Область изобретения
Настоящее изобретение относится к хинолиновым производным, которые являются ингибиторами фермента PDE10A и как таковые применимы для лечения нейродегенеративных и психических расстройств. В частности, настоящее изобретение относится к соединениям, которые являются высоко селективными по отношению к PDE10 по сравнению с другими подтипами PDE. Настоящее изобретение также относится к фармацевтическим композициям, содержащим соединения в соответствии с настоящим изобретением, и к способам лечения расстройств с использованием соединений в соответствии с настоящим изобретением.
Предпосылки изобретения
Циклические нуклеотиды, циклический аденозинмонофосфат (cAMP) и циклический гуанозинмонофосфат (cGMP) функционируют как внутриклеточные вторичные мессенджеры, регулирующие обширный ряд процессов в нейронах. Внутриклеточные cAMP и cGMP образуются аденил - и гуанилциклазами, а разрушаются фасфодиэстеразами (PDE) циклических нуклеотидов путем гидролиза циклических нуклеотидов в соответствующие им нуклеотидмонофосфаты.
Фосфодиэстераза 10A (PDE10A) является фосфодиэстеразой с двойной специфичностью, которая может превращать и сАМР в AMP, и cGMP в GMP (Soderling, S. et al. Proc. Natl. Acad. Sci. 1999, 96, 7071-7076). PDE10A первоначально экспрессируется в нейронах в полосатом теле, прилежащем ядре и в обонятельном бугорке (Kotera, J. et al. Biochem. Biophys. Res. Comm. 1999, 261, 551-557, и Seeger, T.F. et al. Brain Research, 2003, 985, 113-126).
Исследования показывают, что в головном мозге PDE10 экспрессируется на высоких уровнях срединными шипиковыми нейронами (MSN) хвостатого ядра, прилежащего ядра и соответствующими нейронами обонятельного бугорка. MSN экспрессирует два функциональных класса нейронов: класс экспрессирующий дофаминовые рецепторы D1, и класс D2, экспрессирующий дофаминовые рецепторы D2. Класс D1 нейронов является частью "прямого" стриарного пути выхода, который в целом функционирует с облегчением поведенческих реакций. Класс D2 нейронов является частью "непрямого" стриарного пути выхода, который функционирует с подавлением поведенческих реакций, которые конкурируют с реакциями, облегчаемыми "прямым" путем.
Антагонизм дофаминовых рецепторов D2 хорошо доказан при лечении шизофрении. С 1950-х годов антагонизм дофаминовых рецепторов D2 является основным в лечении психозов, и все эффективные антипсихотические лекарственные средства антагонизируют рецепторы D2. Действия D2, вероятно, опосредуются, прежде всего, нейронами в полосатом теле, прилежащем ядре и в обонятельном бугорке, поскольку эти зоны получают плотные дофаминергические проекции и характеризуются сильнейшей экспрессией рецепторов D2 (Konradi, С. and Heckers, S. Society of Biological Psychiatry, 2001, 50, 729-742).
Поскольку PDE10A в данном контексте характеризуется желаемым профилем экспрессии с высокой и относительно специфичной экспрессией в нейронах в полосатом теле, прилежащем ядре и в обонятельном бугорке, ингибирование PDE10A, вероятно, характеризуется эффектами, подобными антагонизму рецепторов D2, и поэтому обладает антипсихотическими эффектами.
Тогда как предполагается, что ингибирование PDE10A отчасти имитирует антагонизм рецепторов D2, ожидается, что оно характеризуется другим профилем. Рецептор D2 содержит компоненты передачи сигнала кроме cAMP (Neve, К.A. et al. Journal of Receptors and Signal Transduction 2004, 24, 165-205), в связи с чем взаимодействие с сАМР посредством ингибирования PDE10A может снижать риск экстрапирамидальных побочных эффектов, которые заметны при сильном антагонизме D2. И наоборот, ингибирование PDE10A может обладать некоторыми эффектами, не заметными при антагонизме рецепторов D2. Также PDE10A экспрессируется в стриарных нейронах, экспрессирующих рецепторы D1 (Seeger, Т.F. et al. Brain Research, 2003, 985, 113-126).
Кроме того, поскольку агонизм рецепторов D1 приводит к стимуляции аденилатциклазы и к повышению в результате уровней сАМР, ингибирование PDE10A, вероятно, также характеризуется эффектами, которые имитируют агонизм рецепторов D1.
Наконец, ингибирование PDE10A будет не только повышать уровни сАМР в клетках, но также, как предполагается, может повышать уровни cGMP, поскольку PDE10A является фосфодиэстеразой с двойной специфичностью. cGMP активирует ряд целевых белков в клетках, подобно сАМР, а также взаимодействует с путями передачи сигнала сАМР.
В заключение, ингибирование PDE10A, вероятно, отчасти имитирует антагонизм рецептора D2 и поэтому обладает антипсихотическим эффектом, но профиль может отличаться от профиля, наблюдаемого с классическими антагонистами рецепторов D2.
Ингибитор PDE10A папаверин, как показано, является активным в нескольких антипсихотических моделях. Папаверин потенциировал каталептический эффект антагониста рецептора D2 галоперидола у крыс, но сам по себе не вызывал каталепсии (WO 03/093499). Папаверин снижал гиперактивность у крыс, индуцированную PCP, тогда как снижение индуцированной амфетамином гиперактивности было незначительным (WO 03/093499). Эти модели подтверждают, что ингибирование PDE10A обладает классическим антипсихотическим потенциалом, который можно было бы предположить из вышеприведенных теоретических соображений. Кроме того, в WO 03/093499 раскрывается применение селективных ингибиторов PDE10 для лечения ассоциированных неврологических и психических расстройств. Более того, ингибирование PDE10A отменяет субхронические индуцированые PCP дефициты в сдвиге внимания у крыс (Rodefer et al. Eur. J. Neurosci. 2005, 4, 1070-1076). Эта модель подтверждает, что ингибирование PDE10A может облегчать когнитивные дефициты, ассоциированные с шизофренией.
Тканевое распределение PDE10A показывает, что ингибиторы PDE10A можно использовать для повышения уровней сАМР и/или cGMP в клетках, которые экспрессируют фермент PDE10A, особенно, в нейронах, которые составляют базальные ганглии, и поэтому ингибиторы PDE10A в соответствии с настоящим изобретением будут применимыми в лечении ряда ассоциированных нейропсихических состояний, вовлекающих базальные ганглии, таких как неврологические и психические расстройства, шизофрения, биполярное расстройство, психоз, обсессивно-компульсивное расстройство и зависимость, и может обладать преимуществом, заключающимся в отсутствии нежелательных побочных эффектов, которые ассоциированы с имеющимися на рынке терапевтическими средствами.
Кроме того, последние публикации (WO 2005/120514, WO 2005012485, Cantin et al, Bioorganic & Medicinal Chemistry Letters 17 (2007) 2869-2873) подтверждают, что ингибиторами PDE10A могут быть применимыми для лечения ожирения и инсулинонезависимого сахарного диабета.
Кроме того, последние публикации подтверждают, что ингибиторы PDE10A могут быть применимыми для лечения болезни Хантингтона (Giampa et al. PLoS One 2010, 5(10), Giampa et al. Neurobiology of Disease (2009), 34(3), 450-456, Hebb et al. Current Opinion in Pharmacology 2007, 7(1), 86-92.)
Пирролодигидроизохинолины и их варианты раскрываются как ингибиторы PDE10 в WO 05/03129 и WO 05/02579. Замещенные пиперидинилом хиназолины и изохинолины, которые служат ингибиторами PDE10, раскрываются в WO 05/82883. В WO 06/11040 раскрываются замещенные хиназолиновые и изохинолиновые соединения, которые служат ингибиторами PDE10. В US 20050182079 раскрываются замещенные тетрагидроизохинолиниловые производные хиназолина и изохинолина, которые служат эффективными ингибиторами фосфодиэстеразы (PDE). В частности, US 20050182079 относится к указанным соединениям, которые являются селективными ингибиторами PDE10. Аналогичным образом, в US 20060019975 раскрываются пиперидиновые производные хиназолина и изохинолина, которые служат эффективными ингибиторами фосфодиэстеразы (PDE). Также US 20060019975 относится к соединениям, которые являются селективными ингибиторами PDE10. В WO 06/028957 раскрываются циннолиновые производные как ингибиторы PDE10 для лечения психических и неврологических синдромов. В WO 09/152825 раскрываются фенилимидазольные производные как соединения, которые служат ингибиторами PDE10.
Однако, эти раскрытия не имеют отношения к соединениям в соответствии с настоящим изобретением, которые структурно не являются родственными каким-либо из известных ингибиторов PDE10 (Kehler, J. et al. Expert Opin. Ther. Patents 2007, 17, 147-158), и которые, как было обнаружено авторами настоящего изобретения, являются высоко активными и селективными ингибиторами фермента PDE10A.
Настоящее изобретение относится к соединениям, которые являются ингибиторами фермента PDE10A и, таким образом, применимы для лечения нейродегенеративных и/или психических расстройств, которое не является эффективным для всех пациентов. Однако сохраняется потребность в альтернативных способах лечения.
Краткое описание изобретения
Цель настоящего изобретения заключается в обеспечении соединений, которые являются селективными ингибиторами фермента PDE10A.
Следующая цель настоящего изобретения заключается в обеспечении соединений, которые обладают такой активностью и которые обладают улучшенной растворимостью, метаболической стабильностью и/или биодоступностью по сравнению с известными из уровня техники соединениями.
Другая цель настоящего изобретения заключается в обеспечении эффективного лечения, в частности, долговременного лечения, пациентов-людей без возникновения побочных эффектов, как правило, ассоциированных с имеющимися терапевтическими средствами против неврологических и психических расстройств.
Следующие цели настоящего изобретения станут понятны при прочтении данного описания.
