ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Данное изобретение относится к соединениям, пригодным для лечения аутоиммунных и воспалительных заболеваний, связанных с киназой, ассоциированной с рецептором интерлейкина-1 (IRAK), и, более конкретно, к соединениям, которые модулируют функцию IRAK4.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Протеинкиназы представляют собой семейство ферментов, которые катализируют фосфорилирование конкретных остатков в белках и широко подразделены на тирозин- и серин/треонинкиназы. Как полагают, ненадлежащая активность в результате дисрегуляции определенных киназ посредством различных механизмов лежит в основе причин многих заболеваний, включая, но без ограничения ими, рак, сердечно-сосудистые заболевания, аллергии, астму, респираторные заболевания, аутоиммунные заболевания, воспалительные заболевания, заболевания костей, метаболические расстройства и неврологические и нейродегенеративные заболевания. В связи с этим ведутся поиски эффективных и селективных ингибиторов киназ в качестве потенциальных средств лечения различных заболеваний человека.
Существует значительный интерес к нацеливанию врожденной иммунной системы в лечении аутоиммунных заболеваний и стерильного воспаления. Рецепторы врожденной иммунной системы обеспечивают первую линию защиты от бактериальных и вирусных воздействий. Эти рецепторы распознают бактериальные и вирусные продукты, а также провоспалительные цитокины и таким образом инициируют сигнальный каскад, который в конце концов приводит к апрегуляции воспалительных цитокинов, таких как TNFα (фактор некроза опухоли-альфа), IL6 (интерлекин-6) и интерфероны. Недавно стало очевидно, что самостоятельно вырабатываемые лиганды, такие как нуклеиновые кислоты, и продукты воспаления, такие как белок В1 группы высокой подвижности (HMGB1), и конечные продукты усиленного гликозилирования (AGE), являются лигандами Толл-подобных рецепторов (TLR), которые являются ключевыми рецепторами врожденной иммунной системы ( 2003, Kanzler et al., 2007, Wagner 2006). Это демонстрирует роль TLR в инициации и сохранении воспаления из-за аутоиммунитета.
2003, Kanzler et al., 2007, Wagner 2006). Это демонстрирует роль TLR в инициации и сохранении воспаления из-за аутоиммунитета.
Киназа 4, ассоциированная с рецептором интерлейкина-1 (IRAK4), представляет собой повсеместно экспрессируемую серин/треонинкиназу, вовлеченную в регуляцию врожденного иммунитета (Suzuki & Saito 2006). IRAK4 отвечает за инициацию сигнального пути от TLR и представителей семейства рецепторов IL-1/18. Сообщалось, что киназа-инактивирующие нокины и направленные делеции IRAK4 у мышей вызывали снижение TLR- и IL-1-индуцированных провоспалительных цитокинов (Kawagoe et al., 2007; Fraczek et al., 2008; Kim et al., 2007). Было также показано, что мыши с нокином, утратившие функцию киназы IRAK4, были резистентны к индуцированному воспалению суставов в моделях антиген-индуцированного артрита (AIA) и вызванного переливанием сыворотки (K/BxN) артрита (Koziczak-Holbro 2009). Аналогично, оказалось, что люди с дефектом IRAK4 также демонстрируют неспособность реагировать на провокацию Толл-лигандами и IL-1 (Hernandez & Bastian 2006). Однако иммунодефицитный фенотип субъектов с отсутствием IRAK4 узко ограничен провокацией грамположительными бактериями, но не грамотрицательными бактериями, вирусами или грибами. Такая грамположительная чувствительность также снижается с возрастом, что влечет за собой резервные или компенсаторные механизмы врожденного иммунитета в отсутствии IRAK4 (Lavine et al., 2007).
Эти данные показывают, что ингибиторы активности киназы IRAK4 должны иметь терапевтическое значение в лечении цитокин-индуцированных аутоиммунных заболеваний, обладая при этом минимальными иммуносупрессивными побочными эффектами. Дополнительные недавние исследования свидетельствуют о том, что нацеливание на IRAK4 может быть полезным в других воспалительных патологиях, таких как атеросклероз и диффузная крупноклеточная В-клеточная лимфома (Rekhter et al., 2008; Ngo et al., 2011). Следовательно, ингибиторы активности киназы IRAK4 являются потенциальными терапевтическими средствами для широкого круга заболеваний, включая, но без ограничения ими, аутоиммунитет, воспаление, сердечнососудистые заболевания, рак и метаболические заболевания. Дополнительную информацию см. в следующих ссылках: N. Suzuki и Т. Saito, Trends in Immunology, 2006, 27, 566. Т. Kawagoe, S. Sato, A. Jung, M. Yamamoto, K. Matsui, H. Kato, S. Uematsu, O. Takeuchi and S. Akira, Journal of Experimental Medicine, 2007, 204, 1013. J. Fraczek, T. W. Kim, H. Xiao, J. Yao, Q. Wen, Y. Li, J.-L. Casanova, J. Pryjma and X. Li, Journal of Biological Chemistry, 2008, 283, 31697. T. W. Kim, K. Staschke, K. Bulek, J. Yao, K. Peters, K.-H. Oh, Y. Vandenburg, H. Xiao, W. Qian, T. Hamilton, B. Min, G. Sen, R. Gilmour and X. Li, Journal of Experimental Medicine, 2007, 204, 1025. M. Koziczak-Holbro, A. Littlewood-Evans, B. Pollinger, J. Kovarik, J. Dawson, G. Zenke, C. Burkhart, M. Muller and H. Gram, Arthritis & Rheumatism, 2009, 60, 1661. M. Hernandez and J. F. Bastian, Current Allergy and Asthma Reports, 2006, 6, 468. E. Lavine, R. Somech, J. Y. Zhang, A. Puel, X. Bossuyt, C. Picard, J. L. Casanova and С.M. Roifman, Journal of Allergy and Clinical Immunology, 2007,120, 948. M. Rekhter, K. Staschke, T. Estridge, P. Rutherford, N. Jackson, D. Gifford-Moore, P. Foxworthy, C. Reidy, X.-d. Huang, M. Kalbfleisch, K. Hui, M.-S. Kuo, R. Gilmour and C. J. Vlahos, Biochemical and Biophysical Research Communications, 2008, 367, 642. O'Neill, L. A. (2003). "Therapeutic targeting of Tolllike receptors for inflammatory and infectious diseases." Curr Opin Pharmacol 3(4): 396. Kanzler, H. et al., (2007) "Therapeutic targeting of innate immunity with toll-like receptor agonists and antagonists." Nature Medicine 13:552. Wagner, H. (2006) "Endogenous TLR ligands and autoimmunity" Advances in Immunol 91: 159. Ngo, V.N. et al. (2011) "Oncogenically active MyD88 mutations in human lymphoma" Nature 470: 115.
В находящейся одновременно на рассмотрении заявке на патент US 14/678114, поданной компанией Pfizer Inc 3 апреля 2015 года, и в находящейся одновременно на рассмотрении заявке на патент US 62/204521, поданной 13 августа 2015 года, описаны ингибиторы IRAK4, и они включены в данную заявку посредством ссылки во всей своей полноте для всех целей.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
В изобретении предложены соединения Формулы I,

где
каждый из X, X', Y и Y' независимо представляет собой СН или N; Z представляет собой С или N; при условии, что не более чем три из X, X', Z, Y и Y' представляют собой N;
R1 представляет собой C1-С6алкил или -(C1-С6алкил)n(C1-С6циклоалкил), где алкил или циклоалкил возможно замещен дейтерием, галогеном, CN, ОН, или C1-С6алкокси;
R2 представляет собой водород или метил;
R3 представляет собой водород, дейтерий, галоген, нитрил, -(CH2)tNR8aR8b, -(СН2)t(6-10-членный арил) или -(СН2)t(5-10-членный гетероарил), имеющий от одного до трех гетероатомов, выбранных из N, О или S, где указанный арил или гетероарил возможно замещен одной-тремя группами C1-С6алкил, дейтерий, галоген, CN, ОН, гидроксиC1-С6алкил или C1-С6алкокси; где алкил возможно замещен гидроксилом, галогеном, CN или C1-С3алкокси;
каждый из R4a и R4b представляет собой водород, фтор, ОН, C1-С3алкокси, или CH2OR7, где R7, взятый вместе с R1, представляет собой C1-С4алкилен, возможно замещенный галогеном или алкилом;
каждый из R5a и R5b представляет собой водород, C1-С3алкил или C1-С3-алкокси, где указанный алкил или алкокси возможно замещен одной-тремя группами дейтерий, галоген, ОН или CN; или R5a и R5b, взятые вместе с атомом, к которому они присоединены, образуют C3-С7циклоалкил или C3-С7гетероциклоалкил, где указанный циклоалкил или гетероциклоалкил возможно замещен одной-тремя группами дейтерий, галоген, ОН, CN или C1-С3алкил;
R6 представляет собой водород или C1-С3алкил; или R5b и R6, взятые вместе с атомами, к которым они присоединены, образуют C3-С7циклоалкил или C3-С7гетероциклоалкил, где указанный циклоалкил или гетероциклоалкил возможно замещен одной-тремя группами дейтерий, галоген, ОН, CN или C1-С3алкил;
каждый из R8a и R8b независимо представляет собой водород, -S(O)2R9 или -C(O)R9;
R9 представляет собой C1-С6алкил, C1-С6циклоалкил, 6-10-членный арил или 5-10-членный гетероарил, имеющий от одного до трех гетероатомов, где указанный алкил, циклоалкил, арил или гетероарил возможно замещен одной-тремя группами C1-С6алкил, галоген, CN, ОН, C1-С6алкокси или C1-С6гидрокси;
n равен 0 или 1;
t равен 0, 1, 2 или 3;
или фармацевтически приемлемая соль указанного соединения, или таутомер указанного соединения или указанной соли.
В изобретении также предложены фармацевтические композиции, содержащие эти соединения, способы применения этих соединений, комбинированные терапии с использованием этих соединений и других терапевтических агентов и способы получения этих соединений. В изобретении также предложены промежуточные соединения, полезные в получении соединений по изобретению.
В частности, новые бициклические соединения Формулы I по настоящему изобретению играют терапевтическую роль в ингибирования IRAK4, полезную в области заболеваний и/или расстройств, которые включают, но без ограничения ими, раковые заболевания, аллергические заболевания, аутоиммунные заболевания, воспалительные заболевания, и/или расстройства, и/или состояния, ассоциированные с воспалением и болью, пролиферативные заболевания, гематопоэтические расстройства, гемобластозы, костные расстройства, фиброзные заболевания и/или расстройства, метаболические расстройства, мышечные заболевания и/или расстройства, респираторные заболевания, легочные расстройства, заболевания генетического развития, неврологические и нейродегенеративные заболевания и/или расстройства, хронические воспалительные демиелинизирующие невропатии, сердечнососудистые, сосудистые или сердечные заболевания, офтальмологические/глазные заболевания, заживление ран, инфекционные и вирусные заболевания. Следовательно, ингибирование IRAK4 будет эффективным для множества терапевтических показаний в широком диапазоне неудовлетворенных потребностей.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение может быть легче понято со ссылкой на следующее подробное описание иллюстративных воплощений изобретения и примеры, включенные в него. Следует понимать, что данное изобретение не ограничено конкретными способами синтеза, которые, конечно, могут различаться. Также следует понимать, что терминология, использованная в данном описании изобретения, предназначена только для описания конкретных воплощений и ее не следует считать ограничивающей.
Все патенты, патентные заявки и источники, на которые даны ссылки в данном описании изобретения, включены в данную заявку посредством ссылки во всей своей полноте.
Другие аспекты и преимущества данного изобретения будут очевидны из данного описания изобретения и прилагаемой формулы изобретения, которая описывает изобретение. Данное изобретение имеет много аспектов, которые не обязательно полностью охвачены формулой изобретения. Однако следует понимать, что все такие новые объекты являются частью изобретения.
Определения
Если иное не определено в данном описании изобретения, научные и технические термины, используемые в связи с настоящим изобретением, имеют значение, обычно подразумеваемое специалистами обычной квалификации в данной области. При использовании в описании изобретения и прилагаемой формуле изобретения, формы единственного числа включают множественные определяемые объекты, если контекст явно не указывает на иное.
Термин "примерно" относится к относительному термину, обозначающему аппроксимацию от плюс или минус 10% от номинального значения, к которому он относится, в одном воплощении до плюс или минус 5%, в другом воплощении до плюс или минус 2%. Для области данного описания изобретения этот уровень аппроксимации является подходящим, если конкретно указанное значение не требует более узкого диапазона.
Термин "алкил" относится к линейной или разветвленной насыщенной углеводородной группировке, состоящей исключительно из атомов углерода и водорода. В одном воплощении от одного до шести атомов углерода; и в другом воплощении от одного до четырех атомов углерода; и в еще одном воплощении от одного до трех атомов углерода. Неограничивающие примеры таких заместителей включают метил, этил, пропил (включая н-пропил и изопропил), бутил (включая н-бутил, изобутил, втор-бутил и трет-бутил), пентил, изоамил, гексил и тому подобное. Если целесообразно, алкил возможно может быть замещен по каждому углероду, как определено в формуле изобретения. Типичное замещение включает, без ограничения ими, фтор, хлор, ОН, циано, алкил (возможно замещенный), циклоалкил и тому подобное.
В некоторых случаях число атомов углерода в углеводородном заместителе (т.е. алкиле, циклоалкиле и т.д.) указано посредством префикса "Сх-Су-" или "Сх-у", где x представляет собой минимальное, а у представляет собой максимальное число атомов углерода в этом заместителе. Таким образом, например, "C1-С6-алкил" или "C1-С6алкил" относится к алкильному заместителю, содержащему от 1 до 6 атомов углерода. Иллюстрируя далее, C3-С6-циклоалкил или C3-С6-циклоалкил относится к насыщенному циклоалкилу, содержащему от 3 до 6 кольцевых атомов углерода.
Если не указано иное, "алкилен" сам по себе или как часть другого термина относится к насыщенному, разветвленному, или прямоцепочечному, или циклическому углеводородному бирадикалу с указанным количеством атомов углерода, обычно с 1-6 атомами углерода, и имеющему два одновалентных радикальных центра, полученных путем удаления двух атомов водорода от одного и того же или двух разных атомов углерода родительского алкана. Типичные алкиленовые радикалы включают, но без ограничения ими, метилен (-СН2-), 1,2-этилен (-СН2СН2-), 2,2-диметилен, 1,3-пропилен (-СН2СН2СН2-), 2-метилпропилен, 1,4-бутилен (-СН2СН2СН2СН2-) и тому подобное; возможно замещенный, при необходимости, 1-5 подходящими заместителями, как определено выше, такими как фтор, хлор, дейтеро, циано, трифторметил, (C1-С6)алкокси, (C6-С10)арилокси, трифторметокси, дифторметокси или (C1-С6)алкил. Когда соединения по изобретению содержат C2-6алкенильную группу, это соединение может существовать в виде чистой E-(entgegen, напротив) формы, чистой Z-(zusammen, вместе) формы или в виде любой их смеси.
"Алкилиден" или "алкенил" относится к двухвалентной группе, образованной из алкана путем удаления двух атомов водорода от одного того же атома углерода, свободные валентности которого являются частью двойной связи, возможно замещенной, как описано в данном описании изобретения. Термин "алкилиден" также включает "аллены", где один атом углерода имеет двойные связи с каждым из его двух соседних углеродных центров, такие как, например, пропадиен. Если целесообразно, алкенил возможно может быть замещен по каждому атому углерода, как определено в формуле изобретения, возможно замещен, если целесообразно, 1-5 подходящими заместителями, как определено выше и в данном описании изобретения, такими как фтор, хлор, дейтеро, циано, трифторметил, (C1-С6)алкокси, (C6-С10)арилокси, трифторметокси, дифторметокси или (C1-С6)алкил.
"Алкинил" относится к алифатическому углеводороду, имеющему по меньшей мере одну углерод-углеродную тройную связь, включающему прямоцепочечные, разветвленные или циклические группы, имеющие по меньшей мере одну углерод-углеродную тройную связь, возможно замещенные, как описано в данном описании изобретения. Предпочтительно, он представляет собой низший алкинил, имеющий от 2 до 6 атомов углерода. Например, при использовании в данном описании изобретения, термин "C2-С6алкинил" используется в данном описании изобретения для обозначения прямого или разветвленного углеводородного алкинильного радикала, как определено выше, имеющего от 2 до 6 атомов углерода и одну тройную связь. При необходимости, алкинил возможно может быть замещен по каждому атому углерода, как определено в формуле изобретения. Типичное замещение включает, но без ограничения ими, возможное замещение, при необходимости, 1-5 подходящими заместителями, как определено выше и в данном описании изобретения, такими как фтор, хлор, дейтеро, циано, трифторметил, (C1-С6)алкокси, (C6-С10)арилокси, трифторметокси, дифторметокси или (C1-С6)алкил.
Термин "циклоалкил" относится к неароматическому кольцу, содержащему от 3 до 10 атомов углерода, которое полностью гидрировано, состоящему из моно-, би- или трициклических колец. Соответственно, циклоалкил может представлять собой одно кольцо, которое обычно содержит от 3 до 7 кольцевых атомов. Примеры включают, но без ограничения ими, циклопропил, циклобутил, циклопентил, и циклогексил. Альтернативно, 2 или 3 кольца могут быть конденсированными вместе, например бициклодеканил и декалинил. Термин "циклоалкил" также включает мостиковые бициклоалкильные системы, такие как, но без ограничения ими, бицикло[2.2.1]гептан и бицикло[1.1.1]пентан. Циклоалкильная группа возможно может быть замещена, как описано в данном описании изобретения, если это целесообразно, 1-5 подходящими заместителями, как определено выше, такими как фтор, хлор, дейтеро, циано, трифторметил, (C1-С6)алкокси, (C6-С10)арилокси, трифторметокси, дифторметокси или (C1-С6)алкил.
Термин "гетероциклоалкил" означает одновалентную насыщенную группировку, состоящую из одного до трех колец, включающих один, два, три или четыре гетероатома (выбранных из N, О или S) и от трех до 10 атомов углерода. Гетероциклоалкил возможно может быть замещен, как определено в данном описании изобретения. Примеры гетероциклоалкильных группировок включают, но без ограничения ими, возможно замещенный пиперидинил, пиперазинил, гомопиперазинил, азепинил, пирролидинил, пиразолидинил, имидазолинил, имидазолидинил, пиридинил, пиридазинил, пиримидинил, оксазолидинил, изоксазолидинил, морфолинил, тиазолидинил, изотиазолидинил, хинуклидинил, хинолинил, изохинолинил, бензимидазолил, тиадиазолидинил, бензотиазолидинил, бензоазолидинил, дигидрофурил, тетрагидрофурил, дигидропиранил, тетрагидропиранил, тиаморфолинил, тиаморфолинилсульфоксид, тиаморфилинилсульфон, дигидрохинолинил, тетрагидрохинолинил, тетрагидроизохинолинил и тому подобное. Гетероциклоалкилы возможно могут быть замещены, если это целесообразно, 1-5 подходящими заместителями, как определено в данном описании изобретения, такими как фтор, хлор, дейтеро, циано, трифторметил, (C1-С6)алкокси, (C6-С10)арилокси, трифторметокси, дифторметокси или (C1-С6)алкил.
Если не указано иное, термин "гетероалкил" сам по себе или в комбинации с другим термином, если не указано иное, означает насыщенный, прямой или разветвленный углеводородный радикал, состоящий из указанного числа атомов углерода и из одного-трех гетероатомов, выбранных из группы, состоящей из О, N и S, и где атомы азота и серы возможно могут быть окислены, и атом азота возможно может быть кватернизирован. Гетероатом(ы) О, N и S могут быть помещены в любое внутреннее положение гетероалкильной группы. Гетероатом S может быть помещен в любое положение гетероалкильной группы, включая положение, в котором алкильная группа присоединена к остатку молекулы. Вплоть до двух гетероатомов могут быть последовательными.
Если не указано иное, термин "гетероалкилен" сам по себе или как часть другого заместителя означает двухвалентную группу, полученную из гетероалкила (как определено выше). В гетероалкиленовых группах гетероатомы также могут занимать любой один или оба конца цепи.
Термин "алкокси" и "алкилокси", который может быть использован взаимозаменяемо, относится к группировке формулы -OR, где R представляет собой прямоцепочечнную насыщенную алкильную или разветвленную насыщенную алкильную группировку, как определено в данном описании изобретения, присоединенную через атом кислорода. Группа алкокси возможно может быть замещена, как определено в данном описании изобретения. Неограничивающие примеры таких групп алкокси представляют собой метокси, этокси, пропокси, изопропокси, бутокси, изобутокси, трет-бутокси, пентокси и тому подобное.
пентокси и тому подобное.
Термин "арил" означает карбоциклическую ароматическую систему, содержащую один или два кольца, где такие кольца могут быть конденсированными. Если кольца конденсированы, то одно из колец должно быть полностью ненасыщенным, и конденсированное(ые) кольцо(кольца) может(гут) быть полностью насыщенными, частично ненасыщенными или полностью ненасыщенными. Термин "конденсированный" означает, что присутствует второе кольцо (т.е. присоединенное или образованное), имеющее два общих (т.е. обобществленных) соседних атомов с первым кольцом. Термин "конденсированный" эквивалентен термину "сочлененный". Арильная группа возможно может быть замещена, как определено в данном описании изобретения. Термин "арил" охватывает ароматические радикалы, такие как фенил, нафтил, тетрагидронафтил, инданил, бифенил, бензо[b][1,4]оксазин-3(4Н)-онил, 2,3-дигидро-1H-инденил и 1,2,3,4-тетрагидронафталинил. Арилы возможно могут быть замещены, при необходимости, 1-5 подходящими заместителями, как определено выше, такими как фтор, хлор, дейтеро, циано, трифторметил, (C1-С6)алкокси, (C6-С10)арилокси, трифторметокси, дифторметокси или (C1-С6)алкил.
Термин "гетероарил" относится к ароматической кольцевой структуре, содержащей от 5 до 6 кольцевых атомов, в которой по меньшей мере один из кольцевых атомов представляет собой гетероатом (т.е. кислород, азот или серу), где остальные кольцевые атомы независимо выбраны из группы, состоящей из углерода, кислорода, азота и серы. Примеры гетероарильных заместителей включают 6-членные кольцевые заместители, такие как пиридил, пиразил, пиримидинил и пиридазинил; и 5-членные кольцевые заместители, такие как триазолил, имидазолил, фуранил, тиофенил, пиразолил, оксазолил, изоксазолил, тиазолил, 1,2,3-, 1,2,4-, 1,2,5- или 1,3,4-оксадиазолил и изотиазолил. В группе, которая имеет гетероарильный заместитель, кольцевой атом этого гетероарильного заместителя, который связан с группой, может представлять собой один из гетероатомов, или он может представлять собой кольцевой атом углерода. Аналогично, если гетероарильный заместитель в свою очередь замещен группой или заместителем, эта группа или заместитель может быть связан(а) с одним из гетероатомов или может быть связан(а) с кольцевым атомом углерода. Термин "гетероарил" также включает пиридил-N-оксиды и группы, содержащие пиридин-N-оксидное кольцо.
Другие примеры включают фурил, тиенил, оксазолил, тиазолил, имидазолил, пиразолил, триазолил, тетразолил, изоксазолил, изотиазолил, оксадиазолил, тиадиазолил, пиридинил, пиридазинил, пиримидинил, пиразинил, пиридин-2(1H)-онил, пиридазин-2(1H)-онил, пиримидин-2(1H)-онил, пиразин-2(1H)-онил, имидазо[1,2-α]пиридинил, пиразоло[1,5-α]пиридинил, 5,6,7,8-тетрагидроизохинолинил, 5,6,7,8-тетрагидрохинолинил, 6,7-дигидро-5H-циклопента[b]пиридинил, 6,7-дигидро-5H-циклопента[с]пиридинил, 1,4,5,6-тетрагидроциклопента[с]пиразолил, 2,4,5,6-тетрагидроциклопента[с]пиразолил, 5,6-дигидро-4H-пирроло[1,2-b]пиразолил, 6,7-дигидро-5H-пирроло[1,2-b][1,2,4]триазолил, 5,6,7,8-тетрагидро-[1,2,4]триазоло[1,5-α]пиридинил, 4,5,6,7-тетрагидропиразоло[1,5-α]пиридинил, 4,5,6,7-тетрагидро-1H-индазолил и 4,5,6,7-тетрагидро-2H-индазолил. Гетероарил возможно может быть замещен, при необходимости, 1-5 подходящими заместителями, как определено в данном описании изобретения, такими как фтор, хлор, дейтеро, циано, трифторметил, (C1-С6)алкокси, (C6-С10)арилокси, трифторметокси, дифторметокси или (C1-С6)алкил.
Примеры однокольцевых гетероарилов и гетероциклоалкилов включают фуранил, дигидрофуранил, тетрагидрофуранил, тиофенил, дигидротиофенил, тетрагидротиофенил, пирролил, изопирролил, пирролинил, пирролидинил, имидазолил, изоимидазолил, имидазолинил, имидазолидинил, пиразолил, пиразолинил, пиразолидинил, триазолил, тетразолил, дитиолил, оксатиолил, оксазолил, изоксазолил, тиазолил, изотиазолил, тиазолинил, изотиазолинил, тиазолидинил, изотиазолидинил, тиаоксадиазолил, оксатиазолил, оксадиазолил (включая оксадиазолил, 1,2,4-оксадиазолил, 1,2,5-оксадиазолил или 1,3,4-оксадиазолил), пиранил (включая 1,2-пиранил или 1,4-пиранил), дигидропиранил, пиридинил, пиперидинил, диазинил (включая пиридазинил, пиримидинил, пиперазинил), триазинил (включая s-триазинил, as-триазинил и v-триазинил), оксазинил (включая 2H-1,2-оксазинил, 6H-1,3-оксазинил или 2Н-1,4-оксазинил), изоксазинил (включая о-изоксазинил или n-изоксазинил), оксазолидинил, изоксазолидинил, оксатиазинил (включая 1,2,5-оксатиазинил или 1,2,6-оксатиазинил), оксадиазинил (включая 2H-1,2,4-оксадиазинил или 2H-1,2,5-оксадиазинил) и морфолинил.
Термин "гетероарил" также включает конденсированные кольцевые системы, имеющие одно или два кольца, где такие кольца могут быть сконденсированы, где конденсированный является таким, как определено выше. Следует понимать, что если карбоциклическая или гетероциклическая группировка может быть связана или другим образом присоединена к обозначенному субстрату через разные кольцевые атомы, без обозначения конкретной точки присоединения, тогда рассматриваются все возможные точки, либо через атом углерод, либо, например, трехвалентный атом азота. Например, термин "пиридил" означает 2-, 3- или 4-пиридил, термин "тиенил" означает 2- или 3-тиенил и так далее.
В некоторых случаях число атомов в циклическом заместителе, содержащем один или более гетероатомов (т.е. гетероариле или гетероциклоалкиле), указано префиксом "х-у-членный", где х представляет собой минимальное, а у представляет собой максимальное число атомов, образующих циклическую группировку заместителя. Таким образом, например, "5-6-членный гетероарил" относится к гетероарилу, содержащему от 5 до 6 атомов, включающему один или более гетероатомов, в циклической группировке гетероарила. Гетероатомы по данному изобретению выбраны из азота, кислорода и серы.
Соединения по настоящему изобретению могут содержать основные атомы азота (например алкиламины или гетероциклы, такие как пиридин, и т.д.), которые могут быть превращены в N-оксиды путем обработки окислителем (например МСРВА (3-хлорпербензойная кислота) и/или перекисями водорода) с получением других соединений по данному изобретению. Таким образом, все азотсодержащие соединения, которые могут быть превращены в N-оксидные (N→O или -N+-O-) производные, являются частью данного изобретения.
Специалисту в данной области понятно, что метаболиты могут быть образованы как часть естественного биохимического процесса разложения и выведения соединений.
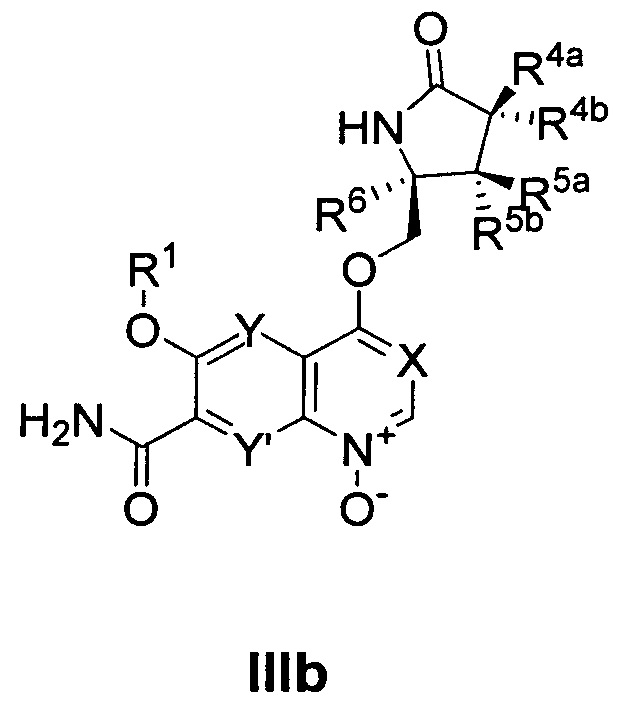
Например, некоторые соединения по изобретению могут естественно образовывать N-оксид, как изображено ниже для соединения Формулы IIIa и IIIb или в других частях соединения Формулы Ia. Метаболиты, такие как эти или другие, образованные как часть естественного биохимического процесса, входят в объем данного изобретения.

Если заместители описаны как "независимо" имеющие более чем одну переменную, в каждом случае заместитель выбран независимо от другого заместителя из списка доступных переменных. Следовательно, каждый заместитель может быть идентичен другому(им) заместителю(ям) или отличен от него(них).
"Пациент" или "субъект" относится к теплокровным животным, таким как, например морские свинки, мыши, крысы, песчанки, кошки, кролики, собаки, крупный рогатый скот, козы, овцы, лошади, обезьяны, шимпанзе и люди.
Термин "фармацевтически приемлемый" означает, что вещество или композиция должны быть совместимыми, химически и/или токсикологически, с другими ингредиентами, составляющими препарат, и/или с млекопитающим, которого им лечат.
Термин "терапевтически эффективное количество" означает количество соединения по настоящему изобретению, которое 1) лечит или предупреждает конкретное заболевание, состояние или расстройство, 2) ослабляет, облегчает или снимает один или более симптомов конкретного заболевания, состояния или расстройства, или 3) предупреждает или задерживает начало одного или более симптомов конкретного заболевания, состояния или расстройства, описанных в данном описании изобретения.
Термин "лечение", при использовании в данном описании изобретения, если не указано иное, означает обращение вспять, облегчение, ингибирование развития, задержку развития, задержку начала или предупреждение расстройства или состояния, к которому применяется такой термин, или одного или более симптомов такого расстройства или состояния. Термин "лечение", при использовании в данном описании изобретения, если не указано иное, относится к акту лечения, где "лечение" определено непосредственно выше. Термин "лечение" также включает адъювантное или неоадъювантное лечение субъекта. Во избежание сомнений, ссылка в данном описании изобретения на "лечение" включает ссылку на куративное, паллиативное и профилактическое лечение, и на введение лекарственного средства для применения в таком лечении.
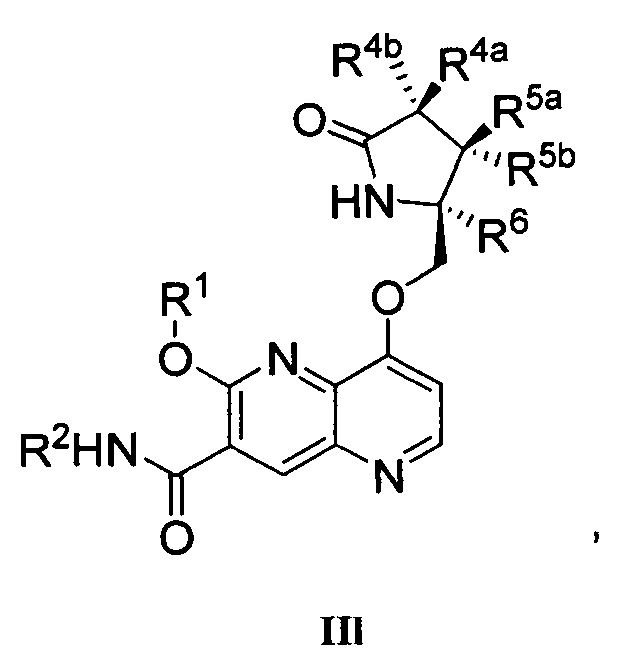
При использовании в данном описании изобретения, термины "Формула I", "Формула Ia", "Формула IIa-IIy", "Формула IIIa" и "Формула IIIb" здесь ниже могут упоминаться как "соединение(я) по изобретению", "настоящее изобретение" и коллективно "соединение Формулы I". Соответственно, термин "соединение Формулы I" включает соединения Формулы Ia, IIa-IIy, IIIa и IIIb. Такие термины также по определению включают все формы соединения Формулы I, включая его гидраты, сольваты, изомеры, кристаллические и некристаллические формы, изоморфы, полиморфы, таутомеры и метаболиты. Например, соединения по изобретению или их фармацевтически приемлемые соли могут существовать в несольватированной и сольватированной формах. Когда растворитель или вода прочно связаны, этот комплекс будет иметь вполне определенную стехиометрию независимо от влажности. Однако когда растворитель или вода слабо связаны, как в канальных сольватах и гигроскопичных соединениях, содержание воды/растворителя будет зависеть от влажности и условий сушки. В таких случаях нормой будет нестехиометричность.
Соединения по изобретению имеют асимметрические атомы углерода. Углерод-углеродные связи соединений по изобретению могут быть обозначены в данном описании изобретения с использованием сплошной линии ( ), сплошного клина (
), сплошного клина ( ) или пунктирного клина (
) или пунктирного клина ( ). Подразумевается, что использование сплошной линии для обозначения связей с асимметрическими атомами указывает, что включены все возможные стереоизомеры (например конкретные энантиомеры, рацемические смеси и т.д.) по этому атому углерода. Подразумевается, что использование или сплошного, или пунктирного клина для обозначения связей с асимметрическими атомами углерода указывает, что включен только один показанный стереоизомер. Возможно, соединения Формулы Ia могут содержать более одного асимметрического атома углерода. В этих соединениях подразумевается, что использование сплошной линии для обозначения связей с асимметрическими атомами углерода указывает, что включены все возможные стереоизомеры. Например, если не указано иное, предполагается, что соединения Формулы I могут существовать в виде энантиомеров и диастереомеров или в виде рацематов и их смесей. Подразумевается, что использование сплошной линии для изображения связей с одним или более асимметрическими атомами углерода в соединении Формулы I и использование сплошного или пунктирного клина для изображения связей с другими асимметрическими атомами углерода в том же соединении указывает, что присутствует смесь диастереомеров.
). Подразумевается, что использование сплошной линии для обозначения связей с асимметрическими атомами указывает, что включены все возможные стереоизомеры (например конкретные энантиомеры, рацемические смеси и т.д.) по этому атому углерода. Подразумевается, что использование или сплошного, или пунктирного клина для обозначения связей с асимметрическими атомами углерода указывает, что включен только один показанный стереоизомер. Возможно, соединения Формулы Ia могут содержать более одного асимметрического атома углерода. В этих соединениях подразумевается, что использование сплошной линии для обозначения связей с асимметрическими атомами углерода указывает, что включены все возможные стереоизомеры. Например, если не указано иное, предполагается, что соединения Формулы I могут существовать в виде энантиомеров и диастереомеров или в виде рацематов и их смесей. Подразумевается, что использование сплошной линии для изображения связей с одним или более асимметрическими атомами углерода в соединении Формулы I и использование сплошного или пунктирного клина для изображения связей с другими асимметрическими атомами углерода в том же соединении указывает, что присутствует смесь диастереомеров.
Стереоизомеры Формулы I включают цис- и транс-изомеры, оптические изомеры, такие как R- и S-энантиомеры, диастереомеры, геометрические изомеры, вращательные изомеры, конформационные изомеры и таутомеры соединений по изобретению, включая соединения, демонстрирующие более чем один тип изомерии; и их смеси (такие как рацематы и диастереомерные пары). Также включены соли присоединения кислоты или основания, где противоион является оптически активным, например D-лактат или L-лизин, или рацемическим, например DL-тартрат или DL-аргинин.
Некоторые из соединений по изобретению, такие как 23, 27 и 66, могут демонстрировать явление таутомерии. Например, соединение, приведенное в примере 23, может существовать в нескольких таутомерных формах, включая пирролидин-2-оновую форму, Пример 23 и 5-гидрокси-3,4-дигидро-2Н-пиррольную форму, Пример 23а. Все такие таутомерные формы включены в объем соединений Формулы I и в объем изобретения. Обычный специалист в данной области должен осознавать и признавать, что многие из Примеров, описанных в данном описании изобретения, могут демонстрировать таутомерию и входят в объем соединения Формулы I, Ia, IIa-IIy, IIIa и IIIb. Таутомеры существуют в виде смесей таутомерного набора в растворе. В твердой форме обычно преобладает один таутомер. Даже если может быть описан один таутомер, настоящее изобретение включает все таутомеры соединений по изобретению и их соли. Примеры таутомеров описаны в Примерах 32 и 32а.


Когда любой рацемат кристаллизуется, возможны кристаллы двух разных типов. Первый тип представляет собой рацемическое соединение (истинный рацемат), упоминаемый выше, где образуется одна гомогенная форма кристалла, содержащая оба энантиомера в эквимолярных количествах. Второй тип представляет собой рацемическую смесь или конгломерат, где образуются две формы кристаллов в эквимолярных количествах, где каждая содержит один энантиомер.
Соединения по данному изобретению можно использовать в форме солей, полученных из неорганических и органических кислот. В зависимости от конкретного соединения, соль этого соединения может быть предпочтительной из-за одного или более физических свойств соли, таких как улучшенная фармацевтическая стабильность при разных температурах и влажностях, или желательная растворимость в воде или масле. В некоторых случаях соль соединения также может быть использована в качестве вспомогательного средства в выделении, очистке и/или повторном растворении соединения.
Когда соль предназначена для введения пациенту (в отличие, например, от использования в условиях in vitro), соль предпочтительно является фармацевтически приемлемой. Термин "фармацевтически приемлемая соль" относится к соли, полученной путем объединения соединения Формулы I с кислотой, анион которой, или с основанием, катион которого, обычно считается подходящим для потребления человеком. Фармацевтически приемлемые соли является особенно полезными в качестве продуктов способов по настоящему изобретению из-за их большей растворимости в воде по сравнению с родительским соединением. Для применения в медицине соли соединений по данному изобретению являются нетоксичными "фармацевтически приемлемыми солями". Соли, охватываемые термином "фармацевтически приемлемые соли", относятся к нетоксичным солям соединений по изобретению, которые обычно получают путем взаимодействия свободного основания с подходящей органической или неорганической кислотой.
Подходящие фармацевтически приемлемые соли присоединения кислоты для соединений по настоящему изобретению, когда это возможно, включают соли, полученные из неорганических кислот, таких как соляная, бромистоводородная, фтористоводородная, борная, фторборная, фосфорная, метафосфорная, азотная, угольная, сульфоновая и серная кислоты, и органических кислот, таких как уксусная, бензолсульфоновая, бензойная, лимонная, этансульфоновая, фумаровая, глюконовая, гликолевая, изотионовая, молочная, лактобионовая, малеиновая, яблочная, метансульфоновая, трифторметансульфоновая, янтарная, толуолсульфоновая, винная и трифторуксусная кислоты. Подходящие органические кислоты обычно включают, например, алифатические, циклоалифатические, ароматические, аралифатические, гетероциклические, карбоновые и сульфоновые классы органических кислот.
Конкретные примеры подходящих органических кислот включают ацетат, трифторацетат, формиат, пропионат, сукцинат, гликолят, глюконат, диглюконат, лактат, малат, тартрат, цитрат, аскорбат, глюкуронат, малеат, фумарат, пируват, аспартат, глутамат, бензоат, антранилат, стеарат, салицилат, n-гидроксибензоат, фенилацетат, манделат, эмбонат (памоат), метансульфонат, этансульфонат, бензолсульфонат, пантотенат, толуолсульфонат, 2-гидроксиэтансульфонат, сульфанилат, циклогексиламиносульфонат, алгенат, β-гидроксибутират, галактарат, галактуронат, адипат, альгинат, бутират, камфорат, камфорсульфонат, циклопентанпропионат, додецилсульфат, гликогептаноат, глицерофосфат, гептаноат, гексаноат, никотинат, 2-нафталинсульфонат, оксалат, пальмоат, пектинат, 3-фенилпропионат, пикрат, пивалат, тиоцианат и ундеканоат.
Кроме того, когда соединения по изобретению несут кислотную группировку, их подходящие фармацевтически приемлемые соли могут включать соли щелочных металлов, например соли натрия или калия; соли щелочноземельных металлов, например соли кальция или магния; и соли, образованные подходящими органическими лигандами, например четвертичные аммониевые соли. В еще одном воплощении основные соли образованы из оснований, которые образуют нетоксичные соли, включающие соли алюминия, аргинина, бензатина, холина, диэтиламина, диоламина, глицина, лизина, меглумина, оламина, трометамина и цинка.
Органические соли могут быть получены из вторичных, третичных и четвертичных аминных солей, таких как трометамин, диэтиламин, N,N'-дибензилэтилендиамин, хлорпрокаин, холин, диэтаноламин, этилендиамин, меглумин (N-метилглюкамин) и прокаин. Основные азотсодержащие группы могут быть кватернизированы агентами, такими как низшие алкил(С1-С6)галогениды (например метил-, этил-, пропил- и бутилхлориды, -бромиды и -йодиды), диалкилсульфаты (например диметил-, диэтил-, дибутил- и диамилсульфаты), длинноцепочечные галогениды (например децил-, лаурил-, миристил- и стеарилхлориды, -бромиды и -йодиды), арилалкилгалогениды (например бензил- и фенетилбромиды) и другие.
В одном воплощении также могут быть образованы гемисоли кислот и оснований, например гемисульфатные и гемикальциевые соли.
Также в объем настоящего изобретения входят так называемые "пролекарства" соединения по изобретению. Так, некоторые производные соединения по изобретению, которые сами по себе могут обладать небольшой фармакологической активностью или не иметь ее, при введении в организм или на тело могут превращаться в соединение по изобретению, имеющее нужную активность, например посредством гидролитического расщепления. Такие производные называются "пролекарства". Дополнительную информацию по применению пролекарств можно найти в "Pro-drugs as Novel Delivery Systems, Vol. 14, ACS Symposium Series (T. Higuchi and V. Stella) и "Bioreversible Carriers in Drug Design," Pergamon Press, 1987 (ed. E.B. Roche, American Pharmaceutical Association). Пролекарства по изобретению могут быть получены, например, путем замены подходящих функциональных групп, присутствующих в соединениях любой Формулы Ia, определенными группировками, известными специалистам в данной области как "про-группировки", как описано, например, в "Design of Prodrugs", Н. Bundgaard (Elsevier, 1985).
Настоящее изобретение также включает изотопно меченые соединения, которые идентичны соединениям, описанным в Формуле Ia, кроме того, что один или более атомов заменены атомом, имеющим атомную массу или массовое число, отличные от атомной массы или массового числа, обычно встречающихся в природе. Примеры изотопов, которые могут быть включены в соединения по настоящему изобретению, включат изотопы водорода, углерода, азота, кислорода, серы, фтора и хлора, такие как 2Н, 3Н, 13С, 11С, 14С, 15N, 18O, 17O, 32Р, 35S, 18F и 36С1 соответственно. Соединения по настоящему изобретению, их пролекарства и фармацевтически приемлемые соли указанных соединений или указанных пролекарств, которые содержат вышеуказанные изотопы и/или другие изотопы других атомов, входят в объем данного изобретения. Некоторые изотопно меченые соединения по настоящему изобретению, например соединения, в которые включены радиоактивно меченые изотопы, такие как 3Н и 14С, являются полезными в анализах распределения лекарственного средства и/или субстрата в тканях. Тритиевые, т.е. 3Н, и углерод-14, т.е. 14С, изотопы являются особенно предпочтительными из-за легкости их получения и обнаружения. Кроме того, замещение более тяжелыми изотопами, такими как дейтерий, т.е. 2Н, может обеспечивать некоторые терапевтические преимущества в результате большей метаболической стабильности, например увеличенного времени полувыведения in vivo или пониженных требований к дозировке, и, следовательно, может быть предпочтительным в некоторых обстоятельствах. В соединениях по изобретению, как заявлено в формуле изобретения, может быть конкретно определено замещение дейтеро или дейтерием. Отсутствие термина дейтеро, дейтерон или дейтерий, все из которых используют взаимозаменяемо, в замещающей группе не следует понимать как исключение дейтеро.
Изотопно меченые соединения Формулы Ia по данному изобретению и их пролекарства обычно могут быть получены путем осуществления процедур, раскрытых в Схемах и/или в Примерах и Подготовительных примерах ниже, путем замещения легко доступным изотопно меченым реагентом не меченного изотопами реагента.
Все патенты и публикации, указанные в данном описании изобретения, включены в данную заявку посредством ссылки во всей своей полноте и для всех целей.
Соединения по изобретению
В одном воплощении, как описано выше и более полно в данном разделе, изобретение относится к соединению Формулы I

