Настоящее изобретение относится к способу переработки минеральных смесей, в частности к способу отделения металлосодержащего минерала от нежелательной пустой породы. Изобретение также относится к определенным новым полимерам.
Распространенная проблема в области переработки минерального сырья заключается в отделении ценного минерального содержимого (минерала, представляющего промышленную ценность) от содержимого с небольшим содержанием минерала, представляющего промышленную ценность (пустой породы). Безусловно, наиболее широко используется общепринятый флотационный метод (Wills' Mineral Processing Technology, 7th Edition, Eds. BA Wills and T Napier-Munn, Butterworth-Heinemann, 2006; содержание указанного источника полностью включено в настоящую заявку посредством ссылки). Руду, содержащую минерал, мелко измельчают и вводят во флотационную камеру в виде пульпы, содержащей руду в виде частиц в воде. К пульпе добавляют химические вещества - «коллекторы», которые адсорбируются на поверхности минералов, обеспечивая их гидрофобность. Пульпу подвергают воздействию воздуха таким образом, что во флотационной камере образуются пузырьки воздуха, которые поднимаются к поверхности пульпы с образованием пены. Присутствие химического соединения-коллектора является крайне важным, так как оно селективно адсорбируется на поверхности минералов, представляющих промышленную ценность, обеспечивая гидрофобность частиц указанных ценных минералов и, таким образом, облегчая их присоединение к пузырькам воздуха. Частицы минералов, представляющих промышленную ценность, присоединенные к пузырькам воздуха, транспортируются в слой пены. Таким образом, отделение минералов, представляющих промышленную ценность, от пустой породы достигается с помощью образования пены, которая обогащена частицами ценного минерала и может легко быть отделена от пульпы.
Несмотря на то, что флотационный процесс на протяжении многих лет является основным методом разделения, в частности, для разделения рудной массы на месте на руднике, существует много аспектов, требующих определенных усовершенствований указанного метода. В связи с ценностью конечного продукта даже небольшое улучшение выхода извлеченного продукта приводит к значительным экономическим преимуществам. Выход извлеченного продукта для флотационных процессов зависит от размера частиц измельченной руды. В частности, выход извлеченного продукта снижается при превышении оптимального значения размера частиц. Указанное оптимальное значение зависит от природы руды и конкретного используемого процесса флотации, но для извлечения меди из халькопиритовой руды оптимальный размер частиц, вероятно, будет составлять в диапазоне от 80 до 150 микрон. Без ограничения какой-либо конкретной теорией или предположением, указанный эффект, возможно, является гравитационным по своей природе в связи с тем, что масса частиц минерала большего размера преодолевает силы адгезии между частицей и пузырьком. Независимо от причины указанного эффекта существует потребность в обеспечении способов извлечения частиц большего размера с более высокой эффективностью. Другим ограничением является то, что извлечение материала из пульпы посредством флотации может происходить по трем разным механизмам, только один из которых представляет собой селективное присоединение минерала, представляющего промышленную ценность, к пузырькам воздуха с помощью химических веществ-коллекторов (также известный как «истинная флотация»). Другие возможные механизмы представляют собой удержание в воде, которая проходит через пену, и «агрегация» или физическое захватывание между частицами в пене, которые присоединены к пузырькам воздуха. Механизмы удержания и агрегации могут приводить к извлечению с пеной материала пустой породы. В связи с этим, как правило, принято избегать использования единственного флотационного этапа с необходимостью проведения нескольких этапов флотации. Другое ограничение заключается в том, что обычно после флотации из богатого металлом минерала металл извлекают путем выплавления. Указанный процесс приводит к разрушению коллекторных химических веществ. Было бы желательно обеспечить способ, предоставляющий возможность восстановления, а не разрушения, используемых для разделения материалов.
Настоящее изобретение в по меньшей мере некоторых вариантах его реализации направлено на устранение описанных выше проблем и ограничений. Согласно настоящему изобретению предложена возможность внедрения в существующий флотационный процесс или для внедрения каким-либо другим образом.
Согласно первому аспекту изобретения предложен способ переработки минеральной смеси, включающий следующие этапы:
(a) обеспечение минеральной смеси, которая содержит металлосодержащий минерал и одну или более нежелательных пустых пород;
(b) приведение в контакт минеральной смеси и полимерного материала, содержащего минерал-связывающий фрагмент, который селективно связывается с металлосодержащим минералом; и
(c) разделение пустой породы и полимерного материала, с которым связан металлосодержащий минерал.
Предпочтительно металлосодержащий минерал содержит медь. Примеры минералов, содержащих медь, которые можно обрабатывать согласно изобретению, включают халькопирит и борнит.
Альтернативно, металлосодержащий минерал может содержать по меньшей мере один из следующих металлов: литий, цинк, железо, золото, серебро, молибден, кобальт, платина, уран, другие благородные металлы, другие редкие металлы, мышьяк, ртуть, кадмий, теллур и свинец.
Минерал-связывающий фрагмент может содержать по меньшей мере один атом серы.
Согласно конкретным вариантам реализации изобретения полимерный материал включает полимер, который инкапсулирует минерал-связывающий фрагмент. Во избежание неоднозначности толкования необходимо уточнить, что термин «инкапсулирует» при использовании в настоящей заявке не ограничивается полным заключением минерал-связывающего фрагмента внутри полимерной матрицы. Указанный термин скорее относится к полимеру, который частично инкапсулирует или другим образом заключает минерал-связывающий фрагмент внутри полимерной матрицы, оставляя по меньшей мере некоторую часть указанного минерал-связывающего фрагмента на поверхности полимера. Без ограничения какой-либо конкретной теорией или предположением считается, что такие «высвобожденные» минерал-связывающие фрагменты могут быть особо эффективными для связывания с металлосодержащими минералами в рудной массе в виде частиц. Предпочтительно инкапсулированный минерал-связывающий фрагмент представляет собой химическое соединение-коллектор минералов, относящееся к типу соединений, которые, как известно, применяются или подходят для применения в стандартном флотационном процессе. Классы минерал-связывающих фрагментов включают тио-, сульфатные, сульфонатные или карбоксильные соединения или анионы. Тио-соединения или анионы являются особо предпочтительными; их примеры включают ксантат, дитиофосфат, тиофосфат, дитиокарбамат; тионокарбамат, дитиофосфинат, тиофосфинат, ксантогенформиат, тиокарбанилид (дифенилтиомочевину) или тиоловые соединения или анионы. Дополнительную информацию по химическим соединениям-коллекторам минералов, которые могут применяться согласно настоящему изобретению, можно найти в следующих источниках: Wills' Mineral Processing Technology, 7th Edition, DE Nagaraj, Cl Basilio and RH Yoon, 118th SME/AIME Annual Meeting, February 27 - March 2, 1989, полное содержание которых включено в настоящую заявку посредством ссылки.
Согласно другим вариантам реализации изобретения полимерный материал представляет собой полимерную структуру, содержащую повторяющиеся звенья, в состав которых входит минерал-связывающий фрагмент. Минерал-связывающий фрагмент может содержать по меньшей мере одну функциональную группу, выбранную из амина, тиола, сложного эфира, краун-эфира, аза-краун-эфира, органической кислоты, порфирина, тиоциклоалкана, мочевины, тиомочевины, фталоцианина, тионокарбамата, тиофосфата или ксантогенформиата. Во избежание неоднозначности толкования, следует уточнить, что термины «тиомочевина» и «тиономочевина» при использовании в настоящей заявке относятся к одному фрагменту.
Могут использоваться многочисленные полимерные материалы. Полимерный материал может включать полимер, образованный путем полимеризации предшественника полимера, который содержит группу подформулы (I)

где R1 представляет собой i) CRa, где Ra представляет собой водород или алкил, ii) группу N+R13 (Zm-)1/m, S(O)pR14 или SiR15, где R13 представляет собой водород, галоген, нитро или гидрокарбил, необязательно содержащий в качестве заместителя или включающий в свою структуру функциональные группы, R14 и R15 независимо выбраны из водорода или гидрокарбила, Z представляет собой анион, имеющий заряд m, р равен 0, 1 или 2, и q равен 1 или 2, iii) C(O)N, C(S)N, S(O)2N, C(O)ON, CH2ON или CH=CHRcN, где Rc представляет собой электроноакцепторную группу или iv) ОС(O)СН, С(O)ОСН или S(O)2CH; где R12 выбран из водорода, галогена, нитро, гидрокарбила, необязательно содержащего в качестве заместителя или включающего в свою структуру функциональные группы или группу 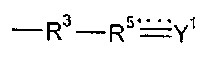 ;
;
R2 и R3 независимо выбраны из (CR7R8)n или группы CR9R10,
CR7R8CR9R10 или CR9R10CR7R8, где n равен 0, 1 или 2, R7 и R8 независимо выбраны из водорода или алкила, и один из R9 или R10 представляет собой водород, а другой представляет собой электроноакцепторную группу, или R9 и R10 вместе образуют электроноакцепторную группу;
R4 и R5 независимо выбраны из СН или CR11, где CR11 представляет собой электроноакцепторную группу,
пунктирные линии обозначают присутствие или отсутствие связи, X1 представляет собой группу СХ2Х3, где обозначенная пунктирной линией связь, к которой присоединена указанная группа, отсутствует, и группу СХ2, где обозначенная пунктирной линией связь, к которой присоединена указанная группа, присутствует, Y1 представляет собой группу CY2Y3, где обозначенная пунктирной линией связь, к которой присоединена указанная группа, отсутствует, и группу CY2, где обозначенная пунктирной линией связь, к которой присоединена указанная группа, присутствует, и X2, X3, Y2 и Y3 независимо выбраны из водорода, фтора или других заместителей.
Во избежание неоднозначности толкования следует уточнить, что термин «предшественник полимера» включает мономеры и также форполимеры, полученные путем частичной полимеризации или форполимеризации одного или более мономеров.
В полимеры указанного типа с помощью различных способов можно успешно включать минерал-связывающие фрагменты; указанные полимеры могут легко подвергаться полимеризации и обработке и обладать рядом полезных свойств.
Предпочтительно предшественник полимера полимеризуют путем подвергания ультрафиолетовому облучению. Альтернативные способы полимеризации включают применение тепла (которое может быть в форме ИК-излучения), при необходимости в присутствии инициатора, применение других видов инициаторов, таких как химические инициаторы, или инициацию с использованием электронного луча. Выражение «химический инициатор» при использовании в настоящей заявке относится к соединениям, которые могут инициировать полимеризацию, таким как свободнорадикальные инициаторы и ионные инициаторы, такие как катионные или анионные инициаторы, принятые в данной области техники. Приемлемо осуществлять вызванную облучением или электронным лучом полимеризацию по существу в отсутствие растворителя. При использовании в настоящей заявке выражение «по существу в отсутствие растворителя» означает, что растворитель отсутствует или присутствует в количестве, недостаточном для полного растворения реагентов; малое количество растворителя может присутствовать для обеспечения текучести реагентов.
Согласно предпочтительным вариантам реализации изобретения, где полимеризацию мономера обеспечивают путем подвергания ультрафиолетовому облучению, полимеризация может происходить либо самопроизвольно, либо в присутствии подходящего инициатора. Примеры подходящих инициаторов включают 2,2'-азобисизобутиронитрил (AIBN), ароматические кетоны, такие как бензофеноны, в частности ацетофенон; хлорированные ацетофеноны, такие как ди- или трихлорацетофенон; диалкоксиацетофеноны, такие как диметоксиацетофеноны (продаваемые под торговым названием «Irgacure 651»), диалкилгидроксиацетофеноны, такие как диметилгидроксиацетофенон (продаваемый под торговым названием «Darocure 1173»); замещенные диалкилгидроксиацетофеноналкиловые эфиры, такие как соединения формулы
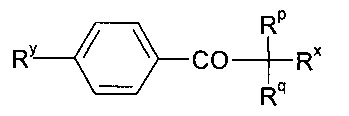
где Ry представляет собой алкил, в частности 2,2-диметилэтил, Rx представляет собой гидроксил или галоген, такой как хлор, Rp и Rq независимо выбраны из алкила или галогена, такого как хлор (например, инициаторы, которые продаются под торговым названиями «Darocure 1116» и «Trigonal P1»); 1-бензоилциклогексанол-2 (продаваемый под торговым названием «Irgacure 184»); бензоин или производные, такие как бензоинацетат, бензоиналкиловые эфиры, в частности бензоинбутиловый эфир, диалкоксибензоины, такие как диметоксибензоин или дезоксибензоин; дибензилкетон; сложные ацилоксимэфиры, такие как сложные метиловые или этиловые эфиры ацилоксима (продаваемые под торговым названием «Quantaqure PDO»); ацилфосфиноксиды, ацилфосфонаты, такие как диалкилацилфосфонат, кетосульфиды, например, следующей формулы
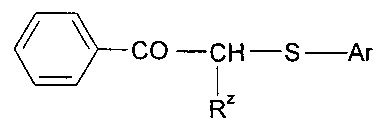
где Rz представляет собой алкил, и Ar представляет собой арильную группу; дибензоилдисульфиды, такие как 4,4'-диалкилбензоилдисульфид; дифенилдитиокарбонат; бензофенон; 4,4'-бис (N,N-диалкиламино)бензофенон; флуоренон; тиоксантон; бензил; или соединение формулы
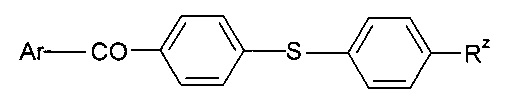
где Ar представляет собой арильную группу, такую как фенил, и Rz представляет собой алкил, такой как метил (соединение продается под торговым названием «Speedcure BMDS»).
При использовании в настоящей заявке термин «алкил» относится к алкильной группе с прямой или разветвленной углеводородной цепью, приемлемо содержащей до 20 и предпочтительно до 6 атомов углерода. Подразумевается, что термин «алкил» при использовании в настоящей заявке включает поливалентные радикалы, такие как бивалентные алкиленовые радикалы, а также моновалентные радикалы. Термины «алкенил» и «алкинил» относятся к ненасыщенным прямым или разветвленным углеводородным цепям, содержащим, например, от 2 до 20 атомов углерода, например от 2 до 6 атомов углерода. Указанные цепи могут содержать одну или более двойных или тройных связей соответственно. Кроме того, термин «арил» относится к ароматическим группам, таким как фенил или нафтил.
Термин «гидрокарбил» относится к любой структуре, содержащей атомы углерода и водорода. Например, гидрокарбил может представлять собой алкил, алкенил, алкинил, арил, такой как фенил или нафтил, арилалкил, циклоалкил, циклоалкенил или циклоалкинил. Они содержат приемлемо до 20 и предпочтительно до 10 атомов углерода. Термин «гетероциклил» включает ароматические или неароматические кольца, содержащие, например, от 4 до 20, приемлемо от 5 до 10 атомов кольца, по меньшей мере один из которых представляет собой гетероатом, такой как атом кислорода, серы или азота. Примеры таких групп включают фурил, тиенил, пирролил, пирролидинил, имидазолил, триазолил, тиазолил, тетразолил, оксазолил, изоксазолил, пиразолил, пиридил, пиримидинил, пиразинил, пиридазинил, триазинил, хинолинил, изохинолинил, хиноксалинил, бензотиазолил, бензоксазолил, бензотиенил или бензофурил.
Термин «функциональная группа» относится к реактивным группам, таким как галоген, циано, нитро, оксо, C(O)nRa, ORa, S(O)tRa, NRbRc, OC(O)NRbRc, C(O)NRbRc, OC(O)NRbRc, -NR7C(O)nR6, -NRaCONRbRc, - C=NORa, -N=CRbRc, S(O)tNRbRc, C(S)nRa, C(S)ORa, C(S)NRbRc или -NRbS(O)tRa, где Ra, Rb и Rc независимо выбраны из водорода или необязательно замещенного гидрокарбила, или Rb и Rc вместе образуют необязательно замещенное кольцо, которое необязательно содержит дополнительные гетероатомы, такие как S(O)s, атом кислорода и азота, n представляет собой целое число 1 или 2, t равен 0 или представляет собой целое число от 1 до 3. В частности, функциональные группы представляют собой такие группы, как галоген, циано, нитро, оксо, C(O)nRa, ORa, S(O)tRa, NRbRc, OC(O)NRbRc, C(O)NRbRc, OC(O)NRbRc, -NR7C(O)nR6, -NRaCONRbRc, -NRaCSNRbRc, C=NORa, -N=CRbRc, S(O)tNRbRc или -NRbS(O)tRa, где Ra, Rb и Rc, n и t являются такими, как определено выше.
Термин «гетероатом» при использовании в настоящей заявке относится к атомам, представляющим собой не атомы углерода, таким как атомы кислорода, азота или серы. Если присутствуют атомы азота, то они в целом являются частью аминоостатка, таким образом, что они содержат в качестве заместителя, например, водород или алкил.
Термин «амид» в целом относится к группе формулы C(O)NRaRb, где Ra и Rb представляют собой атомы водорода или необязательно замещенную гидрокарбильную группу. Подобным образом, термин «сульфонамид» относится к группе формулы S(O)2NRaRb. Подходящие группы Ra включают водород или метил, в частности водород.
Природа любой электроноакцепторной группы или групп, дополнительных к аминному фрагменту, используемых в любом конкретном случае, зависит от их положения и отношения к двойной связи, для активации которой они требуются, а также природы любых других функциональных групп в соединении. Термин «электроноакцепторная группа» включает атомарные заместители, такие как галоген, например фтор, хлор и бром, а также молекулярные заместители, такие как нитрил, трифторметил, ацил, такой как ацетил, нитро или карбонил.
В группе подформулы (I) X1 и Y1 (если присутствует) предпочтительно представляют собой СХ2 X3 и CY2 Y3 соответственно, и связи, обозначенные пунктирными линиями, отсутствуют.
Предпочтительно R14 и R15 (если присутствуют) представляют собой алкильные группы, наиболее предпочтительно C1-С3 алкильные группы.
Предпочтительно Rc (если присутствует) представляет собой карбонильную группу или фенил, содержащий в качестве заместителя в орто- и/или пара-положении электроноакцепторный заместитель, такой как нитро.
Когда R1 представляет собой CH=CHRdNR16-, Rd может представлять собой карбонильную группу или фенил, содержащий в качестве заместителя в орто- и/или пара-положении электроноакцепторный заместитель, такой как нитро.
Предпочтительно R7 и R8 независимо выбраны из фтора, хлора или алкила, или Н. В случае алкила метил является наиболее предпочтительным.
