ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к ингибиторам фермента фосфодиэстеразы 4 (PDE4). Более конкретно, изобретение относится к соединениям, которые являются производными 1-фенил-2-пиридинилалкильных спиртов, к способам получения таких соединений, к композициям, содержащим их, и к их терапевтическому применению.
УРОВЕНЬ ТЕХНИКИ
Обструкция дыхательных путей характеризует рядом серьезных респираторных заболеваний, включая астму и хроническое обструктивное заболевание легких (ХОЗЛ). События, приводящие к обструкции дыхательных путей, включают отек стенок дыхательных путей, повышенную продукцию слизи и воспаление.
Лекарственные средства для лечения респираторных заболеваний, таких как астма и ХОЗЛ, в настоящее время вводят путем ингаляции. Одно из преимуществ ингаляторного пути по сравнению с системным путем введения - это возможность доставки лекарственного средства напрямую к месту действия, снижая системные побочные эффекты, таким образом, получая более быстрый клинический ответ и высокий терапевтический коэффициент.
Кортикостероиды, вводимые путем ингаляции, в настоящее время являются поддерживающей терапией для астмы и вместе с бета2- агонистами используются для уменьшения острых симптомов, они являются основными средствами современной терапии этого заболевания. На данный момент контроль за ХОБЛ является главным образом симптоматическим за счет бронходилятирующей терапии ингаляционными антихолинергическескими препаратами и ингаляционными агонистами бета2-адренорецепторов. Однако кортикостероиды не снижают воспалительный ответ при ХОБЛ, как они делают это в случае астмы.
Другим классом терапевтических средств, который широко исследовался с учетом его противовоспалительных эффектов для лечения воспалительных респирательных заболеваний, такие как астма и ХОБЛ, являются ингибиторы ферментов фосфодиэстеразы (PDE), в частности, фосфодиэстеразы типа 4 (далее в настоящем документе обозначена как PDE4).
В уровне техники были описаны различные соединения, действующие как ингибиторы PDE4. Однако эффективность некоторых ингибиторов PDE4 первого поколения, таких как ролипрам и пикламиласт, было ограничено из-за их нежелательных побочных эффектов. Указанные эффекты включают тошноту и рвоту в связи с их действием на PDE4 центральной нервной системы и на секрецию желудочного сока в связи с их действием на PDE4 в париетальных клетках кишечника.
Причина указанных побочных эффектов была широко исследована.
Было обнаружено, что PDE4 существует в двух различных формах, имеющих различные конформации, которые были обозначены как высокоаффинный связывающий сайт ролипрама или HPDE4, как правило, присутствующий в центральной нервной системе и в париетальных клетках, и как низкоаффинный связывающий сайт ролипрама или LPDE4 (Jacobitz, S et al. Mol. Pharmacol, 1996, 50, 891-899), который находится в иммунных и воспалительных клетках. Хотя обе эти формы по-видимому проявляют каталитическую активность, они отличаются по чувствительности к ингибиторам. В частности, соединения с более высокой аффинностью к LPDE4 менее склонны вызывать побочные эффекты, такие как тошнота, рвота и повышенная желудочная секреция.
Попытки нацеливания на LPDE4 привели к незначительному улучшению селективности ингибиторов PDE4 второго поколения, таких как рофлумиласт. Тем не менее, рофлумиласт имеет пониженную дозу для достижения приемлемых характеристик побочных эффектов.
В уровне техники были описаны другие классы соединений, действующих как ингибиторы PDE4.
Например, в EP 1634606 описаны, наряду с другими, кетоновые производные, такие как производные бензофурана или 1,3-бензодиоксола.
В WO 9402465 описано, наряду с другими, кетоновые производные общей формулы
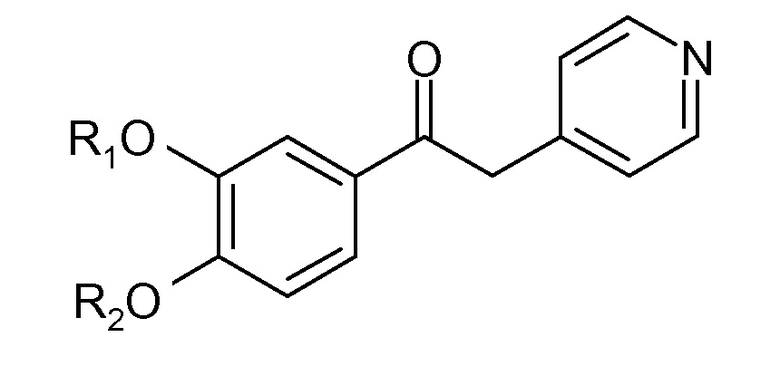
где R1 представляет собой низший алкил, а R2 может быть алкилом, алкенилом, циклоалкилом, циклоалкилом, циклоалкенилом, циклотиоалкилом или циклотиоалкенилом.
WO 9535281, Celltech Therapeutics, касается тризамещенных фенильных производных.
В WO2009/018909 в качестве ингибиторов фермента фосфодиэстеразы 4 (PDE4) описаны производные 1-фенил-2-пиридинилалкильных спиртов, которые имеют общую формулу, показанную ниже
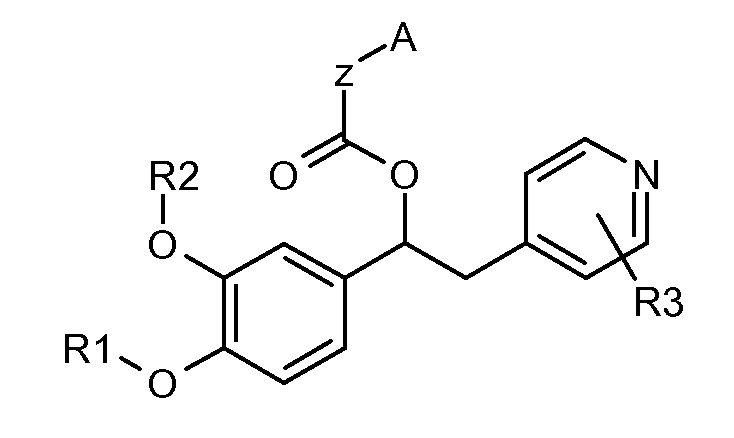
В WO2009/077068 в качестве ингибиторов фермента фосфодиэстеразы 4 (PDE4) описаны другие производные 1-фенил-2-пиридинилалкильных спиртов, которые имеют общую формулу, показанную ниже
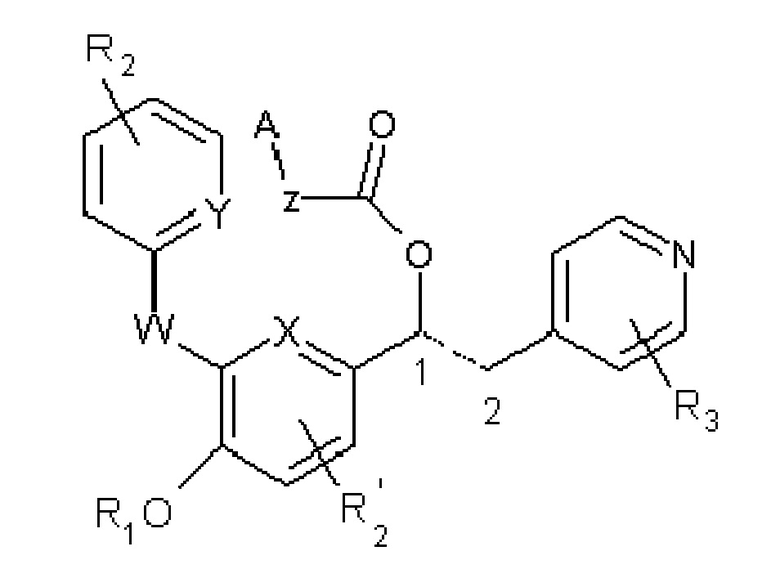
В WO2010/089107 в качестве ингибиторов фермента фосфодиэстеразы 4 (PDE4) описаны другие производные 1-фенил-2-пиридинилалкильных спиртов, которые имеют общую формулу, показаную ниже
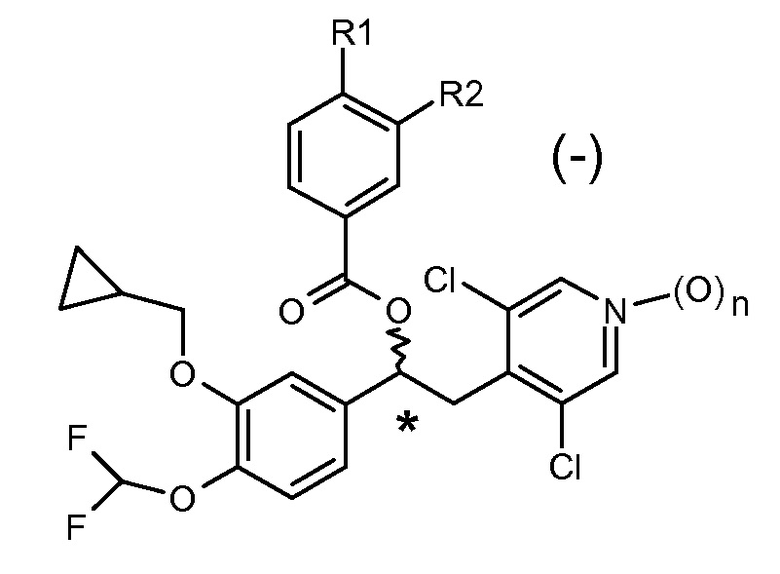
Несмотря на то, что были описаны вышеприведенные ингибиторы PDE4, до сих пор существует необходимость в дополнительных ингибиторах PDE4. В частности, до сих пор существует необходимость в дополнительных ингибиторах PDE4, обеспечивающих высокую аффинность в отношении фермента PDE4. Особенно предпочтительным было бы обнаружение дополнительных ингибиторов PDE4, обеспечивающих высокую аффинность в отношении фермента PDE4 и способных продемонстрировать подходящие характеристики эффективности при ингаляторном лечении, например, пониженные побочные эффекты.
Такое снижение побочных эффектов может быть достигнуто, например, при низком системном воздействии лекарственного препарата; таким образом, ключевыми для этой цели могут быть подходящие параметры фармакокинетических характеристик, особенно, метаболического клиренса.
Настоящее изобретение адресовано вышеуказанной необходимости и предлагает соединения по изобретению.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Изобретение относится к соединениямм, действующим в качестве ингибиторов фермента фосфодиэстеразы 4 (PDE4), к способам получения указанных соединений, к композициям, их содержащим, и к их терапевтическому применению.
В частности изобретение относится к производным 1-фенил-2-пиридинилалкильных спиртов общей формулы (I)
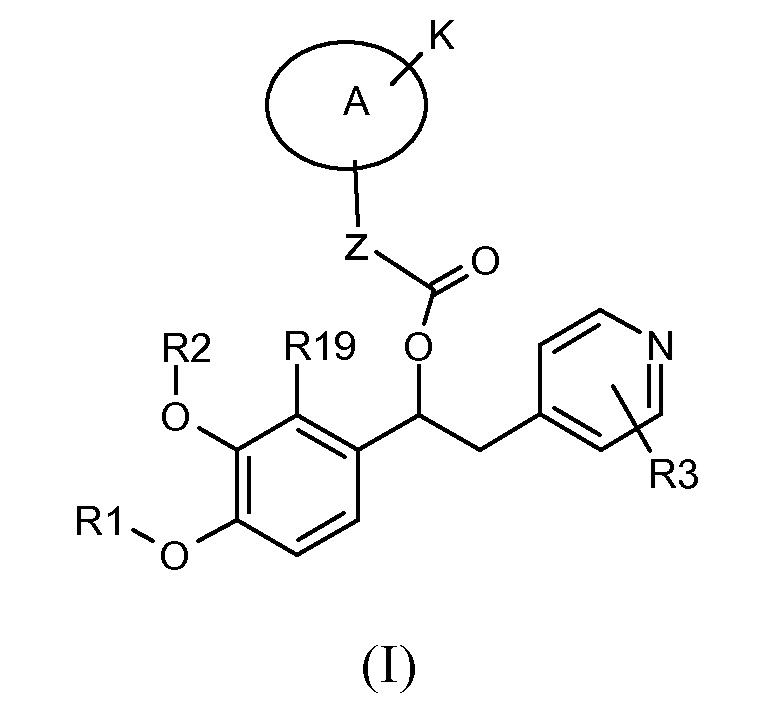 ,
,
где:
R1 выбран из группы, состоящей из:
- H;
- (C3-C7) циклоалкилкарбонила;
- (C1-C6) алкила, необязательно замещенного одним или несколькими заместителями, выбранными из (C3-C7) циклоалкила или (C5-C7) циклоалкенила;
- (C1-C6) галогеналкила;
- (C3-C7) циклоалкила;
- (C5-C7) циклоалкенила;
- (C2-C6) алкенила; и
- (C2-C6) алкинила;
R2 выбран из группы, состоящей из:
- H;
- (C3-C7) циклоалкилкарбонила;
- (C1-C6) алкила, необязательно замещенного одним или несколькими заместителями, выбранными из (C3-C7) циклоалкила или (C5-C7) циклоалкенила;
- (C1-C6) галогеналкила;
- (C3-C7) циклоалкила;
- (C5-C7) циклоалкенила;
- (C2-C6) алкенила; и
- (C2-C6) алкинила;
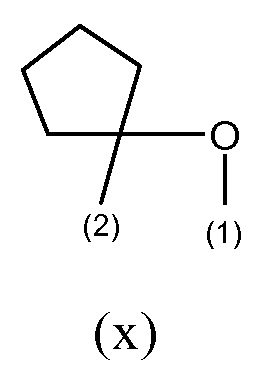
или, если R19 отличается от водорода, то R2 вместе с R19 образует группу формулы (x), как представлено ниже;
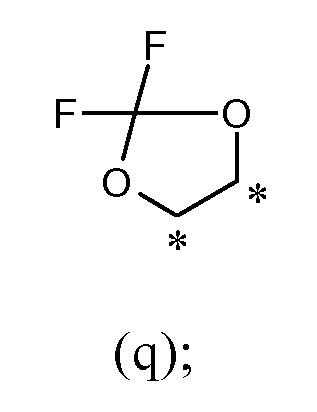
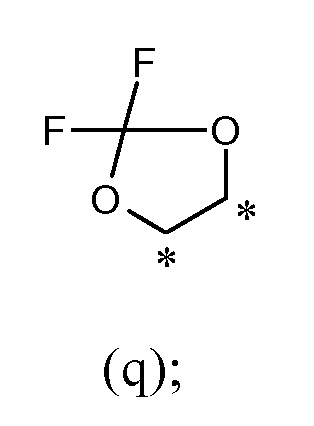
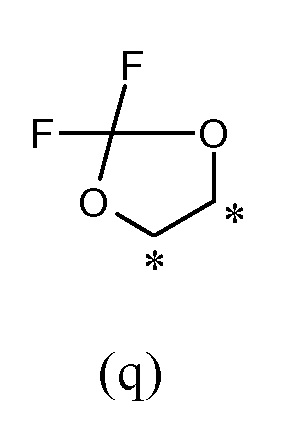
или R1 и R2, вместе с соединяющими их атомами, образуют 2,2-дифтор-1,3-диоксолановое кольцо формулы (q), конденсированное с фенильной группой, которая несет группы -OR1 и -OR2, где звездочки указывают на атомы углерода, совместно используемые таким фенильным кольцом:
R19 представляет собой водород или, если отличается от водорода, образует вместе с R2 группу формулы (x), где связи, отмеченные (1) и (2), указывают места присоединения группы (x) к атомам, несущим группы R19 и R2, соответственно,
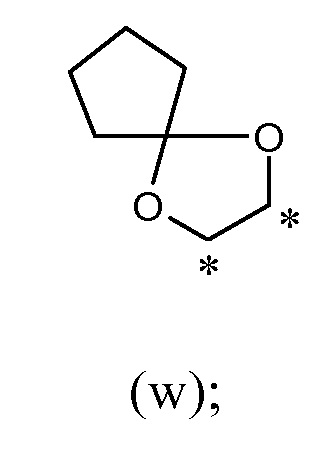
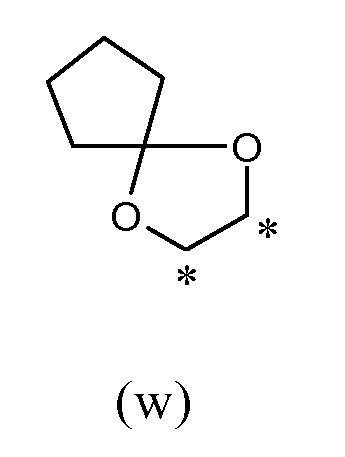
таким образом, что R2 и R19, вместе с соединяющими их атомами, образуют кольцо формулы (w), которое конденсировано с фенильным кольцом, которое несет группы -OR2 и R19, где звездочки указывают на атомы углерода, совместно используемые таким фенильным кольцом:
R3 является одним или несколькими заместителями, независимо выбранными из группы, состоящей из H, CN, NO2, CF3 и атомов галогена;
Z представляет собой группу -(CH2)n-, где n равно 0 или 1;
A представляет собой насыщенную и моноциклическую (C3-C7)гетероциклоалкиленовую группу;
K выбран из группы, состоящей из:
- -(CH2)mC(O)R4, где m может быть равно 0 или 1;
- -C(O)(CH2)jR4, где j может быть равно 1 или 2;
- -SO2 (CH2)pR4, где p может быть равно нулю, 1 или 2;
- -(CH2)ySO2R4, где y может быть равно 1 или 2;
- -(CH2)zR4, где z может быть равно 1 или 2; и
- C(O)(CH2)2SO2R4;
R4 представляет собой кольцевую систему, которая представляет собой моно- или бициклическое кольцо, которое может быть насыщенным, частично ненасыщенным или полностью ненасыщенным, таким как арильное, (C3-C8) циклоалкильное, (C3-C7) гетероциклоалкильное или гетероарильное, такое кольцо необязательно замещено одной или нескольими группами R5, которые могут быть одинаковыми или различными, и которые независимо выбраны из группы, состоящей из:
- (C1-C6) алкила, необязательно замещенного одной или несколькими группами, независимо выбранными из списка, состоящего из: (C3-C7) циклоалкила, -OH и группы -NR18C(O)(C1-C4) алкила, где R18 представляет собой водород или (C1-C4) алкил;
- (C3-C7) гетероциклоалкила;
- 5,6-членного гетероарила, который необязательно замещен одной или двумя группами (C1-C4) алкила;
- (C1-C6) галогеналкила;
- (C3-C7) гетероциклоалкил(C1-C4) алкила;
- группы -OR6, где R6 выбран из группы, состоящей из
- H;
- (C1-C6) галогеналкила;
- группы -SO2R7, где R7 представляет собой (C1-C4) алкил;
- группы -C(O)R7, где R7 представляет собой (C1-C4) алкил;
- (C1-C10) алкила, необязательно замещенного одним или несколькими (C3-C7) циклоалкилами или группой -NR8R9, как представлено ниже; и
- (C3-C7) циклоалкила;
- группы -SR20, где R20 выбран из группы, состоящей из
- H;
- (C1-C6) галогеналкила;
- группы -C(O)R7, где R7 представляет собой (C1-C4) алкил;
- (C1-C10) алкила, необязательно замещенного одним или несколькими (C3-C7) циклоалкилами или группой -NR8R9; и
- (C3-C7) циклоалкила;
- атомов галогена;
- CN;
- NO2;
- NR8R9, где R8 и R9 являются одинаковыми или различными и независимо выбраны из группы, состоящей из:
- H;
- (C1-C4) алкилен-NR13R14, где R13 и R14 являются одинаковыми или различными и независимо выбраны из группы, состоящей из: H и (C1-C6) алкила, который необязательно замещен (C3-C7) циклоалкилом или (C3-C7) гетероциклоалкилом; или они образуют с атомом азота, с которым они связаны, насыщенное или частично насыщенное (C3-C7) гетероциклическое кольцо;
- (C1-C6) алкила, необязательно замещенного (C3-C7) циклоалкилом, (C3-C7) гетероциклоалкилом, группой -OH или (C1-C6) алкоксилом;
- группы -SO2R15, где R15 выбран из группы, состоящей из: (C1-C4) алкила, необязательно замещенного (C3-C7) циклоалкилом или (C3-C7) гетероциклоалкилом; (C3-C7) гетероциклоалкила; и фенила, необязательно замещенного одним или несколькими (C1-C6) алкилом, галогеном или группой -OH;
- группы -C(O)R16, где R16 выбран из группы, состоящей из: (C1-C6) алкила, необязательно замещенного (C3-C7) циклоалкилом или (C3-C7) гетероциклоалкилом; (C3-C7) гетероциклоалкила; фенила, необязательно замещенного одним или несколькими (C1-C6) алкилом, галогеном или -OH; и группы -NH2;
- группы -C(O)OR17, где R17 выбран из группы, состоящей из: (C1-C6) алкила, необязательно замещенного (C3-C7) циклоалкилом или (C3-C7) гетероциклоалкилом; (C3-C7) гетероциклоалкила; фенила, необязательно замещенного одним или несколькими (C1-C6) алкилом, галогеном или -OH; и группы -NH2;
или они образуют с атомом азота, к которому они присоединены, насыщенное или частично насыщенное гетероциклическое кольцо, которое необязательно замещено одним или несколькими (C1-C6) алкилом или оксогруппой;
- (C1-C4) алкилен-NR8R9, как указано выше;
- COR10, где R10 представляет собой фенил или (C1-C6) алкил;
- оксо;
- -SO2R11, где R11 представляет собой (C1-C4) алкил, OH или NR8R9, где R8 и R9 такие, как указано выше;
- -COOR12, где R12 представляет собой H, (C1-C4) алкил или (C1-C4) алкилен-NR8R9, где R8 и R9 такие, как указано выше; и
- -CONR8R9, где R8 и R9 такие, как указано выше;
где группы R6, R7, R8, R9, R10, R11, R12, R13, R14, R15, R16, R17, R18, R19 и R20 в каждом случае могут иметь одинаковое или разное значение, если присутствуют в количестве более одной группы;
к их N-оксидам на пиридиновом кольце, и к их фармацевтически приемлемым солям или сольватами.
В предпочтительном варианте осуществления изобретение относится к производным 1-фенил-2-пиридинилалкильных спиртов общей формулы (IG)
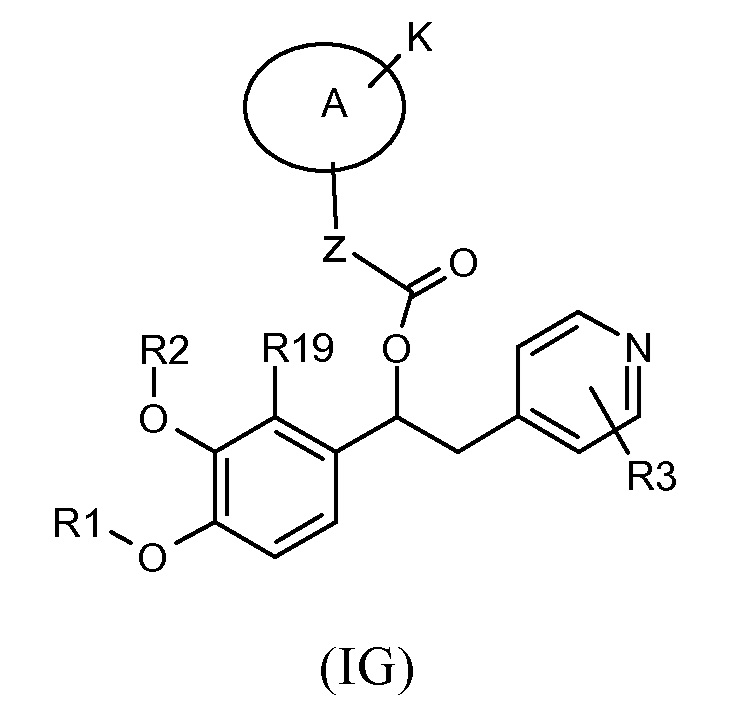
где:
R1 выбран из группы, состоящей из:
- H;
- (C3-C7) циклоалкилкарбонила;
- (C1-C6) алкила, необязательно замещенного одним или несколькими заместителями, выбранными из (C3-C7) циклоалкила или (C5-C7) циклоалкенила;
- (C1-C6) галогеналкила;
- (C3-C7) циклоалкила;
- (C5-C7) циклоалкенила;
- (C2-C6) алкенила; и
- (C2-C6) алкинила;
R2 выбран из группы, состоящей из:
- H;
- (C3-C7) циклоалкилкарбонила;
- (C1-C6) алкила, необязательно замещенного одним или несколькими заместителями, выбранными из (C3-C7) циклоалкила или (C5-C7) циклоалкенила;
- (C1-C6) галогеналкила;
- (C3-C7) циклоалкила;
- (C5-C7) циклоалкенила;
- (C2-C6) алкенила; и
- (C2-C6) алкинила;
или R1 и R2, вместе с соединяющими их атомами, образуют 2,2-дифтор-1,3-диоксолановое кольцо формулы (q), конденсированное с фенильной группой, которая несет группы -OR1 и -OR2, где звездочки указывают на атомы углерода, совместно используемые таким фенильным кольцом:
R19 обозначает водород;
R3 представляет собой один или несколько заместителей, независимо выбранных из группы, состоящей из: H, CN, NO2, CF3 и атомов галогена;
Z представляет собой группу -(CH2)n-, где n равно 0 или 1;
A представляет собой насыщенную и моноциклическую (C3-C7) гетероциклоалкиленовую группу;
K выбран из группы, состоящей из:
- -(CH2)mC(O)R4, где m может быть равно 0 или 1;
- -C(O)(CH2)jR4, где j может быть равно 1 или 2;
- -SO2(CH2)pR4, где p может быть равно 0улю, 1 или 2;
- -(CH2)ySO2R4, где y может быть равно 1 или 2;
- -(CH2)zR4, где z может быть равно 1 или 2; и
- -C(O)(CH2)2SO2R4;
R4 представляет собой кольцевую систему, которая представляет собой моно- и бициклическое кольцо, которое может быть насыщенным, частично ненасыщенным или полностью ненасыщенным, такое как арильное, (C3-C8) циклоалкильное, (C3-C7) гетероциклоалкильное или гетероарильное, такое кольцо необязательно замещено одной или несколькими группами R5, которые могут быть одинаковыми или различными, и которые независимо выбраны из группы, состоящей из:
- (C1-C6) алкила, необязательно замещенного одним или несколькими группами, независимо выбранными из списка, состоящего из: (C3-C7) циклоалкила, -OH и группы -NR18C(O)(C1-C4) алкила, где R18 представляет собой водород или (C1-C4) алкил;
- (C3-C7) гетероциклоалкила;
- 5,6-членного гетероарила, который необязательно замещен одной или двумя группами (C1-C4) алкила;
- (C1-C6) галогеналкила;
- (C3-C7) гетероциклоалкил(C1-C4) алкила;
- группы -OR6, где R6 выбран из группы, состоящей из
- H;
- (C1-C6) галогеналкила;
- группы -SO2R7, где R7 представляет собой (C1-C4) алкил;
- группы -C(O)R7, где R7 представляет собой (C1-C4) алкил;
- (C1-C10) алкила, необязательно замещенного одним или несколькими (C3-C7)циклоалкилом или группой -NR8R9; и
- (C3-C7) циклоалкила;
- группы -SR20, где R20 выбран из группы, состоящей из:
- H;
- (C1-C6) галогеналкила;
- группы -C(O)R7, где R7 представляет собой (C1-C4) алкил;
- (C1-C10) алкила, необязательно замещенного одним или несколькими (C3-C7)циклоалкилом или группой -NR8R9, как представлено ниже; и
- (C3-C7) циклоалкила;
- атомов галогена;
- CN;
- NO2;
- NR8R9, где R8 и R9 являются одинаковыми или различными и независимо выбраны из группы, состоящей из:
- H;
- (C1-C4) алкилен-NR13R14, где R13 и R14 являются одинаковыми или различными и независимо выбраны из группы, состоящей из: H и (C1-C6) алкила, который необязательно замещен (C3-C7)циклоалкилом или (C3-C7) гетероциклоалкилом; или образуют с атомом азота, к которому они присоединены, насыщенное или частично насыщенное (C3-C7) гетероциклическое кольцо;
- (C1-C6) алкила, необязательно замещенного (C3-C7) циклоалкилом, (C3-C7) гетероциклоалкилом, группой -OH или (C1-C6) алкоксилом;
- группы -SO2R15, где R15 выбран из группы, состоящей из: (C1-C4) алкила, необязательно замещенного (C3-C7)циклоалкилом или (C3-C7) гетероциклоалкилом; (C3-C7) гетероциклоалкила; и фенила, необязательно замещенного одним или несколькими (C1-C6) алкилом, галогеном или группой -OH;
- группы -C(O)R16, где R16 выбран из группы, состоящей из: (C1-C6) алкила, необязательно замещенного (C3-C7)циклоалкилом или (C3-C7) гетероциклоалкилом; (C3-C7) гетероциклоалкила; фенила, необязательно замещенного одним или несколькими (C1-C6) алкилом, галогеном или -OH; и группы -NH2;
- группы -C(O)OR17, где R17 выбран из группы, состоящей из: (C1-C6) алкила, необязательно замещенного (C3-C7)циклоалкилом или (C3-C7) гетероциклоалкилом; (C3-C7) гетероциклоалкила; фенила, необязательно замещенного одним или несколькими (C1-C6) алкилом, галогеном или -OH; и группы -NH2;
или они образуют с атомом азота, к которому они присоединены, насыщенное или частично насыщенное гетероциклическое кольцо, которое необязательно замещено одним или несколькими (C1-C6) алкилом или оксогруппами;
- (C1-C4) алкилен-NR8R9, как указано выше;
- COR10, где R10 представляет собой фенил или (C1-C6) алкил;
- оксо;
- SO2R11, где R11 представляет собой (C1-C4) алкил, OH или NR8R9, где R8 и R9 такие, как указано выше;
- COOR12, где R12 представляет собой H или (C1-C4) алкил или (C1-C4) алкилен-NR8R9, где R8 и R9 такие, как указано выше; и
- -CONR8R9, где R8 и R9 такие, как указано выше,
где группы R6, R7, R8, R9, R10, R11, R12, R13, R14, R15, R16, R17, R18, R19 и R20 в каждом случае могут иметь одинаковое или разное значение, если присутствуют в количестве более одной группы;
к их N-оксидам на пиридиновом кольце, и к их фармацевтически приемлемым солям или сольватам.
В другом предпочтительном варианте осуществления изобретение относится к производным 1-фенил-2-пиридинилалкильных спиртов общей формулы (IL)
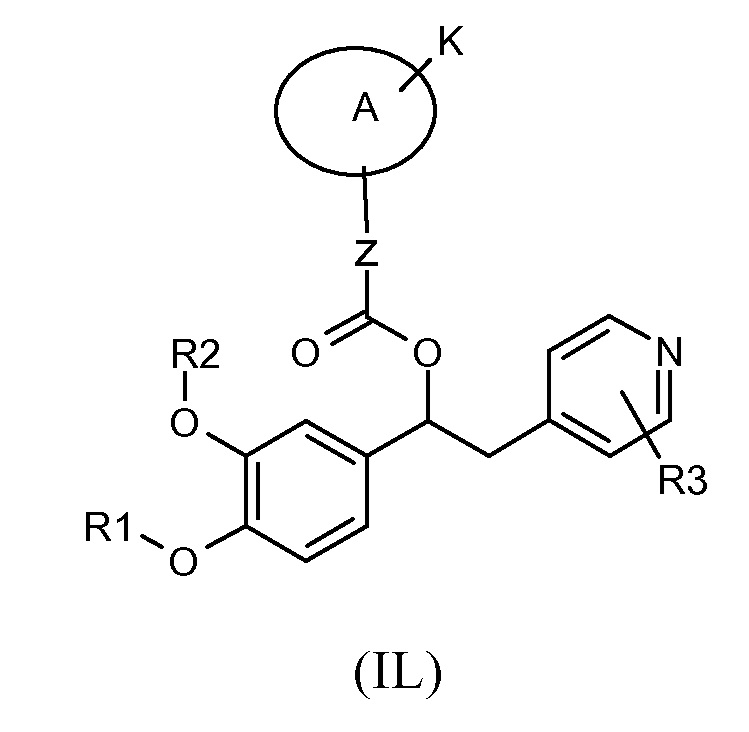
где:
R1 и R2 являются одинаковыми или различными и независимо выбраны из группы, состоящей из:
- H;
- (C3-C7) циклоалкилкарбонила;
- (C1-C6) алкила, необязательно замещенного одним или несколькими заместителями, выбранными из (C3-C7) циклоалкила или (C5-C7) циклоалкенила;
- (C1-C6) галогеналкила;
- (C3-C7) циклоалкила;
- (C5-C7) циклоалкенила;
- (C2-C6) алкенила; и
- (C2-C6) алкинила;
R3 представляет собой один или несколько заместителей, независимо выбранных из группы, состоящей из: H, CN, NO2, CF3 и атомов галогена;
Z представляет собой группу -(CH2)n-, где n равно 0 или 1;
A представляет собой насыщенную и моноциклическую (C3-C7) гетероциклоалкиленовую группу;
K выбран из группы, состоящей из:
- -(CH2)mC(O)R4, где m может быть равно 0 или 1;
- -C(O)(CH2)R4;
- -SO2(CH2)pR4, где p может быть равно нулю или 1;
- -CH2SO2R4; и
- -CH2R4;
R4 представляет собой кольцевую систему, которая представляет собой моно- и бициклическое кольцо, которое может быть насыщенным, частично ненасыщенным или полностью ненасыщенным, такое как арильное, (C3-C8) циклоалкильное, (C3-C7) гетероциклоалкильное или гетероарильное, такое кольцо необязательно замещено одной или несколькими группами R5, которые могут быть одинаковыми или различными, и которые независимо выбраны из группы, состоящей из:
- (C1-C6) алкила, необязательно замещенного одним или несколькими (C3-C7) циклоалкилом;
- (C3-C7) гетероциклоалкила;
- (C3-C7) гетероциклоалкил(C1-C4) алкила;
- группы -OR6, где R6 выбран из группы, состоящей из
- H;
- (C1-C6) галогеналкила;
- группы -SO2R7, где R7 представляет собой (C1-C4) алкил;
- группы -C(O)R7, где R7 представляет собой (C1-C4) алкил;
- (C1-C10) алкила, необязательно замещенного одним или несколькими (C3-C7)циклоалкилом или группой -NR8R9; и
- (C3-C7) циклоалкила;
- группы -SR20, где R20 выбран из группы, состоящей из:
- H;
- (C1-C6) галогеналкила;
- группы -C(O)R7, где R7 представляет собой (C1-C4) алкил;
- (C1-C10) алкила, необязательно замещенного одним или несколькими (C3-C7) циклоалкилом или группой -NR8R9, как представлено ниже; и
- (C3-C7) циклоалкила;
- атомов галогена;
- CN;
- NO2;
- NR8R9, где R8 и R9 являются одинаковыми или различными и независимо выбраны из группы, состоящей из:
- H;
- (C1-C4) алкилен-NR13R14, где R13 и R14 являются одинаковыми или различными и независимо выбраны из группы, состоящей из: H и (C1-C6) алкила, который необязательно замещен (C3-C7) циклоалкилом или (C3-C7) гетероциклоалкилом; или образуют с атомом азота, к которому они присоединены, насыщенное или частично насыщенное (C3-C7) гетероциклическое кольцо;
- (C1-C6) алкила, необязательно замещенного (C3-C7) циклоалкилом, (C3-C7) гетероциклоалкилом, группой -OH или (C1-C6) алкоксилом;
- группы -SO2R15, где R15 выбран из группы, состоящей из: (C1-C4) алкила, необязательно замещенного (C3-C7) циклоалкилом или (C3-C7) гетероциклоалкилом; (C3-C7) гетероциклоалкила; и фенила, необязательно замещенного одним или несколькими (C1-C6) алкилом, галогеном или группой -OH;
- группы -C(O)R16, где R16 выбран из группы, состоящей из: (C1-C6) алкила, необязательно замещенного (C3-C7)циклоалкилом или (C3-C7) гетероциклоалкилом; (C3-C7) гетероциклоалкила; фенила, необязательно замещенного одним или несколькими (C1-C6) алкилом, галогеном или -OH; и группы -NH2;
- группы -C(O)OR17, где R17 выбран из группы, состоящей из: (C1-C6) алкила, необязательно замещенного (C3-C7)циклоалкилом или (C3-C7) гетероциклоалкилом; (C3-C7) гетероциклоалкила; фенила, необязательно замещенного одним или несколькими (C1-C6) алкилом, галогеном или -OH; и группы -NH2;
или образуют с атомом азота, к которому они присоединены, насыщенное или частично насыщенное гетероциклическое кольцо, которое необязательно замещено одним или несколькими (C1-C6) алкилом или оксогруппами;
- (C1-C4) алкилен-NR8R9, как указано выше;
- COR10, где R10 представляет собой фенил или (C1-C6) алкил;
- оксо;
- -SO2R11, где R11 представляет собой (C1-C4) алкил, OH или NR8R9, где R8 и R9 такие, как указано выше;
- -COOR12, где R12 представляет собой H или (C1-C4) алкил или (C1-C4) алкилен-NR8R9, где R8 и R9 такие, как указано выше; и
- -CONR8R9, где R8 и R9 такие, как указано выше,
где группы R6, R7, R8, R9, R10, R11, R12, R13, R14, R15, R16, R17 и R20 в каждом случае могут иметь одинаковое или разное значение, если присутствует более одной группы;
к их N-оксидам на пиридиновом кольце, и к их фармацевтически приемлемым солям или сольватам.
Изобретение также относится к соответствующим N-оксидам на пиридиновом кольце соединений формулы (I).
Изобретение также относится к их фармацевтически приемлемым солям и/или сольватам.
Термин «фармацевтически приемлемые соли», как используется в настоящем описании, относится к производным соединений формулы (I) или к их соответствующим N-оксидам на пиридиновом кольце, где родительское соединение подходящим образом модифицировано путем преобразования любой свободной кислотной или основной группы, если присутствует, до соответствующей соли добавления основания или кислоты, обычно предназначенных в качестве фармацевтически приемлемых.
Подходящие примеры указанных солей могут, таким образом, включать соли добавления неорганических или органических кислот основных остатков, таких как аминогруппы, а также остатки неорганических или органических кислот, таких как карбоксильные группы.
Катионы неорганических оснований, которые могут удобно использоваться для получения солей по изобретению, включают ионы щелочных или щелочноземельных металлов, таких как калий, натрий, кальций или магний.
Соли, полученные путем взаимодействия основного соединения, действующего как основание, с неорганической или органической кислотой, включают, например, соли соляной кислоты, серной кислоты, фосфорной кислоты, метансульфоновой кислоты, камфорсульфоновой кислоты, щавелевой кислоты, малеиновой кислоты, янтарной кислоты и лимонной кислоты.
Специалистам в области органической химии будет понятно, что большое число органических соединений могут образовывать комплексы с растворителями, в которых они взаимодействуют или из которых они осаждаются или кристаллизуются. Эти комплексы известны как «сольваты». Фармацевтически приемлемые сольваты соединения по изобретению входят в объем настоящего изобретения.
Также в объем настоящего изобретения входят полиморфы и кристаллические формы соединений формулы (I), их N-оксидов на пиридиновом кольце, или их фармацевтически приемлемых солей или сольватов.
Далее в настоящем документе соединения формулы (I), (IG), (IL), соответствующие N-оксиды на пиридиновом кольце, их варианты осуществления, энантиомеры, диастереоизомеры, их фармацевтически приемлемые соли и сольваты, и их полиморфы или кристаллические формы, определенные в любом из аспектов настоящего изобретения (кроме промежуточных соединений, описанных в химических процессах), обозначают как «соединения по настоящему изобретению».
Изобретение также относится к способу получения соединений по настоящему изобретению.
Настоящее изобретение также относится к фармацевтическим композициям соединений по изобретению, либо самостоятельно, либо в сочетании, в смеси с одним или несколькими фармацевтически приемлемыми носителями.
В дополнительном аспекте настоящее изобретение относится к применению соединений по изобретению в качестве лекарственного средства.
В одном из аспектов настоящее изобретение относится к применению соединений по изобретению для получения лекарственного средства.
В частности, настоящее изобретение относится к применению соединений по изобретению для профилактики и/или лечения любого заболевания, характеризующегося сверхактивностью фосфодиэстеразы 4 (PDE4) и/или при котором желательно ингибировать активность PDE4.
В частности, соединения по изобретению, самостоятельно или в сочетании с другими активными ингредиентами, могут вводиться для профилактики и/или лечения заболевания респираторного тракта, характеризующегося обструкцией дыхательных путей, такого как астма и ХОЗЛ.
В дополнительном аспекте настоящее изобретение относится к применению соединений по изобретению для получения лекарственного средства для профилактики и/или лечения любого заболевания, характеризующегося сверхактивностью фосфодиэстеразы 4 (PDE4) и/или при котором желательно ингибировать активность PDE4.
Кроме того, настоящее изобретение относится к способу профилактики и/или лечения любого заболевания, при котором желательно ингибировать PDE4, указанный способ включает введение нуждающемся в таком лечении пациенту терапевтически эффективного количества соединения по изобретению.
ОПРЕДЕЛЕНИЯ
Термин «атомы галогена», как используется в настоящем описании, включает фтор, хлор, бром и йод, предпочтительно, хлор.
Как используется в настоящем описании, термин «(C1-Cx) алкил», где x равен целому числу выше 1, относится к алкильным группам с прямой и разветвленной цепью, где число составляющих их атомов углерода составляет от 1 до x. Конкретные алкильные группы представляют собой метил, этил, н-пропил, изопропил и трет-бутил.
По аналогии, термин «(C1-Cx)алкилен», относится к двухвалентному радикалу (C1-Cx)алкилу, где (C1-Cx)алкил такой, как указано выше.
Термин «(C1-Cx)алкоксил», где x равен целому числу выше 1, относится к алкоксигруппам с прямой или разветвленной цепью, где число составляющих их атомов углерода составляет от 1 до x. Конкретные алкильные группы представляют собой метоксил, этоксил, н-пропоксил, изопропоксил и трет-бутоксил.
Выражение «(C1-Cx)галогеналкил» относится к определенной выше «(C1-Cx)алкильной» группе, где один или более атомов водорода заменены одним или более атомами галогена, которые могут быть одинаковыми или отличаться друг от друга.
Примеры указанных (C1-C6)галогеналкильных групп могут, таким образом, включать галогенированные, поли-галогенированные и полностью галогенированные алкильные группы, где все атомы водорода замещены атомами галогена, например, группы трифторметил или дифторметил.
Термин «(C3-Cy) циклоалкил», где y равно целому числу, больше или равному 3, относится к насыщенным циклическим углеводородным группам, состоящим из от 3 до y кольцевых атомов углерода. Примеры включают циклопропил, циклобутил, циклопентил, циклогексил и циклогептил.
Его производное выражение «(C3-Cy)гетероциклоалкил» относится к моноциклической (C3-Cy)циклоалкильной группе, в которой по меньшей мере один кольцевой атом углерода заменен на гетероатом (например, N, NH, S или O). Неограничивающие примеры (C3-Cy)гетероциклоалкила представляют собой: пирролидинил, тиазолидинил, пиперазинил, пиперидинил, морфолинил, тиоморфолинил, азетидинил.
По аналогии, термин «(C3-Cy)гетероциклоалкилен» относится к двухвалентному радикалу (C3-Cy)гетероциклоалкил, где (C3-Cy)гетероциклоалкил такой, как указано выше.
Выражение «(C3-Cy)циклоалкилкарбонил» относится к (C3-Cy)циклоалкилCO- группам, где группа «(C3-Cy)циклоалкил» имеет значение, определенное выше.
Термин «(C2-C6)алкенил» относится к прямым или разветвленным, конъюгированным или неконъюгированным углеродным цепям с одной или несколькими двойными связями, в цис- или транс- конфигурации, где число атомов равно от 2 до 6.
Термин «(C5-Cz) циклоалкенил», где z равно целому числу, больше или равному 5, относится к циклическим углеводородным группам, состоящим из от 5 до z кольцевых атомов углерода и одной или более двойных связей.
Термин «(C2-C6)алкинил» относится к прямым или разветвленным углеродным цепям с одной или более тройными связями, где число атомов равно от 2 до 6.
Термин «(C3-Cy)гетероциклоалкил(C1-Cx) алкил» относится к вышеуказанной группе «(C1-Cx)алкил», где один или более атомов водорода замещены одной или более группами «(C3-Cy)гетероциклоалкил».
Как используется в настоящем описании, выражение «циклическая система» относится к моно- или бициклическим кольцевым системам, которые могут быть насыщенными, частично ненасыщенными или ненасыщенными, таким как арил, (C3-C8) циклоалкил, (C3-C7) гетероциклоалкил или гетероарил, имеющими от 5 до 11 кольцевых атомов, в которых по меньшей мере один кольцевой атом является гетероатомом (например, N, S или O).
Выражение «арил» относится к моно- или бикольцевым системам, которые имеют от 6 до 10 кольцевых атомов, где по меньшей мере одно кольцо является ароматическим.
Выражение «гетероарил» относится к моно- или бикольцевым системам с 5-11 кольцевыми атомами, в которых по меньшей мере одно кольцо является ароматическим и в которых по меньшей мере один кольцевой атом является гетероатомом (например, N, NH, S или O).
Примеры подходящих арильных или 5,6-членных гетероарильных моноциклических систем включают, например, радикалы фенил, тиофен, бензол, пиррол, пиразол, имидазол, изоксазол, оксазол, изотиазол, тиазол, пиридин, имидазолидин, фуран и тому подобное.
Примеры подходящих арильных или гетероарильных бициклических систем включают радикалы нафталин, бифенилен, пурин, птеридин, бензотриазол, хинолин, изохинолин, индол, изоиндол, бензотиофен, дигидробензодиоксин, дигидробензодиоксепин, бензооксазин и тому подобное.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Изобретение относится к классу соединений, действующих как ингибиторы фермента фосфодиэстеразы 4 (PDE4).
Указанный класс соединений ингибирует преобразование циклических нуклеотидов, в частности, циклического аденозинмонофосфата (цАМФ), в их неактивные 5''-мононуклеотидные формы.
В дыхательных путях физиологические ответы на повышенные внутриклеточные уровни циклических нуклеотидов, в частности, цАМФ, приводят к подавлению активности иммунных и провоспалительных клеток, таких как тучные клетки, макрофаги, T-лимфоциты, эозинофилы и нейтрофилы, что приводит к снижению высвобождения медиаторов воспалителения, которые включают цитокины, такие как IL-1, IL-3, и фактор некроза опухоли-альфа (TNF-α).
Это также приводит к расслаблению гладких мышц дыхательных путей и к снижению отека.
Настоящее изобретение относится к производным 1-фенил-2-пиридинилалкильных спиртов общей формулы (I), к N-оксидам пиридинового кольца и к их фармацевтически приемлемым солям или сольватам,
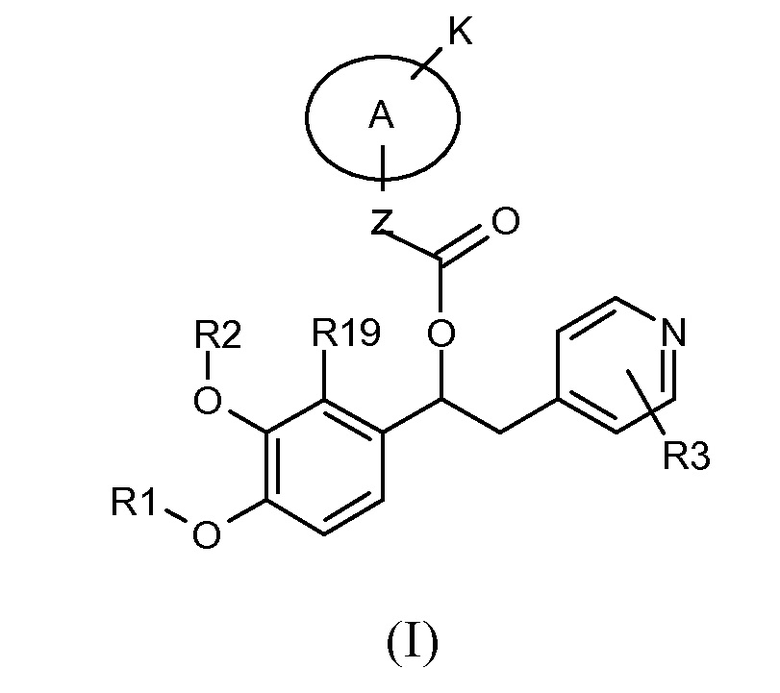
где R1, R2, R3, R19, Z, A и K такие, как указано выше.
Специалистам в данной области будет очевидно, что соединения общей формулы (I) по меньшей мере содержат один стереогенный центр, а именно, представленный атомом углерода (1) со звездочкой внизу, и, таким образом, существуют в виде оптических стереоизомеров.
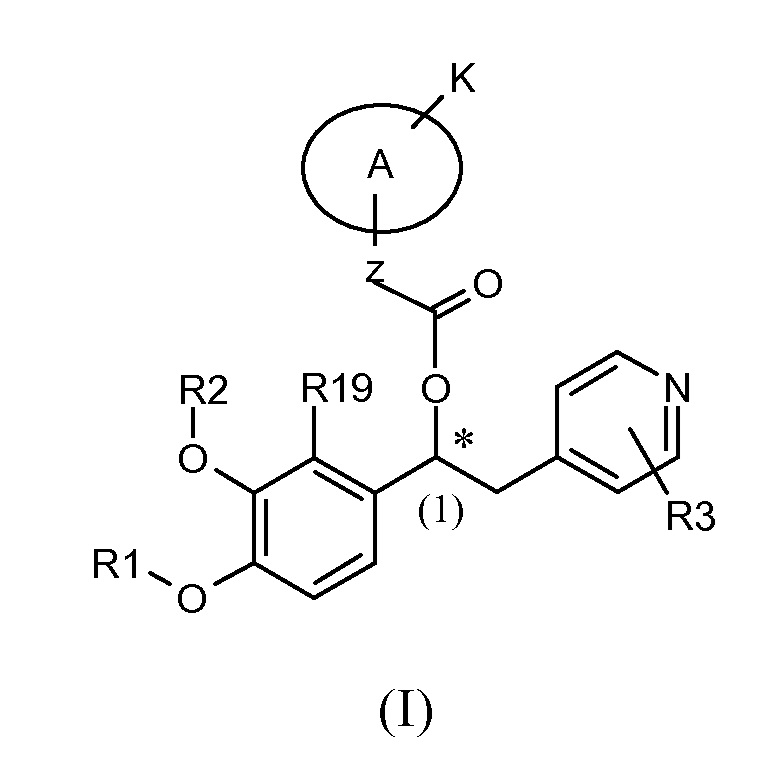
Если соединения по изобретению имеют по меньшей мере один стереогенный центр, то они могут таким образом существовать в виде энантиомеров. Если соединения по изобретению обладают двумя или более стереогенными центрами, то они могут дополнительно существовать в виде диастереоизомеров. Следует понимать, что все такие изомеры и их смеси в любой пропорции входят в объем настоящего изобретения.
В предпочтительном варианте осуществления настоящее изобретение относится к соединениямм формулы (I)', которые представляют собой соединения формулы (I), как указано выше, в которых абсолютная конфигурация углерода (1) такая, как показано ниже:
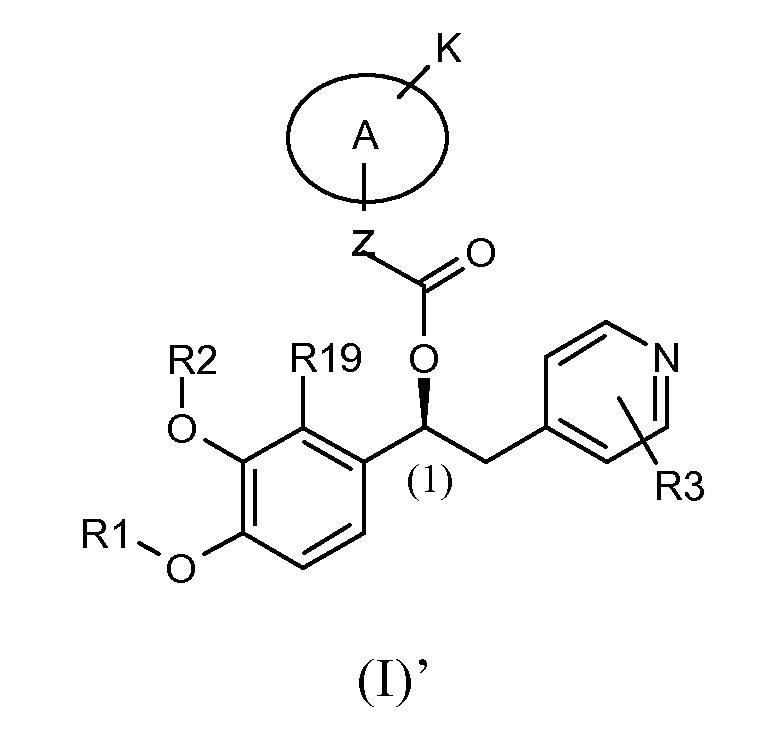
Абсолютная конфигурация углерода (1) задана на основании номенклатуры Кана-Ингольда-Прелога по приоритетам групп.
В одном из предпочтительных вариантов осуществления для соединений формулы (I) абсолютной конфигурацией при углероде (1) является (S).
Если соединения формулы (I) имеют второй стереогенный центр, а именно при углероде (2), показанный еще одной звездочной ниже, то они существуют в виде по меньшей мере четырех диастереоизомеров:
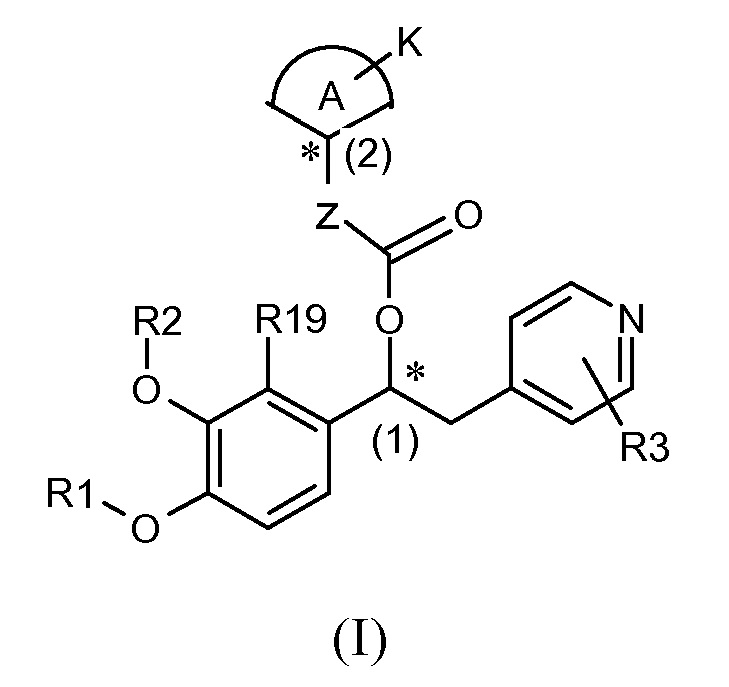
Четыре их диастереоизомера представлены ниже:
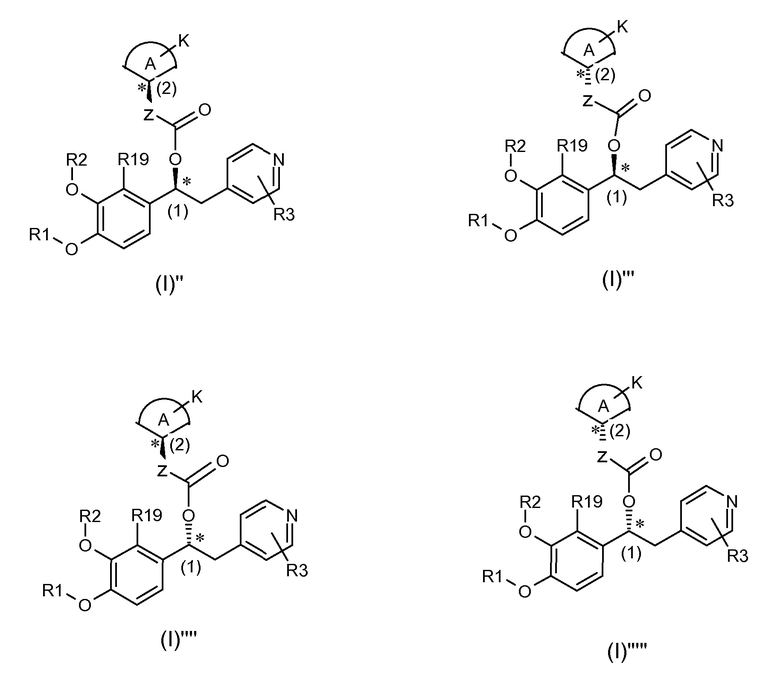
и входят в объем настоящего изобретения.
В предпочтительном варианте осуществления настоящее изобретение относится к соединениям формулы (I)'', которые представляют собой соединения формулы (I)', как указано выше, где абсолютная конфигурация углерода (2) такая, как показано ниже:
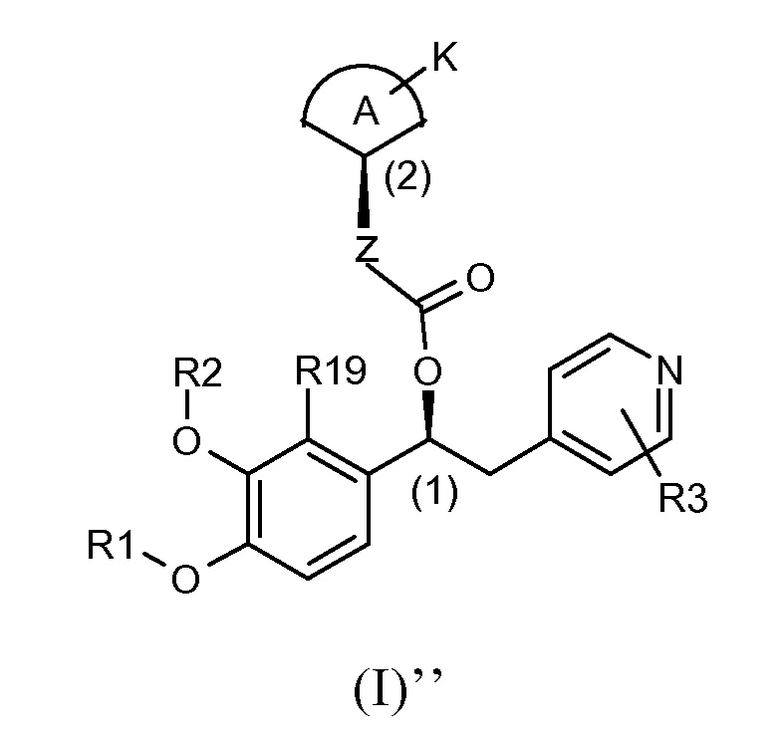
В другом предпочтительном варианте осуществления настоящее изобретение относится к соединениям формулы (I)''', которые представляют собой соединения формулы (I)', как указано выше, где абсолютная конфигурация углерода (2) такая, как показано ниже:
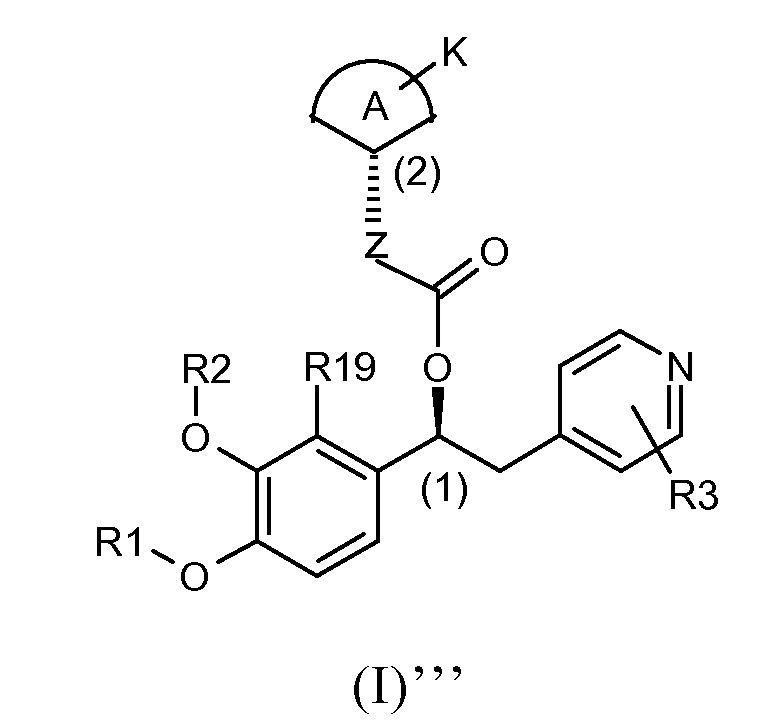
Для соединений формулы (I)'' и (I)''' абсолютная конфигурация углерода (1) и (2) задана на основании номенклатуры Кана-Ингольда-Прелога по приоритетам групп.
Следует понимать, что все предпочтительные группы или варианты осуществления, описанные ниже и выше для соединений формулы (I), могут быть объединены друг с другом и давать соединения формулы (IG), (IL), (I)', (I)'', (I)''', (I)'''' и (I)''''', а также mutatis mutandis.
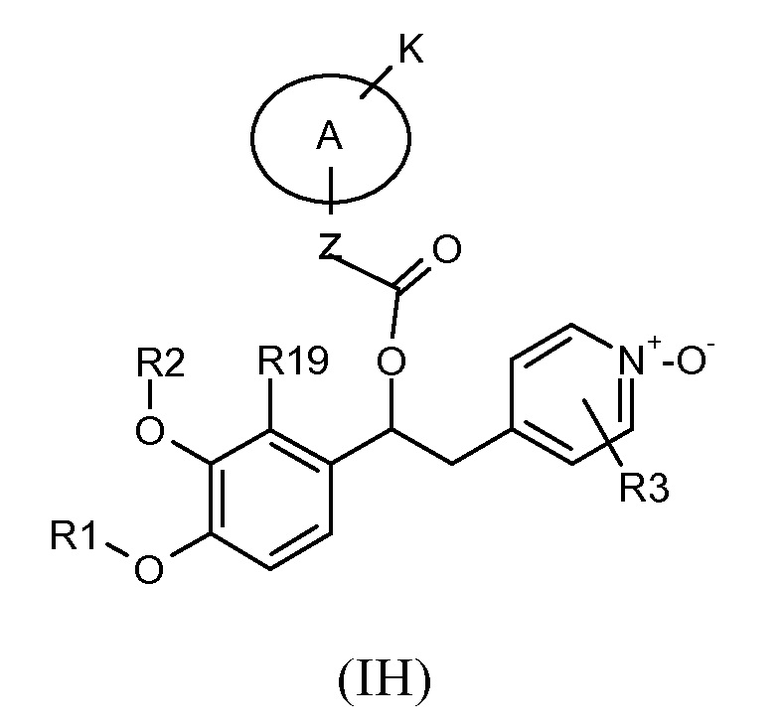
В предпочтительном варианте осуществления изобретение относится к соединениям формулы (IH), которые представляют собой N-оксидные производные пиридинового кольца соединений формулы (I), или к их фармацевтически приемлемым солям:
В предпочтительном варианте осуществления 2-пиридинильное кольцо имеет два заместителя R3, которые являются атомом галогена. В другом предпочтительном варианте осуществления такие заместители R3 представляют собой два атома хлора в положениях 3 и 5 пиридинового кольца.
В одном из предпочтительных вариантов осуществления R1 представляет собой (C1-C6) галогеналкил или (C1-C6) алкил.
В одном из предпочтительных вариантов осуществления R2 представляет собой (C1-C6) алкил, который необязательно замещен (C3-C7) циклоалкилом или представляет собой (C3-C7) циклоалкил.
В другом предпочтительном варианте осуществления R1 и R2, вместе с соединяющими их атомами, образуют 2,2-дифтор-1,3-диоксолановое кольцо формулы (q), конденсированное с фенильной группой, которая несет группы -OR1 и -OR2, где звездочки указывают атомы углерода, совместно используемые таким фенильным кольцом:
В еще одном предпочтительном варианте осуществления R1 представляет собой (C1-C6) галогеналкил, и R2 представляет собой (C1-C6) алкил, который замещен (C3-C7) циклоалкилом.
В другом предпочтительном варианте осуществления R1 представляет собой (C1-C6)алкил, и R2 представляет собой (C1-C6) алкил.
В предпочтительном варианте осуществления R19 обозначает водород.
В еще одном предпочтительном варианте осуществления R19 обозначает водород, R1 представляет собой (C1-C6) галогеналкил, и R2 представляет собой (C1-C6) алкил, который замещен (C3-C7) циклоалкилом.
В другом предпочтительном варианте осуществления R19, если он отличается от водорода, образует, вместе с R2, группу формулы (x), где связи, отмеченные как (1) и (2), указывают на места присоединения группы (x) к атомам, несущим группы R19 и R2, соответственно,
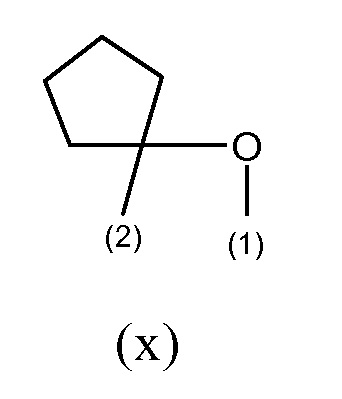
таким образом, что R2 и R19, вместе с соединяющими их атомами, образуют кольцо формулы (w), которое конденсировано с фенильным кольцом, которое несет группы -R2 и R19, где звездочки указывают на атомы углерода, совместно используемые таким фенильным кольцом:
Предпочтительной группой соединений общей формулы (I) является группа, в которой 2-пиридинильное кольцо замещено в 3 и 5 положении двумя атомами хлора, в соответствии с общей формулой (IA)
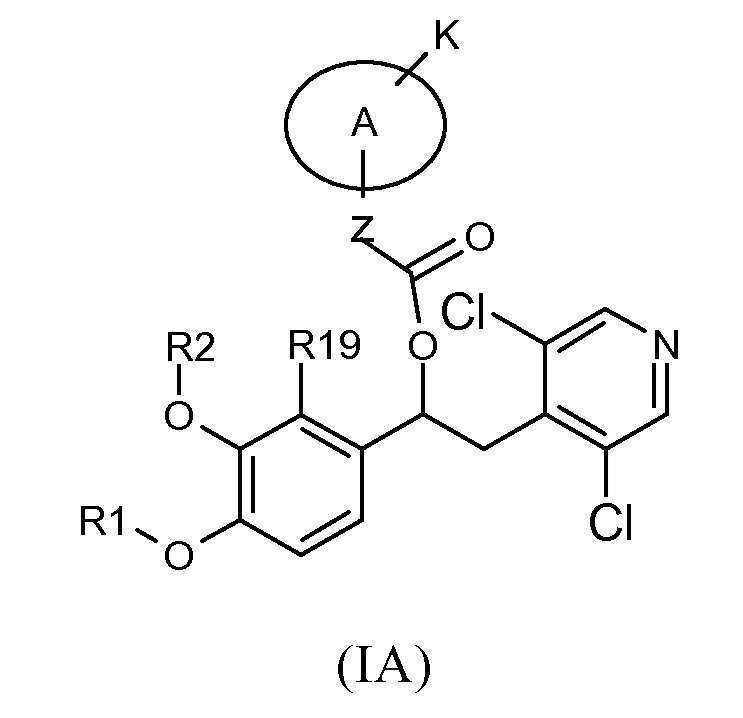
где R1, R2, R19, K, z и A такие, как определено выше, для соединений формулы (I); и соответствующий N-оксид на пиридиновом кольце, или их фармацевтически приемлемая соль.
Другой предпочтительной группой соединений формулы (I) является группа, показанная ниже, в соответствии с общей формулой (IB):
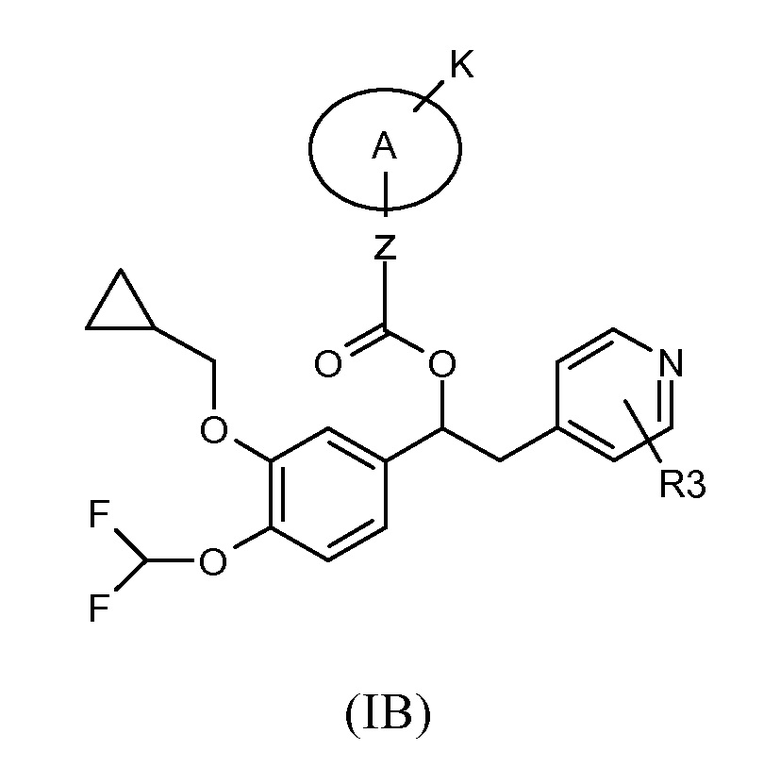
где R3, K, Z и A такие, как определено выше для соединений формулы (I); и соответствующий N-оксид на пиридиновом кольце, или их фармацевтически приемлемая соль.
Другой предпочтительной группой соединений формулы (I) является группа, показанная ниже, в соответствии с общей формулой (IC):
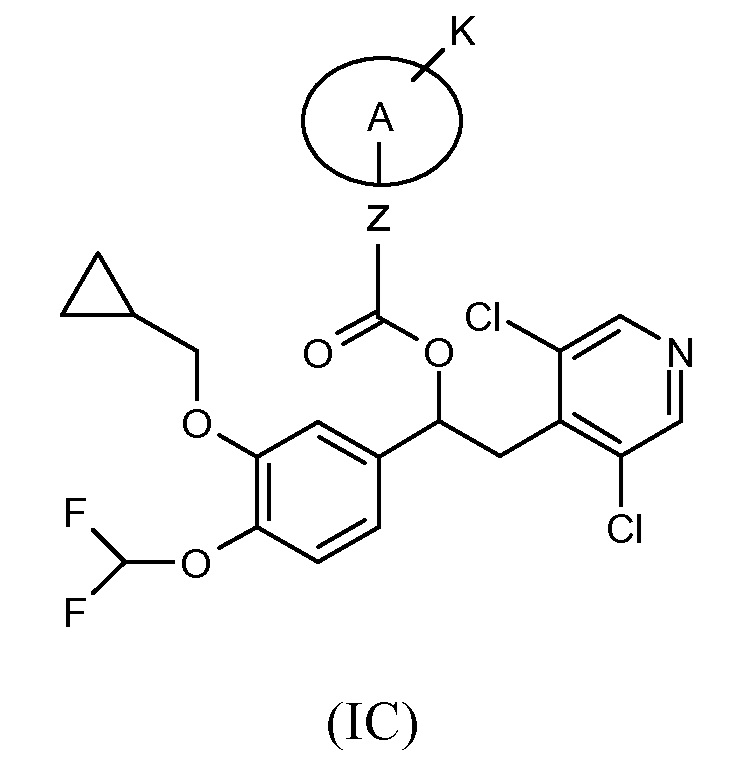
где K, Z и A такие, как определено выше для соединений формулы (I); и соответствующий N-оксид на пиридиновом кольце, или их фармацевтически приемлемая соль.
В одном из предпочтительных вариантов осуществления A представляет собой (C3-C7) гетероциклоалкиленовую группу, содержащую атом азота, который представляет собой место присоединения к группе K, как показано ниже:
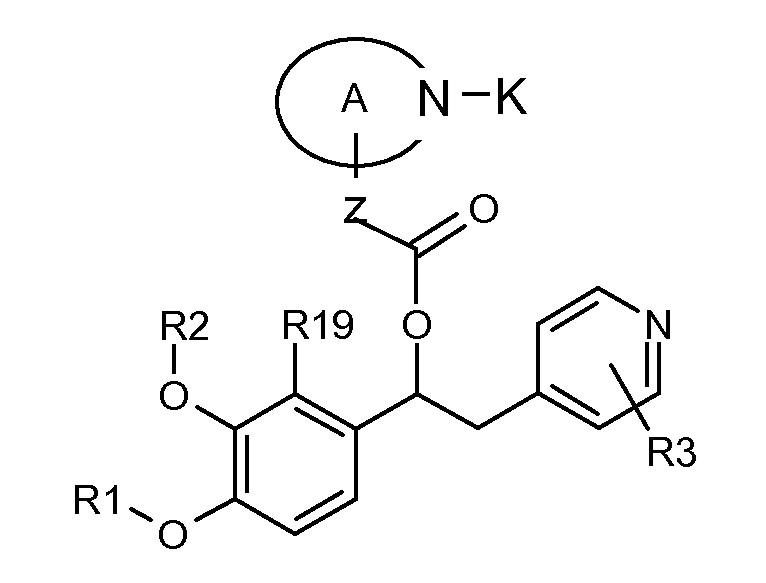
В другом предпочтительном варианте осуществления A выбран из списка двухвалентных радикалов, приведенных ниже:
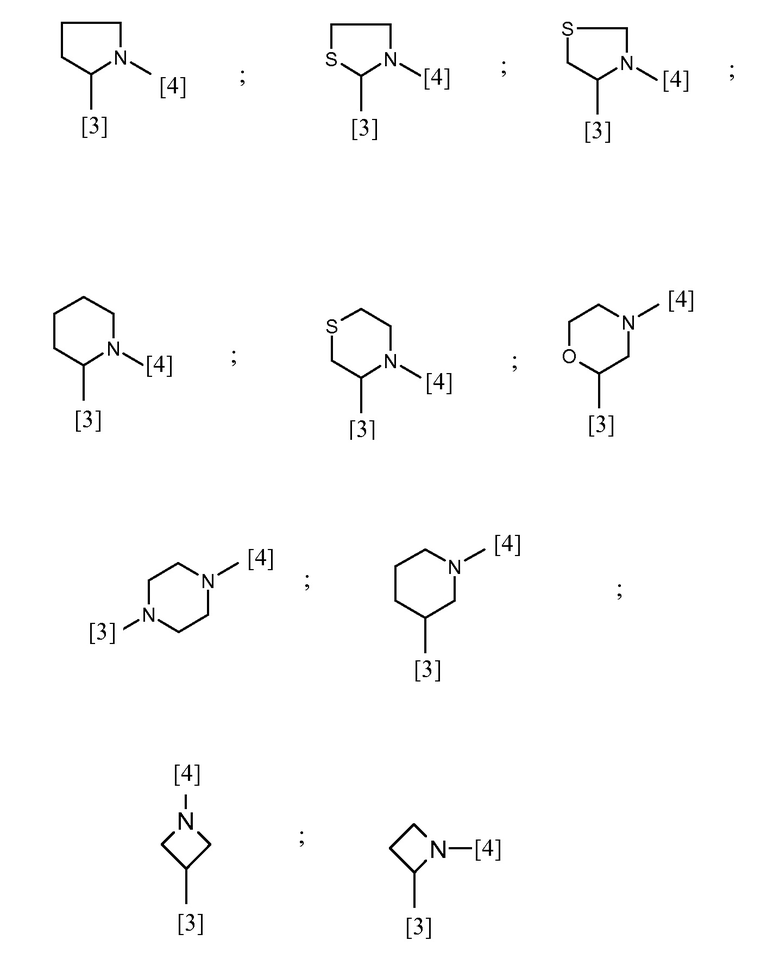
где символы [3] и [4] указывают места связывания группы A с, соответственно, группами Z и K.
В еще одном предпочтительном варианте осуществления A выбран из списка двухвалентных радикалов, приведенных ниже:
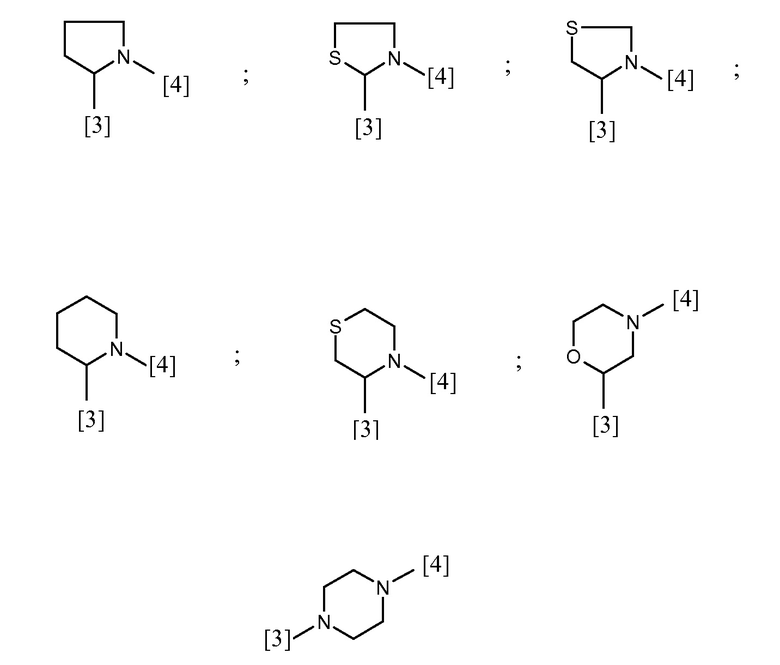
где символы [3] и [4] указывают места связывания группы A с, соответственно, группами Z и K.
В дополнительном предпочтительном варианте осуществления A представляет собой группу
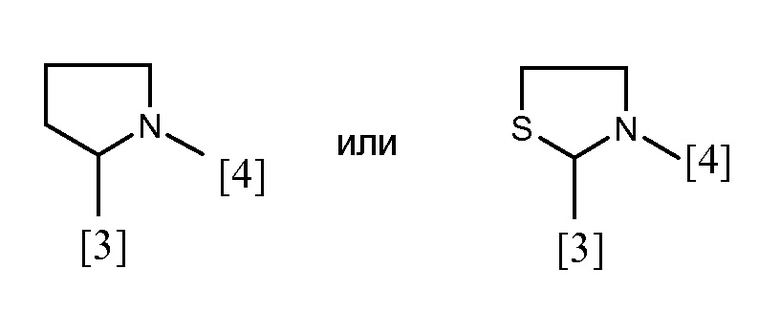
где символы [3] и [4] указывают места связывания группы A с, соответственно, группами Z и K.
В предпочтительном варианте осуществления Z равно нулю.
