ПРАВО ПРИОРИТЕТА
Данная заявка истребует приоритет патента США № 62/210,264, поданной 26 августа 2015 года, который полностью включен в настоящий документ в силу ссылки.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Нейротрофная рецепторная тирозинкиназа (NTRK) типов 1, 2 и 3 относится к рецепторным тирозинкиназам (RTK), которые активируют множественные нисходящие сигнальные пути, связанные с пролиферацией и выживаемостью клеток. Различные генетические слияния, возникающие из-за транслокаций аберрантных хромосом, в генах, кодирующих эти RTK, связаны с этиологией множества видов онкологических заболеваний, включая глиому высокой и низкой степени злокачественности, холангиокарциному, папиллярную карциному щитовидной железы, рак толстой кишки и немелкоклеточный рак легкого. Проведенный геномный анализ разнообразных киназных слияний идентифицировал наличие слияний NTRK при широком спектре дополнительных типов онкологических заболеваний, включая плоскоклеточный рак головы и шеи, аденокарциному поджелудочной железы, саркому и меланому; тем самым обеспечивая дальнейшее терапевтическое обоснование внедрения ингибиторов данных киназ для лечения множества онкологических состояний.
Идентификация слияний NTRK в качестве основной причины некоторых видов онкологических заболеваний привела к открытию и дальнейшей клинической разработке нескольких ингибиторов киназы NTRK для лечения опухолей, содержащих гибридный белок NTRK. Ранние клинические данные подтверждают жизнеспособность подобного подхода в предоставлении преимуществ пациентам с определенными злокачественными опухолями человека. Однако в конечном счете, несмотря на явные признаки клинической активности, онкологическое заболевание большинства пациентов станет устойчивым к терапии ингибиторами киназы, что приведет к рецидиву и прогрессированию заболевания. Киназная реактивация посредством внутренней мутации является частым механизмом устойчивости. При возникновении устойчивости варианты лечения пациента часто очень ограничены. Таким образом, существует потребность в соединениях, которые ингибируют NTRK, а также ее устойчивые мутантные формы.
ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Изобретение включает соединения и фармацевтические композиции, содержащие соединения по формуле (I) или их фармацевтически приемлемые соли, где:
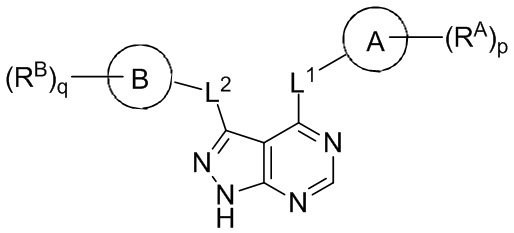
Формула (I)
Каждое из колец A и B независимо выбрано из следующих соединений: арил, гетероарил, циклоалкил и гетероциклил;
каждое L1 и L2 независимо выбрано из связи или следующих соединений: -C(O)-, N(R1), N(R1) C(O)-, -C(O)-N(R1), (C1-C6 алкилен) N(R1), N(R1) (C1-C6 алкилен), N(R1) C(O)-(C1-C6 алкилен) и -C(O)-N(R1) (C1-C6 алкилен) ; и при этом каждый алкилен независимо замещен 0-5 соединениями R';
каждое RA и RB независимо выбрано из следующих соединений: гидроксил, C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C1-C6 алкоксил, галоген, C1-C6 гетероалкил, C1-C6 галогеналкил, C1-C6 галогеналкоксил, C1-C6 гидроксиалкил, циклоалкил, арил, гетероарил, арилокси, аралкил, гетероциклил, гетероциклилалкил, нитро, циано, -C(O)R1, -OC(O)R1, -C(O)OR1, (C1-C6 алкилен)-C(O)R1, -SR1, -S(O)2R1, -S(O)2-N(R1)(R1), (C1-C6 алкилен)-S(O)2R1, (C1-C6 алкилен)-S(O)2-N(R1)(R1), -N(R1)(R1), -C(O)-N(R1)(R1), -N(R1)-C(O)R1, -N(R1)-C(O)OR1, -(C1-C6 алкилен) N(R1) C(O)R1, -N(R1)S(O)2R1, и -P(O)(R1)(R1); и при этом каждое из следующих соединений: алкил, алкенил, алкинил, алкоксил, гетероалкил, галогеналкил, галогеналкоксил, гидроксиалкил, циклоалкил, арил, гетероарил, арилокси, аралкил, гетероциклил и гетероциклилалкил, независимо замещено 0-5 соединениями Ra; или 2 RA или 2 RB вместе с атомом(-ами) углерода, к которому(-ым) они присоединены, формируют циклоалкильное или гетероциклильное кольцо, независимо замещенное 0-5 соединениями Ra;
каждое R1 независимо выбрано из следующих соединений: водород, гидроксил, галоген, тиол, C1-C6 алкил, C1-C6 тиоалкил, C1-C6 алкоксил, C1-C6 галогеналкил, C1-C6 гидроксиалкил, C1-C6 гетероалкил, циклоалкил, циклоалкилалкил, гетероарилалкил, гетероциклил и гетероциклилалкил, и при этом каждое из следующих соединений: алкил, тиоалкил, алкоксил, галогеналкил, гидроксиалкил, гетероалкил, циклоалкил, циклоалкилалкил, гетероарилалкил, гетероциклил и гетероциклилалкил, независимо замещено 0-5 соединениями Rb или 2 R1 вместе с атомом(-ами), к которому(-ым) они присоединены, формируют циклоалкильное или гетероциклильное кольцо, независимо замещенное 0-5 соединениями Rb;
каждое Ra и Rb независимо выбрано из следующих соединений: C1-C6 алкил, галоген, гидроксил, C1-C6 галогеналкил, C1-C6 гетероалкил, C1-C6 гидроксиалкил, C1-C6 алкоксил, циклоалкил, гетероциклил, и циано, и при этом каждое из следующих соединений: алкил, галогеналкил, гетероалкил, гидроксиалкил, алкоксил, циклоалкил и гетероциклил независимо замещено 0-5 соединениями R';
каждое R' независимо выбрано из следующих соединений: C1-C6 алкил, C1-C6 гетероалкил, галоген, гидроксил, C1-C6 галогеналкил, C1-C6 гидроксиалкил, циклоалкил и циано; или 2 R'вместе с атомом(-ами), к которому(-ым) они присоединены, формируют циклоалкильное или гетероциклильное кольцо;
p равно 0, 1, 2, 3, 4 или 5; и
q равно 0, 1, 2, 3 или 4.
Любое из соединений, раскрытых в данном документе, может использоваться отдельно или в сочетании с другим терапевтическим агентом для лечения любого из заболеваний, описанных в данном документе.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
На фиг. 1 показана структура различных примеров соединений в соответствии с настоящим изобретением, а также их пики и массы ЯМР, определенные посредством ЖХ-МС.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Определения
При использовании в данном документе термины «пациент», «субъект», «индивидуум» и «хозяин» относятся к человеку или животному, не являющемуся человеком, у которого имеется или подозревается заболевание или расстройство, связанное с аберрантной экспрессией NTRK (т. е. с увеличенной активностью NTRK, вызванной сигнализацией посредством NTRK) или биологической активностью.
Термины «лечить» и «лечение» в отношении заболевания или расстройства относится к улучшению по меньшей мере одного симптома заболевания или расстройства. Данные термины, когда они используются в связи с состоянием, таким как онкологическое заболевание, относятся к одному или более из следующих: препятствие росту онкологического поражения, уменьшение массы или объема онкологического поражения, продление ожидаемого времени выживания пациента, ингибирование роста опухоли, уменьшение массы опухоли, уменьшение размера или количества метастатических поражений, препятствие развитию новых метастатических поражений, продление выживания, продление выживаемости без прогрессирования, продление времени до прогрессирования и/или улучшение качества жизни.
Термин «предотвращение» при использовании в отношении состояния или заболевания, такого как онкологическое, относится к уменьшению частоты или задержке появления симптомов состояния или заболевания. Таким образом, предотвращение рака включает, например, снижение количества обнаруживаемых раковых опухолей в популяции пациентов, получающих профилактическое лечение, по сравнению с контрольной популяцией, не получающей лечения, и/или замедление появления обнаруживаемых раковых опухолей в популяции, получающей лечение, по сравнению с контрольной популяцией, не получающей лечения, например, на статистически и/или клинически значимое количество.
Термин «терапевтический эффект» относится к полезному локальному или системному эффекту у животных, особенно млекопитающих и, в частности, людей, вызванному введением соединения или композиции в соответствии с изобретением. Фраза «терапевтически эффективное количество» означает количество соединения или композиции в соответствии с изобретением, которое является эффективным для лечения заболевания или состояния, вызванного чрезмерной экспрессией NTRK или аберрантной биологической активностью NTRK при разумном соотношении преимущества/риска. Терапевтически эффективное количество такого вещества будет варьироваться в зависимости от субъекта и состояния заболевания, которое подвергается лечению, массы тела и возраста субъекта, тяжести заболевания, способа введения и тому подобного, что легко может быть определено специалистом в данной области.
Используемый в данном документе термин «развитие устойчивости» означает, что когда препарат вводят пациенту впервые, симптомы пациента улучшаются, что определяется по уменьшению объема опухоли, уменьшению числа новых поражений или посредством использования иных параметров, которые применяет врач для оценки прогрессирования заболевания; однако в какой-то момент времени эти симптомы перестают улучшаться или даже ухудшаются. В таком случае говорят, что у пациента выработалась устойчивость к препарату.
Термин «алифатическая группа» означает неразветвленную, разветвленную или циклическую углеводородную группу и включает насыщенные и ненасыщенные группы, такие как алкильная группа, алкенильная группа и алкинильная группа.
Термин «алкилен» относится к двухвалентному радикалу алкильной группы, например, -CH2-, -CH2CH2- и CH2CH2CH2-.
Термин «алкенил» означает алифатическую группу, содержащую по меньшей мере одну двойную связь.
Термин «алкоксил» или «алкокси» означает алкильную группу с присоединенным к ней кислородным радикалом. Типичные алкоксильные группы включают метокси, этокси, пропилокси, трет-бутокси и им подобные. Термин «галогеналкокси» относится к алкоксилу, в котором один или более атомов водорода замещены галогеном, и включает алкоксильные группы, в которых все атомы водорода замещены галогеном (например, перфторалкокси).
Термин «алкил» относится к одновалентному радикалу насыщенного прямого или разветвленного углеводорода, такого как линейная или разветвленная группа с 1 12, 1 10 или 1 6 атомами углерода, называемая в данном документе C1 C12 алкил, C1 C10 алкил, и C1 C6 алкил соответственно. Примеры алкильных групп включают, но не ограничиваются ими: метил, этил, пропил, изопропил, 2 метил 1 пропил, 2 метил 2 пропил, 2 метил 1 бутил, 3 метил 1 бутил, 2 метил 3 бутил, 2,2 диметил 1 пропил, 2 метил 1 пентил, 3 метил 1 пентил, 4 метил 1 пентил, 2 метил 2 пентил, 3 метил 2 пентил, 4 метил 2 пентил, 2,2 диметил 1 бутил, 3,3 диметил 1 бутил, 2 этил 1 бутил, бутил, изобутил, трет бутил, пентил, изопентил, неопентил, гексил, гептил, октил и т. п.
Термин «алкенилен» относится к алкенильной группе, имеющей две точки соединения. Например, «этенилен» представляет собой группу -CH=CH-. Алкениленовые группы также могут присутствовать в незамещенной форме или в замещенной форме с одним или более заместителями.
Термин «алкинил» относится к линейной или разветвленной углеводородной цепи, содержащей 2-12 атомов углерода и характеризующейся наличием одной или более тройных связей. Примеры алкинильных групп включают, но не ограничиваются ими, этинил, пропаргил и 3-гексинил. Один из атомов тройной связи может необязательно быть точкой присоединения алкинильного заместителя.
Термин «алкинилен» относится к алкинилу, имеющему две точки соединения. Например, «этинилен» представляет собой группу -C≡C-. Алкиниленовые группы также могут находиться в незамещенной форме или замещенной форме с одним или более заместителями.
Термины «гидроксиалкилен» или «гидроксиалкил» относятся к алкиленовому или алкильному фрагменту, в котором алкиленовый или алкильный атом водорода замещен гидроксильной группой. Гидроксиалкилен или гидроксиалкил включает группы, в которых более одного атома водорода замещено гидроксильной группой.
Термин «ароматическая кольцевая система» известен в данной области и относится к моноциклической, бициклической или полициклической углеводородной кольцевой системе, где по меньшей мере одно кольцо является ароматическим
Термин «арил» относится к одновалентному радикалу ароматической кольцевой системы. Типичные арильные группы включают полностью ароматические кольцевые системы, такие как фенил, нафтил и антраценил, и кольцевые системы, в которых ароматическое углеродное кольцо конденсировано с одним или более неароматическими углеродными кольцами, такими как, например, инданил, фталимидил, нафтимидил или тетрагидронафтил и тому подобное.
Термин «арилалкил» или «аралкил» относится к алкильному фрагменту, в котором алкильный атом водорода замещен арильной группой. Аралкил включает группы, в которых более одного атома водорода замещено арильной группой. Примерами «арилалкила» или «аралкила» являются группы бензила, 2-фенилэтила, 3-фенилпропила, 9-флуоренила, бензгидрила и тритила.
Термин «арилокси» относится к соединению вида -O-(арил), в котором гетероарильный фрагмент соответствует определению, приведенному в данном документе.
Термин «галоген» относится к радикалу любого галогена, например, F, Cl, Br или I.
Термины «галогеналкил» и «галогеналкокси» относятся к алкильной и алкоксильной структурам, которые замещены одной или более галогенными группами или их комбинациями. Например, термины «фторалкил» и «фторалкокси» включают галогеналкильные и галогеналкоксильные группы соответственно, в которых галоген представляет собой фтор. Термин «галоалкилен» относится к двухвалентному алкилу, например, -CH2-, -CH2CH2- и -CH2CH2CH2-, в котором один или более атомов водорода замещены галогеном и включают алкильные фрагменты, в которых все атомы водорода заменены галогеном.
Термин «гетероалкил» относится к необязательно замещенному алкилу, в составе которого один или более атомов основной цепи выбраны из атомов, отличных от углерода, таких как, например, кислород, азот, сера, фосфора или их комбинации. Можно указать числовой диапазон, например, гетероалкил C1-C6, что относится к числу атомов углерода в цепи, в данном примере цепь содержит от 1 до 6 атомов углерода. Например, радикал -CH2OCH2CH3 обозначается как гетероалкил «C3». Соединение с остальной частью молекулы может осуществляться с привлечением гетероатома или углерода в гетероалкильной цепи. Термин «гетероалкилен» относится к двухвалентному необязательно замещенному алкилу, в составе которого один или более атомов основной цепи выбраны из атомов, отличных от углерода, таких как, например, кислород, азот, сера, фосфора или их комбинации.
Термин «карбоциклическая кольцевая система» относится к моноциклической, бициклической или полициклической углеводородной кольцевой системе, в которой каждое кольцо либо является полностью насыщенным, либо содержит одно или более ненасыщенных звеньев в каких-либо положениях в неароматическом кольце.
Термин «карбоциклил» относится к одновалентному радикалу карбоциклической кольцевой системы. Типичные карбоциклические группы включают циклоалкильные группы (например, циклопентил, циклобутил, циклопентил, циклогексил и т. п) и циклоалкенильные группы (например, циклопентенил, циклогексенил, циклопентадиенил и т. п).
Термин «циклоалкил» относится к циклическим, бициклическим, трициклическим или полициклическим неароматическим углеводородным группам, имеющим от 3 до 12 атомов углерода. Любой замещаемый кольцевой атом может быть замещен (например, одним или более заместителями). Циклоалкильные группы могут содержать конденсированные кольца или спирокольца. Конденсированными кольцами являются кольца, содержащие общий атом углерода. Примеры циклоалкильных остатков включают, но не ограничиваются ими, циклопропил, циклогексил, метилциклогексил, адамантил и норборнил. В некоторых вариантах осуществления циклоалкил представляет собой бицикло[3.1.0]гексанил.
Термин «циклоалкилалкил» относится к -(циклоалкил)-алкильному радикалу, где циклоалкил и алкил соответствует определениям, приведенным в данном документе. «Циклоалкилалкил» связывается с исходной молекулярной структурой посредством циклоалкильной группы.
Термин «гетероароматическая кольцевая система» известен в данной области и относится к моноциклической, бициклической или полициклической кольцевой системе, в которой по меньшей мере одно кольцо является ароматическим и одновременно содержит по меньшей мере один гетероатом (например, N, О или S); и где никакие другие кольца не являются гетероциклилами (как определено ниже). В некоторых случаях ароматическое кольцо, содержащее гетероатом, содержит 1, 2, 3 или 4 кольцевых гетероатома в таком кольце.
Термин «гетероарил» относится к одновалентному радикалу гетероароматической кольцевой системы. Типичные гетероарильные группы включают кольцевые системы, где (i) каждое кольцо содержит гетероатом и является ароматическим, например, представляет собой имидазолил, оксазолил, тиазолил, триазолил, пирролил, фуранил, тиофенил пиразолил, пиридинил, пиразинил, пиридазинил, пиримидинил, индолизинил, пуринил, нафтиридинил, и птеридинил; (ii) каждое кольцо является ароматическим или карбоциклилом, по меньшей мере одно ароматическое кольцо содержит гетероатом и по меньшей мере одно другое кольцо представляет собой гидрокарбоновое кольцо или, например, индолил, изоиндолил, бензотиенил, бензофуранил, дибензофуранил, индазолил, бензимидазолил, бензотаизолил, хинолил, изохинолил, циннолинил, фталазинил, хиназолинил, хиноксалинил, карбазолил, акридинил, феназинил, фенотиазитнил, феноксазинил, пиридо[2,3 b] 1,4 оксазин 3-(4H) он, 5,6,7,8 тетрагидрохинолинил и 5,6,7,8 тетрагидроизохинолинил; и (iii) каждое кольцо является ароматическим или карбоциклилом, и по меньшей мере одно ароматическое кольцо имеет в голове моста один общий с другим ароматическим кольцом гетероатом, например, 4H хинолизинил.
Термин «гетероциклическая кольцевая система» относится к моноциклическим, бициклическим и полициклическим кольцевым системам, в которых по меньшей мере одно кольцо является насыщенным или частично ненасыщенным (но не ароматическим) и содержит по меньшей мере один гетероатом. Гетероциклическая кольцевая система может быть присоединена к соответствующей боковой группе посредством любого гетероатома или атома углерода, что приводит к стабильной структуре, и любой из атомов кольца может быть необязательно замещен.
Термин «гетероциклил» относится к одновалентному радикалу гетероциклической кольцевой системы. Примеры гетероциклилов включают кольцевые системы, в которых (i) каждое кольцо является неароматическим и по меньшей мере одно кольцо содержит гетероатом, например, тетрагидрофуранил, тетрагидропиранил, тетрагидротиенил, пирролидинил, пирролидонил, пиперидинил, пирролинил, декагидрохинолинил, оксазолидинил, пиперазинил, диоксанил, диоксоланил, диазепинил, оксазепинил, тиазепинил, морфолинил и хинуклидинил; (ii) по меньшей мере одно кольцо является неароматическим и содержит гетероатом и по меньшей мере одно другое кольцо является ароматическим углеродным кольцом, например, 1,2,3,4 тетрагидрохинолинил, 1,2,3,4 тетрагидроизохинолинил; и (iii) по меньшей мере одно кольцо является неароматическим и содержит гетероатом, и по меньшей мере одно другое кольцо является ароматическим и содержит гетероатом, например, 3,4 дигидро 1H пиранo[4,3 c]пиридин и 1,2,3,4 тетрагидро 2,6 нафтиридин.
Термин «гетероциклилалкил» относится к алкильной группе, замещенной гетероциклильной группой.
Термин «циано» относится к радикалу -CN.
Термин «нитро» относится к -NO2.
Термин «гидрокси» или «гидроксил» относится к -OH.
Термин «гидроксиалкилен» относится к двухвалентному алкилу, например, -CH2-, -CH2CH2- и -CH2CH2CH2-, в котором один или более атомов водорода замещены гидрокси и включают алкильные фрагменты, в которых все атомы водорода заменены гидрокси.
Термин «замещенный», которому предшествует или не предшествует термин «необязательно», означает, что один или более атомов водорода в обозначенном фрагменте замещены подходящим заместителем. Если не указано иное, «необязательно замещенная» группа может иметь подходящий заместитель в каждом замещаемом положении группы, и когда более чем одно положение в любой данной структуре может быть замещено более чем одним заместителем, выбранным из указанной группы, заместители в каждом из положений могут быть одинаковыми или разными. Комбинации заместителей, предусмотренных в настоящем изобретении, предпочтительно представляют собой соединения, которые приводят к образованию стабильных или химически приемлемых соединений. Используемый в данном документе термин «стабильный» относится к соединениям, которые по существу не изменяются при воздействии условий, обеспечивающих их получение, обнаружение и, в некоторых вариантах осуществления, их извлечение, очистку и использование для одной или более целей, описанных в данном документе.
При использовании в данном документе определение каждого выражения, например, алкил, m, n и т. д., при указании его более одного раза в любой формулировке, должно считаться независимым от его определения в другом месте в той же формулировке.
Некоторые соединения по настоящему изобретению могут существовать в определенных геометрических или стереоизомерных формах. В настоящем изобретении рассматриваются все такие соединения, включая цис и транс изомеры, R и S энантиомеры, диастереомеры, (D) изомеры, (L) изомеры, их рацемические смеси и другие их смеси, входящие в объем изобретения. Дополнительные асимметричные атомы углерода могут присутствовать в заместителе, таком как алкильная группа. Все подобные изомеры, а также их смеси, предназначены для включения в настоящее изобретение.
Если, например, желателен конкретный энантиомер соединения по настоящему изобретению, его можно получить путем асимметричного синтеза или путем получения с использованием хирального вспомогательного вещества, при котором полученную диастереомерную смесь разделяют и вспомогательную группу расщепляют для получения чистого желаемого энантиомера. Альтернативно, когда молекула содержит основную функциональную группу, такую как аминогруппа, или кислотную функциональную группу, такую как карбоксил, диастереомерные соли образуются с соответствующей оптически активной кислотой или основанием с последующим разделением образующихся диастереомеров посредством фракционной кристаллизации или хроматографических способов, хорошо известные в данной области, с последующим извлечением чистых энантиомеров.
Если не указано иное, в случае, когда соединение в соответствии с раскрытием настоящего изобретения названо или изображено в виде структуры без указания стереохимии и имеет один или более хиральных центров, подразумевается, что они представляют собой все возможные стереоизомеры соединения, а также их энантиомерные смеси.
Значения «энантиомерного избытка» или «выраженного в процентах энантиомерного избытка» композиции можно рассчитать, используя приведенное ниже уравнение. В приведенном ниже примере композиция содержит 90% одного энантиомера, например, S-энантиомера, и 10% другого энантиомера, то есть R-энантиомера.
ee=(90-10)/100=80%.
Таким образом, композиция, содержащая 90% одного энантиомера и 10% другого энантиомера, имеет энантиомерный избыток 80%.
Соединения или композиции, описанные в данном документе, могут содержать энантиомерный избыток по меньшей мере 50%, 75%, 90%, 95% или 99% одной формы соединения, например, S-энантиомера. Другими словами, такие соединения или композиции содержат энантиомерный избыток S-энантиомера над R-энантиомером.
Соединения, описанные в данном документе, могут также содержать неестественные пропорции атомных изотопов в одном или более атомах, которые составляют такие соединения. Например, соединения могут быть радиоактивно мечены радиоактивными изотопами, такими как, например, дейтерий (2Н), тритий (3Н), углерод-13 (13C) или углерод-14 (14C). Все изотопные вариации соединений, описанных в данном документе, независимо от того, являются они радиоактивными или нет, предназначены для охвата объемом настоящего изобретения. Кроме того, все таутомерные формы соединений, описанных в данном документе, предназначены для охвата объемом изобретения.
Соединение может использоваться в виде свободного основания или в виде соли. Типичные соли включают гидробромид, гидрохлорид, сульфат, бисульфат, фосфат, нитрат, ацетат, валерат, олеат, пальмитат, стеарат, лаурат, бензоат, лактат, фосфат, тозилат, цитрат, малеат, фумарат, сукцинат, тартрат, нафтилат, мезилат, глюкогептонат, лактобионат, лаурилсульфонатные соли и тому подобное. (См, например, Berge et al. (1977) «Pharmaceutical Salts», J. Pharm. Sci. 66:1-19.)
Соединения
Изобретение включает соединения по формуле (I) или стереоизомер, энантиомер, таутомер, изотопически меченую форму или фармацевтически приемлемую соль любого из вышеуказанного, где:
Формула (I)
Каждое из колец A и B независимо выбрано из следующих соединений: арил, гетероарил, циклоалкил и гетероциклил;
каждое L1 и L2 независимо выбрано из связи или следующих соединений: -C(O)-, N(R1), N(R1) C(O)-, -C(O)-N(R1), (C1-C6 алкилен) N(R1), N(R1) (C1-C6 алкилен), N(R1) C(O)-(C1-C6 алкилен) и -C(O)-N(R1) (C1-C6 алкилен) ; и при этом каждый алкилен независимо замещен 0-5 соединениями R';
каждое RA и RB независимо выбрано из следующих соединений: гидроксил, C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C1-C6 алкоксил, галоген, C1-C6 гетероалкил, C1-C6 галогеналкил, C1-C6 галогеналкоксил, C1-C6 гидроксиалкил, циклоалкил, арил, гетероарил, арилокси, аралкил, гетероциклил, гетероциклилалкил, нитро, циано, -C(O)R1, -OC(O)R1, -C(O)OR1, (C1-C6 алкилен)-C(O)R1, -SR1, -S(O)2R1, -S(O)2-N(R1)(R1), (C1-C6 алкилен)-S(O)2R1, (C1-C6 алкилен)-S(O)2-N(R1)(R1), -N(R1)(R1), -C(O)-N(R1)(R1), -N(R1)-C(O)R1, -N(R1)-C(O)OR1, -(C1-C6 алкилен) N(R1) C(O)R1, -N(R1)S(O)2R1, и -P(O)(R1)(R1); и при этом каждое из следующих соединений: алкил, алкенил, алкинил, алкоксил, гетероалкил, галогеналкил, галогеналкоксил, гидроксиалкил, циклоалкил, арил, гетероарил, арилокси, аралкил, гетероциклил и гетероциклилалкил, независимо замещено 0-5 соединениями Ra; или 2 RA или 2 RB вместе с атомом(-ами) углерода, к которому(-ым) они присоединены, формируют циклоалкильное или гетероциклильное кольцо, независимо замещенное 0-5 соединениями Ra;
каждое R1 независимо выбрано из следующих соединений: водород, гидроксил, галоген, тиол, C1-C6 алкил, C1-C6 тиоалкил, C1-C6 алкоксил, C1-C6 галогеналкил, C1-C6 гидроксиалкил, C1-C6 гетероалкил, циклоалкил, циклоалкилалкил, гетероарилалкил, гетероциклил и гетероциклилалкил, и при этом каждое из следующих соединений: алкил, тиоалкил, алкоксил, галогеналкил, гидроксиалкил, гетероалкил, циклоалкил, циклоалкилалкил, гетероарилалкил, гетероциклил и гетероциклилалкил, независимо замещено 0-5 соединениями Rb или 2 R1 вместе с атомом(-ами), к которому(-ым) они присоединены, формируют циклоалкильное или гетероциклильное кольцо, независимо замещенное 0-5 соединениями Rb;
каждое Ra и Rb независимо выбрано из следующих соединений: C1-C6 алкил, галоген, гидроксил, C1-C6 галогеналкил, C1-C6 гетероалкил, C1-C6 гидроксиалкил, C1-C6 алкоксил, циклоалкил, гетероциклил, и циано, и при этом каждое из следующих соединений: алкил, галогеналкил, гетероалкил, гидроксиалкил, алкоксил, циклоалкил и гетероциклил независимо замещено 0-5 соединениями R';
каждое R' независимо выбрано из следующих соединений: C1-C6 алкил, C1-C6 гетероалкил, галоген, гидроксил, C1-C6 галогеналкил, C1-C6 гидроксиалкил, циклоалкил и циано; или 2 R'вместе с атомами, к которым они присоединены, формируют циклоалкильное или гетероциклильное кольцо; и
p равно 0, 1, 2, 3, 4 или 5; и
q равно 0, 1, 2, 3 или 4.
В некоторых вариантах осуществления изобретения кольцо A представляет собой циклоалкил. В некоторых вариантах осуществления изобретения кольцо A представляет собой 5-членное или 6-членное циклоалкильное кольцо. В некоторых вариантах осуществления изобретения кольцо A представляет собой циклопентил или циклогексил. В некоторых вариантах осуществления изобретения кольцо A представляет собой гетероциклил. В некоторых вариантах осуществления изобретения кольцо A представляет собой 5-членный или 6-членный гетероциклил. В некоторых вариантах осуществления изобретения кольцо A представляет собой тетрагидропиранил, тетрагидрофуранил или пирролидинил. В некоторых вариантах осуществления изобретения кольцо A представляет собой циклоалкенильное кольцо. В некоторых вариантах осуществления изобретения кольцо A представляет собой циклопентенил.
В некоторых вариантах осуществления изобретения кольцо B представляет собой арил. В некоторых вариантах осуществления изобретения кольцо B представляет собой фенил. В некоторых вариантах осуществления изобретения кольцо B представляет собой гетероарил. В некоторых вариантах осуществления изобретения кольцо B представляет собой пиридил. В некоторых вариантах осуществления изобретения кольцо B представляет собой гетероциклил. В некоторых вариантах осуществления изобретения кольцо B представляет собой пирролидинил.
В некоторых вариантах осуществления изобретения L1 представляет собой связь, -C(O)- или N(R1) ; и L2 представляет собой N(R1) C(O)-(C1-C6 алкилен) или -C(O)-N(R1) (C1-C6 алкилен). В некоторых вариантах осуществления изобретения L1 представляет собой -NH- и L2 представляет собой -C(O)-NH-CH(CH2OH)-*, -C(O)-N(CH3)-CH2-*, -C(O)-N(CH3)-CH(CH3)-*, -C(O)N(CH2CH3)CH2-*, -C(O)NHCH(CH3)-*, -C(O)N(CD3)CH2-*, -C(O)NHCH(CF3)-* и 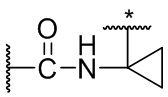 , в которых «*» представляет собой часть L2, связанную с кольцом B. В некоторых вариантах осуществления изобретения L1 представляет собой -NH-, L2 представляет собой -C(O)-, и кольцо B представляет собой пирролидинил. В некоторых вариантах осуществления изобретения L1 представляет собой -NH-, L2 представляет собой -C(O)-, и кольцо B представляет собой пирролидин-1-ил.
, в которых «*» представляет собой часть L2, связанную с кольцом B. В некоторых вариантах осуществления изобретения L1 представляет собой -NH-, L2 представляет собой -C(O)-, и кольцо B представляет собой пирролидинил. В некоторых вариантах осуществления изобретения L1 представляет собой -NH-, L2 представляет собой -C(O)-, и кольцо B представляет собой пирролидин-1-ил.
В некоторых вариантах осуществления изобретения каждое R1 независимо выбрано из водорода и C1-C6 алкила, замещенного 0-5 соединениями Rb. В некоторых вариантах осуществления изобретения каждое R1 независимо выбрано из водорода или -CH3.
В некоторых вариантах осуществления изобретения каждое RA и RB независимо выбрано из следующих соединений: гидроксил, C1-C6 алкил, C1-C6 алкоксил, галоген, C1-C6 гетероалкил, C1-C6 галогеналкил, C1-C6 галогеналкоксил, C1-C6 гидроксиалкил, циклоалкил, арил, гетероарил, нитро, циано, -C(O)R1, -OC(O)R1, -C(O)OR1, -SR1, -S(O)2R1, -S(O)2-N(R1)(R1), -N(R1)(R1), -C(O)-N(R1)(R1), -N(R1)-C(O)R1, -N(R1)-C(O)OR1 и -N(R1)S(O)2R1; и при этом каждое из следующих соединений: алкил, алкоксил, гетероалкил, галогеналкил, галогеналкоксил, гидроксиалкил, циклоалкил, арил и гетероарил, независимо замещено 0-5 соединениями Ra; или 2 RA или 2 RB вместе с атомами углерода, к которым они присоединены, формируют циклоалкильное или гетероциклильное кольцо, независимо замещенное 0-5 соединениями Ra.
В некоторых вариантах осуществления изобретения каждое RA независимо выбрано из следующих соединений: гидроксил, C1-C6 алкил, C1-C6 алкоксил, галоген, -C(O)-N(R1)(R1), -C(O)OR1, -S(O)2R1 и C1-C6 галогеналкил. В некоторых вариантах осуществления изобретения каждое RAдополнительно и независимо выбрано из следующих соединений: -CN, оксэтанил и C1-C6 гидроксиалкил, или два RA, связанные с соседними кольцевыми атомами углерода в кольце A, взяты вместе для формирования C3-C6 циклоалкила, конденсированого с кольцом A. В некоторых вариантах осуществления изобретения каждое RA независимо выбрано из следующих соединений: гидроксил, фтор, оксэтан-3-ил, -CHF2, CH2CH3, -C(CH3)2OH, -OCH3, -C(O)N(CH3)2, -C(O)OCH3, -S(O)2CH3; или два RA связанные с соседними кольцевыми атомами углерода в кольце A, взяты вместе для формирования циклопропила, конденсированого с кольцом A.
В некоторых вариантах осуществления изобретения каждое RB независимо выбрано из следующих соединений: галоген, C1-C6 алкил, циано, C1-C6 алкоксил, арил, гетероарил и C1-C6 галогеналкокси. В некоторых вариантах осуществления изобретения каждое RB дополнительно выбрано из оксо.
В некоторых вариантах осуществления изобретения кольцо B представляет собой пирролидинил и по меньшей мере одно RB является необязательно замещенным арилом или гетероарилом. В некоторых вариантах осуществления изобретения кольцо B представляет собой пирролидинил и по меньшей мере одно RB является необязательно замещенным фенилом или пиридилом.
В некоторых вариантах осуществления изобретения кольцо B представляет собой пирролидинил, и по меньшей мере одно RB выбрано из следующих соединений:
2,3,5-трифторфенил, 2,3-дифторфенил, 2,5-дифторфенил, 2-хлор-5-фторфенил, 2-хлор-5-фторпиридин-3-ил, 2-циано-5-фторфенил, 2-фтор-5-хлорфенил, 2-метокси-3,5-дифторфенил, 2-метокси-5-фторпиридин-3-ил, 2-трифторметокси-5-фторфенил, 3,5-дифторфенил, 3-хлор-5-фторфенил, 3-циано-5-фторфенил, 3-дифторметокси-5-фторфенил, 3-фторфенил, 5-фторпиридин-3-ил и фенил.
В некоторых вариантах осуществления изобретения кольцо B представляет собой пирролидинил и одно дополнительное RB, при его наличии, представляет собой фтор.
В некоторых вариантах осуществления изобретения кольцо B представляет собой соединение, отличное от пирролидинила, и каждое RB независимо выбрано из хлора, фтора, оксо, -CH3, -CF3, -CN, -OCH3, -OCF3 и -OCHF2.
В другом аспекте в объеме изобретения представлены соединения по формуле (Ia):
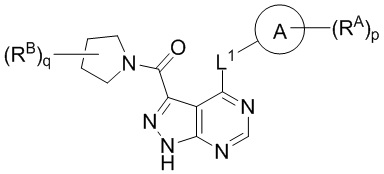 (Ia)
(Ia)
или стереоизомер, энантиомер, таутомер или изотопически меченая форма такого соединения, или фармацевтически приемлемая соль любого из вышеуказанных, где:
кольцо A выбрано из арила, гетероарила, циклоалкила и гетероциклила;
L1 выбрано из связи или следующих соединений: -C(O)-, N(R1), N(R1) C(O)-, -C(O)-N(R1), (C1-C6 алкилен) N(R1), N(R1) (C1-C6 алкилен), N(R1) C(O)-(C1-C6 алкилен) и -C(O)-N(R1) (C1-C6 алкилен) ; и при этом каждый алкилен независимо замещен 0-5 соединениями R';
каждое RA и RB независимо выбрано из следующих соединений: гидроксил, C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C1-C6 алкоксил, галоген, C1-C6 гетероалкил, C1-C6 галогеналкил, C1-C6 галогеналкоксил, C1-C6 гидроксиалкил, циклоалкил, арил, гетероарил, арилокси, аралкил, гетероциклил, гетероциклилалкил, нитро, циано, -C(O)R1, -OC(O)R1, -C(O)OR1, (C1-C6 алкилен)-C(O)R1, -SR1, -S(O)2R1, -S(O)2-N(R1)(R1), (C1-C6 алкилен)-S(O)2R1, (C1-C6 алкилен)-S(O)2-N(R1)(R1), -N(R1)(R1), -C(O)-N(R1)(R1), -N(R1)-C(O)R1, -N(R1)-C(O)OR1, -(C1-C6 алкилен) N(R1) C(O)R1, -N(R1)S(O)2R1, и -P(O)(R1)(R1); и при этом каждое из следующих соединений: алкил, алкенил, алкинил, алкоксил, гетероалкил, галогеналкил, галогеналкоксил, гидроксиалкил, циклоалкил, арил, гетероарил, арилокси, аралкил, гетероциклил и гетероциклилалкил, независимо замещено 0-5 соединениями Ra; или 2 RA или 2 RB вместе с атомом(-ами) углерода, к которому(-ым) они присоединены, формируют циклоалкильное или гетероциклильное кольцо, независимо замещенное 0-5 соединениями Ra;
каждое R1 независимо выбрано из следующих соединений: водород, гидроксил, галоген, тиол, C1-C6 алкил, C1-C6 тиоалкил, C1-C6 алкоксил, C1-C6 галогеналкил, C1-C6 гидроксиалкил, C1-C6 гетероалкил, циклоалкил, циклоалкилалкил, гетероарилалкил, гетероциклил и гетероциклилалкил, и при этом каждое из следующих соединений: алкил, тиоалкил, алкоксил, галогеналкил, гидроксиалкил, гетероалкил, циклоалкил, циклоалкилалкил, гетероарилалкил, гетероциклил и гетероциклилалкил, независимо замещено 0-5 соединениями Rb или 2 R1 вместе с атомом(-ами), к которому(-ым) они присоединены, формируют циклоалкильное или гетероциклильное кольцо, независимо замещенное 0-5 соединениями Rb;
каждое Ra и Rb независимо выбрано из следующих соединений: C1-C6 алкил, галоген, гидроксил, C1-C6 галогеналкил, C1-C6 гетероалкил, C1-C6 гидроксиалкил, C1-C6 алкоксил, циклоалкил, гетероциклил, и циано, и при этом каждое из следующих соединений: алкил, галогеналкил, гетероалкил, гидроксиалкил, алкоксил, циклоалкил и гетероциклил независимо замещено 0-5 соединениями R';
каждое R' независимо выбрано из следующих соединений: C1-C6 алкил, C1-C6 гетероалкил, галоген, гидроксил, C1-C6 галогеналкил, C1-C6 гидроксиалкил, циклоалкил и циано; или 2 R'вместе с атомами, к которым они присоединены, формируют циклоалкильное или гетероциклильное кольцо; и
p равно 0, 1, 2, 3, 4 или 5; и
q равно 0, 1, 2, 3 или 4.
В некоторых вариантах осуществления изобретения кольцо A представляет собой циклоалкил. В некоторых вариантах осуществления изобретения кольцо A представляет собой 5-членное или 6-членное циклоалкильное кольцо. В некоторых вариантах осуществления изобретения кольцо A представляет собой циклопентил или циклогексил. В некоторых вариантах осуществления изобретения кольцо A представляет собой гетероциклил. В некоторых вариантах осуществления изобретения кольцо A представляет собой 5-членный или 6-членный гетероциклил. В некоторых вариантах осуществления изобретения кольцо A представляет собой тетрагидропиран, тетрагидрофуран или пирролидинил. В некоторых вариантах осуществления изобретения кольцо A представляет собой циклоалкенильное кольцо. В некоторых вариантах осуществления изобретения кольцо A представляет собой циклопентенил.
В некоторых вариантах осуществления изобретения L1 представляет собой связь, -C(O)- или N(R1). В некоторых вариантах осуществления изобретения L1 представляет собой NH.
В некоторых вариантах осуществления изобретения каждое R1 независимо выбрано из водорода и C1-C6 алкила, замещенного 0-5 соединениями Rb. В некоторых вариантах осуществления изобретения каждое R1 независимо выбрано из водорода или -CH3.
В некоторых вариантах осуществления изобретения каждое RA и RB независимо выбрано из следующих соединений: гидроксил, C1-C6 алкил, C1-C6 алкоксил, галоген, C1-C6 гетероалкил, C1-C6 галогеналкил, C1-C6 галогеналкоксил, C1-C6 гидроксиалкил, циклоалкил, арил, гетероарил, нитро, циано, -C(O)R1, -OC(O)R1, -C(O)OR1, -SR1, -S(O)2R1, -S(O)2-N(R1)(R1), -N(R1)(R1), -C(O)-N(R1)(R1), -N(R1)-C(O)R1, -N(R1)-C(O)OR1 и -N(R1)S(O)2R1; и при этом каждое из следующих соединений: алкил, алкоксил, гетероалкил, галогеналкил, галогеналкоксил, гидроксиалкил, циклоалкил, арил и гетероарил, независимо замещено 0-5 соединениями Ra; или 2 RA или 2 RB вместе с атомами углерода, к которым они присоединены, формируют циклоалкильное или гетероциклильное кольцо, независимо замещенное 0-5 соединениями Ra.
В некоторых вариантах осуществления изобретения каждое RA независимо выбрано из следующих соединений: гидроксил, C1-C6 алкил, C1-C6 алкоксил, галоген, -C(O)-N(R1)(R1), -C(O)OR1, -S(O)2R1 и C1-C6 галогеналкил. В некоторых вариантах осуществления изобретения каждое RAдополнительно и независимо выбрано из следующих соединений: -CN, оксэтанил и C1-C6 гидроксиалкил, или два RA, связанные с соседними кольцевыми атомами углерода в кольце A, взяты вместе для формирования C3-C6 циклоалкила, конденсированого с кольцом A. В некоторых вариантах осуществления изобретения каждое RA независимо выбрано из следующих соединений: гидроксил, фтор, оксэтан-3-ил, -CHF2, CH2CH3, -C(CH3)2OH, -OCH3, -C(O)N(CH3)2, -C(O)OCH3, -S(O)2CH3; или два RA связанные с соседними кольцевыми атомами углерода в кольце A, взяты вместе для формирования циклопропила, конденсированого с кольцом A.
В некоторых вариантах осуществления изобретения каждое RB независимо выбрано из следующих соединений: галоген, C1-C6 алкил, циано, C1-C6 алкоксил, арил, гетероарил и C1-C6 галогеналкокси. В некоторых вариантах осуществления изобретения каждое RB дополнительно выбрано из оксо.
В некоторых вариантах осуществления изобретения, когда кольцо B представляет собой пирролидинил, по меньшей мере одно из RB выбрано из следующих соединений: 2,3,5-трифторфенил, 2,3-дифторфенил, 2,5-дифторфенил, 2-хлор-5-фторфенил, 2-хлор-5-фторпиридин-3-ил, 2-циано-5-фторфенил, 2-фтор-5-хлорфенил, 2-метокси-3,5-дифторфенил, 2-метокси-5-фторпиридин-3-ил, 2-трифторметокси-5-фторфенил, 3,5-дифторфенил, 3-хлор-5-фторфенил, 3-циано-5-фторфенил, 3-дифторметокси-5-фторфенил, 3-фторфенил, 5-фторпиридин-3-ил и фенил.
В некоторых вариантах осуществления изобретения, когда кольцо B представляет собой пирролидинил, одно дополнительное RB, при его наличии, представляет собой фтор.
В некоторых вариантах осуществления p равно 0, 1 или 2.
В некоторых вариантах осуществления q равно 1, 2 или 3.
В другом аспекте в объеме изобретения представлены соединения по формуле (II):
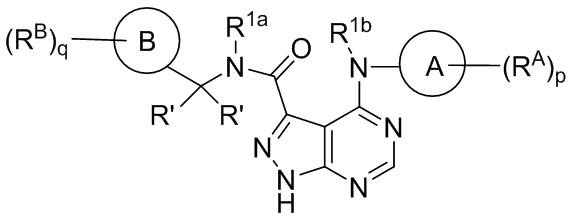 (II),
(II),
или стереоизомер, энантиомер, таутомер или изотопически меченая форма такого соединения, или фармацевтически приемлемая соль любого из вышеуказанных, где:
Каждое из колец A и B независимо выбрано из следующих соединений: арил, гетероарил, циклоалкил и гетероциклил;
каждое RA и RB независимо выбрано из следующих соединений: гидроксил, C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C1-C6 алкоксил, галоген, C1-C6 гетероалкил, C1-C6 галогеналкил, C1-C6 галогеналкоксил, C1-C6 гидроксиалкил, циклоалкил, арил, гетероарил, арилокси, аралкил, гетероциклил, гетероциклилалкил, нитро, циано, -C(O)R1, -OC(O)R1, -C(O)OR1, (C1-C6 алкилен)-C(O)R1, -SR1, -S(O)2R1, -S(O)2-N(R1)(R1), (C1-C6 алкилен)-S(O)2R1, (C1-C6 алкилен)-S(O)2-N(R1)(R1), -N(R1)(R1), -C(O)-N(R1)(R1), -N(R1)-C(O)R1, -N(R1)-C(O)OR1, -(C1-C6 алкилен) N(R1) C(O)R1, -N(R1)S(O)2R1, и -P(O)(R1)(R1); и при этом каждое из следующих соединений: алкил, алкенил, алкинил, алкоксил, гетероалкил, галогеналкил, галогеналкоксил, гидроксиалкил, циклоалкил, арил, гетероарил, арилокси, аралкил, гетероциклил и гетероциклилалкил, независимо замещено 0-5 соединениями Ra; или 2 RA или 2 RB вместе с атомом(-ами) углерода, к которому(-ым) они присоединены, формируют циклоалкильное или гетероциклильное кольцо, независимо замещенное 0-5 соединениями Ra;
каждое R1 независимо выбрано из следующих соединений: водород, гидроксил, галоген, тиол, C1-C6 алкил, C1-C6 тиоалкил, C1-C6 алкоксил, C1-C6 галогеналкил, C1-C6 гидроксиалкил, C1-C6 гетероалкил, циклоалкил, циклоалкилалкил, гетероарилалкил, гетероциклил и гетероциклилалкил, и при этом каждое из следующих соединений: алкил, тиоалкил, алкоксил, галогеналкил, гидроксиалкил, гетероалкил, циклоалкил, циклоалкилалкил, гетероарилалкил, гетероциклил и гетероциклилалкил, независимо замещено 0-5 соединениями Rb или 2 R1 вместе с атомом(-ами), к которому(-ым) они присоединены, формируют циклоалкильное или гетероциклильное кольцо, независимо замещенное 0-5 соединениями Rb;
R1a выбрано из водорода, C1-C6 алкила и дейтерированного C1-C6алкила; R1b выбрано из водорода и C1-C6алкила;
каждое Ra и Rb независимо выбрано из следующих соединений: C1-C6 алкил, галоген, гидроксил, C1-C6 галогеналкил, C1-C6 гетероалкил, C1-C6 гидроксиалкил, C1-C6 алкоксил, циклоалкил, гетероциклил, и циано, и при этом каждое из следующих соединений: алкил, галогеналкил, гетероалкил, гидроксиалкил, алкоксил, циклоалкил и гетероциклил независимо замещено 0-5 соединениями R';
каждое R' независимо выбрано из следующих соединений: C1-C6 алкил, C1-C6 гетероалкил, галоген, гидроксил, C1-C6 галогеналкил, C1-C6 гидроксиалкил, циклоалкил и циано; или 2 R'вместе с атомами, к которым они присоединены, формируют циклоалкильное или гетероциклильное кольцо; и
p равно 0, 1, 2, 3, 4 или 5; и
q равно 0, 1, 2, 3 или 4.
В некоторых вариантах осуществления изобретения кольцо A представляет собой циклоалкил. В некоторых вариантах осуществления изобретения кольцо A представляет собой 5-членное или 6-членное циклоалкильное кольцо. В некоторых вариантах осуществления изобретения кольцо A представляет собой циклопентил или циклогексил. В некоторых вариантах осуществления изобретения кольцо A представляет собой гетероциклил. В некоторых вариантах осуществления изобретения кольцо A представляет собой 5-членный или 6-членный гетероциклил. В некоторых вариантах осуществления изобретения кольцо A представляет собой тетрагидропиран, тетрагидрофуран или пирролидинил. В некоторых вариантах осуществления изобретения кольцо A представляет собой циклоалкенильное кольцо. В некоторых вариантах осуществления изобретения кольцо A представляет собой циклопентенил.
В некоторых вариантах осуществления изобретения кольцо B представляет собой арил. В некоторых вариантах осуществления изобретения кольцо B представляет собой фенил. В некоторых вариантах осуществления изобретения кольцо B представляет собой гетероарил. В некоторых вариантах осуществления изобретения кольцо B представляет собой пиридил.
В некоторых вариантах осуществления изобретения каждое R1 независимо выбрано из водорода и C1-C6 алкила, замещенного 0-5 соединениями Rb.
В некоторых вариантах осуществления изобретения R1a представляет собой водород, -CH3, -CD3 или -CH2CH3.
В некоторых вариантах осуществления изобретения R1b представляет собой водород.
В некоторых вариантах осуществления изобретения каждое RA и RB независимо выбрано из следующих соединений: гидроксил, C1-C6 алкил, C1-C6 алкоксил, галоген, C1-C6 гетероалкил, C1-C6 галогеналкил, C1-C6 галогеналкоксил, C1-C6 гидроксиалкил, циклоалкил, арил, гетероарил, нитро, циано, -C(O)R1, -OC(O)R1, -C(O)OR1, -SR1, -S(O)2R1, -S(O)2-N(R1)(R1), -N(R1)(R1), -C(O)-N(R1)(R1), -N(R1)-C(O)R1, -N(R1)-C(O)OR1 и -N(R1)S(O)2R1; и при этом каждое из следующих соединений: алкил, алкоксил, гетероалкил, галогеналкил, галогеналкоксил, гидроксиалкил, циклоалкил, арил и гетероарил, независимо замещено 0-5 соединениями Ra; или 2 RA или 2 RB вместе с атомами углерода, к которым они присоединены, формируют циклоалкильное или гетероциклильное кольцо, независимо замещенное 0-5 соединениями Ra.
В некоторых вариантах осуществления изобретения каждое RA независимо выбрано из следующих соединений: гидроксил, C1-C6 алкил, C1-C6 алкоксил, галоген, -C(O)-N(R1)(R1), -C(O)OR1, -S(O)2R1 и C1-C6 галогеналкил. В некоторых вариантах осуществления изобретения каждое RA дополнительно и независимо выбрано из следующих соединений: -CN, оксэтанил и C1-C6 гидроксиалкил, или два RA, связанные с соседними кольцевыми атомами углерода в кольце A, взяты вместе для формирования C3-C6 циклоалкила, конденсированного с кольцом A. В некоторых вариантах осуществления изобретения каждое RA независимо выбрано из следующих соединений: гидроксил, фтор, оксэтан-3-ил, -CHF2, CH2CH3, -C(CH3)2OH, -OCH3, -C(O)N(CH3)2, -C(O)OCH3, -S(O)2CH3; или два RA связанные с соседними кольцевыми атомами углерода в кольце A, взяты вместе для формирования циклопропила, конденсированного с кольцом A.
В некоторых вариантах осуществления изобретения каждое RB независимо выбрано из следующих соединений: галоген, C1-C6 алкил, циано, C1-C6 алкоксил, арил, гетероарил и C1-C6 галогеналкокси. В некоторых вариантах осуществления изобретения каждое RB дополнительно выбрано из оксо. В некоторых вариантах осуществления изобретения каждое RB независимо выбрано из следующих соединений: хлор, фтор, оксо, -CH3, -CF3, -CN, -OCH3, -OCF3, и -OCHF2.
В некоторых вариантах осуществления изобретения каждое R' независимо выбрано из следующих соединений: C1-C6 алкил, C1-C6 галогеналкил и C1-C6 гидроксиалкил; или 2 R' вместе с атомами, к которым они присоединены, формируют циклоалкильное или гетероциклильное кольцо. В некоторых вариантах осуществления изобретения одно R' представляет собой водород, и другое R' выбрано из водорода, C1-C6 алкила, C1-C6 галогеналкила и C1-C6 гидроксиалкила; или 2 R' вместе с атомами, к которым они присоединены, формируют циклоалкильное кольцо. В некоторых вариантах осуществления изобретения одно R' представляет собой водород, и другое R' выбрано из водорода, -CH2OH, -CH3 или -CF3 или 2 R' вместе с атомами, к которым они присоединены, формируют циклопроп-1,1-дииловое кольцо.
В некоторых вариантах осуществления p равно 0, 1 или 2.
В некоторых вариантах осуществления изобретения q равно 0, 1, 2 или 3.
Хотя, как указано выше, различные варианты осуществления изобретения и его аспекты для переменной в формулах (I), (Ia) или (II) могут быть выбраны из группы химических фрагментов, изобретение также охватывает дополнительные варианты и аспекты подобных ситуаций, в которых такая переменная: а) выбрана из любой подгруппы химических групп в такой группе; и b) любого отдельного члена такой группы.
Хотя различные варианты осуществления и их аспекты излагаются (или подразумеваются, как обсуждалось в предыдущем параграфе) индивидуально для каждой переменной в формулах (I), (Ia) и (II), изобретение охватывает все возможные комбинации различных вариантов осуществления и аспектов для каждой из переменных в формулах (I), (Ia) и (II).
Структуры, а также данные ЯМР и ЖХМС приведенных в качестве примеров соединений в соответствии с изобретением показаны на фигуре 1. В некоторых вариантах осуществления соединение по настоящему изобретению выбрано из группы, состоящей из любого из соединений на фигуре 1, а также фармацевтически приемлемых солей, сольватов, гидратов, таутомеров, стереоизомеров и изотопически меченых производных таких соединений.
Изобретение также относится к фармацевтическим композициям, содержащим фармацевтически приемлемый носитель и любое соединение по формулам (I), (Ia) и (II).
Фармацевтически приемлемые соли таких соединений также рассматриваются для описанных в данном документе применений.
Термин «фармацевтически приемлемая соль» относится к любой соли соединения в соответствии с изобретением, которая сохраняет свои биологические свойства и которая не является токсичной или не обладает иными свойствами, делающими ее нежелательной для фармацевтического применения. Фармацевтически приемлемые соли могут быть получены из различных органических и неорганических противоионов, хорошо известных в данной области, и включают следующие. Такие соли включают: (1) кислотно-аддитивные соли, образованные с органическими или неорганическими кислотами, такими как хлористоводородная, бромистоводородная, серная, азотная, фосфорная, сульфаминовая, уксусная, трифторуксусная, трихлоруксусная, пропионовая, гексановая, циклопентилпропионовая, гликолевая, глутаровая, пировиноградная, молочная, сорбиновая, аскорбиновая, яблочная, малеиновая, фумаровая, винная, лимонная, бензойная, 3-(4-гидроксибензоил)бензойная, пикриновая, коричная, миндальная, фталевая, лауриновая, метансульфоновая, этансульфоновая, 1,2-этандисульфоновая, 2-гидроксиэтансульфоновая, бензолсульфоновая, 4-хлорбензолсульфоновая, 2-нафталинсульфоновая, 4-толуолсульфоновая, камфорическая, камфорсульфоновая, 4-метилбицикло[2.2.2]-окт-2-ен-1-карбоновая, глюкогептоновая, 3-фенилпропионовая, триметилуксусная, трет-бутилуксусная, лаурилсульфоновая, глюконовая, бензойная, глутаминовая, гидроксинафтоиновая, салициловая, стеариновая, циклогексилсульфаминовая, хиновая, муконовая кислота и подобные кислоты; или (2) соли, образующиеся в случае, когда кислотный протон, присутствующий в исходном соединении, либо (а) замещается ионом металла, например, ионом щелочного металла, ионом щелочноземельного металла или ионом алюминия, или гидроксидом щелочных или щелочноземельных металлов, таких как гидроксид натрия, калия, кальция, магния, алюминия, лития, цинка и бария, аммиак или (б) координаты с органическим основанием, таким как алифатические, алициклические или ароматические органические амины, такие как аммиак, метиламин, диметиламин диэтиламин, пиколин, этаноламин, диэтаноламин, триэтаноламин, этилендиамин, лизин, аргинин, орнитин, холин, N, N'-дибензилэтилендиамин, хлорпрокаин, диэтаноламин, прокаин, N-бензилфенэтиламин, N- метилглюкаминпиперазин, трис (гидроксиметил) аминометан, гидроксид тетраметиламмония и тому подобное. Фармацевтически приемлемые соли дополнительно включают, только в качестве примера, соли натрия, калия, кальция, магния, аммония, тетраалкиламмония и т. п., а в случае, когда соединение содержит основную функциональную группу, также соли нетоксичных органических или неорганических кислот, такие как гидрохлорид, гидробромид, тартрат, мезилат, безилат, ацетат, малеат, оксалат и тому подобное.
Фармацевтические композиции
Фармацевтические композиции в соответствии с изобретением содержат одно или более соединений в соответствии с изобретением и один или более физиологически или фармацевтически приемлемый носитель. Термин «фармацевтически приемлемый носитель» относится к фармацевтически приемлемому материалу, композиции или носителю, такому как жидкий или твердый наполнитель, разбавитель, эксципиент, растворитель или инкапсулирующий материал, участвующий в переноске или транспортировке любой предметной композиции или ее компонента. Каждый носитель должен быть «приемлемым» в смысле совместимости с композицией состава и его компонентов и отсутствия вреда для пациента. Некоторые примеры материалов, которые могут служить в качестве фармацевтически приемлемых носителей, включают следующие вещества: (1) сахара, такие как лактоза, глюкоза и сахароза; (2) крахмалы, такие как кукурузный крахмал и картофельный крахмал; (3) целлюлоза и ее производные, такие как натриевая соль карбоксиметилцеллюлозы, этилцеллюлоза и ацетат целлюлозы; (4) порошкообразный трагакант; (5) солод; (6) желатин; (7) тальк; (8) эксципиенты, такие как масло какао и суппозиторные воски; (9) масла, такие как арахисовое масло, хлопковое масло, сафлоровое масло, кунжутное масло, оливковое масло, кукурузное масло и соевое масло; (10) гликоли, такие как пропиленгликоль; (11) полиолы, такие как глицерин, сорбит, маннит и полиэтилэтенгликоль; (12) сложные эфиры, такие как этилолеат и этиллаурат; (13) агар; (14) буферные агенты, такие как гидроксид магния и гидроксид алюминия; (15) альгиновая кислота; (16) вода, не содержащая пирогенов; (17) изотонический физиологический раствор; (18) раствор Рингера; (19) этиловый спирт; (20) фосфатные буферные растворы; и (21) другие нетоксичные совместимые вещества, используемые в фармацевтических препаратах.
Композиции в соответствии с изобретением можно вводить перорально, парентерально, в виде ингаляционного спрея, местно, ректально, назально, буккально, вагинально или через имплантированный резервуар. Используемый здесь термин «парентеральный» включает подкожную, внутривенную, внутримышечную, внутрисуставную, внутрисиневиальную, внутригрудинную, интратекальную, внутрипеченочную, внутриочаговую и внутричерепную инъекцию или методы инфузии. В некоторых вариантах осуществления композиции в соответствии с изобретением вводят перорально, внутрибрюшинно или внутривенно. Стерильные инъекционные формы композиций в соответствии с изобретением могут представлять собой водную или масляную суспензию. Эти суспензии могут быть составлены в соответствии со способами, известными в данной области, с использованием подходящих диспергирующих или смачивающих агентов и суспендирующих агентов. Стерильный препарат для инъекций может также представлять собой стерильный раствор для инъекций или суспензию в нетоксичном разбавителе или растворителе, пригодном для парентерального введения, например, в виде раствора в 1,3-бутандиоле. В числе приемлемых носителей и растворителей, которые могут быть использованы, могут использоваться вода, раствор Рингера и изотонический раствор хлорида натрия. Кроме того, стерильные нелетучие масла обычно используют в качестве растворителя или суспендирующей среды.
Для этой цели можно использовать любое мягкое фиксированное масло, включая синтетические моно- или диглицериды. Жирные кислоты, такие как олеиновая кислота и ее глицеридные производные, пригодны для приготовления инъекционных материалов, также как и натуральные фармацевтически приемлемые масла, такие как оливковое масло или касторовое масло, особенно в их полиоксиэтилированных версиях. Подобные масляные растворы или суспензии могут также содержать длинноцепочечный спиртовой разбавитель или диспергент, такой как карбоксиметилцеллюлоза или аналогичные диспергирующие агенты, которые обычно используются в составе фармацевтически приемлемых лекарственных форм, включая эмульсии и суспензии. Другие общеупотребительные поверхностно-активные вещества, такие как Tween, Spans и другие эмульгирующие агенты или энхансеры биодоступности, которые обычно используются при изготовлении фармацевтически приемлемых твердых, жидких или других лекарственных форм, также могут быть использованы в технологии изготовления препарата.
Фармацевтически приемлемые композиции по настоящему изобретению можно вводить перорально в любой приемлемой для перорального применения лекарственной форме, включая, но не ограничиваясь ими, капсулы, таблетки, водные суспензии или растворы. В случае таблеток для перорального применения обычно используемые носители включают лактозу и кукурузный крахмал. Также обычно добавляют смазывающие агенты, такие как стеарат магния. Для перорального введения в форме капсулы применяемые разбавители включают лактозу и сухой кукурузный крахмал. При необходимости изготовления водных суспензий для перорального применения, активный ингредиент объединяют с эмульгирующими и суспендирующими агентами. При желании могут быть добавлены некоторые подслащивающие, ароматизирующие или красящие вещества.
Альтернативно, фармацевтически приемлемые композиции по настоящему изобретению можно вводить в виде суппозиториев для ректального введения. Их можно приготовить путем смешивания агента с подходящим не раздражающим эксципиентом, который является твердым при комнатной температуре, но жидким при ректальной температуре и, следовательно, расплавится в прямой кишке с высвобождением лекарственного средства. Такие материалы включают масло какао, пчелиный воск и полиэтиленгликоли.
Фармацевтически приемлемые композиции по настоящему изобретению также могут вводиться местно, особенно когда мишень лечения включает области или органы, легко доступные посредством местного применения, включая заболевания глаз, кожи или нижней части ЖКТ. Подходящие составы для местного применения легко изготовить для каждой из этих областей или органов. Местное применение для нижнего отдела ЖКТ может быть осуществлено в виде ректального суппозитория (см. выше) или при использовании препарата, изготовленного для введения посредством клизмы. Также могут быть использованы трансдермальные пластыри для местного применения.
Фармацевтически приемлемые композиции для местного применения могут быть приготовлены в виде подходящей мази, содержащей активный компонент, суспендированный или растворенный в одном или более носителях. Носители для местного введения соединений по настоящему изобретению включают, но не ограничиваются ими, минеральное масло, жидкий вазелин, белый вазелин, пропиленгликоль, полиоксиэтилен, полиоксипропиленовое соединение, эмульгирующий воск и воду. Альтернативно, фармацевтически приемлемые композиции могут быть приготовлены в виде подходящего лосьона или крема, содержащего активные компоненты, суспендированные или растворенные в одном или более фармацевтически приемлемых носителях. Подходящие носители включают, но не ограничиваются ими, минеральное масло, сорбитанмоностеарат, полисорбат 60, воск цетиловых эфиров, цетеариловый спирт, 2-октилдодеканол, бензиловый спирт и воду.
Фармацевтически приемлемые композиции по данному изобретению также могут вводиться посредством назального аэрозоля или ингаляции. Такие композиции получают в соответствии со способами, хорошо известными в сфере технологий изготовления фармацевтической композиции, и могут быть получены в виде растворов в физиологическом растворе с использованием бензилового спирта или других подходящих консервантов, промоторов абсорбции для повышения биодоступности, фторуглеродов и/или других обычных солюбилизирующих или диспергирующих агентов.
Количество соединений по настоящему изобретению, которые могут быть объединены с материалами-носителями для получения композиции в однодозовой лекарственной форме, будет варьироваться в зависимости от подлежащего обработке хозяина, конкретного способа введения. Предпочтительно, композиции должны быть составлены таким образом, чтобы пациенту, получающему эти композиции, вводили дозу ингибитора, составляющую от 0,01 до 100 мг/кг массы тела/день.
Дозировки
Токсичность и терапевтическая эффективность соединений в соответствии с изобретением, включая фармацевтически приемлемые соли и дейтерированные варианты, могут быть определены посредством стандартных фармацевтических процедур в культурах клеток или с использованием экспериментальных животных. Доза LD50 - доза, летальная для 50% популяции. ED50 - доза, терапевтически эффективная для 50% популяции. Терапевтический показатель - соотношение доз между токсическими и терапевтическими эффектами (LD50/ED50). Соединения, которые проявляют большие терапевтические показатели, являются предпочтительными. В то время как соединения, которые проявляют токсические побочные эффекты, могут быть использованы, следует позаботиться о разработке системы доставки, которая нацеливает такие соединения на сайт пораженной ткани, чтобы свести к минимуму потенциальный ущерб неинфицированным клеткам и, таким образом, уменьшить побочные эффекты.
Данные, полученные с помощью анализов клеточной культуры и исследований на животных, могут быть использованы при разработке диапазона дозировок для применения у людей. Дозировка таких соединений может находиться в пределах диапазона циркулирующих концентраций, которые включают ED50 с небольшой токсичностью или без нее. Доза может варьироваться в пределах данного диапазона в зависимости от используемой лекарственной формы и используемого способа введения. Для любого соединения терапевтически эффективную дозу можно оценить первоначально на основании анализов клеточной культуры. Доза может быть составлена на животных моделях для достижения диапазона концентрации циркулирующей плазмы, который включает IC50 (то есть, концентрацию испытуемого соединения, которая достигает половины максимального ингибирования симптомов), как определено на основании клеточной культуры. Такая информация может использоваться для более точного определения полезных доз у людей. Уровни в плазме можно измерять, например, с помощью высокоэффективной жидкостной хроматографии.
Следует также понимать, что конкретная дозировка и режим лечения для любого конкретного пациента будут зависеть от множества факторов, включая активность конкретного используемого соединения, возраст, массу тела, общее состояние здоровья, пол, питание, время введения, скорости выделения, комбинацию лекарств, суждения лечащего врача и тяжести конкретного излечиваемого заболевания. Количество соединения по настоящему изобретению в композиции также будет зависеть от конкретного соединения в композиции.
Лечение
Слияния NTRK были непосредственно связаны с несколькими типами онкологических заболеваний. Данные слияния содержат интактный домен киназы NTRK, который идентичен нативной или дикой форме рецептора; поэтому при использовании в данном документе любой белок NTRK (NTRK1, 2 или 3) с таким же доменом киназы, как и у NTRK дикого типа, будет называться «NTRK дикого типа». Мутации могут происходить в области киназного домена NTRK, что приведет к появлению мутантов, которые устойчивы к терапии ингибиторами киназы. Такие устойчивые мутации могут быть предсказаны с использованием структурной биологии и вычислительных анализов, а также путем изучения последовательностей кодонов, в которых изменение последовательности приводит к возникновению кодона для другой аминокислоты. Альтернативно, мутации устойчивости для данного ингибитора могут быть идентифицированы экспериментально путем введения данного ингибитора (например, известного ингибитора дикого типа NTRK) и воздействия на клетки агента, способствующего мутации, например, ENU. Клетки промывают, затем высевают с увеличением концентрации (2-100X пролиферации IC50) выбранного соединения. Затем лунки с разросшимися клетками собирают через 3-4 недели. В частности, с помощью обоих методов была идентифицирована мутация в положении аминокислоты 595 в слиянии NTRK (счет от NTRK1 дикого типа), влияющая на изменения остатка с глицина на аргинин (ранее обозначенного «G595R»). Впоследствии эта мутация продемонстрировала значительную устойчивость к двум ингибиторам NTRK, клиническая оценка которых проводится (см. таблицу ниже). Как показано в таблице, соединения активны против NTRK дикого типа, но заметно менее активны против мутантной формы G595R слияния NTRK.
Ферментный анализ
IC50 (нМ)
Клеточный
GI50 (нМ)
Клеточный
GI50 (нМ)
Соответственно, в другом аспекте изобретение относится к способу лечения субъекта, страдающего от состояния, опосредованного аберрантной активностью нейротрофной рецепторной тирозинкиназы (NTRK), включающему введение субъекту терапевтически эффективного количества соединения или фармацевтической композиции описанного здесь соединения.
Изобретение относится к соединениям, которые ингибируют как NTRK дикого типа, так и устойчивые мутанты G595R NTRK.
В другом аспекте изобретение относится к способу лечения субъекта, у которого развилась устойчивость к лечению онкологического заболевания, включающему введение субъекту терапевтически эффективного количества соединения или фармацевтической композиции описанного здесь соединения.
Кроме того, ингибиторы могут быть селективными для NTRK дикого типа по сравнению с другими киназами, что приводит к снижению токсичности, связанной с ингибированием других киназ. Из-за их активности против NTRK дикого типа и мутантной NTRK, описанные в данном документе соединения могут быть использованы для лечения пациента с состоянием, связанным с аберрантной активностью NTRK. Они также могут использоваться для лечения различных видов онкологических заболеваний. В некоторых вариантах осуществления рак выбран из немелкоклеточного рака легкого, рака молочной железы, меланомы, глиомы низкой и высокой степени злокачественности, глиобластомы, педиатрической астроцитомы, колоректального рака, папиллярной карциномы щитовидной железы, аденокарциномы поджелудочной железы, рака головы и шеи, холангиокарциномы, острого миелогенного лейкоза, секреторного рака молочной железы, рака слюнных желез и эпителиоидного невуса.
Соединения могут также использоваться для лечения пациента, у которого развилась устойчивость к ингибитору NTRK дикого типа, или пациенту с мутантной формой NTRK, такой как мутант G595R. Способ включает стадию введения соединения или композиции в соответствии с изобретением, эффективной против мутанта, устойчивого к NTRK. Под «активным» подразумевается, что соединение имеет IC50 менее 1 мкМ, 500 нМ, 250 нМ, 100 нМ, 75 нМ, 50 нМ, 25 нМ, 10 нМ или 5 нМ, измеренное в результате проведения биохимического анализа, по сравнению с по меньшей мере одним устойчивым мутантом.
Соединения и композиции, описанные в данном документе, могут вводиться отдельно или в комбинации с другими соединениями, включая другие модулирующие NTRK соединения или другие терапевтические агенты. В некоторых вариантах осуществления соединение или композицию в соответствии с изобретением можно вводить в комбинации с одним или более соединениями, выбранными из Кабозатиниба (COMETRIQ), Вандетаниба (CALPRESA), Сорафениба (NEXAVAR), Сунитиниба (SUTENT), Регофарениба (STAVARGA), Ронатиниба (ICLUSIG), Бевацизумаба (AVASTIN), Кризотиниба (XALKORI) или Гефитиниба (IRESSA). Соединение или композицию в соответствии с изобретением можно вводить одновременно или последовательно с другим терапевтическим агентом посредством одинаковых или разных способов введения. Соединение в соответствии с изобретением может быть включено в единую композицию с другим терапевтическим агентом или соединение и агент могут быть в различных композициях.
Синтез
Соединения в соответствии с изобретением, включая их соли и N-оксиды, могут быть получены с использованием известных методов органического синтеза и могут быть синтезированы в соответствии с любым из многочисленных возможных путей синтеза, например, указанных на схемах ниже. Реакции для получения соединений в соответствии с изобретением могут быть проведены в подходящих растворителях, которые могут быть легко выбраны специалистом в области органического синтеза. Подходящие растворители могут быть по существу нереакционноспособными с исходными веществами (реагентами), промежуточными продуктами или продуктами при температурах, при которых проводят реакции, например, при температурах, которые могут варьироваться от температуры замораживания растворителя до температуры кипения растворителя. Данную реакцию можно проводить в одном растворителе или смеси более чем одного растворителя. В зависимости от конкретной стадии реакции подходящие растворители для конкретной стадии реакции могут быть выбраны специалистом в данной области техники.
Получение соединений в соответствии с изобретением может включать защиту различных химических групп и снятие защиты с различных химических групп. Специалист в данной области техники может легко определить необходимость защиты и снятие защиты, а также необходимость выбора соответствующих защитных групп. Химия защитных групп может быть найдена, например, в документе Wuts и Greene, Protective Groups in Organic Synthesis, 4th ed., John Wiley & Sons: New Jersey, (2006 г.), который включен в настоящее описание посредством ссылки в полном объеме.
Реакции можно контролировать в соответствии с любым подходящим способом, известным в данной области. Например, образование продукта может контролироваться с помощью спектроскопических средств, таких как спектроскопия ядерного магнитного резонанса (ЯМР) (например, 1H или 13C), инфракрасная (ИК) спектроскопия, спектрофотометрия (например, УФ-видимая), масс-спектрометрия (МС) или же посредством хроматографических способов, таких как высокоэффективная жидкостная хроматография (ВЭЖХ) или тонкослойная хроматография (ТСХ). Далее представлены аналитические инструменты и методы для составной характеристики.
ЖХ-МС. Если не указано иное, все данные жидкостной хроматографии-масс-спектрометрии (ЖХ-МС) (образец, проанализированный на предмет чистоты и идентичности) были получены с помощью системы Agilent model-1260 LC с использованием масс-спектрометра Agilent model 6120 с использованием ионизации ES-API, оснащенной Agilent Poroshel 120 (EC-C18, размер частиц 2,7 мкм, размеры 3,0×50 мм) с колонкой с обращенной фазой при 22,4 градусах Цельсия. Подвижная фаза состояла из смеси раствора 0,1% муравьиной кислоты в воде и 0,1% муравьиной кислоты в ацетонитриле. Использовали постоянный градиент от 95% водной /5% органической подвижной фазы до 5% водной /95% органической подвижной фазы в течение 4 минут. Скорость потока была постоянной при 1 мл/мин.
Препаративная ЖХ-М. Препаративную ВЭЖХ проводили на подготовительной системе Shimadzu Discovery VP®, снабженной колонкой с обращенной фазой Luna 5u C18 (2) 100A c AXIA, колонкой с обращенной фазой 250×21,2 мм при 22,4 градусах Цельсия. Подвижная фаза состояла из смеси раствора 0,1% муравьиной кислоты в воде и 0,1% муравьиной кислоты в ацетонитриле. Использовали постоянный градиент от 95% водной /5% органической подвижной фазы до 5% водной/95% органической подвижной фазы в течение 25 минут. Скорость потока была постоянной при 20 мл/мин. Реакции, проводимые под воздействием микроволн, проводили в приборе для микроволнового воздействия Biotage Initiator.
Хиральная ВЭЖХ. Препаративную ВЭЖХ для растворения хиральных смесей проводили на приборе Thar SFC Pre-80, оборудованном колонкой Chiralpak AS-H (5 мм, 3,0 см id x 25 см л). Мобильные фазы состоят из SFC CO2 (A) и MeOH/0,1% NH4OH (B). Постоянный градиент от 67% до 33% (В) поддерживался со скоростью потока 65 г/мин при противодавлении системы 100 бар. Прогресс разделения контролировался УФ-детектированием на длине волны 220 нм.
Хроматография на силикагеле. Хроматографию на силикагеле проводили либо на блоке Teledyne Isco CombiFlash® Rf, либо на блоке Biotage® Isolera Four.
Протонный ЯМР. Если не указано иное, все спектры 1H-ЯМР были получены с использованием прибора Varian 400 МГц Unity 400 МГц ЯМР (время обнаружения=3,5 секунды с задержкой в 1 секунду, от 16 до 64 сканирований). При наличии характеристики, отчетные данные по всем все протонам в растворителе ДМСО-d6 выражены в виде частей на миллион (ч./млн) по отношению к остаточному ДМСО (2,50 м.д.).
ПРИМЕРЫ
Следующие примеры являются иллюстративными и никоим образом не предназначены для ограничения.
Нижеследующие схемы предназначены для предоставления общего руководства в вопросах, касающихся приготовления соединений в соответствии с изобретением. Специалист в данной области поймет, что препараты, показанные на схемах, могут быть модифицированы или оптимизированы с использованием общих знаний об органической химии для получения различных соединений в соответствии с изобретением.
Общий синтез 1
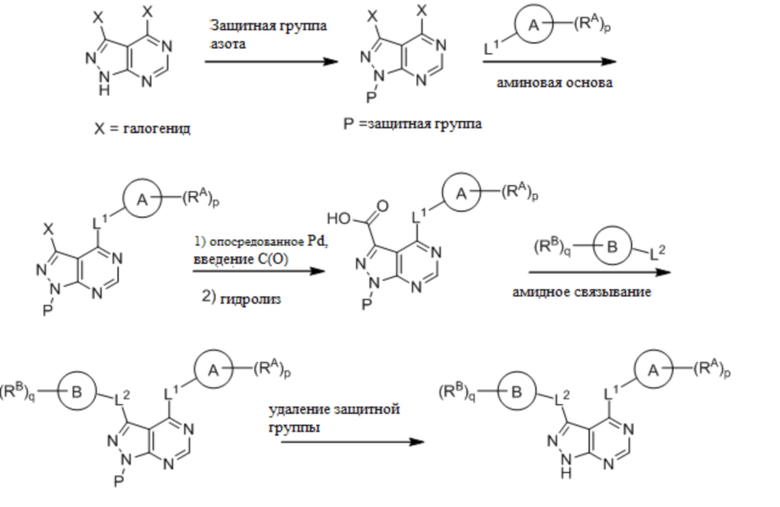
Для некоторых соединений общий синтез начинается с соответствующей азотной защиты (P) замещенного дигалогенидом 1H-пиразоло [3,4-d] пиримидина. Бицикл с защищенным азотом может быть замещен на галогениде в пиримидиновом кольце соответствующим образом замещенным кольцом А в подходящих условиях, например, в условиях реакции нуклеофильного ароматического замещения с использованием основания, такого как диизопропилэтиламин (DIPEA), в полярном растворителе, таком как диоксан, для получения бицикла, замещенного кольцом А. Галогенид пиразольного кольца может быть замещен в условиях опосредованной палладием реакции введения карбонильной группы с последующим гидролизом с получением в результате карбоновой кислоты. Карбоновую кислоту можно подвергнуть взаимодействию с кольцом B в соответствующих условиях связывания, например, в условиях реакции амидного связывания, с получением соединения с защищенным азотом, замещенного кольцами A и B. Удаление защитной группы может давать соединения по формуле I.
Протокол синтеза 1.
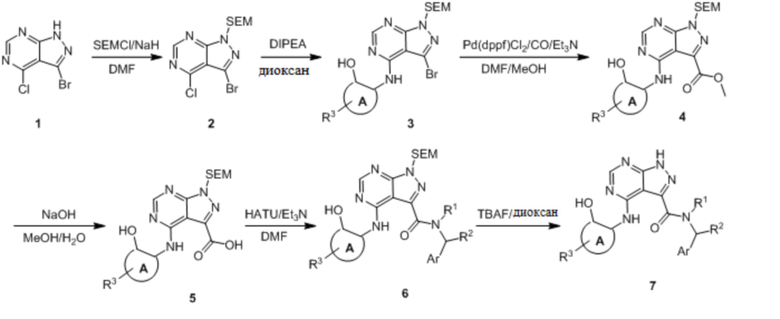
Несколько более конкретная версия показанной выше схемы общего синтеза 1 описана в протоколе синтеза 1. Протокол синтеза начинается с защиты SEM-группой 3-бром-4-хлор-1Н-пиразоло [3,4-d] пиримидина 1. Защищенный SEM-группой гетероцикл 2 может быть замещен аминоспиртом в условиях реакции нуклеофильного ароматического замещения с использованием основания, такого как диизопропилэтиламин (DIPEA), в полярном растворителе, таком как диоксан, с получением аминозамещенного гетероцикла 3. 3-бромпиразолопиримидин 3 подвергают взаимодействию с опосредованной палладием реакцией введения карбонильной группы в смеси растворителей DMF-MeOH с получением сложного метилового эфира 4. После гидролиза сложного эфира посредством обработки NaOH, карбоновую кислоту подвергают 5 взаимодействию с бензиламином или пирролидином в условиях реакции амидного связывания с получением защищенного SEM соединения 6. Защитную группу SEM можно удалить с использованием TBAF или в кислых условиях с получением конечного соединения 7. Соединения, описанные ниже, были получены с использованием общего синтеза 1, 2 или 3, как дополнительно описано в синтетическом протоколе 1, 2 или 3, соответственно.
Общий синтез 2
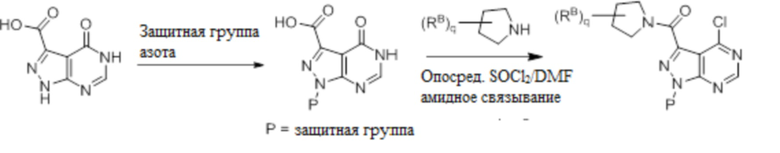
Для некоторых соединений общий синтез начинается с соответствующей азотной защиты (P) 4-оксо-4,5-дигидро-1H-пиразоло[3,4-d]пиримидин-3-карбоновой кислоты. Бицикл с защищенным азотом может быть хлорирован и подвергнут реакции связывания с амином в присутствии хлорирующего реагента, такого как тионилхлорид. Полученное соединение может быть замещено на галогениде в пиримидиновом кольце соответствующим образом замещенным кольцом А в подходящих условиях, например, в условиях реакции нуклеофильного ароматического замещения с использованием основания, такого как диизопропилэтиламин (DIPEA), в полярном растворителе, таком как диоксан, для получения бицикла, замещенного кольцом А. После удаления защитной группы могут быть получены соединения по формуле I. Соединения, описанные ниже, могут быть получены с использованием данного общего синтеза. Кроме того, хиральная ВЭЖХ может быть использована для разделения хиральных смесей соединений по формулам I, (Ia), (Ia-1), (Ia-2), (Ib), (Ib-1, (Ib-2), II, (IIa), (IIb), (IIc).
Протокол синтеза 2.
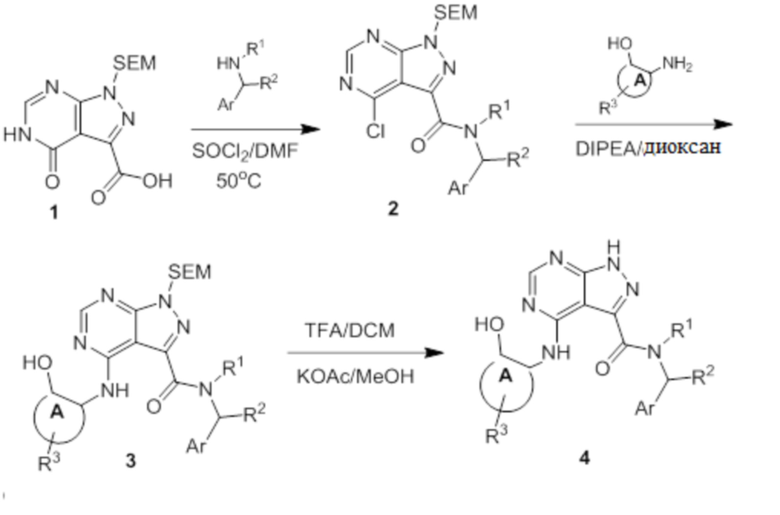
Несколько более конкретная версия показанной выше схемы общего синтеза 2 описана в протоколе синтеза 2. Синтетический протокол начинается с SEM-защищенной 4-оксо-4,5-дигидро-1Н-пиразоло[3,4-d]пиримидин-3-карбоновой кислоты 1, которая может быть хлорирована тионилхлоридом/ДМФ и затем связана с бензиламином или пирролидином при умеренном нагревании с получением SEM-защищенного соединения 2. Защищенный SEM-группой гетероцикл 2 может быть замещен аминоспиртом в условиях реакции нуклеофильного ароматического замещения с использованием основания, такого как диизопропилэтиламин (DIPEA), в полярном растворителе, таком как диоксан, с получением аминозамещенного гетероцикла 3. Защитную группу SEM можно удалить с использованием TBAF или в кислых условиях с получением конечного соединения 4.
Общий синтез 3
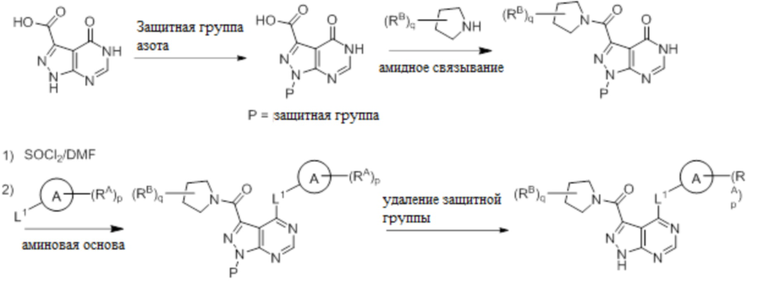
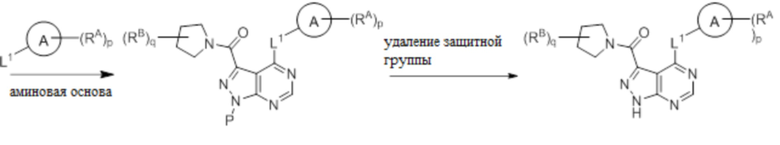
Для некоторых соединений общий синтез начинается с соответствующей азотной защиты (P) 4-оксо-4,5-дигидро-1H-пиразоло[3,4-d]пиримидин-3-карбоновой кислоты. Карбоновую кислоту можно связывать с амином, используя условия амидного связывания. Полученное соединение может быть хлорировано с использованием тионилхлорида с последующим замещением на хлориде в пиримидиновом кольце соответствующим образом замещенным кольцом А в подходящих условиях, например, в условиях реакции нуклеофильного ароматического замещения с использованием основания, такого как диизопропилэтиламин (DIPEA), в полярном растворителе, таком как диоксан, для получения бицикла, замещенного кольцом А. После удаления защитной группы могут быть получены соединения по формуле I. Соединения, описанные ниже, могут быть получены с использованием данного общего синтеза. Кроме того, хиральная ВЭЖХ может быть использована для разделения хиральных смесей соединений по формулам I, (Ia), (Ia-1), (Ia-2), (Ib), (Ib-1, (Ib-2), II, (IIa), (IIb), (IIc).
Протокол синтеза 3.
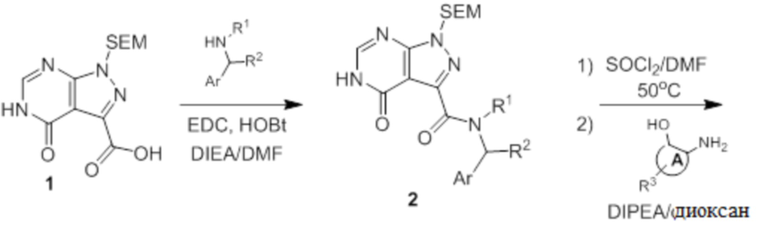
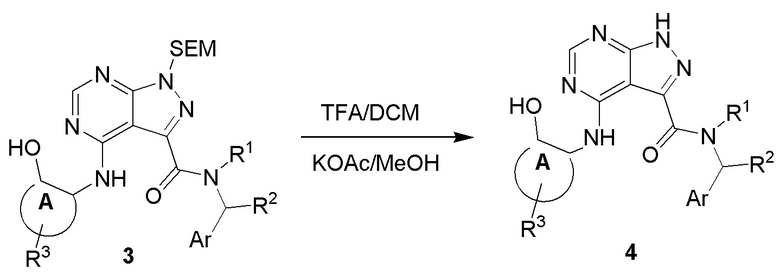
Несколько более конкретная версия показанной выше схемы общего синтеза 3 описана в протоколе синтеза 3. Синтетический протокол начинается с SEM-защищенной 4-оксо-4,5-дигидро-1Н-пиразоло [3,4-d] пиримидин-3-карбоновой кислоты 1, которая может связываться с бензиламином или пирролидином в условиях амидной связи. Защищенный SEM-группой гетероцикл 2 может быть хлорирован тионилхлоридом/ДМФ и затем замещен аминоспиртом в условиях реакции нуклеофильного ароматического замещения с использованием основания, такого как диизопропилэтиламин (DIPEA), в полярном растворителе, таком как диоксан, с получением аминозамещенного гетероцикла 3. Защитную группу SEM можно удалить с использованием TBAF или в кислых условиях с получением конечного соединения 4.
Все соединения, представленные на фиг.1, а также другие соединения в соответствии с изобретением, были получены с использованием одной из трех схем общего синтеза и протоколов, описанных выше. Кроме того, хиральная ВЭЖХ может быть использована для разделения хиральных смесей соединений по формулам I, (Ia), (Ia-1), (Ia-2), (Ib), (Ib-1), (Ib-2), II, (IIa), (IIb), (IIc). Некоторые конкретные примеры синтеза изложены в примерах.
Пример 1. Синтез соединения 45

