РОДСТВЕННЫЕ ЗАЯВКИ НА ПАТЕНТ
Эта заявка испрашивает приоритет на основании заявки на патент Индии №715/MUM/2013, поданной 8 марта 2013 г., раскрытие которой включено в данный документ посредством ссылки в полном ее объеме, как если бы была полностью включена в данный документ. Все ссылочные материалы, включая патенты, заявки на патенты и литературу, приведенную в описании изобретения, явным образом полностью включены в данный документ посредством ссылок.
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
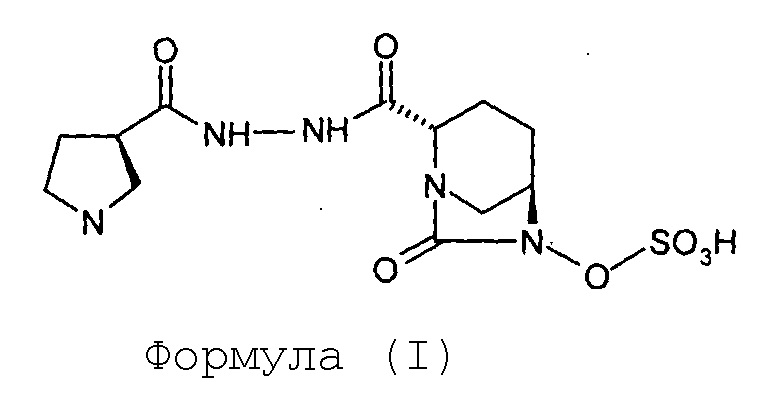
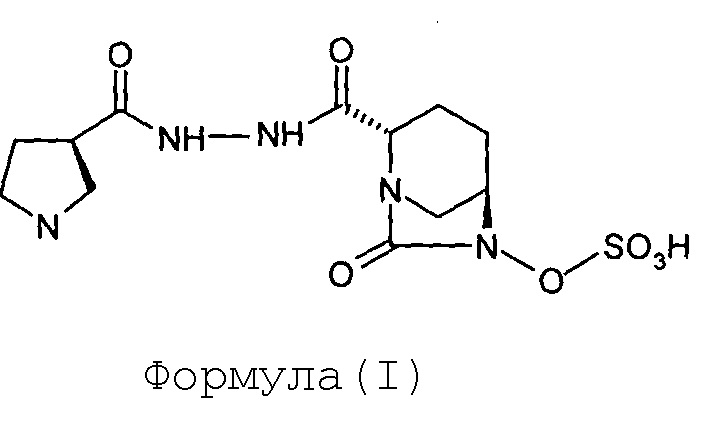
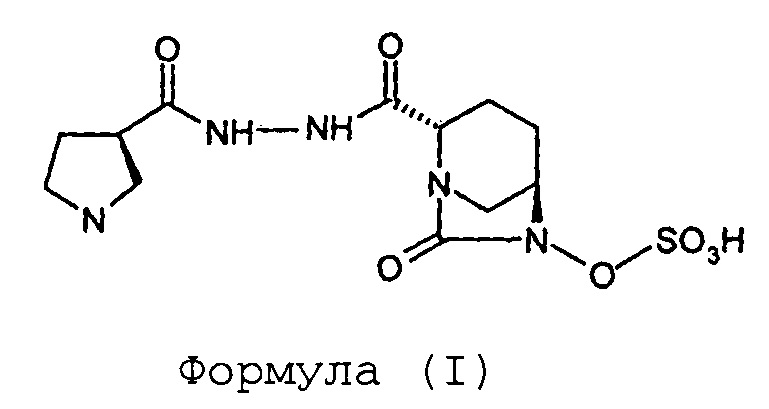
Изобретение относится к способу получения (2S,5R)-7-оксо-6-сульфоокси-2-[((3R)-пирролидин-3-карбонил)гидразинокарбонил]-1,6-диазабицикло[3.2.1]октана.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Соединение Формулы (I), имеющее химическое название (2S,5R)-7-оксо-6-сульфоокси-2-[((3R)-пирролидин-3-карбонил)гидразинокарбонил]-1,6-диазабицикло[3.2.1]октан, обладает антибактериальными свойствами и раскрыто в международной заявке на патент согласно PCT № PCT/IB2012/054290.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
В одном общем аспекте предоставляют способ получения соединения Формулы (I), включающий:
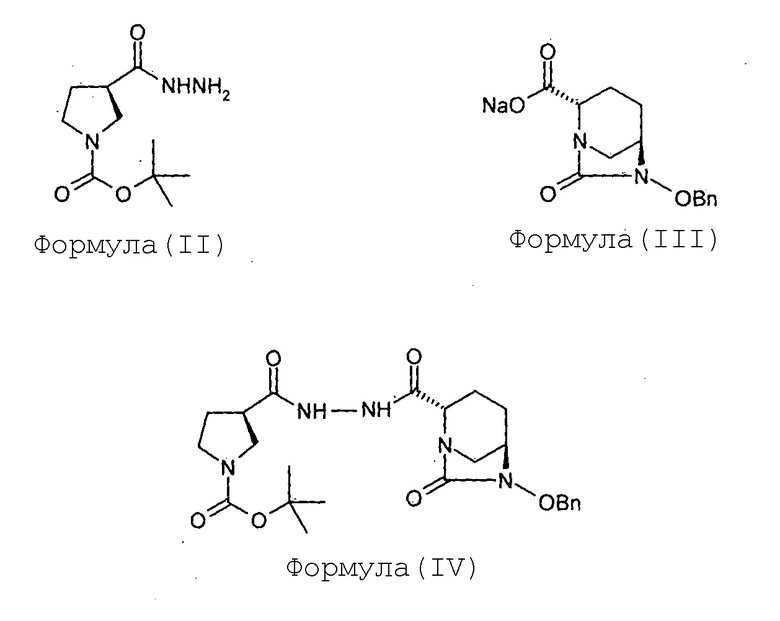
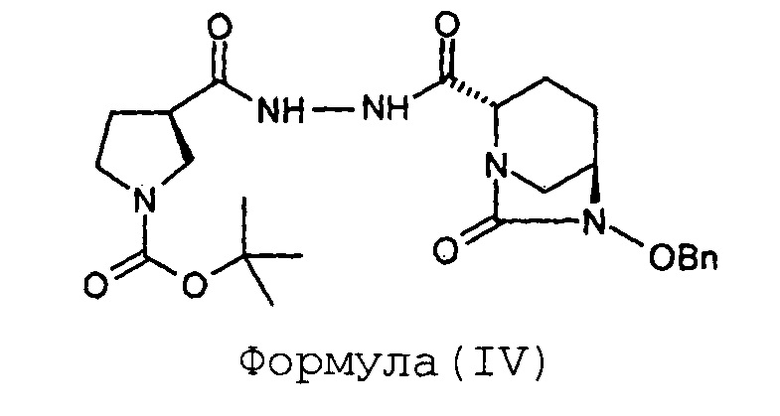
(а) проведение реакции между соединением Формулы (II) и соединением Формулы (III) с получением соединения Формулы (IV);
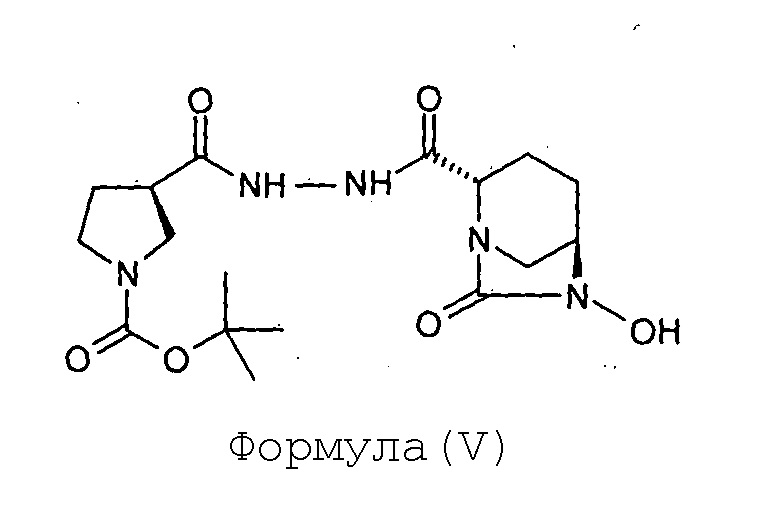
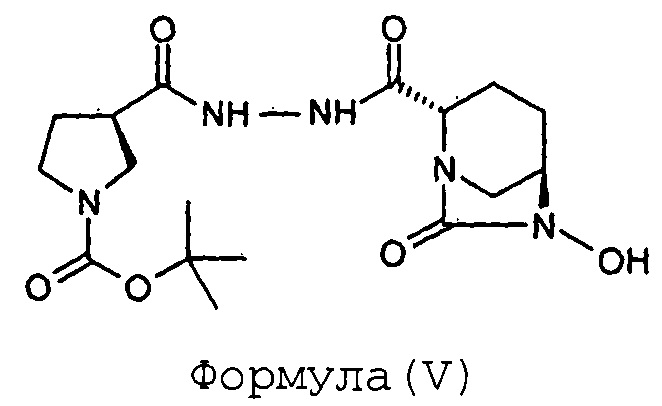
(b) гидрогенолиз соединения Формулы (IV) с получением соединения Формулы (V);
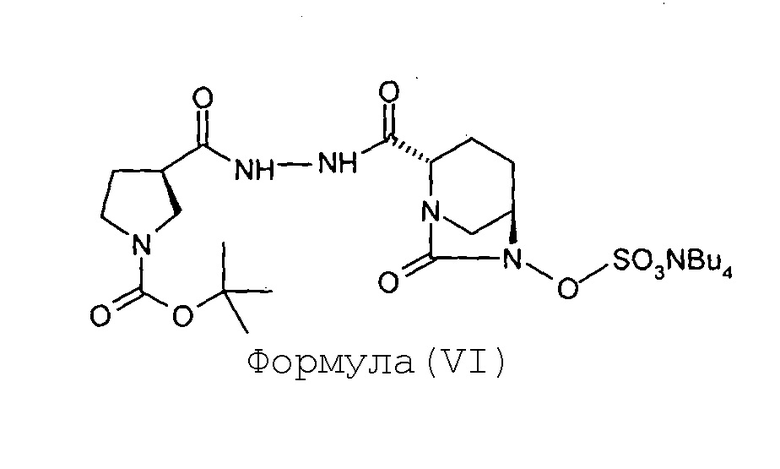
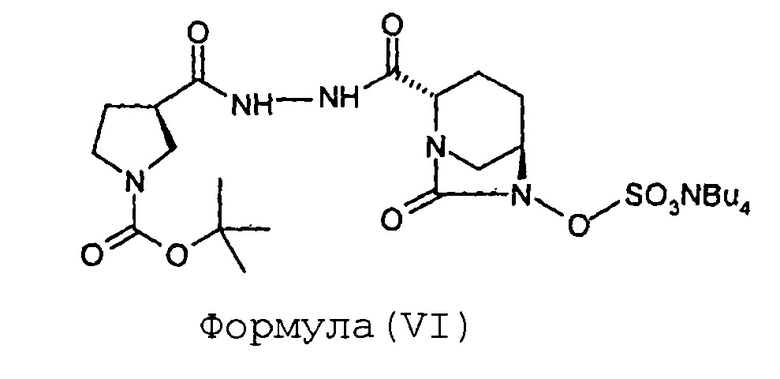
(с) сульфонирование соединения Формулы (V) с получением соединения Формулы (VI);
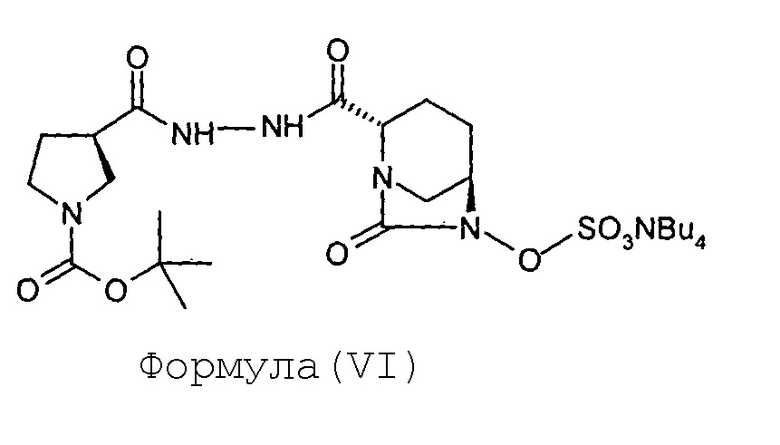
и
(d) превращение соединения Формулы (VI) в соединение Формулы (I).
Подробности одного или более вариантов осуществления изобретения изложены в описании изобретения ниже. Другие признаки, цели и преимущества изобретения будут очевидны из приведенного ниже описания изобретения, в том числе из формулы изобретения.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Далее будет сделана ссылка на примеры осуществления изобретения, и для описания такового будет использован в данном документе особый язык. Тем не менее, следует понимать, что ограничение объема изобретения в результате этого не предполагается. Следует полагать, что изменения и дополнительные модификации признаков изобретения, проиллюстрированных в данном документе, и дополнительные применения принципов изобретения, проиллюстрированных в данном документе, которые мог бы придумать специалист в релевантной области, обладающий