Родственные патентные заявки
По настоящей заявке испрашивается приоритет согласно индийской патентной заявке № 718/MUM/2013, поданной 8 марта 2013 года, содержание которой полностью включено в настоящее описание посредством ссылки, как если бы было в полном объеме описано в настоящем описании. Все ссылки, включая патенты, патентные заявки и литературу, процитированные в описании, полностью включены в настоящее описание посредством ссылки.
Область техники
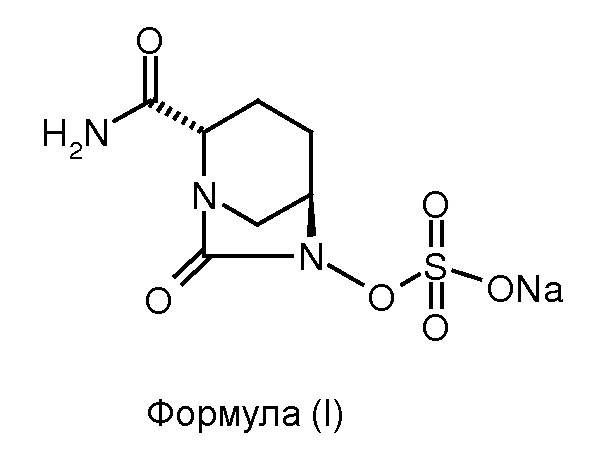
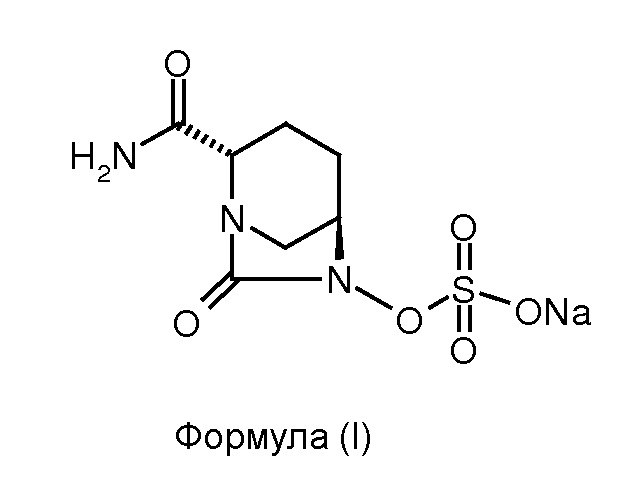
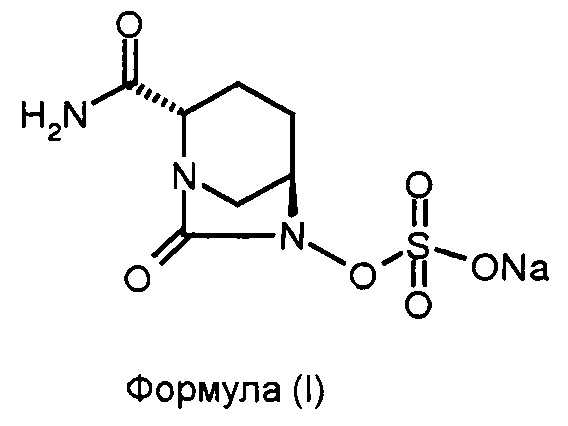
Изобретение относится к способу получения натриевой соли (2S,5R)-2-карбоксамидо-7-оксо-6-сульфоокси-1,6-диазабицикло[3.2.1]октана.