Другой предпочтительной группой соединений формулы (I) является группа, показанная ниже, в соответствии с общей формулой (ID):
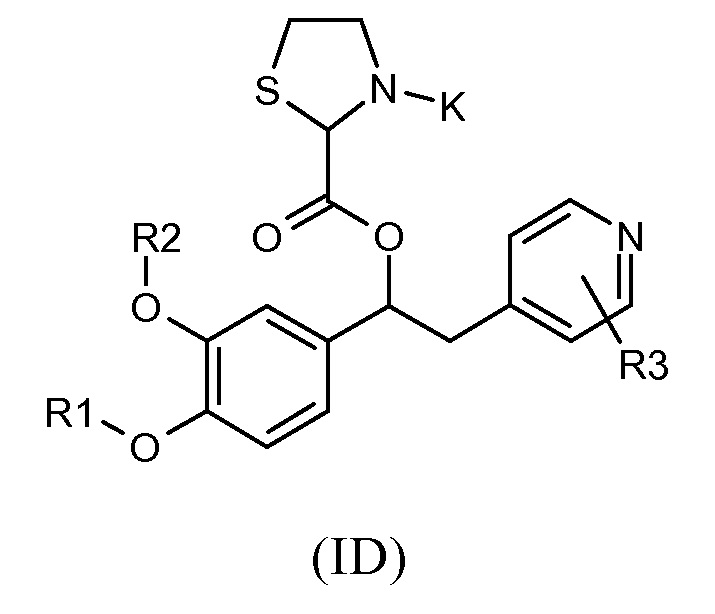
где R1, R2, R3 и K такие, как определено выше для соединений формулы (I), R19 представляет собой водород, Z представляет собой связь, и A представляет собой тиазолидиновую двухвалентную группу радикала, представленную выше; и соответствующий N-оксид на пиридиновом кольце, или их фармацевтически приемлемые соли.
Другая предпочтительная группа соединений формулы (I) представляет собой группу, показанную ниже, в соответствии с общей формулой (ID'''):
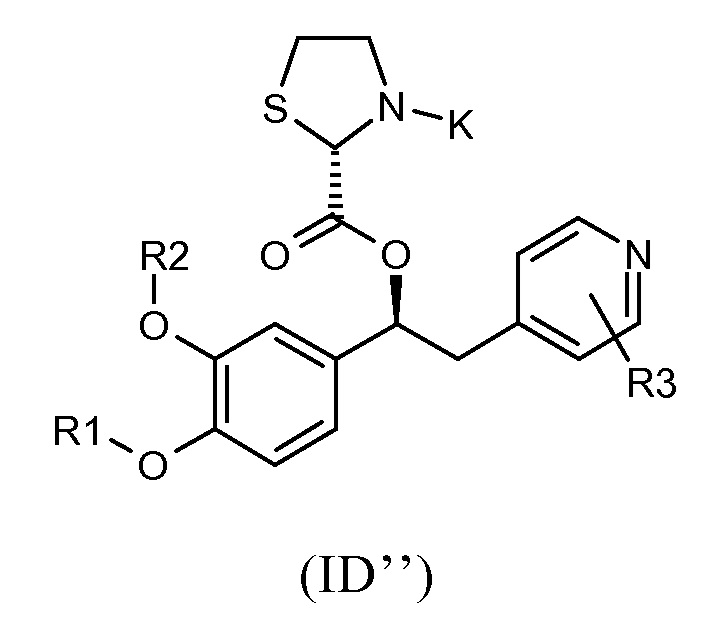
где R1, R2, R3 и K такие, как определено выше для соединений формулы (I), R19 представляет собой водород, Z представляет собой связь, A представляет собой тиазолидиновую двухвалентную группу радикала, и стереогенный центр имеет абсолютную конфигурацию, как представлено ниже; и соответствующий N-оксид на пиридиновом кольце, или их фармацевтически приемлемые соли.
В одном из вариантов осуществления соединений формулы (ID) или (ID'''), R1 представляет собой (C1-C6) галогеналкил, R2 представляет собой (C1-C6) алкил, который замещен (C3-C7) циклоалкилом, 2-пиридинильное кольцо замещено в положениях 3 и 5 двумя группами хлора R3, и K представляет собой группу
Другие предпочтительные группы соединений формулы (I) представляют собой группы, показанные ниже в соответствии с общей формулой (IE):
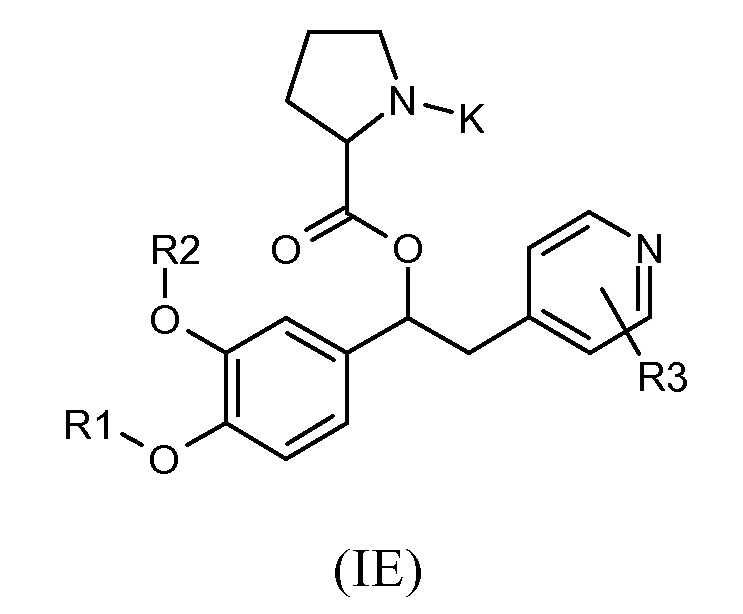
где R1, R2, R3 и K такие, как определено выше, для соединений формулы (I), Z представляет собой связь, R19 представляет собой водород, и A представляет собой пирролидиновую двухвалентную группу радикала, как показано выше; и соответствующий N-оксид на пиридиновом кольце, или их фармацевтически приемлемые соли.
Другая предпочтительная группа соединений формулы (I) такая, как показана ниже, в соответствии с общей формулой (IE'''):
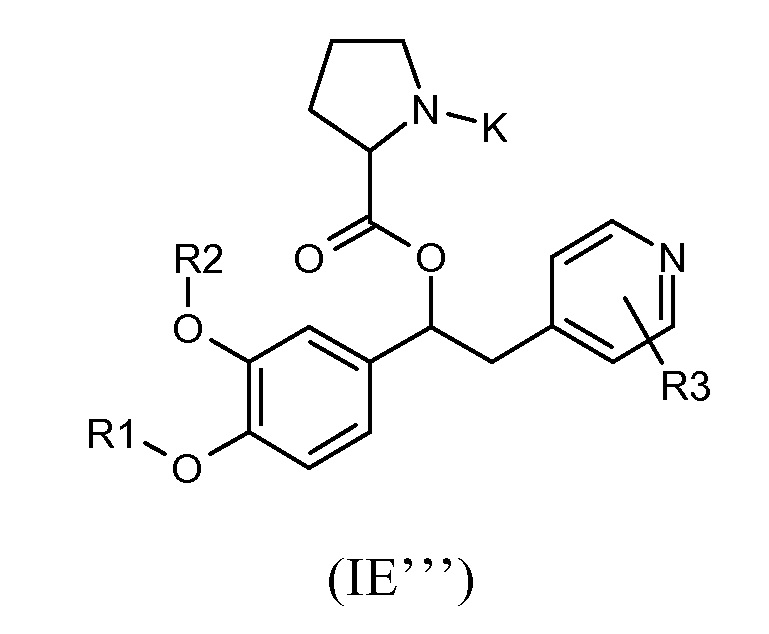
где R1, R2, R3 и K такие, как определено выше, для соединения формулы (I), Z представляет собой связь, R19 представляет собой водород, A представляет собой пирролидиновую двухвалентную группу радикала, и стереогенный центр имеет абсолютную конфигурацию, как указано выше; и соответствующий N-оксид на пиридиновом кольце, или их фармацевтически приемлемые соли.
В одном из вариантов осуществления для соединений формулы (IE) или (IE'''), R1 представляет собой (C1-C6) галогеналкил, R2 представляет собой (C1-C6) алкил, который замещен (C3-C7) циклоалкилом, 2-пиридинильное кольцо замещено в положениях 3 и 5 двумя группами хлора R3, и K представляет собой группу
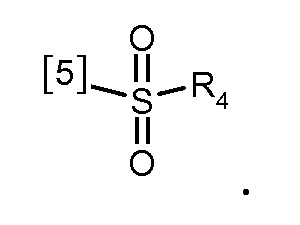
В одном из предпочтительных вариантов осуществления K выбран из списка групп, показанных ниже:
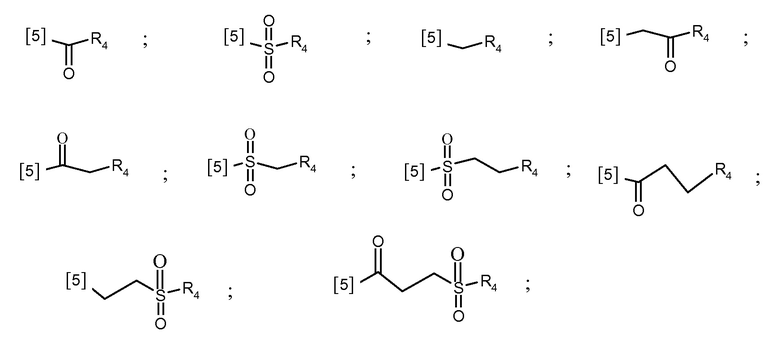
где символ [5] указывает на место соединения группы К с группой А.
В другом предпочтительном варианте осуществления K выбран из из списка групп, показанных ниже:
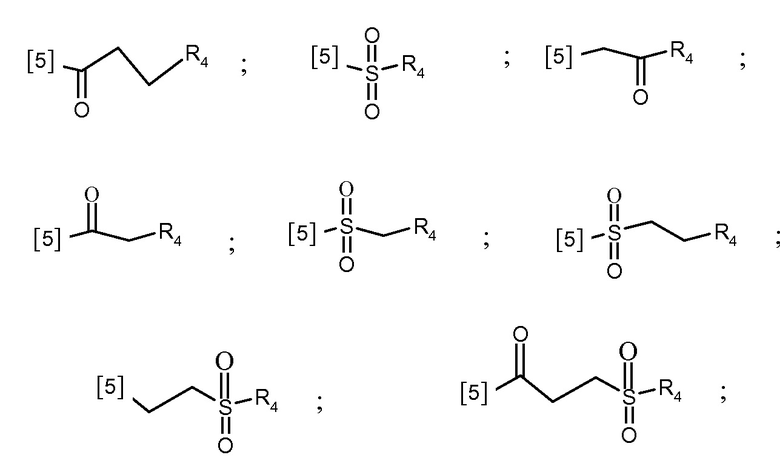
где символ [5] указывает на место соединения группы К с группой А.
В еще одном предпочтительном варианте осуществления K выбран из списка групп, показанных ниже:
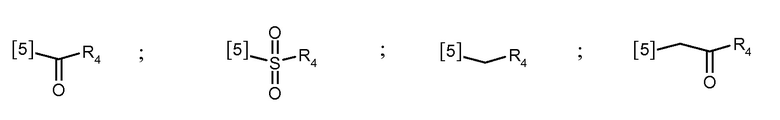
где символ [5] указывает на место соединения группы К с группой А.
В другом предпочтительном варианте осуществления K представляет собой группу
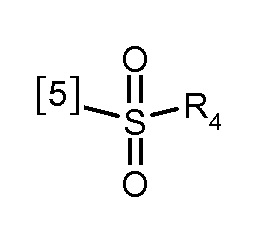
где символ [5] указывает на место соединения группы К с группой А.
В предпочтительном варианте осуществления R4 выбран из группы, состоящей из: фенильной группы, 5,6-членной гетероарильной группы, моноциклического (C3-C7) гетероциклоалкила и бициклической кольцевой системы; и каждый из которых необязательно замещен одной или несколькими группами R5.
В одном из предпочтительных вариантов осуществления R4 представляет собой фенильную группу или 5,6-членную гетероарильную группу, каждая из которых необязательно замещена одной или несколькими группами R5.
В еще одном предпочтительном варианте осуществления R4 представляет собой фенильную группу, которая необязательно замещена одной или несколькими группами R5.
В еще одном предпочтительном варианте осуществления R4 представляет собой 5,6-членную гетероарильную группу, которая необязательно замещена одной или несколькими группами R5.
В другом предпочтительном варианте осуществления R4 представляет собой моноциклический (C3-C7) гетероциклоалкил, необязательно замещенный одной или несколькими группами R5.
В еще одном предпочтительном варианте осуществления R4 представляет собой бициклическую кольцевую систему, необязательно замещенную одной или несколькими группами R5.
В одном из предпочтительных вариантов осуществления число заместителей R5 равно нулю, 1 или 2. В еще одном предпочтительном варианте осуществления такое число равно 1.
В одном из предпочтительных вариантов осуществления R5 независимо выбран из группы, состоящей из:
- (C1-C6) алкила, необязательно замещенного одним или нескольким группами, независимо выбранными из списка, состоящего из: (C3-C7) циклоалкила, -OH и группы -NR18C(O)(C1-C4) алкила, где R18 представляет собой водород или (C1-C4) алкил - (C3-C7) гетероциклоалкил;
- 5,6-членного гетероарила, который необязательно замещен одной или двумя группами (C1-C4) алкила;
- (C1-C6) галогеналкила;
- (C3-C7) гетероциклоалкил(C1-C4) алкила;
- группы -OR6, где R6 выбран из группы, состоящей из:
- (C1-C6) галогеналкила;
- (C1-C10) алкила, необязательно замещенного одним или несколькими (C3-C7) циклоалкилами;
- группы -SO2R7, где R7 представляет собой (C1-C4) алкил;
- атомов галогена;
- циано;
- NR8R9, где R8 и R9 являются одинаковыми или различными и независимо выбраны из группы, состоящей из:
- H;
- (C1-C6) алкила, необязательно замещенного (C3-C7) циклоалкилом, (C3-C7) гетероциклоалкилом;
- группы -SO2R15, где R15 представляет собой (C1-C4) алкил;
или они образуют с атомом азота, к которому они присоединены, насыщенное или частично насыщенное гетероциклическое кольцо, который необязательно замещено одним или несколькими (C1-C6) алкильными или оксогруппами;
- (C1-C4) алкилен-NR8R9;
- COR10, где R10 представляет собой фенил или (C1-C6) алкил;
- оксо;
- SO2R11, где R11 представляет собой NR8R9, где R8 и R9 такие, как указано выше;
- -COOR12, где R12 представляет собой H или (C1-C4) алкил или (C1-C4) алкилен-NR8R9, где R8 и R9 такие, как указано выше; и
- -CONR8R9, где R8 и R9 такие, как определено выше.
В другом предпочтительном варианте осуществления R5 независимо выбран из группы, состоящей из:
- (C1-C6) алкила;
- (C3-C7) гетероциклоалкила;
- (C3-C7) гетероциклоалкил(C1-C4) алкила;
- группы -OR6, где R6 выбран из группы, состоящей из:
- (C1-C6) галогеналкила;
- (C1-C10) алкила, необязательно замещенного одним или несколькими (C3-C7) циклоалкилами;
- группы -SO2R7, где R7 представляет собой (C1-C4) алкил;
- атомов галогена;
- NR8R9, где R8 и R9 являются одинаковыми или различными и независимо выбраны из группы, состоящей из:
- H;
- (C1-C6) алкила, необязательно замещенного (C3-C7) циклоалкилом, (C3-C7) гетероциклоалкилом;
- группы -SO2R15, где R15 представляет собой (C1-C4) алкил;
или они образуют вместе с атомом азота, к которому они присоединены, насыщенное или частично насыщенное гетероциклическое кольцо, которое необязательно замещено одной или несколькими (C1-C6) алкильными или оксогруппами;
- (C1-C4) алкилен-NR8R9;
- COR10, где R10 представляет собой фенил или (C1-C6) алкил;
- оксо;
- -SO2R11, где R11 представляет собой NR8R9, где R8 и R9 такие, как указано выше;
- -COOR12, где R12 представляет собой H или (C1-C4) алкил или (C1-C4) алкилен-NR8R9, где R8 и R9 такие, как указано выше; и
- -CONR8R9, где R8 и R9 такие, как определено выше.
В другом предпочтительном варианте осуществления R5 независимо выбран из группы, состоящей из:
- (C1-C6) алкила;
- (C3-C7) гетероциклоалкил(C1-C4) алкила;
- группы -OR6, где R6 представляет собой (C1-C10) алкил, необязательно замещенный одним или несколькими (C3-C7) циклоалкилами;
- атомов галогена;
- NR8R9, где R8 и R9 являются одинаковыми или различными и независимо выбраны из группы, состоящей из:
- H;
- (C1-C6) алкила, необязательно замещенного (C3-C7) циклоалкилом, (C3-C7) гетероциклоалкилом;
- группы -SO2R15, где R15 представляет собой (C1-C4) алкил;
или они образуют вместе с атомом азота, к которому они присоединены, насыщенное или частично насыщенное гетероциклическое кольцо, который необязательно замещено (C1-C6) алкилом или оксо;
- -COOR12, где R12 представляет собой H или (C1-C4) алкил или (C1-C4) алкилен-NR8R9, где R8 и R9 такие, как указано выше и
- -CONR8R9, где R8 и R9 такие, как определено выше.
В еще одном предпочтительном варианте осуществления R5 выбран из группы, состоящей из:
- -(C1-C6) алкил;
- -NR8R9, где R8 и R9 являются одинаковыми или различными и независимо выбраны из группы, состоящей из:
- H;
- (C1-C6) алкил; и
- CONR8R9, где R8 и R9 такие, как определено выше.
Дополнительная предпочтительная группа соединений формулы (I) такая, как показано ниже в соответствии с общей формулой (IF):
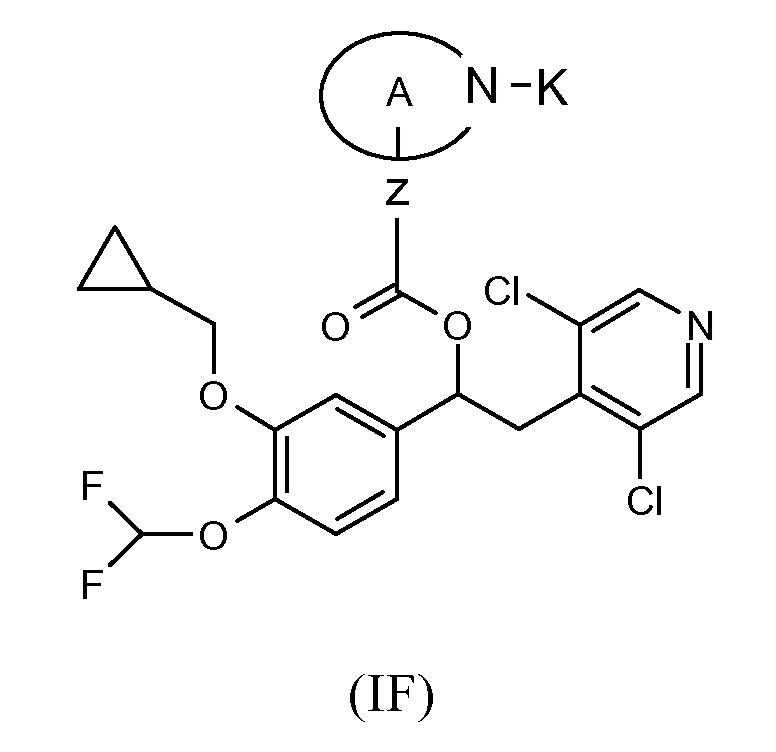
где Z представляет собой связь, R19 представляет собой водород, A представляет собой группу (C3-C7) гетероциклоалкилена, содержащую атом азота, который представляет собой место присоединения к группе K, где K выбран из перечня групп, состоящего из:

R4 представляет собой фенильную группу или 5,6-членную гетероарильную группу, каждая из которых необязательно замещена одной или несколькими группами R5; и соответствующий N-оксид на пиридиновом кольце, или их фармацевтически приемлемые соли.
В соответствии с предпочтительным вариантом осуществления, настоящее изобретение относится к соединениям, указанным в таблице А (см. в конце описания), или их фармацевтически приемлемые соли или сольваты.
В другом предпочтительном варианте осуществления изобретения соединения по изобретению выбраны из группы, состоящей из:
1-оксида 4-((S)-2-((S)-3-(4-аминофенилсульфонил)тиазолидин-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)-3,5-дихлорпиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-1-(3-(диметилкарбамоил)фенилсульфонил)пирролидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((R)-3-(3-(диметилкарбамоил)фенилсульфонил)тиазолидин-4-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(3-(диметилкарбамоил)фенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-1-(3-(диметилкарбамоил)фенилсульфонил)-пиперидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-1-(3-(диметилкарбамоил)-4-метоксифенилсульфонил)-пиперидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(3-(4-метилпиперазин-1-карбонил)фенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(пиридин-3-илсульфонил)тиазолидин-2-карбонилокси)этил)-пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(3-(диметилкарбамоил)-4-метоксифенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(фенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(1-метил-1H-имидазол-2-илсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(3-сульфамоилфенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(4-(метилсульфонил)фенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(3,4-диметоксифенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(3-(N,N-диметилсульфамоил)фенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(4-метоксифенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(5-(пирролидин-1-карбонил)-1H-пиррол-3-илсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(1-метил-5-(метилкарбамоил)-1H-пиррол-3-илсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(1,3-диметил-1H-пиразоло[3,4-b]пиридин-5-илсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(3-метилизоксазоло[5,4-b]пиридин-5-илсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(3-(гидроксиметил)фенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(3-уреидофенилсульфонил)тиазолидин-2-карбонилокси)этил)-пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(1-метил-2-оксоиндолин-5-илсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-((S)-3-(4-цианофенилсульфонил)тиазолидин-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)-пиридина;
и их фармацевтически приемлемые соли или сольваты.
В одном из аспектов настоящего изобретения приведен способ получения соединений по изобретению, в соответствии с общими путями синтеза, представленными на схеме 1 ниже, где сделана ссылка на специфические схемы синтеза, которые более подробно описаны в нижеследующих параграфах.
Способы, которые можно использовать и которые описаны ниже и изображены на схемах, не следует рассматривать как ограничивающие рамки способов синтеза, доступных для получения соединений по изобретению.
Схема 1
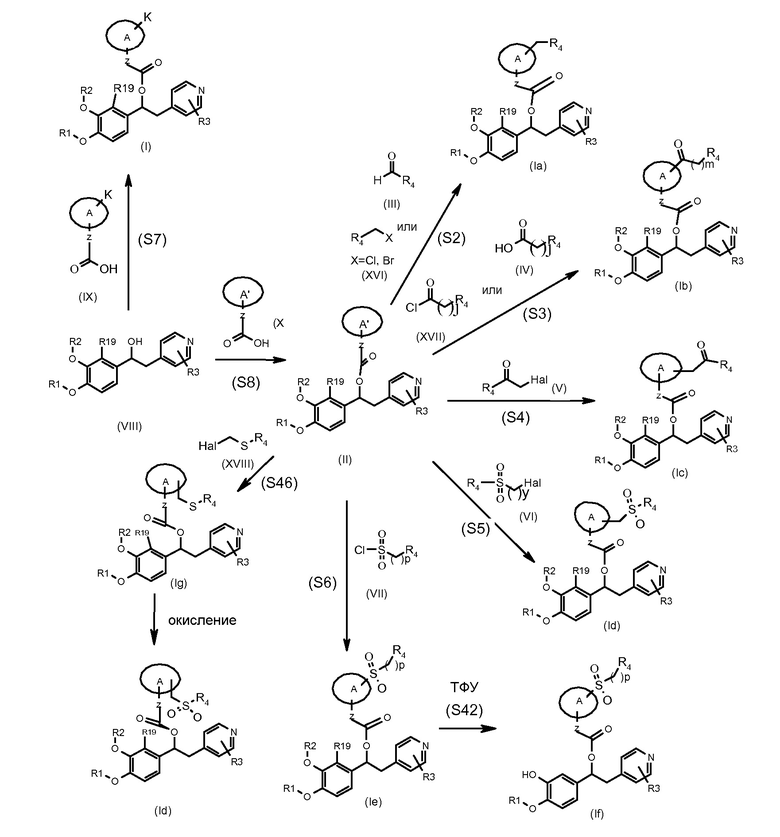
На нижеприведенных схемах для соединений формулы (II) - (XIX), если не указано иного, группы R1-R20, Z, A и K имеют те же значения, которые описаны для соединений формулы (I) выше.
Соединения формулы (Ia), т.е. соединения формулы (I), где K представляет собой группу -(CH2)R4, могут быть получены в соответствии со схемой 2a ниже, где представлена реакция соединения формулы (II), где A' представляет собой (C3-C7) гетероциклоалкиленовую группу, содержащую группу -NH-, с соответствующим соединением формулы (III).
Схема 2 (S2a)
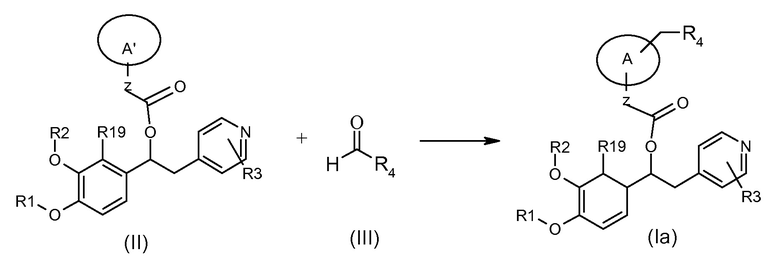
Обычные условия реакции включают взаимодействие соединения формулы (II) с соединением формулы (III) в подходящем биполярном растворителе, таком как ТГФ, метанол, этанол или ДХМ, в присутствии подходящего восстановителя, такого как триацетоксиборгидрид натрия, цианоборгидрид натрия или боргидрид натрия, и подходящей кислоты, такой как уксусная кислота, HCl в метаноле или ацетат аммония. Может быть эффективным получение имина перед добавлением восстановителя. Реакционную смесь плавно проводили при комнатной температуре в течение от 1 до 12 часов.
Альтернативно соединения формулы (Ia), т.е. соединения формулы (I), в которых K представляет собой группу -(CH2)R4, могут быть получены в соответствии со схемой 2b ниже, на которой показана реакция соединения формулы (II), где A' представляет собой (C3-C7) гетероциклоалкиленовую группу, содержащую группу -NH-, с подходящим соединением формулы (XVI).
Схема 2b (S2b)
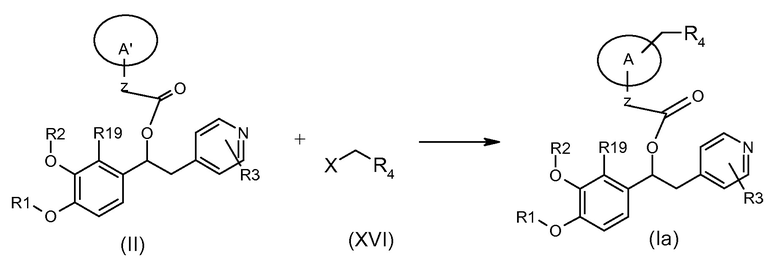
Обычные условия реакции включают взаимодействие соединения формулы (XVI), где X представляет собой уходящую группу, такую как Cl или Br, с соединением формулы (II) в подходящем полярном апротонном растворителе, таком как ацетонитрил или ДМФ, в присутствии подходящего основания, такого как K2CO3, бикарбонат щелочного металла, ТЭА или DIPEA, при температуре в диапазоне от комнатной температуры до 70°C.
Соединения формулы (Ib), т.е. соединения формулы (I), где K представляет собой группу -C(O)(CH2)jR4, могут быть получены в соответствии со схемой 3a ниже, на которой показана реакция соединения формулы (II), как указано выше, с подходящим соединением формулы (IV).
Схема 3 (S3a)
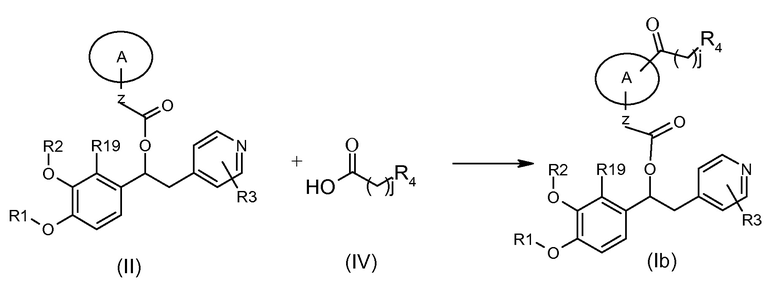
Обычные условия реакции включают взаимодействие соединения формулы (II) с соединением формулы (IV) в подходящем биполярном апротонном растворителе, таком как ДМФ, хлороформ или ДХМ, в присутствии подходящего конденсирующего агента, такого как EDC, DCC, HOBT, HOAT или CDI, и, если необходимо, подходящего агента, такого как DMAP, HOBT, 4-пирролидинопиридина (4-PPY) или другого 4-алкиламинопиридина, при комнатной температуре.
Альтернативно, соединения формулы (Ib), т.е. соединения формулы (I), в которой K представляет собой группу -C(O)(CH2)jR4, могут быть получены в соответствии со схемой 3b ниже, на которой показана реакция соединения формулы (II), как указано выше, с подходящим соединением формулы (XVII).
Схема 3b (S3b)
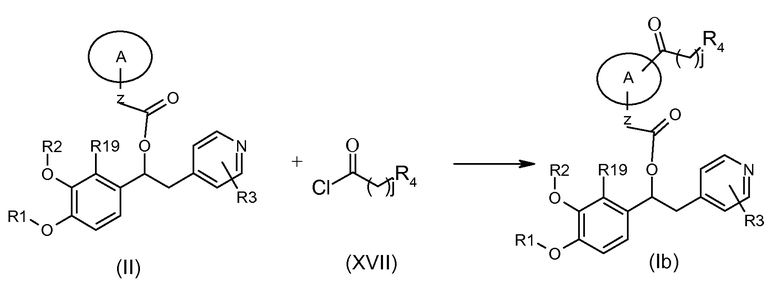
Обычные условия реакции включают взаимодействие соединения формулы (II) с соединением формулы (XVII) в подходящем растворителе, таком как пиридин или ДХМ, и в присутствии, если необходимо, подходящего основания, такого как ТЭА, DIPEA, DBU или другого органического основания при температуре в диапазоне от 0°C до комнатной.
Соединения формулы (Ic), т.е. соединения формулы (I), где K представляет собой группу -(CH2)mC(O)R4, могут быть получены в соответствии со схемой 4 ниже, на которой показана реакция соединения формулы (II), как указано выше, с подходящим соединением формулы (V), где Hal представляет собой подходящую удаляемую группу галогена.
Схема 4 (S4)

Обычные условия реакции включают взаимодействие соединения формулы (II) с соединением формулы (V) в подходящем полярном апротонном растворителе, таком как ДМФ или ацетонитрил, в присутствии подходящего основания, такого как K2CO3, бикарбонат щелочного металла, ТЭА или DIPEA, при температуре в диапазоне от комнатной до 50°C.
Соединения формулы (Id), т.е. соединения формулы (I), где K представляет собой группу -(CH2)ySO2R4, могут быть получены в соответствии со схемой 5 ниже, на которой показана реакция соединения формулы (II), как указано выше, с подходящим соединением формулы (VI), где Hal представляет собой подходящую удаляемую группу галогена.
Схема 5 (S5)
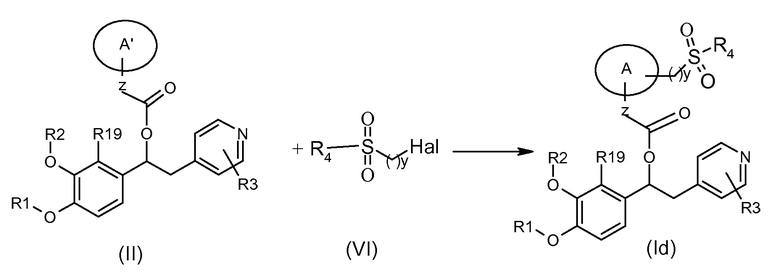
Обычные условия реакции включают взаимодействие соединения формулы (II) с соединением формулы (VI) в подходящем полярном апротонном растворителе, таком как ДМФ или ацетонитрил, в присутствии подходящего основания, такого как K2CO3, бикарбонат щелочного металла, ТЭА или DIPEA, при температуре в диапазоне от комнатной до 50°C.
Соединения формулы (Id), т.е. соединения формулы (I), где K представляет собой группу -(CH2)ySO2R4 и y равно 1, также могут быть получены в соответствии со схемой 46 ниже, на которой показана реакция соединения формулы (II), как указано выше, с подходящим соединением формулы (XVIII), где Hal представляет собой подходящую удаляемую группу галогена.
Схема 46 (S46)
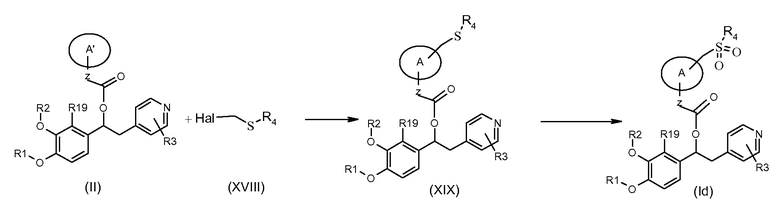
Обычные условия реакции включают взаимодействие соединения формулы (II) с соединением формулы (XVIII) в подходящем полярном апротонном растворителе, таком как ДМФ или ацетонитрил, в присутствии подходящего основания, такого как K2CO3, бикарбонат щелочного металла, ТЭА или DIPEA, при температуре в диапозоне от комнатной до 50°C. Соединение (XIX), полученное таким образом, последовательно взаимодействовало с подходящим окислителем, таким как MCPBA или перекись водорода, в подходящем полярном растворителе, таком как ДХМ, хлороформ, EtOH или MeOH, при температуре в диапазоне от комнатной до 60°C.
Соединения формулы (Ie), т.е. соединения формулы (I), где K представляет собой группу -SO2(CH2)pR4, могут быть получены в соответствии со схемой 6 ниже, на которой показана реакция соединения формулы (II), как указано выше, с подходящим соединением формулы (VII).
Схема 6 (S6)
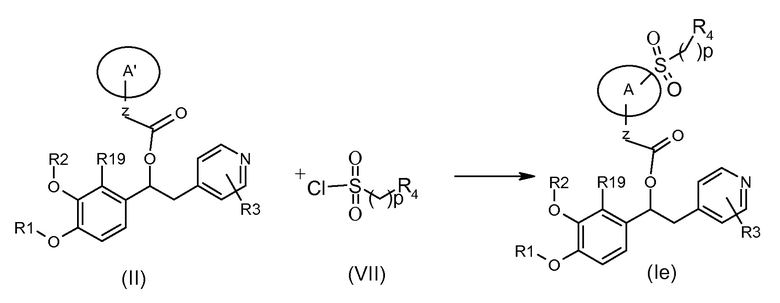
Обычные условия реакции включают взаимодействие соединения формулы (II) с соединением формулы (VII) в подходящем растворителе, таком как пиридин или ДХМ, и в присутствии, если необходимо, подходящего основания, такого как ТЭА, DIPEA, DBU, или другого органического основания, при температуре в диапазоне от 0°C до комнатной температуры.
Альтернативно, соединения формулы (I) могут быть получены в соответствии со схемой 7 ниже, на которой показана реакция соединения формулы (VIII) с подходящим соединением формулы (IX).
Схема 7 (S7)
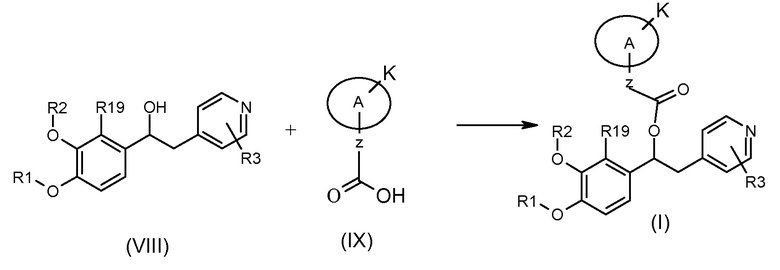
Обычные условия реакции включают взаимодействие соединения формулы (VIII) с соединением формулы (IX) в подходящем полярном апротонном растворителе, таком как ДМФ, ТГФ, хлороформ или ДХМ, в присутствии подходящего конденсирующего агента, такого как EDC, DCC или CDI, и подходящего агента, такого как DMAP, HOBT, 4-пирролидинопиридин (4-PPY) или другого 4-алкиламинопиридина, при комнатной температуре; удаление возможно присутствующей защитной группы осуществляли в условиях, известных специалисту в данной области или описанных в «Protection groups in organic synthesis» T.W. Green и P.Wutz (Wiley-Interscience publication, 1999).
Соединения формулы (II), как указано выше, могут быть получены в соответствии со схемой 8 ниже, на которой показана реакция соединения формулы (X), где A'' представляет собой (C3-C7) гетероциклоалкиленовую группу, содержащую группу -N-, которая защищена подходящей защитной группой, с подходящим соединением формулы (XI), а затем путем удаления N-защитной группы в подходящих условиях.
Схема 8 (S8)
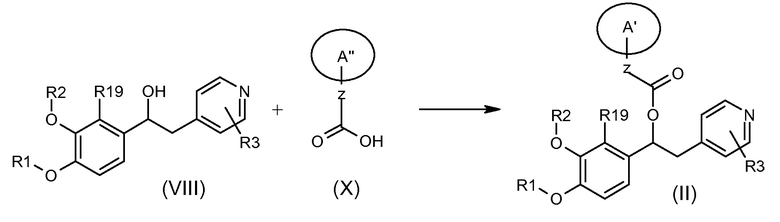
Обычные условия для реакции связывания включают реакцию соединения формулы (VIII) с соединением формулы (X) в подходящем полярном апротонном растворителе, таком как ДМФ, ТГФ, хлороформ или ДХМ, в присутствии подходящего (связывающего) конденсирующего агента, такого как EDC, DCC или CDI, и подходящего агента, такого как DMAP, HOBT, 4-пирролидинопиридин (4-PPY) или другого 4-алкиламинопиридина, при комнатной температуре; удаление защитной группы проводят в условиях, известных специалисту в данной области или описанных в «Protection groups in organic synthesis» T.W. Green и P.Wutz (Wiley-Interscience publication, 1999), например, если защитная группа представляет собой трет-бутоксикарбонильную группу, то удаление защиты может быть подходящим способом осуществлено в кислых условиях (таких как HCl в диоксане или в AcOEt или ТФУ в CH2Cl2).
Альтернативно, может быть получен соответствующий ацилхлорид реакцией соединения (X) с оксалилхлоридом или тионилхлоридом или другими реагентами, хорошо известными специалистам в данной области, в подходящем апротонном растворителе, таком как ДХМ при 0 градусе, в присутствии, если необходимо, каталитического количества ДМФ, и с постепенным добавлением соединения (VIII), и подходящем основании, таком как ТЭА или DIPEA.
Соединения формулы (If), т.е. соединения формулы (I), где K представляет собой группу -SO2(CH2)pR4, и R2 представляет собой водород, могут быть получены в соответствии со схемой 42 ниже, на которой показана реакция соединения формулы (Ie), как указано выше, где R2 представляет собой (C1-C6) алкил, необязательно замещенный одним (C3-C7) циклоалкилом в подходящих условиях.
Схема 42 (S42)
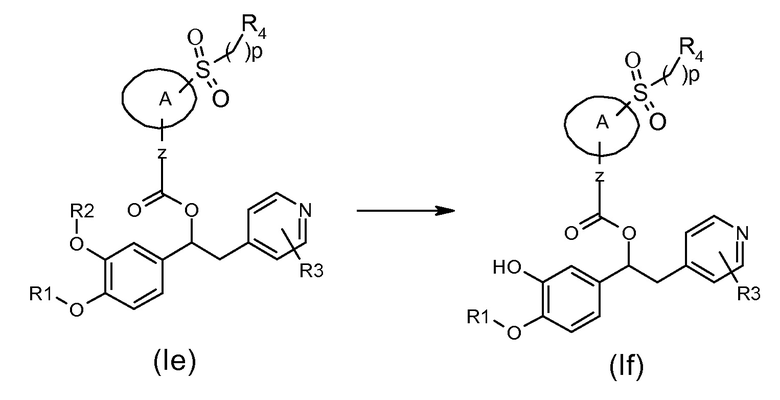
Обычные условия реакции включают взаимодействие соединения формулы (Ie), как указано выше, с подходящей кислотой, такой как ТФУ или BBr3 или BCl3, при температуре в диапазоне от комнатной до 40 градусов.
N-оксиды на 2-пиридинильном кольце соединений общей формулы (I) и их варианты осуществления могут быть получены в соответствии со способами, известными из литературы, и хорошо известны специалистам в данной области. Например, они могут быть получены путем растворения соединения общей формулы (I) или их варианты осуществления в CH2Cl2 или CHCl3, затем добавляя окислитель, такой как м-хлорпербензойная кислота (mCPBA), в полученный раствор. Другие окислительные агенты, которые могут быть использованы, представляют собой перекись водорода, пербензойную кислоту и перуксусную кислоту.
Альтернативно, в частности, для тех соединений, в которых A или A' представляют собой кольцо, замещенное функциональной группой, чувствительной к окислению, соответствующие N-оксиды получают путем проведения стадии окисления перед введением дополнительных функциональных групп, например, на соединения формулы (II) или (VIII).
В предпочтительном варианте осуществления способ получения соединений формулы (I) или их вариантов осуществления проводят, начиная с N-оксида на пиридиновом кольце соединения формулы (VIII), таким образом, обеспечивая получение соединения формулы (I) или их вариантов осуществления в форме N-оксидов на пиридиновом кольце.
Соединения общей формулы (III), (IV), (V), (VI), (VII), (VIII), (IX), (XVI), (XVII), (XVIII), (XIX) и (X) могут быть коммерчески доступны, их препараты могут быть специфически описаны в литературе или они могут быть получены в соответствии со способами, доступными из литературы и известными специалисту в данной области.
В частности, соединения формулы (VIII) и соответствующие N-оксиды на пиридиновом кольце также могут быть получены, как описано в международной патентной заявке WO2009/018909 или WO2010/089107.
В одном из вариантов осуществления предпочтительный способ получения соединений формулы (IDa), т.е. N-оксидных производных на пиридиновом кольце соединений формулы (ID), где R9 представляет собой водород, K представляет собой группу -SO2(CH2)pR4 и где абсолютная конфигурация при стереогенных центрах показана ниже, представлен на схеме 43, приведенной ниже:
Схема 43 (S43)
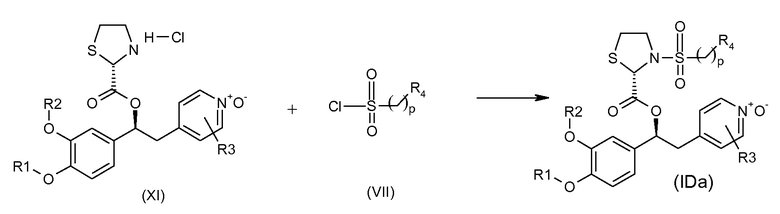
Обычные условия реакции способа, описанного на схеме 43, включают: a) добавление раствора соединения формулы (VII) в пиридине (3-30 об., предпочтительно 8 об.) в охлажденный раствор соединения формулы (XI), в пиридине (3-30 об., предпочтительно 8 об.), перемешивая полученный раствор при комнатной температуре; c) выливание раствора в водный раствор HCl в избытке; d) фильтрование осажденного вещества и промывку водой или d') экстрагирование водной фазы AcOEt, промывку водным раствором 1 M HCl, насыщенным солевым раствором и упаривание полученной органической фазы; и, необязательно, e) растворение твердого вещества, полученного на стадии d) или d'), в AcOEt, помещая его на подушки из силикагеля, элюируя AcOEt/MeOH [100:0 - (90:10)] и упаривая под вакуумом полученный раствор, или e') очистку продукта флэш-хроматографией, элюируя ДХМ/i-PrOH.
В более предпочтительном варианте осуществления соединения формулы (IDa), полученные как указано выше в соответствии со схемой 43, кристаллизуют способом, включающим: f) растворение соединений в EtOH (8 об.); g) энергичное перемешивание в течение ночи при комнатной температуре; h) фильтрацию образовавшегося твердого вещества; и, необязательно, i) промывку твердого вещества, полученного на стадии h) EtOH (2 об.) и l) сушки твердого вещества под вакуумом.
В дополнительном предпочтительном варианте осуществления стадию 1) схемы 43 проводят путем сушки первого твердого вещества под вакуумом при комнатной температуре, а затем путем сушки под вакуумом при 60°C.
В одном из вариантов осуществления предпочтительный способ предназначен для получения соединений формулы (XI), как указано выше, в соответствии со схемой 44, как указано ниже:
Схема 44 (S44)
Обычные условия реакции для способа, описанного на схеме 44, включают: a) добавление при перемешивании раствора концентрированной HCl (приблизительно 5M; с большим избытком) в сухом AcOEt (9 об.) к раствору соединения формулы (XII) в AcOEt (6 об.) при комнатной температуре; b) перемешивание; c) фильтрацию осажденного твердого веществ; необязательно, d) промывку полученного твердого вещества AcOEt; и, необязательно, e) сушку твердого вещества, полученного под вакуумом при комнатной температуре.
В одном из вариантов осуществления предпочтительный способ предназначен для получения соединений формулы (XII), как указано выше, в соответствии со схемой 45, как указано ниже:
Схема 45 (S45)
Обычные условия реакции для способа, описанного на схеме 45, включают: a) добавление соединения формулы (XIV), DMAP и EDC к раствору соединения формулы (XV) в ДМФ; b) перемешивание смеси, предпочтительно, в течение ночи; c) выливание смеси в холодную воду; d) фильтрование остатка; необязательно, e) растворение остатка в ДХМ, промывая раствор водой, высушивая и упаривая растворитель; и, необязательно, f) растворение твердого вещества, полученного на стадии d) или e), в кипящем MTBE (3,5 об.), добавляя петролейный эфир (4 об.) при перемешивании, перемешивая при комнатной температуре, фильтруя полученное твердое вещество и высушивая его при комнатной температуре под вакуумом.
В предпочтительном варианте осуществления способы в соответствии со схемами 43, 44 и 45 осуществляют последовательно с получением кристаллического соединения формулы (IDa).
В предпочтительном варианте осуществления способ предназначен для получения соединений формулы (IDaa), т.е. соединение формулы (IDa), где R1 представляет собой (C1-C6) галогеналкил, R2 представляет собой (C1-C6) алкил, который замещен (C3-C7) циклоалкилом, 2-пиридинильное кольцо замещено в 3 и 5 положении двумя хлорами групп R3, K представляет собой группу
и R4 представляет собой фенильную группу, которая необязательно замещена одной или несколькими группами R5; где способ включает последовательное проведение реакций, как указано на схемах 43, 44 и 45 выше.
В одном из аспектов настоящего изобретения соединения формулы (II), их N-оксиды на пиридиновом кольце или их соли представлены в виде промежуточных соединений в способе получения соединений формулы (I)
где A' представляет собой (C3-C7) гетероциклоалкиленовую группу содержащую группу -NH-.
В одном из вариантов осуществления соединения формулы (XI), как указано выше, представлены в виде промежуточных соединений в способе получения соединений формулы (IDa). В дополнительном предпочтительном варианте осуществления для соединений формулы (XI), R1 представляет собой (C1-C6) галогеналкил, и R2 представляет собой (C1-C6) алкил, который замещен (C3-C7) циклоалкилом, и пиридиновое кольцо замещено двумя группами R3, расположенными в положении 3 и 5.
В другом аспекте настоящее изобретение относится к соединениям формулы (II), как указано выше, которые действуют как ингибиторы фермента фосфодиэстеразы 4 (PDE4), таким образом удовлетворяя указанную выше потребность в обнаружении дополнительных ингибиторов PDE4, обеспечивающих высокую аффинность в отношении фермента PDE4 и способных продемонстрировать подходящие характеристики эффективности при ингаляторном лечении, например, пониженные побочные эффекты.
Также настоящее изобретение относится к композициям, содержащим соединения формулы (II), и к их терапевтическому применению.
Если возможно, то предпочтительные варианты осуществления и группы, описанные выше для соединений формулы (I), применимы для соединений формулы (II), а также mutatis mutandis.
Описанный способ особенно эффективен, так как он допускает соответствующее модулирование с помощью любого подходящего варианта, известного специалисту в данной области, таким образом, с получением любого желаемого соединения по изобретению. Такие варианты входят в объем настоящего изобретения.
Из вышеуказанного специалисту в данной области будет ясно, что любая из описанных групп может быть представлена сама по себе или в любой подходящей защищенной форме.
В частности, функциональные группы, присутствующие в соединениях формулы (II), (III), (IV), (V), (VI), (VII), (VIII), (IX), (X), (XI), (XII), (XIII), (XIV), (XV), (XVI), (XVII), (XVIII) и (XIX) и которые могут дать нежелательную побочную реакцию и побочные продукты, должны быть соответствующим образом защищены перед алкилированием, ацилированием, реакцией присоединения, окислением или сульфонилированием. Также последующее снятие защиты с групп с этими защитными группами может происходить после завершения указанных реакций.
В настоящем изобретение, если не указано иного, термин «защитная группа» означает защитную группу, предназначенную для защиты функциональной группы, к которой она присоединена. Как правило, защитные группы используют для защиты амино, гидроксильной или карбоксильной функциональной группы. Подходящие защитные группы могут таким образом включать, например, бензил, бензилоксикарбонил, трет-бутоксикарбонил, алкиловый или бензиловый эфиры или тому подобное, которые хорошо известны специалистам в данной области [см., в качестве общей информации, T.W. Green; Protective Groups in Organic Synthesis (Wiley, N.Y. 1999)].
Также селективная защита и снятия защиты любой указанной группы, например, включая карбонильную, гидроксильную или аминогруппы, может быть осуществлена в соответствии с широко известными способами, обычно используемыми в химии органического синтеза.
Необязательное солеобразование соединений формулы (I) или их N-оксидов на пиридиновом кольце может быть осуществлено соответствующим преобразованием любой свободной кислотной или аминогрупп до соответствующих фармацевтически приемлемых солей. В этом случае также были использованы рабочие условия для необязательного образования соли соединений по изобретению также находятся в пределах обычных знаний специалиста в данной области.
Из вышеуказанного специалисту в данной области будет ясно, что вышеописанный способ, объединяющий любой вариант получения подходящих соединений по изобретению, может быть модифицирован так, чтобы он был адаптирован для условий реакций с учетом конкретных потребностей, например, выбирая подходящие конденсирующие агенты, растворители и защитные группы, для каждого случая.
Настоящее изобретение также относится к фармацевтическим композициям соединений по изобретению или соединений формулы (II) в смеси с одним или несколькими фармацевтически приемлемыми носителями, например, описанными в Remington's Pharmaceutical Sciences Handbook, XVII Ed., Mack Pub., N.Y., U.S.A.
Введение соединений по настоящему изобретению или соединений формулы (II) может быть осуществлено в соответствии с нуждами пациента, например, перорально, назально, парентерально (подкожно, внутривенно, внутримышечно, интрастернально и путем инфузии), с помощью ингаляции, ректально, вагинально, местно, трансдермально и с помощью введения в глаз. Различные твердые лекарственные формы для перорального введения могут быть использованы для введения соединений по изобретению, включая твердые формы, такие как таблетки, желатиновые капсулы, капсулы, каплеты, гранулы, таблетки-леденцы и порошкообразное вещество. Соединения по настоящему изобретению или соединения формулы (II) могут быть введены самостоятельно или в сочетании с различными фармацевтически приемлемыми носителями, разбавителями (такими как сахароза, маннит, лактоза, крахмалы) и эксципиентами, известными в данной области, включая в качестве неограничивающих примеров суспендирующие средства, солюбилизаторы, буферные средства, связывающие средства, дезинтегранты, консерванты, красители, ароматизаторы, смазочные средства и тому подобное. Капсулы, таблетки и гели с замедленным высвобождением также имеют преимущества при введении соединений по настоящему изобретению или соединений формулы (II).
Различные жидкие пероральные лекарственные формы также можно использовать для введение соединений по изобретению или соединений формулы (II), включая водный и неводные растворы, эмульсии, суспензии, сиропы и эликсиры. Такие лекарственные формы также могут содержать подходящие инертные разбавители, известные в данной области, такие как вода, и подходящие эксципиенты, известные в данной области, такие как консерванты, средства для смачивания, подсластители, ароматизаторы, а также средства для эмульгирования и/или суспендирования соединений по изобретению или соединений формулы (II). Соединения по настоящему изобретению или соединения формулы (II) могут быть инъецированы, например, внутривенно, в форме изотонического стерильного раствора. Другие препараты также возможны.
Суппозитории для ректального введения соединений по настоящему изобретению или соединений формулы (II) могут быть получены путем смешивания соединения с подходящим эксципиентом, таким как масло какао, салицилаты и полиэтиленгликоли.
Составы для вагинального введения могут быть в форме крема, геля, пасты, пены или спрея, содержащего, кроме активного ингредиента, такие подходящие носители, которые известны в данной области.
Для местного введения фармацевтические композиции могут быть в форме кремов, мазей, линиментов, лосьонов, эмульсий, суспензий, гелей, растворов, паст, порошков, спреев и капель, подходящих для нанесения на кожу, введения в глаза, уши или нос. Местное применение также может включать трансдермальное введение посредством средств, таких как трансдермальные пластыри.
Для лечения заболеваний дыхательных путей соединения по изобретению или соединения формулы (II) предпочтительно вводят путем ингаляции.
Препараты для ингаляции включают порошки для ингаляции, пропеллент-содержащие дозируемые аэрозоли или составы для ингаляции без пропеллента.
Для введения сухого порошка можно использовать единичные или мультидозируемые ингаляторы, известные из уровня техники. В этом случае порошок может быть наполнен в желатиновую, пластиковую или другие капсулы, картриджы или блистерные упаковки или в резервуар.
К порошкообразным соединениям по изобретению или соединениям формулы (II) может быть добавлен разбавитель или носитель, обычно нетоксичный и химически инертный в отношении соединения по изобретению, например, лактоза или любая другая добавка, подходящая для улучшения фракции для вдыхания.
Аэрозоли для ингаляции, содержащие газ пропеллент, такой как гидрофторалканы, могут содержать соединения по изобретению или соединения формулы (II) либо в форме раствора, либо в диспергируемой форме. Составы, вводимые с помощью пропеллента, могут также содержать другие ингредиенты, такие как сорастворители, стабилизаторы и необязательно другие эксципиенты.
Ингалируемые составы без пропеллента, включающие соединения по изобретению или соединения формулы (II), могут быть в форме растворов или суспензий в водной, спиртовой или водно-спиртовой среде и их можно доставлять с помощью струйных или ультразвуковых небулайзеров, известных из уровня техники, или с помощью ингалятора мягкого тумана, такого как Respimat®.
Соединения по изобретению или соединения формулы (II) могут вводиться как самостоятельное активное средство, так и в комбинации с другими фармацевтически активными ингредиентами, включая те, которые используются в настоящее время для лечения респираторных заболеваний, например, бета2-агонисты, антимускариновые средства, кортикостероиды, ингибиторы митоген-активируемых протеинкиназ (P38 MAPкиназа), ингибиторы ядерного фактора субъединицы каппа-B киназы бета (IKK2), ингибиторы эластазы нейтрофилов человека (HNE), ингибиторы фосфодиэстеразы 4 (PDE4), модуляторы лейкотриенов, нестероидные противовоспалительные средства (NSAID) и регуляторы секреции слизи.
Настоящее изобретение также относится к комбинациям соединения по изобретению или соединений формулы (II) с β2-агонистом, выбранным из группы, состоящей из: кармотерола, GSK-642444, индакатерола, милветерола, арформотерола, формотерола, сальбутамола, левалбутерола, тербуталина, AZD-3199, BI-1744-CL, LAS-100977, бамбутерола, изопротеренола, прокатерола, кленбутерола, репротерола, фенотерола и ASF-1020 и их соли.
Настоящее изобретение также относится к комбинациям соединения по изобретению или соединения формулы (II) с кортикостероидами, выбранными из группы, состоящей из: флутиказона пропионата, флутиказона фуроата, мометазона фуроата, беклометазона дипропионата, циклесонида, будесонида, GSK 685698, GSK 870086.
Настоящее изобретение также относится к комбинациям соединения по изобретению или соединения формулы (II) с антимускариновым средством, выбранным из группы, состоящей из: солей аклидиния, тиотропия, ипратропия, троспия, гликопиррония и окситропия.
Настоящее изобретение также относится к комбинациям соединения по изобретению или соединения формулы (II) с ингибитором PDE4, выбранными из группы, состоящей из: AN-2728, AN-2898, CBS-3595, апремиласта, ELB-353, KF-66490, K-34, LAS-37779, IBFB-211913, AWD-12-281, ципамфиллина, циломиласта, рофлумиласта, BAY19-8004 и SCH-351591, AN-6415, INDUS-82010, TPI-PD3, ELB-353, CC-11050, GSK-256066, оглемиласта, OX-914, тетомиласта, MEM-1414 и RPL-554.
Настоящее изобретение также относится к комбинациям соединения по изобретению или соединения формулы (II) с ингибитором P38 MAPкиназы, выбранным из группы, состоящей из: семапимода, талмапимода, пирфенидона, PH-797804, GSK-725, минокина и лосмапимода и их солей.
В предпочтительном варианте осуществления настоящее изобретение относится к комбинациями соединения по изобретению или соединения формулы (II) с ингибитором IKK2.
Изобретение также относится к комбинациям соединения по изобретению или соединения формулы (II) с ингибитором HNE, выбранным из группы, состоящей из: AAT, ADC-7828, аэрива, TAPI, AE-3763, KRP-109, AX-9657, POL-6014, AER-002, AGTC-0106, респрива, AZD-9668, земаира, AAT IV, PGX-100, элафина, SPHD-400, проластина C и проластина для ингаляций.
Изобретение также относится к комбинациям соединения по изобретению или соединения формулы (II) с модулятором лейкотриена, выбранным из группы, состоящей из: монтелукаста, зафирлукаста и пранлукаста.
Изобретение также относится к комбинациям соединения по изобретению или соединения формулы (II) с NSAID, выбранными из группы, состоящей из: ибупрофена и кетопрофена.
Изобретение также относится к комбинациям соединения по изобретению или соединения формулы (II) с регуляторами образования слизи, выбранными из группы, состоящей из: INS-37217, диквафозола, сибенадета, CS-003, талнетанта, DNK-333, MSI-1956 и гефинитиба.
Дозы соединений по настоящему изобретению зависят от различных факторов, включая конкретное заболевание, на которое направлено лечение, тяжесть симптомов, путь введения, частоту интервалов введения доз, конкретное используемое соединение, эффективность, токсикологический профиль и фармакокинетический профиль соединения.
Наиболее эффективно соединения по изобретению или соединения формулы (II) могут вводиться, например, в дозе от 0,001 до 1000 мг/день, предпочтительно от 0,1 до 500 мг/день.
Если их вводят путем ингаляции, то доза соединений по изобретению или соединений формулы (II) предпочтительно составляет от 0,01 до 20 мг/день, предпочтительно от 0,1 до 10 мг/день.
Предпочтительно, соединения по изобретению или соединения формулы (II), самостоятельно или в комбинации с другими активными ингредиентами, могут вводиться для профилактики и/или лечения любого обструктивного респираторного заболевания, такого как астма, хронический бронхит и хроническое обструктивное заболевание легких (ХОБЛ).
Однако соединения по изобретению или соединения формулы (II) можно вводить для профилактики и/или лечения любых заболеваний, когда требуется ингибитор PDE4. Указанные заболевание включают аллергические болезненные состояния, такие как атопический дерматит, крапивница, аллергический ринит, аллергический конъюнктивит, весенний конъюнктивит, эозинофильную гранулему, псориаз, воспалительный артрит, ревматоидный артрит, септический шок, язвенный колит, болезнь Крона, реперфузионное повреждение миокарда и головного мозга, хронический гломерулонефрит, эндотоксический шок, кистозный фиброз, артериальный рестеноз, атеросклероз, кератоз, ревматоидный спондилит, остеоартрит, пирозис, сахарный диабет, пневмокониоз, токсическую и аллергическую контактную экзему, атопическую экзему, себоррейную экзему, атопический дерматит, солнечный ожог, зуд в аногенитальной области, очаговую алопецию, гипертрофические шрамы, дискоидную красную волчанку, системную красную волчанку, фолликулярную и распространенную пиодермию, эндогенное и экзогенное акне, розовые угри, Болезнь Бехчета, анафилактоидную пурпуру, нефрит, воспалительное заболевание кишечника, лейкоз, рассеянный склероз, желудочно-кишечные заболевания, аутоиммунные заболевания и тому подобное.
К ним также относятся неврологические и психические нарушения, такие как болезнь Альцгеймера, рассеянный склероз, боковой амиотрофический склероз (ALS), множественная системная атрофия (MSA), шизофрения, болезнь Паркинсона, болезнь Хантингтона, болезнь Пика, депрессия, инсульт и повреждение спинного мозга.
Настоящее изобретение теперь будет дополнительно описано с помощью следующих неограничивающих примеров.
ПРИМЕРЫ
Химические названия соединений были присвоены с помощью программного обеспечения Structure To Name Enterprise 10.0 Cambridge.
Аббревиатуры
EDC= 1-этил-3-(3-диметиламинопропил)карбодиимид)гидрохлорид;
DMAP = 4-диметиламинопиридин;
ДМФ = диметилформамид;
EtOAc или AcOEt= этилацетат;
RT = комнатная температура;
ТГФ = тетрагидрофуран;
ДХМ = дихлорметан;
Et2O = диэтиловый эфир;
MeOH = метиловый спирт;
н-BuOH = н-бутиловый спирт;
EtOH = этиловый спирт;
IprOH или IPA = изопропиловый спирт;
(Ipr)2O = диизопропиловый эфир;
MIK = метил изобутил кетон;
MEK = метил этил кетон;
MTBE = метил трет-бутиловый эфир;
AcOH = уксусная кислота;
об. = объемы;
об./масс. = соотношение объема/массы;
масс./масс. = соотношение масса/масса;
Общие экспериментальные элементы
Характеристика ЯМР:
Спектр 1Н-ЯМР записывали на спектрометре Varian AS400 с 400 МГц. Химический сдвиг указывали как значения δ в м.д. относительно триметилсилана (TMS) в качестве внутреннего стандарта. Константы присоединения (значения J) давали в Герцах (Гц) и мультиплетность указывали, используя следующие аббревиатуры (с=синглет, d=дуплет, t=триплет, q=квартет, m=мультиплет, ушир.= уширенный, н.о.= не определен).
или
Спектр 1Н-ЯМР записывали на спектрометре Bruker ARX300 при 300,13 МГц (1Н), используя дейтериевые растворители, такие как дейтерированный диметилсульфоксид (ДМСО-d6) или дейтерированный хлороформ (CDCl3). Прибор оснащали многоядерным обратным зондом и температурным контроллером. Химические сдвиги выражали в миллионных долях (м.д.), используя тетраметилсилан (единицы d). Мультиплетность указывали следующим образом: (с) синглет, (д) дуплет, (дд) двойной дуплет, (ддд) тройной дуплет, (т) триплет, (дт) дуплет треплет, (q) квартет, (m) мультиплет, (ушир.с) уширенный сигнал. Константы присоединения J выражали в единицах Герца (Гц).
LC/UV/MS Аналитические способы
При расчете времени удержания в LC/MS принимали во внимание экспериментальную ошибку ±0,5 мин.
LC/UV/MS - Способ 1
Прибор LC: HPLC Alliance Waters (или эквивалентный)
Колонка: Kinetex 2,6u C18 100A 100×4,6 мм (Phenomenex)
Температура колонки (°C): 50,0
Подвижная фаза: HCOONH4 0,025M pH3 (A); ацетонитрил (B)
Поток (мл/мин): 2,0 (делили в MS 1:10)
Время остановки (мин): 17,0
Градиент:
Детекция УФ: канал 1 245 нм; канал 2 254 нм
Объем впрыска (мкл): 5,00
Растворитель образца: ацетонитрил
Прибор MS: Waters Quattro Micro API (или эквивалентный)
Полярность ES+
Капилляр (кВ) 3,20
Конус (V) 20,00
Экстрактор (V) 2,00
Объектив RF (V) 0,3
Полярность ES-
Капилляр (кВ) 3,20
Конус (V) 20,00
Экстрактор (V) 3,00
Объектив RF (V) 0,3
Температура источника (°C) 110
Температура растворения (°C) 210
Поток газа конуса (л/час) 150
Поток газа при растворении (л/час) 650
Длительность сканирования (сек): 1,00
Задержка между сканированием (сек): 0,10
Область масс: 125 до 1000
LC/UV/MS - Способ 2
Прибор LC: Acquity Waters UPLC (или эквивалентный)
Колонка: Kinetex 1,7u XB-C18 100A 100×2,1 мм (Phenomenex)
Температура колонки (°C) 50,0
Подвижные фазы: HCOONH4 0,025M pH3 (A); Ацетонитрил + 0,1% муравьиная кислота (B)
Поток (мл/мин) 0,65 (делили в MS 1:3)
Время остановки (мин) 10,0
Градиент:
Детекция УФ: длина волны 254 нм
Объем впрыска (мкл) - 2,00
Растворители образца: ацетонитрил
Прибор MS: Waters ZQ (или эквивалентный)
Полярность ES+
Капилляр (кВ) 3,00
Конус (V) 20,00
Экстрактор (V) 3,00
Объектив RF (V) 1,0
Полярность ES-
Капилляр (кВ) 3,00
Конус (V) 20,00
Экстрактор (V) 3,00
Объектив RF (V) 1,0
Температура источника (°C) 110
Температура растворения (°C) 210
Поток газа конуса (л/час) 150
Поток газа при растворении (л/час) 650
Область масс: 100 до 950
Время сканирования (сек): 0,32
LC/UV/MS - Способ 3
Прибор LC: Acquity Waters UPLC (или эквивалентный) с интерфейсом детектора 2996 PDA
Колонка: Acquity UPLC BEH C18 1,7 мкм 50×2,1 мм
Температура колонки (°C) 40,0
Подвижные фазы: 95:5 H2O:ACN+(0,1% ТФУ) (A); 5:95 H2O:ACN+(0,1% ТФУ) (B)
Поток (мл/мин) 0,6 (делили в MS 1:6)
Время остановки (мин) 8,5
Градиент:
Детекция УФ: детекция BPI (стартовая длина волны нм 210, конечная длина волны нм 400, спектральная выборка/сек = 20)
Объем впрыска (мкл) - 1,00
Растворители образца: ДМСО:MeOH:ACN в соотношении 1:3:3
Прибор MS: Waters ZQ (или эквивалентный)
Полярность ES
Капилляр (кВ) 3,20
Конус (V) 25,00
Экстрактор (V) 3,00
Объектив RF (V) 0,1
Полярность ES-
Капилляр (кВ) 3,00
Конус (V) 20,00
Экстрактор (V) 3,00
Объектив RF (V) 0,3
Температура источника (°C) 150
Температура растворения (°C) 350
Поток газа конуса (л/час) 110
Поток газа при растворении (л/час) 800
Область масс: 60 до 1200
Время сканирования (сек): 0,4
LC/UV/MS - Способ 4
Прибор LC: Acquity Waters UPLC (или эквивалентный) с интерфейсом детектора 2996 PDA
Колонка: Kinetex C18 1,7 мкм 50×2,1 мм (Phenomenex)
Температура колонки (°C): 40,0
Подвижные фазы: 95:5 H2O:ACN+(0,1% ТФУ) (A); 5:95 H2O:ACN+(0,1% ТФУ) (B)
Поток (мл/мин): 0,5 (не делили в MS)
Время остановки (мин): 4,40
Градиент:
Детекция UV: детекция BPI (стартовая длина волны нм 200, конечная длина волны нм 400, спектральная выборка/сек = 20)
Объем впрыска (мкл): 4,00
Растворитель образца: ацетонитрил
Прибор MS: ZQ (или эквивалентный)
Полярность ES
Капилляр (кВ) 3,25
Конус (V) 27,00
Экстрактор (V) 3,00
Объектив RF (V) 0,4
Температура источника (°C) 120
Температура растворения (°C) 400
Поток газа конуса (л/час) 100
Поток газа при растворении (л/час) 800
Время сканирования (сек): 0,42
Область масс: 100 до 800
Условия для препаративная ВЭЖХ с обратной фазой
Препаративная ВЭЖХ - Способ 1
Waters Micromass ZQ/Sample manager 2767
Фотодетектор на диодной матрице 2996;
Колонка: Колонка XTerra Prep MS C18 (5 мкм, 19×150 мм, Waters)
Скорость потока: 20 мл/мин с детекцией MS
Длина волны UV: 254 нм.
Подвижная фаза: растворитель A (вода:MeCN:HCOOH 95:5:0,05); растворитель B (вода:MeCN:HCOOH 5:95:0,05)
Градиент:
Препаративная ВЭЖХ - Способ 2
Колонка: Waters Symmetry Prep C18 17 мкм 19×300
Поток: 20 мл/мин
Подвижная фаза: 90% H2O, 10% ацетонитрил, 0,05% ТФУ (A); 10% H2O, 90% ацетонитрил, 0,05% ТФУ (B)
Градиент:
Препаративная ВЭЖХ - Способ 3
Waters Micromass ZQ/Sample manager 2767
Фотодетектор на диодной матрице: 2996
Колонка: XTERRA Prep MS C18 10 мкм 19×300
Поток: 20 мл/мин
Подвижные фазы: H2O, 0,1% ТФУ (A); ацетонитрил, 0,1% ТФУ (B)
Градиент:
Условия:
Хиральная ВЭЖХ:
Энантиомерную чистоту определяли на системе ВЭЖХ Hewlett Packard 1050, используя колонку Chiracel OD (5μ 4,6×250 мм), элюируя изократической смесью гексана и изопропанола в различных соотношениях, как указано в каждом конкретном примере.
Поток = 0,8 мл/мин
Детекция UV = 230 нм.
Определение оптического вращения (активности)
Специфические вращения соединений измеряли с помощью поляриметра Perkin Elmer model 241 или 341.
Температура (°C) 25
Длина пути (дм) 1
Длина волны Натрий D-линии (589 нм)
Эксперименты, требующие нагревания в микроволновой печи, проводили, используя прибор Biotage Initiator Sixty.
Методики образования соли
Если не указано иначе, соли, описанные в экспериментальной части, получали в соответствии с одной из методик, описанных в настоящем документе:
Формиатные соли: если указаны в колонке «название соли», соединения, содержащие один или более основных центров и очищенные на ВЭЖХ с обратной фазой (способ 1), давали соли муравьиной кислоты, однократно очищенные фракции, собранные из хроматографии, упаривали при пониженном давлении без какой-либо дополнительной обработки основания.
Трифторацетатные соли: если указаны в колонке «название соли», соединения, содержащие один или более основных центров и очищенные на ВЭЖХ с обратной фазой (способ 2 и 3), давали соли 2,2,2-трифторуксусной кислоты, однократно очищенные фракции, собранные из хроматографии, упаривали при пониженном давлении без какой-либо дополнительной обработки основания.
Гидрохлоридные соли: если указаны в колонке «название соли», соединения, содержащие один или более основных центров, с которых снимали защиту Boc в кислых условиях, без какой-либо дополнительной обработки основания, давали гидрохлоридные соли.
Любую другую соль получали путем обработки основания раствором соответствующих кислот в условиях, известных специалистам в данной области.
Стехиометрию соли определяли, при необходимости, с помощью ЯМР.
В методиках, которые приведены ниже, после каждого исходного вещества, как правило, приведена ссылка на номер соединения. Эта ссылка дана просто для удобства специалиста-химика. Исходное вещество может, необязательно, быть получено из указанной партии.
Если ссылка приведена для использования «похожей» или «аналогичной» процедуры, то, как понятно специалистам в данной области, такая методика может предусматривать незначительные изменения, например, температуру реакции, количество реагента/растворителя, время реакции, рабочие условия или условия хроматографической очистки.
Стереогенные центры, которые указаны с помощью неопределенной линии связи, представляют собой центры соединений, которые дают как единичные диастереоизомеры, так и энантиомеры, но чья абсолютная конфигурация так или иначе не была определена.
Большинство соединений, описанных в нижеследующих примерах, были получены из стереохимически чистого исходного вещества, например, 95% ее.
Стереохимия соединений в примерах, если указано, была определена на предположении о том, что абсолютная конфигурация разрешенных стереогенных центров исходных материалов сохраняется на всем протяжении любого последующего условия реакции.
Абсолютная конфигурация некоторых описанных соединений была подтверждена как правильная рентгеноструктурным или VCD (вибрационный круговой дихроизм) анализом кристаллического вещества.
При расчете диастереоизомерного соотношения LC/UV/MS, если указано, принимают во внимание экспериментальную ошибку ±1%. Альтернативно, диастереоизомерное соотношение определяют с помощью 1Н-ЯМР и его считают как >95:5, если единичный диастереоизомер детектировали, используя анализ ЯМР.
Подробные синтетические пути и процедуры приведены на схемах 9-41 и 46, ниже. Синтез спиртовых промежуточных соединений, перечисленных в таблице 12, описан в указанных патентных заявках.
Промежуточные соединения, использованные в нижеописанных процедурах, являются коммерчески доступными, могут быть получены специалистом с помощью подходов синтеза, хорошо известных в данной области, или могут быть получены в соответствии с процедурами синтеза, описанными ниже.
Пример 17
1-Оксид (S)-3,5-дихлор-4-(2-(4-(дифторметокси)-3-метоксифенил)-2-гидроксиэтил)пиридина (164)
Схема 23
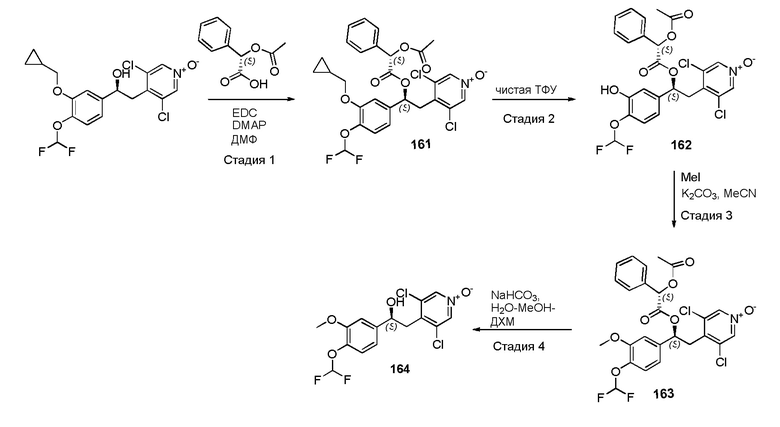
Стадия 1: 1-оксид 4-((S)-2-((S)-2-ацетокси-2-фенилацетокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)-3,5-дихлорпиридина (161)
Смесь (S)-2-ацетокси-2-фенилуксусной кислоты (0,924 г, 4,76 ммоль), 1-оксида (S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-гидроксиэтил)пиридина (1,0 г, 2,380 ммоль), EDC (0,684 г, 3,57 ммоль) и DMAP (0,436 г, 3,57 ммоль) в ДХМ (150 мл) перемешивали при комнатной температуре в течение 24 часов. Добавляли дополнительную порцию (S)-2-ацетокси-2-фенилуксусной кислоты (0,350 г, 1,802 ммоль), EDC (0,456 г, 2,380 ммоль) и DMAP (0,300 г, 2,456 ммоль) и продолжали перемешивать в течение 3 часов до полного преобразования. Реакционную смесь дважды промывали водной 1 н. HCl, а затем водным 1 M K2CO3; органический слой сушили над Na2SO4 и упаривали досуха. Остаток растирали в порошок с iPrOH (30 мл) и фильтровали с получением целевого продукта (1,27 г, 2,129 ммоль, 89% выход). MS/ESI+ 596,18 [MH]+
Стадия 2: 1-оксид 4-((S)-2-((S)-2-ацетокси-2-фенилацетокси)-2-(4-(дифторметокси)-3-гидроксифенил)этил)-3,5-дихлорпиридина (162)
1-оксид 4-((S)-2-((S)-2-ацетокси-2-фенилацетокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)-3,5-дихлорпиридина (1,27 г, 2,129 ммоль) обрабатывали трифторуксусной кислотой (15 мл, 195 ммоль), и полученный раствор перемешивали при комнатной температуре в течение 20 часов. Реакционную смесь разбавляли ДХМ и дважды промывали водой; органический слой сушили над Na2SO4 и упаривали досуха. Остаток очищали с помощью хроматографии на силикагеле (ДХМ/EtOAc = 3:2 до 1:1). Смешанные фракции объединяли и растирали со смесью iPr2O/Et2O (10:1). Собранное твердое вещество затем объединяли с чистыми фракциями из хроматографии с получением целевого соединения (1,08 г, 1,991 ммоль, 94% выход); MS/ESI+ 542,11 [MH]+
1Н ЯМР (300 МГц, ДМСО-d6) δ м.д. 8,56 (с, 2H), 7,27-7,50 (м, 5H), 6,98 (д, 1H), 6,81 (д, 1H), 7,00 (т, 1H), 6,54 (дд, 1H), 5,89 (дд, 1H), 5,84 (с, 1H), 3,40 (дд, 1H), 3,18 (дд, 1H), 2,13 (с, 3H)
Стадия 3: 1-оксид 4-((S)-2-((S)-2-ацетокси-2-фенилацетокси)-2-(4-(дифторметокси)-3-метоксифенил)этил)-3,5-дихлорпиридина (163)
Суспензию 1-оксида 4-((S)-2-((S)-2-ацетокси-2-фенилацетокси)-2-(4-(дифторметокси)-3-гидроксифенил)этил)-3,5-дихлорпиридина (1,080 г, 1,991 ммоль), метилйодида (0,162 мл, 2,59 ммоль) и карбоната калия (0,550 г, 3,98 ммоль) в CH3CN (40 мл) энергично перемешивали при комнатной температуре в течение 20 часов. Реакционную смесь разделяли между ДХМ и водой, и органический слой сушили над Na2SO4. Растворитель удаляли под вакуумом с получением целевого соединения (0,984 г, 1,769 ммоль, 89% выход). MS/ESI+ 556,17 [MH]+. Сырое соединение использовали без дальнейшей очистки.
Стадия 4: 1-оксид (S)-3,5-дихлор-4-(2-(4-(дифторметокси)-3-метоксифенил)-2-гидроксиэтил)пиридина (164)
1-оксид 4-((S)-2-((S)-2-ацетокси-2-фенилацетокси)-2-(4-(дифторметокси)-3-метоксифенил)этил)-3,5-дихлорпиридина (984 мг, 1,769 ммоль) растворяли в смеси MeOH (50 мл) и ДХМ (10 мл). Добавляли насыщенный водный раствор NaHCO3 (10 мл, 11,00 ммоль), и полученную суспензию перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь разделяли между водой и ДХМ; органический слой сушили над Na2SO4 и упаривали досуха с получением целевого соединения (650 мг, 1,71 ммоль, 97% выход). MS/ESI+ 380,03 [MH].
Соединение, указанное в таблице 13, получали по аналогичной методике, описанной на схеме 23, с помощью подходящего реагента алкилирования, и выполняя стадию 3 при 65°C.