таким раскрытием, попадают в рамки объема изобретения. Следует отметить, что используемые в этом описании изобретения и в прилагаемых пунктах формулы формы единственного числа включают соотносимые объекты во множественном числе, если содержание ясно не диктует иное. Все ссылки, включая патенты, заявки на патенты и литературу, приведенную в описании изобретения, явным образом включены в данный документ посредством ссылки в полном их объеме, как если бы они были полностью переписаны в данный документ.
Термин "HOBt", используемый в данном документе, относится к 1-гидроксибензотриазолу.
Термин "EDC", используемый в данном документе, относится к 1-этил-3-(3-диметиламинопропил)карбодиимиду.
В одном общем аспекте предоставляют способ получения соединения Формулы (I), включающий:
(а) проведение реакции между соединением Формулы (II) и соединением Формулы (III) с получением соединения Формулы (IV);
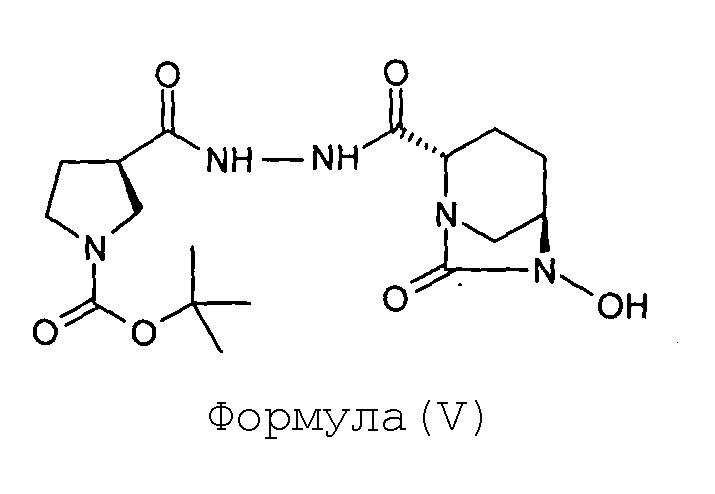
(b) гидрогенолиз соединения Формулы (IV) с получением соединения Формулы (V);
(с) сульфонирование соединения Формулы (V) с получением соединения Формулы (VI);
и
(d) превращение соединения Формулы (VI) в соединение Формулы (I).
Соединение Формулы (IV) получают путем проведения реакции между соединением Формулы (II) и соединением Формулы (III). В некоторых вариантах осуществления реакцию выполняют в присутствии 1-гидроксибензотриазола. В некоторых других вариантах осуществления соединение Формулы (IV) получают в результате проведения реакции между соединением Формулы (II) и соединением Формулы (III) в присутствии 1-гидроксибензотриазола и гидрохлорида 1-этил-3-(3-диметиламинопропил)карбодиимида. В некоторых вариантах осуществления реакцию проводят в воде, взятой в качестве реакционного растворителя.
Соединение Формулы (V) получают путем гидрогенолиза соединения Формулы (IV). Реакция гидрогенолиза может быть проведена с использованием подходящего агента гидрогенолиза. В некоторых вариантах осуществления гидрогенолиз соединения Формулы (IV) с получением соединения Формулы (V) проводят в присутствии катализатора на основе переходного металла и источника водорода. В некоторых вариантах осуществления катализатор на основе переходного металла представляет собой палладий на углеродном носителе, а источник водорода представляет собой газ водород. В некоторых вариантах осуществления реакцию гидрогенолиза проводят в присутствии подходящего растворителя, такого как спирт (например, метанол). В некоторых вариантах осуществления гидрогенолиз соединения Формулы (IV) с получением соединения Формулы (V) проводят с использованием катализатора на основе 10% палладия на углеродном носителе, в присутствии газа водорода, в метаноле, взятом в качестве растворителя.