Шаг 1: синтез 3-бром-4-хлор-1-((2-(триметилсилил)этокси)метил)-1H-пиразоло[3,4-d]пиримидина
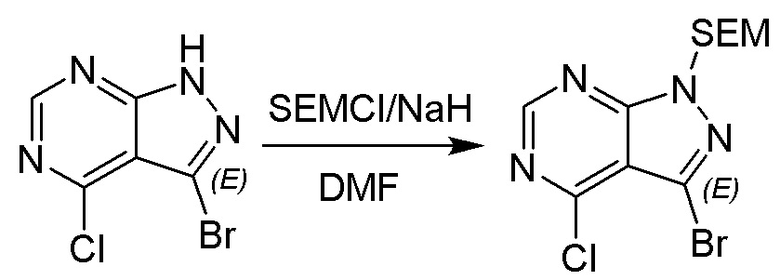
К раствору 3-бром-4-хлор-1H-пиразоло[3,4-d]пиримидина (10,00 г, 42,84 ммоль) в DMF (50,00 мл) добавляли NaH (2,57 г, 64,25 ммоль) порционно при 0 °C. После перемешивания в течение 0,5 часа, SEM-Cl (8,57 г, 51,40 ммоль) добавляли по каплям к реакции при 0°C на протяжении 0,5 часа. Реакцию медленно нагревали до 25°C и перемешивали в течение 16 часов. После того как ТСХ (PE:EtOAc =1:1, Rf=0,88) показала завершение реакции, реакцию медленно гасили 50 мл H2O. Смесь экстрагировали с использованием EtOAc (50 мл*3) и органические слои промывали солевым раствором (20 мл*3), сушили над Na2SO4 и концентрировали. Осадок очищали посредством колоночной хроматографии на силикагеле (PE:EtOAc=20:1) для получения 3-бром-4-хлор-1-((2-(триметилсилил)этокси)метил)-1H-пиразоло[3,4-d]пиримидина (6,20 г, выход: 39,80%) в виде бесцветной маслянистой жидкости. 1H-ЯМР (400 МГц, CDCl3) δ м. д. 8,83 (с, 1H), 5,82 (с, 2H), 3,70 (т, 2H, J=8,4 Гц), 0,97 (т, 2H, J=8,4 Гц), 0,00 (с, 9H).
Шаг 2: синтез (1R,2R)-2-((3-бром-1-((2-(триметилсилил)этокси)метил)-1H-пиразоло[3,4-d]пиримидин-4-ил)амино)циклопентан-1-ола
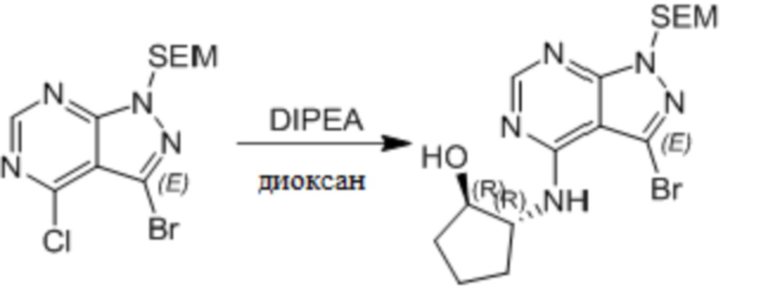
К смеси 3-бром-4-хлор-1-((2-(триметилсилил)этокси)метил)-1H-пиразоло[3,4-d]пиримидина (2,50 г, 6,87 ммоль) и (1R, 2R)-2-аминоциклопентанол гидрохлорида (945,38 мг, 6,87 ммоль) в диоксане (15 мл) добавляли DIPEA (1,78 г, 13,74 ммоль), реакционную смесь оставляли при перемешивании при 70°C в течение 16 часов. Как только ТСХ (PE:EtOAc=5:1) показала, что исходный материал полностью прореагировал, смесь концентрировали в вакууме и очищали посредством колоночной хроматографии на силикагеле (PE:EtOAc=30:1~10:1) для получения (1R,2R)-2-((3-бром-1-((2-(триметилсилил)этокси)метил)-1H-пиразоло[3,4-d]пиримидин-4-ил)амино)циклопентан-1-ола (2,10 г, 4,41 ммоль, выход: 64,22%) в виде маслянистой жидкости желтого цвета. 1H-ЯМР (400 МГц, CD3OD) δ м. д. 8,41 (с, 1H), 6,26 (с, 1H), 5,70 (с, 2H), 4,13-4,12 (м, 2H), 3,69-3,65 (м, 2H), 2,40-2,39 (м, 1H), 2,20-2,18 (м, 1H), 1,95-1,84 (м, 2H), 1,72-1,61 (м, 2H), 0,97 (д, 2H, J=4,0 Гц), 0,00 (с, 9H).
Шаг 3: синтез метил 4-(((1R,2R)-2-гидроксициклопентил)амино)-1-((2-(триметилсилил)этокси)метил)-1H-пиразоло[3,4-d]пиримидин-3-карбоксилата
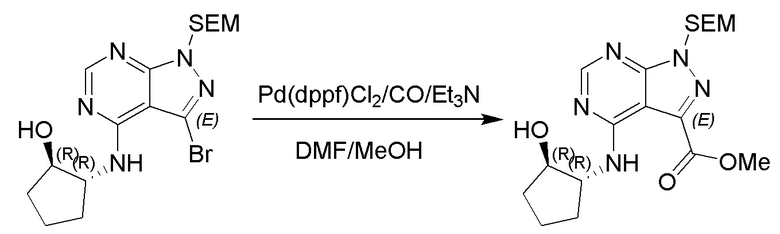
К смеси (1R,2R)-2-((3-бром-1-((2-(триметилсилил)этокси)метил)-1H-пиразоло[3,4-d]пиримидин-4-ил)амино)циклопентан-1-ола (2,10 г, 4,90 ммоль) в DMF (10 мл) и MeOH (15 мл) добавляли Pd(dppf)Cl2 (717,07 мг, 980,00 мкмоль) и Et3N (1,49 г, 14,70 ммоль) в один прием, реакционную смесь оставляли при перемешивании при температуре 70°C в течение 30 часов в атмосфере CO (50 фунтов/кв. дюйм). Как только ТСХ (PE:EtOAc =1:1) и ЖХМС показали, что исходный материал полностью прореагировал, смесь фильтровали и фильтрат концентрировали в вакууме для получения метил 4-(((1R,2R)-2-гидроксициклопентил)амино)-1-((2-(триметилсилил)этокси)метил)-1H-пиразоло[3,4-d]пиримидин-3-карбоксилата (2,70 г, в неочищенном виде) в виде маслянистой жидкости желтого цвета, который использовали непосредственно без дальнейшей очистки. 1H-ЯМР (400 МГц, CDCl3) δ м. д. 8,85 (с, 1H), 8,45 (с, 1H), 5,83 (с, 2H), 4,14 (уш. с., 2H), 4,10 (с, 3H), 3,70 (т, 2H, J=8,4 Гц), 2,40-2,38 (м, 1H), 2,20-2,17 (м, 1H), 1,93-1,79 (м, 4H), 0,98 (т, 2H, J=8,4 Гц), 0,00 (с, 9H).
Шаг 4: синтез 4-(((1R,2R)-2-гидроксициклопентил)амино)-1-((2-(триметилсилил)этокси)метил)-1H-пиразоло[3,4-d]пиримидин-3-карбоновой кислоты

К смеси метил 4-(((1R,2R)-2-гидроксициклопентил)амино)-1-((2-(триметилсилил)этокси)метил)-1H-пиразоло[3,4-d]пиримидин-3-карбоксилата (2,70 г, 6,63 ммоль) в MeOH (10 мл) добавляли H2O (10 мл), после чего добавляли NaOH (530,01 мг, 13,25 ммоль) в один прием. Реакционную смесь после этого оставляли при перемешивании при температуре 26°C в течение 16 часов. Как только ЖХМС и ТСХ (PE:EtOAc =1:1) показали, что исходный материал полностью прореагировал, MeOH удаляли посредством концентрации в вакууме и осадок промывали EtOAc (8 мл*2). Затем добавляли водный раствор HCl (1 N) до получения значения pH < 7, и наблюдалось выпадение осадка белого цвета. Твердую фракцию собирали фильтрацией и высушивали под вакуумом для получения 4-(((1R,2R)-2-гидроксициклопентил)амино)-1-((2-(триметилсилил)этокси)метил)-1H-пиразоло[3,4-d]пиримидин-3-карбоновой кислоты (1,20 г, выход: 37,89%) в виде твердого вещества белого цвета. 1H-ЯМР (400 МГц, CD3OD) δ м. д. 8,40 (с, 1H), 5,80 (с, 2H), 4,37-4,32 (м, 1H), 4,22-4,19 (м, 1H), 3,75 (т, 2H, J=8,0 Гц), 2,39-2,36 (м, 1H), 2,11-2,06 (м, 1H), 1,96-1,91 (м, 2H), 1,78-1,73 (м, 2H), 0,95 (т, 2H, J=8,0 Гц), 0,00 (с, 9H).
Шаг 5: синтез ((R)-2-(2,5-дифторфенил)пирролидин-1-ил)(4-(((1R,2R)-2-гидроксициклопентил)амино)-1-((2-(триметилсилил)этокси)метил)-1H-пиразоло[3,4-d]пиримидин-3-ил)метанона
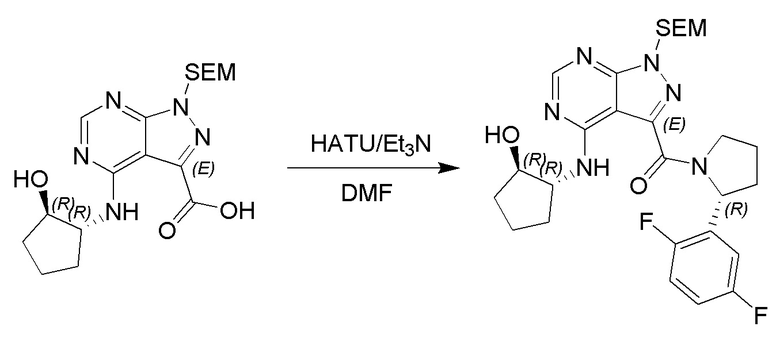
К смеси 4-(((1R,2R)-2-гидроксициклопентил)амино)-1-((2-(триметилсилил)этокси)метил)-1H-пиразоло[3,4-d]пиримидин-3-карбоновой кислоты (100,00 мг, 254,12 мкмоль) и (2R)-2-(2, 5-дифторфенил)пирролидина (55,87 мг, 304,95 мкмоль) в DMF (2 мл) добавляли HATU (144,94 мг, 381,18 мкмоль) и Et3N (128,57 мг, 1,27 ммоль) при температуре 20°C и реакцию перемешивали при температуре 20°C в течение 16 часов. После того как ЖХМС показала завершение реакции, H2O (5 мл) добавляли к смеси, реакцию экстрагировали EtOAc (10 мл*3) и промывали солевым раствором (5 мл*3). Затем органический слой сушили над Na2SO4 и концентрировали. Осадок очищали посредством препаративной ТСХ (PE:EtOAc=1:1, Rf=0,5) для получения ((R)-2-(2,5-дифторфенил)пирролидин-1-ил)(4-(((1R,2R)-2-гидроксициклопентил)амино)-1-((2-(триметилсилил)этокси)метил)-1H-пиразоло[3,4-d]пиримидин-3-ил)метанона (20,00 мг, выход: 14,09%) в виде бесцветной маслянистой жидкости.
Шаг 6: синтез ((R)-2-(2,5-дифторфенил)пирролидин-1-ил)(4-(((1R,2R)-2-гидроксициклопентил)амино)-1H-пиразоло[3,4-d]пиримидин-3-ил)метанона
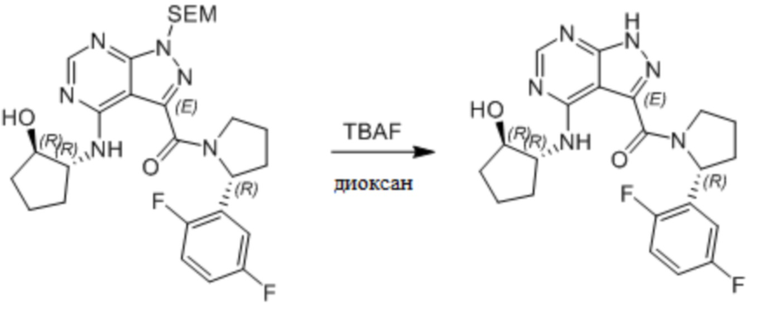
К раствору ((R)-2-(2,5-дифторфенил)пирролидин-1-ил)(4-(((1R,2R)-2-гидроксициклопентил)амино)-1-((2-(триметилсилил)этокси)метил)-1H-пиразоло[3,4-d]пиримидин-3-ил)метанона (20,00 мг, 35,80 мкмоль) в диоксане (20 мл) добавляли TBAF (80,61 мг, 358,00 мкмоль) при температуре 20°C и реакцию нагревали при температуре 80°C в течение 16 часов. После того как ТСХ (EtOAc, Rf=0,1) показала завершение реакции, раствор концентрировали и к осадку добавляли 10 мл H2O. Раствор экстрагировали с использованием EtOAc (10 мл*3), органический слой высушивали над Na2SO4 и концентрировали. Осадок очищали посредством кислотной препаративной ВЭЖХ (смесь растворителей MeOH/H2O/TFA) для получения ((R)-2-(2,5-дифторфенил)пирролидин-1-ил)(4-(((1R,2R)-2-гидроксициклопентил)амино)-1H-пиразоло[3,4-d]пиримидин-3-ил)метанона (11,10 мг, выход: 72,37%) в виде твердого вещества коричневого цвета.
Пример 2. Синтез соединения 97 и соединения 98
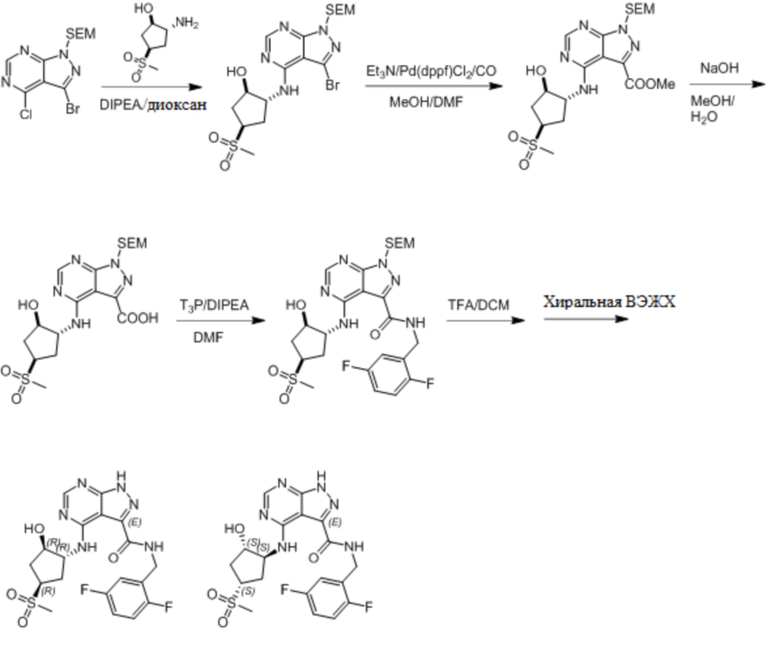
Шаг 1: синтез (1R,2R,4R)-2-((3-бром-1-((2-(триметилсилил)этокси)метил)-1H-пиразоло[3,4-d]пиримидин-4-ил)амино)-4-(метилсульфонил)циклопентан-1-ола
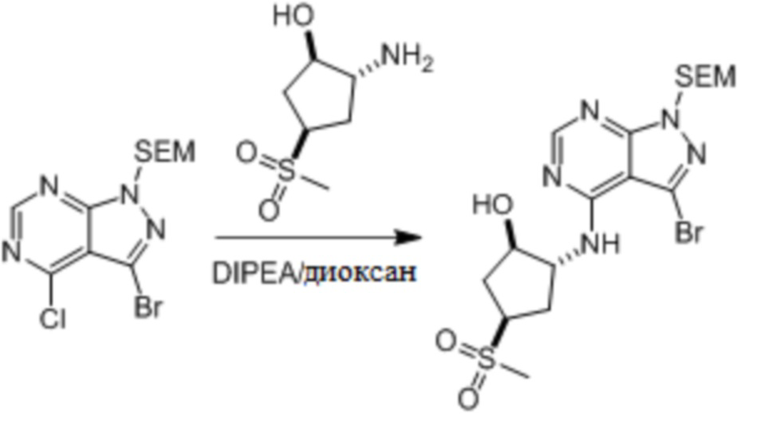
К смеси 3-бром-4-хлор-1-((2-(триметилсилил)этокси)метил)-1H-пиразоло[3,4-d]пиримидина (300,00 мг, 824,83 мкмоль) в диоксане (10,00 мл) добавляли DIPEA (319,80 мг, 2,47 ммоль) и (1R,2R,4R)-2-амино-4-(метилсульфонил)циклопентан-1-ол (195,71 мг, 907,31 мкмоль), и смесь перемешивали при температуре 90°C в течение 32 часов. После того как ЖХМС показала завершение реакции, смесь концентрировали для удаления 1,4-диоксана и осадок растворяли в DCM (20 мл). Органический слой промывали водой (10 мл*4), сушили над Na2SO4 и концентрировали с получением (1R,2R,4R)-2-((3-бром-1-((2-(триметилсилил)этокси)метил)-1H-пиразоло[3,4-d]пиримидин-4-ил)амино)-4-(метилсульфонил)циклопентан-1-ола (350,00 мг, выход: 83,78%) в виде твердого вещества белого цвета. 1H-ЯМР (400 МГц, CDCl3) δ м. д. 8,43 (с, 1H), 6,23 (уш. с., 1H), 5,71 (с, 2H), 4,50 (уш. с., 1H), 4,26-4,24 (м, 1H), 3,70-3,66 (м, 3H), 2,97 (уш. с., 4H), 2,65 (м, 1H), 2,37-2,20 (м, 2H), 0,97 (т, 2H, J=8,4 Гц), 0,00 (с, 9H).
Шаг 2: синтез метил 4-(((1R,2R,4R)-2-гидрокси-4-(метилсульфонил)циклопентил)амино)-1-((2-(триметилсилил)этокси)метил)-1H-пиразоло[3,4-d]пиримидин-3-карбоксилата
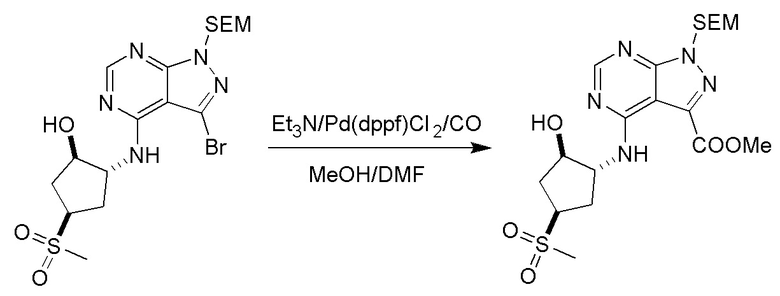
К смеси (1R,2R,4R)-2-((3-бром-1-((2-(триметилсилил)этокси)метил)-1H-пиразоло[3,4-d]пиримидин-4-ил)амино)-4-(метилсульфонил)циклопентан-1-ола (350,00 мг, 691,03 мкмоль) в MeOH (10,00 мл)/DMF (2,00 мл) добавляли Et3N (139,85 мг, 1,38 ммоль) и Pd(dppf)Cl2 (25,28 мг, 34,55 мкмоль). После добавления смесь перемешивали при температуре 75°C в течение 16 часов в атмосфере CO (50 фунтов/кв. дюйм). Как только ЖХМС показала завершение реакции, смесь концентрировали для получения неочищенного продукта, который очищали посредством препаративной ТСХ (PE:EtOAc=0:1) для получения метил 4-(((1R,2R,4R)-2-гидрокси-4-(метилсульфонил)циклопентил)амино)-1-((2-(триметилсилил)этокси)метил)-1H-пиразоло[3,4-d]пиримидин-3-карбоксилата (250,00 мг, выход: 74,50%) в виде твердого вещества красного цвета.
Шаг 3: синтез 4-(((1R,2R,4R)-2-гидрокси-4-(метилсульфонил)циклопентил)амино)-1-((2-(триметилсилил)этокси)метил)-1H-пиразоло[3,4-d]пиримидин-3-карбоновой кислоты
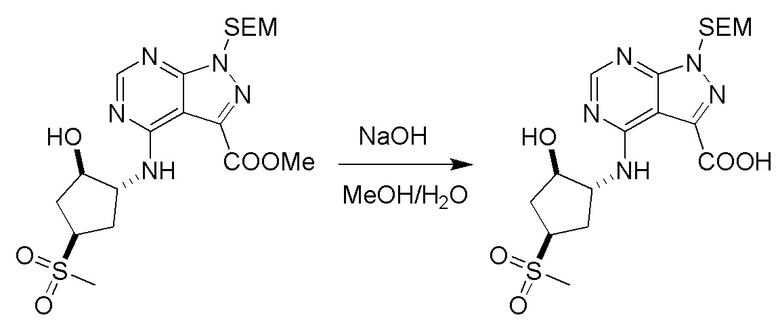
К смеси метил 4-(((1R,2R,4R)-2-гидрокси-4-(метилсульфонил)циклопентил)амино)-1-((2-(триметилсилил)этокси)метил)-1H-пиразоло[3,4-d]пиримидин-3-карбоксилата (250,00 мг, 514,80 мкмоль) в MeOH (10,00 мл)/H2O (5,00 мл) добавляли NaOH (41,18 мг, 1,03 ммоль), который перемешивали при температуре 20°C в течение 16 часов. Как только ЖХМС показала завершение реакции, смесь концентрировали для удаления MeOH. Водный слой промывали EtOAc (3 мл*2) и подкисляли HCl (1 M) до получения значения pH=4, после чего смесь фильтровали и отфильтрованный осадок высушивали под вакуумом для получения 4-(((1R,2R,4R)-2-гидрокси-4-(метилсульфонил)циклопентил)амино)-1-((2-(триметилсилил)этокси)метил)-1H-пиразоло[3,4-d]пиримидин-3-карбоновой кислоты (180,00 мг, выход: 74,14%) в виде твердого вещества черно-коричневого цвета. 1H-ЯМР (400 МГц, CDCl3) δ м. д. 8,47 (с, 1H), 5,77 (с, 2H), 4,43-4,40 (м, 1H), 4,17-4,12 (м, 1H), 3,85-8,82 (м, 1H), 3,69 (т, 2H, J=8,4 Гц), 3,03 (с, 3H), 2,45-2,41 (м, 2H), 1,99-1,91 (м, 2H), 0,92 (т, 2H, J=8,4 Гц), 0,00 (с, 9H).
Шаг 4: синтез N-(2,5-дифторбензил)-4-(((1R,2R,4R)-2-гидрокси-4-(метилсульфонил)циклопентил)амино)-1-((2-(триметилсилил)этокси)метил)-1H-пиразоло[3,4-d]пиримидин-3-карбоксамида
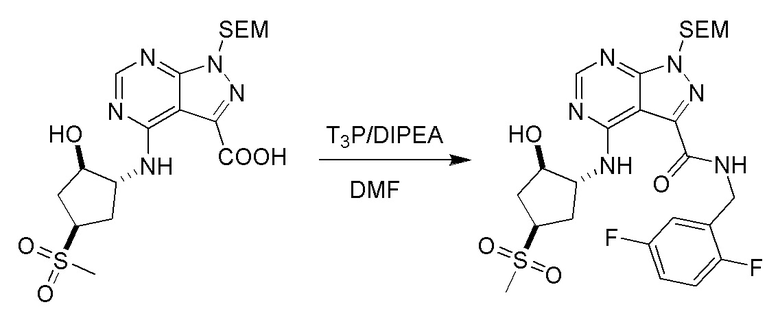
К смеси 4-(((1R,2R,4R)-2-гидрокси-4-(метилсульфонил)циклопентил)амино)-1-((2-(триметилсилил)этокси)метил)-1H-пиразоло[3,4-d]пиримидин-3-карбоновой кислоты (60,00 мг, 127,23 мкмоль) в DMF (2,00 мл) добавляли DIPEA (16,44 мг, 127,23 мкмоль), (2,5-дифторфенил)метанамин (36,42 мг, 254,46 мкмоль) и T3P (40,48 мг, 127,23 мкмоль). После добавления смесь перемешивали при температуре 20°C в течение 1 часа, после чего ЖХМС показала завершение реакции. Смесь добавляли в воду (4 мл) и экстрагировали с использованием EtOAc (5 мл*3), органический слой высушивали над Na2SO4 и концентрировали с получением N-(2,5-дифторбензил)-4-(((1R,2R,4R)-2-гидрокси-4-(метилсульфонил)циклопентил)амино)-1-((2-(триметилсилил)этокси)метил)-1H-пиразоло[3,4-d]пиримидин-3-карбоксамида (50,00 мг, в неочищенном виде) в виде маслянистой жидкости красного цвета.
Шаг 5: синтез N-(2,5-дифторбензил)-4-(((1R,2R,4R)-2-гидрокси-4-(метилсульфонил)циклопентил)амино)-1H-пиразоло[3,4-d]пиримидин-3-карбоксамида
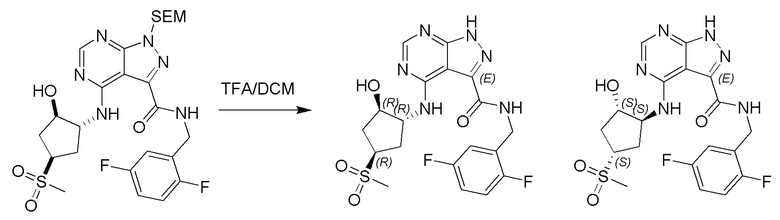
N-(2,5-дифторбензил)-4-(((1R,2R,4R)-2-гидрокси-4-(метилсульфонил)циклопентил)амино)-1-((2-(триметилсилил)этокси)метил)-1H-пиразоло[3,4-d]пиримидин-3-карбоксамид (50,00 мг, 83,79 мкмоль) в DCM (5,00 мл) перемешивали в смеси TFA (5,00 мл) при температуре 20°C в течение 16 часов, после чего ЖХМС показала завершение реакции. Смесь концентрировали для получения неочищенного продукта, который очищали посредством препаративной ВЭЖХ (TFA) и хиральной ВЭЖХ (времена удерживания разделенных изомеров составляли 7,46 мин. и 9,20 мин. соответственно). N-(2,5-дифторбензил)-4-(((1R,2R,4R)-2-гидрокси-4-(метилсульфонил)циклопентил)амино)-1H-пиразоло[3,4-d]пиримидин-3-карбоксамид (2,80 мг, выход: 7,16%) и N-(2,5-дифторбензил)-4-(((1S,2S,4S)-2-гидрокси-4-(метилсульфонил)циклопентил)амино)-1H-пиразоло[3,4-d]пиримидин-3-карбоксамид (4,00 мг, выход: 10,23%) получали в виде твердых веществ белого цвета. Условия ЖХ-МС для данных соединений были следующими: скорость потока=0,8 мл·мин.-1, подвижная фаза: от 99% [вода+0,375‰ об./об. TFA] и 1% [CH3CN+0,188‰ об./об. TFA], в данных условиях 0,4 мин., после чего условия изменяли на 10% [вода+0,375‰ об./об. TFA] и 90% [CH3CN+0,188‰ об./об. TFA] в течение 3,0 мин., после чего условия изменяли на 100% [CH3CN+0,188‰ об./об. TFA] в течение 0,45 мин., в конце условия изменяли на 99% [вода+0,375‰ об./об. TFA] и 1% [CH3CN+0,188‰ об./об. TFA] в течение 0,01 мин., после чего в данных условиях 0,64 мин.; чистота 98,887% и 96,551% соответственно.
Пример 3. Синтез соединения 20 и соединения 21
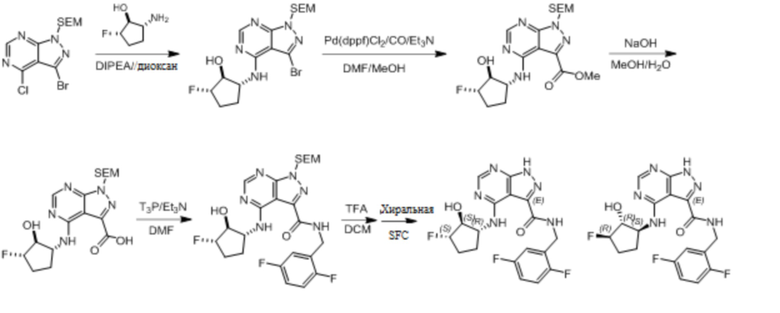
Шаг 1: (1S,2R,5S)-2-((3-бром-1-((2-(триметилсилил)этокси)метил)-1H-пиразоло[3,4-d]пиримидин-4-ил)амино)-5-фторциклопентан-1-ол
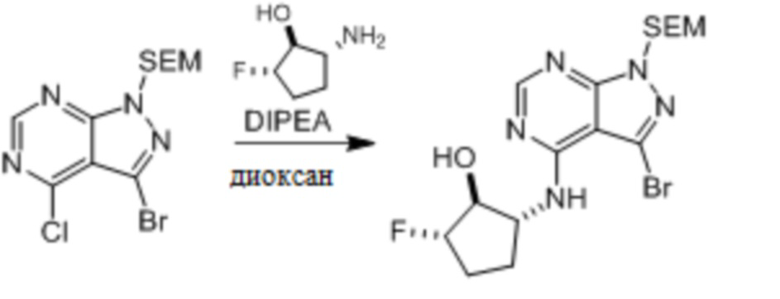
К смеси 3-бром-4-хлор-1-((2-(триметилсилил)этокси)метил)-1H-пиразоло[3,4-d]пиримидина (600,00 мг, 1,65 ммоль) и 2-амино-5-фтор-циклопентанола (196,58 мг, 1,65 ммоль) в диоксане (15 мл) добавляли DIPEA (426,49 мг, 3,30 ммоль). Смесь перемешивали при 110°C в течение 16 часов, после чего ТСХ (PE/EtOAc =1:1) показала завершение реакции. Смесь охлаждали до 25°C и концентрировали при пониженном давлении при температуре 50°C. К осадку добавляли EtOAc (50 мл) и органическую фазу промывали H2O (20 мл*3), сушили над Na2SO4, фильтровали и концентрировали в вакууме для получения (1S,2R,5S)-2-((3-бром-1-((2-(триметилсилил)этокси)метил)-1H-пиразоло[3,4-d]пиримидин-4-ил)амино)-5-фторциклопентан-1-ола (600,00 мг, в неочищенном виде). Осадок использовали напрямую на следующей стадии реакции без дополнительной очистки.
Шаг 2: метил 4-(((1R,2S,3S)-3-фтор-2-гидроксициклопентил)амино)-1-((2-(триметилсилил)этокси)метил)-1H-пиразоло[3,4-d]пиримидин-3-карбоксилат
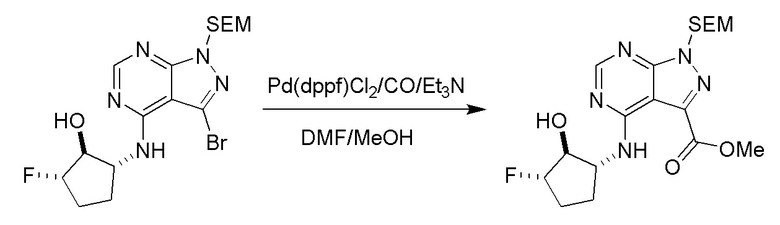
К раствору (1S,2R,5S)-2-((3-бром-1-((2-(триметилсилил)этокси)метил)-1H-пиразоло[3,4-d]пиримидин-4-ил)амино)-5-фторциклопентан-1-ола (600,00 мг, 1,34 ммоль) в MeOH/DMF (20 мл, v:v=2/1) добавляли Pd(dppf)Cl2 (49,17 мг, 67,21 мкмоль) и Et3N (408,03 мг, 4,03 ммоль) в атмосфере N2. Суспензию дегазировали под вакуумом и продували CO несколько раз. Смесь перемешивали в атмосфере CO (50 фунтов/кв. дюйм) при температуре 70°C в течение 16 часов, после чего ТСХ (PE/EtOAc =1:1) показала, что исходный материал полностью прореагировал. Реакционную смесь фильтровали и фильтрат концентрировали для получения метил 4-(((1R,2S,3S)-3-фтор-2-гидроксициклопентил)амино)-1-((2-(триметилсилил)этокси)метил)-1H-пиразоло[3,4-d]пиримидин-3-карбоксилата (700,00 мг, в неочищенном виде). Неочищенный продукт использовали непосредственно без очистки.
Шаг 3: 4-(((1R,2S,3S)-3-фтор-2-гидроксициклопентил)амино)-1-((2-(триметилсилил)этокси)метил)-1H-пиразоло[3,4-d]пиримидин-3-карбоновая кислота
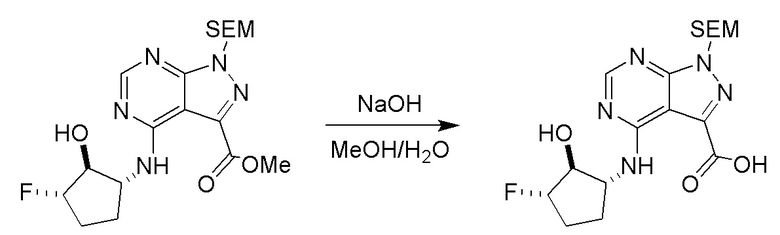
К раствору метил 4-(((1R,2S,3S)-3-фтор-2-гидроксициклопентил)амино)-1-((2-(триметилсилил)этокси)метил)-1H-пиразоло[3,4-d]пиримидин-3-карбоксилата (700,00 мг, 1,65 ммоль) в MeOH/H2O (15 мл, об./об.=2/1) добавляли NaOH (132,00 мг, 3,30 ммоль) в один прием, который перемешивали при температуре 25°C в течение 2 часов. После того как ЖХМС показала завершение реакции, смесь концентрировали при пониженном давлении при температуре 40°C. Водную фазу доводили до показателя pH=4 и фильтровали для получения 4-(((1R,2S,3S)-3-фтор-2-гидроксициклопентил)амино)-1-((2-(триметилсилил)этокси)метил)-1H-пиразоло[3,4-d]пиримидин-3-карбоновой кислоты (700,00 мг, в неочищенном виде) в виде твердого вещества белого цвета.
Шаг 4: N-(2,5-дифторбензил)-4-(((1R,2S,3S)-3-фтор-2-гидроксициклопентил)амино)-1-((2-(триметилсилил)этокси)метил)-1H-пиразоло[3,4-d]пиримидин-3-карбоксамид
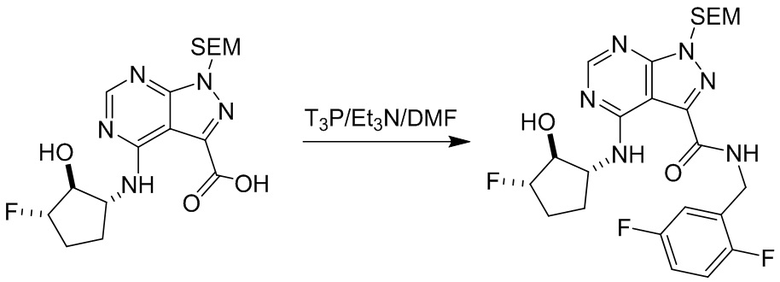
К смеси 4-(((1R,2S,3S)-3-фтор-2-гидроксициклопентил)амино)-1-((2-(триметилсилил)этокси)метил)-1H-пиразоло[3,4-d]пиримидин-3-карбоновой кислоты (100,00 мг, 243,01 мкмоль) и T3P (231,97 мг, 729,04 мкмоль) добавляли Et3N (49,18 мг, 486,03 мкмоль) в DMF (2,00 мл) при температуре 25°C, с последующим добавлением (2,5-дифторфенил)метанамина (69,57 мг, 486,03 мкмоль) в один прием через 10 мин. Смесь перемешивали при 25°C в течение 16 часов. После того как ЖХМС показала завершение реакции, смесь концентрировали при сниженном давлении при температуре 60°C для получения N-(2,5-дифторбензил)-4-(((1R,2S,3S)-3-фтор-2-гидроксициклопентил)амино)-1-((2-(триметилсилил)этокси)метил)-1H-пиразоло[3,4-d]пиримидин-3-карбоксамида (200 мг, в неочищенном виде). Осадок не очищали и использовали в том виде, в котором он был получен.
Шаг 5: N-(2,5-дифторбензил)-4-(((1R,2S,3S)-3-фтор-2-гидроксициклопентил)амино)-1H-пиразоло[3,4-d]пиримидин-3-карбоксамид и N-(2,5-дифторбензил)-4-(((1S,2R,3R)-3-фтор-2-гидроксициклопентил)амино)-1H-пиразоло[3,4-d]пиримидин-3-карбоксамид
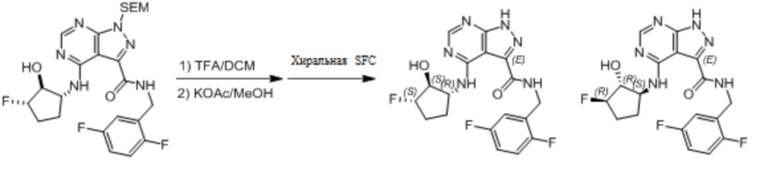
Смесь N-(2,5-дифторбензил)-4-(((1R,2S,3S)-3-фтор-2-гидроксициклопентил)амино)-1-((2-(триметилсилил)этокси)метил)-1H-пиразоло[3,4-d]пиримидин-3-карбоксамида (200,00 мг, 372,70 мкмоль) в TFA/DCM (15,00 мл, об./об.=1/1) перемешивали при температуре 25°C в течение 3 часов, после чего концентрировали при сниженном давлении при температуре 30 oC. К осадку добавляли MeOH (20 мл) и KOAc (100 мг) и смесь перемешивали в течение 16 часов при температуре 25°C. Как только ЖХМС показала завершение реакции, смесь концентрировали при сниженном давлении при температуре 30°C. Осадок очищали посредством кислотной препаративной ВЭЖХ с последующей хиральной препаративной ВЭЖХ для получения N-(2,5-дифторбензил)-4-(((1R,2S,3S)-3-фтор-2-гидроксициклопентил)амино)-1H-пиразоло[3,4-d]пиримидин-3-карбоксамида (25,00 мг, выход: 16,51%) в виде твердого вещества белого цвета и N-(2,5-дифторбензил)-4-(((1S,2R,3R)-3-фтор-2-гидроксициклопентил)амино)-1H-пиразоло[3,4-d]пиримидин-3-карбоксамида (30,00 мг, выход: 19,81%) в виде твердого вещества серого цвета. Условия ЖХ-МС для данных соединений были следующими: скорость потока=0,8 мл·мин.-1, подвижная фаза: от 95% [вода+10 мМ NH4HCO3] и 5% CH3CN, в данных условиях 0,4 мин., после чего условия изменяли на 10% [вода+10 мМ NH4HCO3] и 90% CH3CN в течение 2,6 мин., после чего условия изменяли на 100% CH3CN в течение 0,85 мин., в конце условия изменяли на 95% [вода+10 мМ NH4HCO3] и 5% CH3CN в течение 0,01 мин., после чего в данных условиях 0,64 мин. Чистота составляла 97,125% и 97,690% соответственно.
Синтез промежуточных аминов
Пример 4. Синтез (1R,2S,3R)-3-аминоциклопентан-1,2-диола
Шаг 1: (3aS,4S,6aR)-2,2-диметилтетрагидро-4H-циклопента[d][1,3]диоксол-4-ол
К раствору (3aR,6aR)-2,2-диметил-3a,6a-дигидро-4H-циклопента[d][1,3]диоксол-4-она (92,00 г, 596,78 ммоль, 1,00 экв.) в MeOH (2,00 л) добавляли Pd-C (10%, 12 г). Суспензию дегазировали под вакуумом и продували H2 несколько раз. Смесь перемешивали в атмосфере H2 (30 фунтов/кв. дюйм) при температуре 20°C в течение 4 часов, на момент окончания ТСХ (PE:EtOAc=3:1) показала, что исходный материал полностью прореагировал. Реакционную смесь фильтровали, и добавляли к фильтрату NaBH4 (34,09 г, 901,14 ммоль, 1,51 экв.) порционно при 0 °C. Полученную смесь перемешивали при температуре 20°C в течение 0,5 часа. После этого смесь концентрировали и к осадку добавляли H2O (500 мл). Смесь экстрагировали с использованием EtOAc (500 мл*3), сушили над Na2SO4 и концентрировали с получением (3aS,4S,6aR)-2,2-диметилтетрагидро-4H-циклопента[d][1,3]диоксол-4-ола (84,00 г, выход: 88,98%) в виде маслянистой жидкости желтого цвета. 1H-ЯМР (400 МГц, CDCl3) δ м. д. 4,60 (т, 1H, J=5,2 Гц), 4,39 (т, 1H, J=5,6 Гц), 3,83 (уш. с., 1H), 2,37-2,35 (м, 1H), 1,88-1,76 (м, 2H), 1,65-1,56 (м, 1H), 1,48 (с, 3H), 1,45-1,36 (м, 1H), 1,33 (с, 3H).
Шаг 2: 2-((3aS,4R,6aR)-2,2-диметилтетрагидро-4H-циклопента[d][1,3]диоксол-4-ил)изоиндолин-1,3-дион
К перемешанной смеси (3aS,4S,6aR)-2,2-диметилтетрагидро-4H-циклопента[d][1,3]диоксол-4-ола (70,00 г, 442,51 ммоль, 1,00 экв.), изоиндолин-1,3-диона (80,00 г, 543,74 ммоль, 1,23 экв.) и PPh3 (175,00 г, 667,20 ммоль, 1,51 экв.) в сухом толуоле (1,00 л) в атмосфере N2 добавляли DIAD (135,00 г, 667,62 ммоль, 1,51 экв.) по каплям. Полученную смесь перемешивали при температуре 80°C в течение 20 часов в атмосфере N2. После того как ТСХ (PE:EtOAc=3:1) показала, что исходный материал полностью прореагировал, смесь концентрировали и осадок очищали посредством колоночной хроматографии на силикагеле (PE:EtOAc=80:1/50:1/20:1/10:1) для получения 2-((3aS,4R,6aR)-2,2-диметилтетрагидро-4H-циклопента[d][1,3]диоксол-4-ил)изоиндолин-1,3-диона (90,00 г, выход: 70,79%) в виде твердого вещества белого цвета. 1H-ЯМР (400 МГц, CDCl3) δ м. д. 7,82-7,80 (м, 2H), 7,72-7,70 (м, 2H), 5,03-4,96 (м, 2H), 4,61-4,60 (м, 1H), 2,28-2,20 (м, 2H), 1,94-1,85 (м, 2H), 1,50 (с, 3H), 1,31 (с, 3H).
Шаг 3: 3aS,4R,6aR)-2,2-диметилтетрагидро-4H-циклопента[d][1,3]диоксол-4-амин
Смесь 2-((3aS,4R,6aR)-2,2-диметилтетрагидро-4H-циклопента[d][1,3]диоксол-4-ил)изоиндолин-1,3-диона (90,00 г, 313,25 ммоль, 1,00 экв.) и NH2NH2.H2O (32,00 г, 626,50 ммоль, 2,00 экв.) в EtOH (600,00 мл) перемешивали при температуре 80°C в течение 16 часов. После того как ТСХ (PE:EtOAc=3:1) показала, что исходный материал полностью прореагировал, смесь фильтровали и концентрировали, и к осадку добавляли EtOH (500 мл). После концентрации для удаления растворителя, PE (1000 мл) добавляли и смесь фильтровали и концентрировали с получением (3aS,4R,6aR)-2,2-диметилтетрагидро-4H-циклопента[d][1,3]диоксол-4-амин (41,00 г, в неочищенном виде)в виде маслянистой жидкости желтого цвета, которую отверждали отстаиванием с получением кристаллов желтого цвета. 1H-ЯМР (400 МГц, CDCl3) δ м. д. 4,72 (т, 1H, J=5,2 Гц), 4,18 (д, 1H, J=5,6 Гц), 3,39 (д, 1H, J=4,0 Гц), 2,01-1,93 (м, 2H), 1,78-1,77 (м, 1H), 1,40 (с, 3H), 1,38-1,35 (м, 1H), 1,26 (с, 3H), 1,10 (уш. с., 2H).
Шаг 4: (1R,2S,3R)-3-аминоциклопентан-1,2-диол гидрохлорид
Смесь (3aS,4R,6aR)-2,2-диметилтетрагидро-4H-циклопента[d][1,3]диоксол-4-амина (10,00 г, 63,61 ммоль, 1,00 экв.) в H2O (55,00 мл) и HCl (5,00 мл, 12 M) перемешивали при температуре 20°C в течение 2 часов. ТСХ (EtOAc:MeOH=10:1) показала, что исходный материал полностью прореагировал. Смесь концентрировали для получения гидрохлорида (1R,2S,3R)-3-аминоциклопентан-1,2-диола (9,20 г, выход: 94,15%) в виде твердого вещества желтого цвета. 1H-ЯМР (400 МГц, CD3OD) δ м. д. 4,03 (уш. с., 1H), 3,90 (дд, 1H, J=8,4, 4,4 Гц), 3,48-3,41 (м, 1H), 2,25-2,19 (м, 1H), 2,05-2,02 (м, 1H), 1,75-1,65 (м, 1H), 1,58-1,56 (м, 1H).
Пример 5. Синтез (1R,2R,3S,4R,5S)-4-аминобицикло[3.1.0]гексан-2,3-диола
Шаг 1: (3aS,4S,6aR)-2,2-диметил-3a,6a-дигидро-4H-циклопента[d][1,3]диоксол-4-ол
К перемешанной смеси (3aR,6aR)-2,2-диметил-3a,6a-дигидро-4H-циклопента[d][1,3]диоксол-4-она (90,00 г, 583,81 ммоль, 1,00 экв.) и CeCl3,7H2O (240,00 г, 644,16 ммоль, 1,10 экв.) в MeOH (2,00 л) (температура смеси 0°C) добавляли NaBH4 (44,00 г, 1,16 моль, 1,99 экв.) порционно в течение 0,5 часа. После добавления смесь перемешивали при температуре 18°C в течение 0,5 часа. ТСХ (PE:EtOAc=3:1) показала, что исходный материал полностью прореагировал. Смесь концентрировали, к осадку добавляли EtOAc (2000 мл) и раствор перемешивали при температуре 18°C в течение 0,5 часа. Смесь после чего фильтровали, фильтрат сушили над Na2SO4 и концентрировали с получением неочищенного продукта (3aS,4S,6aR)-2,2-диметил-3a,6a-дигидро-4H-циклопента[d][1,3]диоксол-4-ола (79,00 г, в неочищенном виде) в виде маслянистой жидкости светло-желтого цвета, который непосредственно использовали на следующем этапе без дополнительной очистки. 1H-ЯМР (400 МГц, CDCl3) δ м. д. 5,88 (с, 2 H), 5,01 (д, 1H, J=5,6 Гц), 4,74 (т, 1H, J=5,6 Гц), 4,55 (дд, 1H, J=9,6, 5,6 Гц), 2,76 (д, 1H, J=9,6 Гц), 1,43 (с, 3H), 1,40 (с, 3H).
Шаг 2: 3aR,3bR,4aS,5S,5aS)-2,2-диметилгексагидроциклопропа[3,4]циклопента[1,2-d][1,3]диоксол-5-ол
К перемешанной смеси (3aS,4S,6aR)-2,2-диметил-3a,6a-дигидро-4H-циклопента[d][1,3]диоксол-4-ола (40,00 г, 256,11 ммоль, 1,00 экв.) в DCM (50,00 мл) (температура смеси 0 °C) добавляли по каплям ZnEt2 (1 M, 1,00 л, 3,90 экв.). Через 15 мин. в смесь добавляли CH2I2 (550,00 г, 2,05 моль, 8,02 экв.) и смесь перемешивали при температуре 20°C в течение 16 часов. ТСХ (PE:EtOAc=1:1) показала полное реагирование всего исходного материал. Смесь гасили насыщенным раствором NH4Cl (200 мл) с последующим добавлением H2O (500 мл). Смесь экстрагировали с использованием DCM (500 мл*5), объединенные органические слои сушили над Na2SO4 и концентрировали с получением неочищенного продукта. Неочищенный продукт очищали посредством колоночной хроматографии на силикагеле (PE:EtOAc=0:1/100:1/80:1/ 50:1/20:1/10:1/5:1) для получения (3aR,3bR,4aS,5S,5aS)-2,2-диметилгексагидроциклопропа[3,4]-циклопента[1,2-d][1,3]диоксол-5-ола (18,00 г, выход: 41,29%) в виде маслянистой жидкости светло-желтого цвета. 1H-ЯМР (400 МГц, CDCl3) δ м. д. 4,87 (т, 1H, J=6,0 Гц), 4,53-4,45 (м, 2H), 2,34 (уш. с., 1H), 1,85-1,82 (м, 1H), 1,64-1,62 (м, 1H), 1,54 (с, 3H), 1,28 (с, 3H), 0,98-0,94 (м, 1H), 0,63-0,60 (м, 1H).
Шаг 3: 2-((3aR,3bR,4aS,5R,5aS)-2,2-диметилгексагидроциклопропа[3,4]циклопента[1,2-d][1,3]диоксол-5-ил)изоиндолин-1,3-дион
К перемешанной смеси (3aR,3bR,4aS,5S,5aS)-2,2-диметилгексагидроциклопропа[3,4]-циклопента[1,2-d][1,3]диоксол-5-ола (10,00 г, 58,75 ммоль, 1,00 экв.), изоиндолин-1,3-диона(12,00 г, 81,56 ммоль, 1,39 экв.) и PPh3 (24,00 г, 91,50 ммоль, 1,56 экв.) в сухом толуоле (500,00 мл) в атмосфере N2 добавляли DIAD (20,00 г, 98,91 ммоль, 1,68 экв.) по каплям. Полученную смесь перемешивали при температуре 80°C в течение 20 часов в атмосфере N2. ТСХ (PE:EtOAc=3:1) показала, что исходный материал полностью прореагировал. Смесь концентрировали и осадок очищали посредством колоночной хроматографии на силикагеле (PE:EtOAc=80:1/50:1/30:1/20:1/10:1) для получения 2-((3aR,3bR,4aS,5R,5aS)-2,2-диметилгексагидроциклопропа[3,4]-циклопента[1,2-d][1,3]диоксол-5-ил)изоиндолин-1,3-диона (14,00 г, выход: 79,61%) в виде маслянистой жидкости светло-желтого цвета. 1H-ЯМР (400 МГц, CDCl3) δ м. д. 7,86-7,83 (м, 2H), 7,74-7,72 (м, 2H), 5,37-5,34 (м, 1H), 4,78-4,76 (м, 1H), 4,73 (с, 1H), 2,01-1,95 (м, 1H), 1,51 (с, 3H), 1,47-1,42 (м, 1H), 1,24 (с, 3H), 0,85-0,79 (м, 2H).
Шаг 4: (3aR,3bR,4aS,5R,5aS)-2,2-диметилгексагидроциклопропа[3,4]циклопента[1,2-d][1,3]диоксол-5-амин
Смесь 2-((3aR,3bR,4aS,5R,5aS)-2,2-диметилгексагидроциклопропа[3,4]-циклопента[1,2-d][1,3]диоксол-5-ил)изоиндолин-1,3-диона (14,00 г, 46,77 ммоль, 1,00 экв.) и NH2NH2.H2O (4,78 г, 93,54 ммоль, 2,00 экв.) в EtOH (200,00 мл) перемешивали при температуре 70°C в течение 16 часов. ТСХ (PE:EtOAc=3:1) показала, что исходный материал полностью прореагировал. Смесь фильтровали, фильтрат концентрировали и к осадку добавляли EtOAc (20 мл). Затем смесь фильтровали и концентрировали с получением (3aR,3bR,4aS,5R,5aS)-2,2-диметилгексагидроциклопропа[3,4]циклопента[1,2-d][1,3]диоксол-5-амина (7,00 г, выход: 88,45%) в виде маслянистой жидкости желтого цвета. 1H-ЯМР (400 МГц, CDCl3) δ м. д. 5,06-5,03 (м, 1H), 4,30 (д, 1H, J=6,8 Гц), 3,45 (с, 1H), 1,73-1,71 (м, 1H), 1,48 (с, 3H), 1,43-1,39 (м, 1H), 1,25 (с, 3H), 0,74-0,67 (м, 2H).
Шаг 5: (1R,2R,3S,4R,5S)-4-аминобицикло[3.1.0]гексан-2,3-диол*HCl
Смесь (3aR,3bR,4aS,5R,5aS)-2,2-диметилгексагидроциклопропа[3,4]циклопента[1,2-d][1,3]диоксол-5-амина (1,00 г, 63,61 ммоль, 1,00 экв.) в H2O (5,500 мл) и HCl (0,5 мл, 12 M) перемешивали при температуре 15°C в течение 2 часов. ТСХ (EtOAc:MeOH=10:1) показала, что исходный материал полностью прореагировал. Смесь концентрировали для получения (1R,2R,3S,4R,5S)-4-аминобицикло[3.1.0]гексан-2,3-диола*HCl (780 мг г, выход: 79,9%) в виде твердого вещества желтого цвета.
Пример 6. Синтез (1S,2S,4S)-2-амино-4-фторциклопентан-1-ола (относительная стереохимия)
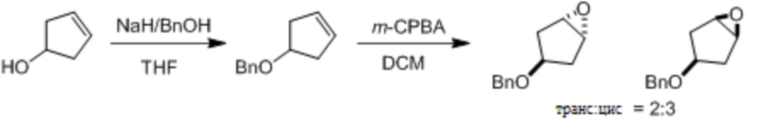
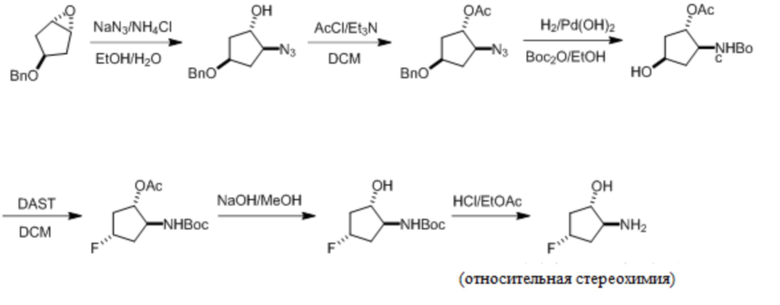
Шаг 1: ((циклопент-3-ен-1-илокси)метил)бензол
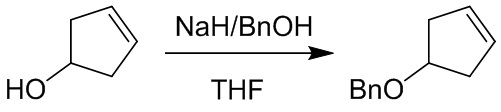
К смеси циклопент-3-ен-1-ол (60,00 г, 713,27 ммоль) в ТГФ (600,00 мл) добавляли NaH (37,09 г, 927,25 ммоль) порционно при 0 oC. После окончания выделения газа, бромметилбензол (158,59 г, 927,25 ммоль) добавляли по каплям при 0°C в течение периода 45 мин., после чего нагревали до 25°C и перемешивали в течение 16 часов. ТСХ (PE/EtOAc=50/1) показала завершение реакции. Излишки NaH гасили MeOH (120 мл) при температуре ниже 5°C. Смесь нагревали до 25°C, разбавляли H2O (600,00 мл) и разделяли два слоя. Водную фазу экстрагировали с использованием этилацетата (200 мл*3). Объединенную органическую фазу промывали солевым раствором (200 мл), сушили над Na2SO4, фильтровали и концентрировали в вакууме. Осадок очищали посредством хроматографии на силикагеле (PE/EtOAc=1/0) для получения ((циклопент-3-ен-1-илокси)метил)бензола (120,00 г, выход: 96,56%) в виде маслянистой жидкости желтого цвета. 1H-ЯМР (400 МГц, CDCl3) δ м. д. 7,38-7,28 (м, 5H), 5,72 (с, 2H), 4,52 (с, 2H), 4,35-4,30 (м, 1H), 2,64-2,59 (м, 2H), 2,50-2,46 (м, 2H).
Шаг 2: (1R,3S,5S)-3-(бензилокси)-6-оксабицикло[3.1.0]гексан и (1R,3r,5S)-3-(бензилокси)-6-оксабицикло[3.1.0]гексан
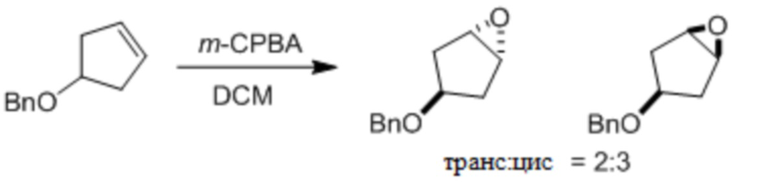
К смеси ((циклопент-3-ен-1-илокси)метил)бензола (120,00 г, 688,71 ммоль) в DCM (600,00 мл) добавляли m-CPBA (297,68 г, 1,38 моль) в один прием при 0 oC. Смесь перемешивали при 25°C в течение 16 часов. ТСХ (PE:EtOAc=20:1) показала завершение реакции. Смесь фильтровали и излишки m-CPBA снижали добавлением насыщенного водн. Na2SO3 до момента наблюдения негативного результата йоднокрахмального теста. Смесь фильтровали и концентрировали в вакууме. Осадок очищали посредством хроматографии на силикагеле (PE:EtOAc=1:0, 20:1) для получения (1R,3R,5S)-3-(бензилокси)-6-оксабицикло[3.1.0]гексан (56,00 г, выход: 42,74%) и (1R,3S,5S)-3-(бензилокси)-6-оксабицикло[3.1.0]гексан (37,00 г, выход: 28,24%) в виде маслянистой жидкости желтого цвета. Спектральный анализ (1R,3S,5S)-3-(бензилокси)-6-оксабицикло[3.1.0]гексана. 1H-ЯМР (400 МГц, CDCl3) δ м. д. 7,31-7,20 (м, 5H), 4,36 (с, 2H), 3,84-3,74 (м, 1H), 3,43 (с, 2H), 2,51-2,35 (м, 2H), 1,66-1,57 (м, 2H). Спектральный анализ (1R,3R,5S)-3-(бензилокси)-6-оксабицикло[3.1.0]гексана1H-ЯМР (400 МГц, CDCl3) δ м. д. 7,31-7,15 (м, 5H), 4,36 (с, 2H), 3,85-3,74 (м, 1H), 3,43 (с, 2H), 2,51-2,35 (м, 2H), 1,66-1,60 (м, 2H).
Шаг 3: (1S,2S,4R)-2-азидо-4-(бензилокси)циклопентан-1-ол
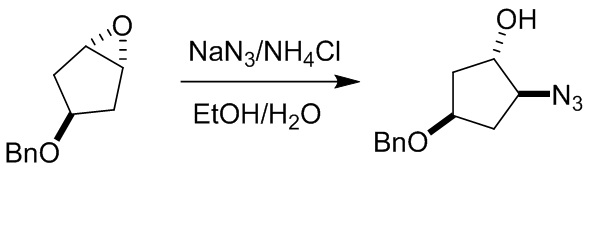
К смеси (1R,3S,5S)-3-(бензилокси)-6-оксабицикло[3.1.0]гексана (20,00 г, 105,13 ммоль) в EtOH (760,00 мл) и H2O (230,00 мл) добавляли NH4Cl (20,98 г, 392,13 ммоль), NaN3 (24,00 г, 369,17 ммоль) в один прием при температуре 25°C. Смесь нагревали до 80°C и перемешивали в течение 16 часов. ТСХ (PE:EtOAc=10:1) показала завершение реакции. Смесь охлаждали до 25°C и EtOH удаляли с использованием N2, водную фазу экстрагировали с использованием DCM (100 мл*3). Объединенную органическую фазу промывали H2O (30 мл*3), сушили над Na2SO4, фильтровали и концентрировали в вакууме для получения (1S,2S,4R)-2-азидо-4-(бензилокси)циклопентан-1-ола (23,00 г, выход: 93,79%) в виде маслянистой жидкости желтого цвета. 1H-ЯМР (400 МГц, CDCl3) δ м. д. 7,43-7,28 (м, 5H), 4,58-4,47 (м, 2H), 4,27-4,25 (м, 1H), 4,11-4,08 (м, 1H), 3,66-3,61 (м, 1H), 2,49-2,44 (м, 2H), 2,16-2,13 (м, 1H), 1,89 (уш. с., 1H), 1,87-1,80 (м, 2H).
Шаг 4: (1S,2S,4R)-2-азидо-4-(бензилокси)циклопентил ацетат
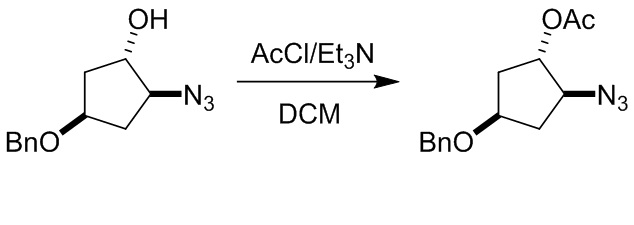
К раствору (1S,2S,4R)-2-азидо-4-(бензилокси)циклопентан-1-ола (22,90 г, 98,17 ммоль), Et3N (59,60 г, 589,02 ммоль) в DCM (550 мл) добавляли раствор ацетилхлорида (38,53 г, 490,85 ммоль) в DCM (50 мл) по каплям при 0°C на протяжении 30 мин. в атмосфере N2, на протяжении чего температура поддерживалась на уровне ниже 5°C. После этого реакционную смесь нагревали до 25°C и перемешивали в течение 16 часов. ТСХ (PE:tOAc=10:1) показала, что исходный материал полностью прореагировал. Реакцию гасили медленным добавлением H2O (100 мл). Органическую фазу промывали насыщенным солевым раствором (50 мл), сушили над Na2SO4, фильтровали и концентрировали под вакуумом. Осадок очищали посредством колоночной хроматографии на силикагеле (PE: EtOAc=100:1, 50:1) для получения (1S,2S,4R)-2-азидо-4-(бензилокси)циклопентил ацетата (17,00 г, выход: 62,90%) в виде маслянистой жидкости желтого цвета. 1H-ЯМР (400 МГц, CDCl3) δ м. д. 7,37-7,28 (м, 5H), 5,24-5,12 (м, 1H), 4,51 (с, 2H), 4,14-4,11 (м, 1H), 3,88-3,85 (м, 1H), 2,45-2,40 (м, 1H), 2,36-2,32 (м, 1H), 2,07 (с, 3H), 1,95-1,88 (м, 2H).
Шаг 5: (1S,2S,4R)-2-((трет-бутоксикарбонил)амино)-4-гидроксициклопентил ацетат
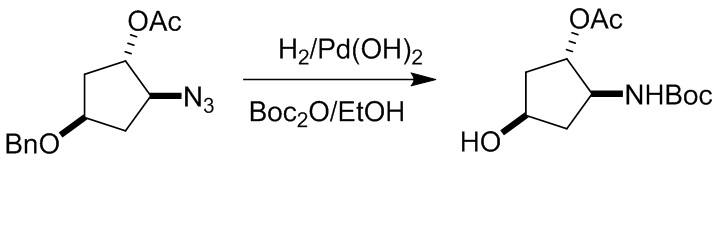
К раствору (1S,2S,4R)-2-азидо-4-(бензилокси)циклопентил ацетата (8,80 г, 31,97 ммоль) в EtOH (100,00 мл) добавляли Pd(OH)2 (4,42 г, 31,97 ммоль) в атмосфере N2. Суспензию дегазировали под вакуумом и продували H2 несколько раз. Смесь перемешивали в атмосфере H2 (50 фунтов/кв. дюйм) при температуре 70°C в течение 32 часов. ТСХ (PE: EtOAc=2:1) показала, что исходный материал полностью прореагировал. Реакционную смесь фильтровали и фильтрат концентрировали. Неочищенный продукт очищали посредством хроматографии на силикагеле (PE:EtOAc=10:1, 2:1) для получения (1S,2S,4R)-2-((трет-бутоксикарбонил)амино)-4-гидроксициклопентилацетата (3,80 г, выход: 45,84%) в виде твердого вещества желтого цвета.
Шаг 6: (1S,2S,4S)-2-((трет-бутоксикарбонил)амино)-4-фторциклопентил ацетат
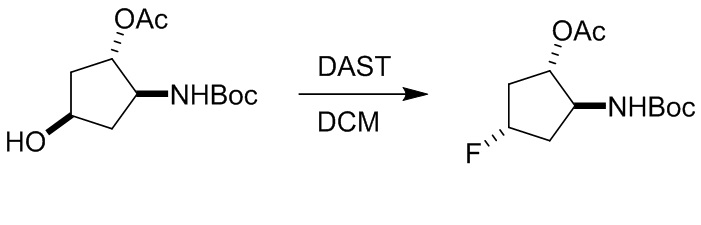
К смеси (1S,2S,4R)-2-((трет-бутоксикарбонил)амино)-4-гидроксициклопентил ацетата (2,57 г, 9,91 ммоль) в DCM (150,00 мл) добавляли DAST (2,40 г, 14,87 ммоль) по каплям при -70°C в атмосфере N2. Смесь перемешивали при -70°C в течение 30 минут. ТСХ (PE:EtOAc=2:1) показала завершение реакции. Смесь охлаждали до 0°C, добавляли водн. NaHCO3 (5 мл, 10%) и оставляли при перемешивании 10 мин. Водную фазу экстрагировали с использованием EtOAc (15 мл*2) и объединенную органическую фазу промывали солевым раствором (10 мл), сушили над Na2SO4, фильтровали и концентрировали в вакууме. Неочищенный продукт очищали посредством хроматографии на силикагеле (PE:AtOAc=20:1, 10:1) для получения (1S,2S,4S)-2-((трет-бутоксикарбонил)амино)-4-фторциклопентилацетата (700,00 мг, выход: 27,03%) в виде маслянистой жидкости желтого цвета. 1H-ЯМР (400 МГц, CDCl3) δ м. д. 5,14 (с, 0,5H), 5,00 (уш. с., 1H), 4,71 (с, 0,5H), 4,14-4,13 (м, 1H), 2,49-2,47 (м, 2H), 2,07-1,94 (м, 3H), 1,81-1,74 (м, 2H), 1,43-1,41 (м, 9H).
Шаг 7: трет-бутил((1S,2S,4S)-4-фтор-2-гидроксициклопентил)карбамат
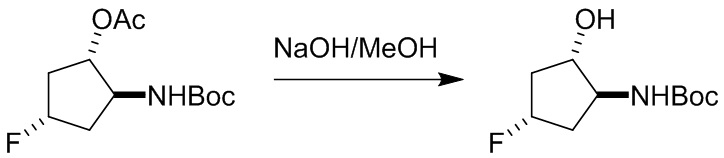
К смеси (1S,2S,4S)-2-((трет-бутоксикарбонил)амино)-4-фторциклопентил ацетата (700,00 мг, 2,68 ммоль) в MeOH (20,00 мл) добавляли NaOH (160,80 мг, 4,02 ммоль) в один прием. Смесь перемешивали при 25°C в течение 1 часа. ТСХ (PE:EtOAc=3:1) показала завершение реакции. Смесь концентрировали при сниженном давлении при температуре 30°C для получения трет-бутил((1S,2S,4S)-4-фтор-2-гидроксициклопентил)карбамата (650,00 мг, в неочищенном виде) в виде твердого вещества белого цвета.
Шаг 8: (1S,2S,4S)-2-амино-4-фторциклопентан-1-ол (относительная стереохимия)
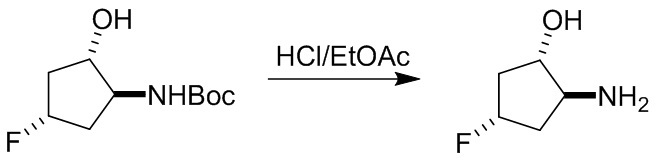
Смесь трет-бутил((1S,2S,4S)-4-фтор-2-гидроксициклопентил)карбамата (650,00 мг, 2,96 ммоль) в MeOH/HCl (20,00 мл, 4 M) перемешивали в течение 1 часа при температуре 25°C. ТСХ (PE:EtOAc=2:1) показала завершение реакции. Смесь концентрировали при сниженном давлении при температуре 30°C для получения (1S,2S,4S)-2-амино-4-фторциклопентан-1-ола (относительная стереохимия) (400,00 мг, выход: 86,85%) в виде твердого вещества белого цвета. 1H-ЯМР (400 МГц, DMSO-d6) δ м. д. 8,38 (уш. с., 3H), 5,09 (д, 1H, J=53,6 Гц), 4,09-4,03 (м, 1H), 3,34-3,30 (м, 1H), 2,44-2,39 (м, 1H), 2,21-2,16 (м, 1H), 1,95-1,87 (м, 1H), 1,75-1,66 (м, 1H).
Пример 7. Синтез (1R,2R,4R)-2-амино-4-(метилсульфонил)циклопентан-1-ола (относительная стереохимия)
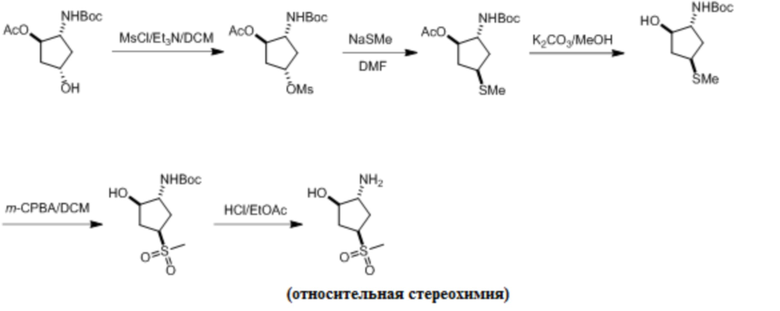
Шаг 1: приготовление (1R,2R,4S)-2-((трет-бутоксикарбонил)амино)-4-((метилсульфонил)окси)циклопентилацетата
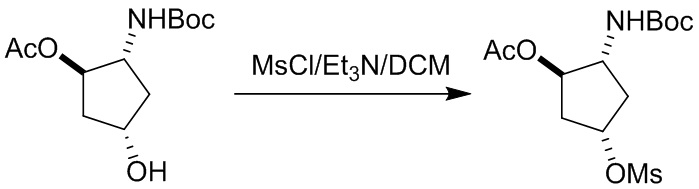
К смеси (1R,2R,4S)-2-((трет-бутоксикарбонил)амино)-4-гидроксициклопентилацетата (3,00 г, 11,57 ммоль) и Et3N (4,68 г, 46,28 ммоль) в DCM (50,00 мл) добавляли по каплям MsCl (3,98 г, 34,71 ммоль) при 0 °C, после чего смесь перемешивали при температуре 20°C в течение 3 часов. ТСХ (PE:EtOAc=1:1) показала завершение реакции. Смесь промывали водой (100 мл*3), после чего органический слой высушивали над Na2SO4 и концентрировали с получением (1R,2R,4S)-2-((трет-бутоксикарбонил)амино)-4-((метилсульфонил)окси)циклопентилацетата (3,5 г, в неочищенном виде: 100%) в виде твердого вещества желтого цвета.
Шаг 2: (1R,2R,4R)-2-((трет-бутоксикарбонил)амино)-4-(метилтио)циклопентилацетат

