ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение в общем относится к гетероароматическим соединениям, которые являются допаминовыми D1 лигандами, например допаминовыми D1 агонистами или частичными агонистами.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
Допамин действует на нейроны через два семейства допаминовых рецепторов, D1-подобные рецепторы (D1R) и D2-подобные рецепторы (D2R). Семейство D1-подобных рецепторов состоит из D1 и D5 рецепторов (D1), которые в высокой степени экспрессируются во многих областях головного мозга. D1 мРНК была обнаружена в полосатом теле и прилежащем ядре (nucleus accumbens), смотри, например, Missale С, Nash SR, Robinson SW, Jaber M, Caron MG "Dopamine receptors: from structure to function", Physiological Reviews 78:189-225 (1998).
Фармакологические исследования сообщают о том, что D1 и D5 рецепторы (D1/D5), а именно Dl-подобные рецепторы, связаны со стимуляцией аденилилциклазы, тогда как D2, D3 и D4 рецепторы, а именно D2-подобные рецепторы, связаны с ингибированием продукции сАМР (циклический аденозинмонофосфат), смотри, например, Jose PA, et. al, "Dopamine D1 receptor regulation of phospholipase С", Hypertension Research 18 Suppl 1:S39-42 (1995).
Допаминовые D1 рецепторы вовлечены в многочисленные нейрофармакологические и нейробиологические функции. Например, D1 рецепторы вовлечены в различные типы функций памяти и синаптическую пластичность, смотри например, Goldman-Rakic PS, Castner SA, Svensson TH, Siever LJ, Williams GV "Targeting dopamine D1 receptor in schizophrenia: insights for cognitive dysfunction", Psychopharmacology 174(1):3-16 (2004); Castner SA, Williams GV "Tuning engine of cognition: focus on NMDA/D1 receptor interactions in prefrontal cortex", Brain Cognition 63(2):94-122 (2007). Кроме того, D1 рецепторы вовлечены в разнообразие психиатрических, неврологических, связанных с неврологическим развитием, нейродегенеративных, поведенческих, мотивационных, метаболических, сердечно-сосудистых, почечных, офтальмологических, эндокринных и/или других расстройств, раскрытых в данном описании, включая шизофрению (например когнитивные и негативные симптомы при шизофрении), нарушение познавательной способности, ассоциированное с терапией D2 антагонистами, ADHD, импульсивность, расстройство аутического спектра, умеренное ухудшение познавательной способности (MCI), связанное с возрастом ухудшение познавательной способности, деменцию Альцгеймера, болезнь Паркинсона, хорею Гентингтона, депрессию, тревогу, устойчивую к лечению депрессию (TRD), биполярное расстройство, хроническую апатию, ангедонию, хроническую утомляемость, пост-травматическое стрессовое расстройство, сезонное аффективное расстройство, социальное тревожное расстройство, послеродовую депрессию, серотониновый синдром, токсикоманию и лекарственную зависимость, синдром Туретта (Tourette), позднюю дискинезию, вялость, сексуальную дисфункцию, мигрень, системную красную волчанку (SLE), гипергликемию, дислипидемию, ожирение, диабет, сепсис, пост-ишемический тубулярный некроз, почечную недостаточность, устойчивый отек, нарколепсию, гипертензию, застойную сердечную недостаточность, послеоперационную гипотонию глаза, нарушения сна, боль и другие расстройства у млекопитающего, смотри, например, Goulet М, Madras BK "D(1) dopamine receptor agonists are more effective in alleviating advanced than mild parkinsonism in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridin-treated monkeys", Journal of Pharmacology and Experimental Therapy 292(2):714-24 (2000); Surmeier DJ, et. al, "Role of dopamin in modulating structure and function of striatal circuits", Prog. Brain Research 183:149-67 (2010); Umrani DN, Goyal RK "Fenoldopam treatment improves peripheral insulin sensitivity and renal function in STZ-induced type2 diabetic rats", Clin. Exp. Hypertension 25(4):221-233 (2003); Bina KG et al., "Dopaminergic agonists normalize elevated hypothalamic neuropeptide Y and corticotropin-releasing hormone, body weight gain, and hyperglycemia in ob/ob mice", Neuroendocrinology 71(1).68-78 (2000).
Рецепторы, сопряженные с G-белком (GPCR, включая D1R), десенсибилизируют посредством общего механизма, включающего фосфорилирование рецепторной киназой, сопряженной с G-белком (GRK), с последующим связыванием β-аррестина, что предотвращает связывание с G-белком (и тем самым активацию G-белка), смотри Louis М. Luttrell et. al., "Role of β-arrestins in termination and transduction of G-protein-coupled receptor signals"; J. Cell Sci., 115, 455-465 (2002). Например, десенсибилизация D1 рецептора включает индуцированное агонистом фосфорилирование рецептора (то есть преимущественное фосфорилирование рецепторов, которые находятся в оккупированной агонистом конформации) и рекрутмент β-аррестина (связывание β-аррестин-рецептор), что предотвращает связывание с G белком и в свою очередь приводит к десенсибилизации D1 рецепторных канонических метаболического пути/активации передачи сигнала G белка [что может быть измерено, например, по аккумулированию/продуцированию циклического аденозинмонофосфата (сАМР)], смотри М.М. Lewis et al, "Homologous Desensitization of D1A Dopamine Receptor: Efficacy in Causing Desensitization Dissociates from Both Receptor Occupancy and Functional Potency"; JPET 286: 345-353, 1998.
В дополнение к их хорошо установленной роли в десенсибилизации GPCR, β-аррестины могут также быть способны к GPCR-опосредованной "аррестинергической" передаче сигнала посредством функционирования в качестве остовов для эффекторных в прямом направлении молекул, таких как внеклеточные регулируемые киназы (ERK), смотри Nikhil М Urs, et al, "А Dopamine D1 Receptor-Dependent β-Arrestin Signaling Complex Potentially Regulates Morphine-Induced Psychomotor Activation but not Reward in Mice," Neuropsychopharmacology (2011) 36, 551-558; Reiter E, et. al, "Molecular mechanism of beta-arrestin-biased agonism at seven-transmembrane receptors," Annual review of pharmacology и toxicology. 2012; 52:179-97; и Allen JA, et al. "Discovery of beta-arrestin-biased dopamine D2 ligands for probing signal transduction pathways essential for antipsychotic efficacy," Proceedings of National Academy of Sciences of United States of America. 2011; 108(45):18488-93.
Новые или улучшенные агенты, которые модулируют (например агонизируют или частично агонизируют) D1, необходимы для создания новых и более эффективных фармацевтических средств для лечения заболеваний или состояний, ассоциированных с разрегулированной активацией D1, таких как раскрытые в данном описании.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Согласно настоящему изобретению предложено, в частности, соединение формулы I:
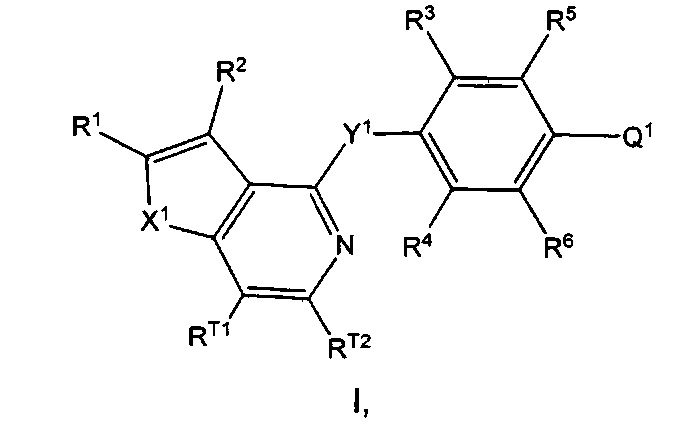
или его N-оксид, или фармацевтически приемлемая соль указанного соединения или указанного N-оксида,
где: X1 представляет собой О или S;
Y1 представляет собой О, S или NRN;
Q1 представляет собой N-содержащий 5-10-членный гетероциклоалкил, N-содержащий 5-10-членный гетероарил или фенил, где гетероциклоалкил или гетероарил возможно замещен 1, 2, 3, 4 или 5 независимо выбранными R7; и фенил возможно замещен 1, 2, 3, 4 или 5 независимо выбранными R7a;
каждый из RT1 и RT2 независимо выбран из группы, состоящей из Н, C1-3алкила, C1-3фторалкила, циклопропила, фторциклопропила, C1-3алкокси, C1-3галогеналкокси, групп -С(=O)-O-(C1-3алкил) и -С(=O)ОН;
R1 выбран из группы, состоящей из Н, F, групп -С(=O)ОН, -C(=O)-O-(C1-3алкил), C1-3алкила, C1-3фторалкила, C1-3циклоалкила и C1-3фторциклоалкила, где указанный C3-6циклоалкил возможно замещен 1, 2, 3, 4 или 5 заместителями, каждый из которых независимо выбран из галогена, C1-4алкила, C1-4галогеналкила, C1-4алкокси и C1-4галогеналкокси;
R2 выбран из группы, состоящей из Н, галогена (например F, Cl, Br или I), -CN, групп -ОН, С(=O)ОН, С(=O)-O-(C1-3алкил), C1-3алкокси, C1-3галогеналкокси, -N(R8)(R9), C1-3алкила, C1-3фторалкила, C3-6циклоалкила, C3-6фторциклоалкила, C2-6алкенила и C2-6алкинила, где указанный C3-6циклоалкил возможно замещен 1, 2, 3, 4 или 5 заместителями, каждый из которых независимо выбран из галогена, C1-4алкила, C1-4галогеналкила, C1-4алкокси и C1-4галогеналкокси;
каждый из R3 и R4 независимо выбран из группы, состоящей из Н, C1-6алкила, C1-6галогеналкила, C1-6алкокси C1-6галогеналкокси, -CN, C1-6циклоалкила, групп -С(=O)ОН, С(=O)-O-(C1-4алкил) и галогена, где каждый из указанных C1-6алкила и C3-6циклоалкила возможно замещен 1, 2, 3, 4 или 5 заместителями, каждый из которых независимо выбран из галогена, -ОН, -CN, C1-4алкила, C1-4галогеналкила, C1-4алкокси и C1-4галогеналкокси;
каждый из R5 и R6 независимо выбран из группы, состоящей из Н, галогена, -ОН, -NO2, -CN, C1-6алкила, C1-6галогеналкила, C1-6галогеналкокси, C2-6алкенила, C2-6алкинила, C3-7циклоалкила, 4-10-членного гетероциклоалкила, групп -N(R8)(R9), -N(R10)(C(=O)R11), -C(=O)-N(R8)(R9), -C(=O)-R12, -C(=O)-OR12 и -OR13, где каждый из указанных C1-6алкила, C3-7циклоалкила и гетероциклоалкила возможно замещен 1, 2 или 3 заместителями, каждый из которых независимо выбран из группы, состоящей из галогена, -CN, -ОН, C1-3алкила, C1-3алкокси, C1-3галогеналкила, C1-3галогеналкокси, C3-6циклоалкила, групп -N(R14)(R15), -N(R16)(C(=O)R17), -C(=O)-OR18, -C(=O)H, -C(=O)R18, -C(=O)N(R14)(R15) и -OR19;
или R5 и R3 вместе с двумя атомами углерода, к которым они присоединены, образуют конденсированный N-содержащий 5- или 6-членный гетероарил, конденсированный N-содержащий 5- или 6-членный гетероциклоалкил, конденсированный 5- или 6-членный циклоалкил или конденсированное бензольное кольцо, каждый(ое) из которых возможно замещен(о) 1, 2 или 3 заместителями, каждый из которых независимо выбран из группы, состоящей из галогена, -CN, -ОН, C1-3алкила, C1-3алкокси, C1-3галогеналкила и C1-3галогеналкокси;
каждый из R7 и R7a независимо выбран из группы, состоящей из галогена, -ОН, -CN, -NO2, оксо, C1-6алкила, C1-6галогеналкила, C1-6гидроксилалкила, C1-6алкокси, C1-6галогеналкокси, C3-7циклоалкила, C2-6алкенила, C2-6алкинила, C6-10арила, 4-10-членного гетероциклоалкила, 5-10-членного гетероарила, циклоалкилалкила, гетероциклоалкилалкила, арилалкила, гетероарилалкила, гетероарилалкенила, групп -CH=N-O-(C1-3алкил), -N(R14)(R15), -N(R16)(C(=O)R17), -S(=O)2N(R14)(R15), -C(=O)N(R14)(R15), -C(=O)-R12, -C(=O)-OR18 и -OR19, где каждая из указанных групп C1-6алкил, C3-7циклоалкил, циклоалкил а л кил, гетероциклоалкилалкил, арилалкил, гетероарилалкил, гетероарилалкенил, С6-10арил, гетероциклоалкил и гетероарил возможно замещена 1, 2, 3 или 4 заместителями, каждый из которых независимо выбран из группы, состоящей из галогена, ОН, -CN, -NO2, C1-4алкила, C1-4гидроксилалкила, C1-4алкокси, -N(R14)(R15), групп -S-(C1-3алкил), -S(=O)2-(C1-4алкил), арилокси, арилалкилокси, возможно замещенного 1 или 2 C1-4алкилами, оксо, групп -С(=O)Н, -С(=O)-C1-4алкил, -С(=O)O-C1-4алкил, -C(=O)NH2, -NHC(=O)H, -NHC(=O)-(C1-4алкил), C3-7циклоалкил, 5- или 6-членный гетероарил, C1-4галогеналкил и C1-4галогеналкокси;
или два соседних R7a вместе с двумя атомами углерода, к которым они присоединены, образуют конденсированный 5- или 6-членный циклоалкил, конденсированный 5- или 6-членный гетероциклоалкил или конденсированное бензольное кольцо, каждый(ое) из которых возможно замещен(о) 1, 2, 3 или 4 R7b, где каждый R7b независимо выбран из группы, состоящей из галогена, -CN, -NO2, -NH2, групп -NH(C1-4алкил), -N(C1-4алкил)2, азетидин-1-ил, пирролидин-1-ил, пиридин-1-ил, ОН, оксо, C1-4алкил, C1-4алкокси, C1-4гидроксилалкил, C1-4галогеналкил и C1-4галогеналкокси;
каждый из R8 и R9 независимо выбран из группы, состоящей из Н, C1-6алкила, C1-6галогеналкила, C3-10циклоалкила, 4-10-членного гетероциклоалкила, циклоалкилалкила, гетероциклоалкилалкила, арилалкила и гетероарилалкила, где каждая из указанных групп C1-6алкил, C3-10циклоалкил, 4-10-членный гетероциклоалкил, циклоалкилалкил, арилалкил и гетероарилалкил возможно замещена 1, 2, 3 или 4 заместителями, каждый из которых независимо выбран из группы, состоящей из -ОН, -CN, C1-3алкила, C3-7циклоалкила, C1-3гидроксилалкила, групп -S-C1-3алкил, -C(=O)H, -C(=O)-C1-3алкил, -С(=O)-O-C1-3алкил, -C(=O)-NH2, -С(=O)-N(C1-3алкил)2, C1-3галогеналкила, C1-3алкокси и C1-3галогеналкокси;
или R8 и R9 вместе с атомом N, к которому они присоединены, образуют 4-10-членный гетероциклоалкил или гетероарил, возможно замещенный 1, 2, 3 или 4 заместителями, каждый из которых независимо выбран из группы, состоящей из галогена, -ОН, оксо, групп -С(=O)Н, -С(=O)ОН, -С(=O)-C1-3алкил, -C(=O)-NH2, -С(=O)-N(C1-3алкил)2, -CN, C1-3алкила, C1-3алкокси, C1-3гидроксилалкила, C1-3галогеналкила и C1-3галогеналкокси;
R10 выбран из группы, состоящей из Н, C1-3алкила и C3-7циклоалкила;
R11 выбран из группы, состоящей из C1-6алкила, C3-7циклоалкила, 4-14-членного гетероциклоалкила, C6-10арила, 5-10-членного гетероарила, циклоалкилалкила, гетероциклоалкилалкила, арилалкила и гетероарилалкила, каждый из которых возможно замещен 1, 2 или 3 заместителями, каждый из которых независимо выбран из группы, состоящей из галогена, -CF3, -CN, -ОН, оксо, группы -S- C1-3алкил, C1-6алкила, C1-6галогеналкила, C2-6алкенила, C2-6алкинила, C3-7циклоалкила, C1-6алкокси и C1-6галогеналкокси;
R12 представляет собой Н или выбран из группы, состоящей из C1-10алкила, C3-7циклоалкила, 4-14-членного гетероциклоалкила, C6-10арила, 5-10-членного гетероарила, циклоалкилалкила, гетероциклоалкилалкила, арилалкила и гетероарилалкила, каждый из которых возможно замещен 1, 2 или 3 заместителями, каждый из которых независимо выбран из группы, состоящей из галогена, -CF3, -CN, -ОН, -С(=O)ОН, C1-6алкила, C1-6галогеналкила, C2-6алкенила, C2-6алкинила, C3-7циклоалкила, C1-6алкокси и C1-6галогеналкокси;
R13 выбран из группы, состоящей из C1-10алкила, C1-6галогеналкила, C3-7циклоалкила, 4-14-членного гетероциклоалкила, C6-10арила, 5-10-членного гетероарила, циклоалкилалкила, гетероциклоалкилалкила, арилалкила и гетероарилалкила, каждый из которых возможно замещен 1, 2, 3 или 4 заместителями, каждый из которых независимо выбран из группы, состоящей из галогена, групп -N(R14)(R15), -C(=O)N(R14)(R15), -N(R16)(C(=O)R17), -C(=O)H, -C(=O)N(R16)(OR18), -C(=O)-R18, -C(=O)-OR18, -O-C(=O)R18, -CF3, -CN, -OH, -O-(C1-6гидроксилалкил), C1-6алкила, оксо, C1-6гидроксилалкила, C1-6галогеналкила, C2-6алкенила, C2-6алкинила, C3-7циклоалкила, C1-6алкокси и C1-6галогеналкокси;
каждый из R14 и R15 независимо выбран из группы, состоящей из Н, C1-6алкила, C2-6алкенила, C3-10циклоалкила, 4-14-членного гетероциклоалкила, циклоалкилалкила, гетероциклоалкилалкила, арилалкила и гетероарилалкила, где каждый из указанных C1-6алкила, C3-7циклоалкила, циклоалкилалкила, арилалкила и гетероарилалкила возможно замещен 1, 2 или 3 заместителями, каждый из которых независимо выбран из группы, состоящей из -ОН, -CN, оксо, групп NHC(=O)-(C1-3алкил), -С(=О)N(C1-3алкил)2, -O-(C1-6гидроксилалкил), -S(=O)2- C1-3алкил, -S-C1-3алкил, C1-3алкила, C3-7циклоалкила, C1-3гидроксилалкила, 5-10-членного гетероарила, C1-3алкокси, C1-3галогеналкила и C1-3галогеналкокси;
или R14 и R15 вместе с атомом N, к которому они присоединены, образуют 4-10-членный гетероциклоалкил или 5-10-членный гетероарил, возможно замещенный 1, 2 или 3 заместителями, каждый из которых каждый независимо выбран из группы, состоящей из галогена, оксо, -ОН, C1-3алкила, C1-3алкокси, C1-3-галогеналкила,C1-3галогеналкокси,C1-3гидроксилалкила, C2-4алкоксиалкила, оксо, 5-6-членного гетероарила, -NH2, групп -N(C1-3алкил)2, -S(=O)2-C1-3алкил, -S-C1-3алкил, -C(=O)H, -С(=O)ОН, -C(=O)NH2 и -С(=O)-C1-3алкил;
R16 выбран из группы, состоящей из Н, C1-3алкила и C3-7циклоалкила;
R17 выбран из группы, состоящей из C1-6алкила, C3-7циклоалкила, 4-14-членного гетероциклоалкила, C6-10арила, 5-10-членного гетероарила, циклоалкилалкила, гетероциклоалкилалкила, арилалкила и гетероарилалкила, каждый из которых возможно замещен 1, 2 или 3 заместителями, каждый из которых независимо выбран из группы, состоящей из галогена, -CF3, -CN, -ОН, C1-6алкила, C1-6галогеналкила, C2-6алкенила, C2-6алкинила, C3-7циклоалкила, C1-6алкокси и C1-6галогеналкокси;
R18 представляет собой Н или выбран из группы, состоящей из C1-6алкила, C3-7циклоалкила, 4-14-членного гетероциклоалкила, C6-10арила, 5-10-членного гетероарила, циклоалкилалкила, гетероциклоалкилалкила, арилалкила и гетероарилалкила, каждый из которых возможно замещен 1, 2 или 3 заместителями, каждый из которых независимо выбран из группы, состоящей из галогена, -CF3, -CN, -ОН, C1-6алкила, C1-6галогеналкила, C2-6алкенила, C2-6алкинила, C3-7циклоалкила, C1-6алкокси и C1-6галогеналкокси;
R19 выбран из группы, состоящей из C1-6алкила, C1-6галогеналкила, C3-7циклоалкила, 4-14-членного гетероциклоалкила, C6-10арила, 5-10-членного гетероарила, циклоалкилалкила, гетероциклоалкилалкила, арилалкила и гетероарилалкила, каждый из которых возможно замещен 1, 2 или 3 заместителями, каждый из которых независимо выбран из группы, состоящей из галогена, групп -N(R14)(R15), -C(=O)N(R14)(R15), -N(R16)(C(=O)R17), -C(=O)-R18, -C(=O)-OR18, -CF3, -CN, -OH, C1-6алкила, C1-6галогеналкила, C2-6алкенила, C2-6алкинила, C3-7циклоалкила, C1-6алкокси и C1-6галогеналкокси; и
RN выбран из группы, состоящей из Н, C1-6алкила, C3-6циклоалкила, C3-6фторциклоалкила, гетероарилалкила и арилалкила, где каждый из указанных C3-6циклоалкила, гетероарилалкила и арилалкила возможно замещен 1, 2, 3, 4 или 5 заместителями, каждый из которых независимо выбран из галогена, C1-4алкила, C1-4галогеналкила, C1-4алкокси и C1-4галогеналкокси.
Как он используется в данном описании, термин "соседний" при описании относительных положений двух групп-заместителей на кольцевой структуре относится к двум группам-заместителям, которые соответственно присоединены к двум образующим кольцо атомам одного и того же кольца, где эти два образующих кольцо атома напрямую соединены посредством химической связи. Например, в каждой из следующих структур:

любая из двух групп R70 является соседней по отношению к группе R60.
Как он используется в данном описании, термин "n-членный", где п представляет собой целое число, обычно описывает число образующих кольцо атомов в группировке, где число образующих кольцо атомов составляет n. Например, пиридин представляет собой пример 6-членного гетероарильного кольца, а тиофен представляет собой пример 5-членной гетероарильной группы.
В разных местах настоящего описания заместители соединений по изобретению описаны в группах или в диапазонах. Специально оговаривается, что изобретение включает любую и каждую индивидуальную подкомбинацию членов таких групп и диапазонов. Например, термин "C1-6алкил" конкретно предполагается как включающий метил, этил, C3алкил, C4алкил, C5алкил и C6алкил. В качестве другого примера, термин "5-10-членная гетероарильная группа" конкретно предполагается как включающий любую 5-, 6-, 7-, 8-, 9- или 10-членную гетероарильную группу.
Как он используется в данном описании, термин "алкил" определен как включающий насыщенные алифатические углеводороды, включая прямые цепи и разветвленные цепи. В некоторых воплощениях алкильная группа имеет от 1 до 10, например от 1 до 6, атомов углерода. Например, как он используется в данном описании, термин " C1-6алкил", также как и алкильные группировки других групп, на которые делается ссылка в данном описании (например C1-6алкокси), относится к линейным или разветвленным радикалам из 1-6 атомов углерода (например метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-пентил или н-гексил), возможно замещенным 1 или более (например от 1 до 5) подходящими заместителями. Термин " C1-4алкил" относится к линейным или разветвленным радикалам из 1-4 атомов углерода (например метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил). Термин " C1-3алкил" относится к линейным или разветвленным радикалам из 1 - 3 атомов углерода.
Как он используется в данном описании, термин "алкенил" относится к алифатическим углеводородам, имеющим по меньшей мере одну углерод-углеродную двойную связь, включая прямые цепи и разветвленные цепи, имеющие по меньшей мере одну углерод-углеродную двойную связь. В некоторых воплощениях алкенильная группа имеет от 2 до 6 атомов углерода. В некоторых воплощениях алкенильная группа имеет от 2 до 4 атомов углерода. Например, как он используется в данном описании, термин "C2-6алкенил" означает ненасыщенные радикалы с прямой или разветвленной цепью из 2-6 атомов углерода, включая, но не ограничиваясь ими, этенил, 1-пропенил, 2-пропенил (аллил), изопропенил, 2-метил-1-пропенил, 1-бутенил, 2-бутенил и тому подобное, возможно замещенные 1-5 подходящими заместителями. Когда соединения формулы I содержат алкенильную группу, алкенильная группа может существовать как чистая Е форма, чистая Z форма или любая их смесь.
Как он используется в данном описании, термин "алкинил" относится к алифатическим углеводородам, имеющим по меньшей мере одну углерод-углеродную тройную связь, включая прямые цепи и разветвленные цепи, имеющие по меньшей мере одну углерод-углеродную тройную связь. В некоторых воплощениях алкинильная группа имеет от 2 до 6 атомов углерода. Например, как он используется в данном описании, термин " C2-6алкинил" означает алкинильные радикалы с прямой или разветвленной цепью, как они определены выше, имеющие от 2 до 6 атомов углерода и одну тройную связь, возможно замещенные 1 или более (например от 1 до 5) подходящими заместителями.
Как он используется в данном описании, термин "циклоалкил" относится к насыщенным или ненасыщенным, неароматическим, моноциклическим или полициклическим (таким как бициклическим) углеводородным кольцам (например моноциклическим, таким как циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклооктил, циклононил, или бициклическим, включая спиро-, конденсированные или мостиковые системы (такие как бицикло[1.1.1]пентанил, бицикло[2.2.1]гептанил, бицикло[3.2.1]октанил или бицикло[5.2.0]нонанил, декагидронафталинил и тому подобное), возможно замещенным 1 или более (например от 1 до 5) подходящими заместителями. Циклоалкильная группа имеет от 3 до 15 атомов углерода. В некоторых воплощениях циклоалкил возможно может содержать одну, две или более некумулятивных неароматических двойных или тройных связей и/или от одной до трех оксо-групп. В некоторых воплощениях бициклоалкильная группа имеет от 6 до 15 атомов углерода. Например, термин "C3-7циклоалкил" относится к насыщенным или ненасыщенным, неароматическим, моноциклическим или полициклическим (например бициклическим) углеводородным кольцам из 3-7 образующих кольцо атомов углерода (например циклопропил, циклобутил, циклопентил, циклогексил, или бицикло[1.1.1]пентанил). Термин " C3-6циклоалкил" относится к насыщенным или ненасыщенным, неароматическим, моноциклическим или полициклическим (например бициклическим) углеводородным кольцам из 3-6 образующих кольцо атомов. Также в определение циклоалкила включены группировки, которые имеют одно или более ароматических колец (включая арил и гетероарил), конденсированных с циклоалкильным кольцом, например бензо- или тиенильные производные циклопентана, циклопентена, циклогексана и тому подобное (например 2,3-дигидро-1Н-инден-1-ил или 1Н-инден-2(3H)-он-1-ил). Циклоалкильная группа возможно замещена 1 или более (например от 1 до 5) подходящими заместителями.
Как он используется в данном описании, термин "арил" относится к состоящим полностью из углерода моноциклическим или конденсированным кольцевым полициклическим ароматическим группам, имеющим конъюгированную пи-электронную систему. Арильная группа имеет 6, 8 или 10 атомов углерода в кольце(ах). В более конкретном виде, арильная группа имеет 6 или 10 атомов углерода в кольце(ах). В наиболее частном виде, арильная группа имеет 6 атомов углерода в кольце. Например, как он используется в данном описании, термин "C6-10арил" означает ароматические радикалы, содержащие от 6 до 10 атомов углерода, такие как фенил, нафтил, тетрагидронафтил, инданил и тому подобное. Арильная группа возможно замещена 1 или более (например от 1 до 5) подходящими заместителями.
Как он используется в данном описании, термин "гетероарил" относится к моноциклическим или конденсированным кольцевым полициклическим ароматическим гетероциклическим группам с одним или более гетероатомами в качестве членов кольца (атомы, образующие кольцо), каждый из которых независимо выбран из О, S и N, в по меньшей мере одном кольце. Гетероарильная группа имеет от 5 до 14 атомов, образующих кольцо, включая от 1 до 13 атомов углерода и от 1 до 8 гетероатомов, выбранных из О, S и N. В некоторых воплощениях гетероарильная группа имеет от 5 до 10 атомов, образующих кольцо, включая от одного до четырех гетероатомов. Гетероарильная группа также может содержать от одной до трех оксо-групп. В некоторых воплощениях гетероарильная группа имеет от 5 до 8 атомов, образующих кольцо, включая один, два или три гетероатома. Примеры моноциклических гетероарилов включают гетероарилы с 5 атомами, образующими кольцо, включая от одного до трех гетероатомов, или гетероарилы с 6 атомами, образующими кольцо, включая один или два гетероатома азота. Примеры конденсированных бициклических гетероарилов включают два конденсированных 5- и/или 6-членных моноциклических кольца, включающих от одного до четырех гетероатомов.
Примеры гетероарильных групп включают пиридинил, пиразинил, пиримидинил, пиридазинил, тиенил, фурил, имидазолил, пирролил, оксазолил (например 1,3-оксазолил, 1,2-оксазолил), тиазолил (например 1,2-тиазолил, 1,3-тиазолил), пиразолил, тетразолил, триазолил (например 1,2,3-триазолил, 1,2,4-триазолил), оксадиазолил (например 1,2,3-оксадиазолил), тиадиазолил (например 1,3,4-тиадиазолил), хинолил, изохинолил, бензотиенил, бензофурил, индолил, пиридон, пиримидон, пиразинон, пиримидинон, 1H-имидазол-2(3Н)-он, 1H-пиррол-2,5-дион и тому подобное. Гетероарильная группа возможно замещена 1 или более (например от 1 до 5) подходящими заместителями.
Как он используется в данном описании, термин "N-содержащий", когда он используется в сочетании с гетероарилом или гетероциклоалкилом, означает, что гетероарил или гетероциклоалкил содержит по меньшей мере один образующий кольцо атом азота (N) и возможно один или более (например 1,2,3 или 4) образующих кольцо гетероатомов, каждый из которых независимо выбран из О, S и N. Термин "N-содержащий 5-10-членный гетероарил" относится к 5-10-членной гетероарильной группе (включая моноциклические или бициклические), содержащей по меньшей мере один образующий кольцо атом азота (N) и возможно один или более (например 1, 2, 3 или 4) образующих кольцо гетероатомов, каждый из которых независимо выбран из О, S и N. Термин "N-содержащий 5- или 6-членный гетероарил" относится к 5-6-членной гетероарильной группе, содержащей по меньшей мере один образующий кольцо атом азота (N) и возможно один или более (например 1, 2, 3 или 4) образующих кольцо гетероатомов, каждый из которых независимо выбран из О, S и N. Примеры N-содержащих 5-10-членных гетероарильных групп включают пиридинил, пиразинил, пиримидинил, пиридазинил, имидазолил, пирролил, оксазолил (например 1,3-оксазолил, 1,2-оксазолил), тиазолил (например 1,2-тиазолил, 1,3-тиазолил), пиразолил, тетразолил, триазолил (например 1,2,3-триазолил, 1,2,4-триазолил), оксадиазолил (например 1,2,3-оксадиазолил), тиадиазолил (например 1,3,4-тиадиазолил), хинолил, изохинолил, пиридон, пиримидон, пиразинон, пиримидинон, 1Н-имидазол-2(3Н)-он, 1H-пиррол-2,5-дион и тому подобное. Примеры N-содержащих 5- или 6-членных гетероарильных групп включают пиридинил, пиразинил, пиримидинил, пиридазинил, имидазолил, пирролил, оксазолил (например 1,3-оксазолил, 1,2-оксазолил), тиазолил (например 1,2-тиазолил, 1,3-тиазолил), пиразолил, тетразолил, триазолил (например 1,2,3-триазолил, 1,2,4-триазолил), оксадиазолил (например 1,2,3-оксадиазолил) и тиадиазолил (например 1,3,4-тиадиазолил). N-содержащая 5-10-членная гетероарильная группа или N-содержащий 5- или 6-членный гетероарил возможно замещены 1 или более (например от 1 до 5) подходящими заместителями.
Как он используется в данном описании, термин "гетероциклоалкил" относится к моноциклической или полициклической [включая 2 или более колец, которые конденсированы вместе, включая спиро-, конденсированные или мостиковые системы, например бициклическую кольцевую систему], насыщенной или ненасыщенной, неароматической 3-15-членной кольцевой системе (такой как 4-14-членная кольцевая система, 4-10-членная кольцевая система или 5-10-членная кольцевая система), включающей от 1 до 14 образующих кольцо атомов углерода и от 1 до 10 образующих кольцо атомов гетероатомов, каждый из которых независимо выбран из О, S и N. Гетероциклоалкильная группа может также включать от одной до трех оксо-групп. Примеры таких гетероциклоалкильных колец включают азетидинил, тетрагидрофуранил, имидазолидинил, пирролидинил, пиперидинил, пиперазинил, оксазолидинил, тиазолидинил, пиразолидинил, тиоморфолинил, тетрагидротиазинил, тетрагидротиадиазинил, морфолинил, оксетанил, тетрагидродиазинил, оксазинил, оксатиазинил, индолинил, изоиндолинил, хинуклидинил, хроманил, изохроманил, бензоксазинил, 2-азабицикло[2.2.1]гептанонил, 3-азабицикло[3.1.0]гексанил, 3-азабицикло[4.1.0]гептанил и тому подобное. Другие примеры гетероциклоалкильных колец включают тетрагидрофуран-2-ил, тетрагидрофуран-3-ил, имидазолидин-1-ил, имидазолидин-2-ил, имидазолидин-4-ил, пирролидин-1-ил, пирролидин-2-ил, пирролидин-3-ил, пиперидин-1-ил, пиперидин-2-ил, пиперидин-3-ил, пиперидин-4-ил, пиперазин-1-ил, пиперазин-2-ил, 1,3-оксазолидин-3-ил, 1,4-оксазепан-1-ил, изотиазолидинил, 1,3-тиазолидин-3-ил, 1,2-пиразолидин-2-ил, 1,2-тетрагидротиазин-2-ил, 1,3-тиазинан-3-ил, 1,2-тетрагидродиазин-2-ил, 1,3-тетрагидродиазин-1-ил, 1,4-оксазин-4-ил, оксазолидинонил и тому подобное. Также в определение гетероциклоалкила включены группировки, которые имеют одно или более ароматических колец (включая арильное и гетероарильное), конденсированных с неароматическим гетероциклоалкильным кольцом, например пиридинил, пиримидинил, тиофенил, пиразолил, фталимидил, нафталимидил, и бензо-производные гетероциклов, такие как группы индол, изоиндол, изоиндолин-1-он-3-ил, 5,7-дигидро-6H-пирроло[3,4-d]пиридин-6-ил, 6,7-дигидро-5Н-пирроло[3,4-d]пиримидин-6-ил, 4,5,6,7-тетрагидротиено[2,3-с]пиридин-5-ил, 5,6-дигидротиено[2,3-с]пиридин-7(4H)-он-5-ил, 1,4,5,6-тетрагидропирроло[3,4-с]пиразол-5-ил и 3,4-дигидроизохинолин-1(2H)-он-3-ил. Гетероциклоалкильная группа возможно замещена 1 или более (например от 1 до 5) подходящими заместителями. Примеры гетероциклоалкильных групп включают 5- или 6-членные моноциклические кольца и 9- или 10-членный конденсированные бициклические кольца.
Как он используется в данном описании, термин "N-содержащий 5-10-членный гетероциклоалкил" относится к 5-10-членной гетероциклоалкильной группе, содержащей по меньшей мере один образующий кольцо атом азота (N) и возможно один или более образующих кольцо гетероатомов, каждый из которых независимо выбран из О, S и N. Термин "N-содержащий 5- или 6-членный гетероциклоалкил" относится к 5- или 6-членной гетероциклоалкильной группе, содержащей меньшей мере один образующий кольцо атом азота (N) и возможно один или более образующих кольцо гетероатомов, каждый из которых независимо выбран из О, S и N. Примеры N-содержащих 5 - 10-членных гетероциклоалкильных групп включают пиперидин-1-ил, пиперидин-4-ил, пиперазин-1-ил, 1,3-тиазинан-3-ил, 1,4,5,6-тетрагидропирроло[3,4-с]пиразол-5-ил и 3,4-дигидроизохинолин-1(2Н)-он-3-ил. Примеры N-содержащих 5- или 6-членных гетероциклоалкильных групп включают пиперидин-1-ил, пиперидин-4-ил, пиперазин-1-ил, 1,3-тиазинан-3-ил и морфолино. N-содержащий 5-10-членный гетероциклоалкил или N-содержащий 5- или 6-членный гетероциклоалкил возможно замещен 1 или более (например от 1 до 5) подходящими заместителями.
Как он используется в данном описании, термин группа "галогено" или "галоген" определен как включающий фтор, хлор, бром или иод.
Как он используется в данном описании, термин "галогеналкил" относится алкильной группе, имеющей один или более галогено-заместителей (вплоть до пергалогеналкила, то есть когда каждый атом водорода алкильной группы заменен атомом галогена), например, термин "C1-6галогеналкил" относится к C1-6алкильной группе, имеющей один или более галогено-заместителей (вплоть до пергалогеналкила, то есть когда каждый атом водорода алкильной группы заменен атомом галогена). Термин "C1-4галогеналкил" относится к C1-4алкильной группе, имеющей один или более галогено-заместителей (вплоть до пергалогеналкила, то есть когда каждый атом водорода алкильной группы заменен атомом галогена). Термин "C1-3згалогеналкил" относится к C1-3алкильной группе, имеющей один или более галогено-заместителей (вплоть до пергалогеналкила, то есть когда каждый атом водорода алкильной группы заменен атомом галогена). Примеры галогеналкильных групп включают CF3, C2F5, CHF2, CH2F, CH2CF3, CH2Cl и тому подобное.
Как он используется в данном описании, термин "алкокси" или "алкилокси" относится к группе -О-алкил. Термин "C1-6алкокси" или "C1-6алкилокси" относится к группе -O-(C1-6алкил). Термин "C1-4лкокси" или "C1-4алкилокси" относится к группе -O-(С1-4алкил). Термин "C1-3алкокси" или " C1-3алкилокси" относится к группе -O-(C1-3алкил). Примеры алкокси включают метокси, этокси, пропокси (например, н-пропокси и изопропокси), трет-бутокси, и тому подобное.
Как он используется в данном описании, термин "галогеналкокси" относится к группе -О-галогеналкил. Термин "C1-6галогеналкокси" относится к группе -O-(C1-6галогеналкил). Термин "C1-4галогеналкокси" относится к группе -O-(C1-4галогеналкил). Термин "C1-3галогеналкокси" относится к группе -O-(C1-3галогеналкил). Примером группы галогеналкокси является -OCF3.
Как он используется в данном описании, термин "арилокси" относится к группе -О-(C6-10арил). Примером группы арилокси является -О-фенил [то есть фенокси].
Как он используется в данном описании, термин "арилалкилокси" или "арилалкокси" относится к группе -О-C1-6алкил-C6-10арил. Примеры группы арилалкилокси включают -О-C1-4алкил-C6-10арил, -О-C1-2алкил-C6арил, или -О-СН2-фенил [то есть бензилокси].
Как он используется в данном описании, термин "фторалкил" относится к алкильной группе, имеющей один или более заместителей фтор (вплоть до перфторалкила, то есть когда каждый атом водорода алкильной группы заменен фтором). Например, термин "C1-6фторалкил" относится к C1-6алкильной группе, имеющей один или более заместителей фтор (вплоть до перфторалкила, то есть когда каждый атом водорода C1-6алкильной группы заменен фтором). Термин "C1-4фторалкил" относится к C1-4алкильной группе, имеющей один или более заместителей фтор (вплоть до перфторалкила, то есть когда каждый атом водорода C1-6алкильной группы заменен фтором). Термин "C1-3фторалкил" относится к C1-3алкильной группе, имеющей один или более заместителей фтор (вплоть до перфторалкила, то есть когда каждый атом водорода C1-3алкильной группы заменен фтором). Термин "C1фторалкил" относится к C1алкильной группе (то есть метилу), имеющей один или более заместителей фтор (вплоть до перфторметила, то есть CF3). Примеры фторалкильных групп включают CF3, C2F5, CH2CF3, CHF2, CH2F и тому подобное.
Как он используется в данном описании, термин "фторциклоалкил" относится к циклоалкильной группе, имеющей один или более заместителей фтор (вплоть до перфторциклоалкила, то есть когда каждый атом водорода циклоалкильной группы заменен фтором). Например, термин " C3-6фторциклоалкил" относится к C3-6циклоалкильной группе, имеющей один или более заместителей фтор (вплоть до C3-6перфторциклоалкила, то есть когда каждый атом водорода C3-6циклоалкильной группы заменен фтором). Примеры фторциклоалкильных групп включают фторциклопропил [то есть циклопропильную группу, имеющую один или более заместителей фтор (вплоть до перфторциклопропила, то есть когда каждый атом водорода циклопропильной группы заменен фтором), например 2-фтор-циклопропан-1-ил или 2,3-дифторциклопропан-1-ил] и фторциклобутил [то есть циклобутильную группу, имеющей один или более заместителей фтор (вплоть до перфторциклобутила, то есть когда каждый атом водорода циклобутильной группы заменен фтором)].
Как он используется в данном описании, термин "гидроксилалкил" или "гидроксиалкил" относится к алкильной группе, имеющей один или более (например, 1, 2 или 3) заместителей ОН. Термин "C1-6гидроксилалкил" или "C1-6гидроксиалкил" относится к C1-6алкильной группе, имеющей один или более (например, 1, 2 или 3) заместителей ОН. Примерами гидроксилалкильных групп являются -СН2ОН и -СН2СН2ОН.
Как он используется в данном описании, термин "алкоксиалкил" относится к алкильной группе, имеющей один или более алкокси (например, 1, 2 или 3) заместителей. Термин "C2-4алкоксиалкил" относится к C1-3алкильной группе, замещено группой C1-3алкокси, где общее количество углеродов алкильной и алкокси-группировок алкоксиалкила составляет 2, 3 или 4. Одним из примеров гидроксилалкильной группы является -СН2ОСН3.
Как он используется в данном описании, термин "цианоалкил" относится к алкильной группе, имеющей один или более (например, 1, 2 или 3) заместителей циано. Термин "C1-6цианоалкил" относится к C1-6алкильной группе, имеющей один или более (например, 1, 2 или 3) заместителей CN. Термин "C1-3цианоалкил" относится к C1-3алкильной группе, имеющей один или более (например, 1, 2 или 3) заместителей CN. Одним примером цианоалкильной группы является -CH2CN.
Как он используется в данном описании, термин "гетероарилалкенил" относится к группе -C2-6алкенил-(гетероарил). Примеры такой гетероарилалкенильной группы включают 2-(тиофен-2-ил)-этен-1-ил и 1-(пиридин-2-ил)-проп-1-ен-3-ил.
Как он используется в данном описании, термин "арилалкил" относится к группе -C1-6алкил-C6-10арил, а "циклоалкилалкил" относится к группе -C1-6алкил-C3-14циклоалкил. Примеры арилалкильных групп включают -C1-4алкил-C6-10арил, -C1-2алкил-C6-10арил и бензил. Примеры циклоалкилалкильных групп включают -C1-4алкил-C3-7циклоалкил, -C1-2алкил-C3-6циклоалкил и циклопропилметил-.
Как он используется в данном описании, термин "гетероарилалкил" относится к группе -C1-6алкил-(5-14-членный гетероарил), а термин "гетероциклоалкилалкил" относится к группе -C1-6алкил-(3-14-членный гетероциклоалкил). Примеры гетероарилалкильных групп включают группы -C1-4алкил-(5-14-членный гетероарил), -C1-2алкил-(5-10-членный гетероарил), -C1-2алкил-(5-6-членный гетероарил) и (пиридин-2-ил)-метил-. Примеры гетероциклоалкилалкильных групп включают группы -C1-4алкил-(3-14-членный гетероциклоалкил), -C1-2алкил-(3-10-членный гетероциклоалкил) и 2-(пиперидин-4-ил)-этил-.
Как он используется в данном описании, термин "оксо" относится к группе=O. Когда оксо является заместителем на атоме углерода, они вместе образуют карбонильную группировку [-С(=O)-]. Когда оксо является заместителем на атоме серы, они вместе образуют сульфинильную группировку [-S(=O)-]; когда две оксо-группы являются заместителями на атоме серы, они вместе образуют сульфонильную группировку [-S(=O)2-].
Как он используется в данном описании, термин "возможно замещенный" означает, что замещение является возможным, и поэтому включает как незамещенные, так и замещенные атомы и группировки. "Замещенный" атом или группировка указывает на то, что любой водород обозначенных атоме или группировке может быть заменен группой, выбранной из указанных групп-заместителей (вплоть до того, что каждый атом водорода на обозначенных атоме или группировке заменен группой, выбранной из указанных групп-заместителей), при условии, что нормальная валентность обозначенных атома или группировки не превышена, и что замещение имеет результатом стабильное соединение. Например, если метильная группа (то есть CH3) возможно замещена, тогда вплоть до 3 атомов водорода на атоме углерода могут быть заменены группами-заместителями.
Как используется в данном описании, если не уточнено иначе, точка присоединения заместителя может быть из любого подходящего положения заместителя, например, пиперидинил может представлять собой пиперидин-1-ил (присоединенный через атом N пиперидинила), пиперидин-2-ил (присоединенный через атом С в положении 2 пиперидинила), пиперидин-3-ил (присоединенный через атом С в положении 3 пиперидинила) или пиперидин-4-ил (присоединенный через атом С в положении 4 пиперидинила). В качестве другого примера, пиридинил (или пиридил) может представлять собой 2-пиридинил (или пиридин-2-ил), 3-пиридинил (или пиридин-3-ил) или 4-пиридинил (или пиридин-4-ил).
Когда связь с заместителем показана пересекающей связь, соединяющую два атома в кольце, тогда такой заместитель может быть связан с любым из образующих кольцо атомов, которые способны быть замещенными (то есть связаны с одним или более атомами водорода). Например, как показано в формуле а-101 ниже, R7 может быть связан с амидным атомом азота или одним из двух кольцевых атомов углерода, каждый из которых связан с атомом водорода. В качестве другого примера, как показано в формуле а-102 ниже (когда связь с заместителем показана пересекающей связь у каждого из двух колец в бициклической кольцевой системе), R7 может быть связан с любым образующим кольцо атомом, который способен быть замещенным (то есть связан с одним или более атомами водорода) либо в бензольном кольце, либо в пиразольном кольце индазола. В качестве еще одного примера, как показано в формуле а-103 ниже, замещение R7a располагается на бензольном кольце, а замещение R7b располагается на 5-членном кольце.
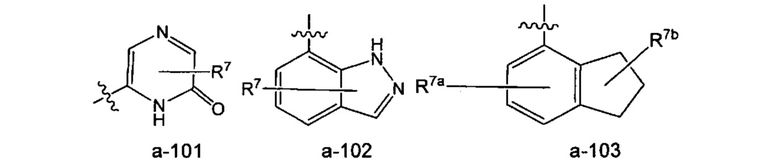
Когда заместитель указан без указания атома, посредством которого такой заместитель связан с остальной частью соединения данной формулы, тогда такой заместитель может быть связан посредством любого атома в таком заместителе. Например, заместитель на арилалкиле может быть связан с любым атомом на алкильной части или на арильной части арилалкила. Комбинации заместителей и/или переменных допустимы, только если такие комбинации имеют результатом стабильные соединения.
Как отмечалось выше, соединения формулы I могут существовать в форме фармацевтически приемлемых солей таких как, например, соли присоединения кислоты и/или соли присоединения основания соединений формулы I. Фраза "фармацевтически приемлемая(ые) соль(и)", как она используется в данном описании, если не указано иного, включает соли присоединения кислоты или основания, которые могут быть представлены в соединениях формулы I.
Фармацевтически приемлемые соли соединений, описанных выше, включают соли присоединения кислоты и соли с основаниями.
Подходящие соли присоединения кислоты могут быть образованы с кислотами, которые образуют нетоксичные соли. Примеры могут охватывать соли ацетат, адипат, аспартат, бензоат, безилат, бикарбонат/карбонат, бисульфат/сульфат, борат, камзилат, цитрат, цикламат, эдисилат, эзилат, формиат, фумарат, глуцептат, глюконат, глюкуронат, гексафторфосфат, гибензат, гидрохлорид/хлорид, гидробромид/бромид, гидройодид/йодид, изетионат, лактат, малат, малеат, малонат, мезилат, метилсульфат, нафтилат, напсилат, никотинат, нитрат, оротат, оксалат, пальмитат, памоат, фосфат/гидрофосфат/дигидрофосфат, пироглутамат, сахарат, стеарат, сукцинат, таннат, тартрат, тозилат, трифторацетат и ксинофоат.
Подходящие соли с основаниями могут быть образованы с основаниями, которые образуют нетоксичные соли. Примеры могут охватывать соли алюминия, соли с аргинином, с бензатином, соли кальция, соли с холином, с диэтиламином, с диоламином, с глицином, с лизином, соли магния, соли с меглумином, с оламином, соли калия, натрия, соли с трометамином и соли цинка.
Также могут быть образованы гемисоли с кислотами и основаниями, например, соль гемисульфат и гемикальциевая соль.
Обзор по подходящим солям смотри в "Handbook of Pharmaceutical Salts: Properties, Selection, and Use" by Stahl and Wermuth (Wiley-VCH, Weinheim, Germany, 2002). Способы получения фармацевтически приемлемых солей соединения формулы I известны специалисту в данной области.
Как они используются в данном описании, термины "формула I", "формула I или ее фармацевтически приемлемые соли", "фармацевтически приемлемые соли соединения или соль [формулы I]" определены как включающие все формы соединения формулы I, включая его гидраты, сольваты, изомеры (включая, например, вращательные стереоизомеры), кристаллические и не-кристаллические формы, изоморфы, полиморфы, метаболиты и пролекарства.
Как известно специалисту в данной области, аминные соединения (то есть те соединения, которые содержат один или более атомов азота), например третичные амины, могут образовывать N-оксиды (также известные как оксиды аминов или аминные N-оксиды). N-оксид имеет формулу (R100R200R300)N+-O-, где родительский амин (R100R200R300)N может представлять собой, например, третичный амин (например, каждый из R100, R200, R300 независимо представляет собой алкил, арилалкил, арил, гетероарил и тому подобное), гетероциклический или гетероароматический амин [например, (R100R200R300)N вместе образует 1-алкилпиперидин, 1-алкилпирролидин, 1-бензилпирролидин, или пиридин]. В частности, иминный азот, особенно гетероциклический или гетероароматический иминный азот, или атом азота пиридинного типа  [такой как атом азота в пиридине, пиридазине или пиразине], может быть N-окисленным с образованием N-оксида, содержащего группу
[такой как атом азота в пиридине, пиридазине или пиразине], может быть N-окисленным с образованием N-оксида, содержащего группу  . Таким образом, соединение согласно настоящему изобретению, содержащее один или более атомов азота (например, иминный атом азота), например, как часть Q1 формулы I, может быть способно образовывать его N-оксид (например, моно-N-оксиды, бис-N-оксиды или мульти-N-оксиды, или их смеси в зависимости от количества атомов азота, подходящих для образования стабильных N-оксидов). Как он используется в данном описании, термин "N-оксид(ы)" относится ко всем возможным и, в частности, всем стабильным N-оксидным формам аминного соединения (например соединения, содержащего один или более иминных атомов азота), раскрытого в данном описании, таким как моно-N-оксиды (включая разные изомеры, когда более чем один атом азота аминного соединения может образовывать моно-N-оксид) или мульти-N-оксиды (например бис-N-оксиды), или их смеси в любом соотношении.
. Таким образом, соединение согласно настоящему изобретению, содержащее один или более атомов азота (например, иминный атом азота), например, как часть Q1 формулы I, может быть способно образовывать его N-оксид (например, моно-N-оксиды, бис-N-оксиды или мульти-N-оксиды, или их смеси в зависимости от количества атомов азота, подходящих для образования стабильных N-оксидов). Как он используется в данном описании, термин "N-оксид(ы)" относится ко всем возможным и, в частности, всем стабильным N-оксидным формам аминного соединения (например соединения, содержащего один или более иминных атомов азота), раскрытого в данном описании, таким как моно-N-оксиды (включая разные изомеры, когда более чем один атом азота аминного соединения может образовывать моно-N-оксид) или мульти-N-оксиды (например бис-N-оксиды), или их смеси в любом соотношении.
Соединения формулы I могут быть превращены, возможно, в их N-оксиды, например, в присутствии подходящего окисляющего реагента в подходящем растворителе, например в присутствии перекиси водорода в метаноле или в присутствии м-хлорпероксибензойной кислоты в дихлорметане. Специалист в данной области легко определит реакционные условия, подходящие для осуществления реакций N-окисления.
Соединения формулы I, раскрытые в данном описании (соединения по изобретению), включают их N-оксиды и фармацевтически приемлемые соли соединений или N-оксидов.
Соединения по изобретению могут существовать в континууме твердых состояний в диапазоне от полностью аморфного состояния до полностью кристаллического состояния. Термин "аморфный" относится к состоянию, в котором у вещества отсутствует дальний порядок на молекулярном уровне, и, в зависимости от температуры, вещество может проявлять физические свойства твердого вещества или жидкости. Обычно такие вещества не дают характерных картин дифракции рентгеновских лучей и, хотя они и проявляют свойства твердого вещества, их более формально описывают как жидкость. При нагревании происходит изменение свойств от свойств твердого вещества до свойств жидкости, которое характеризуется изменением состояния, обычно второго порядка ("стеклование"). Термин "кристаллический" относится к твердой фазе, в которой вещество имеет регулярную упорядоченную внутреннюю структуру на молекулярном уровне и дает картину дифракции рентгеновских лучей с характерными пиками. Такие вещества при достаточном нагревании также будут проявлять свойства жидкости, но переход от твердого состояния в жидкое состояние характеризуется фазовым превращением, обычно первого порядка ("точка плавления").
Соединения формулы I могут существовать в несольватированных и сольватированных формах. Когда растворитель или вода прочно связаны, комплекс будет иметь хорошо определенную стехиометрию независимо от влажности. Когда, однако, растворитель или вода связаны слабо, как в канальных сольватах и гигроскопичных соединениях, содержание воды/растворителя будет зависеть от влажности и условий сушки. В таких случаях нестехиометрия является нормой.
Соединения формулы I могут существовать как клатраты или другие комплексы (например со-кристаллы). В объем изобретения включены комплексы, такие как клатраты, комплексы включения лекарственное средство-хозяин, где лекарственное средство и хозяин присутствуют в стехиометрических или нестехиометрических количествах. Также включены комплексы соединения формулы I, содержащие два или более органических и/или неорганические компонента, которые могут находиться в стехиометрических или нестехиометрических количествах. Полученные комплексы могут быть ионизированными, частично ионизированными или неионизированными. Обзор таких комплексов представлен в J. К. Haleblian, J. Pharm. Sci. 1975, 64, 1269-1288. Сокристаллы обычно определяют как кристаллические комплексы нейтральных молекулярных составляющих, которые связаны вместе посредством нековалентных взаимодействий, но которые также могут представлять собой комплекс нейтральной молекулы с солью. Сокристаллы могут быть получены в результате кристаллизации из расплава, перекристаллизации из растворителей или совместного физического измельчения компонентов (смотри О. Almarsson and М. J. Zaworotko, Chem. Commun., 2004, 17, 1889-1896. Общую информацию по таким многокомпонентным комплексам смотри в Haleblian, J. Pharm. Sci., 1975, 64, 1269-1288.
Соединения по изобретению также могут существовать в мезоморфном состоянии (мезофаза или жидкий кристалл) под воздействием соответствующих условий. Мезоморфное состояние является промежуточным состоянием между истинным кристаллическим состоянием и истинным жидким состоянием (либо расплав, либо раствор). Мезоморфизм, возникающий в результате изменения температуры, описывают как "термотропный", а мезоморфизм, являющийся результатом добавления второго компонента, такого как вода или другой растворитель, описывают как "лиотропный". Соединения, способные образовывать лиотропные мезофазы, описывают как "амфифильные", и они состоят из молекул, которые обладают ионной (например -COO-Na+, -COO-K+ или 
Изобретение также относится к пролекарствам соединений формулы I. Таким образом, некоторые производные соединений формулы (I), которые сами по себе могут иметь небольшую фармакологическую активность или не иметь ее, могут быть превращены в соединения формулы (I), имеющие желаемую активность, например гидролитическим расщеплением, при введении в или на организм. Такие производные упоминаются как "пролекарства". Дополнительную информацию о применении пролекарств можно найти в Prodrugs as Novel Delivery Systems, Vol. 14, ACS Symposium Series (T. Higuchi и W. Stella) и Bioreversible Carriers in Drug Design, Pergamon Press, 1987 (Ed. E. B. Roche, American Pharmaceutical Association).
Пролекарства в соответствии с изобретением можно, например, получать заменой подходящих функциональных групп, присутствующих в соединениях формулы (I), определенными группировками, известными специалистам как "про-группировки", как описано, например, в Design of Prodrugs, Н. Bundgaard (Elsevier, 1985).
Некоторые неограничивающие примеры пролекарств в соответствии с этим изобретением включают в себя:
(1) когда соединение формулы I содержит функциональную группу карбоновой кислоты, она функционализируется в подходящую метаболически лабильную группу (сложные эфиры, карбаматы и так далее);
(2) когда соединение формулы I содержит спиртовую функциональную группу, она функционализируется в подходящую метаболически лабильную группу (простые эфиры, сложные эфиры, фосфонаты, сульфонаты, карбаматы, ацетали, кетали и так далее);
(3) когда соединение формулы I содержит первичную или вторичную аминную функциональную группу или амид, она функционализируется в подходящую метаболически лабильную группу, например, гидролизуемую группу (амиды, карбаматы и так далее).
Другие примеры групп замещения в соответствии с вышеуказанными примерами и примеры других типов пролекарств могут быть найдены в вышеуказанных ссылках.
Кроме того, некоторые соединения формулы I могут сами действовать в качестве пролекарств других соединений формулы I.
Также в объем изобретения включены метаболиты соединений формулы I, то есть соединения, образующиеся in vivo при введении лекарственного средства.
В некоторых воплощениях соединения формулы I включают их N-оксиды и фармацевтически приемлемые соли соединений или N-оксидов.
Соединения формулы I включают все стереоизомеры и таутомеры. Стереоизомеры формулы I включают цис и транс изомеры, оптические изомеры, такие как R и S энантиомеры, диастереомеры, геометрические изомеры, вращательные изомеры, атропоизомеры и конформационные изомеры соединений формулы I, включая соединения, существующие более чем в одном типе изомерии; и их смеси (такие как рацематы и диастереомерные пары). Также включены соли присоединения кислот или соли присоединения оснований, где противоион является оптически активным, например, D-лактат или L-лизин, или рацемическим, например, DL-тартрат или DL-аргинин.
В некоторых воплощенияя соединения формулы I могут иметь ассимметричные атомы углерода. Углерод-углеродные связи соединений формулы I могут быть представлены в данном описании, используя сплошную линию  , сплошной клин
, сплошной клин  или пунктирный клин
или пунктирный клин  . Использование сплошной линии для изображения связей с асимметричными атомами углерода предполагается для указания на то, что включены все возможные стереоизомеры (например конкретные энантиомеры, рацемические смеси и так далее) по этому атому углерода. Использование сплошного или пунктирного клина для изображения связей с асимметричными атомами углерода предназначено для того, чтобы указать на то, что включенным считается только показанный стереоизомер. Возможно, что соединения формулы I могут содержать более чем один асимметричный атом углерода. В таких соединениях использование сплошной линии для изображения связей с асимметричными атомами углерода предназначено для того, чтобы указать на то, что все возможные стереоизомеры подразумеваются как включенные. Например, если не указано иное, предполагается, что соединения формулы I могут существовать как энантиомеры и диастереомеры или как рацематы и их смеси. Использование сплошной линии для изображения связей с одним или более асимметричных атомов углерода в соединении формулы I и использование сплошного или пунктирного клина для изображения связей с другими асимметричными атомами углерода в том же соединении означает, что присутствует смесь диастереомеров.
. Использование сплошной линии для изображения связей с асимметричными атомами углерода предполагается для указания на то, что включены все возможные стереоизомеры (например конкретные энантиомеры, рацемические смеси и так далее) по этому атому углерода. Использование сплошного или пунктирного клина для изображения связей с асимметричными атомами углерода предназначено для того, чтобы указать на то, что включенным считается только показанный стереоизомер. Возможно, что соединения формулы I могут содержать более чем один асимметричный атом углерода. В таких соединениях использование сплошной линии для изображения связей с асимметричными атомами углерода предназначено для того, чтобы указать на то, что все возможные стереоизомеры подразумеваются как включенные. Например, если не указано иное, предполагается, что соединения формулы I могут существовать как энантиомеры и диастереомеры или как рацематы и их смеси. Использование сплошной линии для изображения связей с одним или более асимметричных атомов углерода в соединении формулы I и использование сплошного или пунктирного клина для изображения связей с другими асимметричными атомами углерода в том же соединении означает, что присутствует смесь диастереомеров.
В некоторых воплощениях соединения формулы I могут существовать и/или могут быть выделены как атропоизомеры (например один или более аторопэнантиомеров). Для специалиста в данной области будет понятно, что атропизомеризм может существовать в соединении, которое имеет два или более ароматических колец (например два ароматических кольца, связанные посредством простой связи), смотри, например, Freedman, Т. В. et al. Absolute Configuration Determination of Chiral Molecules in Solution State Using Vibrational Circular Dichroism. Chirality 2003, 15, 743-758; и Brngomann, G. et al. Atroposelective Synthesis Axially Chiral Biaryl Compounds. Angew. Chem., Int. Ed. 2005, 44: 5384-5427.
Когда любой рацемат кристаллизуется, возможны кристаллы двух разных типов. Первый тип представляет собой рацемическое соединение (истинный рацемат), указанное выше, где получается одна гомогенная форма кристалла, содержащая оба энантиомеры в эквимолярных количествах. Второй тип представляет собой рацемическую смесь или конгломерат, где получаются обе формы кристалла в эквимолярных количествах, причем каждая содержит отдельный энантиомер.
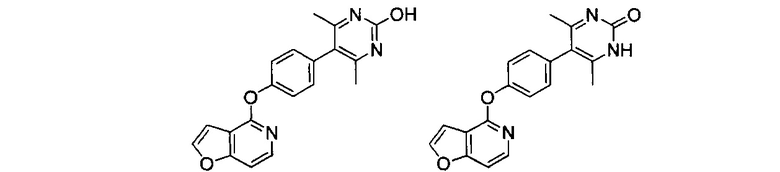
Соединения формулы I могут проявлять феномены таутомеризма и структурного изомеризма. Например, соединения формулы I могут существовать в нескольких таутомерных формах, включая енольную и иминную форму, и кето и енаминную форму, и геометрические изомеры и их смеси. Все такие таутомерные формы включены в объем соединений формулы I. Таутомеры могут существовать в виде смесей таутомерной совокупности в растворе. В твердой форме обычно превалирует один таутомер. Даже несмотря на то, что описан может быть один таутомер, настоящее изобретение включает все таутомеры соединений формулы I. Например, когда один из следующих двух таутомеров по изобретению раскрыт в экспериментальном разделе в данном описании, специалист в данной области с очевидностью признает, что изобретение также включает и другой.
Настоящее изобретение включает в себя все фармацевтически приемлемые меченные изотопом соединения формулы I, где один или более чем один атом заменен атомом, имеющим то же атомное число, но атомная масса или массовое число которого отличается от атомной массы или массового числа, преобладающих в природе.
Примеры изотопов, подходящих для включения в соединения по изобретению, включают изотопы водорода, такие как 2Н и 3Н, углерода, такие как 11С, 13С и 14С, хлора, такие как 36Cl, фтора, такие как 18F, йода, такие как 123I и 125I, азота, такие как 13N и 15N, кислорода, такие как 15O, 17O и 18O, фосфора, такие как 32Р, и серы, такие как 35S.
Некоторые меченные изотопом соединения формулы I, например с включенным радиоактивным изотопом, являются полезными в исследованиях распределения лекарственного средства и/или субстрата в тканях. Радиоактивные изотопы тритий, то есть 3Н, и углерод-14, то есть 14C, являются особенно полезными для этой цели ввиду простоты их введения и готовых средств детектирования.
Замещение более тяжелыми изотопами, такими как дейтерий, то есть 2Н, может обеспечивать терапевтические преимущества по причине большей метаболической стабильности, например большего периода полувыведения in vivo или пониженных требований к дозировке и, следовательно, могут быть предпочтительными в некоторых обстоятельствах.
Замещение позитрон-испускающими изотопами, такими как 11C, 18F, 15O и 13N, может быть полезным в исследованиях с использованием позитроно-эмиссионной томографии (PET) для изучения занятости рецепторов субстратом.
Меченные изотопом соединения формулы I (или их фармацевтически приемлемые соли либо N-оксиды соединений или солей) обычно могут быть получены общепринятыми способами, известными специалистам, или способами, аналогичными описанным в прилагаемых Примерах и Полготовительных примерах, с использованием подходящего меченного изотопом реагента вместо используемого ранее немеченого реагента.
Конкретные воплощения соединений формулы I включают их N-оксиды и фармацевтически приемлемые соли соединений или N-оксидов.
Воплощением настоящего изобретения является соединение формулы I, где Y1 представляет собой О.
Воплощением настоящего изобретения является соединение формулы I, где Y1 представляет собой S.
Воплощением настоящего изобретения является соединение формулы, где Y1 представляет собой NH или N(CH3). В другом воплощении Y1 представляет собой NH. В другом дополнительном воплощении Y1 представляет собой N(CH3).
Воплощением настоящего изобретения является соединение формулы I, где X1 представляет собой О.
Воплощением настоящего изобретения является соединение формулы I, где X1 представляет собой S.
Воплощением настоящего изобретения является соединение формулы I, где Q1 представляет собой N-содержащий 5-10-членный гетероциклоалкил или N-содержащий 5-10-членный гетероарил, где каждый из образующих кольцо атомов гетероциклоалкила или гетероарила независимо выбран из N и С; и гетероциклоалкил или гетероарил возможно замещены 1,2,3 или 4 независимо выбранными R7. В дополнительном воплощении Q1 представляет собой N-содержащий 5-10-членный гетероциклоалкил, возможно замещенный 1, 2, 3 или 4 независимо выбранными R7, и где каждый из образующих кольцо атомов гетероциклоалкила независимо выбран из N и С.
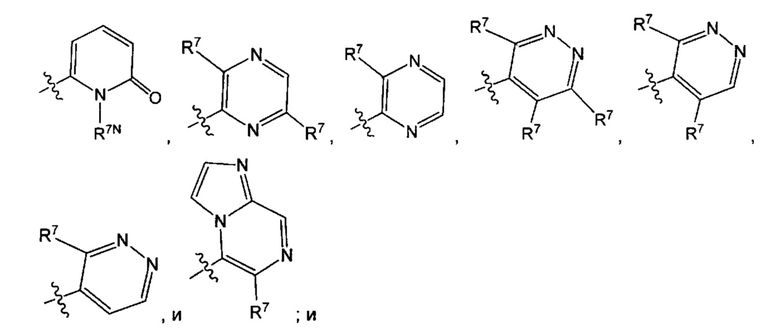
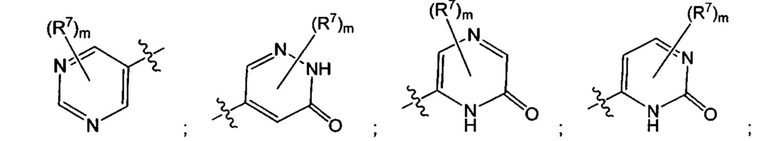
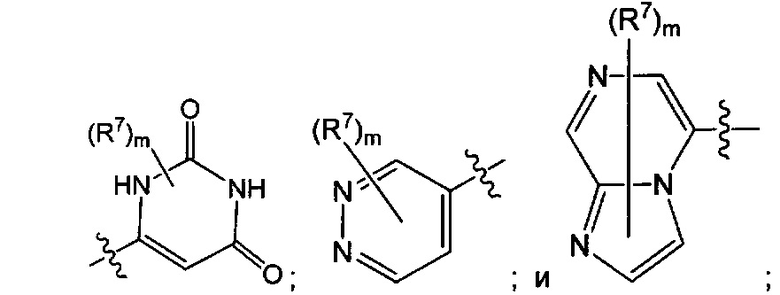
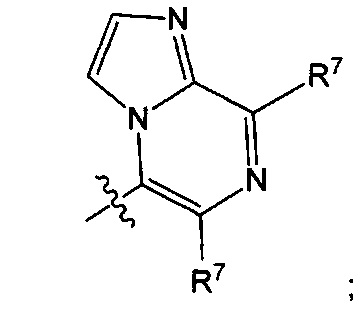
Воплощением настоящего изобретения является соединение формулы I, где Q1 представляет собой N-содержащий 5-10-членный гетероарил, возможно замещенный 1, 2, 3 или 4 независимо выбранными R7, и где каждый из образующих кольцо атомов гетероарила независимо выбран из N и С. В другом конкретном воплощении Q1 выбран из групп хинолинил, изохинолинил, 1Н-имидазо[4,5-с]пиридинил, имидазо[1,2-а]пиридинил, 1Н-пирроло[3,2-с]пиридинил, имидазо[1,2-а]пиразинил, имидазо[2,1-с][1,2,4]triazinyl, имидазо[1,5-а]пиразинил, имидазо[1,2-а]пиримидинил, 1Н-индазолил, 9Н-пуринил, пиримидинил, пиразинил, пиридинил, пиридазинил, 1H-пиразолил, 1Н-пирролил, 4H-пиразолил, 4Н-имидазолил, имидазо[1,2-а]пиримидинил, [1,2,4]триазоло[1,5-а]пиримидинил, [1,2,4]триазоло[4,3-b]пиридазинил, 1Н-имидазолил, 3-оксо-2Н-пиридазинил, 1Н-2-оксо-пиримидинил, 1H-2-оксо-пиридинил, 2,4(1Н,3Н)-диоксо-пиримидинил и 1Н-2-оксо-пиразинил, каждая из которых возможно замещена 1, 2, 3 или 4 независимо выбранными R7.
Воплощением настоящего изобретения является соединение формулы I, где Q1 выбран из 1Н-пиразолила, 1H-имидазолила, пиридинила, пиримидинила, пиридазинила, пиразинила, 3-оксо-2Н-пиридазинила, 1Н-2-оксо-пиримидинила, 1Н-2-оксо-пиразинила, 2,4(1Н,3H)-диоксо-пиримидинила, 1H-2-оксо-пиридинила, изохинолинила, 1Н-имидазо[4,5-с]пиридинила, имидазо[1,2-а]пиридинила, имидазо[1,2-а]пиримидинила, [1,2,4]триазоло[4,3-b]пиридазинила и имидазо[1,2-а]пиразинила, каждый из которых возможно замещен 1, 2, 3 или 4 независимо выбранными R7.
Воплощением настоящего изобретения является соединение формулы I, где Q1 выбран из:
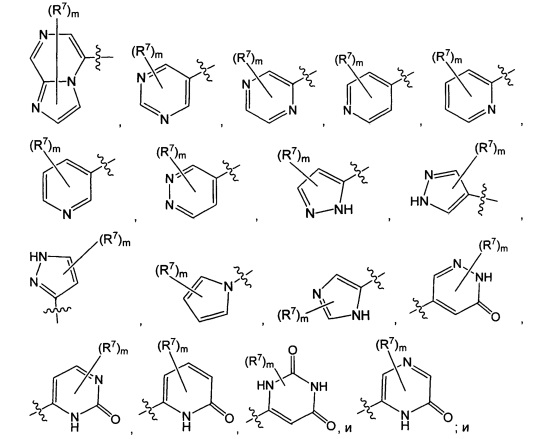
каждый m независимо представляет собой 0, 1, 2 или 3.
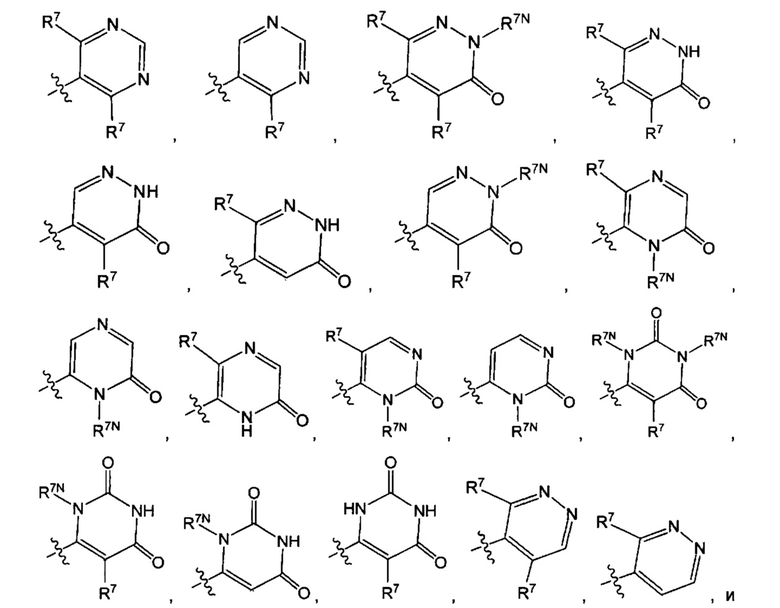
Воплощением настоящего изобретения является соединение формулы I, где Q1 выбран из:
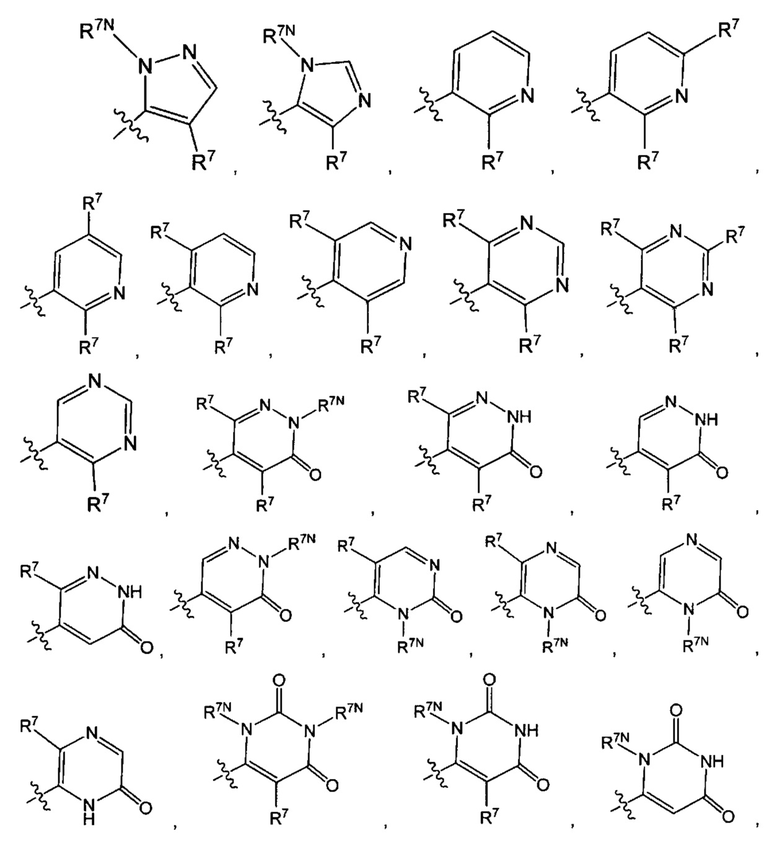
каждый R7N представляет собой Н или C1-3алкил, где C1-3алкил возможно замещен 1, 2, 3, 4 или 5 заместителями, каждый из которых независимо выбран из галогена (например F), ОН, C1-4алкокси, -NH2, групп -NH(C1-4алкил), -N(C1-4алкил)2 и -N(R14)(R15); и где R14 и R15 вместе с N атомом, к которому они присоединены, образуют 4-10-членный гетероциклоалкил, возможно замещенный 1, 2 или 3 заместителями, каждый из которых независимо выбран из группы, состоящей из галогена, оксо, -ОН, C1-4алкила, C1-4алкокси, C1-4галогеналкила, C1-4галогеналкокси и C1-4гидроксилалкила. В другом воплощении каждый R7N представляет собой Н или C1-3алкил, где C1-3алкил возможно замещен 1, 2, 3, 4 или 5 заместителями, каждый из которых независимо выбран из галогена (например F), ОН, C1-4алкокси, -NH2, групп -NH(C1-4алкил), -N(C1-4алкил)2, азетидин-1-ил, пирролидин-1-ил и пиридин-1-ил.
Воплощением настоящего изобретения является соединение формулы I, где Q1 представляет собой пиримидинил, пиразинил, 3-оксо-2Н-пиридазинил, 1H-2-оксо-пиразинил, 2,4(1Н,3H)-диоксо-пиримидинил, 1Н-2-оксо-пиримидинил или имидазо[1,2-а]пиразинил, каждый из которых возможно замещен 1, 2 или 3 независимо выбранными R7. В дополнительном воплощении каждый R7 независимо представляет собой C1-3алкил, возможно замещенный 1, 2, 3, 4 или 5 заместителями, каждый из которых независимо выбран из галогена (например F), ОН, C1-4алкокси, -NH2, групп -NH(C1-4алкил), -N(C1-4алкил)2 и -N(R14)(R15), где R14 и R15 вместе с N атомом, к которому они присоединены, образуют 4-10-членный гетероциклоалкил, возможно замещенный 1, 2 или 3 заместителями, каждый из которых независимо выбран из группы, состоящей из галогена, оксо, -ОН, группы C1-4алкил, C1-4алкокси, C1-4галогеналкил, C1-4галогеналкокси и C1-4гидроксилалкил. В еще одном воплощении каждый R7 независимо представляет собой C1-3алкил, возможно замещенный 1, 2, 3, 4 или 5 заместителями, каждый из которых независимо выбран из галогена (например F), ОН, групп C1-4алкокси, -NH2, -NH(C1-4алкил), -N(C1-4алкил)2, азетидин-1-ил, пирролидин-1-ил и пиридин-1-ил. В еще одном дополнительном воплощении каждый R7 представляет собой метил.
Воплощением настоящего изобретение является соединение формулы I, где Q1 выбран из:
 и m представляет собой 1, 2 или 3. В дополнительном воплощении каждый R7 независимо представляет собой C1-3алкил, возможно замещенный 1, 2, 3, 4 или 5 заместителями, каждый из которых независимо выбран из галогена (например F), ОН, C1-4алкокси, -NH2, групп -NH(C1-4алкил), -N(C1-4алкил)2 и -N(R14)(R15), где R14 и R15 вместе с N атомом, к которому они присоединены, образуют 4-10-членный гетероциклоалкил, возможно замещенный 1, 2 или 3 заместителями, каждый из которых независимо выбран из группы, состоящей из галогена, оксо, -ОН, C1-4алкила, C1-4алкокси, C1-4галогеналкила, C1-4галогеналкокси и C1-4гидроксилалкила. В еще одном дополнительном воплощении каждый R7 независимо представляет собой C1-3алкил, возможно замещенный 1, 2, 3, 4 или 5 заместителями, каждый из которых независимо выбран из галогена (например, F), ОН, C1-4алкокси, -NH2, групп -NH(C1-4алкил), -N(C1-4алкил)2, азетидин-1-ил, пирролидин-1-ил и пиридин-1-ил. В еще одном дополнительном воплощении m представляет собой 1 или 2. В еще одном дополнительном воплощении каждый R7 представляет собой метил.
и m представляет собой 1, 2 или 3. В дополнительном воплощении каждый R7 независимо представляет собой C1-3алкил, возможно замещенный 1, 2, 3, 4 или 5 заместителями, каждый из которых независимо выбран из галогена (например F), ОН, C1-4алкокси, -NH2, групп -NH(C1-4алкил), -N(C1-4алкил)2 и -N(R14)(R15), где R14 и R15 вместе с N атомом, к которому они присоединены, образуют 4-10-членный гетероциклоалкил, возможно замещенный 1, 2 или 3 заместителями, каждый из которых независимо выбран из группы, состоящей из галогена, оксо, -ОН, C1-4алкила, C1-4алкокси, C1-4галогеналкила, C1-4галогеналкокси и C1-4гидроксилалкила. В еще одном дополнительном воплощении каждый R7 независимо представляет собой C1-3алкил, возможно замещенный 1, 2, 3, 4 или 5 заместителями, каждый из которых независимо выбран из галогена (например, F), ОН, C1-4алкокси, -NH2, групп -NH(C1-4алкил), -N(C1-4алкил)2, азетидин-1-ил, пирролидин-1-ил и пиридин-1-ил. В еще одном дополнительном воплощении m представляет собой 1 или 2. В еще одном дополнительном воплощении каждый R7 представляет собой метил.
Воплощением настоящего изобретения является соединение формулы I, где Q1 выбран из:
 каждый R7 независимо представляет собой Н или - C1-3алкил (например метил или этил); и каждый R7N представляет собой Н или C1-3алкил, где C1-3алкил возможно замещен 1, 2, 3, 4 или 5 заместителями, каждый из которых независимо выбран из галогена (например, F), ОН, C1-4алкокси, -NH2, групп -N(C1-4алкил), -N(C1-4алкил)2 и -N(R14)(R15), и где R14 и R15 вместе с атомом N, к которому они присоединены, образуют 4-10-членный гетероциклоалкил, возможно замещенный 1, 2 или 3 заместителями, каждый из которых независимо выбран из группы, состоящей из галогена (например F), оксо, -ОН, C1-4алкила, C1-4алкокси, C1-4галогеналкила, C1-4галогеналкокси и C1-4гидроксилалкила. В другом воплощении каждый R7 независимо представляет собой Н, метил или этил; и каждый R7N представляет собой C1-3алкил, возможно замещенный 1, 2, 3, 4 или 5 заместителями, каждый из которых независимо выбран из галогена (например F), ОН, C1-4алкокси, -NH2, групп -NH(C1-4алкил), -N(C1-4алкил)2, азетидин-1-ил, пирролидин-1-ил и пиридин-1-ил. В другом воплощении каждый R7 представляет собой метил или этил; и каждый R7N представляет собой C1-3алкил, возможно замещенный 1, 2, 3, 4 или 5 заместителями, каждый из которых независимо выбран из галогена (например F), ОН, C1-4алкокси, -NH2, групп -NH(C1-4алкил), -N(C1-4алкил)2, азетидин-1-ил, пирролидин-1-ил и пиридин-1-ил. В еще одном воплощении каждый R7 представляет собой метил, и каждый R7N представляет собой метил.
каждый R7 независимо представляет собой Н или - C1-3алкил (например метил или этил); и каждый R7N представляет собой Н или C1-3алкил, где C1-3алкил возможно замещен 1, 2, 3, 4 или 5 заместителями, каждый из которых независимо выбран из галогена (например, F), ОН, C1-4алкокси, -NH2, групп -N(C1-4алкил), -N(C1-4алкил)2 и -N(R14)(R15), и где R14 и R15 вместе с атомом N, к которому они присоединены, образуют 4-10-членный гетероциклоалкил, возможно замещенный 1, 2 или 3 заместителями, каждый из которых независимо выбран из группы, состоящей из галогена (например F), оксо, -ОН, C1-4алкила, C1-4алкокси, C1-4галогеналкила, C1-4галогеналкокси и C1-4гидроксилалкила. В другом воплощении каждый R7 независимо представляет собой Н, метил или этил; и каждый R7N представляет собой C1-3алкил, возможно замещенный 1, 2, 3, 4 или 5 заместителями, каждый из которых независимо выбран из галогена (например F), ОН, C1-4алкокси, -NH2, групп -NH(C1-4алкил), -N(C1-4алкил)2, азетидин-1-ил, пирролидин-1-ил и пиридин-1-ил. В другом воплощении каждый R7 представляет собой метил или этил; и каждый R7N представляет собой C1-3алкил, возможно замещенный 1, 2, 3, 4 или 5 заместителями, каждый из которых независимо выбран из галогена (например F), ОН, C1-4алкокси, -NH2, групп -NH(C1-4алкил), -N(C1-4алкил)2, азетидин-1-ил, пирролидин-1-ил и пиридин-1-ил. В еще одном воплощении каждый R7 представляет собой метил, и каждый R7N представляет собой метил.
Воплощением по изобретению является соединение формулы I, где Q1 выбран из:
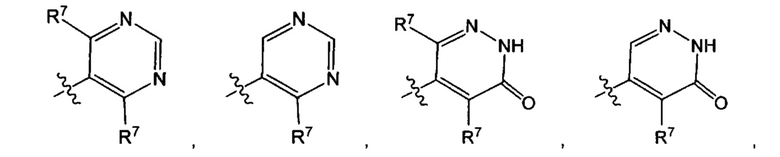
 и каждый R7 независимо представляет собой C1-3алкил (например метил или этил). В другом воплощении каждый R7 независимо представляет собой метил или этил. В еще одном воплощении каждый R7 представляет собой метил.
и каждый R7 независимо представляет собой C1-3алкил (например метил или этил). В другом воплощении каждый R7 независимо представляет собой метил или этил. В еще одном воплощении каждый R7 представляет собой метил.