Предпочтительно все из X2, X3, Y2 и Y3 представляют собой водород. Необязательно, по меньшей мере один и необязательно все из X2, X3, Y2 и Y3 представляют собой заместитель, отличный от водорода или фтора. Предпочтительно по меньшей мере один и необязательно все из X2, X3, Y2 и Y3 представляют собой необязательно замещенную гидрокарбильную группу. Согласно указанным вариантам реализации изобретения предпочтительно по меньшей мере один и наиболее предпочтительно все из X2, X3, Y2 и Y3 представляют собой необязательно замещенную алкильную группу. Особо предпочтительные примеры представляют собой C1-С4 алкильные группы, в частности метил или этил. Варианты реализации, согласно которым X2, X3, Y2 и/или Y3 представляют собой алкильные группы, способны полимеризоваться при подвергании облучению в отсутствие инициатора. Альтернативно, по меньшей мере один и предпочтительно все из X2, X3, Y2 и Y3 представляют собой арил и/или гетероциклическое соединение, такое как пиридил, пиримидинил, или группу, содержащую пиридин или пиримидин.
Согласно предпочтительным вариантам реализации изобретения R12 представляет собой 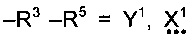 , и Y1 представляют собой группы СХ2Х3 и CY1Y2 соответственно, и пунктирные линии обозначают отсутствие связи. Согласно указанным вариантам реализации изобретения процесс полимеризации может продолжаться реакцией циклополимеризации.
, и Y1 представляют собой группы СХ2Х3 и CY1Y2 соответственно, и пунктирные линии обозначают отсутствие связи. Согласно указанным вариантам реализации изобретения процесс полимеризации может продолжаться реакцией циклополимеризации.
Предпочтительно группа предшественников полимера для применения в способе согласно изобретению представляет собой соединения формулы (II)
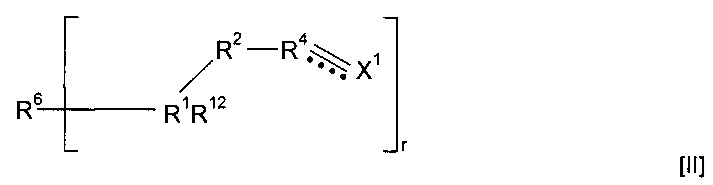
где r представляет собой целое число, составляющее 1 или более, и R6 представляет собой одну или более мостиковых групп, необязательно замещенную гидрокарбильную группу, пергалогеналкильную группу, силоксановую группу, амид или частично полимеризованную цепь, содержащую повторяющиеся звенья.
Предпочтительно r равен 1, 2, 3 или 4. Наиболее предпочтительно r равен 1 или 2.
Предпочтительно предшественник полимера представляет собой соединение, имеющее структуру (III)
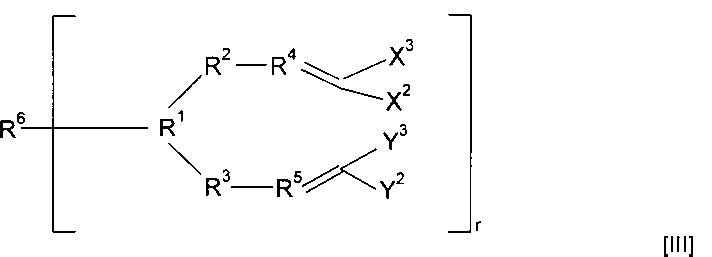
Когда в соединениях формулы (II) r равен 1, указанные соединения можно легко полимеризовать с получением полимеров различных типов в зависимости от природы группы R6.
Когда в соединениях формулы (II) r больше одного, полимеризация может приводить к образованию полимерных сеток. При полимеризации указанных соединений образуются сетки, свойства которых можно выбирать в зависимости от конкретной природы группы R6, количества присутствующего обрывающего цепь агента и используемых условий полимеризации. Некоторые примеры мостиковых групп можно найти в публикации международной заявки WO 00/06610.
Предпочтительно R6 представляет собой гидрокарбильную группу с прямой или разветвленной углеводородной цепью, необязательно содержащую в качестве заместителя или включающую в свою структуру функциональные группы. Предпочтительно R6 представляет собой алкильную группу с прямой или разветвленной углеводородной цепью, содержащую от 1 до 30 атомов углерода, необязательно содержащую в качестве заместителя или включающую в свою структуру функциональные группы. Предпочтительно R6 содержит от двух до двадцати атомов углерода, предпочтительно от двух до двенадцати атомов углерода.
Согласно другим вариантам реализации изобретения R15 представляет собой водород или гидрокарбил, и соединение формулы (I), таким образом, не содержит группу -R3-R5=Y1.
В международных публикациях WO 00/06610, WO 00/06533, WO 00/06658, WO 01/36510, WO 01/40874, WO 01/74919 и WO 2008/001102, содержание которых полностью включено в настоящую заявку посредством ссылки, описан класс полимеров, полученных в результате полимеризации ряда соединений, которые содержат одну или более диенильных групп. Согласно международной публикации WO 01/74919 также описаны полимеры, образованные из четвертичных аммониевых соединений, содержащих одну группу типа винила.
Одним из способов включения в полимерный материал минерал-связывающего фрагмента является полимеризация предшественника полимера, который содержит минерал-связывающий фрагмент в своей структуре. В случае предшественников полимера на основе подформулы (I) указанное включение можно достигать путем использования предшественников полимера, где R6 содержит в качестве заместителя или включает в состав минерал-связывающий фрагмент.
R6 может содержать в качестве заместителя или включать в свою структуру по меньшей мере одну функциональную группу, выбранную из функциональной группы амина, тиола, сложного эфира, краун-эфира, аза-краун-эфира, органической кислоты, порфирина, тиоциклоалкана, мочевины, тиомочевины, фталоцианина, тионокарбамата, тиофосфата или ксантогенформиата. Функциональные группы указанных типов могут взаимодействовать с различными металлами.
Предпочтительно R1 представляет собой N+R13(Zm-)1/m. Четвертичные аммониевые предшественники полимера указанного типа могут включать минерал-связывающий фрагмент в соответствии с рядом применимых схем.
Согласно одной схеме анион Zm- представляет собой минерал-связывающий фрагмент. Например, Zm- может представлять собой диалкилтиофосфатный анион или диалкоксидитиофосфатный анион, в котором алкильные группы содержат между 1 и 6 атомов углерода, такой как диэтилтиофосфатанион. Zm- также может представлять собой другой анион, являющийся коллектором минерала. Функциональные анионы указанного типа могут вводиться в катионный четвертичный аммониевый полимер непосредственно во время синтеза или путем ионного обмена. Предпочтительно предшественник полимера может представлять собой «ионную жидкость», которая является жидкой при комнатной температуре или имеет низкую температуру плавления, что позволяет обрабатывать указанный предшественник полимера без необходимости в присутствии растворителя.
Согласно другой схеме полимер, образованный путем полимеризации предшественника полимера, инкапсулирует минерал-связывающий фрагмент. Полимеры, образованные путем полимеризации предшественников полимера, в которых R1 представляет собой N+R13(Zm-)1/m, являются особо эффективными для инкапсулирования минерал-связывающего фрагмента. Согласно международным публикациям WO 2009/063211 и WO 2007/012860, содержание которых полностью включено в настоящую заявку посредством ссылки, описаны различные способы инкапсулирования с использованием полимеров указанного типа. Можно получать большой диапазон размеров, форм и структур, включая микросферы диаметром в диапазоне 1-100 микрон и частицы, гранулы, блоки и другие структуры большего размера, от миллиметров до метров. Также возможно покрывать различные субстраты тонкой пленкой.
Когда Zm- представляет собой не минерал-связывающий фрагмент, анионы предпочтительно представляют собой ионы галоидоводородной кислоты, предпочтительно Br-, тозилат, трифлат, ион бората, PF6- или анион сложного эфира карбоновой кислоты.
Согласно предпочтительным вариантам реализации изобретения предшественник полимера представляет собой мономер формулы (IV)
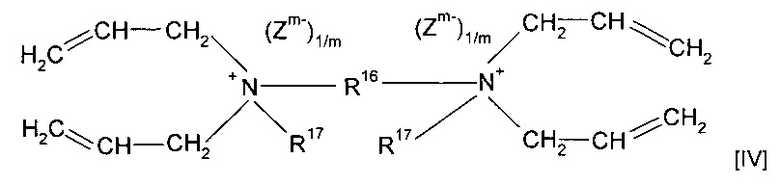
где R16 представляет собой алкильную группу с прямой или разветвленной углеводородной цепью, предпочтительно содержащую от одного до двадцати атомов углерода, наиболее предпочтительно содержащую от двух до двенадцати атомов углерода; и
R17 представляет собой водород или алкильную группу с прямой или разветвленной углеводородной цепью, предпочтительно содержащую от одного до пяти атомов углерода, наиболее предпочтительно метил или этил;
или форполимер, полученный в результате форполимеризации указанного мономера.
Согласно предпочтительному варианту реализации изобретения предшественник полимера представляет собой мономер формулы (V)
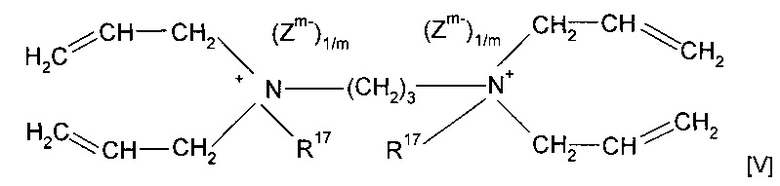
где R17 предпочтительно представляет собой метил или форполимер, полученный в результате форполимеризации указанного мономера.
Согласно другому предпочтительному варианту реализации изобретения предшественник полимера представляет собой мономер формулы (VI)
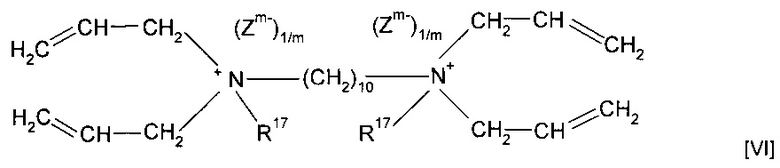
где R17 предпочтительно представляет собой метил или форполимер, полученный в результате форполимеризации указанного мономера.
Альтернативно, предшественник полимера может представлять собой диаллильный эквивалент тетрааллильных мономеров, показанных в формулах (IV) - (VI), такой как N,N-диаллилбутанметильное четвертичное аммониевое соединение с подходящим анионом, таким как тозилат.
Согласно другим предпочтительным вариантам реализации изобретения R13 и R6 вместе с кватернизированным атомом N, к которому они присоединены, образуют гетероциклическую структуру. Предпочтительно R13 и R6 вместе с кватернизированным атомом N, к которому они присоединены, образуют необязательно замещенную гетероциклическую структуру, содержащую четырех - восьмичленное кольцо. Необязательно замещенная гетероциклическая структура может представлять собой пяти- или шестичленное кольцо. Наиболее предпочтительно R13 и R6 вместе с кватернизированным N, к которому они присоединены, образуют необязательно замещенное пиперидиновое кольцо. Согласно патенту США 3912693, содержание которого полностью включено в настоящую заявку посредством ссылки, описаны способы получения и полимеризации типа мономеров, в которых R13 и R6 вместе с кватернизированным атомом N, к которому они присоединены, образуют гетероциклическую структуру. Однако согласно указанной публикации не предполагается возможность использования типа переработки минерального сырья, описанного в настоящей заявке.
Мономер может представлять собой соединение формулы (VII)
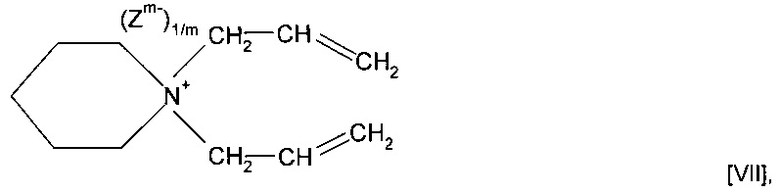
также можно использовать форполимер, полученный посредством форполимеризации указанного мономера.
Гетероциклическая структура может содержать по меньшей мере один дополнительный гетероатом помимо кватернизированного N, к которому присоединены группы R13 и R6. Дополнительный гетероатом может представлять собой N, О или S. Предпочтительно гетероциклическая структура включает по меньшей мере два гетероатома, представляющих собой атомы N; в указанном случае мономер может представлять собой соединение формулы (VIII)
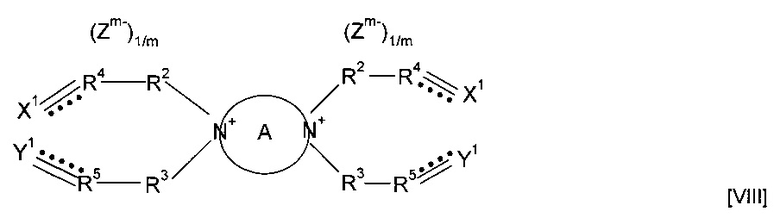
где А представляет собой четырех-шестичленное гетероциклическое кольцо, и кватернизированные атомы азота находятся в любой подходящей паре положений в кольце; также можно использовать форполимер, полученный посредством форполимеризации указанного мономера. Предпочтительно А представляет собой пяти- или шестичленное гетероциклическое кольцо. Согласно вариантам реализации изобретения, в которых А представляет собой шестичленное гетероциклическое кольцо, указанное кольцо представлять собой N-замещенное кольцо в положениях 1,2, 1,3 или 1,4.
Предпочтительно А представляет собой необязательно замещенное пиперазиновое кольцо. Мономер может представлять собой соединение формулы (IX)
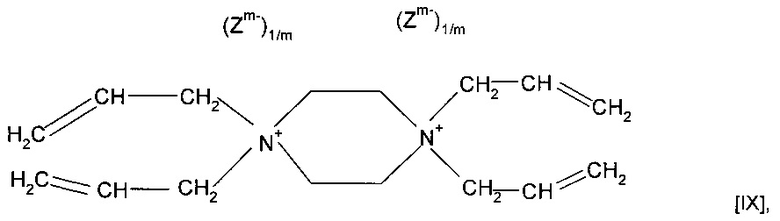
также можно использовать форполимер, полученный посредством форполимеризации указанного мономера.
Согласно вариантам реализации изобретения, в которых кватернизированный атом N не образует часть гетероциклической структуры, R1 может представлять собой Н, алкильную группу, предпочтительно содержащую менее 3 атомов углерода, наиболее предпочтительно метил или группу 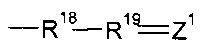 , где R18 и R19 независимо выбраны из (CR7R8)n или группы CR9R10, CR7R8CR9R10 или CR9R10CR7R8, где n равен 0, 1 или 2, R7 и R8 независимо выбраны из водорода, галогена или гидрокарбила, и один из R9 или R10 представляет собой водород, а другой представляет собой электроноакцепторную группу, или R9 и R10 вместе образуют электроноакцепторную группу; пунктирные линии обозначают присутствие или отсутствие связи, и Z1 представляет собой группу CZ2Z3, где обозначенная пунктирной линией связь, к которой присоединена указанная группа, отсутствует, и группу CZ2, где обозначенная пунктирной линией связь, к которой присоединена указанная группа, присутствует, и Z2, Z3 независимо выбраны из водорода, фтора или других заместителей.
, где R18 и R19 независимо выбраны из (CR7R8)n или группы CR9R10, CR7R8CR9R10 или CR9R10CR7R8, где n равен 0, 1 или 2, R7 и R8 независимо выбраны из водорода, галогена или гидрокарбила, и один из R9 или R10 представляет собой водород, а другой представляет собой электроноакцепторную группу, или R9 и R10 вместе образуют электроноакцепторную группу; пунктирные линии обозначают присутствие или отсутствие связи, и Z1 представляет собой группу CZ2Z3, где обозначенная пунктирной линией связь, к которой присоединена указанная группа, отсутствует, и группу CZ2, где обозначенная пунктирной линией связь, к которой присоединена указанная группа, присутствует, и Z2, Z3 независимо выбраны из водорода, фтора или других заместителей.
Согласно другим предпочтительным вариантам реализации предшественников полимера, которые содержат группу подформулы (I), R1 представляет собой C(O)N или C(S)N. Минерал-связывающий фрагмент может быть включен в структуру «ядра» полимеров указанного типа.
Предпочтительно предшественник полимера представляет собой соединение, имеющее структуру [X]
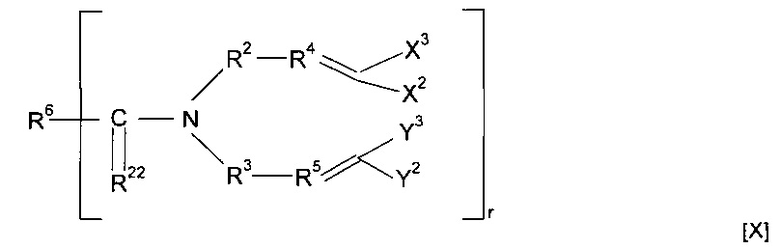
где R22 представляет собой О или S, и R6 содержит минерал-связывающий фрагмент или вместе с C=R22 образует минерал-связывающий фрагмент.
Минерал-связывающий фрагмент может представлять собой функциональную группу, содержащую тионокарбонат, тиомочевину, тиол, тиоциклоалкан, тиофосфат или ксантогенформиат.
Предшественник полимера может представлять собой соединение, имеющее структуру [XI]
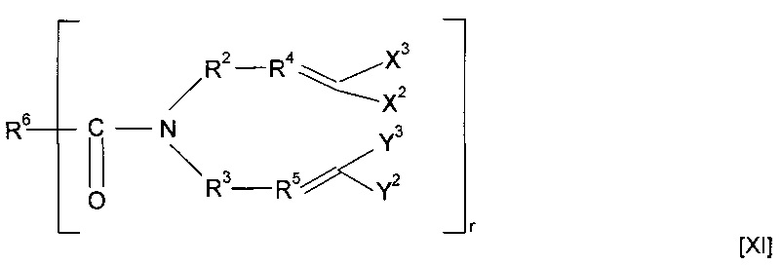
где R6 содержит группу -NHC(S)O-, -C(O)NHC(S)O- или -O-C(S)SC(O)O-. Предпочтительно предшественник полимера представляет собой соединение, имеющее структуру [XII]
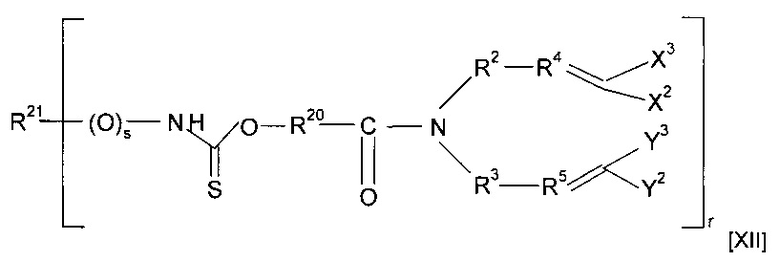
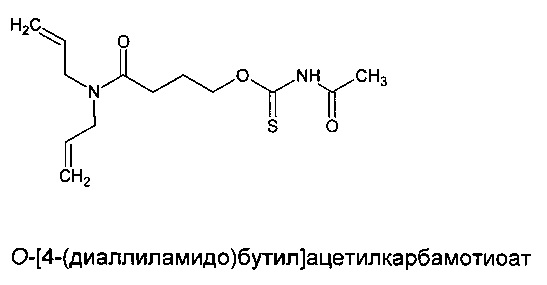
где каждый из R20 и R21 независимо представляет собой алкильную группу, необязательно содержащую в качестве заместителя или включающую в свою структуру функциональные группы, предпочтительно содержащую от одного до двадцати атомов углерода, наиболее предпочтительно содержащую от двух до двенадцати атомов углерода, s равен 0 или 1, и r предпочтительно равен 1 или 2; или форполимер, полученный посредством форполимеризации указанного соединения. Примеры соединений, имеющих структуру [XII], включают O-[4-(диаллиламидо)бутил]бутилкарбамотиоат (r=1, R20=CH2CH2CH2, R21=CH2CH2CH2CH3 и s=0) и O-[4-(диаллиламидо)бутил]ацетилкарбамотиоат (r=1, R20=CH2CH2CH2, R21=CH3 и s=1).
