Перекрестная ссылка на родственную заявку
Настоящая заявка заявляет приоритет к временной заявке США, серийный № 61/265563, поданной 1 декабря 2009 года, и временной заявке США, серийный № 61/364116, поданной 14 июля 2010 года, содержание которых включено в настоящую заявку.
Предпосылки изобретения
Настоящее изобретение обеспечивает новый класс соединений, фармацевтические композиции, включающие такие соединения, и способы применения таких соединений для лечения или профилактики заболеваний или расстройств, связанных с аномальной или нерегулируемой киназной активностью, в частности, заболеваний или расстройств, которые включают аномальную активацию Jak1, Jak2, Jak3, Tyk2, KDR, Flt-3, CDK2, CDK4, TANK, Trk, FAK, Abl, Bcr-Abl, cMet, b-RAF, FGFR3, c-kit, PDGF-R, Syk, BTK, CSF1R, PKC киназ или киназ Aurora.
Протеинкиназы представляют собой большое семейство белков, которые играют центральную роль в регуляции широкого ряда клеточных процессов и поддержании клеточной функции. Частичный, но не ограничивающий, перечень этих киназ включает: не-рецепторные тирозиновые киназы, такие как семейство Janus киназ (Jak1, Jak2, Jak3 и Tyk2); гибридные киназы, такие как BCR-Abl, киназа фокальной адгезии(FAK), Fes, Lck и Syk; рецепторные тирозиновые киназы, такие как киназа рецептора ростового фактора, продуцируемого тромбоцитами (PDGF-R), киназа рецептора для фактора стволовых клеток, c-kit, рецептор ростового фактора гепатоцитов, c-Met, и рецептор ростового фактора фибробластов, FGFR3; и сериновые/треониновые киназы, такие как b-RAF, митоген-активируемые протеинкиназы (например, MKK6) и SAPK2β. Аберрантную киназную активность наблюдали во многих болезненных состояниях, включая доброкачественные и злокачественные пролиферативные расстройства, а также заболевания, являющиеся результатом неправильной активации иммунной и нервной систем. Новые соединения по настоящему изобретению ингибируют активность одной или нескольких протеинкиназ, и поэтому ожидают, что они могут быть полезными для лечения киназа-опосредованных заболеваний.
Краткое описание изобретения
В первом варианте воплощения настоящее изобретение обеспечивает соединение формулы (I)

Формула (I)
его фармацевтически приемлемые соли, пролекарства, биологически активные метаболиты, стереоизомеры и изомеры, где
T представляет собой N, U представляет собой N, X представляет собой CR3, и Y представляет собой N; или
T представляет собой CR6, U представляет собой N, X представляет собой CR3, и Y представляет собой N; или
T представляет собой N, U представляет собой CR4, X представляет собой CR3, и Y представляет собой N; или
T представляет собой CR6, U представляет собой CR4, X представляет собой CR3, и Y представляет собой N; или
T представляет собой CR6, U представляет собой N, X представляет собой NR3, и Y представляет собой C; или
T представляет собой O, U представляет собой N, X представляет собой CR3, и Y представляет собой C; или
T представляет собой NR6, U представляет собой N, X представляет собой CR3, и Y представляет собой C; или
T представляет собой CR6, U представляет собой CR4, X представляет собой NR3, и Y представляет собой C; или
T представляет собой S, U представляет собой N, X представляет собой CR3, и Y представляет собой C; или
T представляет собой N, U представляет собой CR4, X представляет собой NR3, и Y представляет собой C; или
T представляет собой N, U представляет собой N, X представляет собой NR3, и Y представляет собой C;
R1, R2 и R5, каждый независимо, представляют собой водород, дейтерий, -N(Ra)(Rb), галоген, -ORa, -SRa, -S(O)Ra, -S(O)2Ra, -NO2, -C(O)ORa, -CN, -C(O)N(Ra)(Rb), -N(Ra)C(O)(Rb), -C(O)Ra, -C(OH)RaRb, -N(Ra)S(O)2-Rb, -S(O)2N(Ra)(Rb), -CF3, -OCF3, необязательно замещенный (C1-C6)алкил, необязательно замещенный (C2-C6)алкенил, необязательно замещенный (C2-C6)алкинил, необязательно замещенный (C3-C10)циклоалкил, необязательно замещенный (C1-C10)гетероарил, необязательно замещенный (C1-C10) гетероциклил или необязательно замещенный (C6-C10)арил;
где в фрагменте, содержащем -N(Ra)(Rb), азот, Ra и Rb могут образовывать кольцо, таким образом, -N(Ra)(Rb) представляет собой необязательно замещенный (C2-C10)гетероциклил или необязательно замещенный (C1-C10)гетероарил, связанный через азот;
R3 представляет собой водород, необязательно замещенный связанный мостиковой связью (C5-C12)циклоалкил, необязательно замещенный связанный мостиковой связью (C2-C10)гетероциклил, необязательно замещенный (C1-C8)алкил, необязательно замещенный (C3-C10)циклоалкил, необязательно замещенный (C3-C8)циклоалкенил, необязательно замещенный (C6-C10)арил, необязательно замещенный (C1-C10)гетероарил, необязательно замещенный (C2-C10)гетероциклил; или
R3 представляет собой -A-D-E-G, где:
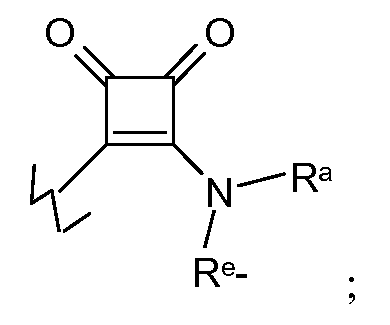
A представляет собой связь, -C(O)-, необязательно замещенный (C1-C6)алкилен, необязательно замещенный (C2-C6)алкенилен, необязательно замещенный (C2-C6)алкинилен, необязательно замещенный (C3-C12)циклоалкилен, необязательно замещенный (C2-C6)гетероциклилен, -C(O)N(Ra)-Re-, -N(Ra)C(O)-Re-, -O-Re-, -N(Ra)-Re-, -S-Re-, -S(O)2-Re-, -S(O)Re-, -C(O-Ra)(Rb)-Re-, -S(O)2N(Ra)-Re-, -N(Ra)S(O)2-Re- или -N(Ra)C(O)N(Rb)-Re-;
D представляет собой необязательно замещенный (C1-C8)алкилен, необязательно замещенный связанный мостиковой связью (C5-C12)циклоалкилен, необязательно замещенный (C3-C10)циклоалкилен, необязательно замещенный связанный мостиковой связью (C5-C10)циклоалкенилен, необязательно замещенный (C3-C10)циклоалкенилен, необязательно замещенный (C6-C10)арилен, необязательно замещенный (C1-C10)гетероарилен, необязательно замещенный связанный мостиковой связью (C2-C10)гетероциклилен или необязательно замещенный (C2-C10)гетероциклилен;
E представляет собой связь, -Re-, -Re-C(=NCN)-Re-, -Re-C(O)-Re-, -Re-C(O)C(O)-Re-, -Re-C(O)O-Re-, -Re-C(O)C(O)N(Ra)-Re-, -Re-N(Ra)-C(O)C(O)-Re-, -Re-O-Re-, -Re-S(O)2-Re-, -Re-S(O)-Re-, -Re-S-Re-, -Re-N(Ra)-Re-, =N-Re-, -Re-N(Ra)C(O)-Re-, -ReC(O)N(Ra)Re-, -Re-OC(O)N(Ra)-Re-, -Re-N(Ra)C(O)ORe-, -Re-OC(O)-Re, -Re-OC(O)-O-Re, -Re-N(Ra)C(O)N(Rb)-Re-, -Re-N(Ra)S(O)2-Re-, -Re-S(O)2N(Ra)-Re- или -Re-N(Ra)S(O)2N(Ra)-Re-; или
Е представляет собой

где во всех случаях, E связан либо с атомом углерода либо с атомом азота в D;
G представляет собой водород, дейтерий, -N(Ra)(Rb), галоген, -ORa, -SRa, -S(O)Ra, -S(O)2Ra, -NO2, -C(O)ORa, -CN, -C(O)N(Ra)(Rb), -N(Ra)C(O)Rb, -N(Ra)C(O)ORb, -OC(O)N(Ra), -N(Ra)C(O)N(Rb)2, -C(O-Ra)(Rb)2, -C(O)Ra, -CF3, -OCF3, -N(Ra)S(O)2Rb, -S(O)2N(Ra)(Rb), -S(O)2N(Ra)C(O)Rb, необязательно замещенный -(C1-C6)алкил, необязательно замещенный -(C2-C6)алкенил, необязательно замещенный -(C2-C6)алкинил, необязательно замещенный -(C3-C10)циклоалкил, необязательно замещенный -(C1-C10)гетероарил, необязательно замещенный -(C1-C10) гетероциклил, необязательно замещенный -(C6-C10)арил;
где в фрагменте, содержащем -N(Ra)(Rb), азот, Ra и Rb могут образовывать кольцо, таким образом, -N(Ra)(Rb) представляет собой необязательно замещенный (C2-C10)гетероциклил или необязательно замещенный (C1-C10) гетероарил, связанный через азот;
R4 и R6, каждый независимо, представляют собой водород, галоген, дейтерий, CF3, CHF2, CH2F, CH2CF3, C(O)OH, C(O)OCH3, CN, необязательно замещенную связанную мостиковой связью (C5-C12)циклоалкильную группу, необязательно замещенный связанный мостиковой связью (C2-C10)гетероциклильную группу, необязательно замещенный (C1-C8)алкил, необязательно замещенный (C3-C10)циклоалкил, необязательно замещенный (C3-C8)циклоалкенил, необязательно замещенный (C6-C10)арил, необязательно замещенный (C1-C10)гетероарил, необязательно замещенный (C2-C10)гетероциклил или -J-L-M-Q;
где:
J представляет собой связь, -C(O)-, необязательно замещенный (C1-C6)алкилен, необязательно замещенный (C2-C6)алкенилен, необязательно замещенный (C2-C6)алкинилен, необязательно замещенный (C3-C12)циклоалкилен, необязательно замещенный (C2-C6)гетероциклилен, -C(O)N(Ra)-Re-, -N(Ra)C(O)-Re-, -O-Re-, -N(Ra)-Re-, -S-Re-, -S(O)2-Re-, -S(O)Re-, -C(O-Ra)(Rb)-Re-, -S(O)2N(Ra)-Re-, -N(Ra)S(O)2-Re- или -N(Ra)C(O)N(Rb)-Re-;
L представляет собой связь, необязательно замещенный (C1-C8)алкилен, необязательно замещенный связанный мостиковой связью (C5-C12)циклоалкилен, необязательно замещенный (C3-C10)циклоалкилен, необязательно замещенный связанный мостиковой связью (C5-C10)циклоалкенилен, необязательно замещенный (C3-C10)циклоалкенилен, необязательно замещенный (C6-C10)арилен, необязательно замещенный (C1-C10)гетероарилен, необязательно замещенный связанный мостиковой связью (C2-C10)гетероциклилен или необязательно замещенный (C2-C10)гетероциклилен;
M представляет собой связь, -Re-, -Re-C(O)-Re-, -Re-C(O)C(O)-Re-, -Re-C(O)O-Re-, -Re-OC(O)-Re, -Re-C(O)C(O)N(Ra)-Re-, -Re-N(Ra)-C(O)C(O)-Re-, -Re-O-Re-, -Re-S(O)2-Re-, -Re-S(O)-Re-, -Re-S-Re-, -Re-N(Ra)-Re-, -Re-N(Ra)C(O)-Re-, -Re-C(O)N(Ra)Re-, -Re-OC(O)N(Ra)-Re-, -Re-N(Ra)C(O)ORe-, -Re-N(Ra)C(O)N(Rb)-Re-, -Re-N(Ra)S(O)2-Re-, или -Re-S(O)2N(Ra)-Re-; или
М представляет собой
где во всех случаях, M связан либо с атомом углерода либо с атомом азота в L;
Q представляет собой водород, дейтерий, -N(Ra)(Rb), галоген, -ORa, -SRa, -S(O)Ra, -S(O)2Ra, -NO2, -C(O)ORa, -CN, -C(O)N(Ra)(Rb), -N(Ra)C(O)Rb, -N(Ra)C(O)ORb, -N(Ra)C(O)N(Rb)2, -C(O-Ra)(Rb)2, -C(O)Ra, -CF3, -OCF3, -N(Ra)S(O)2Rb, -S(O)2N(Ra)(Rb), -S(O)2N(Ra)C(O)Rb, необязательно замещенный (C1-C6)алкил, необязательно замещенный (C2-C6)алкенил, необязательно замещенный (C2-C6)алкинил, необязательно замещенный (C3-C10)циклоалкил, необязательно замещенный (C1-C10)гетероарил, необязательно замещенный (C1-C10) гетероциклил, необязательно замещенный (C6-C10)арил;
где в фрагменте, содержащем -N(Ra)(Rb), азот, Ra и Rb могут образовывать кольцо, таким образом, -N(Ra)(Rb) представляет собой необязательно замещенный (C2-C10)гетероциклил или необязательно замещенный (C1-C10) гетероарил, связанный через азот;
Ra и Rb, каждый независимо, представляют собой водород, дейтерий, CN, необязательно замещенный (C1-C10)алкил, необязательно замещенный (C2-C10)алкенил, необязательно замещенный (C2-C10)алкинил, необязательно замещенный (C1-C10)алкил-O-(C1-C10)алкил, необязательно замещенный (C3-C10)циклоалкил, необязательно замещенный (C6-C10)арил, необязательно замещенный (C1-C10)гетероарил, необязательно замещенный (C1-C10)гетероциклил, необязательно замещенный -(C1-C6)алкилен-(C3-C10)циклоалкил, необязательно замещенный -(C1-C6)алкилен-(C6-C10)арил, необязательно замещенный -(C1-C6)алкилен-(C1-C10)гетероарил или необязательно замещенный -(C1-C6)алкилен-(C1-C10)гетероциклил; и
Re для каждого случая независимо представляет собой связь, необязательно замещенный (C1-C10)алкилен, необязательно замещенный (C2-C10)алкенилен, необязательно замещенный (C2-C10)алкинилен, необязательно замещенную -(C1-C10)алкилен-O-(C1-C10)алкиленовую группу, необязательно замещенный (C3-C10)циклоалкилен, необязательно замещенный (C6-C10)арилен, необязательно замещенный (C1-C10)гетероарилен или необязательно замещенный (C1-C10)гетероциклилен;
при условии, что когда T представляет собой N, U представляет собой CR4, X представляет собой NR3 и Y представляет собой C, R4 является отличным от OH;
при условии, что когда T представляет собой N, U представляет собой CR4, X представляет собой NR3 и Y представляет собой C, R1 представляет собой H;
при условии, что когда соединение представляет собой
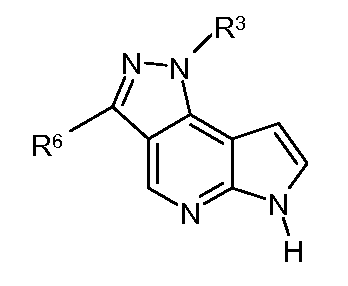
R3 имеет значение, определенное выше, и R6 не связан с пиразольным кольцом посредством атома азота или кислорода; и
при условии, что когда соединение представляет собой
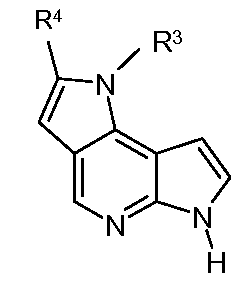
когда R3 представляет собой H, CH3 или -C(O)OH, тогда R4 является отличным от H, -C(O)OCH2CH3, -C(O)NH-необязательно замещенного фенила, -NHC(O)-необязательно замещенного фенила или -S(O)2-фенила.
Во втором варианте воплощения настоящее изобретение обеспечивает соединение в соответствии с первым вариантом воплощения, где R3 представляет собой -A-D-E-G, и A представляет собой связь, необязательно замещенный (C1-C6)алкилен, необязательно замещенный (C3-C12)циклоалкилен или необязательно замещенный (C2-C6)гетероциклилен.
В третьем варианте воплощения настоящее изобретение обеспечивает соединение в соответствии с любым из представленных выше вариантов воплощения, где R3 представляет собой -A-D-E-G, и D представляет собой необязательно замещенный (C1-C8)алкилен, необязательно замещенный (C3-C10)циклоалкилен, необязательно замещенный связанный мостиковой связью (C5-C10)циклоалкенилен, необязательно замещенный (C3-C10)связанный мостиковой связью гетероциклилен или необязательно замещенный (C2-C10)гетероциклилен.
В четвертом варианте воплощения настоящее изобретение обеспечивает соединение в соответствии с любым из представленных выше вариантов воплощения 3, где D представляет собой необязательно замещенный (C1-C6)алкилен, необязательно замещенный (C3-C6)циклоалкилен, необязательно замещенный бицикло[2.2.2]октан-1-ил, необязательно замещенный 2,5-диазабицикло[2.2.1]гептан, необязательно замещенный 2,6-диазабицикло[3.2.1]октан, необязательно замещенный октагидропирроло[3,4-c]пиррол, необязательно замещенный октагидропирроло[3,2-b]пиридин, необязательно замещенный 1,4-диазепан, необязательно замещенный кубан, необязательно замещенный 1,4-диоксан-спиро[4.4]нонан, необязательно замещенный 2,5-диазаспиро[3.5]нонан, необязательно замещенный пиперидин, необязательно замещенный пиперазин, необязательно замещенный пирролидин, необязательно замещенный тетрагидрофуран или необязательно замещенный тетрагидропиран.
В пятом варианте воплощения настоящее изобретение обеспечивает соединение в соответствии с любым из представленных выше вариантов воплощения, где R3 представляет собой -A-D-E-G, и E представляет собой связь, -Re-, -Re-C(O)-Re-, -Re-O-Re-, -Re-S(O)2-Re-, -Re-N(Ra)-Re-, =N-Re-, -Re-N(Ra)C(O)-Re-, -Re-N(Ra)C(O)O-Re-, -Re-N(Ra)C(O)N(Rb)-Re-, -ReC(O)N(Ra)Re-, -Re-N(Ra)S(O)2-Re-, -Re-S(O)2N(Ra)-Re-, -Re-N(Ra)S(O)2N(Ra)-Re-, -Re-OC(O)N(Ra)-Re, -Re-C(O)O-Re, -Re-OC(O)-Re; или 
где
Ra для каждого случая независимо представляет собой водород, CN, необязательно замещенный (C1-C10)алкил или необязательно замещенный -(C1-C6)алкилен-(C3-C10)циклоалкил; и
Re для каждого случая независимо представляет собой связь, необязательно замещенный (C1-C10)алкилен, необязательно замещенный (C3-C10)циклоалкилен, необязательно замещенный (C6-C10)арилен, необязательно замещенный (C1-C10)гетероарилен или необязательно замещенный (C1-C10)гетероциклилен.
В шестом варианте воплощения настоящее изобретение обеспечивает соединение в соответствии с любым из представленных выше вариантов воплощения, где R3 представляет собой -A-D-E-G, и G представляет собой водород, дейтерий, -N(Ra)(Rb), галоген, -ORa, -S(O)2Ra, -CN, -C(O)N(Ra)(Rb), -N(Ra)C(O)Rb, -CF3, -S(O)2N(Ra)(Rb), необязательно замещенный -(C1-C6)алкил, необязательно замещенный -(C3-C10)циклоалкил, необязательно замещенный -(C1-C10)гетероарил, необязательно замещенный -(C1-C10) гетероциклил или необязательно замещенный -(C6-C10)арил;
где в фрагменте, содержащем -N(Ra)(Rb), азот, Ra и Rb могут образовывать кольцо, таким образом, -N(Ra)(Rb) представляет собой необязательно замещенный (C2-C10)гетероциклил или необязательно замещенный (C1-C10) гетероарил, связанный через азот;
Ra представляет собой независимо водород, CN, необязательно замещенный (C1-C10)алкил, необязательно замещенный (C3-C10)циклоалкил или необязательно замещенный (C6-C10)арил.
В седьмом варианте воплощения настоящее изобретение обеспечивает соединение в соответствии с любым из представленных выше вариантов воплощения, где G представляет собой водород, дейтерий, -N(Ra)(Rb), галоген, -ORa, -S(O)2Ra, -CN, -C(O)N(Ra)(Rb), -N(Ra)C(O)Rb, -CF3, -S(O)2N(Ra)(Rb), необязательно замещенный -(C1-C4)алкил, необязательно замещенный -(C3-C6)циклоалкил, необязательно замещенный азепанил, необязательно замещенный азетидинил, необязательно замещенный бензо[d]изоксазолил, необязательно замещенный 4,5-дигидроизоксазолил, необязательно замещенный изотиазолидинил, необязательно замещенный изотиазолил, необязательно замещенный изоксазолил, необязательно замещенный морфолинил, необязательно замещенный оксадиазолил, необязательно замещенный оксазолил, необязательно замещенный оксетанил, необязательно замещенный фенил, необязательно замещенный пиперазинил, необязательно замещенный пиперидинил, необязательно замещенный пиразинил, необязательно замещенный пиразолил, необязательно замещенный пиридазинил, необязательно замещенный пиридинил, необязательно замещенный пиримидинил, необязательно замещенный пирролидинил, необязательно замещенный пирролил, необязательно замещенный тетрагидрофуранил, необязательно замещенный тетрагидропиранил, необязательно замещенный тетрагидротиопиранил, необязательно замещенный тиенил, необязательно замещенный тиоморфолинил, необязательно замещенный 1,1-диоксо-тиоморфолинил, необязательно замещенный тиазолил или необязательно замещенный триазолил.
В восьмом варианте воплощения настоящее изобретение обеспечивает соединение в соответствии с любым из представленных выше вариантов воплощения, где R3 представляет собой водород, необязательно замещенный (C1-C8)алкил, необязательно замещенный (C3-C10)циклоалкил или необязательно замещенный (C2-C10)гетероциклил.
В девятом варианте воплощения настоящее изобретение обеспечивает соединение в соответствии с любым из представленных выше вариантов воплощения, где R6 представляет собой -J-L-M-Q, и J представляет собой связь, необязательно замещенный (C1-C6)алкилен или необязательно замещенный (C2-C6)алкенилен.
В десятом варианте воплощения настоящее изобретение обеспечивает соединение в соответствии с любым из представленных выше вариантов воплощения, где R6 представляет собой -J-L-M-Q, и L представляет собой связь или необязательно замещенный (C1-C8)алкилен.
В одиннадцатом варианте воплощения настоящее изобретение обеспечивает соединение в соответствии с любым из представленных выше вариантов воплощения, где R6 представляет собой -J-L-M-Q, и M представляет собой связь, -Re-, -Re-C(O)-Re-, -Re-O-Re-, -Re-S(O)2-Re-, -Re-S(O)-Re-, -Re-S-Re-, -Re-N(Ra)-Re-, -Re-N(Ra)C(O)-Re-, -Re-C(O)N(Ra)Re-, -Re-N(Ra)C(O)N(Rb)-Re-, -Re-N(Ra)S(O)2-Re-, или -Re-S(O)2N(Ra)-Re-; где во всех случаях, M связан либо с атомом углерода либо с атомом азота в L.
В двенадцатом варианте воплощения настоящее изобретение обеспечивает соединение в соответствии с любым из представленных выше вариантов воплощения, где R6 представляет собой -J-L-M-Q, и Q представляет собой водород, дейтерий, -N(Ra)(Rb), галоген, -ORa, -SRa, -S(O)Ra, -S(O)2Ra, -NO2, -C(O)ORa, -CN, -C(O)N(Ra)(Rb), -N(Ra)C(O)Rb, -N(Ra)C(O)ORb, -N(Ra)C(O)N(Rb)2, -C(O-Ra)(Rb)2, -C(O)Ra, -CF3, -OCF3, -N(Ra)S(O)2Rb, -S(O)2N(Ra)(Rb), -S(O)2N(Ra)C(O)Rb, необязательно замещенный (C1-C6)алкил, необязательно замещенный (C3-C10)циклоалкил, необязательно замещенный (C1-C10)гетероарил, необязательно замещенный (C1-C10) гетероциклил, необязательно замещенный (C6-C10)арил;
где в фрагменте, содержащем -N(Ra)(Rb), азот, Ra и Rb могут образовывать кольцо, таким образом, -N(Ra)(Rb) представляет собой необязательно замещенный (C2-C10)гетероциклил или необязательно замещенный (C1-C10) гетероарил, связанный через азот;
Ra и Rb, каждый независимо, представляют собой водород, дейтерий, необязательно замещенный (C1-C6)алкил, необязательно замещенный (C2-C10)алкенил, необязательно замещенный (C3-C6)циклоалкил, необязательно замещенный (C6-C10)арил, необязательно замещенный (C1-C10)гетероарил или необязательно замещенный (C1-C10)гетероциклил.
В тринадцатом варианте воплощения настоящее изобретение обеспечивает соединение в соответствии с любым из представленных выше вариантов воплощения, где T представляет собой N, U представляет собой N, X представляет собой CR3, и Y представляет собой N с образованием соединения формулы (Ia)

Формула (Ia)
В четырнадцатом варианте воплощения настоящее изобретение обеспечивает соединение в соответствии с тринадцатым вариантом воплощения, где соединение представляет собой
N-(1-((6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)метил)циклобутил)циклопропансульфонамид;
N-(1-((6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)метил)циклобутил)-2-цианоацетамид;
(S)-1-((1-(циклопропилсульфонил)пирролидин-3-ил)метил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин;
N-((1S,3R,4R)-4-этил-3-фтор-3-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)циклопропансульфонамид;
N-((1R,3S,4S)-4-этил-3-фтор-3-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)циклопропансульфонамид;
N-((1R,3R,4S)-4-этил-3-фтор-3-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)циклопропансульфонамид;
N-((1S,3S,4R)-4-этил-3-фтор-3-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)циклопропансульфонамид;
(1S,3R)-1-[3-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-изотиазолидин-2-ил-1,1-диоксид]циклопентан;
N-((1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)-N-метилциклопропансульфонамид;
1-((1S,2R,4S)-2-этил-4-(4-метоксибензилокси)циклопентил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин;
(S)-5-(3-((6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)метил)пирролидин-1-ил)пиразин-2-карбонитрил;
N-(циклопропилметил)-N-((1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)циклопропансульфонамид;
N-((1S,3R)-3-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)-2-(4-цианофенил)ацетамид;
N-((1S,3R)-3-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)циклопропанкарбоксамид;
N-((1S,3R)-3-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)-2-циклопропилацетамид;
N-((1S,3R)-3-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)-4-цианобензамид;
N,N-диэтил-1-(3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)метансульфонамид;
1-((1S,2S,4R)-4-((азетидин-1-илсульфонил)метил)-2-этилциклопентил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин;
1-((1R,2R,4S)-4-((азетидин-1-илсульфонил)метил)-2-этилциклопентил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин;
1-((1R,2S,4R)-4-((азетидин-1-илсульфонил)метил)-2-этилциклопентил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин;
1-((1S,2R,4S)-4-((азетидин-1-илсульфонил)метил)-2-этилциклопентил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин;
N-((1S,3R,4S)-3-этил-4-(7-метил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)циклопропансульфонамид;
N-((1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)-N-(2-гидроксиэтил)циклопропансульфонамид;
5-((1R,3R)-3-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентиламино)пиразин-2-карбонитрил
N-((1R,3R)-3-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)-4-метиланилин;
1-((1R,3S)-3-(1H-пиррол-1-ил)циклопентил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин;
1-((1S,3R)-3-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)-1H-пиррол-3-карбонитрил;
N-((1R,3R)-3-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)анилин;
N-((1-((6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)метил)циклобутил)метил)-2-цианоацетамид;
N-((1R,3R)-3-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)-4-фторанилин;
N-((1R,3R)-3-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)-4-хлоранилин;
N-((1R,3R)-3-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)-3,4-дихлоранилин;
N-((1R,3R)-3-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)-4-метоксианилин;
N-((1R,3R)-3-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)-4-метокси-N-(4-метоксифенил)анилин;
3-((3R,4R)-4-метил-3-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)пиперидин-1-ил)-3-оксопропаннитрил;
1-метил-N-((1S,3R,4S)-3-метил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)-1H-пиразол-4-сульфонамид;
3-((1R,3R)-3-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентиламино)бензонитрил;
N-((1S,3R,4S)-3-метил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)анилин;
4-((1S,2R,4S)-4-(бензилокси)-2-этилциклопентил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин;
4-((1R,2S,4R)-4-(бензилокси)-2-этилциклопентил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин;
5-метил-N-((1S,3R,4S)-3-метил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)изоксазол-4-сульфонамид;
N-(4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)бицикло[2.2.2]октан-1-ил)циклобутансульфонамид;
6-((1R,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентилокси)никотинонитрил;
N-(4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)бицикло[2.2.2]октан-1-ил)пирролидин-1-карбоксамид;
4-((1R,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентилокси)бензонитрил;
4-((1S,3S,4R)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентилокси)бензонитрил;
4-метил-N-((1S,3R,4S)-3-метил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)анилин;
4-хлор-N-((1S,3R,4S)-3-метил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)анилин;
3-((1S,3R,4S)-3-метил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентиламино)бензонитрил;
4-фтор-N-((1S,3R,4S)-3-метил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)анилин;
N-((1S,3S,4R)-3-метил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)анилин;
N-((1R,3R,4S)-3-метил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)анилин;
5-((1S,3R,4S)-3-метил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентиламино)пиразин-2-карбонитрил;
6-((1S,3R,4S)-3-метил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентиламино)никотинонитрил;
6-((1R,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентилокси)никотинонитрил;
6-((1S,3S,4R)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентилокси)никотинонитрил;
1-((1S,2S,4R)-2-этил-4-(4-метоксибензилокси)циклопентил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин;
1-((1R,2R,4S)-2-этил-4-(4-метоксибензилокси)циклопентил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин;
N-((1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)-3,3,3-трифторпропан-1-сульфонамид;
5-((1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентиламино)пиразин-2-карбонитрил;
6-((1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентиламино)никотинонитрил;
2-((1S,3R,4S)-3-метил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентиламино)тиазол-5-карбонитрил;
N-((1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)азетидин-1-сульфонамид;
N-((1R,3R,4R)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)циклопропансульфонамид;
N-((1S,3S,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)циклопропансульфонамид;
3-циано-N-((1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)азетидин-1-сульфонамид;
N-((1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)-3,3-дифторазетидин-1-сульфонамид;
5-((1R,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентилокси)пиразин-2-карбонитрил;
5-((1S,3S,4R)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентилокси)пиразин-2-карбонитрил;
6-((1S,3S,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентилокси)никотинонитрил;
6-((1R,3R,4R)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентилокси)никотинонитрил;
2-((1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентиламино)тиазол-5-карбонитрил;
5-((1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентилокси)пиразин-2-карбонитрил;
5-((1R,3S,4R)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентилокси)пиразин-2-карбонитрил;
N-(4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)бицикло[2.2.2]октан-1-ил)пирролидин-1-сульфонамид;
5-(((1S,3R)-3-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)метиламино)пиразин-2-карбонитрил;
(S)-N-((1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)-2-(трифторметил)пирролидин-1-сульфонамид;
N-((1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)-3,3-дифторпирролидин-1-сульфонамид;
N-((1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)-4,4-дифторпиперидин-1-сульфонамид;
N-((1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)-1-метилциклопропан-1-сульфонамид;
N-(4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)бицикло[2.2.2]октан-1-ил)-1-метилциклопропан-1-сульфонамид;
N-(4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)бицикло[2.2.2]октан-1-ил)азетидин-1-сульфонамид;
6-((1S,3R,4S)-3-метил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентиламино)никотинонитрил;
N-((1S,3R,4R)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)циклопентансульфонамид;
5-(((1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)метокси)пиразин-2-карбоксамид;
((1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)метанол;
((1R,3S,4R)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)метанол;
5-(((1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)метокси)пиразин-2-карбонитрил;
5-(((1R,3S,4R)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)метокси)пиразин-2-карбонитрил;
N-(4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)бицикло[2.2.2]октан-1-ил)-3,3-дифторазетидин-1-сульфонамид;
N-(3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)анилин;
1-((1S,2R,4R)-2-этил-4-(5-(трифторметил)пиридин-2-илокси)циклопентил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин;
1-((1R,2S,4S)-2-этил-4-(5-(трифторметил)пиридин-2-илокси)циклопентил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин;
5-((1R,3S,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентилокси)пиразин-2-карбонитрил;
5-((1S,3R,4R)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентилокси)пиразин-2-карбонитрил;
N-((1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)-2,2,2-трифторэтансульфонамид;
N-((1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)-4-метилпиперазин-1-сульфонамид;
4-((1S,3S,4R)-3-метил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентилокси)бензонитрил;
4-((1R,3R,4S)-3-метил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентилокси)бензонитрил;
3-(((1R,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентилокси)метил)бензонитрил;
3-(((1S,3S,4R)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентилокси)метил)бензонитрил;
4-(((1R,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентилокси)метил)бензонитрил;
4-(((1S,3S,4R)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентилокси)метил)бензонитрил;
1-этил-N-((1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)циклопропан-1-сульфонамид;
N-(((1R,3S,4R)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)метил)циклопропансульфонамид;
N-(((1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)метил)циклопропансульфонамид;
4-((1R,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентилокси)-2-фторбензонитрил;
4-((1R,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентилокси)-3-фторбензонитрил;
3-((1R,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентилокси)бензонитрил;
N-((1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)-2-морфолиноэтансульфонамид;
1-бутил-N-((1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)циклопропан-1-сульфонамид;
2-(3,3-дифторпирролидин-1-ил)-N-((1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)этансульфонамид;
2-(((1S,3R)-3-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)метиламино)изоникотинонитрил;
N-((1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)-2-метилпропан-2-сульфонамид;
N-((1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)-2-(1H-1,2,4-триазол-1-ил)этансульфонамид;
2-(4,4-дифторпиперидин-1-ил)-N-((1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)этансульфонамид;
N-((1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)-2-(2H-1,2,3-триазол-2-ил)этансульфонамид;
N-((1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)-2-(1H-1,2,3-триазол-1-ил)этансульфонамид;
(1R,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентанол;
1-((1S,2R,4R)-2-этил-4-(3,3,3-трифторпропокси)циклопентил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин;
N-((1S,3R,4S)-3-этил-4-(8-иод-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)циклопропансульфонамид;
(1S,3R,4S)-N-(2-(3,3-дифторпирролидин-1-илсульфонил)этил)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентанамин;
N-циано-N-((1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)циклопропансульфонамид;
N-((1S,3R,4S)-3-этил-4-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)циклопентил)циклопропансульфонамид;
N-((1S,3R,4S)-3-этил-4-(8-метил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)-N-(гидроксиметил)циклопропансульфонамид;
N-((1S,3S,4R)-3-(8-циано-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-4-этилциклопентил)циклопропансульфонамид;
1-((1S,2R,4S)-4-(циклопропилметокси)-2-этилциклопентил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин;
1-((1S,2R,4S)-4-(циклопропилметокси)-2-метилциклопентил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин;
1-((1S,2R,4S)-2-этил-4-(2,2,2-трифторэтилсульфонил)циклопентил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин;
1-((1S,2R,4S)-2-этил-4-(тетрагидро-2H-пиран-4-илокси)циклопентил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин;
1-((1S,2R,4S)-2-этил-4-((тетрагидро-2H-пиран-4-ил)метокси)циклопентил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин;
1-((1R,2R,4S)-2-этил-4-((тетрагидро-2H-пиран-4-ил)метокси)циклопентил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин;
1-((1S,2R,4R)-4-(циклопропилметокси)-2-этилциклопентил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин;
1-((1S,2R,4R)-2-этил-4-(тетрагидро-2H-пиран-4-илокси)циклопентил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин;
2-(4-циано-1H-пиразол-1-ил)-N-((1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)этансульфонамид;
1-((1S,2R,4S)-2-этил-4-(2-(тетрагидро-2H-пиран-4-ил)этокси)циклопентил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин;
1-((1R,2R,4S)-2-этил-4-(2-(тетрагидро-2H-пиран-4-ил)этокси)циклопентил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин;
1-((1S,2R,4R)-2-этил-4-((тетрагидро-2H-пиран-4-ил)метокси)циклопентил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин;
1-((1S,2R,4S)-2-этил-4-(2-метоксиэтокси)циклопентил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин;
1-((1R,2R,4S)-2-этил-4-(2-метоксиэтокси)циклопентил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин;
1-((1S,2R,4R)-2-этил-4-изопропоксициклопентил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин;
N-((3R,5R)-1-этил-5-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)пирролидин-3-ил)циклопропансульфонамид;
(3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентанон;
1-((7S,8R)-8-этил-1,4-диоксаспиро[4.4]нонан-7-ил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин;
(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-N-(2,2,2-трифторэтил)циклопентанамин;
(3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентанон O-циклопропилметил оксим;
(3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентанон O-2-(метилсульфонил)этил оксим;
(3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентанон O-циклобутилметил оксим;
1-((1S,2R,4R)-4-(4,4-диметилциклогексилокси)-2-этилциклопентил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин;
N-((1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)-2-метоксиэтансульфонамид;
N-((3R,5R)-1-ацетил-5-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)пирролидин-3-ил)циклопропансульфонамид;
1-((3S,4R)-1-(циклопропилметилсульфонил)-4-этилпирролидин-3-ил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин;
2-((1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)уксусная кислота;
N-циклопропил-2-((1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)ацетамид;
3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентанон O-тетрагидро-2H-пиран-4-ил оксим;
1-((1S,2R,4S)-4-(3,3-дифторазетидин-1-ил)-2-этилциклопентил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин;
1-((1S,2R,4S)-4-(3,3-дифторпирролидин-1-ил)-2-этилциклопентил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин;
(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентиловый эфир диметилкарбаминовой кислоты;
{3-[(1S,3R,4S)-3-Этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентиламино]-оксетан-3-ил}-ацетонитрил;
цианометил-[(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентил]-амид циклопропансульфоновой кислоты;
1-[(3R,4S)-4-Этил-1-(2-морфолин-4-ил-этил)-пирролидин-3-ил]-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил;
[(3R,5R)-1-(2,2-дифтор-этил)-5-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-пирролидин-3-ил]-амид циклопропансульфоновой кислоты;
1-[(3R,4S)-4-Этил-1-(3,3,3-трифтор-пропан-1-сульфонил)-пирролидин-3-ил]-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил;
3-[(1R,3R,4S)-3-Этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентилокси]-пропионитрил;
1-[(3R,4S)-4-Этил-1-(3,3,3-трифтор-пропил)-пирролидин-3-ил]-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил;
2-Циклопропил-1-[(3S,4R)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-пирролидин-1-ил]-этанон;
1-[(3S,4R)-3-Этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-пирролидин-1-ил]-2-(тетрагидро-пиран-4-ил)-этанон;
циклопропилметиламид (3S,4R)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-пирролидин-1-карбоновой кислоты;
[(3R,5R)-1-этил-5-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-пирролидин-3-ил]-метиламид циклопропансульфоновой кислоты;
(3S,4R)-3-Этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-пирролидин-1-карбоновой кислоты (тетрагидро-пиран-4-илметил)-амид;
[(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентил]-амид 3,3-дифтор-циклобутансульфоновой кислоты;
[(1S,4S)-3,3-диметил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентил]-амид циклопропансульфоновой кислоты;
[(1R,4R)-3,3-диметил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентил]-амид циклопропансульфоновой кислоты;
1-[(1S,2R,4R)-4-(4,4-Дифтор-циклогексилокси)-2-этил-циклопентил]-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил;
1-[(1R,2R,4R)-4-(4,4-Дифтор-циклогексилокси)-2-этил-циклопентил]-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил;
6-[(3S,4R)-3-Этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-пирролидин-1-ил]-никотинонитрил;
1-[(3R,4S)-1-(3,3-Дифтор-циклобутансульфонил)-4-этил-пирролидин-3-ил]-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил;
[(1S,3R,4S)-3-Этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентил]-бис-(4,4,4-трифтор-бутил)-амин;
1-[(1S,2R,4R)-2-Этил-4-(4-трифторметил-циклогексилокси)-циклопентил]-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил;
4-[(3S,4R)-3-Этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-пирролидин-1-илметил]-бензонитрил;
3-[(3S,4R)-3-Этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-пирролидин-1-ил]-3-оксо-пропионитрил;
1-[(1S,2R,4R)-2-Этил-4-(4-трифторметил-циклогексилокси)-циклопентил]-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил;
1-[(1R,2R,4R)-2-Этил-4-(4-трифторметил-циклогексилокси)-циклопентил]-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил;
1-[(1R,2R,4R)-2-Этил-4-(4-трифторметил-циклогексилокси)-циклопентил]-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил;
{3-[(3S,4R)-3-Этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-пирролидин-1-ил]-оксетан-3-ил}-ацетонитрил;
3-[(1S,3R,4R)-3-Этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентилокси]-пропионитрил;
3-[(1S,3R,4S)-3-Этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентилокси]-пропионитрил;
(2-циано-этил)-[(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентил]-амид циклопропансульфоновой кислоты;
4-[(1R,3R,4S)-3-Этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентилокси]-циклогексанкарбонитрил;
4-[(1R,3R,4S)-3-Этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентилокси]-циклогексанкарбонитрил;
1-((3R,4S)-1-Циклопропансульфонил-4-этил-пирролидин-3-ил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил;
N-[(1S,3R,4S)-3-Этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентил]-N-(4,4,4-трифтор-бутил)-ацетамид;
(1R,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентиловый эфир циклопропил-карбаминовой кислоты;
(1R,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентиловый эфир 3,3-дифтор-азетидин-1-карбоновой кислоты;
(1R,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентиловый эфир цианометил-карбаминовой кислоты;
N-[(1S,3R,4S)-3-Этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентил]-N-(тетрагидро-пиран-4-илметил)-ацетамид;
3-[(1S,3R,4S)-3-Этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентил]-1,1-диметил-мочевина;
(1R,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентиловый эфир диметил-карбаминовой кислоты;
(1S,3R,4S)-3-Этил-1-(морфолин-4-сульфонилметил)-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентанол;
(1R,3R,4R)-3-Этил-1-(морфолин-4-сульфонилметил)-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентанол;
(1S,3R,4R)-3-Этил-1-(морфолин-4-сульфонилметил)-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентанол;
N-Циклопропилметил-N-[(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентил]-ацетамид;
1-[(1S,3R,4R)-3-Этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентил]-2-метил-пропан-2-ол;
1-[(1R,3R,4S)-3-Этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентил]-2-метил-пропан-2-ол;
1-[(1S,3R,4S)-3-Этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентил]-2-метил-пропан-2-ол;
1-[(1R,2R,4S)-4-(3-Циклопропил-[1,2,4]оксадиазол-5-илметил)-2-этил-циклопентил]-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил;
1-[(1S,2R,4S)-4-(3-Циклопропил-[1,2,4]оксадиазол-5-илметил)-2-этил-циклопентил]-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил;
1-[(1S,2R,4R)-4-(3-Циклопропил-[1,2,4]оксадиазол-5-илметил)-2-этил-циклопентил]-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил;
1-[(1S,2R,4R)-2-Этил-4-(5-метил-изоксазол-3-илметокси)-циклопентил]-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил;
(1R,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентиловый эфир оксетан-3-ил-карбаминовой кислоты;
(1R,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентиловый эфир циклобутил-карбаминовой кислоты;
[(1R,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентил]-амид циклопропансульфоновой кислоты;
{3-[(1R,3R,4S)-3-Этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентиламино]-оксетан-3-ил}-ацетонитрил;
изопропиловый эфир [(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентил]-карбаминовой кислоты;
[(1S,3R,4S)-3-Этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентил]-оксетан-3-иламин;
1-((3R,4S)-1-Бензил-4-изопропил-пирролидин-3-ил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил;
[(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентил]-амид 3-фтор-пропан-1-сульфоновой кислоты;
[(1S,3R,4S)-3-Этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентил]-(3-метил-оксетан-3-ил)-амин;
1-[(1S,2R,4R)-2-Этил-4-(2-морфолин-4-ил-этокси)-циклопентил]-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил;
(1R,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентиловый эфир карбамоилметил-карбаминовой кислоты;
(1R,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентиловый эфир 4-гидрокси-пиперидин-1-карбоновой кислоты;
(1R,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентиловый эфир (2,2,2-трифторэтил)-карбаминовой кислоты;
Циклопропилметил-[(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентил]-оксетан-3-иламин;
Пентан-2-сульфоновой кислоты [(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентил]-амид;
[(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентил]-амид 3-фенил-пропан-1-сульфоновой кислоты;
[(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентил]-амид 4,4,4-трифтор-бутан-1-сульфоновой кислоты;
[(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентил]-амид 2-этил-циклопропансульфоновой кислоты;
[(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентил]-амид 2-метил-пропан-1-сульфоновой кислоты;
[(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентил]-амид 2-фенил-этансульфоновой кислоты;
C-Циклогексил-N-[(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентил]-метансульфонамид;
[(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентил]-амид бутан-1-сульфоновой кислоты;
[(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентил]-амид пропан-2-сульфоновой кислоты;
N-[(1S,3R,4S)-3-Этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентил]-C-фенил-метансульфонамид;
[(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентил]-амид пропан-1-сульфоновой кислоты;
[(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентил]-амид 3-метил-бутан-1-сульфоновой кислоты;
N-[(1S,3R,4S)-3-Этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентил]-C,C-дифтор-метансульфонамид;
[(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентил]-амид 4-циано-бутан-1-сульфоновой кислоты;
[(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентил]-амид 2-этокси-этансульфоновой кислоты;
N-[(1S,3R,4S)-3-Этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентил]-C-(тетрагидро-фуран-2-ил)-метансульфонамид;
[(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентил]-амид тетрагидро-пиран-4-сульфоновой кислоты;
[(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентил]-амид 3-циано-пропан-1-сульфоновой кислоты;
N-[(1S,3R,4S)-3-Этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентил]-C-(5-метил-изоксазол-3-ил)-метансульфонамид;
N-[(1S,3R,4S)-3-Этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентил]-C-(тетрагидро-пиран-2-ил)-метансульфонамид;
[(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентил]-амид 2-пиридин-2-ил-этансульфоновой кислоты;
C-(2,2-Дихлор-циклопропил)-N-[(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентил]-метансульфонамид;
циклобутиламид (3S,4R)-3-изопропил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-пирролидин-1-карбоновой кислоты;
(1S,3R,4S)-3-Этил-1-метил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентанол;
(1S,3R,4S)-3-этил-4-[6-(толуол-4-сульфонил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил]-циклопентиловый эфир 4-нитро-фениловый эфир угольной кислоты;
(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентиловый эфир циклобутил-карбаминовой кислоты;
(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентиловый эфир 4-гидрокси-пиперидин-1-карбоновой кислоты;
3-(Циклопропилметил-амино)-4-[(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентиламино]-циклобут-3-ен-1,2-дион;
3-[(1S,3R,4S)-3-Этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентиламино]-4-(оксетан-3-иламино)-циклобут-3-ен-1,2-дион;
3-[(1S,3R,4S)-3-Этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентиламино]-4-(3,3,3-трифтор-пропиламино)-циклобут-3-ен-1,2-дион;
[(1S,3R,4S)-3-Этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентил]-метил-оксетан-3-ил-амин;
[(1S,3R,4S)-3-Этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентил]-(3-метил-оксетан-3-илметил)-амин;
3-Циклопропиламино-4-[(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентиламино]-циклобут-3-ен-1,2-дион;
(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентиловый эфир цианометил-карбаминовой кислоты;
(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентиловый эфир циклопропил-карбаминовой кислоты;
(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентиловый эфир (2,2,2-трифтор-этил)-карбаминовой кислоты;
(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентиловый эфир3,3-дифтор-азетидин-1-карбоновой кислоты;
(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентиловый эфир 4-циано-пиперидин-1-карбоновой кислоты;
(1-циано-циклопропил)-амид (3S,4R)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-пирролидин-1-карбоновой кислоты;
(1-циано-циклопропил)-амид (3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-пирролидин-1-карбоновой кислоты;
циклобутиламид (3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-пирролидин-1-карбоновой кислоты;
циклобутиламид (3S,4R)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-пирролидин-1-карбоновой кислоты;
(3-метил-изотиазол-5-ил)-амид (3S,4R)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-пирролидин-1-карбоновой кислоты;
(3-метил-изотиазол-5-ил)-амид (3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-пирролидин-1-карбоновой кислоты;
цианометил-амид (3S,4R)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-пирролидин-1-карбоновой кислоты;
цианометил-амид (3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-пирролидин-1-карбоновой кислоты;
(2-Циклопропил-этил)-[(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентил]-оксетан-3-ил-амин;
Циклопропилметил-[(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентил]-(3-метил-оксетан-3-ил)-амин;
(оксазол-4-илметил)-амид (3S,4R)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-пирролидин-1-карбоновой кислоты;
(оксазол-4-илметил)-амид (3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-пирролидин-1-карбоновой кислоты;
(2,2,2-трифтор-этил)-амид (3S,4R)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-пирролидин-1-карбоновой кислоты;
(2,2,2-трифтор-этил)-амид (3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-пирролидин-1-карбоновой кислоты;
(2-Циклопропил-этил)-[(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентил]-(3-метил-оксетан-3-ил)-амин;
(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентиловый эфир 3-циано-азетидин-1-карбоновой кислоты;
(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентиловый эфир бензил-карбаминовой кислоты;
(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентиловый эфир оксетан-3-ил-карбаминовой кислоты;
(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентиловый эфир (1-циано-циклопропил)-карбаминовой кислоты;
(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентиловый эфир(3-метил-оксетан-3-ил)-карбаминовой кислоты;
(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентиловый эфир (R)-3-гидрокси-пирролидин-1-карбоновой кислоты;
(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентиловый эфир (S)-3-гидрокси-пирролидин-1-карбоновой кислоты;
(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентиловый эфир 4-фтор-пиперидин-1-карбоновой кислоты;
(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентиловый эфир (2,2-дифтор-этил)-карбаминовой кислоты;
(3,3-Дифтор-азетидин-1-ил)-[(3S,4R)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-пирролидин-1-ил]-метанон;
1-[(1S,2R,4R)-2-Этил-4-(пиразол-1-илокси)-циклопентил]-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил;
(3,3-Дифтор-азетидин-1-ил)-[(3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-пирролидин-1-ил]-метанон;
{2-[(3S,4R)-3-Этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-пирролидин-1-ил]-4,5-дигидро-оксазол-4-ил}-метанол;
оксетан-3-иламид (3S,4R)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-пирролидин-1-карбоновой кислоты;
оксетан-3-иламид (3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-пирролидин-1-карбоновой кислоты;
(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентиловый эфир 3-фтор-азетидин-1-карбоновой кислоты;
(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентиловый эфир (1-метил-циклобутил)-карбаминовой кислоты;
(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентиловый эфир (1-гидрокси-циклопропилметил)-карбаминовой кислоты;
(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентиловый эфир метил-оксетан-3-ил-карбаминовой кислоты;
(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентиловый эфир (3-метил-оксетан-3-илметил)-карбаминовой кислоты;
(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентиловый эфир фенил-карбаминовой кислоты;
[(3S,4R)-3-Этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-пирролидин-1-ил]-((R)-3-гидрокси-пирролидин-1-ил)-метанон;
[(3R,4S)-3-Этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-пирролидин-1-ил]-((R)-3-гидрокси-пирролидин-1-ил)-метанон;
(1R,3R,4S)-3-Этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентанкарбонитрил;
[(3S,4R)-3-Этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-пирролидин-1-ил]-((S)-3-гидрокси-пирролидин-1-ил)-метанон;
[(3R,4S)-3-Этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-пирролидин-1-ил]-((S)-3-гидрокси-пирролидин-1-ил)-метанон;
(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентиловый эфир трет-бутил-карбаминовой кислоты;
(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентиловый эфир (2,2-диметил-пропил)-карбаминовой кислоты;
(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентиловый эфир (2-метокси-этил)-карбаминовой кислоты;
(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентиловый эфир (3,5-бис-трифторметил-бензил)-карбаминовой кислоты;
(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентиловый эфир(2-диметиламино-этил)-метил-карбаминовой кислоты; соединение с трифторуксусной кислотой;
(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентиловый эфир (3-диметиламино-пропил)-метил-карбаминовой кислоты; соединение с трифторуксусной кислотой;
(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентиловый эфир бензил-изопропил-карбаминовой кислоты;
(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентиловый эфир (R)-3-гидрокси-пиперидин-1-карбоновой кислоты;
(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентиловый эфир 4-метил-пиперазин-1-карбоновой кислоты; соединение с трифторуксусной кислотой;
(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентиловый эфир 4-ацетил-пиперазин-1-карбоновой кислоты;
(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентиловый эфир 4-(2-фтор-фенил)-пиперазин-1-карбоновой кислоты; соединение с трифторуксусной кислотой;
(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентиловый эфир пиридин-2-илметил-карбаминовой кислоты; соединение с трифторуксусной кислотой;
(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентиловый эфир пиридин-3-илметил-карбаминовой кислоты; соединение с трифторуксусной кислотой;
(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентиловый эфир пиридин-4-илметил-карбаминовой кислоты; соединение с трифторуксусной кислотой;
(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентиловый эфир изобутил-карбаминовой кислоты;
(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентиловый эфир [(S)-1-(тетрагидро-фуран-2-ил)метил]-карбаминовой кислоты;
(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентиловый эфир [(R)-1-(тетрагидро-фуран-2-ил)метил]-карбаминовой кислоты;
(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентиловый эфир (2-циано-этил)-циклопропил-карбаминовой кислоты;
(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентиловый эфир диизобутил-карбаминовой кислоты;
(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентиловый эфир азетидин-1-карбоновой кислоты;
(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентиловый эфир (2-метокси-этил)-метил-карбаминовой кислоты;
(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентиловый эфир морфолин-4-карбоновой кислоты;
(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентиловый эфир тиоморфолин-4-карбоновой кислоты;
(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентиловый эфир (2-диметиламино-этил)-карбаминовой кислоты; соединение с трифторуксусной кислотой;
(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентиловый эфир (3-диметиламино-пропил)-карбаминовой кислоты; соединение с трифторуксусной кислотой;
(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентиловый эфир (2-пирролидин-1-ил-этил)- карбаминовой кислоты; соединение с трифторуксусной кислотой;
(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентиловый эфир (3-пирролидин-1-ил-пропил)-карбаминовой кислоты; соединение с трифторуксусной кислотой;
(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентиловый эфир (2-пиперидин-1-ил-этил)-карбаминовой кислоты; соединение с трифторуксусной кислотой;
(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентиловый эфир (3-пиперидин-1-ил-пропил)-карбаминовой кислоты; соединение с трифторуксусной кислотой;
(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентиловый эфир (2-морфолин-4-ил-этил)-карбаминовой кислоты; соединение с трифторуксусной кислотой;
(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентиловый эфир (3-морфолин-4-ил-пропил)-карбаминовой кислоты; соединение с трифторуксусной кислотой;
1-[(1S,2R,4S)-4-(2,2-Дифтор-этокси)-2-этил-циклопентил]-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил;
1-[(1S,3R,4S)-3-Этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентилокси]-2-метил-пропан-2-ол;
(2-Циклопропил-этил)-[(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентил]-(2,2,2-трифтор-этил)-амин;
Циклопропилметил-[(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентил]-(2,2,2-трифтор-этил)-амин;
Циклопропилметил-[(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентил]-(2,2,2-трифтор-этил)-амин;
1-((7S,8R)-8-Этил-1,4-диокса-спиро[4.4]нон-7-ил)-6-(толуол-4-сульфонил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил;
1-[(1R,3R,4R)-3-Этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентил]-2-метил-пропан-2-ол;
(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентиловый эфир уксусной кислоты;
циклопропилметил-амид (3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-пирролидин-1-сульфоновой кислоты;
циклопропилметил-амид (3S,4R)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-пирролидин-1-сульфоновой кислоты;
(2-циклопропил-этил)-амид (3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-пирролидин-1-карбоновой кислоты;
(2-циклопропил-этил)-амид (3S,4R)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-пирролидин-1-карбоновой кислоты;
оксетан-3-иламид (3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-пирролидин-1-сульфоновой кислоты;
оксетан-3-илами (3S,4R)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-пирролидин-1-сульфоновой кислоты;
[(1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентил]-оксетан-3-ил-(4,4,4-трифтор-бутил)-амин;
циклопропиламид (3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентансульфоновой кислоты;
2-[(1S,3R,4S)-3-Этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентил]-этанол;
2-[(1R,3R,4S)-3-Этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентил]-этанол;
циклобутиламид (3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-пирролидин-1-сульфоновой кислоты;
циклобутиламид (3S,4R)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-пирролидин-1-сульфоновой кислоты;
1-[(1S,2R,4S)-2-Этил-4-(3-метоксиметил-[1,2,4]оксадиазол-5-илметил)-циклопентил]-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил;
амид (3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-пирролидин-1-сульфоновой кислоты;
амид (3S,4R)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-пирролидин-1-сульфоновой кислоты;
4-[(1S,3R,4S)-3-Этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентил]-бутиронитрил;
4-[(1R,3R,4S)-3-Этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентил]-бутиронитрил;
[(1R,3R,4S)-3-Этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентил]-ацетонитрил;
[(1S,3R,4S)-3-Этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентил]-ацетонитрил;
[(1S,3R,4S)-3-Этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентил]-(5-метил-изоксазол-3-илметил)-оксетан-3-ил-амин;
{5-[(1S,3R,4S)-3-Этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентилметил]-[1,2,4]оксадиазол-3-ил}-метанол;
1-[(1S,2R,4S)-4-(3-Циклопропил-пиразол-1-ил)-2-этил-циклопентил]-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил;
1-[(1S,2R,4S)-4-(5-Циклопропил-пиразол-1-ил)-2-этил-циклопентил]-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил;
(2,2,2-трифтор-этил)-амид (3S,4R)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-пирролидин-1-сульфоновой кислоты;
(3R,4S)-3-Этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-пирролидин-1-сульфоновой кислот (2,2,2-трифтор-этил)-амид;
1-[(1S,2R,4S)-4-(3-Циклопропил-[1,2,4]триазол-1-ил)-2-этил-циклопентил]-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил;
1-[(1S,2R,4S)-4-(5-Циклопропил-[1,2,4]триазол-1-ил)-2-этил-циклопентил]-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил; или
[(1S,3R,4S)-3-Этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентил]-оксетан-3-ил-(3,3,3-трифтор-пропил)-амин.
В пятнадцатом варианте воплощения настоящее изобретение обеспечивает соединение в соответствии с вариантами воплощения с первого по двенадцатый, где T представляет собой CR6, U представляет собой N, X представляет собой CR3 и Y представляет собой N, с образованием соединения формулы (Ib)
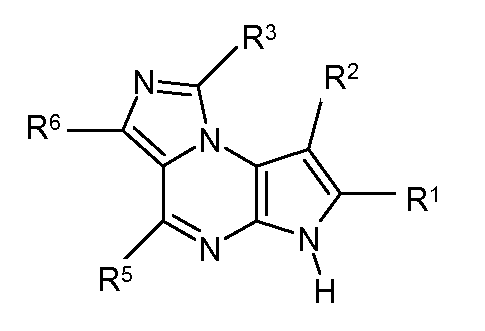
Формула (Ib)
В шестнадцатом варианте воплощения настоящее изобретение обеспечивает соединение в соответствии с пятнадцатым вариантом воплощения, где соединение представляет собой
((цис)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-ил)(2,4-дифторфенил)метанон;
((цис)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-ил)(4-(трифторметил)фенил)метанон;
((цис)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-ил)(пиридин-3-ил)метанон;
((цис)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-ил)(3-(трифторметил)фенил)метанон;
((цис)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-ил)(пиразин-2-ил)метанон;
((цис)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-ил)(пиримидин-5-ил)метанон;
1-((цис)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-ил)-2-циклопропилэтанон;
((цис)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-ил)(фенил)метанон;
1-((цис)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-ил)-2-циклобутилэтанон;
1-((цис)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-ил)-3-циклобутилпропан-1-ол;
((цис)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-ил)(1H-пиразол-4-ил)метанон;
((цис)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-ил)(1H-пиразол-3-ил)метанон;
1-((цис)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-ил)пропан-1-ол;
N-((1S,3R,4S)-3-этил-4-(3-(3-гидроксипропил)-6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)циклопентил)циклопропансульфонамид;
1-((цис)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-карбонил)циклопропанкарбонитрил;
3-((3S,4S)-4-этил-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)пиперидин-1-ил)-3-оксопропаннитрил;
3-((3R,4R)-4-этил-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)пиперидин-1-ил)-3-оксопропаннитрил;
N-((1S,3R,4S)-3-этил-4-(3-(гидроксиметил)-6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)циклопентил)циклопропансульфонамид;
N-((1S,3R,4S)-3-этил-4-(3-(2-гидроксиэтил)-6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)циклопентил)циклопропансульфонамид;
((цис)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-ил)(1-метил-1H-пиразол-4-ил)метанон;
((цис)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-ил)(пиридин-4-ил)метанон;
1-((цис)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-ил)-2-(3-метилизоксазол-5-ил)этанон;
1-((цис)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-ил)-2-(2,4-дифторфенил)этанон;
6-((цис)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-ил)пиридазин-3-карбонитрил;
5-((цис)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-ил)пиразин-2-карбонитрил;
2-((цис)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-ил)тиазол-5-карбонитрил;
6-((цис)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-ил)никотинонитрил;
((цис)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-ил)(пирролидин-1-ил)метанон;
1-((цис)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-карбонил)азетидин-3-карбонитрил;
(цис)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-N,N,4-триметилпиперидин-1-карбоксамид;
1-((цис)-1-(циклопропилсульфонил)-4-метилпиперидин-3-ил)-6H-имидазо[1,5-a]пирроло[2,3-e]пиразин;
(цис)-N-(цианометил)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-карбоксамид;
((цис)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-ил)(изоксазол-5-ил)метанон;
1-((3R,4R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-ил)-3,3,3-трифторпропан-1-он;
1-((3R,4R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-ил)-3-гидрокси-3-метилбутан-1-он;
1-((3R,4R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-ил)-2-метоксиэтанон;
1-((3R,4R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-ил)-3-метоксипропан-1-он;
1-((3R,4R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-ил)пент-4-ин-1-он;
1-((3R,4R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-ил)-2-(4-хлорфенил)этанон;
1-((3R,4R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-ил)-2-(3-хлорфенил)этанон;
4-((3R,4R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-карбонил)бензонитрил;
1-((3R,4R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-ил)-3-(3-хлоризоксазол-5-ил)пропан-1-ол;
3-(2-((3R,4R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-ил)-2-оксоэтил)бензонитрил;
4-(2-((3R,4R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-ил)-2-оксоэтил)бензонитрил;
1-((3R,4R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)- 4-метилпиперидин-1-ил)-2-(1H-пиррол-2-ил)этанон;
1-((3R,4R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-ил)-2-(пиразин-2-ил)этанон;
1-((3R,4R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-ил)-2-(тетрагидро-2H-пиран-4-ил)этанон;
1-((3R,4R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-ил)-2-(пиримидин-2-ил)этанон;
5-((1S,3S,4R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилциклопентиламино)пиразин-2-карбонитрил;
N-(4-(3-аллил-6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)бицикло[2.2.2]октан-1-ил)циклопропансульфонамид;
N-(1-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)пирролидин-3-ил)циклопропансульфонамид;
N-(4-(3-пропил-6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)бицикло[2.2.2]октан-1-ил)циклопропансульфонамид;
2-((3R,4R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-ил)тиазол-5-карбонитрил;
N-(4-(3-(2,3-дигидроксипропил)-6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)бицикло[2.2.2]октан-1-ил)циклопропансульфонамид;
1-((3R,4R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-карбонил)пирролидин-3-карбонитрил;
(3R,4R)-N-(4-(цианометил)фенил)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-карбоксамид;
((3R,4R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-ил)(морфолино)метанон;
((3R,4R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-ил)(4-метилпиперазин-1-ил)метанон;
((3R,4R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-ил)(пиперидин-1-ил)метанон;
(3R,4R)-N-(2,4-дифторфенил)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-карбоксамид;
(3R,4R)-N-(3-цианофенил)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-карбоксамид;
(R)-1-((3R,4R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-карбонил)пирролидин-2-карбонитрил;
(S)-1-((3R,4R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-карбонил)пирролидин-2-карбонитрил;
((3R,4R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-ил)((R)-2-(трифторметил)пирролидин-1-ил)метанон;
((3R,4R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-ил)((S)-2-(трифторметил)пирролидин-1-ил)метанон;
((3R,4R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-ил)(3,3-дифторазетидин-1-ил)метанон;
2-((3R,4R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-ил)бензо[d]оксазол;
N-((1S,3R,4S)-3-этил-4-(3-(2-(метилсульфонил)этил)-6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)циклопентил)циклопропансульфонамид;
((3R,4R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-ил)(азетидин-1-ил)метанон;
(3R,4R)-N-(4-цианофенил)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-карбоксамид;
((3R,4R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-ил)((R)-3-фторпирролидин-1-ил)метанон;
((3R,4R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-ил)(3,3-дифторпирролидин-1-ил)метанон;
1-((3R,4R)-4-метил-1-(пирролидин-1-илсульфонил)пиперидин-3-ил)-6H-имидазо[1,5-a]пирроло[2,3-e]пиразин;
(R)-N-(1-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)пирролидин-3-ил)циклопропансульфонамид;
(S)-N-(1-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)пирролидин-3-ил)циклопропансульфонамид;
3-((3R,4R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-(трифторметил)пиперидин-1-ил)-3-оксопропаннитрил;
3-((3S,4S)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-(трифторметил)пиперидин-1-ил)-3-оксопропаннитрил;
N-(3-((3R,4R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-ил)-3-оксопропил)ацетамид;
((3R,4R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-ил)(тетрагидрофуран-2-ил)метанон;
((3R,4R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-ил)(тетрагидрофуран-3-ил)метанон;
((3R,4R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-ил)(3-метоксициклогексил)метанон;
1-((3R,4R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-ил)-3-гидроксипропан-1-он;
1-((3R,4R)-1-бензил-4-метилпиперидин-3-ил)-6H-имидазо[1,5-a]пирроло[2,3-e]пиразин;
1-((3R,4R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)- 4-метилпиперидин-1-ил)-4,4,4-трифторбутан-1-он;
((3R,4R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-ил)(тетрагидро-2H-пиран-4-ил)метанон;
((3R,4R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-ил)(тетрагидро-2H-пиран-3-ил)метанон;
4-((3R,4R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-ил)-4-оксобутаннитрил;
((3R,4R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-ил)(тетрагидро-2H-пиран-2-ил)метанон;
((3R,4R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-ил)((R)-2-(гидроксиметил)пирролидин-1-ил)метанон;
((3R,4R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-ил)(3-метилпирролидин-1-ил)метанон;
((3R,4R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-ил)(3-фторазетидин-1-ил)метанон;
((3R,4R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-ил)((S)-3-фторпирролидин-1-ил)метанон;
((3R,4R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-ил)((R)-2-метилпирролидин-1-ил)метанон;
((3R,4R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-ил)((R)-морфолин-3-ил)метанон;
1-((3R,4R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-ил)-3-(метилсульфонил)пропан-1-он;
((3R,4R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-ил)(1,4-диоксан-2-ил)метанон;
((3R,4R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-ил)(тетрагидротиофен-3-ил-1,1-диоксид)метанон;
((3R,4R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-ил)(3,3-дифторциклобутил)метанон;
N-((1S,3R,4S)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилциклопентил)анилин;
N-((1R,3S,4R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилциклопентил)анилин;
3-бром-1-циклогексил-6H-имидазо[1,5-a]пирроло[2,3-e]пиразин;
(R)-(3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)пиперидин-1-ил)(3,3-дифторазетидин-1-ил)метанон;
(R)-(3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)пиперидин-1-ил)(3,3-дифторпирролидин-1-ил)метанон;
(R)-(3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)пиперидин-1-ил)(4,4-дифторпиперидин-1-ил)метанон;
(R)-1-(3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)пиперидин-1-карбонил)азетидин-3-карбонитрил;
(3R,4R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метил-N-(пиримидин-2-ил)пиперидин-1-карбоксамид;
(3R,4R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метил-N-(пиридин-2-ил)пиперидин-1-карбоксамид;
(3R,4R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метил-N-(пиримидин-4-ил)пиперидин-1-карбоксамид;
(3R,4R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метил-N-(пиразин-2-ил)пиперидин-1-карбоксамид;
1-циклогексил-3-фенил-6H-имидазо[1,5-a]пирроло[2,3-e]пиразин;
N-((3S,5R)-1-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)- 5-метилпирролидин-3-ил)циклопропансульфонамид;
((3R,4R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-ил)(1-метилпирролидин-3-ил)метанон;
((3R,4R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-ил)(1-метилпиперидин-4-ил)метанон;
(3R,4R)-фенил 3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-карбоксилат;
((R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)пиперидин-1-ил)((R)-2-(трифторметил)пирролидин-1-ил)метанон;
(R)-1-(1-(пирролидин-1-илсульфонил)пиперидин-3-ил)-6H-имидазо[1,5-a]пирроло[2,3-e]пиразин;
(R)-(3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)пиперидин-1-ил)(пирролидин-1-ил)метанон;
3-(1-циклогексил-6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-3-ил)пропановая кислота;
(S)-1-((R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)пиперидин-1-карбонил)пирролидин-3-карбонитрил;
(R)-циклопентил 3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)пиперидин-1-карбоксилат;
(E)-N-(((3R,4R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-ил)(пирролидин-1-ил)метилен)цианамид;
4-((1R,3R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)циклопентиламино)бензонитрил;
(R)-(3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)пиперидин-1-ил)(3,3-дифторциклобутил)метанон;
5-((1S,3R,4S)-3-этил-4-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)циклопентиламино)пиразин-2-карбонитрил;
N-((1S,3S,4R)-3-(3-бром-6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-этилциклопентил)циклопропансульфонамид;
((3R,4R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-ил)(4,4-дифторциклогексил)метанон;
(R)-(3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)пирролидин-1-ил)(3,3-диметилпирролидин-1-ил)метанон;
(R)-(3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)пиперидин-1-ил)(3,3-дифторпиперидин-1-ил)метанон;
(R)-1-(3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)пиперидин-1-карбонил)пиперидин-4-карбонитрил;
(R)-(3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)пиперидин-1-ил)(тиоморфолино-1,1-диоксид)метанон;
((3R,4R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-ил)(азепан-1-ил)метанон;
(R)-(3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)пиперидин-1-ил)(4,4-диметилпиперидин-1-ил)метанон;
(R)-(3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)пиперидин-1-ил)(4-хлорпиперидин-1-ил)метанон;
5-(((1S,3R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)циклопентил)метиламино)пиразин-2-карбонитрил;
5-(((1S,3S)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)циклопентил)метиламино)пиразин-2-карбонитрил;
1-((R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)пиперидин-1-карбонил)пиперидин-3-карбонитрил;
N-((3S,5R)-5-этил-1-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)пирролидин-3-ил)циклопропансульфонамид;
1-(3,3-дифторциклобутил)-6H-имидазо[1,5-a]пирроло[2,3-e]пиразин;
N-(1-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)пирролидин-3-ил)циклопропансульфонамид;
(E)-3-(1-циклогексил-6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-3-ил)акриловая кислота;
N-((1S,3S,4R)-3-(3-хлор-6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-этилциклопентил)циклопропансульфонамид;
4-(((цис)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)циклобутокси)метил)бензонитрил;
5-((3S,5R)-5-этил-1-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)пирролидин-3-иламино)пиразин-2-карбонитрил;
N-((3S,5R)-5-этил-1-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)пирролидин-3-ил)-3,3,3-трифторпропан-1-сульфонамид;
4-((1R,3R,4S)-3-этил-4-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)циклопентилокси)бензонитрил;
N-((1S,3S,4R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилциклопентил)-1-метилциклопропан-1-сульфонамид;
1-((1S,4S)-5-(3,3,3-трифторпропилсульфонил)-2,5-диазабицикло[2.2.1]гептан-2-ил)-6H-имидазо[1,5-a]пирроло[2,3-e]пиразин;
N-((1S,3S,4R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилциклопентил)-3,3-дифторазетидин-1-сульфонамид;
N-((1S,3S,4R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилциклопентил)-3,3,3-трифторпропан-1-сульфонамид;
N-((1S,3S,4R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилциклопентил)-3,3-дифторпирролидин-1-сульфонамид;
(S)-N-((1S,3S,4R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилциклопентил)-2-(трифторметил)пирролидин-1-сульфонамид;
N-(((1S,3S)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)циклопентил)метил)циклопропансульфонамид;
N-(((1S,3R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)циклопентил)метил)-3,3,3-трифторпропан-1-сульфонамид;
N-(((1S,3S)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)циклопентил)метил)-3,3,3-трифторпропан-1-сульфонамид;
N-((1S,3S,4R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилциклопентил)-1-этилциклопропан-1-сульфонамид;
1-((3aR,6aS)-5-(3,3,3-трифторпропилсульфонил)гексагидропирроло[3,4-c]пиррол-2(1H)-ил)-6H-имидазо[1,5-a]пирроло[2,3-e]пиразин;
1-(6-фтор-4-(3,3,3-трифторпропилсульфонил)-1,4-диазепан-1-ил)-6H-имидазо[1,5-a]пирроло[2,3-e]пиразин;
4-([4-(6H-имидазо[1,5-a]рпирроло[2,3-e]пиразин-1-ил)кубанил]метокси)бензонитрил;
N-((3R,4S)-4-метил-1-(3,3,3-трифторпропилсульфонил)пиперидин-3-ил)-6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-амин;
1-(2-(3,3,3-трифторпропилсульфонил)-2,5-диазаспиро[3.5]нонан-5-ил)-6H-имидазо[1,5-a]пирроло[2,3-e]пиразин;
1-((3aS,7aR)-4-(3,3,3-трифторпропилсульфонил)октагидро-1H-пирроло[3,2-b]пиридин-1-ил)-6H-имидазо[1,5-a]пирроло[2,3-e]пиразин;
1-(7-метил-4-(3,3,3-трифторпропилсульфонил)-1,4-диазепан-1-ил)-6H-имидазо[1,5-a]пирроло[2,3-e]пиразин;
1-(5-(3,3,3-трифторпропилсульфонил)-2,5-диазаспиро[3.5]нонан-2-ил)-6H-имидазо[1,5-a]пирроло[2,3-e]пиразин;
N-(1-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)пиперидин-3-ил)-3,3,3-трифторпропан-1-сульфонамид;
1-((1R,5S)-2-(3,3,3-трифторпропилсульфонил)-2,6-диазабицикло[3.2.1]октан-6-ил)-6H-имидазо[1,5-a]пирроло[2,3-e]пиразин;
1-циклогексил-3-(4-(метилсульфонил)фенил)-6H-имидазо[1,5-a]пирроло[2,3-e]пиразин;
N-(4-(1-циклогексил-6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-3-ил)фенил)метансульфонамид;
N-((1S,3S,4R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилциклопентил)-3-хлорбензолсульфонамид;
[(1S,3R,4R)-3-этил-4-(3-трифторметил-6H-2,5,6,8b-тетрааза-ас-индацен-1-ил)-циклопентил]-амид циклопропансульфоновой кислоты;
[(1S,3R,4S)-3-этил-4-(3-трифторметил-6H-2,5,6,8b-тетрааза-ас-индацен-1-ил)-циклопентил]-амид циклопропансульфоновой кислоты;
1-((1S,2R,4S)-4-Циклопропансульфониламино-2-этил-циклопентил)-6H-2,5,6,8b-тетрааза-ас-индацен-3-карбоновая кислота;
1-((1R,2R,4S)-4-Циклопропансульфониламино-2-этил-циклопентил)-6H-2,5,6,8b-тетрааза-ас-индацен-3-карбоновая кислота;
[(1S,3R,4S)-3-метил-4-(3-трифторметил-6H-2,5,6,8b-тетрааза-ас-индацен-1-ил)-циклопентил]-амид циклопропансульфоновой кислоты;
1-[(1R,3R,4S)-3-Этил-4-(3-трифторметил-6H-2,5,6,8b-тетрааза-ас-индацен-1-ил)-циклопентил]-2-метил-пропан-2-ол;
{(1S,3R,4S)-3-этил-4-[3-(2,2,2-трифтор-этил)-6H-2,5,6,8b-тетрааза-ас-индацен-1-ил]-циклопентил}-амид циклопропансульфоновой кислоты;
этиловый эфир [(1R,3R,4S)-3-этил-4-(3-трифторметил-6H-2,5,6,8b-тетрааза-ас-индацен-1-ил)-циклопентил]-уксусной кислоты или
1-[(1S,2R,4R)-2-Этил-4-(3-метоксиметил-[1,2,4]оксадиазол-5-илметил)-циклопентил]-3-трифторметил-6H-2,5,6,8b-тетрааза-ас-индацен.
В семнадцатом варианте воплощения настоящее изобретение обеспечивает соединение в соответствии с вариантами воплощения с первого по двенадцатый, где T представляет собой N, U представляет собой CR4, X представляет собой CR3, и Y представляет собой N, с образованием соединения формулы (Ic)

Формула (Ic)
В восемнадцатом варианте воплощения настоящее изобретение обеспечивает соединение в соответствии с семнадцатым вариантом воплощения, где соединение представляет собой
3-((3S,4S)-3-(3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)-4-метилпиперидин-1-ил)-3-оксопропаннитрил;
5-(3-(3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)пиперидин-1-ил)пиразин-2-карбонитрил;
(S)-1-(3-(3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)пиперидин-1-карбонил)циклопропанкарбонитрил;
N-((1S,3R,4R)-3-этил-4-(3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)циклопентил)циклопропансульфонамид;
N-((1R,3S,4S)-3-этил-4-(3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)циклопентил)циклопропансульфонамид;
(S)-6-(3-(3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)пиперидин-1-ил)никотинонитрил;
(R)-6-(3-(3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)пиперидин-1-ил)никотинонитрил;
(S)-2-(3-(3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)пиперидин-1-ил)тиазол-5-карбонитрил;
(R)-2-(3-(3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)пиперидин-1-ил)тиазол-5-карбонитрил;
(R)-(3-(3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)пиперидин-1-ил)(3,3-дифторазетидин-1-ил)метанон;
(S)-(3-(3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)пиперидин-1-ил)(3,3-дифторазетидин-1-ил)метанон;
5-((1R,3S,4S)-3-(3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)-4-метилциклопентиламино)пиразин-2-карбонитрил;
5-((1S,3R,4R)-3-(3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)-4-метилциклопентиламино)пиразин-2-карбонитрил;
5-((1R,3R,4S)-3-(3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)-4-метилциклопентиламино)пиразин-2-карбонитрил;
5-((1S,3S,4R)-3-(3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)-4-метилциклопентиламино)пиразин-2-карбонитрил;
N-(4-(3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)бицикло[2.2.2]октан-1-ил)циклопропансульфонамид;
(R)-(3-(3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)пиперидин-1-ил)(3,3-дифторциклобутил)метанон;
(R)-(3-(3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)пиперидин-1-ил)(3,3-дифторпирролидин-1-ил)метанон;
(R)-(3-(3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)пиперидин-1-ил)(4,4-дифторпиперидин-1-ил)метанон;
N-((1S,3S,4R)-3-(3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)-4-метилциклопентил)-3,3,3-трифторпропан-1-сульфонамид;
N-((1R,3R,4S)-3-(3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)-4-метилциклопентил)-3,3,3-трифторпропан-1-сульфонамид;
N-((1R,3S,4S)-3-(3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)-4-метилциклопентил)-3,3,3-трифторпропан-1-сульфонамид;
N-((1S,3R,4R)-3-(3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)-4-метилциклопентил)-3,3,3-трифторпропан-1-сульфонамид;
((R)-3-(3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)пиперидин-1-ил)((R)-2-(трифторметил)пирролидин-1-ил)метанон;
N-((3S,5R)-5-этил-1-(3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)пирролидин-3-ил)циклопропансульфонамид;
1-циклогексил-1,6-дигидроимидазо[4,5-d]пирроло[2,3-b]пиридин;
N-((3S,5R)-5-этил-1-(3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)пирролидин-3-ил)-3,3,3-трифторпропан-1-сульфонамид;
3-(3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)-4-метилциклопентанамин;
N-((1R,3S,4R)-3-этил-4-(3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)циклопентил)-3,3,3-трифторпропан-1-сульфонамид;
N-((1S,3R,4S)-3-этил-4-(3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)циклопентил)-3,3,3-трифторпропан-1-сульфонамид;
N-((1S,3S,4R)-3-(3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)-4-метилциклопентил)циклопропансульфонамид;
N-((1S,3R,4S)-3-этил-4-(3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)циклопентил)циклопропансульфонамид;
N-((1S,3R,4S)-3-этил-4-(3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)циклопентил)-3,3-дифторазетидин-1-сульфонамид;
3-хлор-N-((1S,3R,4S)-3-этил-4-(3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)циклопентил)-4-фторбензолсульфонамид;
N-((1S,3S,4R)-3-(3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)-4-метилциклопентил)-3,3-дифторазетидин-1-сульфонамид;
N-(((1S,3R,4S)-3-этил-4-(3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)циклопентил)метил)-3,3,3-трифторпропан-1-сульфонамид;
N-(((1R,3S,4R)-3-этил-4-(3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)циклопентил)метил)-3,3,3-трифторпропан-1-сульфонамид;
N-(((1S,3S,4R)-3-этил-4-(3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)циклопентил)метил)-3,3,3-трифторпропан-1-сульфонамид;
N-(((1R,3R,4S)-3-этил-4-(3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)циклопентил)метил)-3,3,3-трифторпропан-1-сульфонамид;
N-((1S,3S,4R)-3-(3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)-4-метилциклопентил)морфолин-4-сульфонамид;
[(2S,4S,5R)-4-метил-5-(3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)-тетрагидро-фуран-2-илметил]-амид 3,3,3-трифтор-пропан-1-сульфоновой кислоты;
[(2R,4R,5S)-4-метил-5-(3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)-тетрагидро-фуран-2-илметил]-амид 3,3,3-трифтор-пропан-1-сульфоновой кислоты;
метил-[(1S,3R,4S)-3-метил-4-(3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)-циклопентил]-амид 3,3,3-трифтор-пропан-1-сульфоновой кислоты;
[(1S,3R,4S)-3-метил-4-(3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)-циклопентил]-амид азетидин-1-сульфоновой кислоты;
{3-[(1S,3R,4S)-3-Метил-4-(3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)-циклопентиламино]-оксетан-3-ил}-ацетонитрил;
[(1S,3R,4S)-3-метил-4-(3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)-циклопентил]-амид 3,3-дифтор-циклобутансульфоновой кислоты;
8-[(1S,2R,4S)-2-Метил-4-(тетрагидро-пиран-4-илокси)-циклопентил]-3H-3,4,6,8a-тетрааза-ас-индацен;
8-[(1R,2R)-2-Метил-4-(тетрагидро-пиран-4-илокси)-циклопентил]-3H-3,4,6,8a-тетрааза-ас-индацен;
[(1S,3R,4S)-3-метил-4-(3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)-циклопентил]-амид 3-фтор-азетидин-1-сульфоновой кислоты;
[(1S,3R,4S)-3-метил-4-(3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)-циклопентил]-амид 3-фтор-пропан-1-сульфоновой кислоты;
[(1S,3R,4S)-3-метил-4-(7-метил-3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)-циклопентил]-амид циклопропансульфоновой кислоты;
[(1R,3S,4R)-3-метил-4-(7-метил-3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)-циклопентил]-амид циклопропансульфоновой кислоты;
2-Циано-N-[(1S,3R,4S)-3-метил-4-(3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)-циклопентил]-ацетамид;
8-[(1S,2R,4R)-2-Метил-4-(тетрагидро-пиран-4-илокси)-циклопентил]-3H-3,4,6,8a-тетрааза-ас-индацен;
(2-Циклопропил-этил)-[(1S,3R,4S)-3-метил-4-(3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)-циклопентил]-оксетан-3-ил-амин;
Циклопропилметил-[(1S,3R,4S)-3-метил-4-(3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)-циклопентил]-оксетан-3-ил-амин;
(2,2,2-трифтор-этил)-амид (3R,4S)-3-этил-4-(3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)-пирролидин-1-карбоновой кислоты; или
(2,2,2-трифтор-этил)-амид (3S,4R)-3-этил-4-(3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)-пирролидин-1-карбоновой кислоты.
В девятнадцатом варианте воплощения настоящее изобретение обеспечивает соединение в соответствии с вариантами воплощения с первого по двенадцатый, где T представляет собой CR6, U представляет собой CR4, X представляет собой CR3 и Y представляет собой N, с образованием соединения формулы (Id)

Формула (Id)
В двадцатом варианте воплощения настоящее изобретение обеспечивает соединение в соответствии с девятнадцатым вариантом воплощения, где соединение представляет собой
N-(4-(3H-дипирроло[1,2-a:2',3'-e]пиразин-8-ил)бицикло[2.2.2]октан-1-ил)циклопропансульфонамид.
В двадцать первом варианте воплощения настоящее изобретение обеспечивает соединение в соответствии с вариантами воплощения с первого по двенадцатый, где T представляет собой CR6, U представляет собой N, X представляет собой NR3 и Y представляет собой C, с образованием соединения формулы (Ie)

Формула (Ie)
В двадцать втором варианте воплощения настоящее изобретение обеспечивает соединение в соответствии с двадцать первым вариантом воплощения, где соединение представляет собой
(R)-1-(3-(пиразоло[3,4-d]пирроло[2,3-b]пиридин-1(6H)-ил)пиперидин-1-карбонил)циклопропанкарбонитрил; или
(S)-1-(3-(пиразоло[3,4-d]пирроло[2,3-b]пиридин-1(6H)-ил)пиперидин-1-карбонил)циклопропанкарбонитрил.
В двадцать третьем варианте воплощения настоящее изобретение обеспечивает соединение в соответствии с вариантами воплощения с первого по двенадцатый, где T представляет собой O, U представляет собой N, X представляет собой CR3 и Y представляет собой C, с образованием соединения формулы (If)
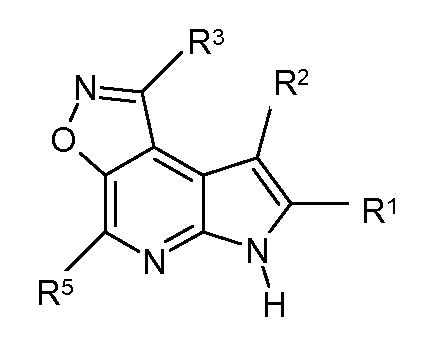
Формула (If)
В двадцать четвертом варианте воплощения настоящее изобретение обеспечивает соединение в соответствии с вариантами воплощения с первого по двенадцатый, где T представляет собой NR6, U представляет собой N, X представляет собой CR3 и Y представляет собой C, с образованием соединения формулы (Ig)
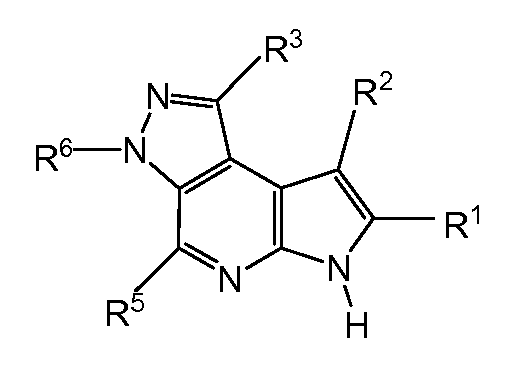
Формула (Ig)
В двадцать пятом варианте воплощения настоящее изобретение обеспечивает соединение в соответствии с двадцать четвертым вариантом воплощения, где соединение представляет собой
1-((1R,2R,4S)-2-этил-4-(4-метоксибензилокси)циклопентил)-3,6-дигидропиразоло[4,3-d]пирроло[2,3-b]пиридин;
1-((1S,2S,4R)-2-этил-4-(4-метоксибензилокси)циклопентил)-3,6-дигидропиразоло[4,3-d]пирроло[2,3-b]пиридин; или
N-(4-(3,6-дигидропиразоло[4,3-d]пирроло[2,3-b]пиридин-1-ил)бицикло[2.2.2]октан-1-ил)циклопропансульфонамид.
В двадцать шестом варианте воплощения настоящее изобретение обеспечивает соединение в соответствии с вариантами воплощения с первого по двенадцатый, где T представляет собой CR6, U представляет собой CR4, X представляет собой NR3 и Y представляет собой C, с образованием соединения формулы (Ih)

Формула (Ih)
В двадцать седьмом варианте воплощения настоящее изобретение обеспечивает соединение в соответствии с двадцать шестым вариантом воплощения, где соединение представляет собой
1-циклогексил-1,6-дигидроимидазо[4,5-d]пирроло[2,3-b]пиридин;
1-циклогексил-2-метил-1,6-дигидроимидазо[4,5-d]пирроло[2,3-b]пиридин; или
1-циклогексил-2-(трифторметил)-1,6-дигидроимидазо[4,5-d]пирроло[2,3-b]пиридин.
В двадцать восьмом варианте воплощения настоящее изобретение обеспечивает соединение в соответствии с вариантами воплощения с первого по двенадцатый, где T представляет собой S, U представляет собой N, X представляет собой CR3, и Y представляет собой C, с образованием соединения формулы (Ii)

Формула (Ii)
В двадцать девятом варианте воплощения настоящее изобретение обеспечивает соединение в соответствии с вариантами воплощения с первого по двенадцатый, где T представляет собой N, U представляет собой CR4, X представляет собой NR3, и Y представляет собой C, с образованием соединения формулы (Ij)
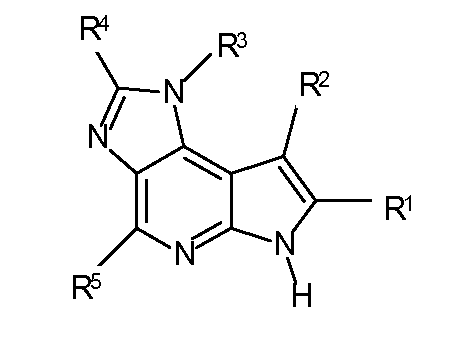
Формула (Ij)
В тридцатом варианте воплощения настоящее изобретение обеспечивает соединение в соответствии с двадцать девятым вариантом воплощения, где соединение представляет собой
N-((1S,3R,4S)-3-этил-4-(имидазо[4,5-d]пирроло[2,3-b]пиридин-1(6H)-ил)циклопентил)циклопропансульфонамид;
N-((1S,3S,4R)-3-(имидазо[4,5-d]пирроло[2,3-b]пиридин-1(6H)-ил)-4-метилциклопентил)циклопропансульфонамид;
N-((1S,3S,4R)-3-(2-циклопропилимидазо[4,5-d]пирроло[2,3-b]пиридин-1(6H)-ил)-4-метилциклопентил)циклопропансульфонамид;
N-((1S,3R,4S)-3-метил-4-(2-метилимидазо[4,5-d]пирроло[2,3-b]пиридин-1(6H)-ил)циклопентил)циклопропансульфонамид;
[(1S,3R,4S)-3-метил-4-(2-трифторметил-имидазо[4,5-d]пирроло[2,3-b]пиридин-1(6H)-ил)-циклопентил]-амид циклопропансульфоновой кислоты;
[(1S,3R,4S)-3-этил-4-(2-трифторметил-имидазо[4,5-d]пирроло[2,3-b]пиридин-1(6H)-ил)-циклопентил]-амид циклопропансульфоновой кислоты;
[(1S,3S,4R)-3-(2-дифторметил-имидазо[4,5-d]пирроло[2,3-b]пиридин-1(6H)-ил)-4-этил-циклопентил]-амид циклопропансульфоновой кислоты;
[(1S,3R,4S)-3-этил-4-(2-метил-имидазо[4,5-d]пирроло[2,3-b]пиридин-1(6H)-ил)-циклопентил]-амид циклопропансульфоновой кислоты;
[(1S,3S,4R)-3-(2-амино-имидазо[4,5-d]пирроло[2,3-b]пиридин-1(6H)-ил)-4-этил-циклопентил]-амид циклопропансульфоновой кислоты
в соответствии с вариантами воплощения с первого по двенадцатый, где Т представляет собой N, U представляет собой N, X представляет собой NR3, и Y представляет собой C, с образованием соединения формулы (Ik)

Формула (Ik)
В тридцать втором варианте воплощения настоящее изобретение обеспечивает соединение в соответствии с тридцать первым вариантом воплощения, где соединение представляет собой
[(1S,3R,4S)-3-этил-4-(6H-[1,2,3]триазоло[4,5-d]пирроло[2,3-b]пиридин-1-ил)-циклопентил]-амид циклопропансульфоновой кислоты.
В тридцать третьем варианте воплощения настоящее изобретение обеспечивает соединение формулы (II), где соединение представляет собой
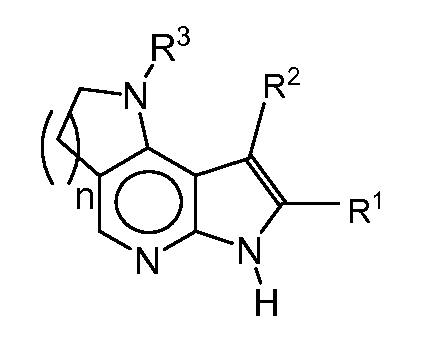
Формула (II)
его фармацевтически приемлемые соли, пролекарства, биологически активные метаболиты, стереоизомеры и изомеры, где
R1 и R2 независимо представляют собой водород, дейтерий, -N(Ra)(Rb), галоген, -ORa, -SRa, -S(O)Ra, -S(O)2Ra, -NO2, -C(O)ORa, -CN, -C(O)N(Ra)(Rb), -N(Ra)C(O)(Rb), -C(O)Ra, -C(OH)RaRb, -N(Ra)S(O)2-Rb, -S(O)2N(Ra)(Rb), -CF3, -OCF3, необязательно замещенный (C1-C6)алкил, необязательно замещенный (C2-C6)алкенил, необязательно замещенный (C2-C6)алкинил, необязательно замещенный (C3-C10)циклоалкил, необязательно замещенный (C1-C10)гетероарил, необязательно замещенный (C1-C10) гетероциклил или необязательно замещенный (C6-C10)арил;
где в фрагменте, содержащем -N(Ra)(Rb), азот, Ra и Rb могут образовывать кольцо, таким образом, -N(Ra)(Rb) представляет собой необязательно замещенный (C2-C10)гетероциклил или необязательно замещенный (C1-C10)гетероарил, связанный через азот;
R3 представляет собой водород, необязательно замещенный связанный мостиковой связью (C5-C12)циклоалкил, необязательно замещенный связанный мостиковой связью (C2-C10)гетероциклил, необязательно замещенный (C1-C8)алкил, необязательно замещенный (C3-C10)циклоалкил, необязательно замещенный (C3-C8)циклоалкенил, необязательно замещенный (C6-C10)арил, необязательно замещенный (C1-C10)гетероарил, необязательно замещенный (C2-C10)гетероциклил; или
R3 представляет собой -A-D-E-G, где:
A представляет собой связь, -C(O)-, необязательно замещенный (C1-C6)алкилен, необязательно замещенный (C2-C6)алкенилен, необязательно замещенный (C2-C6)алкинилен, необязательно замещенный (C3-C12)циклоалкилен, необязательно замещенный (C2-C6)гетероциклилен, -C(O)N(Ra)-Re-, -N(Ra)C(O)-Re-, -O-Re-, -N(Ra)-Re-, -S-Re-, -S(O)2-Re-, -S(O)Re-, -C(O-Ra)(Rb)-Re-, -S(O)2N(Ra)-Re-, -N(Ra)S(O)2-Re- или -N(Ra)C(O)N(Rb)-Re-;
D представляет собой необязательно замещенный (C1-C8)алкилен, необязательно замещенный связанный мостиковой связью (C5-C12)циклоалкилен, необязательно замещенный (C3-C10)циклоалкилен, необязательно замещенный связанный мостиковой связью (C5-C10)циклоалкенилен, необязательно замещенный (C3-C10)циклоалкенилен, необязательно замещенный (C6-C10)арилен, необязательно замещенный (C1-C10)гетероарилен, необязательно замещенный связанный мостиковой связью (C2-C10)гетероциклилен или необязательно замещенный (C2-C10)гетероциклилен;
E представляет собой связь, -Re-, -Re-C(=NCN)-Re-, -Re-C(O)-Re-, -Re-C(O)C(O)-Re-, -Re-C(O)O-Re-, -Re-C(O)C(O)N(Ra)-Re-, -Re-N(Ra)-C(O)C(O)-Re-, -Re-O-Re-, -Re-S(O)2-Re-, -Re-S(O)-Re-, -Re-S-Re-, -Re-N(Ra)-Re-, =N-Re-, -Re-N(Ra)C(O)-Re-, -ReC(O)N(Ra)Re-, -Re-OC(O)N(Ra)-Re-, -Re-N(Ra)C(O)ORe-, -Re-OC(O)-Re, -Re-OC(O)-O-Re, -Re-N(Ra)C(O)N(Rb)-Re-, -Re-N(Ra)S(O)2-Re-, -Re-S(O)2N(Ra)-Re- или -Re-N(Ra)S(O)2N(Ra)-Re-; или
Е представляет собой

где во всех случаях E связан либо с атомом углерода либо с атомом азота в D;
G представляет собой водород, дейтерий, -N(Ra)(Rb), галоген, -ORa, -SRa, -S(O)Ra, -S(O)2Ra, -NO2, -C(O)ORa, -CN, -C(O)N(Ra)(Rb), -N(Ra)C(O)Rb, -N(Ra)C(O)ORb, -OC(O)N(Ra), -N(Ra)C(O)N(Rb)2, -C(O-Ra)(Rb)2, -C(O)Ra, -CF3, -OCF3, -N(Ra)S(O)2Rb, -S(O)2N(Ra)(Rb), -S(O)2N(Ra)C(O)Rb, необязательно замещенный -(C1-C6)алкил, необязательно замещенный -(C2-C6)алкенил, необязательно замещенный -(C2-C6)алкинил, необязательно замещенный -(C3-C10)циклоалкил, необязательно замещенный -(C1-C10)гетероарил, необязательно замещенный -(C1-C10) гетероциклил, необязательно замещенный -(C6-C10)арил;
где в фрагменте, содержащем -N(Ra)(Rb), азот, Ra и Rb могут образовывать кольцо, таким образом, -N(Ra)(Rb) представляет собой необязательно замещенный (C2-C10)гетероциклил или необязательно замещенный (C1-C10) гетероарил, связанный через азот;
Ra и Rb, каждый независимо, представляют собой водород, дейтерий, CN, необязательно замещенный (C1-C10)алкил, необязательно замещенный (C2-C10)алкенил, необязательно замещенный (C2-C10)алкинил, необязательно замещенный (C1-C10)алкил-O-(C1-C10)алкил, необязательно замещенный (C3-C10)циклоалкил, необязательно замещенный (C6-C10)арил, необязательно замещенный (C1-C10)гетероарил, необязательно замещенный (C1-C10)гетероциклил, необязательно замещенный -(C1-C6)алкилен-(C3-C10)циклоалкил, необязательно замещенный -(C1-C6)алкилен-(C6-C10)арил, необязательно замещенный -(C1-C6)алкилен-(C1-C10)гетероарил или необязательно замещенный -(C1-C6)алкилен-(C1-C10)гетероциклил; и
Re для каждого случая независимо представляет собой связь, необязательно замещенный (C1-C10)алкилен, необязательно замещенный (C2-C10)алкенилен, необязательно замещенный (C2-C10)алкинилен, необязательно замещенную -(C1-C10)алкилен-O-(C1-C10)алкиленовую группу, необязательно замещенный (C3-C10)циклоалкилен, необязательно замещенный (C6-C10)арилен, необязательно замещенный (C1-C10)гетероарилен или необязательно замещенный (C1-C10)гетероциклилен.
В тридцать четвертом варианте воплощения настоящее изобретение обеспечивает соединение в соответствии с тридцать третьим вариантом воплощения, где соединение представляет собой
1-Циклогексил-2,3,4,7-тетрагидро-1H-пирроло[2,3-h][1,6]нафтиридин;
[(1S,3R,4S)-3-этил-4-(3,6,7,8-тетрагидро-3,4,9-триаза-циклопента[a]нафталин-9-ил)-циклопентил]-амид циклопропансульфоновой кислоты; или
[(1S,3S,4R)-3-(3,6-дигидро-2H-дипирроло[2,3-b;2',3'-d]пиридин-1-ил)-4-этил-циклопентил]-амид циклопропансульфоновой кислоты.
В тридцать пятом варианте воплощения настоящее изобретение обеспечивает фармацевтическую композицию, включающую соединение формулы (I) или формулы (II), определенное в любом из описанных выше вариантов воплощения