Пример 18
Синтез 1-оксида (S)-3,5-дихлор-4-(2-(3,4-диметоксифенил)-2-гидроксиэтил)пиридина (170)
Схема 24
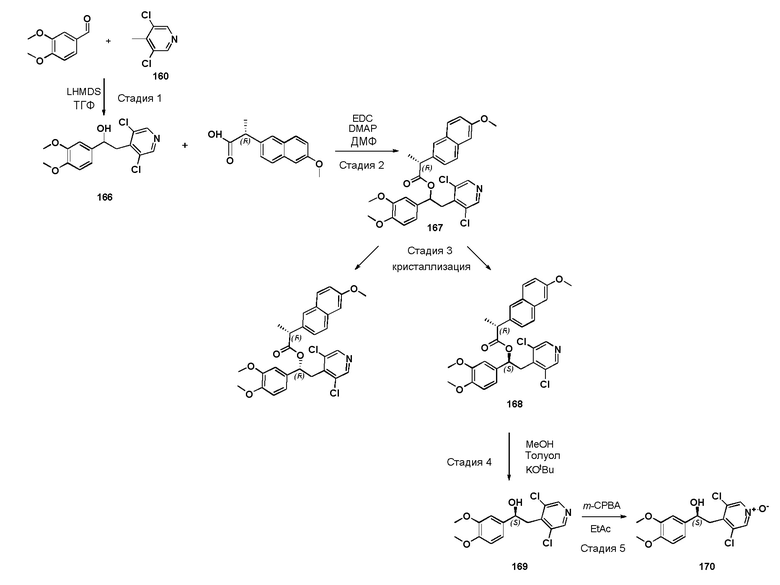
Стадия 1: Синтез 2-(3,5-дихлорпиридин-4-ил)-1-(3,4-диметоксифенил)этанола (166)
3,5-Дихлор-4-метилпиридин (160) (54 г, 331 ммоль) растворяли в сухом ТГФ (480 мл) в атмосфере аргона и охлаждали при -78°C на бане сухой лед/ацетон. По каплям добавляли раствор 1 н. LHMDS в ТГФ (331 мл, 331 ммоль), поддерживая температуру -78°С. Смесь перемешивали при -78°С в течение 1 часа. Затем по каплям добавляли раствор 3,4-диметоксибензальдегида (50 г, 301 ммоль) в сухом ТГФ (120 мл), поддерживая температуру при -78°C. После завершения добавления смесь оставляли нагреваться при комнатной температуре.
Реакционную смесь выливали в лед и воду (1 л), и смесь перемешивали до выпадения обильного осадка. Твердое вещество фильтровали и растворяли в этилацетате (500 мл), сушили над Na2SO4, и растворитель упаривали под вакуумом. Сырое вещество кристаллизовали в CHCl3/гексане. Осадок фильтровали, промывали гексаном и сушили под вакуумом при 40°C в течение 8 часов с получением 55 г (выход 45%). Маточный раствор упаривали под вакуумом при 40°C, растворяли в этилацетате (200 мл) и экстрагировали 200 мл воды. Органический раствор сушили над Na2SO4, и растворитель упаривали под вакуумом при 40°C. Сырое вещество кристаллизовали в CHCl3/гексан и получали дополнительную порцию 15 г целевого продукта (166) (общий выход 70%).
Стадия 2: Синтез ((R)-2-(3,5-дихлорпиридин-4-ил)-1-(3,4-диметоксифенил)этил) 2-(6-метоксинафтален-2-ил)пропаноата (167)
Промежуточное соединение 166 (50 г, 152 ммоль), (R)-2-(6-метоксинафтален-2-ил)пропановую кислоту (38,6 г, 168 ммоль), DMAP (20,5 г, 168 ммоль) и EDC (43,8 г, 229 ммоль) растворяли в ДМФ (300 мл), и реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Затем добавляли воду (500 мл), раствор перемешивали до выпадения осадка. Твердое вещество фильтровали и растворяли в ДХМ (500 мл). Органический раствор промывали водным HCl 1 н. (2×500 мл), насыщенным водным раствором NaHCO3 (500 мл) и сушили над Na2SO4. Растворитель упаривали под вакуумом, и твердое вещество обрабатывали ультразвуком в EtOH (300 мл) и растирали в течение 1 часа. Полученный осадок собирали фильтрацией и сушили под вакуумом при 40°C в течение 4 часов с получением 79 г (выход 99%) соединения 167 в виде диастереоизомерной смеси.
Стадия 3: Синтез (R)-((S)-2-(3,5-дихлорпиридин-4-ил)-1-(3,4-диметоксифенил)этил) 2-(6-метоксинафтален-2-ил)пропаноата (168)
Промежуточное соединение 167 (79 г, 146 ммоль) растворяли в CHCl3 (100 мл) и медленно добавляли MeOH (30 мл) до получения устойчивой опалесценции, и смесь оставляли при комнатной температуре в течение 2 часов. Образовавшееся твердое вещество собирали фильтрацией и перекристаллизовывали с помощью системы растворителей CHCl3/MeOH (70 мл/20 мл) с получением 35 г соединения 168 (выход 88%, ee 98%).
В хиральной ВЭЖХ Rt=42,33 мин (быстрый изомер); элюент: гексан:изопропанол 97:3
1Н ЯМР (600 МГц, ХЛОРОФОРМ-d) δ м.д. 8,04 (с, 2H), 7,67 (д, J=8,79 Гц, 1H), 7,58 (д, J=8,52 Гц, 1H), 7,53 (м, 1H), 7,12-7,20 (м, 3H), 6,95 (дд, J=8,24, 1,92 Гц, 1H), 6,78-6,88 (м, 2H), 6,14 (дд, J=10,44, 4,12 Гц, 1H), 3,95 (с, 3H), 3,88 (с, 3H), 3,78-3,81 (м, 4H), 3,55 (дд, J=13,73, 10,44 Гц, 1H), 3,14 (дд, J=13,60, 4,26 Гц, 1H), 1,44 (д, J=7,14 Гц, 3H).
Стадия 4: Синтез (S)-2-(3,5-дихлорпиридин-4-ил)-1-(3,4-диметоксифенил)этанола (169)
Промежуточное соединение 168 (30 г, 56 ммоль) растворяли в MeOH и медленно добавляли толуол. К суспензии медленно добавляли трет-бутоксид калия. Смесь перемешивали в течение 24 час при комнатной температуре. Реакционную смесь разбавляли водой (500 мл), и водную смесь экстрагировали CHCl3 (500 мл). Органический слой сушили над Na2SO4, и растворитель упаривали под вакуумом. Твердое вещество кристаллизовали из CHCl3 (100 мл) и гексана (20 мл, до устойчивой опалесценции). Маточный раствор концентрировали и перекристаллизовывали таким же образом, с получением второй порции целевого соединения. Всего было получено 16 г соединения 169 (выход 87%).
В хиральной ВЭЖХ Rt= 58,03 мин; элюент: гексан:изопропанол 95:5. [α]D20 = +10,21 (c=0,506, Метанол)
1Н ЯМР (400 МГц, ацетон) δ м.д. 8,47 (с, 2H), 6,96-7,15 (м, 1H), 6,87 (м, 2H), 4,93-5,21 (м, 1H), 4,50 (д, J=3,97 Гц, 1H), 3,78 (с, 6H), 3,44 (дд, J=12,79, 8,38 Гц, 1H), 3,22 (дд, J=13,01, 5,51 Гц, 1H).
MS/ESI+ [MH]+: 328,19
Стадия 5: Синтез 1-оксида (S)-3,5-дихлор-4-(2-(3,4-диметоксифенил)-2-гидроксиэтил)пиридина (170)
Соединение 169 (4 г, 12 ммоль) растворяли в этилацетате, и к раствору добавляли m-CPB кислоту. Смесь перемешивали при комнатной температуре в течение 5 час. Образовавшееся твердое вещество собирали фильтрацией, промывали этилацетатом и сушили под вакуумом с получением 1,72 г указанного в заголовке соединения (выход 41%). В хиральной ВЭЖХ Rt= 22,16 мин; элюент: гексан:изопропанол 6:4. [α]D20 = +68,91 (c = 0,253, Метанол/CHCl3 1:1). MS/ESI+ [MH]+: 344,19
1Н ЯМР (400 МГц, ХЛОРОФОРМ-d) δ м.д. 8,15 (с, 2H), 6,99 (м, 1H), 6,79-6,88 (м, 2H), 5,03 (дд, J=8,50, 5,32 Гц, 1H), 3,75-3,98 (м, 6H), 3,42 (дд, J=13,57, 8,56 Гц, 1H), 3,19 (дд, J=13,51, 5,32 Гц, 1H), 2,06-2,15 (м, 1H).
Пример 36
Синтез 1-оксида 3,5-дихлор-4-(2-(2,2-диметилбензо[d][1,3]диоксол-5-ил)-2-гидроксиэтил)пиридина (172)
Схема 25
Стадия 1: Синтез 2-(3,5-дихлорпиридин-4-ил)-1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)этанола (171)
3,5-Дихлор-4-метилпиридин (166) (4,37 г, 0,016 моль) растворяли в сухом ТГФ (40 мл) в атмосфере аргона, и смесь охлаждали при -78°C на бане сухой лед/ацетон. По каплям добавляли раствор LHMDS 1 н. ТГФ (28 мл, 28 ммоль), поддерживая температуру при -78°С. Смесь перемешивали при -78°С в течение 1 часа. Затем по каплям добавляли раствор 2,3-дифтор-3,4-бензодиоксолокарбоксальдегида (5 г, 0,026 моль) в сухом ТГФ (10 мл), поддерживая температуру при -78°C. После завершения добавления смесь оставляли нагреваться при комнатной температуре.
Реакционную смесь выливали в лед и воду, и водную фазу экстрагировали этилацетатом (3×). Объединенные органические фазы сушили над Na2SO4, и растворитель упаривали под вакуумом. Сырое вещество кристаллизовали в петролейном эфире/гексане 1/1. Осадок фильтровали, промывали гексаном и сушили под вакуумом при 40°C в течение 8 часов с получением 6,4 г (выход 45%).
MS/ESI+ 349,14 [MH]+; 1Н ЯМР (200 МГц, ХЛОРОФОРМ-d) δ м.д. 8,71 (с, 2H), 7,48-7,68 (м, 2H), 6,85-7,07 (м, 1H), 4,61 (м, 1H), 4,11-4,44 (м, 2H).
Стадия 2: Синтез 1-оксида 3,5-дихлор-4-(2-(2,2-диметилбензо[d][1,3]диоксол-5-ил)-2-гидроксиэтил)пиридина (172)
2-(3,5-Дихлорпиридин-4-ил)-1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)этанол (2 г, 5,74 ммоль) растворяли в EtOAc (40 мл), и раствор охлаждали до 0°C. Добавляли m-CPBA (5,29 г, 22,98 ммоль), и полученную смесь перемешивали при комнатной температуре в течение 24 часов. Реакционную смесь охлаждали до 0°C, и осадок в виде твердого вещества белого цвета фильтровали и дважды промывали холодным ДХМ с получением целевого продукта (1,738 г, 4,77 ммоль, 83%); MS/ESI+ 364,03 [MH]+.
Пример 19:
Синтез 1-оксида 3,5-дихлор-4-(2-гидрокси-2-(4-метоксиспиро-[бензо[d][1,3]диоксол-2,1'-циклопентан]-7-ил)этил)пиридина (174)
Схема 26

Стадия 1: 1-оксид 3,5-дихлор-4-(2-(4-метоксиспиро[бензо[d][1,3]диоксол-2,1'-циклопентан]-7-ил)-2-оксоэтил)пиридина (173)
К раствору 2-(3,5-дихлорпиридин-4-ил)-1-(4-метоксиспиро[бензо[d][1,3]диоксол-2,1'-циклопентан]-7-ил)этанона (получен как описано в EP1535920, 4,75 г, 12,05 ммоль) в EtOAc (125 мл), охлажденного до 0°C, добавляли m-CPBA (11,09 г, 48,2 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 24 часов. Добавляли еще m-CPBA (5,54 г, 24,10 ммоль) и продолжали перемешивать в течение дополнительных 24 часов. Смесь несколько раз промывали 1 M водным раствором K2CO3, и органический слой сушили над Na2SO4 и упаривали досуха. Остаток очищали с помощью хроматографии на силикагеле (ДХМ/EtOAc = 3/1 до 1/2) с получением 1-оксида 3,5-дихлор-4-(2-(4-метоксиспиро[бензо[d][1,3]диоксол-2,1'-циклопентан]-7-ил)-2-оксоэтил)пиридина (2,14 г, 5,22 ммоль, 43,3% выход); MS/ESI+ 410,10 [MH]+.
Стадия 2: 1-оксид 3,5-дихлор-4-(2-гидрокси-2-(4-метоксиспиро[бензо[d][1,3]диоксол-2,1'-циклопентан]-7-ил)этил)пиридина (174)
К раствору 1-оксида 3,5-дихлор-4-(2-(4-метоксиспиро[бензо[d][1,3]диоксол-2,1'-циклопентан]-7-ил)-2-оксоэтил)пиридина (2,14 г, 5,22 ммоль) в MeOH (100 мл), порциям добавляли твердый боргидрид натрия (0,197 г, 5,22 ммоль), и смесь перемешивали при комнатной температуре в течение ночи. В течение 2 часов добавляли дополнительную порцию боргидрида натрия (0,394 г, 10,44 ммоль) и продолжали перемешивать в течение еще 12 часов. Смесь концентрировали под вакуумом, разбавляли EtOAc и дважды промывали водным раствором 1 н. NaOH. Органический слой сушили над Na2SO4, растворитель удаляли при пониженном давлении, и остаток очищали с помощью хроматографии на силикагеле (EtOAc) с получением 1-оксида 3,5-дихлор-4-(2-гидрокси-2-(4-метоксиспиро[бензо[d][1,3]диоксол-2,1'-циклопентан]-7-ил)этил)пиридина (650 мг, 1,577 ммоль, 30,2% выход); MS/ESI+ 412,10 [MH]+.
Пример 1
Синтез гидрохлорида 1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-пирролидин-2 карбонилокси)этил)пиридина (3)
Схема 9
Стадия 1: 1-оксид 4-((S)-2-((S)-1-(трет-бутоксикарбонил)пирролидин-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)-3,5-дихлорпиридина (2)
1-Оксид (S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-гидроксиэтил)пиридина (1) (550 мг, 1,309 ммоль), (S)-1-(трет-бутоксикарбонил)пирролидин-2-карбоновую кислоту (282 мг, 1,309 ммоль), EDC (251 мг, 1,309 ммоль) и DMAP (160 мг, 1,309 ммоль) растворяли в ДМФ (5 мл). Реакционную смесь перемешивали при комнатной температуре в течение 48 часов до завершения реакции. По прошествии этого времени реакционную смесь гасили 1 M HCl и экстрагировали EtOAc. Органический экстракт промывали 1 M HCl (×3) и 5% K2CO3 (×3), а затем сушили над Na2SO4 и концентрировали под вакуумом с выходом 800 мг целевого продукта (выход 99%). MS/ESI+ 617,16 [MH]+
Стадия 2: Гидрохлорид 1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-пирролидин-2-карбонилокси)этил)пиридина (3)
1-Оксид 4-((S)-2-((S)-1-(трет-бутоксикарбонил)пирролидин-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)-3,5-дихлорпиридина (2) (300 мг, 0,486 ммоль) растворяли в смеси диоксан/HCl (4 M, 2 мл) и перемешивали при комнатной температуре в течение 8 часов. По прошествии этого времени растворитель удаляли на роторном испарителе при пониженном давлении и сушили в вакуумной печи в течение ночи с выходом желаемого продукта в виде гидрохлорида (200 мг; выход 80%).
MS/ESI+ 517,2 [MH]+; tR/мин (Способ 1) = 3,75; Соотношение диастереомеров= >99:1; [αD] = -32,80 (c = 0,25; CHCl3)
1Н ЯМР (400 МГц, МЕТАНОЛ-d4) δ м.д. 8,49 (с, 2H), 7,18 (д, J=7,94 Гц, 1H), 7,11 (д, J=1,76 Гц, 1H), 7,04 (д, J=1,76 Гц, 1H), 6,78 (т, J=75,00 Гц, 1H), 6,10-6,17 (м, 1H), 4,38-4,51 (м, 1H), 3,85-3,99 (м, 2H), 3,66 (м, 2H), 3,39-3,51 (м, 1H), 2,40-2,60 (м, 1H), 1,89-2,17 (м, 4H), 1,15-1,36 (м, 1H), 0,64 (дд, J=7,94, 1,32 Гц, 2H), 0,33-0,47 (м, 2H).
Соединения перечислены в таблице 1 (см. в конце описания) с использованием подходящих исходных веществ.
Соединения, полученные в виде свободных оснований, подвергали основной обработке в месте удаления растворителя при пониженном давлении, описано выше (например: насыщенный раствор NaHCO3), с последующей экстракцией полярным органическим растворителем (например: AcOEt) для удаления образовавшихся солей при взаимодействии с HCl, которые спонтанно возникают при осуществлении стадии 2 (схема 9).
Соединения 6, 7, 181, 183 получали путем взаимодействия соответствующего Boc-защищенного предшественника с AcOEt/HCl (5M), а затем фильтрацией при комнатной температуре гидрохлоридной соли, которая спонтанно выпадает в осадок из реакционной смеси.
Соединения 177, 178 и 179 получали при осуществлении стадии 2 с HCl в EtOAc и удаляя растворитель без нагревания.
Пример 2
Синтез 1-оксида 3,5-дихлор-4-((2S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(1-((4-(метоксикарбонил)-5-метилфуран-2-ил)метил)пирролидин-2-карбонилокси)этил)пиридина (15)
Схема 10
1-Оксид 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-пирролидин-2-карбонилокси)этил)пиридина (3) (40 мг, 0,077 ммоль) растворяли в ТГФ (1 мл) и добавляли метил 5-формил-2-метилфуран-3-карбоксилат (13,00 мг, 0,077 ммоль) и уксусную кислоту (4,64 мг, 0,077 ммоль). Перед добавлением триацетоксигидробората натрия (16,39 мг, 0,077 ммоль) реакционную смесь перемешивали при комнатной температуре в течение 30 мин. По прошествии этого времени реакционную смесь перемешивали еще 2 часа до завершения реакции. Растворитель удаляли, и остаток разделяли между EtOAc и 1 M водным HCl. Органический слой затем промывали водным 5% K2CO3 и сушили над Na2SO4. Растворитель удаляли под вакуумом с получением прозрачного масла (35 мг; выход 68%), которое очищали с помощью препаративной ВЭЖХ с выходом 18 мг диастеромерной смеси (47,5: 52,5) целевого продукта в виде прозрачного масла (выход 36%). MS/ESI+ 668,9 [MH]+; tR=5,97; 6,15 мин (способ 1); соотношение диастереомеров= 47:53.
Соединения, перечисленные в таблице 3 (см. в конце описания), получали аналогичной процедурой, описанной в стадии 1; на схеме 10 и реакцией предшественника (5) с подходящими реагентами.
Диастереомерную смесь, полученную из (5), разделяли с помощью препаративной ВЭЖХ (способ 1, раздел «Общие экспериментальные элементы») с получением двух диастереомеров, (16) и (17,) идентифицированных как быстрый и медленный изомер, соответственно, согласно наблюдаемому времени их удерживания при хроматографических условиях, описанных в разделе «Общие экспериментальные элементы» (Способ 1).
Пример 3
Синтез 1-оксида 3,5-дихлор-4-((S)-2-((S)-1-(4-(циклопропилметокси)-3-(метилсульфонамидо)бензоил)пирролидин-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси) фенил)этил)пиридина (19)
Схема 11
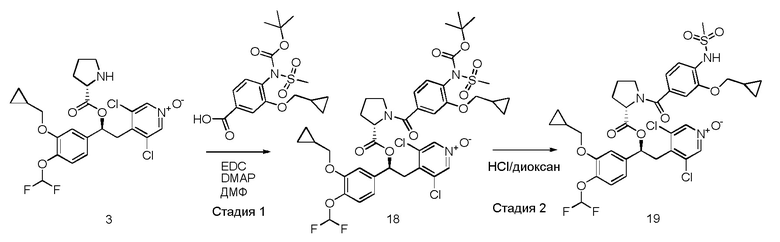
Стадия 1: 1-оксид 4-((S)-2-((S)-1-(3-(N-(трет-бутоксикарбонил)метилсульфонамидо)-4-(циклопропилметокси)бензоил)пирролидин-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)-3,5-дихлорпиридина (18)
1-Оксид 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-пирролидин-2-карбонилокси)этил)пиридина (3) (40 мг; 0,077 ммоль) помещали в 50 мл круглодонную колбу и растворяли в ДМФ (2 мл). В реакционную смесь добавляли 3-(N-(трет-бутоксикарбонил)метилсульфонамидо)-4-(циклопропилметокси)бензойную кислоту (40 мг, 0,104 ммоль, полученную, как описано в WO2010/089107), затем добавляли EDC (20,0 мг, 0,104 ммоль) и DMAP (15,0 мг, 0,123 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 6 часов перед тем как реакцию завершали и гасили, добавляя 20 мл водного HCl 1 M. Водный слой экстрагировали EtOAc и промывали HCl 1 M (×3) и водным раствором 5% K2CO3 (×3). Полученный органический экстракт обезвоживали с помощью Na2SO4, фильтровали на фильтровальной бумаге, и растворитель удаляли на роторном испарителе при пониженном давлении. Маслянистое твердое вещество очищали с помощью препаративной ВЭЖХ (способ 1) с выходом 30 мг целевого продукта (выход 44%). MS/ESI+ 884,1 [MH]+
1Н ЯМР (400 МГц, ацетон) δ м.д. 8,28 (с, 2H), 7,33 (д, J=7,94 Гц, 1H), 7,14-7,23 (м, 4H), 6,68-7,13 (м, 2H), 6,16 (дд, J=9,92, 4,19 Гц, 1H), 4,52 (дд, J=7,94, 6,17 Гц, 1H), 3,88-4,12 (м, 6H), 3,56-3,74 (м, 3H), 3,53 (с, 3H), 3,34 (дд, J=14,11, 4,41 Гц, 1H), 1,74-1,93 (м, 2H), 1,47 (с, 9H), 1,25-1,35 (м, 2H), 0,51-0,69 (м, 4H), 0,27-0,48 (м, 4H).
Стадия 2: 1-оксид 3,5-дихлор-4-((S)-2-((S)-1-(4-(циклопропилметокси)-3-(метилсульфонамидо)бензоил)пирролидин-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)пиридина (19)
1-Оксид 4-((S)-2-((S)-1-(3-(N-(трет-бутоксикарбонил)метилсульфонамидо)-4-(циклопропилметокси)бензоил)пирролидин-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)-3,5-дихлорпиридина (18) (30 мг, 0,034 ммоль) растворяли в растворе HCl/EOAc (4 M; 2 мл) и перемешивали в течение 10 час при комнатной температуре до завершения реакции. Реакционную смесь гасили, добавляя K2CO3 5%, и экстрагировали EtOAc. Полученный органический экстракт сушили над Na2SO4, фильтровали на фильтровальной бумаге, и растворитель удаляли на роторном испарителе при пониженном давлении. Твердое вещество перекристаллизовывали из EtOH:гексана (1:3) с выходом белого твердого вещества (18 мг; выход 68%) соединения, указанного в заголовке. MS/ESI+ 784,1 [MH]+; tR=5,72 (способ 1); [αD]= -48,92 (c = 3,7; ДХМ); Соотношение диастереомеров >99:1.
1Н ЯМР (400 МГц, ХЛОРОФОРМ-d) δ м.д. 8,16 (шир.с, 2H), 7,53 (м, 1H), 7,09-7,20 (м, 3H), 7,05 (м, 1H), 6,96 (м, 2H), 6,41-6,63-6,84 (т, 1H, CHF2), 6,08-6,17 (м, 1H), 4,53-4,66 (м, 1H), 3,81-3,98 (м, 4H), 3,61 (м, 3H), 3,22-3,39 (м, 1H), 3,02 (с, 3H), 2,20-2,34 (м, 1H), 1,78-2,02 (м, 3H), 1,27 (м, 2H), 0,59-0,75 (м, 4H), 0,28-0,42 (м, 4H).
Соединения, перечисленные в таблице 4, получали аналогичным способом, который описан выше в примере 3, на схеме 11, реакцией соответствующих предшественников, перечисленных с подходящими реагентами, а затем стадией очистки, как указано в таблице ниже, вместо перекристаллизации, как указано выше.
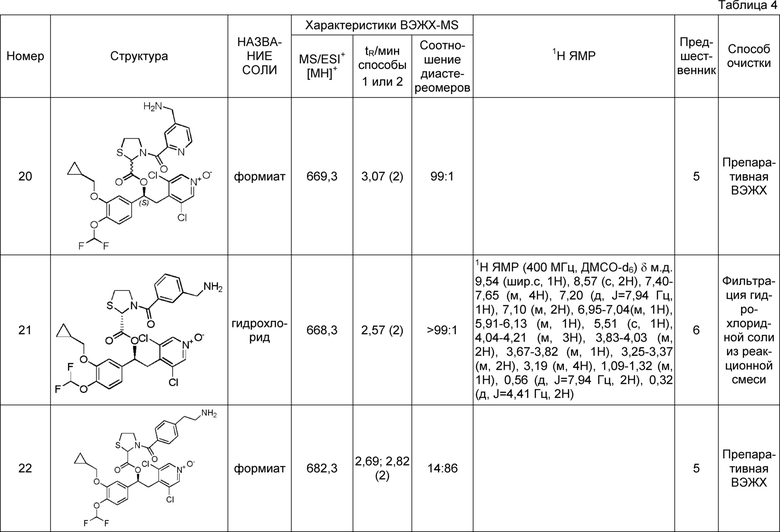
Пример 4
Синтез 1-оксида 4-((2S)-2-(3-(4-аминобензоил)тиазолидин-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)-3,5-дихлорпиридина (26)
Схема 12
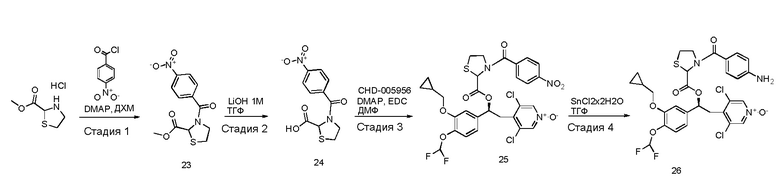
Стадия 1: метил 3-(4-нитробензоил)тиазолидин-2-карбоксилат (23)
Гидрохлорид метил тиазолидин-2-карбоксилата (200 мг, 1,089 ммоль) растворяли в ДХМ (2 мл). Добавляли DMAP (173 мг, 1,416 ммоль) и хлорид 4-нитробензоила (263 мг, 1,416 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 2 часов до завершения реакции. Реакционную смесь разбавляли DMC и экстрагировали водным раствором 1 M HCl. Органическую фазу промывали 1 н. HCl и насыщенным солевым раствором, сушили над Na2SO4 и концентрировали под вакуумом с получением метил 3-(4-нитробензоил)тиазолидин-2-карбоксилата (220 мг, 0,742 ммоль, 68% выход). MS/ESI+ 297,05 [MH]+
Стадия 2: 3-(4-нитробензоил)тиазолидин-2-карбоновая кислота (24)
Метил 3-(4-нитробензоил)тиазолидин-2-карбоксилат (220 мг, 0,742 ммоль) растворяли в ТГФ (2 мл). Добавляли LiOH 1 M (1 мл, 1,000 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 6 час до завершения реакции. Реакционную смесь разбавляли 1 н. HCl и экстрагировали EtOAc. Органическую фазу сушили над Na2SO4 и концентрировали под вакуумом с получением 3-(4-нитробензоил)тиазолидин-2-карбоновой кислоты (180 мг, 0,638 ммоль, 86% выход). MS/ESI+ 283,03 [MH]+
Стадия 3: 1-оксид 3,5-дихлор-4-((2S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(3-(4-нитробензоил)тиазолидин-2-карбонилокси)этил)пиридина (25)
1-Оксид (S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-гидроксиэтил)пиридина (100 мг, 0,238 ммоль), 3-(4-нитробензоил)тиазолидин-2-карбоновую кислоту (134 мг, 0,476 ммоль), DMAP (34,9 мг, 0,286 ммоль) и EDC (137 мг, 0,714 ммоль) растворяли в ДМФ (1,5 мл). Реакционную смесь перемешивали при комнатной температуре в течение 2 час до завершения реакции. Реакционную смесь разбавляли водой, и остаток промывали водой, растворяли в EtOAc и экстрагировали 1 н. HCl, насыщенным раствором Na2CO3 и насыщенным солевым раствором. Органическую фазу сушили над Na2SO4 и концентрировали под вакуумом с получением 1-оксида 3,5-дихлор-4-((2S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(3-(4-нитробензоил)тиазолидин-2-карбонилокси)этил)пиридина (130 мг, 0,190 ммоль, 80% выход).
MS/ESI+ 684,07 [MH]+
Стадия 4: 1-оксид 4-((2S)-2-(3-(4-аминобензоил)тиазолидин-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)-3,5-дихлорпиридина (26)
1-Оксид 3,5-дихлор-4-((2S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(3-(4-нитробензоил)тиазолидин-2-карбонилокси)этил)пиридина (130 мг, 0,190 ммоль) растворяли в ТГФ (3 мл). Добавляли дигидрат хлорида олова(II) (257 мг, 1,140 ммоль), и смесь перемешивали при комнатной температуре в течение 4 суток. Растворитель удаляли под вакуумом, и сырой продукт растворяли в EtOAc и разбавляли насыщенным раствором Na2CO3. К двух фазам добавляли диатомовую землю, и смесь фильтровали на прокладке из диатомовой земли. Органическую фазу промывали насыщенным раствором Na2CO3, насыщенным солевым раствором, сушили над Na2SO4 и концентрировали под вакуумом. Сырой продукт растирали в Et2O с получением 1-оксида 4-((2S)-2-(3-(4-аминобензоил)тиазолидин-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)-3,5-дихлорпиридина (30 мг, 0,046 ммоль, 24% выход). MS/ESI+ 653,8 [MH]+; tR=5,80; 6,00 (способ 1); Соотношение диастереомеров 30:70.
Пример 5
Синтез 1-Оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-1-(4-метокси-3-(метилсульфонилокси)бензоил)пирролидин-2-карбонилокси)этил)пиридина (30)
Схема 13
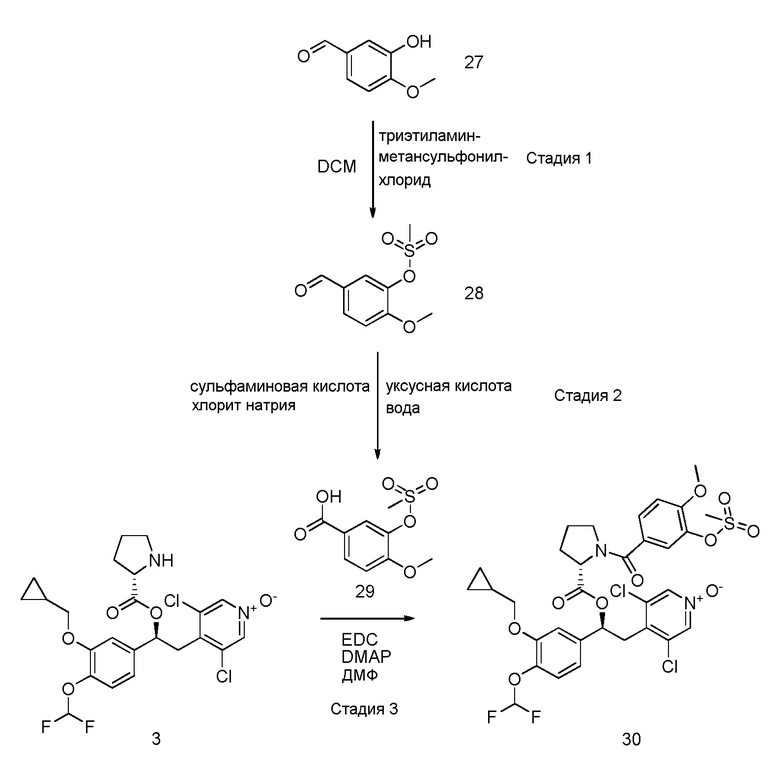
Стадия 1: метансульфонат 5-формил-2-метоксифенила (28)
3-Гидрокси-4-метоксибензальдегид (27) (0,5 г, 3,291 ммоль) растворяли в ДХМ и добавляли хлорид метансульфонила (0,376 г, 3,29 ммоль), а затем триэтиламин (0,499 г, 4,931 ммоль). Реакционный раствор перемешивали при комнатной температуре в течение 2 часов, по прошествии этого времени его гасили водным раствором 1 M HCl и экстрагировали EtOAc. Органический экстракт затем промывали водным раствором 5% K2CO3, удаляли воду над Na2SO4, и растворитель удаляли на роторном испарителе. Целевой продукт получали в виде белого порошка (0,750 г, выход 99%). MS/ESI+ 231,02 [MH]+
Стадия 2: 4-метокси-3-(метилсульфонилокси)бензойная кислота (29)
Метансульфонат 5-формил-2-метоксифенила (28) (0,750 г, 3,261 ммоль) и сульфаминовую кислоту (0,316 г, 3,261 ммоль) растворяли в уксусной кислоте (10 мл) и охлаждали на ледяной бане. Хлорит натрия (0,589 г, 6,521 ммоль) растворяли в воде (4 мл) и медленно добавляли к холодному реакционному раствору. Реакционную смесь оставляли нагреваться при комнатной температуре и перемешивали в течение приблизительно 2 часов. По прошествии этого времени в анализе СЭЖХ-MS наблюдали полное преобразование. Добавление воды (15 мл) вызывало осаждение белого твердого вещества, которое фильтровали и промывали несколько раз водой (0,700 г, выход 87%).
MS/ESI+ 247,02 [MH]+
Стадия 3: 1-оксид 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-1-(4-метокси-3-(метилсульфонилокси)бензоил)пирролидин-2-карбонилокси)этил)пиридина (30)
1-оксид 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-1-(4-метокси-3-(метилсульфонилокси)бензоил)пирролидин-2-карбонилокси)этил)пиридина (30) получали в соответствии со способом, аналогичным способу, описанному в стадии 1 (пример 3). MS/ESI+ 745,0 [MH]+; [αD] = 31,30 (c=0,34; CHCl3), tR=5,57 (способ 1); Соотношение диастереомеров >99:1. 1Н ЯМР (400 МГц, ХЛОРОФОРМ-d) δ м.д. 8,17 (с, 2H), 7,56 (м, 2H), 7,13 (м, 4H), 6,60 (т, J=75,00 Гц, 1H), 6,04-6,21 (м, 1H), 4,46-4,75 (м, 1H), 3,95 (с, 3H), 3,85 (м, 2H), 3,50-3,70 (м, 3H), 3,25-3,31 (м, 1H), 3,22 (с, 3H), 2,19-2,41 (м, 1H), 1,74-2,03 (м, 3H), 1,14-1,34 (м, 1H), 0,64 (д, J=7,09 Гц, 2H), 0,34 (д, J=4,16 Гц, 2H).
Соединения, перечисленные в таблице 5 (см. в конце описания), получали в соответствии со способом, аналогичным способу, описанному в стадии 3 (схема 13) и реакцией соответствующих перечисленных предшественников с подходящими коммерческими реагентами, а затем проводили соответствующую стадию очистки вместо препаративной ВЭЖХ, как указано ниже.
Соединения, перечисленные в таблице 14 (см. в конце описания), получали в соответствии со способом, аналогичным способу, описанному на стадии 3 (схема 13) и реакцией соответствующих перечисленных предшественников с подходящими коммерческими реагентами, используя ДХМ вместо ДМФ в качестве растворителя, а затем проводили соответствующую стадию очистки, как указано ниже. Соединения 193, 194 и 195 получали, начиная с промежуточного соединения 3 или 179, полученных как свободные основания после основной обработки гидрохлоридных солей водным раствором 1 M NaHCO3, а затем экстракцией ДХМ.
Пример 20
Синтез 1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(2-(3-(диметилкарбамоил)фенил)ацетил)тиазолидин-2-карбонилокси)этил)пиридина (198)
Схема 27
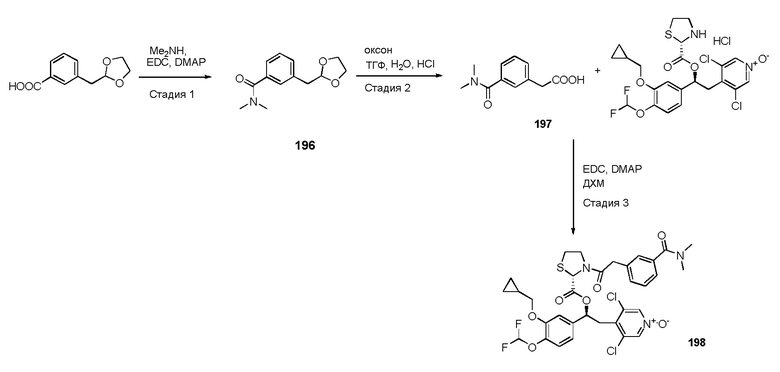
Стадия 1: 3-((1,3-диоксолан-2-ил)метил)-N,N-диметилбензамид (196)
3-((1,3-диоксолан-2-ил)метил)бензойную кислоту (250 мг, 1,201 ммоль), гидрохлорид диметиламина (147 мг, 1,801 ммоль), EDC (345 мг, 1,801 ммоль) и DMAP (513 мг, 4,20 ммоль) растворяли в ДХМ (30 мл), и раствор перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь дважды промывали 1 н. HCl, и органический слой сушили над Na2SO4. Растворитель удаляли при пониженном давлении с получением целевого продукта (244 мг, 1,037 ммоль, 86% выход). MS/ESI+ 236,18 [MH]+.
Стадия 2: 2-(3-(диметилкарбамоил)фенил)уксусная кислота (197)
К раствору 3-((1,3-диоксолан-2-ил)метил)-N,N-диметилбензамида (244 мг, 1,037 ммоль) в ТГФ (30 мл), добавляли воду (20 мл), оксон (1913 мг, 3,11 ммоль) и водный раствор 37% HCl (2 мл, 23,92 ммоль), и смесь перемешивали при комнатной температуре в течение 24 часа. Добавляли дополнительную порцию оксона (1,0 г, 1,627 ммоль) и водный раствор 37% HCl (1 мл, 11,96 ммоль) и продолжали перемешивать при комнатной температуре в течение еще 24 часов. Реакционную смесь разбавляли водой (100 мл) и дважды экстрагировали ДХМ; (2×70 мл); объединенные органические слои сушили над Na2SO4 и упаривали досуха с получением целевого продукта (200 мг, 0,965 ммоль, 93% выход). MS/ESI+ 208,20 [MH]+.
Стадия 3: 1-оксид 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(2-(3-(диметилкарбамоил)фенил)ацетил)тиазолидин-2-карбонилокси)этил)пиридина (198)
1-Оксид 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(2-(3-(диметилкарбамоил)фенил)ацетил)тиазолидин-2-карбонилокси)этил)пиридина получали в соответствии со способом, аналогичным способу, описанному в стадии 3 (схема 13) примера 5, используя ДХМ в качестве растворителя. Его очищали обработкой полимером на подложке с изоцианатом с последующей препаративной ВЭЖХ (способ 2) (10% выход).
MS/ESI+ 724,28 [MH]+, tR=3,82 мин (способ 3); Соотношение диастереомеров >95:5 (1Н ЯМР);
1Н ЯМР (300 МГц, ДМСО-d6) δ м.д. 8,52 (с, 2H), 7,23-7,48 (м, 4H), 7,15 (д, 1H), 7,06 (д, 1H), 6,91 (дд, 1H), 6,79-7,43 (м, 1H), 5,93 (дд, 1H), 5,41 (с, 1H), 3,93 (дд, 2H), 3,88 (д, 2H), 3,82 (с, 2H), 3,41 (дд, 1H), 3,27 (дд, 1H), 3,00-3,22 (м, 2H), 2,96 (шир.с, 3H), 2,90 (шир.с, 3H), 1,06-1,39 (м, 1H), 0,46-0,63 (м, 2H), 0,06-0,41 (м, 2H)
Соединение, указанное в таблице 15, получали в соответствии со способом, аналогичным способу, описанному на схеме 27, и реакцией соответствующего предшественника (полученного в виде свободного основания с помощью основной обработки гидрохлоридой соли водным раствором NaHCO3, а затем экстракцией ДХМ), а затем путем соответствующей стадии очистки, как указано ниже.
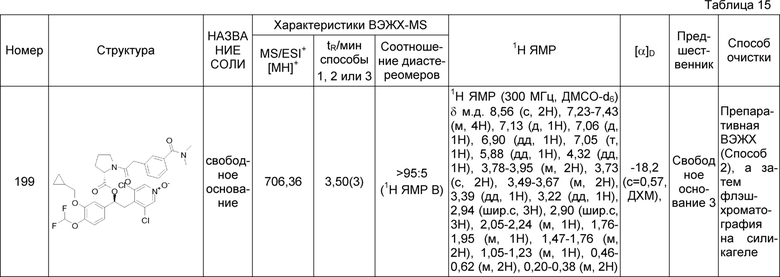
Пример 6
Синтез формиата 1-оксида 3,5-дихлор-4-((2S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(3-(3-(циклопропилметокси)-5-(N-(2-морфолиноэтил)метилсульфонамидо)бензоил)тиазолидин-2-карбонилокси)этил)пиридина (49)
Схема 14
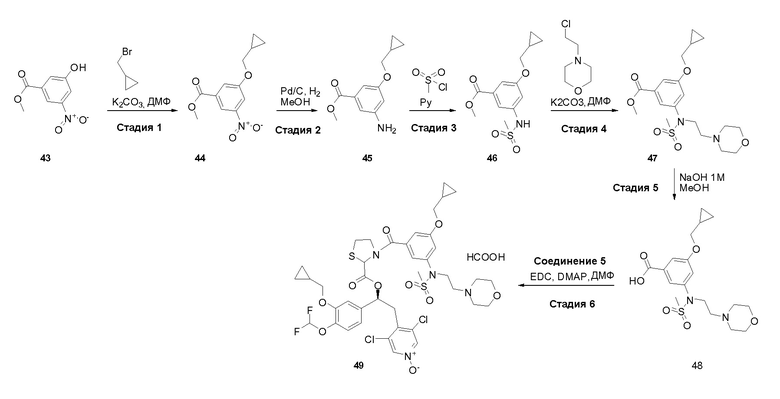
Стадия 1: метил 3-(циклопропилметокси)-5-нитробензоат (44)
Метил 3-гидрокси-5-нитробензоат (43) (1,6 г, 8,1 ммоль) растворяли в ДМФ (15 мл). Добавляли (бромметил)циклопропан (2,2 г, 16,2 ммоль) и K2CO3 (1,7 г, 12,2 ммоль), и смесь перемешивали при 80°C в течение 2 часов. Реакционную смесь охлаждали при комнатной температуре, разбавляли водой и фильтровали. Остаток растворяли в этилацетате, и органическую фазу сушили над Na2SO4 и упаривали под вакуумом с получением 1,65 г целевого продукта (выход 81%).
MS/ESI+ 252,08 [MH]+
Стадия 2: метил 3-амино-5-(циклопропилметокси)бензоат (45)
Метил 3-(циклопропилметокси)-5-нитробензоат (44) (4,9 г, 19,5 ммоль) растворяли в MeOH (200 мл) и добавляли Pd/C 5% (1,5 г, 0,7 ммоль). Раствор трясли в атмосфере водорода на аппарате Парра при 40 фунт/кв.дюйм в течение 1 часа. Катализатор фильтровали на прокладке из диатомовой земли, и растворитель упаривали под вакуумом с получением 3,67 г целевого продукта (выход 85%).
MS/ESI+ 222,11 [MH]+
Стадия 3: метил 3-(циклопропилметокси)-5-(метилсульфонамидо)бензоат (46)
Метил 3-амино-5-(циклопропилметокси)бензоат (45) (1,3 г, 5,9 ммоль) растворяли в пиридине (4 мл). Медленно при 0°C добавляли хлорид метансульфонила (0,6 мл, 7,7 ммоль), и смесь перемешивали при комнатной температуре в течение 2,5 часов. Реакционную смесь разбавляли водным раствором 1 н. HCl, и продукт экстрагировали этилацетатом. Органическую фазу промывали 1 н. HCl, сушили над Na2SO4 и упаривали под вакуумом с получением 1,7 г целевого продукта (выход 97%). MS/ESI+ 300,08 [MH]+
Стадия 4: метил 3-(циклопропилметокси)-5-(N-(2-морфолиноэтил)метилсульфонамидо)бензоат (47)
Метил 3-(циклопропилметокси)-5-(метилсульфонамидо)бензоат (46) (3 г, 10,02 ммоль) растворяли в ДМФ (25 мл). Добавляли 4-(2-хлорэтил)морфолин (4,5 г, 30,1 ммоль) и K2CO3 (2,1 г, 15,03 ммоль), и смесь перемешивали при 60°C в течение 2 часов. Реакционную смесь разбавляли водой и экстрагировали этилацететом. Органическую фазу промывали водой, сушили над Na2SO4 и упаривали под вакуумом с получением 3 г целевого продукта (выход 73%).
MS/ESI+ 413,17 [MH]+
Стадия 5: 3-(циклопропилметокси)-5-(N-(2-морфолиноэтил)метилсульфонамидо)бензойной кислоты (48)
Метил 3-(циклопропилметокси)-5-(N-(2-морфолиноэтил)-метилсульфонамидо)бензоат (47) (3 г, 7,3 ммоль) растворяли в MeOH (45 мл). Добавляли водный раствор 1 н. NaOH (9 мл), и смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавляли водным раствором 1 н. HCl (9 мл), и растворитель удаляли под вакуумом с выходом 3,7 г целевого продукта (количественный выход).
MS/ESI+ 399,15 [MH]+
Стадия 6: формиат 1-оксида 3,5-дихлор-4-((2S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(3-(3-(циклопропилметокси)-5-(N-(2-морфолиноэтил)метилсульфонамидо)бензоил)тиазолидин-2-карбонилокси)этил)пиридина (49)
Соединение (49) получали в соответствии со способом, аналогичным способу, описанному в примере 3, стадии 1, начиная с соединения 5. Сырой продукт очищали с помощью препаративной ВЭЖХ с получением соединения (49) в виде формиатной соли.
MS/ESI+ 915,3 [MH]+; tR=3,37; 3,43 (способ 2); Соотношение диастереомеров = 47:53.
Пример 7
Синтез формиата 1-оксида 3,5-дихлор-4-((2S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(3-(5-((диметиламино)метил)тиофен-2-карбонил)тиазолидин-2-карбонилокси)этил)пиридина (51)
Схема 15
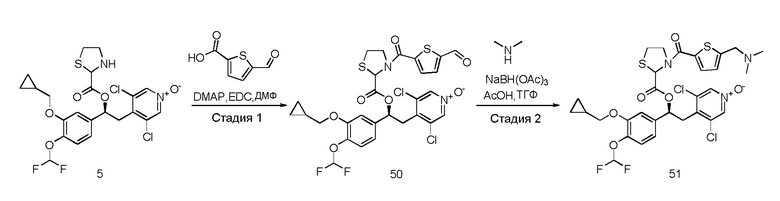
Стадия 1: 1-оксид 3,5-дихлор-4-((2S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(3-(5-формилтиофен-2-карбонил)тиазолидин-2-карбонилокси)этил)пиридина (50)
1-Оксид 3,5-дихлор-4-((2S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(тиазолидин-2-карбонилокси)этил)пиридина (5) (200 мг, 0,374 ммоль), 5-формилтиофен-2-карбоновую кислоту (233 мг, 1,494 ммоль), DMAP (100 мг, 0,822 ммоль) и EDC (358 мг, 1,868 ммоль) растворяли в ДМФ (2 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи, затем смесь разбавляли водой, и остаток промывали водой, растворяли в AcOEt и промывали водным раствором 1 н. HCl, водным насыщенным раствором Na2CO3 и насыщенным солевым раствором. Органическую фазу сушили над Na2SO4 и концентрировали под вакуумом с получением 1-оксида 3,5-дихлор-4-((2S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(3-(5-формилтиофен-2-карбонил)тиазолидин-2-карбонилокси)этил)пиридина (150 мг, выход 60%).
MS/ESI+ 673,04 [MH]+
Стадия 2: 1-оксид 3,5-дихлор-4-((2S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(3-(5-((диметиламино)метил)тиофен-2-карбонил)тиазолидин-2-карбонилокси)этил)пиридина (51)
1-Оксид 3,5-дихлор-4-((2S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(3-(3-формилбензоил)тиазолидин-2-карбонилокси)этил)пиридина (50) (50 мг, 0,075 ммоль) растворяли в ТГФ (1 мл). Добавляли уксусную кислоту (8,58 мкл, 0,150 ммоль) и диметиламин 2M в ТГФ (7,95 мкл, 0,150 ммоль), и смесь перемешивали при комнатной температуре в течение 30 минут. Добавляли триацетоксиборгидрид натрия (32 мг, 0,150 ммоль), и смесь перемешивали при комнатной температуре в течение 3 часов до завершения реакции. Реакционную смесь разбавляли водой и экстрагировали AcOEt. Органическую фазу сушили над Na2SO4 и концентрировали под вакуумом. Сырой продукт очищали полупрепаративной ВЭЖХ (способ 1) с получением желаемого продукта в качестве формиатной соли (16 мг, выход 31%). MS/ESI+ 702,2 [MH]+; tR=2,54 (способ 2); 2,68 мин; Соотношение диастереомеров = 21:79
Пример 8
Синтез 1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(3-(диметилкарбамоил)фенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина (52)
Схема 16
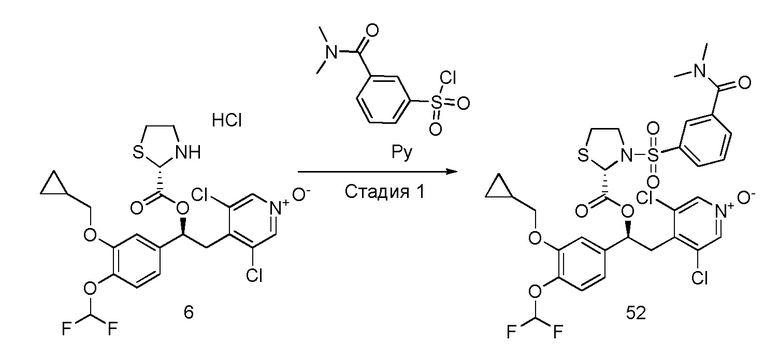
Гидрохлорид 1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-тиазолидин-2-карбонилокси)этил)пиридина (6) (300 мг, 0,525 ммоль) растворяли в Py (3 мл, 37,1 ммоль). Добавляли хлорид 3-(диметилкарбамоил)бензол-1-сульфонила (156 мг, 0,630 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 4 часов до завершения реакции. Реакционную смесь разбавляли водным раствором 1 н. HCl и экстрагировали этилацетатом. Органическую фазу промывали водным раствором 1 н. HCl и насыщенным солевым раствором, сушили над Na2SO4 и концентрировали под вакуумом. Сырой продукт очищали флэш-хроматографией (ДХМ/IsoPrOH 98/2) с получением 1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(3-(диметилкарбамоил)фенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина (250 мг, 0,335 ммоль, 63,8% выход).
MS/ESI+ 746,2 [MH]+; [αD] = -43,30 (c=0,51; CHCl3), tR=3,66 (способ 1); Соотношение диастереомеров= >99/1; 1Н ЯМР (400 МГц, ДМСО-d6) δ м.д. 8,58 (с, 2H), 7,88-7,97 (м, 1H), 7,75-7,82 (м, 1H), 7,68-7,74 (м, 1H), 7,14-7,21 (м, 1H), 7,09-7,13 (м, 1H), 7,08 (т, J=75,00 Гц, 1H), 6,92-6,99 (м, 1H), 5,91-6,10 (м, 1H), 5,54 (с, 1H), 3,79-3,94 (м, 3H), 3,60-3,71 (м, 1H), 3,41-3,51 (м, 1H), 3,26-3,32 (м, 1H), 3,02 (с, 3H), 2,92-3,00 (м, 1H), 2,89 (с, 3H), 2,56-2,70 (м, 1H), 1,20-1,27 (м, 1H), 0,53-0,60 (м, 1H), 0,29-0,36 (м, 1H).
Соединения, перечисленные в таблице 6 (см. в конце описания), получали в соответствии со способом, аналогичным способу, который описан для схемы 16, реакцией соответствующих перечисленных предшественников с коммерческими подходящими реагентами, а затем соответствующей стадией очистки, как описано ниже, если необходимо. Нагревание при МВ облучении (50°C, 30 мин) использовали для синтеза соединения 224.
Некоторые соединения, описанные в таблице 6, дополнительно кристаллизовали в условиях, описанных в таблице 7 (см. в конце описания), с получением одной или нескольких кристаллических форм.
Если в таблице 7 дана ссылка на условие кристаллизации (А), (B), (C), (D) или (Е), то используются следующие рабочие условия:
Эксперименты с циклическими температурами - (А)
Взвесь веществ получали в каждой из выбранных систем растворителей. Приблизительно 10 мг вещества суспендировали в приблизительно 200 мкл растворителя (если материал растворяли, то использовали прозрачный раствор). Взвеси подвергали воздействию циклической температуры при 40°С в 4 часовых циклах в течение 3 суток (уровни охлаждения/нагревания через 4 часовых периодов повышались до около 1°С/мин). Любые твердые вещества выделяли и оставляли сушить при температуре окружающей среды до анализа.
Эксперименты с медленным охлаждением (В)
Эксперимент проводили путем помещения насыщенных растворов вещества в каждой из выбранных систем растворителей при температуре 2°С в течение приблизительно 3 дней. Получали насыщенный раствор и подвергали его соответствующим экспериментальным условиям. Любое твердое вещество затем выделяли и оставляли сушиться при условиях окружающей среды перед анализом.
Эксперименты по быстрому охлаждению - (С)
Эксперимент проводили путем помещения насыщенных растворов вещества в каждой из выбранных систем растворителей при температуре -18°С в течение приблизительно 3 дней. Получали насыщенный раствор и подвергали его соответствующим экспериментальным условиям. Любое твердое вещество затем выделяли и оставляли сушиться при условиях окружающей среды перед анализом.
Эксперименты по упариванию - (D)
Эксперименты по упариванию проводили на насыщенных смесях, как описано выше. Эксперименты проводили, оставляя растворители свободно испаряться в условиях окружающей среды. Любое твердое вещество затем выделяли и оставляли сушиться при условиях окружающей среды перед анализом.
Эксперименты по добавлению антирастворителя - (Е)
Эксперименты по добавлению антирастворителя проводили на насыщенных растворах вещества в каждой из соответствующих систем растворителй и добавляя антирастворитель до осаждения. Любое твердое вещество затем выделяли и оставляли сушиться при условиях окружающей среды перед анализом.
Пример 9
Синтез 1-оксида 3,5-дихлор-4-((2S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(3-(3-(диметилкарбамоил)-4-метоксифенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина (123)
Схема 17
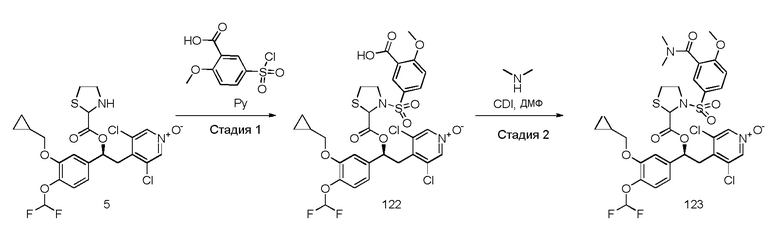
Стадия 1: 1-оксид 4-((2S)-2-(3-(3-карбокси-4-метоксифенилсульфонил)тиазолидин-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)-3,5-дихлорпиридина (122)
1-Оксид 3,5-дихлор-4-((2S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(тиазолидин-2-карбонилокси)этил)пиридина (5) (60 мг, 0,112 ммоль) растворяли в пиридине (1 мл), затем добавляли 5-(хлорсульфонил)-2-метоксибензойную кислоту (56 мл, 0,224 ммоль) при 0°C, и смесь перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь гасили 1 н. HCl, и продукт экстрагировали AcOEt. Органическую фазу промывали 1 н. HCl (×2) и насыщенным солевым раствором, затем сушили над Na2SO4. Растворитель удаляли с получением 70 мг целевого соединения (выход 83%). MS/ESI+ 749,0 [MH]+; tR=5,94; 6,02 (способ 1); Соотношение диастереомеров= 32:68.
Стадия 2: 1-оксид 3,5-дихлор-4-((2S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(3-(3-(диметилкарбамоил)-4-метоксифенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина (123)
1-Оксид 4-((2S)-2-(3-(3-карбокси-4-метоксифенилсульфонил)тиазолидин-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)-3,5-дихлорпиридина (122) (70 мг, 0,093 ммоль) растворяли в ДМФ (1 мл). Добавляли CDI (18 мг, 0,112 ммоль), и смесь перемешивали в течение 30 мин при комнатной температуре. Затем добавляли 2M диметиламин в ТГФ (300 мкл, 0,600 ммоль), и смесь перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь гасили водой, и продукт экстрагировали AcOEt. Органический слой промывали водой (2×) и насыщенным раствором NaCl, сушили над Na2SO4 и упаривали под вакуумом. Сырой продукт очищали с помощью препаративной ВЭЖХ (способ 1) с получением 70 мг целевого соединения (выход 97%). MS/ESI+ 776,1 [MH]+; tR=6,40; 6,50 мин (способ 1); Соотношение диастереомеров = 36:64.
Соединения, перечисленные в таблице 8 (см в конце описания), получали в соответствии со способом, аналогичным способу, описанному на схеме 17, и реакцией соответствующих перечисленных предшественников с подходящими реагентами, а затем соответствующей методикой очистки, как указано ниже.
Соединение, указанное в таблице 9, получали в условиях, аналогичных условиям, указанным на схеме 17, реакцией соответствующего указанного предшественника с подходящими реагентами и используя спиртовой нуклеофил на стадии 2 вместо амина, а затем стадией очистки, как указано ниже.
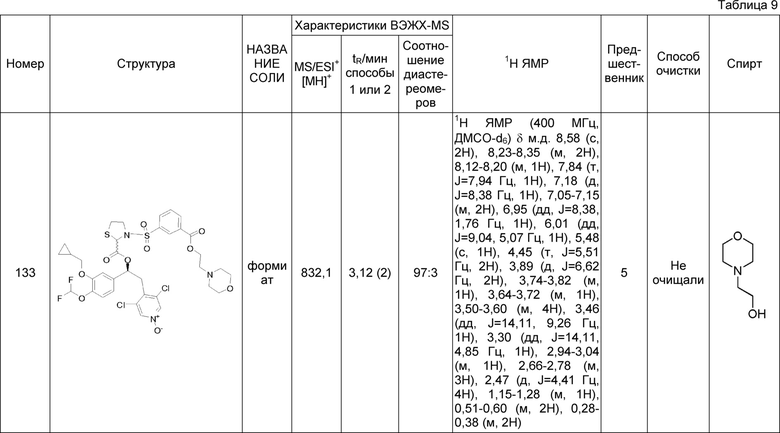
Пример 10
Синтез 1-оксида 4-((2S)-2-(3-(4-аминофенилсульфонил)тиазолидин-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)-3,5-дихлорпиридина (135)
Схема 18
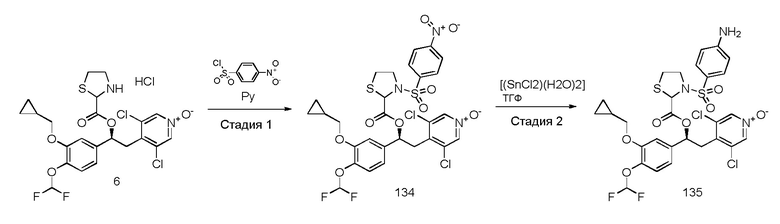
Стадия 1: 1-оксид 3,5-дихлор-4-((2S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(3-(4-нитрофенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина (134)
Гидрохлорид 1-оксида 3,5-дихлор-4-((2S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(тиазолидин-2-карбонилокси)этил)пиридина (6) (1,2 г, 2,24 ммоль) растворяли в пиридине (6 мл), затем при 0°C добавляли хлорид 4-нитробензол-1-сульфонила (596 мг, 2,69 ммоль), и смесь перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь гасили 1 н. HCl, и продукт экстрагировали AcOEt. Органическую фазу промывали 1 н. HCl (×2) и насыщенным солевым раствором, затем сушили над Na2SO4. Растворитель удаляли с выходом 1,5 г целевого соединения (выход 93%). MS/ESI+ 720,04 [MH]+
Стадия 2: 1-оксид 4-((2S)-2-(3-(4-аминофенилсульфонил)тиазолидин-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)-3,5-дихлорпиридина (135)
1-Оксид 3,5-дихлор-4-((2S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(3-(4-нитрофенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина (134) (1 г, 1,388 ммоль) растворяли в ТГФ (10 мл). Добавляли дигидрат хлорида олова(II) (3,13 г, 13,881 ммоль), и смесь перемешивали при комнатной температуре в течение 2 суток. Растворитель удаляли под вакуумом, и сырой продукт растворяли в AcOEt и разбавляли 1 н. HCl. К эмульсии добавляли диатомовую землю, и смесь фильтровали на прокладке из диатомовой земли. Органическую фазу промывали 1 н. HCl, насыщенным солевым раствором, сушили над Na2SO4 и концентрировали под вакуумом с получением 1,5 г сырого вещества, которое очищали флэш-хроматографией (ДХМ/IPA 97:3) с получением 780 мг конечного соединения (1,130 ммоль, выход 81%). MS/ESI+ 689,9 [MH]+; tR=6,30; 6,40 (Способ 1); Соотношение диастереомеров = 38:62
Соединения, перечисленные в таблице 10 (см в конце описания), получали способами, аналогичными способам, описанным для схемы 18, и реакцией соответствующих указанных предшественников с подходящими реагентами, а затем стадией очистки, как указано ниже.
Пример 11
Синтез формиата 1-оксида 3,5-дихлор-4-((2S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(3-(4-(N-(2-морфолиноэтил)-метилсульфонамидо)фенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина (143)
Схема 19
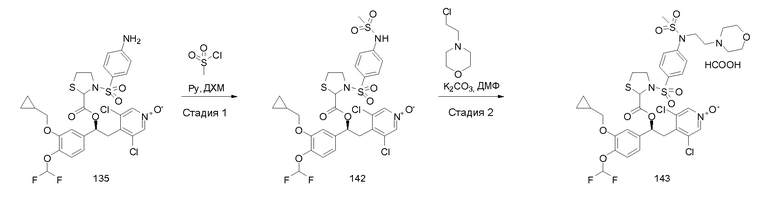
Стадия 1: 1-оксид 3,5-дихлор-4-((2S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(3-(4-(метилсульфонамидо)фенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина (142)
1-Оксид 4-((2S)-2-(3-(4-аминофенилсульфонил)тиазолидин-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)-3,5-дихлорпиридина (135) (128 мг, 0,185 ммоль), полученный в соответствии с аналогичной методикой, описанной в примере 9, растворяли в ДХМ (1,5 мл). В реакционную смесь добавляли пиридин (29,3 мг, 0,371 ммоль) и хлорид метансульфонила (36,1 мг, 0,315 ммоль), смесь перемешивали при комнатной температуре в течение 3 часов до завершения реакции. Реакционную смесь разбавляли ДХМ и экстрагировали 1 н. HCl. Органическую фазу промывали 1 н. HCl и насыщенным солевым раствором, сушили над Na2SO4 и концентрировали под вакуумом. Сырой продукт очищали с помощью препаративной ВЭЖХ (способ 1) с получением 80 мг конечного соединения (выход 56%). MS/ESI+ 767,9 [MH]+, tR=6,25; 6,35 мин (способ 1); Соотношение диастереомеров: 40:60.
Стадия 2: формиат 1-оксида 3,5-дихлор-4-((2S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(3-(4-(N-(2-морфолиноэтил)метилсульфонамидо)фенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина (143)
1-Оксид 3,5-дихлор-4-((2S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(3-(4-(метилсульфонамидо)фенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина (142) (80 мг, 0,104 ммоль) растворяли в ДМФ (1,5 мл). Добавляли 4-(2-хлорэтил)морфолин (78 мг, 0,520 ммоль) и K2CO3 (17,26 мг, 0,125 ммоль), и реакционную смесь перемешивали при 45°C в течение 6 часов до завершения реакции. Реакционную смесь разбавляли водой и экстрагировали AcOEt. Органическую фазу сушили над Na2SO4 и концентрировали под вакуумом. Сырой продукт очищали с помощью препаративной ВЭЖХ (способ 1) с получением 40 мг конечного соединения в виде соли муравьиной кислоты (выход 44%).
MS/ESI+ 880,9 [MH]+; 5 tR=5,15; 5,28 мин (способ 1); Соотношение диастереомеров= 37:63.
Пример 12
Синтез 1-оксида 3,5-дихлор-4-((2S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(3-(2-оксо-2-(тиофен-2-ил)этил)тиазолидин-2-карбонилокси)этил)пиридина (144)
Схема 20
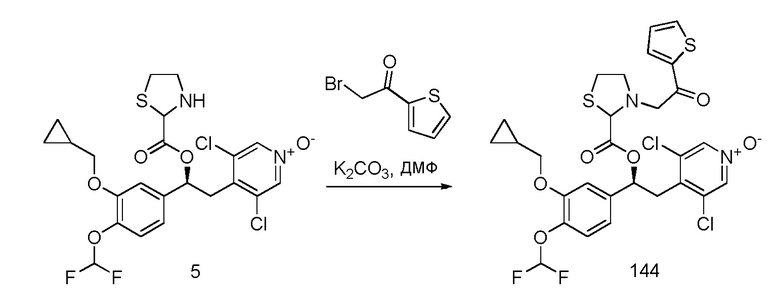
1-Оксид 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(тиазолидин-2-карбонилокси)этил)пиридина (5) (70 мг, 0,131 ммоль) растворяли в ДМФ (1 мл). К раствору добавляли K2CO3 (22 мг, 0,157 ммоль) и 2-бром-1-(тиофен-2-ил)этанон (80 мг, 0,392 ммоль), реакционную смесь перемешивали при 45°C в течение 3 часов до завершения реакции. Реакционную смесь разбавляли водой и экстрагировали AcOEt. Органическую фазу сушили над Na2SO4 и концентрировали под вакуумом. Сырой продукт очищали с помощью препаративной ВЭЖХ (Способ 1) с получением 50 мг конечного продукта (выход 58%). MS/ESI+ 658,9 [MH]+; tR=7,10; 7,24 мин (способ 2); Соотношение диастереомеров = 44:56
Соединения, перечисленные в таблице 16 (см в конце описания), получали в соответствии со способом, аналогичным способу, описанному на схеме 20 и путем взаимодействия соответствующих перечисленных предшественников (полученных как свободное основание после основной обработки гидрохлоридной соли насыщенным водным раствором NaHCO3 с последующей экстракцией ДХМ) с подходящими коммерческими реагентами, используя в качестве растворителя CH3CN вместо ДМФ и нагреванием при 70°C, с последующей соответствующей стадией очистки, как указано ниже.
Пример 21
Синтез 1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(2-(3-(диметилкарбамоил)фенил)-2-оксоэтил)тиазолидин-2-карбонилокси)этил)пиридина (261)
Схема 28
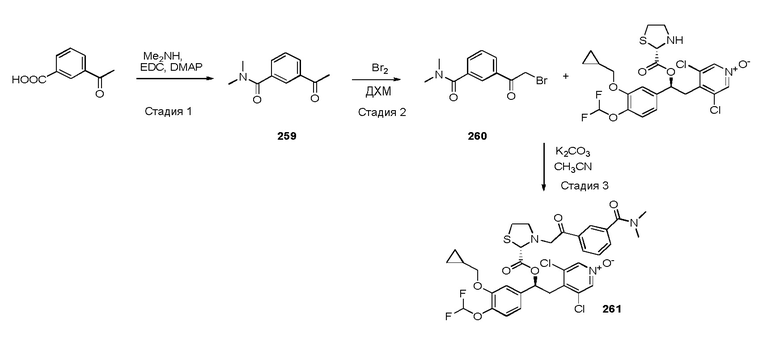
Стадия 1: Ацетил-N,N-диметилбензамид (259)
3-Ацетилбензойную кислоту (400 мг, 2,437 ммоль), гидрохлорид диметиламина (238 мг, 2,92 ммоль), EDC (701 мг, 3,66 ммоль) и DMAP (447 мг, 3,66 ммоль) растворяли в ДХМ (80 мл), полученный раствор перемешивали при комнатной температуре в течение ночи. Реакционную смесь дважды промывали водным раствором 1 н. HCl и насыщенным солевым раствором; органическую фазу сушили над Na2SO4, фильтровали и упаривали с получением целевого продукта (450 мг, 2,353 ммоль, 97% выход); MS/ESI+ 192,12 [MH]+.
Стадия 2: 3-(2-бромацетил)-N,N-диметилбензамид (260)
К раствору 3-ацетил-N,N-диметилбензамида (450 мг, 2,353 ммоль) в ДХМ (20 мл) по каплям добавляли бром (0,121 мл, 2,353 ммоль). Полученный темный раствор перемешивали при комнатной температуре в течение 24 час. Добавляли дополнительную порцию брома (60 мл, 1164 ммоль) и продолжали перемешивать в течение 4 час. Раствор дважды промывали насыщенным водным раствором NaHCO3, и органический слой сушили над Na2SO4. Растворитель удаляли под вакуумом с получением целевого продукта (526 мг, 1,947 ммоль, 83% выход), который использовали на следующей стадии без дальнейшей очистки. MS/ESI+ 269,96 [MH]+
Стадия 3: 1-оксид 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(2-(3-(диметилкарбамоил)фенил)-2-оксоэтил)тиазолидин-2-карбонилокси)этил)пиридина (261)
1-оксид 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(2-(3-(диметилкарбамоил)фенил)-2-оксоэтил)тиазолидин-2-карбонилокси)этил)пиридина получали в соответствии с аналогичным способом, описанным на схеме 20 (пример 12) из промежуточного соединения (6), полученного в виде свободного основания после обработки гидрохлоридной соли водным раствором NaHCO3, а затем экстракцией ДХМ с использованием в качестве растворителя CH3CN и нагревания при 60°C. Его очищали с помощью препаративной ВЭЖХ (способ 2) (41% выход); MS/ESI+ 724,24 [MH]+; tR=3,82 мин (способ 3); соотношение диастереомеров >95:5 (1Н ЯМР); [αD] = -40,6 (c=0,38, ДХМ).
1Н ЯМР (B) (300 МГц, ДМСО-d6) δ м.д. 8,54 (с, 2H), 8,00 (дт, 1H), 7,96 (т, 1H), 7,67 (дт, 1H), 7,60 (т, 1H), 7,18 (д, 1H), 7,13 (д, 1H), 6,96 (дд, 1H), 6,81-7,41 (м, 1H), 5,98 (дд, 1H), 4,90 (с, 1H), 4,13 (д, 1H), 4,02 (д, 1H), 3,92 (д, 2H), 3,45-3,55 (м, 1H), 3,40 (дд, 1H), 3,28 (м, 0H), 3,13-3,23 (м, 1H), 3,01 (шир.с, 3H), 2,86-2,94 (м, 4H), 2,82-3,05 (м, 2H), 1,07-1,39 (м, 1H), 0,45-0,70 (м, 2H), 0,18-0,44 (м, 2H)
Соединение, указанное в таблице 17, получали в соответствии с аналогичной методикой, как описано на схеме 28, и реакцией соответствующего указанного предшественника (полученный в виде свободного основания после основной обработки гидрохлоридной соли насыщенным водным раствором NaHCO3 с последующей экстракцией ДХМ), нагревая при 70°C на стадии 3, а затем соответствующей стадией очистки, как указано ниже.
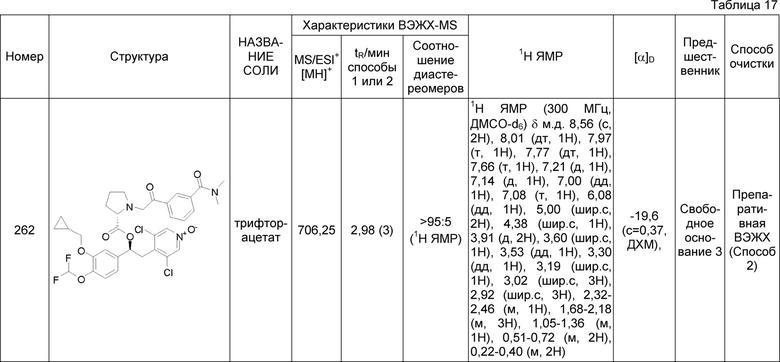
Пример 13
Синтез 1-оксида 4-((S)-2-((S)-3-(4-аминофенилсульфонил)тиазолидин-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)-3,5-дихлорпиридина (150)
Схема 21
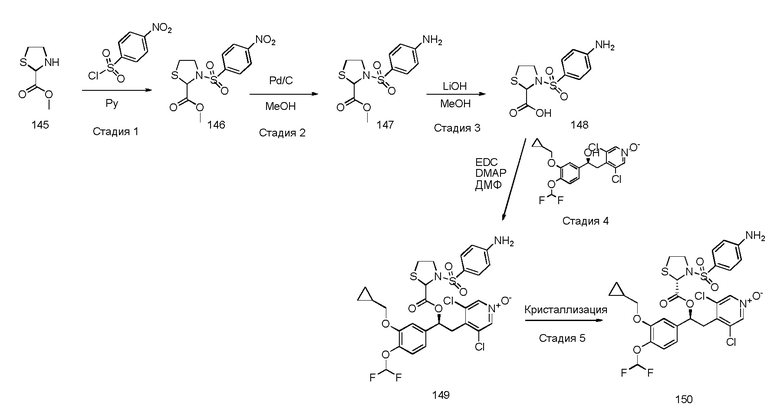
Стадия 1: метил 3-(4-нитрофенилсульфонил)тиазолидин-2-карбоксилат (146)
Гидрохлорид метил тиазолидин-2-карбоксилата (145) (20 г; 109 ммоль) растворяли в пиридине (100 мл), затем добавляли хлорид 4-нитробензол-1-сульфонила (27 г, 124 ммоль) при 0°C, и смесь перемешивали в течение 3 часов. По прошествии этого времени реакционную смесь гасили 1 н. HCl для осаждения твердого вещества, которое фильтровали через фрит и несколько раз промывали водой. Твердое вещество оранжевого цвета растирали в ацетоне, промывали ацетоном (×2) и сушили под вакуумом с выходом 27 г (76%).
MS/ESI+ 333,01 [MH]+
Стадия 2: гидрохлорид метил 3-(4-аминофенилсульфонил)тиазолидин-2-карбоксилата (147)
Смесь метил 3-(4-нитрофенилсульфонил)тиазолидин-2-карбоксилата (146) (5,74 г; 17,27 ммоль) и 5% Pd/C (16 г; 7,547 ммоль) в 600 мл MeOH и 400 мл 1 н. HCl гидрогенизировали в течение 10 часов под давлением 50 фунт/кв.дюйм. По прошествии этого времени реакционную смесь фильтровали через целит, промывали MeOH, и раствор концентрировали под вакуумом с выходом целевого продукта (10,47 г; 72%). MS/ESI+ 312,04 [MH]+
Стадия 3: 3-(4-аминофенилсульфонил)тиазолидин-2-карбоновая кислота (148)
К раствору гидрохлорида метил 3-(4-аминофенилсульфонил)тиазолидин-2-карбоксилата (147) (7,43 г, 24,57 ммоль) в MeOH (62 мл) добавляли 1 M LiOH (62 мл). Раствор перемешивали при комнатной температуре в течение 1 часа, затем pH доводили до 6, с помощью 1 M HCl. MeOH упаривали при пониженном давлении, добавляли 1 M HCl до pH=3, смесь охлаждали при 0 градусов в течение 3 часов до полного осаждения. Твердое вещество фильтровали, промывали водой и сушили под вакуумом при 45 градусах с получением 6,44 г целевого продукта (выход 91%).
MS/ESI+ 289,02 [MH]+
Стадия 4: 1-оксид 4-((2S)-2-(3-(4-аминофенилсульфонил)тиазолидин-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)-3,5-дихлорпиридина (149)
Раствор 1-оксида (S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-гидроксиэтил)пиридина (1,55 г, 3,69 ммоль) растворяли в ДМФ (20 мл), EDC (2,83 г, 14,77 ммоль) и добавляли 3-(4-аминофенилсульфонил)тиазолидин-2-карбоновую кислоту (148) (1,81 г, 6,28 ммоль). Смесь охлаждали до 0 градусов, и добавляли DMAP (0,541 г, 4,43 ммоль). Смесь перемешивали при -20 градусах в течение 20 минут, затем ее оставляли нагреваться до комнатной температуры и перемешивали в течение 3 часов. Реакционную смесь выливали в воду (500 мл), и остаток отделяли фильтрацией. Отфильтрованное твердое вещество промывали водой (150 мл) и сушили под вакуумом при 45 градусах с получением 2,49 г целевого продукта (выход 98%). MS/ESI+ 689,8 [MH]+
Стадия 5: 1-оксид 4-((S)-2-((S)-3-(4-аминофенилсульфонил)тиазолидин-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)-3,5-дихлорпиридина (150)
1-Оксид 4-((2S)-2-(3-(4-аминофенилсульфонил)тиазолидин-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)-3,5-дихлорпиридина (149) (2,49 г, 3,61 ммоль) растворяли в EtOH (35 мл) при 50 градусах, добавляли раствор метансульфоновой кислоты в EtOH 10% масс./масс. (3,47 г, 3,61 ммоль) для солеобразования. Метансульфонатную соль отфильтровывали и вновь кристаллизовали из горячего EtOH (25 мл). Соль растворяли в CH2Cl2 (40 мл), и к раствору добавляли насыщенную соль NaHCO3 (30 мл). Смесь перемешивали в течение 30 минут при комнатной температуре, затем два фазы разделяли. Органическую фазу сушили над Na2SO4 и упаривали при пониженном давлении с получением твердого вещества, которое вновь кристаллизовали из горячего EtOH (25 мл) с выходом 800 мг целевого продукта. MS/ESI+ 689,8 [MH]+; tR=6,37 мин (способ 1); [αD] = +38,57 (c=0,49; CHCl3). 1Н ЯМР (400 МГц, ДМСО-d6) δ м.д. 8,58 (с, 2H), 7,42-7,52 (м, 2H), 7,14-7,20 (м, 1H), 7,05-7,12 (м, 2H), 6,90-6,97 (м, 1H), 6,60-6,69 (м, 2H), 6,15-6,24 (с, 2H), 5,96-6,05 (м, 1H), 5,29 (с, 1H), 3,86-3,95 (м, 2H), 3,68-3,80 (м, 1H), 3,52 (д, J=47,63 Гц, 1H), 3,38-3,48 (м, 1H), 3,29 (м, 1H), 2,84-2,99 (м, 1H), 2,53-2,60 (м, 1H), 1,22-1,36 (м, 1H), 0,46-0,63 (м, 2H), 0,28-0,40 (м, 2H).
Соединение, указанное в таблице 11, получали согласно аналогичным способам, описанным на схеме 21 (стадии 1-4), с последующей очисткой соединения (131), посредством флэш-хроматографии, элюируя смесью ДХМ/н-гексан/i-PrOH/EtOH=55/40/4/1.
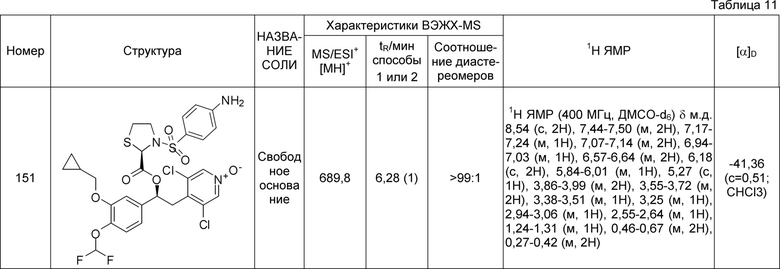
Пример 14
Синтез 1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-1-(1-метил-1H-имидазол-2-илсульфонил)пирролидин-2-карбонилокси)этил)пиридина (155)
Схема 22
Стадия 1: (S)-метил 1-(1-метил-1H-имидазол-2-илсульфонил)пирролидин-2-карбоксилат (153)
(S)-метил пирролидин-2-карбоксилат (152) (50 мг; 0,387 ммоль) растворяли в пиридине (1 мл), а затем добавляли хлорид 1-метил-1H-имидазол-2-сульфонил (70 мг, 0,387 ммоль) при 0°C, и смесь перемешивали в течение 3 часов. По прошествии этого времени реакционную смесь гасили 1 н. HCl и дважды экстрагировали AcOEt. Органическую фазу сушили над Na2SO4 и упаривали под вакуумом с выходом 50 мг (выход 47%).