Воплощением настоящего изобретения является соединение формулы I, где Q1 представляет собой
 и каждый R7 независимо представляет собой C1-3алкил (например метил или этил). В другом воплощении каждый R7 представляет собой метил.
и каждый R7 независимо представляет собой C1-3алкил (например метил или этил). В другом воплощении каждый R7 представляет собой метил.
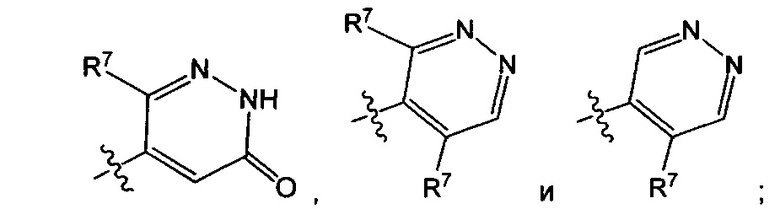
Воплощением настоящего изобретения является соединение формулы I, где Q1 представляет собой
 R7 представляет собой Н или C1-3алкил (например метил или этил); и R7N представляет собой C1-3алкил, возможно замещенный 1, 2, 3, 4 или 5 заместителями, каждый из которых независимо выбран из галогена (например F), ОН, C1-4алкокси, -NH2, групп -NH(C1-4алкил), -N(C1-4алкил)2 и -N(R14)(R15); и где R14 и R15 вместе с атомом N, к которому они присоединены, образуют 4-10-членный гетероциклоалкил, возможно замещенный 1, 2 или 3 заместителями, каждый из которых независимо выбран из группы, состоящей из галогена, оксо, -ОН, C1-4алкила, C1-4алкокси, C1-4галогеналкила, C1-4галогеналкокси и C1-4гидроксилалкила. В другом воплощении R7 представляет собой метил или этил; и R7N представляет собой C1-3алкил, возможно замещенный 1, 2, 3, 4 или 5 заместителями, каждый из которых независимо выбран из галогена (например F), ОН, C1-4алкокси, -NH2, групп -NH(C1-4алкил), -N(C1-4алкил)2, азетидин-1-ил, пирролидин-1-ил и пиридин-1-ил. В еще одном дополнительном воплощении R7 представляет собой метил, и R7N представляет собой метил.
R7 представляет собой Н или C1-3алкил (например метил или этил); и R7N представляет собой C1-3алкил, возможно замещенный 1, 2, 3, 4 или 5 заместителями, каждый из которых независимо выбран из галогена (например F), ОН, C1-4алкокси, -NH2, групп -NH(C1-4алкил), -N(C1-4алкил)2 и -N(R14)(R15); и где R14 и R15 вместе с атомом N, к которому они присоединены, образуют 4-10-членный гетероциклоалкил, возможно замещенный 1, 2 или 3 заместителями, каждый из которых независимо выбран из группы, состоящей из галогена, оксо, -ОН, C1-4алкила, C1-4алкокси, C1-4галогеналкила, C1-4галогеналкокси и C1-4гидроксилалкила. В другом воплощении R7 представляет собой метил или этил; и R7N представляет собой C1-3алкил, возможно замещенный 1, 2, 3, 4 или 5 заместителями, каждый из которых независимо выбран из галогена (например F), ОН, C1-4алкокси, -NH2, групп -NH(C1-4алкил), -N(C1-4алкил)2, азетидин-1-ил, пирролидин-1-ил и пиридин-1-ил. В еще одном дополнительном воплощении R7 представляет собой метил, и R7N представляет собой метил.
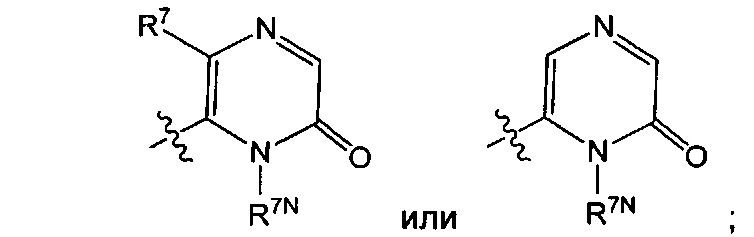
Воплощением настоящего изобретения является соединение формулы I, где Q1 представляет собой
 R7 представляет собой Н или C1-3алкил (например метил или этил); R7N представляет собой C1-3алкил, возможно замещенный 1, 2, 3, 4 или 5 заместителями, каждый из которых независимо выбран из галогена (например F), ОН, C1-4алкокси, -NH2, групп -NH(C1-4алкил), -N(C1-4алкил)2 и -N(R14)(R15); и R14 и R15 вместе с атомом N, к которому они присоединены, образуют 4-10-членный гетероциклоалкил, возможно замещенный 1, 2 или 3 заместителями, каждый из которых независимо выбран из группы, состоящей из галогена, оксо, -ОН, C1-4алкила, C1-4алкокси, C1-4галогеналкила, C1-4галогеналкокси и C1-4гидроксилалкила. В другом воплощении R7 представляет собой метил или этил; и R7N представляет собой C1-3алкил, возможно замещенный 1, 2, 3, 4 или 5 заместителями, каждый из которых независимо выбран из галогена (например F), ОН, C1-4алкокси, -NH2, групп -NH(C1-4алкил), -N(C1-4алкил)2, азетидин-1-ил, пирролидин-1-ил и пиридин-1-ил. В еще одном дополнительном воплощении R7 представляет собой метил, и R7N представляет собой метил.
R7 представляет собой Н или C1-3алкил (например метил или этил); R7N представляет собой C1-3алкил, возможно замещенный 1, 2, 3, 4 или 5 заместителями, каждый из которых независимо выбран из галогена (например F), ОН, C1-4алкокси, -NH2, групп -NH(C1-4алкил), -N(C1-4алкил)2 и -N(R14)(R15); и R14 и R15 вместе с атомом N, к которому они присоединены, образуют 4-10-членный гетероциклоалкил, возможно замещенный 1, 2 или 3 заместителями, каждый из которых независимо выбран из группы, состоящей из галогена, оксо, -ОН, C1-4алкила, C1-4алкокси, C1-4галогеналкила, C1-4галогеналкокси и C1-4гидроксилалкила. В другом воплощении R7 представляет собой метил или этил; и R7N представляет собой C1-3алкил, возможно замещенный 1, 2, 3, 4 или 5 заместителями, каждый из которых независимо выбран из галогена (например F), ОН, C1-4алкокси, -NH2, групп -NH(C1-4алкил), -N(C1-4алкил)2, азетидин-1-ил, пирролидин-1-ил и пиридин-1-ил. В еще одном дополнительном воплощении R7 представляет собой метил, и R7N представляет собой метил.
Воплощением настоящего изобретения является соединение формулы I, где Q1 представляет собой фенил, возможно замещенный 1, 2, 3, 4 или 5 независимо выбранными R7a.
Воплощением настоящего изобретения является соединение формулы I, где:
Q1 представляет собой группировку 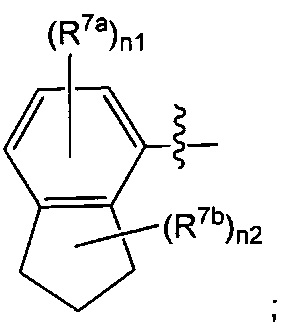 n1 представляет собой 0, 1 или 2; и n2 представляет собой 0, 1, 2 или 3.
n1 представляет собой 0, 1 или 2; и n2 представляет собой 0, 1, 2 или 3.
Воплощением настоящего изобретения является соединение формулы I, где каждый из RT1 и RT2 независимо выбран из группы, состоящей из Н, C1-3алкила и C1-3фторалкила. В другом воплощении каждый из RT1 и RT2 независимо выбран из группы, состоящей из Н, метила и C1фторалкила. В еще одном дополнительном воплощении каждый из RT1 и RT2 независимо выбран из группы, состоящей из Н и метила. В еще одном дополнительном воплощении оба RT1 и RT2 представляют собой Н.
Воплощением настоящего изобретения является соединение формулы I, где R1 представляет собой Н или C1-3алкил (например метил). В другом воплощении R1 представляет собой Н.
Воплощением настоящего изобретения является соединение формулы I, где R2 представляет собой Н, -CN, Br, C1-3алкил (например метил) или циклопропил. В другом воплощении R2 представляет собой Н, -CN или Br. В еще одном дополнительном воплощении R2 представляет собой Н или -CN. В еще одном дополнительном воплощении R2 представляет собой Н. В другом дополнительном воплощении R2 представляет собой -CN. В другом дополнительном воплощении R2 представляет собой Br.
Воплощением настоящего изобретения является соединение формулы I, где каждый из R3 и R4 независимо выбран из группы, состоящей из Н, F, Cl и C1-3алкила. В другом воплощении каждый из R3 и R4 независимо выбран из группы, состоящей из Н, метила и F. В еще одном дополнительном воплощении один из R3 и R4 представляет собой Н; и другой из R3 и R4 выбран из группы, состоящей из Н, метила и F. В еще одном дополнительном воплощении оба R3 и R4 представляют собой Н.
Воплощением настоящего изобретения является соединение формулы I, где каждый из R3 и R4 независимо представляет собой Н или F. В другом воплощении один из R3 и R4 представляет собой Н; и другой из R3 и R4 представляет собой Н или F.
Воплощением настоящего изобретения является соединение формулы I, где каждый из R5 и R6 независимо выбран из группы, состоящей из Н, галогена, ОН, -CN, C1-6алкила, C1-6галогеналкила, 4-10-членного гетероциклоалкила, -N(R8)(R9), -N(R10)(C(=O)R11), -C(=O)-N(R8)(R9), -C(=O)-OR12 и -OR13, где каждый из указанных C1-6алкила и 4-10-членного гетероциклоалкила возможно замещен 1, 2 или 3 заместителями, каждый из которых независимо выбран из группы, состоящей из галогена, -CN, -ОН, -N(R14)(R15), -N(R16)(C(=O)R17), -C(=O)-OR18, -C(=O)H, -C(=O)R18 и -C(=O)N(R14)(R15). В другом воплощении каждый из R5 и R6 независимо выбран из группы, состоящей из Н, ОН, -CN, Cl, F, метила, этила, C1фторалкила, C1-3цианоалкила, -ОСН3, C1фторалкокси, -N(R8)(R9) и -OR13, где каждый из указанных метила или этила возможно замещен группой -N(R14)(R15). В еще одном дополнительном воплощении один из R5 и R6 представляет собой Н, F или метил; и другой из R5 и R6 выбран из группы, состоящей из Н, -ОН, -CN, Cl, F, метила, этила, C1фторалкила, C1-3цианоалкила, -ОСН3, С1фторалкокси, -N(R8)(R9) и -OR13, где каждый из указанных метила или этила возможно замещен группой -N(R14)(R15).
Воплощение настоящего изобретения составляет соединение формулы I, где один из R5 и R6 представляет собой Н, F или метил; а другой из R5 и R6 выбран из группы, состоящей из Н, ОН, -CN, Cl, F, метила, этила, С1фторалкила (например CF3 или CH2F), C1-3цианоалкила, -ОСН3, C1фторалкокси (например -OCF3) и NH2. В еще одном воплощении один из R5 и R6 представляет собой Н, F или метил; а другой из R5 и R6 выбран из группы, состоящей из Н, -ОН, -CN, Cl, F, метила, этила, CF3, CH2F и - ОСН3. В еще одном воплощении один из R5 и R6 представляет собой Н; а другой из R5 и R6 выбран из группы, состоящей из Н, -ОН, -CN, Cl, F, метила, этила, CF3, CH2F и - ОСН3.
Воплощение настоящего изобретения составляет соединение формулы I, где один из R5 и R6 представляет собой Н, F или метил; а другой из R5 и R6 выбран из группы, состоящей из Н, -CN, F, метила и -ОСН3. В другом воплощении один из R5 и R6 представляет собой Н или F; а другой из R5 и R6 выбран из группы, состоящей из Н, -CN, F, метила и -ОСН3. В еще одном воплощении один из R5 и R6 представляет собой Н; а другой из R5 и R6 выбран из группы, состоящей из Н, -CN, F, метила и - ОСН3. В еще одном дополнительном воплощении один из R5 и R6 представляет собой Н; а другой из R5 и R6 представляет собой -CN.
Воплощением настоящего изобретения является соединение формулы I, где один из R5 и R6 представляет собой Н; а другой из R5 и R6 представляет собой -OR13.
Воплощением настоящего изобретения является соединение формулы I, где один из R5 и R6 представляет собой Н; а другой из R5 и R6 выбран из группы, состоящей из -N(R8)(R9) и -CH2-N(R14)(R15).
Воплощением настоящего изобретения является соединение формулы I, где каждый из R4 и R6 независимо выбран из группы, состоящей из Н, F и C1-3алкила; и R5 и R3 вместе с двумя атомами углерода, к которым они присоединены, образуют конденсированный N-содержащий 5- или 6-членный гетероарил, конденсированный N-содержащий 5- или 6-членный гетероциклоалкил или конденсированное бензольное кольцо, где каждый из конденсированного гетероарила, конденсированного гетероциклоалкила и конденсированного бензольного кольца возможно замещен 1, 2, или 3 заместителями, каждый из которых независимо выбран из группы, состоящей из C1-3алкила,C1-3алкокси, C1-3галогеналкила и C1-3галогеналкокси.
Воплощением настоящего изобретения является соединение формулы I, где оба из R6 и R4 представляют собой Н; и R5 и R3 вместе с двумя атомами углерода, к которым они присоединены, образуют конденсированное бензольное кольцо, где это конденсированное бензольное кольцо возможно замещено 1, 2 или 3 заместителями, каждый из которых независимо выбран из группы, состоящей из галогена, C1-3алкила,C1-3алкокси,C1-3галогеналкила и C1-3галогеналкокси.
Воплощением настоящего изобретения является соединение формулы I, где оба из R6 и R4 представляют собой Н; и R5 и R3 вместе с двумя атомами углерода, к которым они присоединены, образуют конденсированный N-содержащий 5- или 6-членный гетероарил, где этот конденсированный гетероарил возможно замещен 1, 2 или 3 заместителями, каждый из которых независимо выбран из группы, состоящей из C1-3алкила, C1-3алкокси, C1-3галогеналкила и C1-3галогеналкокси.
Воплощением настоящего изобретения является соединение формулы I, где оба из R6 и R4 представляют собой Н; и R5 и R3 вместе с двумя атомами углерода, к которым они присоединены, образуют конденсированный N-содержащий 5- или 6-членный гетероциклоалкил, где этот конденсированный гетероциклоалкил возможно замещен от 1 до 2 заместителями, каждый из которых независимо выбран из группы, состоящей из C1-3алкила.
Воплощением настоящего изобретения является соединение формулы I, где каждый из R7 и R7a независимо выбран из группы, состоящей из галогена, оксо, -ОН, -CN, C1-6алкила, C1-6галогеналкила, C1-6алкокси, C3-7циклоалкила, 4-10-членного гетероциклоалкила, 5-10-членного гетероарила, арилалкила, гетероарилалкила и -N(R14)(R15), где C1-6алкил возможно замещен 1, 2, 3, 4 или 5 заместителями, каждый из которых независимо выбран из галогена (например F), ОН, C1-4алкокси и -N(R14)(R15), и где каждый из указанных C3-7циклоалкила, гетероциклоалкила, гетероарила, арилалкила и гетероарилалкила возможно замещен 1, 2 или 3 заместителями, каждый из которых независимо выбран из группы, состоящей из галогена, -ОН, C1-4алкила и C1-4алкокси.
Воплощением настоящего изобретения является соединение формулы I, где каждый из R7 и R7a независимо выбран из группы, состоящей из галогена, оксо, -ОН, -CN, C1-6алкила, C1-6галогеналкила, C1-6алкокси, C3-7циклоалкила, 4-10-членного гетероциклоалкила, 5-10-членного гетероарила, арилалкила, гетероарилалкила и -N(R14)(R15), где C1-6алкил возможно замещен 1, 2, 3, 4 или 5 заместителями, каждый из которых независимо выбран из группы, состоящей из галогена (например F), ОН, C1-4алкокси, -NH2, групп -NH(C1-4алкил), -N(C1-4алкил)2, азетидин-1-ил, пирролидин-1-ил и пиридин-1-ил, и где каждый из указанных C3-7циклоалкила, гетероциклоалкила, гетероарила, арилалкила и гетероарилалкила возможно замещен 1, 2 или 3 заместителями, каждый из которых независимо выбран из группы, состоящей из галогена, -ОН, C1-4алкила и C1-4алкокси. В дополнительном воплощении каждый из R7 и R7a независимо выбран из группы, состоящей из галогена, -ОН, -CN, C1-6алкила, C1-6галогеналкила, C1-6алкокси, C3-7циклоалкила, 4-10-членного гетероциклоалкила, 5-10-членного гетероарила, арилалкила, гетероарилалкила и -N(R14)(R15), где C1-6алкил возможно замещен 1, 2, 3, 4 или 5 заместителями, каждый из которых независимо выбран из группы, состоящей из галогена (например F), ОН, C1-4алкокси, -NH2, групп -NH(C1-4алкил), -N(C1-4алкил)2, азетидин-1-ил, пиррол идин-1-ил и пиридин-1-ил, и где каждый из указанных C3-7циклоалкила, гетероциклоалкила, гетероарила, арилалкила и гетероарилалкила возможно замещен 1, 2 или 3 заместителями, каждый из которых независимо выбран из группы, состоящей из галогена, -ОН, C1-4алкила и C1-4алкокси.
Воплощением настоящего изобретения является соединение формулы I, где каждый из R7 и R7a независимо выбран из группы, состоящей из C1-4алкила, C1-4галогеналкила, оксо, -ОН, C1-4алкокси, C1-4галогеналкокси, галогена, -CN, -NH2, групп -NH(C1-4алкил), -N(C1-4алкил)2 и -N(R14)(R15), где C1-4алкил возможно замещен 1, 2, 3, 4 или 5 заместителями, каждый из которых независимо выбран из группы, состоящей из галогена (например F), ОН, C1-4алкокси, -NH2, групп -NH(C1-4алкил), -N(C1-4алкил)2, азетидин-1-ил, пирролидин-1-ил и пиридин-1-ил; где R14 и R15 вместе с атомом N, к которому они присоединены, образуют 4-10-членный гетероциклоалкил или 5-10-членный гетероарил, возможно замещенный 1, 2 или 3 заместителями, каждый из которых независимо выбран из группы, состоящей из галогена, оксо, -ОН, -CN, -NH2, групп -NH(C1-4алкил), -N(C1-4алкил)2, C1-4алкил, C1-4алкокси, C1-4галогеналкил, C1-4галогеналкокси и C1-4 C1-4гидроксилалкил. В другом воплощении R14 и R15 вместе с атомом N, к которому они присоединены, образуют 4-10-членный гетероциклоалкил, возможно замещенный 1, 2 или 3 заместителями, каждый из которых независимо выбран из группы, состоящей из галогена, оксо, -ОН, C1-4алкила, C1-4алкокси, C1-4галогеналкила, C1-4галогеналкокси и C1-4гидроксилалкила.
Воплощением настоящего изобретения является соединение формулы I, где каждый из R7 и R7a независимо выбран из группы, состоящей из C1-4алкила, C1-4фторалкила, оксо, -ОН, C1-4алкокси и C1-4галогеналкокси, где C1-4алкил возможно замещен 1, 2, 3, 4 или 5 заместителями, каждый из которых независимо выбран из группы, состоящей из галогена (например F), ОН, C1-4алкокси, -NH2, групп -NH(C1-4алкил), -N(C1-4алкил)2, азетидин-1-ил, пирролидин-1-ил и пиридин-1-ил.
Воплощением настоящего изобретения является соединение формулы I, где каждый R7 независимо выбран из группы, состоящей из C1-4алкила, C1-4фторалкила, оксо, ОН, C1-4алкокси, C1-4галогеналкокси, галогена, -CN, -NH2, групп -NH(C1-4алкил), -N(C1-4алкил)2 и азетидинил, где C1-4алкил в R7 возможно замещен 1, 2, 3, 4 или 5 заместителями, каждый из которых независимо выбран из группы, состоящей из галогена (например F), ОН, C1-4алкокси, -NH2, групп -NH(C1-4алкил), -N(C1-4лкил)2, азетидин-1-ил, пирролидин-1-ил и пиридин-1-ил, и где указанный азетидинил в R7 возможно замещен 1, 2 или 3 заместителями, каждый из которых независимо выбран из группы, состоящей из F, C1-4алкила, C1-4гидроксилалкила и оксо.
Воплощением настоящего изобретения является соединение формулы I, где каждый R7 независимо выбран из группы, состоящей из C1-4алкила, C1-4фторалкила, оксо, ОН, C1-4алкокси и C1-4галогеналкокси, где C1-4алкил возможно замещен 1, 2, 3, 4 или 5 заместителями, каждый из которых независимо выбран из группы, состоящей из галогена (например F), ОН, C1-4алкокси, -NH2, групп -NH(C1-4алкил), -N(C1-4алкил)2, азетидин-1-ил, пирролидин-1-ил и пиридин-1-ил. В дополнительном воплощении каждый R7 независимо выбран из группы, состоящей из C1-4алкила, C1-4фторалкила и оксо, где C1-4алкил возможно замещен 1, 2, 3, 4 или 5 заместителями, каждый из которых независимо выбран из группы, состоящей из галогена (например F), ОН, C1-4алкокси, -NH2, групп -NH(C1-4алкил), -N(C1-4алкил)2, азетидин-1-ил, пирролидин-1-ил и пиридин-1-ил. В еще одном воплощении каждый R7 независимо выбран из группы, состоящей из C1-4алкила (например метила) и оксо, где C1-4алкил возможно замещен 1, 2, 3, 4 или 5 заместителями, каждый из которых независимо выбран из группы, состоящей из галогена (например F), ОН, C1-4алкокси, -NH2, групп -NH(C1-4алкил), -N(C1-4алкил)2, азетидин-1-ил, пиррол идин-1-ил и пиридин-1-ил.
Воплощением настоящего изобретения является соединение формулы I, где каждый R7a независимо выбран из группы, состоящей из C1-4алкила, C1-4фторалкила, ОН, C1-4алкокси, C1-4галогеналкокси, галогена, -CN, -NH2, групп -NH(C1-4алкил), -N(C1-4алкил)2, азетидинил, пирролидинил, 1,4,5,6-тетрагидропирроло[3,4-с]пиразолил, 2,5-дигидро-1Н-пирролил, тиоморфолино, пиперидинил, и пиперазинил, где каждый из указанных азетидинила, пирролидинила, 1,4,5,6-тетрагидропирроло[3,4-с]пиразолила, 2,5-дигидро-1Н-пирролила, тиоморфолино, пиперидинила и пиперазинила возможно замещен 1, 2, или 3 заместителями, каждый из которых независимо выбран из группы, состоящей из F, C1-4алкила, C1-4гидроксилалкила и оксо.
Воплощением настоящего изобретения является соединение формулы I, где Y1 представляет собой О, и X1 представляет собой О. Воплощением настоящего изобретения является соединение формулы I, где Y1 представляет собой О; X1 представляет собой О; каждый из RT1, RT2 и R1 представляет собой Н; и R2 представляет собой Н или -CN. Воплощением настоящего изобретения является соединение формулы I, где Y1 представляет собой О; X1 представляет собой О; каждый из RT1, RT2 и R1 представляет собой Н; R2 представляет собой Н или -CN; и каждый из R3 и R4 независимо представляет собой Н или F. В другом воплощении один из R3 и R4 представляет собой Н; а другой из R3 и R4 представляет собой Н или F. В еще одном воплощении R2 представляет собой Н; в еще одном дополнительном воплощении R2 представляет собой -CN.
Воплощением настоящего изобретения является соединение формулы I, где Y1 представляет собой О; X1 представляет собой О; каждый из RT1, RT2 и R1 представляет собой Н; R2 представляет собой Н или -CN; один из R3 и R4 представляет собой Н, а другой из R3 и R4 представляет собой Н или F; и один из R5 и R6 представляет собой Н или F, а другой из R5 и R6 выбран из группы, состоящей из Н, -CN, F, метила и -ОСН3. В дополнительном воплощении один из R5 и R6 представляет собой Н. В еще одном дополнительном воплощении R2 представляет собой Н; в другом дополнительном воплощении R2 представляет собой -CN.
Воплощением настоящего изобретения является соединение формулы I, где Y1 представляет собой О; X1 представляет собой О; каждый из RT1, RT2 и R1 представляет собой Н; R2 представляет собой Н или -CN; один из R3 и R4 представляет собой Н, а другой из R3 и R4 представляет собой Н или F; один из R5 и R6 представляет собой Н или F, а другой из R5 и R6 выбран из группы, состоящей из Н, -CN, F, метила и -ОСН3; и Q1 представляет собой пиримидинил, пиразинил, 3-оксо-2Н-пиридазинил, 1Н-2-оксо-пиразинил, 1Н-2-оксо-пиримидинил или имидазо[1,2-а]пиразинил, каждый из которых возможно замещен 1, 2 или 3 независимо выбранными R7. В еще одном дополнительном воплощении R2 представляет собой Н; в другом дополнительном воплощении R2 представляет собой -CN.
Воплощением настоящего изобретения является соединение формулы I, где Y1 представляет собой О; X1 представляет собой О; каждый из RT1, RT2 и R1 представляет собой Н; R2 представляет собой Н или -CN; один из R3 и R4 представляет собой Н, а другой из R3 и R4 представляет собой Н или F; один из R5 и R6 представляет собой Н или F, а другой из R5 и R6 выбран из группы, состоящей из Н, -CN, F, метила и -ОСН3; и Q1 представляет собой пиримидинил, пиразинил, 3-оксо-2H-пиридазинил, 1H-2-оксо-пиразинил, 1Н-2-оксо-пиримидинил или имидазо[1,2-а]пиразинил, каждый из которых возможно замещен 1, 2 или 3 C1-3алкилами. В другом воплощении Q1 представляет собой пиримидинил, пиразинил, 3-оксо-2Н-пиридазинил, 1Н-2-оксо-пиразинил, 1Н-2-оксо-пиримидинил или имидазо[1,2-а]пиразинил, каждый из которых возможно замещен 1, 2, или 3 метилами.
Воплощением настоящего изобретения является соединение формулы I, где Y1 представляет собой О; X1 представляет собой О; каждый из RT1, RT2 и R1 представляет собой Н; R2 представляет собой Н или -CN; один из R3 и R4 представляет собой Н, а другой из R3 и R4 представляет собой Н или F; один из R5 и R6 представляет собой Н или F, а другой из R5 и R6 выбран из группы, состоящей из Н, -CN, F, метила и -ОСН3; и Q1 выбран из:
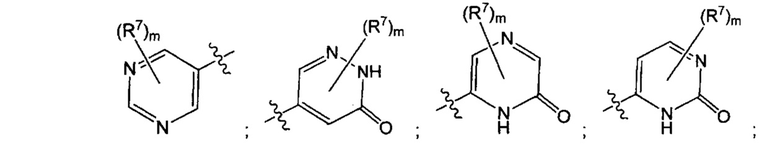
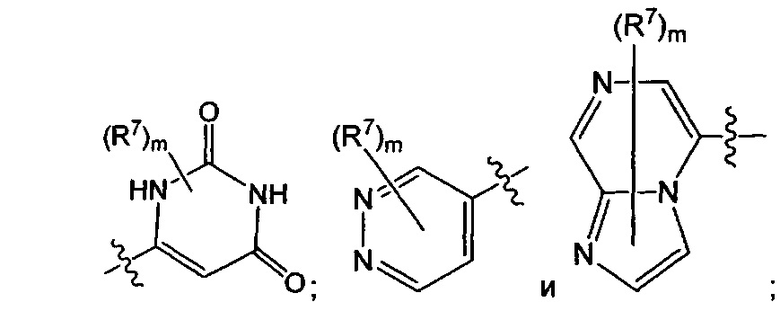 и m представляет собой 1, 2 или 3. В другом воплощении каждый R независимо представляет собой C1-3алкил, возможно замещенный 1, 2, 3, 4 или 5 заместителями, каждый из которых независимо выбран из группы, состоящей из галогена (например F), ОН, C1-4алкокси, -NH2, групп -NH(C1-4алкил), -N(C1-4алкил)2, азетидин-1-ил, пирролидин-1-ил и пиридин-1-ил. В другом дополнительном воплощении каждый R7 представляет собой метил. В еще одном дополнительном воплощении R2 представляет собой Н; в еще одном дополнительном воплощении R2 представляет собой -CN.
и m представляет собой 1, 2 или 3. В другом воплощении каждый R независимо представляет собой C1-3алкил, возможно замещенный 1, 2, 3, 4 или 5 заместителями, каждый из которых независимо выбран из группы, состоящей из галогена (например F), ОН, C1-4алкокси, -NH2, групп -NH(C1-4алкил), -N(C1-4алкил)2, азетидин-1-ил, пирролидин-1-ил и пиридин-1-ил. В другом дополнительном воплощении каждый R7 представляет собой метил. В еще одном дополнительном воплощении R2 представляет собой Н; в еще одном дополнительном воплощении R2 представляет собой -CN.
Воплощением настоящего изобретения является соединение формулы I, где Y1 представляет собой О; X1 представляет собой О; каждый из RT1, RT2 и R1 представляет собой Н; R2 представляет собой Н или -CN; один из R3 и R4 представляет собой Н, а другой из R3 и R4 представляет собой Н или F; один из R5 и R6 представляет собой Н или F, а другой из R5 и R6 выбран из группы, состоящей из Н, -CN, F, метила и -ОСН3; и Q1 выбран из:
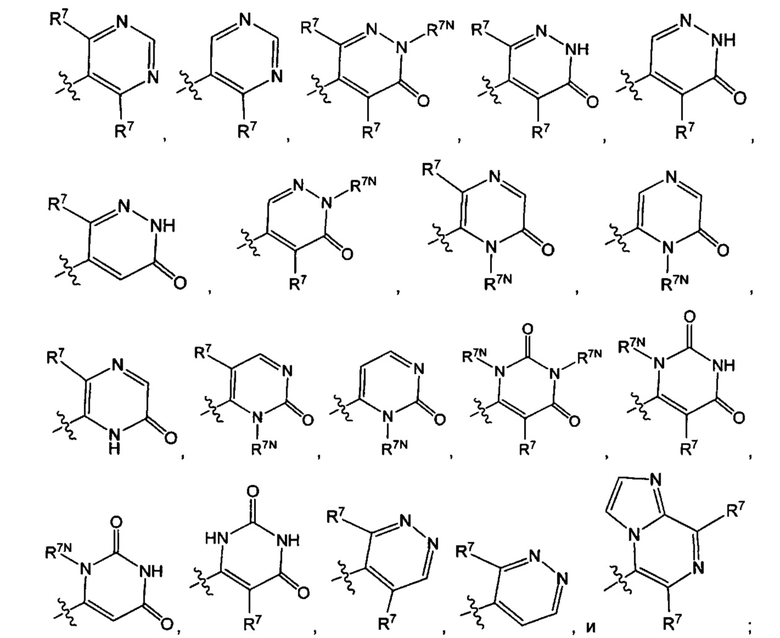
каждый R7 независимо представляет собой Н или C1-3алкил (например метил или этил); и каждый R7N представляет собой Н или C1-3алкил, где C1-3алкил возможно замещен 1, 2, 3, 4 или 5 заместителями, каждый из которых независимо выбран из галогена (например F), ОН, C1-4алкокси, -NH2, групп -NH(C1-4алкил), -N(C1-4алкил)2, азетидин-1-ил, пирролидин-1-ил и пиридин-1-ил. В дополнительном воплощении каждый R7 представляет собой метил или этил, а каждый R7N представляет собой C1-3алкил, возможно замещенный 1, 2, 3, 4 или 5 заместителями, каждый из которых независимо выбран из галогена (например F), ОН, C1-4алкокси, -NH2, групп -NH(C1-4алкил), -N(C1-4алкил)2, азетидин-1-ил, пирролидин-1-ил и пиридин-1-ил. В еще одном воплощении каждый R7 представляет собой метил, и каждый R7N представляет собой метил. В еще одном другом воплощении R2 представляет собой Н; в другом еще одном воплощении R2 представляет собой -CN.
В одном воплощении изобретения также предложены одно или более из соединений, описанных как соединения из Примеров 1-216 в разделе «Примеры» настоящей заявки, их N-оксиды и фармацевтически приемлемые соли соединений или N-оксидов.
В другом воплощении изобретение относится к соединениям формулы I, выбранным из группы, состоящей из:
4-[4-(4,6-диметилпиримидин-5-ил)-3-метилфенокси]фуро[3,2-с]пиридина;
2-(4,6-диметилпиримидин-5-ил)-5-(фуро[3,2-с]пиридин-4-илокси)-бензонитрила;
5-[2-фтор-4-(фуро[3,2-с]пиридин-4-илокси)фенил]-4,6-диметилпиридазин-3(2Н)-она;
5-[4-(фуро[3,2-с]пиридин-4-илокси)фенил]-4,6-диметилпиридазин-3(2H)-она;
(+)-5-[4-(фуро[3,2-с]пиридин-4-илокси)-2-метилфенил]-4,6-диметил-пиридазин-3(2Н)-она;
(-)-5-[4-(фуро[3,2-с]пиридин-4-илокси)-2-метилфенил]-4,6-диметил-пиридазин-3(2H)-она;
5-[4-(фуро[3,2-с]пиридин-4-илокси)-2-метилфенил]-4,6-диметил-пиридазин-3(2H)-она;
(+)-5-[4-(фуро[3,2-с]пиридин-4-илокси)-2-метилфенил]-6-метил-имидазо[1,2-а]-пиразина;
(-)-5-[4-(фуро[3,2-с]пиридин-4-илокси)-2-метилфенил]-6-метил-имидазо[1,2-а]-пиразина;
5-[4-(фуро[3,2-с]пиридин-4-илокси)-2-метилфенил]-6-метилимидазо[1,2-а]-пиразина;
4-[4-(4,6-диметилпиримидин-5-ил)-3-фторфенокси]фуро[3,2-с]пиридина;
4-[4-(4,6-диметилпиримидин-5-ил)фенокси]фуро[3,2-с]пиридина;
(-)-6-[4-(фуро[3,2-с]пиридин-4-илокси)-2-метилфенил]-1,5-диметил-пиразин-2(1H)-она;
(+)-6-[4-(фуро[3,2-с]пиридин-4-илокси)-2-метилфенил]-1,5-диметил-пиразин-2(1H)-она;
6-[4-(фуро[3,2-с]пиридин-4-илокси)-2-метилфенил]-1,5-диметилпиразин-2(1Н)-она;
6-[4-(фуро[3,2-с]пиридин-4-илокси)-2-метилфенил]-1,5-диметил-пиримидин-2(1H)-она;
4-[4-(4,6-диметилпиримидин-5-ил)-2-фторфенокси]фуро[3,2-с]пиридина;
5-[4-(фуро[3,2-с]пиридин-4-илокси)-2-метилфенил]-2,4,6-триметил-пиридазин-3(2Н)-она;
5-[4-(фуро[3,2-с]пиридин-4-илокси)-2-метилфенил]-4-метилпиридазин-3(2Н)-она;
(+)-4-[4-(3,5-диметилпиридазин-4-ил)-3-метилфенокси]фуро[3,2-с]-пиридина;
(-)-4-[4-(3,5-диметилпиридазин-4-ил)-3-метилфенокси]фуро[3,2-с]-пиридина;
4-[4-(3,5-диметилпиридазин-4-ил)-3-метилфенокси]фуро[3,2-с]пиридина;
4-[4-(3,5-диметил-6-оксо-1,6-дигидропиридазин-4-ил)фенокси]фуро[3,2-с]-пиридин-3-карбонитрила;
(-)-4-[4-(3,5-диметилпиридазин-4-ил)-3-метоксифенокси]фуро[3,2-с]-пиридина;
(+)-4-[4-(3,5-диметилпиридазин-4-ил)-3-метоксифенокси]фуро[3,2-с]-пиридина;
4-[4-(3,5-диметилпиридазин-4-ил)-3-метоксифенокси]фуро[3,2-с]-пиридина;
6-[4-(фуро[3,2-с]пиридин-4-илокси)-2-метилфенил]-1,5-диметил-пиримидин-2,4(1Н,3H)-диона;
(-)-6-[4-(фуро[3,2-с]пиридин-4-илокси)-2-метилфенил]-1,5-диметил-пиримидин-2,4(1Н,3Н)-диона;
(+)-6-[4-(фуро[3,2-с]пиридин-4-илокси)-2-метилфенил]-1,5-диметил-пиримидин-2,4(1Н,3Н)-диона и
6-[4-(фуро[3,2-с]пиридин-4-илокси)фенил]-1,5-диметилпиримидин-2,4(1Н,3Н)-диона,
или их N-оксида или фармацевтически приемлемой соли соединения или N-оксида.
В настоящем изобретении также предложены композиции (например фармацевтические композиции), содержащие соединение формулы I (включая его N-оксид или фармацевтически приемлемую соль соединения или N-оксида). Соответственно, в одном воплощении изобретения предложена фармацевтическая композиция, содержащая (терапевтически эффективное количество) соединение формулы I (его N-оксид или фармацевтически приемлемую соль соединения или N-оксида) и возможно содержащая фармацевтически приемлемый носитель. В одном дополнительном воплощении изобретения предложена фармацевтическая композиция, содержащая (терапевтически эффективное количество) соединение формулы I (его N-оксид или фармацевтически приемлемую соль соединения или N-оксида), возможно содержащая фармацевтически приемлемый носитель и, возможно, по меньшей мере один дополнительный медицинский или фармацевтический агент (такой как антипсихотический агент или агент против шизофрении, описанные ниже). В одном воплощении дополнительный медицинский или фармацевтический агент представляет собой агент против шизофрении, как описано ниже.
Фармацевтически приемлемый носитель может представлять собой любой традиционный фармацевтический носитель или эксципиент. Подходящие фармацевтические носители включают инертные разбавители или наполнители, воду и различные органические растворители (такие как гидраты и сольваты). Фармацевтические композиции могут, если желательно, содержать дополнительные ингредиенты, такие как корригенты, связующие агенты, эксципиенты и тому подобное. Так, для перорального введения таблетки, содержащие различные эксципиенты, такие как лимонная кислота, могут использоваться вместе с различными разрыхлителями, такими как крахмал, альгиновая кислота и некоторые сложные силикаты, и со связующими агентами, такими как сахароза, желатин и аравийская камедь. Кроме того, в процессе таблетирования часто являются полезными смазывающие агенты, такие как стеарат магния, лаурилсульфат натрия и тальк. Твердые композиции сходного типа могут также быть использованы в мягких и твердых заполненных желатиновых капсулах. Неограничивающие примеры материалов, таким образом, включают лактозу молочный сахар и высокомолекулярные политиленгликоли. Когда для перорального введения желательны водные суспензии или эликсиры, активное соединение может быть объединено с различными подсластителями или корригентами, красящими веществами или красителями и, если желательно, эмульгирующими агентами или суспендирующими агентами, вместе с разбавителями, такими как вода, этанол, пропиленгликоль, глицерин или их комбинации.
Фармацевтическая композиция может, например, быть в форме, подходящей для перорального введения, такой как таблетка, капсула, пилюля, порошок, форма отсроченного высвобождения, раствор или суспензия, для парентерального введения, такой как стерильный раствор, суспензия или эмульсия, для местного введения, такой как мазь или крем, или для ректального введения, такой как суппозиторий.
Примеры форм для парентерального ведения включают растворы или суспензии активных соединений в стерильных водных растворах, например, водных растворах пропиленгликоля или декстрозы. Такие лекарственные формы могут быть подходящим образом забуферены, если желательно.
Фармацевтическая композиция может быть в формах стандартной дозы, подходящих для разового введения точных дозировок. Для специалиста очевидно, что композиция может быть представлена в суб-терапевтической дозировке, так что предусматриваются множественные дозы.
В одном воплощении композиция содержит терапевтически эффективное количество соединения формулы I (или его N-оксида или фармацевтически приемлемой соли соединения или N-оксида) и фармацевтически приемлемый носитель.
Соединения формулы I (включая их N-оксиды или фармацевтически приемлемые соли соединения или N-оксида) являются D1 модуляторами. В некоторых воплощениях соединение формулы I представляет собой D1 агонист [то есть связывается (обладает аффинностью) и активирует D1 рецепторы]. В некоторых воплощениях, при использовании допамина как эталонного полного D1 агониста, соединение формулы I является суперагонистом (то есть соединением, которое способно продуцировать больший максимальный ответ, чем эндогенный D1 агонист, допамин, в отношении D1 рецептора, и тем самым демонстрировать эффективность более чем примерно 100%, например 120%). В некоторых воплощениях, при использовании допамина как эталонного полного агониста, соединение формулы I является полным D1 агонистом (то есть обладает эффективностью примерно 100%, например, 90%-100%, по сравнению с эффективностью допамина). В некоторых воплощениях, при использовании допамина как эталонного полного агониста, соединение формулы I является частичным агонистом [то есть соединением, имеющим только частичную эффективность (то есть менее чем 100%, например 10%-80% или 50%-70%) в отношении D1 рецептора относительно полного агониста, допамина, хотя оно связывается с D1 рецептором и активирует его]. D1 агонист (включая суперагонист, полный агонист и частичный агонист) может агонизировать или частично агонизировать активность D1. В некоторых воплощениях ЕС50 соединения формулы I в отношении D1 составляет менее чем примерно 10 мкМ, 5 мкМ, 2 мкМ, 1 мкМ, 500 нМ, 200 нМ, 100 нМ, 50, 40, 30, 20, 10, 5, 2 или 1 нМ.
Как он используется в данном описании в отношении соединения, термин "D1 модулятор" или "D1 агонист" (включая супер D1 агонист, полный D1 агонист или частичный D1 агонист) относится к соединению, которое является модулятором D1-подобного рецептора или агонистом D1-подобного рецептора, соответственно (то есть необязательно селективным между/среди субтипов D1-подобных рецепторов), смотри Lewis, JPET 286:345-353, 1998. D1R включают, например, D1 и D5 у людей и D1A и D1B у грызунов.
В настоящем изобретении дополнительно предложен способ модулирования (такого как агонизирование или частичное агонизирование) активности D1 рецептора (in vitro или in vivo), включающий приведение в контакт (включая инкубирование) D1 рецептора с соединением формулы I (таким как соединение, выбранное из Примеров 1-216), или его N-оксидом или фармацевтически приемлемой солью соединения или N-оксида.
Другое воплощение изобретения включает способ лечения D1-опосредованного (или D1-ассоциированного) расстройства, включающий введение нуждающемуся в этом млекопитающему (например человеку) количества соединения формулы I (включая его фармацевтически приемлемую соль или N-оксид соединения или соли), эффективного в модулировании (например агонизировании или частичном агонизировании) D1.
Соединения формулы I, используемые для лечения D1-опосредованного расстройства, включают их N-оксиды или фармацевтически приемлемые соли соединений или N-оксидов.
D1-опосредованные (или D1-ассоциированные) расстройства включают неврологические расстройства [такие как синдром Туретта (Tourette); поздняя дискинезия; болезнь Паркинсона; расстройства познавательной способности {включая амнезию, сенильную деменцию, ухудшение познавательной способности, связанное с возрастом, ВИЧ (вирус иммунодефицита) ассоциированную деменцию, деменцию, ассоциированную с болезнью Альцгеймера, деменцию, ассоциированную с болезнью Хантингтона, деменцию телец Леви (Lewy), васкулярную деменцию, деменцию, связанную с приемом лекарственных средств (например ухудшение познавательной способности, ассоциированное с терапией D2 антагонистом), делирий и умеренное нарушение познавательной способности}; хорея/болезнь Гентингтона], психиатрические расстройства [такие как тревога (включая острое стрессовое расстройство, генерализованное тревожное расстройство, социальное тревожное расстройство, паническое расстройство, посттравматическое стрессовое расстройство и обсессивно-компульсивное расстройство); симулятивное расстройство (включая острую галлюциногенную манию); расстройство побуждений/импульсивность (включая компульсивное влечение к азартным играм и перемежающееся взрывное поведение); расстройства настроения (биполярное расстройство I, биполярное расстройство II, мания, смешанное аффективное состояние, депрессия, включая большую депрессию, хроническую депрессию, сезонную депрессию, психотическую депрессию, послеродовую депрессию и устойчивую к лечению депрессию (TRD)); психомоторные расстройства; психотические расстройства [включая шизофрению (включая, например, когнитивные и негативные симптомы при шизофрении), шизоаффективное расстройство, шизофрениформное расстройство и бредовое расстройство]; токсикоманию и лекарственную зависимость (включая наркотическую зависимость, алкоголизм, амфетаминовую зависимость, кокаиновую аддикцию, никотиновую зависимость и синдром отмены лекарства); расстройства приема пищи (включая анорексию, булимию, компульсивное переедание, гиперфагию и пагофагию); расстройство аутического спектра (например аутизм); хроническую апатию, ангедонию, синдром хронической усталости, сезонное аффективное расстройство и педиатрические психиатрические расстройства (включая расстройство с дефицитом внимания, синдром дефицита внимания с гиперактивностью (ADHD), расстройство поведения и аутизм)], эндокринные нарушения (такие как гиперпролактинемия) или другие расстройства, включая сонливость, сексуальную дисфункцию, боль, мигрень, системную красную волчанку (SLE), гипергликемию, дислипидемию, ожирение, диабет, сепсис, постишемический тубулярный некроз, почечную недостаточность, устойчивый отек, нарколепсию, сердечно-сосудистое заболевание (например гипертензию), застойную сердечную недостаточность, послеоперационную гипотонию глаза, расстройства сна, серотониновый синдром.
В другом воплощении изобретения предложен способ лечения неврологических расстройств [таких как синдром Туретта (Tourette); поздняя дискинезия; болезнь Паркинсона; расстройства познавательной способности {включая амнезию, сенильную деменцию, ухудшение познавательной способности, связанное с возрастом, ВИЧ-ассоциированную деменцию, деменцию, ассоциированную с болезнью Альцгеймера, деменцию, ассоциированную с болезнью Гентингтона, деменцию телец Леви (Lewy), васкулярную деменцию, деменцию, связанную с приемом лекарственных средств (например ухудшение познавательной способности, ассоциированное с терапией D2 антагонистом), делирий и умеренное ухудшение познавательной способности}; хорея/болезнь Гентингтона], психиатрических расстройств [таких как тревога (включая острое стрессовое расстройство, генерализованное тревожное расстройство, социальное тревожное расстройство, паническое расстройство, пост-травматическое стрессовое расстройство и обсессивно-компульсивное расстройство); симулятивное расстройство (включая острую галлюциногенную манию); расстройство побуждений/импульсивность (включая компульсивное влечение к азартным играм и перемежающееся взрывное поведение); расстройства настроения (биполярное расстройство I, биполярное расстройство II, мания, смешанное аффективное состояние, депрессия, включая большую депрессию, хроническую депрессию, сезонную депрессию, психотическую депрессию и послеродовую депрессию); психомоторные расстройства; психотические расстройства [включая шизофрению, шизоаффективное расстройство, шизофрениформное расстройство и бредовое расстройство]; лекарственная зависимость (включая наркотическую зависимость, алкоголизм, амфетаминовую зависимость, кокаиновую аддикцию, никотиновую зависимость и синдром отмены лекарства); расстройства приема пищи (включая анорексию, булимию, компульсивное переедание, гиперфагию и пагофагию); и педиатрические психиатрические расстройства (включая расстройство с дефицитом внимания, синдром дефицита внимания с гиперактивностью (ADHD), расстройство поведения и аутизм)] или эндокринных нарушений (таких как гиперпролактинемия) у млекопитающего, включающий введение указанному млекопитающему терапевтически эффективного количества соединения формулы I.
Другое воплощение изобретения включает способ лечения расстройства у млекопитающего (например человека), включающий введение указанному млекопитающему терапевтически эффективного количества соединения формулы I, где расстройство выбрано из шизофрении (например когнитивных и негативных симптомов при шизофрении), нарушения познавательной способности [например нарушения познавательной способности, ассоциированного с шизофренией, нарушения познавательной способности, ассоциированного с AD, нарушения познавательной способности, ассоциированного с PD, нарушения познавательной способности, ассоциированного с фармакотерапией (например терапией D2 антагонистом)], синдрома дефицита внимания с гиперактивностью (ADHD), импульсивности, компульсивного влечения к азартным играм, переедания, расстройства аутического спектра, умеренного ухудшения познавательной способности (MCI), ухудшения познавательной способности, связанного с возрастом, деменции (например сенильной деменции, ВИЧ-ассоциированной деменции, деменции Альцгеймера, деменции телец Леви, васкулярной деменции или фронтотемпоральной деменции), синдрома усталых ног (RLS), болезни Паркинсона, хореи Гентингтона, тревоги, депрессии (например связанной с возрастом депрессии), большого депрессивного расстройства (MDD), устойчивой к лечению депрессии (TRD), биполярного расстройства, хронической апатии, ангедонии, синдрома хронической усталости, пост-травматического стрессового расстройства, сезонного аффективного расстройства, социального тревожного расстройства, послеродовой депрессии, серотонинового синдрома, токсикомании и лекарственной зависимости, обострения токсикомании, синдрома Туретта, тардивной дискинезии, гиперсомнии, чрезмерной сонливости в дневное время, кахексии, невнимательности, двигательного расстройства [например дискинезии (например хореи, дискинезии, индуцированной леводопой или тардивной дискинезии), тикового расстройства (например синдрома Туретта) или тремора], индуцированного терапией двигательного расстройства [например индуцированной терапией дискинезии (например индуцированной леводопой дискинезии ("LID")) или связанного с терапией дискнезического тремора (SSRI (селективные ингибиторы обратного захвата серотонина)-индуцированного постурального тремора)], сексуальной дисфункции (например эректильной дисфункции или пост-SSRI сексуальной дисфункции), мигрени, системной красной волчанки (SLE), гипергликемии, атеросклероза, дислипидемии, ожирения, диабета, сепсиса, пост-ишемического тубулярного некроза, почечной недостаточности, гипонатриемии, устойчивого отека, нарколепсии, гипертензии, застойной сердечной недостаточности, послеоперационной гипотонии глаза, растройств сна и боли.
Другое воплощение изобретения включает способ лечения расстройства у млекопитающего (например человека), включающий введение указанному млекопитающему терапевтически эффективного количества соединения формулы I, где расстройство выбрано из шизофрении (например когнитивных и негативных симптомов при шизофрении или нарушения познавательной способности, ассоциированного с шизофренией), нарушения познавательной способности, ассоциированного с терапией D2 антагонистом, синдрома дефицита внимания с гиперактивностью (ADHD), импульсивности, компульсивного влечения к азартным играм, расстройства аутического спектра, умеренного ухудшения познавательной способности (MCI), ухудшения познавательной способности, связанного с возрастом, деменции Альцгеймера, деменции телец Леви, васкулярной деменции, болезни Паркинсона, хореи Гентингтона, депрессии, тревоги, устойчивой к лечению депрессии (TRD), биполярного расстройства, хронической апатии, ангедонии, синдрома хронической усталости, посттравматического стрессового расстройства, сезонного аффективного расстройства, социального тревожного расстройства, послеродовой депрессии, серотонинового синдрома, токсикомании и лекарственной зависимости, синдрома Туретта, тардивной дискинезии, гиперсомнии, сексуальной дисфункции, мигрени, системной красной волчанки (SLE), гипергликемии, дислипидемии, ожирения, диабета, сепсиса, пост-ишемического тубулярного некроза, почечной недостаточности, устойчивого отека, нарколепсии, гипертензии, застойной сердечной недостаточности, послеоперационной гипотонии глаза, расстройств сна и боли.
Другое воплощение изобретения включает способ лечения депрессии у млекопитающего, например человека, включающий введение указанному млекопитающему (например человеку) терапевтически эффективного количества соединения формулы I.
Другое воплощение изобретения включает способ лечения болезни Паркинсона у млекопитающего, например человека, включающий введение указанному млекопитающему (например человеку) терапевтически эффективного количества соединения формулы I.
Другое воплощение изобретения включает способ лечения шизофрении (например когнитивных и негативных симптомов при шизофрении или ухудшения познавательной способности, ассоциированного с шизофренией) или психоза у млекопитающего, например человека, включающий введение указанному млекопитающему (например человеку) терапевтически эффективного количества соединения формулы I.
Другое воплощение изобретения включает способ лечения шизофрении (например когнитивных и негативных симптомов при шизофрении или ухудшения познавательной способности, ассоциированного с шизофренией) у млекопитающего, например человека, включающий введение указанному млекопитающему (например человеку) терапевтически эффективного количества соединения формулы I.
Другое воплощение изобретения включает способ лечения ухудшения познавательной способности, ассоциированного с щизофренией, у млекопитающего, например человека, включающий введение указанному млекопитающему (например человеку) терапевтически эффективного количества соединения формулы I.
Термин "терапевтически эффективное количество" как он используется в данном описании, относится к такому количеству соединения (включая его фармацевтически приемлемую соль или N-оксид соединения или соли), которое при введении будет облегчать в некоторой степени один или более симптомов подлежащего лечению расстройства. В случае лечения D1-опосредованного расстройства (например шизофрении), терапевтически эффективное количество относится к такому количеству, которое оказывает воздействие в виде облегчения в некоторой степени (или, например, элиминирования) одного или более симптомов D1-опосредованного расстройства (например шизофрении, или когнитивных и негативных симптомов при шизофрении, или нарушения познавательной способности, ассоциированного с шизофренией).
Термин "лечить", как он используется в данном описании, если не указано иное, означает реверсирование, ослабление, ингибирование развития или предупреждение расстройства или состояния, к котором такой термин применяется, или одного или более симптомов такого расстройства или состояния. Термин "лечение", как он используется в данном описании, если не указано иное, относится к акту лечения в том смысле, как термин "лечить" определен в данном описании. Термин "лечить" также включает адъювантное и нео-адъювантное лечение субъекта.
Введение соединений формулы I может быть осуществлено любым методом, который делает возможной доставку соединений в место их действия. Эти методы включают, например, парентеральные пути (например пероральные пути, буккальные пути, сублабиальные пути, сублингвальные пути), интраназальные пути, ингаляционные пути, интрадуоденальные пути, парентеральную инъекцию (включая внутривенную, подкожную, внутримышечную, внутриглазную инъекцию или инфузию), интратекальные пути, эпидуральные пути, интрацеребральные пути,
интрацеребровентрикулярные пути, местное и ректальное введение.
В одном воплощении настоящего изобретения соединения формулы I могут быть введены/задействованы пероральными путями.
Режимы дозирования могут быть скорректированы для обеспечения оптимального желаемого ответа. Например, может быть введен отдельный болюс, в течение промежутка времени может быть введено несколько разделенных доз или доза может быть пропорционально снижена или увеличена в зависимости от потребностей терапевтической ситуации. Может быть удобно готовить парентеральные композиции в форме стандартной дозы для легкости введения и единообразия дозировок. Форма стандартной дозы, как этот термин используется в данном описании, относится к физически дискретным единицам, подходящим в качестве однократных дозировок для субъектов - млекопитающих, подлежащих лечению; где каждая единица содержит предопределенное количество активного соединения, рассчитанное таким образом, чтобы продуцировать желаемый терапевтический эффект, в сочетании с требующимся фармацевтическим носителем. Спецификации для форм стандартной дозы по изобретению диктуются разнообразием факторов, таких как уникальные характеристики терапевтического агента и конкретный терапевтический или профилактический эффект, который должен быть достигнут.В одном воплощении настоящего изобретения соединения формулы I могут быть использованы для лечения людей.
Следует иметь в виду, что величины дозировок могут варьировать в зависимости от типа и тяжести ослабляемого состояния, и могут включать однократные и множественные дозы. Также следует понимать, что для любого конкретного субъекта специфические режимы дозирования следует корректировать с течением времени согласно индивидуальной потребности и профессиональному усмотрению специалиста, осуществляющего введение или наблюдающего за введением композиций, и что диапазоны доз, указанные в данном описании, являются только примерными и не предназначены для ограничения объема или практики заявленной композиции. Например, дозы могут быть скорректированы на основании фармакокинетических или фармакодинамических параметров, что может включать клинические эффекты, такие как токсические эффекты и/или лабораторные величины. Таким образом, настоящее изобретение охватывает увеличение дозы для одного пациента по определению специалиста в данной области. Определение соответствующих дозировок и режимов введения химиотерапевтического агента хорошо известно в релевантной области, и будет понято как охватываемое специалистом в данной области при знакомстве с информацией, раскрытой в данном описании.
Вводимое количество соединения формулы I будет зависеть от подлежащего лечению субъекта, тяжести расстройства или состояния, скорости введения, распределения соединения и усмотрения лечащего врача. Однако, эффективная дозировка находится в диапазоне от примерно 0,0001 до примерно 50 мг на кг массы тела в сутки, например от примерно 0,01 до примерно 5 мг/кг/сутки, в однократной или разделенных дозах. Для человека массой 70 кг это будет количество от примерно 0,7 мг до примерно 3500 мг/сутки, например от примерно 5 мг до примерно 2000 мг/сутки. В некоторых обстоятельствах уровни дозирования ниже нижнего предела вышеуказанного диапазона могут быть более чем адекватными, тогда как в других случаях могут быть использованы еще большие дозы, не вызывая никакого вредного побочного эффекта при условии, что такие большие дозы сначала делят на несколько маленьких доз для введения на протяжении суток.
Как он используется в данном описании, термин "комбинационная терапия" относится к введению соединения формулы I вместе с по меньшей мере одним дополнительным фармацевтическим или терапевтическим агентом (например агентом против шизофрении), либо последовательно, либо одновременно.
Настоящее изобретение включает применение комбинации соединения формулы I и одного или более дополнительных фармацевтически активных агентов. Если вводят комбинацию активных агентов, то их можно вводить последовательно или одновременно, в отдельных лекарственных формах или объединенными в единую лекарственную форму. Соответственно, настоящее изобретение также включает фармацевтические композиции, содержащие определенное количество: (а) первого агента, содержащего соединение формулы I (включая его N-оксид или фармацевтически приемлемую соль соединения или N-оксида); (b) второй фармацевтически активный агент; и (с) фармацевтически приемлемый носитель, наполнитель или разбавитель.
Для применения в сочетании с соединениями формулы I могут быть выбраны различные фармацевтически активные агенты, в зависимости от заболевания, расстройства или состояния, подлежащего лечению. Фармацевтически активные агенты, которые могут быть использованы в комбинации с композициями по настоящему изобретению, включают, без ограничения ими:
(1) ингибиторы ацетилхолинэстеразы, такие как гидрохлорид донепезила (ARICEPT, МЕМАС); или антагонисты аденозинового А2А рецептора, такие как преладенант (SCH 420814) или SCH 412348;
(2) амилоид-β (или его фрагменты), такой как Aβ1-15, конъюгированный с pan HLA DR-связывающим эпитопом (PADRE) и АСС-001 (Elan/Wyeth);
(3) антитела к амилоиду-β (или его фрагментам), такие как бапинеузумаб (также известный как ААВ-001) и ААВ-002 (Wyeth/Elan);
(4) агенты, понижающие уровень или ингибирующие амилоид (включая те, которые снижают продуцирование амилоида, его накопление и фибриллизацию), такие как колостринин и биснорцимсерин (также известный как BNC);
(5) агонисты альфа-адренергических рецепторов, такие как клонидин (CATAPRES);
(6) агенты, блокирующие бета-адренергические рецепторы (бета-блокаторы), такие как картеолол;
(7) антихолинэргические агенты, такие как амитриптилин (ELAVIL, ENDEP);
(8) противосудорожные агенты, такие как карбамазепин (TEGRETOL, CARBATROL);
(9) антипсихотические агенты, такие как луразидон (также известный как SM-13496; Dainippon Sumitomo);
(10) блокаторы кальциевых каналов, такие как нилвадипин (ESCOR, NIVADIL);
(11) ингибиторы катехол-О-метилтрансферазы (СОМТ), такие как толкапон (TASMAR);
(12) стимуляторы центральной нервной системы, такие как кофеин;
(13) кортикостероиды, такие как преднизон (STERAPRED, DELTASONE);
(14) агонисты допаминовых рецепторов, такие как апоморфин (APOKYN);
(15) антагонисты допаминовых рецепторов, такие как тетрабеназин (NITOMAN, XENAZINE);
(16) ингибиторы обратного захвата допамина, такие как малеат номифензина (MERITAL);
(17) агонисты рецепторов гамма-аминомасляной кислоты (GABA), такие как баклофен (LIORESAL, KEMSTRO);
(18) антагонисты гитамина 3 (H3), такие как ципроксифан;
(19 иммуномодуляторы, такие как ацетат глатирамера (также известный как кополимер-1; COPAXONE);
(20) иммуносупрессанты, такие как метотрексат (TREXALL, RHEUMATREX);
(21) интерфероны, включая интерферон бета-1а (AVONEX, REBIF) и интерферон бета-1b (BETASERON, BETAFERON);
(22) леводопа (или ее метиловый или этиловый сложный эфир), сама по себе или в комбинации с ингибитором DOPA декарбоксилазы (например карбидопа (SINEMET, CARBILEV, PARCOPA));
(23) антагонисты рецепторов N-метил-D-аспартата (NMDA), такие как мемантин (NAMENDA, AXURA, EBIXA);
(24) ингибиторы моноаминоксидазы (МАО) такие как селегилин (EMSAM);
(25) антагонисты мускариновых рецепторов (в частности подтипа М1), такие как хлорид бетанехола (DUVOID, URECHOLINE);
(26) нейропротективные лекарственные средства, такие как оксим 2,3,4,9-тетрагидро-1 H-карбазол-3-она;
(27) агонисты никотиновых рецепторов, такие как эпибатидин;
(28) ингибиторы обратного захвата норэпинефрина (норадреналина), такие как атомоксетин (STRATTERA);
(29) ингибиторы PDE9 (фосфодиэстераза 9), такие как BAY 73-6691 (Bayer AG);
(30) ингибиторы фосфодиэстеразы (PDE), включая (а) ингибиторы PDE1 (например винпоцетин), (б) ингибиторы PDE2 (например эритро-9-(2-гидрокси-3-нонил)аденина (EHNA)), (в) ингибиторы PDE4 (например ролипрам), и (г) ингибиторы PDE5 (например силденафил (VIAGRA, REVATIO));
(31) хинолины, такие как хинин (включая его соли гидрохлорид, дигидрохлорид, сульфат, бисульфат и глюконат);
(32) ингибиторы В-секретазы, такие как WY-25105;
(33) ингибиторы у-секретазы, такие как LY-411575 (Lilly);
(34) антагонисты рецепторов серотонина (5-гидрокситриптамина) 1А (5-HT1A), такие как спиперон;
(35) агонисты рецепторов серотонина (5-гидрокситриптамина) 4 (5-НТ4), такие как PRX-03140 (Epix);
(36) антагонисты рецепторов серотонина (5-гидрокситриптамина) 6 (5-НТ6), такие как миансерин (TORVOL, BOLVIDON, NORVAL);
(37) ингибиторы обратного захвата серотонина (5-НТ), такие как алапроклат, циталопрам (CELEXA, CIPRAMIL);
(38) трофические факторы, такие как фактор роста нервов (NGF), основной фактор роста фибробластов (bFGF; ERSOFERMIN), нейротрофин-3 (NT-3), кардиотрофин-1, мозговой нейротрофический фактор (BDNF), нейбластин, метеорин и глиальный нейротрофический фактор (GDNF), и агенты, которые стимулируют продуцирование трофических факторов, такие как пропентофиллин;
и тому подобное.
Соединение формулы I возможно использовать в комбинации с другим активным агентом. Такой активный агент может представлять собой, например, атипический антипсихотический агент, или агент против болезни Паркинсона, или агент против болезни Альцгеймера. Соответственно, в другом воплощении изобретения предложены способы лечения D1-опосредованного расстройства (например неврологического и психиатрического расстройства, ассоциированного с D1), включающие введение млекопитающему эффективного количества соединения формулы I (включая его N-оксид или фармацевтически приемлемую соль соединения или N-оксида) и дополнительно включающие введение другого активного агента.
Как он используется в данном описании, термин "другой активный агент" относится к любому терапевтическому агенту, отличному от соединения формулы I (включая его N-оксид или фармацевтически приемлемую соль соединения или N-оксида), который полезен для лечения расстройства у субъекта. Примеры дополнительных терапевтических агентов включают антидепрессанты, антипсихотические агенты (такие как агенты против шизофрении), обезболивающие агенты, агенты против болезни Паркинсона, агенты против LID, агенты против болезни Альцгеймера и против тревоги. Примеры конкретных классов антидепрессантов, которые могут быть использованы в комбинации с соединениями по изобретению, включают ингибиторы обратного захвата норэпинефрина, селективные ингибиторы обратного захвата серотонина (SSRI), антагонисты рецепторов NK-1, ингибиторы моноаминоксидазы (MAOI), обратимые ингибиторы моноаминоксидазы (RIMA), ингибиторы обратного захвата серотонина и норадреналина (SNRI), антагонисты фактора высвобождения кортикотропина (CRF), антагонисты α-адренорецепторов и атипичные антидепрессанты. Подходящие ингибиторы обратного захвата норэпинефрина включает третичные аминные трициклы и вторичные аминные трициклы. Примеры подходящих третичных аминных трициклов и вторичных аминных трициклов включают амитриптилин, кломипрамин, доксепин, имипрамин, тримипрамин, дотиепин, бутриптилин, иприндол, лоферпрамин, нортриптилин, протриптилин, амоксапин, десипрамин и мапротилин. Примеры подходящих селективных ингибиторов обратного захвата серотонина включают флуоксепин, флувоксамин, пароксетин и сертралин. Примеры ингибиторов моноаминоксидазы включают изокарбоксазин и транилциклопрамин. Примеры подходящих обратимых ингибиторов моноаминоксидазы включают моклобемид. Примеры подходящих ингибиторов обратного захвата серотонина и норадреналина для применения в настоящем изобретению включают венлафаксин. Примеры подходящих атипичных антидепрессантов включают бупропион, литий, нефазодон, тразодон и вилоксазин. Примеры агентов против болезни Альцгеймера включают димебон, антагонисты NMDA рецепторов, такие как мемантин; и ингибиторы холинэстеразы, такие как донепезил и галантамин. Примеры подходящих классов агентов против тревоги, которые могут быть использованы в комбинации с соединениями по изобретению, включают бензодиазепины и агонисты или антагонисты серотонина 1А (5-НТ1А), особенно 5-НТ1А частичные агонисты, и антагонисты кортикотропин-высвобождающего фактора (CRF). Подходящие бензодиазепины включают алпразолам, хлордиазероксид, клоназепам, хлоразепат, диазепам, галазепам, лоразепам, оксазепам и празепам. Подходящие агонисты или антагонисты 5-НТ1А рецепторов включают буспирон, флесиноксан, геприрон и ипсапирон. Подходящие атипичные антипсихотические агенты включают палиперидон, бифепрунокс, зипразидон, рисперидон, арипипразол, оланзапин и кветиапин. Подходящие агонисты никотина-ацетилхолина включают испрониклин, варенициклин и MEM 3454. Обезболивающие агенты включают прегабалин, габапентин, клонидин, неостигмин, баклофен, мидазолам, кетамин и зиконотид. Примеры подходящих агентов против болезни Паркинсона включают L-DOPA (или ее метиловый или этиловый сложный эфир), ингибитор DOPA декарбоксилазы (например карбидопа (SINEMET, CARBILEV, PARCOPA), антагонист аденозинового A2A рецептора [например преладенант (SCH 420814) или SCH 412348], бенсеразид (MADOPAR), α-метилдопа, монофторметилдопа, дифторметилдопа, брокрезин или м-гидроксибензил гидразин), агонист допамина [такой как апоморфин (APOKYN), бромкриптин (PARLODEL), каберголин (DOSTINEX), дигидрекседин, дигидроэргокриптин, фенолдопам (CORLOPAM), лисурид (DOPERGIN), перголид (PERMAX), пирибедил (TRIVASTAL, TRASTAL), прамипексол (MIRAPEX), хинпирол, ропинирол (REQUIP), ротиготин (NEUPRO), SKF-82958 (GlaxoSmithKline) и саризотан], ингибитор моноаминоксидазы (МАО) [такой как селегилен (EMSAM), гидрохлорид селегелена (L-депренил, ELDEPRYL, ZELAPAR), диметилселегилен, брофаромин, фенелзин (NARDIL), транилципромин (PARNATE), моклобемид (AURORIX, MANERIX), бефлоксатон, сафинамид, изокарбоксазид (MARPLAN), ниаламид (NIAMID), расагилин (AZILECT), ипрониазид (MARSILID, IPROZID, IPRONID), CHF-3381 (Chiesi Farmaceutici), ипроклозид, толоксатон (HUMORYL, PERENUM), бифемелан, дезоксипеганин, гармин (также известный как телепатин или банастерин), гармалин, линезолид (ZYVOX, ZYVOXID) и паргилин (EUDATIN, SUPIRDYL)], ингибитор катехол-О-метилтрансферазы (СОМТ) [такой как толкапон (TASMAR), энтакапон (COMTAN) и трополон], антагонист рецептора N-метил-D-аспартата (NMDA) [такой как амантадин (SYMMETREL)], антихолинэргические агенты [такие как амитриптилин (ELAVIL, ENDEP), бутриптилин, мезилат бензтропина (COGENTIN), тригексифенидил (ARTANE), дифенгидрамин (BENADRYL), орфенадрин (NORFLEX), гиосциамин, атропин (ATROPEN), скополамин (TRANSDERM-SCOP), метилбромид скополамина (PARMINE), дицикловерин (BENTYL, BYCLOMINE, DIBENT, DILOMINE, толтеродин (DETROL), оксибутинин (DITROPAN, LYRINEL XL, OKCMTROL), бромид пентиената, пропантелин (PRO-BANTHINE), циклизин, гидрохлорид имипрамина (TOFRANIL), малеат имипрамина (SURMONTIL), лоферамин, дезипрамин (NORPRAMIN), доксепин (SINEQUAN, ZONALON), тримипрамин (SURMONTIL) и гликопирролат (ROBINUL)] или их комбинацию. Примеры агентов против шизофрении включают зипразидон, рисперидон, оланзапин, кветиапин, арипипразол, азенапин, блонансерин или илоперидон. Примеры ряда дополнительных "других активных агентов" включают ривастигмин (Exelon), клозапин, ловодопа, ротиготин, арицепт, метилфенидат, мемантин, милнаципран, гуанфацин, бупропион и атомоксетин.
Как указано выше, соединения формулы I (включая их N-оксиды и фармацевтически приемлемые соли соединения или соли) могут быть использованы в комбинации с одним или более описанными в данном описании дополнительными агентами против шизофрении. При использовании комбинационной терапии один или более дополнительных агентов против шизофрении могут быть введены последовательно или одновременно с соединением по изобретению. В одном воплощении дополнительный агент против шизофрении вводят млекопитающему (например человеку) перед введением соединения по изобретению. В другом воплощении дополнительный агент против шизофрении вводят млекопитающему после введения соединения по изобретению. В другом воплощении дополнительный агент против шизофрении вводят млекопитающему (например человеку) одновременно с введением соединения по изобретению (или его N-оксидом или фармацевтически приемлемой солью вышеуказанного).
В изобретении также предложена фармацевтическая композиция для лечения шизофрении у млекопитающего, включая человека, которая содержит некоторое количество соединения формулы I (или его N-оксида или фармацевтически приемлемой соли вышеуказанного), как оно определено выше (включая гидраты, сольваты и полиморфы указанного соединения или его фармацевтически приемлемой соли), в комбинации с одним или более (например от одного до трех) агентов против шизофрении, таких как зипразидон, рисперидон, оланзапин, кветиапин, арипиппразол, азенапин, блонасерин или илоперидон, где количества активного агента и комбинации в целом являются терапевтически эффективными для лечения шизофрении.
Следует понимать, что соединения формулы I, раскрытые выше, не ограничены конкретным показанным энантиомером, но также включают все стереоизомеры и их смеси.
Во втором аспекте изобретения предложен D1 агонист с пониженной D1R десенсибилизацией. D1 агонист с пониженной D1R десенсибилизацией десенсибилизирует D1R сАМР передачу сигнала менее чем примерно на 25% относительно контроля, как измерено посредством анализа, сходного (или такого же) с примером ЕЕ, как представлено в данном описании. В некоторых воплощениях D1 агонист с пониженной D1R десенсибилизацией десенсибилизирует D1R сАМР передачу сигнала менее чем примерно на 20%, примерно на 18%, примерно на 15%, примерно на 10% или примерно 5% относительно контроля как измерено посредством анализа, сходного (или такого же) с примером ЕЕ, как представлено в данном описании. В другом воплощении D1 агонист с пониженной D1R десенсибилизацией не является производным катехола. В еще одном воплощении D1 агонист с пониженной D1R десенсибилизацией не является производным допамина.
Как этот термин используется в данном описании, D1R десенсибилизация в связи с D1 агонистами по настоящему изобретению является гомологичной десенсибилизацией.
D1R рецепторная гомологичная десенсибилизация относится к потере (частичной или полной) реактивности после воздействия агонистом, смотри JPET286: 345-353, 1998. D1 агонисты с пониженной D1R десенсибилизацией по настоящему изобретению обеспечивают пролонгированный и/или менее сниженный уровень эффективности/эффектов D1 агонистов (то есть лекарственного эффекта) после воздействия на D1R в течение определенного периода времени по сравнению с D1 агонистами без пониженной десенсибилизации (например D1 агонисты, представляющие собой производное катехола, такое как допамин, SKF-38393, дигидрексидин и SKF-81297). В этом отношении D1 агонист с пониженной D1R десенсибилизацией по настоящему изобретению может поддерживать терапевтический эффект в течение более длительного периода времени и избегать потери эффективности, вызываемой десенсибилизацией (известной как тахифилаксия), и поэтому может требоваться меньшее количество и/или менее частое дозирование для его терапевтического применения в лечении D1-опосредованного/ассоциированного расстройства. Он может также снижать или элиминировать токсикоманию/зависимость.
В некоторых воплощениях D1 агонист с пониженной D1R десенсибилизацией представляет собой полный D1 агонист или супер D1 агонист. В другом воплощении D1 агонист с пониженной D1R десенсибилизацией представляет собой полный D1 агонист.
В некоторых воплощениях D1 агонист с пониженной D1R десенсибилизацией представляет собой частичный D1 агонист.
Как этот термин используется в данном описании, производное катехола относится к соединению или его соли, где структура соединения включает следующую группировку DD-1:
 У производного катехола фенильное кольцо в DD-1 может быть дополнительно возможно замещено или заключено в полициклическое кольцо (которое также может быть возможно замещенным). Некоторые примеры производного катехола включают допамин, SKF-38393, SKF-77434, дигидрексидин и SKF-81297:
У производного катехола фенильное кольцо в DD-1 может быть дополнительно возможно замещено или заключено в полициклическое кольцо (которое также может быть возможно замещенным). Некоторые примеры производного катехола включают допамин, SKF-38393, SKF-77434, дигидрексидин и SKF-81297:
Как этот термин используется в данном описании, производное допамина относится к соединению или его соли, где структура соединения включает следующую группировку DD-2:
 У производного допамина фенильное кольцо в DD-2 может быть дополнительно возможно замещено или включено в поли-циклическую кольцевую систему (которая также может быть возможно замещенной), и/или каждый из атомов углерода этиленовой группы и N атом DD-2 могут быть дополнительно возможно замещены или включены в полициклическую кольцевую систему (которая также может быть возможно замещенной). Некоторые примеры производного допамина включают SKF-38393, SKF-77434, дигидрексидин и SKF-81297.
У производного допамина фенильное кольцо в DD-2 может быть дополнительно возможно замещено или включено в поли-циклическую кольцевую систему (которая также может быть возможно замещенной), и/или каждый из атомов углерода этиленовой группы и N атом DD-2 могут быть дополнительно возможно замещены или включены в полициклическую кольцевую систему (которая также может быть возможно замещенной). Некоторые примеры производного допамина включают SKF-38393, SKF-77434, дигидрексидин и SKF-81297.
В третьем аспекте изобретения предложен способ лечения расстройства у человека, включающий введение указанному человеку терапевтически эффективного количества соединения или его соли, где соединение или его соль представляет собой D1 агонист с пониженной D1R десенсибилизацией во втором аспекте, и где расстройство выбрано из шизофрении (например когнитивных и негативных симптомов при шизофрении), нарушения познавательной способности [например нарушения познавательной способности, ассоциированного с шизофренией, нарушения познавательной способности, ассоциированного с AD, нарушения познавательной способности, ассоциированного с PD, нарушения познавательной способности, ассоциированного с фармакотерапией (например терапией D2 антагонистом)], синдрома дефицита внимания с гиперактивностью (ADHD), импульсивности, компульсивного влечения к азартным играм, переедания, расстройства аутического спектра, умеренного ухудшения познавательной способности (MCI), ухудшения познавательной способности, связанного с возрастом, деменции (например сенильной деменции, ВИЧ-ассоциированной деменции, деменции Альцгеймера, деменции телец Леви, васкулярной деменции или фронтотемпоральной деменции), синдрома усталых ног (RLS), болезни Паркинсона, хореи Гентингтона, тревоги, депрессии (например связанной с возрастом депрессии), большого депрессивного расстройства (MDD), устойчивой к лечению депрессии (TRD), биполярного расстройства, хронической апатии, ангедонии, синдрома хронической усталости, посттравматического стрессового расстройства, сезонного аффективного расстройства, социального тревожного расстройства, послеродовой депрессии, серотонинового синдрома, нарколепсии и лекарственной зависимости, обострения симптомов нарколепсии, синдрома Туретта, тардивной дискинезии, гиперсомнии, чрезмерной сонливости в дневное время, кахексии, невнимательности, двигательного расстройства [например дискинезии (например хореи, дискинезии, индуцированной леводопой или тардивной дискинезии), тикового расстройства (например синдрома Туретта) или тремора], индуцированного терапией двигательного расстройства [например индуцированной терапией дискинезии (например индуцированной леводопой дискинезии ("LID")) или связанного с терапией дискнезического тремора (SSRI-индуцированного постурального тремора)], сексуальной дисфункции (например эректильной дисфункции или пост-SSRI сексуальной дисфункции), мигрени, системной красной волчанки (SLE), гипергликемии, атеросклероза, дислипидемии, ожирения, диабета, сепсиса, пост-ишемического тубулярного некроза, почечной недостаточности, гипонатриемии, устойчивого отека, нарколепсии, гипертензии, застойной сердечной недостаточности, послеоперационной гипотонии глаза, расстройств сна и боли.
В четвертом аспекте изобретения предложен D1 агонист с пониженной относительно допамина активностью в отношении рекрутмента β-аррестина. D1R, после связывания с D1 агонистом с пониженной активностью в отношении рекрутмента β-аррестина, осуществляет рекрутмент менее чем примерно 60% β-аррестина относительно D1R связывания с допамином, как измерено анализом, сходным (или таким же) с Примером СС, представленным в данном описании (используя показатель либо общая интенсивность/клетку, либо общая площадь/клетку). В некоторых воплощениях D1R, после связывания с D1 агонистом с пониженной активностью в отношении рекрутмента р-аррестина, осуществляет рекрутмент менее чем примерно 55%, примерно 50%, примерно 45%, примерно 40%, 35%, или примерно 30% β-аррестина относительно D1R, связавшегося с допамином. В другом воплощении D1 агонист с пониженной активностью в отношении рекрутмента β-аррестина не является производным катехола. В еще одном воплощении D1 агонист с пониженной активностью в отношении рекрутмента β-аррестина не является производным допамина.
При механизме D1R гомологичной десенсибилизации пониженная активность в отношении рекрутмента β-аррестина ведет к пониженной десенсибилизации D1R. Соответственно, D1 агонист с пониженной активностью в отношении рекрутмента β-аррестина также является D1 агонистом с пониженной D1R десенсибилизацией и тем самым обеспечивает пролонгированный и/или менее сниженный уровень эффектов действия D1 агониста (то есть лекарственного эффекта) после воздействия на D1R в течение определенного периода времени по сравнению с D1 агонистами без пониженной десенсибилизации. Более того, D1 агонист с пониженной активностью в отношении рекрутмента β-аррестина может обеспечить другие преимущества или уникальные свойства. Например комплекс передачи сигнала β-arr2/pERK, опосредованный активацией D1 рецептора, может потенциально играть определенную роль в регулировании индуцированной морфином локомоции, смотри Nikhil М Urs, et. al, "А Оорамин D1 Receptor-Dependent β-Arrestin Signaling Complex Potentially Regulates Morphine-Induced Psychomotor Activation but not Reward in Mice," Neuropsychopharmacology (2011) 36, 551-558. Пониженная активность в отношении рекрутмента β-аррестина D1 агониста по настоящему изобретению может нарушать D1 опосредованную "аррестинергическую" передачу сигнала (такую как передачу сигнала комплексом β-arr2/pERK, опосредованную активацией D1 рецептора), что может быть использовано для дополнительных терапевтических преимуществ относительно D1 агониста, который не обладает пониженной активностью в отношении рекрутмента β-аррестина.
В некоторых воплощениях D1 агонист с пониженной активностью в отношении рекрутмента β-аррестина десенсибилизирует D1R сАМР передачу сигнала менее чем примерно на 25% (например примерно на 20%, примерно на 18%, примерно на 15%, примерно на 10% или примерно на 5%) относительно контроля.
В некоторых воплощениях D1 агонист с пониженной D1R десенсибилизацией является полным D1 агонистом или супер D1 агонистом. В некоторых других воплощениях D1 агонист с пониженной D1R десенсибилизацией является полным D1 агонистом.
В некоторых воплощениях D1 агонист с пониженной D1R десенсибилизацией является частичным D1 агонистом.
В пятом аспекте изобретения предложен способ лечения расстройства у человека, включающий введение указанному человеку терапевтически эффективного количества соединения или его соли, где соединение или его соль представляет собой D1 агонист с пониженной активностью в отношении рекрутмента β-аррестина в четвертом аспекте, и где расстройство выбрано из шизофрении (например когнитивных и негативных симптомов при шизофрении), нарушения познавательной способности [например нарушения познавательной способности, ассоциированного с шизофренией, нарушения познавательной способности, ассоциированного с AD, нарушения познавательной способности, ассоциированного с PD, нарушения познавательной способности, ассоциированного с фармакотерапией (например терапией D2 антагонистом)], синдрома дефицита внимания с гиперактивностью (ADHD), импульсивности, компульсивного влечения к азартным играм, переедания, расстройства аутического спектра, умеренного ухудшения познавательной способности (MCI), ухудшения познавательной способности, связанного с возрастом, деменции (например сенильной деменции, ВИЧ-ассоциированной деменции, деменции Альцгеймера, деменции телец Леви, васкулярной деменции или фронтотемпоральной деменции), синдрома усталых ног (RLS), болезни Паркинсона, хореи Гентингтона, тревоги, депрессии (например связанной с возрастом депрессии), большого депрессивного расстройства (MDD), устойчивой к лечению депрессии (TRD), биполярного расстройства, хронической апатии, ангедонии, синдрома хронической усталости, посттравматического стрессового расстройства, сезонного аффективного расстройства, социального тревожного расстройства, послеродовой депрессии, серотонинового синдрома, злоупотребления лекарственными средствами и лекарственную зависимость, обострения симптомов нарколепсии, синдрома Туретта, тардивной дискинезии, гиперсомнии, чрезмерной сонливости в дневное время, кахексии, невнимательности, двигательного расстройства [например дискинезии (например хореи, дискинезии, индуцированной леводопой или тардивной дискинезии), тикового расстройства (например синдрома Туретта) или тремора], индуцированного терапией двигательного расстройства [например индуцированной терапией дискинезии (например индуцированной леводопой дискинезии ("LID")) или связанного с терапией дискнезического тремора (SSRI-индуцированного постурального тремора)], сексуальной дисфункции (например эректильной дисфункции или пост-SSRI сексуальной дисфункции), мигрени, системной красной волчанки (SLE), гипергликемии, атеросклероза, дислипидемии, ожирения, диабета, сепсиса, пост-ишемического тубулярного некроза, почечной недостаточности, гипонатриемии, устойчивого отека, нарколепсии, гипертензии, застойной сердечной недостаточности, послеоперационной гипотонии глаза, растройств сна и боли.
В шестом аспекте изобретения предложен D1 агонист, который в значительной степени взаимодействует с Ser188 в D1R при связывании с D1R. В другом воплощении D1 агонист, взаимодействующий в значительной степени с Ser188 в D1R, не является производным катехола. В еще одном воплощении D1 агонист, взаимодействующий в значительной степени с Ser188 в D1R, не является производным допамина.
Как оно используется в данном описании, понятие "взаимодействующий в значительной степени с Ser188" относится к кратности сдвига EC50 («fold shift»), большей, чем примерно 7,0, как измерено посредством S188I мутантного исследования, сходного с представленным в данном описании. В некоторых воплощениях D1 агонист может взаимодействовать в значительной степени с Ser188 в D1R, когда связывание с D1R имеет кратность сдвига EC50 большую, чем примерно 8,0 или 9,0, как измерено посредством S188I мутантного исследования, сходного с тем, которое представлено в данном описании.
В другом воплощении изобретения предложен D1 агонист, который взаимодействует в значительной степени с Ser188, но не в значительной степени с Ser202 в D1R при связывании с D1R. В другом воплощении D1 агонист, взаимодействующий в значительной степени с Ser188, но не в значительной степени с Ser202 в D1R, не является производным катехола. В еще одном воплощении D1 агонист, взаимодействующий в значительной степени с Ser188, но не в значительной степени с Ser202 в D1R, не является производным допамина.