Предшественник полимера может представлять собой соединение, имеющее структуру [XIII]
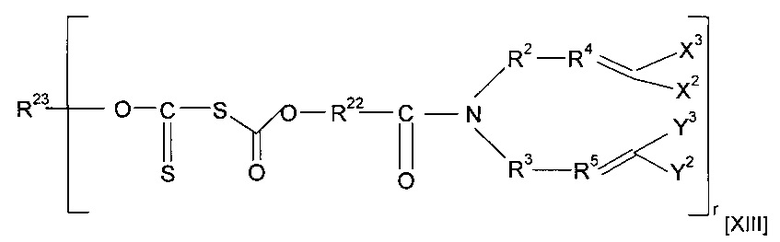
где каждый из R22 и R23 независимо представляет собой алкильную группу, необязательно содержащую в качестве заместителя или включающую в свою структуру функциональные группы, предпочтительно включающую в свою структуру атом О и предпочтительно содержащую от одного до двадцати атомов углерода, наиболее предпочтительно от двух до двенадцати атомов углерода, и r предпочтительно равен 1 или 2, или форполимер, полученный посредством форполимеризации указанного соединения.
Предшественник полимера может представлять собой соединение, имеющее структуру [XIV]
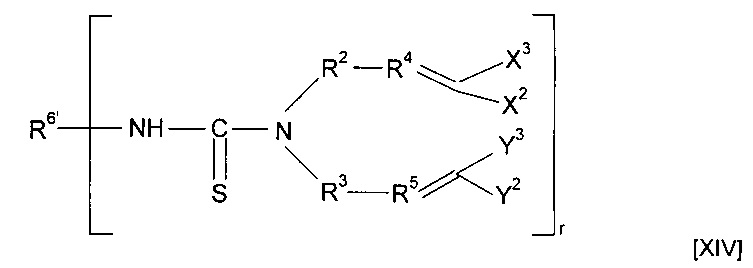
где R6'-NH образует R6, и R6' в комбинации с -NH-CS образует минерал-связывающий фрагмент.
Предшественник полимера может представлять собой соединение, имеющее структуру [XV]
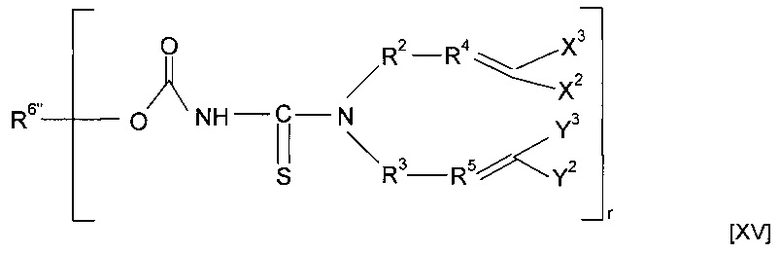
где R6''-OC(O)-NH образует R6, и R6'' вместе с -OC(O)-NH-CS образует минерал-связывающий фрагмент. Полимеризация предшественника полимера может приводить к получению гомополимера. Альтернативно, этап полимеризации предшественника полимера может приводить к получению сополимера, причем указанный предшественник полимера смешивают с одним или более другими предшественниками полимеров. Другие предшественники полимеров могут соответствовать любой из формул, описанных в настоящей заявке. Альтернативно, сомономер может представлять собой другой класс соединений. Предшественник полимера может подвергаться сополимеризации с кросс-линкером. Согласно указанным вариантам реализации изобретения предшественник полимера может подвергаться реакции с соединением формулы (XVI)
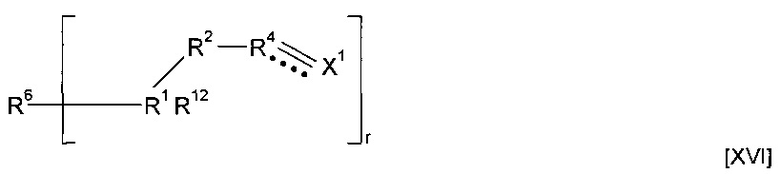
где R1, R2, R4, R12 и X1 являются такими, как определено для соединения формулы (I), r представляет собой целое число, составляющее 2 или более, и R6 представляет собой мостиковую группу, имеющую валентность r, или связь. Предпочтительно r равен 2. Применение соединения формулы (XVI) является особо предпочтительным, когда предшественник полимера не содержит группу 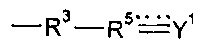 . Однако варианты реализации предшественников полимера, которые содержат группу
. Однако варианты реализации предшественников полимера, которые содержат группу 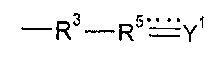 , также могут подвергаться реакции с соединением формулы (XVI).
, также могут подвергаться реакции с соединением формулы (XVI).
Соединение формулы (XVI) может представлять собой соединение формулы (XVII)
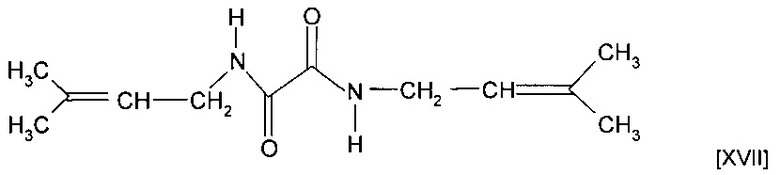
Мономер или сомономеры могут форполимеризоваться с получением форполимера. Как правило, используют термический инициатор, и форполимеризация осуществляется при температуре, превышающей комнатную температуру.
Полимерный материал может представлять собой метакрилатный или силановый полимер. Метакрилатный полимер может быть получен из 2-гидроксиметакрилата, который можно подвергать реакции с тиоизоцианатом с получением тиокарбамата. Амино-функционализированный силан можно использовать для получения содержащего тиомочевину мономера. Альтернативно, минерал-связывающий фрагмент может быть инкапсулирован полимером.
Полимерный материал может включать полимер на основе акрилата, полиуретана или стирола. Указанный полимер может инкапсулировать минерал-связывающий фрагмент или содержать указанный минерал-связывающий фрагмент внутри своей полимерной структуры.
Согласно другим вариантам реализации изобретения полимерный материал включает полимерный субстрат, к поверхности которого присоединен минерал-связывающий фрагмент. Полимерный материал может включать полимерные цепи, привитые к поверхности полимерного субстрата, где указанные полимерные цепи содержат минерал-связывающий фрагмент. В целом, предполагаются и другие формы присоединения, такие как физическая адсорбция или ионные связи. Полимерный субстрат может представлять собой эпоксид или диизоцианат, к которому привиты полимерные цепи. Могут применяться полимерные субстраты, содержащие поверхностные гидроксильные или аминные фрагменты. Подходящие схемы реакций включают реакции указанных полимерных субстратов с содержащими амин или гидроксил полимерами с получением полимерных цепей, что очевидно специалисту в данной области техники. Однако многие другие схемы реакций и варианты кандидатов полимерных субстратов и полимерных цепей специалист в данной области техники может найти в обширной и хорошо известной литературе, которая существует по теме полимерной прививки.
Полимерные цепи могут включать полиимин, предпочтительно полиэтиленимин, функционализированный путем присоединения минерал-связывающего фрагмента. Альтернативно, полимерные цепи могут включать полимер, содержащий гидроксил, такой как поливинилспирт (PVA), функционализированный путем присоединения минерал-связывающего фрагмента.
Минерал-связывающий фрагмент может представлять собой тиономочевину. Указанная тиономочевина может быть образована в результате реакции изотиоцианата с содержащей амин полимерной цепью, такой как полиимин. Альтернативно, минерал-связывающий фрагмент может представлять собой тиокарбамат. Указанный тиокарбамат может быть образован в результате реакции изотиоцианата с содержащей гидроксилполимерной цепью, такой как поливиниловый спирт (ПВС). Другие минерал-связывающие фрагменты, такие как фрагменты, описанные в настоящей заявке, можно присоединять к полимерной цепи с использованием схем реакций, которые хорошо известны в данной области техники.
Согласно другим вариантам реализации изобретения этап b) включает следующие подэтапы:
i) приведение соединения-коллектора во взаимодействие с минеральной смесью, где указанное соединение-коллектор содержит минерал-связывающий фрагмент и фрагмент, присоединяющийся к полимеру;
ii) селективное связывание соединения-коллектора с металлосодержащим минералом; и
iii) присоединение соединения-коллектора к полимеру с помощью фрагмента, присоединяющегося к полимеру.
В подэтапе iii) соединение-коллектор присоединяется к полимеру с помощью ковалентной связи, образованной в результате реакции между фрагментом, присоединяющимися к полимеру, и поверхностной группой полимера. В целом, предполагаются и другие формы присоединения, такие как физическая адсорбция или ионная связь. В случаях, когда образуется ковалентная связь, реакция может представлять собой SN2-нуклеофильную реакцию. Ковалентная связь может представлять собой C-N или С-O связь. Согласно некоторым вариантам реализации изобретения либо фрагмент, присоединяющийся к полимеру, представляет собой аминную функциональную группу или гидроксил, а поверхностная группа представляет собой уходящую группу, либо фрагмент, присоединяющийся к полимеру, представляет собой уходящую группу, а поверхностная группа представляет собой аминную функциональную группу или гидроксил. Полимеры, содержащие аминные или гидроксильные поверхностные группы, легче поддаются повторной обработке после их использования, например, путем шлифования. Полимер может представлять собой целлюлозу или гидроксиметакрилатный полимер, необязательно модифицированный путем превращения поверхностных гидроксильных групп в уходящую группу с улучшенными свойствами, такую как сложный тозилэфир. Может использоваться 2-гидроксиметакрилатный полимер.
Минерал-связывающий фрагмент может представлять собой изотиоцианатный фрагмент, такой как алкоксикарбонилизотиоцианатный фрагмент. Другие возможные минерал-связывающие фрагменты описаны в другом месте в настоящей заявке.
Полимерный материал можно получать в различных формах. Предпочтительно получают структуру, содержащую полимерный материал, где указанный полимерный материал подвергается взаимодействию с минеральной смесью, что позволяет непосредственно отделять металлосодержащий минерал от пустой породы, например, путем удаления полимерного материала из минеральной смеси, или наоборот. Можно использовать любую подходящую структуру, такую как мембрана, необязательно присоединенная к субстрату. Альтернативно, может быть получена пористая структура таким образом, что минеральная смесь проходит через указанную структуру, при этом металлосодержащий минерал селективно связывается минерал-связывающим фрагментом и, таким образом, отделяется от материала пустой породы, который выходит из структуры. Согласно указанным вариантам реализации изобретения структура может представлять собой губку и/или листовой материал, такой как сито или фильтр. Сито может представлять собой тканый материал или другую пористую сетчатую структуру.
Структура может быть образована из структуры субстрата, покрытой полимерным материалом.
Альтернативно, полимерный материал может присутствовать в виде частиц. Как правило, применение полимерного материала в виде частиц обеспечивает относительно большую площадь поверхности, доступную для связывания с металлосодержащим минералом. Отделение пустой породы можно легко достигать несколькими способами, например, посредством удаления частиц полимерного материала или удаления пустой породы через фильтр или путем декантирования.
Этапы от (а) до (с) можно осуществлять как часть флотационного процесса для отделения пустой породы от металлосодержащего минерала. Таким образом, изобретение может быть включено в стандартные флотационные процессы. Могут использоваться частицы полимерного материала, полученные таким образом, чтобы они всплывали, например, путем включения воздуха в полимерную структуру.
Как правило, минеральная смесь присутствует в виде пульпы, содержащей минералы в виде частиц в воде.
Способ может включать дополнительный этап высвобождения металлосодержащего минерала из полимерного материала. Указанное высвобождение может легко достигаться при использовании многих полимеров, образованных путем полимеризации предшественника полимера подформулы (I) с полимером, который восстанавливают для повторного применения. Высвобождение может достигаться физическими способами, такими как взбалтывание или обработка ультразвуком, или химическими способами, такими как повышение или снижение рН путем добавления щелочи или кислоты или добавление химического вещества, такого как депрессор. Термин «депрессор» согласно предшествующему уровню техники относится к химическому соединению, которое можно использовать для удаления химического соединения-коллектора от металлосодержащего фрагмента. Например, кислый сернистый натрий представляет собой депрессор, который используется для удаления ксантатов от сульфидов меди и который может применяться в соответствии с настоящим изобретением.
Способ может включать дополнительный этап получения некоторого количества металла из металлосодержащего минерала. Указанное получение может достигаться с использованием процесса плавки. Предпочтительно металлосодержащий минерал высвобождают из полимерного материала до этапа получения некоторого количества металла из металлосодержащего минерала. Однако возможно осуществлять дополнительный этап получения некоторого количества металла из металлосодержащего минерала без предварительного высвобождения указанного металлосодержащего минерала из полимерного материала.
Преимуществом является то, что изобретение может осуществляться на месте на руднике.
Согласно второму аспекту изобретения предложен металлосодержащий минерал или металл, полученный с помощью способа в соответствии с первым аспектом изобретения.
Согласно третьему аспекту изобретения предложено применение полимерного материала, который содержит минерал-связывающий фрагмент, в процессе переработки минеральной смеси для отделения металлосодержащего минерала от материала пустой породы.
Согласно четвертому аспекту изобретения предложен полимер, полученный посредством полимеризации предшественника полимера, который содержит группу подформулы [XVIII]
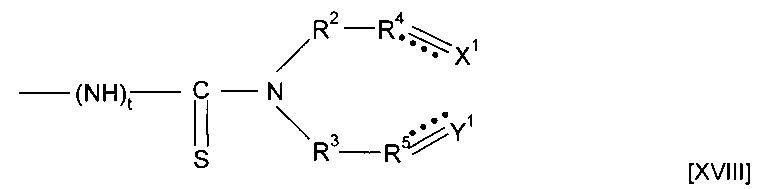
где t равен 0 или 1, R2 и R3 независимо выбраны из (CR7R8)n, или группы CR9R10, CR7R8CR9R10 или CR9R10CR7R8, где n равен 0, 1 или 2, R7 и R8 независимо выбраны из водорода или алкила, и один из R9 или R10 представляет собой водород, а другой представляет собой электроноакцепторную группу, или R9 и R10 вместе образуют электроноакцепторную группу;
R4 и R5 независимо выбраны из СН или CR11, где CR11 представляет собой электроноакцепторную группу, пунктирные линии обозначают присутствие или отсутствие связи, X1 представляет собой группу СХ2Х3, где обозначенная пунктирной линией связь, к которой присоединена указанная группа, отсутствует, и группу СХ2, где пунктирная линия, к которой присоединена указанная группа, присутствует, Y1 представляет собой группу CY2Y3, где обозначенная пунктирной линией связь, к которой присоединена указанная группа, отсутствует, и группу CY2, где обозначенная пунктирной линией связь, к которой присоединена указанная группа, присутствует, и X2X3,Y2 и Y3 независимо выбраны из водорода, фтора или других заместителей.
Предшественник полимера может представлять собой соединение, имеющее структуру [XIX]
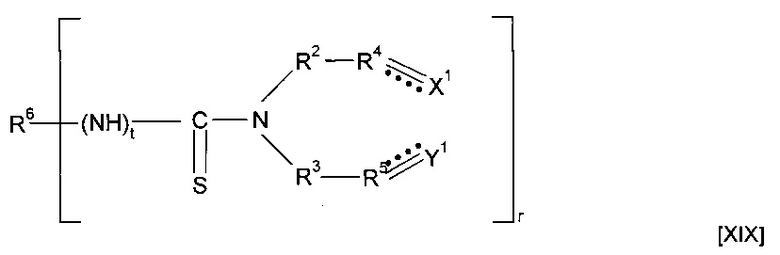
где r представляет собой целое число, составляющее 1 или более, R6 представляет собой одну или более мостиковых групп, необязательно замещенную гидрокарбильную группу, пергалогеналкильную группу, силоксановую группу, амид или частично полимеризованную цепь, содержащую повторяющиеся звенья.
Предшественник полимера может представлять собой мономер, имеющий структуру [XX]
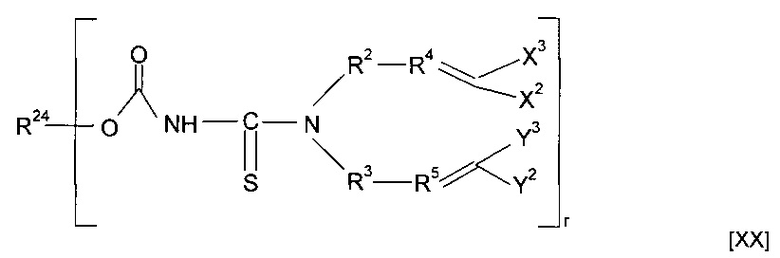
где R24 представляет собой гидрокарбильную группу, необязательно содержащую в качестве заместителя или включающую в свою структуру функциональные группы, или форполимер, полученный посредством форполимеризации указанного мономера.
Предшественник полимера может представлять собой мономер, имеющий структуру [XXI]
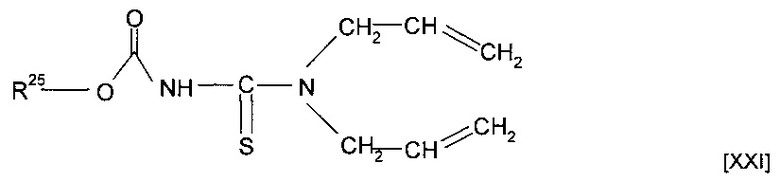
где R25 представляет собой алкильную группу, необязательно содержащую в качестве заместителя или включающую в свою структуру функциональные группы, предпочтительно содержащую от одного до двадцати атомов углерода, наиболее предпочтительно от двух до двенадцати атомов углерода, или форполимер, полученный посредством форполимеризации указанного мономера.