Уровень техники
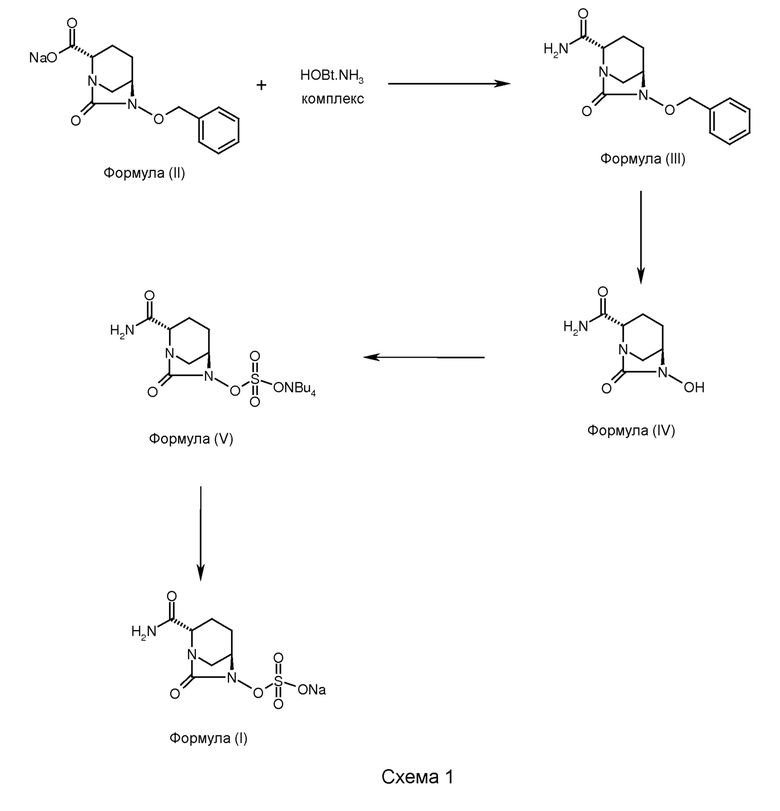
Соединение формулы (I), известное в химии как натриевая соль (2S,5R)-2-карбоксамидо-7-оксо-6-сульфоокси-1,6-диазабицикло[3.2.1]октана, обладает антибактериальными свойствами. Соединение формулы (I) также известно как «Авибактам» или «NXL-104» и раскрыто в патенте США № 7112592.
Сущность изобретения
В первом общем аспекте предоставляется способ получения соединения формулы (I), включающий
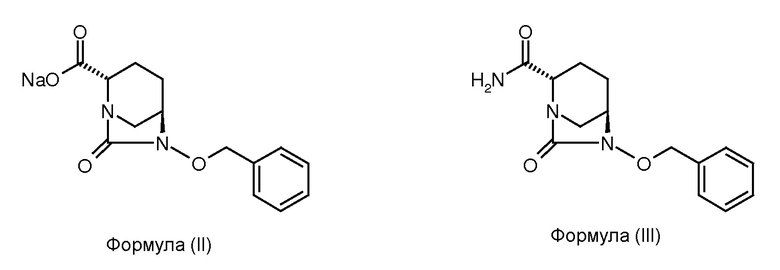
(а) взаимодействие соединения формулы (II) с амидирующим агентом для получения соединения формулы (III);
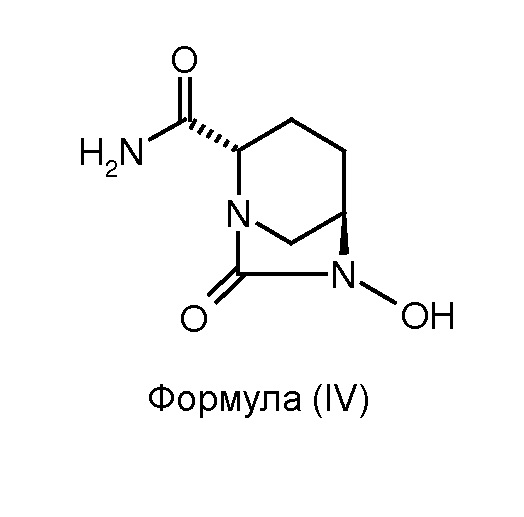
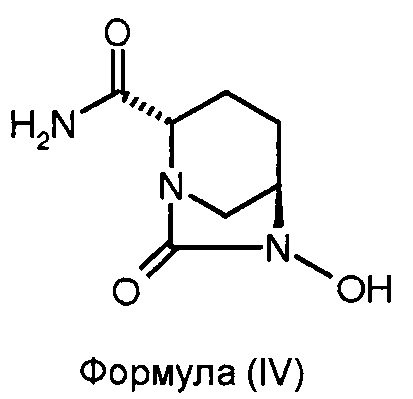
(b) гидрогенолиз соединения формулы (III) для получения соединения формулы (IV);

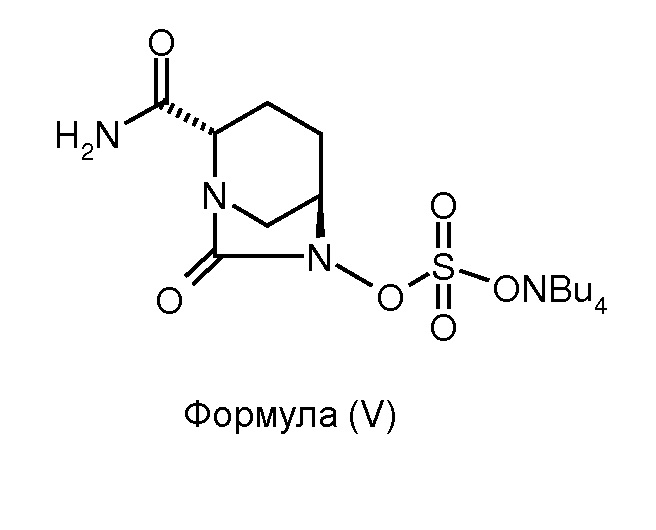
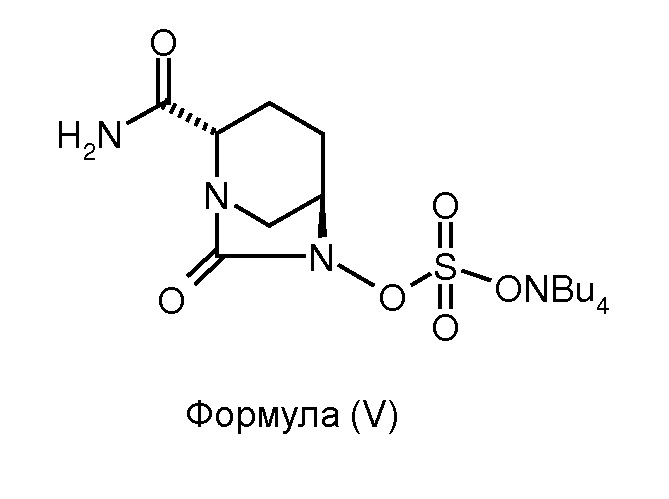
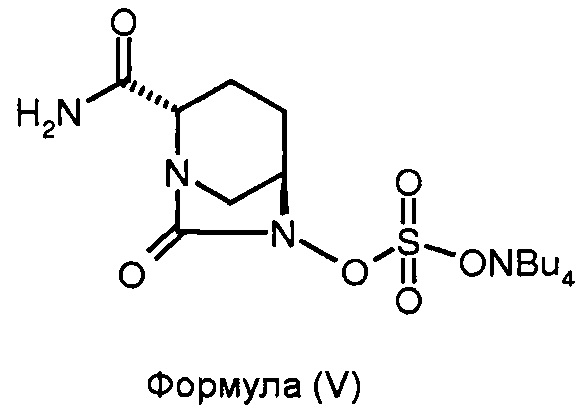
(c) сульфонирование соединения формулы (IV) для получения соединения формулы (V); и
(d) преобразование соединения формулы (V) до соединения формулы (I).
Детали одного или более вариантов осуществления изобретения изложены в описании ниже. Другие признаки, цели и преимущества изобретения станут очевидными из следующего описания, включая формулу изобретения.
Подробное описание изобретения