К смеси (1R,2R,4S)-2-((трет-бутоксикарбонил)амино)-4-((метилсульфонил)окси)циклопентилацетата (3,50 г, 10,37 ммоль) в DMF (30,00 мл) добавляли NaSMe (4,36 г, 12,45 ммоль). Смесь после чего перемешивали при температуре 90°C в течение 2 часов, и ТСХ (PE:EtOAc=2:1) показала завершение реакции. Смесь концентрировали для получения в неочищенном виде (1R,2R,4R)-2-((трет-бутоксикарбонил)амино)-4-(метилтио)циклопентил ацетата (3,00 г, в неочищенном виде 100%) в виде твердого вещества желтого цвета.
Шаг 3: трет-бутил((1R,2R,4R)-2-гидрокси-4-(метилтио)циклопентил)карбамат
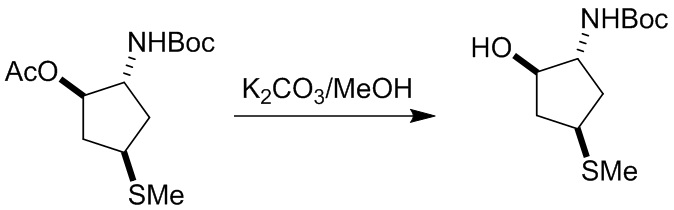
К смеси (1R,2R,4R)-2-((трет-бутоксикарбонил)амино)-4-(метилтио)циклопентилацетата (3,00 г, 10,37 ммоль) в MeOH (100,00 мл) добавляли K2CO3 (2,87 г, 20,74 ммоль). Смесь перемешивали при 25°C в течение 16 часов. ТСХ (PE:EtOAc=2:1) показала завершение реакции. Смесь концентрировали и очищали посредством колоночной хроматографии на силикагеле (PE:EtOAc=5:1-1:1) для получения трет-бутил((1R,2R,4R)-2-гидрокси-4-(метилтио)-циклопентил)карбамата (1,60 г, выход: 62,38%) в виде твердого вещества белого цвета. 1H-ЯМР (400 МГц, CDCl3) δ м. д. 4,10-3,99 (м, 2H), 3,85 (уш. с., 1H), 3,14-3,11 (м, 1H), 2,50-2,46 (м, 1H), 2,17-2,10 (м, 4H), 1,86-1,82 (м, 1H), 1,81-1,67 (м, 1H), 1,45 (с, 9H).
Шаг 4: трет-бутил((1R,2R,4R)-2-гидрокси-4-(метилсульфонил)циклопентил)карбамат
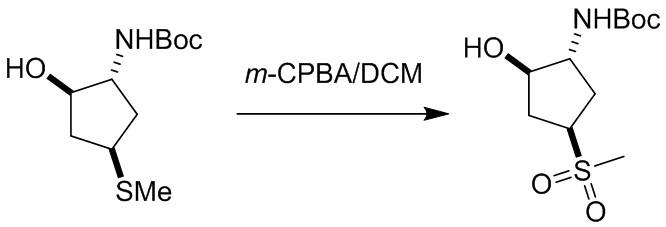
К смеси трет-бутил((1R,2R,4R)-2-гидрокси-4-(метилтио)циклопентил)карбамата (1,60 г, 6,47 ммоль) в DCM (100,00 мл) добавляли m-CPBA (3,49 г, 16,18 ммоль). Смесь перемешивали при 25°C в течение 16 часов. После того как ТСХ (PE:EtOAc=2:1) показала, что исходный материал полностью прореагировал, смесь промывали насыщенным Na2SO3 (водн. 20 мл) и насыщенным NaHCO3 (водн. 20 мл*3). Органический слой после чего сушили над Na2SO4, концентрировали, промывали PE (10 мл), фильтровали, и отфильтрованный осадок высушивали под вакуумом для получения трет-бутил((1R,2R,4R)-2-гидрокси-4-(метилсульфонил)циклопентил)карбамата (1,70 г, выход: 94,06%) в виде твердого вещества белого цвета. 1H-ЯМР (400 МГц, CDCl3) δ м. д. 4,62 (уш. с., 1H), 4,07-4,04 (м, 1H), 3,81-3,79 (м, 1H), 3,48-3,45 (м, 1H), 2,82 (с, 3H), 2,57-2,40 (м, 2H), 2,11-2,07 (м, 1H), 1,90-1,87 (м, 2H), 1,38 (с, 9H).
Шаг 5: (1R,2R,4R)-2-амино-4-(метилсульфонил)циклопентан-1-ол (относительная стереохимия)
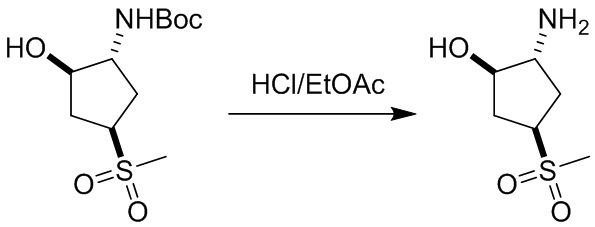
Смесь трет-бутил((1R,2R,4R)-2-гидрокси-4-(метилсульфонил)циклопентил)карбамата (1,2 г, 4,30 ммоль) в HCl/MeOH (10,00 мл) перемешивали при температуре 25°C в течение 16 часов, после чего ЖХМС показала завершение реакции, и смесь концентрировали с получением (1R,2R,4R)-2-амино-4-(метилсульфонил)циклопентан-1-ола (1,0 г, в неочищенном виде: 100%) в виде твердого вещества желтого цвета. 1H-ЯМР (400 МГц, CD3OD) δ м. д. 4,11-4,07 (м, 2H), 3,76 (уш. с., 1H), 2,94 (с, 3H), 2,53 (уш. с., 2H), 1,99 (уш. с., 2H).
Пример 8. Синтез (1S,2R,5R)-2-амино-5-фторциклопентан-1-ол гидрохлорида (относительная стереохимия)

Шаг 1: циклопент-2-ен-1-ол
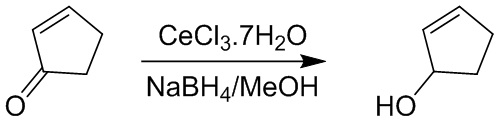
К смеси CeCl3.7H2O (24,00 г, 64,42 ммоль) в MeOH (120,00 мл) добавляли циклопент-2-ен-1-он (5,00 г, 60,90 ммоль) при температуре 15 oC. Через 5 мин. в смесь добавляли NaBH4 (4,61 г, 121,80 ммоль) порционно при 0 oC. Полученную смесь перемешивали при температуре 25°C в течение 1 часа, после чего ТСХ (PE:EtOAc=5:1) показала проявление нескольких пятен и остаток части исходного материала. Реакцию гасили H2O (100 мл) и органический растворитель концентрировали в вакууме. К осадку добавляли H2O (300 мл), с последующим экстрагированием с использованием MTBE (200 мл*3). Объединенные органические слои сушили над Na2SO4 и концентрировали под вакуумом для получения неочищенного продукта циклопент-2-ен-1-ола (3,00 г, в неочищенном виде) в виде маслянистой жидкости коричневого цвета. Его непосредственно использовали на следующем этапе без дополнительной очистки. 1H-ЯМР (400 МГц, CDCl3) δ м. д. 5,91 (д, 1H, J=4,8 Гц), 5,77-5,76 (м, 1H), 4,79 (д, 1H, J=3,6 Гц), 2,47-2,42 (м, 1H), 2,21-2,15 (м, 2H), 1,64-1,59 (м, 1H).
Шаг 2: ((циклопент-2-ен-1-илокси)метил)бензол
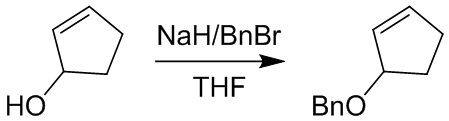
К смеси циклопент-2-ен-1-ол (9,00 г, 106,99 ммоль) в ТГФ (200,00 мл) добавляли NaH (6,80 г, 170,11 ммоль) порционно при 0 °C. После добавления смесь перемешивали при температуре 20°C в течение 0,5 часа, после чего BnBr (20,00 г, 116,94 ммоль) добавляли в смесь по каплям при 0°C. Полученную смесь перемешивали при температуре 20°C в течение 2 часов. ТСХ (PE:EtOAc=20:1) показала образование новых видов (Rf=0,6, 254 нм). В этот момент добавляли H2O (20 мл) с последующим экстрагированием с использованием EtOAc (20 мл*3). Объединенные органические слои сушили над Na2SO4 и концентрировали с получением неочищенного продукта, который очищали посредством колоночной хроматографии на силикагеле (PE:EtOAc=1:0/100:1/80:1) для получения ((циклопент-2-ен-1-илокси)метил)бензола (8,00 г, выход: 42,91%) в виде маслянистой жидкости желтого цвета. 1H-ЯМР (400 МГц, CDCl3) δ м. д. 7,34-7,25 (м, 5H), 6,02 (уш. с., 1H), 5,88 (уш. с., 1H), 4,66 (уш. с., 1H), 4,55-4,47 (м, 2H), 2,52-4,48 (м, 1H), 2,27-2,24 (м, 1H), 2,16-2,13 (м, 1H), 1,87-1,84 (м, 1H).
Шаг 3: (1S,2S,5S)-2-(бензилокси)-6-оксабицикло[3.1.0]гексан
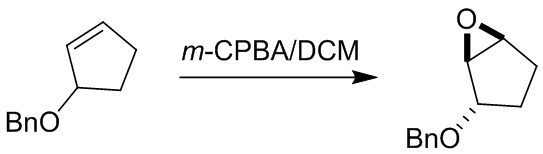
К смеси ((циклопент-2-ен-1-илокси)метил)бензола (8,50 г, 29,27 ммоль) в DCM (50,00 мл) добавляли m-CPBA (13,50 г, 58,67 ммоль) порционно при 0°C. Смесь перемешивали при 25°C в течение 4 часов. Как только ТСХ (PE:EtOAc=10:1) показала, что исходный материал полностью прореагировал, смесь фильтровали, фильтрат концентрировали и очищали посредством колоночной хроматографии на силикагеле (PE:EtOAc=1:0/100:1/80:1/50:1) для получения неочищенного продукта. Затем добавляли DCM (20 мл), смесь фильтровали, и к фильтрату добавляли H2O (20 мл) и Na2CO3 (500 мг), после чего перемешивали смесь при температуре 25°C в течение 0,5 часа. Смесь после чего экстрагировали с использованием DCM (20 мл*3), сушили над Na2SO4 и концентрировали с получением (1S,2S,5S)-2-(бензилокси)-6-оксабицикло[3.1.0]гексана (2,40 г, выход: 43,10%) в виде бесцветной маслянистой жидкости, состав был подтвержден посредством NOE. 1H-ЯМР (400 МГц, CDCl3) δ м. д. 7,37-7,26 (м, 5H), 4,61-4,51 (м, 2H), 4,09 (д, 1H, J=5,2 Гц), 3,55 (уш. с., 1H), 3,49 (уш. с., 1H), 1,99-1,95 (м, 1H), 1,87-1,75 (м, 2H), 1,54-1,52 (м, 1H).
Шаг 4: (1S,2R,5S)-2-азидо-5-(бензилокси)циклопентан-1-ол
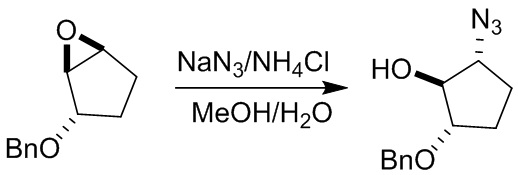
К смеси (1S,2S,5S)-2-(бензилокси)-6-оксабицикло[3.1.0]гексана (2,40 г, 12,62 ммоль) и NH4Cl (1,55 г, 29,03 ммоль) в H2O (3,00 мл) и MeOH (24,00 мл) добавляли NaN3 (4,10 г, 63,10 ммоль), который перемешивали при температуре 80°C в течение 16 часов. После того как ТСХ (PE:EtOAc=10:1) показала полное реагирование всего исходного материала, органический растворитель высушивали с использованием N2, осадок разбавляли H2O (20 мл) и экстрагировали с использованием DCM (20 мл*3). Объединенные органические фазы промывали H2O (10 мл*3), сушили над Na2SO4 и концентрировали с получением (1S,2R,5S)-2-азидо-5-(бензилокси)циклопентан-1-ола (2,60 г, выход: 88,32%) в виде маслянистой жидкости коричневого цвета. 1H-ЯМР (400 МГц, CDCl3) δ м. д. 7,42-7,31 (м, 5H), 4,64-4,55 (м, 2H), 4,02-3,99 (м, 1H), 3,82-3,80 (м, 1H), 3,66-3,63 (м, 1H), 2,25 (уш. с., 1H), 2,07-2,01 (м, 2H), 1,80-1,77 (м, 2H).
Шаг 5: (((1S,2R,5S)-2-азидо-5-(бензилокси)циклопентил)окси)(трет-бутил)диметилсилан
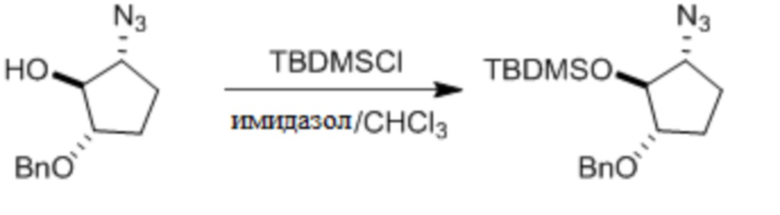
Смесь (1S,2R,5S)-2-азидо-5-(бензилокси)циклопентан-1-ол (2,50 г, 10,72 ммоль), имидазола (1,61 г, 23,69 ммоль) и TBDMSCl (2,42 г, 16,08 ммоль) в CHCl3 (5,00 мл) перемешивали при температуре 80°C в течение 16 часов. Как только ТСХ (PE:EtOAc=10:1) показала полное реагирование всего исходного материала, смесь концентрировали и очищали посредством колоночной хроматографии на силикагеле (PE:EtOAc =1:0/100:1/80:1) для получения (((1S,2R,5S)-2-азидо-5-(бензилокси)циклопентил)окси)(трет-бутил)диметилсилана (3,00 г, выход: 80,52%) в виде бесцветной маслянистой жидкости. 1H-ЯМР (400 МГц, CDCl3) δ м. д. 7,24-7,15 (м, 5H), 4,40 (д, 2H, J=2,4 Гц), 3,88-3,86 (м, 1H), 3,63-3,61 (м, 1H), 3,47-3,43 (м, 1H), 1,94-1,82 (м, 2H), 1,70-1,65 (м, 2H), 0,79 (с, 9H), 0,03 (с, 3H), 0,00 (с, 3H).
Шаг 6: трет-бутил((1R,2S,3S)-2-((трет-бутилдиметилсилил)окси)-3-гидроксициклопентил)карбамат
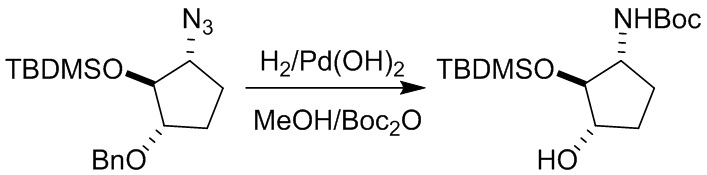
К смеси (((1S,2R,5S)-2-азидо-5-(бензилокси)циклопентил)окси)(трет-бутил)диметилсилана (2,90 г, 8,34 ммоль) и Boc2O (2,20 г, 10,10 ммоль) в MeOH (50,00 мл) добавляли Pd(OH)2 (1,50 г, 5,42 ммоль), который перемешивали при температуре 50°C в атмосфере H2 (50 фунтов/кв. дюйм) в течение 20 часов. После того как ТСХ (PE:EtOAc=3:1) показала, что исходный материал полностью прореагировал, смесь фильтровали, фильтрат концентрировали и очищали посредством колоночной хроматографии на силикагеле (PE:EtOAc=10:1/8:1/5:1) для получения трет-бутил((1R,2S,3S)-2-((трет-бутилдиметилсилил)окси)-3-гидроксициклопентил)карбамата (2,10 г, выход: 75,95%) в виде твердого вещества белого цвета. 1H-ЯМР (400 МГц, CDCl3) δ м. д. 4,81 (уш. с., 1H), 3,89-3,88 (м, 1H), 3,70-3,67 (м, 2H), 2,06-1,89 (м, 2H), 1,58-1,56 (м, 2H), 1,35 (с, 9H), 0,79 (с, 9H), 0,02 (с, 3H), 0,00 (с, 3H).
Шаг 7: трет-бутил((1R,2S,3R)-2-((трет-бутилдиметилсилил)окси)-3-фторциклопентил)карбамат
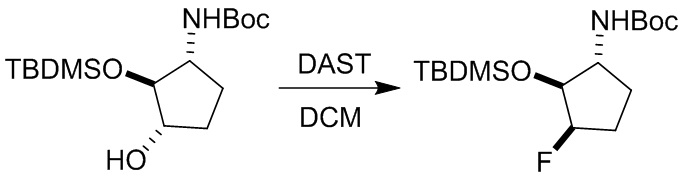
К смеси трет-бутил((1R,2S,3S)-2-((трет-бутилдиметилсилил)окси)-3-гидроксициклопентил)карбамата (1,10 г, 3,32 ммоль) в DCM (50,00 мл) добавляли DAST (1,61 г, 9,96 ммоль) при температуре -70 °C. Реакционную смесь перемешивали при -70°C в течение 1 часа и при 25°C в течение 1 часа. После того как ТСХ (PE:EtOAc=5:1, Rf=0,6) показала завершение реакции, к реакции добавляли ледяную воду (5 мл). Раствор экстрагировали с использованием DCM (20 мл*3) и промывали солевым раствором (30 мл). Органический слой высушивали над Na2SO4 и концентрировали. Осадок очищали посредством колоночной хроматографии на силикагеле (PE:EtOAc=100:1) для получения трет-бутил((1R,2S,3R)-2-((трет-бутилдиметилсилил)окси)-3-фторциклопентил)карбамата (100,00 мг, выход: 9,03%) в виде твердого вещества белого цвета. 1H-ЯМР (400 МГц, CDCl3) δ м. д. 4,64 (д, 1H, J=54,4 Гц), 4,26 (уш. с., 1H), 3,90-3,86 (м, 1H), 3,76-3,68 (м, 1H), 2,11-1,79 (м, 4H), 1,34 (с, 9H), 0,81 (с, 9H), 0,00 (с, 6H).
Шаг 8: (1S,2R,5R)-2-амино-5-фторциклопентан-1-ол гидрохлорид (относительная стереохимия)
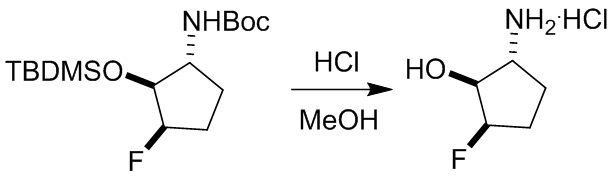
Раствор трет-бутил((1R,2S,3R)-2-((трет-бутилдиметилсилил)окси)-3-фторциклопентил)карбамата (100,00 мг, 299,84 мкмоль) в HCl/MeOH (20,00 мл, 4 M) перемешивали при температуре 25°C в течение 16 часов. ТСХ (PE:EtOAc=5:1, Rf=0) показала завершение реакции. Раствор высушивали с использованием N2 и получали (1S,2R,5R)-2-амино-5-фторциклопентан-1-ол гидрохлорид (относительная стереохимия)(45,00 мг, выход: 96,45%) в виде твердого вещества желтого цвета. 1H-ЯМР (400 МГц, CD3OD) δ м. д. 4,93-4,89 (м, 1H), 3,98-3,88 (м, 1H), 3,52-3,47 (м, 1H), 2,30-2,00 (м, 3H), 1,66-1,62 (м, 1H).
Пример 9. Синтез (1S,2R,5S)-2-амино-5-фторциклопентан-1-ола (относительная стереохимия)
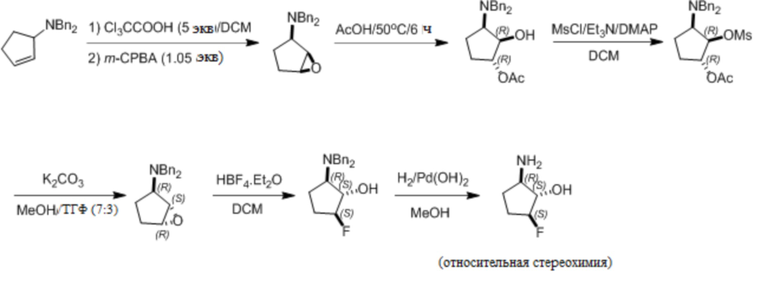
Шаг 1: (1R,2R,5S)-N,N-дибензил-6-оксабицикло[3.1.0]гексан-2-амин
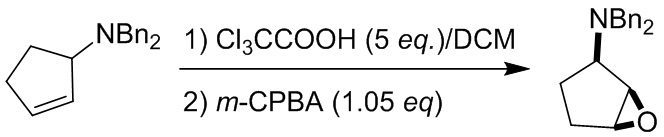
Cl3CCOOH (154,72 г, 949,20 ммоль) добавляли при перемешивании к раствору N,N-дибензилциклопент-2-ен-1-амина (50,00 г, 189,84 ммоль) в DCM (640 мл), и полученную смесь перемешивали при температуре 20°C в течение 0,1 часа. m-CPBA (43,00 г, 199,33 ммоль) добавляли в один прием и реакционную смесь оставляли при постоянном перемешивании при температуре 20°C в течение 16 часов. После того как ТСХ (PE:EtOAc=10:1) показала завершение реакции, смесь разбавляли DCM (500 мл) и насыщ. водн. Na2SO3 добавляли до тех пор, пока йодокрахмальная бумага не показывала полное отсутствие m-CPBA. Насыщ. водн. NaHCO3 (500 мл) добавляли к смеси и слои разделяли. Органический слой промывали водн. NaHCO3 (200 мл*2) после чего высушивали, концентрировали, и очищали посредством колоночной хроматографии на силикагеле (PE:EtOAc=100:1-50:1) для получения (1R,2R,5S)-N,N-дибензил-6-оксабицикло[3.1.0]гексан-2-амина (30,00 г, выход: 56,56%) в виде твердого вещества белого цвета. 1H-ЯМР (400 МГц, CDCl3) δ м. д. 7,41 (д, 4H, J=7,2 Гц), 7,31 (т, 4H, J=7,6 Гц), 7,25-7,22 (м, 2H), 3,86-3,70 (м, 4H), 3,44 (с, 1H), 3,32 (с, 1H), 3,28-3,24 (м, 1H), 2,04-2,01 (м, 1H), 1,54-1,45 (м, 3H).
Шаг 2: (1R,2R,3R)-3-(дибензиламино)-2-гидроксициклопентил ацетат
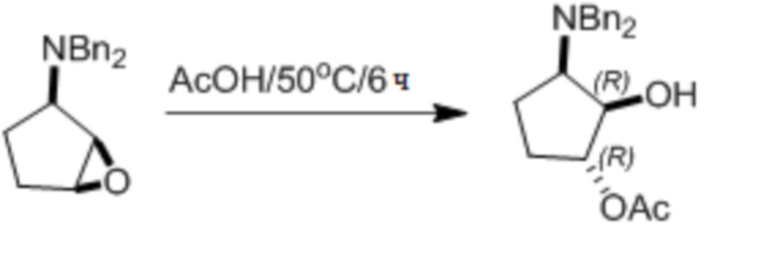
Раствор (1R,2R,5S)-N,N-дибензил-6-оксабицикло[3.1.0]гексан-2-амина (30,00 г, 107,38 ммоль, 1,00 экв.) в AcOH (200 мл) перемешивали при температуре 50°C в течение 16 часов. После того как ТСХ (PE:EtOAc=10:1) показала завершение реакции, смесь концентрировали для удаления AcOH, осадок растворяли в DCM (100 мл), органический слой промывали водн. NaHCO3 (100 мл*3), сушили над Na2SO4 и концентрировали. Осадок очищали посредством колоночной хроматографии на силикагеле (PE:EtOAc=100:1-50:1) для получения (1R,2R,3R)-3-(дибензиламино)-2-гидроксициклопентил ацетата (20,00 г, выход: 54,87%) в виде твердого вещества белого цвета. 1H-ЯМР (400 МГц, CDCl3) δ м. д. 7,36-7,28 (м, 10H), 5,05-5,02 (м, 1H), 4,05 (д, 1H, J=4,0 Гц), 3,81-3,69 (м, 4H), 3,27-3,24 (м, 1H), 2,40-2,36 (м, 1H), 2,07 (с, 3H), 1,97-1,94 (м, 1H), 1,78-1,73 (м, 1H), 1,60-1,54 (м, 1H).
Шаг 3: (1R,2R,3R)-3-(дибензиламино)-2-((метилсульфонил)окси)циклопентил ацетат
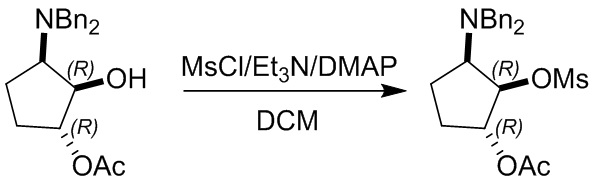
MsCl (8,10 г, 70,71 ммоль) добавляли по каплям к смеси (1R,2R,3R)-3-(дибензиламино)-2-гидроксициклопентил ацетата (20,00 г, 58,92 ммоль), Et3N (18,48 г, 182,66 ммоль) и DMAP (719,83 мг, 5,89 ммоль) в DCM (200 мл). После добавления смесь перемешивали при температуре 20°C в течение 16 часов. После того как ТСХ (PE:EtOAc=10:1 ) показала завершение реакции, смесь промывали водой (100 мл*2), органический слой высушивали над Na2SO4 и очищали посредством колоночной хроматографии на силикагеле (PE:EtOAc=80:1-60:1) для получения (1R,2R,3R)-3-(дибензиламино)-2-((метилсульфонил)окси)циклопентил ацетата (15,00 г, выход: 60,97%) в виде твердого вещества белого цвета. 1H-ЯМР (400 МГц, CDCl3) δ м. д. 7,41 (д, 4H, J=7,6 Гц), 7,33 (т, 4H, J=6,8 Гц), 7,31-7,25 (м, 2H), 5,16-5,13 (м, 1H), 5,01-5,00 (м, 1H), 3,92-3,81 (м, 4H), 3,37-3,32 (м, 1H), 3,15 (с, 3H), 2,33-2,30 (м, 1H), 2,01 (с, 3H), 1,98-1,91 (м, 2H), 1,56-1,53 (м, 1H).
Шаг 4: (1S,2R,5R)-N,N-дибензил-6-оксабицикло[3.1.0]гексан-2-амин
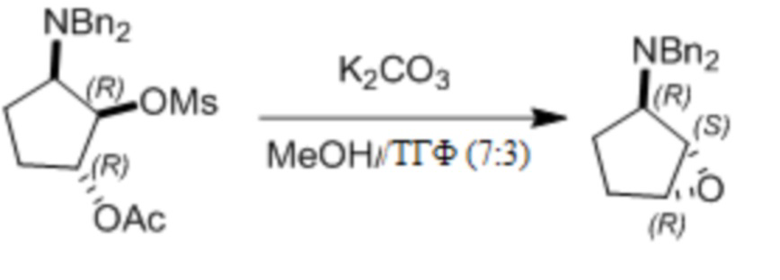
K2CO3 (5,96 г, 43,11 ммоль) добавляли к смеси (1R,2R,3R)-3-(дибензиламино)-2-((метилсульфонил)окси)циклопентил ацетата (15,00 г, 35,93 ммоль) в MeOH (70 мл)/THF (30 мл). Смесь перемешивали при 20°C в течение 16 часов. После того как ТСХ (PE:EtOAc=10:1) показала завершение реакции, смесь концентрировали для удаления MeOH и THF. После этого смесь растворяли в DCM (20 мл), органический слой промывали водой (10 мл*2), сушили над Na2SO4 и концентрировали с получением (1S,2R,5R)-N,N-дибензил-6-оксабицикло[3.1.0]гексан-2-амина (10,00 г, выход: 99,62%) в виде твердого вещества белого цвета. 1H-ЯМР (400 МГц, CDCl3) δ м. д. 7,37 (д, 4H, J=7,6 Гц), 7,32 (д, 4H, J=7,2 Гц), 7,29-7,23 (м, 2H), 3,73-3,69 (м, 2H), 3,53-3,41 (м, 5H), 2,06-2,00 (м, 1H), 1,91-1,90 (м, 1H), 1,87-1,76 (м, 1H), 1,51-1,48 (м, 1H).
Шаг 5: (1S,2R,5S)-2-(дибензиламино)-5-фторциклопентан-1-ол
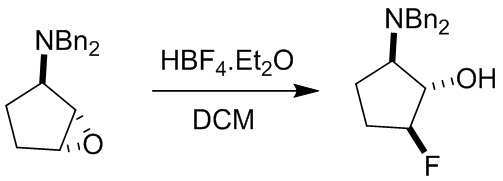
К смеси (1S,2R,5R)-N,N-дибензил-6-оксабицикло[3.1.0]гексан-2-амина (6,50 г, 23,27 ммоль) в DCM (200 мл) добавляли HBF4/Et2O (7,54 г, 46,53 ммоль), и смесь оставляли при перемешивании 30°C в течение 0,2 часа. После того как ТСХ показала завершение реакции, смесь добавляли к Na2CO3 (100 мл) и экстрагировали с использованием DCM (200 мл*2). Органический слой сушили над Na2SO4, концентрировали, и очищали посредством колоночной хроматографии на силикагеле (PE:EtOAc=от 60:1 до 20:1) для получения (1S,2R,5S)-2-(дибензиламино)-5-фторциклопентан-1-ола (1,80 г, выход: 25,84%) в виде твердого вещества белого цвета. 1H-ЯМР (400 МГц, CDCl3) δ м. д. 7,38-7,22 (м, 10H), 4,81-4,67 (м, 1H), 4,23-4,15 (м, 1H), 3,83-3,55 (м, 4H), 3,04-2,98 (м, 1H), 1,93-1,79 (м, 4H).
Шаг 6: (1S,2R,5S)-2-амино-5-фторциклопентан-1-ол(относительная стереохимия)
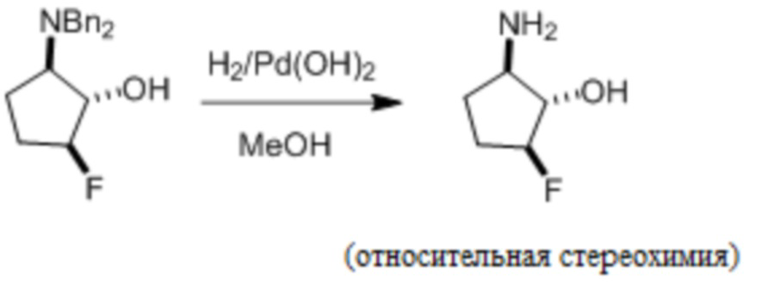
К смеси (1S,2R,5S)-2-(дибензиламино)-5-фторциклопентан-1-ол (1,60 г, 5,34 ммоль) в MeOH (10 мл) добавляли Pd(OH)2 (500,00 мг, 3,61 ммоль), и смесь оставляли при помешивании при температуре 30°C в течение 16 часов в атмосфере H2 (30 фунтов/кв. дюйм). После того как ТСХ показала завершение реакции, смесь фильтровали через целит и фильтрат концентрировали с получением (1S,2R,5S)-2-амино-5-фторциклопентан-1-ола (600,00 мг, выход: 94,31%) в виде твердого вещества белого цвета. 1H-ЯМР (400 МГц, CDCl3) δ м. д. 4,93-4,77 (м, 1H), 3,92-3,84 (м, 1H), 3,10-3,04 (м, 1H), 2,07-1,98 (м, 3H), 1,64-1,61 (м, 1H).
Пример 10. Синтез 3-фтор-5-((2R,4S)-4-фторпирролидин-2-ил)пиридина
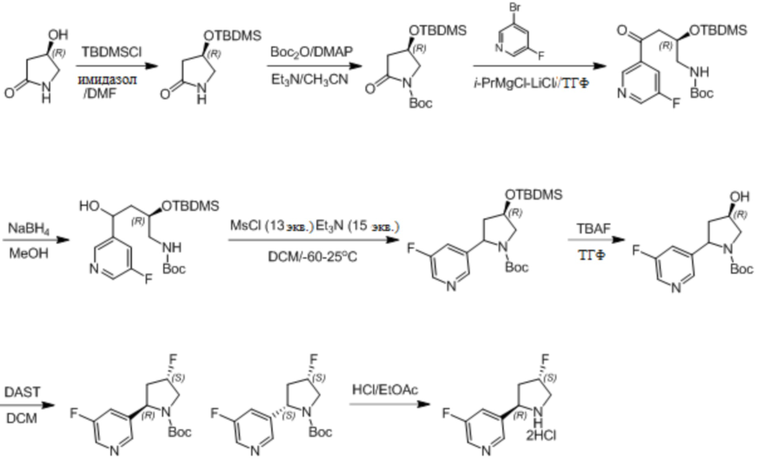
Шаг 1: (R)-4-((трет-бутилдиметилсилил)окси)пирролидин-2-он
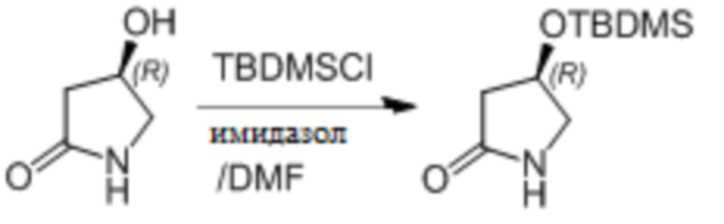
К смеси (R)-4-гидроксипирролидин-2-она (9,0 г, 89,1 ммоль) в DMF (50 мл) добавляли имидазол (9,09 г, 134 ммоль) и TBDMSCl (14,1 г, 93,6 ммоль) в один прием при 0oC. Реакционную смесь перемешивали при 25°C в течение 3 ч. ТСХ (DCM/MeOH=10/1, Rf=0,8) показала завершение реакции, после чего добавляли воду (200 мл), выпавший осадок собирали фильтрацией и высушивали под вакуумом для получения (R)-4-((трет-бутилдиметилсилил)окси)пирролидин-2-она (15,5 г, выход: 80,7%) в виде твердого вещества белого цвета. 1H-ЯМР (400 МГц, CDCl3) δ м. д. 5,90 (уш. с., 1H), 4,55-4,53 (м, 1H), 3,60-3,56 (м, 1H), 3,24-3,21 (м, 1H), 2,56-2,50 (м, 1H), 2,28-2,23 (м, 1H), 0,87-0,85 (м, 9H), 0,06-0,00 (м, 6H).
Шаг 2: приготовление трет-бутил (R)-4-((трет-бутилдиметилсилил)окси)-2-оксопирролидин-1-карбоксилата

К смеси (R)-4-((трет-бутилдиметилсилил)окси)пирролидин-2-она (15,5 г, 72,0 ммоль) в CH3CN (150 мл) добавляли Et3N (8,72 г, 86,4 ммоль), DMAP (4,39 г, 36 ммоль) и Boc2O (20,4 г, 93,7 ммоль) в один прием при 0 oC. Реакционную смесь перемешивали при 25°C в течение 10 часов. ТСХ (PE/EtOAc=3/1) показала завершение реакции, после чего добавляли воду (600 мл), выпавший осадок собирали фильтрацией и высушивали под вакуумом для получения трет-бутил (R)-4-((трет-бутилдиметилсилил)окси)-2-оксопирролидин-1-карбоксилата (19,2 г, выход: 84,6%) в виде твердого вещества розового цвета. 1H-ЯМР (400 МГц, CDCl3) δ м. д. 4,33-4,30 (м, 1H), 3,81-3,77 (м, 1H), 3,56-3,54 (м, 1H), 2,67-2,61 (м, 1H), 2,41-2,37 (м, 1H), 1,46 (с, 9H), 0,80 (с, 9H), 0,00 (с, 6H).
Шаг 3: трет-бутил((2R)-2-((трет-бутилдиметилсилил)окси)-4-(5-фторпиридин-3-ил)-4-гидроксибутил)карбамат
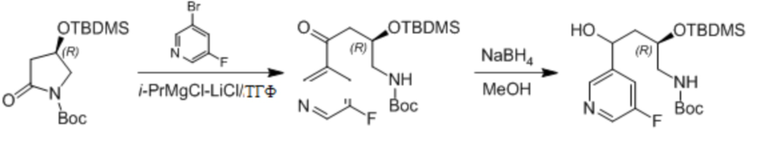
К смеси 3-бром-5-фтор-пиридина (3,35 г, 19,02 ммоль, 1,20 экв.) в ТГФ (40,00 мл) добавляли i-PrMgCl-LiCl (1,3 M, 17,56 мл, 1,44 экв.) по каплям при 0°C на протяжении 30 мин. (экзотермическая). После добавления температуру повышали до 25°C на протяжении 1 часа и перемешивали при температуре 25°C в течение 30 минут. ТСХ (PE/EtOAc =10/1) показала появление нового пятна, что указывает на успешное изготовление реагента Mg. Затем трет-бутил (R)-4-((трет-бутилдиметилсилил)окси)-2-оксопирролидин-1-карбоксилат (5,00 г, 15,85 ммоль, 1,00 экв.) в ТГФ (50 мл) добавляли по каплям к раствору при температуре -78°C на протяжении 30 мин.. Смесь оставляли нагреваться до 25°C на протяжении 1 часа, после чего перемешивали при температуре 25°C в течение 16 часов. ТСХ (PE/EtOAc=3/1) показала, что исходный материал полностью прореагировал и был обнаружен желаемый продукт трет-бутил (R)-(2-((трет-бутилдиметилсилил)окси)-4-(5-фторпиридин-3-ил)-4-оксобутил)карбамат. Реакцию гасили добавлением MeOH (50 мл) при 0 °C. NaBH4 (1,20 г, 31,70 ммоль, 2,00 экв.) добавляли при 0 °C, после чего смесь перемешивали при температуре 25°C в течение 4 часов. ТСХ (PE/EtOAc=2/1) и ЖХМС показала завершение реакции. Объединенную реакционную смесь (4 параллельные реакции) гасили водным NH4Cl (400 мл) и экстрагировали с использованием EtOAc (600 мл*3). Объединенные органические слои сушили над Na2SO4 и концентрировали под вакуумом, осадок очищали посредством ВЭЖХ для получения трет-бутил((2R)-2-((трет-бутилдиметилсилил)окси)-4-(5-фторпиридин-3-ил)-4-гидроксибутил)карбамата (1,24 г, выход: 18,91%) в виде маслянистой жидкости желтого цвета. 1H-ЯМР (400 МГц, CDCl3) δ м. д. 8,26-8,22 (м, 2H), 7,37 (д, 1H, J=8,8 Гц), 4,95-4,88 (м, 2H), 4,69 (уш. с., 1H), 4,00-3,98 (м, 2H), 3,23-3,10 (м, 2H), 1,73 (уш. с., 2H), 1,32 (с, 9H), 0,80-0,79 (м, 9H), 0,00 (с, 6H).
Шаг 4: трет-бутил (4R)-4-((трет-бутилдиметилсилил)окси)-2-(5-фторпиридин-3-ил)пирролидин-1-карбоксилат
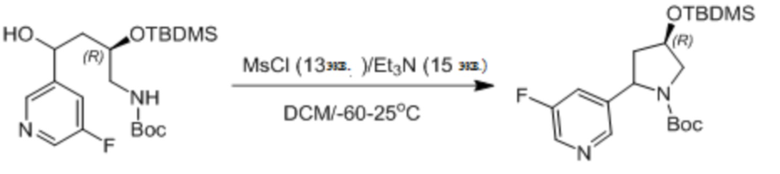
К смеси трет-бутил((2R)-2-((трет-бутилдиметилсилил)окси)-4-(5-фторпиридин-3-ил)-4-гидроксибутил)карбамата (8,70 г, 20,98 ммоль, 1,00 экв.) и Et3N (31,84 г, 314,70 ммоль, 15,00 экв.) в DCM (500,00 мл) добавляли по каплям MsCl (31,24 г, 272,74 ммоль, 13,00 экв.) при температуре -60°C на протяжении 0,5 часа. Смесь затем перемешивали при температуре -60°C в течение 1 часа, реакционную смесь оставляли нагреваться до 25°C и перемешивали в течение 18 часов. ЖХМС показала, что исходный материал полностью прореагировал. Смесь затем промывали H2O (200 мл*3) и водную фазу экстрагировали с использованием DCM (200 мл*4). Объединенные органические слои сушили над Na2SO4 и концентрировали под вакуумом для получения неочищенного продукта трет-бутил (4R)-4-((трет-бутилдиметилсилил)окси)-2-(5-фторпиридин-3-ил)пирролидин-1-карбоксилата (8,30 г, в неочищенном виде) в виде маслянистой жидкости черно-коричневого цвета, который использовали непосредственно без очистки.
Шаг 5: трет-бутил (4R)-2-(5-фторпиридин-3-ил)-4-гидроксипирролидин-1-карбоксилат
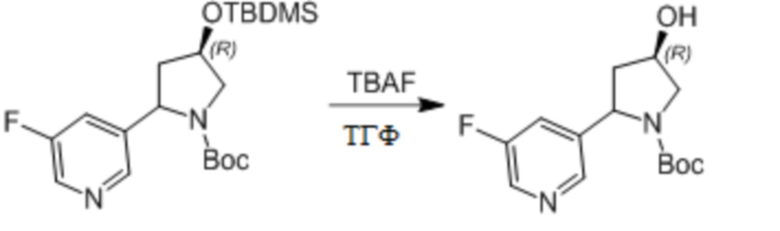
К смеси трет-бутил (4R)-4-((трет-бутилдиметилсилил)окси)-2-(5-фторпиридин-3-ил)пирролидин-1-карбоксилата (8,30 г, 20,93 ммоль, 1,00 экв.) в ТГФ (250,00 мл) добавляли TBAF (9,43 г, 41,86 ммоль, 2,00 экв.) при температуре 25 °C. Смесь перемешивали при 25°C в течение 16 часов. После того как ТСХ (PE/EtOAc=1/1) показала завершение реакции, смесь концентрировали, осадок растворяли в EtOAc (600 мл), промывали водой (200 мл*5), сушили над Na2SO4 и концентрировали. Неочищенный продукт очищали посредством PLC для получения трет-бутил (4R)-2-(5-фторпиридин-3-ил)-4-гидроксипирролидин-1-карбоксилата (4,70 г, 16,65 ммоль, выход: 79,54%) в виде маслянистой жидкости черно-коричневого цвета. 1H-ЯМР (400 МГц, CDCl3) δ м. д. 8,37-8,33 (м, 2H), 7,48 (уш. с., 1H), 5,09-4,89 (м, 1H), 4,56-4,54 (м, 1H), 3,80-3,65 (м, 2H), 2,63-2,43 (м, 1H), 2,03-1,96 (м, 1H), 1,56-1,20 (м, 9H).
Шаг 6: трет-бутил (2R,4S)-4-фтор-2-(5-фторпиридин-3-ил)пирролидин-1-карбоксилат
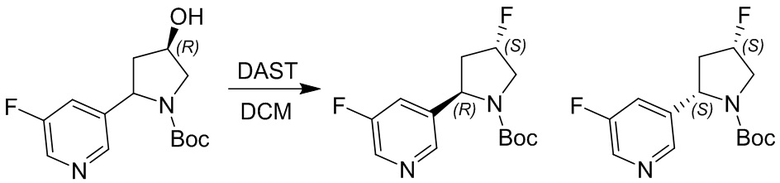
К смеси трет-бутил (4R)-2-(5-фторпиридин-3-ил)-4-гидроксипирролидин-1-карбоксилата (4,70 г, 16,65 ммоль, 1,00 экв.) в DCM (150,00 мл) добавляли DAST по каплям (29,52 г, 183,15 ммоль, 11,00 экв.) при температуре -78°C на протяжении 0,5 часа. Реакционную смесь перемешивали при -78°C в течение 2 часов, после чего оставляли нагреваться до 25°C и перемешивали в течение 20 часов. После того как ТСХ (PE/EtOAc=0/1) показала, что исходный материал полностью прореагировал, смесь охлаждали до 0°C и гасили, добавляя по каплям насыщенный раствор NaHCO3 (100 мл) по каплям. Органическую фазу отделяли и сушили над Na2SO4, концентрировали до выпадения осадка, после чего очищали посредством колоночной хроматографии на силикагеле (PE:EtOAc от 10:1, 8:1 до 5:1, после чего 3:1) для получения трет-бутил (2R,4S)-4-фтор-2-(5-фторпиридин-3-ил)пирролидин-1-карбоксилата (1,38 г, 4,85 ммоль, выход: 29,15%, Rf=0,53) в виде твердого вещества белого цвета и трет-бутил (2S,4S)-4-фтор-2-(5-фторпиридин-3-ил)пирролидин-1-карбоксилата (1,36 г, 4,78 ммоль, выход: 28,73%, Rf=0,43) в виде маслянистой жидкости желтого цвета. 1H-ЯМР (400 МГц, CDCl3) δ м. д. 8,31-8,27 (м, 2H), 7,20-7,18 (м, 1H), 5,18 (д, 1H, J=51,6 Гц), 4,97-4,88 (м, 1H), 4,04-4,00 (м, 1H), 3,64 (дд, 1H, J=38,8, 12,8 Гц), 2,67 (дд, 1H, J=15,6, 6,8 Гц), 1,97-1,67 (м, 1H), 1,56-1,12 (м, 9H).
Шаг 7: 3-фтор-5-((2R,4S)-4-фторпирролидин-2-ил)пиридин
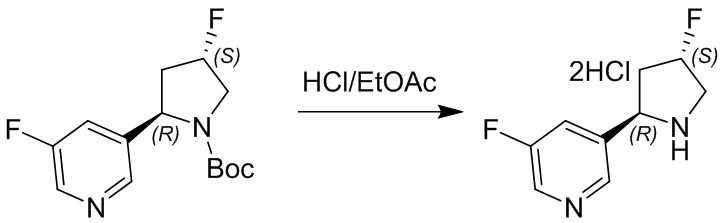
К смеси трет-бутил (2R,4S)-4-фтор-2-(5-фторпиридин-3-ил)пирролидин-1-карбоксилата (1,38 г, 4,85 ммоль, 1,00 экв.) в EtOAc (10 мл) добавляли по каплям HCl/EtOAc (40,00 мл, 4 M) при 0 °C. Смесь оставляли нагреваться до 25°C и перемешивали в течение 3 часов. После того как ТСХ (PE:EtOAc =1:1) показала завершение реакции, растворитель испаряли для получения 3-фтор-5-((2R,4S)-4-фторпирролидин-2-ил)пиридина (1,25 г, 4,86 ммоль, выход: 100,00%) в виде твердого вещества коричневого цвета. 1H-ЯМР (400 МГц, CD3OD) δ м. д. 8,84-8,81 (м, 2H), 8,31 (д, 1H, J=9,2 Гц), 5,62 (дт, 1H, J=52,0, 2,4 Гц), 5,23-5,18 (м, 1H), 4,00-3,95 (м, 1H), 3,88-3,71 (м, 1H), 2,67 (тд, 1H, J=16,0,6,0 Гц), 1,69-1,59 (м, 1H).
Пример 11. Синтез (3R,4S,5R)-5-аминотетрагидро-2H-пиран-3,4-диола
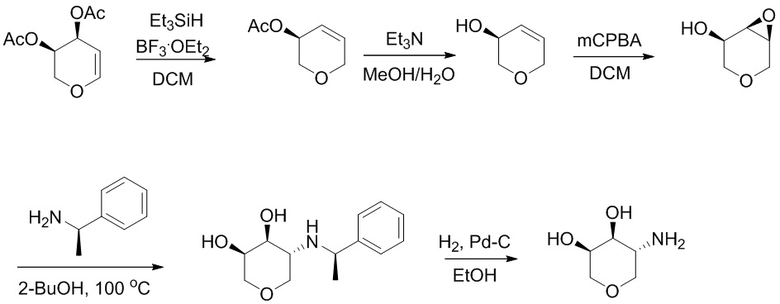
Шаг 1: (с)-3,6-дигидро-2H-пиран-3-ил ацетат
(3R,4S)-3,4-дигидро-2H-пиран-3,4-диил диацетат (2,9 г, 14,49 ммоль) растворяли в DCM (15 мл) и перемешивали в атмосфере N2 при комнатной температуре. Добавляли триэтилсилан (2,55 мл, 15,93 ммоль) и перемешивали в течение 5 минут. Добавляли по каплям BF3.OEt2 (1,836 мл, 14,49 ммоль) и продолжали перемешивание в течение 30 минут. Реакционную смесь гасили насыщенным бикарбонатом (30 мл) и слои разделяли. Объединенные органические слои сушили над сульфатом натрия и удаляли растворитель. Осадок очищали посредством флэш-хроматографии (0-30% гекс/EtOAc). Был получен (с)-3,6-дигидро-2H-пиран-3-ил ацетат (1,9 г, 92% выход) в виде прозрачной маслянистой жидкости. 1H ЯМР (400 МГц, DMSO-d6) δ 6,10 (дддт, J=10,2, 3,2, 2,1, 1,0 Гц, 1H), 5,84 (ддт, J=10,1, 4,3, 2,1 Гц, 1H), 4,96 (дтд, J=4,3, 2,7, 1,5 Гц, 1H), 4,16-3,89 (м, 2H), 3,73 (т, J=2,9 Гц, 2H), 2,01 (д, J=0,9 Гц, 3H).
Шаг 2: (с)-3,6-дигидро-2H-пиран-3-ол
Ацетат (с)-3,6-дигидро-2H-пиран-3-ила (1,9 г, 13,37 ммоль) растворяли в MeOH (30 мл) и воде (20 мл). Добавляли триэтиламин (7 мл, 50,2 ммоль) и перемешивали при комнатной температуре в течение 30 минут. Растворитель удаляли при пониженном давлении. Затем оставшуюся воду трижды экстрагировали с использованием EtOAc. Органические слои комбинировали, сушили над сульфатом натрия и удаляли растворитель. Был получен (с)-3,6-дигидро-2H-пиран-3-ол (1,1 г, 10,99 ммоль, 82% выход) в виде прозрачной маслянистой жидкости. Неочищенный продукт подвергали дальнейшей обработке без последующей очистки.
Шаг 3: (1S,5R,6R)-3,7-диоксабицикло[4,1,0]гептан-5-ол
(с)-3,6-дигидро-2H-пиран-3-ол (1,1 г, 10,99 ммоль) растворяли в CH2Cl2 (20 мл) и охлаждали до 0 °C. mCPBA (4,55 г, 13,18 ммоль) добавляли порционно. Реакционную смесь перемешивали при нагревании до комнатной температуры в течение ночи. Белый осадок реакционной смеси отфильтровывали, элюент сохраняли, растворитель удаляли и растирали с диэтилэфиром. Данную стадию повторили. Осадок, (1S,5R,6R)-3,7-диоксабицикло[4,1,0]гептан-5-ол (1,2 г, 100% выход), подвергали дальнейшей обработке без последующей очистки.
Шаг 4: (3R,4S,5R)-5-(((R)-1-фенилэтил)амино)тетрагидро-2H-пиран-3,4-диол
(1S,5R,6R)-3,7-диоксабицикло[4,1,0]гептан-5-ол (1,26 г, 10,85 ммоль) и (R)-1-фенилэтанамин (1,658 мл, 13,02 ммоль) растворяли в 2-BuOH (15 мл). Реакционную смесь нагревали до 100°C в течение 18 часов. Реакционную смесь охлаждали до комнатной температуры, растворитель удаляли, после чего осадок очищали на ISCO 0-100% EtOAc. Фракции объединяли, растворитель удаляли, после чего осадок обрабатывали MTBE и перемешивали в течение ночи. Белый осадок органической смеси отфильтровывали. Получали (3R,4S,5R)-5-(((R)-1-фенилэтил)амино)тетрагидро-2H-пиран-3,4-диол (0,350 г, 14% выход). 1H ЯМР (400 МГц, DMSO-d6) δ 7,38-7,24 (м, 4H), 7,22-7,12 (м, 1H), 4,61 (д, J=5,6 Гц, 1H), 4,46 (д, J=4,8 Гц, 1H), 3,88 (q, J=6,5 Гц, 1H), 3,64 (тт, J=5,0, 2,8 Гц, 1H), 3,47 (дд, J=11,4, 4,7 Гц, 1H), 3,39 (ддд, J=8,4, 5,6, 3,1 Гц, 1H), 3,31 (с, 2H), 3,29 (т, J=3,0 Гц, 1H), 3,25 (д, J=2,5 Гц, 0H), 2,79 (дд, J=11,1, 7,8 Гц, 1H), 2,57 (тд, J=7,8, 3,9 Гц, 1H), 1,86 (с, 1H), 1,21 (д, J=6,6 Гц, 3H).
Шаг 5: (3R,4S,5R)-5-аминотетрагидро-2H-пиран-3,4-диол
(3R,4S,5R)-5-(((R)-1-фенилэтил)амино)тетрагидро-2H-пиран-3,4-диол (0,350 г, 1,475 ммоль) растворяли в EtOH (3 мл) и Pd-C (0,031 г, 0,295 ммоль) добавляли. Реакционную смесь перемешивали под баллоном H2 в течение ночи. Реакционную смесь фильтровали через целит и удаляли растворитель для получения (3R,4S,5R)-5-аминотетрагидро-2H-пиран-3,4-диола (0,190 г, 1,427 ммоль, 97% выход) в виде твердого вещества грязно-белого цвета. Неочищенный продукт подвергали дальнейшей обработке без последующей очистки. ЖХМС (м+H) 134.
Пример 12. (R)-3-(4,4-дифторпирролидин-2-ил)-5-фторпиридин
Шаг 1: трет-бутил (2R,4R)-4-((трет-бутилдиметилсилил)окси)-2-(5-фторпиридин-3-ил)пирролидин-1-карбоксилат
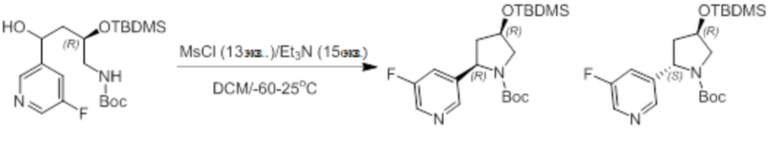
К смеси трет-бутил((2R)-2-((трет-бутилдиметилсилил)окси)-4-(5-фторпиридин-3-ил)-4-гидроксибутил)карбамата (6,80 г, 16,40 ммоль) и Et3N (24,89 г, 246,00 ммоль) в DCM (500,00 мл) добавляли MsCl (24,42 г, 213,20 ммоль) по каплям при -60°C в течение 30 минут. Смесь перемешивали при -60°C в течение 1 часа. Реакционную смесь оставляли нагреваться до 25°C и перемешивали в течение дополнительных 18 часов. Смесь промывали H2O (200 мл*3). Водную фазу экстрагировали с использованием DCM (200 мл*4). Объединенные органические слои сушили над Na2SO4 и концентрировали под вакуумом. Осадок очищали посредством хроматографии на силикагеле (PE:EtOAc=50/1, 20/1, 10/1) для получения трет-бутил (2S,4R)-4-((трет-бутилдиметилсилил)окси)-2-(5-фторпиридин-3-ил)пирролидин-1-карбоксилата (2,70 г, выход: 41,52%) и трет-бутил (2R,4R)-4-((трет-бутилдиметилсилил)окси)-2-(5-фторпиридин-3-ил)пирролидин-1-карбоксилата (2,40 г, выход: 36,89%) в виде маслянистой жидкости коричневого цвета. 1H-ЯМР (400 МГц, CDCl3) δ м. д. 8,40 (уш. с., 2H), 7,56-7,45 (м, 1H), 5,11-4,94 (м, 2H), 4,53 (уш. с., 1H), 3,85-3,79 (м, 1H), 3,66-3,53 (м, 1H), 2,62-2,58 (м, 1H), 2,04-2,01 (м, 1H), 1,56 (с, 3H), 1,32 (с, 6H), 0,99-0,88 (м, 9H), 0,18-0,00 (м, 6H).
Шаг 2: трет-бутил (2R,4R)-2-(5-фторпиридин-3-ил)-4-гидроксипирролидин-1-карбоксилат
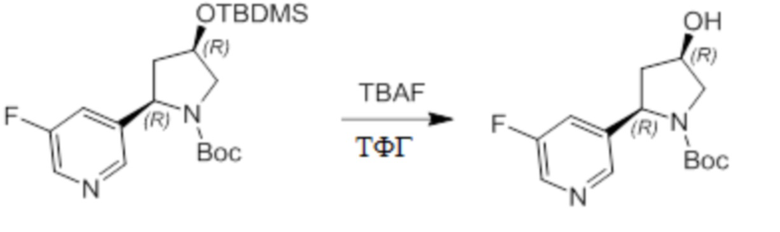
К смеси трет-бутил (2R,4R)-4-((трет-бутилдиметилсилил)окси)-2-(5-фторпиридин-3-ил)пирролидин-1-карбоксилата (2,40 г, 6,05 ммоль) в ТГФ (60,00 мл) добавляли TBAF (3,16 г, 12,10 ммоль) в один прием при температуре 25 °C. Смесь концентрировали при сниженном давлении при температуре 50 °C. Осадок добавляли в воду (20 мл). Водную фазу экстрагировали с использованием этилацетата (30 мл*3). Объединенную органическую фазу промывали насыщенным солевым раствором (20 мл*2), сушили над Na2SO4, фильтровали и концентрировали под вакуумом. Осадок очищали посредством хроматографии на силикагеле (PE:EtOAc=20/1, 10/1,1/3) для получения трет-бутил (2R,4R)-2-(5-фторпиридин-3-ил)-4-гидроксипирролидин-1-карбоксилата (1,30 г, выход: 76,11%) в виде твердого вещества желтого цвета. 1H-ЯМР (400 МГц, CDCl3) δ м. д. 8,26 (д, 2H, J=12,8 Гц), 7,39 (уш. с., 1H), 4,95-4,81 (м, 1H), 4,48-4,47 (м, 1H), 3,73 (уш. с., 1H), 3,56-3,53 (м, 1H), 2,55 (уш. с., 1H), 1,97-1,98 (м, 1H), 1,65-1,16 (м, 9H).
Шаг 3: трет-бутил (R)-2-(5-фторпиридин-3-ил)-4-оксопирролидин-1-карбоксилат
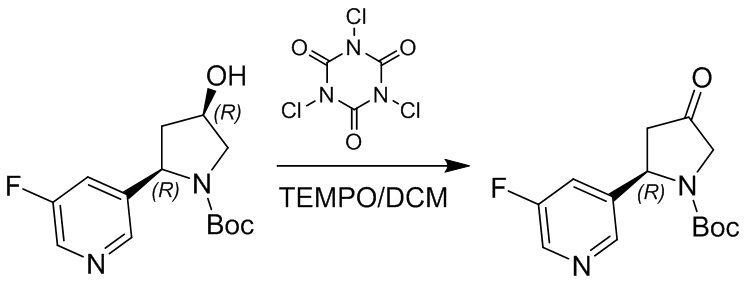
К смеси трет-бутил (2R,4R)-2-(5-фторпиридин-3-ил)-4-гидроксипирролидин-1-карбоксилата (1,30 г, 4,60 ммоль) и трихлоризоциануровой кислоты (1,10 г, 4,60 ммоль) добавляли TEMPO (72,41 мг, 460,49 мкмоль) при температуре -10 °C. Смесь перемешивали при -10°C в течение 15 мин., после чего нагревали до 25°C и перемешивали в течение 1 часа. ТСХ (EtOAc) показала завершение реакции. Органическую фазу промывали NaHCO3 (20 мл*2), сушили над Na2SO4, фильтровали и концентрировали в вакууме. Осадок очищали посредством хроматографии на силикагеле (петролейный эфир/этилацетат=50/1, 10/1) для получения трет-бутил (R)-2-(5-фторпиридин-3-ил)-4-оксопирролидин-1-карбоксилата (1,10 г, выход: 85,32%) в виде маслянистой жидкости коричневого цвета.
Шаг 4: трет-бутил (R)-4,4-дифтор-2-(5-фторпиридин-3-ил)пирролидин-1-карбоксилат
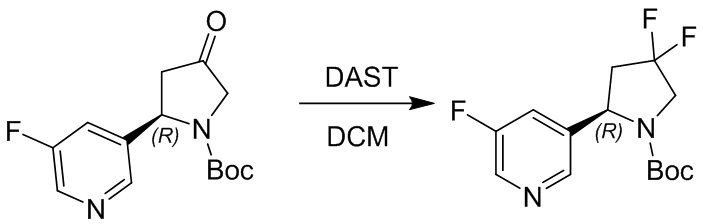
К смеси трет-бутил (R)-2-(5-фторпиридин-3-ил)-4-оксопирролидин-1-карбоксилата (1,00 г, 3,57 ммоль) в DCM (100,00 мл) добавляли DAST (14,39 г, 89,25 ммоль) по каплям при -70°C в атмосфере N2. Смесь перемешивали при -70°C в течение 30 минут. После этого смесь перемешивали при температуре 25°C в течение 16 часов. Реакционную смесь медленно гасили насыщенным водн. NaHCO3 при 0°C и водную фазу экстрагировали с использованием DCM (50 мл*4). Объединенную органическую фазу промывали насыщенным солевым раствором (30 мл), сушили над Na2SO4, фильтровали и концентрировали под вакуумом. Осадок очищали посредством хроматографии на силикагеле (петролейный эфир/этилацетат=100/1, 30/1) для получения трет-бутил (R)-4,4-дифтор-2-(5-фторпиридин-3-ил)пирролидин-1-карбоксилата (1,00 г, выход: 92,66%) в виде маслянистой жидкости коричневого цвета. 1H-ЯМР (400 МГц, CDCl3) δ м. д. 8,40 (с, 1H), 8,34 (с, 1H), 7,30-7,21 (м, 1H), 5,06 (уш. с., 1H), 4,14-3,85 (м, 2H), 2,91-2,84 (м, 1H), 2,39-2,32 (м, 1H), 1,43-1,14 (м, 9H).
Шаг 5: (R)-3-(4,4-дифторпирролидин-2-ил)-5-фторпиридин
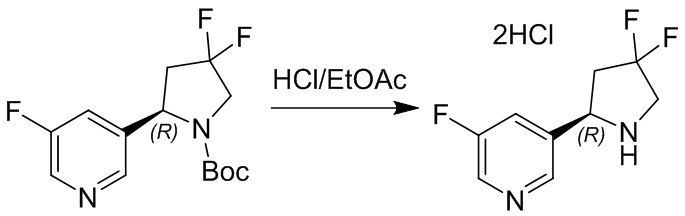
Смесь трет-бутил (R)-4,4-дифтор-2-(5-фторпиридин-3-ил)пирролидин-1-карбоксилата (1,00 г, 3,31 ммоль) в HCl/EtOAc (50,00 мл, 4 M) перемешивали в течение 2 часов при температуре 25 °C. Смесь концентрировали при сниженном давлении при температуре 30°C для получения (R)-3-(4,4-дифторпирролидин-2-ил)-5-фторпиридина (840,00 мг, выход: 92,25%) в виде твердой соли бис-HCl белого цвета. 1H-ЯМР (400 МГц, MeOD) δ м. д. 8,68-8,63 (м, 1H), 7,97 (д, 1H, J=9,2 Гц), 5,26-5,21 (м, 1H), 4,03-3,90 (м, 2H), 3,13-2,92 (м, 2H).
Пример 13. (3S,5R)-5-(2,5-дифторфенил)пирролидин-3-карбонитрил
Шаг 1: трет-бутил((2R)-2-((трет-бутилдиметилсилил)окси)-4-(2,5-дифторфенил)-4-гидроксибутил)карбамат
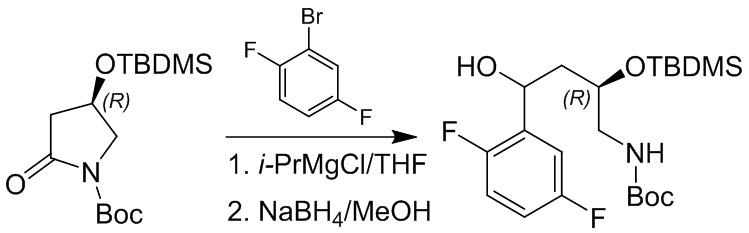
К раствору 2-бром-1,4-дифтор-бензола (3,01 г, 15,60 ммоль, 1,20 экв.) в ТГФ (15 мл) добавляли комплекс хлорида изопропилмагния (2,27 г, 15,60 ммоль, 1,20 экв.) при 0°C по каплям в атмосфере N2. Реакцию перемешивали при температуре 15°C в течение 1 часа для приготовления бромида (2, 5-дифторфенил) магния (23 мл). К раствору трет-бутил (R)-4-((трет-бутилдиметилсилил)окси)-2-оксопирролидин-1-карбоксилата (4,10 г, 13,00 ммоль, 1,00 экв.) в ТГФ (50 мл) добавляли бромид (2,5-дифторфенил)магния (23 мл) по каплям при температуре 0°C в течение 30 мин. Реакционную смесь перемешивали при 0°C в течение 1 часа. К смеси добавляли метанол (20 мл), после чего добавляли NaBH4 (738 мг, 19,50 ммоль, 1,50 экв.) при 0oC. Смесь перемешивали при 0°C в течение 1 часа после чего вливали в 10% водный NH4Cl. Смесь экстрагировали с использованием EtOAc (20 мл*2), объединенные органические слои промывали солевым раствором, сушили над Na2SO4, фильтровали и концентрировали. Неочищенный продукт очищали посредством жидкостной хроматографии среднего давления (ЖХСД) для получения трет-бутил((2R)-2-((трет-бутилдиметилсилил)окси)-4-(2,5-дифторфенил)-4-гидроксибутил)карбамата (2,22 г, 5,14 ммоль, 39,6% выход). 1H-ЯМР (400 МГц, CDCl3) δ м. д. 7,17-7,15 (м, 1H), 6,86-6,79 (м, 2H), 5,11-5,06 (м, 1H), 4,70 (уш. с., 1H), 4,02-3,98 (м, 1H), 3,69 (уш. с., 0,5H), 3,46 (уш. с., 0,5H), 3,33-3,14 (м, 2H), 1,80-1,69 (м, 2H), 1,35 (с, 9H), 0,84-0,82 (9H, m), 0,04-0,03 (6H, m).
Шаг 2: трет-бутил (4R)-4-((трет-бутилдиметилсилил)окси)-2-(2,5-дифторфенил)пирролидин-1-карбоксилат
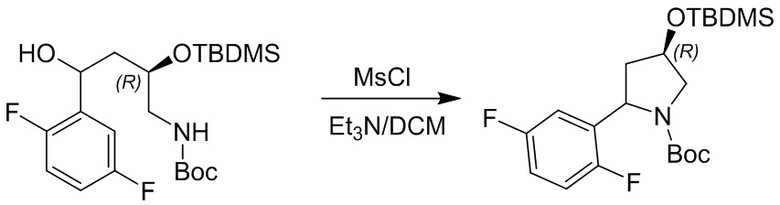
К раствору трет-бутил((2R)-2-((трет-бутилдиметилсилил)окси)-4-(2,5-дифторфенил)-4-гидроксибутил)карбамата (13,40 г, 31,05 ммоль, 1,00 экв.) и Et3N (9,43 г, 93,14 ммоль, 3,00 экв.) в DCM (50 мл) добавляли по каплям метансульфонил хлорид (5,33 г, 46,57 ммоль, 1,50 экв.) при температуре -60°C в атмосфере N2. Смесь перемешивали при температуре -60°C в течение 2 часов и 15°C в течение 16 часов. ЖХМС показала, что исходный материал полностью прореагировал. Реакционную смесь экстрагировали с использованием DCM (30 мл*2) и объединенные органические продукты промывали солевым раствором (50 мл), сушили над Na2SO4, фильтровали и концентрировали с получением трет-бутил (4R)-4-((трет-бутилдиметилсилил)окси)-2-(2,5-дифторфенил)пирролидин-1-карбоксилата (12,00 г, 26,11 ммоль, выход: 84,10%, 90% чистота) который использовали непосредственно без дальнейшей очистки.
Шаг 3: трет-бутил (2R,4R)-2-(2,5-дифторфенил)-4-гидроксипирролидин-1-карбоксилат
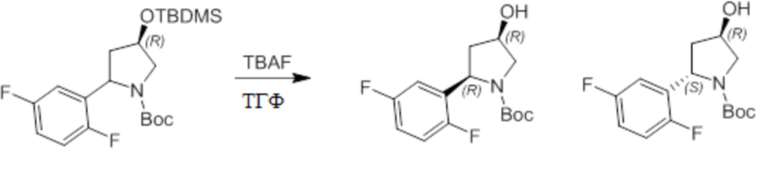
К раствору трет-бутил (4R)-4-((трет-бутилдиметилсилил)окси)-2-(2,5-дифторфенил)пирролидин-1-карбоксилата (4,50 г, 10,88 ммоль, 1,00 экв.) в ТГФ (30 мл) добавляли TBAF/THF (1 M, 14,15 мл, 1,30 экв.) при температуре 15oC. Смесь перемешивали при температуре 15°C в течение 16 часов. ТСХ (PE:EtOAc=3:1) показала, что исходный материал полностью прореагировал. Реакционную смесь гасили H2O (50 мл), экстрагировали с использованием EtOAc (30 мл*2) и объединенные органические продукты промывали солевым раствором (10 мл), сушили над Na2SO4, фильтровали и концентрировали под вакуумом. Осадок очищали посредством нейтральной препаративной ВЭЖХ для получения трет-бутил (2R,4R)-2-(2,5-дифторфенил)-4-гидроксипирролидин-1-карбоксилата (1,00 г, 3,34 ммоль, выход: 30,70%) в виде твердого вещества белого цвета. 1H-ЯМР (400 МГц, CDCl3) δ м. д. 7,04-6,80 (м, 3H), 5,10-5,00 (м, 1H), 4,43 (с, 1H), 3,75 (уш. с., 1H), 3,53-3,49 (м, 1H), 2,53 (уш. с., 1H), 1,93-1,90 (м, 1H), 1,40-1,16 (м, 9H).
Шаг 4: трет-бутил (2R,4R)-2-(2,5-дифторфенил)-4-((метилсульфонил)окси)пирролидин-1-карбоксилат
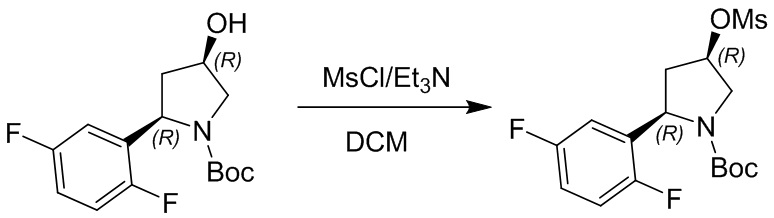
К смеси трет-бутил (2R,4R)-2-(2,5-дифторфенил)-4-гидроксипирролидин-1-карбоксилата (3,00 г, 10,02 ммоль, 1,00 экв.) и Et3N (2,03 г, 20,04 ммоль, 2,00 экв.) в DCM (80,00 мл) добавляли MsCl (1,61 г, 14,03 ммоль, 1,40 экв.) по каплям при 0 °C. Смесь перемешивали при 18°C в течение 2 часов. Смесь гасили H2O (30 мл). Водную фазу экстрагировали с использованием DCM (50 мл*3). Объединенные органические слои сушили над Na2SO4 и концентрировали при пониженном давлении. Трет-бутил (2R,4R)-2-(2,5-дифторфенил)-4-((метилсульфонил)окси)пирролидин-1-карбоксилат (3,60 г, 9,54 ммоль, выход: 95,20%) получали в виде твердого вещества коричневого цвета.
Шаг 5: трет-бутил (2R,4S)-4-циано-2-(2,5-дифторфенил)пирролидин-1-карбоксилат
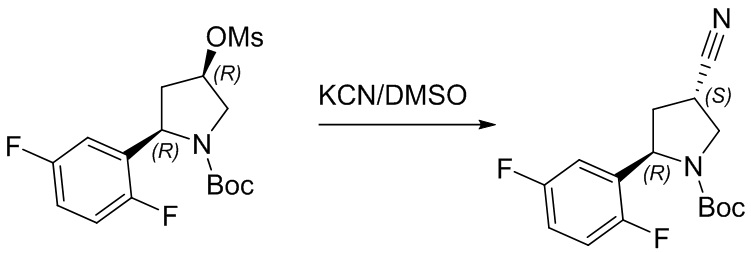
К смеси трет-бутил (2R,4R)-2-(2,5-дифторфенил)-4-((метилсульфонил)окси)пирролидин-1-карбоксилата (3,60 г, 9,54 ммоль, 1,00 экв.) в ДМСО (20,00 мл) добавляли KCN (745,49 мг, 11,45 ммоль, 1,20 экв.) в один прием. Смесь перемешивали при 90°C в течение 3 ч. К смеси добавляли 80 мл H2O и смесь экстрагировали с использованием EtOAc (80 мл*4). Объединенные органические слои концентрировали при пониженном давлении. Осадок очищали посредством хроматографии на силикагеле (PE/EtOAc=40:1, 30:1, 10:1). Трет-бутил (2R,4S)-4-циано-2-(2,5-дифторфенил)пирролидин-1-карбоксилат (1,60 г, 5,19 ммоль, выход: 54,40%) получали в виде жидкости светло-зеленого цвета.
Шаг 6: (3S,5R)-5-(2,5-дифторфенил)пирролидин-3-карбонитрил
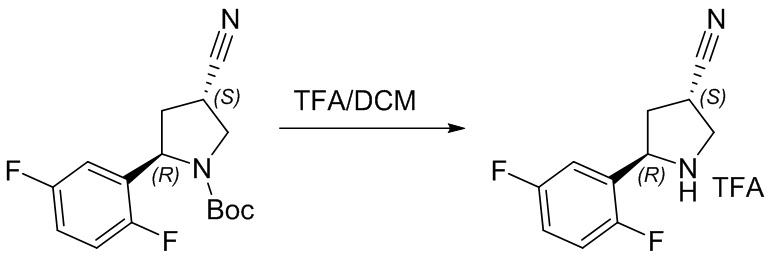
Смесь трет-бутил (2R,4S)-4-циано-2-(2,5-дифторфенил)пирролидин-1-карбоксилата (800,00 мг, 2,59 ммоль, 1,00 экв.) в TFA (4,00 мл)/DCM (20,00 мл) перемешивали при температуре 18°C в течение 3 ч. Смесь высушивали в атмосфере N2. (3S,5R)-5-(2,5-дифторфенил)пирролидин-3-карбонитрил (780,00 мг, 2,42 ммоль, выход: 93,44%) в виде твердого вещества светло-желтого цвета.
Пример 14. 3-фтор-5-((2R,4S)-4-фторпирролидин-2-ил)бензамид
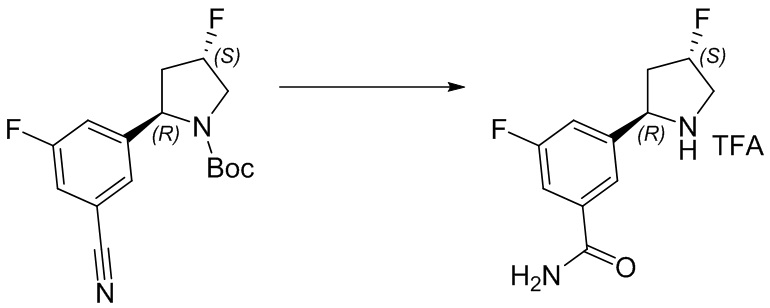
3-фтор-5-((2R,4S)-4-фторпирролидин-2-ил)бензонитрил (0,050 г, 0,240 ммоль) (приготовлен в соответствии с описанным в международной заявке на патент № 2012/034095) растворяли в TFA (0,800 мл, 10,38 ммоль) и H2SO4 (0,200 мл, 3,75 ммоль) и перемешивали в течение ночи при комнатной температуре. Реакционную смесь разбавляли ледяной водой (3 мл), твердое вещество изолировали фильтрацией и использовали непосредственно.
Пример 15. 2-хлор-5-фтор-3-((2R,4S)-4-фторпирролидин-2-ил)пиридин
Шаг 1: (с,Z)-N-((2-хлор-5-фторпиридин-3-ил)метилен)-2-метилпропан-2-сульфинамид
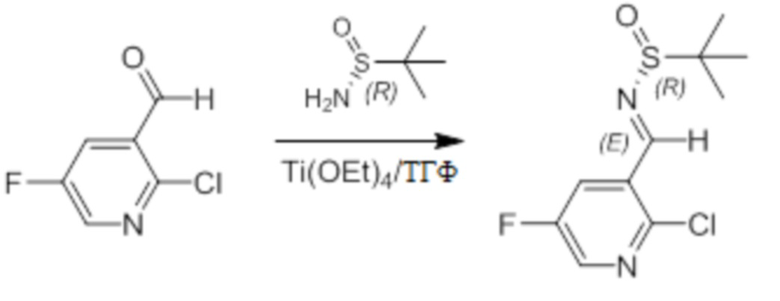
2-хлор-5-фторникотинальдегид (20 г, 125 ммоль) растворяли в ТГФ (150 мл) при 0 °C.Добавляли(R)-2-метилпропан-2-сульфинамид (16,71 г, 138 ммоль), после чего дополнительно добавляли по каплям титантетраэтанолат (22,88 мл, 150 ммоль). Реакционную смесь перемешивали при нагревании до КТ. Через 3 часа реакционную смесь охлаждали до 0 °C, добавляли 150 мл солевого раствора и перемешивали в течение 20 минут. Смесь фильтровали через целит. Водный слой отделяли и удаляли. Органический слой высушивали над Na2SO4 и удаляли растворитель для получения (S,Z)-N-((2-хлор-5-фторпиридин-3-ил)метилен)-2-метилпропан-2-сульфинамида (32 г, 122 ммоль, 97% выход), который подвергали дальнейшей обработке без последующей очистки. ЖХМС: 263 M+H.
Шаг 2: (R)-N-((R)-1-(2-хлор-5-фторпиридин-3-ил)бут-3-ен-1-ил)-2-метилпропан-2-сульфинамид
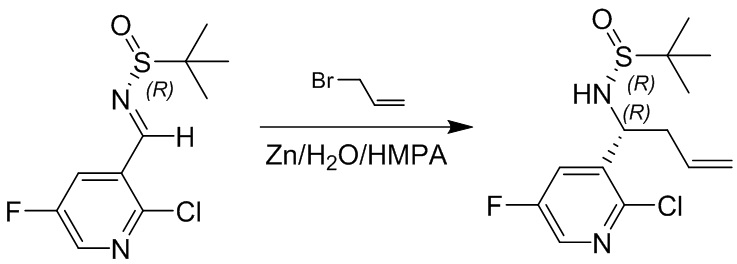
(R,E)-N-((2-хлор-5-фторпиридин-3-ил)метилен)-2-метилпропан-2-сульфинамид (32,9 г, 125 ммоль) растворяли в HMPA (100 мл) и охлаждали до 0 °C. Добавляли цинк (16,37 г, 250 ммоль), аллилбромид (21,67 мл, 250 ммоль) и воду (2,256 мл, 125 ммоль) при 0°C и реакционную смесь оставляли нагреваться до КТ в течение ночи. ЖХМС показала полное превращение в желаемый продукт. Добавляли 100 мл воды при КТ и перемешивали в течение 30 минут. Добавляли 30 мл MBTE, после чего добавляли 60 мл 10% лимонной кислоты и реакционную смесь перемешивали в течение 30 минут. Смесь фильтровали через целит и промывали MTBE. Органический слой промывали 10% лимонной кислотой, водой и солевым раствором. Растворитель удаляли под вакуумом для получения (R)-N-((R)-1-(2-хлор-5-фторпиридин-3-ил)бут-3-ен-1-ил)-2-метилпропан-2-сульфинамида (14,5 г, 47,6 ммоль, 38,0% выход) в виде маслянистой жидкости оранжевого цвета. ЖХМС: 305 M+H.
Шаг 3: (R)-1-(2-хлор-5-фторпиридин-3-ил)бут-3-ен-1-амин, HCl
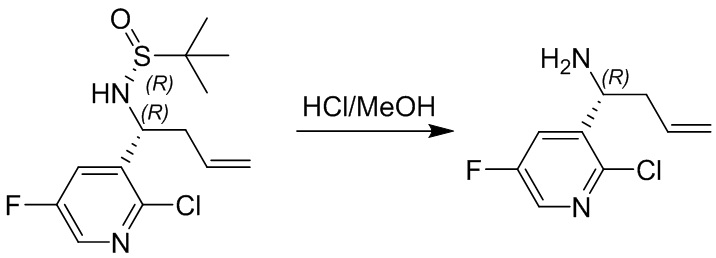
(R)-N-((R)-1-(2-хлор-5-фторпиридин-3-ил)бут-3-ен-1-ил)-2-метилпропан-2-сульфинамид (7,5 г, 24,61 ммоль) растворяли в 10 мл MeOH. HCl (4M в диоксане) (30,8 мл, 123 ммоль) добавляли и перемешивали при КТ в течение 1 ч. Растворитель удаляли под вакуумом и осадок растворяли в DCM и промывали насыщенным водным NaHCO3. Слои разделяли, органический слой сушили с использованием Na2SO4 и удаляли растворитель под вакуумом. Получали (R)-1-(2-хлор-5-фторпиридин-3-ил)бут-3-ен-1-амин, HCl (5,83 г, 24,59 ммоль, 100% выход) в виде твердого вещества. ЖХМС: 201 M+H.
Шаг 4: (R)-N-(1-(2-хлор-5-фторпиридин-3-ил)бут-3-ен-1-ил)ацетамид
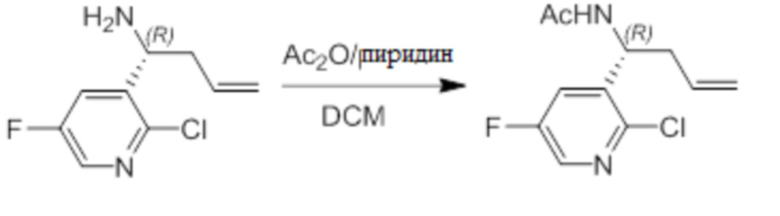
К (R)-1-(2-хлор-5-фторпиридин-3-ил)бут-3-ен-1-амину⋅HCl (5,83 г, 24,59 ммоль) в DCM (70,3 мл) при 0°C добавляли TEA (4,11 мл, 29,5 ммоль) и уксусный ангидрид (2,320 мл, 24,59 ммоль). Смесь перемешивали в течение 2 часов. Реакционную смесь вливали в насыщенный водный NaHCO3 и экстрагировали с использованием DCM. Органический слой промывали солевым раствором, сушили над MgSO4 и испаряли при пониженном давлении. Получали (R)-N-(1-(2-хлор-5-фторпиридин-3-ил)бут-3-ен-1-ил)ацетамид (5,97 г, 24,60 ммоль, 100% выход) и подвергали его дальнейшей обработке без последующей очистки. ЖХМС: 243 M+H.
Шаг 5: (5R)-5-(2-хлор-5-фторпиридин-3-ил)пирролидин-3-ил ацетат
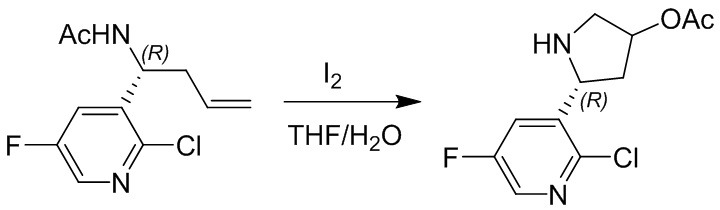
(R)-N-(1-(2-хлор-5-фторпиридин-3-ил)бут-3-ен-1-ил)ацетамид (5,97 г, 24,60 ммоль) растворяли в ТГФ (56,2 мл) и воде (14,06 мл) с последующим добавлением I2 (18,73 г, 73,8 ммоль) и перемешивали в течение ночи при КТ. Реакцию в неочищенном виде разбавляли насыщенными растворами NaHCO3 и Na2S2O3 и дважды экстрагировали с EtOAc. Водный слой подщелачивали насыщенным водным NaHCO3 и экстрагировали с использованием EtOAc для получения (5R)-5-(2-хлор-5-фторпиридин-3-ил)пирролидин-3-ил ацетата (5,9 г, 22,81 ммоль, 93% выход) в виде маслянистой жидкости светло-желтого цвета. ЖХМС: 259 M+H.
Шаг 6: (2R)-трет-бутил 4-ацетокси-2-(2-хлор-5-фторпиридин-3-ил)пирролидин-1-карбоксилат
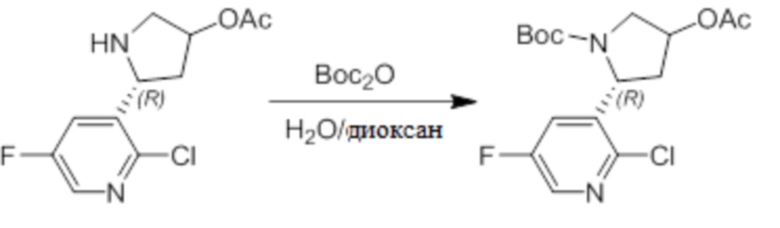
К раствору (5R)-5-(2-хлор-5-фторпиридин-3-ил)пирролидин-3-ил ацетат (5,9 г, 22,81 ммоль) в диоксане (76 мл) и воде (76 мл) добавляли BOC-ангидрид (7,94 мл, 34,2 ммоль) с последующим аккуратным добавлением 2N NaOH (7 мл) до достижения pH ~9. Реакционную смесь перемешивали в течение 1 часа при КТ. Реакционную смесь разбавляли водой и экстрагировали с использованием EtOAc трижды. Органический слой высушивали над Na2SO4 и удаляли растворитель под вакуумом для получения (2R)-трет-бутил 4-ацетокси-2-(2-хлор-5-фторпиридин-3-ил)пирролидин-1-карбоксилата (3,5 г, 9,75 ммоль, 42,8% выход), который подвергали дальнейшей обработке без последующей очистки. ЖХМС: 359 M+H.
Шаг 7: (2R)-трет-бутил 2-(2-хлор-5-фторпиридин-3-ил)-4-гидроксипирролидин-1-карбоксилат
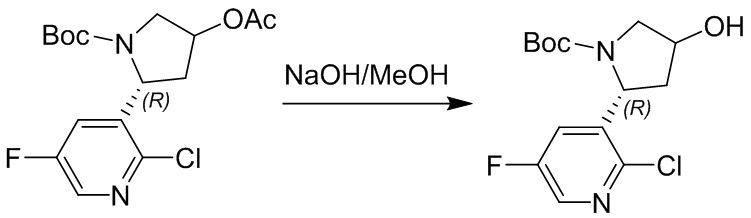
(2R)-трет-бутил 4-ацетокси-2-(2-хлор-5-фторпиридин-3-ил)пирролидин-1-карбоксилат (3,5 г, 9,75 ммоль) растворяли в MeOH (48,8 мл) с последующим добавлением 2M NaOH (5,37 мл, 10,73 ммоль), и реакционную смесь перемешивали при КТ в течение 2 часов. Растворитель удаляли под вакуумом и водный слой нейтрализовывали 1Н HCl и трижды экстрагировали с использованием EtOAc. Объединенные органические слои сушили над Na2SO4. Растворитель удаляли под вакуумом и осадок очищали посредством хроматографии на силикагеле (0-70% гекс/EtOAc) для получения (2R)-трет-бутил 2-(2-хлор-5-фторпиридин-3-ил)-4-гидроксипирролидин-1-карбоксилата (2,1 г, 6,63 ммоль, 68,0% выход). ЖХМС: 317 M+H.
Шаг 8: (R)-трет-бутил 2-(2-хлор-5-фторпиридин-3-ил)-4-оксопирролидин-1-карбоксилат
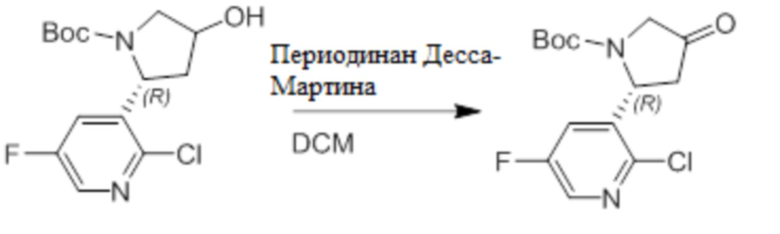
(2R)-трет-бутил 2-(2-хлор-5-фторпиридин-3-ил)-4-гидроксипирролидин-1-карбоксилат (2,1 г, 6,63 ммоль) растворяли в DCM (66,3 мл) и добавляли NaHCO3 (0,557 г, 6,63 ммоль), после чего добавляли периодинан Десса-Мартина (8,44 г, 19,89 ммоль). Реакционную смесь перемешивали в течение ночи. Добавляли воду (0,119 мл, 6,63 ммоль), после чего добавляли периодинан Десса-Мартина (8,44 г, 19,89 ммоль) и перемешивали в течение 18 часов. Значение pH доводили до ~7 посредством насыщенного водного NaHCO3 и экстрагировали с использованием DCM x3. Органические слои комбинировали, сушили над Na2SO4 и удаляли растворитель под вакуумом. Осадок очищали посредством флэш-хроматографии (0-70% гекс/EtOAc) для получения (R)-трет-бутил 2-(2-хлор-5-фторпиридин-3-ил)-4-оксопирролидин-1-карбоксилата (1,6 г, 5,08 ммоль, 77% выход). ЖХМС: 315 M+H.
Шаг 9: (2R,4R)-трет-бутил 2-(2-хлор-5-фторпиридин-3-ил)-4-гидроксипирролидин-1-карбоксилат
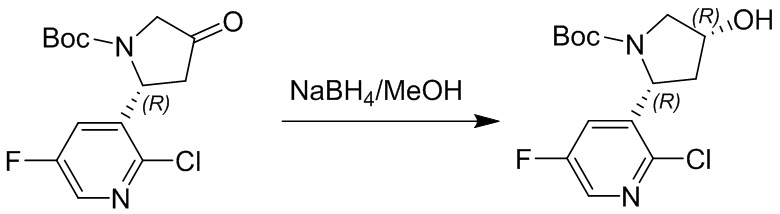
(R)-трет-бутил 2-(2-хлор-5-фторпиридин-3-ил)-4-оксопирролидин-1-карбоксилат (1,6 г, 5,08 ммоль) суспендировали в этаноле (33,9 мл) и охлаждали до 0 °C. Порционно добавляли NaBH4 (0,096 г, 2,54 ммоль) и перемешивали в течение 45 минут при 0 °C. Реакцию медленно гасили насыщенным NH4Cl, оставляли нагреваться до КТ и раствор экстрагировали с использованием DCM x3. Органические слои объединяли и высушивали над Na2SO4. Осадок очищали посредством флэш-хроматографии (0-70% гекс/EtOAc) для получения (2R,4R)-трет-бутил 2-(2-хлор-5-фторпиридин-3-ил)-4-гидроксипирролидин-1-карбоксилата (1,446 г, 4,57 ммоль, 90% выход). ЖХМС: 317 M+H.
Шаг 10: (2R,4S)-трет-бутил 2-(2-хлор-5-фторпиридин-3-ил)-4-фторпирролидин-1-карбоксилат
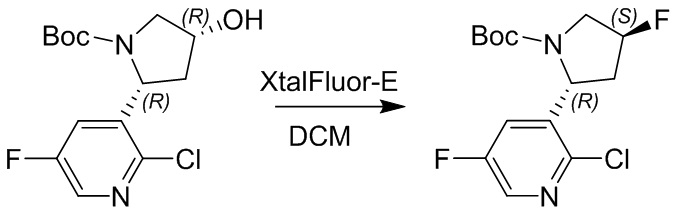
(2R,4R)-трет-бутил 2-(2-хлор-5-фторпиридин-3-ил)-4-гидроксипирролидин-1-карбоксилат (1,0 г, 3,16 ммоль) растворяли в DCM (25 мл) и охлаждали до -78 °C. Добавляли TEA-HF (1,098 мл, 9,47 ммоль) и перемешивали в течение 10 минут. Добавляли XtalFluor-E (1,446 г, 6,31 ммоль) и через 10 минут реакционную смесь переносили на ледяную баню и оставляли нагреваться до 0 °C. Через 2 часа реакционную смесь разбавляли DCM и гасили насыщенным водным NaHCO3. Органические слои разделяли и удаляли растворитель под вакуумом. Осадок очищали посредством ISCO (0-50% гекс/EtOAc; колонка 12 г) для получения (2R,4S)-трет-бутил 2-(2-хлор-5-фторпиридин-3-ил)-4-фторпирролидин-1-карбоксилата (0,805 г, 2,53 ммоль, 80% выход) в виде твердого вещества белого цвета. ЖХМС: 319 M+H.
Шаг 11: 2-хлор-5-фтор-3-((2R,4S)-4-фторпирролидин-2-ил)пиридин, HCl
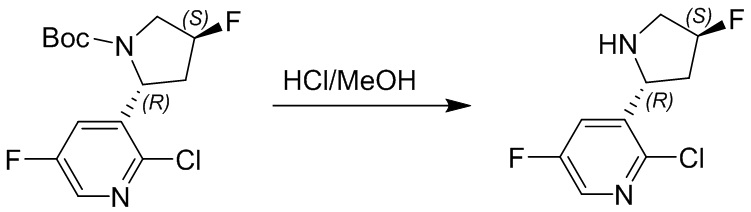
(2R,4S)-трет-бутил 2-(2-хлор-5-фторпиридин-3-ил)-4-фторпирролидин-1-карбоксилат (0,805 г, 2,53 ммоль, 80% выход) растворяли в EtOAc (5 мл) и добавляли 4Н HCl/диоксан (3 мл). Реакционную смесь перемешивали при КТ в течение 1 часа. Осадок отфильтровывали, промывали эфиром, и высушивали под высоким вакуумом в течение ночи для получения 2-хлор-5-фтор-3-((2R,4S)-4-фторпирролидин-2-ил)пиридина, HCl (0,612 г, 2,399 ммоль, 76% выход) в виде твердого вещества грязно-белого цвета. ЖХМС: 219 M+H.
Пример 16. 5-фтор-3-((2R,4S)-4-фторпирролидин-2-ил)-2-метоксипиридин
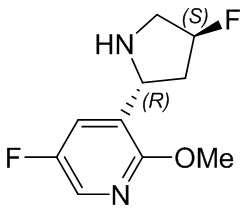
5-фтор-3-((2R,4S)-4-фторпирролидин-2-ил)-2-метоксипиридин изготавливали тем же способом, что и 3-фтор-5-((2R,4S)-4-фторпирролидин-2-ил)бензамид с использованием 5-фтор-2-метоксиникотинальдегида вместо 2-хлор-5-фторникотинальдегида.
Пример 17. Метил(1R,3R,4R)-3-амино-4-гидроксициклопентан-1-карбоксилат
Шаг 1: (1S,2R,4S,5R)-3-окса-6-азатрицикло[3,2,1,02,4]октан-7-он
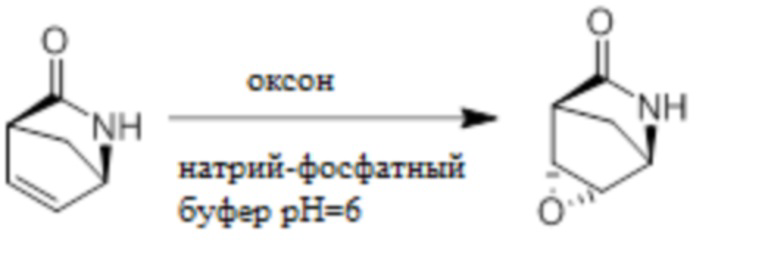
К раствору (1R,4S)-2-азабицикло[2,2,1]гепт-5-ен-3-она (30,00 г, 274,90 ммоль, 1,00 экв.) в NaH2PO4 (395,00 мл, 0,2M) и Na2HPO4 (55,00 мл, 0,2 M) добавляли H2O (450,00 мл) и оксон (669,31 г, 4,40 моль, 16,00 экв.) при 0°C порционно на протяжении 5 часов, поддерживая значение pH=6 добавлением водн. NaOH (12 M) и удерживая температуру на уровне 0°C. После добавления смесь перемешивали при температуре 0°C в течение еще 2 часов, ТСХ (PE:EtOAc=1:1) показала, что исходный материал полностью прореагировал, смесь фильтровали и водную фазу экстрагировали с использованием DCM (400 мл*5), объединенные органические слои сушили над Na2SO4, концентрировали в вакууме для получения (1S,2R,4S,5R)-3-окса-6-азатрицикло[3,2,1,02,4]октан-7-она (9,00 г, 71,93 ммоль, выход: 26,16%) в виде твердого вещества желтого цвета. 1H-ЯМР (400 МГц, CDCl3) δ м. д. 5,96 (уш. с., 1H), 3,86 (с, 1H), 3,62 (1H, d, J=3,2 Гц), 3,53 (1H, d, J=2,8 Гц), 2,86 (с, 1H), 1,82 (д, 1H, J=9,6 Гц), 1,64 (д, 1H, J=10,0 Гц).
Шаг 2: трет-бутил (1S,2R,4S,5R)-7-оксо-3-окса-6-азатрицикло[3,2,1,02,4]октан-6-карбоксилат