фармацевтически приемлемый носитель и эксципиент и второе терапевтическое средство, выбранное из группы, включающей цитокин-супрессивные противовоспалительные лекарственные средства, антитела к, или антагонисты, другим человеческим цитокинам или ростовым факторам, IL-1, IL-2, IL-3, IL-4, IL-5, IL-6, IL-7, IL-8, IL-12, IL-15, IL-16, IL-21, IL-23, интерфероны, EMAP-II, GM-CSF, FGF, PDGF, CTLA или их лиганды, включая CD154, HUMIRATM, REMICADETM, SIMPONITM (голимумаб), CIMZIATM, ACTEMRATM, CDP 571, растворимые p55 или p75 TNF рецепторы, ENBRELTM, Ленерсепт, ингибиторы TNFα-превращающего фермента, ингибиторы IL-1, Интерлейкин 11, антагонисты IL-18, антагонисты IL-12, антитела к IL-12, растворимые IL-12 рецепторы, IL-12-связывающие белки, неистощающие анти-CD4 ингибиторы FK506, рапамицин, микофенолят мофетил, лефлуномид, НСПВЛС(нестероидные противовоспалительные лекарственные средства), ибупрофен, кортикостероиды, ингибиторы фосфодиэстеразы, агонисты аденозина, антитромботические средства, ингибиторы комплемента, адренергические средства, ингибиторы IL-1β превращающего фермента, ингибиторы киназы T-клеточной передачи сигнала, ингибиторы металлопротеиназы, сульфасалазин, 6-меркаптопурины, производные p75TNFRIgG, sIL-1RI, sIL-1RII, sIL-6R, целекоксиб, гидроксихлорохин сульфат, рофекоксиб, инфликсимаб, напроксен, валдекоксиб, сульфасалазин, мелоксикам, ацетат, золото-натрия тиомалат, аспирин, триамцинолон ацетонид, пропоксифен напсилат/апап, фолат, набуметон, диклофенак, пироксикам, этодолак, диклофенак натрий, оксапрозин, оксикодон HCl, гидрокодон битартрат/апап, диклофенак натрий/мисопростол, фентанил, анакинра, трамадол HCl, салсалат, сулиндак, цианокобаламин/фа/пиридоксин, ацетаминофен, алендронат натрий, морфин сульфат, лидокаин гидрохлорид, индометацин, глюкозамин сульф/хондроитин, амитриптилин HCl, сульфадиазин, оксикодон HCl/ацетаминофен, олопатадин HCl мисопростол, напроксен натрий, омепразол, циклофосфамид, ритуксимаб, IL-1 TRAP, MRA, CTLA4-IG, IL-18 BP, анти-IL-12, анти-IL15, VX-740, Рофлумиласт, IC-485, CDC-801, агонисты S1P1, FTY720, ингибиторы PKC семейства, Рубоксистаурин, AEB-071, Мезопрам, метотрексат, лефлуномид, кортикостероиды, буденозид, дексаметазон, сульфасалазин, 5-аминосалициловую кислоту, олсалазин, ингибиторы IL-1β превращающего фермента, IL-1ra, ингибиторы передачи Т-клеточного сигнала, ингибиторы тирозиновой киназы, 6-меркаптопурины, IL-11, месаламин, преднизон, азатиоприн, меркаптопурин, инфликсимаб, метилпреднизолон натрий сукцинат, дифеноксилат/атроп сульфат, лоперамид гидрохлорид, омепразол, фолат, ципрофлоксацин/декстроза-воду, гидрокодон, битартрат/апап, тетрациклин гидрохлорид, флуоцинонид, метронидазол, тимеросал/борную кислоту, холестирамин/сахарозу, ципрофлоксацин гидрохлорид, гиосциамин сульфат, меперидин гидрохлорид, мидазолам гидрохлорид, оксикодон HCl/ацетаминофен, прометазин гидрохлорид, фосфат натрия, сульфаметоксазол/тримтеоприм, поликарбофил, пропоксифен напсилат, гидрокортизон, поливитамины, балсалазид динатрий, кодеин фосфат/апап, колесевелам HCl, цианокобаламин, фолиевую кислоту, левофлоксацин, натализумаб, интерферон-гамма, метилпреднизолон, азатиоприн, циклофосфамид, циклоспорин, метотрексат, 4-аминопиридин, тизанидин, интерферон-β1a, AVONEX®, интерферон-β1b, BETASERON®, интерферон α-n3, интерферон-α, интерферон β1A-IF, Пэгинтерферон α 2b, Сополимер 1, COPAXONE®, кислород под повышенным давлением, внутривенный иммуноглобулин, кладрибин, циклоспорин, FK506, микофенолят мофетил, лефлуномид, НСПВЛС, кортикостероиды, преднизолон, ингибиторы фосфодиэстеразы, агонисты аденозина, антитромботические средства, ингибиторы комплемента, адренергические средства, противовоспалительные цитокины, интерферон-β, IFNβ1a, IFNβ1b, копаксон, кортикостероиды, ингибиторы каспазы, ингибиторы каспазы-1, антитела к CD40 лиганду и CD80, алемтузумаб, дронабинол, даклизумаб, митоксантрон, ксалипроден гидрохлорид, фампридин, глатирамер ацетат, натализумаб, синнабидол, α-иммунокин NNSO3, ABR-215062, AnergiX.MS, антагонисты хемокиновых рецепторов, BBR-2778, калагуалин, CPI-1189, инкапсулированный в липосомах митоксантрон, THC.CBD, каннабиноид агонисты, MBP-8298, мезопрам, MNA-715, анти-IL-6 рецептор антитело, нейровакс, пирфенидон аллотрап 1258 (RDP-1258), sTNF-R1, талампанел, терифлуномид, TGF-beta2, типлимотид, VLA-4 антагонисты, интерферон гамма антагонисты, IL-4 агонисты, диклофенак, мисопростол, напроксен, мелоксикам, индометацин, диклофенак, метотрексат, азатиоприн, миноциклин, преднизон, этанерсепт, рофекоксиб, сульфасалазин, напроксен, лефлуномид, метилпреднизолон ацетат, индометацин, гидроксихлорохин сульфат, преднизон, сулиндак, бетаметазон дипроп-усиленный, инфликсимаб, метотрексат, фолат, триамцинолон ацетонид, диклофенак, диметилсульфоксид, пироксикам, диклофенак натрия, кетопрофен, мелоксикам, метилпреднизолон, набуметон, толметин натрий, кальципотриен, циклоспорин, диклофенак натрия/мисопростол, флуоцинонид, глюкозамин сульфат, золото натрия тиомалат, гидрокодон битартрат/апап, риседронат натрий, сульфадиазин, тиогуанин, валдекоксиб, алефасепт и эфализумаб, диклофенак, напроксен, ибупрофен, пироксикам, индометацин, ингибиторы COX2, рофекоксиб, валдекоксиб, гидроксихлорохин, стероиды, преднизолон, буденозид, дексаметазон, цитотоксические средства, азатиоприн, циклофосфамид, микофенолят мофетил, ингибиторы PDE4, ингибитор синтеза пурина, сульфасалазин, 5-аминосалициловую кислоту, олсалазин, Имуран®, CTLA-4-IgG, антитела против B7 семейства, антитела против PD-1 семейства, анти-цитокиновые антитела, фонолизумаб, анти-IFNg антитело, антитела к рецептору против рецептора, антитело против IL-6 рецептора, антитела к B-клеточноповерхностным молекулам, LJP 394, Ритуксимаб, анти-CD20 антитело и лимфостат-B.
Подробное описание изобретения
Протеинкиназы представляют собой широкий и многообразный класс, охватывающий более 500 ферментов, которые включают онкогены, рецепторы ростовых факторов, медиаторы сигнальной трансдукции, связанные с апоптозом киназы и циклин-зависимые киназы. Они являются ответственными за перенос фосфатной группы к специфическому тирозиновому, сериновому или треониновому аминокислотному остатку, и широко классифицируются как тирозиновые и сериновые/треониновые киназы как результат их специфичности в отношении субстрата.
Семейство Jak киназ (Jak1, Jak2, Jak3 и Tyk2) включает цитоплазматические тирозиновые киназы, которые связываются со связанными с мембраной цитокиновыми рецепторами. Связывание цитокинов с их рецептором инициирует активацию Jak киназы через процессы транс и автофосфорилирования. Активированные Jak киназы фосфорилируют остатки на цитокиновых рецепторах, образуя сайты связывания фосфотирозина для SH2 домен-содержащих белков, таких как факторы сигнальной трансдукции и активаторы транскрипции (STAT) и другие регуляторы сигнальной трансдукции, такие как супрессор цитокиновых сигнальных (SOCS) белков и SH2 домен-содержащие инозит 5'-фосфатазы(SHIP). Активация STAT факторов через этот процесс приводит к их димеризации, нуклеарной транслокации и новой мРНК транскрипции, приводя к экспрессии факторов пролиферации и выживания иммуноцитов, а также дополнительных цитокинов, хемокинов и молекул, которые способствуют внутриклеточному транспорту (см. Journal of Immunology, 2007, 178, p. 2623). Jak киназы осуществляют передачу сигналов для многих различных семейств цитокинов и, следовательно, потенциально играют роли в заболеваниях с самыми различными патологиями, включая следующие далее примеры, но не ограничиваясь этим. Как Jak1 так и Jak3 контролируют передачу сигнала так называемых общих цитокинов гамма цепи (IL2, IL4, IL7, IL9, IL15 и IL21), следовательно, одновременное ингибирование либо Jak1 либо Jak3, как можно предсказать, влияет на Th1-опосредованные заболевания, такие как ревматоидный артрит, через блокаду передачи сигнала IL2, IL7 и IL15. С другой стороны, недавно было показано, что передача сигнала IL2 является существенной для развития и гомеостаза T-регуляторных клеток (Malek TR et al., Immunity, 2002, 17(2), p.167-78). Таким образом, на основании генетических данных, можно предсказать, что блокада передачи сигнала IL2, взятая отдельно, приводит к автоиммунитету (Yamanouchi J et al., Nat Genet., 2007, 39(3), p.329-37, и Willerford DM et al., Immunity, 1995, 3(4), p.521-30). Th2-опосредованные заболевания, такие как астма или атопический дерматит, через блокаду передачи сигнала IL4 и IL9. Jak1 и Tyk2 опосредуют передачу сигнала IL13 (см. Int. Immunity, 2000, 12, p. 1499). Следовательно, также можно предсказать, что их блокада может иметь терапевтический эффект при астме. Также считается, что эти две киназы опосредуют передачу сигнала интерферона Типа I; поэтому можно предсказать, что их блокада может уменьшить тяжесть системной красной волчанки (SLE). Tyk2 и Jak2 опосредуют передачу сигнала IL12 и IL23. Действительно, блокада этих цитокинов с использованием моноклональных антител оказалась эффективной в лечении псориаза. Поэтому можно предсказать, что блокада этого пути с использованием ингибиторов этих киназ также может быть эффективной и при псориазе. Обобщая вышесказанное, настоящее изобретение описывает соединения, представляющие собой малые молекулы, которые ингибируют, регулируют и/или модулируют активность киназ семейства Jak, которая является основной для некоторых механизмов, которые считаются критическими для прогрессирования аутоиммунных заболеваний, включая, но не ограничиваясь этим, ревматоидный артрит (RA), системную красную волчанку (SLE), рассеянный склероз (MS), болезнь Крона, псориаз и астму.
Передача сигнала некоторых патологически существенных цитокинов происходить только через Jak1 (Guschin D, et al., EMBO J. 1995 Apr 3;14(7):1421-9; Parganas E, et al., Cell. 1998 May 1;93(3):385-95; Rodig S.J., et al., Cell. 1998 May 1; 93(3):373-83). Было показано, что блокада одного из них, IL6, с использованием IL6R-нейтрализующего антитела существенно улучшает оценку заболевания у людей, являющихся пациентами, страдающими ревматоидным артритом (Nishimoto N. et al., Ann Rheum Dis., 2007, 66(9), p.1162-7). Подобным образом, блокированная передача сигнала GCSF, которая также опосредована только Jak1, с использованием нейтрализующих моноклональных антител или делеции гена-мишени защищает мышей от экспериментального артрита (Lawlor K.E. et al., Proc Natl Acad Sci U.S.A., 2004, 101(31), p.11398-403). Соответственно, идентификация соединений, представляющих собой малые молекулы, которые ингибируют, регулируют и/или модулируют сигнальную трансдукцию киназ, таких как Jak1, является желательным средством для профилактики или лечения аутоиммунных заболеваний или других заболеваний, связанных с аберрантной функцией Jak1.
Jak2 также активируется в широком ряде раковых заболеваний человека, таких как рак предстательной железы, толстой кишки, яичника и молочной железы, меланома, лейкоз и другие гематопоэтические опухоли. Кроме того, было определено, что соматическая точечная мутация Jak2 гена тесно связана с классическими миелопролиферативными расстройствами (MPD) и в некоторых случаях с другими миелоидными расстройствами. Конститутивная активация активности Jak2 также вызвана хромосомной транслокацией в гематопоэтических опухолях. Также было показано, что ингибирование Jak/STAT пути и, в частности, ингибирование Jak2 активности, приводит к анти-пролиферативным и проапоптическим эффектам, преимущественно за счет ингибирования фосфорилирования STAT. Кроме того, фармакологическая модуляция или ингибирование активности Jak2 могла бы эффективно блокировать рост опухоли и индуцировать апоптоз путем уменьшения STAT фосфорилирования в клеточной культуре и человеческих опухолевых ксенотрансплантатах in vivo. Соответственно, идентификация соединений, представляющих собой малые молекулы, которые ингибируют, регулируют и/или модулируют сигнальную трансдукцию киназ, в частности Jak2, является желательной в качестве средства для лечения или профилактики заболеваний и состояний, связанных с раком.
Jak киназы также осуществляют передачу сигналов, регулирующих существенно важные физиологические процессы, ингибирование которых было бы нежелательным. Например, Jak2 опосредует передачу сигнала Эритропоэтина (Epo) и Гранулоцитарного/Моноцитарного Колониестимулирующего Фактора (GM-CSF). У субъектов с генетическими, врожденными или приобретенными дефектами в этих сигнальных путях могут развиться осложнения с потенциальной угрозой для жизни, такие как анемия и нейтрофильная дисфункция. Соответственно, один неограничивающий аспект настоящего изобретения также относится к способу идентификации соединений, которые могут иметь благоприятный профиль безопасности в результате того, что селективно избегается ингибирование Jak2.
Семейство протеинкиназ C представляет собой группу сериновых/треониновых киназ, которая включает двенадцать родственных изоферментов. Его члены кодируются различными генами и далее подразделяются в соответствии с их требованиями для активации. Для классических ферментов (cPKC) требуется диацилглицерин (DAG), фосфатидилсерин (PS) и кальций для активации. Новые PKC's (nPKC) требуют DAG и PS, но являются кальций-независимыми. Атипичные PKC's (aPKC) не требуют кальция или DAG.
PKCтета представляет собой член субсемейства nPKC (Baier, G., et al., J. Biol. Chem., 1993, 268, 4997). Он имеет ограниченную картину экспрессии, преимущественно обнаруженную в T клетках и скелетных мышцах (Mischak, H. et al., FEBS Lett., 1993, 326, p. 51), при этом сообщалось о некоторой экспрессии в тучных клетках (Liu, Y. et al., J. Leukoc. Biol., 2001, 69, p. 831) и эндотелиальных клетках (Mattila, P. et al., Life Sci., 1994, 55, p. 1253).
При активации T-клеток, образуется супрамолекулярный активирующий комплекс (SMAC) на участке контакта между T-клеткой и антиген-презентирующей клеткой (APC). PKCтета представляет собой единственную PKC изоформу, которая, как было обнаружено, локализуется у SMAC (Monks, C. et al., Nature, 1997, 385, 83), помещая его в непосредственной близости с другими осуществляющими передачу сигнала ферментами, которые опосредуют процессы T-клеточной активации.
В другом исследовании (Baier-Bitterlich, G. et al., Mol. клетк. Biol., 1996, 16, 842) была подтверждена роль PKCтета в активации AP-1, транскрипционного фактора, играющего важную роль в активации IL-2 гена. В нестимулированных T-клетках конститутивно активный PKCтета стимулировал AP-1 активность, тогда как в клетках с доминирующим отрицательным PKCтета, не происходила индукция AP-1 активности при активации посредством PMA.
Другие исследования показали, что PKCтета, через активацию IĸB киназы бета, опосредует активацию NF-ĸB, индуцируемую ко-стимуляцией T-клеточного рецептора/CD28 (N. Coudronniere et al., Proc. Nat. Acad. Sci. U.S.A., 2000, 97, p. 3394; и Lin, X. et al., Mol. Cell. Biol., 2000, 20, p. 2933).
Пролиферация периферических T-клеток у мышей с PKCтета «нокаутом» в ответ на стимуляцию T-клеточного рецептора (TCR)/CD28 была существенно меньше по сравнению с T-клетками мышей дикого типа. Кроме того, количество IL-2, высвобождаемого из T-клеток, также существенно снижалось (Sun, Z. et al., Nature, 2000, 404, p. 402). Также было показано, что PKCтета-дефицитные мыши показывают ослабленное легочное воспаление и гиперчувствительность дыхательных путей (AHR) в мышиной модели Th2-зависимой астмы, с отсутствием дефектов вирусного клиренса и Th1-зависимой цитотоксической T-клеточной функции (Berg-Brown, N.N. et al., J. Exp. Med., 2004, 199, p. 743; Marsland, B.J. et al., J. Exp. Med., 2004, 200, p. 181). Ослабленный Th2 клеточный ответ приводит к пониженным уровням IL-4 и иммуноглобулина E (IgE), способствующим AHR и воспалительной патофизиологии. Иными словами, мыши с PKCтета «нокаутом» казались нормальными и фертильными.
Также существует доказательство того, что PKCтета участвует в IgE рецептор (FcɛRI)-опосредованный ответ тучных клеток (Liu, Y. et al., J. Leukoc. Biol., 2001, 69, p. 831). В человеческих культивированных тучных клетках (HCMC), как было показано, PKC киназная активность быстро локализуется к мембране после FcɛRI перекрестного сшивания (Kimata, M. et al., Biochem. Biophys. Res. Commun., 1999, 257(3), p. 895). Недавно проведенное исследование in vitro активности тучных клеток костного мозга (BMMC), выделенных у дикого типа и PKCтета-дефицитных мышей, показывает, что при FceRI перекрестном сшивании BMMCs PKCтета-дефицитных мышей снижали уровни IL-6, фактора некроза опухоли альфа (TNFα) и IL-13 по сравнению с BMMCs мышей дикого типа, что говорит о потенциальной роли PKCтета в продукции цитокинов тучными клетками, в дополнение к T-клеточной активации (Ciarletta, A.B. et al., poster presentation at the 2005 American Thoracic Society International Conference).
Исследования, указанные выше, и другие исследования подтверждают критическую роль PKCтета в T-клеточной активации и в передачи сигнала тучными клетками (MC). Таким образом, ингибитор PKCтета был бы терапевтически полезным в лечении иммунологических расстройств и других заболеваний, опосредованных несоответствующей активацией T-клеток и передачи сигнала MC.
Было обнаружено, что многие из киназ, независимо от того являются они рецепторными или нерецепторными тирозиновыми киназами или S/T киназами, вовлечены в клеточные сигнальные пути, вовлеченные в различные патогенные состояния, включая иммуномодуляцию, воспаление или пролиферативные расстройства, такие как рак.
Многие аутоиммунные заболевания и заболевание, связанное с хроническим воспалением, а также сильные ответные реакции связывают с чрезмерной или нерегулируемой продукцией или активностью одного или нескольких цитокинов.
Соединения по настоящему изобретению также являются полезными для лечения сердечнососудистых расстройств, таких как острый инфаркт миокарда, острый коронарный синдром, хроническая сердечная недостаточность, инфаркт миокарда, атеросклероз, вирусный миокардит, отторжение сердечного аллотрансплантата и связанная с сепсисом сердечная дисфункция. Кроме того, соединения по настоящему изобретению также являются полезными для лечения расстройств центральной нервной системы, таких как менингококковый менингит, болезнь Альцгеймера и болезнь Паркинсона.
Соединения по настоящему изобретению также являются полезными для лечения глазного расстройства, рака, солидной опухоли, саркомы, фибросаркомы, остеомы, меланомы, ретинобластомы, рабдомиосаркомы, глиобластомы, нейробластомы, тератокарциномы, аллергических реакций, гиперкинетических двигательных расстройств, аллергического пневмонита, гипертензии, гипокинетических двигательных расстройств, аневризмы аорты и периферических сосудов, анализ гипоталамо-гипофизарно-надпочечниковой оси, расслоение аорты, артериальной гипертензии, артериоосклероза, артериовенозной фистулы, атаксии, дегенераций спинного мозга и мозжечка, стрептококкового миозита, структурных поражений мозжечка, субострого склерозирующего панэнцефалита, обморочного состояния, сифилиса сердечнососудистой системы, системной анафилаксии, синдрома системного воспалительного ответа, системного юношеского ревматоидного артрита, T-клеточного или FAB ALL, телеангиэктазии, облитерирующего тромбоангиита, трансплантатов, травмы/кровотечения, аллергических реакций типа III, аллергических реакций типа IV, нестабильной стенокардии, уремии, уросепсиса, крапивницы, порока сердечных клапанов, варикозного расширения вен, васкулита, заболеваний вен, венозного тромбоза, фибрилляции предсердия, вирусных и грибковых инфекций, витального энцефалита/асептического менингита, витально-ассоциируемого гемафагоцитарного синдрома, синдрома Вернике-Корсакова, болезни Вильсона, отторжения ксенотрансплантата любого органа или ткани, отторжения сердечного трансплантата, гемахроматоза, гемодиализа, гемолитического уремического синдрома/тромболитической тромбоцитопенической пурпуры, кровоизлияния, идиопатического легочного фиброза, антитело-опосредованной цитотоксичности, астении, детской спинально-мышечной атрофии, воспаления аорты, гриппа A, воздействия ионизирующей радиации, иридоциклита/увеита/глазного неврита, юношеской спинально-мышечной атрофии, лимфомы, миеломы, лейкоза, злокачественного асцита, гематопоэтического рака, диабетического состояния, такого как глаукома, связанная с инсулин-зависимым сахарным диабетом, диабетическая ретинопатия или микроангиопатия, серповидноклеточной анемии, хронического воспаления, гломерулонефрит, отторжения трансплантата, болезни Лайма, болезни Гиппеля-Линдау, пемфигоида, болезни Паджета, фиброза, саркоидоза, цирроза, тиреоидита, синдрома повышенной вязкости, болезни Рандю-Вебера-Ослера, хронического окклюзивного легочного заболевания, астмы или отека после ожогов, травмы, облучения, удара, гипоксии, ишемии, синдрома гиперстимуляции яичников, пост-перфузионного синдрома, пост-нагнетательного синдрома, пост-MI кардиотомического синдрома, преэклампсии, менометроррагии, эндометриоза, легочной гипертензии, детской гемангиомы или инфекции, вызванной вирусом Herpes simplex, Herpes Zoster, вирусом иммунодефицита человека, парапоксвирусом, протозойной инфекции или токсоплазмоза, прогрессивного супрануклеарного паралича, первичной легочной гипертензии, лучевой терапии, Виброболезни, болезни Рейно, болезни Рефсума, регулярной узкой QRS тахикардии, реноваскулярной гипертензии, рестриктивной кардиомиопатии, саркомы, старческой хореи, старческой деменции по типу телец Леви, шока, кожного аллотрансплантата, синдрома изменений кожи, глазного или макулярного отека, глазного неоваскулярного заболевания, склерита, радиальной кератотомии, увеита, витрита, близорукости, врожденных ямок в диске зрительного нерва, хронического отторжения сетчатки, осложнений после лечения лазером, конъюнктивита, болезни Старгардта, болезни Илса, ретинопатии, дегенерации желтого пятна, рестеноза, ишемического/реперфузионного поражения, ишемического инсульта, окклюзии сосуда, обструктивного заболевания сонной артерии, язвенного колита, воспалительного заболевания кишечника, диабета, сахарного диабета, инсулин-зависимого сахарного диабета, аллергических заболеваний, склеродермического дерматита, болезни “трансплантат-против-хозяина”, отторжения трансплантата органа (включая, но не ограничиваясь этим, отторжение трансплантата костного мозга и твердого органа), острого или хронического иммунного заболевания, связанного с трансплантацией органа, саркоидоза, диссеминирующей внутрисосудистой коагуляции, болезни Кавасаки, нефротического синдрома, синдром хронической усталости, грануломатоза Вегенера, пурпуры Геноха-Шенлейна, микроскопического васкулита почек, хронического активного гепатита, спетического шока, синдрома токсического шока, синдрома сепсиса, кахексии, инфекционных заболеваний, паразитических болезней, синдрома приобретенного иммунодефицита, острого поперечного миелита, хореи Гентингтона, удара, первичного билиарного цирроза, гемолитической анемии, злокачественных опухолей, болезни Аддисона, идиопатической болезни Аддисона, спорадического заболевания, полигландулярного дефицита типа I и полигландулярного дефицита типа II, синдрома Шмидта, (острого) респираторного дистресс-синдрома взрослых, алопеции, гнездной алопеции, серонегативной артропатии, артропатии, болезни Рейтера, псориатической артропатии, язвенной колитной артропатии, энтеропатического синовит, вызванной хламидиями, йерсиниями и сальмонеллой артропатии, атероматозного заболевания/артериоосклероза, атопической аллергии, аутоиммунного буллезного заболевания, пузырчатки обыкновенной, пузырчатки листовидной, пемфигоида, заболевания, связанного с линейным IgA, аутоиммунной гемолитической анемии, положительной гемолитической анемии Кумбса, приобретенной пернициозной анемии, юношеской пернициозной анемии, расстройств периферических сосудов, перитонита, пернициозной анемии, миалгического энцефалита/болезни Royal Free, хронического слизисто-кожного кандидоза, гигантоклеточного артерита, первичного склерозирующего гепатита, криптогенного аутоиммунного гепатита, Синдрома Приобретенного Иммунодефицита, Заболеваний, Связанных с Синдромом Приобретенного Иммунодефицита, Гепатита A, Гепатита B, Гепатита C, аритмий, связанных с пучком Гиса, ВИЧ инфекции/ВИЧ невропатии, вариабельного неклассифицируемого иммунодефицита (вариабельной неклассифицируемой гипогаммаглобулинемии), расширенной кардиомиопатии, женского бесплодия, угасания функции яичников, предждевременного угасания функции яичников, фиброзного легочного заболевания, заживления хронических ран, криптогенного фиброзного альвеолита, пост-воспалительного интерстициального легочного заболевания, интерстициального пневмонита, вызванной pneumocystis carinii пневмонии, пневмонии, интерстициального легочного заболевания, связанного с заболеванием соединительной ткани, смешанного заболевания соединительной ткани, ассоциированного легочного заболевания, связанного с системным склерозом интерстициального легочного заболевания, связанного с ревматоидным артритом интерстициального легочного заболевания, связанного с системной красной волчанкой легочного заболевания, связанного с дерматомиозитом/полимиозитом легочного заболевания, связанного с болезнью Шегрена легочного заболевания, связанного с анкилоизирующим спондилитом легочного заболевания, васкулитного диффузного легочного заболевания, связанного с гемосидерозом легочного заболевания, лекарственно-индуцированного интерстициального легочного заболевания, лучевого фиброза, облитерирующего бронхиолита, хронической эозинофильной пневмонии, лимфоцитарного инфильтрирующего легочного заболевания, постинфекционного интерстициального легочного заболевания, подагрического артрита, аутоиммунного гепатита, аутоиммунного гепатита типа-1 (классический аутоиммунный или люпозный гепатит), аутоиммунного гепатита типа-2 (гепатит с анти-LKM антителом), аутоиммунно-опосредованной гипогликемии, типа B инсулинорезистентности с acanthosis nigricans, гипопаратиреоидизма, острого иммунного заболевания, связанного с трансплантацией органа, хронического иммунного заболевания, связанного с трансплантацией органа, остеоартрита, первичного склерозирующего холангита, псориаза типа 1, псориаза типа 2, идиопатической лейкопении, аутоиммунной нейтропении, почечного заболевания NOS, гломерулонефрита, микроскопического васкулита почек, болезни Лайма, дискоидной красной волчанки, мужского бесплодия идиопатического или NOS, спермаутоиммунитета, рассеянного склероза (всех подтипов), симпатической офтальмии, легочной гипертензии, вторичному заболеванию соединительной ткани, острой и хронической боли (различных форм боли), синдрома Гудпасчура, легочного проявления узелкового полиартерита, острой ревматической лихорадки, ревматоидного спондилита, болезни Стилла, системного склероза, синдрома Шегрена, болезни Такаясу/артерита, аутоиммунной тромбоцитопении, токсичности, трансплантатов и заболеваний, связанных с аномальной васкуляризацией, например, диабетической ретинопатии, юношеской ретинопатии, хороидальной неоваскуляризации из-за возрастной дегенерации желтого пятна и детских гемангиом у людей. Кроме того, такие соединения могут быть полезными для лечения расстройств, такие как асцит, выпоты и экссудаты, включая например, макулярный отек, церебральный отек, острое легочное поражение, респираторный дистресс-синдром взрослых (ARDS), пролиферативные расстройства, такие как рестеноз, фиброзные расстройства, такие как цирроз печени и атеросклероы, мезангиальные клеточнопролиферативные расстройства, такие как диабетическая нефропатия, злокачественный нефросклероз, синдромы тромботической микроангиопатии и гломерулопатии, миокардиальный ангиогенез, коронарный и церебральный коллатерали, ангиогенез ишемических конечностей, ишемическое/реперфузионное поражение, заболевания, связанные с пептической язвой, вызванной Helicobacter, вирусно-индуцированные ангиогенные расстройства, преэклампсия, менометроррагия, лихорадка, вызванная инфекцией, передаваемой кошками через царапины, покраснение радужной оболочки, неоваскулярная глаукома и ретинопатии, например, связанные с диабетической ретинопатией, юношеской ретинопатией или возрастной дегенерацией желтого пятна. Кроме того, эти соединения можно использовать в качестве активных средств против гиперпролиферативных расстройств, таких как гиперплазия щитовидной железы (в частности, болезню Грейва) и кисты (такие как гиперваскулярность стромы яичников, характерная для синдрома поликистозного яичника (синдром Штейна-Левенталя), и поликистозное почечное заболевание, поскольку такие заболевания требуют пролиферации клеток кровеносных сосудов для роста и/или метастазов.
Соединения формулы (I) или формулы (II) по настоящему изобретению можно использовать отдельно или в сочетании с дополнительным средством, например, терапевтическим средством, при этом указанное дополнительное средство выбирает специалист в данной области для предполагаемого применения. Например, дополнительное средство может представлять собой терапевтическое средство признанное как полезное для лечения заболевания или состояния, которое можно лечить соединением по настоящему изобретению. Дополнительное средство также может представлять собой средство, которое придает выгодные свойства терапевтической композиции, например, средство, которое влияет на вязкость композиции.
Также должно быть понятно, что комбинации, которые могут быть включены в настоящее изобретение, представляют собой такие комбинации, которые полезны для их предполагаемого назначения. Средства, описанные ниже, являются иллюстративными для таких назначений, и их не следует рассматривать как ограничение. Комбинации, которые являются частью настоящего изобретения, могут представлять собой соединения по настоящему изобретению и, по меньшей мере, одно дополнительное средство, выбранное из перечисленных ниже. Такая комбинация также может включать более одного дополнительного средства, например, два или три дополнительных средства, если комбинация представляют собой такую, что образуемая композиция способна выполнять свою предполагаемую функцию.
Предпочтительные комбинации включают нестероидное противовоспалительное лекарственное средство(средства), также указываемое как НСПВЛС, которое включает такие лекарственные средства как ибупрофен. Другие предпочтительные комбинации включают кортикостероиды, включая преднизолон; хорошо известные побочные эффекты применения стероидов можно уменьшить или даже устранить путем постепенного уменьшения дозы стероида, необходимой при лечении пациентов в сочетании с соединениями по настоящему изобретению. Неограничивающие примеры терапевтических средств для лечения ревматоидного артрита, с которыми можно сочетать соединение формулы (I) или формулы (II) по настоящему изобретению, включают следующие: цитокин-супрессивное противовоспалительное лекарственное средство(средства) (CSAIDs); антитела к (или антагонисты) другим человеческим цитокинам или ростовым факторам, например, TNF, LT, IL-1, IL-2, IL-3, IL-4, IL-5, IL-6, IL-7, IL-8, IL-12, IL-15, IL-16, IL-21, IL-23, интерферонам, EMAP-II, GM-CSF, FGF и PDGF. Соединения по настоящему изобретению можно использовать в сочетании с антителами к клеточноповерхностным молекулам, таким как CD2, CD3, CD4, CD8, CD25, CD28, CD30, CD40, CD45, CD69, CD80 (B7.1), CD86 (B7.2), CD90, CTLA или их лигандам, включая CD154 (gp39 или CD40L).
Предпочтительные комбинации терапевтических средств могут влиять на различные точки в аутоиммунном и последующем воспалительном каскаде; предпочтительные примеры включают антагонисты TNF, такие как химерные, гуманизированные или человеческие антитела к TNF, D2E7 (Патент США № 6090382, HUMIRATM), CA2 (REMICADETM), SIMPONITM (голимумаб), CIMZIATM, ACTEMRATM , CDP 571 и растворимые p55 или p75 TNF рецепторы, их производные (p75TNFR1gG (ENBRELTM) или p55TNFR1gG (Ленерсепт), а также ингибиторы TNFα превращающего фермента (TACE); подобным образом, ингибиторы IL-1 (ингибиторы Интерлейкин-1-превращающего фермента, IL-1RA и т.п.) могут быть эффективными по тем же причинам. Другие предпочтительные комбинации включают Интерлейкин 11. Следующие предпочтительные комбинации включают других ключевых игроков аутоиммунного ответа, которые могут действовать параллельно, зависимо от, или сообща с IL-18 функцией; особенно предпочтительными являются IL-12 антагонисты, включая антитела к IL-12 или растворимые IL-12 рецепторы или IL-12-связывающие белки. Было показано, что IL-12 и IL-18 имеют перекрывающиеся, но индивидуальные особые функции, и комбинация антагонистов с обоими этими средствами может быть наиболее эффективной. Еще одна предпочтительная комбинация включает неистощающие анти-CD4 ингибиторы. Следующие предпочтительные комбинации включают средства, являющиеся антагонистами ко-стимуляторного пути CD80 (B7.1) или CD86 (B7.2), включая антитела, растворимые рецепторы или антагонистические лиганды.
Соединение формулы (I) или формулы (II) по настоящему изобретению также можно использовать в сочетании со средствами, такими как метотрексат, 6-меркаптопурин, азатиоприн сульфасалазин, месалазин, олсалазин хлорхинин/ гидроксихлорохин, пенцилламин, ауротиомалат (внутримышечный и пероральный), азатиоприн, кохицин, кортикостероиды (пероральные, для ингаляции и местной инъекции), агонисты бета-2 адренорецептора (салбутамол, тербуталин, салметерал), ксантины (теофиллин, аминофиллин), хромогликат, недокромил, кетотифен, ипратропиум и окситропиум, циклоспорин, FK506, рапамицин, микофенолят мофетил, лефлуномид, НСПВЛС, например, ибупрофен, кортикостероиды, такие как преднизолон, ингибиторы фосфодиэстеразы, агонисты аденозина, антитромботические средства, ингибиторы комплемента, адренергические средства, средства, которые препятствуют передаче сигнала или IL-1 (например, ингибиторы NIK, провоспалительными цитокинами, такими как TNFα IKK, p38 или MAP киназы), ингибиторы IL-1β превращающего фермента, ингибиторы передачи сигнала T-клетками, такие как ингибиторы киназы, ингибиторы металлопротеиназы, сульфасалазин, 6-меркаптопурины, ингибиторы ангиотензин-превращающего фермента, растворимые рецепторы цитокинов и их производные (например, растворимые p55 или p75 TNF рецепторы и производные p75TNFRIgG (EnbrelTM) и p55TNFRIgG (Ленерсепт), sIL-1RI, sIL-1RII, sIL-6R), противовоспалительные цитокины (например, IL-4, IL-10, IL-11, IL-13 и TGFβ), целекоксиб, фолиевая кислота, гидроксихлорохин сульфат, рофекоксиб, этанерсепт, инфликсимаб, напроксен, валдекоксиб, сульфасалазин, метилпреднизолон, мелоксикам, метилпреднизолон ацетат, золото натрий тиомалат, аспирин, триамцинолон ацетонид, пропоксифен напсилат/апап, фолат, набуметон, диклофенак, пироксикам, этодолак, диклофенак натрий, оксапрозин, оксикодон HCl, гидрокодон битартрат/апап, диклофенак натрий/мисопростол, фентанил, анакинра, трамадол HCl, салсалат, сулиндак, цианокобаламин/fa/иридоксин, ацетаминофен, алендронат натрия, преднизолон, морфин сульфат, лидокаин гидрохлорид, индометацин, глюкозамин сульф/хондроитин, амитриптилин HCl, сульфадиазин, оксикодон HCl/ацетаминофен, олопатадин HCl мисопростол, напроксен натрий, омепразол, циклофосфамид, ритуксимаб, IL-1 TRAP, MRA, CTLA4-IG, IL-18 BP, анти-IL-12, Анти-IL15, BIRB-796, SCIO-469, VX-702, AMG-548, VX-740, Рофлумиласт, IC-485, CDC-801, S1P1 агонисты (такие как FTY720), ингибиторы PKC семейства (такие как Рубоксистаурин или AEB-071) и Мезопрам. Предпочтительные комбинации включают метотрексат или лефлуномид и, в случаях средней тяжести или тяжелого ревматоидного артрита, циклоспорин и анти-TNF антитела, указанные выше.
Неограничивающие примеры терапевтических средств для лечения воспалительного заболевания кишечника, с которыми можно сочетать соединение формулы (I) или формулы (II) по настоящему изобретению, включают следующие: буденозид; эпидермальный ростовый фактор; кортикостероиды; циклоспорин, сульфасалазин; аминосалицилаты; 6-меркаптопурин; азатиоприн; метронидазол; ингибиторы липоксигеназы; месаламин; олсалазин; балсалазид; антиоксиданты; ингибиторы тромбоксана; антагонисты IL-1 рецептора; анти-IL-1β моноклональные антитела; анти-IL-6 моноклональные антитела; ростовые факторы; ингибиторы эластазы; пиридинил-имидазольные соединения; антитела к (или антагонисты) другим человеческим цитокинам или ростовым факторам, например, TNF, LT, IL-1, IL-2, IL-6, IL-7, IL-8, IL-12, IL-15, IL-16, IL-23, EMAP-II, GM-CSF, FGF и PDGF; клеточноповерхностные молекулы, такие как CD2, CD3, CD4, CD8, CD25, CD28, CD30, CD40, CD45, CD69, CD90 или их лиганды; метотрексат; циклоспорин; FK506; рапамицин; микофенолят мофетил; лефлуномид; НСПВЛС, например, ибупрофен; кортикостероиды, такие как преднизолон; ингибиторы фосфодиэстеразы; агонисты аденозина; антитромботические средства; ингибиторы комплемента; адренергические средства; средства, которые препятствуют передаче сигнала провоспалительными или IL-1 (например, ингибиторы NIK, IKK или MAP цитокинами, такими как TNFα киназы); ингибиторы IL-1β-превращающего фермента; ингибиторы TNFα-превращающего фермента; ингибиторы передачи сигнала T-клетками, такие как ингибиторы киназы; ингибиторы металлопротеиназы; сульфасалазин; азатиоприн; 6-меркаптопурины; ингибиторы ангиотензин-превращающего фермента; растворимые рецепторы цитокинов и их производные (например, растворимые p55 или p75 TNF рецепторы, sIL-1RI, sIL-1RII, sIL-6R) и противовоспалительные цитокины (например, IL-4, IL-10, IL-11, IL-13 и TGFβ). Предпочтительные примеры терапевтических средств для лечения болезни Крона, в комбинации с которыми можно использовать соединение формулы (I) или формулы (II), включают следующие: антагонисты TNF, например, анти-TNF антитела, D2E7 (Патент США № 6090382, HUMIRATM), CA2 (REMICADETM), CDP 571, TNFR-Ig конструкции, ингибиторы (p75TNFRIgG (ENBRELTM) и p55TNFRIgG (ЛЕНЕРСЕПТTM) и ингибиторы PDE4. Соединение формулы (I) или формулы (II) можно использовать в сочетании с кортикостероидами, например, такими как буденозид и дексаметазон; сульфасалазин, 5-аминосалициловая кислота; олсалазин; и средствами, которые препятствуют синтезу или действию провоспалительных цитокинов, таких как IL-1, например, такими как ингибиторы IL-1β-превращающего фермента и IL-1ra; ингибиторы передачи сигнала Т-клетками, например, ингибиторы тирозиновой киназы; 6-меркаптопурин; IL-11; месаламин; преднизон; азатиоприн; меркаптопурин; инфликсимаб; метилпреднизолон натрия сукцинат; дифеноксилат/атроп сульфат; лоперамид гидрохлорид; метотрексат; омепразол; фолат; ципрофлоксацин/декстроза-воды; гидрокодон битартрат/апап; тетрациклин гидрохлорид; флуоцинонид; метронидазол; тимеросал/борная кислота; холестирамин/сахарозы; ципрофлоксацин гидрохлорид; гиосциамин сульфат; меперидин гидрохлорид; мидазолам гидрохлорид; оксикодон HCl/ацетаминофен; прометазин гидрохлорид; фосфат натрия; сульфаметоксазол/тримтеоприм; целекоксиб; поликарбофил; пропоксифен напсилат; гидрокортизон; поливитамины; балсалазид динатрий; кодеин фосфат/апап; колесевелам HCl; цианокобаламин; фолиевая кислота; левофлоксацин; метилпреднизолон; натализумаб и интерферон-гамма.
Неограничивающие примеры терапевтических средств для лечения рассеянного склероза, в комбинации с которыми можно использовать соединение формулы (I) или формулы (II), включают следующие: кортикостероиды; преднизолон; метилпреднизолон; азатиоприн; циклофосфамид; циклоспорин; метотрексат; 4-аминопиридин; тизанидин; интерферон-β1a (AVONEX®; Biogen); интерферон-β1b (БЕТАSERON®; Chiron/Berlex); интерферон α-n3) (Интерферон Sciences/Fujimoto), интерферон-α (Alfa Wassermann/J&J), интерферон β1A-IF (Serono/Inhale Терапевтическs), Пэгинтерферон α 2b (Enzon/Schering-Plough), Сополимер 1 (Cop-1; КОПАКСОН®; Teva Pharmaceutical Industries, Inc.); кислород под повышенным давлением; внутривенный иммуноглобулин; кладрибин; антитела к (или антагонисты) других человеческих цитокинов или ростовых факторов и их рецепторов, например, TNF, LT, IL-1, IL-2, IL-6, IL-7, IL-8, IL-12, IL-23, IL-15, IL-16, EMAP-II, GM-CSF, FGF и PDGF. Соединение формулы (I) или формулы (II) можно использовать в сочетании с антителами к клеточноповерхностным молекулам, таким как CD2, CD3, CD4, CD8, CD19, CD20, CD25, CD28, CD30, CD40, CD45, CD69, CD80, CD86, CD90, или их лигандам. Соединение формулы (I) или формулы (II) также можно использовать в сочетании со средствами, такими как метотрексат, циклоспорин, FK506, рапамицин, микофенолят мофетил, лефлуномид, S1P1 агонист, НСПВЛС, например, ибупрофен, кортикостероиды, такие как преднизолон, ингибиторы фосфодиэстеразы, агонисты аденозина, антитромботические средства, ингибиторы комплемента, адренергические средства, средства, которые препятствуют передаче сигнала провоспалительными цитокинами, такими как TNFα или IL-1 (например, ингибиторы NIK, IKK, p38 или MAP киназ), ингибиторы IL-1β TNFα превращающего фермента, ингибиторы TACE, ингибиторы передачи сигнала Т-клетками, такие как ингибиторы киназы, ингибиторы металлопротеиназы, сульфасалазин, азатиоприн, 6-меркаптопурины, ингибиторы ангиотензин-превращающего фермента, растворимые рецепторы цитокинов и их производные (например, растворимые p55 или p75 TNF рецепторы, sIL-1RI, sIL-1RII, sIL-6R) и противовоспалительные цитокины (например, IL-4, IL-10, IL-13 и TGFβ).
Предпочтительные примеры терапевтических средств для лечения рассеянного склероза, которые можно использовать в комбинации с соединением формулы (I) или формулы (II), включают интерферон-β, например, IFNβ1a и IFNβ1b; копаксон, кортикостероиды, ингибиторы каспазы, например, ингибиторы каспазы-1, ингибиторы IL-1, ингибиторы TNF и антитела к CD40 лиганду и CD80.
Соединение формулы (I) или формулы (II) также можно использовать в сочетании со средствами, такими как алемтузумаб, дронабинол, даклизумаб, митоксантрон, ксалипроден гидрохлорид, фампридин, глатирамер ацетат, натализумаб, синнабидол, α-иммунокин NNSO3, ABR-215062, AnergiX.MS, антагонисты хемокиновых рецепторов, BBR-2778, калагуалин, CPI-1189, LEM (инкапсулированный в липосомах митоксантрон), THC.CBD (агонист каннабиноида), MBP-8298, мезопрам (PDE4 ингибитор), MNA-715, анти-IL-6 рецептор антитело, нейровакс, пирфенидон аллотрап 1258 (RDP-1258), sTNF-R1, талампанел, терифлуномид, TGF-бета2, типлимотид, антагонисты VLA-4 (например, TR-14035, VLA4 Ultrahaler, Antegran-ELAN/Biogen), антагонисты интерферона гамма и агонисты IL-4.
Неограничивающие примеры терапевтических средств для лечения анкилоизирующего спондилита, в комбинации с которыми можно использовать соединение формулы (I) или формулы (II), включают следующие: ибупрофен, диклофенак, мисопростол, напроксен, мелоксикам, индометацин, диклофенак, целекоксиб, рофекоксиб, сульфасалазин, метотрексат, азатиоприн, миноциклин, преднизон и анти-TNF антитела, D2E7 (Патент США № 6090382; HUMIRATM), CA2 (REMICADETM), CDP 571, TNFR-Ig конструкции, (p75TNFRIgG (ENBRELTM) и p55TNFRIgG (ЛЕНЕРСЕПТTM)
Неограничивающие примеры терапевтических средств для лечения астмы, в комбинации с которыми можно использовать соединение формулы (I) или формулы (II), включают следующие: албутерол, салметерол/флутиказон, монтелукаст натрий, флутиказон пропионат, будесонид, преднизон, салметерол ксинафоат, левалбутерол HCl, албутерол сульфат/ипратропиум, преднизолон натрий фосфат, триамцинолон ацетонид, беклометазон дипропионат, ипратропиум бромид, азитромицин, пирбутерол ацетат, преднизолон, теофиллин безводный, метилпреднизолон натрий сукцинат, кларитромицин, зафирлукаст, формотерол фумарат, противогриппозная вакцина, амоксициллин тригидрат, флунизолид, противоаллергическое средство для инъекций, хромолин натрий, фексофенадин гидрохлорид, флунизолид/ментол, амоксициллин/клавуланат, левофлоксацин, устройство для ингаляции, гвайфенесин, дексаметазон натрий фосфат, моксифлоксацин HCl, доксициклин гиклат, гвайфенесин/d-меторфан, p-эфедрин/cod/хлорфенил, гатифлоксацин, цетиризин гидрохлорид, мометазон фуроат, салметерол ксинафоат, бензонатат, цефалексин, пе/гидрокодон/хлорфенил, цетиризин HCl/псевдоэфед, фенилефрин/код/прометазин, кодеин/прометазин, цефпрозил, дексаметазон, гвайфенесин/псевдоэфедрин, хлорфениламин/гидрокодон, недокромил натрий, тербуталин сульфат, эпинефрин, метилпреднизолон, анти-IL-13 антитело и метапротеренол сульфат.
Неограничивающие примеры терапевтических средств для лечения COPD, в комбинации с которыми можно использовать соединение формулы (I) или формулы (II), включают следующие: албутерол сульфат/ипратропиум, ипратропиум бромид, салметерол/флутиказон, албутерол, салметерол ксинафоат, флутиказон пропионат, преднизон, теофиллин безводный, метилпреднизолон натрий сукцинат, монтелукаст натрий, будесонид, формотерол фумарат, триамцинолон ацетонид, левофлоксацин, гвайфенесин, азитромицин, беклометазон дипропионат, левалбутерол HCl, флунизолид, цефтриаксон натрий, амоксициллин тригидрат, гатифлоксацин, зафирлукаст, амоксициллин/клавуланат, флунизолид/ментол, хлорфениламин/гидрокодон, метапротеренол сульфат, метилпреднизолон, мометазон фуроат, п-эфедрин/код/хлорфенил, пирбутерол ацетат, п-эфедрин/лоратадин, тербуталин сульфат, тиотропиум бромид, (R,R)-формотерол, TgAAT, циломиласт и рофлумиласт.
Неограничивающие примеры терапевтических средств для лечения HCV, в комбинации с которыми можно использовать соединение формулы (I) или формулы (II), включают следующие: Интерферон-альфа-2α, Интерферон-альфа-2β, Интерферон-альфа con1, Интерферон-альфа-n1, пэгилированный интерферон-альфа-2α, пэгилированный интерферон-альфа-2β, рибавирин, пэгинтерферон альфа-2b + рибавирин, урсодезоксихолевая кислота, глицирризиновая кислота, тимальфасин, Максамин, VX-497 и любые соединения, которые используют для лечения HCV через вмешательство в следующие мишени: HCV полимераза, HCV протеаза, HCV геликаза и HCV IRES (внутренний сайт входа в рибосому).
Неограничивающие примеры терапевтических средств для лечения Идиопатического Легочного Фиброза, в комбинации с которыми можно использовать соединение формулы (I) или формулы (II), включают следующие: преднизон, азатиоприн, албутерол, колхицин, албутерол сульфат, дигоксин, гамма интерферон, метилпреднизолон натрий сукцинат, лоразепам, фуросемид, лисиноприл, нитроглицерин, спиронолактон, циклофосфамид, ипратропиум бромид, актиномицин d, альтеплаза, флутиказон пропионат, левофлоксацин, метапротеренол сульфат, морфин сульфат, оксикодон HCl, хлорид калия, триамцинолон ацетонид, такролимус безводный, кальций, интерферон-альфа, метотрексат, микофенолят мофетил и интерферон-гамма-1β.
Неограничивающие примеры терапевтических средств для лечения инфаркта миокарда, в комбинации с которыми можно использовать соединение формулы (I) или формулы (II), включают следующие: аспирин, нитроглицерин, метопролол тартрат, эноксапарин натрий, гепарин натрий, клопидогрел бисульфат, карведилол, атенолол, морфин сульфат, метопролол сукцинат, варфарин натрий, лисиноприл, изосорбид мононитрат, дигоксин, фуросемид, симвастатин, рамиприл, тенектеплаза, эналаприл малеат, торсемид, ретаваза, лосартан калий, хинаприл гидрохлорид/карбонат магния, буметанид, альтеплаза, эналаприлат, амиодарон гидрохлорид, тирофибан HCl м-гидрат, дилтиазем гидрохлорид, каптоприл, ирбесартан, валсартан, пропранолол гидрохлорид, фосиноприл натрий, лидокаин гидрохлорид, эптифибатид, цефазолин натрий, атропин сульфат, аминокапроновая кислота, спиронолактон, интерферон, соталол гидрохлорид, хлорид калия, докусат натрий, добутамин HCl, алпразолам, правастатин натрий, аторвастатин кальций, мидазолам гидрохлорид, меперидин гидрохлорид, изосорбид динитрат, эпинефрин, допамин гидрохлорид, бивалирудин, росувастатин, эзетимиб/симвастатин, авасимиб и карипорид.
Неограничивающие примеры терапевтических средств для лечения псориаза, в комбинации с которыми можно использовать соединение формулы (I) или формулы (II), включают следующие: кальципотриен, клобетасол пропионат, триамцинолон ацетонид, галогенбетасол пропионат, тазаротен, метотрексат, флуоцинонид, бетаметазон дипроп-усиленный, флуоцинолон ацетонид, ацитретин, содержащий деготь шампунь, бетаметазон валерат, мометазон фуроат, кетоконазол, прамоксин/флуоцинолон, гидрокортизон валерат, флурандренолид, мочевина, бетаметазон, клобетасол пропионат/эмолл, флутиказон пропионат, азитромицин, гидрокортизон, увлажняющая формула, фолиевая кислота, десонид, пимекролимус, каменноугольная смола, дифлоазон диацетат, этанерсепт фолат, молочная кислота, метоксален, hc/висмут субгал/знокс/резор, метилпреднизолон ацетат, преднизон, солнцезащитный крем, галцинонид, салициловая кислота, антралин, клокортолон пивалат, угольный экстракт, каменноугольная смола/салициловая кислота, каменноугольная смола/салициловая кислота/сера, дезоксиметазон, диазепам, мягчительное средство, флуоцинонид/мягчительное средство, минеральное масло/касторовое масло/na lact, минеральное масло/арахисовое масло, петролейный эфир/изопропилмиристат, псорален, салициловая кислота, мыло/трибромсалан, тимеросал/борная кислота, целекоксиб, инфликсимаб, циклоспорин, алефасепт, эфализумаб, такролимус, пимекролимус, PUVA, UVB, сульфасалазин, ABT-874 и устекинамаб.
Неограничивающие примеры терапевтических средств для лечения псориатического артрита, в комбинации с которыми можно использовать соединение формулы (I) или формулы (II), включают следующие: метотрексат, этанерсепт, рофекоксиб, целекоксиб, фолиевую кислот, сульфасалазин, напроксен, лефлуномид, метилпреднизолон ацетат, индометацин, гидроксихлорохин сульфат, преднизон, сулиндак, бетаметазон дипроп-усиленный, инфликсимаб, метотрексат, фолат, триамцинолон ацетонид, диклофенак, диметилсульфоксид, пироксикам, диклофенак натрий, кетопрофен, мелоксикам, метилпреднизолон, набуметон, толметин натрий, кальципотриен, циклоспорин, диклофенак натрий/мисопростол, флуоцинонид, глюкозамин сульфат, золото натрий тиомалат, гидрокодон битартрат/апап, ибупрофен, риседронат натрий, сульфадиазин, тиогуанин, валдекоксиб, алефасепт, D2E7 (Патент США № 6090382, HUMIRATM) и эфализумаб.
Неограничивающие примеры терапевтических средств для рестеноза, в комбинации с которыми можно использовать соединение формулы (I) или формулы (II), включают следующие: сиролимус, паклитаксел, эверолимус, такролимус, ABT-578 и ацетаминофен.
Неограничивающие примеры терапевтических средств для лечения ишиаса, в комбинации с которыми можно использовать соединение формулы (I) или формулы (II), включают следующие: гидрокодон битартрат/апап, рофекоксиб, циклобензаприн HCl, метилпреднизолон, напроксен, ибупрофен, оксикодон HCl/ацетаминофен, целекоксиб, валдекоксиб, метилпреднизолон ацетат, преднизон, кодеин фосфат/апап, трамадол hcl/ацетаминофен, матаксалон, мелоксикам, метокарбамол, лидокаин гидрохлорид, диклофенак натрий, габапентин, дексаметазон, карисопродол, кеторолак трометамин, индометацин, ацетаминофен, диазепам, набуметон, оксикодон HCl, тизанидин HCl, диклофенак натрий/мисопростол, пропоксифен н-пап, аза/оксикод/оксикодон тер, ибупрофен/гидрокодон бит, трамадол HCl, этодолак, пропоксифен HCl, амитриптилин HCl, карисопродол/кодеин фос/аза, морфин сульфат, поливитамины, напроксен натрий, орфенадрин цитрат и темазепам.
Предпочтительные примеры терапевтических средств для лечения SLE (волчанки), в комбинации с которыми можно использовать соединение формулы (I) или формулы (II), включают следующие: НСПВЛС, например, диклофенак, напроксен, ибупрофен, пироксикам, индометацин; ингибиторы COX2, например, целекоксиб, рофекоксиб, валдекоксиб; противомалярийные средства, например, гидроксихлорохин; стероиды, например, преднизон, преднизолон, буденозид, дексаметазон; цитотоксические средства, например, азатиоприн, циклофосфамид, микофенолят мофетил, метотрексат; ингибиторы PDE4 или ингибитор синтеза пурина, например, Cellcept®. Соединение формулы (I) или формулы (II) также можно использовать в сочетании со средствами, такими как сульфасалазин, 5-аминосалициловая кислота, олсалазин, Имуран® и средства, которые препятствуют синтезу, продукции или действию провоспалительных цитокинов, такие как IL-1, например, ингибиторы каспазы, такие как ингибиторы IL-1β-превращающего фермента и IL-1ra. Соединение формулы (I) или формулы (II) также можно использовать с ингибиторами передачи сигнала Т-клетками, например, ингибиторами тирозиновой киназы; или молекулами, которые прицельно действуют на молекулы T-клеточной активации, например, CTLA-4-IgG или антитела против B7 семейства, антитела против PD-1 семейства. Соединение формулы (I) или формулы (II) можно использовать в сочетании с IL-11 или анти-цитокиновыми антителами, например, фонолизумаб (анти-IFNg антитело) или антитела к рецептору против рецептора, например, анти-IL-6 рецептор антитело и антитела к B-клеточноповерхностным молекулам. Соединение формулы (I) или формулы (II) также можно использовать с LJP 394 (абетимус), средствами, которые истощают или инактивируют B-клетки, например, Ритуксимаб (анти-CD20 антитело), лимфостат-B (анти-BlyS антитело), антагонисты TNF, например, анти-TNF антитела, D2E7 (Патент США № 6090382; HUMIRATM), CA2 (REMICADETM), CDP 571, TNFR-Ig конструкции, (p75TNFRIgG (ENBRELTM) и p55TNFRIgG (ЛЕНЕРСЕПТTM).
В настоящем изобретении применимы следующие определения:
“Терапевтически эффективное количество” представляет собой такое количество соединения формулы (I) или формулы (II) или комбинации двух или более таких соединений, которое ингибирует, полностью или частично, развитие состояния или облегчает, по меньшей мере, частично один или несколько симптомов этого состояния. Терапевтически эффективное количество также может представлять собой такое количество, которое является профилактически эффективным. Количество, которое является терапевтически эффективным, зависит от размера и пола пациента, состояния, подлежащего лечению, тяжести состояния и результата, который хотят получить. Для конкретного пациента терапевтически эффективн количество можно определить способами, известными специалистам в данной области.
Термин “фармацевтически приемлемые соли” относится к тем солям, которые сохраняют биологическую эффективность и свойства свободных оснований и которые получают путем взаимодействия с неорганическими кислотами, например, хлористоводородной кислотой, бромистоводородной кислотой, серной кислотой, азотной кислотой и фосфорной кислотой, или органическими кислотами, например, сульфоновой кислотой, карбоновой кислотой, органической фосфорной кислотой, метансульфоновой кислотой, этансульфоновой кислотой, п-толуолсульфоновой кислотой, лимонной кислотой, фумаровой кислотой, малеиновой кислотой, янтарной кислотой, бензойной кислотой, салициловой кислотой, молочной кислотой, винной кислотой (например, (+) или (-)-винной кислотой или их смесями), аминокислотами (например, (+) или (-)-аминокислотами или их смесями) и подобными. Эти соли можно получить способами, известными специалистам в данной области.
Некоторые соединения формулы (I) или формулы (II), которые содержат кислотные заместители, могут существовать в виде солей с фармацевтически приемлемыми основаниями. Настоящее изобретение включает такие соли. Примеры таких солей включают натриевые соли, калиевые соли, соли лизина и соли аргинина. Эти соли можно получить способами, известными специалистам в данной области.
Некоторые соединения формулы (I) или формулы (II) и их соли могут существовать в более чем одной кристаллической форме, и настоящее изобретение включает каждую кристаллическую форму и их смеси.
Некоторые соединения формулы (I) или формулы (II) и их соли также могут существовать в форме сольватов, например гидратов, и настоящее изобретение включает каждый сольват и их смеси.
Некоторые соединения формулы (I) или формулы (II) могут содержать один или несколько хиральных центров и существовать в различных оптически активных формах. Когда соединения формулы (I) или формулы (II) содержат один хиральный центр, соединения существуют в двух энантиомерных формах, и настоящее изобретение включает оба энантиомера и смеси энантиомеров, такие как рацемические смеси. Энантиомеры можно разделить способами, известными специалистам в данной области, например, путем образования диастереоизомерных солей, которые можно разделить, например, путем кристаллизации; путем образования диастереоизомерных производных или комплексов, которые можно разделить, например, путем кристаллизации, газо-жидкостной или жидкостной хроматографии; путем селективного взаимодействия одного энантиомера с энантиомер-специфическим реагентом, например, путем ферментативной этерификации; или путем газо-жидкостной или жидкостной хроматографии в хиральной среде, например, на хиральном носителе, например, диоксиде кремния со связанным хиральным лигандом или в присутствии хирального растворителя. Должно быть понятно, что, когда желаемый энантиомер преобразуют в другую химическую единицу при помощи одной из процедур разделения, описанных выше, необходима следующая стадия для высвобождения желаемой энантиомерной формы. Альтернативно, специфические энантиомеры можно синтезировать методом асимметрического синтеза с использованием оптически активных реагентов, субстратов, катализаторов или растворителей или путем преобразования одного энантиомера в другой путем асимметрического преобразования.
Когда соединение формулы (I) или формулы (II) содержит более чем один хиральный центр, оно могут существовать в диастереоизомерных формах. Диастереоизомерные соединения можно разделить способами, известными специалистам в данной области, например, хроматографией или кристаллизацией, и индивидуальные энантиомеры можно разделить, как описано выше. Настоящее изобретение включает каждый диастереоизомер соединений формулы (I) или формулы (II) и их смеси.
Некоторые соединения формулы (I) или формулы (II) могут существовать в различных таутомерных формах или в виде различных геометрических изомеров, и настоящее изобретение включает каждый таутомер и/или геометрический изомер соединений формулы (I) или формулы (II) и их смеси.
Некоторые соединения формулы (I) или формулы (II) могут существовать в различных стабильных конформационных формах, которые можно разделить. Торсионная асимметрия из-за ограниченного вращения вокруг асимметрической простой связи, например, из-за пространственного затруднения или деформации кольца делает возможным разделение различных конформеров. Настоящее изобретение включает каждый конформационный изомер соединений формулы (I) или формулы (II) и их смеси.
Некоторые соединения формулы (I) или формулы (II) могут существовать в цвиттерионной форме, и настоящее изобретение включает каждую цвиттерионную форму соединений формулы (I) или формулы (II) и их смеси.
Как он используется в настоящей заявке, термин "пролекарство" относится к средству, которое преобразуется в исходное лекарственное средство in vivo посредством некоторого физиологического химического процесса (например, пролекарство при доведении до физиологического pH преобразуется в желаемое лекарственное средство). Пролекарства часто являются полезными, поскольку, в некоторых ситуациях, они могут быть более удобными для введения, чем исходное лекарственное средство. Они, например, могут быть биодоступны при пероральном введении, тогда как исходное лекарственное средство нет. Пролекарство также может иметь улучшенную растворимость в фармакологических композициях по сравнению с исходным лекарственным средством. Примером пролекарства, без ограничения, может быть соединение по настоящему изобретению, когда его вводят в форме сложного эфира ("пролекарство"), что способствует его транспорту через клеточную мембрану, где водорастворимость не является полезной, но затем оно метаболически гидролизуется до карбоновой кислоты сразу после проникновения внутрь клетки, где водорастворимость является полезной.
Пролекарства имеют много полезных свойств. Например, пролекарство может быть более водорастворимым, чем конечное лекарственное средство, способствуя, таким образом, внутривенному введению лекарственного средства. Пролекарство также может иметь более высокий уровень пероральной биодоступности по сравнению с конечным лекарственным средством. После введения пролекарство ферментативно или химически расщепляется, обеспечивая доставку конечного лекарственного средства в кровь или ткань.
Иллюстративные пролекарства при расщеплении высвобождают соответствующую свободную кислоту, и такие гидролизуемые эфир-образующие остатки соединений по настоящему изобретению включают, но не ограничиваются этим, карбоновокислотные заместители, где свободный водород замещен группой (C1-C4)алкил, (C1-C12)алканоилоксиметил, (C4-C9)1-(алканоилокси)этил, 1-метил-1-(алканоилокси)-этил, содержащий от 5 до 10 атомов углерода, алкоксикарбонилоксиметил, содержащий от 3 до 6 атомов углерода, 1-(алкоксикарбонилокси)этил, содержащий от 4 до 7 атомов углерода, 1-метил-1-(алкоксикарбонилокси)этил, содержащий от 5 до 8 атомов углерода, N-(алкоксикарбонил)аминометил, содержащий от 3 до 9 атомов углерода, 1-(N-(алкоксикарбонил)амино)этил, содержащий от 4 до 10 атомов углерода, 3-фталидил, 4-кротонолактонил, гамма-бутиролактон-4-ил, ди-N,N-(C1-C2)алкиламино(C2-C3)алкил (такой как β-диметиламиноэтил), карбамоил-(C1-C2)алкил, N,N-ди(C1-C2)-алкилкарбамоил-(C1-C2)алкил и пиперидино-, пирролидино- или морфолино(C2-C3)алкил.
Другие иллюстративные пролекарства высвобождают спирт формулы (I) или формулы (II), где свободный водород гидроксильного заместителя (например, R группа содержит гидроксил) замещен группой (C1-C6)алканоилоксиметил, 1-((C1-C6)алканоилокси)этил, 1-метил-1-((C1-C6)алканоилокси)этил, (C1-C12)алкоксикарбонилоксиметил, N-(C1-C6)алкоксикарбониламино-метил, сукциноил, (C1-C6)алканоил, α-амино(C1-C4)алканоил, арилактил и α-аминоацил или α-аминоацил-α-аминоацил, где указанные α-аминоацильные группы независимо выбраны из любой из природных L-аминокислот, обнаруженных в белках, P(O)(OH)2, -P(O)(O(C1-C6)алкил)2 или гликозил (радикал, образованный в результате отщепления гидроксила гемиацеталя углевода).
Как он используется в настоящей заявке, термин "связанная мостиковой связью (C5-C12) циклоалкильная группа” означает насыщенную или ненасыщенную, бициклическую или полициклическую связанную мостиковой связью углеводородную группу, содержащую два или три C3-C10 циклоалкильных кольца. Не связанные мостиковой связью циклоалкилы исключаются. Связанный мостиковой связью циклический углеводород может включать, например бицикло[2.1.1]гексил, бицикло[2.2.1]гептил, бицикло[2.2.2]октил, бицикло[3.2.1]октил, бицикло[4.3.1]децил, бицикло[3.3.1]нонил, борнил, борненил, норборнил, норборненил, 6,6-диметилбицикло [3.1.1]гептил, трициклобутил и адамантил.
Как он используется в настоящей заявке, термин “связанный мостиковой связью (C2-C10) гетероциклил” означает бициклические или полициклические связанные аза-мостиковой связью углеводородные группы, и может включать азанорборнил, хинкулидинил, изохинкулидинил, тропанил, азабицикло[3.2.1]октанил, азабицикло[2.2.1]гептанил, 2-азабицикло[3.2.1]октанил, азабицикло[3.2.1]октанил, азабицикло[3.2.2]нонанил, азабицикло[3.3.0]нонанил и азабицикло [3.3.1]нонанил.
Термин “гетероциклический”, “гетероциклил” или “гетероциклилен”, как он используется в настоящей заявке, включает неароматические кольцевые системы, включая, но не ограничиваясь этим, моноциклические, бициклические, трициклические и спироциклические кольца, которые могут быть полностью насыщенными или которые могут содержать одну или несколько единиц ненасыщенности (во избежание каких-либо сомнений, степень ненасыщенности не приводит к ароматической кольцевой системе) и содержат от 5 до 12 атомов, включая по меньшей мере один гетероатом, такой как азот, кислород или сера. Для целей иллюстрации, что не следует рассматривать как ограничение объема настоящего изобретения, далее представлены примеры гетероциклических колец: азепинил, азетидинил, индолинил, изоиндолинил, морфолинил, пиперазинил, пиперидинил, пирролидинил, хинуклидинил, тиоморфолинил, тетрагидропиранил, тетрагидрофуранил, тетрагидроиндолил, тиоморфолинил и тропанил.
Термин “гетероарил” или “гетероарилен”, как они используются в настоящей заявке, включают ароматические кольцевые системы, включая, но не ограничиваясь этим, моноциклические, бициклические и трициклические кольца, и содержат 5 до 12 атомов, включая, по меньшей мере, один гетероатом, такой как азот, кислород или сера. В целях иллюстрации, что не следует рассматривать как ограничение объема настоящего изобретения: азаиндолил, бензо(b)тиенил, бензимидазолил, бензофуранил, бензоксазолил, бензотиазолил, бензотиадиазолил, бензоксадиазолил, фуранил, имидазолил, имидазопиридинил, индолил, индазолил, изоксазолил, изотиазолил, оксадиазолил, оксазолил, пуринил, пиранил, пиразинил, пиразолил, пиридинил, пиримидинил, пирролил, пирроло[2,3-d]пиримидинил, пиразоло[3,4-d]пиримидинил, хинолинил, хиназолинил, триазолил, тиазолил, тиофенил, тетразолил, тиадиазолил или тиенил.
“Гетероциклоалкильная” группа, как это используется в настоящей заявке, представляет собой гетероциклическую группу, которая связана с соединением посредством алифатической группы, содержащей от одного до около восьми атомов углерода. Например, гетероциклоалкильная группа представляет собой морфолинометильную группу.
Как это используется в настоящей заявке, “алкил“, “алкилен” или условные обозначения, такие как “(C1-C8)” включают линейные или разветвленные углеводороды, которые являются полностью насыщенными. Примерами алкилов являются метил, этил, пропил, изопропил, бутил, пентил, гексил и их изомеры. Как это используется в настоящей заявке, “алкенил”, “алкенилен”, “алкинилен” и “алкинил” означают C2-C8 и включают линейные или разветвленные углеводороды, которые содержат одну или несколько единиц ненасыщенности, одну или несколько двойных связей для алкенила и одну или несколько тройных связей для алкинила.
Как это используется в настоящей заявке, “ароматические” группы (или “арильные” или “ариленовые” группы) включают ароматические карбоциклические кольцевые системы (например, фенил) и конденсированные полициклические ароматические кольцевые системы (например, нафтил, бифенил и 1,2,3,4-тетрагидронафтил).
Как это используется в настоящей заявке, “циклоалкил” или “циклоалкилен” означают C3-C12 моноциклические или полициклические (например, бициклические, трициклические, спироциклические и т.п.) углеводороды, которые являются полностью насыщенными или содержат одну или несколько ненасыщенных связей, до не достигают ароматической группы. Примерами циклоалкильной группы являются циклопропил, циклобутил, циклопентил, циклопентенил, циклогексил и циклогексенил.
Как это используется в настоящей заявке, многие группы или заместители указаны как являющиеся либо “замещенными” либо “необязательно замещенными”. Когда группа модифицирована одним из этих терминов, если не указано иное, это означает, что любая часть этой группы, которая, как это известно специалистам в данной области, является доступной для замещения, может быть замещена и может включать один или несколько заместителей, при этом в случае присутствия более чем одного заместителя каждый заместитель выбран независимо. Такие способы замещения хорошо известны из уровня техники и/или описаны в настоящем раскрытии. Для целей иллюстрации, которые не следует рассматривать как ограничение объема настоящего изобретения, некоторые примеры групп, которые являются заместителями, включают: (C1-C8)алкильные группы, (C2-C8)алкенильные группы, (C2-C8)алкинильные группы, (C3-C10)циклоалкильные группы, галоген (F, Cl, Br или I), галогенированные (C1-C8)алкильные группы (например, но не ограничиваясь этим, -CF3), -O-(C1-C8)алкильные группы, -OH, -S-(C1-C8)алкильные группы, -SH, -NH(C1-C8)алкильные группы, -N((C1-C8)алкил)2 группы, -NH2, -C(O)NH2, -C(O)NH(C1-C8)алкильные группы, -C(O)N((C1-C8)алкил)2, -NHC(O)H, -NHC(O)(C1-C8)алкильные группы, -NHC(O)(C3-C8)циклоалкильные группы, -N((C1-C8)алкил)C(O)H, -N((C1-C8)алкил)C(O)(C1-C8)алкильные группы, -NHC(O)NH2, -NHC(O)NH(C1-C8)алкильные группы, -N((C1-C8)алкил)C(O)NH2 группы, -NHC(O)N((C1-C8)алкил)2 группы, -N((C1-C8)алкил)C(O)N((C1-C8)алкил)2 группы, -N((C1-C8)алкил)C(O)NH((C1-C8)алкил), -C(O)H, -C(O)(C1-C8)алкильные группы, -CN, -NO2, -S(O)(C1-C8)алкильные группы, -S(O)2(C1-C8)алкильные группы, -S(O)2N((C1-C8)алкил)2 группы, -S(O)2NH(C1-C8)алкильные группы, -S(O)2NH(C3-C8)циклоалкильные группы, -S(O)2NH2 группы, -NHS(O)2(C1-C8)алкильные группы, -N((C1-C8)алкил)S(O)2(C1-C8)алкильные группы, -(C1-C8)алкил-O-(C1-C8)алкильные группы, -O-(C1-C8)алкил-O-(C1-C8)алкильные группы, -C(O)OH, -C(O)O(C1-C8)алкильные группы, NHOH, NHO(C1-C8)алкильные группы, -O-галогенированные (C1-C8)алкильные группы (например, но не ограничиваясь этим, -OCF3), -S(O)2-галогенированные (C1-C8)алкильные группы (например, но не ограничиваясь этим, -S(O)2CF3), -S-галогенированные (C1-C8)алкильные группы (например, но не ограничиваясь этим, -SCF3), -(C1-C6) гетероцикл (например, но не ограничиваясь этим, пирролидин, тетрагидрофуран, пиран или морфолин), -(C1-C6) гетероарил (например, но не ограничиваясь этим, тетразол, имидазол, фуран, пиразин или пиразол), -фенил, -NHC(O)O-(C1-C6)алкильные группы, -N((C1-C6)алкил)C(O)O-(C1-C6)алкильные группы, -C(=NH)-(C1-C6)алкильные группы, -C(=NOH)-(C1-C6)алкильные группы или -C(=N-O-(C1-C6)алкил)-(C1-C6)алкильные группы.
 в Формуле (I) представляет собой ароматическое кольцо.
в Формуле (I) представляет собой ароматическое кольцо.
Одно или несколько соединений по настоящему изобретению можно вводить пациенту-человеку как таковые или в фармацевтических композициях, где они смешаны с биологически подходящими носителями или эксципиентом(эксципиентами) в дозах, подходящих для лечения или облегчения заболевания или состояния, описанного в настоящей заявке. Смеси этих соединений также можно вводить пациенту в виде простой смеси или в подходяще сформулированных фармацевтических композициях. Терапевтически эффективная доза относится к такому количеству соединения или соединений, которое является достаточным для обеспечения профилактики или облегчения заболевания или состояния, описанного в настоящей заявке. Способы формулирования и введения соединений, описанных в настоящей заявке, можно найти в справочниках, хорошо известных специалистам в данной области, таких как "Remington's Pharmaceutical Sciences," Mack Publishing Co., Easton, PA, последнее издание.
Подходящие пути введения, например, могут включать пероральный, закапывание в глаз, ректальный, через слизистую оболочку, местное или кишечное введение; парентеральную доставку, включая внутримышечные, подкожные, интрамедуллярные инъекций, а также интратекальные, прямые интравертрикулярные, внутривенные, интраперитонеальные, интраназальные или внутриглазные инъекции.
Альтернативно, можно вводить соединение местным, а не системным путем, например, через инъекцию соединения непосредственно в область отека, часто в виде депо препарата или композиции замедленного высвобождения.
Кроме того, можно вводить лекарственное средство с использованием прицельно направленной системы доставки лекарственного средства, например, в липосомах, покрытых специфическим в отношении эндотелиальных клеток антителом.
Фармацевтические композиции по настоящему изобретению можно получить способом, известным per se, например, обычными способами смешивания, растворения, гранулирования, формирования драже, растирания в порошок, эмульгирования, инкапсулирования, заключения в оболочку или лиофилизации.
Фармацевтические композиции для применения в соответствии с настоящим изобретением, таким образом, могут быть сформулированы традиционным способом с использованием одного или нескольких физиологически приемлемых носителей, включающих эксципиенты и вспомогательные вещества, которые способствуют переработке активных соединений в препараты, которые можно использовать фармацевтически. Подходящая композиция зависит от выбранного пути введения.
Для инъекции, средства по настоящему изобретению могут быть сформулированы в виде водных растворов, предпочтительно в физиологически совместимых буферах, таких как раствор Хэнкса, раствор Рингера или физиологический солевой буфер. Для введения через слизистую оболочку, в композиции используют смачивающие вещества, подходящие для проникновения через определенный барьер. Такие смачивающие вещества хорошо известны из уровня техники.
Для перорального введения, соединения можно легко сформулировать путем объединения активных соединений с фармацевтически приемлемыми носителями, хорошо известными из уровня техники. Такие носители позволяют сформулировать соединения по настоящему изобретению в виде таблеток, пилюль, драже, капсул, жидкостей, гелей, сиропов, взвесей, суспензий и т.п. для перорального введения пациенту, подлежащему лечению. Фармацевтические препараты для перорального применения можно получить путем объединения активного соединения с твердым эксципиентом, необязательного измельчения полученной смеси и переработки смеси гранул, после добавления подходящих вспомогательных веществ, если это желательно, с получением таблеток или сердцевины драже. Подходящими эксципиентами, в частности, являются наполнители, такие как сахара, включая лактозу, сахарозу, маннит или сорбит; целлюлозные препараты, такие как, например, кукурузный крахмал, пшеничный крахмал, рисовый крахмал, картофельный крахмал, желатин, трагакантовая камедь, метилцеллюлоза, гидроксипропилметил-целлюлоза, натрий карбоксиметилцеллюлоза, и/или поливинилпирролидон (PVP). Если это желательно, можно добавить разрыхлители, такие как сшитый поливинилпирролидон, агар или альгиновая кислота или ее соль, такая как альгинат натрия.
Для сердцевины драже предусмотрены подходящие покрытия. Для этих целей, можно использовать концентрированные растворы сахаров, которые, необязательно, могут содержать аравийскую камедь, тальк, поливинилпирролидон, карбополовый гель, полиэтиленгликоль и/или диоксид титана, растворы для глазурирования и подходящие органические растворители или смеси растворителей. Красители или пигменты могут быть добавлены к покрытиям таблеток или драже для идентификации или для характеристики различных комбинаций доз активных соединений.
Фармацевтические препараты, которые можно использовать перорально, включают заполняемые капсулы из желатина, а также мягкие герметичные капсулы, выполненные из желатина и пластификатора, такого как глицерин или сорбит. Заполняемые капсулы могут содержать активные ингредиенты в смеси с наполнителем, таким как лактоза, связующими, такими как крахмалы, и/или смазывающими веществами, такими как тальк или стеарат магния, и, необязательно, стабилизаторы. В мягких капсулах активные соединения могут быть растворены или суспендированы в подходящих жидкостях, таких как жирные масла, жидкий парафин или жидкие полиэтиленгликоли. Кроме того, можно добавить стабилизаторы. Все композициии для перорального введения должны присутствовать в дозах, подходящих для такого введения.
Для буккального введения, композиции могут иметь форму таблеток или лепешек, сформулированных традиционным образом.
Для введения путем ингаляции, соединения для применения в соответствии с настоящим изобретением удобным образом можно доставить в форме аэрозольного спрея из находящихся под давлением упаковок или недулайзера, с использованием подходящего пропеллента, например, дихлордифторметана, трихлорфторметана, дихлортетрафторэтана, диоксида углерода или другого подходящего газа. В случае находящегося под давлением аэрозольного препарата, для определения единицы дозирования предусматривается клапан для доставки отмеренного количества. Могут быть сформулированы капсулы и картриджи, например, из желатина для применения в ингаляторе или инсуффляторе, содержащие порошкообразную смесь соединения и подходящую основу для порошка, такую как лактоза или крахмал.
Соединения могут быть сформулированы для парентерального введения путем инъекции, например, болюсной инъекции или непрерывной инфузии. Композициии для инъекций могут быть представлены в содержащей стандартные дозы лекарственной форме, например, в ампулах или в многодозовых контейнерах, с добавленным консервантом. Композиции могут иметь такие формы как суспензии, растворы или эмульсии в масляных или водных носителях и могут содержать средства для формулирования, такие как суспендирующие, стабилизирующие и/или диспергирующие средства.
Фармацевтические композициии для парентерального введения включают водные растворы активных соединений в водорастворимой форме. Кроме того, суспензии активных соединений можно получить в виде подходящих масляных суспензий для инъекций. Подходящие липофильные растворители или носители включают жирные масла, такие как кунжутное масло или синтетические сложные эфиры жирных кислот, такие как этилолеат или триглицериды, или липосомы. Водные суспензии для инъекций могут содержать вещества, которые повышают вязкость суспензии, такие как натрий карбоксиметилцеллюлоза, сорбит или декстран. Необязательно, суспензия также может содержать подходящие стабилизаторы или средства, которые повышают растворимость соединений, что дает возможность получить высококонцентрированные растворы.
Альтернативно, активный ингредиент может быть в форме порошка для реструктурирования подходящим носителем, например, стерильной апирогенной водой, перед использованием.
Соединения также могут быть сформулированы в ректальные композиции, такие как суппозитории или удерживаемые клизьмы, например, содержащие традиционные основы для суппозиториев, такие как масло какао или другие глицериды.
Помимо композиций, описанных выше, соединения также могут быть сформулированы в виде депо препаратов. Такие долго действующие композиции можно вводить путем имплантации (например, подкожно или внутримышечно или путем внутримышечной инъекции). Таким образом, например, соединения могут быть сформулированы с подходящими полимерными или гидрофобными веществами (например, в виде эмульсии в приемлемом масле) или ионообменными смолами или в виде плохорастворимых производных, например, плохорастворимой соли.
Примером фармацевтического носителя для гидрофобных соединений по настоящему изобретению является система сорастворителей, включающая бензиловый спирт, неполярное поверхностно-активное вещество, смешиваемый с водой органический полимер и водную фазу. Система сорастворителей может представлять собой VPD систему сорастворителей. VPD представляет собой раствор 3% масс/об бензилового спирта, 8% масс/об неполярного поверхностно-активного вещества полисорбата 80 и 65% масс/об полиэтиленгликоля 300, с доведением до объема в абсолютном этаноле. Система сорастворителей VPD (VPD:5W) состоит из VPD разбавленного 1:1 5% раствором декстрозы в воде. Эта система сорастворителей хорошо растворяет гидрофобные соединения и сама является низкотоксичной при системном введении. Действительно, пропорции системы сорастворителей могут существенно варьироваться, без нарушения характеристи ее растворимости и токсичности. Кроме того, сорастворители, являющиеся компонентами этой системы, могут варьировать: например, можно использовать другие низкотоксичные неполярные поверхностно-активные вещества вместо полисорбата 80; размер фракции полиэтиленгликоля может варьироваться; вместо полиэтиленгликоля можно использовать другие биосовместимые полимеры, например, поливинилпирролидон; и вместо декстрозы можно использовать другие сахара или полисахариды.
Альтернативно, можно использовать другие системы доставки для гидрофобных фармацевтических соединений. Липосомы и эмульсии являются хорошо известными примерами наполнителей или носителей для доставки гидрофобных лекарственных средств. Некоторые органические растворители, такие как диметилсульфоксид, также можно использовать, хотя обычно за счет более высокой токсичности. Кроме того, можно обеспечить доставку соединений с использованием системы замедленного высвобождения, таких как полупроницаемые матрицы из твердых гидрофобных полимеров, содержащие терапевтическое средство. Различные вещества замедленного высвобождения были установлены и хорошо известны специалистам в данной области. Капсулы замедленного высвобождения могут, в зависимости от их химической природы, высвобождать соединения в течение от нескольких часов до нескольких дней. В зависимости от химической природы и биологической стабильности терапевтического средства, можно использовать дополнительные стратегии для стабилизации белка.
Фармацевтические композиции также могут содержать подходящие твердые или находящиеся в фазе геля носители или эксципиенты. Примеры таких носителей или эксципиентов включают, но не ограничиваются этим, карбонат кальция, фосфат кальция, различные сахара, крахмалы, производные целлюлозы, желатин и полимеры, такие как полиэтиленгликоли.
Многие соединения по настоящему изобретению могут быть представлены в виде солей с фармацевтически совместимыми противоионами. Фармацевтически совместимые соли могут быть образованы со многими кислотами, включая, но не ограничиваясь этим, хлористоводородной, серной, уксусной, молочной, винной, яблочной, янтарной и т.п. Соли обычно являются более растворимыми в водных или других протонных растворителях, чем соответствующие формы свободного основания.
Фармацевтические композиции, подходящие для применения в настоящем изобретении, включают композиции, где активные ингредиенты содержатся в количестве эффективном для достижения предполагаемого назначения. Более конкретно, терапевтически эффективное количество означает количество эффективное для предотвращения развития или для облегчения существующих симптомов у субъекта, которого лечат. Определение эффективных количеств может легко определить специалист в данной области.
Для любого соединения, используемого в способе по настоящему изобретению, терапевтически эффективную дозу изначально можно определить на основании клеточных анализов. Например, дозу можно сформулировать в клеточных и животных моделях для достижения концентрации в кровотоке в пределах, включающих значение IC50, определенное в клеточных анализах (т.е. концентрация испытываемого соединения, которая достигает полумаксимального ингибирования активности данной протеинкиназы). В некоторых случаях является подходящим определение IC50 в присутствии 3-5% сывороточного альбумина, поскольку такое определение моделирует эффекты связывания плазменного белка на соединение. Такую информацию можно использовать для более точного определения полезных доз для человека. Кроме того, наиболее предпочтительные соединения для системного введения эффективно ингибируют передачу сигнала протеинкиназы в интактных клетках на уровнях, которые являются безопасно достижимыми в плазме.
Терапевтически эффективная доза относится к такому количеству соединения, которое приводит к облегчению симптомов у пациента. Токсичность и терапевтическую эффективность таких соединений можно определить с использованием стандартных фармацевтических процедур в клеточных культурах или с использованием экспериментальных животных, например, для определения максимально переносимой дозы (MTD) и ED50 (эффективная доза для 50% максимального ответа). Отношение доз токсического и терапевтического эффектов представляет собой терапевтический индекс, и это можно выразить как отношение между MTD и ED50. Соединения, которые демонстрируют высокие терапевтические индексы, являются предпочтительными. Данные, полученные из анализов этих клеточных культур и исследований животных, можно использовать в формулировании диапазона доз для человека. Доза таких соединений предпочтительно находится в пределах циркулирующих концентраций, которые включают ED50 с низкой токсичностью или отсутствием токсичности. Доза может варьировать в этих пределах в зависимости от используемой лекарственной формы и используемого пути введения. Точную композицию, путь введения и дозу может выбрать лечащий врач с учетом состояния пациента (см. например, Fingl et al., 1975, "The Pharmacological Basis of Therapeutics", Ch. 1 p. 1). Для лечения кризиса, введение сильного болюса или инфузии, приближающихся к MTD, может быть необходимым для получения быстрого ответа.
Размер дозы и интервал можно регулировать индивидуально для обеспечения уровней активной группы в плазме, которые являются достаточными для поддержания киназа-модулирующих эффектов или минимальной эффективной концентрации (MEC). MEC варьирует для каждого соединения, но ее можно определить на основании in vitro данных; например, концентрации, необходимой для достижения 50-90% ингибирования протеинкиназы, с использованием анализов, описанных в настоящей заявке. Дозы, необходимые для достижения MEC, зависят от индивидуальных характеристик и пути введения. Однако ВЭЖХ анализы или биоанализы можно использовать для определения концентраций в плазме.
Интервалы между введением доз также можно определить с использованием MEC значения. Соединения следует вводить с использованием режима, который поддерживает уровни в плазме выше MEC в течение 10-90% времени, предпочтительно 30-90%, и наиболее предпочтительноо 50-90%, до достижения желаемого облегчения симптомов. В случаях местного введения или селективного поглощения, эффективная местная концентрация лекарственного средства может не быть связана с концентрацией в плазме.
Количество вводимой композиции конечно зависит от субъекта, которого лечат, от массы тела субъекта, тяжести поражения, пути введения и мнения врача, предписывающего такое лечение.
Композиции, если это желательно, могут быть представлены в упаковке или распределяющем устройстве, которые могут содержать одну или несколько стандартных лекарственных форм, содержащих активный ингредиент. Упаковка, например, может включать металлическую или пластиковую фольгу, например, блистерная упаковка. Упаковка или распределяющее устройство могут быть снабжены инструкциями для введения. Композиции, включающие соединение по настоящему изобретению, сформулированное в совместимом фармацевтическом носителе, также могут быть получены, помещены в подходящий контейнер и промаркированы для лечения указанного состояния.
В некоторых композициях может быть полезно использовать соединения по настоящему изобретению в форме частиц очень малого размера, например, полученных путем гидроэнергетического измельчения.
Применение соединений по настоящему изобретению для получения фармацевтических композиций проиллюстрировано далее в описании. В этом описании термин "активное соединение" означает любое соединение по настоящему изобретению, но особенно любое соединение, которое является конечным продуктом одного из следующих Примеров.
a) Капсулы
При получении капсул, можно объединить и смешать вместе 10 частей массовых активного соединения и 240 частей массовых лактозы. Смесью можно заполнить твердые желатиновые капсулы, при этом каждая капсула должна содержать стандартную дозу или часть стандартной дозы активного соединения.
b) Таблетки
Таблетки можно получить, например, из следующих ингредиентов.
Частей массовых
Активное соединение, лактозу и некоторое количество крахмала можно объединить, смешать вместе и полученную смесь можно гранулировать с раствором поливинилпирролидона в этаноле. Сухой гранулят можно смешать со стеаратом магния и оставшимся количеством крахмала. Смесь затем прессуют в машине для таблетирования с получением таблеток, каждая из которых содержит стандартную дозу или часть стандартной дозы активного соединения.
c) Таблетки с энтеросолюбильным покрытием
Таблетки можно получить способом, описанным в (b) выше. Таблетки могут иметь энтеросолюбильное покрытие, нанесенное традиционным способом с использованием раствора 20% ацетата целлюлозы фталата и 3% диэтилфталата в этаноле:дихлорметане (1:1).
d) Суппозитории
При получении суппозиториев, например, 100 частей массовых активного соединения можно включить в 1300 частей массовых триглицеридной основы для суппозиториев, и смесь формируют в суппозитории, каждый из которых содержит терапевтически эффективное количество активного ингредиента.
В композициях по настоящему изобретению активное соединение, если это желательно, может находиться в ассоциации с другими совместимыми фармакологически активными ингредиентами. Например, соединения по настоящему изобретению можно вводить в сочетании с другим терапевтическим средством, которое известно как средство для лечения заболевания или состояния, описанного в настоящей заявке. Например, с одним или несколькими дополнительными фармацевтическими средствами, которые ингибируют или препятствуют продукции VEGF или ангиопоэтинов, ослабляют внутриклеточные ответы на VEGF или ангиопоэтины, блокируют внутриклеточную сигнальную трансдукцию, ингибируют сосудистую гиперпроницаемость, снижают воспаление или ингибируют или препятствуют образованию отека или неоваскуляризации. Соединения по настоящему изобретению можно вводить до введения, после введения или одновременно с дополнительным фармацевтическим средством, при этом любой курс введения является подходящим. Дополнительные фармацевтические средства включают, но не ограничиваются этим, противоотечные стероиды, НСПВЛС, ингибиторы ras, анти-TNF средства, анти-IL1 средства, антигистамины, PAF-антагонисты, ингибиторы COX-1, ингибиторы COX-2, ингибиторы синтазы NO, ингибиторы Akt/PTB, ингибиторы IGF-1R, ингибиторы PKC, ингибиторы PI3 киназы, ингибиторы кальциневрина и иммуносупрессанты. Соединения по настоящему изобретению и дополнительные фармацевтические средства действуют либо аддитивно либо синергически. Таким образом, введение таких комбинаций веществ, которые ингибируют ангиогенез, сосудистую гиперпроницаемость и/или ингибируют образование отека, может обеспечить большее облегчение от вредных эффектов гиперпролиферативного расстройства, ангиогенеза, сосудистой гиперпроницаемости или отека, чем введение любого такого вещества отдельно. Для лечения злокачественных расстройств сочетания с антипролиферативными или цитотоксическими химиотерапевтическими средствами или лучевой терапией включены в объем настоящего изобретения.
Настоящее изобретение также включает применение соединения формулы (I) или формулы (II) в качестве лекарстенного средства.
Следующий аспект настоящего изобретения обеспечивает применение соединения формулы (I) или формулы (II) или его соли для получения лекарстенного средства для лечения сосудистой гиперпроницаемости, ангиогенез-зависимых расстройств, пролиферативных заболеваний и/или расстройств иммунной системы у млекопитающих, в частности, у человека.
Настоящее изобретение также обеспечивает способ лечения сосудистой гиперпроницаемости, аномальной неоваскуляризации, пролиферативных заболеваний и/или расстройств иммунной системы, которые включает введение терапевтически эффективного количества соединения формулы (I) или формулы (II) млекопитающему, в частности, человеку.
Аббревиатуры
АНАЛИЗЫ
In vitro Jak1 киназную активность измеряли методом переноса резонансной энергии флуоресценции с разрешением по времени (trFRET)
Меняющиеся концентрации ингибитора добавляли в лунку для осуществления анализа, содержащую: Jak1 фермент (aa 845-1142; экспрессируемый в SF9 клетках как GST гибридный белок и очищенный глутатион-аффинной хроматографией; 4 нМ), пептидный субстрат (биотин-TYR2, Последовательность: Биотин-(Ahx)-AEEEYFFLFA-амид; 2 мкМ), MOPSO pH 6,5 (50 мМ), MgCl2 (10 мМ), MnCl2 (2 мМ), DTT (2,5 мМ), BSA (0,01% масс/об), Na3VO4 (0,1 мМ) и АТФ (0,001 мМ). После примерно 60 мин инкубации при комнатной температуре реакцию гасили путем добавления EDTA (конечная концентрация: 100 мМ) и проявляли путем добавления реагента для проявления (конечные приблизительные концентрации: 30 мМ HEPES pH 7,0, 0,06% BSA, 0,006% Tween-20, 0,24 M KF, 80 нг/мл PT66K (меченное европием анти-фосфотирозиновое антитело cat #61T66KLB Cisbio, Bedford, MA) и 3,12 мкг/мл SAXL (Phycolink стрептавидин-аллофикоцианиновый акцептор, cat #PJ52S, Prozyme, San Leandro, CA). Проявленную реакционную смесь инкубировали в темноте либо при около 4°C в течение около 14 часов, либо в течение около 60 минут при комнатной температуре, затем считывали через детектор флуоресценции с разрешением по времени (Rubystar, BMG) с использованием 337 нм лазера для возбуждения и длины волны излучения 665 нм. В линейном диапазоне анализа наблюдаемый сигнал при 665 нм непосредственно относится к фосфорилированному продукту, и его использовали для расчета IC50 значений.
In vitro Jak3 киназная активность, измеренная методом переноса резонансной энергии флуоресценции с разрешением по времени (trFRET)
Меняющиеся концентрации ингибитора добавляли в лунку для осуществления анализа, содержащую: Jak3 фермент (aa 811-1103; экспрессируемый в SF9 клетках как GST гибридный белок и очищенный глутатион-аффинной хроматографией; 3 нМ), пептидный субстрат (биотин-TYR2, Последовательность: Биотин-(Ahx)-AEEEYFFLFA-амид; 2 мкМ), MOPSO pH 6,5 (50 мМ), MgCl2 (10 мМ), MnCl2 (2 мМ), DTT (2,5 мМ), BSA (0,01% масс/об), Na3VO4 (0,1 мМ) и АТФ (0,001 мМ). После приблизительно 60-минутной инкубации при комнатной температуре реакцию гасили путем добавления EDTA (конечная концентрация: 100 мМ) и проявляли путем добавления реагента для проявления (конечные приблизительные концентрации: 30 мМ HEPES pH 7,0, 0,06% BSA, 0,006% Tween-20, 0,24 M KF, 80 нг/мл PT66K (меченное европием анти-фосфотирозиновое антитело cat #61T66KLB Cisbio, Bedford, MA) и 0,8 мкг/мл SAXL (Phycolink стрептавидин-аллофикоцианиновый акцептор, cat #PJ52S, Prozyme, San Leandro, CA). Проявленную реакционную смесь инкубировали в темноте либо при около 4°C в течение около 14 часов, либо в течение около 60 минут при комнатной температуре, затем считывали через детектор флуоресценции с разрешением по времени (Rubystar, BMG) с использованием 337 нм лазера для возбуждения и длины волны излучения 665 нм. В линейном диапазоне анализа, наблюдаемый сигнал при 665 нм непосредственно относится к фосфорилированному продукту, и его использовали для расчета IC50 значений.
In vitro Syk киназную активность измеряли методом переноса резонансной энергии флуоресценции с разрешением по времени (trFRET)
0,3 нМ Syk каталитический домен (aa356-635, очистку осуществляли в Abbott Bioreseach Center) смешивали с 0,1 мкМ пептидного субстрата (биотин-TYR1, Последовательность: Биотин-(Ahx)-GAEEEIYAAFFA-COOH) при меняющихся концентрациях ингибитора в реакционном буфере: 50 мМ MOPSO pH 6,5, 10 мМ MgCl2, 2 мМ MnCl2, 2,5 мМ DTT, 0,01% BSA, 0,1 мМ Na3VO4 и 0,001 мМ АТФ. После приблизительно 60-минутной инкубации при комнатной температуре реакцию гасили путем добавления EDTA (конечная концентрация: 100 мМ) и проявляли путем добавления реагента для проявления (конечные приблизительные концентрации: 30 мМ HEPES pH 7,0, 0,06% BSA, 0,006% Tween-20, 0,24 M KF, 90 нг/мл PT66K (меченное европием анти-фосфотирозиновое антитело cat #61T66KLB Cisbio, Bedford, MA) и 0,6 мкг/мл SAXL (Phycolink стрептавидин-аллоофикоцианиновый акцептор, cat #PJ52S, Prozyme, San Leandro, CA). Проявленную реакционную смесь инкубировали в темноте либо при около 4°C в течение около 14 часов, либо в течение около 60 минут при комнатной температуре, затем считывали через детектор флуоресценции с разрешением по времени (Rubystar, BMG) с использованием 337 нм лазера для возбуждения и длины волны излучения 665 нм. В линейном диапазоне анализа, наблюдаемый сигнал при 665 нм непосредственно относится к фосфорилированному продукту, и его использовали для расчета IC50 значений.
Другие in vitro киназные анализы методом переноса резонансной энергии флуоресценции с разрешением по времени (trFRET)
Другие киназные анализы осуществляли с использованием аналогичного протокола. Дополнительные очищенные ферменты Tyk2 (aa 880-1185 с N-концевой гистидиновой меткой и C-концевой FLAG меткой; очищали внутри фирмы методом аффинной хроматографии с иммобилизованными металлическими ионами), RET (aa 711-1072 с N-концевой гистидиновой меткой; очищали методом аффинной хроматографии с иммобилизованными металлическими ионами), Syk (aa356-635 с C-концевой гистидиновой меткой; очищали методом аффинной хроматографии с иммобилизованными металлическими ионами), и KDR (aa 792-1354 с N-концевой гистидиновой меткой; очищали внутри фирмы методом аффинной ионообменной хроматографии с иммобилизованными металлическими ионами) экспрессировали в SF9 клетках, и Aurora 1/B (aa1-344 с N-концевой гистидиновой меткой и очищенный методом аффинной хроматографии с иммобилизованными металлическими ионами) экспрессировали в E. coli. Другие используемые ферменты были доступны из коммерческих источников. Ферменты смешивали с биотинилированными субстратами при меняющихся концентрациях ингибитора в различных реакционных буферах (см. Таблицу A). После приблизительно 60-минутной инкубации при комнатной температуре реакцию гасили путем добавления EDTA и проявляли путем добавления реагента для проявления (конечные приблизительные концентрации: 30 мМ HEPES pH 7,0, 0,06% BSA, 0,006% Tween-20, 0,24 M KF, меняющиеся количества донорных меченных европием антител и акцепторного стрептавидин-меченного аллофикоцианина (SAXL)). Проявленные реакционные смеси инкубировали в темноте либо при около 4°C в течение около 14 часов, либо в течение около 60 минут при комнатной температуре, затем считывали в детекторе флуоресценции с разрешением по времени (Rubystar, BMG Labtech), как описано выше.
Специальные условия (в расчете на 40 мкл ферментативной реакции) для различных ферментов подробно описаны ниже:
фермента (нг/лунка)
0,39 (мкг/лунка) SAXL
0,078 (мкг/лунка) SAXL
0,078 (мкг/лунка) SAXL
0,078 (мкг/лунка) SAXL
0,34 (мкг/лунка) SAXL
0,078 (мкг/лунка) SAXL
0,6 (мкг/лунка) SAXL
0,6 (мкг/лунка) SAXL
0,078 (мкг/лунка) SAXL
0,34 (мкг/лунка) SAXL
0,34 (мкг/лунка) SAXL
0, 045 (мкг/лунка) SAXL
Реакционные буферы:
MOPSO буфер содержит: 50 мМ MOPSO pH 6,5, 10 мМ MgCl2, 2 мМ MnCl2, 2,5 мМ DTT, 0,01% BSA и 0,1 мМ Na3VO4
HEPES буфер содержит: 50 мМ HEPES pH 7,1, 2,5 мМ DTT, 10 мМ MgCl2, 2 мМ MnCl2, 0,01% BSA и 0,1 мМ Na3VO4
MOPS буфер содержит: 20 мМ MOPS pH 7,2, 10 мМ MgCl2, 5 мМ EGTA, 5 мМ Бета-фосфоглицерина, 1 мМ Na3VO4, 0,01% Triton-X-100 и 1 мМ DTT
Субстраты:
Биотин-ATF2-пептид последовательность: Биотин-(Ahx)-AGAGDQTPTPTRFLKRPR-амид
Биотин-TYR1-пептид последовательность: Биотин-(Ahx)-GAEEEIYAAFFA-COOH
Биотин-TYR2-пептид последовательность: Биотин-(Ahx)-AEEEYFFLFA-амид
Биотин-MBP-пептид последовательность: Биотин-(Ahx)-VHFFKNIVTPRTPPPSQGKGAEGQR-амид
Биотин-полиGluTyr пептид закупали у компании Cisbio (cat #61GT0BLA, Bedford, MA)
KinEASE S2 и S3 пептиды закупали у компании Cisbio (cat #62ST0PEB, Bedford, MA)
Реагенты для детекции:
Анти-pATF2-Eu, специальное мечение было выполнено Cisbio (Bedford, MA)
Анти-pMBP-Eu, специальное мечение было выполнено Cisbio (Bedford, MA)
PT66K закупали у компании Cisbio (cat #61T66KLB, Bedford, MA)
SAXL закупали у компании Prozyme (cat #PJ25S, San Leandro, CA)
Клеточный анализ с использованием человеческих T-бластов IL-2 pSTAT5
Материалы:
Фитогемаглютинин T-бласты получали из Leukopacks, закупленных у компании Biological Specialty Corporation, Colmar, PA 18915, и криоконсервировали в 5% DMSO/среде до осуществления анализа.
Для этого анализа клетки оттаивали в среде для осуществления анализа со следующей композицией: RPMI 1640 среда (Gibco 11875093) с 2 мМ L-глутамина (Gibco 25030-081), 10 мМ HEPES (Gibco 15630-080), 100 мкг/мл Пенициллина/Стрептомицина (Gibco 15140-122) и 10% термоинактивированной FBS (Gibco 10438026). Другие вещества, используемые в анализе: DMSO (Sigma D2650), 96-луночные планшеты для разведений (полипропилен) (Corning 3365), 96-луночные аналитические планшеты (белые, Ѕ площади, 96-луночные) (Corning 3642), D-PBS (Gibco 14040133), IL-2 (R&D 202-IL-10 (10 мкг)), набор Alphascreen pSTAT5 (Perkin Elmer TGRS5S10K) и набор Alphascreen белка (Perkin Elmer 6760617M)
Методы:
T-Бласты оттаивали и культивировали в течение около 24 часов без IL-2 до осуществления анализа. Испытываемые соединения или контроли растворяли и осуществляли серийные разведения в 100% DMSO. Исходные растворы DMSO затем разбавляли 1:50 в клеточной культуральной среде для получения 4× исходных растворов соединения (содержащих 2% DMSO). С использованием Corning белого 96-луночного Ѕ площади планшета клетки высевали при плотности 2×105/10 мкл/лунка в 10 мкл среде с последующим добавлением 5 мкл 4× испытываемого соединения в двух повторах. Клетки инкубировали с соединением в течение около 0,5 часов при около 37°C. Затем 5 мкл исходного раствора IL-2 добавляли при конечной концентрации 20 нг/мл. IL-2 хранили в виде 4 мкг/мл исходного раствора, в соответствии с указаниями изготовителя, при около -20°C в аликвотах и разбавляли 1:50 средой для осуществления количественного анализа (до 80 нг/мл) непосредственно перед использованием. Содержимое лунок смешивали, осторожно постукивая стенки планшета(планшетов) несколько раз, с последующей инкубацией при около 37°C в течение около 15 минут. Анализ завершали путем добавления 5 мкл 5x Alphascreen лизисного буфера и встряхивания на орбительном встряхивающем устройстве в течение около 10 минут при комнатной температуре. Смесь акцепторных шариков Alphascreen восстанавливали следуя протоколу Perkin Elmer. Добавляли 30 мкл/лунка восстановленной смеси акцепторных шариков Alphascreen, накрывали фольгой затем встряхивали на орбительном встряхивающем устройстве в течение около 2 минут с высокой интенсивностью затем около 2 часов с низкой интенсивностью. Смесь донорных шариков восстанавливали, следуя протоколу Perkin Elmer Alphascreen; добавляли 12 мкл/лунка, накрывали фольгой, затем встряхивали в течение около 2 минут с высокой интенсивностью и около 2 часов с низкой интенсивностью. Планшеты считывали на EnVision считывающем устройстве, следуя инструкциям протокола Perkin Elmer's Alphascreen.
Клеточный анализ TF-1 IL-6 pSTAT3
Материалы:
TF-1 клетки (ATCC #CRL-2003). Культуральная среда: DMEM среда (Gibco 11960-044) с 2 мМ L-глутамина (Gibco 25030-081), 10 мМ HEPES (Gibco 15630-080), 100 мкг/мл Пенициллина/Стрептомицина (Gibco 15140-122), 1,5 г/л бикарбоната натрия (Gibco 25080-094), 1 мМ пирувата натрия (Gibco 11360-070), 10% термоинактивированной FBS (Gibco 10437-028) и 2 нг/мл GM-CSF (R&D 215-GM-010). Другие вещества, используемые в анализе: DMSO (Sigma D2650), 96-луночные планшеты для разведений (полипропилен) (Corning 3365), 96-луночные аналитические планшеты (белые, Ѕ площади, 96-луночные) (Corning 3642), D-PBS (Gibco 14040133 ), IL-6 (R&D 206-IL/CF-050 (50 мкг)), набор Alphascreen pSTAT3 (Perkin Elmer TGRS3S10K) и набор Alphascreen белка (Perkin Elmer 6760617M).
Методы:
Перед осуществлением анализа клетки культивировали в течение около 18 часов в культуральной среде без GM-CSF. Испытываемые соединения или контроли растворяли и осуществляли серийные разведения в 100% DMSO. Исходные растворы DMSO затем разбавляли 1:50 в клеточной культуральной среде для получения 4× исходных растворов соединений (содержащих 2% DMSO). С использованием Corning белого 96-луночного планшета, Ѕ площади, клетки высевали при плотности 2×107/10 мкл/лунка в 10 мкл среды с последующим добавлением 5 мкл 4x исходного раствора испытываемого соединения в двух повторах. Клетки инкубировали с соединением в течение около 0,5 часов при около 37°C с последующим добавлением 5 мкл 400 нг/мл IL-6. IL-6 хранили в 10 мкг/мл аликвотах с использованием D-PBS(0,1% BSA), не содержащего эндотоксин, при около -20°C. До осуществления анализа IL-6 разбавляли до 400 нг/мл в культуральной среде и наносили (5 мкл/лунка) на все лунки, за исключением лунок с отрицательным контролем, в которые добавляли 5 мкл/лунка среды. Содержимое лунок смешивали, осторожно постукивая стенки планшета несколько раз. Планшеты инкубировали при около 37°C в течение около 30 минут. Клетки лизировали путем добавления 5 мкл 5X Alphascreen буфера для лизиса клеток во все лунки, встряхивали в течение около 10 минут при комнатной температуре, затем анализировали. Альтернативно, аналитические планшеты можно заморозить при около -80°C и в последствии оттаивать при комнатной температуре. С использованием набора pSTAT3 SureFire Assay (Perkin Elmer #TGRS3S10K), смесь акцепторных шариков восстанавливали, следуя инструкциям протокола Perkin Elmer Alphascreen. Добавляли 30 мкл на лунку, затем планшет накрывали фольгой и встряхивали на орбительном встряхивающем устройстве в течение около 2 минут с высокой интенсивностью, затем около 2 часов с низкой интенсивностью при комнатной температуре. Смесь донорных шариков восстанавливали, следуя инструкциям протокола Perkin Elmer Alphascreen. Добавляли 12 мкл на лунку, затем накрывали фольгой и встряхивали на орбительном встряхивающем устройстве в течение около 2 минут с высокой интенсивностью, затем около 2 часов с низкой интенсивностью при около 37°C. Планшеты считывали на считывающем устройстве EnVision, следуя инструкциям протокола Perkin Elmer Alphascreen, при комнатной температуре.
Клеточный анализ UT7/EPO pSTAT5
Материалы:
Осуществляли пассаж UT7/EPO клеток с эритропоэтином (EPO), расщепляли два раза в неделю и свежую культуральную среду оттаивали и добавляли в момент расщепления. Культуральная среда: DMEM среда (Gibco 11960-044) с 2 мМ L-глутамина (Gibco 25030-081), 10 мМ HEPES (Gibco 15630-080), 100 Ед./мл Пенициллина/Стрептомицина (Gibco 15140-122), 10% термоинактивированной FBS (Gibco 10437-028), EPO (5 мкл/мл = 7,1 мкл 7 мкг/мл исходного раствора на мл среды). Среда для осуществления количественного анализа: DMEM, 2 мМ L-глутамина, 5% FBS, 10 мМ HEPES. Другие вещества, используемые в анализе: DMSO (Sigma D2650), 96-луночные планшеты для разведений (полипропилен) (Corning 3365), 96-луночные аналитические планшеты (белые, Ѕ площади, 96-луночные) (Corning 3642), D-PBS (Gibco 14040133), IL-2 (R&D 202-IL-10 (10 мкг)), набор Alphascreen pSTAT5 (Perkin Elmer TGRS5S10K) и набор Alphascreen белка (Perkin Elmer 6760617M).
Методы:
Клетки культивировали в течение около 16 часов без EPO до осуществления анализа. Испытываемые соединения или контроли растворяли и осуществляли серийные разведения в 100% DMSO. Исходные растворы DMSO затем разбавляли 1:50 в клеточной культуральной среде для получения 4x исходных растворов соединений (содержащих 2% DMSO). С использованием Corning белого 96-луночного планшета, Ѕ площади, клетки высевали при плотности 2×105/10 мкл/лунка в 10 мкл среды с последующим добавлением 5 мкл 4× исходного раствора испытываемого соединения в двух повторах. Клетки инкубировали с соединением в течение около 0,5 часов при около 37°C. После инкубации добавляли 5 мкл EPO с получением конечной концентрации 1 нМ EPO. Содержимое лунок смешивали, осторожно постукивая стенки планшета несколько раз, с последующей инкубацией при около 37°C в течение около 20 минут. Добавляли 5 мкл 5x Alphascreen лизисного буфера, с последующим встряхиванием на орбительном встряхивающем устройстве в течение около 10 минут при комнатной температуре. Добавляли 30 мкл/лунка акцепторных шариков после восстановления, следуя протоколу Perkin Elmer Alphascreen, накрывали фольгой и встряхивали на орбительном встряхивающем устройстве в течение около 2 минут с высокой интенсивностью, затем около 2 часов с низкой интенсивностью. Донорные шарики восстанавливали, следуя инструкциям протокола Perkin Elmer Alphascreen, с последующим добавлением 12 мкл/лунка, накрывали фольгой и встряхивали на орбительном встряхивающем устройстве в течение около 2 минут с высокой интенсивностью, около 2 часов с низкой интенсивностью. Планшеты считывали на считывающем устройстве EnVision, следуя инструкциям протокола Perkin Elmer Alphascreen.
Антиген-индуцированная дегрануляция RBL-2H3 клеток:
RBL-2H3 клетки поддерживали в T75 колбах при около 37°C и 5% CO2 и осуществляли пересев каждые 3-4 дня. Для сбора клеток использовали 20 мл PBS для промывки колбы один раз и затем добавляли 3 мл Трипсин-EDTA и инкубировали при около 37°C в течение около 2 минут. Клетки переносили в пробирку с 20 мл среды, центрифугировали при 1000 об/мин при комнатной температуре в течение около 5 минут и ресуспендировали при 1×106 клеток/мл. Клетки сенсибилизировали путем добавления DNP-специфического мышиного IgE до конечной концентрации 0,1 мкг/мл. 50 мкл клеток добавляли в каждую лунку 96-луночного плоскодонного планшета (50×103 клеток/лунка) и инкубировали в течение ночи при около 37°C в 5% CO2. На следующий день получали растворы соединений в 100% DMSO при 10 мМ. Затем осуществляли серийные разведения каждого соединения 1:4 шесть раз в 100% DMSO. Каждое разведение соединений затем разбавляли 1:20 и затем 1:25, оба раза в буфере Тирода. Среду аспирировали из содержащих клетки планшетов и клетки промывали два раза 100 мкл буфера Тирода (предварительно нагретый до около 37°C). 50 мкл соединений, разбавленных в буфере Тирода, добавляли в каждую лунку и планшеты инкубировали в течение около 15 минут при около 37°C в 5% CO2. Затем в каждую лунку добавляли 50 мкл 0,2 мкг/мл DNP-HSA в буфере Тирода и планшеты инкубировали в течение около 30 минут при около 37°C в 5% CO2. Конечные концентрации различных компонентов в смеси для инкубации были следующими: 0,002-10 мкМ соединения, 0,1% DMSO и 0,1 мкг/мл DNP-HSA. В качестве одного контроля, в ряд лунок добавляли 0,2% DMSO (без соединения) в буфере Тирода для определения максимального стимулированного высвобождения. В качестве второго контроля, добавляли буфер Тирода без DNP-HSA в ряд лунок, содержащих 0,2% DMSO, без соединения, для определения нестимулированного высвобождения. Каждое условие (соединения и контроли) повторяли в лунках три раза. По окончании 30-минутной инкубации 50 мкл супернатанта переносили в новый 96-луночный планшет. Оставшийся супернатант в содержащих клетки планшетах аспирировали и заменяли на 50 мкл 0,1% Triton X-100 в буфере Тирода для лизиса клеток. 50 мкл свежеполученного 1,8 мМ 4-Нитрофенил N-ацетил-β-D-глюкозаминида (pNAG) затем добавляли в каждую лунку с супернатантом и клеточным лизатом и планшеты инкубировали в течение около 60 минут при около 37°C в 5% CO2. 100 мкл 7,5 мг/мл бикарбоната натрия добавляли в каждую лунку для прекращения реакции. Планшеты затем считывали при 405 нм на считывающем устройстве для планшетов Molecular Devices SpectraMax 250.
Подсчет результатов
1) Планшетное фоновое значение OD405, полученное из лунок, содержащих буфер Тирода и pNAG (отсутствие супернатанта или лизата), вычитали из OD405 значений для каждой лунки, содержащей супернатант или лизат.
2) Высвобождение для каждой лунки выражали как процент от общего высвобождения для этой лунки, где общее высвобождение представляет собой умноженной на два высвобождение в супернатанте плюс высвобождение в клеточном лизате. Этот расчет скорректирован на изменение количества клеток в каждой лунке.
3) Максимальный ответ представляет собой средний ответ в лунках, содержащих DNP-HSA, но не содержащих соединение.
4) Минимальный ответ представляет собой средний ответ в лунках, не содержащих DNP-HSA и не содержащих соединение.
5) Ответ в каждой содержащей соединение лунке расчитывали как процент от максимального ответа (выраженный как % контроля), где максимальный ответ составляет 100%, и минимальный ответ составляет 0%.
6) Кривую доза-ответ строили для каждого соединения, и значение IC50 рассчитывали из кривой с использованием программы Prism GraphPad и нелинейного метода наименьших квадратов.
Срочное in vivo измерение ингибирования JAK соединениями осуществляли с использованием:
Конкавалин (Con A)-индуцированной продукции цитокинов у крыс Lewis
Испытываемое соединение было сформулировано в инертном носителе (например, таком как, но не ограничиваясь этим, 0,5% гидроксипропилметилцеллюлоза (Sigma, cat # H3785)/0,02% Tween 80 (Sigma, cat # 4780) в воду) при желаемой концентрации для достижения доз в пределах 0,01-100 мг/кг. Шестинедельным самцам крыс Lewis (125г-150г) (Charles River Laboratories) вводили дозы соединения перорально, во время ноль (0 мин). Примерно через 30 минут крысам вводили путем внутривенной инъекции (в.в.) 10 мг/кг Конкавалина (Con A, AmershamBioscience, cat #17-0450-01), растворенного в PBS (Invitrogen, cat # 14190). Примерно через 4 часа у крыс выпускали кровь через сердечную пункцию и их плазму анализировали для определения уровней IL-2 (ELISA набор: R&D Systems cat #R2000) и IFN-γ (ELISA набор: R&D Systems cat #RIF00).
Срочное in vivo измерение ингибирования соединениями сигнала Fcγ рецептора осуществляли следующими методами:
Модель обратимого пассивного Arthus
В день 0, подготавливали OVA при концентрации 17,5 мг/мл в PBS, осторожно качая пробирку до образования раствора. Затем добавляли 2% раствор Эванса синего (Sigma Aldrich, cat # E2129) для увеличения в два раза объема до конечной концентрации 8,75 мг/мл OVA и 1% красителя Эванса синего. Анти-OVA антитело (Abazyme), исходная концентрация 10 мг/мл, оттаивали и получали раствор с PBS 400 мкг/100 мкл. Соединения подготавливали путем добавления носителя, 0,5% HPMC с 0,02% Tween 80 и осуществляли вихревое встряхивание в течение около 15 секунд с последующей гомогенизацией в течение минимум около 2 минут при 28000 об/мин вплоть до получения тонкодисперсной суспензии без каких-либо слипшихся кусков соединения. Крыс взвешивали и вводили дозу соединения с предварительно определенным t-max на основании фармакокинетических исследований. Животных затем помещали под общую анестезию с использованием смеси 5% изофлоурана и кислорода и выбривали. С использованием 1/2 мл инсулинового шприца в двух местах вводили инъекцию, внутрикожно, на 1 участке 100 мкл 400 мкг/100 мкл анти-OVA антитела, и на 1 участке 100 мкл стерильного PBS. Каждый участок затем обводили кружком с использованием перманентного маркера для последующей эксплантации. Сразу после внутрикожных инъекций животным вводили инъекцию 200 мкл смеси OVA (10 мг/кг)/Эванса синего в.в., с использованием 1/2 мл инсулинового шприца. Примерно через четыре часа после инъекции животных умерщвляли, осуществляли сбор крови через сердечную пункцию и кровь собирали с использованием пробирки для отделения плазмы. Образцы крови хранили на льду вплоть до центрифугирования (в течение около 2 часов после сбора). Каждый участок инъекции удаляли с использованием одноразового дерматома (Acuderm Acu-Punch Disposable 12 мм), разрезали на четыре части и помещали в предварительно помеченную 2-мл пробирку Эппендорфа. В каждую пробирку для биопсии добавляли один мл DMF и помещали в нагревательный блок примерно на 24 часа при около 50oC. Примерно через 24 часа после инкубации 100 мкл каждого образца добавляли в 96-луночный плоскодонный планшет. Образцы считывали при 620 нм на считывающем устройстве для планшетов с использованием программы Softmax для измерения уровней синего красителя Эванса. Фоновое значение удаляли путем вычитания OD участка PBS инъекции из OD участка анти-OVA инъекции для каждого индивидуального животного. Образцы плазмы центрифугировали в микроцентрифуге в течение около 5 минут при 16,1 rcf. 200 мкл плазмы помещали в 1,7-мл пробирку Эппендорфа для измерения уровня лекарственного средства и пробирки хранили при -80°C до осуществления измерений.
Хронические in vivo эффекты соединений на модель артрита измеряли с использованием:
Адъювант-индуцированного артрита (AIA) у крыс Lewis
Самок крыс Lewis, (возраста 6 недель, с массой тела 125 г - 150 г от Charles River Laboratories) иммунизировали внутрикожно (в.к.) в правую заднюю подошву с использованием 100 мкл суспензии минерального масла (Sigma, cat # M5905) и содержащей 200 мкг M. tuberculosis, H37RA (Difco, cat # 231141). Воспаление возникало в контралатеральной (левой) задней лапе через семь дней после первичной иммунизации. Через семь дней после иммунизации, соединение формулировали в инертном носителе (например, но не ограничиваясь этим, 0,5% гидроксипропилметилцеллюлоза (Sigma, cat #H3785)/0,02% Tween 80 (Sigma, cat # 4780) в воде) и вводили перорально один или два раза в день в течение, по меньшей мере, 10 дней. Исходный объем лапы определяли в день 0 с использованием плетизмографа с вытеснением воды (Vgo Basile North America Inc. PA 19473, Модель # 7140). Крыс подвергали легкой анестезии с использованием анестетика для ингаляции (изофлуран) и контралатеральную (левую) заднюю лапу погружали в плетизмограф и объем лапы регистрировали. Крыс оценивали через день вплоть до дня 17 после иммунизации. В день 17 после иммунизации всех крыс обескровливали путем сердечной пункции под анестезией изофлураном и левую заднюю лапу брали для оценки влияния на эрозию кости с использованием микро-CT сканов (SCANCO Medical, Southeastern, PA, Модель # µCT 40) при размере воксела 18 мкм, порогового значения 400, сигма-гаусс 0,8, держатель-гаусс 1,0. Объем кости и плотность для 360 мкм (200 срез) вертикальной секции, охватывающей тарзальную секцию лапы. 360 мкм секцию анализировали от основания плюсневых костей до верхней части большеберцовой кости, с нижней контрольной точкой, установленной на тибиоталярном соединении. Воздействие лекарственного средства определяли в плазме с использованием LC/MS.
или:
Коллаген-индуцированного артрита (CIA) у крыс Lewis
В день -1 Коллаген типа II (CII), растворимый из бычьей носовой перегородки (Elastin Products, Cat #CN276) отвешивали для получения дозы 600 мкг/крыса, добавляли 0,01M уксусной кислоты (150 мкл HOAc USP степени чистоты. J.T.Baker, order# 9522-03 и 250 мл Milli Q Water) для получения концентрации 4 мг/мл. Сосуд накрывали алюминиевой фольгой и помещали на качающееся устройство при около 4°C в течение ночи. В день 0 коллагеновый исходный раствор разводили 1:1 неполным адъювантом Фрейнда (IFA) (Difco labs, cat #263910) с использованием стеклянного шприца Гамильтона с наконечником Люэра (SGE Syringe Perfection VWR cat # 007230), конечная концентрация 2 мг/мл. Самки крыс Lewis (Charles River Laboratories), которым давали аклиматизироваться в течение 7 дней, во время иммунизации весили приблизительно 150 г, и их анестезировали в камере для анестезии с использованием изофлурана (5%) и кислорода. После того как крыс полностью анестезировали, их переносили в передний конусообразный отсек для поддержания анестезии при введении инъекций. Крыс выбривали у основания хвоста, вводили 300 мкл коллагена путем внутрикожной инъекции в седалищную область крысы, n=9 на группу. 100 мкл в трех местах с использованием 500 мкл шприца с наконечником Люэра и 27 g иглы. IFA контрольным крысам вводили инъекцию таким же образом (n=6). IFA представлял собой 1:1 эмульсию 0,01M уксусной кислоты. Бустер-дозу вводили в день 6 испытания. Крыс в этот день не выбривали и инъекции вводили таким же образом как для иммунизации. Воспаление появлялось в обеих задних лапах через 10 дней после первичной иммунизации. Через 10 дней после иммунизации соединение формулировали в инертном носителе (например, но не ограничиваясь этим, 0,5% гидроксипропилметилцеллюлоза (Sigma, cat # H3785)/0,02% Tween 80 (Sigma, cat # 4780) в воде) и вводили дозу перорально один или два раза день в течение, по меньшей мере, в течение 9 дней. Исходный объем лапы определяли в день 7 с использованием плетизмографа с вытеснением воды (Vgo Basile North America Inc. PA 19473, Модель # 7140). Крыс подвергали легкой анестезии с использованием анестетика для ингаляции (изофлуран) и обе задние лапы погружали в плетизмограф и объем лапы регистрировали. Крыс оценивали 2-3 раза в неделю вплоть до дня 18 после иммунизации. В день 18 после иммунизации всех крыс обескровливали путем сердечной пункции под анестезией изофлураном и задние лапы брали для оценки влияния на эрозию кости с использованием микро-CT сканов (SCANCO Medical, Southeastern, PA, Модель # µCT 40) при размере воксела 18 мкм, порогового значения 400, сигма-гаусс 0,8, держатель-гаусс 1,0. Объем кости и плотность для 360 мкм (200 срез) вертикальной секции, охватывающей тарзальную секцию лапы. 360 мкм секцию анализировали от основания плюсневых костей до верхней части большеберцовой кости, с нижней контрольной точкой, установленной на тибиоталярном соединении. Воздействие лекарственного средства определяли в плазме с использованием LC/MS.
Хронические in vivo эффекты соединений на модель астмы измеряли с использованием:
Модели OVA-индуцированной астмы у крыс
Самок крыс Brown Norway (возраста 7-9 недель) сенсибилизировали в день 0 и 7 при помощи 40 мкг овальбумина (OVA) (Sigma-Aldrich, St. Louis, MO) в 20 мг/мл раствора Alum Imject (Pierce, Rockford, IL). Затем крысам в день 19 и 20 вводили внутритрахеально 1,5 мкг OVA в 50 мкл PBS. Введение ингибитора начинали в день 18 и продолжали до дня 22. В день 22, через 48 часов после второго введения, крыс подвергали анестезии и испытанию ограниченной функции легких. Гиперреактивность дыхательных путей (AHR) оценивали с использованием плетизмографии всего тела. Вкратце, хирургическую плоскость анестезии индуцировали путем интраперитонеальной инъекции 60 мг/кг кетамина и 5 мг/кг ксилазина (Henry Schein, Inc., Melville, NY). Хирургическим путем вставляли трахеальную канюлю между 3 и 4 трахеальными кольцами. Спонтанное дыхание прекращали путем инъекции в яремную вену 0,12 мг/кг панкуроний бромида (Sigma-Aldrich, St Louis, MO). Животных помещали в плетизмограф, вмещающий животное целиком (Buxco Electronics, Inc., Wilmington, NC) и осуществляли механическую вентиляцию с 0,2 мл комнатного воздуха при 150 вдохов в минуту с использованием дыхательного аппарата с контролируемым объемом (Harvard Apparatus, Framingham, MA). Давление в легком и поток в плетизмографе измеряли с использованием трансдукторов и сопротивление легких рассчитывали как давление/поток с использованием программы Biosystem Xa (Buxco Electronics). Сопротивление воздушного потока измеряли на базовой линии и после введения 3, 10 и 30 мг/мл метахолина (Sigma Aldrich, St. Louis, MO) доставку осуществляли при помощи встроенного в линию ультразвукового небулайзера. После завершения испытания легочной функции легкие промывали 3 раза при помощи 1 мл стерильного PBS. Объем из первой промывки центрифугировали при 2000 об/мин в течение 5 минут и супернатант хранили для последующего анализа. Объем промывок 2-3 добавляли к осадку после центрифугирования первой промывки и затем обрабатывали для оценки клеточного инфильтрата методом проточной цитометрии. Плазму собирали из крови, которую брали из полой вены, и использовали для оценки концентраций лекарственного средства.
Раскрытия всех ссылочных документов, включая журнальные статьи, патенты и опубликованные патентные заявки, включены в настоящую заявку посредством ссылки во всей их полноте.
Следующие примеры предназначены для иллюстративных целей и не должны рассматриваться как ограничивающие объем настоящего изобретения.
ОБЩИЕ СХЕМЫ СИНТЕЗА
Соединения по настоящему изобретению можно получить с использованием синтетических преобразований, проиллюстрированных на Схемах I-XXVIII. Исходные вещества являются коммерчески доступными, могут быть получены с использованием процедур, описанных в настоящей заявке, с использованием процедур, описанных в литературе, или с использованием процедур, которые должны быть хорошо известны специалистам в области органической химии. Способы получения пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразиновых соединений по настоящему изобретению проиллюстрированы на Схеме I. На Схеме I, стадия a, коммерчески доступный 3,5-дибромпиразин-2-амин 1 подвергают взаимодействию с (триметилсилил)ацетиленом посредством реакции перекрестного связывания по методу Соногашира с использованием способов, известных специалистам в данной области (например, Пример #1 или WO2006058120A1), с получением алкина 2. Алкин 2 можно подвергнуть циклизации (Схема I, стадия b) с получением защищенного пирроло[2,3-b]пиразина 3 с использованием способов, известных специалистам в данной области (например, Пример #1 или WO2006058120A1). На Схеме I, стадия c, замещенный гидразин вводят путем взаимодействия с пирролопиразинами 3 в условиях аминирования по методу Бухвальда-Хартвига (например, Пример #1 или Advanced Synthesis & Catalysis 2004, 346, 1599-1626) с получением пирролопиразинов 4. Когда R'' представляет собой такую группу, что пирролопиразины 4 содержат гидразид (R''= -C(O)R'''), вещество непосредственно можно подвергнуть циклизации с пирролотриазолопиразинами 7 (Схема I, стадия h) с использованием условий, таких как условия, описанные в Общих Процедурах B или ZZZZ, или способами, известными специалистам в данной области (например, Bioorganic & Medicinal Chemistry Letters 2007, 17(12), 3373-3377 или Journal of Medicinal Chemistry 1990, 33(9), 2326-2334). Когда R'' представляет собой защитную группу, удаление защиты у соединений 4 (Схема I, стадия d) с получением гидразинилпирролопиразинов 5 можно осуществить с использованием условий, таких как условия, описанные в Общих Процедурах E, E.1, F, F.1, Y или BB; или Greene, T.W. and Wuts, P.G.M. "Protective Groups in Organic Synthesis, 3rd Edition", 1999, Wiley-Interscience. Например, защитную группу, такую как трет-бутоксикарбонильная (Boc) группа, можно удалить при помощи кислоты с использованием условий, таких как условия, описанные в Примере #1, Общих Процедурах E и E.1 или способами, известными специалистам в данной области (например, печатные издания Larock, R.C. "Comprehensive Organic Transformations: Guide to Functional Group Preparations, 2nd edition", 1999, Wiley-VCH или Greene, T.W. and Wuts, P.G.M., указанные выше). Образование гидразидов 6 из гидразинилпирролопиразинов 5 (Схема I, стадия e) можно осуществить различными способами, известными специалистам в данной области, включая in situ условия, такие как условия, описанные в Примере #1, Общей Процедуре А или стандартных способах пептидного связывания, таких которые можно найти в Larock, R.C., указанном выше. Гидразиды 6 можно подвергнуть циклизации с пирролотриазолопиразинами 7 с использованием условий, таких как условия, описанные в Примере #1, Общих Процедурах B, OO, OO.1 или ZZZZ, или способами, известными специалистам в данной области (например, Bioorganic & Medicinal Chemistry Letters 2007, 17(12), 3373-3377 или Journal of Medicinal Chemistry 1990, 33(9), 2326-34). Дальнейшую функционализацию гидразидов 6 или пирролотриазолопиразинов 7 можно осуществить, если это желательно, с использованием реакций, известных специалистам в данной области (например, Larock, R.C., указанный выше). Например, образование амидов, мочевин, сульфонамидов, ариламинов, гетероариламинов, сульфонилмочевин, замещенных аминов, скварамидов или гуанидинов можно осуществить из пирролотриазолопиразинов 7, содержащих первичный или вторичный амин (например, Общие Процедуры G, H, I, J, J.1, XXX, EEEE, K, K.1, L, DD, QQ, RR, YY, ZZ, затем AAA, CCC, YYY, X, X.1, TTTT или EEEEE). Также, удаление защиты у гидразидов 6 или пирролотриазолопиразинов 7, содержащих защищенный первичный или вторичный амин, можно осуществить с использованием условий, таких как условия, описанные в Greene, T.W. and Wuts, P.G.M., указанном выше, или в Общих Процедурах E, E.1, F, F.1 или BB. Например, для R''', содержащего защитную группу, такую как бензилоксикарбонильная (Cbz) группа, защитную группу можно удалить с получением незащищенного амина (например, Общие Процедуры F, F.1 и DDDDD), и соединения 7 с удаленной защитой затем можно подвергнуть дальнейшему взаимодействию, как описано выше. В некоторых случаях также могут происходить дополнительные реакции без выделения исходных пирролотриазолопиразинов 7, как можно видеть в Общей Процедуре C. Альтернативно, гидразинилпирролопиразины 5 непосредственно можно подвергнуть циклизации с пирролотриазолопиразинами 7 (Схема I, стадия i) с использованием условий, таких как условия, описанные в Общей Процедуре BBBBB. Удаление сульфонамидной защитной группы у пирролотриазолопиразинов 7 можно осуществить с использованием условий, таких как условия, описанные в Примере #1, Общих Процедурах D, XXX, AAAA, BBBB или CCCC, или способами, известными специалистам в данной области (например, печатные издания Larock, R.C. или Greene, T.W. and Wuts, P.G.M., указанные выше), с получением пирролотриазолопиразинов 8 (Схема I, стадия g). Дальнейшую функционализацию R''' группы в пирролотриазолопиразинах 8 можно осуществить, если это желательно, с использованием реакций, известных специалистам в данной области (например, Larock, R.C., указанный выше). Например, амиды, мочевины, сульфонамиды, ариламины, гетероариламины, сульфонилмочевины, замещенные амины, скварамиды или гуанидины можно получить из пирролотриазолопиразинов 8 с R''', содержащим первичный или вторичный амин (например, Примеры #8-9 или Общие Процедуры G, H, I, J, J.1, XXX, EEEE, K, K.1, L, DD, QQ, RR, YY, ZZ, затем AAA, CCC, YYY, X, X1, TTTT или EEEEE). Также, удаление защиты у R''' группы в пирролотриазолопиразинах 8 с получением незащищенного соединения можно осуществить с использованием условий, таких как условия, описанные в Greene, T.W. and Wuts, P.G.M., указанном выше, или в Общих Процедурах E, E.1, F, F.1, Y или BB. Например, защитную группу, такую как бензилоксикарбонильная (Cbz) группа, можно удалить из защищенного амина с получением незащищенного амина (например, Общие Процедуры F, F.1 и DDDDD), и соединения с удаленной защитой 8 затем можно подвергнуть дальнейшему взаимодействию, как описано выше.
Схема I
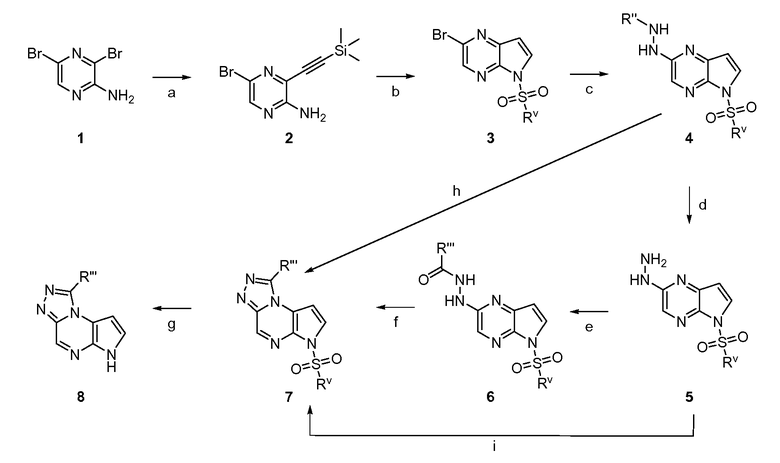
Способы получения имидазо[1,2-a]пирроло[2,3-e]пиразиновых соединений по настоящему изобретению проиллюстрированы на Схеме II. На стадии a, Pd-опосредованное карбонилирование пирролопиразинов 3 дает сложные эфиры 9 с использованием способов, известных специалистам в данной области, таких как способы, описанные в Примере #3; U.S. Pat. Appl. Publ., US 2007293509; или U.S. Pat. Appl. Publ., US 2008248537. Гидролиз сложных эфиров 9 дает кислоты 10 (Схема II, стадия b) с использованием хорошо известных условий, таких как условия, описанные в Примере #3 или Общей Процедуре Z. Перегруппировку Курциуса использовали для получения карбаматов 11, как показано на Схеме II, стадия c, с использованием условий, таких как условия, описанные в Примере #3 или указанные в Li, J.J. "Name Reactions. Collection of Detailed Reaction Mechanisms, 2nd edition", 2003, Springer: New York. Алкилирование пирролопиразин-2-илкарбаматов 11 с использованием подходяще замещенных 2-галогенметилкетонов (которые можно получить через процедуры, описанные в Общих Процедурах R и LLLL; Tetrahedron Letters, 1992, (33), 309-312) способами, известными специалистам в данной области (например, Общие Процедуры S или S.1; Tetrahedron Letters, 2006, 47(34), 6113-6115; или Journal of Medicinal Chemistry, 2005, 48(14), 4535-4546) дает пирролопиразины 12 (Схема II, стадия d). Удаление защиты у пирролопиразинов 12 с получением пирролопиразинов 13 (Схема II, стадия e) осуществляют с использованием условий, таких как условия, описанные в Общих Процедурах E и E.1 или в Greene, T.W. and Wuts, P.G.M., указанном выше. Как показано на Схеме II, стадия f, циклизацию пирролопиразинов 13 с получением имидазопирролопиразинов 14 можно осуществить способами, известными специалистам в данной области (например, Общие Процедуры T или KKKK; Пример #3, European Journal of Medicinal Chemistry, 2001, 36(3), 255-264; или Bioorganic and Medicinal Chemistry Letters, 2007, 17(5), 1233-1237). Дальнейшую функционализацию R''' группы в имидазопирролопиразинах 14 можно осуществить, если это желательно, с использованием реакций, известных специалистам в данной области (например, Larock, R.C., указанный выше). Например, амиды, мочевины, сульфонамиды, ариламины, гетероариламины, сульфонилмочевины, замещенные амины, скварамиды или гуанидины можно получить из имидазопирролопиразинов 14 с R''' группой, содержащей первичный или вторичный амин (например, Пример #3, Пример #7 или Общие Процедуры G, H, I, J, J.1, XXX, EEEE, K, K.1, L, DD, QQ, RR, YY, ZZ, затем AAA, CCC, YYY, X, X.1, TTTT или EEEEE). Также, удаление защиты R''' группы в имидазопирролопиразинах 14 с получением соединений 14, у которых удалена защита, можно осуществить с использованием условий, таких как условия, описанные в Greene, T.W. and Wuts, P.G.M., указанном выше, или в Общих Процедурах E, E.1, F, F.1, Y или BB, и соединения 14 с удаленной защитой затем можно подвергнуть дальнейшему взаимодействию, как описано выше. Удаление сульфонамидной защитной группы у имидазопирролопиразинов 14 можно осуществить с использованием условий, таких как условия, описанные в Примере #3, Общих Процедурах D, XXX, AAAA, BBBB или CCCC, или способами, известными специалистам в данной области (например, печатные издания Larock, R.C. или Greene, T.W. and Wuts, P.G.M., указанные выше), с получением имидазопирролопиразинов 15 (Схема II, стадия g).
Схема II

Способы получения имидазо[1,5-a]пирроло[2,3-e]пиразиновых соединений по настоящему изобретению проиллюстрированы на Схеме III. На стадии a, Pd-опосредованное цианирование бромидов 3 дает соответствующие нитрилы 16 (например, Пример #5 или Tetrahedron Letters 1999, 40(47), 8193-8195). Последующее восстановление нитрилов 16 дает амины 17 (Схема III, стадия b) с использованием способов, известных специалистам в данной области (например, Пример #5 или Journal of Medicinal Chemistry 2003, 46(4), 461-473). Сочетание аминов 17 с кислотами обеспечивает амиды 18 (Схема III, стадия c) с использованием хорошо известных условий, таких как указанные в Примере #5 или Общей Процедуре H. Как показано на Схеме III, стадия d, циклизацию амидов 18 можно осуществить путем преобразования в тиоамид с последующей обработкой активатором (таким как соль ртути, соль серебра или соль меди) с получением имидазо[1,5-a]пирроло[2,3-e]пиразинов 19 (например, Пример #5 или Общая Процедура Q). Альтернативно, когда R''' содержит азот, и, таким образом, соединения 18 представляют собой мочевины вместо амидов, тогда циклизацию до имидазо[1,5-a]пирроло[2,3-e]пиразинов 19 можно осуществить с использованием POCl3, как описано в Общей Процедуре OO или OO.1. Удаление защиты у сульфонамидных соединений 19 с получением имидазо[1,5-a]пирроло[2,3-e]пиразинов 20 (Схема III, стадия e) можно осуществить с использованием условий, таких как условия, описанные в Greene, T.W. and Wuts, P.G.M. "Protective Groups in Organic Synthesis, 3rd Edition", 1999, Wiley-Interscience, Общих Процедурах D, XXX, AAAA, BBBB или CCCC или Примере #5. Дальнейшую функционализацию R''' группы в имидазо[1,5-a]пирроло[2,3-e]пиразинах 19 или имидазо[1,5-a]пирроло[2,3-e]пиразинах 20 можно осуществить, если это желательно, с использованием реакций, известных специалистам в данной области (например, Larock, R.C., указанный выше). Например, образование амидов, мочевин, сульфонамидов, ариламинов, гетероариламинов, сульфонилмочевин, замещенных аминов, скварамидов или гуанидинов можно осуществить из соединений 19 или 20 с R''' группой, содержащей первичный или вторичный амин (например, Пример #6 или Общие Процедуры G, H, I, J, J.1, XXX, EEEE, K, K.1, L, DD, QQ, RR, YY, ZZ, затем AAA, CCC, YYY, X, X.1, TTTT или EEEEE). Также, удаление защиты R''' группы в соединениях 19 или 20 с получением незащищенного соединения можно осуществить с использованием условий, таких как условия, описанные в Greene, T.W. and Wuts, P.G.M., указанном выше, или в Общих Процедурах E, E.1, F или F.1, и соединения с удаленной защитой затем можно подвергнуть дальнейшему взаимодействию, как описано выше.
Схема III
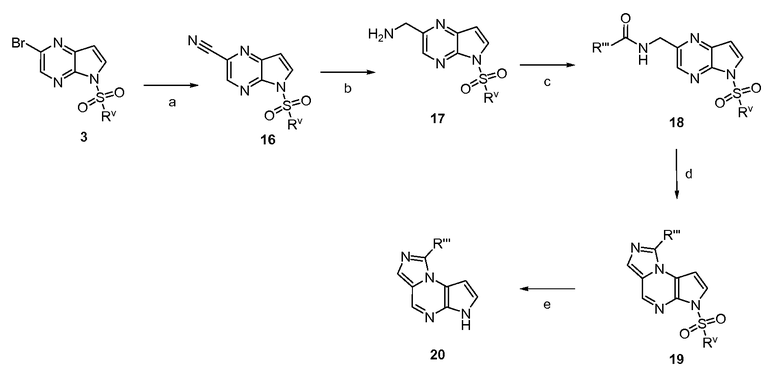
Способы получения 4-замещенных пиперидин-3-карбоновокислотных соединений по настоящему изобретению проиллюстрированы на Схеме IV. На стадии a, 4-замещенные никотиновые кислоты 21 можно сделать полностью насыщенными с использованием способов, которые известны специалистам в данной области (например, Общая Процедура O или Bioorganic and Medicinal Chemistry Letters 2004, 14(17), 4453-4459). Полученные пиперидины 22 могут быть защищены подходящей амино-защитной группой (Схема IV, стадия b), например, описанной в Greene, T.W. and Wuts, P.G.M. "Protective Groups in Organic Synthesis, 3rd Edition", 1999, Wiley-Interscience; Larock, R.C. "Comprehensive Organic Transformations: Guide to Functional Group Preparations, 2nd edition", 1999, Wiley-VCH или в Общих Процедурах M, M.1 или N, с получением защищенных пиперидинов 23.
Схема IV