где
каждый из X, X', Y и Y' независимо представляет собой СН или N; Z представляет собой С или N; при условии, что не более чем три из X, X', Z, Y и Y' представляют собой N;
R1 представляет собой С1-С6алкил или -(С1-С6алкил)n(С1-С6циклоалкил), где алкил или циклоалкил возможно замещен дейтерием, галогеном, CN, ОН, или С1-С6алкокси;
R2 представляет собой водород или метил;
R3 представляет собой водород, дейтерий, галоген, нитрил, -(CH2)tNR8aR8b, -(СН2)t(6-10-членный арил) или -(СН2)t(5-10-членный гетероарил), имеющий от одного до трех гетероатомов, выбранных из N, О или S, где указанный арил или гетероарил возможно замещены одной-тремя группами С1-С6алкил, дейтерий, галоген, CN, ОН, гидроксиС1-С6алкил или С1-С6алкокси; где алкил возможно замещен гидроксилом, галогеном, CN или С1-С3алкокси;
каждый из R4a и R4b независимо представляет собой водород, фтор, ОН, С1-С3алкокси или CH2OR7, где R7, взятый вместе с R1, представляет собой С1-С4алкилен, возможно замещенный галогеном или алкилом;
каждый из R5a и R5b независимо представляет собой водород, С1-С3алкил или С1-С3-алкокси, где указанный алкил или алкокси возможно замещен одной-тремя группами дейтерий, галоген, ОН или CN; или R5a и R5b, взятые вместе с атомом, к которому они присоединены, образуют С3-С7циклоалкил или С3-С7гетероциклоалкил, где указанный циклоалкил или гетероциклоалкил возможно замещен одной-тремя группами дейтерий, галоген, ОН, CN или С1-С3алкил;
R6 представляет собой водород или С1-С3алкил; или R5b и R6, взятые вместе с атомами, к которым они присоединены, образуют С3-С7циклоалкил или С3-С7гетероциклоалкил, где указанный циклоалкил или гетероциклоалкил возможно замещен одной-тремя группами дейтерий, галоген, ОН, CN или С1-С3алкил
каждый из R8a и R8b независимо представляет собой водород, -S(O)2R9 или -C(O)R9;
R9 представляет собой С1-С6алкил, С1-С6циклоалкил, 6-10-членный арил или 5-10-членный гетероарил, имеющий от одного до трех гетероатомов, где указанный алкил, циклоалкил, арил или гетероарил возможно замещен одной-тремя группами С1-С6алкил, галоген, CN, ОН, С1-С6алкокси или С1-С6гидрокси;
n равен 0 или 1;
t равен 0, 1, 2 или 3;
или фармацевтически приемлемую соль указанного соединения или таутомер указанного соединения или указанной соли.
В другом воплощении, изобретение направлено на соединения, где X представляет собой N, Z представляет собой С, X', Y и Y' представляют собой СН; альтернативно, X' представляет собой N, Z представляет собой С, X, Y и Y' представляют собой СН; альтернативно, X, X', Z, Y и Y' представляют собой СН; альтернативно, Y представляет собой N, Z представляет собой С, X, X' и Y' представляют собой СН; альтернативно, Z представляет собой С, X и Y' представляют собой N, X' и Y представляют собой СН; альтернативно Z представляет собой С, Y' представляет собой N, Y, X, и X' представляют собой СН; альтернативно, X и Z представляют собой N, С, X', Y и Y' представляют собой СН; альтернативно, X' и Z представляют собой N, Z представляет собой С, X, Y и Y' представляют собой СН; альтернативно, Z и Y' представляют собой N, Y, X, и X' представляют собой СН; альтернативно, Y и Z представляют собой N, X, X' и Y' представляют собой СН; альтернативно, Z представляет собой N, X, X', Y и Y' представляют собой СН; или фармацевтически приемлемую соль указанного соединения или таутомер указанного соединения или указанной соли.
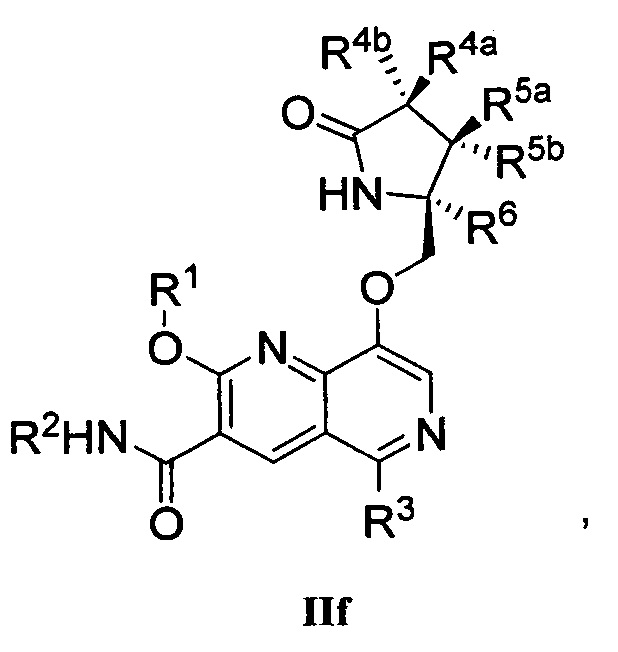
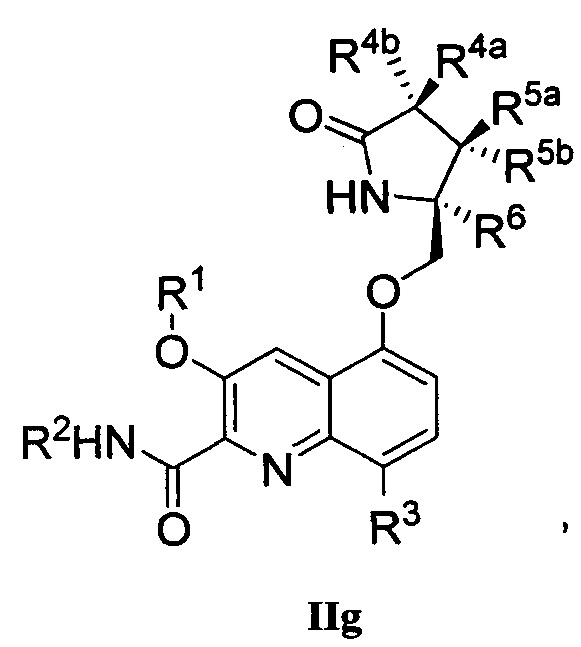
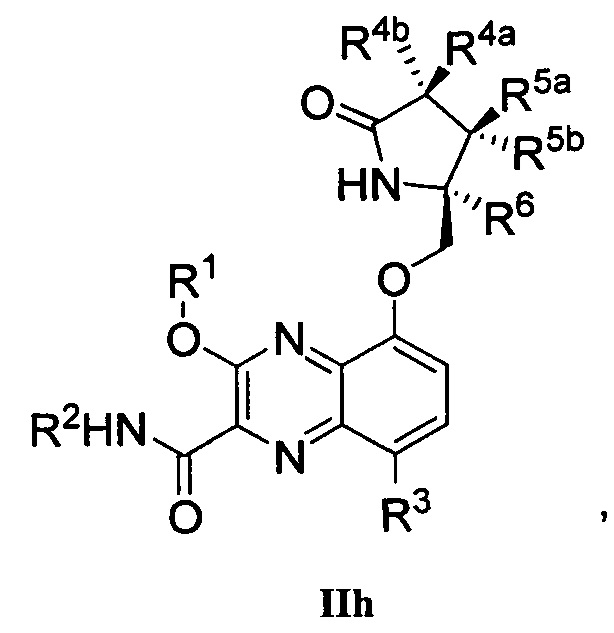
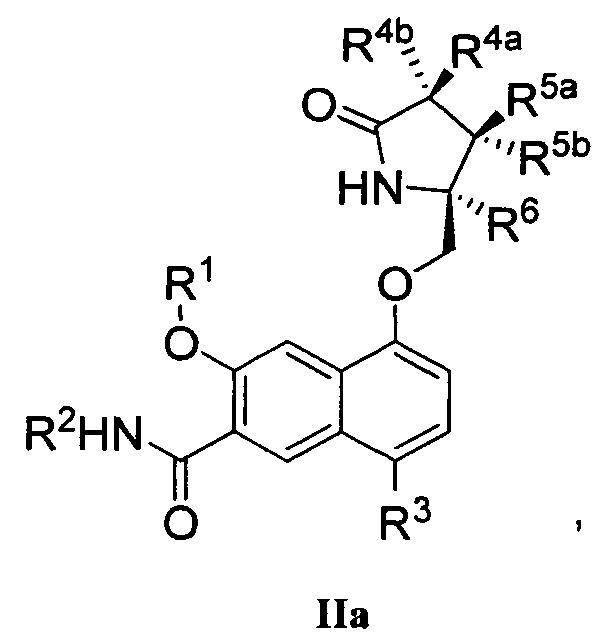
В другом аспекте изобретение направлено на соединение Формулы IIa, IIb, IIc, IId, IIe, IIf, IIg, IIh, IIi, IIj, IIk, III, IIm, IIn, IIo, IIp, IIq, IIr, IIs, IIt, IIu, IIv, IIw, IIx или IIy, как изображено ниже:

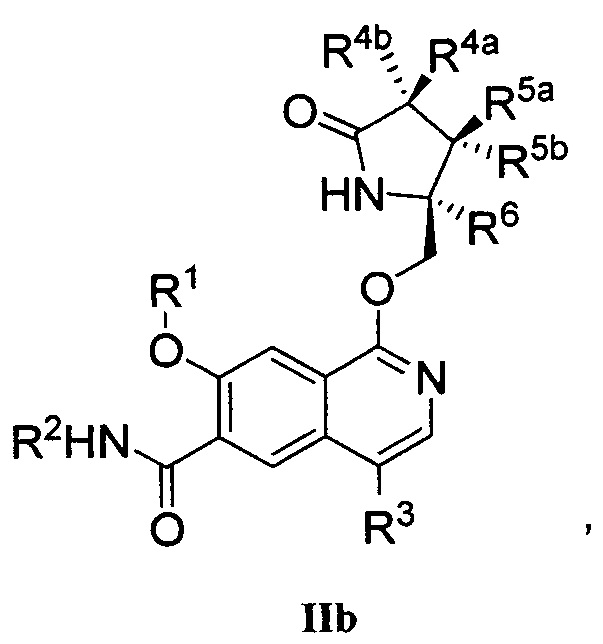


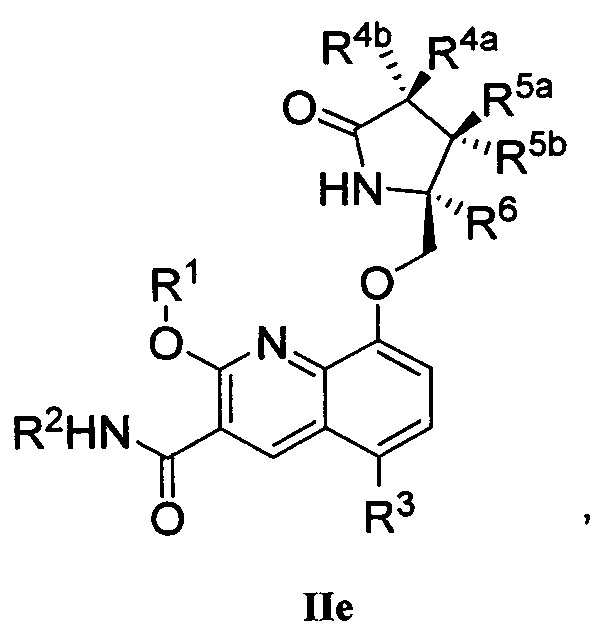





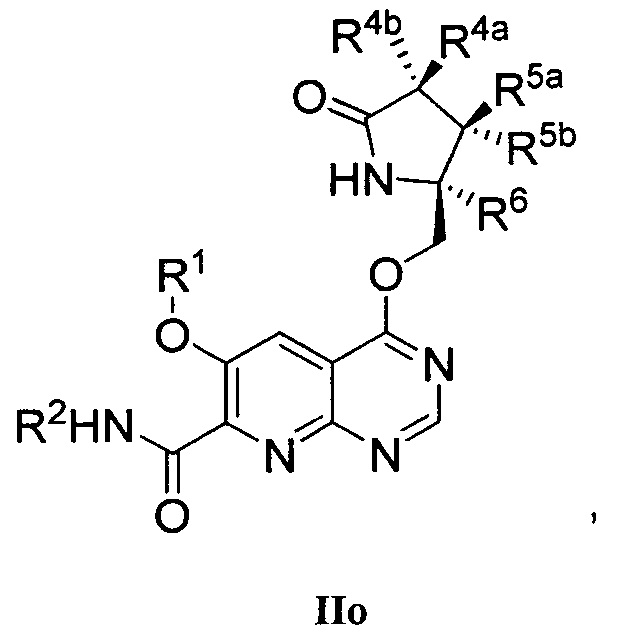




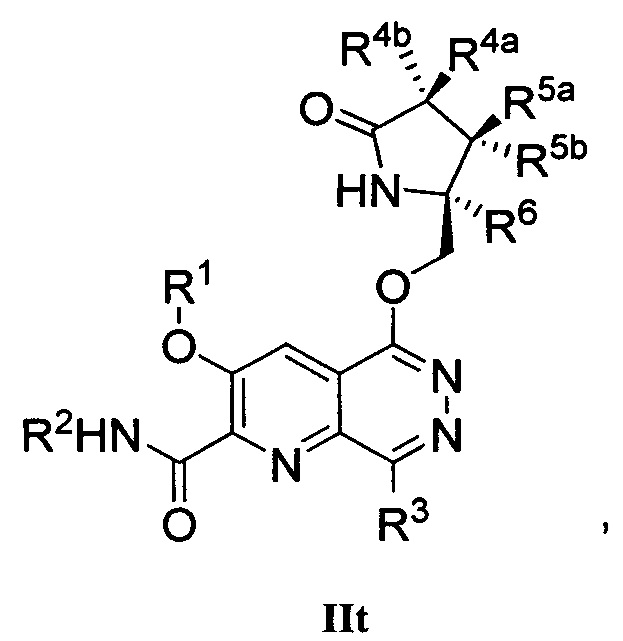



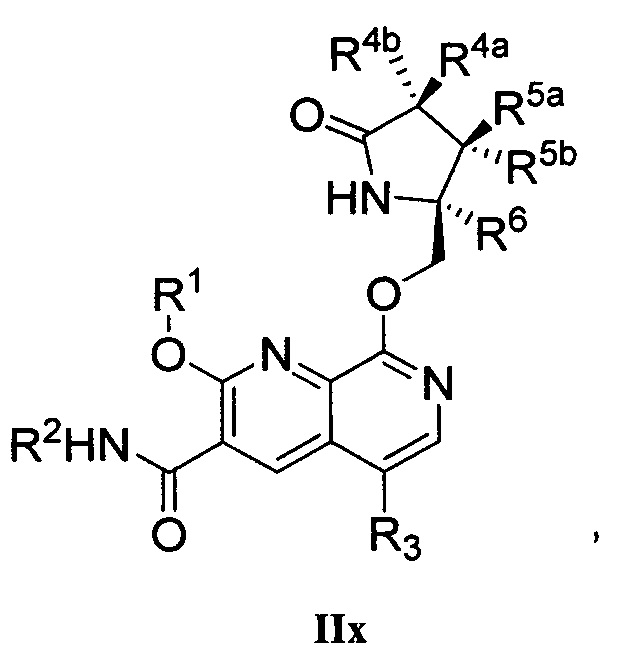
или

где
R1 представляет собой С1-С6алкил или -(С1-С6алкил)n(С1-С6циклоалкил), где алкил или циклоалкил возможно замещен дейтерием, галогеном, CN, ОН или С1-С6алкокси;
R2 представляет собой водород;
R3 представляет собой водород, дейтерий, галоген, нитрил, -(CH2)tNR8aR8b, -(CH2)t(6-10-членный арил) или -(CH2)t(5-10-членный гетероарил), имеющий от одного до трех гетероатомов, выбранных из N, О или S, где указанный арил или гетероарил возможно замещен одной-тремя группами С1-С6алкил, дейтерий, галоген, CN, ОН, гидроксиС1-С6алкил или С1-С6алкокси;
каждый из R4a и R4b независимо представляет собой водород, фтор, ОН, С1-С3алкокси или CH2OR7, где R7, взятый вместе с R1, представляет собой С1-С4алкилен, возможно замещенный галогеном или алкилом;
R5a и R5b независимо представляют собой водород, С1-С3алкил или С1-С3-алкокси, где указанный алкил или алкокси возможно замещен одной-тремя группами дейтерий, галоген, ОН или CN; или R5a и R5b, взятые вместе с атомом, к которому они присоединены, образуют С3-С7циклоалкил или С3-С7гетероциклоалкил, где указанный циклоалкил или гетероциклоалкил возможно замещен одной-тремя группами дейтерий, галоген, ОН, CN или С1-С3алкил;
R6 представляет собой водород или С1-С3алкил; или R5b и R6, взятые вместе с атомами, к которым они присоединены, образуют С3-С7циклоалкил или С3-С7гетероциклоалкил, где указанный циклоалкил или гетероциклоалкил возможно замещен одной-тремя группами дейтерий, галоген, ОН, CN или С1-С3алкил;
каждый из R8a и R8b независимо представляет собой водород, -S(O)2R9 или -C(O)R9;
R9 представляет собой С1-С6алкил, С1-С6циклоалкил, 6-10-членный арил или 5-10-членный гетероарил, имеющий от одного до трех гетероатомов, где указанный алкил, циклоалкил, арил или гетероарил возможно замещен одной-тремя группами С1-С6алкил, галоген, CN, ОН, С1-С6алкокси или С1-С6гидрокси;
n равен 0 или 1; t равен 0, 1, 2 или 3.
В другом воплощении R1 представляет собой С1-С6алкил; R2 представляет собой водород; R3 представляет собой водород, дейтерий, -(CH2)tNR8aR8b, -(СН2)t(6-10-членный арил) или -(СН2)t(5-10-членный гетероарил), имеющий от одного до трех гетероатомов, выбранных из N, О или S, где указанный арил или гетероарил возможно замещен одной-тремя группами С1-С6алкил, дейтерий, галоген, CN, ОН, гидроксиС1-С6алкил или С1-С6алкокси;
R6 представляет собой водород; каждый из R8a и R8b независимо представляет собой водород, -S(O)2R9 или -C(O)R9; R9 представляет собой С1-С6алкил, С1-С6циклоалкил, 6-10-членный арил или 5-10-членный гетероарил, имеющий от одного до трех гетероатомов, где указанный алкил, циклоалкил, арил или гетероарил возможно замещен одной-тремя группами С1-С6алкил, галоген, CN, ОН, С1-С6алкокси или С1-С6гидрокси; и t равен 0 или 1.
В другом воплощении арил и гетероарил из R3 выбран из фенила, пиразолила, имидазолила и оксазолила, возможно замещенного одним или двумя С1-С6алкилами или С1-С6гидроксиалкилами; R3 представляет собой водород, дейтерий или -(CH2)tNR8aR8b; каждый из R8a и R8b независимо представляет собой водород или -S(O)2R9; R9 представляет собой С1-С6алкил, С1-С6циклоалкил, 6-10-членный арил или 5-10-членный гетероарил, имеющий от одного до трех гетероатомов, где указанный алкил, циклоалкил, арил или гетероарил возможно замещены одной-тремя группами С1-С6алкил, галоген, CN, ОН, С1-С6алкокси или С1-С6гидрокси; и t равен 0 или 1.
В другом аспекте изобретение направлено на соединение, выбранное из