К раствору (1S,2R,4S,5R)-3-окса-6-азатрицикло[3,2,1,02,4]октан-7-он (9,00 г, 71,93 ммоль, 1,00 экв.) в DCM (100,00 мл) добавляли Boc2O (17,27 г, 79,12 ммоль, 1,10 экв.), Et3N (8,73 г, 86,32 ммоль, 1,20 экв.) и DMAP (878,71 мг, 7,19 ммоль, 0,10 экв.), смесь перемешивали при температуре 25°C в течение 16 часов, ЖХМС показала, что исходный материал полностью прореагировал, смесь промывали NH4Cl водн. (100 мл*3), объединенные органические слои сушили над Na2SO4, концентрировали в вакууме, неочищенный продукт очищали посредством колоночной хроматографии на силикагеле (PE:EtOAc=5:1~1:1) для получения трет-бутил (1S,2R,4S,5R)-7-оксо-3-окса-6-азатрицикло[3,2,1,02,4]октан-6-карбоксилата (12,00 г, 53,28 ммоль, выход: 74,07%) в виде твердого вещества желтого цвета. 1H-ЯМР (400 МГц, CDCl3) δ м. д. 4,56 (с, 1H), 3,71 (д, 1H, J=2,8 Гц), 3,54 (д, 1H, J=2,8 Гц), 3,00 (с, 1H), 1,75 (д, 1H, J=10,0 Гц), 1,57 (д, 1H, J=10,8 Гц), 1,46 (с, 9H).
Шаг 3: метил (3R,4R)-4-((трет-бутоксикарбонил)амино)-3-гидроксициклопент-1-ен-1-карбоксилат
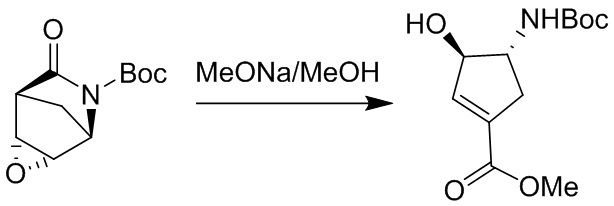
Na (3,37 мг, 146,50 мкмоль, 0,01 экв.) добавляли к MeOH (10,00 мл) при 0°C, после чего раствор перемешивали при температуре 0°C в течение 0,5 часа, раствор добавляли к трет-бутил (1S,2R,4S,5R)-7-оксо-3-окса-6-азатрицикло[3,2,1,02,4]октан-6-карбоксилату (3,30 г, 14,65 ммоль, 1,00 экв.) в MeOH (30,00 мл), после чего смесь перемешивали при температуре 16°C в течение 13,5 часов, ЖХМС показала, что исходный материал полностью прореагировал, реакцию гасили уксусной кислотой (5 мл), после чего промывали NaHCO3 (20 мл*3), органический слой высушивали над Na2SO4 и концентрировали в вакууме, неочищенный продукт промывали PE (20 мл) для получения метил (3R,4R)-4-((трет-бутоксикарбонил)амино)-3-гидроксициклопент-1-ен-1-карбоксилата (2,10 г, 8,16 ммоль, выход: 55,72%) в виде твердого вещества белого цвета. 1H-ЯМР (400 МГц, CDCl3) δ м. д. 6,65 (с, 1H), 4,98 (с, 1H), 4,81 (д, 1H, J=2,8 Гц), 4,46 (с, 1H), 3,98-3,92 (м, 1H), 3,75 (с, 3H), 3,07-3,01 (м, 1H), 2,36-2,29 (м, 1H), 1,45 (с, 9H).
Шаг 4: метил (1R,3R,4R)-3-((трет-бутоксикарбонил)амино)-4-гидроксициклопентан-1-карбоксилат
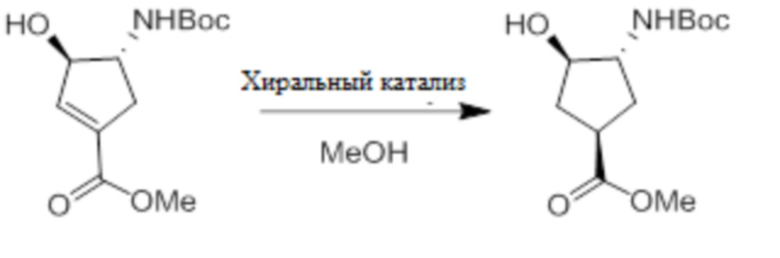
Смесь метил (3R,4R)-4-((трет-бутоксикарбонил)амино)-3-гидроксицикломент-1-ен-1-карбоксилата (2,00 г, 7,77 ммоль, 1,00 экв.), (1Z,5Z)-циклоокта-1,5-диена; (2S,5S)-1-[2-[(2S,5S)-2,5-диметилфосфолан-1-ил]этил]-2,5-диметил-фосфолана; родия(1+); трифторметансульфоната (48,08 мг, 77,74 мкмоль, 0,01 экв.) в MeOH (50,00 мл) дегазировали и трижды продували H2, после чего смесь перемешивали при температуре 55°C в течение 16 часов в атмосфере H2 (40 фунтов/кв. дюйм), ТСХ (PE:EtOAc=1:1) показала полное реагирование всего исходного материала, смесь концентрировали в вакууме, и после чего растворяли в EtOAc (5 мл), после чего к смеси добавляли PE (20 мл), формировалось твердое вещество белого цвета, осадок собирали, высушивали в вакууме, твердое вещество растворяли в MeOH (7 мл) и очищали посредством кислотной препаративной ВЭЖХ (HCl) для получения метил (1R,3R,4R)-3-((трет-бутоксикарбонил)амино)-4-гидроксициклопентан-1-карбоксилата (750,00 мг, 2,89 ммоль, выход: 37,23%) в виде маслянистой жидкости желтого цвета, структура была подтверждена хиральной ВЭЖХ и 1H-ЯМР. 1H-ЯМР (400 МГц, CDCl3) δ м. д. 4,04-3,99 (м, 1H), 3,80-3,77 (м, 1H), 3,70 (с, 3H), 2,93-2,91 (м, 1H), 2,48-2,44 (м, 1H), 2,43-2,35 (м, 1H), 1,92-1,89 (м, 1H), 1,87-1,69 (м, 1H), 1,45 (с, 9H).
Пример 18. Синтез метил (1R,3R,4R)-3-амино-4-гидроксициклопентан-1-карбоксилата
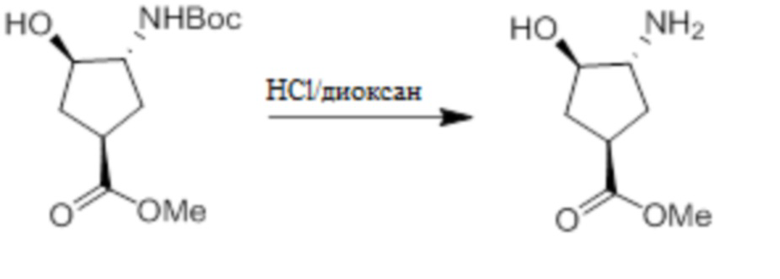
Метил(1R,3R,4R)-3-((трет-бутоксикарбонил)амино)-4-гидроксициклопентан-1-карбоксилат (700,00 мг, 2,70 ммоль, 1,00 экв.) в HCl/диоксане (10,00 мл, 4 M) перемешивали при температуре 19°C в течение 5 часов, ЖХМС показала полное реагирование всего исходного материала, смесь концентрировали в вакууме для получения метил (1R,3R,4R)-3-амино-4-гидроксициклопентан-1-карбоксилата (500,00 мг, 2,56 ммоль, выход: 94,66%) в виде маслянистой жидкости желтого цвета. 1H-ЯМР (400 МГц, CD3OD) δ м. д. 4,09-4,05 (м, 1H), 3,75-3,66 (м, 3H), 3,37-3,33 (м, 1H), 3,06-3,04 (м, 1H), 2,45-2,37 (м, 2H), 1,87-1,81 (м, 2H).
ЖХ-МС (подвижная фаза: от 95% [вода+0,375‰ об./об. TFA] и 5% [CH3CN+0,188‰ об./об. TFA], в данных условиях 0,25 мин., после чего условия изменяли на 15% [CH3CN+0,188‰ об./об. TFA] в течение 10,0 мин., в данных условиях 5 мин., в конце условия изменяли на 95% [вода+0,375‰ об./об. TFA] и 5% [CH3CN+0,188‰ об./об. TFA] в течение 0,01 мин., после чего в данных условиях 5 мин. Поток все время составлял 1,0 мл·мин.-1), чистота составляла 98,803%, Rt=0,893 мин., МС рассчетн.: 159,2, МС получен. 160,1 ([M+1]+).
Пример 19. (3aS,4R,6aR)-2,2-диметил-3a,6a-дигидро-4H-циклопента[d][1,3]диоксол-4-амин
Шаг 1: (3aS,4S,6aR)-2,2-диметил-3a,6a-дигидро-4H-циклопента[d][1,3]диоксол-4-ол
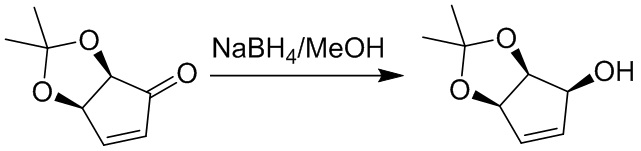
(3aR,6aR)-2,2-диметил-3a,6a-дигидро-4H-циклопента[d][1,3]диоксол-4-он (50,00 г, 324,34 ммоль, 1,00 экв.) растворяли в MeOH (1,00 л), после чего добавляли CeCl3,7H2O (120,84 г, 324,34 ммоль, 30,83 мл, 1,00 экв.). Смесь охлаждали до 0 °C. После этого NaBH4 (24,54 г, 648,68 ммоль, 2,00 экв.) добавляли порционно при 0°C в течение 1,5 часов. После добавления завершенность реакции проверяли посредством ТСХ (PE/EtOAc=5/1). Реакцию гасили насыщенным NH4Cl (1000 мл) и экстрагировали с использованием DCM (500 мл*5). Объединенные органические слои сушили над Na2SO4 и концентрировали под вакуумом при температуре 45 °C. Получили (3aS,4S,6aR)-2,2-диметил-3a,6a-дигидро-4H-циклопента[d][1,3]диоксол-4-ол (50,66 г, 324,37 ммоль, выход: 100,00%) в виде желтой жидкости, который использовали непосредственно без очистки.
Шаг 2: 2-((3aS,4R,6aR)-2,2-диметил-3a,6a-дигидро-4H-циклопента[d][1,3]диоксол-4-ил)изоиндолин-1,3-дион
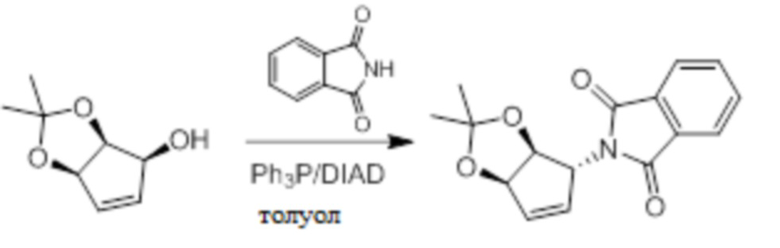
К смеси (3aS,4S,6aR)-2,2-диметил-3a,6a-дигидро-4H-циклопента[d][1,3]диоксол-4-ола (8,00 г, 51,22 ммоль, 1,00 экв.) и изоиндолин-1,3-диона (9,04 г, 61,46 ммоль, 1,20 экв.) в толуоле (250,00 мл) добавляли PPh3 (20,15 г, 76,83 ммоль, 1,50 экв.) при температуре 20 °C. После этого DIAD (15,54 г, 76,83 ммоль, 1,50 экв.) добавляли по каплям к смеси при 0 °C. После добавления смесь оставляли нагреваться до 80°C и перемешивали в течение 16 часов. ТСХ (PE/EtOAc=5/1) показала завершение реакции. Смесь концентрировали. Осадок очищали посредством колоночной хроматографии на силикагеле (PE/EtOAc=25/1 до 15/1). Был получен продукт в необработанном виде в виде маслянистой жидкости желтого цвета с несколькими полярными пятнами на ТСХ. Далее добавляли 80 мл MeOH и образовавшийся белый осадок собирали путем фильтрации. Получали 2-((3aS,4R,6aR)-2,2-диметил-3a,6a-дигидро-4H-циклопента[d][1,3]диоксол-4-ил)изоиндолин-1,3-дион (9,60 г, 33,65 ммоль, выход: 65,70%) в виде твердого вещества белого цвета.
Шаг 3: (3aS,4R,6aR)-2,2-диметил-3a,6a-дигидро-4H-циклопента[d][1,3]диоксол-4-амин
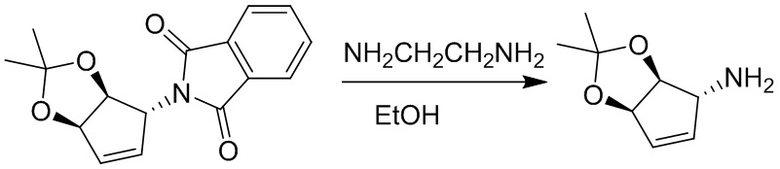
К смеси 2-((3aS,4R,6aR)-2,2-диметил-3a,6a-дигидро-4H-циклопента[d][1,3]диоксол-4-ил)изоиндолин-1,3-диона (9,52 г, 33,37 ммоль, 1,00 экв.) в EtOH (300,00 мл) добавляли этан-1,2-диамин (4,01 г, 66,74 ммоль, 2,00 экв.). Полученную смесь перемешивали при температуре 80°C в течение 16 часов. Выпало большое количество белого осадка. ТСХ (PE/EtOAc=5/1) показала, что исходный материал полностью прореагировал. Выпавший осадок отфильтровали. К фильтрату добавляли 300 мл NaOH (0,5 M). Смесь экстрагировали с использованием DCM (200 мл*5), сушили над Na2SO4 и концентрировали. Получали (3aS,4R,6aR)-2,2-диметил-3a,6a-дигидро-4H-циклопента[d][1,3]диоксол-4-амин (4,90 г, 31,57 ммоль, выход: 94,62%) в виде маслянистой жидкости желтого цвета.
Пример 20. (1R,3R,4R)-3-амино-4-гидроксициклопентан-1-карбонитрил
Шаг 1: (1R,3R,4R)-3-((трет-бутоксикарбонил)амино)-4-гидроксициклопентан-1-карбоновая кислота
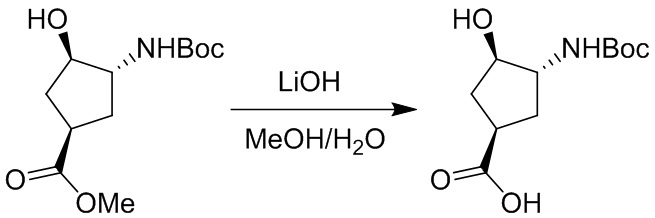
Смесь метил(1R,3R,4R)-3-((трет-бутоксикарбонил)амино)-4-гидроксициклопентан-1-карбоксилата (5,00 г, 19,28 ммоль, 1,00 экв.), LiOH.H2O (2,43 г, 57,85 ммоль, 3,00 экв.) в MeOH (10,00 мл) и H2O (10,00 мл) перемешивали при температуре 15°C в течение 16 часов, ТСХ (PE:EtOAc=1:1) показала завершение реакции, к смеси добавляли разведенный HCl (1 M) до достижения значения pH=6 и концентрировали в вакууме, после чего смесь растворяли в DCM (15 мл) и EtOAc (5 мл), смесь фильтровали и фильтрат концентрировали в вакууме для получения продукта (1R,3R,4R)-3-((трет-бутоксикарбонил)амино)-4-гидроксициклопентан-1-карбоновой кислоты (6,50 г, в неочищенном виде) в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, CD3OD) δ м. д. 3,95-3,90 (м, 1H), 3,74-3,69 (м, 1H), 2,91-2,87 (м, 1H), 2,32-2,22 (м, 2H), 1,82-1,72 (м, 2H), 1,45 (с, 9H).
Шаг 2: трет-бутил((1R,2R,4R)-4-карбамоил-2-гидроксициклопентил)карбамат
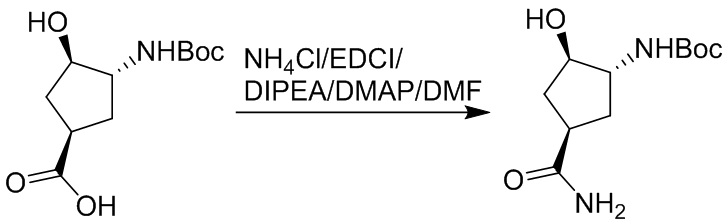
К смеси (1R,3R,4R)-3-((трет-бутоксикарбонил)амино)-4-гидроксициклопентан-1-карбоновой кислоты (3,00 г, 12,23 ммоль, 1,00 экв.) в DMF (40,00 мл) добавляли HATU (6,05 г, 15,90 ммоль, 1,30 экв.), DIPEA (4,74 г, 36,69 ммоль, 3,00 экв.) и NH4Cl (1,96 г, 36,69 ммоль, 3,00 экв.), после чего смесь перемешивали при температуре 15°C в течение 32 часов, ЖХМС показала завершение реакции, смесь концентрировали в вакууме для получения трет-бутил((1R,2R,4R)-4-карбамоил-2-гидроксициклопентил)карбамата (11 г, в неочищенном виде) (2 две партии быль получены и очищены вместе). 1H-ЯМР (400 МГц, CD3OD) δ м. д. 4,58 (уш. с., 1H), 3,96-3,94 (м, 1H), 3,77-3,75 (м, 1H), 2,25-2,20 (м, 2H), 1,79-1,75 (м, 2H), 1,44 (с, 9H).
Шаг 3: трет-бутил((1R,2R,4R)-4-циано-2-гидроксициклопентил)карбамат
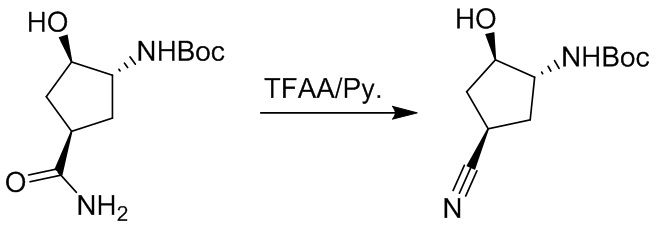
К смеси трет-бутил((1R,2R,4R)-4-карбамоил-2-гидроксициклопентил)карбамата (2,00 г, 8,19 ммоль, 1,00 экв.), пиридина (1,94 г, 24,56 ммоль, 3,00 экв.) в ТГФ (3,00 мл) добавляли TFAA (2,58 г, 12,28 ммоль, 1,50 экв.) по каплям при 0 °C, далее смесь перемешивали при температуре 0°C в течение 0,5 часа, после чего к смеси добавляли Et3N (2,49 г, 24,56 ммоль, 3,00 экв.) при температуре 15 °C, смесь перемешивали при температуре 15°C в течение 0,5 часа, к смеси добавляли TFAA (2,58 г, 12,28 ммоль, 1,50 экв.) и смесь перемешивали при температуре 15°C в течение 0,5 часа, ЖХМС показала завершение реакции, смесь концентрировали в вакууме, очищали посредством препаративной ВЭЖХ (TFA, MS) для получения трет-бутил((1R,2R,4R)-4-циано-2-гидроксициклопентил)карбамата (380,00 мг, 1,68 ммоль, выход: 20,51%) в виде бесцветной маслянистой жидкости. 1H-ЯМР (400 МГц, CD3OD) δ м. д. 4,07-3,97 (м, 1H), 3,79-3,78 (м, 0,5H), 3,51-3,49 (м, 0,5H), 3,19-3,05 (м, 1H), 2,44-2,35 (м, 0,5H), 2,33-2,28 (м, 1,5H), 1,97-1,94 (м, 1H), 1,92-1,82 (м, 1H), 1,41 (с, 9H).
Шаг 4: (1R,3R,4R)-3-амино-4-гидроксициклопентан-1-карбонитрил
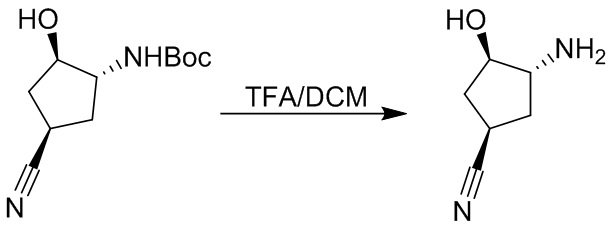
Смесь трет-бутил((1R,2R,4R)-4-циано-2-гидроксициклопентил)карбамата (800,00 мг, 3,54 ммоль, 1,00 экв.) в TFA (5,00 мл) и DCM (5,00 мл) перемешивали при температуре 20°C в течение 2 часов, ЖХМС показала завершение реакции, смесь концентрировали в вакууме для получения (1R,3R,4R)-3-амино-4-гидроксициклопентан-1-карбонитрила (545,00 мг, 2,27 ммоль, выход: 64,10%) в виде бесцветной маслянистой жидкости. 1H-ЯМР (400 МГц, CD3OD) δ м. д. 4,12-4,06 (м, 1H), 3,51-3,47 (м, 1H), 3,23-3,20 (м, 1H), 2,48-2,42 (м, 2H), 2,11-2,10 (м, 1H), 1,94-1,90 (м, 1H).
Пример 21. (3aS,4R,6aS)-6,6-дифтор-2,2-диметилтетрагидро-4H-циклопента[d][1,3]диоксол-4-амин
Шаг 1: 2-((3aS,4R,6S,6aR)-6-гидрокси-2,2-диметилтетрагидро-4H-циклопента[d][1,3]диоксол-4-ил)изоиндолин-1,3-дион
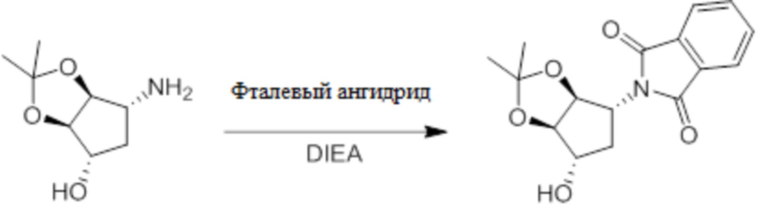
Смесь (3aR,4S,6R,6aS)-6-амино-2,2-диметилтетрагидро-4H-циклопента[d][1,3]диоксол-4-ола (0,43 г, 2,46 ммоль, 1,00 экв.), фталевого ангидрида (0,36 г, 2,46 ммоль, 1 экв.) и DIEA (0,65 мл, 3,7 ммоль, 1,5 экв.) в толуоле (6,2 мл) перемешивали при температуре 100°C в течение 9 часов. ЖХМС показала завершение реакции. К реакционной смеси добавляли EtOAc, после чего промывали насыщенным водным раствором бикарбоната натрия (15 мл). Объединенные органические слои промывали насыщенным солевым раствором, сушили над Na2SO4 и концентрировали под вакуумом. Осадок очищали посредством колоночной хроматографии на силикагеле (гексаны/EtOAc) для получения продукта 2-((3aS,4R,6S,6aR)-6-гидрокси-2,2-диметилтетрагидро-4H-циклопента[d][1,3]диоксол-4-ил)изоиндолин-1,3-диона (0,62 г, 83%) в виде твердого вещества белого цвета.
Шаг 2: 2-((3aS,4R,6aS)-2,2-диметил-6-оксотетрагидро-4H-циклопента[d][1,3]диоксол-4-ил)изоиндолин-1,3-дион
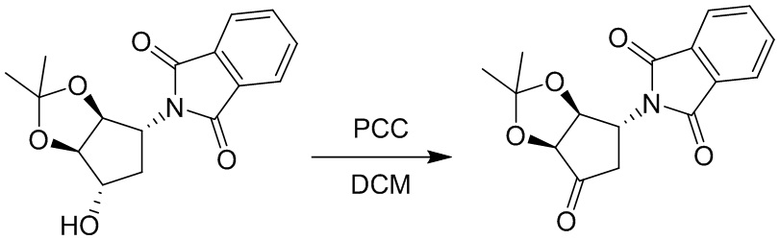
К раствору 2-((3aS,4R,6S,6aR)-6-гидрокси-2,2-диметилтетрагидро-4H-циклопента[d][1,3]диоксол-4-ил)изоиндолин-1,3-диона (0,20 г, 0,68 ммоль, 1,00 экв.) в DCM (4,5 мл) добавляли PCC (0,29 г, 1,35 ммоль, 2 экв.) и раствор перемешивали при температуре 23°C в течение 16 часов. Добавляли еще одну аликвоту PCC (0,15g, 0,67 ммоль) и реакция продолжалась в течение еще 16 часов. ЖХМС показала завершение реакции. К реакционной смеси добавляли EtOAc, после чего фильтровали через слой целита. Осадок концентрировали, после чего очищали посредством колоночной хроматографии на силикагеле (гексаны/EtOAc) для получения продукта 2-((3aS,4R,6aS)-2,2-диметил-6-оксотетрагидро-4H-циклопента[d][1,3]диоксол-4-ил)изоиндолин-1,3-диона (0,19 г, 94%) в виде твердого вещества грязно-белого цвета.
Шаг 3: 2-((3aS,4R,6aS)-6,6-дифтор-2,2-диметилтетрагидро-4H-циклопента[d][1,3]диоксол-4-ил)изоиндолин-1,3-дион
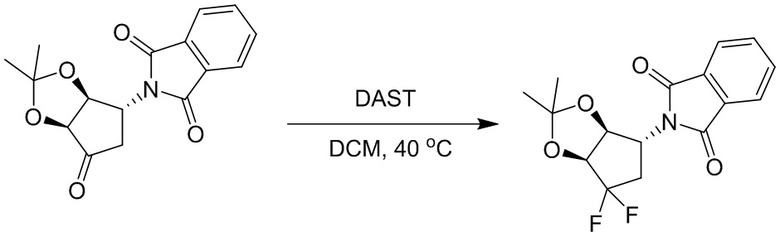
К раствору 2-((3aS,4R,6aS)-2,2-диметил-6-оксотетрагидро-4H-циклопента[d][1,3]диоксол-4-ил)изоиндолин-1,3-диона (0,16 г, 0,53 ммоль, 1,00 экв.) в DCM (3,5 мл) добавляли DAST (0,42 г, 2,64 ммоль, 5 экв.) и раствор перемешивали с обратным холодильником в течение 16 часов. Добавляли еще одну аликвоту DAST (0,42 г, 2,64 ммоль, 5 экв.) и реакция продолжалась в течение еще 16 часов при температуре 23 oC. Реакционную смесь разбавляли DCM и после чего промывали насыщенным водным раствором бикарбоната натрия. Объединенные органические слои промывали насыщенным солевым раствором, сушили над Na2SO4 и концентрировали под вакуумом. Осадок очищали посредством колоночной хроматографии на силикагеле (гексаны/EtOAc) для получения продукта 2-((3aS,4R,6aS)-6,6-дифтор-2,2-диметилтетрагидро-4H-циклопента[d][1,3]диоксол-4-ил)изоиндолин-1,3-диона (0,065 г, 38%).
Шаг 4: (3aS,4R,6aS)-6,6-дифтор-2,2-диметилтетрагидро-4H-циклопента[d][1,3]диоксол-4-амин
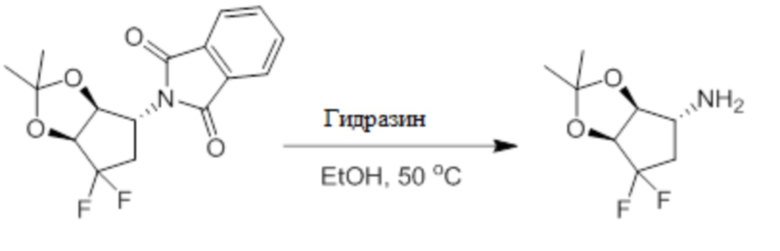
К раствору 2-((3aS,4R,6aS)-6,6-дифтор-2,2-диметилтетрагидро-4H-циклопента[d][1,3]диоксол-4-ил)изоиндолин-1,3-диона (0,065 г, 0,2 ммоль, 1,00 экв.) в этаноле (1,8 мл) добавляли моногидрат гидразина (0,015 мл, 0,3 ммоль, 1,5 экв.) и раствор перемешивали при температуре 50°C в течение 2 часов, после чего при температуре 70°C в течение еще 2 часов. Гетерогенную реакционную смесь фильтровали с использованием минимального объема этанола. Затем фильтрат концентрировали и выделенный неочищенный продукт (3aS,4R,6aS)-6,6-дифтор-2,2-диметилтетрагидро-4H-циклопента[d][1,3]диоксол-4-амин использовали на следующем шаге без дополнительной очистки.
Пример 22. Синтез (1R,3R,4R)-4-аминоциклогексан-1,3-диол
Шаг 1: (1r,4r)-4-(бензилокси)циклогексанол и (1s,4s)-4-(бензилокси)циклогексанол
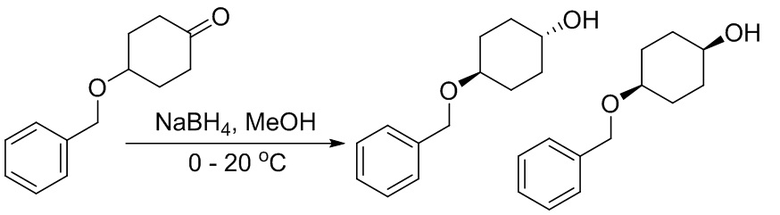
К охлажденному на ледяной бане раствору 4-(бензилокси)циклогексанона (31,0 г, 152 ммоль) в 500 мл метанола добавляли борогидрид натрия (5,78 г, 153 ммоль) несколькими порциями в течение 10 мин., после чего раствор перемешивали при температуре 20°C в течение 2 ч. После этого смесь гасили насыщенным водным раствором хлорида аммония (50 мл), концентрировали, осадок растворяли в 200 мл воды и экстрагировали с использованием этилацетата (200 мл x 3), объединенную органическую фазу сушили над сульфатом натрия, после чего концентрировали под вакуумом для получения искомого продукта (1r,4r)-4-(бензилокси)циклогексанол и (1s,4s)-4-(бензилокси)циклогексанола в виде маслянистой жидкости бледно-желтого цвета (31,0 г, в неочищенном виде), который непосредственно использовали на следующем этапе без дополнительной очистки. MS (ES+) C13H18O2 треб.: 206, получен.: 207[M+H]+.
Шаг 2: ((циклогекс-3-енилокси)метил)бензол
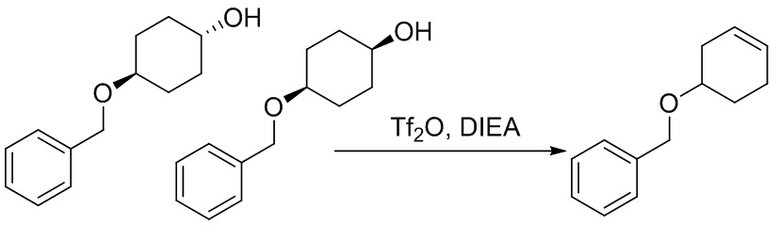
К охлажденному на ледяной бане раствору (1r,4r)-4-(бензилокси)циклогексанола, (1s,4s)-4-(бензилокси)циклогексанола (30,0 г, 145 ммоль) и N,N-диизопропилэтиламина (28,1 г, 218 ммоль) в 1200 мл дихлорметана добавляли по каплям в течение 30 мин. трифторметансульфоновый ангидрид (30,7 г, 109 ммоль), после чего раствор перемешивали при температуре 25°C в течение 18 ч. После этого смесь концентрировали под вакуумом, осадок очищали посредством колоночной хроматографии на силикагеле и элюировали с использованием петролейного эфира:этилацетата=12:1 для получения искомого соединения (28,0 г, выход 100%) в виде маслянистой жидкости желтого цвета. MS (ES+) C13H16O треб.: 188, получен.: 189 [M+H]+.
Шаг 3: (1R,3R,6S)-3-(бензилокси)-7-окса-бицикло[4,1,0]гептан
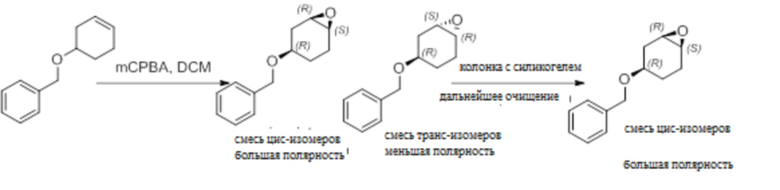
Раствор ((циклогекс-3-енилокси)метил)бензола (12,0 г, 63,7 ммоль) в дихлорметане (200 мл) обрабатывали при 0°C мета-хлорпероксибензоевой кислотой (21,9 г, 127 ммоль). Реакционную смесь перемешивали в течение 2 ч. при 0°C и после чего 15 мин. при комнатной температуре. Испарение промытого (10% водным раствор сульфита натрия, 5% водным раствором гидроксида натрия, после чего водой) органического раствора привело к появлению жидкого осадка, который отделяли посредством колоночной хроматографии на силикагеле, элюировали с использованием гексана:изопропилового эфира:этилацетата=65:28:7 для получения искомого соединения (4,18 г, выход 32%) в виде маслянистой жидкости желтого цвета. Транс-(1S,3R,6R)-3-(бензилокси)-7-окса-бицикло[4,1,0]гептан показал немного меньшую полярность на ТСХ и был элюирован первым. Цис-(1R,3R,6S)-3-(бензилокси)-7-окса-бицикло[4,1,0]гептан был элюирован вторым. MS (ES+) C13H16O2 треб.: 204, получен.: 205 [M+H]+.
Шаг 4: (1R,2R,5R)-5-(бензилокси)-2-((S)-1-фенилэтиламино)циклогексанол
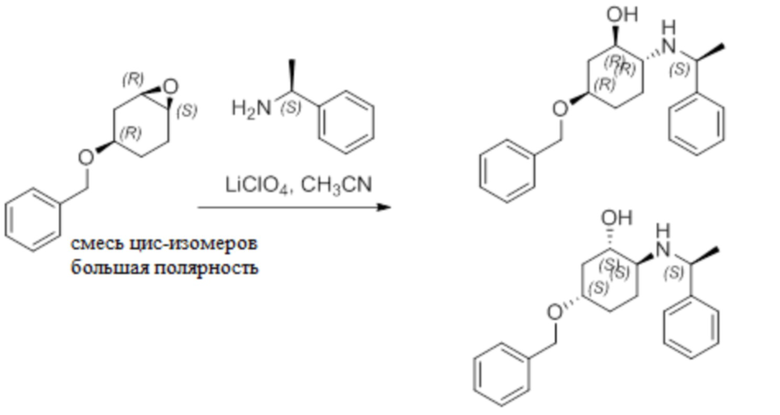
Перхлорат лития (7,27 г, 68,4 ммоль) добавляли к охлажденному на ледяной бане перемешиваемому раствору (1R,3R,6S)-3-(бензилокси)-7-окса-бицикло[4,1,0]гептана (7,0 г, 34,2 ммоль) в 120 мл 4A-MS сухого ацетонитрила, снимали с бани и добавляли по каплям (с)-1-фенилэтанамин (5,58 г, 46,1 ммоль) в течение 15 мин., после чего раствор перемешивали при температуре 25°C в течение 18 ч. После этого смесь растворяли в 200 мл воды и экстрагировали с использованием этилацетата (200 мл x 3), объединенную органическую фазу сушили над сульфатом натрия, после чего концентрировали и осадок очищали посредством колоночной хроматографии на силикагеле, элюировали с использованием петролейного эфира:этилацетата:триэтиламина=98:0:2 ~ 49:49:2 для получения искомого соединения (3,5 г, выход 31%) в виде маслянистой жидкости желтого цвета. (1S,2S,5S)-5-(бензилокси)-2-((S)-1-фенилэтиламино)циклогексанол показал немного меньшую полярность на ТСХ и был элюирован первым. (1R,2R,5R)-5-(бензилокси)-2-((S)-1-фенилэтиламино)циклогексанол был элюирован вторым. MS (ES+) C21H27NO2 треб.: 325, получен.: 326[M+H]+.
Шаг 5: (1R,2R,4R)-4-(бензилокси)-2-(трет-бутилдиметилсилилокси)-N-((S)-1-фенилэтил)циклогексанамин

Трифторметансульфонат трет-бутилдиметилсилила (13,0 г, 49,5 ммоль) добавляли к охлажденному на ледяной бане перемешанному раствору (1R,2R,5R)-5-(бензилокси)-2-((S)-1-фенилэтиламино)циклогексанола (5,4 г, 16,5 ммоль) и триэтиламина (5,0 г, 49,5 ммоль) в 100 мл высушенного дихлорметана. Через 30 мин. полученное промывали насыщенным водным раствором бикарбоната натрия и сушили над сульфатом натрия. Растворитель удаляли и осадок очищали посредством колоночной хроматографии на силикагеле, элюировали с использованием петролейного эфира:этилацетата=100:0~70:30 для получения искомого соединения (5,4 г, выход 71%) в виде маслянистой жидкости желтого цвета. MS (ES+) C27H41NO2Si треб.: 439, получен.: 440 [M+H]+.
Шаг 7: (1R,2R,5R)-5-(бензилокси)-2-((S)-1-фенилэтиламино)циклогексанол
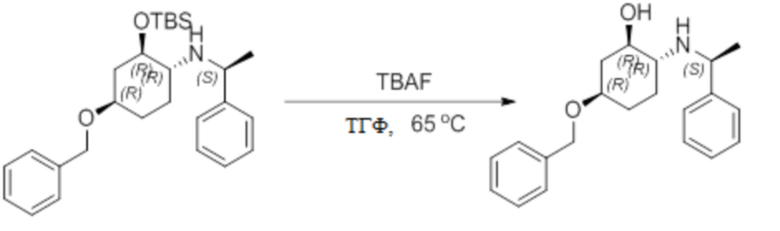
Фторид тетрабутиламмония (2,66 г, 10,2 ммоль) добавляли к перемешанному раствору (1R,2R,4R)-4-(бензилокси)-2-(трет-бутилдиметилсилилокси)-N-((S)-1-фенилэтил)циклогексанамина (1,5 г, 3,41 ммоль) в 50 мл сухого оксолана при комнатной температуре. После этого данный раствор перемешивали при температуре 65°C в течение 2 ч. После этого смесь концентрировали под вакуумом, осадок растворяли в 200 мл воды и экстрагировали с использованием этилацетата (200 мл x 3), объединенную органическую фазу промывали водой и насыщенным водным раствором хлорида натрия и сушили над сульфатом натрия. Растворитель удаляли и осадок очищали посредством колоночной хроматографии на силикагеле, элюировали с использованием петролейного эфира:этилацетата=100:0~70:30 для получения искомого соединения (0,75 г, выход 68%) в виде бесцветной маслянистой жидкости. MS (ES+) C21H27NO2 треб.: 325, получен.: 326[M+H]+.
Шаг 8: (1R,3R,4R)-4-аминоциклогексан-1,3-диол
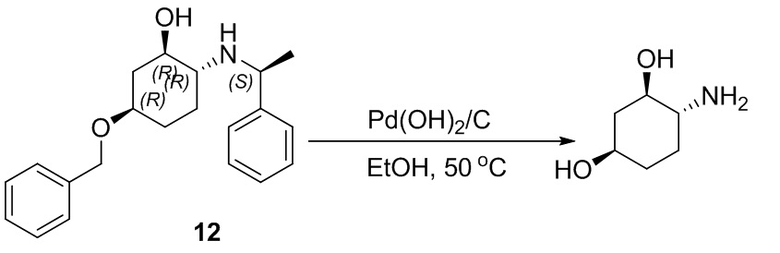
10% гидроксид палладия в активированном угле (697 мг, катализатор) добавляли к раствору (1R,2R,5R)-5-(бензилокси)-2-((S)-1-фенилэтиламино)циклогексанола (650 мг, 1,99 ммоль) в 15 мл этанола при комнатной температуре. После этого данный раствор перемешивали при температуре 50°C в течение 20 ч. в атмосфере водорода. После этого смесь охлаждали и фильтровали через целит, отфильтрованный осадок промывали метанолом:дихлорметаном=1:10, фильтрат концентрировали под вакуумом и осадок растворяли в 20 мл раствора метанола:дихлорметана=1:10, концентрировали, высушивали под высоким вакуумом, после чего охлаждали при температуре -20°C для получения искомого соединения (240 мг, выход 92%) в виде кристаллов белого цвета. MS (ES+) C6H13NO2 треб.: 131, получен.: 132[M+H]+. 1H-ЯМР (400 МГц, 6d-DMSO) δ м. д. 4,62-4,49 (м, 2H), 3,42-3,33 (м, 2H, J=3,2 Гц), 2,93-2,86 (м, 1H), 2,25-2,18 (м, 1H), 1,98-1,92 (м, 1H), 1,72-1,59 (м, 3H), 1,13-1,03 (м, 2H), 0,97-0,90 (м, 1H).
Пример 23. Синтез трет-бутил (1R,2R)-2-(трет-бутилдиметилсилилокси)-4-оксоциклогексилкарбамата
Шаг 1: трет-бутил (1R,2R,4R)-2-(трет-бутилдиметилсилилокси)-4-гидроксициклогексилкарбамат
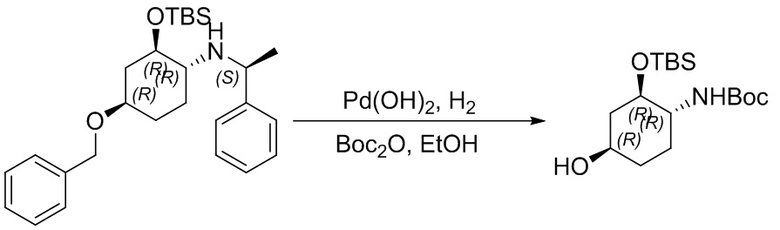
10% гидроксид палладия в активированном угле (1,9 г, катализатор) добавляли к раствору (1R,2R,4R)-4-(бензилокси)-2-(трет-бутилдиметилсилилокси)-N-((S)-1-фенилэтил)циклогексанамина из предыдущего примера (2,0 г, 4,54 ммоль) и ди-трет-бутил дикарбоната (3,95 г, 18,1 ммоль) в 60 мл этанола при комнатной температуре. После этого данный раствор перемешивали при температуре 50°C в течение 20 ч. в атмосфере водорода. Затем смесь охлаждали и фильтровали через целит, отфильтрованный осадок промывали метанолом:дихлорметаном=1:10, фильтрат концентрировали под вакуумом и осадок растворяли в 20 мл раствора метанола:дихлорметана=1:10, концентрировали, высушивали под высоким вакуумом для получения искомого соединения (1,2 г, выход 77%) в виде бесцветной маслянистой жидкости. MS (ES+) C17H35NO4Si треб.: 345, получен.: 346 [M+H]+.
Шаг 2: трет-бутил (1R,2R)-2-(трет-бутилдиметилсилилокси)-4-оксоциклогексилкарбамат
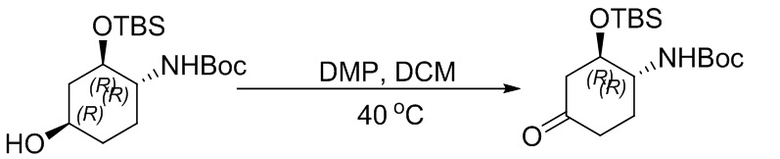
1,1,1-триацетокси-1,1-дигидро-1,2-бензиодоксол-3(1H)-он (периодинан Десса-Мартина, 4,11 г, 9,71 ммоль) добавляли к раствору трет-бутил(1R,2R,4R)-2-(трет-бутилдиметилсилилокси)-4-гидроксициклогексилкарбамата (1,4 г, 4,05 ммоль) в 50 мл дихлорметана при комнатной температуре. После этого данный раствор перемешивали при температуре 40°C в течение 3 ч. в атмосфере азота. После этого смесь концентрировали под вакуумом и осадок очищали посредством колоночной хроматографии на силикагеле, элюировали с использованием петролейного эфира:этилацетата=100:0 ~ 95:5 для получения искомого соединения (1,2 г, выход 86%) в виде маслянистой жидкости желтого цвета. MS (ES+) C17H33NO4Si треб.: 343, получен.: 344[M+H]+. 1H-ЯМР (400 МГц, CDCl3) δ м. д. 4,70-4,49 (уш., 1H), 4,10-3,90 (уш. с, 1H), 3,79-3,65 (уш., 1H), 2,64 (дд, 1H, J=14,4, 4,0 Гц), 2,42-2,28 (м, 4H), 1,46 (с, 9H), 0,87 (с, 9H), 0,08 (д, 6H, J=6,8 Гц).
Пример 23. Синтез трет-бутил (1R,2R)-2-(трет-бутилдиметилсилилокси)-4-оксоциклогексилкарбамата и (1R,3R,4R)-4-аминоциклогексан-1,3-диола
Шаг 1: (1R,4R)-4-(бензилокси)циклогексанол и (1S,4S)-4-(бензилокси)циклогексанол:
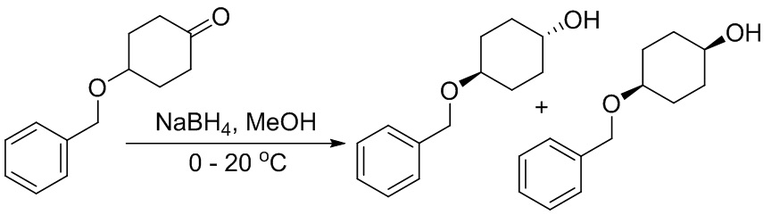
К охлажденному на ледяной бане раствору 4-(бензилокси)циклогексанона (31,0 г, 152 ммоль) в 500 мл метанола добавляли борогидрид натрия (5,78 г, 153 ммоль) несколькими порциями в течение 10 мин., после чего раствор перемешивали при температуре 20°C в течение 2 ч. После этого смесь гасили насыщенным водным раствором хлорида аммония (50 мл), концентрировали, осадок растворяли в 200 мл воды и экстрагировали с использованием этилацетата (200 мл x 3), объединенную органическую фазу сушили над сульфатом натрия, после чего концентрировали под вакуумом для получения искомого продукта (1R,4R)-4-(бензилокси)циклогексанола и (1S,4S)-4-(бензилокси)циклогексанола в виде маслянистой жидкости бледно-желтого цвета (31,0 г, в неочищенном виде), который непосредственно использовали на следующем этапе без дополнительной очистки. MS (ES+) C13H18O2 треб.: 206, получен.: 207[M+H]+.
Шаг 2: ((циклогекс-3-энилокси)метил)бензол.
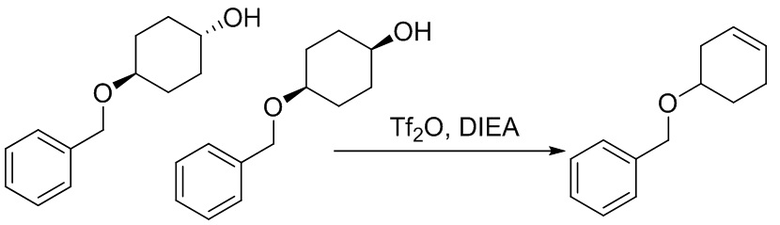
К охлажденному на ледяной бане раствору (1R,4R)-4-(бензилокси)циклогексанола и (1S,4S)-4-(бензилокси)циклогексанола (30,0 г, 145 ммоль) и N,N-диизопропилэтиламина (28,1 г, 218 ммоль) в 1200 мл дихлорметана добавляли трифторметансульфоновый ангидрид (30,7 г, 109 ммоль) по каплям в течение 30 мин., после чего раствор перемешивали при температуре 25°C в течение 18 ч. После этого смесь концентрировали под вакуумом и осадок очищали посредством колоночной хроматографии на силикагеле, элюировали с использованием петролейного эфира:этилацетата=12:1 для получения искомого соединения (28,0 г, выход 100%) в виде маслянистой жидкости желтого цвета. MS (ES+) C13H16O треб.: 188, получен.: 189 [M+H]+.
Шаг 3: (1R,3R,6S)-3-(бензилокси)-7-окса-бицикло[4,1,0]гептан:
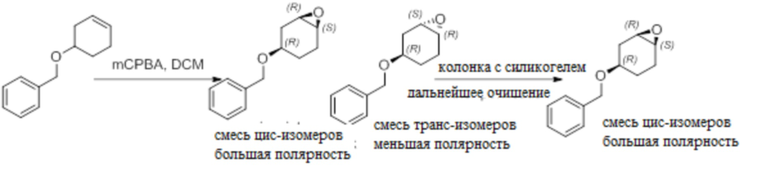
Раствор ((циклогекс-3-енилокси)метил)бензола (12,0 г, 63,7 ммоль) в дихлорметане (200 мл) обрабатывали при 0°C мета-хлорпероксибензоевой кислотой (21,9 г, 127 ммоль). Реакционную смесь перемешивали в течение 2 ч. при 0°C и после чего 15 мин. при комнатной температуре. Испарение промытого (10% водным раствор сульфита натрия, 5% водным раствором гидроксида натрия, после чего водой) органического раствора привело к появлению жидкого осадка, который отделяли посредством колоночной хроматографии на силикагеле, элюировали с использованием гексана:изопропилового эфира:этилацетата=65:28:7 для получения искомого соединения (4,18 г, выход 32%) в виде маслянистой жидкости желтого цвета. Транс-(1S,3R,6R)-3-(бензилокси)-7-окса-бицикло[4,1,0]гептан показал немного меньшую полярность на ТСХ и был элюирован первым. Цис-(1R,3R,6S)-3-(бензилокси)-7-окса-бицикло[4,1,0]гептан был элюирован вторым. MS (ES+) C13H16O2 треб.: 204, получен.: 205 [M+H]+.
Цис-(1R,3R,6S)-3-(бензилокси)-7-окса-бицикло[4,1,0]гептан: 1H-ЯМР (400 МГц, CDCl3) δ м. д. 7,35-7,27 (м, 5H), 4,56-4,45 (м, 2H), 3,35-3,29 (м, 1H), 3,12-3,09 (м, 2H), 2,37-2,32 (м, 1H), 2,25-2,20 (м, 1H), 1,89-1,68 (м, 3H), 1,49-1,44 (м, 1H).
Транс-(1S,3R,6R)-3-(бензилокси)-7-окса-бицикло[4,1,0]гептан: 1H-ЯМР (400 МГц, CDCl3) δ м. д. 7,35-7,27 (м, 5H), 4,48 (дд, 2H, J=28,0, 12,4 Гц), 3,56-3,52 (м, 1H), 3,19-3,17 (м, 2H), 2,24-2,18 (м, 1H), 2,15-2,07 (м, 1H), 2,00-1,91 (м, 2H), 1,64-1,53 (м, 2H).
Шаг 3: (1R,2R,5R)-5-(бензилокси)-2-((S)-1-фенилэтиламино)циклогексанол
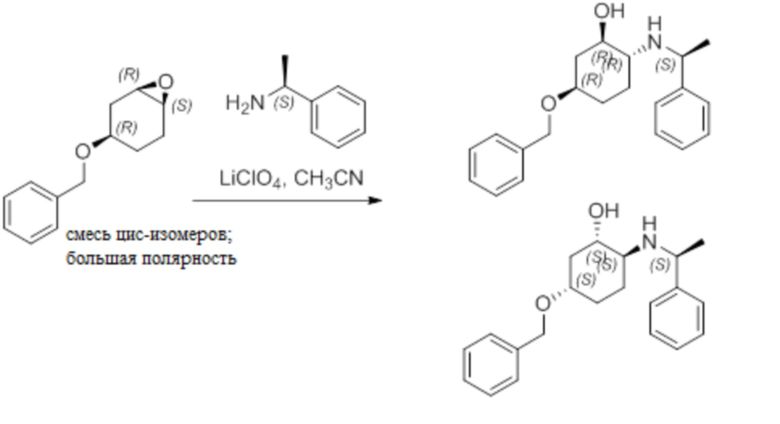
Перхлорат лития (7,27 г, 68,4 ммоль) добавляли к охлажденному на ледяной бане перемешанному раствору (1R,3R,6S)-3-(бензилокси)-7-окса-бицикло[4,1,0]гептана (7,0 г, 34,2 ммоль) в 120 мл 4A-MS сухого ацетонитрила, снимали с бани и добавляли по каплям (с)-1-фенилэтанамин (5,58 г, 46,1 ммоль) в течение 15 мин., после чего раствор перемешивали при температуре 25°C в течение 18 ч. После этого смесь растворяли в 200 мл воды и экстрагировали с использованием этилацетата (200 мл x 3), объединенную органическую фазу сушили над сульфатом натрия, после чего концентрировали и осадок очищали посредством колоночной хроматографии на силикагеле, элюировали с использованием петролейного эфира:этилацетата:триэтиламина=98:0:2~49:49:2 для получения искомого соединения (3,5 г, выход 31%) в виде маслянистой жидкости желтого цвета. (1S,2S,5S)-5-(бензилокси)-2-((S)-1-фенилэтиламино)циклогексанол показал немного меньшую полярность на ТСХ и был элюирован первым. (1R,2R,5R)-5-(бензилокси)-2-((S)-1-фенилэтиламино)циклогексанол был элюирован вторым. MS (ES+) C21H27NO2 треб.: 325, получен.: 326[M+H]+.
(1R,2R,5R)-5-(бензилокси)-2-((S)-1-фенилэтиламино)циклогексанол: 1H-ЯМР (400 МГц, CDCl3) δ м. д. 7,35-7,24 (м, 10H), 4,52 (д, 2H, J=2,0 Гц), 3,97 (q, 1H, J=6,8 Гц), 3,42-3,34 (м, 1H), 3,19-3,12 (м, 1H), 2,39 (дд, 1H, J=12,0, 2,4 Гц), 2,16 (дд, 1H, J=12,0, 3,6 Гц), 2,09-2,00 (м, 2H), 1,65-1,49 (м, 1H), 1,35 (д, 3H, J=6,4 Гц), 1,28-1,15 (м, 2H), 0,90 (qd, 1H, J =13,2, 3,6 Гц).
(1S,2S,5S)-5-(бензилокси)-2-((S)-1-фенилэтиламино)циклогексанол: 1H-ЯМР (400 МГц, CDCl3) δ м. д. 7,36-7,22 (м, 10H), 4,54 (д, 2H, J=3,2 Гц), 3,90 (q, 1H, J=6,4 Гц), 3,44-3,35 (м, 1H), 3,15-3,09 (м, 1H), 2,51-2,45 (м, 1H), 2,43-2,36 (м, 1H), 2,04-1,99 (м, 1H), 1,95-1,90 (м, 1H), 1,47-1,29 (м, 3H), 1,34 (д, 3H, J=6,4 Гц), 0,82 (qd, 1H, J =13,2, 3,2 Гц).
Шаг 4: синтез (1R,2R,4R)-4-(бензилокси)-2-(трет-бутилдиметилсилилокси)-N-((S)-1-фенилэтил)циклогексанамина