MS/ESI+ 274,08 [MH]+
Стадия 2: (S)-1-(1-метил-1H-имидазол-2-илсульфонил)пирролидин-2-карбоновая кислота (154)
Реакция раствора (S)-метил 1-(1-метил-1H-имидазол-2-илсульфонил)пирролидин-2-карбоксилата (153) (50 мг, 0,183 ммоль) в смеси HCl/диоксана 4 M (1 мл) протекала под микроволновым облучением при 100 градусах в течение 30 мин. Затем диоксан упаривали при пониженном давлении с выходом 40 мг целевого соединения (выход 84%).
MS/ESI+ 260,06 [MH]+
Стадия 3: 1-оксид 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-1-(1-метил-1H-имидазол-2-илсульфонил)пирролидин-2-карбонилокси)этил)пиридина (155)
К раствору 1-оксида (S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-гидроксиэтил)пиридина (20 мг, 0,048 ммоль) растворенного в ДМФ (1 мл) добавляли EDC (9 мг, 0,048 ммоль) и 3-(4-аминофенилсульфонил)тиазолидин-2-карбоновую кислоту (154) (40 мг, 0,154 ммоль). Смесь охлаждали при 0 градусов и добавляли DMAP (6 мг. 0,048 ммоль). Смесь перемешивали при -20 градусах в течение 20 минут, затем ее оставляли нагреваться до комнатной температуры и перемешивали в течение 3 часов. Реакционную смесь выливали в воду и экстрагировали AcOEt (×3). Органическую фазу сушили над Na2SO4, упаривали под вакуумом с получением 30 мг целевого продукта (выход 95%). MS/ESI+ 661,3 [MH]+; tR=3,60 мин (способ 2); Соотношение диастереомеров = 99:1; 1Н ЯМР (400 МГц, ДМСО-d6) δ м.д. 8,51 (с, 2H), 7,47 (м, 1H), 7,18 (д, J=7,94 Гц, 1H), 7,13 (д, J=1,76 Гц, 1H), 7,08 (д, J=4,41 Гц, 2H), 6,98 (дд, J=8,38, 1,76 Гц, 1H), 6,00 (дд, J=9,48, 4,63 Гц, 1H), 4,47 (дд, J=8,82, 4,41 Гц, 1H), 3,92 (дд, J=7,06, 1,76 Гц, 2H), 3,84 (с, 3H), 3,37-3,55 (м, 3H), 3,24 (дд, J=14,11, 4,41 Гц, 1H), 2,22-2,37 (м, 1H), 1,83-1,95 (м, 1H), 1,65-1,82 (м, 2H), 1,12-1,30 (м, 1H), 0,49-0,64 (м, 2H), 0,28-0,42 (м, 2H).
Пример 22
Синтез 1-оксида 3,5-дихлор-4-((S)-2-(4-(дифторметокси)-3-гидроксифенил)-2-((S)-3-(3-(диметилкарбамоил)фенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина (263)
Схема 47
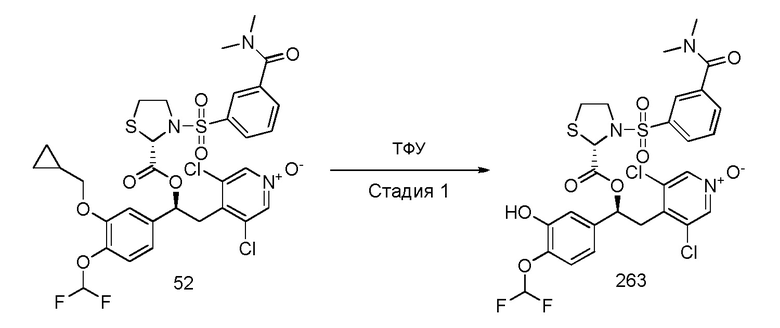
1-оксид 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(3-(диметилкарбамоил)фенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина (Соединение 52) (200 мг, 0,268 ммоль) растворяли в 2,2,2-трифторуксусной кислоте (2 мл, 0,268 ммоль), и раствор перемешивали в течение ночи при комнатной температуре. Реакционную смесь разбавляли ДХМ и концентрировали под вакуумом (2×) с получением сырого вещества, которое очищали посредством препаративного ВЭЖХ (способ 1) с получением 1-оксида 3,5-дихлор-4-((S)-2-(4-(дифторметокси)-3-гидроксифенил)-2-((S)-3-(3-(диметилкарбамоил)фенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина (90 мг, 0,130 ммоль, 49% выход). MS/ESI+ 661,3 [MH]+; tR=2,49 (способ 2); Соотношение диастереомеров = 99:1; 1Н ЯМР (400 МГц, ДМСО-d6) δ м.д. 10,05 (с, 1H), 8,58 (с, 2H), 7,85 7,97 (м, 2H), 7,67-7,80 (м, 2H), 7,11 (д, J=8,38 Гц, 1H), 7,04 (т, J=75,00 Гц, 1H), 6,94 (д, J=2,21 Гц, 1H), 6,83 (м, 1H), 5,86-6,02 (м, 1H), 5,47 (с, 1H), 3,78-3,92 (м, 1H), 3,56-3,68 (м, 1H), 3,37-3,49 (м, 1H), 3,20-3,28 (м, 1H), 2,93-3,09 (м, 4H), 2,89 (с, 3H), 2,66 (м, 1H).
Пример 23
Синтез 1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(циклопропилметил)тиазолидин-2-карбонилокси)этил)пиридина (264)
Схема 30
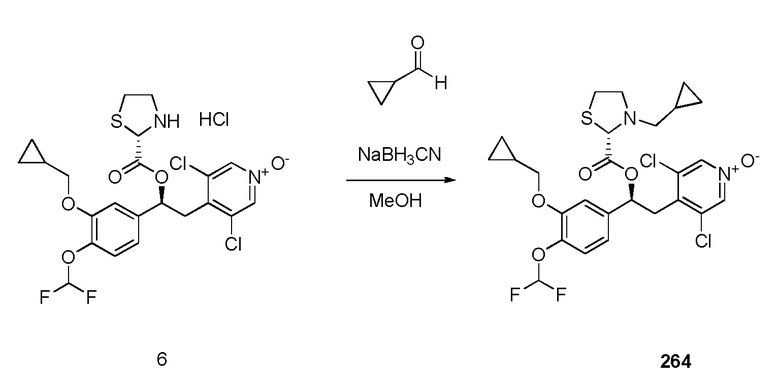
Гидрохлорид 1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-тиазолидин-2-карбонилокси)этил)пиридина (6) (150 мг, 0,262 ммоль) растворяли в MeOH (10 мл) и циклопропанкарбальдегиде (19,60 мкл, 0,262 ммоль) с последующим добавлением цианоборгидрида натрия (33,0 мг, 0,525 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 2 часов. Летучие компоненты удаляли при пониженном давлении, и остаток очищали с помощью препаративной ВЭЖХ (Способ 2). Дальнейшая очистка с помощью флэш-хроматографии на силикагеле (ДХМ/MeOH = 99/1) потребовалась для получения 1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(циклопропилметил)тиазолидин-2-карбонилокси)этил)пиридина (25 мг, 0,042 ммоль, 16,17% выход). MS/ESI+ 588,94 [MH]+; tR=3,54 (способ 3); Соотношение диастереомеров = >95:5 (1Н ЯМР); [αD] = -30,1 (c=0,31, ДХМ);
1Н ЯМР (B) (300 МГц, ДМСО-d6) δ м.д. 8,58 (с, 2H), 7,19 (д, 1H), 7,13 (д, 1H), 6,97 (дд, 1H), 7,08 (т, 1H), 5,95 (дд, 1H), 4,96 (с, 1H), 3,93 (д, 2H), 3,38-3,56 (м, 1H), 3,40 (дд, 1H), 3,26 (дд, 1H), 2,98-3,17 (м, 1H), 2,76-2,95 (м, 2H), 2,31 (дд, 1H), 2,17 (дд, 1H), 1,14-1,35 (м, 1H), 0,72-0,98 (м, 1H), 0,53-0,66 (м, 2H), 0,43-0,53 (м, 2H), 0,26-0,42 (м, 2H), -0,03-0,19 (м, 2H)
Соединения, перечисленные в таблице 18 (см. в конце описания), получали в соответствии с аналогичным способом, описанным на схеме 30, и реакцией соответствующих перечисленных предшественников с коммерчески подходящими реагентами, и последующей соответствующей стадией очистки, как ниже указано.
Пример 24
Синтез 1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(3-уреидофенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина (269)
Схема 31
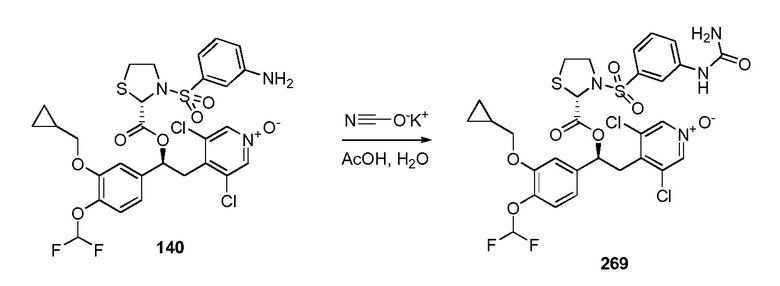
1-Оксид 4-((S)-2-((S)-3-(3-аминофенилсульфонил)тиазолидин-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)-3,5-дихлорпиридина (140) (140, полученного, как описано в примере 10, 210 мг, 0,304 ммоль), растворяли в смеси AcOH (4 мл) и воды (2 мл) при 10°C, немедленно добавляли цианат калия (99 мг, 1,216 ммоль). Реакционную смесь перемешивали в течение 1 часа при 10°C. Добавляли воду (30 мл), осадок собирали фильтрацией и очищали с помощью препаративной ВЭЖХ (способ 2) с получением 1-оксид 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(3-уреидофенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина (136 мг, 0,185 ммоль, 61,0% выход). MS/ESI+ 733,05 [MH]+; tR=5,39 (способ 3); Соотношение диастереомеров >95:5 (1Н ЯМР); [αD] = -78,85 (c=0,4, ДХМ);
1Н ЯМР (B) (300 МГц, ДМСО-d6) δ м.д. 8,95 (с, 1H), 8,58 (с, 2H), 8,08 (т, 1H), 7,62 (ддд, 1H), 7,49 (т, 1H), 7,36 (дт, 1H), 7,18 (д, 1H), 7,12 (д, 1H), 6,96 (дд, 1H), 7,08 (т, 1H), 6,02 (дд, 1H), 6,01 (шир.с, 2H), 5,30 (с, 1H), 3,91 (д, 2H), 3,61-3,82 (м, 2H), 3,49-3,54 (м, 1H), 3,31 (дд, 1H), 2,99 (дт, 1H), 2,73 (дт, 1H), 1,09-1,38 (м, 1H), 0,48-0,62 (м, 2H), 0,21-0,45 (м, 2H)
Пример 25
Синтез 1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(3-(гидроксиметил)фенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина (271)
Схема 32
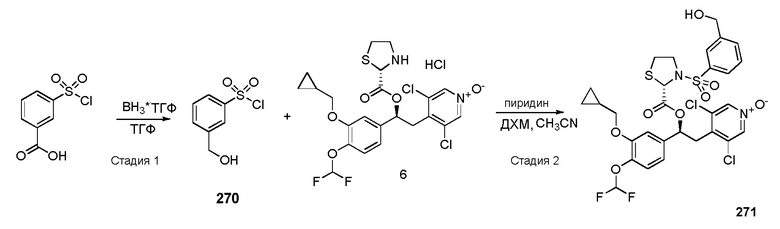
Стадия 1: хлорид 3-(гидроксиметил)бензол-1-сульфонила (270)
К раствору 3-(хлорсульфонил)бензойной кислоты (0,300 г, 1,360 ммоль) в сухом ТГФ (6 мл), охлажденному до 0°C, добавляли комплекс 1 M BH3*ТГФ в ТГФ (5,44 мл, 5,44 ммоль), и полученную смесь оставляли нагреваться до комнатной температуры, перемешивая в течение ночи. Добавляли дополнительную порцию комплекса 1 M BH3*ТГФ в ТГФ (1,360 мл, 1,360 ммоль) и продолжали перемешивать в течение 3 суток при комнатной температуре. Смесь осторожно гасили 2M HCl, разбавляли насыщенным солевым раствором и дважды экстрагировали EtOAc. Объединенные органические слои промывали насыщенным солевым раствором и сушили над сульфатом натрия. Растворитель удаляли, и сырое вещество очищали фильтрацией через картридж с силикагелем (петролейный эфир:EtOAc = 70:30) с получением хлорида 3-(гидроксиметил)бензол-1-сульфонил (0,086 г, 0,416 ммоль, 30,6% выход). MS/ESI+ не поддаются обнаружению [MH]+.
Стадия 2: 1-оксид 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(3-(гидроксиметил)фенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина (271)
К раствору гидрохлорида 1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-тиазолидин-2-карбонилокси)этил)пиридина (6) (0,183 г, 0,320 ммоль) в ДХМ (5 мл), CH3CN (2 мл) и пиридина (0,078 мл, 0,960 ммоль), охлажденному при 0°C, добавляли раствор хлорида 3-(гидроксиметил)бензол-1-сульфонила (0,086 г, 0,416 ммоль) в ДХМ (2 мл), полученную смесь нагревали до комнатной температуры и перемешивали. Летучие компоненты удаляли под вакуумом; остаток очищали с помощью препаративной ВЭЖХ (способ 2 в нейтральной среде, без ТФУ), и собранные фракции лиофилизировали с получением 1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(3-(гидроксиметил)фенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина (0,100 г, 0,142 ммоль, 44,3% выход); MS/ESI+ 705,14 [MH]+; tR=3,78 (способ 3); Соотношение диастереомеров >95:5 (1Н ЯМР); [αD] = -65,67 (c=0,42; MeOH);
1Н ЯМР (B) (300 МГц, ДМСО-d6) δ м.д. 8,58 (с, 2H), 7,83 (т, 1H), 7,73 (дт, 1H), 7,68 (дт, 1H), 7,61 (т, 1H), 7,19 (д, 1H), 7,12 (д, 1H), 6,97 (дд, 1H), 7,08 (т, 1H), 6,03 (дд, 1H), 5,44 (т, 1H), 5,38 (с, 1H), 4,62 (д, 2H), 3,91 (д, 2H), 3,76 (дт, 1H), 3,67 (дт, 1H), 3,47 (дд, 1H), 3,31 (дд, 1H), 2,98 (дт, 1H), 2,68 (дт, 1H), 1,03-1,38 (м, 1H), 0,49-0,67 (м, 2H), 0,15-0,45 (м, 2H).
Пример 26
Синтез 1-оксида 4-((2S)-2-(2-(3-бензоилтиазолидин-2-ил)ацетокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)-3,5-дихлорпиридина (274)
Схема 33
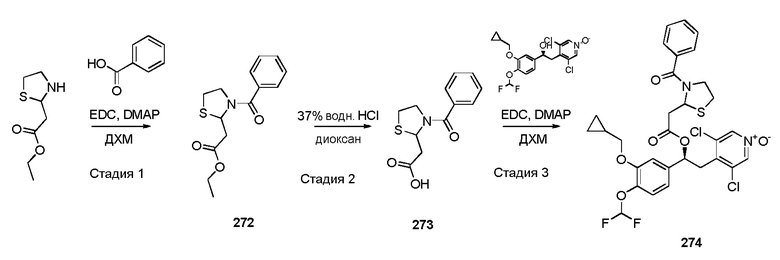
Стадия 1: этил 2-(3-бензоилтиазолидин-2-ил)ацетата (272)
Смесь этил 2-(тиазолидин-2-ил)ацетата (0,100 г, 0,571 ммоль) (получали по следующему синтетическому протоколу, описанному в J. Chem. Soc. Perkin Trans. I, 1987, 1845-1851), бензойной кислоты (0,084 г, 0,685 ммоль), EDC (0,219 г, 1,141 ммоль) и DMAP (0,070 г, 0,571 ммоль) в ДХМ (10 мл) перемешивали при комнатной температуре в течение 1 часа. Смесь разбавляли ДХМ и промывали водным раствором 1 н. HCl, 1 н. NaHCO3 и насыщенным солевым раствором; органическую фазу сушили над сульфатом натрия, и растворитель удаляли. Остаток очищали флэш-хроматографией на силикагелевом картридже (петролейный эфир:EtOAc = 80:20 до 70: 30) с получением этил 2-(3-бензоилтиазолидин-2-ил)ацетата (0,085 г, 0,304 ммоль, 53% выход). MS/ESI+ 280,0 [MH]+.
Стадия 2: 2-(3-бензоилтиазолидин-2-ил)уксусная кислота (273)
К раствору этил 2-(3-бензоилтиазолидин-2-ил)ацетата (0,083 г, 0,297 ммоль) в диоксане (4 мл) добавляли водную 37% HCl (4 мл), и смесь перемешивали при комнатной температуре в течение 20 часов. Летучие компоненты удаляли под вакуумом с выходом сырого вещества 2-(3-бензоилтиазолидин-2-ил)уксусной кислоты (0,074 г, 0,294 ммоль, 99% выход), которое использовали без очистки. MS/ESI+ 251,9 [MH]+.
Стадия 3: 1-оксид 4-((2S)-2-(2-(3-бензоилтиазолидин-2-ил)ацетокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)-3,5-дихлорпиридина (274)
Смесь 1-оксида 2-(3-бензоилтиазолидин-2-ил)уксусной кислоты (0,074 г, 0,294 ммоль), (S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-гидроксиэтил)пиридина (0,103 г, 0,245 ммоль), EDC (0,141 г, 0,736 ммоль) и DMAP (0,030 г, 0,245 ммоль) в ДХМ (10 мл) перемешивали при комнатной температуре в течение 2 часов. Смесь разбавляли ДХМ и промывали 1 н. HCl, 1 н. NaHCO3 и насыщенным солевым раствором; органическую фазу сушили над сульфатом натрия и растворитель удаляли. Сырое вещество очищали с помощью препаративной ВЭЖХ (способ 2), а затем флэш-хроматографией на силикагелевом картридже (ДХМ:MeOH = 99:1) с получением указанного в заголовке соединения в виде диастеромерной смеси (0,065 г, 0,099 ммоль, 40,5% выход); MS/ESI+ 653,19 [MH]+; tR=3,92 (способ 3); соотношение диастереомеров = 1:1 (1Н ЯМР).
Соединение, указанное в таблице 19, получали в соответствии с аналогичным способом, как описано в схеме 33, и используя подходящие реагенты, с последующей соответствующей стадией очистки, как описано ниже.
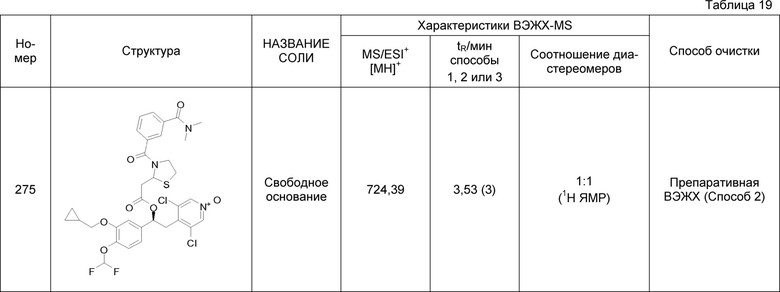
Пример 27
Синтез 1-оксида 3,5-дихлор-4-((2S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(2-(3-(фенилсульфонил)тиазолидин-2-ил)ацетокси)этил)пиридина (278)
Схема 48
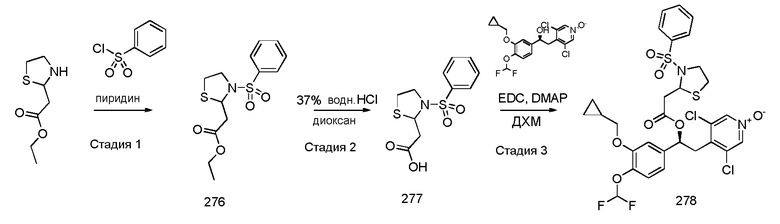
Стадия 1: этил 2-(3-(фенилсульфонил)тиазолидин-2-ил)ацетат (276)
К раствору этил 2-(тиазолидин-2-ил)ацетата (0,100 г, 0,571 ммоль) (полученного в соответствии с синтетическим протоколом, описанным в J. Chem. Soc. Perkin Trans. I, 1987, 1845-1851) в пиридине (4 мл), охлажденному до 0°C, добавляли бензолсульфонилхлорид (0,088 мл, 0,685 ммоль), и реакционную смесь перемешивали в течение 2 часов при комнатной температуре. Смесь разделяли между EtOAc и 1 н. HCl; органическую фазу промывали 1 н. HCl и насыщенным солевым раствором и сушили над сульфатом натрия. Растворитель удаляли, и сырое вещество очищали флэш-хроматографией на силикагеле (петролейный эфир: EtOAc = 90:10 до 80:20) с получением этил 2-(3-(фенилсульфонил)тиазолидин-2-ил)ацетата (0,096 г, 0,304 ммоль, 53,3% выход). MS/ESI+ 316,0 [MH]+.
Стадия 2: 2-(3-(фенилсульфонил)тиазолидин-2-ил)уксусная кислота (277)
К раствор этил 2-(3-(фенилсульфонил)тиазолидин-2-ил)ацетата (0,096 г, 0,304 ммоль) в диоксане (5 мл) добавляли водную 37% HCl (5 мл), и смесь перемешивали при комнатной температуре в течение 25 часов. Летучие компоненты удаляли под вакуумом с получением 2-(3-(фенилсульфонил)тиазолидин-2-ил)уксусной кислоты (0,083 г, 0,289 ммоль, 95% выход), которую использовали без дополнительной очистки. MS/ESI+ 310,0 [Mna]+.
Стадия 3: 1-оксид 3,5-дихлор-4-((2S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(2-(3-(фенилсульфонил)тиазолидин-2-ил)ацетокси)этил)пиридина (278)
Смесь 2-(3-(фенилсульфонил)тиазолидин-2-ил)уксусной кислоты (0,083 г, 0,289 ммоль), 1-оксида (S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-гидроксиэтил)пиридина (0,101 г, 0,241 ммоль), EDC (0,046 г, 0,241 ммоль) и DMAP (0,029 г, 0,241 ммоль) в ДХМ (10 мл) перемешивали при комнатной температуре в течение 1 часа. Смесь разбавляли ДХМ и промывали 1 н. HCl, 1 н. NaHCO3 и насыщенным солевым раствором; органическую фазу сушили над сульфатом натрия, и растворитель удаляли. Сырое вещество очищали с помощью препаративной ВЭЖХ (способ 2) с получением 1-оксида 3,5-дихлор-4-((2S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(2-(3-(фенилсульфонил)тиазолидин-2-ил)ацетокси)этил)пиридина (0,122 г, 0,177 ммоль, 73,5% выход); MS/ESI+ 689,14 [MH]+; tR=4,09 (способ 3); Соотношение диастереомеров = 1:1 (1Н ЯМР).
Соединение, указанное в таблице 20, получали в соответствии со способом, аналогичным способу, описанному на схеме 48, и используя подходящие реагенты, с последующей соответствующей стадией очистки, как описано ниже.
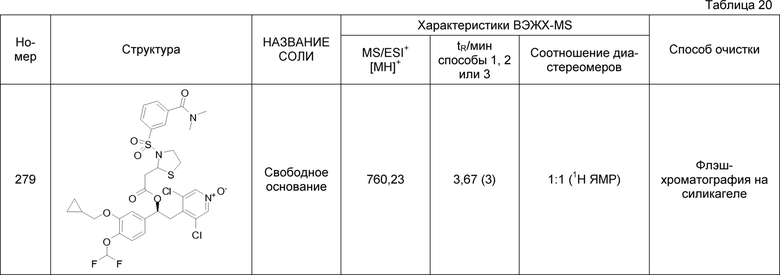
Пример 28
1-Оксид (S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(1-(3-(диметилкарбамоил)фенилсульфонил)азетидин-3-карбонилокси)этил)пиридина (281)
Схема 34
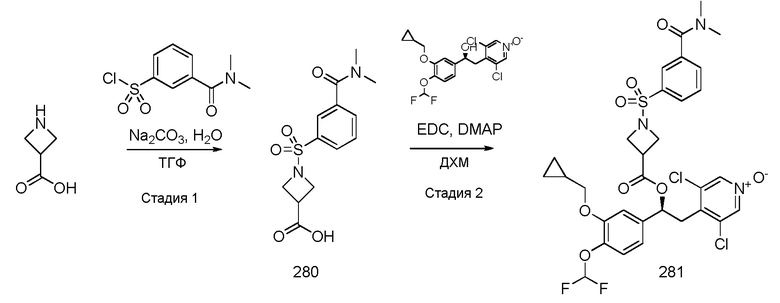
Стадия 1: 1-(3-(диметилкарбамоил)фенилсульфонил)азетидин-3-карбоновая кислота (280)
К суспензии азетидин-3-карбоновой кислоты (100 мг, 0,989 ммоль) в смеси ТГФ (6 мл) и водного 1 M Na2CO3 (6 мл, 6,00 ммоль), охлажденного до 0°C, добавляли хлорид 3-(диметилкарбамоил)бензол-1-сульфонила (269 мг, 1,088 ммоль), и реакционную смесь перемешивали при 0°C в течение 1 часа. Смесь экстрагировали Et2O, и органический слой удаляли. Водный слой осторожно подкисляли, добавляя твердый KHSO4 (pH=3) и дважды экстрагировали EtOAc. Объединенные органические слои сушили над Na2SO4 и упаривали досуха с выходом сырого вещества в виде 1-(3-(диметилкарбамоил)фенилсульфонил)азетидин-3-карбоновой кислоты (255 мг, 0,816 ммоль, 83% выход), которую использовали без дальнейшей очистки.
MS/ESI+ 313,12 [MH]+
Стадия 2: 1-оксид (S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(1-(3-(диметилкарбамоил)фенилсульфонил)азетидин-3-карбонилокси)этил)пиридина (281)
Раствор 1-(3-(диметилкарбамоил)фенилсульфонил)азетидин-3-карбоновой кислоты (255 мг, 0,816 ммоль), 1-оксида (S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-гидроксиэтил)пиридина (175 мг, 0,416 ммоль), EDC (96 мг, 0,500 ммоль) и DMAP (61,0 мг, 0,500 ммоль) в ДХМ (30 мл) перемешивали при комнатной температуре в течение ночи. Дополнительно добавляли EDC (80 мг, 0,416 ммоль) и DMAP (61,0 мг, 0,500 ммоль) и продолжали перемешивать еще 2 часа. Реакционную смесь дважды промывали водной 1 н. HCl затем водным 1 M Na2CO3, органическую фазу сушили над Na2SO4, и растворитель удаляли при пониженном давлении. Твердое вещество очищали с помощью препаративной ВЭЖХ (Способ 2) с получением 1-оксида (S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(1-(3-(диметилкарбамоил)фенилсульфонил)азетидин-3-карбонилокси)этил)пиридина (192 мг, 0,269 ммоль, 64,5% выход); MS/ESI+ 714,16 [MH]+; [αD] = -14,3 (c=0,37, ДХМ);
1Н ЯМР (B) (300 МГц, ДМСО-d6) δ м.д. 8,56 (с, 2H), 7,68-7,90 (м, 4H), 7,16 (д, 1H), 7,02 (д, 1H), 6,85 (дд, 1H), 7,07 (т, 1H), 5,82 (дд, 1H), 3,85-4,06 (м, 2H), 3,89 (д, 2H), 3,71 (дд, 1H), 3,56 (дд, 1H), 3,42-3,49 (м, 2H), 3,14 (дд, 1H), 3,01 (шир.с, 3H), 2,86 (шир.с, 3H), 1,02-1,37 (м, 1H), 0,50-0,78 (м, 2H), 0,07-0,50 (м, 2H)
Соединения, перечисленные в таблице 21 (см. в конце описания), получали в соответствии с аналогичными методиками, как описано на схеме 34, и реакцией соответствующего перечисленного аминокислотного предшественника совместно с коммерчески подходящиим реагентами, и, если это необходимо, с последующей соответствующей стадией очистки, как описано ниже. Для получения соединения 283 и соединения 284 стадию 1 проводили, используя воду в качестве растворителя.
Пример 29
Синтез 1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(2-морфолиноэтилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина (287)
Схема 35
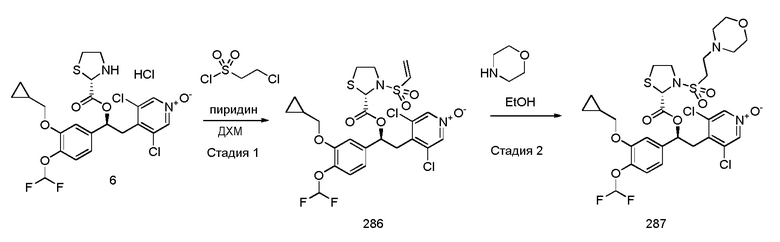
Стадия 1: 1-оксид 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(винилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина (286)
К раствору гидрохлорида 1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-тиазолидин-2-карбонилокси)этил)пиридина (6) (500 мг, 0,874 ммоль) в ДХМ (10 мл), охлажденного до 0°C, добавляли пиридин (212 мкл, 2,62 ммоль) и хлорид 2-хлорэтансульфонила (137 мкл, 1,312 ммоль), смесь оставляли нагреваться до комнатной температуры и перемешивали в течение 2 часов. Добавляли дополнительную порцию пиридина (707 мкл, 8,74 ммоль) и хлорид 2-хлорэтансульфонила (137 мкл, 1,312 ммоль) при 0°C, смесь оставляли реагировать в течение 6 часов при комнатной температуре. Смесь разбавляли ДХМ и промывали 1 н. HCl и насыщенным солевым раствором; органический слой сушили над Na2SO4, и растворитель удаляли под вакуумом. Полученное сухое вещество очищали флэш-хроматографией на силикагеле (ДХМ/MeOH = 98/2) с выходом 1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(винилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина (360 мг, 0,576 ммоль, 66% выход); MS/ESI+ 624,9 [MH]+.
Стадия 2: 1-оксид 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(2-морфолиноэтилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина (287)
К раствору 1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(винилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина (110 мг, 0,176 ммоль) в EtOH (5 мл) добавляли морфолин (35,2 мкл, 0,352 ммоль), и смесь оставляли реагировать при комнатной температуре в течение 1 часа. Растворитель удаляли при пониженном давлении, и сырое вещество очищали с помощью препаративной ВЭЖХ (способ 3) с выходом 1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(винилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина в виде трифторацетатной соли; MS/ESI+ 712,14 [MH]+; tR=3,16 (способ 3); Соотношение диастереомеров >95:5 (1Н ЯМР); [αD] = -126,6 (c=0,23 ДХМ);
1Н ЯМР (B) (300 МГц, ДМСО-d6) δ м.д. 8,55 (с, 2H), 7,19 (д, 1H), 7,11 (д, 1H), 6,97 (дд, 1H), 7,08 (т, 1H), 6,03 (дд, 1H), 5,58 (с, 1H), 3,94-4,02 (м, 1H), 3,91 (д, 2H), 3,61-3,74 (м, 1H), 3,45 (дд, 1H), 3,31 (дд, 1H), 3,06-3,81 (м, 12H), 3,00-3,25 (м, 2H), 1,03-1,35 (м, 1H), 0,47-0,70 (м, 2H), 0,10-0,45 (м, 2H)
Соединения, перечисленные в таблице 22, получали в соответствии с аналогичными методиками, описанными на схеме 35, и реакцией соответствующего перечисленного предшественника совместно с коммерчески подходящими реагентами, с последующей соответствующей стадией очистки.
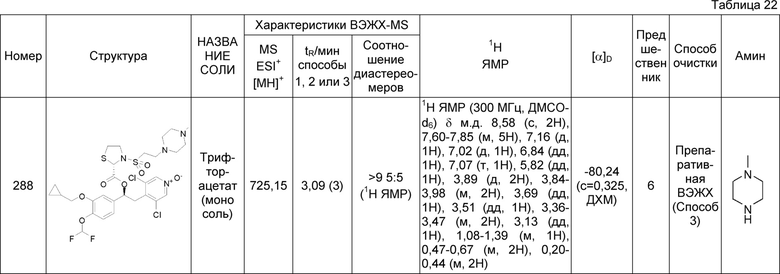
Пример 30:
1-оксид 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-1-(2-(фенилсульфонил)этил)пирролидин-2-карбонилокси)этил)пиридина (291)
Схема 36
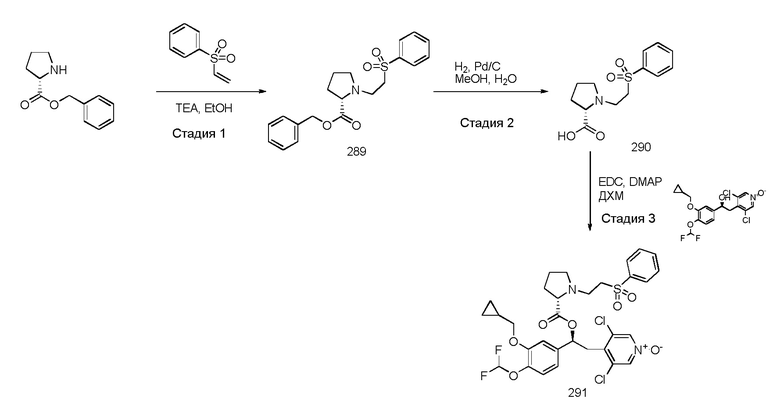
Стадия 1: (S)-бензил 1-(2-(фенилсульфонил)этил)пирролидин-2-карбоксилат (289)
Раствор гидрохлорида (S)-бензил пирролидин-2-карбоксилата (250 мг, 1,034 ммоль), винилсульфонилбензола (261 мг, 1,551 ммоль) и ТЭА (0,216 мл, 1,551 ммоль) в EtOH (15 мл) перемешивали при комнатной температуре в течение 24 часов. Затем реакционную смесь разбавляли ДХМ (50 мл) и дважды промывали насыщенным водным раствором NH4Cl. Органический слой сушили над Na2SO4 и растворитель удаляли под вакуумом. Остаток растворяли в ДХМ (15 мл) и EtOH (15 мл), и добавляли PS-трисамин (свободная -NH2 группа: 4,7 ммоль/г, 0,5 г, 0,35 ммоль). Суспензию перемешивали при комнатной температуре в течение 3 суток. Смолу отфильтровывали, и раствор упаривали досуха с получением целевого продукта (379 мг, 1,01 ммоль, 98% выход). MS/ESI+ 374,10 [MH]+
Стадия 2: (S)-1-(2-(фенилсульфонил)этил)пирролидин-2-карбоновая кислота (290)
Суспензию 10% Pd/C (10,80 мг, 0,101 ммоль) в воде (2 мл) добавляли к раствору (S)-бензил 1-(2-(фенилсульфонил)этил)пирролидин-2-карбоксилата (379 мг, 1,015 ммоль) в MeOH (15 мл). Смесь гидрогенизировали в аппарате Парра под давлением 30 фунт/кв.дюйм в течение 1 часа при комнатной температуре. Катализатор отфильтровывали, и полученный в результате прозрачный раствор упаривали досуха с получением целевого продукта (272 мг, 0,860 ммоль, 95% выход). MS/ESI+ 284,02 [MH]+.
Стадия 3: 1-оксид 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-1-(2-(фенилсульфонил)этил)пирролидин-2-карбонилокси)этил)пиридина (291)
Раствор 1-оксида (S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-гидроксиэтил)пиридина (150 мг, 0,357 ммоль), (S)-1-(2-(фенилсульфонил)этил)пирролидин-2-карбоновой кислоты (152 мг, 0,535 ммоль), EDC (103 мг, 0,535 ммоль) и DMAP (21,80 мг, 0,178 ммоль) в ДХМ (30 мл) перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь дважды промывали 1 н. HCl, сушили над Na2SO4 и упаривали досуха. Остаток очищали с помощью препаративной ВЭЖХ (способ 2), и собранные фракции упаривали досуха, еще раз растворяли ДХМ и элюировали через PL-HCO3 картридж (200 мг, 0,36 моль). Элюированный раствор упаривали досуха, с получением указанного в заголовке соединения (110 мг, 0,160 ммоль, 45% выход); [αD] = -33,8 (c=0,44, ДХМ); MS/ESI+ 685,42 [MH]+; tR=3,15 мин (способ 3); Соотношение диастереомеров >95:5 (1Н ЯМР);
1Н ЯМР (B) (300 МГц, ДМСО-d6) δ м.д. 8,55 (с, 2H), 7,83-7,98 (м, 2H), 7,69-7,82 (м, 1H), 7,53-7,69 (м, 2H), 7,17 (д, 1H), 7,06 (д, 1H), 6,93 (дд, 1H), 7,06 (т, 1H), 5,92 (дд, 1H), 3,80-4,04 (м, 2H), 3,43-3,58 (м, 1H), 3,41 (дд, 1H), 3,23 (дд, 1H), 2,61-3,09 (м, 5H), 2,25-2,44 (м, 1H), 1,84-2,08 (м, 1H), 1,37-1,78 (м, 3H), 1,09-1,33 (м, 1H), 0,48-0,73 (м, 2H), 0,25-0,43 (м, 2H)
Пример 31:
1-оксид 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-1-(2-фенилацетил)пирролидин-2-карбонилокси)этил)пиридина (292)
Схема 37
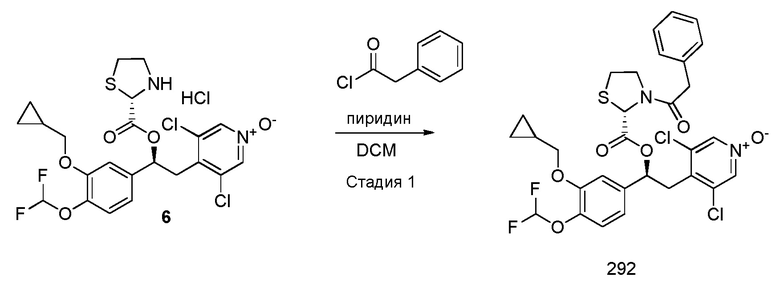
К раствору гидрохлорида 1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-пирролидин-2-карбонилокси)этил)пиридина (6) (200 мг, 0,361 ммоль) и пиридина (292 мкл, 3,61 ммоль) в сухом ДХМ (5 мл), по каплям добавляли раствор хлорида фенил-ацетила (84 мг, 0,542 ммоль) в сухом ДХМ (1 мл) при 0°C, смесь оставляли нагреваться до комнатной температуры, перемешивая в течение 2 часов. Добавляли EtOAc (20 мл) и смесь промывали 5% водным раствором лимонной кислоты и насыщенным солевым раствором; органическую фазу сушили над Na2SO4, фильтровали и упаривали. Остаток очищали при помощи флэш-хроматографии на силикагеле (ДХМ/MeOH = 95/5). Дальнейшая очистка с помощью препаративной ВЭЖХ (способ 2) требовалась для получения 1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-1-(2-фенилацетил)пирролидин-2-карбонилокси)этил)пиридина (35 мг, 0,055 ммоль, 15,25% выход); MS/ESI+ 635,23 [MH]+; tR=3,91 мин (способ 3); Соотношение диастереомеров >95:5 (1Н ЯМР); [αD] = -28,26 (c=0,23, MeOH);
1Н ЯМР (B) (300 МГц, ДМСО-d6 353K) δ м.д. 8,40 (с, 2H), 7,16-7,39 (м, 5H), 7,13 (д, 1H), 7,08 (д, 1H), 6,89-6,95 (м, 1H), 6,97 (т, 1H), 5,85-6,08 (м, 1H), 4,26-4,45 (м, 1H), 3,91 (д, 2H), 3,66 (шир.с, 2H), 3,47-3,63 (м, 3H), 3,27 (дд, 1H), 2,01-2,25 (м, 1H), 1,83-1,99 (м, 1H), 1,49-1,83 (м, 2H), 1,07-1,26 (м, 1H), 0,44-0,69 (м, 2H), 0,19-0,40 (м, 2H)
Пример 32:
1-оксид 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(3-(диметилкарбамоил)бензилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина (295)
Схема 38
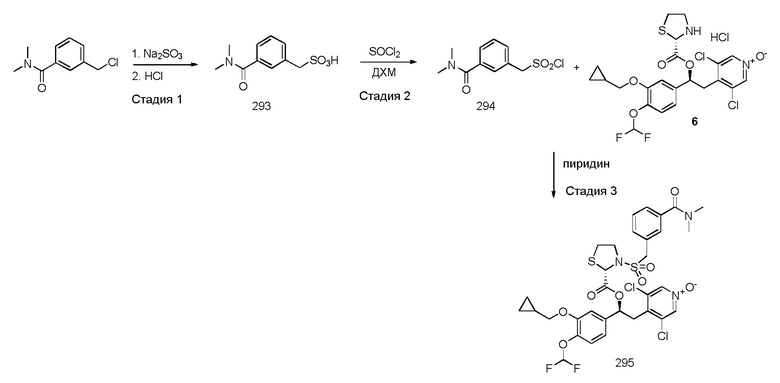
Стадия 1: (3-(диметилкарбамоил)фенил)метансульфоновая кислота (293)
К суспензии 3-(хлорметил)-N,N-диметилбензамида (1,15 г, 5,82 ммоль) в воде (30 мл) добавляли сульфит натрия (1,100 г, 8,73 ммоль), и смесь нагревали при 100°C в течение 1 часа. Растворитель удаляли под вакуумом, и остаток суспендировали в MeOH (40 мл). Добавляли HCl 4 M в диоксане (5 мл), и нерастворимые неорганические соли отфильтровывали. Фильтрат упаривали досуха и остаток очищали с помощью нескольких растираний с CH3CN с получением (3-(диметилкарбамоил)фенил)метансульфоновой кислоты (1,04 г, 4,27 ммоль, 73,5% выход). MS/ESI+ 243,95 [MH]+.
Стадия 2: хлорид (3-(диметилкарбамоил)фенил)метансульфонила (294)
К суспензии (3-(диметилкарбамоил)фенил)метансульфоновой кислоты (500 мг, 2,055 ммоль) в ДХМ (40 мл) добавляли хлорид тионила (0,900 мл, 12,33 ммоль) и полученную смесь перемешивали при комнатной температуре в течение 20 часов. Реакционную смесь выливали в дробленый лед, и органическую фазу отделяли и сушили над Na2SO4. Растворитель удаляли под вакуумом с получением целевого продукта (357 мг, 1,364 ммоль, 66% выход). MS/ESI+ 261,96 [MH]+
Стадия 3: 1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(3-(диметилкарбамоил)бензилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина (295)
1-Оксид 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(3-(диметилкарбамоил)бензилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина получали в соответствии с аналогичной методикой, описанной на схеме 16 (пример 8). Его очищали путем обработки поглотителем на основе подложки из полимерного изоцианата, с последующей препаративной ВЭЖХ (способ 2) (20% выход); MS/ESI+ 760,17 [MH]+, tR=3,78 мин (способ 3); Соотношение диастереомеров >95:5 (1Н ЯМР); [αD] = -15,8 (c=3,0, ДХМ);
1Н ЯМР (B) (300 МГц, ДМСО-d6) δ м.д. 8,53 (с, 2H), 7,38-7,55 (м, 4H), 7,17 (д, 1H), 7,08 (д, 1H), 6,94 (дд, 1H), 7,07 (т, 1H), 6,00 (дд, 1H), 5,27 (с, 1H), 4,67 (д, 1H), 4,61 (д, 1H), 3,85-3,98 (м, 2H), 3,74-3,85 (м, 1H), 3,51-3,66 (м, 1H), 3,36-3,50 (м, 2H), 3,20-3,26 (м, 1H), 3,04-3,14 (м, 1H), 2,99 (шир.с, 3H), 2,92 (шир.с, 3H), 1,04-1,42 (м, 1H), 0,46-0,66 (м, 2H), 0,20-0,41 (м, 2H)
Соединение, указанное в таблице 23, получали в соответствии с аналогичными методиками, для схемы 38, и реакцией соответствующего предшественника, полученного в виде свободного основания после основной обработки гидрохлоридной солью с насыщенным водным раствором NaHCO3, а затем экстракцией ДХМ. Стадия очистки описана ниже.
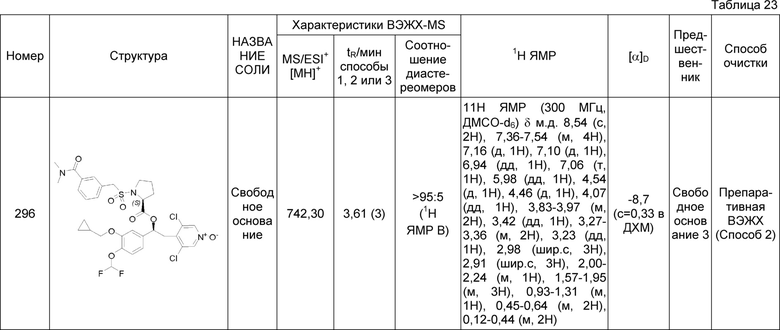
Пример 33:
Синтез 1-оксида 3,5-дихлор-4-((S)-2-(4-(дифторметокси)-3-метоксифенил)-2-((S)-3-(3-(диметилкарбамоил)фенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина (299)
Схема 39
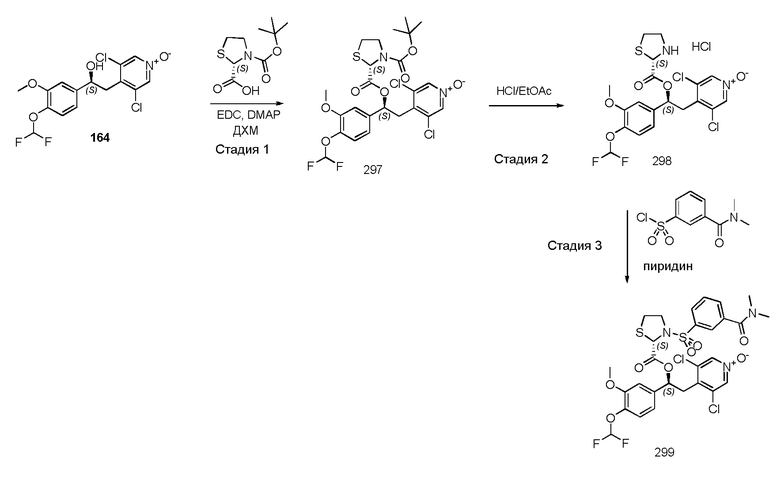
Стадия 1: 1-оксид 4-((S)-2-((S)-3-(трет-бутоксикарбонил)тиазолидин-2-карбонилокси)-2-(4-(дифторметокси)-3-метоксифенил)этил)-3,5-дихлорпиридина (297)
Раствор (S)-3-(трет-бутоксикарбонил)тиазолидин-2-карбоновой кислоты (479 мг, 2,052 ммоль), 1-оксид (S)-3,5-дихлор-4-(2-(4-(дифторметокси)-3-метоксифенил)-2-гидроксиэтил)пиридина (164) (полученного в соответствии с аналогичной методикой, описанной на схеме 23), (650 мг, 1,710 ммоль), EDC (492 мг, 2,56 ммоль) и DMAP (313 мг, 2,56 ммоль) в ДХМ (60 мл) перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь разбавляли ДХМ и дважды промывали водной 1 н. HCl; органический слой сушили над Na2SO4 и упаривали досуха с получением целевого соединения (количественный выход). MS/ESI+ 595,24 [MH]+.
Стадия 2: гидрохлорид 1-оксида 3,5-дихлор-4-((S)-2-(4-(дифторметокси)-3-метоксифенил)-2-((S)-тиазолидин-2-карбонилокси)этил)пиридина (298)
К раствору 1-оксида 4-((S)-2-((S)-3-(трет-бутоксикарбонил)тиазолидин-2-карбонилокси)-2-(4-(дифторметокси)-3-метоксифенил)этил)-3,5-дихлорпиридина (1,710 ммоль) в EtOAc (10 мл), охлажденному до 0°C, добавляли HCl, 4 M раствор в EtOAc (10 мл, 40,0 ммоль), и полученную смесь перемешивали при комнатной температуре в течение 2 часов. Дополнительно добавляли HCl, 4 M раствора в EtOAc (10 мл, 40,0 ммоль), и раствор перемешивали при 0°C еще 2 часа до достижения полного преобразования. Раствор концентрировали до 10 мл при пониженном давлении (температура бани: 10°C; парциальное давление: 8 фунт/кв.дюйм), затем добавляли iPr2O (20 мл), и продукт осаждали в виде липкого клейкого твердого вещества. Затем твердое вещество оставляли стабилизироваться, растворитель удаляли отсасыванием. Твердое вещество сушили под вакуумом при комнатной температуре с получением гидрохлорида 1-оксида 3,5-дихлор-4-((S)-2-(4-(дифторметокси)-3-метоксифенил)-2-((S)-тиазолидин-2-карбонилокси)этил)пиридина (0,890 г, 1,674 ммоль, 97% выход), который использовали на следующей стадии без дополнительной очистки. MS/ESI+ 494,97 [MH].
Стадия 3: 1-оксид 3,5-дихлор-4-((S)-2-(4-(дифторметокси)-3-метоксифенил)-2-((S)-3-(3-(диметилкарбамоил)фенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина (299)
К раствору гидрохлорида 1-оксида 3,5-дихлор-4-((S)-2-(4-(дифторметокси)-3-метоксифенил)-2-((S)-тиазолидин-2-карбонилокси)этил)пиридина (350 мг, 0,658 ммоль) в пиридине (6 мл), охлажденному до 0°C, по каплям добавляли раствор хлорида 3-(диметилкарбамоил)бензол-1-сульфонила (245 мг, 0,987 ммоль) в ДХМ (3 мл), и реакционную смесь перемешивали при 0°C в течение 1 часа. Смесь разбавляли ДХМ (30 мл) и дважды промывали водной 1 н. HCl; органический слой сушили над Na2SO4 и растворитель удаляли под вакуумом. Остаток очищали с помощью препаративной ВЭЖХ (способ 2) с последующей флэш-хроматографией на силикагеле (ДХМ/MeOH = 97/3) с получением 1-оксида 3,5-дихлор-4-((S)-2-(4-(дифторметокси)-3-метоксифенил)-2-((S)-3-(3-(диметилкарбамоил)фенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина (147 мг, 0,208 ммоль, 31,6% выход); MS/ESI+ 705,97 [MH]+, tR=3,28 мин (способ 3); Соотношение диастереомеров = 95:5 (1Н ЯМР); [αD] = -43,1 (c=0,57, ДХМ);
1Н ЯМР (300 МГц, ДМСО-d6) δ м.д. 8,57 (с, 2H), 7,94 (дт, 1H), 7,90 (т, 1H), 7,77 (дт, 1H), 7,71 (т, 1H), 7,18 (д, 1H), 7,15 (д, 1H), 6,97 (дд, 1H), 7,07 (т, 1H), 6,04 (дд, 1H), 5,54 (с, 1H), 3,84 (с, 3H), 3,75-3,94 (м, 1H), 3,66 (дт, 1H), 3,48 (дд, 1H), 3,33 (дд, 1H), 3,02 (шир.с, 3H), 2,98 (дд, 1H), 2,90 (шир.с, 3H), 2,66 (дт, 1H)
Соединения, перечисленные в таблице 24 (см. в конце описания), получали в соответствии с аналогичными методиками, описанными для схемы 39, и реакцией соответствующего спирта из списка коммерчески подходящих реагентов с последующей соответствующей стадией очистки, как описано ниже. Соединение 303 получали в виде второго элюированного диастереоизомера из смеси диастереоизомеров.
Пример 34
Синтез 1-оксида 3,5-дихлор-4-((S)-2-(4-(дифторметокси)-3-метоксифенил)-2-((S)-1-(фенилсульфонил)пирролидин-2-карбонилокси)этил)пиридина (Соединение 305)
Схема 40
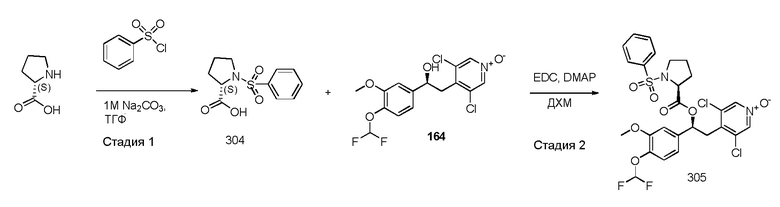
Стадия 1: (S)-1-(фенилсульфонил)пирролидин-2-карбоновая кислота (304)
Бензолсульфонилхлорид (4,03 мл, 31,3 ммоль) добавляли к охлажденной суспензии (0°C) (S)-пирролидин-2-карбоновой кислоты (3 г, 26,1 ммоль) в ТГФ (50 мл) и водном 1 M Na2CO3 (60 мл, 60,0 ммоль), реакционную смесь перемешивали при 0°C в течение 1 часа. Смесь дважды экстрагировали Et2O, и органические слои удаляли. Водную фазу осторожно подкисляли, добавляя твердый KHSO4 до pH=3, и дважды экстрагировали EtOAc. Объединенные органические слои сушили над Na2SO4 и упаривали досуха с получением (S)-1-(фенилсульфонил)пирролидин-2-карбоновой кислоты (6,1 г, 23,89 ммоль, 92% выход). MS/ESI+ 256,10 [MH]+
Стадия 2: 1-оксид 3,5-дихлор-4-((S)-2-(4-(дифторметокси)-3-метоксифенил)-2-((S)-1-(фенилсульфонил)пирролидин-2-карбонилокси)этил)пиридина (305)
Смесь 1-оксида (S)-3,5-дихлор-4-(2-(4-(дифторметокси)-3-метоксифенил)-2-гидроксиэтил)пиридина (164) (55 мг, 0,145 ммоль), (S)-1-(фенилсульфонил)пирролидин-2-карбоновой кислоты (111 мг, 0,434 ммоль), EDC (83 мг, 0,434 ммоль) и DMAP (53,0 мг, 0,434 ммоль) в ДХМ (20 мл) перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь дважды промывали водной 1 н. HCl, а затем водным 1 M K2CO3; органический слой сушили над Na2SO4, и растворитель удаляли при пониженном давлении. Остаток очищали с помощью препаративной ВЭЖХ (Способ 2) с получением 1-оксида 3,5-дихлор-4-((S)-2-(4-(дифторметокси)-3-метоксифенил)-2-((S)-1-(фенилсульфонил)пирролидин-2-карбонилокси)этил)пиридина (45 мг, 0,073 ммоль, 50,4% выход); MS/ESI+ 617,15 [MH]+, tR=3,71 мин (способ 3); Соотношение диастереомеров >95:5 (1Н ЯМР); [αD] = -60,3 (c=0,39, ДХМ);
1Н ЯМР (B) (300 МГц, ДМСО-d6) δ м.д. 8,61 (с, 2H), 7,75-7,85 (м, 2H), 7,69-7,76 (м, 1H), 7,56-7,69 (м, 2H), 7,19 (д, 1H), 7,18 (д, 1H), 7,00 (дд, 1H), 7,07 (т, 1H), 6,05 (дд, 1H), 4,16 (дд, 1H), 3,86 (с, 3H), 3,49 (дд, 1H), 3,39-3,44 (м, 1H), 3,29 (дд, 1H), 3,09-3,23 (м, 1H), 1,83-2,10 (м, 1H), 1,59-1,82 (м, 2H), 1,44-1,59 (м, 1H)
Соединения, перечисленные в таблице 25 (см в конце описания), получали в соответствии с аналогичными методиками, как описано на схеме 40, используя коммерчески подходящие реагенты и реакцией соответствующих перечисленных спиртов, с последующей соответствующей стадией очистки, как описано ниже. Соединение 315 получали в виде второго элюированного диастереоизомера из смеси диастереоизомеров.
Пример 35:
1-оксид 3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-метоксифенил)-2-((S)-3-(3-(диметилкарбамоил)фенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина (320)
Схема 41
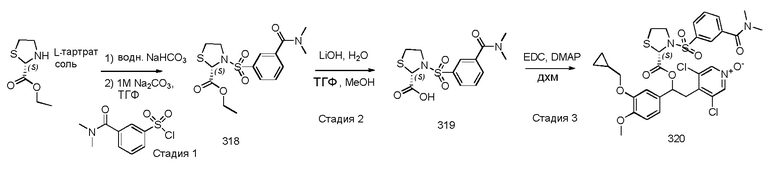
Стадия 1: (S)-этил 3-(3-(диметилкарбамоил)фенилсульфонил)тиазолидин-2-карбоксилат (318)
(S)-этил тиазолидин-2-карбоксилат (2R,3R)-2,3-дигидроксисукцинат (получен, как описано в Bull. Korean Chem. Soc. 2010, 31, 2709), (4 г, 12,85 ммоль) выливали в делительную воронку, содержащую водный раствор NaHCO3 (50 мл, 55,0 ммоль) и Et2O (100,0 мл), предварительно охлажденном до 0°C на ледяной бане. Смесь взбалтывали до растворения твердого вещества; фазы разделяли, и водный слой вновь экстрагировали Et2O (100 мл). Объединенные органические слои сушили над Na2SO4 и упаривали досуха без нагрева. Остаток растворяли в ТГФ (50 мл), раствор охлаждали до 0°C и добавляли водный раствор NaHCO3 (50 мл, 55,0 ммоль). Раствор хлорида 3-(диметилкарбамоил)бензол-1-сульфонила (3,18 г, 12,85 ммоль) в ТГФ (50 мл) добавляли к двухфазной смеси при 0°C при интенсивном перемешивание и реакционную смесь оставляли при комнатной температуре в течение 4 часов. Смесь разделяли между EtOAc и водой, и водный слой экстрагировали EtOAc. Объединенные органические слои промывали водной 1 н. HCl и насыщенным солевым раствором, сушили над Na2SO4 и упаривали досуха с получением (S)-этил 3-(3-(диметилкарбамоил)фенилсульфонил)тиазолидин-2-карбоксилата (3,79 г, 10,18 ммоль, 79% выход); MS/ESI+ 373,04 [MH]+
Стадия 2: (S)-3-(3-(диметилкарбамоил)фенилсульфонил)тиазолидин-2-карбоновая кислота (319)
(S)-этил 3-(3-(диметилкарбамоил)фенилсульфонил)тиазолидин-2-карбоксилат (3,79 г, 10,18 ммоль) растворяли в смеси MeOH (30 мл), ТГФ (30,0 мл) и воды (30,0 мл). Добавляли LiOH (0,487 г, 20,35 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 30 минут. Смесь подкисляли водным раствором 1 н. HCl (pH=1), разбавляли водой и дважды экстрагировали EtOAc. Объединенные органические слои сушили над Na2SO4 и упаривали досуха. Остаток растирали со смесью Et2O (15 мл) и петролейным эфиром (25 мл) с выходом после фильтрации (S)-3-(3-(диметилкарбамоил)фенилсульфонил)тиазолидин-2-карбоновой кислоты (1,86 г, 5,40 ммоль, 53,1% выход); MS/ESI+ 344,94 [MH]+; [αD] = -36,1 (c=1,67, MeOH).
Для определения энантиомерной чистоты, это промежуточное соединение объединяли со спиртом 1 (EDC, DMAP, ДХМ) с получением соединения 52: Соотношение диастереомеров = 95:5.
Стадия 3: 1-оксид 3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-метоксифенил)-2-((S)-3-(3-(диметилкарбамоил)фенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина (320)
К раствору 1-оксида 3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-метоксифенил)-2-гидроксиэтил)пиридина (156) (160 мг, 0,416 ммоль), EDC (160 мг, 0,833 ммоль) и DMAP (102 мг, 0,833 ммоль) в ДХМ добавляли (50 мл), (S)-3-(3-(диметилкарбамоил)фенилсульфонил)тиазолидин-2-карбоновую кислоту (172 мг, 0,500 ммоль), полученную смесь перемешивали при комнатной температуре в течение 24 часов. Реакционную смесь дважды промывали водной 1 н. HCl и органический слой сушили над Na2SO4. Растворитель удаляли под вакуумом и остаток очищали с помощью препаративной ВЭЖХ (способ 2) с получением 1-оксида 3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-метоксифенил)-2-((S)-3-(3-(диметилкарбамоил)фенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина (122 мг, 0,172 ммоль, 41% выход); MS/ESI+ 710,25 [MH]+, tR=3,25 мин (Способ 3); Соотношение диастереомеров 1:1 (1Н ЯМР).
Соединения, перечисленные в таблице 26 (см. в конце описания), получали в соответствии со схемой 41, используя коммерчески подходящие реагенты и реакцией с соответствующими перечисленными спиртами, с последующей соответствующей стадией очистки, как описано ниже. Энантиомерная чистота промежуточного соединения была получена с использованием хлорида бензолсульфонила, определенного в описанной выше стадии 2 (эталонное соединение: 89; Соотношение диастереомеров = 92:8).
ФАРМАКОЛОГИЧЕСКАЯ АКТИВНОСТЬ СОЕДИНЕНИЙ ПО ИЗОБРЕТЕНИЮ
ПРИМЕР 15
In vitro определение ингибирующей PDE4 активности в бесклеточном анализе
Активность PDE4 определяли в супернатантах клеточных лизатов моноцитов клеток U937. Клетки культивировали, и собирали фракцию супернатанта, полученного по существу так, как описано в Torphy TJ et al J. Pharmacol. Exp. Ther. 1992; 263:1195-1205.
Клетки U937 (Cell Bank, Interlab Cell Line Collection, ICLC HTL94002) выращивали при 37°C, 5% CO2 в RPMI 1640 со средой GlutaMAX™-I с добавлением 10% эмбриональной телячьей сыворотки и 100 мкг/мл Pen-strep (Gibco).
Клетки собирали и дважды промывали центрифугированием (150×g, 8 мин) в холодном PBS. Промытые клетки ресуспендировали в холодном буфере Кребс-Рингера-Хенселейта при конечной концентрации 20×106 клеток/мл и обрабатывали ультразвуком. После центрифугирования при 15000×g в течение 20 мин супернатанты объединяли, разделяли на аликвоты и хранили при -80°C.
Активность PDE4 определяли в супернатанах клеток путем анализа исчезновения цАМФ из инкубирующих смесей.
Концентрирование тестируемых соединений находилось в области от 10-12 M до 10-6 M. Реакции останавливали ферментативной инактивацией при нагревании (2,5 минут при 100°C) и остаточный компонент цАМФ определяли, используя 'анализ цАМФ LANCE' из PerkinElmer следуя приведенным инструкциям.
Результаты тестируемых соединений, характерных для изобретения, выражали как среднее ± стандартное отклонение нМ-концентрации тестируемого соединения, что давало 50% ингибирование исчезания цАМФ (IC50), как показано в нижеприведенной таблице:

В таблице выше активность связывания PDE4 (значения IC50) указана следующим образом: >10 нМ '+'; 10-1 нМ '++'; 1-0,1 нМ '+++'; <0,1 нМ '++++'.
Процент ингибирования PDE4-активности рассчитывали, обозначая исчезновение цАМФ в отсутствие ингибиторов как 100%, а исчезновение цАМФ в инактивированных нагреванием образцах как 0%.
Аналогично, результаты для тестированных соединений формулы (II) (выражали как среднее ± стандартное отклонение нМ-концентрации тестируемого соединения, которое давало 50% ингибирование исчезновения цАМФ) (IC50), как показано в следующей таблице:
В таблице выше активность связывания PDE4 (значения IC50) указана следующим образом: >10 нМ '+'; 10-1 нМ '++'; 1-0,1 нМ '+++'; <0,1 нМ '++++'.
ПРИМЕР 16
In vitro определение ингибирующей активности PDE4 в анализе мононуклеарных клеток периферической крови (PBMC)
Анализ, который основан на известной ингибирующей активности, проявляемой ингибиторами PDE4 в отношении липопролисахарид (LPS)-индуцируемого фактора некроза опухоли-альфа (TNF-α высвобождаются в мононуклеарные клетки периферической крови (PBMC)), проводили в соответствии с предварительно описанным способом (Hatzelmann A et al J. Pharmacol. Exp. Ther. 2001; 297:267-279; Draheim R et al J. Pharmacol. Exp. Ther. 2004; 308:555-563.
Криоконсервированные PBMC человека (100 мкл/лунка) инкубировали в 96-луночных планшетах (105 клеток/лунка) в течение 30 мин в присутствии или в отсутствие (50 микрол.) тестируемых соединений, концентрация которых изменялась от 10-12 M до 10-6 M или от 10-13 M до 10-7 M. Затем добавляли LPS (3 нг/мл).
Через 18 часов инкубирования при 37°C в инкубаторе с влажностью в атмосфере 95% воздуха и 5% CO2, среду для культивирования собирали и TNF-α измеряли с помощью ELISA.
Результаты тестируемых соединений, характерных для изобретения, выраженные как ±95% доверительные интервалы молярного концентрирования тестируемого соединения, которое давало 50% ингибирование LPS-индуцированного высвобождения TNF-α (IC50), показаны в следующей таблице:
В таблице выше активность связывания PDE4 (значения IC50) указана следующим образом: >10 нМ '+'; 10-1 нМ '++'; 1-0,1 нМ '+++'; <0,1 нМ '++++'.
Эффекты тестируемых соединений рассчитывали как процент ингибирования TNF-α высвобождения, обозначая LPS-индуцируемое высвобождение TNF-α в отсутствие ингибиторного соединения как 100%, а основную TNF-α продукцию PBMC в отсутствии LPS как 0%.