Как оно используется в данном описании, понятие "взаимодействующий в значительной степени с Ser202" относится к кратности сдвига EC50 большей, чем примерно 7,0, как измерено посредством S202A мутантного исследования, сходного с представленным в данном описании. В некоторых воплощениях D1 агонист может взаимодействовать в значительной степени с Ser202 в D1R, когда связывание с D1R имеет кратность сдвига ЕС50 менее чем примерно 7,0, 6,0, 5,0 или 4,0, как измерено посредством S202A мутантного исследования, сходного с тем, которое представлено в данном описании.
В некоторых воплощениях D1 агонист, взаимодействующий в значительной степени с Ser188 в D1R, является полным D1 агонистом или супер D1 агонистом. В некоторых воплощениях D1 агонист, взаимодействующий в значительной степени с Ser188, но не в значительной степени с Ser202 в D1R, является полным D1 агонистом или супер D1 агонистом.
В некоторых воплощениях D1 агонист, взаимодействующий в значительной степени с Ser188 в D1R, является частичным D1 агонистом. В некоторых воплощениях D1 агонист, взаимодействующий в значительной степени с Ser188, но не в значительной степени с Ser202 в D1R, является частичным D1 агонистом.
В некоторых воплощениях D1 агонист, взаимодействующий в значительной степени с Ser188, но не в значительной степени с Ser202 в D1R, также является D1 агонистом с пониженной D1R десенсибилизацией во втором аспекте.
В некоторых воплощениях D1 агонист, взаимодействующий в значительной степени с Ser188, но не в значительной степени с Ser202 в D1R, является D1 агонистом с пониженной активностью в отношении рекрутмента β-аррестина в четвертом аспекте.
В некоторых воплощениях D1 агонист, взаимодействующий в значительной степени с Ser188, но не в значительной степени с Ser202 в D1R, является также D1 агонистом с пониженной D1R десенсибилизацией во втором аспекте и D1 агонистом с пониженным активностью в отношении рекрутмента β-аррестина в четвертом аспекте.
В некоторых воплощениях настоящего изобретения предложен D1 агонист, который взаимодействует менее сильно с Asp103 в D1R. В некоторых воплощениях настоящего изобретения предложен D1 агонист, который взаимодействует в значительной степени с Ser188, но не в значительной степени с Ser202 в D1R, где D1 агонист взаимодействует менее сильно с Asp103 в D1R.
Как оно используется в данном описании, понятие "который взаимодействует менее сильно с Asp103" относится к кратности сдвига EC50 меньшей, чем примерно 100, как измерено посредством D103A мутантного исследования, сходного (или такого же) с исследованием, представленным в настоящем описании. В некоторых воплощениях D1 агонист, который взаимодействует менее сильно с Asp 103 в D1R при связывании с D1R, имеет кратность сдвига ЕС50 меньшую, чем примерно 95, 90, 85, или 80, как измерено посредством D103A мутантного исследования, сходного с тем, которое представлено в настоящем описании.
В некоторых воплощениях настоящего изобретения предложен полный D1 агонист или супер D1 агонист, который взаимодействует менее сильно с Ser198 в D1R. В некоторых дополнительных воплощениях настоящего изобретения предложен полный D1 агонист или суперагонист D1, который взаимодействует менее сильно с Ser198 в D1R и взаимодействует менее сильно с Asp103 в D1R.
В некоторых воплощениях настоящего изобретения предложен полный D1 агонист или супер D1 агонист, который взаимодействует в значительной степени с Ser188, но не в значительной степени с Ser202 в D1R, где полный D1 агонист взаимодействует менее сильно с Ser198 в D1R. В другом воплощении полный D1 агонист или супер D1 агонист взаимодействует менее сильно с Asp103 в D1R. В еще одном воплощении D1 агонист, взаимодействующий в значительной степени с Ser188, но не в значительной степени с Ser202, взаимодействующий менее сильно с Ser198 и взаимодействующий менее сильно с Asp103 в D1R, также является D1 агонистом с пониженной D1R десенсибилизацией во втором аспекте. В еще одном другом воплощении D1 агонист, взаимодействующий в значительной степени с Ser188, но не в значительной степени с Ser202, взаимодействующий менее сильно с Ser198 и взаимодействующий менее сильно с Asp103 в D1R, является D1 агонистом с пониженной активностью в отношении рекрутмента β-аррестина в четвертом аспекте.
Как оно используется в данном описании, понятие "взаимодействует менее сильно с Ser198" относится к кратности сдвига ЕС50, составляющей менее чем примерно 25, как измерено посредством S198A мутантного исследования, сходного с (или такого же) предложенным в данном описании. В некоторых воплощениях D1 агонист, который взаимодействует менее сильно с Ser198 в D1R при связывании с D1R, имеет кратность сдвига ЕС50 меньшую, чем примерно 22, 20, 18, или 15, как измерено посредством S198A мутантного исследования, сходного с (или такого же) с предложенным в данном описании.
В седьмом аспекте изобретения предложен способ лечения расстройства у человека, включающий введение указанному человеку терапевтически эффективного количества соединения или его соли, где соединение или его соль является D1 агонистом, взаимодействующим в значительной степени с Ser188 (возможно, но не значительно с Ser202) в D1R в шестом аспекте, и где расстройство выбрано из шизофрении (например когнитивных и негативных симптомов при шизофрении), нарушения познавательной способности [например нарушения познавательной способности, ассоциированного с шизофренией, нарушения познавательной способности, ассоциированного с AD, нарушения познавательной способности, ассоциированного с PD, нарушения познавательной способности, ассоциированного с фармакотерапией (например терапией D2 антагонистом)], синдрома дефицита внимания с гиперактивностью (ADHD), импульсивности, компульсивного влечения к азартным играм, переедания, расстройства аутического спектра, умеренного ухудшения познавательной способности (MCI), ухудшения познавательной способности, связанного с возрастом, деменции (например сенильной деменции, ВИЧ-ассоциированной деменции, деменции Альцгеймера, деменции телец Леви, васкулярной деменции или фронтотемпоральной деменции), синдрома усталых ног (RLS), болезни Паркинсона, хореи Гентингтона, тревоги, депрессии (например связанной с возрастом депрессии), большого депрессивного расстройства (MDD), устойчивой к лечению депрессии (TRD), биполярного расстройства, хронической апатии, ангедонии, синдрома хронической усталости, посттравматического стрессового расстройства, сезонного аффективного расстройства, социального тревожного расстройства, послеродовой депрессии, серотонинового синдрома, токсикомании и лекарственной зависимости, обострения симптомов токсикомании, синдром Туретта, тардивной дискинезии, гиперсомнии, чрезмерной сонливости в дневное время, кахексии, невнимательности, двигательного расстройства [например дискинезии (например хореи, дискинезии, индуцированной леводопой или тардивной дискинезии), тикового расстройства (например синдрома Туретта) или тремора], индуцированного терапией двигательного расстройства [например индуцированной терапией дискинезии (например индуцированной леводопой дискинезии ("LID")) или связанного с терапией дискнезического тремора (SSRI-индуцированного постурального тремора)], сексуальной дисфункции (например эректильной дисфункции или пост-SSRI сексуальной дисфункции), мигрени, системной красной волчанки (SLE), гипергликемии, атеросклероза, дислипидемии, ожирения, диабета, сепсиса, пост-ишемического тубулярного некроза, почечной недостаточности, гипонатриемии, устойчивого отека, нарколепсии, гипертензии, застойной сердечной недостаточности, послеоперационной гипотонии глаза, растройств сна и боли.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Соединения по изобретению, включая N-оксиды и соли соединений или N-оксидов, могут быть получены с помощью известных методик органического синтеза и могут быть синтезированы согласно многочисленным возможным путям синтеза.
Реакции для получения соединений по изобретению могут быть осуществлены в подходящих растворителях, которые могут легко быть выбраны специалистом в области органического синтеза. Подходящие растворители могут по существу не взаимодействовать с исходными материалами (реагентами), промежуточными соединениями или продуктами при температурах, при которых проводятся взаимодействия, например при температурах, которые могут варьироваться от температуры замерзания растворителя до температуры кипения растворителя. Данные взаимодействия могут быть осуществлены в одном растворителе или смеси более чем одного растворителя. В зависимости от конкретной стадии реакции специалистом могут быть выбраны подходящие растворители для конкретной стадии реакции.
Получение соединений по изобретению может включать в себя введение и снятие защиты с различных химических групп. Необходимость введения и снятия защиты и выбор соответствующих защитных групп легко могут быть определены специалистом в данной области. С химией защитных групп можно познакомиться, например, в документе Т. W. Greene and P. G. M. Wuts, Protective Groups in Organic Synthesis, 3rd Ed., Wiley & Sons, Inc., New York (1999), который включен в данное описание посредством ссылки во всей полноте.
За ходом реакций можно следить в соответствии с любым подходящим методом, известным в данной области. Например, за образованием продукта можно следить посредством спектроскопии, такой как спектроскопия ядерного магнитного резонанса (например 1Н или 13С), инфракрасная спектроскопия, спектрофотометрия (например УФ (ультрафиолет)-видимая), масс-спектрометрия, или с помощью хроматографических методов, таких как высокоэффективная жидкостная хроматография (HPLC) или тонкослойная хроматография (ТСХ).
Соединения формулы I и их промежуточные соединения могут быть получены в соответствии со следующими реакционными схемами и сопровождающим их обсуждением. Если не указано иного, R1, R2, R3, R4, R5, R6, R7, RT1, RT2, Q1, X1 и Y1, и структурная формула I на реакционных схемах и в следующем далее обсуждении являются такими, как определено выше. В общем соединения по данному изобретению могут быть получены способами, которые включают способы, аналогичные способам, известные специалистам в области химии, особенно в свете содержащегося в данном описании их раскрытия. Некоторые способы получения соединений по данному изобретению и их промежуточных соединений представляют собой дополнительные аспекты изобретения и проиллюстрированы следующими далее реакционными схемами. Другие способы раскрыты в экспериментальной части. Представленные в данном описании схемы и примеры (включая соответствующе описание) представлены только в иллюстративных целях и не предназначены для ограничения объема настоящего изобретения.
Схема 1 относится к получению соединений формулы I. В отношении Схемы 1, соединения формулы 1-1 [где Lg1 представляет собой подходящую уходящую группу, такую как триазолил или галоген (например Cl или Br)] или 1-2 [где Z1 представляет собой галоген (Cl, Br, или I)] имеются в продаже или могут быть получены способами, раскрытыми в данном описании, или другими способами, хорошо известными специалистам в данной области. Соединение формулы 1-3 может быть получено посредством сочетания соединения формулы 1-1 с соединением формулы 1-2, например, посредством нагревания смеси соединения формулы 1-1 с соединением формулы 1-2 в присутствии основания, такого как Cs2CO3, в соответствующем растворителе, таком как DMSO, при температурах между 50°C и 120°C в течение примерно от 20 минут до 48 часов. Альтернативно, для осуществления вышеуказанного сочетания может быть проведено катализируемое металлом (таким как палладиевый или медный катализатор) сочетание. В этом варианте сочетания смесь соединения формулы 1-1 и соединения формулы 1-2 может быть нагрета при температурах, варьирующих в диапазоне между 50°C и 120°C, в присутствии основания [такого как CS2CO3], металлического катализатора [такого как палладиевый катализатор, например Pd(OAc)2] и лиганда [такого как BINAP (2,2'-бис(дифенилфосфино)-1,1'-бинафтил)], в соответствующем растворителе, таком как 1,4-диоксан, в течение примерно от 30 минут до 48 часов. Соединение формулы 1-3 может быть затем подвергнуто взаимодействию с соединением формулы Q1-Z2 [где Z2 может представлять собой Br; В(ОН)2; B(OR)2, где каждый R независимо представляет собой Н или С1-6алкил, или где две группы (OR), вместе с атомом В, к которому они присоединены, образуют 5-10-членный гетероциклоалкил или гетероарил, возможно замещенный одним или более C1-6алкилами; группировку триалкилолова; или тому подобное] посредством катализируемой металлом (таким как палладий) реакции сочетания с получением соединения формулы I. Соединения формулы Q1-Z2 имеются в продаже или могут быть получены способами, аналогичными описанным в области химии.
Альтернативно, соединение формулы 1-3 может быть превращено в соединение формулы 1-4 [где Z2 является таким, как определено выше]. Например, соединение формулы 1-3 (где Z1 представляет собой галоген, такой как Br) может быть превращено в соединение формулы 1-4 [где Z2 представляет собой В(ОН)2; B(OR)2, где каждый R независимо представляет собой Н или C1-6алкил, или где две группы (OR) вместе с атомом В, к которому они присоединены, образуют 5-10-членный гетероциклоалкил или гетероарил, возможно замещенный одним или более С1-6алкилами] способами, раскрытыми в данном описании, или другими способами, хорошо известными специалистам в данной области. В этом примере реакция может быть осуществлена, например, посредством взаимодействия соединения формулы 1-3 (где Z1 представляет собой галоген, такой как Br) с 4,4,4',4',5,5,5',5'-октааметил-2,2'-би-1,3,2-диоксабороланом, подходящим основанием [таким как ацетат калия] и палладиевым катализатором [таким как [1,1'-бис(дифенилфосфино)-ферроцен]дихлорпалладия(II)] в подходящем растворителе, таком как 1,4-диоксан. В другом примере соединение формулы 1-3 (где Z1 представляет собой галоген, такой как Br) может быть превращено в соединение формулы 1-4 [где Z2 представляет собой группировку триалкилолова] альтернативными способами, раскрытыми в данном описании, или другими способами, хорошо известными специалистам в данной области. В этом примере реакция может быть осуществлена, например, посредством взаимодействия соединения формулы 1-3 (где Z1 представляет собой галоген, такой как Br) с гексаалкилдистаннаном [таким как гексаметилдистаннан] и палладиевым катализатором [таким как тетракис(трифенилфосфин)палладий(0)] в подходящем растворителе, таком как 1,4-диоксан. Соединение формулы 1-4 затем может быть подвергнуто взаимодействию с соединением формулы Q1-Z1 [где Z1 является таким, как определено выше] посредством катализируемой металлом (таким как палладий) реакции сочетания с получением соединения формулы I.
Соединения формулы Q1-Z1 имеются в продаже или могут быть получены способами, аналогичными раскрытым в области химии. Тип используемой реакции зависит от выбора Z1 и Z2. Например, когда Z1 представляет собой галоген или трифлат, и реагент Q1-Z2 представляет собой бороновую кислоту или эфир бороновой кислоты, может быть использована реакция Сузуки (Suzuki) [A. Suzuki, J. Organomet. Chem. 1999, 576, 147-168; N. Miyaura and A. Suzuki, Chem. Rev. 1995, 95, 2457-2483; A. F. Littke et al., J. Am. Chem. Soc. 2000, 122, 4020-4028]. В некоторых конкретных воплощениях ароматический йодид, бромид или трифлат формулы 1-3 объединяют с 1-3 эквивалентами арильной или гетероарильной бороновой кислоты или эфира бороновой кислоты формулы Q1-Z2 и подходящим основанием, таким как 2-5 эквивалентов фосфата калия, в подходящем органическом растворителе, таком как THF. Добавляют палладиевый катализатор, такой как 0,01 эквивалента S-Phos предкатализатора {также известного как аддукт хлор(2-дициклогексилфосфино-2',6'-диметокси-1,1'-бифенил)[2-(2-аминоэтилфенил)]-палладий(II) - трет-бутил-метиловый эфир} и реакционную смесь нагревают до температур, варьирующих от 60 до 100°C, в течение от 1 до 24 часов. Альтернативно, когда Z1 представляет собой галоген или трифлат, и Z2 представляет собой триалкилолово, может быть осуществлено сочетание Стилла (Stille) [V. Farina et al., Organic Reactions 1997, 50, 1-652]. Более конкретно, соединение формулы 1-3 [где Z1 представляет собой бромид, йодид или трифлат] может быть объединено с 1,5-3 эквивалентами соединения формулы Q1-Z2 [где соединение Q1-Z2 представляет собой соединение Q1 станнан] в присутствии палладиевого катализатора, такого как 0,05 эквивалентов дихлорбис(трифенилфосфин)палладия(II), в подходящем органическом растворителе, таком как толуол, и реакционная смесь может быть нагрета до температур, варьирующих от 100°C до 130°C, в течение от 12 до 36 часов. Если Z1 представляет собой Br, I или трифлат, и Z2 представляет собой Br или I, может быть использовано сочетание Неглиши (Negishi) [Е. Erdik, Tempahedron 1992, 48, 9577-9648]. Более конкретно, соединение формулы 1-3 [где Z1 представляет собой бромид, йодид или трифлат] может быть подвергнуто трансметаллированию посредством обработки от 1 до 1,1 эквивалентов алкиллитиевого реагента, а затем раствором от 1,2 до 1,4 эквивалентов хлорида цинка в соответствующем растворителе, таком как тетрагидрофуран, при температуре, варьирующей от -80°C до -65°C. После нагревания до температуры между 10°C и 30°C реакционная смесь может быть обработана соединением формулы Q1-Z2 (где Z2 представляет собой Br или I) и нагрето при температуре от 50 до 70°C с добавлением катализатора, такого как тетракис(трифенилфосфин)палладий(0). Реакция может проводиться в течение промежутка времени, варьирующего от 1 до 24 часов. Ни одна из этих реакций не ограничена использованием описанного выше растворителя, основания или катализатора, поскольку могут быть использованы многочисленные другие условия.
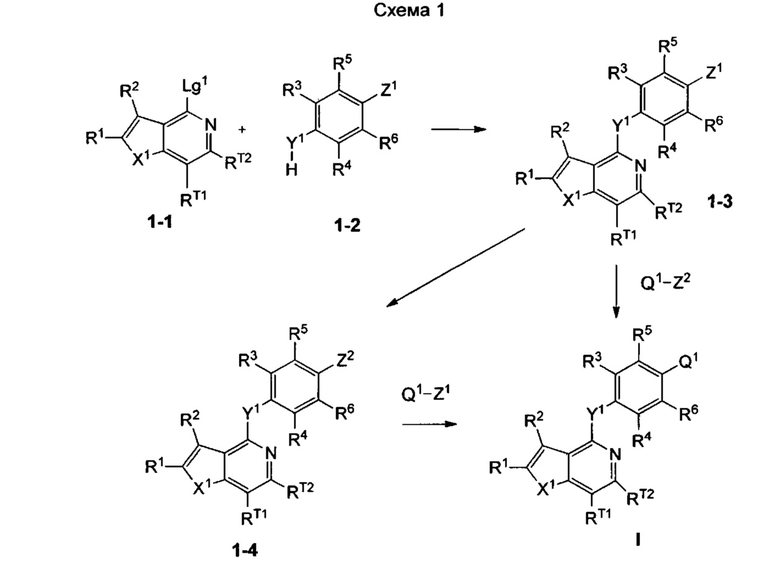
Схема 2 также относится к получению соединений формулы I. В отношении Схемы 2, соединения формулы I могут быть получены с применением химических превращений, аналогичных тем, которые описаны на Схеме 1, но с другим порядком стадий. Соединения формулы 2-1 [где Pg представляет собой подходящую защитную группу, такую как Boc или Cbz, когда Y1 представляет собой NH или метил, или Pg представляет собой бензил, когда Y1 представляет собой О] имеются в продаже или могут быть получены способами, раскрытыми в данном описании, или другими способами, хорошо известными специалистам в данной области. Соединение формулы 2-1 может быть превращено в соединение формулы 2-2 либо непосредственно, либо после превращения в соединение формулы 2-3, используя способы, аналогичные тем, которые раскрыты на Схеме 1. С соединения формулы 2-2 может затем быть удалена защита с использованием подходящих условий в зависимости от выбора группы Pg с получением соединения формулы 2-4, которое в свою очередь может быть подвергнуто сочетанию с соединением формулы 1-1 со Схемы 1 с получением соединения формулы I. Используемые условия сочетания могут быть аналогичными тем, которые раскрыты для получения соединения формулы 1-3 на Схеме 1.
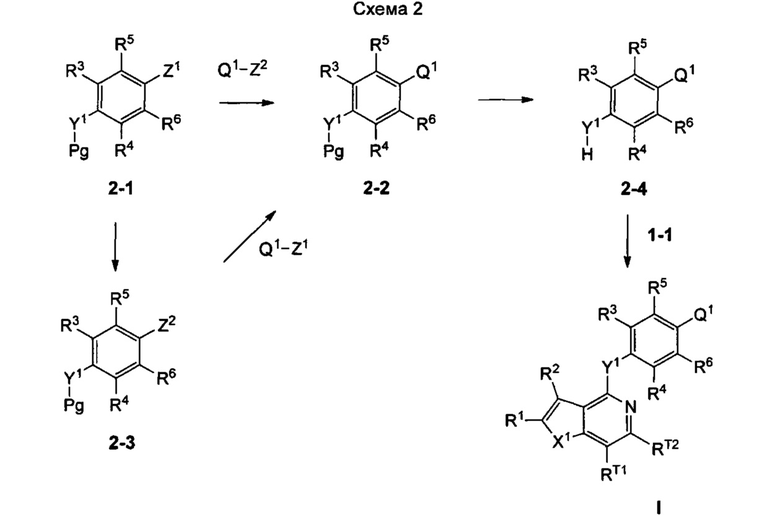
Схема 3 относится к получению соединения формулы 3-3 [где А1 представляет собой либо Pg, как определено выше, либо группировку формулы А1а]. Когда А1 представляет собой Pg, соединение формулы 3-3 является примером соединения формулы 2-2. Когда А1 представляет собой А1а, соединение формулы 3-3 является примером соединения формулы I. В отношении Схемы 3, соединения формулы 3-1 имеются в продаже или могут быть получены способами, раскрытыми в данном описании, или другими способами, хорошо известными специалистам в данной области. Соединение формулы 3-1 может быть подвергнуто взаимодействию с 4-хлор-3-нитропиридином и исходный продукт может быть затем восстановлен с получением соединения формулы 3-2. Примеры подходящих реакционных условий для сочетания соединения формулы 3-1 с 4-хлор-3-нитропиридином включают смешивание двух реагентов с подходящим основанием, таким как триэтиламин, в подходящем реакционном растворителе, таком как этанол, при температурах типично между 0°C и 100°C в течение примерно от 20 минут до 48 часов. Последующее восстановление нитро-группы с получением соединения формулы 3-2 может быть осуществлено посредством, например, гидрирования в присутствии катализатора, такого как палладий на углероде, в подходящем растворителе, таком как метанол, под давлением водорода, типично между 1 атм (101,3 КПа) и 4 атм (405,2 КПа). Соединение формулы 3-2 может затем быть подвергнуто взаимодействию с уксусным ангидридом и триэтилортоформиатом при температурах между примерно 100°C и 150°C в течение примерно от 1 часа до 48 часов с получением соединения формулы 3-3.
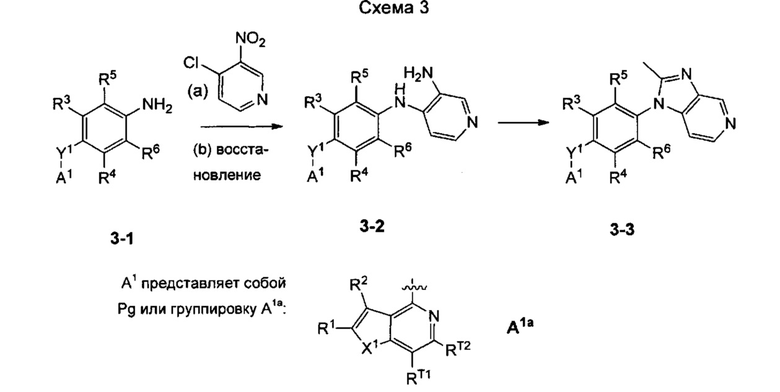
Схема 4 относится к получению соединения формулы 4-3 [где каждый R77 независимо представляет собой Н или R7 (такой как C1-3алкил, например метил)]. Когда А1 представляет собой Pg, соединение формулы 4-3 является примером соединения формулы 2-2. Когда А1 представляет собой А1а, соединение формулы 4-3 является примером соединения формулы I. В отношении Схемы 4, соединения формулы 4-1 имеются в продаже или могут быть получены способами, раскрытыми в данном описании, или другими способами, хорошо известными специалистам в данной области. Соединение формулы 4-2 может быть получено посредством взаимодействия арилкетона формулы 4-1 с диметилацеталем N,N-диметилформамида (DMF-DMA) в подходящем растворителе, таком как N,N-диметилформамид (DMF, который также является реагентом), при температурах типично между 0°C и 160°C, в течение примерно от 1 часа до 24 часов. Пиразолы формулы 4-3 могут быть получены посредством взаимодействия соединения формулы 4-2 с гидразином формулы R77-NH-NH2 в подходящем растворителе, таком как DMF или 1,4-диоксан, при температурах типично между 0°C и 100°C, в течение примерно от 1 часа до 24 часов.
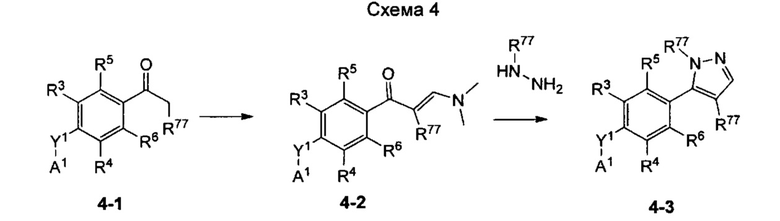
Схема 5 относится к получению соединения формулы 5-4 или 5-5 [где R77 представляет собой Н или R7 (такой как C1-3алкил, например метил)]. Когда А1 представляет собой Pg, соединение формулы 5-4 или 5-5 является примером соединения формулы 2-2. Когда А1 представляет собой А1а, соединение формулы 5-4 или 5-5 является примером соединения формулы I. В отношении Схемы 5, соединения формулы 5-1 имеются в продаже или могут быть получены способами, раскрытыми в данном описании, или другими способами, хорошо известными специалистам в данной области. Соединения формулы 5-2 могут быть получены посредством взаимодействия арилкетона формулы 5-1 с алкилнитрилом (например изоамилнитрилом) в присутствии кислоты (такой как соляная кислота) при температурах типично между 0°C и 100°C в течение примерно от 1 часа до 24 часов. Полученный оксим формулы 5-2 может быть превращен в дикетон формулы 5-3 посредством обработки формальдегидом (или его эквивалентом, таким как метаформальдегид или полиформальдегид) в присутствии кислоты (такой как водный раствор соляной кислоты) при температурах типично между 0°C и 50°C в течение примерно от 1 часа до 24 часов. Дикетоны формулы 5-3 могут быть подвергнуты взаимодействию с глицинамидом или его солью [такой как соль уксусной кислоты] в присутствии основания, такого как гидроксид натрия, с получением пиразинонов формулы 5-4. Алкилирование азота пиразинона с получением соединения формулы 5-5 может быть осуществлено посредством обработки соединения формулы 5-4 основанием [таким как LDA (диизопропиламид лития), LHMDS (гексаметилдисилазид лития) и тому подобное] и соединением формулы R7Z3 (где Z3 представляет собой подходящую уходящую группу, такую как Cl, Br, I, метансульфонат и тому подобное), в подходящем растворителе, таком как DMF, 1,4-диоксан или THF, при температурах типично между 0°C и 50°C в течение примерно от 1 часа до 24 часов.
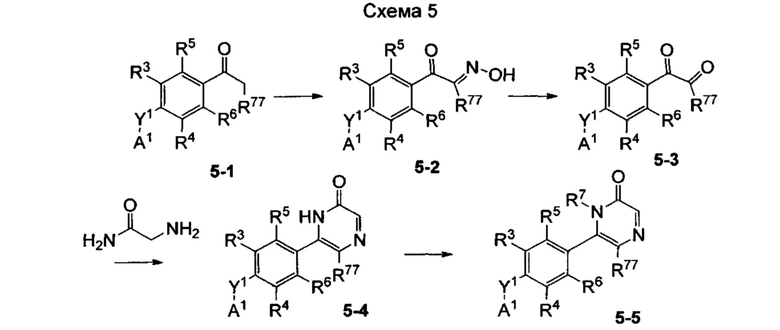
Схема 6 относится к получению соединения формулы 6-5 [где каждый R77 независимо представляет собой Н или R7 (такой как C1-6залкил, например метил)]. Когда А1 представляет собой Pg, соединение формулы 4-3 является примером соединения формулы 6-5. Когда А1 представляет собой А1а, соединение формулы 6-5 является примером соединения формулы I. В отношении Схемы 6, соединения формулы 6-1 имеются в продаже или могут быть получены способами, раскрытыми в данном описании, или другими способами, хорошо известными специалистам в данной области. Соединения формулы 6-3 могут быть получены посредством сочетания формулы 6-1 с енолтрифлатом формулы 6-2. Соединения формулы 6-2 могут быть получены способами, раскрытыми в данном описании, или другими способами, хорошо известными специалистам в данной области. Вышеуказанное сочетание может быть осуществлено посредством взаимодействия соединения формулы 6-1 с 1-3 эквивалентами трифлата формулы 6-2 в присутствии подходящего основания [такого как карбонат калия], подходящего катализатора [такого как ацетат палладия(II)], подходящего лиганда [такого как трициклогексилфосфин] и возможно подходящего катализатора фазового переноса, такого как хлорид тетрабутиламмония, в подходящем растворителе, таком как полярный апротонный растворитель (например1,4-диоксан или THF), при температурах типично между 20°C и 80°C, в течение примерно от 1 часа до 24 часов. Соединение формулы 6-3 может быть подвергнуто взаимодействию с 1-5 эквивалентами подходящего основания [такого как DBU (1,8-диазабицикло[5.4.0]ундец-7-ен)] в атмосфере кислорода с получением соединения формулы 6-4, в подходящем растворителе, таком как полярный апротонный растворитель (например DMF, 1,4-диоксан или THF), при температурах типично между 20°C и 80°C, в течение примерно от 12 часов до 48 часов. Соединение формулы 6-5 может быть получено взаимодействием соединения формулы 6-4 с гидразином в подходящем растворителе, таком как 1-бутанол, при температурах типично между 20°C и 120°C, в течение примерно от 1 часа до 24 часов.
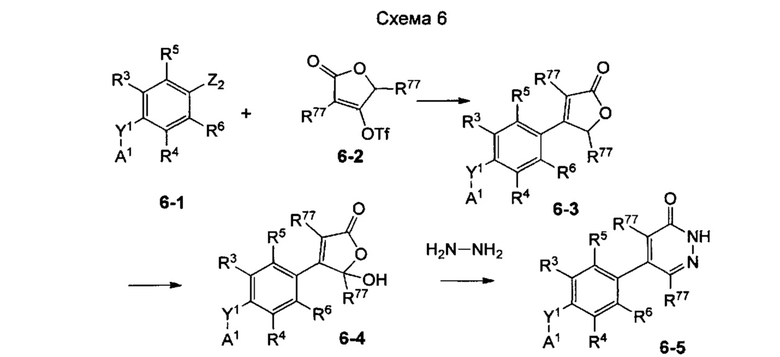
Схема 7 относится к получению соединения формулы 7-6 [где R77 представляет собой Н или R7 (такой как C1-3алкил, например метил)]. Когда А1 представляет собой Pg, соединение формулы 7-6 является примером соединения формулы 2-2. Когда А1 представляет собой А1а, соединение формулы 7-6 является примером соединения формулы I. В отношении Схемы 7, соединения формулы 7-1 имеются в продаже могут быть получены способами, раскрытыми в данном описании, или другими способами, хорошо известными специалистам в данной области. Соединение формулы 7-3 может быть получено сочетанием соединения формулы 7-1 с соединением формулы 7-2 [где Pg3 представляет собой подходящую защитную группу, такую как 2-тетрагидропиранил (ТНР)]. Соединение формулы 7-2 может быть получено способами, раскрытыми в данном описании, или другими способами, хорошо известными специалистам в данной области. Вышеуказанное сочетание может быть осуществлено взаимодействием соединения формулы 7-1 с 1-3 эквивалентами соединения формулы 7-2 в присутствии подходящего основания [такого как карбонат цезия] и подходящего катализатора [такого как [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий(II)], в подходящем растворителе, таком как полярный апротонный растворитель (например 1,4-диоксан или THF), при температурах типично между 50°C и 120°C, в течение примерно от 1 часа до 24 часов. Соединение формулы 7-4 может быть получено удалением защитной группы Pg3, например обработкой соединения формулы 7-3 (где Pg3 представляет собой, например, ТНР) с HCl в алкогольном растворителе [таком как 2-пропанол] при температурах, варьирующих от 20°C до 80°C. Обработка соединения формулы 7-4 оксихлоридом фосфора может дать соединение формулы 7-5 при температурах типично между 50°C и 120°C, в течение примерно от 20 минут до 24 часов. Соединение формулы 7-5 может быть реактивным промежуточным соединением в многочисленных химических трансформациях для получения соединения формулы 7-6. Например, соединение формулы 7-5 может быть подвергнуто взаимодействию с 1 - 3 эквивалентами триметилалюминия и 0,05-0,1 эквивалентами подходящего палладиевого катализатора [такого как тетракис(трифенилфосфин)палладий(0)] в 1,4-диоксане с получением соединения формулы 7-6 [где вновь вводимый R7 представляет собой метил], при температурах типично между 50°C и 120°C примерно от 30 минут до 12 часов.
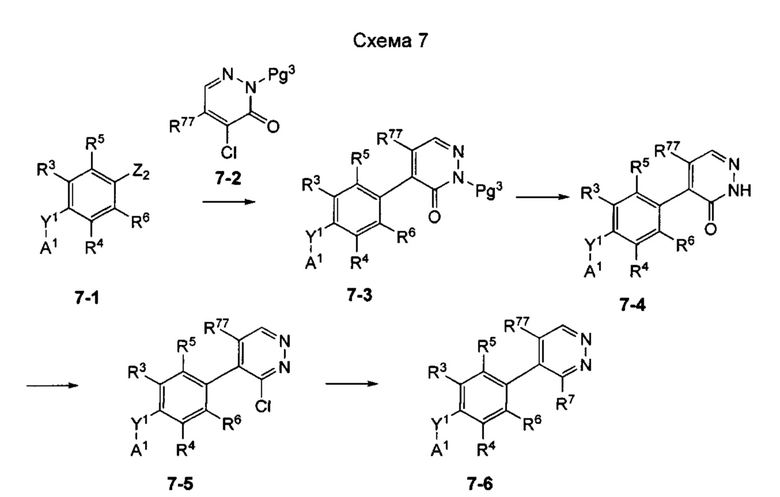
Схема 8 относится к получению соединения формулы 8-4 [где R77 представляет собой Н или R7 (такой как C1-3алкил, например метил)], которое является примером соединения формулы I. В отношении Схемы 8, соединения формулы 8-1 могут быть получены согласно способами, раскрытыми на Схеме 1. Соединение формулы 8-2 может быть получено взаимодействием соединения формулы 8-1 с трибромидом бора при температурах типично между -50°C и 50°C в течение примерно от 1 часа до 24 часов. Соединение формулы 8-3 может быть получено взаимодействием соединения формулы 8-2 с оксихлоридом фосфора при температуре типично от 50°C до 120°C в течение примерно от 20 минут до 24 часов. Соединение формулы 8-3 может быть подвергнуто взаимодействию с 1-3 эквивалентами подходящего амина HNR14R15, от 1 до 5 эквивалентов основания [такого как триэтиламин, диизопропилэтиламин и тому подобного] и каталитического количества фторида цезия с получением соединения формулы 8-4 в подходящем растворителе, таком как полярный апротонный растворитель (например 1,4-диоксан, DMF или диметилсульфоксид), при температурах типично между 50°C и 150°C в течение примерно от 1 часа до 24 часов.
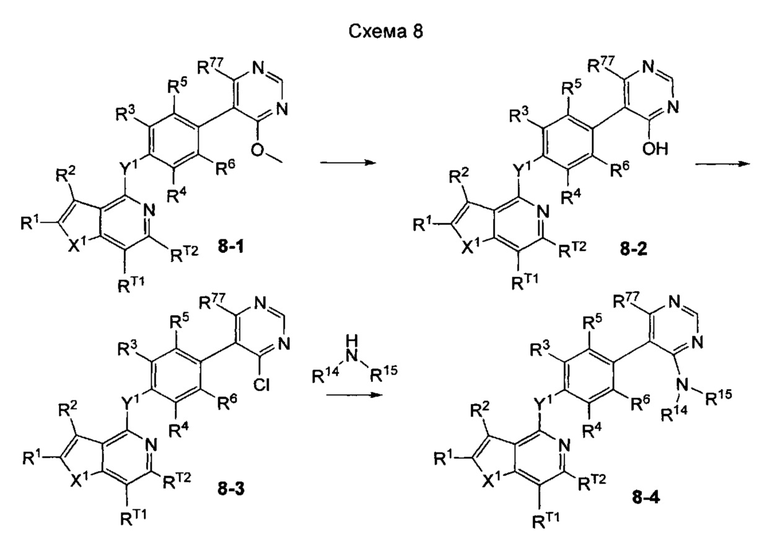
Схема 9 относится к получению соединения формулы 9-3 и/или 9-4, которое может быть использовано на Схемах 1 и/или 2. Например, когда А1 представляет собой Pg, соединение формулы 9-3 или 9-4 является примером соединения формулы 2-1. Когда А1 представляет собой А1а, соединение формулы 9-3 или 9-4 является примером соединения формулы 1-3. В отношении Схемы 9, соединения формулы 9-1 имеются в продаже или могут быть получены способами, раскрытыми в данном описании, или другими способами, хорошо известными специалистам в данной области. Соединение формулы 9-2 может быть получено обработкой соединения формулы 9-1 подходящим основанием [таким как диизопропиламид лития] и затем взаимодействием полученного аниона с N,N-диметилформамидом в подходящем растворителе, таком как полярный апротонный растворитель (например 1,4-диоксане или THF), при температурах типично между -78°C и 0°C в течение примерно от 1 часа до 24 часов. Соединение формулы 9-2 может быть подвергнуто взаимодействию с метилгидразином с получением смеси соединений формулы 9-3 и формулы 9-4 в подходящем растворителе, таком как 1,4-диоксан, при температурах типично между 50°C и 150°C, в течение примерно от 1 часа до 24 часов.
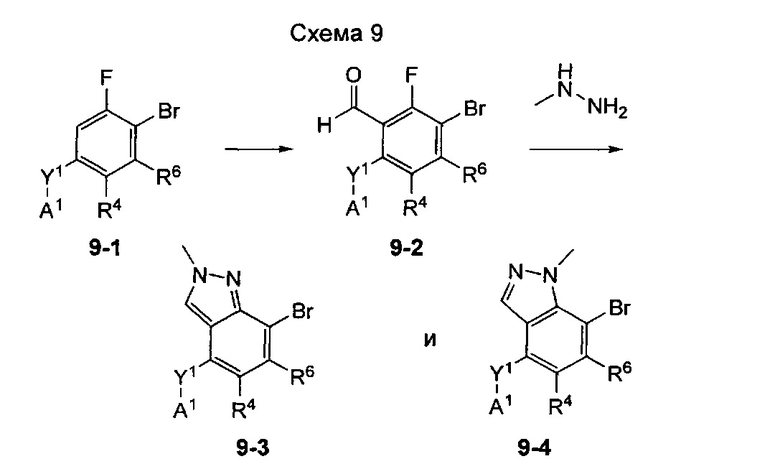
Схема 10 относится к получению соединения формулы 10-3, которое может быть использовано на Схемах 1 и/или 2. Например, когда А1 представляет собой Pg, соединение формулы 10-3 является примером соединения формулы 2-1. Когда А1 представляет собой А1а, соединение формулы 10-3 является примером соединения формулы 1-3. В отношении Схемы 10, соединения формулы 10-1 имеются в продаже или могут быть получены способами, раскрытыми в данном описании, или другими способами, хорошо известными специалистам в данной области. Соединение формулы 10-2 может быть получено обработкой соединения формулы 10-1 N-бромсукцинимидом в подходящем растворителе [таком как ацетонитрил] при температурах типично между 0°C и 20°C в течение примерно от 30 минут до 6 часов. Соединение формулы 10-2 может быть подвергнуто взаимодействию с дийодметаном и подходящим основанием [таким как карбонат цезия] с получением соединения формулы 10-3.
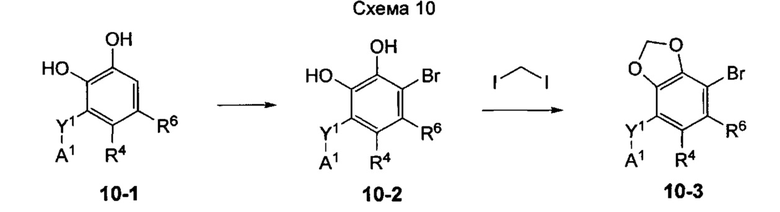
Схема 11 относится к получению соединения формулы 11-2. Когда А1 представляет собой Pg, соединение формулы 11-2 является примером соединения формулы 2-2. Когда А1 представляет собой А1а, соединение формулы 11-2 является примером соединения формулы I. В отношении Схемы 11, соединения формулы 11-1 могут быть получены согласно способам, раскрытым на Схеме 5. Соединение формулы 11-1 может быть подвергнуто взаимодействию с 2-гидразинил-1H-имидазолом в подходящем растворителе, таком как DMF, с получением соединения формулы 11-2 при температурах между примерно 80°C и 120°C.
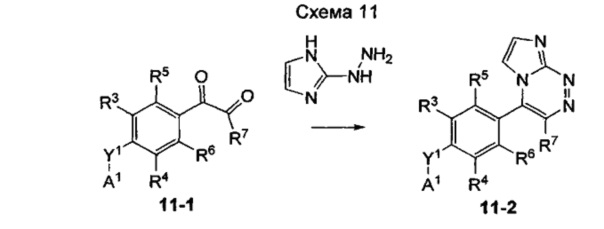
Схема 12 относится к получению соединения формулы 12-2 [где каждый R77 независимо представляет собой Н или R7 (такой как C1-3алкил, например метил)], которое является примером соединения формулы I. В отношении Схемы 12, соединение формулы 12-1 может быть получено способами, раскрытыми на Схеме 1. Соединение формулы 12-1 может быть подвергнуто взаимодействию с хлорацетальдегидом с получением соединения формулы 12-2 при температурах типично между 80°C и 120°C в течение примерно от 1 часа до 24 часов.
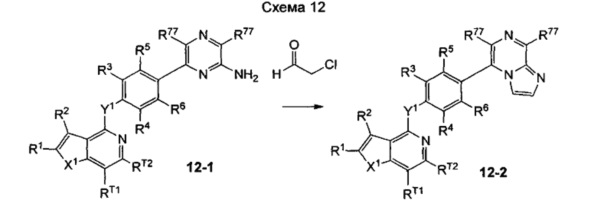
Схема 13 относится к получению соединений формулы 13-3 [где R77 представляет собой Н или R7 (такой как C1-3алкил, например метил)], которое является примером соединения формулы I. В отношении Схемы 13, соединение формулы 13-1 может быть получено согласно способам, раскрытым на Схеме 7. Соединение формулы 13-2 может быть получено взаимодействием соединения формулы 13-1 с гидразином в подходящем растворителе, таком как этанол, при температурах типично между 60°C и 100°C в течение примерно от 12 до 24 часов. Соединение формулы 13-2 может быть подвергнуто взаимодействию с 1,1-карбонилдиимидазолом в растворителе, таком как ацетонитрил, с получением соединения формулы 13-3.
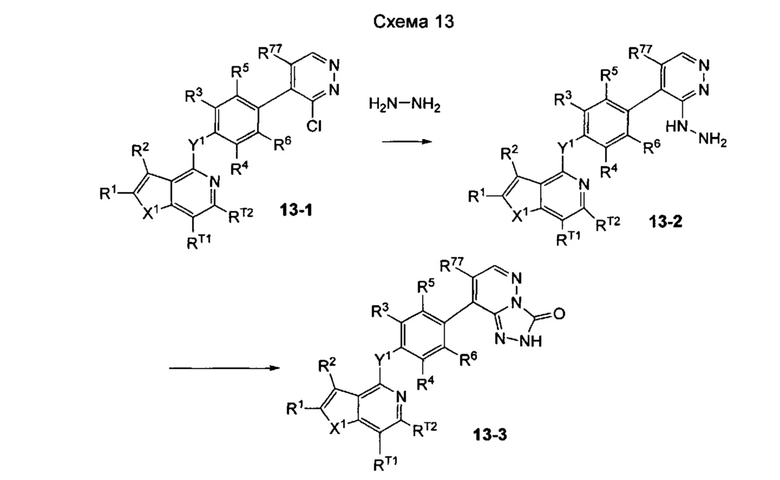
Дополнительно, соединение формулы I может также быть получено посредством ферментативной модификации [такой как микробиологическое окисление] родственного соединения формулы I. Например, как показано на Схеме 14, инкубирование соединения формулы I [например, где Q1 представляет собой группировку, которая может быть окислена, такую как возможно замещенный пиридазинил в соединении формулы 14-1 (где каждый R77 независимо представляет собой Н или R7 (такой как C1-3алкил, например метил))] с Pseudomonas putida в течение времени реакции между 24 и 96 часами в подходящем буфере может обеспечить альтернативное соединение формулы I (например, где Q1 представляет собой возможно замещенный пиридазинонил в соединении формулы 14-2).
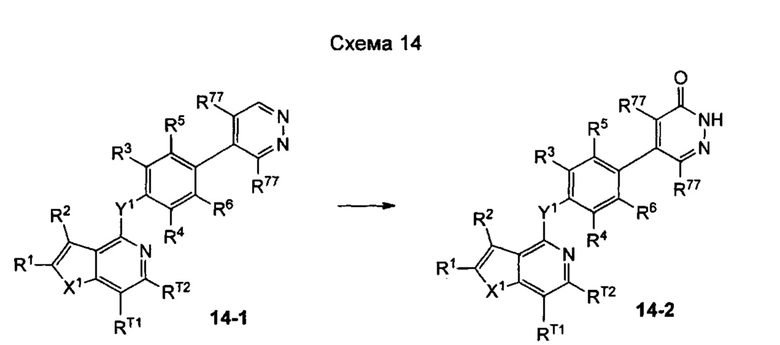
Дополнительные исходные материалы и промежуточные соединения, полезные для получения соединений по настоящему изобретению могут быть получены от химических фирм-поставщиков, таких как Sigma-Aldrich, или могут быть получены согласно способам, раскрытым в уровне техники.
Для специалиста в данной области может являться понятным, что на всех Схемах, раскрытых в данном описании, если имеются функциональные (реакционноспособные) группы, присутствующие на части структуры соединения, такие как защитная группа, например R1, R2, R3, R4, R5, R6, R7, X1, Y1, Q1 и так далее, может быть осуществлена дополнительная модификация, если необходимо и/или желательно, используя способы, хорошо известные специалистам в данной области. Например, группа -CN может быть подвергнута гидролизу с получением амидной группы; группа карбоновой кислоты может быть превращена в амид; группа карбоновой кислоты может быть превращена в сложный эфир, который в свою очередь может быть восстановлен до спирта, который в свою очередь может быть модифицирован далее. В качестве другого примера, группа ОН может быть превращена в более удобную уходящую группу, такую как мезилат, который в свою очередь является подходящим для нуклеофильного замещения, такого как посредством цианидного иона (CN-). В качестве еще одного примера, -S- может быть окислен до -S(=O)- и/или -S(=O)2-. В еще одном примере ненасыщенная связь, такая как С=С или С≡С, может быть восстановлена до насыщенной связи посредством гидрирования. В некоторых воплощениях группировка первичного амина или вторичного амина (присутствующая на группе-заместителе, такой как R2, R5 и так далее) может быть превращена в группировку амида, сульфонамида, мочевины или тиомочевины посредством взаимодействия с соответствующим реагентом, таким как соединение хлорангидрид, сульфонилхлорид, изоцианат или тиоизоцианат. Для специалиста очевидными будут другие такие модификации. Так, соединение формулы I, имеющее заместитель, который содержит функциональную группу, может быть превращено в другое соединение формулы I, имеющее другую группу-заместитель.
Аналогично, для специалиста в данной области также понятно, что на всех схемах, раскрытых в данном описании, в случае, если имеются функциональные (реакционноспособные) группы, присутствующие на группах-заместителях, таких как R3, R5 и так далее, эти функциональные группы могут быть подвергнуты введению защиты/снятию защиты в ходе реализации схем синтеза, раскрытых в данном описании, если целесообразно и/или желательно. Например, ОН группа может быть защищена бензильной, метильной или ацетильной группой, а затем подвергнута снятию защиты с превращением обратно в ОН группу на более поздней стадии способа синтеза. В качестве еще одного примера, группа NH2 может быть защищена бензилоксикарбонильной (Boc) группой, и затем подвергнута снятию защиты с превращением снова в группу NH2 на более поздней стадии способа синтеза.
Как он используется в данном описании, термин "взаимодействие" (или "реакция" или "подвергнутый взаимодействию") относится к приведению в контакт указанных химических реагентов так, что имеет место химическое превращение с образованием соединения, отличного от любого соединения, изначально введенного в систему. Реакции могут иметь место в присутствии или в отсутствие растворителя.
Соединения формулы I, раскрытые в данном описании, включают соединения формулы I, их N-оксиды и соли соединений и N-оксидов.
Соединения формулы I могут существовать как стереоизомеры, такие как атропизомеры, рацематы, энантиомеры или диастереомеры. Традиционные методики получения/выделения индивидуальных энантиомеров включают хиральный синтез из подходящего оптически чистого предшественника или разделение рацемата с помощью, например, хиральной жидкостной хроматографии высокого давления (HPLC). Альтернативно, рацемат (или рацемический предшественник) может быть подвергнут взаимодействию с подходящим оптически активным соединением, например спиртом или, в случае, когда соединение содержит кислотную или основную группировку, кислотой или основанием, таким как винная кислота или 1-фенилэтиламин. Полученная диастереомерная смесь может быть разделена посредством хроматографии и/или фракционной кристаллизации и один или оба диастереоизомера могут быть превращены в соответствующий(ие) чистый(ые) энантиомер(ы) посредством средств, хорошо известных специалисту в данной области. Хиральные соединения формулы I (и их хиральные предшественники) могут быть получены в энантиомерно обогащенной форме с помощью хроматографии, обычно ЖХВД, на ассимметричной смоле с подвижной фазой, состоящей из углеводорода, обычно гептана или гексана, содержащей от 0% до 50% 2-пропанола, обычно от 2% до 20%, и от 0% до 5% алкиламина, обычно 0,1% диэтиламина. Концентрирование элюата дает обогащенную смесь. Стереоизомерные конгломераты могут быть разделены обычными методами, известными специалистам в данной области, смотри, например, Stereochemistry of Organic Compounds by E. L. Eliel and S. H. Wilen (Wiley, New York, 1994), описание которого включено в данное описание посредством ссылки во всей его полноте. Подходящие стереоселективные методы хорошо известны специалистам в данной области.
Когда соединения формулы I содержат алкенильную или алкениленовую (алкилиденовую) группу, возможны геометрические цис/транс (или Z/E) изомеры. Цис/транс изомеры могут быть разделены обычными методиками, хорошо известными специалистам в данной области, например, хроматографией и фракционной кристаллизацией. Соли по настоящему изобретению могут быть получены в соответствии со способами, известными специалистам в данной области.
Соединения формулы I, которые являются основными по своей природе, способны образовывать широкое разнообразие солей с различными неорганическими и органическими кислотами. Хотя такие соли должны быть фармацевтически приемлемыми для введения животным, часто является желательным на практике сначала выделить соединение по настоящему изобретению из реакционной смеси в виде фармацевтически неприемлемой соли и затем просто превратить обратно в свободное основание соединения путем обработки щелочным реагентом и затем превратить это свободное основание в соль присоединения фармацевтически приемлемой кислоты. Соли присоединения кислоты соединений по данному изобретению могут быть получены путем обработки основного соединение по существу эквивалентным количеством выбранной минеральной или органической кислоты в среде водного растворителя или в подходящем органическом растворителе, таком как метанол или этанол. После выпаривания растворителя получают желаемую твердую соль. Желаемая соль с кислотой также может быть осаждена из раствора свободного основания в органическом растворителе путем добавления соответствующей минеральной или органической кислоты к раствору.
Если соединение по изобретению представляет собой основание, желаемая фармацевтически приемлемая соль может быть получена любым подходящим методом, известным в данной области, например обработкой свободного основания неорганической кислотой, такой как соляная кислота, бромистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и тому подобное, или органической кислотой, такой как уксусная кислота, малеиновая кислота, янтарная кислота, миндальная кислота, фумарования кислота, малоновая кислота, пировиноградная кислота, щавелевая кислота, гликолевая кислота, салициловая кислота, изоникотиновая кислота, молочная кислота, пантотеновая кислота, бивинная кислота, аскорбиновая кислота, 2,5-дигидроксибензойная кислота, глюконовая кислота, сахариновая кислота, муравьиная кислота, метансульфоновая кислота, этансульфоновая кислота, бензолсульфоновая кислота, п-толуолсульфоновая кислота и помоевая [то есть 1,1'-метилен-бис-(2-гидрокси-3-нафтоат)] кислота, пиранозидильная кислота, такая как глюкуроновая кислота или галактоуроновая кислота, альфа-гидроксикислота, такая как лимонная кислота или винная кислота, аминокислота, такая как аспарагиновая кислота или глутаминовая кислота, ароматическая кислота, такая как бензойная кислота или циннамовая кислота, сульфоновая кислота, такая как этансульфоновая кислота, или тому подобное.
Те соединения формулы I, которые являются кислотными по своей природе, способны образовывать соли с основаниями с различными фармакологически приемлемыми катионами. Примеры таких солей включают соли щелочных и щелочно-земельных металлов и в частности соли натрия и калия. Все эти соли получают обычными методами. Химическими основаниями, которые используют в качестве реагентов для получения фармацевтически приемлемых солей с основаниями по данному изобретению, являются те основания, которые образуют нетоксичные соли с основаниями с кислотными соединениями формулы I. Эти соли могут быть получены любым подходящим методом, например обработкой свободной кислоты неорганическим или органическим основанием, таким как амин (первичный, вторичный или третичный), гидроксид щелочного металла или гидроксид щелочно-земельного металла или тому подобным. Эти соли также могут быть получены обработкой сответствующих кислотных соединений водным раствором, содержащим желаемые фармакологически приемлемые катионы, и затем выпариванием досуха полученного раствора, например при пониженном давлении. Альтернативно, они также могут быть получены смешиванием растворов кислотных соединений в растворах в низших спиртах и желаемого алкоксида щелочного металла и затем выпаривания досуха полученного раствора таким же образом, что и выше. В любом случае используют, например, стехиометрические количества реагентов, чтобы быть уверенными в завершении взаимодействий и получении максимальных выходов желаемого конечного продукта.
Фармацевтически приемлемые соли соединения формулы I (включая соединения формулы 1а или 1b) могут быть получены одним или более из трех следующих способов:
(1) посредством взаимодействия соединения формулы I с требуемой кислотой или основанием;
(2) посредством удаления лабильной под действием кислоты или основания защитной группы из подходящего предшественника соединения формулы I или посредством раскрытия кольца подходящего циклического предшественника, например лактона или лактама, используя требуемую кислоту или основание; или
(3) посредством превращения одной соли соединения формулы I в другую посредством взаимодействия с соответствующими кислотой или основанием или с помощью подходящей ионообменной колонки.
Все три взаимодействия обычно проводят в растворе. Полученная соль может быть осаждена и собрана фильтрованием или может быть выделена посредством выпаривания растворителя. Степень ионизации в полученной соли может варьировать от полностью ионизированной до почти неионизированной.
Полиморфы могут быть получены согласно методикам, хорошо известным специалистам в данной области, например посредством кристаллизации.
Когда любой рацемат кристаллизуется, возможны кристаллы двух разных типов. Первый тип представляет собой рацемическое соединение (истинный рацемат), упоминавшееся выше, где получают одну гомогенную форму кристалла, содержащую оба энантиомера в эквимолярных количествах. Второй тип представляет собой рацемическую смесь или конгломерат, где получают в эквимолярных количествах две формы кристалла, каждая из которых содержит отдельный энантиомер.
Хотя обе кристаллические формы, присутствующие в рацемической смеси, имеют идентичные физические свойства, они могут иметь другие физические свойства по сравнению с истинным рацематом. Рацемические смеси могут быть разделены обычными методиками, известными специалистам в данной области - смотри, например, Stereochemistry of Organic Compounds by E. L. Eliel и S. H. Wilen (Wiley, New York, 1994).
Изобретение также включает меченные изотопами соединения формулы I, где один или более атомов заменены атомом, имеющим тот же атомный номер, но атомную массу или массовое число, отличное от атомной массы или массового числа, обычно обнаруживаемого в природе. Меченные изотопами соединения формулы I (или их фармацевтически приемлемые соли или N-оксид) могут в общем быть получены обычными методами, известными специалистам в данной области, или с помощью способов, аналогичных раскрытым в данном описании, используя меченный соответствующим изотопом реагент вместо используемого иначе немеченного изотопом реагента.
Пролекарства согласно изобретению могут, например, быть получены посредством замены соответствующих функциональных групп, присутствующих в соединениях формулы I, определенными группировками, известными специалистам в данной области как 'про-группировки', как раскрыто, например, в Design of Prodrugs by H. Bundgaard (Elsevier, 1985).
Соединения формулы I должны быть оценены в отношении их биофармацевтических свойств, таких как растворимость и стабильность в растворе (при разных величинах рН), проникающая способность и так далее, для того, чтобы выбрать наиболее подходящие лекарственные формы и пути введения для лечения по предполагаемому показанию.
Соединения по изобретению, предназначенные для фармацевтического применения, могут вводиться как кристаллические или аморфные продукты. Они могут быть получены, например, в виде твердых брикетов, порошков или пленок такими методами, как осаждение, кристаллизация, сушка замораживанием, распылительная сушка или сушка выпариванием. Для этой цели может быть использована сушка под действием микроволнового излучения или радиочастотного излучения.
Они могут вводиться сами по себе или в комбинации с одним или более другими соединениями по изобретению или в комбинации с одним или более другими лекарственными средствами (или в виде любой их комбинации). В общем их будут вводить в виде композиции в сочетании с одним или более фармацевтически приемлемыми эксципиентами. Термин "эксципиент" используется в данном описании для описания любого ингредиента, иного нежели соединение(я) по изобретению. Выбор эксципиента будет в значительной степени зависеть от таких факторов, как конкретный способ введения, влияние эксципиента на растворимость и стабильность и природа лекарственной формы.
Фармацевтические композиции, подходящие для доставки соединений по настоящему изобретению (или их фармацевтически приемлемых солей), и способы их изготовления будут ясны специалистам в данной области техники. Такие композиции и способы их изготовления могут быть обнаружены, например, в Remington's Pharmaceutical Sciences, 19th Edition (Mack Publishing Company, 1995).
Соединения по изобретению (или их фармацевтически приемлемые соли) могут быть введены перорально. Пероральное введение может включать прием внутрь таким образом, что соединение попадает в желудочно-кишечный тракт, и/или трансбуккальное, лингвальное или сублингвальное введение, посредством которого соединение проникает в кровоток непосредственно из полости рта.
Композиции, подходящие для перорального введения, включают твердые, полутвердые и жидкие системы, такие как таблетки; мягкие или твердые капсулы, содержащие мульти- или наночастицы, жидкости или порошки; пастилки (включая наполненные жидкостью); жевательные таблетки; гели; быстродиспергируемые лекарственные формы; пленки; овули; спреи и буккальные/мукоадгезивные пластыри.
Жидкие композиции включают суспензии, растворы, сиропы и эликсиры. Такие композиции могут быть использованы в качестве наполнителей в мягких или твердых капсулах (изготовленных, например, из желатина или гидроксипропилметилцеллюлозы), и они обычно содержат носитель, например воду, этанол, полиэтиленгликоль, пропиленгликоль, метил целлюлозу или подходящее масло, и один или более чем один эмульгатор и/или суспендирующий агент. Жидкие композиции могут также быть изготовлены растворением твердого вещества, например, из саше.
Соединения по изобретению могут также быть использованы в быстрорастворимых, быстрораспадающихся лекарственных формах, таких как формы, описанные в Liang and Chen, Expert Opinion in Therapeutic Patents, 2001, 11, 981-986.
Для таблетированных лекарственных форм в зависимости от дозы лекарственное средство может составлять от 1 масс. % до 80 масс. % лекарственной формы, более типично от 5 масс. % до 60 масс. % лекарственной формы. В дополнение к лекарственному средству таблетки в большинстве случаев содержат разрыхлитель. Примеры разрыхлителей включают крахмалгликолят натрия, натрий-карбоксиметилцеллюлозу, кальций-карбоксиметилцеллюлозу, натрий-кроскармелозу, кросповидон, поливинилпирролидон, метилцеллюлозу, микрокристаллическую целлюлозу, гидроксипропилцеллюлозу, замещенную низшими алкилами, крахмал, прежелатинизированный крахмал и альгинат натрия. В большинстве случаев разрыхлитель будет составлять от 1 масс. % до 25 масс. %, например от 5 масс. % до 20 масс. % лекарственной формы.
Для придания таблетированной композиции когезионных свойств в большинстве случаев используют связывающие агенты. Подходящие связывающие агенты включают микрокристаллическую целлюлозу, желатин, сахара, полиэтиленгликоль, природные и синтетические смолы, поливинилпирролидон, прежелатинизированный крахмал, гидроксипропилцеллюлозу и гидроксипропилметилцеллюлозу. Таблетки могут также содержать разбавители, такие как лактоза (моногидрат, моногидрат, высушенный распылительной сушкой, безводная форма и тому подобное), маннит, ксилит, декстроза, сахароза, сорбит, микрокристаллическая целлюлоза, крахмал и дигидрат гидрофосфата кальция.
Таблетки могут также, возможно, содержать поверхностно-активные вещества, такие как лаурилсульфат натрия и полисорбат 80, и скользящие вещества, такие как диоксид кремния и тальк. В случае присутствия поверхностно-активные вещества могут составлять от 0,2 масс. % до 5 масс. % таблетки, и скользящие вещества могут составлять от 0,2 масс. % до 1 масс. % таблетки.
Таблетки также в большинстве случаев содержат смазывающие вещества, такие как стеарат магния, стеарат кальция, стеарат цинка, стеарилфумарат натрия и смеси стеарата магния с лаурилсульфатом натрия. В большинстве случаев смазывающие вещества составляют от 0,25 масс. % до 10 масс. %, например от 0,5 масс. % до 3 масс. % таблетки.
Другие возможные ингредиенты включают антиоксиданты, красители, корригенты, консерванты и вещества, маскирующие вкус лекарственного средства.
Типичные таблетки содержат до приблизительно 80% лекарственного средства, от приблизительно 10 масс. % до приблизительно 90 масс. % связывающего агента, от приблизительно 0 масс. % до приблизительно 85 масс. % разбавителя, от приблизительно 2 масс. % до приблизительно 10 масс. % разрыхлителя и от приблизительно 0,25 масс. % до приблизительно 10 масс. % смазывающего вещества.
Таблетки могут быть изготовлены из таблеточных смесей прямым прессованием или с использованием валика. Перед таблетированием таблеточные смеси или части смесей могут альтернативно быть подвергнуты влажному гранулированию, сухому гранулированию или гранулированию плавлением, затвердеванию из расплава или экструзии. Конечная композиция может содержать один или более чем один слой и может иметь покрытие или не иметь покрытия; она может даже быть инкапсулирована.
Изготовление таблеток обсуждается в Pharmaceutical Dosage Forms: Tablets, Vol. 1, by H. Lieberman and L. Lachman (Marcel Dekker, New York, 1980).
Годные к употреблению пероральные пленки для применения у людей или животных обычно представляют собой мягкие растворимые в воде или набухающие в воде тонкие пленчатые лекарственные формы, способные быстро растворяться или являющиеся мукоадгезивными, и обычно содержащие соединение формулы I, пленкообразующий полимер, связывающий агент, растворитель, увлажнитель, пластификатор, стабилизатор или эмульгатор, агент, модифицирующий вязкость, и растворитель. Некоторые компоненты композиции могут выполнять более чем одну функцию.
Соединение формулы I (или его фармацевтически приемлемые соли или N-оксиды), может быть растворимым в воде или нерастворимым в воде. Водорастворимое соединение обычно содержит от 1 масс. % до 80 масс. %, более типично от 20 масс. % до 50 масс. % растворенных веществ. Менее растворимые соединения могут составлять меньшую часть композиции, обычно вплоть до 30 масс. % растворенных веществ. Альтернативно соединение формулы (I) может быть представлено в форме гранул из множества частиц.
Пленкообразующий полимер может быть выбран из природных полисахаридов, белков или синтетических гидроколлоидов, и обычно представлен в количестве от 0,01 до 99 масс. %, более типично в количестве от 30 до 80 масс. %.
Другие возможные ингредиенты включают антиоксиданты, красители, корригенты и усилители вкуса и запаха, консерванты, агенты, стимулирующие слюноотделение, охлаждающие агенты, сорастворители (включая масла), смягчающие средства, наполнители, пеногасители, поверхностно-активные вещества и вещества, маскирующие вкус лекарственного средства.
Пленки по изобретению обычно изготавливают сушкой выпариванием тонких водных пленок, нанесенных на легкоотслаивающуюся подложку или бумагу. Это может быть выполнено в сушильном шкафу или камере, обычно в комбинированной сушилке для нанесения покрытий, или лиофильной сушкой, или вакуумной сушкой.
Твердые композиции для перорального введения могут быть изготовлены для немедленного и/или модифицированного высвобождения. Композиции с модифицированным высвобождением включают замедленное, длительное, импульсное, регулируемое, направленное и программируемое высвобождение.
Подходящие композиции с модифицированным высвобождением для целей изобретения описаны в патенте США №6106864. Подробности о других подходящих технологиях высвобождения, таких как высокоэнергетические дисперсии, и осмотические частицы, и частицы с покрытием представлены в Verma et al., Pharmaceutical Technology On-line, 25(2), 1-14, (2001). Применение жевательной резинки для достижения регулируемого высвобождения описано в WO 00/35298.
Соединения по изобретению (или их фармацевтически приемлемые соли или N-оксиды) могут также быть введены непосредственно в кровоток, в мышцу или во внутренний орган. Подходящие способы парентерального введения включают внутривенный, внутриартериальный, внутрибрюшинный, интратекальный, внутрижелудочковый, интрауретральный, внутригрудинный, интракраниальный, внутримышечный, внутрисуставной и подкожный. Подходящие устройства для парентерального введения включают игольные (включая микроигольные) инъекторы, безыгольные инъекторы и методики инфузии.
Композиции для парентерального применения обычно представляют собой водные растворы, которые могут содержать эксципиенты, такие как соли, углеводы и буферные агенты (предпочтительно до pH от 3 до 9), но, для некоторых применений, они могут быть более подходяще изготовлены в виде стерильного неводного раствора или в виде высушенной формы для применения в сочетании с подходящим носителем, таким как стерильная апирогенная вода.
Изготовление композиций для парентерального применения в стерильных условиях, например, лиофилизацией, может легко быть осуществлено с применением стандартных фармацевтических методик, хорошо известных специалистам в данной области техники.
Растворимость соединений формулы I, используемых в изготовлении растворов для парентерального применения, может быть увеличена применением подходящих методик изготовления, таких как включение агентов, увеличивающих растворимость.
Композиции для парентерального введения могут быть изготовлены для немедленного и/или модифицированного высвобождения. Композиции с модифицированным высвобождением включают замедленное, длительное, импульсное, регулируемое, направленное и программируемое высвобождение. Таким образом, соединения по изобретению могут быть приготовлены в виде суспензии или в виде твердой, полутвердой лекарственной формы или тиксотропной жидкости для введения в виде имплантируемого депо, обеспечивающего модифицированное высвобождение активного соединения. Примеры таких композиций включают покрытые лекарственным средством стенты и полутвердые лекарственные формы или суспензии, содержащие нагруженные лекарственным средством микросферы сополимера D- и L-изомеров молочной и гликолевой кислот (PGLA).
Соединения по изобретению (или их фармацевтически приемлемые соли или N-оксиды) могут также быть введены местно, (внутри)кожно или чрескожно на кожу или слизистую оболочку. Типичные композиции для этой цели включают гели, гидрогели, лосьоны, растворы, кремы, мази, присыпки, перевязочный материал, пены, пленки, трансдермальные пластыри, прокладки, имплантаты, губки, волокна, повязки и микроэмульсии. Также могут быть использованы липосомы. Типичные носители включают спирт, воду, минеральное масло, вазелиновое масло, медицинский вазелин, глицерин, полиэтиленгликоль и пропиленгликоль. Могут быть включены вещества, усиливающие проникновение; см., например, Finnin and Morgan, J. Pharm. Sci., 1999, 88, 955-958.
Другие способы местного введения включают доставку посредством электропорации, ионофореза, фонофореза, сонофореза и микроигольной или безыгольной (например, Powderject™, Bioject™ и тому подобное) инъекции.
Композиции для местного введения могут быть изготовлены для немедленного и/или модифицированного высвобождения. Композиции с модифицированным высвобождением включают замедленное, длительное, импульсное, регулируемое, направленное и программируемое высвобождение.
Соединения по изобретению (или их фармацевтически приемлемые соли) могут также быть введены интраназально или ингаляцией, обычно в форме сухого порошка (либо сами по себе, либо в виде смеси, например, в сухой смеси с лактозой, или в виде смешанной компонентной частицы, например, смешанной с фосфолипидами, такими как фосфатидилхолин) из ингалятора сухого порошка, в виде аэрозольного спрея из герметизированного контейнера, насоса, спрея, аэрозольного ингалятора (предпочтительно аэрозольного ингалятора с использованием электрогидродинамики для образования мелкодисперсного аэрозоля), или небулайзера, с применением или без применения подходящего пропеллента, такого как 1,1,1,2-тетрафторэтан или 1,1,1,2,3,3,3-гептафторпропан, или в виде капель в нос. Для интраназального применения порошок может содержать биоадгезивный агент, например, хитозан или циклодекстрин.
Герметизированный контейнер, насос, спрей, аэрозольный ингалятор или небулайзер содержит раствор или суспензию соединения (соединений) по изобретению, содержащую, например, этанол, водный этанол или подходящий альтернативный агент для диспергирования, солюбилизации или продления высвобождения активного соединения, пропеллент (пропелленты) в качестве растворителя и, возможно, поверхностно-активное вещество, такое как сорбитантриолеат, олеиновая кислота или олигомолочная кислота.
Перед применением в сухом порошке или суспензионной композиции лекарственное средство микронизируют до размера, подходящего для доставки ингаляцией (обычно менее 5 мкм). Это может быть осуществлено любым подходящим способом измельчения, таким как измельчение в спиральной струе, измельчение в струе псевдоожиженного слоя, обработка сверхкритической жидкостью с образованием наночастиц, гомогенизация под высоким давлением или распылительная сушка.
Капсулы (изготовленные, например, из желатина или гидроксипропилметилцеллюлозы), блистеры и картриджи для использования в ингаляторе или инсуффляторе могут быть изготовлены таким образом, чтобы содержать порошковую смесь соединения по изобретению, подходящего порошкового основания, такого как лактоза или крахмал, и вещество, модифицирующее свойства, такое как L-лейцин, маннит или стеарат магния. Лактоза может быть безводной или в форме моногидрата. Другие подходящие эксципиенты включают декстран, глюкозу, мальтозу, сорбит, ксилит, фруктозу, сахарозу и трегалозу.
Подходящая композиция в виде раствора для использования в аэрозольном ингаляторе с применением электрогидродинамики для образования мелкодисперсного аэрозоля может содержать от 1 мкг до 20 мг соединения по изобретению на срабатывание, и объем срабатывания может варьировать от 1 мкл до 100 мкл. Типичная композиция может содержать соединение формулы I или его фармацевтически приемлемую соль, пропиленгликоль, стерильную воду, этанол и хлорид натрия. Альтернативные растворители, которые могут быть использованы вместо пропиленгликоля, включают глицерин и полиэтиленгликоль.
Подходящие корригенты, такие как ментол и левоментол, или подсластители, такие как сахарин или сахарин-натрий, могут быть добавлены к тем композициям по изобретению, которые предназначены для ингаляционного/интраназального введения.
Композиции для ингаляционного/интраназального введения могут быть изготовлены для немедленного и/или модифицированного высвобождения, с использованием, например, PGLA. Композиции с модифицированным высвобождением включают замедленное, длительное, импульсное, регулируемое, направленное и программируемое высвобождение.
В случае ингаляторов сухого порошка и аэрозолей единицу дозы определяют посредством клапана, доставляющего дозированное количество. Единицы в соответствии с изобретением обычно объединены для введения дозированного количества или «пшика», содержащего от 0,01 до 100 мг соединения формулы I. Общая суточная доза будет обычно составлять от 1 мкг до 200 мг, которые могут быть введены в одной дозе, или, более обычно, как отдельные дозы в течение всего дня.
Соединения по изобретению могут быть введены ректально или вагинально, например, в форме суппозитория, пессария или клизмы. Масло какао является обычной основой суппозиториев, но по необходимости могут быть использованы различные альтернативы.
Композиции для ректального/вагинального введения могут быть изготовлены для немедленного и/или модифицированного высвобождения. Композиции с модифицированным высвобождением включают замедленное, длительное, импульсное, регулируемое, направленное и программируемое высвобождение.
Соединения по изобретению могут также быть введены непосредственно в глаз или ухо, обычно в виде капель микронизированной суспензии или раствора в изотоническом стерильном физиологическом растворе с откорректированным рН. Другие композиции, подходящие для глазного или ушного введения включают мази, гели, биодеградируемые (например, губки с абсорбируемыми гелями, коллаген) и небиодеградируемые (например, силикон) имплантаты, облатки, линзы и системы на основе частиц или везикул, таких как ниосомы или липосомы. Полимер, такой как поперечно сшитая полиакриловая кислота, поливиниловый спирт, гиалуроновая кислота, целлюлозный полимер, например гидроксипропилметилцеллюлоза, гидроксиэтилцеллюлоза или метил целлюлоза, или гетерополисахаридный полимер, например гелановая камедь, могут быть включены совместно с консервантом, таким как хлорид бензалкония. Такие композиции могут также быть доставлены ионофорезом.
Композиции для глазного/ушного введения могут быть изготовлены для немедленного и/или модифицированного высвобождения. Композиции с модифицированным высвобождением включают замедленное, длительное, импульсное, регулируемое, направленное или программируемое высвобождение.
Соединения по изобретению можно объединять с растворимыми макромолекулярными веществами, такими как циклодекстрин и его подходящие производные или полиэтиленгликоль-содержащие полимеры, с целью улучшения их растворимости, скорости растворения, коррекции вкуса, биодоступности и/или стабильности для применения в любом из вышеупомянутых способов введения.
Например, было обнаружено, что комплексы циклодекстрин-лекарственное средство, в целом применимы для большинства лекарственных форм и путей введения. Могут быть использованы как комплексы включения, так и комплексы без включения. В качестве альтернативы прямому комплексообразованию с лекарственным средством циклодекстрин может быть использован в качестве вспомогательной добавки, то есть, в качестве носителя, разбавителя или солюбилизатора. Для этих целей чаще всего используют альфа-, бета- и гамма-циклодекстрины, примеры которых могут быть обнаружены в международных заявках на патент №WO 91/11172, WO 94/02518 и WO 98/55148.
Поскольку настоящее изобретение имеет аспект, который относится к лечению раскрытого в данном описании заболевания/состояний комбинацией активных ингредиентов, которые могут быть введены по-раздельности, изобретение также относится к комбинированию отдельных фармацевтических композиций в форме набора. Набор содержит две раздельные фармацевтическая композиции: соединение формулы I, его пролекарство или соль такого соединения или пролекарства и второе соединение, как описано выше. Набор содержит средства для раздельного хранения указанных композиций, такие как контейнер, разделенная бутыль или разделенный пакет из фольги. Обычно набор содержит указания для введения отдельных компонентов. Форма набора является особенно благоприятной, когда отдельные компоненты вводят, например, в разных лекарственных формах (например, пероральной и парентеральной), вводят с разными интервалами дозирования, или когда титрование индивидуальных компонентов комбинации желательно для лечащего врача.
Примером такого набора является так называемая блистерная упаковка. Блистерные упаковки хорошо известны в упаковочной промышленности и широко используются для упаковки фармацевтических стандартных лекарственных форм (таблеток, капсул и тому подобного). Блистерные упаковки обычно состоят из слоя относительно плотного материала, покрытого пленкой из прозрачного пластика. Во время процесса упаковки в полимерной пленке формируют углубления. Эти углубления имеют размер и форму таблеток или капсул, которые подлежат упаковке. Затем таблетки или капсулы помещают соответственно в углубления и запечатывают слоем относительно плотного материала с лицевой стороны полимерной пленки, которая противоположна тому направлению, в котором формировали углубления. В результате, таблетки или капсулы запечатывают в углублениях между полимерной пленкой и этим слоем. В некоторых воплощениях прочность слоя такова, что таблетки или капсулы можно извлечь из блистерной упаковки, надавливая рукой на углубления, в силу чего на слое напротив углубления образуется отверстие. Затем таблетка или капсула может быть извлечена через указанное отверстие.
Может быть желательным приложение к набору памятки, например в форме цифр рядом с таблетками или капсулами, где цифры соответствуют дням схемы приема лекарства, когда следует проглотить таким образом обозначенные таблетки или капсулы. Другим примером такой памятки является календарь, напечатанный на карточке, например следующим образом: "первая неделя, понедельник, вторник," и так далее "вторая неделя, понедельник, вторник," и так далее. Другие варианты памяток будут достаточно очевидными. "Суточная доза" может представлять собой одну таблетку или капсулу, или несколько таблеток или капсул, которые надо принимать в определенный день. Также суточная доза соединения формулы I может состоять из одной таблетки или капсулы, тогда как суточная доза второго соединения может состоять из нескольких таблеток или капсул и наоборот. Памятка должна это отражать.
В другом конфетном воплощении изобретения предложен дозатор, разработанный для распределения суточных доз в количестве одной дозы за один раз в порядке их предполагаемого применения. Например, дозатор снабжают памяткой, чтобы дополнительно облегчить соблюдение схемы приема лекарства. Примером такой памятки является механический счетчик, который указывает количество суточных доз, которые уже распределены. Другим примером такой памятки является микрочип памяти с питанием от батарейки, связанный с жидкокристаллическим индикатором, или слышимый напоминающий сигнал, который, например, зачитывает вслух дату, когда принята последняя суточная доза, и/или напоминает, когда надо принять следующую дозу.
Изобретение будет далее описано более детально с помощью конкретных примеров. Слдеующие далее примеры служат только для иллюстративных целей и не предназанчены для ограничения каким-либо образом изобретения. Для специалиста в данной области будут с легкостью очевидны разнообразные некритические параметры, которые могут быть изменены или модифицированы с получением по существу того же самого результата. В следующих Примерах и Подготовительных примерах, "DMSO" означает диметилсульфоксид, "н." там, где он относится к концентрации, означает «нормальный, "М" означает «молярный», "мл" означает миллилитр, "ммоль" означает миллимоль, "мкмоль" означает микромоль, "экв." означает эквивалент, "°C" means градусы Цельсия, "МГц" означает мегагерцы, "HPLC" означает высокоэффективную жидкостную хроматографию.
ПРИМЕРЫ
Эксперименты в общем проводили в инертной атмосфере (азот или аргон), особенно в случаях, когда использовали реагенты или промежуточные соединения, чувствительные к действию кислорода или влаги. Коммерческие растворители и реагенты в общем использовали без дополнительной очистки, включая безводные растворители, как целесообразно (в основном Sure-Seal™ продукты от Aldrich Chemical Company, Milwaukee, Wisconsin). Продукты как правило сушили под вакуумом перед тем, как подвергать их дальнейшим взаимодействиям или биологическому тестированию. Данные масс-спектрометрии получены с помощью оборудования для осуществления сочетания жидкостная хроматография-масс-спектрометрия (ЖХ-МС), химической ионизации при атмосферном давлении (ХИАД) или сочетания газовая хроматография-масс-спектрометрия (ГХ-МС). Данные по химическим сдвигам для ядерного магнитного резонанса (ЯМР) представлены в миллионных долях (м.д., δ) и соотносятся с остаточными пиками используемых дейтерированных растворителей. В некоторых примерах хиральные разделения осуществляли на отдельные атропизомеры (или атропэнантиомеры) ряда соединений по изобретению. Оптическое вращение атропизомера измеряли с помощью поляриметра. Согласно полученным с его помощью данным по вращению (или его специфических данных по вращению), атропизомер (или атропэнантиомер) с вращением по часовой стрелке был обозначен как (+)-атропизомер [или (+) атропэнантиомер], а атропизомер (или атропэнантиомер) с вращением против часовой стрелки обозначали как (-)-атропизомер [или (-) атропэнантиомер].
Реакционные условия (длительность взаимодействия и температура) процедур, относящихся к синтезу, в других Примерах или методах могут варьировать. В общем, за ходом реакций следили с помощью тонкослойной хроматографии или масс-спектрометрии и подвергали соответствующей обработке, когда целесообразно. Процедуры очистки могут варьировать между экспериментами: в общем, растворители и соотношения растворителей, используемых для элюирования/ градиентов выбирали так, чтобы обеспечить соответствующие величины RfS или времени удерживания.
Пример 1
4-[4-(4,6-диметилпиримидин-5-ил)-3-метилфенокси]фуро[3,2-с]пиридин (1)
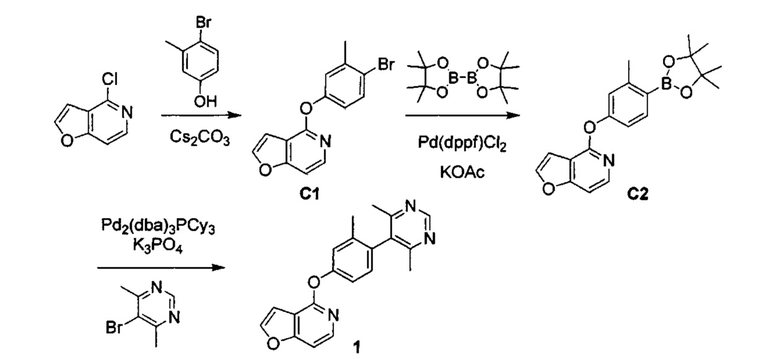
Стадия 1. Синтез 4-(4-бром-3-метилфенокси)фуро[3,2-с]пиридин (С1)
К раствору 4-хлорфуро[3,2-с]пиридина (120 г, 781 ммоль) в диметилсульфоксиде (1,56 л) добавляли карбонат цезия (509 г, 1,56 моль) и 4-бром-3-метилфенол (161 г, 861 ммоль) и реакционную смесь нагревали до 125°C в течение 16 часов. В этой точке реакционную смесь охлаждали до комнатной температуры, вливали в воду (5 л) и экстрагировали этилацетатом (2×2,5 л). Объединенные органические экстракты промывали водой (2,5 л), промывали насыщенным водным раствором хлорида натрия (2,5 л), сушили над безводным сульфатом натрия, фильтровали и концентрировали в вакууме. Очистка хроматографией на силикагеле (элюент: 2%-ный этилацетат в петролейном эфире) дала продукт в виде бледно-желтого твердого вещества. Выход: 205 г, 674 ммоль, 86%. ЖХ-МС m/z 304,0, 306,0 (М+Н). 1Н ЯМР (400 МГц, CDCl3) δ 8.00 (d, J=6,2 Гц, 1Н), 7.64 (d, J=2,1 Гц, 1Н), 7.55 (d, J=8,3 Гц, 1Н), 7.20 (dd, J=5,8, 0,8 Гц, 1Н), 7.12 (d, J=2,9 Гц, 1Н), 6.93 (dd, J=8,5, 2,7 Гц, 1Н), 6.88 (dd, J=2,5, 0,8 Гц, 1Н), 2.41 (s, 3Н).
Стадия 2. Синтез 4-[3-метил-4-(4,4,5,5-тетраметил-1,3,2-диокса6оролан-2-ил)фенокси]фуро[3,2-с]пиридина (С2)
К перемешиваемому раствору 4-(4-бром-3-метилфенокси)фуро[3,2-с]пиридина (С1) (50,0 г, 164 ммоль) в 1,4-диоксане (1,02 л) добавляли 4,4,4',4',5,5,5',5',-октаметил-2,2'-би-1,3,2-диоксаборолан (41,76 г, 164,4 ммоль), ацетат калия (64,6 г, 658 ммоль) и [1,1'-бис(дифенилфосфино)ферроцен]-дихлорпалладий(II) (6,0 г, 8,2 ммоль) и реакционную смесь нагревали при 85°C в течение 16 часов. После охлаждения до комнатной температуры смесь фильтровали через набивку целита и набивку промывали этилацетатом. Объединенные фильтраты концентрировали в вакууме и остаток очищали хроматографией на силикагеле (элюент: 2%-ный этилацетат в петролейном эфире) с получением продукта в виде белого твердого вещества. Выход: 40,0 г, 114 ммоль, 70%. ЖХ-МС m/z 352,2 (М+Н). 1Н ЯМР (400 МГц, CDCl3) δ 8.02 (d, J=5,8 Гц, 1Н), 7.84 (d, J=7,5 Гц, 1Н), 7.61 (d, J=2,1 Гц, 1Н), 7.19 (d, J=5,8 Гц, 1Н), 7.00 (m, 2Н), 6.80 (m, 1Н), 2.56 (s, 3Н), 1.34 (s, 12Н).