Трифторметансульфонат трет-бутилдиметилсилила (13,0 г, 49,5 ммоль) добавляли к охлажденному на ледяной бане перемешиваемому раствору (1R,2R,5R)-5-(бензилокси)-2-((S)-1-фенилэтиламино)циклогексанола (5,4 г, 16,5 ммоль) и триэтиламина (5,0 г, 49,5 ммоль) в 100 мл высушенного дихлорметана. Через 30 мин. полученное промывали насыщенным водным раствором бикарбоната натрия и сушили над сульфатом натрия. Растворитель удаляли и осадок очищали посредством колоночной хроматографии на силикагеле, элюировали с использованием петролейного эфира:этилацетата=100:0~70:30 для получения искомого соединения (5,4 г, выход 71%) в виде маслянистой жидкости желтого цвета. MS (ES+) C27H41NO2Si треб.: 439, получен.: 440 [M+H]+.
Шаг 5: (1R,2R,5R)-5-(бензилокси)-2-((S)-1-фенилэтиламино)циклогексанол
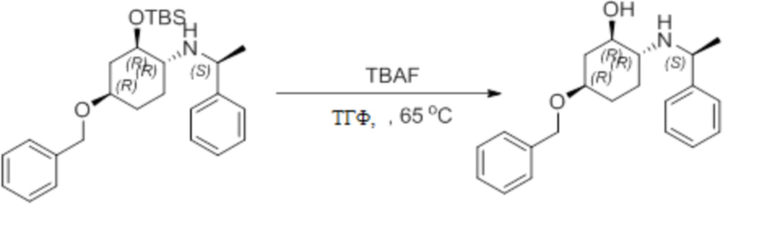
Фторид тетрабутиламмония (2,66 г, 10,2 ммоль) добавляли к перемешиваемому раствору (1R,2R,4R)-4-(бензилокси)-2-(трет-бутилдиметилсилилокси)-N-((S)-1-фенилэтил)циклогексанамина (1,5 г, 3,41 ммоль) в 50 мл сухого оксолана при комнатной температуре. После этого данный раствор перемешивали при температуре 65°C в течение 2 ч. После этого смесь концентрировали под вакуумом, осадок растворяли в 200 мл воды и экстрагировали с использованием этилацетата (200 мл x 3), объединенную органическую фазу промывали водой и насыщенным водным раствором хлорида натрия и сушили над сульфатом натрия. Растворитель удаляли и осадок очищали посредством колоночной хроматографии на силикагеле, элюировали с использованием петролейного эфира:этилацетата=100:0~70:30 для получения искомого соединения (0,75 г, выход 68%) в виде бесцветной маслянистой жидкости. MS (ES+) C21H27NO2 треб.: 325, получен.: 326[M+H]+.
Шаг 6: (1R,3R,4R)-4-аминоциклогексан-1,3-диол
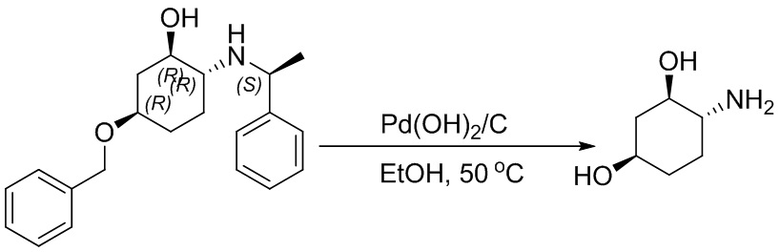
10% гидроксид палладия в активированном угле (697 мг, катализатор) добавляли к раствору (1R,2R,5R)-5-(бензилокси)-2-((S)-1-фенилэтиламино)циклогексанола (650 мг, 1,99 ммоль) в 15 мл этанол при комнатной температуре. После этого данный раствор перемешивали при температуре 50°C в течение 20 ч. в атмосфере водорода. После этого смесь охлаждали и фильтровали через целит, отфильтрованный осадок промывали метанолом:дихлорметаном=1:10, фильтрат концентрировали под вакуумом и осадок растворяли в 20 мл раствора метанола:дихлорметана=1:10, концентрировали, высушивали под высоким вакуумом, после чего охлаждали при температуре -20°C для получения искомого соединения (240 мг, выход 92%) в виде кристаллов белого цвета. MS (ES+) C6H13NO2 треб.: 131, получен.: 132[M+H]+.
(1R,3R,4R)-4-аминоциклогексан-1,3-диол: 1H-ЯМР (400 МГц, 6d-DMSO) δ м. д. 4,62-4,49 (м, 2H), 3,42-3,33 (м, 2H, J=3,2 Гц), 2,93-2,86 (м, 1H), 2,25-2,18 (м, 1H), 1,98-1,92 (м, 1H), 1,72-1,59 (м, 3H), 1,13-1,03 (м, 2H), 0,97-0,90 (м, 1H).
Пример 24. Синтез соединения 232
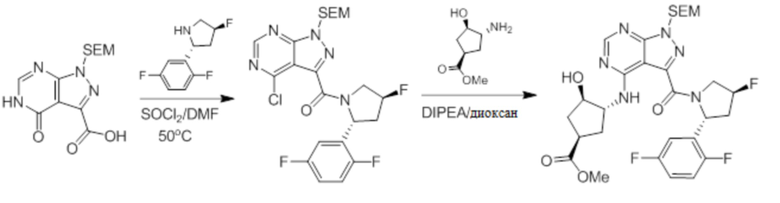
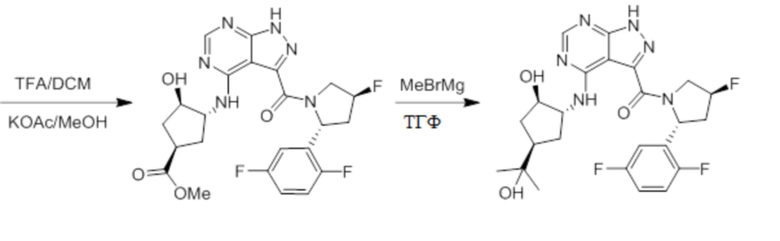
Шаг 1: (4-хлор-1-((2-(триметилсилил)етокси)метил)-1H-пиразоло[3,4-d]пиримидин-3-ил)((2R,4S)-2-(2,5-дифторфенил)-4-фторпирролидин-1-ил)метанон
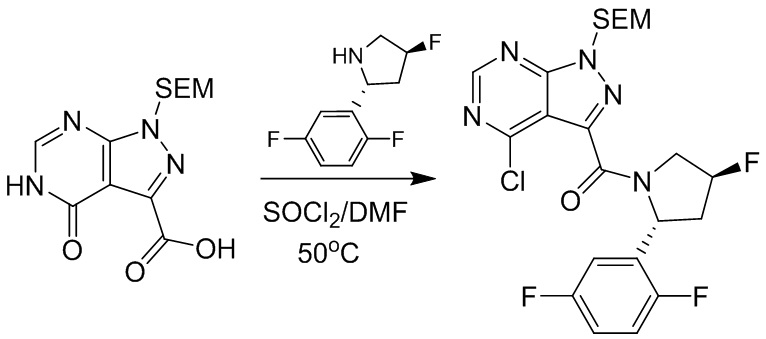
К раствору 4-оксо-1-((2-(триметилсилил)этокси)метил)-4,5-дигидро-1H-пиразоло[3,4-d]пиримидин-3-карбоновой кислоты (1,00 г, 3,22 ммоль, 1,00 экв.) в SOCl2 (164,00 г, 1,38 моль, 428,11 экв.) добавляли DMF (235,49 мг, 3,22 ммоль, 1,00 экв.) при температуре 15 °C. Реакцию нагревали при температуре 50°C в течение 16 часов. ТСХ (PE:EtOAc=3:1, Rf=0,8 и 0,7) показала завершение реакции. Смесь концентрировали. Осадок охлаждали до -10°C и растворяли в DCM (25,00 мл). К реакции добавляли Et3N (1,63 г, 16,10 ммоль, 5,00 экв.) и (2R,4S)-2-(2,5-дифторфенил)-4-фтор-пирролидин (497,40 мг, 2,09 ммоль, 0,65 экв., HCl). Реакцию перемешивали при температуре 0°C в течение 0,2 часа. ТСХ (PE:EtOAc=3:1, Rf=0,38) показала завершение реакции. Раствор промывали солевым раствором (5 мл), сушили над Na2SO4 и концентрировали. Осадок очищали посредством препаративной ТСХ (PE:EtOAc=10:1) для получения (4-хлор-1-((2-(триметилсилил)этокси)метил)-1H-пиразоло[3,4-d]пиримидин-3-ил)((2R,4S)-2-(2,5-дифторфенил)-4-фторпирролидин-1-ил)метанона (400,00 мг, выход: 24,26%) в виде твердого вещества желтого цвета.
Шаг 2: метил(1R,3R,4R)-3-((3-((2R,4S)-2-(2,5-дифторфенил)-4-фторпирролидин-1-карбонил)-1-((2-(триметилсилил)этокси)метил)-1H-пиразоло[3,4-d]пиримидин-4-ил)амино)-4-гидроксициклопентан-1-карбоксилат
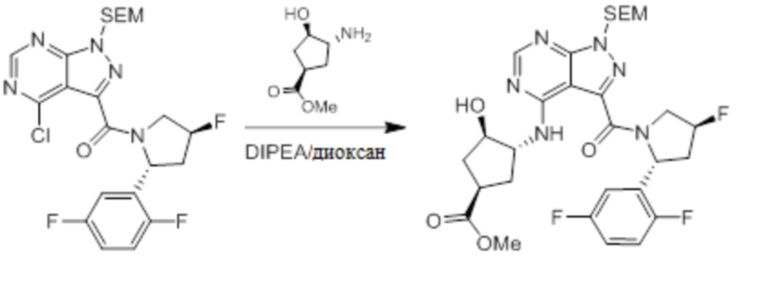
К раствору (4-хлор-1-((2-(триметилсилил)этокси)метил)-1H-пиразоло[3,4-d]пиримидин-3-ил)((2R,4S)-2-(2,5-дифторфенил)-4-фторпирролидин-1-ил)метанона (200,00 мг, 390,63 мкмоль, 1,00 экв.) и метил(1R,3R,4R)-3-амино-4-гидрокси-циклопентанкарбоксилата (80,24 мг, 410,16 мкмоль, 1,05 экв., HCl) в диоксане (10,00 мл) добавляли DIPEA (151,46 мг, 1,17 ммоль, 3,00 экв.). Реакцию нагревали при температуре 90°C в течение 5 часов. ЖХМС показала завершение реакции. Раствор концентрировали. Осадок очищали посредством препаративной ТСХ (PE:EtOAc=2:1) для получения метил (1R,3R,4R)-3-((3-((2R,4S)-2-(2,5-дифторфенил)-4-фторпирролидин-1-карбонил)-1-((2-(триметилсилил)этокси)метил)-1H-пиразоло[3,4-d]пиримидин-4-ил)амино)-4-гидроксициклопентан-1-карбоксилата (120,00 мг, выход: 48,40%) в виде твердого вещества желтого цвета.
Шаг 3: метил (1R,3R,4R)-3-((3-((2R,4S)-2-(2,5-дифторфенил)-4-фторпирролидин-1-карбонил)-1H-пиразоло[3,4-d]пиримидин-4-ил)амино)-4-гидроксициклопентан-1-карбоксилат
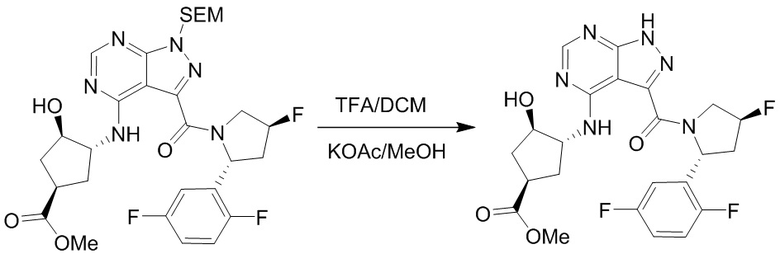
К раствору метил (1R,3R,4R)-3-((3-((2R,4S)-2-(2,5-дифторфенил)-4-фторпирролидин-1-карбонил)-1-((2-(триметилсилил)этокси)метил)-1H-пиразоло[3,4-d]пиримидин-4-ил)амино)-4-гидроксициклопентан-1-карбоксилата (120,00 мг, 189,06 мкмоль, 1,00 экв.) в DCM (3,00 мл) добавляли TFA (3,00 мл) при температуре 15°C. Реакцию перемешивали при температуре 15°C в течение 16 часов. ЖХМС показала, что исходный материал полностью прореагировал. Было обнаружено небольшое количество (1R,3R,4R)-метил 3-((3-((2R,4S)-2-(2,5-дифторфенил)-4-фторпирролидин-1-карбонил)-1-(гидроксиметил)-1H-пиразоло[3,4-d]пиримидин-4-ил)амино)-4-гидроксициклопентанкарбоксилата. Реакцию концентрировали. Остаток растворяли в MeOH (20,00 мл). К реакции добавляли KOAc (185,54 мг, 1,89 ммоль, 10,00 экв.). Реакцию нагревали при температуре 50°C в течение 16 часов. ЖХМС показала завершение реакции. Раствор концентрировали. Осадок растворяли в EtOAc (20 мл), промывали солевым раствором (10 мл), сушили над Na2SO4 и концентрировали с получением метил (1R,3R,4R)-3-((3-((2R,4S)-2-(2,5-дифторфенил)-4-фторпирролидин-1-карбонил)-1H-пиразоло[3,4-d]пиримидин-4-ил)амино)-4-гидроксициклопентан-1-карбоксилата (80,00 мг, в неочищенном виде) в виде твердого вещества красного цвета, который использовался на следующем шаге без очистки.
Шаг 4: ((2R,4S)-2-(2,5-дифторфенил)-4-фторпирролидин-1-ил)(4-(((1R,2R,4R)-2-гидрокси-4-(2-гидроксипропан-2-ил)циклопентил)амино)-1H-пиразоло[3,4-d]пиримидин-3-ил)метанон
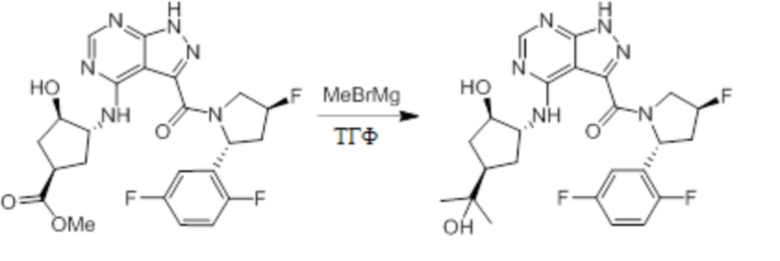
К раствору метил (1R,3R,4R)-3-((3-((2R,4S)-2-(2,5-дифторфенил)-4-фторпирролидин-1-карбонил)-1H-пиразоло[3,4-d]пиримидин-4-ил)амино)-4-гидроксициклопентан-1-карбоксилата (80,00 мг, 158,59 мкмоль, 1,00 экв.) в ТГФ (20,00 мл) добавляли MeMgBr (3 M, 1,59 мл, 30,00 экв.) при температуре -70°C. Реакцию медленно нагревали до 15°C и перемешивали в течение 2 часов. ТСХ (EtOAc, Rf=0,24) и ЖХМС показали завершение реакции. Раствор нейтрализовывали 1Н водн. HCl до значения pH=7. Реакционную смесь концентрировали. Осадок очищали посредством нейтральной препаративной ВЭЖХ. для получения ((2R,4S)-2-(2,5-дифторфенил)-4-фторпирролидин-1-ил)(4-(((1R,2R,4R)-2-гидрокси-4-(2-гидроксипропан-2-ил)циклопентил)амино)-1H-пиразоло[3,4-d]пиримидин-3-ил)метанона (17,40 мг, выход: 21,75%) в виде твердого вещества желтого цвета.
Для данного соединения и указанного ниже соединения 156 условия ЖХ-МС были следующими: (подвижная фаза: от 99% [вода+0,375‰ об./об. TFA] и 1% [CH3CN+0,188‰ об./об. TFA], в данных условиях 0,4 мин., после чего условия изменяли на 10% [вода+0,375‰ об./об. TFA] и 90% [CH3CN+0,188‰ об./об. TFA] в течение 3,0 мин., после чего условия изменяли на 100% [CH3CN+0,188‰ об./об. TFA] в течение 0,45 мин., в конце условия изменяли на 99% [вода+0,375‰ об./об. TFA] и 1% [CH3CN+0,188‰ об./об. TFA] в течение 0,01 мин., после чего в данных условиях 0,64 мин. Скорость потока все время составляла 0,8 мл·мин.-1.) Чистота составляла 99,870%
Пример 25. Синтез соединения 229
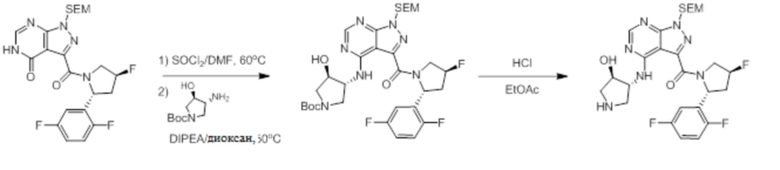
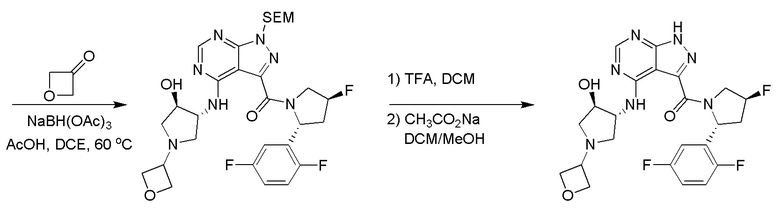
Шаг 1: трет-бутил (3R,4R)-3-((3-((2R,4S)-2-(2,5-дифторфенил)-4-фторпирролидин-1-карбонил)-1-((2-(триметилсилил)этокси)метил)-1H-пиразоло[3,4-d]пиримидин-4-ил)амино)-4-гидроксипирролидин-1-карбоксилат
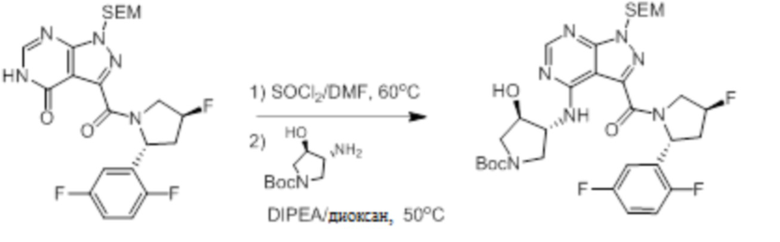
Раствор 3-((2R,4S)-2-(2,5-дифторфенил)-4-фторпирролидин-1-карбонил)-1-((2-(триметилсилил)этокси)метил)-1,5-дигидро-4H-пиразоло[3,4-d]пиримидин-4-она (0,085 г, 0,17 ммоль, 1 экв.) перемешивали с тионилхлоридом (0,031 мл, 0,43 ммоль, 2,5 экв.) и несколькими каплями DMF в DCM (0,7 мл) при температуре 50°C в течение 3 часов. ЖХМС показала полное реагирование SM с образованием хлор-гетероциклического промежуточного вещества. Реакционную смесь охлаждали на льду и добавляли диоксан (0,7 мл), после чего добавляли DIEA (0,21 мл, 1,21 ммоль, 7 экв.) и трет-бутил(3R,4R)-3-амино-4-гидроксипирролидин-1-карбоксилат (0,05g, 0,26 ммоль, 1,5 экв.). После этого реакционную смесь перемешивали при температуре 70°C в течение 3 ч. ЖХМС показала завершение реакции. Реакционную смесь после чего разбавляли DCM и пр омывали насыщенным водным раствором бикарбоната натрия. Объединенные органические слои промывали насыщенным солевым раствором, сушили над Na2SO4 и концентрировали под вакуумом. Осадок очищали посредством колоночной хроматографии на силикагеле (гексаны/EtOAc) для получения продукта трет-бутил (3R,4R)-3-((3-((2R,4S)-2-(2,5-дифторфенил)-4-фторпирролидин-1-карбонил)-1-((2-(триметилсилил)этокси)метил)-1H-пиразоло[3,4-d]пиримидин-4-ил)амино)-4-гидроксипирролидин-1-карбоксилата (0,084 г, 72%).
Шаг 2: ((2R,4S)-2-(2,5-дифторфенил)-4-фторпирролидин-1-ил)(4-(((3R,4R)-4-гидроксипирролидин-3-ил)амино)-1-((2-(триметилсилил)этокси)метил)-1H-пиразоло[3,4-d]пиримидин-3-ил)метанон
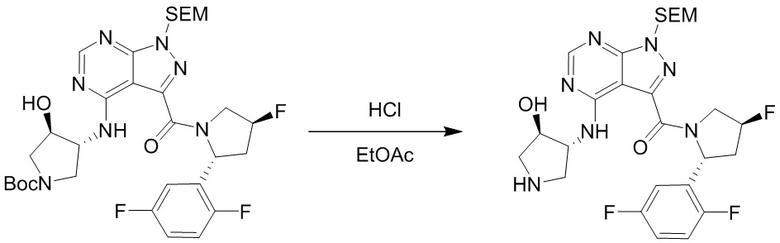
Раствор (3R,4R)-3-((3-((2R,4S)-2-(2,5-дифторфенил)-4-фторпирролидин-1-карбонил)-1-((2-(триметилсилил)этокси)метил)-1H-пиразоло[3,4-d]пиримидин-4-ил)амино)-4-гидроксипирролидин-1-карбоксилата (0,084 г, 0,12 ммоль, 1 экв.) в EtOAc (1,25 мл) обрабатывали HCl в диоксане (4M, 0,9 мл, 3,72 ммоль, 30 экв.). После перемешивания при температуре 23°C в течение 4 часов ЖХМС показала завершение реакции. Реакционную смесь разбавляли EtOAc и промывали насыщенным водным раствором бикарбоната натрия. Объединенные органические слои промывали насыщенным солевым раствором, сушили над Na2SO4 и концентрировали под вакуумом. Неочищенный продукт ((2R,4S)-2-(2,5-дифторфенил)-4-фторпирролидин-1-ил)(4-(((3R,4R)-4-гидроксипирролидин-3-ил)амино)-1-((2-(триметилсилил)этокси)метил)-1H-пиразоло[3,4-d]пиримидин-3-ил)метанон использовали на следующем шаге без дополнительной очистки.
Шаг 3: ((2R,4S)-2-(2,5-дифторфенил)-4-фторпирролидин-1-ил)(4-(((3R,4R)-4-гидрокси-1-(оксэтан-3-ил)пирролидин-3-ил)амино)-1-((2-(триметилсилил)этокси)метил)-1H-пиразоло[3,4-d]пиримидин-3-ил)метанон
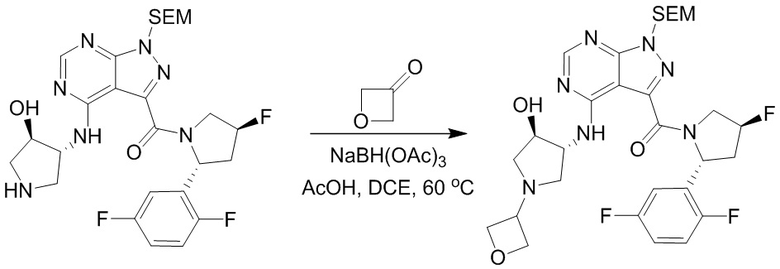
К раствору ((2R,4S)-2-(2,5-дифторфенил)-4-фторпирролидин-1-ил)(4-(((3R,4R)-4-гидроксипирролидин-3-ил)амино)-1-((2-(триметилсилил)этокси)метил)-1H-пиразоло[3,4-d]пиримидин-3-ил)метанона (0,036 г, 0,062 ммоль, 1 экв.) в DCE (0,5 мл) добавляли несколько капель уксусной кислоты (2 μл, 0,031 ммоль, 0,5 экв.), после чего добавляли оксэтан-3-он (0,015 мл, 0,21 ммоль, 3,3 экв.) и реакционную смесь нагревали при температуре 60°C в течение 2 часов. Добавляли триацетоксиборогидрид натрия (0,033 г, 0,16 ммоль, 2,5 экв.) и раствор перемешивали при температуре 23°C в течение 24 часов. ЖХМС показала завершение реакции. Реакционную смесь разбавляли EtOAc и промывали насыщенным водным раствором бикарбоната натрия. Объединенные органические слои промывали насыщенным солевым раствором, сушили над Na2SO4 и концентрировали под вакуумом. После этого осадок очищали посредством колоночной хроматографии на силикагеле (DCM/MeOH) для отделения продукта ((2R,4S)-2-(2,5-дифторфенил)-4-фторпирролидин-1-ил)(4-(((3R,4R)-4-гидрокси-1-(оксэтан-3-ил)пирролидин-3-ил)амино)-1-((2-(триметилсилил)этокси)метил)-1H-пиразоло[3,4-d]пиримидин-3-ил)метанона (0,030 г, 75%).
Шаг 4: ((2R,4S)-2-(2,5-дифторфенил)-4-фторпирролидин-1-ил)(4-(((3R,4R)-4-гидрокси-1-(оксэтан-3-ил)пирролидин-3-ил)амино)-1H-пиразоло[3,4-d]пиримидин-3-ил)метанон
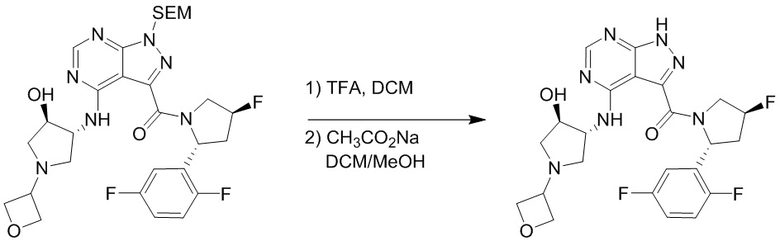
Раствор ((2R,4S)-2-(2,5-дифторфенил)-4-фторпирролидин-1-ил)(4-(((3R,4R)-4-гидрокси-1-(оксэтан-3-ил)пирролидин-3-ил)амино)-1-((2-(триметилсилил)этокси)метил)-1H-пиразоло[3,4-d]пиримидин-3-ил)метанона (0,060 г, 0,095 ммоль, 1 экв.) в DCM (1 мл) обрабатывали TFA (0,73 мл, 9,5 ммоль, 100 экв.) в течение 16 часов. Реакционную смесь разбавляли DCM и промывали насыщенным водным раствором бикарбоната натрия. Объединенные органические слои промывали насыщенным солевым раствором, сушили над Na2SO4 и концентрировали под вакуумом. К промежуточному соединению в DCM/MeOH (1/1, 1 мл) добавляли ацетат натрия (0,016 г, 0,19 ммоль, 2 экв.) и реакцию перемешивали при температуре 23°C в течение 2 часов. Реакционную смесь разбавляли DCM и после чего промывали насыщенным водным раствором бикарбоната натрия. Объединенные органические слои промывали насыщенным солевым раствором, сушили над Na2SO4 и концентрировали под вакуумом. Осадок затем очищали сначала посредством колоночной хроматографии на силикагеле (DCM/MeOH, содержащий 10% NH4OH) и после чего посредством препаративной-ТСХ для отделения продукта ((2R,4S)-2-(2,5-дифторфенил)-4-фторпирролидин-1-ил)(4-(((3R,4R)-4-гидрокси-1-(оксэтан-3-ил)пирролидин-3-ил)амино)-1H-пиразоло[3,4-d]пиримидин-3-ил)метанона (0,034 г, 70%).
Пример 26. Синтез соединения 156
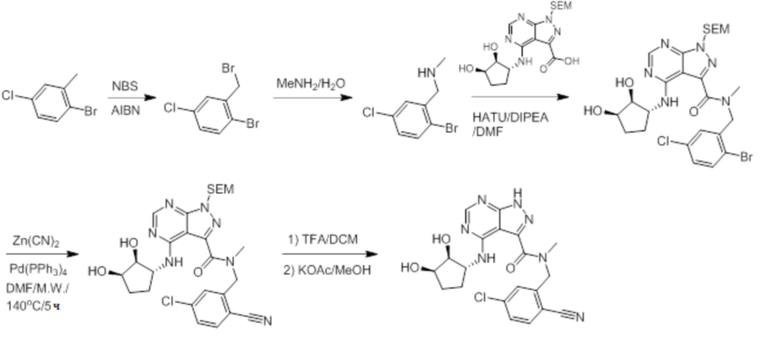
Шаг 1: 1-бром-2-(бромметил)-4-хлорбензол
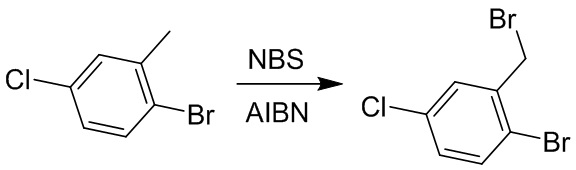
Раствор 1-бром-4-хлор-2-метилбензол (3,00 г, 14,60 ммоль, 1,00 экв.), NBS (2,34 г, 13,14 ммоль, 0,90 экв.) и AIBN (239,75 мг, 1,46 ммоль, 0,10 экв.) в CCl4 (20,00 мл) перемешивали при температуре 90°C в течение 12 часов. Раствор концентрировали под вакуумом для получения неочищенного продукта 1-бром-2-(бромметил)-4-хлорбензола (5,40 г, в неочищенном виде) в виде твердого вещества желтого цвета, которое использовали на следующей стадии непосредственно, без дополнительной очистки.
Шаг 2: 1-(2-бром-5-хлорфенил)-N-метилметанамин
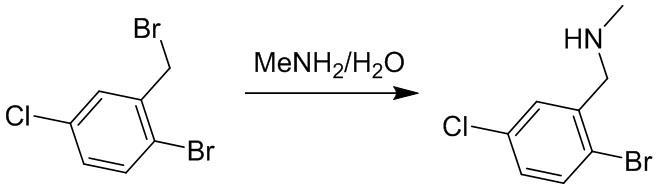
Раствор 1-бром-2-(бромметил)-4-хлорбензола (5,40 г, 18,99 ммоль, 1,00 экв.) в MeNH2/H2O (30,00 мл) перемешивали при температуре 25°C в течение 15 часов. После завершения реакции продукт экстрагировали с использованием EtOAc (50 мл*3) и объединенные органические слои концентрировали под вакуумом для получения неочищенного продукта. Неочищенный продукт очищали посредством колоночной хроматографии на силикагеле (PE:EtOAc=от 5:1 до 1:1) для получения 1-(2-бром-5-хлорфенил)-N-метилметанамина (1,00 г, выход: 22,45%) в виде твердого вещества грязно-белого цвета. 1H-ЯМР (400 МГц, CDCl3) δ м. д. 7,45 (д, 1H, J=8,8 Гц), 7,40-7,39 (м, 1H), 7,10 (дд, 1H, J=8,4, 2,4 Гц), 3,79 (с, 2H), 2,46 (с, 3H).
Шаг 3: N-(2-бром-5-хлорбензил)-4-(((1R,2S,3R)-2,3-дигидроксициклопентил)амино)-N-метил-1-((2-(триметилсилил)этокси)метил)-1H-пиразоло[3,4-d]пиримидин-3-карбоксамид

К раствору 1-(2-бром-5-хлорфенил)-N-метилметанамина (500,00 мг, 1,22 ммоль, 1,00 экв.) и 4-(((1R,2S,3R)-2,3-дигидроксициклопентил)амино)-1-((2-(триметилсилил)этокси)метил)-1H-пиразоло[3,4-d]пиримидин-3-карбоновой кислоты (314,98 мг, 1,34 ммоль, 1,10 экв.) в DMF (5,00 мл) добавляли DIPEA (315,35 мг, 2,44 ммоль, 2,00 экв.) и HATU (556,66 мг, 1,46 ммоль, 1,20 экв.), полученную смесь перемешивали при температуре 25°C в течение 15 часов. После завершения реакции добавляли H2O (20 мл), продукт экстрагировали с использованием EtOAc (20 мл*3) и объединенные органические слои концентрировали под вакуумом для получения неочищенного продукта. Неочищенный продукт очищали посредством препаративной ТСХ (EtOAc) для получения N-(2-бром-5-хлорбензил)-4-(((1R,2S,3R)-2,3-дигидроксициклопентил)амино)-N-метил-1-((2-(триметилсилил)этокси)метил)-1H-пиразоло[3,4-d]пиримидин-3-карбоксамида (520,00 мг, выход: 81,20%) в виде маслянистой жидкости красного цвета. 1H-ЯМР (400 МГц, CDCl3) δ м. д. 9,77 (уш. с., 0,5H), 9,49 (уш. с., 0,5H), 8,45 (д, 1H, J=11,4 Гц), 7,63 (дд, 1H, J=8,4, 4,4 Гц), 7,33-7,31 (м, 1H), 7,26-7,25 (м, 1H), 5,87 (с, 1H), 5,67 (с, 1H), 5,46 (с, 1H), 4,98 (с, 1H), 4,45-4,44 (м, 1H), 4,31-4,29 (м, 1H), 4,03-4,02 (м, 1H), 3,79 (т, 1H, J=8,0 Гц), 3,71 (с, 1,5H), 3,55 (т, 1H, J=8,0 Гц), 3,29 (с, 1,5H), 3,16 (д, 1H, J=6,0 Гц), 2,65-2,61 (м, 1H), 2,05-1,83 (м, 2H), 1,05 (т, 1H, J=8,4 Гц), 0,90 (т, 1H, J=8,0 Гц), 0,07 (с, 4,5H), 0,00 (с, 4,5H).
Шаг 4: N-(5-хлор-2-цианобензил)-4-(((1R,2S,3R)-2,3-дигидроксициклопентил)амино)-N-метил-1-((2-(триметилсилил)этокси)метил)-1H-пиразоло[3,4-d]пиримидин-3-карбоксамид
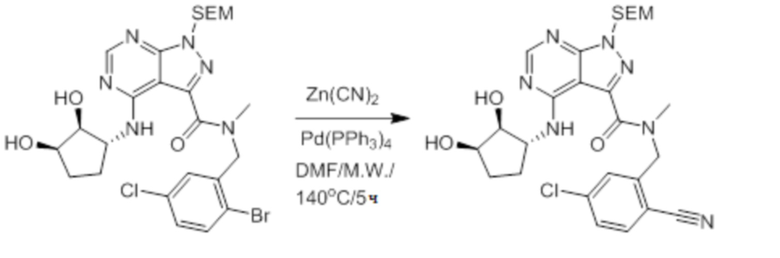
Смесь N-(2-бром-5-хлорбензил)-4-(((1R,2S,3R)-2,3-дигидроксициклопентил)амино)-N-метил-1-((2-(триметилсилил)этокси)метил)-1H-пиразоло[3,4-d]пиримидин-3-карбоксамида (420,00 мг, 670,91 мкмоль, 1,00 экв.), Zn(CN)2 (630,22 мг, 5,37 ммоль, 340,66 мкл, 8,00 экв.) и Pd(PPh3)4 (77,53 мг, 67,09 мкмоль, 0,10 экв.) растворяли в DMF (3,00 мл) в запечатанной пробирке, облучали микроволновым излучением при температуре 140°C в течение 3 ч. Через 3 часа ЖХМС показала неполное реагирование исходного материала, после чего добавляли еще Zn(CN)2 (2 экв.) и Pd(PPh3)4 (0,1 экв.), облучали микроволновым излучением при температуре 150°C в течение еще 2 часов. После завершения реакции добавляли H2O (15 мл) и продукт экстрагировали с использованием EtOAC (20 мл*3). Объединенные органические слои сушили над безводным Na2SO4 и концентрировали с получением неочищенного продукта. Неочищенный продукт очищали посредством препаративной ТСХ (EtOAc) для получения N-(5-хлор-2-цианобензил)-4-(((1R,2S,3R)-2,3-дигидроксициклопентил)амино)-N-метил-1-((2-(триметилсилил)этокси)метил)-1H-пиразоло[3,4-d]пиримидин-3-карбоксамида (520,00 мг, в неочищенном виде, включая PPh3O) получали в виде маслянистой жидкости желтого цвета. 1H-ЯМР (400 МГц, CDCl3) δ м. д. 9,72 (уш. с., 0,5H), 9,44 (уш. с., 0,5H), 8,45 (д, 1H, J=6,4 Гц), 7,79-7,54 (м, 3H), 5,86 (с, 1H), 5,70 (с, 1H), 5,67 (с, 1H), 5,11 (с, 1H), 4,45-4,44 (м, 1H), 4,30-4,29 (м, 1H), 4,03-4,02 (м, 1H), 3,81-3,76 (м, 2,5H), 3,59 (т, 1H, J=8,0 Гц), 3,32 (с, 1,5H), 3,18-3,16 (м, 1H), 2,19-1,86 (м, 3H), 1,04 (т, 1H, J=8,0 Гц), 0,92 (т, 1H, J=8,0 Гц), 0,06 (с, 4,5H), 0,00 (с, 4,5H).
Шаг 5: N-(5-хлор-2-цианобензил)-4-(((1R,2S,3R)-2,3-дигидроксициклопентил)амино)-N-метил-1H-пиразоло[3,4-d]пиримидин-3-карбоксамид
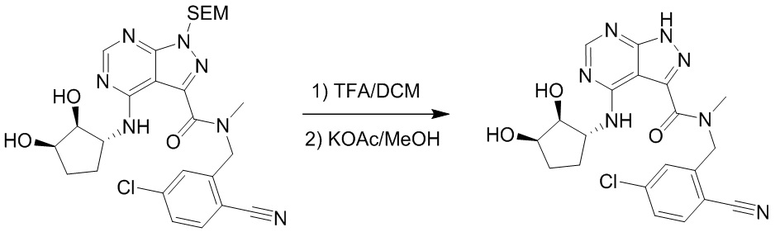
К смеси растворителей TFA (10,00 мл) и DCM (10,00 мл) добавляли N-(5-хлор-2-цианобензил)-4-(((1R,2S,3R)-2,3-дигидроксициклопентил)амино)-N-метил-1-((2-(триметилсилил)этокси)метил)-1H-пиразоло[3,4-d]пиримидин-3-карбоксамид (520,00 мг, 908,88 мкмоль, 1,00 экв.), полученную смесь перемешивали при температуре 25°C в течение 1 часа. Растворитель испаряли с использованием N2 для получения неочищенного продукта. Неочищенный продукт растворяли в MeOH (15,00 мл), доводили до значения pH=7-8 посредством NaHCO3 и добавляли KOAc (178,39 мг, 1,82 ммоль, 2,00 экв.), смесь перемешивали при температуре 50°C в течение 2 часов. После завершения реакции смесь концентрировали под вакуумом для получения неочищенного продукта который растворяли в EtOAc (50 мл) и промывали H2O (15 мл*3). Органический слой концентрировали под вакуумом для получения неочищенного продукта который очищали посредством кислотной препаративной ВЭЖХ (TFA) для получения N-(5-хлор-2-цианобензил)-4-(((1R,2S,3R)-2,3-дигидроксициклопентил)амино)-N-метил-1H-пиразоло[3,4-d]пиримидин-3-карбоксамида (171,00 мг, выход: 33,85%, TFA) в виде твердого вещества белого цвета.
Пример 27. Синтез соединения 226
Шаг 1: Метил-(1R,3R,4R)-3-((трет-бутоксикарбонил)амино)-4-((трет-бутилдифенилсилил)окси) циклопентан-1-карбоксилат
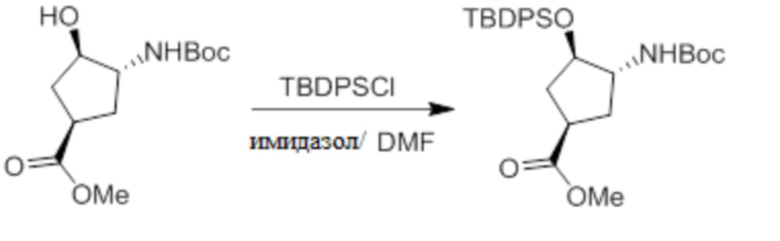
К раствору метил-(1R,3R,4R)-3-((трет-бутоксикарбонил)амино)-4-гидроксициклопентан-1-карбоксилата (1,50 г, 5,78 ммоль, 1,00 экв.) и имидазола (590,74 мг, 8,67 ммоль, 1,50 экв.) в DMF (10,00 мл) добавляли TBDPSCl (1,67 г, 6,07 ммоль, 1,05 экв.) при 0 °C. Реакцию перемешивали при температуре 15°C в течение 16 часов. ТСХ (PE:EtOAc=5:1, Rf=0,43) показала завершение реакции. Раствор вливали в воду (20 мл) и экстрагировали с использованием EtOAc (10 мл * 3). Органический слой высушивали над Na2SO4 и концентрировали. Осадок перекристаллизовывали из PE (1 мл) для получения метил (1R,3R,4R)-3-((трет-бутоксикарбонил)амино)-4-((трет-бутилдифенилсилил)окси)циклопентан-1-карбоксилата (2,80 г, выход: 97,40%) в виде твердого вещества белого цвета. 1H-ЯМР (400 МГц, CDCl3) δ м. д. 7,69-7,64 (м, 4H), 7,43-7,37 (м, 6H), 4,10 (уш. с., 1H), 3,92-3,97 (м, 2H), 3,66 (с, 3H), 2,75-2,71 (м, 1H), 2,46-2,43 (м, 1H), 2,01-1,95 (м, 2H), 1,65-1,61 (м, 1H), 1,40 (с, 9H), 1,05 (с, 9H).
Шаг 2: трет-бутил((1R,2R,4R)-2-((трет-бутилдифенилсилил)окси)-4-(гидроксиметил)циклопентил) карбамат

К раствору метил (1R,3R,4R)-3-((трет-бутоксикарбонил)амино)-4-((трет-бутилдифенилсилил)окси)циклопентан-1-карбоксилата (2,80 г, 5,63 ммоль, 1,00 экв.) в ТГФ (30,00 мл) добавляли LiAlH4 (427,32 мг, 11,26 ммоль, 2,00 экв.) при температуре -30 °C. Реакцию медленно нагревали до 15°C и перемешивали в течение 2 часов. ТСХ (PE:EtOAc=3:1, Rf=0,24) показала завершение реакции. Реакцию гасили 0,43 мл H2O и 0,43 мл 10% водн. NaOH при 0 °C. Смесь фильтровали и фильтрат концентрировали. Осадок промывали PE (5 мл). Твердую фракцию собирали. Получали трет-бутил((1R,2R,4R)-2-((трет-бутилдифенилсилил)окси)-4-(гидроксиметил)циклопентил)карбамат (2,30 г, выход: 86,98%) в виде твердого вещества белого цвета. 1H-ЯМР (400 МГц, CDCl3) δ м. д. 7,69-7,65 (м, 4H), 7,43-7,38 (м, 6H), 4,11-4,10 (м, 1H), 3,89 (уш. с., 2H), 3,54 (уш. с., 2H), 2,14-2,10 (м, 1H), 1,97-1,89 (м, 2H), 1,62-1,58 (м, 1H), 1,39 (с, 9H), 1,06 (с, 9H).
Шаг 3: трет-бутил((1R,2R,4R)-2-((трет-бутилдифенилсилил)окси)-4-формилциклопентил)карбамат
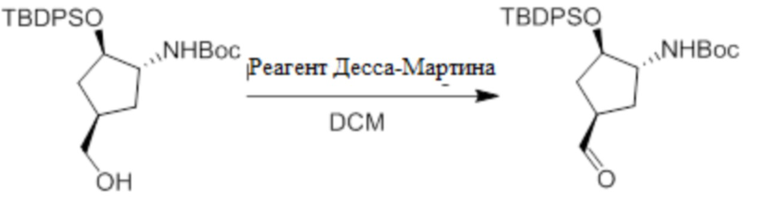
К раствору трет-бутил((1R,2R,4R)-2-((трет-бутилдифенилсилил)окси)-4-(гидроксиметил)циклопентил)карбамата (2,30 г, 4,90 ммоль, 1,00 экв.) в DCM (50,00 мл) добавляли периодинан Десса-Мартина (3,12 г, 7,35 ммоль, 1,50 экв.) при 0 °C. Реакцию перемешивали при температуре 15°C в течение 16 часов. ТСХ (PE:EtOAc=3:1, Rf=0,7) показала завершение реакции. Реакцию гасили насыщ. водн. NaHCO3 (30 мл) при 0°C и экстрагировали с использованием DCM (30 мл). Органический слой высушивали над Na2SO4 и концентрировали. Осадок очищали посредством колоночной хроматографии на силикагеле (PE:EtOAc=30:1). Получали трет-бутил((1R,2R,4R)-2-((трет-бутилдифенилсилил)окси)-4-формилциклопентил)карбамат (900,00 мг, выход: 39,27%) в виде маслянистой жидкости желтого цвета. 1H-ЯМР (400 МГц, CDCl3) δ м. д. 9,55 (с, 1H), 7,61-7,56 (м, 4H), 7,37-7,31 (м, 6H), 4,08 (д, 1H, J=7,2 Гц), 3,93-3,89 (м, 1H), 3,79 (уш. с., 1H), 2,62-2,55 (м, 1H), 2,44-2,40 (м, 1H), 1,86-1,83 (м, 2H), 1,57-1,52 (м, 1H), 1,34 (с, 9H), 0,97 (с, 9H).
Шаг 4: трет-бутил((1R,2R,4R)-2-((трет-бутилдифенилсилил)окси)-4-(дифторметил)циклопентил) карбамат
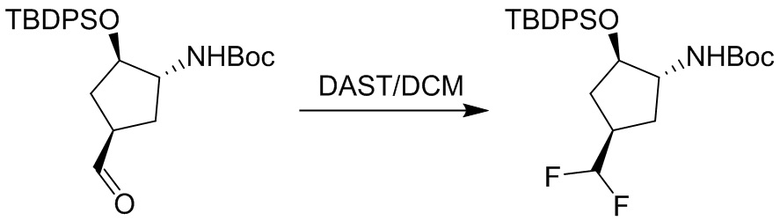
К раствору трет-бутил((1R,2R,4R)-2-((трет-бутилдифенилсилил)окси)-4-формилциклопентил)карбамата (900,00 мг, 1,92 ммоль, 1,00 экв.) в DCM (50,00 мл) добавляли DAST (928,45 мг, 5,76 ммоль, 3,00 экв.) при 0 °C. Реакцию перемешивали при температуре 15°C в течение 5 часов. ТСХ (PE:EtOAc=3:1, Rf=0,6) показала завершение реакции. Раствор гасили насыщ. водн. NaHCO3 (20 мл) и экстрагировали с использованием DCM (20 мл). Органический слой высушивали над Na2SO4 и концентрировали. Осадок очищали посредством колоночной хроматографии на силикагеле (PE:EtOAc=30:1~20:1). Получали трет-бутил((1R,2R,4R)-2-((трет-бутилдифенилсилил)окси)-4-(дифторметил)циклопентил)карбамат (250,00 мг, выход: 26,59%) в виде маслянистой жидкости желтого цвета. 1H-ЯМР (400 МГц, CDCl3) δ м. д. 7,62-7,56 (м, 4H), 7,34-7,31 (м, 6H), 5,76-5,43 (м, 1H), 3,88-3,82 (м, 2H), 2,30-2,24 (м, 1H), 2,07-2,04 (м, 1H), 1,80-1,75 (м, 1H), 1,65-1,57 (м, 1H), 1,33 (с, 9H), 0,99 (с, 9H).
Шаг 5: (1R,2R,4R)-2-((трет-бутилдифенилсилил)окси)-4-(дифторметил)циклопентан-1-амин
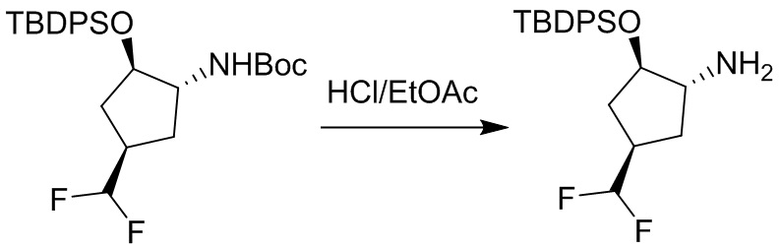
К раствору трет-бутил((1R,2R,4R)-2-((трет-бутилдифенилсилил)окси)-4-(дифторметил)циклопентил)карбамата (50,00 мг, 102,11 мкмоль, 1,00 экв.) в EtOAc (2,00 мл) добавляли HCl/EtOAc (10,00 мл, 4 M) при температуре 15 °C. Реакцию перемешивали при температуре 15°C в течение 1 часа. ТСХ (PE:EtOAc=3:1, Rf=0,05) показала завершение реакции. Растворитель до высушивания продували N2. Осадок не подвергали дальнейшей очистке. Получали (1R,2R,4R)-2-((трет-бутилдифенилсилил)окси)-4-(дифторметил)циклопентан-1-амин (40,00 мг, выход: 91,95%, HCl) в виде маслянистой жидкости желтого цвета. 1H-ЯМР (400 МГц, CDCl3) δ м. д. 7,70-7,68 (м, 4H), 7,49-7,43 (м, 6H), 5,81 (тд, 1H, J=57,2, 4,8 Гц), 4,25 (дд, 1H, J=12,0, 5,6 Гц), 3,50 (дд, 1H, J=13,2, 6,4 Гц), 2,22-2,19 (м, 1H), 1,80-1,74 (м, 2H), 1,66-1,63 (м, 1H), 1,08 (с, 9H).
Шаг 6: (4-(((1R,2R,4R)-2-((трет-бутилдифенилсилил)окси)-4-(дифторметил)циклопентил)амино)-1-((2-(триметилсилил)этокси)метил)-1H-пиразоло[3,4-d]пиримидин-3-ил)((2R,4S)-2-(2,5-дифторфенил)-4-фторпирролидин-1-ил)метанон
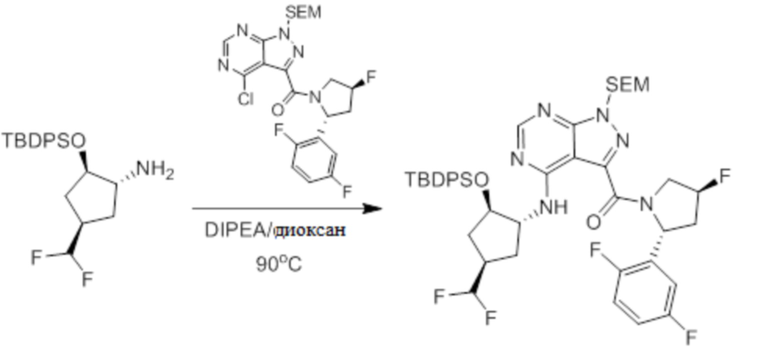
К раствору (1R,2R,4R)-2-((трет-бутилдифенилсилил)окси)-4-(дифторметил)циклопентан-1-амина (40,00 мг, 93,89 мкмоль, 1,00 экв., HCl) и (4-хлор-1-((2-(триметилсилил)этокси)метил)-1H-пиразоло[3,4-d]пиримидин-3-ил)((2R,4S)-2-(2,5-дифторфенил)-4-фторпирролидин-1-ил)метанона (48,07 мг, 93,89 мкмоль, 1,00 экв.) в диоксане (10,00 мл) добавляли DIPEA (60,67 мг, 469,45 мкмоль, 5,00 экв.). Реакцию нагревали при температуре 90°C в течение 0,5 часа. ЖХМС показала завершение реакции. Раствор концентрировали. Осадок очищали посредством препаративной ТСХ (PE:EtOAc=3:1). Получали (4-(((1R,2R,4R)-2-((трет-бутилдифенилсилил)окси)-4-(дифторметил)циклопентил)амино)-1-((2-(триметилсилил)этокси)метил)-1H-пиразоло[3,4-d]пиримидин-3-ил)((2R,4S)-2-(2,5-дифторфенил)-4-фторпирролидин-1-ил)метанон (38,00 мг, выход: 46,78%) в виде маслянистой жидкости желтого цвета.
Шаг 7: (4-(((1R,2R,4R)-4-(дифторметил)-2-гидроксициклопентил)амино)-1H-пиразоло[3,4-d]пиримидин-3-ил)((2R,4S)-2-(2,5-дифторфенил)-4-фторпирролидин-1-ил)метанон (соединение 226)
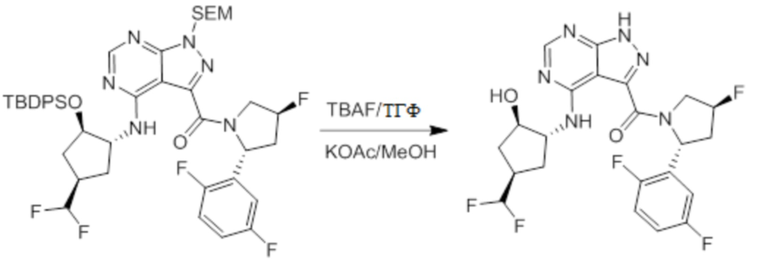
Раствор (4-(((1R,2R,4R)-2-((трет-бутилдифенилсилил)окси)-4-(дифторметил)циклопентил)амино)-1-((2-(триметилсилил)этокси)метил)-1H-пиразоло[3,4-d]пиримидин-3-ил)((2R,4S)-2-(2,5-дифторфенил)-4-фторпирролидин-1-ил)метанона (38,00 мг, 43,93 мкмоль, 1,00 экв.) в TBAF/THF (5,00 мл) нагревали при температуре 50°C в течение 2 часов. ЖХМС показала наличие остатков (4-(((1R,2R,4R)-4-(дифторметил)-2-гидроксициклопентил)амино)-1-(гидроксиметил)-1H-пиразоло[3,4-d]пиримидин-3-ил)((2R,4S)-2-(2,5-дифторфенил)-4-фторпирролидин-1-ил)метанона. Реакционную смесь концентрировали. Осадок растворяли в EtOAc (20 мл) и промывали солевым раствором (10 мл*2). Органический слой концентрировали и растворяли в MeOH (20,00 мл). К реакции добавляли KOAc (21,56 мг, 219,65 мкмоль, 5,00 экв.). Реакцию нагревали при температуре 50°C в течение 16 часов. ЖХМС показала завершение реакции. Раствор концентрировали. Осадок очищали посредством препаративной ВЭЖХ (система MeOH/TFA). Получали (4-(((1R,2R,4R)-4-(дифторметил)-2-гидроксициклопентил)амино)-1H-пиразоло[3,4-d]пиримидин-3-ил)((2R,4S)-2-(2,5-дифторфенил)-4-фторпирролидин-1-ил)метанон (13,50 мг, выход: 50,34%, TFA) в виде твердого вещества желтого цвета.
Пример 28. Синтез других соединений
Дополнительные соединения по изобретению синтезировали с использованием аналогичных методов, описанных в приведенных выше примерах. В приведенной ниже таблице показан конкретный пример («Пример»), на котором основывается синтез каждого соединения («Соед.»), а также соответствующий аминоспирт и амин, которые использовались для синтеза каждого конкретного соединения.
Таблица 1. Протокол и промежуточные соединения, используемые для синтеза синтетических образцов.
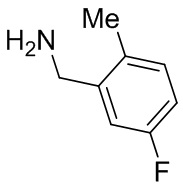
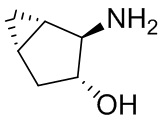
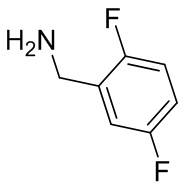
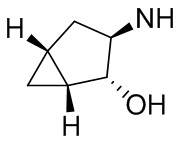
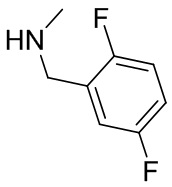
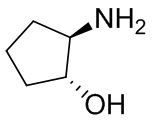
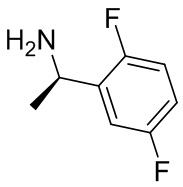
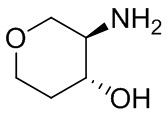
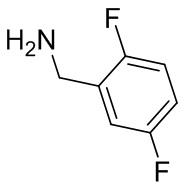
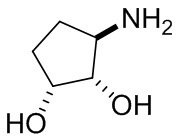
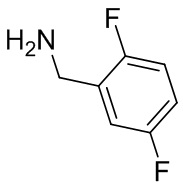
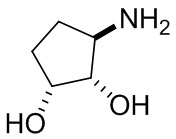
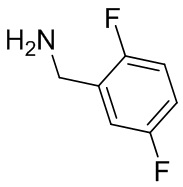
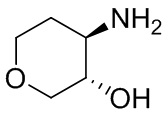
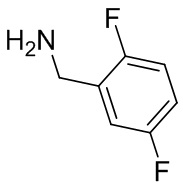
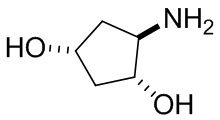
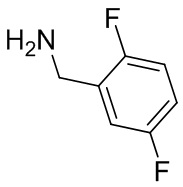
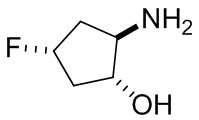
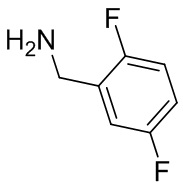
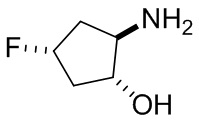



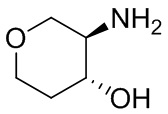
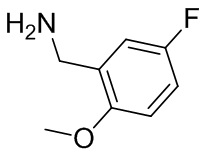
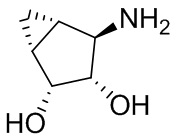
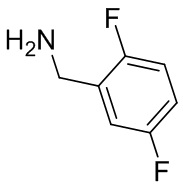
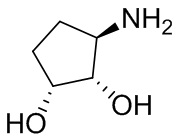
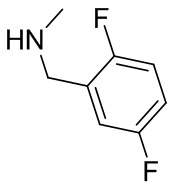
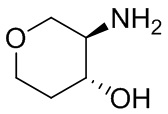
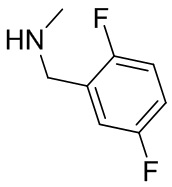
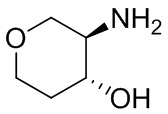
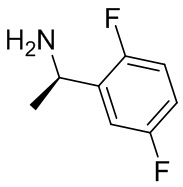
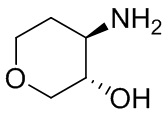
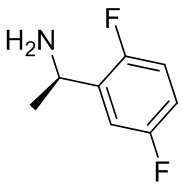

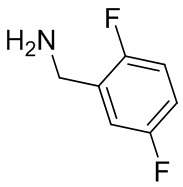
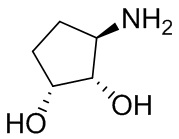
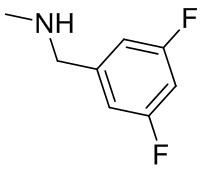
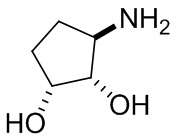
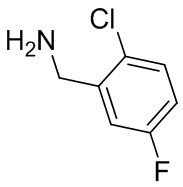
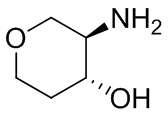
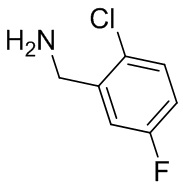
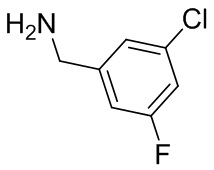
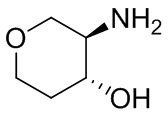
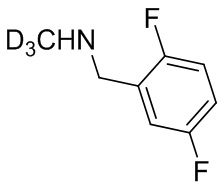
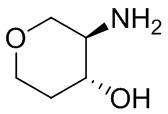
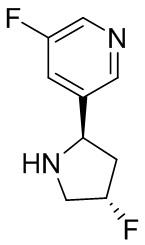
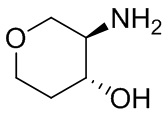
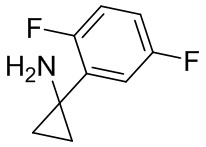
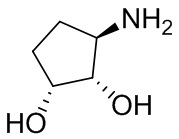
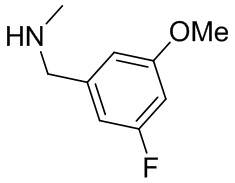
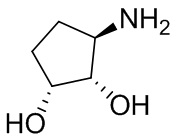
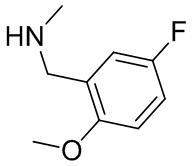
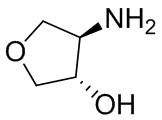
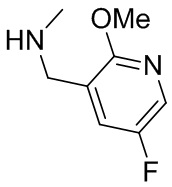
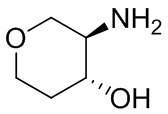

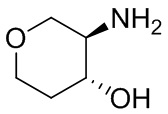
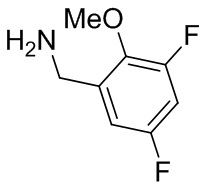
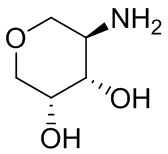
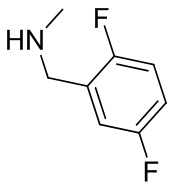
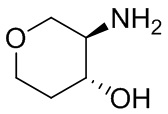
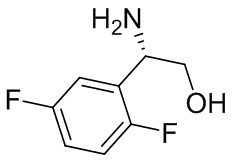
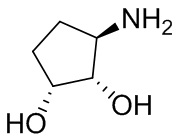
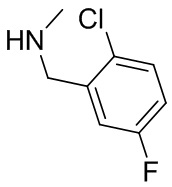
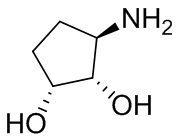
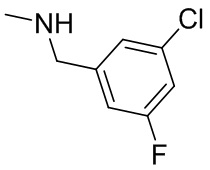
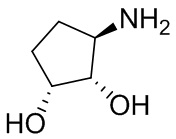
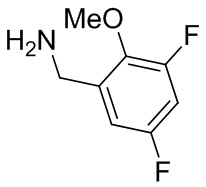
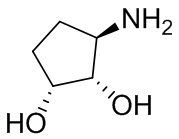
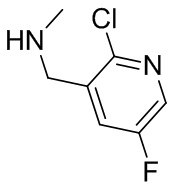

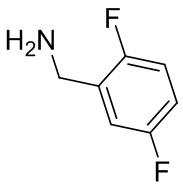
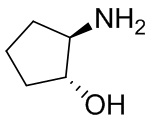
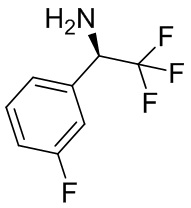
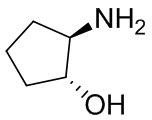
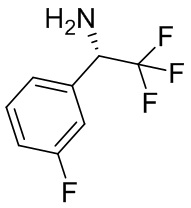
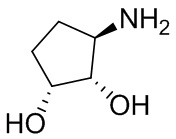
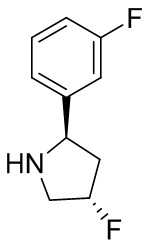
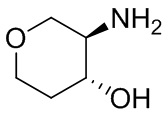
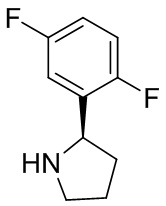
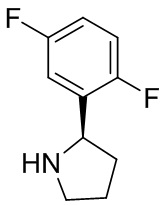
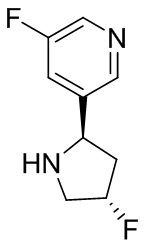
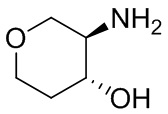
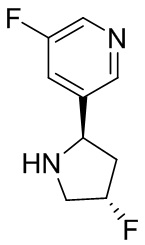
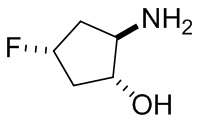
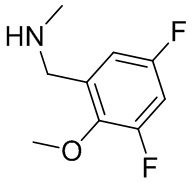
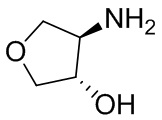
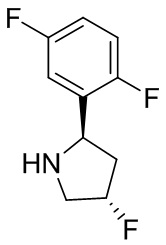
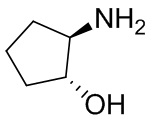
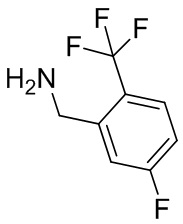
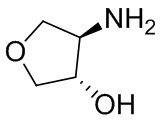
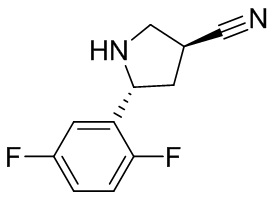
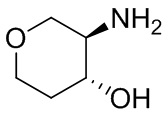

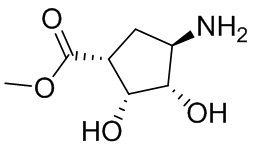
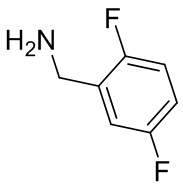
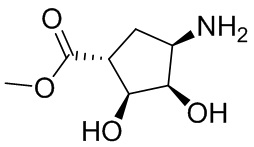
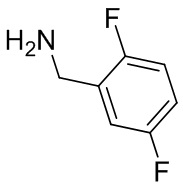
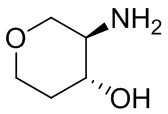
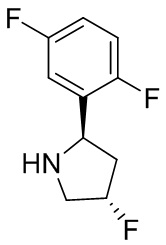
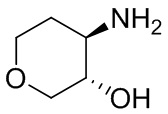
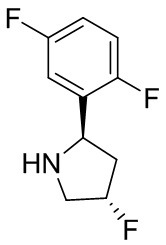

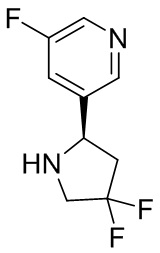
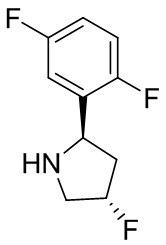
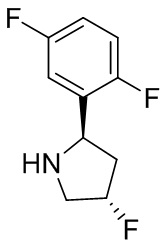
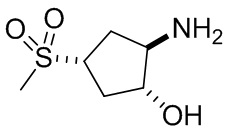
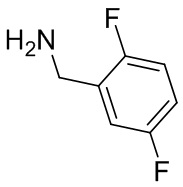
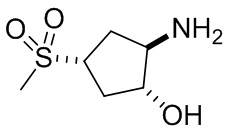
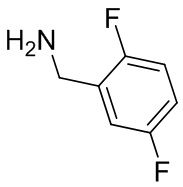
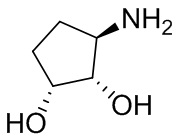
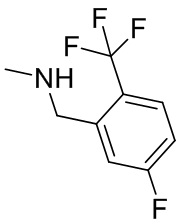
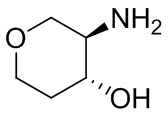
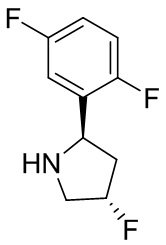
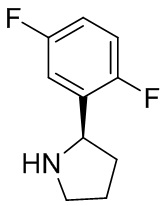

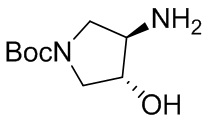
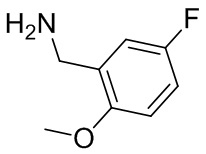
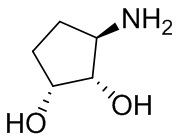
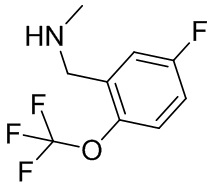
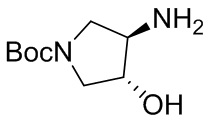
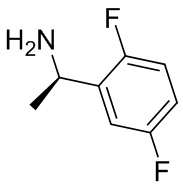
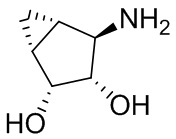
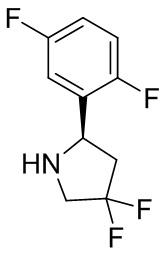
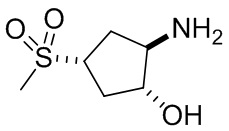
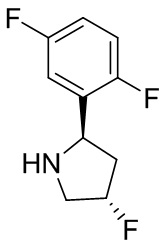
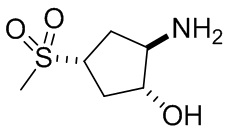
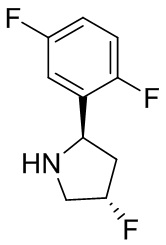





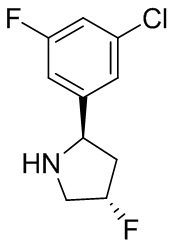
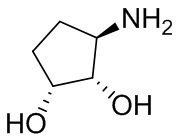
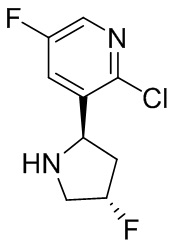
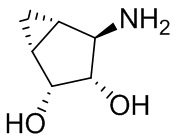
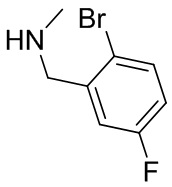
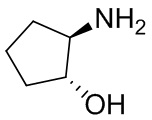
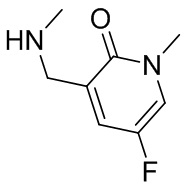
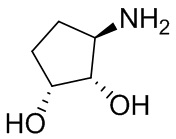
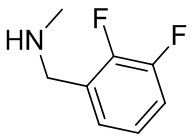
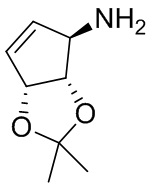
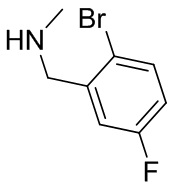
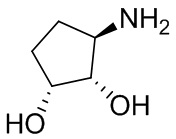

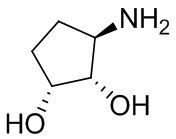
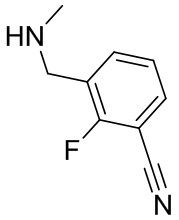
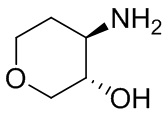
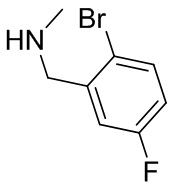
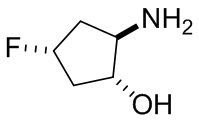
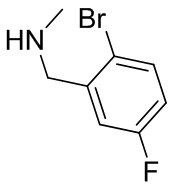
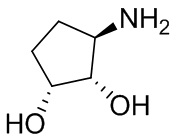
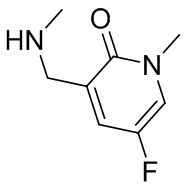
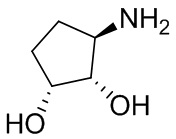
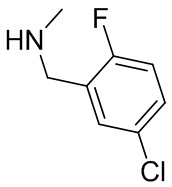
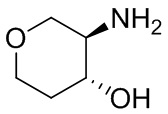
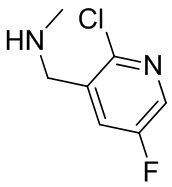
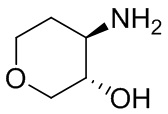
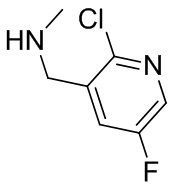
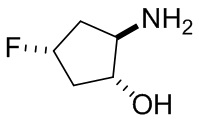
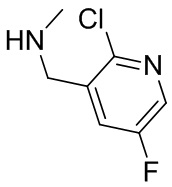
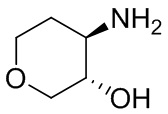
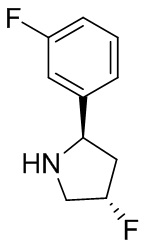
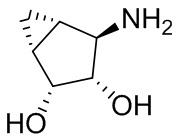
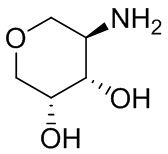
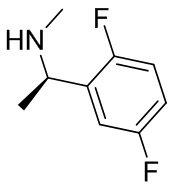
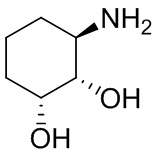
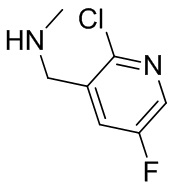
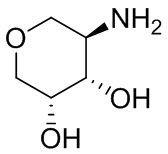
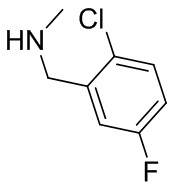
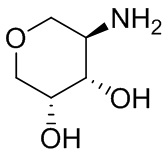
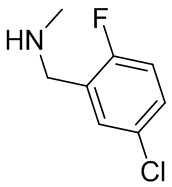
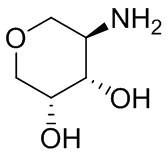
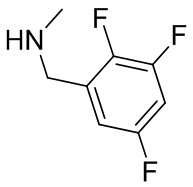
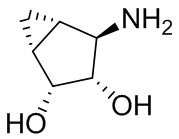
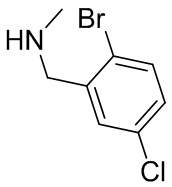
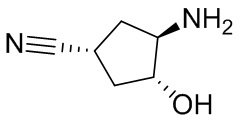
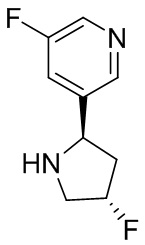
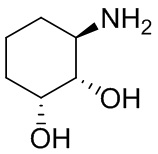
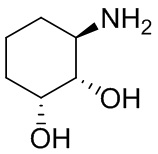
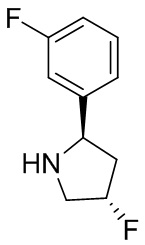
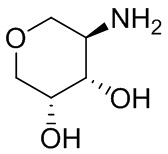
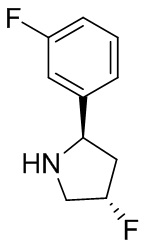
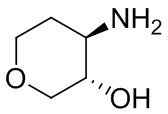
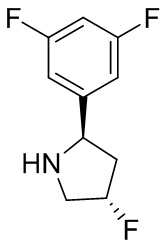
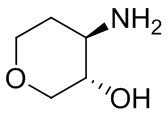
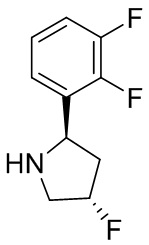
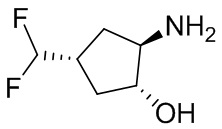
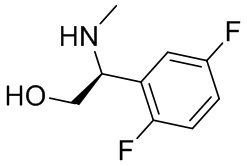
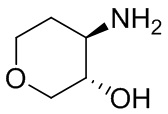
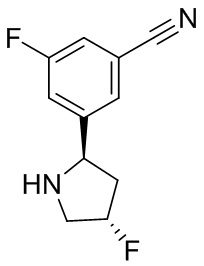
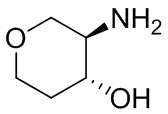
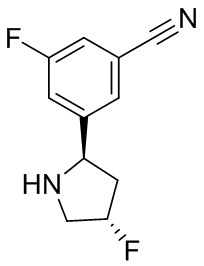
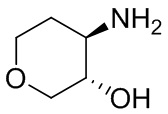
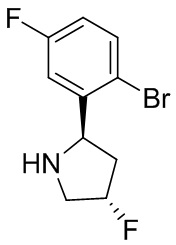
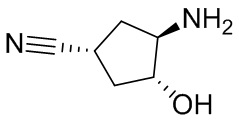
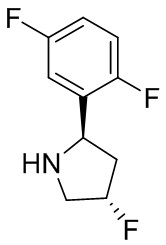
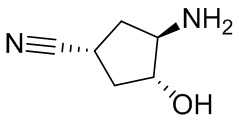
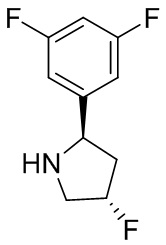
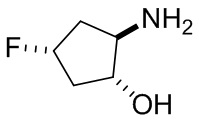
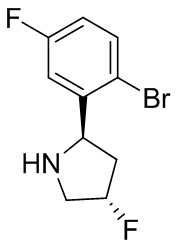

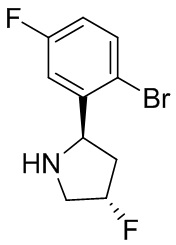
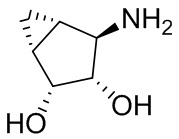

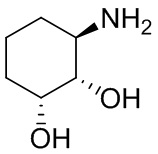
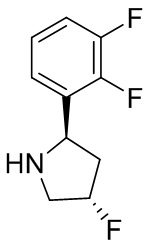
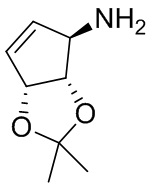
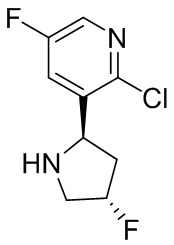
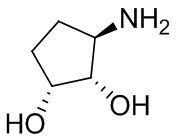
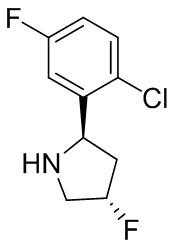
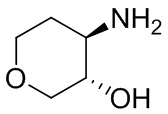
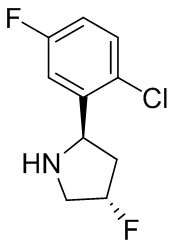
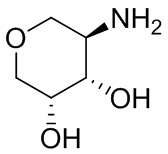
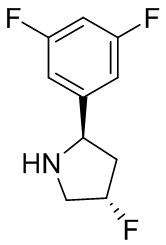
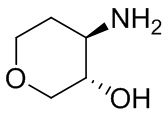
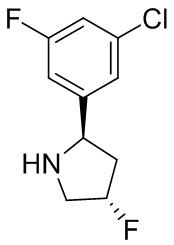
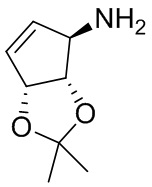
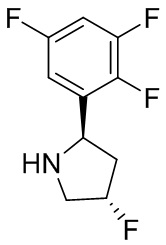
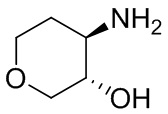
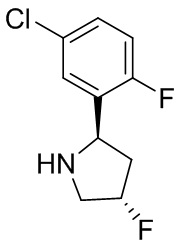
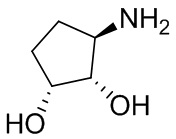
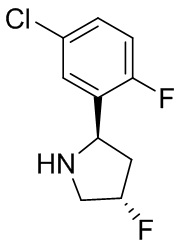
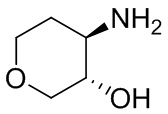
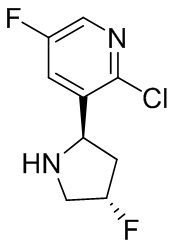
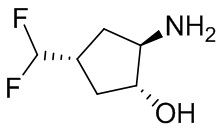
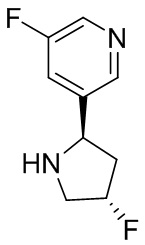
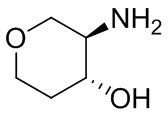
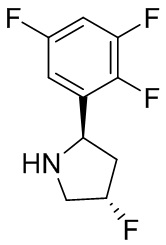
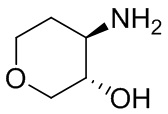
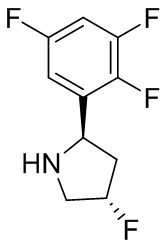
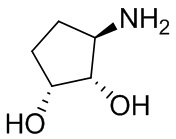
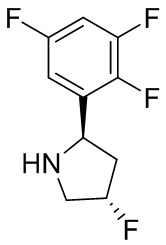
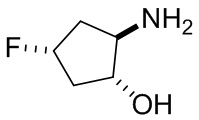
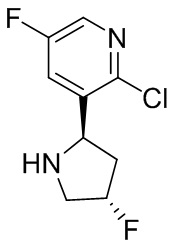
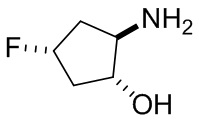
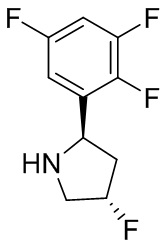
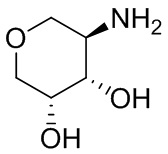
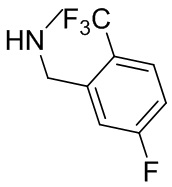
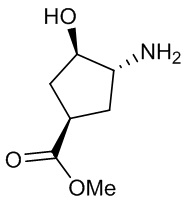
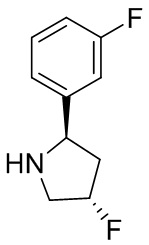
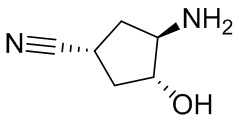
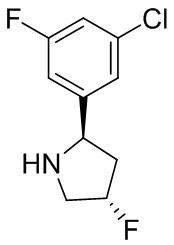
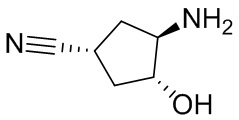
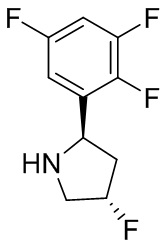
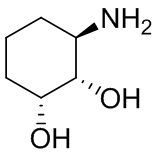
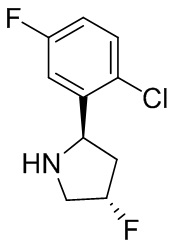
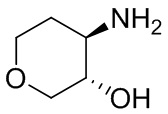
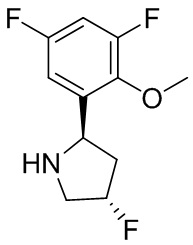
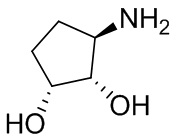
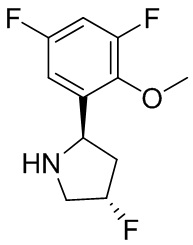
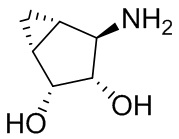
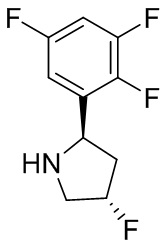
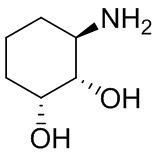
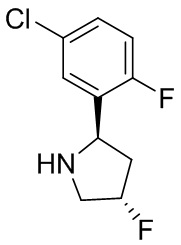
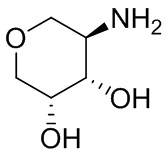
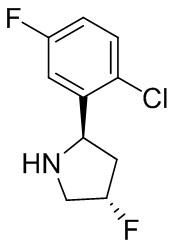
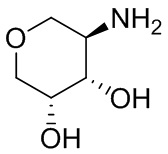
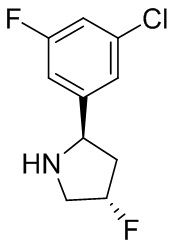
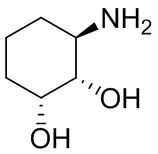
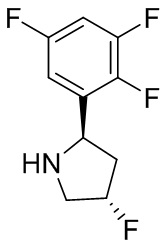
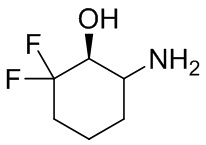
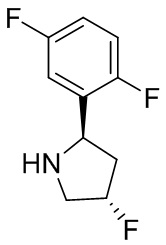
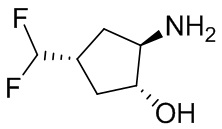
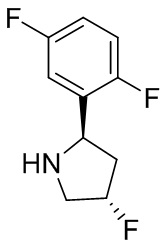
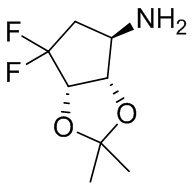
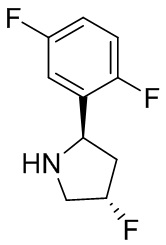
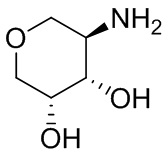
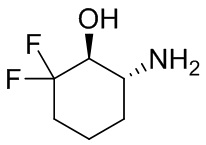
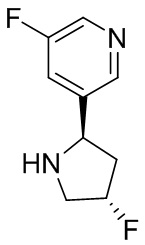
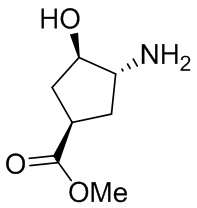

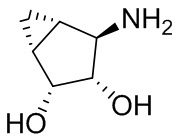
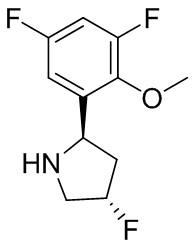
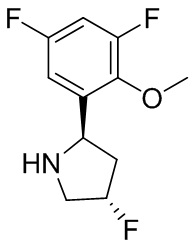
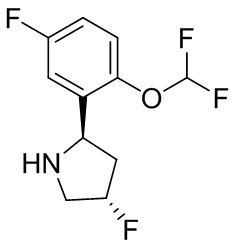
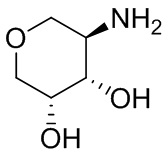
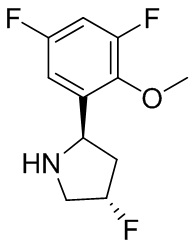
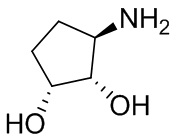
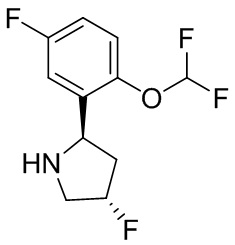
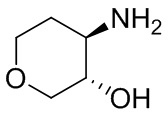
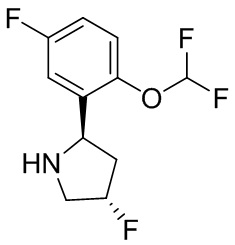
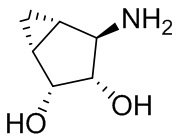
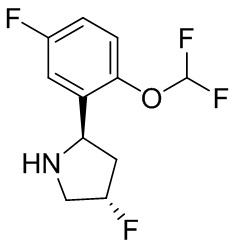
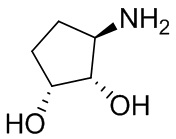
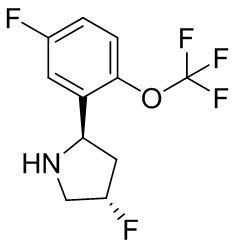
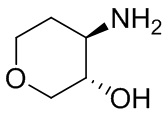