Другие предшественники полимеров, содержащие группу подформулы [XVIII], могут быть получены коммерческим путем или синтезированы из коммерчески доступных соединений с использованием способов, описанных в междунароных публикацих WO 00/06610, WO 00/06533, WO 00/06658, WO 01/36510, WO 01/40874, WO 01/74919 и WO 2008/001102. Согласно указанным публикациям также предложены дополнительные соединения, являющиеся кандидатами фрагментов R6, R24 и R25, описанных в Формулах [XIX]-[XXI],
Согласно пятому аспекту изобретения предложен способ переработки минеральной смеси, включающий следующие этапы:
(a) обеспечение минеральной смеси, которая содержит металлосодержащий минерал и одну или более нежелательных пустых пород;
(b) приведение соединения-коллектора во взаимодействие с минеральной смесью, где указанное соединение-коллектор содержит минерал-связывающий фрагмент, который селективно связывается с металлосодержащим минералом, при этом указанное соединение-коллектор также содержит фрагмент, присоединяющийся к полимеру;
(c) присоединение соединения-коллектора к полимеру с помощью фрагмента, присоединяющегося к полимеру; и
(d) разделение пустой породы и полимера, к которому присоединено соединение-коллектор и металлосодержащий минерал.
Несмотря на то, что настоящее изобретение было описано выше, оно включает любую комбинацию и подкомбинацию признаков согласно изобретению, изложенных выше или в следующем далее описании или формуле изобретения. Изобретение включает также любые соединения, полимеры и полимерные материалы согласно изобретению, описанные в настоящей заявке.
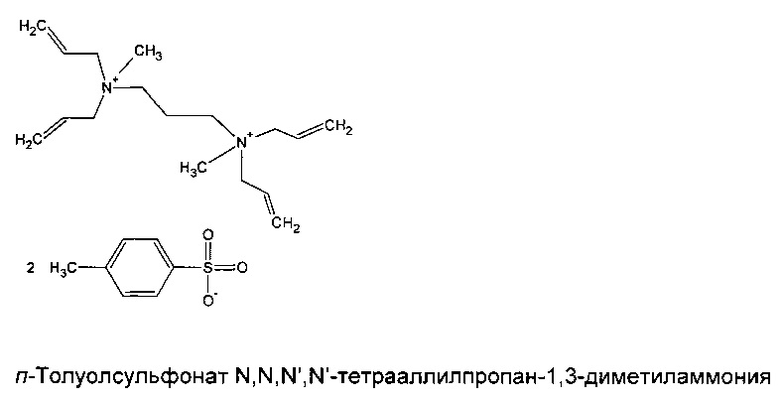
Пример 1. Привлечение сульфида меди, халькопирита, на поверхность четвертичного тетрааллиламмониевого полимера, содержащего химическое вещество-коллектор O,O-диэтилтиофосфат
Способ
Мономерный п-толуолсульфонат N,N,N',N'-тетрааллилпропан-1,3-диметиламмония (>99%, 0,965 г) синтезировали в соответствии со способом, описанным в Примере 7 (детали синтеза также можно найти в более ранней международной публикации заявителя WO 2009/063211) и растворяли в деионизированной воде (0,080 г) при слабом нагревании и интенсивном перемешивании. Затем добавляли фотоинициатор «Irgacure 2022» (Ciba SC, 0,0280 г) с последующим добавлением химического соединения-коллектора О,О-диэтилтиофосфата калия (Sigma Aldrich, 90%, 0,0285 г), которые тщательно размешивали в жидкости.
Затем маленькую каплю указанной смеси помещали в планшет на основе политетрафторэтилена (ПТФЭ), затем отверждали с помощью УФ-лампы высокой интенсивности FusionUV LH6 с D-колбой при 100% интенсивности и скорости 2 м/минуту с использованием одного цикла с получением жесткого прозрачного твердого вещества.
Также получали образец, не содержащий химического соединения-коллектора, с использованием таких же материалов и в таких же отношениях, как выше, но в отсутствие О,О-диэтилтиофосфата. Указанный образец также отверждали в виде гранулы такого же размера с использованием идентичных условий отверждения.
Готовили две пробирки, каждая из которых содержала примерно 4 г деионизированной воды и 50 мг порошка халькопирита, отшлифованного от куска кристалла халькопирита большего размера с использованием наждачной бумаги с размером абразивного зерна 100 (Р100) с получением порошка темно-серого цвета. Гранулу полимера, содержащую коллектор, помещали в одну пробирку и гранулу полимера, не содержащую коллектор, помещали в другую пробирку, обе пробирки плотно закрывали и встряхивали, позволяя порошку халькопирита суспендироваться в воде и затем равномерно оседать на гранулы.
Образцы оставляли на 4 часа, после чего гранулы вынимали и помещали в отдельный стакан с водой (200 мл), затем воду осторожно перемешивали для удаления любых неприкрепившихся к поверхности частиц минерала. Гранулы затем вынимали и помещали на ПЭТФ-планшет для проведения анализа.
Также в деионизированную воду добавляли на 4 часа другую гранулу референсного образца, не содержащую коллектор, для проверки изменения окраски самого полимера в воде.
Гранула, содержащая химическое соединение-коллектор О,O-диэтилтиофосфат, внешне была темнее по сравнению с референсным образцом, не содержащим коллектор, и намного темнее по сравнению с гранулой полимера, содержащей коллектор, который не помещали в воду с халькопиритом.
Через 4 часа после помещения только в деионизированную воду другой гранулы референсного образца того же полимера, не содержащей коллектор, не наблюдали внешних изменений, что указывает на то, что потемнение может быть обусловлено накоплением халькопирита на поверхности полимера.
Пример 2. Привлечение сульфида меда, халькопирита, на поверхность четвертичного тетрааллиламмониевого полимера, содержащего химическое вещество-коллектор O,O-диэтилтиофосфат, после более длительного воздействия халькопирита
Способ
Повторяли эксперимент 1, за исключением того, что гранулу полимера, содержащую коллектор, и референсный образец, не содержащий коллектор, помещали в смесь халькопирита и деионизированной воды на 24 часа.
Результаты
Гранула полимера, содержащая коллектор, внешне была еще темнее по сравнению с гранулой, которую оставляли на 4 часа. Внешняя разница между гранулой, содержащей коллектор, и референсной гранулой без коллектора была еще больше, чем указанная разница при воздействии в течение 4 часов.
Пример 3. Удаление минерала, содержащего медь, с поверхности четвертичного тетрааллиламмониевого соединения с использованием обработки ультразвуком
Способ
Мономерный п-толуолсульфонат N,N,N',N'-тетрааллилпропан-1,3-диметиламмония (>99%, 1,47 г) растворяли в деионизированной воде (0,28 г) при слабом нагревании. В полученной смеси растворяли химическое соединение-коллектор O,O-диэтилтиофосфат калия (Sigma Aldrich, 90%, 0,13 г) с последующим добавлением фотоинициатора «Irgacure 2022» (Ciba SC, приблизительно 40 мг) при интенсивном перемешивании.
Затем часть полученной смеси помещали между двумя предметными стеклами и отверждали с использованием УФ-лампы высокой интенсивности FusionUV LH6 с D-лампочкой, при 100% интенсивности с использованием двух циклов при скорости 4 м/мин с получением прозрачного твердого вещества.
Далее полимерную пленку снимали с предметного стекла, затем помещали в смесь, содержащую примерно 200 мг порошка каждого из следующих веществ: сульфид Cu(I) (-325 мешей), сульфид Cu(II) (-100 мешей), оксид Cu(I) (<5 микрон) и металлопорошки Cu (10-425 микрон) в деионизированной воде (100 мл). Полученную смесь осторожно перемешивали для суспендирования минералов, позволяя сохраняться равномерному слою на полимерной пленке.
Через 2 часа пленку удаляли из смеси и помещали в стакан с деионизированной водой (200 мл) и осторожно перемешивали для удаления любых неприкрепившихся к поверхности минералов. Затем пленку вынимали и помещали в стакан, содержащий примерно 100 мл воды, и обрабатывали на ультразвуковой бане в течение 3 секунд.
Результаты
Наблюдали, что почти весь минерал, содержащий медь, сразу отсоединялся от пленки после начала обработки ультразвуком.
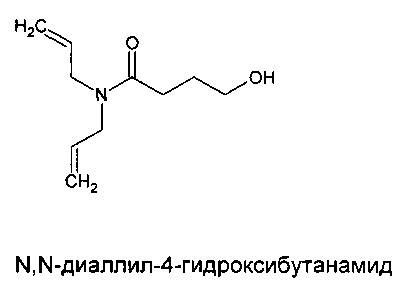
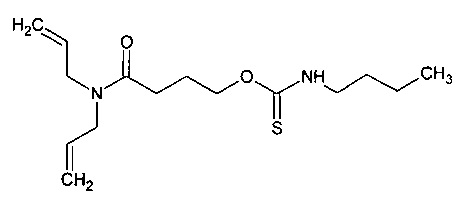
Пример 4. Синтез O-[4-(диаллиламидо)-бутил]бутилкарбамотиоата (мономера диаллиламида, содержащего алкилтионокарбаматную группу)
Получение промежуточного соединения, амидоспирта, N,N-диаллил-4-гидроксибутанамида
Гамма-бутиролактон (171,0 г, 1,99 моль) и диаллиламин (490,0 г, 5,04 моль) смешивали и нагревали до 120°С. Полученную смесь перемешивали при указанной температуре в течение 33 ч. Порцию (200 г) очищали, снижая температуру до 110°С в вакууме (30 мбар), что приводило к удалению диаллиламина, но не гамма-бутиролактона.
Фурье-ИК-спектроскопия (тонкая пленка): 3420, 3082, 1773, 1630, 1196, 993, 927 см-1.
От материала, очищенного при 110°С в вакууме, отбирали 70 г в этилацетате (200 мл), сушили (MgSO4), затем пропускали через пластину с силикагелем, промывая дополнительной порцией этилацетата (2×200 мл). Растворитель удаляли в вакууме.
Амидоспирт, содержащий небольшое количество гамма-бутиролактона (13,2 г, ~0,06 моль), смешивали с водопроводной водой (260 мл) в колбе. К указанной смеси добавляли гидроксид натрия (1,4 г, 0,035 моль). Смесь нагревали до 70°С в течение 16 часов. Температуру повышали до температуры обратной конденсации и поддерживали при указанной температуре в течение 2 часов. Реакционной смеси позволяли охлаждаться до комнатной температуры. В колбу наливали дихлорметан (100 мл). Слои разделяли. Водный слой экстрагировали дихлорметаном (100 мл). Слои разделяли и органические слои объединяли, сушили (MgSO4) и концентрировали в вакууме. В результате получали 6,0 г (восстановление 45%).
Фурье-ИК-спектроскопия (тонкая пленка): 3419, 3083, 1629, 1196, 993, 926 см-1.
Синтез 0-[4-(диаллиламидо)-бутил]бутилкарбамотиоата
N,N-диаллил-4-гидроксибутанамид, содержащий гамма-бутиролактон (15,0 г, ~0,07 моль), наливали в высушенную над огнем колбу. Через капельную воронку по каплям добавляли бутилизотиоцианат (14,7 г, 0,08 моль). Полученную смесь нагревали до 60°С и оставляли перемешиваться при указанной температуре в течение 18 ч. Смеси позволяли охлаждаться до комнатной температуры. Добавляли по каплям дилаурат дибутилолова (0,25 г, 0,4 ммоль). Полученную смесь нагревали до 60°С и оставляли перемешиваться в течение 64 часов. После этого температуру реакционной смеси повышали до 101°С в течение 42 ч. Смеси позволяли охлаждаться до комнатной температуры. Остаточный бутилизотиоцианат выделяли из реакционной смеси в вакууме. В результате получали коричневое масло (21,9 г, выход неочищенного продукта 92%).
Фурье-ИК-спектроскопия (тонкая пленка): 3326, 3082, 1774, 1716, 16, 1546, 1196, 993, 925 см-1.
Пример 5. Синтез O-[4-(диаллиламидо)бутил]ацетилкарбамотиоата (мономера диаллиламида, содержащего алкилкарбонилтионокарбаматную группу)
N,N-диаллил-4-гидроксибутанамид (5,8 г, 0,03 моль) наливали в высушенную над огнем колбу. Добавляли по каплям ацетилизотиоцианат (3,2 г, 0,03 моль) в атмосфере азота. С помощью водяной бани температуру реакционной смеси поддерживали ниже 30°С. Реакционную смесь нагревали до 30°С и перемешивали при указанной температуре в течение 18 часов. Наливали дополнительную порцию N,N-диаллил-4-гидроксибутанамида (0,5 г, 0,02 моль) и полученную смесь перемешивали в течение 5 часов. Затем реакционную смесь нагревали в вакууме (91°С/30 мбар) в течение 2,5 часов.
Порцию реакционной смеси (2,8 г, ~0,01 моль) отбирали и растворяли в тетрагидрофуране (25 мл). К указанному раствору наливали гидроксид натрия (0,11 г, 0,003 моль) и теплую водопроводную воду (25 мл). Смесь оставляли перемешиваться при комнатной температуре в течение ночи. К указанной смеси наливали дихлорметан (100 мл). Слои разделяли и водный слой далее экстрагировали дихлорметаном (2×50 мл). Объединенные органические слои сушили (MgSO4), отбирали в этилацетате (50 мл) и пропускали через пластину с силикагелем. Этилацетат удаляли в вакууме и полученное масло очищали с помощью колоночной флэш-хроматографии на силикагеле (элюент: 40-60°С бензин/этилацетат, 3:1). В результате получали желтое масло (0,48 г, восстановление 17%, в объеме 5,6%), которое было чистым на 95% по результатам 1Н ЯМР-анализа.
Фурье-ИК-спектроскопия (пленка): 3459, 3082, 1738, 1651, 1546, 1196, 994, 928 см-1.
1Н ЯМР (CDCl3): 1,7 (br, 0,6Н), 1,95 (m, 1,9Н), 2,05 (s, 2,9Н), 2,3 (s, 0,8Н), 2,4 (t, 1,9Н), 3,85 (d, 2,1Н), 3,95 (d, 2,1Н), 4,1 (t, 2,0Н), 5,15 (m, 4,2Н), 5,7 (m, 2,0Н) ppm.
Пример 6. Сбор порошка халькопирита (CuFeS2) на полимерную пленку, состоящую из сополимера поли(N,N,N',N'-тетрааллилэтандиамид-со-O-[4-диаллиламидо)бутил]ацетилкарбамотиоата)
Получали смесь бифункционального мономера N,N,N',N'-тетрааллилэтандиамида и монофункционального мономера O-[4-(диаллиламидо)бутил]ацетилкарбамотиоата) в отношении 3:1 по массе соответственно. Затем добавляли фотоинициатор Irgacure 2022 (Ciba SC) (3 масс.%) и тщательно перемешивали при слабом нагревании. Полученную смесь затем наносили в виде тонкой пленки на субстрат, представляющий собой непластифицированный поливинилхлорид (НПВХ), и затем полимеризовали до получения твердого сополимера с использованием УФ-лампы высокой интенсивности (Fe-активированная ртутная лампочка, 200 Вт/см2, 2 цикла при скорости 2 метра в минуту).
Также получали референсный образец, не содержащий тионокарбаматных групп в полимере; получали смесь мономеров N,N,N',N'-тетрааллилэтандиамида и N,N-диаллилгексанамида в отношении 3:1 по массе соответственно. N,N-диаллилгексанамид синтезировали в соответствии с Примером 10. Затем добавляли фотоинициатор Irgacure 2022 (Ciba SC, 3 масс.%) и тщательно перемешивали при слабом нагревании. Полученную смесь отверждали таким же образом, как и вышеуказанную смесь, содержащую тионокарбамат-функционализированный мономер.
Оба образца очищали в деионизированной воде и затем помещали в отдельные суспензии, каждая из которых содержала 50 мг халькопирита, отшлифованного от большого кристалла с использованием шлифовальной бумаги Р100, и 50 мл деионизированной воды, на 18 часов.
Результаты
Каждый образец полимера удаляли из суспензии. Образец полимера, содержащий O-[4-(диаллиламидо)бутил]ацетилкарбамотиоат, присоединял больше халькопирита по сравнению с референсным образцом, на что указывала его более темная окраска; указанный халькопирит затем смывали под струей воды с получением свободного порошка халькопирита.
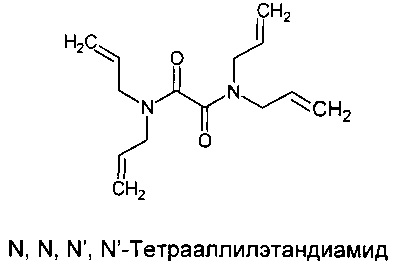
Синтез N,N,N',N'-тетрааллилэтандиамида
Свежий безводный оксалоилхлорид (ClOOCCOOCl) (200 ммоль) помещали в 3-горлую круглодонную (КД) колбу, содержащую 200 мл безводного дихлорметана. Свежеперегнанный диаллиламин (400 ммоль) добавляли к триэтиламину (400 ммоль), дополнительно разводили в безводном дихлорметане (1:1 по объему), затем добавляли в капельную воронку и помещали в реакционную колбу. Газообразный азот прокачивали через сосуд через два других горла. Для нейтрализации полученной HCl остаточный газ барботировали через раствор CaCO3. Реакционный сосуд затем помещали на баню соленая вода/лед и, как только содержимое охлаждалось, к раствору хлорангидрида добавляли по каплям смесь диаллиламин/триэтиламин/ДХМ при непрерывном перемешивании смеси с помощью магнитной мешалки. За температурой следили, поддерживая указанную температуру между 5-10°С. Добавление по каплям диаллиламина и триэтиламина останавливали через три часа и реакционную смесь оставляли перемешиваться в течение дополнительного часа.
Для наблюдения за реакцией путем сравнения количества исходного материала и продукта использовали тонкослойную хроматографию с использованием этилацетата и оксида алюминия. Для проявления планшета использовали йод, продукт реакции можно было наблюдать в виде пятна, которое выходило значительно позже, чем исходный материал.
Для удаления аминохлорида и избытка диаллиламина реакционный раствор промывали 3М раствором HCl. Мономер оставался во фракции ДХМ и его выделяли с использованием разделительной воронки. Использовали две промывки 100 мл HCl. Растворитель затем удаляли в ротационном испарителе.
Продукт добавляли к дихлорметану (1:1 по объему) и пропускали через колонку с силикагелем (силикагель 60 для хроматографии, Merck) с использованием дихлорметана в качестве элюента.