Подробное описание изобретения
Варианты осуществления настоящего изобретения
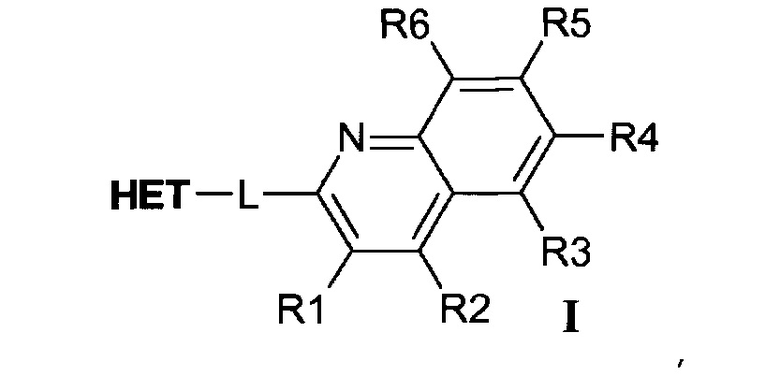
В первом варианте осуществления (E1) настоящее изобретение относится к соединениям формулы I:
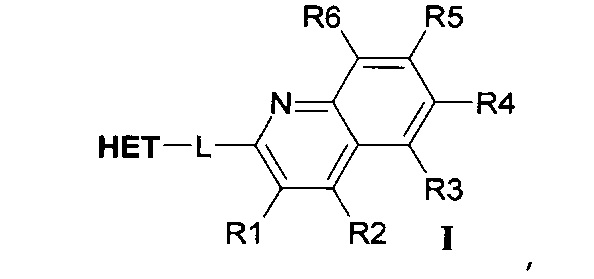
где R1, R2, R3, R4, R5 и R6 по отдельности выбраны из группы, состоящей из водорода, гидроксила, циано, C1-C6алкила; C1-C6алкокси, галогена, метилендиокси, дифторметилендиокси и этилендиокси;
где -L- представляет собой линкер, выбранный из -CH2-CH2- и -CH=CH-;
где НЕТ выбран из группы, состоящей из
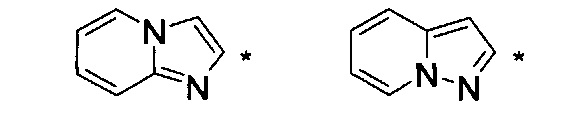
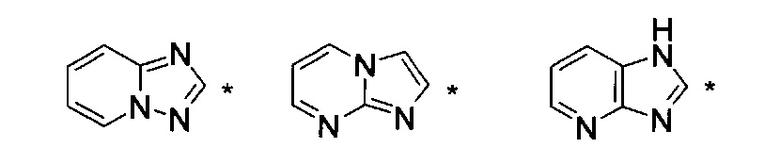
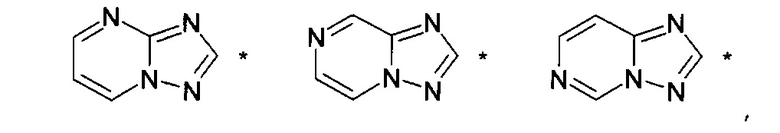
где один или несколько связанных с углеродом водородов в НЕТ необязательно могут быть замещены не более чем тремя заместителями R7, R8 и R9, по отдельности выбранными из C1-C6алкила; галогена; циано, галоген(C1-C6)алкила; арила, алкокси и C1-C6гидроксиалкила;
и где * означает точку присоединения,
а также к их таутомерам и фармацевтически приемлемым солям и их полиморфным формам.
В варианте осуществления (E2) варианта осуществления (E1) один или несколько из R1-R6 выбраны из группы, состоящей из C1-C3алкила, замещенного одним или несколькими F, и незамещенного C1-C3алкила.
В варианте осуществления (E3) варианта осуществления (E1) или (E2) один или несколько из R1-R6 выбраны из группы, состоящей из метила, этила, пропила, изопропила, монофторметила, дифторметила и трифторметила.
В варианте осуществления (E4) варианта осуществления (E1) один или несколько из R1-R6 выбраны из группы, состоящей из метокси, дифторметокси и трифторметокси.
В варианте осуществления (E5) варианта осуществления (E1) один или несколько из R1-R6 выбраны из группы, состоящей из фтора и хлора.
В варианте осуществления (E6) какого-либо из вариантов осуществления (E1) - (E5) -L- представляет собой -CH2-CH2-.
В варианте осуществления (E7) какого-либо из вариантов осуществления (E1) - (E5) -L- представляет собой -CH=CH-.
В варианте осуществления (E8) какого-либо из вариантов осуществления (E1) - (E7) НЕТ выбран из группы, состоящей из

где НЕТ необязательно замещен одним или несколькими из R7-R9, и
где * означает точку присоединения.
В варианте осуществления (E9) какого-либо из вариантов осуществления (E1) - (E8) НЕТ замещен одним заместителем R7, выбранным из группы, состоящей из C1-C6алкила, такого как метил; галогена, такого как хлор или бром; циано; галоген(C1-C6)алкила, такого как трифторметил; арила, такого как фенил; и C1-C6гидроксиалкила, такого как CH2CH2OH.
В варианте осуществления (E10) какого-либо из вариантов осуществления (E1) - (E8) НЕТ замещен двумя заместителями R7 и R8, по отдельности выбранными из C1-C6алкила, такого как метил; галогена, такого как хлор или бром; циано; галоген(C1-C6)алкила, такого как трифторметил; арила, такого как фенил; и C1-C6гидроксиалкила, такого как CH2CH2OH.
В варианте осуществления (E11) какого-либо из вариантов осуществления (E1) - (E8) НЕТ замещен тремя заместителями R7, R8 и R9, по отдельности выбранными из C1-C6алкила, такого как метил; галогена, такого как хлор или бром; циано; галоген(C1-C6)алкила, такого как трифторметил; арила, такого как фенил; и C1-C6гидроксиалкила, такого как CH2CH2OH.
В варианте осуществления (E12) какого-либо из вариантов осуществления (E1) - (E8) НЕТ является незамещенным.
В варианте осуществления (E13) какого-либо из вариантов осуществления (E1), (E9), (E10) и (E11) НЕТ замещен по меньшей мере одним C1-C6алкилом, таким как метил.
В варианте осуществления (E14) какого-либо из вариантов осуществления (E1) - (E11) НЕТ выбран из группы, состоящей из (5,7-диметил-имидазо[1,2-a]пиримидин-2-ила), (5,7-диметил-[1,2,4]триазоло[1,5-a]пиримидин-2-ила), (5,8-диметил-[1,2,4]триазоло[1,5-a]пиразин-2-ила), (8-метокси-5-метил-[1,2,4]триазоло[1,5-a]пиридин-2-ила) и (5,8-диметил-[1,2,4]триазоло[1,5-a]пиридин-2-ила).
В конкретном варианте осуществления (E15) варианта осуществления (E1) соединение выбрано из группы соединений, приведенных в таблице 1.
В варианте осуществления (E16) какого-либо из вариантов осуществления (E1) - (E15) настоящее изобретение относится к соединению формулы I или к его фармацевтически приемлемой соли для применения в качестве медицинского препарата.
В варианте осуществления (E17) какого-либо из вариантов осуществления (E1) - (E15) настоящее изобретение относится к фармацевтической композиции, содержащей терапевтически эффективное количество соединения формулы I и фармацевтически приемлемый носитель, разбавитель или наполнитель.
В варианте осуществления (E18) какого-либо из вариантов осуществления (E1) - (E15) настоящее изобретение относится к применению соединения формулы I или его фармацевтически приемлемой соли для получения медицинского препарата для лечения нейродегенеративного или психического расстройства.
Кроме того, в варианте осуществления (E19) какого-либо из вариантов осуществления (E1) - (E15) настоящее изобретение относится к способу лечения субъекта, страдающего от нейродегенеративного расстройства, предусматривающему введение субъекту терапевтически эффективного количества соединения формулы I.
В варианте осуществления (E20) какого-либо из вариантов осуществления (E1) - (E15) настоящее изобретение относится к способу лечения субъекта, страдающего от психического расстройства, предусматривающему введение субъекту терапевтически эффективного количества соединения формулы I.
В варианте осуществления (E21) какого-либо из вариантов осуществления (E1) - (E15) настоящее изобретение относится к способу лечения субъекта, страдающего от наркотической зависимости, такой как алкогольная, амфетаминовая, кокаиновая или опиатная зависимость.
В варианте осуществления (E22) настоящее изобретение относится к соединениям формулы I:
где R1-R6 и НЕТ описаны в каком-либо из предыдущих вариантов осуществления (E1) - (E14), и -L- представляет собой линкер,
выбранный из -S-CH2-, -CH2-S- и  .
.
Определение заместителей
Используемые в контексте настоящего изобретения термины "галогено" и "галоген" используются взаимозаменяемо и относятся к фтору, хлору, брому или йоду.
Термин "C1-C6алкил" относится к насыщенному углеводороду с прямой или разветвленной цепью, содержащему от одного до шести атомов углерода включительно. Примеры таких групп включают без ограничения метил, этил, 1-пропил, 2-пропил, 1-бутил, 2-бутил, 2-метил-2-пропил, 2-метил-1-бутил и н-гексил. Выражение "C1-C6гидроксиалкил" относится к C1-C6 алкильной группе, определенной выше, которая замещается одной гидроксигруппой. Термин "галоген(C1-C6)алкил" относится к C1-C6 алкильной группе, определенной выше, которая замещается не более чем тремя атомами галогена, такой как трифторметил.
Выражение "C1-C6алкокси" относится к насыщенной алкоксигруппе с прямой или разветвленной цепью, содержащей от одного до шести атомов углерода включительно, с открытой валентностью на кислороде. Примеры таких групп включают без ограничения метокси, этокси, н-бутокси, 2-метил-пентокси и н-гексилокси. Алкокси необязательно может быть замещен не более чем тремя атомами галогена, например, трифторметокси.
Термин "C3-C8циклоалкил", как правило, относится к циклопропилу, циклобутилу, циклопентилу, циклогексилу, циклогептилу или циклооктилу. Выражение "C1-C6алкил(C3-C8)циклоалкил" относится к C3-C8циклоалкилу, определенному выше, который замещается C1-C6алкилом с прямой или разветвленной цепью. Примеры таких групп включают без ограничения циклопропилметил.
Термин "гетероциклоалкил" относится к четырех-восьмичленному кольцу, содержащему атомы углерода и до трех атомов N, О или S, при условии, что четырех-восьмичленное кольцо не содержит соседние атомы О или соседние атомы S. Открытая валентность находится либо на гетероатоме, либо на атоме углерода. Примеры таких групп включают без ограничения азетидинил, оксетанил, пиперазинил, морфолинил, тиоморфолинил и [1,4]диазепанил. Термин "гидроксигетероциклоалкил" относится к гетероциклоалкилу, определенному выше, который замещается одной гидроксигруппой. Термин "C1-C6алкил-гетероциклоалкил" относится к гетероциклоалкилу, определенному выше, который замещается C1-C6 алкильной группой. Примеры таких групп включают без ограничения тетрагидропиран-4-ил-метил и 2-морфолин-4-ил-этил.
Термин "арил" относится к фенильному кольцу, необязательно замещенному галогеном, C1-C6алкилом, C1-C6алкокси или галоген(C1-C6)алкилом, определенными выше. Примеры таких групп включают без ограничения фенил и 4-хлорфенил.
