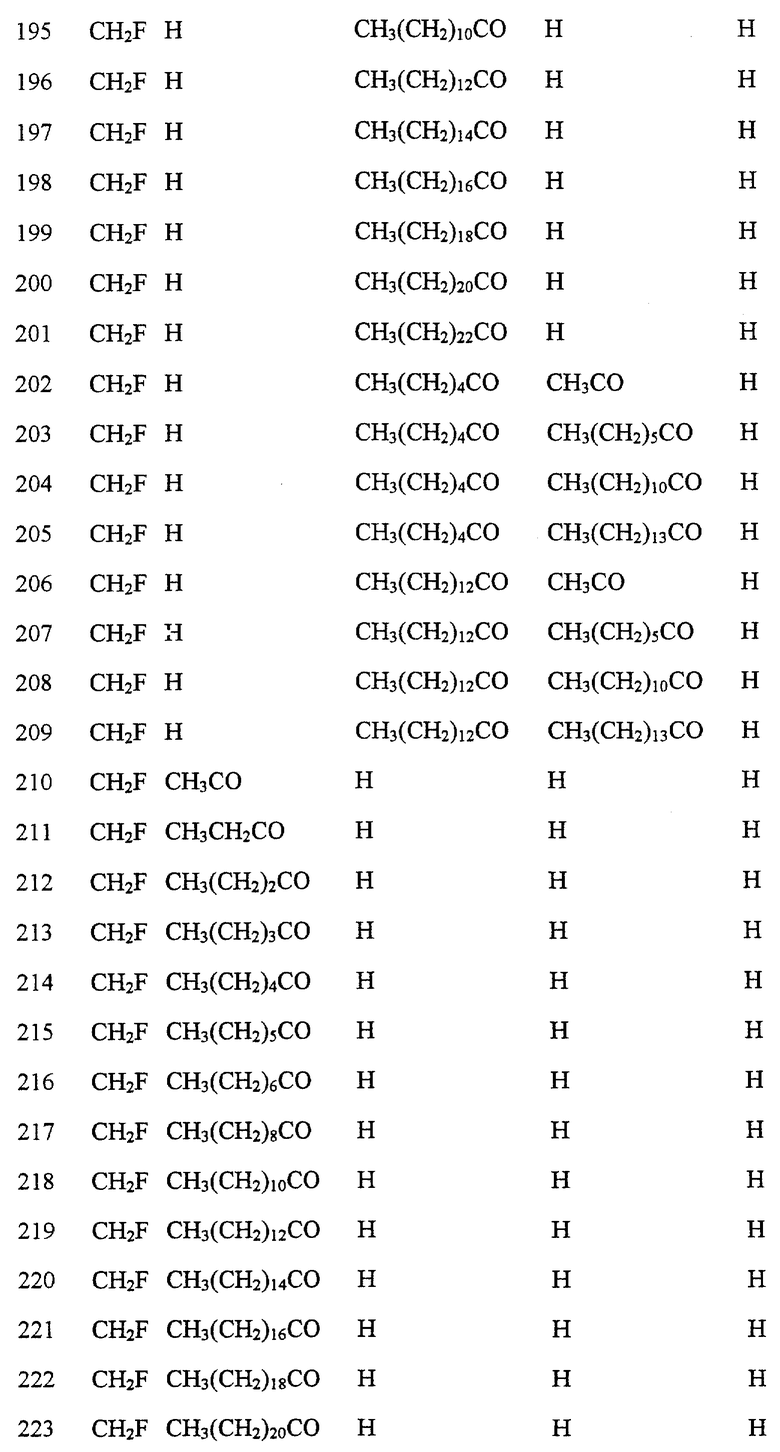
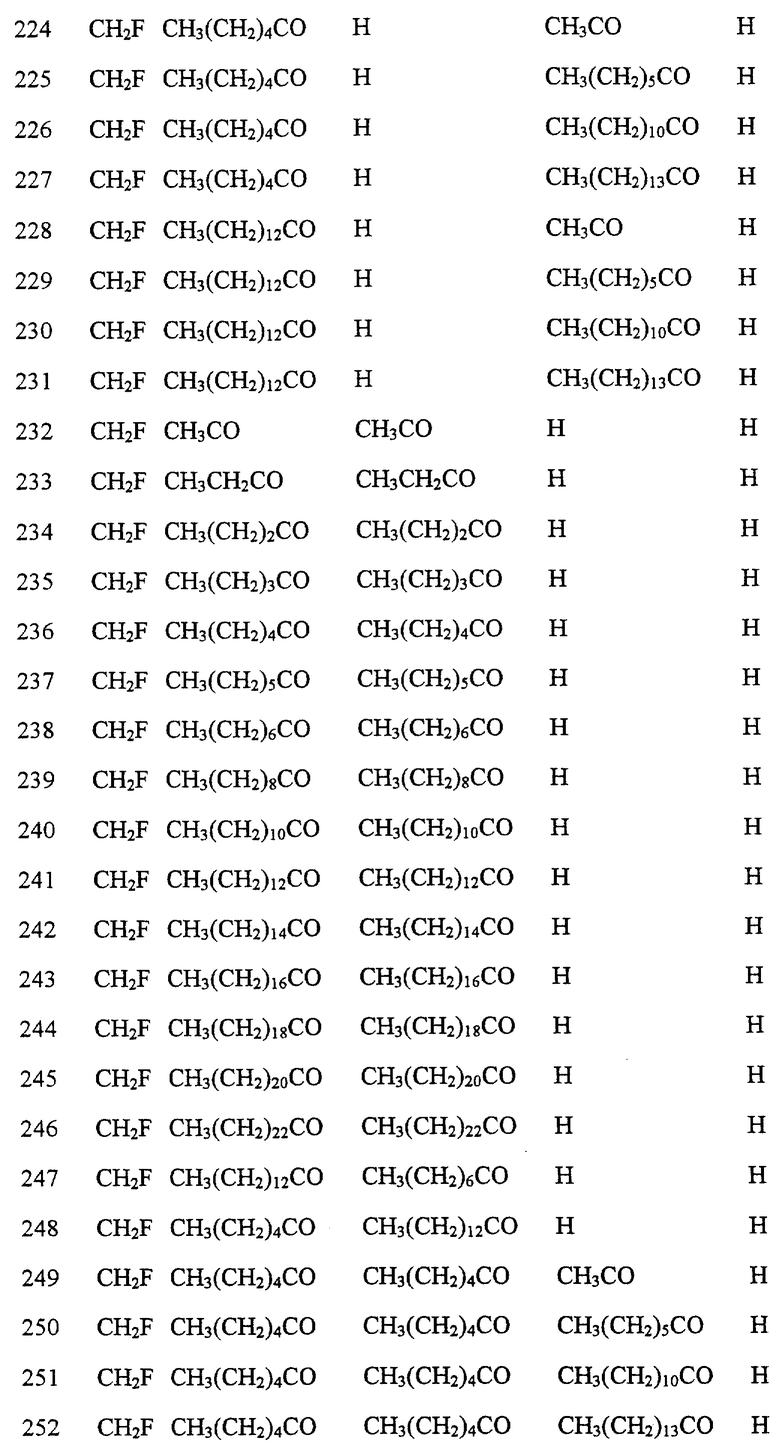
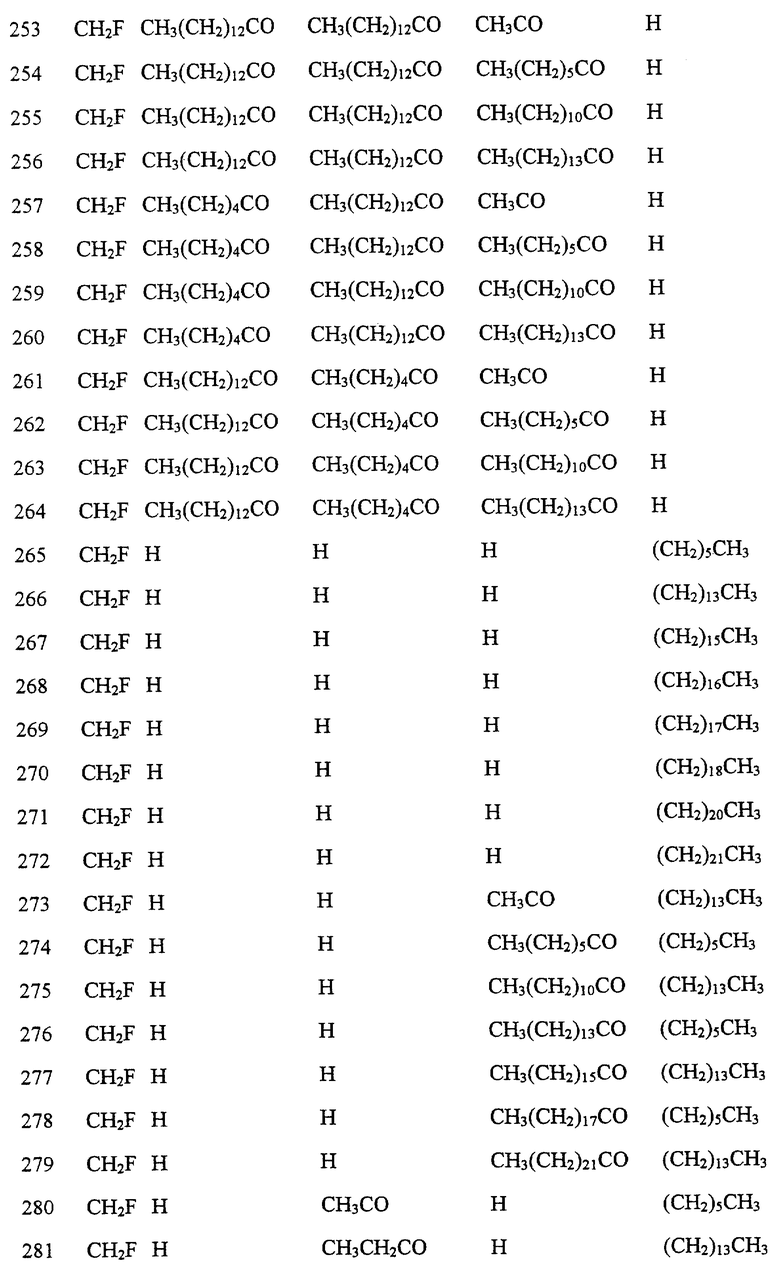
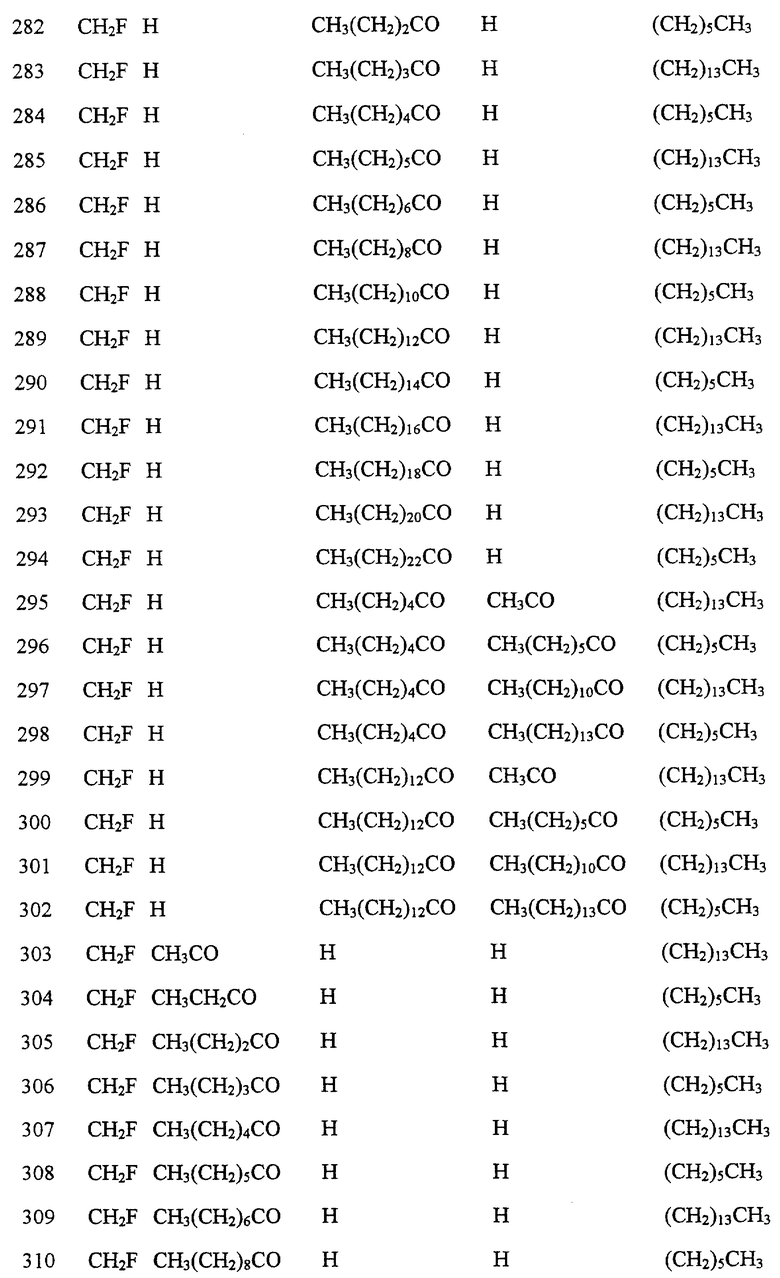