Пример 7. Синтез дитиосфосфата N,N,N',N'-тетрааллилпропандиметиламмония (четвертичного аммониевого мономера, содержащего группу-коллектор в виде аниона)
Синтез диаминного промежуточного соединения А:
Смесь 1,3-дибромпропана (99%, 150,0 г, 0,7429 моль), карбоната натрия (97%, 456 г, 3,2996 молей) и 2-пропанола (400 мл) добавляли в круглодонную реакционную колбу и доводили до температуры обратной конденсации при перемешивании. К реакционной смеси постепенно добавляли диаллиламин (99%, 160,5 г, 1,6519 моль) в течение часа и температуру обратной конденсации поддерживали в течение 120 часов, затем охлаждали до комнатной температуры. Смесь затем фильтровали и летучие вещества удаляли в вакууме. Получали желтое масло, которое далее очищали с помощью колоночной хроматографии с использованием силикагеля (60Å) и ДХМ в качестве элюента. После удаления ДХМ получали бледно-желтое масло (плотность = 0,86 г/см3, выход = 80%).
Синтез дитиосфосфата N,N,N',N'-тетрааллилпропандиметаминия
Диаминное промежуточное соединение А (6,4 г) добавляли к безводному 2-пропанолу (200 мл) и перемешивали при комнатной температуре с последующим добавлением О,O-дитиофосфата (9,213 г) в течение 30 минут с получением соли четвертичного аммония (рН=6,5). 2-пропанол затем удаляли в вакууме с получением мономерного четвертичного диаллиламмония. Выход ~95%.
Мономер можно полимеризовать с использованием способов, описанных в Примере 1.
Пример 8. Сбор обогащенной халькопиритом руды с использованием сополимера, состоящего из поли(N,N-диаллилэтоксикарбонилтиономочевина-со-N,N,N',N'-тетрааллилэтандиамида)
Синтез N,N-диаллилэтоксикарбонилтиономочевины (этилди(аллил)карбамотиоил]карбамата)
К смеси свежедистиллированного диаллиламина (4,0 г) и дихлорметана (50 мл) добавляли по каплям в течение примерно 30 минут этоксикарбонилизотиоцианат (98%, 5,00 г) при непрерывном перемешивании в течение примерно 30 минут. При добавлении изотиоцианата наблюдали экзотермический эффект, и температуре позволяли повышаться от комнатной температуры до температуры обратной конденсации (40°С). Смесь оставляли реагировать в течение дополнительных 90 минут, после чего указанную смесь добавляли к этилацетату (150 мл) и пропускали через короткую колонку с силикагелем (высота 6 см) в неполном вакууме. Раствор затем фильтровали и обрабатывали в ротационном испарителе для удаления всех летучих веществ. Выход = 89%.
1Н ЯМР (500МГц, CDCl3) δ/ppm=1,3(t), 4,2(q), 4,5(m), 5,2(d), 5,85(m), 7,3(s)
N,N,N',N'-тетрааллилэтандиамид синтезировали в соответствии с Примером 6.
Добавляли N,N-диаллилэтоксикарбонилтиономочевину и N,N,N',N'-тетрааллилэтандиамидный кросс-линкер вместе в виде смеси 1:1 (по массе), также добавляли фотоинициатор Irgacure 2022 в отношении 3,5% по массе от общей массы смеси мономеров. Полученную смесь тщательно смешивали и наносили на плоский кусок поли(карбоната) размером ~10 см × 15 см с помощью поролонового валика до тех пор, пока не получали равномерное покрытие массой примерно 3 г/м2. Образец пропускали под фокусированной УФ-лампой высокой интенсивности (FusionUV LH6, D-колба, 100% интенсивность, 5 циклов при скорости 3,5 м/мин).
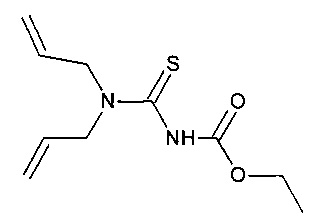
N,N-диаллилэтоксикарбонилтиономочевина
Панель с покрытием помещали в горизонтальный испытательный прибор, который мог подвергать образец взаимодействию с суспензией по площади ~112 см2, с глубиной 2,0 см. Рудное тело, содержащее халькопирит в качестве основного компонента (42% по массе), с остатком, представляющим собой главным образом смесь сульфидов железа (пирротин, 20% по массе, пирит, 16% по массе), измельчали на шаровой мельнице до получения фракции с размером частиц менее 106 мкм (распределение размера частиц: D10[5,68 мкм] D50[37,29 мкм], D90[106,9 мкм]). 2,0 г указанного минерального порошка добавляли в 200 мл деионизированной воды с получением суспензии, которую тщательно размешивали перед добавлением в испытательный прибор, который содержал панель образцов. Указанный испытательный прибор оставляли в неподвижном состоянии на 20 минут, после чего избыточный минерал выливали и минерал, присоединенный к поверхности полимера, собирали посредством отфильтровывания из шлиха. Собранный минерал тщательно высушивали и взвешивали. Указанный эксперимент повторяли несколько раз и среднее значение массы минерала, собранного на единицу площади поверхности полимера, сравнивали со значением для референсного полимера, который не содержал группу тиономочевины (см. референсный образец).
Масса минерала, собранного с помощью образца, содержащего коллекторную группу тиономочевины, превышала на 32% массу минерала, собранного с помощью референсного полимера (Пример 10), полученного с использованием N,N-диаллилгексанамида вместо N.N-диаллилтиономочевины.
Пример 9. Сбор обогащенной халькопиритом руды с использованием сополимера, состоящего из поли(2-{2-[2-(2-этилэтоксиксантогенформиат)этокси]этокси}этил-N,N-диаллилкарбамат-со-N,N,N',N'-тетрааллилэтандиамида)
Синтез мономерного 2-{2-[2-(2-этилэтоксиксантогенформиат)этокси]этокси}этил-N,N-диаллилкарбамата
Триэтиленгликольбисхлорформат (97%, Alfa-Aesar, 275,08 г), безводный тетрагидрофуран (43,5 г) и триэтиламин (101,2 г) смешивали при температуре 25°С при непрерывном перемешивании. К перемешанной смеси добавляли по каплям диаллиламин (97,16 г) в течение 30 минут таким образом, чтобы экзотермический эффект не приводил к повышению температуры выше 30°С, при этом реакции позволяли протекать в течение дополнительного часа. Затем в реакционную смесь наливали этилксантогенформиат калия (96%, Aldrich, 160,3 г) в течение 15 минут и реакционную смесь поддерживали при температуре 25°С в течение 1 часа при непрерывном перемешивании. Температуру повышали до 50°С и поддерживали при указанном значении в течение еще одного часа. После охлаждения смесь фильтровали, затем промывали 2×100 мл воды. Остаточную воду удаляли с помощью безводного MgSO4 перед повторным фильтрованием, после которого образец дополнительно очищали путем удаления кристаллических остатков. Растворитель затем удаляли с использованием ротационного испарителя.
1Н ЯМР (CDCl3) δ/ppm=1,1 (t), 1,3(weak, t), 1,4(weak, m), 3,3(m), 3,55(m), 3,65(m), 3,75(m), 4,2(weak, m), 4,3(s), 4,7(s), 5,2(m), 5,8(m)
N,N,N',N'-тетрааллилэтандиамид синтезировали в соответствии с Примером 6.
Содержащий ксантогенформиат мономер (2-{2-[2-(2-этилэтоксиксантогенформиат)этокси]этокси}этил-N,N-диаллилкарбамат и кросс-линкер N,N,N',N'-тетрааллилэтандиамид вместе добавляли в виде смеси 1:1 (по массе), также добавляли фотоинициатор Irgacure 2022 в отношении 3,5% по массе от общей массы смеси мономеров. Полученную смесь перемешивали и наносили на плоский кусок поли(карбоната) размером ~10 см × 15 см с помощью поролонового валика до тех пор, пока не получали равномерное покрытие массой примерно 3 г/м2. Образец пропускали под фокусированной УФ-лампой высокой интенсивности (FusionUV LH6, D-колба, 100% интенсивность, 5 циклов при скорости 3,5 м/мин).
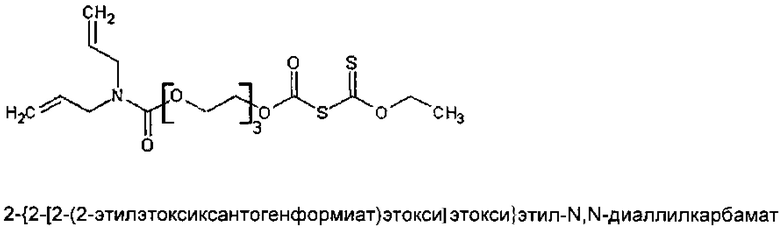
Панель с покрытием помещали в горизонтальный испытательный прибор, который мог подвергать образец взаимодействию с суспензией на площади ~112 см2, с глубиной 2,0 см. Рудное тело, содержащее халькопирит в качестве основного компонента (42% по массе), с остаточным материалом, представляющим собой главным образом смесь сульфидов железа (пирротин, 20% по массе, пирит, 16% по массе), измельчали на шаровой мельнице до получения фракции с размером частиц менее 106 мкм (распределение размера частиц: D10[5,68 мкм] D50[37,29 мкм], D90[106,9 мкм]). 2,0 г указанного минерального порошка добавляли к 200 мл деионизированной воды с получением суспензии, которую тщательно размешивали перед добавлением в испытательный прибор, который содержал панель образцов. Указанный испытательный прибор оставляли в неподвижном состоянии на 20 минут, после чего избыточный минерал выливали и минерал, присоединенный к поверхности полимера, собирали посредством отфильтровывания из шлиха. Собранный минерал тщательно высушивали и взвешивали. Указанный эксперимент повторяли несколько раз, и среднее значение массы минерала, собранного на единицу площади поверхности полимера, сравнивали со значением для референсного полимера, в котором ксантогенформиатная группа была замещена на алкильную группу.
Масса минерала, собранного с помощью образца, содержащего ксантогенформиатную группу-коллектор, превышала на 139% массу минерала, собранного с помощью референсного полимера, полученного с использованием N,N-диаллилгексанамида (Пример 10) вместо ксантогенформиат-модифицированного мономера.
Пример 10. Сбор обогащенной халькопиритом руды с использованием сополимера, состоящего из поли(N,N-диаллилгексанамид-со-N,N,N',N'-тетрааллилэтандиамида) в качестве референсного образца
N,N-диаллилгексанамид и N,N,N',N'-тетрааллилэтандиамидный кросс-линкер добавляли вместе в виде смеси 1:1 (по массе), добавляли фотоинициатор Irgacure 2022 в отношении 3,5% по массе от общей массы смеси мономера. Полученную смесь тщательно перемешивали и наносили на плоский кусок поли(карбоната) размером ~10 см × 15 см с помощью поролонового валика до тех пор, пока не получали равномерное покрытие массой примерно 3 г/м2. Образец пропускали под фокусированной УФ-лампой высокой интенсивности (FusionUV LH6, D-колба, 100% интенсивность, 4 цикла при скорости 3,5 м/мин).
Панель с покрытием помещали в горизонтальный испытательный прибор, который мог подвергать образец взаимодействию с суспензией по площади ~112 см2, с глубиной 2,0 см. Рудное тело, содержащее халькопирит в качестве основного компонента (42% по массе), с остаточным материалом, представляющим собой главным образом смесь сульфидов железа (пирротин, 20% по массе, пирит, 16% по массе), измельчали на шаровой мельнице до получения фракции с размером частиц менее 106 мкм (распределение размера частиц: D10[5,68 мкм] D50[37,29 мкм], D90[106,9 мкм]). 2,0 г указанного минерального порошка добавляли к 200 мл деионизированной воды с получением суспензии, которую тщательно размешивали перед добавлением в испытательный прибор, который содержал панель образцов. Указанный испытательный прибор оставляли в неподвижном состоянии на 20 минут, после чего избыточный минерал выливали и минерал, присоединенный к поверхности полимера, собирали посредством отфильтровывания из шлиха. Собранный минерал тщательно высушивали и взвешивали. Указанный эксперимент повторяли несколько раз, с усреднением по массе, собранной на единицу площади поверхности полимера (20,4 г/м2).
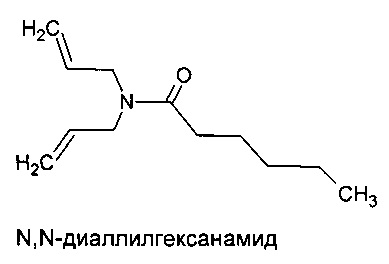
Синтез N,N-диаллилгексанамида
Диаллиламин (99%, 37,0 г), триэтиламин (99%, 40,0 г) и дихлорметан (99+%, 50 мл) смешивали и добавляли по каплям к охлажденной (0°С) смеси гексаноилхлорида (99%+, 50,0 г) в дихлорметане (99+%, 200 мл). Температуру поддерживали между 0 и 10°С при непрерывном перемешивании в течение нескольких часов для возможности добавления всей порции смеси диаллиламина. Реакционную смесь затем оставляли нагреваться до комнатной температуры.
Реакционную смесь затем промывали в разбавленном растворе HCl (3М, 500 мл) и отделяли органический слой. Органический слой повторно промывали водой или слабым солевым раствором, с последующим высушиванием органического слоя с помощью безводного сульфата магния. После этого удаляли в вакууме дихлорметан и другие летучие вещества с получением бледно-желтой жидкости, которую затем дополнительно очищали с помощью колоночной хроматографии на силикагеле (60Å) с использованием дихлорметана в качестве элюента с получением почти бесцветного масла. Выход ~70%.
1Н ЯМР (CDCl3) δ/ppm: 0,85(t), 1,25(m), 1,6(m), 2,25(t), 3.8(d), 3.9(d), 5,1 (m), 5,7(m)
Пример 11. Сбор обогащенной халькопиритом руды с использованием сополимера, состоящего из тозилата поли(N,N,N',N'-тетрааллилпропан-1,3-диметиламмония-со-тозилата N,N-диаллилбутанметиламмония) и коллектора O,O-диэтилтиофосфата (O,O-диэтилтиофосфата калия), инкапсулированного полимером
Синтез тозилата N,N-диаллилбутанметиламмония
(i) Получение N,N-диаллилбутан-1-аминного промежуточного соединения
В круглодонную колбу, снабженную термометром, конденсатором и магнитной мешалкой, наливали диаллиламин (563,9 г, 5,8 моль) и деионизированную воду (875 мл). Постепенно добавляли по каплям н-бутилбромид (194,3 г, 1,4 моль). Реакционную смесь нагревали до 60°С и поддерживали при указанной температуре в течение 24 часов. Указанную реакционную смесь охлаждали до 40°С и медленно наливали гидроксид калия (188 г, раствор 50 масс.%, 3,3 моль). Прекращали перемешивать и реакционной смеси позволяли осаждаться с образованием слоев. Верхний слой удаляли. Нижний слой экстрагировали дихлорметаном (ДХМ, 3×400 мл). Объединенные экстракты ДХМ очищали в виде фракции, при этом вторая фракция представляла собой фракцию неочищенного продукта. Неочищенный продукт дистиллировали (Toil=50°С до 87°С, ~30 мбар) с получением прозрачного масла (165,6 г, 76%).
Фурье-ИК-спектроскопия (пленка): 3078, 1643, 995, 917 см-1.
1Н ЯМР (CDCl3): 50,85 (m, 1,1Н, imp), 0,95 (t, 3,2Н), 1,25 (m, 2,8Н), 1,45 (m, 2,2Н), 1,65 (br, 2,2Н), 2,4 (m, 2Н), 3,1 (d, 4Н), 3,25 (m, 0,3H, imp), 5,1 (m, 4,2H), 6,85 (m, 2,1 H).
(ii) Получение продукта
N,N-диаллилбутан-1-амин (162,7 г, 1,06 моль) и толуол (732 мл) наливали в реактор, снабженный механической мешалкой, термометром, конденсатором и входным отверстием для азота. Смесь нагревали до температуры обратной конденсации. В реактор постепенно наливали сульфонат метил-пара-толуола (186 г, 1 моль) в течение 1 часа 20 минут. Еще через 2 часа нагревания при температуре обратной конденсации смесь охлаждали до комнатной температуры. Реакционную смесь наливали в разделительную воронку и слой неочищенного продукта сливали. Неочищенный продукт постепенно очищали в вакууме (~30 мбар), постепенно повышая температуру масляной бани до 150°С. Неочищенный продукт поддерживали в указанных условиях в течение 3,5 часов, затем охлаждали до комнатной температуры при продувании азотом. Получали вязкое золотисто-коричневое масло (293 г, 86%).
Фурье-ИК-спектроскопия (пленка): 3700-3100 (br), 3088, 3029, 2964, 2875, 1644, 1478, 1215, 1191, 1122, 1035, 1012, 683 см-1.
1Н ЯМР (CDCl3): 50,85 (t, 2,7Н), 1,25 (m, 1,8Н), 1,65 (m, 1,8Н), 2,3 (s, 3,1Н), 2,45 (br, 0,9Н), 2,9 (m, 0,2Н, imp), 3,1 (2s, 3Н), 3,2 (m, 1,6Н), 3,65 (m, 0,4Н, imp), 4,0 (m, 3,3Н), 4,05 (m, 0,3Н), 5,45 (m, 0,4Н), 5,6 (2d, 3,6Н), 5,85 (m, 1,7Н), 6,0 (m, 0,3Н), 7,1 (t, 2Н), 7,75 (t, 2Н), 10,15 (m, 0,07Н, imp).
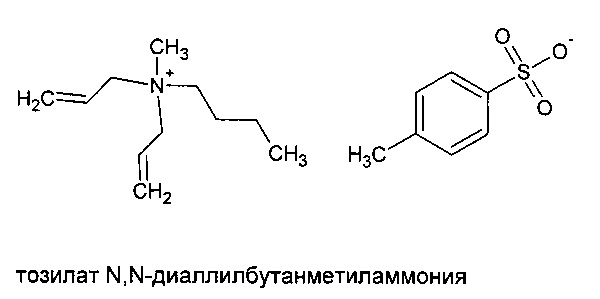
Мономерный тозилат N,N,N',N'-тетрааллилпропан-1,3-диметиламмония синтезировали в соответствии со способом, описанным в Примере 7. Подробности синтеза также можно найти в более ранней международной публикации заявителя WO 2009/063211.
Смесь, содержащая мономеры тозилата N,N,N',N'-тетрааллилпропан-1,3-диметиламмония (14,037 г) и тозилата N,N-диаллилбутанметиламмония (21,070 г) с O,O-диэтилтиофосфатом калия (0,848 г) и деионизированной водой (0,889 г), нагревали до 80°С в течение нескольких часов при обработке ультразвуком, способствующей растворению О,О-диэтилтиофосфата калия. Образец охлаждали и добавляли фотоинициатор Irgacure 2022 (0,732 г), при этом указанный образец снова нагревали и смешивали таким же образом, с получением вязкой жидкости, которую наносили на поликарбонатную панель (10 см × 15 см, 2 мм толщиной) в виде однородного слоя толщиной 1-2 мм на площадь 8 см × 8 см. Указанный слой отверждали путем пропускания под УФ-лампой высокой интенсивности 3 раза при скорости 2,0 м/мин (Fusion UV LH6, D-колба, 100% мощность) с получением твердой пленки.