Способы получения амино-замещенных циклопентилкарбоновых кислот 32 для применения в получении соединений по настоящему изобретению проиллюстрированы на Схеме V. На стадии a, β-кетоэфиры 24 могут быть конденсированы с алкил 4-хлорацетоацетатом 25 с получением солей циклического β-кетоэфиренолята 26 (например, Общая Процедура U). Декарбоксилирование соединений 26 с получением α,β-ненасыщенных кетонов 27 (Схема V, стадия b) осуществляют стандартными способами, известными специалистам в данной области (например, Общая Процедура V). Как показано на стадии c, гидрирование α,β-ненасыщенных кетонов 27 обеспечивает насыщенные кетоны 28 (например, Общие Процедуры W или W.1). Восстановительное аминирование кетонов 28 при помощи дибензиламина дает соединения 29 (Схема V, стадия d) с использованием условий, таких как условия, описанные в Общих Процедурах X или X.1. Дебензилирование соединений 29 можно осуществить через гидрирование, как описано в Общей Процедуре Y, с получением аминов 30 (Схема V, стадия e). Альтернативные условия можно использовать для получения аминов 30 из кетонов 28, например, как описано в Larock, R.C. "Comprehensive Organic Transformations: Guide to Functional Group Preparations, 2nd edition", 1999, Wiley-VCH (Схема V, стадия h). Амины 30 можно подвергнуть дальнейшей функционализации с использованием реакций, известных специалистам в данной области (например, Larock, R.C., указанный выше). Например, амиды, мочевины, сульфонамиды, ариламины, гетероариламины, сульфонилмочевины, замещенные амины, скварамиды или гуанидины можно получить из аминов 30 (например, Общие Процедуры G, H, I, J, J.1, XXX, EEEE, K, K.1, L, DD, QQ, RR, YY, ZZ, затем AAA, CCC, YYY, X, X.1, TTTT или EEEEE) с получением соединения 31 (Схема V, стадия f). Соединения 31 в форме сложного эфира могут быть гидролизованы в водно-щелочных или кислотных условиях с получением желаемых карбоновых кислот 32 (Схема V, стадия g) с использованием условий, таких как условия, описанные в Общих Процедурах Z или TT или Larock, R.C., указанном выше). Альтернативно, соединения 29 в форме сложных эфиров могут быть гидролизованы с получением промежуточных карбоновокислотных соединений 32', как показано на Схеме V, стадия i, с использованием водно-щелочных или кислотных условий (например, Получение #TT.1).
Схема V
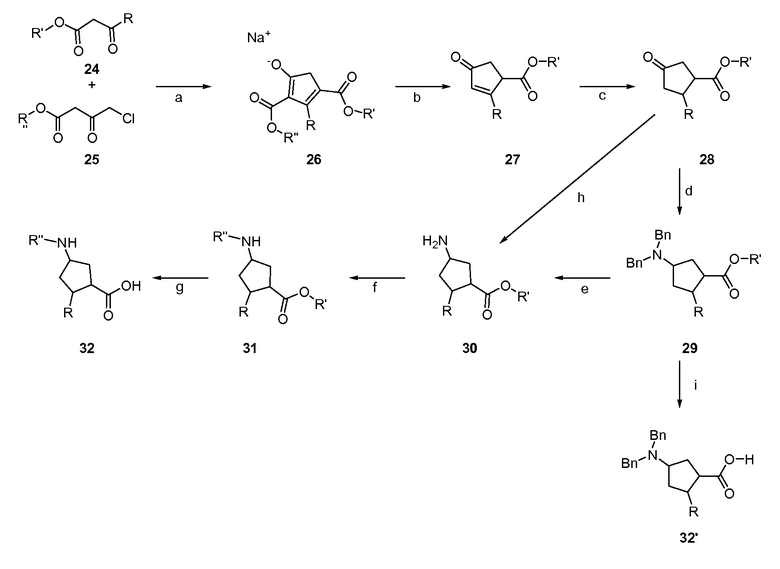
Способы получения эфир-замещенных 1-циклопентил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразиновых соединений по настоящему изобретению проиллюстрированы на Схеме VI. Как показано на стадии a, восстановление α,β-ненасыщенных кетонов 27 при сопутствующем восстановлении кетона обеспечивает насыщенные спирты 33 (например, хиральные восстановительные условия, описанные в Примере #4). Альтернативные условия можно использовать для получения спиртов 33 из кетонов 28 через восстановление (Схема VI, стадия b), как описано в Общей Процедуре P или в Larock, R.C. "Comprehensive Organic Transformations: Guide to Functional Group Preparations, 2nd edition", 1999, Wiley-VCH. Спирты 33 можно подвергнуть взаимодействию с получением эфиров 34 (Схема VI, стадия c) с использованием условий, таких как условия, описанные в Общей Процедуре EE (для этого сначала может потребоваться получение 2,2,2-трихлоримидата, как описано в Общей Процедуре UU), II, JJ или VV, с последующим осуществлением Общей Процедуры FFF, или способов, известных специалистам в данной области (например, Tet. Lett. 1983, 24(48), 5363 или Greene, T.W. and Wuts, P.G.M. "Protective Groups in Organic Synthesis, 3rd Edition", 1999, Wiley-Interscience). Соединения 34 в форме сложных эфиров могут быть гидролизованы в водно-щелочных или кислотных условиях с получением желаемых карбоновых кислот 35 (Схема VI, стадия d) с использованием условий, таких как условия, описанные в Общей Процедуре Z или TT или в Larock, R.C., указанном выше. Образования гидразидов 36 из гидразинилпирролопиразинов 5 и карбоновых кислот 35 (Схема VI, стадия e) можно осуществить различными способами, известными специалистам в данной области, например, описанными в Общей Процедуре А, или стандартными способами пептидного связывания, такими которые можно найти в Larock, R.C., указанном выше. Гидразиды 36 можно подвергнуть циклизации с получением пирролотриазолопиразинов 37 (Схема VI, стадия f) с использованием условий, таких как условия, описанные в Общих Процедурах B или ZZZZ, или способами, известными специалистам в данной области (например, Bioorganic & Medicinal Chemistry Letters 2007, 17(12), 3373-3377 или Journal of Medicinal Chemistry 1990, 33(9), 2326-2334). Удаление сульфонамидной защитной группы в пирролотриазолопиразинах 37 можно осуществить с использованием условий, таких как условия, описанные в Общих Процедурах D, AAAA, BBBB или CCCC, или способами, известными специалистам в данной области (например, печатные издания Larock, R.C. или Greene, T.W. and Wuts, P.G.M., указанные выше), с получением пирролотриазолопиразинов 38 в качестве конечных продуктов или промежуточных соединений (Схема VI, стадия g). Пирролотриазолопиразины 38 могут быть SEM-защищены (Схема VI, стадия h) с использованием условий, таких как условия, описанные в Общей Процедуре KK или описанные в Greene, T.W. and Wuts, P.G.M., указанном выше. Когда R'' группа в пирролотриазолопиразинах 37 или 39 представляет собой защитную группу, защита может быть удалена с получением спиртов 40 (Схема VI, стадия k) или 43 (Схема VI, стадия i), соответственно, с использованием условий, таких как условия, описанные в Greene, T.W. and Wuts, P.G.M., указанном выше. Например, защитную группу, такую как п-метоксибензильная (PMB) группа, можно удалить из PMB-эфира с получением незащищенного спирта (например, Общая Процедура FF), и соединения с удаленной защитой 40 или 43 затем можно подвергнуть дальнейшему взаимодействию. Реакцию Мицунобу спиртов 40 или 43 можно использовать для получения простых эфиров или сложных эфиров 41 (Схема VI, стадия l), 44 (Схема VI, стадия j) или 45 (Схема VI, стадия n) с инверсией в реакционном центре с использованием условий, таких как условия, описанные в Общей Процедуре II, или способов, известных специалистам в данной области, которые можно найти в Larock, R. C., указанном выше. Кроме того, простые эфиры 44 можно получить из спиртов 43 через алкилирование с использованием условий, таких как условия, описанные в Общей Процедуре HHHH. Альтернативно, спирты 40 или 43 можно преобразовать в карбаматы 41 или 44 с использованием хорошо известных условий, таких как условия, описанные в Общих Процедурах OOO, WWW и PPPP. Удаление сульфонамидной защитной группы у пирролотриазолопиразинов 41 можно осуществить с использованием условий, таких как условия, описанные в Общих Процедурах D, AAAA, BBBB, CCCC или PPPP, или способами, известными специалистам в данной области (например, печатные издания Larock, R.C. или Greene, T.W. and Wuts, P.G.M., указанные выше), с получением пирролотриазолопиразинов 42 (Схема VI, стадия m). Можно осуществить расщепление сложноэфирной группы сложных эфиров 45 с получением незащищенных спиртов 46 (Схема VI, стадия o) с использованием условий, таких как условия, описанные в Общей Процедуре SS. Спирты 46 можно подвергнуть дальнейшему взаимодействию с образованием эфиров 44 (Схема VI, стадия p) через химическую процедуру Мицунобу (как описано для Схемы VI, стадия j) или с использованием условий, таких как условия, описанные в Общей Процедуре EE (для этого сначала может потребоваться получение 2,2,2-трихлоримидата, как описано в Общей Процедуре UU) или JJ или способами, известными специалистам в данной области (например, печатное издание Larock, R.C., указанное выше). Защитную группу SEM в пирролотриазолопиразинах 44 можно удалить способами, такими как способы, описанные в Общих Процедурах LL и LL.1, или с использованием условий, таких как описанные в Greene, T.W. and Wuts, P.G.M., указанном выше, с получением пирролотриазолопиразинов 42 (Схема VI, стадия q).
Схема VI
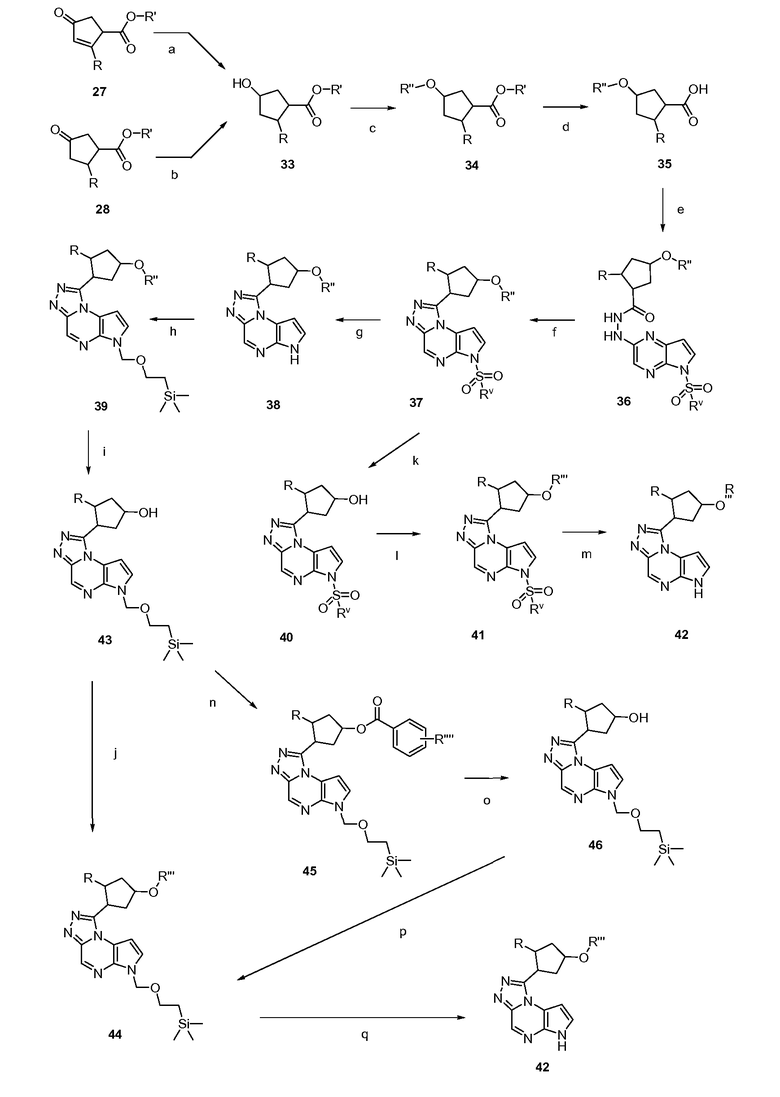
Альтернативный способ получения эфир-замещенных 1-циклопентил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразиновых соединений по настоящему изобретению проиллюстрирован на Схеме VII. Как показано на стадии a, соединения 33 в форме сложных эфиров могут быть гидролизованы в водно-щелочных или кислотных условиях с получением желаемых карбоновых кислот 47, с использованием условий, таких как условия, описанные в Общих Процедурах Z или TT или в Larock, R.C., указанном выше. Образование лактонов 48 из карбоновых кислот 47 (Схема VII, стадия b) можно осуществить способами, такими как способы, описанные в Примере #4, Общей Процедуре GG, или способами, известными специалистам в данной области, которые можно найти в Larock, R.C., указанном выше. Образование гидразидов 49 из гидразинилпирролопиразинов 5 и лактонов 48 (Схема VII, стадия c) можно осуществить различными способами, известными специалистам в данной области, например, описанными в Примере #4 или Общей Процедуре HH. Спирты 49 можно подвергнуть взаимодействию с образованием эфиров 50 (Схема VII, стадия d) с использованием условий, таких как условия, описанные в Общих Процедурах VV (для этого сначала может потребоваться получение 2,2,2-трихлоримидата, как описано в Общей Процедуре UU) или JJ, или способами, известными специалистам в данной области (например, Tet. Lett. 1983, 24(48), 5363). Реакцию Мицунобу спиртов 49 можно использовать для получения эфиров 51 (Схема VII, стадия f) с инверсией в реакционном центре с использованием условий, таких как условия, описанные в Примере #4, Общей Процедуре II, или способами, известными специалистам в данной области, которые можно найти в Larock, R. C., указанном выше. Гидразиды 50 или 51 можно подвергнуть циклизации с получением пирролотриазолопиразинов 37 (Схема VII, стадия e) или 41 (Схема VII, стадия g) с использованием условий, таких как условия, описанные в Примере #4, Общих Процедурах B или ZZZZ, или способами, известными специалистам в данной области (например, Bioorganic & Medicinal Chemistry Letters 2007, 17(12), 3373-3377 или Journal of Medicinal Chemistry 1990, 33(9), 2326-2334). Дальнейшее преобразование соединения 37 или 41 можно осуществить, как описано на Схеме VI.
Схема VII
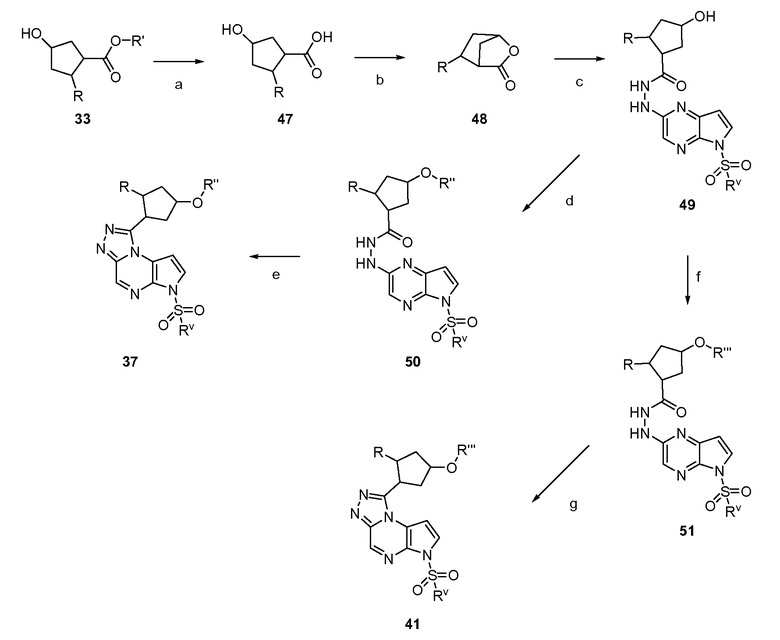
Способы получения 1,6-дигидроимидазо[4,5-d]пирроло[2,3-b]пиридиновых соединений по настоящему изобретению проиллюстрированы на Схеме VIII. Как показано на стадии a, 4-хлор-3-иодпиридин-2-амин 52 может быть нитрирован с получением 4-хлор-3-иод-5-нитропиридин-2-амина 53, как описано в Примере #21 или в Larock, R.C. "Comprehensive Organic Transformations: Guide to Functional Group Preparations, 2nd edition", 1999, Wiley-VCH. 4-Хлор-3-иод-5-нитропиридин-2-амин 53 подвергают взаимодействию с (триметилсилил)ацетиленом посредством реакции перекрестного связывания по методу Соногашира с использованием способов, известных специалистам в данной области (например, Пример #21 или WO2006058120A1), с получением 4-хлор-5-нитро-3-((триметилсилил)этинил)пиридин-2-амина 54 (Схема VIII, стадия b). Как показано на стадии c, 4-хлор-5-нитро-3-((триметилсилил)этинил)пиридин-2-амин 54 подвергают процедуре удаления защиты с получением 4-хлор-3-этинил-5-нитропиридин-2-амина 55, как описано в Примере #21, или с использованием способов, известных специалистам в данной области (например, печатные издания Greene, T.W. and Wuts, P.G.M. "Protective Groups in Organic Synthesis, 3rd Edition", 1999, Wiley-Interscience или Larock, R.C., указанные выше). 4-Хлор-3-этинил-5-нитропиридин-2-амин 55 можно подвергнуть циклизации, как показано на стадии d, с получением 4-хлор-5-нитро-1H-пирроло[2,3-b]пиридина 56, как описано в Примере #21, или способами, известными специалистам в данной области (например, как описано в WO2008004117). Как показано на стадии e, амино-замещенные 1H-пирроло[2,3-b]пиридины 57 получают с использованием способов, известных специалистам в данной области (например, Пример #21 или Larock, R.C., указанный выше). Диамино-замещенные 1H-пирроло[2,3-b]пиридины 58 (Схема VIII, стадия f) получают в результате восстановления нитро-содержащих 1H-пирроло[2,3-b]пиридинов 57 с использованием способов, известных специалистам в данной области (например, Пример #21, Общая Процедура BBB или Larock, R.C., указанный выше). Как показано на стадии g, диамино-замещенные 1H-пирроло[2,3-b]пиридины 58 можно подвергнуть циклизации, как описано в Примере #21 или в Общей Процедуре DDD, с получением 1,6-дигидроимидазо[4,5-d]пирроло[2,3-b]пиридинов 59. Дальнейшую функционализацию R группы в 1,6-дигидроимидазо[4,5-d]пирроло[2,3-b]пиридинах 59 можно осуществить, если это желательно, с использованием реакций, известных специалистам в данной области (например, Larock, R.C., указанный выше). Например, амиды, мочевины, сульфонамиды, ариламины, гетероариламины, сульфонилмочевины, замещенные амины, скварамиды или гуанидины можно получить из 1,6-дигидроимидазо[4,5-d]пирроло[2,3-b]пиридина 59 с R группой, содержащей первичный или вторичный амин (например, Общие Процедуры G, H, I, J, J.1, XXX, EEEE, K, K.1, L, DD, QQ, RR, YY, ZZ, затем AAA, CCC, YYY, X, X.1, TTTT или EEEEE). Также, удаление защиты R группы в 1,6-дигидроимидазо[4,5-d]пирроло[2,3-b]пиридинах 59 с получением незащищенного соединения можно осуществить с использованием условий, таких как условия, описанные в Greene, T.W. and Wuts, P.G.M., указанном выше, или в Общих Процедурах E, E.1, F, F.1, Y или BB. Например, защитную группу, такую как бензилоксикарбонильная (Cbz) группа, можно удалить из защищенного амина с получением незащищенного амина (например, Общие Процедуры F, F.1 или Y), и соединение 59 с удаленной защитой затем можно подвергнуть дальнейшему взаимодействию, как описано выше. Альтернативно, промежуточные соединения 56 или 57 могут быть сульфонамид- защищены с использованием реакций, известных специалистам в данной области (например, Greene, T.W. and Wuts, P.G.M., указанные выше, или Общая Процедура K.1), с получением сульфонамидов 134 и 135, соответственно (Схема VII, стадии h и m). Как показано на стадии i, амино-замещенные 1H-пирроло[2,3-b]пиридины 135 также можно получить из хлор-замещенных 1H-пирроло[2,3-b]пиридинов 134 с использованием способов, известных специалистам в данной области (например, Пример #23 или Larock, R.C., указанный выше). Диамино-замещенные 1H-пирроло[2,3-b]пиридины 136 получают в результате восстановления нитро-содержащих 1H-пирроло[2,3-b]пиридинов 135 с использованием способов, известных специалистам в данной области (например, Пример #23, Общая Процедура BBB или Larock, R.C., указанный выше). Как показано на стадии k, диамино-замещенные 1H-пирроло[2,3-b]пиридины 136 можно подвергнуть циклизации, как описано в Примере #23 или Общей Процедуре DDD, с получением сульфонамид-защищенных 1,6-дигидроимидазо[4,5-d]пирроло[2,3-b]пиридинов 137. Удаление защиты у сульфонамидных соединений 137 с получением 1,6-дигидроимидазо[4,5-d]пирроло[2,3-b]пиридинов 59 (Схема VIII, стадия l) можно осуществить с использованием условий, таких как условия, описанные в Greene, T.W. and Wuts, P.G.M. "Protective Groups in Organic Synthesis, 3rd Edition", 1999, Wiley-Interscience, Общие Процедуры D, XXX, AAAA, BBBB или CCCC или Пример #23.
Схема VIII

Способы получения замещенных циклопентиламинов 61 для применения в получении соединений по настоящему изобретению проиллюстрированы на Схеме IX. На стадии a, карбоновые кислоты 32 подвергают перегруппировке Курциуса, как описано в Общей Процедуре NNN, с образованием изоцианатов 60. Гидролиз изоцианатов 60 дает амины 61 (например, Общая Процедура OOO).
Схема IX

Способы получения 4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)цикло-пентанонов и их производных в качестве соединений по настоящему изобретению проиллюстрированы на Схеме X. На стадии a, кетоны 28 защищают в виде кеталей 62 с использованием условий, описанных в Общей Процедуре WW, или как описано в Greene, T.W. and Wuts, P.G.M., указанном выше. Соединения в форме сложных эфиров 62 могут быть гидролизованы в водно-щелочных условиях с получением желаемых карбоновых кислот 63 (Схема X, стадия b) с использованием условий, таких как условия, описанные в Общей Процедуре Z или в Larock, R.C., указанном выше. Образование гидразидов 64 из гидразинилпирролопиразинов 5 и карбоновых кислот 63 (Схема X, стадия c) можно осуществить различными способами, известными специалистам в данной области, например, описанных в Общей Процедуре А, или стандартными способами пептидного связывания, такими которые можно найти в Larock, R.C., указанном выше. Гидразиды 64 можно подвергнуть циклизации с получением пирролотриазолопиразинов 65 (Схема X, стадия d) с использованием условий, таких как условия, описанные в Общих Процедурах B или ZZZZ, или способами, известными специалистам в данной области (например, Bioorganic & Medicinal Chemistry Letters 2007, 17(12), 3373-3377 или Journal of Medicinal Chemistry 1990, 33(9), 2326-2334). Кетали 65 можно подвергнуть процедуре удаления защиты с получением кетонов 66, как описано в Получении #25 или в Greene, T.W. and Wuts, P.G.M., указанном выше. Удаление сульфонамидной защитной группы в пирролотриазолопиразинах 66 можно осуществить с использованием условий, таких как условия, описанные в Общих Процедурах D, AAAA, BBBB или CCCC, или способами, известными специалистам в данной области (например, Greene, T.W. and Wuts, P.G.M., указанный выше), с получением пирролотриазолопиразинов 67 в качестве конечных продуктов или промежуточных соединений (Схема X, стадия f). Например, стадия g иллюстрирует образование оксимэфиров 68 из кетонов 67, и это можно осуществить с использованием условий, таких как условия, описанные в Общей Процедуре PPP или в Larock, R.C., указанном выше.
Схема Х

Способы получения уксусной кислоты и ацетамидных производных из 4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентанонов в качестве соединений по настоящему изобретению проиллюстрированы на Схеме XI. Как показано на стадии a, реакцию кетонов 66 по методу Хорнера-Вадсворта-Эммонса с получением алкенов 69 можно осуществить с использованием процедур, известных специалистам в данной области, например, описанных в Общей Процедуре III. Удаление сульфонамидной защитной группы в пирролотриазолопиразинах 69 можно осуществить с использованием условий, таких как условия, описанные в Общих Процедурах D, AAAA, BBBB или CCCC, или способами, известными специалистам в данной области (например, Greene, T.W. and Wuts, P.G.M., указанный выше), с получением пирролотриазолопиразинов 70 (Схема XI, стадия b). Гидрирование алкенов 70, как описано в Общих Процедурах W или W.1, дает пирролотриазолопиразины 71 (Схема XI, стадия c). Гидролиз сложных эфиров 71 обеспечивает кислоты 72 (Схема XI, стадия d) с использованием хорошо известных условий, таких как условия, описанные в Общей Процедуре Z. Кислоты 72 можно подвергнуть дальнейшему взаимодействию с получением амидов 73, как показано на стадии e, с использованием условий, таких как условия, описанные в Общей Процедуре H.
Схема XI
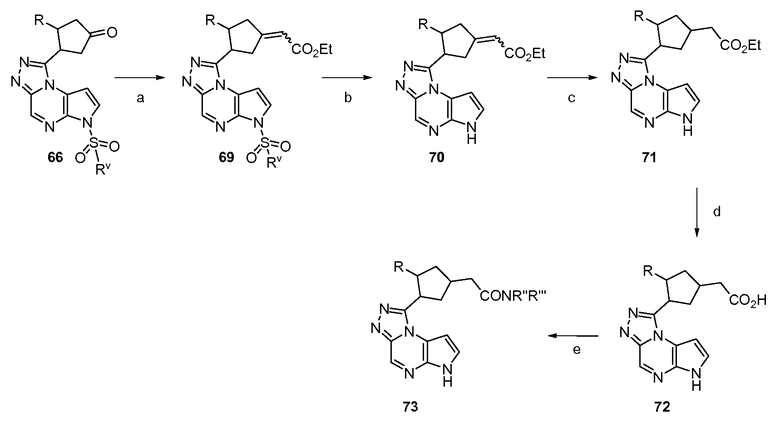
Способы получения 4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)цикло-пентиламинов в качестве соединений по настоящему изобретению проиллюстрированы на Схеме XII. Как показано на стадии a, восстановительное аминирование кетонов 66 с получением аминов 74 можно осуществить с использованием хорошо известных условий, таких как условия, описанные в Общих Процедурах X или X.1. Удаление сульфонамидной защитной группы в пирролотриазолопиразинах 74 можно осуществить с использованием условий, таких как условия, описанные в Общих Процедурах D, AAAA, BBBB или CCCC, или способами, известными специалистам в данной области (например, Greene, T.W. and Wuts, P.G.M., указанный выше), с получением пирролотриазолопиразинов 75 (Схема XII, стадия b).
Схема XII
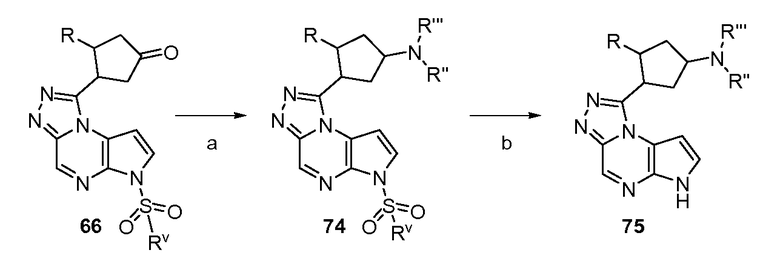
Способы получения дигидропиразоло[4,3-d]пирроло[2,3-b]пиридиновых соединений по настоящему изобретению проиллюстрированы на Схеме XIII. На стадии a, взаимодействие 5-хлор-4-иод-1-(триизопропилсилил)-1H-пирроло[2,3-b]пиридина 76 с замещенным альдегидом обеспечивает спирты 77 с использованием процедур, таких как описанные в Примере #29, Примере #30 или в WO2009152133. Получение кетонов 78 (стадия b) можно осуществить путем обработки спиртов 77 окислителем способами, известными специалистам в данной области (например, Пример #29, Пример # 30 или Larock, R.C., указанный выше). Кетоны 78 затем можно преобразовать в гидразоны 79 с потерей TIPS защитной группы через взаимодействие с гидразином с использованием условий, таких как условия, описанные в Примере #29, Примере #30 или Общей Процедуре XXXX. Циклизацию гидразонов 79 с получением дигидропиразоло[4,3-d]пирроло[2,3-b]пиридинов 80 можно осуществить через внутримолекулярную циклизацию по методу Бухвальда-Хартвига (например, Общая Процедура XX или Organic Letters, 2008, 10(18), 4109-4112). Дальнейшую функционализацию R''' группы в дигидропиразоло[4,3-d]пирроло[2,3-b]пиридинах 80 можно осуществить, если это желательно, с использованием реакций, известных специалистам в данной области (например, Larock, R.C., указанный выше). Например, амиды, мочевины, сульфонамиды, ариламины, гетероариламины, сульфонилмочевины, замещенные амины, скварамиды или гуанидины можно получить из дигидропиразоло[4,3-d]пирроло[2,3-b]пиридинов 80 с R''' группой, содержащей первичный или вторичный амин (например, Общие Процедуры G, H, I, J, J.1, XXX, EEEE, K, K.1, L, DD, QQ, RR, YY, ZZ, затем AAA, CCC, YYY, X, X,1, TTTT или EEEEE). Также, удаление защиты R''' группы в дигидропиразоло[4,3-d]пирроло[2,3-b]пиридинах 80 с получением соединения 80 с удаленной защитой можно осуществить с использованием условий, таких как условия, описанные в Greene, T.W. and Wuts, P.G.M., указанном выше, или в Общих Процедурах E, E.1, F, F.1, Y или BB, и соединения 80 с удаленной защитой затем можно подвергнуть дальнейшему взаимодействию, как описано выше.
Схема XIII
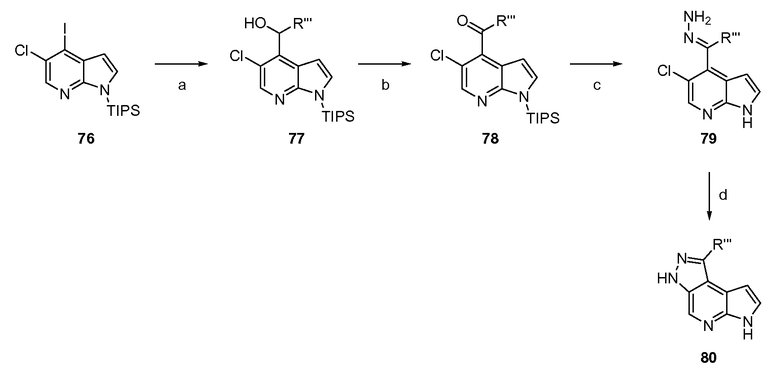
Способы получения 2,3,4,7-тетрагидро-1H-пирроло[2,3-h][1,6]нафтиридиновых соединений по настоящему изобретению проиллюстрированы на Схеме XIV. На стадии a, o-литиирование 4-хлор-1-(триизопропилсилил)-1H-пирроло[2,3-b]пиридина 81 с последующим захватом аниона при помощи этилхлорформиата дает этил 4-хлор-1-(триизопропилсилил)-1H-пирроло[2,3-b]пиридин-5-карбоксилат 82 с использованием условий, описанных в Примере #28. Удаление TIPS группы в соединении 82 можно осуществить, как показано на стадии b, с получением этил 4-хлор-1H-пирроло[2,3-b]пиридин-5-карбоксилата 83 с использованием условий, хорошо известных из литературы (например, Greene, T.W. and Wuts, P.G.M., указанный выше, или Пример #28). На стадии c, сульфонамид защищенные соединения 84 получают с использованием реакций, известных специалистам в данной области (например, Greene, T.W. and Wuts, P.G.M., указанный выше, или Пример #28). Как показано на стадии d, амино-замещенные 1H-пирроло[2,3-b]пиридины 85 получают с использованием способов, известных специалистам в данной области (например, Пример #28 или Larock, R.C., указанный выше). Восстановление сложных эфиров 85 до спиртов 86 (Схема XIV, стадия e) можно осуществить с использованием условий, хорошо известных из литературы, (например, Пример #28 или Larock, R.C., указанный выше). На стадии f, спирты 86 окисляют до альдегидов 87 с использованием способов, известных специалистам в данной области (например, Пример #28 или Larock, R.C., указанный выше). Реакция Виттига альдегидов 87 с ((1,3-диоксолан-2-ил)метил)трифенилфосфонийбромидом (Схема XIV, стадия g) обеспечивает алкены 88 с использованием условий, таких как условия, описанные в Примере #28. Восстановление алкенов 88 можно осуществить с использованием условий, таких как условия, описанные в Примере #28, или Общие Процедуры W или W.1 (Схема XIV, стадия h). Циклизацию аминоацеталей 89 с получением защищенных 2,3,4,7-тетрагидро-1H-пирроло[2,3-h][1,6]нафтиридинов 90 осуществляют с использованием условий, описанных в Примере #28 (Схема XIV, стадия i). Удаление сульфонамидной защитной группы у 2,3,4,7-тетрагидро-1H-пирроло[2,3-h][1,6]нафтиридинов 90 можно осуществить с использованием условий, таких как условия, описанные в Общих Процедурах D, AAAA, BBBB или CCCC, или способами, известными специалистам в данной области (например, Greene, T.W. and Wuts, P.G.M., указанный выше), с получением 2,3,4,7-тетрагидро-1H-пирроло[2,3-h][1,6]нафтиридинов 91 (Схема XIV, стадия j). Дальнейшую функционализацию R' группы в 2,3,4,7-тетрагидро-1H-пирроло[2,3-h][1,6]нафтиридинах 91 можно осуществить, если это желательно, с использованием реакций, известных специалистам в данной области (например, Larock, R.C., указанный выше). Например, амиды, мочевины, сульфонамиды, ариламины, гетероариламины, сульфонилмочевины, замещенные амины, скварамиды или гуанидины можно получить из 2,3,4,7-тетрагидро-1H-пирроло[2,3-h][1,6]нафтиридинов 91 с R' группой, содержащей первичный или вторичный амин (например, Общие Процедуры G, H, I, J, J.1, XXX, EEEE, K, K.1, L, DD, QQ, RR, YY, ZZ, затем AAA, CCC, YYY, X, X.1, TTTT или EEEEE). Также, удаление защиты R' группы в 2,3,4,7-тетрагидро-1H-пирроло[2,3-h][1,6]нафтиридинах 91 с получением соединений 91 с удаленной защитой можно осуществить с использованием условий, таких как условия, описанные в Greene, T.W. and Wuts, P.G.M., указанном выше, или в Общих Процедурах E, E.1, F, F.1, Y или BB, и соединения 91 с удаленной защитой затем можно подвергнуть дальнейшему взаимодействию, как описано выше.
Схема XIV

Способы получения замещенных имидазо[1,5-a]пирроло[2,3-e]пиразиновых соединений по настоящему изобретению проиллюстрированы на Схеме XV. Как показано на стадии a, имидазо[1,5-a]пирроло[2,3-e]пиразины 19 можно подвергнуть галогенированию с использованием условий, таких как условия, описанные в Общей Процедуре ММ, с получением 3-галоген-имидазо[1,5-a]пирроло[2,3-e]пиразинов 92. 3-Галоген-имидазо[1,5-a]пирроло-[2,3-e]пиразины 92 можно подвергнуть различным реакциям, известным специалистам в данной области (например, Larock, R.C., указанный выше), включая, но не ограничиваясь этим, цианирование, как описано в Общей Процедуре HHH (Схема XVI, стадия c), или реакции сочетания по методу Сузуки, такие как реакции, описанные в Общих Процедурах UUU или VVV (Схема XV, стадия b). Удаление сульфонамидной защитной группы у имидазо[1,5-a]пирроло[2,3-e]пиразинов 93 можно осуществить с использованием условий, таких как условия, описанные в Общих Процедурах D, UUU, AAAA, BBBB или CCCC, или способами, известными специалистам в данной области (например, Greene, T.W. and Wuts, P.G.M., указанный выше), с получением имидазо[1,5-a]пирроло[2,3-e]пиразинов 94 (Схема XV, стадия c).
Схема XV
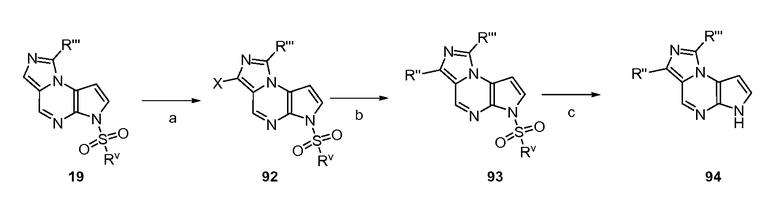
Способы получения пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразиновых соединений по настоящему изобретению проиллюстрированы на Схеме XVI. Пирролотриазолопиразины 8 можно подвергнуть галогенированию с использованием условий, таких как условия, описанные в Общих Процедурах GGG или GGG.1, с получением 8-галогенпирролотриазоло-пиразинов 95 (Схема XVI, стадия a). На стадии b, 8-галогенпирролотриазолопиразины 95 могут быть защищены SEM группой с использованием условий, известных из литературы, которые можно найти в Greene, T.W. and Wuts, P.G.M., указанном выше, или как в Общей Процедуре KK. Полученные SEM-защищенные 8-галогенпирролотриазолопиразины 96 можно подвергнуть различным реакциям, известным специалистам в данной области (например, Larock, R.C., указанный выше), включая, но не ограничиваясь этим, цианирование, как описано в Общей Процедуре HHH (Схема XVI, стадия c), реакцию сочетания по методу Сузуки, как описано в Получении #23, образование сложного эфира карбоновой кислоты, как описано в Общей Процедуре AAAAA, или реакции сочетания по методу Стилле, как описано в Общей Процедуре CCCCC (Схема XVI, стадия e). Полученные продукты 97 или 99 можно подвергнуть процедуре удаления защиты с использованием условий, таких как условия, описанные в Общей Процедуре LL, LL.1, или способами, известными специалистам в данной области (например, Greene, T.W. and Wuts, P.G.M., указанный выше), с получением пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразинов 98 или 100 (Схема XV, стадии d и f, соответственно). Кроме того, соединения 99 и 100 можно подвергнуть дальнейшей функционализации, если это желательно, с использованием реакций, известных специалистам в данной области (например, Larock, R.C., указанный выше). Например, для R = CO2Et, соединение может быть гидролизовано с использованием условий, таких как условия, описанные в Общей Процедуре D, и затем его можно подвергнуть процедуре образования амидной связи, как описано в Общей Процедуре H.
Схема XVI
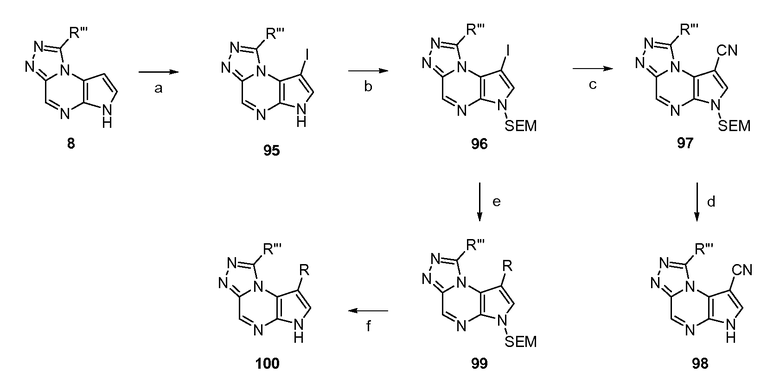
Способы получения замещеных 4-(сульфонамидометил)циклопентанкарбоновых кислот 110 для применения в получении соединений по настоящему изобретению проиллюстрированы на Схеме XVII. На стадии a, 5-замещенные-бицикло[2,2,1]гепт-2-ены 101 окисляют до дикарбоновых кислот 102 с использованием известных условий, таких как условия, описанные в Получении #11, Bioorganic & Medicinal Chemistry, 2007, 15, 7581 или Journal of Organic Chemistry, 1993, 58, 4745. Образование моноэфиров 103 осуществляют через циклический ангидрид, как описано в Получении #11 (Схема XVII, стадия b). Трет-бутиловые эфиры 104 получают на стадии c, используя стандартные условия, например, описанные в Получени #11 или в Larock, R.C., указанном выше. Восстановление формы метилового эфира соединений 104 до спиртов 105 осуществляют с использованием хорошо известных условий, таких которые можно найти в Получении #21 (Схема XVII, стадия d). Мезилаты 106 получают, как описано в Получении #21, или способами, известными специалистам в данной области (Схема XVII, стадия e). Как показано на стадии f, мезилаты 106 можно использовать для образования азидов 107 с использованием хорошо известных условий, таких как условия, описанные в Получении #21 или в Larock, R.C., указанном выше (Схема XVII, стадия f). Восстановление азидов 107 до аминов 108 включает стандартное преобразование, которое можно осуществить, как описано в Получении #21, или с использованием условий, таких как условия, описанные в Общей Процедуре TTT или в Larock, R.C., указанном выше (Схема XVII, стадия g). Стадия h показывает образование сульфонамидов 109 из аминов 108, которое осуществляют, как описано в Общих Процедурах K или K.1, или способами, известными специалистам в данной области (например, Greene, T.W. and Wuts, P.G.M., указанный выше). Кислотное расщепление трет-бутиловых эфиров 109 с получением 4-(сульфонамидометил)цикло-пентанкарбоновых кислот 110 (Схема XVII, стадия i) можно осуществить с использованием условий, описанных в Общей Процедуре QQQ, или способами, известными специалистам в данной области (например, печатные издания Larock, R.C. или Greene, T.W. and Wuts, P.G.M., указанные выше).
Схема XVII

Способы получения 4-((дибензиламино)метил)-2-замещенных-циклопентанкарбоновых кислот 115 для применения в получении соединений по настоящему изобретению проиллюстрированы на Схеме XVIII. Восстановление формы метилового эфира соединений 103 до спиртов 111 осуществляют с использованием хорошо известных условий, таких которые можно найти в Получении #22 или в Larock, R.C., указанном выше (Схема XVIII, стадия a). Стадия b иллюстрирует образование сложных эфиров 112, которое осуществляют, как описано в Получении #22 или в Larock, R.C., указанном выше. На стадии c, спирты 112 окисляют до альдегидов 113 с использованием известных условий, таких как условия, описанные в Получении #22 или в Larock, R.C., указанном выше. Восстановительное аминирование альдегидов 113 с использованием условий, таких как условия, описанные в Общих Процедурах X или X.1, дает амины 114 (Схема XVIII, стадия d). На стадии e, сложные эфиры 114 гидролизуют с получением 4-((дибензиламино)метил)-2-замещенный-циклопентан-карбоновых кислот 115 с использованием условий, таких как условия, описанные в Общих Процедурах Z или TT, или известные специалистам в данной области (например, печатные издания Larock, R.C. или Greene, T.W. and Wuts, P.G.M., указанные выше).
Схема XVIII

Способы получения 3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-аминовых соединений по настоящему изобретению проиллюстрированы на Схеме XIX. Алкилирование пирролопиразин-2-илкарбаматов 11 с использованием трет-бутил 2-бромацетата, способами, известными специалистам в данной области (например, Общие Процедуры S или S.1), дает пирролопиразины 116 (Схема XIX, стадия a). Двойное удаление защиты у пирролопиразинов 116 с получением аминоуксусных кислот 117 можно осуществить с использованием условий, таких как условия, описанные в Общих Процедурах E, E.1 или QQQ (Схема XIX, стадия b). Сочетание кислот 117 с аминами обеспечивает амиды 118 (Схема XIX, стадия c) с использованием хорошо известных условий, таких как указанные в Общей Процедуре H или в Larock, R.C., указанном выше. Как показано на стадии d, циклизацию амидов 118 до имидазопирролопиразин-8-аминов 119 можно осуществить с использованием условий, таких как условия, описанные в Общих Процедурах OO или OO.1. Дальнейшую функционализацию R' или R'' группы в имидазопирролопиразин-8-аминах 119 можно осуществить, если это желательно, с использованием реакций, известных специалистам в данной области (например, Larock, R.C., указанный выше). Например, амиды, мочевины, сульфонамиды, ариламины, гетероариламины, сульфонилмочевины, замещенные амины, скварамиды или гуанидины можно получить из имидазопирролопиразин-8-аминов 119 с R' или R'' группой, содержащей первичный или вторичный амин (например, Общие Процедуры G, H, I, J, J.1, XXX, EEEE, K, K.1, L, DD, QQ, RR, YY, ZZ, затем AAA, CCC, YYY, X, X.1, TTTT или EEEEE). Также, удаление защиты R' или R'' группы в имидазопирролопиразин-8-аминах 119 с получением соединений 119, у которых удалена защита, можно осуществить с использованием условий, таких как условия, описанные в Greene, T.W. and Wuts, P.G.M., указанном выше, или в Общих Процедурах E, E.1, F, F.1, Y или BB, и соединения 119 с удаленной защитой затем можно подвергнуть дальнейшему взаимодействию, как описано выше. Удаление сульфонамидной защитной группы в имидазопирролопиразин-8-аминах 119 можно осуществить с использованием условий, таких как условия, описанные в Общих Процедурах D, XXX, AAAA, BBBB или CCCC, или способами, известными специалистам в данной области (например, печатные издания Larock, R.C. или Greene, T.W. and Wuts, P.G.M., указанные выше), с получением имидазопирролопиразин-8-аминов 120 (Схема XIX, стадия e).
Схема XIX
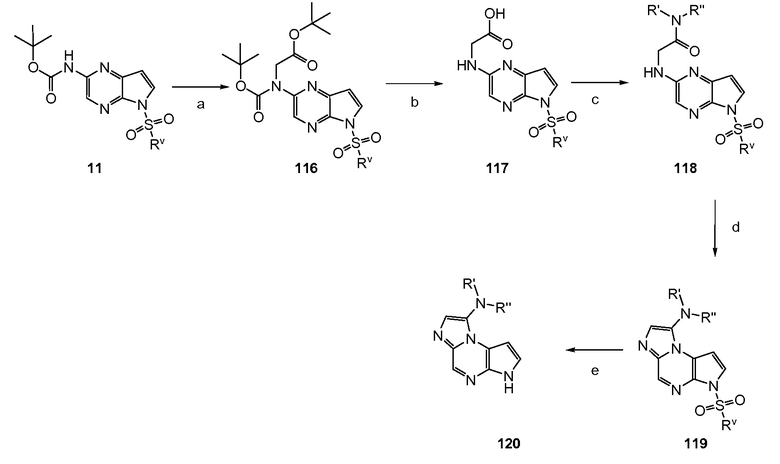
Способы получения пирролидин-3-карбоновых кислот 125 для применения в получении соединений по настоящему изобретению проиллюстрированы на Схеме XX. На стадии a, алкины 121 восстанавливают до алкенов 122, как описано в Общей Процедуре RRR, или с использованием способов, известных специалистам в данной области (например, Larock, R.C., указанный выше). 1,3-диполярное циклоприсоединение алкенов 122 и N-замещенный-1-метокси-N-((триметилсилил)метил)метанамина 123 с получением пирролидинов 124 (Схема XX, стадия b) можно осуществить способами, известными специалистам в данной области (например, Общая Процедура SSS или Journal of Medicinal Chemistry, 2009, 52(24), 7946-7949). Соединения 124 в форме сложных эфиров могут быть гидролизованы в водно-щелочных или кислотных условиях с получением карбоновых кислот 125 (Схема XX, стадия c) с использованием условий, таких как условия, описанные в Общих Процедурах Z или TT или в Larock, R.C., указанном выше.
Схема ХХ
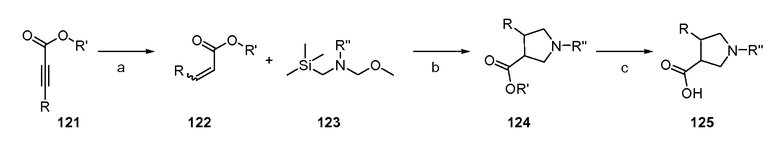
Способы получения сульфон-замещенных 1-циклопентил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразиновых соединений по настоящему изобретению проиллюстрированы на Схеме XXI. Как показано на стадии a, реакция Мицунобу спиртов 46 с подходящими тиолами дает сульфиды 126 при использовании условий, таких как условия, описанные в Общей Процедуре MMM, или способами, известными специалистам в данной области (например, Larock, R.C., указанный выше). Окисление сульфидов 126 до сульфонов 127 (Схема XXI, стадия b) осуществляют, как описано в Общей Процедуре LLL, или способами, известными специалистам в данной области (например, Larock, R.C., указанный выше). SEM защитную группу пирролотриазолопиразинов 127 можно удалить способами, такими как способы, описанные в Общих Процедурах LL и LL.1, или с использованием условий, таких как описанные в Greene, T.W. and Wuts, P.G.M., указанном выше, с получением пирролотриазолопиразинов 128 (Схема XXI, стадия c).
Схема XXI

Способы получения сульфонилхлоридов 133 для применения в получении соединений по настоящему изобретению проиллюстрированы на Схеме XXII. На стадии a, сульфонаты 130 получают их сульфонилхлоридов 129 с использованием известных реакционных условий, таких как условия, описанные в Получении #6 Стадия A, WO2007014011 или WO2009018238. Дополнительный заместитель добавляют к сульфонатам 130 с получением сульфонатов 131 через алкилирование, как описано в Общей Процедуре KKK, WO2007014011 или WO2009018238 (Схема XXII, стадия b). На стадии c, сульфонаты калия 132 получают из сульфонатов 131 при помощи водного цианата калия с использованием условий, таких как описанные в Общей Процедуре JJJ, WO2007014011 или WO2009018238. Сульфонаты калия 132 преобразовывают в сульфонилхлориды 133 (Схема XXII, стадия d) с использованием тионилхлорида, как описано в Общей Процедуре EEE, WO2007014011 или WO2009018238.
Схема XXII

Способы получения имидазопирроло[2,3-e]пиразиновых соединений по настоящему изобретению проиллюстрированы на Схеме XXIII. Имидазопирролопиразины 15 [T=N, U=CH] или 20 [T=CH, U=N] можно подвергнуть галогенированию с использованием условий, таких как условия, описанные в Общих Процедурах GGG или GGG.1, с получением 8-галогенимидазопирролопиразинов 138 (Схема XXIII, стадия a). На стадии b, 8- галогенимидазопирролопиразины 138 могут быть защищены SEM группой с использованием условий, известных из литературы, которые можно найти в Greene, T.W. and Wuts, P.G.M., указанном выше, или как в Общей Процедуре KK. Полученные SEM-защищенные 8-галогенимидазопирролопиразины 139 можно подвергнуть различным реакциям, известным специалистам в данной области (например, Larock, R.C., указанный выше), включая, но не ограничиваясь этим, цианирование, как описано в Общей Процедуре HHH (Схема XXIII, стадия c), или реакции сочетания по методу Сузуки, как описано в Общей Процедуре VVV, или реакции сочетания по методу Стилле, как описано в Общей Процедуре CCCCC (Схема XXIII, стадия e). Полученные продукты 140 или 142 можно подвергнуть процедуре удаления защиты с использованием условий, таких как условия, описанные в Общих Процедурах LL и LL.1, или способами, известными специалистам в данной области (например, Greene, T.W. and Wuts, P.G.M., указанный выше), с получением имидазопирроло[2,3-e]пиразинов 141 или 143 (Схема XXIII, стадии d и f, соответственно).
Схема XXIII
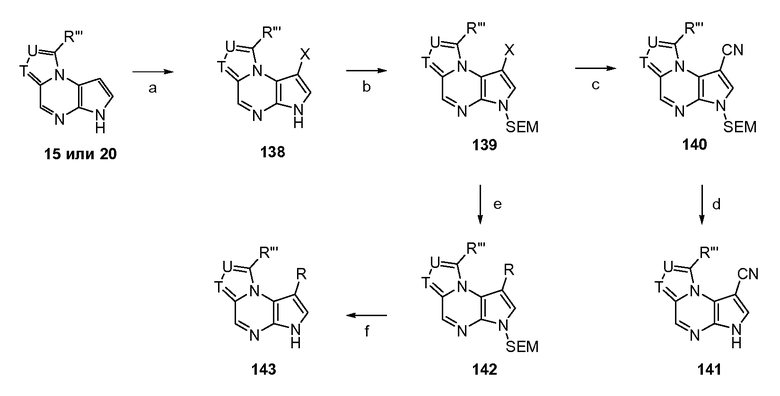
Способы получения соединений по настоящему изобретению из обычного кетонового промежуточного соединения проиллюстрированы на Схеме XXIV. Как показано на Схеме XXIV, стадия a, кетоны 144 можно подвергнуть взаимодействию алкиллитиевым реагентом или реагентом Гриньяра с получением спиртов 145 с использованием условий, известных специалистам в данной области (например, Larock, R.C., указанный выше, или Общая Процедура ZZZ). Альтернативно, кетоны 144 можно подвергнуть реакции Хорнера-Вадсворта-Эммонса с реагентом, таким как этил 2-(диэтоксифосфорил)ацетат (R''' = CO2Et) или диэтилцианометилфосфонат (R''' = CN), как описано в Общей Процедуре III, с получением алкенов 146 (Схема XXIV, стадия b). Алкены 146 можно подвергнуть гидрированию до алканов 147 с использованием хорошо известных условий, таких как условия, описанные в Общей Процедуре W и W.1 (Схема XXIV, стадия c). R''' группу можно подвергнуть дальнейшей функционализации с использованием различных реакций, таких как описанные в Larock, R.C., указанном выше. Например, для R''' = CO2Et, спирты 148 можно получить, как описано в Общей Процедуре ZZZ (Схема XXIV, стадия d), или оксадиазолы 149 можно получить, как описано в Общей Процедуре DDDD (Схема XXIV, стадия e). Как показано на Схеме XXIV, стадия f, кетоны 144 также могут быть восстановлены до спиртов 150, как описано в Общей Процедуре P или в Larock, R.C., указанном выше. Мезилаты 151 получают из спиртов 150 с использованием условий, известных специалистам в данной области, например, описанных в Общей Процедуре IIII (Схема XXIV, стадия g), и можно подвергнуть взаимодействию с различными нуклеофилами (Nu), как описано в Общей Процедуре JJJJ (Схема XXIV, стадия h), с получением соединений 152. В зависимости от используемого нуклеофила, можно осуществить дальнейшую функционализацию с получением соединений 153 (Схема XXIV, стадия i). Эти функционализации можно осуществить с использованием способов, таких как описанные в Larock, R.C., указанном выше, или в Общих Процедурах QQQQ или UUUU.
Схема XXIV
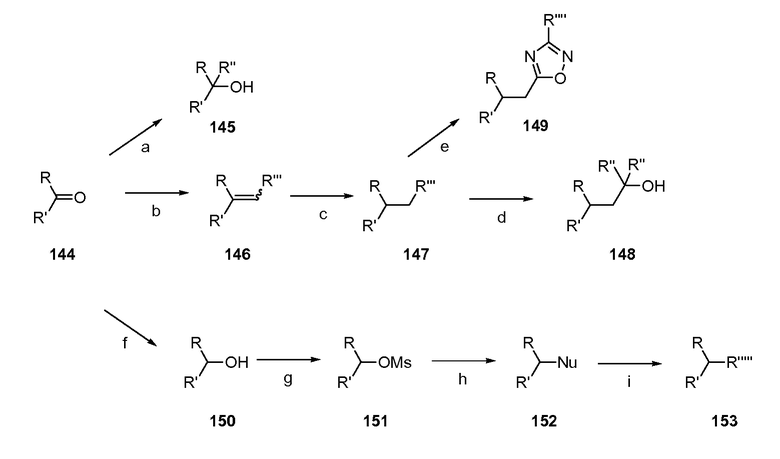
Способы получения 1,6-дигидроимидазо[4,5-d]пирроло[2,3-b]пиридин-2-аминовых соединений по настоящему изобретению проиллюстрированы на Схеме XXV. Диамины 136 (Схема XVIII) можно подвергнуть взаимодействию с цианогенбромидом, как описано в Общей Процедуре RRRR (Схема XXV, стадия a). Полученные 1,6-дигидроимидазо[4,5-d]пирроло[2,3-b]пиридин-2-амины 154 можно подвергнуть дальнейшей функционализации, если это желательно, с использованием реакций, известных специалистам в данной области (см., например, Larock, R.C. выше), с получением 1,6-дигидроимидазо[4,5-d]пирроло[2,3-b]пиридин-2-аминов 155 (Схема XXV, стадия b). Удаление сульфонамидной защитной группы в 1,6-дигидроимидазо[4,5-d]пирроло[2,3-b]пиридин-2-аминах 155 или 154 можно осуществить с использованием условий, таких как условия, описанные в Общих Процедурах D, AAAA, BBBB или CCCC, или способами, известными специалистам в данной области (например, печатные издания Larock, R.C. или Greene, T.W. and Wuts, P.G.M., указанные выше), с получением 1,6-дигидроимидазо[4,5-d]пирроло[2,3-b]пиридин-2-аминов 156 или 157, соответственно (Схема XXV, стадии c и d).
Схема XXV
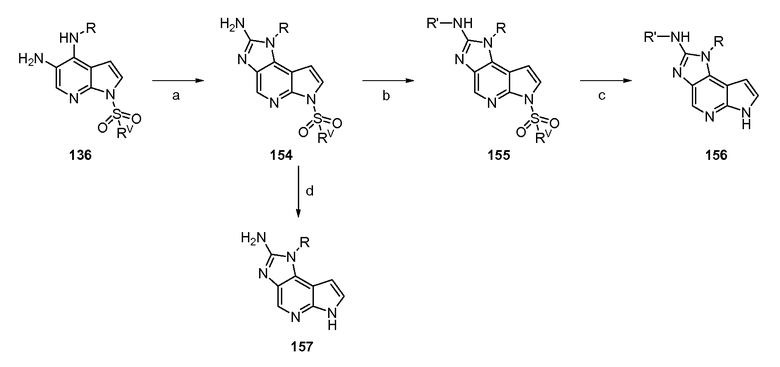
Способы получения 1,6-дигидропирроло[2,3-b][1,2,3]триазоло[4,5-d]пиридиновых соединений по настоящему изобретению проиллюстрированы на Схеме XXV. Диамины 136 (Схема XVIII) можно подвергнуть взаимодействию с нитритом натрия, как описано в Общей Процедуре SSSS, с получением 1,6-дигидропирроло[2,3-b][1,2,3]триазоло[4,5-d]пиридинов 158 (Схема XXVI, стадия a). Удаление сульфонамидной защитной группы в 1,6-дигидроимидазо[4,5-d]пирроло[2,3-b]пиридин-2-аминах 158 можно осуществить с использованием условий, таких как условия, описанные в Общих Процедурах D, AAAA, BBBB или CCCC, или способами, известными специалистам в данной области (например, печатные издания Larock, R.C. или Greene, T.W. and Wuts, P.G.M., указанные выше), с получением 1,6-дигидропирроло[2,3-b][1,2,3]триазоло[4,5-d]пиридинов 159 (Схема XXV, стадия b).
Схема XXVI
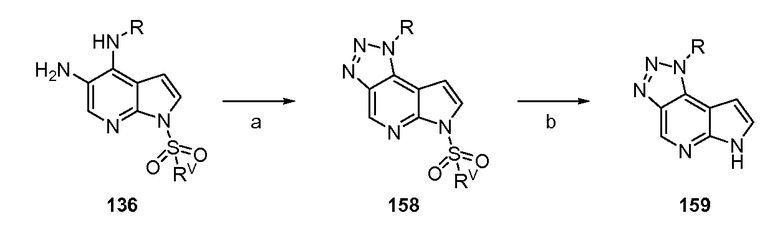
Альтернативные способы получения кетоновых промежуточных соединений, используемых для получения дигидропиразоло[4,3-d]пирроло[2,3-b]пиридиновых соединений по настоящему изобретению, проиллюстрированы на Схеме XXVII. На стадии a, карбоновые кислоты 160 преобразовывают в соответствующие хлорангидриды кислот 161 с использованием условий, хорошо известных специалистам в данной области, например, описанных в Общей Процедуре WWWW. Хлорангидриды кислот 161 подвергают взаимодействию с 5-хлор-1-(триизопропилсилил)-1H-пирроло[2,3-b]пиридином 162, как описано в Общей Процедуре VVVV (Схема XXVII, стадия b), с получением кетонов 78, которые можно подвергнуть дальнейшему взаимодействию, как описано на Схеме XIII.
Схема XXVII

Альтернативные способы получения имидазо[1,2-a]пирроло[2,3-e]пиразиновых соединений по настоящему изобретению проиллюстрированы на Схеме XXVIII. На стадии a, карбоновые кислоты 160 преобразовывают в соответствующие илиды сульфоксония 163 с использованием условий, таких как условия, описанные в Общей Процедуре FFFFF или в J. Org. Chem. 2004, 69, 1629. Пирролопиразин-2-амины 164 можно получить из пирролопиразин-2-илкарбаматов 11 (Схема II) с использованием условий, известных специалистам в данной области, например, описанных в Общей Процедуре E или в Greene, T.W. and Wuts, P.G.M., указанном выше (Схема XXVIII, стадия b). Илиды сульфоксония 163 подвергают взаимодействию с пирролопиразин-2-аминами 164 с использованием условий, таких как условия, описанные в Общей Процедуре GGGGG или в Org. Lett. 2009, 11, 3566, с получением пирролопиразинов 13, которые можно подвергнуть дальнейшему взаимодействию, как описано на Схеме II (Схема XXVIII, стадия c). Альтернативно, как показано на стадии d, пирролопиразин-2-амины 164 можно подвергнуть взаимодействию с α-галогенальдегидом с использованием условий, таких как описанные в Общей Процедуре YYYY, с получением имидазопирролопиразинов 14, которые можно подвергнуть дальнейшему взаимодействию, как описано на Схеме II.
Схема XXVIII

Если это желательно, хиральное разделение любого из хиральных соединений на Схемах I-XXVIII можно осуществить с использованием способов, известных специалистам в данной области, таких как хиральная препаративная ВЭЖХ или хиральная SFC (например, Общая Процедура AA) или кристаллизация диастереомерной соли, как описано в Примере #5. Дальнейшую функционализацию любой из R групп выше (например, R, R'', R''', R'''' и R''''') можно осуществить, если это желательно, с использованием реакций, известных специалистам в данной области (например, Larock, R.C., указанный выше). Например, образование амидов, мочевин, сульфонамидов, ариламинов, гетероариламинов, сульфонилмочевин, замещенных аминов, скварамидов или гуанидинов можно получить с R группой, содержащей первичный или вторичный амин (например, Общие Процедуры G, H, I, J, J.1, XXX, EEEE, K, K.1, L, DD, QQ, RR, YY, ZZ, затем AAA, CCC, YYY, X, X,1, TTTT или EEEEE). Также, удаление защиты R группы с получением соединений, у которых удалена защита, можно осуществить с использованием условий, таких как условия, описанные в Greene, T.W. and Wuts, P.G.M., указанном выше, или в Общих Процедурах E, E.1, F, F.1, Y или BB, и соединения с удаленной защитой затем можно подвергнуть дальнейшему взаимодействию, как описано выше.
ОБЩИЕ ПРОЦЕДУРЫ И ПРИМЕРЫ
Общие схемы синтеза, которые использовали для образования большинства соединений, раскрытых в настоящей заявке, описаны ниже на Схемах 1-111. Эти схемы представлены только для иллюстративных целей, и их не следует рассматривать как ограничивающие объем настоящего изобретения.
Схема 1. Образование гидразида из карбоновой кислоты (Общая Процедура A)

Схема 2. Циклизация гидразида (Общая Процедура B)

Схема 3. Циклизация гидразида с потерей Boc-защитной группы (Общая Процедура C)

Схема 4. Гидролиз сульфонамида (Общая Процедура D)

Схема 5. Кислотное расщепление Boc-защищенного амина (Общие Процедуры E и E.1)

Схема 6. Удаление защиты у Cbz-защищенного амина с использованием HBr в AcOH (Общие Процедуры F и F.1)
Схема 7. Образование ацетамида (Общая Процедура G)

Схема 8. Образование амида из карбоновой кислоты и амина (Общая Процедура H)

Схема 9. Образование мочевины из амина и карбамоилхлорида (Общая Процедура I)
Схема 10. Образование мочевины (X = O) или тиомочевины (X = S) с использованием CDI или тиокарбонилдиимидазола, соответственно (Общие Процедуры J и J.1)

Схема 11. Образование сульфонамида из амина (Общая Процедура K и K.1)

Схема 12. Замещение арила или гетероарилгалогенида амином (Общая Процедура L)

Схема 13. Boc-защита амина (Общие Процедуры M и M.1)

Схема 14. Cbz-защита амина (Общая Процедура N)
Схема 15. Восстановление пиридина (Общая Процедура O)
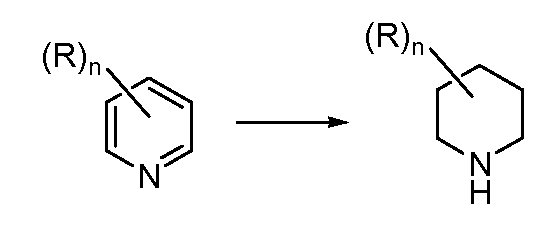
Схема 16. Восстановление карбонила до спирта (Общая Процедура P)

Схема 17. Циклизация амида с использованием дитиафосфетанового реагента (Общая Процедура Q)

Схема 18. Образование бромметилкетона из кислоты (Общая Процедура R)

Схема 19. N-Алкилирование с использованием алкилгалогенида, α-галогенкетона или α-галогенамида (Общие Процедуры S и S.1)

Схема 20. Циклизация кетона с использованием дитиафосфетанового реагента (Общая Процедура T)

Схема 21. Конденсация по методу Кневенагеля с образованием замещенного циклопентадиена (Общая Процедура U)
Схема 22. Декарбоксилирование β-кетоэфиренолята (Общая Процедура V)

Схема 23. Гидрирование алкена (Общие Процедуры W и W.1)

Схема 24. Восстановительное аминирование кетона или альдегида (R' = H) (Общие Процедуры X и X.1)

Схема 25. Гидрирование бензил- или Cbz-защищенного амина (Общая Процедура Y)
Схема 26. Щелочной гидролиз сложного эфира до карбоновой кислоты (Общая Процедура Z)
Схема 27: Разделение стереоизомеров методом хиральной препаративной ВЭЖХ(Общая Процедура AA)

Схема 28: Кислотный гидролиз ацетил-защищенного амина (Общая Процедура BB)

Схема 29: Образование сульфамоилхлорида (Общая Процедура CC)

Схема 30: Образование сульфонилмочевины (Общая Процедура DD)
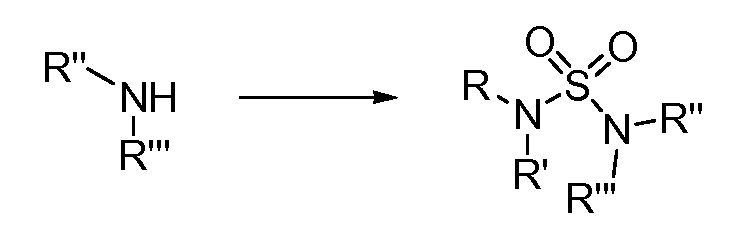
Схема 31: Образование эфира из трихлорацетимидатного производного (Общая Процедура ЕЕ)

Схема 32: Удаление защиты у PMB-защищенного спирта (Общая Процедура FF)

Схема 33: Образование лактона (Общая Процедура GG)
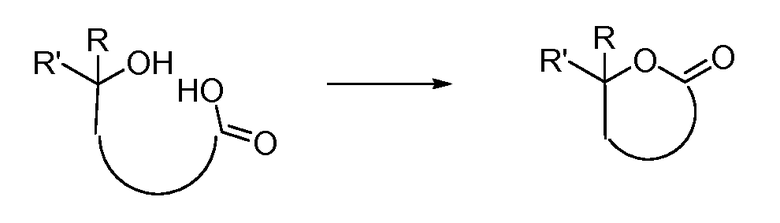
Схема 34: Раскрытие лактона с использованием амина или гидразина (Общая Процедура HH)
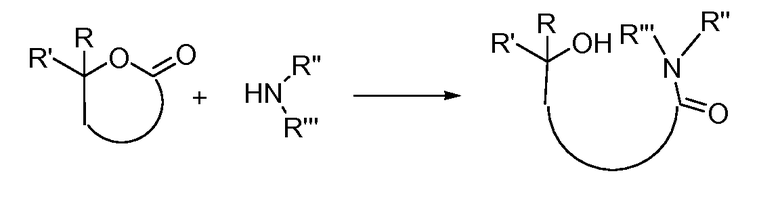
Схема 35: Реакция Мицунобу спирта (Общая Процедура II)

Схема 36: Замещение галогенида спиртом (Общая Процедура JJ)
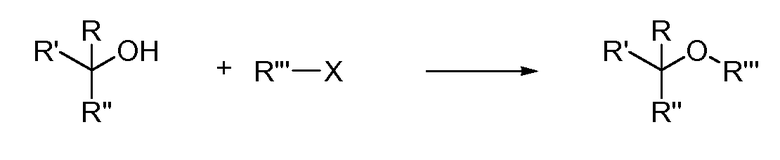
Схема 37: SEM защита азота (Общая Процедура KK)

Схема 38: Удаление SEM-защиты азота (Общие Процедуры LL и LL.1)

Схема 39: Галогенирование имидазола (Общая Процедура ММ)

Схема 40: Образование амида из карбоновой кислоты и амин с потерей сульфонамидной защитной группы (Общая Процедура NN)

Схема 41: Циклизация с POCl3 (Общие Процедуры OO и OO.1)
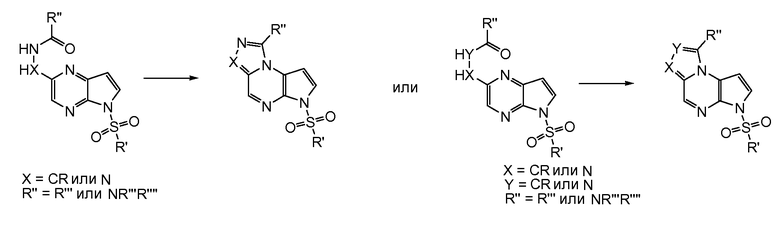
Схема 42: Взаимодействие амина с арилбороновой кислотой (Общая Процедура PP)

Схема 43: Образование мочевины из амина и изоцианата (Общая Процедура QQ)

Схема 44: Образование мочевины из амина, гетероариламина и фенилхлорформиата (Общая Процедура RR)

Схема 45: Гидролиз сложного эфира до спирта (Общая Процедура SS)

Схема 46: Кислотно-опосредованное преобразование сложного эфира в карбоновую кислоту (Общая Процедура TT)
Схема 47: Образование 2,2,2-трихлорацетимидата (Общая Процедура UU)
Схема 48: Образование TBDMS-защищенного спирта (Общая Процедура VV)
Схема 49: Образование кеталя (Общая Процедура WW)
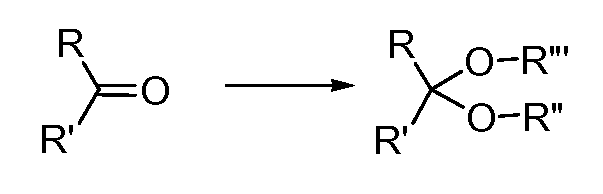
Схема 50: Катализируемое палладием связывание гидразона (Общая Процедура XX)

Схема 51: Присоединение по Михаэлю амина к α,β-ненасыщенному сульфонамиду (Общая Процедура YY)

Схема 52: Образование оксазолидинонсульфономочевины (Общая Процедура ZZ)

Схема 53: Образование сульфонилмочевины из оксазолидинонсульфономочевины (Общая Процедура AAA)
Схема 54: Восстановление нитрогрупы (Общая Процедура BBB)
Схема 55: Образование амида (Общая Процедура CCC)

Схема 56: Циклизация с образованием конденсированного имидазола (Общая Процедура DDD)

Схема 57: Образование сульфонилхлорида (Общая Процедура EEE)

Схема 58: Образование эфира в восстановительных условиях ((Общая Процедура FFF)

Схема 59: Иодирование, хлорирование или бромирование гетероцикла или галогенирование гетероцикла (Общие Процедуры GGG и GGG.1)

Схема 60: Цианирование гетероцикла (Общая Процедура HHH)

Схема 61: Взаимодействие кетона методом Хорнера-Вадсворта-Эммонса (Общая Процедура III)

Схема 62: Образование сульфоната калия (Общая Процедура JJJ)

Схема 63: Алкилирование сульфоната (Общая Процедура KKK)

Схема 64: Окисление тиоэфира до сульфона (Общая Процедура LLL)

Схема 65: Реакция Мицунобу с использованием тиола (Общая Процедура MMM)

Схема 66: Реакция Курциуса с образованием изоцианата (Общая Процедура NNN)

Схема 67: Гидролиз изоцианата (Общая Процедура OOO)
Схема 68: Образование оксимэфира из кетона (Общая Процедура PPP)

Схема 69: TFA-опосредованное преобразование сложного эфира до карбоновой кислоты (Общая Процедура QQQ)
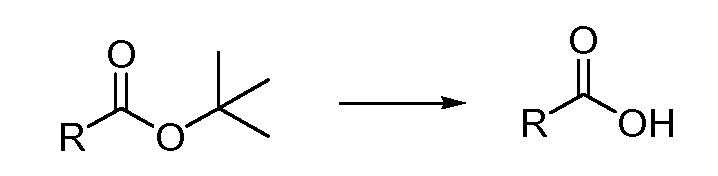
Схема 70: Восстановление алкина до алкена (Общая Процедура RRR)

Схема 71: 1,3-диполярное циклоприсоединение с образованием пирролидина (Общая Процедура SSS)
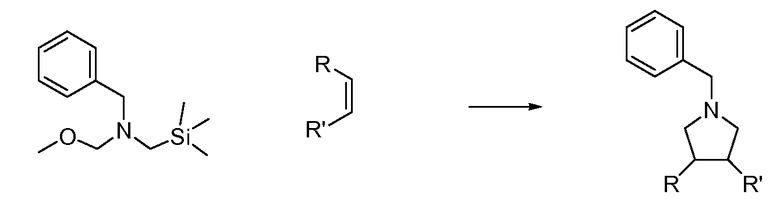
Схема 72: Гидрирование азида до амина (Общая Процедура TTT)
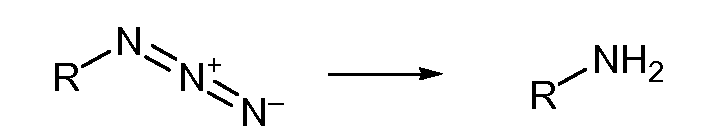
Схема 73: Взаимодействие арил или гетероарилгалогенида с бороновой кислотой или боронатным эфиром с последующим удалением тозильной защитной группы (Общая Процедура UUU)

Схема 74: Взаимодействие арил или гетероарилгалогенида с бороновой кислотой или боронатным эфиром (Общая Процедура VVV)
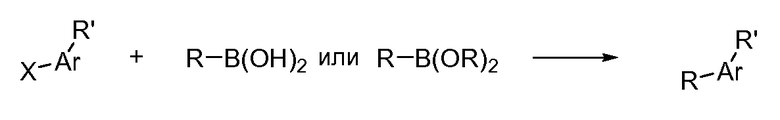
Схема 75: Образование карбамата (Общая Процедура WWW)
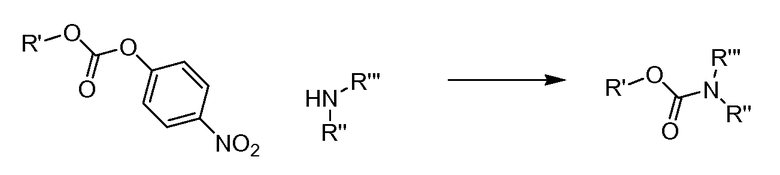
Схема 76: Образование мочевины с потерей защитной группы (Общая Процедура XXX)

Схема 77: Присоединение по Михаэлю (Общая Процедура YYY)

Схема 78: Добавление реагента Гриньяра или алкиллития к карбонил-содержащему соединению (Общая Процедура ZZZ)

Схема 79: Удаление защитной группы сульфонамида с использованием DBU (Общая Процедура AAAA)

Схема 80: Удаление защитной группы сульфонамида с использованием TBAF (Общая Процедура BBBB)

Схема 81: Удаление защитной группы сульфонамида с использованием KCN (Общая Процедура CCCC)

Схема 82: Образование оксадиазола (Общая Процедура DDDD)
Схема 83: Образование мочевины с использованием фосгена (Общая Процедура EEEE)
Схема 84: Образование амида из сложного эфира (Общая Процедура FFFF)
Схема 85: Образование нитрила из первичного амида (Общая Процедура GGGG)

Схема 86: O-алкилирование с использованием KOH или NaOH и TBAB (Общая Процедура HHHH)
Схема 87: Образование мезилата (Общая Процедура IIII)

Схема 88: Замещение алкилмезилата, тозилата или галогенида нуклеофилом (Общая Процедура JJJJ)

Схема 89: Циклизация кетона с использованием TFAA или PFPAA (Общая Процедура KKKK)
Схема 90: Образование бромкетона (Общая Процедура LLLL)

Схема 91: Образование кетона из амида Вайнреба (Общая Процедура MMMM)

Схема 92: Образование β-гидроксисульфонамида из кетона (Общая Процедура NNNN)

Схема 93: Образование фенилкарбоната (Общая Процедура OOOO)

Схема 94: Образование карбамата с последующим гидролизом сульфонамида (Общая Процедура PPPP)

Схема 95: Окисление алкилтиоацетата до алкилсульфоновой кислоты (Общая Процедура QQQQ)

Схема 96: Циклизация диамина с использованием цианогенбромида (Общая Процедура RRRR)

Схема 97: Циклизация диамина с использованием NaNO2 (Общая Процедура SSSS)
Схема 98: Образование скварамида (Общая Процедура TTTT)
Схема 99: Восстановление азида до амина (Общая Процедура UUUU)
Схема 100: Образование кетона из гетероарилгалогенида (Общая Процедура VVVV)
Схема 101: Образование хлорангидрида кислоты (Общая Процедура WWWW)

Схема 102: Образование гидразона (Общая Процедура XXXX)
Схема 103: Циклизация с использованием α-галогенальдегида (Общая Процедура YYYY)

Схема 104: Циклизация гидразида с последующим гидролизом сульфонамида (Общая Процедура ZZZZ)

Схема 105: Образование карбоновой кислоты или сложного эфира из арилгалогенида (Общая Процедура AAAAA)

Схема 106: Циклизация с использованием ортоформиата (Общая Процедура BBBBB)
Схема 107: Реакция Стилле сочетания арила или гетероарилгалогенида (Общая Процедура CCCCC)
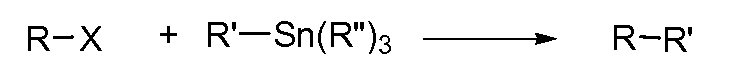
Схема 108: Удаление защиты у Cbz-защищенного амина с использованием триэтилсилана (Общая Процедура DDDDD)

Схема 109: Образование гуанидина (Общая Процедура EEEEE)

Схема 110: Образование сульфоксоний илида (Общая Процедура FFFFF)

Схема 111: Взаимодействие сульфоксоний илида с амином (Общая Процедура GGGGG)
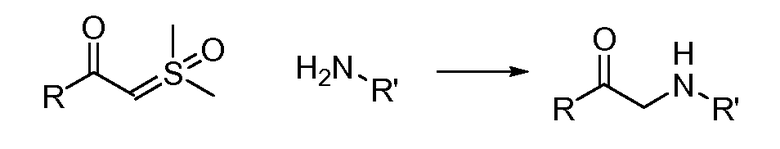
ПЕРЕЧЕНЬ ОБЩИХ ПРОЦЕДУР
Следующие далее примеры представлены в порядке, соответствующем конечной общей процедуре, используемой в их получении. Пути синтеза к любым новым промежуточным соединениям подробно указаны последующим перечислением общих процедур (буквенные коды) в скобках после их названия, при необходимости с указанием дополнительных реагирующих веществ или реагентов. Рабочий пример этого протокола представлен ниже с использованием Получения #Z.1 в качестве неограничивающей иллюстрации. Получение #Z.1 представляет собой (1S,2R,4S)-4-(циклопропансульфонамидо)-2-этилциклопентан-карбоновую кислоту, которую получали из (1S,2R,4S)-этил 4-(циклопропансульфонамидо)-2-этилциклопентанкарбоксилата с использованием Общей Процедуры Z, как представлено на Схеме A.
Схема A
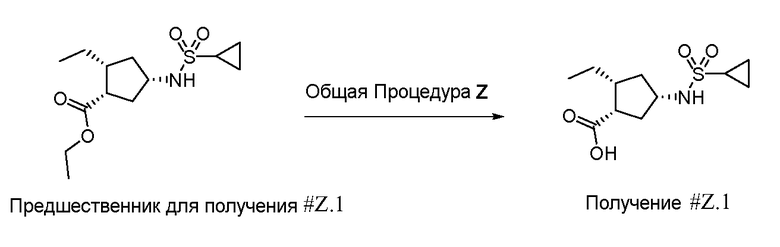
Предшественник для Получения #Z.1, (1S,2R,4S)-этил 4-(циклопропансульфонамидо)-2-этилциклопентан-карбоксилат, получали (как показано на Схеме B) сначала путем взаимодействия этил 4-амино-2-этилциклопентанкарбоксилата (Получение #Y.1) с коммерчески доступным циклопропансульфонилхлоридом, следуя условиям, указанным в Общей Процедуре K, с получением этил 4-(циклопропансульфонамидо)-2-этилциклопентан-карбоксилата в виде смеси стереоизомеров. Эту смесь стереоизомеров разделяли, как описано в Общей Процедуре AA, с использованием условий Способа 1 в Таблице 2, с получением предшественника для Получения #Z.1, (1S,2R,4S)-этил 4-(циклопропансульфонамидо)-2-этилциклопентан-карбоксилата в виде индивидуального энантиомера с временем удерживания 9,5 минут и отрицательным оптическим вращением. Последовательность реакций для синтеза предшественника для Получения #Z.1, (1S,2R,4S)-этил 4-(циклопропансульфонамидо)-2-этилциклопентан-карбоксилата, (подробно описан выше), следовательно, переносится в раздел “Получения и Примеры” для: (1S,2R,4S)-этил 4-(циклопропансульфонамидо)-2-этилциклопентан-карбоксилата (получен с использованием K из Получения #Y.1 и циклопропансульфонилхлорида, AA [Таблица 2, Способ 1, Rt = 9,5 минут, or = отрицательное]). Следовательно, получение #Z.1 должно быть представлено следующим образом: Получение #Z.1 получали из (1S,2R,4S)-этил 4-(циклопропансульфонамидо)-2-этилциклопентанкарбоксилата (получен с использованием K из Получения #Y.1 и циклопропансульфонилхлорида, AA [Таблица 2, Способ 1, Rt = 9,5 мин, or = отрицательное]) с использованием Общей Процедуры Z.
Схема B
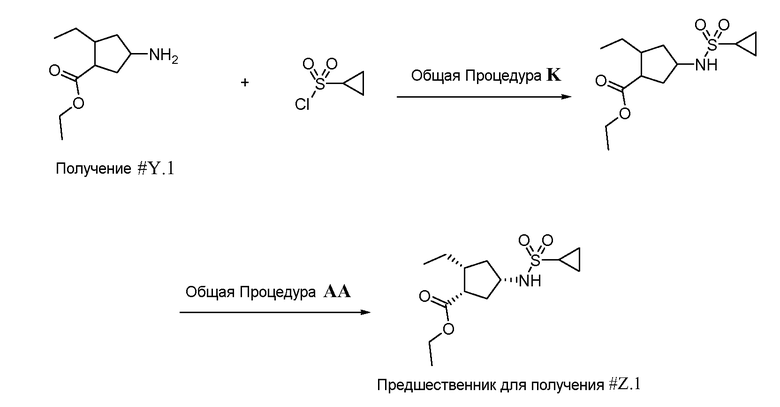
Аналитические методы
Аналитические данные включены в процедуры, описанные ниже, в иллюстрации общих процедур или в таблицы примеров. Если не указано иное, все 1H ЯМР данные собирали с использованием устройства Varian Mercury Plus 400 МГц или Varian Inova 600 МГц, и химические сдвиги представлены в миллионных долях (м.д.). LC/MS и ВЭЖХ данные указаны со ссылкой на таблицу LC/MS и ВЭЖХ условий с использованием буквенного указания способа, представленного ниже в Таблице 1.
Способы LC/MS и ВЭЖХ
Способы хиральной ВЭЖХ
Способы очистки
Для общих процедур, промежуточные соединения и конечные соединения можно очистить любым способом или комбинацией способов, известных специалистам в данной области. Некоторые примеры, которые не являются ограничивающими, включают флэш-хроматографию с твердой фазой (например, силикагель, оксид алюминия и т.п.) и растворителем (или комбинацией растворителей), который элюирует желаемые соединения (например, гептан, EtOAc, DCM, MeOH, MeCN, вода и т.п.); препаративную ТСХ с твердой фазой (например, силикагель, оксид алюминия и т.п.) и растворителем (или комбинацией растворителей), который элюирует желаемые соединения (например, гептан, EtOAc, DCM, MeOH, MeCN, вода и т.п.); обращенно-фазовую ВЭЖХ (см. Таблицу 1 для некоторых неограничивающих условий); перекристаллизацию из подходящего растворителя или комбинации растворителей (например, MeOH, EtOH, IPA, EtOAc, толуол и т.п.) или комбинацию растворителей (например, EtOAc/гептан, EtOAc/MeOH и т.п.); хиральную жидкостную хроматографию (LC) с твердой фазой и подходящим растворителем (см. Таблицу 2 для некоторых неограничивающих условий) для элюирования желаемого соединения; хиральную SFC с твердой фазой и CO2 с подходящим модификатором (например, MeOH, EtOH, IPA с или без дополнительного модификатора, такого как диэтиламин, TFA и т.п.); осаждение из комбинации растворителей (например, DMF/вода, DMSO/DCM, EtOAc/гептан и т.п.); растирание в порошок с подходящим растворителем (например, EtOAc, DCM, MeCN, MeOH, EtOH, IPA, n-IPA и т.п.); экстракции путем растворения соединения в жидкости и промывку подходящей несмешиваемой жидкостью (например, DCM/вода, EtOAc/вода, DCM/насыщенный водный раствор NaHCO3, EtOAc/насыщенный водный раствор NaHCO3, DCM/10% водный раствор HCl, EtOAc/10% водный раствор HCl и т.п.); перегонку (например, простую, фракционную, Kugelrohr и т.п.); газовую хроматографию с использованием подходящей температуры, носителя газа и скорости потока; сублимацию при подходящей температуре и давлении; фильтрование через среду (например, Florosil®, оксид алюминия, Целит®, силикагель и т.п.) с растворителем (например, гептан, гексан, EtOAc, DCM, MeOH и т.п.) или комбинацией растворителей; солеобразование с твердым носителем (на смоле, например, ионообменной) или без носителя. Представляющие интерес соединения можно выделить в виде соли, без применения специфического способа очистки методом солеобразования. Например, в случаях, когда очистку осуществляют при помощи обращенно-фазовой ВЭЖХ с водным TFA буфером, можно выделить TFA соль. Некоторые описания таких способов можно найти в следующих документах: Gordon, A. J. and Ford, R. A.. "The Chemist's Companion”, 1972; Palleros, D. R. “Experimental Organic Chemistry”, 2000; Still, W. C., Kahn and M. Mitra, A. J. Org. Chem. 1978, 43, 2923; Yan, B. “Analysis and Purification Methods in Combinatorial Chemistry”, 2003; Harwood, L. M., Moody, C. J. and Percy, J. M. “Experimental Organic Chemistry: Standard and Microscale, 2nd Edition”, 1999; Stichlmair, J. G. and Fair, J. R. “Distillation; Principles and Practices”, 1998; Beesley, T. E. and Scott, R. P. W. “Chiral Chromatography”, 1999; Landgrebe, J. A. “Theory and Practice in the Organic Laboratory, 4th Ed.”, 1993; Skoog, D. A. and Leary, J. J. “Principles of Instrumental Analysis 4th Ed.”, 1992; G. Subramanian, "Chiral Separation Techniques, 3rd Edition", 2007; Y. Kazakevich, R. Lobrutto, "HPLC for Pharmaceutical Scientists", 2007.
Получения и Примеры
Общие способы синтеза, используемые в каждой Общей Процедуре, описаны далее, и они включают иллюстрацию соединения, которое было синтезировано с использованием указанной Общей Процедуры. Никакие указанные далее специальные условия или реагенты не следует рассматривать как ограничивающие объем настоящего изобретения, и они представлены исключительно в целях иллюстрации. Все исходные вещества являются коммерчески доступными от компании Sigma-Aldrich (включая Fluka и Discovery CPR), если не указано иное после химического названия. Названия реагентов/реагирующих веществ указаны в соответствии с их названиями на коммерческой упаковке или в соответствии с принятыми обозначениями IUPAC, CambridgeSoft® ChemDraw Ultra 9,0,7, CambridgeSoft® Chemistry E-Notebook 9,0,127 или AutoNom 2000. Соединения, указанные как соли (например, гидрохлорид, ацетат), могут содержать более чем один молярный эквивалент соли. Соединения по настоящему изобретению, где определена абсолютная стереохимия с использованием коммерчески доступных энантиомерно чистых исходных веществ или стереохимически определенных промежуточных соединений или методом рентгеновской дифракции, помечены звездочкой после номера примера.
Получение #1: цис-3-(4-цианобензилокси)циклобутанкарбоновая кислота
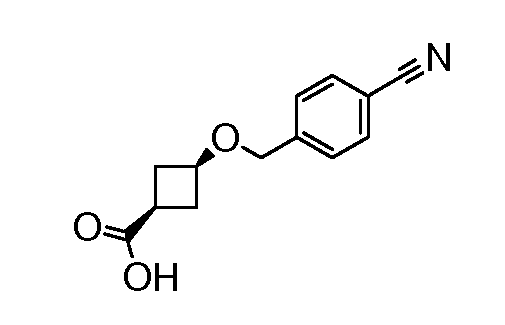
Стадия A: цис-этил 3-гидроксициклобутанкарбоксилат

Раствор этил 3-оксоциклобутанкарбоксилата (2,90 г, 20,4 ммоль, Parkway) в EtOH (30 мл) при температуре окружающей среды обрабатывали при помощи NaBH4 (0,77 г, 20 ммоль). Реакционную смесь перемешивали в течение примерно 1 часа и затем добавляли 2 н водный раствор HCl для доведения pH до около 2. Реакционную смесь концентрировали в вакууме. Реакционную смесь разделяли с использованием DCM (50 мл) и насыщенного солевого раствора (50 мл). Органический слой отделяли, сушили над безводным Na2SO4, фильтровали затем концентрировали в вакууме. Полученный остаток очищали на силикагеле (80 г) с использованием 20-40% EtOAc в DCM с получением цис-этил 3-гидроксициклобутанкарбоксилата (2,75 г, 66%) в виде прозрачного масла: 1H ЯМР (DMSO-d 6) δ 5,17 (д, 1H), 4,09-3,99 (м, 2H), 3,99-3,90 (м, 1H), 2,57-2,47 (м, 1H), 2,42-2,29 (м, 2H), 1,98-1,89 (м, 2H), 1,17 (м, 3H).
Стадия B: цис-этил 3-(4-цианобензилокси)циклобутанкарбоксилат

К раствору цис-этил 3-гидроксициклобутанкарбоксилата (0,17 г, 1,2 ммоль) в DMF (4 мл) добавляли K2CO3 (0,24 г, 1,8 ммоль) с последующим добавлением 4-(бромметил)бензонитрила (0,28 г, 1,4 ммоль). Реакционную смесь перемешивали при около 25°C в течение около 16 часов. Реакционную смесь распределяли между EtOAc (50 мл) и насыщенным солевым раствором (50 мл). Слои разделяли и органический слой промывали дополнительно насыщенным солевым раствором (50 мл). Органический слой затем сушили над безводным Na2SO4, фильтровали и концентрировали в вакууме с получением цис-этил 3-(4-цианобензилокси)-циклобутанкарбоксилата (0,29 г, 95%) в виде масла: 1H ЯМР (DMSO-d 6) δ 7,82 (д, J=8,5 Гц, 2H), 7,63 (д, J=8,4 Гц, 2H), 4,75 (с, 2H), 4,02 (кв., J=7,1 Гц, 2H), 3,93 (м, 1H), 2,58-2,45 (м, 1H), 2,41-2,28 (м, 2H), 1,98-1,85 (м, 2H), 1,20-1,08 (т, J=7,1 Гц, 3H).
Стадия C: цис-3-(4-цианобензилокси)циклобутанкарбоновая кислота

К раствору цис-этил 3-(4-цианобензилокси)циклобутанкарбоксилата (0,44 г, 1,70 ммоль) в 1,4-диоксане (10 мл) добавляли водный раствор NaOH (1 н, 2,0 мл). Реакционную смесь перемешивали при около 25°C в течение около 16 часов. Реакционную смесь распределяли между 10% водным раствором AcOH (20 мл) и EtOAc (25 мл). Слои разделяли и водный слой экстрагировали дополнительным количеством EtOAc (25 мл). Объединенные органические экстракты промывали насыщенным солевым раствором (20 мл), сушили над безводным Na2SO4, фильтровали, затем концентрировали в вакууме с получением цис-3-(4-цианобензилокси)-циклобутанкарбоновой кислоты (0,24 г, 60%): LC/MS (Таблица 1, Способ b) Rt = 1,67 мин; MS m/z: 232 (M+H)+.
Получение #2*: (1S,3R)-1-[3-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-изотиазолидин-2-ил-1,1-диоксид]циклопентан

Стадия A: 3-хлор-N-((1S,3R)-3-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)пропан-1-сульфонамид

К суспензии гидрохлорида (1S,3R)-3-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентанамина (0,05 г, 0,11 ммоль, получали с использованием E из Получения #B.1 и HCl) и TEA (0,03 мл, 0,21 ммоль) в DCM (5 мл) при около 0°C добавляли 3-хлорпропан-1-сульфонилхлорид (0,02 г, 0,11 ммоль) по каплям. Реакционную смесь перемешивали при около 0°C в течение около 1,5 часов. Реакционную смесь разбавляли 5% водным раствором лимонной кислоты (10 мл) и слои разделяли. Органический слой промывали насыщенным водным раствором NaHCO3 (10 мл), водой (10 мл), насыщенным солевым раствором (10 мл), сушили над безводным MgSO4, фильтровали и концентрировали при пониженном давлении с получением 3-хлор-N-((1S,3R)-3-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)пропан-1-сульфонамида (0,052 г, 91%) в виде коричневого остатка: LC/MS (Таблица 1, Способ a) Rt = 2,18 мин; MS m/z: 537 (M+H)+.
Стадия B: (1S,3R)-1-[3-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-изотиазолидин-2-ил-1,1-диоксид]циклопентан
К раствору 3-хлор-N-((1S,3R)-3-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)пропан-1-сульфонамида (0,11 г, 0,21 ммоль) в DMF (5 мл) добавляли 1,8-диазабицикло[5,4,0]ундец-7-ен (0,04 мл, 0,27 ммоль неочищенного вещества). Реакционную смесь перемешивали при температуре окружающей среды в течение около 16 часов. Растворитель удаляли при пониженном давлении с получением (1S,3R)-1-[3-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-изотиазолидин-2-ил-1,1-диоксид]циклопентана (0,106 г, 99%): LC/MS (Таблица 1, Способ a) Rt = 2,04 мин; MS m/z: 501 (M+H)+.
Получение #3*: 1-((1R,3S)-3-(1H-пиррол-1-ил)циклопентил)-6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин

Раствор 2,5-диметокситетрагидрофурана (0,14 г, 1,1 ммоль) в воде (3 мл) нагревали при около 100°C в течение около 1,5 часов. Раствор охлаждали до температуры окружающей среды. Суспензию гидрохлорида (1S,3R)-3-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентанамина (0,10 г, 0,21 ммоль, получали с использованием E из Получения #B.1 и HCl) и NaOAc (0,05 г, 0,61 ммоль) в DCM (5 мл) добавляли к водному раствору. Реакционную смесь перемешивали при температуре окружающей среды в течение примерно 1 часа с последующим добавлением дополнительного количества 2,5-диметокситетрагидрофурана (0,14 г, 1,1 ммоль). Реакционную смесь нагревали до около 40°C в течение около 15 часов. Дополнительно добавляли 2,5-диметокситетрагидрофуран (0,14 г, 1,1 ммоль) и реакционную смесь перемешивали при около 40°C в течение около 8 часов, затем при около 35°C в течение около 48 часов. Реакционную смесь разбавляли DCM (10 мл) и водой (10 мл). Слои разделяли и органический слой промывали водой (2×10 мл) и насыщенным солевым раствором (10 мл), сушили над безводным MgSO4, фильтровали и растворитель удаляли при пониженном давлении с получением 1-((1R,3S)-3-(1H-пиррол-1-ил)циклопентил)-6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразина (0,095 г, 99%) в виде желтого остатка: LC/MS (Таблица 1, Способ a) Rt = 2,42 мин; MS m/z: 447 (M+H)+.
Получение #4*: 1-((1S,3R)-3-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)-1H-пиррол-3-карбонитрил

Стадия A: 1-((1S,3R)-3-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)-1H-пиррол-3-карбальдегид

К суспензии гидрохлорида (1S,3R)-3-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентанамина (0,175 г, 0,373 ммоль, получали с использованием E из Получения #B.1 и HCl) и NaOAc (0,100 г, 1,22 ммоль) в DCM (3 мл) и воде (2 мл) добавляли 2,5-диметокситетрагидрофуран-3-карбальдегид (0,600 г, 3,37 ммоль). Реакционную смесь нагревали до около 40°C в течение около 24 часов. Реакционную смесь разбавляли при помощи DCM (30 мл) и промывали водой (4×20 мл). Органический слой сушили над безводным MgSO4, фильтровали и растворитель удаляли при пониженном давлении с получением коричневого остатка. Неочищенное вещество очищали флэш-хроматографией на силикагеле с градиентным элюированием 20-100% EtOAc в DCM с получением 1-((1S,3R)-3-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)-1H-пиррол-3-карбальдегида (0,059 г, 33%) в виде желтого аморфного твердого вещества: LC/MS (Таблица 1, Способ a) Rt = 2,10 мин; MS m/z: 475 (M+H)+.
Стадия B: 1-((1S,3R)-3-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)-1H-пиррол-3-карбонитрил
К раствору 1-((1S,3R)-3-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)-1H-пиррол-3-карбальдегида (0,050 г, 0,105 ммоль) в ТГФ (2 мл) добавляли иод (0,083 г, 0,327 ммоль) и водный раствор NH4OH (28-30% масс/об, 0,733 мл, 5,27 ммоль). Реакционную смесь перемешивали при температуре окружающей среды в течение около 24 часов. Реакционную смесь разбавляли насыщенным водным раствором Na2SO3 (30 мл) и EtOAc (30 мл). Слои разделяли и органический слой сушили над безводным MgSO4, фильтровали и растворитель удаляли при пониженном давлении с получением 1-((1S,3R)-3-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)-1H-пиррол-3-карбонитрила (0,05 г, 100%): LC/MS (Таблица 1, Способ a) Rt = 2,33 мин; MS m/z: 472 (M+H)+.
Получение #5: 3,3-дифтор-N-(4-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)бицикло[2,2,2]октан-1-ил)азетидин-1-сульфонамид

Стадия A: 1-(1H-имидазол-1-илсульфонил)-3-метил-1H-имидазол-3-ий трифторметансульфонат
К раствору 1,1'-сульфонилдиимидазола (3,50 г, 17,7 ммоль) в DCM (75 мл) при около 0°C добавляли метилтрифторметансульфонат (1,94 мл, 17,7 ммоль). Реакционную смесь перемешивали при около 0°C в течение около 1 часа, затем нагревали до температуры окружающей среды и перемешивали в течение около 5 часов. Твердое вещество собирали вакуумным фильтрованием и промывали при помощи DCM (10 мл) с получением 1-(1H-имидазол-1-илсульфонил)-3-метил-1H-имидазол-3-ий трифторметансульфоната (6,35 г, 98%) в виде белого твердого вещества: LC/MS (Таблица 1, Способ a) Rt = 0,082 мин; MS m/z 213 (M+H)+.
Стадия B: 1-(3,3-дифторазетидин-1-илсульфонил)-1H-имидазол
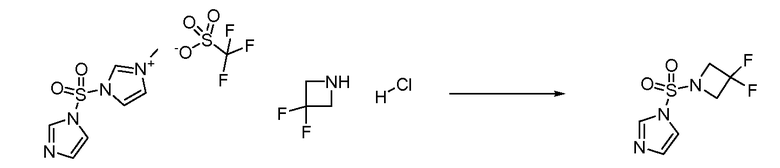
Раствор 3,3-дифторазетидингидрохлорида (1,00 г, 7,72 ммоль) и DIEA (1,5 мл, 8,6 ммоль) в MeCN (5 мл) перемешивали в течение около 5 минут и затем добавляли к раствору 1-(1H-имидазол-1-илсульфонил)-3-метил-1H-имидазол-3-ий трифторметансульфоната (4,20 г, 11,6 ммоль) в MeCN (10 мл) при около 0°C. Реакционную смесь перемешивали при около 0°C в течение примерно 1 часа и затем нагревали до температуры окружающей среды и перемешивали в течение около 16 часов. Реакционную смесь затем концентрировали при пониженном давлении. Неочищенное вещество очищали флэш-хроматографией на силикагеле с градиентным элюированием 5-100% EtOAc в DCM с получением 1-(3,3-дифторазетидин-1-илсульфонил)-1H-имидазола (0,95 г, 55%) в виде желтого твердого вещества: LC/MS (Таблица 1, Способ c) Rt = 1,16 мин; MS m/z 224 (M+H)+.
Стадия C: 1-(3,3-дифторазетидин-1-илсульфонил)-3-метил-1H-имидазол-3-ий трифторметансульфонат

К раствору 1-(3,3-дифторазетидин-1-илсульфонил)-1H-имидазола (0,500 г, 2,24 ммоль) в DCM (5 мл) при около 0°C добавляли метилтрифторметансульфонат (0,27 мл, 2,46 ммоль) по каплям в течение около 3 минут. Реакционную смесь перемешивали при около 0°C в течение около 2 часов. Твердое вещество собирали вакуумным фильтрованием, промывали при помощи DCM (10 мл) и сушили в вакууме с получением 1-(3,3-дифторазетидин-1-илсульфонил)-3-метил-1H-имидазол-3-ий трифторметансульфоната (0,79 г, 90%) в виде белого твердого вещества: LC/MS (Таблица 1, Способ c) Rt = 1,12 мин; MS m/z 238 (M+H)+.
Стадия D: 3,3-дифтор-N-(4-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)бицикло[2,2,2]октан-1-ил)азетидин-1-сульфонамид

К раствору 4-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)бицикло[2,2,2]-октан-1-амина (0,20 г, 0,46 ммоль, Пример #9, Стадия F) в MeCN (5 мл) добавляли 1-(3,3-дифторазетидин-1-илсульфонил)-3-метил-1H-имидазол-3-ий трифторметансульфонат (0,19 г, 0,50 ммоль). Реакционную смесь нагревали до около 70°C в течение около 24 часов. Растворитель удаляли при пониженном давлении. Остаток распределяли между EtOAc (30 мл) и водой (10 мл). Слои разделяли и органический слой промывали водой (10 мл) и насыщенным солевым раствором (2×10 мл), сушили над безводным MgSO4, фильтровали и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали флэш-хроматографией на силикагеле с градиентным элюированием 0-10% MeOH в DCM с получением 3,3-дифтор-N-(4-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)бицикло[2,2,2]октан-1-ил)азетидин-1-сульфонамида (0,119 г, 38%): LC/MS (Таблица 1, Способ a) Rt = 2,32 мин; MS m/z 592 (M+H)+.
Получение #6: 1-метилциклопропан-1-сульфонилхлорид

Стадия A: бутилциклопропансульфонат
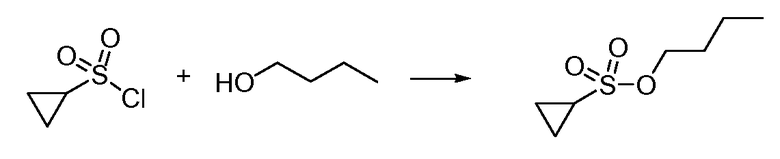
К раствору циклопропансульфонилхлорида (5,00 г, 35,6 ммоль) в n-BuOH (20 мл) при -20°C добавляли по каплям пиридин (5,75 мл, 71,1 ммоль). Полученную смесь перемешивали в течение около 16 часов, при этом медленно нагревая до температуры окружающей среды. Растворители удаляли при пониженном давлении и остаток распределяли между DCM и водой (50 мл каждого). Органическую фазу далее промывали насыщенным солевым раствором (40 мл), сушили над безводным MgSO4 и концентрировали при пониженном давлении с получением бутилциклопропансульфоната (4,7 г, 74%) в виде желтого масло. 1H ЯМР (DMSO-d 6) δ 4,2 (т, 2H), 2,82 (м, 1H), 1,64 (м, 2H), 1,35 (м, 2H), 1,08 (м, 2H), 1,01 (м, 2H), 0,89 (т, 3H).
Стадия B: бутил 1-метилциклопропан-1-сульфонат

К раствору бутилциклопропансульфоната (1,5 г, 8,4 ммоль) в ТГФ (8 мл) при около -78°C добавляли одновременно n-BuLi (1,6 M в гексане, 5,26 мл, 8,42 ммоль) и иодметан (0,684 мл, 10,9 ммоль) и полученную смесь перемешивали при около -78°C в течение около 2 часов и затем при температуре окружающей среды в течение около 2 часов. Реакцию гасили путем добавления насыщенного водного раствора NH4Cl (7 мл) и слои разделяли. Водный слой обратно экстрагировали при помощи EtOAc (15 мл) и объединенные органические экстракты сушили над безводным MgSO4 и концентрировали при пониженном давлении. Остаток подвергали колоночной хроматографии на силикагеле (5 до 25% EtOAc в гептане в течение 30 мин) с получением бутил 1-метилциклопропан-1-сульфоната (0,8 г, 49%) в виде бесцветного масла. 1H ЯМР (DMSO-d 6) δ 4,17 (т, 2H), 1,62 (м, 2H), 1,43 (с, 3H), 1,35 (м, 2H), 1,22 (м, 2H), 0,94 (м, 2H), 0,88 (т, 3H).
Стадия C: 1-метилциклопропан-1-сульфонилхлорид

Смесь бутил 1-метилциклопропан-1-сульфоната (0,80 г, 4,2 ммоль) и тиоцианата калия (0,404 г, 4,16 ммоль) в 1,4-диоксане/воде (1:1, 10 мл) нагревали при температуре кипения с обратным холодильником в течение около 8 часов. Реакционную смесь охлаждали до температуры окружающей среды и растворители концентрировали при пониженном давлении с получением неочищенного 1-метилциклопропан-1-сульфоната калия, который суспендировали в тионилхлориде (7 мл). Добавляли DMF (0,05 мл) и смесь нагревали при температуре кипения с обратным холодильником в течение около 8 часов и затем охлаждали. Летучие вещества удаляли при пониженном давлении и остаток растворяли в DCM (20 мл), промывали водой (15 мл), сушили над безводным MgSO4 и концентрировали при пониженном давлении с получением 1-метилциклопропан-1-сульфонилхлорида (0,56 г, 86%) в виде желтого масла. 1H ЯМР (DMSO-d 6) δ 1,82 (шир.с, 2H), 1,79 (с, 3H), 1,15 (м, 2H).
Получение #7: этил 4-(циклопропансульфонамидо)-2-этил-1-фторциклопентанкарбоксилат

Раствор этил 4-(циклопропансульфонамидо)-2-этилциклопентанкарбоксилата (0,630 г, 2,18 ммоль, получали с использованием K из Получения #Y.1 и циклопропансульфонилхлорида) в ТГФ (14,5 мл) охлаждали до около -78°C и затем к реакционной смеси добавляли по каплям LDA (1,8 M в ТГФ/гексане, 3,63 мл, 6,53 ммоль) в течение около 30 минут. Реакционную смесь перемешивали при около -78°C в течение около 50 минут, затем добавляли раствор N-фтор-N-(фенилсульфонил)бензолсульфонамид (2,06 г, 6,53 ммоль) в ТГФ (7,3 мл) по каплям в течение около 30 минут. Реакционную смесь перемешивали при около -78°C в течение примерно 1 часа и затем нагревали до температуры окружающей среды и перемешивали в течение около 16 часов. Добавляли насыщенный водный раствор NH4Cl (100 мл). Реакционную смесь разделяли с использованием EtOAc (50 мл). Водный слой далее экстрагировали при помощи EtOAc (2×50 мл). Объединенные органические слои концентрировали при пониженном давлении. Остаток очищали хроматографией на силикагеле с градиентным элюированием 0-60% EtOAc в гептане с получением этил 4-(циклопропансульфонамидо)-2-этил-1-фторциклопентанкарбоксилата (0,41 г, 46%) в виде прозрачного масла: LC/MS (Таблица 1, Способ b) Rt = 2,12 мин; MS m/z: 306 (M-H)-.
Получение #8: (1S,2R,4R)-этил 2-метил-4-(фениламино)циклопентанкарбоксилат и (1R,2S,4S)-этил 2-метил-4-(фениламино)циклопентанкарбоксилат
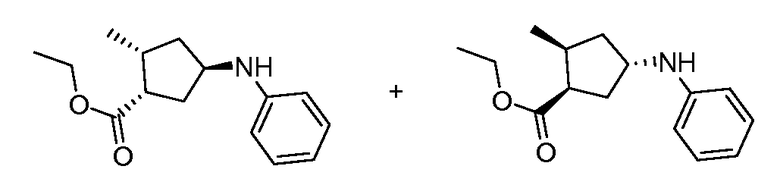
Раствор этил 4-гидрокси-2-метилциклопентанкарбоксилата (1,81 г, 10,5 ммоль, получали с использованием P из Примера #7, стадия G и NaBH4) и пиридина (1,28 мл, 15,8 ммоль) в ТГФ (52,5 мл) охлаждали до около 0°C. Добавляли по каплям метансульфонилхлорид (0,90 мл, 12 ммоль). Реакционную смесь перемешивали при температуре окружающей среды в течение около 16 часов, затем распределяли между водой (50 мл) и DCM (30 мл). Слои разделяли и водный слой экстрагировали при помощи DCM (2×30 мл). Объединенные органические слои сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением белого твердого вещества. Полученное твердое вещество смешивали с анилином (78,0 г, 841 ммоль) и нагревали при около 90°C в течение около 16 часов. Реакционную смесь концентрировали при пониженном давлении и очищали хроматографией на силикагеле с градиентным элюированием 20-100% EtOAc в DCM с получением (1S,2R,4R)-этил 2-метил-4-(фениламино)циклопентанкарбоксилата и (1R,2S,4S)-этил 2-метил-4-(фениламино)цикло-пентанкарбоксилата с 29 моль% анилина в качестве эксципиента (2,73 г, 75%) в виде темного масла: LC/MS (Таблица 1, Способ b) Rt = 2,67 мин; MS m/z: 248 (M+H)+.
Получение #9: 1-трет-бутил 3-этил 4-этил-5,6-дигидропиридин-1,3(2H)-дикарбоксилат

Стадия A: 1-трет-бутил 3-этил 4-(диэтоксифосфорилокси)-5,6-дигидропиридин-1,3(2H)-дикарбоксилат

К раствору 1-трет-бутил 3-этил 4-оксопиперидин-1,3-дикарбоксилата (11,50 г, 42,4 ммоль, ASDI) в MTBE (500 мл) при около -78°C добавляли NaHMDS (1 M в ТГФ, 53,0 мл, 53,0 ммоль). Примерно через 1 час к реакционной смеси добавляли диэтилфосфорохлоридат (7,62 мл, 53,0 ммоль). Примерно через 30 минут реакционной смеси давали нагреться до температуры окружающей среды и перемешивали в течение около 16 часов. Реакционную смесь распределяли между насыщенным водным раствором NH4Cl (100 мл) и EtOAc (50 мл). Слои разделяли. Водный слой далее экстрагировали при помощи EtOAc (2×50 мл). Объединенные органические слои сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении. Неочищенное вещество очищали хроматографией на силикагеле с градиентным элюированием 0-100% EtOAc в гептане с получением 1-трет-бутил 3-этил 4-(диэтоксифосфорилокси)-5,6-дигидропиридин-1,3(2H)-дикарбоксилата (8,55 г, 49%) в виде желтого масла: LC/MS (Таблица 1, Способ b) Rt = 2,35 мин; MS m/z: 408 (M+H)+.
Стадия B: 1-трет-бутил 3-этил 4-этил-5,6-дигидропиридин-1,3(2H)-дикарбоксилат

К суспензии CuI (4,21 г, 22,12 ммоль) в ТГФ (61,4 мл) при около 0°C добавляли этилмагнийбромид (1,0 M в ТГФ, 44,2 мл, 44,2 ммоль) по каплям. Примерно через 30 минут реакционную смесь охлаждали до около -78°C и медленно добавляли раствор 1-трет-бутил 3-этил 4-(диэтоксифосфорилокси)-5,6-дигидропиридин-1,3(2H)-дикарбоксилата (7,51 г, 18,43 ммоль) в ТГФ (61 мл). Реакционную смесь перемешивали при около -78°C в течение примерно 1 часа, затем нагревали до около 0°C. Реакционную смесь перемешивали при около 0°C в течение около 1,5 часов, затем нагревали до температуры окружающей среды и перемешивали в течение около 1 часа. Реакционную смесь охлаждали до около -78°C и медленно добавляли насыщенный водный раствор NH4Cl (100 мл). Реакционной смеси давали нагреться до температуры окружающей среды и перемешивали в течение около 16 часов. Смесь экстрагировали при помощи Et2O (100 мл). Водный слой далее экстрагировали при помощи Et2O (2×50 мл). Органические слои объединяли, промывали насыщенным водным раствором NH4Cl (50 мл), сушили над безводным Na2SO4, фильтровали, концентрировали при пониженном давлении и очищали хроматографией на силикагеле с градиентным элюированием 0-30% EtOAc в гептане с получением 1-трет-бутил 3-этил 4-этил-5,6-дигидропиридин-1,3(2H)-дикарбоксилата (0,785 г, 15%) в виде прозрачного масла: 1H ЯМР (CDCl3) δ 4,23 (д, J=7,1 Гц, 2H), 4,12 (с, 2H), 3,48 (т, J=5,8 Гц, 2H), 2,52 (кв., J=7,5 Гц, 2H), 2,28 (т, J=5,8 Гц, 2H), 1,51 (с, 9H), 1,32 (т, J=7,1 Гц, 3H), 1,09 (т, J=7,5 Гц, 3H).
Получение #10: 1-(1-бензилпиперидин-3-ил)-1,6-дигидропиразоло[3,4-d]пирроло[2,3-b]пиридин
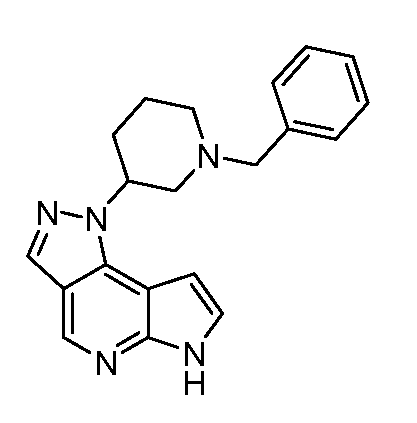
Стадия A: трет-бутил 2-(1-бензилпиперидин-3-ил)гидразинкарбоксилат
Смесь 1-бензилпиперидин-3-он гидрохлорида (1,00 г, 4,10 ммоль), трет-бутил гидразинкарбоксилата (0,596 г, 4,51 ммоль) и AcOH (0,470 мл, 8,21 ммоль) в DCE (20 мл) перемешивали при температуре окружающей среды в течение примерно 1 часа, затем добавляли NaCNBH3 (0,258 г, 4,10 ммоль). Реакционную смесь перемешивали при температуре окружающей среды в течение около 16 часов. Реакцию гасили путем добавления насыщенного водного раствора NaHCO3 (50 мл). Органический слой отделяли, концентрировали при пониженном давлении и очищали при помощи ОФ-ВЭЖХ (Таблица 1, Способ h) с получением трет-бутил 2-(1-бензилпиперидин-3-ил)гидразинкарбоксилата (1,25 г, 100%) в виде прозрачного масла: LC/MS (Таблица 1, Способ b) Rt = 1,66 мин; MS m/z: 306 (M+H)+.
Стадия B: 1-бензил-3-гидразинилпиперидин гидрохлорид
Раствор трет-бутил 2-(1-бензилпиперидин-3-ил)гидразинкарбоксилата (1,25 г, 4,10 ммоль) в водном растворе HCl (6 н, 6,83 мл, 41,0 ммоль) перемешивали при температуре окружающей среды в течение около 8 часов. Растворитель удаляли при пониженном давлении с получением неочищенного 1-бензил-3-гидразинилпиперидин гидрохлорида (1,45 г, 112%) в виде белого твердого вещества, которое использовали без дополнительной очистки: LC/MS (Таблица 1, Способ b) Rt = 0,66 мин; MS m/z: 206 (M+H)+.
Стадия C: 1-(1-бензилпиперидин-3-ил)-1,6-дигидропиразоло[3,4-d]пирроло[2,3-b]пиридин

4-Хлор-1H-пирроло[2,3-b]пиридин-5-карбальдегид (0,40 г, 2,21 ммоль, Adesis) и 1-бензил-3-гидразинилпиперидин гидрохлорид (1,39 г, 4,43 ммоль) суспендировали в n-BuOH (11,1 мл). Смесь нагревали при около 90°C в течение около 3 часов и затем нагревали при около 120°C в течение около 5 часов. Реакционную смесь охлаждали до температуры окружающей среды и растворитель удаляли при пониженном давлении. Остаток очищали хроматографией на силикагеле с градиентным элюированием 0-5% MeOH в DCM с получением 1-(1-бензилпиперидин-3-ил)-1,6-дигидропиразоло[3,4-d]пирроло[2,3-b]пиридина (0,105 г, 14%) в виде коричневого масла: LC/MS (Таблица 1, Способ b) Rt = 1,53 мин; MS m/z: 332 (M+H)+.
Получение #11: цис-3-трет-бутил 1-метил 4-этилциклопентан-1,3-дикарбоксилат

Стадия A: цис-2-этил-4-(метоксикарбонил)циклопентанкарбоновая кислота

Хлорид рутения (III) гидрат (0,203 г, 0,900 ммоль) добавляли к смеси 5-этилбицикло[2,2,1]гепт-2-ена (5,00 г, 40,9 ммоль, ChemSampCo) и периодата натрия (35,0 г, 164 ммоль) в воде (117 мл), MeCN (78 мл) и EtOAc (78 мл). Реакционную смесь перемешивали при температуре окружающей среды в течение около 16 часов. Реакционную смесь фильтровали, экстрагировали при помощи Et2O (2×100 мл). Водный слой далее экстрагировали при помощи Et2O (3×100 мл). Органические слои объединяли, промывали насыщенным солевым раствором (100 мл), сушили над безводным MgSO4, фильтровали и концентрировали при пониженном давлении. Остаток растворяли в Ac2O (20 мл, 24 ммоль) и нагревали при температуре кипения с обратным холодильником в течение около 4 часов. Реакционную смесь охлаждали до температуры окружающей среды и растворитель удаляли при пониженном давлении. Добавляли MeOH (40 мл) и реакционную смесь нагревали при температуре кипения с обратным холодильником в течение около 6 часов. Растворитель удаляли при пониженном давлении с получением цис-2-этил-4-(метоксикарбонил)циклопентанкарбоновой кислоты (4,84 г, 59%) в виде коричневого масла: LC/MS (Таблица 1, Способ b) Rt = 1,91 мин; MS m/z: 201 (M+H)+.
Стадия B: цис-3-трет-бутил 1-метил 4-этилциклопентан-1,3-дикарбоксилат

Смесь цис-2-этил-4-(метоксикарбонил)циклопентанкарбоновой кислоты (4,50 г, 22,47 ммоль) в SOCl2 (8,20 мл, 112 ммоль) перемешивали при температуре окружающей среды в течение около 16 часов. Растворитель удаляли при пониженном давлении. Полученный остаток растворяли в t-BuOH (22,5 мл). Реакционную смесь перемешивали при температуре окружающей среды в течение около 16 часов. Растворитель удаляли при пониженном давлении. Остаток растворяли в воде (50 мл) и DCM (100 мл). Органический слой отделяли, промывали насыщенным водным раствором NaHCO3 (50 мл), сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении с получением цис-3-трет-бутил 1-метил 4-этилциклопентан-1,3-дикарбоксилата (3,94 г, 68%) в виде темно-коричневого масла: LC/MS (Таблица 1, Способ b) Rt = 2,86 мин; MS m/z: 257 (M+H)+.
Получение #12: 1-(5-тозил-5H-пирроло[2,3-b]пиразин-2-ил)бут-3-ен-1-амин гидрохлорид

Стадия A: (E)-2-стирил-5-тозил-5H-пирроло[2,3-b]пиразин

К раствору 2-бром-5-тозил-5H-пирроло[2,3-b]пиразина (3,1 г, 8,8 ммоль, Пример #1. Стадия B), PdCl2(dppf)•DCM (0,719 г, 0,880 ммоль) и (E)-стирилбороновой кислоты (2,60 г, 17,6 ммоль) в ТГФ (3 мл) и воде (2 мл) добавляли Na2CO3 (2,33 г, 22,0 ммоль). Реакционную смесь дегазировали аргоном в течение около 5 минут. Реакционную смесь нагревали при около 50°C. Примерно через 24 часов к реакционной смеси добавляли дополнительное количество PdCl2(dppf)•DCM (0,719 г, 0,880 ммоль), (E)-стирилбороновой кислоты (2,60 г, 17,6 ммоль) и Na2CO3 (2,33 г, 22,0 ммоль). После нагревания при около 50°C в течение около 48 часов реакционную смесь охлаждали до температуры окружающей среды и разбавляли DCM (200 мл) и водой (200 мл). Органический слой отделяли, сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении. Очистка хроматографией на силикагеле с градиентным элюированием 20-60% EtOAc в гептане, содержащем 5% DCM, давала (E)-2-стирил-5-тозил-5H-пирроло[2,3-b]пиразин в виде желтого твердого вещества (1,2 г, 36%): LC/MS (Таблица 1, Способ a) Rt = 2,99 мин; MS m/z: 376 (M+H)+.
Стадия B: 5-тозил-5H-пирроло[2,3-b]пиразин-2-карбальдегид

К раствору (E)-2-стирил-5-тозил-5H-пирроло[2,3-b]пиразина (1,2 г, 3,2 ммоль) в 1,4-диоксане (20 мл) и воде (2,0 мл) добавляли периодат натрия (2,73 г, 12,8 ммоль) с последующим добавлением тетроксид осмия (2,5% масс. в t-BuOH, 4,01 мл, 0,320 ммоль). Реакционную смесь перемешивали примерно в течение 1 дня при температуре окружающей среды и затем добавляли дополнительное количество периодат натрия (2,73 г, 12,78 ммоль) и тетроксид осмия (2,5% масс. в t-BuOH, 4,01 мл, 0,320 ммоль). После перемешивания примерно в течение 2 дней добавляли раствор 10% водного Na2S2O3 (100 мл) и EtOAc (100 мл). Органический слой отделяли, сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении с получением твердого вещества, которое растирали в порошок с гептаном для удаления бензальдегида. Полученное твердое вещество сушили в вакууме с получением 5-тозил-5H-пирроло[2,3-b]пиразин-2-карбальдегида в виде коричневого твердого вещества (0,77 г, 80%): LC/MS (Таблица 1, Способ a) Rt = 2,01 мин; MS m/z: 334 (M+H)+.
Стадия C: 1-(5-тозил-5H-пирроло[2,3-b]пиразин-2-ил)бут-3-ен-1-ол

К раствору 5-тозил-5H-пирроло[2,3-b]пиразин-2-карбальдегида (5,1 г, 17 ммоль) в ТГФ (100 мл) и воде (33,3 мл) добавляли 3-бромпроп-1-ен (2,86 мл, 33,9 ммоль) с последующим добавлением индия (3,89 г, 33,9 ммоль). Реакционную смесь перемешивали примерно в течение 15 часов при температуре окружающей среды и затем добавляли водный раствор HCl (1 н, 150 мл) и EtOAc (150 мл). Органический слой отделяли, сушили над безводным Na2SO4, фильтровали, концентрировали в вакууме и очищали хроматографией на силикагеле с использованием в качестве элюента 20-60% EtOAc в гептане с получением 1-(5-тозил-5H-пирроло[2,3-b]пиразин-2-ил)бут-3-ен-1-ола (4 г, 69%) в виде вязкого масла: LC/MS (Таблица 1, Способ a) Rt = 2,30 мин; MS m/z: 344 (M+H)+.
Стадия D: 2-(1-азидобут-3-енил)-5-тозил-5H-пирроло[2,3-b]пиразин

К раствору 1-(5-тозил-5H-пирроло[2,3-b]пиразин-2-ил)бут-3-ен-1-ола (0,14 г, 0,41 ммоль) в DCM (10 мл) добавляли тионилхлорид (0,045 мл, 0,61 ммоль). Реакционную смесь перемешивали примерно в течение 8 часов при температуре окружающей среды и затем добавляли EtOAc и насыщенный водный раствор NaHCO3 (10 мл каждого). Органический слой отделяли, сушили над безводным Na2SO4, фильтровали и концентрировали в вакууме. Неочищенный хлорид растворяли в DMF (10 мл) и к реакционной смеси добавляли азид натрия (0,159 г, 2,45 ммоль). Реакционную смесь перемешивали примерно в течение 15 часов при температуре окружающей среды и затем к реакционной смеси добавляли EtOAc и насыщенный водный раствор NaHCO3 (10 мл каждого). Органический слой отделяли, концентрировали в вакууме и очищали хроматографией на силикагеле с использованием в качестве элюента 10-60% EtOAc в гептане с получением 2-(1-азидобут-3-енил)-5-тозил-5H-пирроло[2,3-b]пиразина (0,153 г, 87%) в виде масла: LC/MS (Таблица 1, Способ a) Rt = 2,84 мин; MS m/z: 369 (M+H)+.
Стадия E: 1-(5-тозил-5H-пирроло[2,3-b]пиразин-2-ил)бут-3-ен-1-амин гидрохлорид

К раствору 2-(1-азидобут-3-енил)-5-тозил-5H-пирроло[2,3-b]пиразина (3,90 г, 10,6 ммоль) в ТГФ (60 мл) и воде (30 мл) добавляли трифенилфосфин (3,33 г, 12,7 ммоль). Реакционную смесь нагревали до около 50°C примерно в течение 15 часов. Реакционную смесь охлаждали до температуры окружающей среды и концентрировали в вакууме. Остаток растворяли в EtOAc (30 мл) и добавляли HCl (газ) до достижения рН=1 и поддержания на этом уровне, с последующим добавлением Et2O, индуцируя образование осадка. После перемешивания примерно в течение 15 часов осадок собирали фильтрованием с получением 1-(5-тозил-5H-пирроло[2,3-b]пиразин-2-ил)бут-3-ен-1-амин гидрохлорида (2,5 г, 62%) в виде желтовато-коричневого твердого вещества: LC/MS (Таблица 1, Способ a) Rt = 1,80 мин; MS m/z: 343 (M+H)+.
Получение #13: N-((5-тозил-5H-пирроло[2,3-b]пиразин-2-ил)метил)циклогексанкарбоксамид