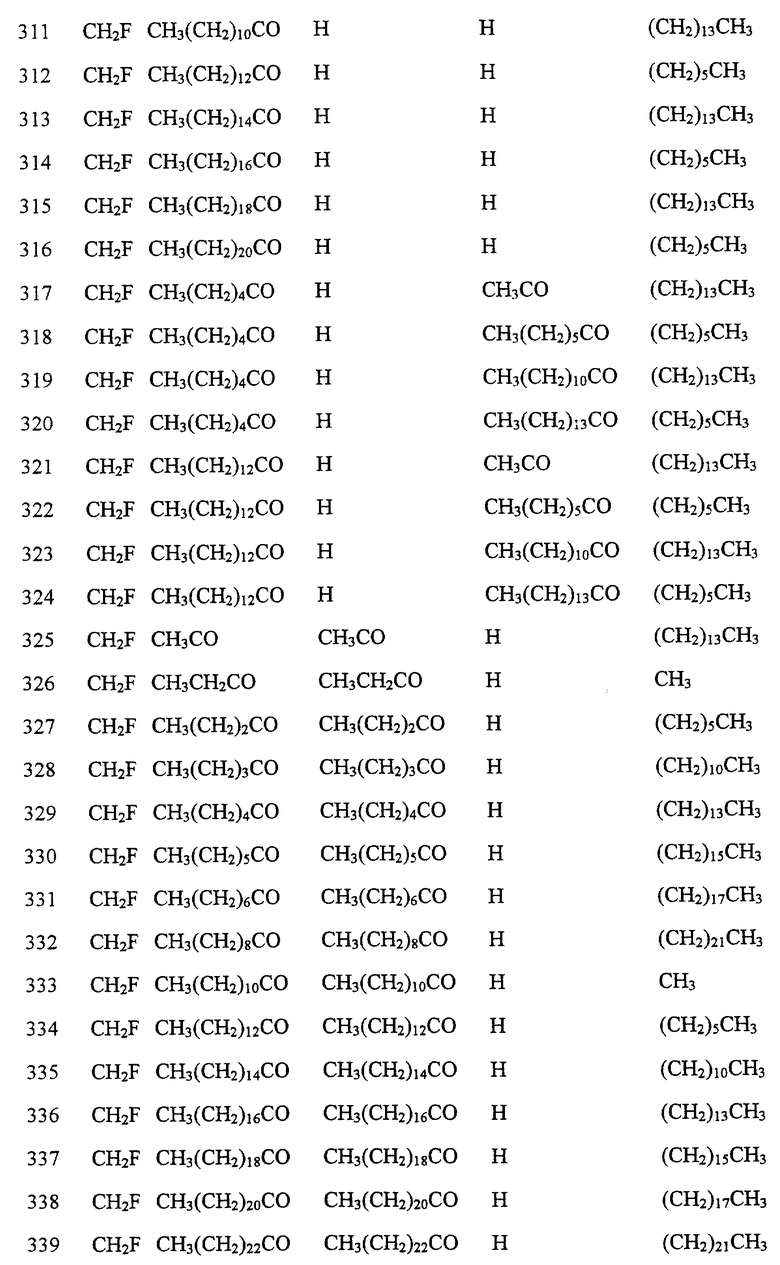
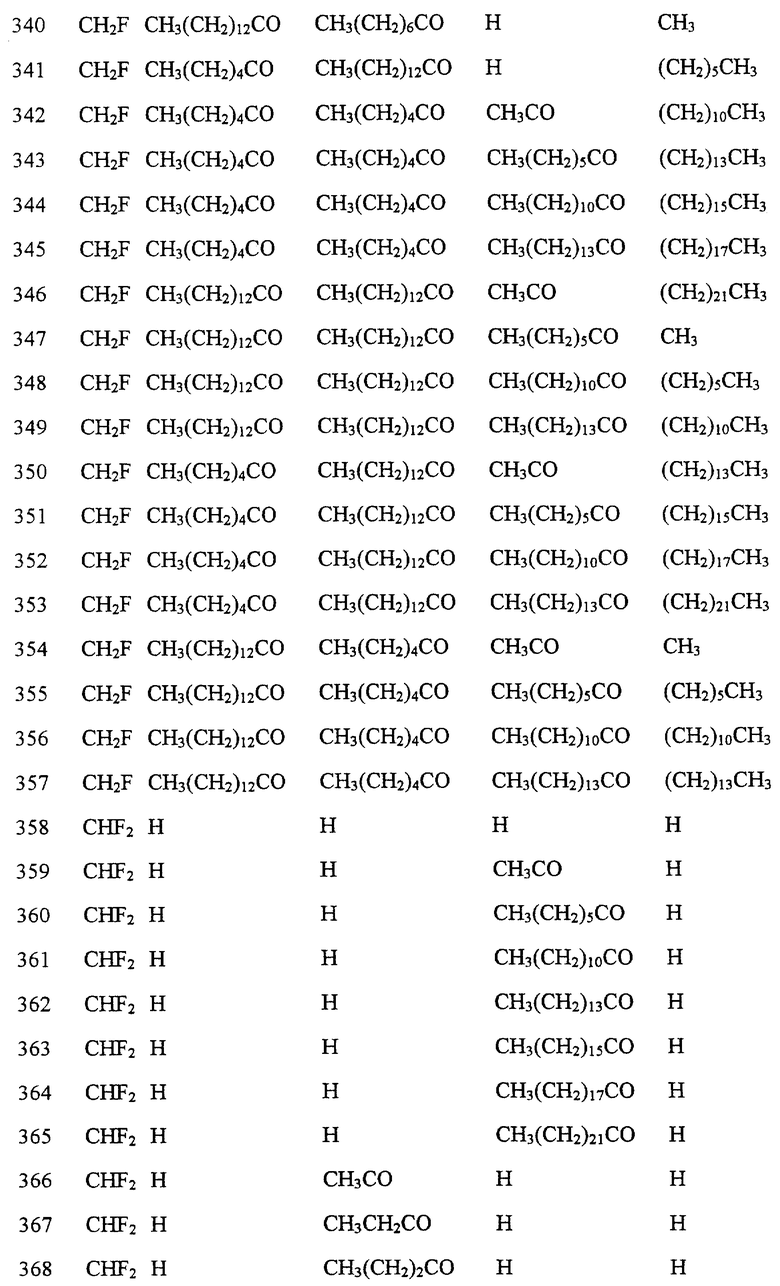
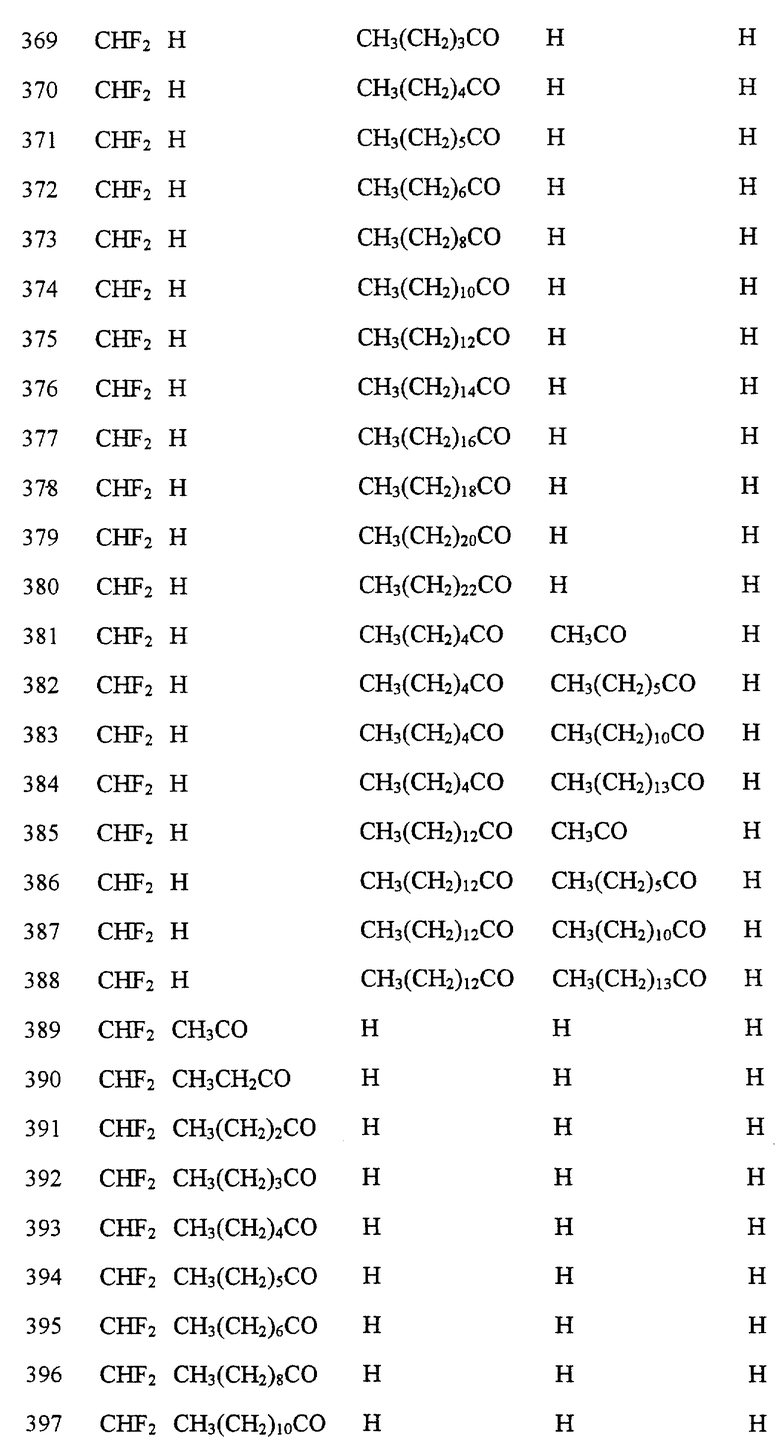
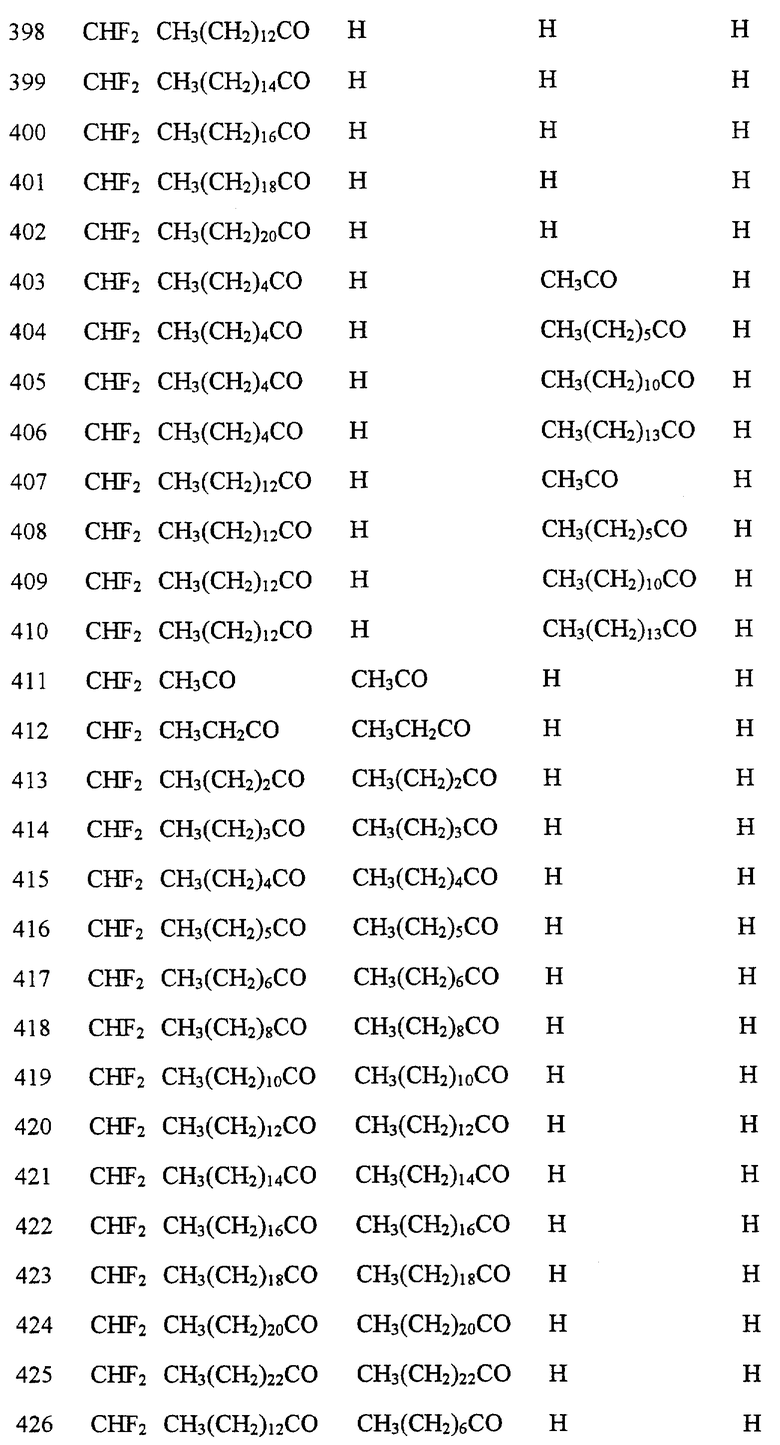