Панель с покрытием помещали в горизонтальный испытательный прибор, который мог содержать суспензию в объеме, соответствующем площади 8 см × 8 см и глубине 1,0 см. Рудное тело, содержащее халькопирит в качестве основного компонента (42% по массе), с остаточным материалом, представляющим собой главным образом смесь сульфидов железа (пирротин, 20% по массе, пирит, 16% по массе), измельчали на шаровой мельнице до получения фракции с размером частиц менее 106 мкм (распределение размера частиц: D10[5,68 мкм] D50[37,29 мкм], D90[106,9 мкм]). 0,3 г указанного минерального порошка добавляли к 30 мл деионизированной воды с получением суспензии, которую тщательно размешивали перед добавлением в испытательный прибор, который содержал панель образцов. Указанный испытательный прибор оставляли в неподвижном состоянии на 20 минут, после чего избыточный минерал выливали и минерал, присоединенный к поверхности полимера, собирали посредством отфильтровывания из шлиха. Собранный минерал тщательно высушивали и взвешивали. Указанный эксперимент повторяли несколько раз, и среднее значение массы минерала, собранного на единицу площади поверхности полимера, сравнивали со значением для референсного полимера, который не содержал O,O-диэтилтиофосфат калия.
Референсный полимер
Образец получали таким же образом, как вышеуказанную панель образцов, за исключением того, что не добавляли O,O-диэтилтиофосфат калия. Указанную панель также анализировали таким же образом, как образцы с О,О-диэтилтиофосфатом калия.
Масса минерала, собранного с помощью образца, содержащего материал-коллектор O,O-диэтилтиофосфат калия, превышала на 24% массу минерала, собранного с помощью референсного полимера.
Пример 12. Сбор обогащенной халькопиритом руды с использованием сополимера, состоящего из поли(тозилата N,N,N',N'-тетрааллилпропан-1,3-диметиламмония-со-тозилата N,N-диаллилбутанметиламмония-со-O,O-диэтилтиофосфат 1,1-диаллилпиперидиния)
Синтез О,О-диэтилтиофосфата 1,1-диаллилпиперидиния
(i) Синтез промежуточного соединения, представляющего собой бромид N,N-диаллилпиперидина
Смесь карбоната натрия (103,66 г), изопропанола (78,50 г) и аллилбромида (133,08 г) наливали в колбу и оставляли перемешиваться при комнатной температуре. Добавляли по каплям пиперидин (42,58 г) в течение 1 часа при непрерывном перемешивании, при этом наблюдалась немедленная экзотермическая реакция. Температуру поддерживали ниже 50°С, при этом наблюдали эпизодические подъемы температуры до 60°С сразу после добавления пиперидина. Реакционную смесь затем доводили до температуры обратной конденсации и поддерживали в течение 24 часов при непрерывном перемешивании. Смесь затем оставляли охлаждаться до примерно 50°С для доведения до необходимого состояния. Теплую реакционную смесь фильтровали для удаления карбоната натрия и осажденной соли, образовавшейся в ходе реакции. Твердые вещества промывали в дихлорметане для удаления остаточного продукта и добавляли к фильтрованному продукту реакции. Для удаления растворителя и летучих веществ проводили ротационное выпаривание до тех пор, пока не оставалось мягкое твердое вещество янтарно-желтого цвета. Затем добавляли толуол (300 мл) для промывки продукта, который затем фильтровали в вакууме с последующей повторной промывкой толуолом до тех пор, пока жидкая фракция толуола не становилась прозрачной. Промывали ацетоном с получением порошка не совсем белого цвета, который затем сушили при 60°С.
Выход 60,4%
1Н ЯМР (CDCl3) δ: 1,8(m), 1,9(m), 3,7(m), 4,25(m), 5,75(m), 5,95(m)
(ii) Получение продукта
Соль O,O-диэтилтиофосфат натрия (10,0 г, 0,048 моль) и метанол (150 мл) наливали в высушенную над огнем колбу. В отдельной высушенной над огнем колбе растворяли бромид 1,1-диаллилпиперидиния (11,8 г, 0,048 моль) в метаноле (30 мл), полученный раствор наливали в первую колбу, промывая метанолом (20 мл). Реакционную смесь нагревали до температуры обратной конденсации и поддерживали при указанной температуре в течение 24 часов и затем охлаждали до комнатной температуры. Растворитель удаляли в вакууме. Оставшуюся суспензию растворяли в хлороформе (60 мл) и твердые вещества удаляли путем декантирования раствора хлороформа. Добавляли дополнительную порцию хлороформа (~20 мл). Раствор хлороформа промывали деионизированной водой (5 мл). Слои разделяли и слой хлороформа промывали дополнительной порцией деионизированной воды (5 мл). Хлороформ удаляли в вакууме с получением чистого желтого масла (14,4 г, 89%). Фурье-ИК-спектроскопия (пленка): 3406, 3085, 1642, 1469, 1165, 1042, 937 см-1.
1Н ЯМР (CDCl3) δ: 1,2 (t, 5,8Н), 1,75 (m, 2Н), 2,7 (br, 1,7Н, imp), 3,6 (t, 4Н), 3,95 (m, 3,8Н), 4,1 (t, 4,0Н), 5,65 (d, 2Н), 5,75 (d, 2,0Н) 5,95 (m, 2Н) ppm.
Синтез тозилата N,N,N',N'-тетрааллилпропан-1,3-диметиламмония и тозилата N,N-диаллилбутанметиламмония описан в Примере 11.
Тозилат N,N,N',N'-тетрааллилпропан-1,3-диметиламмония (5,00 г) нагревали до достижения его плавления и смешивали с тозилатом N,N-диаллилбутанметиламмония (2,50 г) и повторно нагревали до 80°C с периодическим перемешиванием на ультразвуковой бане. Затем к смеси добавляли O,O-диэтилтиофосфат 1,1-диаллилпиперидиния (2,50 г) и полученную смесь поддерживали при температуре 80°С в течение одного часа до полного растворения и диспергировали с периодической обработкой на ультразвуковой бане. Затем добавляли Irgacure 2022 в отношении 2% по массе от общего содержания мономеров с получением вязкой жидкости, которую наносили на поликарбонатную панель (10 см × 15 см, толщиной 2 мм) в виде равномерного слоя толщиной 1-2 мм на площадь 8 см × 8 см. Указанный слой отверждали путем пропускания под УФ-лампой высокой интенсивности 2 раза при скорости 3,0 м/мин (Fusion UV LH6, D-колба, 100% интенсивность) с получением твердой пленки.
Панель с покрытием помещали в горизонтальный испытательный прибор, который мог содержать суспензию в объеме, соответствующем площади 8 см × 8 см, глубине 1,0 см. Рудное тело, содержащее халькопирит в качестве основного компонента (42% по массе), с остаточным материалом, представляющим собой смесь других минералов, содержащих в основном сульфиды железа (пирротин, 20% по массе, пирит, 16% по массе), измельчали на шаровой мельнице до получения фракции с размером частиц менее 106 мкм. (Распределение: D10[5,68 мкм] D50[37,29 мкм], D90[106,9 мкм])). 0,3 г указанного минерального порошка добавляли к 30 мл деионизированной воды с получением суспензии, которую тщательно размешивали перед добавлением в испытательный прибор, который содержал панель образцов. Указанный испытательный прибор оставляли неподвижным на 20 минут, после чего избыточный минерал выливали и минерал, присоединенный к поверхности полимера, собирали посредством отфильтровывания из шлиха собранного минерала и воды. Собранный минерал тщательно высушивали и взвешивали. Указанный эксперимент повторяли несколько раз, и среднее значение массы минерала, собранного на единицу площади поверхности полимера, сравнивали со значением для референсного полимера, который не содержал O,O-диэтилтиофосфат.
Референсная панель
Образец готовили таким же образом как полимер, содержащий тиофосфатную единицу, но O,O-диэтилтиофосфат 1,1-диаллилпиперидиния везде замещали на тозилат N,N-диаллилбутанметиламмония, с получением сополимера поли(тозилата N,N,N',N'-тетрааллилпропан-1,3-диметиламмония-со-тозилта N,N-диаллилбутанметиламмония). Указанную панель также анализировали таким же образом, как образцы с O,O-диэтилтиофосфатом.
Масса минерала, собранного с помощью образца, содержащего коллекторный материал O,O-диэтилтиофосфат, превышала на 14% массу минерала, собранного с помощью референсного полимера.
Пример 13. Сбор обогащенной халькопиритом руды с помощью поверхности полимера, состоящего из функционализированного поли(этиленимина), привитого к поверхности поли(глицидилметакрилата-со-этиленгликольдиметакрилата)
Панель на основе нейлона 6,6 (размером 10 см × 15 см) покрывали тонким слоем смеси, состоящей из глицидилметакрилата (97%, Aldrich, 0,81 г), этиленгликольдиметакрилатного кросс-линкера (98%, Alfa Aesar, 0,20 г) и фотоинициатора Irgacure 2022 (0,025 г), толщиной 2-3 мкм. Указанную смесь отверждали с использованием УФ-лампы высокой интенсивности (FusionUV LH6, D-колба, 100% интенсивность, 6 циклов при скорости 3,5 м/мин).
Поли(этиленимин) («ПЭИ», разветвленный, молекулярная масса 10,000, 99%, Alfa Aesar) наносили в чистом виде в виде тонкого равномерного покрытия поверх метакрилатного покрытия и затем оставляли при 80°С на 1 час. После этого избыток ПЭИ удаляли путем промывки промывочной водой и 2-пропанолом, осторожно протирая поверхность для удаления каких-либо остаточных веществ. После высушивания жесткая поверхность сохранялась, но была значительно более гидрофильной по сравнению с метакрилатным покрытием. С помощью Фурье-ИК-спектроскопии были показаны спектральные изменения, указывающие на добавление ПЭИ.
Для превращения доступных аминогрупп, присутствующих на присоединенных цепях полиэтиленимина (ПЭИ), в коллекторные группы тиономочевины однородное покрытие этоксикарбонилизотиоцианатном (ECITC) распределяли по поверхности панели и оставляли при комнатной температуре на 45 минут. Избыток ECITC вытирали с поверхности и указанную поверхность затем тщательно промывали в 2-пропаноле и сушили.
Панель с покрытием помещали в горизонтальный испытательный прибор, который мог подвергать образец взаимодействию с суспензией по площади ~112 см2, с глубиной 2,0 см. Рудное тело, содержащее халькопирит в качестве основного компонента (42% по массе), с остаточным материалом, представляющим собой главным образом смесь сульфидов железа (пирротин, 20% по массе, пирит, 16% по массе), измельчали на шаровой мельнице до получения фракции с размером частиц менее 106 мкм (распределение размера частиц: D10[5,68 мкм] D50[37,29 мкм], D90[106,9 мкм]). 2,0 г указанного минерального порошка добавляли к 200 мл деионизированной воды с получением суспензии, которую тщательно размешивали перед добавлением в испытательный прибор, который содержал панель образцов. Указанный испытательный прибор оставляли в неподвижном состоянии на 20 минут, после чего избыточный минерал выливали и минерал, присоединенный к поверхности полимера, собирали посредством отфильтровывания из шлиха. Собранный минерал тщательно высушивали и взвешивали. Указанный эксперимент повторяли несколько раз и показали, что среднее значение массы минерала, собранного на единицу площади поверхности полимера, превышало приблизительно на 110% массу минерала, собранного с помощью референсного полимера, полученного из поли(N,N-диаллилгексанамид-со-N,N,N',N'-тетрааллилэтандиамида).
Пример 14. Сбор обогащенной халькопиритом руды с использованием тиокарбамат-функционализированного метакрилатного полимера поли(O-этил-O-(3-метил-2-оксобут-3-ен-1-ил)имидотиодикарбоната)
2-Гидроксиэтилметакрилат (Aldrich, 14,9 г, 0,114 моль) и ТГФ (28 г) наливали в круглодонную колбу, снабженную магнитной мешалкой, конденсатором и впускным отверстием для азота. В указанную колбу наливали 4-метоксифенол (0,23 г, 0,0019 моль). Затем в указанную колбу постепенно наливали этоксикарбонилизотиоцианат (Alfa Aesar, 97% 15,5 г, 0,118 моль). Реакционную смесь нагревали при 62°С в течение 16 часов, затем нагревали при температуре обратной конденсации в течение 3 часов. Наливали дополнительную порцию 2-гидроксиэтилметакрилата (0,5 г, 0,004 моль) и продолжали нагревать при температуре обратной конденсации в течение 4 ч.
Порцию реакционной смеси (14,4 г) обрабатывали водой (80 мл) и гидроксидом натрия (0,07 г, 1,75 ммоль) при 60°С в течение 4 часов. К полученной реакционной смеси добавляли ДХМ (160 мл), затем слои разделяли и водный слой экстрагировали ДХМ (160 мл). Раствор ДХМ сушили (MgSO4), фильтровали и очищали. В результате получали 6,6 г масла (21%). Оставшуюся реакционную смесь (44,5 г) обрабатывали подобным образом водой (247 мл) и гидроксидом натрия (0,2 г). Полученную реакционную смесь экстрагировали ДХМ (2×250 мл), сушили (MgSO4) и очищали. К очищенному маслу добавляли толуол (2×50 мл) и в результате очистки полученной смеси получали мономерный O-этил-O-(3-метил-2-оксобут-3-ен-1-ил)имидотиодикарбонат в виде масла (23,6 г, в общем 30,2 г, 98%).
Фурье-ИК-спектроскопия (пленка): 3517, 3259, 2982, 1770, 1720, 1636, 1521, 1251, 1232, 1171, 1097, 948, 769 см-1.
1Н ЯМР (CDCl3): 1,25 (t, 3,1Н), 1,95 (s, 3Н), 4,2 (q, 1,9Н), 5,15 (m, 4,2Н), 4,3 (m, 0,3H, imp), 4,45 (t, 2,2Н), 2,25 (t, 1,9Н), 5,1 (s, 1Н), 6,15 (s, 1Н), 8,25 (br, 0,9Н).
МС (CH2Cl2): C10H15NO5S; требуется 261,0671; обнаружено 261,0666.
Смесь, содержащую указанный тиокарбамат-функционализированный метакрилатный мономер (0,747 г), этиленгликольдиметакрилат (Alfa-Aesar, 0,752 г) и фотоинициатор Irgacure 2022 (0,039 г), наносили в виде тонкой пленки на несколько поликарбонатных панелей (размером 10 см × 20 см, толщиной 2 мм) с помощью мягкого валика с массой покрытия, составляющей несколько грамм на квадратный метр. Панель с покрытием помещали в горизонтальный испытательный прибор, который мог подвергать образец взаимодействию с суспензией по площади ~15 см2, с глубиной 2,0 см. Рудное тело, содержащее халькопирит в качестве основного компонента (42%, по массе), с остаточным материалом, представляющим собой главным образом смесь сульфидов железа (пирротин, 20%, по массе, пирит, 16%, по массе), измельчали на шаровой мельнице до получения фракции по размеру частиц менее 106 мкм (распределение размера частиц: D10[5,68 мкм], D50[37,29 мкм], D90[106,9 мкм], D3,2[12,36 мкм], D4,3[46,75 мкм]). 2,0 г указанного минерального порошка добавляли к 200 мл деионизированной воды с получением суспензии, которую тщательно размешивали перед добавлением в испытательный прибор, который содержал панель образцов. Указанный испытательный прибор оставляли в неподвижном состоянии на 20 минут, после чего избыточный минерал выливали и минерал, присоединенный к поверхности полимера, собирали посредством отфильтровывания из шлиха. Собранный минерал тщательно высушивали и взвешивали. Указанный эксперимент повторяли несколько раз, и среднее значение массы минерала, собранного на единицу площади поверхности полимера, сравнивали со значением для референсного полимера, который не содержал тиокарбаматную группу.
Масса минерала, собранного с помощью образца, содержащего тиокарбаматную группу-коллектор, составляла 4,18 мг/см2, что превышало на 101% массу минерала, собранного с помощью референсного полимера, полученного из N,N-диаллилгексанамида и N,N-тетрааллилэтандиамида.
Пример 15. Сбор обогащенной халькопиритом руды с использованием функционализированного силанового полимера поли(этил{[3-(триэтоксисилан)пропил]карбамотиоил}карбамата), полученного с помощью «золь-гель» метода
Синтез мономера этил{[3-(триэтоксисилан)пропил]карбаматиол}карбамата
(3-Аминопропил)триэтоксисилан (Sigma-Aldrich, >98%, 23,9 г, 0,108 моль) наливали в высушенную над огнем круглодонную колбу, снабженную магнитной мешалкой, конденсатором и входным отверстием для азота. В указанную колбу наливали 4-метоксифенол (0,23 г, 0,0019 моль). Затем в указанную колбу постепенно наливали этоксикарбонилизотиоцианат (Alfa Aesar, >97%,13,8 г, 0,105 моль). Реакционную смесь нагревали при 45-60°С в течение 5 часов. В результате получали продукт, представляющий собой прозрачное желтое масло (32,8 г, 87%), который имел чистоту 94% по результатам ЯМР-анализа.
Фурье-ИК-спектроскопия (пленка): 3289, 2975, 2927, 2885, 1713, 1547, 1245, 1097, 994, 948, 768 см-1.
1Н ЯМР (CDCl3): 0,15 (m, 2Н), 1,2 (t, 9Н), 1,3 (t, 2,8Н), 1,5 (m, 0,1 Н, sm), 1,75 (quin, 1,9Н), 2,7 (m, 0,1 Н, sm), 3,65 (t, 1,8H), 3,7 (q, 0,3H, sm), 3,8 (q, 5,7H), 4,2 (q, 1,9H), 9,7 (br, 0,9H).
Этил{[3-(триэтоксисилан)пропил]карбамотиоил}карбамат (0,76 г), уксусную кислоту (рН 3,0) (1,01 г) и изопропанол (2,0 г) смешивали вместе и нагревали до 50°С на масляной бане в течение 6 часов при непрерывном перемешивании. Раствор охлаждали до комнатной температуры и оставляли на 24 часа. Затем полученную смесь распределяли в виде слоя толщиной ~1 мм по всей поверхности поли(карбонатной) пластины размером 10 см × 20 см, толщиной 2 мм. Указанную пластину помещали в плоскодонный стеклянный сосуд, указанный сосуд плотно закрывали путем размещения на его верхней части стеклянной крышки и помещали в печь при температуре 50°С еще на 6 часов. Затем образец охлаждали и оставляли при комнатной температуре еще на 18 часов при неполностью закрытой крышке. Образец повторно нагревали до 50°С также в неполностью закрытой емкости в течение дополнительных 6 часов и затем оставляли охлаждаться до комнатной температуры и хранили при указанной температуре в течение 5 дней. Затем указанный образец помещали в стеклянную емкость без крышки еще на 3 часа при 50°С и оставляли охлаждаться с получением твердого бесцветного покрытия.