К суспензии (5-тозил-5H-пирроло[2,3-b]пиразин-2-ил)метанамин гидрохлорида (0,50 г, 1,476 ммоль, Пример #5, Стадия C) в DCM (10 мл) добавляли циклогексанкарбонилхлорид (0,221 мл, 1,623 ммоль) с последующим добавлением DIEA (0,644 мл, 3,69 ммоль). Реакционную смесь перемешивали примерно в течение 4 часов при температуре окружающей среды и затем к реакционной смеси добавляли насыщенный водный раствор NaHCO3 (20 мл) и DCM (20 мл). Органический слой отделяли, концентрировали в вакууме и очищали хроматографией на силикагеле (40 г) с использованием в качестве элюента 20-80% EtOAc в DCM с получением N-((5-тозил-5H-пирроло[2,3-b]пиразин-2-ил)метил)циклогексанкарбоксамида (0,49 г, 80%) в виде бесцветного твердого вещества: LC/MS (Таблица 1, Способ a) Rt = 2,40 мин; MS m/z: 413 (M+H)+.
Получение #14*: (2R,4S)-трет-бутил 4-(циклопропансульфонамидо)-2-метилпирролидин-1-карбоксилат

К суспензии 20% масс. Pd(OH)2 на C (0,605 г, 0,862 ммоль) в EtOH (75 мл) добавляли раствор (2R,4S)-трет-бутил 4-азидо-2-метилпирролидин-1-карбоксилата (3,9 г, 17 ммоль, синтезирован, как описано в Rosen, T.; Chu, D. T. W.; Lico, I. M.; Fernandes, P. B.; Marsh, K.;Shen, L.; Cepa, V. G.; Pernet, A. G. J. Med. Chem. 1988, 31, 1598-1611) в EtOH (25 мл). Реакционную смесь барботировали водородом и атмосферу водорода поддерживали через баллон. Реакционную смесь перемешивали примерно в течение 2 часов при температуре окружающей среды и затем фильтровали и концентрировали в вакууме. Остаток растворяли в DCM (100 мл), охлаждали до около 0°C и добавляли TEA (6,01 мл, 43,1 ммоль) с последующим добавлением циклопропансульфонилхлорида (2,67 г, 19,0 ммоль). Реакционную смесь перемешивали при температуре окружающей среды примерно в течение 15 часов, к реакционной смеси добавляли насыщенный водный раствор NaHCO3 (50 мл) и органический слой отделяли, концентрировали в вакууме и очищали хроматографией на силикагеле (80 г) с использованием в качестве элюента 20-80% EtOAc в гептане с получением (2R,4S)-трет-бутил 4-(циклопропансульфонамидо)-2-метилпирролидин-1-карбоксилата (2,55 г, 48%) в виде масла: LC/MS (Таблица 1, Способ a) Rt = 1,98 минут (ELSD); MS m/z: 305 (M+H)+.
Получение #15*: (2R,4S)-трет-бутил 4-(циклопропансульфонамидо)-2-этилпирролидин-1-карбоксилат
К суспензии 20% масс. Pd(OH)2 на C (0,044 г, 0,062 ммоль) в EtOH (30 мл) добавляли раствор (2R,4S)-трет-бутил 4-азидо-2-этилпирролидин-1-карбоксилата (1,5 г, 6,2 ммоль, синтезирован, как описано в Rosen, T.; Chu, D. T. W.; Lico, I. M.; Fernandes, P. B.; Marsh, K.; Shen, L.; Cepa, V. G.; Pernet, A. G. J. Med. Chem. 1988, 31, 1598-1611) в EtOH (10 мл). Реакционную смесь барботировали водородом и атмосферу водорода поддерживали через баллон. Реакционную смесь перемешивали примерно в течение 4 часов при температуре окружающей среды и затем фильтровали и концентрировали в вакууме. Остаток растворяли в пиридине (30 мл) и добавляли циклопропансульфонилхлорид (1,05 г, 7,49 ммоль). Реакционную смесь перемешивали примерно в течение 15 часов при температуре окружающей среды и затем распределяли между EtOAc (50 мл) и насыщенным водным раствором CuSO4 (50 мл). Органический слой отделяли, промывали насыщенным солевым раствором (30 мл), сушили над безводным Na2SO4, фильтровали, концентрировали в вакууме и очищали хроматографией на силикагеле (80g) с использованием в качестве элюента 20-80% EtOAc в гептане с получением (2R,4S)-трет-бутил 4-(циклопропансульфонамидо)-2-этилпирролидин-1-карбоксилата (0,95 г, 48%) в виде масла: LC/MS (Таблица 1, Способ a) Rt = 2,12 минут (ELSD); MS m/z: 319 (M+H)+.
Получение #16: трет-бутил 1-(6-тозил-6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)пирролидин-3-илкарбамат

К раствору трет-бутил 1-((5-тозил-5H-пирроло[2,3-b]пиразин-2-ил)метилкарбамотиоил)-пирролидин-3-илкарбамата (0,54 г, 1,0 ммоль, Получение #J,1) в ТГФ (15 мл) добавляли DIEA (0,444 мл, 2,54 ммоль) с последующим добавлением трифторацетата ртути (II) (0,478 г, 1,12 ммоль). Реакционную смесь перемешивали при температуре окружающей среды примерно в течение 2 часов и затем добавляли насыщенный водный раствор NaHCO3 (30 мл) и EtOAc (30 мл). Органический слой отделяли, сушили над безводным Na2SO4, фильтровали и концентрировали в вакууме. Неочищенное вещество очищали хроматографией на силикагеле (40 г) с использованием в качестве элюента 10-40% EtOAc в DCM с получением трет-бутил 1-(6-тозил-6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)пирролидин-3-илкарбамата (0,411 г, 81%) в виде желтого стекловидного вещества: LC/MS (Таблица 1, Способ a) Rt = 2,50 мин; MS m/z: 497 (M+H)+.
Получение #17: N-(4-(3-(2,3-дигидроксипропил)-6-тозил-6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)бицикло[2,2,2]октан-1-ил)циклопропансульфонамид

К раствору N-(4-(3-аллил-6-тозил-6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)бицикло-[2,2,2]октан-1-ил)циклопропансульфонамида (0,27 г, 0,47 ммоль, получали с использованием E с 4-(трет-бутоксикарбониламино)бицикло[2,2,2]октан-1-карбоновой кислотой [Prime Organics], K с циклопропилсульфонилхлоридом, H из Получения #12, HATU и DIEA, Q с реагентом Лоуссона и трифторацетатом ртути (II)) в 1,4-диоксане (10 мл) и воде (1 мл) добавляли N-метилморфолин-N-оксид (0,22 г, 1,8 ммоль) с последующим добавлением тетроксида осмия (4% масс. в воде, 0,36 мл, 0,047 ммоль). Реакционную смесь перемешивали примерно в течение 15 часов и затем к реакционной смеси добавляли DCM (20 мл) и воде (10 мл). Органический слой отделяли, концентрировали в вакууме и очищали хроматографией на силикагеле с использованием в качестве элюента 10-50% MeCN в DCM, с получением N-(4-(3-(2,3-дигидроксипропил)-6-тозил-6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)бицикло[2,2,2]октан-1-ил)циклопропансульфонамида (0,009 г, 3%): LC/MS (Таблица 1, Способ a) Rt = 1,90 мин; MS m/z: 612 (M-H)-.
Получение #18: 2-гидразинил-6-метил-5-тозил-5H-пирроло[2,3-b]пиразин

Стадия A: 5-бром-3-(проп-1-инил)пиразин-2-амин

К раствору 3,5-дибромпиразин-2-амина (10,0 г, 39,5 ммоль) в ТГФ (200 мл) добавляли иодид меди (I) (0,377 г, 1,98 ммоль), бис-(трифенилфосфин)палладий (II) дихлорид (1,39 г, 1,98 ммоль) и TEA (16,5 мл, 119 ммоль). Реакционную смесь охлаждали до около 0°C и дегазировали при помощи Ar. Реакционную смесь перемешивали примерно в течение 5 минут и затем реакционную смесь барботировали пропином и атмосферу пропина поддерживали через баллон. Реакционную смесь перемешивали примерно в течение 30 минут при около 0°C и затем давали нагреться до температуры окружающей среды. Реакционную смесь перемешивали примерно в течение 2 часов и затем к реакционной смеси добавляли EtOAc (100 мл) и воде (100 мл). Органический слой отделяли, сушили над безводным Na2SO4, фильтровали и концентрировали в вакууме. Неочищенную смесь очищали хроматографией на силикагеле (120 г) с использованием в качестве элюента 10-60% EtOAc в DCM (с сухой нагрузкой) с получением 5-бром-3-(проп-1-инил)пиразин-2-амина (7,05 г, 84%) в виде желтого твердого вещества: LC/MS (Таблица 1, Способ a) Rt = 1,79 мин; MS m/z: 212, 214 (1:1) (M+H)+.
Стадия B: 2-бром-6-метил-5-тозил-5H-пирроло[2,3-b]пиразин

К суспензии NaH (60% дисперсия в минеральном масле, 2,00 г, 49,9 ммоль) в NMP (100 мл) медленно добавляли раствор 5-бром-3-(проп-1-инил)пиразин-2-амина (7,05 г, 33,2 ммоль) в NMP (20 мл). Реакционную смесь перемешивали при температуре окружающей среды примерно в течение 20 минут и затем добавляли раствор п-толуолсульфонилхлорида (6,97 г, 36,6 ммоль) в NMP (20 мл). Реакционную смесь перемешивали при температуре окружающей среды примерно в течение 20 часов и затем к реакционной смеси добавляли водный раствор HCl (1 н, 100 мл). Полученные твердые вещества собирали фильтрованием. Коричневое твердое вещество растирали в порошок с DCM/EtOAc (1:1, 30 мл) и собирали фильтрованием с получением 2-бром-6-метил-5-тозил-5H-пирроло[2,3-b]пиразина (9,0 г, 74%) в виде коричневого твердого вещества: LC/MS (Таблица 1, Способ a) Rt = 2,68 мин; MS m/z: 366, 368 (1:1) (M+H)+.
Стадия C: трет-бутил 2-(6-метил-5-тозил-5H-пирроло[2,3-b]пиразин-2-ил)гидразинкарбоксилат

Трис(дибензилиденацетон)дипалладий(0) (0,250 г, 0,273 ммоль) и 2-ди-трет-бутилфосфино-2',4',6'-триизопропилбифенил (0,232 г, 0,546 ммоль) объединяли в 1,4-диоксане (15 мл). Из колбы откачивали газ при слабом барботировании растворителя и затем осторожно снова заполняли азотом (3 раза). Азот затем барботировали непосредственно в реакционную смесь. Смесь затем нагревали при около 80°C примерно в течение 10 минут и затем удаляли от источника нагрева. Добавляли 2-бром-6-метил-5-тозил-5H-пирроло[2,3-b]пиразин (1,0 г, 2,73 ммоль), трет-бутил гидразинкарбоксилат (0,541 г, 4,10 ммоль) и NaOt-Bu (0,501 мл, 4,10 ммоль) и реакционную смесь нагревали при около 80°C примерно в течение 1 часа. Реакционную смесь охлаждали до температуры окружающей среды и растворители удаляли при пониженном давлении. Черный остаток затем брали для поглощения в EtOAc (50 мл) и фильтровали. Фильтрат промывали насыщенным водным раствором NH4Cl (50 мл), EDTA (1,0 M водный раствор, 50 мл) и насыщенным водным раствором NaHCO3 (50 мл). Раствор сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении. Вещество очищали хроматографией на силикагеле (80 г) с использованием в качестве элюента 25-100% EtOAc в гептане с получением трет-бутил 2-(6-метил-5-тозил-5H-пирроло[2,3-b]пиразин-2-ил)гидразинкарбоксилата (0,160 г, 14%) в виде коричневого масла: LCMS (Таблица 1, Способ a) Rt = 2,51 мин; MS m/z: 418 (M+H)+.
Стадия D: 2-гидразинил-6-метил-5-тозил-5H-пирроло[2,3-b]пиразин
трет-Бутил 2-(6-метил-5-тозил-5H-пирроло[2,3-b]пиразин-2-ил)гидразинкарбоксилат (0,16 г, 0,38 ммоль) перемешивали в 1,4-диоксане (1,9 мл) в герметично закрытом сосуде с получением коричневого раствора. Добавляли HCl (4 M в 1,4-диоксане, 0,958 мл, 3,83 ммоль) и реакционную смесь перемешивали при температуре окружающей среды примерно в течение 20 часов. Растворители удаляли при пониженном давлении. Остаток распределяли между насыщенным водным раствором NaHCO3 (10 мл) и EtOAc (10 мл). Слои разделяли и водный слой экстрагировали при помощи EtOAc (2×10 мл). Объединенные органические экстракты промывали насыщенным солевым раствором (20 мл), сушили над безводным Na2SO4, фильтровали и концентрировали в вакууме с получением 2-гидразинил-6-метил-5-тозил-5H-пирроло[2,3-b]пиразина (0,089 г, 73%): LC/MS (Таблица 1, Способ a) Rt = 1,92 мин; MS m/z: 318 (M+H)+.
Получение #19:
Получение #19,1: (1S,3R,4S)-3-метил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентанамин
Получение #19,2: (1S,3R,4S)-3-метил-4-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентанамин

К смеси N-((1S,3R,4S)-3-метил-4-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)ацетамида (1,52 г, 3,36 ммоль, получали с использованием Y из Примера #7, стадия H и Pd/C, G, AA [Таблица 2, Способ 3, Rt = 6,1 мин, or = ND], Z с NaOH, с Примером #1 Стадия D, HATU и TEA, и B с TEA) и 1,4-диоксана (25 мл) добавляли водный раствор HCl (6 н, 25 мл, 150 ммоль). Реакционную смесь нагревали при около 100°C примерно в течение 14 часов и затем охлаждали до температуры окружающей среды и концентрировали при пониженном давлении. К полученному коричневому остатку добавляли MeOH (30 мл) и раствор концентрировали при пониженном давлении. К полученному остатку добавляли MeOH (5 мл) с последующим медленным добавлением Et2O (20 мл). Сначала образовался мутный раствор и затем образовалось темное масло/смола, и смесь концентрировали при пониженном давлении. К полученному коричневому остатку добавляли MeOH (30 мл), (1S,3R,4S)-3-метил-4-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентан-амин (1,35 г, 2,50 ммоль, Уф чистота 75%) из отдельной реакции и силикагель (7 г). Смесь концентрировали при пониженном давлении и очищали хроматографией на силикагеле с градиентным элюированием 0-100% (DCM/[2 M NH3 в MeOH] (9:1)) в DCM, колонку дополнительно промывали при помощи MeOH, затем MeOH/водный раствор NH4OH (9:1), с получением (1S,3R,4S)-3-метил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентанамина [Получение #19.1] (0,092 г, 5%) в виде темного коричневого твердого вещества: LC/MS (Таблица 1, Способ a) Rt = 1,35 мин; MS m/z: 257 (M+H)+ и 2,9 г коричневого остатка, который распределяли между DCM и насыщенным водным раствором NaHCO3 (50 мл каждого). Слои разделяли и водный слой экстрагировали дополнительным количеством DCM (2×50 мл). Объединенные органические слои промывали насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали при пониженном давлении с получением (1S,3R,4S)-3-метил-4-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентанамина [Получение #19.2] (1,94 г, 78%) в виде серо-коричневого пенообразного вещества: LC/MS (Таблица 1, Способ a) Rt = 1,80 мин; MS m/z: 411 (M+H)+.
Получение #20: 3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил бензоат
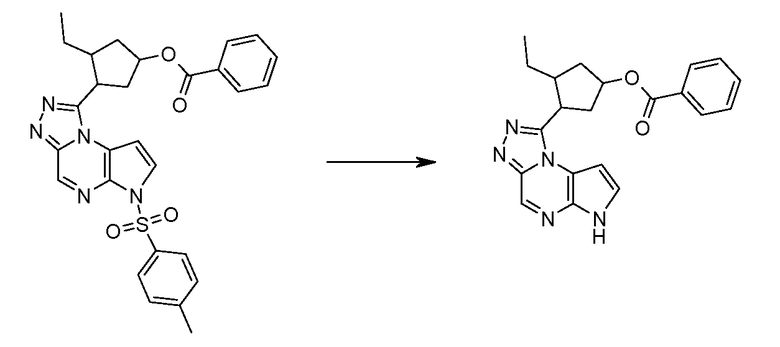
К смеси 3-этил-4-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентилбензоата (5,00 г, 7,84 ммоль, получали из Примера #4, Стадия J, с использованием II с бензойной кислотой и B) в MeOH (16 мл) добавляли раствор цианида калия (0,74 мл, 17,2 ммоль) в MeOH (16 мл). Реакционную смесь перемешивали при температуре окружающей среды примерно в течение 16 часов. Реакционную смесь концентрировали при пониженном давлении с получением остатка. Остаток распределяли между водой (20 мл) и DCM (20 мл). Слои разделяли и водный слой экстрагировали при помощи DCM (3×10 мл). Экстракт затем промывали насыщенным водным раствором NaHCO3, сушили над безводным MgSO4, фильтровали и концентрировали при пониженном давлении с получением неочищенного масла. Неочищенное вещество очищали хроматографией на силикагеле с градиентным элюированием 0-10% MeOH в DCM с получением 3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентилбензоата (2,30 г, 78%) в виде красноватого твердого вещества. LC/MS (Таблица 1, Способ a) Rt = 2,08 мин; MS m/z: 376 (M+H)+.
Получение #21: трет-бутил 4-(аминометил)-2-этилциклопентанкарбоксилат

Стадия A: трет-бутил 2-этил-4-(гидроксиметил)циклопентанкарбоксилат

Раствор 3-трет-бутил 1-метил 4-этилциклопентан-1,3-дикарбоксилата (3,88 г, 15,1 ммоль, Получение #11, Стадия B) в Et2O (150 мл) охлаждали до около -40°C. Добавляли по каплям LAH (2 н в ТГФ, 8,32 мл, 16,6 ммоль). Реакционную смесь перемешивали при около -40°C примерно в течение 1 часа. Реакционную смесь распределяли между насыщенным водным раствором NaHCO3 (50 мл) и EtOAc (3×50 мл). Объединенные органические экстракты концентрировали при пониженном давлении. Неочищенное вещество очищали хроматографией на силикагеле с градиентным элюированием 0-100% EtOAc/гептан с получением трет-бутил 2-этил-4-(гидроксиметил)циклопентанкарбоксилата (1,00 г, 29%) в виде коричневого масла: LC/MS (Таблица 1, Способ a) Rt = 2,37 мин; MS m/z: 229 (M+H)+.
Стадия B: трет-бутил 2-этил-4-((метилсульфонилокси)метил)циклопентанкарбоксилат

К раствору трет-бутил 2-этил-4-(гидроксиметил)циклопентанкарбоксилата (0,220 г, 0,964 ммоль) в DCM (5 мл) добавляли TEA (0,16 мл, 1,15 ммоль) и метансульфонилхлорид (0,083 мл, 1,06 ммоль) при около 0°C. Реакционной смеси давали нагреться до около 25°C и перемешивали при около 25°C примерно в течение 16 часов. Реакционную смесь распределяли между водой (20 мл) и DCM (20 мл). Водный раствор промывали при помощи DCM (2×20 мл). Объединенные органические экстракты сушили над безводным MgSO4, фильтровали и концентрировали при пониженном давлении с получением трет-бутил 2-этил-4-((метилсульфонилокси)метил)циклопентанкарбоксилата (0,295 г, 100%): LC/MS (Таблица 1, Способ b) Rt = 2,55 мин; MS m/z: 307 (M+H)+.
Стадия C: трет-бутил 4-(аминометил)-2-этилциклопентанкарбоксилат

К раствору трет-бутил 2-этил-4-((метилсульфонилокси)метил)циклопентанкарбоксилата (0,295 г, 0,964 ммоль) в DMF (5 мл) добавляли азид натрия (0,313 г, 4,82 ммоль). Реакционную смесь нагревали при около 50°C примерно в течение 16 часов и затем охлаждали до около 15-20°C. К реакционной смеси добавляли воду (40 мл). Водный раствор экстрагировали при помощи DCM (3×30 мл). Объединенные органические экстракты сушили над безводным MgSO4, фильтровали и концентрировали при пониженном давлении с получением темного коричневого масла. Коричневое масло растворяли в ТГФ (6,5 мл) и воде (3,5 мл). Добавляли трифенилфосфин (0,316 г, 1,205 ммоль). Реакционную смесь перемешивали при около 25°C примерно в течение 15 часов. Органический растворитель удаляли при пониженном давлении и остаток распределяли между насыщенным водным раствором NaHCO3 (20 мл) и DCM (20 мл). Органическую фазу концентрировали при пониженном давлении. Полученный остаток очищали с использованием хроматографии на силикагеле с градиентным элюированием 0-20% (20% (7 н раствор аммония в MeOH) в MeOH)) в DCM с получением трет-бутил 4-(аминометил)-2-этилциклопентанкарбоксилата (0,102 г, 46%) в виде коричневого масла: LC/MS (Таблица 1, Способ b) Rt = 1,72 мин; MS m/z: 228 (M+H)+.
Получение #22: этил 2-этил-4-формилциклопентанкарбоксилат
Стадия A: 2-этил-4-(гидроксиметил)циклопентанкарбоновой кислоты
К раствору 2-этил-4-(метоксикарбонил)циклопентанкарбоновой кислоты (8,34 г, 41,7 ммоль, Получение #11, Стадия A) в ТГФ (208 мл) добавляли LiBH4 (0,907 г, 41,7 ммоль) при около -20°C. Реакционную смесь перемешивали при около -20°C примерно в течение 1 часа. Реакционной смеси давали нагреться до около 25°C, затем перемешивали при около 25°C примерно в течение 16 часов. Добавляли дополнительное количество LiBH4 (0,907 г, 41,7 ммоль). Реакционную смесь перемешивали при около 25°C примерно в течение 4 часов. Медленно добавляли воду (10 мл) для гашения реакции. Твердое вещество удаляли вакуумным фильтрованием. Фильтрат концентрировали при пониженном давлении. Полученный остаток распределяли между водой (50 мл) и DCM (3×50 мл). Объединенные органические экстракты сушили над безводным MgSO4, фильтровали и концентрировали при пониженном давлении с получением 2-этил-4-(гидроксиметил)циклопентанкарбоновой кислоты (7,29 г, 100%): LC/MS (Таблица 1, Способ n) Rt = 0,44 мин; MS m/z: 173 (M+H)+.
Стадия B: этил 2-этил-4-(гидроксиметил)циклопентанкарбоксилат

HCl газ барботировали через раствор 2-этил-4-(гидроксиметил)циклопентанкарбоновой кислоты (7,29 г, 42,3 ммоль) в EtOH (60 мл) при около 25°C примерно в течение 10 минут. Реакционную смесь перемешивали при около 25°C примерно в течение 72 часов. Растворитель удаляли при пониженном давлении. Неочищенный остаток распределяли между водой (30 мл) и DCM (3×30 мл). Объединенные органические экстракты концентрировали при пониженном давлении. Неочищенное вещество очищали хроматографией на силикагеле с градиентным элюированием 0-100% EtOAc/гептан с получением этил 2-этил-4-(гидроксиметил)циклопентанкарбоксилата (4,89 г, 58%) в виде желтого масла: 1H ЯМР (CDCl3) δ 4,23-4,02 (м, 2H), 3,74-3,47 (м, 2H), 2,96-2,83 (м, 1H), 2,31-2,17 (м, 1H), 2,15-1,98 (м, 2H), 1,97-1,84 (м, 1H), 1,79-1,66 (м, 1H), 1,65-1,50 (м, 1H), 1,49-1,37 (м, 1H), 1,30-1,21 (м, 5H), 1,04-0,82 (м, 3H).
Стадия C: этил 2-этил-4-формилциклопентанкарбоксилат

К раствору этил 2-этил-4-(гидроксиметил)циклопентанкарбоксилата (4,84 г, 24,2 ммоль) в DCM (100 мл) добавляли хлорхромат пиридиния (10,42 г, 48,3 ммоль). Реакционную смесь перемешивали при около 25°C примерно в течение 3 часов. Добавляли силикагель (1 г). Смесь перемешивали при около 25°C примерно в течение 30 минут. Твердое вещество удаляли вакуумным фильтрованием с промывкой при помощи DCM (100 мл). Фильтрат концентрировали при пониженном давлении. Полученный остаток очищали с использованием хроматографии на силикагеле с градиентным элюированием 0-40% EtOAc/гептан с получением этил 2-этил-4-формилциклопентанкарбоксилата (3,03 г, 63%) в виде прозрачного масла: 1H ЯМР (DMSO-d 6) δ 9,66-9,47 (м, 1H), 4,12-3,94 (м, 2H), 2,94-2,73 (м, 2H), 2,19-1,90 (м, 4H), 1,55-1,65 (м, 1H), 1,37-1,23 (м, 1H), 1,23-1,06 (м, 4H), 0,96-0,82 (м, 3H).
Получение #23.: N-((1S,3R,4S)-3-этил-4-(8-метил-6-((2-(триметилсилил)этокси)метил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)-N-((2-(триметилсилил)этокси)метил)циклопропансульфонамид

Карбонат цезия (0,274 г, 0,841 ммоль), трициклогексилфосфин (20% масс. раствор в толуоле, 0,094 г, 0,067 ммоль), Pd2(dba)3 (0,039 г, 0,042 ммоль) и триметилборат (0,069 г, 0,547 ммоль) добавляли к раствору N-((1S,3R,4S)-3-этил-4-(8-иод-6-((2-(триметилсилил)этокси)метил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)-N-((2-(триметилсилил)этокси)метил)циклопропансульфонамида (0,32 г, 0,421 ммоль, получали с использованием KK из Получения #GGG,1) в 1,4-диоксане (8 мл). Смесь дегазировали и нагревали при около 85°C примерно в течение 2 часов. Растворитель удаляли и остаток распределяли между EtOAc и водой (20 мл каждого). Органическую фазу промывали насыщенным солевым раствором (15 мл), сушили над безводным MgSO4, фильтровали и концентрировали. Полученную смесь очищали флэш-хроматографией на силикагеле (40 до 100% EtOAc в гептане) с получением N-((1S,3R,4S)-3-этил-4-(8-метил-6-((2-(триметилсилил)этокси)метил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)-N-((2-(триметилсилил)этокси)метил)циклопропансульфонамида (0,21 г, 77%) в виде желтого аморфного твердого вещества. LC/MS (Таблица 1, Способ a) Rt = 3,39 мин; MS m/z: 650 (M+H)+.
Получение #24: диэтил 2-(4-(циклопропансульфонамидо)бицикло[2,2,2]октан-1-ил)-2-оксоэтилфосфонат
Стадия A: метил 4-(циклопропансульфонамидо)бицикло[2,2,2]октан-1-карбоксилат

К раствору метил 4-аминобицикло[2,2,2]октан-1-карбоксилата (500 мг, 2,73 ммоль) (Yeh, V. S. C.; Kurukulasuriya, R.; Madar, D.; Patel, J. R.; Fung, S.; Monzon, K.; Chiou, W.; Wang, J.; Jacobson, P.; Sham, Н. L.; Link, J. T. Bioorg. and Med. Chem. Let, 2006, vol. 16, # 20 p. 5408-5413) в DCM (10 мл) при комнатной температуре добавляли TEA (0,76 мл, 5,46 ммоль) и DMAP (50 мг, 0,41 ммоль). Добавляли по каплям циклопропансульфонилхлорид (764 мг, 5,46 ммоль, Matrix) через шприц. Реакционную смесь перемешивали примерно в течение 15 часов при комнатной температуре. Смесь промывали водой (10 мл) и водный слой экстрагировали при помощи DCM (2×10 мл), органические слои объединяли и сушили над безводным Na2SO4, фильтровали и концентрировали в вакууме. Неочищенное вещество очищали хроматографией на силикагеле с градиентным элюированием 20-35% EtOAc в гексане с получением метил 4-(циклопропансульфонамидо)бицикло[2,2,2]октан-1-карбоксилата (410 мг, 52% выход). LC/MS (Таблица 1, Способ p) Rt = 1,68 мин; MS m/z: 288 (M+H)+.
Стадия B: диэтил (4-(циклопропансульфонамидо)бицикло[2,2,2]октан-1-ил)метилфосфонат
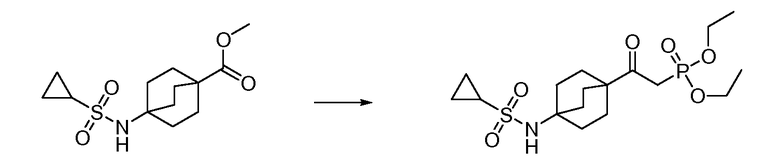
Раствор диэтилметилфосфоната (1,27 г, 8,36 ммоль) растворяли в ТГФ (20 мл) и охлаждали до около -78oC на бане сухой лед-ацетон в атмосфере азота. Затем добавляли по каплям n-BuLi (9,77 ммоль, 3,9 мл, 2,5M в гексане) примерно в течение 5 минут. Реакционную смесь перемешивали примерно в течение 3 часов, поддерживая температуру ниже около -70 oC. Затем добавляли раствор метил 4-(циклопропансульфонамидо)бицикло[2,2,2]октан-1-карбоксилата (800 мг, 2,79 ммоль) в ТГФ (10 мл), поддерживая температуру при около -78oC. Раствор перемешивали примерно в течение 15 часов, давая температуре медленно подняться до комнатной температуры. К реакционной смеси добавляли насыщенный водный раствор NH4Cl (30 мл) и экстрагировали при помощи EtOAc (3×30 мл). Органические слои объединяли и промывали насыщенным солевым раствором, сушили над безводным Na2SO4 и концентрировали с получением диэтил (4-(циклопропансульфонамидо)бицикло[2,2,2]октан-1-ил)метилфосфоната (1,30 г, 100% выход). Неочищенный продукт использовали на следующей стадии без дополнительной очистки. LC/MS (Таблица 1, Способ p) Rt = 1,62 мин; MS m/z: 408 (M+H)+.
Получение #25: 3-этил-4-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентанон

Стадия A: этил 2-этил-4-оксоциклопентанкарбоксилат

В круглодонную колбу загружали этил 2-этил-4-оксоциклопентанкарбоксилат (1,5 г, 8,1 ммоль, Пример #22, Стадия B) в DCM (22 мл). В колбу добавляли этиленгликоль (0,91 мл, 16 ммоль), триэтилортоформиат (2,0 мл, 12 ммоль) и моногидрат п-толуолсульфоновой кислоты (0,31 г, 1,6 ммоль). Реакционную смесь перемешивали при комнатной температуре примерно в течение 24 часов. Раствор концентрировали при пониженном давлении с получением коричневого масла, которое растворяли в EtOAc и очищали флэш-хроматографией на силикагеле (Silicycle 25 г колонка) с градиентным элюированием 0-50% EtOAc в гептане. Содержащие продукт фракции объединяли и концентрировали досуха при пониженном давлении с получением этил 2-этил-4-оксоциклопентанкарбоксилата в виде светло-желтого масла (1,6 г, 83%): LC/MS (Таблица 1, Способ c) MS m/z 229 (M+H)+; 1H ЯМР (CDCl) δ 4,14 (кв., 2H), 3,90 (м, 4H), 2,99 (кв., 1H), 2,32-2,27 (м, 1H), 2,26-2,11 (м, 1H), 2,05-1,99 (м, 1H), 1,96-1,91 (м, 1H), 1,83-1,78 (м, 1H), 1,46-1,39 (м, 1H), 1,31-1,24 (м, 1H), 1,26 (т, 3H), 0,90 (т, 3H).
Стадия B: 8-этил-1,4-диоксаспиро[4,4]нонан-7-карбоновая кислота

В круглодонную колбу загружали этил 8-этил-1,4-диоксаспиро[4,4]нонан-7-карбоксилат (0,32 г, 1,4 ммоль) и 1н водный раствор гидроксида натрия (14,0 мл, 14,0 ммоль). Раствор перемешивали в течение ночи при комнатной температуре. К раствору добавляли DCM (30 мл) с последующим добавлением 20% водного раствора лимонной кислоты (около 20 мл) для достижения pH около 2. Слои разделяли и водный раствор экстрагировали при помощи DCM (2×30 мл) и DCM/EtOAc (1:1, 30 мл). Объединенные экстракты сушили над безводным MgSO4, фильтровали и концентрировали при пониженном давлении с получением 8-этил-1,4-диоксаспиро[4,4]нонан-7-карбоновой кислоты в виде прозрачного бесцветного масла (0,27 г, 96%): LC/MS (Таблица 1, Способ c) Rt = 1,20 мин; MS m/z: 201 (M+H)+.
Стадия C: 8-этил-N'-(5-тозил-5H-пирроло[2,3-b]пиразин-2-ил)-1,4-диоксаспиро[4,4]нонан-7-карбогидразид
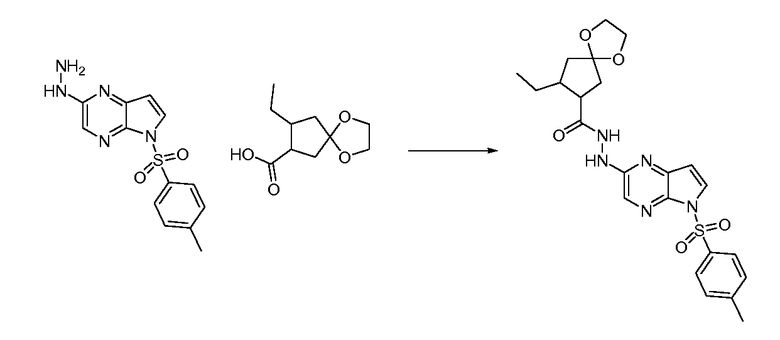
A 50-мл круглодонную колбу загружали 2-гидразинил-5-тозил-5H-пирроло[2,3-b]пиразин (0,350 г, 1,16 ммоль, Пример #1. Стадия D), 8-этил-1,4-диоксаспиро[4,4]нонан-7-карбоновую кислоту (0,250 г, 1,25 ммоль) и DCM (6,0 мл). К реакционной смеси добавляли HATU (0,483 г, 1,27 ммоль) и TEA (0,64 мл, 4,6 ммоль) и полученную желтую суспензию перемешивали при комнатной температуре примерно в течение 3 часов. К реакционному раствору добавляли DCM (25 мл) и раствор промывали водой и насыщенным солевым раствором (20 мл каждого). Органический слой сушили над безводным MgSO4, фильтровали и концентрировали при пониженном давлении с получением коричневого масла. Неочищенный продукт очищали флэш-хроматографией на силикагеле (колонка с 25 г Silicycle) с градиентным элюированием: 0-10% MeOH в DCM в течение 25 минут. Содержащие продукт фракции концентрировали при пониженном давлении с получением 8-этил-N'-(5-тозил-5H-пирроло[2,3-b]пиразин-2-ил)-1,4-диоксаспиро[4,4]нонан-7-карбогидразида в виде пенообразного вещества (0,50 г, 89%): LC/MS (Таблица 1, Способ c) Rt = 1,49 мин; MS m/z: 486 (M+H)+.
Стадия D: 1-(8-этил-1,4-диоксаспиро[4,4]нонан-7-ил)-6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин

В круглодонную колбу загружали 8-этил-N'-(5-тозил-5H-пирроло[2,3-b]пиразин-2-ил)-1,4-диоксаспиро[4,4]нонан-7-карбогидразид (4,90 г, 10,1 ммоль) и 1,4-диоксан (50 мл). В колбу добавляли DIEA (8,81 мл, 50,5 ммоль) с последующим добавлением тионилхлорида (0,770 мл, 10,6 ммоль). Смесь нагревали до около 75°C примерно в течение 90 минут. Добавляли дополнительное количество тионилхлорида (0,074 мл, 1,0 ммоль) и нагревание продолжали примерно в течение 1 часа. Реакционную смесь охлаждали до комнатной температуры и перемешивали в течение ночи. Раствор разбавляли при помощи DCM (75 мл) и промывали водой (50 мл). Слои разделяли и органический слой сушили над безводным MgSO4, фильтровали и концентрировали при пониженном давлении с получением темно-коричневого масла. Неочищенный продукт очищали флэш-хроматографией на силикагеле с градиентным элюированием 0-60% ацетона в гептане с удерживанием при 60% ацетона в гептане. Содержащие продукт фракции объединяли и концентрировали с получением вещества, которое загружали во вторую колонку (Silicycle, 40 г колонка), с градиентным элюированием 0-60% ацетона в гептане. Содержащие продукт фракции объединяли и концентрировали при пониженном давлении с получением 1-(8-этил-1,4-диоксаспиро[4,4]нонан-7-ил)-6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразина в виде желто-коричневого порошка (3,0 г, 64%): LC/MS (Таблица 1, Способ c) Rt = 1,44 мин; MS m/z: 468 (M+H)+.
Стадия E: 3-этил-4-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентанон
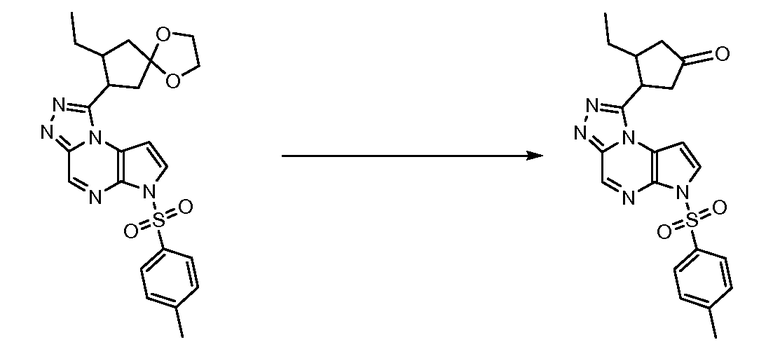
В круглодонную колбу загружали 1-((7S,8R)-8-этил-1,4-диоксаспиро[4,4]нонан-7-ил)-6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин (3,56 г, 7,61 ммоль) и ТГФ (20 мл). К раствору добавляли водный раствор HCl (6н, 3,81 мл, 22,8 ммоль) и смесь перемешивали при комнатной температуре примерно в течение 2 часов. Растворитель удаляли при пониженном давлении и добавляли DCM (75 мл) и воде (50 мл). Слои разделяли и органический раствор сушили над безводным MgSO4, фильтровали и концентрировали при пониженном давлении с получением 3-этил-4-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентанона в виде коричневого пенообразного вещества (2,99 г, 93%): LC/MS (Таблица 1, Способ c) Rt = 1,40 мин; MS m/z: 424 (M+H)+.
Получение #26: 3,3-дифтор-1-(винилсульфонил)пирролидин

Раствор 3,3-дифторпирролидингидрохлорида (0,3 г, 2,1 ммоль, Matrix) и DIEA (0,37 мл, 2,1 ммоль) в MeCN (5 мл) перемешивали при около 50°C примерно в течение 30 минут. Реакционную смесь охлаждали до температуры окружающей среды и концентрировали при пониженном давлении. Твердое вещество растворяли в MeCN (2 мл) и добавляли раствор 2-хлорэтансульфонилхлорида (0,22 мл, 2,1 ммоль) в Et2O (3 мл) при около -78°C и перемешивали примерно в течение 2 часов. К реакционной смеси добавляли DIEA (0,6 мл, 3,4 ммоль) и перемешивали примерно в течение 1 часа. Реакционную смесь нагревали до температуры окружающей среды и растворитель удаляли при пониженном давлении. Остаток распределяли между DCM (5 мл) и водой (2×2 мл). Объединенные органические слои сушили над безводным MgSO4, фильтровали и концентрировали при пониженном давлении с получением неочищенного 3,3-дифтор-1-(винилсульфонил)пирролидина (0,11 г, 27%), который использовали без дополнительной очистки: LC/MS (Таблица 1, Способ b) Rt = 2,04 мин; MS m/z: 198 (M+H)+.
Получение #27: 4-хлор-5-нитро-1H-пирроло[2,3-b]пиридин

Стадия A: 4-хлор-3-иод-5-нитропиридин-2-амин

Раствор 4-хлор-3-иодпиридин-2-амина (0,25 г, 0,982 ммоль, Boa Pharma) в концентрированной H2SO4 (1,95 мл) охлаждали до около 0°C, затем добавляли по порциям нитрат калия (0,21 г, 2,2 ммоль) в течение 10 минут. Реакционную смесь перемешивали примерно в течение 4 часов при около 0°C. Реакционную смесь медленно добавляли через пипетку в раствор гидроксида аммония и колотого льда (10 мл) на ледяной бане. pH реакционной смеси поддерживали выше 9 путем постепенно увеличивающегося добавления гидроксида аммония. Полученный осадок фильтровали и сушили с получением 4-хлор-3-иод-5-нитропиридин-2-амина (0,085 г, 29%) в виде зеленоватого твердого вещества LC/MS (Таблица 1, Способ n) Rt = 0,64 мин; MS m/z: 298 (M-H)-.
Стадия B: 4-хлор-5-нитро-3-((триметилсилил)этинил)пиридин-2-амин
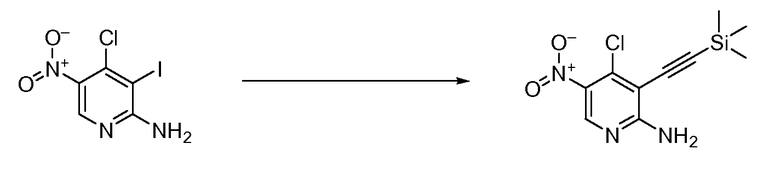
К раствору 4-хлор-3-иод-5-нитропиридин-2-амина (5,30 г, 17,7 ммоль) в ТГФ (90 мл) добавляли TEA (15,0 мл, 108 ммоль). Реакционную смесь дегазировали и продували азотом 3 раза. Креакционной смеси добавляли бис(трифенилфосфин)-палладий(II)дихлорид (0,62 г, 0,88 ммоль, Strem), иодид меди(I) (0,17 г, 0,89 ммоль) и триметилсилилацетилен (5,4 мл, 39 ммоль). Смесь дегазировали и продували 3 раза азотом. Реакционную смесь нагревали при около 60°C примерно в течение 16 часов. Реакционную смесь охлаждали до температуры окружающей среды. Реакционную смесь фильтровали и промывали при помощи ТГФ (200 мл). Фильтрат концентрировали при пониженном давлении. К остатку добавляли DCM (100 мл) и образовавшийся осадок фильтровали и собирали с получением 4-хлор-5-нитро-3-((триметилсилил)этинил)пиридин-2-амина (0,77 г). Остальной фильтрат концентрировали при пониженном давлении и неочищенное вещество очищали флэш-хроматографией на силикагеле с градиентным элюированием 0-100% EtOAc в DCM. Очищенное вещество объединяли с 0,77 г осадка с получением 4-хлор-5-нитро-3-((триметилсилил)этинил)пиридин-2-амина (2,22 г, 47%) в виде желтого твердого вещества: LC/MS (Таблица 1, Способ c) Rt = 1,62 мин; MS m/z 268 (M-H)-.
Стадия C: 4-хлор-3-этинил-5-нитропиридин-2-амин

К раствору 4-хлор-5-нитро-3-((триметилсилил)этинил)пиридин-2-амина (1,98 г, 7,34 ммоль) в DMF (25 мл) добавляли фторид калия на оксиде алюминия (40% масс., 2,67 г, 18,35 ммоль). Суспензию перемешивали при температуре окружающей среды примерно в течение 1 часа. Добавляли активированный уголь (0,3 г) и суспензию фильтровали через Целит® и промывали при помощи DMF (150 мл). Растворитель удаляли при пониженном давлении и неочищенное вещество очищали хроматографией на силикагеле с градиентным элюированием 0-10% MeOH в DCM с получением 4-хлор-3-этинил-5-нитропиридин-2-амина (1,03 г, 71%) в виде желтого твердого вещества: LC/MS (Таблица 1, Способ n) Rt = 0,59 мин; MS m/z: 196 (M-H)-.
Стадия D: 4-хлор-5-нитро-1H-пирроло[2,3-b]пиридин

К раствору 4-хлор-3-этинил-5-нитропиридин-2-амина (0,16 г, 0,81 ммоль) в DMF (3 мл) добавляли хлор(1,5-циклооктадиен)родий (I) димер (0,02 г, 0,04 ммоль) и трис(4-фторфенил)фосфин (0,128 г, 0,405 ммоль). Реакционную смесь дегазировали путем барботирования аргоном в течение 15 минут. Реакционную смесь нагревали при около 80°C примерно в течение 45 минут. Реакционную смесь охлаждали до температуры окружающей среды и растворитель удаляли при пониженном давлении и остаток суспендировали в эфире (10 мл). Осадок собирали фильтрованием и сушили с получением 4-хлор-5-нитро-1H-пирроло[2,3-b]пиридина (0,132 г, 83%, содержит приблизительно 6 моль% DMF и приблизительно 3 моль% трис(4-фторфенил)фосфина) в виде коричневого твердого вещества: LC/MS (Таблица 1, Способ a) Rt = 2,05 мин; MS m/z 198 (M+H)+.
Получение #28*: N-((1S,3R,4S)-3-этил-4-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)-2-метилпропан-2-сульфонамид

К раствору (1S,3R,4S)-3-этил-4-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентанамина (115 мг, 0,271 ммоль, Получение #BB.1*) в DCM (1,5 мл) добавляли DIEA (0,071 мл, 0,406 ммоль) с последующим добавлением хлорангидрида 2-метилпропан-2-сульфиновой кислоты (0,037 мл, 0,298 ммоль). Примерно через 4 часа реакционную смесь разбавляли EtOAc (10 мл) и насыщенным водным раствором NaHCO3 (10 мл). Органический слой отделяли, сушили над безводным Na2SO4, фильтровали и концентрировали в вакууме. Неочищенный остаток растворяли в DCM (1,5 мл) и добавляли свежеполученный раствор м-хлорпербензойной кислоты (0,271 мл, 0,271 ммоль, 1M в DCM). Примерно через 2 часа реакционную смесь разбавляли EtOAc (10 мл) и насыщенным водным раствором NaHCO3 (10 мл). Органический слой отделяли, сушили над безводным Na2SO4, фильтровали и концентрировали в вакууме. Неочищенный остаток очищали хроматографией на силикагеле с использованием в качестве элюента EtOAc с получением N-((1S,3R,4S)-3-этил-4-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)-2-метилпропан-2-сульфонамида (95 мг, 64% выход) в виде масла. LC/MS (Таблица 1, Способ a) Rt = 2,40 мин; MS m/z: 545 (M+H)+.
Получение #29*: 3-((1S,3R,4S)-3-этил-4-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентиламино)-4-метоксициклобут-3-ен-1,2-дион

К раствору (1S,3R,4S)-3-этил-4-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентанамина (0,40 г, 0,942 ммоль, Пример #8 Стадия M) в MeOH (3 мл) добавляли 3,4-диметоксициклобут-3-ен-1,2-дион (0,14 г, 0,98 ммоль) и DIEA (0,18 мл, 1,0 ммоль). Реакционную смесь перемешивали при комнатной температуре примерно в течение 16,5 часов. Затем твердое вещество из реакционной смеси собирали путем вакуумного фильтрования, осуществляя при этом промывку охлажденным MeOH (около 4°C, 10 мл) и сушили в вакуумной печи при около 60°C с получением неочищенного 3-((1S,3R,4S)-3-этил-4-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентиламино)-4-метоксициклобут-3-ен-1,2-диона (0,36 г, 73%, 90% чистота): LC/MS (Таблица 1, Способ a) Rt = 2,13 мин; MS m/z: 535 (M+H)+.
Получение #30: 3,3,3-трифтор-1-(5-тозил-5H-пирроло[2,3-b]пиразин-2-ил)пропан-1-амин гидрохлорид
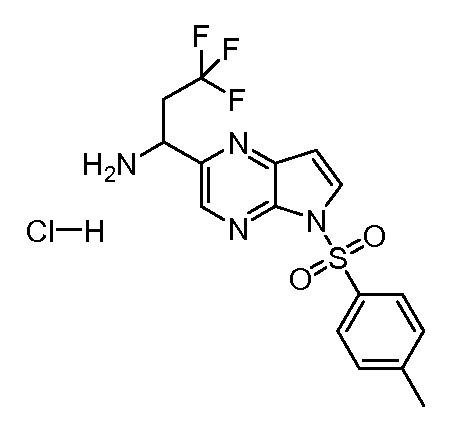
Стадия A: N-(дифенилметилен)-1-(5-тозил-5H-пирроло[2,3-b]пиразин-2-ил)метанамин

К раствору (5-тозил-5H-пирроло[2,3-b]пиразин-2-ил)метанамина (2,00 г, 6,61 ммоль, Пример #5 Стадия C) в DCM (30 мл) добавляли дифенилметанимин (1,16 мл, 6,61 ммоль). Примерно через 2 дня реакционную смесь концентрировали в вакууме с получением N-(дифенилметилен)-1-(5-тозил-5H-пирроло[2,3-b]пиразин-2-ил)метанамина (2,75 г, 89%) в виде пенообразного вещества и использовали без дополнительной очистки. LC/MS (Таблица 1, Способ a) Rt = 3,02 мин; MS m/z: 467 (M+H)+.
Стадия B: 3,3,3-трифтор-1-(5-тозил-5H-пирроло[2,3-b]пиразин-2-ил)пропан-1-амин, гидрохлорид

К раствору N-(дифенилметилен)-1-(5-тозил-5H-пирроло[2,3-b]пиразин-2-ил)метанамина (0,722 г, 1,55 ммоль) в ТГФ (3 мл) при около -78°C добавляли NaHMDS (0,5 M в ТГФ, 1,55 мл, 1,55 ммоль). Примерно через 30 минут к реакционной смеси добавляли 1,1,1-трифтор-2-иодэтан (1,51 мл, 15,5 ммоль). Примерно через 4 часа реакционной смеси давали медленно нагреться до комнатной температуры в течение ночи. Примерно через 15 часов добавляли EtOAc (30 мл) и насыщенный водный раствор NaHCO3 (30 мл). Органический слой отделяли, концентрировали в вакууме и очищали хроматографией на силикагеле с использованием в качестве элюента EtOAc/гептан (20-50%) с получением неочищенного алкилированного имина. Имин растворяли в изопропилацетате (30 мл) и добавляли концентрированную HCl (0,50 мл). Реакционную смесь центрифугировали на роторном испарителе в течение 1 часа перед частичным концентрированием до приблизительно 10 мл. Добавляли дополнительное количество изопропилацетата (30 мл) и растворитель частично удаляли в вакууме до тех пор, пока не осталось приблизительно 10 мл. Добавляли Et2O (30 мл) и раствор оставляли для старения примерно в течение 30 минут. Полученные твердые вещества собирали фильтрованием и сушили в вакууме с получением 3,3,3-трифтор-1-(5-тозил-5H-пирроло[2,3-b]пиразин-2-ил)пропан-1-амина, гидрохлорида, (0,150 г, 23%) в виде бесцветного твердого вещества. LC/MS (Таблица 1, Способ a) Rt = 1,88 мин; MS m/z 385 (M+H)+.
Получение #31: (1S,2R,4R)-4-(2-этокси-2-оксоэтил)-2-этилциклопентанкарбоновая кислота

Стадия A: (3R,4S)-3-этил-4-(гидроксиметил)циклопентанол

К раствору (1S,2R)-этил 2-этил-4-оксоциклопентанкарбоксилата (5 г, 27,1 ммоль, Пример #22 стадия B) в ТГФ (100 мл) при около -78°C добавляли LAH (2 M в ТГФ, 54,3 мл, 109 ммоль). Примерно через 1 час реакционной смеси давали медленно нагреться до комнатной температуры. Примерно через 4 часа к реакционной смеси добавляли воду (4,8 мл), затем водный раствор NaOH (15% масс/об, 4,8 мл), затем воду (9,6 мл). Примерно через 15 часов добавляли безводный Na2SO4 и суспензию фильтровали и концентрировали в вакууме с получением неочищенного (3R,4S)-3-этил-4-(гидроксиметил)циклопентанола (3,9 г, 100%) в виде масла, которое использовали без дополнительной очистки. LC/MS (Таблица 1, Способ a) Rt = 2,40 мин; MS m/z: 145 (M+H)+.
Стадия B: (3R,4S)-3-этил-4-(гидроксиметил)циклопентанон

К раствору (3R,4S)-3-этил-4-(гидроксиметил)циклопентанола (4,00 г, 27,7 ммоль) в MeCN (70 мл) и воде (30,0 мл) добавляли бромат калия (1,487 мл, 29,1 ммоль) и CAN (0,760 г, 1,387 ммоль). Реакционную смесь нагревали до около 80°C. Примерно через 2 часа реакционную смесь охлаждали до комнатной температуры и добавляли Et2O (100 мл). Органический слой отделяли, промывали насыщенным солевым раствором (30 мл), концентрировали в вакууме и очищали хроматографией на силикагеле с использованием в качестве элюента EtOAc/гептан (20-60%) с получением (3R,4S)-3-этил-4-(гидроксиметил)циклопентанона (2,4 г, 61%) в виде масла. 1H ЯМР (400 МГц, CDCl3) δ 3,79 (дд, J=10,5, 5,3 Гц, 1H), 3,70 (дд, J=10,5, 6,5 Гц, 1H), 2,55-2,44 (м, 1H), 2,41-2,25 (м, 4H), 2,15-2,05 (м, 1H), 1,55-1,65 (м, 2H), 1,43-1,30 (м, 1H), 0,97 (т, J=7,3 Гц, 3H).
Стадия C: (3S,4R)-3-((трет-бутилдиметилсилилокси)метил)-4-этилциклопентанон
К раствору (3R,4S)-3-этил-4-(гидроксиметил)циклопентанона (2,60 г, 18,3 ммоль) в DMF (30 мл) добавляли имидазол (1,87 г, 27,4 ммоль) с последующим добавлением трет-бутилхлордиметилсилана (3,03 г, 20,1 ммоль). Примерно через 4 часа добавляли гептан (50 мл). Гептановый слой удаляли и промывали насыщенным солевым раствором. Слой насыщенного солевого раствора объединяли с DMF слоем и экстрагировали при помощи EtOAc/гептан (1:1, 30 мл). Гептановый и EtOAc слои объединяли, концентрировали в вакууме и очищали хроматографией на силикагеле с использованием в качестве элюента EtOAc/гептан (0-30%) с получением (3S,4R)-3-((трет-бутилдиметилсилилокси)метил)-4-этилциклопентанона (3,5 г, 75%) в виде бесцветного масла. 1H ЯМР (400 МГц, CDCl3) δ 3,77 (дд, J=10,5, 4,3 Гц, 1H), 3,64 (дд, J=10,5, 4,0 Гц, 1H), 2,40-2,20 (м, 5H), 2,18-2,02 (м, 1H), 1,65-1,55 (м, 1H), 1,52-1,37 (м, 1H), 0,97 (т, J=7,4 Гц, 3H), 0,87 (с, 9H), 0,43 (с, 3H), 0,03 (с, 3H).
Стадия D: этил 2-((3S,4R)-3-((трет-бутилдиметилсилилокси)метил)-4-этилциклопентилиден)ацетат
К суспензии NaH (60% дисперсия в минеральном масле, 0,608 г, 15,2 ммоль) в ТГФ (50 мл) добавляли этил 2-(диэтоксифосфорил)ацетат (3,25 мл, 16,2 ммоль). Примерно через 30 минут фосфонатный раствор добавляли в колбу, содержащую (3S,4R)-3-((трет-бутилдиметилсилилокси)метил)-4-этилциклопентанон (2,6 г, 10,14 ммоль). Примерно через 20 часов добавляли EtOAc (20 мл) и насыщенный водный раствор NH4Cl (20 мл). Органический слой удаляли, концентрировали в вакууме и очищали хроматографией на силикагеле с использованием в качестве элюента EtOAc/гептан (20-60%) с получением этил 2-((3S,4R)-3-((трет-бутилдиметилсилилокси)метил)-4-этилциклопентилиден)ацетата (3,3 г, 100%) в виде масла. LC/MS (Таблица 1, Способ a) Rt = 3,91, 3,96 мин; MS m/z: 327 (M+H)+.
Стадия E: этил 2-((3R,4S)-3-этил-4-(гидроксиметил)циклопентилиден)ацетат
К раствору этил 2-((3S,4R)-3-((трет-бутилдиметилсилилокси)метил)-4-этилциклопентилиден)ацетата (1,00 г, 3,06 ммоль) в ТГФ (20 мл) добавляли TBAF (1M в ТГФ, 4,59 мл, 4,59 ммоль). Через 6 часов добавляли EtOAc и воду. Органический слой отделяли, концентрировали в вакууме и очищали хроматографией на силикагеле с использованием в качестве элюента EtOAc/гептан с получением этил 2-((3R,4S)-3-этил-4-(гидроксиметил)циклопентилиден)ацетата (0,620 г, 95%) в виде масла. LC/MS (Таблица 1, Способ a) Rt = 1,96, 2,08 мин; MS m/z: 213 (M+H)+.
Стадия F: этил 2-((1R,3R,4S)-3-этил-4-(гидроксиметил)циклопентил)ацетат

К раствору этил 2-((3R,4S)-3-этил-4-(гидроксиметил)циклопентилиден)ацетата (0,160 г, 0,754 ммоль) в DCM (3 мл) добавляли катализатор Крэбтри (0,030 г, 0,038 ммоль). Реакционную смесь барботировали водородом примерно в течение 5 минут и атмосферу водорода поддерживали через баллон. Примерно через 24 часа реакционную смесь концентрировали в вакууме и очищали хроматографией на силикагеле с использованием в качестве элюента EtOAc/гептан (30-80%) с получением этил 2-((1R,3R,4S)-3-этил-4-(гидроксиметил)циклопентил)ацетата (0,140 г, 87%) в виде масла. 1H ЯМР (400 МГц, CDCl3) δ 4,12 (кв., J=7,1 Гц, 2H), 3,71-3,64 (дд, J=10,5, 8,0 Гц, 1H), 3,47 (дд, J=10,5, 8,0 Гц, 1H), 2,55-2,41 (м, 1H), 2,32 (д, J=6,7 Гц, 2H), 2,02-1,89 (м, 1H), 1,88-1,76 (м, 1H), 1,70-1,60 (м, 1H), 1,48-1,33 (м, 4H), 1,26 (т, J=7,1 Гц, 3H), 1,22-1,07 (м, 1H), 0,90 (т, J=7,4 Гц, 3H).
Стадия G: (1S,2R,4R)-4-(2-этокси-2-оксоэтил)-2-этилциклопентанкарбоновая кислота

К раствору этил 2-((1R,3R,4S)-3-этил-4-(гидроксиметил)циклопентил)ацетата (0,140 г, 0,653 ммоль) в MeCN (2 мл), воде (4 мл) и EtOAc (2 мл) добавляли периодат натрия (0,349 г, 1,633 ммоль) с последующим добавлением хлорида рутения(III) гидрата (0,0015 г, 0,0065 ммоль). Примерно через 2 часа реакционную смесь разбавляли EtOAc (20 мл) и водой (10 мл). Органический слой отделяли и экстрагировали водным раствором NaOH (1 н, 10 мл). pH водного слоя доводили до около 1 при помощи концентрированной HCl и экстрагировали при помощи EtOAc (20 мл). Органический слой сушили над безводным MgSO4, фильтровали и концентрировали в вакууме с получением (1S,2R,4R)-4-(2-этокси-2-оксоэтил)-2-этилциклопентанкарбоновой кислоты (0,150 г, 101%) в виде масла, которое использовали без дополнительной очистки. 1H ЯМР (600 МГц, CDCl3) δ 10,68 (шир.с, 1H), 4,13 (кв., J=7,1 Гц, 2H), 2,99-2,95 (м, 1H), 2,76-2,64 (м, 1H), 2,31 (д, J=7,6 Гц, 2H), 2,24 (ддд, J=13,5, 8,7, 4,8 Гц, 1H), 2,18-2,11 (м, 1H), 1,81 (дт, J=13,0, 8,4 Гц, 1H), 1,55-1,45 (м, 3H), 1,31-1,27 (м, 1H), 1,25 (т, J=7,0 Гц, 3H), (т, J=7,4 Гц, 3H).
Получение #32: 2,2,2-трифтор-1-(5-тозил-5H-пирроло[2,3-b]пиразин-2-ил)этанамин гидрохлорид
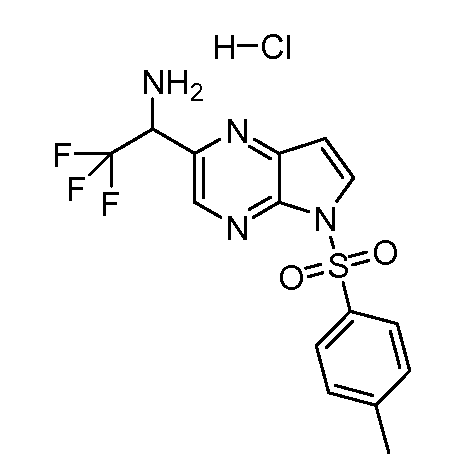
Стадия A: (S,E)-2-метил-N-((5-тозил-5H-пирроло[2,3-b]пиразин-2-ил)метилен)пропан-2-сульфинамид

К раствору 5-тозил-5H-пирроло[2,3-b]пиразин-2-карбальдегид (8,66 г, 28,7 ммоль, Получение #12 Стадия B) и (S)-2-метилпропан-2-сульфинамида (4,18 г, 34,5 ммоль) в DCM (20 мл) при температуре окружающей среды добавляли безводный порошкообразный сульфат меди(II) (13,8 г, 86 ммоль). Примерно через 20 часов реакционную смесь фильтровали и частично концентрировали в вакууме. К раствору добавляли гептан и полученные твердые вещества собирали фильтрованием и сушили в вакууме с получением (S,E)-2-метил-N-((5-тозил-5H-пирроло[2,3-b]пиразин-2-ил)метилен)пропан-2-сульфинамида (11,5 г, 99%) в виде твердого вещества. LC/MS (Таблица 1, Способ a) Rt = 2,50 мин; MS m/z: 405 (M+H)+.
Стадия B: (S)-2-метил-N-(2,2,2-трифтор-1-(5-тозил-5H-пирроло[2,3-b]пиразин-2-ил)этил)пропан-2-сульфинамид

В сухую колбу, содержущую 4 Å молекулярные сита (5 г) и тетраметил-аммонийфторид (0,553 г, 5,93 ммоль), добавляли ТГФ (20 мл). Реакционную смесь перемешивали примерно в течение 30 минут, после чего смесь охлаждали до около -78°C и добавляли раствор (S,E)-2-метил-N-((5-тозил-5H-пирроло[2,3-b]пиразин-2-ил)метилен)пропан-2-сульфинамида (1,20 г, 2,97 ммоль) в ТГФ (10 мл). Примерно через 15 минут к реакционной смеси добавляли триметил(трифторметил)силан (0,877 мл, 5,93 ммоль). Смеси давали нагреться до -35 - -45°C. Примерно через 3 часа реакционную смесь охлаждали до -78°C и добавляли водный раствор NH4Cl. Реакционной смеси давали нагреться до комнатной температуры. Добавляли EtOAc (30 мл) и насыщенный солевой раствор (30 мл). Органический слой отделяли, сушили над безводным Na2SO4, фильтровали и концентрировали в вакууме с получением неочищенного (S)-2-метил-N-(2,2,2-трифтор-1-(5-тозил-5H-пирроло[2,3-b]пиразин-2-ил)этил)пропан-2-сульфинамида (1,4 г, 99%) в виде пенообразного сульфонамида, который использовали без дополнительной очистки. LC/MS (Таблица 1, Способ a) Rt = 2,49 мин; MS m/z 475 (M+H)+.
Стадия C: 2,2,2-трифтор-1-(5-тозил-5H-пирроло[2,3-b]пиразин-2-ил)этанамин гидрохлорид

К раствору (S)-2-метил-N-(2,2,2-трифтор-1-(5-тозил-5H-пирроло[2,3-b]пиразин-2-ил)этил)пропан-2-сульфинамида (1,40 г, 2,95 ммоль) в MeOH (20 мл) добавляли HCl (4 н в 1,4-диоксане, 7,38 мл, 29,5 ммоль). Примерно через 2 часа реакционную смесь частично концентрировали в вакууме и разбавляли при помощи Et2O до начала образования твердых частиц. Примерно через 30 минут полученные твердые вещества собирали фильтрованием и сушили в вакууме с получением 2,2,2-трифтор-1-(5-тозил-5H-пирроло[2,3-b]пиразин-2-ил)этанамин гидрохлорида (0,840 г, 70%) в виде твердого вещества. LC/MS (Таблица 1, Способ a) Rt = 2,16 мин; MS m/z 371 (M+H)+.
Получение #33: (1S,2R,4S)-4-(дибензиламино)-2-метилциклопентанкарбоновая кислота

Стадия A: (1S,2R,4S)-4-(дибензиламино)-2-метилциклопентанкарбоксилат · (R)-1-фенилэтанамин
К раствору 4-(дибензиламино)-2-метилциклопентанкарбоновой кислоты (1240 г, 1499 ммоль, получали с использованием X с Примером #24 Стадия H и дибензиламина и TT) в ТГФ (8,0 L) добавляли (R)-(+)-1-фенилэтиламин (0,193 л, 1499 ммоль). Смесь нагревали до температуры кипения с обратным холодильником для растворения твердых веществ и затем охлаждали до температуры окружающей среды. Примерно через 15 часов реакционную смесь фильтровали, промывали при помощи ТГФ (800 мл) и сушили в вакуумной печи с получением (1S,2R,4S)-4-(дибензиламино)-2-метилциклопентанкарбоксилат · (R)-1-фенилэтанамина (565 г, 85%, 97,5% э.и.): LC/MS (Таблица 2, Способ 70) Rt = 8,49 минут. Маточный раствор концентрировали. Остаток растворяли в ТГФ (1 л), нагревали для растворения твердых веществ и охлаждали до температуры окружающей среды. Примерно через 15 часов реакционную смесь фильтровали, промывали при помощи ТГФ (800 мл) и сушили в вакуумной печи с получением дополнительного количества (1S,2R,4S)-4-(дибензиламино)-2-метилциклопентанкарбоксилат · (R)-1-фенилэтанамина (78,5 г, 12%, 95,2% э.и.): ВЭЖХ (Таблица 2, Способ 70) Rt = 8,57 мин.
Стадия B: (1S,2R,4S)-4-(дибензиламино)-2-метилциклопентанкарбоновая кислота

Фосфорную кислоту (11,40 мл, 196 ммоль) добавляли в колбу, содержащую воду (500 мл). Раствор перемешивали примерно в течение 5 минут. К раствору добавляли (1S,2R,4S)-4-(дибензиламино)-2-метилциклопентанкарбоксилат · (R)-1-фенилэтанамин (83 г, 187 ммоль) небольшими порциями. Добавляли MTBE (500 мл) и содержимое тщательно смешивали, растворяя твердые вещества. Фазы осаждали и разделяли. Водный слой обратно экстрагировали при помощи MTBE (150 мл). Объединенные органические фазы сушили над безводным сульфатом натрия, фильтровали и концентрировали в вакууме с получением (1S,2R,4S)-4-(дибензиламино)-2-метилциклопентанкарбоновой кислоты (60 г, 99%) в виде масла: ВЭЖХ (Таблица 1, Способ x) Rt = 4,57 минут.
Получение #34: 3,3-дифторциклобутан-1-сульфонилхлорид

Стадия A: 3-бром-1,1-дифторциклобутан

К интенсивно перемешиваемому раствору 3-бромциклобутанона (18,0 г, 121 ммоль, получали, как описано в J. Am. Chem. Soc., 1971, 93, 2481) в DCM (375 мл) при около 0°C добавляли по каплям DAST (36,9 мл, 279 ммоль) через капельную воронку примерно в течение 1 часа. Реакционную смесь продолжали перемешивать при около 0°C примерно в течение 2 часов и при температуре окружающей среды примерно в течение 14 часов. Реакционную смесь охлаждали до около -5°C на бане лед/ацетон и добавляли по каплям насыщенный водный раствор NaHCO3 (400 мл) через капельную воронку. Оставшиеся бислои интенсивно перемешивали примерно в течение 1 часа. Слои разделяли и водный слой экстрагировали при помощи DCM (4×200 мл). Объединенные органические слои сушили над безводным MgSO4, фильтровали и растворитель удаляли при пониженном давлении (180 мм Hg максимально, 30°C водяная баня) с получением 3-бром-1,1-дифторциклобутана (15,3 г, 59%) в виде светло-коричневого масла в качестве продукта: 1H ЯМР (400 МГц, CDCl3) δ 4,28-4,14 (м, 1H), 3,35-3,16 (м, 2H), 3,06-2,87 (м, 2H).
Стадия B: S-3,3-дифторциклобутилэтантиоат

К раствору 3-бром-1,1-дифторциклобутана (13,8 г, 64,7 ммоль) в DMSO (24,6 мл) добавляли тиоацетат калия (22,2 г, 194 ммоль). Раствор нагревали при около 45°C примерно в течение 16 часов. Добавляли воду (20 мл) и Et2O (50 мл). Слои разделяли и водный слой экстрагировали при помощи Et2O (7×50 мл). Объединенные органические слои сушили над безводным MgSO4, фильтровали и растворитель удаляли при пониженном давлении (60 мм Hg максимально, 30°C водяная баня) с получением неочищенного S-3,3-дифторциклобутилэтантиоата (13,09 г, 78%) в виде масла: 1H ЯМР (400 МГц, d 6-DMSO) δ 3,84-3,69 (м, 1H), 3,14 (ддд, J=13,0, 7,5, 3,9 Гц, 2H), 2,66-2,55 (м, 2H), 2,33 (с, 3H).
Стадия C: 3,3-дифторциклобутан-1-сульфонат калия
К раствору неочищенного S-3,3-дифторциклобутилэтантиоата (13,0 г, 39,1 ммоль) в уксусной кислоте (100 мл) добавляли H2O2 (24,0 мл, 235 ммоль, 30% в воде). Примерно через 4 часа отмечали экзотерму с генерацией тепла, достаточного для нагревания реакционной смеси до температуры кипения с обратным холодильником. Примерно через 20 часов реакционную смесь разбавляли толуолом (500 мл) и частично концентрировали в вакууме. Этот процесс повторяли (5×). Раствор разбавляли при помощи EtOH (около 500 мл) и к реакционной смеси добавляли KOH (4,4 г, 78 ммоль). Осадок собирали фильтрованием и отбрасывали. К фильтрату добавляли дополнительное количество KOH (4,4 г, 78 ммоль) и осадок собирали фильтрованием. Раствор частично концентрировали в вакууме. Раствор разбавляли при помощи EtOH (приблизительно 500 мл) и частично концентрировали снова (3×). Осадок собирали фильтрованием. Последние 2 собранные твердые вещества сушили в вакууме и объединяли с получением 3,3-дифторциклобутан-1-сульфоната калия (3,5 г, 42,6%). Дополнительное количество KOH (4,39 г, 78 ммоль) и раствор частично концентрировали в вакууме. Раствор разбавляли при помощи EtOH (приблизительно 500 мл) и концентрировали снова (3×). Полученные твердые вещества собирали фильтрованием с получением 3,3-дифторциклобутан-1-сульфоната калия (1,6 г, 19%):1H ЯМР (400 МГц, d 6-DMSO) δ 3,01 (ддд, J=13,5, 6,3, 2,5 Гц, 1H), 2,72-2,59 (м, 4H).
Стадия D: 3,3-дифторциклобутан-1-сульфонилхлорид

К суспензии 3,3-дифторциклобутан-1-сульфоната калия (0,250 г, 1,189 ммоль) в тионилхлориде(2,60 мл, 35,7 ммоль) добавляли DMF (3 капли). Реакционную смесь нагревали до около 60°C примерно в течение 21 часов. Растворитель удаляли при пониженном давлении и остаток использовали в следующей реакции без дополнительной обработки или очистки с получением неочищенного 3,3-дифторциклобутан-1-сульфонилхлорида (0,227 г, 100%) в качестве продукта. 1H ЯМР (400 МГц, CDCl3) δ 4,33-4,17 (м, 1H), 3,28 (дд, J=11,1, 7,6 Гц, 2H), 3,21-3,05 (м, 2H).
Получение #35: изопропил (1S,3R,4S)-3-этил-4-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентилкарбамат

К раствору (1S,3R,4S)-3-этил-4-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентанамина (0,080 г, 0,19 ммоль, Получение #BB.1*) в ТГФ (2 мл) добавляли TEA (0,079 мл, 0,565 ммоль) и раствор перемешивали при температуре окружающей среды примерно в течение 10 минут. К реакционной смеси добавляли изопропилхлорформиат (1 M в толуоле, 0,18 мл, 0,18 ммоль) и реакционную смесь перемешивали примерно в течение 1 часа. Растворитель удаляли при пониженном давлении и добавляли DCM (5 мл) и насыщенный водный раствор NaHCO3 (2 мл). Слои разделяли и органический слой промывали насыщенным солевым раствором (2 мл), сушили над MgSO4, фильтровали и концентрировали при пониженном давлении с получением неочищенного изопропил (1S,3R,4S)-3-этил-4-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентилкарбамата (0,080 г, 60%), который использовали без дополнительной очистки: LC/MS (Таблица 1, Способ b) Rt = 2,33 мин; MS m/z: 511 (M+H)+.
Получение #36: 3-(аминометил)циклобутанкарбоновая кислота
В колбу, содержащую 10% палладий на углероде (0,20 г, 0,19 ммоль), добавляли раствор бензил 3-(азидометил)циклобутанкарбоксилата (2,00 г, 8,15 ммоль, получали с использованием IIII из бензил 3-(гидроксиметил)циклобутанкарбоксилата (Parkway Scientific), JJJJ с азидом натрия) в MeOH (100 мл). Реакционную смесь барботировали водородом и атмосферу водорода поддерживали через баллон. Реакционную смесь перемешивали примерно в течение 4 часов при температуре окружающей среды и затем фильтровали через слой Целита®, промывали при помощи MeOH и концентрировали в вакууме с получением неочищенной 3-(аминометил)циклобутанкарбоновой кислоты (1,08 г, 100%), которую использовали без дополнительной очистки: LC/MS (Таблица 1, Способ r) Rt = 2,41 минут (ELSD); MS m/z: 130 (M+H)+.
Получение #37: Этил 3-тозил-3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-7-карбоксилат
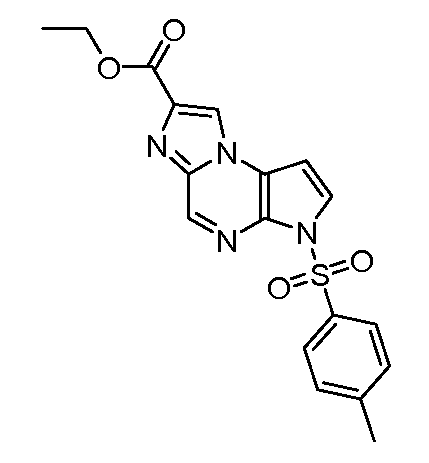
Этиловый эфир 3-бром-2-оксо-пропионовой кислоты (0,090 мл, 0,72 ммоль) добавляли к смеси 5-тозил-5H-пирроло[3,2-b]пиразин-2-амина (0,180 г, 0,624 ммоль, получали с использованием E из Примера #3 Стадия E и HCl) и 1,4-диоксана (3,5 мл) в атмосфере азота. Примерно через 3 дня летучие вещества удаляли при пониженном давлении. Остаток суспендировали в Et2O (5 мл) и затем фильтровали с получением желто-коричневого порошка. Твердое вещество суспендировали в MeCN (3,50 мл) в атмосфере азота. Добавляли PFPAA (0,40 мл, 2,1 ммоль). Примерно через 30 минут летучие вещества удаляли при пониженном давлении. Остаток растворяли в DCM (20 мл) и промывали насыщенным водным раствором NaHCO3/вода (2:1, 20 мл). Водный слой экстрагировали при помощи DCM (20 мл). Объединенные органические слои сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали хроматографией на силикагеле с градиентным элюированием 20-100% EtOAc/гептан с получением этил 3-тозил-3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-7-карбоксилата (0,181 г, 75%): LC/MS (Таблица 1, Способ n) Rt = 0,70 мин; MS m/z: 385 (M+H)+.
Получение #38: 6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-8-карбальдегид

К смеси 6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразина (0,200 г, 1,26 ммоль, получали с использованием D из Получения #BBBBB.1 и NaOH) и гексаметилентетрамина (0,264 г, 1,89 ммоль) добавляли воду (1,0 мл). Добавляли уксусную кислоту (0,5 мл). Реакционный сосуд герметично закрывали и смесь нагревали до около 100°C. Примерно через 8 часов раствору давали охладиться до температуры окружающей среды. После перемешивания примерно в течение 13 часов смесь охлаждали до около 0°C. Полученную смесь разбавляли водой (1 мл) и затем фильтровали промывая водой. Твердое вещество сушили с получением 6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-8-карбальдегида (0,041 г, 18%): LC/MS (Таблица 1, Способ n) Rt = 0,23 мин; MS m/z 188 (M+H)+.
Получение #39: 2-(4-метилпиперазин-1-ил)-4-(трибутилстаннил)пиримидин
1-Метилпиперазин (0,160 мл, 1,44 ммоль) добавляли к раствору 2-(метилсульфонил)-4-(трибутилстаннил)пиримидина (0,250 г, 0,481 ммоль, синтезирован, как описано в Majeed, A. J., et al. Tetrahedron 1989, 45, 993-1006) и 1,4-диоксана (1,0 мл) в атмосфере азота. Примерно через 2 часа раствор нагревали до около 50°C. Примерно через 30 минут раствор нагревали до около 80°C. Примерно через 30 минут подсоединяли обратный холодильник и раствор нагревали до около 100°C. Примерно через 16 часов коричневому раствору давали охладиться до температуры окружающей среды. Добавляли воду (5 мл). Смесь экстрагировали при помощи EtOAc (2×5 мл). Объединенные органические слои сушили над безводным Na2SO4, фильтровали и концентрировали. Остаток очищали хроматографией на силикагеле с градиентным элюированием 2-10% MeOH/DCM с получением 2-(4-метилпиперазин-1-ил)-4-(трибутилстаннил)пиримидина (0,127 г, 56%):1H ЯМР (400 МГц, CDCl3) δ 8,07 (д, J=4,6 Гц, 1H), 6,63 (д, J=4,6 Гц, 1H), 3,98-3,82 (м, 4H), 2,63-2,48 (м, 4H), 2,40 (с, 3H), 1,70-1,43 (м, 6H), 1,42-1,20 (м, 6H), 1,18-0,97 (м, 6H), 0,88 (т, J=7,3 Гц, 9H).
Получение #40: 2-(4-метилпиперазин-1-ил)-4-(трибутилстаннил)хиназолин

Стадия A: 4-хлор-2-(4-метилпиперазин-1-ил)хиназолин

2,4-Дихлорхиназолин (2,00 г, 10,1 ммоль, получали, как описано в Prasad, M., et al. Org. Process Res. Dev. 2004, 8, 330-340) суспендировали в 1,4-диоксане (20,0 мл). Добавляли 1,4-диметилпиперазин (1,44 мл, 10,6 ммоль). Смесь нагревали в микроволновом устройстве CEM при около 150°C примерно в течение 5 минут. Вещество выливали в насыщенный водный раствор NaHCO3/воду (1:1, 150 мл). Смесь экстрагировали при помощи EtOAc (5×100 мл). К объединенным органическим экстрактам добавляли 20 г силикагеля и летучие вещества удаляли при пониженном давлении. Полученное твердое вещество очищали хроматографией на силикагеле с градиентным элюированием 2-10% MeOH/DCM с получением 4-хлор-2-(4-метилпиперазин-1-ил)хиназолина (1,36 г, 52%): LC/MS (Таблица 1, Способ n) Rt = 0,51 мин; MS m/z 263 (M+H)+.
Стадия B: 4-иод-2-(4-метилпиперазин-1-ил)хиназолин
Иодистый водород (55% водный раствор, 4,00 мл, 29,3 ммоль) медленно добавляли к 4-хлор-2-(4-метилпиперазин-1-ил)хиназолину (1,36 г, 5,18 ммоль) в воздушной атмосфере при охлаждении на водяной бане с температурой окружающей среды. Примерно через 5 минут баню удаляли, реакционный сосуд оборачивали алюминиевой фольгой и смесь перемешивали при температуре окружающей среды примерно в течение 5 часов. Добавляли DCM (4,0 мл) и смесь перемешивали примерно в течение 39 часов. Добавляли иодистый водород (55% водный раствор, 8,0 мл, 110 ммоль) и смесь перемешивали примерно в течение 71 часа. Смесь медленно добавляли к насыщенному водному раствору NaHCO3 (200 мл) и EtOAc (200 мл). После завершения гашения слои разделяли. Органические слои промывали насыщенным водным раствором NaHCO3/водой (1:1, 200 мл). Органические слои сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали хроматографией на силикагеле с градиентным элюированием 2-5% MeOH/DCM с получением (1,18 г, 69%) в виде 3:1 смеси 4-иод-2-(4-метилпиперазин-1-ил)хиназолина и 4-хлор-2-(4-метилпиперазин-1-ил)хиназолина. 4-иод-2-(4-метилпиперазин-1-ил)хиназолин: LC/MS (Таблица 1, Способ n) Rt = 0,55 мин; MS m/z 355 (M+H)+.
Стадия C: 2-(4-метилпиперазин-1-ил)-4-(трибутилстаннил)хиназолин
Ацетат бис(трифенилфосфин)палладия(II) (0,063 г, 0,085 ммоль) добавляли к 3:1 смеси 4-иод-2-(4-метилпиперазин-1-ил)хиназолина:4-хлор-2-(4-метилпиперазин-1-ил)хиназолина (0,300 г) в атмосфере азота. Добавляли бис(трибутилолово) (0,855 мл, 1,69 ммоль). Добавляли TBAF (1,0 M раствор в ТГФ, 2,54 мл, 2,54 ммоль). Смесь продували азотом примерно в течение 20 минут и затем перемешивали в атмосфере азота при температуре окружающей среды примерно в течение 7 часов. Добавляли насыщенный водный раствор NaHCO3/воду (1:1, 20 мл) и EtOAc (50 мл). Смесь фильтровали через фильтровальный шприц, слои разделяли и органические слои промывали водой (2×10 мл). Органические слои сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали хроматографией на силикагеле с градиентным элюированием 5-10% MeOH/DCM с получением липкого коричневого твердого вещества. Вещество растворяли в EtOAc (10 мл) и промывали водой (2×5 мл). Органические слои сушили над безводным Na2SO4, фильтровали и концентрировали с получением 1:1 смеси 2-(4-метилпиперазин-1-ил)-4-(трибутилстаннил)хиназолина:4-хлор-2-(4-метилпиперазин-1-ил)хиназолина (0,058 г, 17%). 2-(4-метилпиперазин-1-ил)-4-(трибутилстаннил)хиназолин: 1H ЯМР (400 МГц, DMSO-d 6) δ 7,69-7,64 (м, 1H), 7,61-7,57 (м, 1H), 7,49-7,44 (м, 1H), 7,31-7,26 (м, 1H), 3,95-3,83 (м, 4H), 2,44-2,35 (м, 4H), 2,22 (с, 3H), 1,66-1,48 (м, 6H), 1,37-1,18 (м, 12H), 0,82 (т, J=7,3 Гц, 9H).
Получение #41: 4-(метилсульфонил)морфолин

К раствору морфолина (2,00 мл, 22,96 ммоль) в DCM (40 мл) добавляли TEA (3,20 мл, 22,96 ммоль) при около -20°C, затем добавляли по каплям метансульфонилхлорид (2,68 мл, 34,4 ммоль) при около -20°C. Реакционную смесь перемешивали при около -20°C примерно в течение 2 часов, затем нагревали до комнатной температуры. Смесь разделяли с использованием насыщенного водного раствора NH4Cl (100 мл) и DCM (3×50 мл). Объединенные органические слои концентрировали и очищали хроматографией на силикагеле с градиентным элюированием 0-100% EtOAc/гептан с получением 4-(метилсульфонил)морфолина (3,95 г, 100%) в виде белого твердого вещества: 1H ЯМР (DMSO-d 6) δ 3,70-3,60 (м, 4H), 3,12-3,04 (м, 4H), 2,89 (с, 3H).
Получение #42: метил 5-(хлорметил)-3-метилфуран-2-карбоксилат

К раствору метил 3-метилфуран-2-карбоксилата (8,00 г, 57,1 ммоль) в DCM (285 мл) добавляли хлорид цинка (2,14 г, 15,7 ммоль) и параформальдегид (2,2 мл, 82 ммоль). Раствор нагревали до около 35°C. HCl газ барботировали через реакционную смесь примерно в течение 20 минут. Смесь разделяли с использованием воды (50 мл) и DCM (3×30 мл). Объединенные органические слои концентрировали и очищали хроматографией на силикагеле с градиентным элюированием 0-50% EtOAc/гептан с получением метил 5-(хлорметил)-3-метилфуран-2-карбоксилата (8,24 г, 77%) в виде белого твердого вещества: LC/MS (Таблица 1, Способ n) Rt = 0,69 мин; MS m/z: 189 (M+H)+.
Получение #43: цис-метил 5-((трет-бутоксикарбониламино)метил)-3-метилтетрагидрофуран-2-карбоксилат

Раствор метил 5-(азидометил)-3-метилфуран-2-карбоксилата (3,10 г, 15,88 ммоль, получали с использованием Общей процедуры JJJJ из Получения #42 и азида натрия) в MeOH (50 мл) добавляли к суспензии 5% Rh/C (0,31 г, 3,01 ммоль) и ди-трет-бутил дикарбоната (4,16 г, 19,06 ммоль) в 50-мл сосуде высокого давления. Реакционную смесь перемешивали под давлением водорода 40 ф/дюйм2 (2,812 кг/см2) при около 50°C примерно в течение 3,5 дней. Смесь фильтровали через найлоновую мембрану. Органический растворитель концентрировали при пониженном давлении с получением цис-метил 5-((трет-бутоксикарбониламино)метил)-3-метилтетрагидрофуран-2-карбоксилата (4,19 г, 81%) в виде коричневого масла: 1H ЯМР (CDCl3) δ 5,70 (с, 1H), 4,43-4,46 (д, 1H), 4,28-4,12 (м, 1H), 3,75 (с, 3H), 3,50-3,30 (м, 2H), 2,75-2,55 (м, 1H), 1,95-2,05 (м, 1H), 1,65-1,48 (м, 1H), 1,45 (с, 9H), 1,03-0,97 (д, 3H).
Получение #44: (1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентанамин и (1S,3R,4S)-3-этил-4-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентанамин

К смеси N-((1S,3R,4S)-3-этил-4-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)ацетамида (5,0 г, 10,7 ммоль, Пример #8 Стадия L) и ТГФ (110 мл) добавляли водный раствор HCl (6 н, 63 мл, 375 ммоль). Реакционную смесь нагревали при около 95°C примерно в течение 20 часов и затем охлаждали до температуры окружающей среды и концентрировали при пониженном давлении. К полученному коричневому остатку добавляли DCM (100 мл) и раствор промывали насыщенным раствором NaHCO3 (3×50 мл). Водную часть экстрагировали при помощи DCM (3×50 мл). Объединенные органические слои промывали насыщенным солевым раствором (100 мл), сушили над безводным MgSO4, фильтровали и концентрировали при пониженном давлении. Вещество очищали хроматографией на силикагеле с использованием в качестве элюента 0-100% DCM/MeOH/NH4OH (950:45:5) с получением смеси (1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентанамина и (1S,3R,4S)-3-этил-4-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентанамина (3,2 г, 70%) в 1:10 соотношении согласно данным Н-ЯМР в виде не совсем белого твердого вещества: LC/MS (Таблица 1, Способ a) Rt = 1,75 мин; MS m/z: 425 (M+H)+.
Получение #45: Метил 5-тозил-5H-пирроло[2,3-b]пиразин-2-карбоксилат
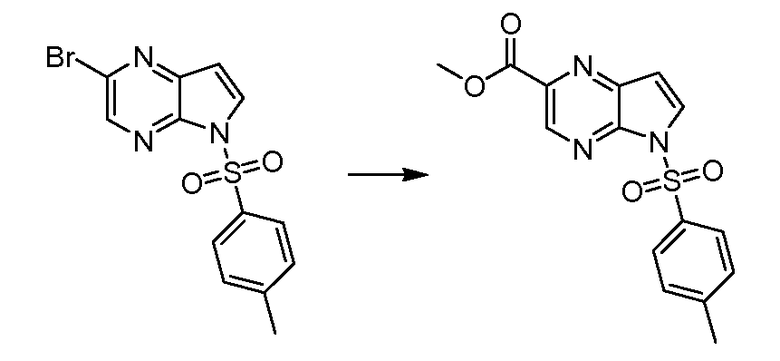
CO барботировали в оранжевый раствор 2-бром-5-тозил-5H-пирроло[2,3-b]пиразина (50,0 г, 142 ммоль, Пример #7, стадия B) в DMF (2,50 L) в 5-л круглодонной колбе примерно в течение 2 минут. Добавляли бис(трифенилфосфин)-палладий(II) дихлорид (9,96 г, 14,2 ммоль), TEA (59 мл, 423 ммоль) и MeOH (173,0 мл, 4259 ммоль) и колбу снабжали баллоном CO. Смесь нагревали при около 95°C в атмосфере CO (1 атмосфера). После перемешивания в течение ночи реакционную смесь охлаждали до температуры окружающей среды в течение ночи и выливали в ледяную воду (3,2 л). Смесь перемешивали примерно в течение 10 минут и осадок собирали фильтрованием, осуществляя при этом промывку водой, и сушили в течение 1 часа. Неочищенное вещество растворяли в DCM, отделяли от остаточной воды, сушили над безводным MgSO4, фильтровали, добавляли силикагель и концентрировали при пониженном давлении для подготовки к хроматографии. Неочищенное вещество очищали колоночной хроматографией на силикагеле с использованием в качестве элюента 0-5% MeOH в DCM с получением метил 5-тозил-5H-пирроло[2,3-b]пиразин-2-карбоксилата с 5 моль% DCM в качестве эксципиента (40,7 г, 86%, 93% чистота): LC/MS (Таблица 1, Способ a) Rt = 2,35 мин; MS m/z 332 (M+H)+.
Получение #46: 5-Тозил-5H-пирроло[2,3-b]пиразин-2-карбоновая кислота
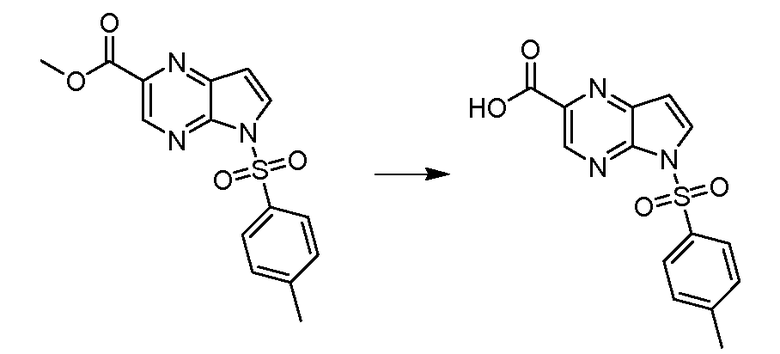
HCl (6 н водный раствор, 714 мл) добавляли к желтому раствору метил 5-тозил-5H-пирроло[2,3-b]пиразин-2-карбоксилата (17,8 г, 53,6 ммоль, Получение #45) в 1,4-диоксане (715 мл) в 2-л круглодонной колбе и смесь нагревали при около 60°C примерно в течение 16 часов. Реакционную смесь охлаждали до температуры окружающей среды. Органический растворитель удаляли при пониженном давлении и осадок собирали, промывали водой и сушили с получением 5-тозил-5H-пирроло[2,3-b]пиразин-2-карбоновой кислоты (14,4 г, 85%) в виде желтого твердого вещества: LC/MS (Таблица 1, Способ a) Rt = 1,63 мин; MS m/z 316 (M-H)-.
Получение #47: трет-Бутил 5-тозил-5H-пирроло[2,3-b]пиразин-2-илкарбамат
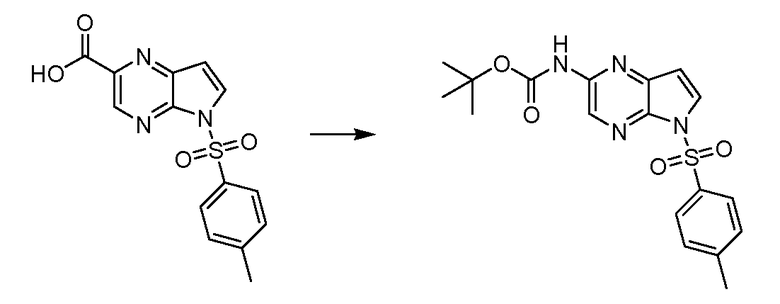
В 500-мл круглодонную колбу добавляли 5-тозил-5H-пирроло[2,3-b]пиразин-2-карбоновую кислоту (14,4 г, 45,3 ммоль, Получение #46), дифенилфосфорилазид (9,78 мл, 45,3 ммоль) и TEA (13,9 мл, 100 ммоль) в t-BuOH (200 мл) с получением оранжевой суспензии. Смесь нагревали при около 70°C примерно в течение 16 часов, охлаждали до температуры окружающей среды и нерастворимые вещества отфильтровывали. Растворитель удаляли при пониженном давлении и неочищенное вещество очищали колоночной хроматографией на силикагеле с элюированием 25-60% EtOAc в гептане в течение 30 минут с получением трет-бутил 5-тозил-5H-пирроло[2,3-b]пиразин-2-илкарбамата (9,75 г, 54%) в виде не совсем белого твердого вещества: LC/MS (Таблица 1, Способ a) Rt = 2,79 мин; MS m/z 389 (M+H)+.
Получение #48: 2-бром-1-(4-(дибензиламино)-2-метилциклопентил)этанон
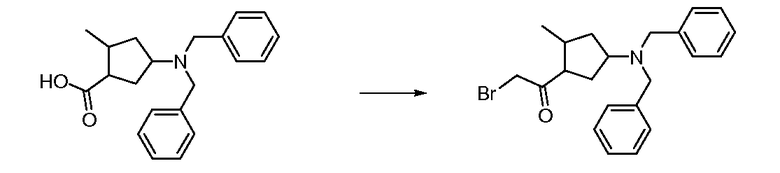
Оксалилхлорид (4,37 мл, 49,9 ммоль) медленно добавляли к раствору 4-(дибензиламино)-2-метилциклопентанкарбоновой кислоты (7,34 г, 22,7 ммоль, Пример #7, стадия I) в DCM (100 мл), (примечание: слабое выделение газа) с последующим добавлением по каплям DMF (0,26 мл, 3,41 ммоль). Смесь перемешивали при температуре окружающей среды примерно в течение 14 часов. Растворитель удаляли при пониженном давлении с получением бежевого аморфного твердого вещества, которое растворяли в ТГФ и MeCN (1:1, 100 мл) и добавляли к раствору триметилсилилдиазометана (2 M в Et2O, 39,7 мл, 79 ммоль) в ТГФ и MeCN (1:1, 100 мл) при около 0°C. Полученную смесь перемешивали при около 0°C примерно в течение 3 часов и затем гасили путем добавления по каплям HBr (48% водный раствор, 25 мл, 221 ммоль). Полученную смесь нейтрализовали путем добавления по каплям насыщенного водного раствора NaHCO3 (300 мл) и слои разделяли. Органический слой сушили над безводным MgSO4 и концентрировали при пониженном давлении. Остаток очищали флэш-хроматографией на силикагеле с использованием в качестве элюента 5% → 45% EtOAc в гептане с получением 2-бром-1-(4-(дибензиламино)-2-метилциклопентил)этанона (6,3 г, 69%) в виде желтого масла: LC/MS (Таблица 1, Способ a) Rt = 2,90 мин; MS m/z 400, 402 (M+H)+.
Получение #49: трет-Бутил 2-(4-(дибензиламино)-2-метилциклопентил)-2-оксоэтил(5-тозил-5H-пирроло[2,3-b]пиразин-2-ил)карбамат

Раствор трет-бутил 5-тозил-5H-пирроло[2,3-b]пиразин-2-илкарбамата (0,59 г, 1,519 ммоль, Пример #7, Стадия C) в DMF (5 мл) добавляли по каплям к суспензии NaH (60% дисперсия в минеральном масле, 0,058 г, 1,45 ммоль) в DMF (5 мл) при около 0°C. Полученную смесь перемешивали при около 0°C примерно в течение 30 минут и затем добавляли по каплям к раствору 2-бром-1-(4-(дибензиламино)-2-метилциклопентил)этанона (0,73 г, 1,8 ммоль) в DMF (10 мл) при около 0°C. Полученную смесь перемешивали при около 0°C в течение примерно 1 часа и растворитель удаляли при пониженном давлении. Остаток распределяли между насыщенным водным раствором NaHCO3 и EtOAc (100 мл каждого). Органическую фазу отделяли, сушили над безводным MgSO4 и концентрировали при пониженном давлении с получением трет-бутил 2-(4-(дибензиламино)-2-метилциклопентил)-2-оксоэтил(5-тозил-5H-пирроло[2,3-b]пиразин-2-ил)карбамата (1,04 г, 97%) в виде желтого аморфного твердого вещества: LC/MS (Таблица 1, Способ a) Rt = 3,30 мин; MS m/z 708 (M+H)+.
Получение #50: 1-(4-(дибензиламино)-2-метилциклопентил)-2-(5-тозил-5H-пирроло[2,3-b]пиразин-2-иламино)этанон

трет-Бутил 2-(4-(дибензиламино)-2-метилциклопентил)-2-оксоэтил(5-тозил-5H-пирроло[2,3-b]пиразин-2-ил)карбамат (6,19 г, 8,75 ммоль, Получение #49) растворяли в HCl (4 н в 1,4-диоксане, 25 мл). Реакционную смесь перемешивали при температуре окружающей среды примерно в течение 2 часов. Растворитель удаляли при пониженном давлении и остаток распределяли между насыщенным водным раствором NaHCO3 и EtOAc (100 мл каждого). Органическую фазу промывали насыщенным солевым раствором (80 мл), сушили над безводным MgSO4 и концентрировали при пониженном давлении с получением 1-(4-(дибензиламино)-2-метилциклопентил)-2-(5-тозил-5H-пирроло[2,3-b]пиразин-2-иламино)этанона (5,2 г, 98%) в виде коричневого аморфного твердого вещества: LC/MS (Таблица 1, Способ a) Rt = 3,00 мин; MS m/z 608 (M+H)+.
Получение #51: N,N-дибензил-3-метил-4-(3-тозил-3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)циклопентанамин

Смесь 1-(4-(дибензиламино)-2-метилциклопентил)-2-(5-тозил-5H-пирроло[2,3-b]пиразин-2-иламино)этанона (5,32 г, 8,75 ммоль, Получение #50) и реагента Лоуссона (1,88 г, 4,64 ммоль) нагревали при около 60°C примерно в течение 2 часов. Добавляли реагент Лоуссона (1,88 г, 4,64 ммоль). Реакционную смесь перемешивали при около 60°C примерно в течение 1 часа. Растворитель удаляли при пониженном давлении и остаток очищали флэш-хроматографией на силикагеле с градиентным элюированием 0-8% MeOH в DCM с получением N,N-дибензил-3-метил-4-(3-тозил-3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)циклопентанамина (4,47 г, 87%) в виде коричневого аморфного твердого вещества: LC/MS (Таблица 1, Способ a) Rt = 2,99 мин; MS m/z 590 (M+H)+.
Получение #52: N,N-дибензил-3-(3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)-4-метилциклопентанамин

N,N-Дибензил-3-метил-4-(3-тозил-3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)циклопентан-амин (4,47 г, 7,58 ммоль, Получение #51) растворяли в 1,4-диоксане (40 мл). Добавляли NaOH (2 н водный раствор, 4 мл) и реакционную смесь нагревали при около 90°C примерно в течение 80 минут. Органический растворитель удаляли при пониженном давлении и остаток обрабатывали насыщенным водным раствором NH4Cl (70 мл) и экстрагировали при помощи DCM (2×60 мл). Объединенные органические экстракты промывали насыщенным солевым раствором (70 мл), сушили над безводным MgSO4 и концентрировали при пониженном давлении. Очистка флэш-хроматографией на силикагеле с градиентным элюированием 0-8% MeOH в DCM давала N,N-дибензил-3-(3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)-4-метилциклопентан-амин (1,84 г, 56%) в виде желтого масла: LC/MS (Таблица 1, Способ a) Rt = 2,31 мин; MS m/z 436 (M+H)+.
Получение #53: 3-(3H-Имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)-4-метилциклопентанамин

К смеси N,N-дибензил-3-(3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)-4-метилцикло-пентанамина (1,84 г, 4,22 ммоль, Получение #52) в EtOH (50 мл) добавляли 20% масс. Pd(OH)2 на C (0,43 г, 0,61 ммоль) и полученную смесь встряхивали под давлением водорода около 50 ф/дюйм2 (3,515 кг/см2) во встряхивающем аппарате Парра при около 50°C примерно в течение 2 часов. Катализатор отфильтровывали с использованием слоя Целита®, добавляли 20% масс. Pd(OH)2 на C (0,43 г, 0,61 ммоль) и смесь встряхивали под давлением водорода около 50 ф/дюйм2 (3,515 кг/см2) во встряхивающем аппарате Парра при около 50°C примерно в течение 16 часов. Катализатор отфильтровывали с использованием слоя Целита®, добавляли 20% масс. Pd(OH)2 на C (0,43 г, 0,61 ммоль) и смесь встряхивали под давлением водорода около 50 ф/дюйм2 (3,515 кг/см2) во встряхивающем аппарате Парра при около 50°C примерно в течение 4 часов. Катализатор отфильтровывали с использованием слоя Целита® и фильтрат концентрировали при пониженном давлении с получением 3-(3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)-4-метилциклопентанамина (0,88 г, 82%) в виде не совсем белого аморфного твердого вещества: LC/MS (Таблица 1, Способ a) Rt = 0,75 минут и 0,87 мин; MS m/z 256 (M+H)+.
Общая процедура A: Образование гидразида из карбоновой кислоты
К смеси 2-гидразинилпирроло[2,3-b]пиразина (предпочтительно 1 экв.) и карбоновой кислоты (1-2 экв., предпочтительно 1,1-1,3 экв.) в органическом растворителе (таком как DCM, DMF или ТГФ, предпочтительно DMF) добавляют связующий агент, такой как EDC•HCl или HATU (1,0-2,0 экв., предпочтительно 1,2-1,6 экв.) с использованием или без органического основания (такого как TEA или DIEA, 2-5 экв., предпочтительно 3-4 экв.). Примерно через 1-72 часа (предпочтительно 2-16 часов) при около 20-60°C (предпочтительно около температуры окружающей среды) реакционную смесь обрабатывают с использованием одного из следующих способов. При использовании DMF в качестве растворителя, реакционную смесь сначала концентрировают при пониженном давлении. Способ 1: Добавляют воду и слои разделяли. Необязательно, смесь можно фильтровать через Целит® до разделения слоев. Водный слой затем экстрагировали органическим растворителем, таким как EtOAc или DCM. Объединенные органические слои, необязательно, промывают насыщенным солевым раствором, сушат над безводным Na2SO4 или MgSO4, фильтруют или декантируют и концентрируют при пониженном давлении. Способ 2: Реакционную смесь разбавляют органическим растворителем, таким как EtOAc или DCM и промывают либо водой, либо насыщенным солевым раствором, либо и тем и другим. Водный слой, необязательно, далее экстрагируют органическим растворителем, таким как EtOAc или DCM. Затем, необязательно, органический слой или объединенные органические слои промывают насыщенным солевым раствором, сушат над безводным Na2SO4 или MgSO4, фильтруют или декантируют и концентрируют при пониженном давлении. Способ 3: Реакционную смесь разбавляют органическим растворителем, таким как EtOAc или DCM, и добавляют воду. Слои разделяют и органический слой концентрируют при пониженном давлении и непосредственно очищают хроматографией.
Иллюстрация Общей Процедуры A
Получение #A.1*: (S)-трет-бутил 3-(2-оксо-2-(2-(5-тозил-5H-пирроло[2,3-b]пиразин-2-ил)гидразинил)этил)пирролидин-1-карбоксилат

К раствору трет-бутилового эфира (S)-3-карбоксиметил-пирролидин-1-карбоновой кислоты (0,756 г, 3,30 ммоль, AstaTech) и 2-гидразинил-5-тозил-5H-пирроло[2,3-b]пиразина (1,0 г, 3,3 ммоль, Пример #1. Стадия D) в DMF (33 мл) добавляли TEA (1,38 мл, 9,89 ммоль) с последующим добавлением HATU (1,25 г, 3,30 ммоль). Полученную смесь перемешивали при температуре окружающей среды примерно в течение 15 часов, затем концентрировали при пониженном давлении. Остаток брали для поглощения в EtOAc (100 мл) и промывали водой (100 мл). Органическую часть отделяли, промывали насыщенным солевым раствором (100 мл), сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении с получением (S)-трет-бутил 3-(2-оксо-2-(2-(5-тозил-5H-пирроло[2,3-b]пиразин-2-ил)гидразинил)этил)пирролидин-1-карбоксилата в виде липкого коричневого твердого вещества (1,90 г, 100%). Это вещество использовали без дополнительной очистки: LC/MS (Таблица 1, Способ c) Rt = 1,38 мин; MS m/z: 515 (M+H)+.
Общая процедура B: Циклизация гидразида
К раствору 2-гидразидил-5H-пирроло[2,3-b]пиразина (предпочтительно 1 экв.) в органическом растворителе (например, 1,4-диоксан) добавляют основание (такое как TEA или DIEA, 1-5 экв., предпочтительно 2-4 экв.) и SOCl2 (1-5 экв., предпочтительно 1-2 экв.). Смесь нагревают при около 60-100°C (предпочтительно около 80°C) примерно в течение 1-16 часов (предпочтительно около 1-2 часов). Реакционную смесь охлаждают до температуры окружающей среды и обрабатывают с использованием одного из следующих способов. Способ 1: добавляют органический растворитель (такой как EtOAc или DCM) и воду. Слои разделяют и водный слой, необязательно, экстрагируют дополнительным количеством органического растворителя. Объединенные органические слои, необязательно, промывают водным раствором основания (такого как NaHCO3) и/или насыщенным солевым раствором, сушат над безводным Na2SO4 или MgSO4, затем декантируют или фильтруют и затем концентрируют при пониженном давлении. Способ 2: добавляют органический растворитель (такой как EtOAc или DCM) и органический слой, необязательно, промывают насыщенным солевым раствором или водой, сушат над безводным MgSO4 или Na2SO4, фильтруют или декантируют и концентрируют при пониженном давлении. Способ 3: Реакционную смесь распределяют между органическим растворителем (таким как EtOAc или DCM) и насыщенным водным раствором NaHCO3 или насыщенным солевым раствором, сушат над безводным Na2SO4 или MgSO4, затем декантируют или фильтруют и затем концентрируют при пониженном давлении.
Иллюстрация Общей Процедуры B
Получение #B.1*: трет-бутил (1S,3R)-3-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентилкарбамат

К раствору трет-бутил (1S,3R)-3-(2-(5-тозил-5H-пирроло[2,3-b]пиразин-2-ил)гидразинкарбонил)циклопентилкарбамата (9,30 г, 18,1 ммоль, получали с использованием из Примера #1 Стадия D и (1R,3S)-3-трет-бутоксикарбониламино)циклопентанкарбоновой кислоты [Peptech]) в 1,4-диоксане (100 мл) добавляли TEA (10,0 мл, 72,3 ммоль) и SOCl2 (2,11 мл, 28,9 ммоль). Смесь нагревали при около 80°C примерно в течение 1,5 часов. Реакционную смесь охлаждали до температуры окружающей среды, добавляли EtOAc (200 мл) и воду (200 мл) и слои разделяли. Водную часть экстрагировали при помощи EtOAc (2×100 мл) и объединенные органические экстракты промывали насыщенным водным раствором NaHCO3 (100 мл) и насыщенным солевым раствором (100 мл), сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении. Неочищенное вещество очищали хроматографией на силикагеле с градиентным элюированием 25-100% EtOAc в DCM с получением трет-бутил-(1S,3R)-3-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентилкарбамата (7,65 г, 85%): LC/MS (Таблица 1, Способ a) Rt = 2,37 мин; MS m/z: 497 (M+H)+.
Общая процедура C: Циклизация гидразида с потерей Boc-защитной группы
К раствору подходяще замещенного 2-гидразидил-5H-пирроло[2,3-b]пиразина, содержащего защитную группу Boc (предпочтительно 1 экв.), и TEA или DIEA (0-6 экв., предпочтительно 4 экв.) в органическом растворителе (таком как 1,4-диоксан или DCM, предпочтительно 1,4-диоксан) добавляют SOCl2 (2,0-6,0 экв., предпочтительно 2,5 экв.). Реакционную смесь нагревают при около 60-120°C (предпочтительно около 80-90°C) примерно в течение 1-8 часов (предпочтительно около 2-4 часов) и затем обрабатывают с использованием одного из следующих способов. Способ 1: Реакционную смесь фильтруют и промывают подходящим органическим растворителем (таким как EtOAc или DCM) с получением целевого соединения без какой-либо дополнительной очистки. Способ 2: Неочищенное вещество разбавляют подходящим органическим растворителем (таким как EtOAc или DCM) и добавляют насыщенный водный раствор NaHCO3, слои разделяют и органическую часть сушат над безводным Na2SO4 или MgSO4, фильтруют и концентрируют при пониженном давлении. Способ 3: Реакционную смесь промывают водно-щелочным раствором (предпочтительно насыщенным водным раствором NaHCO3) и фильтруют с получением целевого соединения с удаленной Boc-защитой, без какой-либо дополнительной очистки. В случае частичного удаления Boc-защиты, фильтрат экстрагируют подходящим органическим растворителем (таким как EtOAc или DCM), слои разделяют и органическую часть сушат над безводным Na2SO4 или MgSO4, фильтруют и концентрируют при пониженном давлении с получением оставшегося Boc-защищенного соединения. Неочищенное Boc-защищенное вещество или частично Boc-защищенное вещество, полученное выше, растворяют в 1,4-диоксане или DCM (предпочтительно в 1,4-диоксане) и добавляют к раствору HCl в органическом растворителе (1-6 н, предпочтительно 4 н HCl в 1,4-диоксане) и нагревают до около 30-60°C (предпочтительно около 50°C) примерно в течение 1-5 часов (предпочтительно около 3 часов). Если образуется осадок, его собирают и затем растворяют в подходящем органическом растворителе (таком как EtOAc или DCM) и промывают водно-щелочным раствором (предпочтительно насыщенным водным раствором NaHCO3). Если никакого осадка не образуется, реакционную смесь промывают водно-щелочным раствором (предпочтительно насыщенным водным раствором NaHCO3). В любом случае, слои разделяют и органическую часть сушат над безводным Na2SO4 или MgSO4, фильтруют и концентрируют при пониженном давлении.
Иллюстрация Общей Процедуры C
Получение #C.1 4-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)бицикло[2,2,2]октан-1-амин

К раствору трет-бутил 4-(2-(5-тозил-5H-пирроло[2,3-b]пиразин-2-ил)гидразинкарбонил)-бицикло[2,2,2]октан-1-илкарбамата (6,1 г, 11,0 ммоль, Пример #9, Стадия E) и TEA (6,1 мл, 44,0 ммоль) в 1,4-диоксане (110 мл) добавляли SOCl2 (2,0 мл, 27,5 ммоль). Реакционную смесь нагревали при около 80°C примерно в течение 2 часов, затем охлаждали до температуры окружающей среды. Реакционную смесь промывали насыщенным водным раствором NaHCO3 (3×50 мл). Слои разделяли и водную часть фильтровали с получением 4-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)бицикло[2,2,2]-октан-1-амина в виде коричневого твердого вещества (1,17 г, 24%): LC/MS (Таблица 1, Способ a) Rt = 1,28 мин; MS m/z: 437 (M+H)+. Остальной фильтрат экстрагировали при помощи EtOAc (10 мл). Объединенный органический слой сушили над безводным MgSO4, фильтровали и концентрировали при пониженном давлении с получением неочищенного трет-бутил 4-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)бицикло[2,2,2]октан-1-илкарбамата (3,5 г). Неочищенное Boc-защищенное вещество растворяли в 1,4-диоксане (38 мл) и добавляли HCl (4 н в 1,4-диоксане, 8 мл). Реакционную смесь нагревали до около 50°C примерно в течение 3 часов. Образовавшийся осадок собирали фильтрованием. Твердое вещество растворяли в DCM (50 мл) и промывали насыщенным водным раствором NaHCO3 (3×20 мл). Слои разделяли и органическую часть сушили над безводным MgSO4, фильтровали и концентрировали при пониженном давлении с получением дополнительного количества 4-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)бицикло[2,2,2]октан-1-амина в виде коричневого твердого вещества (2,3 г, 50% за 2 стадии): LC/MS (Таблица 1, Способ a) Rt = 1,28 мин; MS m/z: 437 (M+H)+.
Общая процедура D: Гидролиз сульфонамида
В колбу, содержащую сульфонамид, например, сульфонил-защищенный пиррол (предпочтительно 1 экв.), в органическом растворителе (таком как 1,4-диоксан, MeOH или ТГФ/MeOH, предпочтительно 1,4-диоксан) добавляют водный раствор основания (такой как водный раствор Na2CO3 или водный раствор NaOH, 1-30 экв., предпочтительно 2-3 экв. для водного раствора NaOH, предпочтительно 15-20 экв. для водного раствора Na2CO3). Смесь перемешивают при около 25-100°C (предпочтительно около 60°C) примерно в течение 1-72 часов (предпочтительно около 1-16 часов). В случаях, когда реакция не достигает завершения, согласно данным ТСХ, LC/MS или ВЭЖХ, добавляют дополнительное количество водного раствора основания (такого как водный раствор Na2CO3, 10-20 экв., предпочтительно 10 экв., или водный раствор NaOH, 1-5 экв., предпочтительно 1-2 экв.) и/или добавляют co-растворитель (такой как EtOH). Реакцию продолжают при около 25-100°C (предпочтительно около 60°C) примерно в течение 0,25-3 часов (предпочтительно около 1-2 часов). В любом случае присутствия дополнительной основно-лабильной группы (например, сложный трифторметиловый эфир или цианогруппа), эта группа также может быть гидролизована. Реакционную смесь обрабатывают с использованием одного из следующих способов. Способ 1. Органический растворитель, необязательно, удаляют при пониженном давлении и водный раствор нейтрализовают путем добавления подходящего водного раствора кислоты (такого как водный раствор HCl). Добавляют подходящий органический растворитель (такой как EtOAc или DCM) и воду, слои разделяют и органический раствор сушат над безводным Na2SO4 или MgSO4, фильтруют и концентрируют досуха при пониженном давлении с получением целевого соединения. Способ 2. Органический растворитель, необязательно, удаляют при пониженном давлении, добавляют подходящий органический растворитель (такой как EtOAc или DCM) и воду, слои разделяют и органический раствор сушат над безводным Na2SO4 или MgSO4, фильтруют и концентрируют досуха при пониженном давлении с получением целевого соединения. Способ 3. Реакционную смесь концентрируют при пониженном давлении и непосредственно очищают одним из описанных ниже способов.
Иллюстрация Общей Процедуры D
Получение #D.1*: (3R,4R)-трет-бутил-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-карбоксилат

К раствору (3R,4R)-трет-бутил 4-метил-3-(6-тозил-6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)пиперидин-1-карбоксилата (40 г, 78 ммоль, Пример #5 Стадия Н) в 1,4-диоксане (160 мл) добавляли NaOH (1 н водный раствор, 157 мл). Реакционную смесь нагревали при около 60°C примерно в течение 1 часа. Реакционной смеси давали охладиться до температуры окружающей среды. Реакционную смесь нейтрализовали водным раствором HCl (4 н, 50 мл). Слои разделяли и экстрагировали при помощи DCM (2×300 мл). Объединенные органические экстракты промывали насыщенным солевым раствором (400 мл), сушили над безводным Na2SO4, фильтровали и затем концентрировали в вакууме. Продукт очищали хроматографией на силикагеле (330 г) с использованием 1-5% MeOH в DCM с получением (3R,4R)-трет-бутил 3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-карбоксилата (30 г, 99%): LC/MS (Таблица 1, Способ b) Rt = 2,00 мин; MS m/z: 356 (M+H)+.
Примеры, полученные с использованием Общей Процедуры D с NaOH
(Таблица 1, Способ)





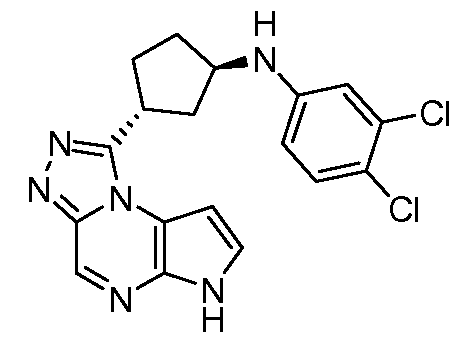

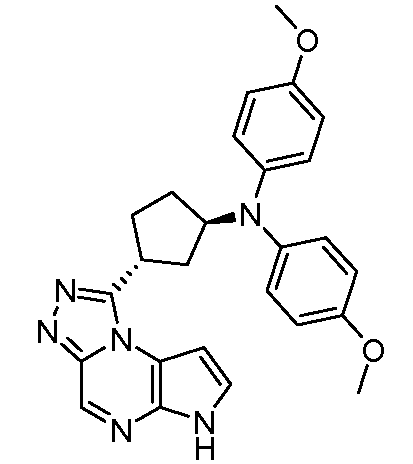






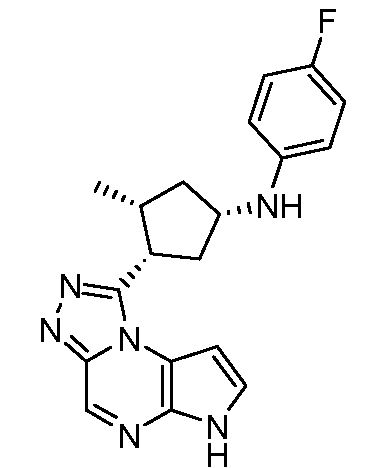




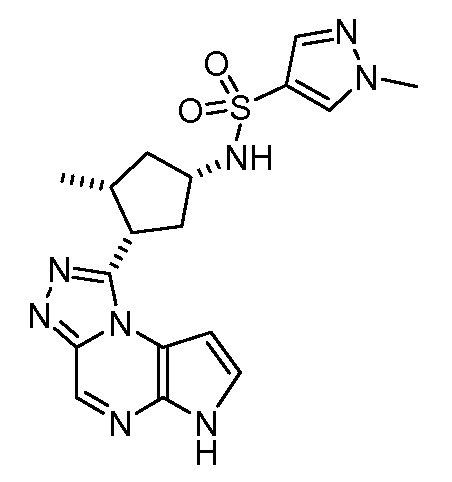



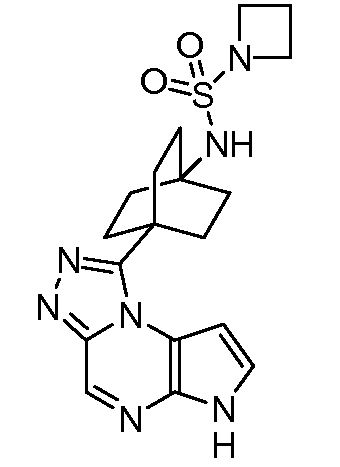

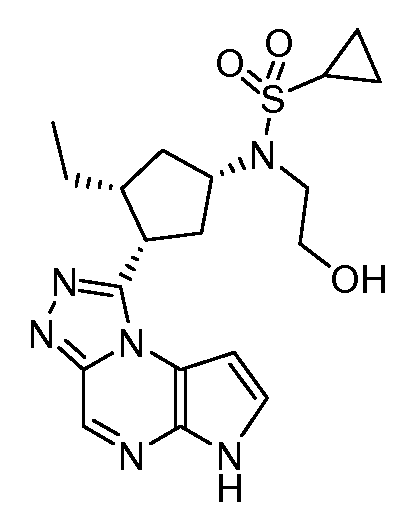

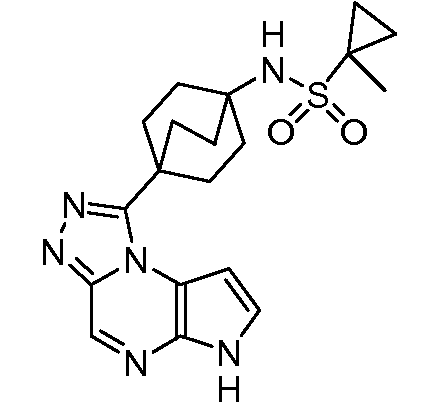










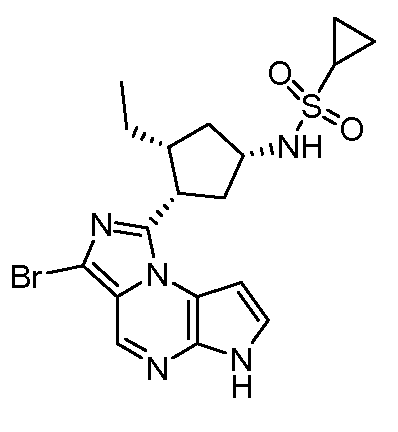


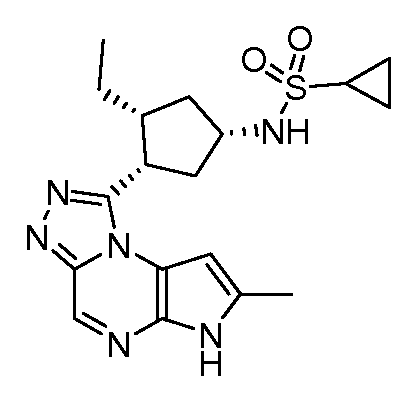
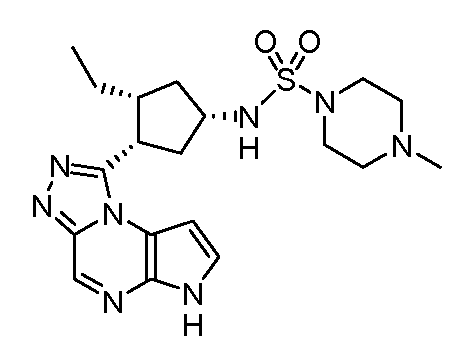








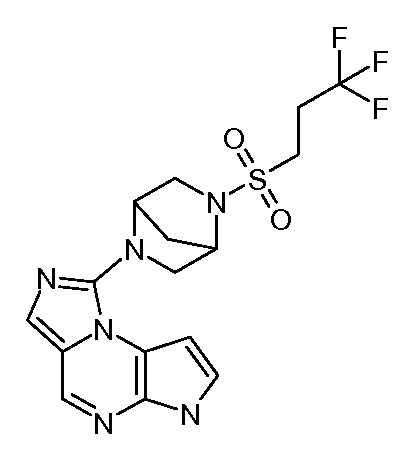

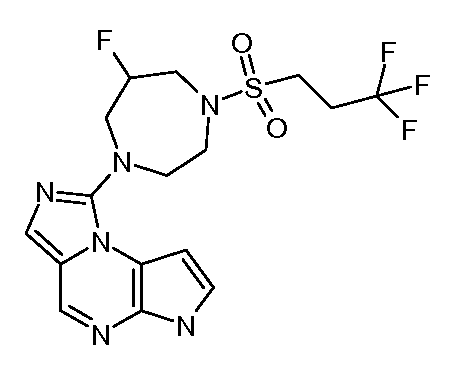
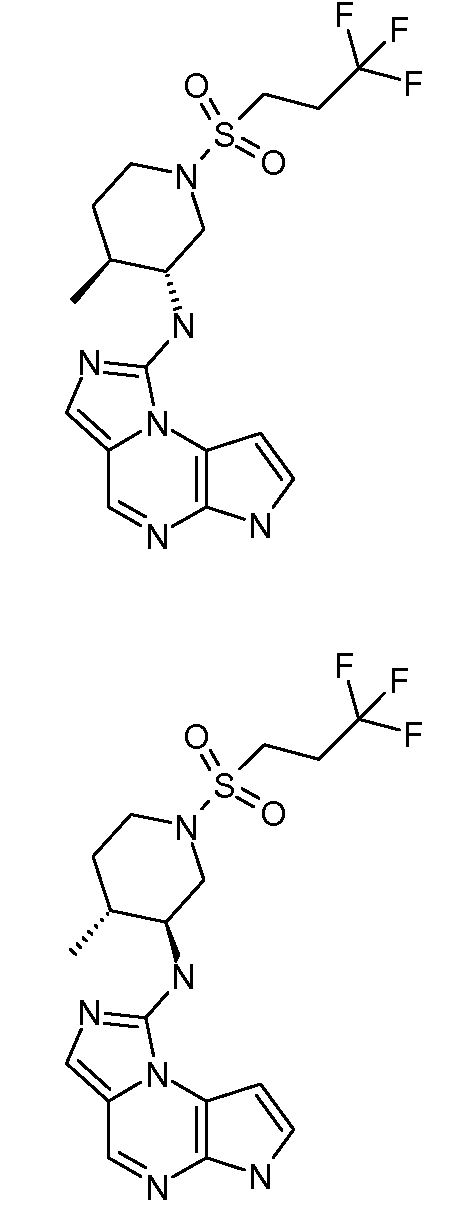