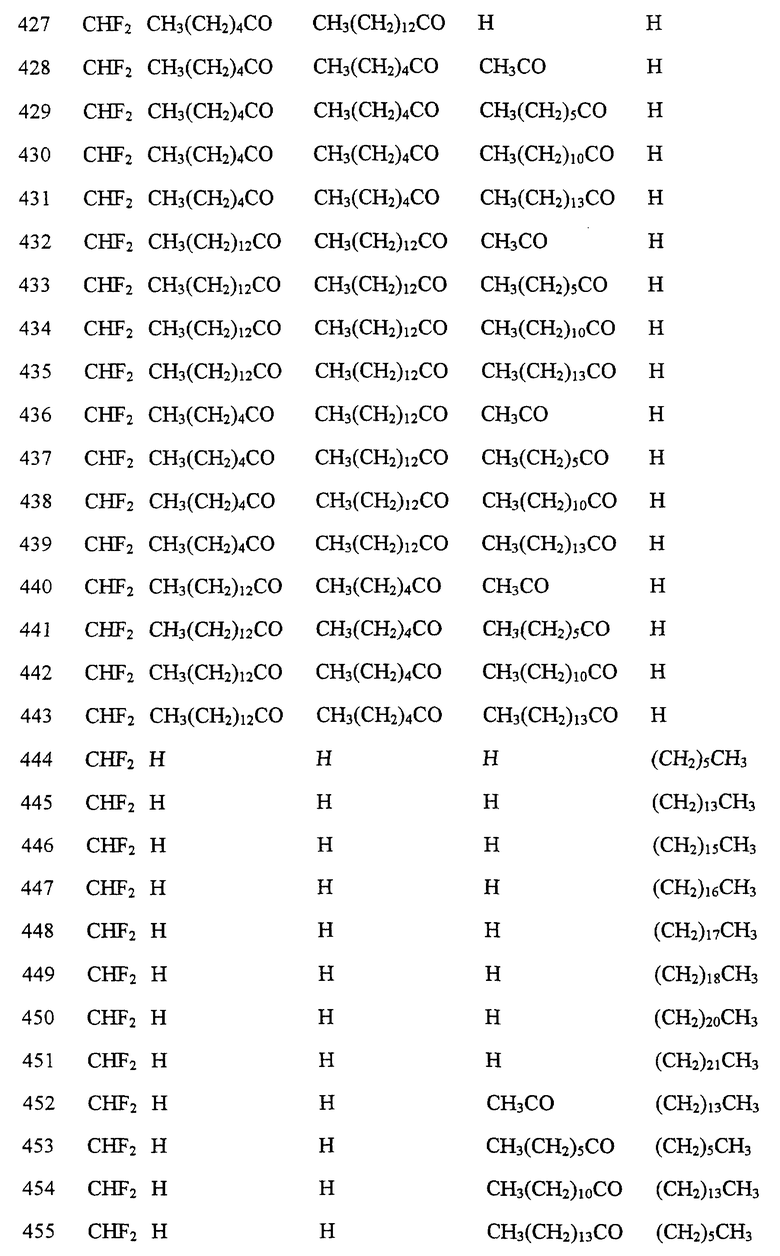


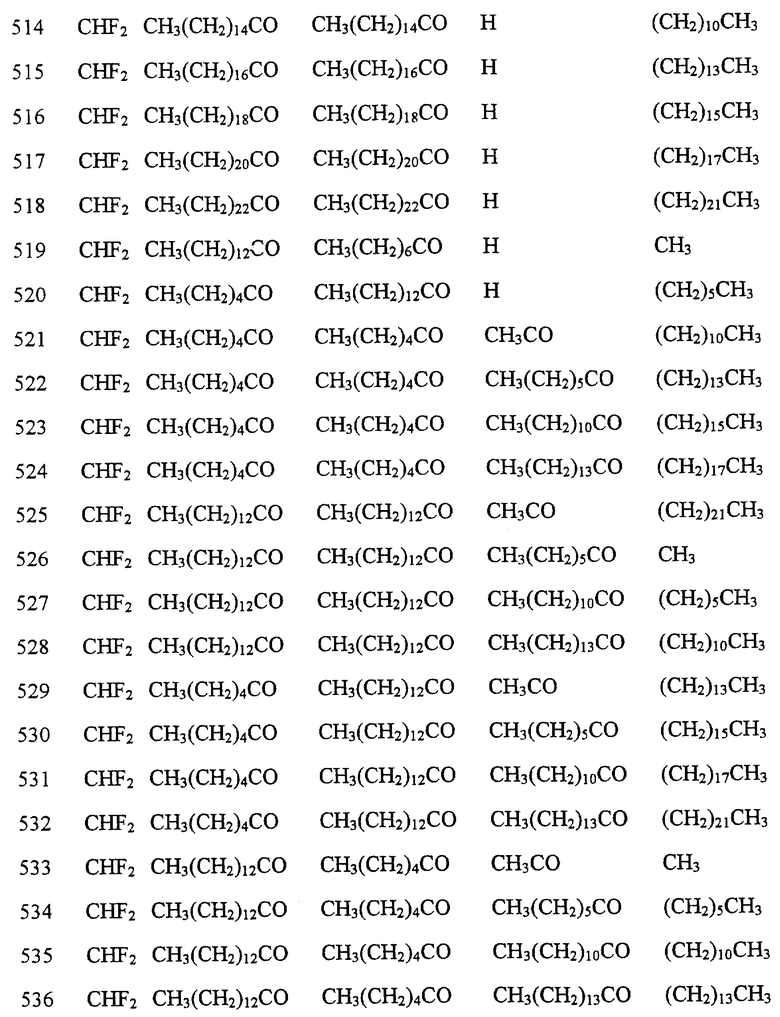


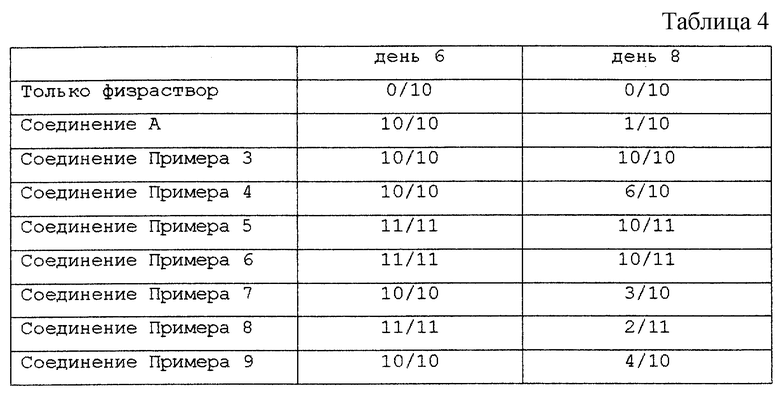
Описываются новые производные нейраминовой кислоты общей формулы (I), в которой R1 - метильная группа; R2, R3 и R4 могут быть одинаковыми или различными и каждый - атом водорода или алифатическая ацильная группа, содержащая 3-25 атомов углерода, и W - атом водорода или сложноэфирный остаток, при условии, что соединения формулы (I), в которой каждый из R2, R3 и R4 и W - атом водорода, исключаются. Соединение нейраминовой кислоты настоящего изобретения проявляет отличную in vivo способность ингибировать сиалидазу и является полезным в качестве терапевтического средства или средства для профилактики вирусного заражения гриппом. Описывается также фармацевтическая композиция и способ лечения и профилактики гриппа. 4 с. и 29 з.п.ф-лы, 4 табл.