Данные ЯМР и LC MS, полученные для раскрытых в данном документе соединений, показаны на фиг. 1.
Пример 29. Анализы
Анализ NTRK1 дикого типа с 1 мМ АТФ
В каждой лунке 384-луночного планшета инкубировали 1-1,5 нМ фермента NTRK1 дикого типа (BPS Bioscience, 40280), в общей сложности было использовано 12,5 мкл буфера (100 мМ HEPES pH 7,5, 0,015% Brij 35, 10 мМ MgCl2, 1 мМ DTT) с добавлением 1-2 мкМ CSKtide (Университет Тафтса или Anaspec, FITC-AHA-KKKKD DIYFFFG-NH2) и 1 мМ АТФ при 25°C в течение 60 минут в присутствии или в отсутствие дозированной серии концентраций соединения (1% конечной концентрации ДМСО). Реакцию останавливали добавлением 70 мкл останавливающего буфера (100 мМ HEPES, pH 7,5, 0,015% Brij 35, 35 мМ ЭДТА и 0,2% покрывающего реагента Coating Reagent 3 (Caliper Lifesciences)). Затем планшет считывали на считывателе EZReader 2 от компании Caliper (параметры протокола: -1,7 фунт/кв.дюйм, напряжение на входе -500 В, напряжение на выходе -3000 В, время после захвата образца 35 с). Данные были нормализованы до 0% и 100% контроля ингибирования, а IC50 рассчитан с использованием четырехпараметрического набора в CORE LIMS.
Протоколы анализа клеточных линий NTRK дикого типа и мутантной клеточной линии G595R
Клеточная линия карциномы толстой кишки дикого типа KM12, содержащая гибридный белок TPM3-NTRK1, была получена из Национального института рака (NCI). Рост и выживание данной линии, как было показано ранее, зависит от активности NTRK, полученной из гибридного белка NTRK. Клеточная линия KM12 Cliff (G595R) была получена путем мутагенеза линии KM12 дикого типа с помощью метилирующего ДНК агента и последующего выбора для клонирования образцов, которые показали устойчивость к хроническому воздействию высокой концентрации известного ингибитора NTRK (Кризотиниб). Клетки сначала помещали в 384-луночные планшеты по 1000 клеток/лунку в полную среду (10% FBS и 1% пен./стрепт.) и инкубировали в течение ночи при 37°С. Затем к клеткам дозировано добавляли испытуемые образцы в различных концентрациях с использованием системы перенесения жидкостей Bravo. Концентрации варьировались от 25 мкМ до 9,5 пМ (четарехкратные разведения, всего 10 концентраций). Каждое соединение анализировали дважды на планшет. На каждый планшет были добавлены ДМСО и стауроспорин (25 мкМ) в качестве отрицательных и положительных контролей ингибирования роста. Через 72 часа после дозирования аналитические планшеты обрабатывали с использованием CellTiter-Glo (Promega), и полученную люминесценцию считывали на считывателе планшетов Envision. Значения IC50 были рассчитаны с использованием алгоритма подбора кривой с 4 параметрами.
В приведенной ниже таблице представлены результаты описанных выше биологических анализов. Следующие обозначения используются для обозначения IC50 в каждом анализе A < 10,00 нМ; B=10,01-100,0 нМ; C=100,01-1000,0 нМ; и D > 1000,1 нМ; «НО»=не определено.
Включение в качестве ссылки
Все публикации и патенты, упомянутые здесь, полностью включены в качестве ссылки во всей их полноте, как если бы каждая отдельная публикация или патент была конкретно и индивидуально указана для включения в качестве ссылки.
Эквиваленты
Специалисты в данной области смогут определить либо будут способны установить с помощью стандартных экспериментов множество эквивалентов конкретных вариантов осуществления изобретения, описанных в настоящем документе. Подразумевается, что такие эквиваленты охватываются следующей формулой изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИТОРЫ НЕПРИЛИЗИНА | 2011 |
|
RU2622288C2 |
| ИМИДАЗОПИРРОЛОПИРАЗИНОВЫЕ ПРОИЗВОДНЫЕ, ПОЛЕЗНЫЕ ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ, ВЫЗВАННЫХ АНОМАЛЬНОЙ АКТИВНОСТЬЮ ПРОТЕИНКИНАЗ Jak1, Jak3 ИЛИ Syk | 2010 |
|
RU2570416C2 |
| ТРИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ИХ ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ ИММУНОЛОГИЧЕСКИХ И ОНКОЛОГИЧЕСКИХ СОСТОЯНИЙ | 2009 |
|
RU2545023C9 |
| Имидазопирролопиразиновые производные, полезные для лечения заболеваний, вызванных аномальной активностью протеинкиназ Jak1, Jak3 или Syk | 2010 |
|
RU2711869C2 |
| КОНДЕНСИРОВАННОЕ ХИНОЛИНОВОЕ ПРОИЗВОДНОЕ И ЕГО ПРИМЕНЕНИЕ | 2005 |
|
RU2384571C2 |
| АРИЛ-ИЛИ ГЕТЕРОАРИЛЗАМЕЩЕННЫЕ БЕНЗОЛЬНЫЕ СОЕДИНЕНИЯ | 2012 |
|
RU2632193C2 |
| НОВОЕ ПИРРОЛОПИРИМИДИНОВОЕ СОЕДИНЕНИЕ ИЛИ ЕГО СОЛЬ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ЕЕ, В ЧАСТНОСТИ, АГЕНТ ДЛЯ ПРЕДОТВРАЩЕНИЯ И/ИЛИ ЛЕЧЕНИЯ ОПУХОЛЕЙ, И ТОМУ ПОДОБНОЕ, НА ОСНОВЕ ИНГИБИТОРНОГО ВОЗДЕЙСТВИЯ НА NAE | 2015 |
|
RU2658008C2 |
| ЗАМЕЩЕННЫЕ ПУРИНОВЫЕ И 7-ДЕАЗАПУРИНОВЫЕ СОЕДИНЕНИЯ | 2011 |
|
RU2606514C2 |
| Соединения циклопентана | 2019 |
|
RU2794997C2 |
| ЗАМЕЩЕННЫЕ АМИНОМАСЛЯНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ НЕПРИЛИЗИНА | 2012 |
|
RU2604522C2 |
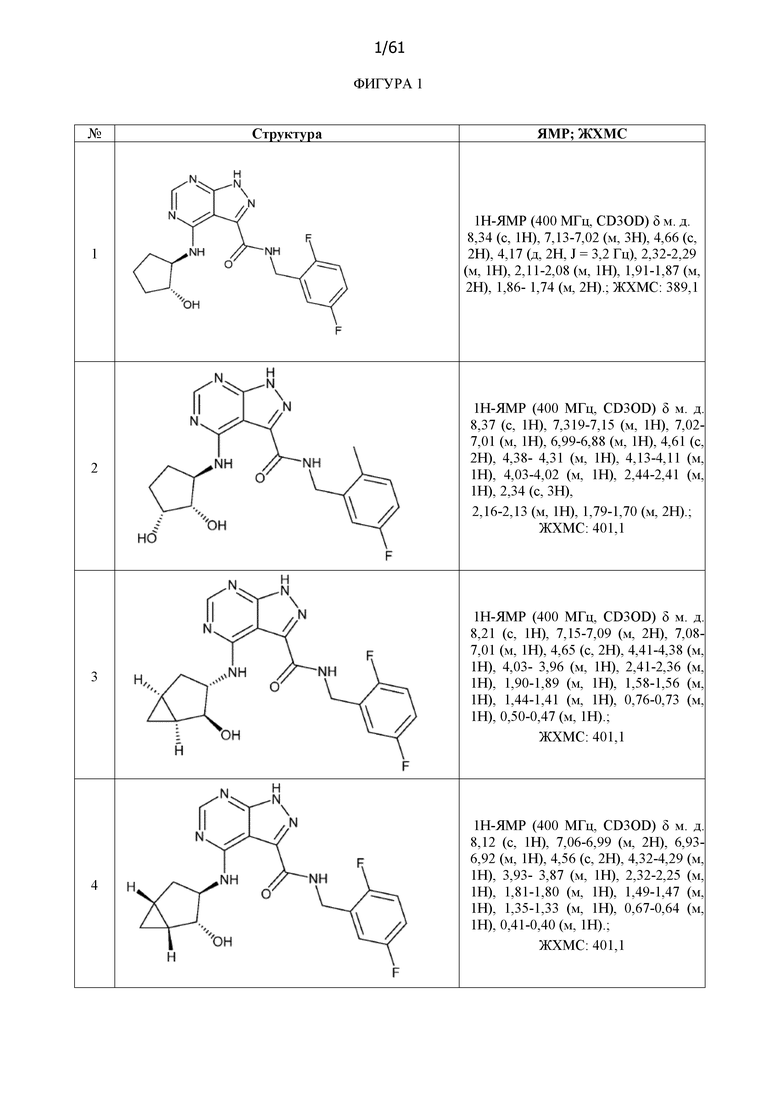
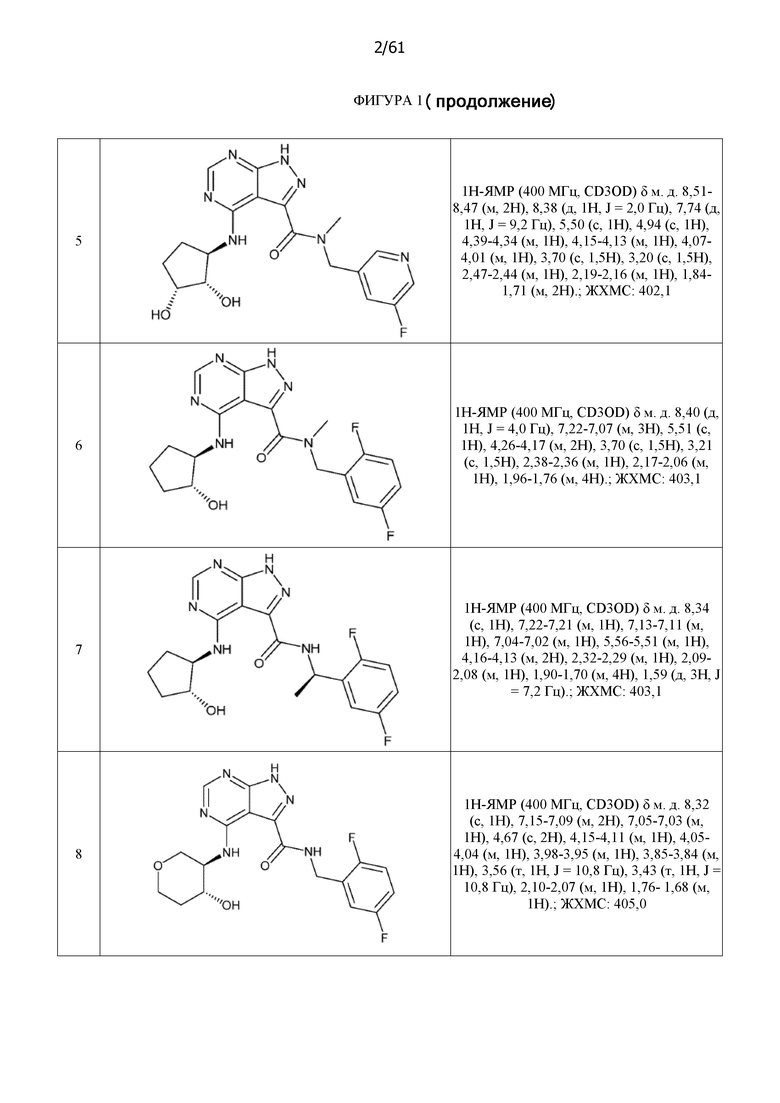
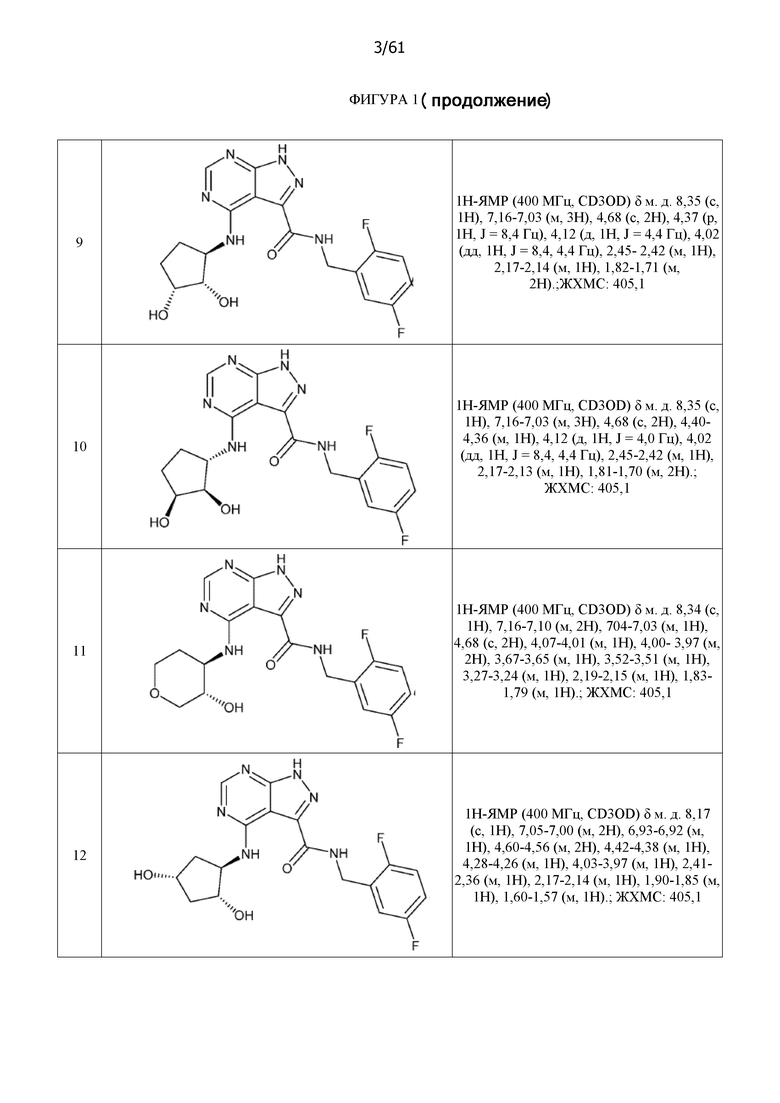
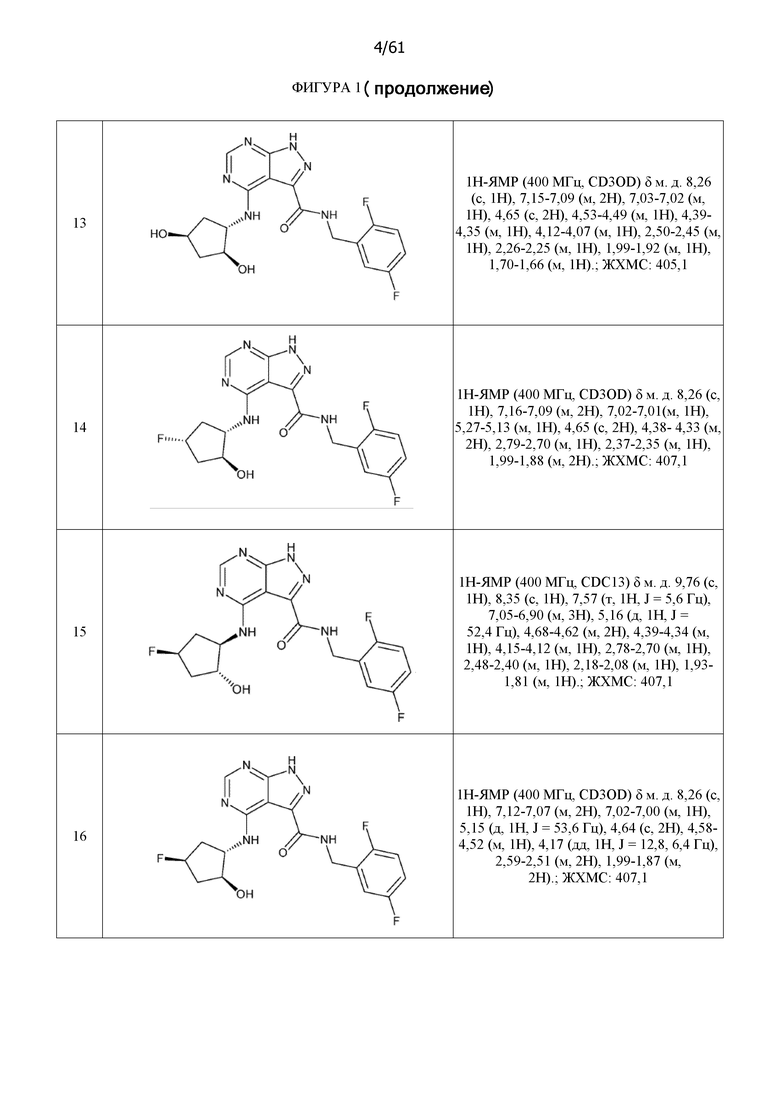
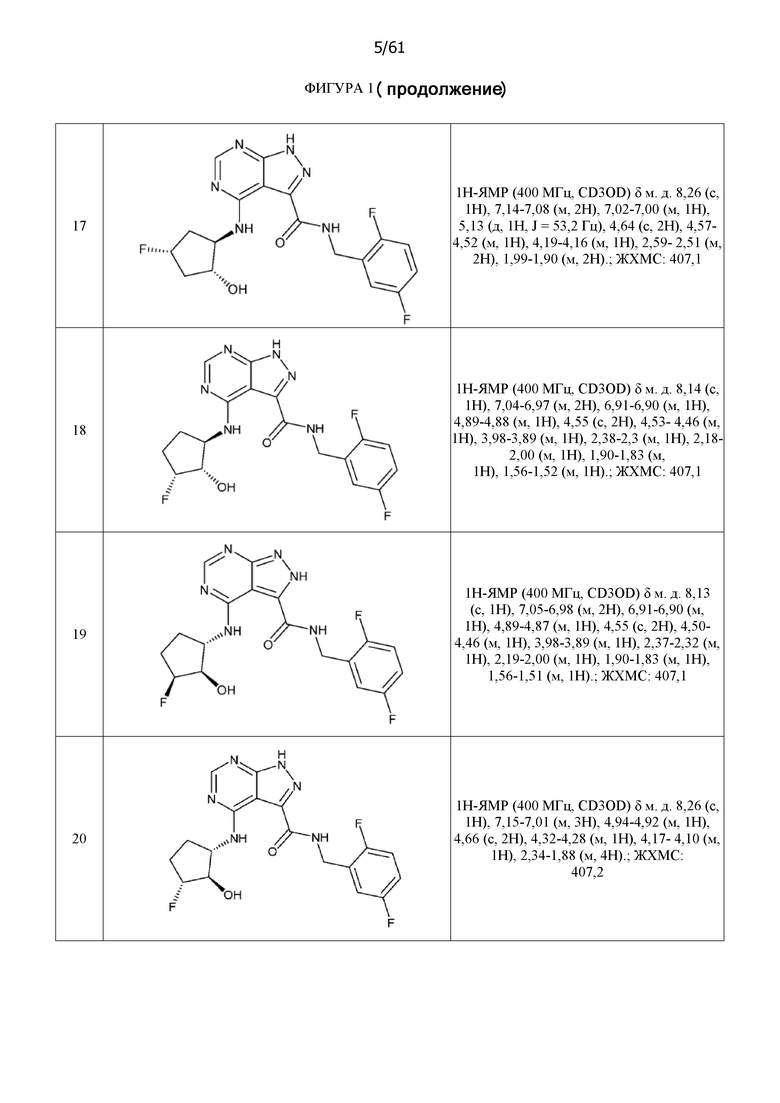
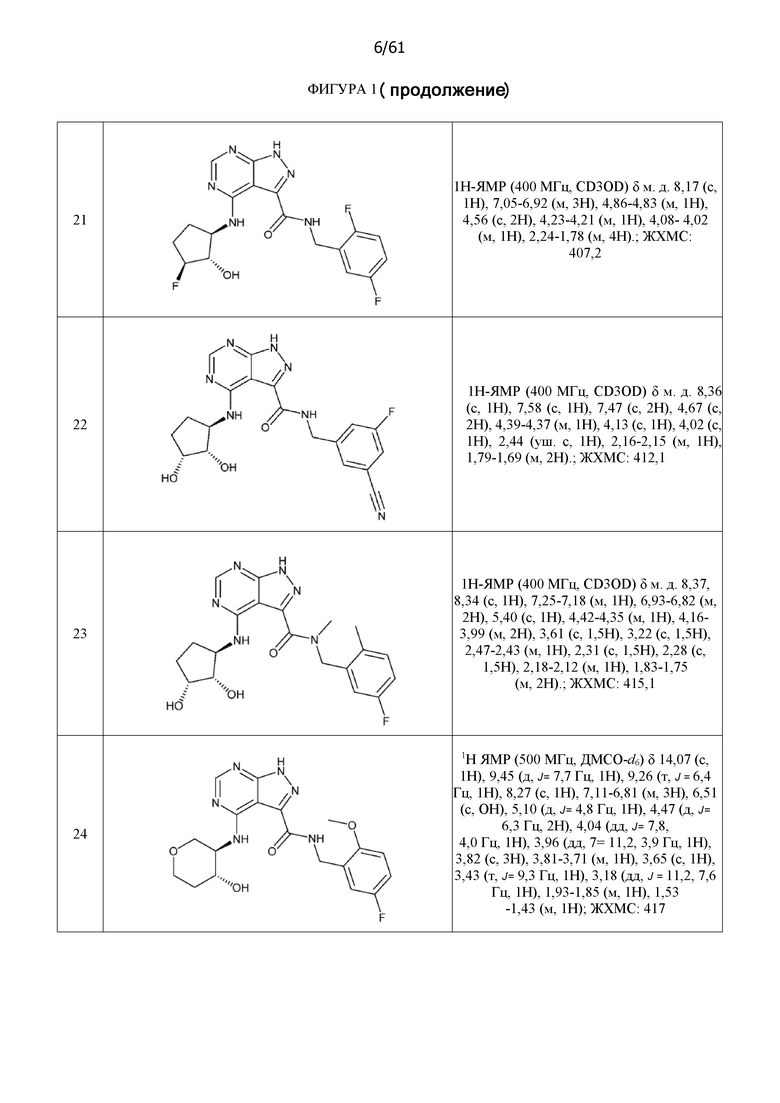
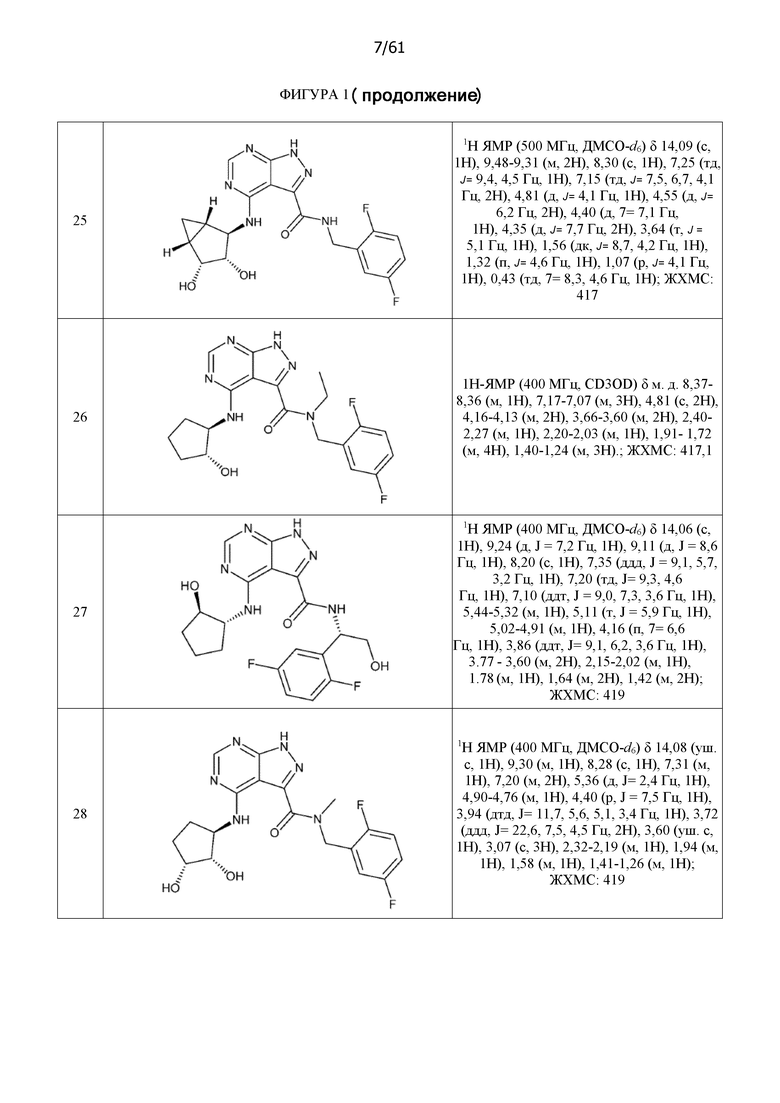
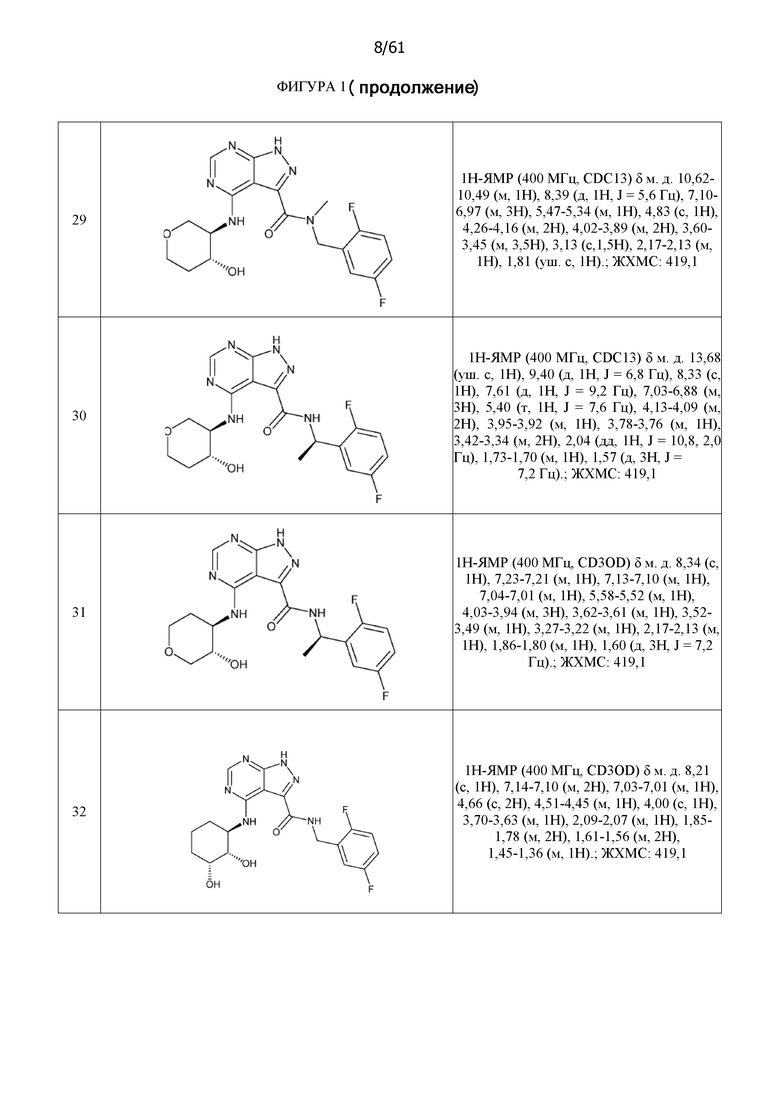
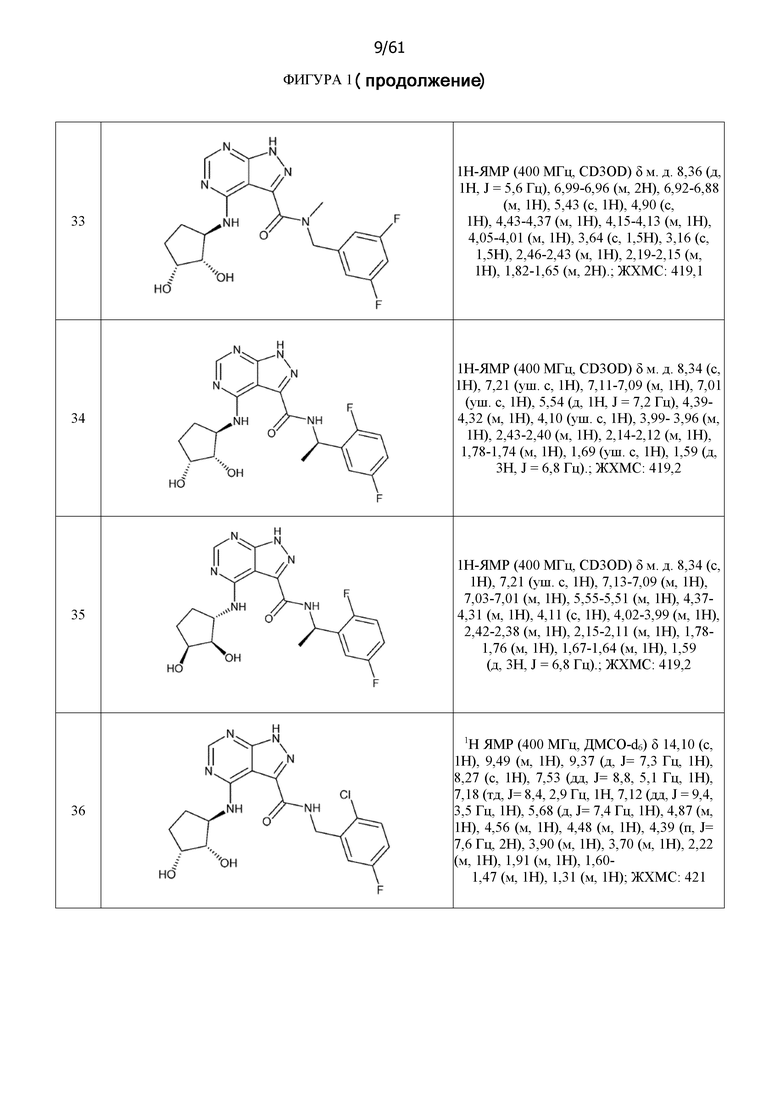
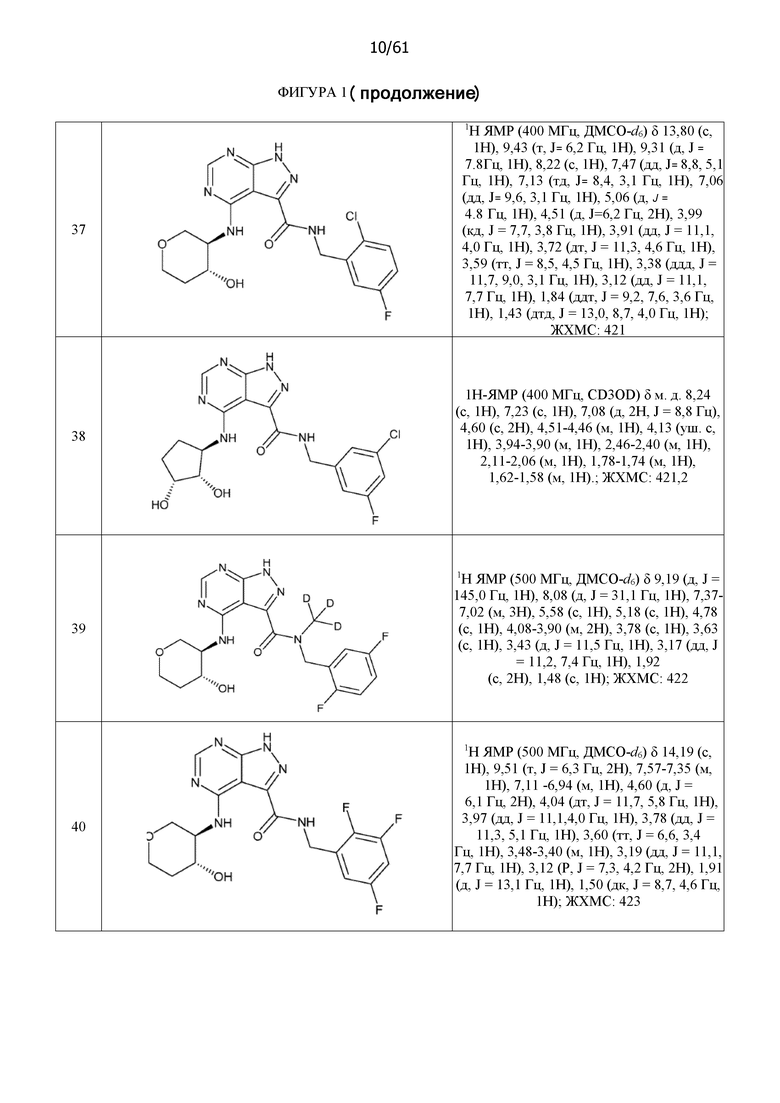
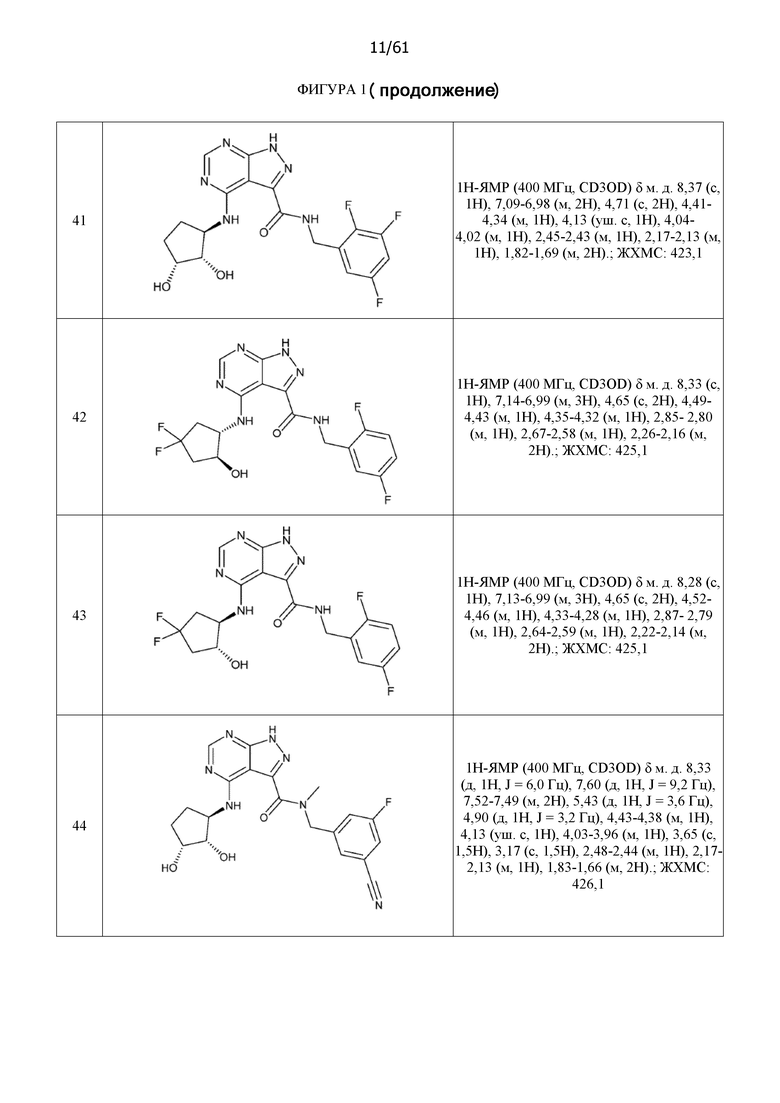
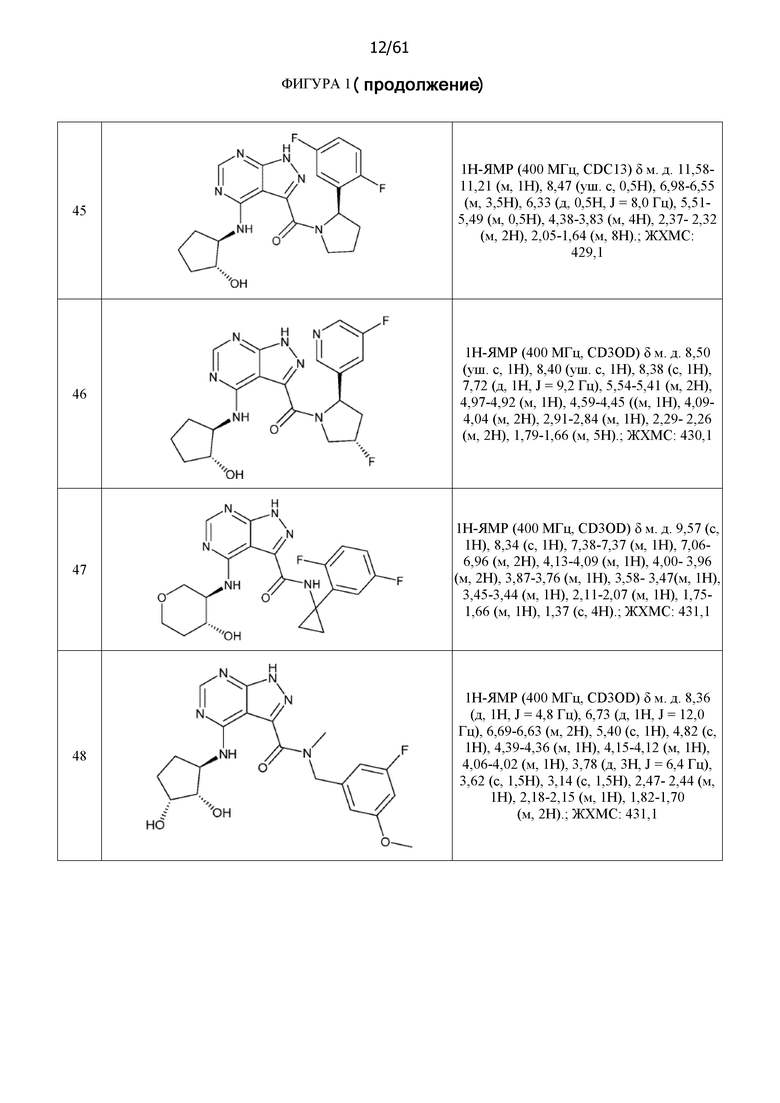
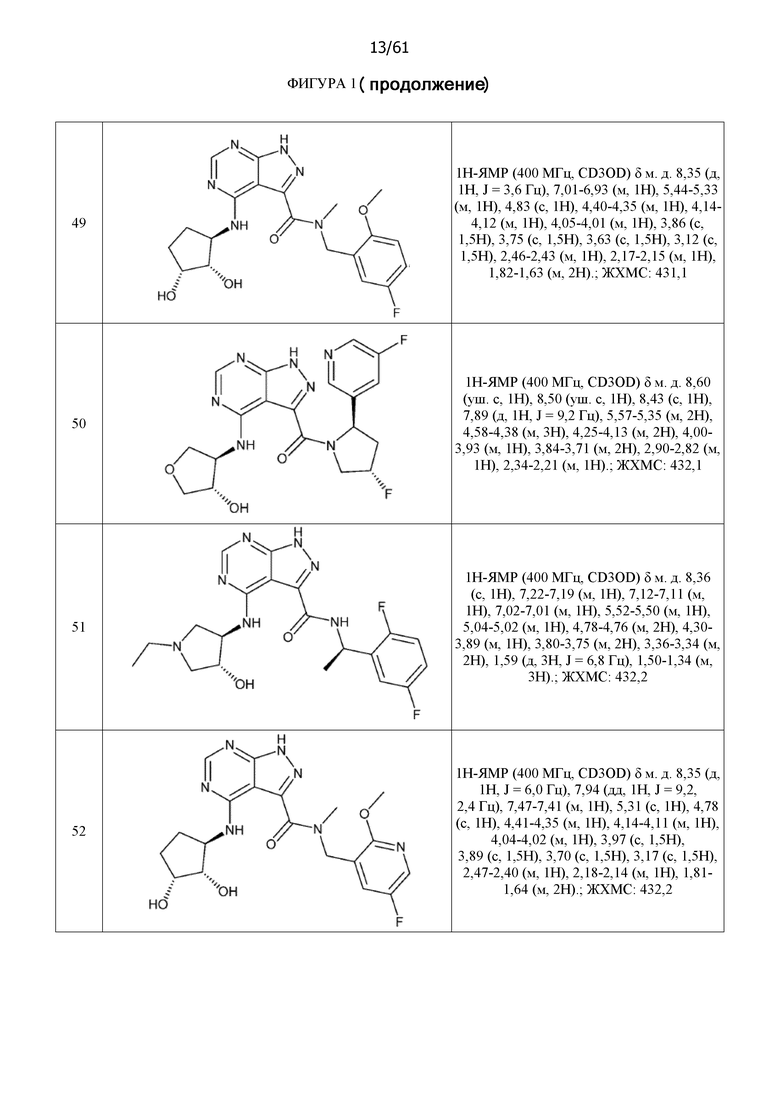
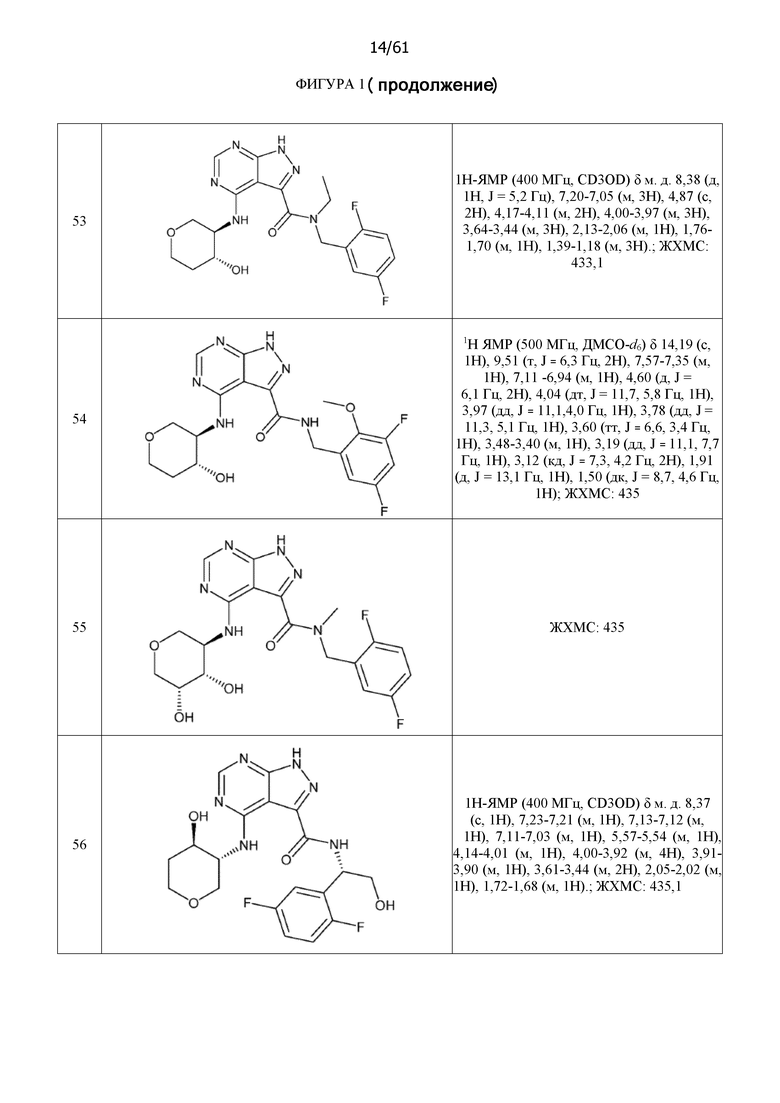
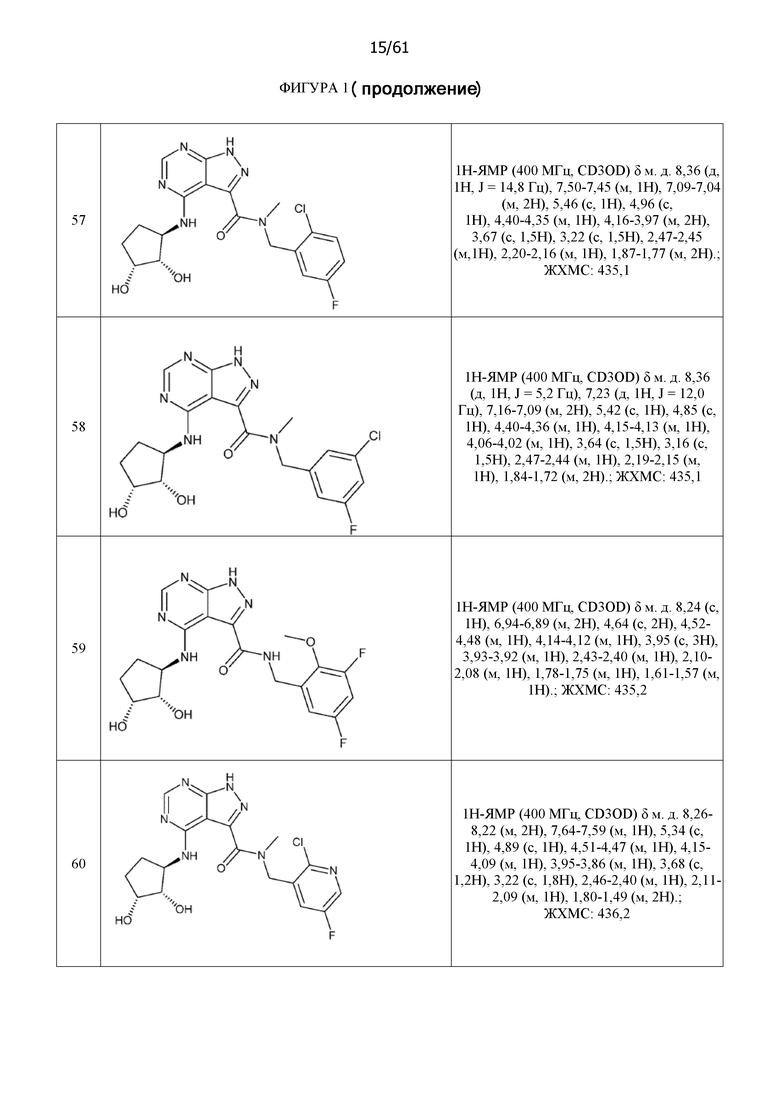
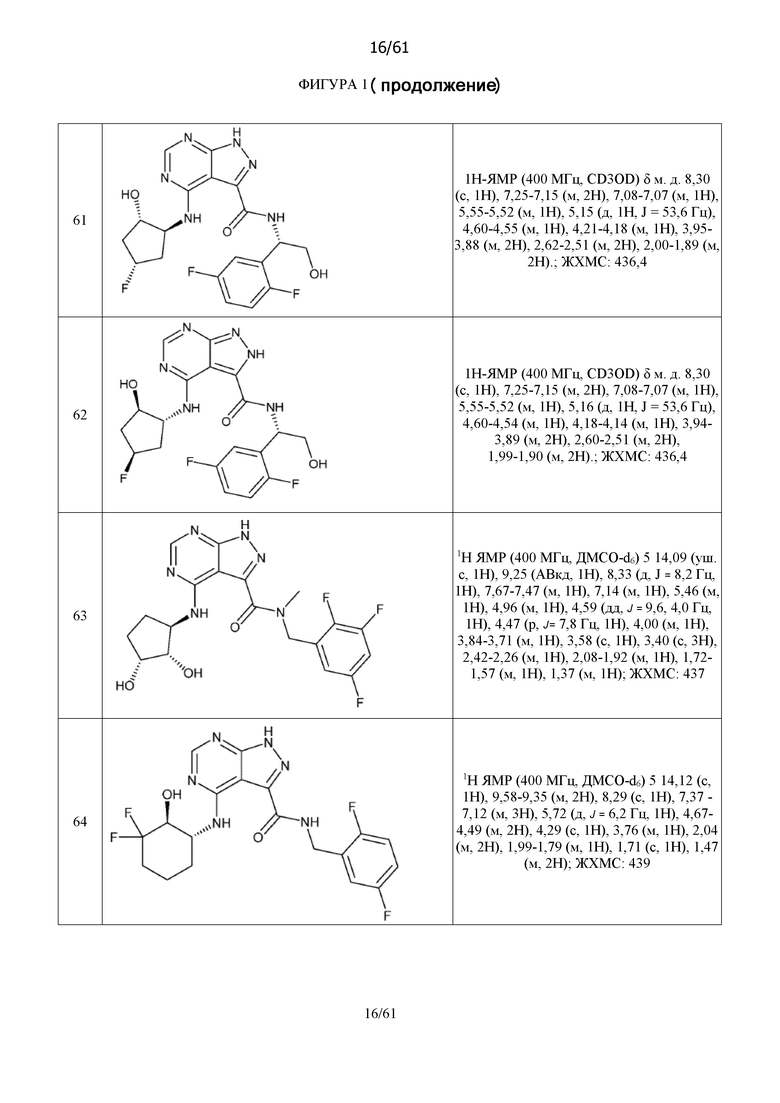
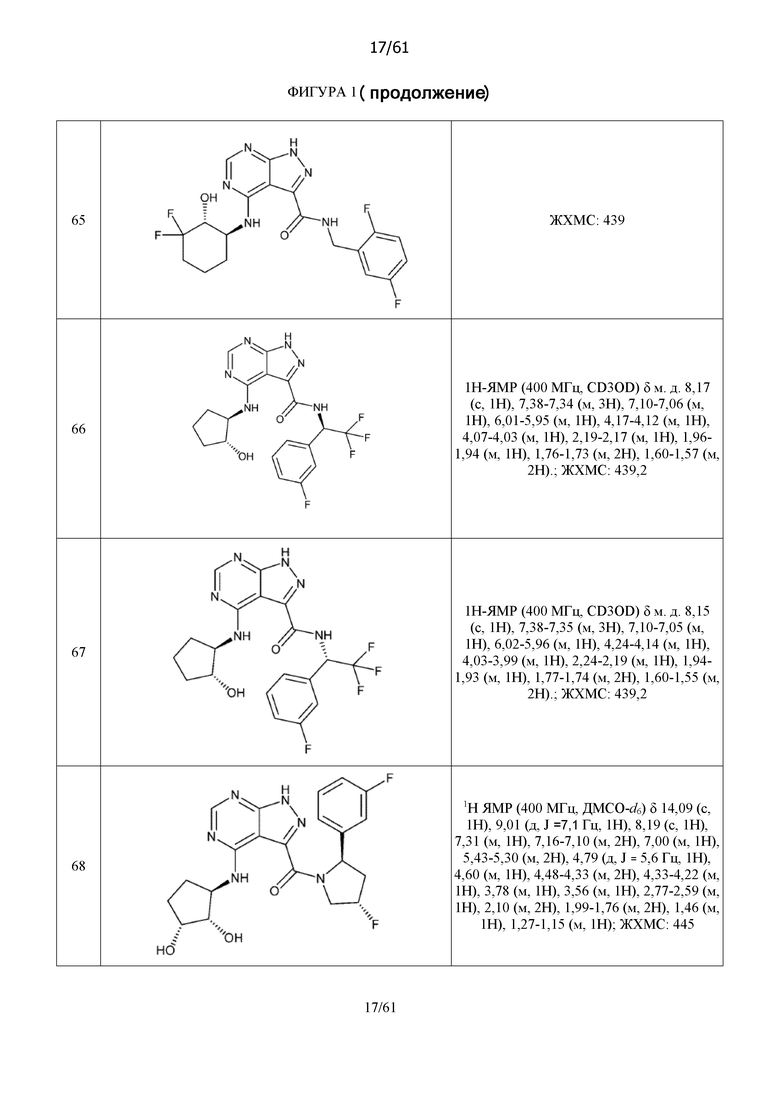
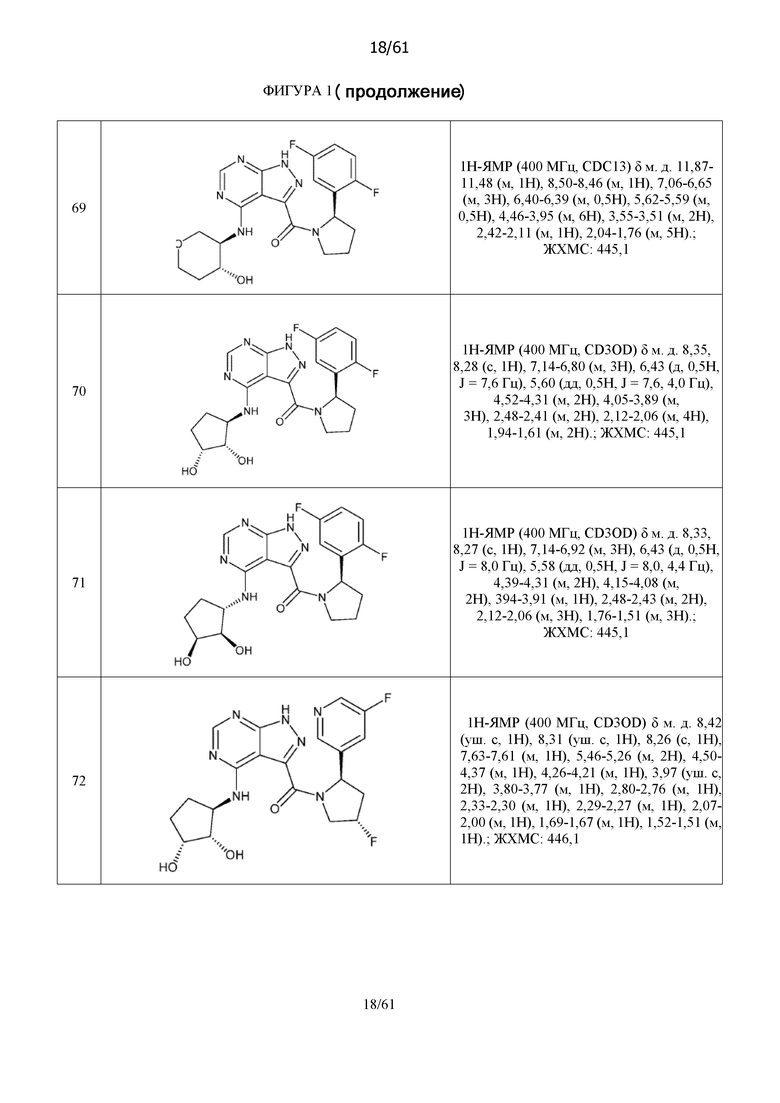

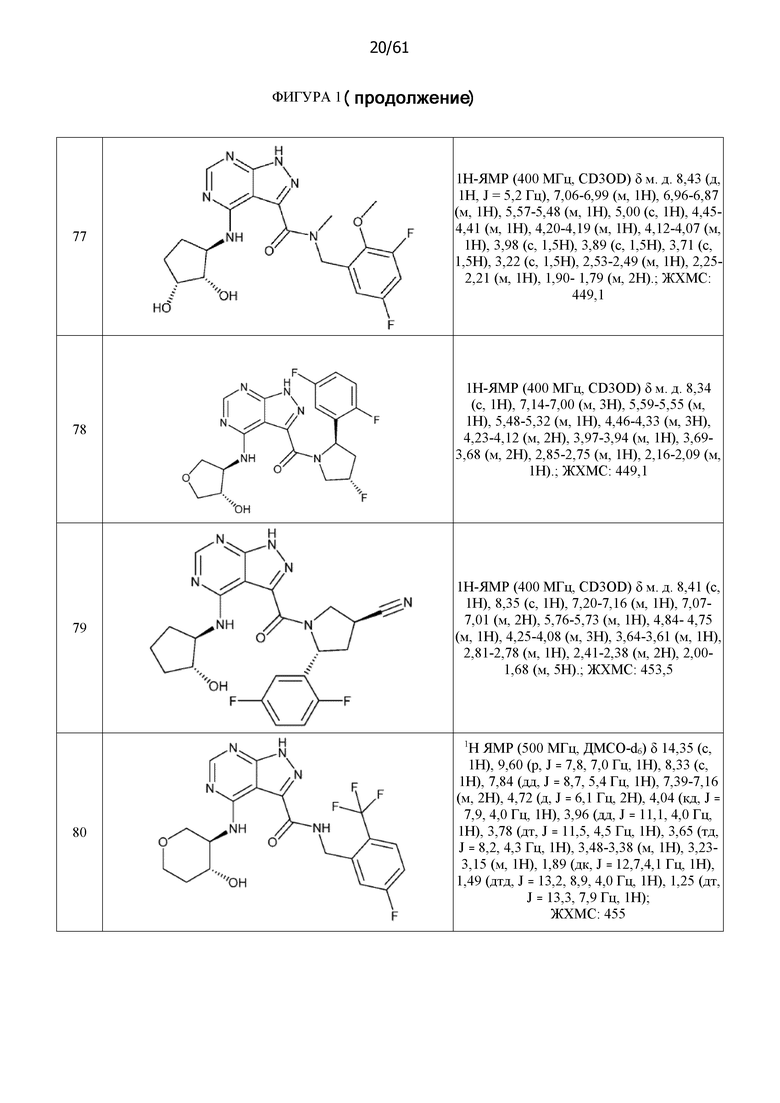
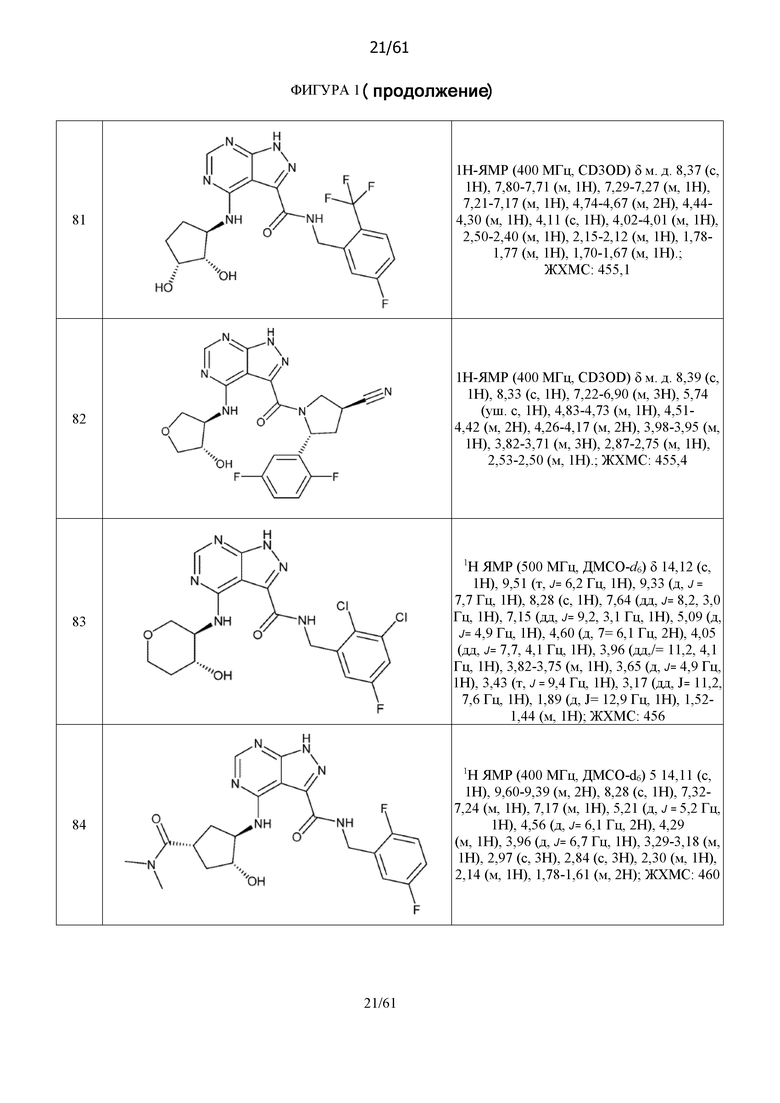

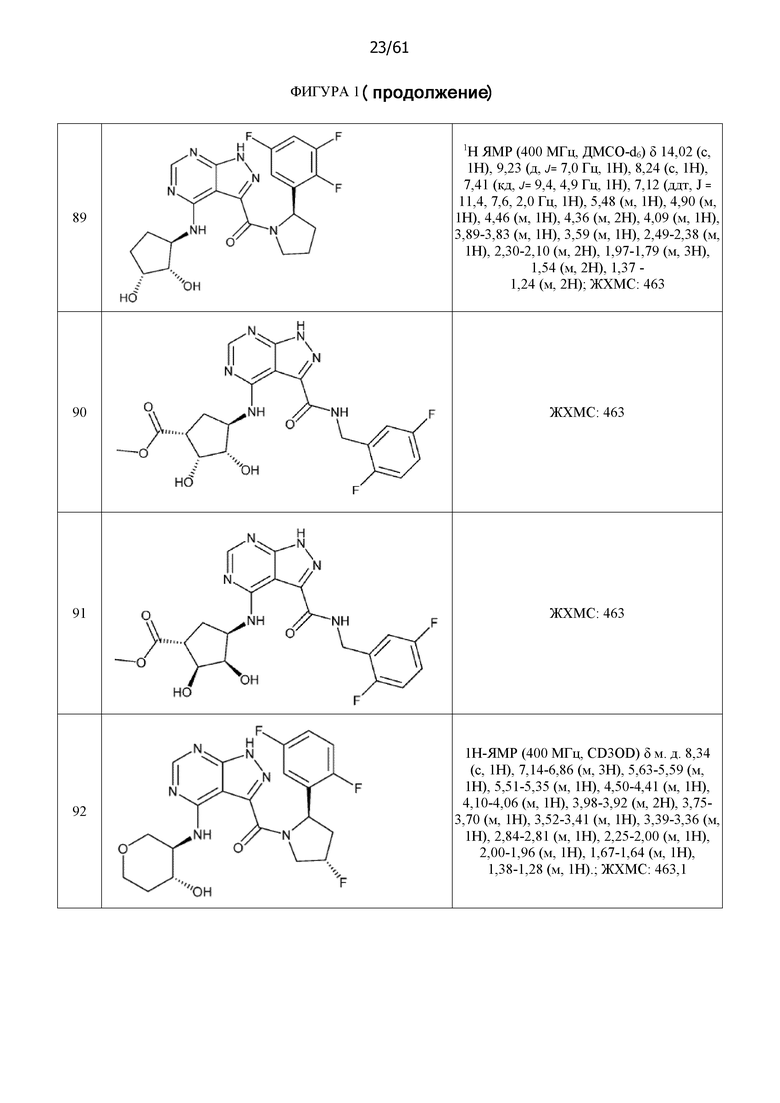
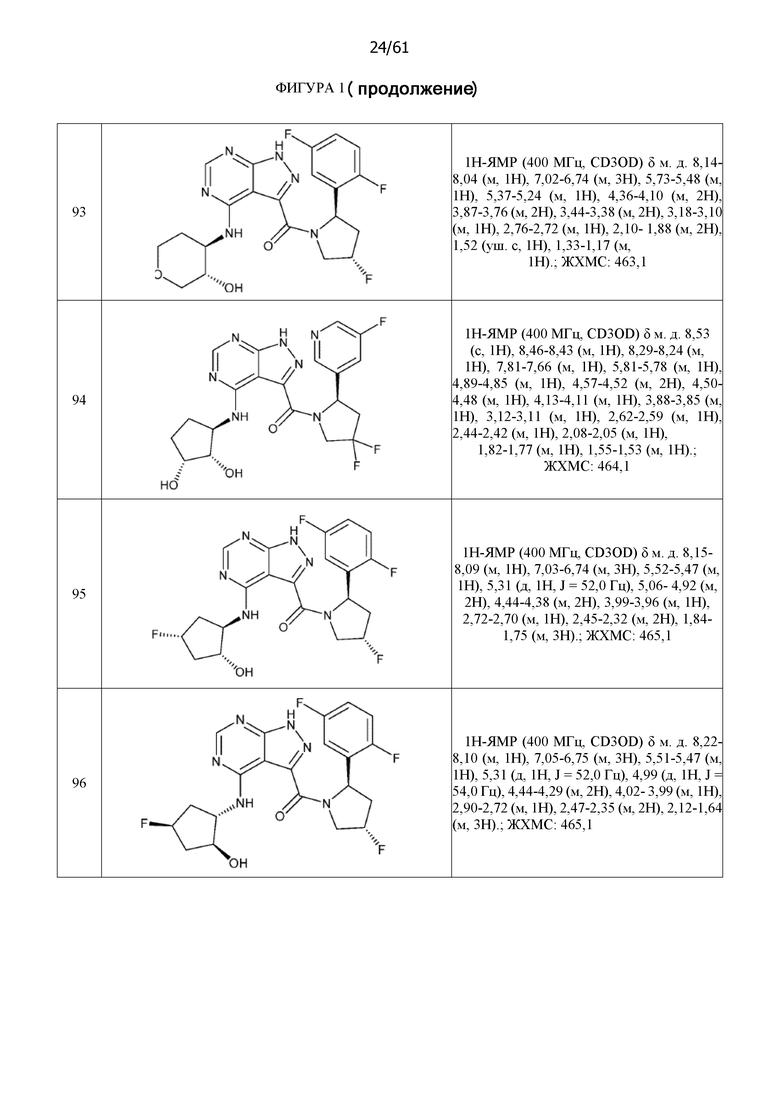
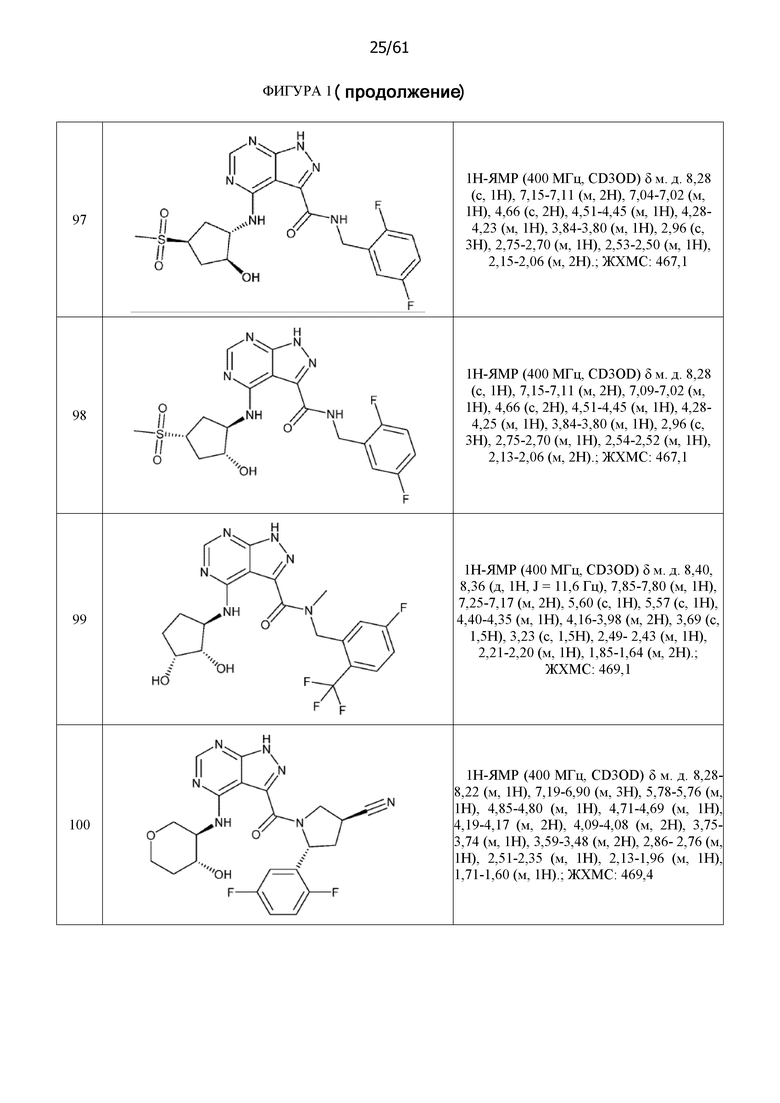

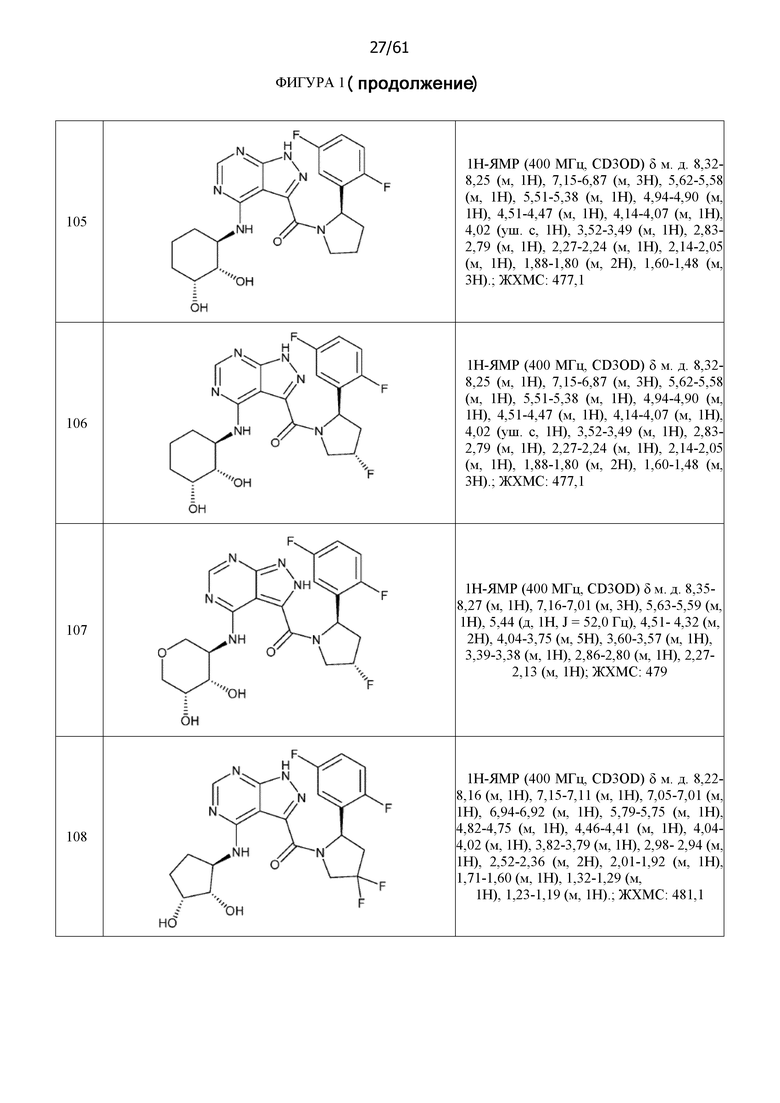
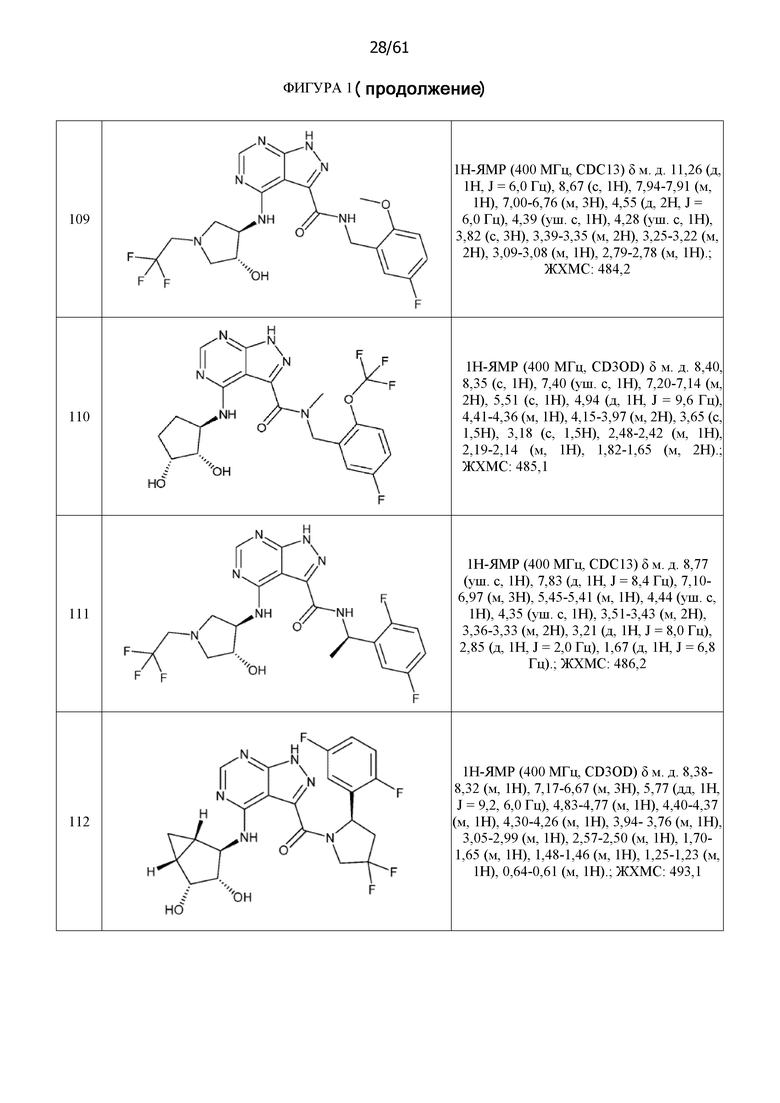
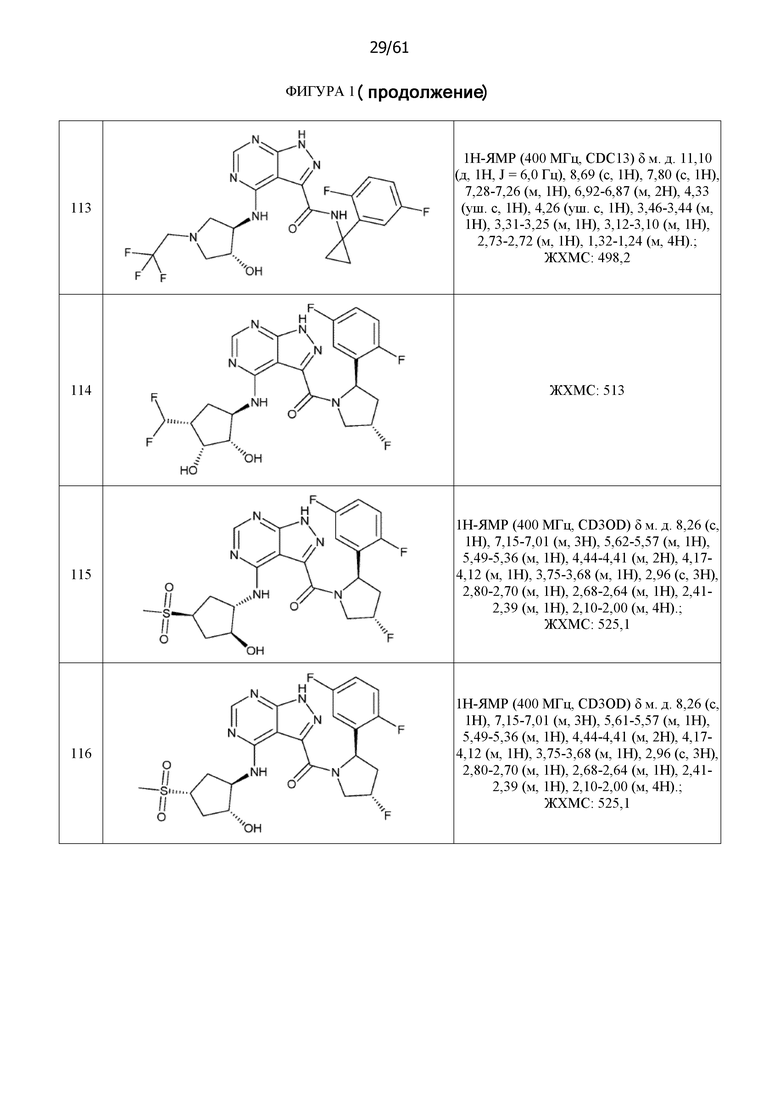

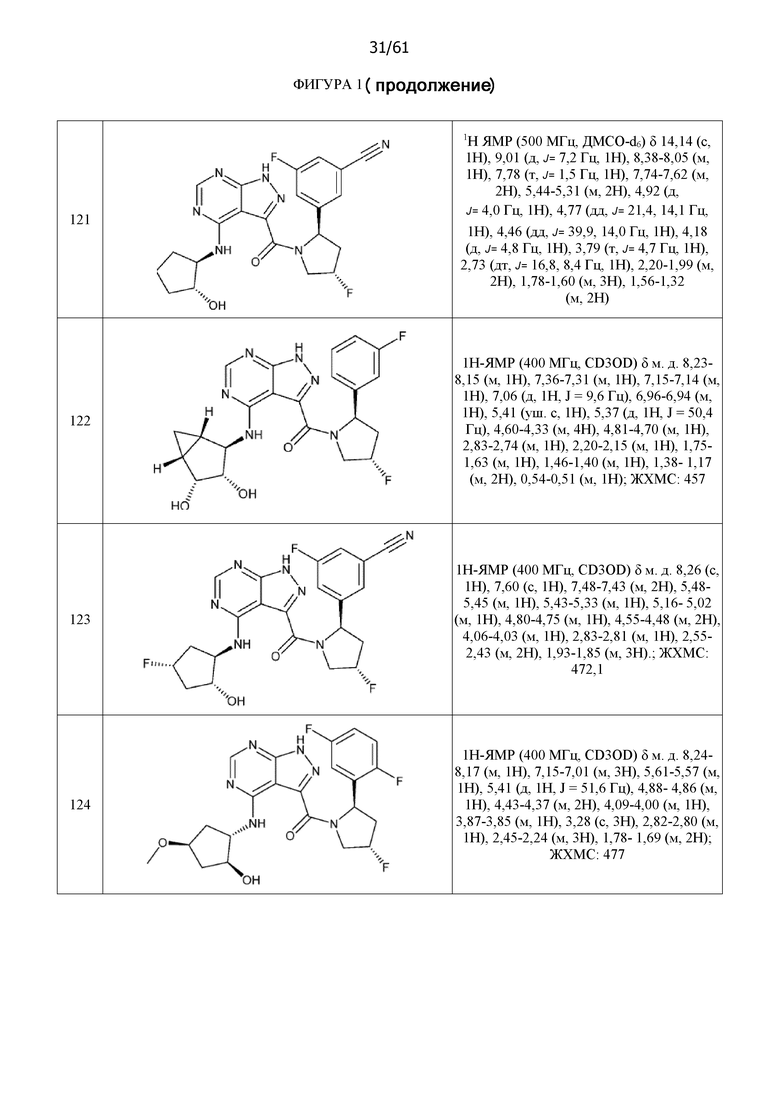
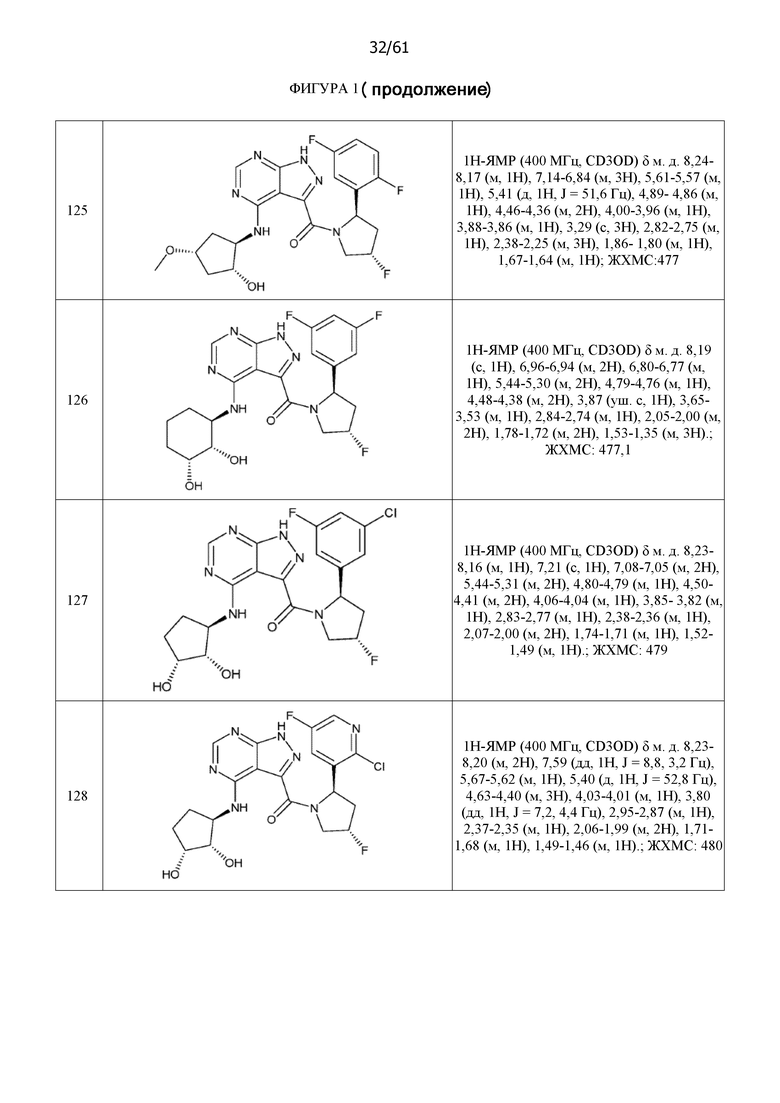
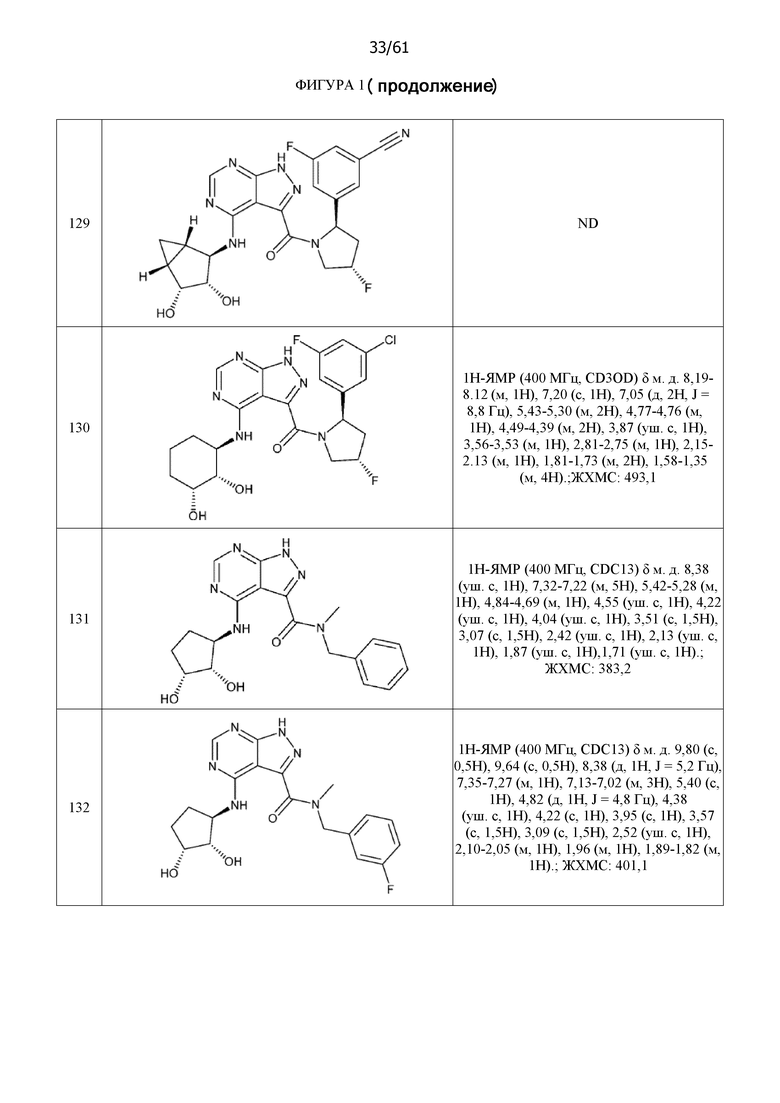
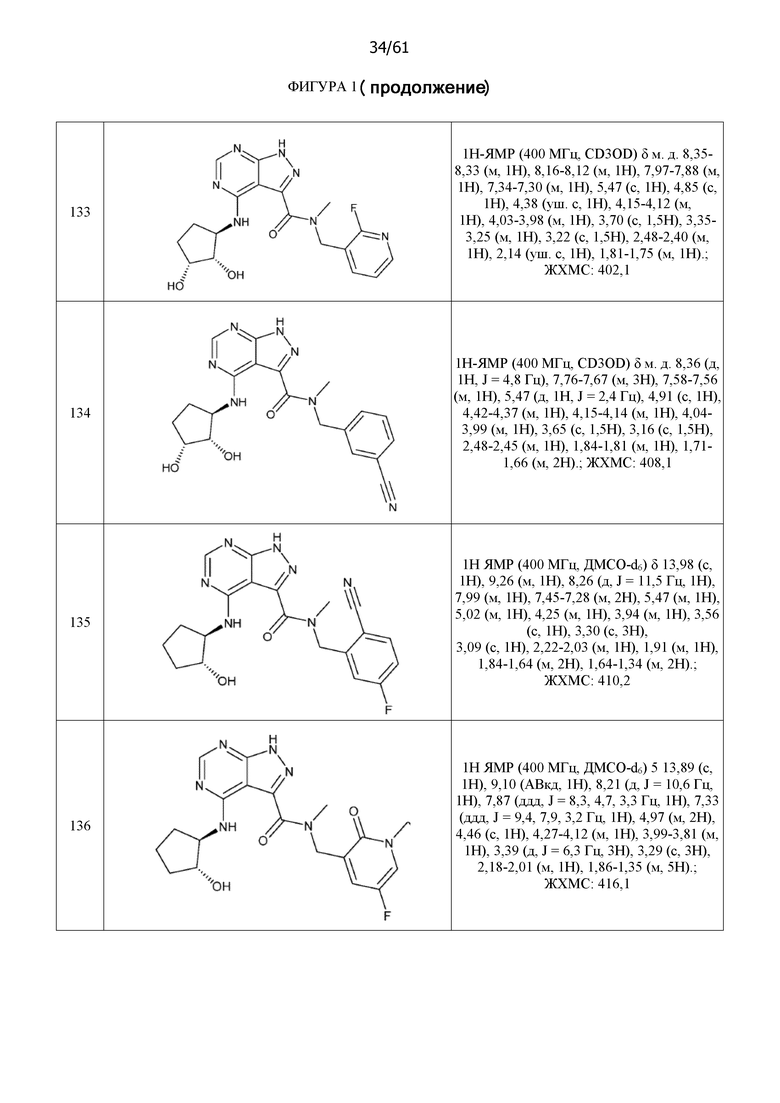