Ниже приводится ссылка на примеры осуществления изобретения, и для описания подобных примеров в настоящем описании будет использоваться специфичный язык. Однако следует понимать, что ограничение объема изобретения таким образом не предполагается. Изменения и дальнейшие модификации признаков изобретения представлены в настоящем описании, а дополнительные заявки на принципы изобретения, представленные в настоящем описании, которые будут использоваться специалистами в соответствующей области техники, и владение сущностью данного изобретения, не считаются объектом изобретения. Все ссылки, включая патенты, патентные заявки и литературные цитаты в настоящем описании, явным образом включены в настоящее описание посредством ссылки как если бы было в полном объеме описано в настоящем описании.
Термин «HOBt» при использовании в настоящем описании относится к 1-гидроксибензотриазолу.
Термин «EDC» при использовании в настоящем описании относится к 1-этил-3-(3-диметиламинопропил)карбодиимиду.
В первом общем аспекте предоставляется способ получения соединения формулы (I), включающий
(а) взаимодействие соединения формулы (II) с амидирующим агентом для получения соединения формулы (III);

(b) гидрогенолиз соединения формулы (III) для получения соединения формулы (IV);
(c) сульфонирование соединения формулы (IV) для получения соединения формулы (V); и
(d) преобразование соединения формулы (V) до соединения формулы (I).