где R1 представляет собой С1-С3алкил, возможно замещенный дейтерием или галогеном; R2 представляет собой водород; R3 представляет собой водород, дейтерий, -NH2 или 5-10-членный гетероарил, имеющий от одного до трех гетероатомов, выбранных из N, О или S, где указанный гетероарил возможно замещен одной-тремя группами С1-С6алкил, дейтерий, галоген, CN, ОН или С1-С6алкокси; каждый из R4a и R4b представляет собой водород, фтор или ОН; R5a и R5b независимо представляют собой водород, С1-С3алкил или С1-С3-алкокси, где указанный алкил или алкокси возможно замещен одной-тремя группами дейтерий, галоген, ОН или CN; или R5a и R5b, взятые вместе с атомом, к которому они присоединены, образуют С3-С7циклоалкил или С3-С7гетероциклоалкил, где указанный циклоалкил или гетероциклоалкил возможно замещен одной-тремя группами дейтерий, галоген, ОН, CN или С1-С3алкил; R6 представляет собой водород или С1-С3алкил; или R5b и R6, взятые вместе с атомами, к которым они присоединены, образуют С3-С7циклоалкил или С3-С7гетероциклоалкил, где указанный циклоалкил или гетероциклоалкил возможно замещен одной-тремя группами дейтерий, галоген, ОН, CN или С1-С3алкил; каждый из R8a и R8b независимо представляет собой водород, -S(O)2R9 или -C(O)R9; R9 представляет собой С1-С6алкил, С1-С6циклоалкил, 6-10-членный арил или 5-10-членный гетероарил, имеющий от одного до трех гетероатомов, где указанный алкил, циклоалкил, арил или гетероарил возможно замещены одной-тремя группами С1-С6алкил, галоген, CN, ОН, С1-С6алкокси или С1-С6гидрокси; или фармацевтически приемлемую соль указанного соединения или таутомер указанного соединения или указанной соли.
В другом воплощении R3 представляет собой водород, -NH2, пиразолил, имидазолил или оксазолил, где указанные гетероарилы возможно замещены одним или двумя С1-С3алкилами; R4a представляет собой водород или фтор; R5a и R5b независимо представляют собой водород, метил или этил; или R5a и R5b, взятые вместе с атомом, к которому они присоединены, образуют циклопропил; и R6 представляет собой водород.
В другом воплощении каждый из R4a и R4b представляет собой водород или фтор; или фармацевтически приемлемая соль указанного соединения или таутомер указанного соединения или соли. В другом аспекте R4a представляет собой фтор, R4b представляет собой водород.
В другом воплощении изобретение направлено на соединения из Таблицы I и те, которые приведены в качестве примеров в данной заявке; или их фармацевтически приемлемые соли или таутомеры указанных соединений или солей.
В еще одном воплощении изобретение направлено на промежуточные соединения, описанные в Схемах синтеза и/или Подготовительных примерах; или фармацевтически приемлемую соль указанного соединения, или таутомер указанного соединения, или указанную соль.
В еще одном воплощении изобретение направлено на способ синтеза и получения промежуточных соединений, описанных в данном описании изобретения, как подробно указано в Схемах и разделе получения, описанных в данном описании изобретения. В другом аспекте изобретение направлено на способ синтеза и получение соединений из Таблиц 1 или 3, как подробно указано в Схемах и разделе получения, описанных в данном описании изобретения.
Показания, связанные с IRAK4
Соединения по изобретению также являются полезными в лечении и/или предупреждении заболевания или состояния, опосредованного или другим образом ассоциированного с ферментом IRAK; где способ включает введение субъекту, нуждающемуся в этом, эффективного количества соединения по изобретению.
Заболевание может представлять собой, без ограничения ими, один из следующих классов: аутоиммунные заболевания, воспалительные заболевания, аллергические заболевания, метаболические заболевания, инфекционные заболевания, заболевания, связанные с повреждением тканей или травмой, фиброзные заболевания, генетические заболевания, заболевания, обусловленные чрезмерной активностью путей IL1, сердечнососудистые заболевания, сосудистые заболевания, заболевания сердца, неврологические заболевания, нейродегенеративные заболевания, респираторные заболевания, легочные заболевания, заболевания дыхательных путей, почечные заболевания, кожные и/или дерматологические заболевания, заболевания печени, желудочно-кишечные заболевания, заболевания ротовой полости, болевые и сенсорные заболевания, гематопоэтические заболевания, заболевания суставов, мышечные заболевания, заболевания костей и офтальмологические и/или глазные заболевания.
Конкретные аутоиммунные заболевания включают, но без ограничения ими: ревматоидный артрит, остеоартрит, псориаз, аллергический дерматит, системную красную волчанку (и возникающие осложнения), синдром Шегрена, рассеянный склероз, астму, гломерулярный нефрит, синдром раздраженного кишечника, воспалительное заболевание кишечника, болезнь Крона, анкилозирующий спондилит, болезнь Бехчета, волчаночный нефрит, склеродермию, системную склеродермию, диабет I типа или ювенильный диабет, генерализованную алопецию, острый диссеминированный энцефаломиелит, болезнь Аддисона, синдром антифосфолипидных антител, атрофический гастрит при пернициозной анемии, аутоиммунную алопецию, аутоиммунную гемолитическую анемию, аутоиммунный гепатит, аутоиммунный энцефаломиелит, аутоиммунную тромбоцитопению, буллезный пемфигоид, болезнь Шагаса, целиакию, хронический гепатит, синдром Когана, дерматомиозит, эндометриоз, синдром Гудпасчера, болезнь Грейвса, синдром Гийена-Барре, болезнь Хашимото (или тиреоидит Хашимото), гемолитическую анемию, гнойный гидрадентит, идиопатическую тромбоцитопеническую пурпуру, интерстициальный цистит, мембранную гломерулопатию, склеродермию, тяжелую миастению, нарколепсию, пемфигус, пернициозную анемию, узелковый полиартериит, полимиозит, первичный билиарный цирроз, синдром Рейтера, шизофрению, симпатическую офтальмию, системный склероз, височный артериит, тиреоидит, васкулит, витилиго, вульводинию, гранулематоз Вегенера, ладонно-подошвенную кератодермию, ювенильный идиопатический артрит с системным началом (SJIA) или показание, указанное в отдельной категории в данном описании изобретения.
Конкретные воспалительные заболевания включают, без ограничения ими: хроническую обструктивную болезнь легких, гиперчувствительность дыхательных путей, кистозный фиброз, острый респираторный дистресс-синдром, синусит, ринит, гингивит, атеросклероз, хронический простатит, гломерулярный нефрит, язвенный колит, увеит, заболевание пародонта или показание, перечисленное в отдельной категории в данной заявке.
Конкретные воспалительные заболевания включают, но без ограничения ими: хронические обструктивные болезни легких, гиперчувствительность дыхательных путей, кистозный фиброз, острый респираторный дистресс-синдром, синусит, ринит, гингивит, атеросклероз, хронический простатит, гломерулярный нефрит, язвенный колит, увеит, заболевание пародонта или показание, указанное в отдельной категории в данном описании изобретения.
Конкретные болевые состояния включают, но без ограничения ими: воспалительную боль, хирургическую боль, висцеральную боль, зубную боль, предменструальную боль, центральную боль, боль из-за ожогов, мигрень или кластерные головные боли, повреждения нервов, интерстициальный цистит, раковую боль, вирусную, паразитарную или бактериальную инфекцию, посттравматическое повреждение, боль, ассоциированную с синдромом раздраженного кишечника, подагру, боль, связанную с любыми другими показаниями, перечисленными в данном описании, или показанием, указанным в отдельной категории в данном описании изобретения.
Конкретные состояния дыхательных путей, респираторные и легочные состояния включают, но без ограничения ими: астму (которая может включать хроническую, позднюю, бронхиальную, аллергическую, внутреннюю, внешнюю или пылевую), хроническую обструктивную болезнь легких, идиопатический легочный фиброз, легочную артериальную гипертензию, кистозный фиброз, интерстициальную болезнь легких, острое повреждение легких, саркоидоз, аллергический ринит, хронический кашель, бронхит, рецидивирующую обструкцию дыхательных путей, эмфизему или бронхоспазм или показание, указанное в отдельной категории заболеваний в данном описании изобретения.
Конкретные желудочно-кишечные (GI) расстройства включают, но без ограничения ими: синдром раздраженного кишечника (IBS), воспалительное заболевание кишечника (IBD), желчные колики и другие желчные расстройства, почечные колики, IBS с преобладанием диареи, боль, ассоциированную с растяжением GI, язвенный колит, болезнь Крона, синдром раздраженного кишечника, целиакию, проктит, эозинофильный гастроэнтерит, мастоцитоз или показание, указанное в отдельной категории заболеваний в данном описании изобретения.
Конкретные аллергические заболевания включают, но без ограничения ими: анафилаксию, аллергический ринит, аллергический дерматит, аллергическую крапивницу, ангионевротический отек, аллергическую астму, аллергические реакции на: пищу, лекарственные средства, укусы насекомых, пыльцу; или показание, указанное в отдельной категории заболеваний в данном описании изобретения.
Конкретные инфекционные заболевания включают, но без ограничения ими: сепсис, септический шок, вирусные заболевания, малярию, болезнь Лайма, глазные инфекции, конъюнктивит, болезнь Уиппла или показание, указанное в отдельной категории заболеваний в данном описании изобретения.
Конкретные состояния, связанные с травмами и повреждением тканей, включают, но без ограничения ими: почечное клубочковое повреждение, реперфузионное повреждение (например сердца, почек, легких), повреждение спинного мозга, рубцевание тканей, спайки тканей, восстановление тканей, отторжение трансплантата (например сердца, легкого, костного мозга, хряща, роговицы, почки, конечности, печени, мышцы, миобласта, поджелудочной железы, панкреатических островков, кожи, нервов, тонкого кишечника, трахеи), гиперчувствительности, или показание, указанное в отдельной категории заболеваний в данном описании изобретения.
Конкретные фиброзные заболевания включают, но без ограничения ими: идиопатический легочный фиброз, фиброз печени, почечный фиброз или показание, указанное в отдельной категории заболеваний в данном описании изобретения.
Конкретные заболевания, которые считаются обусловленными чрезмерной активностью сигнальных путей IL1, включают, но без ограничения ими: криопирин-ассоциированные периодические синдромы, миозит и показания, включенные в следующую обзорную статью: С.A. Dinarello, A. Simon and J. W.M. van der Meer, Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases, Nat Rev Drug Discov, 2012, 11(8), 633-652, http://dx.doi.org/10.1038/nrd3800 и дополнительные показания, содержащиеся в ней, или показание, указанное в отдельной категории заболеваний в данном описании изобретения.
Конкретные офтальмологические/глазные заболевания включают, но без ограничения ими: увеит, возрастную макулярную дегенерацию, диабетический макулярный отек, кератоконъюктивит, увеит, ассоциированный с болезнью Бехчета, весенний конъюнктивит, кератит, индуцированный линзами увеит, герпетический кератит, конический кератит, эпителиальную дистрофию роговицы, глазной пемфигоид, язвенную болезнь Морена, склерит, офтальмопатию Грейвса, синдром Фогта-Коянаги-Харада, сухой кератоконъюнктивит, фликтену, иридоциклит, симпатическую офтальмию, аллергический конъюнктивит, окулярную неоваскуляризацию, синдром сухого глаза или показание, указанное в отдельной категории заболеваний в данном описании изобретения.
Конкретные заболевания суставов, мышц и костей включают, но без ограничения ими: остеоартрит, остеопороз, ревматоидный артрит, ювенильный артрит, псориатический артрит, эрозивный остеоартрит руки, артрофиброз/травматическое повреждение колена, разрыв передней крестообразной коленной связки, рецидивирующий полихондрит, повторяющийся мультифокальный остеомиелит, синдром Маджида, анкилозирующий спондилит, подагру поясничного отдела позвоночника, антисинтетазный синдром, идиопатические воспалительные миопатии, суставной хондрокальциноз, ювенильный идиопатический артрит с системным началом (SJIA), подагру и артрит, ассоциированный с кристаллами пирофосфата, или показание, указанное в отдельной категории заболеваний в данном описании изобретения.
Конкретные кожные/дематологические заболевания включают, но без ограничения ими: псориаз, атопический дерматит, кожную волчанку, акне, дерматомиозит, экзему, зуд, склеродермию, синдром Свита/нейтрофильный дерматоз, нейтрофильный панникулит, акродерматит (форма пустулярного псориаза) или показание, указанное в отдельной категории заболеваний в данном описании изобретения.
Конкретные заболевания почек включают, но без ограничения ими: острое повреждение почек (AKI) (сепсис-AKI, аортокоронарное шунтирование-AKI, кардиохирургия-AKI, несердечная хирургия-AKI, трансплантационная хирургия-AKI цисплатин-AKI, индуцированное контрастным/визуализирующим агентом AKI), гломерулонефрит, IgA-нефропатию, серповидный GN (гломерулонефрит), волчаночный нефрит, ВИЧ-ассоциированную нефропатию, мембранозную нефропатию, С3-гломерулопатию, болезнь плотного осадка, АNСА(антинейтрофильные цитоплазматические антитела)-васкулит, диабетическую нефропатию, гемолитико-уремический синдром, атипичный гемолитическо-уремический синдром, нефротический синдром, нефритический синдром, гипертензивный нефросклероз, АроL1-нефропатию, фокальный сегментный гломерулосклероз, синдром Альпорта, синдром Фанкони, кристаллическую нефропатию, нефролитиаз, нефротический синдром, отторжение почечного трансплантата, амилоидоз, гломерулонефрит при SJIA или показание, указанное в отдельной категории заболеваний в данном описании изобретения.
Конкретные генетические заболевания включают, но без ограничения ими: семейную средиземноморскую лихорадку (FMF), CAPS (FCAS, синдром Маккл-Уэллса, NOMID/CINCA), мужское бесплодие при CAPS, NLRP12 аутовоспалительный синдром или показание, указанное в отдельной категории заболеваний в данном описании изобретения.
Конкретные гематопоэтические заболевания включают, но без ограничения ими: гемолитическую анемию или показание, указанное в отдельной категории заболеваний в данном описании изобретения.
Конкретные заболевания печени включают, но без ограничения ими: фиброз печени, цирроз печени, неалкогольный стеатогепатит (NASH) или показание, указанное в отдельной категории заболеваний в данном описании изобретения.
Конкретные заболевания ротовой полости включают, но без ограничения ими: гингивит, болезнь пародонта или показание, указанное в отдельной категории заболеваний в данном описании изобретения.
Конкретные метаболические заболевания включают, но без ограничения ими: диабет 2 типа (и возникающие осложнения), подагру и гиперурикемию, метаболический синдром, инсулинорезистентность, ожирение или показание, указанное в отдельной категории заболеваний в данном описании изобретения.
Соединения по настоящему изобретению также являются полезными в лечении пролиферативного заболевания, выбранного из доброкачественной или злокачественной опухоли, солидной опухоли, карциномы головного мозга, почки, печени, надпочечника, мочевого пузыря, молочной железы, желудка, опухолей желудка, яичников, толстой кишки, прямой кишки, предстательной железы, поджелудочной железы, легкого, влагалища, шейки матки, яичка, мочеполового тракта, пищевода, гортани, кожи, кости или щитовидной железы, саркомы, глиобластом, нейробластом, множественной миеломы, рака желудочно-кишечного тракта, особенно карциномы толстой кишки или колоректальной аденомы, опухоли шеи и головы, эпидермальной гиперпролиферации, псориаза, гиперплазии предстательной железы, неоплазии, неоплазии эпителиального характера, аденомы, аденокарциномы, кератоакантомы, эпидермоидной карциномы, крупноклеточной карциномы, немелкоклеточной карциномы легкого, лимфом, ходжкинских и неходжкинских, карциномы молочной железы, фолликулярной карциномы, недифференцированной карциномы, папиллярной карциномы, семиномы, меланомы, вялотекущей индолентной множественной миеломы или гемобластозов (включая лейкоз, диффузную крупноклеточную В-клеточную лимфому (DLBCL), ABC DLBCL, хронический лимфоцитарный лейкоз (CLL), хроническую лимфоцитарную лимфому, первичную эффузионную лимфому, лимфому/лейкоз Беркитта, острый лимфоцитарный лейкоз, пролимфоцитарный В-клеточный лейкоз, лимфоплазматическую лимфому, макроглобулинемию Вальденстрема (WM), лимфому маргинальной зоны селезенки, множественную миелому, плазмоцитому, внутрисосудистую крупноклеточную В-клеточную лимфому) или показание, указанное в отдельной категории заболеваний в данном описании изобретения.
Сердечно-сосудистые заболевания включают, но без ограничения ими, коронарную болезнь сердца, острый коронарный синдром, ишемическое заболевание сердца, первичный или рецидивирующий инфаркт миокарда, вторичный инфаркт миокарда, инфаркт миокарда без подъема сегмента ST или инфаркт миокарда с подъемом сегмента ST, внезапную ишемическую смерть, транзиторную ишемическую атаку, окклюзионную болезнь периферических артерий, стенокардию, атеросклероз, гипертензию, сердечную недостаточность (такую как застойная сердечная недостаточность), диастолическую дисфункцию (такую как диастолическая дисфункция левого желудочка, диастолическая сердечная недостаточность и нарушение диастолического наполнения), систолическую дисфункцию (такую как систолическая сердечная недостаточность со снижением фракции выброса), васкулит, ANCA-васкулит, фибрилляцию предсердий при ремоделировании сердца после инфаркта миокарда, аритмию (желудочковую), ишемию, гипертрофическую кардиомиопатию, внезапную сердечную смерть, фиброз миокарда и сосудов, нарушение податливости сосудистой стенки, некротические повреждения миокарда, повреждение сосудов, гипертрофия левого желудочка, снижение фракции выброса, поражения сердца, гипертрофию сосудистой стенки, эндотелиальное утолщение, фибриноидный некроз коронарных артерий, неблагоприятное ремоделирование, инсульт и тому подобное, или показание, указанное в отдельной категории заболеваний в данном описании изобретения. Также включены тромбоз вен, тромбоз глубоких вен, тромбофлебит, артериальная эмболия, тромбоз коронарных артерий, тромбоз артерий головного мозга, эмболия мозга, эмболия почки, эмболия легкого и тромбоз из-за а) искусственных клапанов или других имплантатов, б) постоянных катетеров, в) стентов, г) сердечно-легочного шунтирования, д) гемодиализа или е) других процедур, в которых кровь подвергается воздействию искусственной поверхности, которая способствует тромбозу. Следует отметить, что тромбоз включает окклюзию (например после шунтирования) и реокклюзию (например во время или после чрескожной транслюминальной коронарной ангиопластики).
Сердечно-сосудистые осложнения диабета 2 типа связаны с воспалением, соответственно, соединения по настоящему изобретению можно использовать для лечения диабета и диабетических осложнений, таких как макрососудистое заболевание, гипергликемия, метаболический синдром, нарушение толерантности к глюкозе, гиперурикемия, глюкозурия, катаракты, диабетическая невропатия, диабетическая нефропатия, диабетическая ретинопатия, ожирение, дислипидемия, гипертензия, гиперинсулинемия и синдром резистентности к инсулину или показание, указанное в отдельной категории заболеваний в данном описании изобретения.
Связь врожденного иммунитета и воспаления с заболеванием была продемонстрирована для нейровоспалительных и нейродегенеративных состояний. Следовательно, соединения по настоящему изобретению особенно показаны для применения в лечении нейровоспалительных и нейродегенеративных состояний (т.е. расстройств или заболеваний) у млекопитающих, включая людей, таких как рассеянный склероз, мигрень; эпилепсия; болезнь Альцгеймера; болезнь Паркинсона; травма головного мозга; инсульт; цереброваскулярные заболевания (включая церебральный артериосклероз, церебральную амилоидную ангиопатию, наследственный геморрагический инсульт и гипоксию-ишемию головного мозга); когнитивные расстройства (включаю амнезию, сенильную деменцию, ВИЧ-ассоциированную деменцию, деменцию, ассоциированную с болезнью Альцгеймера, деменцию, ассоциированную с болезнью Хантингтона, деменцию с тельцами Леви, сосудистую деменцию, деменцию, ассоциированную с наркотиками, делирий и легко когнитивное нарушение); умственную неполноценность (включая синдром Дауна и синдром ломкой X-хромосомы); расстройства сна (включая гиперсомнию, расстройство циркадного ритма сна, бессонницу, парасомнию и депривацию сна) и психиатрические расстройства (такие как тревога (включая острое стрессовое расстройство, генерализованное тревожное расстройство, социальное тревожное расстройство, паническое расстройство, посттравматическое стрессовое расстройство и обсессивно-компульсивное расстройство); симулятивное расстройство (включая острую галлюцинаторную манию); расстройства побуждений (включая компульсивное влечение к азартным играм и интермиттирующее эксплозивное расстройство); расстройства настроения (включая биполярное расстройство I типа, биполярное расстройство II типа, манию, смешанное аффективное состояние, большую депрессию, хроническую депрессию, сезонную депрессию, психотическую депрессию и послеродовую депрессию); психомоторное нарушение; психотические расстройства (включая шизофрению, шизоаффективное расстройство, шизофреноформное и бредовое расстройство); зависимость от лекарственных средств (включая наркотическую зависимость, алкоголизм, амфетаминовую зависимость, пристрастие к кокаину, никотиновую зависимость и синдром отмены лекарственных средств); расстройства пищевого поведения (включая анорексию, булимию, компульсивное переедание, гиперфагию и пагофагию); и педиатрические психиатрические расстройства (включая расстройство дефицита внимания, расстройство дефицита внимания/гиперактивности, расстройство поведения и аутизм), боковой амиотрофический склероз, синдром хронической усталости или показание, указанное в отдельной категории заболеваний в данном описании изобретения.
Обычно соединение по изобретению вводят в количестве, эффективном для лечения состояния, как описано в данном описании изобретения. Соединения по изобретению вводят любым подходящим путем в форме фармацевтической композиции, адаптированной к такому пути, и в дозе, эффективной для предполагаемого лечения. Терапевтически эффективные дозы соединений, требующиеся для лечения прогрессирования медицинского состояния, могут быть легко установлены обычным специалистом в данной области с использованием доклинических и клинических подходов, известных в области медицины.
Соединения по изобретению могут быть введены перорально. Пероральное введение может включать проглатывание, так что соединение проникает в желудочно-кишечный тракт, или можно использовать буккальное или подъязычное введение, посредством которого соединение попадает в кровоток прямо изо рта.
В еще одном воплощении соединения по изобретению также могут быть введены непосредственно в кровоток, в мышцу или во внутренний орган. Подходящие способы парентерального введения включают внутривенное, внутриартериальное, внутрибрюшинное, интратекальное, интравентрикулярное, интрауретральное, интрастернальное, интракраниальное, внутримышечное и подкожное. Подходящие устройства для парентерального введения включают игольные (включая микроигольные) инжекторы, безыгольные инжекторы и инфузионные методики.
В еще одном воплощении соединения по изобретению также могут быть введены местно на кожу или слизистую, то есть дермально или трансдермально. В еще одном воплощении соединения по изобретению также могут быть введены интраназально или путем ингаляции. В еще одном воплощении соединения по изобретению могут быть введены ректально или вагинально. В еще одном воплощении соединения по изобретению также могут быть введены непосредственно в глаз или ухо.
Схема приема соединений и/или композиций, содержащих эти соединения, основан на различных факторах, включая тип, возраст, массу, пол и медицинское состояние пациента; тяжесть состояния, путь введения; и активность конкретного используемого соединения. Таким образом, схема приема может сильно варьироваться. Уровни дозировки порядка от примерно 0,01 мг до примерно 100 мг на килограмм массы тела в сутки являются полезными в лечении вышеуказанных состояний. В одном воплощении общая суточная доза соединения по изобретению (введенная в однократной дозе или в дробных дозах) обычно составляет от примерно 0,01 до примерно 100 мг/кг. В еще одном воплощении общая суточная доза соединения по изобретению составляет от примерно 0,1 до примерно 50 мг/кг, и в другом воплощении, от примерно 0,5 до примерно 30 мг/кг (т.е. мг соединения по изобретению на кг массы тела). В одном воплощении дозировка составляет от 0,01 до 10 мг/кг/сутки. В еще одном воплощении дозировка составляет от 0,1 до 1,0 мг/кг/сутки. Композиции со стандартной дозировкой могут содержать такие ее количества или субколичества, чтобы составить суточную дозу. Во многих случаях введение соединения будет повторено несколько раз в сутки (обычно не более 4 раз). Множественные дозы в сутки обычно можно использовать для увеличения общей суточной дозы, если это необходимо.
Для перорального введения могут быть предложены композиции в форме таблеток, содержащие от примерно 0,01 мг до примерно 500 мг активного ингредиента, или в другом воплощении, от примерно 1 мг до примерно 100 мг активного ингредиента. Внутривенные дозы могут варьироваться от примерно 0,1 до примерно 10 мг/кг/минута во время инфузии с постоянной скоростью.
Подходящие субъекты согласно настоящему изобретению включают млекопитающих субъектов. Млекопитающие согласно настоящему изобретению включают, но без ограничения ими, псовых, кошачьих, бычьих, козьих, лошадиных, овечьих, свиней, грызунов, зайцеобразных, приматов и тому подобное, и включают внутриутробных млекопитающих. В одном воплощении подходящими субъектами являются люди. Субъекты-люди могут быть любого пола и находиться на любой стадии развития.
В еще одном воплощении изобретение включает применение одного или более соединений по изобретению для изготовления лекарственного средства для лечения состояний, указанных в данном описании изобретения.
Для лечения состояний, о которых идет речь выше, соединение по изобретению может быть введено в виде соединения самого по себе. Альтернативно, фармацевтически приемлемые соли являются подходящими для медицинских применений вследствие их большей растворимости в воде по сравнению с родительским соединением.
В еще одном воплощении настоящее изобретение включает фармацевтические композиции. Такие фармацевтические композиции содержат соединение по изобретению, представленное с фармацевтически приемлемым носителем. Носитель может быть твердым, жидким или в обоих состояниях, и может быть приготовлен в композиции с соединением в виде композиции на один прием, например таблетки, которая может содержать от 0,05% до 95% по массе активных соединений. Соединение по изобретению может быть соединено с подходящими полимерами в качестве носителей лекарственных средств направленного действия. Также могут присутствовать другие фармакологически активные вещества.
Соединения по настоящему изобретению могут быть введены любым подходящим путем, предпочтительно в форме фармацевтической композиции, адаптированной для такого пути, и в дозе, эффективной для предполагаемого лечения. Например, активные соединения и композиции могут быть введены перорально, ректально, парентерально или местно.
Пероральное введение твердой лекарственной формы может быть представлено, например, отдельными единицами, такими как твердые и мягкие капсулы, пилюли, саше, пастилки или таблетки, каждая из которых содержит предварительно определенное количество по меньшей мере одного соединения по настоящему изобретению. В еще одном воплощении пероральное введение может происходить в форме порошка или гранул. В еще одном воплощении пероральная лекарственная форма является подъязычной, такой как, например, пастилка. В таких твердых лекарственных формах соединения Формулы Ia обычным образом объединяют с одним или более вспомогательными веществами. Такие капсулы или таблетки могут содержать композицию с контролируемым высвобождением. В случае капсул, таблеток и пилюль, лекарственные формы также могут содержать буферные агенты или могут быть изготовлены с кишечнорастворимыми оболочками.
В еще одном воплощении пероральное введение может происходить в жидкой лекарственной форме. Жидкие лекарственные формы для перорального введения включают, например, фармацевтически приемлемые эмульсии, растворы, суспензии, сиропы и эликсиры, содержащие инертные разбавители, обычно используемые в данной области (например воду). Такие композиции также могут содержать вспомогательные вещества, такие как увлажняющие, эмульгирующие, суспендирующие, вкусо-ароматические (например подслащивающие) и/или отдушки.
В еще одном воплощении настоящее изобретение включает парентеральную лекарственную форму. "Парентеральное введение" включает, например, подкожные инъекции, внутривенные инъекции, внутрибрюшинные инъекции, внутримышечные инъекции, интрастернальные инъекции и инфузию. Инъецируемые препараты (например стерильные инъецируемые водные или маслянистые суспензии) могут быть изготовлены согласно способам, известным в данной области техники, с использованием подходящих диспергирующих, увлажняющих агентов и/или суспендирующих агентов.
В еще одном воплощении настоящее изобретение включает местную лекарственную форму. "Местное введение" включает, например, чрескожное введение, такое как посредством чрескожных пластырей или ионтофорезных устройств, внутриглазное введение, или интраназальное или ингаляционное введение. Композиции для местного введения также включают, например, местные гели, спреи, мази и кремы. Местный препарат может содержать соединение, которое усиливает абсорбцию или проникновение активного ингредиента через кожу или другие области воздействия. Когда соединения по данному изобретению вводят при помощи чрескожного устройства, введение будет осуществлено с использованием пластыря или резервуарного типа или типа пористой мембраны, или какой-либо разновидности твердой матрицы. Типичные препараты для этой цели включают гели, гидрогели, лосьоны, растворы, кремы, мази, присыпки, повязки, пены, пленки, кожные пластыри, облатки, импланты, губки, волокна, бинты и микроэмульсии. Также можно использовать липосомы. Типичные носители включают спирт, воду, минеральное масло, жидкий вазелин, белый вазелин, глицерин, полиэтиленгликоль и пропиленгликоль. Могут быть включены усилители проникновения; см., например, J. Pharm. Sci., 88(10), 955-958, Finnin and Morgan (October 1999).
Препараты, подходящие для местного введения в глаз, включают, например, глазные капли, где соединение по данному изобретению растворено или суспендировано в подходящем носителе. Типичный препарат, подходящий для глазного или ушного введения, может быть в форме капель микронизированной суспензии или раствора в изотоническом стерильном растворе со скорректированным рН. Другие препараты, подходящие для глазного и ушного введения, включают мази, биоразлагаемые (например рассасывающиеся гелевые губки, коллаген) и небиоразлагаемые (например силиконовые) имплантаты, облатки, линзы и системы микрочастиц или везикулярные системы, такие как ниосомы или липосомы. Полимер, такой как сшитая полиакриловая кислота, поливиниловый спирт, гиалуроновая кислота, целлюлозный полимер, например гидроксипропилметилцеллюлоза, гидроксиэтилцеллюлоза или метилцеллюлоза, или гетерополисахаридный полимер, например геллановая камедь, может быть включен вместе с консервантом, таким как бензалкония хлорид. Такие препараты также могут быть доставлены посредством ионтофореза.
Для интраназального введения или введения посредством ингаляции активные соединения по изобретению удобным образом доставляют в форме раствора или суспензии из контейнера-пульверизатора, который сжимает или накачивает пациент, или в форме аэрозольного спрея из находящегося под давлением контейнера или небулайзера, с использованием подходящего пропеллента. Препараты, подходящие для интраназального введения, обычно вводят в форме сухого порошка (или отдельно, или в виде смеси, например в сухой смеси с лактозой или в виде частиц из смеси компонентов, например смеси с фосфолипидами, такими как фосфатидилхолин) из ингалятора сухого порошка или в виде аэрозольного спрея из контейнера под давлением, насоса, спрея, распылителя (предпочтительно распылителя с использованием электродинамики для получения мелкодисперсного тумана), или небулайзера, с использованием или без использования подходящего пропеллента, такого как 1,1,1,2-тетрафторэтан или 1,1,1,2,3,3,3-гептафторпропан. Для интраназального применения порошок может содержать биоадгезивный агент, например хитозан или циклодекстрин.
В еще одном воплощении настоящее изобретение содержит ректальную лекарственную форму. Такая ректальная лекарственная форма может находиться в форме, например, суппозитория. Масло какао представляет собой традиционную основу для суппозитория, но при необходимости можно использовать различные альтернативы.
Также можно использовать другие вещества-носители и способы введения, известные в области фармацевтики. Фармацевтические композиции по изобретению могут быть получены при помощи любой из методик, хорошо известных в фармацевтике, такой как эффективные процедуры изготовления в виде препаратов и введения. Вышеуказанные соображения относительно эффективных препаратов и процедур введения хорошо известны в области техники и описаны в стандартных учебниках. Приготовление лекарственных средств в виде препаратов обсуждается, например, в Hoover, John Е.,  Pharmaceutical Sciences, Mack Publishing Co., Easton, Pennsylvania, 1975; Liberman et al., Eds., Pharmaceutical Dosage Forms, Marcel Decker, New York, N.Y., 1980; и Kibbe et al., Eds., Handbook of Pharmaceutical Excipients (3rd Ed.), American Pharmaceutical Association, Washington, 1999.
Pharmaceutical Sciences, Mack Publishing Co., Easton, Pennsylvania, 1975; Liberman et al., Eds., Pharmaceutical Dosage Forms, Marcel Decker, New York, N.Y., 1980; и Kibbe et al., Eds., Handbook of Pharmaceutical Excipients (3rd Ed.), American Pharmaceutical Association, Washington, 1999.
Соединения по настоящему изобретению можно использовать отдельно или в комбинации с другими терапевтическими агентами в лечении различных состояний или заболеваний. Соединение(я) по настоящему изобретению и другой(ие) терапевтический(ие) агент(ы) может(гут) быть введен(ы) одновременно (либо в той же лекарственной форме, либо в отдельных лекарственных формах) или последовательно.
Два или более соединений могут быть введены одновременно, параллельно или последовательно. Кроме того, одновременное введение может быть осуществлено путем смешивания соединений перед введением или путем введения соединений в один и тот же момент времени, но в разные анатомические места или с использованием разных путей введения.
Выражения "параллельное введение", "совместное введение", "одновременное введение" и "введенный одновременно" означают, что соединения вводят в комбинации.
Настоящее изобретение включает применение комбинации соединения, ингибирующего IRAK, как предложено в соединении Формулы I, и одного или более дополнительных фармацевтически активных агентов. Если вводят комбинацию активных агентов, они могут быть введены последовательно или одновременно, в отдельных лекарственных формах или объединенными в одну лекарственную форму. Соответственно, настоящее изобретение также включает фармацевтические композиции, содержащие некоторое количество: а) первого агента, включающего соединение Формулы I или фармацевтически приемлемую соль этого соединения; б) второго фармацевтически активного агента; и в) фармацевтически приемлемого носителя, наполнителя или разбавителя.
Соединения по настоящему изобретению могут быть введены отдельно или в комбинации с одним или более дополнительными терапевтическими агентами. Под словами "введены в комбинации" или "комбинированная терапия" подразумевают, что соединение по настоящему изобретению и один или более дополнительных терапевтических агентов вводят параллельно млекопитающему, которое лечат. При введении в комбинации каждый компонент может быть введен в одно и то же время или последовательно, в любом порядке, в разные моменты времени. Таким образом, каждый компонент может быть введен отдельно, но достаточно близко по времени для того, чтобы обеспечить нужный терапевтический эффект. Таким образом, способы предупреждения и лечения, описанные в данном описании изобретения, включают применение комбинированных агентов.
Комбинированные агенты вводят млекопитающему, включая человека, в терапевтически эффективном количестве. Под "терапевтически эффективным количеством" понимают количество соединения по настоящему изобретению, которое, при введении млекопитающему отдельно или в комбинации с дополнительным терапевтическим агентом, является эффективным для лечения целевого заболевания/состояния, например воспалительного состояния, такого как системная красная волчанка. См. также, Т. Koutsokeras and Т. Healy, Systemic lupus erythematosus and lupus nephritis, Nat Rev Drug Discov, 2014, 13(3), 173-174, в отношении терапевтических агентов, пригодных в лечении волчанки.
В частности, предполагается, что соединения по изобретению могут быть введены со следующими терапевтическими агентами:
Нестероидные противовоспалительные лекарственные средства (NSAID), включая, но без ограничения ими, неселективные ингибиторы СОХ 1/2 (циклооксигеназа 1/2), такие как пироксикам, напроксен, флубипрофен, фенопрофен, кетопрофен, ибупрофен, этодолак (Лодин), мефанаминовую кислоту, сулиндак, апазон, пиразолоны (такие как фенилбутазон), салицилаты (такие как аспирин); селективные ингибиторы СОХ2, такие как: целекоксиб, рофекоксиб, эторикоксиб, валдекоксиб, мелоксикам;
Иммуномодулирующие и/или противовоспалительные агенты, включая, но без ограничения ими, метотрексат, лефлуномид, циклезонид хлорохин, гидроксихлорохин, d-пеницилламин, ауранофин, сульфасалазин, натрия ауротиомалат, циклоспорин, азатиоприн, кромолин, гидроксикарбамид, ретиноиды, фумараты (такие как монометил- и диметилфумарат), глатирамера ацетат, митоксантрон, терифлуномид, суплатаста тозилат, микофенолята мофетил и циклофосфамид, лаквинимод, воклоспорин, PUR-118, AMG 357, AMG811,BCT197;
Противомалярийные агенты, включая, но без ограничения ими, гидроксихлорохин (Плаквенил) и хлорохин (Арален), циклофосфамид (Цитоксан), метотрексат (Ревматрекс), азатиоприн (Имуран), мезаламин (Асакол) и сульфасалазин (Азульфидин);
Антибиотики, включая, но без ограничения ими, флагил или ципрофлоксацин;
Анти-TNFα агенты, включая, но без ограничения ими, инфликсимаб, адалимумаб, цертолизумаба пэгол, голимумаб и этанерцепт;
Aнти-CD20 агенты, включая, но без ограничения ими, ритуксимаб, окрелизумаб, офатумумаб и PF-05280586;
Противодиарейные агенты, такие как дифеноксилат (Ломотил) и лоперамид (Имодиум);
Агенты, связывающие желчные кислоты, такие как холестирамин, алосетрон (Лотронекс) и убипростон (Амитиза);
Слабительные средства, такие как гидроксид магния, полиэтиленгликоль (Миралакс), Дульколакс, Корректол и Сенокот, и антихолинергические средства или спазмолитики, такие как дицикломин (Бентил);
Ингибиторы активации Т-лимфоцитов, включая, но без ограничения ими, абатацепт:
Анти-IL1 агенты, включая, но без ограничения ими, анакинру, рилонацепт, канакинумаб, гевокизумаб, MABp1 и MEDI-8968;
Модуляторы глюкокортикоидных рецепторов, которые могут быть дозированным образом введены перорально, посредством ингаляции, инъекции, местно, ректально, посредством внутриглазного введения, включая, но без ограничения ими, бетаметазон, преднизон, гидрокортизон, преднизолон, флунизолид, триамцинолона ацетамид, беклометазона дипропионат, будесонид, флутиказона пропионат, циклесонид, мометазона фуроат, флуоцинонид, дезоксиметазон, метилпреднизолон или PF-04171327;
Производные аминосалициловой кислоты, включая, но без ограничения ими, сульфасалазин и месалазин;
Агенты против интегрина α4, включая, но без ограничения ими, натализумаб;
α1- или α2-адренергические агонисты, включая, но без ограничения ими: пропилгексидрин, фенилэфрин, фенилпропаноламин, псевдоэфедрин или нафазолина гидрохлорид, оксиметазолина гидрохлорид, тетрагидрозолина гидрохлорид, ксилометазолина гидрохлорид или этилнорэпинефрина гидрохлорид;
β-адренергические агонисты, включая, но без ограничения ими, метапротеренол, изопротенерол, изопреналин, альбутерол, сальбутамол, формотерол, салметерол, тербуталин, орципреналин, ботолтерола мезилат, пирбутерол;
Антихолинергические агенты, включая, но без ограничения ими, ипратропия бромид, тиотропия бромид, окситропия бромид, аклидиния бромид, гликопирролат, пирензипин или телензепин;
Ингаляционные бета-агонисты длительного действия, мускариновые антагонисты длительного действия и кортикостероиды длительного действия, включая, но без ограничения ими, лекарственные средства, включенные в следующую ссылку: Y. Mushtaq, The COPD pipeline, Nat Rev Drug Discov, 2014, 13(4), 253-254. http://dx.doi.org/10.1038/nrd425;
Модуляторы лейкотриенового пути, включая, но без ограничения ими, ингибиторы 5-LO (5-липоксигеназа) (такие как зилеутон), антагонисты FLAP (5-липоксигеназа-активирующий белок) (такие как велифлапон, фибофлапон), антагонисты LTD4 (лейкотриен D4) (такие как монтелукаст, зафирлукаст или пранлукаст);
Антагонисты рецептора H1, включая, но без ограничения ими, цетиризин, лоратадин, дезлоратадин, фексофенадин, астемизол, азеластин или хлорфенирамин;
Ингибиторы PDE4, включая, но без ограничения ими, апремиласт, рофлумиласт или AN2728;
Модуляторы рецептора витамина D, включая, но без ограничения им, парикальцитол;
Активаторы сигнального пути Nrf2, включая, но без ограничения ими, фумараты, сульфорафан и бардоксолон-метил;
Модуляторы семейства родственных RAR (рецептор ретиноевой кислоты) орфанных рецепторов (ROR), в частности RORg;
Модуляторы и/или антагонисты хемокиновых рецепторов, включая, но без ограничения ими, антагонисты CCR2 (такие как ССХ140, BMS-741672, PF-4634817, ССХ-872, NOX-E36), антагонисты CCR2/5 (такие как PF-4634817), CCR9 (такие как верцирнон, ССХ507), модуляторы CCR1, модуляторы CCR4, модуляторы CCR5, модуляторы CCR6, модуляторы CXCR6, модуляторы CXCR7) и модуляторы CXCR2 (такие как данириксин, AZD5069);
Простагландины, включая, но без ограничения им, простациклин;
Ингибиторы PDE5, включая, но без ограничения ими, силденафил, PF-489791, варденафил и тадалафил;
Антагонисты эндотелиновых рецепторов, включая, но без ограничения ими, босентан, амбрисентан, спарсентан, атрасентан, зиботентан и мацитантан;
Активаторы растворимой гуанилатциклазы, включая, но без ограничения им, риоцигуат;
Интерфероны, включая, но без ограничения ими, интерферон бета-1а, интерферон бета-1b;
Модуляторы рецептора сфингозин-1-фосфата, включая, но без ограничения ими, финголимод и понесимод;
Ингибиторы комплементного пути, включая, но без ограничения ими, антагонисты C5aR (такие как ССХ168, РМХ-53, NN8210), ингибиторы С5 (такие как экулизумаб), ингибиторы факторов В и D комплемента, ингибиторы MASP2 (такие как OMS-721) и ARC-1905;
Ингибиторы янус-киназ (одной или более из JAK1, JAK2, JAK3, TYK2), включая, но без ограничения ими, децернотиниб, цердулатиниб, JTE-052, руксолитиниб, тофацитиниб, барицитиниб, пефицитиниб, GLPG-0634, INCB-47986, INCB-039110, PF-04965842, XL-019, АВТ-494, R-348, GSK-2586184, АС-410, BMS-911543 и PF-06263276;
Ингибиторы других противовоспалительных или иммуномодулирующих киназ, включая, но без ограничения ими, ингибиторы тирозинкиназы селезенки (SYK), ингибиторы MAP киназы р38 (такие как PF-3715455, РН-797804, AZD-7624, АКР-001, UR-13870, FX-005, семапимод, пексметиниб, ARRY-797, RV-568, дилмапимод, ралиметиниб), ингибиторы PI3K (такие как GSK-2126458, пиларалисиб, GSK-2269557), ингибиторы PI3Kg и/или PI3Kd (такие как CAL-101/GS-1101, дувелисиб), ингибиторы JNK, ингибиторы ERK1 и/или 2, ингибиторы IKKb, ингибиторы BTK, ингибиторы ITK, ингибиторы ASK1 (такие как GS-4997), ингибиторы PKC (такие как сотрастаурин), антагонисты TrkA (такие как СТ-327), ингибиторы MEK1 (такие как Е6201);
Антиоксиданты, включая, но без ограничения ими, ингибиторы миелопероксидазы (такие как AZD-3241), NOX4 и других ферментов NOX (такие как GKT-137831) и N-ацетилцистеин;
Ингибиторы IL5, включая, но без ограничения ими, меполизумаб, реслизумаб и бенрализумаб;
Ингибиторы IL4, включая, но без ограничения ими, пасколизумаб, алтракинцепт и питракинру;
Ингибиторы IL13, включая, но без ограничения ими, тралокинумаб, анрукинзумаб и лебрикизумаб;
Анти-1b6 агенты, включая, но без ограничения ими, тоцилизумаб, олокизумаб, силтуксимаб, PF-4236921 и сирукумаб;
Ингибиторы/антагонисты IL17/IL17R, включая, но без ограничения ими, секукинумаб, RG-7624, бродалумаб и иксекизумаб;
Антагонисты IL12 и/или IL23, включая, но без ограничения ими, тилдракизумаб, гуселькумаб, MEDI2070 и AMG 139;
Ингибиторы IL33, включая, но без ограничения ими, AMG 282;
Ингибиторы IL9, включая, но без ограничения ими, MEDI-528;
Ингибиторы GM-CSF (гранулоцитарно-макрофагальный колониестимулирующий фактор), включая, но без ограничения ими, МТ203;
Анти-CD4 агенты, включая, но без ограничения ими, трегализумаб и ригеримод;
Антагонисты CRTH2, включая, но без ограничения ими, AZD-1981;
Ингибиторы стимулятора В-лимфоцитов (BLYS; также известного как BAFF), белка, уровень которого часто повышен у пациентов с SLE (системная красная волчанка), включая, но без ограничения ими, белимумаб, табалумаб, блисибимод и атацицепт;
CD22-специфичные моноклональные антитела, включая, но без ограничения им, эпратузумаб;
Ингибиторы интерферона-α, включая, но без ограничения ими, сифалимумаб и ронтализумаб;
Ингибитор рецепторов интерферона I типа, включая, но без ограничения им, MEDI-546;
Агонисты FcγRIIB, включая, но без ограничения им, SM-101;
Модифицированные и/или рекомбинантные варианты белка теплового шока 10 (Hsp10, также известного как шаперонин 10 или EPF), включая, но без ограничения им, INV-103;
Ингибиторы рецептора 12А из надсемейства рецепторов TNF (рецептор TWEAK), включая, но без ограничения ими, BIIB-023, энаватузумаб и RG-7212;
Ингибиторы ксантиноксидазы, включая, но без ограничения ими, аллопуринол, бензбромарон, фебуксостат, топироксостат, тизопурин и инозитолы;
Ингибиторы URAT1 (белок-переносчик уратов - 1) (также известного как SLC22A12), включая, но без ограничения ими, лесинурад, RDEA 3170, UR1102 и левотофиспам;
Дополнительное лечение подагры и/или снижения уровней мочевых кислот, включая, но без ограничения ими, колхицины, пеглотиказу, бензиодарон, изоброминидион, ВСХ4208 и архалофенат;
Ингибиторы Толл-подобных рецепторов (TLR), включая, но без ограничения ими, один или более из TLR7, TLR8, TLR9 (такой как IMO-8400, IMO-3100, DV-1179), TLR2 и/или TLR 4 (такой как VB-201, OPN-305);
Агонисты TLR, включая, но без ограничения ими, TLR7 (такой как GSK2245035, AZD8848), TLR9 (такой как AZD1419);
Активаторы SIRT1, включая, но без ограничения им, SRT2104;
Агонисты рецептора A3, включая, но без ограничения им, CF101;
Другие агенты для применения в лечении псориаза, включая, но без ограничения ими, IDP-118, LAS41004, LEO 80185, LEO 90100, РН-10, WBI-1001, CNT01959, ВТ-061, симзию, устекинумаб, MK-3222/SCH 900222, ACT-128800, АЕВ071, алитретиноин, ASP015K, Apo805K1, BMS-582949, FP187, гекторал (доксеркальциферол), LEO 22811, Ly3009104 (INCB28050), кальципотриеновую пену (STF 115469), тофацитиниб (СР-690,550), М518101 и CycloPsorb™;
Антифибротические агенты, включая, но без ограничения ими: пирфенидон, ингибиторы LOXL2 (гомолог 2 лизилоксидазы) (такие как симтузумаб), FT-011, модуляторы эпирегулина и/или TGFα (трансформирующий ростовой фактор альфа) (такие как LY-3016859), модуляторы TGFβ (такие как LY-2382770, фрезолимумаб);
Ингибиторы пролилгидроксилазы, включая, но без ограничения ими, GSK1278863, FG-2216, ASP-1517/FG-4592, AKB-6548, JTZ-951, BAY-85-3934 и DS-1093;
Ингибиторы гранулоцитарно-макрофагального колониестимулирующего фактора, включая, но без ограничения ими, GSK3196165 (MOR103), PD-0360324 и маврилимумаб;
Ингибиторы MAdCAM (молекула клеточной адгезии слизистой оболочки адрессин) и/или α4β7-интегрина, включая, но без ограничения ими, PF-00547659 и MEDI7183 (абрилумаб);
Ингибиторы фактора роста соединительной ткани (CTGF), включая, но без ограничения им, PF-06473871; ингибиторы катепсина С, включая, но без ограничения им, GSK2793660;
Ингибиторы растворимой эпоксидгидролазы, включая, но без ограничения им, GSK2269557;
Ингибиторы доменного белка TNFR1-ассоциированной гибели, включая, но без ограничения ими, GSK2862277;
Анти-CD19 агенты, включая, но без ограничения ими, MEDI-551 и AMG 729;
Анти-B7RP1 агенты /ингибиторы ICOS-лиганда (индуцируемого костимулирующего лиганда), включая, но без ограничения ими, MEDI5872 и AMG-557;
Ингибиторы тимусного стромального лимфопоэтина, включая, но без ограничения ими, AMG157;
Ингибиторы IL2, включая, но без ограничения ими, даклизумаб;
Ингибиторы лейцин-богатых повторов нейронального белка 6А, включая, но без ограничения им, анти-линго (Biogen);
Ингибиторы интегринов, включая, но без ограничения ими, альфа-V/бета-6 (STX-100) и альфа-V/бета-3 (VPI-2690B);
Анти-CD40L агенты, включая, но без ограничения им, CDP-7657;
Модуляторы дофаминового рецептора D3, включая, но без ограничения им, АВТ-614;
Ингибиторы и/или модуляторы галектина-3, включая, но без ограничения ими, GCS-100 и GR-MD-02;
Агенты для лечения диабетической нефропатии, включая, но без ограничения ими, DA-9801 и ASP-8232;
Агенты для лечения острого почечного повреждения, включая, но без ограничения ими, THR-184, TRC-160334, NX-001, ЕА-230, АВТ-719, СМХ-2043, ВВ-3 и МТР-131;
Модуляторы инфламмасом, включая, но без ограничения ими, ингибиторы NLRP3;
Модуляторы бромодоменов, включая, но без ограничения им, BRD4;
Модуляторы GPR43 (сопряженный с белком G рецептор 43); и
Ингибиторы каналов TRP (каналы с транзиторным рецепторным потенциалом), включая, но без ограничения ими, TRPA1, TRPC3, TRPC5, TRPC6 и TRPC6.
Дополнительные терапевтические агенты включают антикоагулянты или ингибиторы коагуляции, антитромбоцитарные агенты или агенты, ингибирующие тромбоциты, ингибиторы тромбина, тромболитические или фибринолитические агенты, противоаритмические агенты, противогипертензивные агенты, блокаторы кальциевых каналов (L-типа и Т-типа), сердечные гликозиды, диуретики, антагонисты минералокортикоидных рецепторов, агенты-доноры NO, такие как органонитраты, NO-промотирующие агенты, такие как ингибиторы фосфодиэстеразы, агенты, снижающие холестерин/липиды и терапевтические средства липидного профиля, противодиабетические агенты, антидепрессанты, противовоспалительные агенты (стероидные и нестероидные), агенты против остеопороза, гормонозаместительные терапии, пероральные контрацептивы, агенты против ожирения, средства против тревоги, антипролиферативные агенты, противоопухолевые агенты, противоязвенные агенты и агенты против гастроэзофагеальной рефлюксной болезни, гормон роста и/или стимуляторы секреции гормона роста, тиреотропные миметики (включая антагонист рецептора тиреотропного гормона), противоинфекционные агенты, противовирусные агенты, антибактериальные агенты и противогрибковые агенты.
Включены агенты, используемые в ICU (интенсивной терапии), например добутамин, дофамин, эпинефрин, нитроглицерин, нитропруссид и т.д.
Включены комбинированные агенты, пригодные для лечения васкулита, например азатиоприн, циклофосфамид, микофенолят, мофетил, ритуксимаб и т.д.
В еще одном воплощении в настоящем изобретении предложена комбинация, где второй агент представляет собой по меньшей мере один агент, выбранный из ингибитора фактора Ха, антикоагулянта, антитромбоцитарного агента, ингибитора тромбина, тромболитического агента и фибринолитического агента. Типичные ингибиторы фактора Ха включают апиксабан и ривароксабан. Примеры подходящих антикоагулянтов для применения в комбинации с соединениями по настоящему изобретению включают гепарины (например нефракционированные и низкомолекулярные гепарины, такие как эноксапарин и далтепарин).
В еще одном воплощении второй агент представляет собой по меньшей мере один агент, выбранный из варфарина, нефракционированного гепарина, низкомолекулярного гепарина, синтетического пентасахарида, гирудина, аргатробана, аспирина, ибупрофена, напроксена, сулиндака, индометацина, мефенамата, дроксикама, диклофенака, сульфинпиразона, пироксикама, тиклопидина, клопидогрела, тирофибана, эптифибатида, абциксимаба, мелагатрана, дисульфатогирудина, активатора тканевого плазминогена, модифицированного активатора тканевого плазминогена, анистреплазы, урокиназы и стрептокиназы.
В еще одном воплощении агент представляет собой по меньшей мере один антитромбоцитарный агент. Особенно предпочтительными антитромбоцитарными агентами являются аспирин и клопидогрел. Термин "антитромбоцитарные агенты" (или ингибиторы агрегации тромбоцитов), при использовании в данном описании изобретения, означает агенты, которые ингибируют функцию тромбоцитов, например путем ингибирования агрегации, адгезии или гранулярной секреции тромбоцитов. Агенты включают, но без ограничения ими, различные известные нестероидные противовоспалительные лекарственные средства (NSAID), такие как аспирин, ибупрофен, напроксен, сулиндак, индометацин, мефенамат, дроксикам, диклофенак, сульфинпиразон, пироксикам и их фармацевтически приемлемые соли или пролекарства. Из NSAID предпочтительными являются аспирин (ацетилсалициловая кислота или ASA) и ингибиторы СОХ-2, такие как целекоксиб или пироксикам. Другие подходящие агенты для ингибирования агрегации тромбоцитов включают антагонисты IIb/IIIa (например тирофибан, эптифибатид и абциксимаб), антагонисты рецептора тромбоксана А2 (например ифетробан), ингибиторы тромбоксан-А2-синтетазы, ингибиторы PDE-III (например плетал, дипиридамол) и их фармацевтически приемлемые соли или пролекарства.
Термин "антитромбоцитарные агенты" (или агенты, ингибирующие агрегацию тромбоцитов), при использовании в данном описании изобретения, так же, как предполагается, включает антагонисты рецептора ADP (аденозиндифосфат), предпочтительно антагонисты пуринергических рецепторов P2Y1 и P2Y12 где P2Y12 является более предпочтительным. Предпочтительные антагонисты рецептора P2Y12 включают тикагрелор, празугрел, тиклопидин и клопидогрел, включая их фармацевтически приемлемые соли или пролекарства. Клопидогрел является еще более предпочтительным агентом. Тиклопидин и клопидогрел также являются предпочтительными соединениями, так как известно, что они являются щадящими в отношении желудочно-кишечного тракта при применении.
Термин "ингибиторы тромбина" (или антитромбиновые агенты), при использовании в данном описании изобретения, означает ингибиторы сериновой протеазы тромбина. В результате ингибирования тромбина прерываются различные тромбин-опосредованные процессы, такие как тромбин-опосредованная активация тромбоцитов (то есть, например, агрегация тромбоцитов и/или гранулярная секреция ингибитора активатора плазминогена-1 и/или серотонина) и/или образование фибрина. Ряд ингибиторов тромбина известен специалисту в данной области, и эти ингибиторы рассматриваются для применения в комбинации с настоящими соединениями. Такие ингибиторы включают, но без ограничения ими, производные бораргинина, борпептиды, гепарины, гирудин, аргатробан и мелагатран, включая их фармацевтически приемлемые соли и пролекарства. Бораргининовые производные и борпептиды включают N-ацетильные и пептидные производные бороновой кислоты, такие как С-концевые производные альфа-аминобороновой кислоты лизина, орнитина, аргинина, гомоаргинина и их соответствующих тиоурониевых аналогов. Термин "гирудин", при использовании в данном описании изобретения, включает подходящие производные или аналоги гирудина, упоминаемые в данном описании изобретения как гирулоги, такие как дисульфатогирудин. Термин "тромболитические или фибринолитические агенты" (или тромболитики или фибринолитики), при использовании в данном описании изобретения, означает агентов, которые лизируют сгустки крови (тромбы). Такие агенты включают активатор тканевого плазминогена (естественный или рекомбинантный) и его модифицированные формы, анистреплазу, урокиназу, стрептокиназу, тенектеплазу (TNK), ланотеплазу (nPA), ингибиторы фактора VIIa, ингибиторы PAI-1 (т.е. инактиваторы ингибиторов активатора тканевого плазминогена), ингибиторы альфа2-антиплазмина и анизоилированный активаторный комплекс плазминогена и стрептокиназы, включая их фармацевтически приемлемые соли или пролекарства. Термин "анистреплаза", при использовании в данном описании изобретения, относится к анизоилированному активаторному комплексу плазминогена и стрептокиназы, как описано, например, в ЕР 028489, раскрытие которого включено в данную заявку посредством ссылки. Термин "урокиназа", при использовании в данном описании изобретения, означает как двухцепочечную, так и одноцепочечную урокиназу, причем последняя также упоминается в данном описании изобретения как проурокиназа. Примеры подходящих противоаритмических агентов включают: агенты класса I (такие как пропафенон); агенты класса II (такие как метопролол, атенолол, карвадиол и пропранолол); агенты класса III (такие как соталол, дофетилид, амиодарон, азимилид и ибутилид); агенты класса IV (такие как дитиазем и верапамил); агенты, открывающие K+ каналы, такие как ингибиторы IAch и ингибиторы IKur (например такие, как соединения, раскрытые в WO 01/40231).
Соединения по настоящему изобретению можно использовать в комбинации с противогипертензивными агентами, и такая противогипертензивная активность легко определяется специалистами в данной области в результате стандартных исследований (например измерений кровяного давления). Примеры подходящих противогипертензивных агентов включают: альфа-адренергические блокаторы; бета-адренергические блокаторы; блокаторы кальциевых каналов (например дилтиазем, верапамил, нифедипин и амлодипин); вазодилаторы (например гидралазин), диуретики (например хлортиазид, гидрохлортиазид, флуметиазид, гидрофлуметиазид, бендрофлуметиазид, метилхлортиазид, трихлорметиазид, политиазид, бензтиазид, этакриновой кислоты трикринафен, хлорталидон, торасемид, фуросемид, музолимин, буметанид, триамтренен, амилорид, спиронолактон); ингибиторы ренина; ингибиторы АСЕ (ангиотензин-превращающий фермент) (например каптоприл, зофеноприл, фозиноприл, эналаприл, цераноприл, цилазаприл, делаприл, пентоприл, хинаприл, рамиприл, лизиноприл); антагонисты рецептора АТ-1 (рецептор ангиотензина подтип АТ-1) (например лозартан, ирбезартан, валсартан); антагонисты ЕТ (эндотелинового) рецептора (например ситаксентан, атрсентан и соединения, раскрытые в патентах US 5612359 и 6043265); двойной антагонист ЕТ/AII (например соединения, раскрытые в WO 00/01389); ингибиторы нейтральной эндопептидазы (NEP); ингибиторы вазопептидазы (двойные ингибиторы NEP-ACE) (например гемопатрилат и нитраты). Типичным антиангинальным агентом является ивабрадин.
Примеры подходящих блокаторов кальциевых каналов (L-типа или Т-типа) включают дилтиазем, верапамил, нифедипин, и амлодипин, и мибефрадил. Примеры подходящих сердечных гликозидов включают дигиталис и уабаин.
В одном воплощении соединение по изобретению может быть введено совместно с одним или более диуретиками. Примеры подходящих диуретиков включают а) петлевые диуретики, такие как фуросемид (такой как LASIX™), торсемид (такой как DEMADEX™), беметанид (такой как BUMEX™) и этакриновая кислота (такой как EDECRIN™); б) диуретики тиазидного типа, такие как хлортиазид (такой как DIURIL™, ESIDRIX™ или HYDRODIURIL™), гидрохлортиазид (такой как MICROZIDE™ или ORETIC™), бензтиазид, гидрофлуметиазид (такой как SALURON™), бендрофлуметиазид, метилхлортиазид, политиазид, трихлорметиазид и индапамид (такой как LOZOL™); в) диуретики фталимидинового типа, такие как хлорталидон (такой как HYGROTON™) и метолазон (такой как ZAROKCOLYN™); г) диуретики хиназолинового типа, такие как хинетазон; и д) калийсберегающие диуретики, такие как триамтерен (такой как DYRENIUM™) и амилорид (такой как MIDAMOR™ или MODURETIC™). В еще одном воплощении соединение по изобретению может быть введено совместно с петлевым диуретиком. В еще одном воплощении петлевой диуретик выбран из фуросемида и торсемида. В еще одном воплощении одно или более соединений по изобретению может быть введено совместно с фуросемидом. В еще одном воплощении одно или более соединений по изобретению может быть введено совместно с торсемидом, который возможно может представлять собой форму торсемида с контролируемым или модифицированным высвобождением.
В еще одном воплощении соединение по изобретению может быть введено совместно с диуретиком тиазидного типа. В еще одном воплощении диуретик тиазидного типа выбран из группы, состоящей из хлортиазида и гидрохлортиазида. В еще одном воплощении одно или более соединений по изобретению могут быть введены совместно с хлортиазидом. В еще одном воплощении одно или более соединений по изобретению могут быть введены совместно с гидрохлортиазидом. В еще одном воплощении одно или более соединений по изобретению могут быть введены совместно с диуретиком фталимидинового типа. В еще одном воплощении диуретик фталимидинового типа представляет собой хлорталидон.
Примеры подходящей комбинации антагонистов минералокортикоидного рецептора включают сприонолактон и эплеренон. Примеры подходящей комбинации ингибиторов фосфодиэстеразы включают: ингибиторы PDE III (такие как цилостазол); и ингибиторы PDE V (такие как силденафил).
Соединения по настоящему изобретению можно использовать в комбинации с агентами, модулирующими уровень холестерина (включая агенты, снижающие уровень холестерина), такими как ингибитор липазы, ингибитор HMG-CoA-редуктазы (3-гидрокси-3-метилглутарил-кофермент А-редуктаза), ингибитор HMG-CoA-синтазы, ингибитор экспрессии гена HMG-CoA-редуктазы, ингибитор экспрессии гена HMG-CoA-синтазы, ингибитор секреции МТР/Аро В (микросомальный белок-переносчик триглицеридов/аполипопротеин В), ингибитор СЕТР (белок-переносчик эфиров холестерина), ингибитор абсорбции желчных кислот, ингибитор абсорбции холестерина, ингибитор синтеза холестерина, ингибитор скваленсинтетазы, ингибитор скваленэпоксидазы, ингибитор скваленциклазы, комбинированный ингибитор скваленэпоксидазы/скваленциклазы, фибрат, ниацин, ионообменная смола, антиоксидант, ингибитор АСАТ (ацил-КoA-холестерол-ацилтрансфераза) или секвестрант желчных кислот или такой агент, как мипомерсен.
Примеры подходящих агентов, снижающих уровень холестерина/липидов, и терапевтических средств липидного профиля, включают: ингибиторы HMG-CoA-редуктазы (например правастатин, ловастатин, аторвастатин, симвастатин, флувастатин, NK-104 (также известный как итавастатин, или нисвастатин, или нисбастатин) и ZD-4522 (также известный как розувастатин, или атавастатин, или висастатин)); ингибиторы скваленсинтетазы; фибраты; секвестранты желчных кислот (такие как квестран); ингибиторы АСАТ; ингибиторы МТР; ингибиторы липооксигеназы; ингибиторы абсорбции холестерина; и ингибиторы белка-переносчика эфиров холестерина.
Противовоспалительные агенты также включают ингибиторы sPLA2 и lpPLA2 (такие как дарапладиб), ингибиторы 5 LO (такие как атрелеутон) и антагонисты IL-1 и IL-1r (такие как канакинумаб).
Другие атеросклеротические агенты включают агенты, которые модулируют действие PCSK9, например бокоцизумаб.
Сердечно-сосудистые осложнения диабета 2 типа связаны с воспалением, соответственно, соединения по настоящему изобретению можно использовать в комбинации с противодиабетическими агентами, особенно с агентами против диабета 2 типа. Примеры подходящих противодиабетических агентов включают, например, инсулины, метформин, ингибиторы DPPIV (дипептидил-пептидазы IV), агонисты, аналоги и миметики GLP-1, ингибиторы SGLT1 (натрий-зависимый переносчик глюкозы 1) и SGLT2. Подходящие противодиабетические агенты включают ингибиторы ацетил-СоА-карбоксилазы (АСС), такие как агенты, описанные в WO 2009144554, WO 2003072197, WO 2009144555 и WO 2008065508, ингибиторы диацилглицерин-О-ацилтрансферазы 1 (DGAT-1), такие агенты, описанные в WO 09016462 или WO 2010086820, AZD7687 или LCQ908, ингибитор диацилглицерин-О-ацилтрансферазы 2 (DGAT-2), ингибиторы моноацилглицерин-О-ацилтрансферазы, ингибитор фосфодиэстеразы (PDE)-10, активатор АМРК (АМФ-активируемая протеинкиназа), сульфонилмочевина (например ацетогексамид, хлорпропамид, диабинезе, глибенкламид, глипизид, глибурид, глимепирид, гликлазид, глипентид, гликвидон, глизоламид, толазамид и толбутамид), меглитинид, ингибитор α-амилазы (например тендамистат, трестатин и AL-3688), ингибитор α-глюкозидгидролазы (например акарбоза), ингибитор α-глюкозидазы (например адипозин, камиглибоза, эмиглитат, миглитол, воглибоза, прадимицин-Q и салбостатин), агонист PPARγ (активируемый пролифератором пероксисом рецептор гамма) (например балаглитазон, циглитазон, дарглитазон, энглитазон, исаглитазон, пиоглитазон и розиглитазон), агонист PPAR α/γ (например CLX-0940, GW-1536, GW-1929, GW-2433, KRP-297, L-796449, LR-90, МК-0767 и SB-219994), бигуанид (например метформин), модулятор глюкагон-подобного пептида 1 (GLP-1), такой как агонист (например эксендин-3 и эксендин-4), лираглутид, албиглутид, эксенатид (Byetta®), албиглутид, ликсисенатид, дулаглутид, семаглутид, NN-9924, ТТР-054, ингибитор протеинтирозинфосфатазы-1В (РТР-1B) (например тродусквемин, экстракт гиртиозал (экстракт морских губок Hyrtios sp.) и соединения, раскрытые в Zhang, S., et al., Drug Discovery Today, 12(9/10), 373-381 (2007)), ингибитор SIRT-1 (ситртуин-1) (например ресвератрол, GSK2245840 или GSK184072), ингибитор дипептидилпептидазы IV (DPP-IV) (например агенты, раскрытые в WO 2005116014, ситаглиптин, вилдаглиптин, алоглиптин, дутоглиптин, линаглиптин и саксаглиптин), стимулятор секреции инсулина, ингибитор окисления жирных кислот, антагонист А2, ингибитор c-jun-аминоконцевой киназы (JNK), активаторы глюкокиназы (GKa), такие как агенты, описанные в WO 2010103437, WO 2010103438, WO 2010013161, WO 2007122482, ТТР-399, ТТР-355, ТТР-547, AZD1656, ARRY403, МК-0599, ТАК-329, AZD5658 или GKM-001, инсулин, миметик инсулина, ингибитор гликогенфосфорилазы (например GSK1362885), агонист рецептора VPAC2, ингибиторы SGLT2, такие как агенты, описанные в Е.С. Chao et al., Nature Reviews Drug Discovery 9, 551-559 (July 2010), включая дапаглифлозин, канаглифлозин, эмпаглифлозин, тофоглифлозин (CSG452), ASP-1941, THR1474, TS-071, ISIS388626 и LX4211, а также агенты, описанные в WO 2010023594, модулятор глюкагонового рецептора, такой как агенты, описанные в Demong, D.E. et al., Annual Reports in Medicinal Chemistry 2008, 43, 119-137, модуляторы GPR119, в частности агонисты, такие как агенты, описанные в WO 2010140092, WO 2010128425, WO 2010128414, WO 2010106457, Jones, R.M. et al., in Medicinal Chemistry 2009, 44, 149-170 (например MBX-2982, GSK1292263, APD597 и PSN821), производные или аналоги FGF21 (фактор роста фибробластов 21), такие как агенты, описанные в Kharitonenkov, A. et al., Current Opinion in Investigational Drugs 2009, 10(4)359-364, модуляторы рецептора TGR5 (рецептор желчных кислот 5) (также называемого GPBAR1), в частности агонисты, такие как агенты, описанные в Zhong, М., Current Topics in Medicinal Chemistry, 2010, 10(4), 386-396, и INT777, агонисты GPR40 (сопряженный с G-белком рецептор 40), такие как агенты, описанные в Medina, J.C., Annual Reports in Medicinal Chemistry, 2008, 43, 75-85, включая, но без ограничения ими, ТАК-875, GPR120 модуляторы, в частности агонисты, высокоаффинные активаторы рецепторов никотиновой кислоты (НМ74А), и ингибиторы SGLT1, такие как GSK1614235. Другой репрезентативный список противодиабетических агентов, которые можно комбинировать с соединениями по настоящему изобретению, можно найти, например, на странице 28, строка 35, и вплоть до страницы 30, строка 19, WO 2011005611. Предпочтительными противодиабетическими агентами являются метформин и ингибиторы DPP-IV (например ситаглиптин, вилдаглиптин, алоглиптин, дутоглиптин, линаглиптин и саксаглиптин). Другие противодиабетические агенты могут включать ингибиторы или модуляторы ферментов карнитинпальмитоилтрансфераз, ингибиторы фруктоза-1,6-дифосфатазы, ингибиторы альдозаредуктазы, ингибиторы минералокортикоидного рецептора, ингибиторы TORC2, ингибиторы CCR2 и/или CCR5, ингибиторы изоформ PKC (протеинкиназа С) (например PKCα, PKCβ, PKCγ), ингибиторы синтетазы жирных кислот, ингибиторы серинпальмитоилтрансферазы, модуляторы GPR81, GPR39, GPR43, GPR41, GPR105, Kv1.3, ретинолсвязывающий белок 4, глюкокортикоидный рецептор, соматостатиновые рецепторы (например SSTR1, SSTR2, SSTR3 и SSTR5), ингибиторы или модуляторы PDHK2 или PDHK4, ингибиторы MAP4K4 (киназа 4 митоген-активируемой протеинкиназы 4), модуляторы семейства IL1, включая IL1бета, модуляторы RXRaльфa (ретиноидный Х-рецептор альфа). Кроме того, подходящие противодиабетические агенты включают механизмы, перечисленные в Carpino, Р.А., Goodwin, В. Expert Opin. Ther. Pat, 2010, 20(12), 1627-51.
Специалистам в данной области понятно, что соединения по данному изобретению также можно использовать в сочетании с другими сердечно-сосудистыми или цереброваскулярными терапевтическими средствами, включающими PCI (чрескожное коронарное вмешательство), стентирование, стенты, элюирующие лекарственное средство, терапию стволовыми клетками и медицинские устройства, такие как имплантируемые кардиостимуляторы, дефибрилляторы или ресинхронизирующая терапия сердца.
Соединения по настоящему изобретению можно использовать у млекопитающих в комбинации с нейровоспалительными и нейроденегенеративными агентами. Примеры дополнительных нейровоспалительных и нейродегенеративных агентов включают антидепрессанты, антипсихотические средства, средства против боли, агенты против болезни Альцгеймера и противотревожные агенты. Примеры конкретных классов антидепрессантов, которые можно использовать в комбинации с соединениями по изобретению, включают ингибиторы обратного захвата норэпинефрина, селективные ингибиторы обратного захвата серотонина (SSRI), антагонисты рецептора NK-1 (нейрокинин-1), ингибиторы моноаминоксидазы (MAOI), обратимые ингибиторы моноаминоксидазы (RIMA), ингибиторы обратного захвата серотонина и норадреналина (SNRI), антагонисты кортикотропин-рилизинг фактора (CRF) и атипичные антидепрессанты. Подходящие ингибиторы обратного захвата норэпинефрина включают третичные аминные трициклические агенты и вторичные аминные трициклические агенты. Примеры подходящих третичных аминных трициклических агентов и вторичных аминных трициклических агентов включают амитриптилин, кломипрамин, доксепин, имипрамин, тримипрамин, дотиепин, бутриптилин, нортриптилин, протриптилин, амоксапин, дезипрамин и мапротилин. Примеры подходящих SSRI включают флуоксетин, флувоксамин, пароксетин и сертралин. Примеры ингибиторов моноаминоксидазы включают изокарбоксазид, фенелзин и транилциклопрамин. Примеры подходящих обратимых ингибиторов моноаминоксидазы включают моклобемид. Примеры подходящих SNRI для применения в настоящем изобретении включают венлафаксин. Примеры подходящих атипичных антидепрессантов включают бупропион, литий, тразодон и вилоксазин. Примеры агентов против болезни Альцгеймера включают антагонисты рецептора NMDA (N-метил-D-аспартат), такие как мемантин; и ингибиторы холинэстеразы, такие как донепезил и галантамин. Примеры подходящих классов противотревожных агентов, которые можно использовать в комбинации с соединениями по изобретению, включают бензодиазепины и агонисты рецептора серотонина-1А (5-НТ1А) и антагонисты CRF. Подходящие бензодиазепины включают алпразолам, хлордиазепоксид, клоназепам, хлоразепат, диазепам, лоразепам, оксазепам и празепам. Подходящие агонисты рецептора 5-НТ1А включают буспирон и ипсапирон. Подходящие антагонисты CRF включают веруцерфонт. Подходящие атипичные антипсихотические средства включают палиперидон, зипрасидон, рисперидон, арипипразол, оланзапин и кветиапин. Подходящие агонисты никотинацетилхолина включают СР-601927 и варениклин. Средства против боли включают прегабалин, габапентин, клонидин, неостигмин, баклофен, мидазолам, кетамин и зиконотид.
Настоящее изобретение также включает наборы, которые пригодны для применения в осуществлении способов лечения, описанных выше. В одном воплощении набор содержит первую лекарственную форму, содержащую одно или более соединений по настоящему изобретению и контейнер для дозировки, в количествах, достаточных для осуществления способов по настоящему изобретению.
В еще одном воплощении набор по настоящему изобретению содержит одно или более соединений по изобретению.
Настоящее изобретение также включает промежуточные соединения, полезные в синтезе соединений по изобретению, включая их соли и/или таутомеры.
Общие схемы синтеза
Соединения Формулы Ia могут быть получены способами, описанными ниже, вместе со способами синтеза, известными в области органической химии, или модификациями или превращениями, которые знакомы обычным специалистам в данной области. Исходные вещества, использованные в данном изобретении, имеются в продаже или могут быть получены обычными способами, известными в данной области [такими как способы, изложенные в стандартных справочных книгах, таких как Compendium of Organic Synthetic Methods, Vol. I-XII (опубликована Wiley-Interscience)]. Предпочтительные способы включают, но без ограничения ими, способы, описанные ниже.
В ходе любой из следующих последовательностей синтеза может быть необходимо и/или желательно защитить чувствительные или реакционноспособные группы на любой из участвующих молекул. Это может быть достигнуто с помощью традиционных защитных групп, таких как группы, описанные в Т.W. Greene, Protective Groups in Organic Chemistry, John Wiley & Sons, 1981; T.W. Greene and P.G.M. Wuts, Protective Groups in Organic Chemistry, John Wiley & Sons, 1991; и Т.W. Greene and P.G.M. Wuts, Protective Groups in Organic Chemistry, John Wiley & Sons, 1999, которые включены в данную заявку посредством ссылки.
Соединения Формулы Ia или их фармацевтически приемлемые соли могут быть получены согласно реакционным схемам, которые описаны в данном описании изобретения ниже. Если не указано иное, заместители в схемах являются такими, как определено выше. Выделение и очистку продуктов осуществляют с помощью стандартных процедур, которые известны химику обычной квалификации.
Специалисту в данной области следует понимать, что различные символы, верхние и нижние индексы, используемые в схемах, способах и примерах, использованы для удобства представления и/или для отображения порядка, в котором они представлены на схемах, и они не должны обязательно соответствовать символам, верхним или нижним индексам в прилагаемой формуле изобретения. Кроме того, специалисту в данной области понятно, что во многих случаях эти соединения будут представлять собой смеси стереомеров, которые могут быть разделены на разных стадиях схем синтеза с использованием традиционных методик, таких как, но без ограничения ими, кристаллизация, нормально-фазовая хроматография, обращенно-фазовая хроматография и хиральная хроматография, с получением отдельных энантиомеров. Схемы являются репрезентативными для способов, пригодных в синтезе соединений по настоящему изобретению. Они не предназначены каким-либо образом ограничивать объем изобретения.
Способы получения соединений по изобретению аналогичны способам, описанным в PCT/IB2015/052251, поданной 26 марта 2015 года, и соответствующей ей заявке на патент US 14/678114, поданной 3 апреля 2015 года. Эти способы включены в данную заявку во всей своей полноте как способы получения соединений по этому изобретению.
Схема 1


На Схеме 1 показан способ получения соединений Формулы Iа. Соединение Формулы А, в котором Lv представляет собой замещаемую уходящую группу (такую как, например, хлор или фтор), обрабатывают соединением Формулы В (как описано в PCT/IB2015/052251) с получением продукта Формулы Ia. Реакцию обычно осуществляют в присутствии подходящего основания, такого как карбонат цезия, трет-бутилат калия, гидрид натрия или гексаметилдисилазид калия, в подходящем растворителе или смеси растворителей, таком как THF (тетрагидрофуран) или диметилформамид. Соединения Формулы А могут быть получены, как описано в следующих ниже схемах. Соединения Формулы В (R2-OH) могут быть получены от коммерческих поставщиков или получены способами, описанными в химической литературе, или могут быть получены, как описано в следующих ниже схемах.
Если это необходимо, могут быть осуществлены дополнительные трансформации соединений Формулы Ia. Например, соединение Формулы Ia, где R6 представляет собой CN, может быть подвергнуто реакции нитрильного гидролиза с получением соединения Формулы Ia, в котором R6 представляет собой CONH2. Реакция может быть осуществлена различными способами, известными специалисту в данной области, например путем использования кислот или оснований, возможно в присутствии окислителя, такого как перекись водорода, или путем использования химических или ферментных катализаторов. В других случаях соединение Формулы Ia дополнительно может быть обработано реагентами, такими как кислоты, для удаления защитных групп, таких как трет-бутоксикарбонильные группы, и/или другими реагентами с образованием производных функциональных групп, таких как карбоксильные, амино- или гидроксильные группы.
Схема 2