Термин "C1-C6арилалкил" относится к арилу, определенному выше, который замещается C1-C6алкилом с прямой или разветвленной цепью. Примеры таких групп включают без ограничения бензил и 4-хлорбензил.
В следующем варианте осуществления один или несколько атомов водорода соединения формулы I были замещены дейтерием.
Следует учитывать, что в контексте настоящей заявки значения "R1 - R6", "от R1 до R6" и "R1, R2, R3, R4, R5 и R6" являются одинаковыми.
Кроме того, настоящее изобретение также относится к определенным вариантам осуществления настоящего изобретения, которые описываются ниже.
В отдельных вариантах осуществления настоящего изобретения соединение формулы I выбрано из следующих конкретных соединений, приведенных в таблице 1, в форме свободного основания, одного или нескольких их таутомеров или их фармацевтически приемлемой соли. В таблице 1 приводятся соединения в соответствии с настоящим изобретением и соответствующие значения IC50, определенные как описано в разделе "Анализ ингибирования PDE10A". Каждое из соединений представляет собой отдельный вариант осуществления настоящего изобретения.
Следует понимать, что различные аспекты, варианты осуществления, реализации и признаки настоящего изобретения, упомянутые в данном документе, могут быть заявлены отдельно или в любом сочетании, как иллюстрируется следующими неограничивающими примерами.




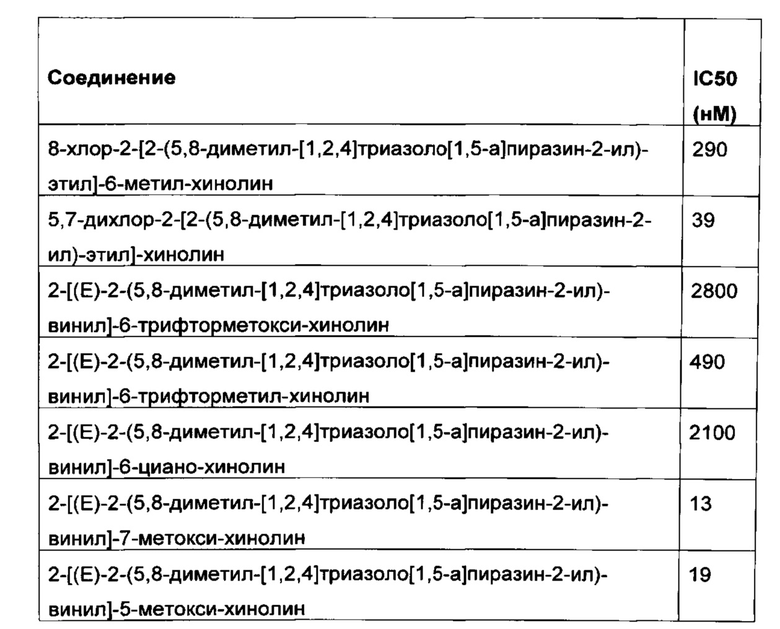
В конкретном варианте осуществления настоящего изобретения соединения в соответствии с настоящим изобретением имеют значение IC50 меньше 50 нМ, например, в диапазоне 0,2-20 нМ, в частности, в диапазоне 0,2-10 нМ, например, в диапазоне 0,2-5 нМ.
Фармацевтически приемлемые соли
Настоящее изобретение также относится к солям соединений, как правило, фармацевтически приемлемым солям. Такие соли включают фармацевтически приемлемые кислотно-аддитивные соли. Кислотно-аддитивные соли включают соли неорганических кислот, а также органических кислот.
Типичные примеры подходящих неорганических кислот включают хлористоводородную, бромистоводородную, йодистоводородную, фосфорную, серную, сульфаминовую, азотную кислоты и т.п. Типичные примеры подходящих органических кислот включают муравьиную, уксусную, трихлоруксусную, трифторуксусную, пропионовую, бензойную, коричную, лимонную, фумаровую, гликолевую, итаконовую, молочную, метансульфоновую, малеиновую, яблочную, малоновую, миндальную, оксалиновую, пикриновую, пировиноградную, салициловую, янтарную, метансульфоновую, этансульфоновую, винную, аскорбиновую, памовую, бисметиленсалициловую, этандисульфоновую, глюконовую, цитраконовую, аспарагиновую, стеариновую, пальмитиновую, EDTA, гликолевую, п-аминобензойную, глутаминовую, бензолсульфоновую, п-толуолсульфоновую кислоты, теофиллин-уксусные кислоты, а также 8-галогентеофиллины, например, 8-бромтеофиллин и т.п. Следующие примеры фармацевтически приемлемых неорганических или органических кислотно-аддитивных солей включают фармацевтически приемлемые соли, приведенные в Berge, S.M. et al., J. Pharm. Sci. 1977, 66, 2, содержание которой тем самым включено в настоящий документ посредством ссылки.
Кроме того, соединения в соответствии с настоящим изобретением может существовать в несольватированной, а также в сольватированной формах с фармацевтически приемлемыми растворителями, такими как вода, этанол и т.п. Как правило, сольватированные формы считаются эквивалентными несольватированным формам для целей настоящего изобретения.
Фармацевтические композиции
Настоящее изобретение, кроме того, относится к фармацевтической композиции, содержащей терапевтически эффективное количество соединения формулы I и фармацевтически приемлемый носитель или разбавитель. Настоящее изобретение также относится к фармацевтической композиции, содержащей терапевтически эффективное количество одного из конкретных соединений, раскрытых в экспериментальном разделе в настоящем документе, и фармацевтически приемлемый носитель или разбавитель.
Соединения в соответствии с настоящим изобретением можно вводить отдельно или в комбинации с фармацевтически приемлемыми носителями, разбавителями или наполнителями либо одной, либо несколькими дозами. Фармацевтические композиции в соответствии с настоящим изобретением можно составлять с фармацевтически приемлемыми носителями или разбавителями, а также любыми другими известными вспомогательными средствами и наполнителями согласно традиционным методикам, таким как раскрытые в Remington: The Science and Practice of Pharmacy, 19th Edition, Gennaro, Ed., Mack Publishing Co., Easton, PA, 1995.
Фармацевтические композиции можно специально составлять для введения любым подходящим путем, таким как пероральный, ректальный, назальный, легочный, местный (в том числе буккальный и подъязычный), чрескожный, интрацистернальный, интраперитонеальный, вагинальный и парентеральный (в том числе подкожный, внутримышечный, интратекальный, внутривенный и внутрикожный) пути. Очевидно, что путь будет зависеть от общего состояния и возраста подлежащего лечению субъекта, природы подлежащего лечению состояния и активного ингредиента.
Фармацевтические композиции для перорального введения включают твердые дозированные формы, такие как капсулы, таблетки, драже, пилюли, пастилки, порошки и гранулы. При необходимости композиции можно получать с покрытиями, такими как энтеросолюбильные покрытия, или же их можно составлять с обеспечением контролированного высвобождения активного ингредиента, такого как замедленное или пролонгированное высвобождение, согласно способам, хорошо известным в данной области. Жидкие дозированные формы для перорального введения включают растворы, эмульсии, суспензии, сиропы и эликсиры.
Фармацевтические композиции для парентерального введения включают стерильные водные и неводные инъекционные растворы, дисперсии, суспензии или эмульсии, а также стерильные порошки, подлежащие восстановлению в стерильных инъекционных растворах или дисперсиях перед применением. Другие подходящие формы введения включают без ограничения суппозитории, аэрозоли, мази, кремы, гели, ингаляторы, кожные пластыри и имплантаты.
Типичные пероральные дозировки варьируют от приблизительно 0,001 до приблизительно 100 мг/кг массы тела в сутки. Типичные пероральные дозировки также варьируют от приблизительно 0,01 до приблизительно 50 мг/кг массы тела в сутки. Типичные пероральные дозировки далее варьируют от приблизительно 0,05 до приблизительно 10 мг/кг массы тела в сутки. Пероральные дозировки обычно вводят одной или несколькими дозировками, как правило, одной - тремя дозировками в сутки. Точная дозировка будет зависеть от частоты и способа введения, пола, возраста, массы и общего состояния принимающего лечение субъекта, природы и тяжести подлежащего лечению состояния и каких-либо сопутствующих подлежащих лечению заболеваний, а также других факторов, очевидных специалистам в данной области.
Составы также могут быть представлены в форме единичной дозировки с помощью способов, известных специалистам в данной области. В качестве примера типичная форма единичной дозировки для перорального введения может содержать от приблизительно 0,01 до приблизительно 1000 мг, от приблизительно 0,05 до приблизительно 500 мг или от приблизительно 0,5 мг до приблизительно 200 мг.
В случае парентеральных путей, таких как внутривенное, интратекальное, внутримышечное и подобное введение, типичные дозы составляют порядка половины дозы, используемой для перорального введения.
Настоящее изобретение также относится к способу получения фармацевтической композиции, предусматривающему смешивание терапевтически эффективного количества соединения формулы I и по меньшей мере одного фармацевтически приемлемого носителя или разбавителя. В варианте осуществления в соответствии с настоящим изобретением соединение, используемое в вышеупомянутом способе, является одним из конкретных соединений, раскрытых в экспериментальном разделе в настоящем документе.
Соединения в соответствии с настоящим изобретением обычно используют как свободное вещество или как его фармацевтически приемлемую соль. Одним примером является кислотно-аддитивная соль соединения, что характеризуется применимостью свободного основания. Если соединение формулы I содержит свободное основание, то такие соли получают традиционным способом путем обработки раствора или суспензии свободного основания формулы I молярным эквивалентом фармацевтически приемлемой кислоты. Выше описаны типичные примеры подходящих органических и неорганических кислот.
Для парентерального введения можно использовать растворы соединений формулы I в стерильном водном растворе, водном пропиленгликоле, водном витамине E или кунжутном или арахисовом масле. Такие водные растворы должны быть подходящим образом забуферены, если необходимо, а жидкий разбавитель сначала делают изотоническим с помощью достаточного количества солевого раствора или глюкозы. Водные растворы являются особенно подходящими для внутривенного, внутримышечного, подкожного и внутриперитонеального введения. Соединения формулы I можно легко включить в известную стерильную водную среду с использованием стандартных методик, известных специалистам в данной области.
Подходящие фармацевтические носители включают инертные твердые разбавители или заполнители, стерильные водные растворы и различные органические растворители. Примеры твердых носителей включают лактозу, сульфат кальция, сахарозу, циклодекстрин, тальк, желатин, агар, пектин, аравийскую камедь, стеарат магния, стеариновую кислоту и низшие алкиловые эфиры целлюлозы. Примеры жидких носителей включают без ограничения сироп, арахисовое масло, оливковое масло, фосфолипиды, жирные кислоты, амины жирных кислот, полиоксиэтилен и воду. Подобным образом, носитель или разбавитель может включать любые материалы для замедленного высвобождения, известные в данной области, такие как глицерилмоностеарат или глицерилдистеарат, отдельно или смешанные с воском. Затем фармацевтические композиции, образованные путем объединения соединений формулы I с фармацевтически приемлемым носителем, легко вводят множеством дозированных форм, подходящих для раскрытых путей введения. При помощи известных в области фармацевтики способов, составы для удобства могут быть представлены в форме единичной дозировки.