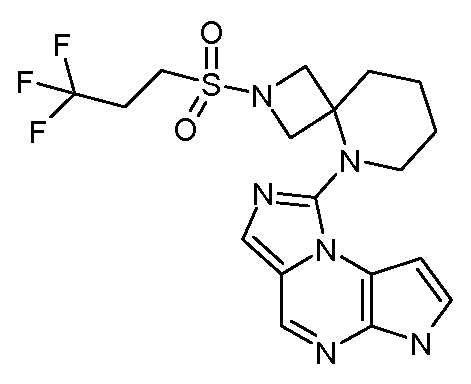
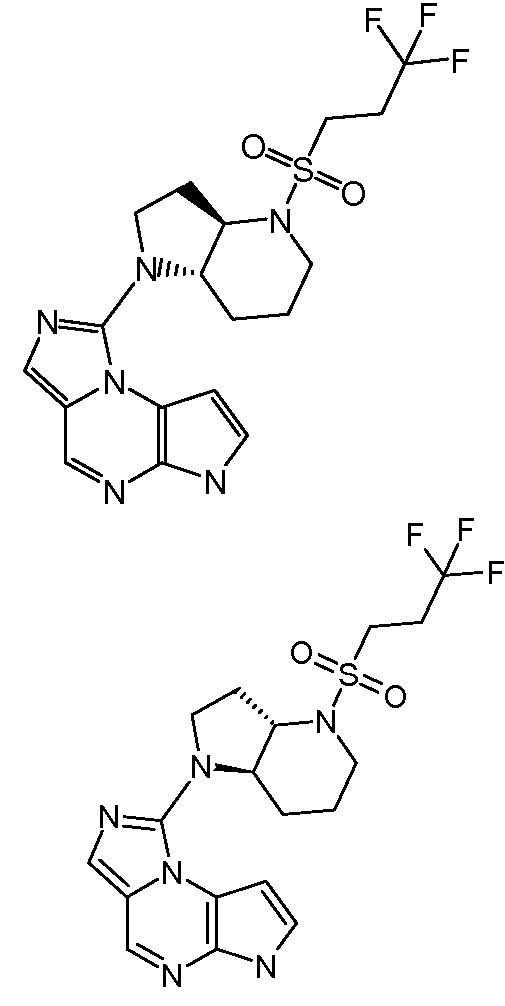
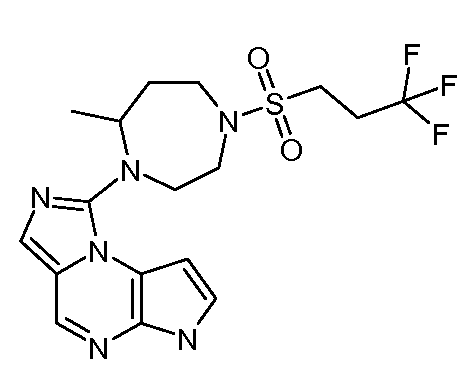
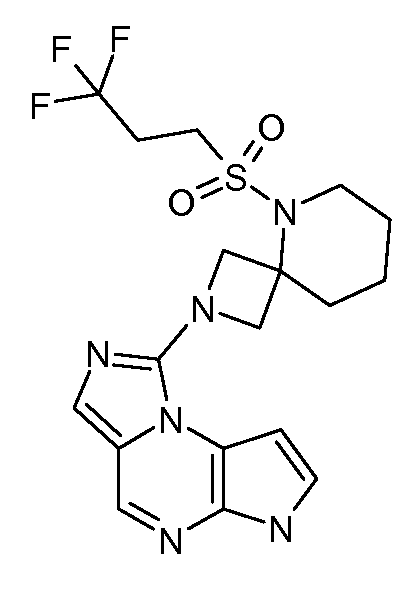
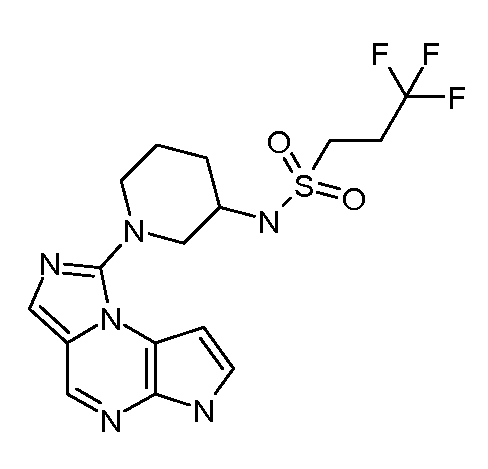


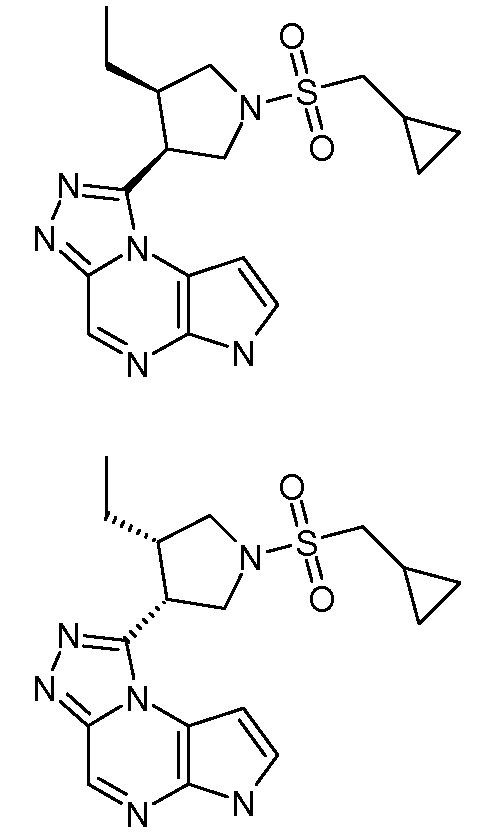


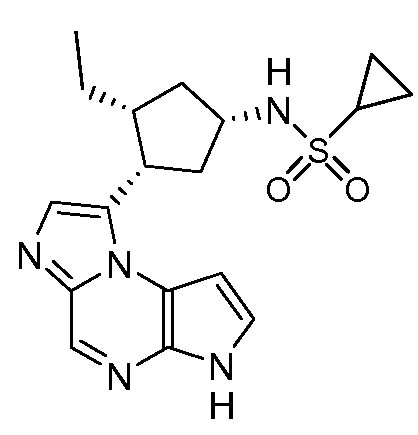



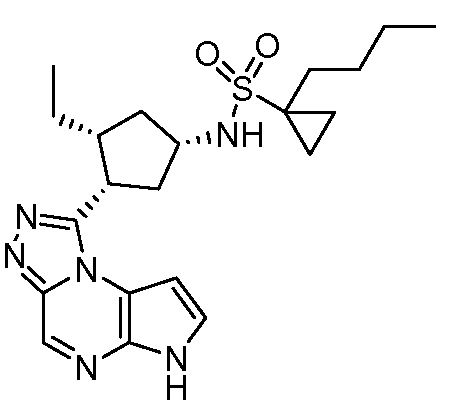





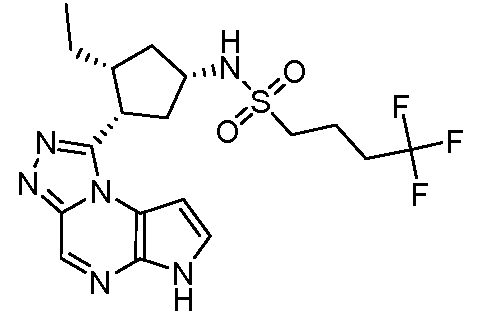

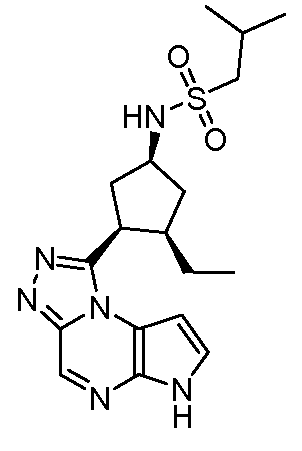




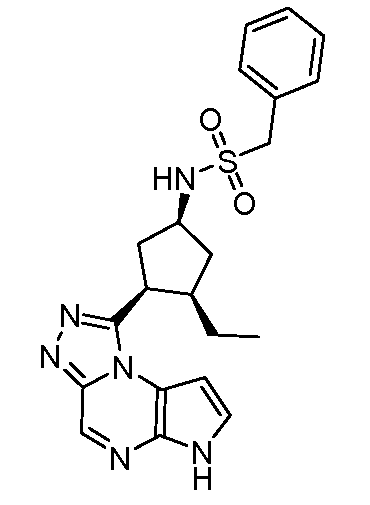


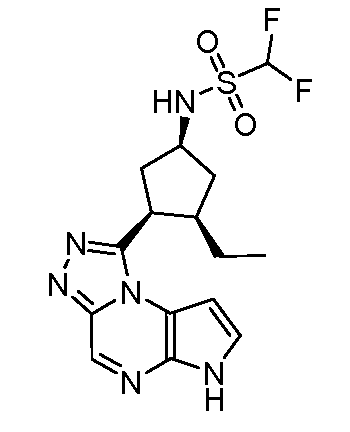





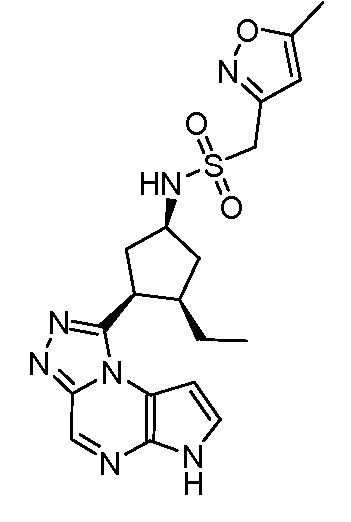


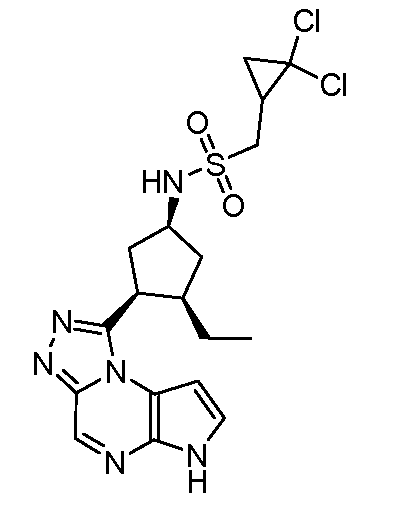

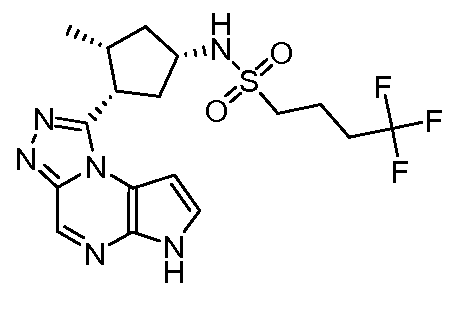
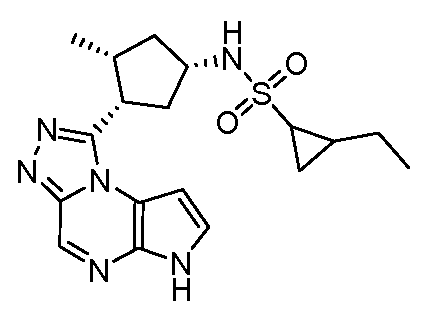

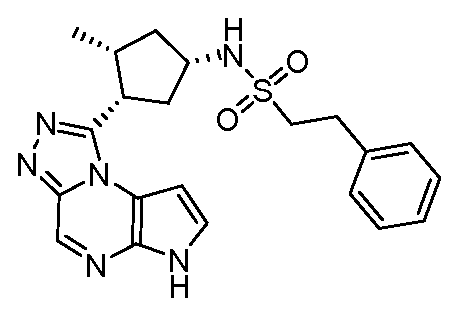






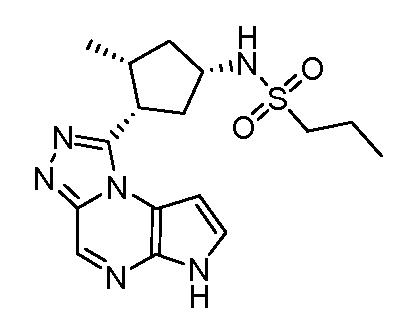

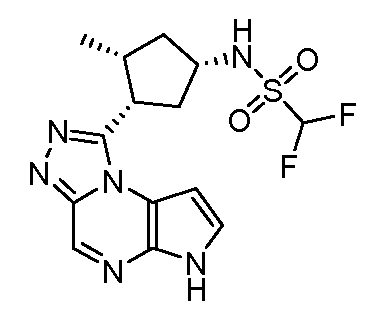


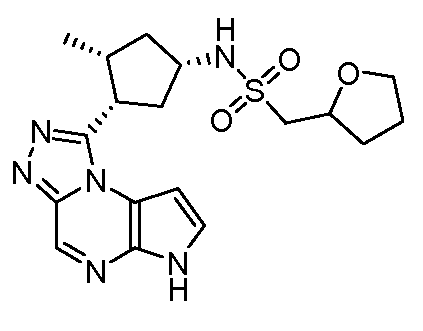
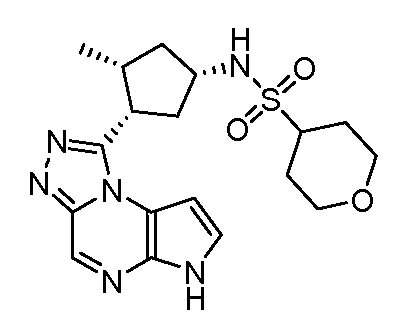






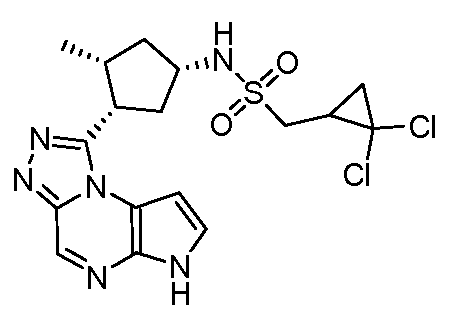

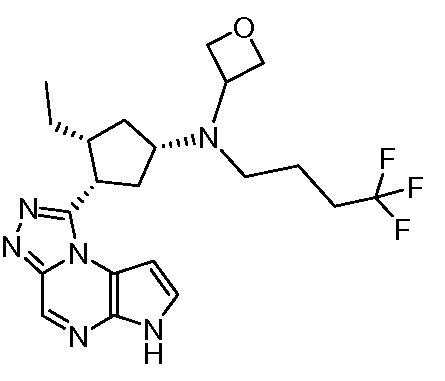
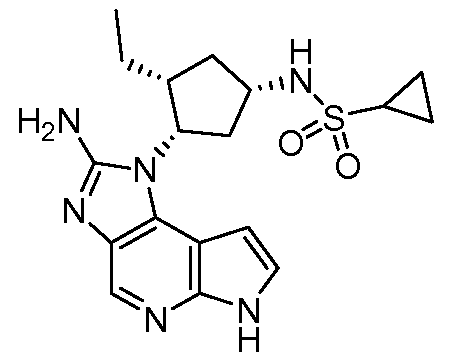
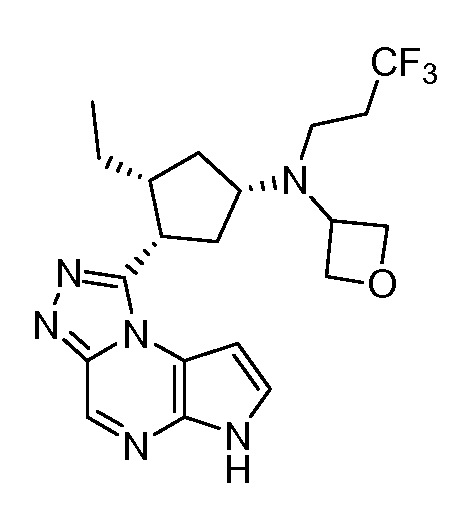


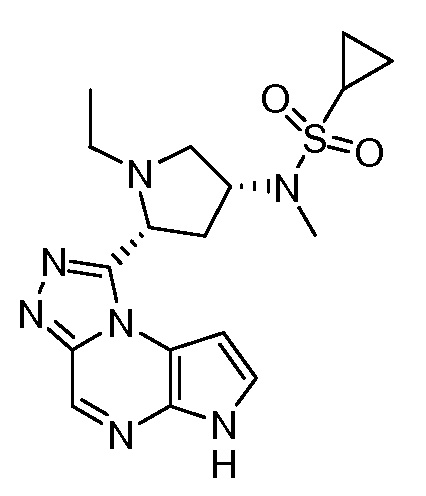

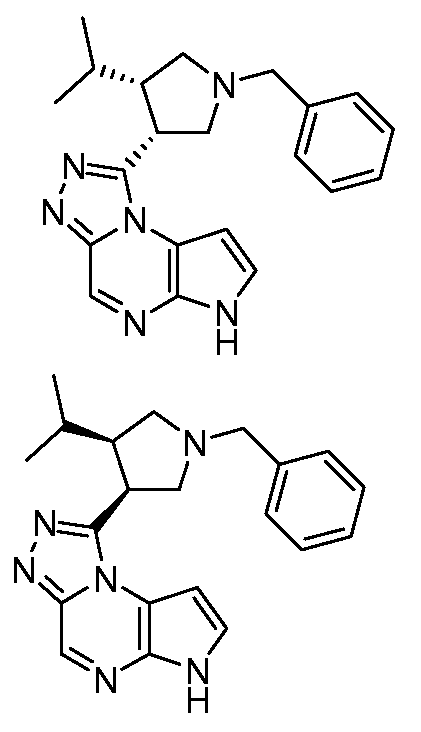


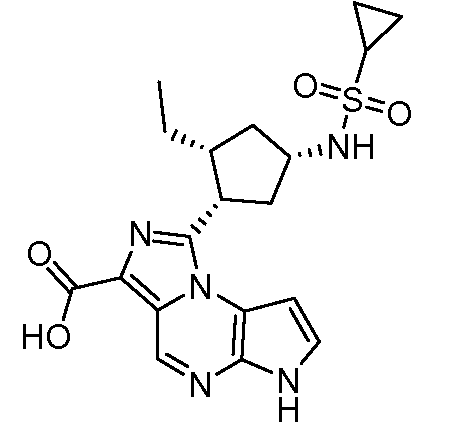






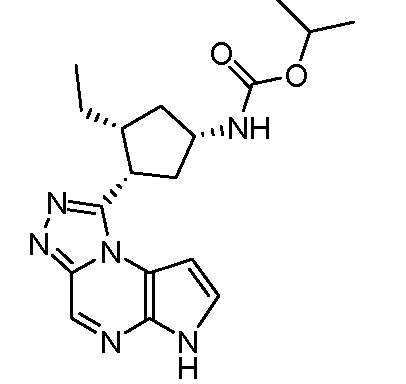

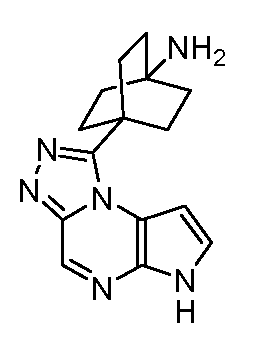

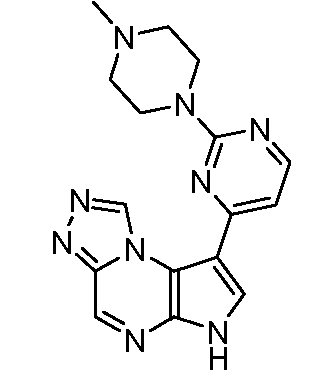


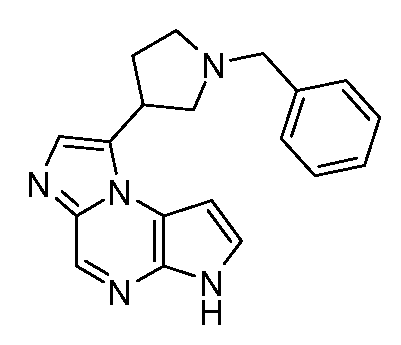
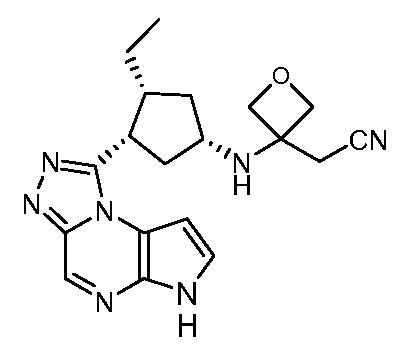


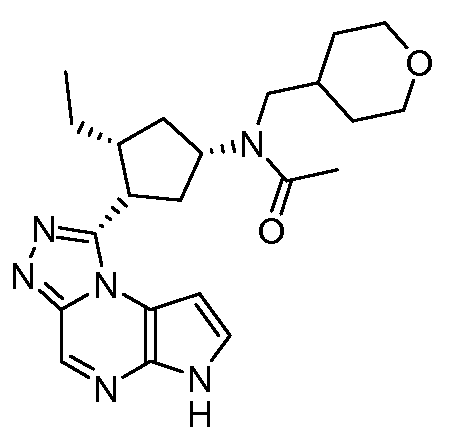
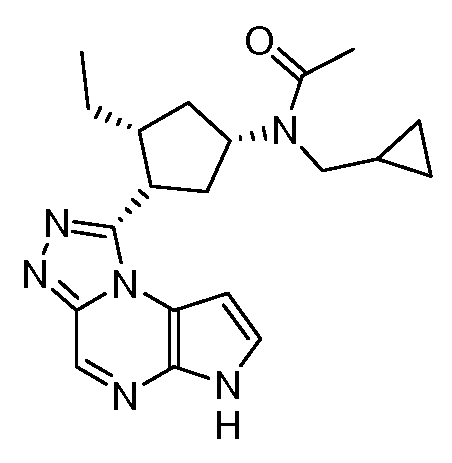





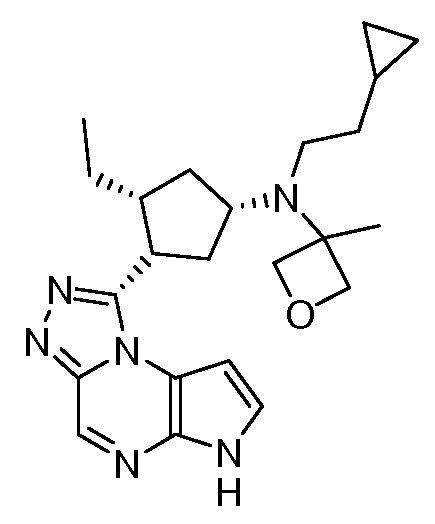

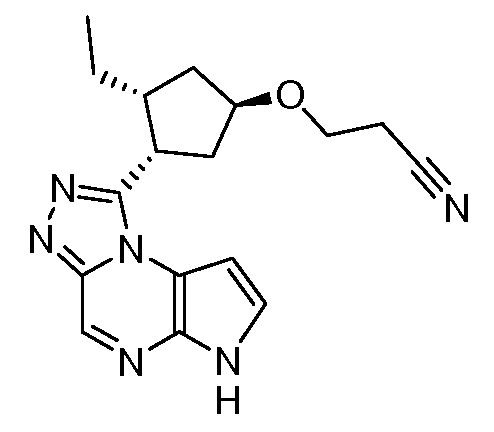
Примеры, полученные с использованием Общей Процедуры D с Na2CO3
(Таблица 1, Способ)










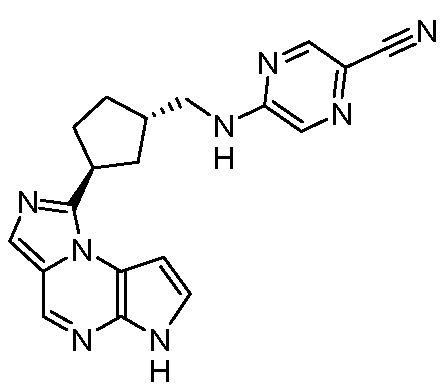

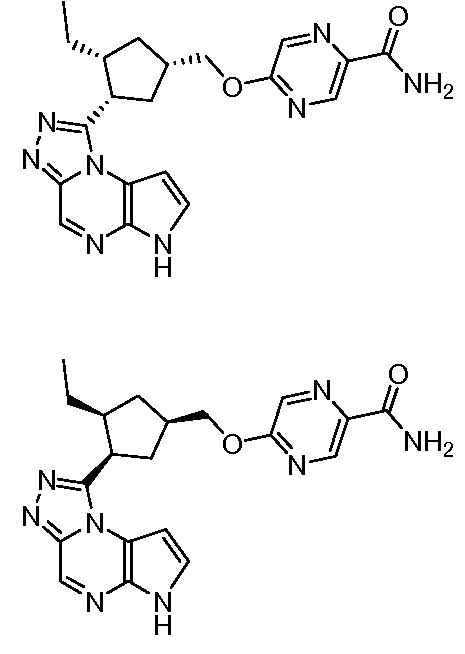

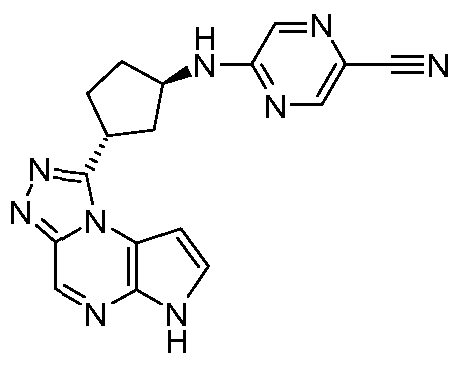








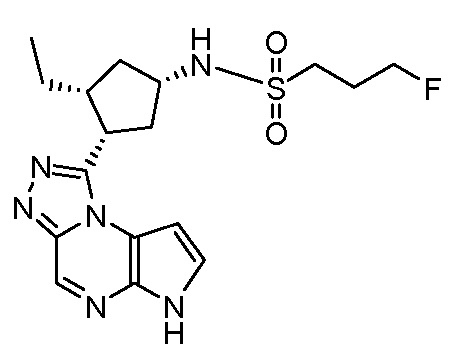



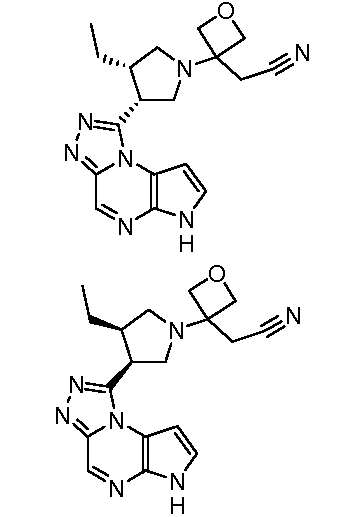

Примеры, полученные с использованием Общей Процедуры D с Na2CO3 с последующим добавлением NaOH
(Таблица 1, Способ)


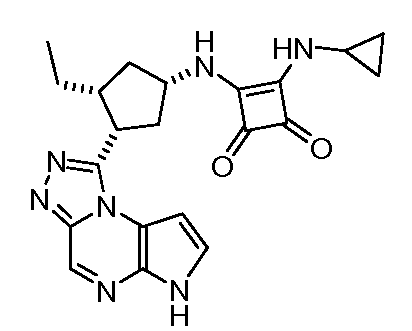
Общая процедура E: кислотное расщепление Boc-защищенного амина
К раствору Boc-защищенного амина (предпочтительно 1 экв.) в органическом растворителе (таком как DCM, 1,4-диоксан или MeOH) добавляют TFA или HCl (предпочтительно 4 н HCl в 1,4-диоксане, 2-35 экв., предпочтительно 2-15 экв.). Реакционную смесь перемешивают при около 20-100°C (предпочтительно от температуры окружающей среды до около 60°C) примерно в течение 1-24 часов (предпочтительно около 1-6 часов). В любом случае присутствия дополнительной кислотно-лабильной группы (например, трет-бутиловый эфир), эту группу также можно расщепить в процессе реакции. В случаях, когда реакция не достигает завершения, согласно данным ТСХ, LC/MS или ВЭЖХ, к реакционной смеси, необязательно, можно добавить дополнительное количество TFA или HCl (предпочтительно 4 н раствор HCl в 1,4-диоксане, 2-35 экв., предпочтительно 2-15 экв.). Когда реакция осуществлена до приемлемой степени, можно осуществить концентрирование реакционной смеси в вакууме с получением амина в виде соли. Альтернативно, реакционную смесь можно распределить между органическим растворителем (таким как EtOAc, DCM или 1,4-диоксан) и водным раствором основания (таким как насыщенный водный раствор NaHCO3 или насыщенный водный раствор Na2CO3, предпочтительно насыщенный водный раствор NaHCO3). Водный слой, необязательно, можно экстрагировать дополнительным количеством органического растворителя, такого как EtOAc или DCM. Объединенные органические слои, необязательно, можно промывать насыщенным солевым раствором, сушить над безводным Na2SO4 или MgSO4, затем декантровать или фильтровать, и затем концентрировать при пониженном давлении с получением целевого соединения.
Получение #E.1: гидрохлорид N-((3S,5R)-5-этилпирролидин-3-ил)циклопропансульфонамида

К раствору (2R,4S)-трет-бутил 4-(циклопропансульфонамидо)-2-этилпирролидин-1-карбоксилата (0,95 г, 2,98 ммоль, Получение #15) в 1,4-диоксане (7,5 мл) добавляли HCl (4 н в 1,4-диоксане, 7,46 мл, 29,8 ммоль). Реакционную смесь нагревали до около 60°C. Примерно через 4 часа реакционную смесь охлаждали до температуры окружающей среды и концентрировали в вакууме с получением неочищенного гидрохлорида N-((3S,5R)-5-этилпирролидин-3-ил)циклопропансульфонамида (0,38 г, 50%) в виде коричневого остатка: LC/MS (Таблица 1, Способ a) Rt = 0,63 мин; MS m/z: 219 (M+H)+.
Примеры, полученные с использованием Общей Процедуры E с HCl
(Таблица 1, Способ)
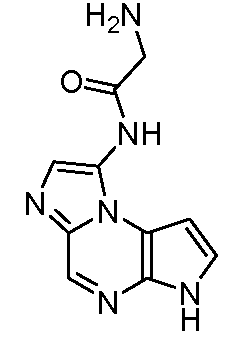




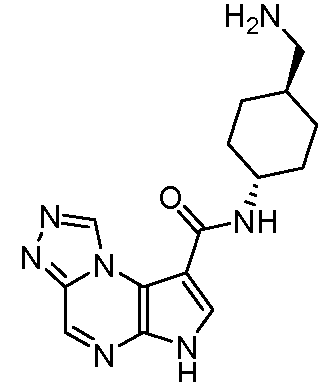


Общая процедура E.1: кислотное расщепление Boc-защищенного амина
К раствору Boc-защищенного амина (предпочтительно 1 экв.) в органическом растворителе (таком как DCM, 1,4-диоксан, MeOH или ТГФ) добавляют кислоту (такую как TFA, HCl или H3PO4 (предпочтительно H3PO4, 1-50 экв., предпочтительно 5-10 экв.). Реакционную смесь перемешивают при около 20-100°C (предпочтительно от температуры окружающей среды до около 65°C) примерно в течение 1-24 часов (предпочтительно около 1-6 часов). В любом случае присутствия дополнительной кислотно-лабильной группы (например, трет-бутиловый эфир), эту группу также можно расщепить в процессе реакции. Необязательно, возможно добавление к реакционной смеси дополнительного количества TFA, HCl или H3PO4 в случаях, когда не достигается завершение реакции, согласно данным ТСХ, LC/MS или ВЭЖХ. Когда реакция осуществлена до приемлемой степени, можно осуществить концентрирование реакционной смеси в вакууме с получением амина в виде соли. Альтернативно, реакционную смесь можно охладить до около -10-25°C перед добавлением водного раствора основания (такого как насыщенный водный раствор NaHCO3, насыщенный водный раствор Na2CO3 или водный раствор K3PO4, предпочтительно водный раствор K3PO4) с необязательным распределением между органическим растворителем (таким как EtOAc, DCM, ТГФ или 1,4-диоксан). Водный слой, необязательно, можно экстрагировать дополнительным количеством органического растворителя, такого как EtOAc или DCM. Объединенные органические слои можно подвергнуть необязательной процедуре промывки насыщенным солевым раствором, сушки над безводным Na2SO4 или MgSO4, затем декантирования или фильтрования перед концентрированием при пониженном давлении с получением целевого соединения.
Получение #E.1.1 5-тозил-5H-пирроло[2,3-b]пиразин-2-амин

К раствору трет-бутил 5-тозил-5H-пирроло[2,3-b]пиразин-2-илкарбамата (12,35 г, 31,8 ммоль, Пример #3 Стадия E) в ТГФ (35 мл) добавляли H3PO4 (20,16 мл, 350 ммоль). Реакционную смесь нагревали до около 65°C. Примерно через 90 минут реакционную смесь охлаждали до около 0°C и добавляли раствор K3PO4 (29,0 мл, 350 ммоль) в воде (100 мл). Белый осадок удаляли фильтрованием. Органический слой отделяли, сушили над безводным MgSO4 и концентрировали в вакууме с получением неочищенного 5-тозил-5H-пирроло[2,3-b]пиразин-2-амина (8,58 г, 94%) в виде желтовато-коричневого твердого вещества: LC/MS (Таблица 1, Способ a) Rt = 1,85 мин; MS m/z: 289 (M+H)+.
Общая процедура F: Удаление защиты у Cbz-защищенного амина с использованием HBr в AcOH
К Cbz-защищенному амину (предпочтительно 1 экв.) добавляют HBr в AcOH (40-400 экв., предпочтительно 70-90 экв. раствора 33% HBr в AcOH) при температуре от около 0°C до 40°C (предпочтительно при температуре окружающей среды) и смесь перемешивают при этой температуре примерно в течение 5-45 минут (предпочтительно около 10 мин). Осадок собирают фильтрованием и тщательно промывают органическим растворителем, таким как Et2O, EtOAc, 1,4-диоксан, ТГФ или MeCN (предпочтительно EtOAc или MeCN), с получением целевого соединения.
Иллюстрация Общей Процедуры F
Получение #F.1: 4-(3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)бицикло[2,2,2]октан-1-амин гидробромид.

Бензил 4-(3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)бицикло[2,2,2]октан-1-илкарбамат (0,22 г, 0,529 ммоль, получали с использованием Z из Получения #N,1 и NaOH, R с диазометаном, S из Примера #3, Стадия E, T с реагентом Лоуссона и D с NaOH) растворяли в HBr (33% в AcOH, 10 мл) и смесь перемешивали примерно в течение 10 минут при температуре окружающей среды. Реакционную смесь затем разбавляли при помощи EtOAc (30 мл) и осадок собирали фильтрованием, тщательно промывали при помощи MeCN и сушили с получением 4-(3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)бицикло[2,2,2]октан-1-амин гидробромида (0,16 г, 83%) в виде желтого твердого вещества: LC/MS (Таблица 1, Способ a) Rt = 1,67 мин; MS m/z 282 (M+H)+.
Примеры, полученные с использованием Общей Процедуры F с HBr в AcOH
(Таблица 1, Способ)



Общая процедура F.1: Удаление защиты у Cbz-защищенного амина с использованием HBr в AcOH
К Cbz-защищенному амину (предпочтительно 1 экв.) добавляют HBr в уксусной кислоте (5-400 экв., 33% HBr в AcOH) при температуре от около 0°C до 40°C (предпочтительно при температуре окружающей среды) и смесь перемешивают при этой температуре примерно в течение 0,5-5 часов (предпочтительно около 1 часа). Реакционную смесь обрабатывают с использованием одного из следующих способов. Способ 1: Осадок собирают фильтрованием и тщательно промывают органическим растворителем, таким как Et2O, EtOAc, 1,4-диоксан, ТГФ или MeCN (предпочтительно EtOAc или MeCN), с получением целевого соединения. Способ 2: Реакционную смесь разбавляют водой и подходящим органическим растворителем (такие как Et2O). Слои перемешивают в течение короткого периода времени и органический слой декантируют. Это повторяют (3-10×) и органический слой отбрасывают. Водный слой подщелачивают водным раствором основания (таким как насыщенный водный раствор NaHCO3 или насыщенный водный раствор Na2CO3, предпочтительно насыщенный водный раствор NaHCO3) и экстрагируют подходящим органическим растворителем (такие как EtOAc, DCM или Et2O). Объединенные органические слои можно подвергнуть либо процедуре промывки насыщенным солевым раствором и концентрирования в вакууме, либо сушки над безводным Na2SO4 или MgSO4 с последующим декантированием или фильтрованием перед концентрированием при пониженном давлении, с получением целевого соединения.
Иллюстрация Общей Процедуры F.1
Получение #F.1.1: 8-((цис)-4-этилпирролидин-3-ил)-3-тозил-3H-имидазо[1,2-a]пирроло[2,3-e]пиразин

К раствору (цис)-бензил 3-этил-4-(3-тозил-3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)пирролидин-1-карбоксилата (0,838 г, 1,541 ммоль, получали с использованием E из Примера #36 Стадия D с TFA, N, R, S.1 с Примером #3 Стадия E, и T с реагентом Лоуссона) добавляли раствор HBr (2,50 мл, 15,19 ммоль, 33% в уксусной кислоте). Реакционную смесь перемешивали при температуре окружающей среды примерно в течение 1 часа. Реакционную смесь разбавляли при помощи Et2O (50 мл) и воде (20 мл). Слои перемешивали примерно в течение 3 минут и органический слой декантировали, затем процедуру повторяли 5 раз. Водный слой охлаждали до около 0°C, подщелачивали насыщенным водным раствором NaHCO3 (10 мл) до около pH 7. Водный слой экстрагировали при помощи EtOAc (3×50 мл), объединяли и сушили над безводным Na2SO4, фильтровали и концентрировали с получением коричневого твердого вещества. Твердое вещество растворяли в DCM (50 мл) и промывали водой (3×20 мл), сушили над безводным Na2SO4, фильтровали и концентрировали с получением 8-((цис)-4-этилпирролидин-3-ил)-3-тозил-3H-имидазо[1,2-a]пирроло[2,3-e]пиразина (0,453, 61%) в виде коричневого остатка: LC/MS (Таблица 1, Способ a) Rt = 1,73 мин; MS m/z: 410 (M+H)+.
Общая процедура G: Образование ацетамида
К раствору амина (предпочтительно 1 экв.) в пиридине (5-25 экв., предпочтительно 10 экв.) при около 0-25°C (предпочтительно около 0°C) добавляют Ac2O (2-10 экв., предпочтительно 5 экв.). Если реакционная смесь охлаждается, перемешивание продолжают при более низкой температуре примерно в течение 5-30 минут (предпочтительно 10-15 мин) и затем нагревают до температуры окружающей среды. Примерно через 1-24 часа (предпочтительно 2-16 часов) реакционную смесь концентрируют при пониженном давлении и распределяют между органическим растворителем, таким как EtOAc или DCM (предпочтительно EtOAc), и водным раствором кислоты, таким как водный раствор HCl (1-6 н, предпочтительно 1 н). Слои разделяют и органический слой, необязательно, промывают водным раствором кислоты, таким как водный раствор HCl (1-6 н, предпочтительно 1 н), водным раствором основания, таким как водный раствор NaHCO3 или водный раствор Na2CO3 (предпочтительно насыщенный водный раствор NaHCO3) и насыщенным солевым раствором. Органический слой затем сушат над безводным MgSO4, фильтруют через слой Florisil®, осуществляя при этом промывку дополнительным количеством органического растворителя, такого как EtOAc или DCM (предпочтительно EtOAc), и концентрируют при пониженном давлении.
Иллюстрация Общей Процедуры G
Получение #G.1*: (1S,2R,4S)-этил 4-ацетамидо-2-этилциклопентанкарбоксилат

Раствор этил 4-амино-2-этилциклопентанкарбоксилата (49,0 г, 264 ммоль, Пример #8, Стадия I) в пиридине (214 мл, 2645 ммоль) охлаждали до около 0°C. Добавляли Ac2O (125 мл, 1322 ммоль) и перемешивание продолжали при около 0°C примерно в течение 15 минут. Полученный раствор нагревали до температуры окружающей среды и перемешивали примерно в течение 12 часов. Реакционную смесь концентрировали при пониженном давлении и добавляли EtOAc (500 мл) и водный раствор HCl (1 н, 200 мл). Слои разделяли и органический слой промывали водным раствором HCl (1 н, 200 мл), насыщенным водным раствором NaHCO3 (2×200 мл) и насыщенным солевым раствором (150 мл), сушили над безводным MgSO4, фильтровали через слой Florisil®, осуществляя при этом промывку при помощи EtOAc (600 мл), и концентрировали при пониженном давлении с получением не совсем белого твердого вещества (52 г), которое очищали с использованием Общей процедуры AA (Таблица 2, Способ 24, Rt = 8,2 мин, or = положительное) с получением (1S,2R,4S)-этил 4-ацетамидо-2-этилциклопентанкарбоксилата (20,3 г, 34%): LC/MS (Таблица 1, Способ a) Rt = 1,82 мин; MS m/z: 228 (M+H)+.
Примеры, полученные с использованием Общей Процедуры G
(Таблица 1, Способ)
Общая процедура H: Образование амида из карбоновой кислоты и амина
К раствору или суспензии карбоновой кислоты (1-5 экв., предпочтительно 1,0 экв.) и амина или соли амина (1-5 экв., предпочтительно 1 экв.) в органическом растворителе (таком как DCM, DCE, ТГФ или 1,4-диоксан, предпочтительно DCM) добавляют реагент для пептидного связывания (такой как BOP-Cl, IBCF, HATU или EDC•HCl, предпочтительно HATU, 1-10 экв., предпочтительно 1-1,5 экв.), основание (такое как TEA, DIEA или пиридин, предпочтительно DIEA, 0-20 экв., предпочтительно 3 экв.). Реакционную смесь затем перемешивают при температуре окружающей среды примерно в течение времени от 15 минут до 24 часов (предпочтительно около 45 минут - 16 часов). Реакционную смесь затем обрабатывают с использованием одного из следующих способов. Способ 1: Реакционную смесь разбавляют водой или насыщенным водным раствором NaHCO3. Слои разделяют. Водный слой, необязательно, экстрагируют дополнительным количеством органического растворителя, такого как EtOAc или DCM. Органический слой (или объединенные слои), необязательно, промывают водой, насыщенным водным раствором NaHCO3 и/или насыщенным солевым раствором, сушат над безводным MgSO4 или Na2SO4, фильтруют или декантируют и концентрируют при пониженном давлении. Способ 2: Неочищенную реакционную смесь фильтруют через слой силикагеля, промывают подходящим растворителем (таким как EtOAc, MeOH или DCM, предпочтительно MeOH) и концентрируют при пониженном давлении. Способ 3: Неочищенную реакционную смесь непосредственно очищают хроматографией без обработки.
Иллюстрация Общей Процедуры H
Получение #H.1*: (3R,4R)-трет-бутил 4-метил-3-((5-тозил-5H-пирроло[2,3-b]пиразин-2-ил)метилкарбамоил)пиперидин-1-карбоксилат

К суспензии (5-тозил-5H-пирроло[2,3-b]пиразин-2-ил)метанамингидрохлорида (34,0 г, 100 ммоль, Пример #5, Стадия C), (3R,4R)-1-(трет-бутоксикарбонил)-4-метилпиперидин-3-карбоновой кислоты (24,43 г, 100 ммоль, Пример #5, Стадия F) и HATU (38,2 г, 100 ммоль) в DCM (700 мл) добавляли DIEA (52,6 мл, 301 ммоль). Реакционную смесь перемешивали при температуре окружающей среды примерно в течение 45 минут. Реакционную смесь промывали насыщенным водным раствором NaHCO3 (300 мл). Органический слой отделяли, сушили над безводным Na2SO4, фильтровали и концентрировали в вакууме. Полученный остаток очищали хроматографией на силикагеле (330 г) с использованием 33-100% EtOAc в гептане с получением (3R,4R)-трет-бутил-4-метил-3-((5-тозил-5H-пирроло[2,3-b]пиразин-2-ил)метилкарбамоил)-пиперидин-1-карбоксилата (53 г, 96%) в виде бледно-желтого пенообразного вещества: LC/MS (Таблица 1, Способ b) Rt = 2,40 мин; MS m/z: 528 (M+H)+.
Примеры, полученные из 1-((3R,4R)-4-метилпиперидин-3-ил)-6H-имидазо[1,5-a]пирроло[2,3-e]пиразин гидрохлорида (Пример #5, Стадия J) с использованием Общей Процедуры H с EDC•HCl и DIEA
(Таблица 1, Способ)

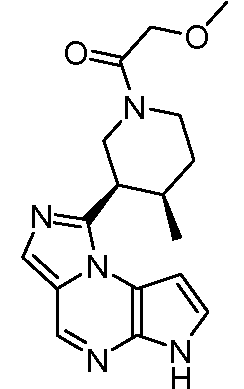

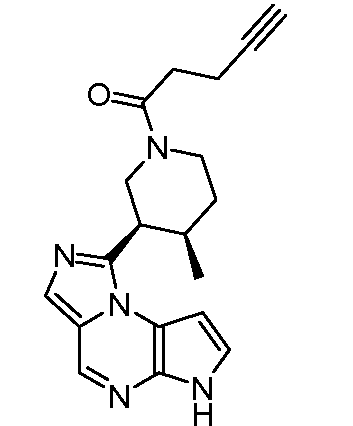




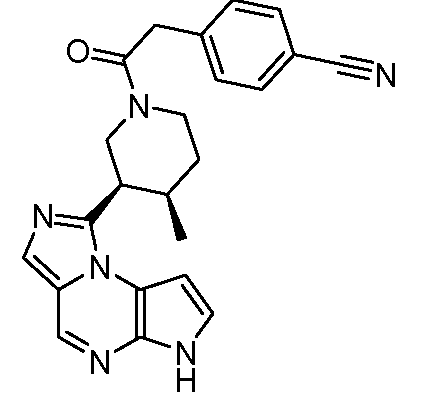
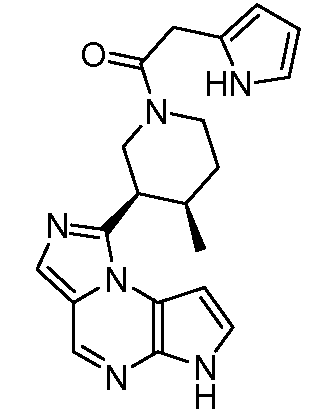










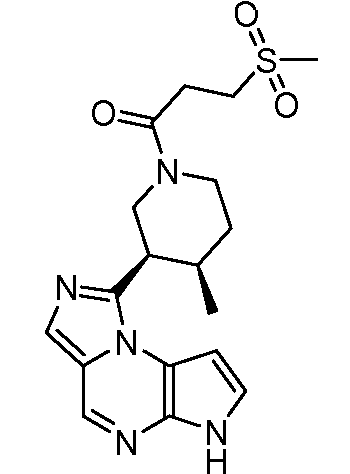




Примеры, полученные из 1-((3R,4R)-4-метилпиперидин-3-ил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин гидрохлорида (получен с использованием A из Примера #1, Стадия D, и Примера #5, Стадия F, HATU и DIEA; B с DIEA; D с NaOH; E с HCl) с использованием Общей Процедуры H с EDC•HCl и DIEA
(Таблица 1, Способ)
Примеры, полученные из 1-(цис)-4-метилпиперидин-3-ил)-6H-имидазо[1,5-a]пирроло[2,3-e]пиразин гидрохлорида (получен с использованием D из Получения #Q,1 с NaOH и E с 4 н HCl в 1,4-диоксане) с использованием Общей Процедуры H с HATU и DIEA
(Таблица 1, Способ)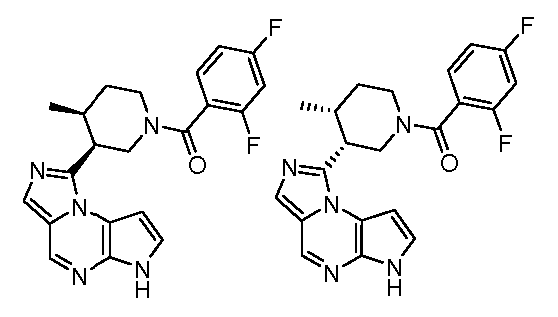



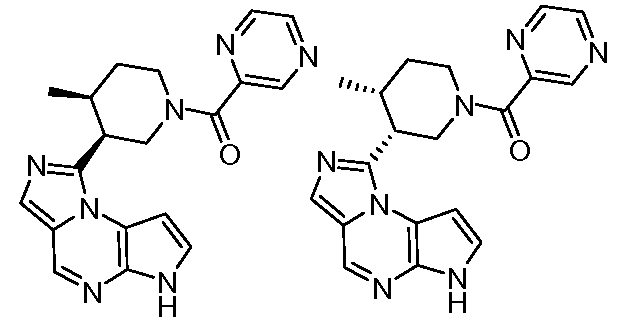

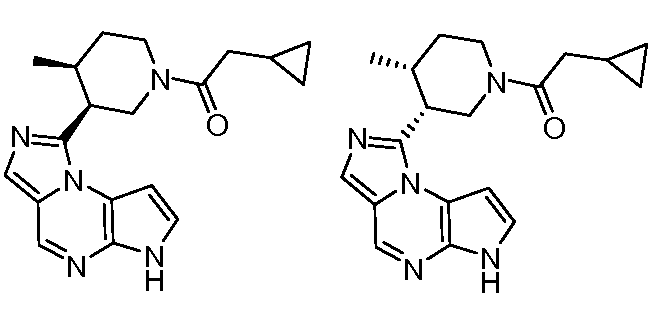



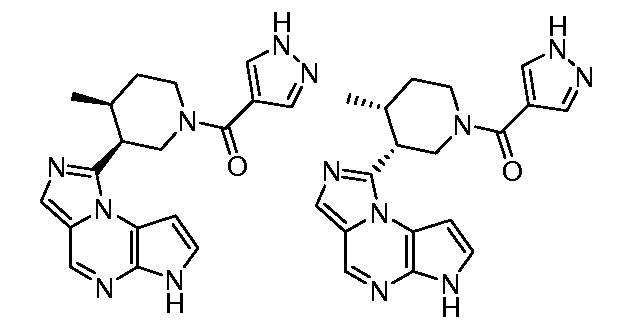


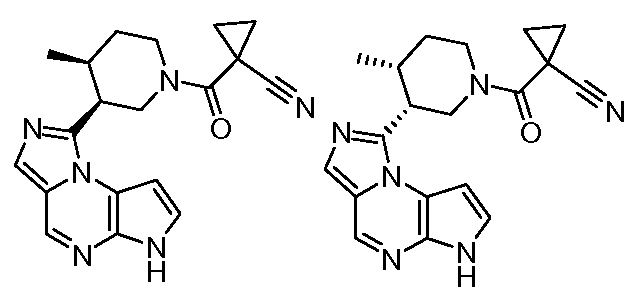


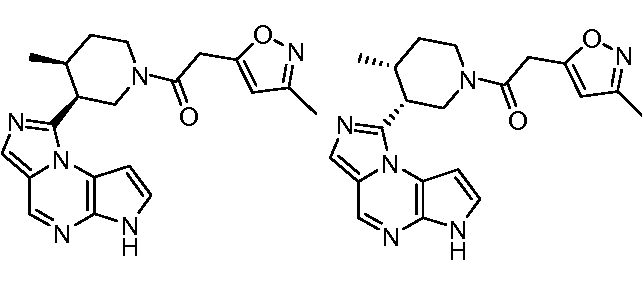


Примеры, полученные из (R)-1-(пиперидин-3-ил)-6H-имидазо[1,5-a]пирроло[2,3-e]пиразин гидрохлорида (Пример #6, Стадия H) с использованием Общей Процедуры H с EDC•HCl и DIEA
(Таблица 1, Способ)
Примеры, полученные из циклопропанамина (Aldrich) с использованием Общей Процедуры H с HATU и TEA
(Таблица 2, Способ а)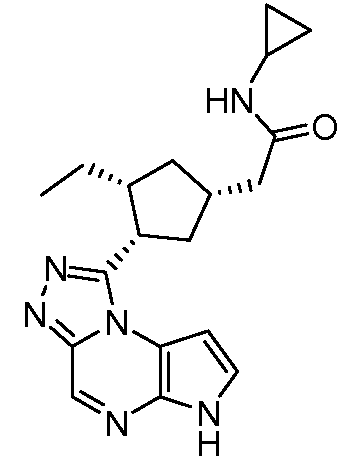
Общая Процедура I: Образование мочевины из амина и карбамоилхлорида
В колбу, содержащую амин или соль амина (1 экв.), в органическом растворителе (таком как ТГФ или 1,4-диоксан, предпочтительно ТГФ) добавляют основание (такое как DIEA или TEA, предпочтительно TEA [3-5 экв., предпочтительно 4 экв.]) и перемешивают при температуре окружающей среды примерно в течение 0-30 минут (предпочтительно около 5 мин), затем добавляют карбамоилхлорид (0,5-2 экв., предпочтительно 0,75 экв.). Смесь перемешивают при около 0-90°C (предпочтительно около 45°C) примерно в течение 2-24 часов (предпочтительно около 18 часов). Реакционную смесь оставляют для достижения температуры окружающей среды. Необязательно, осуществляют удаление органического растворителя при пониженном давлении. Неочищенное вещество можно распределить между органическим растворителем (таким как EtOAc или DCM) и водой, водным раствором основания (такого как насыщенный водный раствор NaHCO3) или насыщенным солевым раствором. Слои разделяют и органический слой необязательно промывают водой, водным раствором основания (таким как насыщенный водный раствор NaHCO3) или насыщенным солевым раствором, сушат над безводным Na2SO4 или MgSO4, фильтруют и концентрируют при пониженном давлении с получением целевого соединения.
Иллюстрация Общей Процедуры I
Пример #I.1.1*: ((3R,4R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-ил)(пиперидин-1-ил)метанон

В круглодонную колбу загружали 1-((3R,4R)-4-метилпиперидин-3-ил)-6H-имидазо[1,5-a]пирроло[2,3-e]пиразин гидрохлорид (0,050 г, 0,17 ммоль, Пример #5, Стадия J), TEA (0,10 мл, 0,69 ммоль) в ТГФ (1,6 мл). Реакционную смесь перемешивали примерно в течение 5 минут при температуре окружающей среды и затем добавляли пиперидин-1-карбонилхлорид (0,019 г, 0,13 ммоль). Реакционную смесь нагревали при около 45°C примерно в течение 18 часов, охлаждали до температуры окружающей среды и концентрировали при пониженном давлении. Неочищенный продукт растворяли в DCM (5 мл) и промывали водой (3 мл), сушили над безводным MgSO4, фильтровали и концентрировали при пониженном давлении. Вещество очищали при помощи ОФ-ВЭЖХ (Таблица 1, Способ f) с получением ((3R,4R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-ил)(пиперидин-1-ил)метанона (0,018 г, 8%): LC/MS (Таблица 1, Способ b) Rt = 1,80 мин; MS m/z 367 (M+H)+.
Примеры, полученные из 1-((3R,4R)-4-метилпиперидин-3-ил)-6H-имидазо[1,5-a]пирроло[2,3-e]пиразингидрохлорида (Пример #5, Стадия J) с использованием Общей Процедуры I с TEA
(Таблица 1, Способ)

Примеры, полученные из 1-(цис-4-метилпиперидин-3-ил)-6H-имидазо[1,5-a]пирроло[2,3-e]пиразингидрохлорида (получен с использованием D из Получения #Q,1 с NaOH и E с 4 н HCl в 1,4-диоксане) с использованием Общей Процедуры I с TEA
(Таблица 1, Способ)

Пример, полученный из (R)-1-(пиперидин-3-ил)-6H-имидазо[1,5-a]пирроло[2,3-e]пиразингидрохлорида (Пример #6, Стадия H) с использованием Общей Процедуры I с TEA
(Таблица 1, Способ)
Общая процедура J: Образование мочевины или тиомочевины с использованием CDI или тиокарбонилдиимидазола, соответственно
К раствору или суспензии амина или соли амина (1-3 экв., предпочтительно 1 экв.) в органическом растворителе, таком как DCM, ТГФ или DMF (предпочтительно DCM), при около -20-40°C (предпочтительно около 0°C) добавляют органическое основание, такое как TEA, DIEA, пиридин (предпочтительно TEA) (1-10 экв., предпочтительно 1-3 экв.), с последующим добавлением CDI или 1,1'-тиокарбонилдиимидазола (0,5-2 экв., предпочтительно 1 экв.). Примерно через 0,5-24 часа (предпочтительно около 0,5-1 часа) добавляют второй амин или соль амина (1-10 экв., предпочтительно 3 экв.) чистый или в виде раствора или суспензии в органическом растворителе, таком как DCM, ТГФ или DMF (предпочтительно DCM). Реакционную смесь поддерживают при около 0°C примерно в течение 10-60 минут (предпочтительно около 15-30 мин) и затем реакционной смеси дают нагреться до температуры окружающей среды. Примерно через 1-48 часов (предпочтительно около 12-16 часов) реакционную смесь распределяют между органическим растворителем (таким как EtOAc, DCM или 1,4-диоксан) и водным раствором основания (таким как насыщенный водный раствор NaHCO3 или насыщенный водный раствор Na2CO3, предпочтительно насыщенный водный раствор NaHCO3). Необязательно, реакционную смесь концентрируют при пониженном давлении и остаток распределяют, как описано выше. В любом случае, водный слой затем необязательно экстрагируют дополнительным количеством органического растворителя, такого как EtOAc или DCM. Объединенные органические слои можно подвергнуть либо процедуре промывки насыщенным солевым раствором и концентрирования в вакууме, либо сушки над безводным Na2SO4 или MgSO4 и затем декантирования или фильтрования перед концентрированием при пониженном давлении, с получением целевого соединения. Промежуточные соединения и конечные соединения, полученные через эту Общую Процедуру, необязательно, можно очистить с использованием одного или нескольких из Способов Очистки, описанных выше.
Получение #J.1: трет-бутил 1-((5-тозил-5H-пирроло[2,3-b]пиразин-2-ил)метилкарбамотиоил)пирролидин-3-илкарбамат

К суспензии (5-тозил-5H-пирроло[2,3-b]пиразин-2-ил)метанамин гидрохлорида (0,50 г, 1,5 ммоль, Пример #5, Стадия C) в DCM (10 мл) при около 0°C добавляли TEA (0,226 мл, 1,62 ммоль). К гомогенной реакционной смеси добавляли раствор 1,1'-тиокарбонилдиимидазола (0,29 г, 1,6 ммоль) в DCM (10 мл). Примерно через 30 минут к реакционной смеси добавляли суспензию трет-бутил пирролидин-3-илкарбамата (0,83 г, 4,4 ммоль, TCI) в DCM (10 мл). После перемешивания примерно в течение 20 минут реакционной смеси давали нагреться до температуры окружающей среды. После перемешивания примерно в течение 15 часов к реакционной смеси добавляли насыщенный водный раствор NaHCO3 (30 мл). Органический слой отделяли, концентрировали в вакууме и очищали хроматографией на силикагеле с использованием в качестве элюента 20-40% EtOAc в DCM с получением трет-бутил 1-((5-тозил-5H-пирроло[2,3-b]пиразин-2-ил)метилкарбамотиоил)пирролидин-3-илкарбамата (0,54 г, 69%) в виде желтого стекловидного вещества: LC/MS (Таблица 1, Способ a) Rt = 2,37 мин; MS m/z: 531 (M+H)+.
Общая процедура J.1: Образование мочевины или тиомочевины с использованием CDI или тиокарбонилдиимидазола, соответственно
К раствору или суспензии амина или соли амина (1-3 экв., предпочтительно 1-2 экв.) в органическом растворителе, таком как DCM, ТГФ или DMF (предпочтительно DMF), при около 20-80°C (предпочтительно около 65°C), необязательно, добавляют органическое основание, такое как TEA, DIEA, пиридин (предпочтительно TEA) (1-10 экв., предпочтительно 1-5 экв.), с последующим добавлением CDI или 1,1'-тиокарбонилдиимидазола (0,5-2 экв., предпочтительно 1 экв.). Примерно через 0,5-24 часов (предпочтительно около 1-3 часов) добавляют второй амин или соль амина (1-10 экв., предпочтительно 1-3 экв.), чистый или в виде раствора или суспензии в органическом растворителе, таком как DCM, ТГФ или DMF (предпочтительно DMF). Реакционную смесь поддерживают при около 20-80°C (предпочтительно около 65°C ) примерно в течение 2-24 часов (предпочтительно около 3 часов). Если реакционную смесь нагревали, ее охлаждают до температуры окружающей среды. Реакционную смесь распределяют между органическим растворителем (таким как EtOAc, DCM или 1,4-диоксан) и водным раствором основания (таким как насыщенный водный раствор NaHCO3 или насыщенный водный раствор Na2CO3, предпочтительно насыщенный водный раствор NaHCO3). Необязательно, реакционную смесь концентрируют при пониженном давлении и остаток распределяли, как описано выше. В любом случае, водный слой затем, необязательно, экстрагируют дополнительным количеством органического растворителя, такого как EtOAc или DCM. Объединенные органические слои можно подвергнуть процедуре, необязательно, промывки насыщенным солевым раствором и концентрирования в вакууме, либо сушки над безводным Na2SO4 или MgSO4 и затем декантирования или фильтрования перед концентрированием при пониженном давлении, с получением целевого соединения. Необязательно, реакционную смесь концентрируют при пониженном давлении и остаток непосредственно очищают.
Иллюстрация Общей Процедуры J.1
Получение #J.1.1: (цис)-N-(2-циклопропилэтил)-3-этил-4-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)пирролидин-1-карбоксамид
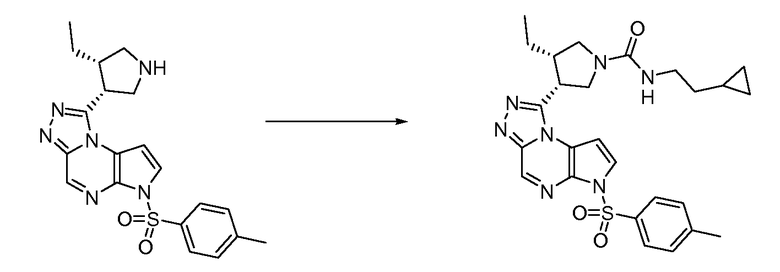
К раствору 2-циклопропилэтанамина (0,068 г, 0,804 ммоль, Oakwood) в DMF (3 мл) добавляли CDI (0,150 г, 0,926 ммоль). Раствор перемешивали при около 65°C примерно в течение 2 часов. Добавляли 1-((цис)-4-этилпирролидин-3-ил)-6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин (0,250 г, 0,609 ммоль, Пример #36, стадия F) и продолжали нагревание реакционной смеси при около 65°C. Примерно через 2 часа реакционную смесь охлаждали до температуры окружающей среды. Растворитель удаляли при пониженном давлении. Неочищенное вещество очищали хроматографией на силикагеле с градиентным элюированием 0-10% MeOH в DCM с получением (цис)-N-(2-циклопропилэтил)-3-этил-4-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)пирролидин-1-карбоксамида (0,238 г, 64%) в качестве продукта: LC/MS (Таблица 1, Способ a) Rt = 2,17 мин; MS m/z: 522 (M+H)+.
Примеры, полученные из 1-(цис)-4-метилпиперидин-3-ил)-6H-имидазо[1,5-a]пирроло[2,3-e]пиразингидрохлорида (получен с использованием D из Получения #Q.1 с NaOH и E с 4 н HCl в 1,4-диоксане) с использованием Общей Процедуры J с CDI
(Таблица 1, Способ)

Примеры, полученные из 1-((3R,4R)-4-метилпиперидин-3-ил)-6H-имидазо[1,5-a]пирроло[2,3-e]пиразингидрохлорида (Пример #5, Стадия J) с использованием Общей Процедуры J с CDI
(Таблица 1, Способ)










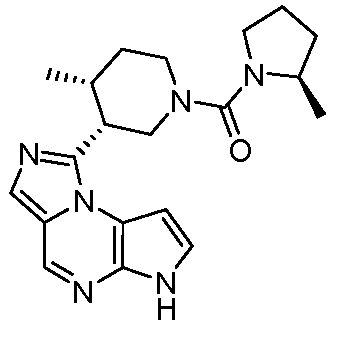


Примеры, полученные из (R)-1-(пиперидин-3-ил)-6H-имидазо[1,5-a]пирроло[2,3-e]пиразингидрохлорида (Пример #6, Стадия H) с использованием Общей Процедуры J с CDI и пиридином
(Таблица 1, Способ)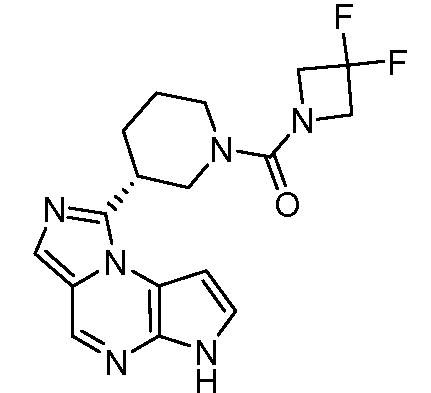








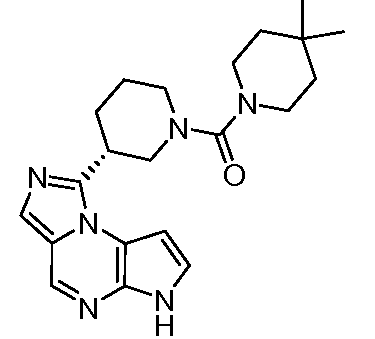
Примеры, полученные из 1-((3R,4S)-4-изопропилпирролидин-3-ил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразина (получен с использованием Y из Примера #D.1.143) с использованием Общей Процедуры J с CDI
(Таблица 1, Способ)
Общая процедура K: Образование сульфонамида из амина
К смеси амина или соли амина (предпочтительно 1 экв.) в органическом растворителе, таком как ТГФ, DMA, DCM или DMF (предпочтительно DMF), добавляют органическое основание, такое как TEA или DIEA (1-10 экв., предпочтительно 2-4 экв.) или водный раствор основания, такой как насыщенный водный раствор NaHCO3 (5-20 экв., предпочтительно 5-10 экв.) (предпочтительно органическое основание) и сульфонилхлорид (0,9-3 экв., предпочтительно 1-1,5 экв.). Реакционную смесь перемешивают при около -10-25°C (предпочтительно при температуре окружающей среды) примерно в течение 0,5-150 часов (предпочтительно около 144 часов). Необязательно, можно добавить дополнительное количество основания (1-10 экв.) и/или сульфонилхлорида (0,4-2 экв.) в любой момент в процессе реакции. Реакционную смесь обрабатывают с использованием одного из следующих способов. Способ 1: Реакционную смесь разбавляют водой и экстрагируют органическим растворителем, таким как DCM или EtOAc. Объединенные органические слои, необязательно, промывают насыщенным солевым раствором, сушат над безводным Na2SO4 или MgSO4, фильтруют или декантируют и концентрируют при пониженном давлении. Способ 2: Неочищенную реакционную смесь очищают методом препаративной ВЭЖХ непосредственно или после добавления органического растворителя, такого как MeOH или DMF или водного буфера, такого как 50 мМ NH4OAc, с осуществлением сначала, или без, концентрирования смеси при пониженном давлении. Способ 3: Растворитель удаляют при пониженном давлении и остаток распределяют между органическим растворителем, таким как DCM или EtOAc (предпочтительно EtOAc) и водой. Слои разделяют и органический слой необязательно промывают насыщенным солевым раствором, сушат над безводным Na2SO4 или MgSO4, фильтруют или декантируют и концентрируют при пониженном давлении. Способ 4: Реакционную смесь разбавляют водой и полученное твердое вещество собирают вакуумным фильтрованием.
Иллюстрация Общей Процедуры K
Пример #K.1: N-((1S,3S,4R)-3-(3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)-4-метилциклопентил)-3,3,3-трифторпропан-1-сульфонамид

3,3,3-Трифторпропан-1-сульфонилхлорид (0,194 г, 0,987 ммоль, Matrix) добавляли по каплям к раствору TEA (0,31 мл, 2,2 ммоль) и 3-(3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)-4-метилциклопентанамина (0,28 г, 1,1 ммоль, Получение #53) в DMF (10 мл). Полученную смесь перемешивали при температуре окружающей среды примерно в течение 144 часов. Растворитель удаляли при пониженном давлении и остаток распределяли между EtOAc и водой (20 мл каждого). Слои разделяли и органический слой промывали насыщенным солевым раствором (30 мл), сушили над безводным MgSO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали с использованием Общей процедуры AA (Таблица 2, Способ 9, Rt = 17,7 мин, or = отрицательное) с получением N-((1S,3S,4R)-3-(3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)-4-метилциклопентил)-3,3,3-трифторпропан-1-сульфонамида (0,021 г, 4,6%) в виде белого твердого вещества: LC/MS (Таблица 1, Способ a) Rt = 1,79 мин; MS m/z 416 (M+H)+.
Примеры, полученные из (1S,3R,4S)-3-метил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентанамина (Получение #19.1) с использованием Общей Процедуры K
(Таблица 1, Способ)
Пример, полученный из 1-(цис)-4-метилпиперидин-3-ил)-6H-имидазо[1,5-a]пирроло[2,3-e]пиразингидрохлорида (получен с использованием D из Получения #Q.1 с NaOH и E с 4 н HCl в 1,4-диоксане) с использованием Общей Процедуры K
(Таблица 1, Способ)
Примеры, полученные из 4-(3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)бицикло[2,2,2]октан-1-амингидробромида (Получение #F.1) с использованием Общей Процедуры K
(Таблица 1, Способ)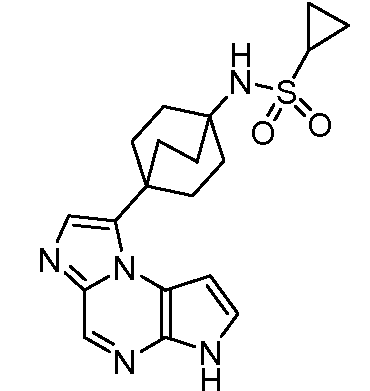
Примеры, полученные из (3S,5R)-5-этил-1-(3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)пирролидин-3-амина (Получение #TTT.1) с использованием Общей Процедуры K
(Таблица 1, Способ)
Примеры, полученные из (1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентанамина (получен из Примера #8 Стадия M с использованием D) с использованием Общей Процедуры K
(Таблица 1, Способ)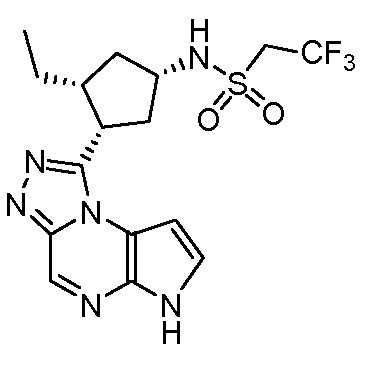
Общая процедура K.1: Образование сульфонамида из амина или азот-содержащего гетероцикла (дополнительное количество условия)
К смеси амина, соли амина или азот-содержащего гетероцикла (предпочтительно 1 экв.) в органическом растворителе, таком как ТГФ, DMA, DCM или DMF (предпочтительно DMF) добавляют органическое основание, такое как TEA или DIEA (1-10 экв., предпочтительно 2-4 экв.) или водный раствор основания, такой как насыщенный водный раствор NaHCO3 (5-20 экв., предпочтительно 5-10 экв.), или неорганическое основание, такое как NaH (1-10 экв., предпочтительно 1-3 экв.), и сульфонилхлорид (0,9-3 экв., предпочтительно 1-1,5 экв.). Реакционную смесь перемешивают при около -10-25°C (предпочтительно при около 0°C) примерно в течение 5 минут - 150 часов (предпочтительно около 90 мин). Необязательно, дополнительное количество основания (1-10 экв.) и/или сульфонилхлорида (0,4-2 экв.) можно добавить в любой момент в процессе реакции. В случаях, когда присутствует галоген, галоген может быть удален, и можно получить алкен. Реакционную смесь обрабатывают с использованием одного из следующих способов. Способ 1: Реакционную смесь разбавляют водой и экстрагируют органическим растворителем, таким как DCM или EtOAc. Объединенные органические слои необязательно промывают насыщенным водным раствором основания и насыщенным солевым раствором, сушат над безводным Na2SO4 или MgSO4, фильтруют или декантируют и концентрируют при пониженном давлении. Способ 2: Неочищенную реакционную смесь очищают методом препаративной ВЭЖХ непосредственно или после добавления органического растворителя, такого как MeOH или DMF, или водного буфера, такого как 50 мМ NH4OAc, с концентрированием сначала смеси при пониженном давлении или без концентрирования. Способ 3: Растворитель удаляют при пониженном давлении и остаток распределяют между органическим растворителем, таким как DCM или EtOAc (предпочтительно EtOAc) и водой. Слои разделяют и органический слой необязательно промывают насыщенным солевым раствором, сушат над безводным Na2SO4 или MgSO4, фильтруют или декантируют и концентрируют при пониженном давлении. Способ 4: Реакционную смесь разбавляют водой и полученное твердое вещество собирают вакуумным фильтрованием.
Иллюстрация Общей Процедуры K.1
Получение #K.1: N-((1S,3R,4S)-3-этил-4-(5-нитро-1-тозил-1H-пирроло[2,3-b]пиридин-4-иламино)циклопентил)циклопропансульфонамид

К раствору N-((1S,3R,4S)-3-этил-4-(5-нитро-1H-пирроло[2,3-b]пиридин-4-иламино)циклопентил)циклопропансульфонамида (Пример #23, Стадия г) (0,123 г, 0,314 ммоль) в DMF (3,0 мл) при около 0°C добавляли NaH (60% в минеральном масле, 0,015 г, 0,37 ммоль). Реакционную смесь перемешивали примерно в течение 5 минут. Добавляли 4-метилбензол-1-сульфонилхлорид (0,060 г, 0,314 ммоль) и реакционную смесь перемешивали примерно в течение 30 минут. Добавляли NaH (60% в минеральном масле, 0,007 г, 0,18 ммоль) и реакционную смесь перемешивали примерно в течение 10 минут. Добавляли NaH (60% в минеральном масле, 0,005 г, 0,12 ммоль) и реакционную смесь перемешивали примерно в течение 15 минут. Добавляли 4-метилбензол-1-сульфонилхлорид (0,012 г, 0,063 ммоль) и реакционную смесь перемешивали примерно в течение 40 минут. Реакционную смесь концентрировали при пониженном давлении. Остаток растворяли в EtOAc (25 мл) и промывали водой (15 мл). Органический слой отделяли, сушили над безводным MgSO4, фильтровали и концентрировали при пониженном давлении с получением N-((1S,3R,4S)-3-этил-4-(5-нитро-1-тозил-1H-пирроло[2,3-b]пиридин-4-иламино)циклопентил)-циклопропансульфонамида (0,218 г) в виде красновато-оранжевого масла, содержащего 40 моль% DMF и 1 экв. EtOAc: LC/MS (Таблица 1, Способ n) Rt = 0,88 мин; MS m/z 548 (M+H)+.
Общая процедура L: Замещение арила или гетероарилгалогенида амином
В микроволновой сосуд или круглодонную колбу добавляют амин или соль амина (предпочтительно 1 экв.), арил или гетероарилгалогенид (1-10 экв., предпочтительно 1,5 экв.), растворитель (такой как MeCN, n-PrOH, n-BuOH, толуол, DMSO, DMF или EtOH, предпочтительно n-PrOH[микроволновой нагрев] или DMF [термонагревание]) и основание (такое как K2CO3, Na2CO3, TEA или DIEA, предпочтительно TEA, DIEA или K2CO3, 1-5 экв., предпочтительно 2-4 экв.). Реакционную смесь нагревают при около 40-220°C термически (предпочтительно около 65°C) примерно в течение 0,5-16 часов (предпочтительно около 8,5 часов) или подвергают микроволновому нагреванию при около 100-200°C (предпочтительно около 130-150°C) примерно в течение 0,5-8 часов (предпочтительно около 0,5-2 часов). В случаях, когда реакция не достигает завершения, согласно данным ТСХ, LC/MS или ВЭЖХ, реакционную смесь можно снова подвергнуть термонагреванию при около 40-220°C (предпочтительно около 65°C) примерно в течение 0,5-8 часов (предпочтительно около 1-2 часов) или микроволновому нагреванию при около 120-200°C (предпочтительно около 130-150°C) еще в течение около 1-8 часов (предпочтительно около 0,5-2 часов) с необязательным добавлением дополнительного количества арила или гетероарилгалогенида (1-10 экв., предпочтительно 1,5 экв.) и/или основания (такого как K2CO3, Na2CO3, TEA или DIEA, предпочтительно TEA, DIEA или K2CO3, 1-5 экв., предпочтительно 2-4 экв.). Этот процесс повторяют вплоть до прекращения развития реакции. После охлаждения до температуры окружающей среды реакционную смесь обрабатывают с использованием одного из следующих способов. Способ 1: Реакционную смесь концентрируют при пониженном давлении. Способ 2: реакционную смесь, содержащую осадок, можно отфильтровать с получением целевого соединения, с необязательной промывкой органическим растворителем или растворителями, такими как Et2O, DCM и/или петролейный эфир. Способ 3: Реакционную смесь разбавляют органическим растворителем, таким как MeOH, добавляют силикагель и смесь концентрируют при пониженном давлении для подготовки к разделению хроматографией с твердой нагрузкой. Способ 4: Реакционную смесь концентрируют при пониженном давлении перед добавлением органического растворителя, такого как EtOAc или DCM, и затем, необязательно, промывают водой и/или насыщенным солевым раствором, сушат над безводным Na2SO4 или MgSO4, фильтруют или декантируют и концентрируют при пониженном давлении. Способ 5: добавляют органический растворитель, такой как EtOAc или DCM, с необязательным добавлением воды или насыщенного солевого раствора и слои разделяют. Водный слой затем, необязательно, экстрагируют дополнительным количеством органического растворителя, такого как EtOAc или DCM. Объединенные органические слои необязательно промывают насыщенным солевым раствором или водой, сушат над безводным MgSO4 или Na2SO4, фильтруют или декантируют и концентрируют при пониженном давлении.
Иллюстрация Общей Процедуры L
Получение #L.1: (S)-5-(3-((6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)метил)пирролидин-1-ил)пиразин-2-карбонитрил
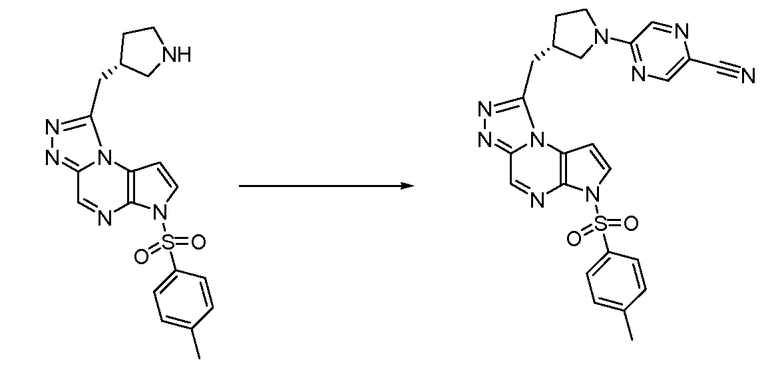
Смесь (S)-1-(пирролидин-3-илметил)-6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразина (0,160 г, 0,404 ммоль, получали с использованием B из Получения #A,1 и E с HCl), 2-хлор-5-цианопиразина (0,084 г, 0,60 ммоль, ArkPharm) и DIEA (0,28 мл, 1,6 ммоль) в n-PrOH (2,0 мл) нагревали в микроволновом устройстве CEM при около 150°C примерно в течение 30 минут (максимальное давление 250 ф/дюйм2 (17,58 кг/см2), максимальное линейное изменение 10 минут, максимальная мощность 200 Ватт). Реакционную смесь охлаждали до температуры окружающей среды и добавляли DCM (20 мл). Раствор промывали водой (20 мл) и насыщенным солевым раствором (20 мл). Органический слой отделяли, сушили над безводным MgSO4, фильтровали и концентрировали при пониженном давлении. Остаток брали для поглощения в DCM (10 мл), адсорбировали на силикагель (1 г) и очищали хроматографией на силикагеле с использованием в качестве элюента 100% EtOAc с получением розового твердого вещества. Вещество растирали в порошок со смесью EtOAc (10 мл) и 10% MeOH в DCM (10 мл). Нерастворимые вещества собирали фильтрованием с получением (S)-5-(3-((6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)метил)пирролидин-1-ил)пиразин-2-карбонитрила в виде не совсем белого твердого вещества (0,056 г, 27%): LC/MS (Таблица 1, Способ c) Rt = 1,34 мин; MS m/z: 500 (M+H)+.
Примеры, полученные из 1-(цис)-4-метилпиперидин-3-ил)-6H-имидазо[1,5-a]пирроло[2,3-e]пиразингидрохлорида (получен с использованием D из Получения #Q.1 с NaOH и E с 4 н HCl в 1,4-диоксане) с использованием Общей Процедуры L
(Таблица 1, Способ)
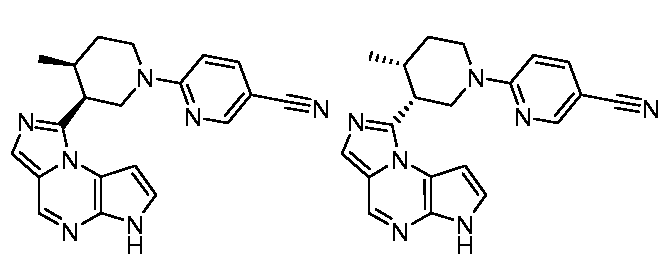

Пример, полученный из 1-((3R,4R)-4-метилпиперидин-3-ил)-6H-имидазо[1,5-a]пирроло[2,3-e]пиразингидрохлорида (Пример #5, Стадия J) с использованием Общей Процедуры L
(Таблица 1, Способ)
Примеры, полученные из (R)-8-(пиперидин-3-ил)-3H-имидазо[1,2-a]пирроло[2,3-e]пиразингидробромида и (S)-8-(пиперидин-3-ил)-3H-имидазо[1,2-a]пирроло[2,3-e]пиразингидробромида (Пример #3, Стадия G) с использованием Общей Процедуры L
(Таблица 1, Способ)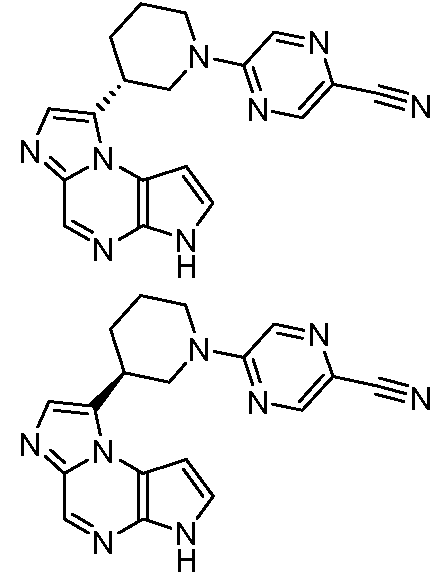
Общая процедура M: Boc-защита амина
К раствору амина или соли амина (предпочтительно 1 экв.) в органическом растворителе (например, MeCN, 1,4-диоксан или ТГФ, предпочтительно ТГФ) добавляют водный раствор основания, такого как Na2CO3, NaOH, K2CO3 или NaHCO3 (2-20 экв., предпочтительно 2-10 экв. Na2CO3) или органическое основание, такое как TEA или DIEA (1-5 экв., предпочтительно 1-2 экв. TEA), с последующим добавлением ди-трет-бутилдикарбоната (1-3,0 экв., предпочтительно 1,2 экв.). Добавление основания необязательно, если соль амина не используют. Реакционную смесь перемешивают при около 10-40°C (предпочтительно температуре окружающей среды) примерно в течение 2-24 часов (предпочтительно около 2-6 часов) и обрабатывают с использованием одного из следующих способов. Способ 1: добавлют органический растворитель (такой как Et2O, EtOAc или DCM) и воду и слои разделяют. Водный слой экстрагируют дополнительным количеством органического растворителя и объединенные органические слои, необязательно, промывают насыщенным солевым раствором, сушат над безводным Na2SO4 или MgSO4 и затем декантируют или фильтруют перед концентрированием при пониженном давлении. Способ 2: Реакционную смесь распределяют между органическим растворителем (таким как Et2O, EtOAc или DCM) и водным раствором кислоты (такой как HCl). Кислотный слой экстрагируют дополнительным количеством органического растворителя и объединенные органические слои, необязательно, промывают насыщенным солевым раствором. Органический слой необязательно сушат над безводным Na2SO4 или MgSO4 и затем декантируют или фильтруют перед концентрированием при пониженном давлении.
Иллюстрация Общей Процедуры M
Получение #M.1*: (1R,3S)-3-((трет-бутоксикарбониламино)метил)циклопентанкарбоновая кислота
К раствору (1R,3S)-3-(аминометил)циклопентанкарбоновой кислоты (0,500 г, 3,49 ммоль, AFID) в ТГФ (4 мл) и воде (4 мл) добавляли Na2CO3 (1,11 г, 10,5 ммоль) и ди-трет-бутилдикарбонат (0,915 г, 4,19 ммоль). Реакционную смесь перемешивали при температуре окружающей среды примерно в течение 4 часов. Добавляли EtOAc (15 мл) и водный раствор HCl (1 н, 15 мл) и слои разделяли. Водный слой экстрагировали при помощи EtOAc (2×10 мл) и объединенные органические слои промывали насыщенным солевым раствором (10 мл). Органический слой сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении с получением (1R,3S)-3-((трет-бутоксикарбониламино)метил) циклопентанкарбоновой кислоты (0,300 г, 35%). 1H ЯМР (DMSO-d 6) δ 11,97 (с, 1H), 6,83 (с, 1H), 2,89-2,86 (т, J=8,0 Гц, 2H), 2,73-2,58 (м, 1H), 2,04-1,87 (м, 2H), 1,82-1,68 (м, 2H), 1,68-1,58 (м, 1H), 1,37 (с, 9H), 1,34-1,19 (м, 2H).
Общая процедура M.1: Boc-защита азот-содержащего соединения
К азот-содержащему соединению (предпочтительно 1 экв.) в органическом растворителе (например, DCM, MeCN, 1,4-диоксан или ТГФ, предпочтительно DCM) добавляют водный раствор основания, такого как Na2CO3, NaOH, K2CO3 или NaHCO3 (предпочтительно Na2CO3, 2-20 экв., предпочтительно 2-10 экв.) или органическое основание, такое как TEA или DIEA (предпочтительно TEA, 1-5 экв., предпочтительно 1-2 экв.) с последующим добавлением ди-трет-бутилдикарбоната (1-3 экв., предпочтительно 1,2 экв.). К реакционной смеси, необязательно, добавляют DMAP (0,1- 2 экв., предпочтительно 0,1 экв.). Реакционную смесь перемешивают при около 10-40°C (предпочтительно при комнатной температуре) примерно в течение 0,5-24 часов (предпочтительно около 1 часа) и обрабатывают с использованием одного из следующих способов. Способ 1: Добавляют органический растворитель (такой как Et2O, EtOAc или DCM) и воду и слои разделяют. Водный слой необязательно экстрагируют дополнительным количеством органического растворителя и объединенные органические слои, необязательно, промывают насыщенным солевым раствором, сушат над безводным Na2SO4 или MgSO4 и затем декантируют или фильтруют перед концентрированием при пониженном давлении. Способ 2: Реакционную смесь распределяют между органическим растворителем (таким как Et2O, EtOAc или DCM) и водным раствором кислоты (такой как HCl). Кислотный слой экстрагируют дополнительным количеством органического растворителя и объединенные органические слои, необязательно, промывают насыщенным солевым раствором. Органический слой необязательно сушат над безводным Na2SO4 или MgSO4 и затем декантируют или фильтруют перед концентрированием при пониженном давлении. Способ 3: Добавляют воду или водный раствор (такой как насыщенный солевой раствор) и слои разделяли. Водный слой, необязательно, экстрагируют дополнительным количеством органического растворителя (такого как Et2O, EtOAc или DCM) и объединенные органические слои, необязательно, промывают насыщенным солевым раствором, сушат над безводным Na2SO4 или MgSO4 и затем декантируют или фильтруют перед концентрированием при пониженном давлении.
Иллюстрация Общей Процедуры M.1
Получение #M.1.1: трет-бутил циклопропилсульфонил(цис-3-метил-4-пропионилциклопентил)карбамат
К раствору N-(цис-3-метил-4-пропионилциклопентил)циклопропансульфонамида (2,70 г, 10,4 ммоль, получали с использованием H из цис-4-(циклопропансульфонамидо)-2-метилциклопентанкарбоновой кислоты (WO2009152133) с N,O-диметилгидроксиламин хлористоводородной кислотой, MMMM с этилмагнийхлоридом) в DCM (52 мл) добавляли TEA (1,60 мл, 11,5 ммоль), ди-трет-бутил дикарбонат (2,90 мл, 12,5 ммоль) и DMAP (0,127 г, 1,04 ммоль). Реакционную смесь перемешивали при комнатной температуре примерно в течение 1 часа. Добавляли воду (50 мл) и слои разделяли. Водный слой экстрагировали при помощи DCM (3×30 мл) и объединенный органический слой концентрировали при пониженном давлении. Продукт очищали хроматографией на силикагеле с градиентным элюированием 0-50% EtOAc в гептане с получением трет-бутил циклопропилсульфонил(цис-3-метил-4-пропионилциклопентил)карбамата (3,71 г, 99%) в виде белого твердого вещества: LC/MS (Таблица 1, Способ b) Rt = 2,62 мин; MS m/z: 360 (M+H)+.
Общая процедура N: Cbz-защита амина
Раствор амина или соли амина (предпочтительно 1 экв.) и основания (например, Na2CO3 или NaOH, 1-3 экв., предпочтительно Na2CO3, 1,6 экв.) в воде или водный раствор органического растворителя (например, вода/1,4-диоксан или вода/MeCN, предпочтительно вода/1,4-диоксан) перемешивают при температуре окружающей среды примерно в течение 1-10 минут (предпочтительно 5 мин). К реакционной смеси добавляют раствор бензил 2,5-диоксопирролидин-1-илкарбоната (1-2 экв., предпочтительно 1,0 экв.) в органическом растворителе, таком как 1,4-диоксан или MeCN. Реакционную смесь перемешивают при температуре окружающей среды примерно в течение 8-144 часов (предпочтительно около 72 часов). Необязательно, реакционную смесь концентрируют при пониженном давлении. Полученный водный раствор разбавляют органическим растворителем (таким как EtOAc или DCM). Органические экстракты, необязательно, промывают водой и/или насыщенным солевым раствором, сушат над безводным Na2SO4 или MgSO4, фильтруют или декантируют и концентрируют при пониженном давлении. Альтернативно, полученный водный раствор подкисляют путем добавления кислоты, такой как водный раствор NH4Cl или HCl, и затем экстрагируют органическим растворителем (таким как EtOAc или DCM).
Иллюстрация Общей Процедуры N
Получение #N.1: метил 4-(бензилоксикарбониламино)бицикло[2,2,2]октан-1-карбоксилат

К раствору метил гидрохлорида 4-аминобицикло[2,2,2]октан-1-карбоксилата (1,16 г, 5,29 ммоль, Prime Organics) в 1,4-диоксане (15 мл) добавляли раствор Na2CO3 (0,90 г, 8,49 ммоль) в воде (15 мл). Реакционную смесь перемешивали примерно в течение 5 минут при температуре окружающей среды. Добавляли бензил 2,5-диоксопирролидин-1-ил карбонат (1,32 г, 5,29 ммоль) и реакционную смесь перемешивали при температуре окружающей среды примерно в течение 72 часов. Реакционную смесь разбавляли при помощи EtOAc (50 мл). Слои разделяли и водный слой экстрагировали при помощи EtOAc (2×20 мл). Объединенные органические слои сушили над безводным MgSO4, фильтровали и концентрировали при пониженном давлении с получением метил 4-(бензилоксикарбониламино)бицикло[2,2,2]октан-1-карбоксилата (1,68 г, 95%): LC/MS (Таблица 1, Способ a) Rt = 2,44 мин; MS m/z: 318 (M+H)+.
Общая процедура O: Восстановление пиридина
Замещенный пиридин (предпочтительно 1 экв.) растворяют в органическом растворителе (таком как AcOH, EtOH или MeOH; предпочтительно AcOH, если используют встряхивающее устройство Парра, или EtOH, если используют H-cube™). Подходящий катализатор, такой как оксид платины (IV) или Pd/C (0,05-0,20 экв., предпочтительно 0,05-0,10 экв. оксида платины (IV) для реакции с использованием встряхивающего устройства Парра или ThalesNano CatCart® картриджи с 10% масс. Pd/C катализатора для H-Cube™) используют для восстановления в атмосфере водорода при около 15-1450 ф/дюйм2 (1,055-115,0 кг/см2) (предпочтительно около 220 ф/дюйм2 (15,5 кг/см2) для встряхивающего устройства Парра, или предпочтительно 1305 ф/дюйм2 (91,5 кг/см2) для H-Cube™). Реакцию осуществляют примерно в течение 1-10 дней (предпочтительно примерно 3-5 дней) при около 20-100°C (предпочтительно около 25°C) для встряхивающего устройства Парра, или при около 1-3 мл/мин (предпочтительно 1 мл/мин) при около 25-100°C (предпочтительно около 80°C) примерно в течение 1-10 часов (предпочтительно около 3 часов) для H-Cube™. Реакционную смесь фильтруют через Целит®, если реакцию осуществляют во встряхивающем устройстве Парра, и концентрируют при пониженном давлении в любом случае.
Иллюстрация Общей Процедуры O
Получение #O.1: цис-4-(трифторметил)пиперидин-3-карбоновая кислота

Раствор 4-(трифторметил)никотиновой кислоты (1,50 г, 7,85 ммоль) в EtOH (78 мл) пропускали через H-cube™, снабженный ThalesNano CatCart® картриджем с 10% масс. Pd/C катализатора, при около 1,0 мл/мин, при около 80°C, под давлением водорода около 1305 ф/дюйм2 (91,5 кг/см2). Примерно через 3 часа растворитель удаляли при пониженном давлении с получением цис-4-(трифторметил)пиперидин-3-карбоновой кислоты (1,55 г, 100% неочищенн): LC/MS (Таблица 1, Способ b) Rt = 0,54 мин; MS m/z: 198 (M+H)+.
Общая процедура P: Восстановление карбонила до спирта
Восстановитель (1,0-3,0 экв., предпочтительно 1,25 экв.), такой как LAH, DIBAL-H, NaBH4 или LiBH4 (предпочтительно DIBAL-H), добавляют либо по порциям в виде твердого вещества, либо по каплям в виде раствора в органическом растворителе (таком как ТГФ, Et2O, EtOH или MeOH, предпочтительно ТГФ) к раствору карбонильного соединения (предпочтительно 1 экв.) в органическом растворителе (таком как ТГФ, Et2O, EtOH или MeOH, предпочтительно MeOH) при около -40-50°C (предпочтительно при температуре окружающей среды). Реакционную смесь перемешивают примерно в течение 1-20 часов (предпочтительно около 16 часов) перед гашением водным раствором (таким как NH4Cl или NaHCO3, предпочтительно насыщенным водным раствором NH4Cl). Реакционную смесь перемешивают примерно в течение 10 минут - 3 часов (предпочтительно около 20-30 мин) и затем раствор разделяют с использованием органического растворителя (такого как EtOAc, Et2O или DCM, предпочтительно Et2O). Органический слой промывают насыщенным солевым раствором, сушат над безводным Na2SO4 или MgSO4, фильтруют и концентрируют при пониженном давлении.
Иллюстрация Общей Процедуры P
Получение #P.1: этил 2-этил-4-гидроксициклопентанкарбоксилат
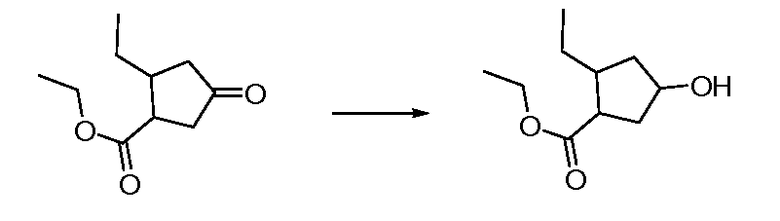
К этил 2-этил-4-оксоциклопентанкарбоксилату (10 г, 54,3 ммоль, Пример #8, Стадия г) в MeOH (143 мл) добавляли NaBH4 (2,57 г, 67,8 ммоль) по порциям. Полученную суспензию перемешивали примерно в течение 16 часов при температуре окружающей среды, затем добавляли насыщенный водный раствор NH4Cl (240 мл). Реакционную смесь перемешивали примерно в течение 20 минут, затем раствор разделяли с использованием Et2O (300 мл). Органический слой отделяли и водный слой промывали при помощи Et2O (2×150 мл). Объединенные органические слои промывали насыщенным солевым раствором (100 мл), сушили над безводным MgSO4, фильтровали и концентрировали при пониженном давлении. Продукт очищали хроматографией на силикагеле (220 г) с градиентным элюированием 30-70% EtOAc в гептане с получением этил 2-этил-4-гидроксициклопентанкарбоксилата (8,51 г, 84%, преимущественно (1S,2R,4S)-этил 2-этил-4-гидроксициклопентанкарбоксилат и (1R,2S,4R)-этил 2-этил-4-гидроксициклопентанкарбоксилат) в виде прозрачного масла: LC/MS (Таблица 1, Способ b) Rt = 2,02 мин; MS m/z: 187 (M+H)+.
Примеры, полученные с использованием Общей Процедуры P с DIBAL-H
(Таблица 1, Способ)
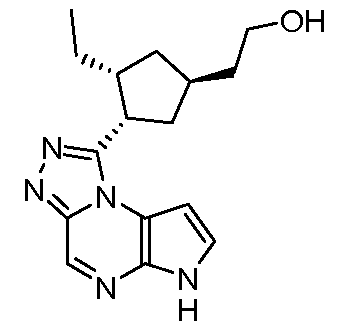
Общая процедура Q: Циклизация амида с использованием дитиадифосфетанового реагента
К раствору амида (предпочтительно 1 экв.) в органическом растворителе (предпочтительно 1,4-диоксан) добавляют дитиадифосфетановый реагент, такой как реагент Лоуссона или реагент Belleau (2,4-бис(4-феноксифенил)-1,3-дитиа-2,4-дифосфетан-2,4-дисульфид) (предпочтительно реагент Лоуссона) (0,5-2,0 экв., предпочтительно 0,6 экв.). Реакционную смесь нагревают при около 25-120°C (предпочтительно около 80°C) примерно в течение 0,5-10 часов (предпочтительно около 1 часа). Реакционную смесь охлаждают до температуры окружающей среды и, необязательно, концентрируют при пониженном давлении с получением остатка. Реакционную смеси или остаток распределяют между органическим растворителем (таким как DCM или EtOAc, предпочтительно EtOAc) и водой, водным раствором основания (такого как насыщенный водный раствор NaHCO3) или насыщенным солевым раствором. Слои разделяют и органический слой, необязательно, промывают водой, водным раствором основания (таким как насыщенный водный раствор NaHCO3) и/или насыщенным солевым раствором, сушат над безводным Na2SO4 или MgSO4, фильтруют и концентрируют при пониженном давлении с получением тиоамида. К раствору тиоамида (предпочтительно 1 экв.) в органическом растворителе (предпочтительно 1,4-диоксан) добавляют кислоту Льюиса, такую как диацетоксиртуть, дихлорид ртути, трифторацетат ртути (II), трифторацетат серебра, нитрат серебра, бромид меди (предпочтительно диацетоксиртуть или трифторацетат ртути (II)) (1-3 экв., предпочтительно 1 экв.). Реакционную смесь перемешивают при около 20-60°C (предпочтительно при температуре окружающей среды) примерно в течение 0,5-4 часов (предпочтительно около 1 часа). Необязательно, добавляли дополнительное количество кислоты Льюиса (предпочтительно диацетоксиртути или трифторацетата ртути (II)) (0,2-1,0 экв., предпочтительно 0,6 экв.) и реакцию продолжалают примерно в течение 10 минут - 3 часов (предпочтительно около 15 минут). Реакционную смесь необязательно разбавляют насыщенным раствором тиосульфата натрия, водой и/или органическим растворителем (предпочтительно EtOAc) и фильтруют, предпочтительно через слой Целита®. Слой Целита® можно промыть дополнительным количеством органического растворителя (предпочтительно EtOAc или DCM). Фильтрат концентрируют при пониженном давлении. Неочищенное вещество, необязательно, распределяют между органическим растворителем (таким как EtOAc или DCM) и промывают насыщенным раствором тиосульфата натрия и/или водой, водным раствором основания (такого как насыщенный водный раствор NaHCO3) и/или насыщенным солевым раствором, сушат над безводным Na2SO4 или MgSO4, фильтруют и концентрируют при пониженном давлении.
Иллюстрация Общей Процедуры Q
Получение #Q.1: цис-трет-бутил 4-метил-3-(6-тозил-6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)пиперидин-1-карбоксилат

В круглодонную колбу загружали цис-трет-бутил 4-метил-3-((5-тозил-5H-пирроло[2,3-b]пиразин-2-ил)метилкарбамоил)пиперидин-1-карбоксилат (5,62 г, 10,6 ммоль, получали с использованием O из 4-метилникотиновой кислоты, M, H из Примера #5, Стадия C, HATU и DIEA) и реагент Лоуссона (3,0 г, 7,4 ммоль) в 1,4-диоксане (100 мл). Реакционную смесь нагревали при около 80°C примерно в течение 1 часа, охлаждали до температуры окружающей среды и концентрировали при пониженном давлении. Неочищенный продукт растворяли в EtOAc (200 мл) и промывали насыщенным водным раствором NaHCO3 (3×100 мл). Органический слой отделяли, сушили над безводным MgSO4, фильтровали и концентрировали при пониженном давлении. Вещество очищали хроматографией на силикагеле с градиентным элюированием 0-5% MeOH в DCM с получением цис-трет-бутил 4-метил-3-((5-тозил-5H-пирроло[2,3-b]пиразин-2-ил)метилкарбамотиоил)пиперидин-1-карбоксилата (5,2 г, 90%): LC/MS (Таблица 1, Способ b) Rt = 2,65 мин; MS m/z: 544 (M+H)+. В круглодонную колбу загружали цис-трет-бутил 4-метил-3-((5-тозил-5H-пирроло[2,3-b]пиразин-2-ил)метилкарбамотиоил)пиперидин-1-карбоксилат (2,6 г, 4,8 ммоль) и трифторацетат ртути (II) (2,1 г, 4,8 ммоль) в 1,4-диоксане (72 мл) и перемешивали при температуре окружающей среды примерно в течение 1 часа. Реакционную смесь фильтровали через слой Целита®. Слой Целита® промывали при помощи DCM (30 мл) и EtOAc (30 мл). Фильтрат концентрировали при пониженном давлении. Остаток растворяли в DCM (50 мл) и промывали насыщенным водным раствором тиосульфата натрия (10 мл) и насыщенным водным раствором NaHCO3 (25 мл), сушили над безводным MgSO4, фильтровали и концентрировали при пониженном давлении. Вещество очищали хроматографией на силикагеле с градиентным элюированием 0-5% MeOH в DCM с получением цис-трет-бутил-4-метил-3-(6-тозил-6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)пиперидин-1-карбоксилата (2,2 г, 90%): LC/MS (Таблица 1, Способ b) Rt = 2,57 мин; MS m/z: 510 (M+H)+.
Общая процедура R: Образование бромметилкетона из кислоты
К раствору карбоновой кислоты (предпочтительно 1 экв.) в органическом растворителе (DCM или DCE, предпочтительно DCM) медленно добавляют оксалилхлорид (1,2-3,0 экв., предпочтительно 2,2 экв.) с последующим добавлением по каплям DMF (0,01-0,20 экв., предпочтительно около 0,15 экв.). Реакционную смесь перемешивают при около 0-40°C (предпочтительно при температуре окружающей среды) примерно в течение 3-24 часов (предпочтительно около 14 часов), затем смесь концентрируют при пониженном давлении до постоянной массы с получением неочищенного хлорангидрида кислоты. Раствор неочищенного хлорангидрида кислоты (предпочтительно 1 экв.) в органическом растворителе (таком как ТГФ, MeCN, Et2O или ТГФ/MeCN, предпочтительно ТГФ/MeCN) добавляют к триметилсилилдиазометану (2,0 M в Et2O) или раствору диазометана в Et2O (получен из Diazald® в соответствии с протоколом Aldrich или J. Chromatogr. Sci. 1991, 29, 8) (2-10 экв., предпочтительно 3,5 экв. триметилсилилдиазометана) при около -20-20°C (предпочтительно около 0°C) в подходящем органическом растворителе, таком как ТГФ, MeCN, Et2O или ТГФ/MeCN (предпочтительно ТГФ/MeCN). Реакционную смесь перемешивают примерно в течение 0,5-5 часов (предпочтительно около 3 часов) при около -20-20°C (предпочтительно около 0°C), затем добавляют по каплям 48% водный раствор HBr (5-40 экв., предпочтительно около 10 экв.). Примерно через 0-30 минут (предпочтительно около 5 мин) реакционную смесь можно концентрировать досуха с получением желаемого продукта, нейтрализовать путем добавления по каплям насыщенного водного раствора NaHCO3 или, необязательно, промыть насыщенным солевым раствором после необязательного добавления органического растворителя (такого как EtOAc или DCM, предпочтительно EtOAc). В случаях, когда реакционную смесь подвергают водной обработке, органический слой сушат над безводным Na2SO4 или MgSO4 (предпочтительно MgSO4), фильтруют и концентрируют при пониженном давлении.
Иллюстрация Общей Процедуры R
Получение #R.1: 2-бром-1-(4-(дибензиламино)-2-метилциклопентил)этанон

К раствору 4-(дибензиламино)-2-метилциклопентанкарбоновой кислоты (7,34 г, 22,7 ммоль, Получение #TT.1) в DCM (100 мл) медленно добавляли оксалилхлорид (4,37 мл, 49,9 ммоль) с последующим добавлением по каплям DMF (0,26 мл, 3,4 ммоль). Смесь перемешивали при температуре окружающей среды примерно в течение 14 часов и растворитель удаляли при пониженном давлении с получением 4-(дибензиламино)-2-метилциклопентанкарбонилхлорида в виде бежевого твердого вещества. Твердое вещество растворяли в ТГФ и MeCN (1:1, 100 мл) и добавляли к раствору триметилсилилдиазометана (2 M в Et2O, 39,7 мл, 79,4 ммоль) в 1:1 смеси ТГФ и MeCN (100 мл) при около 0°C. Полученную смесь перемешивали при около 0°C примерно в течение 3 часов и затем гасили путем добавления по каплям 48% водного раствора HBr (25 мл, 221 ммоль). Полученную смесь нейтрализовали путем добавления по каплям насыщенного водного раствора NaHCO3 (300 мл) и слои разделяли. Органический слой сушили над безводным MgSO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали флэш-хроматографией на силикагеле с градиентным элюированием 5-45% EtOAc в гептане с получением 2-бром-1-(4-(дибензиламино)-2-метилциклопентил)этанона (6,3 г, 69%) в виде желтого масла: LC/MS (Таблица 1, Способ a) Rt = 2,90 мин; MS m/z 400, 402 (1:1) (M+H)+.
Общая процедура S: N-Алкилирование с использованием алкилгалогенида или α-галогенкетона
В круглодонную колбу загружают основание, такое как NaH (60% дисперсия в минеральном масле), K2CO3 или Cs2CO3 (предпочтительно NaH (60% дисперсия в минеральном масле) 0,9-1,5 экв., предпочтительно 0,95 экв.) и органический растворитель (такой как DMF или NMP, предпочтительно DMF). Смесь охлаждают до температуры от около -10°C до 10°C (предпочтительно около 0°C) и добавляют раствор подходяще замещенного амина (предпочтительно 1 экв.) в органическом растворителе (таком как DMF). Реакционную смесь перемешивают примерно в течение 5-90 минут (предпочтительно около 15-30 мин) при температуре от около -10°C до температуры окружающей среды (предпочтительно около 0°C) с последующим добавлением алкилгалогенида или α-галогенкетона (1-2 экв., предпочтительно 1,2 экв.). Альтернативно, раствор амина и основания в органическом растворителе можно добавить к раствору алкилгалогенида или α-галогенкетона в органическом растворителе при около 0°C. Реакционную смесь перемешивают при температуре от около -10°C до температуры окружающей среды (предпочтительно при температуре окружающей среды) примерно в течение 0,5-2 часов (предпочтительно около 1 часа). Органический растворитель удаляют при пониженном давлении. Необязательно, неочищенную смесь можно разбавить водой и органическим растворителем (например, EtOAc или DCM). Слои разделяют и водный слой экстрагируют снова органическим растворителем (такие как EtOAc и/или DCM). Объединенные органические слои необязательно промывают насыщенным солевым раствором, сушат над безводным MgSO4, фильтруют и концентрируют досуха при пониженном давлении.
Иллюстрация Общей Процедуры S
Получение #S.1: трет-бутил 2-(4-(дибензиламино)-2-метилциклопентил)-2-оксоэтил(5-тозил-5H-пирроло[2,3-b]пиразин-2-ил)карбамат

К суспензии NaH (60% дисперсия в минеральном масле, 0,058 г, 1,45 ммоль) в DMF (5 мл) добавляли раствор трет-бутил 5-тозил-5H-пирроло[2,3-b]пиразин-2-илкарбамата (0,59 г, 1,519 ммоль, Пример #3, Стадия E) в DMF (5 мл) по каплям при около 0°C. Полученную смесь перемешивали при этой температуре примерно в течение 30 минут и затем добавляли по каплям к раствору 2-бром-1-(4-(дибензиламино)-2-метилциклопентил)этанона (0,73 г, 1,823 ммоль, Получение #R,1) в DMF (10 мл). Полученную смесь перемешивали при около 0°C в течение примерно 1 часа и растворитель удаляли при пониженном давлении. Остаток распределяли между насыщенным водным раствором NaHCO3 и EtOAc (100 мл каждого). Органическую фазу сушили над безводным MgSO4 и концентрировали при пониженном давлении с получением трет-бутил 2-(4-(дибензиламино)-2-метилциклопентил)-2-оксоэтил(5-тозил-5H-пирроло[2,3-b]пиразин-2-ил)карбамата (1,04 г, 97%) в виде желтого аморфного твердого вещества: LC/MS (Таблица 1, Способ a) Rt = 3,30 мин; MS m/z 708 (M+H)+.
Общая процедура S.1: N-Алкилирование с использованием алкилгалогенид, α-галогенкетон или α-галогенамид
В круглодонную колбу загружают основание, такое как NaH (60% дисперсия в минеральном масле), K2CO3 или Cs2CO3 (предпочтительно NaH (60% дисперсия в минеральном масле) 0,9-1,5 экв., предпочтительно 0,95 экв.) и органический растворитель (такой как DMF, DCM, 1,4-диоксан или NMP, предпочтительно DMF). Смесь охлаждают до температуры от около -10°C до температуры окружающей среды (предпочтительно около 0°C) и добавляют раствор подходяще замещенного амина (предпочтительно 1 экв.) в органическом растворителе (таком как DMF). Альтернативно, основание можно добавить по порциям к раствору амина и органического растворителя при температуре от около 0°C до температуры окружающей среды. Реакционную смесь перемешивают примерно в течение 5-90 минут (предпочтительно около 15-30 мин) при около температуре от -10°C до температуры окружающей среды (предпочтительно около 0°C) с последующим добавлением алкилгалогенида, α-галогенкетона или α-галогенамида (1-2 экв., предпочтительно 1,2 экв.). Альтернативно, раствор амина и основания в органическом растворителе можно добавить к раствору алкилгалогенида, α-галогенкетона или α-галогенамида в органическом растворителе при около 0°C. Реакционную смесь перемешивают при температуре от около -10°C до температуры окружающей среды (предпочтительно при температуре окружающей среды) примерно в течение 0,5-24 часов (предпочтительно около 1 часа). Необязательно, органический растворитель можно удалить при пониженном давлении. Необязательно, реакционную смесь или остаток можно разбавить водой, водным раствором NH4Cl или водным раствором NaHCO3. Если происходит образование осадка, твердое вещество, необязательно, можно собирать путем вакуумного фильтрования с получением целевого соединения. Альтернативно, органический растворитель (такой как EtOAc или DCM) добавляют к водной смеси и слои разделяют. Водный слой, необязательно, можно далее экстрагировать органическим растворителем (таким как EtOAc и/или DCM). Объединенные органические слои необязательно промывают дополнительными водными растворами, такими как насыщенный солевой раствор, сушат над безводным Na2SO4 или MgSO4, фильтруют и концентрируют досуха при пониженном давлении.
Иллюстрация Общей Процедуры S.1
Получение #S.1.1: трет-бутил 2-амино-2-оксоэтил(5-тозил-5H-пирроло[3,2-b]пиразин-2-ил)карбамат

К раствору трет-бутил 5-тозил-5H-пирроло[3,2-b]пиразин-2-илкарбамата (1,00 г, 2,57 ммоль, Пример #3 Стадия E) и DMF (13 мл) в атмосфере азота при около 0°C добавляли NaH (60% дисперсия в минеральном масле, 0,113 г, 2,83 ммоль) одной порцией. Примерно через 30 минут добавляли 2-бромацетамид (0,391 г, 2,83 ммоль) одной порцией. Примерно через 30 минут ледяную баню удаляли и раствор перемешивали при температуре окружающей среды примерно в течение 2 часов. Добавляли насыщенный водный раствор NH4Cl/вода (1:1, 100 мл). После перемешивания примерно в течение 10 минут смесь фильтровали с использованием воды для промывки фильтровальной лепешки. Водную фазу экстрагировали при помощи EtOAc (50 мл). Фильтровальную лепешку растворяли в EtOAc и добавляли к органическому слою. Органический слой сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Вещество очищали хроматографией на силикагеле с градиентным элюированием 20-100% EtOAc/гептан с получением трет-бутил 2-амино-2-оксоэтил(5-тозил-5H-пирроло[3,2-b]пиразин-2-ил)карбамата (0,980 г, 82%): LC/MS (Таблица 1, Способ n) Rt = 0,70 мин; MS m/z 446 (M+H)+.
Общая процедура T: Циклизация кетона с использованием дитиафосфетанового реагента
К раствору кетона (предпочтительно 1 экв.) в органическом растворителе, таком как ТГФ или 1,4-диоксан (предпочтительно 1,4-диоксан) добавляют тиолирующий реагент, такой как реагент Лоуссона или реагент Belleau (2,4-бис(4-феноксифенил)-1,3-дитиа-2,4-дифосфетан-2,4-дисульфид) (0,5-2,0 экв., предпочтительно реагент Лоуссона, 0,5-0,6 экв.). Реакционную смесь нагревают при около 30°C to 120°C (предпочтительно около 60-70°C) примерно в течение 0,5-10 часов (предпочтительно около 1-2 часов). Необязательно, дополнительное количество тиолирующего реагента (0,5-2,0 экв., предпочтительно 0,5-0,6 экв.) можно добавить к реакционной смеси и нагревание можно продолжить примерно в течение 0,5-10 часов (предпочтительно около 1-2 часов). Реакционную смесь концентрируют при пониженном давлении.
Иллюстрация Общей Процедуры T
Получение #T.1: N,N-дибензил-3-метил-4-(3-тозил-3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)циклопентанамин

Смесь 1-(4-(дибензиламино)-2-метилциклопентил)-2-(5-тозил-5H-пирроло[2,3-b]пиразин-2-иламино)этанона (5,32 г, 8,75 ммоль, Получение #50) и реагент Лоуссона (1,88 г, 4,64 ммоль) в 1,4-диоксане (60 мл) нагревали при около 60°C примерно в течение 2 часов. Добавляли реагент Лоуссона (1,88 г, 4,64 ммоль) и перемешивание при около 60°C продолжали примерно в течение 1 часа. Растворитель удаляли и остаток подвергали флэш-хроматографии на силикагеле с градиентным элюированием 0-8% MeOH в DCM с получением N,N-дибензил-3-метил-4-(3-тозил-3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)циклопентанамина (4,47 г, 87%) в виде коричневого аморфного твердого вещества: LC/MS (Таблица 1, Способ a) Rt = 2,99 мин; MS m/z 590 (M+H)+.
Общая процедура U: Конденсация по методу Кневенагеля с образованием замещенного циклопентадиена
В круглодонную колбу загружают органический растворитель (например, ТГФ или диметиловый эфир диэтиленгликоля; предпочтительно ТГФ), с последующим добавлением по порциям NaH (60% дисперсия в минеральном масле, предпочтительно 1 экв.). Необязательно, можно добавить органический растворитель. Реакционную смесь охлаждают до около -15-5°C (предпочтительно около -10-0°C). β-кетоэфир (предпочтительно 1 экв.) добавляют по каплям при такой скорости, чтобы поддерживать внутреннюю температуру ниже около 10°C. Полученную смесь перемешивают при около 0-60°C (предпочтительно около 25°C) примерно в течение 0,1-2 часов (предпочтительно около 0,5 часов), с последующим добавлением по каплям подходяще замещенного α-галогенкетона (предпочтительно 0,45-0,55 экв.). Полученную смесь нагревают до около 40-80°C (предпочтительно около 50°C) примерно в течение 3-24 часов (предпочтительно около 19 часов). Органический растворитель удаляют при пониженном давлении и полученное неочищенное вещество перемешивают с водой при охлаждении на ледяной бане. Полученную суспензию фильтруют примерно через 0,5-3 часов (предпочтительно около 2 часов) и фильтровальную лепешку промывают водой и сушат в вакууме примерно в течение 1-3 часов (предпочтительно около 1 часа). Полученное твердое вещество суспендируют в органическом растворителе (предпочтительно Et2O), собирают вакуумным фильтрованием, промывают органическим растворителем (предпочтительно Et2O) и сушат в вакууме с получением желаемого продукта в виде натриевой соли енолятя. Необязательно, добавляют толуол и воду азеотропно отгоняют. Полученное твердое вещество снова суспендируют в органическом растворителе (предпочтительно Et2O), собирают вакуумным фильтрованием, промывают органическим растворителем (предпочтительно Et2O) и затем сушат в вакууме.
Иллюстрация Общей Процедуры U
Получение #U.1: натрия 4-(этоксикарбонил)-3-этил-2-(метоксикарбонил)циклопента-1,3-диенолят
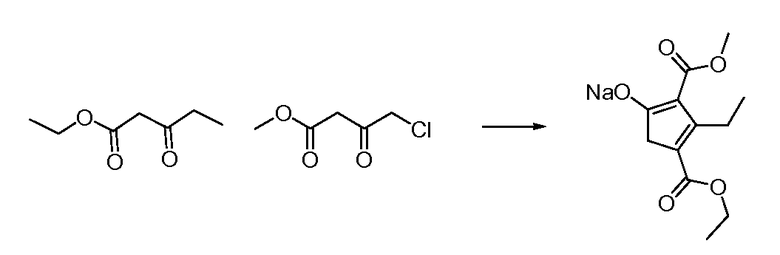
В круглодонную колбу загружали ТГФ (1,5 л) с последующим добавлением по порциям NaH (60% дисперсия в минеральном масле, 70,0 г, 1,75 моль). Добавляли дополнительное количество ТГФ (500 мл) и полученную смесь охлаждали до около -10°C. Добавляли этилпропионилацетат (250 мл, 1,80 моль) по каплям примерно в течение 1 часа для поддержания внутренней температуры ниже около 10°C. Полученную смесь перемешивали при температуре окружающей среды примерно в течение 0,5 часов с получением прозрачного желтого раствора и затем добавляли по каплям метил 4-хлорацетоацетат (100 мл, 0,88 моль) примерно в течение 5 минут. Полученную смесь нагревали при около 50°C примерно в течение 19 часов с получением красновато-оранжевой суспензии. Реакционную смесь охлаждали до температуры окружающей среды, концентрировали при пониженном давлении и полученную жидкость переносили в химический стакан и разбавляли водой (350 мл). Смесь перемешивали на ледяной бане примерно в течение 2 часов. Твердое вещество собирали вакуумным фильтрованием и фильтровальную лепешку промывали водой (150 мл) и сушили в вакууме примерно в течение 1 часа. Твердое вещество суспендировали в Et2O (1,5 л), фильтровали, промывали при помощи Et2O (1,5 л) и сушили в вакууме. Полученное твердое вещество подвергали азеотропной перегонке с толуолом (1 л) с получением твердого вещества, которое снова суспендировали в Et2O (1 л) и собирали вакуумным фильтрованием. Фильтровальную лепешку промывали при помощи Et2O (500 мл) и сушили в вакууме с получением 4-(этоксикарбонил)-3-этил-2-(метоксикарбонил)циклопента-1,3-диенолята натрия (204,2 г, 89%) в виде бежевого твердого вещества: 1H ЯМР (DMSO-d 6) δ 3,94 (кв., J=7,1 Гц, 2H), 3,46 (с, 3H), 3,04 (кв., J=7,2 Гц, 2H), 2,66 (с, 2H), 1,13 (т, J=7,1 Гц, 3H), 0,99 (т, J=7,3 Гц, 3H).
Общая процедура V: Декарбоксилирование β-кетоэфиренолята
В круглодонную колбу загружают подходящий β-кетоэфир или его натрийенолят (предпочтительно 1 экв.), органический растворитель (например, диметиловый эфир диэтиленгликоля или толуол, предпочтительно толуол), AcOH (2-5 экв., предпочтительно 3,5 экв.), NaI или KCl (1-5 экв., предпочтительно 1,4-1,5 экв. KCl) с использованием или без воды (предпочтительно с водой). Реакционную смесь нагревают до температуры кипения с обратным холодильником примерно в течение 1-10 часов (предпочтительно около 3-6 часов). Реакционную смесь охлаждают до температуры окружающей среды и добавляют по каплям в водный раствор NaHCO3 (предпочтительно 8-10% NaHCO3). Полученную смесь экстрагируют органическим растворителем, таким как Et2O или MTBE (предпочтительно MTBE). Объединенные органические слои сушат над безводным Na2SO4 или MgSO4, фильтруют и концентрируют досуха при пониженном давлении.
Иллюстрация Общей Процедуры V
Получение #V.1: этил 2-этил-4-оксоциклопент-2-енкарбоксилат

В 5-л круглодонную колбу загружали 4-(этоксикарбонил)-3-этил-2-(метоксикарбонил)циклопента-1,3-диенолят натрия (316 г, 1205 ммоль, Получение #U,1), KCl (126 г, 1687 ммоль, JT-Baker), AcOH (241 мл, 4218 ммоль, JT-Baker), толуол (1850 мл) и воду (130 мл). Реакционную смесь нагревали при температуре кипения с обратным холодильником примерно в течение 6 часов, затем охлаждали до температуры окружающей среды и добавляли по каплям к 8% водному раствору NaHCO3 (3,5 л). Полученную бифазную смесь экстрагировали при помощи MTBE (2×1,5 л). Объединенные органические слои промывали насыщенным солевым раствором (1 л), сушили над безводным MgSO4 и концентрировали при пониженном давлении с получением 191 г неочищенного вещества, который очищали перегонкой в вакууме (97-99°C, 0,600 мм Hg) с получением этил 2-этил-4-оксоциклопент-2-енкарбоксилата (160 г, 69%):1H ЯМР (CDCl3) δ 6,04 (м, 1H), 4,26-4,15 (м, 2H), 3,76-3,69 (м, 1H), 2,75-2,57 (м, 2H), 2,56-2,44 (м, 2H), 1,32-1,26 (м, 3H), 1,23-1,18 (м, 3H).
Общая процедура W: Гидрирование алкена
В круглодонную колбу загружают 10% масс. Pd/C (около 0,005-0,05 экв., предпочтительно 0,02 экв.). В колбе создают вакуум, затем продувают азотом 2-5 раз (предпочтительно 3 раза), затем, необязательно, охлаждают до около -10-10°C (предпочтительно около 0°C) перед добавлением органического растворителя или смеси растворителей (таких как EtOAc, MeOH, EtOH или MeOH/AcOH, предпочтительно EtOAc или MeOH) в атмосфере азота. Охлаждающую баню удаляют и к смеси добавляют алкен (предпочтительно 1 экв.), чистый или, необязательно, в виде раствора в органическом растворителе или смеси растворителей (таких как EtOAc, MeOH, EtOH или MeOH/AcOH, предпочтительно EtOAc или MeOH). Газообразный водород барботируют через реакционную смесь примерно в течение 1-20 минут (предпочтительно около 5 мин) и смесь перемешивают в атмосфере водорода примерно в течение 12-60 часов (предпочтительно около 48 часов). В случаях, когда реакция не достигает завершения, согласно данным ТСХ, LC/MS или ВЭЖХ, источник водорода удаляют, реакционную смесь барботируют азотом примерно в течение 1-20 минут (предпочтительно около 5 мин) и затем фильтруют через слой Целита® и фильтрат концентрируют при пониженном давлении. Неочищенное вещество снова подвергают описанным выше реакционным условиям примерно в течение 2-20 часов (предпочтительно около 5 часов). Источник водорода удаляют и смесь барботируют азотом примерно в течение 1-20 минут (предпочтительно около 5 мин) и затем фильтруют через слой Целита®. Фильтровальную лепешку промывают органическим растворителем (таким как EtOAc, MeOH или EtOH, предпочтительно используемым в реакции растворителем) и фильтрат концентрируют при пониженном давлении с получением неочищенного продукта.
Иллюстрация Общей Процедуры W
Получение #W.1: этил 2-этил-4-оксоциклопентанкарбоксилат