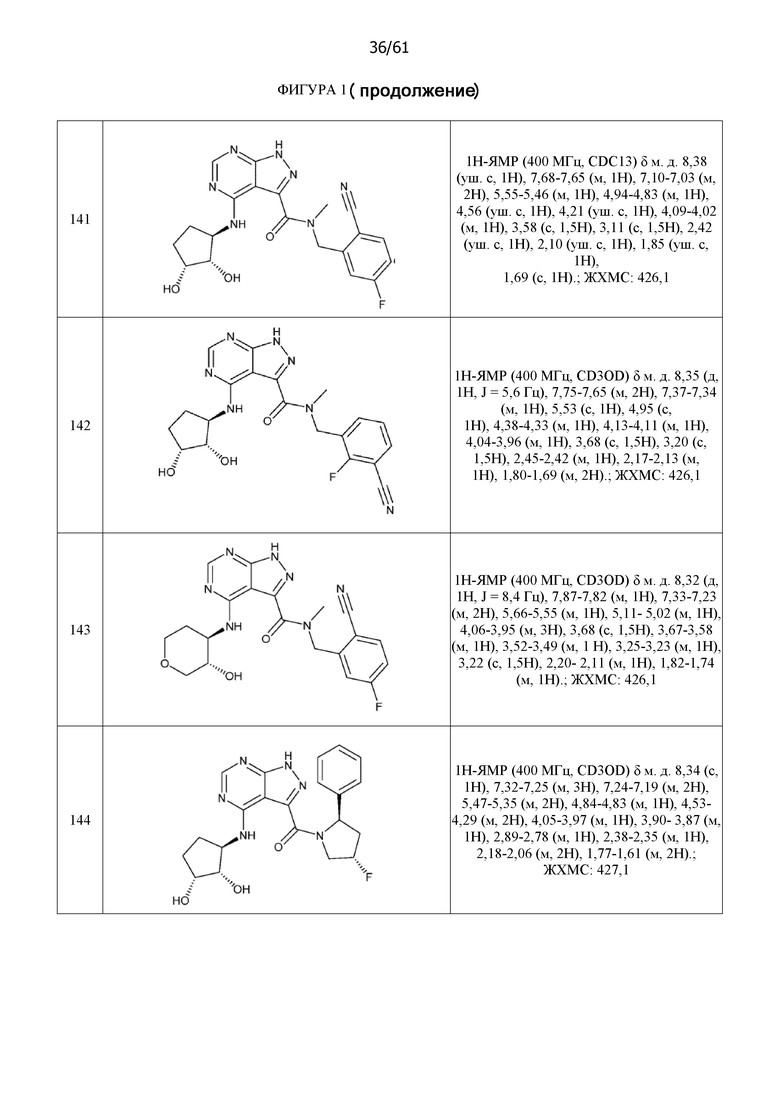
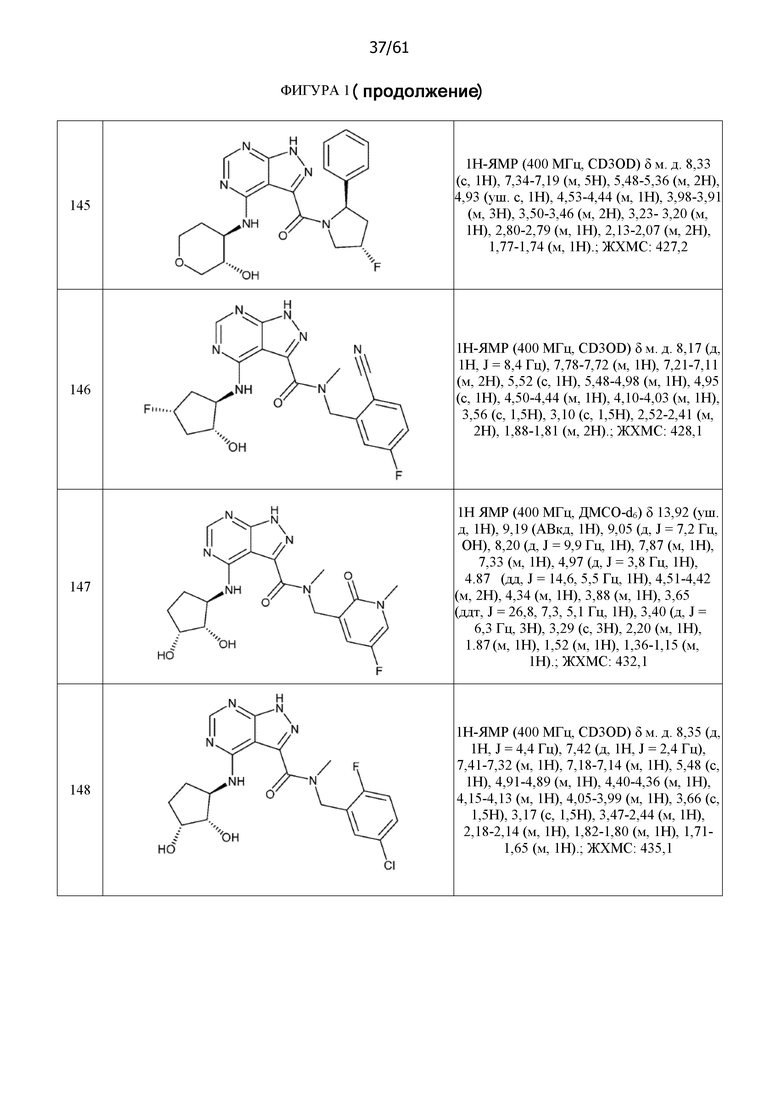
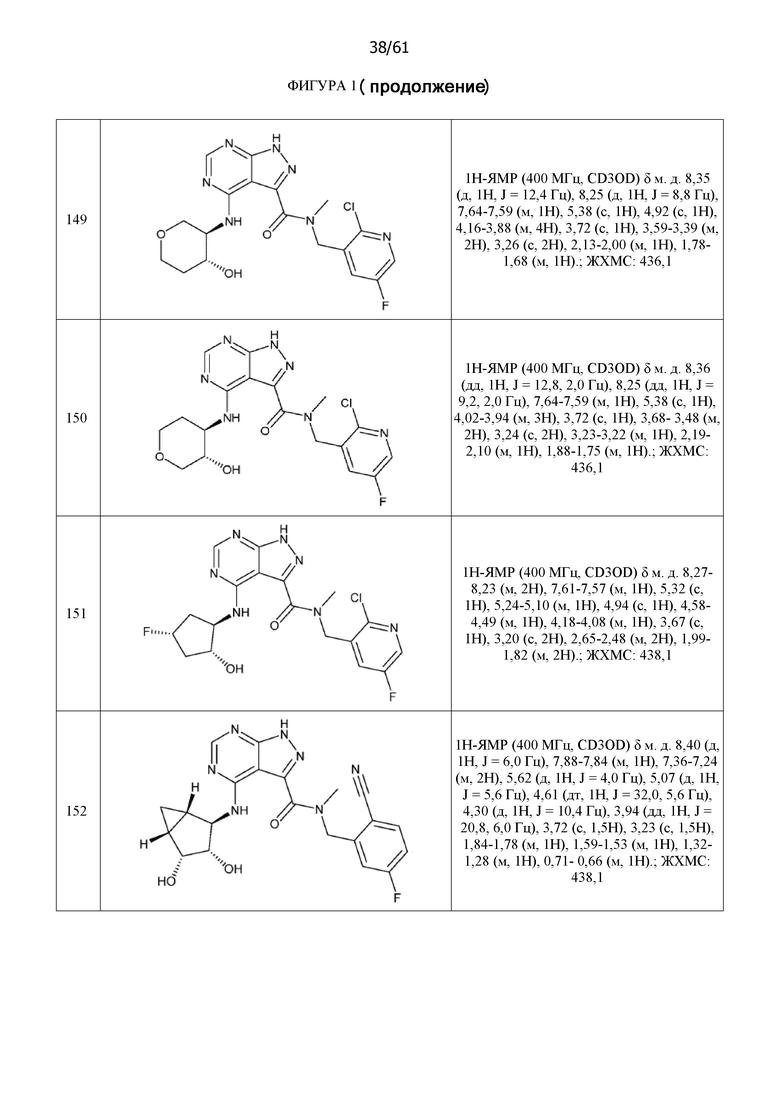
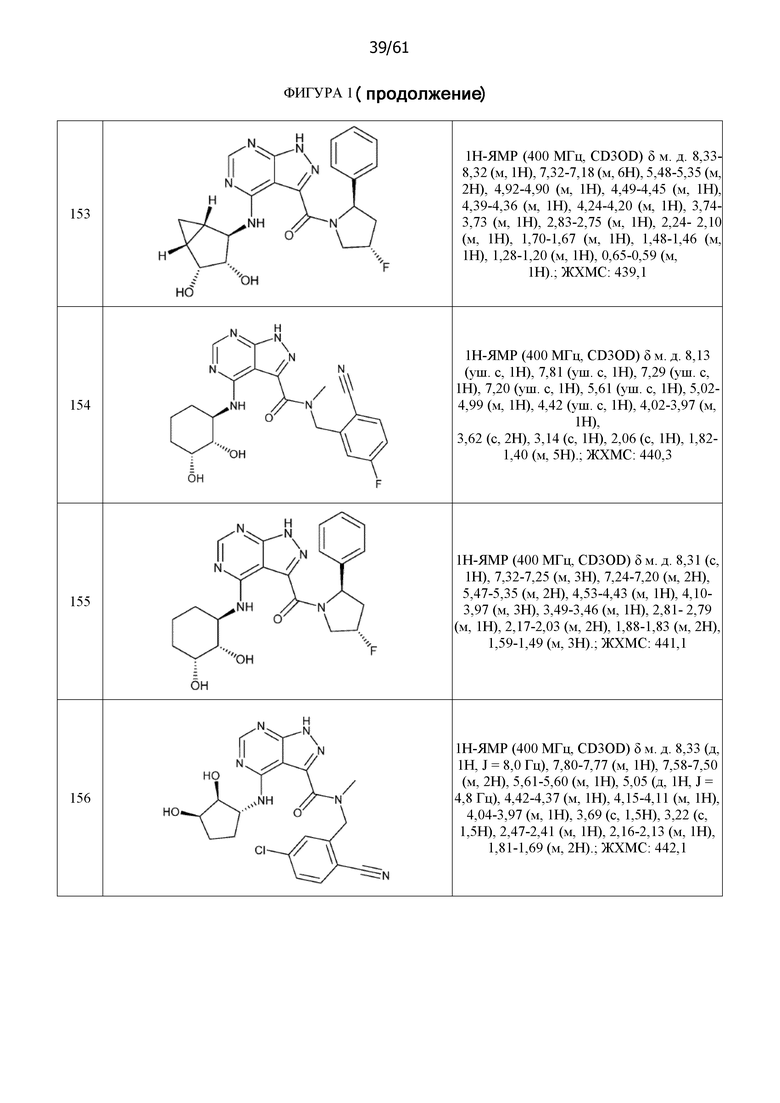
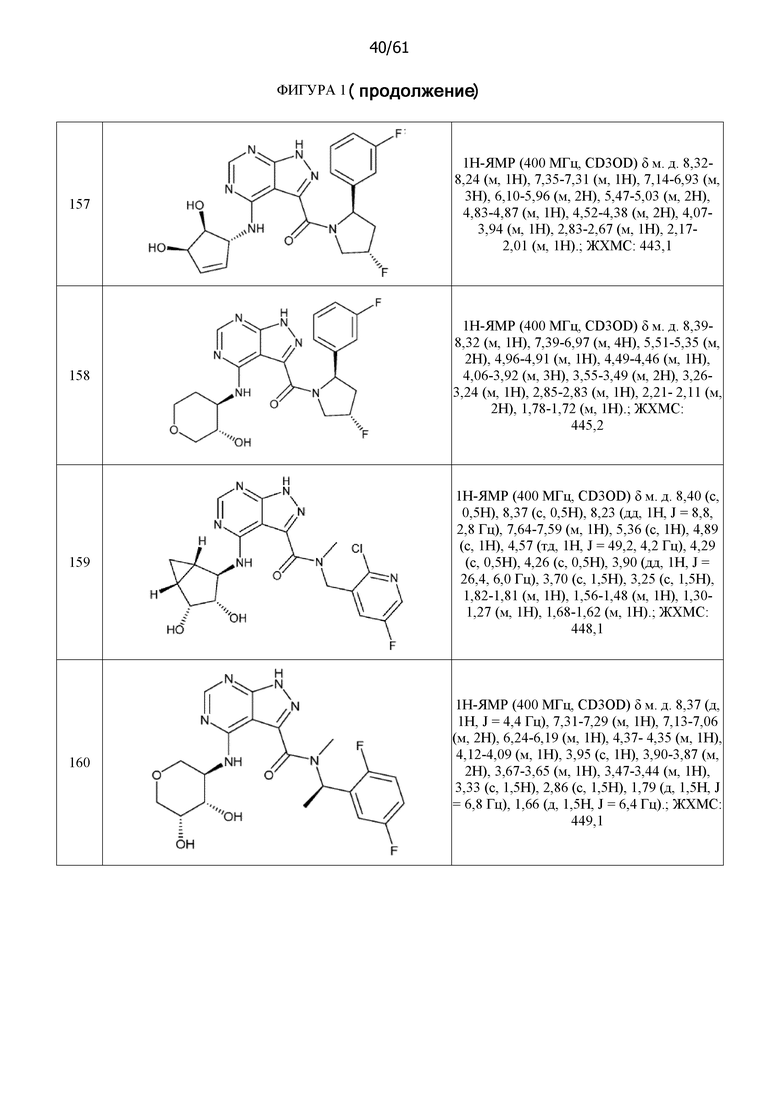
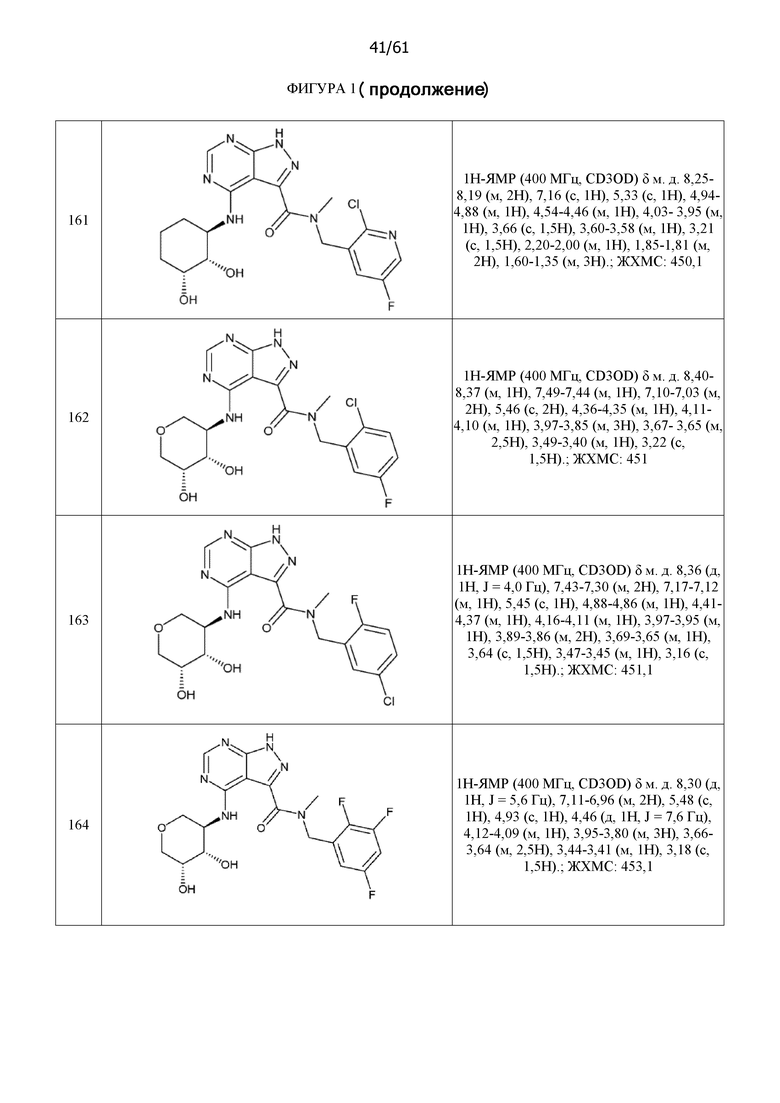
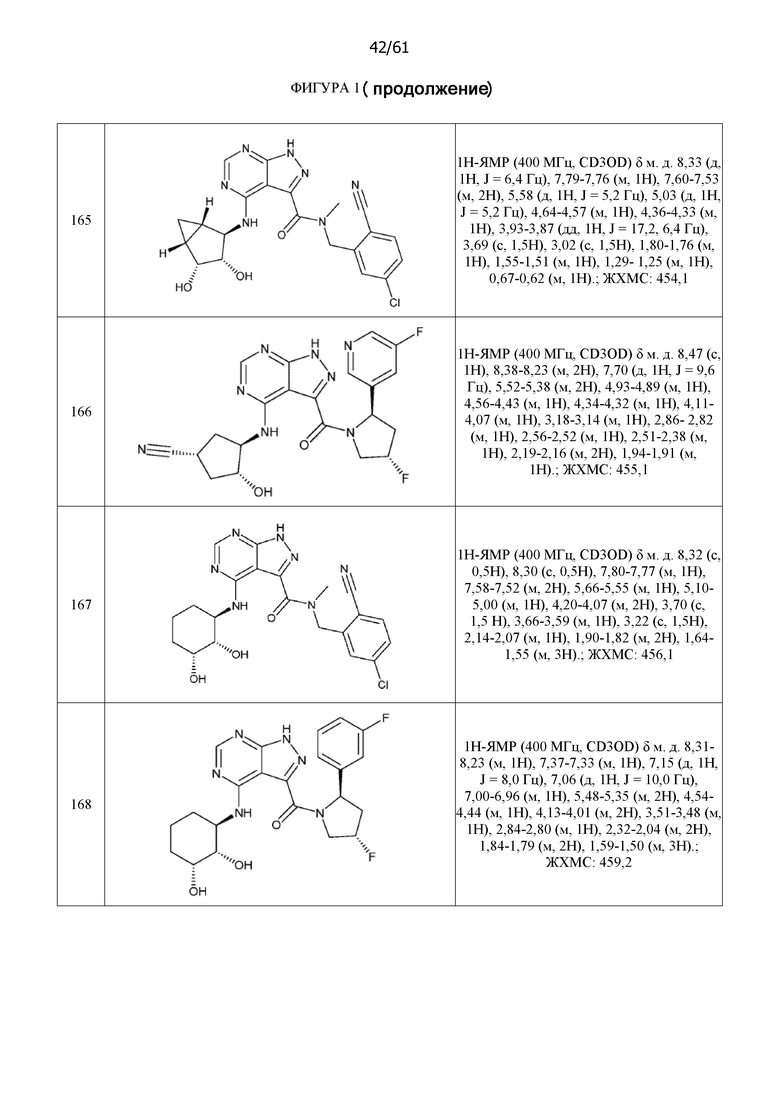
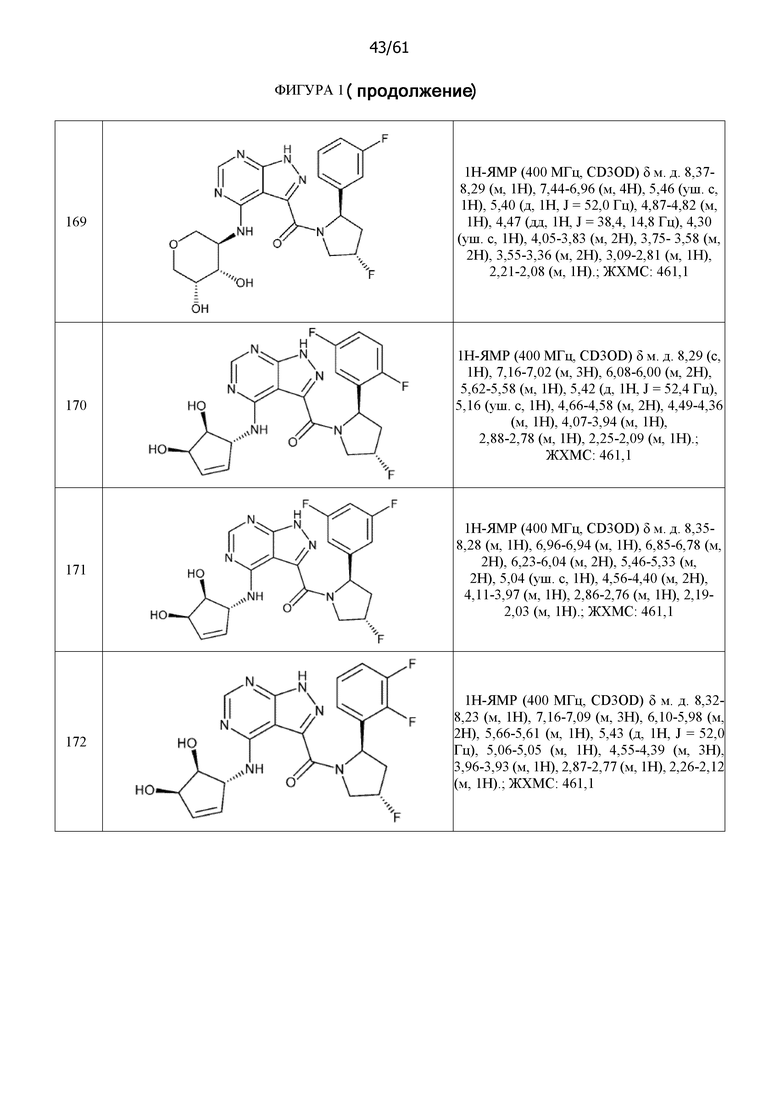
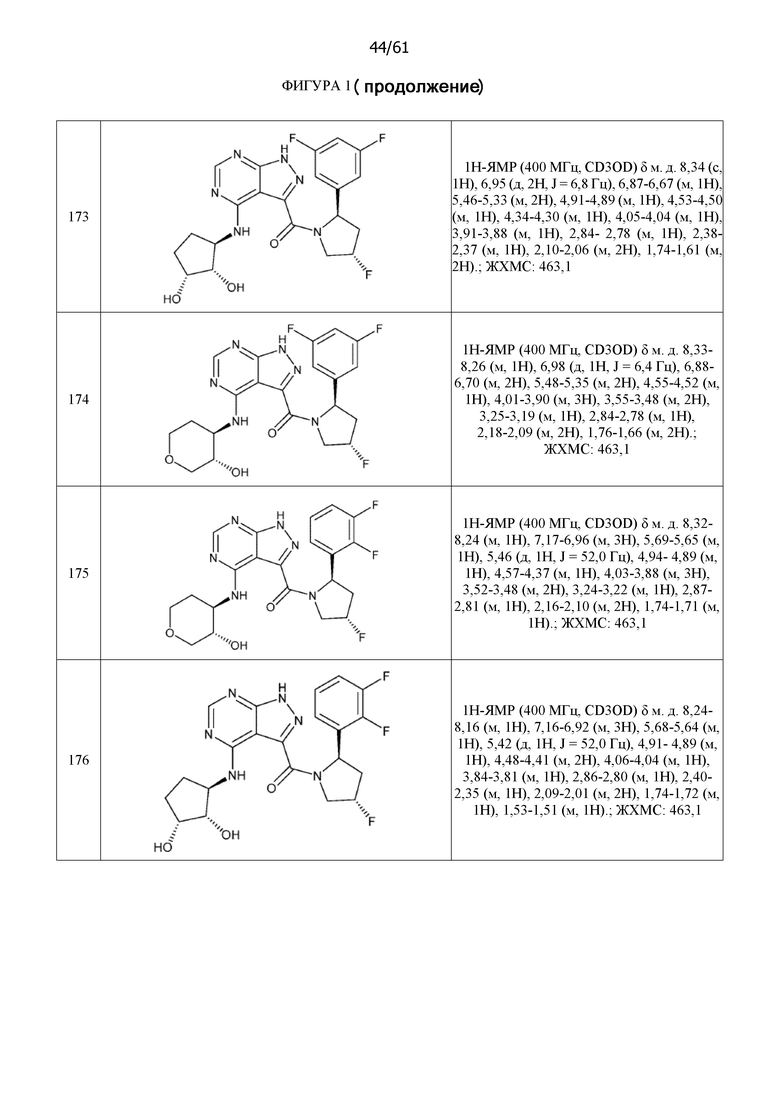
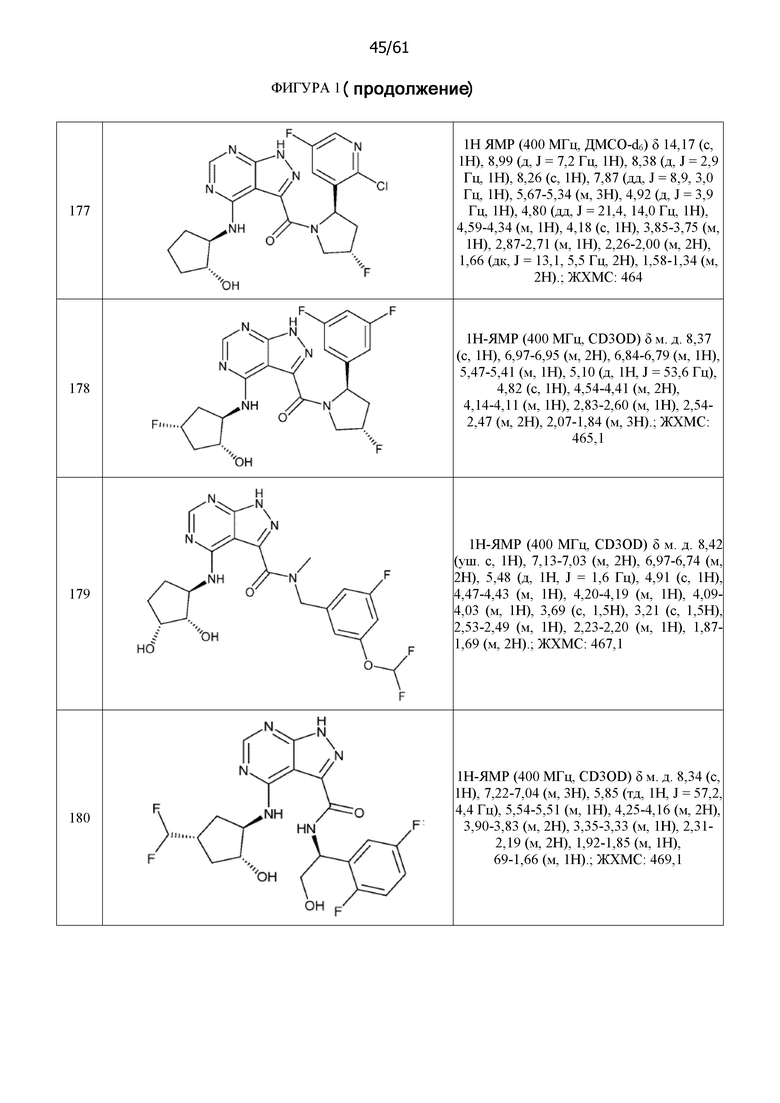

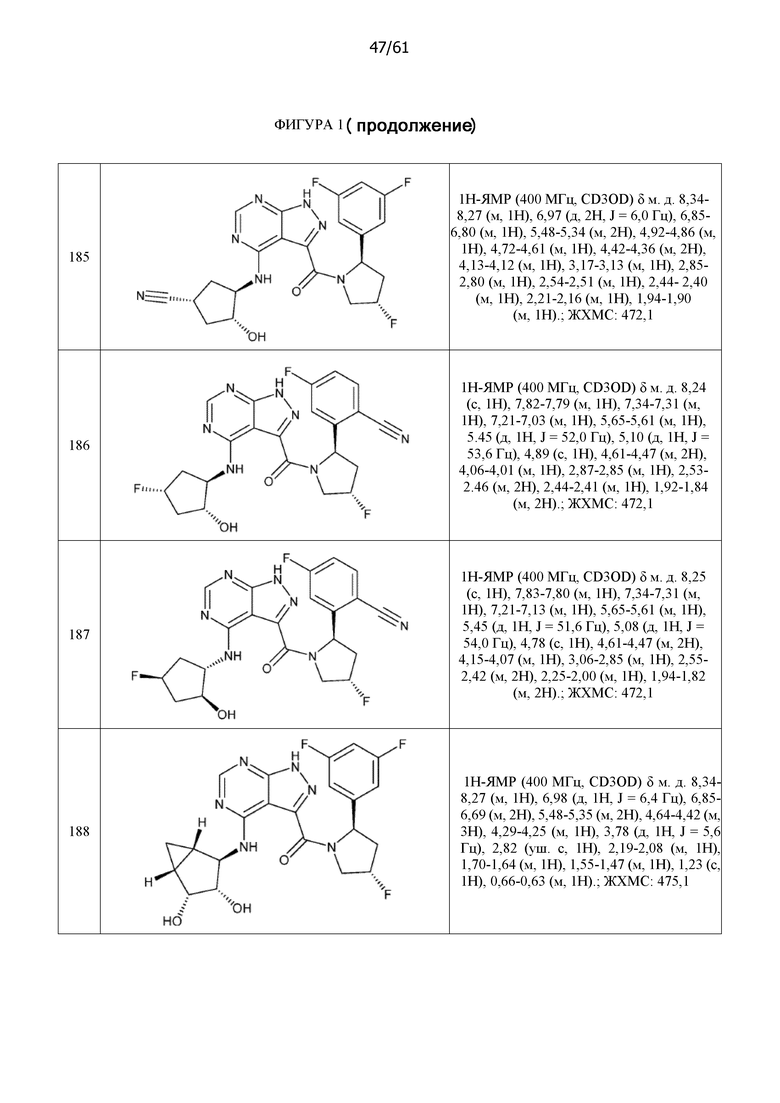
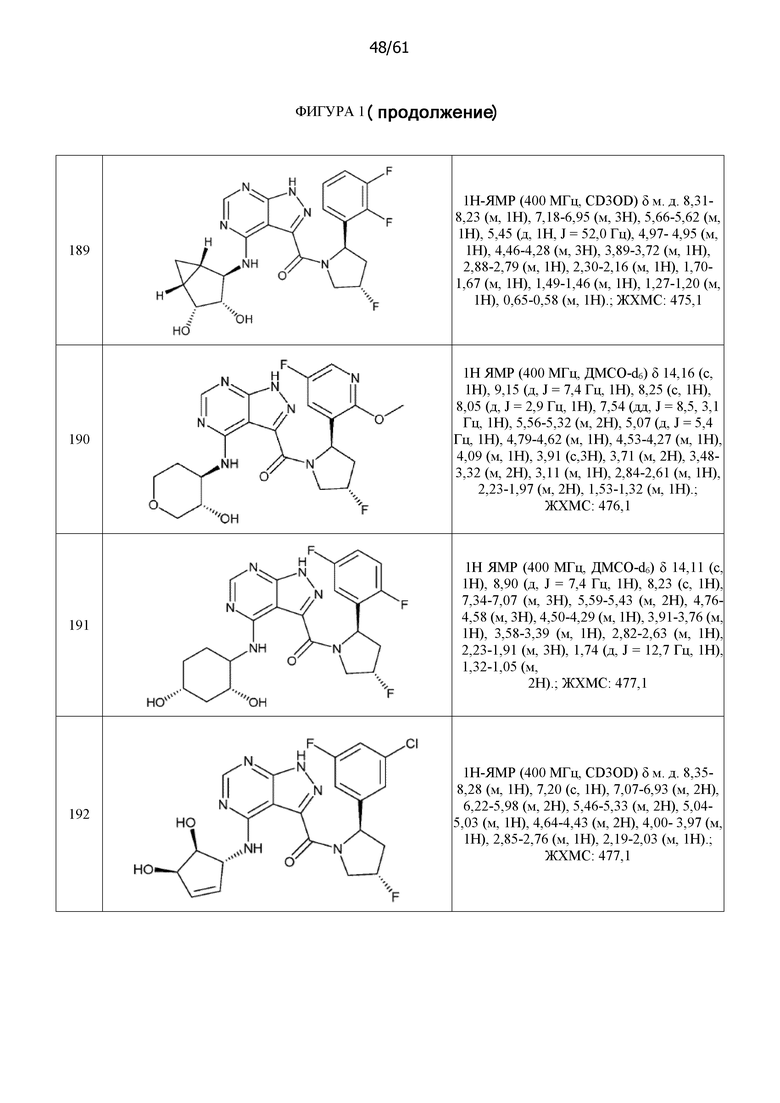

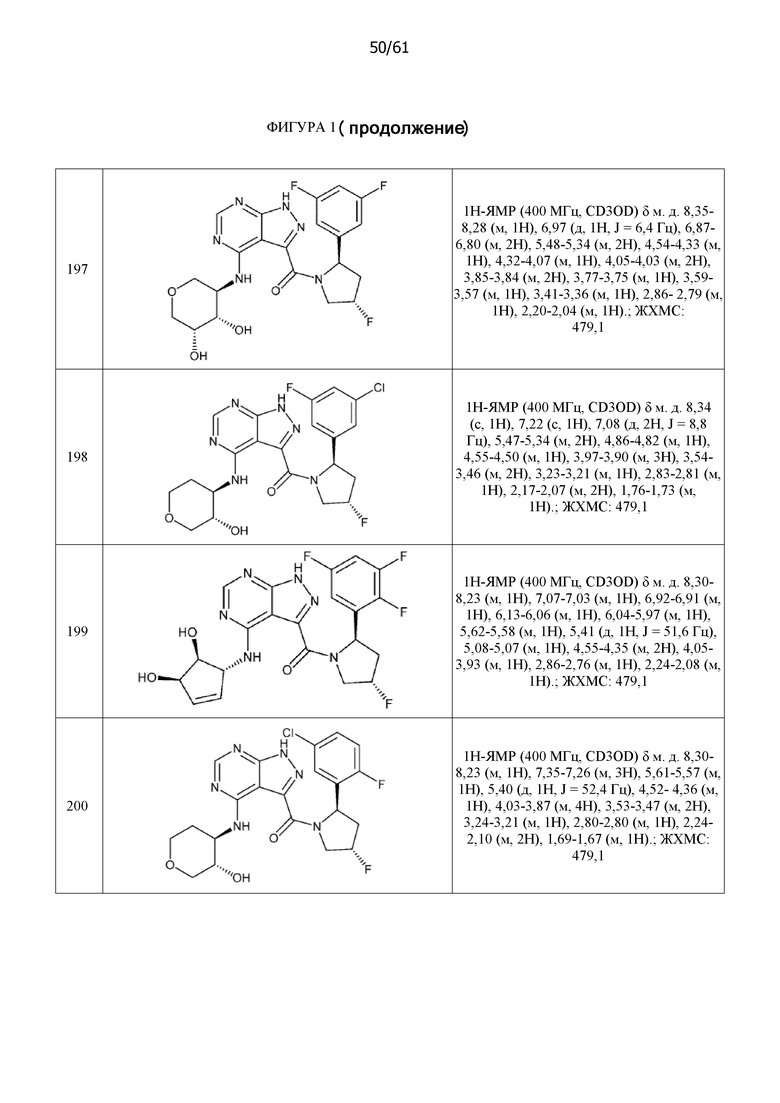
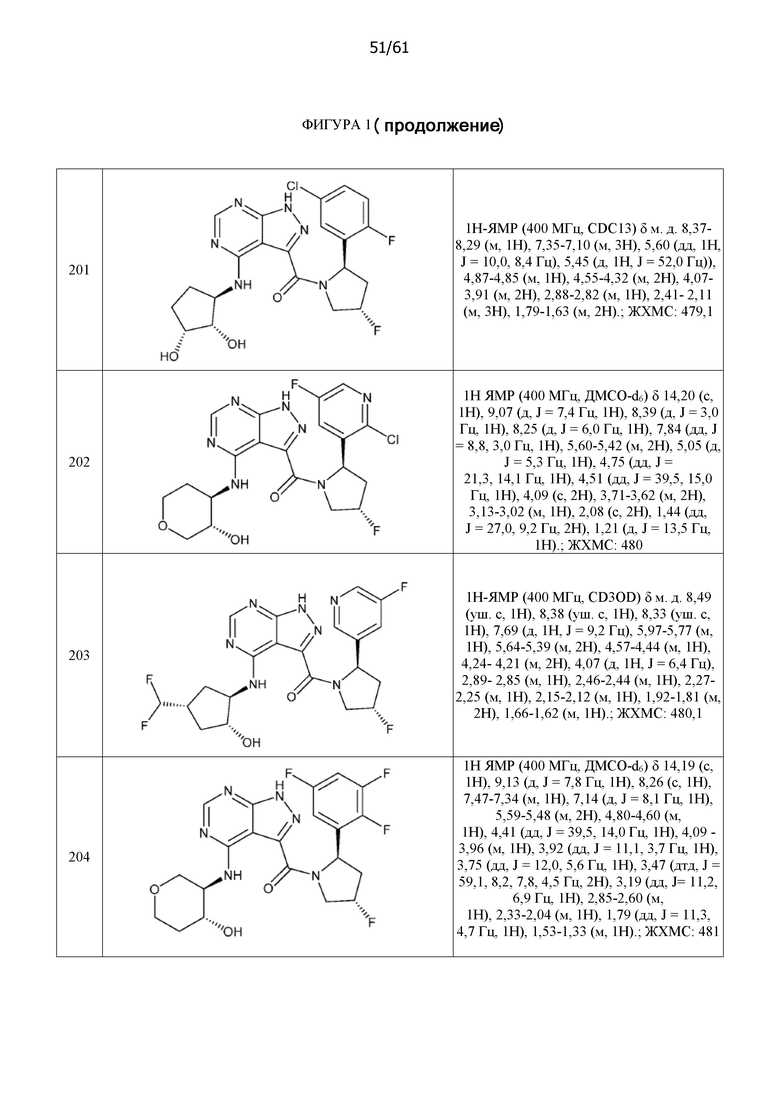
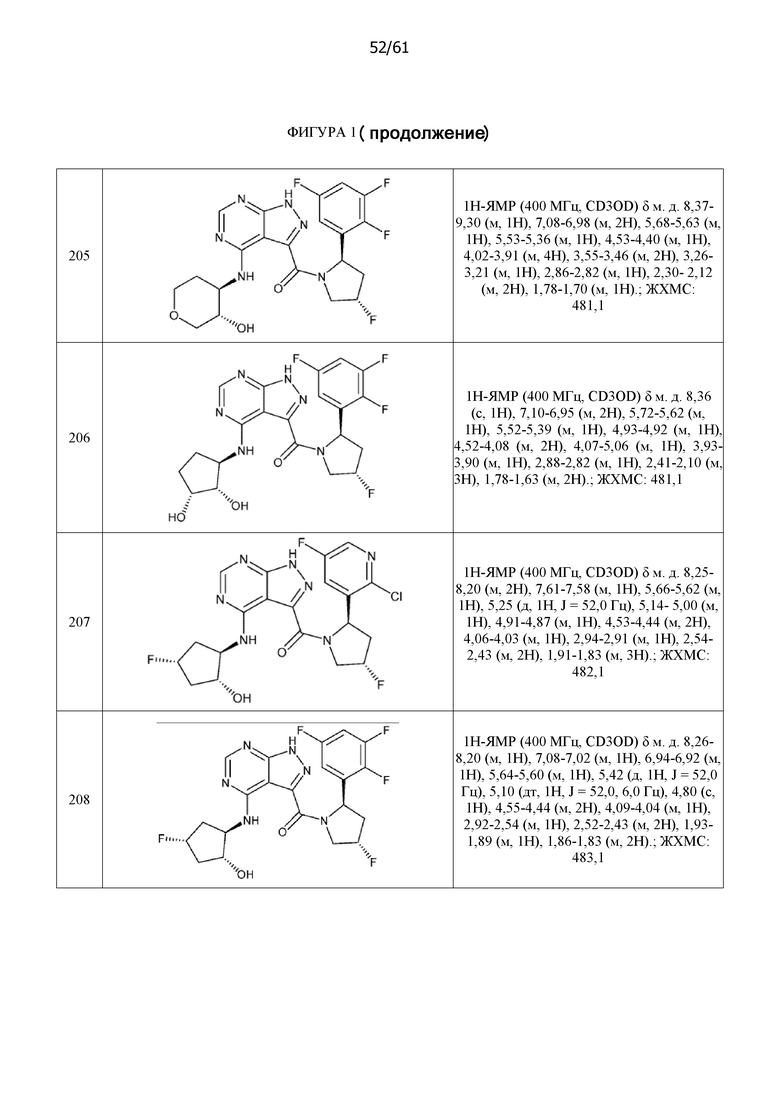
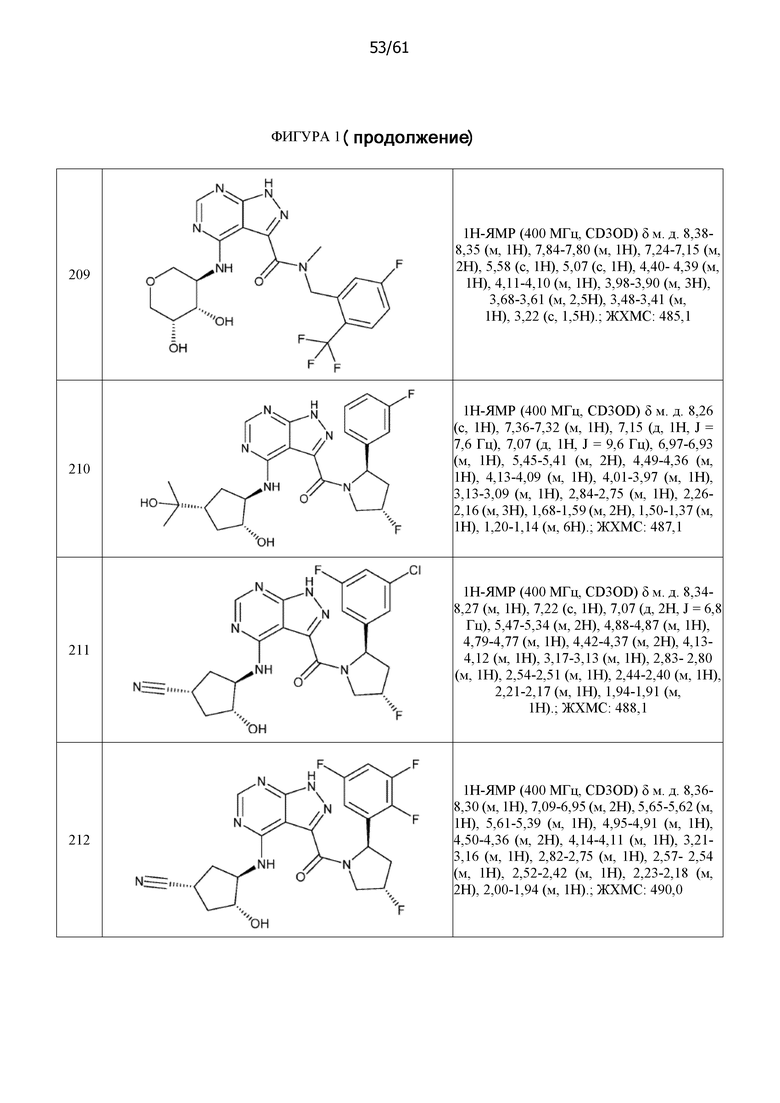
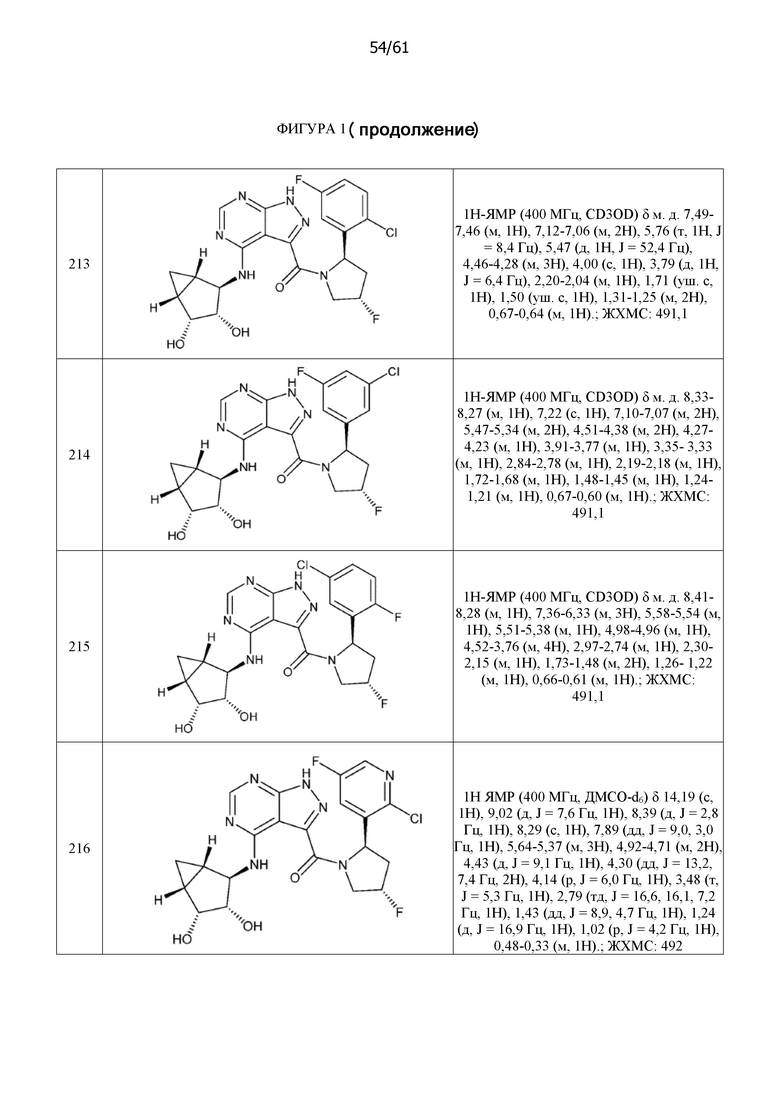

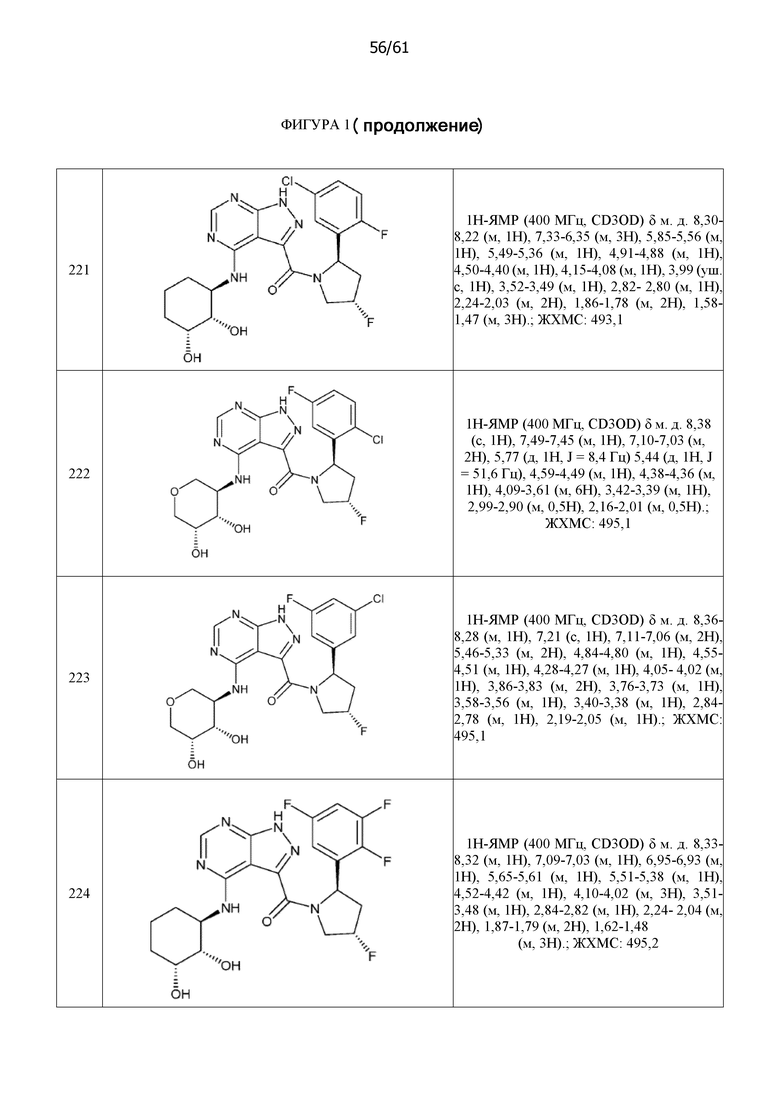
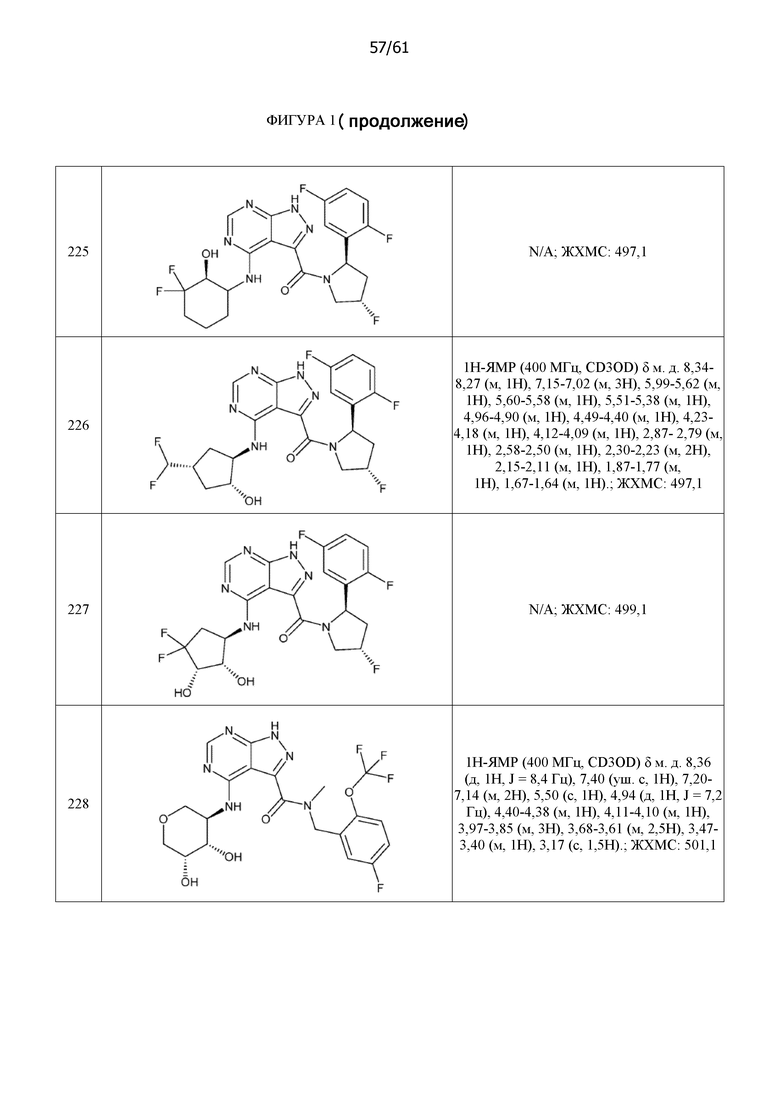
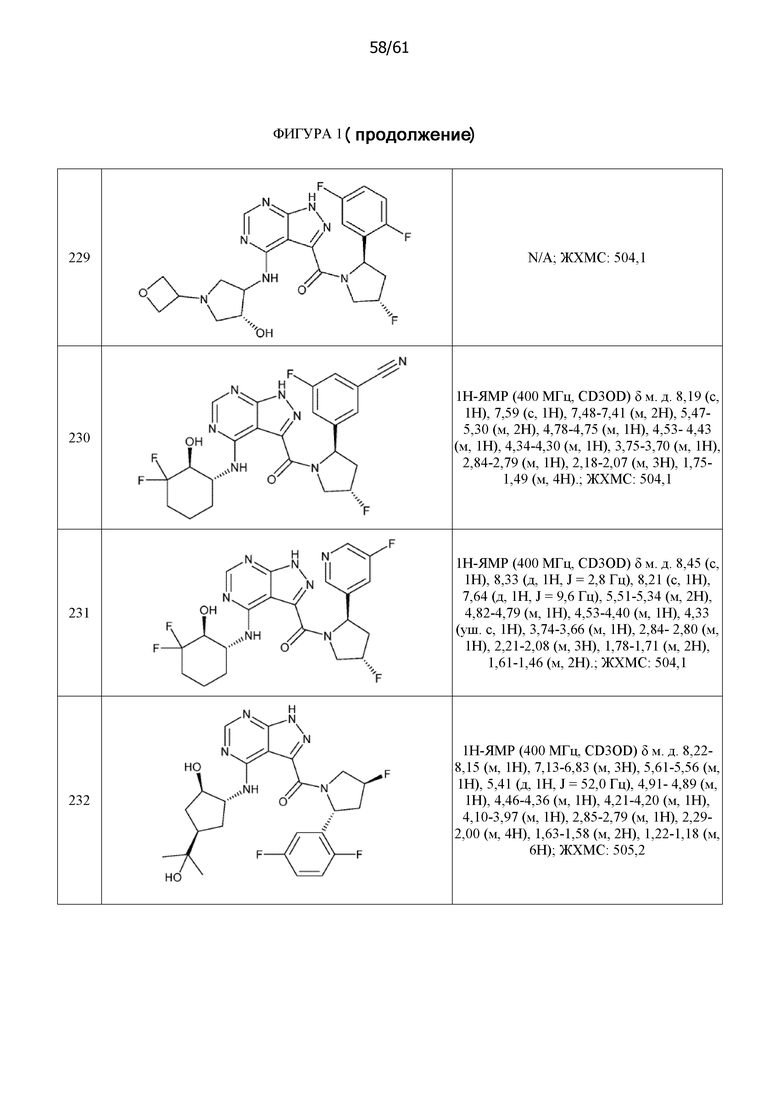
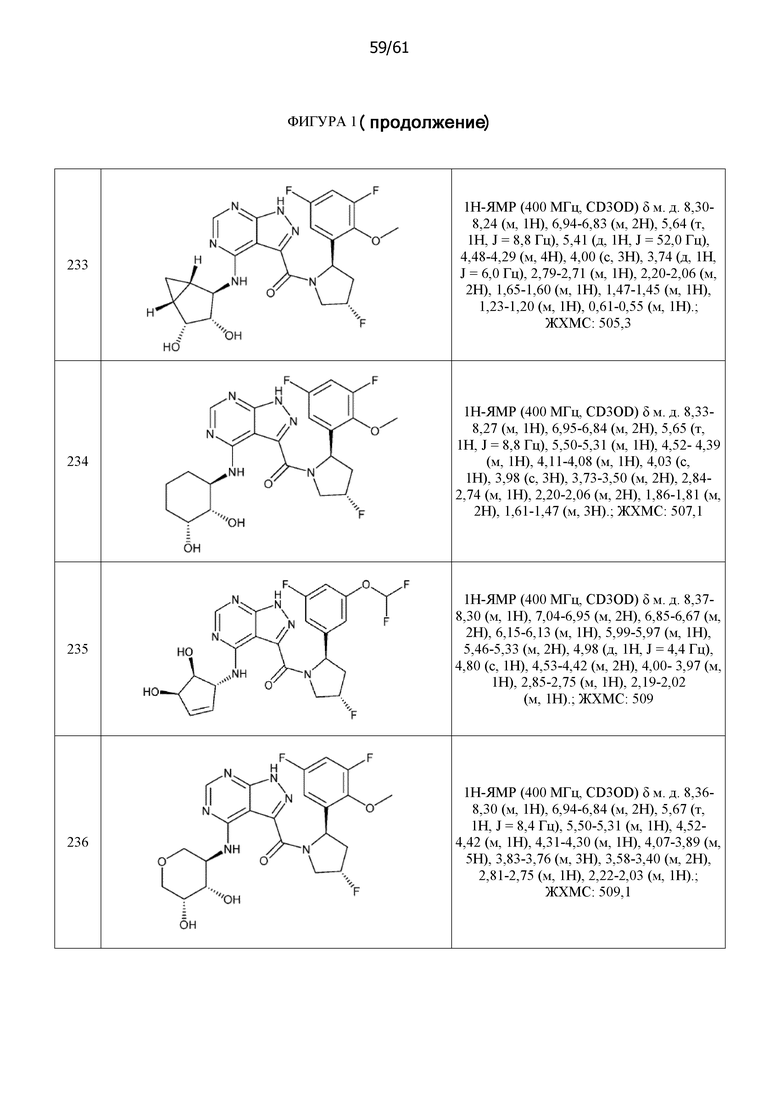
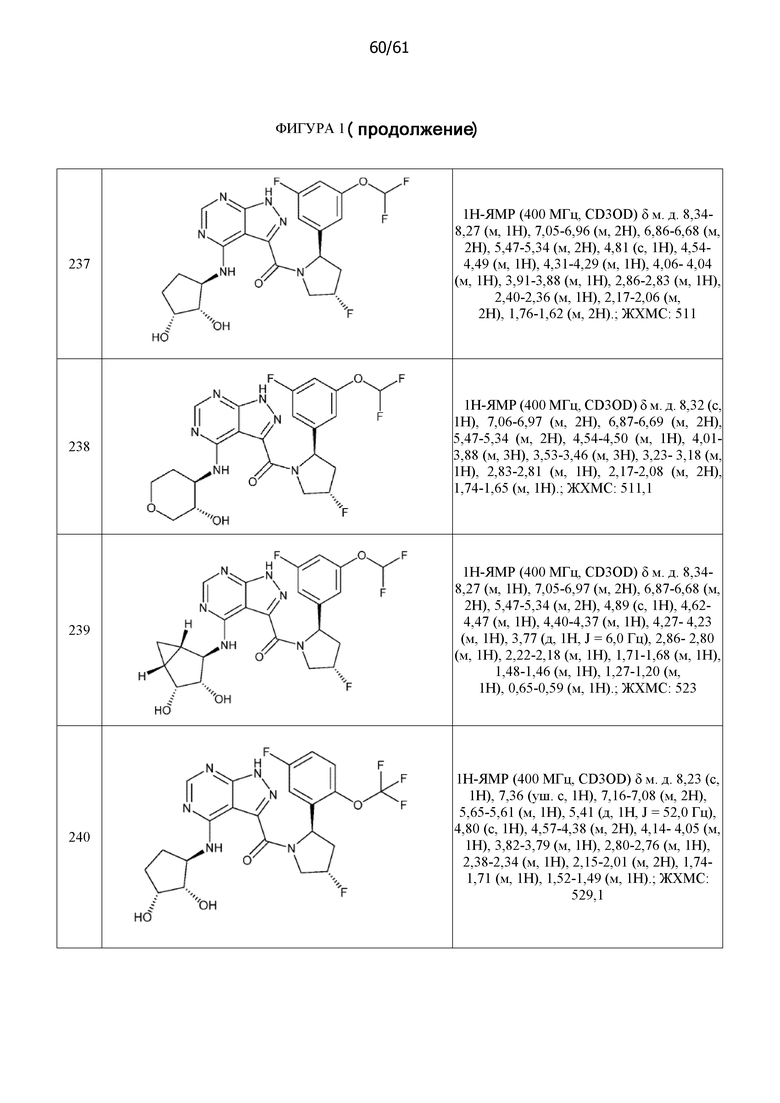
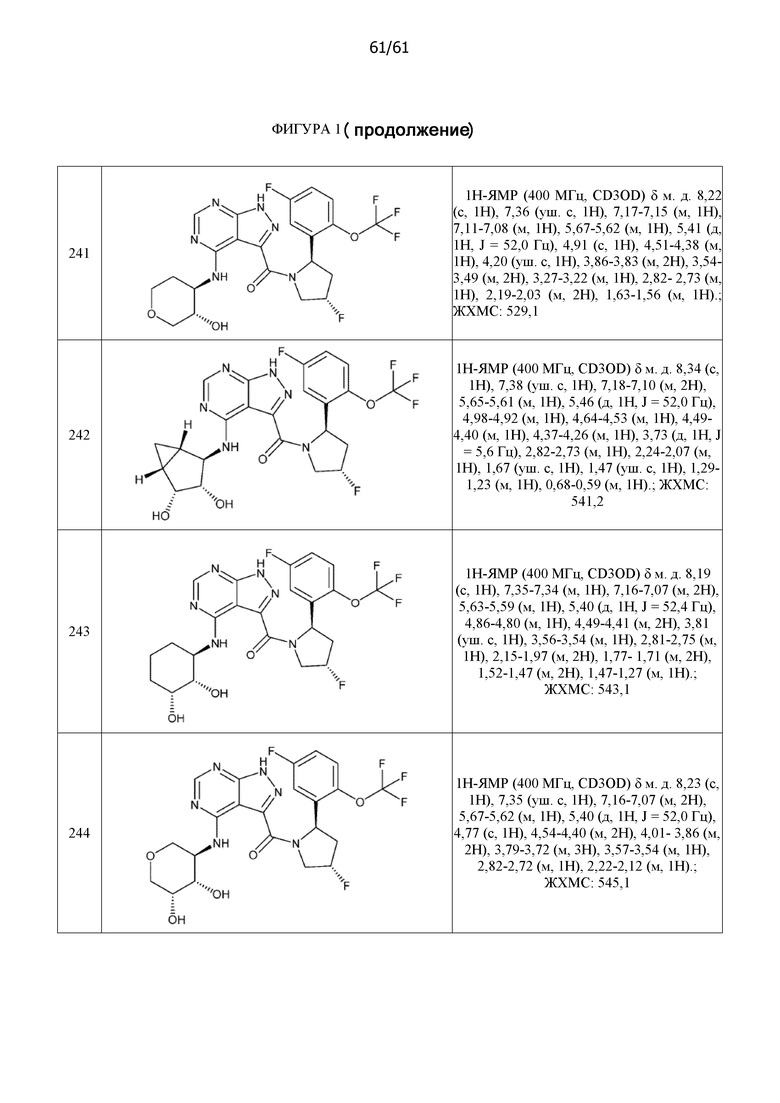
Изобретение относится к новым ингибиторам нейротрофной рецепторной тирозинкиназы (NTRK), к фармацевтической композиции и к способу лечения субъекта, имеющего состояние, опосредованное аберрантной активностью NTRK. 8 н.п. ф-лы, 1 ил., 1 табл., 29 пр.
1. Соединение, выбранное из любого из:
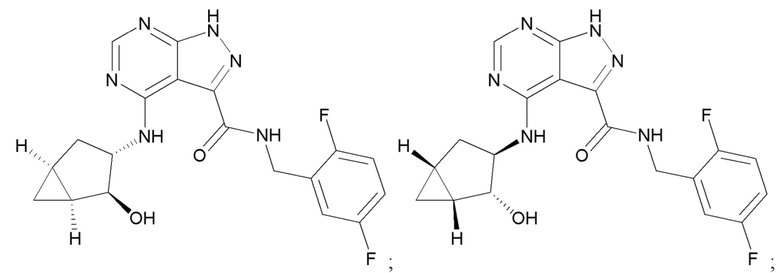
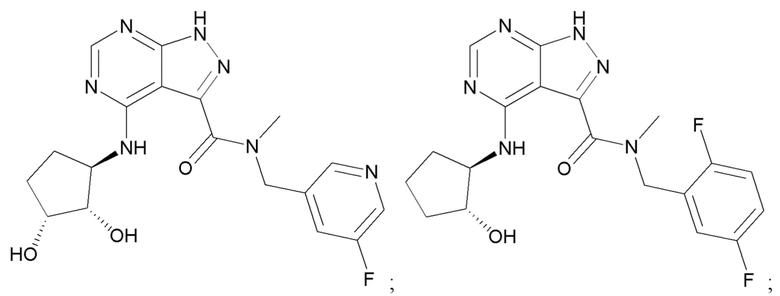
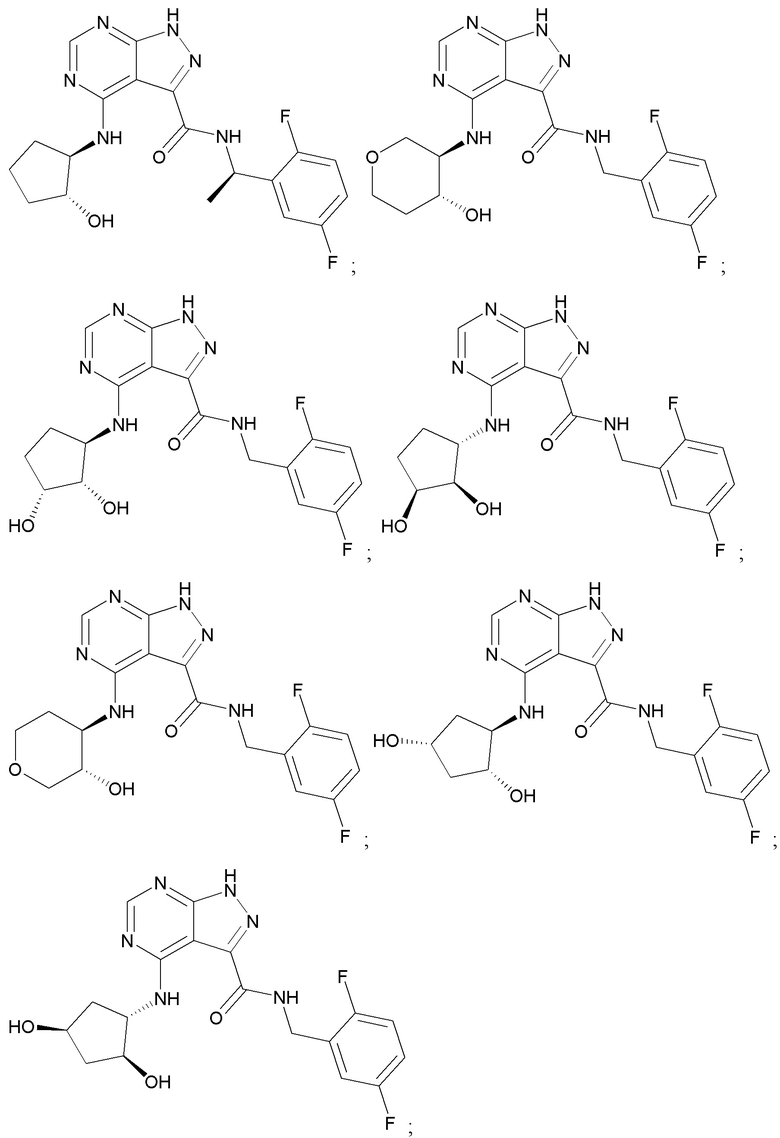
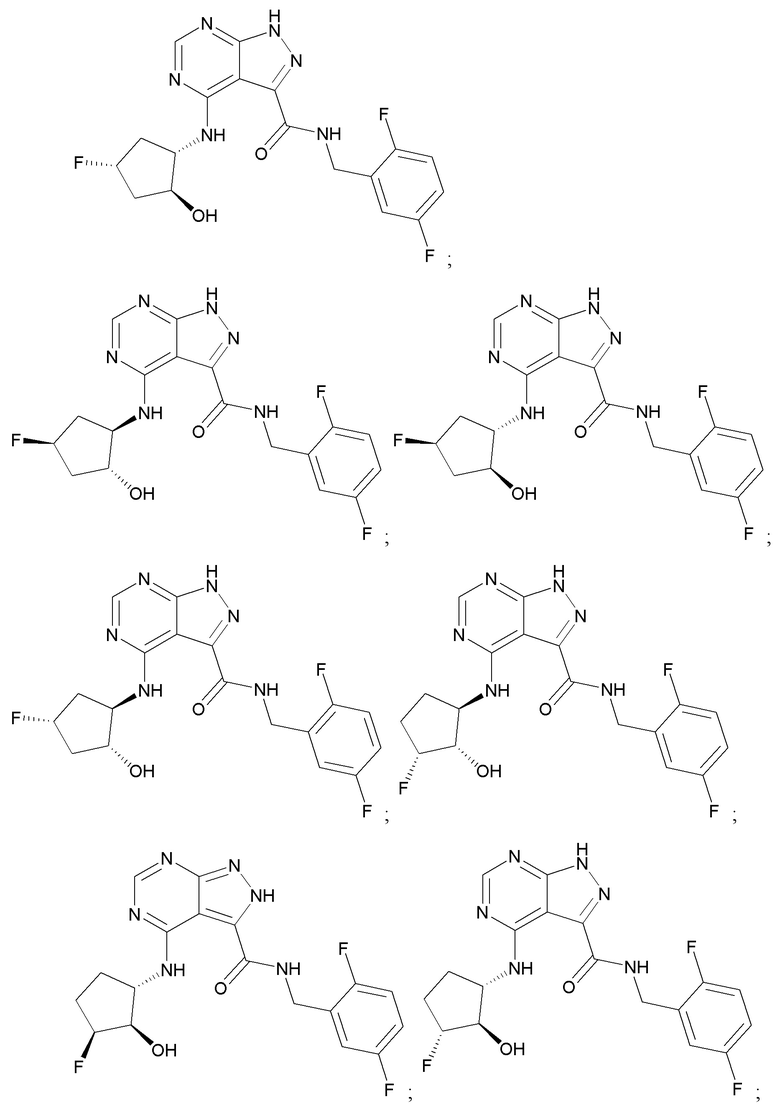