Панель с покрытием помещали в горизонтальный испытательный прибор, который мог подвергать образец взаимодействию с суспензией по площади ~15 см2, глубиной 2,0 см. Рудное тело, содержащее халькопирит в качестве основного компонента (42% по массе), с остаточным материалом, представляющим собой главным образом смесь сульфидов железа (пирротин, 20% по массе, пирит, 16% по массе), измельчали на шаровой мельнице до получения фракции с размером частиц менее 106 мкм (распределение размера частиц: D10[5,68 мкм] D50[37,29 мкм], D90[106,9 мкм]). 2,0 г указанного минерального порошка добавляли к 200 мл деионизированной воды с получением суспензии, которую тщательно размешивали перед добавлением в испытательный прибор, который содержал панель образцов. Указанный испытательный прибор оставляли в неподвижном состоянии на 20 минут, после чего избыточный минерал выливали и минерал, присоединенный к поверхности полимера, собирали посредством отфильтровывания из шлиха. Собранный минерал тщательно высушивали и взвешивали. Указанный эксперимент повторяли несколько раз, и среднее значение массы, собранной на единицу площади поверхности полимера, сравнивали с массой для референсного полимера, который не содержал группу тиономочевины (см. референсный образец).
Масса минерала, собранного с помощью образца, содержащего коллекторную группу тиономочевины, превышала более чем в два раза массу минерала, собранного с помощью референсного полимера, полученного при использовании N,N-диаллилгексанамида вместо N,N,N',N'-тетрааллилэтандиамида.
Пример 16. Сбор сульфида кобальта (CoS) с использованием сополимера, состоящего из поли(N,N-диаллилэтоксикарбонилтиономочевины-со-N,N,N',N'-тетрааллилэтандиамида)
Получали панель с покрытием и помещали в горизонтальный испытательный прибор в соответствии с Примером 8. 2,0 г сульфида кобальта (CoS) со средним размером частиц ~150 мкм (-100 мешей) добавляли к 200 мл деионизированной воды с получением суспензии, которую тщательно размешивали перед добавлением в испытательный прибор, который содержал панель образцов. Указанный испытательный прибор оставляли в неподвижном состоянии на 20 минут, после чего избыточный минерал выливали и минерал, присоединенный к поверхности полимера, собирали посредством отфильтровывания из шлиха. Собранный минерал тщательно высушивали и взвешивали. Указанный эксперимент повторяли несколько раз, с усреднением по массе дисульфида кобальта, собранного на единицу площади поверхности полимера. Полученные данные сравнивали с массой дисульфида кобальта, собранного с помощью референсного полимера, который получали с использованием такого же способа и условий эксперимента, но с использованием N,N-диаллилгексанамида вместо мономера N,N-диаллилтиономочевины.
Масса сульфида кобальта, собранного с помощью образца, содержащего коллекторную группу тиономочевины, превышала на 65% массу сульфида кобальта, собранного с помощью референсного полимера.
Пример 17. Сбор минерала, содержащего дисульфид железа (пирит), с использованием содержащего ксантогенформиат сополимера, поли((2-(2-(2-(2-этилэтоксиксантогенформиат)этокси)этокси)этил-N,N-диаллилкарбамата-со-N,N,N',N'-тетрааллилэтандиамида)
Получали панель с покрытием и помещали в горизонтальный испытательный прибор согласно Примеру 9. 2,0 г дисульфида железа с размером частиц менее 106 мкм добавляли к 200 мл деионизированной воды с получением суспензии, которую тщательно размешивали перед добавлением в испытательный прибор, который содержал панель образцов. Указанный испытательный прибор оставляли в неподвижном состоянии на 20 минут, после чего избыточный минерал выливали и минерал, присоединенный к поверхности полимера, собирали посредством отфильтровывания из шлиха. Собранный минерал тщательно высушивали и взвешивали. С помощью образца было собрано 1,85 мг пирита железа на см2.
Пример 18. Сбор минерала халькопирита с использованием халькопирита, предварительно обработанного амин-функционализированным тионокарбаматным коллектором с последующим реагированием с целлюлозной поверхностью, модифицированной функциональной группой сложного тозилэфира
Описание
В настоящем эксперименте использовали твердую поверхность с различными функциональными химическими группами, соединяющимися с частицей халькопирита, предварительно обработанной реактивным функционализированным коллектором. Таким образом, механизм реакции состоял из следующих этапов:
(1) Присоединение коллектора к халькопириту в растворе (как при флотации вспениванием)
(2) Присоединение присутствующего на халькопирите коллектора к активной группе на коллекторной твердой поверхности
В указанной схеме использовали коллектор, молекула которого на одном конце содержала тионокарбамат для связывания с халькопиритом, а на другом конце - амин для связывания с группой сложного тозилэфира на поверхности модифицированной целлюлозы. Обработку халькопирита коллектором осуществляли отдельно от присоединения минерала к твердой поверхности.
Эксперимент
Получение молекулы коллектора
В 3-горлую колбу на 50 мл наливали этоксикарбонилизотиоцианат (5,01 г) в дихлорметане (10 мл) и охлаждали до 10°С. Добавляли по каплям 2-диметиламиноэтанол (3,68 г) в дихлорметане (10 мл) в течение 10 минут при перемешивании. Реакционной смеси затем позволяли достигать комнатной температуры, после чего добавляли дополнительную порцию дихлорметана (30 мл), при этом продолжали перемешивать еще в течение 2 часов. Затем удаляли летучие вещества с использованием ротационного испарителя с получением вязкого желтого масла. Выход >90%.
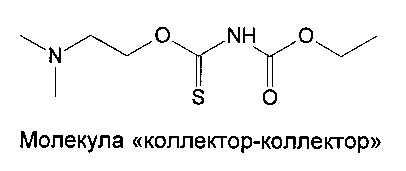
1Н ЯМР (500МГц, CDCl3) δ/ppm = 1,3 (t), 2,85 (s), 2,95 (s), 3,4 (t), 4,2 (q), 4,5 (t)
Получение халькопирита с коллектором
Образец измельченного халькопирита (приблизительно 20 г, ~16% Cu, <106 мкм) вводили в разбавленный раствор указанной выше амин-функционализированной молекулы-коллектора (~0,3 г) в деионизированной воде (200 мл). Полученную смесь нагревали до примерно 40°С и затем осторожно перемешивали в течение 30 минут. Халькопирит фильтровали и затем промывали 4 раза, при этом в ходе каждого этапа очистки указанный халькопирит отбирали и повторно добавляли к 200 мл воды при перемешивании. Обработанный халькопирит затем сушили при 60°C с получением порошка зеленого цвета, внешне сходного с минералом, который использовали изначально.
Получение поверхности модифицированной целлюлозы для улавливания комплекса халькопирит/коллектор
Смесь толуола (100 мл), пиридина (15 мл) и тозилхлорида (0,5 г) нагревали до примерно 80°С в плоскодонной стеклянной емкости. Целлюлозную фильтровальную бумагу (Whatman no. 2, диаметром примерно 8 см) сушили, затем помещали в полученную смесь и емкость плотно закрывали. Бумагу оставляли на 45 минут, при этом раствор периодически осторожно перемешивали.
После этого бумагу извлекали, промывали толуолом и затем тщательно промывали ацетоном для удаления всех остаточных веществ. Затем образец сушили при 55°С в течение 30 минут.
Обработка модифицированной целлюлозы комплексом халькопирит/коллектор
Обработанную целлюлозную фильтровальную бумагу затем помещали в смесь, содержащую 2,0 г обработанного халькопирита в 200 мл воды, помещенную в стеклянный стакан на 2 литра, при этом указанную смесь поддерживали в состоянии суспензии во время добавления бумаги. Бумагу помещали на дно стакана, при этом суспензии позволяли осаждаться на ее поверхность. Указанную смесь затем нагревали до 70-80°С в течение одного часа, после чего бумагу осторожно вынимали из указанной смеси таким образом, что тонкий слой минерала оставался присоединенным к ее поверхности.
Халькопирит, который оставался на фильтровальной бумаге, удаляли путем смывания с указанной бумаги водой и повторного фильтрования, затем полученный халькопирит тщательно сушили и исследовали с помощью рентгенофлуоресцентного анализа (РФА).
Описанный эксперимент повторяли, но при этом оставляли немого больше минерала на бумаге после ее извлечения из смеси.
Результаты
С помощью РФА показали, что среднее содержание Cu в извлеченном из модифицированной целлюлозы минерале составляло 18,03%. При повторном эксперименте с оставленным немного более толстым слоем собранного минерала получали значение 17,33% Cu, которое значительно превышало концентрацию меди в исходном материале первичного минерала, составляющую 16,16%.
Пример 19. Референсный эксперимент для процесса сбора минерала халькопирита на поверхность целлюлозы с использованием халькопирита, предварительно обработанного амин-функционализированным тионокарбаматным коллектором
Описание
Настоящий эксперимент представляет собой референсный анализ для сбора халькопирита на поверхность модифицированный целлюлозы. Эксперимент был идентичен эксперименту, в котором использовали амин-функционализированную тионокарбаматную группу-коллектор, за исключением того, что не проводили обработку поверхности целлюлозы таким образом, чтобы она содержала сложный тозилэфир.
Результаты
С помощью РФА показали, что среднее содержание меди в минерале, извлеченном из немодифицированной целлюлозной бумаги, составляло 15,94%. Результат был сходным с концентрацией меди в исходном материале первичного минерала, составляющей 16,17%.
Пример 20. Сбор халькопирита из смеси раздельно измельченного халькопирита и рудного тела с использованием сополимера, состоящего из поли(N,N-диаллилэтоксикарбонилтиономочевины-со-N,N,N',N'-тетрааллилэтандиамида)
Получали панель с покрытием и помещали в горизонтальный испытательный прибор в соответствии с Примером 8. Минерал использовали для получения суспензии, состоящей из смеси халькопирита (>80% по массе) и измельченного рудного тела, которое состояло главным образом из силикатов с содержанием халькопирита, составляющим приблизительно 1% по массе, в отношении халькопирит : рудное тело, составляющем 60:40 соответственно (распределение размера частиц: D10[5,93 мкм], D50[33,06 мкм], D90[104 мкм] D3,2[15,89 мкм], D4,3[44,55 мкм]). 2,0 г указанного минерального порошка добавляли к 200 мл деионизированной воды с получением суспензии, которую тщательно размешивали перед добавлением в испытательный прибор, который содержал панель образцов. Указанный испытательный прибор оставляли в неподвижном состоянии на 20 минут, после чего избыточный минерал выливали и минерал, присоединенный к поверхности полимера, собирали посредством отфильтровывания из шлиха. Собранный минерал тщательно высушивали и взвешивали. Исследование повторяли несколько раз, с усреднением по массе, собранной на единицу площади поверхности полимера.
С помощью рентгеновской флуоресцентной спектроскопии было показано, что содержание меди в минерале, собранном с помощью полимера, содержащего тиономочевину, было выше на 12,7% по сравнению с исходным материалом первичного минерала, с распределением размера частиц: D10[8,04 мкм], D50[45,03 мкм], D90[112,53 мкм], D3,2[20,45 мкм], D4,3[53,51 мкм].
Пример 21. Сбор халькопирита из смеси раздельно измельченного халькопирита и рудного тела с использованием сополимера, состоящего из поли(2-(2-(2-(2-этилэтоксиксантогенформиат)этокси)этокси)этил-N,N-диаллилкарбамата-со-N,N,N',N'-тетрааллилэтандиамида)
Получали панель с покрытием и помещали в горизонтальный испытательный прибор в соответствии с Примером 9. Минерал использовали для получения суспензии, состоящей из смеси халькопирита (чистота приблизительно 80%) и измельченного рудного тела, которое состояло главным образом из силикатов (только ~1% халькопирита по массе) с отношением халькопирит : рудное тело, составляющим 60:40 соответственно (распределение размера частиц: D10[5,61 мкм] D50[26,68 мкм], D90[96,38 мкм], D3,2[14,82 мкм], D4,3[46,83 мкм]). 2,0 г указанного минерального порошка добавляли к 200 мл деионизированной воды с получением суспензии, которую тщательно размешивали перед добавлением в испытательный прибор, который содержал панель образцов. Указанный испытательный прибор оставляли неподвижным на 20 минут, после чего избыточный минерал выливали и минерал, присоединенный к поверхности полимера, собирали посредством отфильтровывания из шлиха. Собранный минерал тщательно высушивали и взвешивали. Указанный эксперимент повторяли несколько раз, с усреднением по массе, собранной на единицу площади поверхности полимера.
С помощью рентгеновской флуоресцентной спектроскопии было показано, что содержание меди в минерале, собранном с помощью полимера, содержащего ксантогенформиатную группу, было выше на 16,5% по сравнению с исходным материалом первичного минерала, со следующим распределением размера частиц: D10[8,52 мкм] D50[46,50 мкм], D90[112,69 мкм], D3,2[21,36 мкм], D4,3[54,58 мкм].
| название | год | авторы | номер документа |
|---|---|---|---|
| АГЕНТЫ ДЛЯ ОПТИЧЕСКОЙ ВИЗУАЛИЗАЦИИ | 2008 |
|
RU2484111C9 |
| НОВЫЕ ПУТИ К ПОЛИАКРИЛАТАМ | 2011 |
|
RU2588569C2 |
| КОМПОЗИЦИЯ СВЯЗУЮЩЕГО ДЛЯ АГЛОМЕРАЦИИ МЕЛКОДИСПЕРСНЫХ МИНЕРАЛОВ И СПОСОБ ОКАТЫВАНИЯ С ЕЁ ИСПОЛЬЗОВАНИЕМ | 2012 |
|
RU2604546C2 |
| ЦИКЛИЧЕСКИЕ КАРБОНИЛЬНЫЕ СОЕДИНЕНИЯ С БОКОВЫМИ ПЕНТАФТОРФЕНИЛКАРБОНАТНЫМИ ГРУППАМИ, ИХ ПОЛУЧЕНИЕ И ПОЛУЧАЕМЫЕ ИЗ НИХ ПОЛИМЕРЫ | 2011 |
|
RU2572246C2 |
| ГЛИЦЕРИН-СВЯЗАННЫЕ ПЭГИЛИРОВАННЫЕ САХАРА И ГЛИКОПЕПТИДЫ | 2007 |
|
RU2460543C2 |
| МАТЕРИАЛЫ И СПОСОБЫ, СВЯЗАННЫЕ С ЛИНКЕРАМИ ДЛЯ ПРИМЕНЕНИЯ В КОНЪЮГАТАХ ЛЕКАРСТВЕННОГО СРЕДСТВА И БЕЛКА | 2015 |
|
RU2737553C2 |
| МНОГОФАЗНАЯ ДЕТЕРГЕНТНАЯ ТАБЛЕТКА, СПОСОБ ЕЕ ПОЛУЧЕНИЯ И СПОСОБ МЫТЬЯ В МОЕЧНОЙ МАШИНЕ | 1999 |
|
RU2205869C2 |
| НОВЫЕ ПОЛИМЕРНЫЕ ФОТОИНИЦИАТОРЫ | 2011 |
|
RU2584165C2 |
| НОВЫЕ АФФИННЫЕ ЛИГАНДЫ И ИХ ПРИМЕНЕНИЕ | 1996 |
|
RU2175261C2 |
| НОВЫЕ СОЕДИНЕНИЯ И КОМПОЗИЦИИ ДЛЯ ИНГИБИРОВАНИЯ FASN | 2014 |
|
RU2686323C2 |
Изобретение относится к способу переработки минеральной смеси, который заключается в том, что вначале готовят минеральную смесь, которая содержит металлосодержащий минерал и одну или более нежелательных пустых пород. Затем приводят в контакт минеральную смесь с полимерным материалом, содержащим минерал-связывающий фрагмент, который селективно связывается с металлосодержащим минералом. После этого разделяют пустую породу и полимерный материал, к которому присоединен металлосодержащий минерал. Все этапы способа проводят как часть флотационного процесса. Полимерный материал содержит частицы, выполненные с возможностью обеспечения их всплытия. Полимерный материал включает полимер, образованный путем полимеризации предшественника полимера формулы (I), в которой R1 представляет собой i) CRa, где Ra представляет собой водород или алкил, содержащий от 1 до 20 атомов углерода; ii) группу N+R13 (Zm)1/m, S(O)pR14 или SiR15, где R13 представляет собой водород, галоген, нитро или гидрокарбил, содержащий от 1 до 20 атомов углерода, необязательно содержащий в качестве заместителя или включающий в свою структуру функциональные группы, R14 и R15 независимо выбраны из водорода или гидрокарбила, содержащего от 1 до 20 атомов углерода, Z представляет собой анион, имеющий заряд m, и p равен 0, 1 или 2, iii) C(O)N, C(S)N, S(O)2N, C(O)ON, CH2ON или CH=CHRcN, где Rc представляет собой электроноакцепторную группу, или iv) ОС(O)СН, С(O)ОСН или S(O)2CH; где R12 выбран из водорода, галогена, нитро, гидрокарбила, содержащего от 1 до 20 атомов углерода, R2 выбран из (CR7R8)n или группы CR9R10, CR7R8CR9R10 или CR9R10CR7R8, где n равен 0, 1 или 2, R7 и R8 независимо выбраны из водорода или алкила, содержащего от 1 до 20 атомов углерода, и один из R9 или R10 представляет собой водород, а другой представляет собой электроноакцепторную группу, или R9 и R10 вместе образуют электроноакцепторную группу; R4 выбран из СН или CR11, где CR11 представляет собой электроноакцепторную группу, пунктирные линии обозначают присутствие или отсутствие связи, X1 представляет собой группу СХ2Х3, где обозначенная пунктирной линией связь, к которой присоединена указанная группа, отсутствует, и группу СХ2, где обозначенная пунктирной линией связь, к которой присоединена указанная группа, присутствует, X2, X3 независимо выбраны из водорода или фтора. Изобретение позволяет эффективно отделить металлосодержащий минерал от нежелательной пустой породы. 41 з.п. ф-лы, 21 пр.