Соединение Формулы (VI) получают путем сульфонирования соединения Формулы (V). Реакция сульфонирования может быть проведена в присутствии подходящего растворителя. В некоторых вариантах осуществления сульфонирование соединения Формулы (V) с получением соединения Формулы (VI) выполняют путем проведения реакции между соединением Формулы (V) и комплексом триоксид серы-пиридин, с последующей обработкой посредством гидросульфата тетрабутиламмония.
Соединение Формулы (VI) превращают в соединение Формулы (I) в присутствии подходящего реагента. В некоторых вариантах осуществления соединение Формулы (VI) превращают в соединение Формулы (I) путем проведения реакции между соединением Формулы (VI) и трифторуксусной кислотой.
В некоторых вариантах осуществления соединение Формулы (I) получают с использованием способа, описанного на Схеме 1.
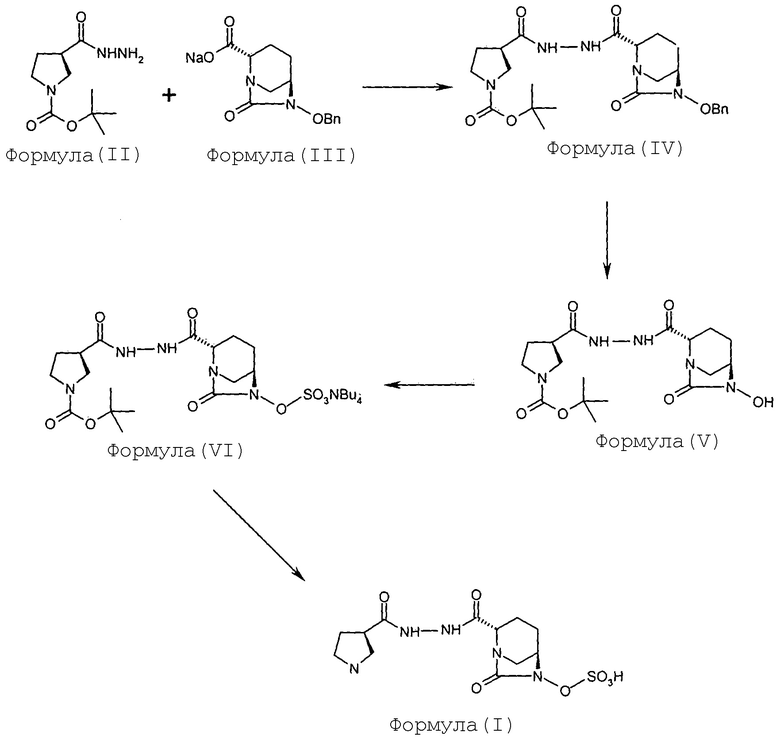
Схема-1
В некоторых вариантах осуществления предоставляют соединение Формулы (I) в кристаллической форме.
В некоторых других вариантах осуществления предоставляют соединение Формулы (I), находящееся в кристаллической форме и имеющее порошковую рентгеновскую дифрактограмму, содержащую пик, выбранный из группы, состоящей из 7,03 (±0,2), 9,17 (±0,2), 13,52 (±0,2), 15,19 (±0,2), 16,28 (±0,2), 16,92 (±0,2), 18,30 (±0,2), 19,10 (±0,2), 20,49 (±0,2), 21,62 (±0,2), 22,01 (±0,2), 22,77 (±0,2), 23,72 (±0,2), 25,05 (±0,2), 25,64 (±0,2), 27,04 (±0,2), 27,96 (±0,2), 29,41 (±0,2), 30,21 (±0,2), 35,68 (±0,2), 36,75 (±0,2) и 37,89 (±0,2) градусов угла рассеяния 2 тета.
В некоторых других вариантах осуществления предоставляют соединение Формулы (I), находящееся в кристаллической форме и имеющее порошковую рентгеновскую дифрактограмму, содержащую пик, выбранный из группы, состоящей из 7,03 (±0,2), 9,17 (±0,2), 15,19 (±0,2), 16,92 (±0,2), 18,30 (±0,2), 19,10 (±0,2), 22,77 (±0,2) и 23,72 (±0,2) градусов угла рассеяния 2 тета.
В некоторых других вариантах осуществления предоставляют соединение Формулы (I), находящееся в кристаллической форме и имеющее порошковую рентгеновскую дифрактограмму, по существу такую же, как показана на Фигуре 1.
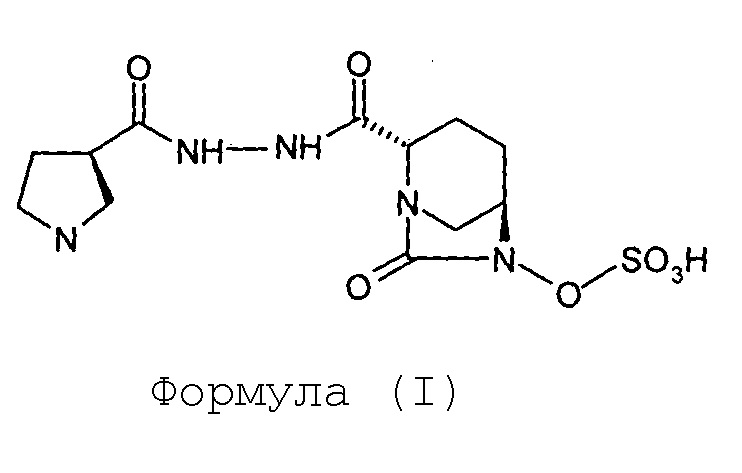
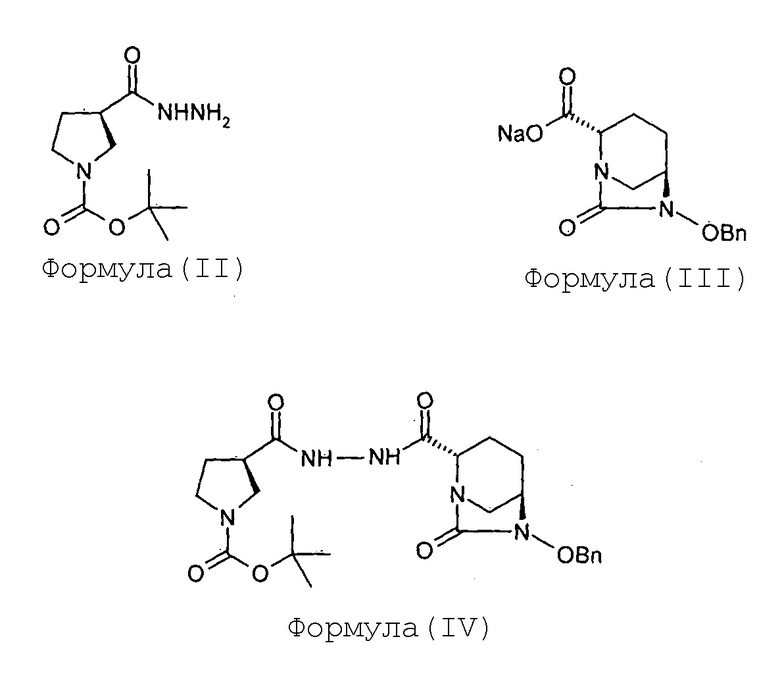
В некоторых вариантах осуществления предоставляют способ получения соединения Формулы (II), включающий:
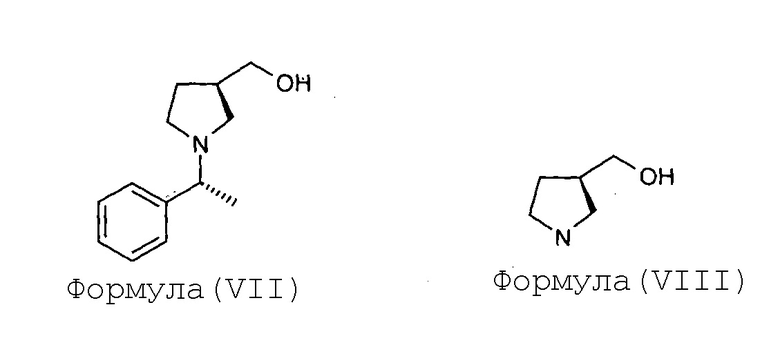
(а) гидрогенолиз соединения Формулы (VII) с получением соединения Формулы (VIII)
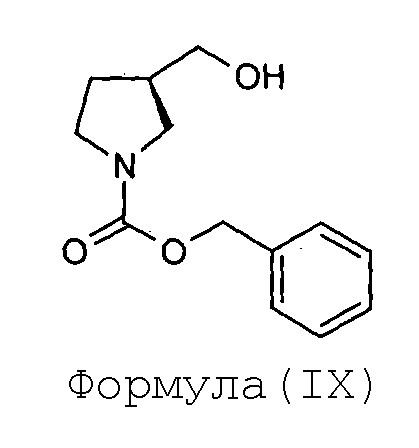
(b) превращение соединения Формулы (VIII) в соединение Формулы (IX)
(с) окисление соединения Формулы (IX) с получением соединения Формулы (X),
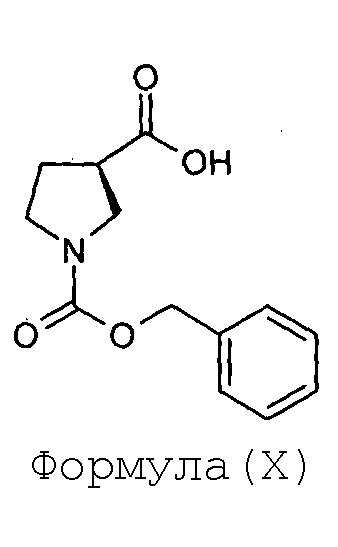
(d) этерификацию соединения Формулы (X) с получением соединения Формулы (XI)
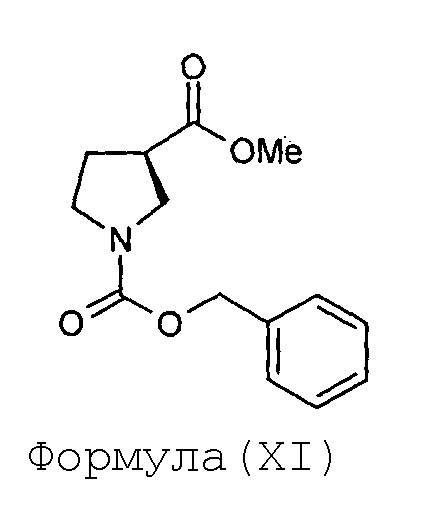
е) гидрогенолиз соединения Формулы (XI) с получением соединения Формулы (XII)
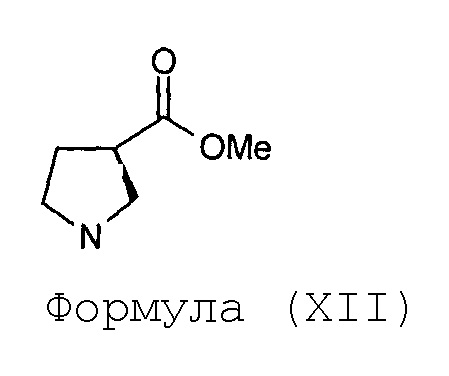
(f) превращение соединения Формулы (XII) в соединение Формулы (XIII),
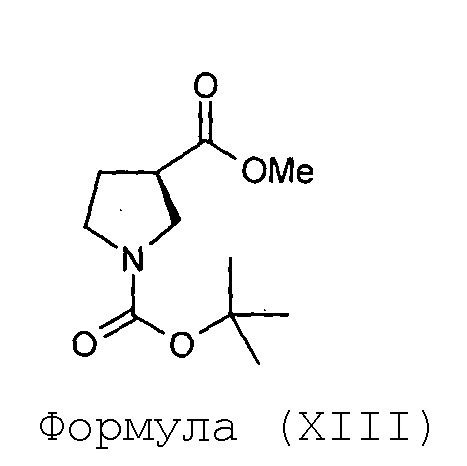
и
(g) превращение соединения Формулы (XIII) в соединение Формулы (II).