Подходящие для перорального введения составы в соответствии с настоящим изобретением могут быть представлены в виде отдельных единиц, таких как капсулы или таблетки, каждая из которых содержит предварительно заданное количество активного ингредиента и необязательно подходящий наполнитель. Кроме того, перорально доступные составы могут иметь форму порошка или гранул, раствора или суспензии в водной или неводной жидкости или жидкой эмульсии масло-в-воде или вода-в-масле.
Если для перорального введения используется твердый носитель, то препарат может быть таблетированным, помещенным в твердую желатиновую капсулу в форме порошка или пеллеты, или же он может быть в форме лепешки или пастилки. Количество твердого носителя будет существенно варьировать, но находиться в диапазоне от приблизительно 25 мг до приблизительно 1 г на дозированную единицу. Если используется жидкий носитель, то препарат может иметь форму сиропа, эмульсии, мягкой желатиновой капсулы или стерильной инъекционной жидкости, такой как водная или неводная жидкая суспензия или раствор.
Фармацевтические композиции в соответствии с настоящим изобретением можно получать традиционными известными в данной области способами. Например, таблетки можно получать путем смешивания активного ингредиента со стандартными вспомогательными средствами и/или разбавителями, а затем прессования смеси в традиционной таблеточной машине. Примеры вспомогательных средств или разбавителей включают кукурузный крахмал, картофельный крахмал, тальк, стеарат магния, желатин, лактозу, камеди и т.п. Можно использовать любые другие вспомогательные средства или добавки, обычно применяемые для таких целей, такие как красители, ароматизаторы, консерванты и т.п., при условии, что они совместимы с активными ингредиентами.
Терапевтически эффективное количество
В данном контексте термин "терапевтически эффективное количество" соединения означает количество, достаточное для излечения, облегчения или частично остановки клинических проявлений данного заболевания и его осложнений при терапевтическом вмешательстве, предусматривающем введение указанного соединения. Количество, адекватное для осуществления этого, определяется как "терапевтически эффективное количество". Эффективные количества для такой цели будут зависеть от тяжести заболевания или повреждения, а также от массы и общего состояния субъекта. Следует понимать, что определения соответствующей дозировки можно достигнуть с использованием общепринятого эксперимента, путем построения матрицы значений и тестирования различных точек в матрице, что находится в компетенции квалифицированного врача.
В данном контексте термины "лечение" и "обработка" означают контроль и уход за пациентом с целью борьбы с состоянием, таким как заболевание или расстройство. Термин предназначен для включения полного спектра видов лечения данного состояния, от которого страдает пациент, таких как введение активного соединения для облегчения симптомов или осложнений, для приостановки развития заболевания, расстройства или состояния, для облегчения или смягчения симптомов и осложнений и/или для излечения или устранения заболевания, расстройства или состояния, а также для предотвращения состояния, при этом предотвращение следует понимать как контроль и наблюдение пациента с целью борьбы с заболеванием, состоянием или расстройством, и он предусматривает введение активных соединений для предотвращения появления симптомов или осложнений. При этом профилактический (превентивный) и терапевтический (клинический) виды лечения являются двумя отдельными аспектами в соответствии с настоящим изобретением. Подлежащим лечению пациентом предпочтительно является млекопитающее, в частности человек.
Лечение расстройств
Как упоминалось выше, соединения формулы I являются ингибиторами фермента PDE10A и как таковые применимы для лечения ассоциированных неврологических и психических расстройств.
Таким образом, настоящее изобретение относится к соединению формулы I или его фармацевтически приемлемой кислотно-аддитивной соли, а также к фармацевтической композиции, содержащей такое соединение, для применения в лечении нейродегенеративного расстройства, психического расстройства или наркозависимости у людей.
В одном варианте осуществления в соответствии с настоящим изобретением нейродегенеративное расстройство или состояние включает нейродегенерацию стриарных средних шипиковых нейронов у человека. В конкретном варианте осуществления в соответствии с настоящим изобретением нейродегенеративным расстройством или состоянием является болезнь Хантингтона. В следующем варианте осуществления расстройством является дискинезия, ассоциированная с терапией дофаминомиметическим препаратом.
В варианте осуществления психическое расстройство выбрано из группы, состоящей из шизофрении, например, параноидного типа, тяжелой формы шизофрении, кататонического, недифференцированного или резидуального типа; шизофрениформного расстройства; шизоаффективного расстройства, например, бредового типа или депрессивного типа; бредового расстройства; индуцированного веществами психического расстройства, например, психоза, индуцированного алкоголем, амфетамином, марихуаной, кокаином, галлюциногенами, средствами для ингаляции, опиоидами или фенциклидином; расстройства личности параноидного типа и расстройства личности шизоидного типа.
Настоящее изобретение, кроме того, относится к способу лечения у человека наркозависимости, например, алкогольной, амфетаминовой, кокаиновой или опиатной зависимости, при этом способ предусматривает введение указанному человеку эффективного для лечения зависимости, такой как наркозависимость, количества соединения формулы I.
Термин "наркозависимость", как используется в настоящем документе, означает патологическое желание наркотического средства и, как правило, характеризуется нарушениями мотивационной сферы, такими как компульсивное побуждение принять желаемое наркотическое средство и эпизоды непреодолимого влечения к нему.
Другими расстройствами, которые можно лечить в соответствии с настоящим изобретением, являются обсессивно/компульсивные расстройства, инсулинонезависимый сахарный диабет (NIDDM), синдром Туретта и другие тиковые расстройства, а также синдром дефицита внимания с гиперактивностью (ADHD).
Соединения формулы I или их фармацевтически приемлемые соли можно использовать в комбинации с одним или несколькими другими лекарственными средствами (в том числе типичным и атипичным антипсихотическим средством) в лечении заболеваний или состояний, при которых полезны соединения в соответствии с настоящим изобретением, при этом объединение лекарственных средств вместе является более безопасным или более эффективным, нежели каждое лекарственное средство отдельно. Кроме того, соединения в соответствии с настоящим изобретением можно использовать в комбинации с одним или несколькими другими лекарственными средствами, которые лечат, предотвращают, регулируют, облегчают или снижают риск побочных эффектов или токсичности соединений в соответствии с настоящим изобретением. Комбинации, применения и способы лечения в соответствии с настоящим изобретением также могут обеспечивать преимущества в лечение пациентов, которые не способны адекватно отвечать или которые устойчивы к другим известным видам лечения.
Другие такие лекарственные средства можно вводить путем и в количестве, обычно применяемых таким образом, одновременно или последовательно с соединениями в соответствии с настоящим изобретением. Следовательно, фармацевтические композиции в соответствии с настоящим изобретением включают содержащие один или несколько других активных ингредиентов в дополнение к соединениям в соответствии с настоящим изобретением. Комбинации можно вводить как часть комбинированного продукта в форме единичной дозировки, или набора, или лечебного протокола, где одно или несколько дополнительных лекарственных средств вводят в отдельных дозированных формах как часть режима лечения.
Термин "нейролептическое средство", используемый в настоящем документе, относится к лекарственным средствам, которые по отношению к когнитивной функции и поведению обладают эффектом антипсихотических лекарственных средств, уменьшающим спутанность сознания, бред, галлюцинации и психомоторное возбуждение у пациентов с психозом. Нейролептические средства, также известные как основные транквилизаторы и антипсихотические лекарственные средства, включают без ограничения типичные антипсихотические лекарственные средства, в том числе фенотиазины, дополнительно разделяемые на алифатические соединения, пиперидины и пиперазины, тиоксантены (например, цисординол), бутирофеноны (например, галоперидол), дибензоксазепины (например, локсапин), дигидроиндолоны (например, молиндон), дифенилбутилпиперидины (например, пимозид), и атипичные антипсихотические лекарственные средства, в том числе бензисоксазолы (например, рисперидон), сертиндол, оланзапин, кветиапин, осанетант и зипрасидон.
Особенно предпочтительными нейролептическими средствами для применения в настоящем изобретении являются сертиндол, оланзапин, рисперидон, кветиапин, арипипразол, галоперидол, клозапин, зипрасидон и осанетант.
Как используется в настоящем документе, и если не указано иное, "нейродегенеративное расстройство или состояние" относится к расстройству или состоянию, которое вызвано дисфункцией и/или гибелью нейронов в центральной нервной системе. Лечение этих расстройств и состояний можно облегчать введением средства, которое предотвращает дисфункцию или гибель нейронов при риске этих расстройств или состояний и/или усиливает функцию поврежденных или здоровых нейронов таким образом, чтобы компенсировать потерю функции, вызванную дисфункцией или гибелью подверженных риску нейронов. Термин "нейротрофическое средство", используемый в настоящем документе, относится к веществу или средству, которое обладает некоторыми или всеми из этих свойств.
Все ссылки, включая публикации, патентные заявки и патенты, приведенные в данном документе, включены посредством ссылки в данный документ во всей своей полноте и в той же степени, как если бы каждая ссылка была индивидуально и конкретно обозначена для включения посредством ссылки и приведена во всей своей полноте (в максимальной степени, допускаемой законом).
Заголовки и подзаголовки используют в настоящем документе исключительно для удобства, и их не следует рассматривать как ограничивающие настоящее изобретение каким-либо образом.
Применение каких-либо и всех примеров или типичной фразы (в том числе "так, например", "например", "к примеру" и "как таковой") в настоящем описании предназначено исключительно для лучшего освещения настоящего изобретения и не является ограничением объема настоящего изобретения, если не указано иное.
Цитирование и включение патентных документов в настоящий документ служит исключительно для удобства и не отражает какую-либо оценку действительности, патентоспособности и/или осуществимости таких патентных документов.
Настоящее изобретение включает все модификации и эквиваленты предмета приложенной формулы изобретения в соответствии с действующим законодательством.
Раскрытое в данном документе настоящее изобретение будет дополнительно проиллюстрировано следующими неограничивающими примерами.
Экспериментальный раздел
Получение соединений в соответствии с настоящим изобретением
Соединения общей формулы I в соответствии с настоящим изобретением можно получать, как описано в следующих схемах реакций.
Соединения формулы I, где L представляет собой -CH=CH- или -CH2-CH2-, можно получать последовательностью реакций, показанной на схеме 1.