В круглодонную колбу загружали 10% масс. Pd/C (10 г, 9,4 ммоль). Колбу охлаждали до около 0°C и добавляли EtOAc (400 мл) в атмосфере азота. Охлаждающую баню удаляли и добавляли этил 2-этил-4-оксоциклопент-2-енкарбоксилата (47,8 г, 263 ммоль, Получение #V.1). Газообразный водород барботировали через смесь примерно в течение 5 минут и смесь затем перемешивали в атмосфере водорода примерно в течение 48 часов. Источник водорода удаляли и смесь барботировали азотом примерно в течение 5 минут и фильтровали через слой Целита®. Фильтровальную лепешку промывали при помощи EtOAc (400 мл). Фильтрат концентрировали при пониженном давлении с получением этил 2-этил-4-оксоциклопентанкарбоксилата (примерно 9:1 смесь цис:транс) (48,0 г, 99%) в виде желтой жидкости: 1H ЯМР (CDCl3) δ 4,23-4,10 (м, 2H), 3,22 (м, 1H), 2,59-2,50 (м, 1H), 2,44-2,28 (м, 3H), 2,26-2,16 (м, 1H), 1,58-1,46 (м, 1H), 1,41-1,30 (м, 1H), 1,30-1,23 (м, 3H), 1,02-0,91 (м, 3H).
Примеры, полученные с использованием Общей Процедуры W
(Таблица 1, Способ)
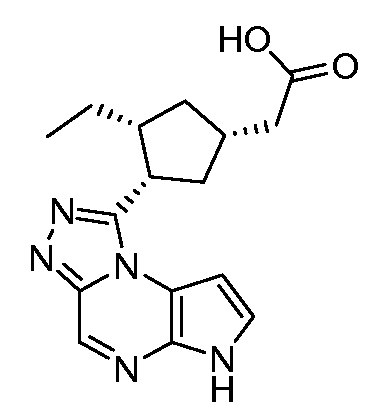
Общая процедура W.1: Гидрирование алкена
В круглодонную колбу загружают суспензию Pd(OH)2 на углероде или Pd/C (около 0,005-0,10 экв., предпочтительно 0,05 экв.) в органическом растворителе или смеси растворителей (таких как ТГФ, EtOAc, MeOH, EtOH или MeOH/AcOH, предпочтительно ТГФ) в атмосфере азота. Смесь добавляют к алкену (предпочтительно 1 экв.), чистому или можно в виде раствора в органическом растворителе или смеси растворителей (таких как ТГФ, EtOAc, MeOH, EtOH или MeOH/AcOH, предпочтительно ТГФ), или возможно добавление алкена к Pd смеси. Реакционную смесь барботируют водородом. Смесь перемешивают или встряхивают (предпочтительно перемешивают, когда используют атмосферный водород, или встряхивают, когда использовали более высокое давление водорода) в атмосфере водорода при давлении около атмосферного - 60 ф/дюйм2 (4,219 кг/см2) (предпочтительно при атмосферном давлении) при около 20-60°C (предпочтительно при температуре окружающей среды) примерно в течение 0,5-5 дней (предпочтительно около 3 дней). Реакционную смесь фильтруют через слой Целита®. Фильтровальную лепешку промывают органическим растворителем (таким как ТГФ, EtOAc, DCM, MeOH или EtOH, предпочтительно используемым в реакции растворителем) и фильтрат концентрируют при пониженном давлении с получением неочищенного продукта.
Иллюстрация Общей Процедуры W.1
Получение #W.1.1 и W.1.2: этил 2-((1R,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)ацетат и 2-((1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)ацетат
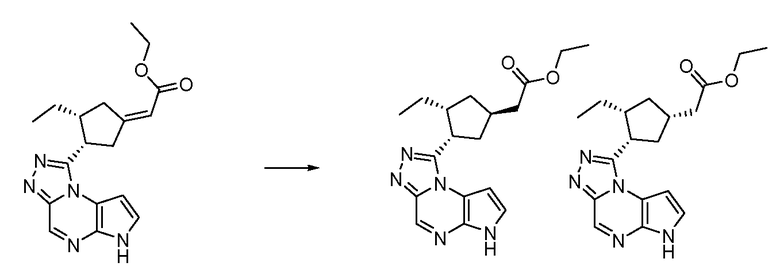
К суспензии 20% масс. Pd(OH)2 на углероде (0,134 г, 0,192 ммоль) в ТГФ (20 мл) добавляли раствор (E)-этил 2-((цис)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентилиден)ацетата (1,3 г, 3,83 ммоль, Пример #38, Стадия G) в ТГФ (5 мл). Реакционную смесь барботировали водородом и атмосферу водорода поддерживали через баллон. Примерно через 3 дня реакционную смесь фильтровали через Целит®, концентрировали при пониженном давлении и очищали флэш-хроматографией на силикагеле с использованием в качестве элюента EtOAc с получением темного коричневого/черного твердого вещества. Соединение подвергали дальнейшей очистке методом хиральной хроматографии (Таблица 2, Способ 47) с получением этил 2-((1R,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)ацетата [W.1.1] (Rt = 12,0 мин, or = отрицательное) (0,400 г, 31%): LC/MS (Таблица 1, Способ a) Rt = 1,85 мин; MS m/z: 342 (M+H)+ и этил 2-((1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)ацетата [W.1.2] (Rt = 13,7 мин, or = отрицательное) (0,420 г, 32%) в виде белого твердого вещества: LC/MS (Таблица 1, Способ a) Rt = 1,85 мин; MS m/z: 342 (M+H)+.
Общая процедура X: Восстановительное аминирование кетона или альдегида
В круглодонную колбу загружают кетон или альдегид (1-40 экв.; предпочтительно 1 экв.) в органическом растворителе (таком как DCE, MeCN, MeOH или MeCN/MeOH; предпочтительно DCE). Смесь необязательно охлаждают до около -10-10°C (предпочтительно около 0°C) и добавляют по каплям AcOH (1-3 экв.; предпочтительно 1,5 экв.) и амин (1-3 экв., предпочтительно 1 экв.) с последующим добавлением по порциям подходящего восстановителя, такого как NaBH(OAc)3, Na(CN)BH3, NaBH4, предпочтительно NaBH(OAc)3 (1-6 экв., предпочтительно 1,5 экв.). Альтернативно, к раствору амина (1-3 экв., предпочтительно 1 экв.) в органическом растворителе (таком как DCE, MeCN или MeOH; предпочтительно DCE) добавляют кетон или альдегид (1-40 экв.; предпочтительно 1 экв.) с последующим добавлением по порциям подходящего восстановителя, такого как NaBH(OAc)3, Na(CN)BH3, NaBH4, предпочтительно NaBH(OAc)3 (1-6 экв., предпочтительно 1,5 экв.). Смесь перемешивают примерно в течение 5-20 минут (предпочтительно около 15 мин) с последующим добавлением по каплям AcOH (1-3 экв.; предпочтительно 1,5 экв.). Если реакционная смесь становится слишком вязкой и трудной для перемешивания, возможно добавление дополнительного количества органического растворителя (такого как DCE, MeCN, MeOH или смесь MeCN/MeOH; предпочтительно DCE) для облегчения перемешивания. Реакционную смесь перемешивают при температуре окружающей среды примерно в течение 1-48 часов (предпочтительно около 20 часов). Реакционную смесь медленно выливают в водный раствор основания (такой как насыщенный водный раствор NaHCO3) с последующим необязательным добавлением твердого NaHCO3 и перемешивают примерно в течение 0,5-3 часов (предпочтительно около 2 часов). Слои разделяют и органический раствор сушат над безводным Na2SO4 или MgSO4, фильтруют и концентрируют досуха при пониженном давлении.
Иллюстрация Общей Процедуры X
Получение #X.1: этил 4-(дибензиламино)-2-этилциклопентанкарбоксилат

В круглодонную колбу загружали этил 2-этил-4-оксоциклопентанкарбоксилат (95,9 г, 521 ммоль, Получение #W.1) и DCE (1,8 л). Раствор охлаждали до около 0°C и добавляли по каплям AcOH (45 мл, 780 ммоль) и дибензиламин (120 мл, 625 ммоль), приводя к образованию вязкой суспензии. Реакционную смесь нагревали до около 10°C и добавляли дополнительное количество DCE (500 мл). Добавляли по порциям NaBH(OAc)3 (166 г, 781 ммоль) и реакционную смесь перемешивали при температуре окружающей среды примерно в течение 20 часов. Реакционную смесь медленно выливали в перемешиваемый насыщенный водный раствор NaHCO3 (1,5 л), с последующим добавлением по порциям твердого NaHCO3 (175 г). Смесь перемешивали примерно в течение 2 часов и органический слой отделяли, сушили над безводным Na2SO4 и концентрировали досуха при пониженном давлении. Неочищенное желтое масло очищали хроматографией на силикагеле с использованием в качестве элюента 0-20% EtOAc в гептане с получением этил 4-(дибензиламино)-2-этилциклопентанкарбоксилата (136,6 г, 72%) в виде белого твердого вещества: LC/MS (Таблица 1, Способ a) Rt = 3,26 мин; MS m/z: 366 (M+H)+
Примеры, полученные из 1-((3R,4R)-4-метилпиперидин-3-ил)-6H-имидазо[1,5-a]пирроло[2,3-e]пиразингидрохлорида (Пример #5, Стадия J) с использованием Общей Процедуры X с NaBH3CN
(Таблица 1, Способ)
Примеры, полученные из ацетальдегида с использованием Общей Процедуры X с NaBH3CN
(Таблица 1, Способ)
Примеры, полученные из 4,4,4-трифторбутиральдегида [Matrix] с использованием Общей Процедуры X с Na(OAc)3BH
(Таблица 1, Способ)
Примеры, полученные из (3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентанона (получен с использованием D из Получения #25 с использованием D) и Общей Процедуры X с Na(OAc)3BH
(Таблица 1, Способ)
Общая процедура X.1: Восстановительное аминирование кетона или альдегида
Кетон или альдегид (1-40 экв.; предпочтительно 1 экв.), либо растворяют либо суспендируют в органическом растворителе или растворителях, таких как DCE, MeCN, MeOH, MeCN/MeOH, EtOH, ТГФ, DMF, AcOH или DCM (предпочтительно DCE). Смесь необязательно охлаждают до около -10-10°C (предпочтительно около 0°C). Необязательно добавляют AcOH (1-3 экв.; предпочтительно 1,5 экв.). Добавляют амин (1-3 экв., предпочтительно 1 экв.), чистый или в виде раствора в органическом растворителе или растворителях, таких как DCE, MeCN, MeOH, EtOH, ТГФ, DMF, AcOH или DCM (предпочтительно DCE). Альтернативно, кетон или альдегид, или раствор кетона или альдегида, можно добавить к амину или раствору амина. Необязательно, можно добавить дегидратирующий реагент, такой как молекулярные сита или тетраизопропоксид титана(IV), или воду можно удалить с использованием ловушки Дина-Старка. Растворитель необязательно удаляют при пониженном давлении, и можно добавить органический растворитель или растворители, такие как DCE, MeCN, MeOH, EtOH, ТГФ, DMF, AcOH или DCM. После перемешивания примерно в течение 5 минут - 24 часов (предпочтительно 15 мин) при 0°C-100°C (предпочтительно при температуре окружающей среды) добавляют по порциям подходящий восстановитель, такой как NaBH(OAc)3, Na(CN)BH3, NaBH4, предпочтительно NaBH(OAc)3 (1-10 экв., предпочтительно 1,5 экв.). Если реакционная смесь становится слишком вязкой и трудной для перемешивания, возможно добавление дополнительного количества органического растворителя для облегчения перемешивания. Реакционную смесь перемешивают при температуре окружающей среды примерно в течение 1-72 часов (предпочтительно около 20 часов). Необязательно, реакционную смесь можно обработать водой и затем отфильтровать, или летучие вещества можно удалить при пониженном давлении. Реакционную смесь медленно выливают в водный раствор основания, воду или водный раствор кислоты (предпочтительно насыщенный водный раствор NaHCO3), или, альтернативно, водный раствор медленно добавляют к реакционной смеси. Можно добавить, но необязательно, дополнительное количество твердого NaHCO3. Смесь интенсивно перемешивают примерно в течение 0,5-20 часов (предпочтительно около 2 часов). Слои разделяют и органический раствор сушат над безводным Na2SO4 или MgSO4, фильтруют и концентрируют досуха при пониженном давлении.
Иллюстрация Общей Процедуры X.1
Получение #X.1.1: трет-бутил (транс-4-((6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-8-ил)метиламино)циклогексил)метилкарбамат

трет-Бутил транс-4-аминоциклогексилметилкарбамат (0,059 г, 0,258 ммоль, AMRI) добавляли к смеси 6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-8-карбальдегида (0,0403 г, 0,215 ммоль, Получение #38) и ТГФ (1,0 мл). Смесь перемешивали при температуре окружающей среды примерно в течение 90 минут. Добавляли триацетоксиборогидрид натрия (0,068 г, 0,32 ммоль). Примерно через 3 часа добавляли DMF (0,500 мл). Примерно через 15 часов добавляли Na(OAc)3BH (0,091 г, 0,43 ммоль). Примерно через 24 часов добавляли Na(OAc)3BH (0,091 г, 0,43 ммоль). Смесь нагревали до около 40°C. Примерно через 22 часа смеси давали охладиться до температуры окружающей среды. Добавляли насыщенный водный раствор NaHCO3/вода (1:1, 2 мл). После интенсивного перемешивания примерно в течение 1 часа раствор разбавляли водой (3 мл) и затем экстрагировали при помощи EtOAc (6×10 мл). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали хроматографии на силикагеле с градиентным элюированием 10-100% [(1% 7 н NH3 в MeOH) в 10% MeOH/DCM]/DCM с получением трет-бутил (транс-4-((6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-8-ил)метиламино)циклогексил)метилкарбамата (0,0476 г, 53%): LC/MS (Таблица 1, Способ a) Rt = 1,24 мин; MS m/z 400 (M+H)+.
Общая процедура Y: Гидрирование бензил- или Cbz-защищенного амина
В сосуд, содержащий бензил- или Cbz-защищенный амин (предпочтительно 1 экв.), добавляют палладиевый катализатор (например, Pd(OH)2 на C или Pd/C; предпочтительно Pd(OH)2 на C) (0,01-0,2 экв., предпочтительно 0,02-0,15 экв.) и органический растворитель (такой как MeOH или EtOH, предпочтительно EtOH). Смесь встряхивают или перемешивают при около 25-60°C (предпочтительно около 50°C) примерно в течение 1-96 часов (предпочтительно около 1,5-3 часов) при давлении водорода около 15-60 ф/дюйм2 (1,055-4,219 кг/см2) (предпочтительно при давлении водорода около 30-50 ф/дюйм2 (2,109-3,515 кг/см2). В случаях, когда реакция не достигает завершения, согласно данным ТСХ, LC/MS или ВЭЖХ, источник водорода удаляют, реакционную смесь барботируют азотом примерно в течение 5-20 минут (предпочтительно около 5 мин) и затем фильтруют через слой Целита® и фильтрат концентрируют при пониженном давлении. Неочищенное вещество снова подвергают описанным выше реакционным условиям примерно в течение 2-20 часов (предпочтительно около 3-5 часов). Когда реакция завершается, согласно данным ТСХ, LC/MS или ВЭЖХ, источник водорода удаляют, вводят атмосферу азот и реакционную смесь фильтруют через слой Целита®. Фильтрат концентрируют при пониженном давлении с получением желаемого продукта.
Иллюстрация Общей Процедуры Y
Получение #Y.1: этил 4-амино-2-этилциклопентанкарбоксилат

В сосуд, содержащий суспензию 20% масс. Pd(OH)2 на C (12,9 г, 18,4 ммоль) в EtOH (1,0 л) добавляли этил 4-(дибензиламино)-2-этилциклопентанкарбоксилат (129 г, 352 ммоль, Получение #X.1). Реакционную смесь встряхивали примерно в течение 90 минут при около 50°C под давлением водорода около 30 ф/дюйм2 (2,109 кг/см2). После удаления источника водорода и введения атмосферы азота полученную смесь фильтровали через слой Целита® и фильтрат концентрировали при пониженном давлении с получением этил 4-амино-2-этилциклопентанкарбоксилата (64,5 г, 99%) в виде желтого сиропа: 1H ЯМР (CDCl3) δ 4,03-3,88 (м, 2H), 3,17 (м, 1H), 2,68 (м, 1H), 2,09-2,02 (м, 2H), 2,02-1,94 (м, 2H), 1,84 (м, 1H), 1,58-1,48 (м, 1H), 1,32-1,18 (м, 1H), 1,09 (м, 3H), 1,03 (м, 2H), 0,78-0,69 (м, 3H).
Примеры, полученные с использованием Общей Процедуры Y
(Таблица 1, Способ)
Общая процедура Z: Щелочной гидролиз сложного эфира до карбоновой кислоты
В колбу, содержащую сложный эфир (предпочтительно 1 экв.), либо чистый либо в органическом растворителе (таком как 1,4-диоксан, MeOH или ТГФ/MeOH, предпочтительно 1,4-диоксан), добавляют водный раствор основания (такой как водный раствор NaOH или LiOH, 1-10 экв., предпочтительно 2-6 экв.). Смесь перемешивают при около 0-100°C (предпочтительно при температуре окружающей среды) примерно в течение 1-48 часов (предпочтительно около 4-8 часов). Реакционную смесь затем подкисляют путем добавления подходящего водного раствора кислоты (такого как водный раствор HCl). Слои разделяют и водный слой необязательно экстрагируют дополнительным количеством органического растворителя (такого как EtOAc или DCM, предпочтительно DCM). Органический слой или слои, необязательно, сушат над безводным Na2SO4 или MgSO4, фильтруют и концентрируют досуха при пониженном давлении с получением неочищенного целевого соединения. Альтернативно, реакционную смесь концентрируют при пониженном давлении с получением неочищенного целевого соединения в виде карбоксилатной соли.
Иллюстрация Общей Процедуры Z
Получение #Z.1*: (1S,2R,4S)-4-(циклопропансульфонамидо)-2-этилциклопентанкарбоновая кислота

В колбу, содержащую (1S,2R,4S)-этил 4-(циклопропансульфонамидо)-2-этилциклопентан-карбоксилат (11,1 г, 38,4 ммоль, получали с использованием K из Получения #Y.1, циклопропансульфонилхлорид и TEA, AA [Таблица 2, Способ 1, Rt = 9,5 мин, or = отрицательное]), добавляли водный раствор NaOH (1 н, 210 мл, 210 ммоль). После перемешивания при температуре окружающей среды примерно в течение 8 часов, реакционную смесь подкисляли до около pH 1 с использованием 6 н водного раствора HCl и экстрагировали при помощи DCM (3×150 мл). Объединенные органические слои промывали насыщенным солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали при пониженном давлении с получением (1S,2R,4S)-4-(циклопропансульфонамидо)-2-этилциклопентанкарбоновой кислоты с 25 моль% DCM в качестве эксципиента (10,7 г, 99%): LC/MS (Таблица 1, Способ a) Rt = 1,71 мин; MS m/z: 260 (M-H)-.
Общая процедура AA: Очистка методом хиральной препаративной ВЭЖХ
Хиральную очистку осуществляют с использованием Varian 218 LC насосов, Varian CVM 500 с переключающими клапанами и нагревателями для автоматического контроля растворителя, колонки и температуры, и устройства для сбора фракций Varian 701. Способы детекции включают Varian 210 детектор с переменной длиной волны, встроенный в линию поляриметр (PDR-хиральный усовершенствованный лазерный поляриметр, модель ALP2002), который использовали для измерения качественного оптического вращения (+/-), и испарительный светорассеивающий детектор (ELSD) (PS-ELS 2100 (Polymer Laboratories)) с использованием 100:1 расщепленного потока. ELSD настройки: испаритель: 46°C, распылитель: 24°C и поток газа: 1,1 SLM. Абсолютную стереохимию очищенных соединений приписывали произвольно и представляли графически как таковую. Соединения по настоящему изобретению, где абсолютная стереохимия определена с использованием коммерчески доступного энантиомерно чистого исходного вещества или стереохимически определенного промежуточного соединения или методом рентгеновской дифракции, помечены звездочкой после номера примера.
Иллюстрация Общей Процедуры AA
Примеры #AA.1.1 и AA.1.2: 3-((3R,4R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-(трифторметил)пиперидин-1-ил)-3-оксопропаннитрил и 3-((3S,4S)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-(трифторметил)пиперидин-1-ил)-3-оксопропаннитрил

Смесь 3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-(трифторметил)пиперидин-1-ил)-3-оксопропаннитрила (0,067 г, 0,18 ммоль, получали с использованием O из 4-(трифторметил)никотиновой кислоты, N, H с Примером #5, Стадия C, HATU и DIEA, Q с реагентом Лоуссона и трифторацетатом ртути (II), D с NaOH, F, H с 2-цианоуксусной кислотой, HATU и DIEA) растворяли в DMSO:MeOH (2:1, 3 мл). Смесь разделяли с использованием Varian 218 LC насосов, Varian CVM 500 с переключающими клапанами и нагревателями для автоматического контроля растворителя, колонки и температуры, и устройства для сбора фракций Varian 701, с использованием Способа 4 (Таблица 2) с получением 3-((3R,4R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-(трифторметил)пиперидин-1-ил)-3-оксопропаннитрила (Rt = 12,2 мин, or = положительное) (0,0284 г, 15%) [AA,1,1] и 3-((3S,4S)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-(трифторметил)пиперидин-1-ил)-3-оксопропаннитрила (Rt = 5,3 мин, or = отрицательное) (0,0282 г, 15%) [AA,1,2]: LC/MS (Таблица 1, Способ b) Rt = 1,55 мин; MS m/z: 377 (M+H)+.
Примеры, полученные с использованием Общей Процедуры AA
[Способ хирального разделения]
(способ)
[Таблица 2, Способ 18, Rt = 14,5 мин or = ND]
[Таблица 2, Способ 2, Rt = 10,4 мин or = отрицательное]
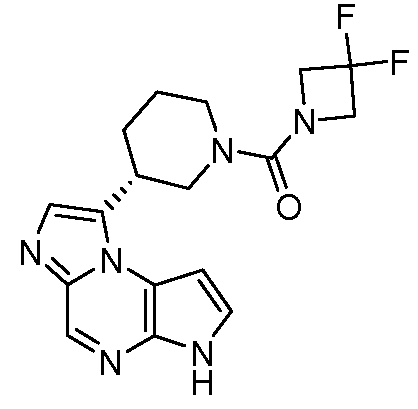
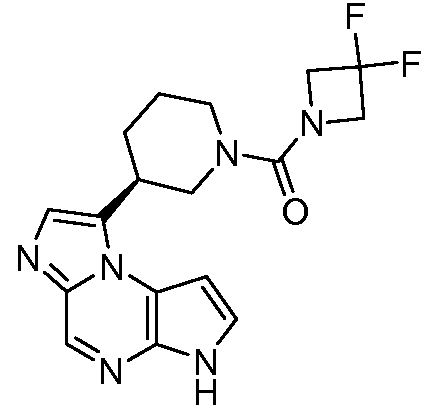

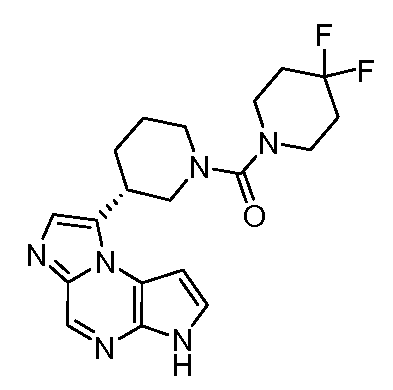
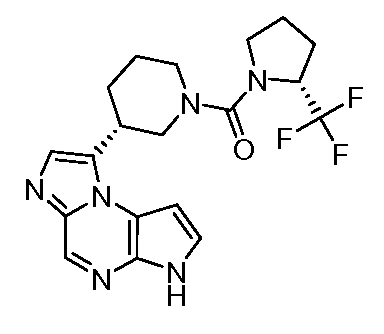






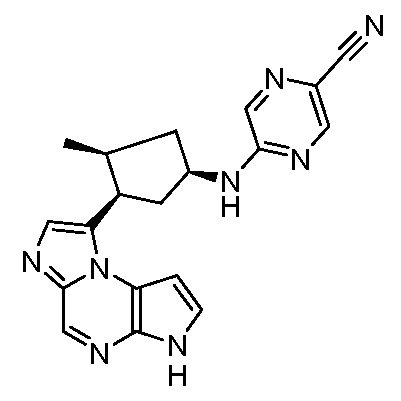
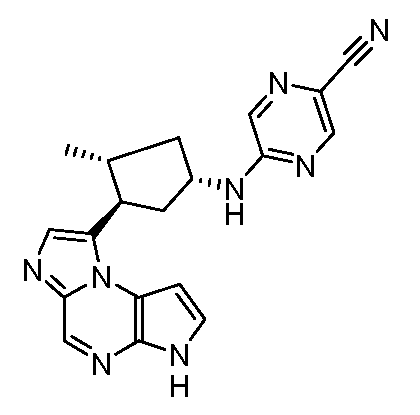
[Таблица 2, Способ 8, Rt = 9,5 мин or = отрицательное]








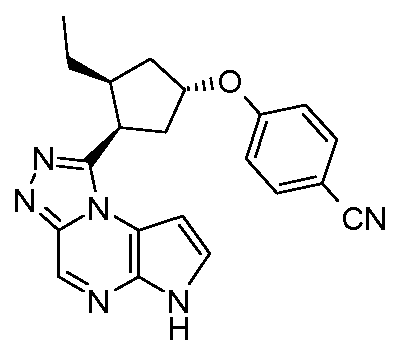

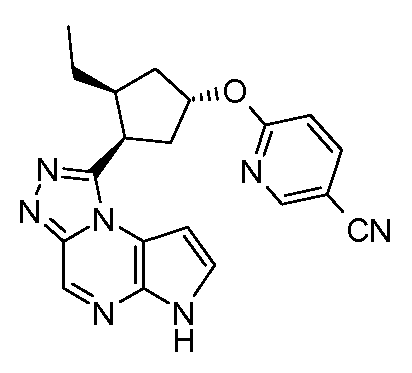
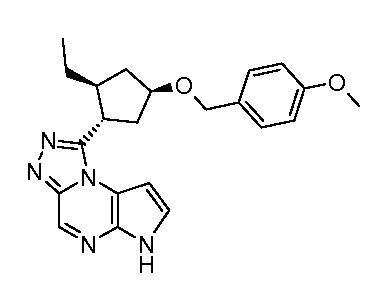
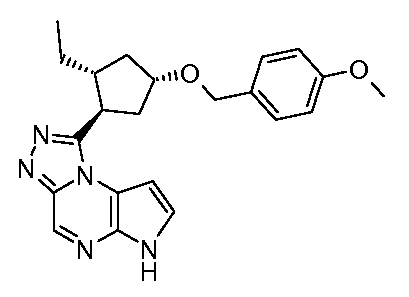


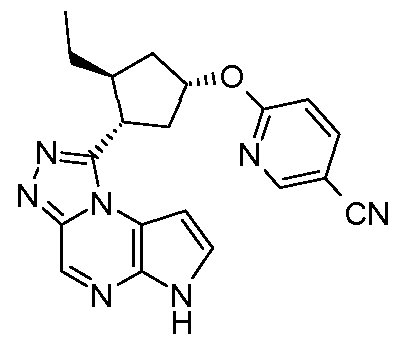
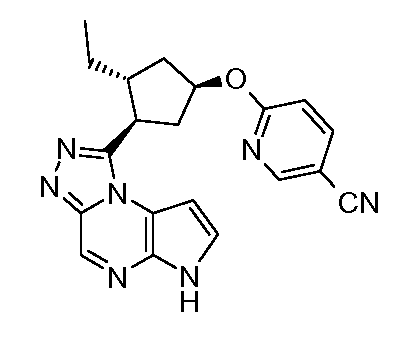

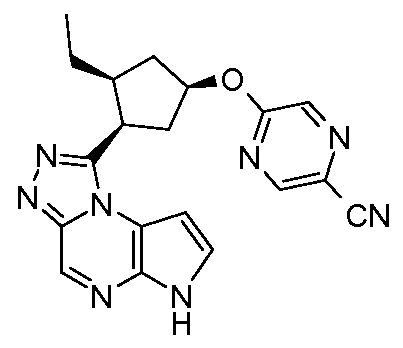
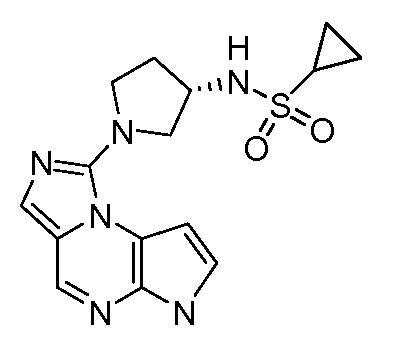

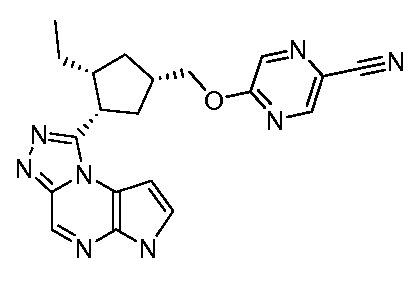



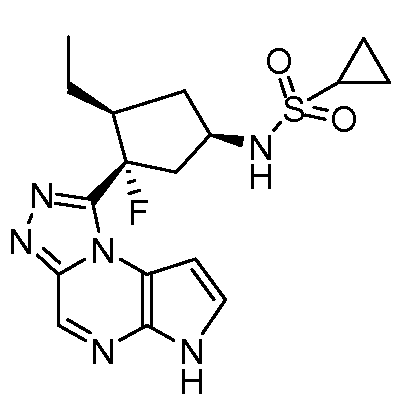

[Таблица 2, Способ 10, Rt = 16,5 мин or = отрицательное]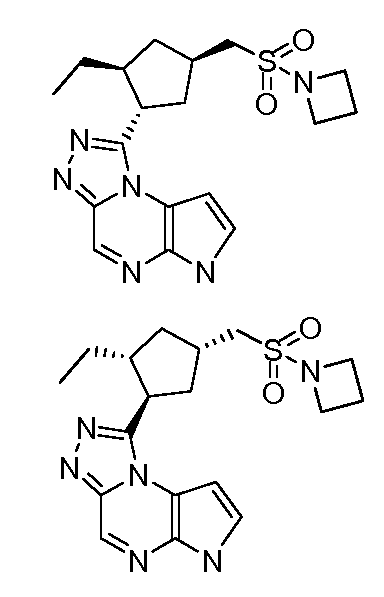




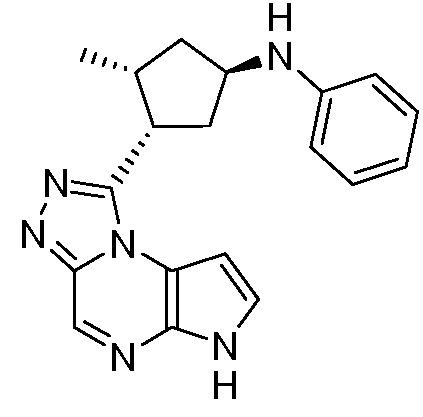







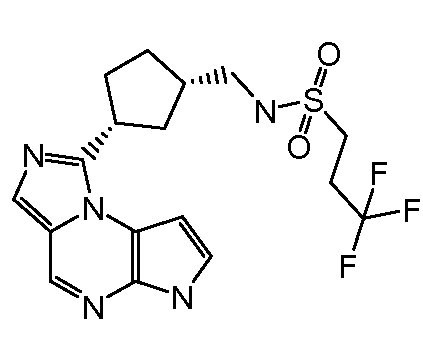
















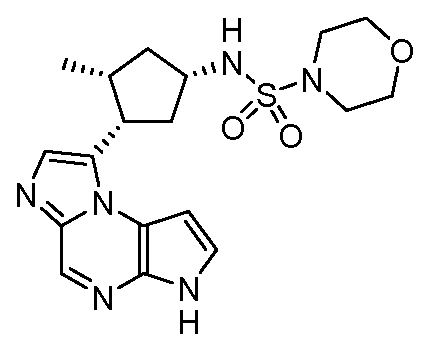


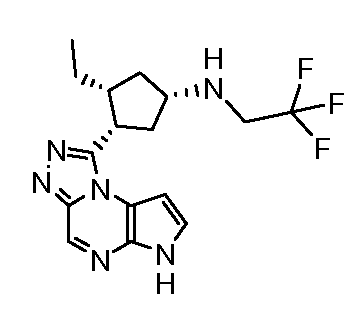




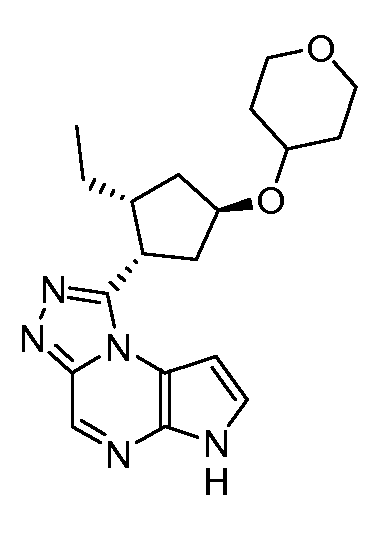
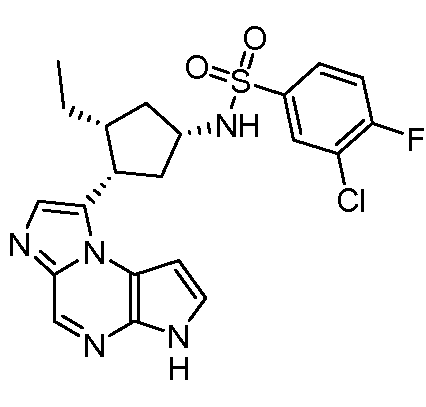

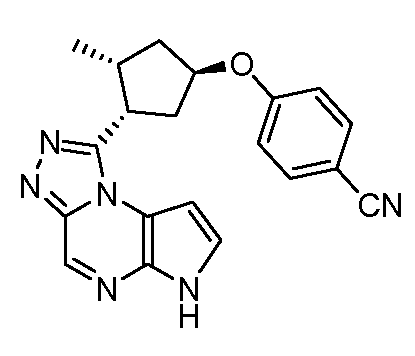

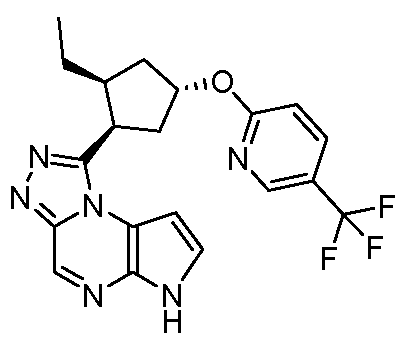





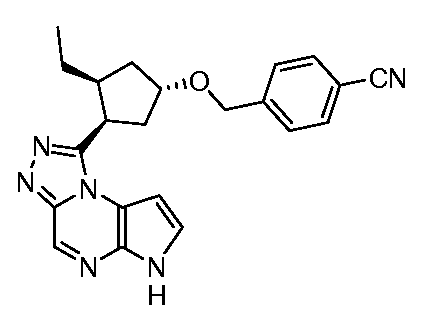

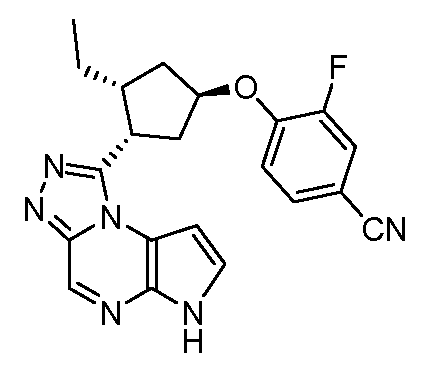
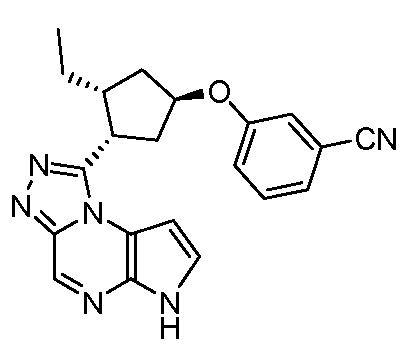



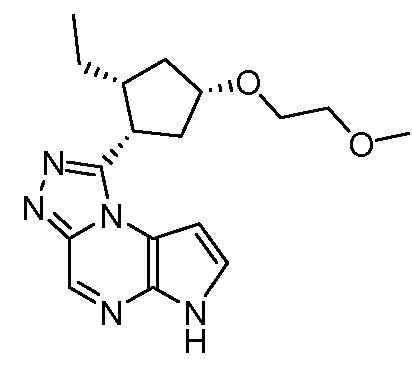









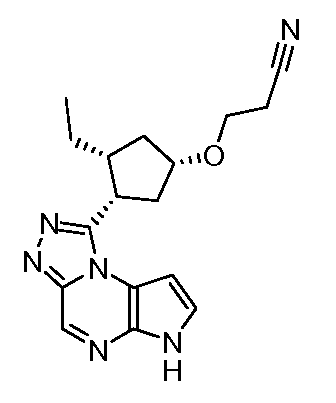



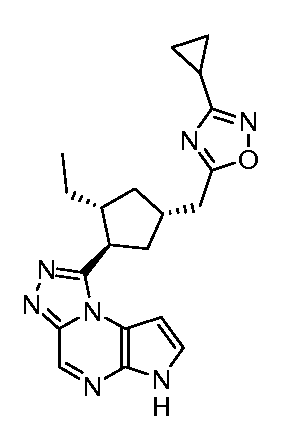


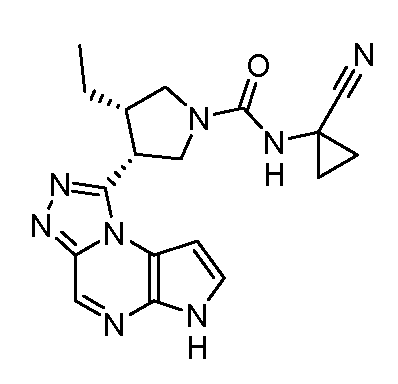






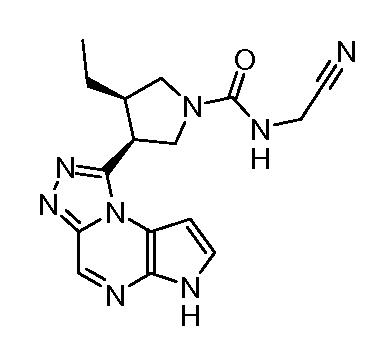

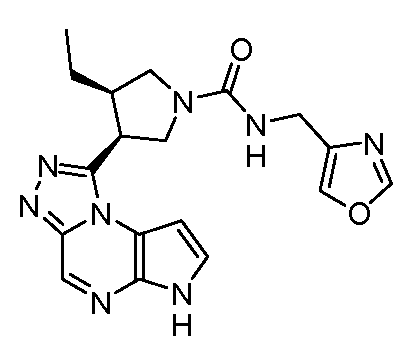

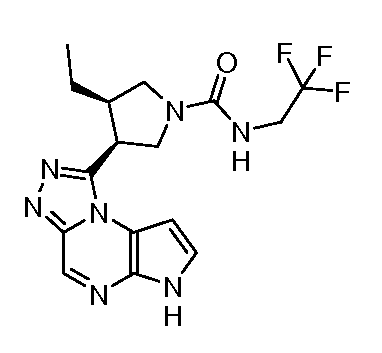
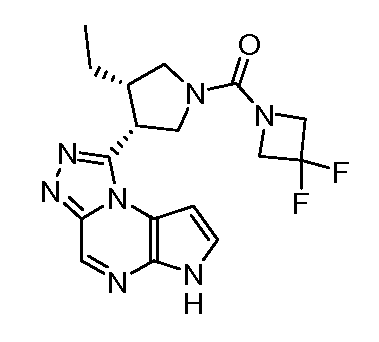
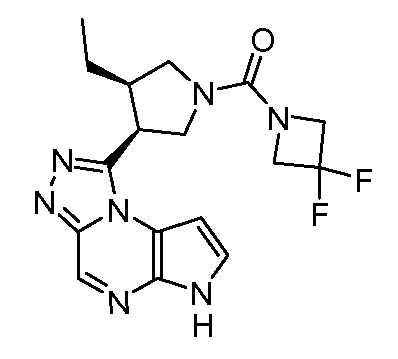
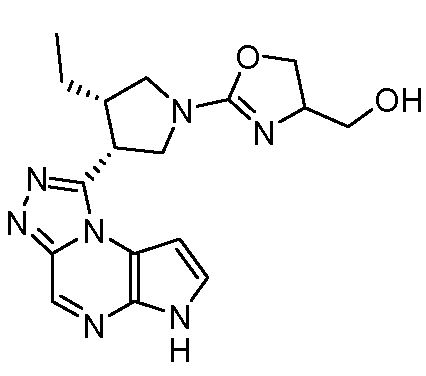
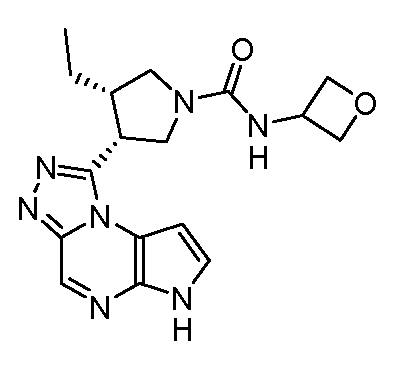







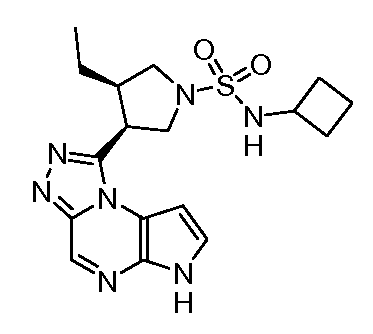

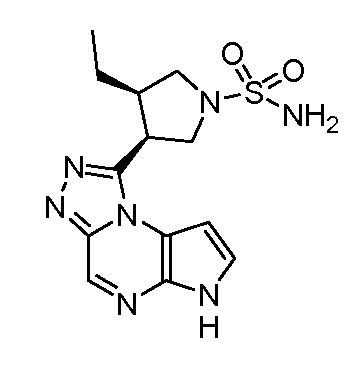




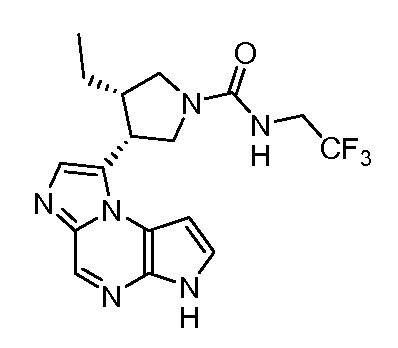

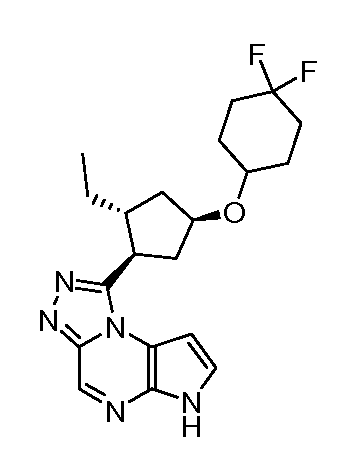









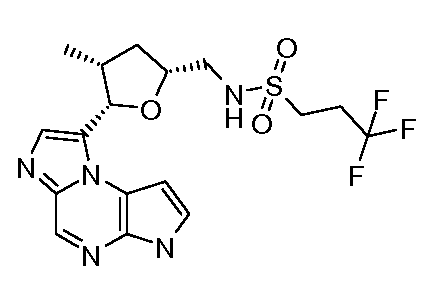











Общая процедура BB: кислотный гидролиз ацетил-защищенного амина
К раствору N-ацетамида (предпочтительно 1 экв.) в органическом растворителе (таком как 1,4-диоксан) добавляют кислоту, такую как 6 н водный раствор HCl (3-100 экв., предпочтительно 30-40 экв.). Реакционную смесь нагревают при около 60-100°C (предпочтительно около 90-100°C) примерно в течение 1-24 часов (предпочтительно около 16 часов). Реакционной смеси дают охладиться до температуры окружающей среды, затем смесь распределяют между органическим растворителем (таким как EtOAc или DCM) и водным раствором основания (такого как NaHCO3, Na2CO3 или NaOH, предпочтительно NaHCO3) и водный слой необязательно экстрагируют дополнительным количеством органического растворителя (такого как EtOAc или DCM). Органический слой сушат над безводным MgSO4 или Na2SO4, фильтруют и концентрируют при пониженном давлении.
Иллюстрация Общей Процедуры BB
Получение #BB.1*: (1S,3R,4S)-3-этил-4-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентанамин

К раствору N-((1S,3R,4S)-3-этил-4-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)ацетамида (6,0 г, 12,86 ммоль, Пример #8, Стадия L) в 1,4-диоксане (78 мл) добавляли водный раствор HCl (6 н, 75 мл, 450 ммоль). Реакционную смесь нагревали при около 95°C примерно в течение 16 часов. Реакционную смесь охлаждали до температуры окружающей среды и растворитель удаляли при пониженном давлении. Остаток разбавляли при помощи DCM (50 мл) и промывали насыщенным водным раствором NaHCO3 (100 мл). Водную часть экстрагировали дополнительным количеством DCM (3×50 мл) и объединенные органические слои сушили над безводным MgSO4, фильтровали и концентрировали при пониженном давлении. Неочищенное вещество очищали хроматографией на силикагеле с градиентным элюированием 0-100% DCM/MeOH/NH4OH (950:45:5) в DCM с получением (1S,3R,4S)-3-этил-4-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентанамина (3,05 г, 56%) в виде желто-коричневого твердого вещества: LC/MS (Таблица 1, Способ a) Rt = 1,85 мин; MS m/z: 425 (M+H)+.
Общая процедура CC: Образование сульфамоилхлорида
В круглодонную колбу загружают амин или соль амина (предпочтительно 1 экв.) в органическом растворителе (например, DCM или толуол или толуол/DCM). Когда используют соль амина, добавляют основание, такое как TEA или DIEA, предпочтительно DIEA (1-10 экв., предпочтительно 2,5 экв.) и реакционную смесь перемешивают примерно в течение 1-20 минут (предпочтительно около 5 мин). Реакционную смесь затем охлаждают до около -50-20°C, (предпочтительно около -30°C) примерно в течение 1-10 минут (предпочтительно около 5 мин). К реакционной смеси добавляют по каплям сульфурилхлорид или раствор сульфурилхлорида (такой как 1 M в DCM), предпочтительно сульфурилхлорид (1-10 экв., предпочтительно 3,5 экв.). Реакционную смесь перемешивают при около -50-0°C (предпочтительно около -30°C) примерно в течение 0,5-4 часов (предпочтительно около 1 часа), затем дают нагреться до температуры окружающей среды и перемешивают примерно в течение 1-24 часов (предпочтительно около 5 часов). Реакционную смесь затем разбавляют органическим растворителем (таким как DCM, EtOAc или толуол) и промывают водным раствором HCl (таким как 0,1-6 M, предпочтительно 1 M). Необязательно, реакционную смесь выливают на колотый лед и слои разделяли. Органические экстракты, необязательно, промывают водой и/или насыщенным солевым раствором, сушат над безводным Na2SO4 или MgSO4, фильтруют или декантируют и концентрируют при пониженном давлении.
Иллюстрация Общей Процедуры CC
Получение #CC.1: азетидин-1-сульфонилхлорид

В высушенную в печи колбу загружали азетидингидрохлорид (2,00 г, 21,38 ммоль), DIEA (5,60 мл, 32,10 ммоль) и DCM (50 мл). Реакционную смесь перемешивали примерно в течение 5 минут при температуре окружающей среды и затем охлаждали до около -30°C на бане сухой лед/MeCN примерно в течение 5 минут. Добавляли по каплям сульфурилхлорид (4,30 мл, 53,60 ммоль, Acros) примерно в течение 5 минут. Реакционную смесь перемешивали при около -30°C примерно в течение 1 часа, затем при температуре окружающей среды примерно в течение 5 часов. Реакционную смесь разбавляли водным раствором HCl (1 н, 15 мл). Слои разделяли и водный слой экстрагировали при помощи DCM (10 мл). Объединенные органические слои промывали водным раствором HCl (1 н, 10 мл) и насыщенным солевым раствором (20 мл). Органические слои сушили над безводным MgSO4, фильтровали и растворитель удаляли при пониженном давлении с получением азетидин-1-сульфонилхлорида (1,86 г, 56%): 1H ЯМР (CDCl3) δ 4,25-4,01 (м, 4H), 2,51-2,29 (м, 2H).
Общая процедура DD: Образование сульфонилмочевины
К раствору амина (предпочтительно 1 экв.) и основания, такого как TEA, DIEA, Na2CO3 или K2CO3 (1-20 экв., предпочтительно 2,5 экв. TEA), в органическом растворителе (таком как DMF, DMA, DCM, ТГФ или 1,4-диоксан, предпочтительно DMF) при температуре от около -10°C до температуры окружающей среды (предпочтительно около 0°C) добавляют сульфамоилхлорид (1-5 экв., предпочтительно 2,2 экв.). Реакционную смесь перемешивают примерно в течение 1-48 часов (предпочтительно около 2-4 часов) при температуре окружающей среды. В случаях, когда реакция не достигает завершения, согласно данным ТСХ, LC/MS или ВЭЖХ, к реакционной смеси добавляют по порциям дополнительное количество сульфамоилхлорида (1-20 экв. всего, предпочтительно 3 экв. на добавление) примерно через каждые 12-72 часа (предпочтительно примерно через каждые 24 часа) и реакционную смесь перемешивают при температуре окружающей среды вплоть до прекращения развития реакции согласно данным ТСХ, LC/MS или ВЭЖХ. Реакционную смесь концентрируют досуха при пониженном давлении и/или разбавляют органическим растворителем (таким как EtOAc или DCM) и водой. Объединенные органические экстракты, необязательно, промывают водой и/или насыщенным солевым раствором, сушат над безводным Na2SO4 или MgSO4, фильтруют или декантируют и концентрируют при пониженном давлении. Необязательно, реакционную смесь разбавляют водой и твердое вещество собирают вакуумным фильтрованием, промывают дополнительным количеством воды и сушат в вакууме.
Иллюстрация Общей Процедуры DD
Получение #DD.1*: N-((1S,3R,4S)-3-этил-4-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)азетидин-1-сульфонамид

В колбу загружали (1S,3R,4S)-3-этил-4-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентанамин (0,200 г, 0,471 ммоль, Пример #8, Стадия M) и DMF (4 мл). Раствор охлаждали до около 0°C с последующим добавлением TEA (0,16 мл, 1,2 ммоль) и азетидин-1-сульфонилхлорида (0,165 г, 1,06 ммоль, Получение #CC.1). Реакционную смесь нагревали до температуры окружающей среды и перемешивали примерно в течение 2 часов. Растворитель удаляли при пониженном давлении и к полученному остатку добавляли DCM (10 мл). Органический раствор промывали водой и насыщенным солевым раствором (5 мл каждого). Объединенные органические слои сушили над безводным MgSO4, фильтровали и концентрировали при пониженном давлении с получением коричневого масла. Неочищенное вещество очищали хроматографией на силикагеле с градиентным элюированием 0-70% EtOAc в DCM с получением N-((1S,3R,4S)-3-этил-4-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-циклопентил)азетидин-1-сульфонамида (0,20 г, 77%) в виде белого твердого вещества: LC/MS (Таблица 1, Способ a) Rt = 2,39 мин; MS m/z: 544 (M+H)+.
Пример, полученный из 1-((3R,4R)-4-метилпиперидин-3-ил)-6H-имидазо[1,5-a]пирроло[2,3-e]пиразингидрохлорида (Пример #5, Стадия J) с использованием Общей Процедуры DD
(Таблица 1, Способ)
Пример, полученный из (R)-1-(пиперидин-3-ил)-6H-имидазо[1,5-a]пирроло[2,3-e]пиразингидрохлорида (Пример #6, Стадия H) с использованием Общей Процедуры DD
(Таблица 1, Способ)
Общая процедура EE: Образование эфира из трихлорацетимидатного производного
К спирту (предпочтительно 1 экв.) в смеси органических растворителей, таких как DCM и циклогексан (1:1 до 1:5, предпочтительно 1:2) при около -10-5°C (предпочтительно около 0°C) добавляют раствор 2,2,2-трихлорацетимидатного производного (1-3 экв., предпочтительно 1,6 экв.) с последующим медленным добавлением кислоты, такой как п-толуолсульфоновая кислота или трифторметансульфоновая кислота (0,05-1 экв., предпочтительно 0,08-0,1 экв.). Реакционную смесь перемешивают при около -10-5°C (предпочтительно около 0°C) примерно в течение 5-60 минут (предпочтительно около 30 мин). Ледяную баню удаляют и реакционную смесь перемешивают при температуре окружающей среды примерно в течение 2-24 часов (предпочтительно около 16 часов). Суспензию выливают в ледяную воду и перемешивают примерно в течение 5-60 минут (предпочтительно около 30 мин). Суспензию либо фильтруют, осуществляя при этом промывку органическим растворителем, таким как DCM, либо разбавляют органическим растворителем, таким как DCM. Слои разделяют и водный слой экстрагируют органическим растворителем, таким как DCM. Объединенные органические слои промывают водой, сушат над безводным Na2SO4 или MgSO4, фильтруют и концентрируют при пониженном давлении.
Иллюстрация Общей Процедуры EE
Получение #EE.1: Этил 2-этил-4-(4-метоксибензилокси)циклопентанкарбоксилат

К смеси этил 2-этил-4-гидроксициклопентанкарбоксилата (37,78 г, 203 ммоль, Получение #P.1) в DCM (100 мл) и циклогексане (200 мл) при около 0°C добавляли 4-метоксибензил 2,2,2-трихлорацетимидат (93,58 г, 331 ммоль) с последующим добавлением по каплям трифторметансульфоновой кислоты (1,6 мл, 18,0 ммоль) примерно в течение 35 минут. Реакционную смесь перемешивали при около 0°C примерно в течение 30 минут. Ледяную баню удаляли и реакционную смесь перемешивали при температуре окружающей среды примерно в течение 16 часов. Суспензию выливали в ледяную воду (500 мл) и перемешивали примерно в течение 30 минут. Твердое вещество удаляли фильтрованием, осуществляя при этом промывку при помощи DCM (100 мл). Слои в фильтрате разделяли и водный слой экстрагировали при помощи DCM (3×200 мл). Объединенные органические слои промывали водой (200 мл), сушили над безводным MgSO4, фильтровали и концентрировали при пониженном давлении. Неочищенное вещество очищали с использованием хроматографии на силикагеле с градиентным элюированием 0-100% DCM:EtOAc (95:5) в DCM с получением этил 2-этил-4-(4-метоксибензилокси)циклопентанкарбоксилата (39,80 г, 64%): LC/MS (Таблица 1, Способ b) Rt = 2,90 мин; MS m/z: 307 (M+H)+.
Общая процедура FF: Удаление защиты у PMB-защищенного спирта
К PMB-защищенному спирту (предпочтительно 1 экв.) в смеси растворителей, таких как DCM и вода (1:1 до 7:1, предпочтительно 5:1), добавляют 2,3-дихлор-5,6-дициано-п-бензохинон (1-2 экв., предпочтительно 1,2 экв.). Реакционную смесь перемешивают при температуре окружающей среды примерно в течение 8-24 часов (предпочтительно около 16 часов). Твердое вещество удаляют фильтрованием, осуществляя при этом промывку органическим растворителем, таким как DCM. Слои в фильтрате разделяют и органический слой промывают насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушат над безводным Na2SO4 или MgSO4, фильтруют и концентрируют при пониженном давлении.
Иллюстрация Общей Процедуры FF
Получение #FF.1: 3-этил-4-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентанон

К 2-этил-4-(4-метоксибензилокси)циклопентил)-6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразину (1,153 г, 2,11 ммоль, получали с использованием Z из Получения #EE.1, А из Примера #1. Стадия D, HATU и TEA, B с DIEA) в DCM (18 мл) и воде (3,5 мл) добавляли 2,3-дихлор-5,6-дициано-п-бензохинон (0,576 г, 2,54 ммоль). Реакционную смесь перемешивали при температуре окружающей среды примерно в течение 16 часов. Твердое вещество удаляли фильтрованием, осуществляя при этом промывку при помощи DCM (150 мл). Органический слой отделяли и промывали насыщенным водным раствором NaHCO3 (2×40 мл) и насыщенным солевым раствором (40 мл), сушили над безводным MgSO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали с использованием хроматографии на силикагеле (40 г) с градиентным элюированием 30-100% EtOAc в DCM с получением 3-этил-4-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентанола (0,672 г, 75%): LC/MS (Таблица 1, Способ b) Rt = 2,09 мин; MS m/z: 426 (M+H)+.
Общая процедура GG: Образование лактона
К соединению γ-спирта и карбоновой кислоты (предпочтительно 1 экв.) в органическом растворителе, таком как DCM, добавляют основание (такое как TEA, 3-5 экв., предпочтительно 3 экв.) и BOP-Cl (1-2 экв., предпочтительно 1,2 экв.). Реакционную смесь перемешивают при температуре окружающей среды примерно в течение 1-5 часов (предпочтительно около 2 часов). Реакционную смесь выливают в органический растворитель (предпочтительно Et2O). Твердое вещество удаляют фильтрованием, осуществляя при этом промывку органическим растворителем, таким как Et2O. Фильтрат концентрируют при пониженном давлении. Альтернативно, фильтрат промывают насыщенным водным раствором NaHCO3, 1 н водным раствором лимонной кислот и насыщенным солевым раствором, сушат над безводным Na2SO4 или MgSO4, фильтруют и концентрируют при пониженном давлении.
Иллюстрация Общей Процедуры GG
Получение #GG.1*: (1S,4S,5R)-5-этил-2-охабицикло[2,2,1]гептан-3-он

К (1S,2R,4S)-2-этил-4-гидроксициклопентанкарбоновой кислоте (0,943 г, 5,96 ммоль, Пример #4, Стадия Н) в DCM (60 мл) добавляли TEA (2,5 мл, 18 ммоль) и BOP-Cl (1,82 г, 7,15 ммоль). Реакционную смесь перемешивали при температуре окружающей среды примерно в течение 2 часов, затем выливали в Et2O (350 мл). Твердое вещество удаляли фильтрованием, осуществляя при этом промывку при помощи Et2O (50 мл). Фильтрат концентрировали при пониженном давлении с получением желтого масла, которое растворяли в DCM (5 мл), и добавляли Et2O с получением твердого вещества. Надосадочную жидкость декантировали и твердое вещество промывали дополнительным количеством Et2O. Объединенные органические экстракты концентрировали при пониженном давлении с получением (1S,4S,5R)-5-этил-2-оксабицикло[2,2,1]гептан-3-она, содержащего около 15% моль ТЕА (0,912 г, 99% сырой)1H ЯМР (CDCl3) δ 4,85 (с, 1H), 2,88 (с, 1H), 2,19 (м, 2H), 2,08 (м, 1H), 1,69 (м, 1H), 1,41 (м, 3H), 0,97 (т, J=5,4, 3H).
Общая процедура HH: Раскрытие лактона с использованием амина или гидразина
К лактону (предпочтительно 1 экв.) в органическом растворителе, таком как 1,4-диоксан или DCM (предпочтительно 1,4-диоксан), добавляют гидразин (1-1,5 экв., предпочтительно 1 экв.). Альтернативно, лактон (предпочтительно 1 экв.) добавляют к раствору HCl соли амина и DIEA (1-1,5 экв., предпочтительно 1 экв.) в органическом растворителе или смеси растворителей (таких как 1,4-диоксан, DCM или DCM/DMF, предпочтительно DCM). Реакционную смесь перемешивают при температуре окружающей среды или нагревают при около 40-100°C (предпочтительно около 80°C, когда используют 1,4-диоксан, температура кипения с обратным холодильником, когда используют DCM) примерно в течение 1-24 часов (предпочтительно около 16 часов). В случае нагревания, реакционную смесь охлаждают до температуры окружающей среды. В случаях, когда реакция не достигает завершения, согласно данным ТСХ, LC/MS или ВЭЖХ, добавляют по каплям триметилалюминий (1-8 экв., предпочтительно 3 экв.), чистый или в растворе (таком как 2 M в хлорбензоле, 2 M в гептане или 2 M в толуоле, предпочтительно 2M в толуоле), после необязательного добавления органического растворителя (такого как 1,4-диоксан, DCM или DMF, предпочтительно 1,4-диоксан), и реакционную смесь перемешивают при температуре окружающей среды примерно в течение 0,25-16 часов (предпочтительно около 0,5 часов). Необязательно, триметилалюминий, чистый или в растворе, как описано выше, можно добавить с самого начала реакции. Добавляют по каплям водный раствор HCl (1 н, 3-10 экв., предпочтительно 8 экв.) и реакционную смесь перемешивают примерно в течение 10-60 минут (предпочтительно около 30 мин). Слои разделяют и водный слой экстрагируют органическим растворителем, таким как EtOAc или DCM (предпочтительно EtOAc). Объединенные органические части промывают водой, насыщенным водным раствором NaHCO3, насыщенным солевым раствором и сушат над безводным Na2SO4 или MgSO4, фильтруют и концентрируют при пониженном давлении.
Иллюстрация Общей Процедуры HH
Получение #HH.1*: (1S,2R,4S)-2-этил-4-гидрокси-N'-(5-тозил-5H-пирроло[2,3-b]пиразин-2-ил)циклопентанкарбогидразид
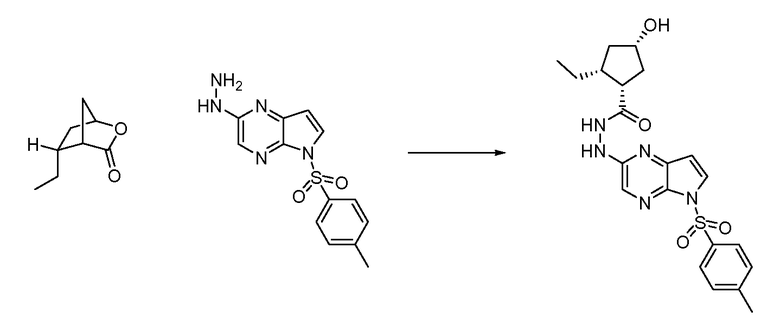
К (1S,4S,5R)-5-этил-2-оксабицикло[2,2,1]гептан-3-ону (0,835 г, 5,96 ммоль, Получение #GG.1) в 1,4-диоксане (12 мл) добавляли 2-гидразинил-5-тозил-5H-пирроло[2,3-b]пиразин (Пример #1. Стадия D, 1,81 г, 5,96 ммоль). Реакционную смесь нагревали при около 80°C примерно в течение 16 часов, затем охлаждали до температуры окружающей среды. Последовательно добавляли 1,4-диоксан (25 мл) и триметилалюминий (2 н раствор в толуоле, 9 мл, 18 ммоль). Реакционную смесь перемешивали при температуре окружающей среды примерно в течение 30 минут. Добавляли по каплям водный раствор HCl (1 н, 50 мл) и реакционную смесь перемешивали примерно в течение 30 минут. Слои разделяли. Водную часть экстрагировали при помощи EtOAc (2×100 мл). Объединенные органические экстракты промывали водой (10 мл), насыщенным водным раствором NaHCO3 (15 мл), насыщенным солевым раствором (15 мл) и сушили над безводным MgSO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали при помощи хроматографии на силикагеле (40 г) с использованием в качестве элюента 100% EtOAc с получением (1S,2R,4S)-2-этил-4-гидрокси-N'-(5-тозил-5H-пирроло[2,3-b]пиразин-2-ил)циклопентан-карбогидразида (1,887 г, 53%): LC/MS (Таблица 1, Способ b) Rt = 2,05 мин; MS m/z: 444 (M+H)+.
Примеры, полученные из 1-((3R,4R)-4-метилпиперидин-3-ил)-6H-имидазо[1,5-a]пирроло[2,3-e]пиразингидрохлорида (Пример #5, Стадия J) с использованием Общей Процедуры HH с DIEA
(Таблица 1, Способ)
Общая процедура II: Реакция Мицунобу для спирта
К спирту (предпочтительно 1 экв.) в органическом растворителе, таком как ТГФ, бензол, толуол или 1,4-диоксан (предпочтительно ТГФ), добавляют подходяще кислый реагент (такой как карбоновая кислота, фенол или гетероариловый спирт, 1-3 экв., предпочтительно 1,5 экв.), с последующим добавлением три-н-бутилфосфина, трифенилфосфина или связанного с полимером трифенилфосфина (предпочтительно связанного с полимером трифенилфосфина, 1-3 экв., предпочтительно 1,5 экв.) и TEA (1-6 экв., предпочтительно 4,5 экв.). Добавляют по каплям TMAD, 1,1'-(азодикарбонил)дипиперидин, DIAD или DEAD (предпочтительно DEAD, 1-3 экв., предпочтительно 1,5 экв.). Реакционную смесь перемешивают при температуре окружающей среды примерно в течение 5-48 часов (предпочтительно около 16 часов). Альтернативно, примерно через 0,1-24 часа (предпочтительно около 1 часа) добавляют дополнительное количество фосфинового реагента (0,2-2 экв., предпочтительно 0,75 экв.) и TMAD, 1,1'-(азодикарбонил)дипиперидин, DIAD или DEAD (0,2-1 экв., предпочтительно 0,75 экв.) для доведения реакции до завершения. Когда используют связанный с полимером реагент, реакционную смесь фильтруют и промывают смесью растворителей, таких как DCM, EtOAc и MeOH (предпочтительно DCM, затем MeOH). Фильтрат концентрируют при пониженном давлении. Когда не используют связанный с полимером реагент, реакционную смесь разбавляют органическим растворителем, таким как DCM или EtOAc, и затем промывают водой, насыщенным водным раствором NaHCO3, насыщенным солевым раствором и сушат над безводным Na2SO4 или MgSO4, фильтруют и концентрируют при пониженном давлении.
Иллюстрация Общей Процедуры II
Получение #II.1*: (1S,2R,4R)-4-(4-цианофенокси)-2-этил-N'-(5-тозил-5H-пирроло[2,3-b]пиразин-2-ил)циклопентанкарбогидразид

К (1S,2R,4S)-2-этил-4-гидрокси-N'-(5-тозил-5H-пирроло[2,3-b]пиразин-2-ил)циклопентан-карбо-гидразиду (0,885 г, 1,99 ммоль, Пример #4, Стадия J) в ТГФ (15 мл) добавляли 4-гидроксибензонитрил (0,357 г, 2,99 ммоль), трифенилфосфин (0,998 г, 2,99 ммоль, связанный с полимером, 3 ммоль/г) и TEA (1,3 мл, 9 ммоль). Добавляли по каплям DEAD (0,47 мл, 2,99 ммоль). Реакционную смесь перемешивали в течение примерно 1 часа, затем добавляли дополнительное количество трифенилфосфина (0,50 г, 1,50 ммоль, связанный с полимером, 3 ммоль/г) и DEAD (0,2 мл, 1,3 ммоль) и реакционную смесь перемешивали при температуре окружающей среды примерно в течение 16 часов. Твердое вещество удаляли фильтрованием, осуществляя при этом промывку при помощи DCM (5×5 мл), затем MeOH (4×5 мл). Фильтрат концентрировали при пониженном давлении и остаток очищали с использованием хроматографии на силикагеле (40 г) с градиентным элюированием 0-40% EtOAc в DCM с получением (1S,2R,4R)-4-(4-цианофенокси)-2-этил-N'-(5-тозил-5H-пирроло[2,3-b]пиразин-2-ил)циклопентанкарбогидразида (0,958 г, 88%) в виде желтого пенообразного вещества: LC/MS (Таблица 1, Способ b) Rt = 2,56 мин; MS m/z: 545 (M+H)+.
Общая процедура JJ: Замещение галогенида спиртом
К спирту (предпочтительно 1 экв.) в органическом растворителе, таком как DMF, ТГФ или 1,4-диоксан (предпочтительно DMF), при около 0-25°C (предпочтительно при температуре окружающей среды) добавляют NaH (60% дисперсия в минеральном масле, 1-4 экв., предпочтительно 1,2 экв.) по порциям. Примерно через 2-60 минут (предпочтительно около 5 мин), добавляют галогенид (1-30 экв., предпочтительно 1,1 экв.). Реакционную смесь нагревают при около 60-80°C (предпочтительно около 70°C) примерно в течение 1-16 часов (предпочтительно около 2 часов). После охлаждения до температуры окружающей среды к реакционной смеси добавляют ледяную воду или реакционную смесь выливают в ледяную воду и затем экстрагируют органическим растворителем, таким как DCM или EtOAc (предпочтительно DCM). Объединенные органические части концентрируют при пониженном давлении. Альтернативно, объединенные органические части промывают водой, насыщенным водным раствором NaHCO3, насыщенным солевым раствором и сушат над безводным Na2SO4 или MgSO4, фильтруют и концентрируют при пониженном давлении.
Иллюстрация Общей Процедуры JJ
Получение #JJ.1: 5-(3-этил-4-(6-((2-(триметилсилил)этокси)метил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентилокси)пиразин-2-карбонитрил

К 3-этил-4-(6-((2-(триметилсилил)этокси)метил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентанолу (0,098 г, 0,24 ммоль, получали с использованием FF из Получения #KK.1) в DMF (1 мл) добавляли NaH (0,012 г, 0,29 ммоль, 60% дисперсия в минеральном масле) по порциям. Примерно через 5 минут добавляли 2-хлор-5-цианопиразин (0,039 г, 0,28 ммоль, ArkPharm). Реакционную смесь нагревали при около 70°C примерно в течение 2 часов. После охлаждения до температуры окружающей среды добавляли ледяную воду (2 мл) и смесь экстрагировали при помощи DCM (3×5 мл). Органические слои объединяли и растворители удаляли при пониженном давлении. Остаток очищали с использованием хроматографии на силикагеле (12 г) с градиентным элюированием 20-80% EtOAc в DCM с получением 5-(3-этил-4-(6-((2-(триметилсилил)этокси)метил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентилокси)пиразин-2-карбонитрила (0,085 г, 69%): LC/MS (Таблица 1, Способ b) Rt = 2,84 мин; MS m/z: 505 (M+H)+.
Общая процедура KK: SEM-защита
К раствору пиррольного производного (предпочтительно 1 экв.) в органическом растворителе (таком как ТГФ, 1,4-диоксан или DMF, предпочтительно 1,4-диоксан) при около 0-40°C (предпочтительно при температуре окружающей среды) добавляют NaH (60% дисперсия в минеральном масле)(1-3 экв., предпочтительно 1,05 экв.) по порциям. Реакционную смесь перемешивают примерно в течение 1-60 минут (предпочтительно около 30 мин). Затем добавляют SEM-Cl (1-3 экв., предпочтительно 1,5 экв.). Примерно через 15 минут - 24 часа (предпочтительно около 30 мин) растворитель удаляют и остаток распределяют между органическим растворителем, таким как EtOAc, и водой. Слои разделяют и органический растворитель удаляют при пониженном давлении с получением целевого соединения. Альтернативно, реакционную смесь медленно выливают в ледяную воду при перемешивании с получением суспензии. Твердые вещества могут быть собраны фильтрованием и высушены с получением целевого соединения. Также, фильтрат можно распределить между органическим растворителем (таким как EtOAc или DCM) и водным раствором основания (таким как насыщенный водный раствор NaHCO3 или насыщенный водный раствор Na2SO4, предпочтительно насыщенный водный раствор NaHCO3). Органическую часть отделяют и концентрируют при пониженном давлении с получением целевого соединения.
Иллюстрация Общей Процедуры KK
Получение #KK.1: 1-(2-этил-4-(4-метоксибензилокси)циклопентил)-6-((2-(триметилсилил)этокси)метил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин

К суспензии 1-(2-этил-4-(4-метоксибензилокси)циклопентил)-6H-пирроло[2,3-e][1,2,4]-триазоло[4,3-a]пиразина (0,323 г, 0,825 ммоль, получали с использованием Z из Получения #EE.1, А из Примера #1. Стадия D, HATU и TEA, B с DIEA, D с NaOH) в 1,4-диоксане (2,5 мл) добавляли NaH (0,035 г, 0,866 ммоль, 60% дисперсия в минеральном масле) по порциям. Реакционную смесь перемешивали при температуре окружающей среды примерно в течение 30 минут. Добавляли SEM-Cl (0,15 мл, 0,83 ммоль). Примерно через 30 минут растворитель удаляли и остаток распределяли между EtOAc (12 мл) и водой (2 мл). Органический слой отделяли и концентрировали при пониженном давлении. Остаток очищали с использованием хроматографии на силикагеле (40 г) с градиентным элюированием 0-60% EtOAc в DCM с получением 1-(2-этил-4-(4-метоксибензилокси)циклопентил)-6-((2-(триметилсилил)этокси)метил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразина (0,372 г, 86%): LC/MS (Таблица 1, Способ b) Rt = 2,96 мин; MS m/z: 522 (M+H)+.
Общая процедура LL: Удаление SEM защиты
К раствору N-SEM-защищенного соединения (предпочтительно 1 экв.) в органическом растворителе (таком как DMF, 1,4-диоксан или DCM, предпочтительно DCM) добавляют TFA (5-70 экв., предпочтительно 50 экв.) и реакционную смесь перемешивают при около 0-40°C (предпочтительно при температуре окружающей среды) примерно в течение 1-20 часов (предпочтительно около 1-4 часов). Можно добавить дополнительное количество TFA (5-20 экв., предпочтительно 10 экв.). Полученную смесь концентрируют при пониженном давлении и остаток растворяют в органическом растворителе, таком как 1,4-диоксан, MeOH или EtOH (предпочтительно 1,4-диоксан). Добавляют водный раствор основания (такого как NaOH или NH4OH, предпочтительно NH4OH, 30-200 экв., предпочтительно 120 экв.) и реакционную смесь нагревают при около 30-100°C (предпочтительно около 60°C) примерно в течение 30 минут - 10 часов (предпочтительно около 30 мин). Реакционную смесь охлаждают до температуры окружающей среды, добавляют воду и продукт выделяют фильтрованием. Альтернативно, смесь можно распределить между органическим растворителем (таким как EtOAc или DCM) и водным раствором основания (таким как насыщенный водный раствор NaHCO3 или насыщенный водный раствор Na2SO4, предпочтительно насыщенный водный раствор NaHCO3). Органическую часть разделяют и концентрируют при пониженном давлении с получением целевого соединения. В некоторых случаях можно выделить промежуточное соединение гидроксиметилсульфонамид.
Иллюстрация Общей Процедуры LL
Получение #LL.1: 5-(-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентилокси)пиразин-2-карбонитрил

К 5-(3-этил-4-(6-((2-(триметилсилил)этокси)метил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентилокси)пиразин-2-карбонитрилу (0,097 г, 0,19 ммоль, Получение #JJ.1) в DCM (2,5 мл) добавляли TFA (0,7 мл, 10 ммоль). Реакционную смесь перемешивали при температуре окружающей среды примерно в течение 1,5 часов. Растворители удаляли при пониженном давлении и остаток растворяли в 1,4-диоксане (1,3 мл). Добавляли гидроксид аммония (28-30% водный раствор аммиака, 2,5 мл, 24 ммоль) и реакционную смесь нагревали при около 60°C примерно в течение 30 минут, затем охлаждали до температуры окружающей среды. Добавляли воду (4 мл) и осадок собирали фильтрованием с получением 5-(-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентилокси)пиразин-2-карбонитрила (0,0628 г, 87%): LC/MS (Таблица 1, Способ b) Rt = 1,99 мин; MS m/z: 375 (M+H)+.
Примеры, полученные с использованием Общей Процедуры LL
(Таблица 1, Способ)








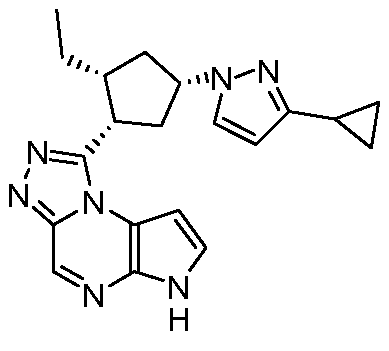


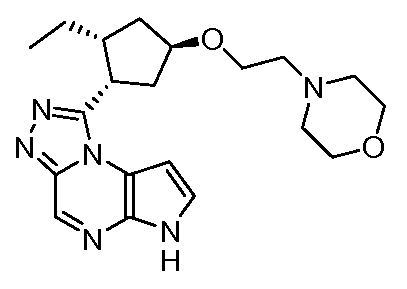

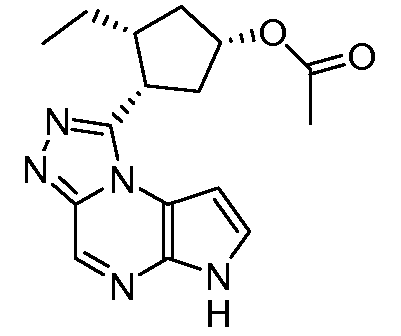


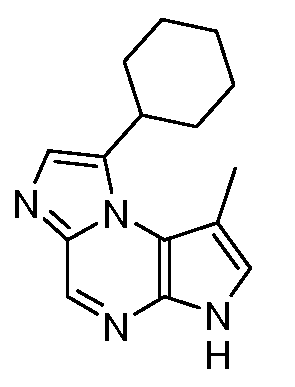

Общая процедура LL.1: Удаление SEM защиты
N-SEM-защищенное соединение растворяют или суспендируют в органическом растворителе (таком как DMF, 1,4-диоксан, ТГФ, MeOH или DCM, предпочтительно DCM). Можно добавить TFA, камфорсульфоновую кислоту или HCl, предпочтительно TFA (5-70 экв., предпочтительно 50 экв.), и реакционную смесь перемешивают при около 0-40°C (предпочтительно при температуре окружающей среды) примерно в течение 1-20 часов (предпочтительно около 1-4 часов). Необязательно, можно добавить дополнительное количество TFA (5-20 экв., предпочтительно 10 экв.). Полученную смесь концентрируют при пониженном давлении. Альтернативно, раствор или суспензию SEM-защищенного вещества можно обработать источником фторида, таким как TBAF или LiBF4, предпочтительно TBAF (1-20 экв., предпочтительно 6 экв.). Необязательно, можно добавить основание, такое как водный раствор NaOH, этилендиамин или водный раствор NH4OH (1-200 экв., предпочтительно этилендиамин, 2 экв.). Реакционную смесь нагревают при около 30-100°C (предпочтительно около 60°C) примерно в течение 30 минут - 72 часов (предпочтительно около 24 часов). Реакционную смесь охлаждают до температуры окружающей среды. Необязательно, летучие вещества удаляют при пониженном давлении. Реакционную смесь обрабатывают с использованием одного из следующих способов. Способ 1: Остаток растворяют в органическом растворителе, таком как 1,4-диоксан, MeOH или EtOH (предпочтительно 1,4-диоксан). Добавляют основание, такое как водный раствор NaOH, этилендиамин или водный раствор NH4OH (предпочтительно водный раствор NH4OH, 1-200 экв., предпочтительно 120 экв.), и реакционную смесь нагревают при около 30-100°C (предпочтительно около 60°C) примерно в течение 5 минут - 10 часов (предпочтительно около 30 мин). Реакционную смесь охлаждают до температуры окружающей среды, добавляют воду и продукт выделяют фильтрованием. Способ 2: Смесь распределяют между органическим растворителем (таким как EtOAc или DCM) и водным раствором основания (таким как насыщенный водный раствор NaHCO3 или насыщенный водный раствор Na2SO4, предпочтительно насыщенный водный раствор NaHCO3). Органическую часть отделяют и концентрируют при пониженном давлении с получением целевого соединения. Способ 3: Добавляют, необязательно, воду, водный раствор NaHCO3 или водный раствор NH4Cl (предпочтительно воду). Продукт можно выделить фильтрованием или смесь можно экстрагировать органическим растворителем (таким как EtOAc или DCM). Органические слои сушат над Na2SO4 или MgSO4, фильтруют и концентрируют при пониженном давлении с получением целевого соединения. В некоторых случаях можно выделить промежуточное соединение гидроксиметилсульфонамид.
Иллюстрация Общей Процедуры LL.1
Получение #LL.1,1: трет-бутил (транс-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-8-карбоксамидо)циклогексил)метилкарбамат

Этилендиамин (0,011 мл, 0,16 ммоль) добавляли к раствору трет-бутил (транс-4-(6-((2-(триметилсилил)этокси)метил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-8-карбоксамидо)циклогексил)метилкарбамата (0,043 г, 0,079 ммоль, получали с использованием Z из Получения #AAAAA,1 и KOH, H с трет-бутил транс-4-аминоциклогексилметилкарбаматом [AMRI], HATU и TEA) в ТГФ (1 мл). Добавляли одной порцией TBAF (1,0 M раствор в ТГФ, 0,470 мл, 0,470 ммоль). Смесь нагревали при около 60°C. Примерно через 24 часа раствору давали охладиться до температуры окружающей среды и перемешивали примерно в течение 40 часов. Летучие вещества удаляли при пониженном давлении. Остаток суспендировали в воде (10 мл) и экстрагировали при помощи EtOAc (4×20 мл). Объединенные органические части промывали насыщенным солевым раствором (10 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали хроматографией на силикагеле с градиентным элюированием 2-10% MeOH/DCM с получением трет-бутил (транс-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-8-карбоксамидо)циклогексил)метилкарбамата (0,0094 г, 29%): LC/MS (Таблица 1, Способ n) Rt = 0,55 мин; MS m/z: 414 (M+H)+.
Общая процедура ММ: Галогенирование имидазола
К раствору имидазола (предпочтительно 1 экв.) в органическом растворителе (таком как DCM, MeOH или ТГФ, предпочтительно ТГФ) добавляют галогенирующий реагент (такой как бром, пиридиний гидробромид пербромид, NCS, NBS или NIS) (0,9-1,1 экв., предпочтительно 1 экв.). Реакционную смесь перемешивают при около -20-150°C (предпочтительно около 0-60°C) примерно в течение 10 минут - 48 часов (предпочтительно около 30 мин). Реакционную смесь затем распределяют между органическим растворителем (таким как EtOAc, DCM или 1,4-диоксан, предпочтительно EtOAc) и водным раствором основания (таким как насыщенный водный раствор NaHCO3 или насыщенный водный раствор Na2CO3, предпочтительно насыщенный водный раствор NaHCO3). Водный слой, необязательно, экстрагируют дополнительным количеством органического растворителя (такого как EtOAc или DCM). Объединенные органические слои можно подвергнуть процедуре, необязательно, промывки насыщенным солевым раствором и концентрирования в вакууме, либо сушки над безводным Na2SO4 или MgSO4 и затем декантирования или фильтрования перед концентрированием при пониженном давлении, с получением целевого продукта и ацетат ртути(II)) в ТГФ (10 мл) при около 0°C добавляли раствор NBS (0,12 г, 0,672 ммоль) в ТГФ (2 мл). Примерно через 30 минут реакционную смесь разбавляли EtOAc (20 мл) и насыщенным водным раствором NaHCO3 (20 мл). Органический слой отделяли, концентрировали в вакууме и очищали хроматографией на силикагеле (40 г) с использованием в качестве элюента EtOAc:DCM:гептан (1:1:2) с получением 3-бром-1-циклогексил-6-тозил-6H-имидазо[1,5-a]пирроло[2,3-e]пиразина (0,27 г, 83%) в виде желтовато-коричневого твердого вещества: LC/MS (Таблица 1, Способ a) Rt = 3,12 мин; MS m/z 473, 475 (1:1) (M+H)+.
Получение #MM.1: 3-бром-1-циклогексил-6-тозил-6H-имидазо[1,5-a]пирроло[2,3-e]пиразин

К раствору 1-циклогексил-6-тозил-6H-имидазо[1,5-a]пирроло[2,3-e]пиразина (0,27 г, 0,67 ммоль, получен с использованием Q из Получения #13, реагента Лоуссона и ацетата ртути(II)) в ТГФ (10 мл) при около 0°C добавляли раствор NBS (0,12 г, 0,672 ммоль) в ТГФ (2 мл). Примерно через 30 минут реакционную смесь разбавляли EtOAc (20 мл) и насыщенным водным раствором NaHCO3 (20 мл). Органический слой отделяли, концентрировали в вакууме и очищали хроматографией на силикагеле (40 г) с использованием в качестве элюента EtOAc:DCM:гептан (1:1:2) с получением 3-бром-1-циклогексил-6-тозил-6H-имидазо[1,5-a]пирроло[2,3-e]пиразина (0,27 g 83%) в виде желтовато-коричневого твердого вещества: LC/MS (Таблица 1, Способ a) Rt = 3,12 мин; MS m/z 473, 475 (1:1) (M+H)+.
Общая процедура NN: Образование амида из карбоновой кислоты и амина с потерей сульфонамидной защитной группы
К смеси 1-замещенного 6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразина с аминогруппой в виде боковой цепи (предпочтительно 1 экв.) и карбоновой кислоты (1-2 экв., предпочтительно 1,5 экв.) в растворителе (таком как DMF или ТГФ, предпочтительно DMF) добавляют связующий агент, такой как EDC•HCl или HATU (1,0-2,0 экв., предпочтительно 1,2 экв.) с органическим основанием (таким как TEA или DIEA, 1-5 экв., предпочтительно 2 экв.). Когда используют EDC•HCl в качестве связывающего реагента, добавляют HOBT (1-3 экв., предпочтительно 1,2 экв.). Примерно через 1-72 часа(предпочтительно около 18 часов) при около 20-60°C (предпочтительно при температуре окружающей среды) добавляют воду и водный слой экстрагируют органическим растворителем, таким как EtOAc или DCM. Объединенные органические слои сушат над безводным Na2SO4 или MgSO4, фильтруют или декантируют и концентрируют при пониженном давлении. Промежуточные соединения и конечные соединения, полученные через эту Общая Процедуру, необязательно, могут быть очищены с использованием одного или нескольких из Способов Очистки, описанных выше.
Иллюстрация Общей Процедуры NN
Пример #NN.1.1: N-((1-((6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)метил)циклобутил)метил)-2-цианоацетамид

К раствору (1-((6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)метил)-циклобутил)метанамина (0,225 г, 0,548 ммоль) (получен с использованием из Примера #1. Стадия D, 2-(1-(трет-бутоксикарбониламино)циклобутил)уксусной кислоты [получали, как описано WO9921824A1], EDC·HCl, B с TEA, E с 4,0 M HCl в 1,4-диоксане) в DMF (10 мл) добавляли цианоуксусную кислоту (0,070 г, 0,822 ммоль), HOBt (0,101 г, 0,658 ммоль), EDC•HCl (0,126 г, 0,658 ммоль) и DIEA (0,190 мл, 1,096 ммоль) с получением коричневого раствора. Смесь перемешивали при температуре окружающей среды примерно в течение 18 часов. Добавляли воду (20 мл) и смесь экстрагировали при помощи EtOAc (3×25 мл). Объединенные органические слои сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Неочищенное вещество очищали флэш-хроматографией на силикагеле с градиентным элюированием 0-10% MeOH в DCM с получением N-((1-((6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)метил)циклобутил)метил)-2-цианоацетамида в виде не совсем белого твердого вещества (0,030 г, 17%): LC/MS (Таблица 1, Способ a) Rt = 1,48 мин; MS m/z: 324 (M+H)+.
Примеры, полученные с использованием Общей Процедуры NN с цианоуксусной кислотой
(Таблица 1, Способ)

Общая процедура OO: Циклизация с использованием POCl 3
К раствору мочевины, амида или гидразида (1-3 экв., предпочтительно 2 экв.), чистого или в органическом растворителе (например, в 1,4-диоксане), добавляют POCl3 (10-200 экв., предпочтительно 100 экв.). Смесь нагревают при около 25-100°C (предпочтительно около 60°C) примерно в течение 1-16 часов (предпочтительно около 1-3 часов). Реакционную смесь охлаждают до температуры окружающей среды и добавляют лед. После растворения pH смеси доводят до около 7 при помощи основания, такого как водный раствор NaOH. В случае осаждения продукта из реакционной смеси, его можно собирать фильтрованием. Альтернативно, продукт можно экстрагировать в органический растворитель (такой как EtOAc или DCM), и органические слои, необязательно, промывают водным раствором основания (таким как насыщенный водный раствор NaHCO3) и/или насыщенным солевым раствором, сушат над безводным Na2SO4 или MgSO4, затем декантируют или фильтруют перед концентрированием при пониженном давлении.
Получение #OO.1: N-((3S,5R)-5-метил-1-(6-тозил-6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)пирролидин-3-ил)циклопропансульфонамид

В колбу загружали (2R,4S)-4-(циклопропансульфонамидо)-2-метил-N-((5-тозил-5H-пирроло[2,3-b]пиразин-2-ил)метил)пирролидин-1-карбоксамид (0,11 г, 0,207 ммоль, получен с использованием E из Получения #14 и J из Примера #5 Стадия C и CDI) и POCl3 (1,9 мл, 21 ммоль). Реакционную смесь нагревали до около 60°C с получением гомогенной смеси. Примерно через 2 часа реакционную смесь охлаждали до температуры окружающей среды и добавляли колотый лед. После того как лед растаял, добавляли 2 н водный раствор NaOH до достижения pH около 7. Полученный осадок собирали фильтрованием и сушили в вакууме с получением N-((3S,5R)-5-метил-1-(6-тозил-6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)пирролидин-3-ил)циклопропан-сульфонамида (0,10 г, 94%) в виде желтовато-коричневого твердого вещества: LC/MS (Таблица 1, Способ a) Rt = 2,14 мин; MS m/z: 515 (M+H)+.
Общая процедура OO.1: Циклизация с использованием POCl 3
К мочевине, амиду или гидразиду, чистому или в органическом растворителе (таком как 1,4-диоксан, DCE или толуол), добавляют POCl3 (3-200 экв., предпочтительно 100 экв.). Смесь нагревают при около 25-110°C (предпочтительно около 100°C) примерно в течение 1-16 часов (предпочтительно около 1-3 часов). Реакционной смеси дают охладиться до температуры окружающей среды. Реакционную смесь можно добавить ко льду, или лед можно добавить. Альтернативно, летучие вещества можно удалить при пониженном давлении. Необязательно, добавляют DCM с последующим медленным добавлением MeOH и затем смесь концентрируют при пониженном давлении. Добавляют водный слой, такой как вода или водный раствор HCl, и можно добавить органический растворитель, такой как 1,4-диоксан, и раствор можно нагреть до около 30-110°C (предпочтительно около 100°C) примерно в течение 0,5-6 часов (предпочтительно около 3 часов). После концентрации при пониженном давлении pH смеси можно довести при помощи основания, такого как водный раствор NaOH или NaHCO3 (предпочтительно до около pH 7) и добавить органический растворитель, такой как EtOAc или DCM. Продукт можно собирать фильтрованием или экстрагировать в органический растворитель (такой как EtOAc или DCM). Органические слои, необязательно, промывают водным раствором основания (таким как насыщенный водный раствор NaHCO3) и/или насыщенным солевым раствором, сушат над безводным Na2SO4 или MgSO4 и затем декантируют или фильтруют перед концентрированием при пониженном давлении.
Получение #OO.1.1: 3-тозил-3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-амин
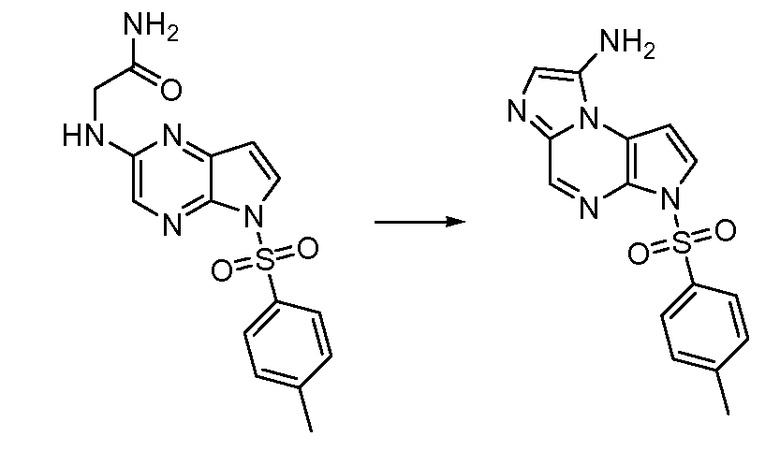
К 2-(5-тозил-5H-пирроло[3,2-b]пиразин-2-иламино)ацетамиду (0,845 г, 2,45 ммоль, получен с использованием E из Получения #S.1.1 и HCl) в атмосфере азота добавляли POCl3 (5,0 мл, 54 ммоль). Примерно через 15 минут подсоединяли обратный холодильник и смесь нагревали до около 100°C. Примерно через 2 часа раствору давали охладиться до температуры окружающей среды. Смесь концентрировали при пониженном давлении. Остаток суспендировали в DCM (10 мл) и медленно обрабатывали при помощи MeOH (10 мл). Реакционную смесь перемешивали примерно в течение 5 минут, затем концентрировали при пониженном давлении. Остаток растворяли в MeOH (20 мл) и затем концентрировали при пониженном давлении. Остаток растворяли в 1,4-диоксане (5 мл) и 2н водном растворе HCl (5 мл). Раствор нагревали до около 100°C примерно в течение 3 часов. Раствору давали охладиться до температуры окружающей среды и летучие вещества удаляли при пониженном давлении. Водную смесь суспендировали в насыщенном водном растворе NaHCO3/воде (1:1, 100 мл) и DCM (50 мл). Твердое вещество собирали фильтрованием, промывали водой и DCM и сушили с получением 3-тозил-3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-амина (0,343 г, 43%): LC/MS (Таблица 1, Способ n) Rt = 0,56 мин; MS m/z: 328 (M+H)+.
Общая процедура PP: Взаимодействие амина с арилбороновой кислотой
К раствору бороновой кислоты (предпочтительно 1-3 экв.) в органическом растворителе (таком как DCM или MeCN) добавляют органическое основание, такое как DIEA (1-5 экв., предпочтительно 1 экв.), неорганический катализатор (такой как ацетат меди (II) моногидрат (0,1 до 0,5 экв., предпочтительно 0,25 экв.), амин (предпочтительно 1 экв.) и осушитель (такой как 4 Å молекулярные сита). Реакционную смесь продувают кислородом (1-5 раз, предпочтительно 3 раза) и нагревают при около 20-60°C (предпочтительно около 40-50°C) примерно в течение 1-24 часов (предпочтительно около 18 часов) в атмосфере кислорода. Когда реакция не достигает завершения, можно добавить дополнительное количество неорганического катализатора (такого как ацетат меди (II) моногидрат (0,1 до 0,5 экв., предпочтительно 0,25 экв.). Реакционной смеси дают охладиться до температуры окружающей среды, затем смесь концентрируют при пониженном давлении
Иллюстрация Общей Процедуры PP
Получение #PP.1*: 3-((1S,3R,4S)-3-метил-4-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентиламино)бензонитрил

К раствору 3-цианофенилбороновой кислоты (0,143 г, 0,974 ммоль) в DCM (4 мл) добавляли ацетат меди (II) моногидрат (0,013 г, 0,122 ммоль) и 4 Å молекулярные сита. Реакционную смесь продували 3 раза кислородом. Добавляли раствор (1S,3R,4S)-3-метил-4-(6-тозил-6H-пирроло[2,3-e][1,2,4]-триазоло[4,3-a]пиразин-1-ил)циклопентанамина (0,20 г, 0,48 ммоль, Получение #19.2) и DIEA (0,085 мл, 0,487 ммоль) в MeCN (1 мл) и реакционную смесь нагревали при около 45°C примерно в течение 18 часов. Добавляли дополнительное количество ацетата меди (II) моногидрата (0,013 г, 0,122 ммоль) и реакционную смесь перемешивали примерно в течение 4 часов. Реакционную смесь фильтровали через слой Целита® и концентрировали при пониженном давлении. Неочищенное вещество очищали хроматографией на силикагеле с градиентным элюированием 0-60% EtOAc в DCM с получением 3-((1S,3R,4S)-3-метил-4-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентиламино)бензонитрила (0,16 г, 48%) в виде темного коричневого твердого вещества: LC/MS (Таблица 1, Способ c) Rt = 1,54 мин; MS m/z: 512 (M+H)+.
Общая процедура QQ: Образование мочевины из амина и изоцианата
В колбу, содержащую амин или соль амина (1 экв.) в органическом растворителе (таком как ТГФ, DCM или MeCN, предпочтительно DCM), добавляют, необязательно, основание (такое как DIEA или TEA, предпочтительно DIEA, 1-3 экв., предпочтительно 1 экв.) и реакционную смесь перемешивают при температуре окружающей среды примерно в течение 0-30 минут (предпочтительно около 5 мин). Добавляют изоцианат (1-5 экв., предпочтительно 1 экв.) и смесь перемешивают при около 10-60°C (предпочтительно при температуре окружающей среды) примерно в течение 1-24 часов (предпочтительно около 18 часов). Органический растворитель необязательно удаляют при пониженном давлении, если только не используют MeCN, в этом случае растворитель предпочтительно удаляют при пониженном давлении. Неочищенное вещество можно распределить между органическим растворителем (таким как EtOAc или DCM) и водой, водным раствором основания (таким как насыщенный водный раствор NaHCO3) или насыщенным солевым раствором. Слои разделяют и водный слой необязательно промывают органическим растворителем (таким как EtOAc или DCM). Объединенные органические экстракты сушат над безводным Na2SO4 или MgSO4, фильтруют и концентрируют при пониженном давлении с получением целевого соединения.
Иллюстрация Общей Процедуры QQ
Пример #QQ.1.1*: (3R,4R)-N-(2,4-дифторфенил)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-карбоксамид

В круглодонную колбу загружали 1-((3R,4R)-4-метилпиперидин-3-ил)-6H-имидазо[1,5-a]пирроло[2,3-e]пиразин гидрохлорид (0,05 г, 0,17 ммоль, Пример #5 Стадия J) и DIEA (0,03 мл, 0,17 ммоль) в DCM (1,6 мл). Реакционную смесь перемешивали примерно в течение 5 минут при температуре окружающей среды, затем добавляли 2,4-дифтор-1-изоцианатобензол (0,02 мл, 0,17 ммоль) и реакционную смесь перемешивали при температуре окружающей среды примерно в течение 18 часов. Реакционную смесь разбавляли при помощи DCM (5 мл) и промывали водой (2 мл). Водный слой обратно экстрагировали в DCM (2 мл). Объединенные органические слои сушили над безводным MgSO4, фильтровали и концентрировали при пониженном давлении. Вещество очищали при помощи ОФ-ВЭЖХ (Таблица 1, Способ e) с получением (3R,4R)-N-(2,4-дифторфенил)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метилпиперидин-1-карбоксамида (0,014 г, 20%): LC/MS (Таблица 1, Способ j) Rt = 1,77 мин; MS m/z 411 (M+H)+.
Примеры, полученные из 1-((3R,4R)-4-метилпиперидин-3-ил)-6H-имидазо[1,5-a]пирроло[2,3-e]пиразингидрохлорида (Пример #5 Стадия J) с использованием Общей Процедуры QQ
(Таблица 1, Способ)

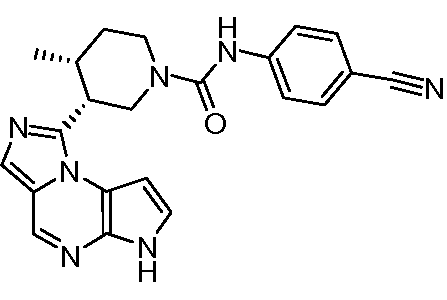
Общая процедура RR: Образование мочевины из амина, гетероариламина и фенилхлорформиата
В колбу, содержащую гетероариламин (1-6 экв., предпочтительно 2,1 экв.) в органическом растворителе или смеси растворителей (таких как ТГФ/MeCN, ТГФ, DCM или MeCN, предпочтительно MeCN), основание, такое как пиридин, DIEA или TEA, предпочтительно TEA (1-6 экв., предпочтительно 2 экв.) и DMAP (0,1-0,6 экв., предпочтительно 0,2 экв.), добавляют фенилхлорформиат (1-6 экв., предпочтительно 2,0 экв.) при около -5-25°C (предпочтительно около 0°C). Реакционную смесь нагревают до температуры окружающей среды и перемешивают примерно в течение 1-4 часов (предпочтительно около 3 часов). Органический растворитель необязательно удаляют при пониженном давлении. Неочищенное вещество можно распределить между органическим растворителем, таким как EtOAc, DCM или Et2O (предпочтительно Et2O), и водой или насыщенным солевым раствором. Слои разделяют и органический слой необязательно промывают водой или насыщенным солевым раствором, сушат над безводным Na2SO4 или MgSO4, фильтруют и концентрируют при пониженном давлении с получением неочищенного карбамата. Неочищенный карбамат растворяют в органическом растворителе, таком как MeCN, ТГФ или DMF (предпочтительно MeCN), и добавляют к раствору амина или соли амина (1-2 экв., предпочтительно 1 экв.) и основания, такого как пиридин, TEA или DIEA (предпочтительно DIEA, 1-2 экв., предпочтительно 1 экв.), в органическом растворителе, таком как MeCN, ТГФ или DMF (предпочтительно MeCN), и перемешивают при около 25-80°C (предпочтительно около 70°C) примерно в течение 0,5-48 часов (предпочтительно около 2-18 часов). Растворитель необязательно удаляют при пониженном давлении. Неочищенное вещество можно распределить между органическим растворителем (таким как EtOAc или DCM) и водой, водным раствором основания (таким как водный раствор NaHCO3) или насыщенным солевым раствором. Слои разделяют и органический слой необязательно промывают водой, водным раствором основания (таким как насыщенный водный раствор NaHCO3) и/или насыщенным солевым раствором, сушат над безводным Na2SO4 или MgSO4, фильтруют и концентрируют при пониженном давлении с получением целевого соединения.
Иллюстрация Общей Процедуры RR
Пример #RR.1.1*: (3R,4R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метил-N-(пиримидин-4-ил)пиперидин-1-карбоксамид

К раствору 4-аминопиримидина (0,04 г, 0,43 ммоль), TEA (0,07 мл, 0,47 ммоль) и DMAP (0,006 г, 0,05 ммоль) в MeCN (1 мл) при около 0°C добавляли фенилхлорформиат (0,05 мл, 0,41 ммоль). Реакционную смесь нагревали до температуры окружающей среды и перемешивали примерно в течение 3 часов. К реакционной смеси добавляли воду (2 мл) и Et2O (5 мл). Органический слой отделяли, промывали водой (2 мл), сушили над безводным MgSO4, фильтровали и концентрировали при пониженном давлении с получением неочищенного карбамата. Карбамат растворяли в MeCN (1 мл) и добавляли раствор 1-((3R,4R)-4-метилпиперидин-3-ил)-6H-имидазо[1,5-a]пирроло[2,3-e]пиразингидрохлорида (0,06 г, 0,21 ммоль, Пример #5 Стадия J) и DIEA (0,04 мл, 0,21 ммоль) в MeCN (1 мл). Реакционную смесь нагревали до около 70°C примерно в течение 2 часов. Растворитель удаляли при пониженном давлении. Неочищенный остаток растворяли в DCM (5 мл) и промывали водой (2 мл), насыщенным солевым раствором (3 мл), сушили над безводным MgSO4, фильтровали и концентрировали при пониженном давлении. Неочищенное вещество очищали при помощи ОФ-ВЭЖХ (Таблица 1, Способ e) с получением (3R,4R)-3-(6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)-4-метил-N-(пиримидин-4-ил)пиперидин-1-карбоксамида (0,007 г, 9%): LC/MS (Таблица 1, Способ b) Rt = 1,40 мин; MS m/z 377 (M+H)+.
Примеры, полученные из 1-((3R,4R)-4-метилпиперидин-3-ил)-6H-имидазо[1,5-a]пирроло[2,3-e]пиразингидрохлорида (Пример #5 Стадия J) с использованием Общей Процедуры RR
(Таблица 1, Способ)
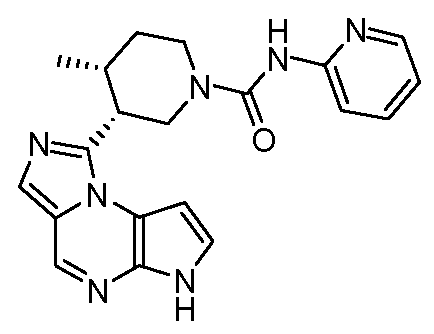

Общая процедура SS: Гидролиз сложного эфира до спирта
Раствор сложного эфира (предпочтительно 1 экв.) в органическом растворителе, таком как ТГФ, MeOH или EtOH (предпочтительно MeOH) добавляют к основанию в органическом растворителе (таком как NaOH в MeOH) или водному раствору основания (такого как Na2CO3 или NaOH) (1-20 экв., предпочтительно 2-10 экв.). Реакционную смесь перемешивают при температуре окружающей среды примерно в течение 1-16 часов (предпочтительно около 3 часов). Смесь распределяют между органическим растворителем (таким как EtOAc или DCM) и водным раствором основания (таким как насыщенный водный раствор NaHCO3 или насыщенный водный раствор Na2CO3, предпочтительно насыщенный водный раствор NaHCO3). Органический слой разделяют, сушат над безводным Na2SO4 или MgSO4, фильтруют и концентрируют при пониженном давлении с получением целевого соединения.
Иллюстрация Общей Процедуры SS
Получение #SS.1: 3-этил-4-(6-((2-(триметилсилил)этокси)метил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентанол

К раствору NaOH (0,088 г, 2,20 ммоль) в MeOH (8 мл) добавляли раствор 3-этил-4-(6-((2-(триметилсилил)этокси)метил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентилбензоата (0,158 г, 0,312 ммоль, получен с использованием KK из Получения #20.2) в MeOH (2 мл). Реакционную смесь перемешивали при температуре окружающей среды примерно в течение 3 часов. Растворитель удаляли при пониженном давлении и добавляли DCM (150 мл). Органический слой промывали водой (5 мл), насыщенным водным раствором NaHCO3 (15 мл), насыщенным солевым раствором (15 мл) и сушили над безводным MgSO4, фильтровали и концентрировали при пониженном давлении с получением 3-этил-4-(6-((2-(триметилсилил)этокси)метил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентанола (0,123 г, 98%) в виде прозрачного масла: LC/MS (Таблица 1, Способ b) Rt = 2,34 мин; MS m/z: 402 (M+H)+.
Общая процедура TT: Кислотно-опосредованное преобразование сложного эфира в карбоновую кислоту
К раствору сложного эфира (предпочтительно 1 экв.) в органическом растворителе, таком как 1,4-диоксан или ТГФ (предпочтительно 1,4-диоксан), добавляют HCl (0,5-12 н, предпочтительно 1-6 н водный раствор; 5-100 экв., предпочтительно 10-20 экв.). Реакционную смесь нагревают при около 30-120°C (предпочтительно около 60°C) примерно в течение 12-120 часов (предпочтительно около 36-72 часов). В любом случае присутствия дополнительной кислотно-лабильной группы (например, Boc группа), эту группу также можно расщепить в процессе реакции. Реакционную смесь концентрируют при пониженном давлении и pH доводят до около 8 водным раствором неорганического основания, такого как NaHCO3 или Na2CO3 (предпочтительно насыщенный водный раствор NaHCO3), и водную фазу экстрагируют органическим растворителем, таким как DCM или EtOAc (предпочтительно EtOAc). Органический экстракт, необязательно, промывают насыщенным солевым раствором, сушат над осушителем, таким как безводный MgSO4 или Na2SO4 (предпочтительно безводный MgSO4), и концентрируют при пониженном давлении с получением целевого соединения.
Иллюстрация Общей Процедуры TT
Получение #TT.1: 4-(дибензиламино)-2-метилциклопентанкарбоновая кислота
Этил 4-(дибензиламино)-2-метилциклопентанкарбоксилат (3,65 г, 10,38 ммоль, Пример #7, стадия Н) растворяли в HCl (6 н водный раствор, 20 мл) и 1,4-диоксане (50 мл) и полученную смесь нагревали при около 60°C примерно в течение 72 часов. Растворители удаляли при пониженном давлении и остаток нейтрализовали путем добавления насыщенного водного раствора NaHCO3 (40 мл) и экстрагировали при помощи EtOAc (50 мл). Органическую фазу промывали насыщенным солевым раствором (40 мл), сушили над безводным MgSO4 и концентрировали при пониженном давлении с получением 4-(дибензиламино)-2-метилциклопентанкарбоновой кислоты (3,3 г, 98%) в виде белого аморфного твердого вещества: LC/MS (Таблица 1, Способ a) Rt = 1,66 мин; MS m/z 324 (M+H)+.
Общая процедура UU: Образование 2,2,2-трихлорацетимидата
К смеси спирта (предпочтительно 1 экв.) в органическом растворителе (таком как Et2O, гептан или DCM, предпочтительно DCM) при температуре от около -20°C до 30°C (предпочтительно около 0°C) добавляют водный раствор основания (такой как водный раствор гидроксида натрия или гидроксида калия, предпочтительно водный раствор гидроксида калия, 1-20 экв., предпочтительно 10 экв.). Добавляют каталитическое количество агента фазового переноса (предпочтительно гидросульфат тетрабутиламмония, 0,01-0,5 экв., предпочтительно 0,1 экв.) с последующим добавлением 2,2,2-трихлорацетонитрила (1-10 экв., предпочтительно 5 экв.). Реакционной смеси дают нагреться до температуры окружающей среды и перемешивают при около 15-60°C (предпочтительно при температуре окружающей среды) примерно в течение 5-48 часов (предпочтительно около 14 часов). Слои разделяют и водный слой экстрагируют органическим растворителем (таким как Et2O, EtOAc или DCM, предпочтительно DCM). Объединенные органические слои промывают водой, водным раствором основания (таким как насыщенный водный раствор Na2CO3 или NaHCO3) или насыщенным солевым раствором, сушат над безводным Na2SO4 или MgSO4, фильтруют и концентрируют при пониженном давлении. Альтернативно, можно использовать органическое основание (такое как DBU) в качестве основания для этой реакции. В этом случае, к смеси спирта (предпочтительно 1 экв.) в органическом растворителе (таком как Et2O, гептан или DCM, предпочтительно DCM) при температуре около -20°C до 30°C (предпочтительно около 0°C) добавляют 2,2,2-трихлорацетонитрил (1-10 экв., предпочтительно 5 экв.), с последующим добавлением органического основания, предпочтительно DBU (0,2-1 экв., предпочтительно около 0,4 экв.). Реакционную смесь перемешивают при около -20-30°C (предпочтительно около 0°C) примерно в течение 0,5-10 часов (предпочтительно около 1 часа), затем концентрируют.
Иллюстрация Общей Процедуры UU
Получение #UU.1: этил 2-этил-4-(2,2,2-трихлор-1-иминоэтокси)циклопентанкарбоксилат

К этил 2-этил-4-гидроксициклопентанкарбоксилату (3,52 г, 18,9 ммоль, Получение #P.1) в DCM (21 мл) при около 0°C добавляли водный раствор гидроксида калия (50%, 21 мл, 189 ммоль), гидросульфат тетрабутиламмония (0,64 г, 1,891 ммоль) и 2,2,2-трихлорацетонитрил (9,5 мл, 95 ммоль). Реакционной смеси давали нагреться до температуры окружающей среды и перемешивали при температуре окружающей среды примерно в течение 14 часов. Слои разделяли и водный слой экстрагировали при помощи DCM (4×60 мл). Объединенные органические слои промывали водой (2×50 мл), насыщенным солевым раствором (60 мл), сушили над безводным MgSO4, фильтровали и концентрировали при пониженном давлении. Вещество очищали хроматографией на силикагеле с градиентным элюированием 15-50% EtOAc в гептане с получением этил 2-этил-4-(2,2,2-трихлор-1-иминоэтокси)циклопентанкарбоксилата (2,80 г, 45%) в виде бесцветного масла: LC/MS (Таблица 1, Способ b) Rt = 2,91 мин; MS m/z: 330 (M+H)+.
Общая процедура VV: TBDMS-защита спирта
К смеси спирта (предпочтительно 1 экв.) в органическом растворителе (предпочтительно DMF) добавляют TBDMS-Cl (1-5 экв., предпочтительно 1,2 экв.) и имидазол (1-10 экв., предпочтительно 2,5 экв.). Реакционную смесь перемешивают при около 10-60°C (предпочтительно при температуре окружающей среды) примерно в течение 1-24 часов (предпочтительно около 3 часов). Добавляют органический растворитель (такой как гептан, гексан или пентан, предпочтительно гептан). Слои разделяют и нижний слой (DMF слой) экстрагируют органическим растворителем (таким как пентан, гексан или гептан, предпочтительно гептан). Объединенные экстракты промывают водой, водным раствором основания (таким как насыщенный водный раствор NaHCO3) или насыщенным солевым раствором, сушат над безводным Na2SO4 или MgSO4, фильтруют и концентрируют при пониженном давлении.
Иллюстрация Общей Процедуры VV
Получение #VV.1: Этил 4-(трет-бутилдиметилсилилокси)-2-этилциклопентанкарбоксилат
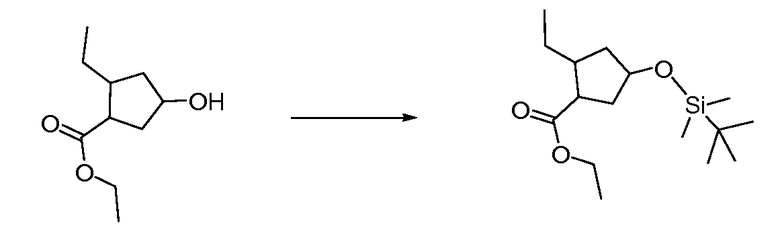
К раствору этил 2-этил-4-гидроксициклопентанкарбоксилата (4,97 г, 26,7 ммоль, получен с использованием II из Получения #P.1, SS с NaOH)) в DMF (9 мл) добавляли TBDMS-Cl (4,83 г, 32,1 ммоль) и имидазол (4,55 г, 66,8 ммоль). Реакционную смесь перемешивали при температуре окружающей среды примерно в течение 3 часов. Добавляли гептан (30 мл). Слои разделяли и нижний слой (DMF слой) экстрагировали гептаном (3×30 мл). Объединенные органические экстракты промывали водой (2×30 мл), насыщенным солевым раствором (30 мл), сушили над безводным MgSO4, фильтровали и концентрировали при пониженном давлении. Вещество очищали хроматографией на силикагеле с градиентным элюированием 0-15% EtOAc в гептане с получением этил 4-(трет-бутилдиметилсилилокси)-2-этилциклопентанкарбоксилата (5,16 г, 64%) в виде бесцветного масла: 1H ЯМР (400 МГц, CDCl3) δ 4,45 (м, 1H), 4,11 (м, 2H), 3,08 (м, 1H), 2,34 (м, 1H), 2,18 (м, 1H), 1,75 (м, 2H), 1,57 (м, 1H), 1,41 (м, 1H), 1,25 (м, 3H), 1,10 (м, 1H), 0,90 (м, 3H), 0,87 (с, 9H), 0,03 (с, 6H).
Общая процедура WW: Образование кеталя
К раствору кетона (предпочтительно 1 экв.), органического растворителя (такого как DCM, DCE или толуол, предпочтительно DCM), диола, такого как этиленгликоль (1-3 экв., предпочтительно 2 экв.) и кислоты, такой как моногидрат п-толуолсульфоновой кислоты (0,1-0,5 экв., предпочтительно 0,2 экв.), необязательно добавляют дегидратирующий агент, такой как триэтилортоформиат или триметилортоформиат (предпочтительно триэтилортоформиат, 1-4 экв., предпочтительно 1,5 экв.). Реакционную смесь перемешивают при температуре от комнатной до около 110°C (предпочтительно при комнатной температуре в присутствии дегидратирующего агента, такого как триэтилортоформиат, или предпочтительно при около 110°C в отсутствие дегидратирующего агента) примерно в течение 16-96 часов (предпочтительно около 24 часов). Если реакционную смесь нагревали, ее охлаждают до комнатной температуры. Реакционную смесь обрабатывают с использованием одного из следующих способов. Способ 1: К реакционной смеси добавляют воду, слои разделяют и органический раствор, необязательно, промывают насыщенным солевым раствором, сушат над безводным Na2SO4 или MgSO4, затем декантируют или фильтруют перед концентрированием при пониженном давлении. Способ 2: Реакционную смесь концентрируют при пониженном давлении и очищают непосредственно.
Иллюстрация Общей Процедуры WW
Получение #WW.1: этил 2-этил-4-оксоциклопентанкарбоксилат

В круглодонную колбу загружали этил 2-этил-4-оксоциклопентанкарбоксилат (1,5 г, 8,1 ммоль, Пример #22, Стадия B) в DCM (22 мл). В колбу добавляли этиленгликоль (0,91 мл, 16 ммоль), триэтилортоформиат (2,0 мл, 12 ммоль) и моногидрат п-толуолсульфоновой кислоты (0,31 г, 1,6 ммоль). Реакционную смесь перемешивали при комнатной температуре примерно в течение 24 часов. Раствор концентрировали при пониженном давлении с получением коричневого масла, которое растворяли в минимальном количестве EtOAc и очищали хроматографией на силикагеле (Silicycle 25 г колонка) с градиентным элюированием 0-50% EtOAc в гептане с получением этил 2-этил-4-оксоциклопентанкарбоксилата (1,6 г, 83%) в виде светло-желтого масла: LC/MS (Таблица 1, Способ c) MS m/z 229 (M+H)+; 1H ЯМР (CDCl3) δ 4,14 (кв., 2H), 3,90 (м, 4H), 2,99 (кв., 1H), 2,32-2,27 (м, 1H), 2,26-2,11 (м, 1H), 2,05-1,99 (м, 1H), 1,96-1,91 (м, 1H), 1,83-1,78 (м, 1H), 1,46-1,39 (м, 1H), 1,31-1,24 (м, 1H), 1,26 (т, 3H), 0,90 (т, 3H).
Общая процедура XX: Катализируемое палладием связывание гидразона
К смеси замещенного 5-хлор-4-(гидразонометил)-1H-пирроло[2,3-b]пиридина (1 экв.) в органическом растворителе (предпочтительно NMP) добавляют основание (такое как K2CO3 или трет-бутоксид натрия, предпочтительно трет-бутоксид натрия [1-4 экв., предпочтительно 2,5 экв.]), палладиевый катализатор (предпочтительно ацетат палладия [0,01-0,2 экв., предпочтительно 0,1 экв.]) и лиганд (предпочтительно (R)-1-[(S)-2-(дициклогексилфосфино)ферроценил]этил-ди-трет-бутилфосфин [0,01-0,2 экв., предпочтительно 0,1 экв.]). Реакционную смесь нагревают термически или в микроволновой печи (предпочтительно в микроволновой печи) при около 100-165°C (предпочтительно 150°C примерно в течение 10 минут - 6 часов (предпочтительно около 2 часов)). Реакционную смесь фильтруют через слой Целита®, промывая органическим растворителем (таким как EtOAc или DCM, предпочтительно EtOAc), и концентрируют при пониженном давлении для удаления промывочного растворителя. Неочищенное вещество, необязательно, снова подвергают реакционным условиям. Затем неочищенное вещество распределяют между органическим растворителем (таким как EtOAc или DCM, предпочтительно EtOAc) и водой и водную фазу экстрагируют органическим растворителем (таким как EtOAc или DCM, предпочтительно EtOAc), промывают водой и/или насыщенным солевым раствором, сушат над безводным MgSO4, фильтруют и концентрируют при пониженном давлении.
Иллюстрация Общей Процедуры XX
Получение #XX.1: трет-бутил бензил(4-(3,6-дигидропиразоло[4,3-d]пирроло[2,3-b]пиридин-1-ил)бицикло[2,2,2]октан-1-ил)карбамат

В микроволновой реакционный сосуд загружали трет-бутил бензил(4-((5-хлор-1H-пирроло[2,3-b]пиридин-4-ил)(гидразоно)-метил)бицикло[2,2,2]октан-1-ил)карбамат (0,700 г, 1,38 ммоль, Пример #29 Стадия G) и NMP (11 мл). Добавляли последовательно трет-бутоксид натрия (0,331 г, 3,44 ммоль), ацетат палладия (0,031 г, 0,138 ммоль) и (R)-1-[(S)-2-(дициклогексилфосфино)ферроценил]этил-ди-трет-бутилфосфин (0,076 г, 0,138 ммоль). Реакционную смесь нагревали в микроволновом устройстве Biotage при около 150°C примерно в течение 2 часов (максимальное давление 250 ф/дюйм2 (17,58 кг/см2), линейное нарастание 1 мин, максимальная мощность 150 Ватт). Реакционную смесь фильтровали через слой Целита®, промывая при помощи EtOAc (около 15 мл), и EtOAc удаляли при пониженном давлении. Оставшееся вещество переносили в микроволновой сосуд и добавляли трет-бутоксид натрия (0,331 г, 3,44 ммоль), ацетат палладия (0,031 г, 0,138 ммоль) и (R)-1-[(S)-2-(дициклогексилфосфино)ферроценил]-этил-ди-трет-бутилфосфин (0,076 г, 0,138 ммоль). Реакционную смесь нагревали в микроволновом устройстве Biotage при около 160°C примерно в течение 2 часов (максимальное давление 250 ф/дюйм2 (17,58 кг/см2), 1 мин линейное нарастание, максимальная мощность 150 Ватт). Реакционную смесь фильтровали через слой Целита® с промывкой при помощи EtOAc (около 20 мл). Добавляли воду (15 мл) и слои разделяли. Водный слой экстрагировали при помощи EtOAc (2×10 мл) и объединенные органические слои промывали водой (3×10 мл) и насыщенным солевым раствором (5×15 мл), сушили над безводным MgSO4, фильтровали и концентрировали при пониженном давлении. Оставшийся темный остаток очищали хроматографией на силикагеле с градиентным элюированием 10-100% EtOAc в гептане с получением трет-бутил бензил(4-(3,6-дигидропиразоло[4,3-d]пирроло[2,3-b]пиридин-1-ил)бицикло[2,2,2]октан-1-ил)карбамата с 0,5 экв. EtOAc в качестве эксципиента (0,281 г, 39,5%) в виде светло-коричневого твердого вещества.: LC/MS (Таблица 1, Способ b) Rt = 2,57 мин; MS m/z: 472 (M+H)+.
Общая процедура YY: Присоединение по Михаэлю амина, соли амина или гетероцикла к α,β-ненасыщенному сульфонамиду
К смеси α,β-ненасыщенного сульфонамида (1-3 экв., предпочтительно 1,0 экв.) и амина, соли амина или гетероцикла (1-10 экв., предпочтительно 4 экв.) в органическом растворителе или смеси растворителей (таких как ТГФ, n-PrOH, вода, EtOH, ТГФ/PrOH, ТГФ/EtOH, предпочтительно n-PrOH), необязательно, добавляют основание (такое как DIEA или TEA 0-25 экв., предпочтительно DIEA 10-20 экв.). Смесь перемешивают при около 25-100°C (предпочтительно около 60-80°C) примерно в течение 2-72 часов (предпочтительно около 18-20 часов). В случаях, когда реакция не достигает завершения, согласно данным LC/MS, ВЭЖХ, и/или ТСХ; можно добавить дополнительное количество амина, соли амина или гетероцикла (1-10 экв., предпочтительно 2 экв.) и/или сорастворитель (такой как EtOH). Реакцию продолжают при около 25-100°C (предпочтительно около 80°C) примерно в течение 1-24 часов (предпочтительно около 1-2 часов). В случаях, когда присутствует основно-лабильная защитная группа (например, тозил), соединение можно подвергнуть процедуре удаления защиты. Реакционную смесь оставляют для достижения температуры окружающей среды и органический растворитель необязательно удаляют при пониженном давлении. Неочищенное вещество можно распределить между органическим растворителем (таким как EtOAc или DCM) и водой, водным раствором основания (такого как насыщенный водный раствор NaHCO3) или насыщенным солевым раствором. Слои разделяют и органический слой необязательно промывают водой, водным раствором основания (таким как насыщенный водный раствор NaHCO3) или насыщенным солевым раствором, сушат над безводным Na2SO4 или MgSO4, фильтруют и концентрируют при пониженном давлении с получением целевого соединения.
Иллюстрация Общей Процедуры YY
Пример #YY.1.1*: 2-(4-циано-1H-пиразол-1-ил)-N-((1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)этансульфонамид