Аналогично, результаты тестируемых соединений для соединений формулы (II), выраженные как ±95% доверительные интервалы молярного концентрирования тестируемого соединения, которое давало 50% ингибирование LPS-индуцированного высвобождения TNF-α (IC50), показаны в следующей таблице:
В таблице выше активность связывания PDE4 (значения IC50) указана следующим образом: >10 нМ '+'; 10-1 нМ '++'; 1-0,1 нМ '+++'; <0,1 нМ '++++'.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ 1-ФЕНИЛ-2-ПИРИДИНИЛ-АЛКИЛОВЫХ СПИРТОВ В КАЧЕСТВЕ ИНГИБИТОРОВ ФОСФОДИЭСТЕРАЗЫ | 2013 |
|
RU2637945C2 |
| Производные 1-фенил-2-пиридинилалкиловых спиртов в качестве ингибиторов фосфодиэстеразы | 2013 |
|
RU2655170C2 |
| ПРОИЗВОДНЫЕ 1-ФЕНИЛ 2-ПИРИДИНИЛАЛКИЛОВЫХ СПИРТОВ В КАЧЕСТВЕ ИНГИБИТОРОВ ФОСФОДИЭСТЕРАЗЫ | 2012 |
|
RU2617401C2 |
| КОНДЕНСИРОВАННЫЕ ТРИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ИМИДАЗОЛА В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2700004C1 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛОПИРИДИНА В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ | 2015 |
|
RU2733400C2 |
| ПРОИЗВОДНЫЕ ИМИДАЗОПИРИДИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2695664C1 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ БЕНЗОДИОКСОЛА ИЛИ БЕНЗОДИОКСЕПИНА В КАЧЕСТВЕ ИНГИБИТОРОВ ФОСФОДИЭСТЕРАЗ | 2011 |
|
RU2583787C2 |
| МОДУЛЯТОРЫ ЯДЕРНЫХ РЕЦЕПТОРОВ | 2016 |
|
RU2715897C2 |
| КОНДЕНСИРОВАННЫЕ ТРИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛОВ В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2689777C1 |
| НОВЫЕ 1,2-БИС-СУЛЬФОНАМИДНЫЕ ПРОИЗВОДНЫЕ КАК МОДУЛЯТОРЫ ХЕМОКИНОВОГО РЕЦЕПТОРА | 2011 |
|
RU2654213C9 |









