Стадия 3. Синтез 4-[4-(4,6-диметилпиримидин-5-ил)-3-метилфенокси]фуро[3,2-с]пиридина (1)
4-[3-Метил-4-(4,4,5,5-тетра метил-1,3,2-диоксаборолан-2-ил)фенокси]-фуро[3,2-с]пиридин (С2) (250 мг, 0,712 ммоль), 5-бром-4,6-диметилпиримидин (160 мг, 0,855 ммоль), трис(дибензилиденацетон)дипалладий(0) (95%, 26,9 мг, 0,142 ммоль), трициклогексилфосфин (79,9 мг, 0,285 ммоль) и фосфат калия (302 мг, 1,42 ммоль) объединяли в смеси 1,4-диоксана и воды в соотношении 3:1 (12 мл) и подвергали облучению в микроволновом реакторе при 120°C в течение 5 часов. Реакционную смесь фильтровали через целит; фильтрат концентрировали при пониженном давлении, переносили в этилацетат, фильтровали через силикагель (1 г) и концентрировали в вакууме. Очистка посредством хроматографии на силикагеле (градиент: от 0% до 100% этилацетата в гептане) дала продукт в виде бесцветного масла. Выход: 123 мг, 0,371 ммоль, 52%. ЖХ-МС m/z 332,1 (М+Н). 1Н ЯМР (500 МГц, CDCl3) δ 8.98 (s, 1Н), 8.07 (d, J=5,9 Гц, 1Н), 7.67 (d, J=2,2 Гц, 1Н), 7.25-7.27 (m, 1Н, предполагаемый; частично скрыт за пиком растворителя), 7.24 (br d, J=2,4 Гц, 1Н), 7.19 (br dd, J=8,3, 2,4 Гц, 1Н), 7.08 (d, J=8,3 Гц, 1Н), 6.90 (dd, J=2,2, 1,0 Гц, 1Н), 2.27 (s,6H), 2.04 (s, 3H).
Пример 2
5-[4-(Фуро[3,2-с]пиридин-4-илокси)-2-метилфенил]-6-метил-[8-2Н]-имидазо[1,2-а]пиразин (2)
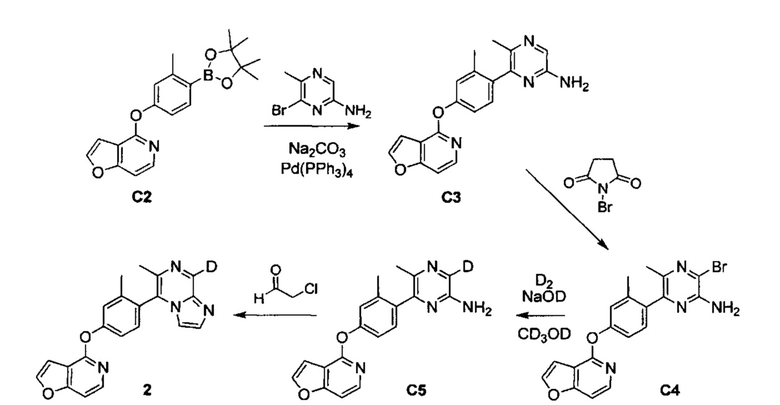
Стадия 1. Синтез 6-[4-(фуро[3,2-с]пиридин-4-илокси)-2-метилфенил]-5-метилпиразин-2-амина (С3)
6-Бром-5-метилпиразин-2-амин (который может быть получен согласно методу N. Sato, J. Heterocycl. Chem. 1980, 171, 143-147) (2,40 г, 12,8 ммоль), 4-[3-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенокси]фуро[3,2-фиридин (С2) (4,48 г, 12,8 ммоль) и тетракис(трифенилфосфин)палладий(0) (95%, 466 мг, 0,383 ммоль) объединяли в сосуде, работающем в условиях давления, и растворяли в 1,4-диоксане (60 мл) и этаноле (20 мл). Добавляли раствор карбоната натрия (2,0 М в воде, 19,1 мл, 38,2 ммоль) и через эту реакционную смесь барботировали аргон в течение 15 минут. Сосуд герметично закрывали и затем нагревали при 140°C в течение 16 часов. Реакционную смесь объединяли со второй идентичной реакционной смесью для обработки. Объединенные реакционные смеси фильтровали; твердые вещества, оставшиеся в реакционных сосудах, суспендировали в воде и фильтровали и осадок на фильтре промывали этанолом. Все органические фильтраты пропускали через набивку целита и набивку целита промывали этанолом. Эти фильтраты концентрировали в вакууме и полученное твердое вещество суспендировали в воде, фильтровали и промывали водой. Твердое вещество затем суспендировали в смеси гептан/диэтиловый эфир в соотношении 1:1, фильтровали и промывали диэтиловым эфиром с получением продукта в виде светло-желтого твердого вещества. Выход: 6,774 г, 20,38 ммоль, 80%. 1Н ЯМР (500 МГц, DMSO-d6) δ 8.14 (d, J=2,2 Гц, 1Н), 8.01 (d, J=5,7 Гц, 1Н), 7.82 (s, 1Н), 7.47 (dd, J=5,8, 0,9 Гц, 1Н), 7.21 (d, J=8,3 Гц, 1Н), 7.15 (br d, J=2,4 Гц, 1H), 7.09 (br dd, J=8,2, 2,4 Гц, 1H), 7.06 (dd, J=2,2, 0,7 Гц, 1H), 6.18 (br s, 2Н), 2.12 (s, 3Н), 2.07 (br s, 3Н).
Стадия 2. Синтез 3-бром-6-[4-(фуро[3,2-с]пиридин-4-илокси)-2-метилфенил]-5-метилпиразин-2-амина (С4)
N-Бромсукцинимид (95%, 609 мг, 3,25 ммоль) добавляли к раствору 6-[4-(фуро[3,2-с]пиридин-4-илокси)-2-метилфенил]-5-метилпиразин-2-амина (С3) (900 мг, 2,71 ммоль) в N,N-диметилформамиде (15 мл) и реакционную смесь нагревали до 60°C в течение 45 минут. Реакционную смесь охлаждали до комнатной температуры, разбавляли этилацетатом и гасили небольшим количеством воды. После адсорбции на силикагеле продукт очищали посредством хроматографии на силикагеле (градиент: от 0% до 50% этилацетата в гептане). Очищенный материал переносили в этилацетат и промывали смесью вода/насыщенный водный раствор бикарбоната натрия в соотношении 1:1, водой и насыщенным водным раствором хлорида натрия для удаления остаточного N,N-диметилформамида. Органический слой сушили над сульфатом натрия и концентрировали в вакууме с получением продукта в виде желтого твердого вещества. Выход: 700 мг, 1,71 ммоль, 63%. ЖХ-МС m/z 412,9 (М+Н). 1Н ЯМР (400 МГц, DMSO-d6) δ 8.14 (d, J=2,2 Гц, 1Н), 8.01 (d, J=5,9 Гц, 1Н), 7.48 (dd, J=5,9, 1,0 Гц, 1Н), 7.26 (d, J=8,2 Гц, 1Н), 7.17 (br d, J=2,3 Гц, 1H), 7.11 (br dd, J=8,3, 2,4 Гц, 1H), 7.07 (dd, J=2,2, 0,9 Гц, 1H), 6.51 (br s, 2H), 2.13 (s, 3H), 2.09 (br s, 3H).
Стадия 3. Синтез 6-[4-(фуро[3,2-с]пиридин-4-илокси)-2-метилфенил]-5-метил-[3-2Н]-пиразин-2-амина (С5)
3-Бром-6-[4-(фуро[3,2-с]пиридин-4-илокси)-2-метилфенил]-5-метилпиразин-2-амин (С4) (575 мг, 1,40 ммоль) растворяли в смеси 2Н4-метанола и 2Н6-ацетона при слабом нагревании. Раствор оставляли стоять на 10 минут, затем концентрировали в вакууме. Остаток растворяли в смеси тетрагидрофуран/2Н4-метанола в соотношении 1:1 (30 мл) и раствора дейтероксида натрия в 2Н4-метаноле (3 мМ, 1,5 эквивалента) и гидрировали при давлении 2Н2 5 фунт/кв.дюйм в течение 2,5 часов при комнатной температуре, используя катализатор 10%-ный палладий на углероде (5%-ная загрузка). Реакционную смесь затем фильтровали для удаления катализатора и концентрировали при пониженном давлении с получением желтого твердого вещества. Это твердое вещество суспендировали в небольшом количестве этилацетата, фильтровали и промывали этилацетатом с получением продукта в виде желтого твердого вещества. Посредством ЖХ-МС анализа обнаружили, что фильтрат содержит дополнительный продукт. Фильтрат концентрировали в вакууме с получением желтого твердого вещества, которое промывали этилацетатом; полученный белый осадок удаляли фильтрованием и отбрасывали. Фильтрат объединяли с первоначально собранным желтым твердым веществом, разбавляли дополнительным количеством этилацетата и промывали водой, насыщенным водным раствором хлорида аммония, насыщенным водным раствором хлорида натрия, сушили над сульфатом натрия и фильтровали. Концентрирование фильтрата при пониженном давлении дало желтое твердое вещество, которое очищали хроматографией на силикагеле (градиент: от 20% до 100% этилацетата в гептане). Получили желтое твердое вещество; после попытки растворения в этилацетате образовалось белое твердое вещество, которое отфильтровывали с получением продукта в виде белого твердого вещества. Выход: 207 мг, 0,621 ммоль, 44%. ЖХ-МС m/z 334,1 (М+Н). 1Н ЯМР (400 МГц, DMSO-d6) δ 8.14 (d, J=2,2 Гц, 1Н), 8.01 (d, J=5,9 Гц, 1Н), 7.47 (dd, J=5,9, 1,0 Гц, 1Н), 7.21 (d, J=8,2 Гц, 1Н), 7.15 (br d, J=2,4 Гц, 1Н), 7.07-7.11 (m, 1Н), 7.06 (dd, J=2,2, 1,1 Гц, 1H), 6.18 (br s, 2H), 2.11 (s, 3Н), 2.07 (br s, 3H).
Стадия 4. Синтез 5-[4-(фуро[3,2-с]пиридин-4-илокси)-2-метилфенил]-6-метил-[8-2Н]-имидазо[1,2-а]пиразина (2)
Хлорацетальдегид (55%-ный раствор в воде, 1,28 мл, 10,9 ммоль) добавляли к смеси 6-[4-(фуро[3,2-с]пиридин-4-илокси)-2-метилфенил]-5-метил-[3-2Н]-пиразин-2-амина (С5) (182 мг, 0,546 ммоль) в воде (2,5 мл) и реакционную смесь нагревали до 100°C в течение 1 часа. После охлаждения до комнатной температуры реакционную смесь разбавляли водой (15 мл) и этилацетатом (15 мл), затем обрабатывали насыщенным водным раствором бикарбоната натрия (от 5 до 10 мл). Водный слой экстрагировали этилацетатом и объединенные органические слои промывали водой, промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Хроматография на силикагеле (градиент: от 0% до 5% метанола в дихлорметане) дала продукт в виде твердого вещества. Выход: 158 мг, 0,442 ммоль, 81%. ЖХ-МС m/z 358,0 (М+Н). 1Н ЯМР (400 МГц, DMSO-d6) δ 8.18 (d, J=2,2 Гц, 1H), 8.08 (d, J=5,9 Гц, 1H), 7.77 (d, J=1,0 Гц, 1H), 7.54 (dd, J=5,8, 0,9 Гц, 1H), 7.46 (d, J=8,4 Гц, 1H), 7.40 (br d, J=2,4 Гц, 1H), 7.30 (br dd, J=8,3, 2,4 Гц, 1H), 7.26 (d, J=1,0 Гц, 1H), 7.12 (dd, J=2,2, 1,0 Гц, 1H), 2.27 (s, 3H), 2.00 (br s, 3H).
Примеры 3 и 4
(+)-5-[4-(Фуро[3,2-с]пиридин-4-илокси)-2-метилфенил]-6-метил-[8-2Н]-имидазо[1,2-а]пиразин (3) и (-)-5-[4-(Фуро[3,2-с]пиридин-4-илокси)-2-метилфенил]-6-метил-[8-2Н]-имидазо[1,2-а]пиразин (4)
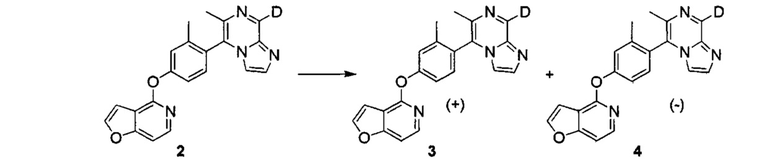
Хиральное разделение 5-[4-(фуро[3,2-с]пиридин-4-илокси)-2-метилфенил]-6-метил-[8-2Н]-имидазо[1,2-а]пиразина (2) (0,158 г) осуществляли с использованием сверхкритической флюидной хроматографии (колонка: Chiralpak AD-H, 5 мкм; элюент: смесь диоксид углерода/метанол в соотношении 3:1) с получением 3 [элюирующийся первым пик, обозначенный как (+)-атропоизомер согласно его обнаруженным данным вращения, 50 мг, 32%] и 4 [элюирующийся вторым пик, обозначенный как (-)-атропоизомер согласно его обнаруженным данным вращения, 55 мг, 34%]. Соединение 3: 1Н ЯМР (400 МГц, CDCl3) δ 8.09 (d, J=5,7 Гц, 1Н), 7.78-7.86 (br m, 1Н), 7.71 (d, J=2,4 Гц, 1Н), 7.35-7.37 (m, 1Н), 7.29-7.34 (m, 3Н), 7.23-7.27 (m, 1Н, предполагаемый; частично скрыт за пиком растворителя), 6.96 (dd, J=2,2, 1,0 Гц, 1Н), 2.44 (s, 3Н), 2.08 (s, 3Н). Соединение 4: 1Н ЯМР (500 МГц, DMSO-d6) δ 8.18 (d, J=2,3 Гц, 1Н), 8.08 (d, J=5,7 Гц, 1Н), 7.77 (d, J=1,0 Гц, 1Н), 7.53 (dd, J=5,8, 0,9 Гц, 1Н), 7.46 (d, J=8,3 Гц, 1Н), 7.40 (d, J=2,4 Гц, 1Н), 7.30 (dd, J=8,2, 2,6 Гц, 1Н), 7.26 (d, J=1,0 Гц, 1Н), 7.12 (dd, J=2,2, 0,8 Гц, 1Н), 2.27 (s, 3Н), 2.00 (s, 3Н).
Пример 5
1-[4-(Фуро[3,2-с]пиридин-4-илокси)-2-метилфенил]-2-метил-1H-имидазо[4,5-с]пиридин (5)

Стадия 1. Синтез N-(4-метокси-2-метилфенил)-3-нитропиридин-4-амина (С6)
Раствор 4-метокси-2-метиланилина (23,8 г, 173 ммоль), 4-хлор-3-нитропиридина (25 г, 160 ммоль) и триэтиламина (33,0 мл, 237 ммоль) в этаноле (250 мл) перемешивали при комнатной температуре в течение 16 часов, затем концентрировали при пониженном давлении. Остаток растворяли в этилацетате (200 мл) и фильтровали через плотную набивку силикагеля (элюент: этилацетат, 1 л). Фильтрат концентрировали в вакууме с получением продукта в виде пурпурного масла, которое затвердевало при стоянии. Этот материал использовали без дополнительной очистки. Выход: 41 г, 160 ммоль, 100%. ЖХ-МС m/z 260,1 (М+Н).
Стадия 2. Синтез N4-(4-метокси-2-метилфенил)пиридин-3,4-диамина (С7)
Палладий на углероде (10%, 3×2,12 г) добавляли к каждой из трех партий N-(4-метокси-2-метилфенил)-3-нитропиридин-4-амина (С6) (каждая приблизительно 10 г; суммарно 31 г, 120 ммоль) в метаноле (3×100 мл). Три суспензии независимо гидрировали под давлением водорода 45 фунт/кв.дюйм при комнатной температуре в смесителе Парра в течение 24 часов. Три реакционных смеси объединяли, фильтровали через набивку целита и концентрировали в вакууме. Очистка хроматографией на силикагеле [градиент: от 2% до 10% (1,7 М аммиака в метаноле) в дихлорметане] дала продукт в виде светло-коричневого твердого вещества. Выход: 24,0 г, 105 ммоль, 88%. ЖХ-МС m/z 230,1 (М+Н). 1Н ЯМР (400 МГц, CDCl3) δ 8.01 (s, 1Н), 7.88 (d, J=5,5 Гц, 1Н), 7.08 (d, J=8,6 Гц, 1Н), 6.84 (br d, J=2,8 Гц, 1H), 6.78 (br dd, J=8,6, 3,0 Гц, 1H), 6.34 (d, J=5,5 Гц, 1H), 5.66 (br s, 1Н), 3.82 (s, 3Н), 2.20 (brs, 3Н).
Стадия 3. Синтез 1-(4-метокси-2-метилфенил)-2-метил-1Н-имидазо[4,5-с]пиридина (С8)
Смесь N4-(4-метокси-2-метилфенил)пиридин-3,4-диамина (С7) (3,95 г, 17,2 ммоль), уксусного ангидрида (1,96 мл, 20,7 ммоль) и триэтилортоацетата (99%, 15,9 мл, 86,4 ммоль) нагревали при 145°C в течение 1 часа, затем при 100°C в течение 48 часов. После охлаждения до комнатной температуры реакционную смесь разбавляли этилацетатом (100 мл), промывали насыщенным водным раствором бикарбоната натрия (30 мл), промывали водой, сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Очистка хроматографией на силикагеле (градиент: от 2% до 5% метанола в дихлорметане) дала продукт в виде светло-розового масла. Выход: 4,10 г, 16,2 ммоль, 94%. ЖХ-МС m/z 254,1 (М+Н). 1Н ЯМР (400 МГц, CDCl3) δ 9.07 (br d, J=0,8 Гц, 1Н), 8.36 (d, J=5,5 Гц, 1Н), 7.15 (d, J=8,6 Гц, 1Н), 6.89-6.97 (m, 3Н), 3.90 (s, 3Н), 2.42 (s, 3Н), 1.94 (br s, 3Н).
Стадия 4. Синтез 3-метил-4-(2-метил-1Н-имидазо[4,5-с]пиридин-1-ил)фенола (С9)
Трибромид бора (1 М раствор в дихлорметане, 44,1 мл, 44,1 ммоль) добавляли по каплям к раствору 1-(4-метокси-2-метилфенил)-2-метил-1Н-имидазо[4,5-с]пиридина (С8) (3,72 г, 14,7 ммоль) в дихлорметане (150 мл) при -78°C. Реакционную смесь перемешивали при -78°C в течение 15 минут, затем охлаждающую баню удаляли и реакционную смесь оставляли постепенно нагреваться до комнатной температуры. Через 20 часов при комнатной температуре реакционную смесь снова охлаждали до -78°C и медленно гасили метанолом (20 мл). В этот момент охлаждающую баню удаляли; смесь оставляли достигать температуры окружающей среды и затем перемешивали в течение 15 минут. Летучие вещества удаляли в вакууме, добавляли метанол (100 мл) и смесь кипятили с обратным холодильником в течение 30 минут. После концентрирования при пониженном давлении полученное твердое вещество переносили непосредственно на следующую стадию. ЖХ-МС m/z 240,1 (М+Н).
Стадия 5. Синтез 1-[4-(фуро[3,2-с]пиридин-4-илокси)-2-метилфенил]-2-метил-1Н-имидазо[4,5-с]пиридина (5)
Смесь 3-метил-4-(2-метил-1H-имидазо[4,5-с]пиридин-1-ил)фенола (С9) (с предыдущей стадии, не более 14,7 ммоль), 4-хлорфуро[3,2-с]пиридина (2,37 г, 15,4 ммоль) и карбоната цезия (99%, 19,3 г, 58,6 ммоль) в диметилсульфоксиде (100 мл) нагревали до 140°C в течение 16 часов. После охлаждения до комнатной температуры реакционную смесь разбавляли этилацетатом (400 мл) и фильтровали через набивку целита. Фильтрат промывали водой, смесью воды и насыщенного водного раствора хлорида натрия в соотношении 1:1 (4×100 мл), сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Остаток очищали хроматографией на силикагеле (градиент: от 2% до 10% метанола в этилацетате) с получением желтого твердого вещества, которое растворяли в трет-бутилметиловом эфире (500 мл), обрабатывали активированным углеродом (5 г) и нагревали до 40°C. Смесь фильтровали с получением бесцветного раствора, который концентрировали при кипячении с обратным холодильником до тех пор, пока он не помутнел (осталось примерно 150 мл трет-бутилметилового эфира). После ступенчатого охлаждения до комнатной температуры образовался осадок. В результате фильтрования и промывки диэтиловым эфиром получили продукт в виде свободно-текучего белого твердого вещества. Выход: 2,02 г, 5,67 ммоль, 39% за 2 стадии. ЖХ-МС m/z 357,1 (М+Н). 1Н ЯМР (500 МГц, CDCl3) δ 9.08 (d, J=1,0 Гц, 1Н), 8.39 (d, J=5,5 Гц, 1Н), 8.08 (d, J=5,9 Гц, 1Н), 7.71 (d, J=2,2 Гц, 1Н), 7.34-7.36 (m, 1Н), 7.30 (dd, J=5,9, 1,0 Гц, 1Н), 7.28-7.29 (m, 2Н), 7.00 (dd, J=5,5, 1,1 Гц, 1Н), 6.97 (dd, J=2,2, 1,0 Гц, 1Н), 2.48 (s, 3Н), 1.99 (brs, 3Н).
Пример 6
4-[3-Метокси-4-(3-метилпиразин-2-ил)фенокси]фуро[3,2-с]пиридин (6)
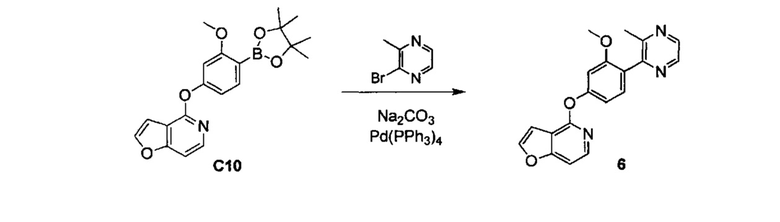
2-Бром-3-метилпиразин (104 мг, 0,600 ммоль), тетракис(трифенилфосфин)палладий(0) (95%, 133 мг, 0,109 ммоль) и карбонат натрия (175 мг, 1,64 ммоль) объединяли с 4-[3-метокси-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенокси]фуро[3,2-с]пиридином [С10, который получили способом, аналогичным получению 4-[3-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенокси]фуро[3,2-с]пиридина (С2) в Примере 1] (200 мг, 0,545 ммоль) в 1,4-диоксане (3 мл) и воде (1 мл). Реакционную смесь нагревали до 130°C в микроволновом реакторе в течение 1 часа. Смесь охлаждали до комнатной температуры и супернатант декантировали в другую колбу. Оставшееся твердое вещество промывали этилацетатом (3×10 мл) и объединенные органические порции концентрировали в вакууме. Очистку осуществляли дважды с использованием хроматографии на силикагеле (первая колонка: элюент: 2% метанола в дихлорметане; вторая колонка: градиент: от 0% до 100% этилацетата в гептане). Бесцветные фракции объединяли и концентрировали при пониженном давлении с получением продукта в виде белого твердого вещества. Выход: 85 мг, 0,25 ммоль, 46%. ЖХ-МС m/z 334,0 (М+Н). 1Н ЯМР (400 МГц, CDCl3) δ 8.47 (АВ квартет, слабополньный дублет уширен, JAB=2,5 Гц, Δνab=14 Гц, 2Н), 8.08 (d, J=5,9 Гц, 1Н), 7.66 (d, J=2,3 Гц, 1Н), 7.36 (d, J=8,0 Гц, 1Н), 7.25-7.28 (m, 1Н, предполагаемый; частично скрыт за пиком растворителя), 6.90-6.96 (m, 2Н), 6.88 (dd, J=2,2, 0,8 Гц, 1Н), 3.79 (s, 3Н), 2.50 (s, 3Н). Желтые фракции снова очищали с получением дополнительного продукта: 55 мг, суммарный выход: 75%.
Пример 7
4-[4-(1-Метил-1Н -пиразол-5-ил)фенокси]тиено[3,2-с]пиридин (7)
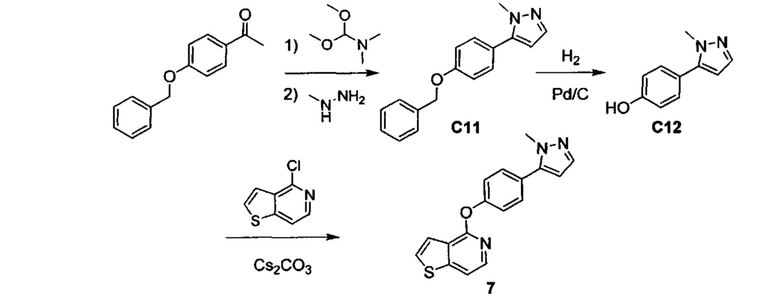
Стадия 1. Синтез 5-[4-(бензилокси)фенил]-1-метил-1Н-пиразола (С11)
N,N-Диметилформамиддиметилацеталь (94%, 19,0 мл, 134 ммоль) добавляли к раствору 1-[4-(бензилокси)фенил]этанона (15,32 г, 67,71 ммоль) в N,N-диметилформамиде (30 мл) и реакционную смесь кипятили с обратным холодильником в течение 18 часов. В этот момент конденсатор системы обратного холодильника заменяли дистилляционной насадкой и дистилляцию осуществляли до тех пор, пока температура дистиллята не достигла 140°C. Материал в реакционном сосуде охлаждали до комнатной температуры, обрабатывали метилгидразином (98%, 7,4 мл, 136 ммоль) и нагревали при 75°C в течение 3 часов. Реакционную смесь охлаждали, разбавляли этилацетатом, промывали четыре раза водным 5%-ным раствором хлорида натрия, сушили над сульфатом магния, фильтровали и концентрировали в вакууме. Очистка хроматографией на силикагеле (градиент: от 2% до 10% этилацетата в дихлорметане) дала продукт в виде светло-желтого твердого вещества. Выход: 13,79 г, 52,17 ммоль, 77%. ЖХ-МС m/z 265,1 (М+Н). 1Н ЯМР (400 МГц, DMSO-d6) характеристические пики, δ 3.81 (s, 3Н), 5.17 (s, 2Н), 6.31 (d, J=1,5 Гц, 1Н), 7.12 (d, J=8,8 Гц, 2Н).
Стадия 2. Синтез 4-(1-метил-1H-пиразол-5-ил)фенола (С12)
5-[4-(Бензилокси)фенил]-1-метил-1H-пиразол (С11) (13,49 г, 51,04 ммоль) смешивали с 10%-ным палладием на углероде (примерно 50% в воде, 1,46 г) и растворяли в этаноле (125 мл). Реакционную смесь гидрировали при комнатной температуре и 1 атмосфере водорода в течение 18 часов, затем фильтровали и концентрировали в вакууме. Остаток растирали с гептаном с получением продукта в виде бесцветного твердого вещества. Выход: 8,74 г, 50,2 ммоль, 98%. ЖХ-МС m/z 175,1 (М+Н). 1Н ЯМР (400 МГц, DMSO-d6) δ 9.73 (br s, 1Н), 7.40 (d, J=1,9 Гц, 1Н), 7.31 (br d, J=8,7 Гц, 2H), 6.86 (br d, J=8,7 Гц, 2H), 6.26 (d, J=1,9 Гц, 1H), 3.79 (s, 3Н).
Стадия 3. Синтез 4-[4-(1-метил-1Н-пиразол-5-ил)фенокси]тиено[3,2-с]пиридина (7)
4-(1-Метил-1Н-пиразол-5-ил)фенол (С12) (123 мг, 0,706 ммоль) и 4-хлортиено[3,2-с]пиридин (100 мг, 0,590 ммоль) объединяли в 1-метилпирролидин-2-оне (2 мл). Добавляли карбонат цезия (99%, 388 мг, 1,18 ммоль) и реакционную смесь нагревали до 135°C в течение 24 часов. После добавления воды (30 мл) слои разделяли и водный слой экстрагировали смесью диэтиловый эфир/гексаны в соотношении 1:1 (4×30 мл). Объединенные органические слои промывали водным раствором гидроксида натрия (1 н., 2×20 мл) и насыщенным водным раствором хлорида натрия (20 мл), затем сушили над сульфатом натрия. После фильтрования и концентрирования при пониженном давлении очистка с использованием хроматографии на силикагеле (элюент: 30% этилацетата в гептане) дала продукт в виде белого твердого вещества. Выход: 78 мг, 0,25 ммоль, 42%. ЖХ-МС m/z 308,3 (М+Н). 1Н ЯМР (500 МГц, CD3OD) δ 7.90 (d, J=5,6 Гц, 1Н), 7.74 (d, J=5,5 Гц, 1Н), 7.69 (dd, J=5,7, 0,7 Гц, 1Н), 7.65 (dd, J=5,5, 0,8 Гц, 1Н), 7.55 (br d, J=8,7 Гц, 2H), 7.51 (d, J=2,0 Гц, 1H), 7.32 (br d, J=8,7 Гц, 2H), 6.39 (d, J=2,0 Гц, 1H), 3.91 (s, 3H).
Пример 8
Трифторацетатная соль 4-{[4-(1-метил-1Н-пиразол-5-ил)фенил]-сульфанил}фуро[3,2-с]пиридина (8)

Стадия 1. Синтез 4-[(4-бромфенил)сульфанил]фуро[3,2-с]пиридина (С13)
Карбонат цезия (99%, 522 мг, 1,59 ммоль) добавляли к смеси 4-хлорфуро[3,2-с]пиридина (146 мг, 0,951 ммоль) и 4-бромбензолтиола (150 мг, 0,793 ммоль) в диметилсульфоксиде (3 мл); реакционную смесь дегазировали и затем нагревали при 80°C в течение 16 часов. Добавляли воду (30 мл) и экстракцию осуществляли смесью этилацетат/гексаны в соотношении 1:1 (4×30 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Очистка хроматографией на силикагеле (градиент: от 5% до 10% этилацетата в гептане) дала бесцветное масло (220 мг); это масло растворяли в диэтиловом эфире (20 мл) и промывали водным раствором гидроксида натрия (1 н., 3×15 мл). Органический слой концентрировали при пониженном давлении с получением продукта, определенного путем 1H ЯМР анализа как загрязненного посторонней фуро[3,2-с]пиридил-активностью. Этот продукт переносили на следующую стадию без дополнительной очистки. ЖХ-МС m/z 308,3 (М+Н). 1Н ЯМР (400 МГц, CDCl3) только пики продукта, δ 8.32 (d, J=5,7 Гц, 1Н), 7.60 (d, J=2,2 Гц, 1Н), 7.47 (br АВ квартет, Jab=8,7 Гц, ΔνАВ=31,2 Гц, 4Н), 7.29 (dd, J=5,8, 1,0 Гц, 1Н), 6.58 (dd, J=2,3, 1,0 Гц, 1Н).
Стадия 2. Синтез трифторацетатной соли 4-{[4-(1-метил-1H-пиразол-5-ил)фенил]сульфанил}-фуро[3,2-с]пиридина (8)
4-[(4-Бромфенил)сульфанил]фуро[3,2-с]пиридин (С13) (210 мг с предыдущей стадии), (1-метил-1H-пиразол-5-ил)бороновую кислоту (104 мг, 0,826 ммоль), трифенилфосфин (21,5 мг, 0,0819 ммоль) и карбонат калия (190 мг, 1,37 ммоль) объединяли в N,N-диметилформамиде (6 мл) и воде (2 мл) и дегазировали азотом в течение 20 минут. Добавляли ацетат палладия(II) (98%, 4,8 мг, 0,021 ммоль) и реакционную смесь нагревали при 80°C в течение 18 часов. После охлаждения до комнатной температуры реакционную смесь разбавляли водой (15 мл) и экстрагировали смесью этилацетат/гексаны в соотношении 1:1 (3×15 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Очистку осуществляли сначала посредством хроматографии на силикагеле (элюент: 80% этилацетата в гептане), а затем посредством HPLC (колонка: Waters XBridge С18, 5 мкм; подвижная фаза А: вода с трифторуксуснокислотным модификатором; подвижная фаза В: ацетонитрил с трифторуксуснокислотным модификатором; градиент: от 40% до 100% В) с получением продукта в виде белого твердого вещества. Выход: 30 мг, 0,071 ммоль, 9% за две стадии. ЖХ-МС m/z 308,0 (М+Н). 1Н ЯМР (400 МГц, CD3OD) δ 8.29 (d, J=5,8 Гц, 1Н), 7.87 (d, J=2,2 Гц, 1Н), 7.61 (br d, J=8,6 Гц, 2H), 7.53 (brd, J=8,7 Гц, 2H), 7.51 (d, J=2,1 Гц, 1H), 7.49 (dd, J=5,8, 1,0 Гц, 1H), 6.66 (dd, J=2,3, 1,1 Гц, 1H), 6.42 (d, J=2,0 Гц, 1H), 3.90 (s, 3Н).
Пример 9
2-(4,6-Диметилпиримидин-5-ил)-5-(фуро[3,2-с]пиридин-4-илокси)-бензонитрил (9)
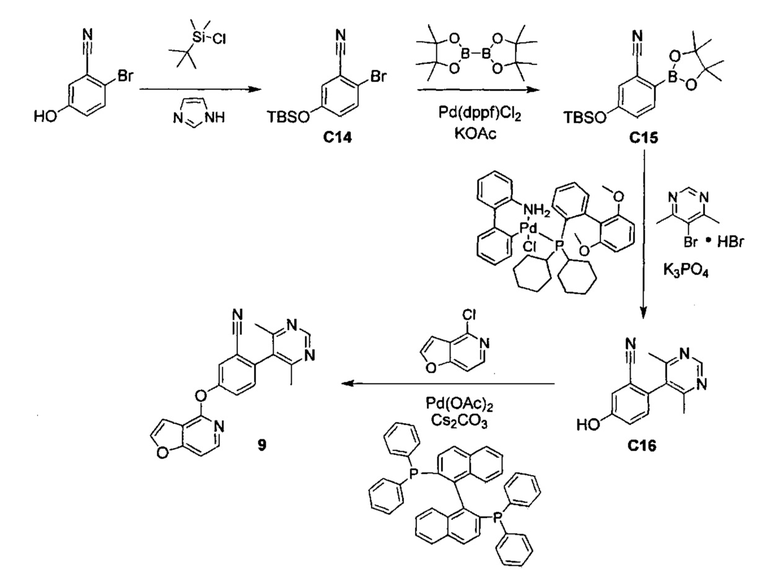
Стадия 1. Синтез 2-бром-5-{[трет-бутил(диметил)силил]окси}-бензонитрила (С14)
1Н-Имидазол (2,14 г, 31,4 ммоль) добавляли порциями при 0°C к раствору 2-бром-5-гидроксибензонитрила (5,65 г, 28,5 ммоль) и трет-бутилдиметилсилилхлорида (4,52 г, 30,0 ммоль) в тетрагидрофуране (56,5 мл). Реакционную смесь оставляли перемешиваться при комнатной температуре в течение 2 часов и затем фильтровали. Фильтрат промывали водой и насыщенным водным раствором хлорида натрия. Водный слой экстрагировали диэтиловым эфиром и объединенные органические слои концентрировали в вакууме с получением продукта в виде оранжевого масла. Выход: 8,87 г, 28,4 ммоль, 99,6%. 1Н ЯМР (400 МГц, CDCl3) δ 7.50 (d, J=8,8 Гц, 1Н), 7.08-7.12 (m, 1Н), 6.90-6.95 (m, 1Н), 0.98 (s, 9Н), 0.22 (s, 6Н).
Стадия 2. Синтез 5-{[трет-6утил(диметил)силил]окси}-2-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензонитрила (С15)
2-Бром-5-{[трет-бутил(диметил)силил]окси}бензонитрил (С14) (8,00 г, 25,6 ммоль), 4,4,4',4',5,5,5',5',-октаметил-2,2'-би-1,3,2-диоксаборолан (6,83 г, 26,9 ммоль) и ацетат калия (10,06 г, 102,5 ммоль) объединяли в дегазированном 1,4-диоксане (160 мл). После добавления [1,1'-бис(дифенилфосфино)ферроцен]-дихлорпалладия(II) (1,05 г, 1,28 ммоль) реакционную смесь нагревали до 80°C в течение 4 часов. После охлаждения ее фильтровали через целит и фильтровальную набивку промывали этилацетатом. Фильтрат концентрировали в вакууме и очищали посредством хроматографии на силикагеле (градиент: от 20% до 50% этилацетата в гептане) с получением продукта в виде бесцветного вязкого масла. Выход: 5,60 г, 15,6 ммоль, 61%. 1Н ЯМР (400 МГц, CDCl3) δ 7.76 (br d, J=8,3 Гц, 1Н), 7.15 (dd, J=2,4, 0,3 Гц, 1Н), 7.02 (dd, J=8,3, 2,3 Гц, 1Н), 1.38 (s, 12Н), 0.98 (s, 9Н), 0.22 (s, 6Н).
Стадия 3. Синтез 2-(4,6-диметилпиримидин-5-ил)-5-гидроксибензонитрила (С16)
5-{[трет-Бутил (диметил)силил]окси}-2-(4,4,5,5-тетра мети л-1,3,2-диоксаборолан-2-ил)бензонитрил (С15) (4,05 г, 11,3 ммоль) объединяли с гидробромидом 5-бром-4,6-диметилпиримидина (7,16 г, 26,7 ммоль) и фосфатом калия (7,03 г, 33,1 ммоль) в 2-метилтетрагидрофуране (20,2 мл) и воде (16,2 мл). Добавляли [2'-(азанидил-kN)бифенил-2-ил-kC2](хлор)-[дициклогексил(2',6'-диметоксибифенил-2-ил)-λ5-фосфанил]палладий (полученный из бифенил-2-амина и дициклогексил(2',6'-диметоксибифенил-2-ил)фосфана (S-Phos) согласно методике S. L. Buchwald et al., J. Am. Chem. Soc. 2010, 132, 14073-14075) (0,20 г, 0,28 ммоль) и реакционную смесь кипятили с обратным холодильником в течение 18 часов. Ее затем охлаждали до комнатной температуры и органический слой экстрагировали водной соляной кислотой (2 н., 2×20 мл). Значение pH объединенных экстрактов доводили до примерно 6-7 с использованием 2 М водного раствора гидроксида натрия и затем экстрагировали этилацетатом. Эти объединенные органические слои сушили над сульфатом магния, фильтровали и концентрировали в вакууме. Полученное твердое вещество растирали с горячим гептаном с получением продукта в виде желто-коричневого твердого вещества. Выход: 1,86 г, 8,26 ммоль, 73%. 1Н ЯМР (400 МГц, DMSO-d6) δ 10.48 (s, 1Н), 8.94 (s, 1Н), 7.36 (d, J=8,4 Гц, 1Н), 7.31 (d, J=2,5 Гц, 1Н), 7.23 (dd, J=8,5, 2,6 Гц, 1Н), 2.18 (s, 6Н).
Стадия 4. Синтез 2-(4,6-диметилпиримидин-5-ил)-5-(фуро[3,2-с]пиридин-4-илокси)бензонитрила (9)
2-(4,6-Диметилпиримидин-5-ил)-5-гидроксибензонитрил (С16) (1,00 г, 4,44 ммоль), 4-хлорфуро[3,2-с]пиридин (750 мг, 4,88 ммоль), ацетат палладия(II) (49,8 мг, 0,22 ммоль), 1,1'-бинафталин-2,2'-диилбис(дифенилфосфан) (96%, 288 мг, 0,444 ммоль) и карбонат цезия (99%, 2,92 г, 8,87 ммоль) объединяли в 1,4-диоксане (25 мл) и через эту смесь в течение 15 минут барботировали азот. Реакционную смесь затем нагревали при 100°C в течение 18 часов, охлаждали до комнатной температуры и фильтровали через целит. Фильтрат распределяли между этилацетатом и водой и водный слой экстрагировали этилацетатом. Объединенные органические слои промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом магния, фильтровали и концентрировали в вакууме. Очистка с использованием хроматографии на силикагеле (градиент: от 75% до 100% этилацетата в гептане) дала продукт в виде вязкого желтого масла, которое медленно затвердевало при стоянии. Дополнительную очистку осуществляли с использованием сверхкритической флюидной хроматографии (колонка: Princeton 2-этилпиридин, 5 мкм; элюент: смесь диоксид углерода/метанол в соотношении 4:1). Выход: 1,5 г, 4,4 ммоль, 99%. ЖХ-МС m/z 343,1 (М+Н). 1Н ЯМР (400 МГц, CDCl3) δ 9.04 (s, 1Н), 8.06 (d, J=5,9 Гц, 1Н), 7.78 (br d, J=2,5 Гц, 1H), 7.72 (d, J=2,2 Гц, 1H), 7.66 (dd, J=8,4, 2,5 Гц, 1H), 7.36 (dd, J=8,4, 0,4 Гц, 1H), 7.33 (dd, J=5,7, 1,0 Гц, 1H) 6.97 (dd, J=2,2, 1,0 Гц, 1H), 2.36 (s, 6Н).
Пример 10
бис-Гидрохлоридная соль 4-[4-(фуро[3,2-с]пиридин-4-илокси)-2-метилфенил]-5-метилпиридазин-3(2Н)-она (10)
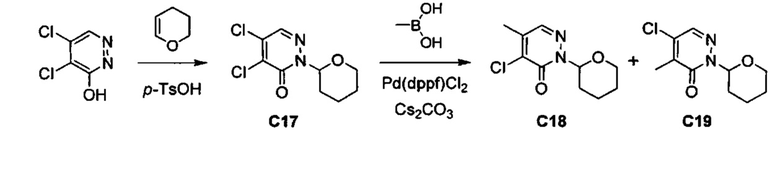
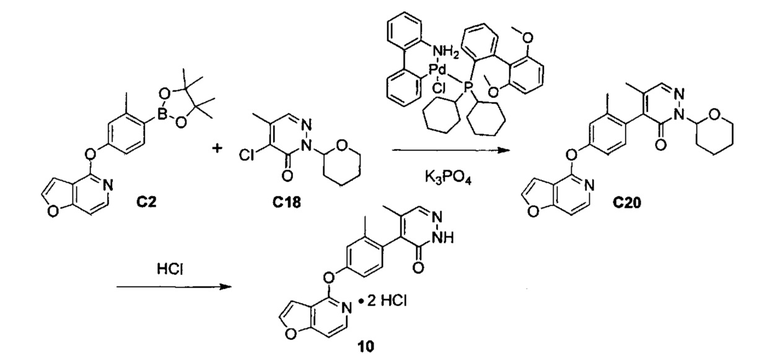
Стадия 1. Синтез 4,5-дихлор-2-(тетрагидро-2Н-пиран-2-ил)пиридазин-3(2H)-она (С17)
Смесь 4,5-дихлорпиридазин-3-ола (42 г, 250 ммоль), 3,4-дигидро-2H-пирана (168 г, 2,00 моль) и пара-толуолсульфоновой кислоты (8,8 г, 51 ммоль) в тетрагидрофуране (2 л) кипятили с обратным холодильником в течение 2 суток. После охлаждения до комнатной температуры смесь концентрировали при пониженном давлении. Остаток очищали хроматографией на силикагеле (градиент: от 3% до 5% этилацетата в петролейном эфире) с получением продукта в виде белого твердого вещества. Выход: 42 г, 170 ммоль, 68%. 1Н ЯМР (400 МГц, CDCl3) δ 7.84 (s, 1Н), 6.01 (br d, J=11 Гц, 1Н), 4.10-4.16 (m, 1Н), 3.70-3.79 (m, 1Н), 1.99-2.19 (m, 2Н), 1.50-1.80 (m, 4Н).
Стадия 2. Синтез 4-хлор-5-метил-2-(тетрагидро-2H-пиран-2-ил)пиридазин-3(2H)-она (С18) и 5-хлор-4-метил-2-(тетрагидро-2H-пиран-2-ил)пиридазин-3(2Н)-она (С19)
К смеси 4,5-дихлор-2-(тетрагидро-2H-пиран-2-ил)пиридазин-3(2Н)-она (С17) (40 г, 0,16 моль), метилбороновой кислоты (9,6 г, 0,16 моль) и карбоната цезия (155 г, 0,476 моль) в смеси 1,4-диоксана (500 мл) и воды (50 мл) добавляли [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий(II) (5 г, 7 ммоль). Реакционную смесь перемешивали при 110°C в течение 2 часов, затем концентрировали при пониженном давлении. Очистка хроматографией на силикагеле (градиент: от 3% до 5% этилацетата в петролейном эфире) дала продукт С18 в виде бледно-желтого твердого вещества (Выход: 9 г, 40 ммоль, 25%) и продукт С19, также в виде бледно-желтого твердого вещества (Выход: 9,3 г, 41 ммоль, 26%). С18: ЖХ-МС m/z 250,8 (M+Na+). 1Н ЯМР (400 МГц, CDCl3) δ 7.71 (s, 1Н), 6.07 (dd, J=10,7, 2,1 Гц, 1Н), 4.10-4.18 (m, 1Н), 3.71-3.81 (m, 1Н), 2.30 (s, 3Н), 1.98-2.19 (m, 2Н), 1.53-1.81 (m, 4Н). С19: ЖХ-МС m/z 250,7 (M+Na+). 1Н ЯМР (400 МГц, CDCl3) δ 7.77 (s, 1Н), 6.02 (dd, J=10,7, 2,1 Гц, 1Н), 4.10-4.17 (m, 1Н), 3.71-3.79 (m, 1Н), 2.27 (s, 3Н), 1.99-2.22 (m, 2Н), 1.51-1.79 (m, 4Н).
Стадия 3. Синтез 4-[4-(фуро[3,2-с]пиридин-4-илокси)-2-метилфенил]-5-метил-2-(тетрагидро-2Н-пиран-2-ил)пиридазин-3(2Н)-она (С20)
Смесь 4-хлор-5-метил-2-(тетрагидро-2H-пиран-2-ил)пиридазин-3(2Н)-она (С18) (457 мг, 2,00 ммоль), 4-[3-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенокси]фуро[3,2-с]пиридина (С2) (702 мг, 2,00 ммоль) и [2'-(азанидил-kN)бифенил-2-ил-kC2)(хлор)(дициклогексил(2',6'-диметоксибифенил-2-ил)-λ5-фосфанил]палладия (29 мг, 0,040 ммоль) подвергали трем циклам вакуумирования с последующим введением азота. Добавляли дегазированный тетрагидрофуран (4 мл), а затем дегазированный раствор фосфата калия (0,5 М, 8,0 мл, 4,0 ммоль) и реакционную смесь оставляли перемешиваться при комнатной температуре в течение 23 часов. Реакционную смесь затем распределяли между этилацетатом (20 мл) и водой (8 мл); органический слой сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Очистка посредством хроматографии на силикагеле (градиент: от 20% до 70% этилацетата в гептане) дала продукт в виде белого твердого вещества. С помощью ЯМР было определено, что он состоит из диастереомерной смеси в виду наличия тетрагидропиранильной группы. Выход: 588 мг, 1,41 ммоль, 70%. ЖХ-МС m/z 418,0 (М+Н). 1Н ЯМР (500 МГц, CDCl3) δ 8.06 (d, J=5,9 Гц, 1Н), 7.82 (d, J=2,8 Гц, 1Н), 7.63 (d, J=2,3 Гц, 1Н), 7.23-7.25 (m, 1Н), 7.16-7.17 (m, 1Н), 7.06-7.13 (m, 2Н), 6.79-6.81 (m, 1Н), 6.10 (dd, J=10,6, 2,2 Гц, 1Н), 4.14-4.20 (m, 1Н), 3.72-3.80 (m, 1Н), 2.15-2.25 (m, 1Н, предполагаемый; частично скрыт за метильной группой), 2.14 и 2.15 (2 s, суммарный 3Н), 2.01-2.08 (m, 1Н, предполагаемый; частично скрыт за метильной группой), 2.03 и 2.04 (2 s, суммарный 3Н), 1.71-1.82 (m, 3Н), 1.55-1.63 (m, 1Н).
Стадия 4. Синтез бис-гидрохлоридной соли 4-[4-(фуро[3,2-с]пиридин-4-илокси)-2-метилфенил]-5-метилпиридазин-3(2Н)-она (10)
4-[4-(Фуро[3,2-с]пиридин-4-илокси)-2-метилфенил]-5-метил-2-(тетрагидро-2H-пиран-2-ил)пиридазин-3(2H)-он (С20) (580 мг, 1,39 ммоль) растворяли в метаноле (3 мл), обрабатывали раствором хлористого водорода в 1,4-диоксане (4 М, 5,0 мл, 20 ммоль) и оставляли перемешиваться при комнатной температуре в течение 3 часов. Удаление растворителя при пониженном давлении дало продукт в виде бледно-желтого твердого вещества, предположительно в виде бис-гидрохлоридной соли. Выход: 550 мг, 1,35 ммоль, 97%. ЖХ-МС m/z 334,0 (М+Н). 1Н ЯМР (400 МГц, DMSO-d6) δ 13.01 (br s, 1Н), 8.15 (d, J=2,3 Гц, 1Н), 8.02 (d, J=5,8 Гц, 1Н), 7.89 (s, 1Н), 7.48 (dd, J=5,8, 1,1 Гц, 1Н), 7.16-7.18 (m, 1Н), 7.08-7.12 (m, 3Н), 2.06 (br s, 3Н), 1.95 (s, 3Н).
Пример 11
4-[4-(3-Хлор-5-метилпиридазин-4-ил)-3-метилфенокси]фуро[3,2-с]пиридин (11)
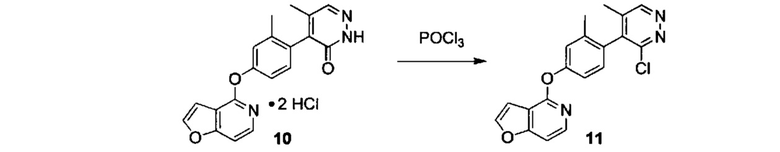
4-[4-(Фуро[3,2-с]пиридин-4-илокси)-2-метилфенил]-5-метилпиридазин-3(2H)-он в виде бис-гидрохлоридной соли (10) (550 мг, 1,35 ммоль) суспендировали в оксихлориде фосфора (6,0 мл, 64 ммоль) и реакционную смесь нагревали при 90°C в течение 2 часов. После удаления оксихлорида фосфора при пониженном давлении остаток распределяли между дихлорметаном (35 мл), водой (10 мл) и насыщенным водным раствором бикарбоната натрия (10 мл). Органический слой сушили над сульфатом натрия, фильтровали и концентрировали в вакууме с получением продукта в виде пенистого бледно-янтарного твердого вещества. Выход: 465 мг, 1,32 ммоль, 98%. ЖХ-МС m/z 352,0 (М+Н). 1Н ЯМР (400 МГц, CDCl3) δ 9.07 (s, 1Н), 8.11 (d, J=5,8 Гц, 1Н), 7.69 (d, J=2,3 Гц, 1Н), 7.31 (dd, J=5,9, 0,9 Гц, 1Н), 7.25-7.28 (m, 1Н, предполагаемый; частично скрыт за пиком растворителя), 7.21-7.24 (m, 1Н), 7.09 (d, J=8,2 Гц, 1Н), 6.84 (dd, J=2,2, 0,8 Гц, 1Н), 2.19 (s, 3Н), 2.08 (br s, 3Н).
Пример 12
4-[4-(3,5-Диметилпиридазин-4-ил)-3-метилфенокси]фуро[3,2-с]пиридин
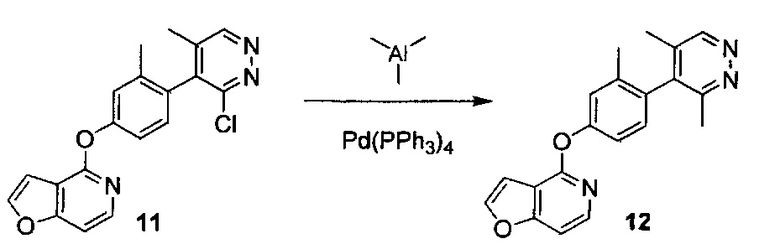
Азот барботировали через смесь тетракис(трифенилфосфин)палладия(0) (31,0 мг, 0,027 ммоль) и 4-[4-(3-хлор-5-метилпиридазин-4-ил)-3-метилфенокси]-фуро[3,2-с]пиридина (11) (427 мг, 1,21 ммоль) в 1,4-диоксане (12 мл) в течение 10 минут. Добавляли раствор триметилалюминия в толуоле (2 М, 1,2 мл, 2,4 ммоль) и реакционную смесь нагревали до 95°C в течение 90 минут, затем охлаждали в ледяной бане и обрабатывали по каплям метанолом (12 мл) {осторожно: выделение газа!}. Смесь фильтровали через целит и осадок на фильтре промывали дополнительным количеством метанола (35 мл); фильтрат концентрировали в вакууме и очищали с использованием хроматографии на силикагеле (элюент: 2,5% метанола в этилацетате) с получением продукта в виде твердого вещества. Выход: 320 мг, 0,966 ммоль, 80%. ЖХ-МС m/z 332,1 (М+Н). 1Н ЯМР (500 МГц, CD3OD) δ 9.05 (s, 1Н), 7.99 (d, J=6,0 Гц, 1Н), 7.90 (d, J=2,2 Гц, 1Н), 7.39 (dd, J=5,9, 0,9 Гц, 1Н), 7.26-7.27 (m, 1Н), 7.19 (br dd, половина АВХ картины, J=8,3, 2,1 Гц, 1Н), 7.15 (d, половина АВ картины, J=8,3 Гц, 1Н), 6.94 (dd, J=2,2, 1,0 Гц, 1Н), 2.42 (s, 3Н), 2.16 (s, 3Н), 2.03 (s, 3Н).
Примеры 13 и 14
(+)-4-[4-(3,5-Диметилпиридазин-4-ил)-3-метилфенокси]фуро[3,2-с]пиридин (13) и (-)-4-[4-(3,5-диметилпиридазин-4-ил)-3-метилфенокси]-фуро[3,2-с]пиридин (14)
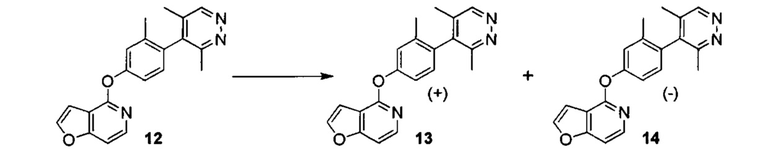
Соединение Примера 12 (4-[4-(3,5-диметилпиридазин-4-ил)-3-метилфенокси]фуро[3,2-с]пиридин) (316 мг) разделяли на его компоненты атропоэнантиомеры с использованием сверхкритической флюидной хроматография (колонка: Chiralpak AS-H, 5 мкм; элюент: смесь диоксид углерода/этанол в соотношении 7:3). Оба были получены в виде твердых веществ. Элюирующийся первым атропоэнантиомер: 13 [обозначенный как (+)-атропоэнантиомер согласно его наблюдаемым данным вращения], выход: 137 мг, 43%. ЖХ-МС m/z 332,3 (М+Н). 1Н ЯМР (400 МГц, CD3OD) δ 9.03 (s, 1Н), 7.99 (d, J=5,8 Гц, 1Н), 7.89 (d, J=2,2 Гц, 1Н), 7.38 (br d, J=5,8 Гц, 1H), 7.24-7.27 (m, 1Н), 7.19 (br dd, половина ABX картины, J=8,3, 2,0 Гц, 1H), 7.14 (d, половина АВ квартет, J=8,2 Гц, 1Н), 6.91-6.94 (m, 1Н), 2.41 (s, 3Н), 2.14 (s, 3Н), 2.02 (s, 3Н). Элюирующийся вторым атропоэнантиомер: 14 [обозначенный как (-)-атропоэнантиомер согласно его наблюдаемым данным вращения], выход: 132 мг, 42%. ЖХ-МС m/z 332,3 (М+Н). 1Н ЯМР (400 МГц, CD3OD) δ 9.04 (s, 1Н), 7.99 (d, J=6,0 Гц, 1Н), 7.89 (d, J=2,2 Гц, 1Н), 7.38 (dd, J=6,0, 1,0 Гц, 1Н), 7.25-7.27 (m, 1Н), 7.19 (br dd, половина ABX картины, J=8,3, 2,2 Гц, 1Н), 7.15 (d, половина АВ квартет, J=8,2 Гц, 1Н), 6.93 (dd, J=2,2, 1,0 Гц, 1Н), 2.41 (s, 3Н), 2.15 (s, 3Н), 2.02 (s, 3Н).
Пример 15
4-[4-(1-трет-Бутил-4-метил-1Н-пиразол-5-ил)-3-метилфенокси]-фуро[3,2-с]пиридин (15)
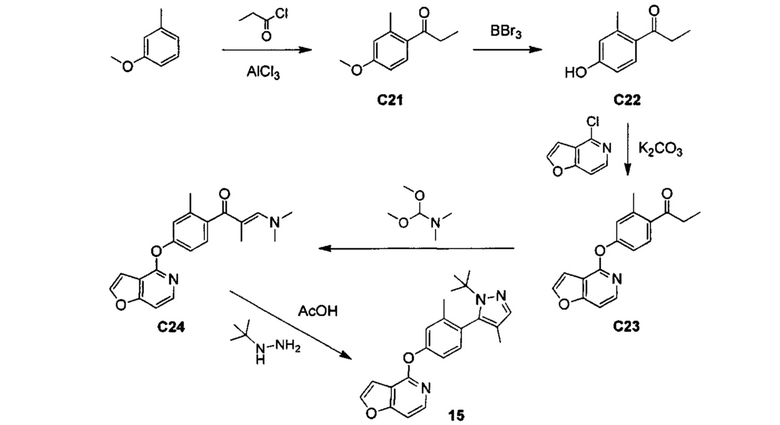
Стадия 1. Синтез 1-(4-метокси-2-метилфенил)пропан-1-она (С21)
К смеси 1-метокси-3-метилбензола (12,2 г, 100 ммоль) и пропаноилхлорида (18,5 г, 200 ммоль) в дихлорметане (200 мл) добавляли одной порцией хлорид алюминия (26,5 г, 199 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 4 часов. Реакционную смесь гасили водной соляной кислотой (1 н., 100 мл) и органический слой сушили над сульфатом магния, фильтровали и концентрировали в вакууме. Остаток очищали хроматографией на силикагеле с получением продукта в виде желтого твердого вещества. Выход: 3,87 г, 21,7 ммоль, 22%.
Стадия 2. Синтез 1-(4-гидрокси-2-метилфенил)пропан-1-она (С22)
Трибромид бора (5,57 г, 22,2 ммоль) добавляли к раствору 1-(4-метокси-2-метилфенил)пропан-1-она (С21) (3,87 г, 21,7 ммоль) в дихлорметане (50 мл) и реакционную смесь перемешивали при комнатной температуре в течение 4 часов. Добавляли воду (20 мл) и органический слой отделяли, сушили над сульфатом магния и концентрировали при пониженном давлении с получением продукта в виде желтого твердого вещества, которое использовали без дополнительной очистки. Выход: 3,77 г, более 100%.
Стадия 3. Синтез 1-[4-(фуро[3,2-с]пиридин-4-илокси)-2-метилфенил]-пропан-1-она (С23)
Смесь 1-(4-гидрокси-2-метилфенил)пропан-1-она (С22) (1,64 г, менее 10,0 ммоль), 4-хлорфуро[3,2-с]пиридина (1,53 г, 9,96 ммоль) и карбоната калия (2,76 г, 20,0 ммоль) в N,N-диметилформамиде (50 мл) кипятили с обратным холодильником в течение 8 часов. Реакционную смесь распределяли между водой (50 мл) и этилацетатом (150 мл); органический слой сушили над сульфатом магния и концентрировали в вакууме с получением продукта в виде желтого масла, которое использовали без дополнительной очистки. Выход: 2,97 г, более 100%.
Стадия 4. Синтез 3-(диметиламино)-1-[4-(фуро[3,2-с]пиридин-4-илокси)-2-метилфенил]-2-метилпроп-2-ен-1-она (С24)
1-[4-(Фуро[3,2-с]пиридин-4-илокси)-2-метилфенил]пропан-1-он (С23) (2,87 г, менее 10,7 ммоль) в смеси N,N-диметилформамид-диметилацеталя (10 мл) и N,N-диметилформамида (10 мл) кипятили с обратным холодильником в течение 30 минут. После удаления растворителя при пониженном давлении остаток промывали этилацетатом с получением продукта в виде желтого твердого вещества. Выход: 1,76 г, 5,23 ммоль, более 49%. ЖХ-МС m/z 337,1 (М+Н). 1Н ЯМР (400 МГц, CD3OD) δ 7.94 (d, J=6,1 Гц, 1Н), 7.87 (d, J=2,2 Гц, 1Н), 7.35 (dd, J=5,9, 1,0 Гц, 1H), 7.14 (d, J=8,2 Гц, 1H), 7.04-7.07 (m, 2Н), 7.00 (br dd, J=8,1, 2,4 Гц, 1H), 6.90 (dd, J=2,3, 1,0 Гц, 1H), 3.15 (s, 6H), 2.24 (s, 3Н), 2.14 (s, 3Н).
Стадия 5. Синтез 4-[4-(1-трет-бутил-4-метил-1H-пиразол-5-ил)-3-метилфенокси]фуро[3,2-с]пиридина (15)
Раствор 3-(диметиламино)-1-[4-(фуро[3,2-с]пиридин-4-илокси)-2-метилфенил]-2-метилпроп-2-ен-1-она (С24) в этаноле (0,125 М, 0,600 мл, 0,075 ммоль) объединяли с раствором трет-бутилгидразина в 0,2 М водной соляной кислоте (0,128 М, 0,700 мл, 0,090 ммоль). Добавляли уксусную кислоту (0,05 мл, 0,9 ммоль) и реакционную смесь встряхивали при 100°C в течение 3 часов. Растворители удаляли в вакууме и остаток очищали посредством HPLC (колонка: Phenomenex Gemini С18, 5 мкм; подвижная фаза А: водный гидроксид аммония, pH 10; подвижная фаза В: ацетонитрил; градиент: от 70% до 90% В) с получением продукта. ЖХ-МС m/z 362 (М+Н). Время удерживания: 3,056 мин (колонка: Welch ХВ-С18, 2,1×50 мм, 5 мкм; подвижная фаза А: 0,0375%-ная трифторуксусная кислота в воде; подвижная фаза В: 0,01875%-ная трифторуксусная кислота в ацетонитриле; градиент: от 25% В за 0,50 минут, от 25% до 100% В за 3,0 минут; скорость потока: 0,8 мл/минуты).
Пример 16
5-(Фуро[3,2-с]пиридин-4-илокси)-2-(имидазо[1,2-а]пиридин-5-ил)анилин (16)
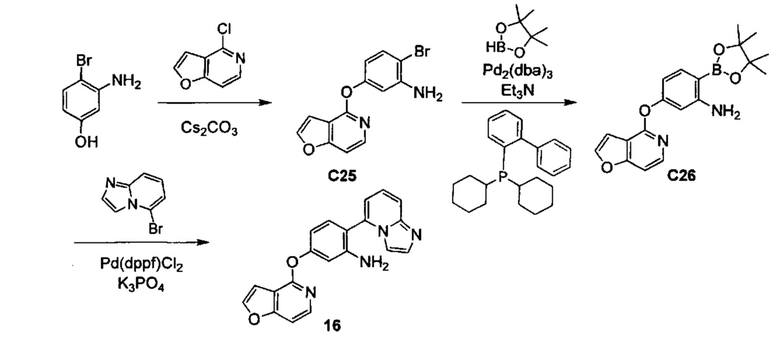
Стадия 1. Синтез 2-бром-5-(фуро[3,2-с]пиридин-4-илокси)анилина (С25)
Эту реакцию осуществляли в двух идентичных партиях. Смесь 3-амино-4-бромфенола (13 г, 69 ммоль), карбоната цезия (45 г, 140 ммоль) и 4 хлорфуро[3,2-с]пиридина (7,0 г, 46 ммоль) в диметилсульфоксиде (200 мл) нагревали до 130°C в течение 18 часов. Две партии охлаждали до комнатной температуры и объединяли и смесь вливали в ледяную воду (800 мл) и экстрагировали этилацетатом (5×1200 мл). Объединенные органические слои промывали насыщенным водным раствором хлорида натрия (500 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Очистка с использованием хроматографии на силикагеле (градиент: от 17% до 25% этилацетата в петролейном эфире) дала продукт в виде белого твердого вещества. Выход: 25 г, 82 ммоль, 89%.
Стадия 2. Синтез 5-(фуро[3,2-с]пиридин-4-илокси)-2-(4,4,5,5-тетраметил-1,3,2-диокса6оролан-2-ил)анилина (С26)
Эту реакцию осуществляли в двух идентичных партиях. К раствору 2-бром-5-(фуро[3,2-с]пиридин-4-илокси)анилина (С25) (10,9 г, 35,7 ммоль), трис(дибензилиденацетон)дипалладия(0) (3,3 г, 3,6 ммоль) и бифенил-2-ил(дициклогексил)фосфана (1,3 г, 3,7 ммоль) в толуоле (250 мл) добавляли триэтиламин (10,9 г, 108 ммоль) и 4,4,5,5-тетраметил-1,3,2-диоксаборолан (13,8 г, 108 ммоль) и реакционную смесь кипятили с обратным холодильником в течение 18 часов. Две партии охлаждали до комнатной температуры и объединяли, затем фильтровали и упаривали досуха. Остаток растворяли в метаноле, фильтровали и концентрировали в вакууме. Очистка посредством хроматографии на силикагеле (градиент: от 9% до 25% этилацетата в петролейном эфире) дала продукт в виде желтого твердого вещества. Выход: 13,5 г, 38,3 ммоль, 54%. 1Н ЯМР (400 МГц, DMSO-d6) δ 8.10 (d, J=2,0 Гц, 1Н), 8.00 (d, J=5,9 Гц, 1Н), 7.47 (dd, J=5,9, 0,8 Гц, 1Н), 7.40 (d, J=8,2 Гц, 1Н), 6.96 (dd, J=2,4, 0,8 Гц, 1Н), 6.36 (d, J=2,0 Гц, 1Н), 6.28 (dd, J=8,2, 2,4 Гц, 1Н), 5.65 (br s, 2Н), 1.29 (s, 12Н).
Стадия 3. Синтез 5-(фуро[3,2-с]пиридин-4-илокси)-2-(имидазо[1,2-а]пиридин-5-ил)анилина (16)
Эту реакцию осуществляли в двух идентичных партиях. Смесь 5-(фуро[3,2-с]пиридин-4-илокси)-2-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилина (С26) (4,5 г, 13 ммоль), тригидрата фосфата калия (9,6 г, 36 ммоль), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия(II) (1,1 г, 1,3 ммоль) и 5-бромимидазо[1,2-а]пиридина (3,8 г, 19 ммоль) в 2-метилтетрагидрофуране (50 мл) и воде (10 мл) нагревали до 75°C в течение 18 часов. Две партии охлаждали до комнатной температуры и объединяли. После фильтрования осадок на фильтре промывали водой и объединенные фильтраты экстрагировали этилацетатом (4×100 мл). Объединенные органические слои промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Остаток объединяли с осадком на фильтре и очищали хроматографией на силикагеле (градиент: от 2% до 5% метанола в дихлорметане) с получением продукта в виде желтого твердого вещества. Выход: 4,2 г, 12 ммоль, 46%. ЖХ-МС m/z 342,9 (М+Н). 1Н ЯМР (400 МГц, DMSO-d6) δ 8.14 (d, J=2,2 Гц, 1Н), 8.06 (d, J=5,9 Гц, 1Н), 7.60 (br d, J=9,0 Гц, 1H), 7.58 (d, J=1,0 Гц, 1H), 7.50 (dd, J=5,9, 0,8 Гц, 1H), 7.33 (dd, J=9,0, 6,8 Гц, 1H), 7.32 (br s, 1H), 7.19 (d, J=8,2 Гц, 1H), 7.07 (dd, J=2,2, 0,9 Гц, 1H), 6.89 (br dd, J=6,8, 0,7 Гц, 1H), 6.65 (d, J=2,4 Гц, 1H), 6.50 (dd, J=8,4, 2,4 Гц, 1H), 5.17 (br s, 2H).
Пример 17
N-[4-(Имидазо[1,2-a]пиридин-5-ил)-3-метилфенил]фуро[3,2-с]пиридин-4-амин (17)
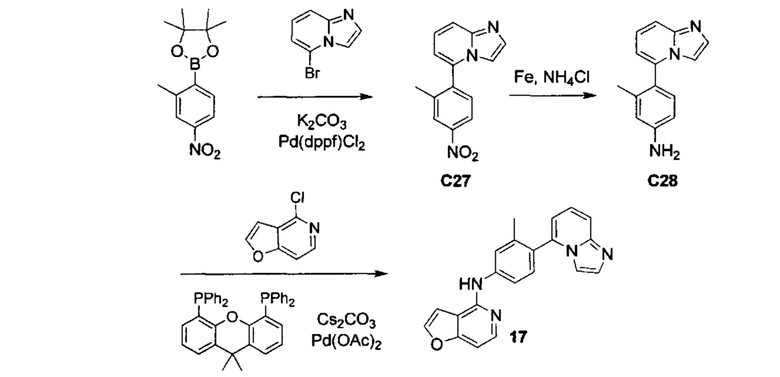
Стадия 1. Синтез 5-(2-метил-4-нитрофенил)имидазо[1,2-а]пиридина (027)
Смесь 4,4,5,5-тетраметил-2-(2-метил-4-нитрофенил)-1,3,2-диоксаборолана (390 мг, 1,48 ммоль), 5-бромимидазо[1,2-а]пиридина (243 мг, 1,23 ммоль), карбоната калия (683 мг, 4,94 ммоль) и [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия(II) (90 мг, 0,12 ммоль) в N,N-диметилформамиде (10 мл) перемешивали при 120°C в течение 1 часа. Реакционную смесь фильтровали и фильтрат концентрировали в вакууме. Очистка хроматографией на силикагеле (элюент: 2% метанола в дихлорметане) дала продукт в виде желтого масла. Выход: 320 мг, 1,26 ммоль, 100%. 1Н ЯМР (400 МГц, CDCl3) δ 8.27 (br s, 1Н), 8.22 (br d, J=8,5 Гц, 1H), 7.73 (d, J=9,0 Гц, 1H), 7.66 (br s, 1H), 7.56 (d, J=8,0 Гц, 1H), 7.31 (dd, J=9,0, 7,0 Гц, 1H), 7.05 (s, 1H), 6.75 (d, J=6,5 Гц, 1H),2.23 (s, 3Н).
Стадия 2. Синтез 4-(имидазо[1,2-а]пиридин-5-ил)-3-метиланилина (С28)
Смесь 5-(2-метил-4-нитрофенил)имидазо[1,2-а]пиридина (С27) (300 мг, 1.18 ммоль), железа (199 мг, 3,56 ммоль) и хлорида аммония (253 мг, 4,73 ммоль) в этаноле (9 мл) и воде (3 мл) кипятили с обратным холодильником в течение 1 часа. Смесь фильтровали и фильтрат концентрировали в вакууме; очистка хроматографией на силикагеле (элюент: 5% метанола в дихлорметане) дала продукт в виде твердого вещества. Выход: 224 мг, 1,00 ммоль, 85%. 1Н ЯМР (400 МГц, CDCl3) δ 7.72 (br d, J=9 Гц, 1Н), 7.61 (br s, 1Н), 7.29-7.36 (m, 1Н), 7.19 (brs, 1Н), 7.12 (d, J=8,3 Гц, 1H), 6.74 (br d, J=6,5 Гц, 1H), 6.67-6.69 (m, 1H), 6.64 (dd, J=8,2 Гц, 1H), 2.01 (s, 3Н).
Стадия 3. Синтез N-[4-(имидазо[1,2-а]пиридин-5-ил)-3-метилфенил]-фуро[3,2-с]пиридин-4-амина (17)
Смесь 4-(имидазо[1,2-а]пиридин-5-ил)-3-метиланилина (С28) (185 мг, 0,828 ммоль), 4-хлорфуро[3,2-с]пиридина (127 мг, 0,827 ммоль), карбоната цезия (810 мг, 2,49 ммоль), ацетата палладия(II) (28 мг, 0,12 ммоль) и 4,5-бис(дифенилфосфино)-9,9-диметилксантина (Xantphos, 72 мг, 0,12 ммоль) в 1,4-диоксане (8 мл) перемешивали при 120°C в течение 2 часов. После фильтрования реакционной смеси фильтрат разбавляли этилацетатом (100 мл), промывали насыщенным водным раствором хлорида натрия и концентрировали в вакууме. Остаток очищали препаративной тонкослойной хроматографии (элюент: 5% метанола в дихлорметане) с получением продукта в виде желтого твердого вещества. Выход: 157 мг, 0,461 ммоль, 56%. ЖХ-МС m/z 341,3 (М+Н). 1Н ЯМР (400 МГц, CDCl3) δ 8.16 (d, J=6,0 Гц, 1Н), 7.58-7.67 (m, 4Н), 7.29 (d, J=8,0 Гц, 1Н), 7.25-7.36 (br m, 1Н, предполагаемый; частично скрыт за пиком растворителя), 7.21 (br s, 1Н), 7.09 (br d, J=6 Гц, 1H), 6.92-7.03 (br m, 1H), 6.72-6.80 (br m, 2H), 2.11 (s, 3Н).
Пример 18
4-[4-(4-Хлор-6-метилпиримидин-5-ил)-3-метилфенокси]фуро[3,2-с]пиридин (18)
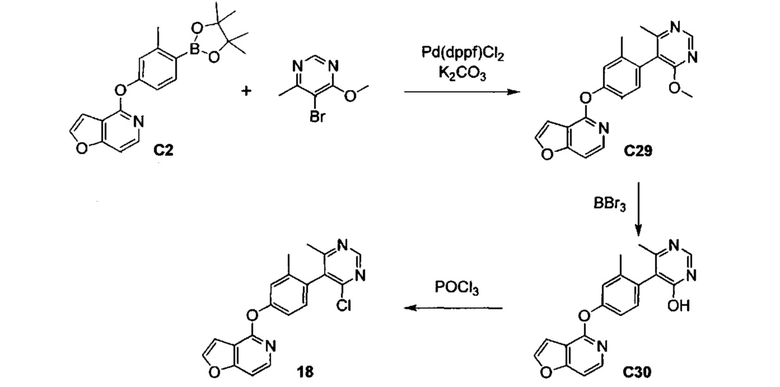
Стадия 1. Синтез 4-[4-(4-метокси-6-метилпиримидин-5-ил)-3-метилфенокси]фуро[3,2-с]пиридина (С29)
Смесь 4-[3-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенокси]фуро[3,2-с]пиридина (С2) (4,0 г, 11 ммоль), 5-бром-4-метокси-6-метилпиримидина (Z. Wang et al., Synthesis 2011, 1529-1531) (2,0 г, 10 ммоль), {1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия(II) (1,1 г, 1,4 ммоль) и карбоната калия (4,0 г, 29 ммоль) в 1,4-диоксане (30 мл), содержащем 5 капель воды, нагревали при 120°C в течение 2 часов. После фильтрования и концентрирования фильтрата при пониженном давлении остаток очищали хроматографией на силикагеле (элюент: 33% этилацетата в петролейном эфире) с получением продукта в виде желтого твердого вещества. Выход: 1,8 г, 5,2 ммоль, 52%. 1H (400 МГц, CDCl3) δ 8.72 (s, 1Н), 8.07 (d, J=6,0 Гц, 1Н), 7.66 (d, J=2,3 Гц, 1Н), 7.25 (dd, J=5,9, 0,9 Гц, 1Н), 7.19-7.21 (m, 1Н), 7.09-7.16 (m, 2Н), 6.88 (dd, J=2,3, 0,8 Гц, 1Н), 3.95 (s, 3Н), 2.29 (s, 3Н), 2.07 (s, 3Н).
Стадия 2. Синтез 5-[4-(фуро[3,2-с]пиридин-4-илокси)-2-метилфенил]-6-метилпиримидин-4-ола (C30)
Трибромид бора (20 г, 80 ммоль) медленно добавляли к раствору 4-[4-(4-метокси-6-метилпиримидин-5-ил)-3-метилфенокси]фуро[3,2-с]пиридина (С29) (1,8 г, 5,2 ммоль) в дихлорметане (150 мл) при -60°C. Реакционную смесь оставляли нагреваться до комнатной температуры и перемешивали в течение 18 часов. Затем добавляли метанол (150 мл) и pH доводили до 6 добавлением твердого бикарбоната натрия. Смесь фильтровали и фильтрат концентрировали в вакууме. Этот остаток смешивали с ацетоном и фильтровали еще раз; концентрирование фильтрата дало продукт в виде желтого твердого вещества. Выход: 1,5 г, 4,5 ммоль, 87%.
Стадия 3. Синтез 4-[4-(4-хлор-6-метилпиримидин-5-ил)-3-метилфенокси]фуро[3,2-с]пиридина (18)
Смесь 5-[4-(фуро[3,2-с]пиридин-4-илокси)-2-метилфенил]-6-метилпиримидин-4-ола (C30) (1,5 г, 4,5 ммоль) и оксихлорида фосфора (100 г, 65 ммоль) кипятили с обратным холодильником в течение 2 часов. После концентрирования при пониженном давлении остаток медленно обрабатывали насыщенным водным раствором бикарбоната натрия (200 мл). Полученную смесь экстрагировали этилацетатом (4×100 мл) и объединенные органические слои сушили, фильтровали и концентрировали в вакууме. Очистка хроматографией на силикагеле (элюент: 50% этилацетата в петролейном эфире) дала продукт в виде желтого твердого вещества. Выход: 750 мг, 2,13 ммоль, 47%. ЖХ-МС т/z 352,1 (М+Н). 1Н ЯМР (400 МГц, CD3OD) δ 8.86 (s, 1Н), 7.99 (br d, J=5,9 Гц, 1H), 7.88 (d, J=2,3 Гц, 1H), 7.38 (dd, J=5,9, 0,9 Гц, 1H), 7.22-7.25 (m, 1H), 7.20 (d, половина AB квартета, J=8,2 Гц, 1H), 7.16 (br dd, половина ABX картины, J=8,3, 2,2 Гц, 1H), 6.88 (dd, J=2,3, 1,0 Гц, 1H), 2.35 (s, 3Н), 2.08 (br s, 3Н).
Пример 19
5-[4-(Фуро[3,2-с]пиридин-4-илокси)-2-метилфенил]-6-метилимидазо[1,2-а]пиразин-8-ол (19)
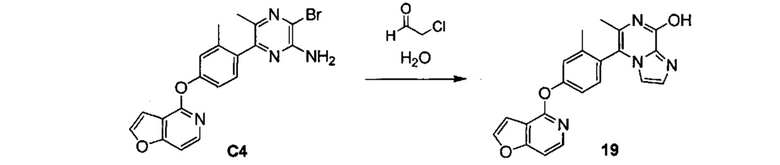
К смеси 3-бром-6-[4-(фуро[3,2-с]пиридин-4-илокси)-2-метилфенил]-5-метилпиразин-2-амина (С4) (1,5 г, 3,6 ммоль) в воде (30 мл) добавляли хлорацетальдегид (0,57 г, 7,3 ммоль) и реакционную смесь кипятили с обратным холодильником в течение 18 часов. После подщелачивания до pH 8 твердым бикарбонатом натрия смесь концентрировали в вакууме. Очистка хроматографией на силикагеле (градиент: от 2% до 5% метанола в дихлорметане) дала продукт в виде желтого твердого вещества. Выход: 255 мг, 0,685 ммоль, 19%. ЖХ-МС m/z 372,8 (М+Н). 1Н ЯМР (400 МГц, CD3OD) δ 7.98 (d, J=5,8 Гц, 1Н), 7.93 (d, J=2,3 Гц, 1Н), 7.46-7.48 (m, 1Н), 7.43 (d, J=8,3 Гц, 1Н), 7.40 (br d, J=5,8 Гц, 1H), 7.31 (d, J=2,3 Гц, 1H), 7.22 (dd, J=8,3, 2,5 Гц, 1H), 7.17-7.18 (m, 1Н), 7.01-7.03 (m, 1Н), 2.16 (s, 3Н), 2.07 (s, 3Н).
Пример 20
[2-(4,6-Диметилпиримидин-5-ил)-5-(фуро[3,2-с]пиридин-4-илокси)фенил]-метанол (20)
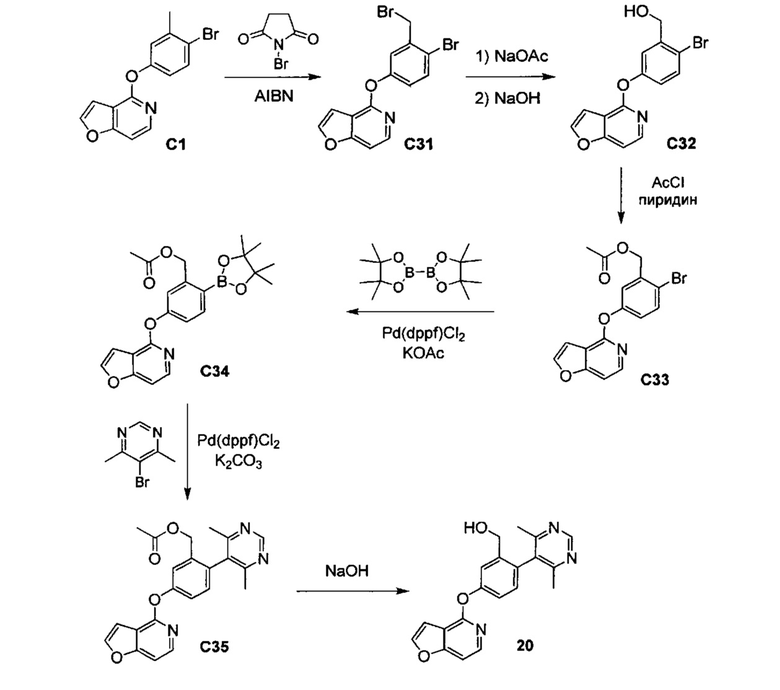
Стадия 1. Синтез 4-[4-бром-3-(бромметил)фенокси]фуро[3,2-с]пиридина (С31)
К раствору 4-(4-бром-3-метилфенокси)фуро[3,2-с]пиридина (С1) (4,00 г, 13,2 ммоль) в тетрахлориде углерода (80 мл) добавляли N-бромсукцинимид (2,34 г, 13,2 ммоль) и 2,2'-азобисизобутиронитрил (AIBN, 108 мг, 0,658 ммоль) при комнатной температуре. Реакционную смесь кипятили с обратным холодильником в течение 3 часов, охлаждали до комнатной температуры и обрабатывали водой (150 мл). Смесь экстрагировали дихлорметаном (3×50 мл) и объединенные органические слои сушили над сульфатом натрия и концентрировали в вакууме с получением неочищенного продукта. Выход: 5,04 г, 13,2 ммоль, 100%. ЖХ-МС m/z 383,7 (М+Н).
Стадия 2. Синтез [2-бром-5-(фуро[3,2-с]пиридин-4-илокси)фенил]-метанола (С32)
К раствору 4-[4-бром-3-(бромметил)фенокси]фуро[3,2-с]пиридина (С31) (5,04 г, 13,2 ммоль) в N,N-диметилформамиде (60 мл) добавляли ацетат натрия (5,40 г, 65,8 ммоль) при комнатной температуре. Реакционную смесь нагревали до 80°C в течение 3 часов, затем охлаждали и распределяли между водой (150 мл) и дихлорметаном (200 мл). Водный слой отделяли и экстрагировали дихлорметаном (3×50 мл). Объединенные органические слои сушили над сульфатом натрия и концентрировали в вакууме; полученный остаток растворяли в метаноле (40 мл) и обрабатывали водным раствором гидроксида натрия (1 н., 13,1 мл, 13,1 ммоль). После перемешивания в течение 1 часа при комнатной температуре реакционную смесь распределяли между водой (100 мл) и дихлорметаном (100 мл). Водный слой отделяли и экстрагировали дихлорметаном (3×50 мл). Объединенные органические слои сушили над сульфатом натрия и концентрировали при пониженном давлении с получением неочищенного продукта. Выход: 4,2 г, 13,1 ммоль, 99%. ЖХ-МС m/z 321,7 (М+Н).
Стадия 3. Синтез 2-бром-5-(фуро[3,2-с]пиридин-4-илокси)-бензилацетата (С33)
[2-Бром-5-(фуро[3,2-с]пиридин-4-илокси)фенил]метанол (С32) (230 мг, 0,718 ммоль), пиридин (170 мг, 2,15 ммоль) и ацетилхлорид (113 мг, 1,44 ммоль) объединяли в тетрагидрофуране (5 мл) при комнатной температуре. Реакционную смесь подвергали микроволновому излучению при 60°C в течение 40 минут, затем вливали в насыщенный водный раствор бикарбоната натрия (30 мл). После экстрагирования дихлорметаном (3×20 мл) объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали в вакууме с получением продукта. Выход: 260 мг, 0,718 ммоль, 100%. 1Н ЯМР (400 МГц, CDCl3) δ 8.00 (d, J=5,8 Гц, 1Н), 7.67 (d, J=2,0 Гц, 1H), 7.62 (d, J=8,5 Гц, 1H), 7.32 (d, J=2,5 Гц, 1H), 7.23 (d, J=6,0 Гц, 1H), 7.10 (dd, J=8,6, 2,6 Гц, 1H), 6.90-6.93 (m, 1Н), 5.20 (s, 2H), 2.14 (s, 3Н).