На Схеме 2 показан другой способ получения соединений Формулы Ia, особенно подходящий для случаев, когда оба из X и Y в соединении Формулы А представляют собой углерод. Этот способ предусматривает алкилирование соединения Формулы А соединением Формулы В (где группа R12O- представляет собой гидроксил или сульфонатный сложный эфир, такой как n-толуолсульфонат или метансульфонат; например, как описано в PCT/IB2015/052251, или в виде, имеющемся в продаже), с использованием способов известных специалистам в данной области, с получением продукта Формулы Ia. Например, эта реакция может быть осуществлена путем обработки соединения Формулы А соединением Формулы В (R12 представляет собой Н) в присутствии трифенилфосфина и азодикарбоксилатного сложного эфира ("реакция Мицунобу") в подходящем растворителе, таком как THF. Альтернативно, алкилирование соединения Формулы А может быть осуществлено с использованием соединения Формулы В (R12O представляет собой TsO или другой сульфонатный сложный эфир) в присутствии основания, такого как карбонат цезия, в подходящем растворителе, таком как THF или диметилформамид.
Если необходимо, могут быть осуществлены дальнейшие превращения соединения Формулы Ia. Например, соединение Формулы Ia, где R6 представляет собой CN, может быть подвергнуто реакции нитрильного гидролиза с получением соединения Формулы Ia, в котором R6 представляет собой CONH2. Реакция может быть осуществлена различными способами, известными специалисту в данной области, например путем использования кислот или оснований, возможно в присутствии окислителя, такого как перекись водорода, или путем использования химических или ферментных катализаторов. В других случаях соединение Формулы Ia дополнительно может быть обработано реагентами, такими как кислоты, для отщепления защитных групп, таких как трет-бутоксикарбонильные группы, и/или другими реагентами для получения производных функциональных групп, таких как карбоксильные, амино- или гидроксильные группы.
Схема 3
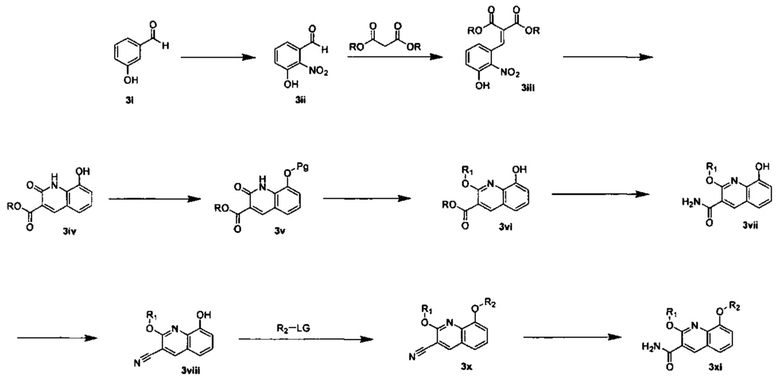
Путь к соединениям Формулы Ia, где Q представляет собой N, и W, X, Z и Y представляют собой СН, показан на Схеме 3. Альдегид, такой как 3i, может быть нитрирован, например, нитратом, таким как изопропилнитрат, в присутствии кислоты, с получением нитро-соединения, такого как 3ii. Конденсация соединения 3ii с малонатом может давать промежуточное соединение, такое как 3iii, которое может быть восстановлено, например с использованием гидросульфита натрия, что приводит к циклизации в пиридин, такой как 3iv. Фенольная группировка может быть защищена подходящей защитной группой, такой как бензильная группа, как описано в литературе [см., например, Wuts, P.G.M. and Greene, Т.W., Greene's Protective Groups in Organic Synthesis, Wiley (2007)] с получением соединения, такого как 3v, где Pg представляет собой подходящую защитную группу. Соединение, такое как 3v, может быть активировано, например, оксихлоридом фосфора, с получением иминохлорида, который затем может быть обработан алкилатом, таким как метилат натрия, с получением продукта, такого как соединение 3vi, где защитная группа была параллельно удалена. Соединение, такое как сложный эфир 3vi, может быть превращено в амид путем обработки, например, аммиаком в метаноле, с получением амида 3vii. Амид 3vii может быть дегидратирован такими реагентами, как пиридин TFAA (трифторуксусный ангидрид), с получением нитрилов, таких как 3viii. Алкилирование фенола 3viii с использованием алкилирующего агента, такого как мезилатное или галогенидное производное, в присутствии основания может давать соединения, представленные 3х. Типичные основания включают, без ограничения им, карбонат цезия. Нитрил 3х может быть гидролизован, например, с использованием перекиси водорода и карбоната калия в DMSO (диметилсульфоксид), с получением амидов, таких как 3xi.
Схема 4
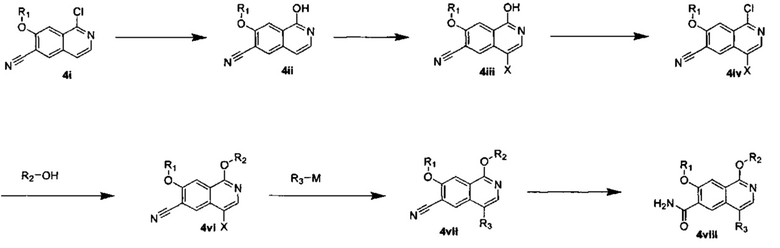
Соединения по изобретению могут быть получены посредством катализируемых металлами реакций кросс-сочетания соединений, таких как галогениды 4vi на Схеме 4. Получение соединений, таких как 4vi, может быть осуществлено следующим образом. Соединения, такие как 4i, могут быть обработаны водной кислотой с получением соединений, таких как 4ii, которые могут быть превращены в галогениды, такие как 4iii, например, путем бромирования с использованием NBS (N-бромсукцинимид). Активация соединений, таких как 4iii, реагентами, такими как оксихлорид фосфора, может давать соединения, такие как хлорид 4iv. Обработка соединения 4iv спиртом и подходящим основанием, таким как NaHMDS, может вызывать превращение в такие соединения, как 4vi. Обработка таких соединений, как 4vi, например, гетероциклическими станнатами, или боронатами, или металлоидами и подходящим катализатором, например палладиевым катализатором, может давать продукты кросс-сочетания, такие как соединение 4vii. Гидрирование нитрила 4vii, например перекисью водорода и карбонатом калия, может давать карбоксамиды, такие как соединение 4viii.
Схема 5

Альтернативно, соединения данного типа могут быть получены при помощи реакций сочетания Сузуки, как изображено на Схеме 5. Например, соединения, такие как 5i, где X представляет собой галоген, могут быть обработаны борным реагентом, таким как бис(пинаколато)дибор, основанием и подходящим палладиевым катализатором с получением боронатного сложноэфирного производного, такого как 5ii. Обработка 5ii гетероциклическим галогенидом, основанием и подходящим палладиевым катализатором дает соединения, такие как 5iii, где R представляет собой гетероцикл. Соединения, такие как 5iii, могут быть гидратированы с использованием, например, перекиси водорода или карбоната калия, с получением таких соединений, как 5iv. В некоторых случаях гетероцикл будет нести защитную группу, и могут быть использованы стандартные способы удаления защитных групп, известные специалистам в данной области.
Схема 6
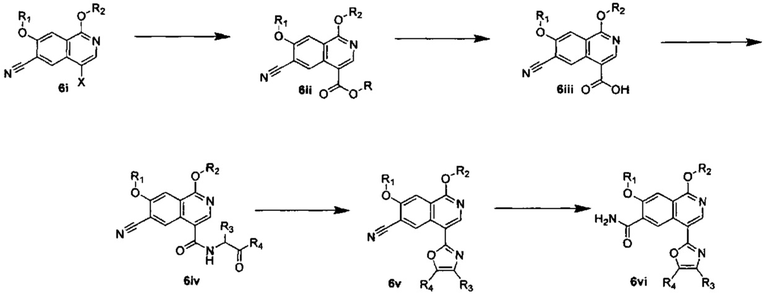
Карбонилирование соединения, такого как 6i, где X представляет собой галоген, такой как Br, с использованием подходящего палладиевого катализатора в атмосфере монооксида углерода, с помощью основания и подходящего спирта может давать соединения, такие как сложный эфир карбоновой кислоты 6ii, который в свою очередь, может быть гидролизован, например, в присутствии гидроксида лития в смеси водный THF/спирт, до карбоновых кислот, таких как 6iii. Карбоновые кислоты, такие как 6iii, могут быть превращены в амиды, такие как 6iv, например, путем обработки амином, основанием и агентом сочетания, таким как HATU. Оксазольные соединения, такие как 6v, могут быть образованы путем замыкания кольца амида 6iv в подходящих условиях реакции, таких как TFAA и аминное основание. Гидролиз нитрила может быть осуществлен с использованием, например, перекиси водорода и карбоната калия в DMSO, с получением карбоксамидов, таких как 6vi.
Схема 7

Сульфонамидные соединения по изобретению могут быть получены традиционными способами. Например, такие соединения, как 7i, могут быть нитрованы с использованием, например, азотной кислоты в уксусной кислоте, с получением продуктов, таких как 7ii, которые могут быть обработаны хлорирующими реагентами, такими как оксихлорид фосфора, с получением хлоридов, таких как 7iii. Хлориды, такие как 7iii, могут быть обработаны спиртом в присутствии основания, такого как карбонат цезия, с получением соединений, таких как 7v. Восстановление нитрогруппы соединений, таких как 7v, может быть осуществлено, например, с помощью цинка и хлорида аммония, с получением таких аминов, как 7vi. Превращение группировки циано в карбоксамид, как в 7vii, может быть осуществлено, например, с помощью перекиси водорода и карбоната калия. Соединения, такие как 7vii, могут быть превращены в сульфонамиды, такие как 7viii, путем реакции с сульфонилхлоридами и подходящим основанием, таким как пиридин. Стадия гидрирования нитрила может быть осуществлена перед или после сульфонилирования с получением такого соединения, как 7viii.
Схема 8

На Схеме 8 показана последовательность получения соединений, где Z представляет собой N. Восстановительное аминирование альдегида, такого как 8i, глицинатным сложным эфиром дает аминное производное 8ii, которое может быть сульфонилировано, например, арилсульфонилхлоридом, таким как n-толуолсульфонилхлорид, в присутствии основания, такого как пиридин, с получением группировок, таких как 8iii. Гидролиз сложного эфира, например с использованием гидроксида лития в смеси водный THF/спирт, дает карбоновые кислоты, такие как 8iv, которые могут быть превращены в хлорангидриды, такие как 8v, с использованием реагентов, таких как тионилхлорид. Ацилирование Фриделя-Крафтса соединений, таких как 8v, может быть осуществлено с использованием кислот Льюиса, таких как трихлорид алюминия, с получением продуктов, таких как 8vi. Обработка соединений, таких как 8vi, основанием, таким как карбонатные или бикарбонатные соли в спирте, таком как этанол, при температуре флегмообразования обеспечивает превращение в фенольные соединения, такие как 8vii, которые могут быть превращены в циано-производные, такие как 8viii, например под действия цианида меди или цинка и палладиевого катализатора в растворителе, таком как DMF. Реакция Мицунобу или реакция О-алкилирования может быть осуществлена, как описано в Схеме 2, с получением простого эфирного соединения, такого как 8ix, и обработка нитрила основной перекисью водорода, как описано в Схеме 1, может давать такие соединения, как 8х.
Схема 9
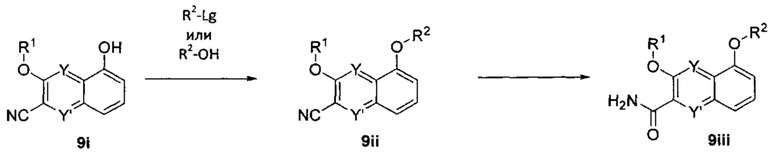
На Схеме 9 описано превращение соединения 9i (например нафтола, если Y=Y'=СН) в простой эфир, такой как 9ii. Например, алкилирование 9i может быть осуществлено с использованием соединения, такого как R2-Lg (например, в котором уходящая группа Lg представляет собой MsO или другие сульфонатные сложные эфиры) в присутствии основания, такого как карбонат цезия, в подходящем растворителе, таком как THF или диметилформамид. Альтернативно, эта реакция может быть осуществлена путем обработки соединения, такого как 9i, спиртом R2-OH в присутствии трифенилфосфина и азодикарбоксилатного сложного эфира ("реакция Мицунобу") в подходящем растворителе, таком как THF. Затем нитрил 9ii может быть превращен в амидное соединение 9iii, как описано ранее, с помощью основания и перекиси водорода в DMSO.
Схема 10

Спиртовое соединение, которое может быть использовано, например, в качестве R2OH (как в Схеме 1) или превращено в R2OR12 (как в Схеме 2), может быть получено с использованием последовательности, изложенной в Схеме 10. Сложный эфир, такой как 10i (R представляет собой Et) (Organic Letters, 2014, 16, 4352.), может быть превращен в производное нитрометана 10ii в присутствии подходящего основания, такого как DBU, и нитрометана. Нитроалкановое производное может быть алкилировано параформальдегидом и основанием, таким как фторид калия, в продукт 10iii. Нитро-группа в 10iii может быть восстановлена до соответствующего амина с использованием подходящего восстановителя, такого как никель Ренея и газообразный водород, в спиртовом растворителе, таком как этанол. Этот неочищенный раствор может быть нагрет и циклизован в указанное лактамное соединение 10iv. Образование аминаля с кеталями, такими как ацетондиметилкеталь (R1=R2=R3=Me), с кислотными катализаторами, такими как n-толуолсульфоновая кислота, может давать соединение 10v. С этого соединения 10v может быт удалена защита, или оно может быть дополнительно функционализировано, например путем депротонирования с помощью сильного основания, такого как лития диизопропиламид или лития гексаметилдисилазид, в растворителе, таком как THF, и затем обработано стандартными фторирующими агентами, такими как N-фторбензолсульфонимид (NFSI), с получением смеси диастереомеров соединения, такого как 10vi. Водная кислота, например, смесь TFA в воде и MeCN, может быть использована для снятия защиты с аминаля с получением смеси диастереомерных спиртовых соединений 10vii, которые могут быть использованы как таковые в различных получениях.
Схема 11

На Схеме 11 изображен способ получения макроциклических соединений по изобретению. Енолят, полученный в результате воздействия подходящего основания, такого как LDC, на защищенный лактам, такой как 11i (например, где R представляет собой Me) взаимодействует с показанным дихлоридом, с получением смеси цис- и транс-замещенных хлорэтилметиловым простым эфиром лактамов, которые после разделения диастереомеров могут давать 11ii, в котором нововведенная группировка находится в syn-положении с заместителем в положении 5 лактама. Удаление кеталя в водных кислотных условиях, таких как трифторуксусная кислота в походящей среде, такой как водный ацетонитрил, дает соединение 11iii. Другие связывающие группы могут быть включены аналогичными способами с получением макроциклических предшественников, родственных 11iii. Соединение 11iv может быть получено путем деалкилирования соединения, где R1 представляет собой iPr, например с помощью трихлорида алюминия, и алкилировано подходящей защитной группой путем О-алкилирования реагентом защитной группы, таким как SEMCl, в присутствии подходящего основания, такого как DIEA, с получением 11iv, где R1 представляет собой SEM. Соединение, такое как 11iv, может подвергаться реакции SNAr со спиртом, таким как 11iii, с получением соединения, такого как 11v. Защитная группа соединения 11v может быть удалена в кислых условиях, например, в случае группы SEM с помощью HCl в МеОН. Внутримолекулярная циклизация может быть индуцирована в разбавленном растворе с использованием основного катализа, например с помощью трет-бутилата калия в присутствии NaI, с получением соединения 11vii. Превращение цианида 11vii в амид 11viii может быть осуществлено, например, с помощью перекиси водорода в DMSO с помощью карбоната калия.
Схема 12

Лактамы, несущие конденсированный циклопропан, например такие соединения, как 12х, могут быть получены, как описано в Схеме 12. Гомоаллиламин 12i может быть защищен подходящей защитной группой, такой как РМВ, например, посредствомвзаимодействия с 4-метоксибензальдегидом в присутствии восстановителя, обычно цианоборгидрида натрия, в протонном растворителе, обычно этаноле, с получением вторичных аминов, таких как 12ii, путем восстановительного аминирования. Соединения, такие как 12ii, могут быть N-ацилированы, например путем обработки основанием и хлорангидридом общей формулы ClC(O)CO2R, где R представляет собой алкильную группу, такую как метил, с получением амидного соединения 12iii. Типичные основания включают, без ограничения им, бикарбонат натрия. Соединения, такие как 12iii, могут быть восстановлены до первичного спирта 12iv путем обработки восстановителем, таким как боргидрид натрия, в протонном растворителе, таком как метанол. Такие соединения, как 12iv, могут быть защищены, как триалкилсилильный простой эфир, например TBS простой эфир, путем обработки основанием и соответствующим триалкилсилилхлоридом, таким как TBSCl. Типичные основания включают, без ограничения им, имидазол. Полученное соединение 12v может претерпевать циклизацию Кулинковича-де Майере с получением бициклического соединения 12vi путем обработки алкилатом титана и реагентом Гриньяра. Типичные алкилаты титана включают, без ограничения им, изопропилат титана, и типичные реагенты Гриньяра включают, без ограничения им, циклопентилмагния бромид. Общее удаление защиты может быть достигнуто путем обработки 12vi с помощью АСЕ-Cl в хлорированном растворителе, обычно 1,2-дихлорметане, с последующим нагреванием промежуточного вещества в метаноле с получением HCl соли соединения 12vii. Для этого превращения температура может варьироваться от комнатной температуры вплоть до температуры флегмообразования в метаноле. Соединение 12vii может быть защищено по азоту, например как трет-бутил-карбамат, путем обработки ди-трет-бутил-дикарбонатом, основанием и 4-диметиламинопиридином. Типичные основания включают, без ограничения ими, третичные амины, такие как триэтиламин. In situ добавление TBSCl и основания, обычно имидазола, может приводить к полностью защищенному аминоспиртовому соединению 12viii. Соединение 12viii может быть окислено до лактама 12ix посредством обработки оксидом металла, обычно гидратом диоксида рутения и перйодатом натрия в двухфазной среде, такой как равнообъемная смесь этилацетата и воды, для осуществления этого окисления. Появились альтернативные способы получения лактамов, таких как 12ix, и они могут применимыми к таким синтезам (например DOI: 10.1002/anie.201505916). Обработка лактама 12ix источником фторидного аниона в эфирном растворителе, обычно THF, может приводить к соединению 12х. Типичные источники фторидного аниона включают, без ограничения им, тетрабутиламмония фторид. (Было показано, что происходит миграция защитной группы в релевантных примерах: см. Org. Lett., 2001, 3 (3), рр 433-435.) Соединения 12х могут быть превращены в 12xii путем реакции SNAr с активированной гетероциклической группировкой, такой как хлорид, представленный 12xi (например, где X и/или Z представляет собой N, и где R1 может представлять собой любой алкильный заместитель; типичные заместители R1 включают, без ограничения ими, метил и изо-пропил) с избытком основания при низкой температуре в полярном растворителе. Типичные основания и растворители включают, без ограничения ими, KHMDS в DMF, соответственно. Температура может варьироваться от -78°С до комнатной температуры и реакцию обычно осуществляют при -10°С. Превращение соединения 12xii в соединение 12xiii может быть достигнуто путем обработки основанием, пероксидом в полярном растворителе. Типичные основания, пероксиды и растворители включают, без ограничения ими, карбонат калия, перекись водорода и DMSO, соответственно. Энантиомеры соединения 12xiii могут быть разделены при помощи хиральной хроматографии.
Схема 13

На Схеме 13 представлен способ получения 1,6-нафтиридиновых производных по изобретению. Производное никотиновой кислоты, такое как 13i, может быть превращено в соответствующий хлорангидрид специалистами в данной области. Типичные условия включают, без ограничения ими, использование оксалилхлорида в присутствии DMF. Затем хлорангидридное промежуточное соединение может взаимодействовать с 1-(аминоокси)-2,2-диметилпропан-1-она трифлатом в присутствии основания с получением таких соединений, как 13ii. Типичные основания включают, без ограничения им, пиридин. Пиридиновые соединения, такие как 13ii, могут быть окислены до соответствующих N-оксидных производных 13iii. Типичные условия окисления включают, без ограничения ими, использование каталитического количества метил(триоксо)рения в присутствии водного раствора перекиси водорода в гетерогенной системе растворителей. Rh-катализируемая активация С-Н в присутствии основания и алкена в протонном растворителе может приводить к таким соединениям, как 13v (J. Am. Chem. Soc. 2013, 135, 14492). Типичные основания и алкены включают, без ограничения ими, ацетат натрия и норборнадиен 13iv. При нагревании может происходить ретрореакция Дильса-Альдера с получением таких соединений, как 13vi. Последние соединения могут быть цианилированы, например путем обработки диметилкарбаминхлоридом в присутствии источника цианида, обычно триметилсиланкарбонитрила, с получением таких соединений, как 13vii. Хлорирование, например, путем обработки фосфорилхлоридом при высокой температуре, обычно между 70°С и 110°С, может давать разработанные 1,6-нафтиридины, представленные 13viii. Превращение цианогруппы 13viii в карбоксамид, как в 13ix, может быть достигнуто путем обработки карбонатом калия, перекисью водорода и DMSO, как описано для других соединений в данной заявке. Реакция SNAr спиртов R2-OH с активированными гетероциклами, такими как 13ix, может быть осуществлена в присутствии избытка основания в полярном растворителе при нагревании с получением таких соединений, как 13х. Типичные основания и растворители включают, без ограничения ими, KHMDS и DMF, соответственно.
Экспериментальные процедуры и рабочие примеры
Ниже проиллюстрован синтез различных соединений по настоящему изобретению. Дополнительные соединения, входящие в объем данного изобретения, могут быть получены с использованием способов, проиллюстрированных в этих Примерах, либо отдельно, либо в комбинации с методиками, общеизвестными в данной области техники.
Следует понимать, что промежуточные соединения по изобретению, изображенные выше, не ограничены конкретным изображенным энантиомером, а также включают все стереоизомеры и их смеси. Также следует понимать, что соединения Формулы Ia могут включать промежуточные соединения соединений Формулы Ia.
Экспериментальные процедуры
Обычно эксперименты проводили в инертной атмосфере (азот или аргон), особенно в случаях, когда использовали реагенты или промежуточные соединения, чувствительные к кислороду или влажности. Имеющиеся в продаже растворители и реагенты обычно использовали без дополнительной очистки, включая безводные растворители, где это необходимо (обычно продукты Sure-Seal™ от Aldrich Chemical Company, Milwaukee, Wisconsin). Обычно продукты сушили в вакууме, после чего использовали в следующих реакциях или передавали на биологическое тестирование. Масс-спектрометрические данные регистрировали на оборудования жидкостной хроматографии-масс-спектрометрии (LCMS) с химической ионизацией при атмосферном давлении (APCI) или газовой хроматографии-масс-спектрометрии (GCMS). Химические сдвиги для данных ядерного магнитного резонанса (ЯМР) выражены в миллионных долях (м.д., δ) относительно остаточных пиков от использованных дейтерированных растворителей.
Для синтезов процедуры со ссылками в других Примерах или Способах условия реакции (длительность реакции и температура) могут варьироваться. Обычно за реакциями следовала тонкослойная хроматография и/или жидкостная хроматография-масс-спектрометрия, и реакционные смеси подвергали обработке, если это было необходимо. Специалисту в данной области понятно, что способы очистки в экспериментах могут варьироваться: обычно сорбенты, растворители и соотношения растворителей, используемых для элюентов/градиентов, выбирали, чтобы обеспечить подходящие значения Rf или времени удерживания. Также специалисту в данной области понятно, что очистки посредством HPLC (высокоэффективная жидкостная хроматография) могут быть осуществлены различными способами, включающими использование нормальных неподвижных фаз, обращенных неподвижных фаз, хиральных неподвижных фаз и сверхкритических элюентов. Выбор подходящих условий для хроматографических и HPLC очисток будет определен специалистом в данной области.
В следующих Подготовительных примерах описано получение некоторых промежуточных соединений, используемых в следующих ниже Способах и Примерах. Следующие ниже Подготовительные примеры, Способы и Примеры предназначены для иллюстрации конкретных воплощений изобретения и их получений, и не предназначены каким-либо образом ограничивать заявку, включая формулу изобретения. Если не указано иное, все реагенты были приобретены у коммерческих поставщиков.
В неограничивающих Примерах и Подготовительных примерах, которые изложены ниже в описании, и в вышеуказанных Схемах могут быть использованы следующие сокращения, определения и аналитические процедуры:
Сокращения
АСЕ-Cl: 1-хлорэтилхлорформиат
Boc: трет-бутоксикарбонил
СО: монооксид углерода
DBU: 1,8-диазабицикло[5.4.0]ундец-7-ен
DCE: дихлорэтан
DCM: дихлорметан
DIEA: диизопропилэтиламин
DMAP: 4-диметиламинопиридин
DMF: диметилформамид
DMSO: диметилсульфоксид
EDCI: 1-этил-3-(3-диметиламинопропил)карбодиимид
EtOAc: этилацетат
EtOH: этиловый спирт
FA: муравьиная кислота
ч.: час
HATU: O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат
HCl: соляная кислота
HNO3: азотная кислота
Н2О:вода
Н2О2: перекись водорода
НОАс: уксусная кислота
НОВТ: гидроксибензотриазол
H2SO4: серная кислота
K2CO3: карбонат калия
KHMDS: калия бис(триметилсилил)амид
LiOH.Н2О: лития гидроксида моногидрат
РМВ: пара-метоксибензил
MeCN: ацетонитрил
МеОН: метанол
MgSO4: сульфат магния
мин: минуты
MS: масс-спектрометрия
Na: натрий
Na2S2O3: гидросульфит натрия
Na2SO4: сульфат натрия
NH4Cl: хлорид аммония
NaHCO3: бикарбонат натрия
NaHMDS: бис(триметилсилил)амид натрия
H-BuLi: н-бутиллитий
NBS: N-бромсукцинимид
Pd(PPh3)4: тетракис(трифенилфосфин)палладий(0)
PdCl2(dppf): [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий(2),
POCl3: оксихлорид фосфора
SNAr: нуклеофильное ароматическое замещение
TBAF: тетрабутиламмония фторид
TBA-HSO4 тетрабутиламмония гидросульфат
TBS: трет-бутилсилил
TBSCl: трет-бутилдиметилсилилхлорид
TEA: триэтиламин
TFA: трифторуксусная кислота
TFAA: трифторуксусный ангидрид
THF: тетрагидрофуран
TLC: тонкослойная хроматография
Zn: цинк.
Спектры 1H-ядерного магнитного резонанса (ЯМР) во всех случаях соответствовали предложенным структурам. Характеристические химические сдвиги (δ) приведены в м.д. в сторону слабого поля от тетраметилсилана (для 1Н-ЯМР) с использованием традиционных сокращений для обозначения основных пиков: например s, синглет; d, дублет; t, триплет; q, квартет; m, мультиплет; br, уширенный. Следующие сокращения были использованы для часто встречающихся растворителей: CDCl3, дейтерохлороформ; d6-DMSO, дейтеродиметилсульфоксид; и CD3OD, дейтерометанол.
Масс-спектры, MS (m/z), регистрировали с использованием либо ионизации электрораспылением (ESI), либо химической ионизации при атмосферном давлении (APCI). Когда это релевантно и если не указано иное, данные m/z указаны для изотопов 19F, 35Cl, 79Br и 127I.
Обозначение стереохимии энантиомеров было основано на устойчивой картине SAR, наблюдаемой для этой серии ингибиторов IRAK4, и допущений в свете стереохимии, установленной в предыдущих сериях, как подробно описано в находящейся одновременно на рассмотрении заявке на патент US 14/678114, поданной Pfizer Inc 3 апреля 2015 года, и в находящейся одновременно на рассмотрении заявке на патент US 62/204521, поданной 13 августа 2015 года.
ПРИМЕРЫ
Пример 1
8-{[(2S,3S,4S)-3-Этил-4-фтор-5-оксопирролидин-2-ил]метокси}-2-метоксихинолин-3-карбоксамид
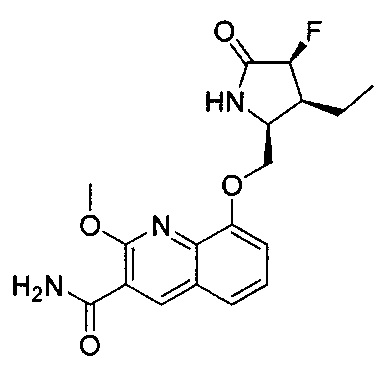
Стадия 1: Получение 3-гидрокси-2-нитробензальдегида 
К 3-гидроксибензальдегиду (5,00 г, 40,9 ммоль) в безводном DCM (100 мл) при комнатной температуре добавляли изопропилнитрат (10,8 г, 102 ммоль), затем добавляли TBA-HSO4 (139 мг, 0,409 ммоль). По каплям добавляли серную кислоту (5 мл). Смесь перемешивали при 15°С в течение 30 мин. Смесь промывали рассолом, и органический слой собирали и сушили над безводным MgSO4, фильтровали и концентрировали. Остаток очищали посредством флэш-хроматографии с использованием 0-99% EtOAc в петролейном эфире с получением указанного в заголовке соединения в виде твердого вещества (3,1 г, 45%-ный выход). 1Н-ЯМР (400 МГц, CDCl3) δ 10.43 (s, 1H), 10.32 (s, 1H), 7.64-7.81 (m, 1H), 7.36-7.43 (m, 1H), 7.30-7.35 (m, 1Н). HPLC: Ultimate ХВ-С18, 3 мкм, 3,0×50 мм, SN:111201514. Подвижная фаза: от 1% MeCN в воде (0,1% TFA) до 5% MeCN в воде (0,1% TFA) в течение 1 мин, затем от 5% MeCN в воде (0,1% TFA) до 100% MeCN (0,1% TFA) в течение 5 мин, выдерживание при 100% MeCN (0,1% TFA) в течение 2 мин и затем снова до 1% MeCN в воде (0,1% TFA) в 8,01 мин, и выдерживание 2 мин. Скорость потока: 1,2 мл/мин. Время удерживания 3,19 мин.
Стадия 2: Получение диметил-2-(3-гидрокси-2-нитробензилиден)малоната
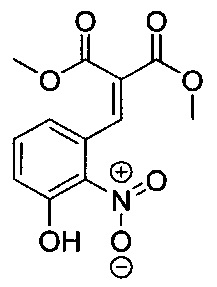
К 3-гидрокси-2-нитробензальдегиду (200 мг, 1.20 ммоль) в МеОН (5 мл) добавляли пиперидин (118 мкл, 1,20 ммоль), затем добавляли диметилмалонат (190 мг, 1,20 ммоль) и НОАс (87,9 мкл, 1,20 ммоль). Полученную коричневую смесь перемешивали при 80°С в течение 20 ч. Смесь концентрировали досуха. Остаток разбавляли EtOAc (100 мл), промывали 0,1 н. HCl, затем добавляли рассол и органический слой сушили над безводным MgSO4, фильтровали и концентрировали. Неочищенный продукт очищали посредством флэш-хроматографии на силикагеле с использованием 0-40% EtOAc в петролейном эфире с получением указанного в заголовке соединения в виде желтого твердого вещества (150 мг, 45%-ный выход). 1Н-ЯМР (400 МГц, CDCl3) δ 10.79 (s, 1Н), 8.16 (s, 1H), 7.52 (t, 1H), 7.22 (d, 1H), 6.86 (d, 1H), 3.90 (s, 3H), 3.62 (s, 3H). HPLC: Ultimate XB-C18, 3 мкм, 3,0×50 мм, SN:111201514. Подвижная фаза: от 1,0% MeCN в воде (0,1% TFA) до 5% MeCN в воде (0,1% TFA) в 1 мин, затем от 5% MeCN в воде (0,1% TFA) до 100% MeCN (0,1% TFA) в 5 мин, выдерживание при 100% MeCN (0,1% TFA) в течение 2 мин, затем обратно до 1% MeCN в воде (0,1% TFA) в 8,01 мин и выдерживание 2 мин. Скорость потока: 1,2 мл/мин. Время удерживания 3,92 мин.
Стадия 3: Получение метил-8-гидрокси-2-оксо-1,2-дигидрохинолин-3-карбоксилата
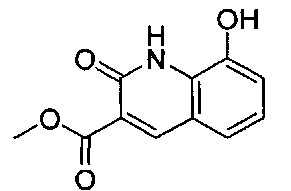
К раствору диметил-2-(3-гидрокси-2-нитробензилиден)малоната (5,0 г, 18 ммоль) в МеОН (240 мл) добавляли Na2S2O4 (12,4 г, 71,1 ммоль). Прозрачный раствор перемешивали в течение 5 ч при 80°С. Смесь фильтровали и фильтраты концентрировали при пониженном давлении. Остаток объединяли с другой партией, полученной с использованием диметил-[(Е)-2-(3-гидрокси-2-нитрофенил)этенил]пропандиоата (3,0 г, 11 ммоль) в МеОН (240 мл) и Na2S2O4 (7,43 г, 42,7 ммоль). Объединенные партии очищали посредством флэш-хроматографии с использованием 0-10% МеОН в DCM с получением указанного в заголовке соединения в виде желтого твердого вещества (2,5 г, 40%). 1Н-ЯМР (400 МГц, МЕТАНОЛ-d4) δ 8.62 (s, 1Н), 7.26 (d, 1H), 7.06-7.16 (m, 2Н), 3.90 (s, 3Н). MS m/z 220 [М+Н]+.
Стадия 4: Получение метил-8-(бензилокси)-2-оксо-1,2-дигидрохинолин-3-карбоксилата
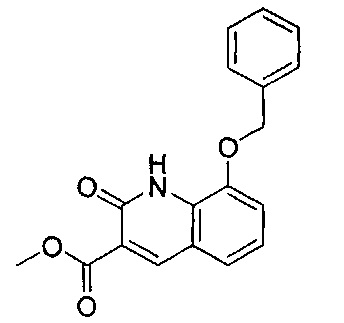
К смеси метил-8-гидрокси-2-оксо-1,2-дигидрохинолин-3-карбоксилата (2000 мг, 9,12 ммоль) в DMF (3,0 мл) добавляли DBU (1390 мг, 9,12 ммоль). Смесь перемешивали в течение 5 мин, после чего добавляли N-бензилбромид (1560 мг, 9,12 ммоль) и смесь нагревали до 70°С в течение 16 ч. Добавляли N-бензилбромид (700 мг, 4 ммоль) и смесь нагревали в течение 4 ч. Смесь охлаждали до температуры окружающей среды. Смесь распределяли между рассолом и EtOAc. Твердые вещества собирали при помощи вакуум-фильтрации. Водную фазу экстрагировали EtOAc. Объединенные органические экстракты промывали рассолом, сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Полученный остаток объединяли с твердым фильтратом от вышеуказанной процедуры, и растирали с 75% EtOAc в гексанах, и фильтровали, и сушили с получением указанного в заголовке соединения в виде не совсем белого твердого вещества (1,15 г, 41%-ный выход). 1Н-ЯМР (400 МГц, DMSO-d6) δ 11.16 (br s, 1H), 8.50 (s, 1H), 7.60 (d, 2Н), 7.36-7.44 (m, 3Н), 7.27-7.36 (m, 2Н), 7.14 (t, 1Н), 5.32 (s, 2Н), 3.82 (s, 3Н). MS m/z 310 [М+Н]+.
Стадия 5: Получение метил-8-гидрокси-2-метоксихинолин-3-карбоксилата

В кругло донную колбу, содержащую метил-8-(бензилокси)-2-оксо-1,2-дигидрохинолин-3-карбоксилат (1000 мг, 3,23 ммоль), добавляли POCl3 (8,0 мл) и DMF (3 капли). Смесь нагревали до 95°С в течение 2 ч и затем концентрировали при пониженном давлении. Толуол (3 мл) добавляли и удаляли при пониженном давлении. К этой смеси добавляли полученный заранее и хранимый в атмосфере азота раствор натрия (850 мг натрия в керосине, 37 ммоль, промывали гексанами для удаления керосина) в МеОН (20 мл). Смесь нагревали до 65°С в течение ночи. Смесь охлаждали до комнатной температуры и распределяли между EtOAc и 1 н. HCl. Слои разделяли и водную фазу три раза экстрагировали EtOAc. Объединенные органические слои сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Остаток очищали посредством флэш-хроматографии на силикагеле с использованием 0-20% EtOAc в гексанах на протяжении 5 объемов колонки, поддерживая 20% EtOAc в течение 4 объемов колонки, затем 20-60% EtOAc в гексанах на протяжении 2 объемов колонки, с получением смеси указанного в заголовке соединения и метил-8-(бензилокси)-2-метоксихинолин-3-карбоксилата (0,512 г). Эту смесь использовали далее без дополнительной очистки. 1Н-ЯМР (400 МГц, CDCl3) δ 8.66 (s, 1H), 8.63 (s, 1H), 7.58 (d, 2Н), 7.37-7.46 (m, 3Н), 7.29-7.36 (m, 3Н), 7.21 (d, 1Н), 5.38 (s, 2Н), 4.23 (s, 3Н), 4.19 (s, 2Н), 3.98 (s, 4Н). Waters Acquity HSS Т3, 2,1×50 мм, С18, 1,7 мкм; Температура колонки 60°С, 0,1%-ная муравьиная кислота в воде (об./об.); Подвижная фаза В: 0,1%-ная муравьиная кислота в MeCN (об./об.) Поток-1,25 мл/мин Исходные условия: А-95%:В-5%; выдерживание в исходных условиях в 0,0-0,1 мин; линейный подъем до А-5%:В-95% в 0,1-1,0 мин; выдерживание при А-5%:В-95% в 1,0-1,1 мин; возвращение к исходным условиям в 1,1-1,5 мин. Время удерживания 0,81 мин. MS m/z 234 [М+Н]+.
Стадия 6: Получение 8-гидрокси-2-метоксихинолин-3-карбоксамида

К смеси метил-8-гидрокси-2-метоксихинолин-3-карбоксилата (463,2 мг, 1,433 ммоль) в сосуде под давлением добавляли 7 н. аммиак в МеОН (2000 мг, 100 ммоль, 20 мл). Сосуд герметично закрывали и смесь нагревали до 70°С в течение ночи. Твердые вещества собирали вакуумной фильтрацией и сушили. Фильтраты концентрировали при пониженном давлении и очищали посредством флэш-хроматографии с использованием 0-100% EtOAc в гексанах в качестве элюента с получением смеси указанного в заголовке соединения и 8-(бензилокси)-2-метоксихинолин-3-карбоксамида (164 мг, 22%-ный выход). Эту смесь использовали далее без дополнительной очистки. 1Н-ЯМР (400 МГц, CDCl3) δ 8.99 (s, 1H), 8.94 (s, 1Н), 7.85 (br s, 1Н), 7.72 (br s, 1Н), 7.50 (d, 1Н), 7.43 (d, 1Н), 7.22-7.38 (m, 6Н), 7.10-7.17 (m, 1Н), 5.81 (br s, 2Н), 5.30 (s, 1Н), 4.21 (s, 2H), 4.18 (s, 3Н). Waters Acquity HSS T3, 2,1×50 мм, С18, 1,7 мкм; Температура колонки 60°С, 0,1%-ная муравьиная кислота в воде (об./об.); Подвижная фаза В: 0,1%-ная муравьиная кислота в MeCN (об./об.) Поток-1,25 мл/мин. Исходные условия: А-95%:В-5%; вьщерживание в исходных условиях при 0,0-0,1 мин; Линейный подъем до А-5%:В-95% в течение 0,1-1,0 мин; выдерживание при А-5%:В-95% в 1,0-1,1 мин; возврат к исходным условиям 1,1-1,5 мин. Время удерживания 0,74 мин. MS m/z 219 [М+Н]+.
Стадия 7: Получение 8-гидрокси-2-метоксихинолин-3-карбонитрила

Колбу, содержащую 8-гидрокси-2-метоксихинолин-3-карбоксамид (164 мг, 0,752 ммоль), закупоривали резиновой пробкой, ненадолго помещали в вакуум и затем продували азотом. Добавляли 1,4-диоксан (2 мл) и пиридин (0,49 мл, 6,01 ммоль). Смесь перемешивали при температуре окружающей среды в течение 10 мин и затем по каплям добавляли TFAA (631 мг, 3,01 ммоль) с получением слабо экзотермической реакции. Смесь перемешивали при комнатной температуре в течение 3 ч. Смесь распределяли между рассолом и EtOAc. Слои разделяли и водную фазу экстрагировали EtOAc. Объединенные органические экстракты промывали рассолом, сушили над Na2SO4 и концентрировали при пониженном давлении с получением смеси неочищенного указанного в заголовке соединения и 8-(бензилокси)-2-метоксихинолин-3-карбонитрила (169,7 мг, >100%-ный выход). Эту смесь использовали далее без дополнительной очистки. 1H-ЯМР (400 МГц, CDCl3) δ 8.48 (s, 1Н), 8.43 (s, 1H), 7.58 (d, 2H), 7.38-7.49 (m, 4H), 7.31-7.38 (m, 3H), 5.39 (s, 1H), 4.25 (s, 2H), 4.23 (s, 3H). Waters Acquity HSS T3, 2,1×50 мм, C18, 1,7 мкм; Температура колонки 60°C, 0,1%-ная муравьиная кислота в воде (об./об.); Подвижная фаза В: 0,1%-ная муравьиная кислота в ацетонитриле (об./об.) Поток-1,25 мл/мин. Исходные условия: А-95%:В-5%; выдерживание в исходных условиях в 0,0-0,1 мин; Линейный подъем до А-5%:В-95% в течение 0,1-1,0 мин; выдерживание при А-5%:В-95% в 1,0-1,1 мин; возврат к исходным условиям 1,1-1,5 мин. Время удерживания 0,89 мин. MS m/z 201 [М+Н]+.
Стадия 8: Получение 8-{[(2S,3S,4S)-3-Этил-4-фтор-5-оксопирролидин-2-ил]метокси}-2-метоксихинолин-3-карбонитрила

К смеси (3S,4S,5S)-4-этил-3-фтор-5-(гидроксиметил)пирролидин-2-она (167 мг, 0,834 ммоль) в DCM (2,0 мл) добавляли DIEA и метансульфонилхлорид (197 мг, 1,71 ммоль). Смесь помещали в атмосферу азота и перемешивали при температуре окружающей среды в течение 2 ч. Смесь упаривали, позволяя DCM выпариваться в токе азота. К остатку добавляли раствор 8-гидрокси-2-метоксихинолин-3-карбонитрила (269 мг, 1,67 ммоль) в DMF (3,0 мл), затем добавляли K2CO3 (346 мг, 2,50 ммоль). Смесь нагревали до 50°С в течение ночи. Добавляли K2CO3 (200 мг, 1,45 ммоль) и смесь нагревали при 50°С в течение ночи. Реакция не прошла неполностью, и потому получали дополнительное количество мезилата для завершения реакции. В круглодоную колбу добавляли (3S,4S,5S)-4-этил-3-фтор-5-(гидроксиметил)пирролидин-2-он (269 мг, 1,67 ммоль) в DCM и смесь охлаждали до 0°С. Добавляли DIEA и метансульфонилхлорид (191 мг, 1,67 ммоль). Смесь перемешивали при 0°С в течение 2 ч, затем в колбу пропускали поток азота для выпаривания DCM. Остаток растворяли в DMF и добавляли в нагретую реакционную смесь, указанную выше, с дополнительным количеством K2CO3 (346 мг, 2,50 ммоль). Смесь нагревали при 50°С в течение ночи до завершения согласно LCMS-анализу. Смесь распределяли между рассолом и EtOAc. Слои разделяли и водную фазу экстрагировали EtOAc. Объединенные экстракты EtOAc четыре раза промывали рассолом, сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Остаток очищали посредством флэш-хроматографии на силикагеле с использованием 0-100% EtOAc в гексанах в качестве элюента, с получением указанного в заголовке соединения в виде не совсем белого твердого вещества (36 мг, 12%-ный выход). 1Н-ЯМР (400 МГц, CDCl3) δ 8.44 (s, 1H), 7.39-7.51 (m, 2Н), 7.28 (dd, 1H), 6.86 (br s, 1H), 4.95 (d, 0.5 Н), 4.82 (d, 0.5Н), 4.40 (d, 1Н), 4.17-4.27 (m, 5H), 2.46-2.67 (m, 1H), 1.57-1.87 (m, 2H), 1.15 (t, 3H). MS rn/z 344 [M+H]+.
Стадия 9: Получение 8-{[(2S,3S,4S)-3-этил-4-фтор-5-оксопирролидин-2-ил]метокси}-2-метоксихинолин-3-карбоксамида
К смеси 8-{[(2S,3S,4S)-3-этил-4-фтор-5-оксопирролидин-2-ил]метокси}-2-метоксихинолин-3-карбонитрила (36 мг, 0,10 ммоль) в DMSO добавляли K2CO3 (72 мг, 0,52 ммоль). Добавляли 30% перекись водорода (83 мг, 0,73 ммоль). Смесь перемешивали при температуре окружающей среды в течение 4,5 ч. Смесь распределяли между рассолом и EtOAc. Слои разделяли и водную фазу экстрагировали EtOAc. Объединенные органические экстракты промывали рассолом пять раз, сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Остаток очищали посредством флэш-хроматографии с использованием 0-5% МеОН в DCM в качестве элюента с получением указанного в заголовке соединения в виде не совсем белого твердого вещества (11 мг, 30%). 1Н-ЯМР (500 МГц, CDCl3) δ 9.00 (s, 1H), 7.88 (br s, 1H), 7.54 (d, 1H), 7.36 (t, 1H), 7.22 (d, 1H), 7.08 (br s, 1H), 6.06 (br s, 1H), 4.92 (d, 0.5H), 4.81 (d, 0.5H), 4.37 (dd, 1H), 4.26 (s, 3H), 4.15-4.24 (m, 2H), 2.42-2.62 (m, 1H), 1.54-1.81 (m, 2H), 1.12 (t, 3H). MS m/z 362 [M+H]+.
Пример 2
4-(1,3-Оксазол-2-ил)-1-{[(2S)-5-оксопирролидин-2-ил]метокси}-7-(пропан-2-илокси)изохинолин-6-карбоксамид

Стадия 1: Получение 1-гидрокси-7-(пропан-2-илокси)изохинолин-6-карбонитрила

К 1-хлор-7-(пропан-2-илокси)изохинолин-6-карбонитрилу (500 мг, 2,03 ммоль) в герметизируемой пробирке добавляли 1,4-диоксан (6,7 мл), затем добавляли концентрированную HCl (3,3 мл) и Н2О (10 мл). Смесь изменялась от прозрачного желтого раствора до вязкой суспензии, и добавление было экзотермическим. Пробирку герметично закрывали и нагревали при 120°С в течение 3 ч. Суспензию разбавляли Н2О и твердые вещества собирали посредством фильтрации и промывали Н2О с получением указанного в заголовке соединения в виде желтого твердого вещества (410 мг, 88,6%). 1Н-ЯМР (400 МГц, DMSO-d6) δ 11.51 (br s, 1H), 8.22 (s, 1H), 7.78 (s, 1H), 7.16 (dd, 1H), 6.56 (d, 1H), 4.90 (spt, 1H), 1.37 (d, 6H). MS m/z 229 [M+H]+.
Стадия 2: Получение 4-бром-1-гидрокси-7-(пропан-2-илокси)изохинолин-6-карбонитрила

Суспензию 1-гидрокси-7-(пропан-2-илокси)изохинолин-6-карбонитрила (7,69 г, 34 ммоль) в MeCN (673 мл) по каплям обрабатывали NBS (7,26 г, 41 ммоль) в течение периода 5 мин и реакционную смесь перемешивали при 15°С в течение 16 ч. Реакционную смесь фильтровали и твердые вещества промывали MeCN и сушили в вакууме с получением указанного в заголовке соединения в виде бледно-зеленого твердого вещества (2,7 г, 26%). 1Н-ЯМР (400 МГц, DMSO-d6) δ 11.82 (d, 1Н), 8.06 (s, 1H), 7.82 (s, 1H), 7.52 (d, 1H), 4.94 (td, 1H), 1.37 (d, 6H). MS m/z 307 [M+H]+.
Стадия 3: Получение 4-бром-1-хлор-7-(пропан-2-илокси)изохинолин-6-карбонитрила

Суспензию 4-бром-1-гидрокси-7-(пропан-2-илокси)изохинолин-6-карбонитрила (5800 мг, 18,9 ммоль) в POCl3 (180 мл) нагревали до температуры флегмообразования в течение 1,5 часов. Затем смесь охлаждали до комнатной температуры и избыток POCl3 удаляли при пониженном давлении. Остаток выливали на лед и гасили путем добавления K2CO3. Затем водный раствор разбавляли DCM и слои разделяли. Водную фазу экстрагировали DCM и объединенную органическую фазу сушили над Na2SO4, фильтровали и концентрировали с получением указанного в заголовке соединения в виде не совсем белого твердого вещества (5,8 г, 94%-ный выход). 1H-ЯМР (400 МГц, CDCl3) δ 8.50 (s, 1H), 8.43 (s, 1H), 7.66 (s, 1H), 4.91 (td, 1Н), 1.54 (d, 6Н). MS m/z 326 [М+Н]+.
Стадия 4: Получение 4-бром-1-{[(2S)-5-оксопирролидин-2-ил]метокси}-7-(пропан-2-илокси)изохинолин-6-карбонитрила
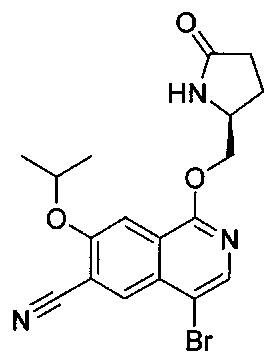
К раствору 4-бром-1-хлор-7-(пропан-2-илокси)изохинолин-6-карбонитрила (2,5 г, 7,68 ммоль) и (S)-5-(гидроксиметил)пирролидин-2-она (1,06 г, 9,21 ммоль) в THF (80 мл) при -15°С добавляли 1 н. NaHMDS (19,2 мл, 19,2 ммоль). Реакционную смесь перемешивали при -15°С в течение 3 ч, затем нагревали до 25°С и перемешивали в течение 16 ч. Смесь гасили насыщенным NH4Cl и смесь экстрагировали EtOAc. Объединенные органические экстракты промывали рассолом, сушили (Na2SO4), фильтровали и концентрировали. Остаток очищали посредством флэш-хроматографии с использованием градиента от 0/100 до 7/93 MeOH/DCM с получением указанного в заголовке соединения в виде желтого твердого вещества (1,24 г, 40%-ный выход). 1Н-ЯМР (400 МГц, CDCl3) δ 8.30-8.45 (m, 1H), 8.07 (s, 1Н), 7.56 (s, 1H), 6.47 (br s, 1H), 4.76-4.94 (m, 1H), 4.63 (dd, 1H), 4.29-4.43 (m, 1H), 4.22 (br s, 1H), 2.29-2.56 (m, 3H), 1.87-2.13 (m, 1H), 1.34-1.56 (m, 6H). MS m/z 404 [M+H]+.
Стадия 5: Получение 2-(трибутилстаннанил)-1,3-оксазола 
Раствор оксазола (1,00 г, 14,5 ммоль) в THF (25 мл) при -78°С обрабатывали н-BuLi (5,79 мл, 14,5 ммоль, 2,5 М бутиллития в гексане). После перемешивания в течение 30 мин добавляли трибутилолова хлорид (3,93 мл, 14,5 ммоль) и раствор оставляли нагреваться до комнатной температуры. Через 1 ч смесь концентрировали при пониженном давлении. Полученный остаток обрабатывали гексаном (50 мл), и полученный осадок отделяли посредством фильтрации через Filtercel. Фильтраты концентрировали при пониженном давлении с получением указанного в заголовке соединения в виде масла (4 г, 80%, 50%-ная чистота согласно ЯМР). Это вещество использовали без дополнительной очистки.
Стадия 6: Получение 4-(1,3-оксазол-2-ил)-1-{[(2S)-5-оксопирролидин-2-ил]метокси}-7-(пропан-2-илокси)изохинолин-6-карбонитрила