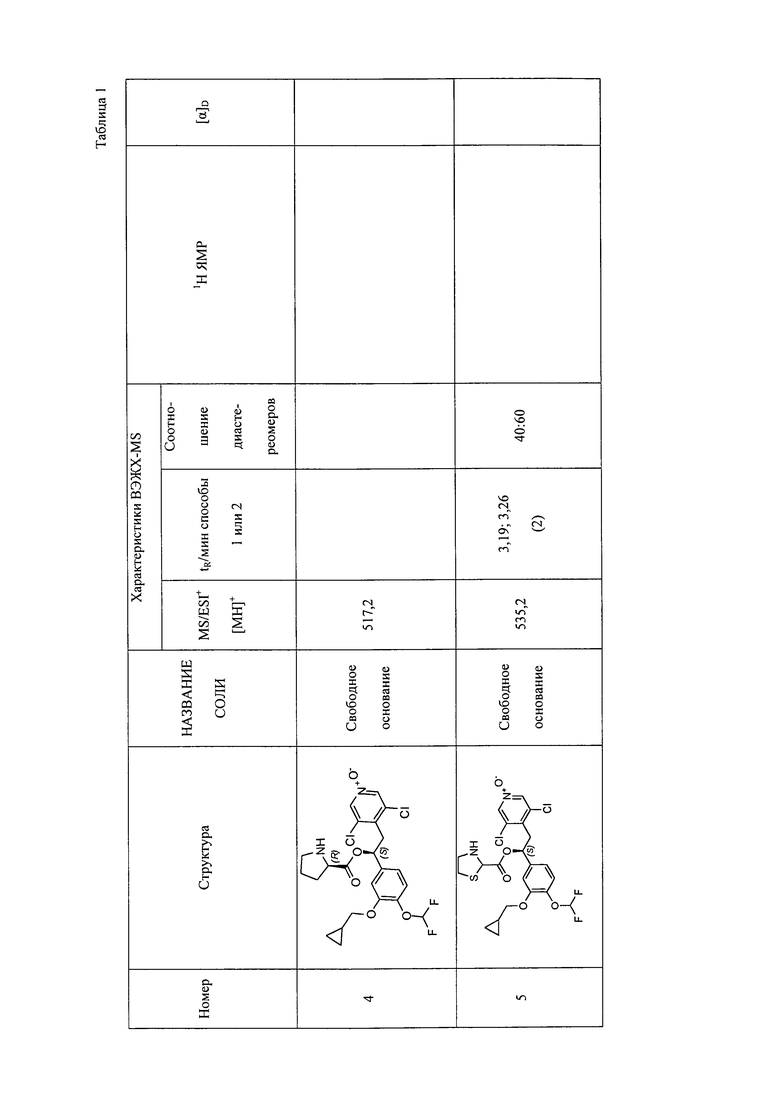
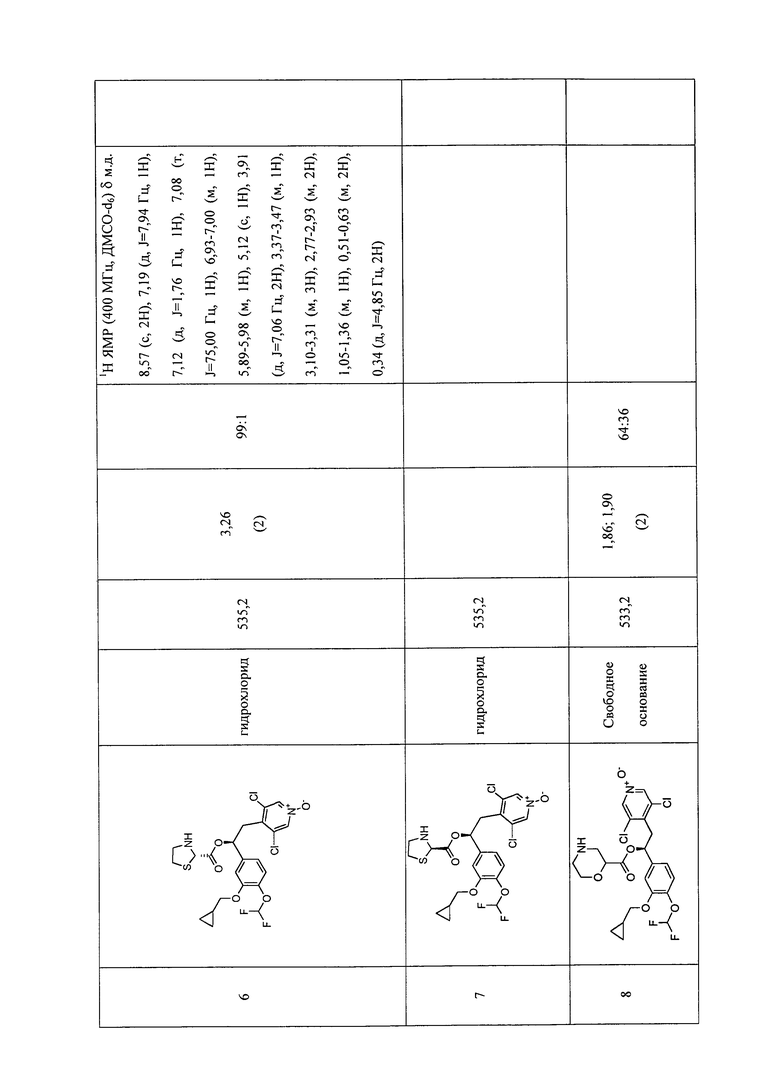
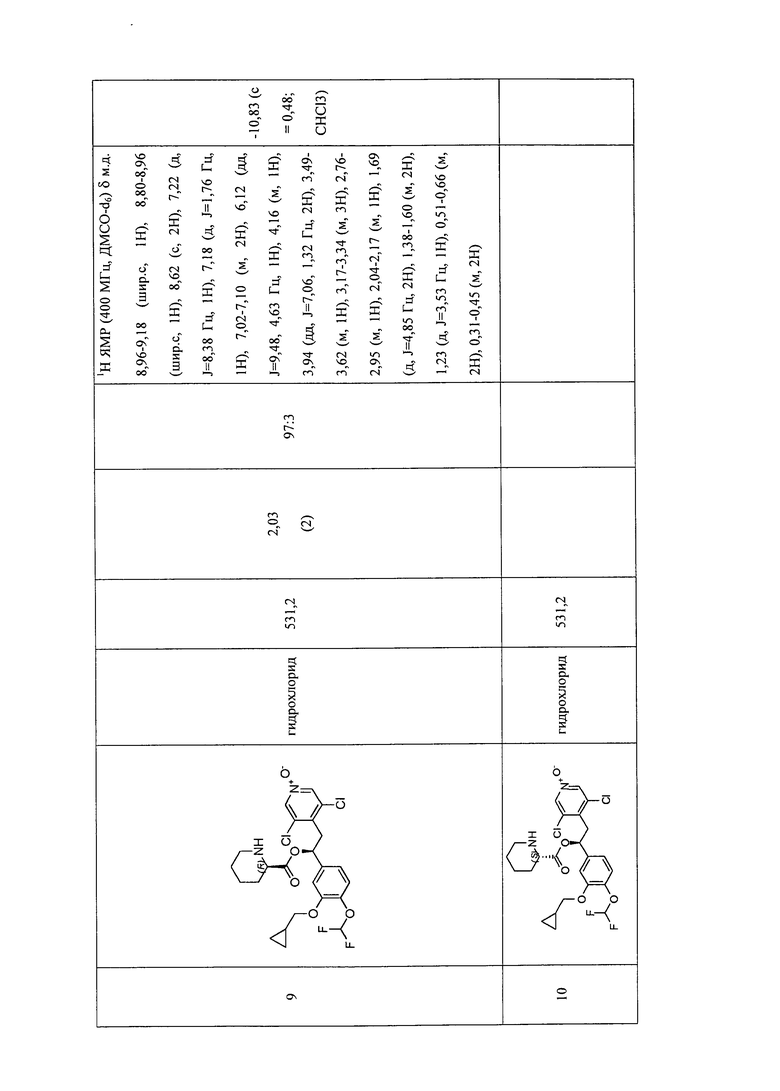
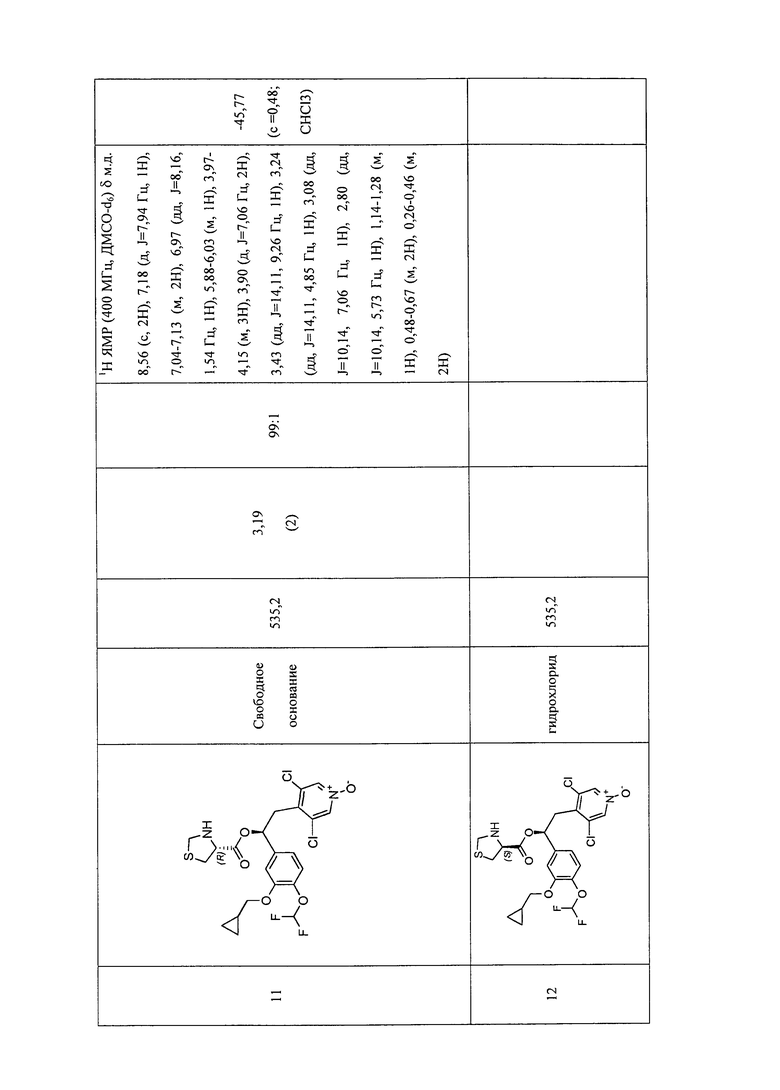
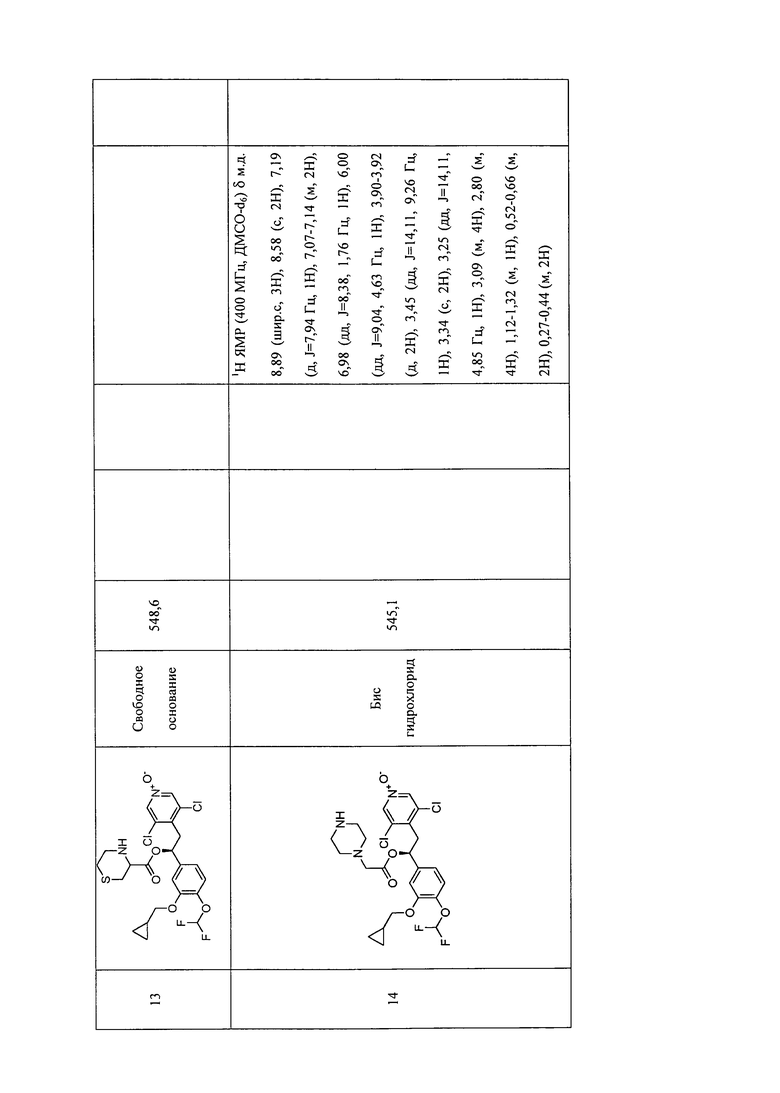
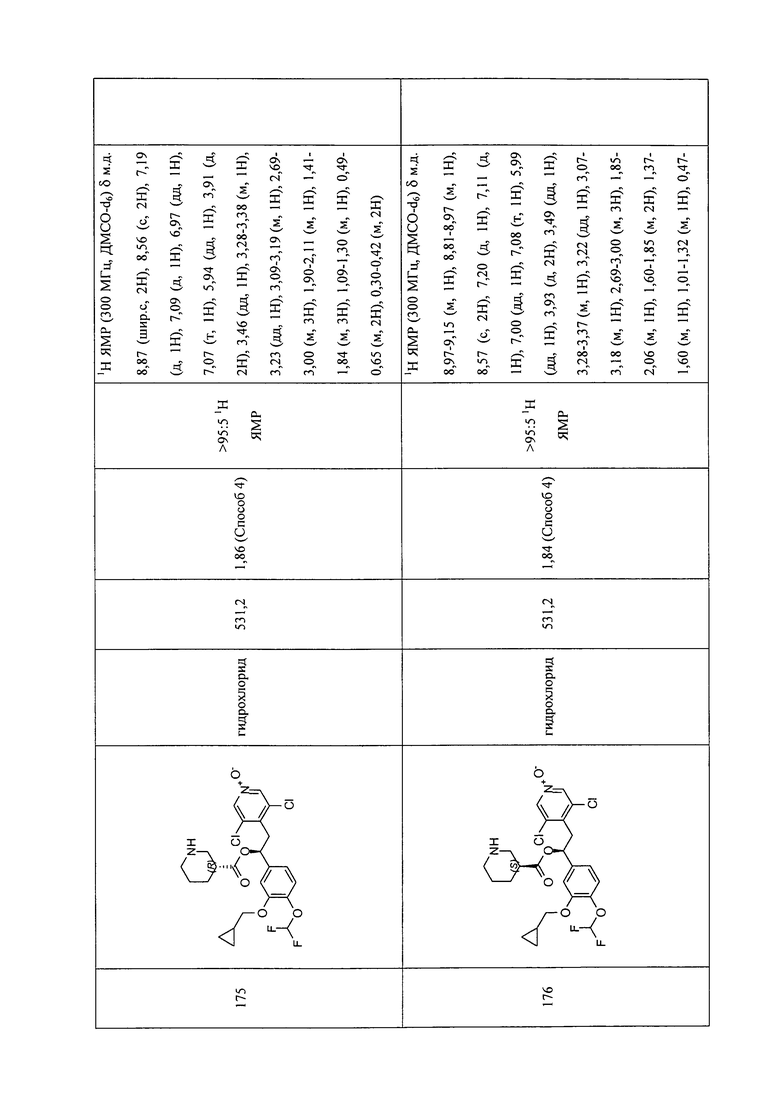
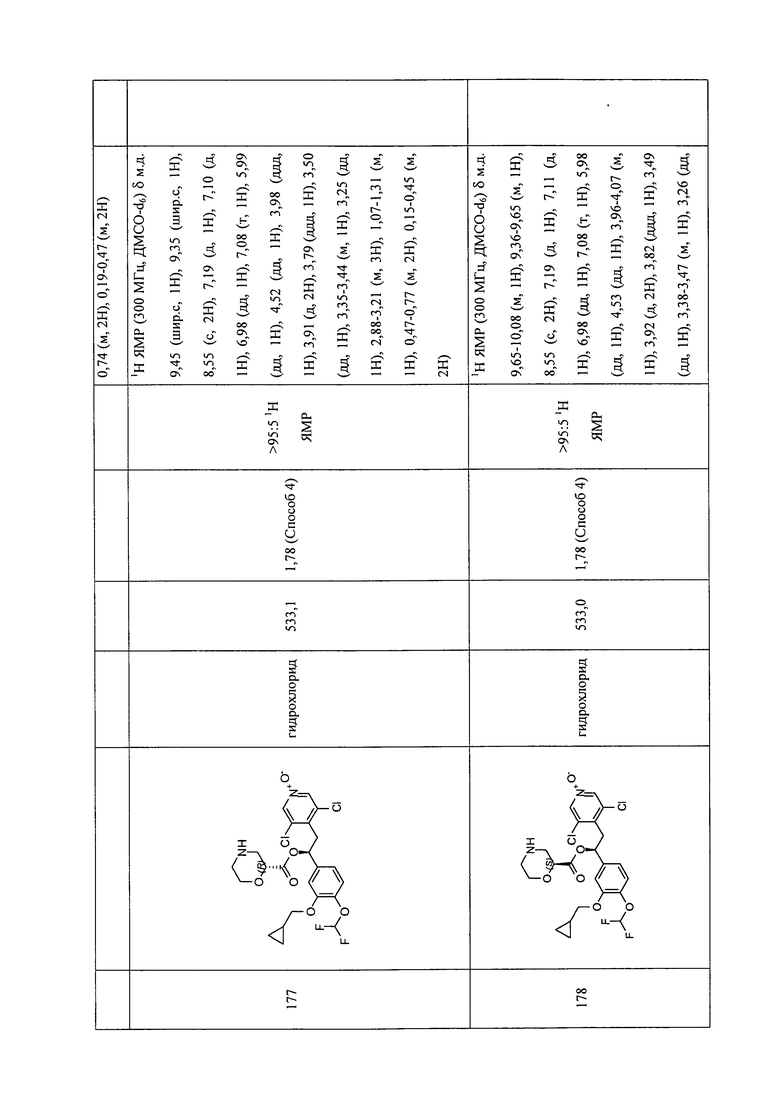
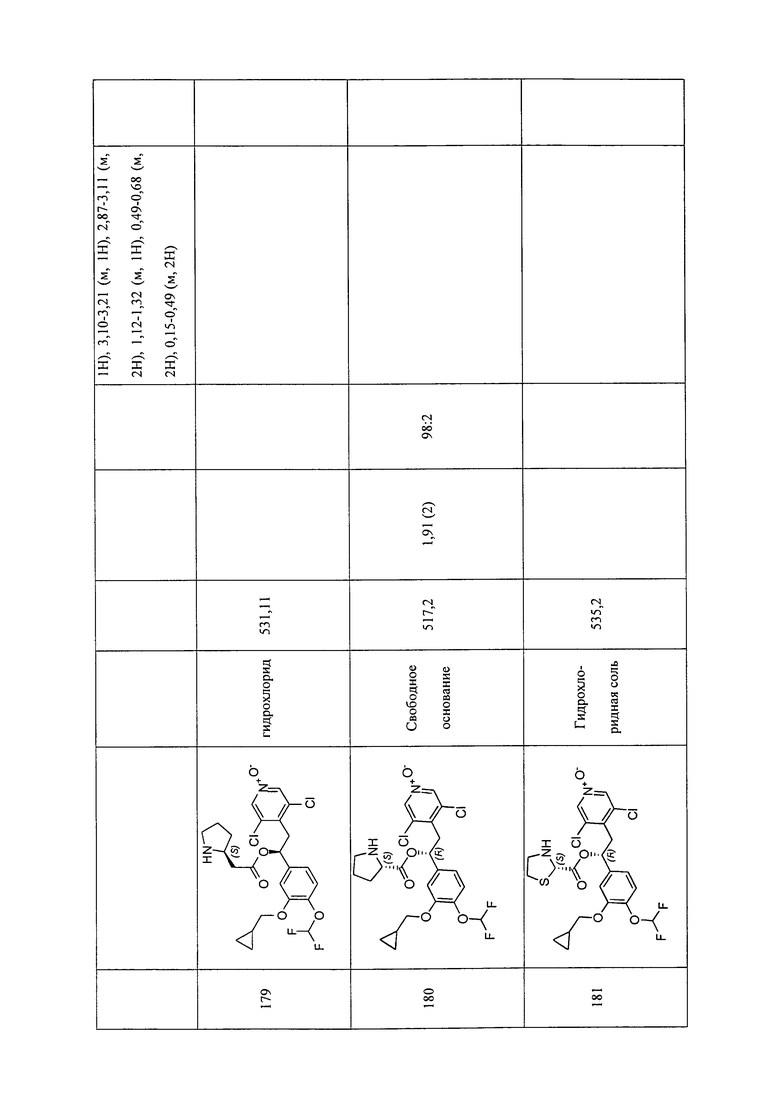
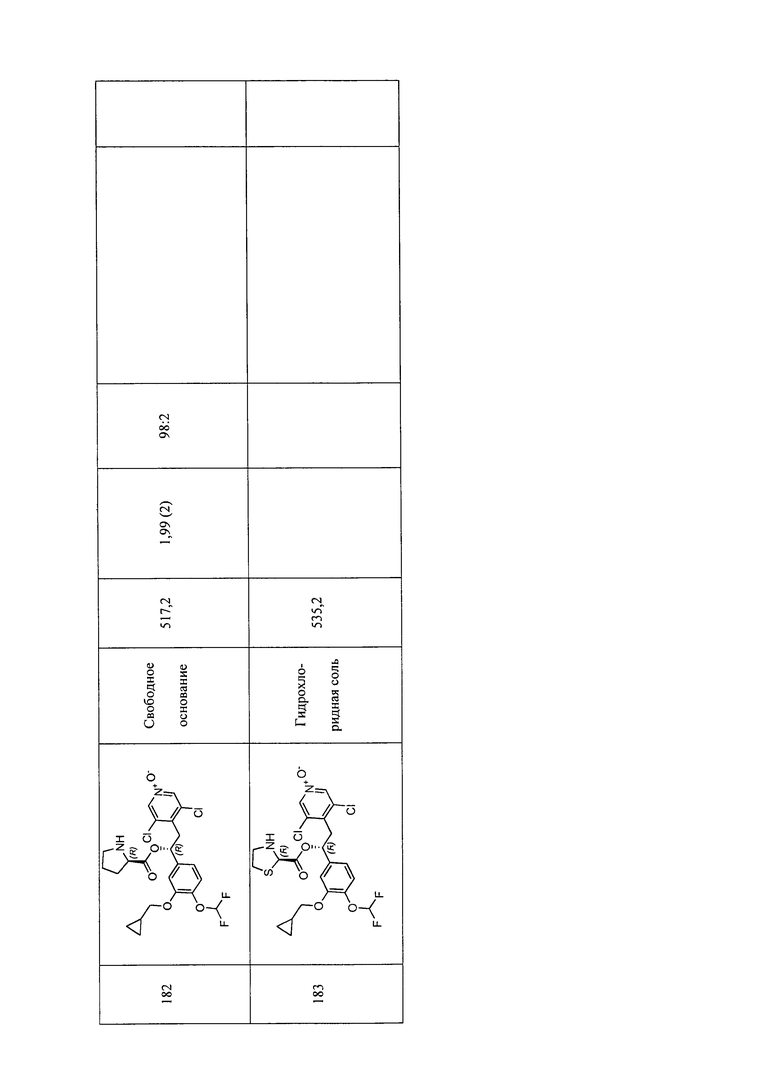
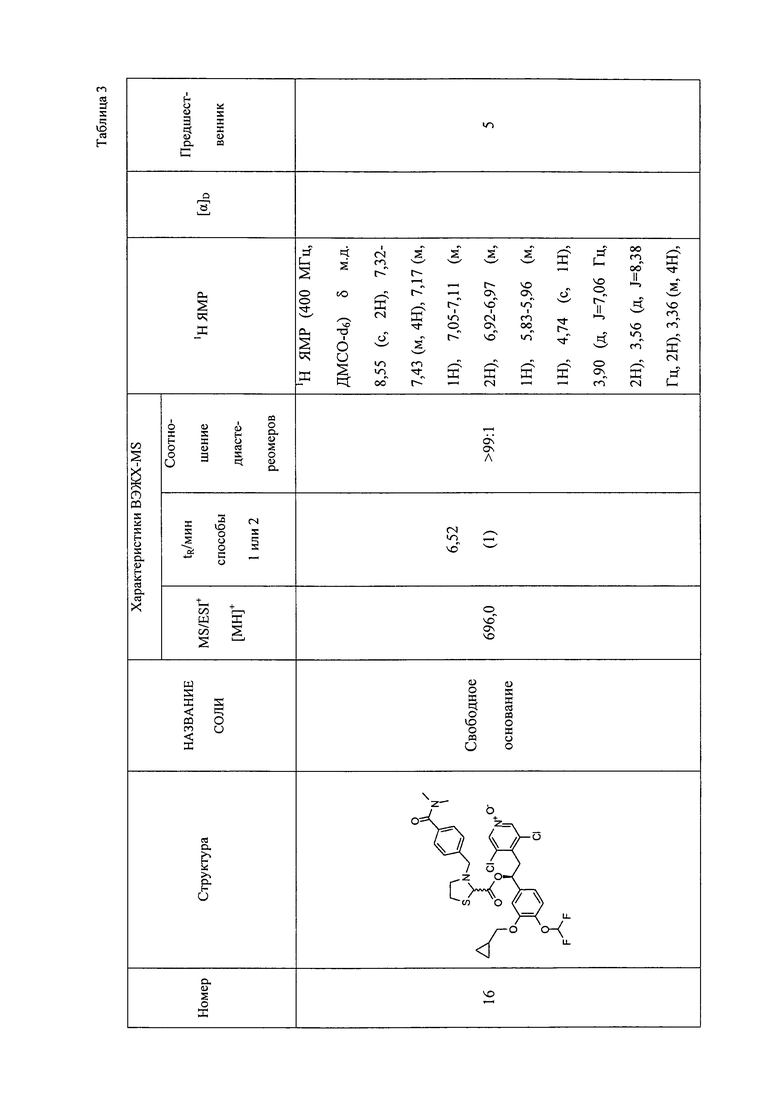
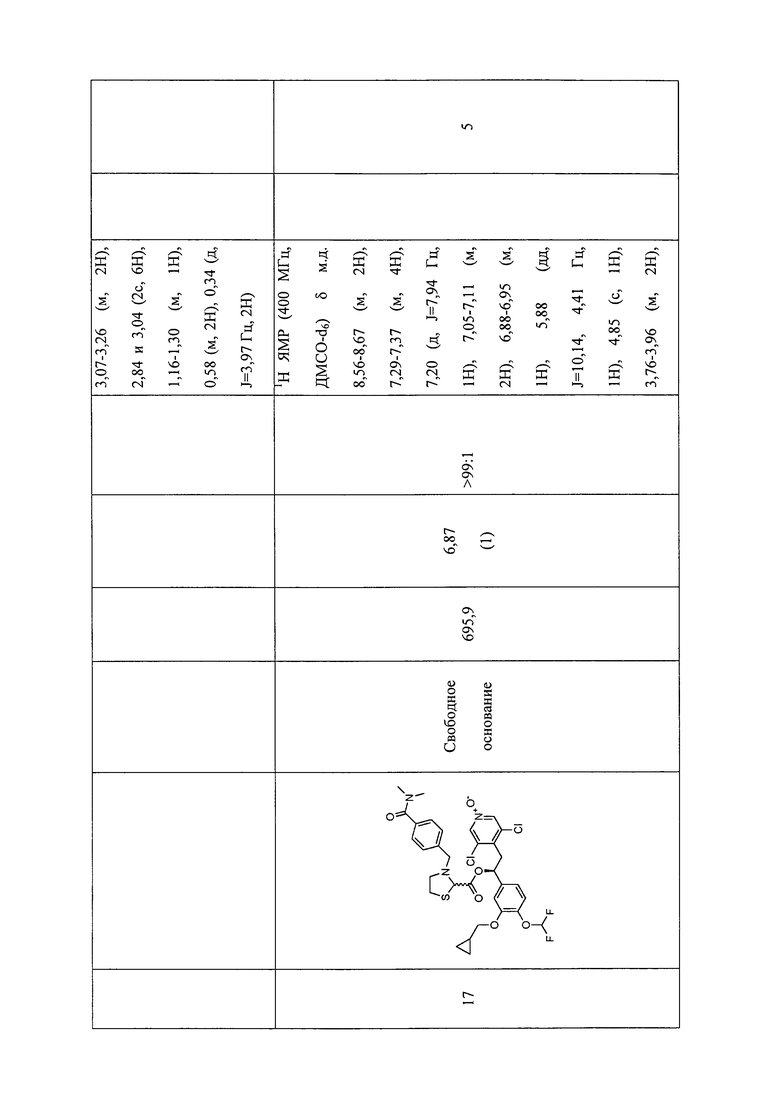

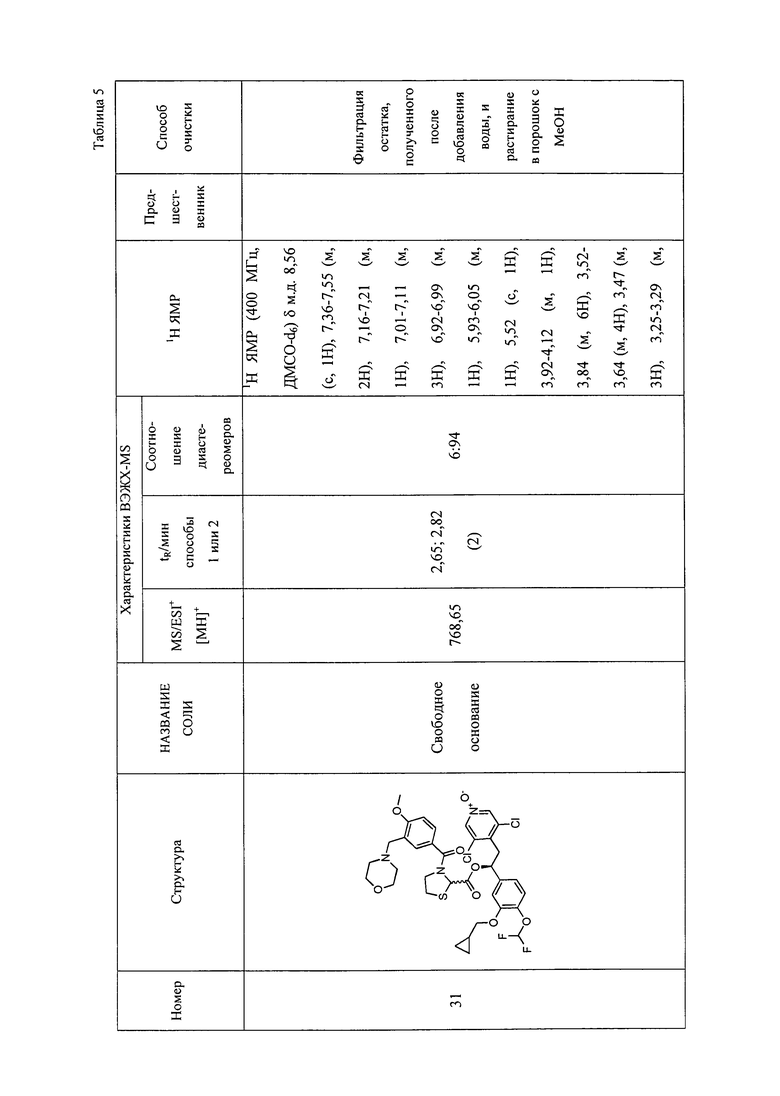
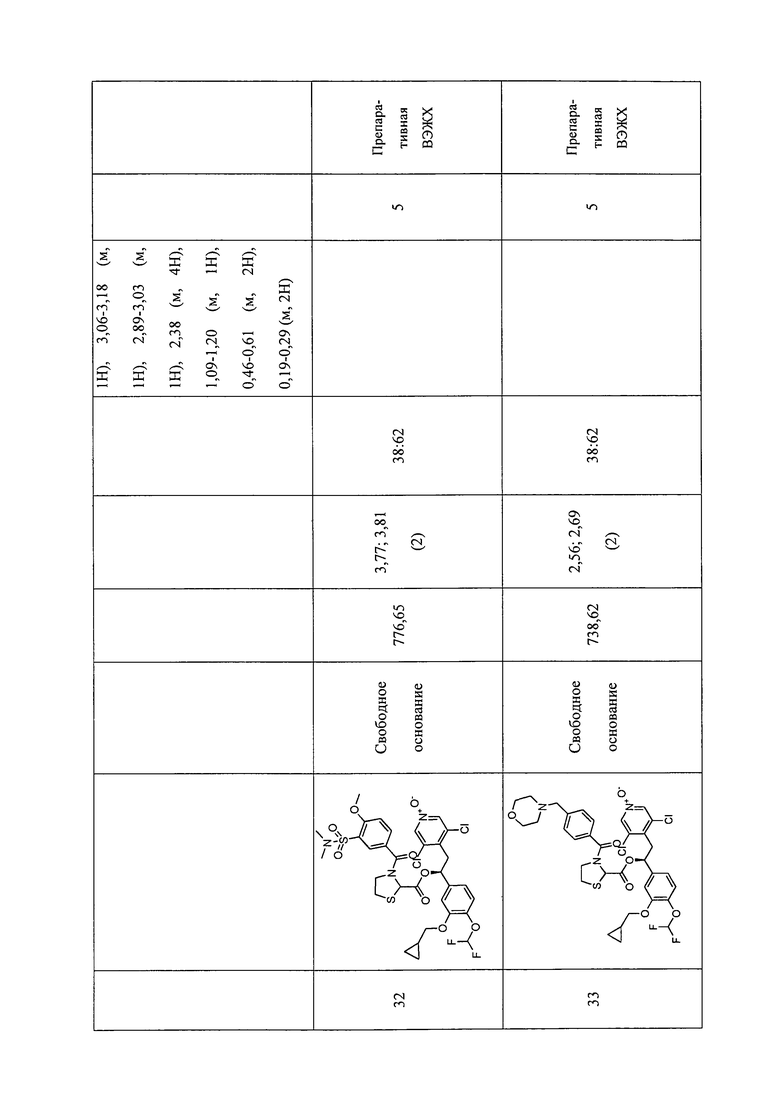
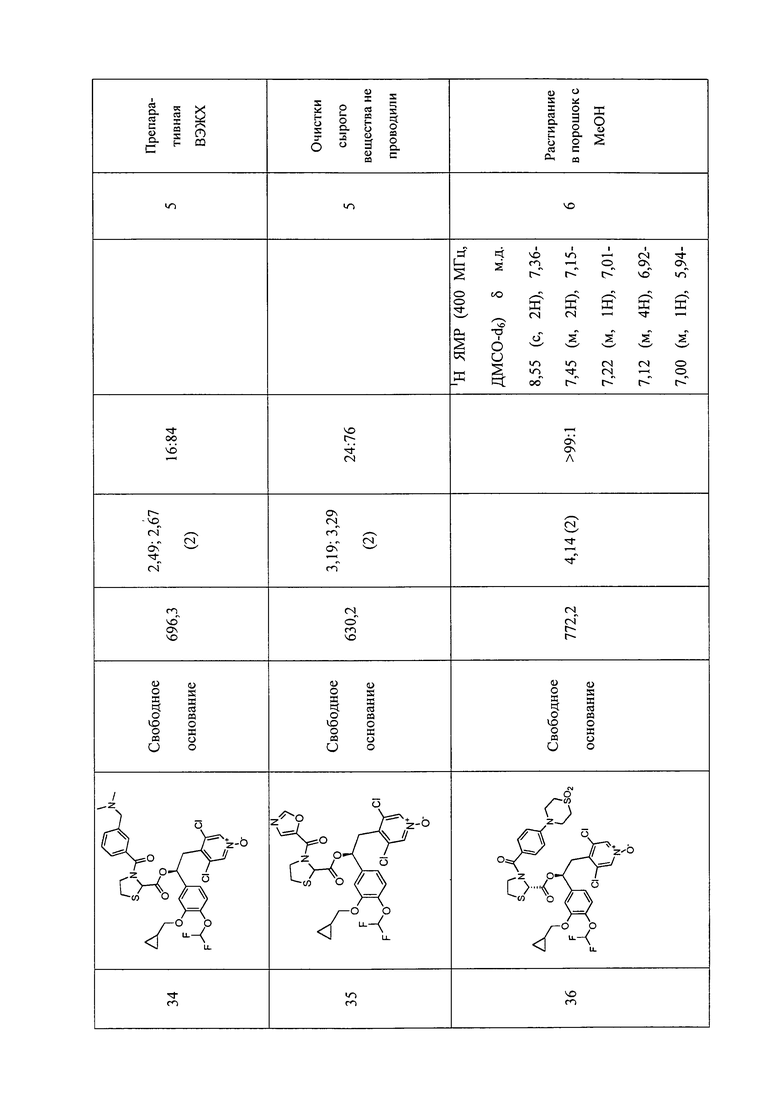
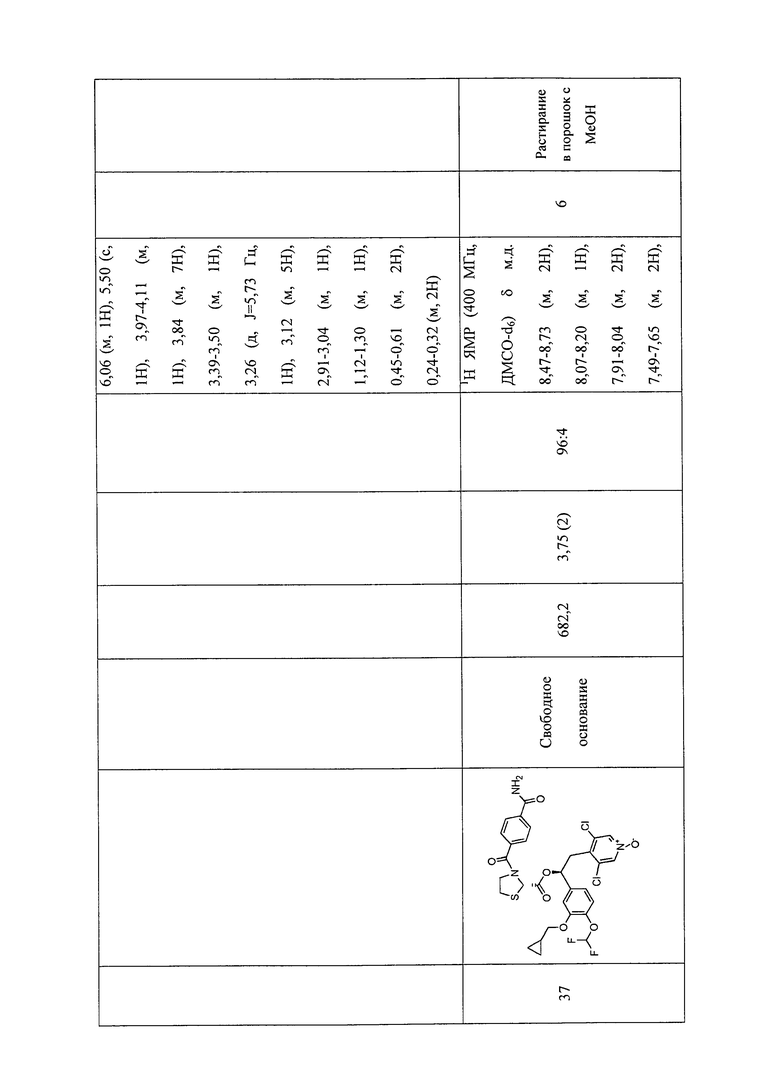

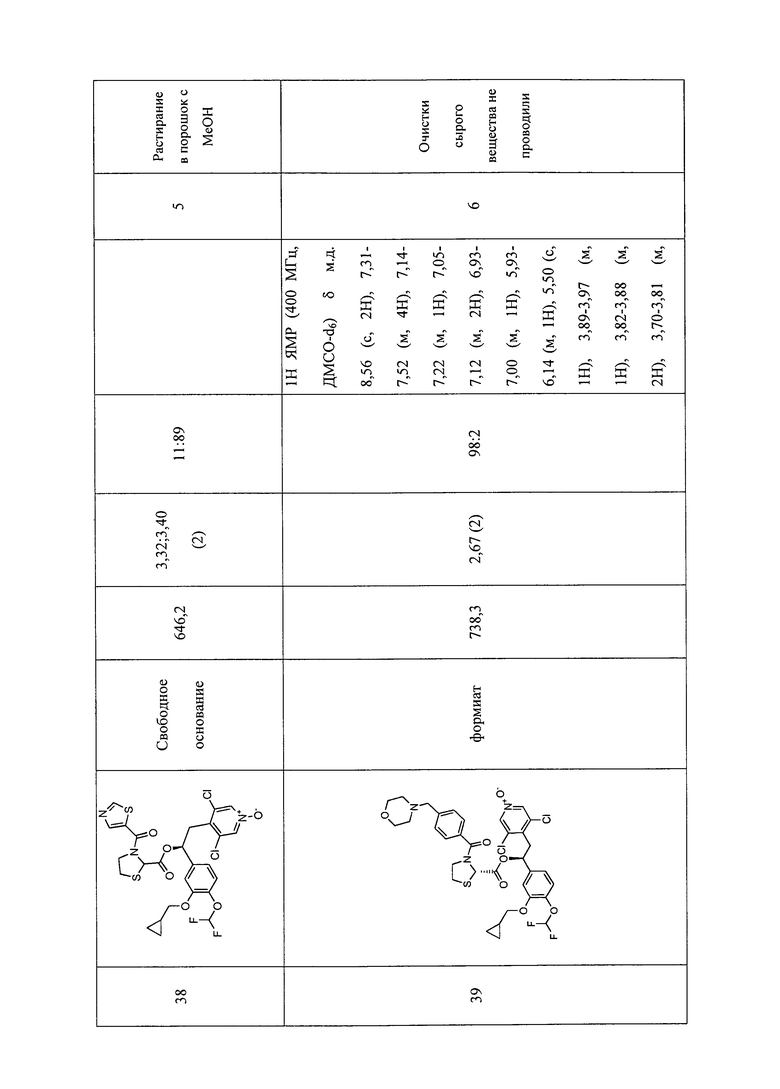
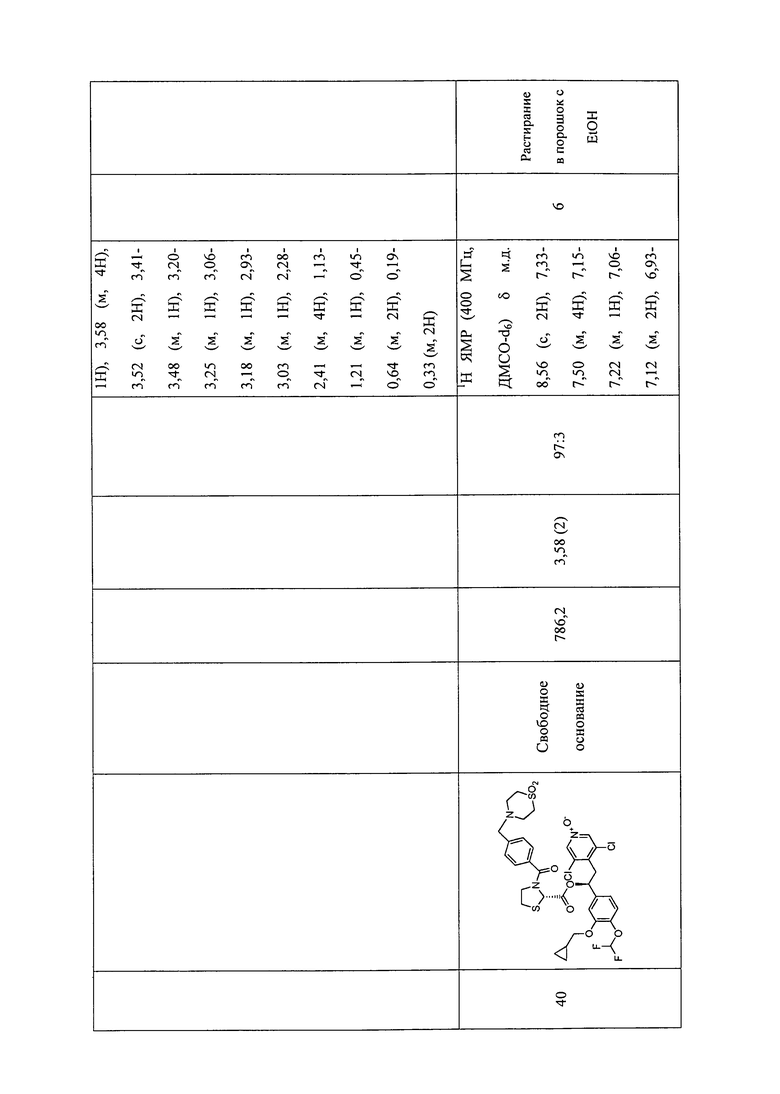
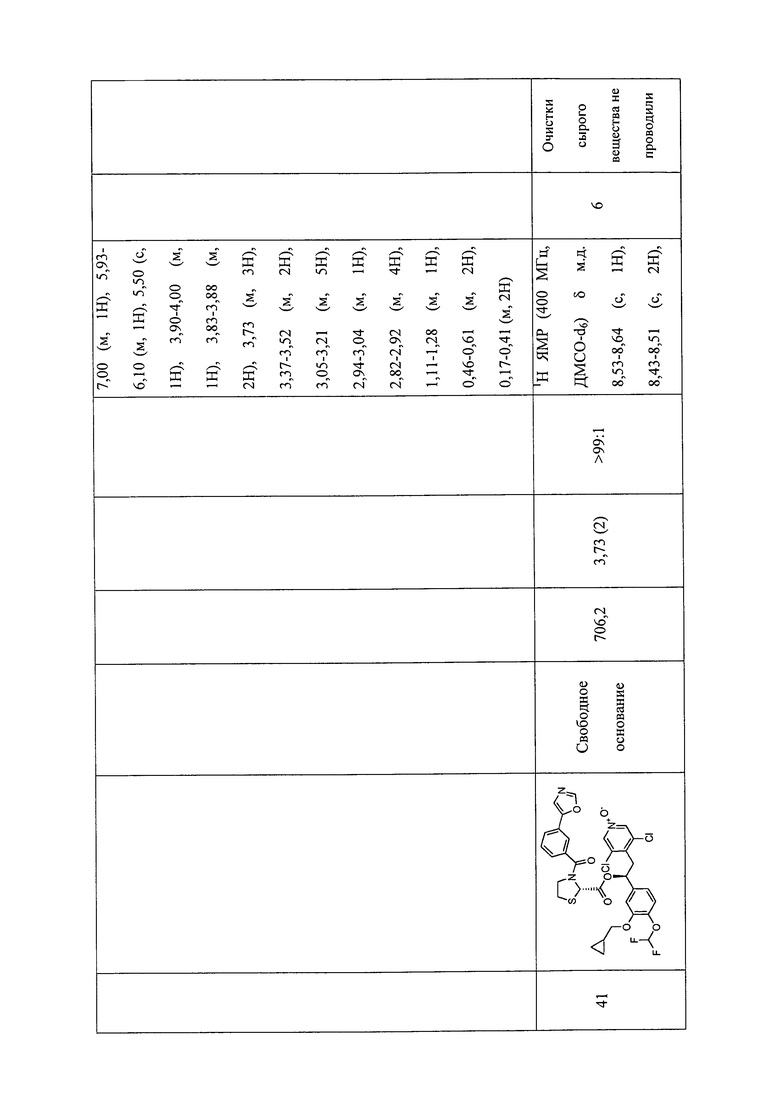

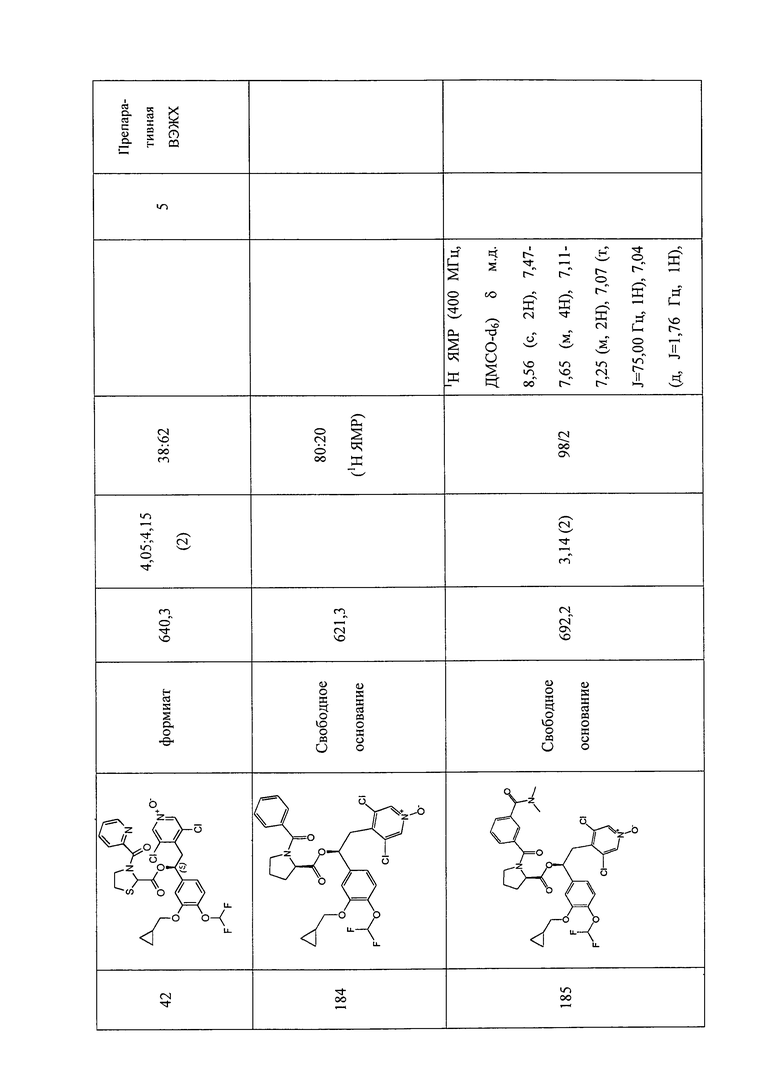
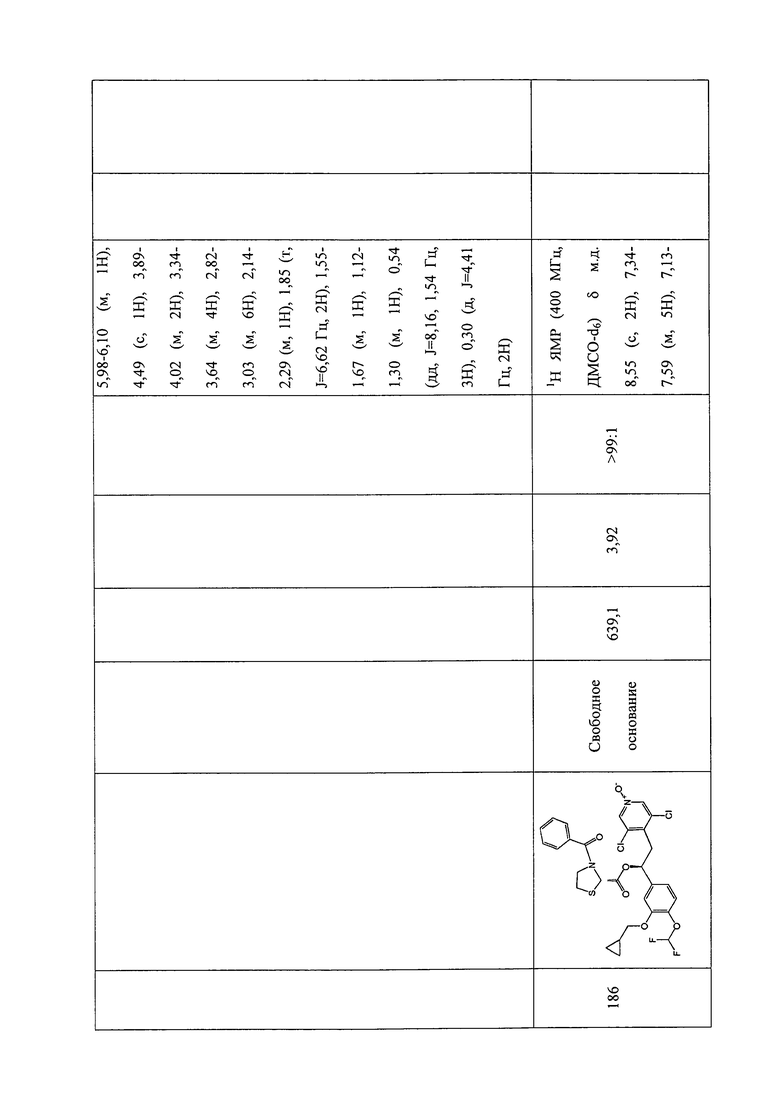

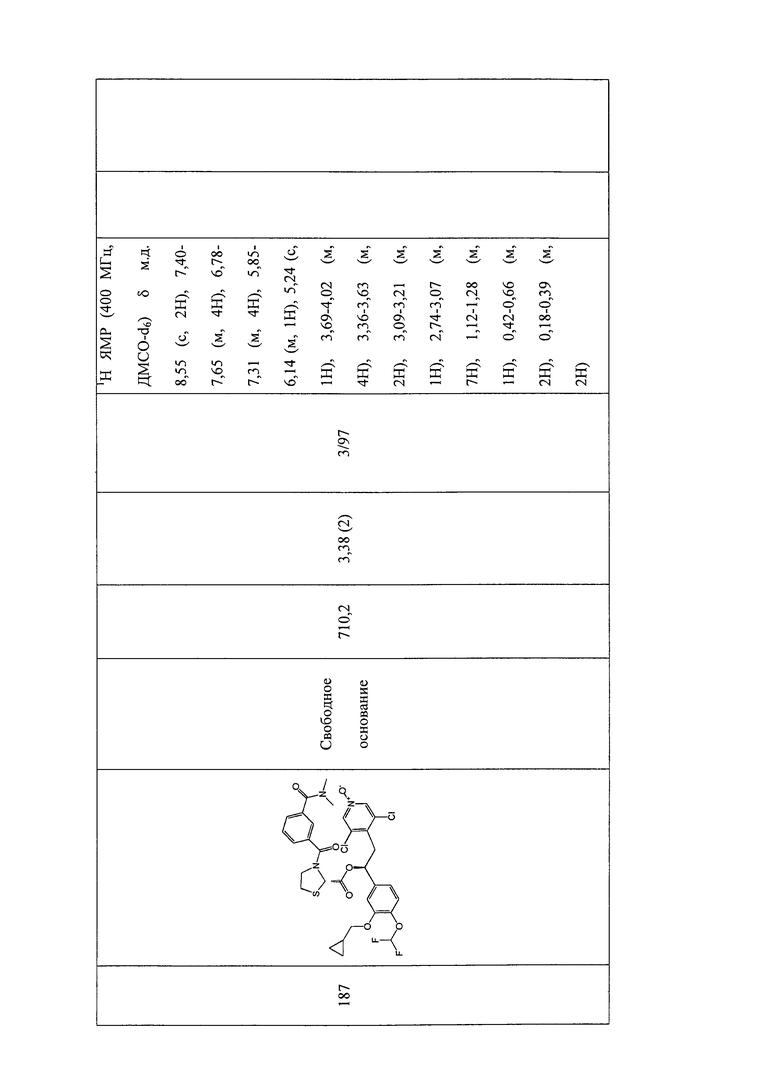
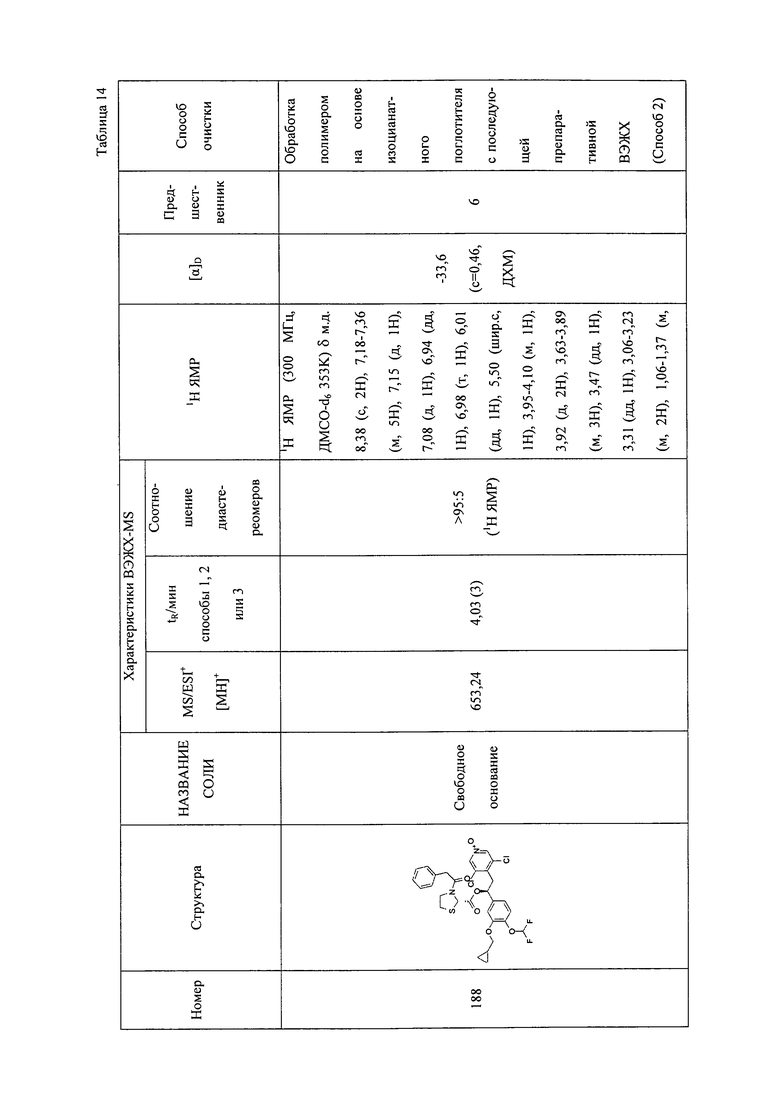
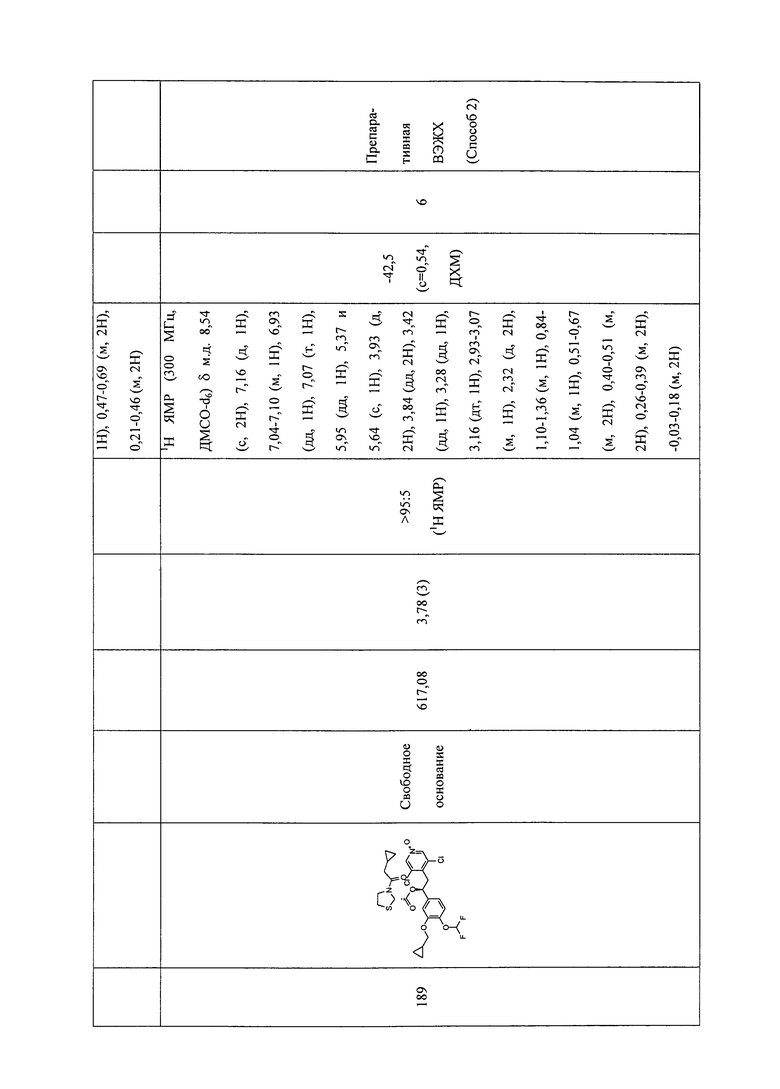
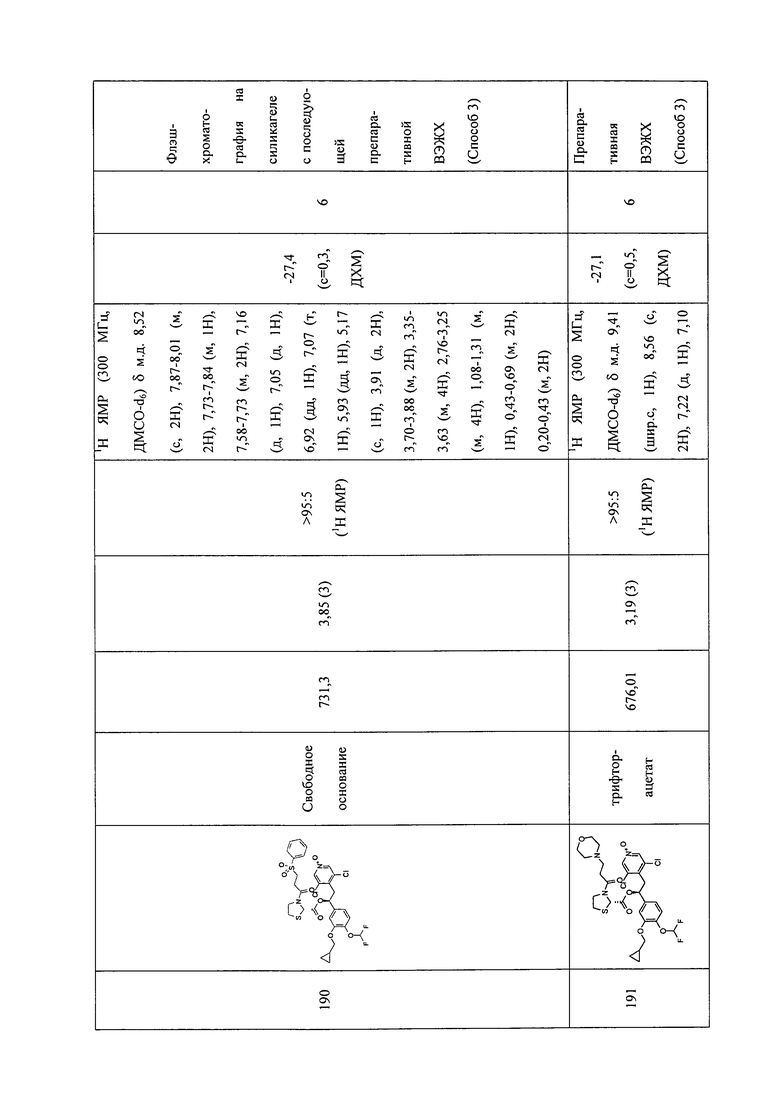
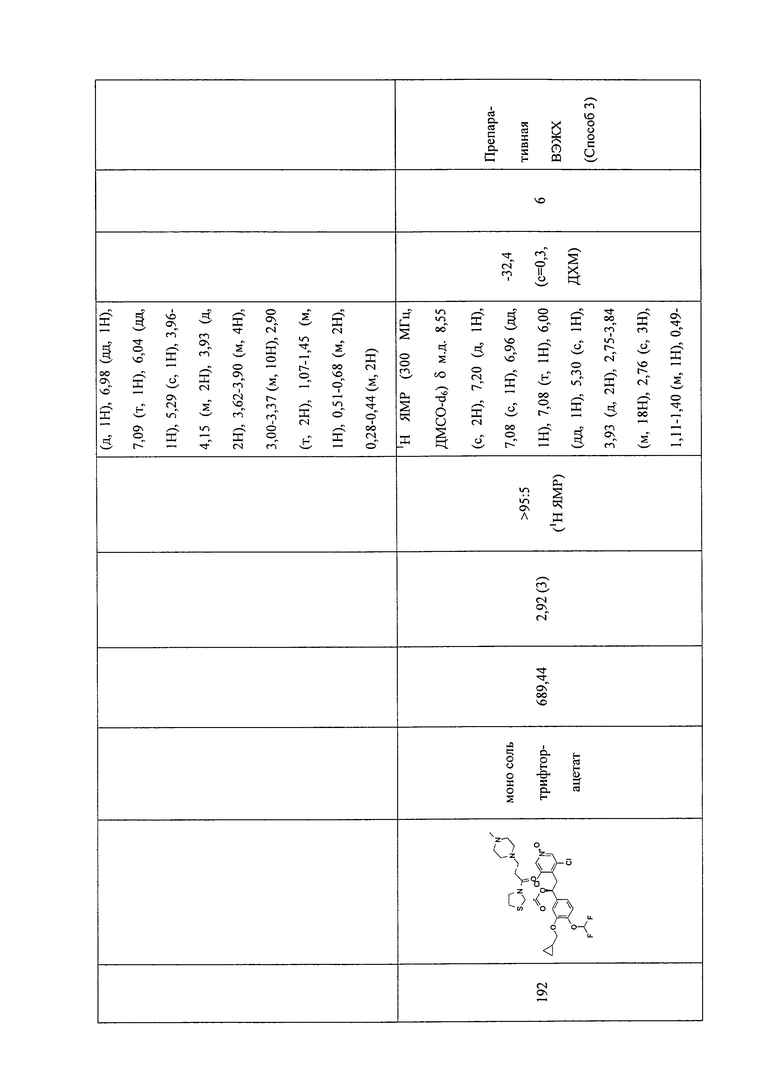
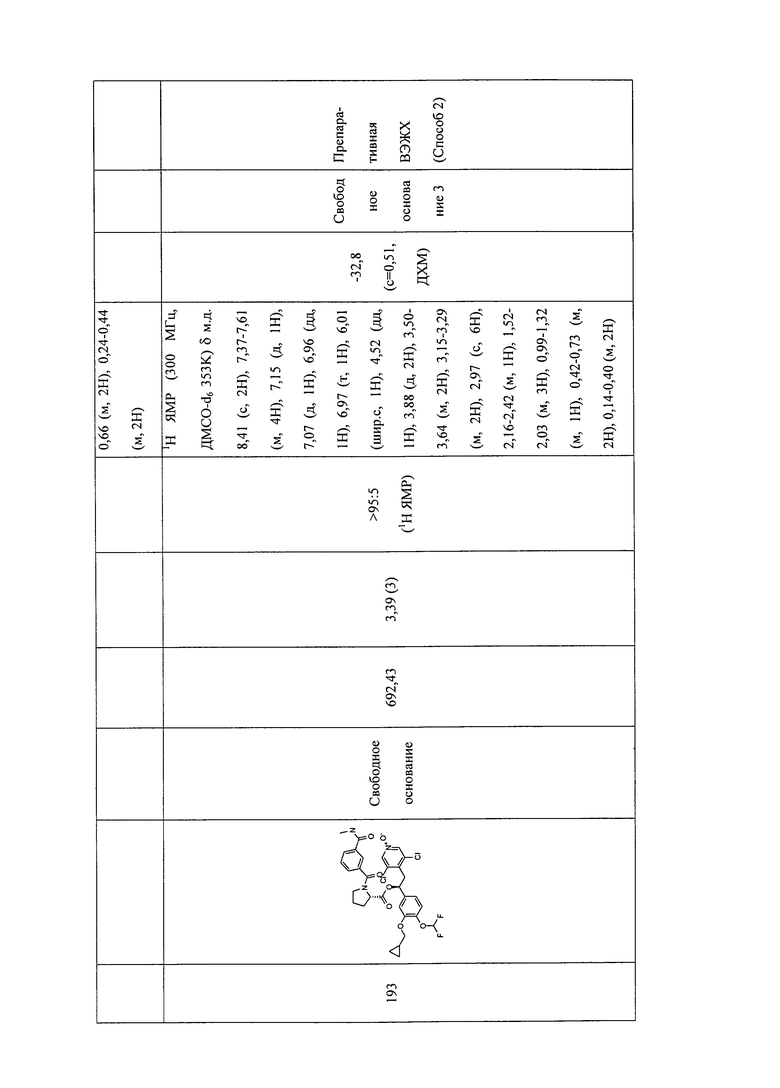
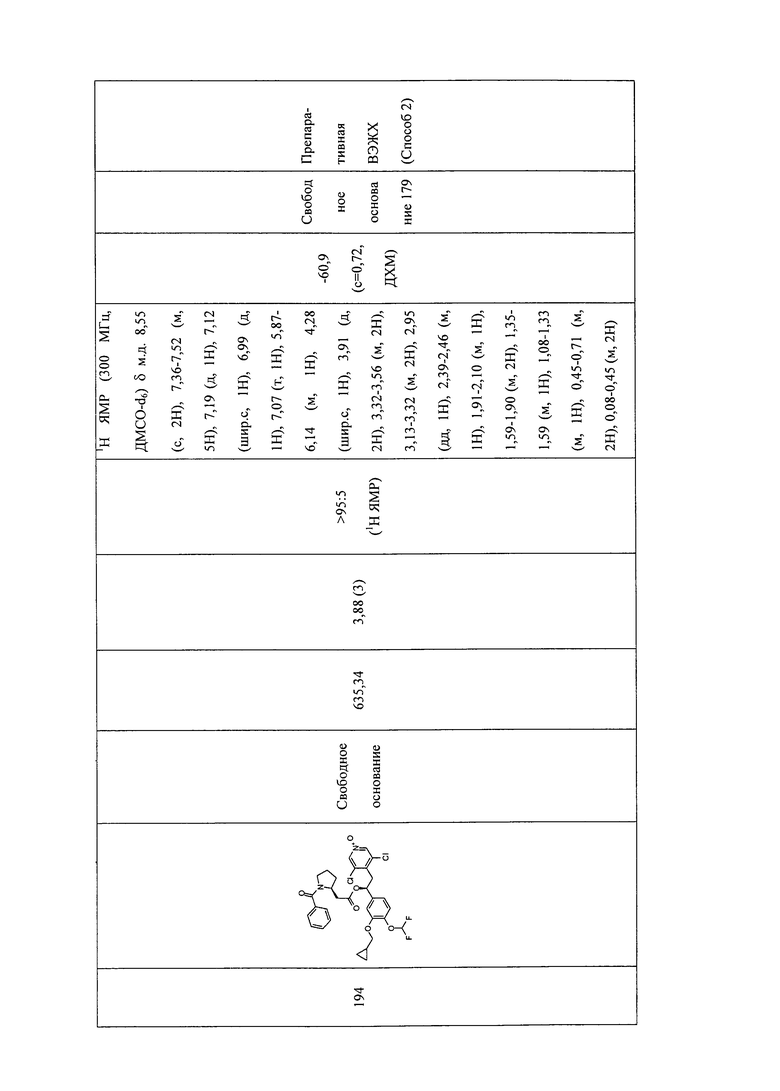
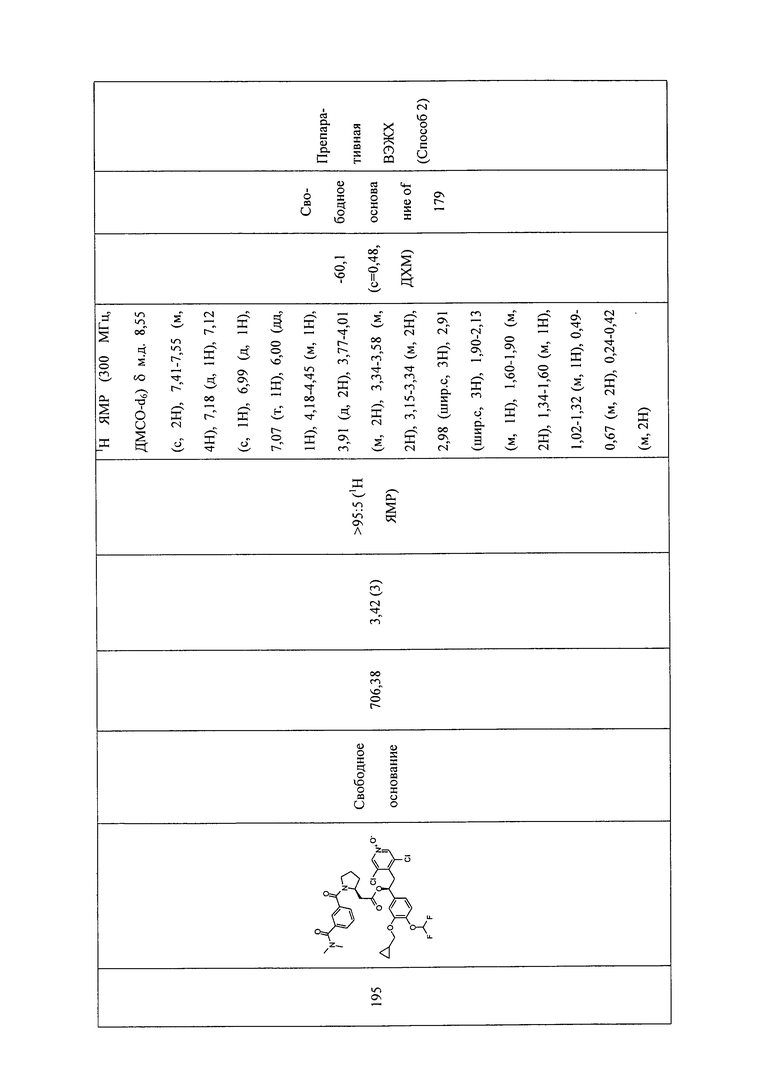
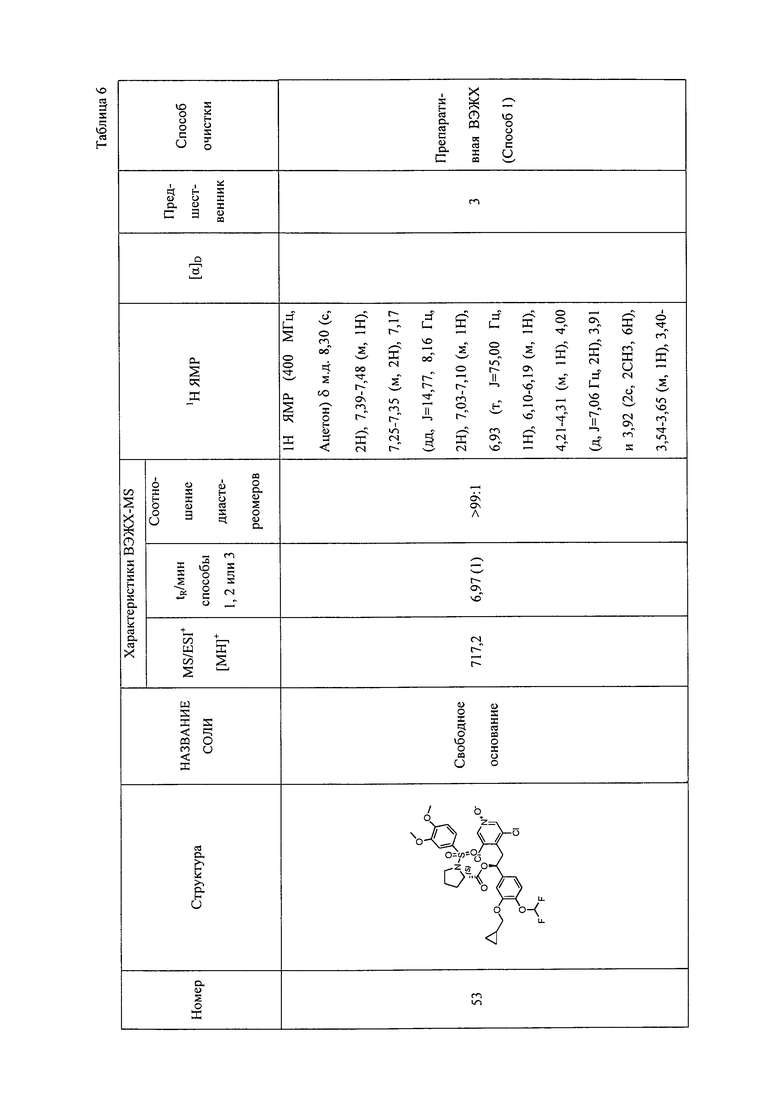
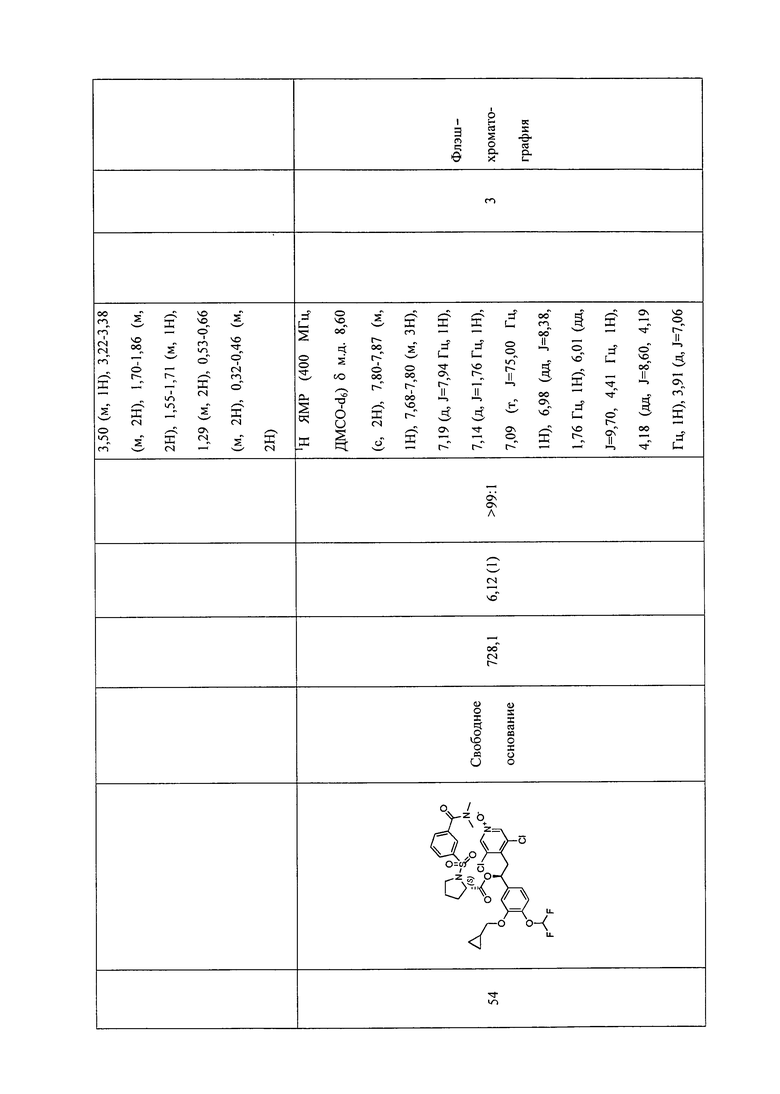
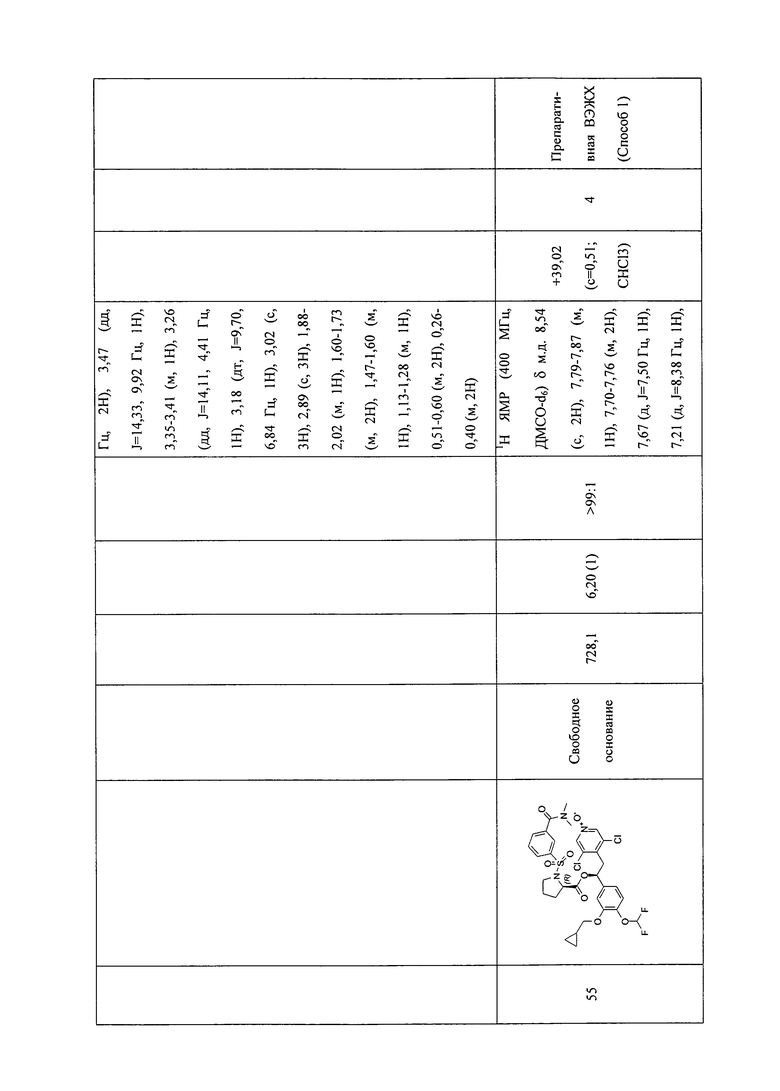

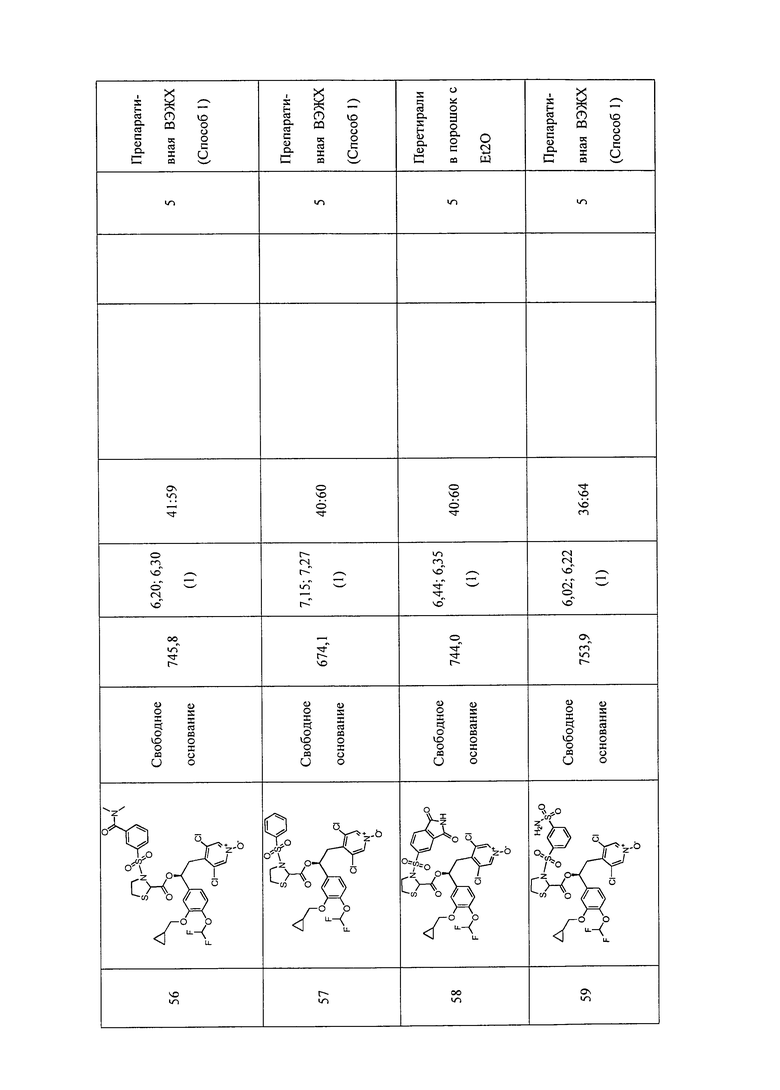
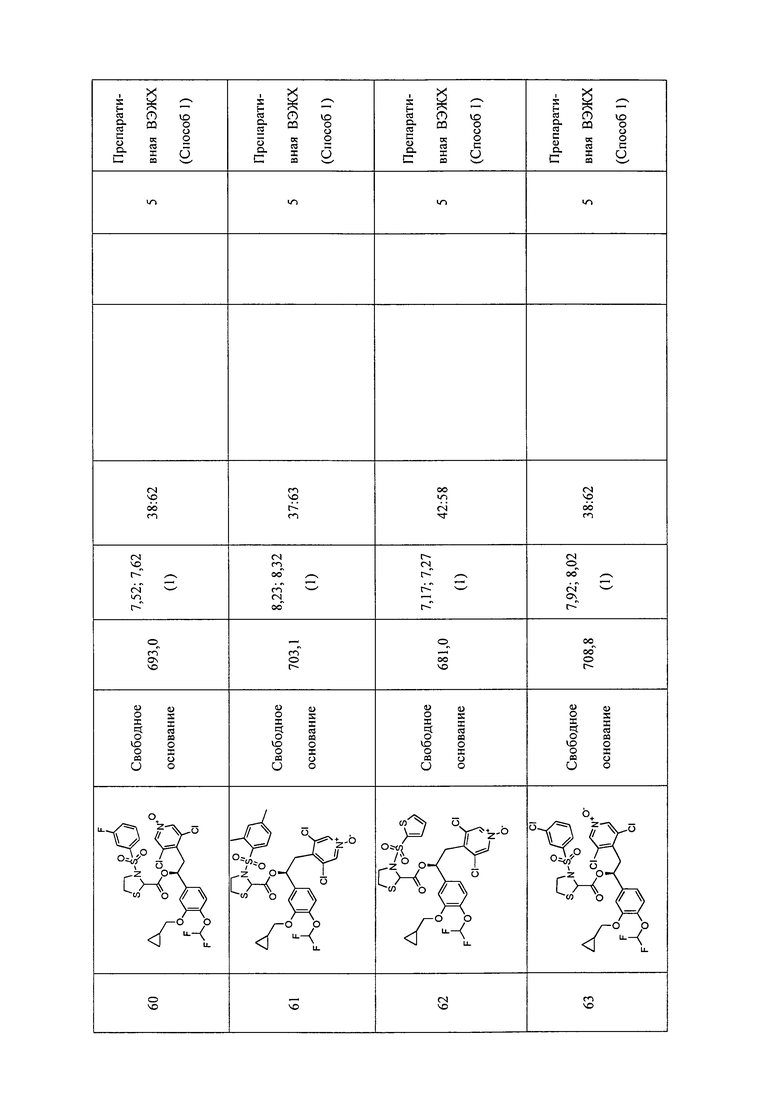
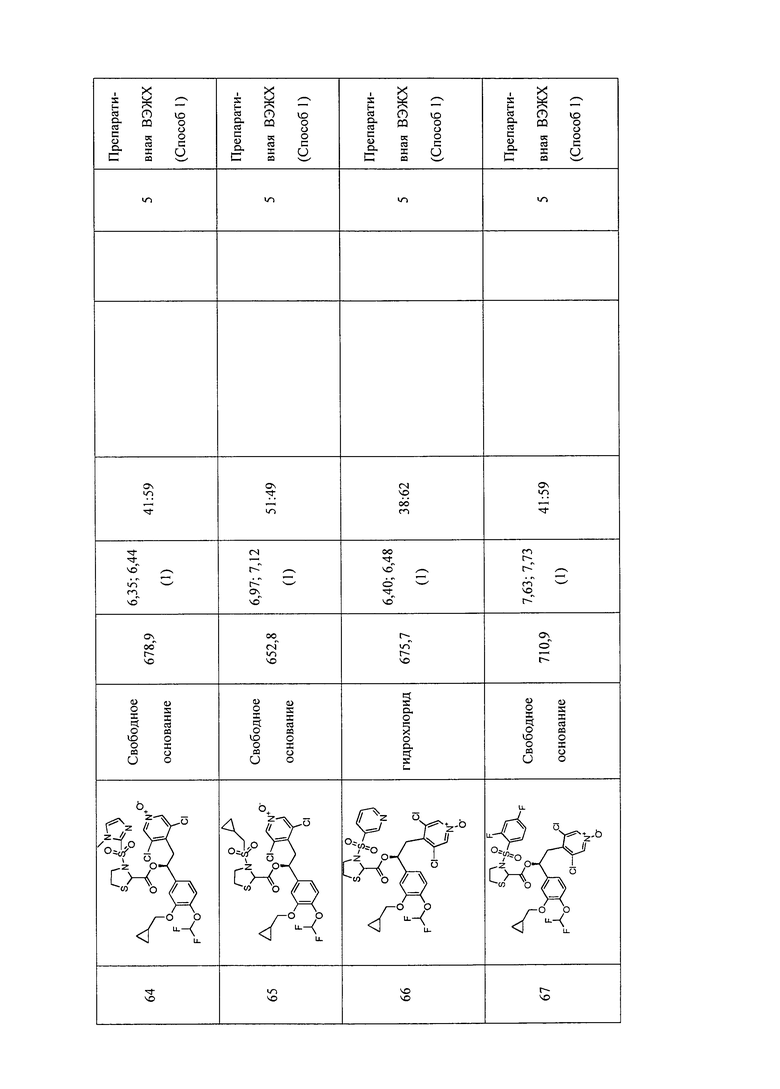
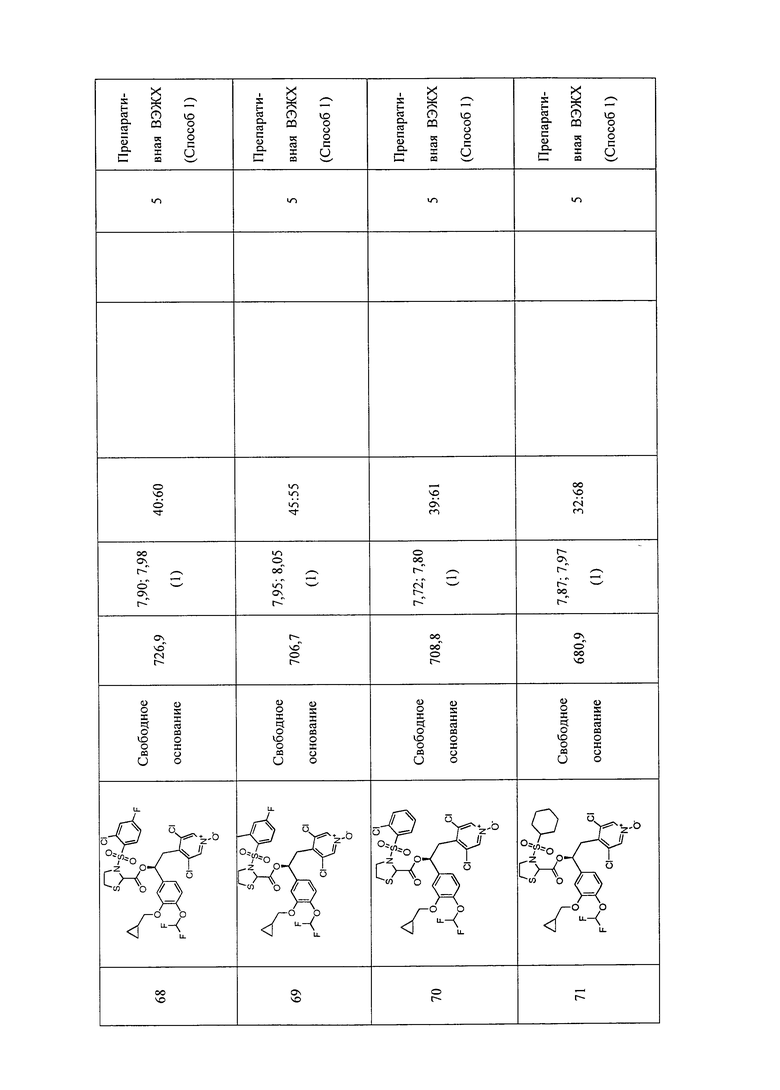
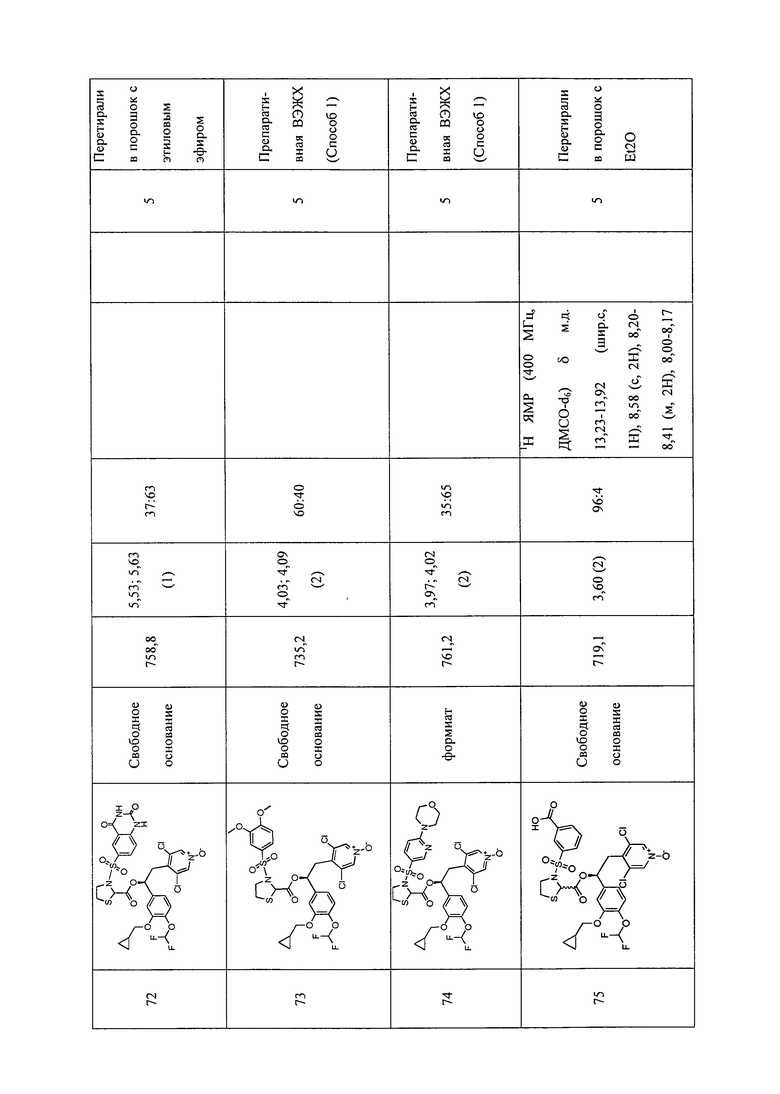
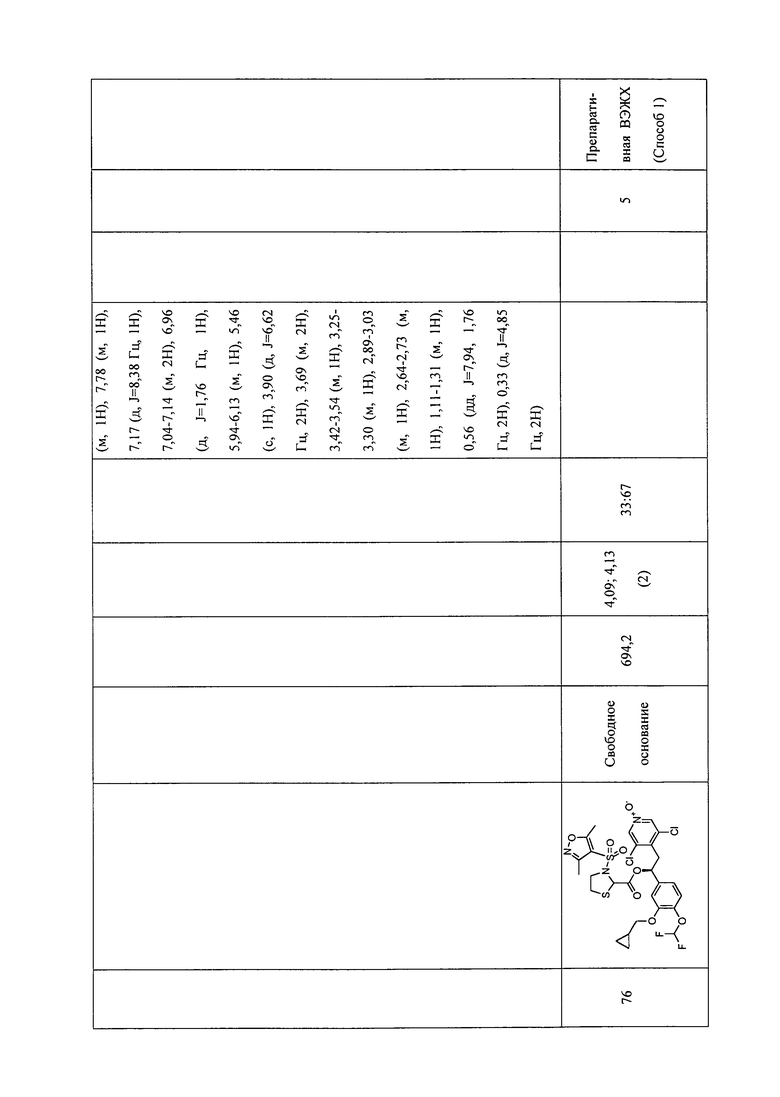
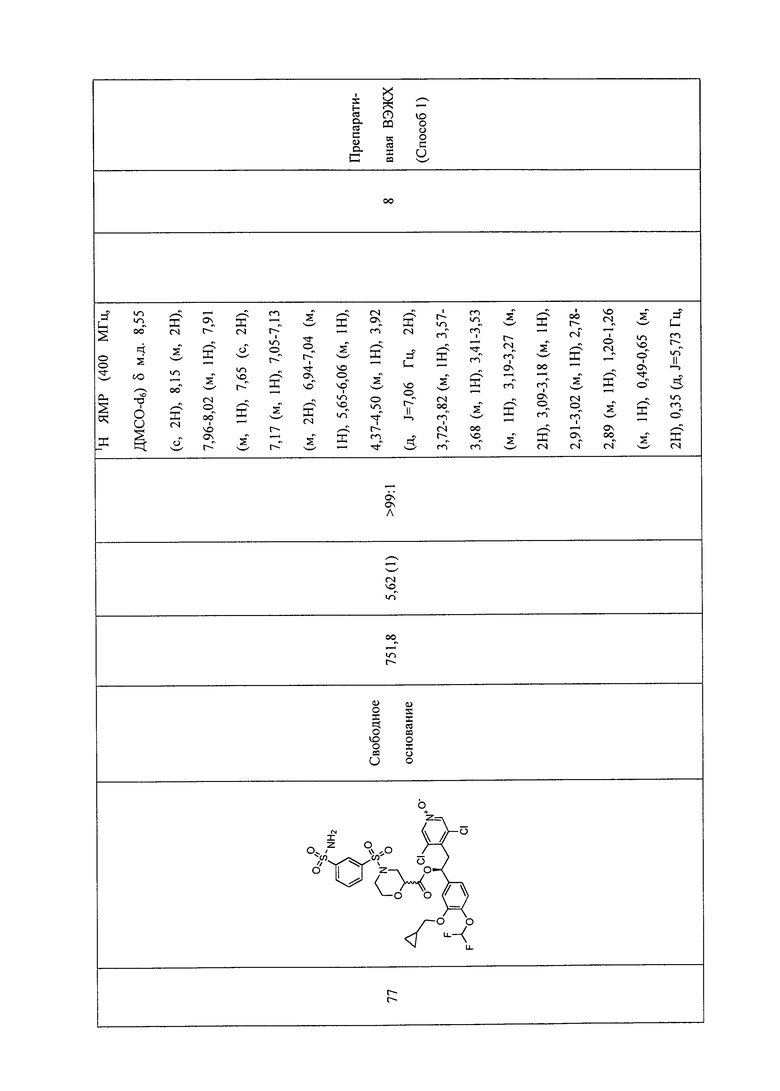
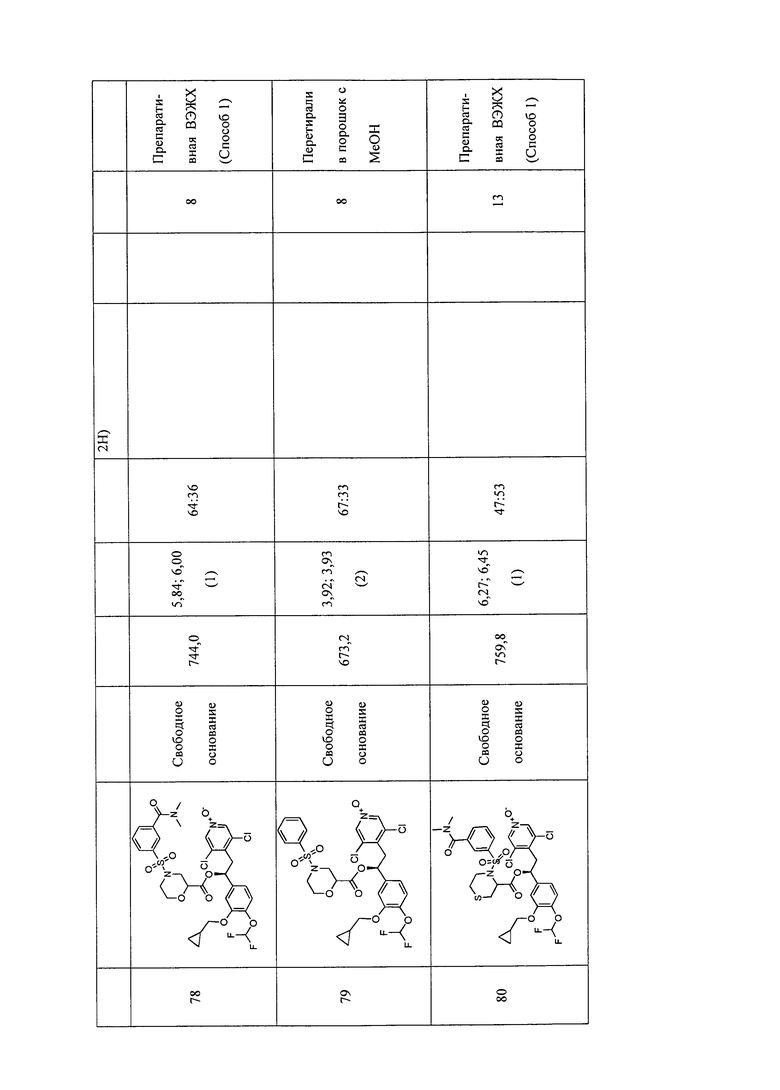
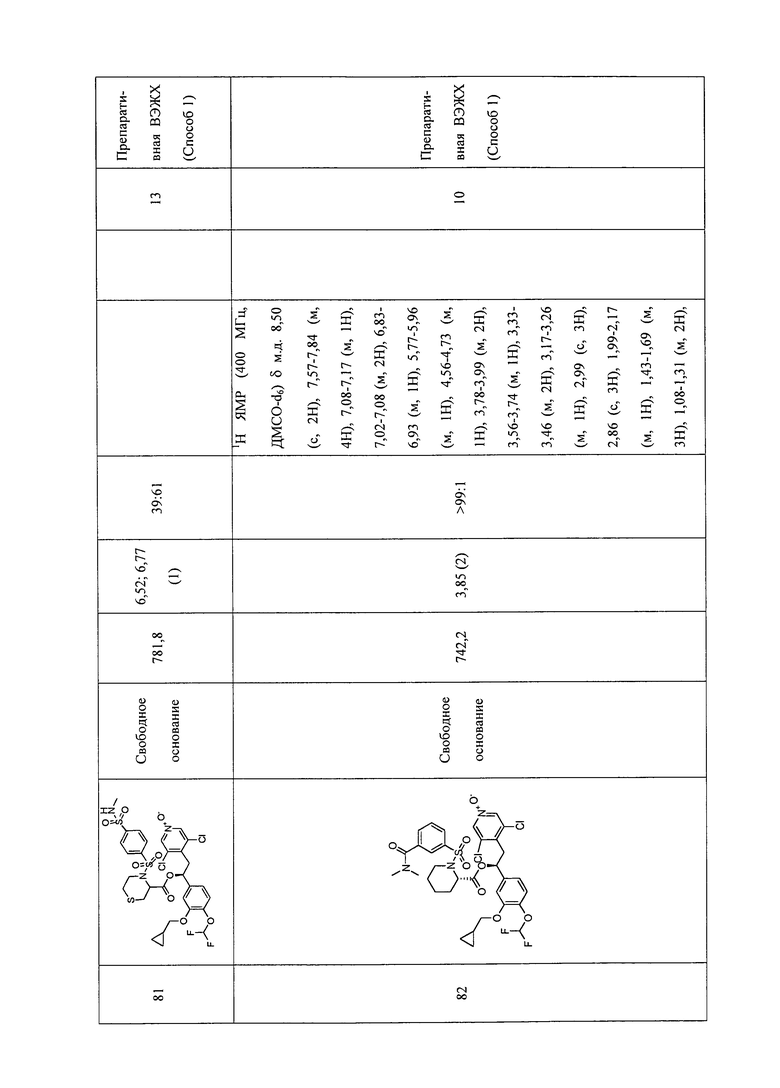
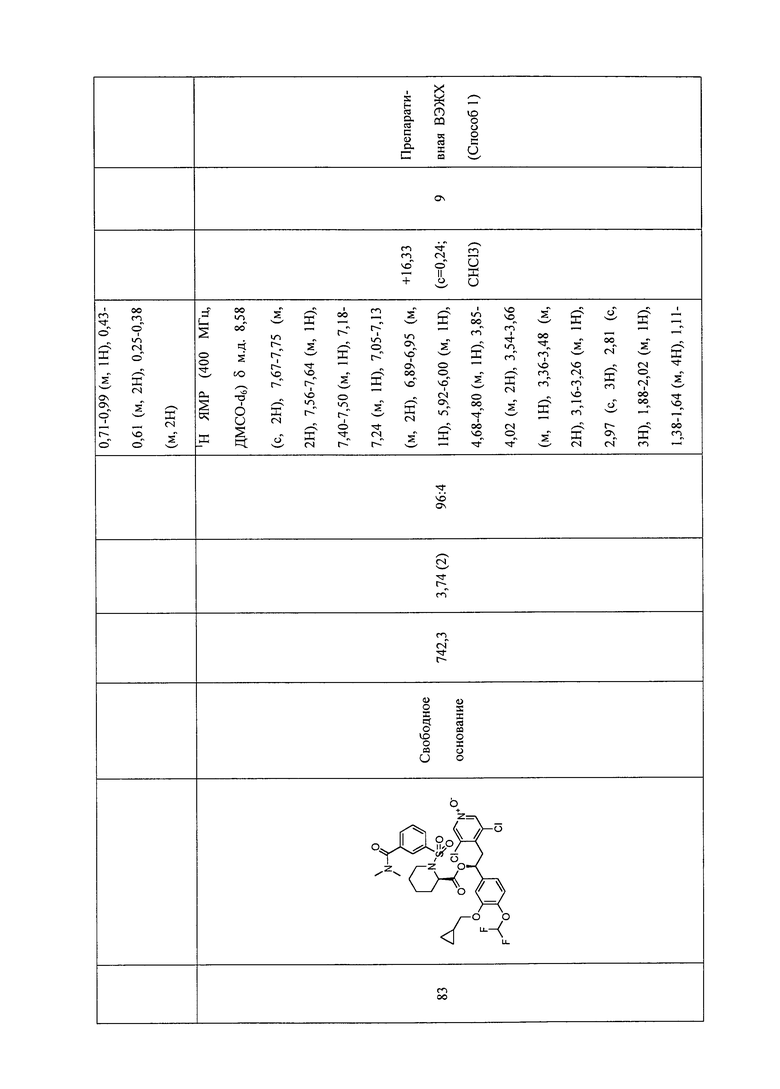