Соединение Формулы (VIII) получают гидрогенолизом соединения Формулы (VII). Реакция гидрогенолиза может быть проведена с использованием подходящего агента гидрогенолиза. В некоторых вариантах осуществления гидрогенолиз соединения Формулы (VII) с получением соединения Формулы (VIII) проводят в присутствии катализатора на основе переходного металла и источника водорода. В некоторых вариантах осуществления катализатор на основе переходного металла представляет собой палладий на углеродном носителе, а источник водорода представляет собой газ водород или формиат аммония. В некоторых вариантах осуществления реакцию гидрогенолиза проводят в присутствии подходящего растворителя, такого как спирт (например, метанол). В некоторых вариантах осуществления гидрогенолиз соединения Формулы (VII) с получением соединения Формулы (VIII) проводят с использованием катализатора на основе 10% палладия на углеродном носителе, в присутствии формиата аммония или газа водорода, в метаноле, взятом в качестве растворителя.
Соединение Формулы (VIII) затем превращают в соединение Формулы (IX) в присутствии подходящего реагента, такого как бензил-хлорформиат. Соединение Формулы (IX) подвергают обработке подходящим окисляющим реагентом (как например, реактивом Джонса) с получением соединения Формулы (X). Соединение Формулы (X) затем подвергают этерификации с использованием подходящего реагента с получением соединения Формулы (XI).
Соединение Формулы (XII) получают гидрогенолизом соединения Формулы (XI). Реакция гидрогенолиза может быть проведена с использованием подходящего агента гидрогенолиза. В некоторых вариантах осуществления гидрогенолиз соединения Формулы (XI) с получением соединения Формулы (XII) проводят в присутствии катализатора на основе переходного металла и источника водорода. В некоторых вариантах осуществления катализатор на основе переходного металла представляет собой палладий на углеродном носителе, а источник водорода представляет собой газ водород или формиат аммония. В некоторых других вариантах осуществления реакцию гидрогенолиза проводят в присутствии подходящего растворителя, такого как спирт (например, метанол). В некоторых вариантах осуществления гидрогенолиз соединения Формулы (XI) с получением соединения Формулы (XII) проводят с использованием катализатора на основе 10% палладия на углеродном носителе, в присутствии формиата аммония или газа водорода, в метаноле, взятом в качестве растворителя.
Соединение Формулы (XII) превращают в соединение Формулы (XIII) в присутствии ди-трет-бутилкарбоната и триэтиламина в дихлорметане. Соединение Формулы (II) получают в результате обработки соединения Формулы (XIII) гидразингидратом в этаноле. Схематическое представление синтеза соединения Формулы (II) дано на Схеме-2.
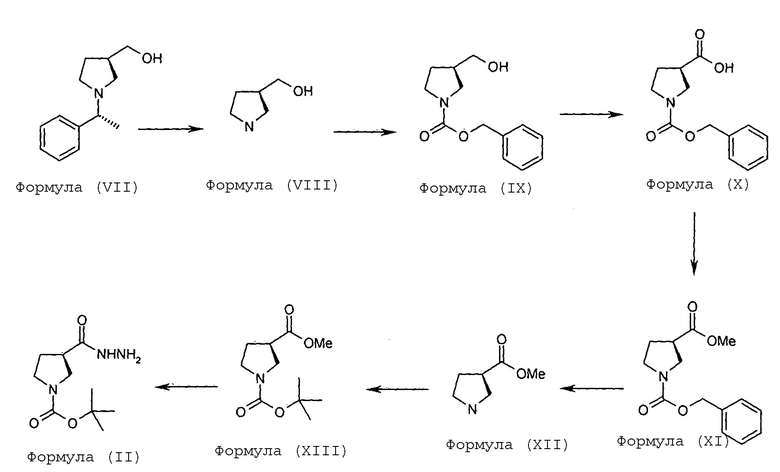
Схема-2
Для специалиста в данной области будет совершенно очевидно, что в изобретении, раскрытом в данном документе, могут быть сделаны различные замены и модификации без отступления от объема и существа изобретения. Например, специалистам в данной области будет ясно, что изобретение может быть использовано на практике с применением ряда различных соединений в рамках рассматриваемых общих описаний.
ПРИМЕРЫ
Следующие примеры иллюстрируют варианты осуществления изобретения, которые в настоящее время являются наиболее известными. Однако следует понимать, что следующее является только примером или иллюстрацией применения принципов настоящего изобретения. Многочисленные модификации и альтернативные композиции, способы и системы могут быть разработаны специалистами в данной области без отступления от существа и объема настоящего изобретения. Прилагаемая формула изобретения, как подразумевается, охватывает такие модификации и расстановки. Таким образом, тогда как настоящее изобретение было выше описано с использованием характерных особенностей, приведенные ниже примеры предоставляют дополнительные подробности, касающиеся, как в настоящее время считается, наиболее используемых на практике и наиболее предпочтительных вариантов осуществления изобретения.