Соединение формулы (III) получено в результате взаимодействия соединения формулы (II) с подходящим амидирующим агентом. В некоторых вариантах осуществления изобретения амидирующий агент содержит 1-этил-3-(3-диметиламинопропил)карбодиимид и комплекс 1-гидроксибензотриазол:аммиак. В некоторых других вариантах осуществления изобретения соединение формулы (III) получено в результате взаимодействия соединения формулы (II) с комплексом 1-гидроксибензотриазол:аммиак в присутствии 1-этил-3-(3-диметиламинопропил)карбодиимида гидрохлорида и 1-гидроксибензотриазола. Реакцию амидирования можно проводить в подходящем растворителе. В некоторых вариантах осуществления изобретения реакцию амидирования проводят в воде в качестве реакционного растворителя. В некоторых вариантах осуществления изобретения соединение формулы (III) получено в результате взаимодействия соединения формулы (II) с 1-этил-3-(3-диметиламинопропил)карбодиимида гидрохлоридом и 1-гидроксибензотриазолом в присутствии аммиачного раствора.
Соединение формулы (IV) получено в результате гидрогенолиза соединения формулы (III). Реакцию гидрогенолиза можно проводить с использованием подходящего агента гидрогенолиза. В некоторых вариантах осуществления изобретения гидрогенолиз соединения формулы (III) для получения соединения формулы (IV) проводят в присутствии катализатора переходного металла и источника водорода. В некоторых других вариантах осуществления изобретения катализатором переходного металла является палладий на углеродном носителе, а источником водорода является газообразный водород. В некоторых других вариантах осуществления изобретения реакцию гидрогенолиза проводят в присутствии подходящего растворителя, такого как, например, смесь N,N-диметилформамида и дихлорметана (объемное соотношение 1:1). В некоторых вариантах осуществления изобретения гидрогенолиз соединения формулы (III) для получения соединения формулы (IV) проводят с использованием 10%-ного палладиевого катализатора на углеродном носителе в присутствии газообразного водорода в смеси N,N-диметилформамид:дихлорметан (объемное соотношение 1:1).
Соединение формулы (V) получено в результате сульфонирования соединения формулы (IV). Реакцию сульфонирования можно проводить в присутствии подходящего растворителя. В некоторых вариантах осуществления изобретения сульфонирование соединения формулы (IV) для получения соединения формулы (V) проводят путем взаимодействия соединения формулы (IV) с комплексом триоксид серы - N,N-диметилформамид с последующей обработкой 10%-ным водным тетрабутиламмония ацетатом.
Соединение формулы (V) преобразовано в соединение формулы (I) в присутствии подходящего реагента. В некоторых вариантах осуществления изобретения соединение формулы (V) преобразовано в соединение формулы (I) в результате взаимодействия соединения формулы (V) с натрий-2-этилгексаноатом.
В некоторых вариантах осуществления изобретения соединение формулы (I) получено с использованием способа, описанного в схеме 1.
В другом общем аспекте способ согласно результатам изобретения в получении соединения формулы (I), характеризующегося чистотой по меньшей мере 97% при определении с помощью ВЭЖХ.
В некоторых вариантах осуществления изобретения предоставляется соединение формулы (I), характеризующееся порошковой рентгенограммой XRPD, включающей пик, выбранный из группы, состоящей из 8,69 (±0,2), 9,65 (±0,2), 11,22 (±0,2), 12,44 (±0,2), 13,01 (±0,2), 16,48 (±0,2), 17,48 (±0,2), 18,58 (±0,2), 19,35 (±0,2), 20,89 (±0,2), 22,27 (±0,2), 25,03 (±0,2), 26,07 (±0,2), 28,14 (±0,2), 29,74 (±0,2), 34,28 (±0,2), 36,01 (±0,2) и 37,18 (±0,2) градусов 2-Тета.
В некоторых вариантах осуществления изобретения соединение формулы (I) характеризуется порошковой рентгенограммой XRPD, практически совпадающей с изображенным на фигуре 1.
Для специалиста в данной области техники очевидно, что варьирование замен и модификаций может иметь место в изобретении, раскрытом в настоящем описании, без отклонения от объема и основного положения изобретения. Например, специалисты в данной области техники признают, что изобретение можно осуществить с использованием множества различных соединений, соблюдая описанные общие описания.