Смесь N-((1S,3R,4S)-3-этил-4-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)этансульфонамида (0,065 г, 0,13 ммоль, получен с использованием K.1 из Примера #8 Стадия M и 2-хлорэтансульфонилхлорида с TEA), DIEA (0,30 мл, 1,7 ммоль) и 1H-пиразол-4-карбонитрила (0,047 г, 0,51 ммоль, American Custom Chemicals Corp) в n-PrOH (2,0 мл) перемешивали примерно в течение 2 часов при около 60°C, затем при около 80°C примерно в течение 18 часов. Реакционную смесь охлаждали до температуры окружающей среды и концентрировали при пониженном давлении. Неочищенный остаток растворяли в DCM (10 мл), промывали насыщенным раствором NaHCO3 (5 мл), сушили над безводным MgSO4, фильтровали и концентрировали при пониженном давлении. Неочищенное вещество очищали хроматографией на силикагеле с градиентным элюированием 0-5% MeOH в DCM с получением 2-(4-циано-1H-пиразол-1-ил)-N-((1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)этансульфонамида (0,024 г, 42%): LC/MS (Таблица 1, Способ b) Rt = 1,63 мин; MS m/z: 454 (M+H)+.
Примеры, полученные из N-((1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)-2-(1H-1,2,4-триазол-1-ил)этансульфонамида (получен с использованием K.1 из Примера #8 Стадия M и 2-хлорэтансульфонилхлорида с TEA) с использованием Общей Процедуры YY
(Таблица 1, Способ)
Общая процедура ZZ: Образование оксазолидинонсульфономочевины
К смеси амина или соли амина (1 экв.) и 2-хлорэтилхлорсульфонилкарбамата (получен, как подробно описано в Bioorg. Med. Chem. Lett., 2006 16, 3367-3370)(1-3 экв., предпочтительно 1 экв.) в органическом растворителе (предпочтительно DCM) добавляют основание (такое как DIEA или TEA, предпочтительно TEA [2-5 экв., предпочтительно 3 экв.]) и необязательно DMAP (1-3 экв., предпочтительно 1 экв.) и перемешивают при температуре окружающей среды примерно в течение 10 минут - 6 часов (предпочтительно около 1 часа). Растворитель удаляют при пониженном давлении. В случаях, когда используют DMAP, неочищенное вещество можно распределить между органическим растворителем (таким как EtOAc или DCM) и водой или насыщенным солевым раствором, с последующей сушкой над безводным MgSO4, фильтрованием и концентрированием при пониженном давлении.
Иллюстрация Общей Процедуры ZZ
Получение #ZZ.1*: N-((1S,3R,4S)-3-этил-4-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)-2-оксооксазолидин-3-сульфонамид
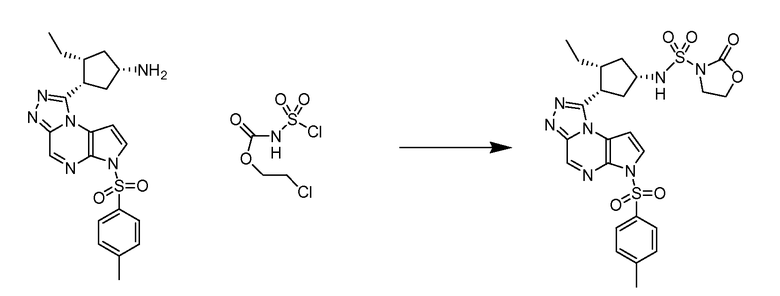
К смеси 2-хлорэтилхлорсульфонилкарбамата (получен, как подробно описано в Bioorg. Med. Chem. Lett., 2006 16, 3367-3370; 0,052 г, 0,236 ммоль) и (1S,3R,4S)-3-этил-4-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентанамина (0,100 г, 0,236 ммоль, Пример #8 Стадия M) в DCM (2,4 мл) добавляли TEA (0,098 мл, 0,71 ммоль) и реакционную смесь перемешивали при температуре окружающей среды примерно в течение 1 часа. Реакционную смесь концентрировали при пониженном давлении с получением N-((1S,3R,4S)-3-этил-4-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)-2-оксооксазолидин-3-сульфонамида (0,098 г, 65%) в виде светло-коричневого твердого вещества: LC/MS (Таблица 1, Способ a) Rt = 2,18 мин; MS m/z 574 (M+H)+.
Общая процедура AAA: Образование сульфонилмочевины из оксазолидинонсульфономочевины
К раствору оксазолидинона (предпочтительно 1 экв.) в органическом растворителе (предпочтительно MeCN) добавляют амин или гидрохлоридную соль амина (1-2 экв., предпочтительно 1,5 экв.) и органическое основание, такое как TEA или DIEA (1-4 экв., предпочтительно 2 экв.). Реакционную смесь подвергают облучению в микроволновой печи при около 100-150°C (предпочтительно 120°C) примерно в течение 0,5-1 часа (предпочтительно 0,5 часа). Реакционную смесь охлаждают до температуры окружающей среды и, необязательно, концентрируют при пониженном давлении с получением остатка. Реакционную смесь или остаток, необязательно, распределяют между органическим растворителем (таким как DCM или EtOAc, предпочтительно EtOAc), водой, водным раствором (таким как насыщенный водный раствор NaHCO3 или насыщенным водным раствором хлорид аммония (предпочтительно насыщенным водным раствором хлорида аммония) или насыщенным солевым раствором. Слои разделяют и органический слой сушат над безводным Na2SO4 или MgSO4, фильтруют и концентрируют при пониженном давлении с получением сульфонилмочевины.
Иллюстрация Общей Процедуры AAA
Получение #AAA.1*: (R)-N-((1S,3R,4S)-3-метил-4-(6-тозил-6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)циклопентил)-2-(трифторметил)пирролидин-1-сульфонамид
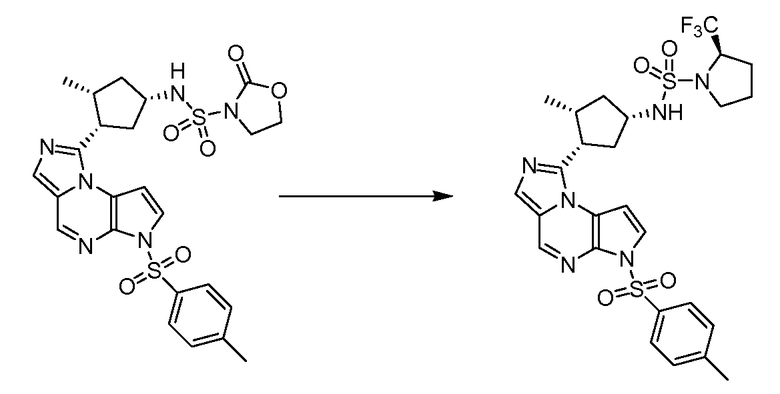
К раствору N-((1S,3R,4S)-3-метил-4-(6-тозил-6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)циклопентил)-2-оксооксазолидин-3-сульфонамида (0,200 г, 0,261 ммоль, получен из 5-тозил-5H-пирроло[2,3-b]пиразин-2-ил)метанамин гидрохлорида (WO2009152133) и (1S,2R,4S)-4-ацетамидо-2-метилциклопентанкарбоновой кислоты [полученной из этил 4-амино-2-метил-циклопентанкарбоксилата (WO2009152133) с использованием г, AA и Z] с использованием Н, OO, BB и ZZ) и (R)-2-трифторметилпирролидина (0,055 г, 0,392 ммоль) в MeCN (1,4 мл) добавляли TEA (0,073 мл, 0,523 ммоль). Реакционную смесь подвергали облучению в микроволновом устройстве CEM при около 120°C примерно в течение 0,5 часов. Реакционную смесь охлаждали до температуры окружающей среды и концентрировали при пониженном давлении с получением остатка. Неочищенное вещество очищали хроматографией на силикагеле с градиентным элюированием 0-70% EtOAc в DCM с получением (R)-N-((1S,3R,4S)-3-метил-4-(6-тозил-6H-имидазо[1,5-a]пирроло[2,3-e]пиразин-1-ил)циклопентил)-2-(трифторметил)пирролидин-1-сульфонамида (0,12 г, 75%, 72% чистоты) в виде не совсем белого твердого вещества: LC/MS (Таблица 1, Способ a) Rt = 2,79 мин; MS m/z: 611 (M+H)+.
Общая процедура BBB: Восстановление нитрогруппы
К раствору нитро-содержащего соединения (предпочтительно 1 экв.) в органическом растворителе (предпочтительно EtOH) добавляют хлорид олова (II) дигидрат (1-3 экв., предпочтительно 1 экв.) и реакционную смесь перемешивают при около 25-80°C (предпочтительно при около 75°C) примерно в течение 0,5-24 часов (предпочтительно около 1-2 часов). Необязательно, к реакционной смеси можно добавить дополнительную порцию хлорида олова(II) дигидрата (1-5 экв., предпочтительно 2 экв.) и нагревание можно продолжить примерно в течение 0,5-24 часов (предпочтительно около 5-14 часов). Реакционную смесь концентрируют при пониженном давлении. Неочищенную смесь можно разбавить органическим растворителем (например, EtOAc или DCM) и водным раствором основания (таким как 1 н раствор NaOH или насыщенный водный раствор NaHCO3). Слои разделяют и водный слой экстрагируют органическим растворителем (таким как EtOAc и/или DCM). Объединенные органические слои необязательно промывают насыщенным солевым раствором, сушат над безводным MgSO4, фильтруют и концентрируют при пониженном давлении.
Иллюстрация Общей Процедуры BBB
Получение #BBB.1: N-4-циклогексил-1-тозил-1H-пирроло[2,3-b]пиридин-4,5-диамин

К смеси N-циклогексил-5-нитро-1-тозил-1H-пирроло[2,3-b]пиридин-4-амина (0,111 г, 0,268 ммоль, получен с использованием K.1 из Примера #21 Стадия D с 4-метилбензол-1-сульфонилхлоридом, L с циклогексиламином) в EtOH (2,5 мл) добавляли хлорид олова (II) дигидрат (0,060 г, 0,268 ммоль). Реакционную смесь нагревали при около 75°C примерно в течение 75 минут. Добавляли хлорид олова (II) дигидрат (0,030 г, 0,134 ммоль) и реакционную смесь нагревали при около 75°C примерно в течение 5 часов. Добавляли дополнительное количество хлорида олова (II) дигидрата (0,060 г, 0,268 ммоль) и реакционную смесь нагревали при около 75°C примерно в течение 14 часов. Растворитель удаляли при пониженном давлении. Остаток разбавляли при помощи EtOAc (25 мл) и промывали насыщенным водным раствором NaHCO3 (25 мл) и насыщенным солевым раствором (25 мл). Органическую часть отделяли. Водную часть экстрагировали при помощи EtOAc (3×25 мл). Органические экстракты объединяли, сушили над MgSO4, фильтровали и концентрировали при пониженном давлении с получением N-4-циклогексил-1-тозил-1H-пирроло[2,3-b]пиридин-4,5-диамина (0,081 г, 79%) в виде коричневого масла: LC/MS (Таблица 1, Способ n) Rt = 0,82 мин; MS m/z 385 (M+H)+.
Общая процедура CCC: Образование амида
К смеси амина или соли амина (1 экв.) в органическом растворителе (например, DCM или ТГФ, предпочтительно DCM) при около 0-25°C (предпочтительно 0°C) добавляют органическое основание (TEA или DIEPA, предпочтительно TEA) (чистое или в виде раствора в органическом растворителе (предпочтительно DCM)), 1-3 экв. (предпочтительно 1 экв.) и ацилирующий агент (например, ангидрид или хлорангидрид кислоты) (предпочтительно ангидрид) (чистый или в виде раствора в органическом растворителе (предпочтительно DCM)), 1-3 экв. (предпочтительно 1 экв.). Реакционную смесь перемешивают при температуре окружающей среды примерно в течение 5 минут - 6 часов (предпочтительно около 10 мин). Реакционную смесь, необязательно, промывают насыщенным водным раствором NaHCO3, водой или насыщенным солевым раствором, сушат над MgSO4, фильтруют и концентрируют при пониженном давлении.
Иллюстрация Общей Процедуры CCC
Получение #CCC.1: N-(4-(циклогексиламино)-1-тозил-1H-пирроло[2,3-b]пиридин-5-ил)-2,2,2-трифторацетамид
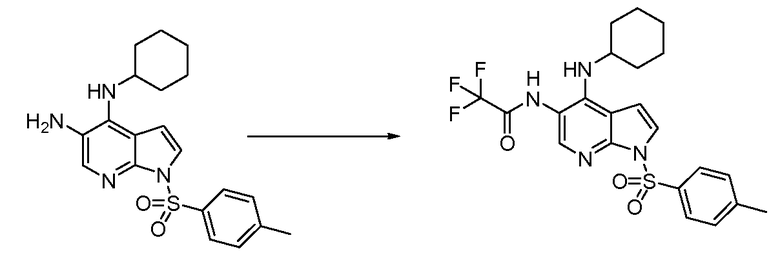
К 0°C раствору N-4-циклогексил-1-тозил-1H-пирроло[2,3-b]пиридин-4,5-диамина (Получение #BBB.1, 0,080 г, 0,208 ммоль) в DCM (2,0 мл) добавляли TEA (2 M в DCM, 0,104 мл, 0,208 ммоль) и TFAA (2 M в DCM, 0,104 мл, 0,208 ммоль). Реакционную смесь перемешивали примерно в течение 10 минут. Реакционную смесь промывали насыщенным водным раствором NaHCO3 (2 мл) и водой (2 мл), сушили над безводным MgSO4, фильтровали и концентрировали при пониженном давлении с получением N-(4-(циклогексиламино)-1-тозил-1H-пирроло[2,3-b]пиридин-5-ил)-2,2,2-трифторацетамида с 40% моль дихлорметана в качестве эксципиента (0,089 г, 83%, 90% чистота) в виде коричневого твердого вещества: LC/MS (Таблица 1, Способ n) Rt = 0,88 мин; MS m/z 481 (M+H)+.
Общая процедура DDD: Циклизация с образованием конденсированного имидазола
К раствору диамина (предпочтительно 1 экв.) в органическом растворителе, таком как DMF, DCM, 1,4-диоксан или MeOH (предпочтительно MeOH), добавляют соответствующий реагент для циклизации, такой как TMOF (1-10 экв., предпочтительно 1-2 экв..). (Когда используют TMOF, к реакционной смеси, необязательно, добавляют каталитическое количество кислоты, такой как TsOH (0,005-0,5 экв., предпочтительно 0,01 экв.)). Альтернативно, раствор орто-замещенного амидоаминоарила или гетероарильного соединения (предпочтительно 1 экв.) можно подвергнуть циклизации в органическом растворителе, таком как DMF или ТГФ, с использованием дегидратирующего агента, такого как TPP, POCl3 или HCl (5-100 экв., предпочтительно 10 экв. TPP). Реакционную смесь нагревают при около 25-120°C (предпочтительно около 65°C) примерно в течение 1-24 часов (предпочтительно около 12-16 часов), охлаждают до температуры окружающей среды и, необязательно, концентрируют при пониженном давлении с получением остатка. Остаток распределяют между органическим растворителем (таким как DCM или EtOAc, предпочтительно EtOAc), водой, водным раствором основания (таким как насыщенный водный раствор NaHCO3) и/или насыщенным солевым раствором. Слои разделяют и органический слой необязательно промывают водой, водным раствором основания (таким как насыщенный водный раствор NaHCO3 ) и/или насыщенным солевым раствором, сушат над безводным Na2SO4 или MgSO4, фильтруют и концентрируют при пониженном давлении.
Иллюстрация Общей Процедуры DDD
Получение #DDD.1: N-(3-этил-4-(6-тозилимидазо[4,5-d]пирроло[2,3-b]пиридин-1(6H)-ил)циклопентил)циклопропансульфонамид
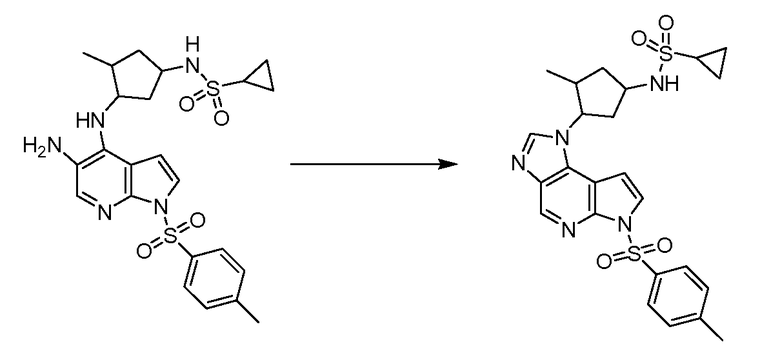
К раствору N-(3-(5-амино-1-тозил-1H-пирроло[2,3-b]пиридин-4-иламино)-4-этилциклопентил)циклопропансульфонамида (0,095 г, чистота 75%, 0,142 ммоль, получен с использованием L из Получения #27 и Получения #OOO.1 с DIEA, K.1 с TsCl и NaH и BBB) и TMOF (0,016 мл, 0,147 ммоль) в MeOH (3,09 мл) добавляли гидрат толуол-4-сульфоновой кислоты (0,0003 г, 0,0015 ммоль). Реакционную смесь нагревали при около 65°C примерно в течение 14 часов. Реакционную смесь охлаждали до температуры окружающей среды и концентрировали при пониженном давлении с получением неочищенного твердого вещества. Твердое вещество растворяли в EtOAc (10 мл) и промывали насыщенным водным раствором NaHCO3 (5 мл), водой (5 мл) и насыщенным солевым раствором (5 мл). Органическую часть отделяли и сушили над безводным MgSO4, фильтровали и концентрировали при пониженном давлении с получением N-(3-этил-4-(6-тозилимидазо[4,5-d]пирроло[2,3-b]пиридин-1(6H)-ил)циклопентил)циклопропансульфонамида (0,075 г, 99%) в виде желтого твердого вещества: LC/MS (Таблица 1, Способ a) Rt = 2,15 мин; MS m/z: 514 (M+H)+.
Общая процедура EEE: Образование сульфонилхлорида
К раствору сульфоновой кислоты или калиевой соли сульфоната (предпочтительно 1 экв.) в тионилхлориде (2-30 экв., предпочтительно 20-25 экв.) добавляют DMF (0,01-0,10 экв., предпочтительно 0,09 экв.). Реакционную смесь нагревают при около 50-100°C (предпочтительно около 80°C) примерно в течение 8-24 часов (предпочтительно около 12-16 часов). Реакционную смесь охлаждают до 0-25°C (предпочтительно около 0°C) и разбавляют водой. Реакционную смесь распределяют между органическим растворителем (таким как DCM или EtOAc) и водой или насыщенным солевым раствором. Слои разделяют и органический слой необязательно промывают водой и/или насыщенным солевым раствором, сушат над безводным Na2SO4 или MgSO4, фильтруют и концентрируют при пониженном давлении.
Иллюстрация Общей Процедуры EEE
Получение #EEE.1: 1-этилциклопропан-1-сульфонилхлорид

К смеси 1-этилциклопропан-1-сульфоната калия (0,420 г, 2,23 ммоль, Получение #JJJ.1) в тионилхлорида (3,58 мл, 49,1 ммоль) добавляли DMF (0,016 мл, 0,20 ммоль). Реакционную смесь нагревали при около 80°C примерно в течение 16 часов. Реакционную смесь охлаждали до 0°C, затем медленно добавляли воду (10 мл). Реакционную смесь разбавляли при помощи DCM (20 мл). Слои разделяли и водную часть экстрагировали при помощи DCM (3×10 мл). Объединенные органические слои разделяли, сушили над безводным MgSO4, фильтровали и концентрировали при пониженном давлении с получением неочищенного 1-этилциклопропан-1-сульфонилхлорида (0,52 г, 83% выход, чистота 60%) в виде оранжевого масла: 1H ЯМР (400 МГц, DMSO) δ 2,09 (кв., J=7,4, 2H), 1,62-1,60 (м, 2H), 1,45-1,39 (м, 2H), 0,91 (т, J=7,5, 3H).
Общая процедура FFF: Образование эфира в восстановительных условиях
К раствору TBDMS эфира (1,0 экв.) в MeCN при температуре окружающей среды добавляют триэтилсилан (1-2 экв. предпочтительно 1,5 экв.) и бромид висмута(III) (0,05-0,2 экв., предпочтительно 0,06 экв.). Реакционную смесь перемешивают при около 25-60°C (предпочтительно около 25°C) примерно в течение 0,5-5 минут (предпочтительно 1-3 мин). К реакционной смеси добавляют альдегид или кетон (1-6 экв., предпочтительно 1,5 экв.), который, необязательно, может быть высушен над безводным Na2SO4 или MgSO4. В случаях, когда реакция не достигает завершения, согласно данным ТСХ, можно добавить дополнительное количество триэтилсилана (1-2 экв. предпочтительно 1,5 экв.) и/или бромида висмута(III) (0,05-0,2 экв., предпочтительно 0,06 экв.) и/или альдегида или кетона (1-6 экв., предпочтительно 1,5 экв.). Реакцию продолжают при около 25-60°C (предпочтительно около 25°C) примерно в течение 15 минут - 24 часов (предпочтительно около 1 часа). Реакционную смесь обрабатывают с использованием одного из следующих способов. Способ 1: Реакционную смесь фильтруют через слой Целита®. Слой Целита® можно промыть дополнительным количеством органического растворителя (предпочтительно гептана или MeCN), и фильтрат концентрируют при пониженном давлении. Способ 2: Реакционную смесь фильтруют через Acrodisc® и фильтрат концентрируют при пониженном давлении.
Иллюстрация Общей Процедуры FFF
Получение #FFF.1: этил 2-этил-4-(тетрагидро-2H-пиран-4-илокси)циклопентанкарбоксилат

К раствору этил 4-(трет-бутилдиметилсилилокси)-2-этилциклопентанкарбоксилата (0,200 г, 0,666 ммоль, Пример #22 Стадия D) в MeCN (4,5 мл) добавляли триэтилсилан (0,160 мл, 1,00 ммоль) и бромид висмута(III) (0,020 г, 0,045 ммоль). Реакционную смесь перемешивали при температуре окружающей среды примерно в течение 1 минуты с последующим добавлением по каплям дигидро-2H-пиран-4(3H)-ола (0,100 г, 0,998 ммоль). Реакционную смесь перемешивали при температуре окружающей среды примерно в течение 15 минут. Реакционную смесь фильтровали через Acrodisc® и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали хроматографией на силикагеле с использованием градиента 10-100% EtOAc в гептане с получением этил 2-этил-4-(тетрагидро-2H-пиран-4-илокси)циклопентанкарбоксилата (0,253 г, 94%) в виде бесцветного масла; 1H ЯМР (400 МГц, CDCl3) δ 4,13 (кв., J=7,1, 2H), 4,05-3,98 (м, 1H), 3,98-3,88 (м, 2H), 3,58-3,47 (м, 1H), 3,46-3,36 (м, 2H), 2,80 (кв., J=8,5, 1H), 2,16 (дт, J=13,3, 7,7, 1H), 2,09-1,93 (м, 3H), 1,90-1,81 (м, 2H), 1,62-1,49 (м, 3H), 1,43 (ддд, J=11,1, 7,4, 5,2, 1H), 1,33-1,22 (м, 4H), 0,92-0,83 (м, 3H).
Общая процедура GGG: Иодирование гетероцикла на основе пиррола
К гетероциклу на основе пиррола (предпочтительно 1 экв.) в органическом растворителе, таком как DMF, добавляют основание, такое как KOH (1-10 экв., предпочтительно 3 экв.), при около 0°C-40°C (предпочтительно при температуре окружающей среды) и смесь перемешивают примерно в течение 2-45 минут (предпочтительно около 5 мин). Добавляют иод (0,95-1,2 экв., предпочтительно 1,0 экв.) небольшими порциями и смесь перемешивают в течение 10-100 минут (предпочтительно около 30 мин). Смесь добавляют по каплям в насыщенный водный раствор хлорида аммония (10 мл для каждого 1 мл используемого DMF) и целевое соединение собирают фильтрованием, промывают дополнительным количеством воды и сушат.
Иллюстрация Общей Процедуры GGG
Получение #GGG.1: N-((1S,3R,4S)-3-этил-4-(8-иод-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)циклопропансульфонамид

К раствору N-((1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)циклопропансульфонамида (0,396 г, 1,06 ммоль, получен, как подробно описано в WO2009152133) в DMF (20 мл) добавляли KOH (0,190 г, 3,38 ммоль). Смесь перемешивали при комнатной температуре в течение 5 минут. Добавляли иод (0,268 г,1,058 ммоль) небольшими порциями и реакционную смесь перемешивали при комнатной температуре в течение 30 минут. Смесь добавляли по каплям в насыщенный водный раствор хлорида аммония (200 мл). Осадок собирали фильтрованием, промывали водой и сушили с получением N-((1S,3R,4S)-3-этил-4-(8-иод-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)циклопропансульфонамида (0,494 г, 93%) в виде не совсем белого твердого вещества: LC/MS (Таблица 1, Способ a) Rt = 1,83 мин; MS m/z 501 (M+H)+.
Общая процедура GGG.1: Иодирование, хлорирование или бромирование гетероцикла на основе пиррола
К гетероциклу на основе пиррола (предпочтительно 1 экв.) в органическом растворителе, таком как DMF, ТГФ, MeCN, MeOH, AcOH, CHCl3 или DCM (предпочтительно DMF), необязательно, добавляют основание, такое как TEA, NaOAc, K2CO3 или KOH (1-10 экв.) при около 0-40°C (предпочтительно при 0°C) и смесь перемешивают примерно в течение 2-45 минут (предпочтительно около 5 мин). Добавляют порциями по каплям источник галогена, такой как I2, Br2, NBS, пиридинийтрибромид, NCS или NIS (0,95-1,2 экв., предпочтительно 1,0 экв.), чистый или в виде раствора в растворителе, таком как DMF. Если используют охлаждение, ледяную баню удаляют и смесь перемешивают примерно в течение 0,1-2 часов (предпочтительно около 40 мин) при комнатной температуре. Необязательно, можно добавить реагент, такой как тиосульфат натрия или бисульфит натрия в виде раствора в воде, или реакционную смесь добавляют к такому раствору и реакционную смесь перемешивают примерно в течение 5-60 минут (предпочтительно около 30 мин). Смесь можно разбавить или добавить в воду или насыщенный водный раствор NH4Cl (предпочтительно с использованием 10 мл воды на 1 мл DMF). Целевое соединение можно собирать фильтрованием или экстрагировать с использованием органического растворителя, такого как EtOAc или DCM, его сушат над Na2SO4 или MgSO4, фильтруют и концентрируют при пониженном давлении.
Иллюстрация Общей Процедуры GGG.1
Получение #GGG.1.1: 8-иод-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин

Раствор 6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразина (0,500 г, 3,14 ммоль, получен с использованием D из Получения #BBBBB.1 и NaOH) и DMF (16 мл) в атмосфере азота охлаждали до около 0°C. Смесь перемешивали примерно в течение 5 минут. Добавляли N-иодсукцинимид (0,707 г, 3,14 ммоль). Примерно через 40 минут добавляли 5% водный раствор тиосульфата натрия (10 мл). Охлаждающую баню удаляли. После перемешивания примерно в течение 30 минут добавляли воду (15 мл). Твердое вещество собирали фильтрованием. Фильтровальную лепешку промывали водой (2×5 мл). Водный слой экстрагировали при помощи EtOAc (4×50 мл). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Остаток суспендировали в воде (10 мл) и затем фильтровали, промывая водой (2×1 мл). Твердое вещество сушили в вакууме с получением коричневого твердого вещества (0,689 г), содержащего моно- и ди-иодированное вещество приблизительно в соотношении 4:1. 8-иод-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин (0,506 г, 57%): LC/MS (Таблица 1, Способ n) Rt = 0,39 мин; MS m/z 286 (M+H)+.
Общая процедура HHH: Цианирование гетероцикла
К раствору гетероарилгалогенида (предпочтительно 1 экв.) в органическом растворителе (таком как 1,4-диоксан, NMP или DMF, предпочтительно DMF) добавляют цианид калия (1-4 экв., предпочтительно 2,5 экв.), иодид меди(I) (1-4 экв., предпочтительно 2,5 экв.), тетракис(трифенилфосфин) палладий(0) (0,01-0,05 экв., предпочтительно 0,01 экв.) и 18-краун-6 (0,01-1,0 экв., предпочтительно 0,06-0,07 экв.). Реакционную смесь нагревают при около 25-120°C (предпочтительно около 110°C) примерно в течение 0,5-10 часов (предпочтительно около 4 часов). Реакционную смесь охлаждают до комнатной температуры и органический растворитель, необязательно, удаляют при пониженном давлении. Неочищенное вещество можно распределить между органическим растворителем (таким как EtOAc или DCM) и водой, водным раствором основания (таким как насыщенный водный раствор NaHCO3) или насыщенным солевым раствором. Слои разделяют и водный слой, необязательно, промывают органическим растворителем (таким как EtOAc или DCM). Объединенные органические экстракты сушат над безводным Na2SO4 или MgSO4, фильтруют и концентрируют при пониженном давлении.
Иллюстрация Общей Процедуры HHH
Получение #HHH.1*: N-((1S,3S,4R)-3-(8-циано-6-((2-(триметилсилил)этокси)метил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-4-этилциклопентил)циклопропансульфонамид

К раствору N-((1S,3R,4S)-3-этил-4-(8-иод-6-((2-(триметилсилил)этокси)метил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)циклопропансульфонамида (0,1 г, 0,16 ммоль, получен с использованием KK из Получения #GGG.1) в DMF (1,2 мл) добавляли цианид калия (0,03 г, 0,40 ммоль), иодид меди(I) (0,076 г, 0,40 ммоль), тетракис(трифенил-фосфин)палладий(0) (0,002 г, 0,002 ммоль) и 18-краун-6 (0,003 г, 0,01 ммоль). Реакционную смесь перемешивали при около 110°C примерно в течение 4 часов и охлаждали до температуры окружающей среды. Растворитель удаляли при пониженном давлении. Остаток распределяли между EtOAc (15 мл) и водой (8 мл). Водный слой далее экстрагировали при помощи EtOAc (15 мл). Объединенные органические слои сушили при помощи MgSO4, фильтровали и концентрировали при пониженном давлении с получением N-((1S,3S,4R)-3-(8-циано-6-((2-(триметилсилил)этокси)метил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-4-этилциклопентил)циклопропансульфонамида (0,069 г, 82%): LC/MS (Таблица 1, Способ b) Rt = 2,50 мин; MS m/z: 530 (M+H)+.
Общая процедура III: Реакция кетона по методу Хорнера-Вадсворта-Эммонса
В колбу, содержащую основание (предпочтительно NaH) (1-5 экв., предпочтительно 1,2 экв.) в органическом растворителе (предпочтительно ТГФ), при около 0-50°C (предпочтительно при комнатной температуре) добавляют бета-кетофосфонат (1-5 экв., предпочтительно 1,25 экв.). После прекращения выделения газообразного водорода добавляют раствор кетона (предпочтительно 1 экв.) в органическом растворителе (предпочтительно ТГФ). Примерно через 1-20 часов (предпочтительно около 4 часов) реакционную смесь распределяют между органическим растворителем (таким как DCM или EtOAc, предпочтительно EtOAc) и водной фазой, такой как насыщенный водный раствор NaHCO3. Органический слой отделяют и, необязательно, промывают насыщенным солевым раствором, сушат над безводным Na2SO4 или MgSO4, фильтруют и концентрируют при пониженном давлении.
Иллюстрация Общей Процедуры III
Получение #III.1: (E)-этил 2-((3R,4S)-3-этил-4-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентилиден)ацетат

К суспензии NaH (0,034 г, 0,85 ммоль) в ТГФ (5 мл) добавляли этил 2-(диэтоксифосфорил)ацетат (0,177 мл, 0,886 ммоль) при комнатной температуре. Примерно через 30 минут добавляли раствор (3R,4S)-3-этил-4-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентанона (0,300 г, 0,708 ммоль, Получение #25) в ТГФ (1 мл). Примерно через 4 часа добавляли EtOAc (20 мл) и насыщенный водный раствор NaHCO3 (20 мл). Органический слой отделяли, концентрировали в вакууме и очищали хроматографией на силикагеле (40 г) с использованием в качестве элюента EtOAc/гептан/DCM (2:1:1) с получением (E)-этил 2-((3R,4S)-3-этил-4-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентилиден)ацетата (0,260 г, 74%). LC/MS (Таблица 1, Способ a) Rt = 2,54 мин; MS m/z: 494 (M+H)+.
Общая процедура JJJ: Образование сульфоната калия
К раствору сульфоната (предпочтительно 1 экв.) в органическом растворителе (предпочтительно 1,4-диоксан) и воде добавляют тиоцианат калия (1-3 экв., предпочтительно 1 экв.). Реакционную смесь нагревают при около 80-100°C (предпочтительно около 100°C) примерно в течение 5-24 часов (предпочтительно около 16 часов). Реакционную смесь охлаждают до температуры окружающей среды и концентрируют при пониженном давлении.
Иллюстрация Общей Процедуры JJJ
Получение #JJJ.1: калия 1-этилциклопропан-1-сульфонат

К раствору бутил 1-этилциклопропан-1-сульфоната (0,46 г, 2,23 ммоль, получен из Получения #6 Стадия и этилиодида с использованием KKK) в 1,4-диоксане (2,79 мл) и воде (2,79 мл) добавляли тиоцианат калия (0,12 мл, 2,23 ммоль). Реакционную смесь нагревали до около 100°C примерно в течение 16 часов. Реакционную смесь охлаждали до температуры окружающей среды и концентрировали при пониженном давлении с получением 1-этилциклопропан-1-сульфоната калия (0,42 г, 100%) в виде белого кристаллического твердого вещества: 1H ЯМР (400 МГц, DMSO) δ 1,70-1,58 (м, 2H), 0,89 (т, J=7,5, 3H), 0,80 (кв., J=3,8, 2H), 0,32 (кв., J=3,8, 2H).
Общая процедура KKK: Алкилирование сульфоната
К раствору сульфоната (предпочтительно 1 экв.) в органическом растворителе (предпочтительно ТГФ), охлажденному до около -78-0°C (предпочтительно -78°C), добавляют органическое основание, такое как n-BuLi, KHMDS или LDA (предпочтительно n-BuLi) (1-3 экв., предпочтительно 1 экв.), и алкилирующий реагент, такой как иодметан, иодэтан или трифторэтилиодид (1-5 экв., предпочтительно 1,2 экв.). Реакционную смесь перемешивают при около -78-25°C (предпочтительно -78°C) примерно в течение 1-24 часов (предпочтительно 2 часов). Необязательно, реакционную смесь нагревают до температуры окружающей среды и перемешивают примерно в течение 1-24 часов (предпочтительно 2 часа). Реакционную смесь гасят путем добавления насыщенного водного раствора хлорида аммония. Реакционную смесь распределяют между органическим растворителем (таким как DCM или EtOAc) и водой или насыщенным солевым раствором. Слои разделяют и органический слой, необязательно, промывают водой и/или насыщенным солевым раствором, сушат над безводным Na2SO4 или MgSO4, фильтруют и концентрируют при пониженном давлении.
Иллюстрация Общей Процедуры KKK
Получение #KKK.1: бутил 1-метилциклопропан-1-сульфонат
К раствору бутилциклопропансульфоната (1,5 г, 8,4 ммоль, Получение #6 Стадия A) в ТГФ (8 мл) при около -78°C одновременно добавляли n-BuLi (1,6 M в гексане, 5,26 мл, 8,42 ммоль) и иодметан (0,684 мл, 10,9 ммоль). Полученную смесь перемешивали при около -78°C примерно в течение 2 часов и затем при температуре окружающей среды примерно в течение 2 часов. Реакцию гасили путем добавления насыщенного водного раствора NH4Cl (7 мл) и слои разделяли. Водный слой экстрагировали при помощи EtOAc (15 мл) и объединенные органические экстракты сушили над безводным MgSO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с использованием в качестве элюента 5 до 25% EtOAc в гептане с получением бутил 1-метилциклопропан-1-сульфоната (0,8 г, 49%) в виде бесцветного масла. 1H ЯМР (DMSO-d 6) δ 4,17 (т, 2H), 1,62 (м, 2H), 1,43 (с, 3H), 1,35 (м, 2H), 1,22 (м, 2H), 0,94 (м, 2H), 0,88 (т, 3H).
Общая процедура LLL: Окисление тиоэфира до сульфона
К раствору тиоэфира (предпочтительно 1 экв.) в органическом растворителе (предпочтительно DCM) добавляют окислитель (такой как m-CPBA, оксон, предпочтительно m-CPBA) (1-4 экв., предпочтительно 2 экв.). Реакционную смесь перемешивают при температуре окружающей среды примерно в течение 0,25-24 часов (предпочтительно около 0,5 часов). Реакционную смесь, необязательно, фильтруют, промывают дополнительным количеством DCM и фильтрат концентрируют при пониженном давлении. Реакционную смесь, необязательно, гасят путем добавления водного раствора основания (такого как насыщенный водный раствор NaHCO3) и распределяют между органическим растворителем (таким как DCM или EtOAc, предпочтительно DCM). Слои разделяют и, необязательно, промывают водой и/или насыщенным солевым раствором, сушат над безводным Na2SO4 или MgSO4, фильтруют и концентрируют при пониженном давлении с получением сульфона.
Иллюстрация Общей Процедуры LLL
Получение #LLL.1: 1-(2-этил-4-(2,2,2-трифторэтилсульфонил)циклопентил)-6-((2-(триметилсилил)этокси)метил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин

К смеси 1-(2-этил-4-(2,2,2-трифторэтилтио)циклопентил)-6-((2-(триметилсилил)этокси)метил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразина (0,100 г, 0,200 ммоль, Получение #MMM.1) в DCM (0,667 мл) добавляли m-CPBA (0,090 г, 0,400 ммоль). Реакционную смесь перемешивали при температуре окружающей среды примерно в течение 0,5 часов. Реакцию гасили путем добавления насыщенного водного раствора NaHCO3 (5 мл). Водную часть экстрагировали при помощи DCM (2×10 мл). Объединенные органические слои разделяли, сушили над безводным MgSO4, фильтровали и концентрировали при пониженном давлении. Неочищенное вещество очищали хроматографией на силикагеле с градиентным элюированием 0-60% EtOAc в DCM с получением 1-2-этил-4-(2,2,2-трифторэтилсульфонил)циклопентил)-6-((2-(триметилсилил)этокси)метил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразина (0,095 г, 89%, 93% чистота) в виде прозрачного масла: LC/MS (Таблица 1, Способ a) Rt = 2,63 мин; MS m/z: 532 (M+H)+.
Общая процедура MMM: Реакция Мицунобу с использованием тиола
К раствору азодикарбоксилата, такого как DIAD, DEAD или TMAD (предпочтительно DIAD) (1-2 экв., предпочтительно 1,2 экв.), в органическом растворителе (предпочтительно ТГФ) добавляют фосфиновый реагент, такой как PPh3 или P(n-Bu)3 (предпочтительно P(n-Bu)3) (1-2 экв., предпочтительно 1,2 экв.), спирт (предпочтительно 1 экв.), TEA (1-2 экв., предпочтительно 1,2 экв.) и тиол (1-1,5 экв., предпочтительно 1,2 экв.). Реакционную смесь перемешивают при температуре окружающей среды примерно в течение 1-24 часов (предпочтительно 16 часов). Реакционную смесь, необязательно, концентрируют при пониженном давлении с получением остатка. Реакционную смесь или остаток распределяют между органическим растворителем (таким как DCM или EtOAc, предпочтительно EtOAc) и водой, водным раствором основания (таким как насыщенный водный раствор NaHCO3) или насыщенным солевым раствором. Слои разделяют и органический слой, необязательно, промывают водой, водным раствором основания (таким как насыщенный водный раствор NaHCO3) и/или насыщенным солевым раствором, сушат над безводным Na2SO4 или MgSO4, фильтруют и концентрируют при пониженном давлении с получением тиоэфира.
Иллюстрация Общей Процедуры MMM
Получение #MMM.1: 1-(2-этил-4-(2,2,2-трифторэтилтио)циклопентил)-6-((2-(триметилсилил)этокси)метил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин
В высушенную в печи колбу загружали DIAD (0,177 мл, 0,896 ммоль) и ТГФ (3,74 мл). Реакционную колбу охлаждали до 0°C перед добавлением P(n-Bu)3 (0,221 мл, 0,896 ммоль), 3-этил-4-(6-((2-(триметилсилил)этокси)метил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентанола (0,300 г, 0,747 ммоль, получен из Получения #20 с использованием KK и SS), TEA (0,125 мл, 0,896 ммоль) и 2,2,2-трифторэтантиола (0,080 мл, 0,896 ммоль). Реакционную смесь перемешивали при температуре окружающей среды примерно в течение 16 часов. Реакционную смесь распределяли между водой (10 мл) и EtOAc (10 мл). Водный слой экстрагировали при помощи EtOAc (3×10 мл). Объединенные органические слои сушили над безводным MgSO4, фильтровали и концентрировали при пониженном давлении с получением неочищенного масла. Неочищенное вещество очищали хроматографией на силикагеле с градиентным элюированием 0-60% EtOAc в DCM с получением 1-(2-этил-4-(2,2,2-трифторэтилтио)циклопентил)-6-((2-(триметилсилил)этокси)метил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразина (0,165 г, 44%) в виде оранжевого твердого вещества: LC/MS (Таблица 1, Способ a) Rt = 3,02 мин; MS m/z: 500 (M+H)+.
Общая процедура NNN: Перегруппировка Курциуса с образованием изоцианата
К раствору карбоновой кислоты (предпочтительно 1 экв.) в органическом растворителе, таком как t-BuOH или толуол (предпочтительно t-BuOH), добавляют DPPA (1-3 экв., предпочтительно 1-1,1 экв.) и органическое основание, такое как TEA (2-4 экв., 2,2 экв.). Реакционную смесь перемешивают при около 25-110°C (предпочтительно около 70°C для t-BuOH и 110°C для толуол) примерно в течение 0,5-16 часов (предпочтительно около 2 часов). Реакционную смесь охлаждают до температуры окружающей среды и, необязательно, концентрируют при пониженном давлении с получением остатка. Реакционную смесь или остаток, необязательно, распределяют между органическим растворителем (таким как DCM или EtOAc) и водой, водным раствором основания (такого как насыщенный водный раствор NaHCO3) или насыщенным солевым раствором. Слои разделяют и органический слой сушат над безводным Na2SO4 или MgSO4, фильтруют и концентрируют при пониженном давлении.
Иллюстрация Общей Процедуры NNN
Получение #NNN.1: N-(3-изоцианато-4-метилциклопентил)циклопропансульфонамид

К раствору 4-(циклопропансульфонамидо)-2-метилциклопентанкарбоновой кислоты (4,10 г, 16,58 ммоль, получена из Примера #24 Стадия I с использованием Y, K и Z) и DPPA (3,58 мл, 16,58 ммоль) в t-BuOH (55 мл) добавляли TEA (5,0 мл, 36,5 ммоль). Реакционную смесь нагревали до около 70°C примерно в течение 2 часов, затем охлаждали до температуры окружающей среды и концентрировали при пониженном давлении с получением неочищенного остатка. Неочищенное вещество очищали хроматографией на силикагеле с градиентным элюированием 0-10% MeOH в DCM с получением N-(3-изоцианато-4-метилциклопентил)циклопропансульфонамида (3,25 г, 80%) в виде белого твердого вещества: LC/MS (Таблица 1, Способ n) Rt = 0,49 мин; MS m/z: 245 (M+H)+.
Общая процедура OOO: Гидролиз изоцианата
К смеси изоцианата (предпочтительно 1 экв.) в органическом растворителе (предпочтительно ТГФ) добавляют водный раствор основания или кислоты (такой как водный раствор NaOH, LiOH или HCl) (10-50 экв., предпочтительно 20 экв.). Реакционную смесь нагревают при около 30-100°C примерно в течение 5-36 часов (предпочтительно около 50°C примерно в течение 16 часов). Реакционную смесь охлаждают до температуры окружающей среды и реакционную смесь распределяют между органическим растворителем (таким как DCM или EtOAc) и водой, водным раствором основания (таким как насыщенный водный раствор NaHCO3) или насыщенным солевым раствором. Слои разделяют и органический слой, необязательно, промывают водой и или насыщенным солевым раствором, сушат над безводным Na2SO4 или MgSO4, фильтруют и концентрируют при пониженном давлении.
Иллюстрация Общей Процедуры OOO
Получение #OOO.1: N-(3-амино-4-метилциклопентил)циклопропансульфонамид

К смеси N-(3-изоцианато-4-метилциклопентил)циклопропансульфонамида (1,00 г, 4,09 ммоль, Получение #NNN.1) в ТГФ (2,0 мл) добавляли водный раствор LiOH (4 н, 20,5 мл, 82 ммоль). Реакционную смесь нагревали до около 50°C примерно в течение 16 часов, охлаждали до температуры окружающей среды и распределяли между водой (5 мл) и EtOAc (10 мл). Органическую часть отделяли и водную часть экстрагировали при помощи DCM (3×20 мл). Объединенные органические экстракты сушили над безводным MgSO4, фильтровали и концентрировали при пониженном давлении с получением неочищенного N-(3-амино-4-метилциклопентил)циклопропансульфонамида (0,66 г, 74%) в виде белого твердого вещества: LC/MS (Таблица 1, Способ n) Rt = 0,020 мин; MS m/z: 219 (M+H)+.
Общая процедура PPP: Образование оксимэфира из кетона
К раствору кетона (предпочтительно 1 экв.) в органическом растворителе (предпочтительно EtOH) добавляют O-алкилгидроксиламин (1-10 экв., предпочтительно около 1 экв.). Когда O-алкилгидроксиламин используют в форме гидрохлоридной соли, добавляют органическое основание, такое как TEA или DIEA (предпочтительно TEA, 1-5 экв., предпочтительно около 1,5 экв.). Реакционную смесь перемешивают при температуре окружающей среды примерно в течение 12-24 часов (предпочтительно около 18 часов). В случаях, когда реакция не достигает завершения, согласно данным ТСХ, LC/MS или ВЭЖХ, можно добавить дополнительное количество O-алкилгидроксиламина (1-10 экв., предпочтительно около 1 экв.). Реакционную смесь перемешивают при температуре окружающей среды примерно в течение 1-24 часов (предпочтительно около 5 часов). Растворитель удаляют при пониженном давлении.
Иллюстрация Общей Процедуры PPP
Пример #PPP.1.1: (3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентанон O-циклопропилметилоксим

К раствору (3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентанона (0,05 г, 0,19 ммоль, Пример #AA.1.59) в EtOH (1 мл) добавляли TEA (0,04 мл, 0,28 ммоль) и O-(циклопропилметил)гидроксиламин гидрохлорид (0,02 г, 0,19 ммоль, Huhu Technologies). Реакционную смесь перемешивали примерно в течение 18 часов при температуре окружающей среды. Добавляли дополнительное количество O-(циклопропилметил)гидроксиламин гидрохлорида (0,02 г, 0,19 ммоль, Huhu Technologies). Реакционную смесь перемешивали при температуре окружающей среды примерно в течение 5 часов. Растворитель удаляли при пониженном давлении. Неочищенное вещество очищали хроматографией на силикагеле с градиентным элюированием 1-10% MeOH в DCM с получением (3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентанон O-циклопропилметилоксима (0,051 г, 80%): LC/MS (Таблица 1, Способ a) Rt = 1,94 мин; MS m/z: 339 (M+H)+.
Примеры, полученные с использованием PPP и (3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентанона (Пример #AA.1.59)
(Таблица 1, Способ)


Общая процедура QQQ: Кислотно-опосредованное преобразование трет-бутилового эфира в карбоновую кислоту с использованием TFA
К трет-бутиловому эфиру (предпочтительно 1 экв.) добавляют TFA (10-400 экв., предпочтительно 200-250 экв.). Реакционную смесь поддерживают при около -20-60°C (предпочтительно около 25°C) примерно в течение 0,5-16 часов (предпочтительно около 1 часа). В любом случае присутствия дополнительной кислотно-лабильной группы (например, Boc группы) эту группу также можно расщепить в процессе реакции. Реакционную смесь концентрируют при пониженном давлении. Полученный остаток можно использовать без дополнительной очистки, или его растворяют в органическом растворителе, таком как DCM или EtOAc (предпочтительно DCM), и промывают водным раствором неорганического основания, такого как NaHCO3 или Na2CO3 (предпочтительно насыщенным водным раствором NaHCO3). Органический слой, необязательно, промывают насыщенным солевым раствором, сушат над безводным MgSO4 или Na2SO4 и концентрируют при пониженном давлении с получением целевого соединения.
Иллюстрация Общей Процедуры QQQ
Получение #QQQ.1: 4-(циклопропансульфонамидометил)-2-этилциклопентанкарбоновая кислота

трет-Бутил 4-(циклопропансульфонамидометил)-2-этилциклопентанкарбоксилат (0,080 г, 0,241 ммоль, получен с использованием K из Получения #21 и циклопропилсульфонилхлорида) в TFA (4 мл, 51,9 ммоль) перемешивали при около 25°C примерно в течение 1 часа. Органический растворитель удаляли при пониженном давлении с получением неочищенной 4-(циклопропансульфонамидометил)-2-этилциклопентанкарбоновой кислоты (0,066 г, 100%): LC/MS (Таблица 1, Способ b) Rt = 1,81 мин; MS m/z: 276 (M+H)+.
Общая процедура RRR: Восстановление алкина до алкена
В колбу, содержащую катализатор гидрирования (предпочтительно катализатор Линдлара) (0,001 до 1 экв., предпочтительно 0,01 экв.), добавляют растворитель (предпочтительно ТГФ) и добавку для предотвращения чрезмерного восстановления (такую как пиридин или хинолин, предпочтительно пиридин) в соотношении 5:1 до 20:1 (предпочтительно 10:1) с последующим добавлением алкина (1 экв.). Реакционную смесь барботируют водородом примерно в течение 5-30 минут (предпочтительно около 10 мин) и атмосферу водорода поддерживают через баллон. Примерно через 1-40 часов (предпочтительно около 15 часов) реакционную смесь фильтруют, разбавляют органическим растворителем (предпочтительно Et2O) и промывают насыщенным водным раствором CuSO4, с последующей промывкой водой. Органический слой отделяют, сушат над безводным Na2SO4 или MgSO4, фильтруют и концентрируют при пониженном давлении.
Иллюстрация Общей Процедуры RRR
Получение #RRR.1: (Z)-этил пент-2-еноат

К суспензии катализатора Линдлара (0,844 г, 0,396 ммоль) в ТГФ (100 мл) и пиридина (10,00 мл) добавляли этил пент-2-иноат (5,22 мл, 39,6 ммоль). Реакционную смесь барботировали водородом примерно в течение 10 минут и атмосферу водорода поддерживали через баллон. Примерно через 15 часов реакционную смесь фильтровали через слой Целита®, разбавляли при помощи Et2O (30 мл) и промывали насыщенным водным раствором CuSO4 (40 мл), с последующей промывкой водой (40 мл). Органический слой отделяли, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме с получением неочищенного (Z)-этил пент-2-еноата (5 г, 98%).1H ЯМР (DMSO-d 6) δ 1,05 (т, 3H), 1,28 (т, 3H), 2,65 (м, 2H), 4,18 (кв., 2H), 5,72 (м, 1H), 6,21 (м, 1H).
Общая процедура SSS: 1,3-Диполярное циклоприсоединение с образованием пирролидина
К раствору 1,3-дипольного предшественника (0,5-3 экв., предпочтительно 1 экв.) и диполярофила (0,5-3 экв., предпочтительно 1 экв.) в органическом растворителе (предпочтительно DCM) при около 0-45°C (предпочтительно при комнатной температуре) добавляют кислоту (предпочтительно TFA) (0,001-1 экв., предпочтительно 0,01 экв.). Примерно через 1-60 часов (предпочтительно около 48 часов) смесь концентрируют в вакууме с получением неочищенного цикло-продукт присоединения.
Иллюстрация Общей Процедуры SSS
Получение #SSS.1: цис-этил 1-бензил-4-этилпирролидин-3-карбоксилат
К раствору N-бензил-1-метокси-N-((триметилсилил)метил)метанамина (9,98 мл, 39,0 ммоль,) и (Z)-этилпент-2-еноата (5 г, 39,0 ммоль, Получение #RRR.1) в DCM (50 мл) добавляли TFA (0,030 мл, 0,390 ммоль) при комнатной температуре. Примерно через 2 дня реакционную смесь концентрировали в вакууме с получением неочищенного цис-этил 1-бензил-4-этилпирролидин-3-карбоксилата (9,8 г, 96%) в виде масла. LC/MS (Таблица 1, Способ a) Rt = 0,51 мин; MS m/z: 262 (M+H)+.
Общая процедура TTT: Гидрирование азида до амина
К азиду (предпочтительно 1 экв.) в EtOH, MeOH, EtOAc или ТГФ (предпочтительно EtOH) добавляют катализатор, такой как 20% масс. гидроксида палладия на углероде или 10% масс. палладия на углероде (предпочтительно гидроксид палладия на углероде, 0,05-0,5 экв., предпочтительно 0,15 экв.) и смесь перемешивают при температуре окружающей среды при атмосферном давлении водорода в течение 1-24 часов, предпочтительно около 2 часов. Катализатор удаляют фильтрованием через слой Целита® и фильтрат концентрируют при пониженном давлении с получением желаемого продукта.
Иллюстрация Общей Процедуры TTT
Получение #TTT.1: (3S,5R)-5-этил-1-(3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)пирролидин-3-амин
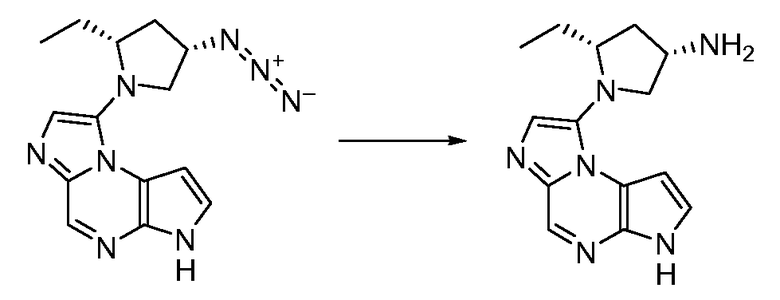
К раствору 8-((2R,4S)-4-азидо-2-этилпирролидин-1-ил)-3H-имидазо[1,2-a]пирроло[2,3-e]пиразина (0,136 г, 0,459 ммоль, получен с использованием S из Примера #3 Стадия E и трет-бутил бромацетата, E с HCl, H с (2R,4S)-4-азидо-2-метилпирролидином (получен из (2R,4S)-трет-бутил 4-азидо-2-метилпирролидин-1-карбоксилата, как подробно описано в Rosen, T.; Chu, D. T. W.; Lico, I. M.; Fernandes, P. B.; Marsh, K.; Shen, L.; Cepa, V. G.; Pernet, A. G. J. Med. Chem. 1988, 31, 1598-1611, затем E с HCl), OO, D с NaOH) в EtOH (15 мл) добавляли 20% гидроксид палладия на углероде (0,05 г, 0,071 ммоль) и реакционную смесь перемешивали при атмосферном давлении водорода в течение 2 часов. Катализатор удаляли фильтрованием через слой Целита® и растворитель удаляли при пониженном давлении с получением (3S,5R)-5-этил-1-(3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)пирролидин-3-амина (0,11 г, 89%) в виде не совсем белого аморфного твердого вещества. LC/MS (Таблица 1, Способ a) Rt = 1,00 мин; MS m/z 271 (M+H)+.
Общая процедура UUU: Взаимодействие арил или гетероарилгалогенида с бороновой кислотой или боронатным эфиром с последующим удалением тозильной защитной группы
К смеси арилгалогенида (предпочтительно 1 экв.), бороновой кислоты или боронатного сложного эфира (1-1,75 экв., предпочтительно 1,1 экв.) и неорганического основания (например, фторид калия, карбонат натрия или карбонат цезия, предпочтительно карбонат цезия (2-16 экв., предпочтительно 2,5 экв.) в растворителе (например, ТГФ, DME, DMF, 1,4-диоксан, DME/вода, 1,4-диоксан/вода, толуол/EtOH/вода, 1,4-диоксан/EtOH/вода, вода; предпочтительно 1,4-диоксан/EtOH/вода) добавляют палладиевый катализатор (например, трис(бензилиденацетон)дипалладий(0), тетракис(трифенилфосфин)палладий(0), бис(ацетато)трифенилфосфинпалладий(II), полимер-связанный FibreCatTM 1032, комплекс (1,1'-бис(дифенилфосфино)ферроцен)дихлорпалладия(II) с DCM или дихлорбис(трифенилфосфин)палладий(II); предпочтительно дихлорбис(трифенилфосфин)палладий(II) (0,01-0,20 экв., предпочтительно 0,1 экв.)). Реакционную смесь нагревают при около 40-120°C (предпочтительно около 60°C) примерно в течение 1-24 часов (предпочтительно около 6 часов) термически или при около 100-200°C (предпочтительно около 120°C примерно в течение 5-60 минут (предпочтительно около 20 мин) в микроволновой печи (предпочтительно время линейного нарастания 5 минут, максимальная мощность 300 Ватт, максимальное давление 250 ф/дюйм2 (17,58 кг/см2)). Реакционной смеси дают охладиться до температуры окружающей среды и обрабатывают с использованием одного из следующих способов. Способ 1. Для реакционных смесей, содержащих воду, реакционную смесь можно разбавить органическим растворителем (таким как DCM или EtOAc). Слои разделяют, органический раствор, необязательно, промывают водой и/или насыщенным солевым раствором, сушат над безводным MgSO4 или Na2SO4, фильтруют и растворитель удаляют при пониженном давлении с получением промежуточных соединений. Способ 2. Реакционную смесь концентрируют при пониженном давлении и, необязательно, очищают с использованием одного или нескольких из Способов очистки, описанных выше, с получением промежуточных соединений. К промежуточным соединениям добавляют органический растворитель (такой как 1,4-диоксан, MeOH или ТГФ/MeOH, предпочтительно 1,4-диоксан) и водный раствор основания (такой как водный раствор Na2CO3 или водный раствор NaOH, 1-30 экв., предпочтительно 2-3 экв. для водного раствора NaOH, предпочтительно 15-20 экв. для водного раствора Na2CO3). Смесь перемешивают при около 25-100°C (предпочтительно около 60°C) примерно в течение 1-72 часов (предпочтительно около 1-16 часов) при термическом нагревании, или при около 80-200°C (предпочтительно около 100°C) примерно в течение 10-60 минут (предпочтительно около 15 мин) при нагревании в микроволновой печи (предпочтительно, время линейного нарастания 5 минут, максимальная мощность 300 Ватт, максимальное давление 250 ф/дюйм2 (17,58 кг/см2)). В случаях, когда реакция не достигает завершения, согласно данным ТСХ, LC/MS или ВЭЖХ, добавляют дополнительное количество водного раствора основания (такого как водный раствор Na2CO3, 10-20 экв., предпочтительно 10 экв., или водный раствор NaOH, 1-5 экв., предпочтительно 1-2 экв.) и/или сорастворитель (такой как EtOH). Реакцию продолжают при около 25-100°C (предпочтительно около 60°C) примерно в течение 0,25-3 часов (предпочтительно около 1-2 часов) при термическом нагревании, или при около 80-100°C (предпочтительно около 100°C) примерно в течение 10-60 минут (предпочтительно около 15 мин) при нагревании в микроволновой печи. В любом случае, когда присутствует дополнительная основно-лабильная группа (например, сложный эфир или цианогруппа), эта группа также может быть гидролизована. Реакционную смесь обрабатывают с использованием одного из следующих способов. Способ 1. Органический растворитель, необязательно, удаляют при пониженном давлении и водный раствор нейтрализуют путем добавления подходящего водного раствора кислоты (такого как водный раствор HCl). Добавляют подходящий органический растворитель (такой как EtOAc или DCM) и воду, слои разделяют и органический раствор сушат над безводным Na2SO4 или MgSO4, фильтруют и концентрируют досуха при пониженном давлении с получением целевого соединения. Способ 2. Органический растворитель, необязательно, удаляют при пониженном давлении, добавляют подходящий органический растворитель (такой как EtOAc или DCM) и воду, слои разделяют и органический раствор сушат над безводным Na2SO4 или MgSO4, фильтруют и концентрируют досуха при пониженном давлении с получением целевого соединения. Способ 3. Реакционную смесь концентрируют при пониженном давлении и непосредственно очищают одним из описанных ниже способов с получением целевого соединения.
Иллюстрация Общей Процедуры UUU
Пример #UUU.1: 1-циклогексил-3-(4-(метилсульфонил)фенил)-6H-имидазо[1,5-a]пирроло[2,3-e]пиразин

В микроволновой сосуд загружали 3-бром-1-циклогексил-6-тозил-6H-имидазо[1,5-a]пирроло[2,3-e]пиразин (0,050 г, 0,11 ммоль, Получение #MM.1), 4-(метилсульфонил)фенилбороновую кислоту (0,023 г, 0,12 ммоль, Acros), карбонат цезия (0,086 г, 0,26 ммоль), дихлорбис(трифенилфосфин)палладий(II) (0,0066 г, 0,0000094 ммоль) и 1,4-диоксан (0,42 мл), EtOH (0,42 мл) и воду (0,21 мл). Сосуд закрывали крышкой и смесь нагревали до около 120°C примерно в течение 20 минут (время линейного нарастания 5 минут, максимальная мощность 300 Ватт, максимальное давление 250 ф/дюйм2 (17,58 кг/см2)) в микроволновой печи. Реакционную смесь концентрировали при пониженном давлении с получением твердого вещества, которое растворяли в 1,4-диоксане (1,0 мл) и переносили в микроволновой сосуд. Добавляли 2 н водный раствор NaOH (0,11 мл, 0,21 ммоль) и сосуд закрывали крышкой. Раствор нагревали до около 100°C примерно в течение 15 минут в микроволновой печи (максимальная мощность 300 Ватт, максимальное давление 250 ф/дюйм2 (17,58 кг/см2), время линейного нарастания 5 минут). К реакционному раствору добавляли DCM (10 мл) и насыщенный водный раствор NH4Cl (5 мл). Слои разделяли и водный раствор экстрагировали дополнительным количеством DCM (5 мл). Объединенные экстракты сушили над безводным MgSO4, фильтровали и концентрировали досуха при пониженном давлении с получением светло-коричневого твердого вещества. При добавлении DCM (1 мл) образовывался желтый осадок, который собирали вакуумным фильтрованием и сушили в течение ночи в воронке Бюхнера с получением 1-циклогексил-3-(4-(метилсульфонил)фенил)-6H-имидазо[1,5-a]пирроло[2,3-e]пиразина (0,017 г, 41%): LC/MS (Таблица 1, Способ a) Rt = 2,39 мин; MS m/z: 395 (M+H)+.
Примеры, полученные из 3-бром-1-циклогексил-6-тозил-6H-имидазо[1,5-a]пирроло[2,3-e]пиразина (Получение #MM.1) с использованием Общей Процедуры UUU
(Таблица 1, Способ)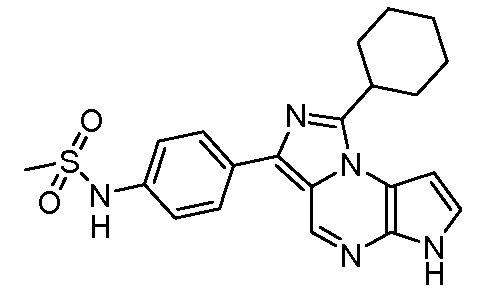
Общая процедура VVV: Взаимодействие арил или гетероарилгалогенида с бороновой кислотой или боронатным эфиром
К смеси арилгалогенида (предпочтительно 1 экв.), бороновой кислоты или боронатного сложного эфира (1-1,75 экв., предпочтительно 1,1 экв.) и неорганического основания (например, фторида калия, карбоната натрия или карбоната цезия, предпочтительно карбоната цезия) (1,1-16 экв., предпочтительно 2 экв.) в растворителе (например, ТГФ, DME, DMF, 1,4-диоксан, DME/вода, 1,4-диоксан/вода, толуол/EtOH/вода, 1,4-диоксан/EtOH/вода или вода; предпочтительно 1,4-диоксан) добавляют палладиевый катализатор (например, трис(бензилиденацетон)дипалладий(0), тетракис(трифенилфосфин)палладий(0), бис(ацетато)трифенилфосфинпалладий(II), полимер-связанный FibreCatTM 1032, комплекс (1,1'-бис(дифенилфосфино)ферроцен)дихлорпалладий(II) с DCM или дихлорбис(трифенилфосфин)палладий(II); предпочтительно трис(бензилиденацетон)дипалладий(0), 0,01-0,20 экв., предпочтительно 0,1 экв.) и, необязательно, добавляют лиганд (например, трициклогексилфосфин, три-трет-бутил-фосфан; предпочтительно трициклогексилфосфин (0,01-1,0 экв., предпочтительно 0,16 экв.)). Реакционную смесь нагревают при около 40-120°C (предпочтительно около 85°C) примерно в течение 1-24 часов (предпочтительно около 2 часов) термически, или при около 100-200°C (предпочтительно около 120°C) примерно в течение 5-60 минут (предпочтительно около 20 мин) в микроволновой печи (предпочтительно, время линейного нарастания 5 минут, максимальная мощность 300 Ватт, максимальное давление 250 ф/дюйм2 (17,58 кг/см2)). Реакционной смеси дают охладиться до температуры окружающей среды и обрабатывают с использованием одного из следующих способов. Способ 1. Для реакционных смесей, содержащих воду, реакционную смесь можно разбавить органическим растворителем (таким как DCM или EtOAc). Слои разделяют, органический раствор, необязательно, промывают водой и/или насыщенным солевым раствором, сушат над безводным MgSO4 или Na2SO4, фильтруют и растворитель удаляют при пониженном давлении с получением желаемого соединения. Способ 2. Реакционную смесь концентрируют при пониженном давлении и, необязательно, очищают с использованием одного или нескольких из Способов Очистки, описанных выше, с получением желаемого соединения. Способ 3. Катализатор удаляют фильтрованием и фильтрат концентрируют при пониженном давлении.
Иллюстрация Общей Процедуры VVV
Получение #VVV.1: 8-циклогексил-1-метил-3-((2-(триметилсилил)этокси)метил)-3H-имидазо[1,2-a]пирроло[2,3-e]пиразин
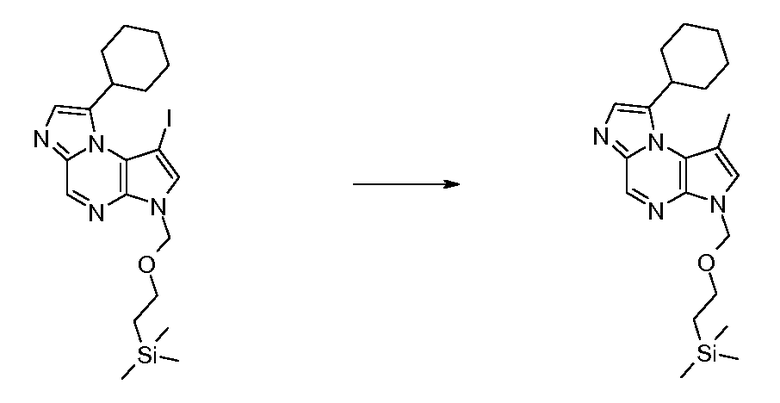
К раствору 8-циклогексил-1-иод-3-((2-(триметилсилил)этокси)метил)-3H-имидазо[1,2-a]пирроло[2,3-e]пиразина (0,100 г, 0,201 ммоль, получен с использованием GGG с 8-циклогексил-3H-имидазо[1,2-a]пирроло[2,3-e]пиразином [WO2009152133A1], KK) в 1,4-диоксане (1 мл) добавляли карбонат цезия (0,131 г, 0,403 ммоль), трициклогексилфосфин (20% масс. раствор в толуоле, 0,045 г, 0,032 ммоль), Pd2(dba)3 (0,018 г, 0,020 ммоль) и триметилборат (0,033 г, 0,262 ммоль). Смесь дегазировали и нагревали при около 85°C примерно в течение 2 часов. Катализатор отфильтровывали. Фильтрат концентрировали и очищали при помощи ОФ-ВЭЖХ (Таблица 1, Способ s) с получением 8-циклогексил-1-метил-3-((2-(триметилсилил)этокси)метил)-3H-имидазо[1,2-a]пирроло[2,3-e]пиразина (0,032 г, 41%) в виде прозрачного масла: LC/MS (Таблица 1, Способ b) Rt = 3,41 мин; MS m/z: 385 (M+H)+.
Общая процедура WWW: Образование карбамата
К амину (2-10 экв., предпочтительно 5 экв.) и DMAP (0-5 экв., предпочтительно 2 экв.) в органическом растворителе (таком как ТГФ или 1,4-диоксан, предпочтительно 1,4-диоксан) при около -20°C-80°C (предпочтительно около 40°C) добавляют карбонат или раствор карбоната (предпочтительно 1 экв.) в органическом растворителе (таком как ТГФ или 1,4 диоксан, предпочтительно 1,4-диоксан). Примерно через 1-16 часов (предпочтительно около 2 часов) реакционную смесь либо концентрируют при пониженном давлении, либо, необязательно, разбавляют органическим растворителем (таким как Et2O, EtOAc или DCM, предпочтительно EtOAc), промывают водой и водным раствором основания (таким как насыщенный водный раствор Na2CO3 или NaHCO3) и насыщенным солевым раствором, сушат над безводным Na2SO4 или MgSO4, фильтруют и концентрируют при пониженном давлении.
Иллюстрация Общей Процедуры WWW
Получение #WWW.1: (1S,3R,4S)-3-этил-4-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил фенилкарбамат

К анилину (0,063 г, 0,677 ммоль) и DMAP (0,033 г, 0,271 ммоль) в 1,4-диоксане (1 мл) при около 40°C добавляли раствор (1S,3R,4S)-3-этил-4-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил 4-нитрофенилкарбоната (0,080 г, 0,135 ммоль, получен из Примера #42 Стадия N) в 1,4-диоксане (1 мл). Примерно через 2 часа растворитель удаляли и остаток очищали хроматографией на силикагеле (12 г) с использованием в качестве элюента 0-40% EtOAc в DCM с получением (1S,3R,4S)-3-этил-4-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентилфенилкарбамата (0,0468 г, 63% ): LC/MS (Таблица 1, Способ b) Rt = 2,58 мин; MS m/z: 545 (M+H)+.
Общая процедура XXX: Образование мочевины с потерей защитной группы
К раствору или суспензии амина или соли амина (1-3 экв., предпочтительно 1-2 экв.) в органическом растворителе, таком как DCM, ТГФ или DMF (предпочтительно ТГФ) при около 20-80°C (предпочтительно около 20°C), необязательно, добавляют органическое основание, такое как TEA, DIEA, пиридин (предпочтительно DIEA) (1-10 экв., предпочтительно 1-5 экв.) с последующим добавлением CDI (1- 5 экв., предпочтительно 1 экв.). Примерно через 0,5-24 часа (предпочтительно около 1-3 часов) добавляют второй амин или соль амина (1-10 экв., предпочтительно 1-3 экв.), чистый или в виде раствора или суспензии в органическом растворителе, таком как DCM, ТГФ или DMF (предпочтительно ТГФ). Реакционную смесь поддерживают при около 20-80°C примерно в течение 2-24 часов (предпочтительно около 16 часов). Если реакция не завершилась, реакционную смесь можно нагреть при около 40-80°C (предпочтительно 55°C). Кроме того, можно добавить дополнительное количество амина или соли амина (1-50 экв., предпочтительно 20 экв.), и/или DMAP (1-10 экв., предпочтительно 1 экв.). Реакционную смесь поддерживают при около 20-80°C примерно в течение 24-96 часов (предпочтительно 72 часа). Это можно повторить, если реакция не завершилась, согласно данным ТСХ, LC/MS или ВЭЖХ. Реакционную смесь охлаждают до температуры окружающей среды. Реакционную смесь, необязательно, распределяют между органическим растворителем (таким как EtOAc или DCM) и водным раствором основания (таким как насыщенный водный раствор NaHCO3 или насыщенный водный раствор Na2CO3, предпочтительно насыщенный водный раствор NaHCO3). Альтернативно, реакционную смесь концентрируют при пониженном давлении и остаток распределяют, как описано выше. В любом случае, водный слой затем, необязательно, экстрагируют дополнительным количеством органического растворителя, такого как EtOAc или DCM. Объединенные органические слои, необязательно, можно промыть насыщенным солевым раствором и концентрировать в вакууме или сушить над безводным Na2SO4 или MgSO4 с последующим декантированием или фильтрованием перед концентрированием при пониженном давлении. Необязательно, реакционную смесь концентрируют при пониженном давлении и остаток непосредственно очищают.
Иллюстрация Общей Процедуры XXX
Получение #XXX.1: (цис)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-N-((тетрагидро-2H-пиран-4-ил)метил)пирролидин-1-карбоксамид
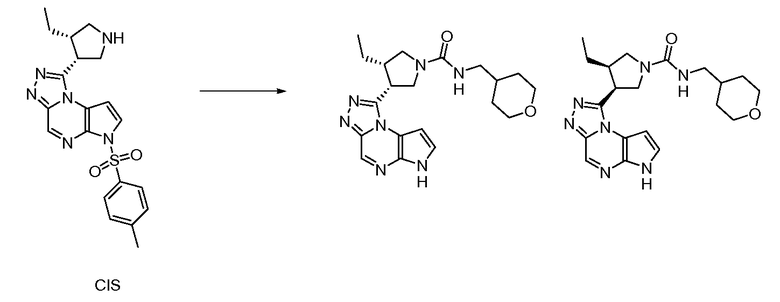
К раствору 1-((цис)-4-этилпирролидин-3-ил)-6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин · гидрохлорида (0,075 г, 0,168 ммоль, Пример #36, стадия F) в ТГФ (1,00 мл) добавляли DIEA (0,150 мл, 0,861 ммоль) и CDI (0,027 г, 0,168 ммоль). Примерно через 1 час добавляли 4-аминометилтетрагидропиран (0,020 г, 0,17 ммоль, Acros) и реакционную смесь перемешивали при температуре окружающей среды примерно в течение 16 часов. Реакционную смесь нагревали при около 55°C примерно в течение 24 часов. Добавляли DMAP (0,021 г, 0,168 ммоль) и продолжали перемешивание при около 55°C примерно в течение 48 часов. Добавляли 4-аминометилтетрагидропиран (0,400 г, 3,47 ммоль, Acros) и продолжали перемешивание при около 55°C примерно в течение 24 часов. Растворитель удаляли при пониженном давлении. Неочищенное вещество очищали при помощи ОФ-ВЭЖХ (Таблица 1, Способ m) с получением (цис)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)-N-((тетрагидро-2H-пиран-4-ил)метил)пирролидин-1-карбоксамида (0,007 г, 10%) в качестве продукта: LC/MS (Таблица 1, Способ a) Rt = 1,32 мин; MS m/z: 398 (M+H)+.
Образование мочевины с потерей защитной группы (получали из Примера #36, стадия F) с использованием Общей Процедуры XXX
(Таблица 1, Способ)
Общая процедура YYY: Присоединение по Михаэлю
К смеси нуклеофила (такого как амин или спирт, предпочтительно 1 экв.) и акцептора Михаэля (0,5-30 экв., предпочтительно 2-5 экв.), необязательно, в органическом растворителе (таком как DMF, EtOH или MeCN, предпочтительно DMF), необязательно, добавляют органическое основание (такое как TEA, DIEA или DBU, предпочтительно DBU, 1-5 экв., предпочтительно 1-2 экв.). Реакционную смесь нагревают при около 20-120°C (предпочтительно около 80°C) примерно в течение 2-60 часов (предпочтительно около 12-16 часов). Необязательно, добавляют дополнительное количество акцептора Михаэля (0,5-30 экв., предпочтительно 2-5 экв.) с последующим необязательным добавлением органического основания (такого как TEA, DIEA или DBU, предпочтительно DBU, 1-5 экв., предпочтительно 1-2 экв.) и реакционную смесь нагревают при около 20-120°C (предпочтительно около 80°C) примерно в течение 2-60 часов (предпочтительно около 2-5 часов). В случаях, когда реакция не достигает завершения, согласно данным ТСХ, LC/MS или ВЭЖХ, реакционную смесь снова подвергают описанным выше условиям. Реакционную смесь затем охлаждают до температуры окружающей среды. Необязательно, добавляют DCM и суспензию фильтруют. Реакционную смесь или необязательно полученный фильтрат концентрируют при пониженном давлении.
Иллюстрация Общей Процедуры YYY
Получение #YYY.1: 2-(3-((1S,3S,4R)-3-(3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)-4-метилциклопентиламино)оксетан-3-ил)ацетонитрил

К раствору (1S,3R,4S)-3-метил-4-(3-((2-(триметилсилил)этокси)метил)-3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)циклопентанамина (0,605 г, 1,569 ммоль, получен с использованием FFFFF из Получения #33, GGGGG с Получением #E.1.1, KKKK с PFPAA, D с NaOH, KK, Y) в DMF (6 мл) добавляли 2-(оксетан-3-илиден)ацетонитрил (0,298 г, 3,14 ммоль, J. Med. Chem. 2010, 53(8), 3227) и реакционную смесь нагревали при около 80°C примерно в течение 15 часов. Добавляли 2-(оксетан-3-илиден)ацетонитрил (0,149 г, 1,569 ммоль) и реакционную смесь нагревали при около 80°C примерно в течение 3,5 часов. Реакционную смесь охлаждали до температуры окружающей среды, концентрировали в вакууме и остаток очищали хроматографией на силикагеле с использованием в качестве элюента 0% → 10% MeOH в DCM. Вторая очистка хроматографией на силикагеле с использованием в качестве элюента 50% → 100% EtOAc в гептане, затем 10% MeOH в DCM давала 2-(3-((1S,3S,4R)-3-(3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)-4-метилциклопентиламино)оксетан-3-ил)ацетонитрил (0,262 г, 33%) в виде липкого коричневого твердого вещества: LC/MS (Таблица 1, Способ n) Rt = 0,75 мин; MS m/z 481 (M+H)+.
Общая процедура ZZZ: Добавление реагента Гриньяра или алкиллитиевого реагента к карбонил-содержащему соединению
Раствор карбонил-содержащего соединения (предпочтительно 1 экв.) в органическом растворителе (таком как ТГФ, 1,4-диоксан, Et2O, предпочтительно ТГФ) охлаждают до около -78°C-50°C (предпочтительно около 0°C) с последующим необязательным добавлением добавки, такой как хлорид лития (1-10 экв., предпочтительно 4 экв.) в органическом растворителе (таком как ТГФ или Et2O, предпочтительно ТГФ). К реакционному раствору добавляют раствор реагента Гриньяра или алкиллития в органическом растворителе (таком как ТГФ или Et2O, предпочтительно Et2O) и полученную смесь перемешивают при около -78°C-50°C примерно в течение 15 минут - 2 часов (предпочтительно около 0°C примерно в течение 20 мин) и затем, необязательно, нагревают до температуры окружающей среды и перемешивают примерно в течение 2-16 часов (предпочтительно около 4 часов). В случаях, когда реакция не достигает завершения, добавляют дополнительную порцию или порции раствора реагента Гриньяра или алкиллития в органическом растворителе (таком как ТГФ или Et2O, предпочтительно Et2O) для доведения реакции до завершения. Реакционную смесь затем, необязательно, охлаждают до около -78°C-0°C (предпочтительно около -78°C) и гасят путем добавления насыщенного водного раствора NH4Cl. Смесь, необязательно, перемешивают примерно в течение 5-30 минут (предпочтительно около 5 мин) с последующим добавлением органического растворителя (такого как EtOAc или DCM). Слои разделяют и органический раствор сушат над безводным MgSO4, фильтруют и концентрируют при пониженном давлении.
Иллюстрация Общей Процедуры ZZZ
Пример #ZZZ.1: 1-((1R,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)-2-метилпропан-2-ол
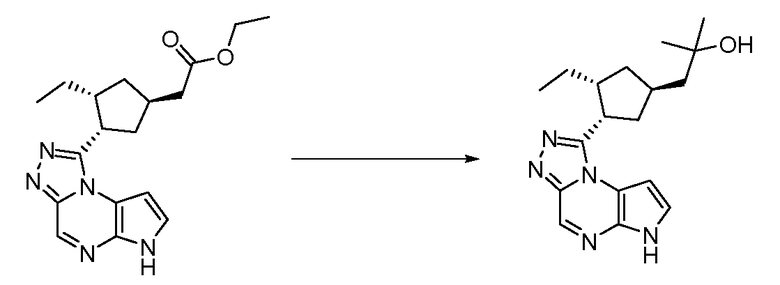
К раствору этил 2-((1R,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)ацетата (0,166 г, 0,486 ммоль, Пример #38, Стадия Н) в ТГФ (4 мл) при около 0°C добавляли хлорид лития в ТГФ (0,5 M, 3,9 мл) с последующим добавлением метилмагнийбромида в Et2O (3,0 M, 0,65 мл). Примерно через 20 минут реакционной смеси давали нагреться до комнатной температуры. Примерно через 4 часа реакционную смесь охлаждали до около -78°C и добавляли насыщенный водный раствор NH4Cl (около 5 мл). Примерно через 5 минут реакционной смеси давали нагреться до комнатной температуры и добавляли EtOAc (около 10 мл). Слои разделяли и органический раствор сушили над безводным MgSO4, фильтровали и концентрировали при пониженном давлении. Неочищенное вещество очищали хроматографией на силикагеле с использованием в качестве элюента EtOAc/MeOH с получением 1-((1R,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)-2-метилпропан-2-ола (0,118 г, 74%) в виде пенообразного вещества : LC/MS (Таблица 1, Способ a) Rt = 1,64 мин; MS m/z 328 (M+H)+.
Примеры, полученные с использованием Общей Процедуры ZZZ с метилмагнийбромидом
(Таблица 1, Способ)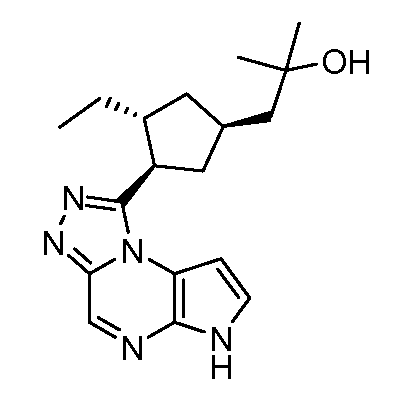

Общая процедура AAAA: Удаление защиты сульфонамида с использованием DBU
В колбу, содержащую сульфонамид, например, сульфонил-защищенный пиррол, (предпочтительно 1 экв.) в органическом растворителе (таком как 1,4-диоксан, MeOH или ТГФ/MeOH, MeCN, предпочтительно MeCN) добавляют DBU (1-30 экв., предпочтительно 5-6 экв.). Смесь перемешивают при около 20-100°C (предпочтительно примерно при комнатной температуре) примерно в течение 1-72 часов (предпочтительно около 24 часов ). В случаях, когда реакция не достигает завершения, согласно данным ТСХ, LC/MS или ВЭЖХ, реакционную смесь нагревают при около 30-100°C (предпочтительно около 45°C) примерно в течение 1-48 часов (предпочтительно около 12-24 часов). В случаях, когда реакция не достигает завершения, согласно данным ТСХ, LC/MS или ВЭЖХ, добавляют дополнительное количество DBU (1-20 экв., предпочтительно 1 экв.). Это можно повторить, если реакция не завершается, согласно данным ТСХ, LC/MS или ВЭЖХ. Реакционную смесь охлаждают до комнатной температуры и обрабатывают с использованием одного из следующих способов. Способ 1. Органический растворитель, необязательно, удаляют при пониженном давлении, добавляют подходящий органический растворитель (такой как EtOAc или DCM) и воду или насыщенный солевой раствор, слои разделяют и органический раствор сушат над безводным Na2SO4 или MgSO4, фильтруют и концентрируют досуха при пониженном давлении с получением целевого соединения. Способ 2. Реакционную смесь концентрируют при пониженном давлении и непосредственно очищают одним из описанных ниже способов.
Иллюстрация Общей Процедуры AAAA
Получение #AAAA.1: (E/Z)-этил 2-((3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентилиден)ацетат

К раствору (E/Z)-этил 2-((3R,4S)-3-этил-4-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентилиден)ацетата (2,00 г, 4,05 ммоль, Получение #III.1) в MeCN (20 мл) добавляли DBU (3,70 мл, 24,51 ммоль). Реакционную смесь перемешивали при комнатной температуре примерно в течение 16 часов. Реакционную смесь нагревали при около 45°C примерно в течение 24 часов. Добавляли DBU (1,00 мл, 6,63 ммоль) и продолжали нагревание при около 45°C примерно в течение 24 часов. Дополнительно добавляли DBU (1,00 мл, 6,63 ммоль) и продолжали нагревание при около 45°C примерно в течение 24 часов. Реакционную смесь охлаждали до комнатной температуры и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали флэш-хроматографией на силикагеле с градиентным элюированием 0-10% MeOH в DCM с получением (E/Z)-этил 2-((3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентилиден)ацетата (0,70 г, 51%) в виде коричневого пенообразного вещества: LC/MS (Таблица 1, Способ b) Rt = 1,90-1,95 мин; MS m/z: 340 (M+H)+.
Общая процедура BBBB: Удаление защиты сульфонамида с использованием TBAF
К раствору сульфонамида (предпочтительно 1 экв.) в органическом растворителе (предпочтительно ТГФ) при около -30 до 65°C (предпочтительно 0°C) добавляют TBAF (1-10 экв., предпочтительно 3 экв.). Можно добавить дополнительное количество TBAF (1-10 экв., предпочтительно 3 экв.) для доведения реакции до завершения. При развитии реакции до приемлемого уровня реакционную смесь распределяют между органическим растворителем (таким как DCM или EtOAc, предпочтительно EtOAc) и водной фазой (такой как вода или насыщенный солевой раствор). Органический слой отделяют и, необязательно, промывают насыщенным солевым раствором, сушат над безводным Na2SO4 или MgSO4, и/или фильтровуют перед концентрированием при пониженном давлении.
Иллюстрация Общей Процедуры BBBB
Получение #BBBB.1*: Этил 2-((3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентилиден)ацетат

К раствору этил 2-((3R,4S)-3-этил-4-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентилиден)ацетата (1,9 г, 3,85 ммоль, Получение #III.1) в ТГФ (30 мл) при около 0°C добавляли раствор TBAF (11,55 мл, 11,55 ммоль, 1M в ТГФ). Примерно через 30 минут добавляли дополнительное количество TBAF (7,70 мл, 7,70 ммоль, 1M в ТГФ). Примерно через 1 час к реакционной смеси добавляли EtOAc и насыщенный солевой раствор. Органический слой отделяли, концентрировали в вакууме и очищали хроматографией на силикагеле с использованием в качестве элюента EtOAc с получением этил 2-((3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентилиден)ацетата (1,3 г, 100%) в виде смеси стереоизомеров. LC/MS (Таблица 1, Способ a) Rt = 1,86 и 1,90 минут.; MS m/z: 340 (M+H)+.
Примеры, полученные с использованием Общей Процедуры BBBB
(Таблица 1, Способ)

Общая процедура CCCC: Удаление защиты сульфонамида с использованием KCN
В колбу содержащей сульфонамид, например, сульфонил-защищенный пиррол, (предпочтительно 1 экв.) в органическом растворителе (таком как 1,4-диоксан, MeOH или ТГФ, предпочтительно MeOH), добавляют KCN (1-3 экв., предпочтительно 2,2 экв.) в виде раствора в органическом растворителе (таком как 1,4-диоксан, MeOH или ТГФ, предпочтительно MeOH) или в виде твердого вещества. Смесь перемешивают при температуре окружающей среды примерно в течение 1-18 часов (предпочтительно около 16 часов). Органический растворитель, необязательно, удаляют при пониженном давлении и добавляют подходящий органический растворитель (такой как EtOAc или DCM) и воду. Слои разделяют и органический раствор сушат над безводным Na2SO4 или MgSO4, фильтруют или декантируют и концентрируют досуха при пониженном давлении и непосредственно очищают одним из описанных ниже способов.
Иллюстрация Общей Процедуры CCCC
Получение #CCCC.1: 3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил бензоат

К смеси 3-этил-4-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил бензоата (5,00 г, 7,84 ммоль, получен с использованием II из Примера #4 Стадия J с бензойной кислотой и B) в MeOH (16 мл) добавляли раствор цианида калия (0,74 мл, 17 ммоль) в MeOH (16 мл). Реакционную смесь перемешивали при температуре окружающей среды примерно в течение 16 часов. Реакционную смесь концентрировали при пониженном давлении с получением остатка. Остаток распределяли между водой (20 мл) и DCM (20 мл). Слои разделяли и водный слой экстрагировали при помощи DCM (3×10 мл). Экстракт затем промывали насыщенным водным раствором NaHCO3, сушили над безводным MgSO4, фильтровали и концентрировали при пониженном давлении с получением неочищенного масла. Неочищенное вещество очищали хроматографией на силикагеле с градиентным элюированием 0-10% MeOH в DCM с получением 3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентилбензоата (2,30 г, 78%) в виде твердого вещества. LC/MS (Таблица 1, Способ a) Rt = 2,08 мин; MS m/z: 376 (M+H)+.
Примеры, полученные с использованием Общей Процедуры D с KCN
(Таблица 1, Способ)
Общая процедура DDDD: Образование оксадиазола
К раствору эфира карбоновой кислоты (предпочтительно 1 экв.) в органическом растворителе (таком как DMF, NMP, ТГФ, MeOH/толуол, п-диоксан или MeOH, предпочтительно MeOH/толуол) добавляют основание (такое как K2CO3 или Cs2CO3, 2-10 экв., предпочтительно 2-4 экв.) и ацетимидамид (1-20 экв., предпочтительно 4-10 экв.). Реакционную смесь нагревают при около 100-160°C (предпочтительно около 130°C) примерно в течение 15 минут - 2 часов (предпочтительно около 45 мин) в условиях микроволнового облучения. В случаях, когда реакция не достигает завершения, согласно данным ТСХ, LC/MS или ВЭЖХ, можно добавить дополнительное количество ацетимидамида (1-20 экв., предпочтительно 3-10 экв.) и/или основания (такого как K2CO3 или Cs2CO3, 2-10 экв., предпочтительно 2-4 экв.). Реакционную смесь нагревают при около 100-160°C (предпочтительно около 130 -140°C) примерно в течение 15 минут - 2 часов (предпочтительно около 45 мин) в условиях микроволнового облучения. Необязательно, используют дополнительное нагревание с добавлением или без ацетимидамида и/или основания. Альтернативно, к раствору ацетимидамида (1-20 экв., предпочтительно 4-10 экв.) в органическом растворителе (таком как ТГФ или п-диоксан, предпочтительно ТГФ) добавляют основание (такое как NaH, 1-5 экв., предпочтительно 3 экв.). Примерно через 0,5-2 часа (предпочтительно около 0,5 часа), добавляют эфир карбоновой кислоты (предпочтительно 1 экв.). Примерно через 0,25-3 часа (предпочтительно около 0,25 часа) реакционную смесь нагревают при около 40-120°C (предпочтительно около 70°C) примерно в течение 1-48 часов (предпочтительно около 4 часов). Если реакционную смесь нагревали, реакционную смесь охлаждают до температуры окружающей среды. Реакционную смесь обрабатывают с использованием одного из следующих способов. Способ 1. Органический растворитель, необязательно, удаляют при пониженном давлении, добавляют подходящий органический растворитель (такой как EtOAc или DCM) и воду, насыщенный солевой раствор или насыщенный раствор NH4Cl, слои разделяют. Органический раствор промывают водой, насыщенным солевым раствором или насыщенным раствором NH4Cl и органический раствор сушат над безводным Na2SO4 или MgSO4, фильтруют и концентрируют досуха при пониженном давлении с получением целевого соединения. Способ 2. Реакционную смесь концентрируют при пониженном давлении и непосредственно очищают.
Иллюстрация Общей Процедуры DDDD
Получение #DDDD.1: (5-(((1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)метил)-1,2,4-оксадиазол-3-ил)метанол
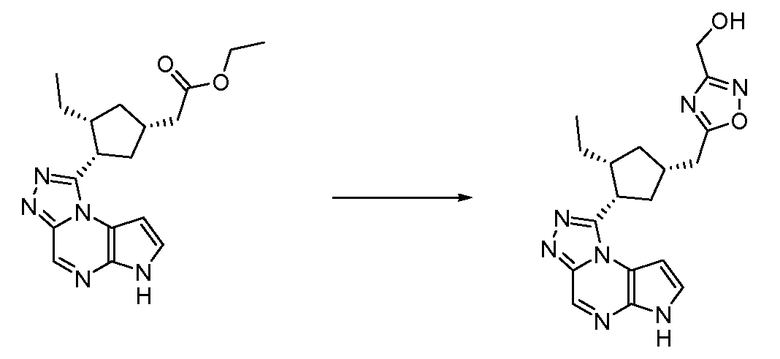
К раствору этил 2-((1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)ацетата (0,195 г, 0,571 ммоль, Пример #38, Стадия Н) в толуоле (1,00 мл) и MeOH (1,000 мл) добавляли (Z)-N',2-дигидроксиацетимидамид (0,515 г, 5,71 ммоль, Tyger) и K2CO3 (0,195 г, 1,41 ммоль). Реакционную смесь нагревали в микроволновом устройстве CEM при около 130°C два раза, каждый раз около 45 минут (максимальное давление 250 ф/дюйм2 (17,58 кг/см2), 1 мин линейное нарастание, максимальная мощность 300 Ватт). Добавляли (Z)-N',2-дигидроксиацетимидамид (0,200 г, 2,22 ммоль, Tyger) и реакционную смесь нагревали в микроволновом устройстве CEM при около 140°C примерно в течение 45 минут (максимальное давление 250 ф/дюйм2 (17,58 кг/см2), 1 мин линейное нарастание, максимальная мощность 300 Ватт). Добавляли (Z)-N',2-дигидроксиацетимидамид (0,200 г, 2,22 ммоль, Tyger) и K2CO3 (0,100 г, 0,725 ммоль) и реакционную смесь нагревали в микроволновом устройстве CEM при около 140°C примерно в течение 45 минут (максимальное давление 250 ф/дюйм2 (17,58 кг/см2), 1 мин линейное нарастание, максимальная мощность 300 Ватт). Растворитель удаляли при пониженном давлении. Остаток растворяли водой (20 мл) и EtOAc (50 мл). Водный слой экстрагировали при помощи EtOAc (5×50 мл). Объединенные органические слои сушили над безводным MgSO4, фильтровали и концентрировали с получением желтого остатка. Неочищенное вещество очищали флэш-хроматографией на силикагеле с градиентным элюированием 0-10% MeOH в EtOAc с получением (5-(((1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)метил)-1,2,4-оксадиазол-3-ил)метанола (0,042 г, 20%) в виде желтого твердого вещества: LC/MS (Таблица 1, Способ a) Rt = 1,67 мин; MS m/z: 368 (M+H)+.
Примеры, полученные с использованием Общей Процедуры DDDD с (Z)-N'-гидрокси-метоксиацетимидамидом
(Таблица 1, Способ)
Общая процедура EEEE: Образование мочевины с использованием фосгена
К раствору фосгена (1-1,5 экв., предпочтительно 1,2 экв., 20% раствор в толуоле) в органическом растворителе (таком как DCM), в инертной атмосфере при около 0°C добавляют раствор или суспензию амина или соли амина (предпочтительно 1 экв.) в органическом растворителе (таком как DCM, ТГФ или 1,4-диоксан, предпочтительно DCM) и органическое основание (такое как TEA, DIEA, пиридин, 1-10 экв., предпочтительно 5 экв., предпочтительно TEA). Примерно через 0,5-24 часа (предпочтительно около 40 мин) при около 0°C добавляют второй амин или соль амина (1-10 экв., предпочтительно 1-3 экв.), чистый или в виде раствора или суспензии в органическом растворителе (таком как DCM, ТГФ или DMF, предпочтительно DCM) и органическое основание (такое как TEA, DIEA, пиридин, 1-10 экв., предпочтительно 5 экв., предпочтительно TEA). Реакционную смесь перемешивают при около 0°C в течение 0,5-24 часов (предпочтительно 45 мин). Добавляют водный раствор основания (такой как водный раствор NH4OH или насыщенный водный раствор Na2CO3) с необязательным добавлением органического растворителя, такого как EtOAc или DCM. Водный слой затем, необязательно, экстрагируют дополнительным количеством органического растворителя (такого как EtOAc или DCM). Объединенные органические слои, необязательно, можно промыть водой или насыщенным солевым раствором и концентрировать в вакууме или сушить над безводным Na2SO4 или MgSO4 с последующим декантированием или фильтрованием перед концентрированием при пониженном давлении с получением целевого соединения.
Иллюстрация Общей Процедуры EEEE
Получение #EEEE.1: (3,3-дифторазетидин-1-ил)((цис)-3-этил-4-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)пирролидин-1-ил)метанон

К раствору фосгена (0,400 мл, 0,761 ммоль, 20% в толуоле) в DCM (1,5 мл) под баллоном N2 при около 0°C добавляли раствор 1-((цис)-4-этилпирролидин-3-ил)-6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразина (0,250 г, 0,609 ммоль, Пример #36, стадия F) в DCM (5,0 мл) и TEA (0,430 мл, 3,08 ммоль). Примерно через 40 минут при около 0°C добавляли по каплям раствор 3,3-дифторазетидин · гидрохлорида (0,095 г, 0,731 ммоль, Matrix) и TEA (0,430 мл, 3,08 ммоль) в DCM (5,0 мл) и перемешивали при около 0°C примерно в течение 45 минут. Добавляли насыщенный водный раствор бикарбоната натрия (2 мл) и слои разделяли. Водный слой экстрагировали при помощи DCM (2×30 мл). Объединенные органические слои промывали водой (25 мл), сушили над безводным MgSO4, фильтровали и концентрировали с получением коричневого остатка. Неочищенное вещество очищали флэш-хроматографией на силикагеле с градиентным элюированием 0-10% MeOH в DCM с получением (3,3-дифторазетидин-1-ил)((цис)-3-этил-4-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)пирролидин-1-ил)метанона (0,208 г, 65%) в виде коричневого остатка: LC/MS (Таблица 1, Способ a) Rt = 2,17 мин; MS m/z: 530 (M+H)+.
Общая процедура FFFF: Образование амида из сложного эфира
В реактор высокого давления загружают сложный эфир (предпочтительно 1 экв.), добавляют раствор аммиака в протонном растворителе (таком как этанол, метанол или вода, предпочтительно метанол). Реактор герметично закрывают и температуру поддерживают от около температуры окружающей среды до около 200°C (предпочтительно около 85°C). Примерно через 1-10 дней (предпочтительно около 2 дней) реакционную смесь охлаждают до комнатной температуры и реакционную смесь концентрировают в вакууме с получением неочищенного амида.
Иллюстрация Общей Процедуры FFFF
Пример #FFFF.1: 4-(3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)бутанамид

Этил 4-(3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)бутаноат (0,080 г, 0,217 ммоль, получен с использованием III из Получения #25 и (E)-этил 4-(диэтоксифосфорил)бут-2-еноата, W) и аммиак (7 н раствор в MeOH, 6,2 мл, 43,3 ммоль). Реакционный сосуд герметично закрывали и нагревали до около 85°C. Примерно через 2 дня пробирку охлаждали до комнатной температуры и реакционную смесь концентрировали в вакууме с получением 4-((3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)бутанамида (0,074 г, 100%) в виде твердого вещества, которое использовали без дополнительной очистки: LC/MS (Таблица 1, Способ c) Rt = 0,50 минут; MS m/z: 341 (M+H)+.
Общая процедура GGGG: Образование нитрила из первичного амида
К раствору первичного амида (предпочтительно 1 экв.) в органическом растворителе (таком как DCM, ТГФ, DCE, предпочтительно DCM) добавляют дегидратирующий реагент (такой как TFAA или SOCl2, предпочтительно TFAA) (1-20 экв., предпочтительно 10 экв.). Примерно через 1-20 часов (предпочтительно около 4 часов) при 10-60°C (предпочтительно при температуре окружающей среды) реакционную смесь концентрировают в вакууме.
Иллюстрация Общей Процедуры GGGG
Пример #GGGG.1: 4-((1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)бутаннитрил и 4-((1R,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)бутаннитрил

К раствору 4-(3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)бутанамида (0,090 г, 0,264 ммоль, Пример #FFFF.1) в DCM (3 мл) добавляли TFAA (0,373 мл, 2,64 ммоль). Примерно через 4 часа при температуре окружающей среды реакционную смесь концентрировали в вакууме и очищали хиральной препаративной ВЭЖХ (Таблица 2, Способ 33) с получением 4-((1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)бутаннитрила (0,013 г, 15%) (rt = 16,1 мин, or = отрицат.) LC/MS (Таблица 1, Способ a) Rt = 1,79 мин; MS m/z: 323 (M+H)+ и 4-((1R,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)бутаннитрила (0,010 г, 12%) (rt = 13,7 мин, or = отрицат.) LC/MS (Таблица 1, Способ a) Rt = 1,79 мин; MS m/z: 323 (M+H)+ в виде твердых веществ.
Общая процедура HHHH: O-алкилирование с использованием KOH или NaOH и TBAB
К спирту (предпочтительно 1 экв.) добавляют водный раствор основания (такой как 50% масс/об KOH или 50% масс/об NaOH, 1-60 экв., предпочтительно 11-24 экв.) и растворитель (такой как 1,4-диоксан или ТГФ, предпочтительно 1,4-диоксан) и реакционную смесь нагревают до около 45-100°C (предпочтительно около 70°C). К реакционной смеси добавляют алкилгалогенид или мезилат (1-30 экв., предпочтительно 8-16 экв.) и TBAB (0,05-2 экв., предпочтительно 0,08-1,6 экв.) и перемешивают примерно в течение 8-48 часов (предпочтительно около 24 часов). Альтернативно, порядок добавления может быть обратным. В случаях, когда реакция не достигает завершения, согласно данным ТСХ, LC/MS или ВЭЖХ, реакционную смесь можно снова подвергнуть нагреванию при около 25-100°C (предпочтительно около 70°C) примерно в течение 2-48 часов (предпочтительно около 8-24 часов) с необязательным добавлением дополнительного количества основания (такого как 50% масс/масс водный раствор KOH или 50% масс/масс водный раствор NaOH, 1-60 экв., предпочтительно 11-24 экв.), растворителя (такого как 1,4-диоксан или ТГФ предпочтительно 1,4-диоксан), алкилгалогенида или мезилата (1-30 экв., предпочтительно 8-16 экв.) и/или TBAB (0,05-2 экв., предпочтительно 0,08-1,5 экв.). Этот процесс повторяют вплоть до прекращения развития реакции. После охлаждения до температуры окружающей среды, реакционную смесь обрабатывают с использованием одного из следующих способов. Способ 1: Добавляют органический растворитель, такой как EtOAc или DCM, с необязательным добавлением воды или насыщенного солевого раствора, и слои разделяют. Водный слой затем, необязательно, экстрагируют дополнительным количеством органического растворителя, такого как EtOAc или DCM. Объединенные органические слои, необязательно, промывают насыщенным солевым раствором или водой, сушат над безводным MgSO4 или Na2SO4, фильтруют или декантируют и концентрируют при пониженном давлении. Способ 2: Можно осуществить фильтрование реакционной смеси, содержащей осадок. К фильтрату добавляют органический растворитель, такой как EtOAc или DCM, с необязательным добавлением воды или насыщенного солевого раствора, и слои разделяют. Водный слой затем, необязательно, экстрагируют дополнительным количеством органического растворителя, такого как EtOAc или DCM. Объединенные органические слои, необязательно, промывают насыщенным солевым раствором или водой, сушат над безводным MgSO4 или Na2SO4, фильтруют или декантируют и концентрируют при пониженном давлении.
Иллюстрация Общей Процедуры HHHH
Получение #HHHH.1: 3-(((1R,3R,4S)-3-этил-4-(6-((2-(триметилсилил)этокси)метил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентилокси)метил)-5-метилизоксазол
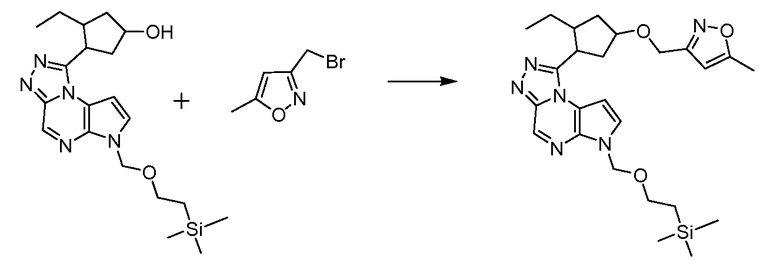
Смесь 3-этил-4-(6-((2-(триметилсилил)этокси)метил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентанола (0,072 г, 0,179 ммоль, Пример #35, Стадия Н), водного раствора KOH (50% масс/об 0,118 г, 2,10 ммоль) и 1,4-диоксана (0,1 мл) нагревали до около 70°C. К реакционной смеси добавляли 3-(бромметил)-5-метилизоксазол (0,063 г, 0,359 ммоль, Maybridge) и TBAB (0,004 г, 0,01 ммоль) и реакционную смесь перемешивали примерно в течение 24 часов. К реакционной смеси добавляли 3-(бромметил)-5-метилизоксазол (0,063 г, 0,36 ммоль, Maybridge) и водный раствор KOH (50% масс/об 0,118 г, 2,10 ммоль) и перемешивание продолжали примерно в течение 24 часов. Реакционную смесь охлаждали до температуры окружающей среды и добавляли EtOAc (10 мл) и воду (5 мл). Слои разделяли и водный слой экстрагировали при помощи EtOAc (10 мл). Объединенные органические слои сушили над безводным MgSO4, фильтровали и концентрировали при пониженном давлении. Неочищенное вещество очищали хроматографией на силикагеле с градиентным элюированием 0-5% MeOH в DCM с получением 3-(((1R,3R,4S)-3-этил-4-(6-((2-(триметилсилил)этокси)метил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентилокси)метил)-5-метилизоксазола (0,064 г, 72%): LC/MS (Таблица 1, Способ b) Rt = 2,58 мин; MS m/z: 497 (M+H)+.
Общая процедура IIII: Образование мезилата
К раствору спирта (предпочтительно 1 экв.) в органическом растворителе, таком как DCM, добавляют органическое основание, так как TEA или основание Хунига (1-4 экв., предпочтительно 2 экв.) при около 0-40°C (предпочтительно при комнатной температуре) с последующим добавлением по каплям, при этой же температуре, мезилхлорида (1-2 экв. предпочтительно 1,1 экв.). В случаях, когда реакционную смесь охлаждают до температуры ниже комнатной, ее перемешивают при этой температуре примерно в течение 1-3 часов (предпочтительно около 2 часов) и затем, необязательно, нагревают до температуры окружающей среды при перемешивании в течение ночи. Продукт можно обработать одним из следующих способов. 1) Реакционную смесь концентрируют. 2) Реакционную смесь промывают насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушат над безводным MgSO4, фильтруют и концентрируют.
Иллюстрация Общей Процедуры IIII
Получение #IIII.1: 3-этил-4-(6-((2-(триметилсилил)этокси)метил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил метансульфонат

Мезилхлорид (0,067 мл, 0,866 ммоль) добавляли по каплям к раствору 3-этил-4-(6-((2-(триметилсилил)этокси)метил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентанона (0,316 г, 0,787 ммоль, Пример #35 Стадия Н) и TEA (0,219 мл, 1,57 ммоль) в DCM (8 мл) и реакционную смесь перемешивали при комнатной температуре в течение ночи. Растворитель удаляли при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле (0 до 60% EtOAc в DCM) с получением 3-этил-4-(6-((2-(триметилсилил)этокси)метил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил метансульфоната (0,29 г, 77%) в виде белого аморфного твердого вещества: LC/MS (Таблица 1, Способ a) Rt = 2,53 мин; MS m/z 480 (M+H)+.
Общая процедура JJJJ: Замещение алкилмезилата, тозилата или галогенида нуклеофилом
В круглодонную колбу загружают алкил мезилат, тозилат или галогенид (предпочтительно 1 экв.) и органический растворитель, такой как DMF, DMA, NMP или DMSO (предпочтительно DMF). В реакционную колбу добавляют натриевую или калиевую соль (предпочтительно натриевую соль) нуклеофила, такого как, но не ограничиваясь этим, азид, цианид, тиоацетат, пиразол и триазол (1-10 экв., предпочтительно 5,0 экв.), по порциям. Когда нуклеофил еще не образовал натриевую или калиевую соль, добавляют основание, такое как 60% NaH в минеральном масле (1-10 экв., предпочтительно количество, эквимолярное используемому нуклеофилу). Смесь перемешивают при около 10-100°C (предпочтительно при температуре окружающей среды) примерно в течение 1-24 часов (предпочтительно около 20 часов). Если реакция не доведена до завершения, согласно данным ВЭЖХ, LC/MS или ТСХ, можно добавить дополнительное количество нуклеофила и/или основания (5-300% от используемого исходного количества, предпочтительно 10%), и реакцию продолжают примерно в течение 0,5-24 часов (предпочтительно около 2 часов). Реакционную смесь распределяют между органическим растворителем, таким как EtOAc или DCM (предпочтительно EtOAc) и водой. Слои разделяют и органический раствор сушат над безводным MgSO4 или Na2SO4, фильтруют и концентрируют при пониженном давлении с получением целевого соединения.
Иллюстрация Общей Процедуры JJJJ
Получение #JJJJ.1: 1-((1S,2R,4R)-4-азидо-2-этилциклопентил)-6-((2-(триметилсилил)этокси)метил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин

В круглодонную колбу загружали 3-этил-4-(6-((2-(триметилсилил)этокси)метил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентилметансульфонат (0,83 г, 1,7 ммоль, Получение #IIII.1) и DMF (7,0 мл). В реакционную колбу добавляли азид натрия (0,56 г, 8,6 ммоль). Смесь перемешивали при температуре окружающей среды примерно в течение 20 часов. Добавляли еще одну порцию азида натрия (0,056 г, 0,86 ммоль) и реакционную смесь перемешивали примерно в течение 2 часов. Реакционную смесь распределяли между EtOAc (20 мл) и водой(20 мл). Слои разделяли и органический раствор сушили над безводным MgSO4, фильтровали и концентрировали при пониженном давлении с получением 1-(-4-азидо-2-этилциклопентил)-6-((2-(триметилсилил)этокси)метил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразина (0,65 г, 88%) в виде коричневого масла: LC/MS (Таблица 1, Способ b) Rt = 2,85 мин; MS m/z: 427 (M+H)+.
Общая процедура KKKK: Циклизация кетона с использованием TFAA или PFPAA
К кетону (предпочтительно 1 экв.), необязательно раствореному в органическом растворителе, таком как ацетонитрил или DCM (предпочтительно ацетонитрил), добавляют TFA/TFAA (2-100 экв./10-60 экв., предпочтительно 2 экв./10 экв.) или PFPAA (2-30 экв., предпочтительно 10 экв.) или 2,2,3,3,3-пентафторпропановую кислоту/PFPAA (1-10 экв./5-50 экв., предпочтительно 2 экв./10 экв.) при около 0°C-50°C (предпочтительно при температуре окружающей среды). Реакционную смесь нагревают и перемешивают при температуре от около 0°C до около 80°C (предпочтительно около 60°C) примерно в течение 0,5-48 часов (предпочтительно около 2-4 часов). Можно добавить дополнительное количество TFAA или PFPAA (2-10 экв.) для завершения реакции. Необязательно, добавляют MeOH для гашения реакции. Реакционную смесь концентрируют при пониженном давлении. Альтернативно, неочищенную смесь можно, но необязательно, концентрировать перед распределением между водным раствором неорганического основания (например, водным раствором NaHCO3 или K2CO3) и органическим растворителем (например, EtOAc или DCM). Слои разделяют и водный слой снова экстрагируют органическим растворителем (таким как EtOAc и/или DCM). Объединенные органические слои, необязательно, промывают насыщенным солевым раствором, сушат над безводным MgSO4 или Na2SO4, фильтруют и концентрируют досуха при пониженном давлении.
Иллюстрация Общей Процедуры KKKK
Получение #KKKK.1: 3-Тозил-8-(2-тозил-2-азаспиро[3,3]гептан-6-ил)-3H-имидазо[1,2-a]пирроло[2,3-e]пиразин

К раствору 2-(5-тозил-5H-пирроло[2,3-b]пиразин-2-иламино)-1-(2-тозил-2-азаспиро[3,3]гептан-6-ил)этанона (0,631 г, 1,089 ммоль, получен с использованием R с 2-тозил-2-азаспиро[3,3]гептан-6-карбоновой кислотой [полученной, как описано в J. Org. Chem, 2010, 75, 5941] и триметилсилилдиазометаном, S с Примером #3 Стадия E, E с TFA) в MeCN (5 мл) добавляли PFPAA (2,15 мл, 10,9 ммоль). Смесь нагревали при около 60°C примерно в течение 2 часов. Реакционную смесь распределяли между DCM (30 мл) и насыщенным водным раствором NaHCO3 (50 мл). Слои разделяли и водный слой снова экстрагировали при помощи DCM (2×30 мл). Объединенные органические слои сушили над безводным MgSO4, фильтровали и концентрировали досуха при пониженном давлении. Неочищенное вещество очищали с использованием флэш-хроматографии на силикагеле с градиентным элюированием 50-100% EtOAc в гептане с получением 3-тозил-8-(2-тозил-2-азаспиро[3,3]гептан-6-ил)-3H-имидазо[1,2-a]пирроло[2,3-e]пиразина (0,467 г, 76%) в виде белого твердого вещества: LC/MS (Таблица 1, Способ b) Rt = 2,53 мин; MS m/z 562 (M+H)+.
Общая процедура LLLL: Образование бромкетона из кетона или альдегида
К кетону или альдегиду (предпочтительно 1 экв.) в органическом растворителе (DCM или DCE, предпочтительно DCM) при температуре от около -20 до 20°C (предпочтительно около 0°C) добавляют органическое основание, такое как TEA или DIEA (предпочтительно DIEA, 1-20 экв., предпочтительно 5-10 экв.), с последующим добавлением триметилсилилтрифторметансульфоната (1- 8 экв., предпочтительно 4,5 экв.). Реакционную смесь перемешивают при такой же температуре примерно в течение 0,5-6 часов (предпочтительно около 1 часа). Необязательно, добавляют подходящий органический растворитель (такой как EtOAc или DCM). Добавляют водный раствор (такой как насыщенный водный раствор NaHCO3 или вода). Слои разделяют, водный слой, необязательно, экстрагируют дополнительным количеством органического растворителя (такого как EtOAc или DCM) и органический слой или объединенные органические слои сушат над безводным Na2SO4 или MgSO4, фильтруют и концентрируют при пониженном давлении с получением TMS-защищенного енольного промежуточного соединения. Промежуточное соединение растворяют в органическом растворителе (DCM или DCE, предпочтительно DCM) при температуре около -20 до 60°C (предпочтительно при комнатной температуре) и добавляют неорганическое основание, такое как NaHCO3 или Na2CO3 (предпочтительно NaHCO3, 1-20 экв., предпочтительно 4 экв.) и NBS (1-3 экв., предпочтительно 1 экв.). Реакционную смесь перемешивают при такой же температуре примерно в течение 1-48 часов (предпочтительно около 18 часов). Добавляют подходящий органический растворитель (такой как EtOAc или DCM) и водный раствор (такой как насыщенный водный раствор NaHCO3 или вода), слои разделяют и органический раствор сушат над безводным Na2SO4 или MgSO4, фильтруют и концентрируют при пониженном давлении.
Иллюстрация Общей Процедуры LLLL
Получение #LLLL.1: трет-бутил-(цис-5-(2-бромацетил)-4-метилтетрагидрофуран-2-ил)метил(3,3,3-трифторпропилсульфонил)карбамат

К раствору трет-бутил (цис-5-ацетил-4-метилтетрагидрофуран-2-ил)метил(3,3,3-трифторпропилсульфонил)карбамата (0,54 г, 1,3 ммоль, получен с использованием M.1 из Получения #MMMM.1) в DCM (5 мл) при около 0°C добавляли DIEA (2,03 мл, 11,6 ммоль) и триметилсилилтрифторметансульфонат (1,06 мл, 5,82 ммоль). Реакционную смесь перемешивали при около 0°C примерно в течение 1 часа. Добавляли насыщенный водный раствор NaHCO3 (10 мл) и слои разделяли. Водный слой экстрагировали при помощи DCM (2×10 мл). Объединенные органические экстракты сушили над безводным MgSO4, фильтровали и концентрировали при пониженном давлении. Остаток растворяли в DCM (5 мл) и добавляли NaHCO3 (0,435 г, 5,17 ммоль) и NBS (0,230 г, 1,294 ммоль). Реакционную смесь перемешивали при температуре окружающей среды примерно в течение 18 часов. Реакционную смесь распределяли между водой (30 мл) и DCM (30 мл). Водный слой экстрагировали при помощи DCM (2×30 мл). Объединенные органические экстракты сушили над безводным MgSO4, фильтровали и концентрировали при пониженном давлении. Продукт очищали хроматографией на силикагеле с градиентным элюированием 0-30% EtOAc в гептане с получением трет-бутил-(цис-5-(2-бромацетил)-4-метилтетрагидрофуран-2-ил)метил(3,3,3-трифторпропилсульфонил)карбамата (0,472 г, 73%) в виде желтого твердого вещества: LC/MS (Таблица 1, Способ b) Rt = 2,76 мин; MS m/z: 494, 496 (M-H)-.
Общая процедура MMMM: Образование кетона из амида Вайнреба
К амиду Вайнреба (предпочтительно 1 экв.) в органическом растворителе (например, DCM, MeCN, 1,4-диоксан или ТГФ, предпочтительно ТГФ) добавляют реагент Гриньяра или алкиллитиевый реагент (1-10,0 экв., предпочтительно 6 экв.) при температуре около -30 до 40°C (предпочтительно около -10°C). Реакционную смесь перемешивают при температуре от около -30 до 40°C (предпочтительно около -10°C) примерно в течение 1-24 часов (предпочтительно около 5 часов). Реакционную смесь гасят водным раствором кислоты (таким как водный раствор HCl) и затем водой, распределяют между органическим растворителем (таким как Et2O, EtOAc или DCM) и водой. Слои разделяют и водный слой экстрагируют дополнительным количеством органического растворителя и объединенные органические слои, необязательно, промывают насыщенным солевым раствором. Органический слой, необязательно, сушат над безводным Na2SO4 или MgSO4 и затем декантируют или фильтруют перед концентрированием при пониженном давлении.
Иллюстрация Общей Процедуры MMMM
Получение #MMMM.1: N-((цис-5-ацетил-4-метилтетрагидрофуран-2-ил)метил)-3,3,3-трифторпропан-1-сульфонамид

К раствору цис-N-метокси-N,3-диметил-5-((3,3,3-трифторпропилсульфонамидо)метил)тетрагидрофуран-2-карбоксамида (0,70 г, 1,9 ммоль, получен с использованием E из Получения #43 с HCl, K с 3,3,3-трифторпропан-1-сульфонилхлоридом (Matrix), Z с NaOH, H с N,O-диметилгидроксиламин хлористоводородной кислотой) в ТГФ (5 мл) добавляли метилмагнийбромид (3 н раствор в Et2O, 3,86 мл, 11,6 ммоль) по каплям при температуре около -10°C. Реакционную смесь перемешивали при температуре около -10°C примерно в течение 5 часов. Добавляли водный раствор HCl (1 н, 9,66 мл, 9,66 ммоль) для гашения реакции. Реакционную смесь распределяли между водой (10 мл) и DCM (20 мл). Слои разделяли и водный слой экстрагировали при помощи DCM (2×20 мл). Объединенные органические экстракты концентрировали при пониженном давлении. Продукт очищали хроматографией на силикагеле с градиентным элюированием 0-100% EtOAc в гептане с получением N-((цис-5-ацетил-4-метилтетрагидрофуран-2-ил)метил)-3,3,3-трифторпропан-1-сульфонамида (0,57 г, 93%) в виде прозрачного масла: LC/MS (Таблица 1, Способ b) Rt = 2,02 мин; MS m/z: 318 (M+H)+.
Общая процедура NNNN: Образование β-гидроксисульфонамида из кетона
К необязательно замещенному метилсульфонамиду (1-8 экв., предпочтительно 1,5 экв.) в органическом растворителе (DCM или ТГФ, предпочтительно ТГФ) при температуре от около -20 до 20°C (предпочтительно около 0°C) добавляют алкиллитиевый реагент (например, n-BuLi, t-BuLi или LDA (предпочтительно n-BuLi, 1-20 экв., предпочтительно 1-2 экв.). Реакционную смесь перемешивают при температуре от около -20 до 20°C (предпочтительно около 0°C) примерно в течение 0,5-72 часов (предпочтительно около 1 часа). Полученный раствор добавляют по каплям к раствору кетона (предпочтительно 1,0 экв.) в органическом растворителе (DCM или ТГФ, предпочтительно ТГФ) при температуре от около -20 до 20°C (предпочтительно около 0-5°C). Реакционную смесь перемешивают при температуре от около -20 до 20°C (предпочтительно около 0-5°C) примерно в течение 1-72 часов (предпочтительно около 48 часов). Добавляют подходящий органический растворитель (такой как EtOAc или DCM) и водный раствор (такого как насыщенный водный раствор NaHCO3 или вода), слои разделяют и органический раствор сушат над безводным Na2SO4 или MgSO4, фильтруют и концентрируют при пониженном давлении.
Иллюстрация Общей Процедуры NNNN
Получение #NNNN.1: 3-Этил-1-(морфолиносульфонилметил)-4-(6-((2-(триметилсилил)этокси)метил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентанол