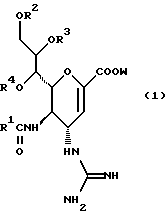
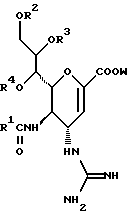
или его фармакологически приемлемая соль,
в которой R1 - метильная группа;
R2, R3 и R4 могут быть одинаковыми или различными и каждый может представлять собой атом водорода или алифатическую ацильную группу, содержащую 3 - 25 атомов углерода;
W - атом водорода или сложноэфирный остаток, при условии, что соединения формулы (I), в которой каждый из R2, R3, R4 и W - атом водорода, исключаются.
5-ацетамидо-2,3,4,5-тетрадеокси-4-гуанидино-9-O-гексаноил-D-глицеро-D-галакто-нон-2-энопиранозойная кислота,
5-ацетамидо-2,3,4,5-тетрадеокси-4-гуанидино-9-O-октаноил-D-глицеро-D-галакто-нон-2-энопиранозойная кислота,
5-ацетамидо-2,3,4,5-тетрадеокси-4-гуанидино-9-O-деканоил-D-глицеро-D-галакто-нон-2-энопиранозойная кислота,
5-ацетамидо-2,3,4,5-тетрадеокси-4-гуанидино-9-O-додеканоил-D-глицеро-D-галакто-нон-2-энопиранозойная кислота,
5-ацетамидо-2,3,4,5-тетрадеокси-4-гуанидино-9-O-миристоил-D-глицеро-D-галакто-нон-2-энопиранозойная кислота,
5-ацетамидо-2,3,4,5-тетрадеокси-4-гуанидино-9-O-пальмитоил-D-глицеро-D-галакто-нон-2-энопиранозойная кислота,
5-ацетамидо-2,3,4,5-тетрадеокси-4-гуанидино-9-O-стеароил-D-глицеро-D-галакто-нон-2-энопиранозойная кислота,
гексил-5-ацетамидо-2,3,4,5-тетрадеокси-4-гуанидино-D-глицеро-D-галакто-нон-2-энопиранозоат,
миристил-5-ацетамидо-2,3,4,5-тетрадеокси-4-гуанидино-D-глицеро-D-галакто-нон-2-энопиранозоат,
цетил-5-ацетамидо-2,3,4,5-тетрадеокси-4-гуанидино-D-глицеро-D-галакто-нон-2-энопиранозоат и
стеарил-5-ацетамидо-2,3,4,5-тетрадеокси-4-гуанидино-D-глицеро-D-галакто-нон-2-энопиранозоат.
| СПОСОБ ПОЛУЧЕНИЯ 2-ФОРМИЛ-2,5- ДИБУТИЛТИО-2,3- ДИГИДРО-4H-ПИРАНА | 1992 |
|
RU2030412C1 |
| RU 2000786 C1, 25.07.1989 | |||
| 2-МЕТИЛ-5-МЕТОКСИ-3-МЕТОКСАЛИЛАЦЕТОБЕНЗОФУРАН, ПРОЯВЛЯЮЩИЙ ПРОТИВОВИРУСНУЮ АКТИВНОСТЬ В ОТНОШЕНИИ ВИРУСА ГРИППА ТИПА А И В | 1991 |
|
RU2032683C1 |
| Прибор для очистки паром от сажи дымогарных трубок в паровозных котлах | 1913 |
|
SU95A1 |
| Испарительная горелка для сжигания жидкого топлива | 1974 |
|
SU539204A1 |
| Домовый номерной фонарь, служащий одновременно для указания названия улицы и номера дома и для освещения прилежащего участка улицы | 1917 |
|
SU93A1 |