Примеры
Следующие примеры иллюстрируют варианты осуществления изобретения, которые в настоящее время наиболее известны. Однако следует понимать, что следующие примеры являются только показательной или иллюстративной заявкой основных принципов настоящего изобретения. Множество модификаций и альтернативных композиций, способов и систем может быть разработано специалистами в данной области техники без отступления от основного направления и объема настоящего изобретения. Прилагаемая формула изобретения направлена на обеспечение таких модификаций и композиций. Таким образом, ввиду того, что настоящее изобретение и его особенности описаны выше, следующие примеры предоставляют дополнительно деталь в связи с тем, что в настоящее время они считаются наиболее осуществимыми и предпочтительными вариантами осуществления изобретения.
Пример 1
Получение натриевой соли (2S,5R)-серной кислоты моно-2-карбоксамидо-7-оксо-1,6-диазабицикло[3.2.1]октана
Стадия 1: Получение (2S,5R)-2-карбоксамидо-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]октана:
Способ 1
Исходное соединение ((2S,5R)-натрия 6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксилат; соединение формулы (II)) получено согласно методике, раскрытой в индийской патентной заявки № 699/MUM/2013. 100 мл круглодонную колбу, оснащенную магнитной мешалкой, наполнили (2S,5R)-натрия 6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксилатом (10,0 г, 0,033 моль), затем свежеприготовленным HOBt. аммиачным комплексом (10,0 г, 0,066 моль), EDC гидрохлоридом (9,62 г, 0,050 моль) и 1-гидроксибензотриазолом (4,51 г, 0,033 моль). К данной смеси твердых фаз добавили воду (30 мл) при приблизительно 35°С и включили мешалку. Выпадение осадка произошло через 30 минут. Реакционную смесь перемешивали дополнительно 20 часов при приблизительно 35°С. Дихлорметан (150 мл) добавили к суспензии, и реакционную массу перемешивали 10 минут. Слои разделили. Водный слой промыли дополнительно дихлорметаном (50 мл). Смешанный органический слой выпарили под вакуумом для получения осадка (21 г). Осадок перемешивали с ацетоном (21 мл) 30 минут и профильтровали под вакуумом для получения (2S,5R)-2-карбоксамидо-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]октана в виде белого осадка в количестве 5,5 г с выходом 60% после осушения под вакуумом при приблизительно 45°С.
Анализ
Н1 ЯМР (ДМСО-d6)
7,35-7,45 (м, 6H), 7,25 (шир.с, 1H), 4,89-4,96 (дд, 2H), 3,68 (д, 1H), 3,62 (с, 1H), 2,90 (с, 2H), 2,04-2,079 (м, 1H), 1,70-1,83 (м, 1H), 1,61-1,66 (м, 2H).
Масса (ES+) C14H17N3O3: 276,1 (М+1)
Чистота: 93,95% при измерении с помощью ВЭЖХ
Удельное вращение: [α]25D - 8,51° (c 0,5%, CHCl3)
Способ 2
В другом варианте упомянутое выше соединение получено с использованием следующего способа. 50 мл круглодонную колбу, оснащенную магнитной мешалкой, наполнили раствором (2S,5R)-натрия 6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксилата (1г, 0,003 моль) в воде (15 мл), затем EDC гидрохлоридом (1 г, 0,005 моль) и 1-гидроксибензотриазолом (0,39 г, 0,003 моль) при 35°С при перемешивании. Реакционную массу перемешивали 1 час для получения белой суспензии. На данном этапе добавили водный раствор аммиака (2 мл, отношение веса к объему 40%) при перемешивании. Реакционную смесь перемешивали дополнительно 5 часов. Суспензию отфильтровали, промыли дополнительно водой (10 мл) для получения (2S,5R)-2-карбоксамидо-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1] после осушения под вакуумом при 45°С в количестве 0,21 г.
Стадия 2: Получение тетрабутиламмониевой соли (2S,5R)-2-карбоксамидо-6-сульфоокси-7-оксо-1,6-диазабицикло[3.2.1]октана.