Пример 1
Получение гидразида (R)-N-трет-бутоксикарбонилпирролидин-3-карбоновой кислоты (II):
Стадия-1: Получение формиатной соли (R)-(пирролидин-3-ил)метанола (VIII):
К раствору (1R,3R)-[1-(1-фенилэтил)пирролидин-3-ил]метанола (VII, 124 г, 0,60 моль) в метаноле (2,5 л) загружают формиат аммония (114 г, 1,81 моль), после этого вводят катализатор на основе 10% палладия на углеродном носителе (37 г, влажность 50%). Суспензию черного цвета перемешивают в течение 5 минут и затем ее нагревают до температуры, при которой проводят кипячение с обратным холодильником в течение 1 часа. Когда метод тонкослойной хроматографии (TLC) (20%-ный раствор метанола в хлороформе) показывает завершение реакции, реакционную смесь оставляют остыть до комнатной температуры. Катализатор отфильтровывают при отсасывании под вакуумом на слое целлита и катализатор промывают метанолом (500 мл). Выпаривание растворителя под вакуумом дает формиатную соль (R)-(пирролидин-3-ил)метанола (VIII) в виде маслянистого сиропа, в количестве 73 грамм с выходом 82%.
Анализ:
Масс-спектрометрия: (М+1): 102,0 для C5H11NO.HCOOH в форме свободного основания.
Стадия-2: Получение (R)-(1-бензилоксикарбонилпирролидин-3-ил)метанола (IX):
К прозрачному раствору формиатной соли (R)-(пирролидин-3-ил)метанола (VIII, 73 г, 0,49 моль) в воде (365 мл) добавляют тетрагидрофуран (365 мл) при перемешивании. К реакционной смеси добавляют 50%-ный раствор бензилоксихлорформиата в толуоле (152 мл, 77 г, 0,44 моль), после этого вводят бикарбонат натрия (125 г, 1,48 моль) в виде твердого вещества. Реакционную смесь перемешивают в течение ночи при 35°С. Метод TLC (20%-ный раствор метанола в хлороформе) показывает завершение реакции. К реакционной смеси добавляют воду (365 мл) и экстрагируют дважды этилацетатом (600 мл и 400 мл). Объединенный органический слой промывают рассолом (500 мл) и органический слой упаривают под вакуумом с получением (R)-(1-бензилоксикарбонилпирролидин-3-ил)метанола (IX) в виде маслянистого сиропа в количестве 90 грамм с выходом 79%.
Анализ:
ЯМР: (CDCl3): 7,25-7,37 (м, 5Н), 5,12 (с, 2Н), 3,44-3,61 (м, 4Н), 3,37-3,42 (м, 1Н), 3,19 (кв, 1Н), 2,39-2,45 (м, 1Н), 1,96-2,02 (м, 1Н), 1,71 (кв, 1Н), 1,65 (с, 1Н).
Масс-спектрометрия: (М+1): 236,2 для C13H17NO3.
Стадия-3: Получение (R)-1-бензилоксикарбонилпирролидин-3-карбоновой кислоты (X):
К раствору (R)-(1-бензилоксикарбонилпирролидин-3-ил)метанола (IX, 80 г, 0,34 моль), растворенного в ацетоне (800 мл), добавляют по каплям при перемешивании реактив Джонса (200 мл, приготовленные в результате растворения 53,4 грамм CrO3 в растворе, приготовленном смешением 46 мл H2SO4 и 140 мл воды, и доведения конечного объема до 200 мл) при 20°С до тех пор пока не будет подвергаться изменению темно-красный цвет раствора и не будет выделяться твердое вещество зеленого цвета. Суспензию перемешивают в течение последующих 30 минут. Когда метод TLC (10%-ный раствор метанола в хлороформе) показывает полное превращение, в реакционную смесь добавляют по каплям изопропиловый спирт (100 мл), до тех пор пока не будет подвергаться изменению зеленый цвет в течение 10 минут. Суспензию фильтруют на слое целлита при отсасывании под вакуумом и твердые вещества промывают свежей порцией ацетона (100 мм, дважды). Фильтрат упаривают под вакуумом и к остатку добавляют насыщенный водный раствор бикарбоната натрия (600 мл) до тех пор, пока не установится рН 8. Получающуюся в результате смесь подвергают экстрагированию этилацетатом (400 мл) и слои разделяют. Значение рН водного слоя доводят до 2 в результате использования 6N-ного водного раствора хлористоводородной кислоты (125 мл). Реакционную смесь подвергают экстрагированию этилацетатом (500 мл × 2) и сушат над сульфатом натрия. Выпаривание растворителя дает (R)-1-бензилоксикарбонилпирролидин-3-карбоновую кислоту (X) в количестве 67 грамм в виде маслянистого сиропа с выходом 79%.
Анализ:
ЯМР: (CDCl3): 9,2 (шир.с, 1Н), 7,25-7,35 (м, 5Н), 5,13 (с, 2Н), 3,62-3,71 (м, 2Н), 3,52-3,54 (м, 1Н), 3,43-3,49 (м, 1Н), 3,07-3,10 (м, 1Н), 2,09-2,18 (м, 2Н).
Масс-спектрометрия: (М-1): 248,1 для C13H15NO4.
Стадия-4: Получение (R)-метил-1-бензилоксикарбонилпирролидин-3-карбоксилата (XI):
Раствор (R)-1-бензилоксикарбонилпирролидин-3-карбоновой кислоты (X, 67 г, 0,26 моль) в метанольном растворе HCl (670 мл) перемешивают в течение 1,5 часа при 35°С. Когда метод TLC (10%-ный раствор метанола в хлороформе) показывает полное превращение, растворитель выпаривают под вакуумом и к остатку загружают насыщенный водный раствор бикарбоната натрия (640 мл) при тщательном перемешивании. Реакционную смесь подвергают экстрагированию этилацетатом (400 мл × 2). Объединенные органические слои сушат над сульфатом натрия и упаривают под вакуумом с получением (R)-метил-1-бензилоксикарбонилпирролидин-3-карбоксилата (XI) в виде маслянистого сиропа в количестве 64 грамм с выходом 91 %.
Анализ:
ЯМР: (CDCl3): 7,25-7,36 (м, 5Н), 5,12 (с, 2Н), 3,70 (с, 3Н), 3,53-3,63 (м, 3Н), 3,42-3,51 (м, 1Н), 3,03-3,42 (м, 1Н), 2,12-2,16 (м, 2Н).
Масс-спектрометрия: (М+1): 264,2 для C14H17NO4.
Стадия-5: Получение гидрохлоридной соли (R)-метил-пирролидин-3-карбоксилата (XII):
Прозрачный раствор (R)-метил-1-бензилоксикарбонилпирролидин-3-карбоксилата (XI, 64 г, 0,24 моль) в метаноле (640 мл) переносят в реактор под давлением и вводят катализатор на основе 10% палладия на углеродном носителе (20 г, влажность 50%). Значение рН реакционной смеси корректируют с доведением до 3-3,5 путем добавления концентрированной хлористоводородной кислоты (25 мл). Реакционную смесь перемешивают под давлением газа водорода 100 фунтов на квадратный дюйм (psi) в течение 1,5 часа. Когда метод TLC (50%-ный раствор этилацетата в гексанах) показывает завершение реакции, катализатор отфильтровывают на слое целлита при отсасывании под вакуумом. Катализатор промывают свежей порцией метанола (100 мл). Выпаривание растворителя под вакуумом предоставляет гидрохлоридную соль (R)-метилпирролидин-3-карбоксилата (XII) в количестве 40 грамм с количественным выходом, которую используют сразу же для следующей реакции.
Анализ:
Масс-спектрометрия: (М+1): 130,0 для C6H11NO2 в форме свободного основания.