Раствор 4-бром-1-{[(2S)-5-оксопирролидин-2-ил]метокси}-7-(пропан-2-илокси)изохинолин-6-карбонитрила (404 мг, 1,0 ммоль), 2-(трибутилстаннанил)-1,3-оксазола (1,43 г, 2,0 ммоль) и Pd(PPh3)2Cl2 (35 мг, 0,05 ммоль) в MeCN (50 мл) перемешивали при 80°С в течение 4 ч. Растворитель упаривали и остаток очищали посредством флэш-хроматографии (MeOH/DCM от 1/100 до 3,8/96,2) с получением указанного в заголовке соединения в виде желтого твердого вещества (0,12 г, 31%-ный выход). 1Н-ЯМР (400 МГц, CDCl3) δ 9.68 (s, 1Н), 8.63 (s, 1H), 7.81 (s, 1H), 7.60 (s, 1H), 7.37 (s, 1H), 6.46 (br s, 1H), 4.78-4.97 (m, 1H), 4.72 (dd, 1H), 4.47 (dd, 1H), 4.25 (br s, 1H), 2.37-2.55 (m, 3H), 2.03 (t, 1H), 1.50 (d, 6H). MS m/z 393 [M+H]+.
Стадия 7: Получение 4-(1,3-оксазол-2-ил)-1-{[(2S)-5-оксопирролидин-2-ил]метокси}-7-(пропан-2-илокси)изохинолин-6-карбоксамида.
Смесь 4-(1,3-оксазол-2-ил)-1-{[(2S)-5-оксопирролидин-2-ил]метокси}-7-(пропан-2-илокси)изохинолин-6-карбонитрила (60 мг, 0,15 ммоль) и K2CO3 (106 мг, 0,76 ммоль) в DMSO (4 мл) перемешивали при 25°С в течение 5 мин. Добавляли Н2О2 (121 мг, 1,07 ммоль) и смесь перемешивали при 25°С в течение 2 ч. Смесь гасили диметилсульфидом (95 мг, 1,53 ммоль) и перемешивали при 25°С в течение 30 мин. Смесь фильтровали и промывали DCM и EtOAc. Фильтрат концентрировали и остаток очищали посредством препаративной HPLC (Колонка: Ultimate ХВ-С18, 3 мкм, 3,0×50 мм. Время удерживания: 3,46 мин. Подвижная фаза: от 1% MeCN в воде (0,05% TFA) до 100% MeCN в воде (0,05% TFA). Скорость потока: 1,2 мл/мин. Длина волны: 220 нм) с получением неочищенного продукта (30 мг, 90%-ная чистота). Неочищенный продукт перемешивали в МеОН (1,5 мл) в течение 2 мин и фильтровали с получением указанного в заголовке соединения в виде белого твердого вещества (20 мг, 32%-ный выход). 1Н-ЯМР (400 МГц, DMSO-d6) δ 9.43 (s, 1H), 8.57 (s, 1H), 8.32 (s, 1H), 8.17 (s, 1H), 7.78 (br s, 2H), 7.75 (s, 1H), 7.53 (s, 1H), 4.96 (td, 1H), 4.56 (dd, 1H), 4.40 (dd, 1H), 4.06 (br s, 1H), 2.17-2.38 (m, 3H), 1.94 (d, 1H), 1.41 (dd, 6H). MS m/z 433 [M+Na]+.
Пример 3
4-(4-Метил-1Н-имидазол-2-ил)-1-{[2S)-5-оксопирролидин-2-ил]метокси}-7-(пропан-2-илокси)изохинолин-6-карбоксамид

Стадия 1: Получение трет-бутил-2-бром-4-метил-1Н-имидазол-1-карбоксилата
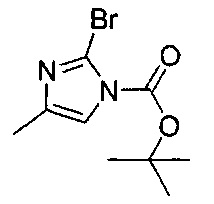
К перемешиваемому раствору 2-бром-4-метил-1Н-имидазола (300 мг, 1,86 ммоль) и DMAP (341 мг, 2,79 ммоль) в сухом THF (12 мл) добавляли ВОС2О (0,43 мл, 1,86 ммоль) и перемешивали при комнатной температуре в течение 16 ч. Смесь упаривали досуха и разбавляли EtOAc. Органическую фазу промывали насыщенным раствором NaHCO3 и затем рассолом, сушили над Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очищали посредством флэш-хроматографии с использованием 8-15% EtOAc в гексане с получением указанного в заголовке соединения в виде не совсем белого твердого вещества (190 мг, 59%-ный выход). 1Н-ЯМР (400 МГц, CDCl3) δ 7.14 (s, 1Н), 2.33 (s, 3Н), 1.63 (s, 9Н). MS m/z 261 [М+Н]+.
Стадия 2: Получение 1-{[(2S)-5-оксопирролидин-2-ил]метокси}-7-(пропан-2-илокси)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)изохинолин-6-карбонитрила

К перемешиваемому раствору 4-бром-1-{[(2S)-5-оксопирролидин-2-ил]метокси}-7-(пропан-2-илокси)изохинолин-6-карбоксамида (4 г, 9,9 ммоль) в 1,4-дикосане (100 мл) добавляли свежевысушенный ацетат калия (2,91 г, 29,7 ммоль) и бис(пинаколатодибор) (3,52 г, 13,9 ммоль). Смесь дегазировали аргоном в течение 20 мин, после чего добавляли тетракистрифенилфосфинпалладий(0) (572 мг, 0,49 ммоль) и смесь нагревали до 100°С в течение 16 ч. Смесь охлаждали до комнатной температуры и фильтровали через целит. Фильтрат упаривали досуха и очищали посредством флэш-хроматографии (10-20%-ный ацетон в DCM) с получением 3 г боронатного сложного эфира, который также содержал трифенилфосфиноксид. Его очищали путем растирания с 20%-ным EtOAc в гексане (3 раза) с получением указанного в заголовке соединения в виде светло-коричневого твердого вещества (2,3 г, 52%-ный выход). 1Н-ЯМР (400 МГц, DMSO-d6) δ 8.70-8.89 (m, 1Н), 8.30 (s, 1H), 8.17 (s, 1H), 7.78 (s, 1H), 5.00 (td, 1H), 4.54 (dd, 1H), 4.32 (dd, 1H), 4.03 (br s, 1H), 2.10-2.37 (m, 3H), 1.81-1.95 (m, 1H), 1.25-1.47 (m, 18H). MS m/z 452 [M+H]+.
Стадия 3: Получение 4-(4-метил-1Н-имидазол-2-ил)-1-{[(2S)-5-оксопирролидин-2-ил]метокси}-7-(пропан-2-илокси)изохинолин-6-карбонитрила

1-{[(2S)-5-Оксопирролидин-2-ил]метокси}-7-(пропан-2-илокси)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)изохинолин-6-карбонитрил (150 мг, 0,33 ммоль), трет-бутил-2-бром-4-метил-1Н-имидазол-1-карбоксилата (104,17 мг, 0,39 ммоль) и K2CO3 (114,74 мг, 0,83 ммоль) растворяли в смеси диоксан/Н2О (3 мл смеси 4:1) и дегазировали аргоном в течение 10 мин. Добавляли Pd(dppf)Cl2⋅DCM (13,57 мг, 0,017 ммоль) и смесь снова дегазировали в течение 5 мин. Смесь нагревали до 100°С в течение 16 ч. Смесь разбавляли EtOAc и промывали водой, рассолом, сушили над Na2SC4 и концентрировали. Неочищенное вещество очищали посредством препаративной TLC (5% MeOH/DCM) с получением указанного в заголовке соединения в виде твердого вещества (42 мг, 31%-ный выход). 1Н-ЯМР (400 МГц, DMSO-d6) δ 12.7 (d, 1H), 9.78 (d, 1H), 8.32 (s, 1H), 8.17 (s, 1H), 7.78 (s, 1H), 7.05 (s, 0.5H),6.88 (s, 0.5H), 5.02 (td, 1H), 4.53 (dd, 1H), 4.32 (dd, 1H), 4.05 (br s, 1H), 2.20-2.32 (m, 6H),1.91 (m, 1H), 1.40-1.44 (m, 6H). MS m/z 406 [M+H]+.
Стадия 4: Получение 4-(4-метил-1Н-имидазол-2-ил)-1-{[(2S)-5-оксопирролидин-2-ил]метокси}-7-(пропан-2-илокси)изохинолин-6-карбоксамида
Перемешиваемый раствор 4-(4-метил-1Н-имидазол-2-ил)-1-{[(2S)-5-оксопирролидин-2-ил]метокси}-7-(пропан-2-илокси)изохинолин-6-карбонитрила (60 мг, 0,15 ммоль) в DMSO (1,0 мл) обрабатывали тонкоизмельченным K2CO3 (81,8 мг, 0,59 ммоль) и полученную смесь нагревали до 45°С. К этому раствору медленно по каплям добавляли 30%-ный Н2О2 (0,19 мл, 1,93 ммоль). Через 45 мин реакционную смесь разбавляли МеОН, фильтровали и промывали МеОН. Фильтрат упаривали при пониженном давлении. Неочищенное вещество очищали посредством препаративной HPLC с получением указанного в заголовке соединения в виде желтого твердого вещества (8 мг, 13%-ный выход). 1Н-ЯМР (400 МГц, DMSO-d6) δ 12.28 (br s, 1H), 9.19-9.42 (m, 2H), 8.15 (d, 2H), 7.72 (br s, 2H), 7.68 (s, 1H), 6.81-7.03 (m, 1H), 4.93 (td, 1H), 4.51 (dd, 1H), 4.36 (dd, 1H), 4.05 (br s, 1H), 2.18-2.31 (m, 5H), 1.89-1.99 (m, 1H), 1.40 (dd, 6H). MS m/z 424 [M+H]+.
Пример 4
4-(1-Метил-1Н-пиразол-3-ил)-1-{[(2S)-5-оксопирролидин-2-ил]метокси}-7-(пропан-2-илокси)изохинолин-6-карбоксамид
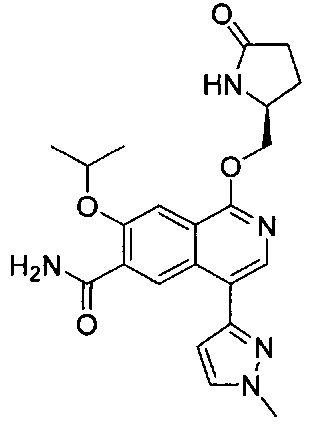
Стадия 1: Получение 4-(1-метил-1Н-пиразол-3-ил)-1-{[(2S)-5-оксопирролидин-2-ил]метокси}-7-(пропан-2-илокси)изохинолин-6-карбонитрила

1-{[(2S)-5-Оксопирролидин-2-ил]метокси}-7-(пропан-2-илокси)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)изохинолин-6-карбонитрил (100 мг, 0,22 ммоль), 3-йод-1-метил-1Н-пиразол (55,34 мг, 0,26 ммоль) и K2CO3 (76,5 мг, 0,54 ммоль) растворяли в смеси диоксан/Н2O (2 мл, 4:1) и дегазировали аргоном в течение 10 мин. Добавляли Pd(dppf)Cl2⋅DCM (9,04 мг, 0,012 ммоль) и реакционную смесь снова дегазировали в течение 5 мин. Смесь нагревали до 100°С в течение 16 ч. Смесь разбавляли EtOAc и промывали водой, рассолом, сушили над Na2SO4 и концентрировали. Неочищенное вещество очищали посредством колоночной хроматографии на силикагеле (4% MeOH/DCM) с получением указанного в заголовке соединения в виде коричневого твердого вещества (90 мг, ~ 100%-ный выход), которое было загрязнено примесью. Это вещество использовали на следующей стадии без дополнительной очистки. MS m/z 406 [М+Н]+.
Стадия 2: Получение 4-(1-метил-1Н-пиразол-3-ил)-1-{[(2S)-5-оксопирролидин-2-ил]метокси}-7-(пропан-2-илокси)изохинолин-6-карбоксамида
Перемешиваемый раствор 4-(1-метил-1Н-пиразол-3-ил)-1-{[(2S)-5-оксопирролидин-2-ил]метокси}-7-(пропан-2-илокси)изохинолин-6-карбонитрила (90,0 мг, 0,22 ммоль) в DMSO (1,0 мл) обрабатывали тонкоизмельченным K2CO3 (122,66 мг, 0,88 ммоль) и смесь нагревали до 45°С. К реакционной смеси медленно по каплям добавляли 30%-ный раствор Н2О2 (0,29 мл, 2,88 ммоль). Через 45 мин реакционную смесь разбавляли МеОН, фильтровали и промывали МеОН. Фильтрат упаривали при пониженном давлении. Неочищенное вещество очищали посредством препаративной HPLC с получением указанного в заголовке соединения в виде не совсем белого твердого вещества (12 мг, 13%-ный выход). 1Н-ЯМР (400 МГц, DMSO-d6) δ 8.88 (s, 1Н), 8.11 (s, 1H), 8.07 (s, 1H), 7.85 (s, 1H), 7.64-7.76 (m, 3H), 6.61 (s, 1H), 4.94 (td, 1H), 4.50 (dd, 1H), 4.36 (dd, 1H), 4.05 (br s, 1H), 3.96 (s, 3H), 2.14-2.36 (m, 3H), 1.88-1.97 (m, 1H), 1.41 (t, 6H). MS m/z 424 [M+H]+.
Пример 5
4-(1-метил-1Н-пиразол-4-ил)-1-([(2S)-5-оксопирролидин-2-ил]метокси}-7-(пропан-2-илокси)изохинолин-6-карбоксамид

Стадия 1: Получение 4-(1-метил-1Н-пиразол-4-ил)-1-{[(2S)-5-оксопирролидин-2-ил]метокси}-7-(пропан-2-илокси)изохинолин-6-карбонитрила

1-{[(2S)-5-Оксопирролидин-2-ил]метокси}-7-(пропан-2-илокси)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)изохинолин-6-карбонитрил (100 мг, 0,22 ммоль), 4-бром-1-метил-1Н-пиразол (42,57 мг, 0,26 ммоль) и K2CO3 (76,49 мг, 0,55 ммоль) растворяли в смеси диоксан/Н2О (2 мл, 4:1) и смесь дегазировали аргоном в течение 10 мин. Добавляли Pd(dppf)Cl2⋅DCM (9,05 мг, 0,01 ммоль) и смеси дегазировали в течение 5 мин. Реакционную смесь нагревали до 100°С в течение 16 ч. Смесь охлаждали до комнатной температуры, разбавляли EtOAc и промывали водой, рассолом, сушили над Na2SO4 и концентрировали. Неочищенное вещество очищали посредством колоночной хроматографии на силикагеле (0-4% MeOH/DCM) с получением указанного в заголовке соединения в виде не совсем белого твердого вещества (75 мг, ~ 84%-ный выход), которое содержало примесь и было использовано на следующей стадии без дополнительной очистки. MS m/z 406 [М+Н]+.
Стадия 2: Получение 4-(1-метил-1Н-пиразол-4-ил)-1-{[(2S)-5-оксопирролидин-2-ил]метокси}-7-(пропан-2-илокси)изохинолин-6-карбоксамида
Перемешиваемый раствор 4-(1-метил-1Н-пиразол-4-ил)-1-{[(2S)-5-оксопирролидин-2-ил]метокси}-7-(пропан-2-илокси)изохинолин-6-карбонитрила (75,0 мг, 0,18 ммоль) в DMSO (1,0 мл) обрабатывали тонкоизмельченным K2CO3 (102 мг, 0,74 ммоль) и нагревали до 45°С. Этот раствор медленно по каплям обрабатывали 30%-ным раствором Н2О2 (0,24 мл, 2,41 ммоль). Через 45 мин реакционную смесь разбавляли метанолом, и фильтровали, и промывали метанолом. Фильтрат упаривали при пониженном давлении. Неочищенное вещество очищали посредством препаративной HPLC с получением указанного в заголовке соединения в виде не совсем белого твердого вещества (14 мг, 18%-ный выход). 1Н-ЯМР (400 МГц, DMSO-d6) δ 8.30 (s, 1Н), 8.11 (s, 1H), 8.02 (s, 1H), 7.85 (s, 1H), 7.74 (br s, 2H), 7.72 (s, 1H), 7.68 (s, 1H), 4.95 (td, 1H), 4.49 (dd, 1H), 4.34 (dd, 1H), 3.95 (s, 3H), 2.18-2.36 (m, 3H), 1.93 (d, 1H), 1.38-1.44 (m, 6H). MS m/z 424 [M+H]+.
Пример 6
4-(4-Метил-1,3-оксазол-2-ил)-1-{[(2S)-5-оксопирролидин-2-ил]метокси}-7-(пропан-2-илокси)изохинолин-6-карбоксамид

Стадия 1: Получение 4-метил-2-(трибутилстаннанил)-1,3-оксазола

Раствор 4-метилоксазола (1,00 г, 12 ммоль) в THF (30 мл) при -78°С обрабатывали н-BuLi (4,81 мл, 12 ммоль, 2,5 М в гексане). Через 30 мин добавляли трибутилолова хлорид (3,92 г, 12 ммоль) и раствор оставляли нагреваться до комнатной температуры. Перемешивание продолжали в течение еще часа, после чего  часть растворителей выпаривали в вакууме. Полученный остаток собирали в гексане (50 мл) и полученный осадок собирали путем фильтрации. Фильтрат упаривали с получением указанного в заголовке соединения в виде масла (4 г, 89%, 60%-ная чистота согласно ЯМР). Это вещество использовали без дополнительной очистки. 1H-ЯМР (400 МГц, CDCl3) δ 7.53 (s, 1H), 2.21 (s, 3Н), 1.68-1.58 (m, 10Н), 1.36-1.32 (m, 9Н). 1.21-1.71 (m, 8Н), 0.92-0.88 (m, 14Н). ЯМР указывает на присутствие трибутилолова хлорида.
часть растворителей выпаривали в вакууме. Полученный остаток собирали в гексане (50 мл) и полученный осадок собирали путем фильтрации. Фильтрат упаривали с получением указанного в заголовке соединения в виде масла (4 г, 89%, 60%-ная чистота согласно ЯМР). Это вещество использовали без дополнительной очистки. 1H-ЯМР (400 МГц, CDCl3) δ 7.53 (s, 1H), 2.21 (s, 3Н), 1.68-1.58 (m, 10Н), 1.36-1.32 (m, 9Н). 1.21-1.71 (m, 8Н), 0.92-0.88 (m, 14Н). ЯМР указывает на присутствие трибутилолова хлорида.
Стадия 2: Получение 4-(4-метил-1,3-оксазол-2-ил)-1-{[(2S)-5-оксопирролидин-2-ил]метокси}-7-(пропан-2-илокси)изохинолин-6-карбонитрила

Раствор 4-бром-1-{[(2S)-5-оксопирролидин-2-ил]метокси}-7-(пропан-2-илокси)изохинолин-6-карбонитрила (300 мг, 0,742 ммоль), 4-метил-2-(трибутилстаннанил)-1,3-оксазола (1,7 г, 2,7 ммоль) и транс-дихлорбис(трифенилфосфин)палладия(II) (52 мг, 0,10 ммоль) в MeCN (50 мл) перемешивали при 80°С в течение 16 часов. Растворитель выпаривали и остаток очищали посредством флэш-хроматографии на силикагеле (MeOH/DCM от 0/100 до 4/96) с получением указанного в заголовке соединения в виде желтого твердого вещества (140 мг, 46%-ный выход). 1Н-ЯМР (400 МГц, CDCl3) δ 9.67 (s, 1H), 8.57 (s, 1H), 7.61 (s, 1H), 7.50 (s, 1H), 6.77 (br s, 1H), 4.88 (td, 1H), 4.69 (dd, 1H), 4.44 (dd, 1H), 4.24 (br s, 1H), 2.37-2.55 (m, 3H), 2.32 (s, 3H), 1.92-2.13 (m, 1H), 1.44-1.57 (m, 6H). MS m/z 429 [M+Na]+.
Стадия 3: Получение 4-(4-метил-1,3-оксазол-2-ил)-1-{[(2S)-5-оксопирролидин-2-ил]метокси}-7-(пропан-2-илокси)изохинолин-6-карбоксамида
Смесь 4-(4-метил-1,3-оксазол-2-ил)-1-{[(2S)-5-оксопирролидин-2-ил]метокси}-7-(пропан-2-илокси)изохинолин-6-карбонитрила (80 мг, 0,20 ммоль) и K2CO3 (136 мг, 0,98 ммоль) в DMSO (4 мл) перемешивали при 25°С в течение 5 мин. Добавляли Н2О2 (156 мг, 1,38 ммоль). Реакционную смесь перемешивали при 25°С в течение 2 ч. Смесь гасили диметилсульфидом (122 мг, 1,97 ммоль) и перемешивали при 25°С в течение 30 мин. Смесь фильтровали и промывали DCM и EtOAc. Осадок на фильтре суспендировали в МеОН (2 мл) и перемешивали в течение 2 ч. Смесь фильтровали и осадок на фильтре суспендировали в смеси MeOH/DCM (1/10, 5 мл) и перемешивали в течение 5 мин. Смесь фильтровали и фильтрат концентрировали с получением указанного в заголовке соединения в виде не совсем белого твердого вещества (23 мг, 28%). 1Н-ЯМР (400 МГц, DMSO-d6) δ 9.34 (s, 1Н), 8.53 (s, 1H), 8.16 (s, 1H), 8.00 (s, 1H), 7.79 (br s, 1H), 7.74 (s, 2H), 4.86-5.02 (m, 1H), 4.55 (d, 1H), 4.39 (dd, 1H), 4.06 (br s, 1H), 2.13-2.40 (m, 6H), 1.93 (br s, 1H), 1.40 (dd, 6H). MS m/z 425 [M+H]+.
Пример 7
4-(4,5-Диметил-1,3-оксазол-2-ил)-1-{[(2S)-5-оксопирролидин-2-ил]метокси}-7-(пропан-2-илокси)изохинолин-6-карбоксамид

Стадия 1: Получение метил-6-циано-1-{[(2S)-5-оксопирролидин-2-ил]метокси}-7-(пропан-2-илокси)изохинолин-4-карбоксилата
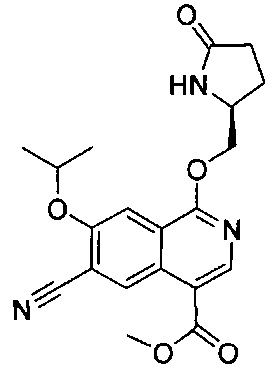
Смесь 4-бром-1-{[(2S)-5-оксопирролидин-2-ил]метокси}-7-(пропан-2-илокси)изохинолин-6-карбонитрила (6,5 г, 16,08 ммоль), TEA (4,88 г, 48,2 ммоль) и Pd(dppf)2Cl2 (1,18 г, 1,61 ммоль) в МеОН (500 мл) перемешивали в атмосфере СО (50 фунтов на кв. дюйм (344,7 кПа)) при 80°С в течение 16 ч. Смесь фильтровали и растворитель выпаривали. Остаток очищали посредством флэш-хроматографии на силикагеле (MeOH/DCM от 0/100 до 5/95) с получением указанного в заголовке соединения в виде желтого твердого вещества (5,3 г, 86%-ный выход). 1Н-ЯМР (400 МГц, CDCl3) δ 9.36 (s, 1Н), 8.70 (s, 1Н), 7.58 (s, 1Н), 6.29 (br s, 1Н), 4.85 (td, 1H), 4.72 (dd, 1H), 4.48 (dd, 1H), 4.23 (br s, 1H), 4.00 (s, 3H), 2.31-2.53 (m, 3H), 1.93-2.15 (m, 1H), 1.49 (d, 6H). MS m/z 406 [M+Na]+.
Стадия 2: Получение 6-циано-1-{[(2S)-5-оксопирролидин-2-ил]метокси}-7-(пропан-2-илокси)изохинолин-4-карбоновой кислоты
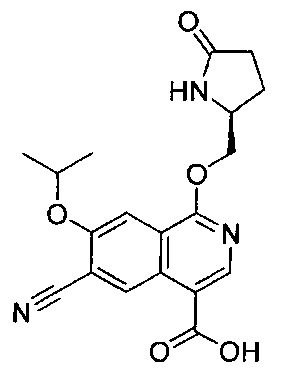
Смесь метил-6-циано-1-{[(2S)-5-оксопирролидин-2-ил]метокси}-7-(пропан-2-илокси)изохинолин-4-карбоксилата (5,2 г, 13.56 ммоль) и LiOH⋅H2O (1,71 г, 40,7 ммоль) в Н2О (20 мл), EtOH (20 мл) и THF (80 мл) перемешивали при 20°С в течение 3 ч. Смесь подкисляли 1 н. HCl до рН 7 и растворитель выпаривали. К остатку добавляли NaHCO3 (2 г) в Н2О (100 мл) и смесь перемешивали в течение 15 мин. Смесь промывали DCM и водную фазу подкисляли 1 н. HCl до рН 6. Смесь фильтровали с получением указанного в заголовке соединения в виде не совсем белого твердого вещества (3,4 г, 68%-ный выход). 1Н-ЯМР (400 МГц, DMSO-d6) δ 9.35 (s, 1H), 8.65 (s, 1H), 8.20 (s, 1H), 7.80 (s, 1H), 4.91-5.14 (m, 1H), 4.52-4.74 (m, 1H), 4.37 (dd, 1H), 3.89-4.18 (m, 1H), 2.12-2.36 (m, 3H), 1.91 (br s, 1H), 1.41 (dd, 6H). MS m/z 370 [M+H]+.
Стадия 3: Получение 6-циано-N-(3-оксобутан-2-ил)-1-{[(2S)-5-оксопирролидин-2-ил]метокси}-7-(пропан-2-илокси)изохинолин-4-карбоксамида
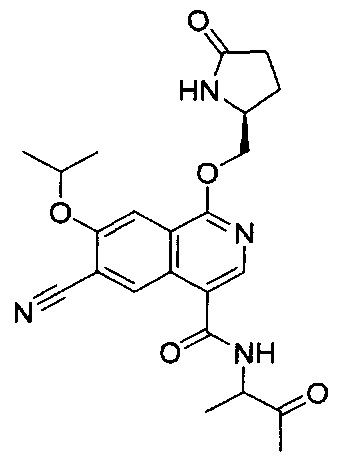
К раствору 6-циано-1-{[(2S)-5-оксопирролидин-2-ил]метокси}-7-(пропан-2-илокси)изохинолин-4-карбоновой кислоты (300 мг, 0,81 ммоль) и DIEA (315 мг, 2,4 ммоль) в DMF (0,5 мл) в DCM (30 мл) добавляли 3-аминобутан-2-он (100 мг, 0,81 ммоль) и HATU (463 мг, 1,2 ммоль). Реакционную смесь перемешивали при 20°С в течение 3 ч. Растворитель выпаривали и остаток очищали посредством флэш-хроматографии на силикагеле (MeOH/DCM от 0/100 до 4/96) с получением указанного в заголовке соединения в виде не совсем белого твердого вещества (200 мг, 56%-ный выход). MS m/z 370 [М+Н]+.
Стадия 4: Получение 4-(4,5-диметил-1,3-оксазол-2-ил)-1-{[(2S)-5-оксопирролидин-2-ил]метокси}-7-(пропан-2-илокси)изохинолин-6-карбонитрила

К смеси 6-циано-N-(3-оксобутан-2-ил)-1-{[(2S)-5-оксопирролидин-2-ил]метокси}-7-(пропан-2-илокси)изохинолин-4-карбоксамида (130 мг, 0,30 ммоль) в 1,2-дихлорэтане (30 мл) при 0°С добавляли DIEA (1 мл) и TFAA (1 мл). Реакционную смесь оставляли нагреваться до 20°С и перемешивали в течение 2 ч. Растворитель выпаривали и остаток очищали посредством флэш-хроматографии на силикагеле (MeOH/DCM от 0/100 до 4/96) с получением указанного в заголовке соединения в виде желтого твердого вещества (110 мг, 88%-ный выход). 1Н-ЯМР (400 МГц, CDCl3) δ 9.65 (s, 1H), 8.54 (s, 1H), 7.59 (s, 1H), 7.04 (br s, 1H), 4.87 (td, 1H), 4.75 (dd, 1H), 4.45 (dd, 1H), 4.21-4.35 (m, 1H), 2.40-2.58 (m, 3H), 2.38 (s, 3H), 2.23 (s, 3H), 1.97-2.09 (m, 1H), 1.50 (dd, 6H). MS m/z 443 [M+Na]+.
Стадия 5: Получение 4-(4,5-диметил-1,3-оксазол-2-ил)-1-{[(2S)-5-оксопирролидин-2-ил]метокси}-7-(пропан-2-илокси)изохинолин-6-карбоксамида
Смесь 4-(4,5-диметил-1,3-оксазол-2-ил)-1-{[(2S)-5-оксопирролидин-2-ил]метокси}-7-(пропан-2-илокси)изохинолин-6-карбонитрила (100 мг, 0,238 ммоль) и K2CO3 (164 мг, 1,19 ммоль) в DMSO (4 мл) перемешивали при 25°С в течение 5 мин. Добавляли Н2О2 (189 мг, 1,66 ммоль) и реакционную смесь перемешивали при 25°С в течение 2 ч. Смесь гасили диметилсульфидом (148 мг, 2,38 ммоль) и перемешивали при 25°С в течение 30 мин. Смесь фильтровали и промывали DCM и EtOAc. Фильтрат концентрировали и остаток очищали посредством препаративной HPLC (Колонка: DIKMA Diamonsil(2) С18 200*20 мм*5 мкм. Подвижная фаза: от 24% MeCN в воде (0,225% FA) до 44% MeCN в воде (0,225% FA). Скорость потока: 30 мл/мин. Длина волны: 220 нм) с получением указанного в заголовке соединения в виде желтого твердого вещества (23 мг, 22%-ный выход). 1Н-ЯМР (400 МГц, DMSO-d6) δ 9.36 (s, 1Н), 8.47 (s, 1H), 8.15 (s, 1H), 7.77 (br s, 1H), 7.72 (s, 1H), 4.90-4.97 (m, 1H), 4.54 (d, 1H), 4.38 (dd, 1H), 4.05 (br s, 1H), 2.36 (s, 3H), 2.19-2.34 (m, 3Н), 2.17 (s, 3Н), 1.94 (d, 1H), 1.40 (dd, 6H). Один NH скрыт. MS m/z 439 [M+H]+.
Пример 8
4-[4-(Гидроксиметил)-1Н-имидазол-2-ил]-1-{[(2S)-5-оксопирролидин-2-ил]метокси}-7-(пропан-2-илокси)изохинолин-6-карбоксамид
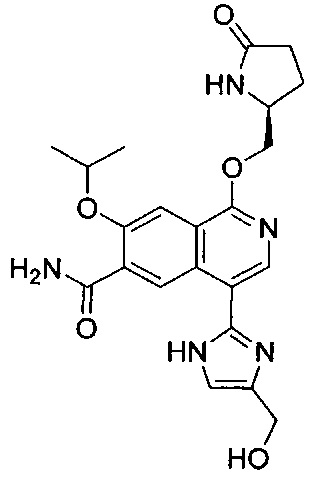
Стадия 1: Получение 2-йод-4-({[три(пропан-2-ил)силил]окси}метил)-1Н-имидазола

К перемешиваемому раствору (2-йод-1Н-имидазол-4-ил)метанола (250 мг, 1,14 ммоль) и имидазола (155 мг, 2,28 ммоль) в DMF (5 мл) добавляли триизопропилсилилхлорид (0,29 мл, 1,37 ммоль). Смесь перемешивали при комнатной температуре в течение 16 ч. Смесь разбавляли EtOAc и промывали водой, рассолом, сушили над Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очищали посредством колоночной хроматографии (10-30% EtOAc в гексане) с получением указанного в заголовке соединения в виде не совсем белого твердого вещества (400 мг, 92%-ный выход). 1Н-ЯМР (400 МГц, DMSO-d6) δ 12.62-12.45 (m, 1H), 7.02-6.75 (m, 1H), 4.64-4.53 (m, 2H), 1.17-1.07 (m, 3H), 1.06-0.95 (m, 18H). MS m/z 382 [M+H]+.
Стадия 2: Получение трет-бутил-2-йод-4-({[три(пропан-2-ил)силил]окси}метил)-1Н-имидазол-1-карбоксилата
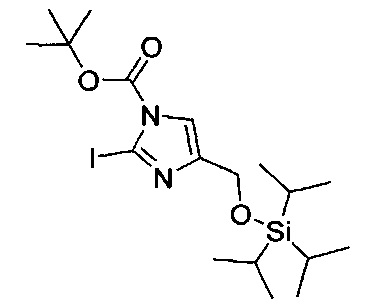
К перемешиваемому раствору 2-йод-4-({[три(пропан-2-ил)силил]окси}метил)-1Н-имидазола (400 мг, 1,05 ммоль) и DMAP (193 мг, 1,58 ммоль) в сухом THF (20 мл) добавляли ВОС2О (0,242 мл, 1,05 ммоль) и смесь перемешивали при комнатной температуре в течение 16 ч. Смесь упаривали досуха и разбавляли EtOAc. Органическую фазу промывали раствором 0,5 н. HCl, насыщенным водным раствором NaHCO3, водой, рассолом, сушили над Na2SO4 и концентрировали с получением указанного в заголовке соединения в виде светло-желтого полутвердого вещества (500 мг, 99%-ный выход). 1Н-ЯМР (400 МГц, DMSO-d6) δ 7.43 (s, 1H), 4.55 (s, 2H), 1.58 (s, 9H), 1.02-0.94 (m, 18H).
Стадия 3: Получение 1-{[(2S)-5-оксопирролидин-2-ил]метокси}-7-(пропан-2-илокси)-4-[4-({[три(пропан-2-ил)силил]окси}метил)-1Н-имидазол-2-ил]изохинолин-6-карбонитрила

1-{[(2S)-5-Оксопирролидин-2-ил]метокси}-7-(пропан-2-илокси)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)изохинолин-6-карбонитрил (200 мг, 0,44 ммоль), трет-бутил-2-йод-4-({[три(пропан-2-ил)силил]окси}метил)-1Н-имидазол-1-карбоксилат (255 мг, 0,53 ммоль) и K2CO3 (153 мг, 1,11 ммоль) растворяли в смеси диоксан/Н2О (3,0 мл, 4:1) и дегазировали аргоном в течение 10 мин. Добавляли Pd(dppf)Cl2⋅DCM (18 мг, 0,02 ммоль) и реакционную смесь дегазировали в течение 5 мин. Реакционную смесь нагревали до 100°С в течение 16 ч. Смесь охлаждали до комнатной температуры и разбавляли EtOAc, промывали водой, рассолом, сушили над Na2SO4 и концентрировали. Неочищенное вещество очищали посредством колоночной хроматографии (2% MeOH/DCM) с получением указанного в заголовке соединения (150 мг, ~ 59%-ный выход). Это вещество также содержало некоторые примеси, наряду с целевым соединением, и его использовали на следующей стадии без дополнительной очистки. MS m/z 578 [М+Н]+.
Стадия 4: Получение 4-[4-(гидроксиметил)-1Н-имидазол-2-ил]-1-{[(2S)-5-оксопирролидин-2-ил]метокси}-7-(пропан-2-илокси)изохинолин-6-карбонитрила

К перемешиваемому раствору 1-{[(2S)-5-оксопирролидин-2-ил]метокси}-7-(пропан-2-илокси)-4-[4-({[три(пропан-2-ил)силил]окси}метил)-1Н-имидазол-2-ил]изохинолин-6-карбонитрила (147 мг, 0,25 ммоль) в THF (2 мл) добавляли TBAF [1М в THF] (0,38 мл, 0,38 ммоль) при 0°С и смесь перемешивали при комнатной температуре в течение 1 ч. Смесь разбавляли EtOAc и промывали водой, рассолом, сушили над Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очищали посредством колоночной хроматографии на силикагеле (5-10% MeOH/DCM) с получением указанного в заголовке соединения в виде коричневого твердого вещества (95 мг, 88%-ный выход). MS m/z 422 [М+Н]+.
Стадия 5: Получение 4-[4-(гидроксиметил)-1Н-имидазол-2-ил]-1-{[(2S)-5-оксопирролидин-2-ил]метокси}-7-(пропан-2-илокси)изохинолин-6-карбоксамида
Перемешиваемый раствор 4-[4-(гидроксиметил)-1Н-имидазол-2-ил]-1-{[(2S)-5-оксопирролидин-2-ил]метокси}-7-(пропан-2-илокси)изохинолин-6-карбонитрила (95 мг, 0,23 ммоль) в DMSO (2 мл) обрабатывали тонкоизмельченным K2CO3 (125 мг, 0,90 ммоль) и нагревали до 45°С. Медленно по каплям добавляли 30%-ный раствор Н2О2 (0,30 мл, 2,93 ммоль). Через 45 мин смесь разбавляли МеОН, фильтровали и промывали МеОН. Фильтрат упаривали при пониженном давлении. Неочищенное вещество очищали посредством препаративной HPLC с получением указанного в заголовке соединения в виде желтого твердого вещества (7 мг, 7%-ный выход). 1Н-ЯМР (400 МГц, МЕТАНОЛ-d4) δ 8.83 (s, 1H), 8.09 (s, 1H), 7.79 (s, 1H), 7.20 (br s, 1H), 4.99 (td, 1H), 4.67 (s, 1H), 4.52-4.66 (m, 2H), 4.24 (br s, 1H), 2.37-2.60 (m, 3H), 2.06-2.18 (m, 1H), 1.50 (t, 6H). Один протон был скрыт пиком растворителя. MS m/z 440 [М+Н]+.
Пример 9
4-(5-Метил-1,3-оксазол-2-ил)-1-{[(2S)-5-оксопирролидин-2-ил]метокси}-7-(пропан-2-илокси)изохинолин-6-карбоксамид

Стадия 1: Получение 6-циано-N-(2-оксопропил)-1-{[(2S)-5-оксопирролидин-2-ил]метокси}-7-(пропан-2-илокси)изохинолин-4-карбоксамида
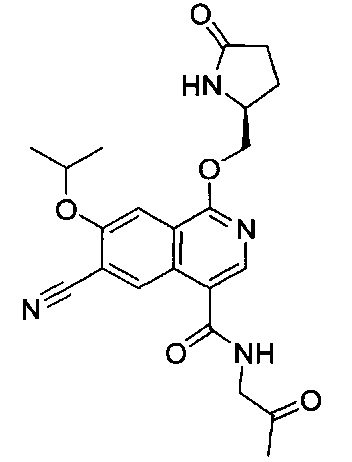
К раствору 6-циано-1-{[(2S)-5-оксопирролидин-2-ил]метокси}-7-(пропан-2-илокси)изохинолин-4-карбоновой кислоты (200 мг, 0,541 ммоль) и DIEA (210 мг, 1,62 ммоль) в DMF (2 мл) и DCM (20 мл) добавляли 1-аминопропан-2-он (59,3 мг, 0,541 ммоль) и HATU (309 мг, 0,812 ммоль). Реакционную смесь перемешивали при 20°С в течение 3 ч. Растворитель выпаривали и остаток очищали посредством флэш-хроматографии на силикагеле (MeOH/DCM от 0/100 до 4/96) с получением указанного в заголовке соединения в виде не совсем белого твердого вещества 10 (150 мг, 65%-ный выход). 1Н-ЯМР (400 МГц, CDCl3) δ 8.81 (s, 1Н), 8.20 (s, 1H), 7.55 (s, 1H), 6.85 (br s, 1H), 6.34 (br s, 1H), 4.84 (td, 1H), 4.68 (dd, 1H), 4.38-4.49 (m, 3H), 4.23 (br s, 1H), 2.37-2.54 (m, 3H), 2.33 (s, 3H), 1.95-2.12 (m, 1H), 1.48 (d, 6H). MS m/z 447 [M+Na]+.
Стадия 2: Получение 4-(5-метил-1,3-оксазол-2-ил)-1-{[(2S)-5-оксопирролидин-2-ил]метокси}-7-(пропан-2-илокси)изохинолин-6-карбонитрила
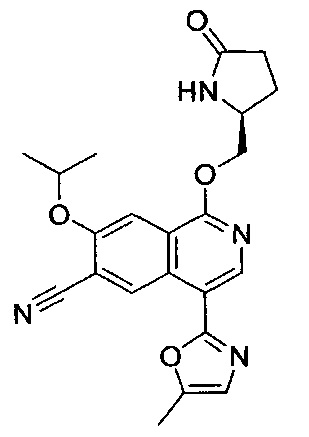
К смеси 6-циано-N-(2-оксопропил)-1-{[(2S)-5-оксопирролидин-2-ил]метокси}-7-(пропан-2-илокси)изохинолин-4-карбоксамида (130 мг, 0,31 ммоль) в 1,2-дихлорэтане (20 мл) при 0°С добавляли TFAA (1 мл) и DIE А (1 мл). Реакционную смесь перемешивали при 0°С в течение 2 ч. Реакционную смесь нагревали до 20°С и перемешивали в течение 18 ч. Добавляли TFAA (1 мл) и DIEA (3 мл) и смесь перемешивали при 20°С в течение 3 ч. Смесь разбавляли DCM (30 мл), промывали насыщенным NaHCO3 (20 мл) и рассолом (20 мл), сушили (Na2SO4), фильтровали и концентрировали. Остаток очищали посредством флэш-хроматографии на силикагеле (MeOH/DCM от 0/100 до 4/96) с получением указанного в заголовке соединения в виде желтого твердого вещества (60 мг, 48%-ный выход). 1Н-ЯМР (400 МГц, CDCl3) δ 9.67 (s, 1Н), 8.56 (s, 1H), 7.58 (s, 1H), 6.97 (s, 1H), 6.40 (br s, 1H), 4.86 (td, 1H), 4.70 (dd, 1H), 4.46 (dd, 1H), 4.25 (br s, 1H), 2.05 (d, 1H), 1.62 (s, 3H), 1.43-1.54 (m, 6H). Некоторые пики были скрыты растворителем. MS m/z 429 [M+Na]+.
Стадия 3: Получение 4-(5-метил-1,3-оксазол-2-ил)-1-{[(2S)-5-оксопирролидин-2-ил]метокси}-7-(пропан-2-илокси)изохинолин-6-карбоксамида
Смесь 4-(5-метил-1,3-оксазол-2-ил)-1-{[(2S)-5-оксопирролидин-2-ил]метокси}-7-(пропан-2-илокси)изохинолин-6-карбонитрила (60 мг, 0,15 ммоль) и K2CO3 (102 мг, 0,74 ммоль) в DMSO (4 мл) перемешивали при 25°С в течение 5 мин. Добавляли Н2О2 (117 мг, 1,03 ммоль) и реакционную смесь перемешивали при 25°С в течение 2 ч. Смесь гасили диметилсульфидом (91,7 мг, 1,48 ммоль) и перемешивали при 25°С в течение 30 мин. Смесь фильтровали и промывали DCM и EtOAc. Фильтрат концентрировали и остаток очищали посредством препаративной HPLC (Колонка: DIKMA Diamonsil(2) С18 200*20 мм*5 мкм. Подвижная фаза: от 20% MeCN в воде (0,225% FA) до 40% MeCN в воде (0,225% FA) Скорость потока: 30 мл/мин. Длина волны: 220 нм) с получением указанного в заголовке соединения в виде желтого твердого вещества (13 мг, 21%-ный выход). 1Н-ЯМР (400 МГц, DMSO-d6) δ 9.44 (s, 1Н), 8.50 (s, 1H), 8.15 (s, 1H), 7.77 (br s, 1H), 7.73 (s, 1H), 7.13 (s, 1H), 4.86-5.04 (m, 1H), 4.50-4.62 (m, 1H), 4.33-4.45 (m, 1H), 4.06 (br s, 1H), 2.43 (s, 3Н), 2.19-2.36 (m, 3Н), 1.94 (br s, 1H), 1.40 (dd, 6H). Протон NH был скрыт. MS m/z 425 [M+H]+.
Пример 10
1-{[(2S,3R)-3-этил-5-оксопирролидин-2-ил]-метокси}-4-[(фенилсульфонил)амино]-7-(пропан-2-илокси)изохинолин-6-карбоксамид

Стадия 1: Получение 4-нитро-1-оксо-7-(пропан-2-илокси)-1,2-дигидроизохинолин-6-карбонитрила
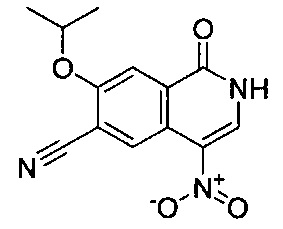
К смеси 1-оксо-7-(пропан-2-илокси)-1,2-дигидроизохинолин-6-карбонитрила (8,3 г, 36,4 ммоль) в АсОН (160 мл) и EtOAc (30 мл) при 0°С добавляли HNO3 (9,17 г, 145 ммоль). Реакционную смесь оставляли нагреваться до комнатной температуры и затем нагревали при 50°С в течение 12 ч. Реакционную смесь вливали в ледяную воду. Смесь фильтровали с получением указанного в заголовке соединения в виде желтого твердого вещества (5,1 г, 51%-ный выход). 1Н-ЯМР (400 МГц, DMSO-d6) δ 11.52 (br s, 1Н), 8.22 (s, 1H), 7.77 (s, 1H), 7.16 (t, 1H), 6.56 (d, 1H), 4.82-5.01 (m, 1H), 1.37 (d, 6H). MS m/z 274 [M+H]+.
Стадия 2: Получение 1-хлор-4-нитро-7-(пропан-2-илокси)изохинолин-6-карбонитрила

К перемешиваемому раствору 4-нитро-1-оксо-7-(пропан-2-илокси)-1,2-дигидроизохинолин-6-карбонитрила (6,2 г, 22,7 ммоль) в POCl3 (50 мл) добавляли TEA (2,3 мг, 22,7 ммоль) и реакционную смесь нагревали до температуры флегмообразования в течение 2 ч. Смесь охлаждали до комнатной температуры и избыток POCl3 выпаривали при пониженном давлении. Остаток гасили водным NaHCO3. Водную фазу экстрагировали DCM. Объединенную органическую фазу промывали насыщенным раствором бикарбоната натрия, водой, затем рассолом, сушили над Na2SO4 и концентрировали. Остаток очищали посредством флэш-хроматографии на силикагеле (петролейный эфир/DCM от 0/100 до 43/57) с получением указанного в заголовке соединения в виде желтого твердого вещества (4,8 г, 73%-ный выход). 1Н-ЯМР (400 МГц, DMSO-d6) δ 12.64 (br s, 1Н), 8.84 (s, 1H), 8.60 (s, 1H), 7.85 (s, 1H), 4.97 (td, 1H), 1.38 (d, 6H). MS m/z 292 [M+H]+.
Стадия 3: Получение 1-{[(2S,3R)-3-этил-5-оксопирролидин-2-ил]метокси}-4-нитро-7-(пропан-2-илокси)изохинолин-6-карбонитрила
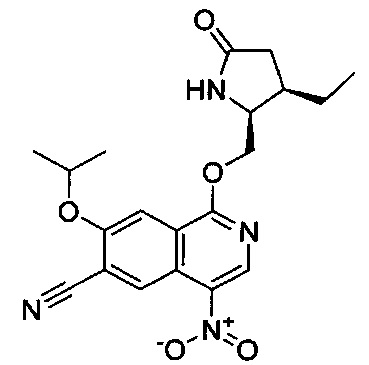
К раствору 1-хлор-4-нитро-7-(пропан-2-илокси)изохинолин-6-карбонитрила (3 г, 10,3 ммоль) и Cs2CO3 (6,7 г, 20,6 ммоль) в 1,4-диоксане (10 мл) добавляли (4R,5S)-4-этил-5-(гидроксиметил)пирролидин-2-он (1,77 г, 12,3 ммоль). Смесь перемешивали при 20°С в течение 16 ч. Реакционную смесь фильтровали через набивку целита и фильтрат выпаривали в вакууме. Остаток очищали посредством хроматографии на силикагеле (от 0% до 30% МеОН в DCM) с получением указанного в заголовке соединения в виде желтого твердого вещества (2,4 г, 59%-ный выход). 1Н-ЯМР (400 МГц, CDCl3) δ 9.12 (s, 1Н), 8.91 (s, 1Н), 7.61 (s, 1H), 6.51 (s, 1H), 4.91 (td, 1H), 4.56-4.78 (m, 2Н), 4.01-4.21 (m, 1Н), 2.60-2.78 (m, 1H), 2.51 (dd, 1H), 2.17 (dd, 1H), 1.62-1.75 (m, 2Н), 1.50 (dd, 6Н), 1.03 (t, 3Н).
Стадия 4: Получение 4-амино-1-{[(2S,3R)-3-этил-5-оксопирролидин-2-ил]метокси}-7-(пропан-2-илокси)изохинолин-6-карбонитрила