Бутыль для смешивания аппарата Парра наполнили (2S,5R)-2-карбоксамидо-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]октаном (7,0 г, 0,025 моль), затем 1:1 смесью N,N-диметилформамида и дихлорметана (35 мл:35 мл). К чистому раствору добавили 10%-ный палладий на углеродном носителе (1,75 г), и давление водорода подняли до 50 psi. Суспензию перемешивали 3 часа при 35°С. Катализатор удалили путем фильтрования реакционной смеси через слой целлита. Слой катализатора промыли дихлорметаном (30 мл). Смешанный фильтрат выпарили под вакуумом при температуре ниже 40°С для получения маслянистого осадка. Маслянистый осадок (4,72 г) растворили в N,N-диметилформамиде (35 мл) и к чистому раствору добавили комплекс триоксид серы. DMF при 10°С при перемешивании в одном сосуде. Смесь перемешивали при 35°С дополнительно 2 часа. Когда ТСХ-анализ показал полное превращение, 10%-ный водный раствор тетрабутиламмония ацетата (9,44 г, 0,031 моль, в 30 мл воды) добавили при перемешивании, и реакционную смесь перемешивали в течение ночи и затем подвергли высокой вакуумной дистилляции на роторном испарителе при температуре, не превышающей 40°С для получения осадка. Ксилен (50 мл) добавили к осадку и аналогичным образом выпарили для уничтожения следов DMF. Сухой осадок, полученный таким образом, перемешали с водой (70 мл) и экстрагировали дихлорметаном (70 мл × 2). Смешанный органический экстракт осушили сульфатом натрия, и растворитель выпарили под вакуумом, при температуре ниже 40°С для получения маслянистого осадка в количестве 7 г в виде неочищенного продукта. Его перемешивали с метилизобутилкетоном (21 мл) 30 минут при приблизительно 35°С для получения белого осадка в количестве 5,9 г в виде тетрабутиламмониевой соли (2S,5R)-2-карбоксамидо-6-сульфоокси-7-оксо-1,6-диазабицикло[3.2.1]октана в чистом виде с выходом 46%.
Анализ
ЯМР (CDCl3)
6,63 (с, 1H), 5,48 (с, 1H), 4,34 (шир. с, 1H), 3,90 (д, 1H), 3,27-3,40 (м, 9H), 2,84 (д, 1H), 2,38 (дд, 1H), 2,21-2,20 (м, 1H), 1,60-1,71 (м, 12H), 1,40-1,50 (м, 8H), 1,00 (т, 12H).
Масса (ES-) C7H10N306S. N(C4H9)4=264,0 (M-1) в виде свободной сульфоновой кислоты.
Чистота: 98,98% при измерении с помощью ВЭЖХ
Удельное вращение: [α]25D - 30,99° (c 0,5%, MeOH)
Стадия 3: Синтез натриевой соли (2S,5R)-2-карбоксамидо-6-сульфоокси-7-оксо-1,6-диазабицикло[3.2.1]октана.
100 мл круглодонную колбу, оснащенную магнитной мешалкой, наполнили тетрабутиламмониевой соли (2S,5R)-2-карбоксамидо-6-сульфоокси-7-оксо-1,6-диазабицикло[3.2.1]октаном (5,5 г, 0,0108 моль), затем этанолом (28 мл) для получения чистого раствора при перемешивании при приблизительно 35°С. К реакционной смеси добавили раствор натрий-2-этилгексаноата (3,6 г, 0,021 моль), растворенного в этаноле (28 мл) в одном сосуде при перемешивании для обеспечения выпадения осадка. Суспензию перемешивали дополнительно 2 часа для осуществления полного выпадения осадка при приблизительно 35°С. Реакционную смесь профильтровали под вакуумом, и отфильтрованный осадок промыли ацетоном (30 мл ×2). Отфильтрованный осадок осушили при 40°С под вакуумом для получения натриевой соли (2S,5R)-2-карбоксамидо-6-сульфоокси-7-оксо-1,6-диазабицикло[3.2.1]октана в виде белого осадка в количестве 2,6 г с выходом 83%.
Анализ
H1 ЯМР (ДМСО-d6)
7,39 (с, 1H), 7,24 (с, 1H), 3,98 (с, 1H), 3,68 (д, 1H), 3,02 (д, 1H), 2,92 (д, 1H), 2,00-2,10 (м, 1H), 2,80-2,90 (м, 1H), 1,55-1,70 (м, 2H).
Масса (ES-) C7H10N306SNa=264,0 (M-1) в качестве свободной сульфоновой кислоты.