К раствору 4-(метилсульфонил)морфолина (0,217 г, 1,314 ммоль, Получение #41) в ТГФ (4 мл) при около 0°C добавляли n-BuLi (2,5 M в гексане, 0,53 мл, 1,3 ммоль). Реакционную смесь перемешивали при около 0°C примерно в течение 1 часа. Полученный раствор добавляли по каплям к раствору 3-этил-4-(6-((2-(триметилсилил)этокси)метил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентанона (0,350 г, 0,876 ммоль, Пример #35 Стадия G) в ТГФ (4 мл) при около 0°C. Реакционную смесь поддерживали при около 4°C в холодильнике примерно в течение 48 часов. Реакционную смесь распределяли между водой (5 мл) и DCM (5 мл). Слои разделяли и водный раствор экстрагировали при помощи DCM (2×5 мл). Объединенные органические экстракты концентрировали при пониженном давлении. Продукт очищали хроматографией на силикагеле с градиентным элюированием 0-2% MeOH/DCM, затем при помощи ОФ-ВЭЖХ (Таблица 1, Способ L) с получением 3-этил-1-(морфолиносульфонилметил)-4-(6-((2-(триметилсилил)этокси)метил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентанола (0,17 г, 34%) в виде желтого масла: LC/MS (Таблица 1, Способ b) Rt = 2,32 & 2,42 мин; MS m/z: 565 (M+H)+.
Общая процедура OOOO: Образование карбоната
К спирту (предпочтительно 1 экв.) в органическом растворителе (предпочтительно пиридин) при температуре от около -20°C до 80°C (предпочтительно при температуре окружающей среды) добавляют DMAP (0,1-5 экв., предпочтительно 0,3 экв.) и хлорформиат (1-10 экв., предпочтительно 2 экв.). Реакционную смесь перемешивают при температуре от около -20°C до 80°C (предпочтительно при температуре окружающей среды) примерно в течение 1-16 часов (предпочтительно около 1 часа). Реакционную смесь либо концентрируют при пониженном давлении, либо, необязательно, фильтруют, разбавляют органическим растворителем (предпочтительно EtOAc), промывают водой и водным раствором основания (таким как насыщенный водный раствор Na2CO3 или NaHCO3) или насыщенным солевым раствором, сушат над безводным Na2SO4 или MgSO4, фильтруют и концентрируют при пониженном давлении.
Иллюстрация Общей Процедуры OOOO
Получение #OOOO.1: (1R,3R,4S)-3-этил-4-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил 4-нитрофенилкарбонат
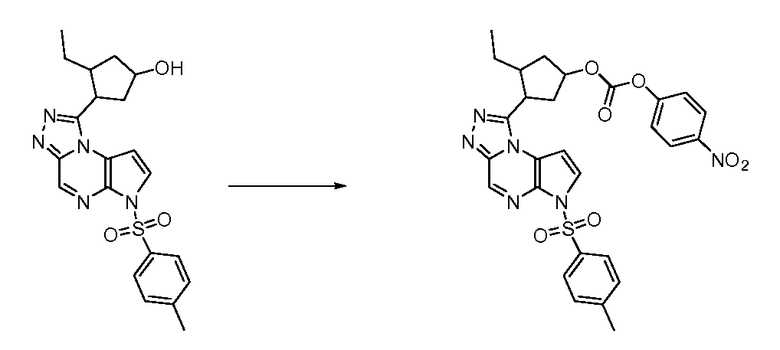
К скалемической смеси, обогащенной (1R,3R,4S)-3-этил-4-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентанолом (1,20 г, 2,82 ммоль, Пример #41, Стадия N), в пиридине (10 мл) добавляли DMAP (0,103 г, 0,846 ммоль) и 4-нитрофенилхлорформиат (0,853 г, 4,23 ммоль). Полученную смесь перемешивали при температуре окружающей среды примерно в течение 1 часа. Реакционную смесь концентрировали и очищали с использованием хроматографии на силикагеле с использованием в качестве элюента 0-30% EtOAc в DCM с получением скалемической смеси, обогащенной (1R,3R,4S)-3-этил-4-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил 4-нитрофенилкарбонатом (0,72 г, 43%): LC/MS (Таблица 1, Способ b) Rt = 2,64 мин; MS m/z: 591 (M+H)+.
Общая процедура PPPP: Образование карбамата с последующим гидролизом сульфонамида
К карбонату (предпочтительно 1 экв.) в органическом растворителе (предпочтительно 1,4-диоксан) при температуре от около -20 до 60°C (предпочтительно при температуре окружающей среды) добавляют амин (2-10 экв., предпочтительно 5 экв.) и, необязательно, DMAP (0-5 экв., предпочтительно 0 экв.). Примерно через 1-16 часов (предпочтительно около 1 часа) добавляют водный раствор гидроксида натрия (1-2 н, предпочтительно 1 н; 1-10 экв., предпочтительно 4 экв.). Реакционную смесь перемешивают при около 25-100°C (предпочтительно около 60°C) примерно в течение 10 минут - 5 часов (предпочтительно около 30 мин) и, если реакционную смесь нагревали, ее охлаждают до температуры окружающей среды. Реакционную смесь либо концентрируют при пониженном давлении, либо слои разделяют и водный слой экстрагируют органическим растворителем (предпочтительно DCM). Объединенные органические экстракты промывают водой, водным раствором основания (таким как насыщенный водный раствор Na2CO3 или NaHCO3) или насыщенным солевым раствором, сушат над безводным Na2SO4 или MgSO4, фильтруют и затем концентрируют при пониженном давлении.
Получение #PPPP.1: (1R,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил циклопропилкарбамат

К скалемической смеси, обогащенной (1R,3R,4S)-3-этил-4-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил 4-нитрофенилкарбонатом (Пример #41 Стадия O, 0,211 г, 0,357 ммоль), в 1,4-диоксане (1,5 мл) добавляли циклопропиламин (0,102 г, 1,79 ммоль). Примерно через 1 час добавляли 1н водный раствор гидроксида натрия (1,5 мл, 1,5 ммоль) и реакционную смесь нагревали при около 60°C примерно в течение 30 минут, затем охлаждали до температуры окружающей среды. Слои разделяли и водный слой экстрагировали при помощи DCM (3×5 мл). Объединенные органические слои концентрировали при пониженном давлении. Остаток очищали хроматографией на силикагеле с использованием в качестве элюента 0-10% MeOH в EtOAc с получением (1R,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентилциклопропилкарбамата (0,085 г, 67% ): LC/MS (Таблица 1, Способ b) Rt = 1,73 мин; MS m/z: 355 (M+H)+.
Примеры, полученные из скалемической смеси, обогащенной (1R,3R,4S)-3-этил-4-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил 4-нитрофенилкарбонатом (Пример #41 Стадия O)
(Таблица 2, Способ)

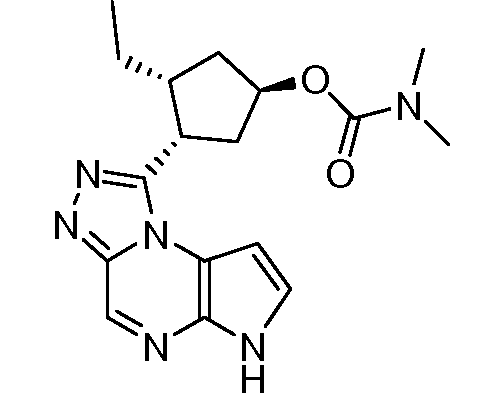
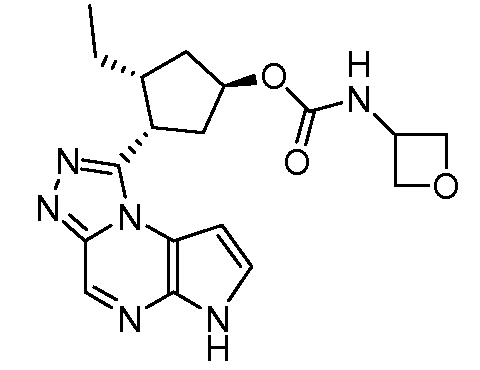

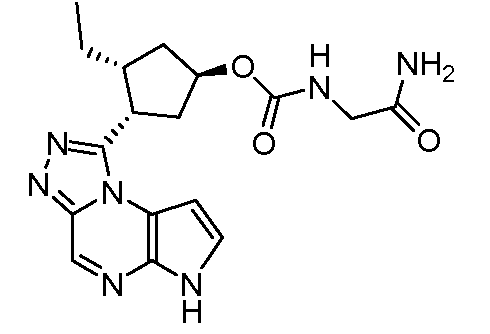
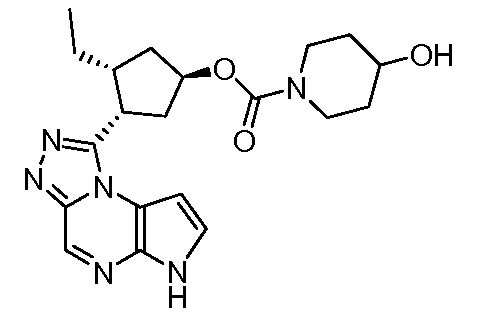

Примеры, полученные из скалемической смеси, обогащенной (1S,3R,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентилциклобутилкарбаматом (Пример #42 Стадия N)
(Таблица 2, Способ)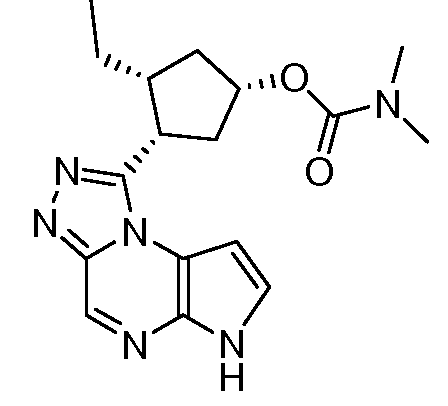










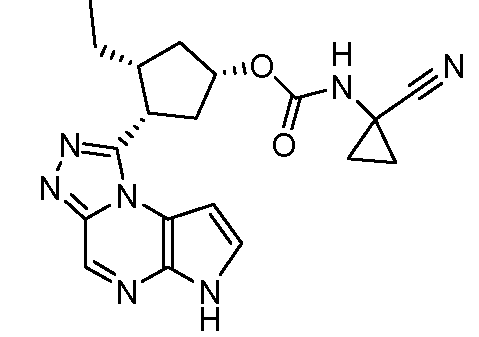

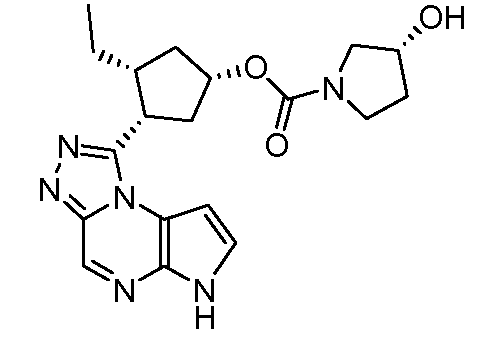

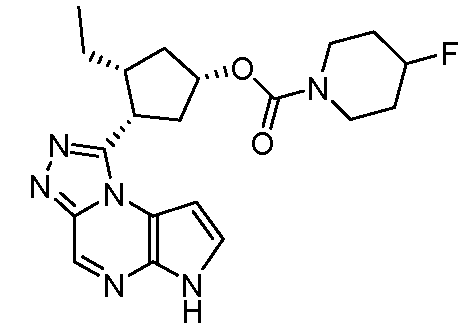








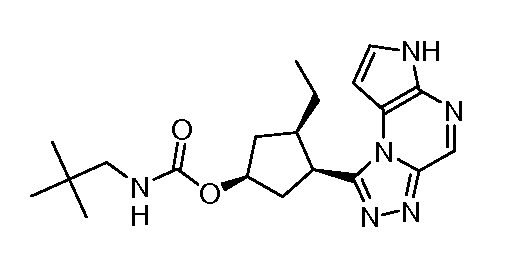





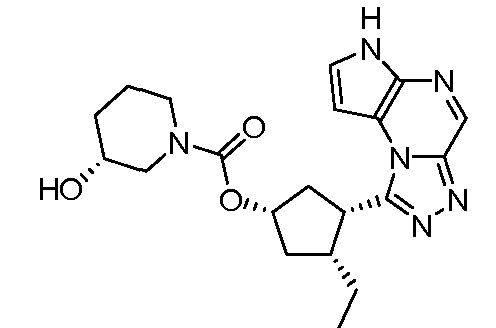








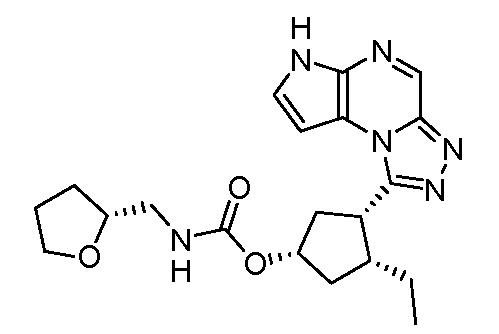



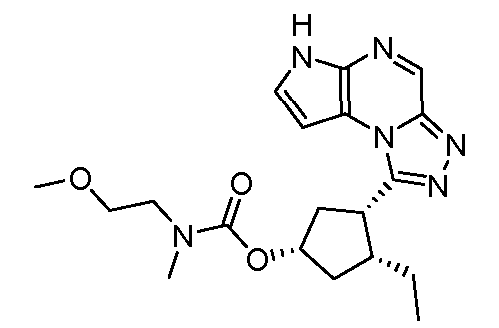
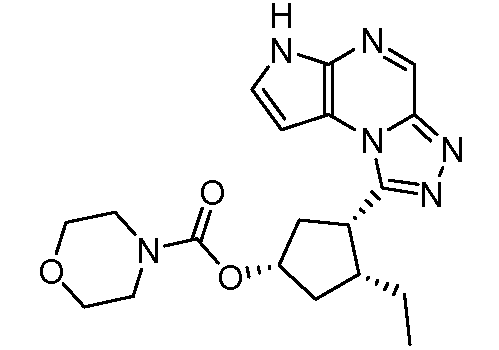

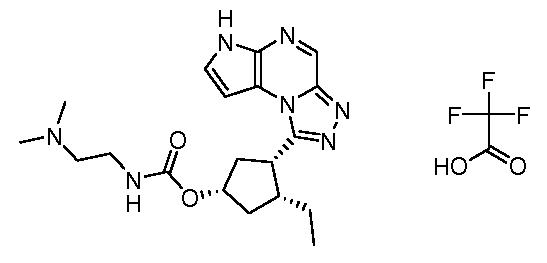


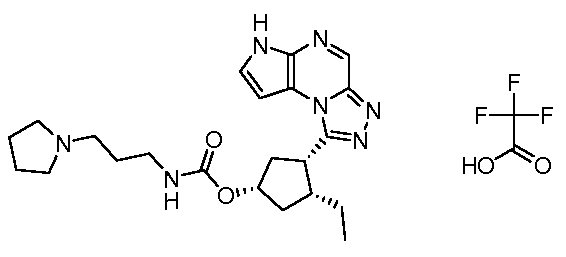

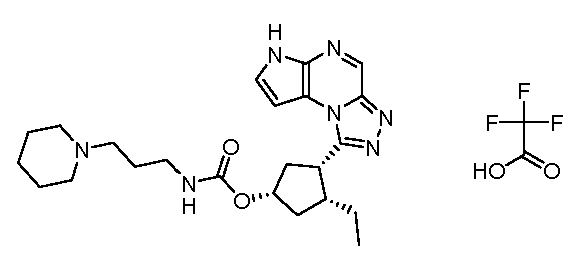

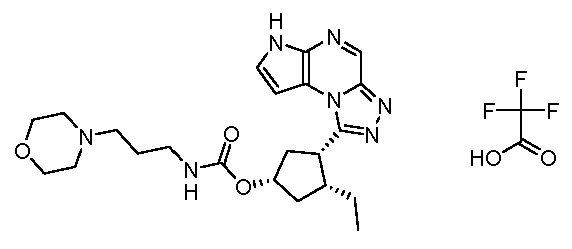
Общая процедура QQQQ: Окисление алкилтиоацетата до алкилсульфоновой кислоты
К смеси алкилтиоацетата (предпочтительно 1 экв.) и муравьиной кислоты (30-100 экв., предпочтительно 36 экв.) и добавляют по каплям водный H2O2 (~30%, 3-10 экв., предпочтительно 5 экв.). Реакционную смесь перемешивают при температуре окружающей среды примерно в течение 1-8 часов (предпочтительно около 2 часов). Реакционную смесь гасят насыщенным водным раствором Na2S2O3 и экстрагируют органическим растворителем, таким как DCM. Органический экстракт концентрируют при пониженном давлении. Полученный остаток, необязательно, распределяют между органическим растворителем, таким как EtOAc, и насыщенным солевым раствором. Водный экстракт концентрируют при пониженном давлении и полученный остаток, необязательно, растирают в порошок в органическом растворителе или в смеси органических растворителей, таких как MeOH, DCM или MeOH/DCM (предпочтительно MeOH/DCM), и фильтруют. Фильтрат концентрируют при пониженном давлении и, необязательно, очищают.
Иллюстрация Общей Процедуры QQQQ
Получение #QQQQ.1: (1S,3R,4S)-3-этил-4-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентан-1-сульфоновая кислота

К смеси S-(1S,3R,4S)-3-этил-4-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентилэтантиоата (0,28 г, 0,58 ммоль, получен с использованием P из Получения #25, Стадия E и DIBAL-H; IIII и JJJJ с тиоацетатом калия) и муравьиной кислоты (0,80 мл, 20,8 ммоль) добавляли по каплям водный раствор H2O2 (~30%, 0,30 мл, 2,9 ммоль). Реакционную смесь перемешивали при температуре окружающей среды примерно в течение 2 часов. Реакцию гасили насыщенным водным раствором Na2S2O3 (25 мл) и экстрагировали при помощи DCM (2×25 мл). Объединенные экстракты концентрировали при пониженном давлении. Полученный остаток распределяли между EtOAc и насыщенным солевым раствором (25 мл каждого). Водный экстракт концентрировали при пониженном давлении. Полученный остаток частично растворяли в MeOH/DCM (1:1, 50 мл), фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали при помощи ОФ-ВЭЖХ (Таблица 1, Способ y) с получением (1S,3R,4S)-3-этил-4-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентан-1-сульфоновой кислоты (0,058 г, 20%) в виде не совсем белого твердого вещества: LC/MS (Таблица 1, Способ b) Rt = 1,60 мин; MS m/z: 490 (M+H)+.
Общая процедура RRRR: Циклизация диамина с использованием цианогенбромида
К смеси замещенного диамина (1 экв.) в органическом растворителе (например, MeOH или EtOH, предпочтительно MeOH) добавляют цианогенбромид или цианогенбромид в MeCN (1-10 экв., предпочтительно 8,0 экв.). Смесь перемешивают при температуре окружающей среды примерно в течение 1-24 часов (предпочтительно около 16 часов) и растворитель удаляют при пониженном давлении.
Иллюстрация Общей Процедуры RRRR
Получение #RRRR.1*: N-((1S,3S,4R)-3-(2-амино-6-тозилимидазо[4,5-d]пирроло[2,3-b]пиридин-1(6H)-ил)-4-этилциклопентил)циклопропансульфонамид

К раствору N-((1S,3S,4R)-3-(5-амино-1-тозил-1H-пирроло[2,3-b]пиридин-4-иламино)-4-этилциклопентил)циклопропансульфонамида (0,200 г, 0,301 ммоль, Пример #23 Стадия I) в MeOH (3,0 мл) добавляли по каплям цианогенбромид (5 M в MeCN, 0,482 мл, 2,41 ммоль). Реакционную смесь перемешивали при температуре окружающей среды примерно в течение 16 часов. Растворитель удаляли при пониженном давлении и остаток очищали хроматографией на силикагеле с градиентным элюированием 0-10% MeOH в DCM с получением N-((1S,3S,4R)-3-(2-амино-6-тозилимидазо[4,5-d]пирроло[2,3-b]пиридин-1(6H)-ил)-4-этилциклопентил)циклопропан-сульфонамида (0,11 г, 67%) в виде коричневого твердого вещества.: LC/MS (Таблица 1, Способ a) Rt = 2,00 мин; MS m/z: 543 (M+H)+.
Общая процедура SSSS: Циклизация диамина с использованием NaNO 2
Смесь диамина (предпочтительно 1 экв.) и кислотного водного раствора (такого как 6 M HCl в воде) охлаждают до около 0°C. Затем добавляют водный раствор NaNO2 (1-5 экв., предпочтительно 1-2 экв.) и реакционную смесь поддерживают при около 0°C примерно в течение 1-6 часов (предпочтительно около 2-3 часов) и затем медленно нагревают до комнатной температуры или дают медленно нагреться до комнатной температуры сразу после добавления. Примерно через 1-18 часов (предпочтительно около 12-16 часов) реакционную смесь фильтруют, осуществляя при этом промывку водой, для сбора твердого вещества.
Иллюстрация Общей Процедуры SSSS
Получение #SSSS.1*: N-((1S,3R,4S)-3-этил-4-(6-тозилпирроло[2,3-b][1,2,3]триазоло[4,5-d]пиридин-1(6H)-ил)циклопентил)циклопропансульфонамид

Смесь N-((1S,3S,4R)-3-(5-амино-1-тозил-1H-пирроло[2,3-b]пиридин-4-иламино)-4-этилциклопентил)циклопропансульфонамида (0,15 г, 0,23 ммоль, Пример #23 Стадия I) и водного раствора HCl (6 н, 1,0 мл, 6,00 ммоль) охлаждали до около 0°C. Добавляли раствор NaNO2 (0,022 г, 0,32 ммоль) в воде (0,2 мл) и реакционную смесь перемешивали при около 0°C. Примерно через 3 часа реакционную смесь нагревали до комнатной температуры. Примерно через 15,5 часов реакционную смесь фильтровали, собирая желтое твердое вещество вакуумным фильтрованием, осуществляя при этом промывку водой (10 мл). Неочищенное твердое вещество очищали хроматографией на силикагеле с использованием в качестве элюента 0-20% EtOAc в DCM с получением N-((1S,3R,4S)-3-этил-4-(6-тозилпирроло[2,3-b][1,2,3]триазоло[4,5-d]пиридин-1(6H)-ил)циклопентил)циклопропансульфонамида (0,088 г, 74%): LC/MS (Таблица 1, Способ a) Rt = 2,44 мин; MS m/z: 529 (M+H)+.
Общая процедура TTTT: Образование скварамида
Смесь 3-амино-4-метоксициклобут-3-ен-1,2-диона (предпочтительно 1 экв.), амина (1-5 экв., предпочтительно 2 экв.), органического основания, такого как DIEA или TEA (1-10 экв., предпочтительно 5-6 экв. DIEA) и подходящего органического растворителя, такого как MeOH или DCE (предпочтительно MeOH) нагревают при около 40-65°C (предпочтительно около 50°C). Примерно через 1-24 часа (предпочтительно около 12-18 часов) реакционную смесь фильтруют, осуществляя при этом промывку водой, для сбора твердого вещества.
Иллюстрация Общей Процедуры TTTT
Получение #TTTT.1*: 3-((1S,3R,4S)-3-этил-4-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентиламино)-4-(3,3,3-трифторпропиламино)циклобут-3-ен-1,2-дион

Смесь 3-((1S,3R,4S)-3-этил-4-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентиламино)-4-метоксициклобут-3-ен-1,2-диона (0,090 г, 0,17 ммоль, Получение #29), 3,3,3-трифторпропан-1-амингидрохлорида (0,050 г, 0,337 ммоль, Fluorochem Limited), DIEA (0,18 мл, 1,0 ммоль) и MeOH (1,2 мл) нагревали при около 50°C. Примерно через 18 часов реакционную смесь охлаждали до комнатной температуры. Твердое вещество собирали путем вакуумного фильтрования, осуществляя при этом промывку при помощи MeOH (около 3-5 мл), и затем сушили в вакуумной печи при около 60°C с получением 3-((1S,3R,4S)-3-этил-4-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентиламино)-4-(3,3,3-трифторпропиламино)циклобут-3-ен-1,2-диона (0,083 г, 79%) в виде не совсем белого твердого вещества: LC/MS (Таблица 1, Способ a) Rt = 2,27 мин; MS m/z: 616 (M+H)+.
Общая процедура UUUU: Восстановление азида до амина
К раствору азида (предпочтительно 1 экв.) в подходящем органическом растворителе (таком как ТГФ или 1,4-диоксан, предпочтительно ТГФ) и воды добавляют трифенилфосфин (1-2 экв., предпочтительно 1,2 экв.). Реакционную смесь перемешивают при температуре около комнатной - 80°C (предпочтительно около 45°C) примерно в течение 1-24 часов (предпочтительно около 7 часов). Если реакционную смесь нагревали, ее охлаждают до комнатной температуры. Реакционную смесь обрабатывают с использованием одного из следующих способов. Способ 1. Реакционную смесь разбавляют в органическом растворителе (таком как DCM или EtOAc) и добавляют воду. Слои разделяют и органический раствор, необязательно, промывают водой и/или насыщенным солевым раствором, сушат над безводным MgSO4 или Na2SO4, фильтруют и растворитель удаляют при пониженном давлении. Способ 2. Реакционную смесь концентрируют при пониженном давлении.
Иллюстрация Общей Процедуры UUUU
Получение #UUUU.1: 3-этил-4-(6-((2-(триметилсилил)этокси)метил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентанамин

В круглодонную колбу загружали 1-(-4-азидо-2-этилциклопентил)-6-((2-(триметилсилил)этокси)метил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин (0,650 г, 1,52 ммоль, получен с использованием D из Получения #25 с NaOH, KK, P с NaBH4, IIII, JJJJ с NaN3), ТГФ (8,0 мл) и воду (1,6 мл). В колбу добавляли трифенилфосфин (0,480 г, 1,83 ммоль). Реакционную смесь нагревали до около 45°C примерно в течение 7 часов. Реакционную смесь охлаждали до комнатной температуры и добавляли EtOAc (20 мл) и воду (15 мл. Слои разделяли и органический раствор сушили над безводным MgSO4, фильтровали и концентрировали при пониженном давлении с получением масла, которое отверждалось при выстаивании. Неочищенное вещество очищали флэш-хроматографией на силикагеле с градиентным элюированием 1-10% DCM/MeOH/DEA (900:90:10) в DCM. Содержащие продукт фракции объединяли и концентрировали при пониженном давлении с получением масла, которое затем сушили при помощи вакуумного насоса в течение ночи с получением 3-этил-4-(6-((2-(триметилсилил)этокси)метил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентанамина в виде липкого масла (0,49 г, 80%): LC/MS (Таблица 1, Способ b) Rt = 1,85 мин; MS m/z 401 (M+H)+.
Общая процедура VVVV: Образование кетона из гетероарилгалогенида.
К раствору гетероарилгалогенида (предпочтительно 1 экв.) в органическом растворителе (например, ТГФ) при температуре около -100oC-0oC (предпочтительно около -78oC) добавляют по каплям алкиллитиевое основание (1-2 экв.) (предпочтительно втор-бутиллитий, 1,3 экв.). Реакционную смесь перемешивают при температуре около -100oC-0oC (предпочтительно около -78oC) примерно в течение 15 минут - 5 часов (предпочтительно около 1 часа). Раствор ацилирующего агента (такого как, например, хлорангидрид кислоты, амид Вайнреба или ацилимидазол, предпочтительно хлорангидрид кислоты, 1-3 экв., предпочтительно 1,5 экв.). Реакционную смесь оставляют для достижения температуры окружающей среды и добавляют воду. Слои разделяют и водный слой затем экстрагируют органическим растворителем, таким как DCM или EtOAc. Объединенные органические слои затем промывают водой и/или насыщенным солевым раствором, сушат над безводным MgSO4 или NaSO4, фильтруют и концентрируют при пониженном давлении.
Иллюстрация Общей Процедуры VVVV:
Получение #VVVV.1: трет-бутил 4-(2-(5-хлор-1-(триизопропилсилил)-1H-пирроло[2,3-b]пиридин-4-ил)-2-оксоэтил)пиперидин-1-карбоксилат

К раствору 5-хлор-1-(триизопропилсилил)-1H-пирроло[2,3-b]пиридина (0,338 г, 1,09 ммоль, Adesis) в ТГФ (5,5 мл) при температуре около -78°C добавляли по каплям втор-бутиллитий (1,015 мл, 1,421 ммоль). Реакционную смесь перемешивали при температуре около -78°C в течение примерно 1 часа, затем добавляли суспензию трет-бутил 4-(2-хлор-2-оксоэтил)пиперидин-1-карбоксилата (0,429 г, 1,64 ммоль, Получение #WWWW.1) в ТГФ (2 мл). Реакционную смесь перемешивали при температуре около -78°C в течение примерно 1 часа, затем оставляли для достижения температуры окружающей среды. Добавляли воду (5 мл) и продукт экстрагировали в DCM (3×10 мл). Объединенные органические экстракты промывали насыщенным солевым раствором и сушили над безводным MgSO4, фильтровали и концентрировали при пониженном давлении. Неочищенное вещество очищали хроматографией на силикагеле с использованием в качестве элюента 0-30% EtOAc в гептане с получением трет-бутил 4-(2-(5-хлор-1-(триизопропилсилил)-1H-пирроло[2,3-b]пиридин-4-ил)-2-оксоэтил)пиперидин-1-карбоксилата (0,147 г, 25%) в виде бесцветного масла: LC/MS (Таблица 1, Способ r) Rt = 3,97 мин; MS m/z: 534/536 (M+H)+
Общая процедура WWWW: Образование хлорангидрида кислоты.
К раствору карбоновой кислоты (предпочтительно 1 экв.) в органическом растворителе (например, DCM или DCE, предпочтительно DCM) добавляют оксалилхлорид (1-5 экв., предпочтительно 1-2 экв.) и N,N-диметилформамид ( 0,05-0,5 экв., предпочтительно 0,1 экв.). Реакционную смесь перемешивают при около 0-50oC (предпочтительно при температуре окружающей среды) примерно в течение 30 минут - 15 часов (предпочтительно 3 часа). Растворитель удаляют при пониженном давлении и остаток используют на следующей стадии без дополнительной очистки.
Иллюстрация Общей Процедуры WWWW:
Получение #WWWW.1: трет-бутил 4-(2-хлор-2-оксоэтил)пиперидин-1-карбоксилат

К раствору 2-(1-(трет-бутоксикарбонил)пиперидин-4-ил)уксусной кислоты (3,84 г, 15,78 ммоль) (получена с использованием M из этил 2-(пиперидин-4-ил)ацетата (Oakwood), Z ) в DCM (79 мл) при температуре окружающей среды добавляли оксалилхлорид (1,658 мл, 18,94 ммоль) и DMF (0,115 г, 1,58 ммоль). Реакционную смесь перемешивали при температуре окружающей среды примерно в течение 3 часов. Растворитель удаляли при пониженном давлении с получением трет-бутил 4-(2-хлор-2-оксоэтил)пиперидин-1-карбоксилата (4,13 г, 100%) в виде светло-желтого твердого вещества. Продукт использовали на следующей стадии без дополнительной очистки.
Общая процедура XXXX: Образование гидразона.
К смеси кетона (предпочтительно 1 экв.) в органическом растворителе (предпочтительно EtOH) добавляют гидразин (5-100 экв., предпочтительно 45-55 экв.) и уксусную кислоту (1-10 экв., предпочтительно 4-6 экв.). Реакционную смесь перемешивают при температуре окружающей среды - температуре кипения с обратным холодильником (предпочтительно при температуре кипения с обратным холодильником) примерно в течение 1-24 часов (предпочтительно около 16 часов). Растворитель удаляют при пониженном давлении и неочищенное вещество берут для поглощения в органический растворитель (такой как DCM) и сушат над безводным MgSO4 или NaSO4. Растворитель удаляют при пониженном давлении.
Иллюстрация Общей Процедуры XXXX:
Получение #XXXX.1: трет-бутил 4-(2-(5-хлор-1H-пирроло[2,3-b]пиридин-4-ил)-2-гидразоноэтил)пиперидин-1-карбоксилат
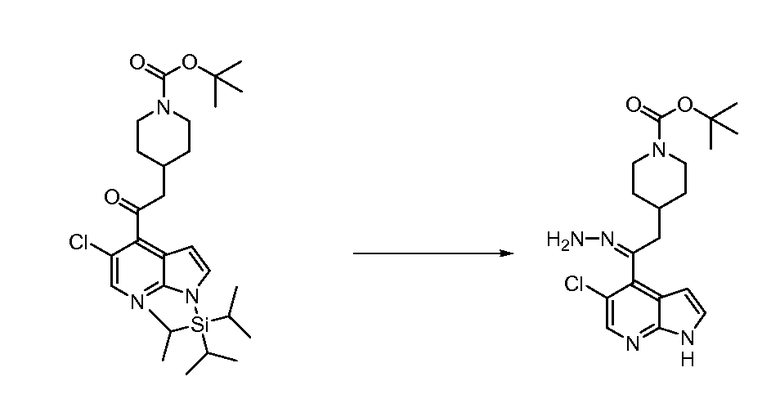
К суспензии трет-бутил 4-(2-(5-хлор-1-(триизопропилсилил)-1H-пирроло[2,3-b]пиридин-4-ил)-2-оксоэтил)пиперидин-1-карбоксилата (1,00 г, 1,87 ммоль) в EtOH (6,4 мл) добавляли безводный гидразин (2,94 мл, 94,0 ммоль) и AcOH (0,536 мл, 9,36 ммоль). Реакционную смесь перемешивали при температуре кипения с обратным холодильником примерно в течение 16 часов. Растворитель удаляли при пониженном давлении и неочищенное вещество брали для поглощения в DCM и сушили над безводным MgSO4. Растворитель удаляли и добавляли DCM (3 мл). Твердое вещество удаляли фильтрованием и фильтрат очищали хроматографией на силикагеле с использованием в качестве элюента 0-10% MeOH в DCM с получением трет-бутил 4-(2-(5-хлор-1H-пирроло[2,3-b]пиридин-4-ил)-2-гидразоноэтил)пиперидин-1-карбоксилата (0,324 г, 44%) в виде белого твердого вещества, состоящего из 1/1 смеси E/Z изомеров: LC/MS (Таблица 1, Способ r) Rt = 1,46 и 1,53 мин; MS m/z: 392/394 и 392/394 (M+H)+.
Общая процедура YYYY: Циклизация с использованием α-галогенальдегида
К α-галогенальдегиду (1-20 экв., предпочтительно 1,5 экв.) и защищенному 2-амино-5H-пирроло[2,3-b]пиразину (предпочтительно 1 экв.), необязательно, добавляют органический растворитель, такой DCE, DMF, 1,4-диоксан, EtOH, н-бутанол или толуол (предпочтительно н-бутанол или 1,4-диоксан) с использованием или без кислотного катализатора, такого как TsOH или серная кислота (0,05-0,2 экв.). Реакционную смесь перемешивают при температуре около комнатной - 150°C (предпочтительно около 90°C) примерно в течение 30 минут - 72 часов (предпочтительно около 48 часов). Необязательно, реакционную смесь подвергают микроволновому нагреванию при около 100-150°C (предпочтительно около 130°C) примерно в течение 30 минут - 15 часов (предпочтительно около 9 часов). В случаях, когда реакция не достигает завершения, согласно данным ТСХ, LC/MS или ВЭЖХ, реакционную смесь можно снова подвергнуть нагреванию при около 25-100°C (предпочтительно около 70°C) примерно в течение 2-48 часов (предпочтительно около 8-24 часов) с необязательным добавлением. В случаях, когда реакция не достигает завершения, согласно данным ТСХ, LC/MS или ВЭЖХ, можно добавить дополнительную порцию или порции α-галогенальдегида (1-20 экв., предпочтительно 2,5 экв.) в органическом растворителе, таком как 1,4-диоксан, и реакцию продолжают при температуре около комнатной - 150°C (предпочтительно около 125°C). Летучие вещества удаляют при пониженном давлении. Необязательно, неочищенную смесь разбавляют водой, водным раствором NH4Cl или водным раствором NaHCO3. Продукт можно выделить фильтрованием или можно добавить органический растворитель (например, EtOAc или DCM). Слои разделяют и водный слой можно снова экстрагировать органическим растворителем (таким как EtOAc и/или DCM). Объединенные органические слои, необязательно, промывают дополнительными водными растворами, такими как водный раствор NH4Cl, водный раствор NaHCO3, вода и/или насыщенный солевой раствор, сушат над безводным Na2SO4 или MgSO4, фильтруют и концентрируют досуха при пониженном давлении.
Иллюстрация Общей Процедуры YYYY
Получение #YYYY.1: этил 3-тозил-3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-карбоксилат

Раствор этил 2-хлор-3-оксопропаноата (1,60 г, 7,65 ммоль, получен, как описано в US2009005359A1) и 1,4-диоксана (10,0 мл) добавляли к 5-тозил-5H-пирроло[3,2-b]пиразин-2-амину (1,45 г, 5,03 ммоль, Получение #E.1.1) в атмосфере азота. Добавляли безводный бутан-1-ол (30,0 мл), подсоединяли обратный холодильник и систему герметично закрывали. Примерно через 30 минут смесь нагревали до около 80°C. Раствору давали охладиться до температуры окружающей среды. Добавляли раствор этил 2-хлор-3-оксопропаноата (2,78 г, 13,3 ммоль) и 1,4-диоксана (5 мл). Примерно через 30 минут реакционную смесь нагревали до около 80°C. Примерно через 30 минут смесь нагревали до около 125°C. Примерно через 48 часов коричневому раствору давали охладиться до температуры окружающей среды. Летучие вещества удаляли при пониженном давлении. Остаток очищали хроматографией на силикагеле с градиентным элюированием 5-50% EtOAc в гептане с получением этил 3-тозил-3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-карбоксилата (1,16 г, 60%): LC/MS (Таблица 1, Способ b) Rt = 2,52 мин; MS m/z 385 (M+H)+.
Общая процедура ZZZZ: Циклизация с использованием SOCl 2
К амиду, мочевине, гидразиду или кетону (предпочтительно 1 экв.), чистому или в виде раствора в органическом растворителе, таком как 1,4-диоксан, DCE или толуол (предпочтительно 1,4-диоксан), необязательно с буферным co-растворителем, таким как пиридин или TEA (предпочтительно TEA), добавляют по каплям SOCl2 (1,3-200 экв., предпочтительно 3 экв.), либо чистый либо в виде раствора в органическом растворителе, таком как 1,4-диоксан, DCE или толуол. Необязательно, реакционный сосуд охлаждают до около -10-25°C (предпочтительно около 0°C) в процессе добавления. Альтернативно, порядок добавления может быть обратным. Реакционную смесь нагревают до около 30-100°C (предпочтительно около 80°C) примерно в течение 0,5-24 часов (предпочтительно около 2 часов). Реакционной смеси дают охладиться до температуры окружающей среды. Летучие вещества, необязательно, удаляют при пониженном давлении и добавляют органический растворитель, такой как DCM, 1,4-диоксан или EtOAc (предпочтительно EtOAc). Органический слой промывают водным раствором, таким как водный раствор HCl, водный раствор NaOH, водный раствор NaHCO3, водный раствор NH4Cl, водный раствор Na2CO3, или водой (предпочтительно водным раствором Na2CO3) с необязательным охлаждением и продукт выделяют с использованием одного или нескольких из Способов Очистки, описанных выше. Необязательно, последующее удаление защитных групп можно осуществить с использованием Общих Процедур, перечисленных выше.
Получение #ZZZZ.1: трет-бутил (транс-4-((6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)метил)циклогексил)метилкарбамат

Тионилхлорид (0,030 мл, 0,41 ммоль ) добавляли по каплям к раствору трет-бутил (транс-4-(2-оксо-2-(2-(5-тозил-5H-пирроло[2,3-b]пиразин-2-ил)гидразинил)этил)циклогексил)метил-карбамата (0,127 г, 0,228 ммоль, получен с использованием M из гидрохлорида транс-(4-аминометилциклогексил)уксусной кислоты [AstaTech], H из Примера #1. Стадия D, HATU, TEA), TEA (0,160 мл, 1,15 ммоль) и 1,4-диоксана (2,3 мл) в атмосфере азота. Подсоединяли обратный холодильник и реакционную смесь нагревали до около 80°C. Примерно через 2 часа раствор охлаждали до температуры окружающей среды и добавляли водный раствор Na2CO3 (2 M, 3,4 мл, 6,8 ммоль) и двухфазную смесь нагревали до около 80°C. Примерно через 2 часа добавляли водный раствор NaOH (2 M, 0,570 мл, 1,14 ммоль) из-за низкой скорости реакции удаления защиты. Примерно через 17 часов смеси давали охладиться до температуры окружающей среды. Реакционный раствор разбавляли водой (5 мл) и затем экстрагировали при помощи EtOAc (2×10 мл). Объединенные органические слои сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали хроматографией на силикагеле с градиентным элюированием 2-8% MeOH в DCM с получением трет-бутил (транс-4-((6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)метил)циклогексил)метилкарбамата (0,0565 г, 63%): LC/MS (Таблица 1, Способ a) Rt = 1,85 мин; MS m/z: 385 (M+H)+.
Общая процедура AAAAA: Образование карбоновой кислоты или сложного эфира из арилгалогенида
Арил- или гетероарилгалогенид (предпочтительно 1 экв.) растворяют или суспендируют в органическом растворителе, таком как DMF, 1,4-диоксан, ТГФ, Et2O или толуол (предпочтительно DMF или ТГФ). Галогенид можно подвергнуть трансметаллированию с использованием основания, такого н-, трет- или втор-бутиллитий (1-3 экв.), или реагента Гриньяра, такого как изопропилмагнийбромид (1-3 экв.), с последующим улавливанием при помощи CO2 с получением карбоновой кислоты после кислотной обработки. Альтернативно, раствор арил- или гетероарилгалогенида можно обработать основанием, таким как Cs2CO3, K2CO3 или TEA (1-10 экв., предпочтительно TEA, 2 экв.). Необязательно, добавляют MeOH, (1-200 экв., предпочтительно 50 экв.). Добавляют источник палладия, такой продукт присоединения [1,1'-бис(дифенилфосфино)ферроцен]-дихлорпалладий(II)-CH2Cl2, [1,1'-бис(дифенилфосфино)ферроцен]-дихлорпалладий(II), бис(трифенилфосфин)дихлорпалладий или тетракис(трифенилфосфинпалладий(0) (0,02-1 экв., предпочтительно продукт присоединения [1,1'-бис(дифенилфосфино)ферроцен]-дихлорпалладий(II)-CH2Cl2, 0,1 экв.). Смесь помещают в атмосферу CO и затем нагревают до около 40-120°C (предпочтительно около 100°C) примерно в течение 0,5-24 часов (предпочтительно около 4,5 часов). Реакционную смесь, необязательно, гасят с использованием метоксида натрия или водного раствора NaOH (1-100 экв.) и добавляют органический растворитель (например, EtOAc или DCM). Слои разделяют и водный слой можно далее экстрагировать органическим растворителем (таким как EtOAc и/или DCM). Объединенные органические слои, необязательно, промывают дополнительными водными растворами, такими как насыщенный солевой раствор, сушат над безводным Na2SO4 или MgSO4, фильтруют и концентрируют досуха при пониженном давлении.
Иллюстрация Общей Процедуры AAAAA
Получение #AAAAA.1: метил 6-((2-(триметилсилил)этокси)метил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-8-карбоксилат

К раствору 8-иод-6-((2-(триметилсилил)этокси)метил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразина (0,050 г, 0,12 ммоль, получен с использованием KK из Получения #GGG.1.1 и NaH), TEA (0,034 мл, 0,24 ммоль), MeOH (0,25 мл, 6,2 ммоль) и DMF (0,6 мл), который продували азотом, добавляли [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий(II) (0,0098 г, 0,012 ммоль). Смесь продували при помощи CO и к реакционному сосуду подсоединяли баллон CO. Смесь нагревали до около 100°C. Примерно через 4,5 часа раствору давали охладиться до температуры окружающей среды. Добавляли воду (5 мл) и смесь экстрагировали при помощи EtOAc (2×10 мл). Объединенные органические слои сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении. Неочищенное вещество очищали хроматографией на силикагеле с градиентным элюированием 25-75% EtOAc/гептан в течение 30 минут с получением метил 6-((2-(триметилсилил)этокси)метил)-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-8-карбоксилата (0,0311 г, 74%): LC/MS (Таблица 1, Способ n) Rt = 0,74 мин; MS m/z: 348 (M+H)+.
Общая процедура BBBBB: Циклизация с использованием ортоэфира
К ортоэфиру (1-20 экв., предпочтительно 10 экв.) и защищенному 2-гидразинил-5H-пирроло[2,3-b]пиразину (предпочтительно 1 экв.), необязательно, добавляют органический растворитель, такой DCE, DMF, 1,4-диоксан или толуол (предпочтительно DMF), с использованием или без кислотного катализатора, такого как TsOH или TFA (0,05-0,2 экв.). Смесь можно оставить при температуре окружающей среды, или ее нагревают при около 30-100°C (предпочтительно около 100°C) примерно в течение 0,5-24 часов (предпочтительно около 17 часов). Летучие вещества можно удалить при пониженном давлении. Необязательно, неочищенную смесь можно разбавить водой, водным раствором NH4Cl или водным раствором NaHCO3. Продукт можно выделить фильтрованием, и можно добавить органический растворитель (например, EtOAc или DCM). Альтернативно, органический растворитель можно добавить непосредственно к водной смеси. Слои разделяют и водный слой можно снова экстрагировать органическим растворителем (таким как EtOAc и/или DCM). Объединенные органические слои, необязательно, промывают дополнительными водными растворами, такими как водный раствор NH4Cl, водный раствор NaHCO3, водой, и/или насыщенным солевым раствором, сушат над безводным Na2SO4 или MgSO4, фильтруют и концентрируют досуха при пониженном давлении.
Иллюстрация Общей Процедуры BBBBB
Получение #BBBBB.1: 6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин

Триэтилортоформиат (76,0 мл, 456 ммоль) добавляли к смеси 2-гидразинил-5-тозил-5H-пирроло[2,3-b]пиразина (13,8 г, 45,4 ммоль, Пример #1. Стадия D) и DMF (45 мл) в атмосфере азота. Подсоединяли обратный холодильник и смесь нагревали до около 100°C. Примерно через 17 часов раствору давали охладиться до температуры окружающей среды. Летучие вещества удаляли при пониженном давлении. Остаток суспендировали в воде (100 мл) и затем фильтровали, промывая водой. Водную фазу экстрагировали при помощи EtOAc (200 мл). Органические слои сушили над безводным Na2SO4, фильтровали и концентрировали. Полученное вещество объединяли с осадком и затем очищали хроматографией на силикагеле с градиентным элюированием 0-5% MeOH в DCM с получением 6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразина (10,4 г, 73%): LC/MS (Таблица 1, Способ n) Rt = 0,59 мин; MS m/z 314 (M+H)+.
Общая процедура CCCCC: Реакция Стилле сочетания арил- или гетероарилгалогенида
К дегазированному раствору арила, гетероарила или винилстаннана (предпочтительно 1,3 экв.) и арил-, гетероарил- или алкенилгалогенида (предпочтительно 1 экв.) в органическом растворителе, таком как DMF, 1,4-диоксан или толуол (предпочтительно DMF), можно добавить основание, такое как Cs2CO3, K2CO3 или TEA (1-10 экв.). Необязательно, можно использовать добавки, такие как LiCl (1-10 экв., предпочтительно 3 экв.), CsF (1-10 экв., предпочтительно 1,5 экв.) и/или CuI (0,05-0,5 экв., предпочтительно 0,2 экв.). Добавляют источник палладия(0), такой тетракис(трифенилфосфин)палладий(0), бис(дибензилиденацетон)палладий, или источник палладия(II), такой как бис(трифенилфосфин)палладий(II) хлорид, или ацетат палладия (0,01-0,2 экв., предпочтительно тетракис(трифенилфосфин)палладий(0), предпочтительно 0,1 экв.). Смесь нагревают при около 40-150°C (предпочтительно около 80°C) либо термически, либо с использованием микроволнового нагрева, примерно в течение 0,5-72 часов (предпочтительно около 4 часов). Раствор охлаждают до комнатной температуры, и летучие вещества можно удалить при пониженном давлении и неочищенную смесь разбавить водой, водным раствором NH4Cl, водным раствором NaHCO3 и органическим растворителем, таким как EtOAc или DCM. Когда присутствует твердое вещество, полученную реакционную смесь фильтруют для его удаления. Образовавшиеся слои фильтрата разделяют, и водный слой можно экстрагировать дополнительным органическим растворителем. Объединенные органические слои, необязательно, промывают дополнительными водными растворами, такими как насыщенный солевой раствор, затем сушат над безводным Na2SO4 или MgSO4, фильтруют и концентрируют досуха при пониженном давлении.
Иллюстрация Общей Процедуры CCCCC
Получение #CCCCC.1: 8-(2-(4-метилпиперазин-1-ил)пиримидин-4-ил)-6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин

В сосуде, содержащем 8-бром-6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин (0,030 г, 0,076 ммоль, получен с использованием D из Получения #BBBBB.1 и NaOH, GGG.1 с NBS, K.1 с TsCl и NaH), 2-(4-метилпиперазин-1-ил)-4-(трибутилстаннил)пиримидин (0,054 г, 0,12 ммоль, Получение #39), LiCl (0,010 г, 0,24 ммоль), CuI (0,003 г, 0,02 ммоль), CsF (0,017 г, 0,12 ммоль) и тетракис(трифенилфосфин)палладий(0) (0,009 г, 0,008 ммоль) в атмосфере азота, создавали вакуум и затем снова заполняли азотом. Добавляли 1,4-диоксан (0,5 мл) и азот барботировали через смесь примерно в течение 30 минут. Реакционный сосуд герметично закрывали и смесь нагревали до около 80°C. Примерно через 4 часа смеси давали охладиться до температуры окружающей среды. Смесь разбавляли водой (5 мл) и EtOAc (5 мл) и затем фильтровали через фильтровальный шприц. Слои разделяли и водную фазу экстрагировали при помощи EtOAc (5 мл). Объединенные органические слои концентрировали при пониженном давлении. Остаток очищали хроматографией на силикагеле с градиентным элюированием 0-10% MeOH в DCM в течение 40 минут с получением 8-(2-(4-метилпиперазин-1-ил)пиримидин-4-ил)-6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразина (0,026 г, 69%): LC/MS (Таблица 1, Способ n) Rt = 0,56 мин; MS m/z 490 (M+H)+.
Общая процедура DDDDD: Удаление защиты у Cbz-защищенного амина с использованием силана
К раствору Cbz-защищенного амина (предпочтительно 1 экв.) и силана (например, триэтилсилан, t-BuMe2SiH (предпочтительно триэтилсилан, 10-500 экв., предпочтительно 100 экв.)) добавляют органическое основание, такое как TEA или DIEA (предпочтительно TEA, 0,1-10 экв., предпочтительно 0,2 экв.) и палладиевый катализатор (например, хлорид палладия(II), ацетат палладия(II), трис(бензилиденацетон)дипалладий(0), бис(ацетато)трифенилфосфинпалладий(II) или дихлорбис(трифенилфосфин)палладий(II); предпочтительно хлорид палладия(II), 0,01-0,20 экв., предпочтительно 0,1 экв.). Реакционную смесь нагревают при около 40-180°C (предпочтительно около 120°C) примерно в течение 1-48 часов (предпочтительно около 8 часов). Катализатор удаляют фильтрованием и фильтрат концентрируют при пониженном давлении. Реакционную смесь, необязательно, обрабатывают путем добавления подходящего органического растворителя (такого как EtOAc или DCM) и воды. Слои разделяют и органический раствор сушат над безводным Na2SO4 или MgSO4, фильтруют и концентрируют при пониженном давлении с получением целевого соединения.
Иллюстрация Общей Процедуры DDDDD
Получение #DDDDD.1: 8-(пиперидин-4-ил)-3-((2-(триметилсилил)этокси)метил)-3H-имидазо[1,2-a]пирроло[2,3-e]пиразин

Раствор бензил 4-(3-((2-(триметилсилил)этокси)метил)-3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)пиперидин-1-карбоксилата (0,580 г, 1,15 ммоль, получен с использованием R из 1-(бензилоксикарбонил)пиперидин-4-карбоновой кислоты (Matrix), S из Примера #3 Стадия E, E с TFA, KKKK с PFPAA, D с NaOH, KK), TEA (0,03 мл, 0,229 ммоль), хлорида палладия(II) (0,020 г, 0,115 ммоль) в триэтилсилане (18,3 мл, 115 ммоль) нагревали при около 120°C примерно в течение 8 часов. Катализатор удаляли фильтрованием и фильтрат концентрировали при пониженном давлении. Продукт очищали хроматографией на силикагеле с градиентным элюированием 0-10% (90:9:1) (MeOH/DCM/DEA) в DCM с получением 8-(пиперидин-4-ил)-3-((2-(триметилсилил)этокси)метил)-3H-имидазо[1,2-a]пирроло[2,3-e]пиразина (0,234 г, 55%) в виде коричневого масла: LC/MS (Таблица 1, Способ b) Rt = 1,93 мин; MS m/z: 372 (M+H)+.
Общая процедура EEEEE: Образование гуанидина
К амину (предпочтительно 1 экв.) в органическом растворителе (например, DMF, MeCN, 1,4-диоксан или ТГФ, предпочтительно DMF) добавляют водный раствор основания (например, водный раствор Na2CO3, NaOH, K2CO3 или NaHCO3; (предпочтительно Na2CO3, 2-20 экв., предпочтительно 2-10 экв.)) или органическое основание, такое как TEA или DIEA (предпочтительно DIEA, 1-5 экв., предпочтительно 4 экв.) и добавляют 1H-пиразол-1-карбоксимидамид гидрохлорид (1-10,0 экв., предпочтительно 3 экв.). Реакционную смесь перемешивают при около 10-40°C (предпочтительно при комнатной температуре) примерно в течение 2-90 часов (предпочтительно около 72 часов) и обрабатывают с использованием одного из следующих способов. Способ 1: Добавляют органический растворитель (такой как Et2O, EtOAc или DCM) и добавляют воду и слои разделяют. Водный слой экстрагируют дополнительным количеством органического растворителя и объединенные органические слои, необязательно, промывают насыщенным солевым раствором, сушат над безводным Na2SO4 или MgSO4 и затем декантируют или фильтруют перед концентрированием при пониженном давлении. Способ 2: Реакционную смесь непосредственно очищают.
Иллюстрация Общей Процедуры EEEEE
Получение #EEEEE.1: 4-(3-((2-(триметилсилил)этокси)метил)-3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)пиперидин-1-карбоксимидамид
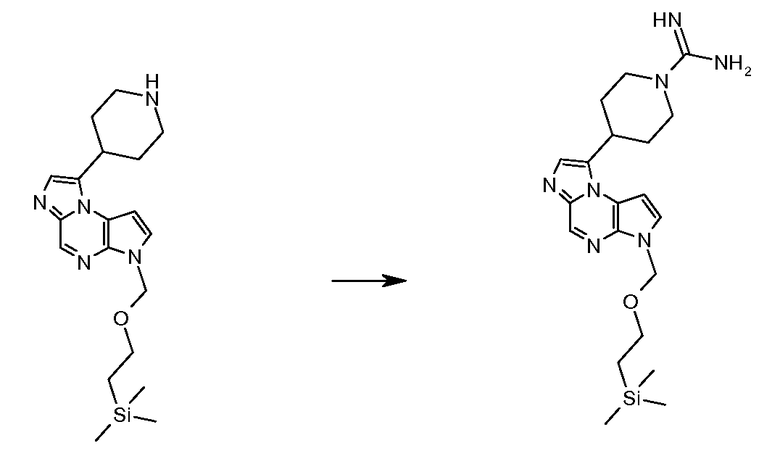
Раствор 8-(пиперидин-4-ил)-3-((2-(триметилсилил)этокси)метил)-3H-имидазо[1,2-a]пирроло[2,3-e]пиразина (0,100 г, 0,269 ммоль, Получение #DDDDD.1), 1H-пиразол-1-карбоксимидамид гидрохлорида (0,118 г, 0,807 ммоль) и DIEA (0,188 мл, 1,08 ммоль) в DMF (2 мл) перемешивали при комнатной температуре примерно в течение 72 часов. Реакционную смесь очищали при помощи ОФ-ВЭЖХ (Таблица 1, Способ I) с получением 4-(3-((2-(триметилсилил)этокси)метил)-3H-имидазо[1,2-a]пирроло[2,3-e]пиразин-8-ил)пиперидин-1-карбоксимидамида (0,037 г, 33%) в виде коричневого масла: LC/MS (Таблица 1, Способ b) Rt = 1,82 мин; MS m/z: 414 (M+H)+.
Общая процедура FFFFF: Образование сульфоксоний илида
К суспензии карбоновой кислоты (предпочтительно 1 экв.) в органическом растворителе (таком как ТГФ, 2-метилтетрагидрофуран или MTBE, предпочтительно ТГФ) добавляют органическое основание, такое как основание Хунига или TEA (предпочтительно TEA) (1,2-3,5 экв., предпочтительно 3,5 экв.), и активирующий агент, такой как DCC или HATU (предпочтительно HATU) (1-1,5 экв., предпочтительно 1,01 экв.). Реакционную смесь перемешивают при 10-40°C, предпочтительно при температуре окружающей среды, примерно в течение 1-20 часов (предпочтительно около 1-2 часов). В отдельной колбе, триметилсульфоксоний хлорид (1,25-5 экв., предпочтительно 3 экв.) добавляют к суспензии основания, такого как трет-бутоксид натрия или трет-бутоксид калия (3-5 экв., предпочтительно 3,15 экв.), в органическом растворителе (таком как ТГФ, 2-метилтетрагидрофуран или MTBE, предпочтительно ТГФ). Реакционную смесь перемешивают при около 60-70°C (предпочтительно 65°C) примерно в течение 2-4 часов (предпочтительно около 3 часов). Суспензию охлаждают до около -5-5°C и полученный выше активированный эфирный раствор добавляют по каплям примерно в течение 20-60 минут. Реакционную смесь перемешивают при температуре около -5-5°C примерно в течение 1-20 часов (предпочтительно около 1-2 часов). Реакционную смесь гасят водой, по каплям, при около 0-40°C (предпочтительно при температуре окружающей среды) примерно в течение 2-50 минут и перемешивают примерно в течение 0,2-20 часов (предпочтительно около 18 часов при температуре окружающей среды. Реакционную смесь можно концентрировать при пониженном давлении для удаления летучих веществ и затем распределить между органическим растворителем (таким как EtOAc) и водой. Водный слой, необязательно, можно экстрагировать дополнительным органическим растворителем, таким как EtOAc. Объединенный органический слой промывают водой, сушат над безводным Na2SO4 или MgSO4, фильтруют, концентрируют при пониженном давлении с получением целевого соединения.
Иллюстрация Общей Процедуры FFFFF
Получение #FFFFF.1: 2-(4-(Дибензиламино)циклогексил)-диметилсульфоксоний-2-оксо-этилид
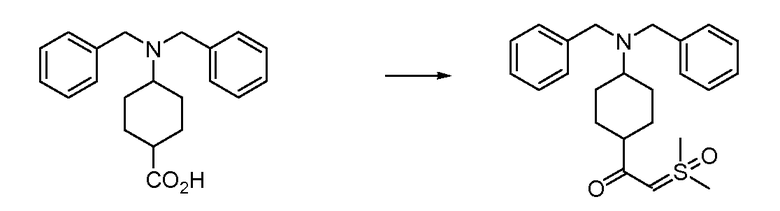
В 250-мл колбу добавляли 4-(дибензиламино)циклогексанкарбоновую кислоту (5,6 г, 17,3 ммоль), HATU (6,75 г, 17,4 ммоль) и TEA (8,45 мл, 60,6 ммоль) в ТГФ (60 мл) с получением белой суспензии. Реакционную смесь перемешивали при температуре окружающей среды примерно в течение 1 часа. В 500-мл колбу добавляли триметилсульфоксоний хлорид (6,82 г, 51,9 ммоль) и трет-бутоксид калия (6,44 г, 54,5 ммоль) в ТГФ (60 мл) с получением другой белой суспензии. Реакционную смесь перемешивали при около 65°C примерно в течение 3 часов. Реакционную смесь охлаждали до около 5°C. Полученный выше активированный эфирный раствор добавляли по каплям примерно в течение 50 минут. Реакционную смесь перемешивали при около 0-5°C примерно в течение 90 минут. Реакцию гасили путем добавления по каплям воды (120 мл) при около 0-5°C примерно в течение 25 минут. Реакционную смесь перемешивали при около 0-5°C примерно в течение 30 минут, затем при температуре окружающей среды примерно в течение 18 часов. Смесь концентрировали при пониженном давлении с получением белой суспензии. Суспензию распределяли между EtOAc (300 мл) и водой (200 мл). Водный слой экстрагировали при помощи EtOAc (2×100 мл). Объединенный органический слой промывали водой (50 мл) и насыщенным солевым раствором (3×40 мл). Органический слой сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Остаток растворяли в горячем MeOH (100 мл) и концентрировали при пониженном давлении. Масло растворяли в горячем MeOH (60 мл) и концентрировали с получением белого твердого вещества. Твердое вещество растворяли в MeOH (36 мл) и воде (12 мл) при около 55°C. Раствор охлаждали до температуры окружающей среды, затем до около 5°C. К суспензии добавляли дополнительное количество смеси 3:1 MeOH/вода (40 мл). Суспензию фильтровали, промывали смесью 1:1 MeOH/вода (20 мл), затем гептаном (20 мл). Полученную мокрую лепешку сушили в нагретой вакуумной печи при около 60°C примерно в течение 72 часов с получением 2-(4-(дибензиламино)циклогексил)-диметилсульфоксоний-2-оксо-этилида (5,44 г, 79%) в виде белого твердого вещества: LC/MS (Таблица 1, Способ a) Rt = 1,42, 1,45 мин; MS m/z 398 (M+H)+.
Общая процедура GGGGG: Взаимодействие сульфоксоний илида с амином
К смеси сульфоксоний илида (предпочтительно 1 экв.) и амина (0,7-2 экв., предпочтительно 1,2 экв.) добавляют катализатор (такой как [Ir(COD)Cl]2, [(COD)Ir(OMe)]2, (COD)Ir(acac), Ir(COD)2BF4, Ir(COD)2BArF, Rh2(OAc)2, Rh2(TFA)4, [Ru(cym)Cl2]2, RuCl2(PPh3)3, RuCl2(DMSO)4, предпочтительно [Ir(COD)Cl]2 (0,01-0,1 экв., предпочтительно 0,04 экв.)). Добавляют дегазированный органический растворитель (такой как DCM, DCE, MeCN, ТГФ, 2-метилтетрагидрофуран, CHCl3, толуол или DMF, предпочтительно DCE). Реакционную смесь продувают при помощи N2 примерно в течение 10-20 минут и перемешивают при около 20-90°C (предпочтительно около 70°C) примерно в течение 1-96 часов (предпочтительно около 3-6 часов). Необязательно, к реакционной смеси можно добавить дополнительное количество катализатора (предпочтительно [Ir(COD)Cl]2 экв.) в случаях, когда реакция не достигает завершения, согласно данным ТСХ, LC/MS или ВЭЖХ. Когда реакция осуществлена до приемлемой степени, можно осуществить концентрирование реакционной смеси в вакууме с получением продукта.
Иллюстрация Общей Процедуры GGGGG
Получение #GGGGG.1: 1-(4-(дибензиламино)циклогексил)-2-(5-тозил-5H-пирроло[2,3-b]пиразин-2-иламино)этанон
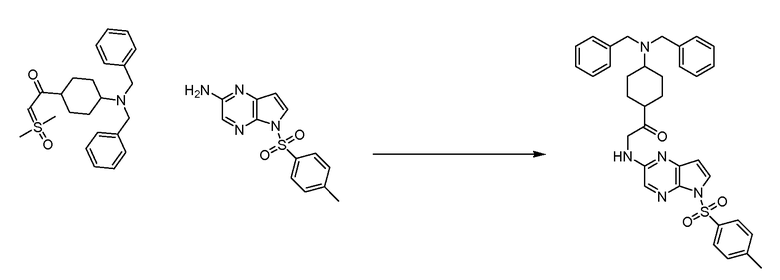
В 100-мл 2-горлую круглодонную колбу добавляли 2-(4-(дибензиламино)циклогексил)-диметилсульфоксоний-2-оксо-этилид (5,4 г, 13,6 ммоль, Получение #FFFFF.1), 5-тозил-5H-пирроло[2,3-b]пиразин-2-амин (4,7 г, 16,3 ммоль, Получение #E.1.1) и [Ir(COD)Cl]2 (0,365 г, 0,543 ммоль, Альфа Aesar). Реакционный сосуд продували при помощи N2 примерно в течение 10 минут. В реакционный сосуд добавляли дегазированный DCE (25 мл) через шприц. Реакционную смесь продували при помощи N2 примерно в течение 10 минут и перемешивали в атмосфере N2 при около 70°C примерно в течение 3 часов. Реакционной смеси давали охладиться до температуры окружающей среды. Растворитель удаляли при пониженном давлении. Остаток очищали флэш-хроматографией на силикагеле с градиентным элюированием 5-70% EtOAc в гептане с получением 1-(4-(дибензиламино)циклогексил)-2-(5-тозил-5H-пирроло[2,3-b]пиразин-2-иламино)этанона (5,8 г, 65%) в виде стекловидного твердого вещества: LC/MS (Таблица 1, Способ a) Rt = 3,24 и 3,26 мин; MS m/z 608 (M+H)+.
Пример #1:
Пример #1.1: N,N-диэтил-1-((1S,3R,4R)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)метансульфонамид,
Пример #1.2: N,N-диэтил-1-((1R,3S,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)метансульфонамид,
Пример #1.3: N,N-диэтил-1-((1S,3S,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)метансульфонамид,
Пример #1.4: N,N-диэтил-1-((1R,3R,4R)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)метансульфонамид,
Пример #1.5: N,N-диэтил-1-((1S,3S,4R)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)метансульфонамид и
Пример #1.6: N,N-диэтил-1-((1R,3S,4R)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)метансульфонамид
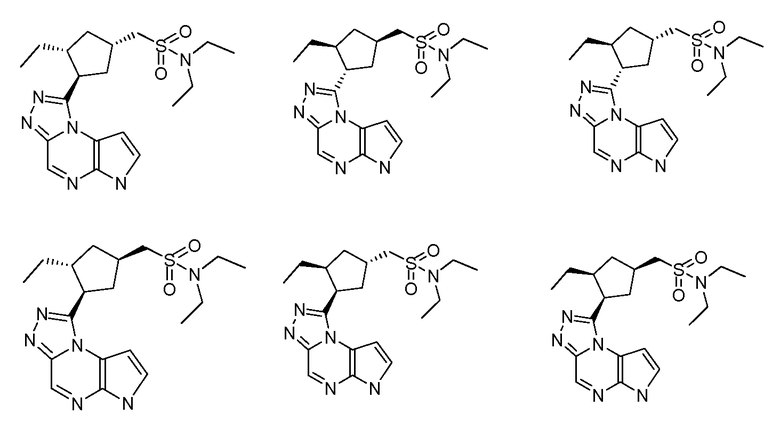
Стадия A: 5-бром-3-((триметилсилил)этинил)пиразин-2-амин

К раствору 3,5-дибромпиразин-2-амина (125 г, 494 ммоль), TEA (207,0 мл, 1483 ммоль) и иодида меди (I) (0,941 г, 4,94 ммоль) в ТГФ (1255 мл) добавляли PdCl2(PPh3)2 (3,47 г, 4,94 ммоль). Реакционную смесь охлаждали при около -5-0°C и добавляли по каплям раствор (триметилсилил)ацетилена (65,0 мл, 470 ммоль) в ТГФ (157 мл) примерно в течение 15 минут. Реакционную смесь перемешивали при около -5-0°C примерно в течение 1,5 часов и затем давали нагреться до комнатной температуры в течение ночи. Реакционную смесь затем фильтровали через слой Целита® и промывали при помощи ТГФ до тех пор, пока продукт больше не выделялся. Фильтрат концентрировали при пониженном давлении с получением коричневого-оранжевого твердого вещества. Твердое вещество растирали в порошок, при обработке ультразвуком, с теплым петролейным эфиром (т.кип. 30-60°C, 400 мл), охлаждали до комнатной температуры, собирали, промывали петролейным эфиром (т.кип. 30-60°C; 2×60 мл) и сушили с получением 5-бром-3-((триметилсилил)этинил)пиразин-2-амина (124 г, 93%, чистота 93%) в виде коричневого твердого вещества: LC/MS (Таблица 1, Способ b) Rt = 2,51 мин; MS m/z: 270, 272 (M+H)+.
Стадия B: 2-бром-5-тозил-5H-пирроло[2,3-b]пиразин

К раствору 5-бром-3-((триметилсилил)этинил)пиразин-2-амина (3,00 г, 11,1 ммоль) в DMF (60 мл) при около 0°C добавляли NaH (60% дисперсия в минеральном масле, 0,577 г, 14,4 ммоль) тремя порциями. Примерно через 15 минут добавляли п-толуолсульфонилхлорид (2,75 г, 14,4 ммоль) и реакционной смеси давали медленно нагреться до температуры окружающей среды. Примерно через 16 часов реакционную смесь выливали на ледяную воду (120 мл) и осадок собирали вакуумным фильтрованием. Неочищенное твердое вещество растворяли в DCM (15 мл) и очищали хроматографией на силикагеле, элюируя при помощи DCM, с получением 2-бром-5-тозил-5H-пирроло[2,3-b]пиразина (2,16 г, 52%): LC/MS (Таблица 1, Способ c) Rt = 1,58 мин; MS m/z: 352, 354 (M+H)+.
Стадия C: трет-бутил 2-(5-тозил-5H-пирроло[2,3-b]пиразин-2-ил)гидразинкарбоксилат и трет-бутил 1-(5-тозил-5H-пирроло[2,3-b]пиразин-2-ил)гидразинкарбоксилат
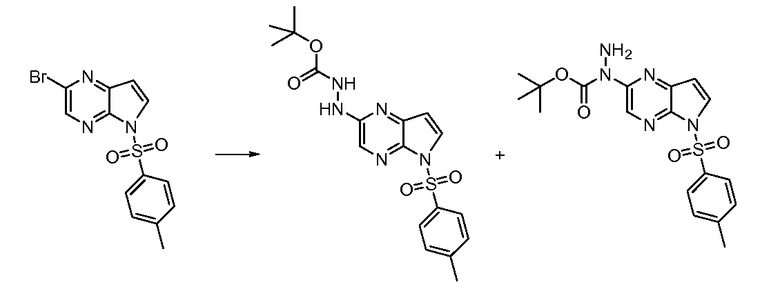
В колбу добавляли Pd2(dba)3 (3,90 г, 4,26 ммоль), ди-трет-бутил-(2',4',6'-триизопропилбифенил-2-ил)фосфан (3,62 г, 8,52 ммоль) и 1,4-диоксан (453 мл). Дегазирование смеси катализатор-лиганд осуществляли через вакуум/продувку азотом (3 раза) и нагревали при около 80°C примерно в течение 10 минут и охлаждали до температуры окружающей среды. Затем добавляли 2-бром-5-тозил-5H-пирроло[2,3-b]пиразин (30,0 г, 85 ммоль), трет-бутил гидразинкарбоксилат (16,9 г, 128 ммоль) и NaOt-Bu (12,28 г, 128 ммоль). После дополнительного цикла вакуум/продувка азотом реакционную смесь нагревали при около 80°C. Примерно через 50 минут реакционную смесь охлаждали до температуры окружающей среды и фильтровали через слой силикагеля (6 см в высоту × 6 см в диаметре), содержащего сверху слой Целита® (1 см в высоту × 6 см в диаметре), осуществляя при этом промывку при помощи EtOAc (3×150 мл). К фильтрату добавляли воду (300 мл) и органический слой отделяли. Водный слой экстрагировали дополнительным количеством EtOAc (3×200 мл). Объединенные органические экстракты промывали насыщенным водным раствором NH4Cl, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором (400 мл каждого), сушили над безводным MgSO4, фильтровали и концентрировали при пониженном давлении с получением темно-коричневого масла (45 г). Коричневое масло растворяли в DCM (250 мл), добавляли силикагель (200 г) и смесь концентрировали при пониженном давлении. Полученную смесь с диоксидом кремния очищали с использованием хроматографии на силикагеле с градиентным элюированием 25-65% EtOAc в гептане с получением смеси трет-бутил 2-(5-тозил-5H-пирроло[2,3-b]пиразин-2-ил)гидразинкарбоксилата [основной региоизомер] и трет-бутил 1-(5-тозил-5H-пирроло[2,3-b]пиразин-2-ил)гидразинкарбоксилата [второстепенный региоизомер] (18,8 г, 50%): LC/MS (Таблица 1, Способ c) Rt = 1,47 мин; MS m/z: 404 (M+H)+.
Стадия D: 2-гидразинил-5-тозил-5H-пирроло[2,3-b]пиразин

К смеси трет-бутил 2-(5-тозил-5H-пирроло[2,3-b]пиразин-2-ил)гидразинкарбоксилата и трет-бутил 1-(5-тозил-5H-пирроло[2,3-b]пиразин-2-ил)гидразинкарбоксилата (49,2 г, 122 ммоль) в 1,4-диоксане (290 мл) добавляли HCl (4 M в 1,4-диоксане, 226 мл, 902 ммоль). Реакционную смесь нагревали при около 60°C примерно в течение 2,5 часов и затем охлаждали до около 15-20°C. Твердое вещество собирали вакуумным фильтрованием, промывали при помощи EtOAc (3×50 мл) и затем растирали в порошок с Et2O (60 мл), собирали вакуумным фильтрованием и сушили до постоянной массы в вакууме с получением 35,6 г твердого вещества. Твердое вещество перемешивали со смесью насыщенного водного раствора NaHCO3 и EtOAc (1:1, 400 мл). Примерно через 1 час твердое вещество собирали вакуумным фильтрованием, промывали ледяной водой (3×30 мл) и EtOAc (3×30 мл) и сушили в вакуумной печи до постоянной массы с получением 2-гидразинил-5-тозил-5H-пирроло[2,3-b]пиразина в виде желтовато-коричневого твердого вещества (21,2 г, 57%): LC/MS (Таблица 1, Способ a) Rt = 1,88 мин; MS m/z: 304 (M+H)+.
Стадия E: натрия 4-(этоксикарбонил)-3-этил-2-(метоксикарбонил)циклопента-1,3-диенолят
В круглодонную колбу загружали ТГФ (1,5 л) с последующим добавлением по порциям NaH (60% дисперсия в минеральном масле, 70,0 г, 1,75 моль). Добавляли дополнительное количество ТГФ (500 мл) и полученную смесь охлаждали до около -10°C и добавляли по каплям этилпропионилацетат (250 мл, 1,80 моль) примерно в течение 1 часа для поддержания внутренней температуры ниже около 10°C. Полученную смесь перемешивали при температуре окружающей среды примерно в течение 0,5 часа с получением прозрачного желтого раствора и добавляли по каплям метил 4-хлорацетоацетат (100 мл, 0,88 моль) примерно в течение 5 минут. Полученную смесь нагревали при около 50°C примерно в течение 19 часов с получением красновато-оранжевой суспензии. Реакционную смесь охлаждали до температуры окружающей среды, концентрировали при пониженном давлении и полученную жидкость переносили в химический стакан и разбавляли водой (350 мл). Смесь перемешивали на ледяной бане примерно в течение 2 часов. Твердое вещество собирали вакуумным фильтрованием и фильтровальную лепешку промывали водой (150 мл) и сушили в вакууме примерно в течение 1 часа. Твердое вещество суспендировали в Et2O (1,5 л), фильтровали, промывали при помощи Et2O (1,5 л) и сушили в вакууме. Полученное твердое вещество подвергали азеотропной перегонке с толуолом (1 л) с получением твердого вещества, которое снова суспендировали в Et2O (1 л) и собирали вакуумным фильтрованием. Фильтровальную лепешку промывали при помощи Et2O (500 мл) и сушили в вакууме с получением 4-(этоксикарбонил)-3-этил-2-(метоксикарбонил)циклопента-1,3-диенолята натрия (204,2 г, 89%) в виде бежевого твердого вещества: 1H ЯМР (DMSO-d 6) δ 3,94 (кв., J=7,1 Гц, 2H), 3,46 (с, 3H), 3,04 (кв., J=7,2 Гц, 2H), 2,66 (с, 2H), 1,13 (т, J=7,1 Гц, 3H), 0,99 (т, J=7,3 Гц, 3H).
Стадия F: этил 2-этил-4-оксоциклопент-2-енкарбоксилат

В 5-л круглодонную колбу загружали 4-(этоксикарбонил)-3-этил-2-(метоксикарбонил)циклопента-1,3-диенолят натрия (316 г, 1205 ммоль), KCl (126 г, 1687 ммоль, JT-Baker), AcOH (241 мл, 4218 ммоль, JT-Baker), толуол (1850 мл) и воду (130 мл). Реакционную смесь нагревали при температуре кипения с обратным холодильником примерно в течение 6 часов, затем охлаждали до температуры окружающей среды и добавляли по каплям к NaHCO3 (8% водный раствор, 3,5 л). Полученную двухфазную смесь экстрагировали при помощи MTBE (2×1,5 л). Объединенные органические слои промывали насыщенным солевым раствором (1 л), сушили над безводным MgSO4 и концентрировали при пониженном давлении с получением 191 г неочищенного вещества, которое очищали перегонкой в вакууме (97-99°C, 0,600 мм Hg) с получением этил 2-этил-4-оксоциклопент-2-енкарбоксилата (160 г, 69%):1H ЯМР (CDCl3) δ 6,04 (м, 1H), 4,26-4,15 (м, 2H), 3,76-3,69 (м, 1H), 2,75-2,57 (м, 2H), 2,56-2,44 (м, 2H), 1,32-1,26 (м, 3H), 1,23-1,18 (м, 3H).
Стадия G: этил 2-этил-4-оксоциклопентанкарбоксилат
В круглодонную колбу загружали 10% масс. Pd/C (10 г, 9,4 ммоль). Колбу охлаждали до около 0°C и добавляли EtOAc (400 мл) в атмосфере азота. Охлаждающую баню удаляли и добавляли этил 2-этил-4-оксоциклопент-2-енкарбоксилат (47,8 г, 263 ммоль). Газообразный водород барботировали через смесь примерно в течение 5 минут и смесь затем перемешивали в атмосфере водорода примерно в течение 48 часов. Источник водорода удаляли и смесь барботировали азотом примерно в течение 5 минут и фильтровали через слой Целита®. Фильтровальную лепешку промывали при помощи EtOAc (400 мл). Фильтрат концентрировали при пониженном давлении с получением этил 2-этил-4-оксоциклопентанкарбоксилата (около 9:1 смесь цис:транс) (48,0 г, 99%) в виде желтой жидкости: 1H ЯМР (CDCl3) δ 4,23-4,10 (м, 2H), 3,22 (м, 1H), 2,59-2,50 (м, 1H), 2,44-2,28 (м, 3H), 2,26-2,16 (м, 1H), 1,58-1,46 (м, 1H), 1,41-1,30 (м, 1H), 1,30-1,23 (м, 3H), 1,02-0,91 (м, 3H).
Стадия H: этил 2-этил-4-метиленциклопентанкарбоксилат

Раствор KOt-Bu (3,65 г, 32,6 ммоль) и метилтрифенилфосфонийбромида (11,6 г, 32,6 ммоль) в ТГФ (69,5 мл) охлаждали до около -10°C. Добавляли по каплям раствор этил 2-этил-4-оксоциклопентанкарбоксилата (4,00 г, 21,7 ммоль) в ТГФ (17,4 мл), поддерживая температуру при около 0°C. Реакционной смеси давали нагреться до температуры окружающей среды и перемешивали примерно в течение 16 часов. Нерастворимые вещества удаляли фильтрованием. Фильтрат концентрировали при пониженном давлении. Полученное вещество очищали хроматографией на силикагеле (120 г) с градиентным элюированием 0-20% EtOAc в гептане с получением этил 2-этил-4-метиленциклопентанкарбоксилата (2,55 г, 64%) в виде бесцветной жидкости: 1H ЯМР (d-DMSO) δ 4,88-4,78 (м, 2H), 4,16-3,96 (м, 2H), 2,66-2,31 (м, 4H), 2,24-1,82 (м, 2H), 1,50 (м, 1H), 1,35-1,22 (м, 1H), 1,18 (т, 3H), 0,85 (м, 3H).
Стадия I: этил 2-этил-4-(меркаптометил)циклопентанкарбоксилат

Этил 2-этил-4-метиленциклопентанкарбоксилат (0,720 г, 3,95 ммоль), трифенилсилантиол (1,329 г, 4,54 ммоль) и 2,2'-азобис(2-метилпропионитрил) (0,195 г, 1,185 ммоль) в толуоле (3,95 мл) нагревали при температуре кипения с обратным холодильником примерно в течение 6 часов. Реакционную смесь охлаждали до температуры окружающей среды и затем концентрировали при пониженном давлении. Вещество очищали хроматографией на силикагеле (40 г) с градиентным элюированием 0-10% EtOAc в гептане с получением бесцветного масла. Полученное масло растворяли в DCM (4 мл) и добавляли TFA (1,52 мл, 19,7 ммоль). После перемешивания при температуре окружающей среды примерно в течение 1 часа растворитель удаляли при пониженном давлении. Вещество очищали хроматографией на силикагеле (40 г) с градиентным элюированием 0-15% EtOAc в гептане с получением этил 2-этил-4-(меркаптометил)циклопентанкарбоксилата (0,620 г, 72%) в виде бесцветного масла: 1H ЯМР (DMSO-d 6) δ 4,13-4,01 (м, 2H), 2,50-2,30 (м, 3H), 2,24 (м, 1H), 2,15-1,87 (м, 3H), 1,66-1,54 (м, 1H), 1,50-1,37 (м, 2H), 1,31-1,23 (м, 2H), 1,17 (т, 3H), 0,83 (м, 3H).
Стадия J: (3-(этоксикарбонил)-4-этилциклопентил)метансульфоновая кислота
К перемешиваемому раствору этил 2-этил-4-(меркаптометил)циклопентанкарбоксилата (2,50 г, 11,6 ммоль) в DCM (50,7 мл) добавляли по каплям этанпероксокислоту (7,29 мл, 34,7 ммоль) при около 0°C. Реакционную смесь нагревали до температуры окружающей среды и перемешивали примерно в течение 16 часов. Раствор концентрировали при пониженном давлении с получением неочищенной (3-(этоксикарбонил)-4-этилциклопентил)метансульфоновой кислоты (3,18 г, 104%) в виде темно-коричневого масла: LC/MS (Таблица 1, Способ b) Rt = 1,39 мин; MS m/z: 265 (M+H)+.
Стадия K: этил 4-((N,N-диэтилсульфамоил)метил)-2-этилциклопентанкарбоксилат
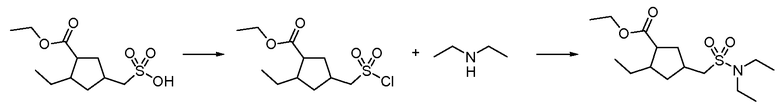
Раствор (3-(этоксикарбонил)-4-этилциклопентил)метансульфоновой кислоты (3,18 г, 12,03 ммоль) в DCM (10 мл) и DMF (10 мл) охлаждали до около 0°C. Добавляли по каплям оксалилхлорид (24,1 мл, 48,1 ммоль) добавляли по каплям, поддерживая температуру при около 0°C. После завершения добавления, реакционную смесь нагревали до температуры окружающей среды и перемешивали примерно в течение 1 часа. Растворитель удаляли при пониженном давлении. Остаток растворяли в DMF (10 мл) и затем добавляли по каплям к раствору TEA (2,51 мл, 18,03 ммоль) и диэтиламина (0,937 мл, 9,02 ммоль) в DMF (10 мл) при около 0°C. Реакционную смесь перемешивали при температуре окружающей среды примерно в течение 16 часов. Растворитель удаляли при пониженном давлении. Вещество очищали хроматографией на силикагеле (120 г) с градиентным элюированием 10-60% EtOAc в гептане с получением этил 4-((N,N-диэтилсульфамоил)метил)-2-этилциклопентанкарбоксилата (0,570 г, 30%) в виде желтого масла: LC/MS (Таблица 1, Способ b) Rt = 2,60 мин; MS m/z: 320 (M+H)+.
Стадия L: 4-((N,N-диэтилсульфамоил)метил)-2-этилциклопентанкарбоновая кислота
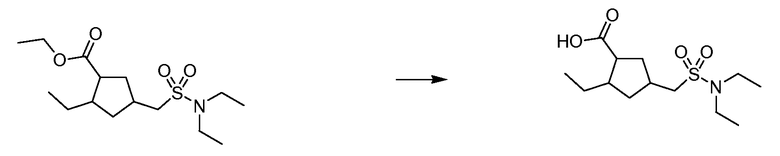
Смесь этил 4-((N,N-диэтилсульфамоил)метил)-2-этилциклопентанкарбоксилата (0,570 г, 1,784 ммоль) в NaOH (1 н водный раствор, 10 мл, 10 ммоль) перемешивали при температуре окружающей среды примерно в течение 72 часов. Смесь разделяли с использованием DCM (10 мл). Водную фазу подкисляли до около pH = 4 путем добавления 6 н водного раствора HCl. Раствор разделяли с использованием DCM (10 мл). Водную фазу промывали при помощи DCM (2×10 мл). Органические слои объединяли, сушили над безводным MgSO4 и концентрировали при пониженном давлении с получением 4-((N,N-диэтилсульфамоил)метил)-2-этилциклопентанкарбоновой кислоты (0,375 г, 72%) в виде желтого масла: LC/MS (Таблица 1, Способ b) Rt = 1,95 мин; MS m/z: 292 (M+H)+.
Стадия M: N,N-диэтил-1-(3-этил-4-(2-(5-тозил-5H-пирроло[2,3-b]пиразин-2-ил)гидразинкарбонил)циклопентил)метансульфонамид

К суспензии 2-гидразинил-5-тозил-5H-пирроло[2,3-b]пиразина (0,390 г, 1,287 ммоль, Пример #1 Стадия D), 4-((N,N-диэтилсульфамоил)метил)-2-этилциклопентанкарбоновой кислоты (0,375 г, 1,287 ммоль) и HATU (0,538 г, 1,416 ммоль) в DCM (6,4 мл) добавляли TEA (0,538 мл, 3,86 ммоль). Реакционную смесь перемешивали при температуре окружающей среды примерно в течение 1 часа. Реакционную смесь распределяли между водой (50 мл) и DCM (50 мл). Водный слой экстрагировали при помощи DCM (2×50 мл). Органические слои объединяли и концентрировали при пониженном давлении. Вещество очищали хроматографией на силикагеле (120 г) с градиентным элюированием 20-100% EtOAc в DCM с получением N,N-диэтил-1-(3-этил-4-(2-(5-тозил-5H-пирроло[2,3-b]пиразин-2-ил)гидразинкарбонил)циклопентил)метансульфонамида (0,730 г, 98%) в виде коричневого твердого вещества: LC/MS (Таблица 1, Способ b) Rt = 2,39 мин; MS m/z: 577 (M+H)+.
Стадия N: N,N-диэтил-1-(3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)метансульфонамид

К смеси N,N-диэтил-1-(3-этил-4-(2-(5-тозил-5H-пирроло[2,3-b]пиразин-2-ил)гидразинкарбонил)циклопентил)метансульфонамида (0,730 г, 1,27 ммоль) и TEA (0,529 мл, 3,80 ммоль) в 1,4-диоксане (12,7 мл) добавляли SOCl2 (0,185 мл, 2,53 ммоль). Реакционную смесь нагревали при около 80°C примерно в течение 2 часов. Реакционную смесь охлаждали до температуры окружающей среды и распределяли между насыщенным водным раствором NaHCO3 (30 мл) и DCM (30 мл). Водный слой промывали при помощи DCM (2×30 мл). Органические слои объединяли, концентрировали при пониженном давлении и очищали хроматографией на силикагеле (80 г) с градиентным элюированием 0-60% MeOH в DCM с получением коричневого твердого вещества. Полученное твердое вещество суспендировали в Na2CO3 (2 M водный раствор, 2 мл), EtOH (2 мл) и 1,4-диоксане (2 мл). Реакционную смесь нагревали при около 60°C примерно в течение 16 часов. Реакционную смесь охлаждали до температуры окружающей среды и очищали при помощи ОФ-ВЭЖХ (Таблица 1, Способ d) с получением N,N-диэтил-1-(3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)метансульфонамида (0,300 г, 58%) в виде желтовато-коричневого твердого вещества: LC/MS (Таблица 1, Способ b) Rt = 1,89 мин; MS m/z: 405 (M+H)+. Твердое вещество подвергали дополнительной очистке с использованием Общей процедуры AA с получением N,N-диэтил-1-((1S,3R,4R)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)метансульфонамида (Таблица 2, Способ 27, Rt = 11,8 мин, or = отрицательное) (0,021 г, 7%) [Пример #1.1]; N,N-диэтил-1-((1R,3S,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)метан-сульфонамида (Таблица 2, Способ 27, Rt = 11,1 мин, or = положительное) (0,018 г, 6%) [Пример #1.2]; N,N-диэтил-1-((1S,3S,4S)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)цикло-пентил)метансульфонамида (Таблица 2, Способ 27, Rt = 10,7 мин, or = положительное) (0,018 г, 6%) [Пример #1.3]; N,N-диэтил-1-((1R,3R,4R)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)метансульфонамида (Таблица 2, Способ 28, Rt = 20,1 мин, or = отрицательное) (0,031 г, 11%) [Пример #1.4]; N,N-диэтил-1-((1S,3S,4R)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)метансульфонамида (Таблица 2, Способ 27, Rt = 12,8 мин, or = положительное) (0,002 г, 1%) [Пример #1.5]; и N,N-диэтил-1-((1R,3S,4R)-3-этил-4-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)метансульфонамида (Таблица 2, Способ 27, Rt = 12,8 мин, or = положительное) (0,001 г, 1%) [Пример #1.6].
Пример #2*: N-((1R,3R)-3-(6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)анилин
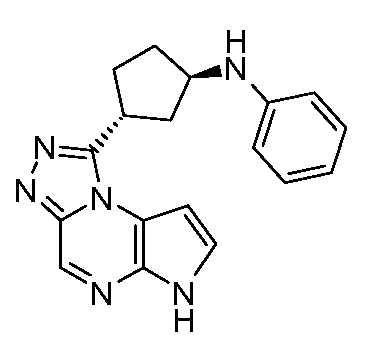
Стадия A: 5-бром-3-((триметилсилил)этинил)пиразин-2-амин

К раствору 3,5-дибромпиразин-2-амина (125 г, 494 ммоль), TEA (207,0 мл, 1483 ммоль) и иодида меди (I) (0,941 г, 4,94 ммоль) в ТГФ (1255 мл) добавляли PdCl2(PPh3)2 (3,47 г, 4,94 ммоль). Реакционную смесь охлаждали при около -5-0°C и добавляли по каплям раствор (триметилсилил)ацетилена (65,0 мл, 470 ммоль) в ТГФ (157 мл) примерно в течение 15 минут. Реакционную смесь перемешивали при около -5-0°C примерно в течение 1,5 часов и затем давали нагреться до комнатной температуры в течение ночи. Реакционную смесь затем фильтровали через слой Целита® и промывали при помощи ТГФ до тех пор, пока продукт больше не выделялся. Фильтрат концентрировали при пониженном давлении с получением коричнево-оранжевого твердого вещества. Твердое вещество растирали в порошок, при обработке ультразвуком, с теплым петролейным эфиром (т.кип. 30-60°C, 400 мл), охлаждали до комнатной температуры, собирали, промывали петролейным эфиром (т.кип. 30-60°C; 2×60 мл) и сушили с получением 5-бром-3-((триметилсилил)этинил)пиразин-2-амина (124 г, 93%, 93% чистота) в виде коричневого твердого вещества: LC/MS (Таблица 1, Способ b) Rt = 2,51 мин; MS m/z: 270, 272 (M+H)+.
Стадия B: 2-бром-5-тозил-5H-пирроло[2,3-b]пиразин

К раствору 5-бром-3-((триметилсилил)этинил)пиразин-2-амина (3,00 г, 11,1 ммоль) в DMF (60 мл) при около 0°C добавляли NaH (60% дисперсия в минеральном масле, 0,577 г, 14,4 ммоль) тремя порциями. Примерно через 15 минут добавляли п-толуолсульфонилхлорид (2,75 г, 14,4 ммоль) и реакционной смеси давали медленно нагреться до температуры окружающей среды. Примерно через 16 часов реакционную смесь выливали на ледяную воду (120 мл) и осадок собирали вакуумным фильтрованием. Неочищенное твердое вещество растворяли в DCM (15 мл) и очищали хроматографией на силикагеле, элюируя при помощи DCM, с получением 2-бром-5-тозил-5H-пирроло[2,3-b]пиразина (2,16 г, 52%): LC/MS (Таблица 1, Способ c) Rt = 1,58 мин; MS m/z: 352, 354 (M+H)+.
Стадия C: трет-бутил 2-(5-тозил-5H-пирроло[2,3-b]пиразин-2-ил)гидразинкарбоксилат и трет-бутил 1-(5-тозил-5H-пирроло[2,3-b]пиразин-2-ил)гидразинкарбоксилат

В колбу добавляли Pd2(dba)3 (3,90 г, 4,26 ммоль), ди-трет-бутил-(2',4',6'-триизопропилбифенил-2-ил)фосфан (3,62 г, 8,52 ммоль) и 1,4-диоксан (453 мл). Дегазирование смеси катализатор-лиганд осуществляли через вакуум/продувку азотом (3 раза) и нагревали при около 80°C примерно в течение 10 минут. Затем добавляли 2-бром-5-тозил-5H-пирроло[2,3-b]пиразин (30,0 г, 85 ммоль), трет-бутил гидразинкарбоксилат (16,9 г, 128 ммоль) и NaOt-Bu (12,28 г, 128 ммоль). После дополнительного цикла вакуум/продувка азотом реакционную смесь нагревали при около 80°C. Примерно через 50 минут реакционную смесь охлаждали до температуры окружающей среды и фильтровали через слой силикагеля (6 см в высоту × 6 см в диаметре), содержащего сверху слой Целита® (1 см в высоту × 6 см в диаметре), осуществляя при этом промывку при помощи EtOAc (3×150 мл). К фильтрату добавляли воду (300 мл) и органический слой отделяли. Водный слой экстрагировали дополнительным количеством EtOAc (3×200 мл). Объединенные органические экстракты промывали насыщенным водным раствором NH4Cl, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором (400 мл каждого), сушили над безводным MgSO4, фильтровали и концентрировали при пониженном давлении с получением темно-коричневого масла (45 г). Коричневое масло растворяли в DCM (250 мл), добавляли силикагель (200 г) и смесь концентрировали при пониженном давлении. Полученную смесь с диоксидом кремния очищали с использованием хроматографии на силикагеле с градиентным элюированием 25-65% EtOAc в гептане с получением смеси трет-бутил 2-(5-тозил-5H-пирроло[2,3-b]пиразин-2-ил)гидразинкарбоксилата [основной региоизомер] и трет-бутил 1-(5-тозил-5H-пирроло[2,3-b]пиразин-2-ил)гидразинкарбоксилата [второстепенный региоизомер] (18,8 г, 50%): LC/MS (Таблица 1, Способ c) Rt = 1,47 мин; MS m/z: 404 (M+H)+.
Стадия D: 2-гидразинил-5-тозил-5H-пирроло[2,3-b]пиразин

К смеси трет-бутил 2-(5-тозил-5H-пирроло[2,3-b]пиразин-2-ил)гидразинкарбоксилата и трет-бутил 1-(5-тозил-5H-пирроло[2,3-b]пиразин-2-ил)гидразинкарбоксилата (49,2 г, 122 ммоль) в 1,4-диоксане (290 мл) добавляли HCl (4 M в 1,4-диоксане, 226 мл, 902 ммоль). Реакционную смесь нагревали при около 60°C примерно в течение 2,5 часов и затем охлаждали до около 15-20°C. Твердое вещество собирали вакуумным фильтрованием, промывали при помощи EtOAc (3×50 мл) и затем растирали в порошок с Et2O (60 мл), собирали вакуумным фильтрованием и сушили до постоянной массы в вакууме с получением 35,6 г твердого вещества. Твердое вещество перемешивали со смесью насыщенного водного раствора NaHCO3 и EtOAc (1:1, 400 мл). Примерно через 1 час твердое вещество собирали вакуумным фильтрованием, промывали ледяной водой (3×30 мл) и EtOAc (3×30 мл) и сушили в вакуумной печи до постоянной массы с получением 2-гидразинил-5-тозил-5H-пирроло[2,3-b]пиразина в виде желтовато-коричневого твердого вещества (21,2 г, 57%): LC/MS (Таблица 1, Способ a) Rt = 1,88 мин; MS m/z: 304 (M+H)+.
Стадия E: трет-бутил (1R,3R)-3-(2-(5-тозил-5H-пирроло[2,3-b]пиразин-2-ил)гидразинкарбонил)циклопентилкарбамат

К (1R,3R)-3-(трет-бутоксикарбониламино)циклопентанкарбоновой кислоте (2,25 г, 9,81 ммоль, Acros) в DCM (98 мл) добавляли 2-гидразинил-5-тозил-5H-пирроло[2,3-b]пиразин (2,98 г, 9,81 ммоль), HATU (3,73 г, 9,81 ммоль) и TEA (5,5 мл, 39 ммоль). Реакционную смесь перемешивали при температуре окружающей среды примерно в течение 4 часов, затем разбавляли при помощи DCM (300 мл). Реакционную смесь промывали водой (2×80 мл), насыщенным водным раствором NaHCO3 (80 мл), сушили над безводным MgSO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали с использованием хроматографии на силикагеле (220 г) с градиентным элюированием 50-100% EtOAc в DCM с получением трет-бутил (1R,3R)-3-(2-(5-тозил-5H-пирроло[2,3-b]пиразин-2-ил)гидразинкарбонил)циклопентилкарбамата (5,03 г, 100%) в виде коричневого твердого вещества: LC/MS (Таблица 1, Способ b) Rt = 2,18 мин; MS m/z: 513 (M-H)-
Стадия F: (1R,3R)-3-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентанамин

К трет-бутил (1R,3R)-3-(2-(5-тозил-5H-пирроло[2,3-b]пиразин-2-ил)гидразинкарбонил)цикло-пентилкарбамату (5,03 г, 9,78 ммоль) в 1,4-диоксане (103 мл) добавляли DIEA (7,2 мл, 41 ммоль) и SOCl2 (2,3 мл, 31 ммоль). Реакционную смесь нагревали при около 80°C примерно в течение 1 часа. Растворитель удаляли при пониженном давлении и остаток очищали с использованием хроматографии на силикагеле (330 г) с градиентным элюированием 0-20% MeOH в DCM с получением (1R,3R)-3-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентанамина (2,65 г, 68%): LC/MS (Таблица 1, Способ b) Rt = 1,55 мин; MS m/z: 397 (M+H)+.
Стадия G: N-((1R,3R)-3-(6-тозил-6H-пирроло[2,3-e][1,2,4]триазоло[4,3-a]пиразин-1-ил)циклопентил)анилин
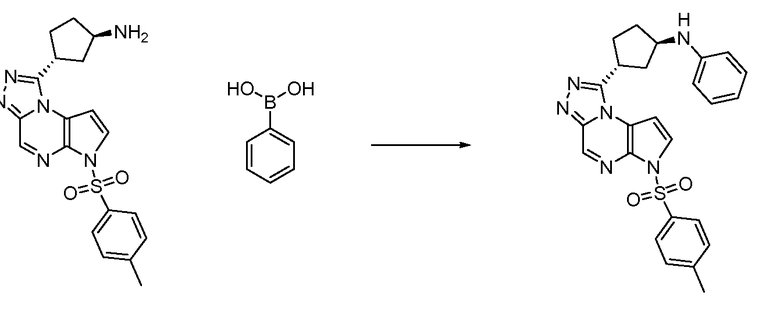
| название | год | авторы | номер документа |
|---|---|---|---|
| Имидазопирролопиразиновые производные, полезные для лечения заболеваний, вызванных аномальной активностью протеинкиназ Jak1, Jak3 или Syk | 2010 |
|
RU2711869C2 |
| ТРИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ИХ ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ ИММУНОЛОГИЧЕСКИХ И ОНКОЛОГИЧЕСКИХ СОСТОЯНИЙ | 2009 |
|
RU2545023C9 |
| СОЕДИНЕНИЯ, ЯВЛЯЮЩИЕСЯ АНТАГОНИСТАМИ А3-АДЕНОЗИНОВОГО РЕЦЕПТОРА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ МЕДИЦИНСКОЕ ПРИМЕНЕНИЕ | 2017 |
|
RU2737157C2 |
| ПЕРВИЧНЫЕ КАРБОКСАМИДЫ В КАЧЕСТВЕ ИНГИБИТОРОВ BТK | 2014 |
|
RU2708395C2 |
| СПОСОБЫ ПОЛУЧЕНИЯ И ОЧИСТКИ ГЕТЕРОАРИЛЬНЫХ СОЕДИНЕНИЙ | 2010 |
|
RU2557242C2 |
| ИНГИБИТОРЫ RMT5 | 2019 |
|
RU2814198C2 |
| СОЕДИНЕНИЯ, ГЕТЕРОБИЦИКЛО-ЗАМЕЩЕННЫЕ-[1,2,4]ТРИАЗОЛО[1,5c]ХИНАЗОЛИН-5-АМИНА, ОБЛАДАЮЩИЕ СВОЙСТВАМИ А2А АНТАГОНИСТОВ | 2013 |
|
RU2671628C2 |
| ГЕТЕРОАРИЛЬНЫЕ СОЕДИНЕНИЯ, СОДЕРЖАЩИЕ ИХ КОМПОЗИЦИИ И СПОСОБЫ ЛЕЧЕНИЯ С ПРИМЕНЕНИЕМ ЭТИХ СОЕДИНЕНИЙ | 2007 |
|
RU2478635C2 |
| ИНГИБИТОРЫ mTOR КИНАЗЫ ДЛЯ ОНКОЛОГИЧЕСКИХ ПОКАЗАНИЙ И ЗАБОЛЕВАНИЙ, СВЯЗАННЫХ С mTOR/PI3K/AKT ПУТЕМ МЕТАБОЛИЗМА | 2009 |
|
RU2692796C2 |
| ИНГИБИТОРЫ mTOR КИНАЗЫ ДЛЯ ОНКОЛОГИЧЕСКИХ ПОКАЗАНИЙ И ЗАБОЛЕВАНИЙ, СВЯЗАННЫХ С mTOR/PI3K/AKT ПУТЕМ МЕТАБОЛИЗМА | 2009 |
|
RU2546658C2 |
Изобретение относится к области органической химии, а именно к новому производному имидазопирролопиразина формулы (Ic) и к его фармацевтически приемлемой соли, стереоизомеру или изомеру, где R1, R2 и R5 представляют собой Н; R3 представляет собой (С3-С6)циклоалкил, замещенный одной группой, независимо выбранной из CH2NH2 или NH2; или R3 представляет собой -A-D-E-G, где: А представляет собой связь или -N(Ra)С(О)-Re-; D представляет собой (С1-С3)алкилен, связанный мостиковой связью (С8)циклоалкилен, (С4-С6)циклоалкилен, замещенный одним заместителем, выбранным из (С1-С3)алкила; (С4-С5)моногетероциклилен, необязательно замещенный одним заместителем, выбранным из (С1-С3)алкила, где моногетероциклилен содержит 1-2 гетероатома, независимо выбранных из атомов азота или кислорода; Е представляет собой связь, -Re, -Re-C(О)-Re-, -Re-O-Re-, -Re-N(Ra)-Re-, -Re-N(Ra)C(O)-Re-, -ReC(O)N(Ra)Re-, -Re-N(Ra)S(O)2-Re- или -Re-N(Ra)S(O)2N(Ra)-Re-; где во всех случаях Е связан либо с атомом углерода, либо с атомом азота в D; G представляет собой водород, -N(Ra)(Rb), -(C1-C6)алкил, необязательно замещенный 1-3 заместителями, независимо выбранными из галогена, гидрокси или циано; (С3-С6)моноциклоалкил, необязательно замещенный 1-2 заместителями, независимо выбранными из галогена, циано, моноциклический (С3-С5)гетероарил, необязательно замещенный одним заместителем, независимо выбранным из циано, где гетероарил содержит 2 гетероатома, независимо выбранных из атомов азота; (С3-С5)моногетероциклил, необязательно замещенный 1 или 2 заместителями, независимо выбранными из метила, -CF3, галогена или CH2CN, где моногетероциклил содержит 1-2 гетероатома, независимо выбранных из атомов азота или кислорода; (С6)арил, необязательно замещенный 1-2 заместителями, независимо выбранными из галогена или циано; где во фрагменте, содержащем -N(Ra)(Rb), азот, Ra и Rb могут образовывать кольцо, таким образом, что -N(Ra)(Rb) представляет собой С4моногетероциклил, где моногетероциклил содержит 1-2 гетероатома, независимо выбранных из азота или кислорода; R4 представляет собой водород или -(С1-С6)алкил; Ra и Rb, каждый независимо, представляют собой водород, (C1-С6)алкил, оксетан; и Re для каждого случая независимо представляет собой связь или -(С1-С4)алкилен. Также изобретение относится к конкретным производным имидазопирролопиразина, фармацевтической композиции на основе заявленного соединения и к применению заявленного соединения. Технический результат: получены новые производные имидазопирролопиразина, полезные при лечении состояний, опосредованных активностью Jak1, Jak3 и Syk протеинкиназ. 16 н. и 7 з.п. ф-лы, 54 табл., 690 пр.

1. Соединение формулы (Ic)
,
его фармацевтически приемлемые соли, стереоизомеры или изомеры, где:
R1 и R2, каждый, представляют собой водород;
R5 представляет собой Н;
R3 представляет собой (С3-С6)циклоалкил, замещенный одной группой, независимо выбранной из CH2NH2 или NH2; или
R3 представляет собой -A-D-E-G, где:
А представляет собой связь или -N(Ra)С(О)-Re-;
D представляет собой (С1-С3)алкилен, связанный мостиковой связью (С8)циклоалкилен, (С4-С6)циклоалкилен, замещенный одним заместителем, выбранным из (С1-С3)алкила; (С4-С5)моногетероциклилен, необязательно замещенный одним заместителем, выбранным из (С1-С3)алкила, где моногетероциклилен содержит 1-2 гетероатома, независимо выбранных из атомов азота или кислорода;
Е представляет собой связь, -Re, -Re-C(О)-Re-, -Re-O-Re-, -Re-N(Ra)-Re-, -Re-N(Ra)C(O)-Re-, -ReC(O)N(Ra)Re-, -Re-N(Ra)S(O)2-Re- или -Re-N(Ra)S(O)2N(Ra)-Re-; где во всех случаях Е связан либо с атомом углерода, либо с атомом азота в D;
G представляет собой водород, -N(Ra)(Rb), -(C1-C6)алкил, необязательно замещенный 1-3 заместителями, независимо выбранными из галогена, гидрокси или циано; (С3-С6)моноциклоалкил, необязательно замещенный 1-2 заместителями, независимо выбранными из галогена, циано, моноциклический (С3-С5)гетероарил, необязательно замещенный одним заместителем, независимо выбранным из циано, где гетероарил содержит 2 гетероатома, независимо выбранных из атомов азота; (С3-С5)моногетероциклил, необязательно замещенный 1 или 2 заместителями, независимо выбранными из метила, -CF3, галогена или CH2CN, где моногетероциклил содержит 1-2 гетероатома, независимо выбранных из атомов азота или кислорода; (С6)арил, необязательно замещенный 1-2 заместителями, независимо выбранными из галогена или циано;
где во фрагменте, содержащем -N(Ra)(Rb), азот, Ra и Rb могут образовывать кольцо, таким образом, что -N(Ra)(Rb) представляет собой С4моногетероциклил, где моногетероциклил содержит 1-2 гетероатома, независимо выбранных из азота или кислорода;
R4 представляет собой водород или -(С1-С6)алкил,
Ra и Rb, каждый независимо, представляют собой водород, (C1-С6)алкил, оксетан; и
Re для каждого случая независимо представляет собой связь или -(С1-С4)алкилен.
2. Соединение по п.1, где R3 представляет собой -A-D-E-G и А представляет собой связь.
3. Соединение по п.1, где R3 представляет собой -A-D-E-G и D представляет собой (С4-С6)циклоалкилен, замещенный одним заместителем, независимо выбранным из (С1-С3)алкила, связанный мостиковой связью (С8)циклоалкенилен, или (С4-С5)моногетероциклилен, необязательно замещенный одним заместителем, независимо выбранным из (С1-С3)алкила, где моногетероциклилен содержит 1-2 гетероатома, независимо выбранных из атомов азота или кислорода.
4. Соединение по п.3, где D представляет собой (С4-С6)циклоалкилен, замещенный одним заместителем, независимо выбранным из (С1-С3)алкила; бицикло[2,2,2]октан-1-ил, пиперидин, необязательно замещенный одним заместителем, независимо выбранным из (С1-С3)алкила, пиперазин, пирролидин, тетрагидрофуран или тетрегидропиран, где пиперазин, пирролидин, тетрагидрофуран или тетрагидропиран являются необязательно замещенными одним заместителем, независимо выбранным из (С1-С3)алкила.
5. Соединение по п.1, где G представляет собой водород, -N(Ra)(Rb), -(С1-С4)алкил, необязательно замещенный 1-3 заместителями, независимо выбранными из галогена, гидрокси или циано; (С3-С6)моноциклоалкил, необязательно замещенный 1-2 заместителями, независимо выбранными из галогена, циано; азетидинил, необязательно замещенный 1 или 2 заместителями, независимо выбранными из метила, -CF3, галогена или CH2CN, 4,5-дигидроизоксазолил, необязательно замещенный 1 или 2 заместителями, независимо выбранным из метила, -CF3, галогена или CH2CN, морфилинил, необязательно замещенный 1 или 2 заместителями, независимо выбранными из метила, -CF3, галогена или CH2CN, оксетанил, необязательно замещенный 1 или 2 заместителями, независимо выбранными из метила, -CF3, галогена или CH2CN, фенил, необязательно замещенный 1-2 заместителями, независимо выбранными из галогена, циано, пиперазинил, необязательно замещенный 1 или 2 заместителями, независимо выбранными из метила, -CF3, галогена или CH2CN, пиперидинил, необязательно замещенный 1 или 2 заместителями, независимо выбранными из метила, -CF3, галогена или CH2CN, пиразинил, необязательно замещенный одним заместителем, независимо выбранным из циано, пиразолил, необязательно замещенный одним заместителем, независимо выбранным из циано, пиридазинил, необязательно замещенный одним заместителем, независимо выбранным из циано, пиримидинил, пирролидинил, необязательно замещенный 1 или 2 заместителями, независимо выбранными из метила, -CF3, галогена или CH2CN, тетрагидрофуранил, необязательно замещенный 1 или 2 заместителями, независимо выбранными из метила, -CF3, галогена или CH2CN, тетрагидропиранил, необязательно замещенный 1 или 2 заместителями, независимо выбранными из метила, -CF3, галогена или CH2CN.
6. Соединение, выбранное из:
3-((3S,4S)-3-(3Н-имидазо[1,2-а]пирроло[2,3-е]пиразин-8-ил)-4-метилпиперидин-1-ил)-3-оксопропаннитрила;
5-(3-(3Н-имидазо[1,2-а]пирроло[2,3-е]пиразин-8-ил)пиперидин-1-ил)пиразин-2-карбонитрила;
(S)-1-(3-(3Н-имидазо[1,2-а]пирроло[2,3-е]пиразин-8-ил)пиперидин-1-карбонил)циклопропанкарбонитрила;
N-((1S,3R,4R)-3-этил-4-(3Н-имидазо[1,2-а]пирроло[2,3-е]пиразин-8-ил)циклопентил)циклопропансульфонамида;
N-((1R,3S,4S)-3-этил-4-(3Н-имидазо[1,2-а]пирроло[2,3-е]пиразин-8-ил)циклопентил)циклопропансульфонамида;
(S)-6-(3-(3Н-имидазо[1,2-а]пирроло[2,3-е]пиразин-8-ил)пиперидин-1-ил)никотинонитрила;
(R)-6-(3-(3Н-имидазо[1,2-а]пирроло[2,3-е]пиразин-8-ил)пиперидин-1-ил)никотинонитрила;
(S)-2-(3-(3Н-имидазо[1,2-а]пирроло[2,3-е]пиразин-8-ил)пиперидин-1-ил)тиазол-5-карбонитрила;
(R)-2-(3-(3Н-имидазо[1,2-а]пирроло[2,3-е]пиразин-8-ил)пиперидин-1-ил)тиазол-5-карбонитрила;
(R)-(3-(3Н-имидазо[1,2-а]пирроло[2,3-е]пиразин-8-ил)пиперидин-1-ил)(3,3-дифторазетидин-1-ил)метанона;
(S)-(3-(3Н-имидазо[1,2-а]пирроло[2,3-е]пиразин-8-ил)пиперидин-1-ил)(3,3-дифторазетидин-1-ил)метанона;
5-((1R,3S,4S)-3-(3Н-имидазо[1,2-а]пирроло[2,3-е]пиразин-8-ил)-4-метилциклопентиламино)пиразин-2-карбонитрила;
5-((1S,3R,4R)-3-(3Н-имидазо[1,2-а]пирроло[2,3-е]пиразин-8-ил)-4-метилциклопентиламино)пиразин-2-карбонитрила;
5-((1R,3R,4S)-3-(3Н-имидазо[1,2-а]пирроло[2,3-е]пиразин-8-ил)-4-метилциклопентиламино)пиразин-2-карбонитрила;
5-((1S,3S,4R)-3-(3Н-имидазо[1,2-а]пирроло[2,3-е]пиразин-8-ил)-4-метилциклопентиламино)пиразин-2-карбонитрила;
N-(4-(3Н-имидазо[1,2-а]пирроло[2,3-е]пиразин-8-ил)бицикло[2.2.2]октан-1-ил)циклопропансульфонамида;
(R)-(3-(3Н-имидазо[1,2-а]пирроло[2,3-е]пиразин-8-ил)пиперидин-1-ил)(3,3-дифторциклобутил)метанона;
(R)-(3-(3Н-имидазо[1,2-а]пирроло[2,3-е]пиразин-8-ил)пиперидин-1-ил)(3,3-дифторпирролидин-1-ил)метанона;
(R)-(3-(3Н-имидазо[1,2-а]пирроло[2,3-е]пиразин-8-ил)пиперидин-1-ил)(4,4-дифторпиперидин-1-ил)метанона;
N-((1S,3S,4R)-3-(3Н-имидазо[1,2-а]пирроло[2,3-е]пиразин-8-ил)-4-метилциклопентил)-3,3,3-трифторпропан-1-сульфонамида;
N-((1R,3R,4S)-3-(3Н-имидазо[1,2-а]пирроло[2,3-е]пиразин-8-ил)-4-метилциклопентил)-3,3,3-трифторпропан-1-сульфонамида;
N-((1R,3S,4S)-3-(3Н-имидазо[1,2-а]пирроло[2,3-е]пиразин-8-ил)-4-метилциклопентил)-3,3,3-трифторпропан-1-сульфонамида;
N-((1S,3R,4R)-3-(3Н-имидазо[1,2-а]пирроло[2,3-е]пиразин-8-ил)-4-метилциклопентил)-3,3,3-трифторпропан-1-сульфонамида;
((R)-3-(3Н-имидазо[1,2-а]пирроло[2,3-е]пиразин-8-ил)пиперидин-1-ил)((R)-2-(трифторметил)пирролидин-1-ил)метанона;
N-((3S,5R)-5-этил-1-(3Н-имидазо[1,2-а]пирроло[2,3-е]пиразин-8-ил)пирролидин-3-ил)циклопропансульфонамида;
3-(3Н-имидазо[1,2-а]пирроло[2,3-е]пиразин-8-ил)-4-метилциклопентанамин;
N-((1R,3S,4R)-3-этил-4-(3Н-имидазо[1,2-а]пирроло[2,3-е]пиразин-8-ил)циклопентил)-3,3,3-трифторпропан-1-сульфонамида;
N-((1S,3R,4S)-3-этил-4-(3Н-имидазо[1,2-а]пирроло[2,3-е]пиразин-8-ил)циклопентил)-3,3,3-трифторпропан-1-сульфонамида;
N-((1S,3S,4R)-3-(3Н-имидазо[1,2-а]пирроло[2,3-е]пиразин-8-ил)-4-метилциклопентил)циклопропансульфонамида;
N-((1S,3R,4S)-3-этил-4-(3Н-имидазо[1,2-а]пирроло[2,3-е]пиразин-8-ил)циклопентил)циклопропансульфонамида;
N-((1S,3R,4S)-3-этил-4-(3Н-имидазо[1,2-а]пирроло[2,3-е]пиразин-8-ил)циклопентил)-3,3-дифторазетидин-1-сульфонамида;
3-хлор-N-((1S,3R,4S)-3-этил-4-(3Н-имидазо[1,2-а]пирроло[2,3-е]пиразин-8-ил)циклопентил)-4-фторбензолсульфонамида;
N-((1S,3S,4R)-3-(3Н-имидазо[1,2-а]пирроло[2,3-е]пиразин-8-ил)-4-метилциклопентил)-3,3-дифторазетидин-1-сульфонамида;
N-(((1S,3R,4S)-3-этил-4-(3Н-имидазо[1,2-а]пирроло[2,3-е]пиразин-8-ил)циклопентил)метил)-3,3,3-трифторпропан-1-сульфонамида;
N-(((1R,3S,4R)-3-этил-4-(3Н-имидазо[1,2-а]пирроло[2,3-е]пиразин-8-ил)циклопентил)метил)-3,3,3-трифторпропан-1-сульфонамида;
N-(((1S,3S,4R)-3-этил-4-(3Н-имидазо[1,2-а]пирроло[2,3-е]пиразин-8-ил)циклопентил)метил)-3,3,3-трифторпропан-1-сульфонамида;
N-(((1R,3R,4S)-3-этил-4-(3Н-имидазо[1,2-а]пирроло[2,3-е]пиразин-8-ил)циклопентил)метил)-3,3,3-трифторпропан-1-сульфонамида;
N-((1S,3S,4R)-3-(3Н-имидазо[1,2-а]пирроло[2,3-е]пиразин-8-ил)-4-метилциклопентил)морфолин-4-сульфонамида;
[(2S,4S,5R)-4-метил-5-(3Н-имидазо[1,2-а]пирроло[2,3-е]пиразин-8-ил)-тетрагидро-фуран-2-илметил]-амида 3,3,3-трифторпропан-1-сульфоновой кислоты;
[(2R,4R,5S)-4-метил-5-(3Н-имидазо[1,2-а]пирроло[2,3-е]пиразин-8-ил)-тетрагидро-фуран-2-илметил]-амида 3,3,3-трифторпропан-1-сульфоновой кислоты;
метил-[(1S,3R,4S)-3-метил-4-(3Н-имидазо[1,2-а]пирроло[2,3-е]пиразин-8-ил)-циклопентил]-амида 3,3,3-трифторпропан-1-сульфоновой кислоты;
[(1S,3R,4S)-3-метил-4-(3Н-имидазо[1,2-а]пирроло[2,3-е]пиразин-8-ил)-циклопентил]-амида азетидин-1-сульфоновой кислоты;
{3-[(1S,3R,4S)-3-метил-4-(3Н-имидазо[1,2-а]пирроло[2,3-е]пиразин-8-ил)-циклопентиламино]-оксетан-3-ил}-ацетонитрила;
[(1S,3R,4S)-3-метил-4-(3Н-имидазо[1,2-а]пирроло[2,3-е]пиразин-8-ил)-циклопентил]-амида 3,3-дифторциклобутансульфоновой кислоты;
8-[(1S,2R,4S)-2-метил-4-(тетрагидропиран-4-илокси)-циклопентил]-3Н-3,4,6,8а-тетрааза-ас-индацена;
8-[(1R,2R)-2-метил-4-(тетрагидропиран-4-илокси)-циклопентил]-3Н-3,4,6,8а-тетрааза-ас-индацена;
[(1S,3R,4S)-3-метил-4-(3Н-имидазо[1,2-а]пирроло[2,3-е]пиразин-8-ил)-циклопентил]-амида 3-фторазетидин-1-сульфоновой кислоты;
[(1S,3R,4S)-3-метил-4-(3Н-имидазо[1,2-а]пирроло[2,3-е]пиразин-8-ил)-циклопентил]-амида 3-фторпропан-1-сульфоновой кислоты;
[(1S,3R,4S)-3-метил-4-(7-метил-3Н-имидазо[1,2-а]пирроло[2,3-е]пиразин-8-ил)-циклопентил]-амида циклопропансульфоновой кислоты;
[(1R,3S,4R)-3-метил-4-(7-метил-3Н-имидазо[1,2-а]пирроло[2,3-е]пиразин-8-ил)-циклопентил]-амида циклопропансульфоновой кислоты;
2-циано-N-[(1S,3R,4S)-3-метил-4-(3Н-имидазо[1,2-а]пирроло[2,3-е]пиразин-8-ил)-циклопентил]-ацетамида;
8-[(1S,2R,4R)-2-метил-4-(тетрагидропиран-4-илокси)-циклопентил]-3Н-3,4,6,8а-тетрааза-ас-индацена;
(2-циклопропилэтил)-[(1S,3R,4S)-3-метил-4-(3Н-имидазо[1,2-а]пирроло[2,3-е]пиразин-8-ил)-циклопентил]-оксетан-3-ил-амина;
циклопропилметил-[(1S,3R,4S)-3-метил-4-(3Н-имидазо[1,2-а]пирроло[2,3-е]пиразин-8-ил)-циклопентил]-оксетан-3-ил-амина;
(2,2,2-трифторэтил)-амида (3R,4S)-3-этил-4-(3Н-имидазо[1,2-а]пирроло[2,3-е]пиразин-8-ил)-пирролидин-1-карбоновой кислоты;
(2,2,2-трифторэтил)-амида (3S,4R)-3-этил-4-(3Н-имидазо[1,2-а]пирроло[2,3-е]пиразин-8-ил)-пирролидин-1-карбоновой кислоты; или
их фармацевтически приемлемые соли или стереоизомеры.
7. Соединение, выбранное из
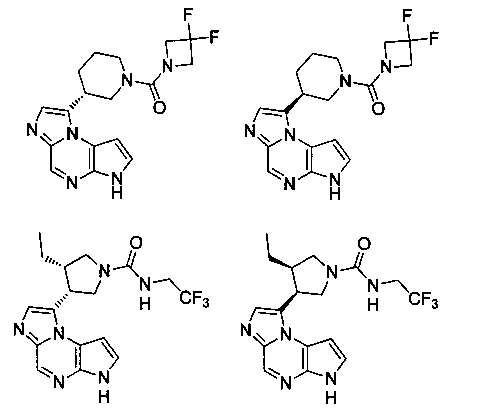 ,
,
или их фармацевтически приемлемые соли или стереоизомеры.
8. Соединение, представленное следующей структурной формулой:
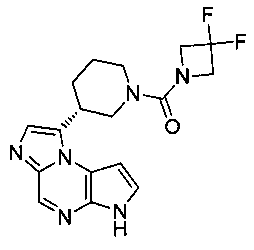 ,
,
или его фармацевтически приемлемые соли или стереоизомеры.
9. Соединение, представленное следующей структурной формулой:
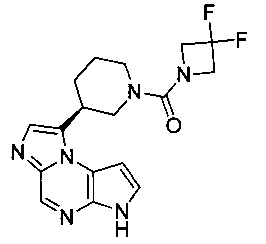 ,
,
или его фармацевтически приемлемые соли или стереоизомеры.
10. Соединение, представленное следующей структурной формулой:
 ,
,
или его фармацевтически приемлемые соли или стереоизомеры.
11. Соединение, представленное следующей структурной формулой:
 ,
,
или его фармацевтически приемлемые соли или стереоизомеры.
12. Соединение, выбранное из

или его фармацевтически приемлемые соли или стереоизомеры.
13. Соединение, представленное следующей структурной формулой:

или его фармацевтически приемлемые соли или стереоизомеры.
14. Соединение, представленное следующей структурной формулой:

или его фармацевтически приемлемые соли или стереоизомеры.
15. Соединение, представленное следующей структурной формулой:

или его фармацевтически приемлемые соли или стереоизомеры.
16. Соединение, выбранное из
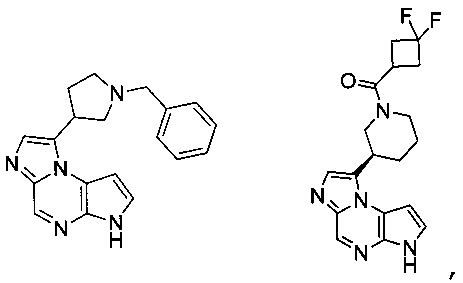
или его фармацевтически приемлемые соли или стереоизомеры.
17. Соединение, представленное следующей структурной формулой:
или его фармацевтически приемлемые соли или стереоизомеры.
18. Соединение, представленное следующей структурной формулой:
или его фармацевтически приемлемые соли или стереоизомеры.
19. Фармацевтическая композиция для лечения заболеваний и расстройств, связанных с аномальной или нерегулируемой киназной активностью Jak1, Jak3 или Syk киназ, содержащая терапевтически эффективное количество соединения по любому из пп.1-18, фармацевтически приемлемый носитель и эксципиент.
20. Применение соединения по любому из пп.1-18 или его фармацевтически приемлемой соли, стереоизомера или изомера в производстве лекарственного средства для лечения состояния, опосредованного активностью Jak1, Jak3 или Syk протеинкиназ.
21. Применение по п.20, где протеинкиназа представляет собой Jak1.
22. Применение по п.20 или 21, где состояние выбирают из неспецифического язвенного колита, ревматоидного артрита, псориаза или болезни Крона.
23. Применение по п.20 или 21, где состояние выбирают из аутоиммунного заболевания; астмы; атопического дерматита; системной красной волчанки (SLE); рассеянного склероза (MS); онкологического заболевания, такого как рак простаты, рак толстой кишки, рак яичников, рак молочной железы, меланомы, лейкемии и других злокачественных расстройств кроветворной системы; классических миелопролиферативных расстройств (MPD); воспаления; воспалительных заболеваний кишечника.
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| МОДУЛЯТОРЫ А3 -РЕЦЕПТОРОВ АДЕНОЗИНА | 1999 |
|
RU2250904C2 |