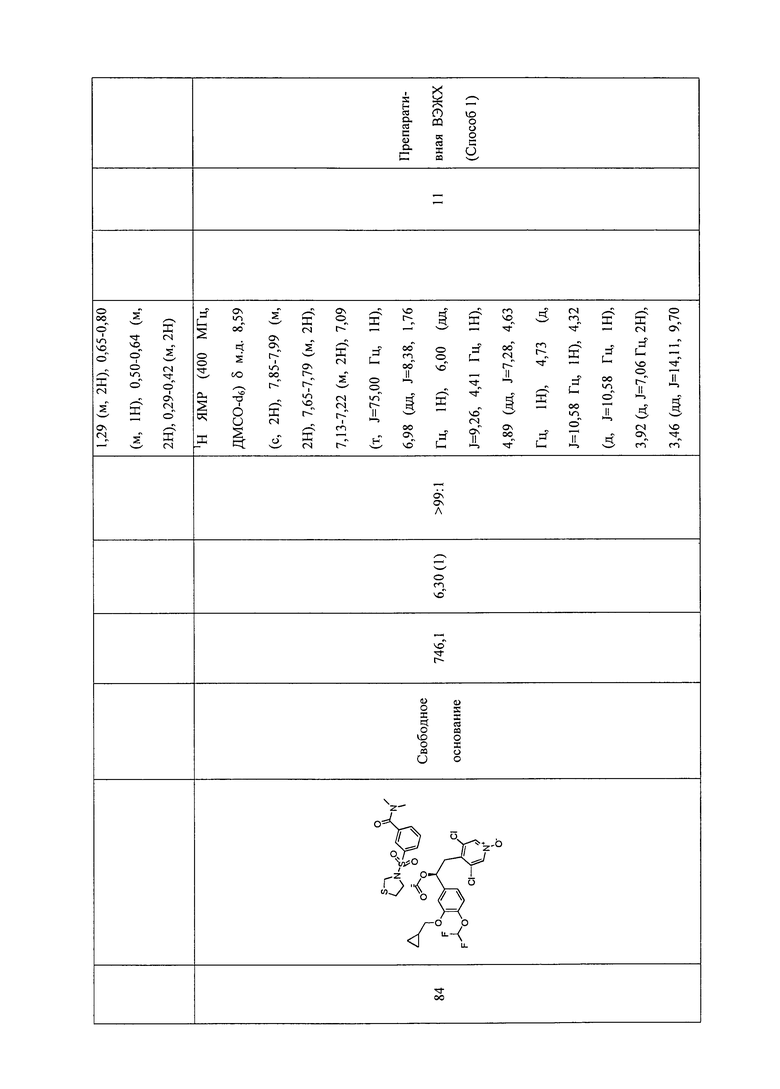
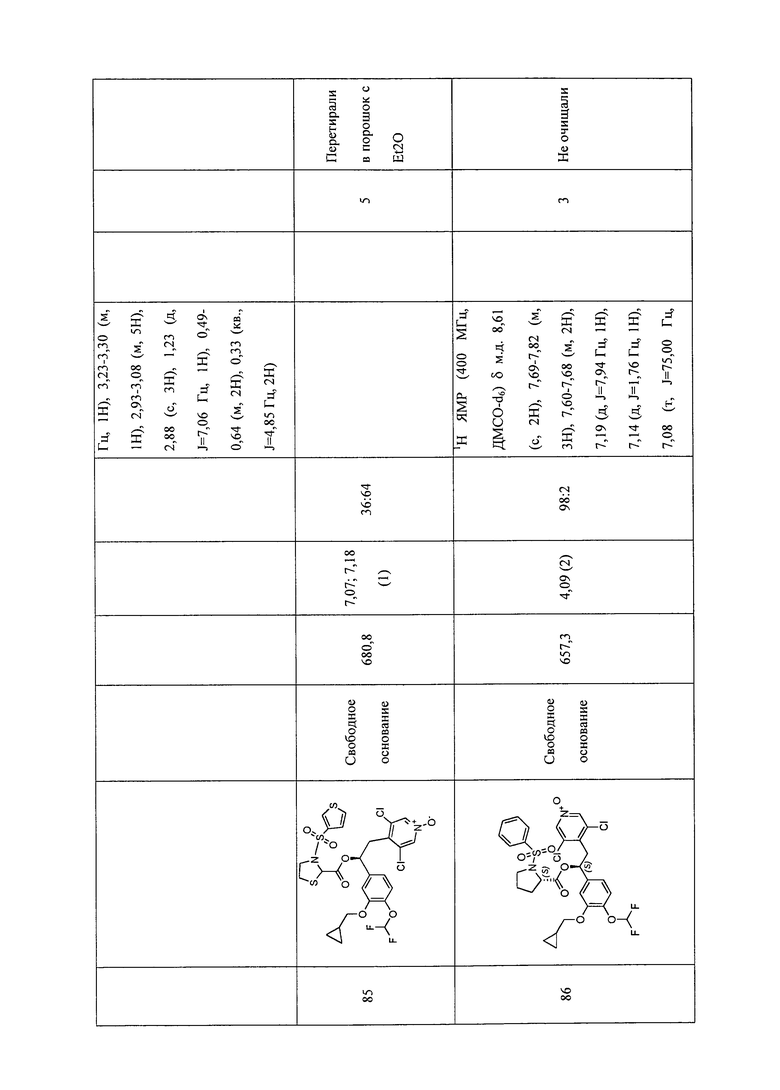

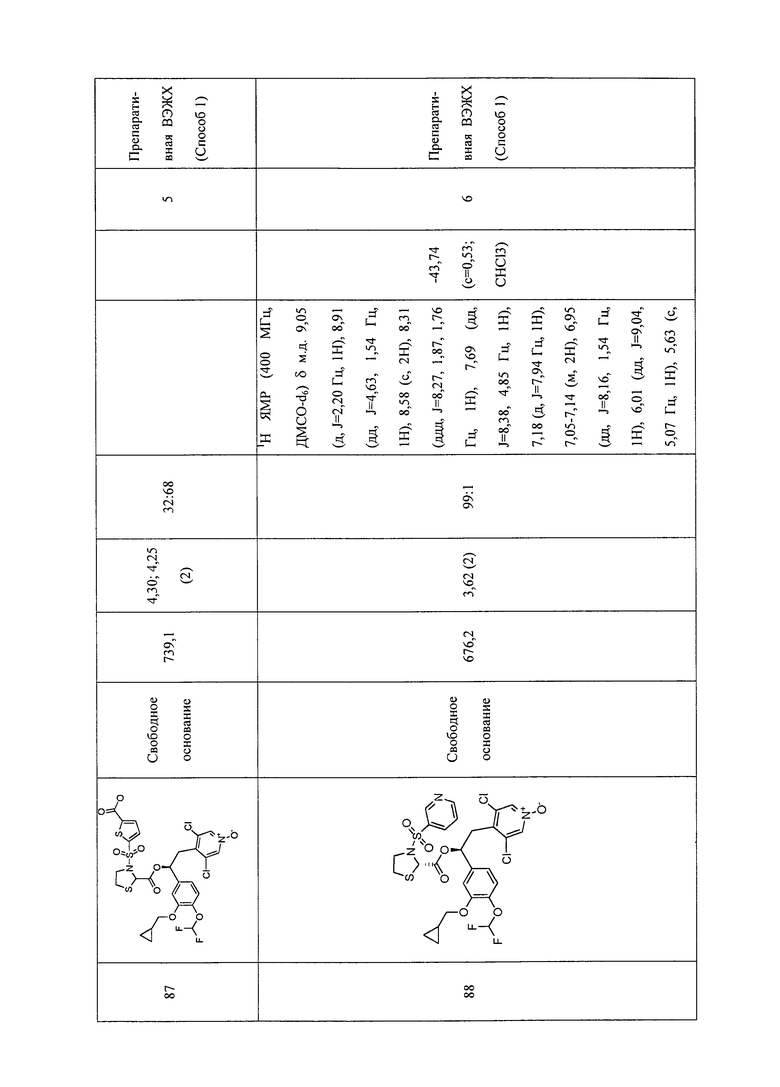
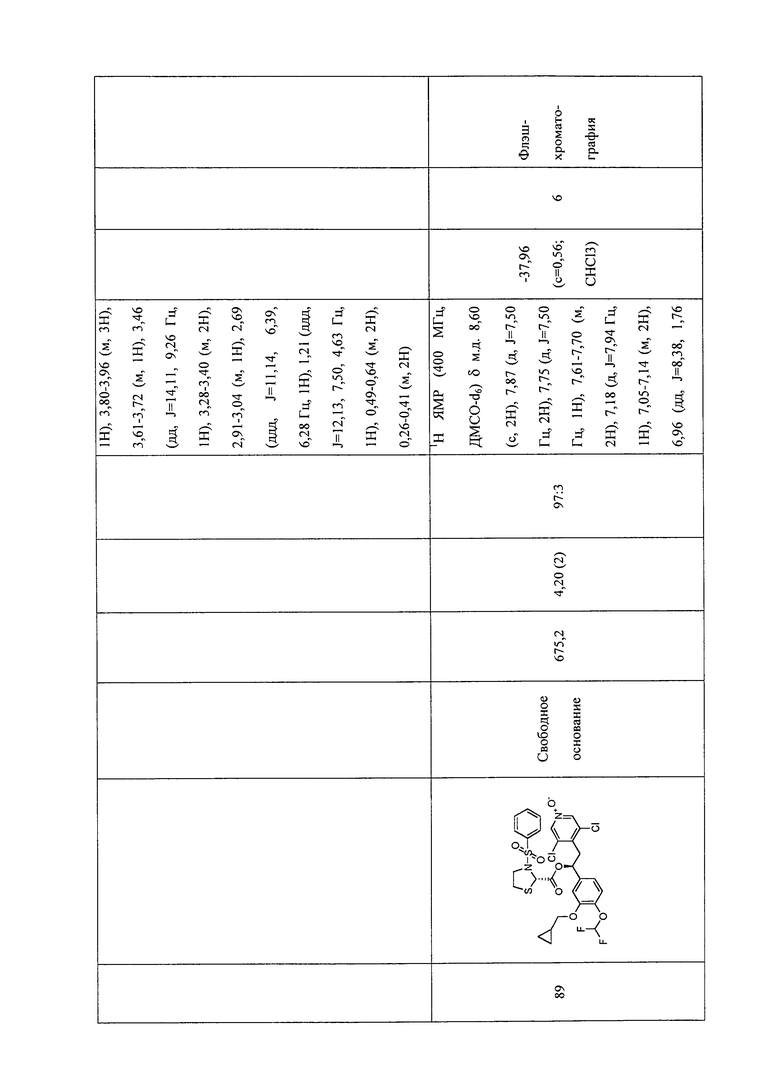
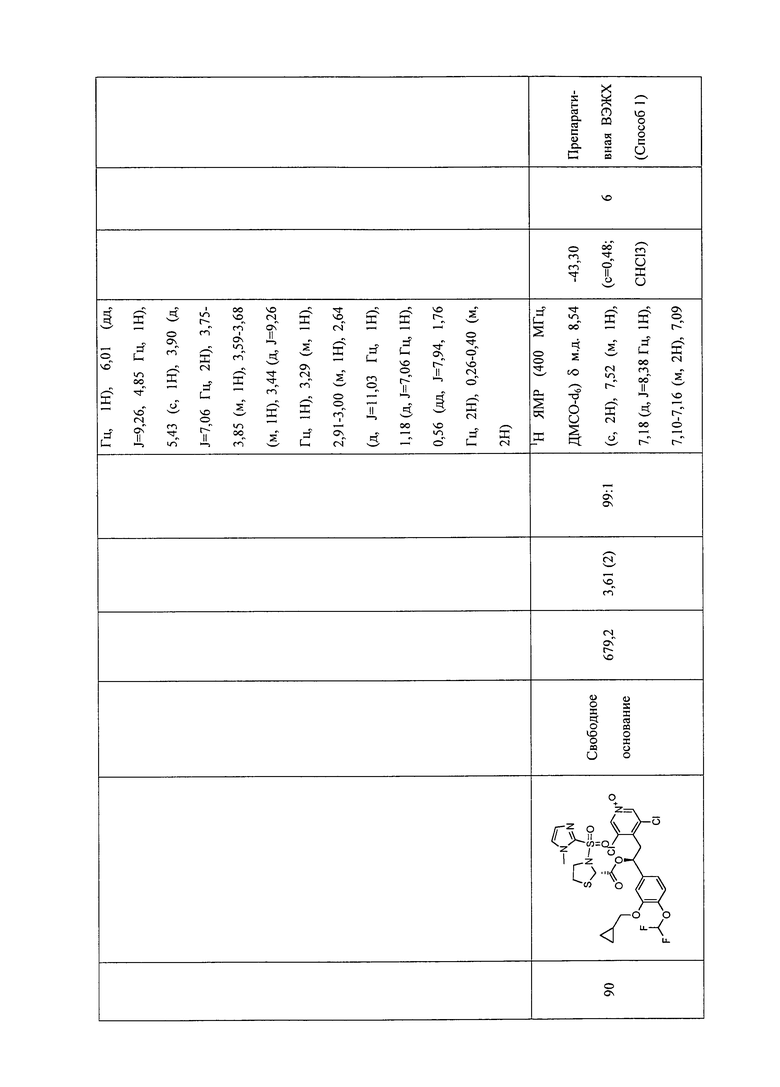
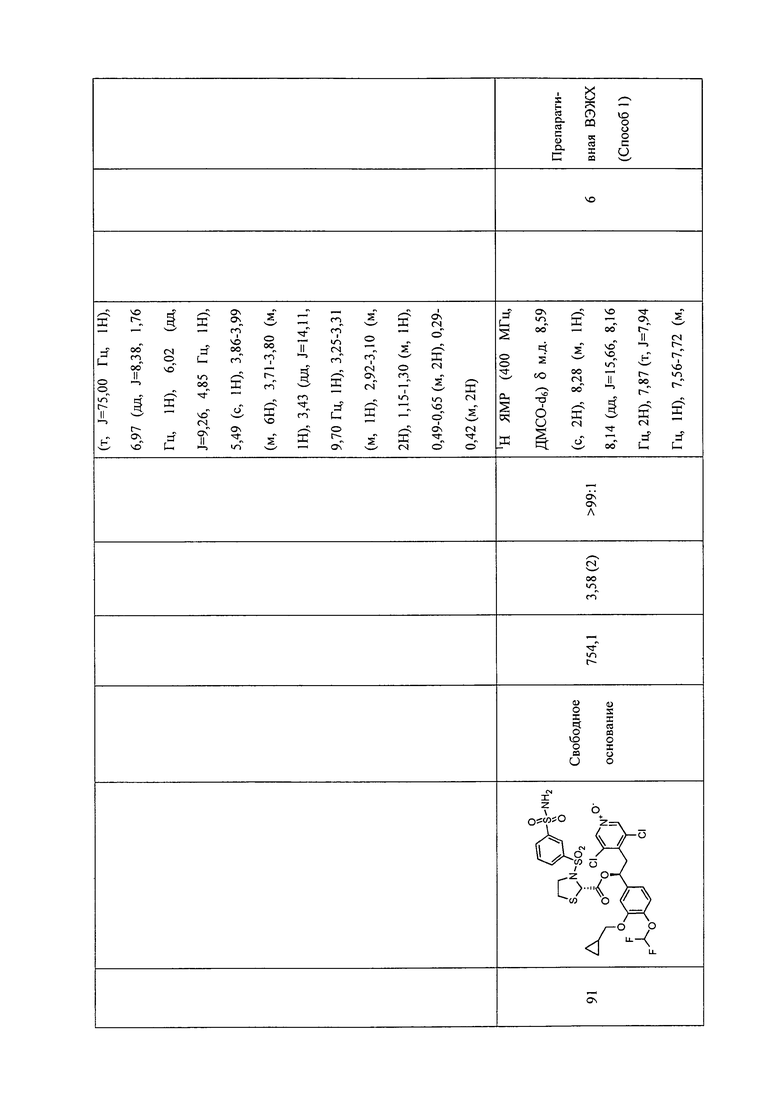

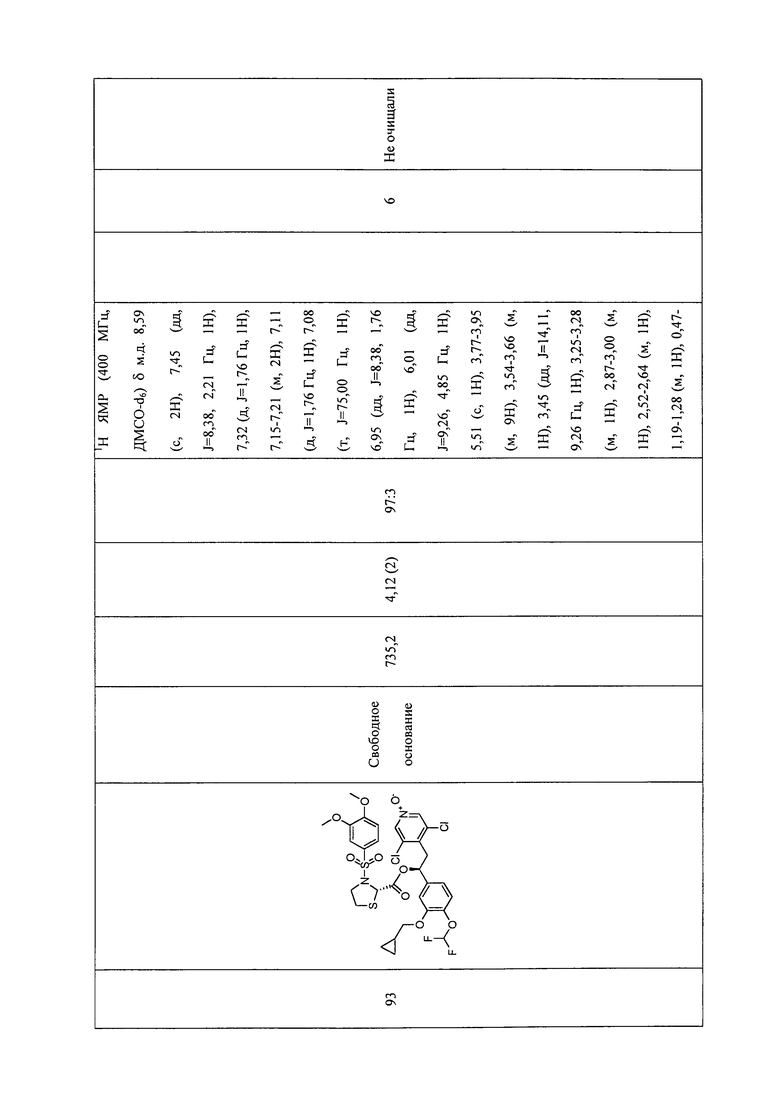
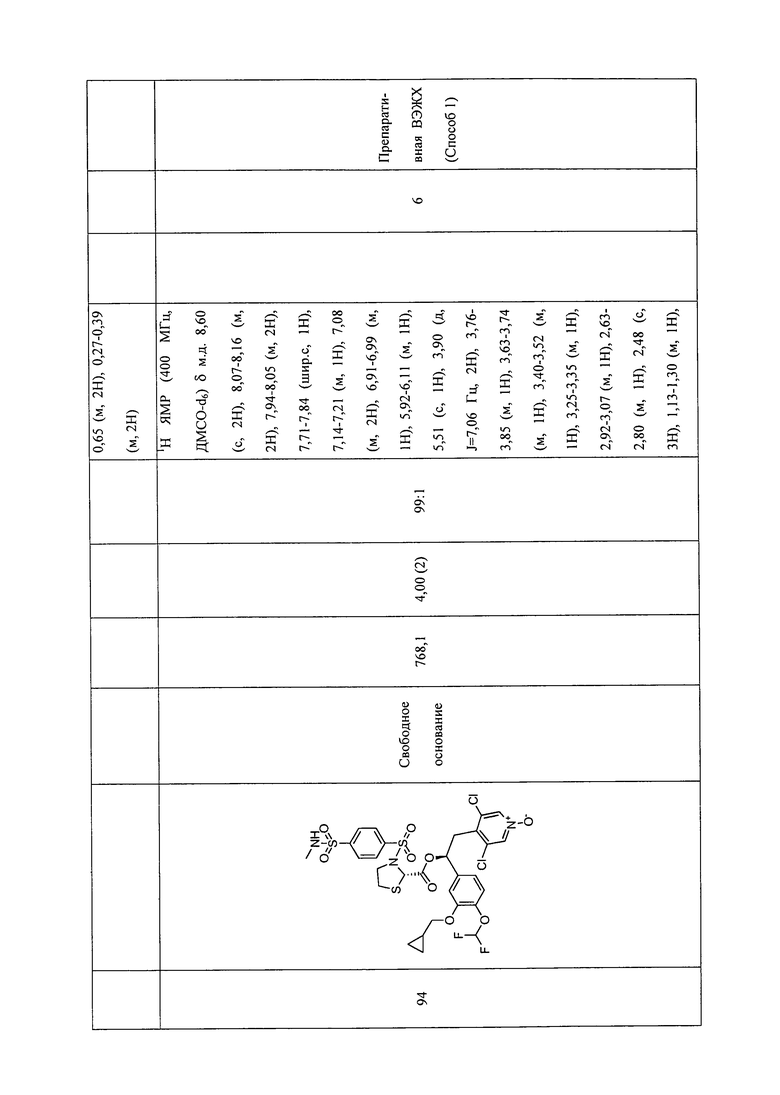

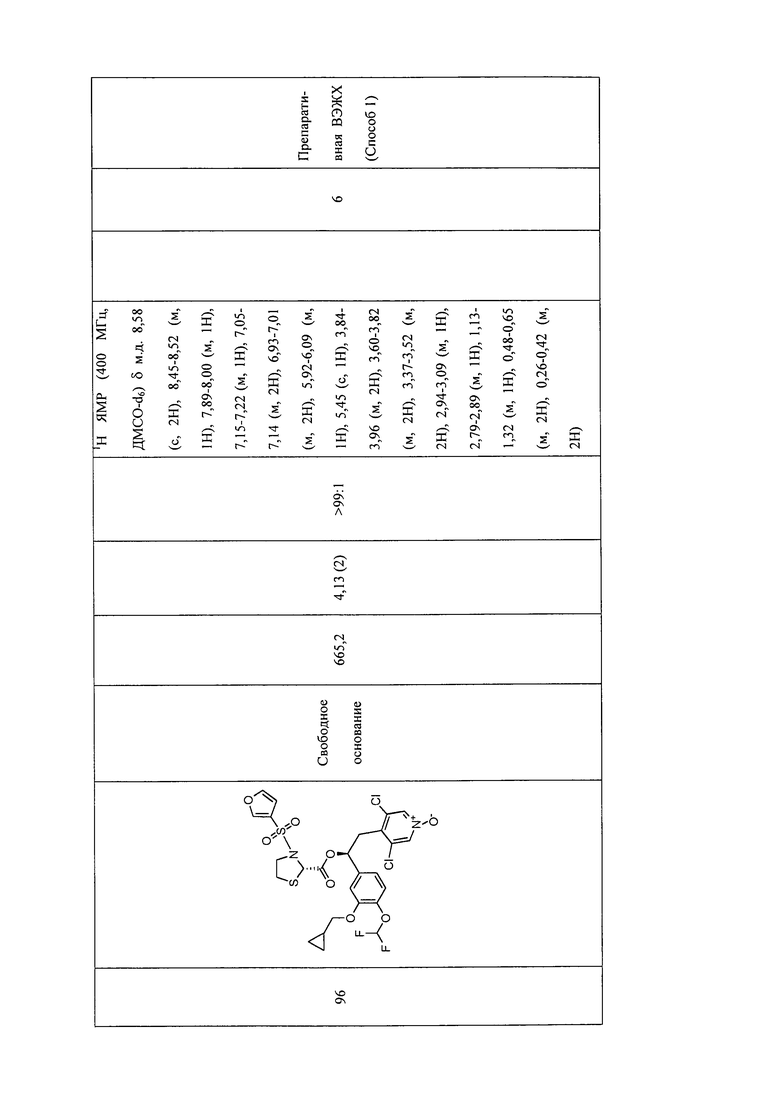
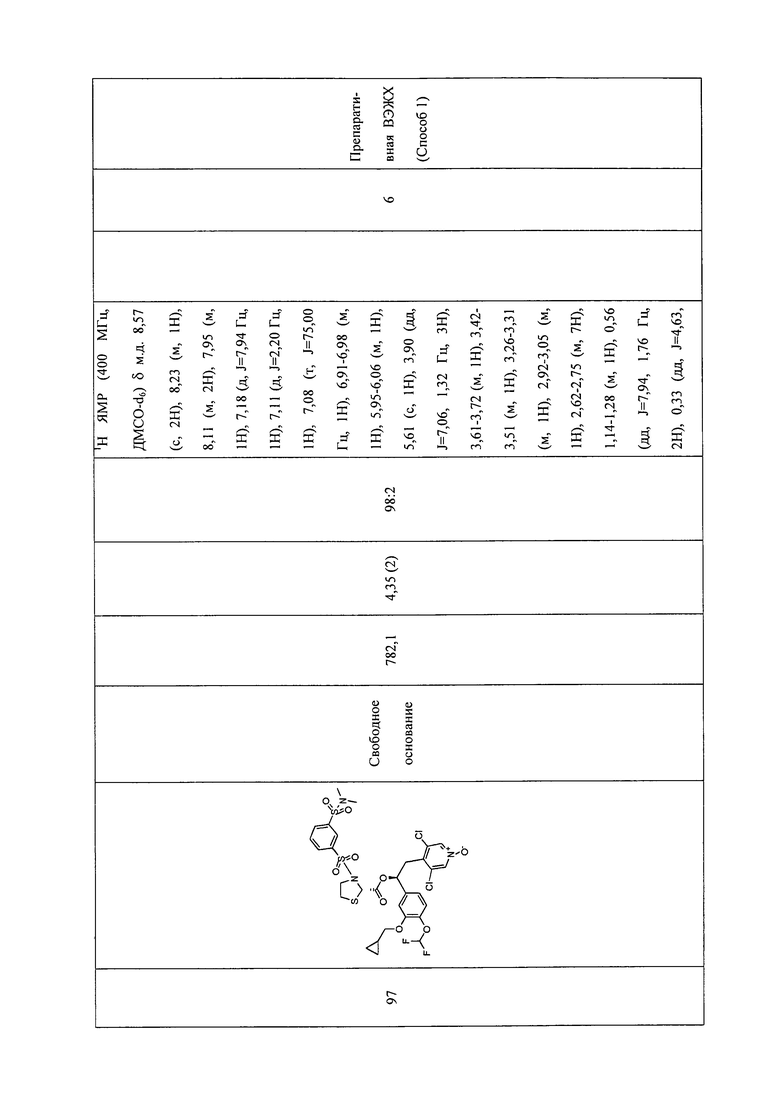
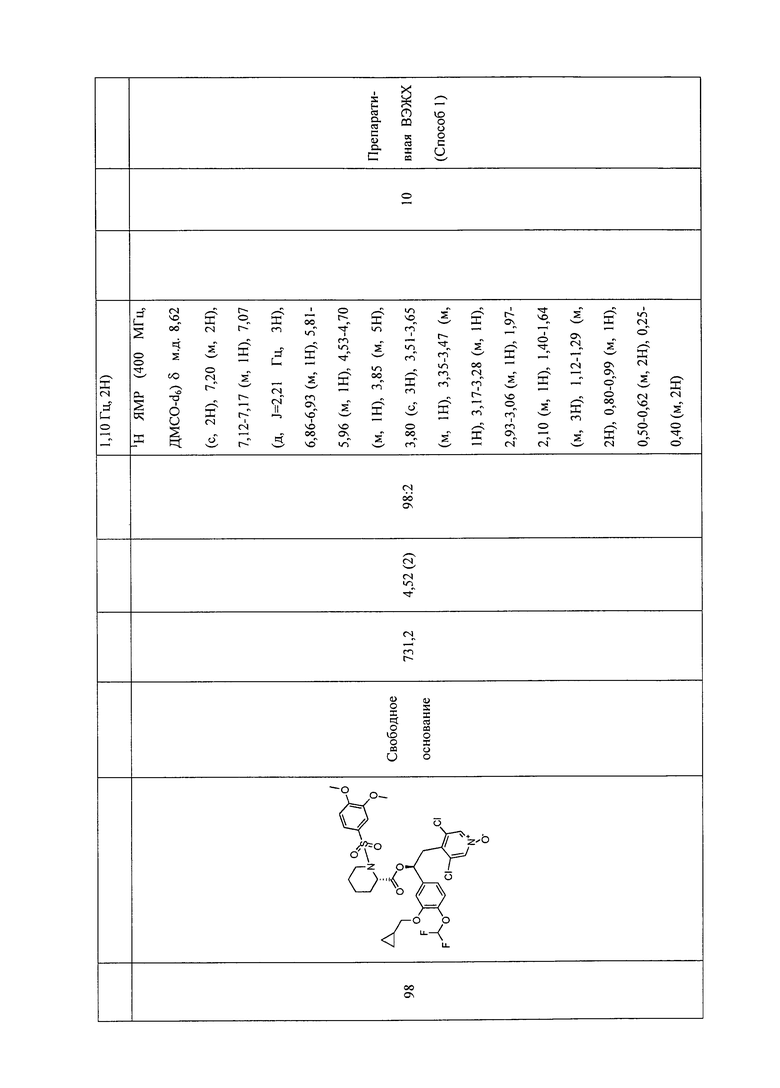
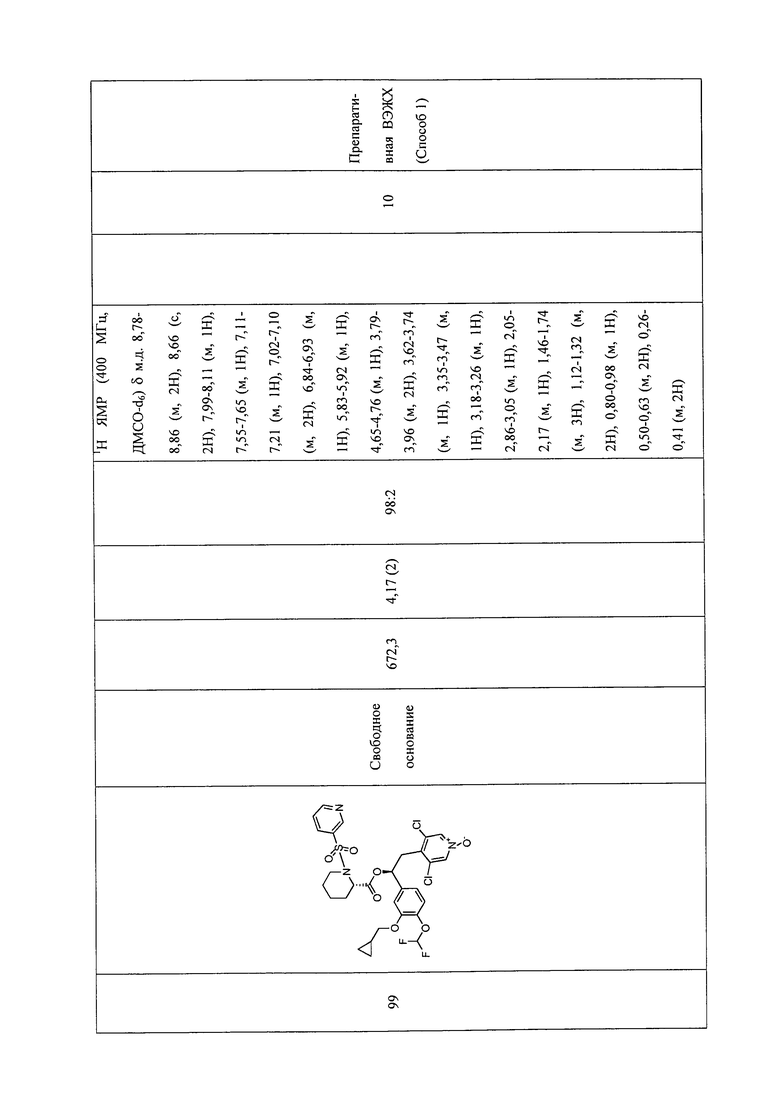
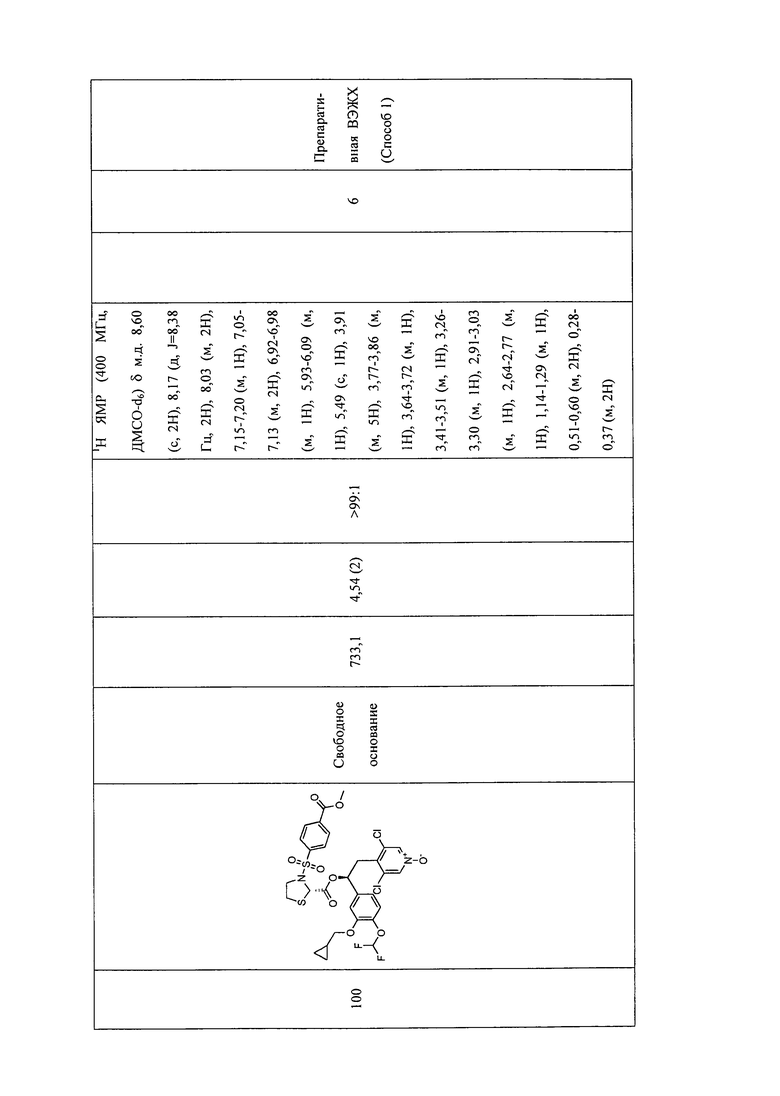
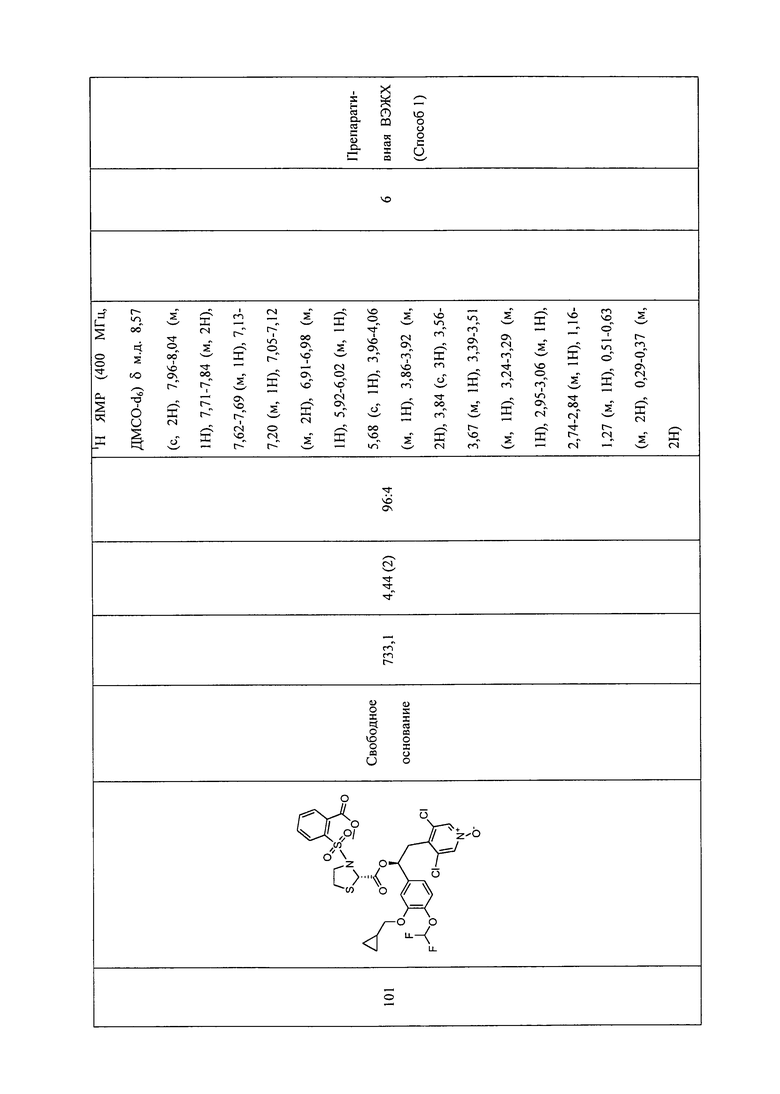
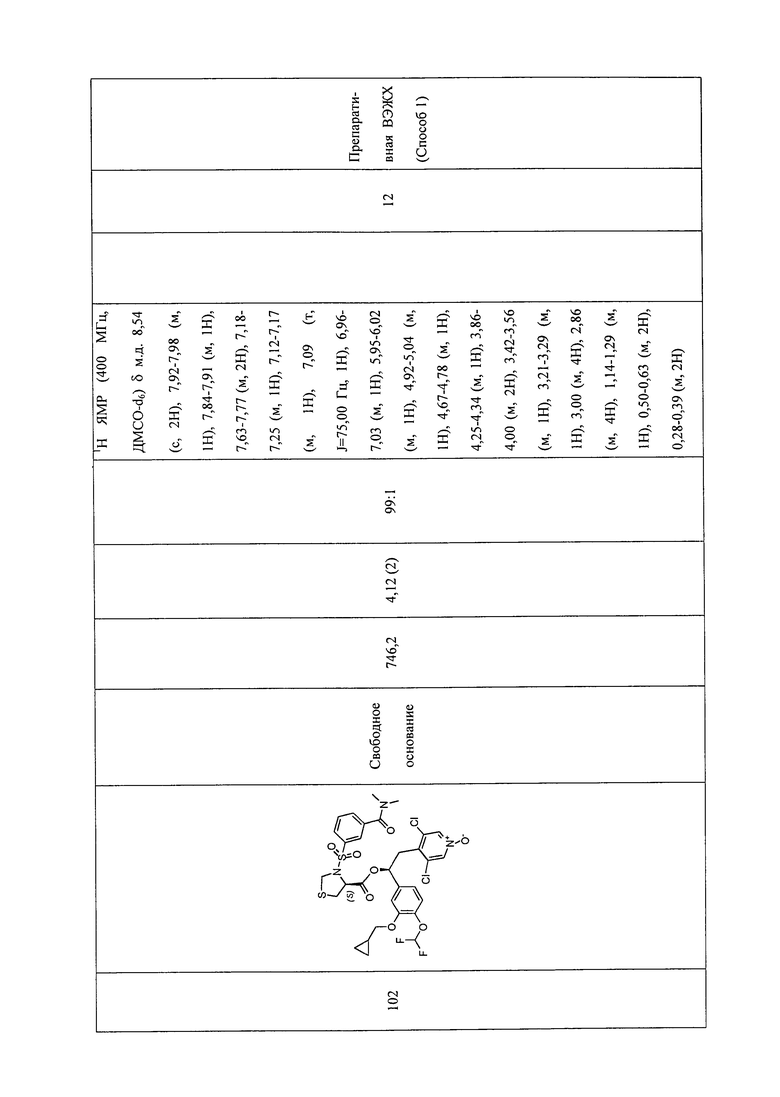
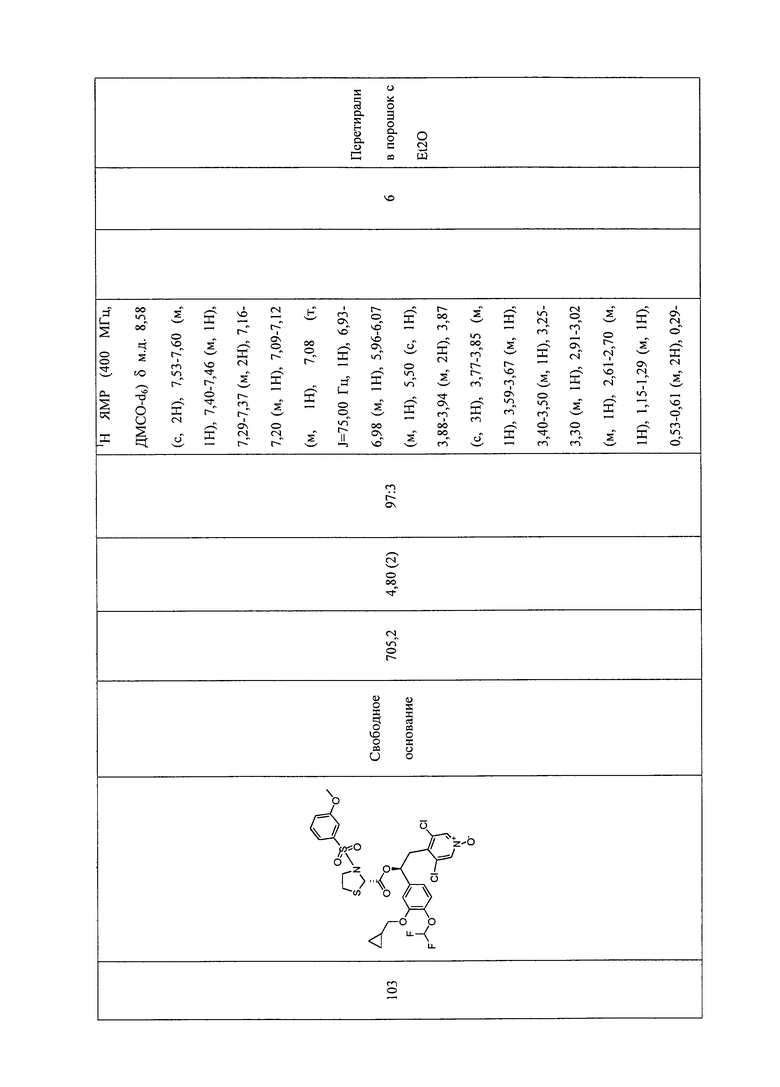
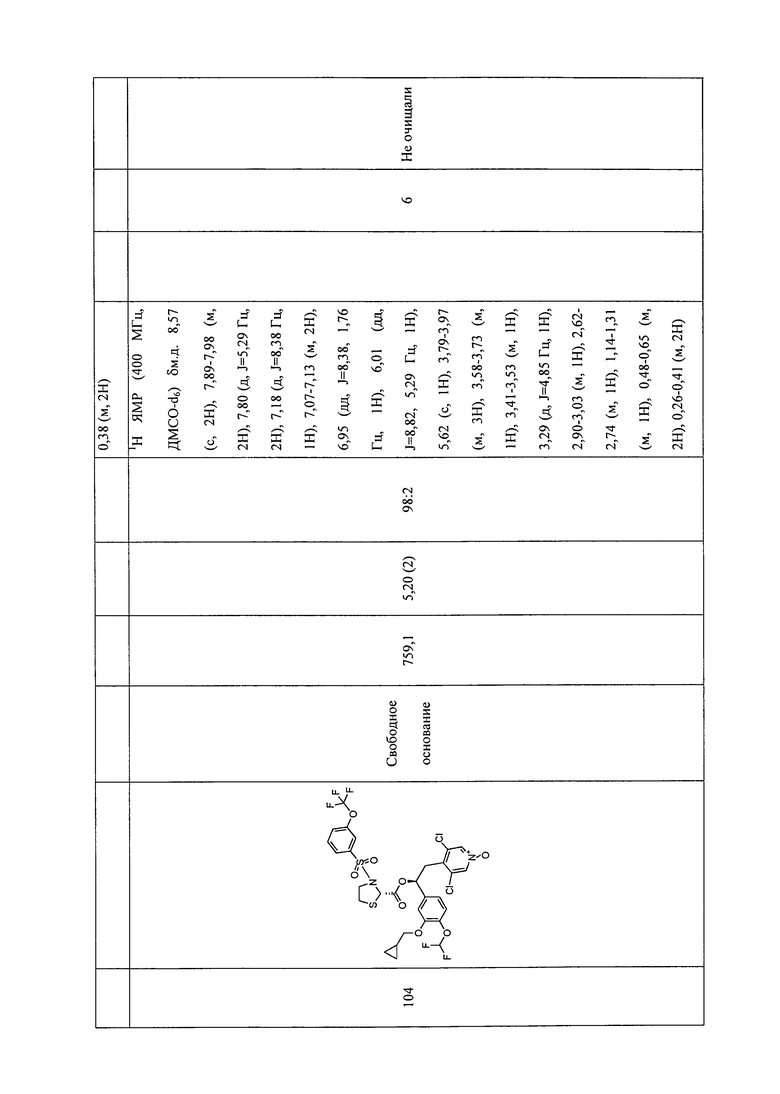
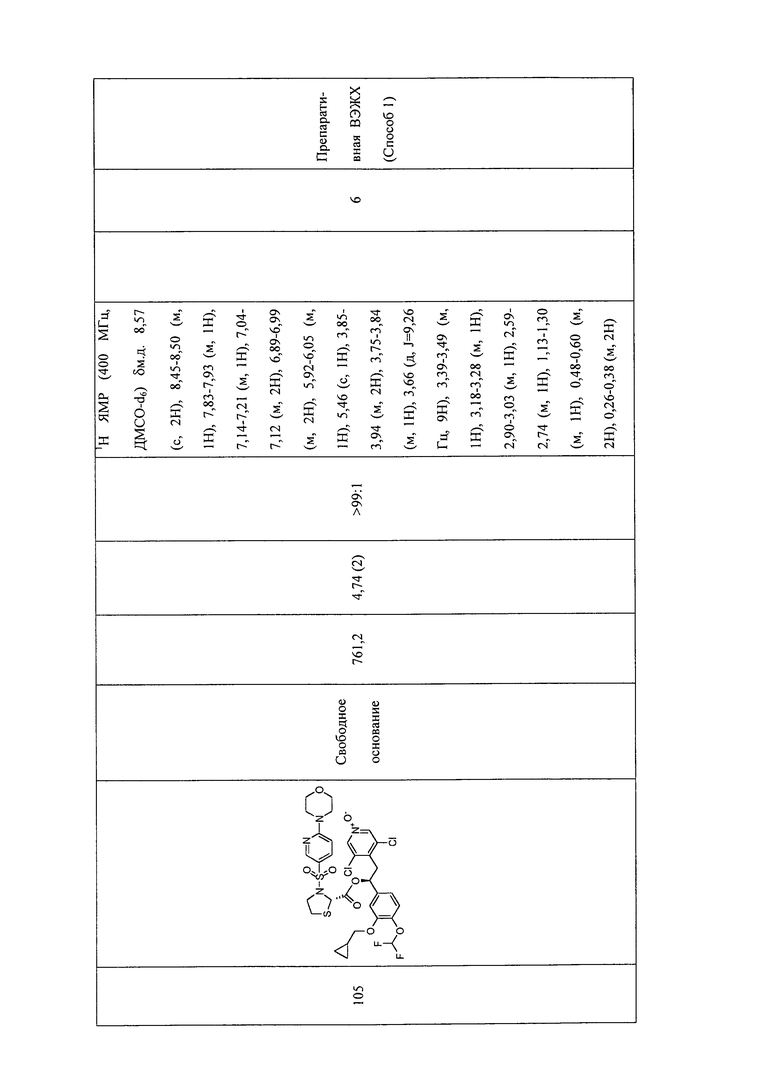
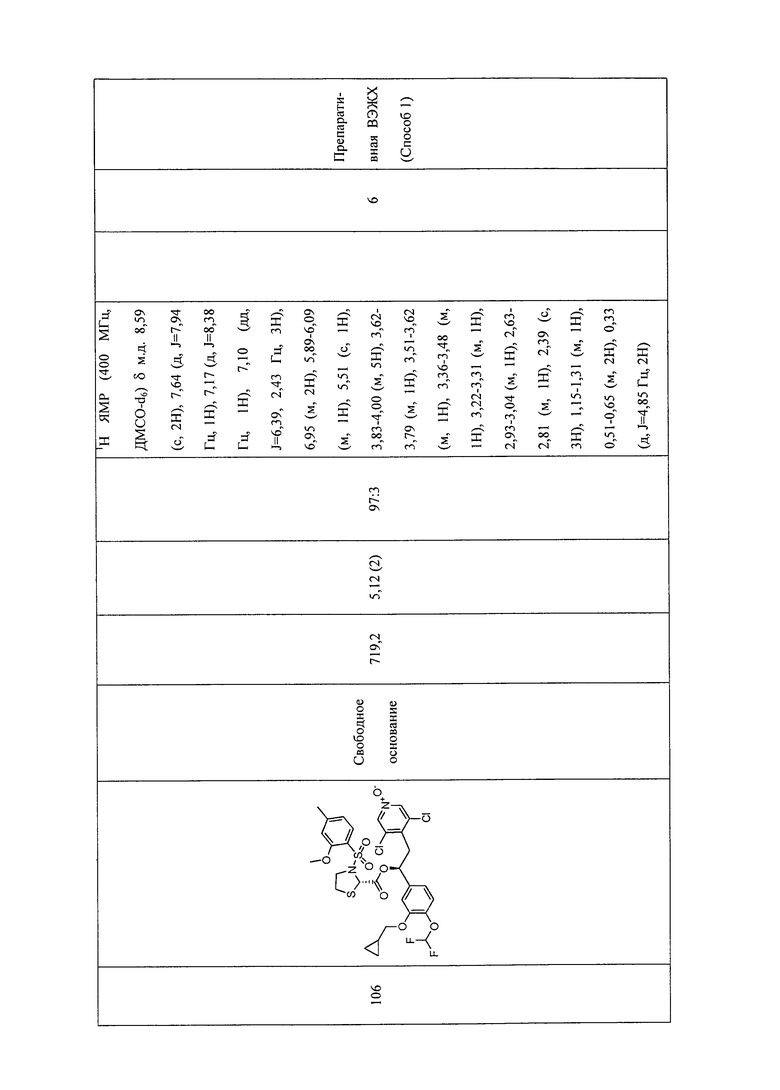
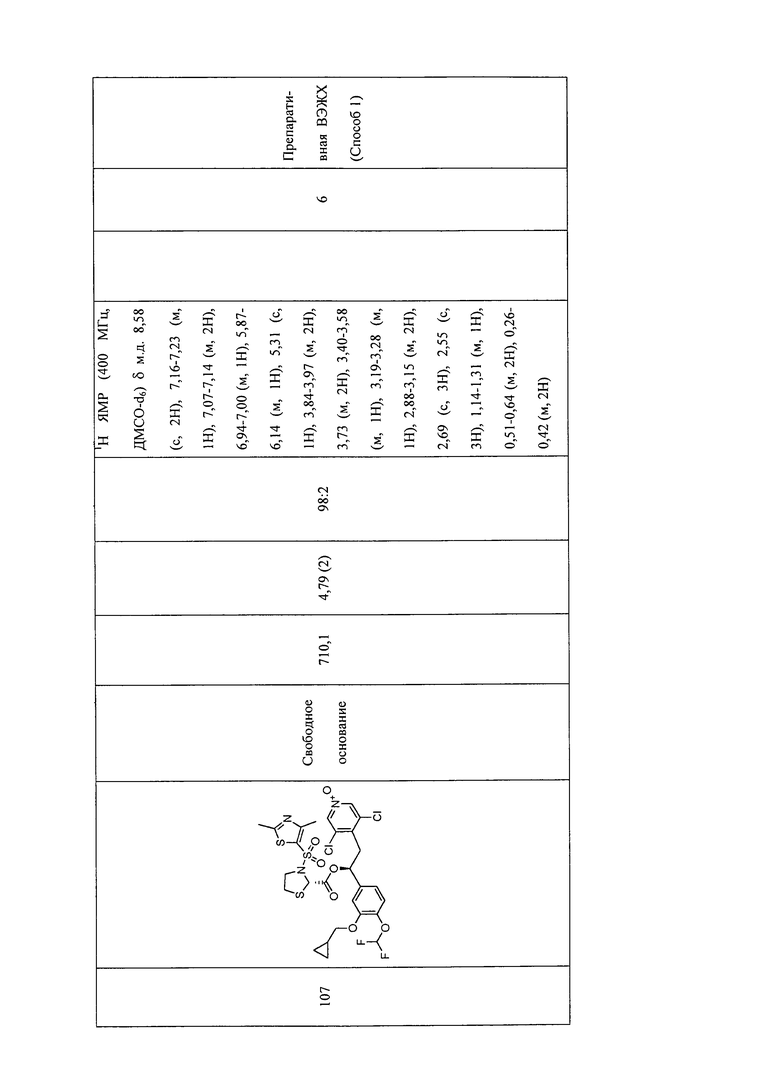
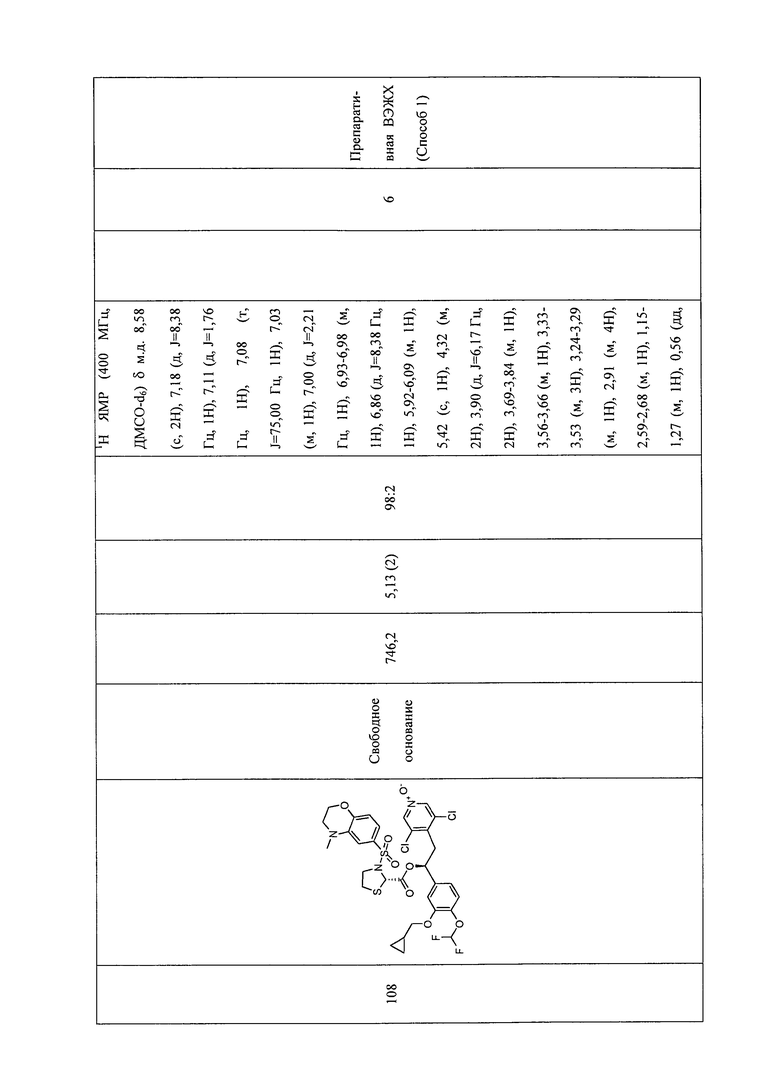
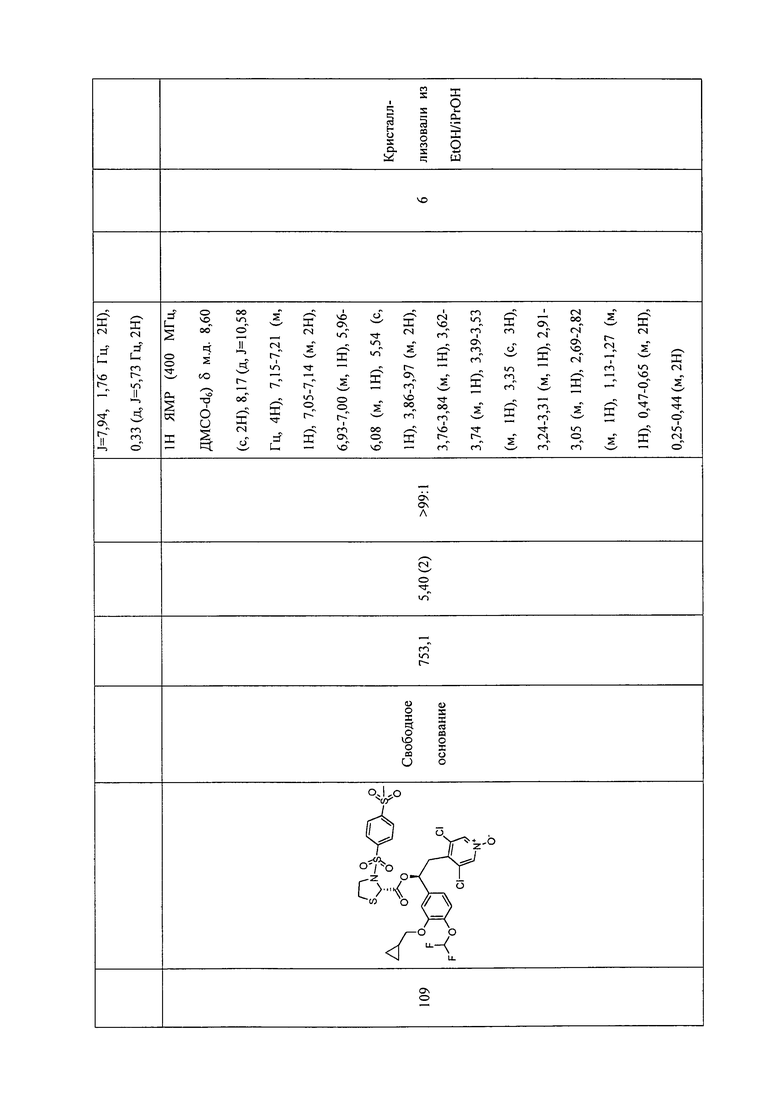
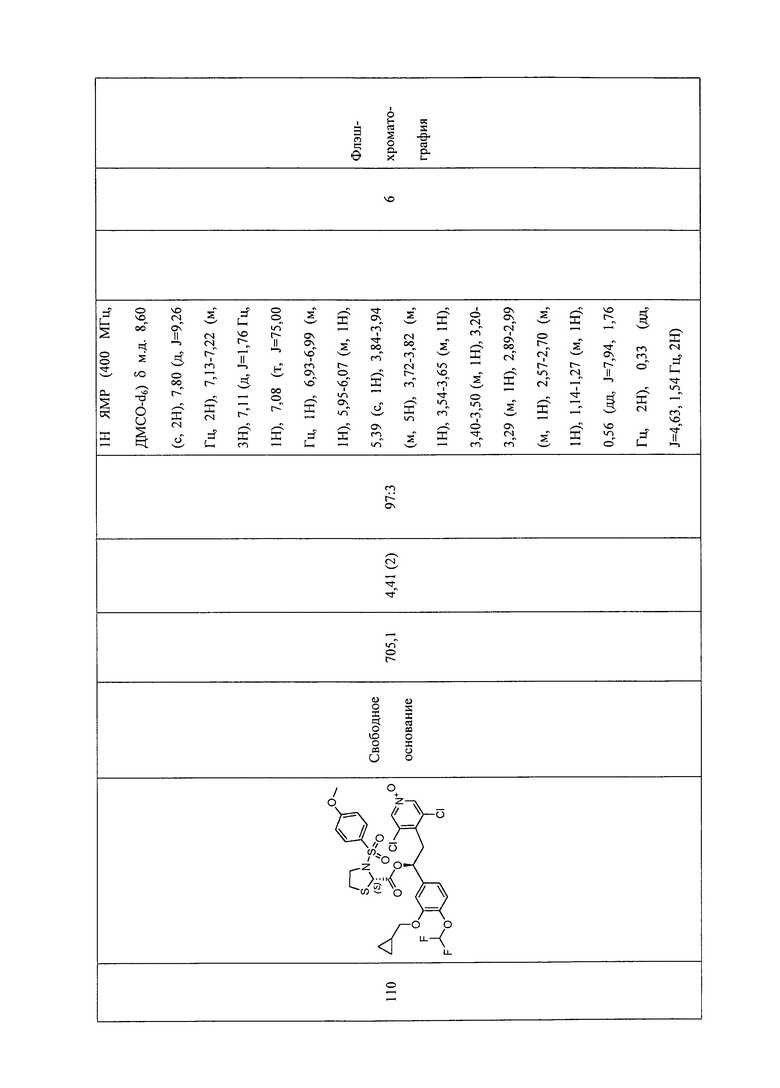
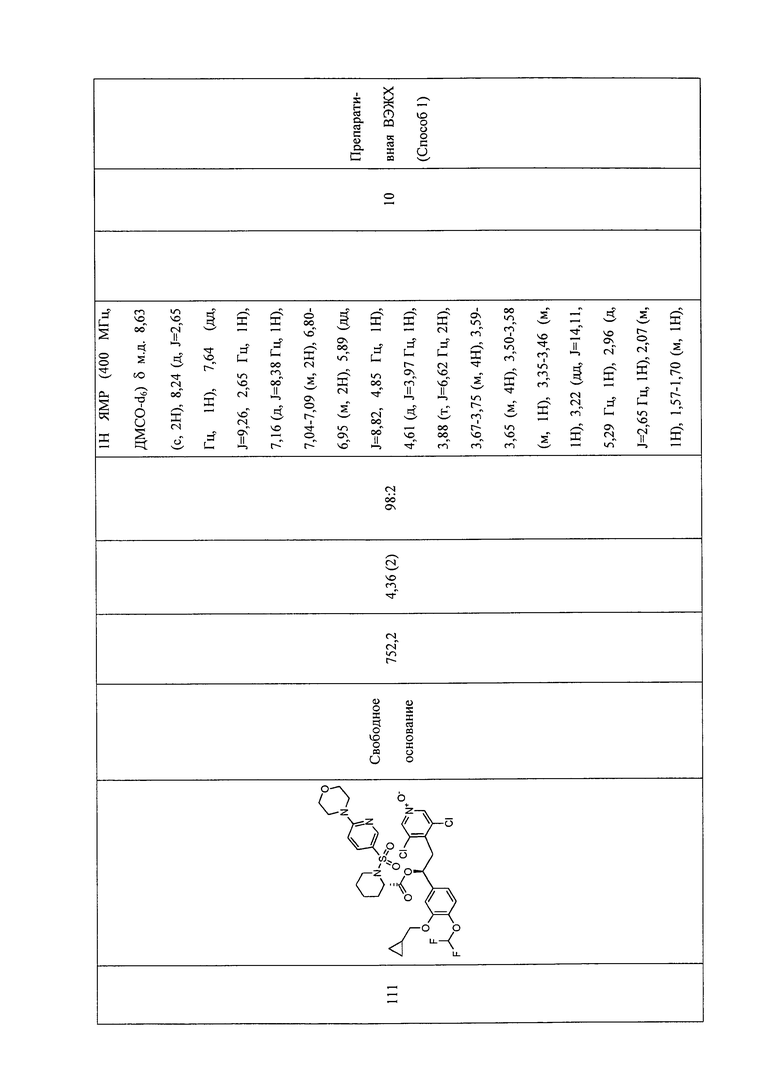
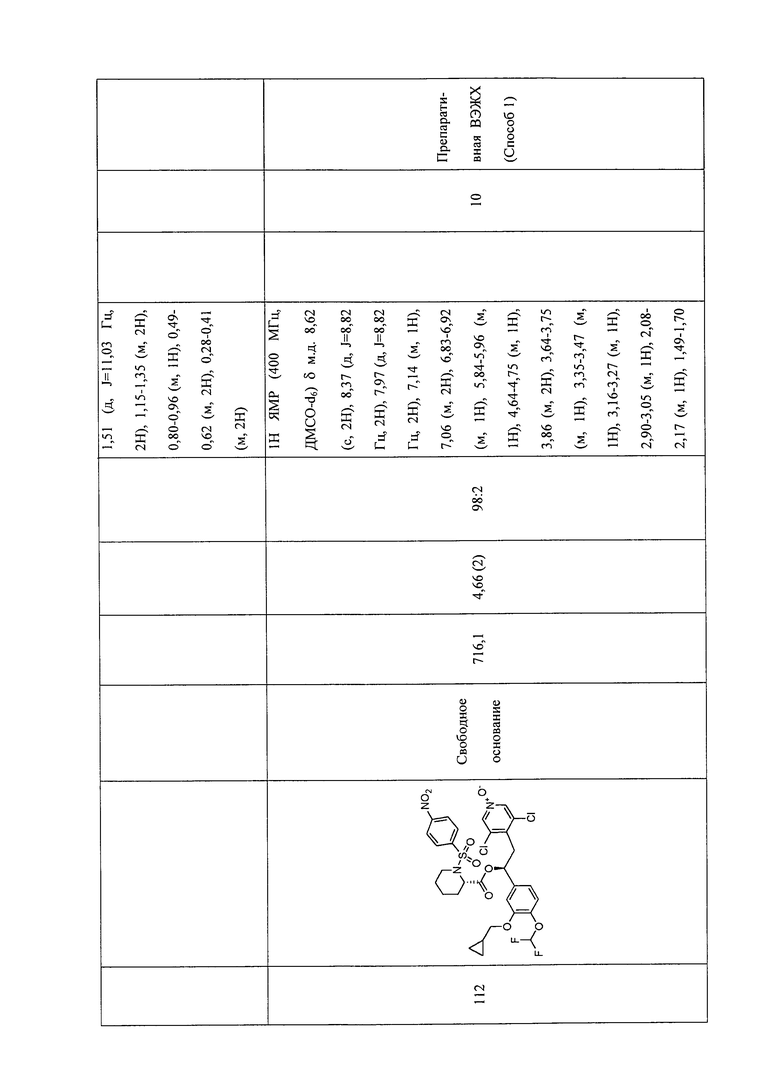
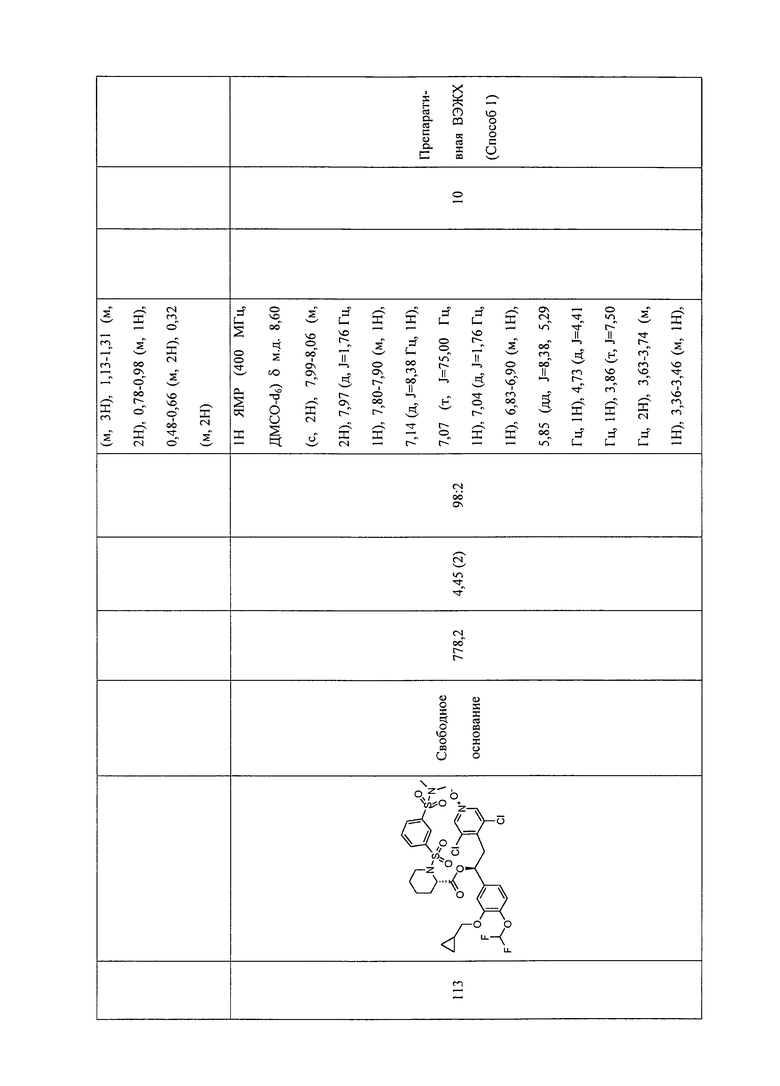
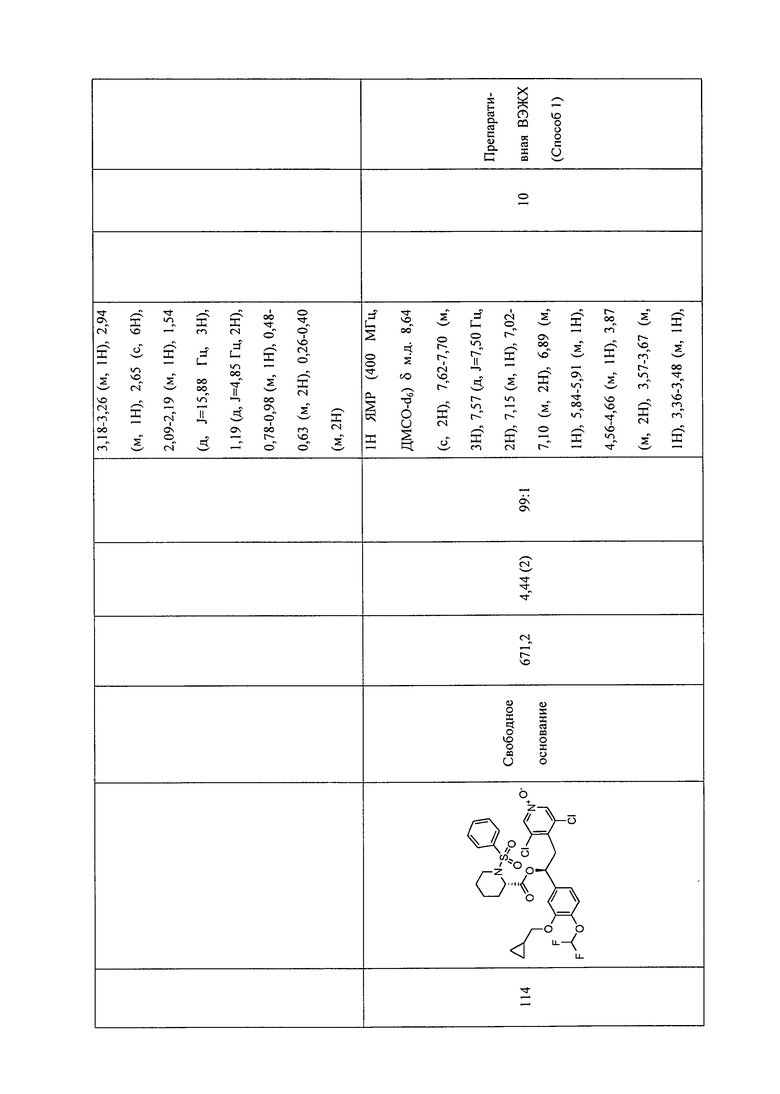
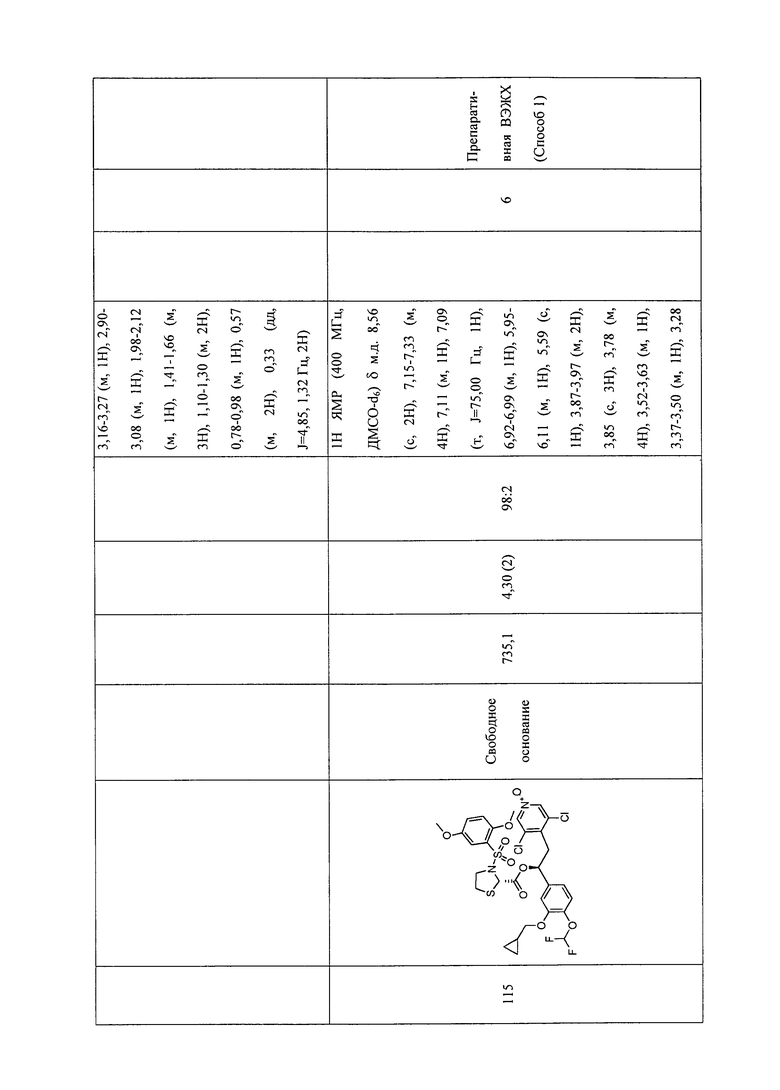
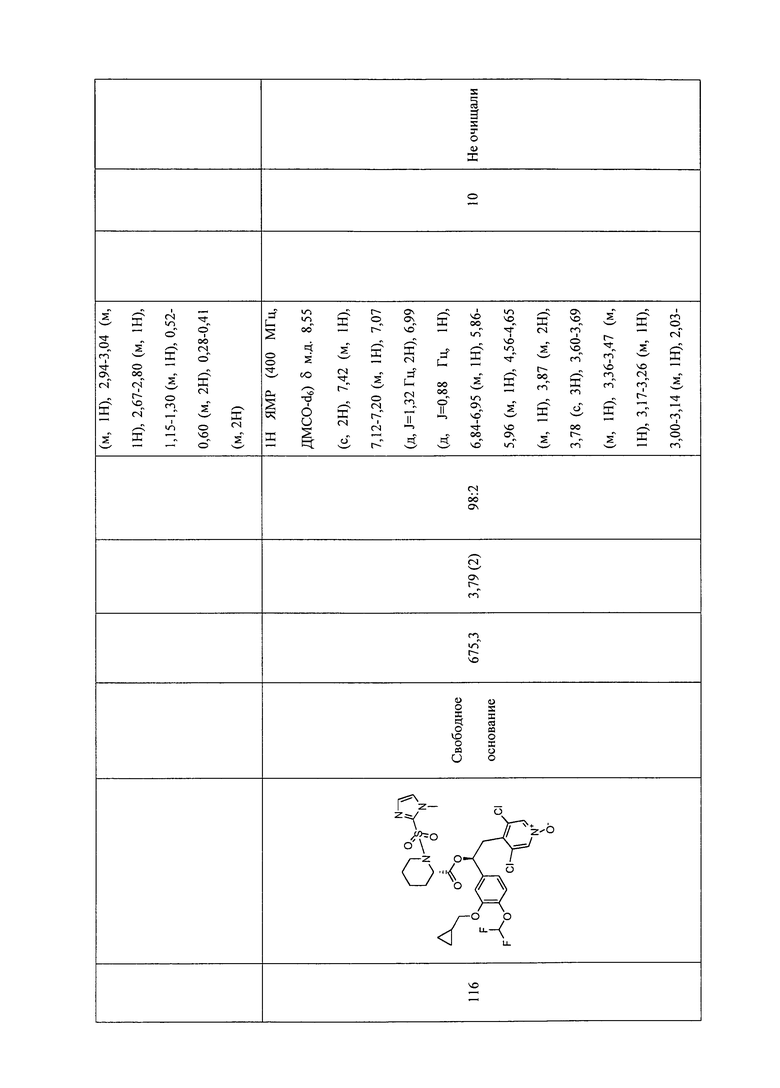
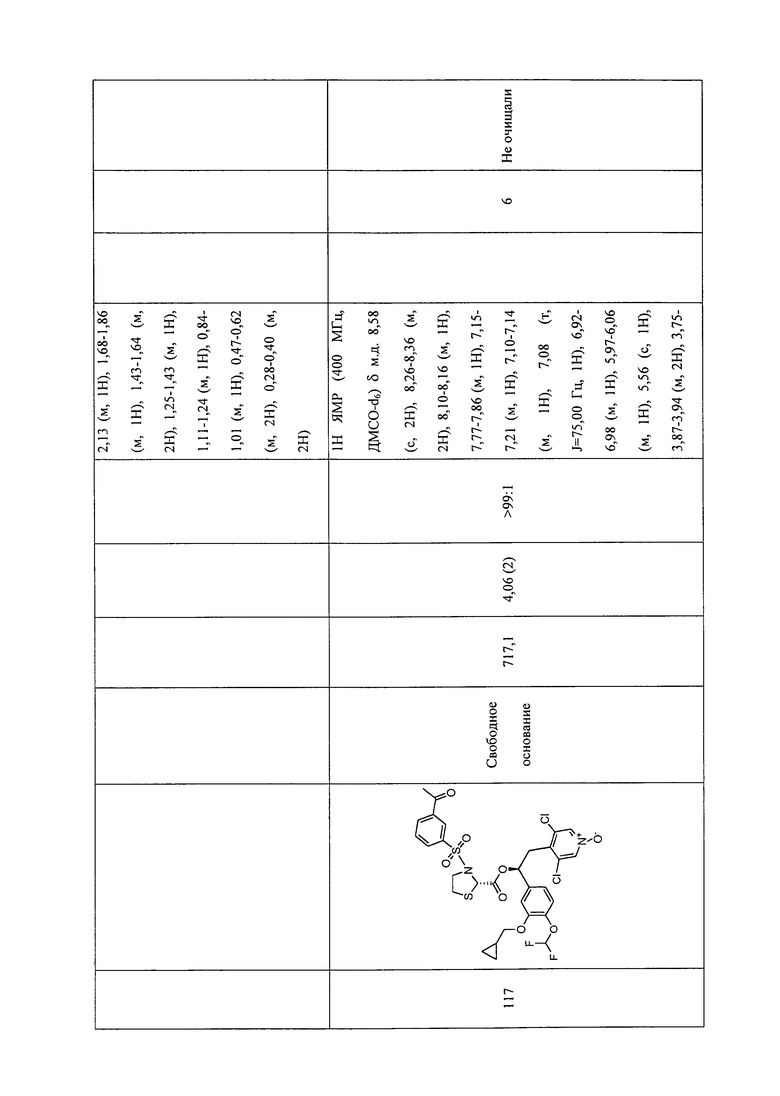
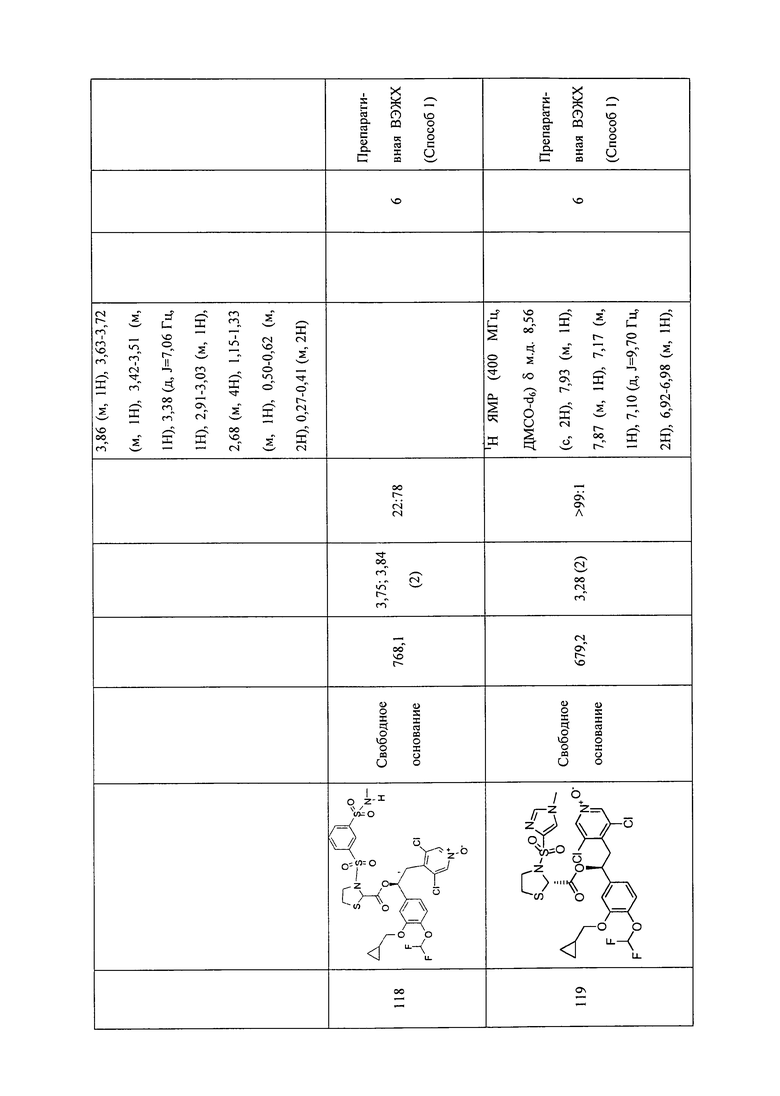
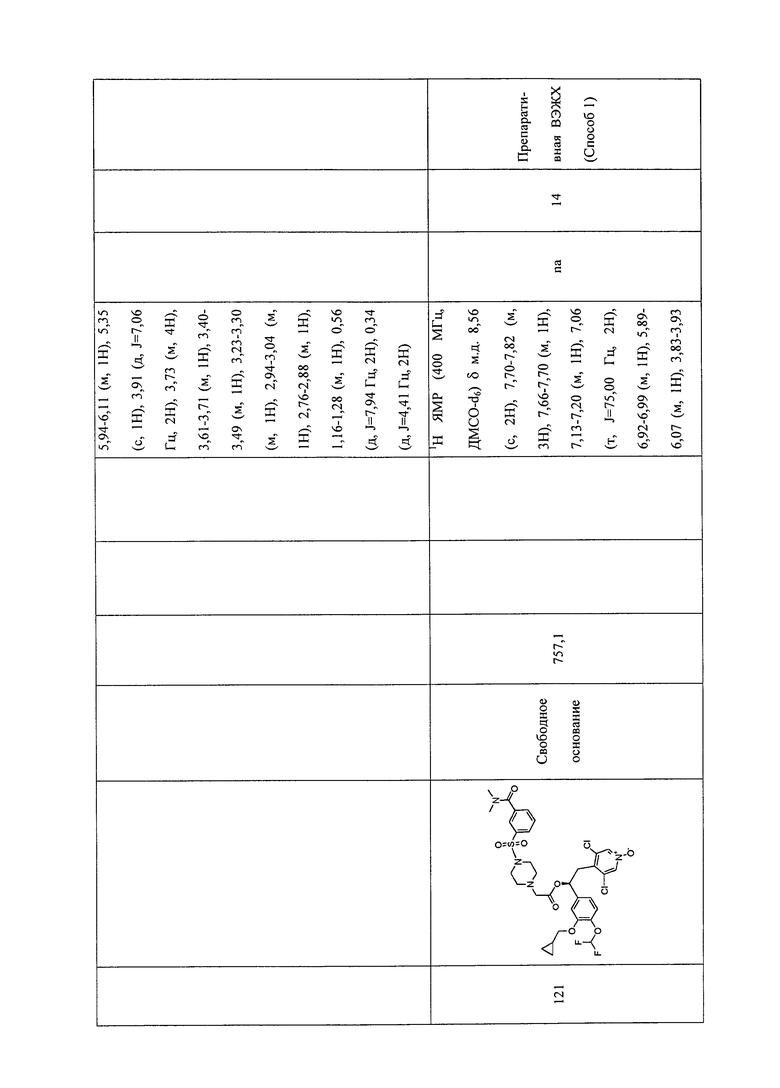
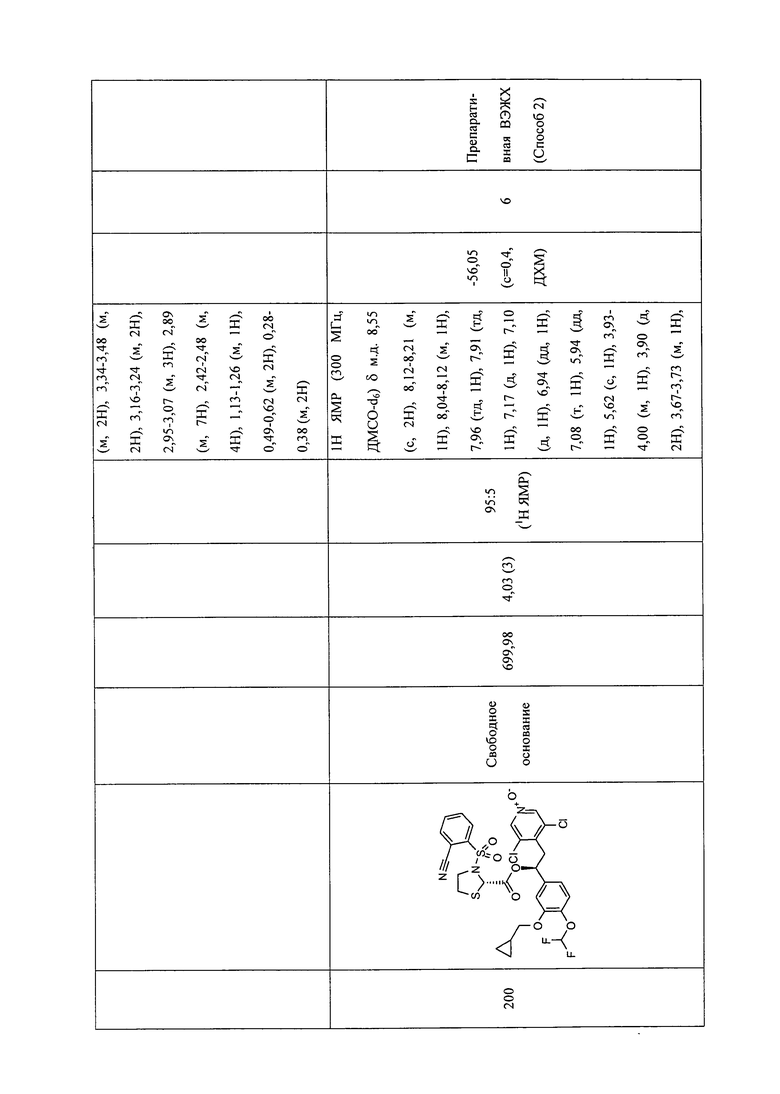
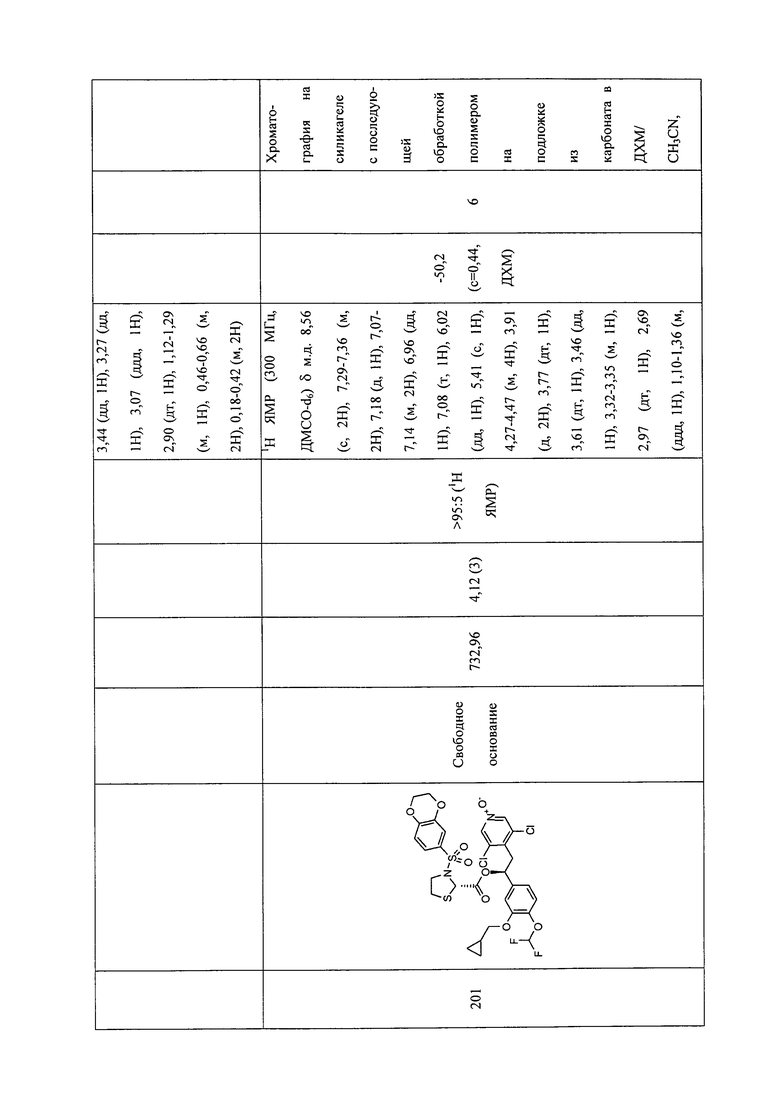
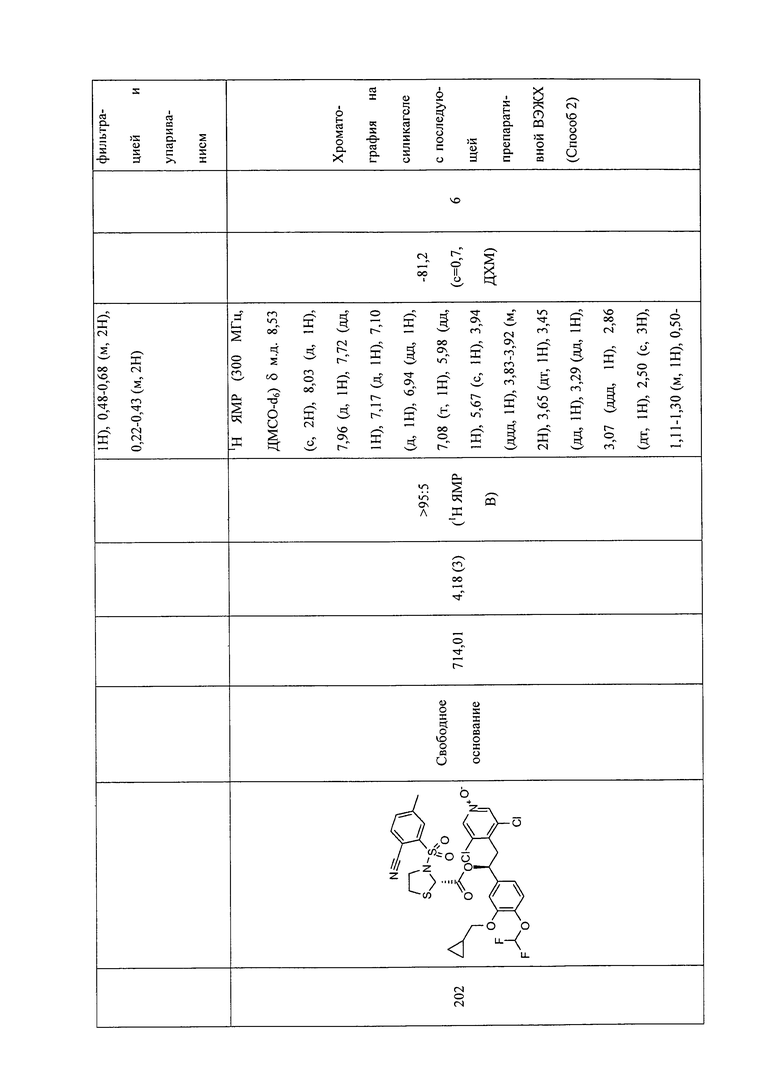
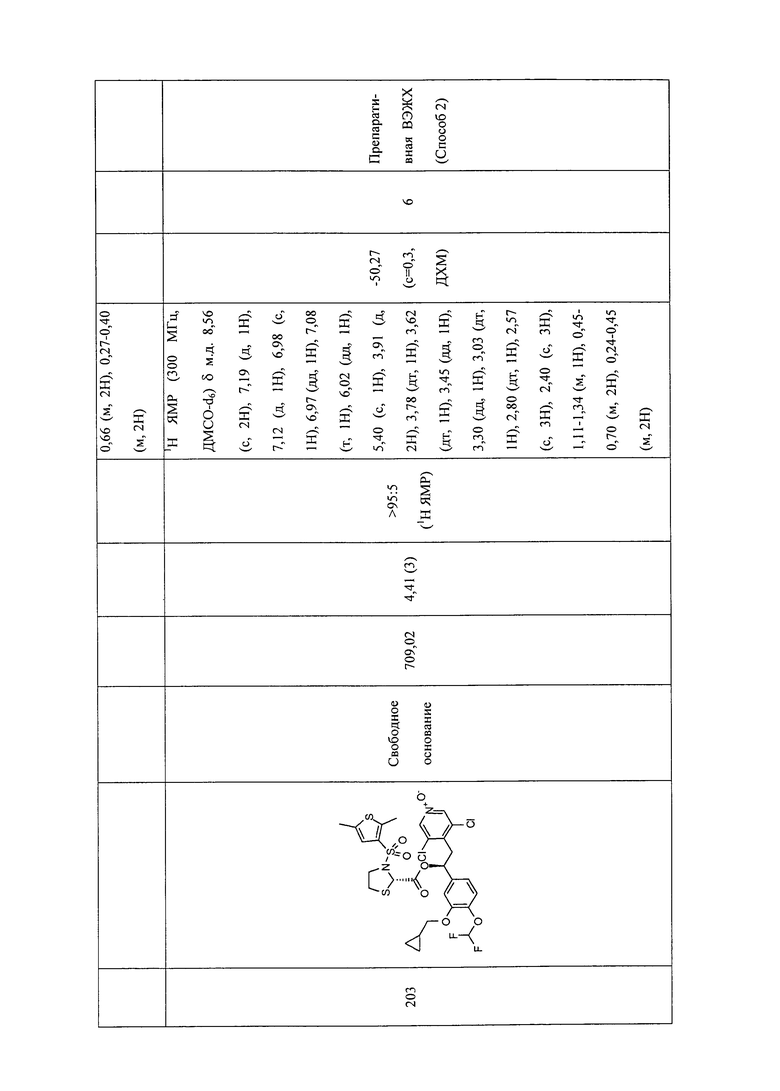
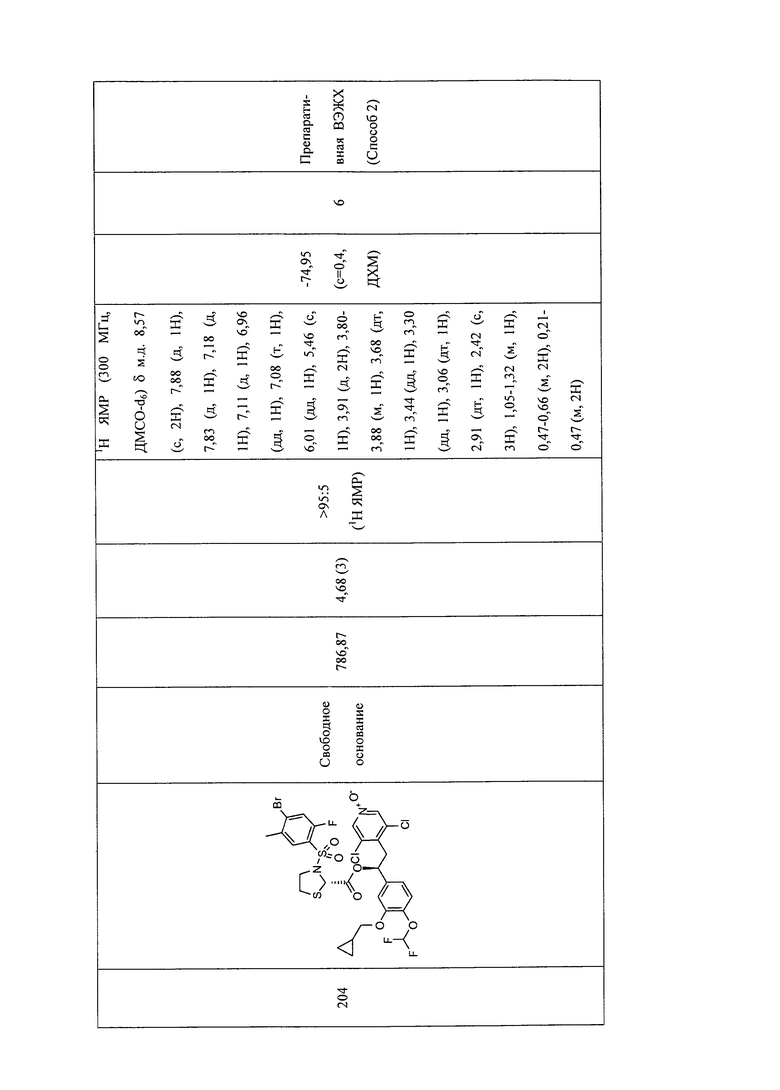
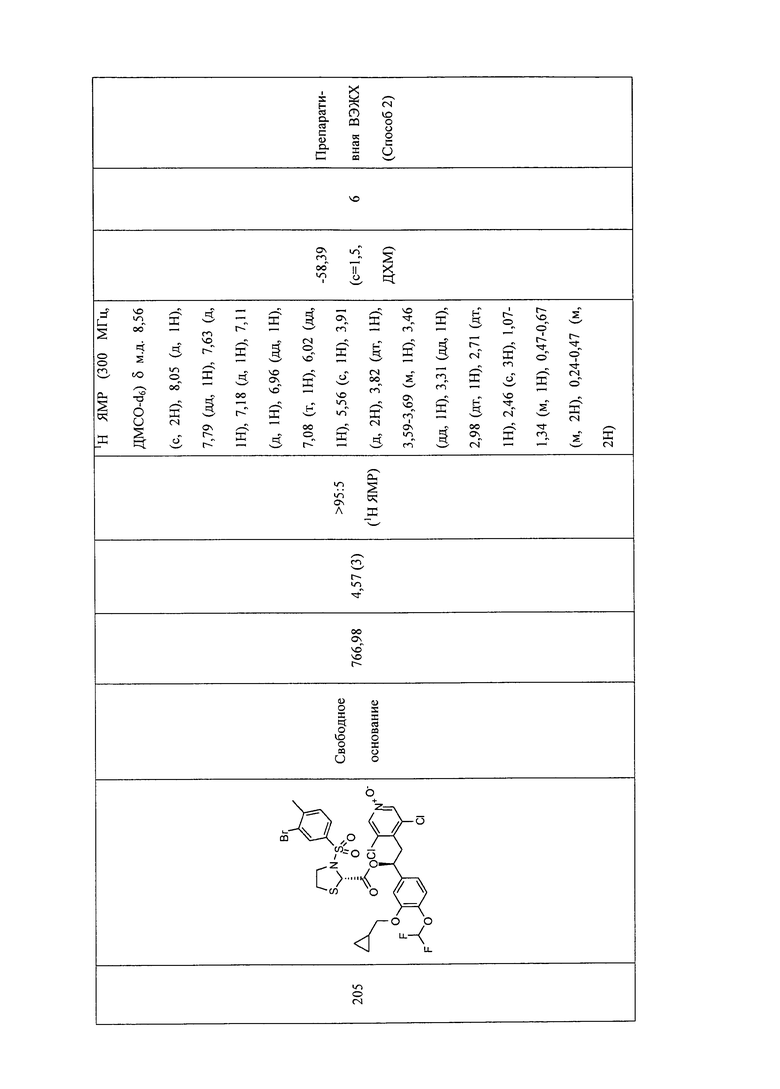
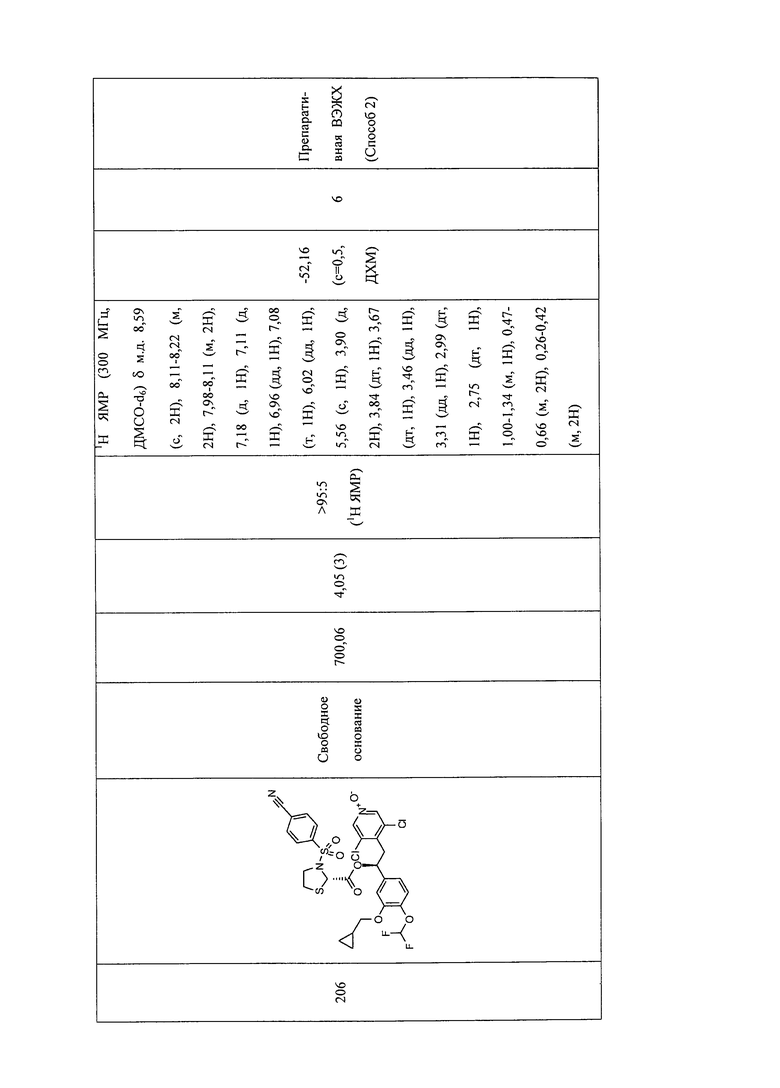
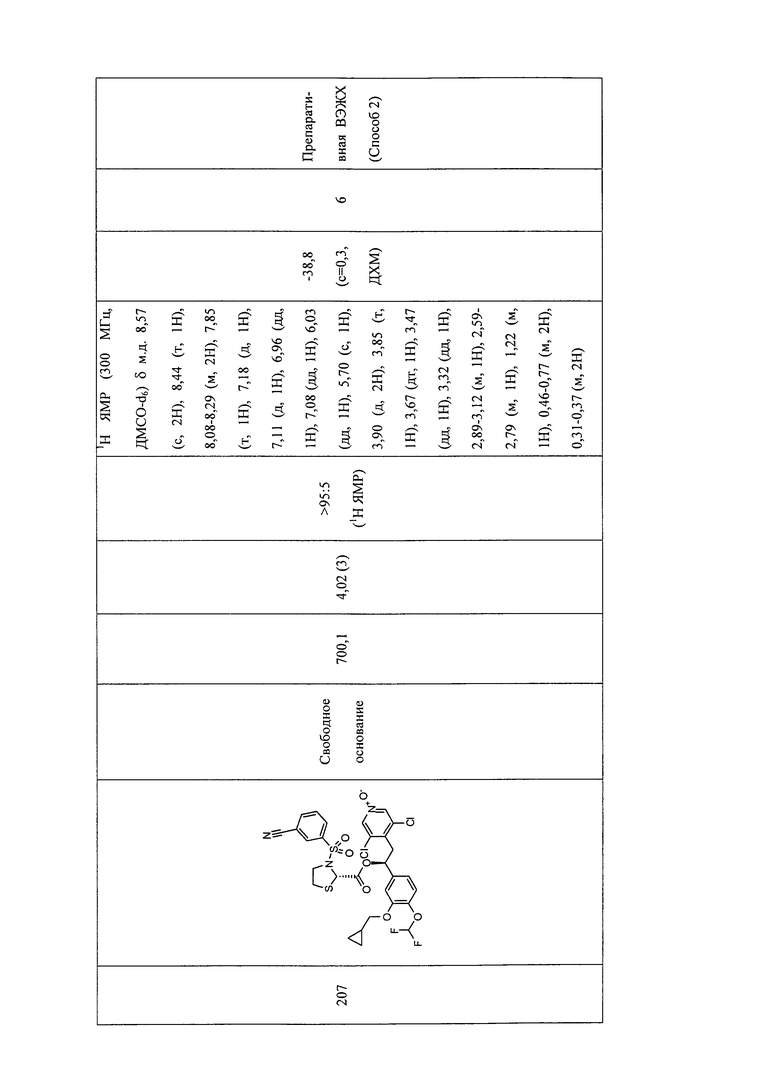
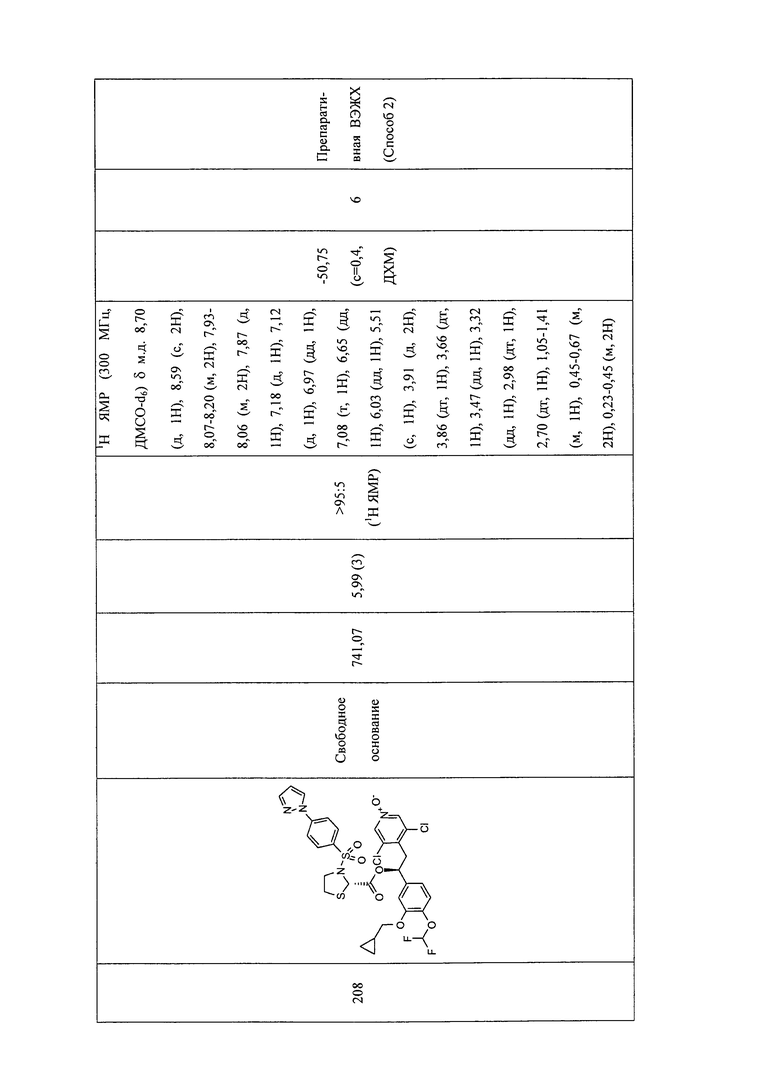
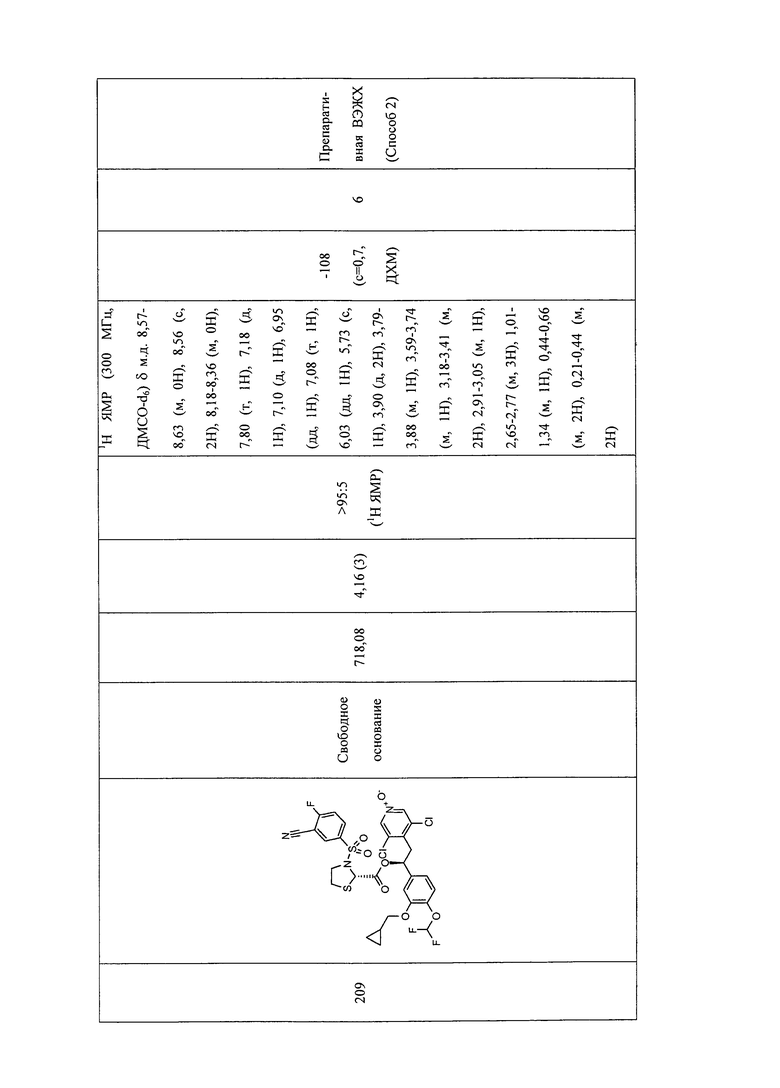