Стадия-6: Получение (R)-метил-1-трет-бутоксикарбонилпирролидин-3-карбоксилата (XIII):
Гидрохлоридную соль(R)-метилпирролидин-3-карбоксилата, полученную так, как указано выше, (XII, 40 г, 0,24 моль) суспендируют в дихлорметане (400 мл) и охлаждают до 0°С, и к этому добавляют ди-трет-бутилкарбонат (55 мл, 0,24 моль), с последующим введением триэтиламина (101 мл, 0,72 моль) при перемешивании. Реакционную смесь перемешивают в течение 1 часа, и, когда метод TLC (50%-ный раствор этилацетата в гексане) показывает завершение реакции, ее разбавляют дихлорметаном (200 мл), после этого водой (400 мл) и суспензию фильтруют на слое целлита и промывают дихлорметаном (200 мл). Слои в фильтрате разделяют и органический слой упаривают под вакуумом с предоставлением остатка, который очищают на короткой колонке с силикагелем с предоставлением (R)-метил-1-трет-бутоксикарбонилпирролидин-3-карбоксилата (XIII) в виде бесцветного масла в количестве 52 грамм с выходом 95%.
Анализ:
ЯМР: (CDCl3): 3,70 (с, 3Н), 3,40-3,60 (м, 3Н), 3,30-3,40 (м, 1Н), 3,00-3,10 (м, 1Н), 2,09-2,20 (шир.м, 2Н), 1,45 (с, 9Н).
Масс-спектрометрия: (М+1): 230,2 для C11H19NO4.
Хиральная чистота по методу высокоэффективной жидкостной хроматографии (HPLC): 99,87%.
Стадия-7: Получение гидразида (R)-N-трет-бутоксикарбонилпирролидин3-карбоновой кислоты (II):
К раствору (R)-метил-1-трет-бутоксикарбонилпирролидин-3-карбоксилата (XIII, 52 г, 0,22 моль) в этаноле (520 мл) добавляют гидразингидрат (57 мл, 1,13 моль). Реакционную смесь перемешивают в течение 2,5 часов при 80°С. Когда метод TLC показывает полное превращение, растворитель выпаривают под вакуумом. К остатку добавляют воду (500 мл) и дважды проводят экстрагирование 10%-ным раствором метанола в хлороформе (400 мл и 300 мл). Объединенный органический слой сушат над сульфатом натрия и упаривают под вакуумом, что позволяет получить гидразид (R)-N-трет-бутоксикарбонилпирролидин-3-карбоновой кислоты (II) в виде масла в количестве 54 граммов с количественным выходом.
Анализ:
ЯМР: (CDCl3): 7,03 (шир.с, 1Н), 3,91 (шир.с, 2Н), 3,41-3,68 (м, 3Н), 3,29-3,40 (м, 1Н), 2,81 (шир.д, 1Н), 2,03-2,14 (м, 2Н), 1,45 (с, 9Н).
Масс-спектрометрия: (М+1): 230,2 для C10H19N3O3.
Хиральная чистота по методу HPLC: 99,88%.
Пример 2
Получение (2S, 5R)-7-оксо-6-сульфоокси-2-[((3R)-пирролидин-3-карбонил)гидразинокарбонил]-1,6-диазабицикло[3.2.1]октана (I):
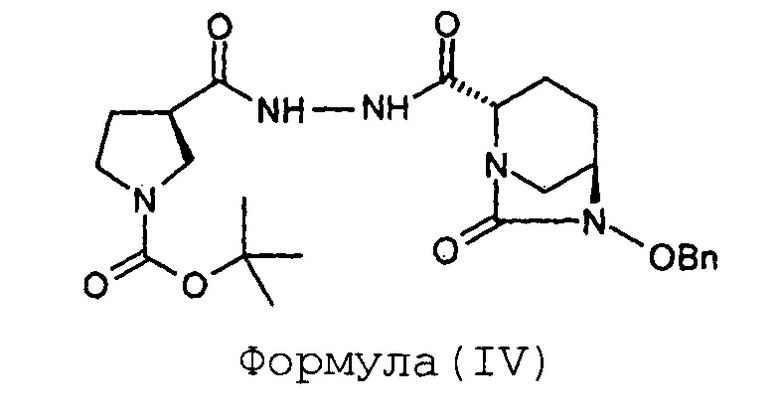
Стадия-1: Получение (2S,5R)-6-бензилокси-7-оксо-2-[((3R)-N-трет-бутоксикарбонилпирролидин-3-карбонил)гидразинокарбонил]-1,6-диазабицикло[3.2.1]октана (IV):
Натрия (2S,5R)-6-бензилокси-7-оксобицикло[3.2.1]-1,6-диазаоктан-2-карбоксилат (III, 67 г, 0,22 моль; полученный с применением способа, раскрытого в заявке на патент Индии № 699/MUM/2013) растворяют в воде (1,0 л) с получением в результате прозрачного раствора при 35°С. К прозрачному раствору последовательно добавляют, гидразид (R)-N-трет-бутоксикарбонилпирролидин-3-карбоновой кислоты (II, 54 г, 0,23 моль), EDC гидрохлорид (65 г, 0,33 моль) и HOBt (30,2 г, 0,22 моль), с последующим введением воды (0,14 л) при перемешивании при 35°С. Получающуюся в результате суспензию перемешивают при 35°С в течение 18 часов. По достижении максимального выпадения осадка метод TLC (метанол: хлороформ 1:9) показывает завершение реакции. Выпавшее в осадок твердое вещество белого цвета отфильтровывают при отсасывании под вакуумом и влажный фильтрационный осадок перемешивают с дополнительной порцией воды (1,0 л) в течение 3 часов. Суспензию фильтруют и фильтрационный осадок промывают водой (200 мл), сушат на воздухе в течение ночи с получением (2S,5R)-6-бензилокси-7-оксо-2-[((3R)-N-трет-бутоксикарбонилпирролидин-3-карбонил)гидразинокарбонил]-1,6-диазабицикло[3.2.1]октана (IV) в виде порошка белого цвета в количестве 95 граммов с выходом 88%.
Анализ:
ЯМР: (CDCl3): 8,61 (шир.с, 1Н), 8,21 (шир.д, 1Н), 7,36-7,42 (м, 5Н), 5,03 (д, 1Н), 4,90 (д, 1Н), 3,98 (д, 1Н), 3,60-3,70 (м, 1Н), 3,48-3,52 (м, 2Н), 3,31-3,35 (м, 2Н), 3,04-3,12 (м, 2Н), 2,98 (т, 1Н), 2,26-2,30 (м, 1Н), 2,11 (шир.с, 2Н), 1,91-1,99 (м, 2Н), 1,59-1,61 (м, 1Н), 1,43 (с, 9Н).
Масс-спектрометрия: (М-1) = 486,3 для C24H33N5O6.
Степень чистоты "для HPLC": 98,89%.
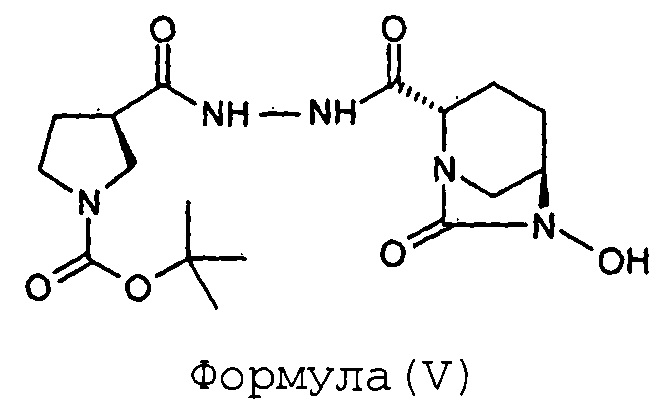
Стадия-2: Получение (2S,5R)-6-гидрокси-7-оксо-2-[((3R)-N-трет-бутоксикарбонилпирролидин-3-карбонил)гидразинокарбонил]-1,6-диазабицикло[3.2.1]октана (V):
Соединение (2S,5R)-6-бензилокси-7-оксо-2-[((3R)-N-трет-бутоксикарбонилпирролидин-3-карбонил)гидразинокарбонил]-1,6-диазабицикло[3.2.1]октан (IV, 87 г, 0,17 моль) растворяют в метаноле (0,87 л) с получением прозрачного раствора. К этому раствору добавляют катализатор на основе 10%-ного Pd на углеродном носителе (17 г, влажность 50%). Суспензию перемешивают в течение 3 часов под давлением водородной атмосферы 100 фунтов на квадратный дюйм (psi) при 35°С. Когда метод TLC (Система для TLC метанол: хлороформ 1:9) показывает завершение реакции, катализатор отфильтровывают через слой целлита при отсасывании под вакуумом. Слой целлита промывают метанолом (200 мл). Фильтрат упаривают под вакуумом при температуре ниже 40°С с получением сырого твердого вещества (2S,5R)-6-гидрокси-7-оксо-2-[((3R)-N-трет-бутоксикарбонилпирролидин-3-карбонил)гидразинокарбонил]-1,6-диазабицикло[3.2.1]октана (V) в количестве 72 граммов с количественным выходом. Этот продукт реакции, являющийся нестабильным, сразу же используют в следующей реакции.