Чистота: 97,98% при измерении с помощью ВЭЖХ
Удельное вращение: [α]25D - 49,37° (c 0,5%, вода)
Порошковая рентгенограмма XRPD: (градусы 2-Тета):
8,69 (±0,2), 9,65 (±0,2), 11,22 (±0,2), 12,44 (±0,2), 13,01 (±0,2), 16,48 (±0,2), 17,48 (±0,2), 18,58 (±0,2), 19,35 (±0,2), 20,89 (±0,2), 22,27 (±0,2), 25,03 (±0,2), 26,07 (±0,2), 28,14 (±0,2), 29,74 (±0,2), 34,28 (±0,2), 36,01 (±0,2) и 37,18 (±0,2).
Стандартный рентгенодифракционный анализ провели следующим образом. Исследуемый материал проносят через фильтр #100 BSS или осторожно толкут в ступке пестом. Исследуемый материал наносят равномерно на держатель для образца, имеющий неровную поверхность с одной стороны, спрессовывают образец и нарезают на тонкие однородные пленки с использованием стеклянной пластинки таким образом, чтобы поверхность образца была гладкая и выровненная. Регистрация порошковой рентгенограммы XRPD с использованием следующих параметров прибора.
Прибор: рентгеновский дифрактометр (PANalytical, модель X'Pert Pro MPD)
Целевой источник: Cu k (α)
Противорассеивающие щели (падающего луча света): 1°
Настраиваемые дивергирующие щели: 10 мм (фиксированно)
Противорассеивающие щели (отклоненного луча): 5,5 мм
Ширина шага: 0,02°
Напряжение: 40 кВ
Сила тока: 40 мА
Время шага: 30 с
Диапазон сканирования: от 3 до 40°
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ (2S,5R)-7-ОКСО-6-СУЛЬФООКСИ-2-[((3R)-ПИПЕРИДИН-3-КАРБОНИЛ)-ГИДРАЗИНОКАРБОНИЛ]-1,6-ДИАЗАБИЦИКЛО[3.2.1]ОКТАНА | 2013 |
|
RU2627700C2 |
| СПОСОБ ПОЛУЧЕНИЯ (2S,5R)-7-ОКСО-6-СУЛЬФООКСИ-2-[((3R)-ПИРРОЛИДИН-3-КАРБОНИЛ)ГИДРАЗИНОКАРБОНИЛ]-1,6-ДИАЗАБИЦИКЛО[3.2.1]ОКТАНА | 2013 |
|
RU2625304C2 |
| СПОСОБ ПОЛУЧЕНИЯ (2S, 5R)-МОНО-{ [(4-АМИНОПИПЕРИДИН-4-ИЛ)КАРБОНИЛ]-7-ОКСО-1, 6-ДИАЗАБИЦИКЛО[3.2.1]ОКТ-6-ИЛ} ОВОГО СЛОЖНОГО ЭФИРА СЕРНОЙ КИСЛОТЫ | 2013 |
|
RU2621051C2 |
| НОВЫЙ ИНГИБИТОР бета-ЛАКТАМАЗЫ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2013 |
|
RU2693898C2 |
| НОВЫЙ ИНГИБИТОР β-ЛАКТАМАЗЫ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2019 |
|
RU2800050C2 |
| ИНГИБИТОРЫ БЕТА-ЛАКТАМАЗ | 2009 |
|
RU2445314C9 |
| СПОСОБ ПОЛУЧЕНИЯ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ, ВКЛЮЧАЯ ТРАНС-7-ОКСО-6-(СУЛЬФОКСИ)-1,6-ДИАЗАБИЦИКЛО[3.2.1]ОКТАН-2-КАРБОКСАМИД И ЕГО СОЛИ | 2012 |
|
RU2769076C2 |
| (2S,5R)-5-[(бензилокси)амино]пиперидин-2-карбоксамид | 2012 |
|
RU2610091C2 |
| АЗОТСОДЕРЖАЩИЕ БИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ | 2020 |
|
RU2835672C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО ДИАЗАБИЦИКЛООКТАНА И ЕГО ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ | 2014 |
|
RU2719480C2 |

Изобретение относится к области органической химии, а именно к способу получения натриевой соли (2S,5R)-2-карбоксамидо-7-оксо-6-сульфоокси-1,6-диазабицикло[3.2.1]октана (формулы (I)), включающий: (а) взаимодействие соединения формулы (II) с комплексом 1-гидроксибензотриазол:аммиак в присутствии воды в качестве растворителя для получения соединения формулы (III); (b) гидрогенолиз соединения формулы (III) для получения соединения формулы (IV); (с) сульфонирование соединения формулы (IV) для получения соединения формулы (V) и (d) преобразование соединения формулы (V) до соединения формулы (I). Технический результат: разработан новый способ получения соединения формулы (I), отличающийся простотой реализации, низкой пожароопасностью. 6 з.п. ф-лы, 1 ил., 1 пр.