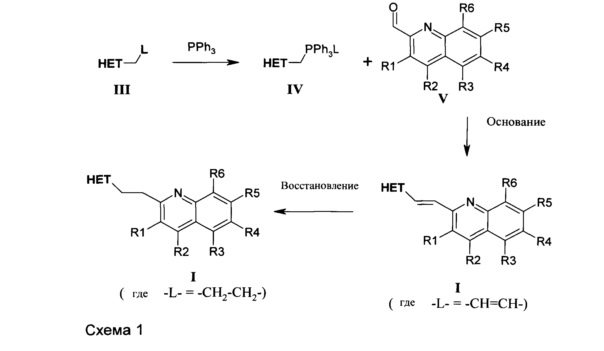
Более определенно, соединения формулы I, где L представляет собой -CH2-CH2-, можно получать восстановлением алкена формулы I, где L представляет собой -CH=CH- гидрогенизацией с использованием катализатора на основе переходного металла, такого как металл палладий, вместе с источником водорода, таким как газообразный водород, гидрокарбонат аммония, или циклогексадиен. Указанные алкены формулы I, где L представляет собой -CH=CH- можно получать реакцией Виттига между фосфониевой солью формулы IV и альдегидом формулы V в подходящем растворителе, таком как тетрагидрофуран, в присутствии подходящего основания, такого как 1,8-диазабицикло[5.4.0]ундец-7-ен. Фосфониевые соли формулы IV легко доступны посредством реакции соединений формулы IV (см. схему 1 выше) с трифенилфосфином с помощью способов, известных химикам, специализирующимся в данной области, и описанных, например, в WO-2011072696, WO-2011072694 и WO-2009152825. Альдегиды формулы V являются коммерчески доступными или получаемыми с помощью способов, описанных в литературе, см., например, Organometallics (2011), 30(5), 1008-1012, Journal of Medicinal Chemistry (2010), 53(24), 8663-8678. Chemical Communications (2010), 46(35), 6554-6556, Journal of Medicinal Chemistry (2010), 53(5), Science of Synthesis (2005), 15 389-549. Journal of the Chemical Society (1932), Journal of the American Chemical Society (1941), 63 2654-5.
Общие способы
Аналитические данные LC-MS получали с использованием следующего способа.
Способ 111
LC-MS выполняли на Sciex API150EX, оснащенном APPI-источником, функционирующем в режиме положительных ионов. HPLC включала насосы Shimadzu LC10-ADvp LC, SPD-M20A PDA УФ-детектор (функционирующий при 254 нМ) и контроллер системы Shimadzu СВМ-20А. Автоматический пробозаборник представлял собой Gilson 215. Колоночный термостат представлял собой Metalox модели 200-С с температурой колонки: 60°C. Инжектор: Gilson модели 841 (1-микролитровая петля).
Детектор ELS представлял собой Sedere Sedex 85.
Условия LC: Колонка представляла собой Waters Symmetry С-18, 4,6×30 мм, 3,5 мкм, функционирующую при 60°C с 3,3 мл/минута бинарного градиента, включающего растворитель A: 100% H2O 0,05% TFA, и растворитель B: 95% ACN 5% H2O 0,035% TFA.
Объем введения: 10 мкл (1 мкл вводили в колонку).
Градиент: 10% B - 100% B за 2,4 минуты,
10% В за 0,4 минуты.
Общее время прогона: 2,8 минуты.
Способ 131
LC-MS выполняли на Sciex API150EX, оснащенном APPI-источником, функционирующем в режиме положительных ионов. HPLC включала насосы Shimadzu LC10-ADvp LC, SPD-M20A PDA УФ-детектор (функционирующий при 254 нМ) и контроллер системы Shimadzu СВМ-20А. Автоматический пробозаборник представлял собой Gilson 215. Колоночный термостат представлял собой Jones Chromatography 7990R с температурой колонки: 60°C.
Детектор ELS представлял собой Sedere Sedex 85.
Условия LC: Колонка представляла собой Waters Symmetry С-18, 4,6×30 мм, 3,5 мкм, функционирующую при 60°C с 3,0 мл/минута бинарного градиента, включающего растворитель A: H2O с 0,05% объем/объем TFA, и растворитель B: метанол с 0,05% TFA.
Объем введения: 10 мкл (1 мкл вводили в колонку).
Градиент:
0,01 минуты 17% B (объем/объем)
0,27 минуты 28% B (объем/объем)
0,53 минуты 39% B (объем/объем)
0,80 минуты 50% B (объем/объем)
1,07 минуты 59% B (объем/объем)
1,34 минуты 68% B (объем/объем)
1,60 минуты 78% B (объем/объем)
1,87 минуты 86% B (объем/объем)
2,14 минуты 93% B (объем/объем)
2,38 минуты 100% B (объем/объем)
2,40 минуты 17% B (объем/объем)
2,80 минуты 17% B (объем/объем)
Общее время прогона: 2,8 минуты.
Способ 132
LC-MS выполняли на Sciex API150EX, оснащенном APPI-источником, функционирующем в режиме положительных ионов. HPLC включала насосы Shimadzu LC10-ADvp LC, SPD-M20A PDA детектор (функционирующий при 254 нМ) и контроллер системы SCL-10A. Автоматический пробозаборник представлял собой Gilson 215. Колоночный термостат представлял собой Jones Chromatography 7990R, и детектор ELS представлял собой Sedere Sedex 85.
Условия LC: Колонка представляла собой Waters Symmetry С-18, 4,6×30 мм, 3,5 мкм, функционирующую при 60°C с 2,5 мл/минута бинарного градиента, включающего воду + 0,05% TFA (A) и метанол + 0,05% TFA (B).
Общее время прогона: 2,8 минуты.
Способ 350
LC-MS выполняли на Sciex API300, оснащенном APPI-источником, функционирующем в режиме положительных ионов. UPLC включала Waters Aquity, в том числе управляющее устройство колонки, управляющее устройство двойного растворителя, подставку для образца, детектор PDA (функционирующий при 254 нМ) и детектор ELS.
Условия LC: Колонка представляла собой Waters Aquity UPLC ВЕН С-18, 2,1×50 мм, 1,7 мкм, функционирующую при 60°C с 1,2 мл/минута бинарного градиента, включающего воду + 0,05% TFA (A) и 95% ацетонитрил, содержащий 5% воды + 0,03% TFA (B).
Общее время прогона 1,15 минуты.
Очистку препаративной LC-MS выполняли на устройстве РЕ Sciex API 150ЕХ с химической ионизацией при атмосферном давлении. Колонка: 50×20 мм YMC ODS-A с размером частиц 5 мкм; способ: элюирование в линейном градиенте с А:В=80:20-0:100 за 7 минут и со скоростью потока 22,7 мл/минута. Сбор фракции выполняли с помощью детектирования MS с разделенным потоком.
1Н ЯМР спектры регистрировали при 500,13 МГц на приборе Bruker Avance AV500 или при 600,16 МГц на приборе Bruker Avance Ultrashield plus. В качестве внутреннего стандарта использовали TMS. Значения химического сдвига выражали в ррт. Для множества сигналов ЯМР использовали следующие сокращения: s = синглет, d = дублет, t = триплет, q = квартет, qui = квинтет, h = гептет, dd = двойной дублет, dt = двойной триплет, dq = двойной квартет, td = триплет дублетов, tt = триплет триплетов, m = мультиплет, br s = широкий синглет и br = широкий сигнал.
Сокращения соответствуют ACS Style Guide: "The ACS Styleguide - A manual for authors and editors" Janet S. Dodd, Ed. 1997, ISBN: 0841234620.
Получение промежуточных соединений
Фосфониевые соли формулы IV, показанные на схеме 1, легко доступны посредством реакции соединений формулы IV (см. схему 1 выше) с трифенилфосфином с помощью способов, известных химикам, специализирующимся в данной области, и описанных, например, в WO-2011072696, WO-2011072694 и WO-2009152825. Альдегиды формулы V являются коммерчески доступными или получаемыми с помощью способов, описанных в литературе, см., например, Organometallics (2011), 30(5), 1008-1012, Journal of Medicinal Chemistry (2010), 53(24), 8663-8678. Chemical Communications (2010), 46(35), 6554-6556, Journal of Medicinal Chemistry (2010), 53(5), Science of Synthesis (2005), 15 389-549. Journal of the Chemical Society (1932), Journal of the American Chemical Society (1941), 63 2654-5.
2-[(E)-2-(5,8-Диметил-[1,2,4]триазоло[1,5-а]пиразин-2-ил)-винил]-хинолин-6-карбонитрил
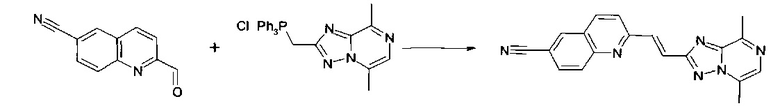
К суспензии (5,8-диметил-[1,2,4]триазоло[1,5-a]пиразин-2-илметил)-трифенил-фосфония хлорида (0,222 г, 0,483 ммоль) и 2-формил-хинолин-6-карбонитрила (80 мг, 0,4 ммоль) в сухом тетрагидрофуране (6 мл, 80 ммоль) добавляли 1,8-диазабицикло[5.4.0]ундец-7-ен (66 мкл, 0,44 ммоль) (реакционная смесь становилась слегка желтой в течение некоторого времени, а характер осадка изменялся) и смесь перемешивали при комнатной температуре в атмосфере аргона в течение ночи. Смесь упаривали при вращении, а также выпаривали THF. Твердое вещество растворяли в DCM и хроматографировали на силикагеле (0-30% МеОН в EtOAc). Очищенный продукт 2-[(E)-2-(5,8-диметил-[1,2,4]триазоло[1,5-a]пиразин-2-ил)-винил]-хинолин-6-карбонитрил осаждали в одной из фракций (10): выделяли фильтрацией. Выход: 6 мг белого твердого вещества.