Анализ:
ЯМР: (DMSO-d6): 9,70-9,90 (м, 2Н), 4,06-4,08 (м, 1Н), 3,76 (д, 1Н), 3,58 (шир.с, 1Н), 3,35-3,50 (м, 1Н), 3,10-3,40 (м, 4Н), 2,97 (шир.д, 2Н), 1,79-2,04 (м, 4Н), 1,73-1,81 (м, 1Н), 1,53-1,71 (м, 1Н), 1,37 (с, 9Н).
Масс-спектрометрия: (М-1): 396,2 для C17H27N5O6.
Степень чистоты "для HPLC": 90,99%.
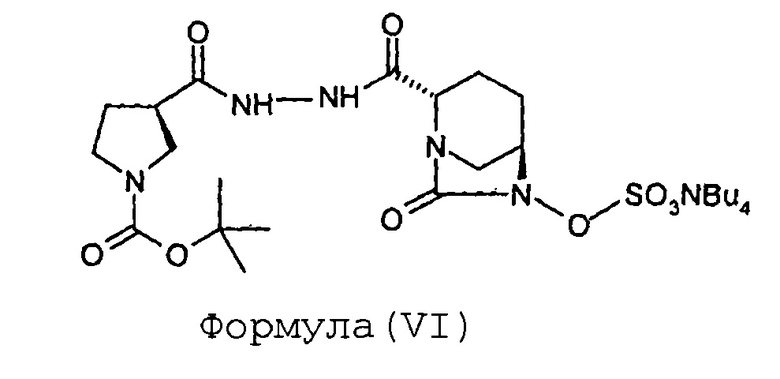
Стадия-3: Получение тетрабутиламмониевой соли (2S,5R)-6-сульфоокси-7-оксо-2-[((3R)-N-трет-бутоксикарбонилпирролидин-3-карбонил)гидразинокарбонил]-1,6-диазабицикло[3.2.1]октана (VI):
В раствор (2S,5R)-6-гидрокси-7-оксо-2-[((3R)-N-трет-бутоксикарбонилпирролидин-3-карбонил)гидразинокарбонил]-1,6-диазабицикло[3.2.1]октана (V, 67 г, 0,16 моль) в пиридине (0,67 л) загружают комплекс пиридин-триоксид серы (134 г, 0,84 моль) при перемешивании при 35°С. Реакционнную смесь перемешивают в течение 16 часов. Суспензию фильтруют через слой целлита и этот слой промывают дихлорметаном (500 мл). Фильтрат упаривают досуха при температуре ниже 40°С с предоставлением остатка. К остатку добавляют 0,5 М-ный водный раствор дигидрофосфата калия (1,7 л). Реакционную смесь перемешивают в течение 15 минут при 35°С и затем экстрагируют дихлорметаном (1 л × 2). Слои разделяют. К водному слою добавляют твердый гидросульфат тетрабутиламмония (51 г, 0,15 моль) и перемешивание продолжают в течение 2 часов при 35°С. Реакционную смесь экстрагируют дихлорметаном (0,7 л × 2). Слои разделяют. Объединенный органический слой упаривают под вакуумом при температуре ниже 40°С с получением тетрабутиламмониевой соли (2S,5R)-6-сульфоокси-7-оксо-2-[((3R)-N-трет-бутоксикарбонилпирролидин-3-карбонил)гидразинокарбонил]-1,6-диазабицикло[3.2.1]октана (VI) в виде твердого вещества белого цвета в количестве 106 г с выходом 87 %.
Анализ
ЯМР: (CDCl3): 8,63-8,70 (м, 2Н), 5,28 (с, 1Н), 4,23 (шир.с, 1Н), 3,97 (д, 1Н), 3,10-3,40 (м, 1Н), 3,49 (т, 2Н), 3,22-3,40 (м, 10Н), 3,09 (шир.с, 2Н), 2,28-2,33 (м, 1Н), 2,20-2,17 (м, 5Н), 1,80-1,90 (м, 2Н), 1,57-1,71 (м, 9Н), 1,33-1,46 (м, 18Н), 0,98 (т, 12Н).
Масс-спектрометрия: (М-1): 476,4 в форме свободной сульфоновой кислоты для C17H26N5O9S.N(C4H9)4.
Степень чистоты "для HPLC": 98,34%.
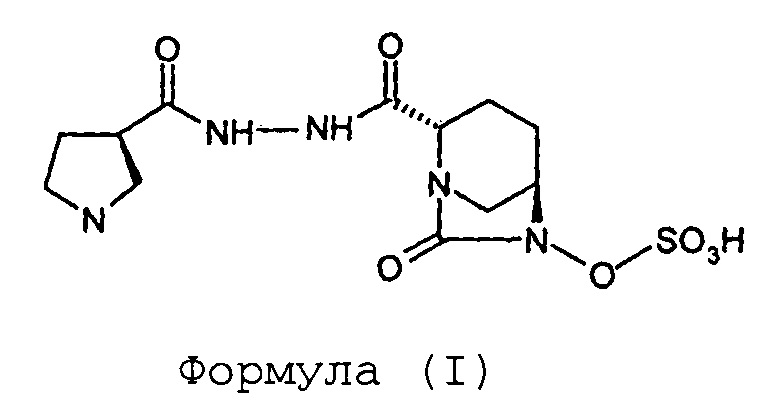
Стадия-4: Синтез (2S,5R)-7-оксо-6-сульфоокси-2-[((3R)-пирролидин-3-карбонил)гидразинокарбонил]-1,6-диазабицикло[3.2.1]октана (I):
Тетрабутиламмониевую соль (2S,5R)-6-сульфоокси-7-оксо-2-[((3R)-N-трет-бутоксикарбонилпирролидин-3-карбонил)гидразинокарбонил]-1,6-диазабицикло[3.2.1]октана (VI, 110 г, 0,15 моль) растворяют в дихлорметане (275 мл) и к прозрачному раствору медленно добавляют трифторуксусную кислоту (275 мл) при температуре от 0 до 5°С. Реакционную смесь перемешивают при температуре от 0 до 5°С в течение дополнительного 1 часа. Растворитель и избыточную трифторуксусную кислоту выпаривают под вакуумом при температуре ниже 40°С до объема, составляющего приблизительно 1/3 от первоначального объема, с обеспечением маслянистого остатка бледно-желтого цвета. Маслянистый остаток перемешивают вместе с диэтиловым эфиром (1,0 л) с предоставлением суспензии. Осадок отфильтровывают при отсасывании под вакуумом, переносят в круглодонную колбу и опять перемешивают с диэтиловым эфиром (1,0 л) в течение 30 минут. Суспензию фильтруют при отсасывании под вакуумом с предоставлением сырого твердого вещества. Сырое твердое вещество загружают в круглодонную колбу и к этому добавляют дихлорметан (1,0 л). Значение рН для суспензии корректируют с доведением до 7,0-7,5 путем добавления триэтиламина. Получающуюся в результате суспензию фильтруют при отсасывании под вакуумом и влажный фильтрационный осадок промывают дихлорметаном (200 мл) с предоставлением сырого твердого вещества. Сырое твердое вещество сушат под вакуумом при температуре ниже 40°С с предоставлением 61 грамма сырой массы. Сырую массу растворяют в воде (61 мл) при перемешивании, и к прозрачному раствору добавляют изопропиловый спирт (580 мл). Суспензию перемешивают в течение 70 часов и фильтруют при отсасывании под вакуумом. Влажный фильтрационный осадок промывают изопропиловым спиртом (100 мл) и сушат под вакуумом при температуре ниже 40°С с получением кристаллического (2S,5R)-7-оксо-6-сульфоокси-2-[((3R)-пирролидин-3-карбонил)гидразинокарбонил]-1,6-диазабицикло[3.2.1]октана (I) в количестве 33 грамм с выходом 60 %.
Анализ:
ЯМР: (DMSO-d6)=9,25 (шир.с, 3Н), 4,00 (шир.с, 1Н), 3,82 (д, 1Н), 3,22-3,37 (м, 5Н), 3,15-3,22 (м, 3Н), 3,05-3,12 (м, 2Н), 2,95-3,05 (м, 1Н), 2,12-2,22 (м, 1Н), 1,94-2,08 (м, 2Н), 1,82-1,90 (шир.с, 1Н), 1,66-1,78 (м, 1Н), 1,54-1,64 (м, 1Н).