К раствору 1-{[(2S,3R)-3-этил-5-оксопирролидин-2-ил]метокси}-4-нитро-7-(пропан-2-илокси)изохинолин-6-карбонитрила (4,1 г, 10,3 ммоль) в THF (51 мл), Н2О (51 мл) и EtOH (25 мл) добавляли Zn (6,73 г, 103 ммоль) и NH4Cl (5,5 г, 103 ммоль). Полученную смесь перемешивали при 25°С в течение 16 ч. Реакционную смесь фильтровали через набивку целита и фильтрат упаривали в вакууме. Остаток очищали посредством хроматографии на силикагеле (от 0% до 12% МеОН в EtOAc) с получением указанного в заголовке соединения в виде желтого твердого вещества (3,4 г, 90%-ный выход). 1Н-ЯМР (400 МГц, CDCl3) δ 8.03 (s, 1H), 7.38 (s, 1H), 7.30 (s, 1Н), 6.93 (s, 1H), 4.71 (td, 1Н), 4.35-4.51 (m, 2Н), 4.01-4.08 (m, 1Н), 2.56-2.69 (m, 1H), 2.42-2.52 (m, 1Н), 2.20 (dd, 1H), 2.05 (s, 1H), 1.44 (dd, 6Н), 0.98 (t, 3Н). MS m/z 369 [М+Н]+.
Стадия 5: Получение 4-амино-1-{[(2S,3R)-3-этил-5-оксопирролидин-2-ил]метокси}-7-(пропан-2-илокси)изохинолин-6-карбоксамида

К раствору 4-амино-1-{[(2S,3R)-3-этил-5-оксопирролидин-2-ил]метокси}-7-(пропан-2-илокси)изохинолин-6-карбонитрила (1,4 г, 3,8 ммоль) и K2CO3 (2,63 г, 19 ммоль) в DMSO (5 мл) добавляли Н2О2 (1,29 г, 38 ммоль). Полученную оранжевую смесь перемешивали при 25°С в течение 16 ч. Добавляли Н2О2 (517 мг, 15,2 ммоль) и полученную смесь перемешивали при 25°С в течение 10 ч. Реакционную смесь вливали в воду и полученные твердые вещества собирали посредством фильтрации и промывали водой. Твердое вещество сушили с получением указанного в заголовке соединения в виде желтого твердого вещества (1,1 г, 75%-ный выход). 1Н-ЯМР (400 МГц, DMSO-d6) δ 8.37 (s, 1H), 7.90 (s, 1H), 7.69 (br s, 2H), 7.45 (s, 1H), 7.25 (s, 1H), 5.23 (s, 2H), 4.83 (td, 1H), 4.17-4.36 (m, 2H), 3.83-3.93 (m, 1H), 3.32 (s, 1H), 2.20-2.31 (m, 1H), 2.06-2.17 (m, 1H), 1.58 (td, 1H), 1.39 (t, 6H), 0.90 (t, 3H). MS m/z 387 [M+H]+.
Стадия 6: Получение 1-{[(2S,3R)-3-этил-5-оксопирролидин-2-ил]метокси}-4-[(фенилсульфонил)амино]-7-(пропан-2-илокси)изохинолин-6-карбоксамида
К раствору 4-амино-1-{[(2S,3R)-3-этил-5-оксопирролидин-2-ил]метокси}-7-(пропан-2-илокси)изохинолин-6-карбоксамида (100 мг, 0,26 ммоль) в пиридине (2 мл) добавляли бензолсульфонилхлорид (55 мг, 0,31 ммоль). Смесь перемешивали при 25°С в течение 5 ч. Добавляли воду (5 мл) и смесь экстрагировали DCM. Объединенную органическую фазу сушили над Na2SO4. Остаток очищали посредством препаративной HPLC (Колонка: Ultimate ХВ-С18 3 мкм, 3,0*50 мм, Время градиента: 11 мин, Подвижная фаза: от 1% MeCN в воде (0,05% TFA) до 100% MeCN в воде (0,05% TFA), Скорость потока: 35 мл/мин, Длина волны: 220 нм) с получением указанного в заголовке соединения в виде белого твердого вещества (78 мг, 57%-ный выход). 1Н-ЯМР (400 МГц, CDCl3) δ 9.30 (br s, 1Н), 8.95 (br s, 1H), 8.17 (br s, 1H), 8.00 (s, 1H), 7.69 (d, 3H), 7.28-7.35 (m, 1H), 7.28-7.39 (m, 2H), 7.18-7.24 (m, 2H), 7.09 (br s, 1H), 4.65-4.78 (m, 1H), 4.56 (d, 1H), 4.33 (d, 1H), 4.05 (d, 1H), 2.60 (d, 1H), 2.46 (dd, 1H), 2.19 (dd, 1H), 1.57 (d, 3H), 1.44 (d, 3H), 1.38 (d, 1H), 0.96 (t, 3H). MS m/z 527 [M+H]+.
Пример 11
1-{[(2S,3R)-3-Этил-5-оксопирролидин-2-ил]метокси}-7-(пропан-2-илокси)-4-[(пиридин-3-илсульфонил)амино)изохинолин-6-карбоксамид
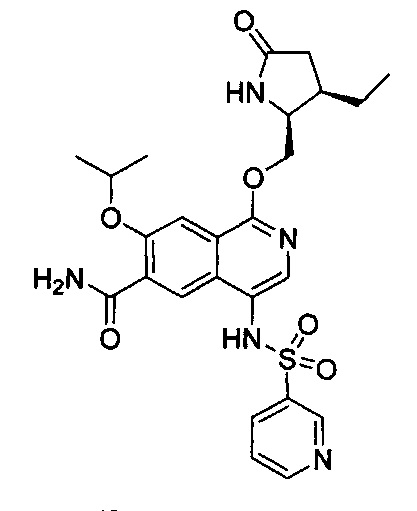
К раствору 4-амино-1-{[(2S,3R)-3-этил-5-оксопирролидин-2-ил]метокси}-7-(пропан-2-илокси)изохинолин-6-карбоксамида (80 мг, 0,21 ммоль) в пиридине (2 мл) добавляли пиридин-3-сульфонилхлорид (53 мг, 0,25 ммоль). Смесь перемешивали при 25°С в течение 5 ч. Добавляли воду (5 мл) и смесь экстрагировали DCM. Объединенный органический растворитель сушили над Na2SO4. Остаток очищали посредством препаративной HPLC (Колонка: Agela durashell С18*21,2 мм*5 мкм, Время градиента: 11 мин, Подвижная фаза: от 30% МеОН в воде (0,225% FA) до 50% МеОН в воде (0,225% FA), Скорость потока: 35 мл/мин, Длина волны: 220 нм) с получением указанного в заголовке соединения в виде белого твердого вещества (67 мг, 61%-ный выход). 1Н-ЯМР (400 МГц, CDCl3) δ 8.83 (br s, 1Н), 8.64 (br s, 1H), 8.51 (d, 1H), 8.26 (br s, 1Н), 7.93-8.13 (m, 3Н), 7.19 (br s, 1H), 7.06 (br s, 1H), 4.71 (br s, 1H), 4.59 1H), 4.33 (br s, 1H), 4.06 (d, 1H), 2.62 (br s, 1H), 2.47 (dd, 1H), 2.19 (dd, 1H), 1.61 (d, 3H), 1.56 (br s, 2H), 1.45 (d, 3H), 1.40 (br s, 1H), 0.97 (t, 3H). MS m/z 528 [M+H]+.
Пример 12
1-{[(2S,3R)-3-Этил-5-оксопирролидин-2-ил]метокси}-4-[(1H-имидазол-4-илсульфонил)амино]-7-(пропан-2-илокси)изохинолин-6-карбоксамид

К раствору 4-амино-1-{[(2S,3R)-3-этил-5-оксопирролидин-2-ил]метокси}-7-(пропан-2-илокси)изохинолин-6-карбоксамида (80 мг, 0,21 ммоль) в пиридине (2 мл) добавляли 1Н-имидазол-4-сульфонилхлорид (138 мг, 0,828 ммоль). Смесь перемешивали при 25°С в течение 5 ч. Добавляли воду (5 мл), смесь экстрагировали DCM. Объединенную органическую фазу сушили над Na2SO4. Остаток очищали посредством препаративной HPLC (Колонка: Agela durashell C18*21,2 мм*5 мкм, Время градиента: 11 мин, Подвижная фаза: от 20% МеОН в воде (0,225% FA) до 40% МеОН в воде (0,225% FA), Скорость потока: 35 мл/мин, Длина волны: 220 нм) с получением указанного в заголовке соединения в виде белого твердого вещества (46 мг, 43%-ный выход). 1Н-ЯМР (400 МГц, DMSO-d6) δ 9.92 (br s, 1Н), 8.35 (s, 1H), 7.96 (s, 1H), 7.84 (s, 1H), 7.73 (br s, 1H), 7.66 (br s, 1H), 7.52 (s, 1H), 7.47 (s, 2H), 4.79-4.88 (m, 1H), 4.38 (d, 2H), 3.90 (br s, 1H), 2.21-2.35 (m, 2H), 2.04-2.15 (m, 1H), 1.51-1.63 (m, 1H), 1.38 (dd, 6H), 0.90 (t, 3Н). Пик был скрыт растворителем. MS m/z 539 [M+Na]+.
Пример 13
1-{[(2S,3R)-3-Этил-5-оксопирролидин-2-ил]метокси}-4-{[(1-метил-1Н-имидазол-4-ил)сульфонил]амино}-7-(пропан-2-илокси)изохинолин-6-карбоксамид

К раствору 4-амино-1-{[(2S,3R)-3-этил-5-оксопирролидин-2-ил]метокси}-7-(пропан-2-илокси)изохинолин-6-карбоксамида (80 мг, 0,21 ммоль) в пиридине (2 мл) добавляли 1-метил-1Н-имидазол-4-сульфонилхлорид (45 мг, 0,25 ммоль). Смесь перемешивали при 25°С в течение 5 ч. Добавляли воду (5 мл) и смесь экстрагировали DCM. Объединенную органическую фазу сушили над Na2SO4. Остаток очищали посредством препаративной HPLC (Колонка: Agela durashell С18*21,2 мм*5 мкм, Время градиента: 11 мин, Подвижная фаза: от 25% МеОН в воде (0,225% FA) до 45% МеОН в воде (0,225% FA), Скорость потока: 35 мл/мин, Длина волны: 220 нм) с получением указанного в заголовке соединения в виде белого твердого вещества (81 мг, 74%-ный выход). 1Н-ЯМР (400 МГц, CDCl3) δ 9.27 (br s, 1Н), 8.78 (s, 1H), 7.99 (br s, 1H), 7.89 (s, 1H), 7.46 (br s, 1H), 7.34 (br s, 1H), 7.25 (br s, 1H), 7.20 (br s, 1H), 7.12 (br s, 1H), 4.76 (br s, 1H), 4.52-4.62 (m, 1H), 4.41 (d, 1H), 4.08 (br s, 1H), 3.57 (s, 3H), 2.61 (br s, 1H), 2.47 (dd, 1H), 2.20 (dd, 1H), 1.59 (d, 2H), 1.51 (d, 3H), 1.44 (d, 3H), 0.98 (t, 3H). MS m/z 531 [M+H]+.
Пример 14
4-{[(1,2-Диметил-1H-имидазол-4-ил)сульфонил]амино}-1-{[(2S,3R)-3-этил-5-оксопирролидин-2-ил]метокси}-7-(пропан-2-илокси)изохинолин-6-карбоксамид

К раствору 4-амино-1-{[(2S,3R)-3-этил-5-оксопирролидин-2-ил]метокси}-7-(пропан-2-илокси)изохинолин-6-карбоксамида (80 мг, 0,21 ммоль) в пиридине (2 мл) добавляли 1,2-диметил-1Н-имидазол-4-сульфонилхлорид (60 мг, 0,31 ммоль). Смесь перемешивали при 25°С в течение 5 ч. Добавляли воду (5 мл) и смесь экстрагировали DCM. Объединенную органическую фазу сушили над Na2SO4. Остаток очищали посредством препаративной HPLC (Колонка: Agela durashell С18*21,2 мм*5 мкм, Время градиента: 11 мин, Подвижная фаза: от 20% МеОН в воде (0,225% FA) до 40% МеОН в воде (0,225% FA), Скорость потока: 35 мл/мин, Длина волны: 220 нм) с получением указанного в заголовке соединения в виде белого твердого вещества (57 мг, 51%-ный выход). 1Н-ЯМР (400 МГц, DMSO-d6) δ 8.18 (s, 1Н), 8.14 (s, 1H), 7.97 (s, 1H), 7.71 (br s, 1H), 7.62 (br s, 1H), 7.60 (s, 1H), 7.46 (s, 1H), 7.41 (s, 1H), 4.83 (td, 1H), 4.39 (d, 2H), 3.89-3.95 (m, 1H), 3.48 (s, 3H), 2.23-2.34 (m, 5H), 2.03-2.16 (m, 2H), 1.51-1.63 (m, 1H), 1.37 (dd, 6H), 0.91 (t, 3H). MS m/z 545 [M+H]+.
Пример 15
4-Амино-1-{[(2S,3S,4S)-3-этил-4-фтор-5-оксопирролидин-2-ил]метокси}-7-метоксиизохинолин-6-карбоксамид
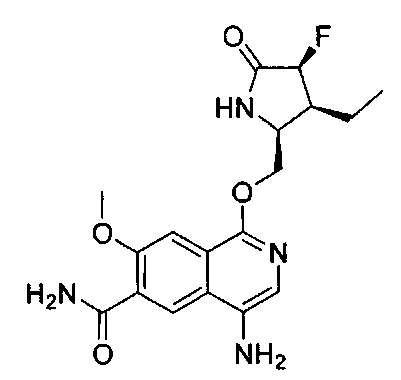
Стадия 1: Получение 1-{[(2S,3S,4S)-3-Этил-4-фтор-5-оксопирролидин-2-ил]метокси}-7-метокси-4-нитроизохинолин-6-карбонитрила

В колбу добавляли 1-хлор-7-метокси-4-нитроизохинолин-6-карбонитрил (0,2 г, 0,76 ммоль), (3S,4S,5S)-4-этил-3-фтор-5-(гидроксиметил)пирролидин-2-он (0,12 г, 0,76 ммоль), карбонат цезия (1,24 г, 3,8 ммоль) и 1,4-диоксан (7,6 мл). Смесь интенсивно перемешивали в течение ночи. Смесь фильтровали через набивку силикагеля и промывали EtOAc. Фильтраты очищали посредством хроматографии на силикагеле с использованием 0-20% MeOH/DCM. Затем остаток очищали на силикагеле, используя 0-100% EtOAc в гептане, с получением указанного в заголовке соединения в виде твердого вещества (135 мг, 46%-ный выход). 'Н-ЯМР (400 МГц, CDC13) δ 9.03-9.20 (m, 1H), 8.92 (s, 1H), 7.84 (s, 1H), 7.41 (br s, 1H), 4.80-5.01 (m, 2H), 4.51 (dd, 1H), 4.16-4.29 (m, 1H), 4.04-4.14 (m, 3H), 2.51-2.75 (m, 1H), 1.74-1.92 (m, 1H), 1.58-1.72 (m, 1H), 1.15 (t, 3H). MS m/z 389 [M+H]+.
Стадия 2: Получение 1-{[(2S,3S,4S)-3-этил-4-фтор-5-оксопирролидин-2-ил]метокси}-7-метокси-4-нитроизохинолин-6-карбоксамида

В круглодонную колбу добавляли 1-{[(2S,3S,4S)-3-этил-4-фтор-5-оксопирролидин-2-ил]метокси}-7-метокси-4-нитроизохинолин-6-карбонитрил (90 мг, 0,23 ммоль) и метансульфоновую кислоту (1,75 мл, 26,8 ммоль). Смесь нагревали до 70°С в течение 18 ч. Смесь гасили во льду. К этой смеси добавляли EtOAc и смесь подщелачивали до рН 10 путем добавления гидроксида аммония. Слои разделяли и водную фазу экстрагировали пять раз EtOAc. Объединенные экстракты EtOAc сушили над безводным Na2SO4. Остаток очищали посредством хроматографии на силикагеле с использованием 0-20%-ного метанола в DCM с получением указанного в заголовке соединения в виде твердого вещества (63 мг, 67%-ный выход). 1Н-ЯМР (400 МГц, CDCl3) δ 9.10 (s, 1H), 8.79 (s, 1H), 7.82 (br s, 1H), 7.63 (s, 1H), 7.56 (br s, 1H), 6.62 (br s, 1H), 5.01 (d, 1H), 4.84-4.95 (m, 1H), 4.58 (d, 1H), 4.30 (br s, 1H), 4.03 (s, 2H), 2.56-2.75 (m, 1H), 2.18 (s, 1H), 1.99-2.10 (m, 1H), 1.64-1.90 (m, 1H), 1.16 (t, 3H). MS m/z 407 [M+H]+.
Стадия 3: Получение 4-амино-1-{[(2S,3S,4S)-3-этил-4-фтор-5-оксопирролидин-2-ил]метокси}-7-метоксиизохинолин-6-карбоксамида
К смеси цинка (91 мг, 1,39 ммоль), хлорида аммония (75 мг, 1,39 ммоль) и 1-{[(2S,3S,4S)-3-этил-4-фтор-5-оксопирролидин-2-ил]метокси}-7-метокси-4-нитроизохинолин-6-карбоксамида (57 мг, 0,14 ммоль) добавляли воду (0,7 мл), тетрагидрофуран (0,7 мл) и этанол (0,35 мл). Смесь перемешивали при температуре окружающей среды в течение 20 мин. Смесь фильтровали через целит и фильтраты очищали посредством хроматографии на силикагеле с использованием 0-20%-ного метанола в DCM с получением указанного в заголовке соединения в виде твердого вещества (37 мг, 71%-ный выход). 1Н-ЯМР (400 МГц, метаноле) δ 8.54 (s, 1H), 7.81 (s, 1H), 7.40 (s, 1H), 4.96 (dd, 1H), 4.51 (dd, 1H), 4.32 (dd, 1H), 4.17 (sext, 1Н). 4.06 (s, 3Н), 2.78-2.61 (m, 1H), 1.84-1.65 (m, 2Н), 1.11 (t, 3Н). MS m/z 377 [М+Н]+.
Пример 16
1-(((4R,7S)-7-фтор-6-оксо-5-азаспиро[2.4]гептан-4-ил)метокси)-7-изопропоксиизохинолин-6-карбоксамид
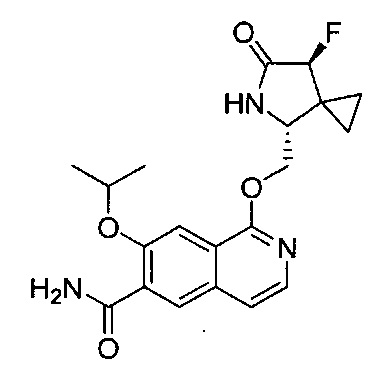
Стадия 1: Получение этил-2-циклопропилиденацетата 
Суспензию (1-этоксициклопропокси)триметилсилана (68 г, 390 ммоль), этил-2-(трифенилфосфанилиден)ацетата (178 г, 507 ммоль) и бензойной кислоты (6,19 г, 50,7 ммоль) в толуоле (1020 мл) перемешивали при 90°С в течение ночи. После охлаждения реакционную смесь концентрировали для удаления толуола. К остатку добавляли эфир (500 мл) и петролейный эфир (250 мл) и смесь перемешивали при комнатной температуре в течение 2 ч. Полученную смесь фильтровали и фильтрат концентрировали с получением неочищенного продукта, который очищали с помощью флэш-колонки (петролейный эфир/EtOAc=10/1) с получением этил-2-циклопропилиденацетата в виде желтого масла (50 г, которое использовали без дополнительной очистки). 1Н-ЯМР (400 МГц, CDCl3) δ=6.40 (s, 1Н), 4.38 (m, 2Н), 1.40-0.69 (m, 7Н).
Стадия 2: Получение этил-2-(1-(нитрометил)циклопропил)ацетата 
Смесь этил-2-циклопропилиденацетата (40 г, 317 ммоль), нитрометана (96,8 г, 1590 ммоль) и DBU (483 г, 317 ммоль) в CH3CN (160 мл) перемешивали при 60°С в течение ночи в атмосфере N2. Реакционную смесь вливали в 1 н. HCl (400 мл) и экстрагировали EtOAc (600 мл×2). Объединенные соли промывали водой и рассолом, затем сушили над безводным Na2SO4. Неочищенный продукт очищали с помощью флэш-колонки (петролейный эфир/EtOAc=10/1) с получением этил-2-(1-(нитрометил)циклопропил)ацетата (32,5 г, 55%-ный выход) в виде бесцветного масла. 1Н-ЯМР (400 МГц, CDCl3) δ=4.43 (s, 2Н), 4.17 (m, 2Н), 2.50 (s, 2Н), 1.28 (m, 3Н), 0.88-0.69 (m, 4Н).
Стадия 3: Получение этил-2-(1-(2-гидрокси-1-нитроэтил)циклопропил)ацетата
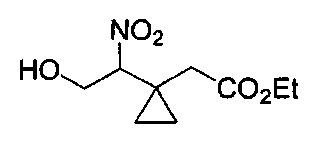
Раствор соединения этил-2-(1-(нитрометил)циклопропил)ацетата (15 г, 80 ммоль) в iPrOH (15 мл) перемешивали с параформальдегидом (4,65 г, 160 ммоль) и KF (466 мг, 8,01 ммоль) при 22°С в течение 7 ч. Полученную смесь обрабатывали EtOAc (500 мл×3) и Н2О (200 мл). Объединенные органические слои промывали рассолом, сушили и концентрировали с получением неочищенного продукта, который очищали посредством колоночной хроматографии (петролейный эфир/этилацетат = 3/1) с получением этил-2-(1-(2-гидрокси-1-нитроэтил)циклопропил)ацетата (9 г, 52%-ный выход) в виде бесцветного масла. Исходное вещество (4 г, 27%-ный выход) выделяли в виде желтого масла. 1Н-ЯМР (400 МГц, CDCl3) δ 4.16-3.95 (m, 3Н), 3.93-3.85 (m, 1H), 3.16 (br s, ОН), 2.81 (d, 1H), 2.33 (m, 1Н), 2.18 (d, 1H), 1.25-1.16 (m, 3Н), 0.97-0.83 (m, 2Н), 0.81-0.69 (m, 1H), 0.67-0.53 (m, 1H).
Стадия 4: Получение 4-(гидроксиметил)-5-азаспиро[2.4]гептан-6-она 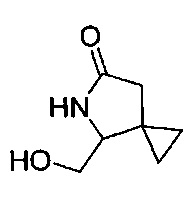
Смесь этил-2-(1-(2-гидрокси-1-нитроэтил)циклопропил)ацетата (5,50 г, 25,3 ммоль) и Ni Ренея (2,0 г) в EtOH (100 мл) перемешивали при 30-40°С в течение 6 ч в атмосфере Н2. Полученную смесь фильтровали и фильтрат перемешивали при 80°С в течение 36 ч. Реакционную смесь концентрировали с получением неочищенного продукта, который очищали с помощью флэш-колонки с получением 4-(гидроксиметил)-5-азаспиро[2.4]гептан-6-она (1,9 г, 53%-ный выход) в виде не совсем белого твердого вещества. 1Н-ЯМР (400 МГц, DMSO-d6) δ 7.72 (br. s., 1H), 4.67 (t, 1H), 3.31 (m, 2H), 3.13-3.00 (m, 1H), 2.39 (d, 1H), 1.88 (d, 1H), 0.86-0.73 (m, 1H), 0.61-0.41 (m, 3H). MS m/z 142,1 [M+H]+.
Стадия 5: Получение 3',3'-диметилдигидро-3'Н-спиро[циклопропан-1,7'-пирроло[1,2-с]оксазол]-5'(6'Н)-она

К перемешиваемому раствору 4-(гидроксиметил)-5-азаспиро[2.4]гептан-6-она (4,50 г, 31,9 ммоль) в толуоле (100 мл) добавляли TsOH.H2O (60,6 мг, 0,319 ммоль), затем добавляли 2,2-диметоксипропан (13,3 г, 128 ммоль). Реакционную смесь нагревали до температуры флегмообразования в течение 2 ч. Реакционную смесь охлаждали до комнатной температуры и упаривали досуха. Остаток растворяли в МТВЕ (500 мл), промывали 1 н. водн. NaOH (50 мл) и водой (50 мл), затем сушили над Na2SO4 с получением 3',3'-диметилдигидро-3'Н-спиро[циклопропан-1,7'-пирроло[1,2-с]оксазол]-5'(6'Н)-она (5,4 г, 93%-ный выход) в виде бесцветного масла, которое использовали на следующей стадии без дополнительной очистки.
Стадия 6: Получение 6'-фтор-3',3'-диметилдигидро-3'Н-спиро[циклопропан-1,7'-пирроло[1,2-с]оксазол]-5'(6'Н)-она

Раствор 3',3'-диметилдигидро-3'Н-спиро[циклопропан-1,7'-пирроло[1,2-с]оксазол]-5'(6'Н)-она (5,4 г, 29,8 ммоль) в сухом THF (130 мл) ненадолго помещали в вакуум, затем продували азотом. Смесь охлаждали в бане сухой лед-ацетон в течение 15 мин, после чего медленно через шприц добавляли LiHMDS (27 мл, 67,5 ммоль). Полученную смесь перемешивали охлажденной в течение 45 мин, после чего эту смесь через трубку добавляли к смеси N-фтордибензолсульфонимида (NFSI) (12,2 г, 38,7 ммоль) в сухом THF (130 мл), предварительно охлажденном до -78°С. Смесь перемешивали при -78°С в течение 15 мин. Охлаждающую баню удаляли и реакционную смесь медленно гасили водой (100 мл). Добавляли EtOAc (200 мл). Органическую фазу перемешивали с 5%-ным водным NaI (13,4 г NaI в 250 мл Н2О) в течение 15 мин. Органическую фазу промывали 0,1 М тиосульфатом натрия (100 мл), 1 н. NaOH (100 мл) и наконец рассолом. Органическую фазу сушили над безводным Na2SO4 и остаток очищали посредством флэш-хроматографии с использованием смеси 40% EtOAc:гептаны с получением 6'-фтор-3',3'-диметилдигидро-3'Н-спиро[циклопропан-1,7'-пирроло[1,2-с]оксазол]-5'(6'Н)-она (3 г, 50%-ный выход), белого твердого вещества, в виде смеси диастереомеров. 1Н-ЯМР (400 МГц, CDCl3) δ 5.19-5.00 (m, 0.5Н), 4.58-4.41 (m, 0.5Н), 4.38 (m, 0.5Н), 4.04 (m, 0.5Н), 3.86 (m, 1H), 3.50-3.36 (m, 1H), 1.73 (m, 3Н), 1.52 (m, 3Н), 1.29-1.10 (m, 1H), 0.99-0.58 (m, 3Н). MS m/z 200.1 [М+Н]+
Стадия 7: Получение 7-фтор-4-(гидроксиметил)-5-азаспиро[2.4]гептан-6-она
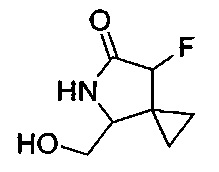
К перемешиваемому раствору 6'-фтор-3',3'-диметилдигидро-3'Н-спиро[циклопропан-1,7'-пирроло[1,2-с]оксазол]-5'(6'Н)-она (1 г, 5,02 ммоль) в смеси ацетонитрил-вода (10 мл: 0,5 мл) добавляли TFA (57,2 мг, 0,50 ммоль) и смесь нагревали до 90°С в течение 1 ч. Смесь концентрировали досуха и три раза азеотропно отгоняли с MeCN (3×10 мл), один раз со смесью MeCN-вода (10 мл + 0,5 мл) и с толуолом (10 мл×3) с получением 7-фтор-4-(гидроксиметил)-5-азаспиро[2.4]гептан-6-она (0,8 г, ~100%) в виде белого твердого вещества в виде ~1:1 смеси диастереомеров, которую использовали без дополнительной очистки. 1Н-ЯМР (400 МГц, DMSO-d6) δ=8.61 (br. s., 0.5Н), 8.34 (br. s., 0.5), 4.98-4.69 (m, 0.5H), 4.57-4.33 (m, 0.5H), 3.55-3.17 (m, 4H), 1.07-0.55 (m, 4H).
Стадия 8: Получение 1-((7-фтор-6-оксо-5-азаспиро[2.4]гептан-4-ил)метокси)-7-изопропоксиизохинолин-6-карбонитрила

К перемешиваемому раствору 1-хлор-7-изопропоксиизохинолин-6-карбонитрила (600 мг, 2,43 ммоль) и 7-фтор-4-(гидроксиметил)-5-азаспиро[2.4]гептан-6-она (426 мг, 2,68 ммоль) в DMF (20 мл) по каплям добавляли KHMDS (6,1 мл, 6,1 ммоль, 1 M в THF) в атмосфере N2 при 0°С. Смесь перемешивали при 0°С в течение 1 ч. Смесь обрабатывали насыщенным водным раствором NH4Cl и экстрагировали EtOAc (100 мл×3), промывали водой, затем рассолом (50 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении.
Остаток очищали посредством флэш-хроматографии на силикагеле (50% EtOAc-гексан) с получением первого элюируемого изомера в виде рацемического 1-(((анти)-7-фтор-6-оксо-5-азаспиро[2.4]гептан-4-ил)метокси)-7-изопропоксиизохинолин-6-карбонитрила (300 мг, 33%-ный выход) в виде желтого твердого вещества. 1Н-ЯМР (400 МГц, DMSO-d6) δ 8.92 (s, 1Н), 8.52 (s, 1H), 7.97 (d, 1H), 7.61 (s, 1H), 7.42 (d, 1H), 5.14-4.96 (d, 1H), 4.96-4.87 (m, 1H), 4.45-4.31 (m, 2H), 3.83 (t, 1H), 1.40 (dd, 6H), 1.11-1.00 (m, 2H), 0.89-0.76 (m, 2H). Эксперимент nOe выявил пространственное взаимодействие между фторсодержащим углеродом С-Н (5.14-4.96 (d, 1Н)) и изопропиловой группой, требующее транс-взаимодействия между фтором и группой CH2O-. MS m/z 388.0 [М+Н]+ и MS m/z 409.9 [M+Na]+.
Второй элюируемый изомер собирали в виде рацемического 1-(((син)-7-фтор-6-оксо-5-азаспиро[2.4]гептан-4-ил)метокси)-7-изопропоксиизохинолин-6-карбонитрила (500 мг, 56%-ный выход) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, DMSO-d6) δ 8.50 (s, 1Н), 8.00-7.92 (m, 2H), 7.81 (s, 1H), 7.40 (d, 1H), 4.90 (td, 1H), 4.72-4.52 (m, 1H), 4.48 (dd, 1H), 4.27-4.17 (m, 1H), 3.73 (d, 1H), 1.38 (t, 6H), 1.09-0.96 (m, 4H).
Стадия 9: Разделение рацемического 1-(((анти)-7-фтор-6-оксо-5-азаспиро[2.4]гептан-4-ил)метокси)-7-изопропоксиизохинолин-6-карбоксамида

К перемешиваемой смеси рацемического 1-(((анти)-7-фтор-6-оксо-5-азаспиро[2.4]гептан-4-ил)метокси)-7-изопропоксиизохинолин-6-карбонитрила (300 мг, 0,812 ммоль) в DMSO (12 мл) добавляли K2CO3 (561 мг, 4,06 ммоль) при 15°С. Реакционную смесь перемешивали при 15°С в течение 5 мин. К реакционной смеси добавляли Н2О2 (0,36 мл) при 15°С. Реакционную смесь перемешивали при 15°С в течение 2 ч. К полученной смеси добавляли Н2О (15 мл) при 0-5°С и смесь перемешивали в течение 1 ч при 15°С. Смесь фильтровали и осадок на фильтре промывали Н2О (40 мл) и сушили в вакууме с получением рацемического 1-(((анти)-7-фтор-6-оксо-5-азаспиро[2.4]гептан-4-ил)метокси)-7-изопропоксиизохинолин-6-карбоксамида (220 мг, 70%-ный выход) в виде белого твердого вещества.
Энантиомеры разделяли посредством препаративной хиральной SFC-хроматографии. Инструмент: SFC-200 Колонка: Chiralpak AS 300×50 мм в.д., 10 мкм. Подвижная фаза: Сверхкритический CO2/МеОН (0,1% NH3H2O)=55/45 при 200 мл/мин, температура колонки: 38°С. Давление форсунки: 100 бар (107 Па). Температура форсунки: 60°С. Температура испарителя: 20°С. Температура триммера: 25°С.

1-(((4R,7S)-7-фтор-6-оксо-5-азаспиро[2.4]гептан-4-ил)метокси)-7-изопропоксиизохинолин-6-карбоксамид. Аналитическая SFC-хроматография: Колонка: Chiralpak AS-H 150×4,6 мм в.д., 5 мкм. Подвижная фаза: метанол (0,05% DEA) в СО2 от 5% до 40%. Скорость потока: 3 мл/мин. Длина волны: 220 нм. Аналитическая SFC-хроматография: Время удерживания 4,265 мин. 1Н-ЯМР (400 МГц, DMSO-d6) δ 8.85 (s, 1Н), 8.20 (s, 1H), 7.89 (d, 1H), 7.72 (br. s., 2H), 7.52 (s, 1H), 7.43 (d, 1H), 5.15-4.94 (m, 1H), 4.90-4.81 (m, 1H), 4.39 (d, 2H), 3.81 (br. s., 1H), 1.40 (dd, 6H), 1.06 (br. s., 2H), 0.83 (d, 2H). MS m/z 388.0 [M+H]+ и MS m/z 409,9 [M+Na]+.
Пример 17
1-(((4S,7R)-7-фтор-6-оксо-5-азаспиро[2.4]гептан-4-ил)метокси)-7-изопропоксиизохинолин-6-карбоксамид

Аналитическая SFC-хроматография: Колонка: Chiralpak AS-H 150×4,6 мм в.д., 5 мкм. Подвижная фаза: метанол (0,05% DEA) в СО2 от 5% до 40%. Скорость потока: 3 мл/мин. Длина волны: 220 нм. Аналитическая SFC-хроматография: Время удерживания 4,678 мин. 1Н-ЯМР (400 МГц, DMSO-d6) δ 8.85 (s, 1H), 8.20 (s, 1H), 7.89 (d, 1H), 7.72 (br. s., 2H), 7.52 (s, 1H), 7.43 (d, 1H), 5.15-4.94 (m, 1H), 4.90-4.81 (m, 1H), 4.39 (d, 2H), 3.81 (br. s., 1H), 1.40 (dd, 6H), 1.06 (br. s., 2H), 0.83 (d, 2H). MS m/z 388.1 [M+H]+ и MS m/z 410,0 [M+Na]+.
Пример 18
Рацемический 1-(((син)-7-фтор-6-оксо-5-азаспиро[2.4]гептан-4-ил)метокси)-7-изопропоксиизохинолин-6-карбоксамид
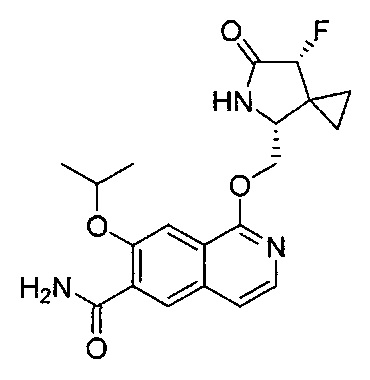
К перемешиваемой смеси рацемического 1-(((син)-7-фтор-6-оксо-5-азаспиро [2.4] гептан-4-ил)метокси)-7-изопропоксиизохинолин-6-карбонитрила (400 мг, 1,08 ммоль) в DMSO (16 мл) добавляли K2CO3 (748 мг, 5,41 ммоль) при 15°С. Реакционную смесь перемешивали при 15°С в течение 5 мин. Добавляли Н2О2 (0,5 мл) при 15°С. Реакционную смесь перемешивали при 15°С в течение 2 ч. Добавляли Н2О (20 мл) при 0-5°С и смесь перемешивали в течение 1 ч при 15°С. Смесь фильтровали и осадок на фильтре промывали Н2О (50 мл) и сушили в вакууме с получением рацемического 1-(((син)-7-фтор-6-оксо-5-азаспиро[2.4]гептан-4-ил)метокси)-7-изопропоксиизохинолин-6-карбоксамида (300 мг, 71%-ный выход) в виде белого твердого вещества.
Энантиомеры разделяли посредством препаративной хиральной SFC-хроматографии. Инструмент: SFC-200, колонка: Chiralpak AS 300×50 мм в.д., 10 мкм. Подвижная фаза: Сверхкритический СО2/МеОН (0,1% NH3H2O)=55/45 при 200 мл/мин. Температура колонки: 38°С. Давление форсунки: 100 бар (107 Па). Температура форсунки: 60°С. Температура испарителя: 20°С. Температура триммера: 25°С.
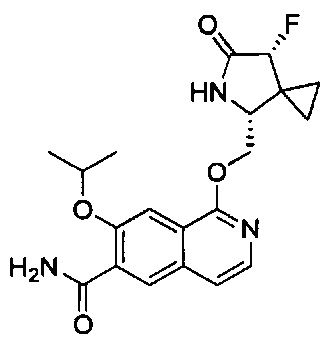
1-(((4R,7R)-7-фтор-6-оксо-5-азаспиро[2.4]гептан-4-ил)метокси)-7-изопропоксиизохинолин-6-карбоксамид. Аналитическая SFC-хроматография: Колонка: Chiralpak AS-H 150×4,6 мм в.д., 5 мкм. Подвижная фаза: метанол (0,05% DEA) в CO2 от 5% до 40%. Скорость потока: 3 мл/мин. Длина волны: 220 нм. Аналитическая SFC-хроматография: Время удерживания: 4,161 мин. 1Н-ЯМР (400 МГц, DMSO-d6) δ 8.96 (s, 1Н), 8.18 (s, 1H), 7.88 (d, 1H), 7.77-7.67 (m, 3H), 7.41 (d, 1H), 4.86 (td, 1H), 4.72-4.52 (m, 1H), 4.46 (dd, 1H), 4.22 (dd, 1H), 3.73 (d, 1H), 1.37 (t, 6H), 1.08-0.93 (m, 4H). MS m/z 388.1 [М+Н]+ и MS m/z 410.0 [M+Na]+.
Пример 19
1-(((4S,7S)-7-фтор-6-оксо-5-азаспиро[2.4]гептан-4-ил)метокси)-7-изопропоксиизохинолин-6-карбоксамид

Аналитическая SFC-хроматография: Колонка: Chiralpak AS-H 150×4,6 мм в.д., 5 мкм. Подвижная фаза: метанол (0,05% DEA) в СО2 от 5% до 40%. Скорость потока: 3 мл/мин. Длина волны: 220 нм. Аналитическая SFC-хроматография: Время удерживания: 6,239 мин. 1Н-ЯМР (400 МГц, DMSO-d6) δ 8.96 (s, 1Н), 8.18 (s, 1H), 7.88 (d, 1H), 7.77-7.67 (m, 3H), 7.41 (d, 1H), 4.86 (td, 1H), 4.72-4.52 (m, 1H), 4.46 (dd, 1H), 4.22 (dd, 1H), 3.73 (d, 1H), 1.37 (t, 6H), 1.08-0.93 (m, 4H). MS m/z [M+H]+ 387.9 и MS m/z [M+Na]+ 409,9.
Пример 20
1-(((4S,7R)-7-фтор-6-оксо-5-азаспиро[2.4]гептан-4-ил)метокси)-7-метоксиизохинолин-6-карбоксамид

Стадия 1: Получение 1-((7-фтор-6-оксо-5-азаспиро[2.4]гептан-4-ил)метокси)-7-метоксиизохинолин-6-карбонитрила

К смеси 1-хлор-7-метоксиизохинолин-6-карбонитрила (650 мг, 2,97 ммоль) и 7-фтор-4-(гидроксиметил)-5-азаспиро[2.4]гептан-6-она (530 мг, 3,33 ммоль) в DMF (15,0 мл) при 0°С по каплям добавляли KHMDS (6,54 мл, 1 M в THF). Полученный раствор перемешивали при 0°С в течение 1 ч. Реакционную смесь оставляли нагреваться до 30°С и перемешивали в течение 2 ч. Реакционную смесь гасили водным NH4Cl (10 мл) и распределяли между смесью Н2О/EtOAc (100 мл/100 мл). Водный слой экстрагировали EtOAc (100 мл×2). К органическому слою добавляли МеОН (100 мл). Органический слой сушили над MgSO4, затем фильтровали и растворитель удаляли при пониженном давлении. Осадок на фильтре суспендировали с 150 мл EtOAc и перемешивали в течение 18 часов при 35°С, затем фильтровали и растворитель выпаривали. Фильтраты объединяли, концентрировали и очищали с помощью флэш-колонки (EtOAc: петролейный эфир от 50% до 70%) с получением двух фракций (140 мг и 657 мг) в виде желтых твердых веществ.
Первые фракции (140 мг) смешивали с другой партией, полученной таким же образом, и очищали с помощью флэш-колонки (EtOAc: петролейный эфир от 40% до 55%) с получением 335 мг ранней фракции (23%-ный выход) в виде желтого твердого вещества. Это вещество превращали в карбоксамид и его энантиомеры разделяли на этой стадии.
Рацемический 1-(((анти)-7-фтор-6-оксо-5-азаспиро[2.4]гептан-4-ил)метокси)-7-метоксиизохинолин-6-карбонитрил

Ранняя фракция: 1Н-ЯМР (400 МГц, DMSO-d6) δ 8.97 (s, 1Н), 8.54 (s, 1H), 7.99 (d, 1H), 7.63 (s, 1H), 7.43 (d, 1H), 5.18-4.96 (m, 1H), 4.47-4.38 (m, 1H), 4.37-4.29 (m, 1H), 4.05 (s, 3H), 3.84 (dd, 1H), 1.11-1.01 (m, 2H), 0.89-0.76 (m, 2H). MS m/z 342,0 [M+H]+.
Рацемический 1-(((син)-7-фтор-6-оксо-5-азаспиро[2.4]гептан-4-ил)метокси)-7-метоксиизохинолин-6-карбонитрил

Вторые фракции (657 мг) смешивали с другой партией, полученной таким же образом, и очищали с получением 500 мг поздней фракции (34%-ный выход) в виде желтого твердого вещества. Это вещество отдельно превращали в карбоксамид и его энантиомеры разделяли на этой стадии. Поздняя фракция 1Н-ЯМР (400 МГц, DMSO-d6) δ 9.03 (s, 1Н), 8.51 (s, 1H), 7.98 (d, 1H), 7.81 (s, 1H), 7.41 (d, 1H), 4.70-4.52 (m, 1H), 4.49 (dd, 1H), 4.21 (dd, 1H), 4.01 (s, 3H), 3.74 (br. s., 1H), 1.10-0.97 (m, 4H). MS m/z 342,0 [M+H]+.
Стадия 2: Получение рацемического 1-(((анти)-7-фтор-6-оксо-5-азаспиро[2.4]гептан-4-ил)метокси)-7-метоксиизохинолин-6-карбоксамида

Желтую смесь рацемического 1-(((анти)-7-фтор-6-оксо-5-азаспиро[2.4]гептан-4-ил)метокси)-7-метоксиизохинолин-6-карбонитрила (230 мг, 0,67 ммоль) со Стадии 1 и K2CO3 (466 мг, 1,46 ммоль) в DMSO (8,0 мл) перемешивали при 30°С в течение 5 мин и затем медленно добавляли Н2О2 (0,46 мл, 15 ммоль). Полученную смесь перемешивали при 30°С в течение 2 ч. К реакционной смеси добавляли Н2О (18 мл) при 0-5°С и смесь перемешивали в течение 1 часа. Смесь фильтровали и осадок на фильтре промывали Н2О (20 мл×4). Остаток сушили при пониженном давлении с получением неочищенного продукта (202 мг, 84%-ный выход) в виде не совсем белого твердого вещества. Энантиомеры разделяли посредством SFC. MS m/z 382.1 [M+Na]+. Энантиомерное разделение: Хиральная SFC-хроматография: Колонка: AD (250 мм×30 мм, 5 мкм). Подвижная фаза: EtOH : CO2=40:60 (0,1% NH4OH). Скорость потока: 50 мл/мин. Длина волны: 220 нм.
1-(((4S,7R)-7-фтор-6-оксо-5-азаспиро[2.4]гептан-4-ил)метокси)-7-метоксиизохинолин-6-карбоксамид
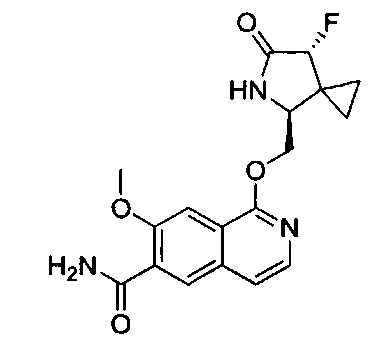
Аналитическая хиральная хроматография: Колонка: AD (250 мм×30 мм, 5 мкм). Подвижная фаза: EtOH : CO2 = 40:60 (0,1% NH4OH). Скорость потока: 50 мл/мин. Длина волны: 220 нм. Время удерживания 6,437 мин. 1Н-ЯМР (400 МГц, DMSO-d6) δ 8.92 (s, 1Н), 8.17 (s, 1H), 7.90 (d, 1H), 7.85 (br. s., 1H), 7.72 (br. s., 1H), 7.53 (s, 1H), 7.44 (d, 1H), 5.18-4.97 (m, 1H), 4.44-4.31 (m, 2H), 3.98 (s, 3H), 3.82 (t, 1H), 1.11-1.01 (m, 2H), 0.90-0.75 (m, 2H). MS m/z 382.1 [M+Na]+.
Пример 21
1-(((4R,7S)-7-фтор-6-оксо-5-азаспиро[2.4]гептан-4-ил)метокси)-7-метоксиизохинолин-6-карбоксамид

Аналитическая хиральная хроматография: Колонка: AD (250 мм×30 мм, 5 мкм). Подвижная фаза: EtOH : CO2 = 40:60 (0,1% NH4OH). Скорость потока: 50 мл/мин. Длина волны: 220 нм. Время удерживания 7,090 мин. 1Н-ЯМР (400 МГц, DMSO-d6) δ 8.90 (s, 1H), 8.17 (s, 1H), 7.90 (d, 1H), 7.84 (br. s., 1H), 7.70 (br. s., 1H), 7.53 (s, 1H), 7.44 (d, 1H), 5.17-4.96 (m, 1H), 4.44-4.33 (m, 2H), 3.98 (s, 3H), 3.85-3.79 (m, 1H), 1.07 (d, 2H), 0.89-0.73 (m, 2H). MS m/z 382.1 [M+Na]+.
Пример 22
1-(((4R,7R)-7-фтор-6-оксо-5-азаспиро[2.4]гептан-4-ил)метокси)-7-метоксиизохинолин-6-карбоксамид