Подобным образом получали следующие промежуточные соединения:
2-[2-(5,7-диметил-имидазо[1,2-a]пиримидин-2-ил)-винил]-хинолин; 2-[2-(5,7-диметил-[1,2,4]триазоло[1,5-a]пиримидин-2-ил)-винил]-хинолин;
2-[2-(5,8-диметил-[1,2,4]триазоло[1,5-a]пиразин-2-ил)-винил]-хинолин;
2-[2-(5,8-диметил-[1,2,4]триазоло[1,5-a]пиразин-2-ил)-винил]-хинолин;
2-[2-(5,7-диметил-[1,2,4]триазоло[1,5-a]пиримидин-2-ил)-винил]-6-метокси-хинолин;
2-[2-(8-метокси-5-метил-[1,2,4]триазоло[1,5-a]пиридин-2-ил)-винил]-хинолин;
2-[2-(8-этил-5-метил-[1,2,4]триазоло[1,5-a]пиридин-2-ил)-винил]-6-фтор-хинолин;
6-фтор-2-[2-(8-метокси-5-метил-[1,2,4]триазоло[1,5-a]пиридин-2-ил)-винил]-хинолин;
2-[2-(5,8-диметил-[1,2,4]триазоло[1,5-a]пиридин-2-ил)-винил]-хинолин;
2-[2-(6-фтор-хинолин-2-ил)-винил]-5-метил-[1,2,4]триазоло[1,5-a]пиридин-8-ол;
2-[2-(5,8-диметил-[1,2,4]триазоло[1,5-a]пиразин-2-ил)-винил]-4-метил-хинолин;
2-[2-(5,8-диметил-[1,2,4]триазоло[1,5-a]пиразин-2-ил)-винил]-4-метокси-хинолин;
4-метокси-2-[2-(5-метил-[1,2,4]триазоло[1,5-a]пиридин-2-ил)-винил]-хинолин;
4-метокси-2-[2-(8-метокси-5-метил-[1,2,4]триазоло[1,5-a]пиридин-2-ил)-винил]-хинолин;
4-метил-2-[2-(5-метил-[1,2,4]триазоло[1,5-a]пиридин-2-ил)-винил]-хинолин;
2-[2-(8-метокси-5-метил-[1,2,4]триазоло[1,5-a]пиридин-2-ил)-винил]-4-метил-хинолин;
4-хлор-8-фтор-2-[(E)-2-(8-метокси-5-метил-[1,2,4]триазоло[1,5-a]пиридин-2-ил)-винил]-хинолин;
8-фтор-2-[2-(8-метокси-5-метил-[1,2,4]триазоло[1,5-a]пиридин-2-ил)-винил]-хинолин;
2-[2-(5,8-диметил-[1,2,4]триазоло[1,5-a]пиразин-2-ил)-винил]-7-фтор-хинолин;
2-[2-(5,8-диметил-[1,2,4]триазоло[1,5-a]пиразин-2-ил)-винил]-6-фтор-хинолин;
2-[2-(5,8-диметил-[1,2,4]триазоло[1,5-a]пиразин-2-ил)-винил]-6-фтор-хинолин;
2-[2-(5,8-диметил-[1,2,4]триазоло[1,5-a]пиразин-2-ил)-винил]-4-фтор-хинолин;
2-[(E)-2-(5,8-диметил-[1,2,4]триазоло[1,5-a]пиразин-2-ил)-винил]-6-фтор-хинолин;
2-[2-(5,8-диметил-[1,2,4]триазоло[1,5-a]пиразин-2-ил)-винил]-7-фтор-4-метокси-хинолин;
2-[2-(5,8-диметил-[1,2,4]триазоло[1,5-a]пиразин-2-ил)-винил]-7-фтор-хинолин-4-ол;
2-[(E)-2-(5,8-диметил-[1,2,4]триазоло[1,5-a]пиразин-2-ил)-винил]-7-трифторметил-хинолин;
2-[2-(5,8-диметил-[1,2,4]триазоло[15-a]пиразин-2-ил)-винил]-6-фтор-4-метокси-хинолин;
2-[2-(5,8-диметил-[1,2,4]триазоло[1,5-a]пиразин-2-ил)-винил]-7-трифторметил-хинолин;
2-[2-(5,8-диметил-[1,2,4]триазоло[1,5-a]пиразин-2-ил)-винил]-6-фтор-хинолин-4-ол;
2-[2-(5,8-диметил-[1,2,4]триазоло[1,5-a]пиразин-2-ил)-винил]-5-фтор-хинолин;
7-хлор-2-[2-(5,8-диметил-[1,2,4]триазоло[1,5-a]пиразин-2-ил)-винил]-хинолин;
2-[2-(5,8-диметил-[1,2,4]триазоло[1,5-c]пиримидин-2-ил)-винил]-6-изопропил-хинолин;
2-[2-(5,8-диметил-[1,2,4]триазоло[1,5-a]пиразин-2-ил)-винил]-5,7-дифтор-хинолин;
2-[2-(5,8-диметил-[1,2,4]триазоло[1,5-a]пиразин-2-ил)-винил]-5,6,8-трифтор-хинолин;
2-[2-(5,8-диметил-[1,2,4]триазоло[1,5-a]пиразин-2-ил)-винил]-6,8-дифтор-хинолин;
6-[2-(5,8-диметил-[1,2,4]триазоло[1,5-a]пиразин-2-ил)-винил]-[1,3]диоксоло[4,5-g]хинолин;
2-[2-(5,8-диметил-[1,2,4]триазоло[1,5-a]пиразин-2-ил)-винил]-6-фтор-8-метил-хинолин;
2-[2-(5,8-диметил-[1,2,4]триазоло[1,5-a]пиразин-2-ил)-винил]-6-фтор-7-метил-хинолин;
6-[2-(5,8-диметил-[1,2,4]триазоло[1,5-a]пиразин-2-ил)-винил]-2,2-дифтор-[1,3]диоксоло[4,5-g]хинолин;
7-хлор-2-[2-(5,8-диметил-[1,2,4]триазоло[1,5-a]пиразин-2-ил)-винил]-хинолин-6-карбонитрил;
7-[2-(5,8-диметил-[1,2,4]триазоло[1,5-a]пиразин-2-ил)-винил]-2,3-дигидро-[1,4]диоксино[2,3-g]хинолин;
6-хлор-2-[2-(5,8-диметил-[1,2,4]триазоло[1,5-a]пиразин-2-ил)-винил]-хинолин;
6-хлор-2-[2-(5,8-диметил-[1,2,4]триазоло[1,5-a]пиразин-2-ил)-винил]-8-фтор-хинолин;
8-хлор-2-[2-(5,8-диметил-[1,2,4]триазоло[1,5-a]пиразин-2-ил)-винил]-6-метил-хинолин;
5,7-дихлор-2-[2-(5,8-диметил-[1,2,4]триазоло[1,5-a]пиразин-2-ил)-винил]-хинолин.
Получение соединений в соответствии с настоящим изобретением
Пример 1: 2-[(E)-2-(5,8-диметил-[1,2,4]триазоло[1,5-a]пиразин-2-ил)-винил]-6-трифторметил-хинолин
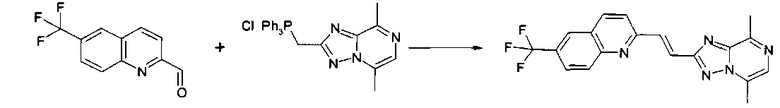
К раствору (5,8-диметил-[1,2,4]триазоло[1,5-a]пиразин-2-илметил)-трифенил-фосфония хлорида (0,48 г, 1,0 ммоль) и 6-трифторметил-хинолин-2-карбальдегида (0,24 г, 1,0 ммоль) в сухом N,N-диметилформамиде (25 мл, 320 ммоль) добавляли 1,8-диазабицикло[5.4.0]ундец-7-ен (0,16 мл, 1,0 ммоль) (реакционная смесь становилась более темной) и смесь перемешивали при комнатной температуре в атмосфере аргона в течение ночи. Через день реакционная смесь показывала осадок.
Осадок отфильтровывали. Промывали водой и диэтиловым эфиром. Сушили на фильтре вакуумом, затем в вакууме в течение 2 часов при 60°C. Осадок на фильтре давал белое твердое вещество, содержащее конечный продукт 2-[(E)-2-(5,8-диметил-[1,2,4]триазоло[1,5-a]пиразин-2-ил)-винил]-6-трифторметил-хинолин. LC-MS: масса/заряд = 369,7 (МН+). Rt = 1,96 минуты, способ = 131.
Подобным образом получали следующие соединения.
2-[(E)-2-(5,7-Диметил-имидазо[1,2-a]пиримидин-2-ил)-винил]-хинолин, LC-MS: масса/заряд = 301,1 (МН+). Rt = 0,55 минуты, способ = 111.
2-[(E)-2-(5,8-Диметил-[1,2,4]триазоло[1,5-a]пиразин-2-ил)-винил]-6-трифторметокси-хинолин, LC-MS: масса/заряд = 386,1, Rt = 1,97 минуты, способ = 131.
2-[(E)-2-(5,8-Диметил-[1,2,4]триазоло[1,5-a]пиразин-2-ил)-винил]-6-циано-хинолин, LC-MS: масса/заряд = 327,3, Rt = 1,97 минуты, способ = 131.
2-[(E)-2-(5,8-Диметил-[1,2,4]триазоло[1,5-a]пиразин-2-ил)-винил]-7-метокси-хинолин, LC-MS: масса/заряд = 332,1, Rt = 1,22 минуты, способ = 131.
2-[(E)-2-(5,8-Диметил-[1,2,4]триазоло[1,5-a]пиразин-2-ил)-винил]-5-метокси-хинолин, LC-MS: масса/заряд = 332,2, Rt = 1,41 минуты, способ = 131.
Пример 2: 6-[2-(5,8-диметил-[1,2,4]триазоло[1,5-a]пиразин-2-ил)-этил]-1,3-диоксоло[4,5-д]хинолин
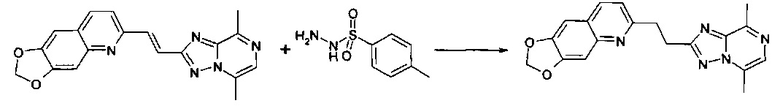
6-[(E)-2-(5,8-Диметил-[1,2,4]триазоло[1,5-a]пиразин-2-ил)-винил]-1,3-диоксоло[4,5-g]хинолин (0,183 г, 0,530 ммоль) растворяли в N,N- диметилформамиде (11 мл, 140 ммоль). [B] Добавляли п-толуолсульфонилгидразид (0,296 г, 1,59 ммоль; поставщик = Avocado) и реакционную смесь перемешивали при 130°C в атмосфере аргона ON. Выполняли LCMS и наблюдали почти полное превращение. Добавляли 0,100 г [B] к смеси, перемешивали 2 дня при 130°C.
DMF выпаривали. Твердое вещество растворяли в 50 мл EtOAc, экстрагировали с помощью 2×5 мл насыщ. NaHCO3 и промывали 50 мл солевого раствора. Органическую фазу упаривали при вращении и хроматографировали на силикагеле с использованием EtOAc:гептан (1:1), а затем 0-30% МеОН в EtOAc. Выход: 40 мг твердого вещества. LC-MS: масса/заряд = 348,4 (МН+). Rt = 0,34 минуты, способ = 350.
Подобным образом получали следующие соединения.
2-[2-(5,7-Диметил-имидазо[1,2-a]пиримидин-2-ил)-этил]-хинолин, LC-MS: масса/заряд = 303,4 (МН+). Rt = 0,34 минуты, способ = 111.
2-[2-(5,7-Диметил-[1,2,4]триазоло[1,5-a]пиримидин-2-ил)-этил]-хинолин, LC-MS: масса/заряд = 304,3 (МН+). Rt = 0,46 минуты, способ = 111.
2-[2-(5,8-Диметил-[1,2,4]триазоло[1,5-a]пиразин-2-ил)-этил]-хинолин, LC-MS: масса/заряд = 304,3 (МН+). Rt = 0,61 минуты, способ = 131.
2-[2-(5,7-Диметил-[1,2,4]триазоло[1,5-a]пиримидин-2-ил)-этил]-6-метокси-хинолин, LC-MS: масса/заряд = 334,5 (МН+). Rt = 0,62 минуты, способ = 131.
2-[2-(8-Метокси-5-метил-[1,2,4]триазоло[1,5-a]пиридин-2-ил)-этил]-хинолин, LC-MS: масса/заряд = 319,1 (МН+). Rt = 0,71 минуты, способ = 131.
2-[2-(8-Этил-5-метил-[1,2,4]триазоло[1,5-a]пиридин-2-ил)-этил]-6-фтор-хинолин, LC-MS: масса/заряд = 335,2 (МН+). Rt = 1,12 минуты, способ = 131.
6-Фтор-2-[2-(8-метокси-5-метил-[1,2,4]триазоло[1,5-a]пиридин-2-ил)-этил]-хинолин, LC-MS: масса/заряд = 337,5 (МН+). Rt = 0,96 минуты, способ = 131.
2-[2-(5,8-Диметил-[1,2,4]триазоло[1,5-a]пиридин-2-ил)-этил]-хинолин, LC-MS: масса/заряд = 303,5 (МН+). Rt = 0,76 минуты, способ = 131.
2-[2-(6-Фтор-хинолин-2-ил)-этил]-5-метил-[1,2,4]триазоло[1,5-a]пиридин-8-ол, LC-MS: масса/заряд = 323,1 (МН+). Rt = 0,41 минуты, способ = 350.
2-[2-(5,8-Диметил-[1,2,4]триазоло[1,5-a]пиразин-2-ил)-этил]-4-метил-хинолин, LC-MS: масса/заряд = 318,2 (МН+). Rt = 0,83 минуты, способ = 131.
2-[2-(5,8-Диметил-[1,2,4]триазоло[1,5-a]пиразин-2-ил)-этил]-4-метокси-хинолин, LC-MS: масса/заряд = 334,5 (МН+). Rt = 0,9 минуты, способ = 131.
4-Метокси-2-[2-(5-метил-[1,2,4]триазоло[1,5-a]пиридин-2-ил)-этил]-хинолин, LC-MS: масса/заряд = 319,2 (МН+). Rt = 0,93 минуты, способ = 131.
4-Метокси-2-[2-(8-метокси-5-метил-[1,2,4]триазоло[1,5-a]пиридин-2-ил)-этил]-хинолин, LC-MS: масса/заряд = 349,1 (МН+). Rt = 1,01 минуты, способ = 131.
4-Метил-2-[2-(5-метил-[1,2,4]триазоло[1,5-a]пиридин-2-ил)-этил]-хинолин, LC-MS: масса/заряд = 303,4 (МН+). Rt = 0,85 минуты, способ = 131.
2-[2-(8-Метокси-5-метил-[1,2,4]триазоло[1,5-a]пиридин-2-ил)-этил]-4-метил-хинолин, LC-MS: масса/заряд = 333,2 (МН+). Rt = 0,95 минуты, способ = 131.
4-Хлор-8-фтор-2-[(E)-2-(8-метокси-5-метил-[1,2,4]триазоло[1,5-a]пиридин-2-ил)-винил]-хинолин, LC-MS: масса/заряд = 369,2 (МН+). Rt = 1,98 минуты, способ = 131.
8-Фтор-2-[2-(8-метокси-5-метил-[1,2,4]триазоло[1,5-a]пиридин-2-ил)-этил]-хинолин, LC-MS: масса/заряд = 337,5 (МН+). Rt = 1,32 минуты, способ = 131.
2-[2-(5,8-Диметил-[1,2,4]триазоло[1,5-a]пиразин-2-ил)-этил]-7-фтор-хинолин, LC-MS: масса/заряд = 321,8 (МН+). Rt = 0,43 минуты, способ = 350.
2-[2-(5,8-Диметил-[1,2,4]триазоло[1,5-a]пиразин-2-ил)-этил]-6-фтор-хинолин, LC-MS: масса/заряд = 322,1 (МН+). Rt = 0,44 минуты, способ = 350.
2-[2-(5,8-Диметил-[1,2,4]триазоло[1,5-a]пиразин-2-ил)-этил]-4-фтор-хинолин, LC-MS: масса/заряд = 321,9 (МН+). Rt = 0,44 минуты, способ = 350.
2-[2-(5,8-Диметил-[1,2,4]триазоло[1,5-a]пиразин-2-ил)-этил]-7-фтор-4-метокси-хинолин, LC-MS: масса/заряд = 352,3 (МН+). Rt = 0,87 минуты, способ = 131.
2-[2-(5,8-Диметил-[1,2,4]триазоло[1,5-a]пиразин-2-ил)-этил]-7-фтор-хинолин-4-ол, LC-MS: масса/заряд = 338,4 (МН+). Rt = 1,08 минуты, способ = 131.
2-[(Е)-2-(5,8-Диметил-[1,2,4]триазоло[1,5-a]пиразин-2-ил)-винил]-7-трифторметил-хинолин, LC-MS: масса/заряд = 370,2 (МН+). Rt = 0,79 минуты, способ = 350.
2-[2-(5,8-Диметил-[1,2,4]триазоло[1,5-a]пиразин-2-ил)-этил]-6-фтор-4-метокси-хинолин, LC-MS: масса/заряд = 352,3 (МН+). Rt = 0,9 минуты, способ = 131.
2-[2-(5,8-Диметил-[1,2,4]триазоло[1,5-a]пиразин-2-ил)-этил]-7-трифторметил-хинолин, LC-MS: масса/заряд = 372,3 (МН+). Rt = 0,61 минуты, способ = 350.
2-[2-(5,8-Диметил-[1,2,4]триазоло[1,5-a]пиразин-2-ил)-этил]-6-фтор-хинолин-4-ол, LC-MS: масса/заряд = 338,1 (МН+). Rt = 1,05 минуты, способ = 131.
2-[2-(5,8-Диметил-[1,2,4]триазоло[1,5-a]пиразин-2-ил)-этил]-5-фтор-хинолин, LC-MS: масса/заряд = 322,1 (МН+). Rt = 1,25 минуты, способ = 131.
7-Хлор-2-[2-(5,8-диметил-[1,2,4]триазоло[1,5-a]пиразин-2-ил)-этил]-хинолин, LC-MS: масса/заряд = 338,3 (МН+). Rt = 1,82 минуты, способ = 132.
2-[2-(5,8-Диметил-[1,2,4]триазоло[1,5-c]пиримидин-2-ил)-этил]-6-изопропил-хинолин, LC-MS: масса/заряд = 346,2 (МН+). Rt = 0,47 минуты, способ = 350.
2-[2-(5,8-Диметил-[1,2,4]триазоло[1,5-a]пиразин-2-ил)-этил]-5,7-дифтор-хинолин, LC-MS: масса/заряд = 339,8 (МН+). Rt = 0,57 минуты, способ = 350.
2-[2-(5,8-Диметил-[1,2,4]триазоло[1,5-a]пиразин-2-ил)-этил]-5,6,8-трифтор-хинолин, LC-MS: масса/заряд = 358,4 (МН+). Rt = 0,67 минуты, способ = 350.
2-[2-(5,8-Диметил-[1,2,4]триазоло[1,5-a]пиразин-2-ил)-этил]-6,8-дифтор-хинолин, LC-MS: масса/заряд = 339,7 (МН+). Rt = 0,6 минуты, способ = 350.
6-[2-(5,8-Диметил-[1,2,4]триазоло[1,5-a]пиразин-2-ил)-этил]-[1,3]диоксоло[4,5-д]хинолин, LC-MS: масса/заряд = 348,4 (МН+). Rt = 0,34 минуты, способ = 350.
2-[2-(5,8-Диметил-[1,2,4]триазоло[1,5-a]пиразин-2-ил)-этил]-6-фтор-8-метил-хинолин, LC-MS: масса/заряд = 336,3 (МН+). Rt = 0,57 минуты, способ = 350.
2-[2-(5,8-Диметил-[1,2,4]триазоло[1,5-a]пиразин-2-ил)-этил]-6-фтор-7-метил-хинолин, LC-MS: масса/заряд = 336,3 (МН+). Rt = 0,41 минуты, способ = 350.
6-[2-(5,8-Диметил-[1,2,4]триазоло[1,5-a]пиразин-2-ил)-этил]-2,2-дифтор-[1,3]диоксоло[4,5-g]хинолин, LC-MS: масса/заряд = 384,2 (МН+). Rt = 1,6 минуты, способ = 131.
7-Хлор-2-[2-(5,8-диметил-[1,2,4]триазоло[1,5-a]пиразин-2-ил)-этил]-хинолин-6-карбонитрил, LC-MS: масса/заряд = 363,2 (МН+). Rt = 1,61 минуты, способ = 131.
7-[2-(5,8-Диметил-[1,2,4]триазоло[1,5-a]пиразин-2-ил)-этил]-2,3-дигидро-[1,4]диоксино[2,3-g]хинолин, LC-MS: масса/заряд = 362,3 (МН+). Rt = 0,82 минуты, способ = 131.
6-Хлор-2-[2-(5,8-диметил-[1,2,4]триазоло[1,5-a]пиразин-2-ил)-этил]-хинолин, LC-MS: масса/заряд = 338,3 (МН+). Rt = 1,31 минуты, способ = 131.
6-Хлор-2-[2-(5,8-диметил-[1,2,4]триазоло[1,5-a]пиразин-2-ил)-этил]-8-фтор-хинолин, LC-MS: масса/заряд = 356,2 (МН+). Rt = 1,72 минуты, способ = 131.
8-Хлор-2-[2-(5,8-диметил-[1,2,4]триазоло[1,5-a]пиразин-2-ил)-этил]-6-метил-хинолин, LC-MS: масса/заряд = 352,4 (МН+). Rt = 1,7 минуты, способ = 131.
5,7-Дихлор-2-[2-(5,8-диметил-[1,2,4]триазоло[1,5-a]пиразин-2-ил)-этил]-хинолин, LC-MS: масса/заряд = 372,0 (МН+). Rt = 2,01 минуты, способ = 131.
Фармакологическое тестирование
Фермент PDE10A
Активный фермент PDE10A получали рядом способом для применения а анализах PDE (Loughney, К. et al. Gene 1999, 234, 109-117; Fujishige, К. et al. Eur J Biochem. 1999, 266, 1118-1127, и Soderling, S. et al. Proc. Natl. Acad. Sci. 1999, 96, 7071-7076). PDE10A может быть экспрессирован как белки полной длины или как усеченные белки, при условии, что они экспрессируют каталитический домен. PDE10A можно получать в различных типах клеток, например, в клетках насекомых или Е. coli. Пример способа получения каталитически активного PDE10A является следующим. Каталитический домен PDE10A человека (аминокислоты 440-779 из последовательности с номером доступа NP 006652) амплифицировали из общей РНК головного мозга человека с помощью стандартной RT-PCR и клонировали в сайты BamH1 и Xho1 вектора рЕТ28а (Novagen). Экспрессию в Е. coli выполняли согласно стандартным протоколам. Вкратце, плазмиды экспрессии трансформировали в штамм Е. coli BL21(DE3) и 50 мл культур, инокулированных клетками, позволяли расти до OD600 0,4-0,6 перед индуцированием экспрессии белка с помощью 0,5 мМ IPTG. После индукции клетки инкубировали в течение ночи при комнатной температуре, после чего клетки собирали центрифугированием. Клетки, экспрессирующие PDE10A, повторно суспендировали в 12 мл (50 мМ TRIS-HCl, pH 8,0, 1 мМ MgCl2 и ингибиторы протеазы). Клетки подвергали лизису путем обработки ультразвуком, а после лизиса всех клеток добавляли TritonX100 согласно протоколам Novagen. PDE10A частично очищали на сефарозе Q и объединяли наиболее активные фракции.
Анализ ингибирования PDE10A
Анализ PDE10A можно выполнять, например, следующим образом. Анализ выполняли в 60 мкл образцов, содержащих фиксированное количество релевантного фермента PDE (достаточного для превращения 20-25% субстрата циклического нуклеотида), буфер (50 мМ HEPES 7.6; 10 мМ MgCl2; 0,02% Tween 20), 0,1 мг/мл BSA, 225 pCi 3H-меченного субстрата циклического нуклеотида, меченный тритием cAMP до конечной концентрации 5 нМ и варьирующие количества ингибиторов. Реакции инициировали путем добавления субстрата циклического нуклеотида и позволяли им протекать в течение одного часа при комнатной температуре до завершения посредством смешивания с 15 мкл 8 мг/мл SPA гранул силиката иттрия (Amersham). Обеспечивали оседание гранул в течение одного часа в темноте до подсчета планшетов на устройстве для подсчета Wallac 1450 Microbeta. Измеренный сигнал может быть преобразован в активность относительно неингибированного контроля (100%), и значения IC50 могут быть рассчитаны с использованием расширения Xlfit к EXCEL.
Индуцированная фенциклидином (РСР) гиперактивность
Использовали самцов мышей (ЯМРI, Charles River) массой 20-25 г. В каждой группе использовали по восемь мышей, которые получали тестируемое соединение (5 мг/кг) с PCP (2,3 мг/кг), включая параллельные контрольные группы, где мыши получали среду тестируемого соединения с PCP или только инъекции среды. Объем инъекции составлял 10 мл/кг. Эксперимент выполняли при нормальных световых условиях в помещении без помех. Тестируемое вещество инъецировали за 60 минут до инъекции PCP, которую водили подкожно.
Сразу же после инъекции PCP мышей помещали по отдельности в специально разработанные тестовые клетки (20 см × 32 см). Активность измеряли с помощью 5×8 источников инфракрасного излучения и фотоэлементов с интервалом 4 см. Световые лучи пересекали клетку на 1,8 см выше дна клетки. Для регистрации единицы счета подвижности требовалось прерывание соседних световых лучей, что позволяло избежать единиц счета, вызванных стационарными движениями мышей.
Моторику регистрировали с 5-минутными интервалами в течение 1 часа. Эффект лекарственного средства рассчитывали по общему числу единиц счета во время 1-часового периода поведенческого теста следующим образом.
В качестве исходных данных использовали среднюю подвижность, индуцированную обработкой средой при отсутствии PCP. 100-процентным эффектом PCP, следовательно, считали общее число единиц счета подвижности минус исходные данные. Ответ в группах, где мыши получали тестируемое соединение, таким образом, определяли по общему числу единиц счета подвижности минус исходные данные, что выражали в проценте подобного результата, зарегистрированного в параллельной контрольной группе, где мыши получали PCP. Процент ответов преобразовывали в процент ингибирования.
| название | год | авторы | номер документа |
|---|---|---|---|
| ГЕТЕРОАРИЛЬНЫЕ СОЕДИНЕНИЯ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2013 |
|
RU2644558C2 |
| Применение 1H-пиразоло[4,3-b]пиридинов в качестве ингибиторов PDE1 | 2017 |
|
RU2759380C2 |
| КОМБИНАЦИИ, СОДЕРЖАЩИЕ ИНГИБИТОРЫ PDE 2, ТАКИЕ КАК 1-АРИЛ-4-МЕТИЛ-[1,2,4]ТРИАЗОЛО[4,3-А]ХИНОКСАЛИНОВЫЕ СОЕДИНЕНИЯ, И ИНГИБИТОРЫ PDE 10, ДЛЯ ПРИМЕНЕНИЯ В ЛЕЧЕНИИ НЕВРОЛОГИЧЕСКИХ ИЛИ МЕТАБОЛИЧЕСКИХ РАССТРОЙСТВ | 2013 |
|
RU2657540C2 |
| БИЦИКЛИЧЕСКИЕ КЕТОНЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2018 |
|
RU2797922C2 |
| ПРОИЗВОДНЫЕ АЗАБИЦИКЛО [3.1.0]ГЕКСАНА, ПРИМЕНИМЫЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ ДОПАМИНОВЫХ РЕЦЕПТОРОВ D3 | 2005 |
|
RU2434011C2 |
| ИМИДАЗОПИРРОЛОПИРАЗИНОВЫЕ ПРОИЗВОДНЫЕ, ПОЛЕЗНЫЕ ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ, ВЫЗВАННЫХ АНОМАЛЬНОЙ АКТИВНОСТЬЮ ПРОТЕИНКИНАЗ Jak1, Jak3 ИЛИ Syk | 2010 |
|
RU2570416C2 |
| Имидазопирролопиразиновые производные, полезные для лечения заболеваний, вызванных аномальной активностью протеинкиназ Jak1, Jak3 или Syk | 2010 |
|
RU2711869C2 |
| ИМИДАЗОПИРАЗИНОНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ PDE1 | 2016 |
|
RU2712219C2 |
| СОЕДИНЕНИЯ, ГЕТЕРОБИЦИКЛО-ЗАМЕЩЕННЫЕ-[1,2,4]ТРИАЗОЛО[1,5c]ХИНАЗОЛИН-5-АМИНА, ОБЛАДАЮЩИЕ СВОЙСТВАМИ А2А АНТАГОНИСТОВ | 2013 |
|
RU2671628C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ ПИРАЗИНА | 2012 |
|
RU2612138C2 |
Изобретение относится к области органической химии, а именно к гетероциклическим соединениям, выбранным из 7-[2-(5,8-диметил-[1,2,4]триазоло[1,5-а]пиразин-2-ил)-этил]-2,3-дигидро-[1,4]диоксино[2,3-g]хинолина и 6-[2-(5,8-диметил-[1,2,4]триазоло[1,5-а]пиразин-2-ил)-этил]-[1,3]диоксоло[4,5-g]хинолина, а также к их фармацевтически приемлемым солям. Также изобретение относится к фармацевтической композиции на основе одного из указанных соединений и применению указанных соединений. Технический результат: получены новые гетероциклические соединения, полезные при лечении нейродегенеративного или психического расстройства. 3 н. и 7 з.п. ф-лы, 1 табл., 2 пр.
1. Соединение, выбранное из группы, состоящей из 7-[2-(5,8-диметил-[1,2,4]триазоло[1,5-а]пиразин-2-ил)-этил]-2,3-дигидро-[1,4]диоксино[2,3-g]хинолина и 6-[2-(5,8-диметил-[1,2,4]триазоло[1,5-а]пиразин-2-ил)-этил]-[1,3]диоксоло[4,5-g]хинолина, или его фармацевтически приемлемая соль.
2. Соединение по п.1 или его фармацевтически приемлемая соль для применения в качестве лекарственного средства для лечения нейродегенеративного или психического расстройства.
3. Соединение по п.2, при этом психическое расстройство выбрано из группы, состоящей из шизофрении, шизофрениформного расстройства, шизоаффективного расстройства, бредового расстройства, индуцированного веществами психического расстройства, расстройства личности параноидного типа и расстройства личности шизоидного типа.
4. Соединение по п.2, при этом нейродегенеративное расстройство выбрано из группы, состоящей из болезни Хантингтона и дискинезии, ассоциированной с терапией дофаминомиметическим препаратом.
5. Фармацевтическая композиция для лечения нейродегенеративного или психического расстройства, содержащая терапевтически эффективное количество соединения по п.1 или его фармацевтически приемлемой соли и фармацевтически приемлемый носитель, разбавитель или наполнитель.
6. Фармацевтическая композиция по п.5, при этом психическое расстройство выбрано из группы, состоящей из шизофрении, шизофрениформного расстройства, шизоаффективного расстройства, бредового расстройства, индуцированного веществами психического расстройства, расстройства личности параноидного типа и расстройства личности шизоидного типа.
7. Фармацевтическая композиция по п.5, при этом нейродегенеративное расстройство выбрано из группы, состоящей из болезни Хантингтона и дискинезии, ассоциированной с терапией дофаминомиметическим препаратом.
8. Применение соединения по п.1 или его фармацевтически приемлемой соли для получения лекарственного средства для лечения нейродегенеративного или психического расстройства.
9. Применение по п.8, при этом психическое расстройство выбрано из группы, состоящей из шизофрении, шизофрениформного расстройства, шизоаффективного расстройства, бредового расстройства, индуцированного веществами психического расстройства, расстройства личности параноидного типа и расстройства личности шизоидного типа.
10. Применение по п.8, при этом нейродегенеративное расстройство выбрано из группы, состоящей из болезни Хантингтона и дискинезии, ассоциированной с терапией дофаминомиметическим препаратом.
| Способ приготовления лака | 1924 |
|
SU2011A1 |
| EA 201170061 A1, 30.08.2011. | |||