Масс-спектрометрия: (М-1): 376,3 для C12H19N5O7S.
Степень чистоты "для HPLC": 96,64%.
Удельное вращение: [α]25D = -47,5° (около 0,5, вода).
Порошковая рентгеновская дифрактограмма, содержащая пик при (значениях угла рассеяния 2 тета): 7,03 (±0,2), 9,17 (±0,2), 13,52 (±0,2), 15,19 (±0,2), 16,28 (±0,2), 16,92 (±0,2), 18,30 (±0,2), 19,10 (±0,2), 20,49 (±0,2), 21,62 (±0,2), 22,01 (±0,2), 22,77 (±0,2), 23,72 (±0,2), 25,05 (±0,2), 25,64 (±0,2), 27,04 (±0,2), 27,96 (±0,2), 29,41 (±0,2), 30,21 (±0,2), 35,68 (±0,2), 36,75 (±0,2) и 37,89 (±0,2) градусов.
Обычный рентгеновский анализ выполняют следующим образом. Пропускают вещество для испытаний через стандартное сито № 100 британской гранулометрической шкалы (BSS) или мягко измельчают его с помощью ступки и пестика. Равномерно помещают вещество для испытаний на держатель для образца, имеющий на одной стороне полостную поверхность, спрессовывают образец и срезают тонкую равномерную пленку с использованием стеклянной пластинки таким образом, чтобы поверхность образца могла быть гладкой и ровной. Записывают рентгеновскую дифрактограмму с применением следующих параметров прибора.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ (2S,5R)-7-ОКСО-6-СУЛЬФООКСИ-2-[((3R)-ПИПЕРИДИН-3-КАРБОНИЛ)-ГИДРАЗИНОКАРБОНИЛ]-1,6-ДИАЗАБИЦИКЛО[3.2.1]ОКТАНА | 2013 |
|
RU2627700C2 |
| СПОСОБ ПОЛУЧЕНИЯ (2S, 5R)-МОНО-{ [(4-АМИНОПИПЕРИДИН-4-ИЛ)КАРБОНИЛ]-7-ОКСО-1, 6-ДИАЗАБИЦИКЛО[3.2.1]ОКТ-6-ИЛ} ОВОГО СЛОЖНОГО ЭФИРА СЕРНОЙ КИСЛОТЫ | 2013 |
|
RU2621051C2 |
| НОВЫЙ ИНГИБИТОР бета-ЛАКТАМАЗЫ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2013 |
|
RU2693898C2 |
| НОВЫЙ ИНГИБИТОР β-ЛАКТАМАЗЫ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2019 |
|
RU2800050C2 |
| ИНГИБИТОРЫ БЕТА-ЛАКТАМАЗ | 2009 |
|
RU2445314C9 |
| СПОСОБ ПОЛУЧЕНИЯ НАТРИЕВОЙ СОЛИ (2S,5R)-2-КАРБОКСАМИДО-7-ОКСО-6-СУЛЬФООКСИ-1,6-ДИАЗАБИЦИКЛО[3.2.1]ОКТАНА | 2013 |
|
RU2632192C2 |
| СПОСОБ ПОЛУЧЕНИЯ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ, ВКЛЮЧАЯ ТРАНС-7-ОКСО-6-(СУЛЬФОКСИ)-1,6-ДИАЗАБИЦИКЛО[3.2.1]ОКТАН-2-КАРБОКСАМИД И ЕГО СОЛИ | 2012 |
|
RU2769076C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО ДИАЗАБИЦИКЛООКТАНА И ЕГО ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ | 2014 |
|
RU2719480C2 |
| (2S,5R)-5-[(бензилокси)амино]пиперидин-2-карбоксамид | 2012 |
|
RU2610091C2 |
| ПРОИЗВОДНЫЕ 1,6-ДИАЗАБИЦИКЛО[3.2.1]ОКТАН-7-ОНА И ИХ ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ БАКТЕРИАЛЬНЫХ ИНФЕКЦИЙ | 2013 |
|
RU2614418C2 |

Изобретение относится к области органической химии, а именно к способу получения соединения формулы (I) ((2S,5R)-7-оксо-6-сульфоокси-2-[((3R)-пирролидин-3-карбонил)гидразинокарбонил]-1,6-диазабицикло[3.2.1]октана), включающий (а) проведение реакции между соединением Формулы (II) и соединением Формулы (III) в присутствии воды, взятой в качестве растворителя, с получением соединения Формулы (IV); (b) гидрогенолиз соединения Формулы (IV) с получением соединения Формулы (V); (с) сульфонирование соединения Формулы (V) с получением соединения Формулы (VI); и (d) превращение соединения Формулы (VI) в соединение Формулы (I). Также изобретение относится к способу получения соединения Формулы (I) в кристаллической форме. Технический результат: разработан новый способ получения соединения формулы (I), отличающийся простотой операций, более безопасными условиями, высоким выходом целевого продукта. 2 н. и 8 з.п. ф-лы, 1 ил., 2 пр.

 ,
, 
1. Способ получения соединения Формулы (I), включающий:
(а) проведение реакции между соединением Формулы (II) и соединением Формулы (III) в присутствии воды, взятой в качестве растворителя, с получением соединения Формулы (IV);

(b) гидрогенолиз соединения Формулы (IV) с получением соединения Формулы (V);
(с) сульфонирование соединения Формулы (V) с получением соединения Формулы (VI);
и
(d) превращение соединения Формулы (VI) в соединение Формулы (I).
2. Способ по п.1, где реакцию соединения Формулы (II) с соединением Формулы (III) с получением соединения Формулы (IV) проводят в присутствии 1-гидроксибензотриазола и гидрохлорида 1-этил-3-(3-диметиламинопропил)карбодиимида.
3. Способ по п.1, где гидрогенолиз соединения Формулы (IV) с получением соединения Формулы (V) проводят в присутствии катализатора на основе переходного металла и источника водорода.
4. Способ по п.3, где катализатор на основе переходного металла представляет собой палладий на углеродном носителе, а источник водорода представляет собой газ водород.
5. Способ по п.1, где сульфонирование соединения Формулы (V) с получением соединения Формулы (VI) выполняют путем проведения реакции между соединением Формулы (V) и комплексом триоксид серы-пиридин, с последующей обработкой гидросульфатом тетрабутиламмония.
6. Способ по п.1, где соединение Формулы (VI) превращают в соединение Формулы (I) путем проведения реакции между соединением Формулы (VI) и трифторуксусной кислотой.
7. Способ получения соединения Формулы (I) в кристаллической форме, включающий:
(a) растворение в воде соединения Формулы (I), полученного способом по п.1, с получением прозрачного раствора;
(b) добавление изопропилового спирта к прозрачному раствору, полученному на стадии (а) при перемешивании; и
(c) выделение соединения Формулы (I) в кристаллической форме.
8. Способ по п.7, где полученное соединение Формулы (I) имеет порошковую рентгеновскую дифрактограмму, содержащую пик, выбранный из группы, состоящей из 7,03 (±0,2), 9,17 (±0,2), 13,52 (±0,2), 15,19 (±0,2), 16,28 (±0,2), 16,92 (±0,2), 18,30 (±0,2), 19,10 (±0,2), 20,49 (±0,2), 21,62 (±0,2), 22,01 (±0,2), 22,77 (±0,2), 23,72 (±0,2), 25,05 (±0,2), 25,64 (±0,2), 27,04 (±0,2), 27,96 (±0,2), 29,41 (±0,2), 30,21 (±0,2), 35,68 (+0,2), 36,75 (±0,2), и 37, 89 (±0,2) градусов 2 тета.
9. Способ по п.7, где полученное соединение Формулы (I) имеет порошковую рентгеновскую дифрактограмму, содержащую пик, выбранный из группы, состоящей из 7,03 (±0,2), 9,17 (±0,2), 15,19 (±0,2), 16,92 (±0,2), 18,30 (±0,2), 19,10 (±0,2), 22,77 (±0,2), и 23,72 (±0,2) градусов 2 тета.
10. Способ по п.7, где полученное соединение Формулы (I) имеет порошковую рентгеновскую дифрактограмму по существу такую же, как показана на Фигуре 1.
| Многоступенчатая активно-реактивная турбина | 1924 |
|
SU2013A1 |
| Колосоуборка | 1923 |
|
SU2009A1 |
| ДИАЗАБИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1994 |
|
RU2124014C1 |