Стадия 4. Синтез 5-(фуро[3,2-с]пиридин-4-илокси)-2-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензилацетата (С34)
К 2-бром-5-(фуро[3,2-с]пиридин-4-илокси)бензилацетату (С33) (260 мг, 0,718 ммоль) в 1,4-диоксане (6 мл) добавляли 4,4,4',4',5,5,5',5'-октаметил-2,2'-би-1,3,2-диоксаборолан (237 мг, 0,933 ммоль), ацетат калия (211 мг, 2,15 ммоль) и [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий(II) (157 мг, 0,215 ммоль) при комнатной температуре. Смесь нагревали до 80°C и перемешивали в течение 3 часов, затем охлаждали и фильтровали. Фильтрат концентрировали в вакууме и очищали хроматографией на силикагеле с получением продукта. Выход: 164 мг, 0,401 ммоль, 56%. 1Н ЯМР (400 МГц, CD3OD) δ 7.97 (d, J=6,0 Гц, 1Н), 7.85-7.89 (m, 2Н), 7.39 (d, J=6,0 Гц, 1Н), 7.20-7.23 (m, 1Н), 7.11-7.15 (m, 1Н), 6.82-6.84 (m, 1Н), 5.36 (s, 2Н), 2.1 (s, 3Н), 1.36 (s, 12Н).
Стадия 5. Синтез 2-(4,6-диметилпиримидин-5-ил)-5-(фуро[3,2-с]пиридин-4-илокси)бензилацетата (С35)
К раствору 5-(фуро[3,2-с]пиридин-4-илокси)-2-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензилацетата (С34) (82 мг, 0,20 ммоль) в 1,4-диоксане (10 мл) добавляли 5-бром-4,6-диметилпиримидин (41 мг, 0,22 ммоль), карбонат калия (83 мг, 0,6 ммоль), [1,1'-бис(дифенилфосфино)ферроцен]-дихлорпалладий(II) (44 мг, 0,060 ммоль) и воду (5 капель) при комнатной температуре. Реакционную смесь дегазировали азотом в течение 5 минут, затем подвергали микроволновому излучению при 120°C в течение 50 минут. После фильтрования реакционной смеси фильтрат концентрировали в вакууме; очистку осуществляли препаративной тонкослойной хроматографией с получением продукта. Выход: 28 мг, 0,072 ммоль, 36%. ЖХ-МС m/z 389,9 (М+Н).
Стадия 6. Синтез [2-(4,6-диметилпиримидин-5-ил)-5-(фуро[3,2-с]пиридин-4-илокси)фенил]метанола (20)
Водный раствор гидроксида натрия (1 н., 0,36 мл, 0,36 ммоль) добавляли к раствору 2-(4,6-диметилпиримидин-5-ил)-5-(фуро[3,2-с]пиридин-4-илокси)-бензилацетата (С35) (28 мг, 0,072 ммоль) в тетрагидрофуране (2 мл) и реакционную смесь перемешивали при комнатной температуре в течение 18 часов. Добавляли насыщенный водный раствор хлорида натрия и смесь экстрагировали тетрагидрофураном (3×10 мл). Объединенные органические слои концентрировали в вакууме и очищали препаративной тонкослойной хроматографией на силикагеле с получением продукта. Выход: 19 мг, 0,055 ммоль, 76%. ЖХ-МС m/z 347,9 (М+Н). 1Н ЯМР (400 МГц, CDCl3), характеристические пики: δ 8.96 (s, 1Н), 8.03 (d, J=5,5 Гц, 1Н), 7.67 (br s, 1Н), 7.53 (br s, 1Н), 7.21-7.34 (m, 2Н, предполагаемый; частично скрыт за пиком растворителя), 7.10 (d, J=8,0 Гц, 1Н), 6.90 (br s, 1Н), 4.33 (s, 2Н), 2.26 (s, 6Н).
Пример 21
4-[4-(4,6-Диметилпиримидин-5-ил)-3-(фторметил)фенокси]фуро[3,2-с]пиридин (21)
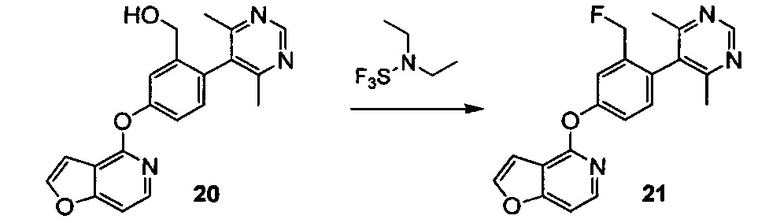
Трифторид (диэтиламино)серы (37 мг, 0,23 ммоль) добавляли к раствору [2-(4,6-диметилпиримидин-5-ил)-5-(фуро[3,2-с]пиридин-4-илокси)фенил]-метанола (20) (20 мг, 0,058 ммоль) в дихлорметане (2 мл) при 0°C. Реакционную смесь перемешивали в течение 30 минут при 40°C, затем концентрировали в вакууме. Очистка препаративной тонкослойной хроматографией на силикагеле дала продукт. Выход: 10 мг, 0,029 ммоль, 50%. ЖХ-МС m/z 350,0 (М+Н). 1Н ЯМР (400 МГц, CDCl3) δ 9.01 (s, 1Н), 8.07 (d, J=5,8 Гц, 1Н), 7.69 (d, J=2,3 Гц, 1Н), 7.49-7.52 (m, 1Н), 7.39-7.43 (m, 1Н), 7.29 (dd, J=5,9, 0,6 Гц, 1Н), 7.18 (br d, J=8,0 Гц, 1H), 6.94 (dd, J=2,0, 0,7 Гц, 1H), 5.04 (d, Jhf=47,4 Гц, 2H), 2.28 (s, 6H).
Пример 22
4-[4-(4,6-Диметилпиримидин-5-ил)-3-метилфенокси]-3-метилфуро[3,2-c]пиридин (22)
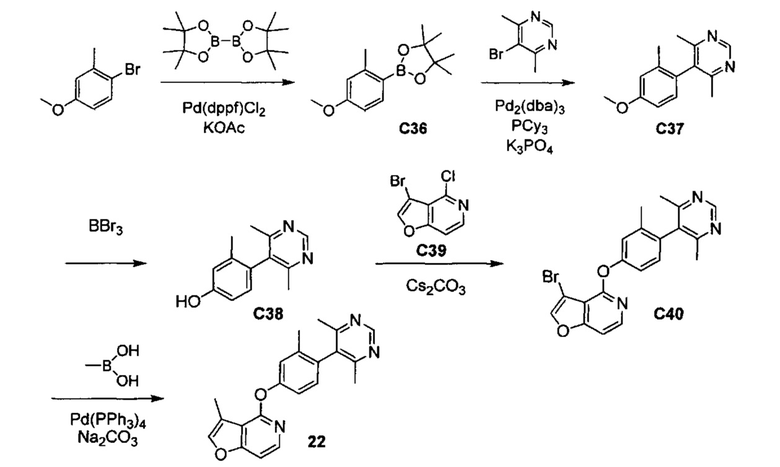
Стадия 1. Синтез 2-(4-метокси-2-метилфенил)-4,4,5,5-тетраметил-1,3,2-диоксаборолана (С36)
Соединение С36 получали из 1-бром-4-метокси-2-метилбензола согласно общей методике синтеза 4-[3-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенокси]фуро[3,2-с]пиридина (С2) в Примере 1. Продукт получили в виде твердого вещества. Выход: 15 г, 60 ммоль, 80%.
Стадия 2. Синтез 5-(4-метокси-2-метилфенил)-4,6-диметилпиримидина (С37)
Продукт получали из 2-(4-метокси-2-метилфенил)-4,4,5,5-тетраметил-1,3,2-диоксаборолана (С36) и 5-бром-4,6-диметилпиримидина согласно общей методике, описанной на стадии 3 Примера 1. Продукт получили в виде твердого вещества. Выход: 3,5 г, 15 ммоль, 75%.
Стадия 3. Синтез 4-(4,6-диметилпиримидин-5-ил)-3-метилфенола (С38)
Трибромид бора (3,8 мл, 40 ммоль) добавляли по каплям к раствору 5-(4-метокси-2-метилфенил)-4,6-диметилпиримидина (С37) (3,0 г, 13 ммоль) в дихлорметане (150 мл) при -70°C. Реакционную смесь перемешивали при комнатной температуре в течение 16 часов, затем pH доводили до 8 насыщенным водным раствором бикарбоната натрия. Водный слой экстрагировали дихлорметаном (3×200 мл) и объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Хроматография на силикагеле (градиент: от 60% до 90% этилацетата в петролейном эфире) дала продукт в виде желтого твердого вещества. Выход: 1,2 г, 5,6 ммоль, 43%. ЖХ-МС m/z 215,0 (М+Н). 1Н ЯМР (400 МГц, CDCl3) δ 8.98 (s, 1Н), 6.89 (d, J=8,0 Гц, 1Н), 6.86 (d, J=2,3 Гц, 1Н), 6.80 (dd, J=8,3, 2,5 Гц, 1Н), 2.24 (s, 6Н), 1.96 (s, 3Н).
Стадия 4. Синтез 3-6ром-4-[4-(4,6-диметилпиримидин-5-ил)-3-метилфенокси]фуро[3,2-с]пиридина (С40)
3-Бром-4-хлорфуро[3,2-с]пиридин (С39, полученный согласно методике Y. Miyazaki et al., Bioorg. Med. Chem. Lett. 2007, 17, 250-254; 430 мг, 1,85 ммоль), 4-(4,6-диметилпиримидин-5-ил)-3-метилфенол (C38) (396 мг, 1,85 ммоль) и карбонат цезия (1,21 г, 3,71 ммоль) объединяли в диметилсульфоксиде (8,0 мл) и нагревали при 120°C в течение 3 часов. Реакционную смесь фильтровали через целит, набивку целита тщательно промывали этилацетатом и объединенные фильтраты промывали дважды смесью воды и насыщенного водного раствора хлорида натрия в соотношении 1:1, затем промывали дважды 1 н. водным раствором гидроксида натрия. Органический слой сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Хроматографическая очистка на силикагеле (градиент: от 50% до 90% этилацетата в гептане) дала продукт в виде белого твердого вещества. Выход: 404 мг, 0,985 ммоль, 53%. ЖХ-МС m/z 412,0 (М+Н). 1Н ЯМР (400 МГц, CDCl3) δ 8.98 (s, 1Н), 8.07 (d, J=5,9 Гц, 1Н), 7.69 (s, 1Н), 7.26-7.28 (m, 1Н, предполагаемый; частично скрыт за пиком растворителя), 7.25 (d, J=5,9 Гц, 1Н), 7.21-7.25 (m, 1Н), 7.09 (br d, J=8,2 Гц, 1Н), 2.28 (s, 6Н), 2.05 (br s, 3Н).
Стадия 5. Синтез 4-[4-(4,6-диметилпиримидин-5-ил)-3-метилфенокси]-3-метилфуро[3,2-с]пиридина (22)
3-Бром-4-[4-(4,6-диметилпиримидин-5-ил)-3-метилфенокси]фуро[3,2-c]пиридин (С40) (89,0 мг, 0,217 ммоль), метилбороновую кислоту (98%, 27 мг, 0,44 ммоль) и тетракис(трифенилфосфин)палладий(0) (15 мг, 0,013 ммоль) объединяли в смеси 1,4-диоксана (2,4 мл) и этанола (0,78 мл) и смесь деоксигенировали путем барботирования через нее азота. Добавляли водный раствор карбоната натрия (2 М, 0,34 мл, 0,68 ммоль) и реакционную смесь подвергали микроволновому излучению при 120°C в течение 2 часов. Поскольку исходные вещества еще обнаруживали на этот момент времени посредством ГХ-МС, добавляли дополнительные количества метилбороновой кислоты (2 эквивалента) и тетракис(трифенилфосфин)палладия(0) (0,06 эквивалентов), реакционную смесь снова продували азотом и затем подвергали воздействию микроволновых условий в течение дополнительных 12 часов при 120°C. Смесь фильтровали через фильтр 0,45 мкм, который затем промывали этилацетатом; объединенные фильтраты концентрировали в вакууме и очищали посредством HPLC (колонка: Phenomenex Lux Cellulose-2, 5 мкм; подвижная фаза А: гептан; подвижная фаза В: этанол; градиент: от 5% до 100% В). Продукт получили в виде желто-оранжевого твердого вещества. Выход: 10,1 мг, 0,0292 ммоль, 13%. ЖХ-МС m/z 345,9 (М+Н). 1Н ЯМР (500 МГц, CDCl3) δ 8.98 (s, 1Н), 8.01 (d, J=5,9 Гц, 1Н), 7.42-7.43 (m, 1Н), 7.23 (br d, J=2,1 Гц, 1Н), 7.18 (d, J=5,9 Гц, 1Н), 7.17-7.20 (m, 1Н), 7.08 (d, J=8,2 Гц, 1Н), 2.44 (d, J=1,3 Гц, 3Н), 2.28 (s, 6Н), 2.04 (s, 3Н).
Пример 23
4-{[4-(4,6-Диметилпиримидин-5-ил)-1Н-индол-7-ил]окси}фуро[3,2-c]пиридин (23)
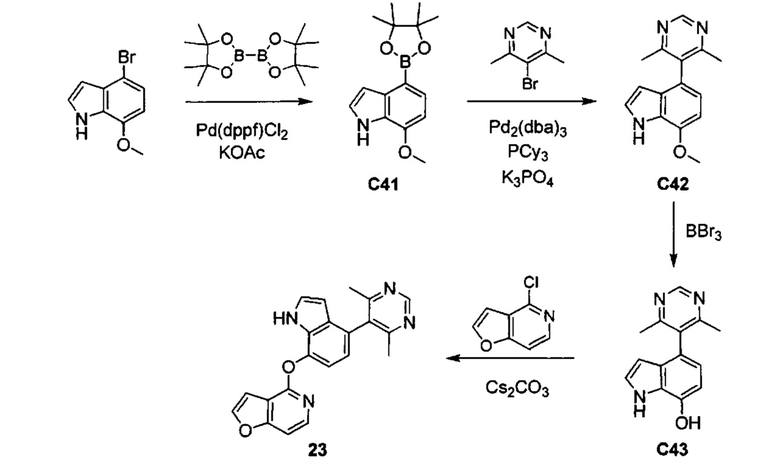
Стадия 1. Синтез 7-метокси-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-индола (С41)
Соединение С41 получали из 4-бром-7-метокси-1Н-индола согласно общей методике синтеза 4-[3-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенокси]фуро[3,2-с]пиридина (С2) в Примере 1, за исключением того, что используемый в реакции растворитель представлял собой 6% воды в 1,4-диоксане. Очистку в данном случае осуществляли посредством хроматографии на силикагеле (градиент: от 90% до 100% дихлорметана в гептане) с получением продукта в виде темно-желтого твердого вещества. Выход: 371 мг, 1,36 ммоль, 62%. GCMS m/z 273 (М+). 1Н ЯМР (400 МГц, CDCl3) δ 7.70 (d, J=8,0 Гц, 1Н), 7.55 (d, J=3,7 Гц, 1Н), 7.10 (d, J=3,5 Гц, 1Н), 6.81 (d, J=8,0 Гц, 1Н), 3.97 (s, 3Н), 1.37 (s, 12Н).
Стадия 2. Синтез 4-(4,6-диметилпиримидин-5-ил)-7-метокси-1H-индола (C42)
Соединение С42 получали из 7-метокси-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-индола (С41) согласно общей методике синтеза 4-[4-(4,6-диметилпиримидин-5-ил)-3-метилфенокси]фуро[3,2-с]пиридина (1) в Примере 1, с получением продукта в виде желтого масла. Выход: 70 мг, 0,28 ммоль, 24%. GCMS m/z 253 (М+). 1Н ЯМР (400 МГц, CDCl3) δ 8.99 (s, 1Н), 7.54 (d, J=3,7 Гц, 1Н), 6.94 (АВ квартет, Jab=8,1 Гц, Δνab=24,6 Гц, 2Н), 6.01 (d, J=3,7 Гц, 1Н), 4.02 (s, 3Н), 2.23 (s, 6Н).
Стадия 3. Синтез 4-(4,6-диметилпиримидин-5-ил)-1H-индол-7-ола (С43)
Соединение С43 получали из 4-(4,6-диметилпиримидин-5-ил)-7-метокси-1Н-индола (С42) согласно общей методике синтеза 3-метил-4-(2-метил-1Н-имидазо[4,5-с]пиридин-1-ил)фенола (С9) в Примере 5. Неочищенный продукт растирали с этилацетатом с получением горчично-желтого твердого вещества, содержащего некоторое количество примесей. Выход: 53 мг, менее 0,22 ммоль, менее 88%. ЖХ-МС m/z 240,1 (М+Н). 1Н ЯМР (400 МГц, CD3OD), только пики продукта: δ 9.29 (s, 1Н), 7.29 (d, J=3,1 Гц, 1Н), 6.75 (АВ квартет, Jab=7,8 Гц, Δνab=38,4 Гц, 2Н), 6.04 (d, J=3,1 Гц, 1Н), 2.49 (s, 6Н).
Стадия 4. Синтез 4-{[4-(4,6-диметилпиримидин-5-ил)-1H-индол-7-ил]окси}фуро[3,2-с]пиридина (23)
4-(4,6-диметилпиримидин-5-ил)-1H-индол-7-ол (С43) (50 мг, 0,21 ммоль), 4-хлорфуро[3,2-с]пиридин (32 мг, 0,21 ммоль) и карбонат цезия (136 мг, 0,417 ммоль) объединяли в диметилсульфоксиде (1 мл) и реакционную смесь нагревали до 120°C в течение 19 часов. После охлаждения до комнатной температуры смесь фильтровали через целит, набивку фильтра промывали этилацетатом и объединенные фильтраты промывали дважды смесью воды и насыщенного водного раствора хлорида натрия в соотношении 1:1, затем дважды промывали водным 1 н. раствором гидроксида натрия. Органический слой сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Очистка посредством хроматографии на силикагеле (градиент: от 50% до 100% этилацетата в гептане) дала продукт в виде не совсем белого твердого вещества. Выход: 3 мг, 0,008 ммоль, 4%. ЖХ-МС m/z 357,2 (М+Н). 1Н ЯМР (500 МГц, CDCl3) δ 9.01 (s, 1Н), 8.67 (br s, 1Н), 8.07 (d, J=5,9 Гц, 1Н), 7.68 (d, J=2,2 Гц, 1Н), 7.29 (br d, J=5,7 Гц, 1H), 7.22 (dd, J=2,9, 2,7 Гц, 1H), 7.17 (d, J=7,8 Гц, 1H), 6.92 (d, J=7,8 Гц, 1H), 6.86-6.87 (m, 1H), 6.12 (dd, J=2,9, 2,2 Гц, 1H), 2.31 (s, 6H).
Пример 24
4-[4-(4-Этокси-6-метилпиримидин-5-ил)-3-метилфенокси]фуро[3,2-с]пиридин (24)
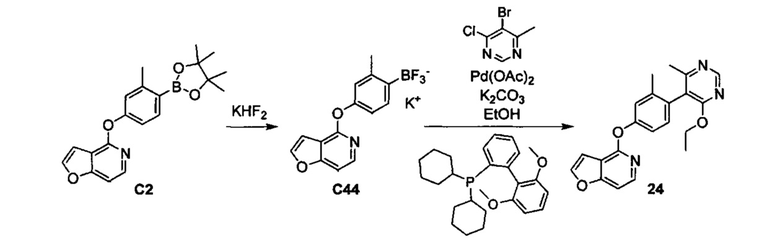
Стадия 1. Синтез трифтор[4-(фуро[3,2-с]пиридин-4-илокси)-2-метилфенил]бората калия (С44)
Раствор гидрофторида калия (124 мг, 1,59 ммоль) в воде (0,50 мл) добавляли к смеси 4-[3-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенокси]фуро[3,2-с]пиридина (С2) (186 мг, 0,530 ммоль) в метаноле (0,50 мл) и ацетоне (0,30 мл). Через 1 час объем реакционной смеси сокращали в вакууме и полученное твердое вещество выделяли посредством фильтрации и промывали небольшим количеством метанола. Продукт получили в виде белого твердого вещества. Выход: 110 мг, 0,332 ммоль, 63%. 1Н ЯМР (400 МГц, DMSO-d6) δ 8.13 (d, J=2,4 Гц, 1Н), 7.97 (d, J=5,9 Гц, 1Н), 7.68 (d, J=8,2 Гц, 1Н), 7.47 (dd, J=5,9, 1,0 Гц, 1H), 7.04 (dd, J=2,2, 1,0 Гц, 1H), 7.03 (br d, J=2,4 Гц, 1H), 6.98 (br dd, J=8,0, 2,4 Гц, 1H), 2.47 (s, 3H).
Стадия 2. Синтез 4-[4-(4-этокси-6-метилпиримидин-5-ил)-3-метилфенокси]фуро[3,2-с]пиридина (24)
5-Бром-4-хлор-6-метилпиримидин (65 мг, 0,31 ммоль), трифтор[4-(фуро[3,2-с]пиридин-4-илокси)-2-метилфенил]борат калия (С44) (110 мг, 0,332 ммоль), карбонат калия (130 мг, 0,941 ммоль), ацетат палладия(II) (0,40 мг, 0,0018 ммоль) и дициклогексил(2',6,-диметоксибифенил-2-ил)фосфан (1,20 мг, 0,0029 ммоль) растворяли в продутом азотом этаноле и реакционную смесь нагревали до 85°C в течение 66 часов. После охлаждения до комнатной температуры реакционную смесь разбавляли метанолом и этилацетатом, фильтровали через целит и концентрировали при пониженном давлении. Очистка хроматографией на силикагеле (градиент: от 0% до 70% этилацетата в гептане) дала продукт в виде бесцветного масла. Выход: 24 мг, 0,066 ммоль, 21%. ЖХ-МС т/z 362,4 (М+Н). 1Н ЯМР (400 МГц, CDCl3) δ 8.67 (s, 1Н), 8.06 (d, J=5,9 Гц, 1Н), 7.63 (d, J=2,0 Гц, 1Н), 7.23 (d, J=5,9 Гц, 1Н), 7.16-7.19 (m, 1Н), 7.13 (dd, половина АВХ картины, J=8,2, 2,0 Гц, 1Н), 7.09 (d, половина АВ картины, J=8,2 Гц, 1Н), 6.80-6.84 (m, 1Н), 4.32-4.52 (m, 2Н), 2.25 (s, 3Н), 2.06 (s, 3Н), 1.28 (t, J=7,0 Гц, 3Н).
Пример 25 и Пример 26
(+)-5-[4-(Фуро[3,2-с]пиридин-4-илокси)-2-метилфенил]-6-метилимидазо[1,2-а]пиразин (25) и (-)-5-[4-(Фуро[3,2-с]пиридин-4-илокси)-2-метилфенил]-6-метилимидазо[1,2-а]пиразин (26)
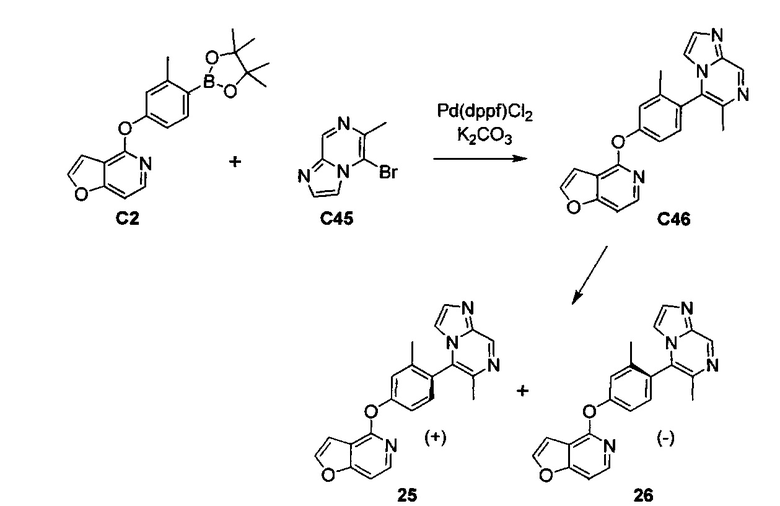
Стадия 1. Синтез 5-[4-(фуро[3,2-с]пиридин-4-илокси)-2-метилфенил]-6-метилимидазо[1,2-а]пиразина (С46)
К раствору 4-[3-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенокси]фуро[3,2-с]пиридина (С2) (13,5 г, 38,4 ммоль) в 1,4-диоксане (200 мл) и воде (10 мл) добавляли 5-бром-6-метилимидазо[1,2-а]пиразин (С45, см. А. R. Harris et al., Tetrahedron 2011, 67, 9063-9066) (8,15 г, 38,4 ммоль), карбонат калия (15,9 г, 115 ммоль) и [1,1'-бис(дифенилфосфино)ферроцен]-дихлорпалладий(II) (2,8 г, 3,8 ммоль) при комнатной температуре. Реакционную смесь дегазировали азотом в течение 5 минут, затем перемешивали в течение 10 часов при дефлегмации. Смесь охлаждали до комнатной температуры и фильтровали; фильтрат концентрировали в вакууме и очищали хроматографией на силикагеле (градиент: от 0% до 50% этилацетата в петролейном эфире) с получением продукта в виде желтого твердого вещества. Выход: 12,4 г, 34,8 ммоль, 91%. ЖХ-МС m/z 357,0 (М+Н). 1Н ЯМР (400 МГц, CD3OD) δ 9.02 (s, 1Н), 8.00 (d, J=6,0 Гц, 1Н), 7.93 (d, J=2,0 Гц, 1Н), 7.79-7.80 (m, 1Н), 7.48-7.51 (m, 1Н), 7.44 (d, J=8,5 Гц, 1Н), 7.41 (dd, 7=6,0, 1,0 Гц, 1Н), 7.36 (br d, J=2,0 Гц, 1Н), 7.28 (br dd, J=8,2 Гц, 1H), 7.02-7.05 (m, 1Н), 2.38 (s, 3Н), 2.07 (s, 3Н).
Стадия 2. Синтез (+)-5-[4-(фуро[3,2-с]пиридин-4-илокси)-2-метилфенил]-6-метилимидазо[1,2-а]пиразина (25) и (-)-5-[4-(фуро[3,2-с]пиридин-4-илокси)-2-метилфенил]-6-метилимидазо[1,2-а]пиразина (26)
5-[4-(Фуро[3,2-с]пиридин-4-илокси)-2-метилфенил]-6-метилимидазо[1,2-а]пиразин (С46) разделяли на его атропоэнантиомеры с использованием сверхкритической флюидной хроматографии (колонка: Chiralpak AD-H, 5 мкм; элюент: смесь диоксид углерода/метанол в соотношении 3:1). Соединение Примера 25 [обозначенное как (+)-атропоэнантиомер согласно его наблюдаемым данным по вращению] было первым элюирующимся изомером, а затем соединение Примера 26. Соединение Примера 26 [обозначенное как (-)-атропоэнантиомер согласно его наблюдаемым данным по вращению] проверяли с помощью спектроскопии вибрационного кругового дихроизма (VCD) [Chiral/R™ VCD спектрометр (BioTools, Inc.)], и на основе этой работы соединение Примера 26 было обозначено как (R).
Пример 25: ЖХ-МС m/z 357,1 (М+Н). 1Н ЯМР (400 МГц, CDCl3) δ 9.10 (s, 1Н), 8.08 (d, J=5,8 Гц, 1Н), 7.73 (d, J=1,0 Гц, 1Н), 7.70 (d, J=2,2 Гц, 1Н), 7.31-7.34 (m, 2Н), 7.26-7.30 (m, 2Н, предполагаемый; частично скрыт за пиком растворителя), 7.16-7.18 (m, 1Н), 6.95 (dd, J=2,2, 1,0 Гц, 1Н), 2.38 (s, 3Н), 2.07 (br s, 3Н).
Пример 26: ЖХ-МС m/z 357,1 (М+Н). 1Н ЯМР (400 МГц, CDCl3) δ 9.10 (s, 1Н), 8.09 (d, J=5,8 Гц, 1Н), 7.73 (d, J=1,0 Гц, 1Н), 7.70 (d, J=2,3 Гц, 1Н), 7.31-7.35 (m, 2Н), 7.26-7.31 (m, 2Н, предполагаемый; частично скрыт за пиком растворителя), 7.16-7.18 (m, 1Н), 6.95 (dd, J=2,2, 0,9 Гц, 1Н), 2.38 (s, 3Н), 2.07 (br s, 3Н).
Пример 27
5-[2-Фтор-4-(фуро[3,2-с]пиридин-4-илокси)фенил]-4,6-диметилпиридазин-3(2H)-он (27)
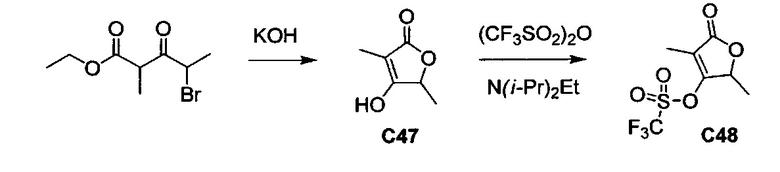
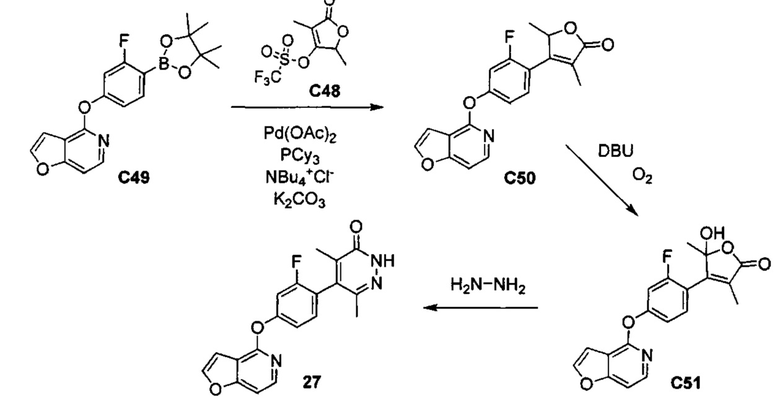
Стадия 1. Синтез 4-гидрокси-3,5-диметилфуран-2(5H)-она (С47)
Метилирование этил-3-оксопентаноата (согласно методике D. Kalaitzakis et al., Tetrahedron: Asymmetry 2007, 18, 2418-2426) дало этил-2-метил-3-оксопентаноат; последующая обработка одним эквивалентом брома в хлороформе дала этил-4-бром-2-метил-3-оксопентаноат. Этот неочищенный материал (139 г, 586 ммоль) медленно добавляли при 0°C к раствору гидроксида калия (98,7 г, 1,76 моль) в воде (700 мл); внутренняя температура реакции поднялась до 30°C при добавлении. Реакционную смесь подвергали интенсивному перемешиванию в течение 4 часов в ледяной бане, после чего ее подкисляли медленным добавлением концентрированной соляной кислоты. После экстрагирования этилацетатом водный слой насыщали твердым хлоридом натрия и экстрагировали три дополнительных раза этилацетатом. Объединенные органические слои промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении с получением смеси масла и твердого вещества (81,3 г). Этот материал суспендировали в хлороформе (200 мл); твердые вещества фильтровали, затем промывали хлороформом (2×50 мл). Объединенные фильтраты концентрировали в вакууме и обрабатывали смесью гептана и диэтилового эфира в соотношении 3:1 (300 мл). Смесь интенсивно взбалтывали, до тех пор пока некоторое количество масла не начало затвердевать, затем концентрировали при пониженном давлении с получением маслянистого твердого вещества (60,2 г). После добавления смеси гептана и диэтилового эфира в соотношении 3:1 (300 мл) и интенсивного перемешивания в течение 10 минут и фильтрования получили продукт в виде не совсем белого твердого вещества. Выход: 28,0 г, 219 ммоль, 37%. 1Н ЯМР (400 МГц, CDCl3) δ 4.84 (br q, J=6,8 Гц, 1Н), 1.74 (br s, 3Н), 1.50 (d, J=6,8 Гц, 3Н).
Стадия 2. Синтез 2,4-диметил-5-оксо-2,5-дигидрофуран-3-ил-трифторметансульфоната (С48)
Трифторметансульфоновый ангидрид (23,7 мл, 140 ммоль) добавляли порциями к раствору 4-гидрокси-3,5-диметилфуран-2(5H)-она (С47) (15,0 г, 117 ммоль) и N,N-диизопропилэтиламина (99%, 24,8 мл, 140 ммоль) в дихлорметане (500 мл) при -20°C с такой скоростью, чтобы поддерживать внутреннюю температуру реакции ниже -10°C. Реакционную смесь перемешивали при -20°C, затем оставляли нагреваться постепенно до 0°C в течение 5 часов. Реакционную смесь пропускали через набивку силикагеля, сушили над сульфатом магния и концентрировали в вакууме. Остаток суспендировали в диэтиловом эфире и фильтровали; фильтрат концентрировали при пониженном давлении. Очистка хроматографией на силикагеле (градиент: от 0% до 17% этилацетата в гептане) дала продукт в виде бледно-желтого масла. Выход: 21,06 г, 80,94 ммоль, 69%. 1Н ЯМР (400 МГц, CDCl3) δ 5.09-5.16 (m, 1Н), 1.94-1.96 (m, 3Н), 1.56 (d, J=6,6 Гц, 3Н).
Синтез 4-[3-фтор-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-фенокси]фуро[3,2-с]пиридина (С49)
Соединение С49 синтезировали, используя способ, описанный для 4-[3-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенокси]фуро[3,2-с]пиридина (С2) в Примере 1, за исключением того, что 4-бром-3-фторфенол использовали вместо 4-бром-3-метилфенола. Продукт получили в виде не совсем белого твердого вещества. Выход: 22,5 г, 63,3 ммоль, 39% за 2 стадии. ЖХ-МС m/z 356,1 (М+Н). 1Н ЯМР (400 МГц, CDCl3) δ 8.04 (d, J=5,9 Гц, 1Н), 7.80 (dd, J=8,2, 6,9 Гц, 1Н), 7.65 (d, J=2,3 Гц, 1Н), 7.25 (dd, J=5,8, 0,9 Гц, 1Н), 7.02 (dd, J=8,3, 2,1 Гц, 1Н), 6.94 (dd, J=10,2, 2,1 Гц, 1Н), 6.85 (dd, J=2,3, 1,0 Гц, 1Н), 1.37 (s, 12Н).
Стадия 3. Синтез 4-[2-фтор-4-(фуро[3,2-с]пиридин-4-илокси)фенил]-3,5-диметилфуран-2(5H)-она (С50)
Раствор 4-[3-фтор-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенокси]фуро[3,2-с]пиридина (С49) (3,20 г, 9,01 ммоль) и 2,4-диметил-5-оксо-2,5-дигидрофуран-3-ил-трифторметансульфоната (С48) (2,46 г, 9,45 ммоль) в 1,4-диоксане (80 мл) продували азотом в течение 5 минут. Добавляли смесь хлорида тетрабутиламмония (99%, 127 мг, 0,452 ммоль), трициклогексилфосфина (99%, 128 мг, 0,452 ммоль) и ацетата палладия(II) (101 мг, 0,450 ммоль), а затем водный раствор карбонат калия (3 М, 9,0 мл, 27,0 ммоль) и реакционную смесь нагревали при 50°C в течение 18 часов. После охлаждения до комнатной температуры реакционную смесь разбавляли этилацетатом, промывали три раза водой, промывали один раз насыщенным водным раствором хлорида натрия и сушили над сульфатом магния. Фильтрование и удаление растворителя при пониженном давлении, а затем хроматографическая очистка на силикагеле (градиент: от 15% до 50% этилацетата в гептане) дала продукт в виде желто-коричневого масла, которое медленно затвердевало при стоянии. Выход: 1,55 г, 4,57 ммоль, 51%. ЖХ-МС т/z 340,3 (М+Н). 1Н ЯМР (400 МГц, CDCl3) δ 8.06 (d, J=5,9 Гц, 1Н), 7.70 (d, J=2,2 Гц, 1Н), 7.33-7.38 (m, 1Н), 7.31 (dd, J=5,9, 1,0 Гц, 1Н), 7.13-7.20 (m, 2Н), 6.94 (dd, J=2,2, 0,9 Гц, 1Н), 5.43-5.51 (m, 1Н), 1.99-2.01 (m, 3Н), 1.38 (d, J=6,6 Гц, 3Н).
Стадия 4. Синтез 4-[2-фтор-4-(фуро[3,2-с]пиридин-4-илокси)фенил]-5-гидрокси-3,5-диметилфуран-2(5H)-она (С51)
Раствор 4-[2-фтор-4-(фуро[3,2-с]пиридин-4-илокси)фенил]-3,5-диметилфуран-2(5H)-она (С50) (5,0 г, 15 ммоль) в тетрагидрофуране (200 мл) и N,N-диметилформамиде (100 мл) обрабатывали 1,8-диазабицикло[5.4.0]ундец-7-еном (6,61 мл, 44,2 ммоль) и продували кислородом в течение 10 минут. Слабое положительное давление кислорода подавали в колбу и реакционную смесь нагревали при 50°C при интенсивном перемешивании в течение 5 часов. После нагревания отмечалось слабое дополнительное давление в колбе при проверке прокладки. ЖХ-МС анализ показал приблизительно 6% оставшегося исходного материала; колбу охлаждали до комнатной температуры, снова загружали кислородом и нагревали при 50°C в течение дополнительных 18 часов. Реакционную смесь охлаждали до комнатной температуры, разбавляли этилацетатом (300 мл) и промывали последовательно водной соляной кислотой (0,25 М, 175 мл) и водой (150 мл). Значение pH объединенных водных слоев доводили от pH 3 до рН примерно 4-5 и водный слой экстрагировали этилацетатом (300 мл). Объединенные органические слои промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом магния, фильтровали и концентрировали в вакууме. Очистка хроматографией на силикагеле (градиент: от 0% до 40% этилацетата в гептане) дала продукт в виде белой пены. Выход: 4,20 г, 11,8 ммоль, 79%. ЖХ-МС m/z 356,4 (М+Н). 1Н ЯМР (400 МГц, CDCl3) δ 8.07 (d, J=5,8 Гц, 1Н), 7.66-7.71 (m, 2Н), 7.31 (br d, J=5,8 Гц, 1H), 7.11-7.17 (m, 2Н), 6.93-6.94 (m, 1Н), 3.95 (br s, 1H), 1.86-1.88 (m, 3H), 1.64 (s, 3H).
Стадия 5. Синтез 5-[2-фтор-4-(фуро[3,2-с]пиридин-4-илокси)фенил]-4,6-диметилпиридазин-3(2H)-она (27)
Безводный гидразин (98,5%, 1,88 мл, 59,0 ммоль) добавляли к раствору 4-[2-фтор-4-(фуро[3,2-с]пиридин-4-илокси)фенил]-5-гидрокси-3,5-диметилфуран-2(5Н)-она (С51) (4,20 г, 11,8 ммоль) в 1-бутаноле (75 мл) и реакционную смесь нагревали при 110°C в течение 2 часов. После охлаждения до комнатной температуры и перемешивания при этой температуре в течение 18 часов реакционную смесь хранили в морозильнике в течение 66 часов. Полученную суспензию фильтровали с получением серого твердого вещества, которое растворяли в горячем этаноле (150-175 мл) и фильтровали через найлоновый шприц-фильтр. Фильтрат концентрировали в вакууме с получением продукта в виде белого твердого вещества. Выход: 1,30 г, 3,70 ммоль, 31%. ЖХ-МС m/z 352,2 (М+Н). 1Н ЯМР (400 МГц, DMSO-d6) δ 12.89 (br s, 1Н), 8.17 (d, J=2,2 Гц, 1H), 8.06 (d, J=5,8 Гц, 1H), 7.54 (br d, J=5,8 Гц, 1H), 7.38-7.46 (m, 2H), 7.25 (br dd, J=8,4, 2,2 Гц, 1H), 7.12-7.14 (m, 1H), 1.99 (s, 3H), 1.85 (s, 3H).
Пример 28
5-[4-(Фуро[3,2-с]пиридин-4-илокси)фенил]-4,6-диметилпиридазин-3(2Н)-он (28)
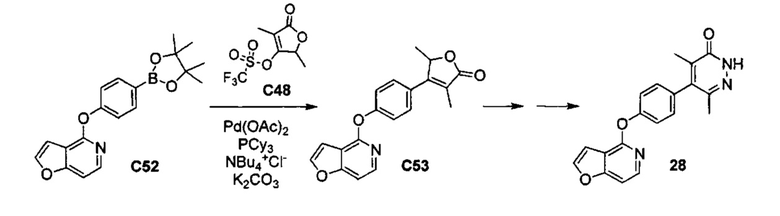
Стадия 1. Синтез 4-[4-(фуро[3,2-с]пиридин-4-илокси)фенил]-3,5-диметилфуран-2(5H)-она (С53)
Продукт получили в виде не совсем белого твердого вещества посредством взаимодействия 2,4-диметил-5-оксо-2,5-дигидрофуран-3-ил-трифторметансульфоната (С48) с 4-[4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенокси]фуро[3,2-с]пиридином (С52) [может быть получено аналогично 4-[3-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенокси]фуро[3,2-c]пиридину (С2) в Примере 1], как описано для синтеза 4-[2-фтор-4-(фуро[3,2-с]пиридин-4-илокси)фенил]-3,5-диметилфуран-2(5H)-она (С50) в Примере 27. Выход: 760 мг, 2,36 ммоль, 80%. ЖХ-МС m/z 322,2 (М+Н). 1Н ЯМР (400 МГц, CDCl3) δ 8.04 (d, J=5,9 Гц, 1Н), 7.69 (d, J=2,2 Гц, 1Н), 7.40 (br АВ квартет, JAB=8,8 Гц, Δνab=27,3 Гц, 4Н), 7.26-7.29 (m, 1Н, предполагаемый; частично скрыт за пиком растворителя), 6.93 (dd, J=2,2, 1,0 Гц, 1Н), 5.43 (qq, J=6J, 1,8 Гц, 1Н), 2.09 (d, J=1,8 Гц, 3Н), 1.43 (d, J=6,6 Гц, 3Н).
Стадия 2. Синтез 5-[4-(фуро[3,2-с]пиридин-4-илокси)фенил]-4,6-диметилпиридазин-3(2Н)-она (28)
4-[4-(Фуро[3,2-с]пиридин-4-илокси)фенил]-3,5-диметилфуран-2(5Н)-он (С53) превращали в продукт аналогично тому, как описано для синтеза 5-[2-фтор-4-(фуро[3,2-с]пиридин-4-илокси)фенил]-4,6-диметилпиридазин-3(2Н)-она (27) в Примере 27. Неочищенный продукт подвергали хроматографии на силикагеле (элюент: 40% этилацетата в дихлорметане), затем перекристаллизовывали из этанола с получением указанного в заголовке продукта в виде белого твердого вещества. Выход: 270 мг, 0,810 ммоль, 35% за 2 стадии. ЖХ-МС m/z 334,0 (М+Н). 1Н ЯМР (400 МГц, DMSO-d6) δ 12.79 (br s, 1Н), 8.15 (d, J=2,4 Гц, 1H), 8.03 (d, J=5,9 Гц, 1H), 7.50 (dd, J=5,9, 1,0 Гц, 1H), 7.31-7.38 (m, 4H), 7.09 (dd, J=2,2, 1,0 Гц, 1H), 1.97 (s, 3Н), 1.83 (s, 3Н).
Пример 29
4-[3,5-Диметил-4-(3-метилпиридин-4-ил)фенокси]фуро[3,2-с]пиридин (29)
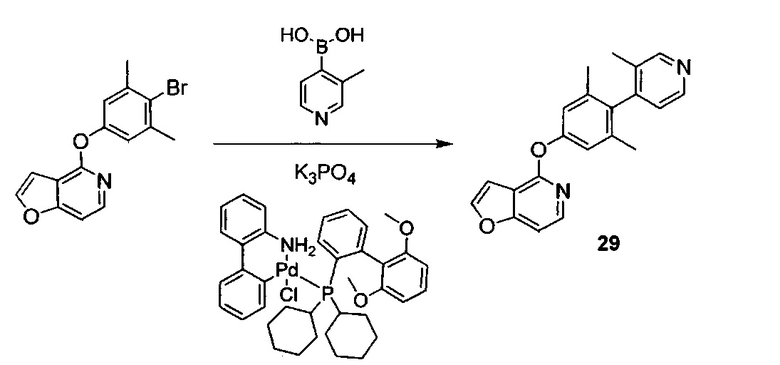
Продукт получали из 4-(4-бром-3,5-диметилфенокси)фуро[3,2-с]пиридина [синтезированного посредством взаимодействия 4-бром-3,5-диметилфенола с 4-хлорфуро[3,2-с]пиридином] и (3-метилпиридин-4-ил)бороновой кислоты, согласно общей методике для синтеза 5-(2-хлор-4-метоксифенил)-4,6-диметилпиримидина (С64) в Подготовительном примере Р7. ЖХ-МС m/z 331,1 (М+Н). 1Н ЯМР (600 МГц, DMSO-d6) δ 8.57 (br s, 1Н), 8.49 (br d, J=4,8 Гц, 1H), 8.13 (d, J=2,2 Гц, 1H), 8.02 (d, J=5,9 Гц, 1H), 7.47 (dd, J=5,8, 1,0 Гц, 1H), 7.10 (br d, J=4,8 Гц, 1H), 7.05 (dd, J=2,2, 0,9 Гц, 1H), 7.02-7.04 (m, 2H), 1.97 (s, 3H), 1.89 (s, 6H).
Пример 30
Трифторацетатная соль 4-{[4-(имидазо[1,2-а]пиридин-5-ил)нафталин-1-ил]окси}фуро[3,2-с]пиридина (30)
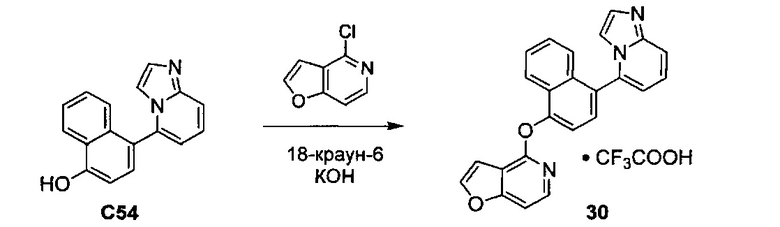
Гидроксид калия гидроксид (112 мг, 1,99 ммоль) и 1,4,7,10,13,16-гексаоксациклооктадекан (18-краун-6; 13,3 мг, 0,050 ммоль) добавляли к раствору 4-(имидазо[1,2-а]пиридин-5-ил)нафталин-1-ола (С54) [полученного реакцией Сузуки между (4-метоксинафталин-1-ил)бороновой кислотой и 5-бромимидазо[1,2-а]пиридином, как описано в Примере 8, с последующим отщеплением метилового эфира, опосредованным бором] (85 мг, 0,25 ммоль) и 4-хлорфуро[3,2-с]пиридина (57,3 мг, 0,373 ммоль) в ксилоле (3 мл) и реакционную смесь нагревали до 140°C в течение 24 часов. Растворитель удаляли в вакууме и неочищенный материал объединяли с неочищенным продуктом от подобной реакции, осуществленной на 30 мг С54. Затем реакционную смесь распределяли между этилацетатом (25 мл) и водой (25 мл), водный слой экстрагировали этилацетатом (3×30 мл) и объединенные органические слои сушили над сульфатом натрия. Очистку сначала осуществляли посредством хроматографии на силикагеле (элюент: этилацетат), а затем HPLC (колонка: XBridge С18, 5 мкм, подвижная фаза А: вода с трифторуксуснокислотным модификатором; подвижная фаза В: ацетонитрил с трифторуксуснокислотным модификатором; градиент: от 30% до 50% В). Продукт получили в виде бесцветной смолы. Выход: 20 мг, 0,041 ммоль, 12%. ЖХ-МС m/z 378,1 (М+Н). 1Н ЯМР (500 МГц, CD3OD) δ 8.17 (dd, половина АВХ картины, J=9,0, 7,1 Гц, 1Н), 8.15 (br d, J=8,0 Гц, 1Н), 8.10 (br d, половина АВ картины, J=9 Гц, 1Н), 7.99-8.01 (m, 2Н), 7.89 (d, J=5,9 Гц, 1Н), 7.83 (d, J=7,8 Гц, 1Н), 7.70 (br d, J=2 Гц, 1Н), 7.67 (dd, J=7,1, 1,0 Гц, 1Н), 7.61 (ddd, J=8,3, 6,8, 1,2 Гц, 1Н), 7.56 (ddd, J=8,3, 6,8, 1,2 Гц, 1Н), 7.54 (d, J=7,6 Гц, 1Н), 7.41-7.44 (m, 2Н), 7.20 (dd, J=2,2, 1,0 Гц, 1Н).
ПОДГОТОВИТЕЛЬНЫЕ ПРИМЕРЫ
Подготовительные примеры Р1-Р15 описывают получения некоторых исходных материалов или промежуточных соединений, используемых для получения некоторых соединений по изобретению.
Подготовительный пример Р1
5-(Фуро[3,2-с]пиридин-4-илокси)-2-(3-метилпиразин-2-ил)фенол (Р1)
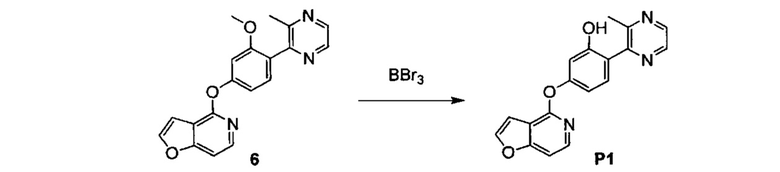
Трибромид бора (1,9 г, 7,6 ммоль) медленно добавляли к раствору 4-[3-метокси-4-(3-метилпиразин-2-ил)фенокси]фуро[3,2-с]пиридина (6) (2,3 г, 6,9 ммоль) в дихлорметане (100 мл) при 0°C. Реакционную смесь перемешивали при 0°C в течение 1 часа, затем гасили водой, перемешивали и фильтровали. Значение рН фильтрата доводили до нейтрального рН насыщенным водным раствором бикарбоната натрия и экстрагировали дихлорметаном (3×50 мл). Объединенные органические слои сушили, фильтровали и концентрировали в вакууме. Хроматография на силикагеле (градиент: от 0% до 2% метанола в дихлорметане) дала продукт. Выход: 1,2 г, 3,8 ммоль, 55%. ЖХ-МС m/z 320,1 (М+Н). 1Н ЯМР (400 МГц, CDCl3) δ 11.83 (s, 1Н), 8.48 (d, J=2,5 Гц, 1Н), 8.36 (d, J=2,5 Гц, 1Н), 8.08 (d, J=5,8 Гц, 1Н), 7.68 (d, J=8,5 Гц, 1Н), 7.66 (d, J=2,3 Гц, 1Н), 7.25-7.28 (m, 1Н, предполагаемый; частично скрыт за пиком растворителя), 6.95 (d, J=2,5 Гц, 1Н), 6.90 (dd, J=2,3, 1,0 Гц, 1Н), 6.86 (dd, J=8,8, 2,5 Гц, 1Н), 2.87 (s, 3Н).
Подготовительный пример Р2
Гидробромидная соль 4-(6-метилимидазо[1,2-а]пиридин-5-ил)фенола (Р2)

Стадия 1. Синтез 5-(4-метоксифенил)-6-метилимидазо[1,2-а]пиридина (С56)
Продукт получали из С55 (смесь 1:1 5-бром-6-метилимидазо[1,2-а]пиридина и 5-хлор-6-метилимидазо[1,2-а]пиридина, см. A. R. Harris et al., Tetrahedron 2011, 67, 9063-9066) (210 мг, 1,00 ммоль) и (4-метоксифенил)бороновой кислоты (116 мг, 0,765 ммоль), используя способ Примера 6. Хроматография на силикагеле (градиент: от 0% до 40% [20%-ный метанол в дихлорметане] в дихлорметане) дала продукт. Выход: 159 мг, 0,667 ммоль, 87%. 1Н ЯМР (500 МГц, CDCl3) δ 7.55 (d, J=9,3 Гц, 1Н), 7.50 (s, 1Н), 7.30 (d, J=8,5 Гц, 2Н), 7.14 (d, J=9,3 Гц, 1Н), 7.12 (s, 1Н), 7.07 (d, J=8,5 Гц, 2Н), 3.89 (s, 3Н), 2.13 (s, 3Н).
Стадия 2. Синтез гидробромидной соли 4-(6-метилимидазо[1,2-а]пиридин-5-ил)фенола (Р2)
Продукт получали из 5-(4-метоксифенил)-6-метилимидазо[1,2-а]пиридина (С56) (159 мг, 0,667 ммоль), как описано для синтеза 6-(4-гидрокси-2-метилфенил)-1,5-диметилпиразин-2(1H)-она (Р8) в Подготовительном примере Р8. В данном случае, после второго добавления метанола смесь концентрировали в вакууме, затем подвергали азеотропной перегонке с гептаном с получением продукта в виде коричневого твердого вещества. Выход: 193 мг, 0,63 ммоль, 95%. ЖХ-МС m/z 225,0 (М+Н). 1Н ЯМР (400 МГц, CD3OD) δ 7.97 (d, J=9,2 Гц, 1Н), 7.91 (d, J=2,2 Гц, 1Н), 7.83 (br d, J=9,4 Гц, 1Н), 7.54 (dd, J=2,2, 0,7 Гц, 1Н), 7.36 (br d, J=8,6 Гц, 2Н), 7.08 (br d, J=8,8 Гц, 2H), 2.31 (s, 3Н).
Подготовительный пример Р3
7-Хлор-6-метил[1,2,4]триазоло[1,5-а]пиримидин (Р3)
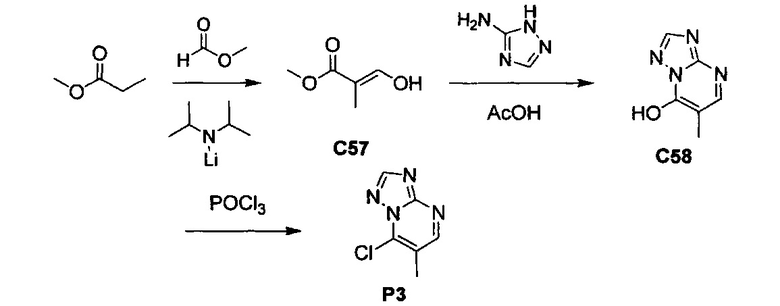
Стадия 1. Синтез метил-3-гидрокси-2-метилпроп-2-еноата (С57)
Метилпропаноат (44 г, 0,50 моль) подвергали взаимодействию с метилформиатом (55,5 г, 0,75 моль) согласно методике F. Kido et al., Tetrahedron 1987, 43, 5467-5474. Очистка дистилляцией (70-104°C) дала соединение С57 в виде бесцветной жидкости. Выход: 23 г, 0,20 моль, 40%. 1Н ЯМР (400 МГц, CDCl3), смесь альдегидной и енольной форм в соотношении приблизительно 1:1: δ 11.24 (d, J=11,5 Гц, 1Н), 9.78 (s, 1Н), 6.99 (d, J=10,5 Гц, 1Н), 3.79 (s, 6Н), 3.41 (q, J=7 Гц, 1Н), 1.68 (s, 3Н), 1.36 (d, J=7 Гц, 3Н).
Стадия 2. Синтез 6-метил[1,2,4]триазоло[1,5-а]пиримидин-7-ола (С58)
Раствор метил-3-гидрокси-2-метилпроп-2-еноата (С57) (95 г, 0,82 моль) и 1H-1,2,4-триазол-5-амина (100 г, 1,19 моль) в смеси этанола (300 мл) и уксусной кислоты (150 мл) нагревали до дефлегмации в течение 12 часов. Реакционную смесь оставляли охлаждаться до температуры окружающей среды и твердые вещества отфильтровывали с получением продукта в виде белого твердого вещества. Выход: 41 г, 27 ммоль, 33%. 1Н ЯМР (400 МГц, DMSO-d6) δ 8.18 (s, 1Н), 7.91 (s, 1Н), 2.00 (s, 3Н).
Стадия 3. Синтез 7-хлор-6-метил[1,2,4]триазоло[1,5-а]пиримидина (Р3)
К перемешиваемой суспензии 6-метил[1,2,4]триазоло[1,5-а]пиримидин-7-ола (С58) (105 г, 0,699 моль) в оксихлориде фосфора (500 мл) при комнатной температуре добавляли по каплям N,N-диизопропилэтиламин (100 мл) и реакционную смесь нагревали до дефлегмации в течение 110 минут. Затем смесь охлаждали до температуры окружающей среды, концентрировали ее почти до сухого состояния в вакууме, вливали в ледяную воду и доводили до рН 9 добавлением карбоната калия. Полученный раствор экстрагировали три раза дихлорметаном (800 мл) и объединенные органические фазы промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом натрия и концентрировали при пониженном давлении. Хроматография на силикагеле (градиент: от 17% до 33% этилацетата в петролейном эфире) дала продукт в виде белого твердого вещества. Выход: 55 г, 330 ммоль, 47%. ЖХ-МС m/z 169,2 (М+Н). 1Н ЯМР (400 МГц, CDCl3) δ 8.70 (s, 1Н), 8.52 (s, 1Н), 2.54 (s, 3Н).
Подготовительный пример Р4 3-Бром-2-метилимидазо[1,2-а]пиразин (Р4)
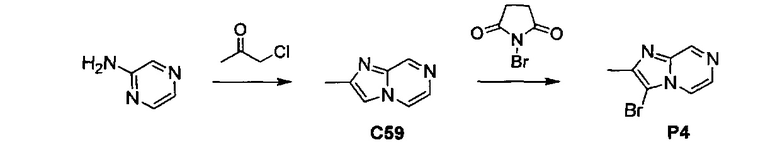
Стадия 1. Синтез 2-метилимидазо[1,2-а]пиразина (С59)
Пиразин-2-амин (1 г, 10 ммоль) растворяли в этаноле (15 мл) и добавляли 1-хлорпропан-2-он (1,2 мл, 14 ммоль). Полученный раствор перемешивали при дефлегмации в течение 2 часов, охлаждали до комнатной температуры и концентрировали в вакууме. Добавляли насыщенный водный раствор бикарбоната натрия (50 мл) и смесь экстрагировали три раза хлороформом (20 мл); объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали. Хроматография на силикагеле (градиент: от 0% до 50% метанола в этилацетате) дала С59 в виде оранжевого твердого вещества. Выход: 122 мг, 0,916 ммоль, 9%. ЖХ-МС m/z 133,9 (М+Н). 1Н ЯМР (400 МГц, CDCl3) δ 8.98 (br s, 1Н), 7.99 (dd, J=4,6, 1,5 Гц, 1H), 7.83 (br d, J=4,5 Гц, 1H), 7.46 (brs, 1Н), 2.53 (s, 3Н).
Стадия 2. Синтез 3-6ром-2-метилимидазо[1,2-а]пиразина (Р4)
2-Метилимидазо[1,2-а]пиразин (С59) (122 мг, 0,916 ммоль) растворяли в хлороформе (2 мл) и обрабатывали N-бромсукцинимидом (189 мг, 1,1 ммоль). Полученную смесь перемешивали при температуре окружающей среды в течение 1,5 часов и затем концентрировали в вакууме. Хроматография на силикагеле (градиент: от 33% до 100% этилацетата в гептане) дала продукт, все еще содержащий некоторое количество сукцинимида. Этот материал растворяли в дихлорметане (25 мл) и промывали водным раствором гидроксида натрия (0,5 М, 3×10 мл). Органический слой сушили над сульфатом натрия, фильтровали и концентрировали в вакууме с получением продукта в виде не совсем белого твердого вещества. Выход: 125 мг, 0,59 ммоль, 64%. ЖХ-МС m/z 213,9 (М+Н). 1Н ЯМР (400 МГц, CDCl3) δ 8.93 (s, 1Н), 7.96 (br s, 2Н), 2.51 (s, 3Н).
Подготовительный пример Р5
4-[4-(4,4,5,5- Тетраметил-1,3,2-диоксаборолан-2-ил)-3-(трифторметил)фенокси]фуро[3,2-с]пиридин (Р5)
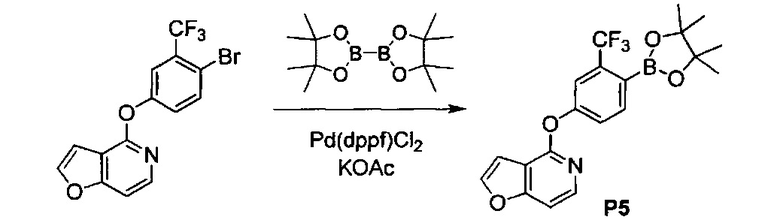
4-[4-Бром-3-(трифторметил)фенокси]фуро[3,2-с]пиридин (3,58 г, 10,0 ммоль) подвергали взаимодействию с 4,4,4',4',5,5,5',5'-октаметил-2,2'-би-1,3,2-диоксабороланом (99%, 3,33 г, 13,0 ммоль), ацетатом калия (95%, 4,13 г, 40,0 ммоль) и [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладием(II) (732 мг, 1,00 ммоль) аналогично синтезу 4-[3-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенокси]фуро[3,2-с]пиридина (С2) в Примере 1. Хроматография на силикагеле (градиент: от 0% до 20% этилацетата в гептане) дала продукт в виде белого твердого вещества. Выход: 2,035 г, 5,022 ммоль, 50%. ЖХ-МС m/z 406,2 (М+Н). 1Н ЯМР (500 МГц, CDCl3) δ 8.00 (d, J=5,9 Гц, 1Н), 7.84 (br d, J=8,0 Гц, 1Н), 7.66 (d, J=2,2 Гц, 1Н), 7.55 (br d, J=2,2 Гц, 1H), 7.39 (br dd, J=8,2, 2,3 Гц, 1H), 7.25 (dd, J=5,9, 1,0 Гц, 1H), 6.87 (dd, J=2,2, 1,0 Гц, 1H), 1.38 (s, 12H).
Подготовительный пример Р6
2,5-Диметил-4-(6-метилимидазо[1,2-а]пиразин-5-ил)фенол (Р6)
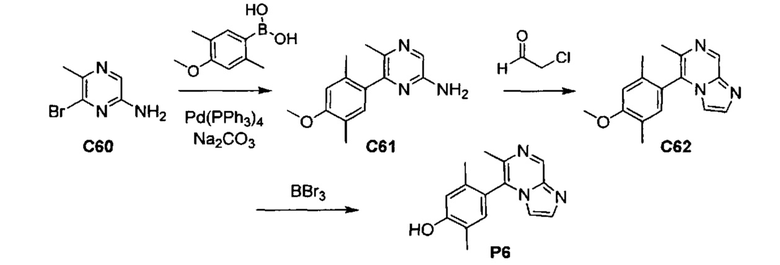
Стадия 1. Синтез 6-(4-метокси-2,5-диметилфенил)-5-метилпиразин-2-амина (С61)
6-Бром-5-метилпиразин-2-амин (С60, см. A. R. Harris et al., Tetrahedron 2011, 67, 9063-9066; 111 мг, 0,590 ммоль), (4-метокси-2,5-диметилфенил)-бороновую кислоту (127 мг, 0,708 ммоль) и тетракис(трифенилфосфин)-палладий(0) (95%, 40 мг, 0,033 ммоль) объединяли в сосуде под давлением и растворяли в 1,4-диоксане (2 мл) и воде (0,6 мл). Добавляли водный раствор карбоната натрия (2,0 М, 0,885 мл, 1,77 ммоль) и реакционную смесь подвергали аналогичным образом синтезу 6-[4-(фуро[3,2-с]пиридин-4-илокси)-2-метилфенил]-5-метилпиразин-2-амина (С3) в Примере 2. Хроматография на силикагеле (градиент: от 0% до 75% этилацетата в гептане) дала продукт. Выход: 116 мг, 0,477 ммоль, 81%. ЖХ-МС m/z 244,1 (М+Н). 1Н ЯМР (400 МГц, CD3CN) δ 7.83 (s, 1Н), 6.90 (s, 1Н), 6.82 (s, 1Н), 4.93 (br s, 2Н), 3.83 (s, 3Н), 2.15 (br s, 3Н), 2.11 (s, 3Н), 2.05 (br s, 3Н).
Стадия 2. Синтез 5-(4-метокси-2,5-диметилфенил)-6-метилимидазо[1,2-а]пиразина (С62)
Хлорацетальдегид (55%-ный раствор в воде, 0,28 мл, 2,38 ммоль) добавляли к смеси 6-(4-метокси-2,5-диметилфенил)-5-метилпиразин-2-амина (С61) (116 мг, 0,477 ммоль) в воде (3,6 мл). Реакционную смесь нагревали до 115°C в течение 2 часов в микроволновом реакторе и затем охлаждали до комнатной температуры, после чего растворитель удаляли в вакууме. Хроматография на силикагеле (градиент: от 0% до 10% метанола в дихлорметане) дала продукт. Выход: 115 мг, 0,43 ммоль, 90%. ЖХ-МС m/z 268,1 (М+Н). 1Н ЯМР (400 МГц, CD3CN) δ 9.45 (s, 1Н), 7.99 (br s, 1Н), 7.37 (br s, 1H), 7.08 (s, 1H), 7.04 (s, 1H), 3.91 (s, 3Н), 2.41 (s, 3Н), 2.20 (br s, 3Н), 2.03 (br s, 3H).
Стадия 3. Синтез 2,5-диметил-4-(6-метилимидазо[1,2-а]пиразин-5-ил)фенола (Р6)
5-(4-Метокси-2,5-диметилфенил)-6-метилимидазо[1,2-а]пиразин (С62) (115 мг, 0,43 ммоль) растворяли в дихлорметане (5 мл) и реакционную смесь охлаждали до -78°C. Медленно по каплям добавляли раствор трибромида бора (1 M в дихлорметане, 2,58 мл, 2,58 ммоль) и полученную смесь перемешивали в течение 15 минут; охлаждающую баню затем удаляли и реакционную смесь перемешивали при комнатной температуре в течение 18 часов. Добавляли метанол (5 мл) и полученную смесь нагревали до слабой дефлегмации в течение 30 минут. Растворитель удаляли в вакууме и полученный желтый остаток растирали три раза с этилацетатом (10 мл) с получением продукта. Выход: 104 мг, 0,410 ммоль, 95%. ЖХ-МС m/z 254,1 (М+Н). 1Н ЯМР (400 МГц, CD3OD) δ 9.40 (s, 1Н), 8.20 (d, J=2,0 Гц, 1Н), 7.60-7.62 (m, 1Н), 7.11 (s, 1Н), 6.91 (s, 1Н), 2.46 (s, 3Н), 2.23 (br s, 3Н), 1.98 (br s, 3Н).
Подготовительный пример Р7
3-Хлор-4-(4,6-диметилпиримидин-5-ил)фенол (Р7)
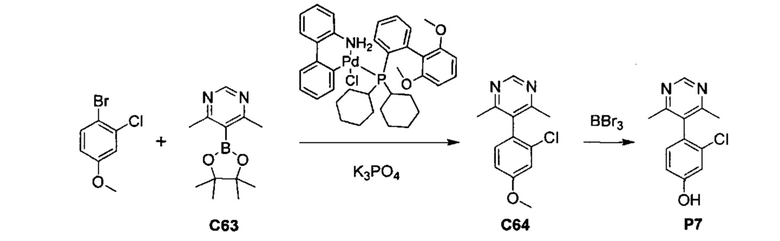
Стадия 1. Синтез 5-(2-хлор-4-метоксифенил)-4,6-диметилпиримидина (С64)
4,6-Диметил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиримидин (С63, полученный из 5-бром-4,6-диметилпиримидина с использованием способа Примера 1, стадия 2) (750 мг, 3,2 ммоль) и 1-бром-2-хлор-4-метоксибензол (1,46 г, 6,41 ммоль) растворяли в тетрагидрофуране (10 мл) и добавляли водный раствор фосфата калия (0,5 М, 12,8 мл). Азот барботировали через эту реакционную смесь в течение 10 минут. Добавляли [2'-(азанидил-kN)бифенил-2-ил-кC2](хлор)[дициклогексил(2',6'-диметоксибифенил-2-ил)-λ5-фосфанил]палладий (116 мг, 0,161 ммоль) и затем барботирование азота продолжали в течение нескольких минут. Реакционный сосуд герметично закрывали и перемешивали при 70°C в течение 18 часов. Реакционную смесь охлаждали до комнатной температуры, разбавляли этилацетатом, промывали водой и насыщенным водным раствором хлорида натрия, сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении. Неочищенный материал очищали хроматографией на силикагеле (элюент: 25% этилацетата в гептане) с получением продукта в виде светло-желтого масла, которое затвердевало при стоянии. Выход: 320 мг, 1,29 ммоль, 40%. 1Н ЯМР (400 МГц, CDCl3) δ 8.93 (s, 1Н), 7.05 (d, J=2,5 Гц, 1Н), 7.02 (d, J=8,6 Гц, 1Н), 6.90 (dd, J=8,6, 2,5 Гц, 1Н), 3.84 (s, 3Н), 2.21 (s, 6Н).
Стадия 2. Синтез 3-хлор-4-(4,6-диметилпиримидин-5-ил)фенола (Р7)
5-(2-Хлор-4-метоксифенил)-4,6-диметилпиримидин (С64) (310 мг, 1,25 ммоль) превращали в продукт согласно общей методике для синтеза 5-[4-(фуро[3,2-с]пиридин-4-илокси)-2-метилфенил]-6-метилпиримидин-4-ола (С30) в Примере 18. Продукт получили в виде оранжевого твердого вещества. Выход: 280 мг, 1,19 ммоль, 95%. 1Н ЯМР (400 МГц, CD3OD) δ 8.82 (s, 1Н), 7.05 (d, J=8,4 Гц, 1Н), 6.98 (d, J=2,3 Гц, 1Н), 6.85 (dd, J=8,4, 2,3 Гц, 1Н), 2.20 (s, 6Н).
Подготовительный пример Р8
6-(4-Гидрокси-2-метилфенил)-1,5-диметилпиразин-2(1H)-он (Р8)
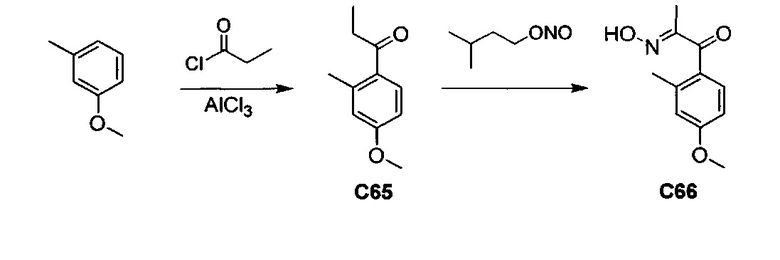
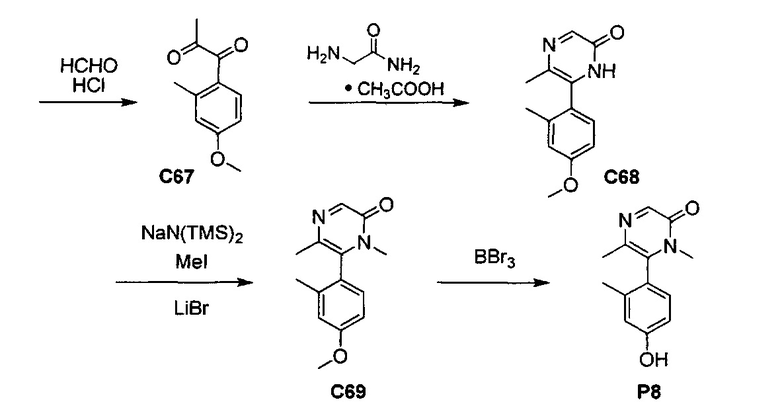
Стадия 1. Синтез 1-(4-метокси-2-метилфенил)пропан-1-она (С65)
Смесь 1-метокси-3-метилбензола (85,5 г, 0,700 моль) и хлорида алюминия (138,6 г, 1,04 моль) в дихлорметане (2,5 л) охлаждали в ледяной бане; пропаноилхлорид (97,1 г, 1,05 моль) добавляли по каплям в течение периода 30 минут. Ледяную баню удаляли и полученную смесь перемешивали при комнатной температуре в течение 20 минут, затем снова охлаждали в ледяной бане. Добавляли по каплям воду (150 мл) с последующим добавлением дополнительного количества воды (500 мл). Органическую фазу отделяли и концентрировали в вакууме. Хроматография на силикагеле (3% этилацетата в петролейном эфире) дала продукт в виде бесцветного масла, которое затвердевало в виде белого твердого вещества при стоянии при комнатной температуре. Посредством ЯМР было обнаружено, что продукт загрязнен небольшим количеством другого изомера. Выход: 100 г, 0,56 моль, 80%. 1Н ЯМР (400 МГц, CDCl3), пики продукта: δ 7.73 (d, J=9,5 Гц, 1Н), 6.73-6.78 (m, 2Н), 3.84 (s, 3Н), 2.91 (q, J=7,3 Гц, 2Н), 2.55 (s, 3Н), 1.19 (t, J=7,3 Гц, 3Н).
Стадия 2. Синтез 2-(гидроксиимино)-1-(4-метокси-2-метилфенил)-пропан-1-она (С66)
К смеси 1-(4-метокси-2-метилфенил)пропан-1-она (С65) (100 г, 0,56 моль) в тетрагидрофуране (2,5 л) медленно добавляли изоамилнитрит (131 г, 1,12 моль) и хлористый водород (4 н. в 1,4-диоксане, 200 мл). Смесь перемешивали при комнатной температуре в течение 24 часов, затем концентрировали в вакууме. Хроматография на силикагеле (градиент: от 3% до 10% этилацетата в петролейном эфире) дала неочищенный продукт (120 г), который дополнительно очищали путем суспендирования в смеси петролейного эфира (1 л) и этилацетата (100 мл) при комнатной температуре в течение 30 минут. Смесь фильтровали с получением продукта в виде твердого вещества. Выход: 75 г, 0,36 моль, 64%. 1Н ЯМР (400 МГц, CDCl3) δ 7.98-8.12 (br m, 1Н), 7.46 (d, J=8,3 Гц, 1H), 6.72-6.79 (m, 2Н), 3.84 (s, 3Н), 2.40 (s, 3Н), 2.16 (s, 3Н).
Стадия 3. Синтез 1-(4-метокси-2-метилфенил)пропан-1,2-диона (С67)
К смеси 2-(гидроксимимно)-1 -(4-метокси-2-метилфенил)пропан-1 -она (С66) (37,5 г, 181 ммоль) в воде (720 мл) медленно добавляли раствор формальдегида (450 мл) и концентрированной соляной кислоты (270 мл). Вторую партию реакционной смеси получали аналогичным образом. Обе смеси перемешивали при комнатной температуре в течение 18 часов. Две партии объединяли и экстрагировали этилацетатом (3×2 л); объединенные органические экстракты концентрировали. Хроматография на силикагеле (5% этилацетата в петролейном эфире) дала продукт в виде желтого масла. Выход: 60 г, 310 ммоль, 86%. 1Н ЯМР (400 МГц, CDCl3) δ 7.66 (d, J=8,5 Гц, 1Н), 6.75-6.83 (m, 2Н), 3.87 (s, 3Н), 2.60 (s, 3Н), 2.51 (s, 3Н).
Стадия 4. Синтез 6-(4-метокси-2-метилфенил)-5-метилпиразин-2(1Н)-она (С68)
1-(4-Метокси-2-метилфенил)пропан-1,2-дион (С67) (4,0 г, 21 ммоль) и ацетат глицинамида (2,79 г, 20,8 ммоль) растворяли в метаноле (40 мл) и охлаждали до -10°C. Добавляли водный раствор гидроксида натрия (12 н., 3,5 мл, 42 ммоль) и полученную смесь медленно нагревали до комнатной температуры. После перемешивания в течение 3 суток реакционную смесь концентрировали в вакууме. Остаток разбавляли водой и добавляли 1 н. водную соляную кислоту до pH приблизительно 7. Водную фазу экстрагировали несколько раз этилацетатом и объединенные органические экстракты промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении. Полученный остаток суспендировали смесью этилацетат/гептан в соотношении 3:1, перемешивали в течение 5 минут и затем фильтровали. Фильтрат концентрировали при пониженном давлении. Хроматография на силикагеле (элюент: этилацетат) дала продукт в виде желто-коричневого твердого вещества, которое содержало 15% нежелательного региоизомера; этот материал использовали без дополнительной очистки. Выход: 2,0 г, 8,7 ммоль, менее 41%. ЖХ-МС m/z 231,1 (М+Н). 1Н ЯМР (400 МГц, CDCl3), пики продукта: δ 8.09 (s, 1Н), 7.14 (d, J=8,2 Гц, 1Н), 6.82-6.87 (m, 2Н), 3.86 (s, 3Н), 2.20 (s, 3Н), 2.11 (s, 3Н).
Стадия 5. Синтез 6-(4-метокси-2-метилфенил)-1,5-диметилпиразин-2(1H)-она (С69)
6-(4-Метокси-2-метилфенил)-5-метилпиразин-2(1Н)-он (С68) (с предыдущей стадии, 1,9 г, менее 8,2 ммоль) растворяли в N,N-диметилформамиде (40 мл). Добавляли бромид лития (0,86 г, 9,9 ммоль) и бис(триметилсилил)амид натрия (95%, 1,91 г, 9,89 ммоль) и реакционную смесь перемешивали в течение 30 минут. Добавляли метилйодид (0,635 мл, 10,2 ммоль) и полученный раствор перемешивали при комнатной температуре в течение 18 часов. Реакционную смесь разбавляли водой и доводили значение pH до приблизительно 7 медленным добавлением по каплям 1 н. водной соляной кислоты. Водный слой экстрагировали этилацетатом и объединенные этилацетатные слои промывали несколько раз водой, сушили над сульфатом магния, фильтровали и концентрировали. Хроматография на силикагеле (градиент: от 75% до 100% этилацетата в гептане) дала продукт в виде вязкого оранжевого масла. Выход: 1,67 г, 6,84 ммоль, 33% за две стадии. ЖХ-МС m/z 245,1 (М+Н). 1Н ЯМР (400 МГц, CDCl3) δ 8.17 (s, 1Н), 7.03 (brd, J=8 Гц, 1Н), 6.85-6.90 (m, 2Н), 3.86 (s, 3Н), 3.18 (s, 3Н), 2.08 (br s, 3Н), 2.00 (s, 3Н).
Стадия 6. Синтез 6-(4-гидрокси-2-метилфенил)-1,5-диметилпиразин-2(1H)-она (Р8)
К охлажденному (-78°C) раствору 6-(4-метокси-2-метилфенил)-1,5-диметилпиразин-2(1Н)-она (С69) (1,8 г, 7,37 ммоль) в дихлорметане добавляли раствор трибромида бора в дихлорметане (1 М, 22 мл, 22 ммоль). Охлаждающую баню удаляли через 30 минут и реакционную смесь оставляли нагреваться до комнатной температуры и перемешивали в течение 18 часов. Реакционную смесь охлаждали до -78°C и медленно добавляли метанол (10 мл); полученную смесь медленно нагревали до комнатной температуры. Реакционную смесь концентрировали в вакууме, добавляли метанол (20 мл) и смесь снова концентрировали при пониженном давлении. Остаток разбавляли этилацетатом (300 мл) и водой (200 мл) и полученный водный слой доводили до pH 7 добавлением по каплям насыщенного водного раствора карбоната натрия. Смесь экстрагировали этилацетатом (3×200 мл). Объединенные органические экстракты промывали водой и насыщенным водным раствором хлорида натрия, сушили над сульфатом магния, фильтровали и концентрировали в вакууме с получением продукта в виде светло-коричневого твердого вещества. Выход: 1,4 г, 6,0 ммоль, 81%. ЖХ-МС m/z 231,1 (М+Н). 1Н ЯМР (400 МГц, CDCl3) δ 8.21 (s, 1Н), 6.98 (d, J=8,2 Гц, 1Н), 6.87-6.89 (m, 1Н), 6.85 (br dd, J=8,2, 2,5 Гц, 1H), 3.22 (s, 3Н), 2.06 (br s, 3Н), 2.03 (s, 3Н).
Подготовительный пример Р9
3-Метил-4-(3-метилимидазо[2,1-с][1,2,4]триазин-4-ил)фенол (Р9)
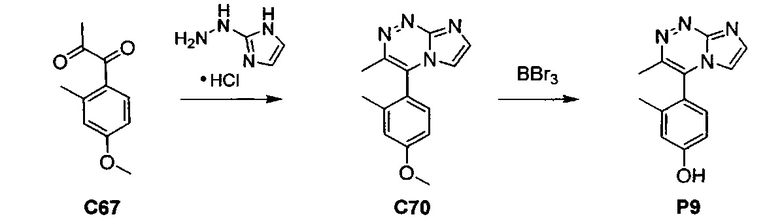
Стадия 1. Синтез 4-(4-метокси-2-метилфенил)-3-метилимидазо[2,1-с][1,2,4]триазина (С70)
Смесь 1-(4-метокси-2-метилфенил)пропан-1,2-диона (С67) (1,0 г, 5,2 ммоль) и гидрохлорида 2-гидразинил-1Н-имидазола (1,05 г, 7,8 ммоль) в N,N-диметилформамиде (8 мл) нагревали до 100°C в микроволновом реакторе в течение 20 минут. Затем развитие реакции оценивали с помощью тонкослойной хроматографии, смесь нагревали до 120°C в течение 20 минут. Растворитель удаляли в вакууме и остаток переносили в этилацетат (30 мл) и воду (10 мл). Добавляли насыщенный водный раствор бикарбоната натрия для доведения pH до примерно 8. Водный слой экстрагировали дополнительным количеством этилацетата (30 мл) и объединенные органические экстракты сушили над сульфатом магния, фильтровали и концентрировали в вакууме. Хроматография на силикагеле (градиент: от 50% до 100% этилацетата в гептане) дала продукт в виде светло-желтого твердого вещества. Выход: 587 мг, 2,31 ммоль, 44%. 1Н ЯМР (400 МГц, CDCl3) δ 8.06 (d, J=0,9 Гц, 1Н), 7.21 (d, J=8,2 Гц, 1Н), 7.15 (d, J=1,1 Гц, 1Н), 6.95-7.00 (m, 2Н), 3.91 (s, 3Н), 2.63 (s, 3Н), 2.03 (br s, 3Н).
Стадия 2. Синтез 3-метил-4-(3-метилимидазо[2,1-с][1,2,4]триазин-4-ил)фенола (Р9)
4-(4-Метокси-2-метилфенил)-3-метилимидазо[2,1-с][1,2,4]триазин (С70) (587 мг, 2,31 ммоль) в дихлорметане (5 мл) подвергали взаимодействию с трибромидом бора (1 M в дихлорметане, 13,1 мл, 13,1 ммоль) как описано в Подготовительном примере Р8. Продукт получили в виде желто-коричневого твердого вещества. Выход: 543 мг, 2,25 ммоль, 97%. ЖХ-МС m/z 241,1 (М+Н). 1Н ЯМР (400 МГц, DMSO-d6) δ 9.99 (s, 1Н), 8.09 (d, J=1,0 Гц, 1Н), 7.43 (d, J=1,2 Гц, 1Н), 7.27 (d, J=8,4 Гц, 1Н), 6.89 (br d, J=2,2 Гц, 1H), 6.83 (br dd, J=8,3, 2,4 Гц, 1H), 2.49 (s, 3Н), 1.91 (br s, 3Н).
Подготовительный пример P10
7-(4,6-Диметилпиримидин-5-ил)-2-метил-2Н-индазол-4-ол (Р10)
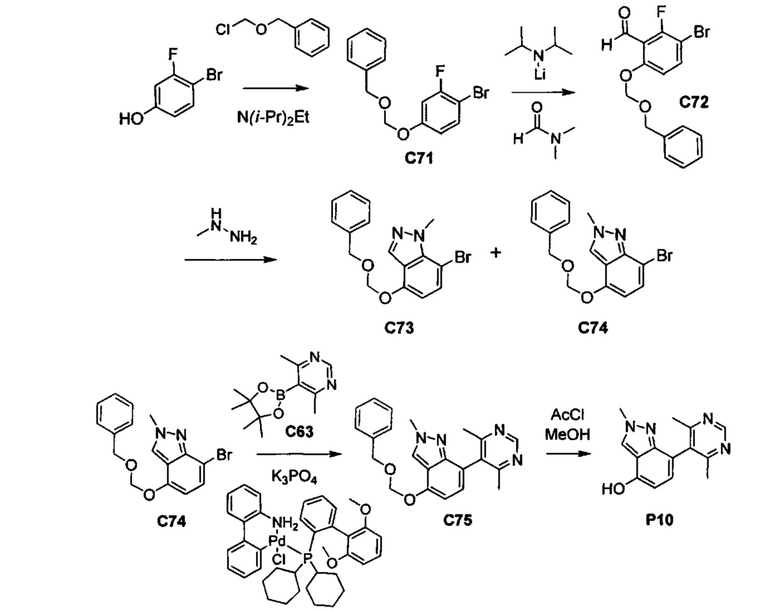
Стадия 1. Синтез 4-[(бензилокси)метокси]-1-бром-2-фторбензола (C71)
Раствор 4-бром-3-фторфенола (1,22 г, 6,39 ммоль), бензилхлорметилового эфира (60%, 2,22 мл, 9,58 ммоль) и диизопропилэтиламина (2,23 мл, 12,8 ммоль) в дихлорметане кипятили с обратным холодильником в течение двух часов. Реакционную смесь затем концентрировали в вакууме и очищали хроматографией на силикагеле (градиент: от 15% до 40% этилацетата в гептане) с получением продукта в виде бесцветного масла. Выход: 2,35 г, более 100%. 1Н ЯМР (400 МГц, CD3OD), характеристические пики: δ 7.48 (dd, J=8,9, 8,1 Гц, 1Н), 6.95 (dd, J=10,6, 2,7 Гц, 1Н), 6.84 (ddd, J=8,9, 2,8, 1,1 Гц, 1Н), 5.31 (s, 2Н), 4.70 (s, 2Н).
Стадия 2. Синтез 6-[(бензилокси)метокси]-3-бром-2-фторбензальдегида (С72)
Раствор 4-[(бензилокси)метокси]-1-бром-2-фторбензола (С71) (с предыдущей стадии, 525 мг, менее 1,69 ммоль) в тетрагидрофуране (20 мл) охлаждали до -78°C в течение 15 минут. Затем добавляли по каплям в течение 15 минут диизопропиламид лития (1,60 М, 1,58 мл, 2,53 ммоль). После одного часа при -78°C добавляли N,N-диметилформамид (0,197 мл, 2,53 ммоль) в тетрагидрофуране (5 мл). Реакционную смесь перемешивали при -78°C в течение 30 минут, гасили 50%-ным насыщенным водным раствором хлорида натрия (30 мл) и затем оставляли достигать комнатной температуры. Реакционную смесь экстрагировали этилацетатом (3х30 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали, концентрировали в вакууме и очищали хроматографией на силикагеле (градиент: от 15% до 40% этилацетата в гептане) с получением продукта в виде светло-желтого масла. Выход: 397 мг, 1,17 ммоль, 82% за две стадии. 1Н ЯМР (400 МГц, CDCl3) δ 10.36 (d, J=1,4 Гц, 1Н), 7.66 (dd, J=9,2, 7,6 Гц, 1H), 7.29-7.38 (m, 5Н), 7.04 (dd, J=9,1. 1,5 Гц, 1Н), 5.42 (s, 2Н), 4.75 (s, 2Н).
Стадия 3. Синтез 4-[(бензилокси)метокси]-7-бром-1-метил-1Н-индазола (С73) и 4-[(бензилокси)метокси]-7-бром-2-метил-2Н-индазола (С74)
Смесь 6-[(бензилокси)метокси]-3-бром-2-фторбензальдегида (С72) (1,40 г, 4,13 ммоль) и метилгидразина (8,69 мл, 165 ммоль) растворяли в 1,4-диоксане (8 мл) в герметичном сосуде и нагревали при 110°C в течение 4 часов, затем при 120°C в течение 16 часов. Смесь подвергали микроволновому излучению при 150°C в течение 90 минут. Реакционную смесь концентрировали в вакууме и очищали хроматографией на силикагеле (градиент: от 15% до 40% этилацетата в гептане) с получением С73 в виде бесцветного масла и С74 в виде желтого масла. Выход: С73, 801 мг, 2,31 ммоль, 56%; С74, 296 мг, 0,852 ммоль, 21%. С73: 1Н ЯМР (400 МГц, CDCl3) δ 8.05 (s, 1Н), 7.41 (d, J=8,2 Гц, 1Н), 7.28-7.38 (m, 5Н), 6.67 (d, J=8,2 Гц, 1H), 5.44 (s, 2Н), 4.76 (s, 2Н), 4.41 (s, 3Н). С74: 1Н ЯМР (400 МГц, CDCl3) δ 8.06 (br s, 1Н), 7.38 (d, J=7,9 Гц, 1H), 7.28-7.38 (m, 5Н), 6.59 (d, J=8,0 Гц, 1H), 5.42 (s, 2Н), 4.76 (s, 2Н), 4.26 (brs, 3Н).
Стадия 4. Синтез 4-[(бензилокси)метокси]-7-(4,6-диметилпиримидин-5-ил)-2-метил-2Н-индазола (С75)
Смесь 4,6-диметил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиримидина (С63) (152 мг, 0,649 ммоль), 4-[(бензилокси)метокси]-7-бром-2-метил-2Н-индазола (С74) (150 мг, 0,432 ммоль), тетрагидрофурана (5 мл) и водного раствора фосфата калия (0,5 М, 2,59 мл, 1,30 ммоль) продували азотом в течение двух минут, затем добавляли [2'-(азанидил-кN)бифенил-2-ил-кC2](хлор)[дициклогексил(2',6'-диметоксибифенил-2-ил)-λ5-фосфанил]палладий (31 мг, 0,043 ммоль). Реакционную смесь нагревали при 70°C в течение 40 часов, затем фильтровали через тонкий слой целита. Фильтрат концентрировали в вакууме и очищали хроматографией на силикагеле (градиент: от 5% до 10% метанола в дихлорметане) с получением продукта в виде темного масла. Выход: 63 мг, 0,17 ммоль, 39%. ЖХ-МС m/z 375,2 (М+Н). 1Н ЯМР (400 МГц, CDCl3) δ 8.98 (s, 1Н), 8.06 (s, 1Н), 7.29-7.41 (m, 5Н), 6.96 (d, J=7,6 Гц, 1H), 6.76 (d, J=7,6 Гц, 1H), 5.50 (s, 2H), 4.83 (s, 2H), 4.17 (s, 3Н), 2.31 (s, 6H).
Стадия 5. Синтез 7-(4,6-диметилпиримидин-5-ил)-2-метил-2Н-индазол-4-ола (Р10)
К раствору ацетилхлорида (98%, 0,122 мл, 1,68 ммоль) в метаноле (2 мл) добавляли раствор 4-[(бензилокси)метокси]-7-(4,6-диметилпиримидин-5-ил)-2-метил-2Н-индазола (С75) (63 мг, 0,17 ммоль) в метаноле (2 мл). Через 16 часов реакционную смесь концентрировали в вакууме и очищали хроматографией на силикагеле (градиент: от 5% до 10% метанола в дихлорметане) с получением продукта в виде стекловидного твердого вещества. Выход: 37 мг, 0,14 ммоль, 82%. ЖХ-МС m/z 255,2 (М+Н). 1Н ЯМР (400 МГц, CD3OD) δ 8.87 (s, 1Н), 8.28 (s, 1Н), 6.97 (d, J=7,6 Гц, 1Н), 6.47 (d, J=7,6 Гц, 1Н), 4.13 (s, 3Н), 2.25 (s, 6Н).
Подготовительный пример Р11
7-(4,6-Диметилпиримидин-5-ил)-1 -метил- 1Н-индазол-4-ол (Р11)
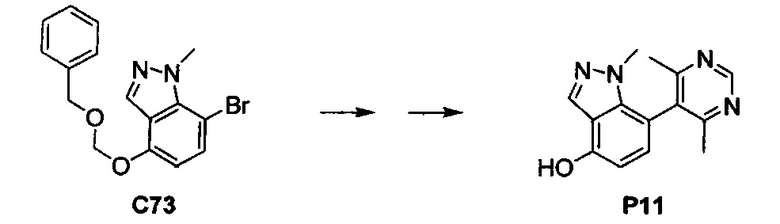
Соединение Р11 получали из 4-[(бензилокси)метокси]-7-бром-1-метил-1Н-индазола (С73) согласно стадиям 4 и 5 синтеза 7-(4,6-диметилпиримидин-5-ил)-2-метил-2Н-индазол-4-ола (Р10) в Подготовительном примере Р10, с получением продукта в виде не совсем белого твердого вещества. Выход: 36 мг, 0,14 ммоль, 64%. ЖХ-МС m/z 255,2 (М+Н). 1Н ЯМР (400 МГц, DMSO-d6) δ 10.40 (br s, 1Н), 8.95 (s, 1Н), 8.09 (s, 1Н), 6.96 (d, J=7,6 Гц, 1Н), 6.53 (d, J=7,8 Гц, 1Н), 3.38 (s, 3Н), 2.15 (s, 6Н).
Подготовительный пример Р12
5-(Фуро[3,2-с]пиридин-4-илокси)-2-(имидазо[1,2-а]пиридин-5-ил)бензойная кислота (Р12)

Стадия 1. Синтез 5-(фуро[3,2-с]пиридин-4-илокси)-2-(триметилстаннанил)бензонитрила (С76)
К раствору 2-бром-5-(фуро[3,2-с]пиридин-4-илокси)бензонитрила (полученного из 2-бром-5-гидроксибензонитрила и 4-йодфуро[3,2-с]пиридина способом стадии 3 в Примере 7; 4-йодфуро[3,2-с]пиридин был синтезирован из 4-хлорфуро[3,2-с]пиридина с использованием ацетилхлорида и йодида натрия в ацетонитриле) (7,0 г, 22 ммоль) в 1,4-диоксане (70 мл) добавляли гексаметилдистаннан (21,8 г, 66,6 ммоль) и тетракис(трифенилфосфин)-палладий(О) (1,28 г, 1,11 ммоль). Полученную смесь нагревали при 120°C в течение 18 часов. Реакционную смесь фильтровали и фильтрат концентрировали с получением неочищенного остатка, который очищали хроматографией на силикагеле (элюент: смесь петролейный эфир/этилацетат в соотношении 400:1) с получением продукта в виде белого твердого вещества. Выход: 6,0 г, 15 ммоль, 67%. 1Н ЯМР (400 МГц, CDCl3) δ 8.01 (d, J=5,9 Гц, 1Н), 7.68 (d, J=2,2 Гц, 1Н), 7.62 (d, J=8,1 Гц, 1Н), 7.55-7.58 (m, 1Н), 7.42 (dd, J=8,0, 2,4 Гц, 1H), 7.26 (dd, J=5,8, 0,9 Гц, 1H), 6.93 (dd, J=2,2, 0,9 Гц, 1H), 0.47 (s, 9Н).
Стадия 2. Синтез 5-(фуро[3,2-с]пиридин-4-илокси)-2-(имидазо[1,2-а]пиридин-5-ил)бензонитрила (С77)
К раствору 5-(фуро[3,2-с]пиридин-4-илокси)-2-(триметилстаннил)-бензонитрила (С76) (8,3 г, 21 ммоль) в тетрагидрофуране (160 мл) добавляли 5-бромимидазо[1,2-а]пиридин (3,9 г, 20 ммоль), хлорид лития (0,67 г, 15,8 ммоль), бромид меди(I) (0,57 г, 4,0 ммоль) и тетракис(трифенилфосфин)палладий(0) (2,27 г, 2,0 ммоль). Смесь кипятили с обратным холодильником в течение 48 часов. Реакционную смесь фильтровали и фильтрат концентрировали с получением неочищенного продукта, который очищали хроматографией на силикагеле (градиент: от 7% до 20% этилацетата в петролейном эфире) с получением продукта в виде коричневого твердого вещества. Выход: 5 г, 13 ммоль, 68%. ЖХ-МС m/z 353,0 (М+Н). 1Н ЯМР (400 МГц, CD3OD, концентрированная HCl), характеристические пики: δ 8.23-8.26 (m, 1Н), 8.12 (br d, половина АВ квартета, J=8 Гц, 1Н), 8.06 (br d, половина АВ квартета, J=8 Гц, 1Н), 7.93 (br d, J=6 Гц, 1Н), 7.77-7.81 (m, 1Н).
Стадия 3. Синтез 5-(фуро[3,2-с]пиридин-4-илокси)-2-(имидазо[1,2-а]пиридин-5-ил)бензойной кислоты (Р12)
К водному раствору гидроксида натрия (15% масс/об., 25 мл) добавляли 5-(фуро[3,2-с]пиридин-4-илокси)-2-(имидазо[1,2-а]пиридин-5-ил)бензонитрил (С77) (4,35 г, 12,3 ммоль) и этанол (25 мл) и реакционную смесь кипятили с обратным холодильником в течение 18 часов. Смесь охлаждали до комнатной температуры и экстрагировали дихлорметаном. pH водного слоя доводили до 7 с помощью 3 н. водной соляной кислоты; полученную смесь фильтровали и осадок на фильтре промывали этилацетатом и дихлорметаном, затем сушили под вакуумом с получением продукта в виде желтого твердого вещества.
Выход: 1,9 г, 5,1 ммоль, 42%. ЖХ-МС m/z 371,9 (М+Н). 1Н ЯМР (400 МГц, DMSO-d6), характеристические пики: δ 8.17 (d, J=2,4 Гц, 1Н), 8.04 (d, J=5,9 Гц, 1Н), 7.52 (d, J=5,9 Гц, 1Н), 6.72 (br d, J=6,7 Гц, 1Н).
Подготовительный пример Р13
4-{[7-(4,4,5,5-Тетраметил-1,3,2-диоксаборолан-2-ил)-1,3-бензодиоксол-4-ил]окси}фуро[3,2-с]пиридин (Р13)
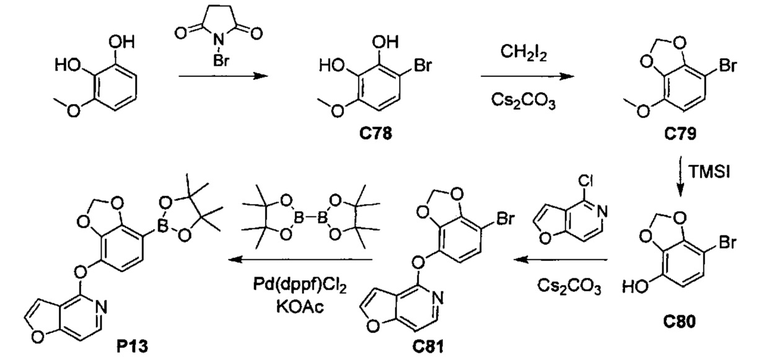
Стадия 1. Синтез 3-бром-6-метоксибензол-1,2-диола (С78)
К смеси 3-метоксибензол-1,2-диола (578 мг, 4,12 ммоль) в ацетонитриле (10 мл) при 0°C медленно добавляли N-бромсукцинимид (95%, 811 мг, 4,33 ммоль) в ацетонитриле (5 мл). Через два часа при 0°C добавляли водный раствор тиосульфата натрия (1 М, 2 мл). Через десять минут реакционную смесь концентрировали в вакууме и очищали хроматографией на силикагеле (градиент: от 20% до 40% этилацетата в гептане) с получением продукта в виде белого твердого вещества. Выход: 858 мг, 0,392 ммоль, 95%. ЖХ-МС m/z 216,8 (М-Н). 1Н ЯМР (400 МГц, CDCl3) δ 7.00 (d, J=9,0 Гц, 1Н), 6.43 (d, J=9,0 Гц, 1Н), 5.54 (s, 1Н), 5.48 (s, 1Н), 3.89 (s, 3Н).
Стадия 2. Синтез 4-бром-7-метокси-1,3-бензодиоксола (С79)
К раствору 3-бром-6-метоксибензол-1,2-диола (С78) (420 мг, 1,92 ммоль) в N,N-диметилформамиде (5 мл) добавляли дийодметан (0,170 мл, 2,11 ммоль) и карбонат цезия (690 мг, 2,1 ммоль). Реакционную смесь перемешивали при 100°C в течение одного часа, затем охлаждали до комнатной температуры и разбавляли этилацетатом (20 мл). Твердое вещество удаляли фильтрованием и промывали этилацетатом (30 мл). Фильтрат промывали 50%-ным насыщенным водным раствором хлорида натрия (4×20 мл), сушили над сульфатом натрия, фильтровали, концентрировали в вакууме и очищали хроматографией на силикагеле (градиент: от 20% до 40% этилацетата в гептане) с получением продукта в виде белого твердого вещества. Выход: 335 мг, 1,45 ммоль, 76%. 1Н ЯМР (400 МГц, CDCl3) δ 6.92 (d, J=9,0 Гц, 1Н), 6.46 (d, J=9,1 Гц, 1Н), 6.05 (s, 2Н), 3.90 (s, 3Н).
Стадия 3. Синтез 7-бром-1,3-бензодиоксол-4-ола (С80)
К раствору 4-бром-7-метокси-1,3-бензодиоксола (С79) (186 мг, 0,805 ммоль) в ацетонитриле (5 мл) добавляли триметилсилилйодид (0,343 мл, 2,42 ммоль). Реакционную смесь нагревали при 85°C в течение 18 часов и очищали хроматографией на силикагеле (градиент: от 30% до 40% этилацетата в гептане) с получением продукта в виде масла. Выход: 59 мг, 0,27 ммоль, 34%. 1Н ЯМР (400 МГц, CDCl3) δ 6.86 (d, J=9,0 Гц, 1Н), 6.44 (d, J=9,0 Гц, 1Н), 6.05 (s, 2Н).
Стадия 4. Синтез 4-[(7-бром-1,3-бензодиоксол-4-ил)окси]фуро[3,2-с]пиридина (С81)
Смесь 7-бром-1,3-бензодиоксол-4-ола (С80) (59 мг, 0,27 ммоль), 4-хлорфуро[3,2-с]пиридина (62,7 мг, 0,408 ммоль) и карбоната цезия (224 мг, 0,687 ммоль) в диметилсульфоксиде (2 мл) нагревали при 140°C в течение 4 часов. Реакционную смесь охлаждали до комнатной температуры и объединяли с аналогичной реакционной смесью, полученной при осуществлении реакции на 16 мг С80. Добавляли этилацетат и твердое вещество удаляли фильтрованием. Фильтрат промывали 50%-ным насыщенным водным раствором хлорида натрия (3×15 мл), концентрировали в вакууме и очищали хроматографией на силикагеле (градиент: от 10% до 30% этилацетата в гептане) с получением продукта в виде масла. Выход: 61 мг, 0,182 ммоль, 53%. ЖХ-МС m/z 335,9 (М+Н).
Стадия 5. Синтез 4-{[7-(4,4,5,5-тетраметил-1,3,2-диокса6оролан-2-ил)-1,3-бензодиоксол-4-ил]окси}фуро[3,2-с]пиридина (Р13)
Смесь 4-[(7-бром-1,3-бензодиоксол-4-ил)окси]фуро[3,2-с]пиридина (С81) (61 мг, 0,18 ммоль), 4,4,4',4',5,5,5',5'-октаметил-2,2'-би-1,3,2-диоксаборолана (99%, 70,3 мг, 0,274 ммоль), 1,1'-бис(дифенилфосфино)ферроцен]-дихлорпалладия(II) (50%, 26,3 мг, 0,018 ммоль) и ацетата калия (55 мг, 0,55 ммоль) объединяли в ацетонитриле (3 мл). После барботирования азота через реакционную смесь в течение пяти минут ее нагревали при 80°C в течение 18 часов. Реакционную смесь затем фильтровали через тонкий слой целита, промывая этилацетатом (20 мл). Фильтрат концентрировали в вакууме и остаток распределяли между водой (15 мл) и этилацетатом (20 мл). Водный слой экстрагировали этилацетатом (3×10 мл); объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Очистка хроматографией на силикагеле (градиент: от 15% до 50% этилацетата в гептане) дала продукт в виде светло-желтой смолы. Выход: 25 мг, 0,066 ммоль, 37%. 1Н ЯМР (400 МГц, CDCl3) δ 8.00 (d, J=5,8 Гц, 1Н), 7.64 (d, J=2,2 Гц, 1Н), 7.30 (d, J=8,4 Гц, 1Н), 7.21 (dd, J=5,8, 1,0 Гц, 1H), 6.92 (dd, J=2,2, 0,9 Гц, 1H), 6.80 (d, J=8,5 Гц, 1H), 6.03 (s, 2Н), 1.37 (s, 12Н).
Подготовительный пример Р14
8-(4,6-Диметилпиримидин-5-ил)изохинолин-5-ол (Р14)
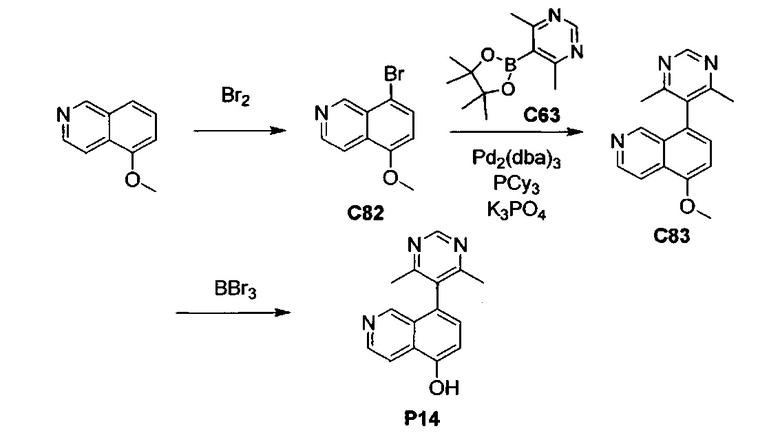
Стадия 1. Синтез 8-бром-5-метоксиизохинолина (С82)
К раствору 5-метоксиизохинолина (1,48 г, 9,30 ммоль) в уксусной кислоте (15 мл) добавляли раствор брома (2,1 г, 13 ммоль) в уксусной кислоте (5 мл). Через трое суток при комнатной температуре реакционную смесь охлаждали до 0°C, гасили насыщенным водным раствором бикарбоната натрия и экстрагировали дихлорметаном (3×50 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали, концентрировали в вакууме и очищали хроматографией на силикагеле (градиент: от 5% до 33% этилацетата в петролейном эфире) с получением продукта в виде твердого вещества. Выход: 1,72 г, 7,22 ммоль, 78%. 1Н ЯМР (400 МГц, DMSO-d6) δ 9.40 (s, 1Н), 8.64 (d, J=6,0 Гц, 1Н), 7.99 (d, J=5,5 Гц, 1Н), 7.90 (d, J=8,5 Гц, 1Н), 7.18 (d, J=8,5 Гц, 1Н), 4.00 (s, 3Н).
Стадия 2. Синтез 8-(4,6-диметилпиримидин-5-ил)-5-метокси-изохинолина (С83)
К раствору 8-бром-5-метоксиизохинолина (С82) (1,72 г, 7,22 ммоль) в 1,4-диоксане (75 мл) и воде (5 мл) добавляли 4,6-диметил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиримидин (С63) (2,20 г, 9,40 ммоль), трис(дибензилиденацетон)дипалладии(0) (659 мг, 0,72 ммоль), трициклогексилфосфин (403 мг, 1,44 ммоль) и фосфат калия (3,07 г, 14,46 ммоль). Реакционную смесь дегазировали азотом в течение пяти минут, затем перемешивали в течение 6 часов при 120°C. Добавляли дополнительное количество 4,6-диметил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-пиримидина (С63) (1,1 г, 4,7 ммоль). Реакционную смесь перемешивали в течение 7 часов при 120°C и затем фильтровали. Фильтрат концентрировали в вакууме и очищали хроматографией на силикагеле (градиент: от 0,5% до 2,5% метанола в дихлорметане) с получением продукта в виде твердого вещества. Выход: 1,0 г, 3,8 ммоль, 53%. 1Н ЯМР (400 МГц, DMSO-d6) δ 9.00 (s, 1Н), 8.56-8.60 (m, 2Н), 8.07 (dd, J=5,8, 0,8 Гц, 1Н), 7.51 (d, J=7,8 Гц, 1Н), 7.36 (d, J=8,0 Гц, 1Н), 4.07 (s, 3Н), 2.08 (s, 6Н).
Стадия 3. Синтез 8-(4,6-диметилпиримидин-5-ил)изохинолин-5-ола (Р14)
К раствору 8-(4,6-диметилпиримидин-5-ил)-5-метоксиизохинолина (С83) (1,0 г, 3,8 ммоль) в дихлорметане (60 мл) медленно добавляли трибромид бора (4,7 г, 19 ммоль) при -78°C. Смесь оставляли нагреваться до комнатной температуры и перемешивали в течение ночи, а затем гасили при -20°C метанолом. Реакционную смесь промывали насыщенным водным раствором бикарбоната натрия; водный слой экстрагировали дихлорметаном (5×50 мл) и этилацетатом (5×50 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали, концентрировали в вакууме и очищали хроматографией на силикагеле (градиент: от 0,5% до 5% метанола в дихлорметане) с получением продукта в виде твердого вещества. Выход: 300 мг, 1,19 ммоль, 31%. 1Н ЯМР (400 МГц, DMSO-d6) δ 10.88 (brs, 1Н), 8.98 (s, 1Н), 8.49-8.55 (m, 2H), 8.04 (br d, J=6 Гц, 1H), 7.36 (d, J=7,8 Гц, 1H), 7.21 (d, J=7,8 Гц, 1H), 2.07 (s, 6H).
Подготовительный пример Р15
4-(3,5-Диметилпиридазин-4-ил)-3-метоксифенол (Р15)
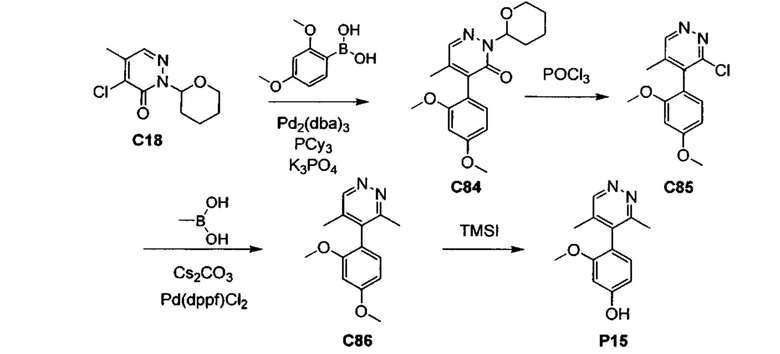
Стадия 1. Синтез 4-(2,4-диметоксифенил)-5-метил-2-(тетрагидро-2Н-пиран-2-ил)пиридазин-3(2H)-она (С84)
Смесь 4-хлор-5-метил-2-(тетрагидро-2Н-пиран-2-ил)пиридазин-3(2Н)-она (С18) (30 г, 130 ммоль), (2,4-диметоксифенил)бороновой кислоты (26 г, 140 ммоль), трис(дибензилиденацетон)дипалладия(0) (9,69 г, 10,6 ммоль), трициклогексилфосфина (7,5 г, 27 ммоль) и моногидрата фосфата калия (69 г, 300 ммоль) в 1,4-диоксане (250 мл) кипятили с обратным холодильником в течение 3 часов и затем охлаждали до комнатной температуры, фильтровали и концентрировали в вакууме. Хроматография на силикагеле (градиент: от 9% до 17% этилацетата в петролейном эфире) дала продукт в виде желтого твердого вещества. Выход: 40 г, 120 ммоль, 92%. 1Н ЯМР (400 МГц, CDCl3), смесь диастереомеров, характеристические пики: δ 7.76 и 7.77 (2 s, суммарный 1Н), [7.10 (d, J=8,3 Гц) и 7.07 (d, J=8,3 Гц), суммарный 1Н], 6.51-6.59 (m, 2Н), 6.06-6.12 (m, 1Н), 4.11-4.20 (m, 1Н), 3.85 (s, 3Н), 3.74 и 3.76 (2 s, суммарный 3Н), 1.99 и 2.00 (2 s, суммарный 3Н).
Стадия 2. Синтез 3-хлор-4-(2,4-диметоксифенил)-5-метилпиридазина (С85)
4-(2,4-Диметоксифенил)-5-метил-2-(тетрагидро-2Н-пиран-2-ил)-пиридазин-3(2Н)-он (С84) (30 г, 91 ммоль) растворяли в оксихлориде фосфора (158 мл) и смесь кипятили с обратным холодильником в течение 5 часов, охлаждали до комнатной температуры и вливали в ледяную воду. Осторожно добавляли карбонат калия для нейтрализации реакционной смеси с последующей экстракцией этилацетатом (3×500 мл). Объединенные органические экстракты концентрировали в вакууме. Хроматография на силикагеле (градиент: от 17% до 50% этилацетата в петролейном эфире) дала продукт в виде оранжевого твердого вещества. Выход: 20 г, 76 ммоль, 83%. ЖХ-МС m/z 264,7 (М+Н). 1Н ЯМР (400 МГц, CD3OD) δ 8.90 (s, 1Н), 6.88 (d, J=8,3 Гц, 1Н), 6.60 (d, J=2,3 Гц, 1Н), 6.53 (dd, J=8,2, 2,1 Гц, 1Н), 3.73 (s, 3Н), 2.36 (s, 3Н), 2.10 (s, 3Н).
Стадия 3. Синтез 4-(2,4-диметоксифенил)-3,5-диметилпиридазина (С86)
Смесь 3-хлор-4-(2,4-диметоксифенил)-5-метилпиридазина (С85) (18 г, 68 ммоль), метилбороновой кислоты (17 г, 280 ммоль), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия(II) (5,2 г, 70 ммоль) и карбоната цезия (46 г, 140 ммоль) в 1,4-диоксане (300 мл) кипятили с обратным холодильником в течение 2,5 часов и затем охлаждали до комнатной температуры, фильтровали и концентрировали в вакууме. Хроматография на силикагеле (градиент: от 17% до 50% этилацетата в петролейном эфире) дала продукт в виде оранжевого твердого вещества. Выход: 14 г, 57 ммоль, 84%). ЖХ-МС m/z 245,0 (М+Н).
Стадия 4. Синтез 4-(3,5-диметилпиридазин-4-ил)-3-метоксифенола (Р15)
Триметилсилилйодид (58 г, 290 ммоль) добавляли к перемешиваемому раствору 4-(2,4-диметоксифенил)-3,5-диметилпиридазина (С86) (12 г, 49 ммоль) в ацетонитриле (100 мл) и смесь кипятили с обратным холодильником в течение 18 часов. Реакционную смесь охлаждали до 0°C, медленно разбавляли метанолом и концентрировали в вакууме. Остаток распределяли между этилацетатом и насыщенным водным раствором тиосульфата натрия. Водный слой экстрагировали этилацетатом (4×150 мл) и объединенные органические экстракты сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Хроматография на силикагеле (градиент: от 50% до 100% этилацетата в петролейном эфире) дала продукт в виде желтого твердого вещества. Выход: 3,0 г, 13 ммоль, 26%. ЖХ-МС m/z 230,7 (М+Н). 1Н ЯМР (400 МГц, CD3OD) δ 8.90 (s, 1Н), 6.88 (d, J=8,0 Гц, 1Н), 6.60 (d, J=2,0 Гц, 1Н), 6.53 (dd, J=8,3, 2,3 Гц, 1Н), 3.73 (s, 3Н), 2.36 (s, 3Н), 2.10 (s, 3Н).
МЕТОДЫ
Методы М1-М7 описывают конкретные методики получения некоторых соединений по изобретению.
Метод М1: Катализируемое палладием взаимодействие фенолов с 4-хлорфуро[3,2-с]пиридинами
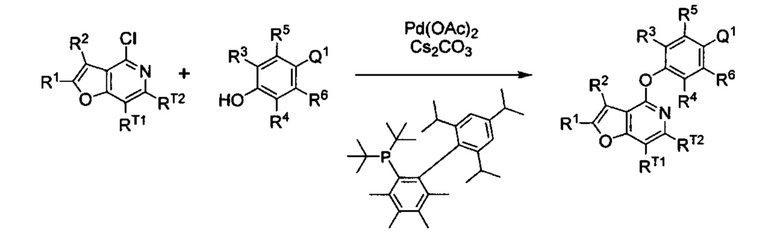
Растворы соответствующего фенола и 4-хлорфуро[3,2-с]пиридина готовили при 0,2 М с использованием дегазированного 1,4-диоксана. Флакон на 2 драхмы (примерно 7 мл) загружали раствором фенола (0,5 мл, 0,1 ммоль) и раствором 4-хлорфуро[3,2-с]пиридина (0,5 мл, 0,1 ммоль). Добавляли карбонат цезия (100 мг, 0,3 ммоль), ацетат палладия(II) (2,5 мг, 0,01 ммоль) и ди-трет-бутил[3,4,5,6-тетраметил-2',4',6'-три(пропан-2-ил)бифенил-2-ил]фосфан (10 мг, 0,02 ммоль). Флакон подвергали трем циклам вакуумного отсасывания с последующим заполнением азотом и полученную смесь встряхивали и нагревали при 100°C в течение 12 часов. Реакционную смесь охлаждали до комнатной температуры, распределяли между водой (1,5 мл) и этилацетатом (2,5 мл), встряхивали и оставляли стоять. Органический слой пропускали через картридж для твердофазной экстракции, заполненный сульфатом натрия (1,0 г); эту процедуру экстракции повторяли дважды и объединенные фильтраты концентрировали в вакууме. Продукты в общем очищали посредством HPLC (колонка: Waters XBridge С18, 5 мкм; подвижная фаза А: 0,03%-ный гидроксид аммония в воде (об./об.); подвижная фаза В: 0,03%-ный гидроксид аммония в ацетонитриле (об./об.); градиент: увеличение процентного содержания В, начиная с 10% или 20% В).
Метод М2: Алкилирование фенолов
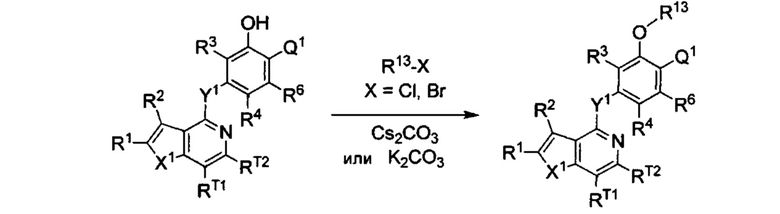
Раствор соответствующего фенола (0,050 ммоль, 1,0 экв.) в безводном N,N-диметилформамид-диметилацетале или N,N-диметилформамиде (0,2 мл) обрабатывали карбонатом цезия или карбонатом калия (0,10 ммоль, 2,0 экв.), йодидом натрия (0,008 ммоль, 0,2 экв.) и соответствующим бромидным или хлоридным реагентом (0,075 ммоль, 1,5 экв.). Реакционный сосуд закрывали крышкой и встряхивали при 80°C в течение 16 часов. Реакционную смесь концентрировали и неочищенный остаток очищали посредством HPLC с обращенной фазой (градиент: увеличение концентрации ацетонитрила в воде, содержащей 0,225%-ную муравьиную кислоту, или ацетонитрила в водном растворе гидроксида аммония с рН 10) с получением конечного соединения.
Метод М3: Образование амида с использованием гексафторфосфата 0-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония
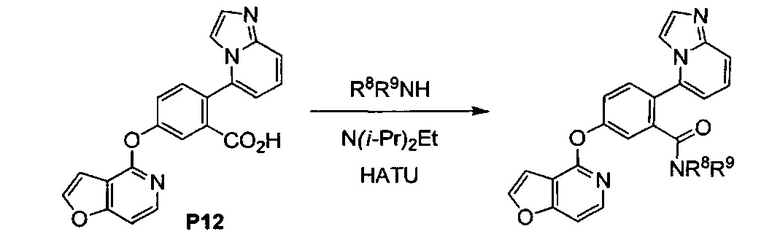
Раствор 5-(фуро[3,2-с]пиридин-4-илокси)-2-(имидазо[1,2-а]пиридин-5-ил)бензойной кислоты (Р12) (0,060 ммоль, 1,0 экв.) в безводном N,N-диметилформамиде (0,2 мл) обрабатывали соответствующим имеющимся в продаже амином (0,090 ммоль, 1,5 экв.), гексафторфосфатом 0-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (HATU, 0,060 ммоль, 1,0 экв.) и диизопропилэтиламином (0,240 ммоль, 4,0 экв.). Реакционный сосуд закрывали крышкой и встряхивали при 30°C в течение 16 часов. Реакционную смесь концентрировали и неочищенный остаток очищали HPLC с обращенной фазой (градиент: увеличение концентрации ацетонитрила в воде, содержащей 0,225%-ную муравьиную кислоту, или ацетонитрила в водном растворе гидроксида аммония с рН 10) с получением конечного соединения.
Метод М4: Реакция Мицунобу с фенолами

Раствор 5-(фуро[3,2-с]пиридин-4-илокси)-2-(2-метилпиридин-3-ил)фенола (полученного посредством отщепления метилового эфира Примера 181) (0,075 ммоль, 1,0 экв.) в смеси тетрагидрофуран/дихлорметан (об./об., 1:1, 1,0 мл) добавляли во флакон, содержащий соответствующий имеющийся в продаже первичный спирт (0,120 ммоль, 1,6 экв.) и трифенилфосфин на полимерной подложке (0,225 ммоль, 3,0 экв.). В реакционный сосуд добавляли диизопропилазодикарбоксилат (DIAD; 0,150 ммоль, 2,0 экв.), который затем закрывали крышкой и встряхивали при 30°C в течение 16 часов. Реакционную смесь концентрировали и неочищенный остаток очищали HPLC с обращенной фазой (градиент: увеличение концентрации ацетонитрила в воде, содержащей 0,225%-ную муравьиную кислоту, или ацетонитрила в водном растворе гидроксида аммония с рН 10) с получением конечного соединения.
Метод М5: Восстановительное аминирование альдегидов
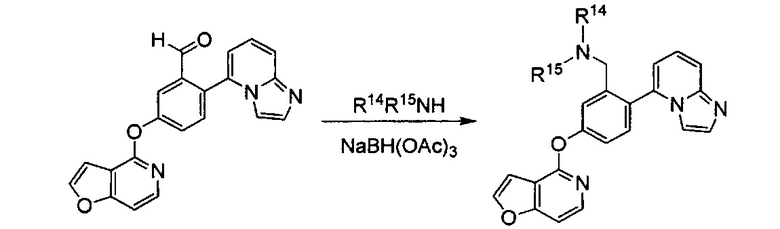
Раствор 5-(фуро[3,2-с]пиридин-4-илокси)-2-(имидазо[1,2-а]пиридин-5-ил)бензальдегида [полученный из 4-бром-3-(1,3-диоксан-2-ил)фенола (см. F. Kaiser et al., J. Org. Chem. 2002, 67, 9248-9256) с использованием методик Примера 1 с последующим снятием защиты с помощью водной соляной кислоты в тетрагидрофуране] (0,094 ммоль, 1,25 экв.) в дихлорметане (1,0 мл) добавляли во флакон, содержащий соответствующий имеющийся в продаже амин (0,075 ммоль, 1,0 экв.). Добавляли бикарбонат натрия (18 мг, 0,225 ммоль, 3,0 экв.) и реакционный сосуд закрывали крышкой и встряхивали при 30°C в течение 16 часов. Добавляли триацетоксиборогидрид натрия (47 мг, 0,225 ммоль, 3,0 экв.) и реакционную смесь встряхивали при 30°C в течение дополнительных 5 часов. Реакционную смесь концентрировали и неочищенный остаток очищали HPLC с обращенной фазой (градиент: увеличение концентрации ацетонитрила в воде, содержащей 0,1%-ную трифторуксусную кислоту) с получением конечного соединения.
Метод М6: Аминозамещение гетероарилхлоридов
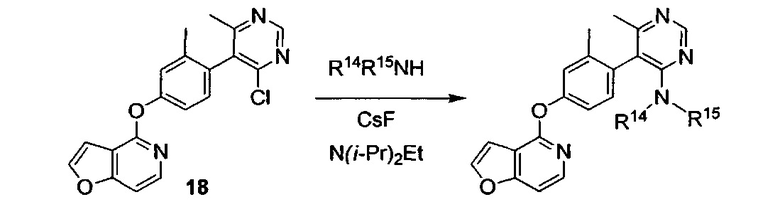
Раствор 4-[4-(4-хлор-6-метилпиримидин-5-ил)-3-метилфенокси]фуро[3,2-c]пиридина (Пример 18) (0,50 ммоль, 1,0 экв.) в безводном диметилсульфоксиде (0,5 мл) добавляли во флакон, содержащий соответствующий имеющийся в продаже амин (0,110 ммоль, 2,2 экв.). Добавляли диизопропилэтиламин (0,170 ммоль, 3,4 экв.) и фторид цезия (15 мг, 0,100 ммоль, 2,0 экв.) и реакционный сосуд закрывали крышкой и встряхивали при 120°C в течение 16 часов. Реакционную смесь концентрировали и неочищенный остаток очищали HPLC с обращенной фазой (градиент: увеличение концентрации ацетонитрила в воде, содержащей 0,225%-ную муравьиную кислоту или 0,1%-ную трифторуксусную кислоту) с получением конечного соединения.
Метод М7: Микробное окисление с использованием Pseudomonas putida
Стадия 1. Получение биокатализатора
Флакон с замороженным посевным материалом, содержащий Pseudomonas putida (АТСС 17453), извлекали из рефрижератора с температурой -80°C, оттаивали и использовали для инокуляции среды IOWA (1 л; среда IOWA состоит из глюкозы [20 г], хлорида натрия [5 г], гидрофосфата калия [5 г], соевой муки [5 г] и дрожжевого экстракта [5 г]; рН смеси доводили до 7,0 перед стерилизацией в автоклаве) в 3-литровой качалочной колбе с дефлекторами (Corning, #431253). Культуры выращивали в течение 2-4 суток, одновременно встряхивая при 30°C и 160 об/мин на 2-дюймовой круговой качалке. Клетки собирали центрифугированием; клеточный осадок замораживали при -80°C.
Стадия 2. Окислительная реакция
Клетки Pseudomonas putida (АТСС 17453) суспендировали в водном буфере фосфата калия (25 мМ, рН 7,0) при концентрации 45 г клеток на 150 мл буфера. Эту суспензию добавляли в 1-литровую качалочную колбу с дефлекторами (Nalge, 4116-1000) и к суспензии добавляли раствор субстрата (30 мг) в диметилсульфоксиде (3 мл). Колбу инкубировали при 30-40°C и 300 об/мин в течение 24-96 часов на 1-дюймовой круговой качалке.
Стадия 3. Обработка реакционной смеси
Реакционную смесь экстрагировали этилацетатом и объединенные органические слои концентрировали в вакууме. Продукт выделяли с использованием хроматографических методик.
Таблица 1. Примеры 31-208
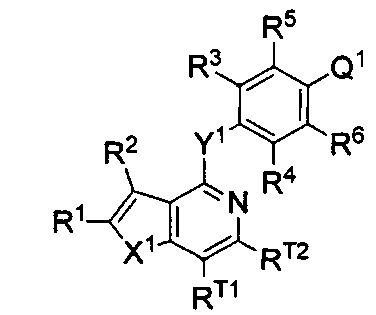
каждый из R1, R2, RT1 и RT2 представляет собой Н; и Х1=0
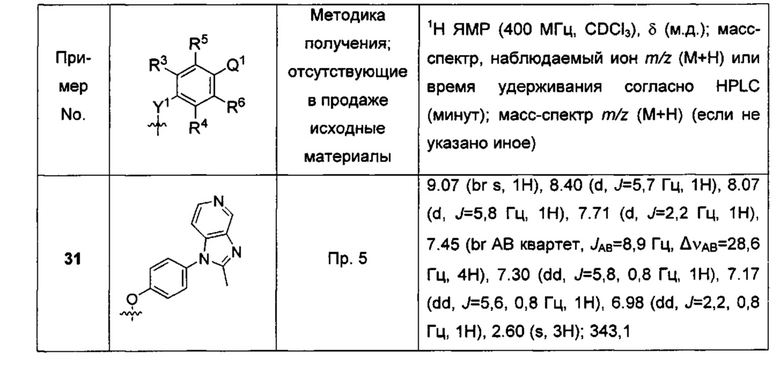
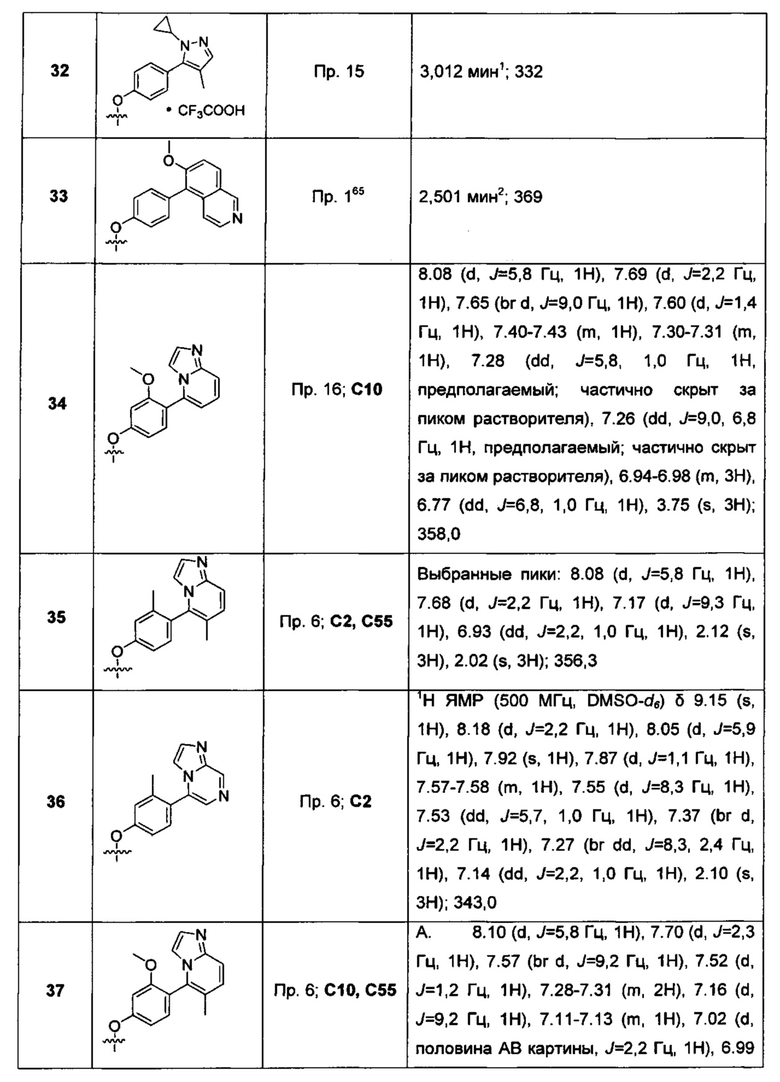
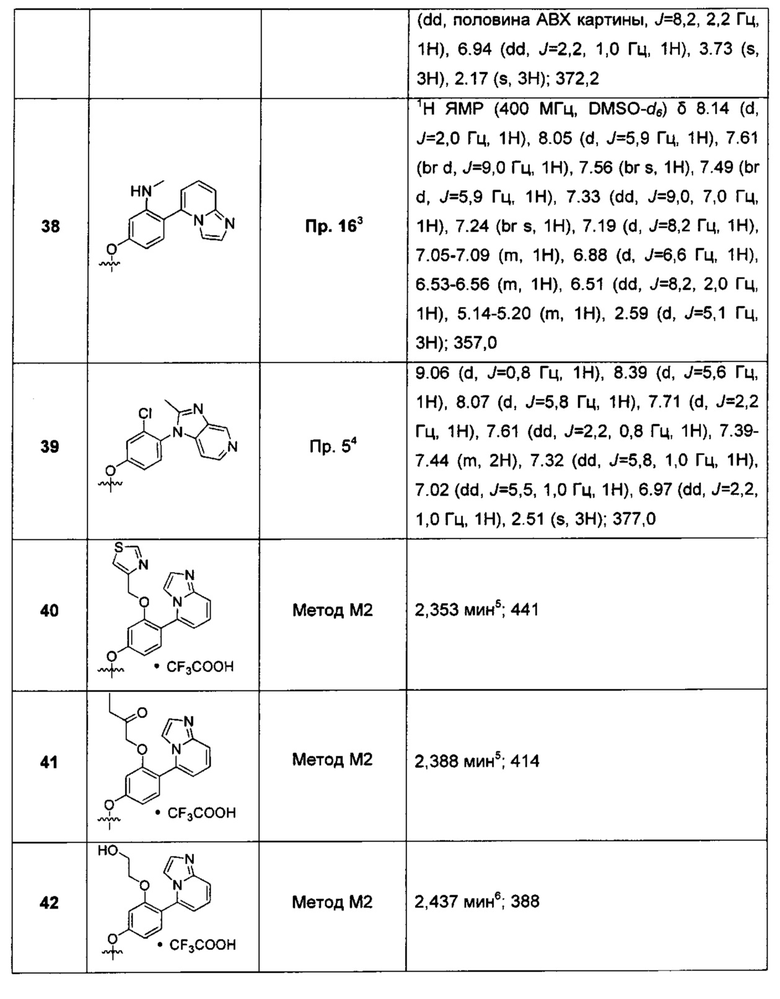
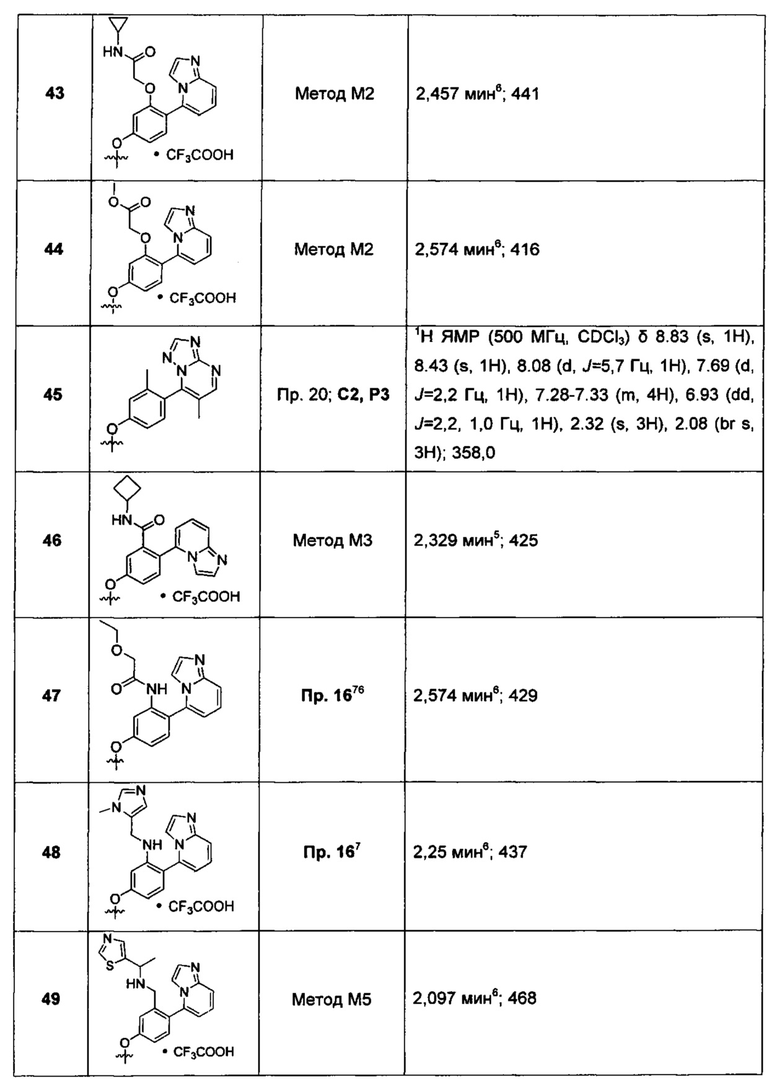
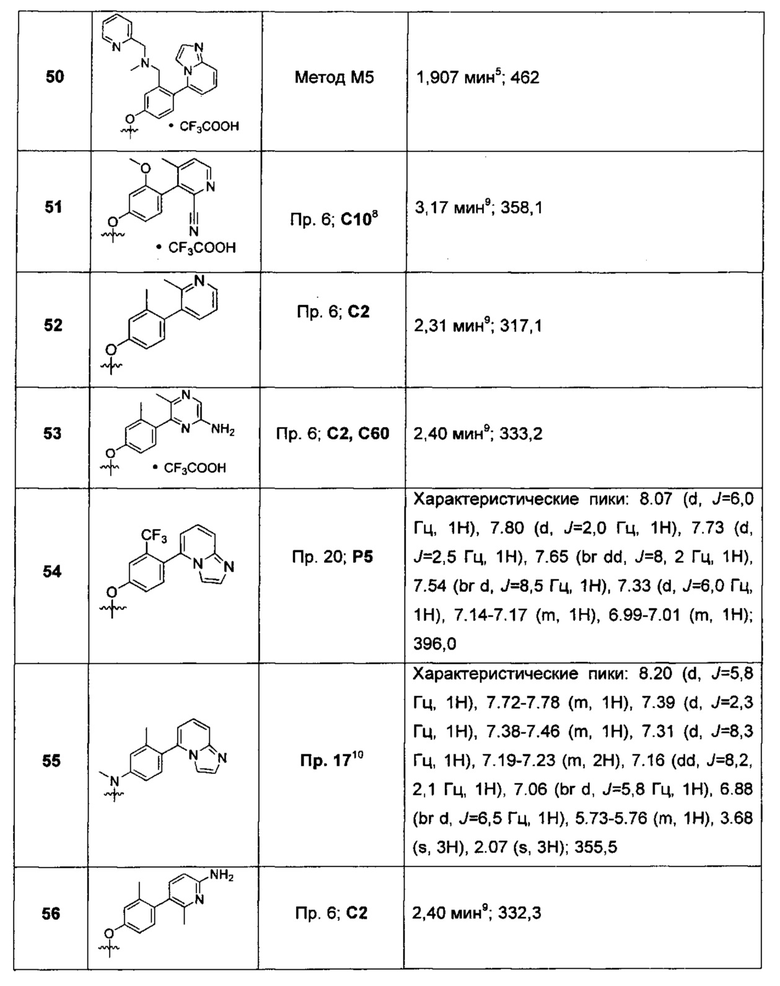
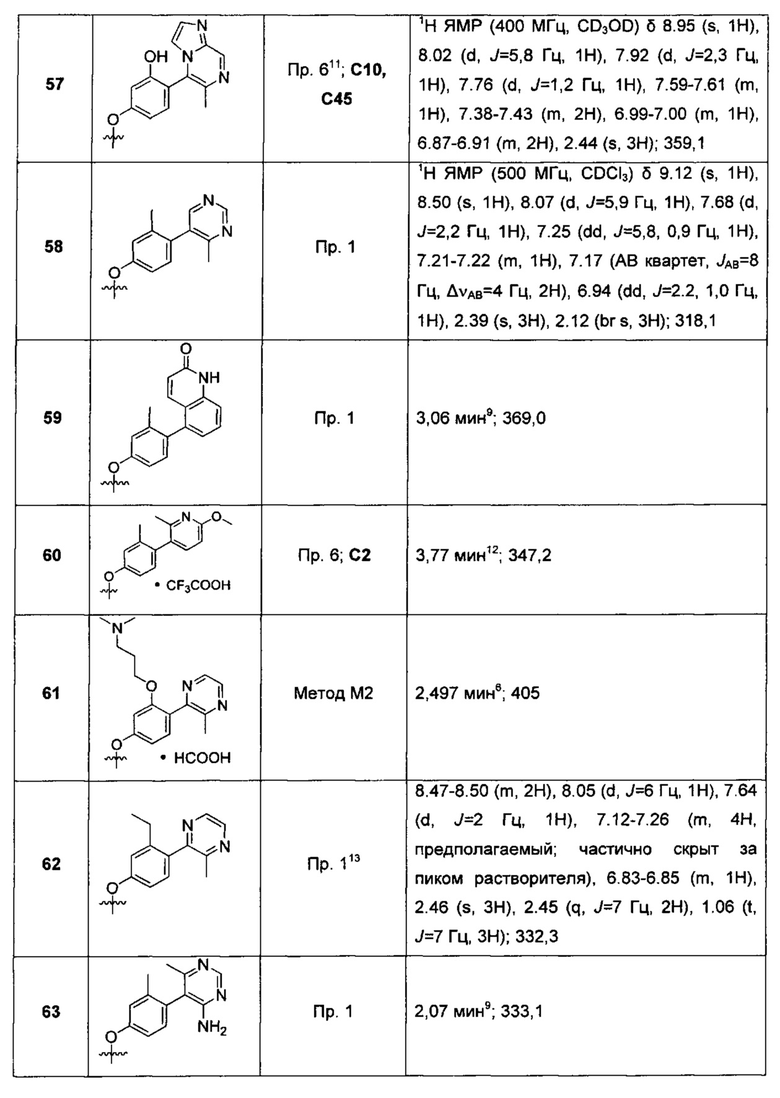
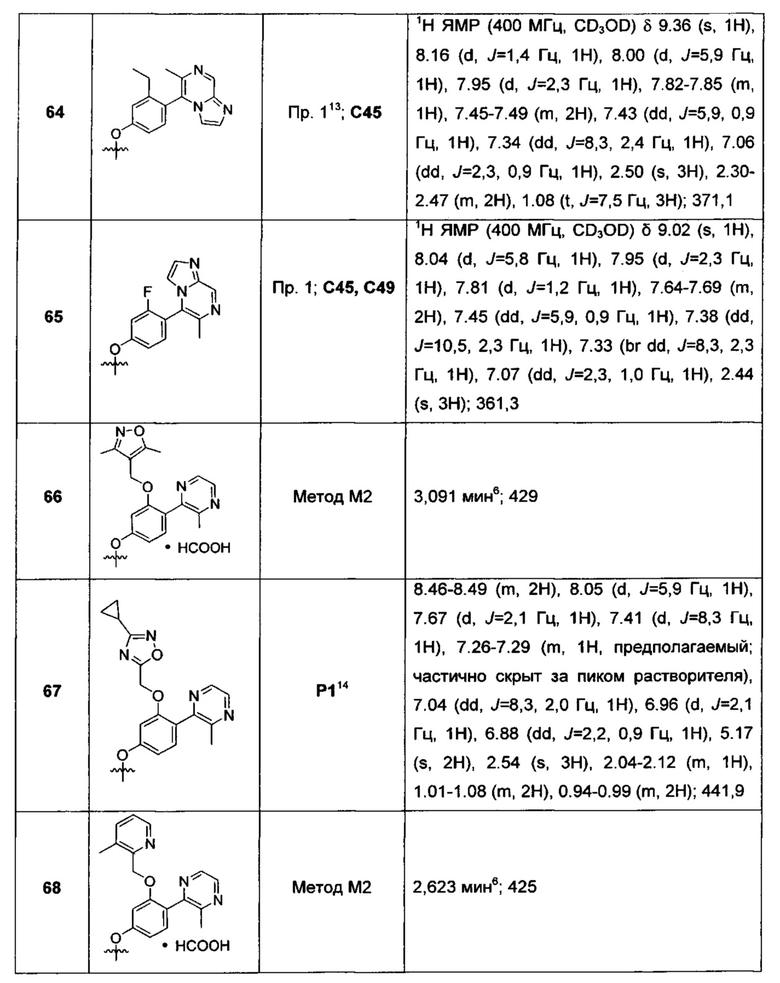
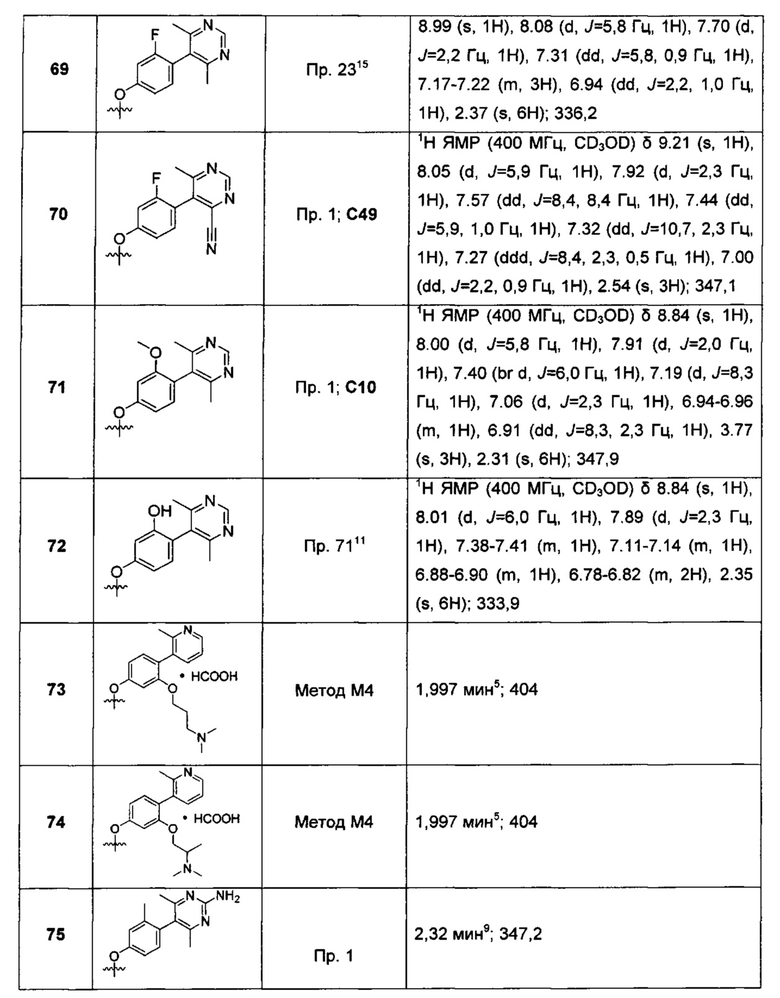
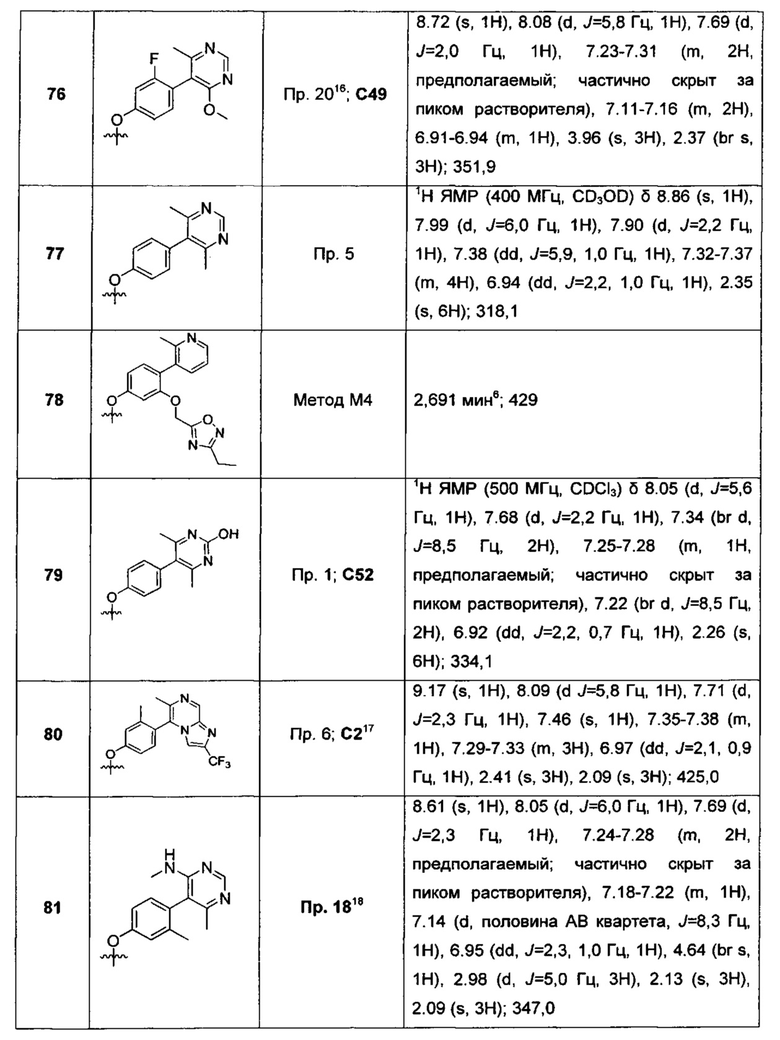
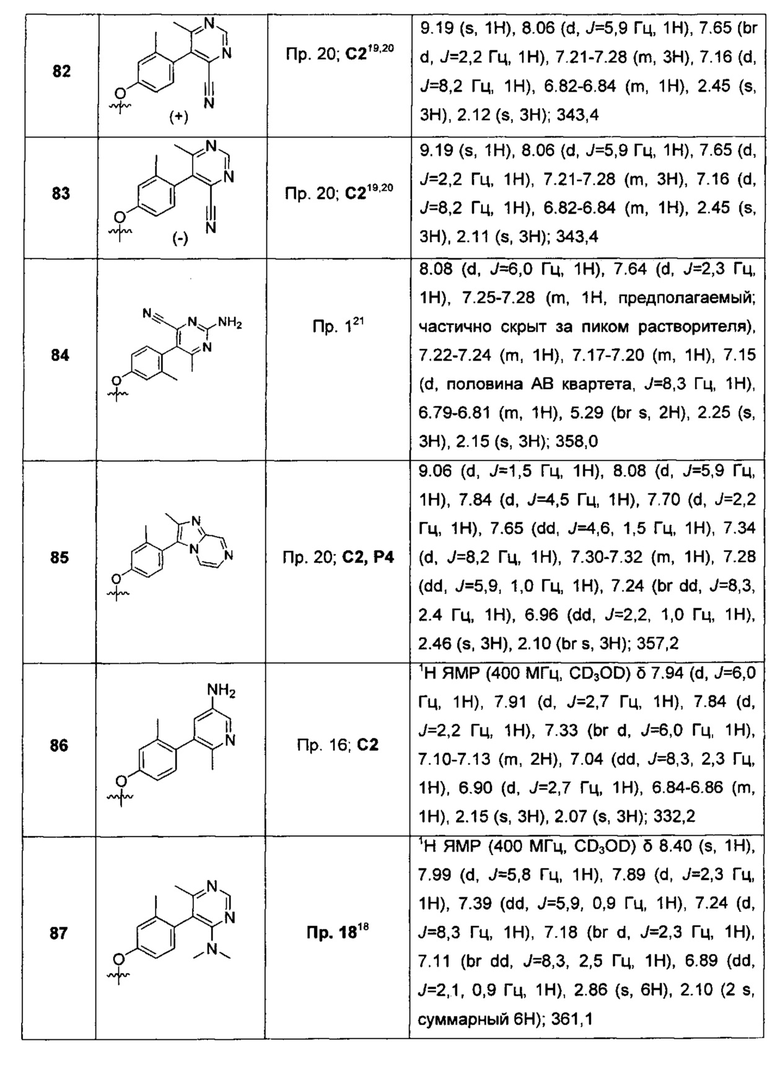
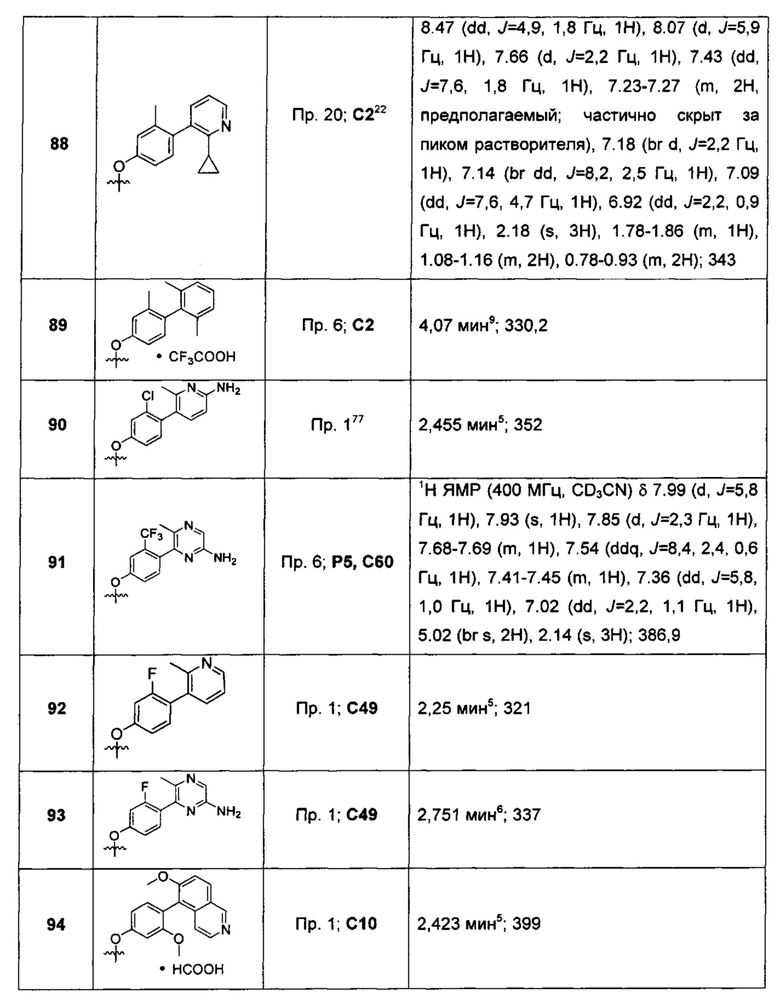
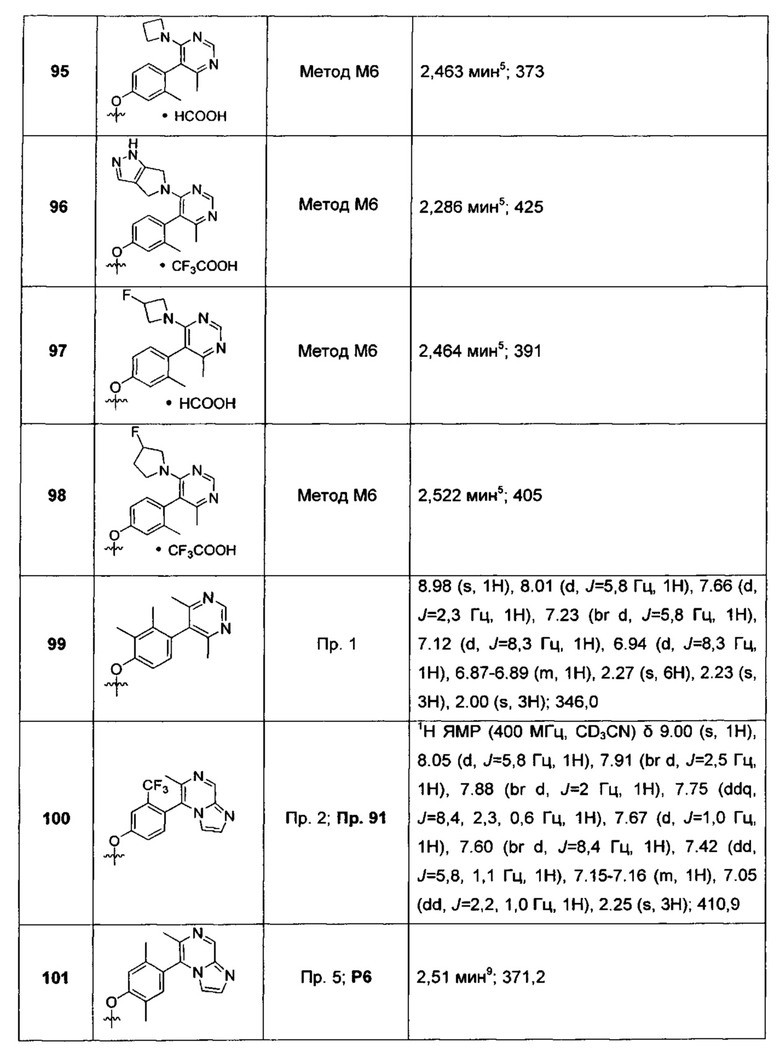
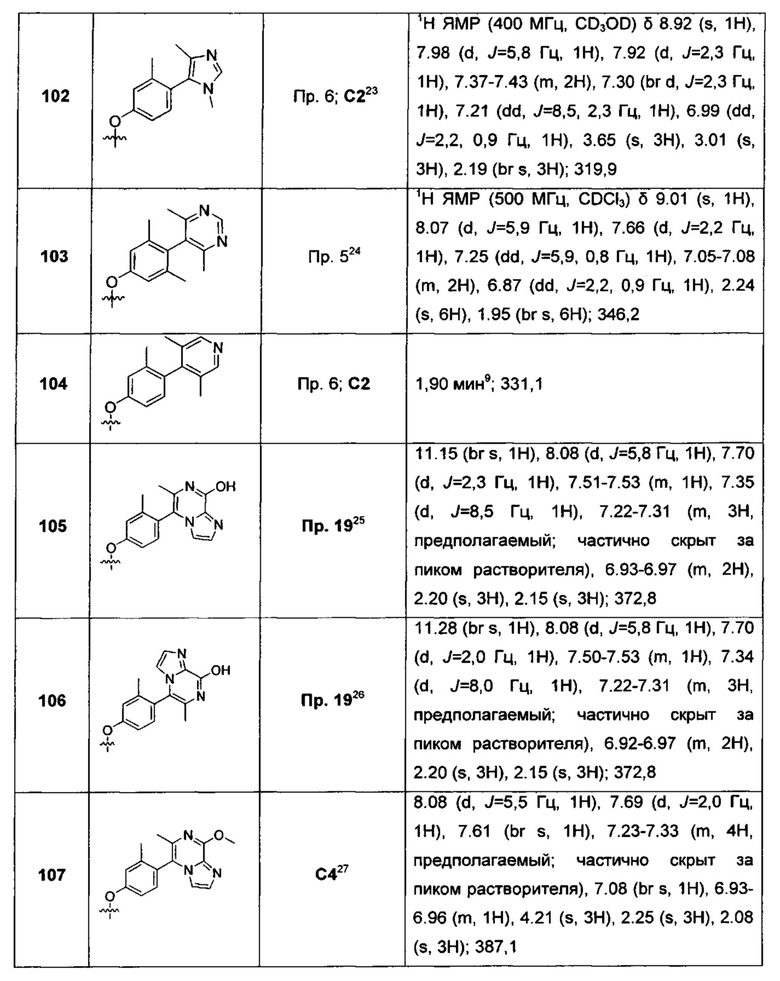


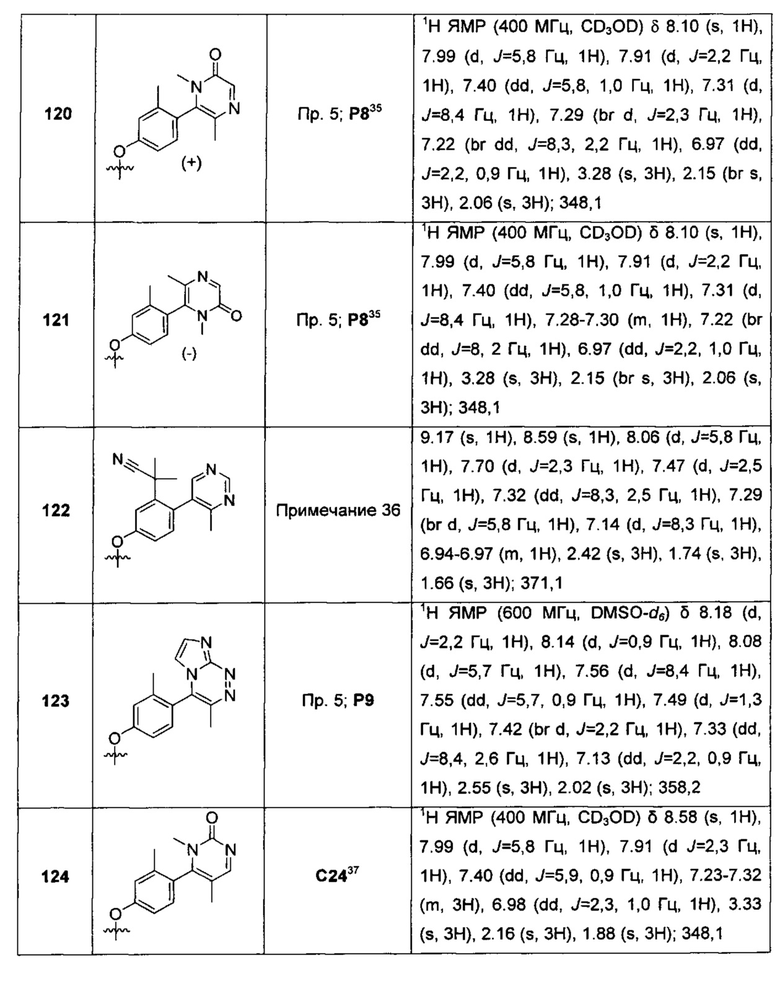
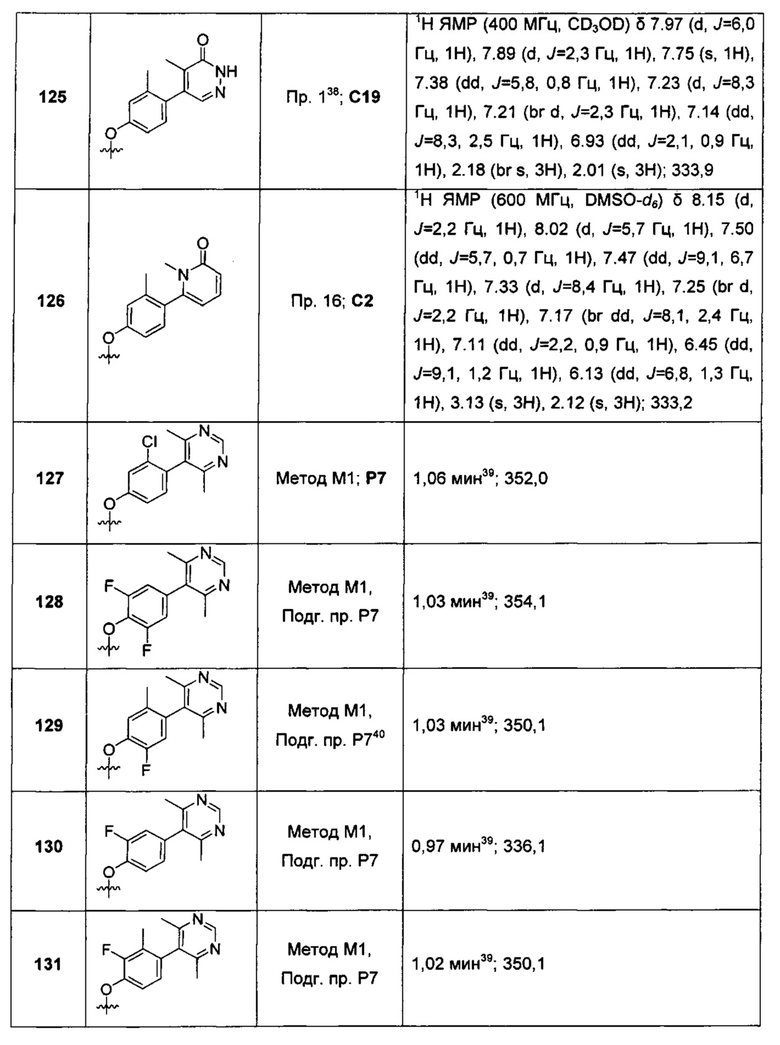
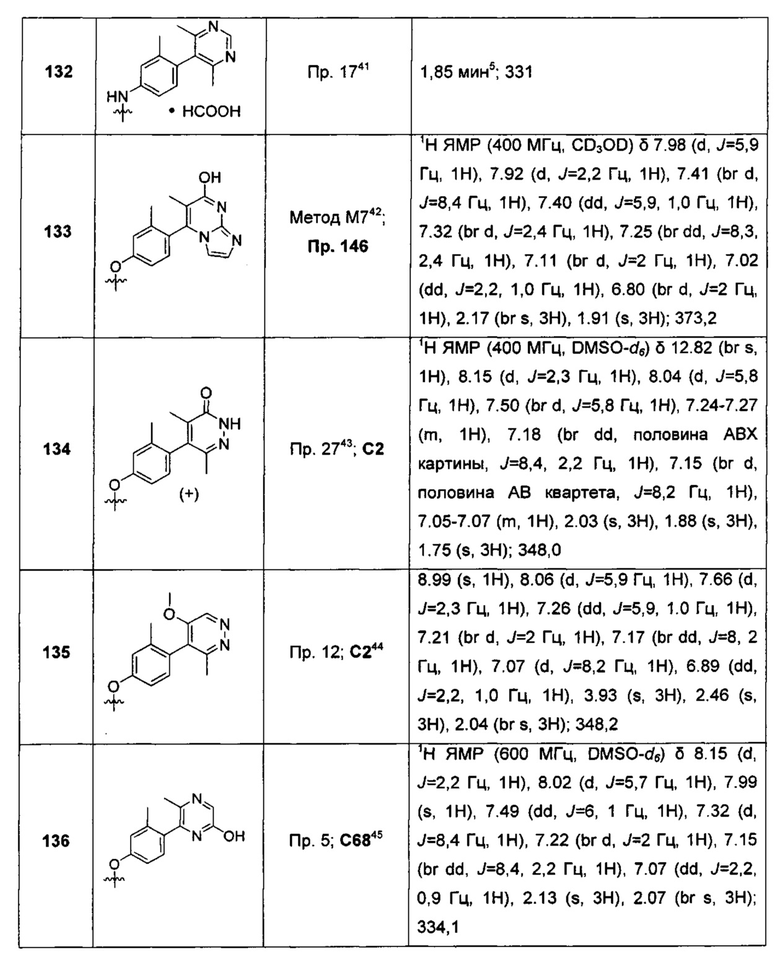
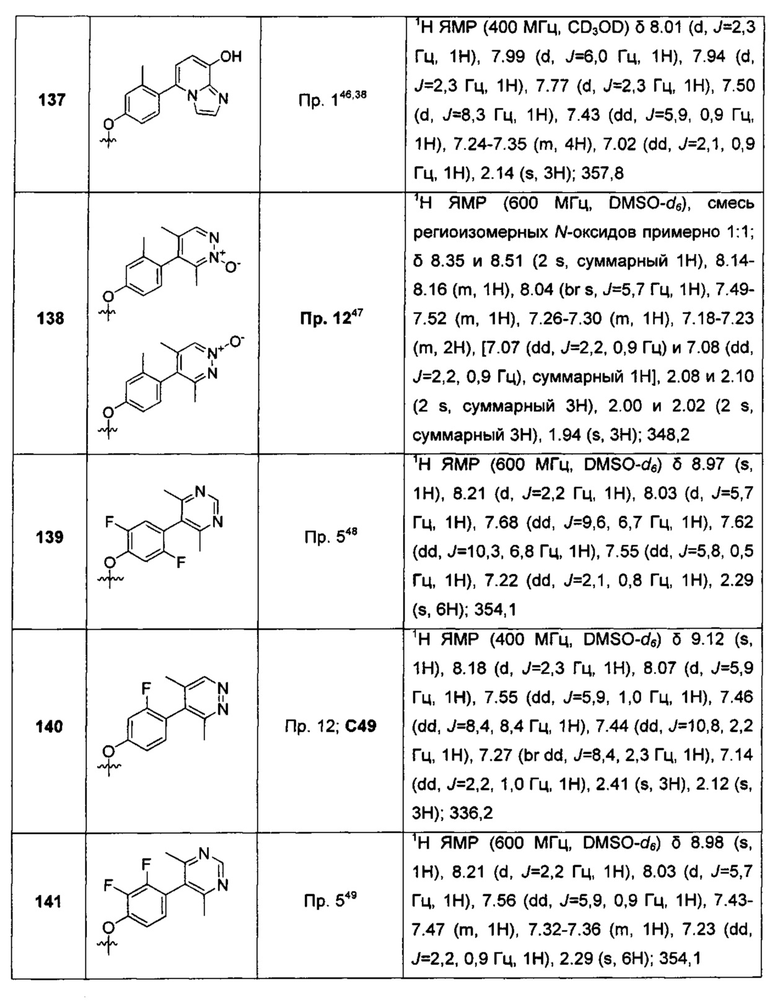
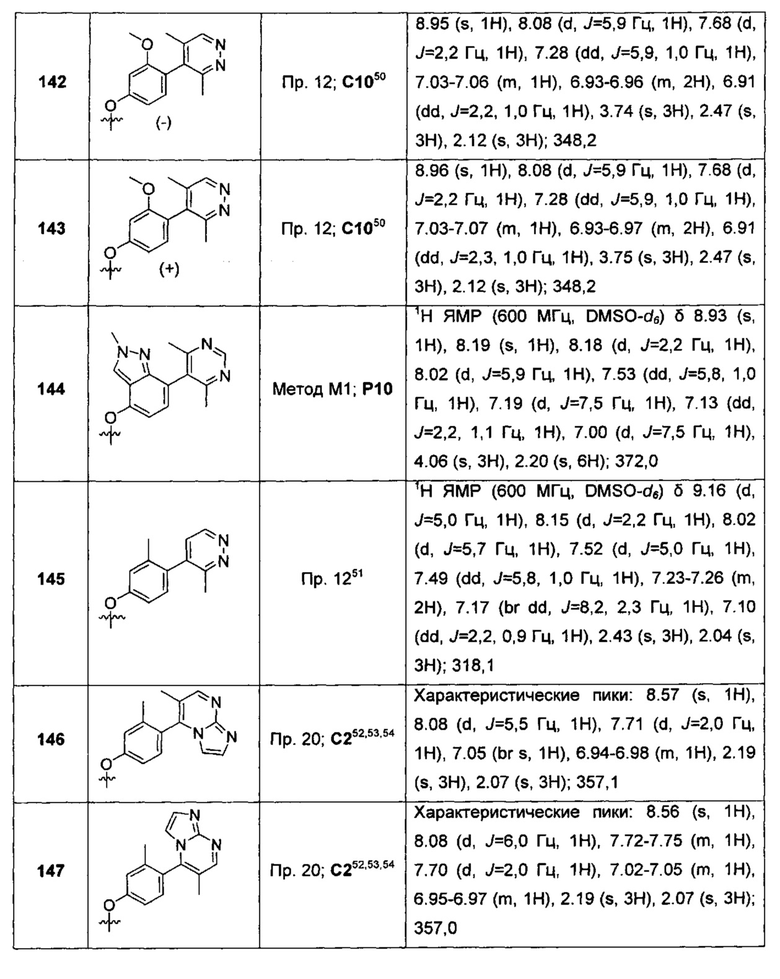

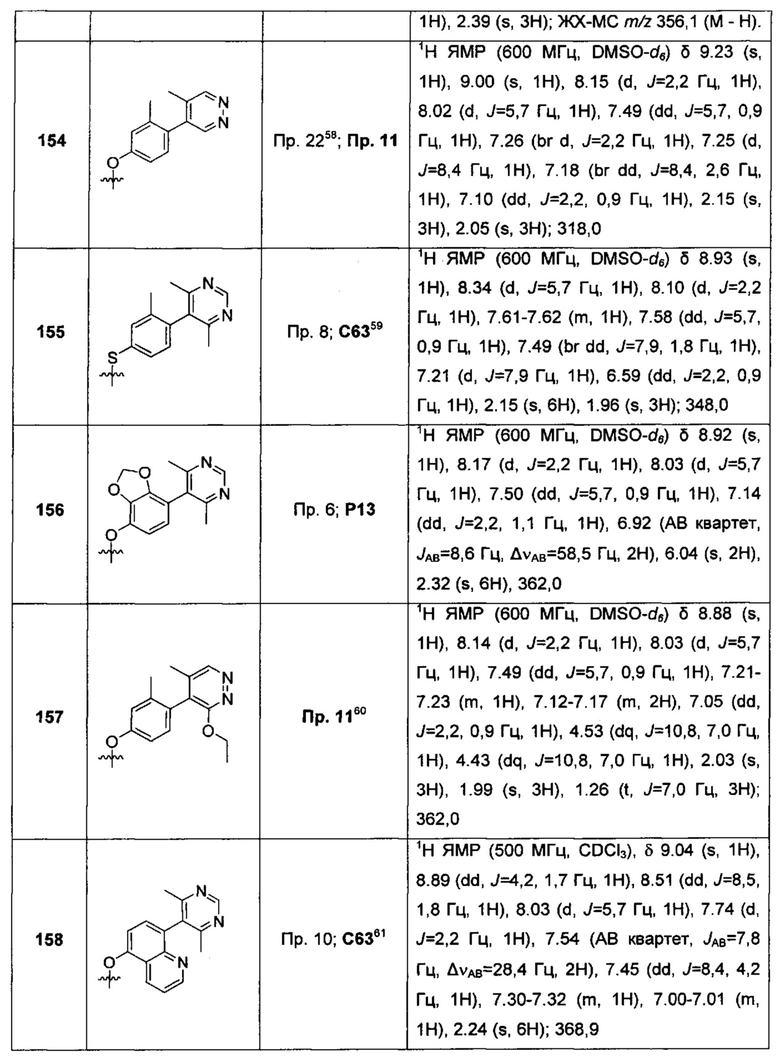
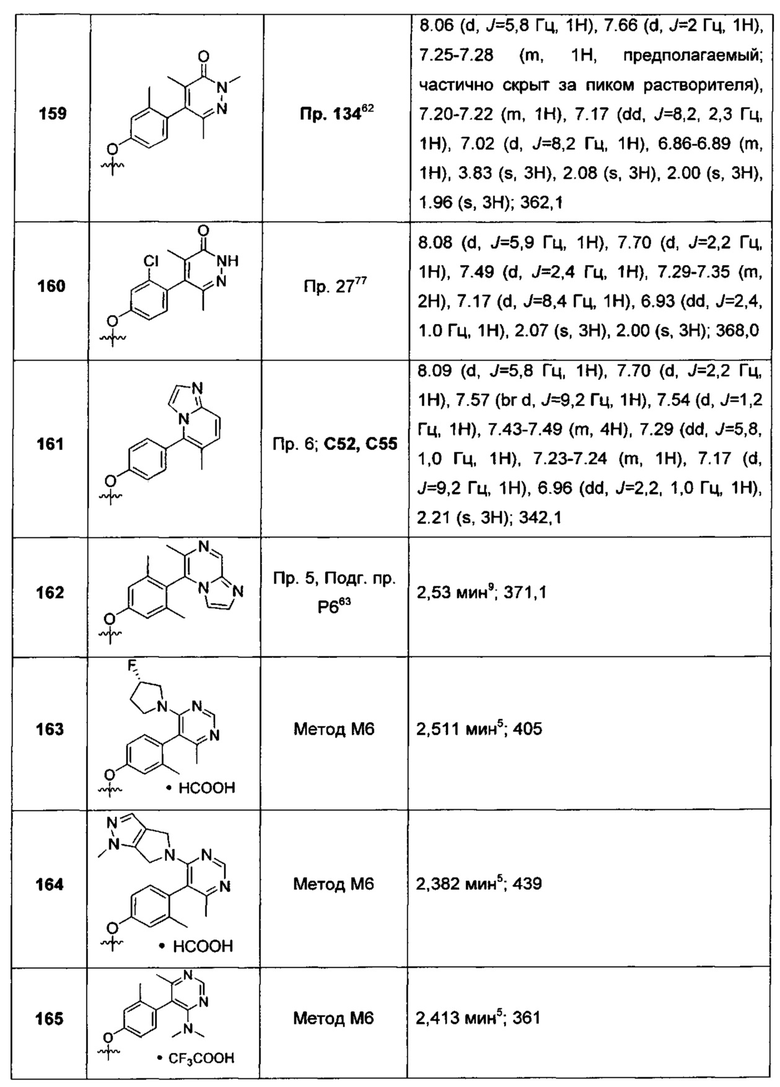
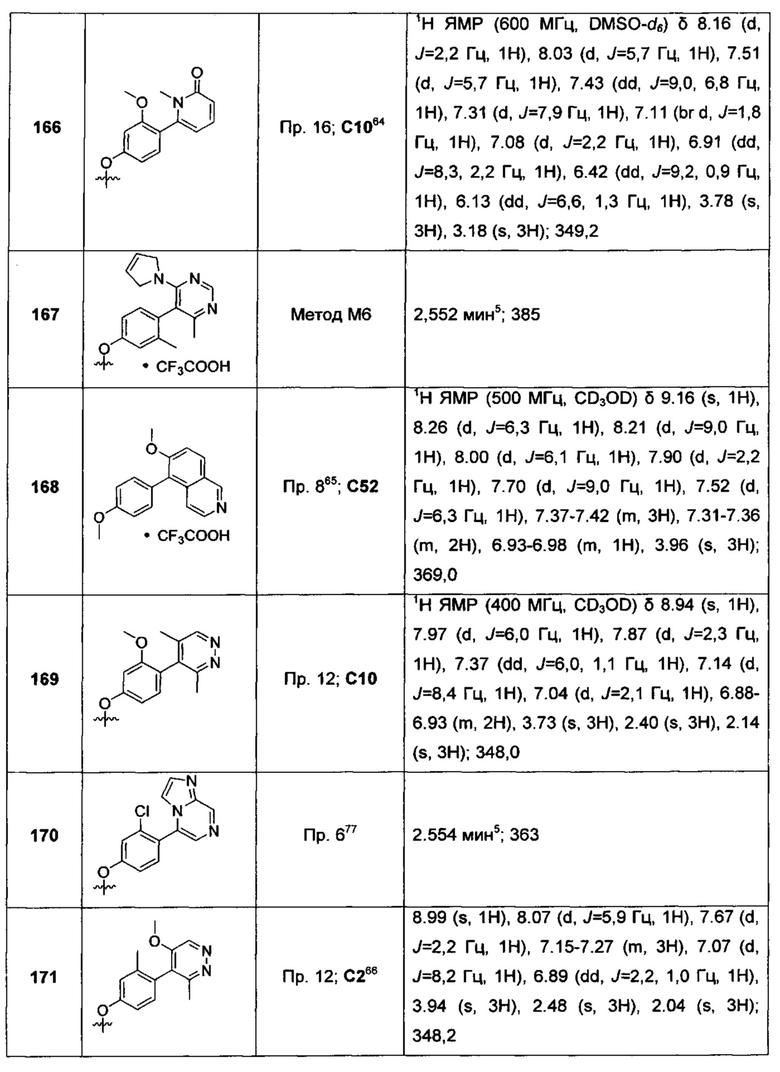
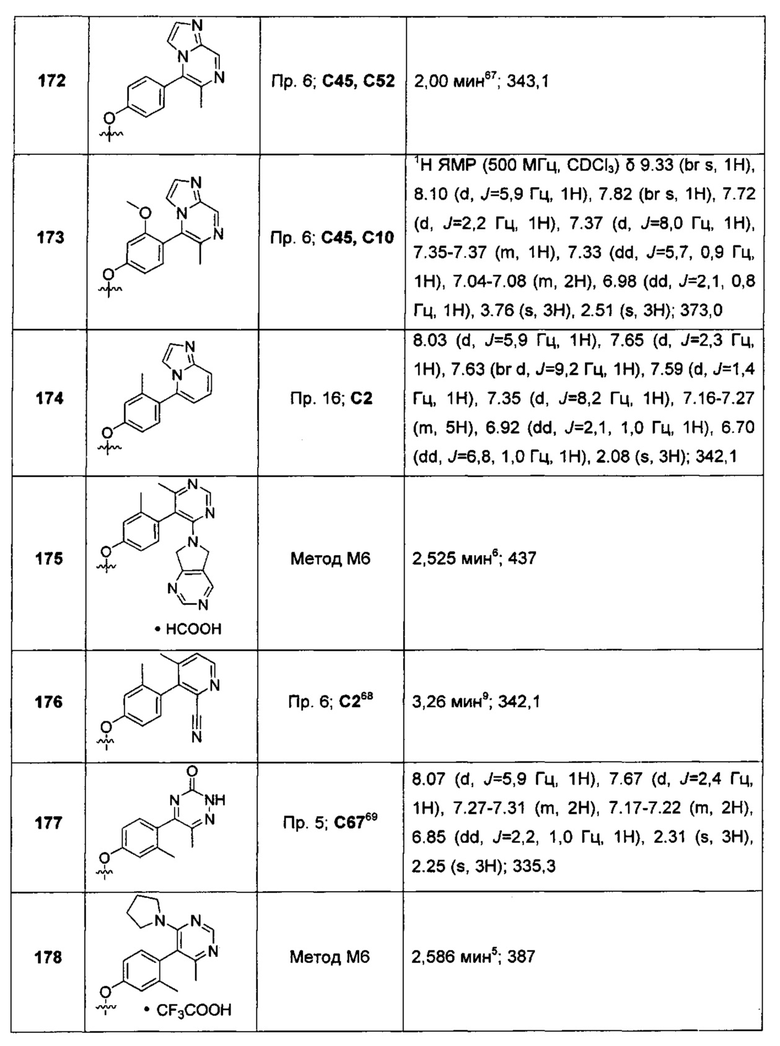
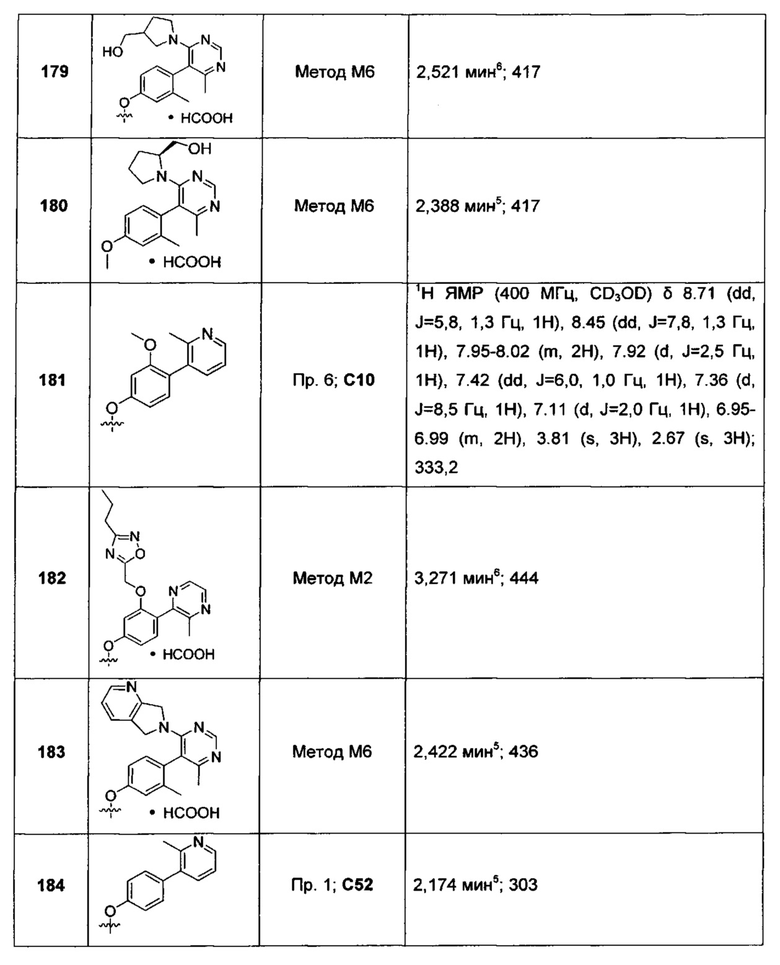
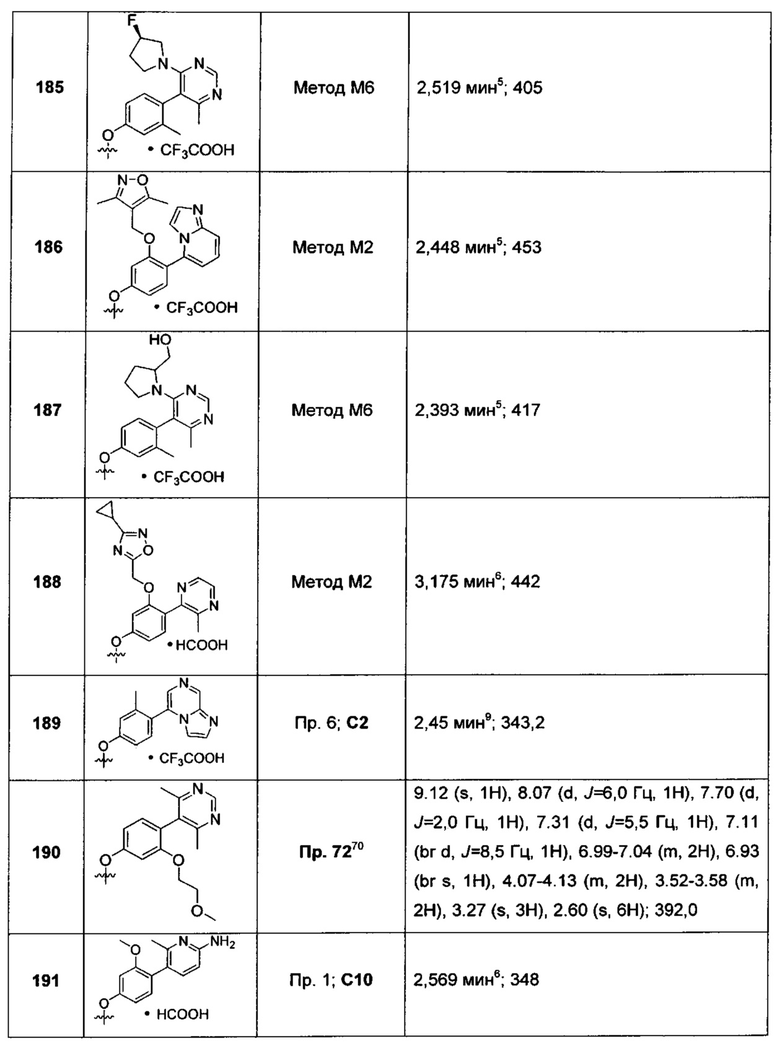


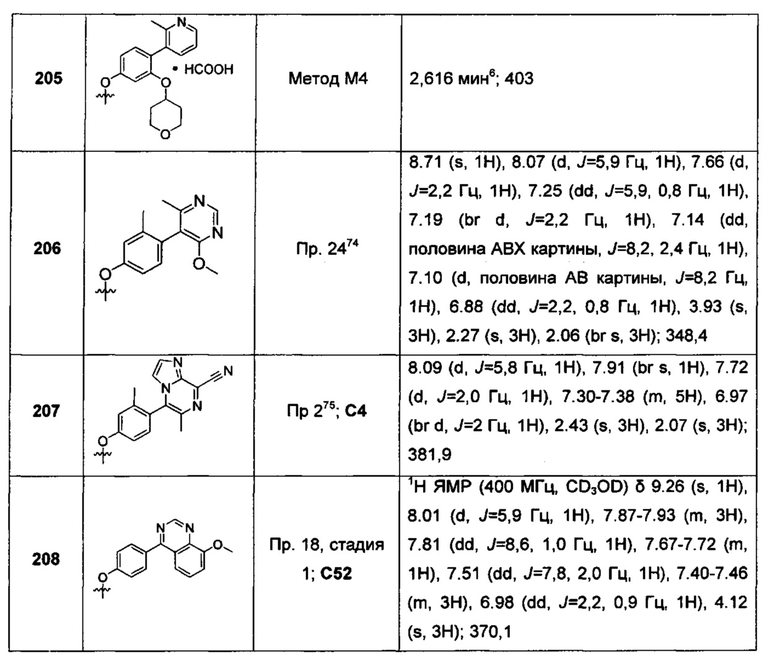
1. Условия HPLC. Колонка: Welch ХВ-С18, 2,1×50 мм, 5 мкм; подвижная фаза А: 0,05%-ный гидроксид аммония в воде (об./об.); подвижная фаза В: ацетонитрил.
2. Условия HPLC. Колонка: Welch ХВ-С18, 2,1×50 мм, 5 мкм; подвижная фаза А: 0,05%-ная трифторуксусная кислота в воде (об./об.); подвижная фаза В: ацетонитрил.
3. Соединение Примера 16 N-формилировали с получением N-[5-(фуро[3,2-с]пиридин-4-илокси)-2-(имидазо[1,2-а]пиридин-5-ил)фенил]-формамида путем нагревания в метилформиате в присутствии гидрида натрия и [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия(II). Восстановление с помощью боран-диметилсульфидного комплекса дало соединение Примера 38.
4. В данном случае 4-амино-3-хлорфенол использовали в качестве исходного материала, и этот фенол в конструкции имидазо[4,5-с]пиридина не был защищен.
5. Условия HPLC. Колонка: Waters XBridge С18, 2,1×50 мм, 5 мкм; подвижная фаза А: 0,0375%-ная трифторуксусная кислота в воде; подвижная фаза В: 0,01875%-ная трифторуксусная кислота в ацетонитриле; градиент: от 10% до 100% В за 4,0 минуты; скорость потока: 0,8 мл/мин.
6. Условия HPLC. Колонка: Waters XBridge С18, 2,1×50 мм, 5 мкм; подвижная фаза А: 0,0375%-ная трифторуксусная кислота в воде; подвижная фаза В: 0,01875%-ная трифторуксусная кислота в ацетонитриле; градиент: от 1% до 5% В за 0,6 минут, затем от 5% до 100% В за 3,4 минут; скорость потока: 0,8 мл/мин.
7. Соединение этого примера получали восстановительным аминированием соединения Примера 16 с 1-метил-1Н-имидазол-5-карбальдегидом.
8. Партнер сочетания 3-бром-4-метилпиридин-2-карбонитрила может быть получен из 3-бром-4-метилпиридина образованием пиридин-N-оксида посредством взаимодействия с пероксидом водорода с последующим цианированием согласно способу Т. Sakamoto et al., Chem. Pharm. Bull. 1985, 33, 565-571.
9. Условия HPLC. Колонка: Waters Atlantis dC18, 4,6×50 мм, 5 мкм; подвижная фаза А: 0,05%-ная трифторуксусная кислота в воде (об./об.); подвижная фаза В: 0,05%-ная трифторуксусная кислота в ацетонитриле (об./об.); градиент: от 5,0% до 95% В за 4,0 минуты, линейный; скорость потока: 2 мл/мин.
10. Соединение Примера 17 подвергали N-метилированию с использованием гидрида натрия и метилйодида.
11. Конечная стадия в синтезе представляла собой расщепление метилового эфира с использованием трибромида бора.
12. Условия HPLC. Колонка: Waters XBridge С18, 4,6×50 мм, 5 мкм; подвижная фаза А: 0,03%-ный гидроксид аммония в воде (об./об.); подвижная фаза В: 0,03%-ный гидроксид аммония в ацетонитриле (об./об.); от 5,0% до 95% В за 4,0 минуты, линейный; скорость потока: 2 мл/мин.
13. В данном случае сочетание Сузуки осуществляли с использованием тетракис(трифенилфосфин)палладия(0) и карбоната калия или карбоната натрия.
14. Исходный материал ал кил провал и с использованием 5-(хлорметил)-3-циклопропил-1,2,4-оксадиазола и карбоната цезия.
15. 1-Бром-2-фтор-4-метоксибензол использовали в качестве исходного материала.
16. 5-Бром-4-метокси-6-метилпиримидин получали путем взаимодействия 5-бром-4-хлор-6-метилпиримидина с метилатом натрия.
17. Требуемый 5-бром-6-метил-2-(трифторметил)имидазо[1,2-а]пиразин получали посредством взаимодействия С60 с 3-бром-1,1,1-трифторпропан-2-оном.
18. Соединение Примера 18 обрабатывали соответствующим амином.
19. Требуемый 5-бром-6-метилпиримидин-4-карбонитрил получали посредством взаимодействия 5-бром-4-хлор-6-метилпиримидина с цианидом тетра-н-бутиламмония.
20. Продукт разделяли на его составляющие атропоэнантиомеры с использованием сверхкритической флюидной хроматографии (колонка: Chiralpak AD-H, 5 мкм; элюент: смесь диоксид углерода/пропанол в соотношении 3:1). Элюирующееся первым соединение представляло собой соединение Примера 83, а элюирующееся вторым соединение представляло собой соединение Примера 82.
21. Требуемый 2-амино-5-бром-6-метилпиримидин-4-карбонитрил может быть получен посредством взаимодействия 5-бром-4-хлор-6-метилпиримидин-2-амина с цианидом тетраэтиламмония и 1,4-диазабицикло[2.2.2]октаном в смеси ацетонитрила и N,N-диметилформамида.
22. Требуемый 3-бром-2-циклопропилпиридин получали посредством взаимодействия 2,3-дибромпиридина с циклопропилбороновой кислотой при 100°C в присутствии ацетата палладия(II), трициклогексилфосфина и фосфата калия.
23. Требуемый 5-бром-1,4-диметил-1Н-имидазол может быть получен посредством метилирования 5-бром-4-метил-1H-имидазола с использованием гидрида натрия и метилйодида.
24. Реакция Сузуки (4-метокси-2,6-диметилфенил)бороновой кислоты с 5-бром-4,6-диметилпиримидином, опосредованная трис(дибензилиденацетон)-дипалладием(0) и дициклогексил(2,6,-диметоксибифенил-2-ил)фосфаном, с последующим отщеплением метилового эфира дала требуемый фенол.
25. Получили в результате сверхкритического жидкостного хроматографического разделения соединения Примера 19 [колонка: Chiralcel AS, 20 мкм; подвижная фаза: смесь 7:3 диоксид углерода/(метанол, содержащий 0,2%-ный диэтиламин)]. Это соединение Примера представляло собой атропоэнантиомер, элюирующийся с колонки вторым.
26. Это соединение представляло собой элюирующийся вторым атропоэнантиомер из разделения, описанного в подпункте 25.
27. Соединение С4 нагревали с водным хлорацетальдегидом при дефлегмации в течение 2 часов с получением 8-бром-5-[4-(фуро[3,2-с]пиридин-4-илокси)-2-метилфенил]-6-метилимидазо[1,2-а]пиразина. Взаимодействие этого промежуточного соединения с метилатом натрия в метаноле дало соединение Примера 107.
28. 8-Бром-промежуточное соединение из подпункта 27 подвергали взаимодействию с триметилбороксином в присутствии [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия(Н) и карбоната калия с получением соединения Примера 109.
29. Взаимодействие хлорацетальдегида с 2-амино-5-метилпиримидин-4-олом дало смесь 6-метилимидазо[1,2-а]пиримидин-5-ола и 6-метилимидазо[1,2-а]пиримидин-7-ола, которую подвергали взаимодействию с оксихлоридом фосфора с получением смеси 5-хлор-6-метилимидазо[1,2-а]пиримидина и 7-хлор-6-метилимидазо[1,2-а]пиримидина. Взаимодействие этой смеси с С2 дало смесь соединений Примеров 110 и 111, которую можно разделить. Структуры этих двух соединений затем устанавливали согласно NOE исследованиям, осуществленным на раздельных промежуточных соединениях: 6-метилимидазо[1,2-а]пиримидин-5-ол и 6-метилимидазо[1,2-a]пиримидин-7-ол.
30. 8-Бром-промежуточное соединение из подпункта 27 подвергали взаимодействию с трет-бутилкарбаматом в присутствии ацетата палладия(II), 1,1'-бинафталин-2,2'-диилбис(дифенилфосфана) и карбоната цезия при 120°C в течение 2 часов с получением соединения Примера 112.
31. Требуемый 4-(4-бром-3,5-дифторфенокси)фуро[3,2-с]пиридин получали из 4-хлорфуро[3,2-с]пиридина и 4-бром-3,5-дифторфенола с использованием общей методики стадии 3 Примера 17.
32. Соединение Примера 11 подвергали взаимодействию с гидразином. Полученный 4-[4-(3-гидразинил-5-метилпиридазин-4-ил)-3-метилфенокси]-фуро[3,2-с]пиридин циклизовали с 1,1'-карбонилдиимидазолом с получением продукта.
33. Соединение Примера 117 выделяли в виде побочного продукта в процессе синтеза соединений Примеров 120 и 121, производных из метилированной с избытком примеси в Р8.
34. Рацемическую модификацию соединения Примера 82 подвергали гидролизу с водным гидроксидом натрия в этаноле с получением продукта.
35. Рацемический продукт разделяли посредством сверхкритической флюидной хроматографии (колонка: Chiralcel OJ-H, 5 мкм; элюент: смесь диоксид углерода/метанол в соотношении 3:1). Соединение Примера 121 элюировалось первым с последующим элюированием соединения Примера 120.
36. (2-Хлор-5-метоксифенил)ацетонитрил (см. С.Pierre and О. Baudoin, Org. Lett. 2011, 13, 1816-1819) может быть диметилирован с использованием гидрида натрия и метилйодида с получением 2-(2-хлор-5-метоксифенил)-2-метилпропаннитрила. После реакции Сузуки с 4-метил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиримидином осуществляли отщепление метилового эфира с помощью натриевой соли этантиола, которое дало требуемый 2-[5-гидрокси-2-(4-метилпиримидин-5-ил)фенил]-2-метилпропаннитрил. Реакция с 4-хлорфуро[3,2-с]пиридином была опосредована трис(дибензилиденацетон)-дипалладием(О), трициклогексилфосфином и карбонатом цезия.
37. Соединение С24 подвергали взаимодействию с 1-метилмочевиной и лара-толуолсульфоновой кислотой с получением продукта.
38. Защитную группу удаляли на последней стадии с использованием раствора хлористого водорода в метаноле.
39. Условия HPLC: колонка: Acquity HSS ТЗ, 2,1×50 мм, 1,8 мкм; подвижная фаза А: 0,05%-ная трифторуксусная кислота в воде (об./об.); подвижная фаза В: 0,05%-ная трифторуксусная кислота в ацетонитриле (об./об.); градиент: от 5,0% до 98% В за 1,6 мин; скорость потока: 1,3 мл/мин.
40. Взаимодействие 1-фтор-2-метокси-4-метилбензола с N-бромсукцинимидом дало требуемый 1-бром-5-фтор-4-метокси-2-метилбензол.
41. В данном случае восстановление нитрогруппы в анилин достигалось путем гидрирования с использованием Pd/C в смеси этанола и метанола в соотношении 1:1. В последней реакции сочетания в качестве источника палладия использовали трис(дибензилиденацетон)дипалладий(0).
42. Неочищенную смесь метаболитов сначала очищали хроматографией на силикагеле (элюент: 10%-ный 2-пропанол в толуоле), затем подвергали HPLC разделению (колонка: Kromasil С18, 10 мкм; элюент: смесь метанол/вода в соотношении 3:2). Фракции продукта концентрировали в вакууме и водный остаток экстрагировали этилацетатом (2×50 мл). Объединенные органические слои концентрировали при пониженном давлении с получением продукта.
43. Рацемический продукт разделяли на атропоэнантиомеры посредством HPLC (колонка: Phenomenex Lux Cellulose-3, 5 мкм; градиент: от 5% до 95% этанола в гептане). Элюирующийся первым атропоэнантиомер представляет собой соединение этого Примера.
44. Соединение С2 подвергали сочетанию с 4-хлор-5-метокси-2-(тетрагидро-2Н-пиран-2-ил)пиридазин-3(2H)-оном, который может быть получен согласно В. Dyck et al„ J. Med. Chem. 2006, 49, 3753-3756, в присутствии [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия(Н) и карбоната цезия. Полученный 4-[4-(фуро[3,2-с]пиридин-4-илокси)-2-метилфенил]-5-метокси-2-(тетрагидро-2Н-пиран-2-ил)пиридазин-3(2H)-он превращали в продукт, используя способы Примеров 10, 11 и 12. Рацемический продукт разделяли на его составные атропоэнантиомеры с использованием сверхкритической флюидной хроматографии (колонка: Chiralpak AS-H, 5 мкм; элюент: смесь диоксид углерода/метанол в соотношении 3:1). Соединение Примера 135 представляло собой элюирующийся первым атропоэнантиомер.
45. Отщепление метилового эфира С68 с использованием трибромида бора дало требуемый 6-(4-гидрокси-2-метилфенил)-5-метилпиразин-2-ол.
46. Взаимодействие 2-амино-6-бромпиридин-3-ола с хлорацетальдегидом с последующим снятием защиты с использованием бензилхлорметилового эфира дало требуемый 8-[(бензилокси)метокси]-5-бромимидазо[1,2-а]пиридин.
47. Соединение Примера 12 подвергали взаимодействию с пероксидом водорода и малеиновым ангидридом с получением смеси в соотношении примерно 1:1 4-[4-(3,5-диметил-2-оксидопиридазин-4-ил)-3-метилфенокси]-фуро[3,2-с]пиридина и 4-[4-(3,5-диметил-1-оксидопиридазин-4-ил)-3-метилфенокси]фуро[3,2-с]пиридина.
48. 4-(4,6-Диметилпиримидин-5-ил)-2,5-дифторфенол получали из (2,5-дифтор-4-метоксифенил)бороновой кислоты и 5-бром-4,6-диметилпиримидина с использованием общей методики Примера 6 с последующим отщеплением метилового эфира.
49. 5-Бром-4,6-диметилпиримидин подвергали взаимодействию с (2,3-дифтор-4-метоксифенил)бороновой кислотой согласно общей методике синтеза 1 в Примере 1. Полученный 5-(2,3-дифтор-4-метоксифенил)-4,6-диметилпиримидин повергали снятию защиты с использованием трибромида бора с получением требуемого 4-(4,6-диметилпиримидин-5-ил)-2,3-дифторфенола.
50. Рацемический продукт разделяли посредством сверхкритической флюидной хроматографии (колонка: Chiralpak AS-H, 5 мкм; элюент: смесь диоксид углерод/метанол в соотношении 4:1). Соединение Примера 143 элюировалось первым, затем элюировалось соединение Примера 142.
51. Исходный материал 4-бром-2-(тетрагидро-2Н-пиран-2-ил)пиридазин-3(2Н)-он получали согласно С.Aciro et al.f РСТ Int. Appl. (2010) WO 2010131147 A1 20101118.
52. 2-Амино-5-метилпиримидин-4-ол подвергали взаимодействию с хлорацетальдегидом с получением 6-метилимидазо[1,2-а]пиримидин-5-ола; его подвергали хлорированию с использованием оксихлорида фосфора с получением требуемого 5-хлор-6-метилимидазо[1,2-а]пиримидина.
53. Хиральное разделение осуществляли с использованием сверхкритической флюидной хроматографии (колонка: Chiralpak AD-H, 5 мкм; элюент: смесь диоксид углерода/этанол в соотношении 65:35).
54. Анализ на Chiralpak AD-H [5 мкм, сверхкритическая флюидная хроматография; градиент: от 5% до 40% (этанол, содержащий 0,05% диэтиламина) в диоксиде углерода], соединение Примера 147 элюировалось первым, затем элюировалось соединение Примера 146.
55. Взаимодействие соединения Примера 152 с оксихлоридом фосфора с последующим замещением метилатом натрия в метаноле дало соединение этого Примера.
56. Соединение Примера 11 подвергали взаимодействию с диметиламином и карбонатом натрия с получением продукта.
57. 5-Бром-4,6-диметилпиримидин-2-ол защищали в виде его триизопропилсилилового эфира и использовали в реакции Сузуки.
58. В данном случае использовали фосфат калия, и катализатор для взаимодействия с метилбороновой кислотой представлял собой бис(три-трет-бутилфосфин)палладий(О). Соединение Примера 154 получили в результате дехлорирования соединения Примера 11.
59. Катализатор, используемый для реакции Сузуки, был таким же, как тот, который использовали в процессе синтеза соединения Примера 10, стадия 3.
60. Продукт синтезировали путем взаимодействия соединения Примера 11 с этилатом натрия в этаноле.
61. Реакцию Сузуки осуществляли с использованием условий Примера 10. Партнер сочетания 8-хлор-5-(фуро[3,2-с]пиридин-4-илокси)хинолина синтезировали следующим образом: реакция Скраупа 2-хлор-5-метоксианилина с пропан-1,2,3-триолом дала 8-хлор-5-метоксихинолин, который деметилировали с использованием водной бромистоводородной кислоты. Полученный 8-хлорхинолин-5-ол затем подвергали взаимодействию с 4-хлорфуро[3,2-с]пиридином, используя карбонат цезия в диметилсульфоксиде.
62. Соединение Примера 134 подвергали взаимодействию с бромидом лития, бис(триметилсилил)амидом натрия и метилйодидом с получением продукта.
63. В данном случае первую стадию осуществляли с использованием [2'-(азанидил-кN)бифенил-2-ил-кC2](хлор){дициклогексил[2',4',6',-три(пропан-2-ил)бифенил-2-ил]-λ5-фосфанил}палладия в качестве катализатора.
64. 6-Бром-1-метилпиридин-2(H)-он использовали в качестве партнера сочетания.
65. Требуемый 5-бром-6-метоксиизохинолин может быть получен согласно P. Chen et al., Bioorg. Med. Chem. Lett. 2003, 13, 1345-1348.
66. В данном случае С17 подвергали взаимодействию с метилатом натрия с получением 4-хлор-5-метокси-2-(тетрагидро-2H-пиран-2-ил)пиридазин-3(2Н)-она до реакции Сузуки.
67. Условия HPLC. Колонка: Waters Sunfire С18, 4,6×50 мм, 5 мкм; подвижная фаза А: 0,05%-ная трифторуксусная кислота в воде (об./об.); подвижная фаза В: 0,05%-ная трифторуксусная кислота в ацетонитриле (об./об.); градиент: от 5,0% до 95% В за 4,0 минуты; скорость потока: 2 мл/мин.
68. Требуемый 3-бром-4-метилпиридин-2-карбонитрил может быть получен из N-оксида 3-бром-4-метилпиридина по методу В. Elman, Tetrahedron 1985, 47,4941-4948.
69. Циклизация С67 с гидразинкарбоксамидом с последующим отщеплением метилового эфира, опосредованным трибромидом бора, дала 5-(4-гидрокси-2-метилфенил)-6-метил-1,2,4-триазин-3(2Н)-он.
70. Соединение Примера 72 подвергали взаимодействию с 2-бромэтил-метиловым эфиром и карбонатом цезия.
71. Требуемый 5-бром-4-этокси-6-метилпиримидин получали из 5-бром-4-хлор-6-метилпиримидина посредством обработки этилатом натрия в этаноле.
72. Условия HPLC: колонка: XBridge С18, 2,1×50 мм, 5 мкм; подвижная фаза А: 0,05%-ный гидроксид аммония в воде; подвижная фаза В: ацетонитрил; градиент: от 5% до 100% В за 3,4 минуты; скорость потока: 0,8 мл/мин.
73. 4-[4-Бром-3-(трифторметил)фенокси]фуро[3,2-с]пиридин подвергали взаимодействию с (1-метил-1Н-пиразол-5-ил)бороновой кислотой.
74. В данном случае последнее взаимодействие осуществляли в метаноле.
75. Соединение С4 превращали в 8-бром-5-[4-(фуро[3,2-с]пиридин-4-илокси)-2-метилфенил]-6-метилимидазо[1,2-а]пиразин посредством взаимодействия с хлорацетальдегидом. Последующее взаимодействие с цианидом калия и 1,4,7,10,13,16-гексаоксациклооктадеканом (18-краун-6) дало продукт.
76. Соединение Примера 16 превращали в продукт путем взаимодействия с этоксиуксусной кислотой и хлоридом 2-хлор-1,3-диметилимидазолиния (DMC) в присутствии N,N-диизопропилэтиламина.
77. Промежуточное соединение 4-[3-хлор-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенокси]фуро[3,2-с]пиридин синтезировали способом Примера 1, но используя 4-бром-3-хлорфенол вместо 4-бром-3-метилфенола.
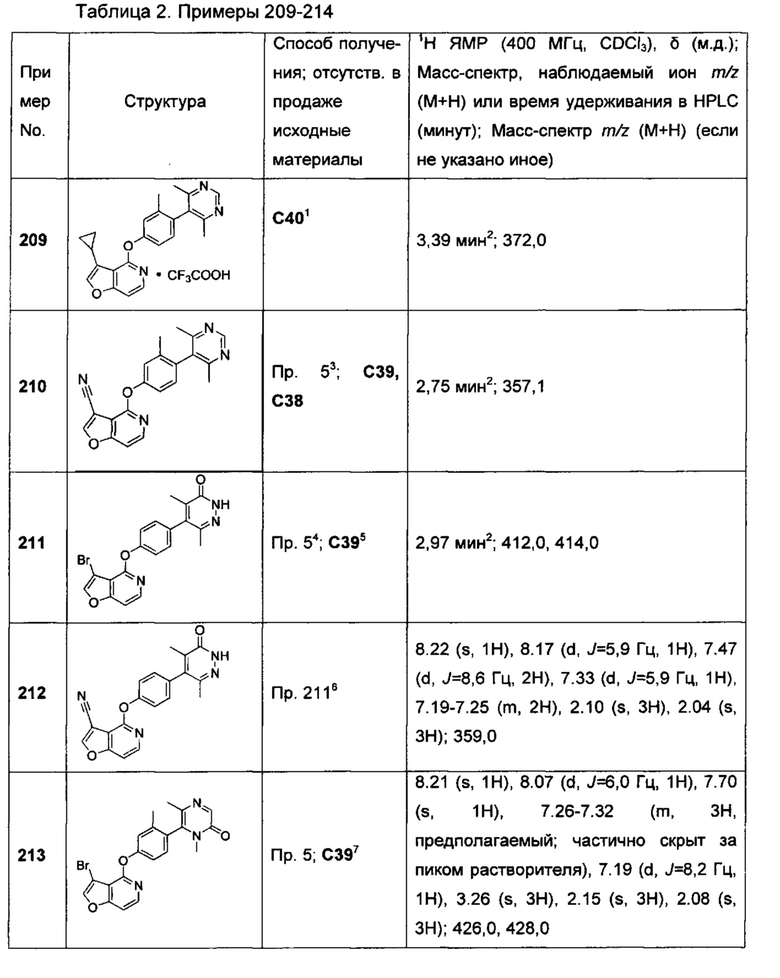
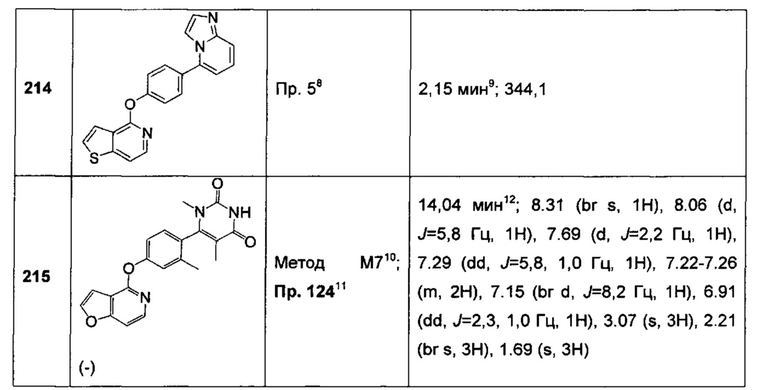
1. Соединение С40 подвергали реакции Сузуки с циклопропилбороновои кислотой с использованием условий, описанных в подпункте 22 к Таблице 1.
2. Условия HPLC: колонка: Waters Atlantis dC18, 4,6×50 мм, 5 мкм; подвижная фаза А: 0,05%-ная трифторуксусная кислота в воде (об./об.); подвижная фаза В: 0,05%-ная трифторуксусная кислота в ацетонитриле (об./об.); градиент: от 5,0% до 95% В за 4,0 минуты, линейный; скорость потока: 2 мл/мин.
3. Замещение бромида цианогруппой осуществляли в качестве последней стадии, используя цианид меди(1) в N,N-диметилформамиде.
4. Защитную группу удаляли на последней стадии с использованием раствора хлористого водорода в метаноле.
5. Требуемый 5-(4-гидроксифенил)-4,6-диметил-2-(тетрагидро-2Н-пиран-2-ил)пиридазин-3(2Н)-он получали следующим образом: (4-{[трет-бутил(диметил)силил]окси}фенил)бороновую кислоту и 2,4-диметил-5-оксо-2,5-дигидрофуран-3-ил-трифторметансульфонат (С48) подвергали взаимодействию согласно Примеру 27 с получением 4-(4-{[трет-бутил(диметил)силил]окси}фенил)-3,5-диметилфуран-2(5Н)-она. Силильную защитную группу удаляли с помощью фторида тетрабутиламмония и заменяли бензильной защитной группой с получением 4-[4-(бензилокси)фенил]-3,5-диметилфуран-2(5H)-она. Его подвергали взаимодействию с кислородом, а затем с гидразином, как описано в Примере 27, с получением 5-[4-(бензилокси)фенил]-4,6-диметилпиридазин-3(2Н)-она. Защита азота с помощью 3,4-дигидро-2H-пирана, как в Примере 10, с последующим гидролизом бензильной группы дала требуемый фенол.
6. Перед кислотным удалением тетрагидропирановой защитной группы в Примере 211 бром заменяли цианогруппой с использованием цианида меди(1) в N,N-диметилформамиде. Удаление защитной группы дало соединение Примера 212.
7. Требуемый 6-(4-гидрокси-2-метилфенил)-1,5-диметилпиразин-2(1Н)-он получали следующим образом: реакция Сузуки между (4-метокси-2-метилфенил)бороновой кислотой и 2-бром-3-метилпиразином дала 2-(4-метокси-2-метилфенил)-3-метилпиразин. После образования N-оксида и перестановки с помощью уксусного ангидрида (см. A. Ohta et al., J. Het. Chem. 1985, 19, 465-473) полученный 6-(4-метокси-2-метилфенил)-5-метилпиразин-2-ол N-метилировали и затем снимали защиту с помощью трибромида бора.
8. 4-(Имидазо[1,2-а]пиридин-5-ил)фенол получали из (4-гидроксифенил)бороновой кислоты и 5-бромимидазо[1,2-а]пиридина, используя способ Примера 6.
9. Условия HPLC. Колонка: Waters Atlantis dC18, 4,6×50 мм, 5 мкм; подвижная фаза А: 0,05%-ная трифторуксусная кислота в воде (об./об.); подвижная фаза В: 0,05%-ная трифторуксусная кислота в ацетонитриле (об./об.); от 15,0% до 95% В, линейный, за 4,0 минуты; скорость потока: 2 мл/мин.
10. В данном случае инкубацию осуществляли в течение 2,25 часов, а не 24-96 часов.
11. Соединение Примера 124 разделяли на его составные атропоэнантиомеры посредством сверхкритической флюидной хроматографии (колонка: Chiralpak AD-H, 5 мкм; элюент: смесь диоксид углерода/пропанол в соотношении 7:3). Элюирующийся вторым энантиомер [(-)-6-[4-(фуро[3,2-с]пиридин-4-илокси)-2-метилфенил]-1,5-диметилпиримидин-2(1Н)-он] использовали в биотрансформации. Неочищенный продукт биотрансформации очищали посредством хроматографии на силикагеле (элюент: 70% этилацетата в гептане).
12. Условия сверхкритической флюидной хроматографии. Колонка: Phenomenex Cellulose-4, 4,6×250 мм, 5 мкм; элюент: смесь диоксид углерода/метанол в соотношении 55:45; скорость потока 2,5 мл/мин.
Пример 216
Трифторацетатная соль 6-[4-(фуро[3,2-с]пиридин-4-илокси)фенил]-1,5-диметилпиримидин-2,4(1H,3Н)-диона (216)
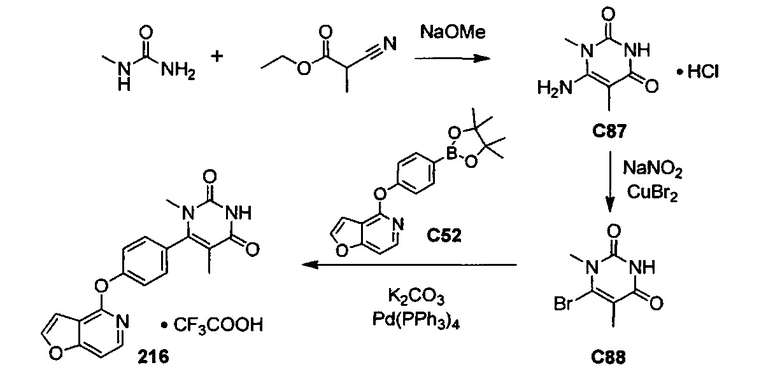
Стадия 1. Синтез гидрохлоридной соли 6-амино-1,5-диметилпиримидин-2,4(1H,3H)-диона (С87)
1-Метилмочевину (98%, 8,26 г, 109 ммоль) и этил-2-цианопропаноат (95%, 13,2 мл, 99,6 ммоль) растворяли в метаноле (75 мл) и обрабатывали метилатом натрия (25%-ный по массе раствор в метаноле, 27 мл, 120 ммоль). Полученную смесь кипятили с обратным холодильником в течение 18 часов. После охлаждения до комнатной температуры реакционную смесь концентрировали при пониженном давлении для удаления избытка метанола. Растворитель затем заменяли повторным добавлением ацетонитрила (3×50 мл) с последующим концентрированием в вакууме. Полученное твердое вещество растворяли в ацетонитриле (100 мл) и воде (100 мл) и 6 М водную соляную кислоту добавляли до достижения рН приблизительно 2. Во время такого подкисления образовывался белый осадок. После перемешивания смеси в течение часа твердое вещество собирали фильтрованием и промывали mpem-бутилметиловым эфиром с получением продукта в виде белого твердого вещества. Выход: 15,2 г, 79,3 ммоль, 80%. ЖХ-МС m/z 156,3 [М+Н]. 1Н ЯМР (400 МГц, DMSO-d6) δ 10.37 (br s, 1Н), 6.39 (br s, 2Н), 3.22 (s, 3Н), 1.67 (s, 3Н).
Стадия 2. Синтез 6-6ром-1,5-диметилпиримидин-2,4(1H,3H)-диона (С88)
Смесь ацетонитрила и воды (60 мл) в соотношении 1:1 добавляли к смеси 6-амино-1,5-диметилпиримидин-2,4(1H,3H)-диона в виде соли гидрохлорида (С87) (5,00 г, 26,1 ммоль), нитрита натрия (98%, 2,76 г, 39,2 ммоль) и бромида меди(Н) (99%, 11,8 г, 52,3 ммоль) {Осторожно: наблюдается образование пузырьков и слабая экзотермическая реакция}, и реакционную смесь оставляли перемешиваться при комнатной температуре в течение 18 часов. После разбавления водной серной кислотой (1 н., 100 мл) и этилацетатом (100 мл) образовывался осадок; его выделяли фильтрованием и промывали водой и этилацетатом с получением продукта в виде твердого вещества (3,65 г). Фильтрат концентрировали в вакууме до приблизительно 25% от его первоначального объема, во время которого наблюдали больше осадка. Фильтрование и промывка этого твердого вещества водой и этилацетатом дали дополнительное количество продукта (0,60 г). Суммарный выход: 4,25 г, 19,4 ммоль, 74%. ЖХ-МС m/z 219,0, 221,0 [М+Н]. 1Н ЯМР (400 МГц, DMSO-d6) δ 11.58 (br s, 1Н), 3.45 (s, 3Н), 1.93 (s, 3Н).
Стадия 3. Синтез трифторацетатной соли 6-[4-(фуро[3,2-с]пиридин-4-илокси)фенил]-1,5-диметилпиримидин-2,4(1H, 3H)-диона (216)
6-Бром-1,5-диметилпиримидин-2,4(1H,3H)-дион (С88) (78,0 мг, 0,356 ммоль), 4-[4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенокси]фуро[3,2-с]пиридин (С52) (60,0 мг, 0,178 ммоль), карбонат калия (99%, 74,5 мг, 0,534 ммоль) и тетракис(трифенилфосфин)палладий(0) (99%, 10,5 мг, 0,0090 ммоль) объединяли в этаноле (5 мл) и нагревали до 80°C в течение 18 часов. Реакционную смесь разбавляли водой, слегка подкисляли добавлением 1,0 М водной соляной кислоты и экстрагировали несколько раз этилацетатом. Объединенные органические слои промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом магния, фильтровали и концентрировали в вакууме. Очистка хроматографией на силикагеле (градиент: от 75% до 100% этилацетата в гептане) с последующей HPLC с обращенной фазой (колонка: Waters Sunfire С18, 5 мкм; подвижная фаза А: 0,05%-ная трифторуксусная кислота в воде (об./об.); подвижная фаза В: 0,05%-ная трифторуксусная кислота в ацетонитриле (об./об.); градиент: от 20% до 100% В) дала продукт в виде твердого вещества. Выход: 20 мг, 0,057 ммоль, 32%. ЖХ-МС m/z 350,0 [М+Н]. 1Н ЯМР (600 МГц, DMSO-d6) δ 8.14 (d, J=2,2 Гц, 1Н), 8.04 (d, J=5,9 Гц, 1H), 7.51 (br d, J=5,9 Гц, 1H), 7.42 (br АВ квартет, JAB=8,8 Гц, ΔνAB=16,7 Гц, 4Н), 7.08 (dd, J=2,2, 0,9 Гц, 1Н), 2.94 (s, 3Н), 1.55 (s, 3Н).
Пример АА: Анализ и данные по связыванию с человеческим D1 рецептором
Аффинность описанных здесь соединений определяли анализами конкурентного связывания, подобными тем, которые описаны в Ryman-Rasmussen et al., "Differential activation of adenylate cyclase and receptor internalization by novel dopamin D1 receptor agonists", Molecular Pharmacology 68(4): 1039-1048 (2005). В этом анализе связывания радиолиганда использовали [3H]-SCH23390, радиоизотоп D1 лиганда, для оценки способности тестируемого соединения конкурировать с радиолигандом при связывании с D1 рецептором.
Анализы связывания D1 осуществляли с использованием сверхэкспрессирующих LTK человеческих клеточных линий. Для определения основных параметров анализа концентрации лиганда определяли из исследований связывания с насыщением, где Kd (константа диссоциации) для [3H]-SCH23390, как было обнаружено, составляет 1,3 нМ. Из исследований кривой концентрации в ткани было определено, что оптимальное количество ткани составляет 1,75 мг/мл на 96-луночный планшет при использовании 0,5 нМ [3H]-SCH23390. Эти концентрации лиганда и ткани использовали в исследованиях кривых в зависимости от времени для определения линейности и равновесных условий для связывания. Связывание было в равновесном состоянии при конкретном количестве ткани в момент времени 30 минут при 37°C. Из этих параметров Ki значения определяли путем гомогенизации конкретного количества ткани для каждого вида в 50 мМ трис (рН 7,4 при 4°C), содержащем 2,0 мМ MgCl2 с использованием политрона и центрифугировали при 40000 х д в течение 10 минут. Осадок ресуспендировали в буфере для анализа (50 мМ трис (рН 7,4 при КТ), содержащем 4 мМ MgS04 и 0,5 мМ EDTA). Инкубации инициировали добавлением 200 мкл ткани в 96-луночные планшеты, содержащие тестируемые лекарственные средства (2,5 мкл) и 0,5 нМ [3Н]-SCH23390 (50 мкл) в конечном объеме 250 мкл. Неспецифическое связывание определяли путем связывания радиолиганда в присутствии насыщающей концентрации (+)-бутакламола (10 мкМ), представляющего собой D1 антагонист. После 30-минутного периода инкубации при 37°C образцы анализа быстро фильтровали через Unifilter-96 GF/B PEI-покрытые фильтр-планшеты и промывали 50 мМ трис-буфером (рН 7,4 при 4°C). Уровни мембранного связывания [3H]-SCH23390 определяли путем жидкостного сцинтилляционного подсчета в Ecolume. Значение IC50 (концентрация, при которой имеет место 50%-ное ингибирование специфического связывания) рассчитывали путем линейной регрессии данных зависимости ответа от концентрации в Microsoft Excel. Значения Ki рассчитывали согласно уравнению Ченга-Прусоффа:
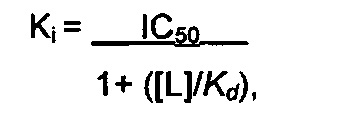
где [L] представляет собой концентрацию свободного радиолиганда, и Kd представляет собой константу диссоциации радиолиганда для D1 рецептора (1,3 нМ для [3H]-SCH23390).
Пример ВВ: Анализ и данные по D1 cAMP HTRF
Анализ D1 сАМР (циклический аденозинмонофосфат) HTRF (гомогенная флуоресценция с разрешением во времени), используемый и описанный здесь, представляет собой конкурентный иммуноанализ между нативным сАМР, продуцируемым клетками, и сАМР, меченным XL-665. Этот анализ использовали для определения способности тестируемого соединения агонизировать (в том числе частично агонизировать) D1. Моноклональное антитело (Мао) против сАМР, меченное криптатом (Cryptate), визуализирует радиоактивный индикатор. Максимальный сигнал достигается, если образцы не содержат свободного сАМР, из-за близости донорных (Eu-криптат) и акцепторных (XL665) объектов. Сигнал, таким образом, обратно пропорционален концентрации сАМР в образце. Разрешение во времени и логометрическое измерение (эм. 665 нм/эм. 620 нм) минимизирует интерференцию со средой. Наборы для анализов сАМР HTRF имеются в продаже, например, от Cisbio Bioassays, IBA group.
Материалы и методы
Материалы: Набор сАМР Dynamic получали от Cisbio International (Cisbio 62AM4PEJ). Multidrop Combi (Thermo Scientific) использовали для дополнительных анализов. Envision (PerkinElmer) ридер использовали для считывания HTRF.
Клеточная культура: Стабильную клеточную линию HEK293T/hD1#1 создавали для внутренних целей (Pfizer Ann Arbor). Клетки выращивали в виде адгерентных клеток в колбах NuncT500
со средой DMEM с высоким содержанием глюкозы (Invitrogen 11995-065), диализированной 10%-ной фетальной бычьей сывороткой (Invitrogen 26400-044), 1×MEM NEAA (Invitrogen 1140, 25 мМ HEPES (Invitrogen 15630), 1×пенициллина (Реп)/стрептомицина (Strep) (Invitrogen 15070-063) и 500 мкг/мл генентицина (Invitrogen 10131-035) при 37°C и 5% CO2. В моменты времени 72 или 96 часов после роста клетки промывали фосфатно-солевым буфером Дульбекко (DPBS) и к отделенным клеткам добавляли 0,25% трипсин-EDTA. Затем добавляли среду, клетки центрифугировали и среду удаляли. Клеточные осадки ресуспендировали в среде для замораживания клеточных культур (Invitrogen 12648-056) при плотности 4е7 клеток/мл. Аликвоты клеток по 1 мл помещали в криофлаконы и замораживали при -80°C для будущего использования в анализе D1 HTRF.
Методика анализа D1 cAMP HTRF: Замороженные клетки быстро оттаивали, ресуспендировали в 50 мл теплой среды и оставляли стоять в течение 5 мин до центрифугирования (1000 об/мин) при комнатной температуре. Среды удаляли и клеточный осадок ресуспендировали в PBS/0,5 мкМ IBMX с получением 2е5 клеток/мл. Используя Multidrop Combi, 5 мкл клеток/лунку добавляли к аналитическому планшету (Greiner 784085), который уже содержал 5 мкл тестируемого соединения. Контроли соединения [5 мкМ допамин (конечная) и 0,5% DMSO (конечная)] также включали в каждый планшет для анализа данных. Клетки и соединения инкубировали при комнатной температуре в течение 30 мин. Рабочие растворы CAMP-D2 и анти-сАМР-криптата готовили согласно инструкциям Cisbio. Используя Multidrop, 5 мкл рабочего раствора CAMP-D2 добавляли к аналитическому планшету, содержащему тестируемое соединение и клетки. Используя Multidrop, 5 мкл рабочих растворов анти-сАМР-криптата добавляли к аналитическому планшету, содержащему тестируемое соединение, клетки и CAMP-D2. Аналитический планшет инкубировали в течение 1 часа при комнатной температуре. Аналитический планшет считывали в аппарате для считывания планшетов Envision с использованием настроек, рекомендованных Cisbio. Стандартную кривую CAMP генерировали с использованием исходного раствора сАМР, предложенного в наборе Cisbio.
Анализ данных: Анализ данных осуществляли с использованием программного обеспечения. Эффекты в процентах рассчитывали в сравнении с контрольными соединениями. Соотношение ЕС50 определяли с использованием необработанных данных, полученных с аппарата для считывания планшетов Envision. Стандартную кривую сАМР использовали в аналитической программе для определения концентраций сАМР из необработанных данных. сАМР ЕС50 определяли с использованием рассчитанных сАМР данных.
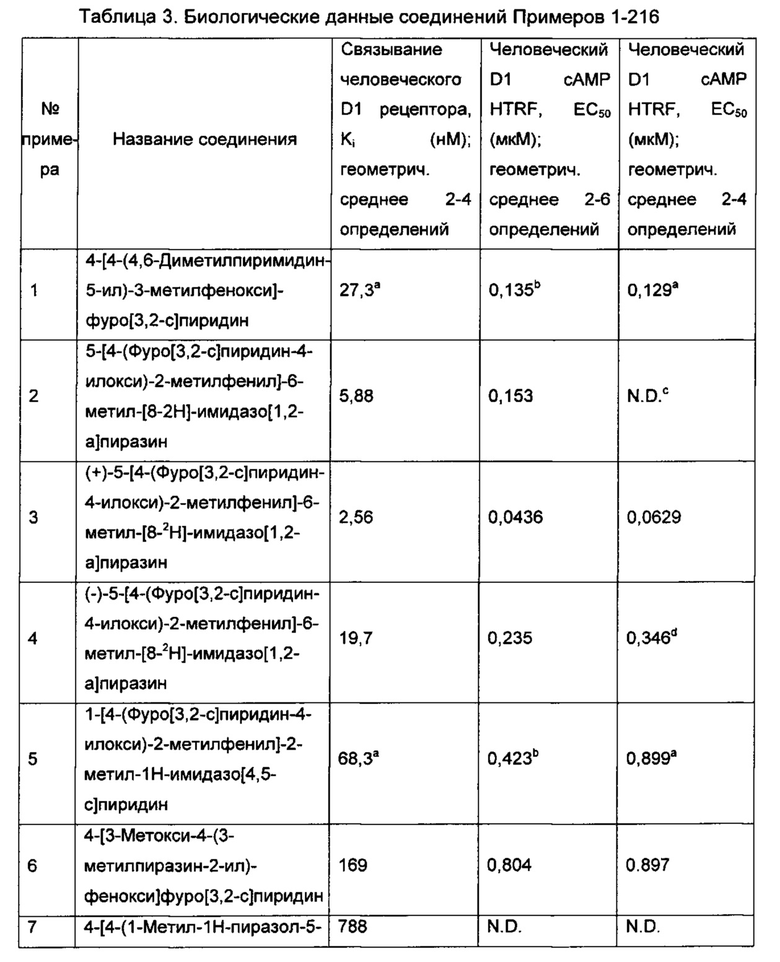
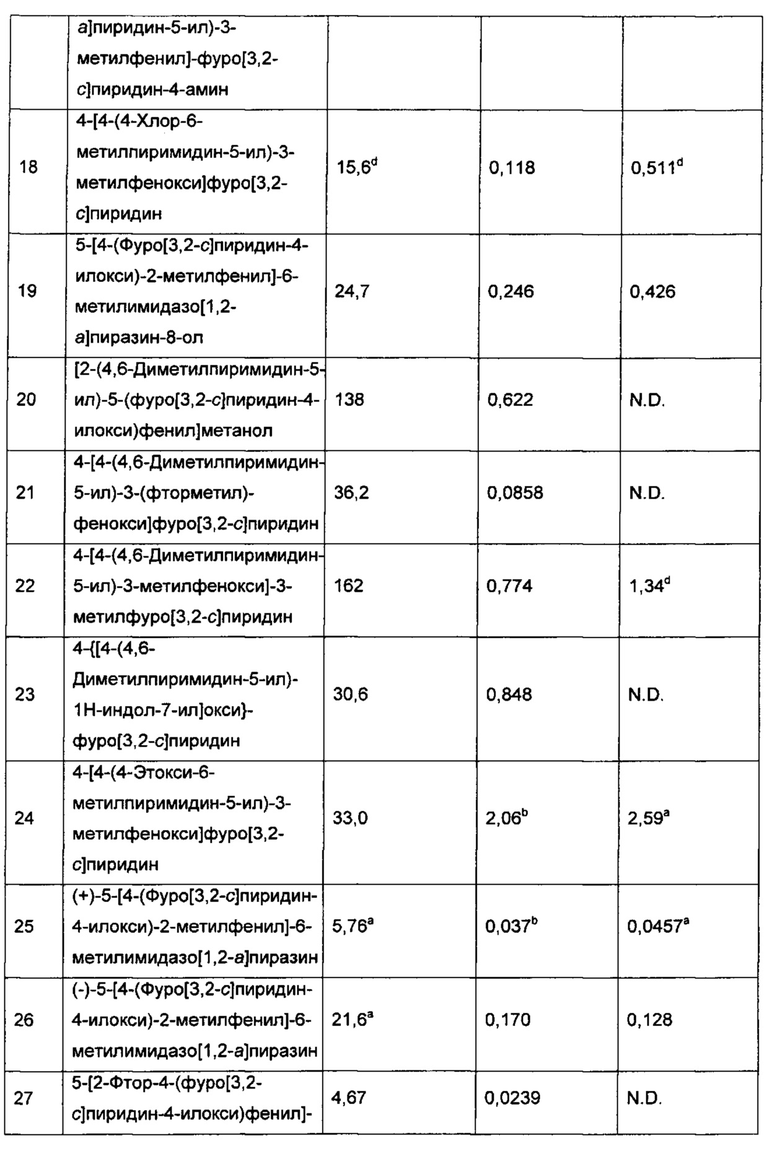
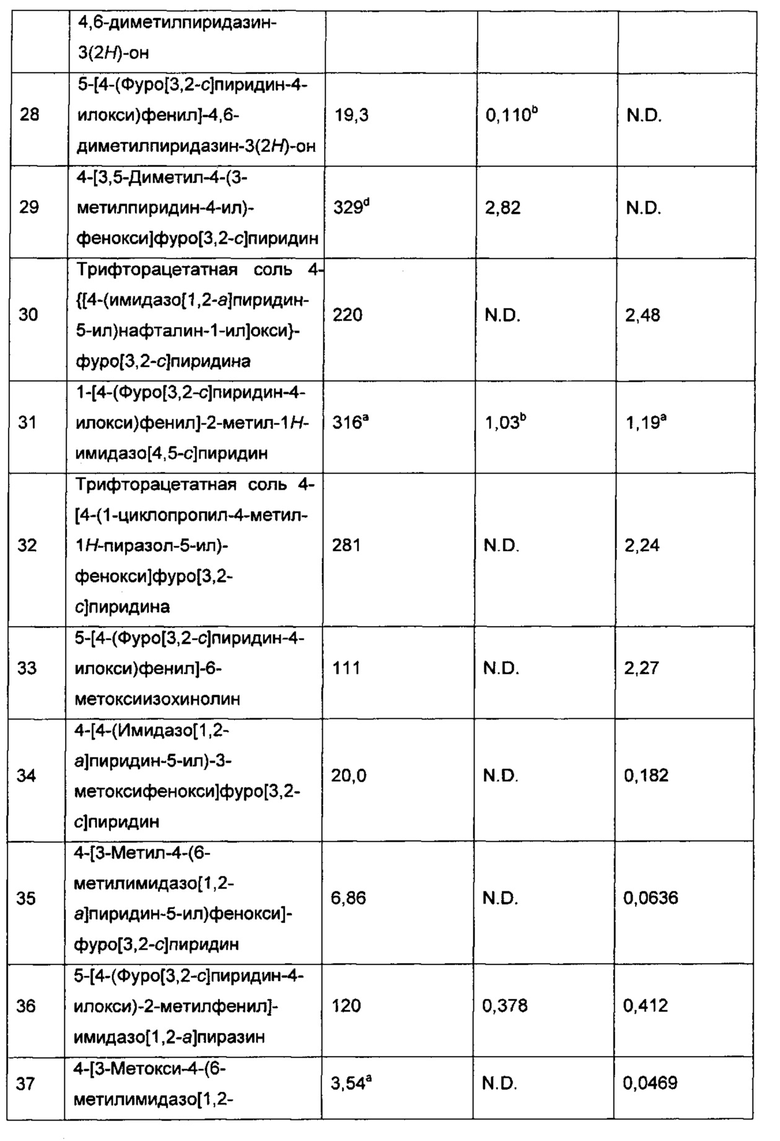
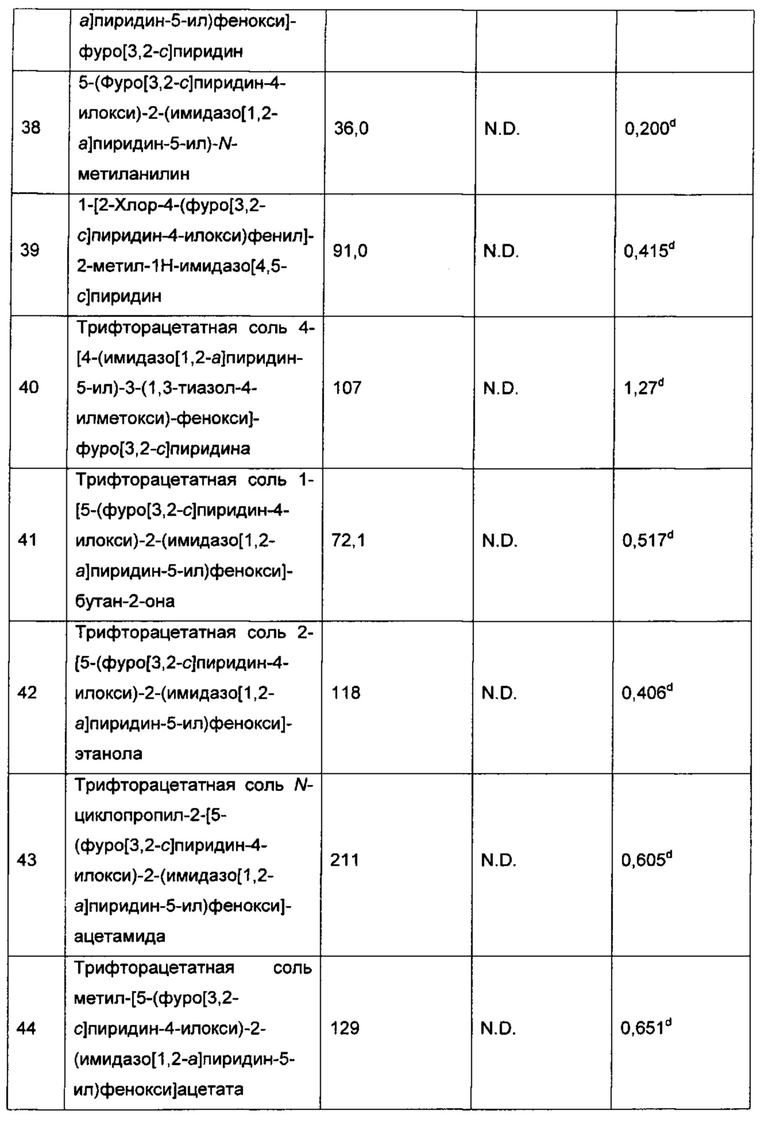
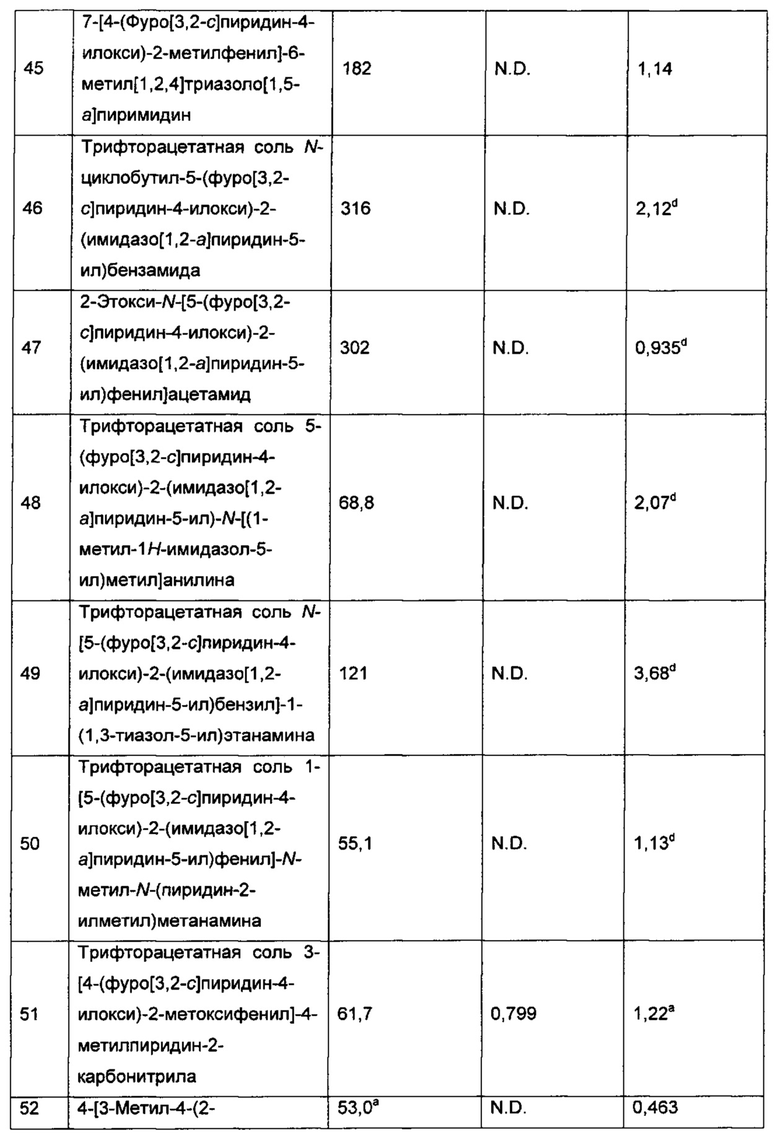
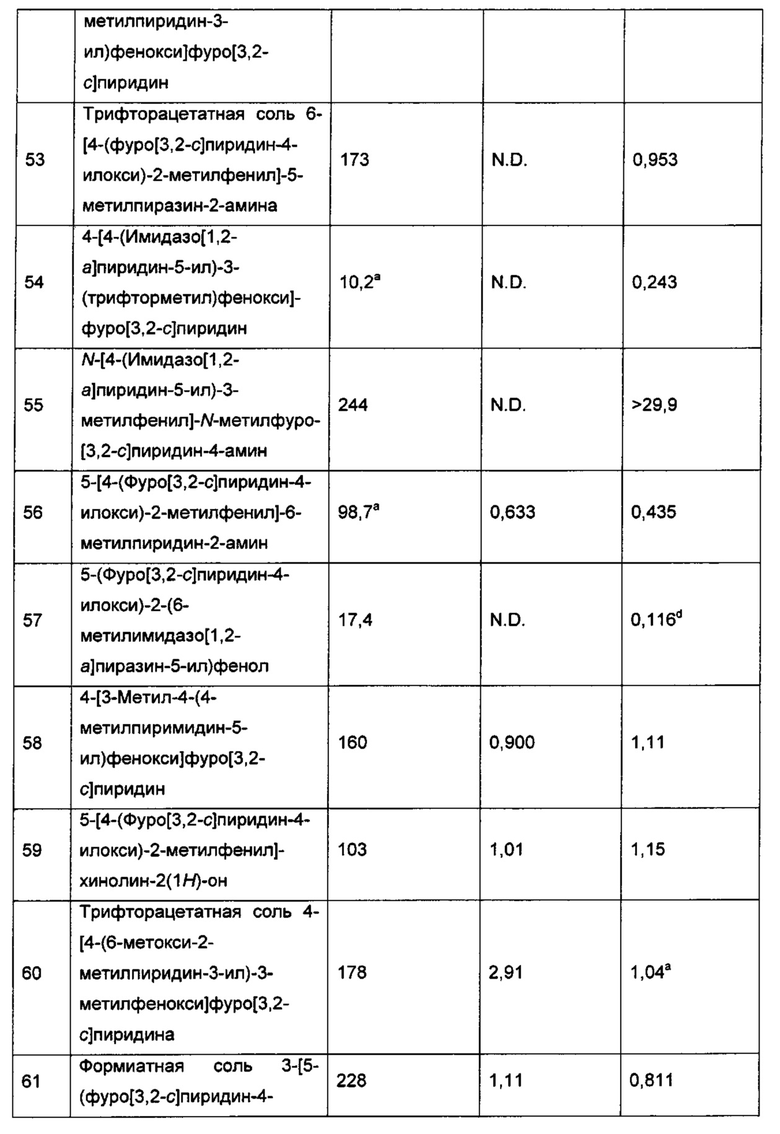
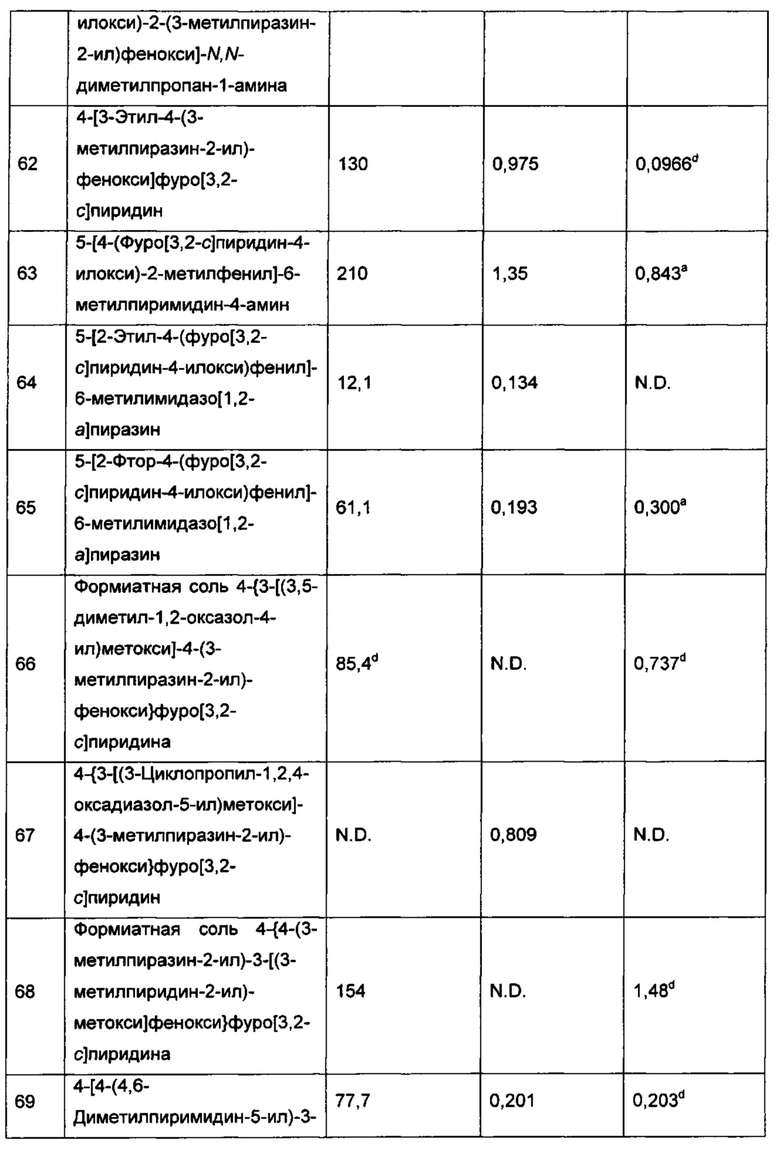
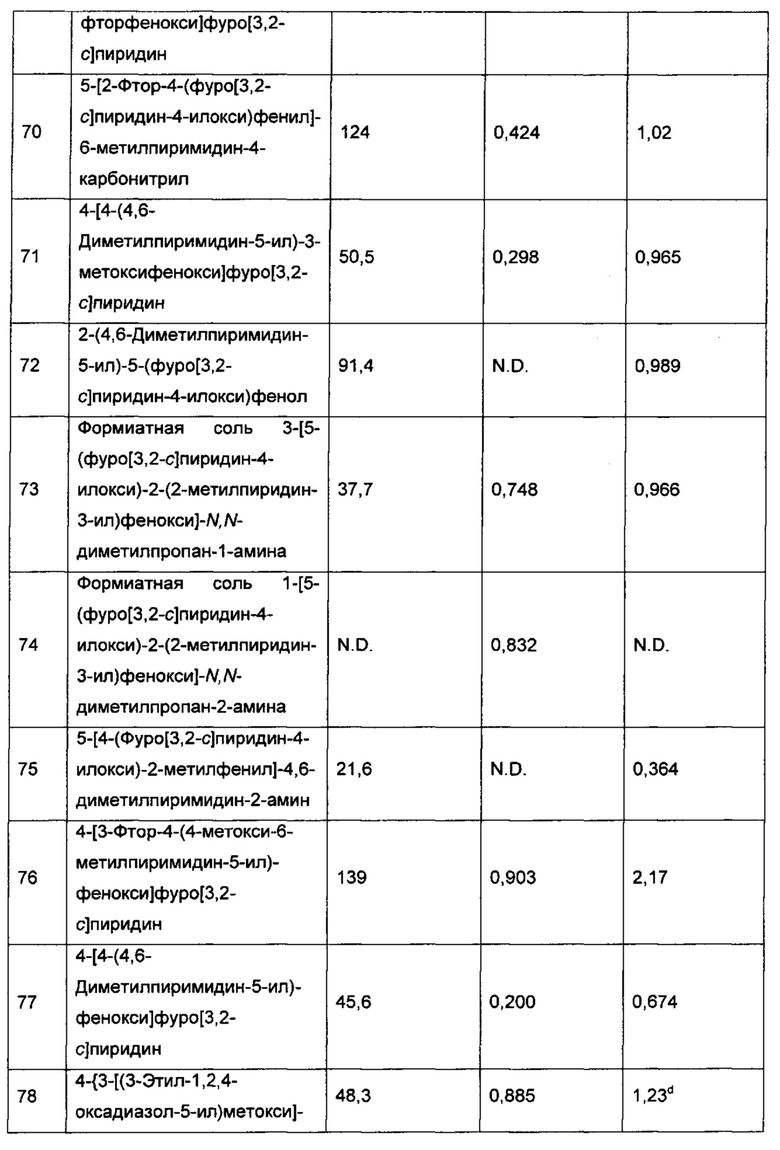
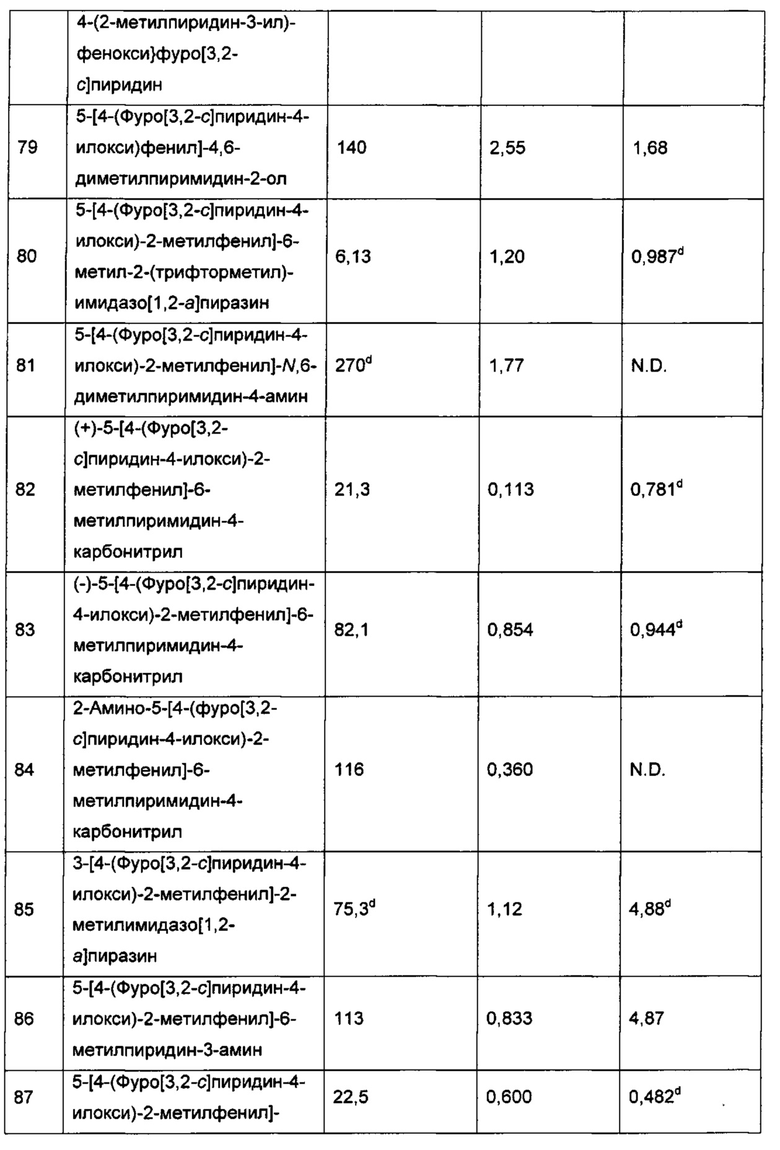
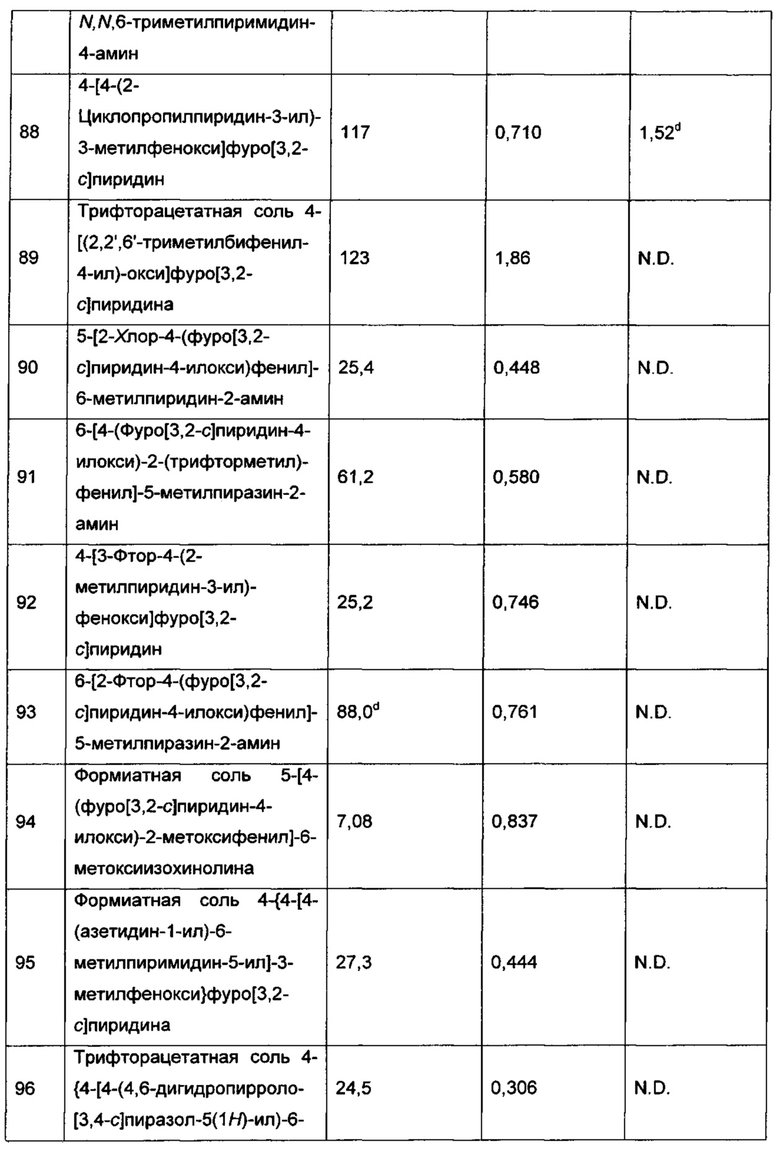
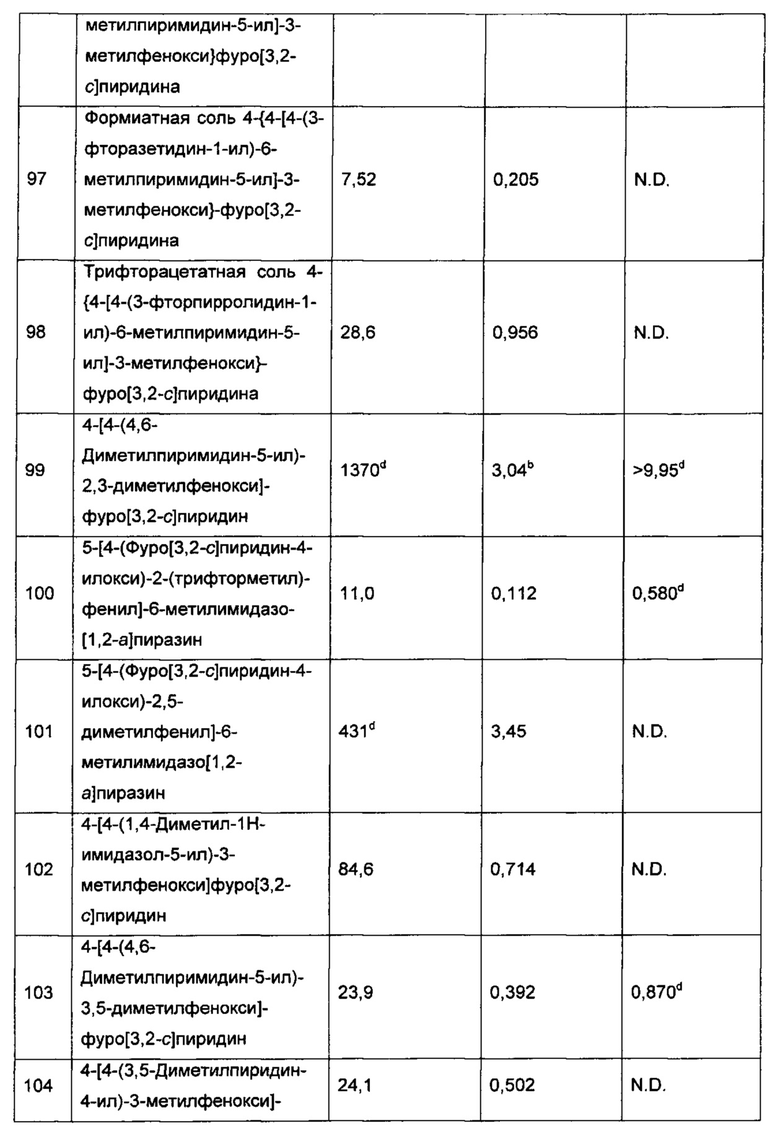
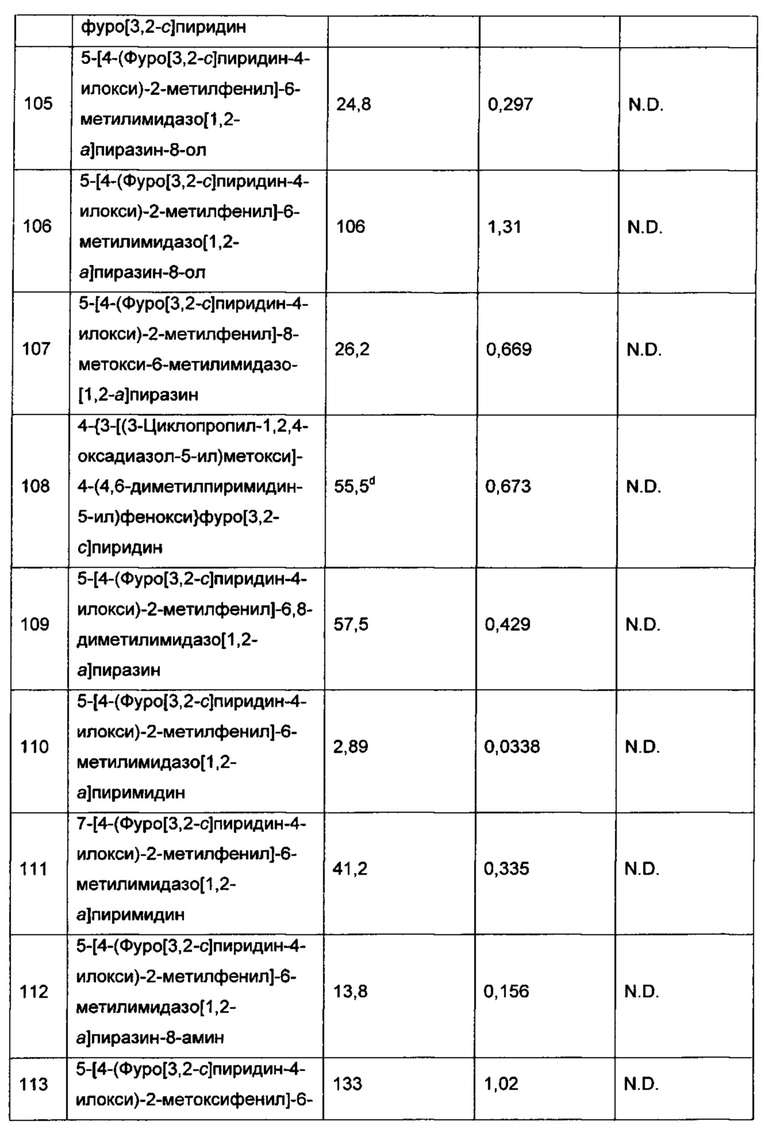
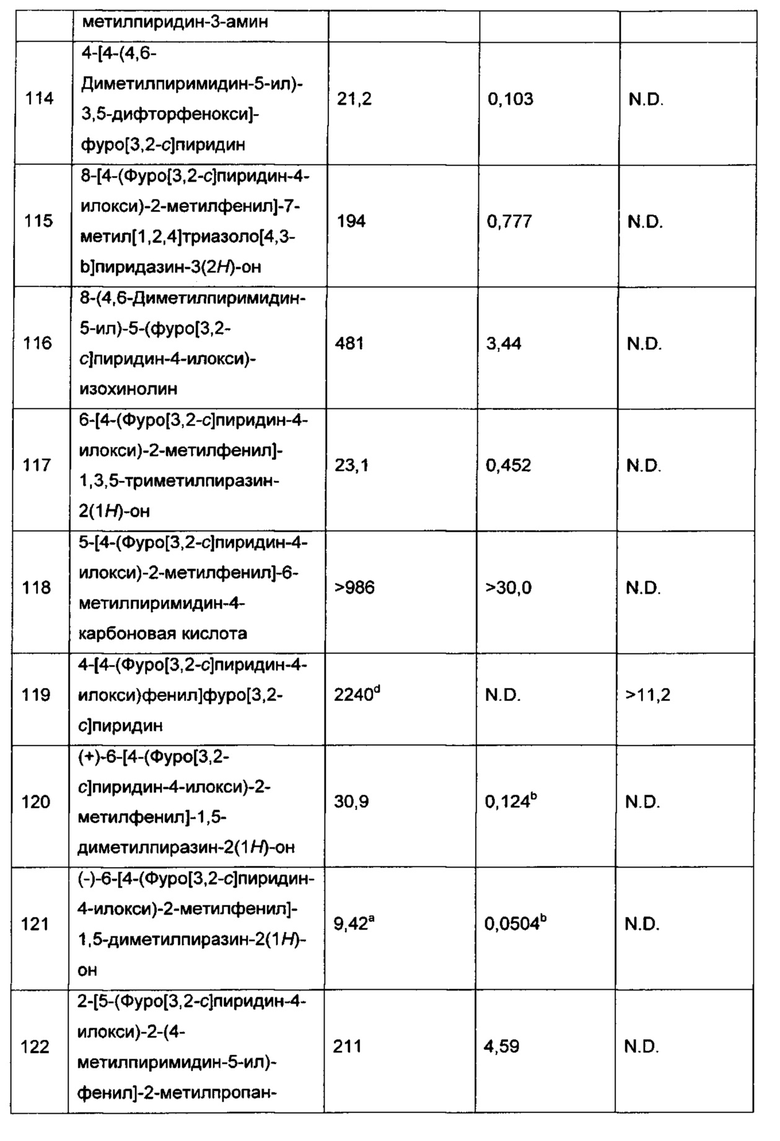
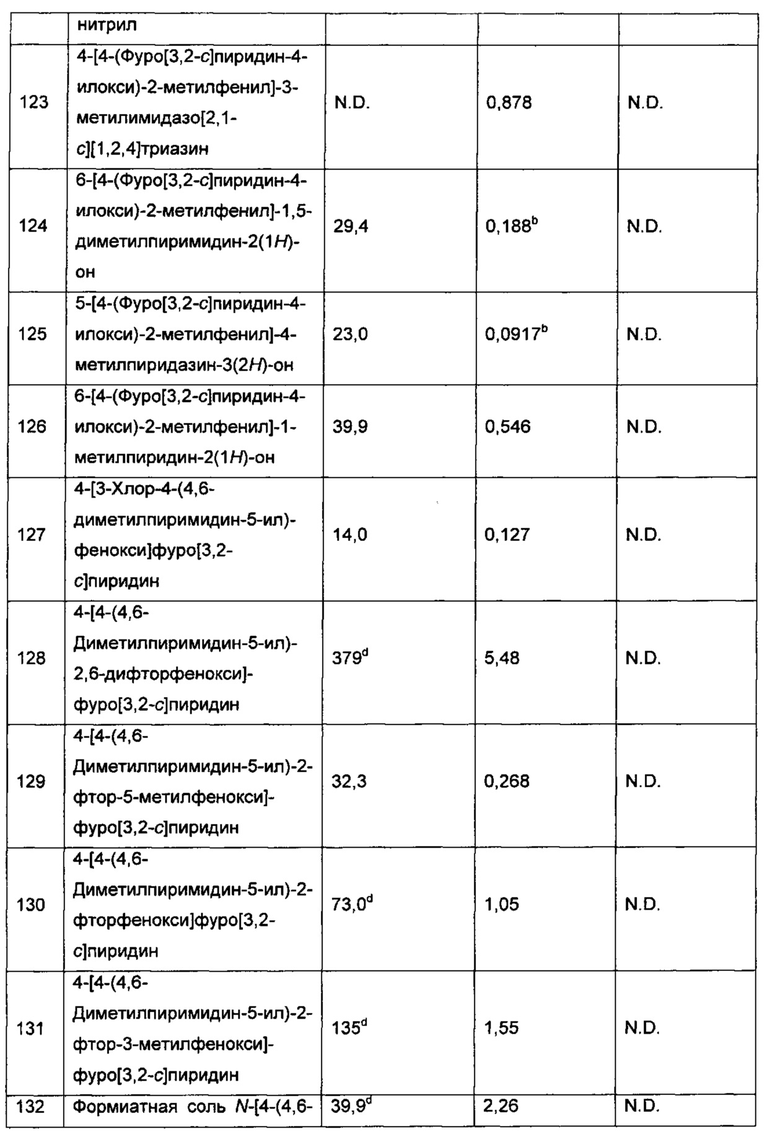
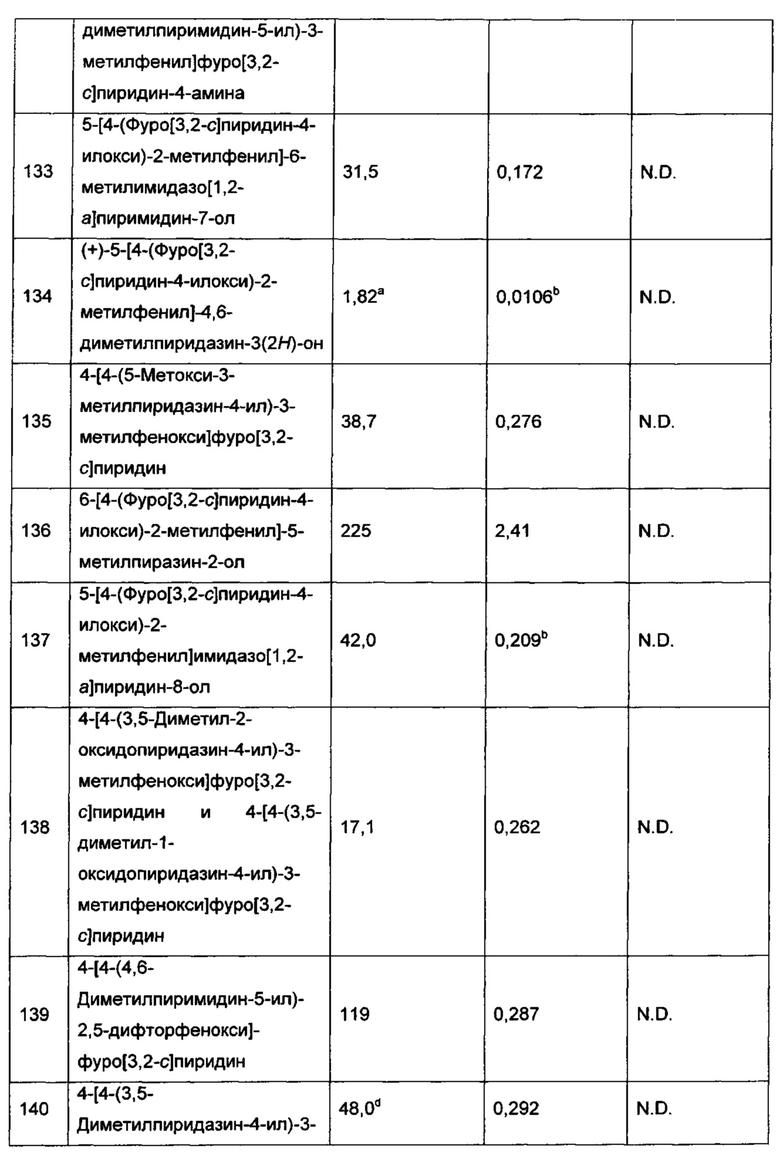
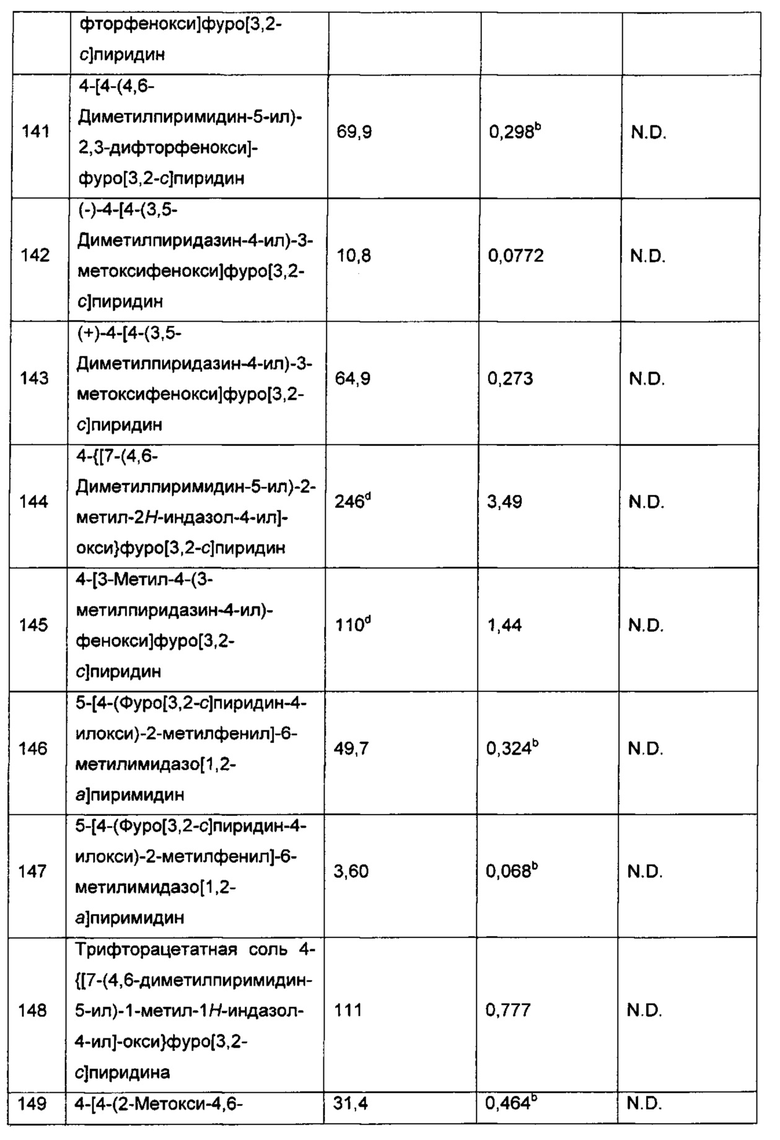
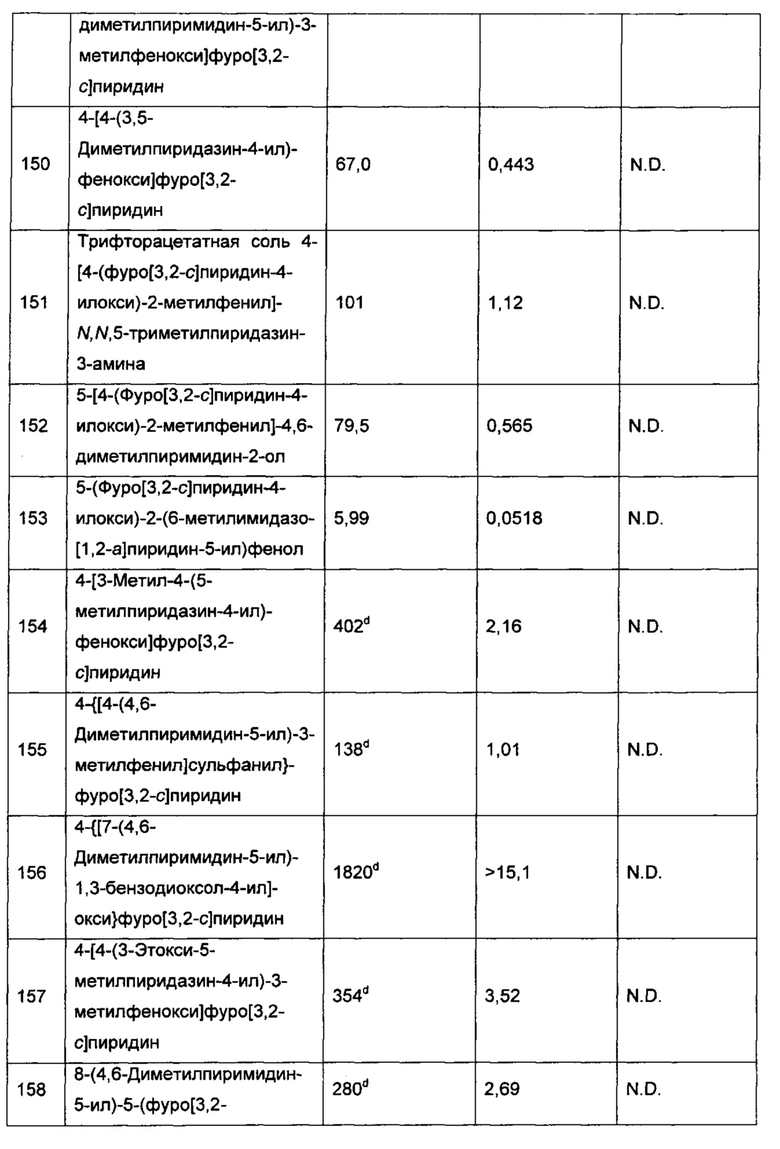
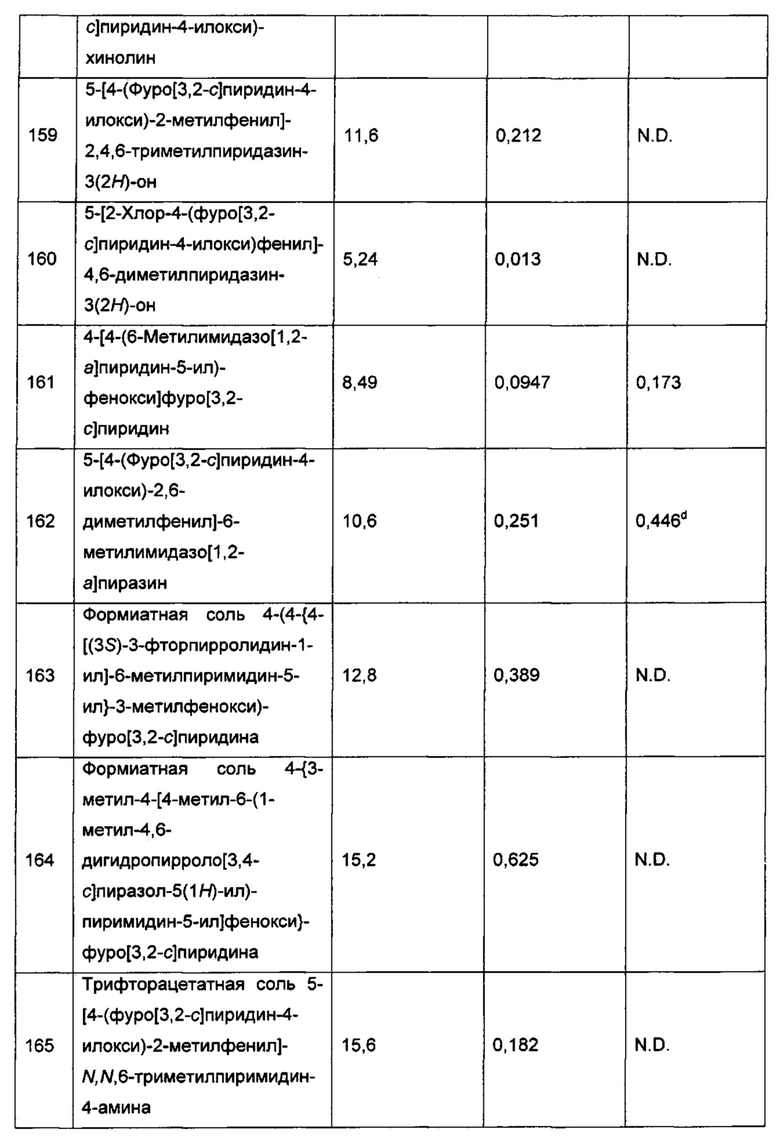
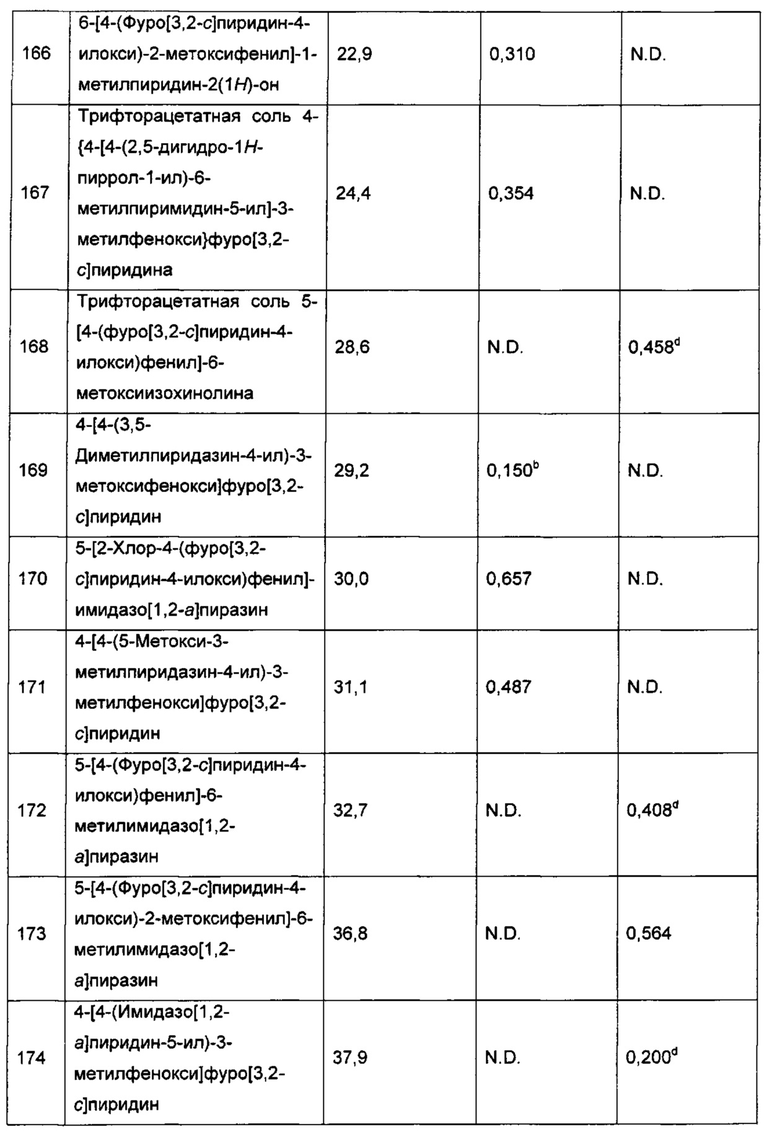
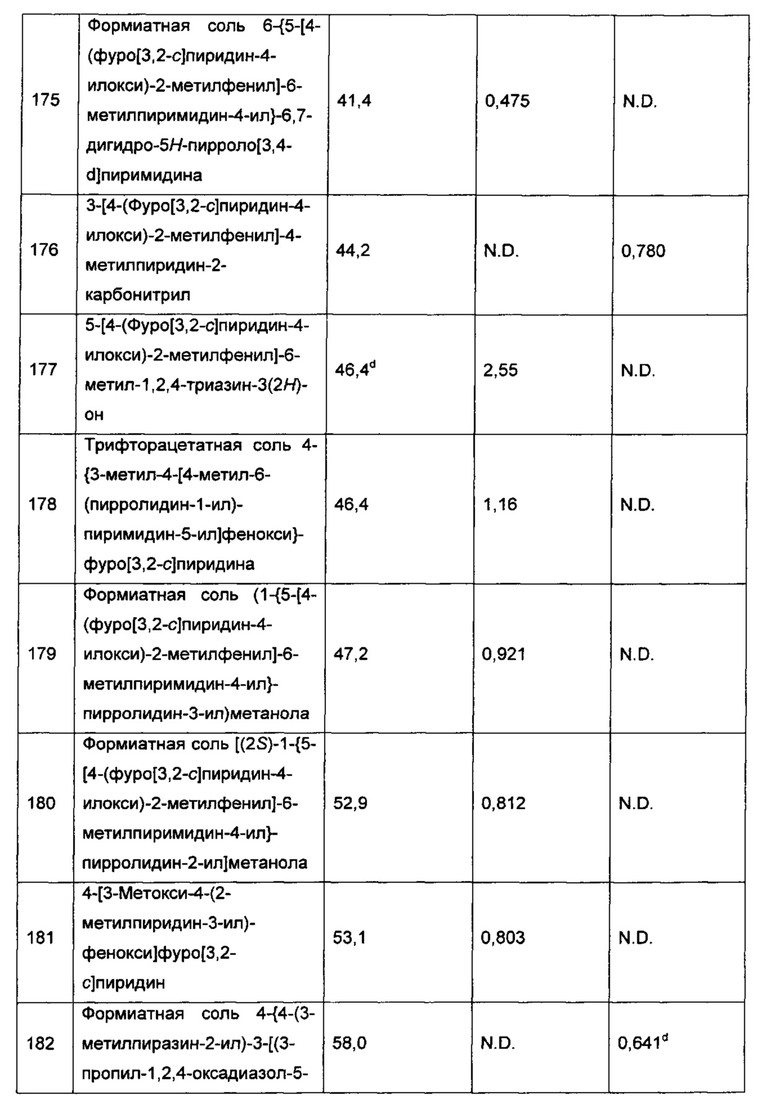


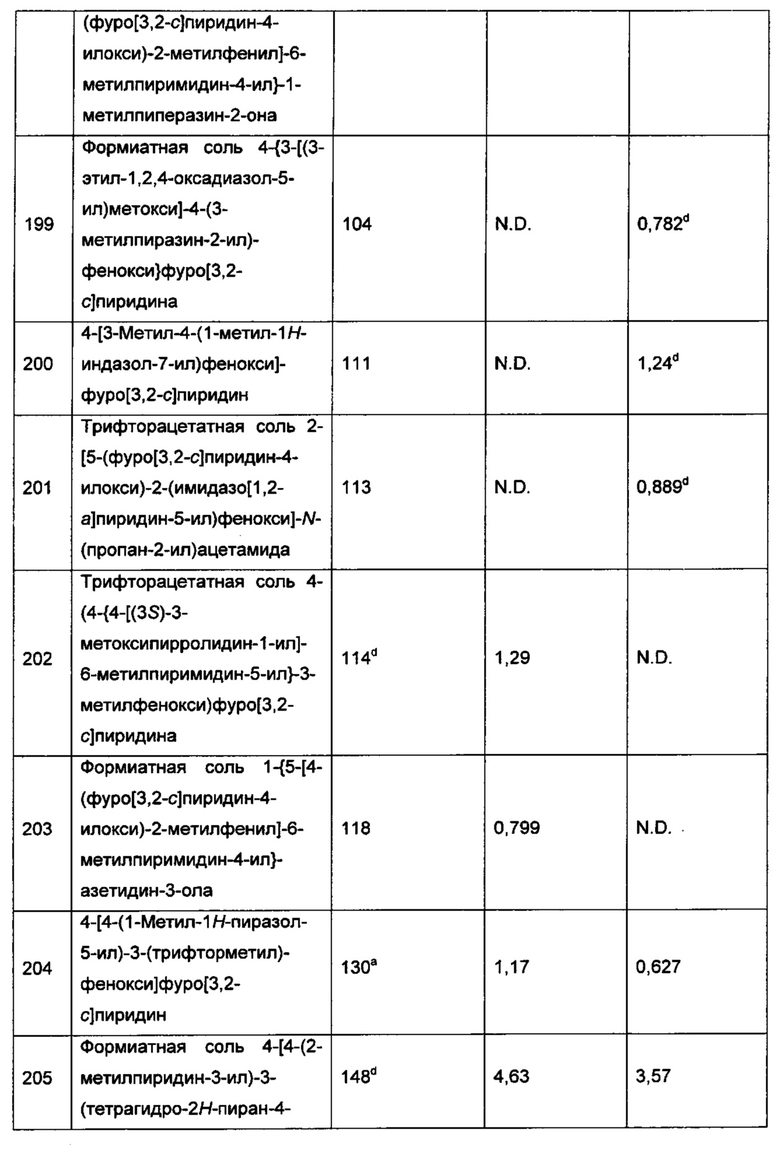
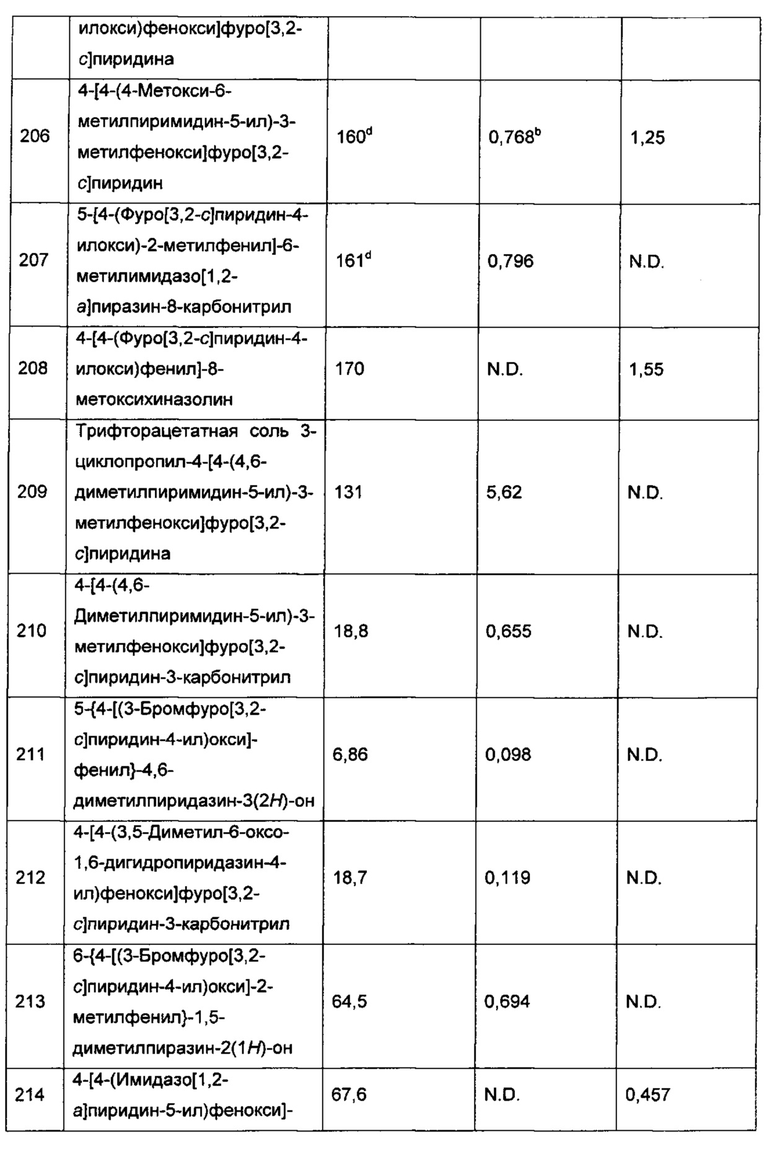
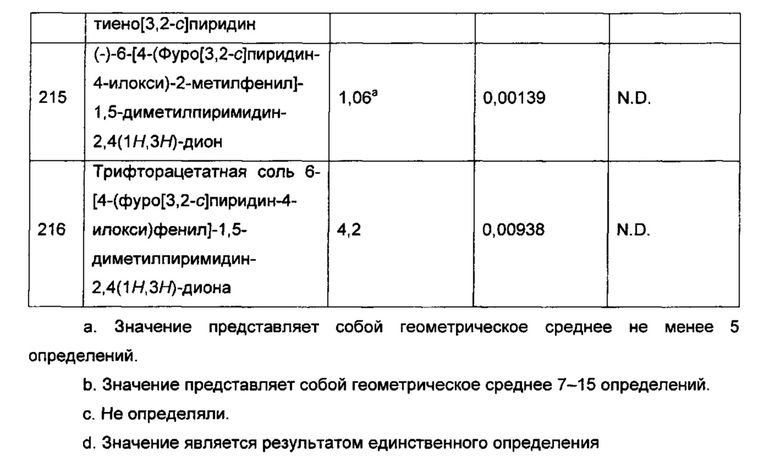
Пример СС: Исследования D1R мутантов
Осуществляли четырнадцать разных мутаций по остаткам сайта потенциального связывания D1R, для того чтобы более точно определить, где связываются агонисты D1 по настоящему изобретению. В общем, имеется очень четкая взаимосвязь между величинами кратности сдвига (fold-shift values) агонистов D1 по настоящему изобретению при сравнении с теми известными производными катехола, которые являются полными (или сверх) агонистами D1 и частичными агонистами; однако, 4 из этих 14 остатков (Ser188, Ser198, Ser202 и Asp103) показали статистически значимые отклонения, и репрезентативные результаты показаны здесь.
Агонистическая активность в отношении человеческого рецептора допамина D1 измеряли с использованием набора Cisbio для динамического обнаружения 3'-5'-циклического аденозинмонофосфата (cAMP) (Cisbio International 62AM4PEJ). cAMP измеряли с использованием конкурентного иммуноанализа гомогенной флуоресценции с разрешением во времени (HTRF) между нативным сАМР и сАМР, меченным красителем d2.
Моноклональное анти-сАМР антитело, меченное криптатом, связывает меченый сАМР. Добавляли донор Europium-криптат и измеряли перенос энергии на d2 акцептор. Максимальный сигнал достигался, если образцы не содержали свободного сАМР, в виду близости Eu-криптатного донорного и d2 акцепторного объектов. Таким образом, сигнал был обратно пропорционален концентрации нативного сАМР в образце. Проводили разрешение во времени и логометрические измерения (эм. 665 нм/эм. 620 нм), которое затем преобразовывали в концентрации сАМР с использованием стандартной кривой. Все сАМР эксперименты проводили в присутствии 500 нМ IBMX для ингибирования активности фосфодиэстеразы (PDE).
сАМР стандартную кривую генерировали с использованием сАМР, предложенного в наборе Cisbio сАМР обнаружения. Получение стандартной кривой состоит в следующем. (1) Получали 2848 нМ сАМР исходный раствор в забуференном фосфатом физиологическом растворе Дульбекко (PBS, от Sigma, Cat#D8537), этот исходный раствор разделяли на аликвоты (40 мкл/флакон) и замораживали при -20°C. 2) В день проведения анализа 40 мкл PBS добавляли в два ряда 96-луночного планшета с соединением (Costar, Cat#3357). 2) В день проведения анализа 40 мкл 2848 нМ сАМР исходного раствора переносили в первую лунку и смешивали с 40 мкл PBS (см. фигуру ниже) и затем осуществляли 16-точечное 2-кратное разведение путем переноса 40 мкл от более высокой конц. в более низкую конц. (3) Вручную переносили 10 мкл/лунку (в трех повторностях) раствора сАМР в аналитический планшет.
Стабильные НЕК293Т клетки, экспрессирующие hD1R (дикого типа или его мутант), выращивали в среде DMEM с высоким содержанием глюкозы (Invitrogen 11995-065), 10%-ной диализированной фетальной бычьей сывороткой (Invitrogen 26400-044), 1Х MEM NEAA (Invitrogen 1140), 25 мМ HEPES (Invitrogen 15630), 1Х пенициллина/стрептомицина (Invitrogen 15070-063) и 500 мкг/мл генентицина (Invitrogen 10131-035) при 37С и 5% CO2. В период от 72 до 96 часов после высевания клетки промывали забуференным фосфатом физиологическим раствором и к отделенным клеткам добавляли 0,25% трипсин-EDTA. Затем добавляли среды и клетки центрифугировали, а среды удаляли. Клеточные осадки ресуспендировали в среде для замораживания клеточных культур (Invitrogen 12648-056) при плотности 40 миллионов клеток/мл. Делили на аликвоты по одному мл клеток в криофлаконы и замораживали при -80°C для применения в hD1 (или его мутант) HTRF сАМР анализе.
Замороженные клетки подвергали быстрому оттаиванию, ресуспендировали в теплой среде и оставляли стоять на 5 мин перед центрифугированием (1000 об/мин) при комнатной температуре. Среду удаляли и клеточный осадок ресуспендировали в PBS, содержащем 500 нМ IBMX. Используя Multidrop Combi (Thermo Scientific), 5 мкл клеток/лунку при клеточной плотности приблизительно 1000 клеток/лунку добавляли в аналитический планшет (Greiner 784085), который содержал 5 мкл тестируемого соединения. Точная плотность клеток могла варьировать в зависимости от концентрации сАМР относительно стандартной кривой. Каждый планшет содержал положительные контроли в виде 5 мкМ допамина (конечная концентрация) и отрицательные контроли в виде 0,5% DMSO (конечная концентрация). Клетки и соединения инкубировали при комнатной температуре в течение 30 мин. Рабочие растворы cAMP-d2 и анти-сАМРкриптата готовили согласно инструкциям Cisbio. Используя Multidrop Combi, 5 мкл cAMP-d2 рабочий раствор добавляли в аналитический планшет, содержащий тестируемое соединение и клетки. Используя Multidrop Combi, 5 мкл рабочих растворов анти-сАМР-криптата добавляли в аналитический планшет, содержащий тестируемое соединение, клетки и cAMP-d2. Аналитические планшеты инкубировали в течение 1 часа при комнатной температуре, затем считывали с использованием аппарата для считывания планшетов Envision (Perkin Elmer), используя рекомендуемые Cisbio установки. сАМР стандартную кривую генерировали с использованием сАМР исходного раствора, предложенного в наборе Cisbio, которую затем использовали для преобразования необработанных логометрических данных в концентрации сАМР. Значения ЕС50 определяли с использованием логарифмической 4-параметрической модели. Эффективность в процентах для каждой кривой определяли по максимуму асимптоты этой построенной кривой и выражали в виде процента максимального ответа, индуцированного положительными контролями (5 мкм допамин) в каждом планшете.
Экспрессирующую конструкцию 3×НА-h D1 дикого типа (в pcDNA3.1+) получали из Missouri S&T cDNA Resource Center. Создавали несколько мутаций с использованием методик мутагенеза (например, используя набор для мутагенеза Stratagene Quick Change). Все мутации подтверждали посредством секвенирования. Экспрессию в клетках дикого типа и НЕК293 мутанте(ах) генерировали (для сАМР анализов) посредством временной трансфекции (48 часов) в клетках Freestyle НЕК 293F (Invitrogen). Число клеток/массу, используемое на точку данных, основывали на относительных уровнях экспрессии, как определено посредством вестерн-блот анализа.
D1R WT относится к дикому типу. Некоторые мутанты обозначены на основе рассчитанной модели гомологии D1, и нумерация мутантов согласуется с ранее опубликованной в литературе, см., например, N J Pollock, et.al, "Serine mutations in transmembrane V of the dopamine D1 receptor affect ligand interactions and receptor activation." J. Biol. Chem. 1992, 267 [25], 17780-17786. Мутанты обозначены по номеру, соответствующему их положению в исходной последовательности и трехбуквенному коду аминокислот. Например, D103A мутант относится к аминокислоте аспартату (D) в 103м положении в исходной последовательности, мутированной до аминокислоты аланин (A); S188I мутант относится к аминокислоте серину (S) в 188м положении в исходной последовательности, мутированной до аминокислоты изолейцин (I); и S198A мутант относится к аминокислоте серину (S) в 198м положении в исходной последовательности, мутированной до аминокислоты аланин (А).
Относительные уровни экспрессии 3×HA-hD1 мутанта нормализовали к уровням hD1 дикого типа посредством вестерн-блот анализа. Растворимые RIPA лизаты временно трансфицированных HEK293F клеток получали путем лизиса клеток при 4°C в течение 30 минут в RIPA буфере (Sigma) с ингибиторами протеазы и фосфатазы (Pierce). Эквивалентные количества суммарных растворимых RIPA лизатов (определено посредством ВСА анализа на суммарное содержание белка, Pierce) подвергали полиакриламидному гель-электрофорезу с додецилсульфатом натрия (SDS-PAGE), переносили на нитроцеллюлозу и вводили в качестве зонда анти-НА, а также анти-GAPDH антитела (Sigma). Суммарную НА иммунореактивность hD1 мутанта количественно сравнивали с GAPDH иммунореактивностью (HA/GAPDH) и наконец нормализовали к 3×HA-hD1 дикого типа (HA/GAPDH), используя программное обеспечение LiCor/Odyssey. Исходя из этих относительных HA/GAPDH соотношений по сравнению с диким типом, относительное количество клеточной массы или число клеток/лунку корректировали для уровней экспрессии каждого мутанта.
Осуществляли первую серию анализов сАМР. Из первой серии было определено, что результаты находились на верхнем конце линейного диапазона (для агонистов) стандартной кривой (диапазон предложен Cisbo), указывая на то, что эта первая серия представляет высшую плотность клеток/лунку. Как правило, высшая плотность клеток/лунку (в пределах линейного диапазона) является подходящей для мутантов, которые проявляют либо более низкую экспрессию, либо низкую активность; но не является такой подходящей для мутантов с более высокой активностью/экспрессией. В Таблице 4 показаны данные по ЕС50 в первой серии анализов сАМР. Осуществляли вторую серию анализов сАМР. Согласно сравнению со стандартной кривой, эта серия анализов показала более низкую плотность клеток/лунку, так как результаты находились в нижнем конце линейного диапазона (для агонистов) стандартной кривой. Как правило, более низкая плотность клеток/лунку (в пределах линейного диапазона) является подходящей для мутантов с более высокой активностью/экспрессией, но менее подходящей для мутантов с более низкой экспрессией/активностью. В Таблице 5 показаны данные по ЕС50 во второй серии анализов сАМР.
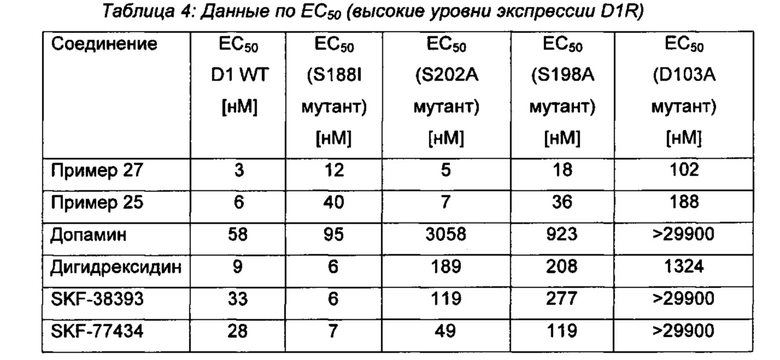
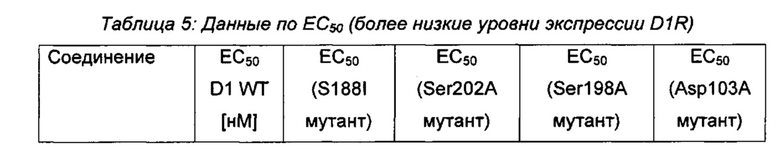
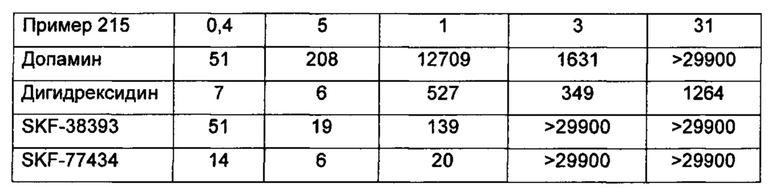
Результаты из обоих серий мутаций выявили, что многие из мутантных рецепторов имеют слабую активность (более высокие значения ЕС50) по сравнению с WT D1, отражая потерю взаимодействия между лигандом и рецептором с мутированной боковой цепью. В попытке определить вклад боковой цепи в активность, количественные значения сдвига активности между мутантным рецептором и WT рецептором, то есть данные по величинам кратности сдвига (Fold Shift), рассчитывали согласно уравнению: Fold Shift=ЕС50(Мутант)/ЕС50 (WT). Данные по величинам кратности сдвига показаны в Таблице 6.
В общем, анализы с большинством мутантных D1 рецепторов дали значения в "определенном в наборе" линейном диапазоне с более низким вариантом клетка/лунку. Однако, S198A показал слабые результаты для серий более низких клеток/лунку. Сравнение средних величин кратности сдвига для каждого тестированного мутанта в обоих сериях выявило, что величины кратности сдвига были более явно выраженными для серии с более низкой активностью примерно в 2,5 раза. Этот коэффициент определили путем регрессии средних значений log(foldshift) между сериями для всех мутантов:
log (fold-shift_lower)=0,3968+1,023*log(fold-shift_higher). (R∧2=0,92)
Свободный член уравнения регрессии 0,3968 отражает ~ 2,5х систематическую разницу между сериями.
Допамин, другой полный D1 агонист-производное катехола (дигидрексидин) и два других частичных D1 агониста-производных катехола (SKF-38393 и SKF-77434) продемонстрировали величину кратности сдвига менее чем примерно 4,0 относительно S188I мутанта, указывая на то, что они не взаимодействуют в значительной степени с Ser188 частью D1R. В противоположность этому, соединения Примеров 215 и 27 (полные D1 агонисты) и Примера 25 (частичный D1 агонист) продемонстрировали величину кратности сдвига более чем примерно 7,0 относительно S188I мутанта, указывая на то, что они взаимодействуют в значительной степени с Ser188 частью D1R.
Допамин и другой полный D1 агонист-производное катехола (дигидрексидин) продемонстрировали величину кратности сдвига более чем примерно 70 относительно S202A мутанта, указывая на то, что они взаимодействуют в значительной степени с Ser202 частью D1R. В противоположность этому, соединения Примеров 215 и 27 (полные D1 агонисты) продемонстрировали величину кратности сдвига менее чем примерно 4,0 относительно S202A мутанта, указывая на то, что они не взаимодействуют в значительной степени с Ser202 частью D1R.
Допамин и 3 других D1 агонистов-производных катехола, а также соединения Примеров 215 и 27 (полные D1 агонисты) и Примера 25 (частичный D1 агонист) продемонстрировали величину кратности сдвига более чем примерно 7,0 относительно D103A мутанта, указывая на то, что они взаимодействуют в значительной степени с Asp103 частью D1R. В среднем, величина кратности сдвига для агониста-производного катехола (более чем 100, 150 или 180) намного больше, чем для соединений Примеров 215 и 27 (полные D1 агонисты) и Примера 25 (частичный D1 агонист), указывая на то, что взаимодействия между D1R и соединением Примеров 215, 27 и 25, не являющимся производным катехола, менее сильные, чем между D1R и агонистами-производными катехола.
Допамин и 3 других D1 агониста-производных катехола, а также соединения Примеров 215 и 27 (полные D1 агонисты) и Примера 25 (частичный D1 агонист) продемонстрировали величину кратности сдвига более чем примерно 7,0 относительно S198A мутанта, указывая на то, что они взаимодействуют в значительной степени с Ser198 частью D1R. Однако, в среднем, величина кратности сдвига для полных агонистов-производных катехола (допамин и дигидрексидин, оба более чем 25, 30 или 35) больше, чем для соединений Примеров 215 и 27 (полные D1 агонисты), указывая на то, что взаимодействия между D1R и не являющимися производными катехола полными агонистами соединениями Примеров 215 и 27 менее сильные чем взаимодействия между D1R и полными агонистами-производными катехола.
% собственной активности каждого из тестированных соединений [то есть, максимальная эффективность в процентах (рассчитанная по максимальной концентрации сАМР) в сравнении с допамином] определяли с использованием сАМР данных из D1 сАМР HTRF анализов, как в Примере ВВ.
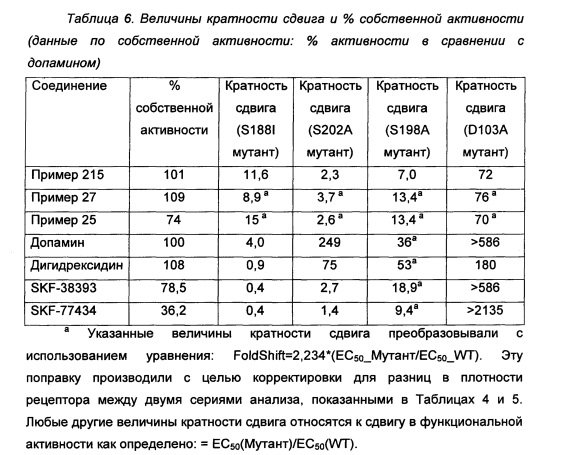
Пример DD: Анализы рекрутмента β-аррестина мембраны и TIRF микроскопия
Для всех исследований β-аррестина использовали стабильную клеточную линию U20S, совместно экспрессирующую человеческие рецепторы допамина D1(D1A) и человеческого β-аррестин-2-зеленого флуоресцентного слитого белка (GFP). Эту клеточную линию и лицензию получали от профессора Marc G. Caron, Duke University, Durham, NC, США. Стабильная клеточная линия U2OS обеспечивает флуоресцентный биосенсор β-аррестин-2-GFP, который может быть использован для оценки передачи сигнала рецептора, сопряженного с G-белком (GPCR), и GPCR-опосредованного рекрутмента В-аррестина мембраны с использованием методик визуализации, таких как флуоресцентная микроскопия (патенты США 7572888 и 7138240)(9); эта технология в настоящее время имеется в продаже как Transfluor Assay (Molecular Devices, USA). Клетки U2OS культивировали с селекцией антибиотика в среде DMEM (Invitrogen), содержащей 25 мМ глюкозу и 4 мМ L-глутамин, дополненной 10%-ной диализированной фетальной бычьей сывороткой, 200 мг/мл генетицина, 100 мг/мл зеоцина и 100 Ед/мл пенициллина/стрептомицина (все от Invitrogen), и инкубировали при 37°C в 5%-ном диоксиде углерода. В этих экспериментах использовали клетки с пассирования от четвертого по десятое. Клетки выращивали в 35 мм чашках для визуализирования со стеклянным дном (Mattek Corp). Клетки инкубировали в течение 1 ч в бессывороточной среде (SFM) и затем обрабатывали в течение 10 минут при 37°C 0,01%-ным DMSO (контроль) или 1 мкМ каждого тестируемого соединения, растворенного в SFM, с последующей непосредственной фиксацией на льду с 4%-ным параформальдегидом/1х забуференным фосфатом физиологическим раствором.
Использовали микроскопию полного внутреннего отражения (TIRFM). TIRFM представляет собой методику микроскопии, которая обеспечивает визуализацию плазматической мембраны и узкой области только внутри клетки, давая возможность визуализировать белки в плазматической мембране клеток, такие как D1 рецепторы и рекрутированный β-аррестин-GFP (см. Yudowski GA, von Zastrow М. "Investigating G protein-coupled receptor endocytosis and trafficking by TIR-FM"; Methods in Molecular Biology. 2011;756:325-32.). Все изображения получали с использованием флуоресцентного микроскопа Zeiss PS.1 Elyra Superresoution, оборудованного TIRF модулем. Изображения клеток получали с использованием TIRF и 100х масляно-иммерсионного объектива и специального 488 нм возбуждающего лазера. Оптимальное время воздействия и мощности лазера определяли с использованием клеток, обработанных допамином, которые проявляли максимальный сигнал β-аррестин-GFP мембраны, и идентичные параметры съемки использовали для всех клеток и условий. Для количественного определения рекрутмента β-аррестин-GFP мембраны идентифицировали индивидуальные клетки в изображениях микроскопа и интересующую область фиксировали для каждой клетки, используя программное обеспечение анализа визуализации ImageJ (Schneider СА, Rasband WS, Eliceiri KW. "NIH Image to ImageJ: 25 years of image analysis". Nature Methods. 2012;9(7):671-5). Порог интенсивности устанавливали путем оценки клеток, обработанных допамином, которые проявляли максимальный сигнал от β-аррестин-GFP плазматической мембраны. Тестировали интервал значений 10, 30, 60, 90 и так далее, и выбирали наименьший возможный порог, в данном случае 60, способный идентифицировать индивидуальную β-аррестин-GFP точку для последующего анализа. Генерировали фрагменты изображений для всех идентифицированных клеток и устанавливали суммарное количество мембранных β-аррестин-GFP точек/клетку, интегральную интенсивность/клетку и суммарную область/клетку. Индивидуальные объекты отбирали по размеру. Минимум из 60 клеток для каждого условия анализировали в трех независимых клеточных получениях и экспериментах. Определяли среднюю интенсивность мембранного β-аррестин-GFP/клетку и область точек/клетку и статистически значимые отличия сравнивали с помощью однопараметрического ANOVA с апостериорным анализом с использованием критерия Даннетта с использованием Graphpad Prism 5.02.
U20S клетки, стабильно экспрессирующие человеческие D1 рецепторы и человеческие β-аррестин-GFP белки, обрабатывали в течение 10 минут 0,01%-ным DMSO в бессывороточной среде (контроль) или 1 мкМ тестируемым соединением).
Тестируемые соединения включают допамин, дигидрексидин, SKF-81297, SKF-38393, SKF-77434, соединение Примера 5 (частичный агонист, 70%-ная собственная активность в отношении человеческого D1R в сравнении с допамином), Примера 9 (полный агонист, 92%-ная собственная активность в отношении человеческого D1R в сравнении с допамином), Примера 13 (частичный агонист, 58%-ная собственная активность в отношении человеческого D1R в сравнении с допамином) и Примера 25 (полный агонист, 88%-ная собственная активность в отношении человеческого D1R в сравнении с допамином). % собственной активности каждого из тестируемых соединений [то есть, максимальная эффективность в процентах (рассчитанная по максимальной концентрации AMP) в сравнении с допамином] определяли с использованием сАМР данных из D1 сАМР HTRF анализа, как в Примере ВВ.
Клетки непосредственно фиксировали и β-аррестин-GFP, локализованный на плазматической мембране, определяли с использованием флуоресцентной микроскопии полного внутреннего отражения (TIRFM).
В Таблицах 7 и 8 представлены количественные данные по β-аррестин-GFP сигналу в плазматической мембране клеток с использованием TIRFM для оценки суммарной интенсивности/клетку и суммарной области/клетку; агонисты D1 рецептора, не являющиеся производными катехола (соединения Примеров 5, 9, 13 и 25), показали значительно сниженные относительно допамина суммарную интенсивность и суммарную область β-аррестин-GFP плазматической мембраны. Все результаты представляют собой средние значения ± стандартное отклонение, усредненное от ≥ 60 клеток/условие, полученные из трех независимых экспериментов (n=3): а, р<0,05 против контроля; b, р<0,05 против допамина.
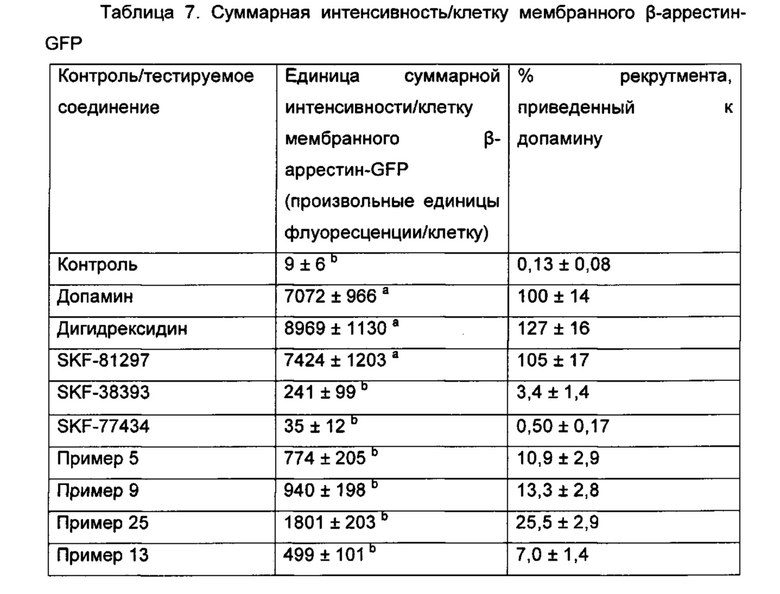
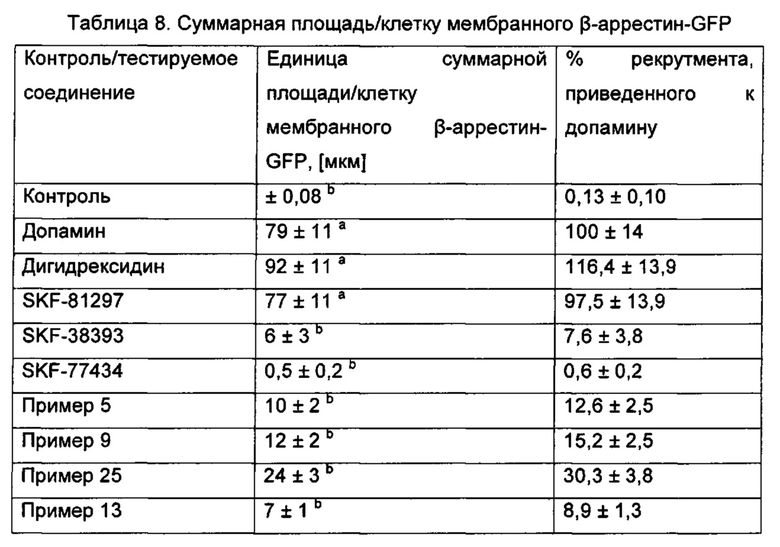
Как показано в Таблицах 7 и 8, допамин и два полных D1 агониста-производных катехола (дигидрексидин и SKF-81297) рекрутировали более чем примерно 95% β-аррестин-GFP в плазматическую мембрану относительно допамина (результат можно также качественно наблюдать из иллюстративных TIRFM изображений клеток, обработанных этими агонистами). В противоположность этому, каждое из соединений Примеров 9 и 25 (полные D1 агонисты, не являющиеся производными катехола) рекрутировало менее чем 60% (или 50%, или 40%, или 30%) β-аррестин-GFP в плазматическую мембрану относительно допамина. Каждое из тестированных частичных D1 агонистов (SKF-38393, SKF-77434 и соединения Примеров 5 и 13) рекрутировало менее чем 60% (или 50%, или 40%, или 30%) β-аррестин-GFP в плазматическую мембрану относительно допамина.
Пример ЕЕ: Анализы сАМР и десенсибилизации рецептора
Исходные стриарные нейроны получали из 18-суточных эмбрионов (Е18) крыс с помощью стандартных методик нейронального извлечения и помещали с плотностью 35000 клеток/лунку в покрытые полиорнитином/ламинином 96-луночные планшеты (BD Falcon). Были выбраны стриарные нейроны, так как они экспрессируют эндогенные D1-подобные рецепторы и представляют физиологически подходящую ткань для изучения десенсибилизации нейротрансмиттерных рецепторов in vitro. Нейроны культивировали в нейробазальной среде, дополненной В27, 1х Glutamax и пенициллином/стрептомицином (100 Ед/мл) (все от Invitrogen), и инкубировали при 37°C в 5%-ном диоксиде углерода в течение 14-16 суток до анализа. Для оценки D1R десенсибилизации нейроны в лунках предварительно обрабатывали в течение 120 минут 0,1%-ным DMSO в бессывороточной среде (Control/SFM) или 10 мкМ тестируемым соединением, растворенным в бессывороточной нейробазальной среде. После предварительной обработки клетки промывали дважды с 5-минутными интервалами свежей нейробазальной средой в концентрации 250 мкл/лунку. Способность D1-подобных рецепторов передавать сигнал затем проверяли путем обработки клеток в течение 30 минут 1 мкМ SKF-81297, представляющим собой D1-подобный селективный полный агонист-производное катехола, в присутствии 500 мкМ изобутилметилксантина. Концентрацию сАМР, накопленного в каждой лунке, определяли с использованием набора для анализа в динамическом диапазоне Cisbio HTRF сАМР (Cisbio) согласно протоколу, предложенному изготовителем. Концентрацию сАМР (нМ) из необработанных лунок интерполировали из сАМР стандартной кривой нелинейной регрессией по методу наименьших квадратов с использованием Graphpad Prism 5.02. Среднее ± стандартное отклонение для сАМР концентраций рассчитывали исходя из результатов, полученных в трех независимых экспериментах (n=3), каждый проанализированный в четырех параллельных определениях. % Десенсибилизации рассчитывали как процентное уменьшение в сАМР относительно контроля. Статистически значимые отличия сравнивали с помощью однопараметрического ANOVA с апостериорным анализом с использованием критерия Даннета с использованием Graphpad Prism 5.02.
Все результаты представляют собой среднее ± стандартное отклонение из трех независимых экспериментов, проанализированных в четырех параллельных определениях (n=3). *, р<0,05 против контроля.



Как показано в Таблице 9, предварительная обработка нейронов допамином, двумя полными D1 агонистами-производными катехола (дигидрексидин и SKF-81297) и двумя частичными D1 агонистами-производными катехола (SKF-38393 и SKF-77434) снижала в значительной степени DIR-опосредованную передачу сигнала сАМР. В противоположность этому, предварительная обработка полными D1 агонистами, не являющимися производными катехола (соединения Примеров 9 и 25), и частичными D1 агонистами, не являющимися производными катехола (соединения Примеров 5 и 13), не снижала в значительной степени DIR-опосредованную передачу сигнала сАМР (ближе к контролю).
Как показано в Таблице 10, допамин, два полных D1 агониста-производных катехола (дигидрексидин и SKF-81297) и два частичных D1 агониста-производных катехола (SKF-38393 и SKF-77434) десенсибилизировали в значительной степени D1R рецепторы (снижение более чем на примерно 30%, 40% или 50% по сравнению с контролем). В противоположность этому, полные D1 агонисты, не являющиеся производными катехола (соединения Примеров 9 и 25), и частичные D1 агонисты, не являющиеся производными катехола (соединения Примеров 5 и 13), демонстрируют пониженную десенсибилизацию (понижение только менее чем на примерно 25%, 20%, 18% или 15% по сравнению с контролем).
Различные модификации изобретения, в дополнение к описанным здесь, будут очевидны специалистам в данной области техники из данного описания изобретения. Такие модификации также предназначены входить в объем прилагаемой формулы изобретения. Каждый источник информации (включая все патенты, заявки на патент, журнальные статьи, книги и любые другие публикации), цитированный в настоящей заявке, включен в данное описание посредством ссылки во всей ее полноте.
| название | год | авторы | номер документа |
|---|---|---|---|
| ФЕНОКСИПИРИДИНИЛАМИДНЫЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ В ЛЕЧЕНИИ PDE4-ОПОСРЕДОВАННЫХ БОЛЕЗНЕННЫХ СОСТОЯНИЙ | 2009 |
|
RU2509077C2 |
| ПРОИЗВОДНЫЕ ФУРО[3,2-В]- И ТИЕНО[3,2-В]ПИРИДИНА В КАЧЕСТВЕ ИНГИБИТОРОВ TBK1 И IKKε | 2013 |
|
RU2622034C2 |
| ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛА, СОДЕРЖАЩИЕ ИХ КОМПОЗИЦИИ, ИХ ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ | 2004 |
|
RU2346938C2 |
| ИНГИБИТОРЫ КИНАЗЫ | 2012 |
|
RU2623734C9 |
| C-3 и C-17 модифицированные тритерпеноиды в качестве ингибиторов ВИЧ-1 | 2017 |
|
RU2716502C2 |
| ПОЛИГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ АНТАГОНИСТОВ МЕТАБОТРОПНОГО РЕЦЕПТОРА ГЛУТАМАТА | 2005 |
|
RU2381226C2 |
| ЛИГАНДЫ НИКОТИНОВЫХ АЦЕТИЛХОЛИНОВЫХ РЕЦЕПТОРОВ | 2007 |
|
RU2441007C2 |
| ИНГИБИТОРЫ АКТИВНОСТИ ПРОТЕИНТИРОЗИНКИНАЗЫ | 2009 |
|
RU2533827C2 |
| ПРОИЗВОДНОЕ НА ОСНОВЕ ДИГИДРОПИРИМИДО-КОЛЬЦА В КАЧЕСТВЕ ИНГИБИТОРА HBV | 2015 |
|
RU2693897C2 |
| Бициклические конденсированные гетероарильные или арильные соединения в качестве модуляторов IRAK4 | 2016 |
|
RU2684324C1 |
Изобретение относится к соединениям формулы I:
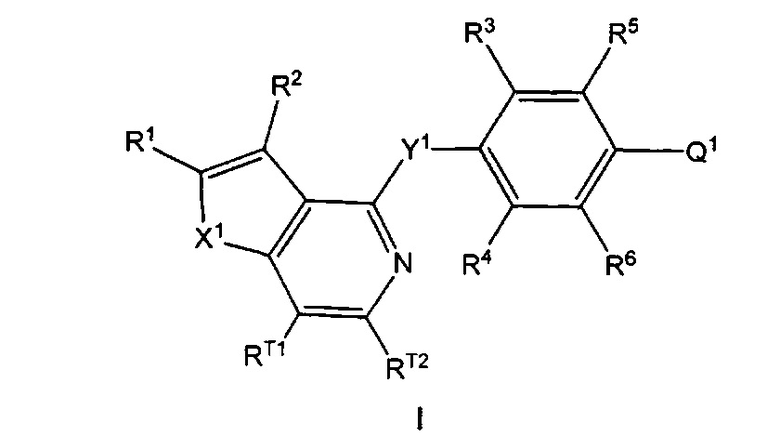 , а также к их фармацевтически приемлемым солям и N-оксидам. Технический результат: предложены новые соединения формулы I, а также фармацевтические композиции на их основе и их применение для агонизирования или частичного агонизирования рецепторов допамина D1. 3 н. и 14 з.п. ф-лы, 10 табл., 216 пр.
, а также к их фармацевтически приемлемым солям и N-оксидам. Технический результат: предложены новые соединения формулы I, а также фармацевтические композиции на их основе и их применение для агонизирования или частичного агонизирования рецепторов допамина D1. 3 н. и 14 з.п. ф-лы, 10 табл., 216 пр.
1. Соединение формулы I:
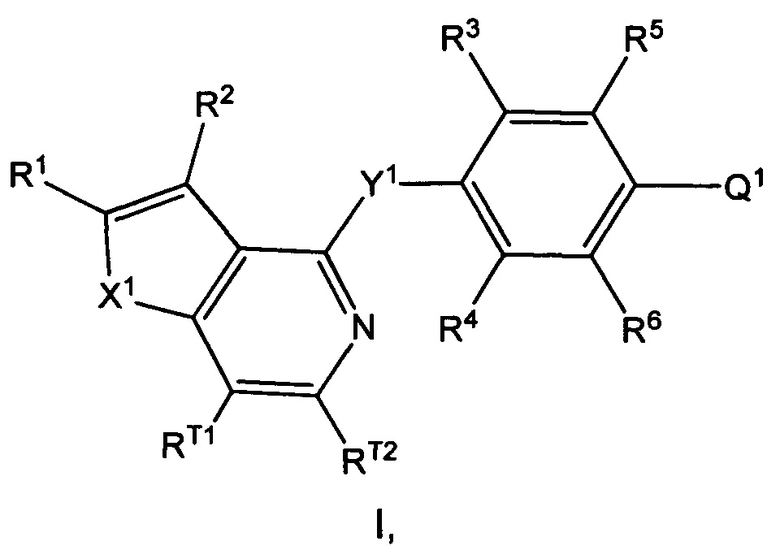
или его N-оксид, или фармацевтически приемлемая соль указанного соединения или указанного N-оксида, где:
X1 представляет собой О или S;
Y1 представляет собой О, S или NRN;
Q1 представляет собой N-содержащий 5-10-членный гетероарил или фенил, где гетероарил возможно замещен 1, 2, 3 или 4 независимо выбранными R7 и фенил возможно замещен 1 или 2 независимо выбранными R7a;
каждый из RT1 и RT2 независимо выбран из Н;
R1 выбран из Н;
R2 выбран из группы, состоящей из Н, галогена, -CN, C1-3алкила и С3-6циклоалкила;
каждый из R3 и R4 независимо выбран из группы, состоящей из Н, C1-6алкила, С3-6циклоалкила и галогена;
каждый из R5 и R6 независимо выбран из группы, состоящей из Н, галогена, -ОН, -CN, C1-6алкила C1-6галогеналкила, С3-7циклоалкила, групп -N(R8)(R9), -N(R10)(C(=O)R11), -C(=O)-N(R8)(R9) и -OR13, где каждый из указанных C1-6алкила и С3-7циклоалкила возможно замещен 1, 2 или 3 заместителями, каждый из которых независимо выбран из группы, состоящей из галогена, -CN, -ОН и группы -N(R14)(R15);
или R5 и R3 вместе с двумя атомами углерода, к которым они присоединены, образуют конденсированный N-содержащий 5- или 6-членный гетероарил, конденсированный 5-членный гетероциклоалкил, содержащий гетероатомы О, или конденсированное бензольное кольцо, каждый(ое) из которых возможно замещен(о) заместителями, выбранными из С1-3алкила;
каждый из R7 и R7a независимо выбран из группы, состоящей из галогена, -ОН, -CN, оксо, C1-6алкила, C1-6галогеналкила, С1-6алкокси, C1-6галогеналкокси, С3-7циклоалкила, 4-10-членного гетероциклоалкила, содержащего до 2 атомов азота, 5-10-членного гетероарила, содержащего от 1 до 3 атомов азота, групп -N(R14)(R15), -C(=O)-OR18 и -OR19, где каждая из указанных групп возможно замещена 1 или 2 заместителями, каждый из которых независимо выбран из группы, состоящей из галогена, ОН, С1-4алкила, С1-4гидроксилалкила, C1-4алкокси, оксо и С3-7циклоалкила;
каждый из R8 и R9 независимо выбран из группы, состоящей из Н, C1-6алкила, С3-10циклоалкила и 5-членного гетероарилалкила, содержащего до 2 атомов азота, где указанный гетероарилалкил возможно замещен C1-3алкилом или С3-7циклоалкилом;
R10 представляет собой Н;
R11 выбран из группы, состоящей из С1-6алкила и С3-7циклоалкила, каждый из которых возможно замещен С1-6алкокси;
R13 выбран из группы, состоящей из С1-10алкила, C1-6галогеналкила, С3-7циклоалкила, 6-членного гетероциклоалкила, содержащего 1 атом кислорода, 5-членного гетероарила, содержащего от 1 до 3 гетероатомов, выбранных из N, О и S, и 5-членного гетероарилалкила, содержащего от 1 до 3 гетероатомов, выбранных из N, О и S, каждый из которых возможно замещен 1, 2, 3 или 4 заместителями, каждый из которых независимо выбран из групп -N(R14)(R15), -C(=O)N(R14)(R15), -C(=O)N(R16)(OR18), -C(=O)-R18, -C(=O)-OR18, -OH, C1-6алкила, C1-6гидроксилалкила и С3-7циклоалкила;
каждый из R14 и R15 независимо выбран из группы, состоящей из Н, C1-6алкила и С3-10циклоалкила, где каждая из указанных групп возможно замещена 1 заместителем, выбранным из 5-6-членного гетероарила, содержащего от 1 до 2 гетероатомов, выбранных из N и S;
R16 представляет собой Н;
R18 представляет собой Н или выбран из группы, состоящей из С1-6алкила и С3-7циклоалкила;
R19 выбран из группы, состоящей из С1-6алкила и С3-7циклоалкила; и
RN выбран из группы, состоящей из Н, С1-6алкила и С3-6циклоалкила.
2. Соединение по п. 1, или его N-оксид, или фармацевтически приемлемая соль указанного соединения или указанного N-оксида, где Y1 представляет собой О.
3. Соединение по п. 1, или его N-оксид, или фармацевтически приемлемая соль указанного соединения или указанного N-оксида, где X1 представляет собой О.
4. Соединение по п. 1, или его N-оксид, или фармацевтически приемлемая соль указанного соединения или указанного N-оксида, где Q1 выбран из хинолинила, изохинолинила, 1H-имидазо[4,5-с]пиридинила, имидазо[1,2-а]пиридинила, 1H-пирроло[3,2-с]пиридинила, имидазо[1,2-а]пиразинила, имидазо[2,1-с][1,2,4]триазинила, имидазо[1,5-а]пиразинила, имидазо[1,2-а]пиримидинила, 1H-индазолила, 9H-пуринила, пиримидинила, пиразинила, пиридинила, пиридазинила, 1H-пиразолила, 1H-пирролила, 4Н-пиразолила, 4H-имидазолила, имидазо[1,2-а]пиримидинила, [1,2,4]триазоло[1,5-а]пиримидинила, [1,2,4]триазоло[4,3-b]пиридазинила, 1H-имидазолила, 3-оксо-2H-пиридазинила, 1H-2-оксо-пиримидинила, 1H-2-оксо-пиридинила, 2,4(1H,3H)-диоксо-пиримидинила и 1H-2-оксо-пиразинила, каждый из которых возможно замещен 1, 2, 3 или 4 независимо выбранными R7.
5. Соединение по п. 1, или его N-оксид, или фармацевтически приемлемая соль указанного соединения или указанного N-оксида, где Q1 выбран из:
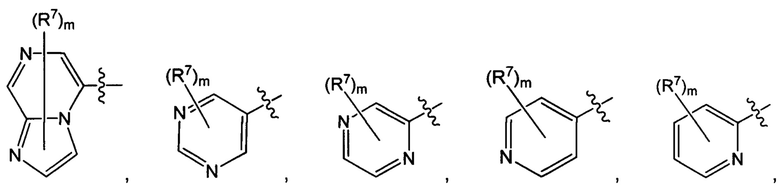
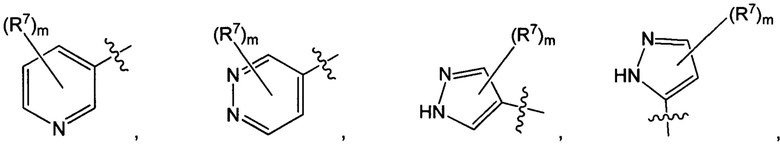
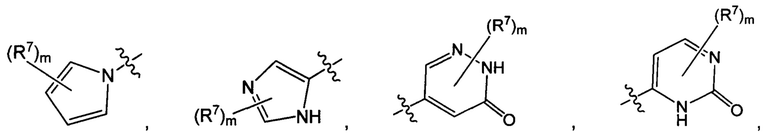
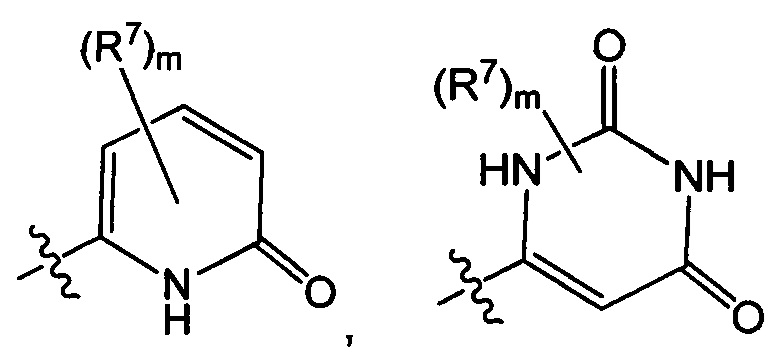 и
и 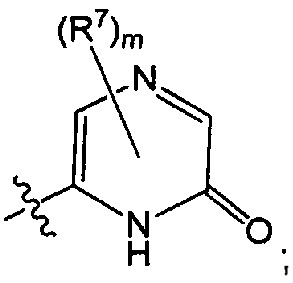 и
и
каждый m независимо представляет собой 0, 1, 2 или 3.
6. Соединение по п. 1, или его N-оксид, или фармацевтически приемлемая соль указанного соединения или указанного N-оксида, где RT1 и RT2 оба представляют собой Н; R1 представляет собой Н и R2 представляет собой Н, -CN, Br, C1-3алкил или циклопропил.
7. Соединение по п. 1, или его N-оксид, или фармацевтически приемлемая соль указанного соединения или указанного N-оксида, где каждый из R3 и R4 независимо выбран из группы, состоящей из Н, F, Cl и C1-3алкила.
8. Соединение по п. 1, или его N-оксид, или фармацевтически приемлемая соль указанного соединения или указанного N-оксида, где один из R5 и R6 представляет собой Н; и другой из R5 и R6 выбран из группы, состоящей из Н, -ОН, -CN, Cl, F, метила, этила, CF3, CH2F и -ОСН3.
9. Соединение по любому из пп. 1-8, или его N-оксид, или фармацевтически приемлемая соль указанного соединения или указанного N-оксида, где каждый из R7 и R7a независимо выбран из группы, состоящей из С1-4алкила, С1-4фторалкила, оксо, -ОН, С1-4алкокси и С1-4галогеналкокси, где С1-4алкил возможно замещен 1 или 2 заместителями, каждый из которых независимо выбран из галогена, ОН и С1-4алкокси.
10. Соединение по п. 1, или его N-оксид, или фармацевтически приемлемая соль указанного соединения или указанного N-оксида, где:
X1 представляет собой О;
Y1 представляет собой О;
Q1 представляет собой 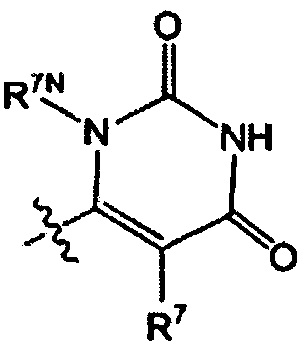 или
или 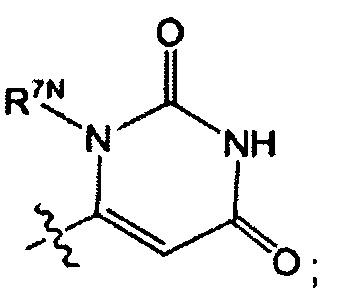
R7 отсутствует или представляет собой C1-3алкил; и
R7N представляет собой С1-3алкил, возможно замещенный 1 или 2 заместителями, каждый из которых независимо выбран из галогена, ОН и C1-4алкокси.
11. Соединение по п. 10, или его N-оксид, или фармацевтически приемлемая соль указанного соединения или указанного N-оксида, где R7 представляет собой метил или этил и R7N представляет собой C1-3алкил, возможно замещенный 1 или 2 заместителями, каждый из которых независимо выбран из галогена (например, F), ОН и С1-4алкокси.
12. Соединение по п. 10, или его N-оксид, или фармацевтически приемлемая соль указанного соединения или указанного N-оксида, где R7 представляет собой метил и R7N представляет собой метил.
13. Соединение по п. 10, или его N-оксид, или фармацевтически приемлемая соль указанного соединения или указанного N-оксида, где
RT1 и RT2 оба представляют собой Н;
R1 представляет собой Н;
R2 представляет собой Н или -CN;
R3 и R4 оба представляют собой Н; и
один из R5 и R6 представляет собой Н, F или метил; а другой из R5 и R6 выбран из группы, состоящей из Н, -ОН, -CN, Cl, F, метила, этила, CF3, CH2F и -ОСН3.
14. Соединение по любому из пп. 10-13, или его N-оксид, или фармацевтически приемлемая соль указанного соединения или указанного N-оксида, где Q1 представляет собой 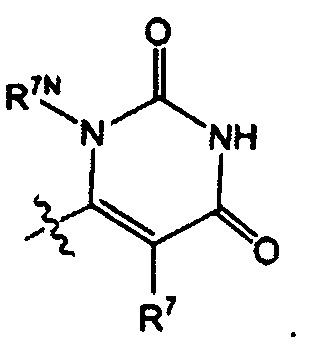
15. Соединение по п. 1, выбранное из:
4-[4-(4,6-диметилпиримидин-5-ил)-3-метилфенокси]фуро[3,2-с]пиридина;
2-(4,6-диметилпиримидин-5-ил)-5-(фуро[3,2-с]пиридин-4-илокси)-бензонитрила;
5-[2-фтор-4-(фуро[3,2-с]пиридин-4-илокси)фенил]-4,6-диметилпиридазин-3(2H)-она;
5-[4-(фуро[3,2-с]пиридин-4-илокси)фенил]-4,6-диметилпиридазин-3(2H)-она;
(+)-5-[4-(фуро[3,2-с]пиридин-4-илокси)-2-метилфенил]-4,6-диметилпиридазин-3(2H)-она;
(+)-5-[4-(фуро[3,2-с]пиридин-4-илокси)-2-метилфенил]-6-метилимидазо[1,2-а]пиразина;
(-)-5-[4-(фуро[3,2-с]пиридин-4-илокси)-2-метилфенил]-6-метилимидазо[1,2-а]пиразина;
5-[4-(фуро[3,2-с]пиридин-4-илокси)-2-метилфенил]-6-метилимидазо[1,2-а]пиразина;
4-[4-(4,6-диметилпиримидин-5-ил)-3-фторфенокси]фуро[3,2-с]пиридина;
(-)-6-[4-(фуро[3,2-с]пиридин-4-илокси)-2-метилфенил]-1,5-диметилпиразин-2(1H)-она;
(+)-6-[4-(фуро[3,2-с]пиридин-4-илокси)-2-метилфенил]-1,5-диметилпиразин-2(1H)-она;
6-[4-(фуро[3,2-с]пиридин-4-илокси)-2-метилфенил]-1,5-диметилпиразин-2(1H)-она;
6-[4-(фуро[3,2-с]пиридин-4-илокси)-2-метилфенил]-1,5-диметилпиримидин-2(1H)-она;
4-[4-(4,6-диметилпиримидин-5-ил)-2-фторфенокси]фуро[3,2-с]пиридина;
5-[4-(фуро[3,2-с]пиридин-4-илокси)-2-метилфенил]-2,4,6-триметилпиридазин-3(2H)-она;
5-[4-(фуро[3,2-с]пиридин-4-илокси)-2-метилфенил]-4-метилпиридазин-3(2H)-она;
(+)-4-[4-(3,5-диметилпиридазин-4-ил)-3-метилфенокси]фуро[3,2-с]-пиридина;
(-)-4-[4-(3,5-диметилпиридазин-4-ил)-3-метилфенокси]фуро[3,2-с]-пиридина;
4-[4-(3,5-диметилпиридазин-4-ил)-3-метилфенокси]фуро[3,2-с]пиридина;
4-[4-(3,5-диметил-6-оксо-1,6-дигидропиридазин-4-ил)фенокси]фуро[3,2-с]-пиридин-3-карбонитрила;
(-)-4-[4-(3,5-диметилпиридазин-4-ил)-3-метоксифенокси]фуро[3,2-с]-пиридина;
(+)-4-[4-(3,5-диметилпиридазин-4-ил)-3-метоксифенокси]фуро[3,2-с]-пиридина;
4-[4-(3,5-диметилпиридазин-4-ил)-3-метоксифенокси]фуро[3,2-с]пиридина;
(-)-6-[4-(фуро[3,2-с]пиридин-4-илокси)-2-метилфенил]-1,5-диметилпиримидин-2,4(1H,3H)-диона; и
6-[4-(фуро[3,2-с]пиридин-4-илокси)фенил]-1,5-диметилпиримидин-2,4(1H,3H)-диона,
или его N-оксид, или фармацевтически приемлемая соль указанного соединения или указанного N-оксида.
16. Фармацевтическая композиция для модулирования рецепторов допамина D1, содержащая эффективное количество соединения по любому из пп. 1-15, или его N-оксида, или фармацевтически приемлемой соли указанного соединения или указанного N-оксида и фармацевтически приемлемый носитель.
17. Применение соединения по любому из пп. 1-15, или его N-оксида, или фармацевтически приемлемой соли указанного соединения или указанного N-оксида для агонизирования или частичного агонизирования рецепторов допамина D1.
| WO 2004009557 A1, 29.01.2004 | |||
| WO 2008020306 A2, 21.02.2008 | |||
| СОЕДИНЕНИЯ КОНДЕНСИРОВАННОГО ФУРАНА | 2004 |
|
RU2302422C2 |