Смесь рацемического 1-(((син)-7-фтор-6-оксо-5-азаспиро[2.4]гептан-4-ил)метокси)-7-метоксиизохинолин-6-карбонитрила (350 мг, 1,02 ммоль) из Примера 21, Стадии 1, и K2CO3 (709 мг, 2,20 ммоль) в DMSO (10,4 мл) перемешивали при 30°С в течение 5 мин и затем медленно добавляли Н2О2 (0,90 мл, 29 ммоль). Полученную белую суспензию перемешивали при 30°С в течение 2 ч. К реакционной смеси добавляли Н2О (30 мл) при 0-5°С и смесь перемешивали в течение 1 ч. Смесь фильтровали и осадок на фильтре промывали Н2О (30 мл×3). Остаток сушили при пониженном давлении с получением неочищенного продукта (350 мг, 95%-ный выход) в виде не совсем белого твердого вещества. Энантиомеры разделяли посредством SFC-хроматографии. MS m/z 360,0 (М+Н)+. Энантиомерное разделение посредством хиральной SFC-хроматографии: Колонка: AD (250 мм×30 мм, 5 мкм). Подвижная фаза: EtOH : CO2 = 40:60 (0,1% NH4OH). Скорость потока: 50 мл/мин. Длина волны: 220 нм.
1-(((4R,7R)-7-фтор-6-оксо-5-азаспиро[2.4]гептан-4-ил)метокси)-7-метоксиизохинолин-6-карбоксамид. Аналитическая хиральная SFC-хроматография: Колонка: AD (250 мм×30 мм, 5 мкм) Подвижная фаза: EtOH : CO2 = 30:70 (0,1% NH4OH). Скорость потока: 60 мл/мин. Длина волны: 220 нм. Время удерживания 6,687 мин. 1Н-ЯМР (400 МГц, DMSO-d6) δ 9.00 (s, 1Н), 8.15 (s, 1H), 7.89 (d, 1H), 7.84 (br. s., 1H), 7.74 (s, 1H), 7.69 (br. s., 1H), 7.42 (d, 1H), 4.70-4.52 (m, 1H), 4.48 (dd, 1H), 4.23 (dd, 1H), 3.95 (s, 3H), 3.74 (br. s., 1H), 1.02 (br. s., 4H). MS m/z 359.9 [M+H]+.
Пример 23
1-(((4S,7S)-7-фтор-6-оксо-5-азаспиро[2.4]гептан-4-ил)метокси)-7-метоксиизохинолин-6-карбоксамид

Аналитическая хиральная SFC-хроматография: Колонка: AD (250 мм×30 мм, 5 мкм). Подвижная фаза: EtOH : СО2 = 30:70 (0,1% NH4OH). Скорость потока: 60 мл/мин. Время удерживания 6,829 мин. 1Н-ЯМР (400 МГц, DMSO-d6) δ 9.00 (s, 1Н), 8.15 (s, 1H), 7.89 (d, 1H), 7.84 (br. s., 1H), 7.74 (s, 1H), 7.69 (br. s., 1H), 7.42 (d, 1H), 4.70-4.52 (m, 3H), 4.48 (d, 2H), 4.22 (br. s., 2H), 3.95 (s, 3H), 3.74 (br. s., 1H), 1.02 (br. s., 4H). MS m/z 360.0 [M+H]+.
Пример 24
4-(((2S,3R)-3-этил-5-оксопирролидин-2-ил)метокси)-6-метоксиизохинолин-7-карбоксамид

Стадия 1: Получение этил-N-(3-бром-4-метоксибензил)глицината

Получение этил-N-(3-бром-4-метоксибензил)глицината осуществляли в пяти параллельных партиях. К раствору 3-бром-4-метоксибензальдегида (5,0 г, 20 ммоль) и этилглицината (8,12 г, 58,1 ммоль, соль HCl) в DCM (120 мл) добавляли TEA (5,12 г, 50,7 ммоль), затем добавляли АсОН (3,07 г, 51,2 ммоль) и NaBH(ОАс)3 (11,8 г, 55,8 ммоль). Смесь перемешивали при 15°С в атмосфере азота в течение ночи. Таким образом получали в общей сложности пять партий, которые объединяли для обработки и очистки. Полученную смесь вливали в насыщенный водный NaHCO3 (500 мл) и экстрагировали DCM. Объединенные органические фазы сушили над Na2SO4, фильтровали и растворитель удаляли с получением неочищенного масла, которое затем очищали посредством хроматографии на силикагеле с использованием смеси EtOAc/петролейный эфир (от 20% до 100%) с получением указанного в заголовке соединения в виде масла (26 г, 74%-ный выход). 1Н-ЯМР (400 МГц, DMSO-d6) δ 7.43-7.59 (m, 1Н), 7.26 (dd, 1H), 7.03 (d, 1H), 4.08 (q, 2H), 3.82 (s, 3H), 3.63 (s, 2H), 3.26 (s, 2H), 1.18 (t, 3H). MS m/z 304 [M+H]+.
Стадия 2: Получение этил-N-(3-бром-4-метоксибензил)-N-[(4-метилфенил)сульфонил]глицината

Получение этил-N-(3-бром-4-метоксибензил)-N-[(4-метилфенил)сульфонил]глицината осуществляли в пяти параллельных партиях. К раствору этил-N-(3-бром-4-метоксибензил)глицината (5000 мг, 16,5 ммоль) и пиридина (6540 мг, 82,7 ммоль) в THF (60 мл) добавляли л-толуолсульфонилхлорид (3150 мг, 16,5 ммоль) при 0°С. Смесь перемешивали при 15°С в течение ночи. К смеси добавляли DMAP (202 мг, 1,65 ммоль) и смесь перемешивали при 15°С в течение ночи. Таким образом получали в общей сложности пять партий, которые объединяли для обработки и очистки. Смесь подкисляли концентрированной HCl до рН 3 и экстрагировали DCM. Объединенные органические слои промывали безводным Na2SO4, концентрировали в вакууме и очищали посредством хроматографии на силикагеле (EtOAc/петролейный эфир от 8% до 20%) с получением указанного в заголовке соединения в виде белого твердого вещества (21 г, 56%-ный выход) (84%-ная чистота согласно LCMS). 1Н-ЯМР (400 МГц, CDCl3) δ 7.76 (d, 2Н), 7.30-7.36 (m, 3Н), 7.19 (dd, 1Н), 6.83 (d, 1Н), 4.40 (s, 2Н), 4.02 (q, 2Н), 3.90 (s, 2Н), 3.88 (s, 3Н), 2.45 (s, 3Н), 1.16 (t, 3Н). MS m/z 477,8 [M+Na]+.
Стадия 3: Получение N-(3-Бром-4-метоксибензил)-N-[(4-метилфенил)сульфонил]глицина
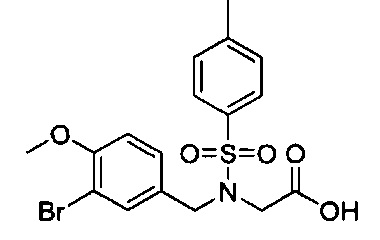
К раствору этил-N-(3-бром-4-метоксибензил)-N-[(4-метилфенил)сульфонил]глицината (10,0 г, 21,9 ммоль) в смеси THF/MeOH (70 мл/70 мл) добавляли раствор LiOH⋅H2O (1840 мг, 43,8 ммоль) в Н2О (50 мл) при 15°С. Смесь перемешивали при этой температуре в течение 4 ч. Смесь концентрировали в вакууме и разбавляли H2O (100 мл), затем подкисляли концентрированным HCl до рН 3. Полученную смесь экстрагировали DCM, сушили над безводным Na2SO4, фильтровали и концентрировали в вакууме с получением указанного в заголовке соединения в виде белого твердого вещества (9 г, 96%-ный выход). 1Н-ЯМР (400 МГц, CDCl3) δ 7.76 (d, 2Н), 7.34 (d, 2Н), 7.28 (d, 1Н), 7.17 (dd, 1H), 6.84 (d, 1H), 4.39 (s, 2H), 3.94 (s, 2H), 3.89 (s, 3H), 2.44-2.50 (m, 3H). MS m/z 449,7 [M+Na]+.
Стадия 4: Получение N-(3-бром-4-метоксибензил)-N-[(4-метилфенил)сульфонил]глицилхлорида
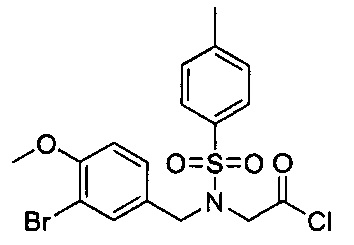
Раствор N-(3-бром-4-метоксибензил)-N-[(4-метилфенил)сульфонил]глицина (3000 мг, 7,00 ммоль) упаривали совместно с сухим толуолом (30 мл×3) для удаления воды и растворяли в сухом DCM (75 мл). К смеси добавляли оксалилхлорид (4450 мг, 35,0 ммоль) и DMF (3 капли) при 15°С в атмосфере азота. Смесь перемешивали в течение 2 ч. Раствор упаривали с получением неочищенного указанного в заголовке соединения в виде желтого твердого вещества (3130 мг, ~100%), которое использовали непосредственно на следующей стадии. MS m/z 465 [M+Na]+.
Стадия 5: Получение 7-бром-6-метокси-2-[(4-метилфенил)сульфонил]-2,3-дигидроизохинолин-4(1Н)-она

К раствору N-(3-бром-4-метоксибензил)-N-[(4-метилфенил)сульфонил]глицилхлорида (3130 мг, 7,01 ммоль) в DCM (15 мл) одной порцией добавляли AlCl3 (2340 мг, 17,5 ммоль) при -65°С. Реакционную смесь перемешивали в течение 1 ч при -65°С, затем медленно нагревали до 0°С и перемешивали в течение 1 ч. Реакционную смесь гасили водой (15 мл). Смесь экстрагировали DCM и сушили над безводным Na2SO4. Неочищенный продукт очищали посредством хроматографии на силикагеле с использованием смеси петролейный эфир : EtOAc (от 20:1 до 3:1) в качестве элюента с получением указанного в заголовке соединения в виде желтого твердого вещества (1,2 г, 41%-ный выход). 1Н-ЯМР (400 МГц, CDCl3) δ 7.63 (d, 2Н), 7.48 (s, 1Н), 7.32 (s, 1Н), 7.27 (t, 2Н), 4.44 (s, 2Н), 4.00 (s, 2Н), 3.90 (s, 3Н), 2.40 (s, 3Н).
Стадия 6: Получение 7-бром-6-метоксиизохинолин-4-ола

К раствору 7-бром-6-метокси-2-[(4-метилфенил)сульфонил]-2,3-дигидроизохинолин-4(1Н)-она (2700 мг, 6,58 ммоль) в EtOH (68 мл) добавляли NaHCO3 (2210 мг, 26,3 ммоль). Смесь нагревали до температуры флегмообразования в течение 3 ч. Смесь фильтровали и осадок на фильтре промывали ацетоном. Фильтрат концентрировали с получением неочищенного продукта, который очищали посредством хроматографии на силикагеле с использованием DCM : MeOH (от 100:1 до 8:1) в качестве элюента с получением указанного в заголовке соединения в виде желтого твердого вещества (1400 мг, 83%-ный выход). 1Н-ЯМР (400 МГц, DMSO-d6) δ 10.46 (br s, 1Н), 8.66 (s, 1H), 8.38 (s, 1H), 8.02 (s, 1H), 7.46 (s, 1H), 4.00 (s, 3H).
Стадия 7: Получение 4-Гидрокси-6-метоксиизохинолин-7-карбонитрила

К раствору 7-бром-6-метоксиизохинолин-4-ола (1400 мг, 5,51 ммоль, 1 экв.) в DMF (65 мл) добавляли Zn(CN)2 (3240 мг, 27,6 ммоль) и Pd(PPh3)4 (637 мг, 0,551 ммоль) при 15°С. Суспензию дегазировали в вакууме и дважды продували азотом. Реакционную смесь перемешивали в течение 10 мин при 15°С, затем в течение 6 ч при 140°С. DMF выпаривали. Остаток очищали посредством хроматографии на силикагеле с использованием смеси DCM : MeOH (от 50:1 до 10:1) в качестве элюента с получением неочищенного продукта, который растирали с DCM и фильтровали с получением указанного в заголовке соединения (750 мг, ~68%-ный выход), загрязненного остаточным DCM. Маточный раствор концентрировали с получением неочищенного указанного в заголовке соединения (785 мг, ~71%-ный выход), загрязненного остаточным DCM. 1Н-ЯМР (400 МГц, DMSO-d6) δ 10.72 (s, 1Н), 8.76 (s, 1Н), 8.70 (s, 1Н), 8.11 (s, 1Н), 7.52 (s, 1Н), 4.04 (s, 3Н). MS m/z 201 [М+Н]+.
Стадия 8: Получение 4-(((2S,3R)-3-этил-5-оксопирролидин-2-ил)метокси)-6-метоксиизохинолин-7-карбонитрила
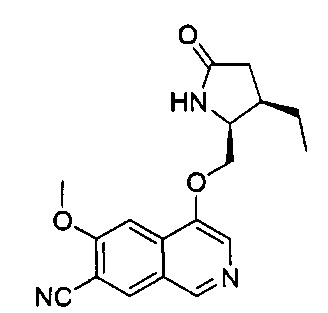
Смесь диизопропилазодикарбоксилата (DIAD) (253 мг, 1,25 ммоль) и трифенилфосфина (328 мг, 1,25 ммоль) в THF (8 мл) перемешивали в течение 10 мин в атмосфере N2. Добавляли 4-гидрокси-6-метоксиизохинолин-7-карбонитрил (100 мг, 0,50 ммоль) и смесь перемешивали в течение примерно 10 мин. К этой смеси добавляли (4R,5S)-4-этил-5-(гидроксиметил)пирролидин-2-он (93 мг, 0,6 ммоль). Смесь перемешивали и нагревали до 65°С в течение 16 ч в атмосфере N2. Реакционную смесь концентрировали при пониженном давлении и очищали посредством флэш-хроматографии на силикагеле (от 0% до 15% МеОН в EtOAc) с получением целевого продукта (106 мг, 57%-ная чистота согласно Н-ЯМР, ~37%-ный выход) в виде бледно-желтого твердого вещества. MS m/е 325,9 [М+Н]+.
Стадия 9: Получение 4-(((2S,3R)-3-этил-5-оксопирролидин-2-ил)метокси)-6-метоксиизохинолин-7-карбоксамида
К раствору 4-(((2S,3R)-3-этил-5-оксопирролидин-2-ил)метокси)-6-метоксиизохинолин-7-карбонитрила (106 мг, 0,19 ммоль) в DMSO (4 мл) добавляли K2CO3 (128 мг, 0,93 ммоль) и Н2О2 (147 мг, 30% масс./масс. раствор в воде, 1,30 ммоль) при 20°С. Реакционную смесь перемешивали при 20°С в течение 2 ч и затем разбавляли водой (20 мл) и экстрагировали смесью 10:1 DCM/MeOH (4×25 мл). Объединенную органическую фазу промывали рассолом, сушили над Na2SO4, фильтровали и концентрировали с получением неочищенного продукта, который очищали посредством HPLC. Колонка: Phenomenex Gemini C18 250×21,2 мм×8 мкм. Время градиента: 10 мин. Подвижная фаза: от 19% MeCN в воде (аммиак) до 39% MeCN в воде (аммиак). Скорость потока: 30 мл/мин. Длина волны: 220 нм; целевые фракции концентрировали при пониженном давлении с получением целевого продукта (20 мг) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, MeOD) δ 8.86 (s, 1Н), 8.59 (s, 1H), 8.08 (s, 1H), 7.54 (s, 1H), 4.46 (dd, 1H), 4.25 (dd, 1H), 4.14-4.07 (m, 2H), 4.11 (s, 3H), 2.78-2.63 (m, 1H), 2.58-2.46 (m, 1H), 2.45-2.33 (m, 1H), 1.80-1.67 (m, 1H), 1.60-1.45 (m, 1H), 1.03 (t, 3H). MS m/e 344,1 [M+H]+.
Пример 25
4-(((2S,3S,4S)-3-этил-4-фтор-5-оксопирролидин-2-ил)метокси)-6-метоксиизохинолин-7-карбоксамид

Стадия 1: Получение 4-(((2S,3S,4S)-3-этил-4-фтор-5-оксопирролидин-2-ил)метокси)-6-метоксиизохинолин-7-карбонитрила
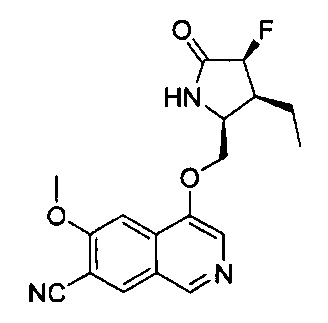
Смесь диизопропилазодикарбоксилата (DIAD) (253 мг, 1,25 ммоль) и трифенилфосфина (328 мг, 1,25 ммоль) в THF (8 мл) перемешивали в течение 10 мин в атмосфере N2. Добавляли 4-гидрокси-6-метоксиизохинолин-7-карбонитрил (100 мг, 0,50 ммоль) и смесь перемешивали в течение примерно 10 мин. Добавляли (3S,4S,5S)-4-этил-3-фтор-5-(гидроксиметил)пирролидин-2-он (161 мг, 0,99 ммоль) и смесь перемешивали при 65°С в течение 16 ч в атмосфере N2. Реакционную смесь концентрировали при пониженном давлении с получением остатка, который очищали посредством флэш-хроматографии на силикагеле (от 100% EtOAc до 15% МеОН в EtOAc) с получением целевого продукта (25 мг, 4%-ный выход) в виде бледно-желтого масла. MS т/е 343,9 [М+Н]+.
Стадия 2: Получение 4-(((2S,3S,4S)-3-этил-4-фтор-5-оксопирролидин-2-ил)метокси)-6-метоксиизохинолин-7-карбоксамида
К раствору 4-(((2S,3S,4S)-3-этил-4-фтор-5-оксопирролидин-2-ил)метокси)-6-метоксиизохинолин-7-карбонитрила (25 мг, 0,051 ммоль) в DMSO (1 мл) добавляли K2CO3 (35,2 мг, 0,255 ммоль) и Н2О2 (40,5 мг, 30% масс./масс. раствора в воде, 0,357 ммоль). Реакционную смесь перемешивали в течение 2 ч. Реакционную смесь разбавляли водой (15 мл) и экстрагировали смесью 10:1 DCM/MeOH (4×20 мл). Объединенную органическую фазу промывали рассолом, сушили над Na2SO4, фильтровали и концентрировали с получением неочищенного продукта, который очищали посредством HPLC. Колонка: Phenomenex Gemini C18 250×21,2 мм×8 мкм. Время градиента: 11 мин. Подвижная фаза: от 19% MeCN в воде (аммиак) до 39% MeCN в воде (аммиак). Скорость потока: 35 мл/мин. Длина волны: 220 нм. Целевые фракции концентрировали при пониженном давлении с получением целевого продукта (15 мг) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, MeOD) δ 8.85 (s, 1H), 8.59 (s, 1Н), 8.04 (s, 1H), 7.80 (s, 1H), 5.07 (d, 0.5H), 4.94 (d, 0.5H), 4.31 (d, 2H), 4.21 (dd, 1H), 2.85-2.67 (m, 1H), 1.88-1.64 (m, 2H), 1.12 (t, 3H). MS m/e 383,9 [M+Na]+.
Пример 26
5-(((2S,3S,4S)-3-этил-4-фтор-5-оксопирролидин-2-ил)метокси)-3-метокси-2-нафтамид

Стадия 1: Получение 5-гидрокси-3-метокси-2-нафтонитрила

К перемешиваемой суспензии 5-гидрокси-3-метокси-2-нафтамида (233 мг, 1,07 ммоль) в 1,4-диоксане (10 мл) по каплям добавляли пиридин (679 мг, 8,58 ммоль). По каплям добавляли TFAA (901 мг, 4,29 ммоль) на протяжении 10 мин в атмосфере N2. Реакционную смесь перемешивали в течение 2 ч в атмосфере N2. Реакционную смесь разбавляли EtOAc (100 мл), промывали водой (2×50 мл), рассолом (50 мл), сушили над Na2SO4, фильтровали и концентрировали с получением 5-гидрокси-3-метокси-2-нафтонитрила (210 мг, 98%-ный выход) в виде оранжевого твердого вещества. 1Н-ЯМР (400 МГц, DMSO-d6) δ 10.43 (s, 1H), 8.43 (s, 1H), 7.57 (s, 1H), 7.40 (d, 1H), 7.33-7.23 (m, 1H), 7.01 (d, 1H), 3.99 (s, 3H).
Стадия 2: Получение [(2S,3S,4S)-3-этил-4-фтор-5-оксопирролидин-2-ил]метил-метансульфоната
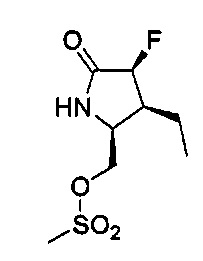
К раствору (3S,4S,5S)-4-этил-3-фтор-5-(гидроксиметил)пирролидин-2-она (400 мг, 2,48 ммоль) в DCM (25 мл) добавляли метансульфонилхлорид (398 мг, 3,47 ммоль) и TEA (502 мг, 4,96 ммоль) при 0°С в атмосфере N2. Реакционную смесь перемешивали в атмосфере N2 в течение 1 ч при 20°С. Смесь разбавляли DCM (80 мл), промывали насыщенным раствором NaHCO3 (40 мл), рассолом (40 мл), сушили над Na2SO4, фильтровали и концентрировали с получением целевого продукта (580 мг, ~98%-ный выход) в виде бледно-желтого масла, которое использовали без дополнительной очистки. 1Н-ЯМР (400 МГц, CDCl3) δ 6.95 (br. s., 1Н), 4.87 (d, 0.5H), 4.74 (d, Hz, 0.5H), 4.40 (dd, Hz, 1H), 4.11 (t, 1H), 4.04-3.94 (m, 1H), 2.57-2.48 (m, 1H), 2.48-2.37 (m, 1H), 1.76-1.48 (m, 1H), 1.63 (s, 3H), 1.09 (t, 3H).
Стадия 3: Получение 5-(((2S,3S,4S)-3-этил-4-фтор-5-оксопирролидин-2-ил)метокси)-3-метокси-2-нафтонитрила
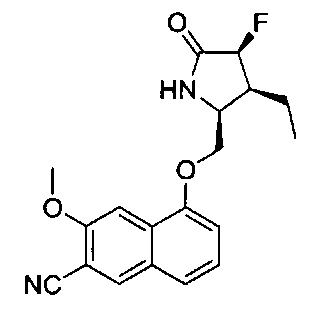
К раствору 5-гидрокси-3-метокси-2-нафтонитрила (350 мг, 1,76 ммоль) в сухом DMF (20 мл) добавляли [(2S,3S,4S)-3-этил-4-фтор-5-оксопирролидин-2-ил]метилметансульфонат (580 мг, 2,42 ммоль) и K2CO3 (486 мг, 3,51 ммоль). Смесь перемешивали при 60°С в течение 6 ч и затем разбавляли EtOAc (160 мл), промывали рассолом (3×60 мл), водой (60 мл) и рассолом (60 мл) и сушили над MgSO4. Неочищенный продукт очищали посредством хроматографии на силикагеле с использованием смеси петролейный эфир/EtOAc (от 2:1 до 1:4) с получением целевого продукта (370 мг, 61%-ный выход) в виде бледно-желтого твердого вещества. 1Н-ЯМР (400 МГц, CDCl3) δ 8.02 (s, 1Н), 7.57 (s, 1H), 7.49 (br. s., 1H), 7.40-7.34 (m, 1H), 7.32-7.28 (m, 1H), 6.84 (d, 1H), 5.00-4.80 (m, 1H), 4.20 (d, 2H), 4.17-4.08 (m, 1H), 3.98 (s, 3H), 2.71-2.47 (m, 1H), 1.86-1.73 (m, 1H), 1.69-1.54 (m, 1H), 1.11 (t, 3H). MS m/e 342,9 [M+H]+.
Стадия 4: Получение 5-{[(2S,3S,4S)-3-этил-4-фтор-5-оксопирролидин-2-ил]метокси}-3-метоксинафталин-2-карбоксамида
К раствору 5-{[(2S,3S,4S)-3-этил-4-фтор-5-оксопирролидин-2-ил]метокси}-3-метоксинафталин-2-карбоксамида (430 мг, 1,26 ммоль) в DMSO (5 мл) добавляли K2CO3 (868 мг, 6,28 ммоль) и Н2О2 (997 мг, 30% масс./масс. раствор в воде, 8,79 ммоль). Через 2 ч реакционную смесь разбавляли DCM (120 мл) и промывали рассолом (2×40 мл), водой (30 мл) и рассолом (30 мл) и сушили над Na2SO4, фильтровали и концентрировали с получением неочищенного продукта, который растирали с DCM (10 мл). Смесь фильтровали и осадок на фильтре промывали водой (2×8 мл) и DCM (6 мл). Осадок собирали и сушили в вакууме с получением целевого продукта (330 мг, 73%-ный выход) в виде не совсем белого твердого вещества. 1Н-ЯМР (400 МГц, DMSO-d6) δ 8.83 (s, 1Н), 8.26 (s, 1H), 7.78 (br. s., 1H), 7.73 (s, 1H), 7.63 (br. s., 1H), 7.52 (d, 1H), 7.29 (t, 1H), 7.02 (d, 1H), 5.03-4.80 (m, 1H), 4.20-4.00 (m, 3H), 3.96 (s, 3H), 2.72-2.53 (m, 1H), 1.71-1.53 (m, 2), 1.01 (t, 3H). MS m/e 360,9 [M+H]+. MS m/e 382,8 [M+Na]+.
Пример 27
(3S,6R)-5-оксо-2,3,4,5,6,7,9,10-октагидро-12,14-(этандиилиден)-3,6-метанопиридо[2,3-1][1,4,11,8]триоксазациклопентадецин-19-карбоксамид

Стадия 1: Получение (6R,7aS)-6-((2-хлорэтокси)метил)-3,3-диметилтетрагидро-3Н,5Н-пирроло[1,2-с]оксазол-5-она

К перемешиваемому раствору (S)-3,3-диметилтетрагидро-3Н,5Н-пирроло[1,2-с]оксазол-5-она (5 г, 32,2 ммоль) в сухом THF (100 мл) добавляли LDA (2 М раствор в смеси THF/гептан/этилбензол, 20 мл, 40,3 ммоль) при -78°С в атмосфере азота. Через 30 мин по каплям добавляли 1-хлор-2-(хлорметокси)этан (3,58 мл, 35,5 ммоль) и перемешивали 10 мин при -78°С. Смесь оставляли нагреваться до комнатной температуры и перемешивали в течение 1 ч и затем гасили смесью EtOAc : вода (1:1). Водный слой экстрагировали EtOAc. Органическую фазу промывали рассолом и сушили над Na2SO4. Остаток очищали посредством колоночной хроматографии на силикагеле (0-20% EtOAc-гексан) с получением (6R,7aS)-6-((2-хлорэтокси)метил)-3,3-диметилтетрагидро-3Н,5Н-пирроло[1,2-с]оксазол-5-она (1,25 г, 16%) и изомера (1,1 г, 14%) и желтой жидкости. 1Н-ЯМР (400 МГц, CDCl3) δ 4.15-4.05 (m, 2Н), 3.76-3.65 (m, 4Н), 3.60-3.57 (m, 2Н), 3.45 (t, 1Н), 3.04-2.98 (m, 1Н), 2.32-2.25 (m, 1Н), 1.87-1.79 (m, 1Н), 1.62 (s, 3Н), 1.45 (s, 3Н). MS m/z 248,2 [М+Н]+.
Стадия 2: Получение (3R,5S)-3-((2-хлорэтокси)метил)-5-(гидроксиметил)пирролидин-2-она

(6R,7aS)-6-((2-хлорэтокси)метил)-3,3-диметилтетрагидро-3Н,5Н-пирроло[1,2-с]оксазол-5-он (734 мг, 2,96 ммоль) растворяли в CH3CN/H2O (9 мл/1 мл). Добавляли n-TsOH (28 мг, 0,15 ммоль) и реакционную смесь нагревали до ~90°С. Через 1 ч реакционную смесь охлаждали до температуры окружающей среды, концентрировали и азеотропно отгоняли с CH3CN. Остаток очищали посредством хроматографии на силикагеле (5-20% MeOH/DCM) с получением указанного в заголовке соединения (581 мг). 1Н-ЯМР (400 МГц, CDCl3) δ 6.22 (br. s., 1H), 3.87-3.67 (m, 5Н), 3.65-3.59 (m, 2Н), 3.59-3.49 (m, 1Н), 2.77-2.64 (m, 1H), 2.44-2.31 (m, 2H), 2.24 (t, 1H), 1.91-1.77 (m, 1H) MS m/z 207,9 [M+H]+.
Стадия 3: Получение 1-хлор-7-гидроксиизохинолин-6-карбонитрила

К перемешиваемому раствору 1-хлор-7-изопропоксиизохинолин-6-карбонитрила (2 г, 8,13 ммоль) в DCM (25 мл) добавляли AlCl3 (3,44 г, 25,8 ммоль) и смесь нагревали на 50°С-ной бане в течение 16 ч. Реакционную смесь упаривали при пониженном давлении и обрабатывали ледяной водой с получением твердого вещества, которое фильтровали, промывали водой и сушили с получением 1-хлор-7-гидроксиизохинолин-6-карбонитрила (1,4 г, 84%-ный выход) в виде светло-желтого твердого вещества. 1Н-ЯМР (400 МГц, DMSOd-6) δ 11.95 (s, 1Н), 8.65 (s, 1H), 8.22 (d, 1H), 7.85 (d, 1H), 7,68 (s, 1H). MS m/z 205,2 [M+H]+.
Стадия 4: Получение 1-хлор-7-((2-(триметилсилил)этокси)метокси)изохинолин-6-карбонитрила

Раствор 1-хлор-7-гидроксиизохинолин-6-карбонитрила (1 г, 4,9 ммоль) в DCM (12 мл) обрабатывали DIEA (1,3 мл, 5,86 ммоль). Через 10 мин по каплям добавляли SEM хлорид (0,99 мл, 5,38 ммоль). Реакционную смесь гасили насыщенным водным раствором NaHCO3, затем вливали в NaHCO3 и дважды экстрагировали EtOAc. Объединенную органическую фазу сушили над MgSO4. Хроматография на силикагеле (градиент 10-30% EtOAc/гептан) давала указанное в заголовке соединение (1,20 г, %-ный выход). 1Н-ЯМР (400 МГц, CDCl3) δ 8.30 (d, 1H), 8.20 (s, 1H), 8.05 (s, 1H), 7.58 (d, 1H), 5.52 (s, 2H), 3.92-3.85 (m, 2H), 1.05-0.96 (m, 2H), 0.02 (s, 9H). MS m/z 334,1 [M+H]+.
Стадия 5: Получение 1-(((2S,4R)-4-((2-хлорэтокси)метил)-5-оксопирролидин-2-ил)метокси)-7-((2-(триметилсилил)этокси)метокси)изохинолин-6-карбонитрила

1-Хлор-7-((2-(триметилсилил)этокси)метокси)изохинолин-6-карбонитрил (1,04 г, 3,09 ммоль) и (3R,5S)-3-((2-хлорэтокси)метил)-5-(гидроксиметил)пирролидин-2-он (710 мг, 3,42 ммоль) растворяли в DMF (10 мл) и охлаждали до 0°С. По каплям добавляли KHMDS (6,81 мл, 1 М раствор в толуоле). Через 15 мин реакционную смесь гасили сначала водой (~7 мл), затем 10%-ным водным раствором NaH2PO4 (~4 мл) и дважды экстрагировали EtOAc. Органическую фазу концентрировали и остаток очищали посредством хроматографии на силикагеле (градиент 50-100% EtOAc/гептан) с получением желтого твердого вещества (689 мг). 1Н-ЯМР (400 МГц, MeOD) δ 8.29 (s, 1Н), 8.01 (s, 1H), 7.96 (d, 1H), 7.34 (d, 1H), 5.60-5.49 (m, 2H), 4.63 (dd, 1H), 4.46 (dd, 1H), 4.23-4.14 (m, 1H), 3.91 (t, 2H), 3.82-3.76 (m, 1H), 3.76-3.67 (m, 3H), 3.66-3.61 (m, 2H), 2.83-2.74 (m, 1H), 2.60-2.49 (m, 1H), 2.09-1.98 (m, 1H), 0.99 (t, 2H), -0.01 (s, 9H). MS m/z 528,2 [M+Na]+.
Стадия 6: Получение 1-(((2S,4R)-4-((2-хлорэтокси)метил)-5-оксопирролидин-2-ил)метокси)-7-гидроксиизохинолин-6-карбонитрила
1-(((2S,4R)-4-((2-хлорэтокси)метил)-5-оксопирролидин-2-ил)метокси)-7-((2-(триметилсилил)этокси)метокси)изохинолин-6-карбонитрил (480 мг, 0,95 ммоль) суспендировали в МеОН (7 мл) и охлаждали до 0°С. Добавляли раствор конц. HCl (1,5 мл) в МеОН (3 мл). Реакционную смесь нагревали до температуры окружающей среды и перемешивали в течение ночи. Реакционную смесь аккуратно гасили насыщенным водным раствором NaHCO3. Смесь частично концентрировали, затем вливали в воду (20 мл) и доводили до рН 6-7 с помощью 1 н. HCl. Этот раствор дважды экстрагировали EtOAc и сушили над MgSO4. Остаток очищали посредством хроматографии на силикагеле, элюируя градиентом 0-5% МеОН/CH2Cl2, с получением указанного в заголовке соединения (323 мг, 91%-ный выход) в виде масла. 1Н-ЯМР (400 МГц, CDCl3) δ=10.03 (br. s., 1Н), 8.03 (s, 1Н), 7.88 (d, 1Н), 7.82 (s, 1H), 7.35 (s, 1Н), 7.18 (d, 1Н), 4.81 (dd, 1Н), 4.27-4.19 (m, 1Н), 4.14 (q, 1Н), 3.86 (dd, 1Н), 3.75-3.65 (m, 3Н), 3.61-3.48 (m, 2Н), 2.86 (tt, 1Н), 2.56-2.44 (m, 1Н), 2.01 (td, 1Н) MS m/z 373.9 [М-Н]+ и 375,9 [М+Н]+.
Стадия 7: Получение (3S,6R)-5-оксо-2,3,4,5,6,7,9,10-октагидро-12,14-(этандиилиден)-3,6-метанопиридо[2,3-1][1,4,11,8]триоксазациклопентадецин-19-карбонитрила.

1-(((2S,4R)-4-((2-Хлорэтокси)метил)-5-оксопирролидин-2-ил)метокси)-7-гидроксиизохинолин-6-карбонитрил (100 мг, 0,266 ммоль) растворяли в THF (90 мл). Добавляли NaI (40,2 мг, 0,266 ммоль). Добавляли KOtBu (0,56 мл, 0,56 ммоль) и через нескольких минут добавляли DMF (10 мл) и смесь нагревали до 50-55°С в течение 24 ч. Реакционную смесь гасили 10%-ным водным NaH2PO4 (~4 мл), затем добавляли воду и THF удаляли в вакууме. Остаток распределяли между водой и EtOAc и экстрагировали EtOAc, и объединенную органическую фазу сушили над MgSO4, фильтровали и концентрировали. Хроматографировали на силикагеле, элюируя градиентом 10-60% ацетон/CH2Cl2, с получением не совсем белого твердого вещества (14,7 мг), которое очищали на силикагеле, элюируя градиентом от 0 до 10% EtOAc/MeOH, с получением указанного в заголовке соединения (5,9 мг, 6,5%-ный выход). 1Н-ЯМР (400 МГц, DMSO-d6) δ=11.45 (br. s., 1Н), 8.71 (s, 1H), 8.43 (s, 1H), 7.97 (d, 1H), 7.65 (s, 1H), 7.39 (d, 1H), 4.84 (d, 1H), 4.71-4.62 (m, 1H), 4.53-4.45 (m, 1H), 4.23 (d, 1H), 3.91 (t, 1H), 3.81 (d, 2H), 3.77 (d, 1H), 3.49 (d, 1H), 2.69-2.57 (m, 1H), 2.19-2.08 (m, 1H). MS m/z 340,2 [M+H]+.
Стадия 8: Получение (3S,6R)-5-оксо-2,3,4,5,6,7,9,10-октагидро-12,14-(этандиилиден)-3,6-метанопиридо[2,3-1][1,4,11,8]триоксазациклопентадецин-19-карбоксамида.
К раствору (3S,6R)-5-оксо-2,3,4,5,6,7,9,10-октагидро-12,14-(этандиилиден)-3,6-метанопиридо[2,3-1][1,4,11,8]триоксазациклопентадецин-19-карбонитрила (5,9 мг, 0,017 ммоль) в DMSO-d6 (1,0 мл) добавляли K2CO3 (9,6 мг, 0,068 ммоль). Суспензию перемешивали в течение ~5 мин, затем добавляли перекись водорода (0,01 мл). Добавляли дополнительно 2 капли раствора H2O2 (~0,03 мл) и добавляли K2CO3 (~15 мг). Через 1,5 ч добавляли дополнительную каплю раствора H2O2 (0,015 мл). Через 1 ч реакционную смесь гасили Me2S и перемешивали в течение примерно 15 мин. Реакционную смесь фильтровали и очищали посредством HPLC с получением указанного в заголовке соединения (1,8 мг, 30%-ный выход). Условия HPLC: Остаток растворяли в DMSO (1 мл) и очищали посредством обращенно-фазной HPLC. Колонка: Waters XBridge C18 19×100, 5 мкм; Подвижная фаза А: 0,03% NH4OH в воде (об./об.); Подвижная фаза В: 0,03% NH4OH в ацетонитриле (об./об.); Градиент: 95,0% Н2О/5,0% ацетонитрил линейный до 60,0% Н2О/40,0% ацетонитрил в течение 10,5 мин, 60,0% Н2О/40,0% ацетонитрил линейный до 0% Н2О/100% ацетонитрил в течение 0,5 мин. Выдерживание при 0% Н2О/100% ацетонитрил от 11,0 до 12,0 мин. Поток: 25 мл/мин. Аналитическая QC Колонка: Waters Atlantis dC18 4,6×50, 5 мкм; Подвижная фаза А: 0,05% TFA в воде (об./об.); Подвижная фаза В: 0,05% TFA в ацетонитриле (об./об.); Градиент: 95,0% Н2О/5,0% ацетонитрил линейный до 5% H2O/95% ацетонитрил в течение 4,0 мин, Выдерживание при 5% H2O/95% Ацетонитрил до 5,0 мин. Поток: 2 мл/мин. Время удерживания 1,75 мин. 1Н-ЯМР (600 МГц, DMSO-d6) δ 8.66 (s, 1Н), 8.13 (s, 1H), 7.86 (d, 1H), 7.84 (br. s., 1H), 7.66 (s, 1H), 7.61 (br. s., 1H), 7.38 (d, 1H), 7.10-7.04 (m, 1H), 4.78 (d, 1H), 4.59 (dd, 1H), 4.43 (dd, 1H), 4.20 (d, 1H), 3.92 (t, 1H), 3.85-3.75 (m, 2H), 3.59-3.52 (m, 1H), 3.49 (dd, 1H), 3.43 (1H скрыт водой), 2.66-2.60 (m, 1H), 2.57-2.51 (m, 1H), 2.18-2.09 (m, 1H). MS m/z 358,1 [M+H]+.
Пример 28
7-метокси-1-[(3-оксо-2-азабицикло[3.1.0]гекс-1-ил)метокси]изохинолин-6-карбоксамид

Стадия 1: Получение N-(4-метоксибензил)бут-3-ен-1-амина 
К раствору бут-3-ен-1-амина (1,89 мл, 20 ммоль) в EtOH (40 мл) добавляли 4-метоксибензальдегид (2,48 мл, 20 ммоль). Через 15 мин одной порцией добавляли NaBH3CN (1,55 г, 24 ммоль). Через 2 ч добавляли другую порцию NaBH3CN (1,55 г, 24 ммоль) и перемешивание продолжали в течение 4 ч. Затем добавляли высушенные в печи порошкообразные  молекулярные сита (3 г) и смесь перемешивали в течение ночи. Смесь фильтровали через Celite® и осадок на фильтре промывали МеОН. Растворитель выпаривали при пониженном давлении и полученное неочищенное желтое масло очищали с использованием колоночной хроматографии на силикагеле, элюируя 10%-ным МеОН в DCM, с получением указанного в заголовке соединения в виде бледно-желтого масла (2,69 г, 70%-ный выход). 1Н-ЯМР (CDCl3, 400 МГц): δ 7.38 (d, 2Н), 6.93 (d, 2Н), 5.71 (ddt, 1Н), 5.21 (dd, 1Н), 5.18-5.16 (m, 1Н), 4.05 (s, 2Н), 3.79 (s, 3Н), 2.92 (t, 2Н), 2.51-2.46 (m, 2Н). MS m/z 192 [М+Н]+
молекулярные сита (3 г) и смесь перемешивали в течение ночи. Смесь фильтровали через Celite® и осадок на фильтре промывали МеОН. Растворитель выпаривали при пониженном давлении и полученное неочищенное желтое масло очищали с использованием колоночной хроматографии на силикагеле, элюируя 10%-ным МеОН в DCM, с получением указанного в заголовке соединения в виде бледно-желтого масла (2,69 г, 70%-ный выход). 1Н-ЯМР (CDCl3, 400 МГц): δ 7.38 (d, 2Н), 6.93 (d, 2Н), 5.71 (ddt, 1Н), 5.21 (dd, 1Н), 5.18-5.16 (m, 1Н), 4.05 (s, 2Н), 3.79 (s, 3Н), 2.92 (t, 2Н), 2.51-2.46 (m, 2Н). MS m/z 192 [М+Н]+
Стадия 2: Получение метил-[бут-3-ен-1-ил(4-метоксибензил)амино](оксо)ацетата

N-(4-Метоксибензил)бут-3-ен-1-амин (6,87 г, 35,92 ммоль) растворяли в DCM (40 мл) и добавляли водный насыщенный NaHCO3 (120 мл). По каплям добавляли метилхлор(оксо)ацетат (13,20 г, 108 ммоль) при интенсивном перемешивании на протяжении 5 мин. Смесь перемешивали в течение 2 ч. Водный слой экстрагировали DCM и объединенные органические экстракты сушили над Na2SO4. Соединение получали в виде бледно-желтого масла (7,18 г, 72%-ный выход) в виде смеси двух ротамеров в соотношении 1:1, и его использовали без очистки. 1H-ЯМР (CDCl3, 400 МГц): δ 7.23 (d, 1Н), 7.21 (d, 1Н), 6.90 (d, 1Н), 6.88 (d, 1Н), 5.79-5.71 (m, 0.5Н), 5.71-5.62 (m, 0.5Н), 5.10-5.06 (m, 1Н), 5.05-5.01 (m, 1Н), 4.58 (s, 1Н), 4.39 (s, 1Н), 3.89 (s, 1.5Н), 3.87 (s, 1.5Н), 3.82 (s, 1.5Н), 3.81 (s, 1.5H), 3.35 (dd, 1H), 3.25-3.21 (m, 1H), 2.35-2.30 (m, 1H), 2.30-2.25 (m, 1H). MS m/z 278 [M+H]+
Стадия 3: Получение N-(бут-3-ен-1-ил)-2-гидрокси-N-(4-метоксибензил)ацетамида

К раствору метил [бут-3-ен-1-ил(4-метоксибензил)амино](оксо)ацетата (4,25 г, 15,3 ммоль) в МеОН (61,3 мл) порциями добавляли боргидрид натрия (3,00 г, 79,2 ммоль). Реакция была экзотермической. После окончания добавления реакционную смесь перемешивали, пока температура не возвращалась к комнатной. МеОН удаляли при пониженном давлении и полученную суспензию распределяли в смеси DCM/насыщенный водный раствор NH4Cl (40 мл, 1:1 об./об.). Затем добавляли воду. Водный слой экстрагировали DCM и объединенные органические экстракты сушили над Na2SO4, фильтровали и упаривали досуха с получением указанного в заголовке соединения в виде бесцветного масла (3,70 г, ~97%-ный выход) в виде смеси двух ротамеров в соотношении 1:1. Это вещество использовали без дополнительной очистки. 1Н-ЯМР (CDCl3, 400 МГц): δ 7.20 (d, 1Н), 7.08 (d, 1Н), 6.90 (d, 1H), 6.87 (d, 1Н), 5.82-5.73 (m, 0.5Н), 5.72-5.63 (m, 0.5Н), 5.10 (d, 1Н), 5.07-5.02 (m, 1Н), 4.62 (s, 1Н), 4.29 (s, 1Н), 4.22 (d, 1Н), 4.21 (d, 1Н), 3.82 (s, 1.5Н), 3.81 (s, 1.5Н), 3.68 (t, 0.5Н), 3.65 (t, 0.5Н), 3.51-3.45 (m, 1Н), 3.13-3.08 (m, 1Н), 2.35-2.30 (m, 1Н), 2.29-2.24 (m, 1Н). MS m/z 250 [М+Н]+
Стадия 4: Получение N-(бут-3-ен-1-ил)-2-{[трет-бутил(диметил)силил]окси}-N-(4-метоксибензил)ацетамида

К раствору N-(бут-3-ен-1-ил)-2-гидрокси-N-(4-метоксибензил)ацетамида (6,00 г, 24,1 ммоль) в DCM (96,3 мл) добавляли имидазол (2,47 г, 36,10 ммоль), затем TBDMSCl (4,49 г, 28,9 ммоль) и реакционную смесь перемешивали в течение ночи. Добавляли воду и водную фазу экстрагировали DCM. Объединенные органические экстракты промывали водой, сушили над Na2SO4 и концентрировали при пониженном давлении. Остаток растворяли в МеОН и концентрировали. Неочищенное масло очищали с использованием колоночной хроматографии на силикагеле, элюируя смесью гептан/EtOAc, с получением указанного в заголовке соединения в виде бесцветного масла (7,72 г, 88%-ный выход) в виде смеси двух ротамеров в соотношении 1:1. 1Н-ЯМР (CDCl3, 400 МГц): δ 7.19 (d, 1Н), 7.12 (d, 1Н), 6.88 (d, 1Н), 6.85 (d, 1Н), 5.80-5.74 (m, 0.5Н), 5.74-5.67 (m, 0.5Н), 5.06 (d, 1Н), 5.04-4.98 (m, 1H), 4.56 (s, 1Н), 4.52 (s, 1Н), 4.37 (s, 1Н), 4.34 (s, 1Н), 3.82 (s, 1.5Н), 3.81 (s, 1.5Н), 3.38 (t, 1Н), 3.29 (t, 1H), 2.33-2.29 (m, 1H), 2.29-2.26 (m, 1H), 0.93 (s, 4.5H), 0.89 (s, 4.5H), 0.14 (s, 3H), 0.09 (s, 3H).
Стадия 5: Получение 1-({[трет-бутил(диметил)силил]окси}метил)-2-(4-метоксибензил)-2-азабицикло[3.1.0]гексана

В высушенную колбу загружали N-(бут-3-eн-1-ил)-2-{[трет-бутил(диметил)силил]окси}-N-(4-метоксибензил)ацетамид (4,00 г, 11,0 ммоль) и помещали в инертную атмосферу. Добавляли сухой THF (110 мл) и к хорошо перемешиваемому раствору добавляли изопропилат титана (4,69 г, 16,5 ммоль), затем с помощью шприцевого насоса по каплям добавляли циклопентилмагния бромид (22,0 мл, 2,0 М в диэтиловом эфире, 44,0 ммоль) на протяжении 60 мин. Через 2 ч реакцию гасили холодным раствором соли Рошели и экстрагировали EtOAc. Объединенные органические экстракты промывали водой и рассолом и сушили над Na2SO4. После фильтрации летучие вещества удаляли при пониженном давлении с получением неочищенного продукта. Неочищенное масло очищали с использованием колоночной хроматографии на силикагеле, элюируя смесью гептан/EtOAc, с получением указанного в заголовке соединения в виде бесцветного масла (2,00 г, 52%-ный выход). 1Н-ЯМР (CDCl3, 400 МГц): δ 7.27 (d, 2Н), 6.84 (d, 2Н), 4.18 (d, 1Н), 4.05 (d, 1Н), 3.80 (s, 3Н), 3.69 (d, 1Н), 3.18 (d, 1Н), 2.89-2.81 (m, 1Н), 1.97-1.84 (m, 2Н), 1.77-1.68 (m, 1Н), 1.29 (td, 1Н), 0.89 (s, 9Н), 0.85 (t, 1Н), 0.49 (dd, 1Н), 0.06 (s, 6Н). MS m/z 348 [М+Н]+
Стадия 6: Получение 2-азабицикло[3.1.0]гекс-1-илметанола гидрохлорида
При 0°С в инертной атмосфере к раствору 1-({[трет-бутил(диметил)силил]окси}метил)-2-(4-метоксибензил)-2-азабицикло[3.1.0]гексана (2,00 г, 5,75 ммоль) в 1,2-дихлорэтане (19,2 мл) добавляли АСЕ-Cl (1,08 г, 7,48 ммоль) и смесь перемешивали при 0°С в течение 30 мин. Летучие вещества удаляли при пониженном давлении и полученное неочищенное вещество солюбилизировали в МеОН (29 мл). Смесь нагревали при 50°С в течение 2 ч и летучие вещества выпаривали при пониженном давлении. Полученную коричневую смолу растирали со смесью DCM/гептан (3:1) и надосадочную жидкость отбрасывали. Эту операцию повторяли 5 раз, и указанный в заголовке продукт получали в виде бледно-коричневого твердого вещества (860 мг, 99%-ный выход) и использовали без дополнительной очистки. 1Н-ЯМР (DMSO-d6, 400 МГц): δ 9.45 (br. s., 1Н), 9.23 (br. s., 1Н), 5.33 (br. s., 1H), 3.83-3.76 (m, 1H), 3.65 (d, 1H), 3.30-3.24 (m, 1H), 2.94-2.81 (m, 1H), 2.08-1.91 (m, 2H), 1.65 (td, 1H), 1.10-1.05 (m, 1H), 0.87-0.80 (m, 1H).
Стадия 7: Получение трет-бутил-1-({[трет-бутил(диметил)силил]окси}метил)-2-азабицикло[3.1.0]гексан-2-карбоксилата