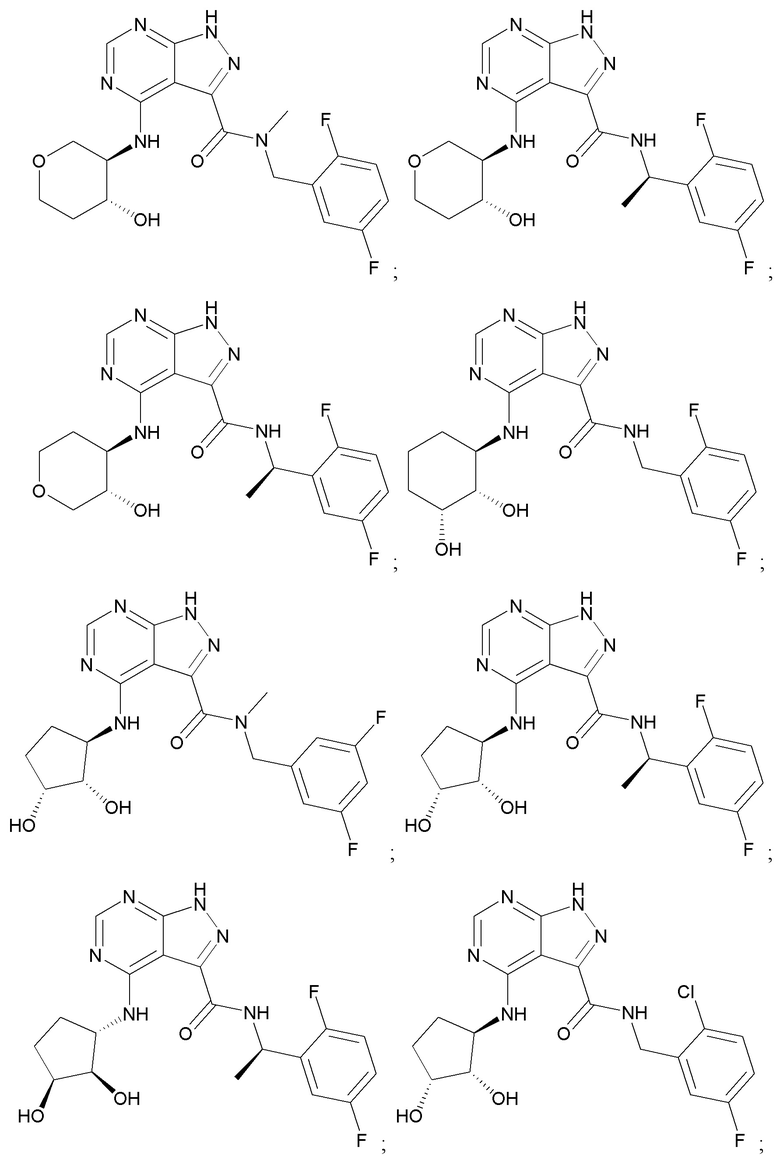
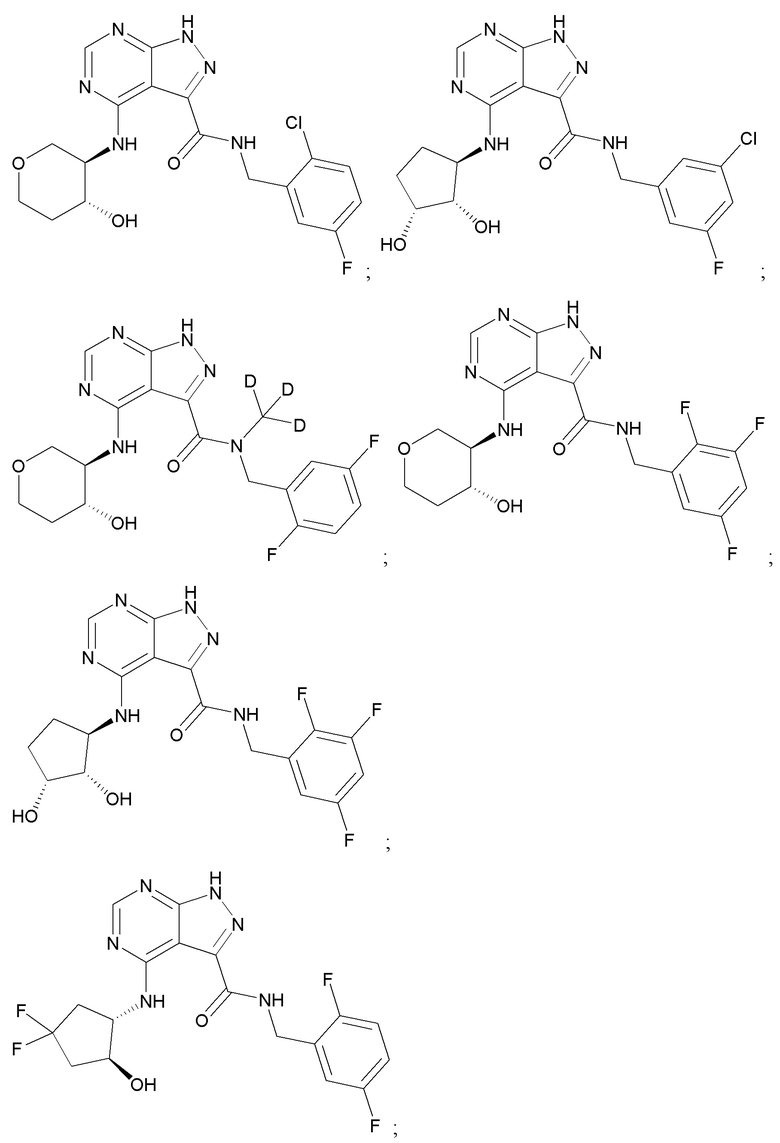
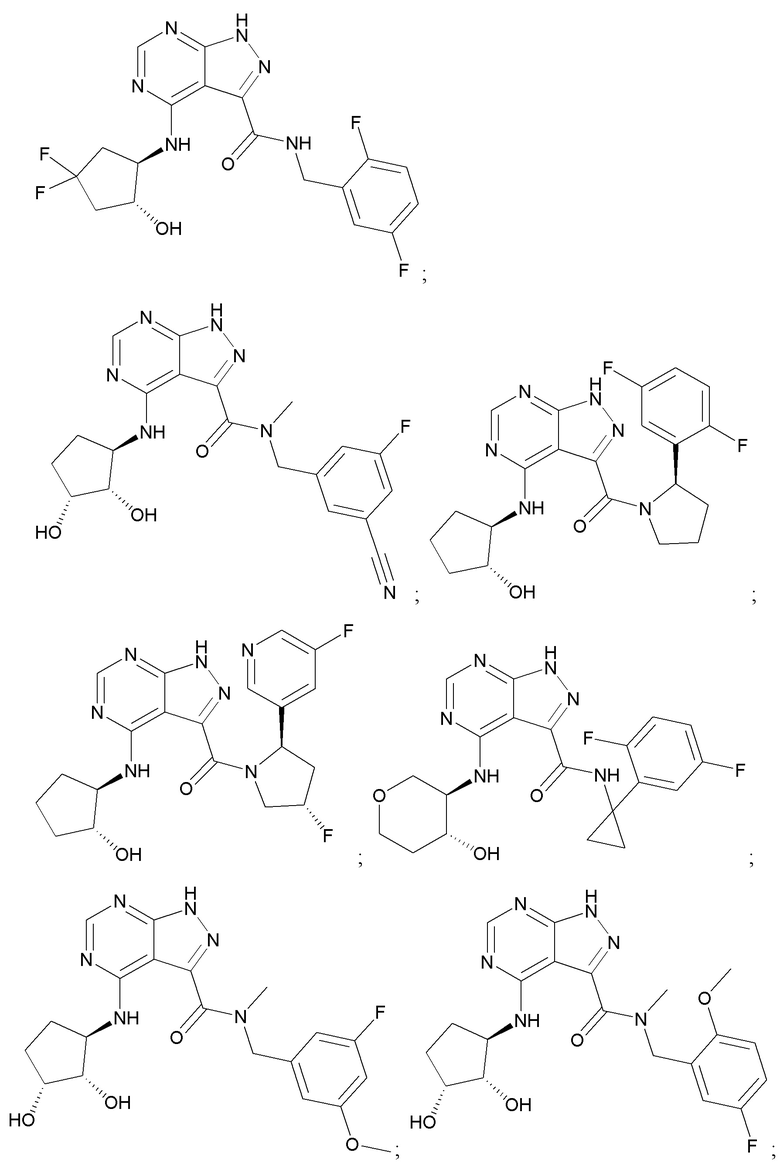
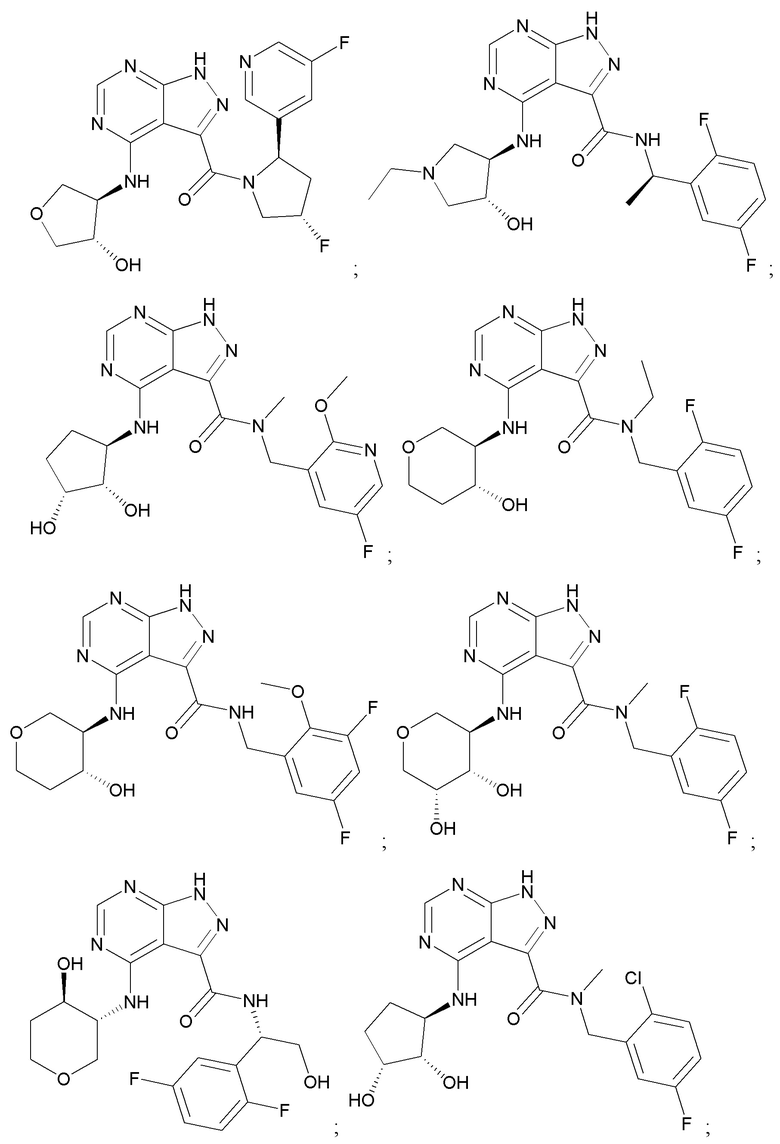
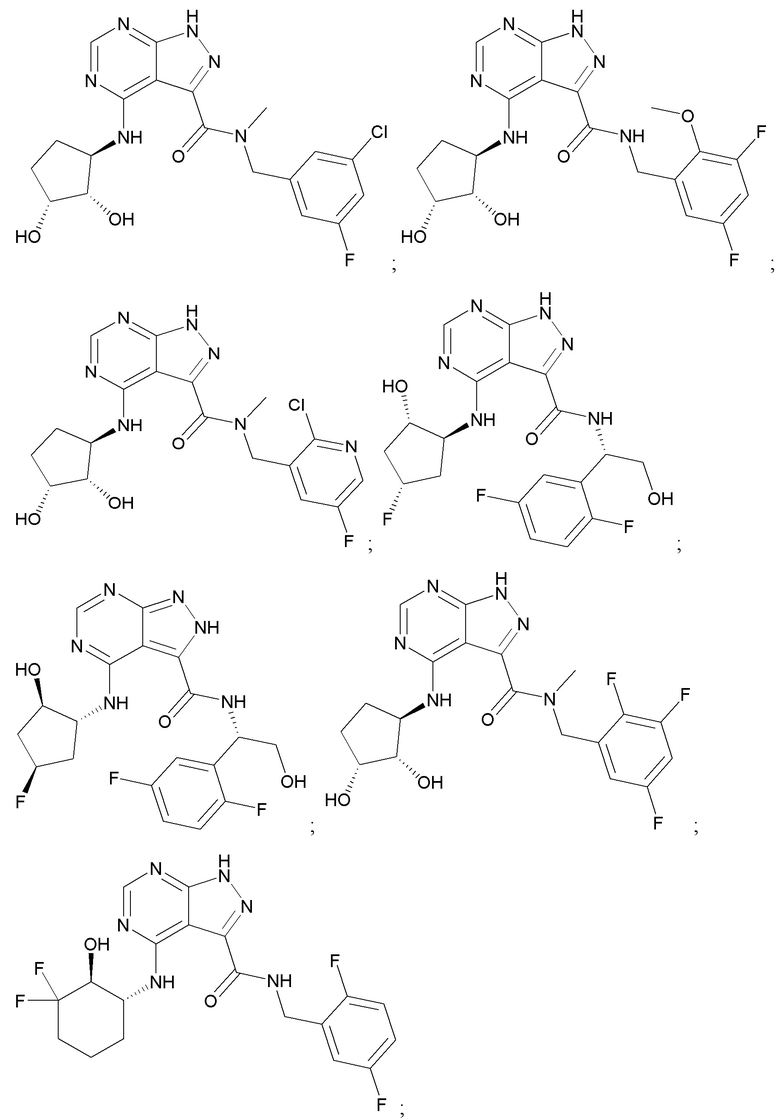
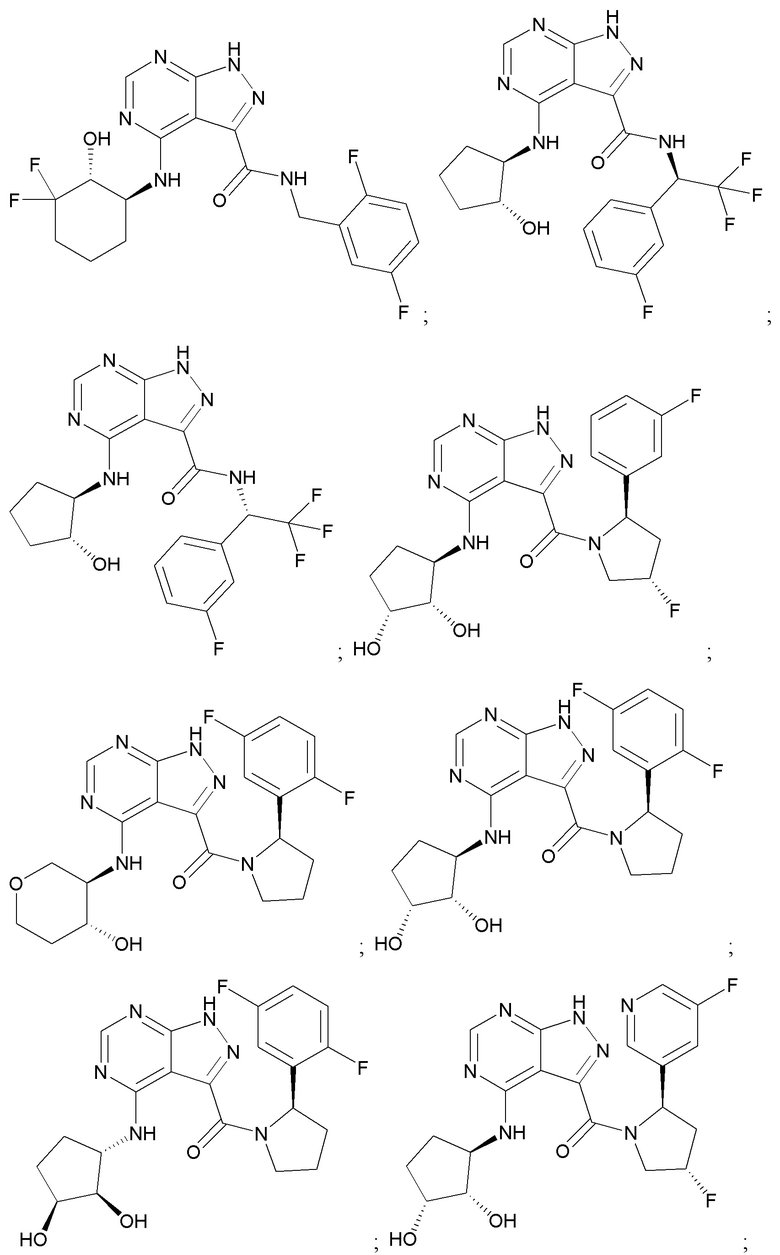
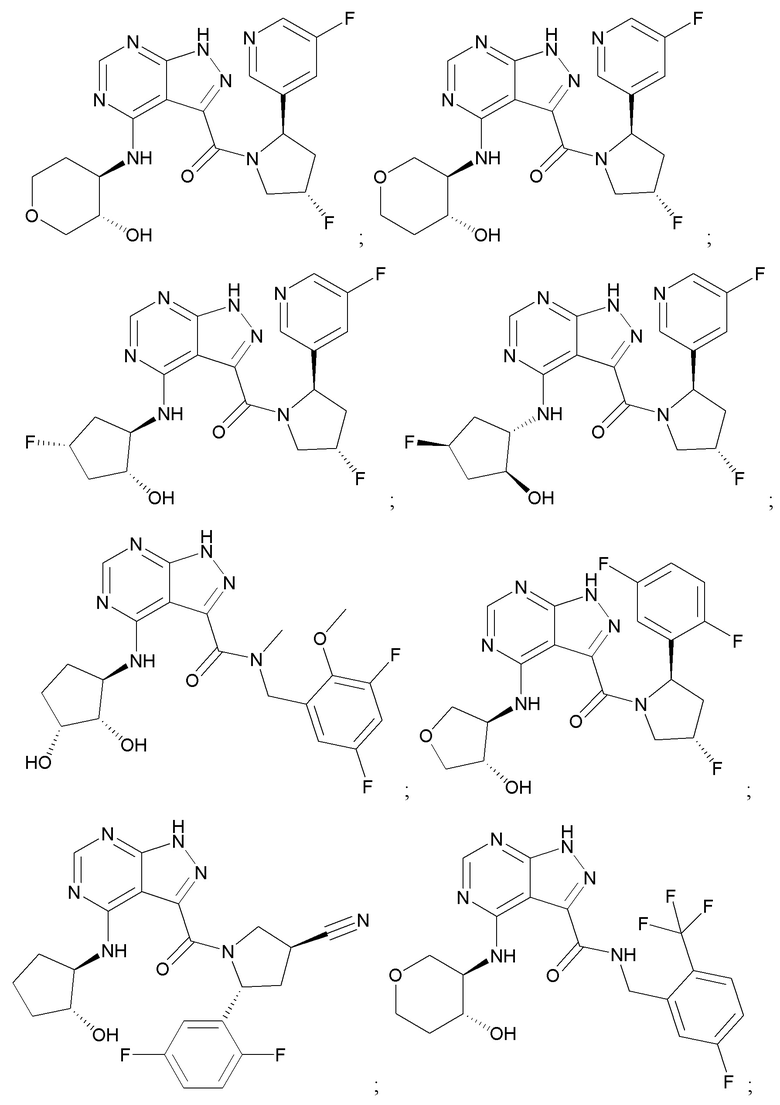
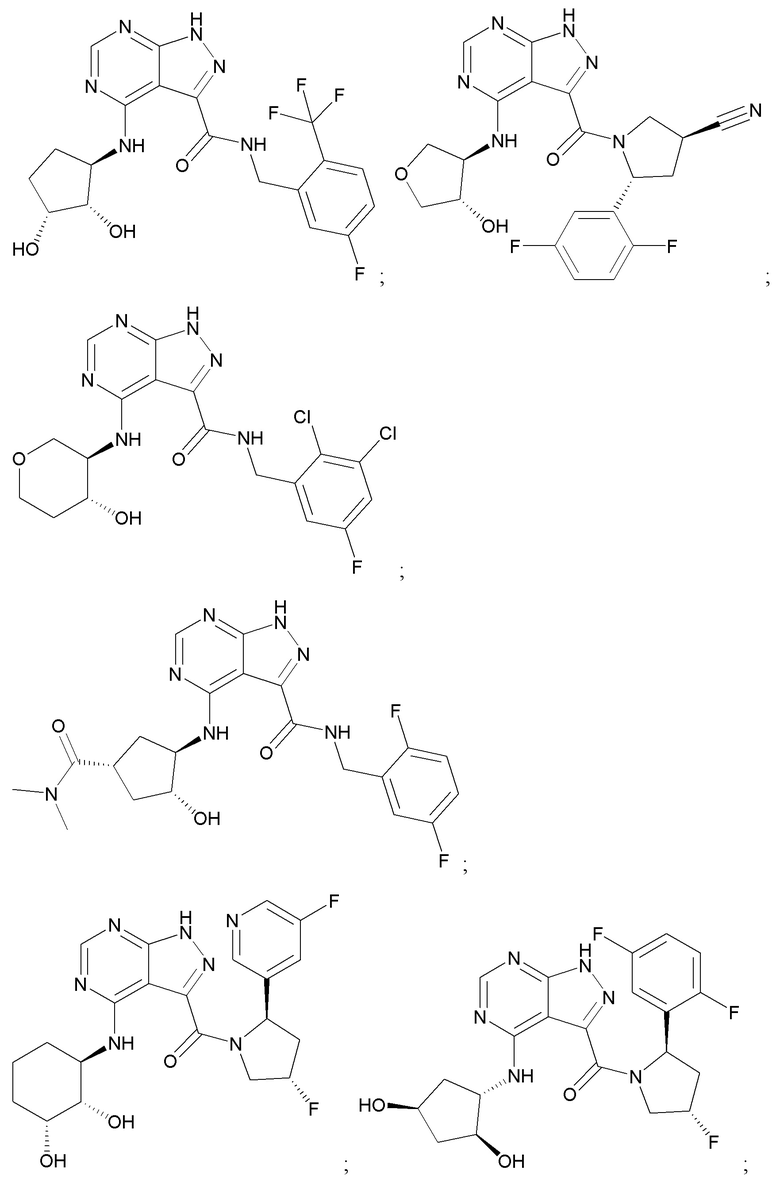
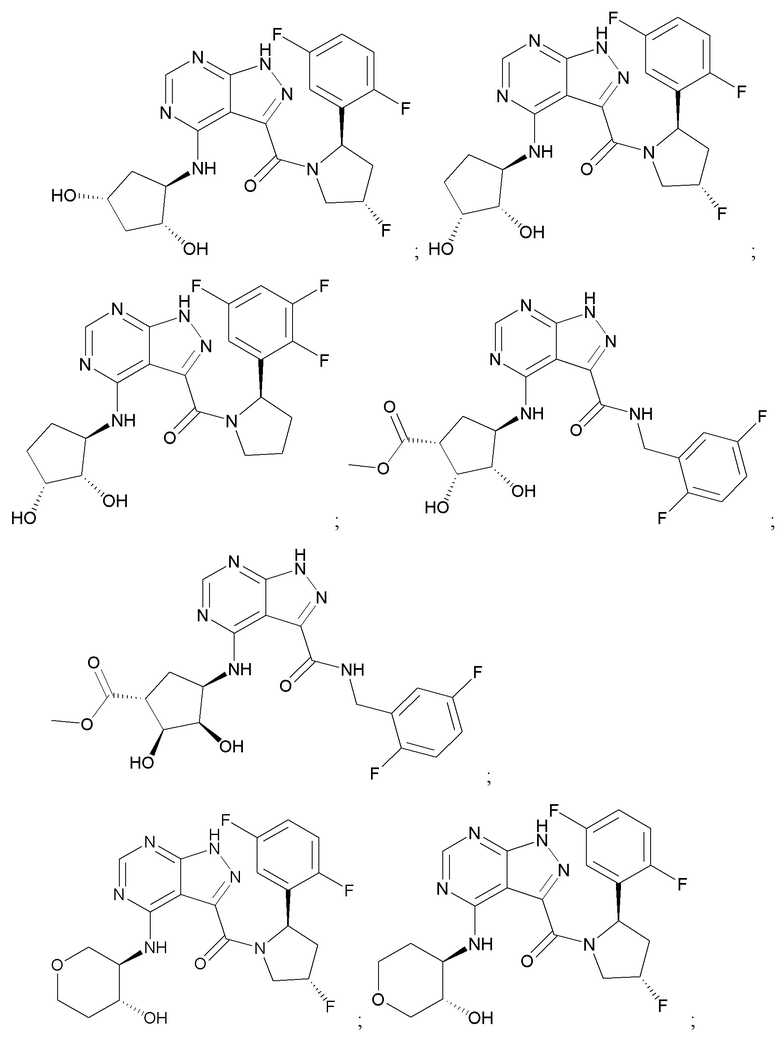
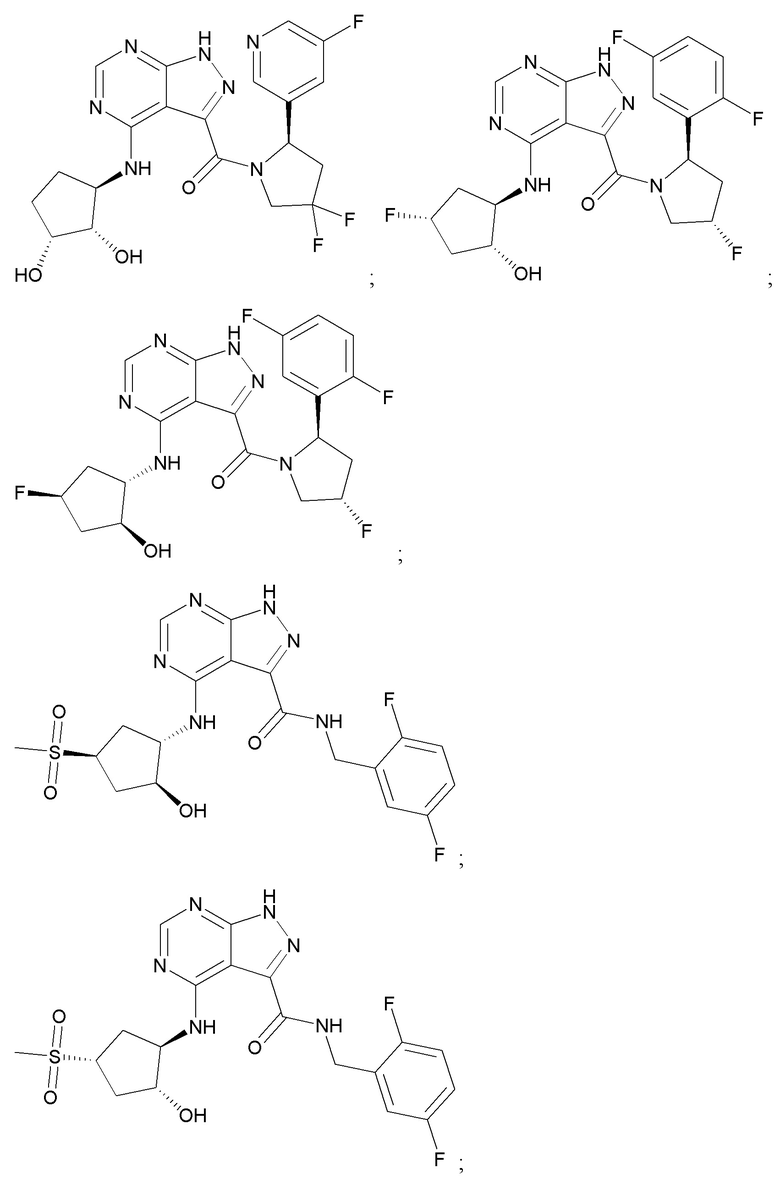
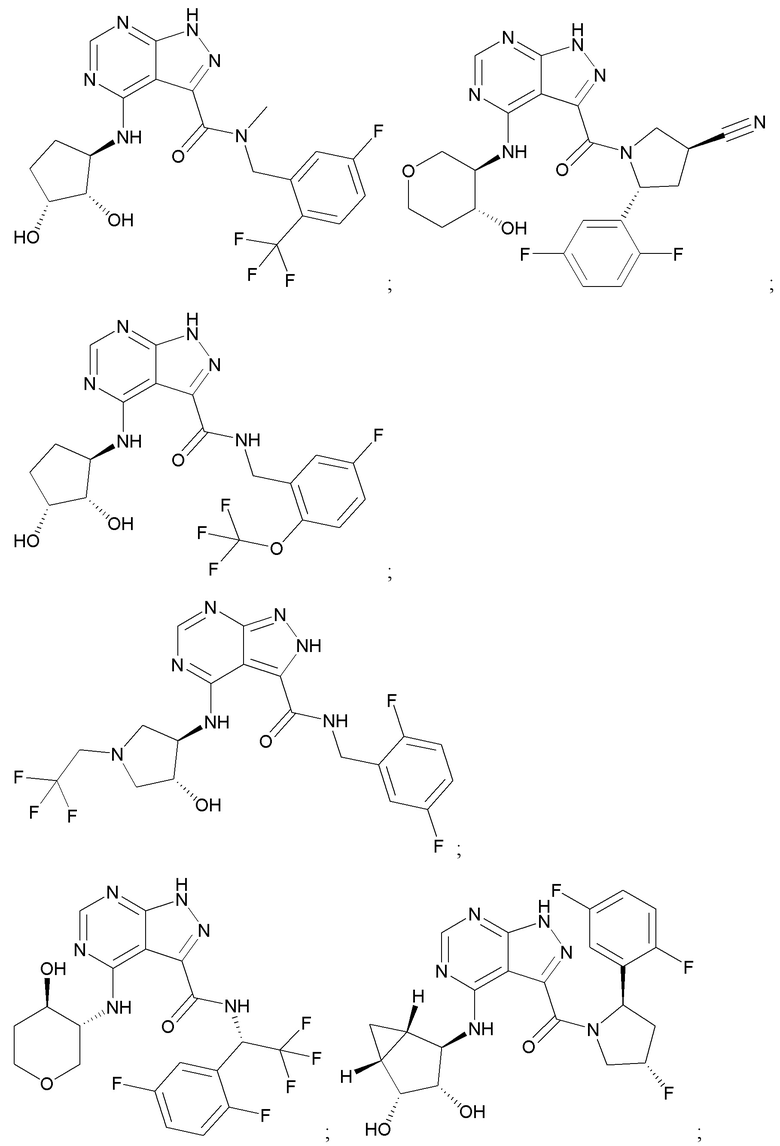
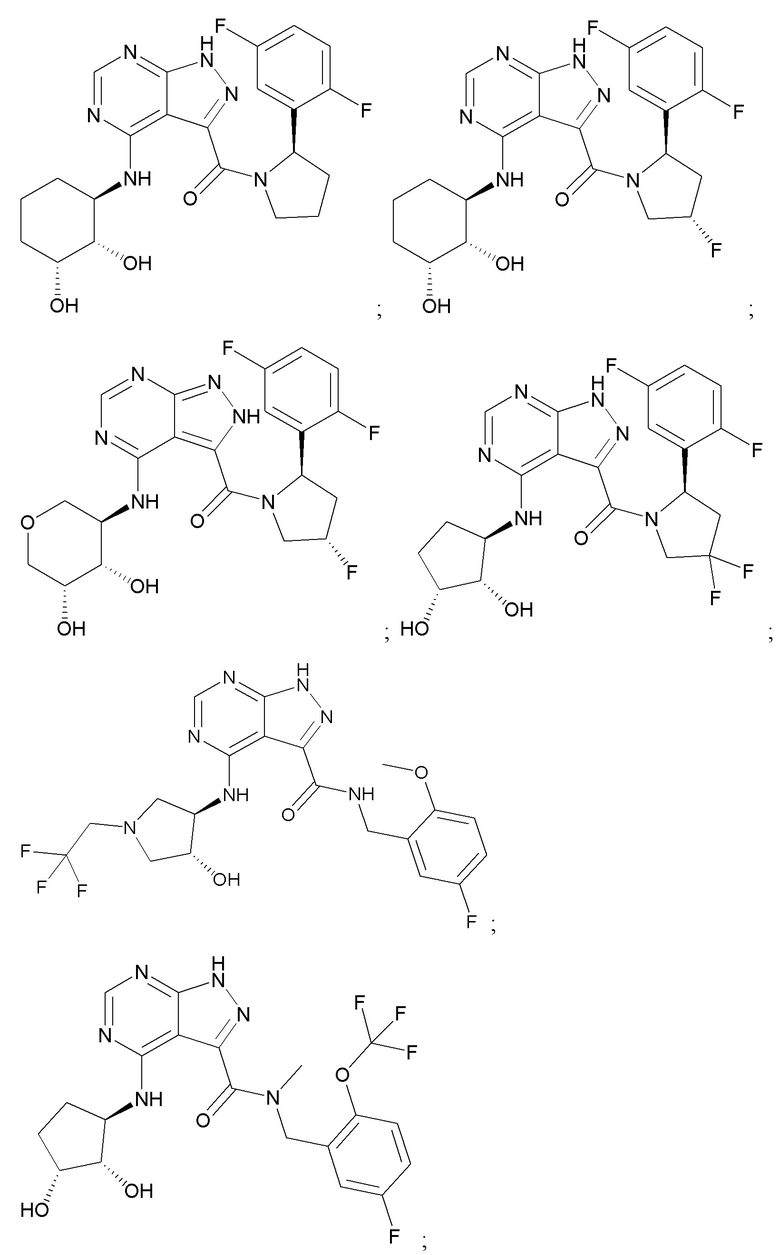
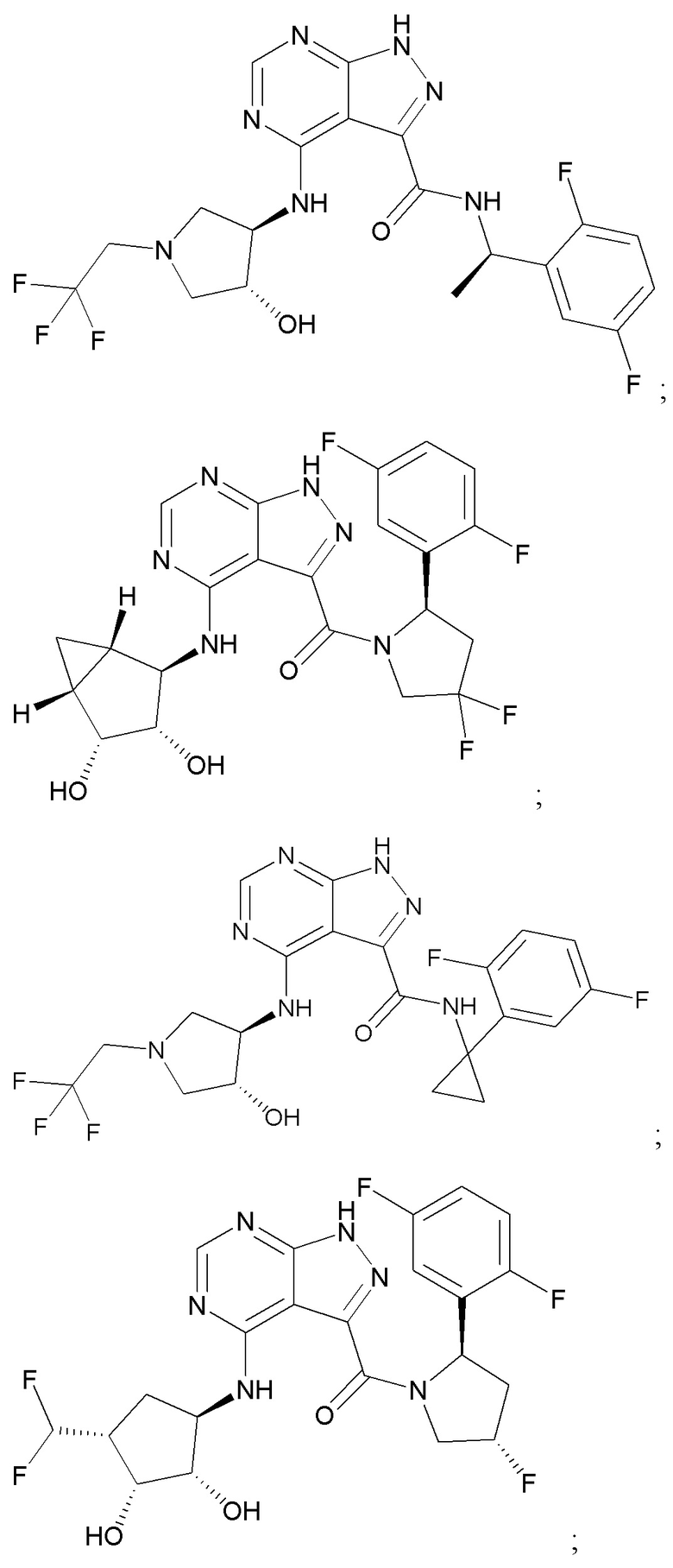
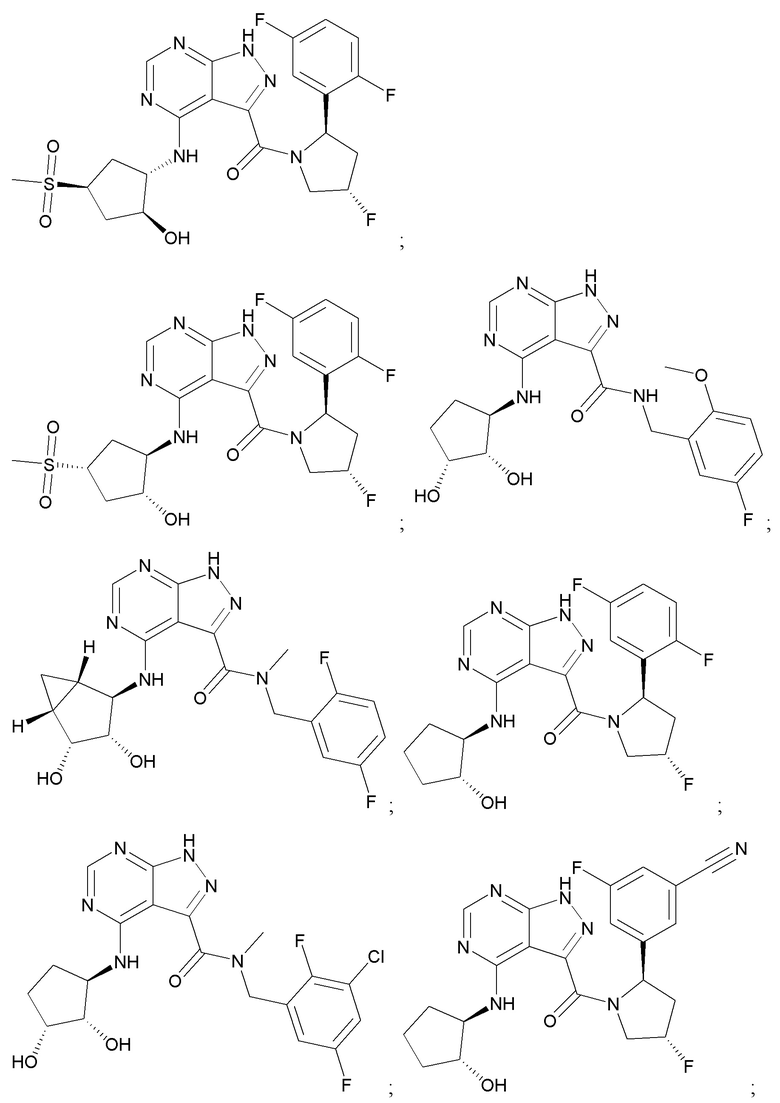
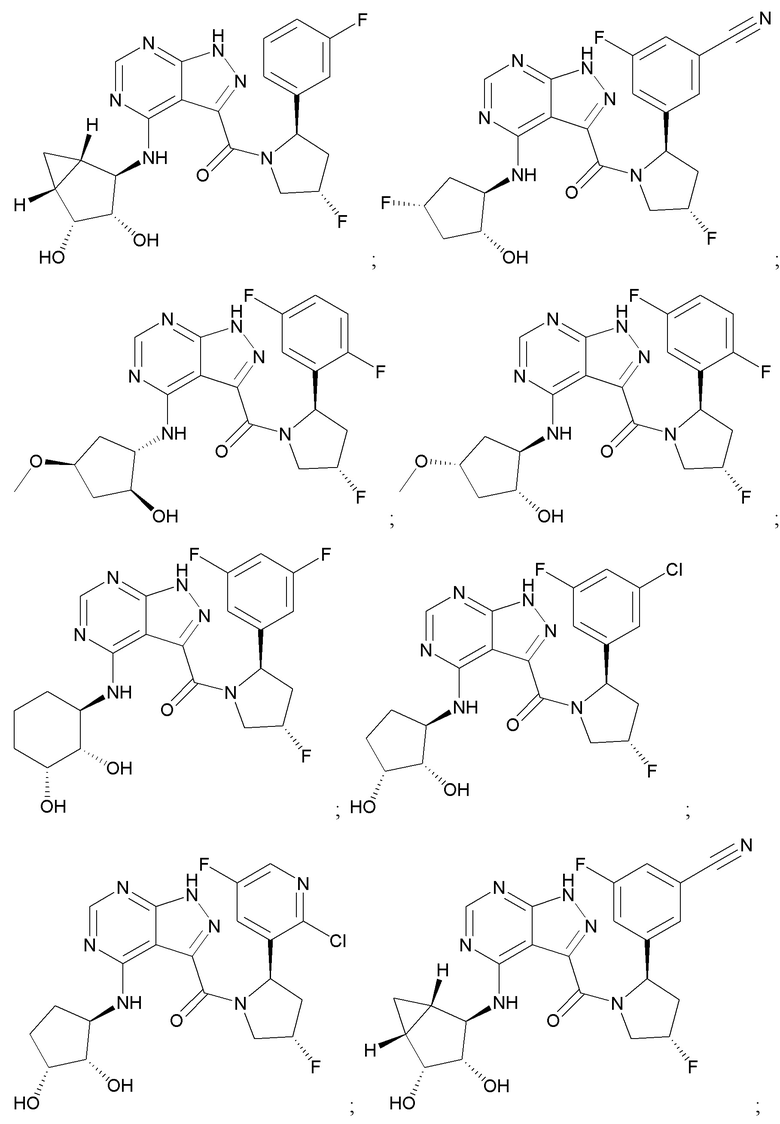
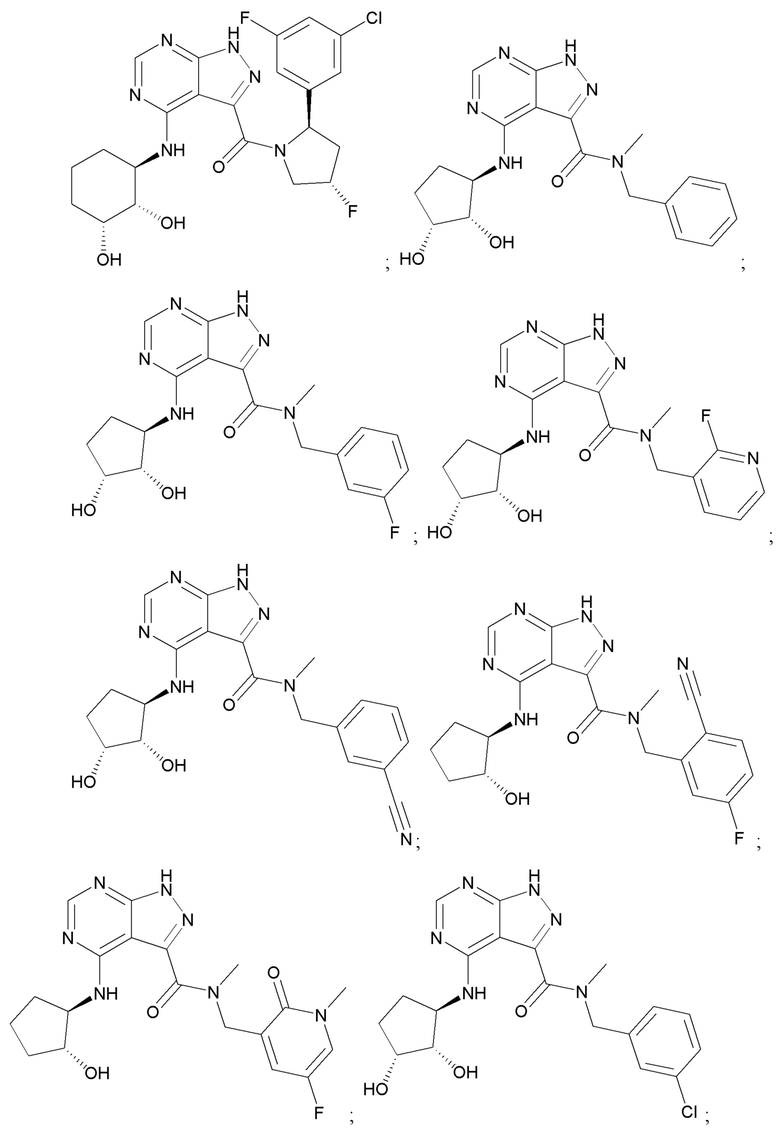
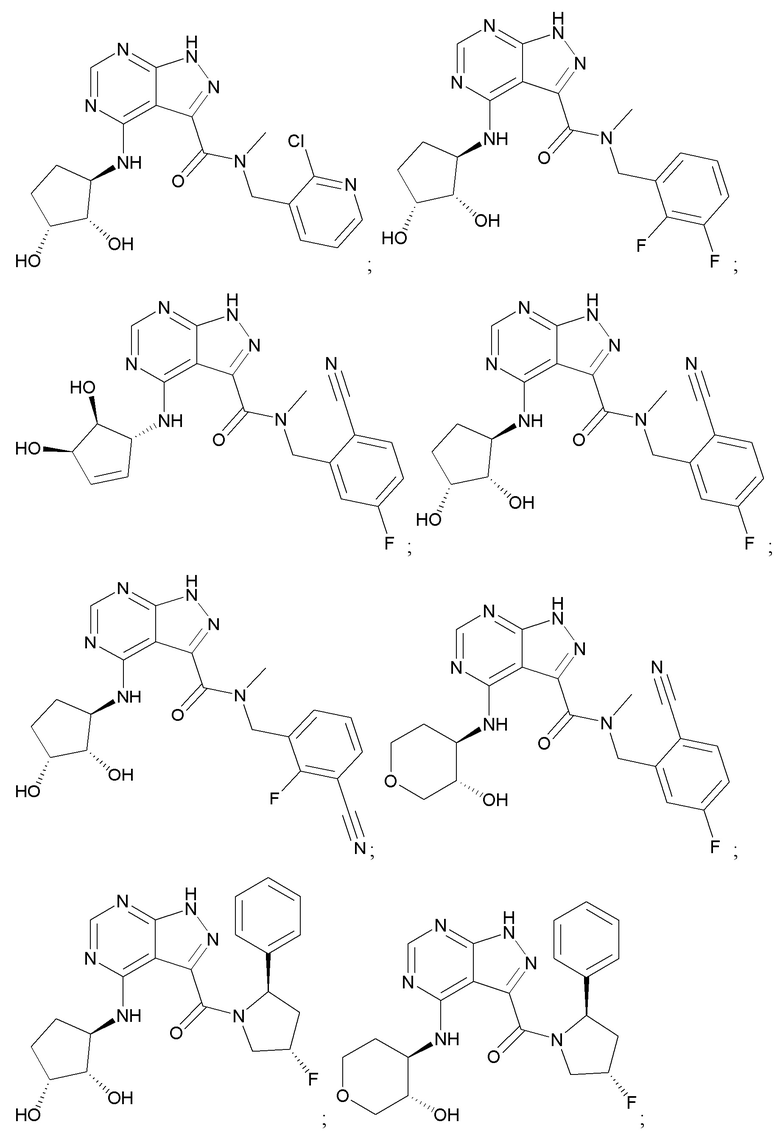
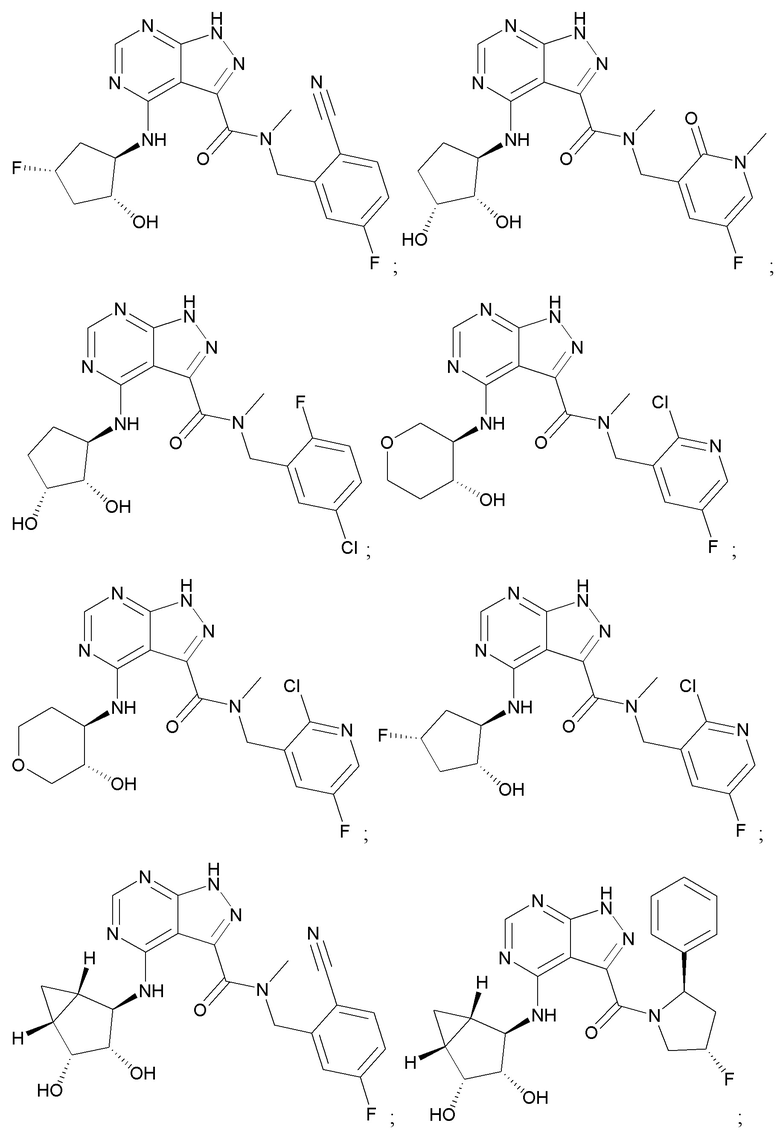
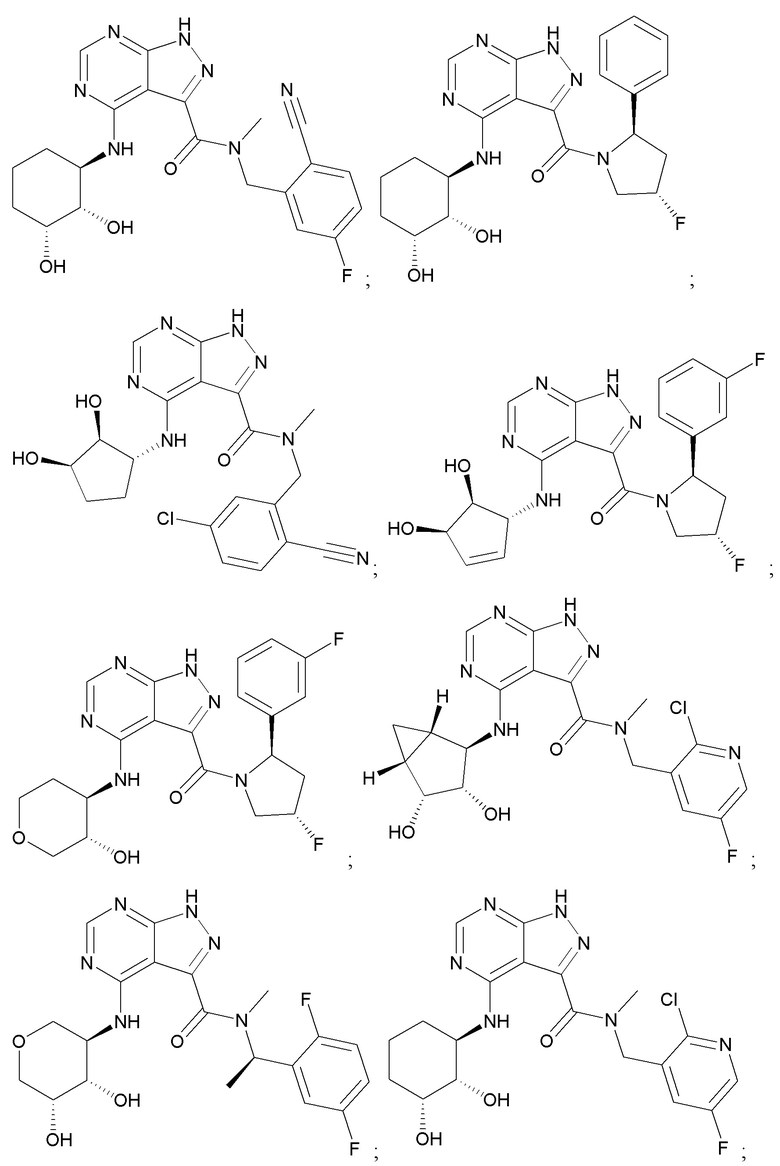
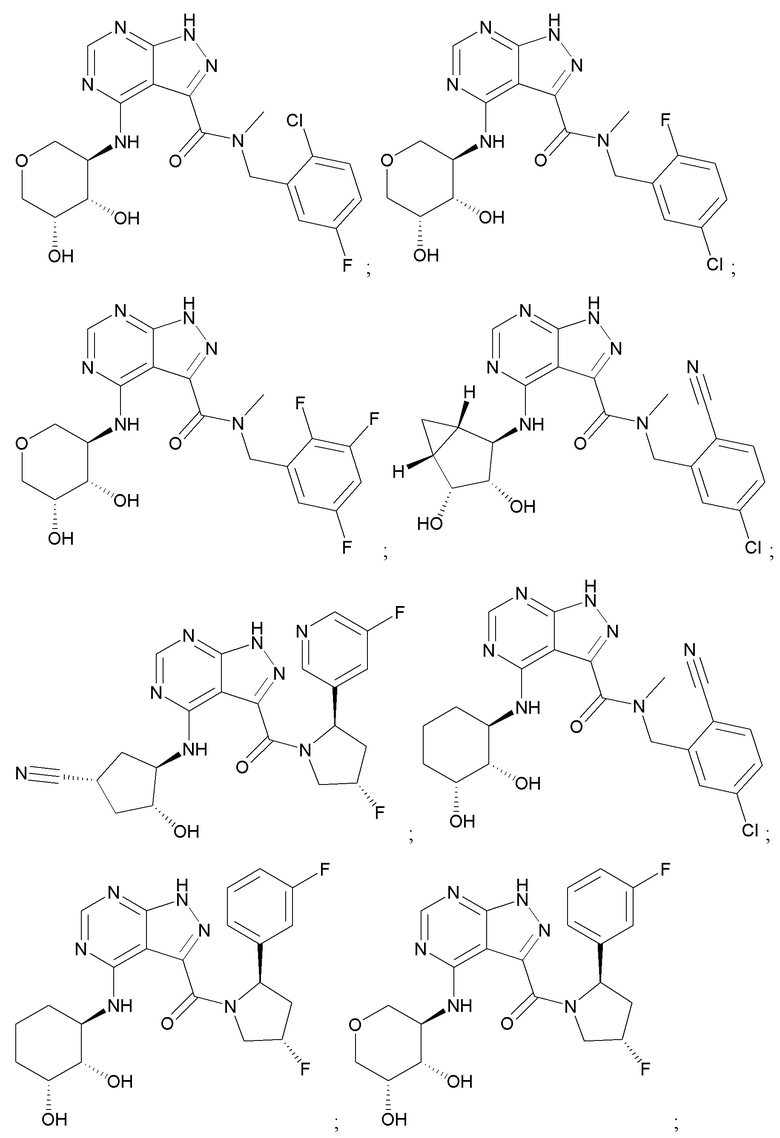
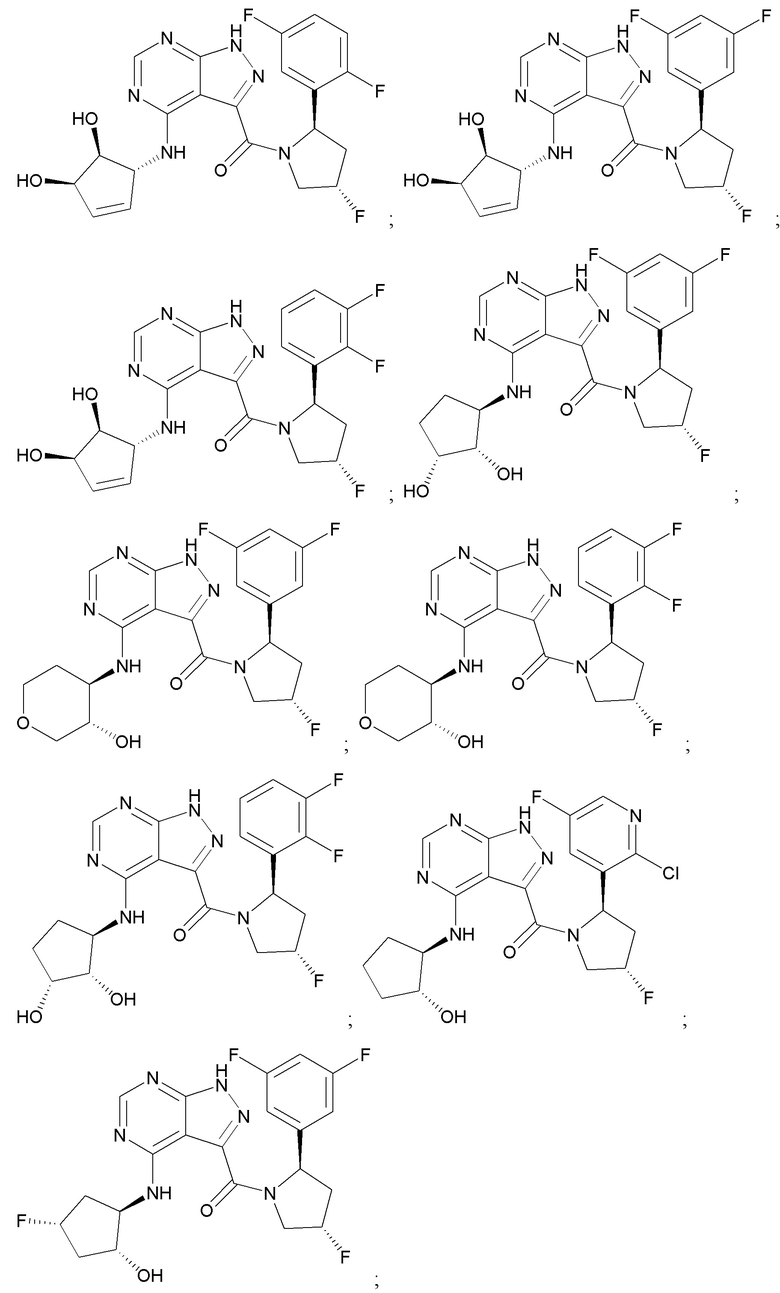
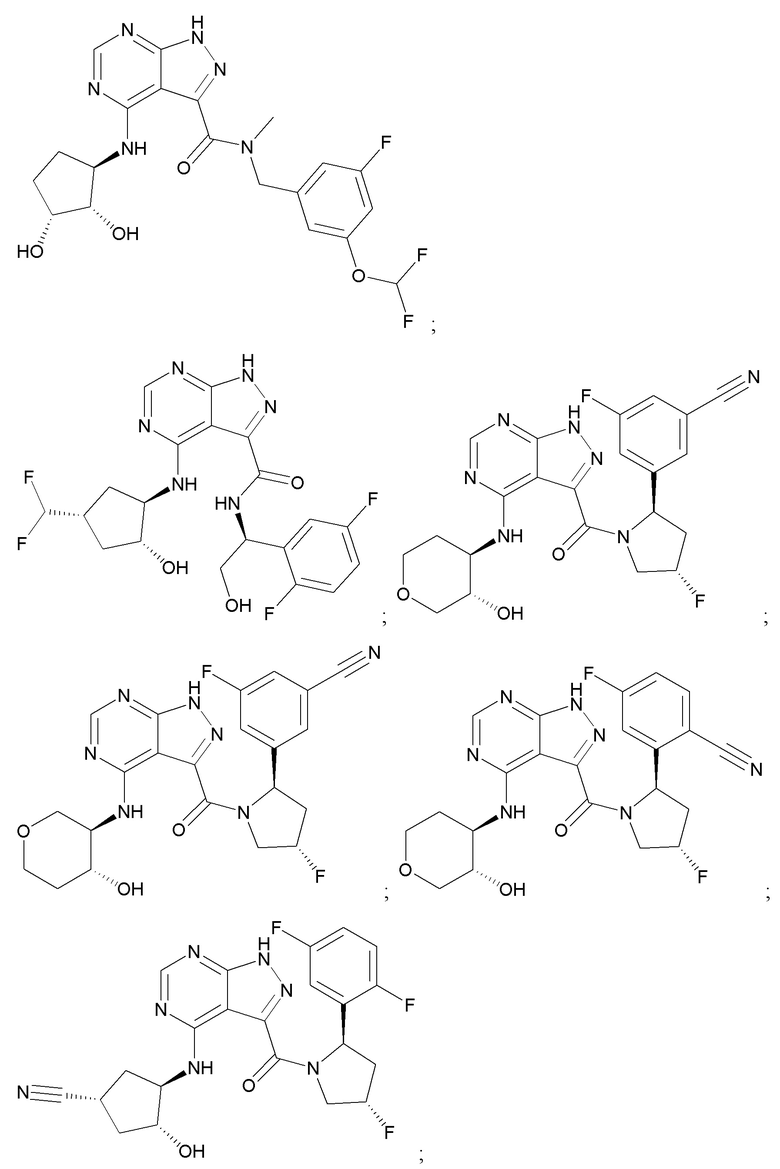
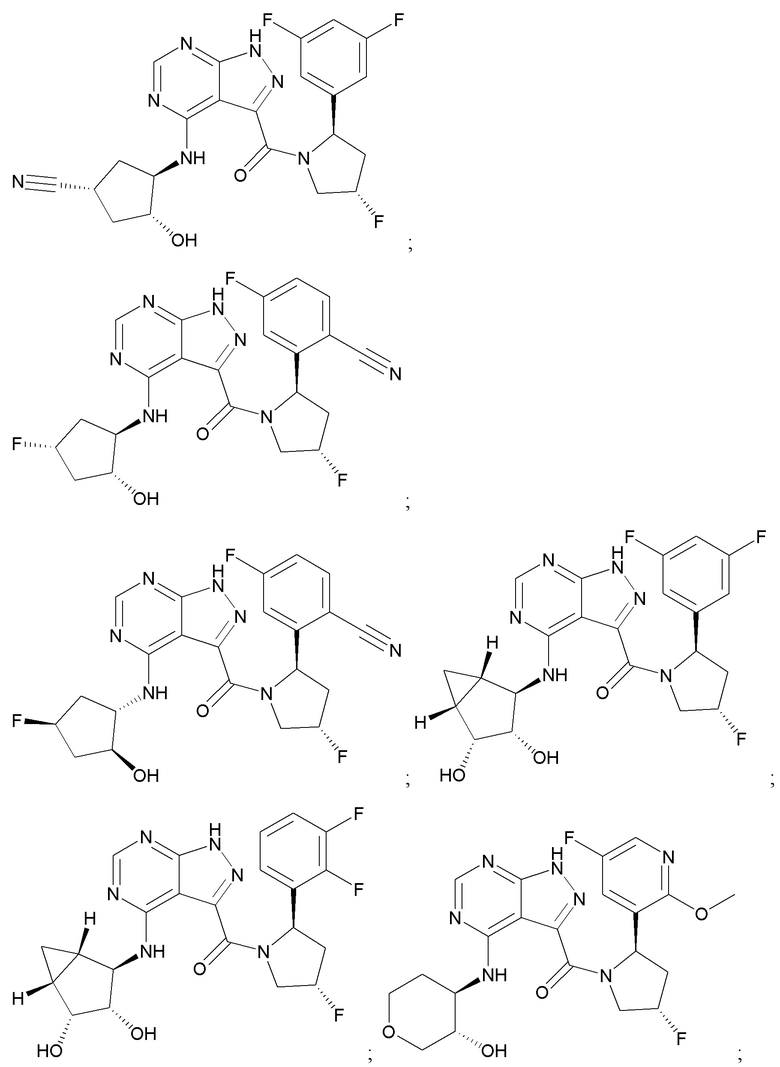
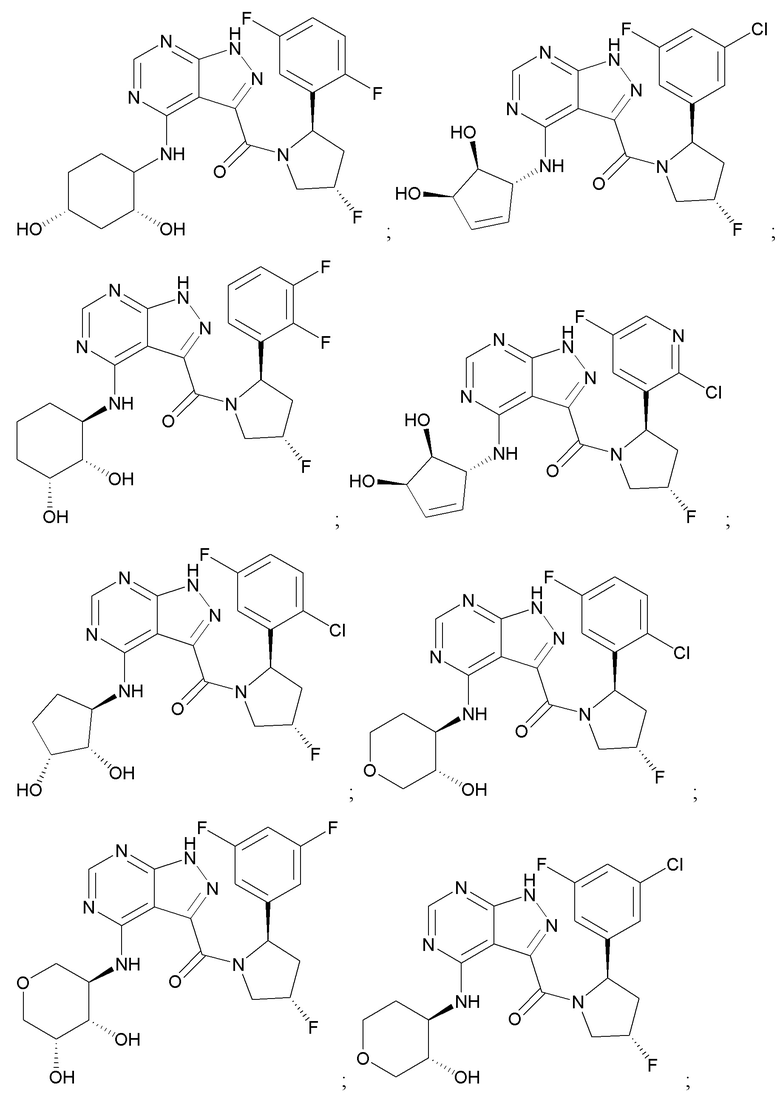
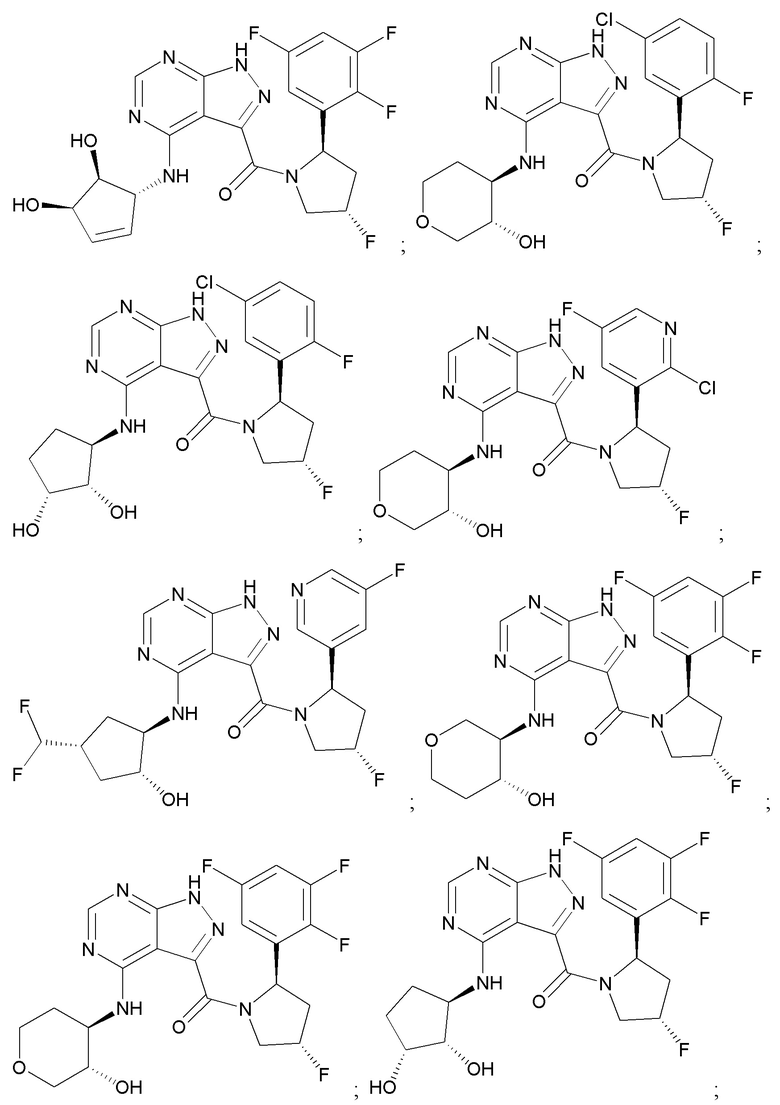
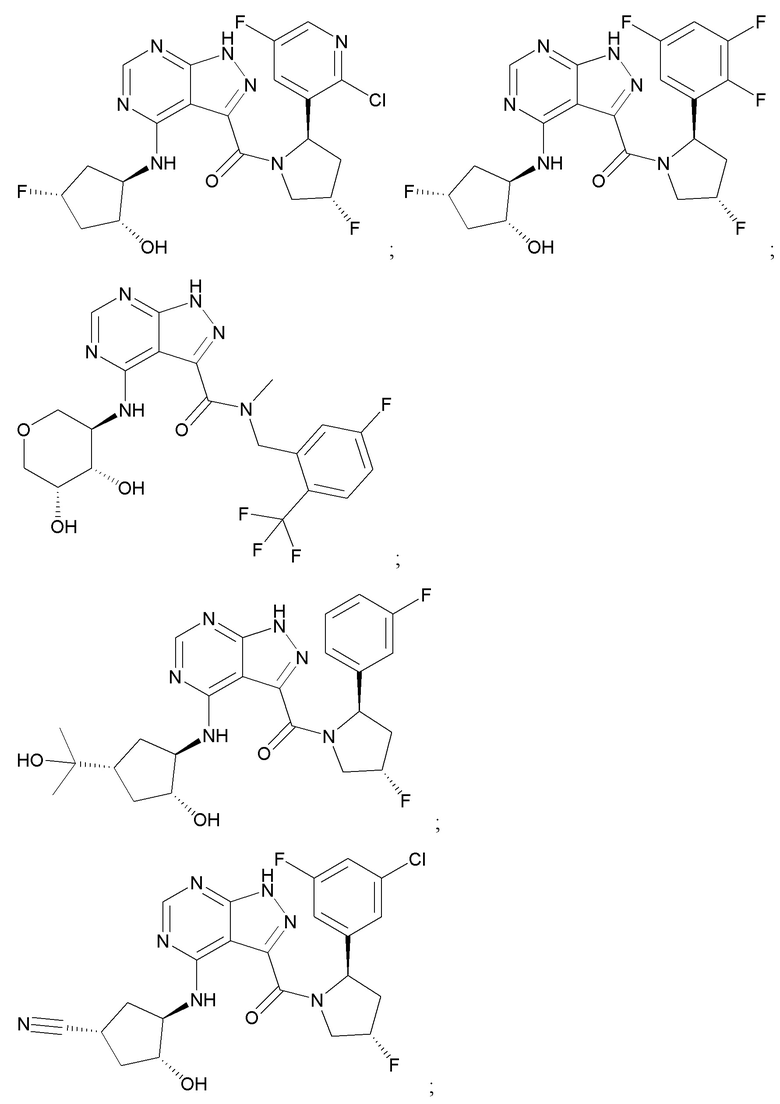
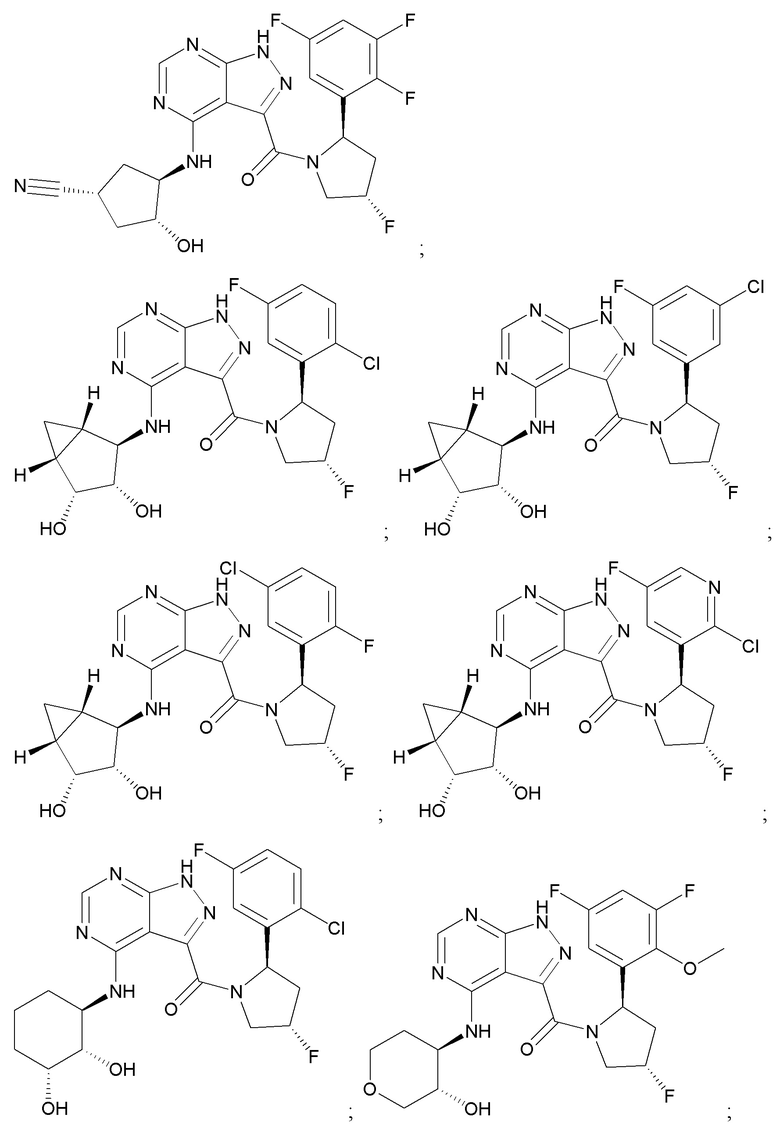

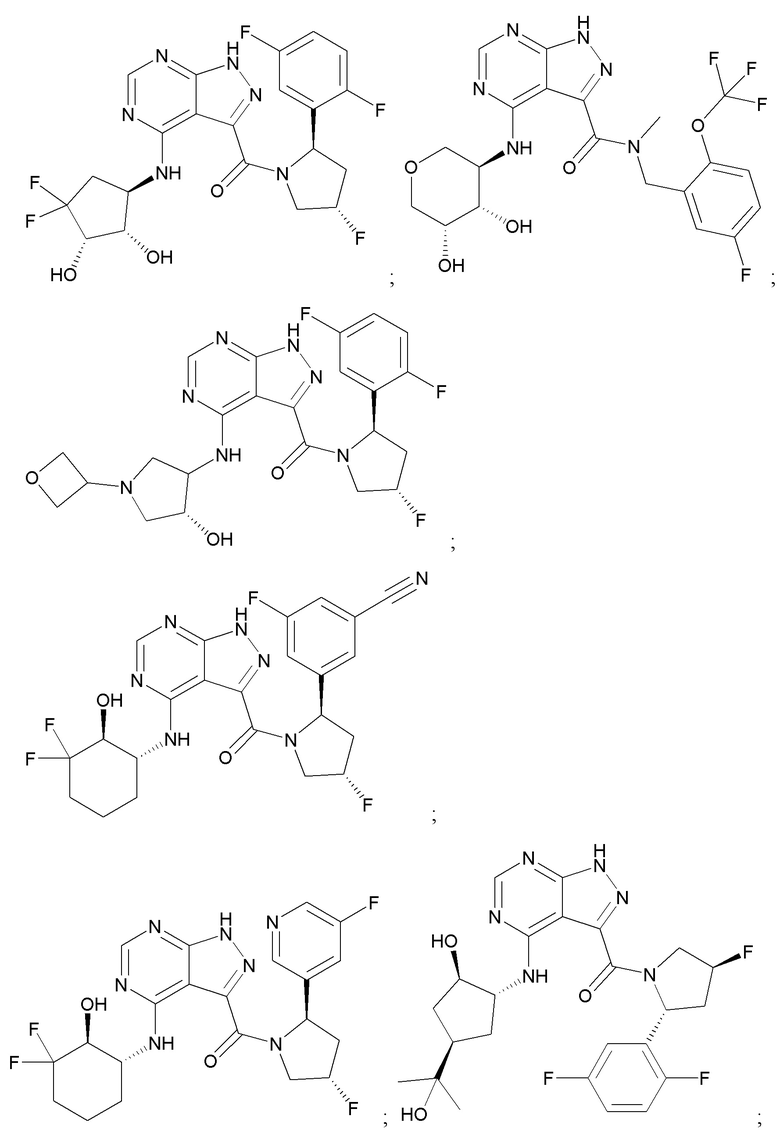
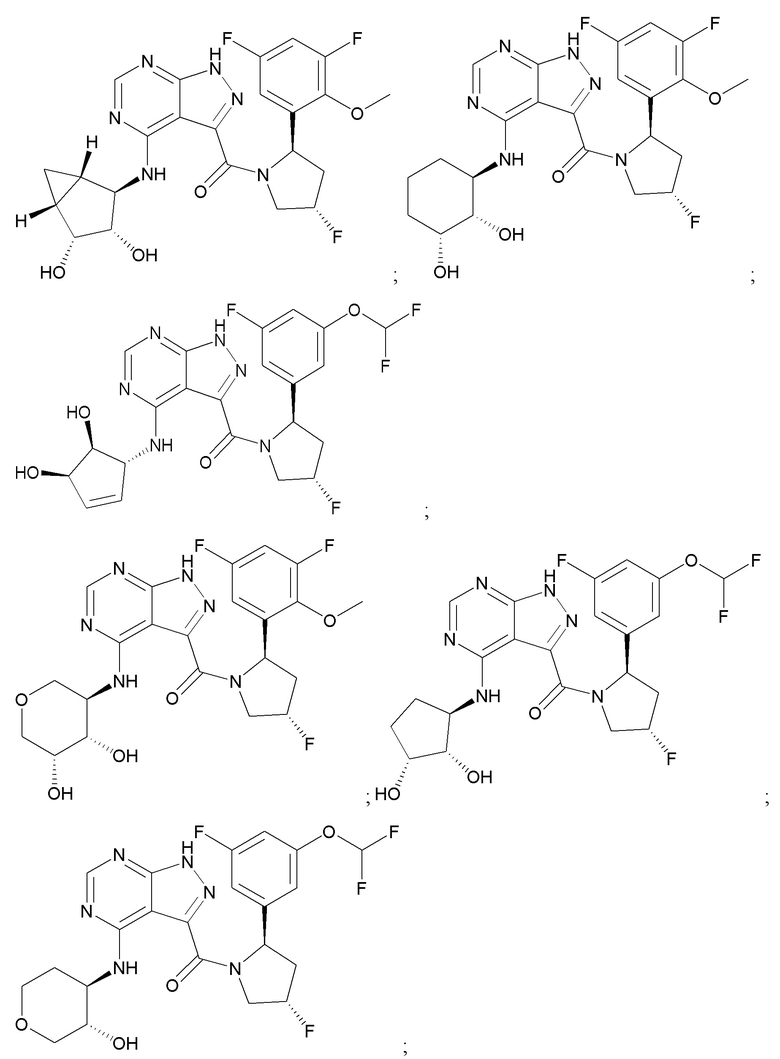
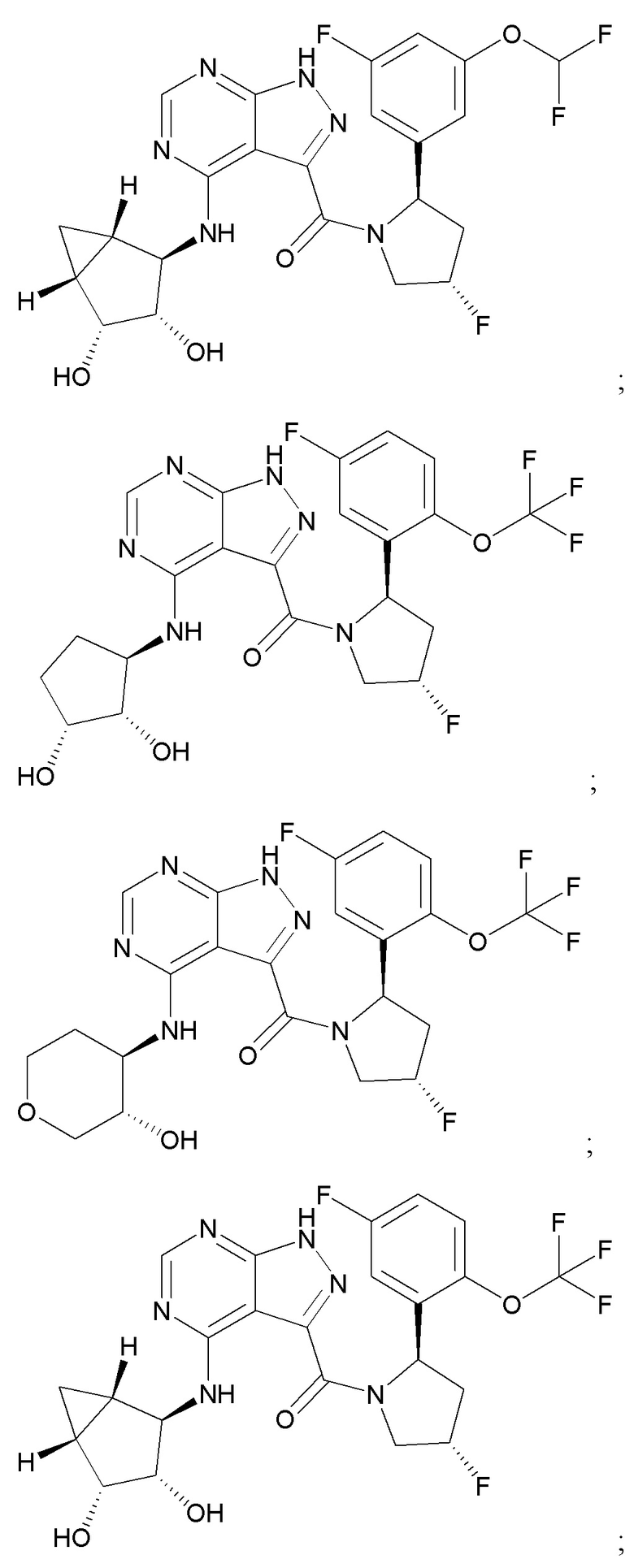
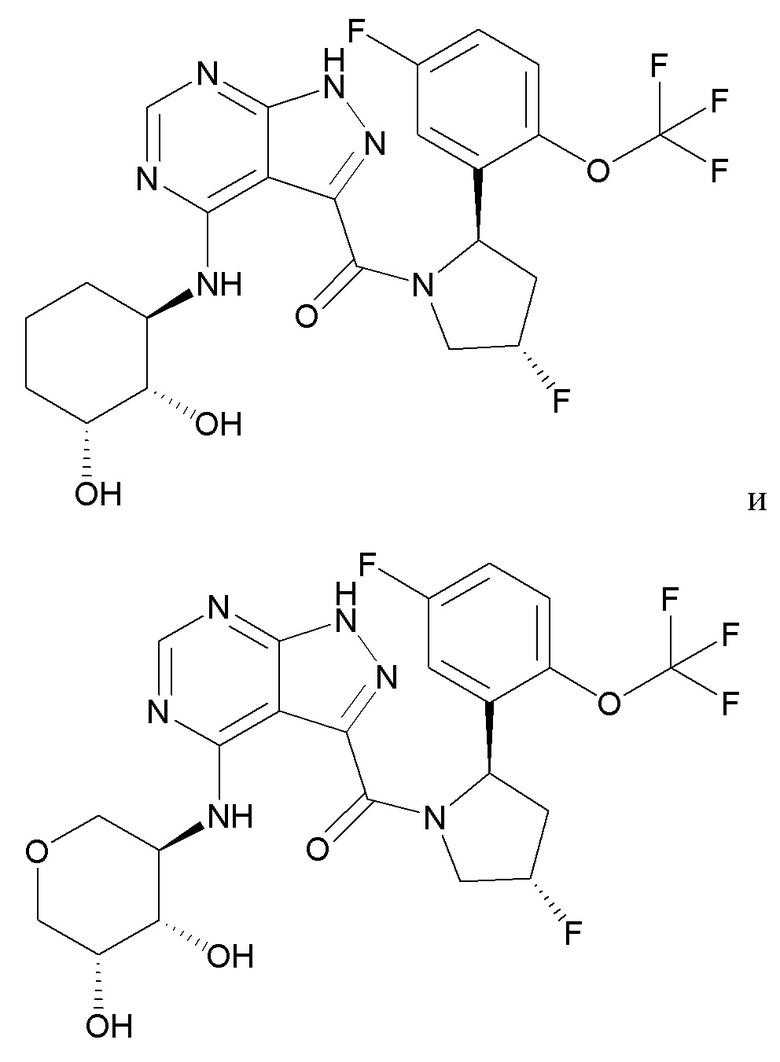
или его фармацевтически приемлемая соль.
2. Соединение, представляющее собой
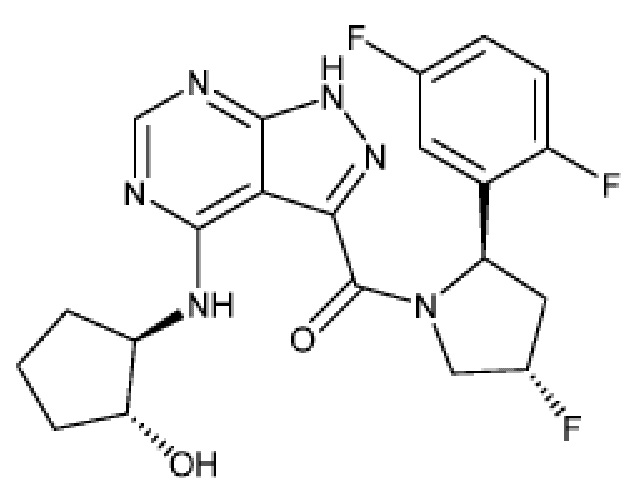
или его фармацевтически приемлемая соль.
3. Соединение, представляющее собой
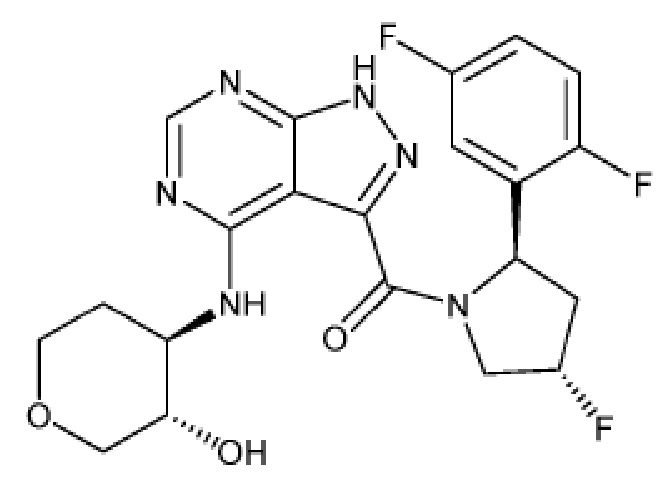
или его фармацевтически приемлемая соль.
4. Соединение, представляющее собой
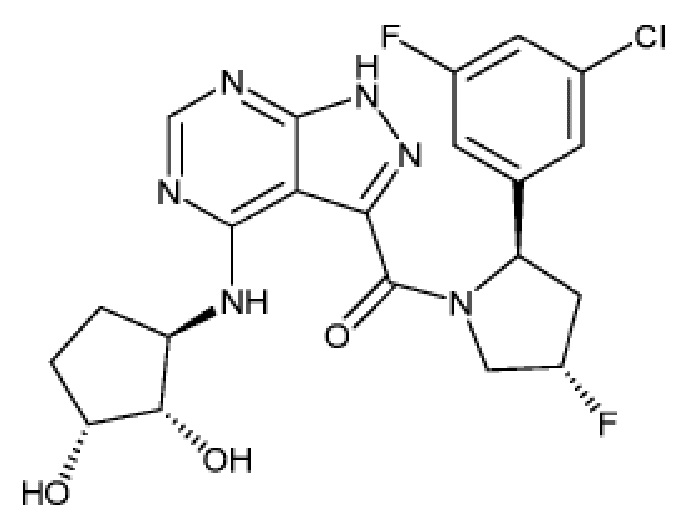
или его фармацевтически приемлемая соль.
5. Соединение, представляющее собой
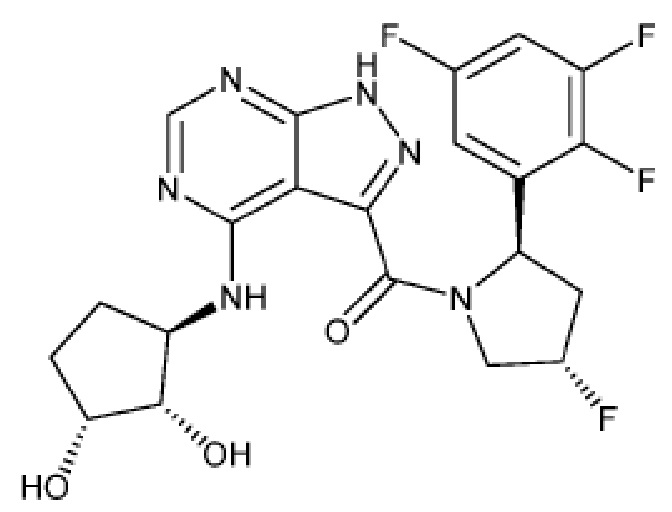
или его фармацевтически приемлемая соль.
6. Соединение, представляющее собой
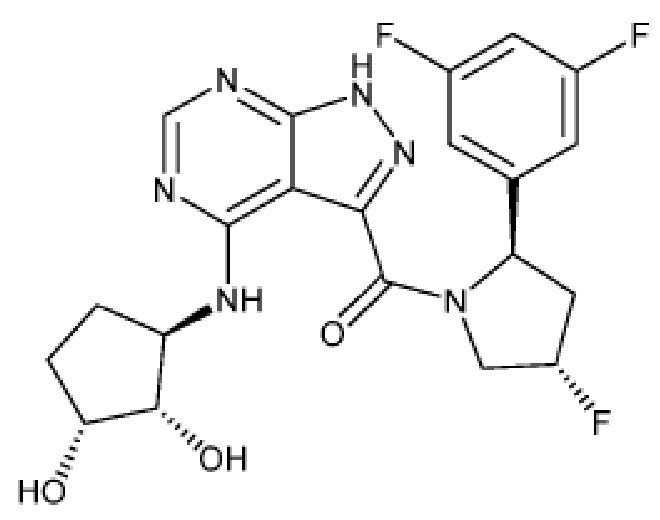
или его фармацевтически приемлемая соль.
7. Фармацевтическая композиция для ингибирования нейротрофной рецепторной тирозинкиназы (NTRK), содержащая фармацевтически приемлемый носитель и терапевтически эффективное количество соединения по любому из пп. 1-6 или его фармацевтически приемлемой соли.
8. Способ лечения субъекта, имеющего состояние, опосредованное аберрантной активностью нейротрофной рецепторной тирозинкиназы (NTRK), включающий введение субъекту терапевтически эффективного количества соединения по любому из пп.1-6 или его фармацевтически приемлемой соли или фармацевтической композиции по п.7.
| P.Traxler et al | |||
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| J.Med.Chem., 1997, 40(22), 3601-3616 | |||
| WO 2005117909 A2, 15.12.2005 | |||
| WO 2014118226 A1, 07.08.2014 | |||
| WO 2011139273 A1, 10.11.2011 | |||
| СПОСОБ ПРОИЗВОДСТВА КОНСЕРВИРОВАННОГО САЛАТА "БОГАТЫРЬ" | 2013 |
|
RU2515785C1 |
| 6, 6-БИЦИКЛИЧЕСКИЕ КОЛЬЦЕВЫЕ ЗАМЕЩЕННЫЕ ГЕТЕРОБИЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ ПРОТЕИНКИНАЗ | 2005 |
|
RU2379308C2 |