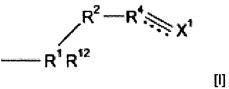
1. Способ переработки минеральной смеси, включающий следующие этапы:
(a) обеспечение минеральной смеси, которая содержит металлосодержащий минерал и одну или более нежелательных пустых пород;
(b) приведение указанной минеральной смеси в контакт с полимерным материалом, содержащим минерал-связывающий фрагмент, который селективно связывается с указанным металлосодержащим минералом; и
(c) разделение пустой породы и полимерного материала, к которому присоединен металлосодержащий минерал;
где указанный полимерный материал включает полимер, образованный путем полимеризации предшественника полимера, содержащего группу подформулы (I):
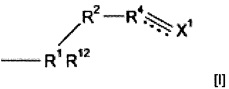
где R1 представляет собой i) CRa, где Ra представляет собой водород или алкил, содержащий от одного до двадцати атомов углерода; ii) группу N+R13 (Zm-)1/m, S(O)pR14 или SiR15, где R13 представляет собой водород, галоген, нитро или гидрокарбил, содержащий от одного до двадцати атомов углерода, необязательно содержащий в качестве заместителя или включающий в свою структуру функциональные группы, R14 и R15 независимо выбраны из водорода или гидрокарбила, содержащего от одного до двадцати атомов углерода, Z представляет собой анион, имеющий заряд m, и p равен 0, 1 или 2, iii) C(O)N, C(S)N, S(O)2N, C(O)ON, CH2ON или CH=CHRcN, где Rc представляет собой электроноакцепторную группу, или iv) ОС(O)СН, С(O)ОСН или S(O)2CH; где R12 выбран из водорода, галогена, нитро, гидрокарбила, содержащего от одного до двадцати атомов углерода, необязательно содержащего в качестве заместителя или включающего в свою структуру функциональные группы, или
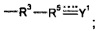
R2 и R3 независимо выбраны из (CR7R8)n или группы CR9R10, CR7R8CR9R10 или CR9R10CR7R8, где n равен 0, 1 или 2, R7 и R8 независимо выбраны из водорода или алкила, содержащего от одного до двадцати атомов углерода, и один из R9 или R10 представляет собой водород, а другой представляет собой электроноакцепторную группу, или R9 и R10 вместе образуют электроноакцепторную группу;
R4 и R5 независимо выбраны из СН или CR11, где CR11 представляет собой электроноакцепторную группу,
пунктирные линии обозначают присутствие или отсутствие связи, X1 представляет собой группу СХ2Х3, где обозначенная пунктирной линией связь, к которой присоединена указанная группа, отсутствует, и группу СХ2, где обозначенная пунктирной линией связь, к которой присоединена указанная группа, присутствует, Y1 представляет собой группу CY2Y3, где обозначенная пунктирной линией связь, к которой присоединена указанная группа, отсутствует, и группу CY2, где обозначенная пунктирной линией связь, к которой присоединена указанная группа, присутствует, и X2, X3, Y2 и Y3 независимо выбраны из водорода или фтора, и
при этом если этапы (а)-(с) проводят как часть флотационного процесса, указанный полимерный материал содержит частицы, выполненные с возможностью обеспечения их всплытия.
2. Способ по п. 1, отличающийся тем, что металлосодержащий минерал содержит медь.
3. Способ по п. 2, отличающийся тем, что металлосодержащий минерал представляет собой халькопирит или борнит.
4. Способ по п. 1, отличающийся тем, что металлосодержащий минерал содержит по меньшей мере один из следующих металлов: литий, цинк, железо, золото, серебро, молибден, кобальт, платина, уран, мышьяк, ртуть, кадмий, теллур и свинец.
5. Способ по любому из пп. 1-4, отличающийся тем, что минерал-связывающий фрагмент содержит по меньшей мере один атом серы.
6. Способ по п. 1, отличающийся тем, что полимерный материал включает полимер, инкапсулирующий указанный минерал-связывающий фрагмент.
7. Способ по п. 6, отличающийся тем, что минерал-связывающий фрагмент представляет собой химическое соединение-коллектор минералов.
8. Способ по п. 6, отличающийся тем, что минерал-связывающий фрагмент представляет собой тио-, сульфатное, сульфонатное или карбоксильное соединение или анион.
9. Способ по п. 8, отличающийся тем, что тиосоединение представляет собой ксантат, дитиофосфат, тиофосфат, дитиокарбамат; тионокарбамат, дитиофосфинат, тиофосфинат, ксантогенформиат, тиокарбанилид или тиоловое соединение или анион.
10. Способ по п. 1, отличающийся тем, что полимерный материал представляет собой полимерную структуру, содержащую повторяющиеся звенья, которые включают указанный минерал-связывающий фрагмент.
11. Способ по п. 10, отличающийся тем, что минерал-связывающий фрагмент содержит по меньшей мере одну функциональную группу, выбранную из амина, тиола, сложного эфира, краун-эфира, аза-краун-эфира, органической кислоты, порфирина, тиоциклоалкана, мочевины, тиомочевины, фталоцианина, тионокарбамата, тиофосфата или ксантогенформиата.
12. Способ по п. 1, отличающийся тем, что предшественник полимера представляет собой соединение, имеющее структуру (II)
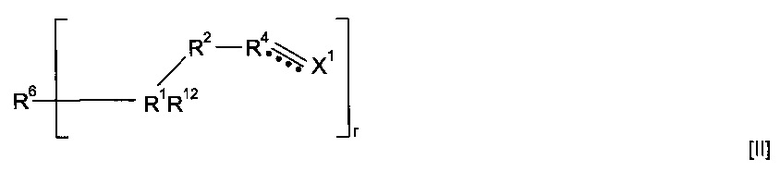
где r представляет собой целое число, составляющее 1 или более, R6 представляет собой одну или более мостиковых групп, необязательно замещенную гидрокарбильную группу, содержащую от одного до двадцати атомов углерода, пергалогеналкильную группу, содержащую от одного до двадцати атомов углерода, силоксановую группу, амид или частично полимеризованную цепь, содержащую повторяющиеся звенья.
13. Способ по п. 12, отличающийся тем, что предшественник полимера представляет собой соединение, имеющее структуру [III]
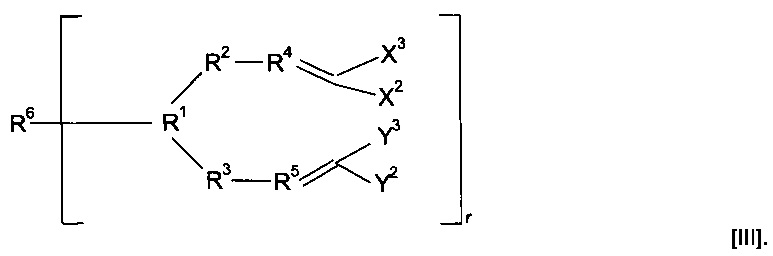
14. Способ по п. 12 или п. 13, отличающийся тем, что R6 содержит гидрокарбильную группу с прямой или разветвленной углеводородной цепью, содержащую от одного до двадцати атомов углерода, необязательно содержащую в качестве заместителя или включающую в свою структуру функциональные группы.
15. Способ по п. 14, отличающийся тем, что R6 содержит от двух до двадцати атомов углерода, предпочтительно от двух до двенадцати атомов углерода.
16. Способ по п. 14, отличающийся тем, что R6 содержит в качестве заместителя или включает в свою структуру указанный минерал-связывающий фрагмент.
17. Способ по п. 16, отличающийся тем, что R6 содержит в качестве заместителя или включает в свою структуру по меньшей мере одну функциональную группу, выбранную из амина, тиола, сложного эфира, краун-эфира, аза-краун-эфира, органической кислоты, порфирина, тиоциклоалкана, мочевины, тиомочевины, фталоцианина, тиофосфата, тионокарбамата или ксантогенформиата.
18. Способ по п. 1, отличающийся тем, что R1 представляет собой N+R13(Zm-)1/m.
19. Способ по п. 18, отличающийся тем, что Zm- представляет собой минерал-связывающий фрагмент.
20. Способ по п. 19, отличающийся тем, что Zm- представляет собой диалкилтиофосфат-анион, где алкильные группы содержат от одного до шести атомов углерода.
21. Способ по п. 19 или п. 20, отличающийся тем, что R13 и R6 вместе с кватернизированным атомом N, к которому они присоединены, образуют гетероциклическую структуру.
22. Способ по пп. 19-20, отличающийся тем, что предшественник полимера представляет собой мономер формулы [IV]
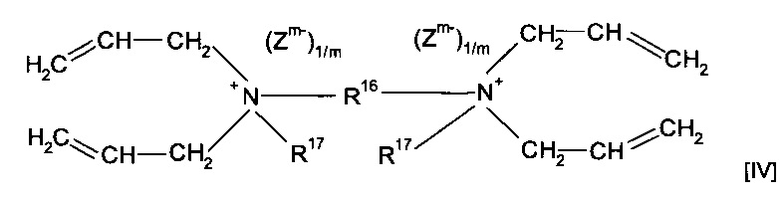
где R16 представляет собой алкильную группу с прямой или разветвленной углеводородной цепью, предпочтительно содержащую от одного до двадцати атомов углерода, наиболее предпочтительно от двух до двенадцати атомов углерода;
и R17 представляет собой водород или алкильную группу с прямой или разветвленной углеводородной цепью, предпочтительно содержащую от одного до пяти атомов углерода, наиболее предпочтительно метил или этил;
или форполимер, полученный посредством форполимеризации указанного мономера.
23. Способ по п. 22, отличающийся тем, что предшественник полимера представляет собой мономер формулы [V]
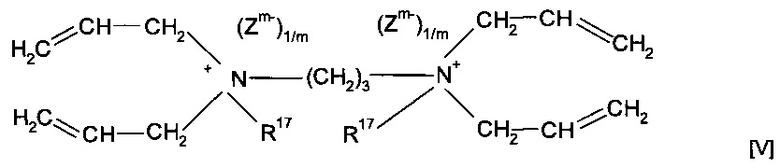
где R17 предпочтительно представляет собой метил, или форполимер, полученный посредством форполимеризации указанного мономера.
24. Способ по пп. 12-13 или 15-17, отличающийся тем, что указанный предшественник полимера представляет собой соединение, имеющее структуру [X]
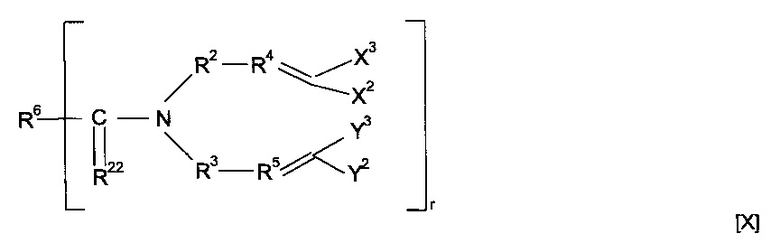
где R22 представляет собой О или S, и R6 содержит минерал-связывающий фрагмент или вместе с C=R22 образует минерал-связывающий фрагмент.
25. Способ по п. 24, отличающийся тем, что минерал-связывающий фрагмент представляет собой функциональную группу, содержащую тионокарбамат, тиомочевину, тиол, тиоциклоалкан, тиофосфат или ксантогенформиат.
26. Способ по п. 25, отличающийся тем, что предшественник полимера представляет собой соединение, имеющее структуру [XI]
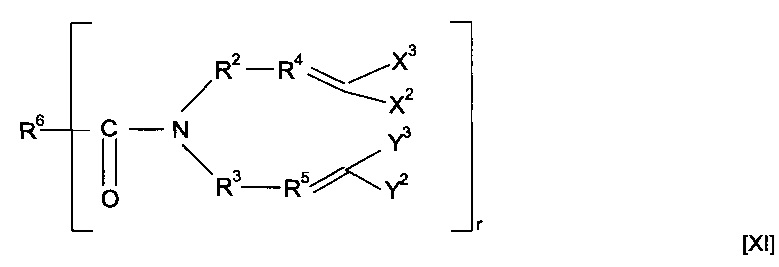
где R6 содержит группу -NHC(S)O-, -C(O)NHC(S)O- или -O-C(S)SC(O)O-.
27. Способ по п. 26, отличающийся тем, что предшественник полимера представляет собой соединение, имеющее структуру [XII]
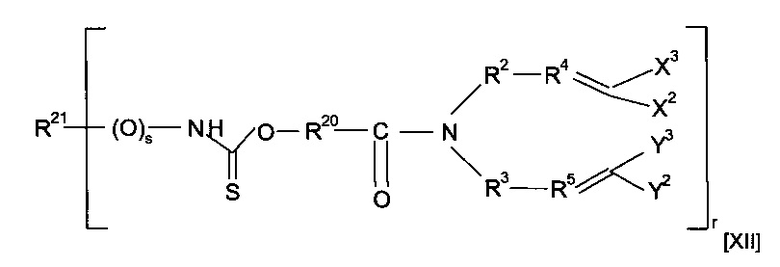
где каждый из R20 и R21 независимо представляет собой алкильную группу, необязательно содержащую в качестве заместителя или включающую в свою структуру функциональные группы, предпочтительно содержащую от одного до двадцати атомов углерода, наиболее предпочтительно от двух до двенадцати атомов углерода, s равен 0 или 1, и r предпочтительно равен 1 или 2; или форполимер, полученный посредством форполимеризации указанного соединения.
28. Способ по п. 26, отличающийся тем, что предшественник полимера представляет собой соединение, имеющее структуру [XIII]
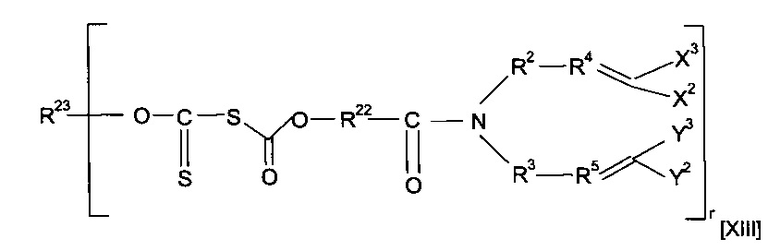
где каждый из R22 и R23 независимо представляет собой алкильную группу, необязательно содержащую в качестве заместителя или включающую в свою структуру функциональные группы, предпочтительно содержащую атом О, и предпочтительно содержащую от одного до двадцати атомов углерода, наиболее предпочтительно от двух до двенадцати атомов углерода, и r предпочтительно равен 1 или 2, или форполимер, полученный посредством форполимеризации указанного соединения.
29. Способ по п. 24, отличающийся тем, что предшественник полимера представляет собой соединение, имеющее структуру [XIV]
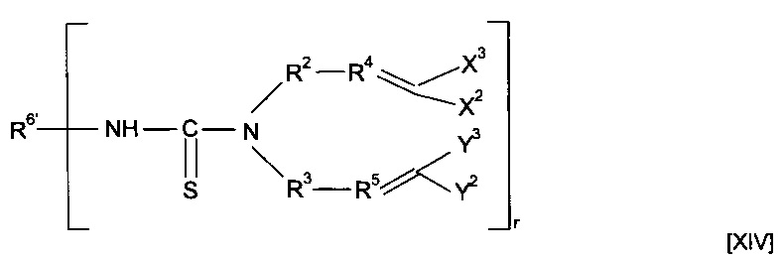
где R6'-NH представляет собой R6, и R6' вместе с группой -NH-CS образует указанный минерал-связывающий фрагмент.
30. Способ по п. 29, отличающийся тем, что предшественник полимера представляет собой соединение, имеющее структуру [XV]
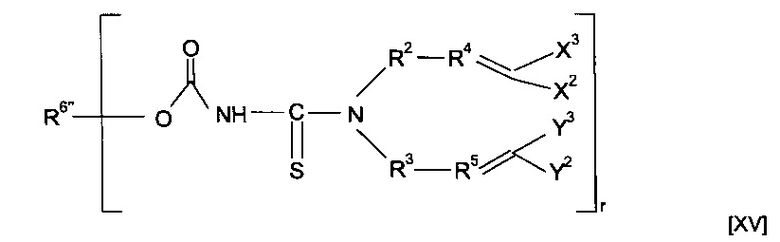
где R6ʺ-OC(O)-NH представляет собой R6, и R6ʺ вместе с группой -OC(O)-NH-CS образует указанный минерал-связывающий фрагмент.
31. Способ по любому из пп. 1-4, 6-13, 15-20, 23, 25-30, отличающийся тем, что указанный полимер, образованный путем полимеризации предшественника полимера, инкапсулирует минерал-связывающий фрагмент.
32. Способ по любому из пп. 1-4, 6-13, 15-20, 23, 25-30, отличающийся тем, что указанный полимер, образованный путем полимеризации предшественника полимера, представляет собой гомополимер.
33. Способ по любому из пп. 1-4, 6-13, 15-20, 23, 25-30, отличающийся тем, что указанный полимер представляет собой сополимер, образованный путем сополимеризации предшественника полимера с одним или более другими полимерными предшественниками и/или кросс-линкером.
34. Способ по любому из пп. 1-4, 6-13, 15-20, 23, 25-30, отличающийся тем, что обеспечивает структуру, которая содержит полимерный материал, с которым взаимодействует указанная минеральная смесь.
35. Способ по п. 34, отличающийся тем, что указанная структура является пористой, и минеральная смесь проходит через указанную структуру, причем металлосодержащий минерал селективно связывается минерал-связывающим фрагментом и, таким образом, отделяется от пустой породы, которая выходит из указанной структуры.
36. Способ по п. 34, отличающийся тем, что указанная структура представляет собой пену и/или листовой материал, такой как сито или фильтр.
37. Способ по любому из пп. 1-4, 6-13, 15-20, 23, 25-30, отличающийся тем, что указанный полимерный материал присутствует в виде частиц.
38. Способ по любому из пп. 1-4, 6-13, 15-20, 23, 25-30, отличающийся тем, что этапы (а)-(с) осуществляют как часть флотационного процесса для отделения пустой породы от металлосодержащего минерала.
39. Способ по любому из пп. 1-4, 6-13, 15-20, 23, 25-30, отличающийся тем, что минеральная смесь присутствует в форме пульпы, содержащей частицы минерала в воде.
40. Способ по любому из пп. 1-4, 6-13, 15-20, 23, 25-30, включающий дополнительный этап высвобождения металлосодержащего минерала из полимерного материала.
41. Способ по любому из пп. 1-4, 6-13, 15-20, 23, 25-30, включающий дополнительный этап получения некоторого количества металла из металлосодержащего минерала.
42. Способ по любому из пп. 1-4, 6-13, 15-20, 23, 25-30, осуществляемый по месту на руднике.
| US 4888106 A, 19.12.1989 | |||
| US 3224582 A, 21.12.1965 | |||
| Способ и приспособление для нагревания хлебопекарных камер | 1923 |
|
SU2003A1 |
| J | |||
| Rubio et al | |||
| The process of separation of fine mineral particles by flotation with hydrophobic polymeric carrier | |||
| International Journal of Mineral Processing, 1993, 37, p.109-122. | |||