К раствору неочищенного 2-азабицикло[3.1.0]гекс-1-илметанола гидрохлорида (860 мг, 5,75 ммоль) в DCM (28,7 мл) добавляли TEA (640 мг, 6,32 ммоль), затем N,N-диметилпиридин-4-амин (353 мг, 2,87 ммоль) и BOC2O (1,42 г, 6,32 ммоль). Смесь перемешивали в течение 24 ч и затем добавляли имидазол (472 мг, 6,90 ммоль), после чего TBDMSC1 (983 мг, 6,32 ммоль). Смесь перемешивали при комнатной температуре в течение ночи. Реакцию гасили насыщенным водным NH4Cl и экстрагировали DCM. Объединенные органические экстракты сушили над Na2SO4. После фильтрации летучие вещества удаляли при пониженном давлении с получением неочищенного продукта. Остаток очищали с использованием колоночной хроматографии на силикагеле, элюируя смесью гептан/EtOAc, с получением указанного в заголовке соединения в виде бледно-желтого масла (1,52 г, 81%-ный выход). 1Н-ЯМР (CDCl3, 400 МГц): δ 4.32 (br. s., 1Н), 3.71-3.60 (m, 2H), 3.45 (br. s., 1H), 2.16-2.05 (m, 1H), 1.82-1.73 (m, 1H), 1.65-1.57 (m, 1H), 1.47 (s, 9H), 1.05 (dd, 1H), 0.88 (s, 9H), 0.64 (t, 1H), 0.04 (s, 6H).
Стадия 8: Получение трет-бутил-1-({[трет-бутил(диметил)силил]окси}метил)-3-оксо-2-азабицикло[3.1.0]гексан-2-карбоксилата
Натрия метаперйодат (989 мг, 4,58 ммоль) растворяли в воде (25 мл) в атмосфере N2 и добавляли рутения диоксида гидрат (70 мг, 0,46 ммоль). Через 5 мин добавляли трет-бутил-1-({[трет-бутил(диметил)силил]окси}метил)-2-азабицикло[3.1.0]гексан-2-карбоксилат (500 мг, 1,53 ммоль) в виде раствора в EtOAc (25 мл) и полученный двухфазный раствор интенсивно перемешивали в течение 5 ч. Реакционную смесь экстрагировали EtOAc. Объединенные органические слои несколько раз промывали NaHSO3 до получения прозрачного бесцветного органического слоя. Органический слой еще раз промывали рассолом и сушили над Na2SO4. Остаток очищали с использованием колоночной хроматографии на силикагеле, элюируя смесью гептан/EtOAc, с получением указанного в заголовке соединения в виде бесцветного масла (260 мг, 50%-ный выход). 1Н-ЯМР (CDCl3, 400 МГц): δ 4.40 (d, 1Н), 3.58 (d, 1Н), 2.89 (dd, 1Н), 2.49 (d, 1Н), 1.55 (s, 9Н), 1.53-1.45 (m, 1Н), 1.10 (dd, 1Н), 0.88 (s, 9Н), 0.66 (t, 1Н), 0.05 (s, 6Н). MS m/z 242 [М-Вос+Н]+ (Удаление защитной группы Boc в условиях LCMS).
Стадия 9: Получение трет-бутил-(3-оксо-2-азабицикло[3.1.0]гекс-1-ил)метилкарбоната

К раствору трет-бутил-1-({[трет-бутил(диметил)силил]окси}метил)-3-оксо-2-азабицикло[3.1.0]гексан-2-карбоксилата (155 мг, 0,45 ммоль) в THF (0,76 мл) при комнатной температуре добавляли TBAF (0,73 мл, 1,0 М в THF, 0,73 ммоль) и смесь перемешивали в течение 30 мин, затем разбавляли EtOAc и водой. Водный слой экстрагировали EtOAc и объединенные органические слои сушили над Na2SO4 и концентрировали с получением указанного в заголовке соединения в виде бледно-желтого масла (103 мг, 99%), которое использовали без дополнительной очистки. 1Н-ЯМР (CDCl3, 400 МГц): δ 6.03 (br. s., 1Н), 4.34 (d, 1H), 4.14 (d, 1H), 2.77 (dd, 1H), 2.35 (d, 1H), 1.67-1.57 (m, 1H), 1.51 (s, 9H), 1.14 (dd, 1H), 0.72 (t, 1H). MS m/z 228 [M+H]+
Стадия 10: Получение 7-метокси-1-[(3-оксо-2-азабицикло[3.1.0]гекс-1-ил)метокси]изохинолин-6-карбонитрила
К раствору трет-бутил-(3-оксо-2-азабицикло[3.1.0]гекс-1-ил)метилкарбоната (103 мг, 0,453 ммоль) в DMF (3,5 мл) добавляли KHMDS (1,36 мл, 1,0 М в THF, 1,36 ммоль) и смесь перемешивали при -10°С в течение 15 мин. Затем раствор 1-хлор-7-метоксиизохинолин-6-карбонитрила (104 мг, 0,48 ммоль) в DMF (1,0 мл) и смесь перемешивали при -10°С в течение 2 ч. Смесь гасили насыщенным водным NH4Cl и разбавляли DCM. Водный слой экстрагировали DCM и объединенные органические слои промывали рассолом и сушили над Na2SO4. Неочищенное вещество очищали с использованием колоночной хроматографии на силикагеле, элюируя смесью DCM/EtOAc, с получением указанного в заголовке соединения в виде желтого твердого вещества (50 мг, 36%-ный выход). 1Н-ЯМР (CDCl3, 400 МГц): δ 8.08 (s, 1Н), 7.95 (d, 1Н), 7.58 (s, 1Н), 7.23 (d, 1Н), 6.60 (br. s., 1Н), 4.93 (d, 1Н), 4.58 (d, 1Н), 4.08 (s, 3Н), 2.82 (dd, 1Н), 2.41 (d, 1Н), 1.79-1.72 (m, 1H), 0.89 (t, 1Н), 0.74 (t, 1Н). MS m/z 310 [М+Н]+
Стадия 11: Получение 7-метокси-1-[(3-оксо-2-азабицикло[3.1.0]гекс-1-ил)метокси]изохинолин-6-карбоксамида

Раствор 7-метокси-1-[(3-оксо-2-азабицикло[3.1.0]гекс-1-ил)метокси]изохинолин-6-карбонитрила (50 мг, 0,16 ммоль) в DMSO (1,6 мл) обрабатывали K2CO3 (112 мг, 0,81 ммоль). Полученную смесь перемешивали в течение 5 мин, после чего к реакционной смеси добавляли перекись водорода (0,064 мл, 50% масс./масс. в воде, 1,13 ммоль). Перемешивание продолжали в течение 5 ч. Реакционную смесь гасили Me2S (80,3 мг, 1,29 ммоль) и перемешивали при комнатной температуре в течение 30 мин, после чего реакционную смесь фильтровали через Celite®. Осадок на фильтре промывали DCM и EtOAc и фильтрат концентрировали при пониженном давлении с получением раствора в DMSO, который сушили потоком азота при 45°С в течение ночи. Неочищенное вещество очищали с использованием колоночной хроматографии на силикагеле, элюируя смесью DCM/MeOH, с получением указанного в заголовке соединения в виде бледно-желтого твердого вещества (33 мг, 62%-ный выход). 1Н-ЯМР (CDCl3 - с одной каплей CD3OD - 400 МГц): δ 8.42 (s, 1Н), 7.76 (d, 1Н), 7.56 (br. s., 1Н), 7.18 (d, 1H), 4.70 (d, 1H), 4.55-4.46 (m, 1H), 4.03-3.98 (m, 3H), 2.68 (dd, 1H), 2.27 (d, 1H), 1.65 (br. s., 1H), 1.22-1.10 (m, 1H), 0.67-0.59 (m, 1H). MS m/z 328 [M+H]+
Это рацемическое вещество (29 мг) разделяли посредством хиральной хроматографии, что приводило к двум энантиомерам.
Энантиомер 1: бледно-желтое твердое вещество, 12 мг (100%-ный э.и. (энантиомерный избыток)), MS m/z 350,1 [M+Na]+. Энантиомер 2: бледно-желтое твердое вещество, 13 мг (99,5%-ный э.и.). MS m/z, 328,1 [М+Н]+. 1Н-ЯМР (400 МГц, DMSO-d6) d=8.58 (s, 1Н), 8.17 (s, 1H), 7.89 (d, 1H), 7.84 (br. s., 1H), 7.69 (br. s, 2H), 7.42 (d, 1H), 4.70 (d, 1H), 4.58 (d, 1H), 4.00 (s, 3H), 2.70-2.62 (m, 1H), 1.74-1.65 (m, 1H), 1.17 (dd1H), 0.61 (t, 1H) один протон был скрыт, предположительно, перекрывался водой.
Пример 29 (Энантиомер 1)
7-метокси-1-([(1S,5S)-3-оксо-2-азабицикло[3.1.0]гекс-1-ил]метокси)изохинолин-6-карбоксамид

Получали в виде бледно-желтого твердого вещества (12 мг). 1Н-ЯМР (CDCl3 - с одной каплей CD3OD - 400 МГц): δ 8.41 (s, 1Н), 7.77 (d, 1Н), 7.56 (br. s., 1Н), 7.18 (d, 1H), 4.71 (d, 1H), 4.57-4.47 (m, 1H), 4.05-3.99 (m, 3H), 2.68 (dd, 1H), 2.26 (d, 1H), 1.63 (br. s., 1H), 1.22-1.11 (m, 1H), 0.66-0.58 (m, 1H). MS m/z 350 [M+Na]+.
Пример 30 (Энантиомер 2)
7-метокси-1-{[(1R,5R)-3-оксо-2-азабицикло[3.1.0]гекс-1-ил]метокси}изохинолин-6-карбоксамид

Получали в виде бледно-желтого твердого вещества (13 мг). 1Н-ЯМР (CDCl3 - с одной каплей CD3OD - 400 МГц): δ 8.39 (s, 1Н), 7.76 (d, 1Н), 7.54 (br. s., 1Н), 7.17 (d, 1H), 4.73 (d, 1H), 4.56-4.47 (m, 1H), 4.03-3.97 (m, 3H), 2.69 (dd, 1H), 2.26 (d, 1H), 1.64 (br. s., 1H), 1.23-1.11 (m, 1H), 0.66-0.59 (m, 1H). MS m/z 328 [M+H]+.
Пример 31
5-{[(2S,3S,4S)-3-этил-4-фтор-5-оксопирролидин-2-ил]метокси}-3-метокси-1,6-нафтиридин-2-карбоксамид
Стадия 1: Получение 1-(аминоокси)-2,2-диметилпропан-1-она трифлата

На воздухе трет-бутил-гидроксикарбамат (10,68 г, 80,21 ммоль) взвешивали в реакционной колбе, снабженной механической мешалкой. Последовательно добавляли CHCl3 (201 мл) и 2,2-диметилпропановый ангидрид (17,9 г, 96,3 ммоль), затем трубку герметично закрывали. Реакционную смесь перемешивали при 80°С в течение 16 ч. Реакционную смесь выливали в насыщенный водный раствор NaHCO3 и органический слой отделяли, промывали насыщенным водным NaHCO3, сушили над MgSO4 и упаривали с получением белого твердого вещества. Это твердое вещество загружали в круглодонную колбу, снабженную механической мешалкой. Добавляли диэтиловый эфир (201 мл) и колбу закрывали мембраной и охлаждали до 0°С. Одной порцией добавляли трифторметансульфокислоту (12,00 г, 80,2 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь разбавляли гептаном (400 мл): образовывался осадок, и его собирали путем фильтрации на фриттованной воронке с получением указанного в заголовке соединения в виде белого твердого вещества (11,9 г, 55%-ный выход). 1Н-ЯМР (DMSO-d6, 400 МГц): δ 9.44-8.84 (m, 3Н), 1.21 (s, 9Н). 19F-ЯМР (DMSO-d6, 376 МГц): δ-77.0.
Стадия 2: Получение N-[(2,2-диметилпропаноил)окси]-5-метоксипиридин-3-карбоксамида

К раствору 5-метоксипиридин-3-карбоновой кислоты (5,00 г, 32,6 ммоль) в DCM (54,4 мл) и DMF (1,1 мл) добавляли оксалилхлорид (4,35 г, 34,3 ммоль) при комнатной температуре в инертной атмосфере. Через 3 ч при помощи шприца добавляли раствор 1-(аминоокси)-2,2-диметилпропан-1-она трифлата (8,90 г, 33,3 ммоль) в DCM (27,2 мл) и пиридине (5,68 г, 71,8 ммоль) (полученный в атмосфере азота с обработкой ультразвуком) и полученную смесь перемешивали при комнатной температуре в течение 3 ч. Затем реакцию гасили насыщенным водным раствором NH4Cl и водный слой экстрагировали DCM. Объединенные органические экстракты сушили над Na2SO4. Остаток очищали с использованием колоночной хроматографии на силикагеле, элюируя смесью DCM/EtOAc, с получением указанного в заголовке соединения в виде белого твердого вещества (5,3 г, 64%-ный выход). 1Н-ЯМР (DMSO-d6, 400 МГц): δ 12.49 (s, 1Н), 8.57 (s, 1Н), 8.49 (s, 1Н), 7.70 (br. s., 1Н), 3.89 (s, 3Н), 1.29 (s, 9Н). MS m/z 253 [М+Н]+.
Стадия 3: Получение N-[(2,2-диметилпропаноил)окси]-5-метоксипиридин-3-карбоксамид-1-оксида

В колбу, содержащую N-[(2,2-диметилпропаноил)окси]-5-метоксипиридин-3-карбоксамид (3,30 г, 13,1 ммоль), загружали метил(триоксо)рений (32,6 мг, 0,131 ммоль), затем добавляли DCM (17,4 мл). К реакционной смеси добавляли 30%-ный водную Н2О2 (2,94 мл, 28,8 ммоль), и ее перемешивали при комнатной температуре в течение 5 ч. Добавляли водный тиосульфат натрия (4 мл) и смесь перемешивали при комнатной температуре в течение 15 мин. Реакционную смесь разбавляли DCM (30 мл). Органический слой сушили над MgSO4, фильтровали и концентрировали при пониженном давлении с получением вязкого масла. Масло растворяли в iPrOH (20 мл) и концентрировали при пониженном давлении с получением указанного в заголовке соединения в виде белого твердого вещества (3,33 г, 95%-ный выход). 1Н-ЯМР (DMSO-d6, 400 МГц): δ 12.63 (s, 1Н), 8.27 (s, 1Н), 8.18 (s, 1Н), 7.33 (s, 1Н), 3.88 (s, 3Н), 1.28 (s, 9Н). MS m/z 269.0 [М+Н]+
Стадия 4: Получение 3-метокси-6а,7,10,10а-тетрагидро-7,10-метанобензо[h][1,6]нафтиридин-5(6Н)-он-1-оксида

В сосуд загружали N-[(2,2-диметилпропаноил)окси]-5-метоксипиридин-3-карбоксамид-1-оксид (530 мг, 1,98 ммоль), NaOAc (81,0 мг, 0,99 ммоль) и бис(пентаметилциклопентадиенил)дихлорродий (30,5 мг, 0,049 ммоль). Добавляли МеОН (10 мл), затем бицикло[2.2.1]гепта-2,5-диен (273 мг, 3,0 ммоль). Сосуд герметично закрывали и перемешивали при 50°С в течение 2 ч. Смесь охлаждали до комнатной температуры и фильтровали. Белое твердое вещество промывали холодным МеОН и тщательно сушили при пониженном давлении. После сушки получали указанное в заголовке соединение в виде белого твердого вещества (435 мг, 85%-ный выход) и использовали без дополнительной очистки. 1Н-ЯМР (DMSO-d6, 400 МГц): δ 8.52 (br. s., 1Н), 8.32 (s, 1H), 7.41 (s, 1H), 6.45-6.41 (m, 1H), 6.19-6.16 (m, 1H), 3.86 (s, 3H), 3.61 (d, 1H), 3.22 (br. s., 1H), 3.07 (d, 1H), 2.92 (br. s., 1H), 1.38-1.33 (m, 1H), 1.29-1.24 (m, 1H). MS m/z 259 [M+H]+
Стадия 5: Получение 3-метокси-1,6-нафтиридин-5(6Н)-он-1-оксида

Суспензию 3-метокси-6а,7,10,10а-тетрагидро-7,10-метанобензо[h][1,6]нафтиридин-5(6Н)-он-1-оксида (44,0 мг, 0,17 ммоль) в толуоле (0,6 мл) и МеОН (0,6 мл) нагревали в герметичной колбе при 130°С в течение 1 ч при микроволновом излучении. Крышку удаляли и реакцию контролировали с помощью LCMS. Эту операцию повторяли пять раз, после чего LCMS показывала полное расходование исходного вещества. Полученный раствор концентрировали при пониженном давлении с получением таким образом указанного в заголовке соединения в виде бледно-желтого твердого вещества (33 мг, 99%-ный выход). 1Н-ЯМР (DMSO-d6, 400 МГц): δ 11.78 (br. s., 1Н), 8.51 (s, 1H), 7.52 (s, 1H), 7.34 (t, 1H), 6.99 (d, 1H), 3.91 (s, 3H). MS m/z 193 [M+H]+
Стадия 6: Получение 3-метокси-5-оксо-5,6-дигидро-1,6-нафтиридин-2-карбонитрила
К раствору 3-метокси-1,6-нафтиридин-5(6Н)-он-1-оксида (150 мг, 0,776 ммоль) в DCM (2,59 мл) добавляли хлорангидрид диметилкарбаминовой кислоты (125 мг, 1,16 ммоль), затем добавляли TMSCN (154 мг, 1,55 ммоль). Добавляли DMF (0,2 мл) и смесь перемешивали при 50°С в течение 5 ч. Летучие вещества удаляли при пониженном давлении и остаток очищали с использованием колоночной хроматографии на силикагеле, элюируя смесью DCM/MeOH, с получением указанного в заголовке соединения в виде желтого твердого вещества (118 мг, 76%-ный выход). 1Н-ЯМР (DMSO-d6, 400 МГц): δ 11.73 (br. s., 1H), 8.21 (s, 1Н), 7.41 (t, 1Н), 6.63 (d, 1H), 4.08 (s, 3Н). MS m/z 202 [М+Н]+
Стадия 7: Получение 5-хлор-3-метокси-1,6-нафтиридин-2-карбонитрила

В сухой сосуд загружали 3-метокси-5-оксо-5,6-дигидро-1,6-нафтиридин-2-карбонитрил (150 мг, 0,746 ммоль), пиридиния гидрохлорид (86 мг, 0,746 ммоль) и фосфорилхлорид (2,76 мл). Реакционную смесь нагревали при 90°С в течение 1 ч и затем охлаждали до комнатной температуры. Раствор аккуратно выливали в стакан, содержащий перемешанную смесь водного раствора Na2HPO4 и льда. Осадок фильтровали, промывали водой и сушили в вакууме. Указанное в заголовке соединение получали в виде бежевого твердого вещества (111 мг, 68%-ный выход) и использовали без дополнительной очистки. 1Н-ЯМР (DMSO-d6, 400 МГц): δ 8.53 (d, 1Н), 8.15 (s, 1Н), 8.01 (d, 1Н), 4.18 (s, 3Н). MS m/z 220 [М+Н]+
Стадия 8: Получение 5-хлор-3-метокси-1,6-нафтиридин-2-карбоксамида

Раствор 5-хлор-3-метокси-1,6-нафтиридин-2-карбонитрила (149 мг, 0,678 ммоль) в DMSO (6,78 мл) обрабатывали K2CO3 (469 мг, 3,39 ммоль). Полученную смесь перемешивали в течение 5 мин, после чего добавляли водный раствор 50%-ного перекиси водорода (269 мкл, 4,75 ммоль). Через 5 мин реакционную смесь гасили диметилдисульфидом (337 мг, 5,43 ммоль) и перемешивали при комнатной температуре в течение 30 мин, затем реакционную смесь фильтровали через целит. Осадок на фильтре промывали DCM, затем EtOAc и фильтрат концентрировали при пониженном давлении с получением раствора в DMSO, который сушили потоком азота при 45°С в течение ночи. Неочищенное вещество очищали с использованием колоночной хроматографии на силикагеле, элюируя смесью DCM/MeOH, с получением указанного в заголовке соединения в виде бледно-желтого твердого вещества (62 мг, 38%-ный выход). 1Н-ЯМР (DMSO-d6, 400 МГц): δ 8.45 (d, 1Н), 8.10 (br. s., 1Н), 7.93 (d, 1H), 7.90 (s, 1H), 7.84 (br. s., 1H), 4.04 (s, 3H). MS m/z 238 [M+H]+
Стадия 9: Получение 5-{[(2S,3S,4S)-3-этил-4-фтор-5-оксопирролидин-2-ил]метокси}-3-метокси-1,6-нафтиридин-2-карбоксамида
К раствору (3S,4S,5S)-4-этил-3-фтор-5-(гидроксиметил)пирролидин-2-она (38 мг, 0,236 ммоль) и 5-хлор-3-метокси-1,6-нафтиридин-2-карбоксамида (59 мг, 0,25 ммоль) в DMF (1,24 мл) добавляли KHMDS (0,95 мл, 1,0 М в THF, 0,95 ммоль) при комнатной температуре. Смесь нагревали при 50°С и перемешивали в течение 2 ч. Затем реакционную смесь охлаждали и гасили насыщенным водным раствором NH4Cl и разбавляли DCM. Водный слой экстрагировали DCM и объединенные органические слои промывали рассолом и сушили над Na2SO4. Неочищенное вещество очищали с использованием колоночной хроматографии на силикагеле, элюируя смесью DCM/MeOH, с получением указанного в заголовке соединения в виде не совсем белого твердого вещества (24 мг, 27%-ный выход). 1Н-ЯМР (DMSO-d6, 500 МГц): δ 8.93 (s, 1Н), 8.11 (d, 1Н), 8.06 (s, 1Н), 8.02 (br. s., 1Н), 7.73 (br. s., 1Н), 7.44 (dd, 1H), 4.97-4.83 (m, 1H), 4.61 (dd, 1H), 4.23 (dd, 1H), 4.14-4.07 (m, 1H), 3.95 (s, 3H), 2.68-2.56 (m, 1H), 1.63-1.56 (m, 2H), 1.02 (t, 3H). MS m/z 364 [M+H]+.
Пример 32
1-(((2S,3S,4S)-4-фтор-3-метил-5-оксопирролидин-2-ил)метокси)-7-метокси-N-метилизохинолин-6-карбоксамид
Стадия 1: Получение 1-(((2S,3S,4S)-4-фтор-3-метил-5-оксопирролидин-2-ил)метокси)-7-метоксиизохинолин-6-карбоновой кислоты

1-(((2S,3S,4S)-4-фтор-3-метил-5-оксопирролидин-2-ил)метокси)-7-метоксиизохинолин-6-карбоксамид (250 мг, 0,72 ммоль) растворяли в TFA (5 мл) при комнатной температуре, затем охлаждали до 0°С. Через 5 мин смесь обрабатывали NaNO2 (497 мг, 7,20 ммоль) и перемешивали в течение 15 мин. Реакционную смесь выливали в стакан с ледяной водой (60 г) при перемешивании. Водный слой экстрагировали EtOAc (60 мл×3) и органический слой сушили над Na2SO4 с получением неочищенной 1-(((2S,3S,4S)-4-фтор-3-метил-5-оксопирролидин-2-ил)метокси)-7-метоксиизохинолин-6-карбоновой кислоты. 100% э.и. (Колонка: Chiralpak AD-H 250×4,6 мм в.д., 5 мкм. Подвижная фаза: изопропанол в СО2 от 5% до 40% Скорость потока: 2,5 мл/мин. Время удерживания: 7,8 мин. Длина волны: 220 нм. MS m/e 348,8 [М+Н]+. Это вещество использовали без дополнительной очистки в следующей реакции.
Стадия 2: Получение 1-(((2S,3S,4S)-4-фтор-3-метил-5-оксопирролидин-2-ил)метокси)-7-метокси-N-метилизохинолин-6-карбоксамида
К раствору 1-(((2S,3S,4S)-4-фтор-3-метил-5-оксопирролидин-2-ил)метокси)-7-метоксиизохинолин-6-карбоновой кислоты (70 мг, 0,20 ммоль) в DCM (4 мл) добавляли EDCI (62 мг, 0,32 ммоль) и НОВТ (46 мг, 0,34 ммоль), затем добавляли метиламина гидрохлорид (41 мг, 0,60 ммоль) и DIPEA (130 мг, 1,00 ммоль). Бледно-желтую реакционную смесь перемешивали в течение 16 ч при комнатной температуре. Реакционную смесь разбавляли 30 мл EtOAc и промывали 20 мл насыщенного водного раствора NaHCO3. Двухфазную смесь фильтровали и промывали 3×8 мл воды и 3×6 мл МТВЕ. Осадок на фильтре собирали и сушили в вакууме с получением 1-(((2S,3S,4S)-4-фтор-3-метил-5-оксопирролидин-2-ил)метокси)-7-метокси-N-метилизохинолин-6-карбоксамида (49 мг, 67%-ный выход) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, DMSO-d6) δ 8.76 (br. s., 1Н), 8.35 (br. s., 1H), 8.12 (s, 1H), 7.91 (d, 1H), 7.71 (s, 1H), 7.43 (d, 1H), 5.03-4.82 (m, 1H), 4.57 (d, 1H), 4.33-4.22 (m, 1H), 4.04 (br. s., 1H), 3.96 (s, 3H), 2.91 (m, 1H), 2.82 (d, 3H), 1.09 (d, 3H). 19F-ЯМР (376 МГц, DMSO-d6,) -200.80 (br. s., 1F). MS m/e 362,0 [M+H]+.
Биологическая активность:
Ферментативный анализ IRAK4 с помощью DELFIA, Протокол А. Он представляет собой анализ in vitro для измерения ферментативной активности IRAK4 с использованием платформы DELFIA (Усиленный диссоциацией лантанидный флуоресцентный иммуноанализ, Perkin-Elmer), с конструкцией домена человеческой киназы IRAK4 (полного размера) для характеристики ингибитора IRAK4 и контрольных соединений при 600 мкМ АТР (КМ). Конечное количество фермента в анализе составляет 0,1 нМ IRAK4 FL, конечная концентрация субстрата составляет 50 нМ, и конечная концентрация DMSO составляет 2,5%.
Тестируемое соединение солюбилизировали в DMSO до концентрации исходного раствора 30 мМ. Планшеты для анализа зависимости ответа от дозы готовили с первичной концентрацией соединения 4 мМ (40-кратная концентрация относительно конечной концентрации в анализе) и разбавляли DMSO четырехкратными сериями для получения суммарно 11 точек данных. 1 мкл из планшета для разбавления соединений помещали в суперпрозрачные полипропиленовые 384-луночные планшеты с U-образным дном (Corning Life Sciences).
Для начала анализа 19 мкл реакционной смеси, содержащей 20 мМ HEPES, рН=7,5, 5 мМ MgCl2, 0,0025% Brij-35, 600 мкм ATP, 0,21 пМ полноразмерного фосфорилированного рекомбинантного человеческого IRAK4 (GenBank ID AF445802) аликвотировали в полипропиленовые 384-луночные планшеты с U-образным дном, содержиащие 1 мкл тестируемого соединения, кратковременно смешивали и инкубировали в течение 20 минут при комнатной температуре (к.т.). Затем добавляли 20 мкл 20 мМ HEPES рН=7,5, 5 мМ MgCl2, 0,0025% Brij-35, 600 мкм ATP и 100 нМ ERM-биотинилированного пептида (AGAGPJDKYKTLRQIR) для начала реакции. Реакционную смесь инкубировали в течение 60 минут при комнатной температуре и реакцию останавливали путем добавления 20 мкл 0,3M EDTA.
50 мкл реакционной смеси переносили в покрытый стрептавидином планшет для детектирования (DELFIA стрептавидин-покрытые планшеты, 384-луночные, белые планшеты, Perkin-Elmer Life Sciences) и инкубировали в течение 30 минут при к.т. Планшеты 4 раза промывали 75 мкл на лунку PBS, содержащего 0,05% Tween-20. Затем планшеты инкубировали с 50 мкл на лунку коктейля анти-pERM антитела с концентрацией 0,125 мкг/мл (Cell Signaling Technology) с добавлением меченого EuN1 антитела против кроличьего IgG в количестве 0,25 мкг/мл (Perkin-Elmer Life Sciences) в растворе 10 мМ MOPS рН=7,5, 150 мМ NaCl, 0,05% Tween-20, 0,02% NaN3, 1% BSA, 0,1% желатин в течение 45 минут. Затем планшеты промывали как ранее. К планшету добавляли по 50 мкл на лунку усиливающего раствора DELFIA (Perkin-Elmer Life Sciences) и инкубировали в течение 15 минут при к.т., после чего считывали на многоканальном ридере Envision Model 2104 с использованием возбуждения на длине волны 340 нм и детектирования испускания при 665 нм.
Ферментативный анализ IRAK4 с помощью DELFIA, Протокол А. Он представляет собой анализ in vitro для измерения ферментативной активности IRAK4 с использованием платформы DELFIA (Усиленный диссоциацией лантанидный флуоресцентный иммуноанализ, Perkin-Elmer), с неактивной нефосфорилированной (0-phos) конструкцией человеческой IRAK4 FL (полной длины) для характеристики ингибитора IRAK4 и контрольных соединений при 600 мкМ АТР (KМ). Конечное количество фермента в анализе составляет 0,1 нМ неактивной, 0-phos, IRAK4 FL, конечная концентрация субстрата составляет 50 нМ, и конечная концентрация DMSO составляет 2,5%.
Тестируемое соединение солюбилизировали в DMSO до концентрации исходного раствора 30 мМ. Планшеты для анализа зависимости ответа от дозы готовили с первичной концентрацией соединения 4 мМ, серийно разбавляли в DMSO и помещали (1 мкл) в 384-луночные полипропиленовые планшеты, как ранее.
Для начала анализа 19 мкл реакционной смеси, содержащей 20 мМ HEPES рН=7,5, 5 мМ MgCl2, 0,0025% Brij-35, 600 мкм АТР, 0,21 нМ неактивной, 0-phos, полноразмерной рекомбинантной человеческой IRAK4 (GenBank ID AF445802) аликвотировали в полипропиленовые планшеты, содержащие 1 мкл тестируемого соединения, как ранее. Добавляли 20 мкл 20 мМ HEPES рН=7,5, 5 мМ MgCl2, 0,0025% Brij-35, 600 мкМ АТР, и 100 нМ ERM-биотинилированного пептида (AGAGRDKYKTLRQIR) для начала реакции, которую осуществляли в течение 90 минут при к.т. и останавливали путем добавления 20 мкл 0,3 М EDTA.
50 мкл реакционной смеси переносили в покрытый стрептавидином планшет для детектирования (покрытые стрептавидином планшеты DELFIA, 384-луночные, белые планшеты, Perkin-Elmer Life Sciences) и инкубировали в течение 30 минут при к.т. Планшеты 4 раза промывали с 75 мкл на лунку PBS, содержащего 0,05% Tween-20. Затем планшеты инкубировали с 50 мкл на лунку коктейля анти-pERM антитела с концентрацией 0,125 мкг/мл (Cell Signaling Technology) с добавлением меченого EuN1 антитела против кроличьего IgG в количестве 0,25 мкг/мл (Perkin-Elmer Life Sciences) в растворе 10 мМ MOPS рН=7,5, 150 мМ NaCl, 0,05% Tween-20, 0,02% NaN3, 1% BSA, 0,1% желатин в течение 45 минут. Затем планшеты промывали, как ранее. К планшету добавляли по 50 мкл на лунку усиливающего раствора DELFIA (Perkin-Elmer Life Sciences) и инкубировали в течение 15 минут при к.т., после чего считывали на многоканальном ридере Envision Model 2104 с использованием возбуждения на длине волны 340 нм и детектирования испускания при 665 нм.
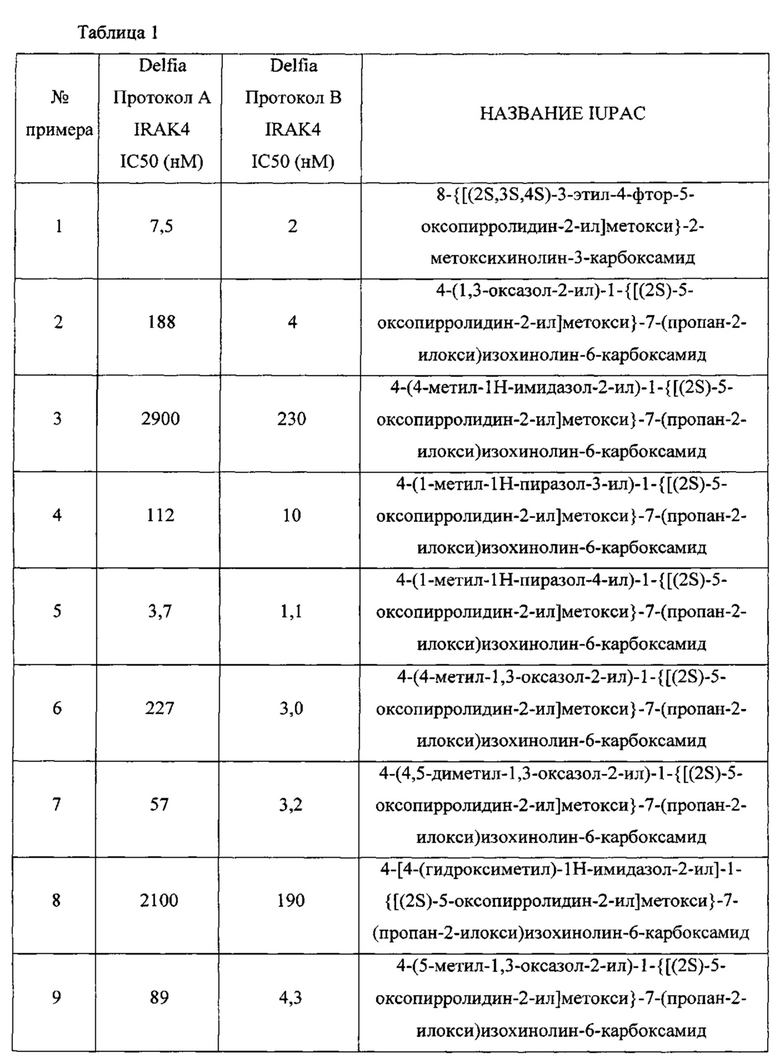

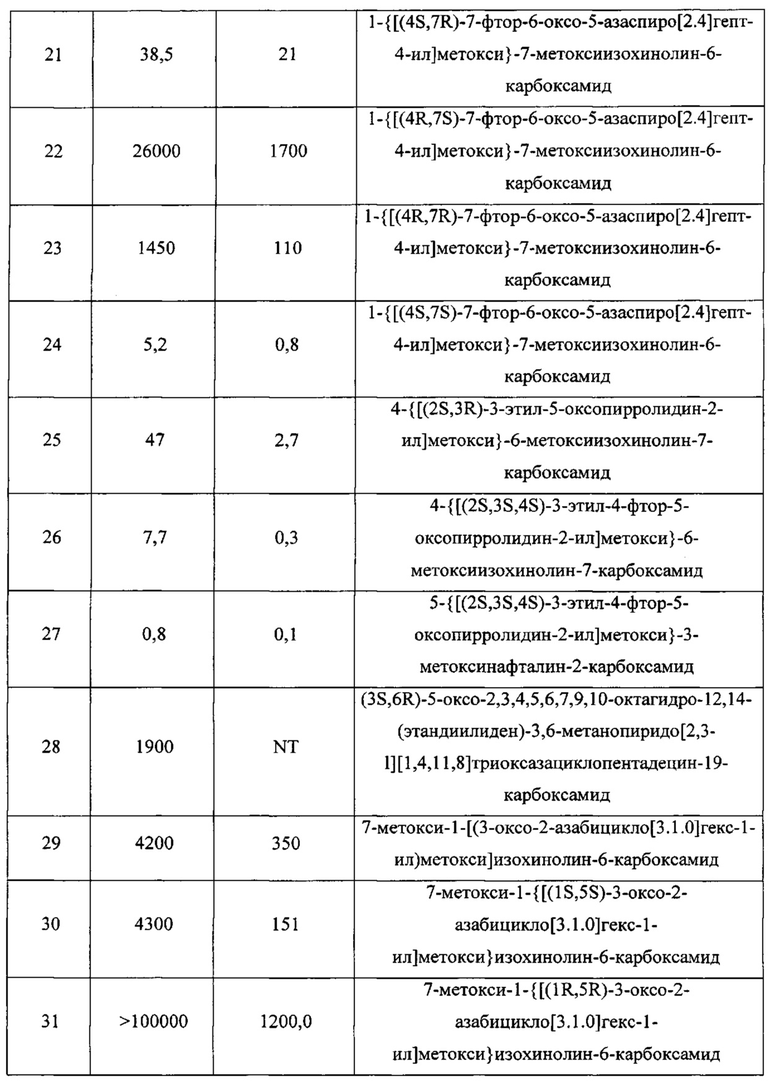

| название | год | авторы | номер документа |
|---|---|---|---|
| ФЕНОКСИПИРИДИНИЛАМИДНЫЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ В ЛЕЧЕНИИ PDE4-ОПОСРЕДОВАННЫХ БОЛЕЗНЕННЫХ СОСТОЯНИЙ | 2009 |
|
RU2509077C2 |
| Гетероциклические ингибиторы МСТ4 | 2017 |
|
RU2771875C2 |
| Спироконденсированные пирролидиновые производные в качестве ингибиторов деубиквитилирующих ферментов (DUB) | 2017 |
|
RU2730552C2 |
| Новые соединения | 2016 |
|
RU2734256C2 |
| Соединения циклопентана | 2019 |
|
RU2794997C2 |
| Производные N-пиперидин-3-илбензамида для лечения сердечно-сосудистых заболеваний | 2014 |
|
RU2618628C1 |
| БИАРИЛЬНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ БЕЛОК-БЕЛКОВОГО ВЗАИМОДЕЙСТВИЯ YAP/TAZ-TEAD | 2021 |
|
RU2830596C1 |
| ПРОИЗВОДНЫЕ 7-ОКСО-1,6-ДИАЗАБИЦИКЛО[3.2.1]ОКТ-3-ЕНА, ПОЛЕЗНЫЕ ДЛЯ ЛЕЧЕНИЯ БАКТЕРИАЛЬНЫХ ИНФЕКЦИЙ | 2013 |
|
RU2645678C2 |
| ИМИДАЗОПИРАЗИНОНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ PDE1 | 2016 |
|
RU2712219C2 |
| ИЗОИНДОЛИНОНОВЫЕ ИНГИБИТОРЫ ВЗАИМОДЕЙСТВИЯ MDM2-P53, ОБЛАДАЮЩИЕ ПРОТИВОРАКОВОЙ АКТИВНОСТЬЮ | 2016 |
|
RU2797295C1 |
Изобретение относится к гетероциклическому соединению формул IIс или IIе, где R1 представляет собой C1-С6алкил; R2 представляет собой водород; R3 представляет собой водород; каждый из R4a и R4b независимо представляет собой водород или фтор; R5a и R5b независимо представляют собой водород или C1-С3алкил; и R6 представляет собой водород. Также изобретение относится к конкретным соединениям и фармацевтической композиции на основе указанных соединений. Технический результат: получены новые гетероциклические соединения, обладающие IRAK4-ингибирующей активностью. 3 н.п. ф-лы, 1 табл., 33 пр.
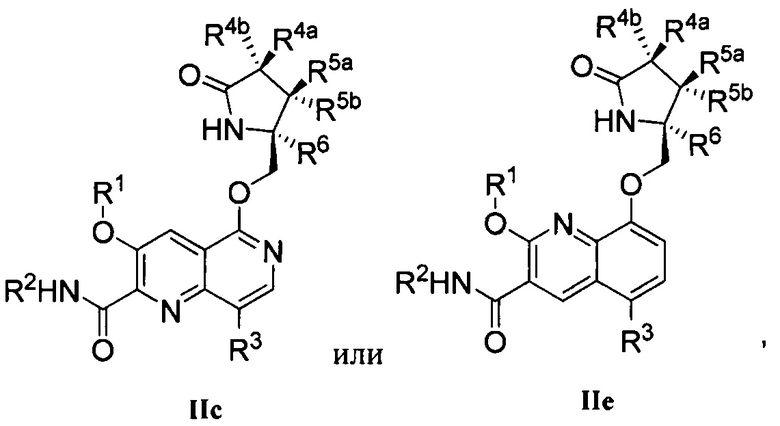
1. Соединение Формулы IIc или IIe:

где
R1 представляет собой C1-С6алкил;
R2 представляет собой водород;
R3 представляет собой водород;
каждый из R4a и R4b независимо представляет собой водород или фтор;
R5a и R5b независимо представляют собой водород или C1-С3алкил; и
R6 представляет собой водород.
2. Соединение, выбранное из:
8-{[(2S,3S,4S)-3-этил-4-фтор-5-оксопирролидин-2-ил]метокси}-2-метоксихинолин-3-карбоксамида;
4-(1,3-оксазол-2-ил)-1-{[(2S)-5-оксопирролидин-2-ил]метокси}-7-(пропан-2-илокси)изохинолин-6-карбоксамида;
4-(4-метил-1Н-имидазол-2-ил)-1-{[(2S)-5-оксопирролидин-2-ил]метокси}-7-(пропан-2-илокси)изохинолин-6-карбоксамида;
4-(1-метил-1Н-пиразол-3-ил)-1-{[(2S)-5-оксопирролидин-2-ил]метокси}-7-(пропан-2-илокси)изохинолин-6-карбоксамида;
4-(1-метил-1Н-пиразол-4-ил)-1-{[(2S)-5-оксопирролидин-2-ил]метокси}-7-(пропан-2-илокси)изохинолин-6-карбоксамида;
4-(4-метил-1,3-оксазол-2-ил)-1-{[(2S)-5-оксопирролидин-2-ил]метокси}-7-(пропан-2-илокси)изохинолин-6-карбоксамида;
4-(4,5-диметил-1,3-оксазол-2-ил)-1-{[(2S)-5-оксопирролидин-2-ил]метокси}-7-(пропан-2-илокси)изохинолин-6-карбоксамида;
4-[4-(гидроксиметил)-1Н-имидазол-2-ил]-1-{[(2S)-5-оксопирролидин-2-ил]метокси}-7-(пропан-2-илокси)изохинолин-6-карбоксамида;
4-(5-метил-1,3-оксазол-2-ил)-1-{[(2S)-5-оксопирролидин-2-ил]метокси}-7-(пропан-2-илокси)изохинолин-6-карбоксамида;
1-{[(2S,3R)-3-этил-5-оксопирролидин-2-ил]метокси}-4-[(фенилсульфонил)амино]-7-(пропан-2-илокси)изохинолин-6-карбоксамида;
1-{[(2S,3R)-3-этил-5-оксопирролидин-2-ил]метокси}-7-(пропан-2-илокси)-4-[(пиридин-3-илсульфонил)амино]изохинолин-6-карбоксамида;
1-{[(2S,3R)-3-этил-5-оксопирролидин-2-ил]метокси}-4-[(1Н-имидазол-4-илсульфонил)амино]-7-(пропан-2-илокси)изохинолин-6-карбоксамида;
1-{[(2S,3R)-3-этил-5-оксопирролидин-2-ил]метокси}-4-{[(1-метил-1Н-имидазол-4-ил)сульфонил]амино}-7-(пропан-2-илокси)изохинолин-6-карбоксамида;
4-{[(1,2-диметил-1Н-имидазол-4-ил)сульфонил]амино}-1-{[(2S,3R)-3-этил-5-оксопирролидин-2-ил]метокси}-7-(пропан-2-илокси)изохинолин-6-карбоксамида;
4-амино-1-{[(2S,3S,4S)-3-этил-4-фтор-5-оксопирролидин-2-ил]метокси}-7-метоксиизохинолин-6-карбоксамида;
1-{[(4R,7S)-7-фтор-6-оксо-5-азаспиро[2.4]гепт-4-ил]метокси}-7-(пропан-2-илокси)изохинолин-6-карбоксамида;
1-{[(4S)-7-фтор-6-оксо-5-азаспиро[2.4]гепт-4-ил]метокси}-7-(пропан-2-илокси)изохинолин-6-карбоксамида;
1-{[(4R,7R)-7-фтор-6-оксо-5-азаспиро[2.4]гепт-4-ил]метокси}-7-(пропан-2-илокси)изохинолин-6-карбоксамида;
1-(((4S,7R)-7-фтор-6-оксо-5-азаспиро[2.4]гептан-4-ил)метокси)-7-изопропоксиизохинолин-6-карбоксамида;
1-{[(4S,7R)-7-фтор-6-оксо-5-азаспиро[2.4]гепт-4-ил]метокси}-7-метоксиизохинолин-6-карбоксамида;
1-{[(4R,7S)-7-фтор-6-оксо-5-азаспиро[2.4]гепт-4-ил]метокси}-7-метоксиизохинолин-6-карбоксамида;
1-{[(4R,7R)-7-фтор-6-оксо-5-азаспиро[2.4]гепт-4-ил]метокси}-7-метоксиизохинолин-6-карбоксамида;
1-{[(4S,7S)-7-фтор-6-оксо-5-азаспиро[2.4]гепт-4-ил]метокси}-7-метоксиизохинолин-6-карбоксамида;
4-{[(2S,3R)-3-этил-5-оксопирролидин-2-ил]метокси}-6-метоксиизохинолин-7-карбоксамида;
4-{[(2S,3S,4S)-3-этил-4-фтор-5-оксопирролидин-2-ил]метокси}-6-метоксиизохинолин-7-карбоксамида;
5-{[(2S,3S,4S)-3-этил-4-фтор-5-оксопирролидин-2-ил]метокси}-3-метоксинафталин-2-карбоксамида;
(3S,6R)-5-оксо-2,3,4,5,6,7,9,10-октагидро-12,14-(этандиилиден)-3,6-метанопиридо[2,3-l][1,4,11,8]триоксазациклопентадецин-19-карбоксамида;
7-метокси-1-[(3-оксо-2-азабицикло[3.1.0]гекс-1-ил)метокси]изохинолин-6-карбоксамида;
7-метокси-1-{[(1S,5S)-3-оксо-2-азабицикло[3.1.0]гекс-1-ил]метокси}изохинолин-6-карбоксамида;
7-метокси-1-{[(1R,5R)-3-оксо-2-азабицикло[3.1.0]гекс-1-ил]метокси}изохинолин-6-карбоксамида;
5-{[(2S,3S,4S)-3-этил-4-фтор-5-оксопирролидин-2-ил]метокси}-3-метокси-1,6-нафтиридин-2-карбоксамида; и
1-{[(2S,3S,4S)-4-фтор-3-метил-5-оксопирролидин-2-ил]метокси}-7-метокси-N-метилизохинолин-6-карбоксамида.
3. Фармацевтическая композиция, обладающая IRAK4-ингибирующей активностью, содержащая терапевтически эффективное количество соединения по любому из пп.1 и 2 и фармацевтически приемлемый носитель, разбавитель или наполнитель.
| US 2013029949 A1, 31.01.2013 | |||
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЛЕЧЕНИЯ | 1994 |
|
RU2135478C1 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |