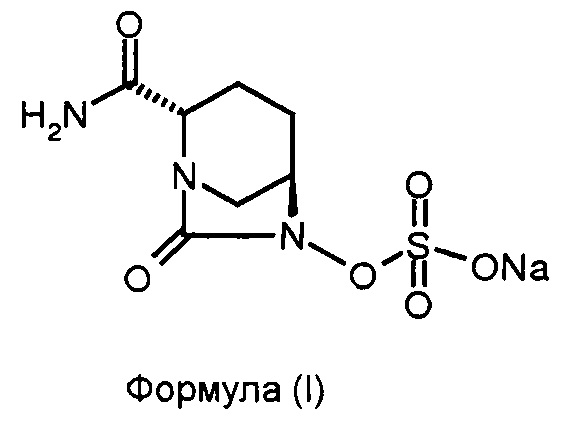


1. Способ получения соединения формулы (I)
 ,
,
включающий:
(а) взаимодействие соединения формулы (II) с комплексом 1-гидроксибензотриазол:аммиак в присутствии воды в качестве растворителя для получения соединения формулы (III);

(b) гидрогенолиз соединения формулы (III) для получения соединения формулы (IV);
(с) сульфонирование соединения формулы (IV) для получения соединения формулы (V); и
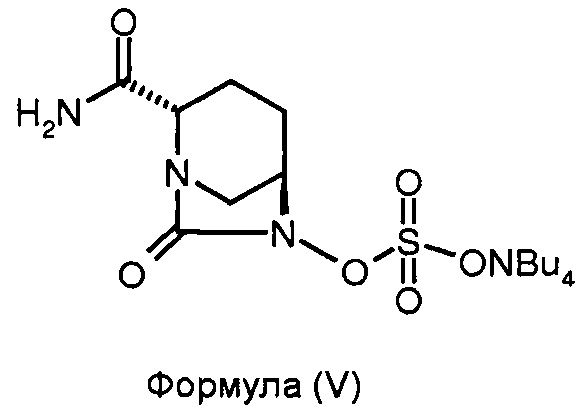
(d) преобразование соединения формулы (V) до соединения формулы (I).
2. Способ по п.1, где реакция на стадии (а) протекает в присутствии 1-этил-3-(3-диметиламинопропил)карбодиимида и гидроксибензотриазола.
3. Способ по п.1, где гидрогенолиз соединения формулы (III) для получения соединения формулы (IV) проводят в присутствии катализатора переходного металла и источника водорода.
4. Способ по п.3, где катализатором переходного металла является палладий на углеродной подложке, и источником водорода является газообразный водород.
5. Способ по п.3 или 4, где гидрогенолиз соединения формулы (III) для получения соединения формулы (IV) проводят в присутствии N,N-диметилформамида и дихлорметана (объемное соотношение 1:1) в качестве реакционного растворителя.
6. Способ по п.1, где сульфонирование соединения формулы (IV) для получения соединения формулы (V) проводят путем взаимодействия соединения формулы (IV) с комплексом триоксид серы - N,N-диметилформамид с последующей обработкой 10%-ным водным тетрабутиламмония ацетатом.
7. Способ по п.1, где соединение формулы (V) преобразуют в соединение формулы (I) путем взаимодействия соединения формулы (V) с натрий-2-этилгексаноатом.
| Соединения формул (I) и (A), фармацевтическая композиция, лекарственное средство, применение и способ получения соединения формулы (I) | 2018 |
|
RU2822758C2 |
| Устройство для подачи пара в тепловые аккумуляторы типа аккумуляторов Рутса | 1926 |
|
SU4920A1 |
| Колосоуборка | 1923 |
|
SU2009A1 |