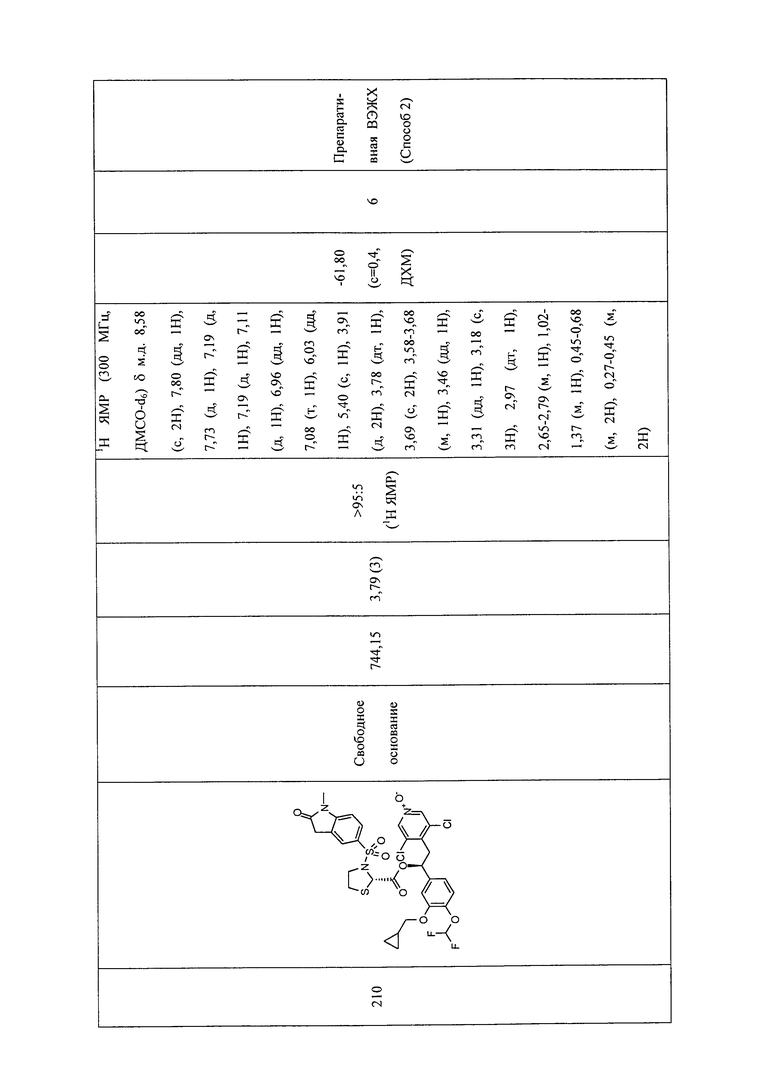
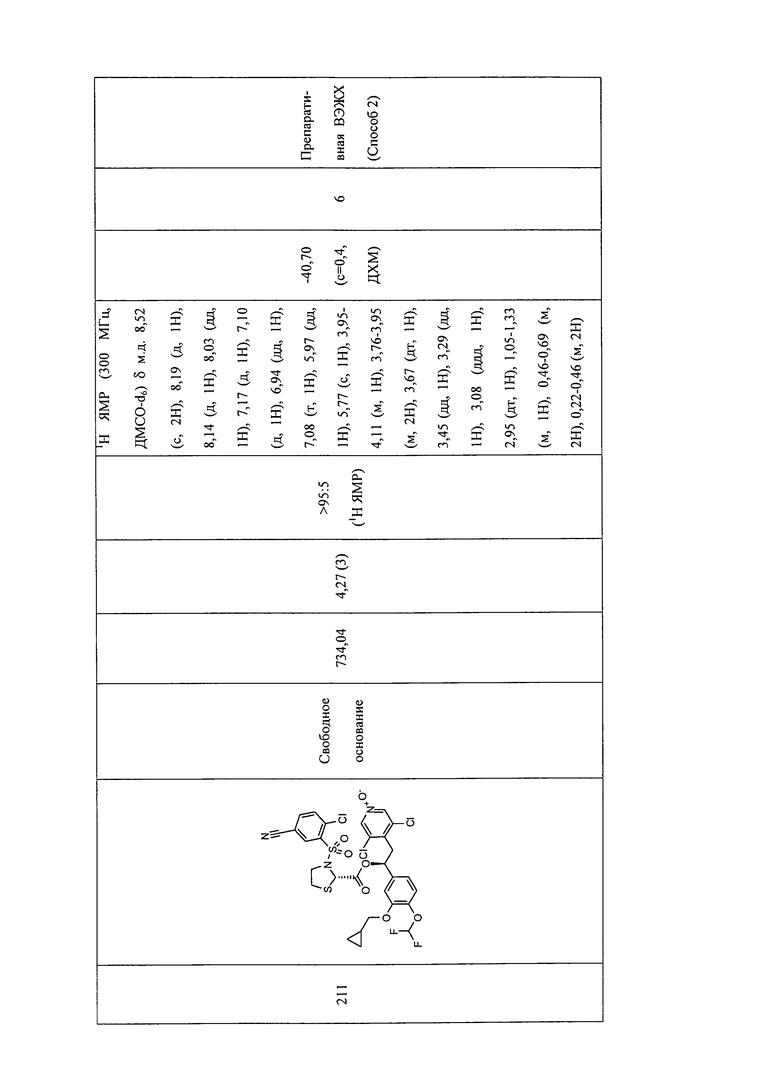
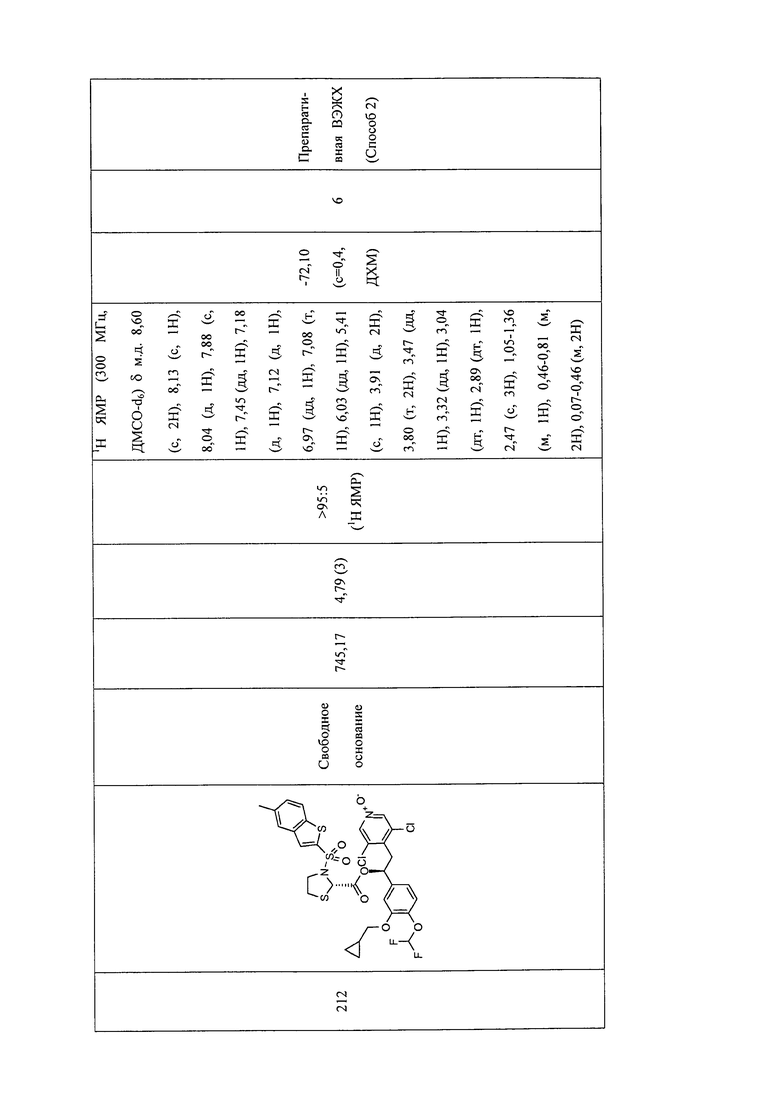
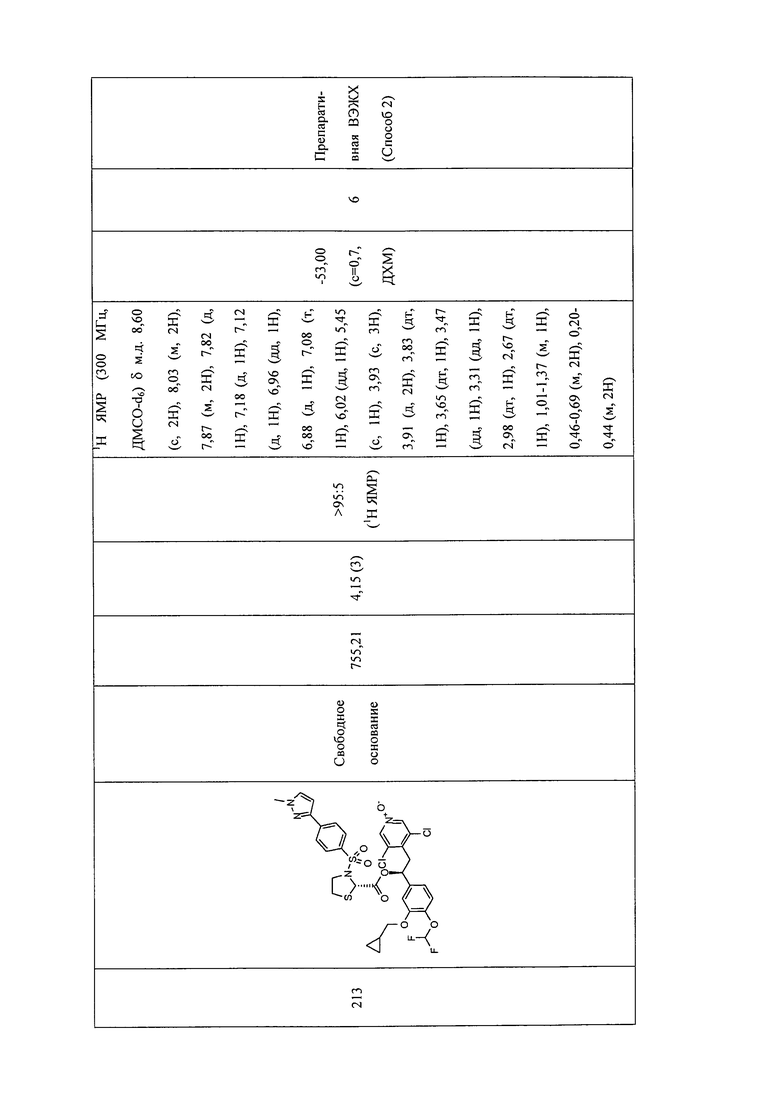
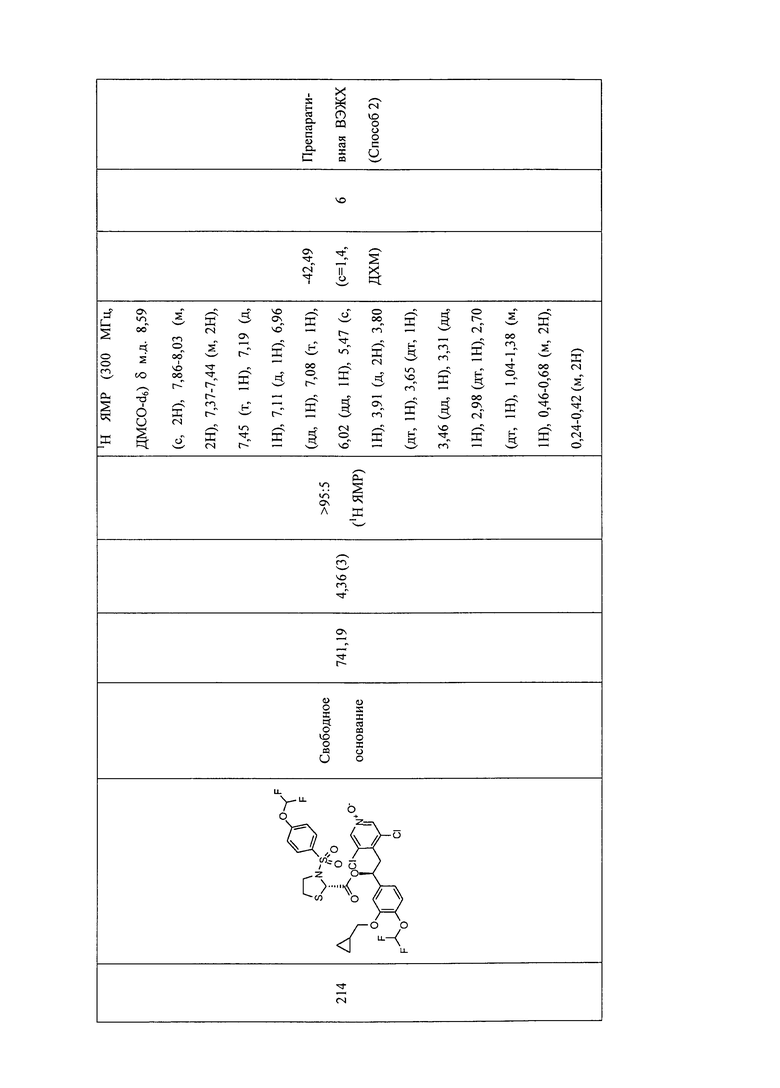
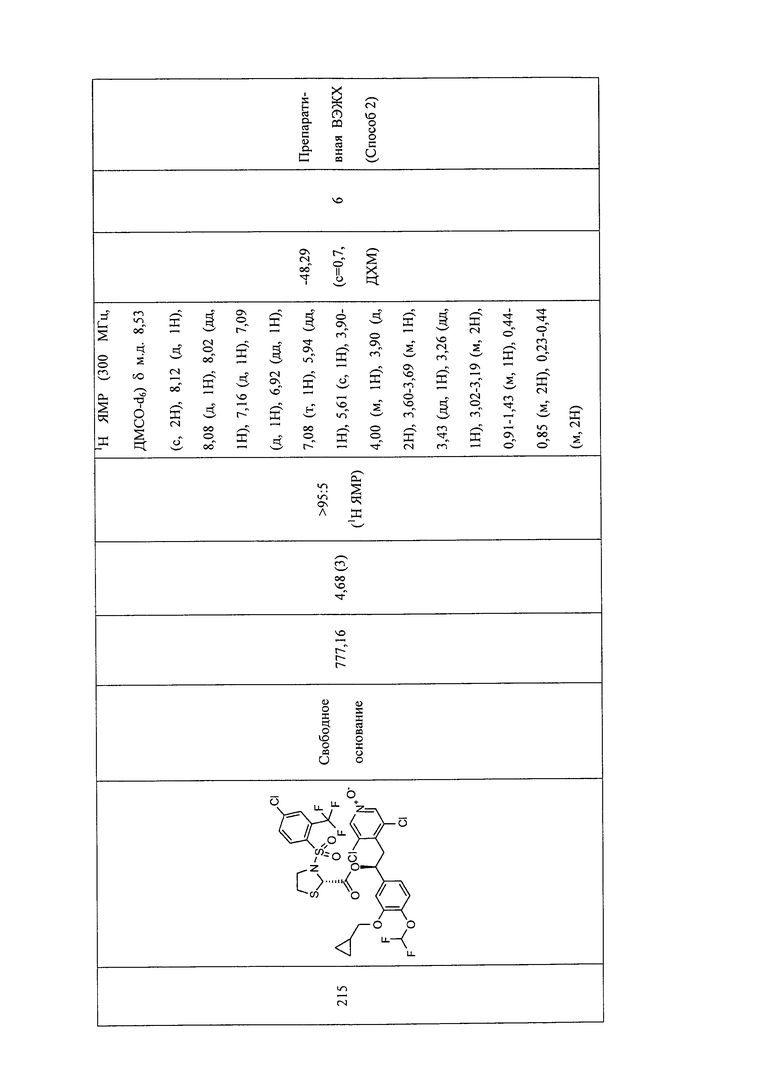
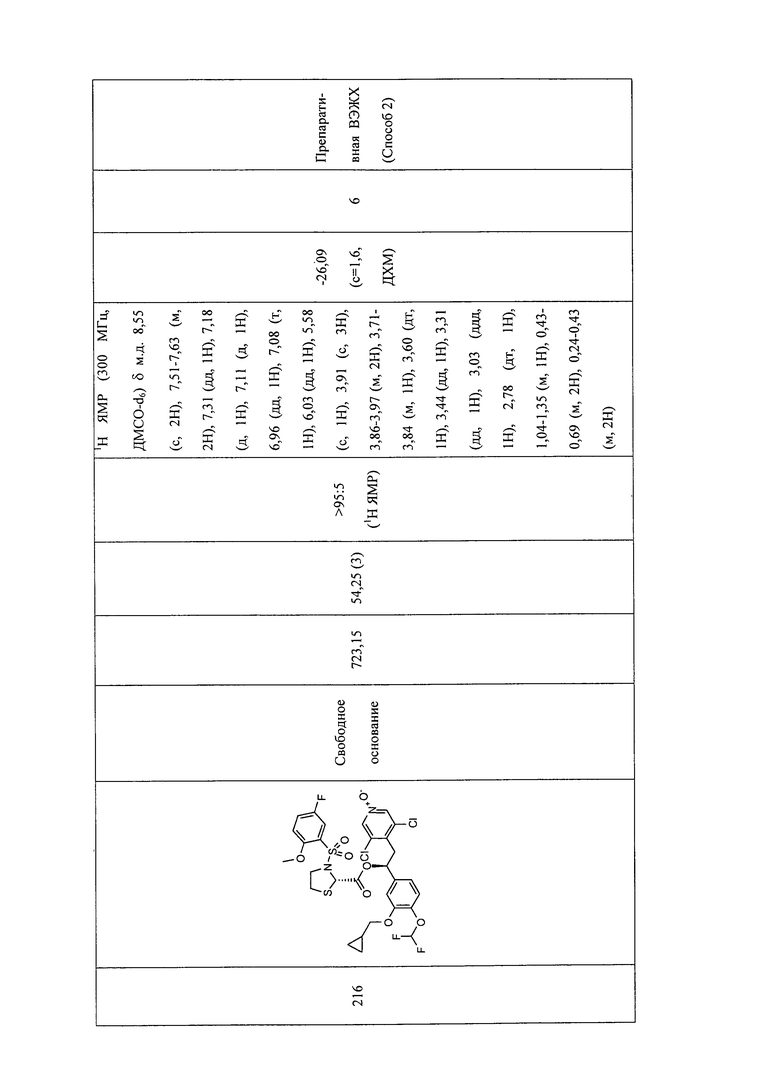
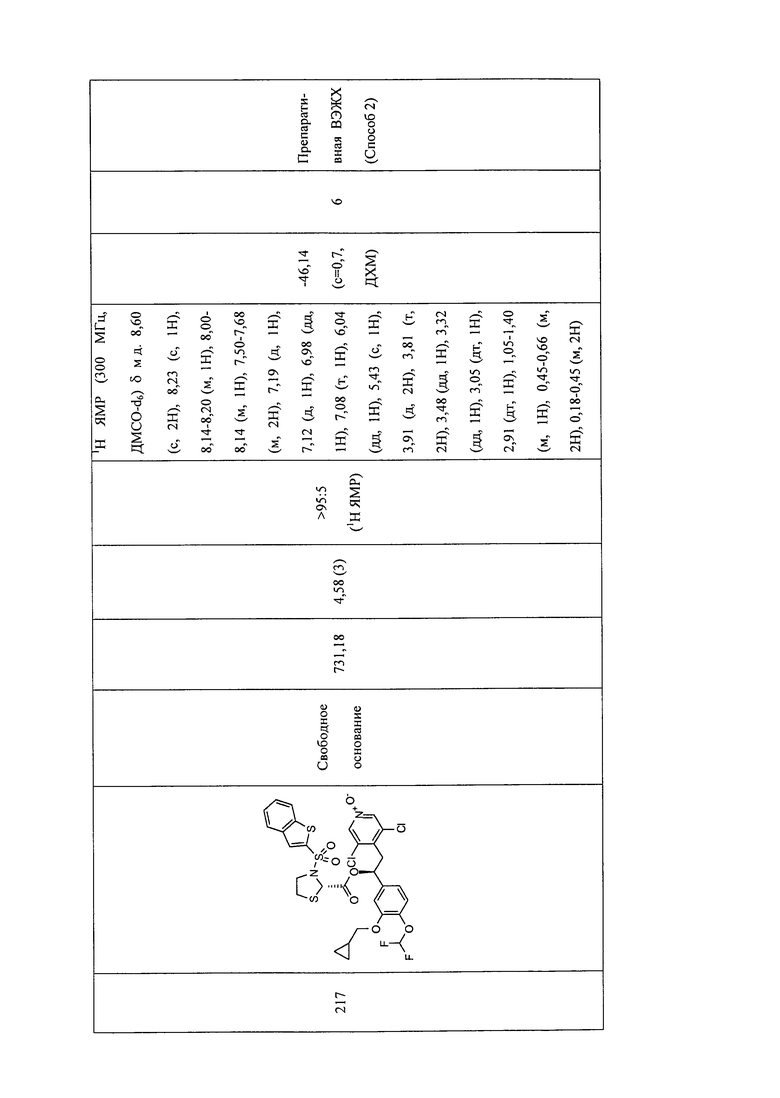
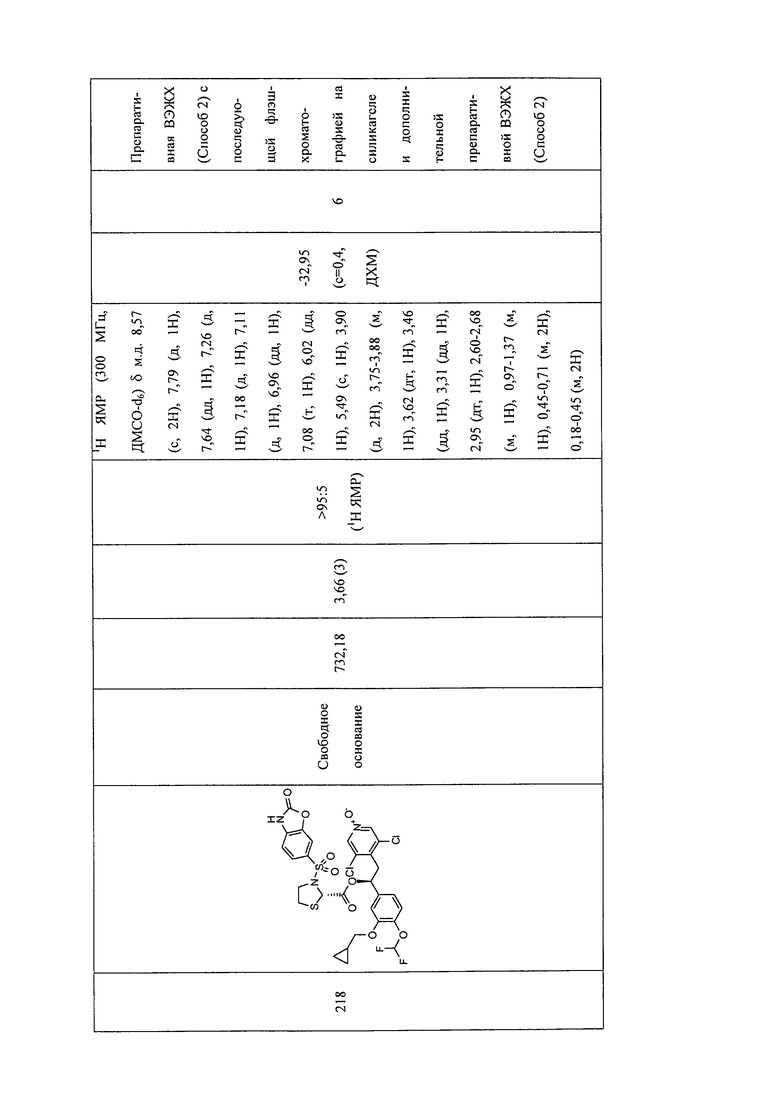
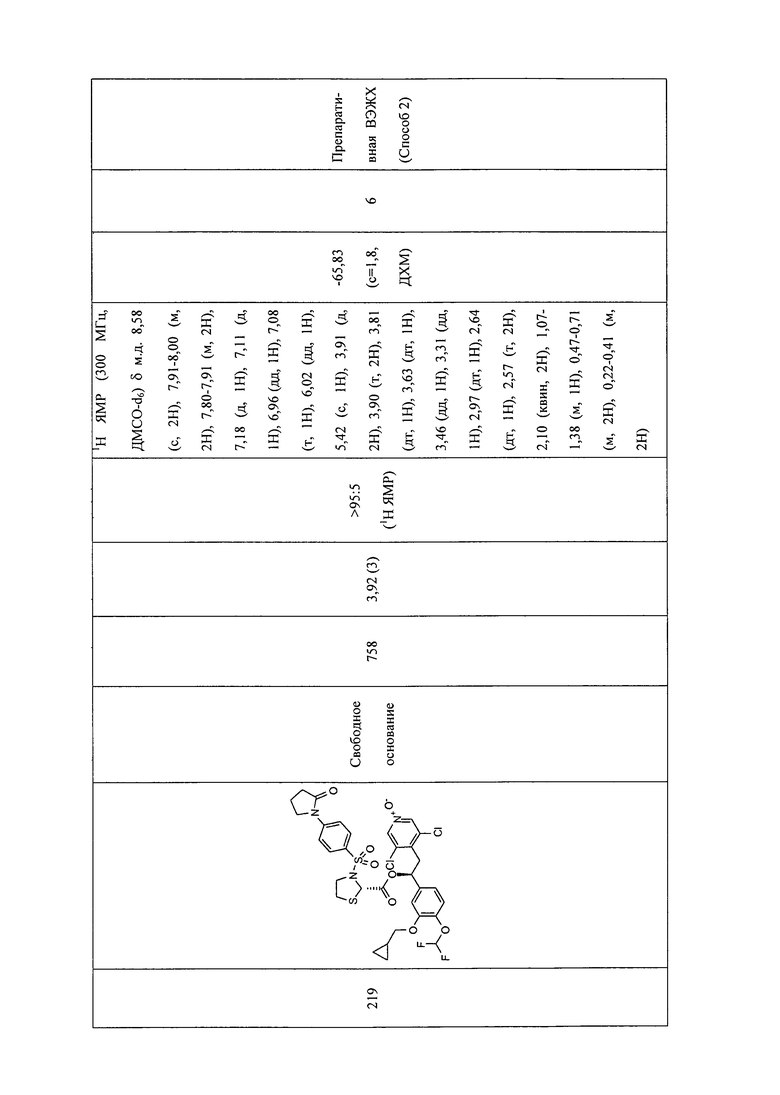
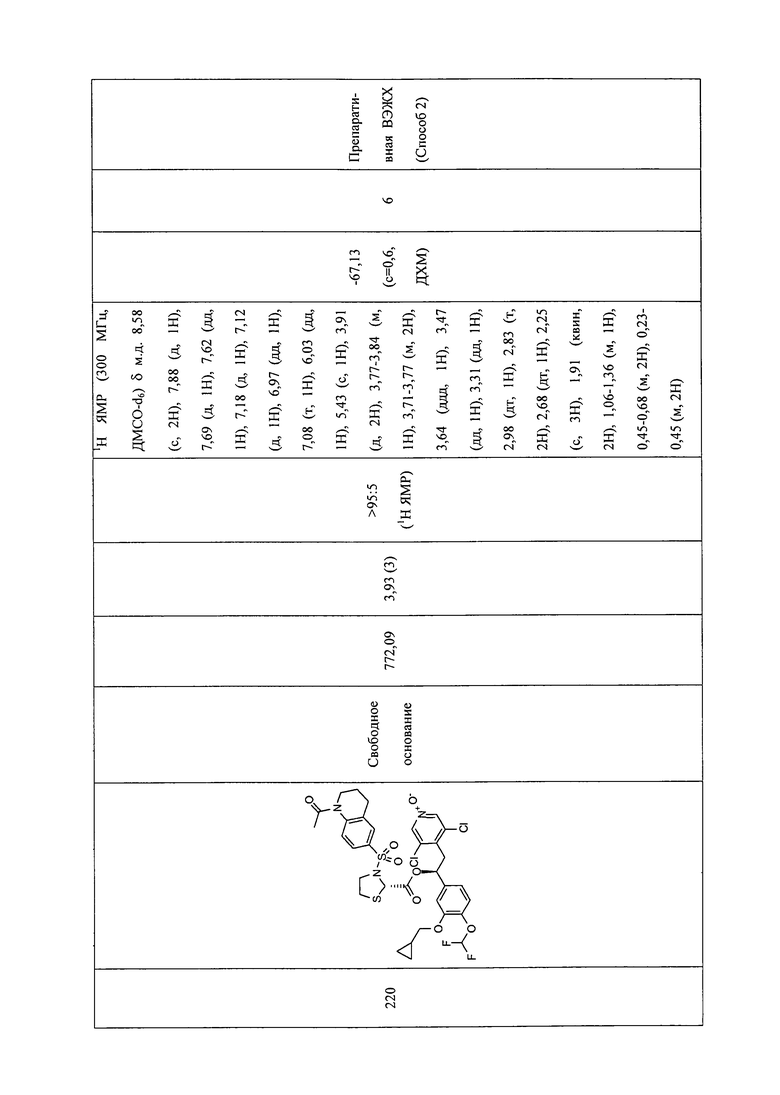
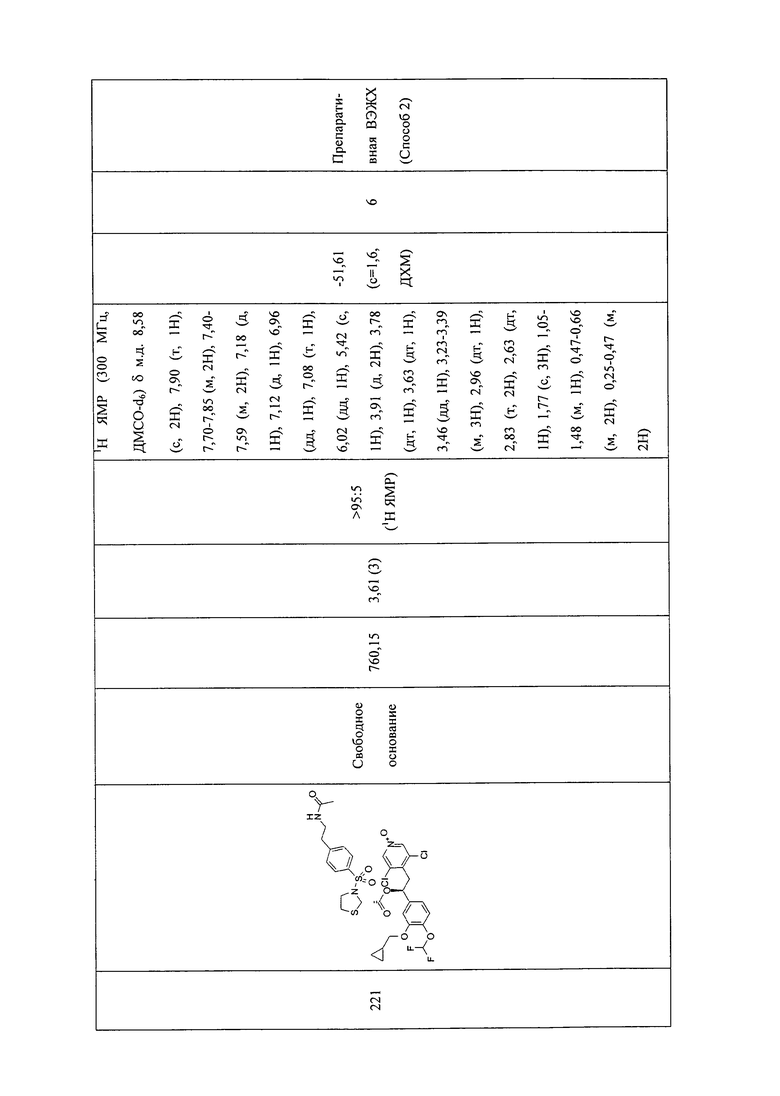
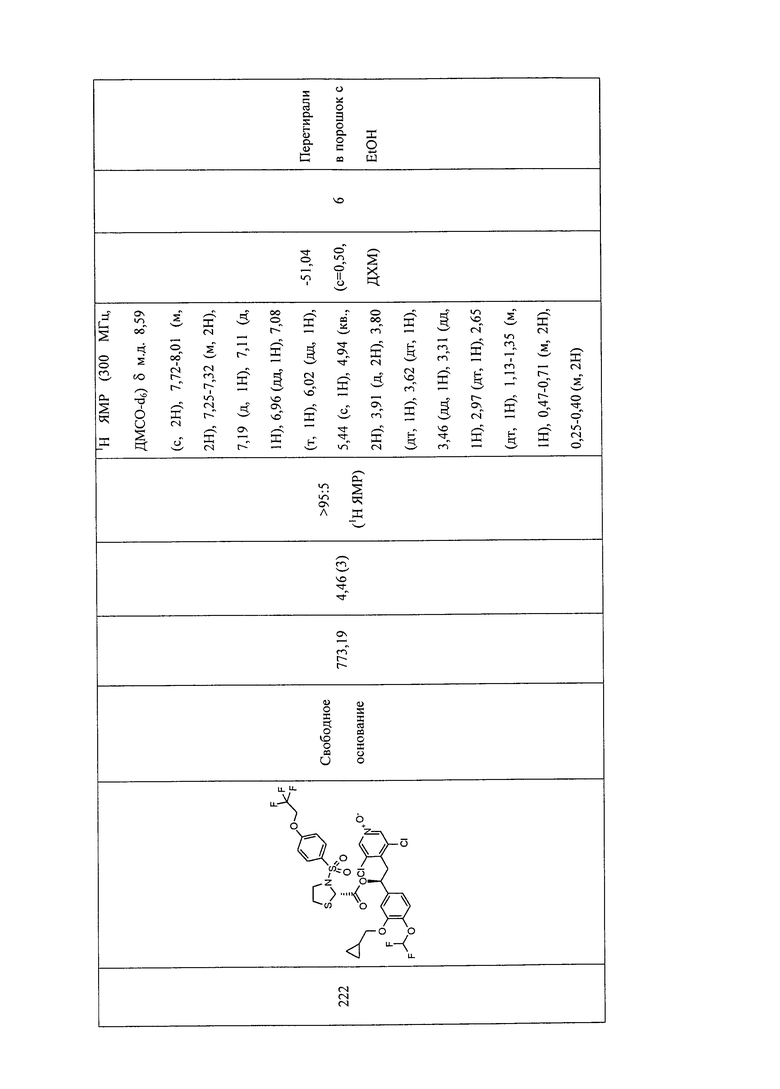
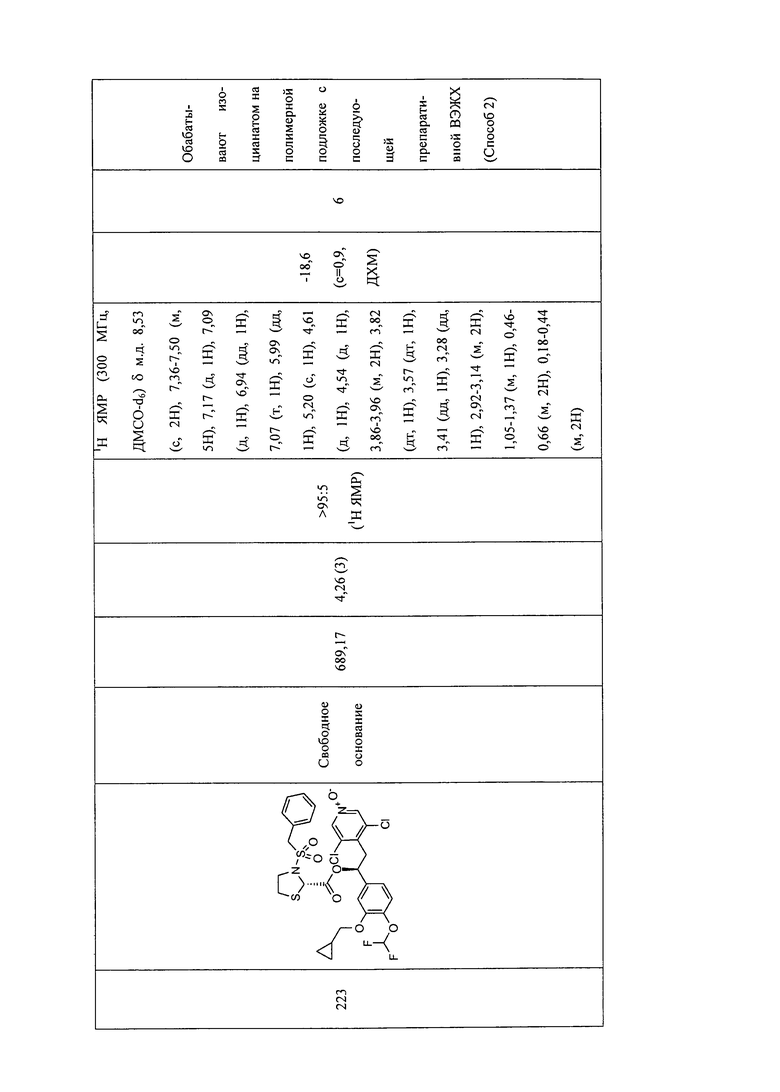
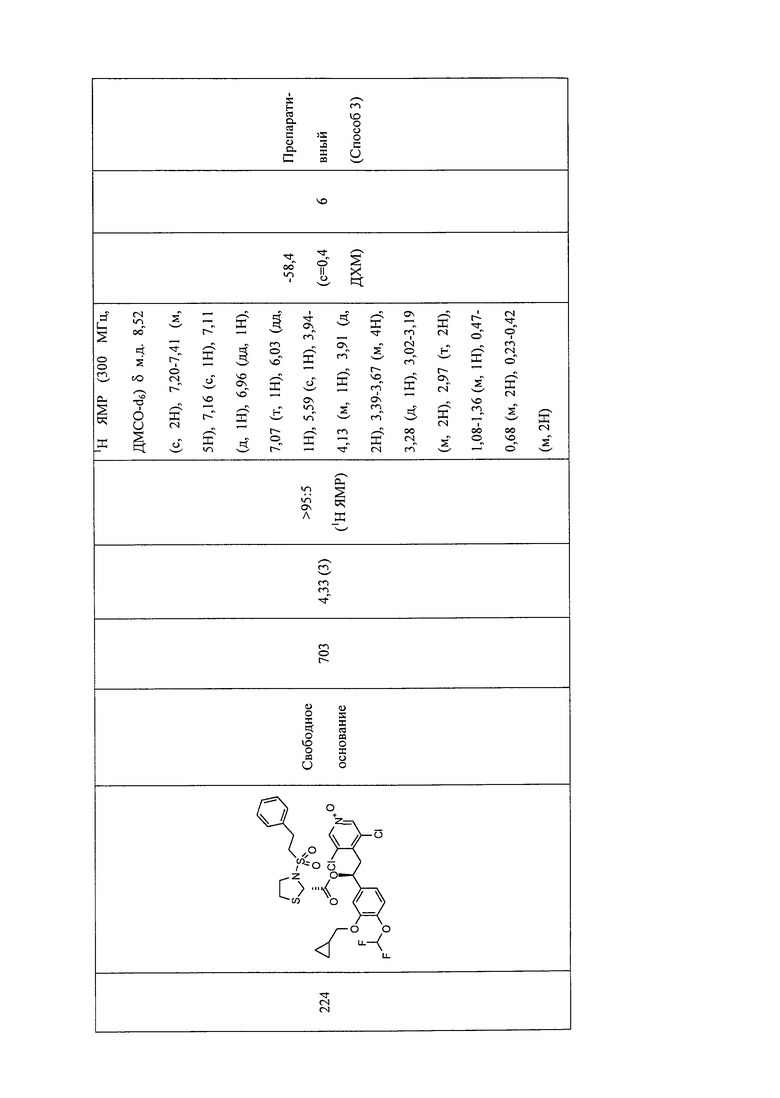
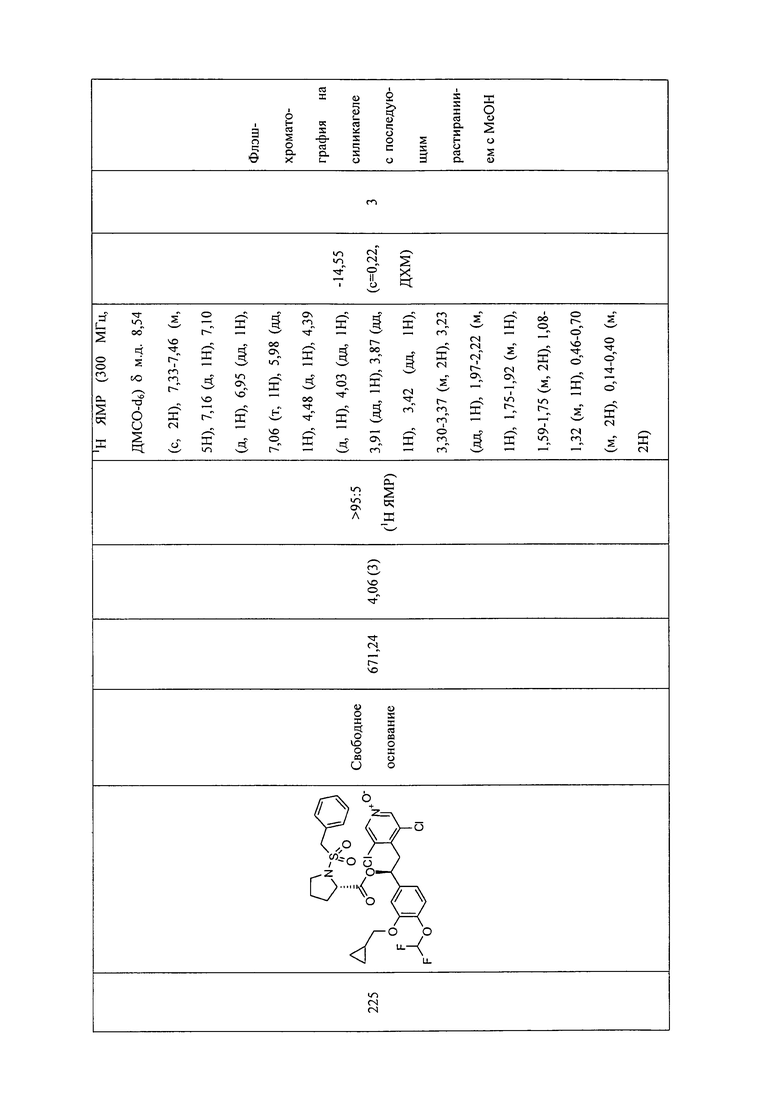
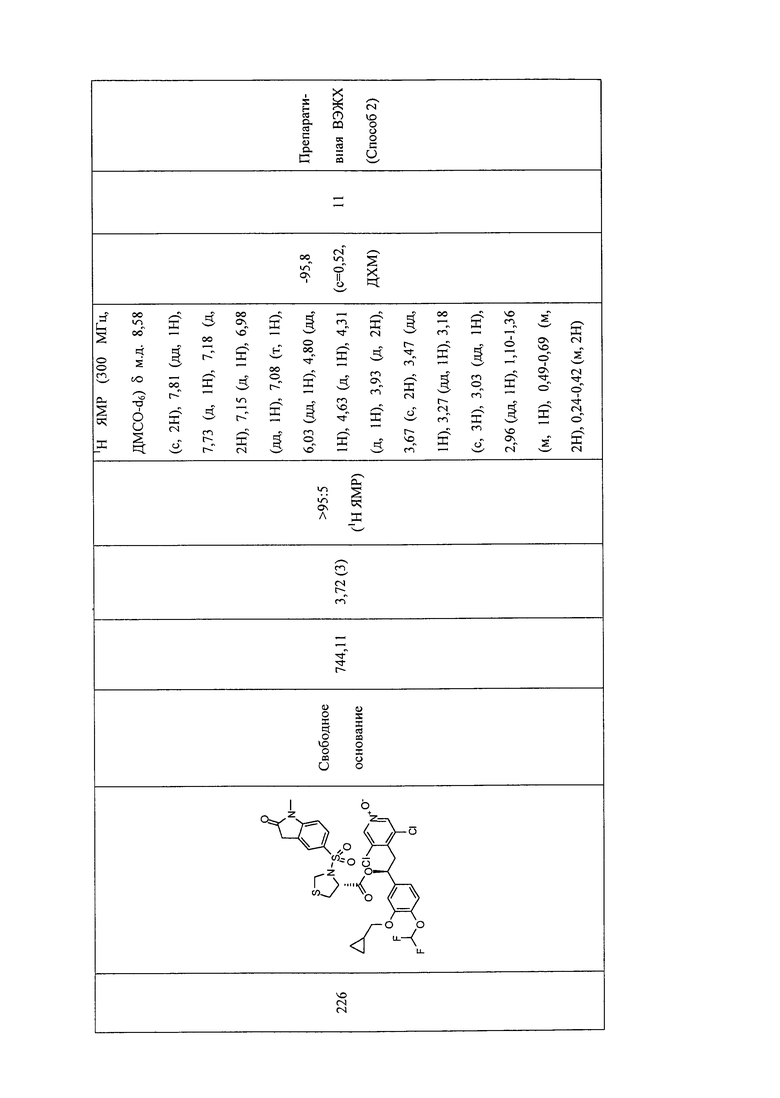
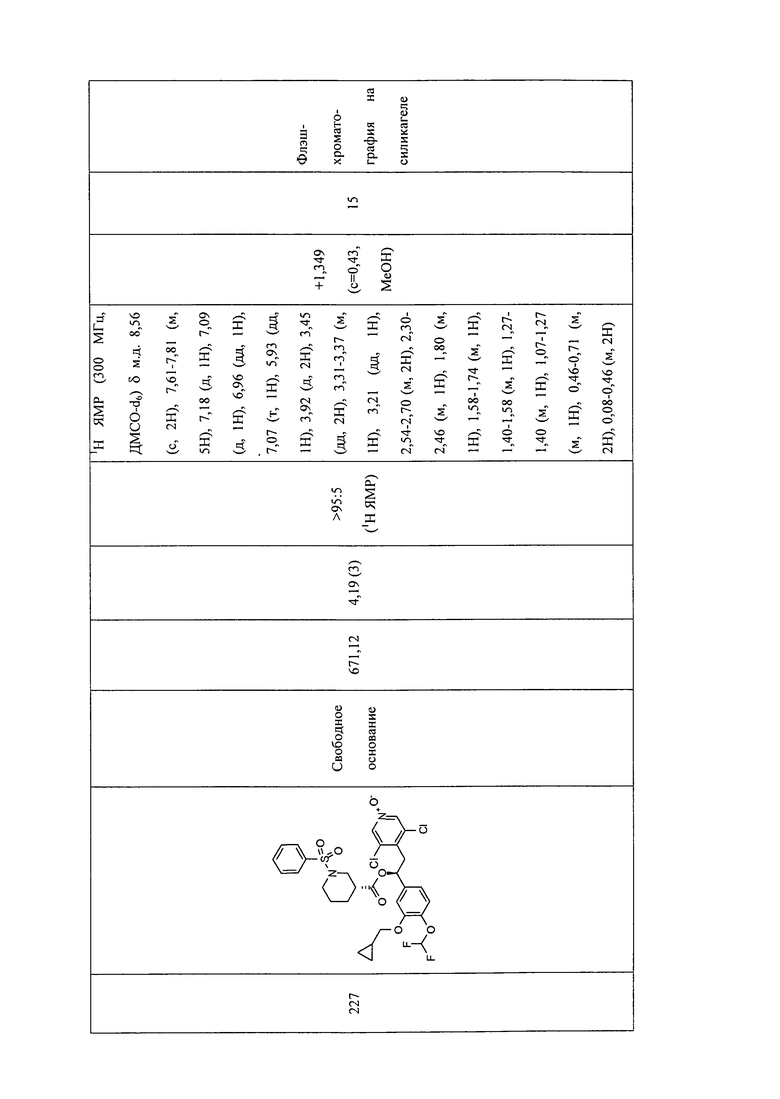
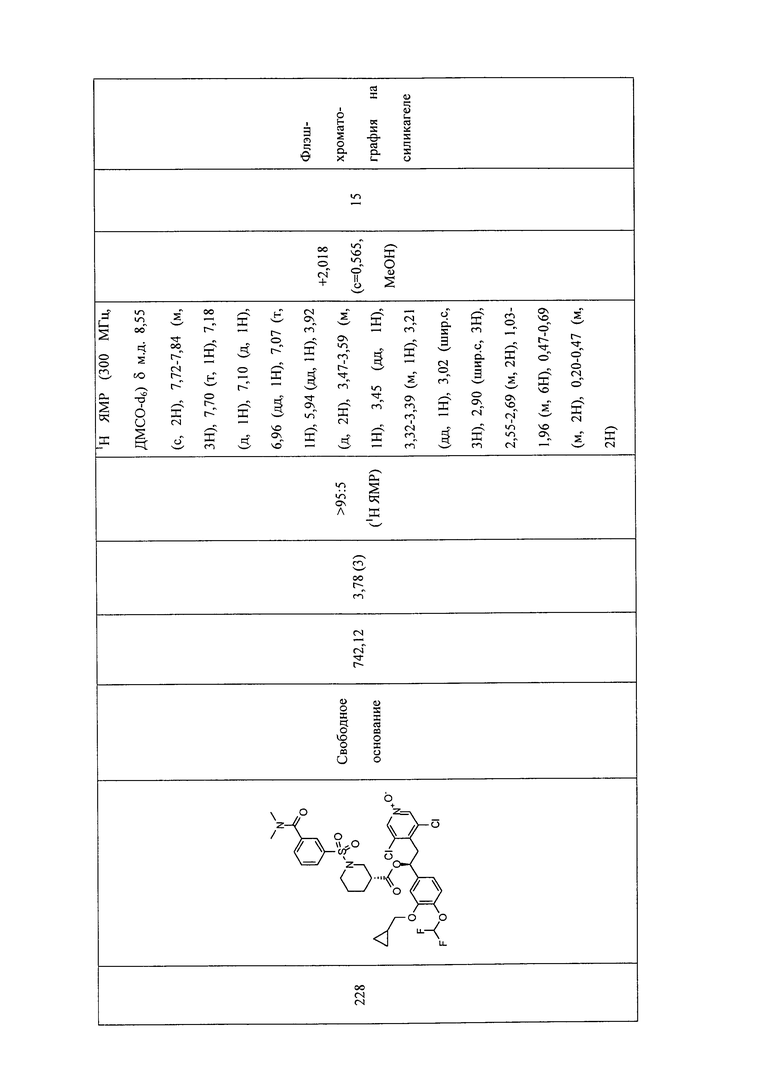
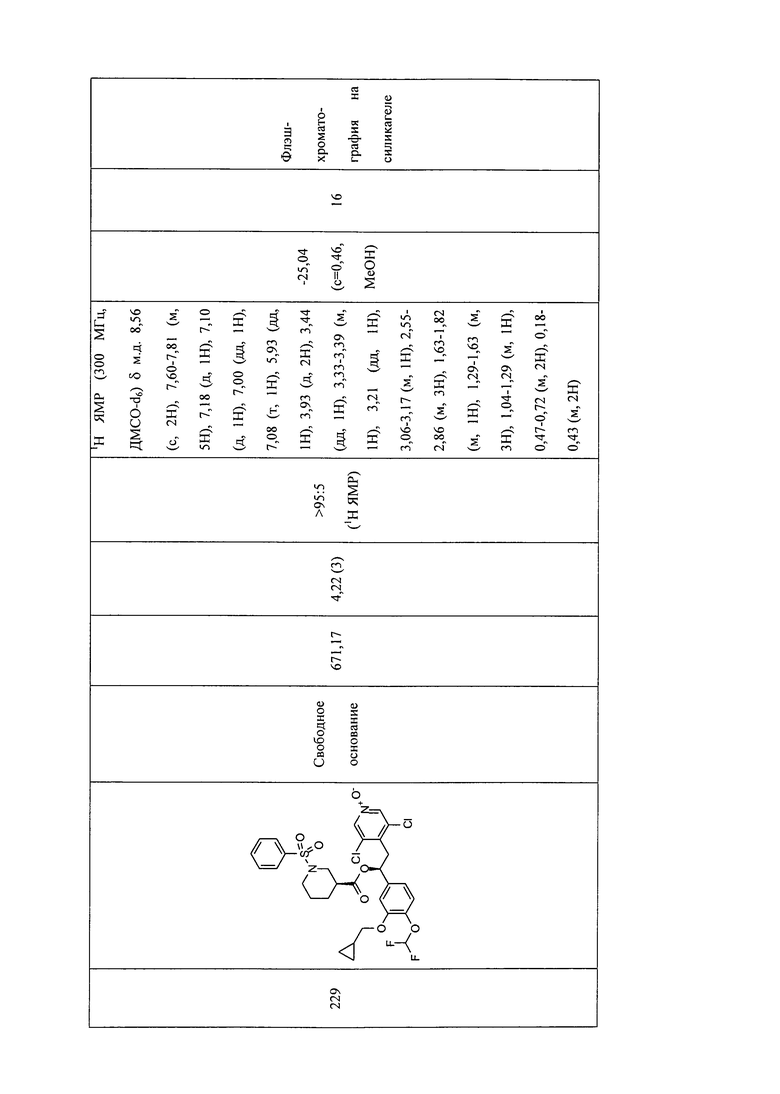
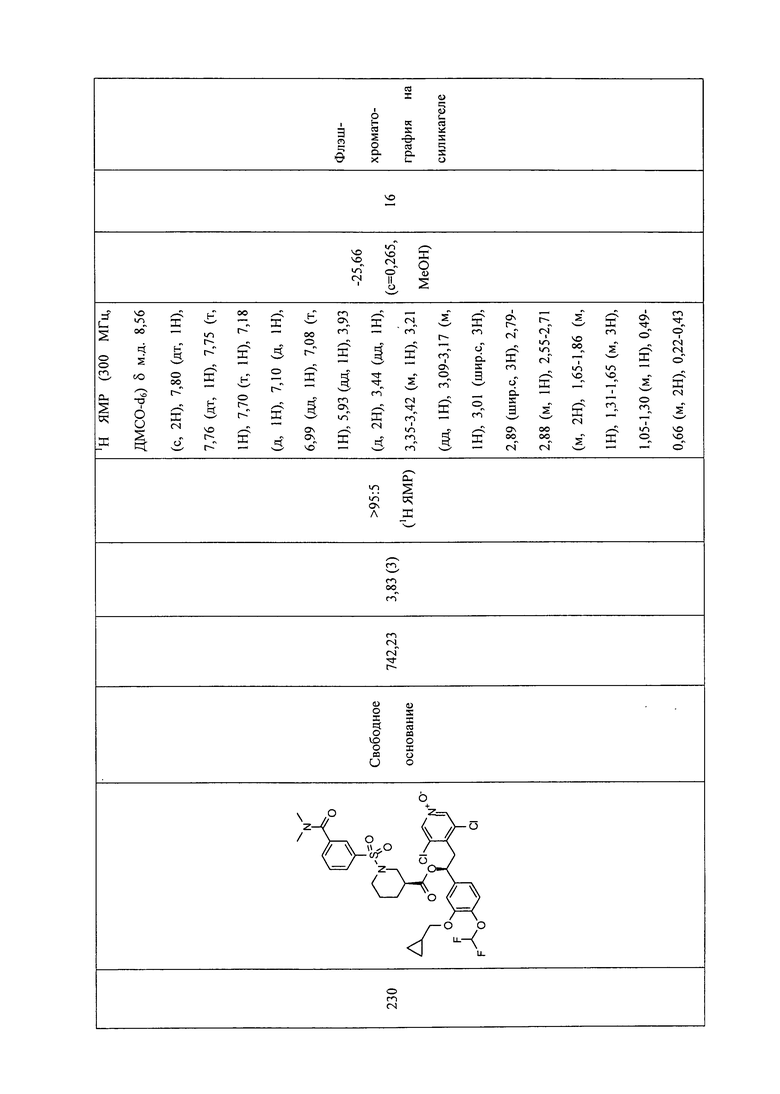
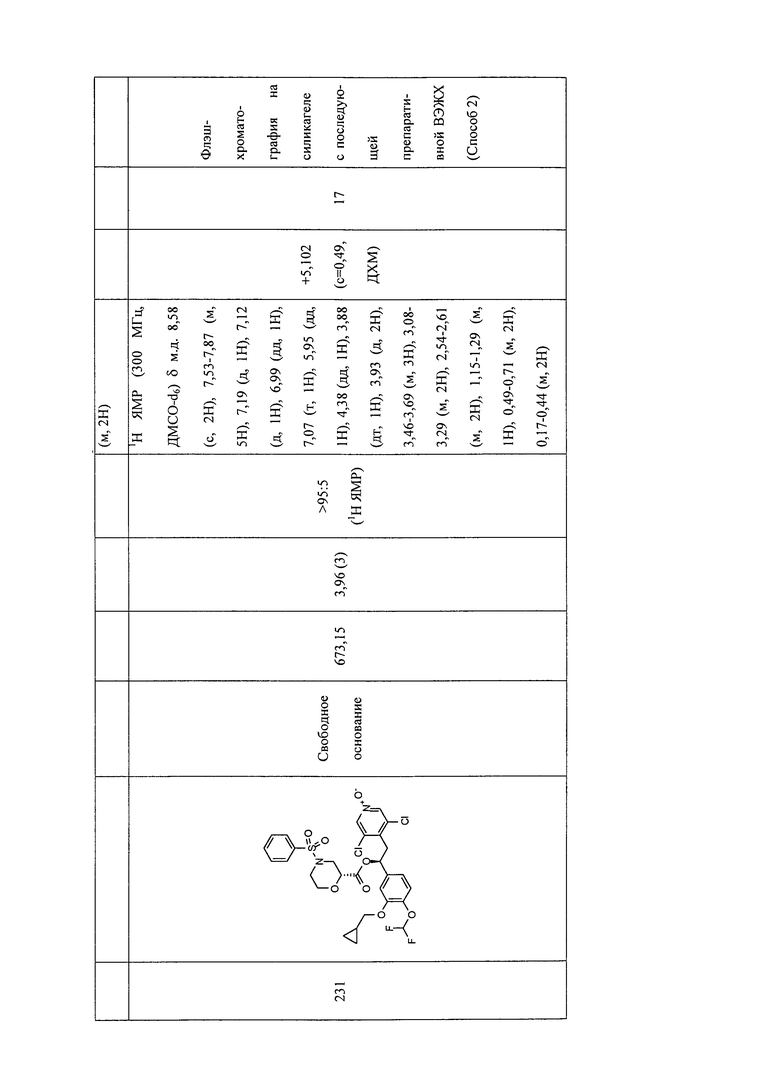
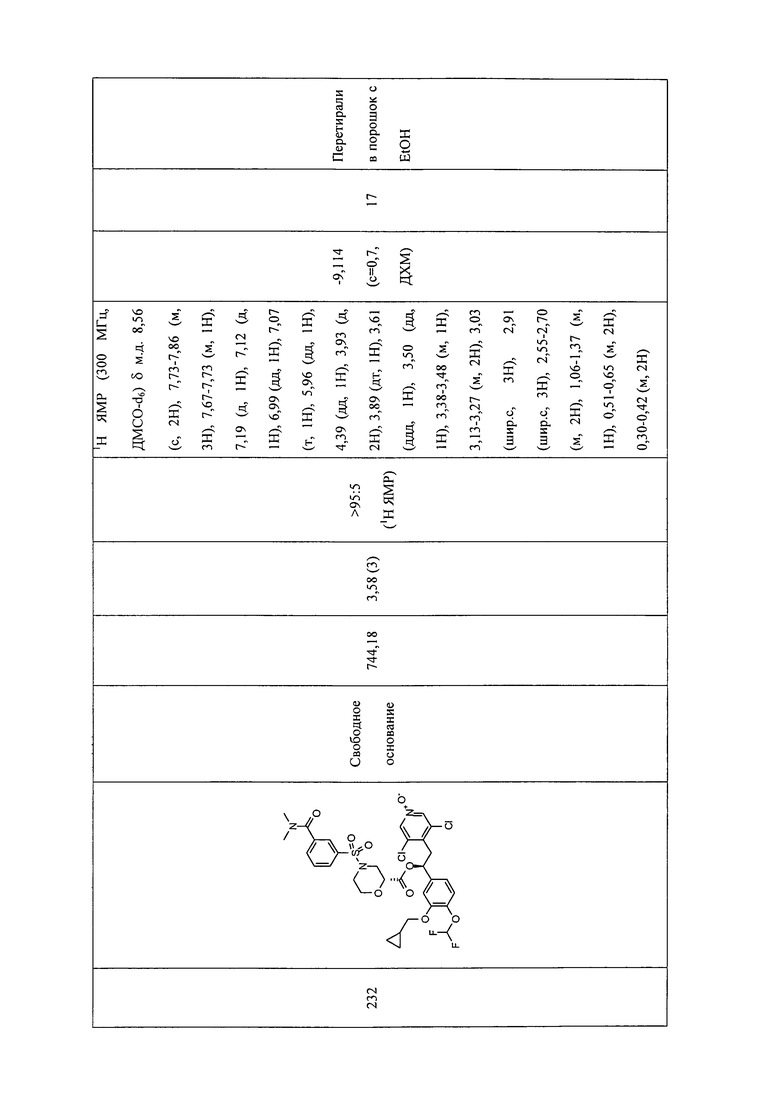
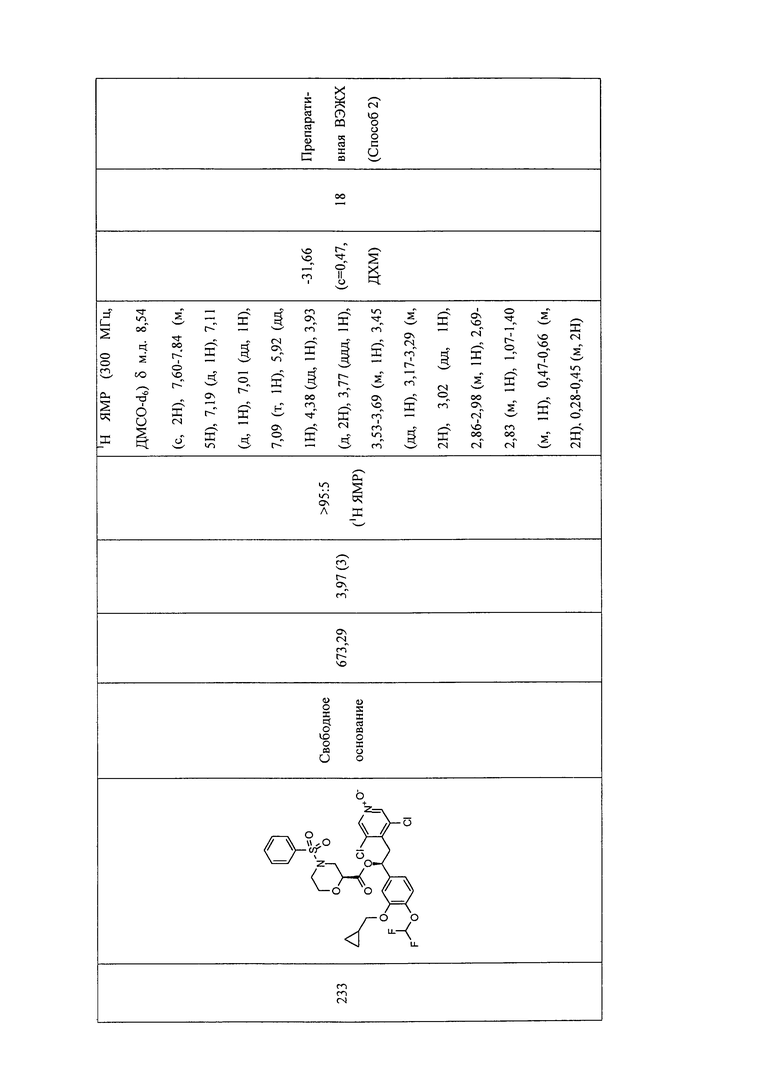
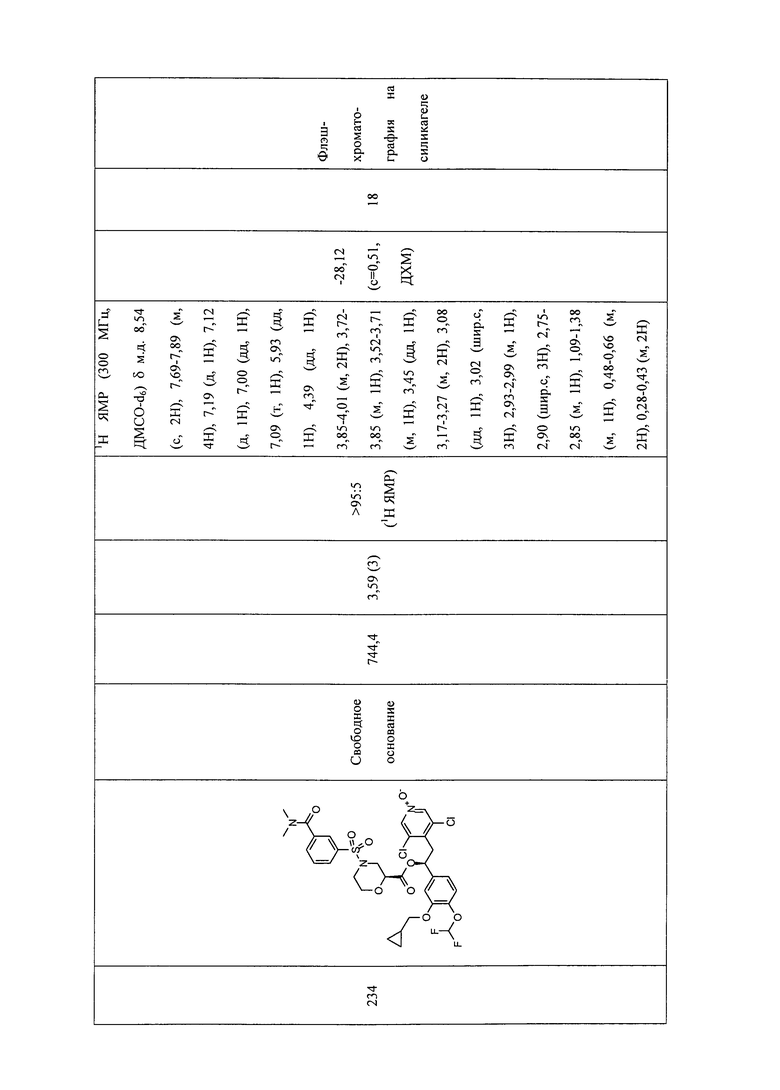
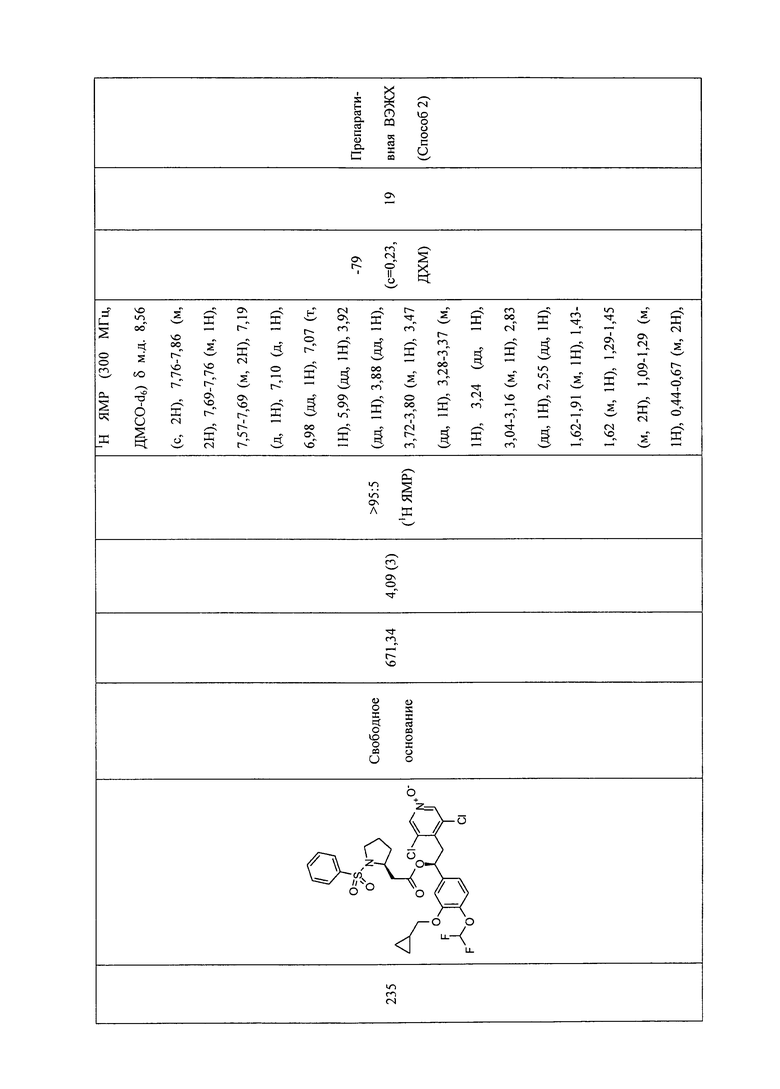
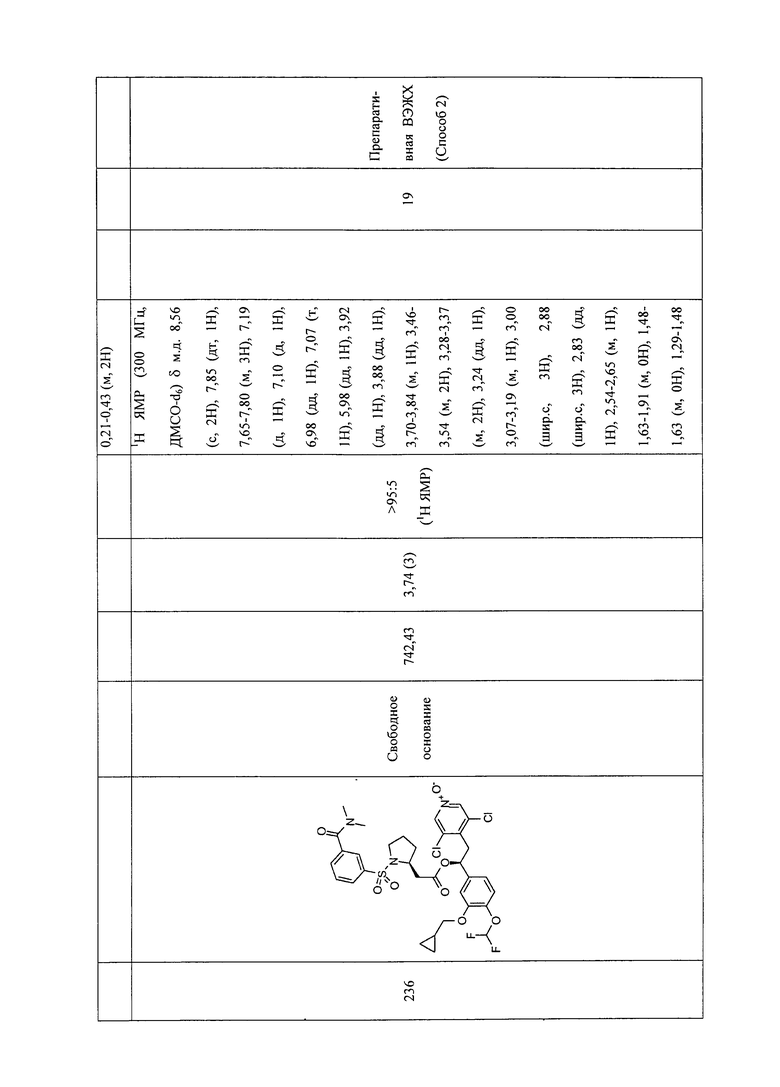
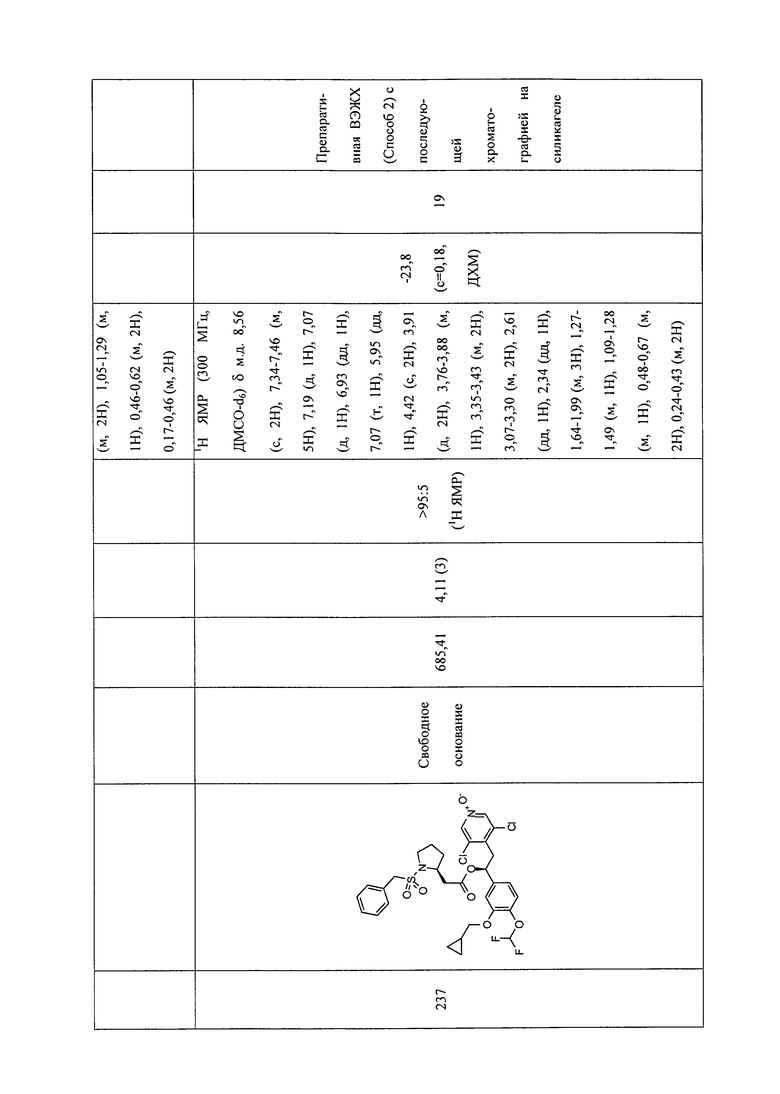
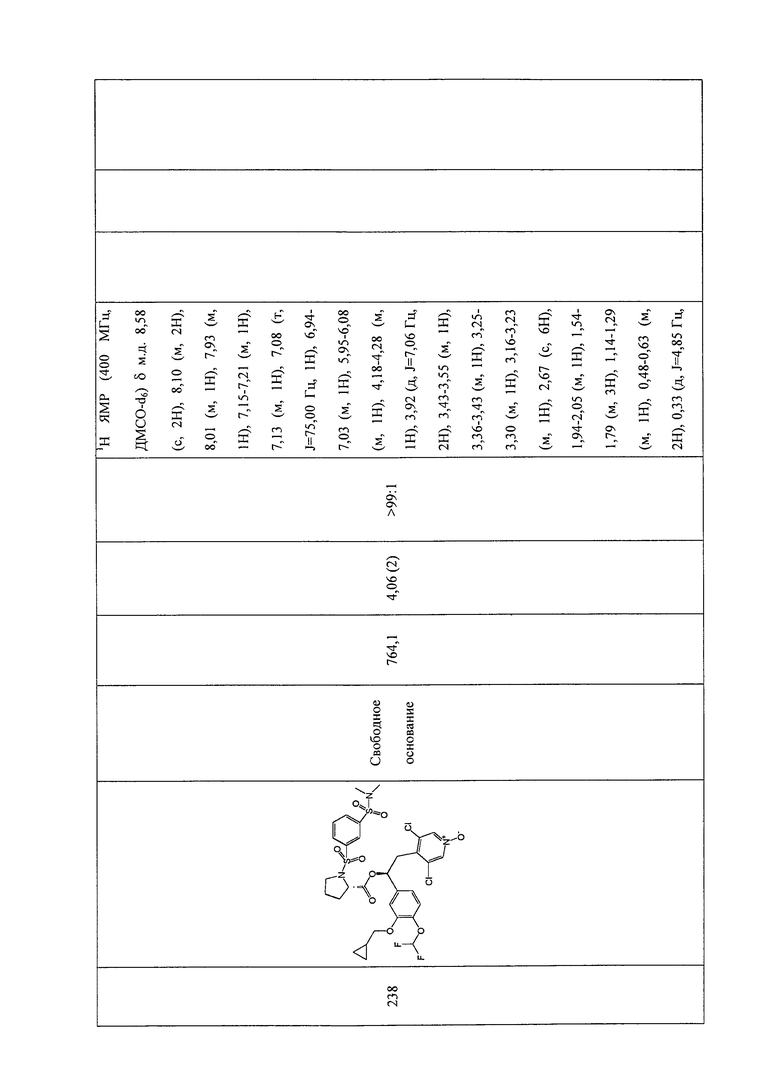
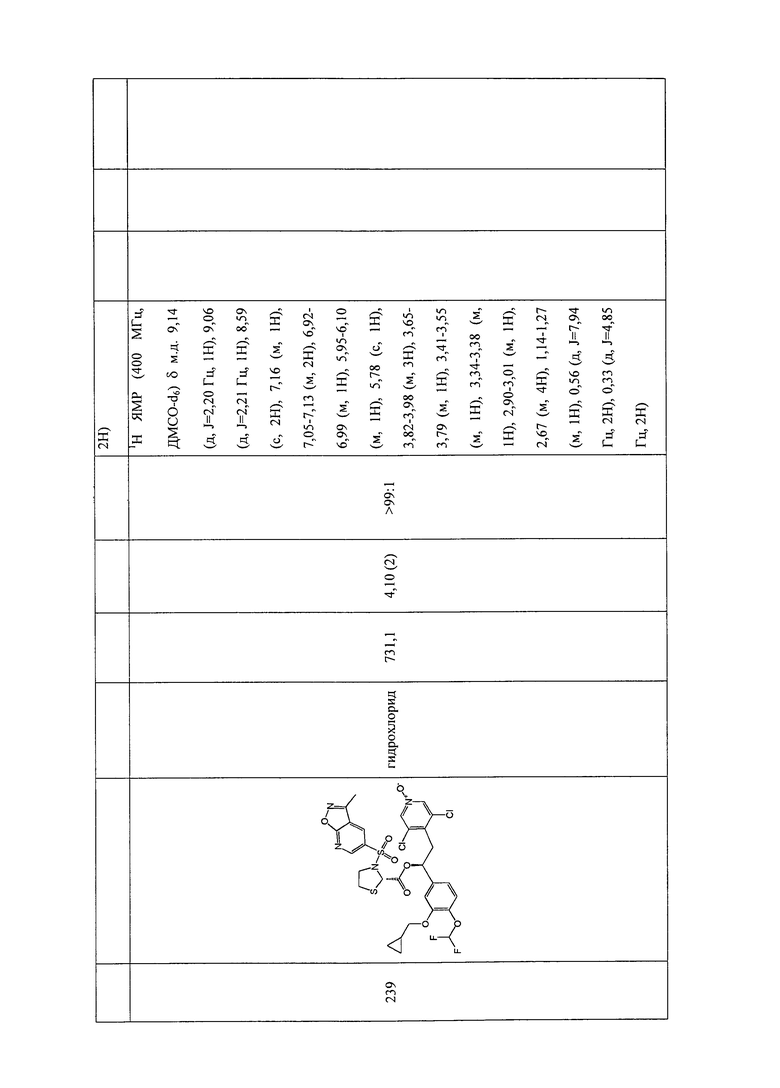
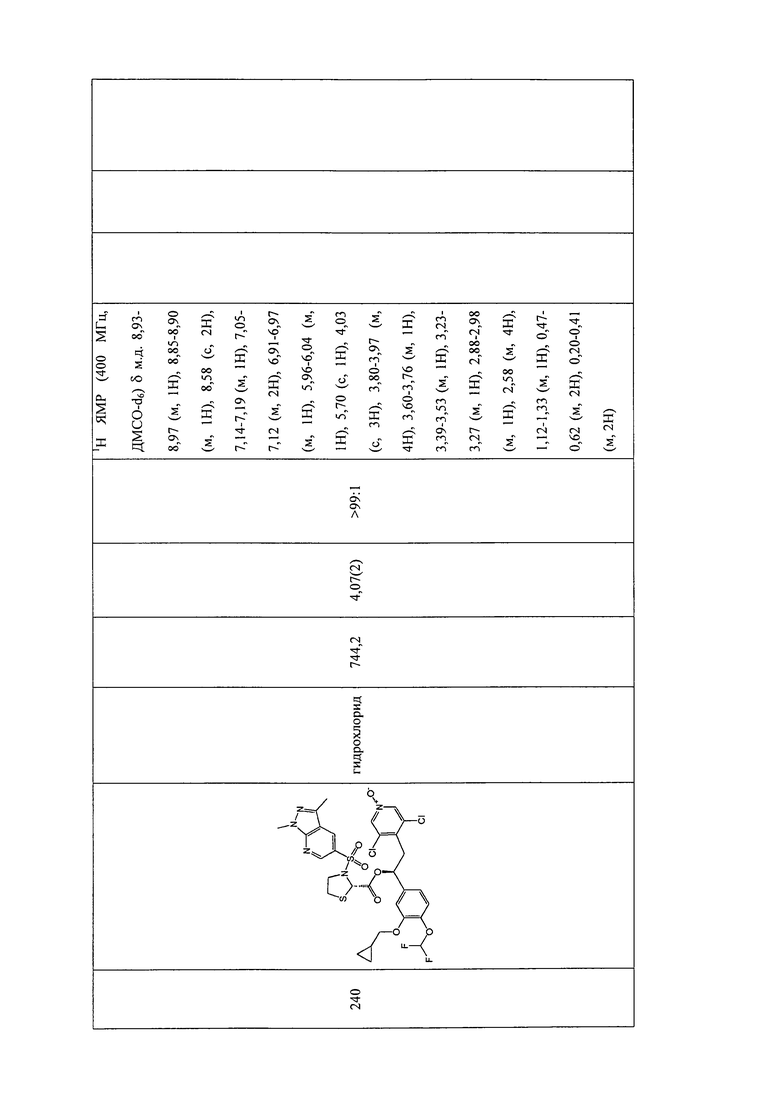
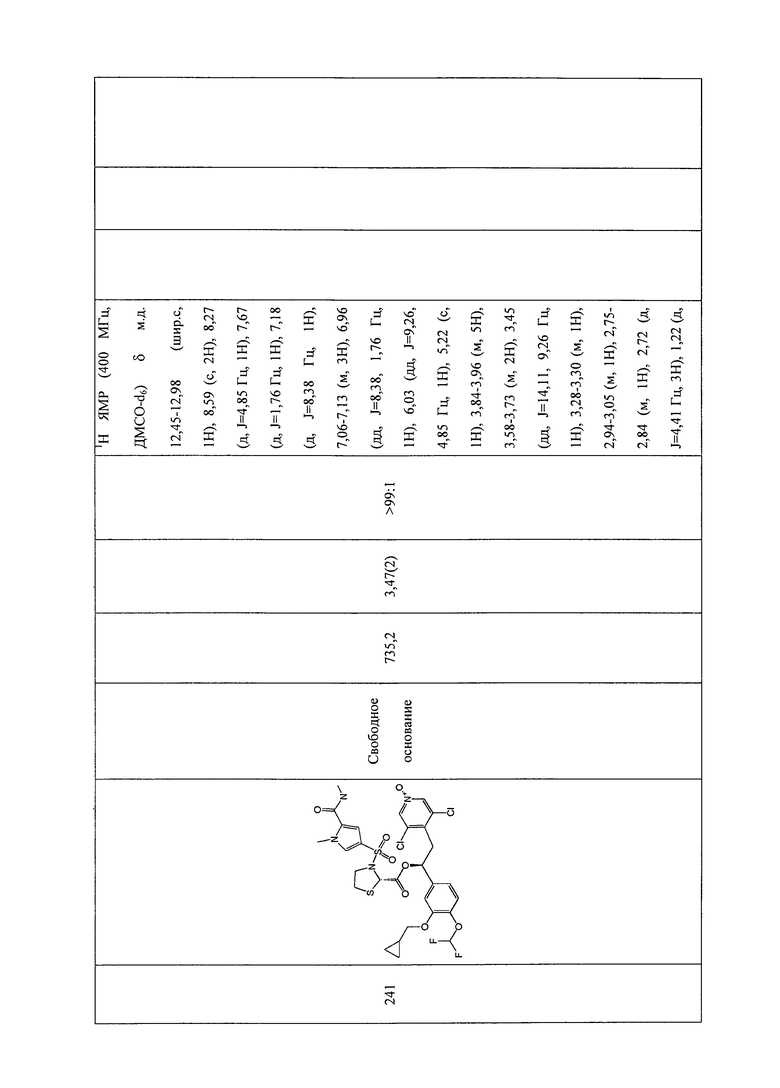
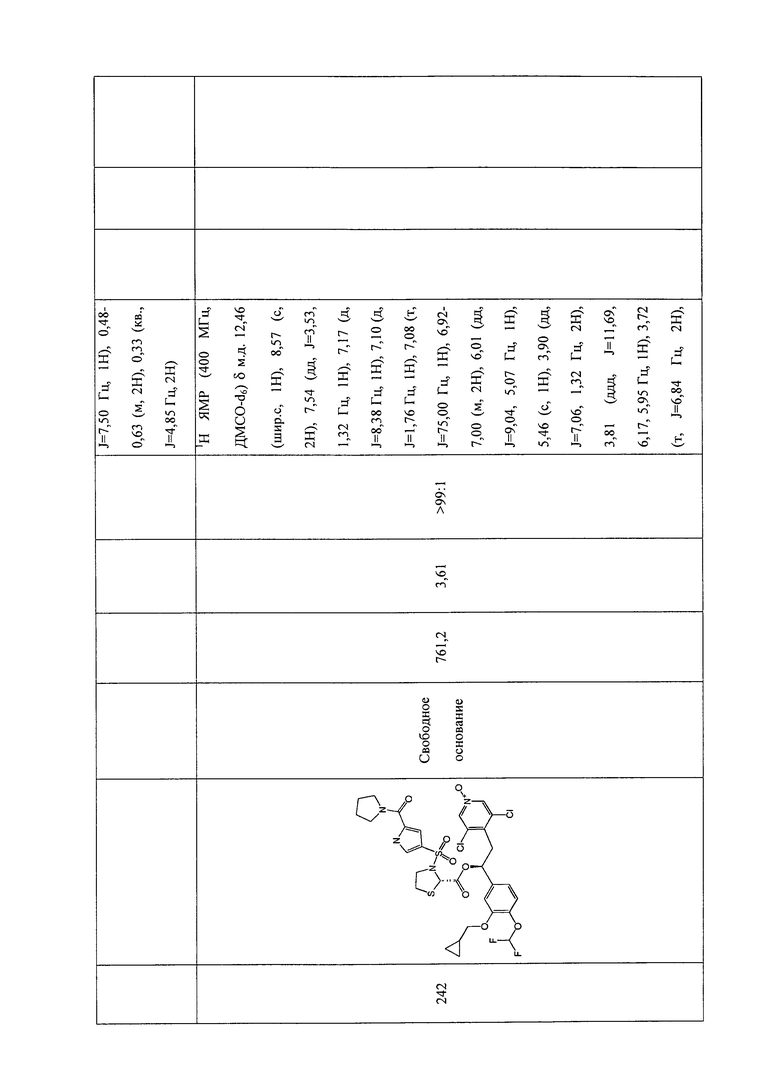
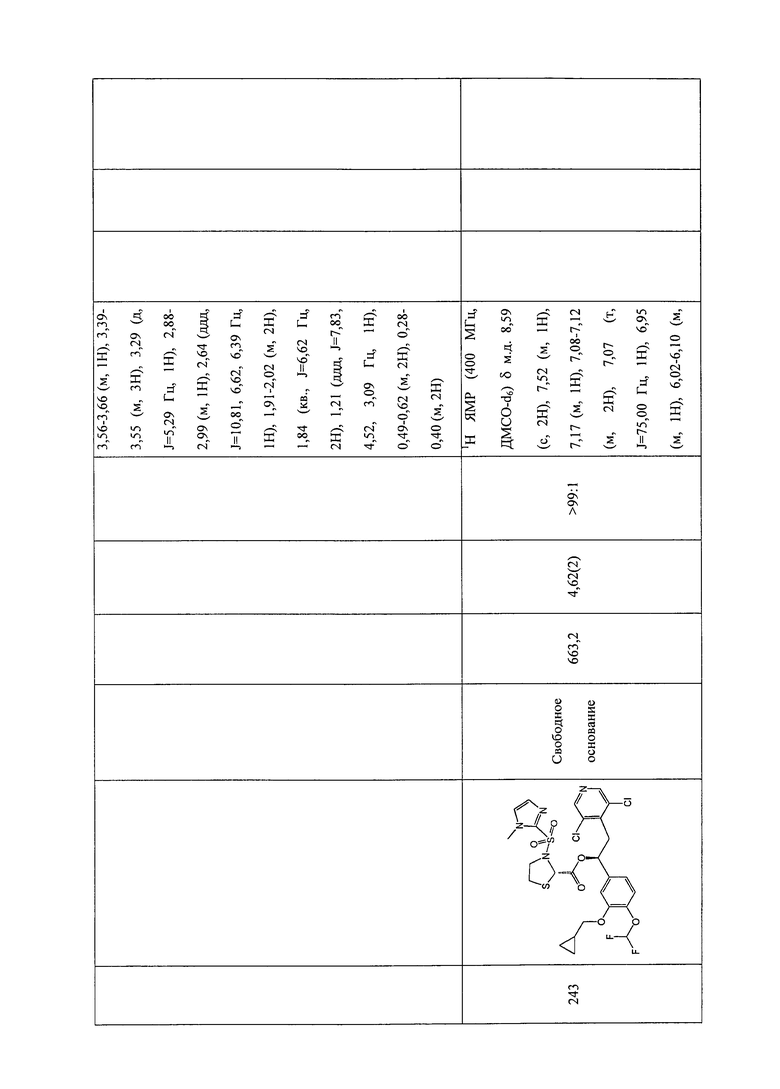
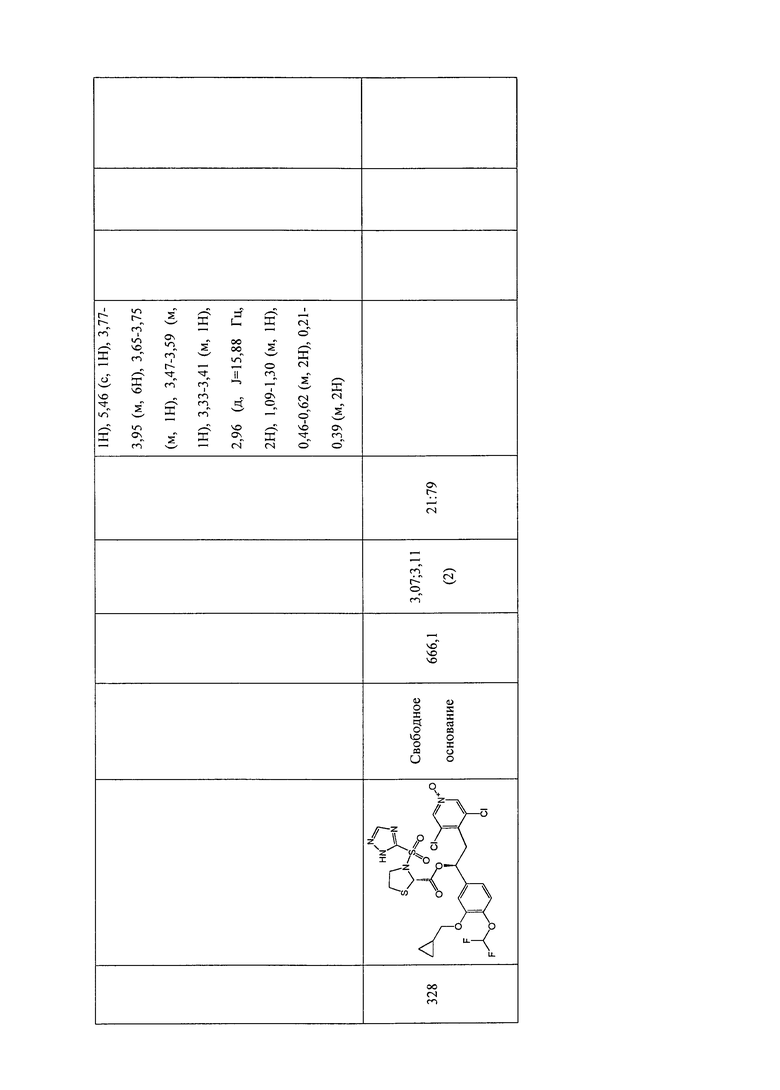
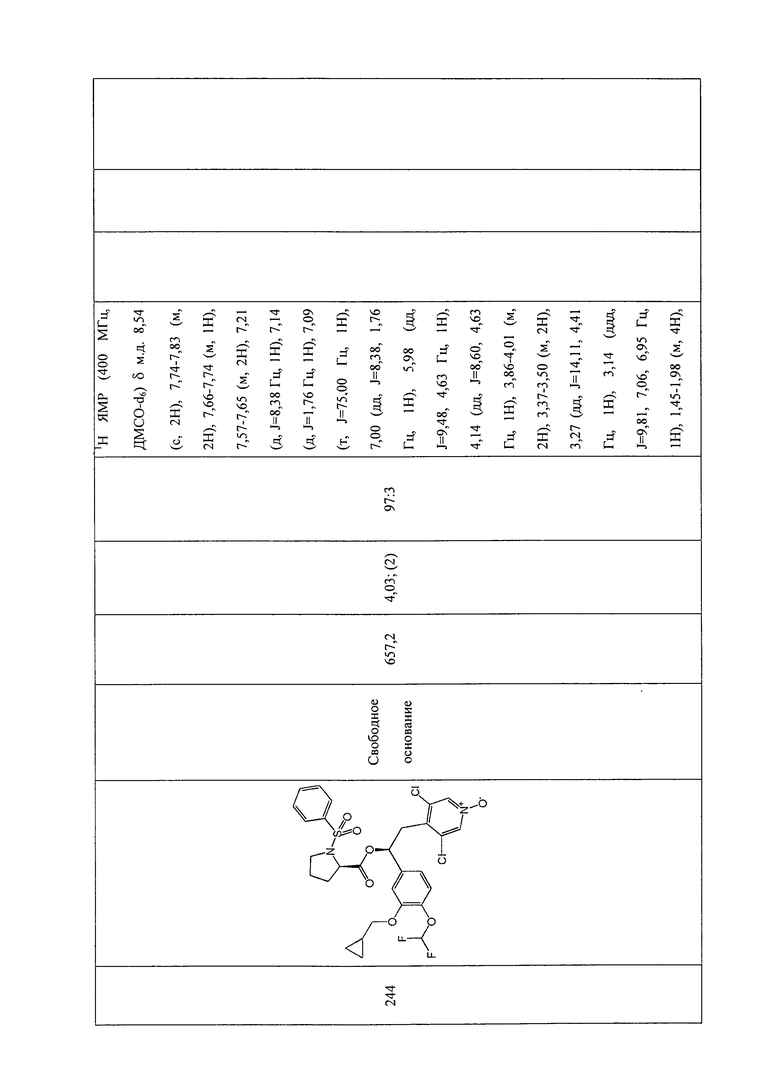
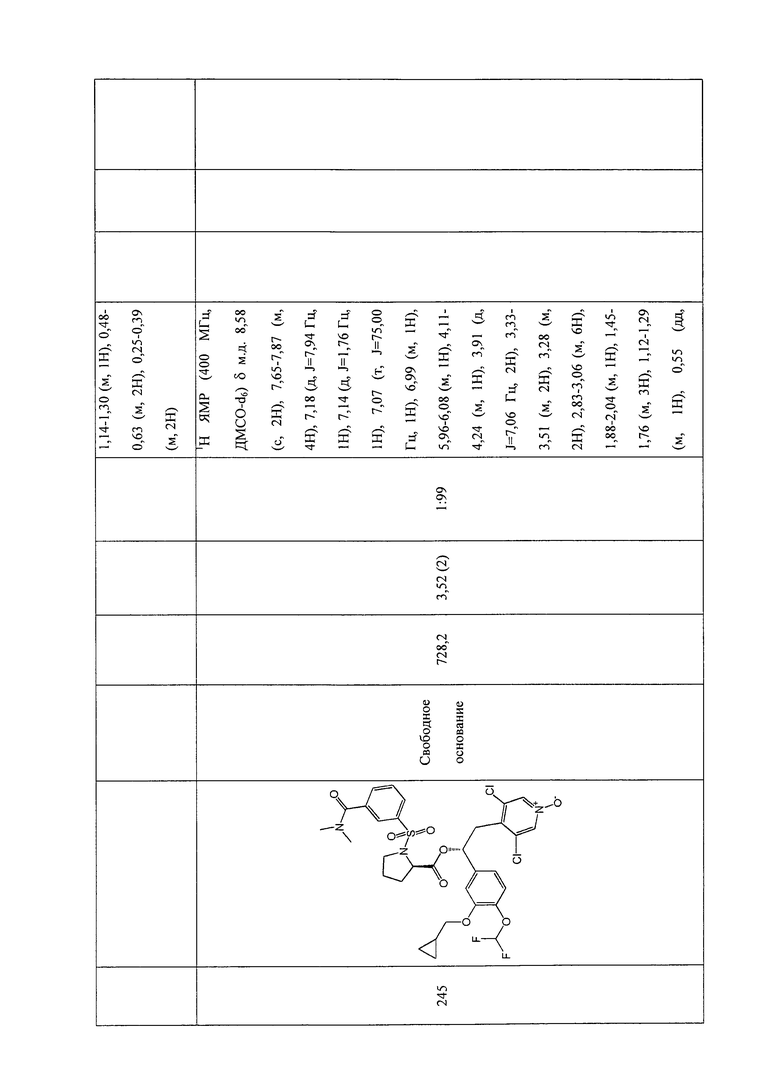
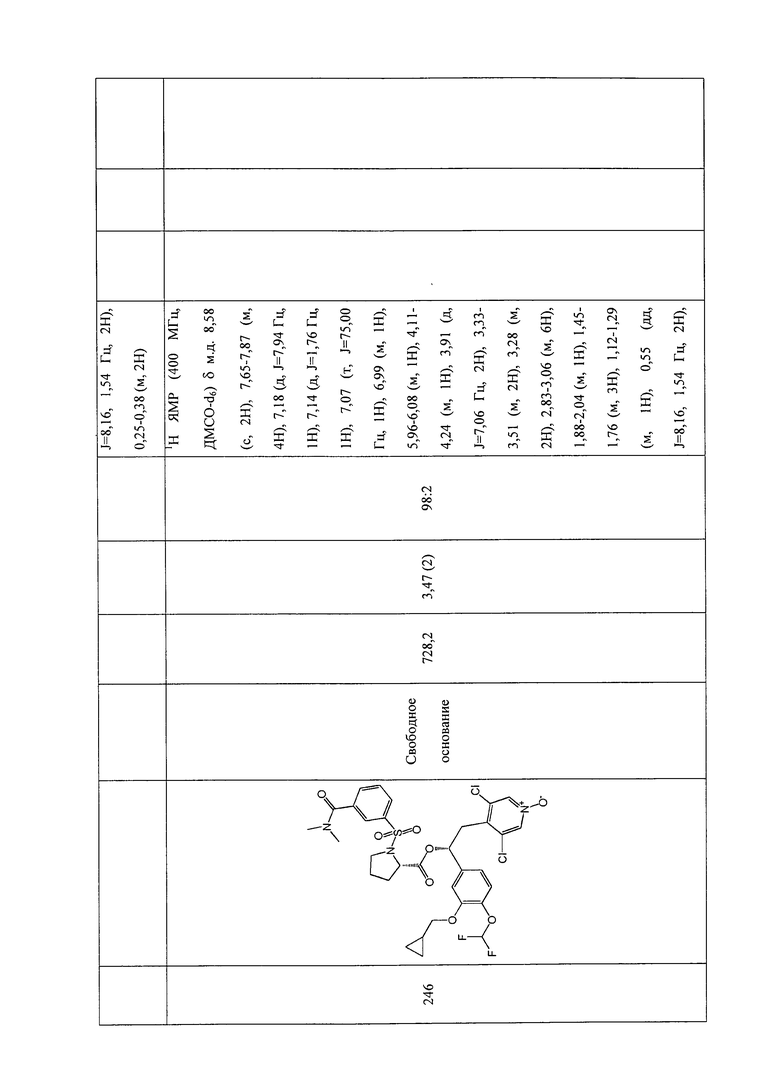
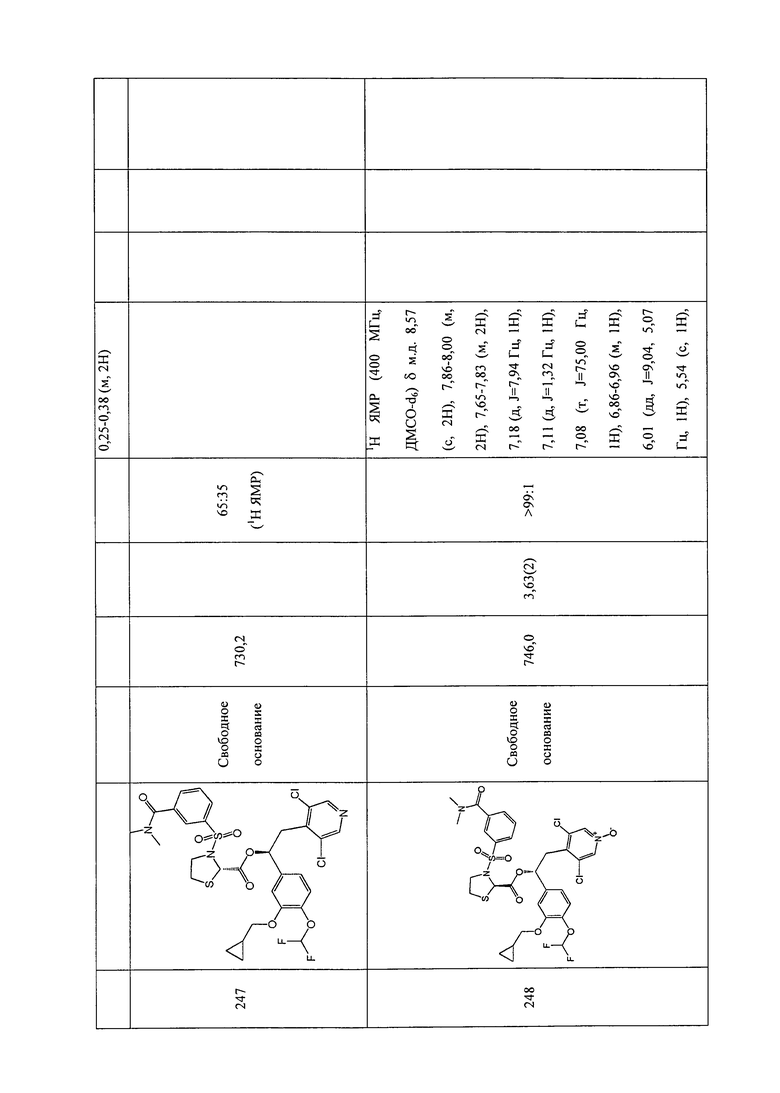
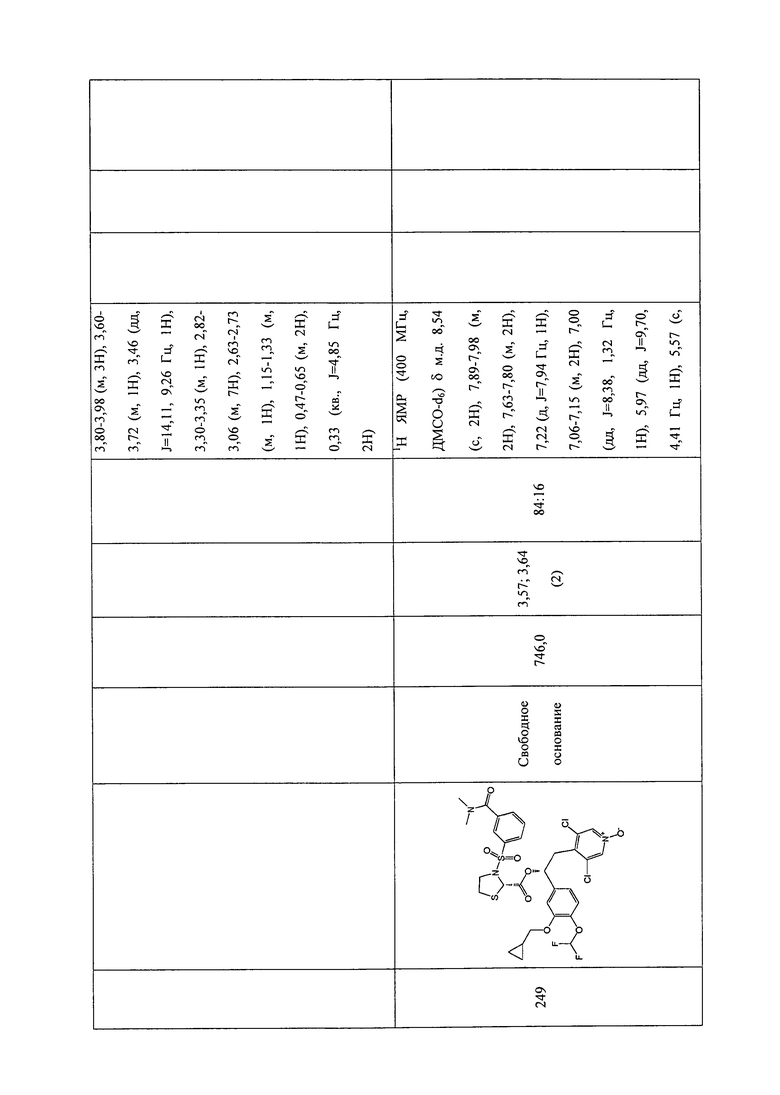
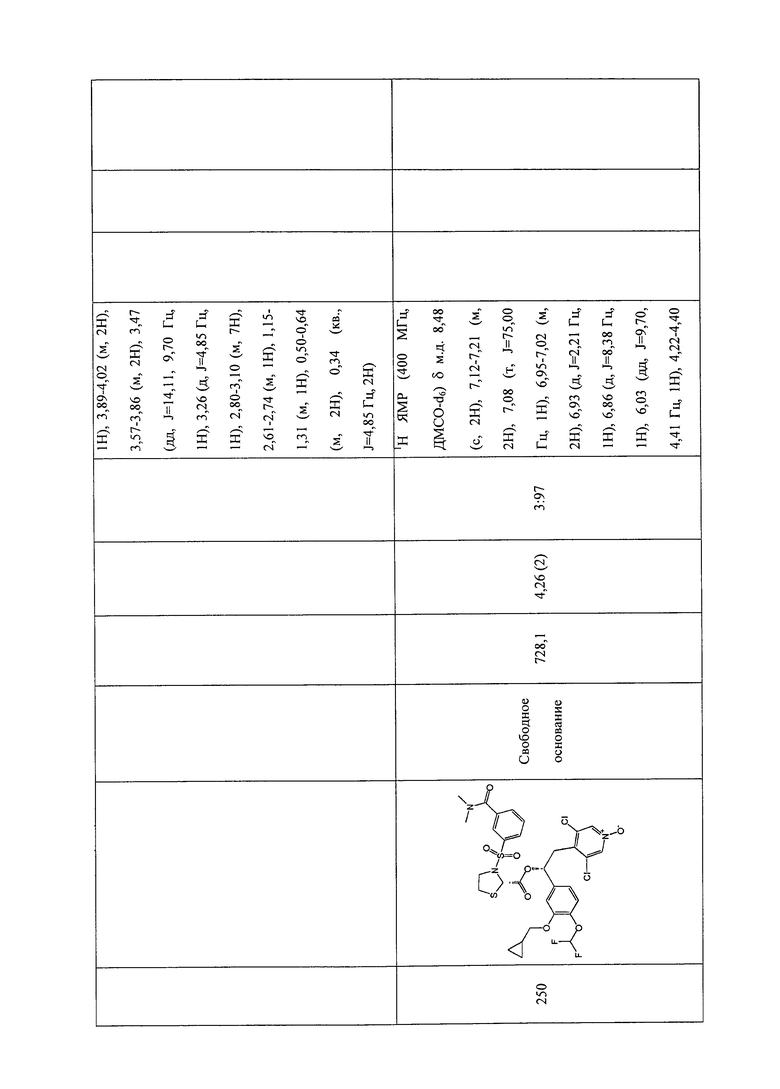
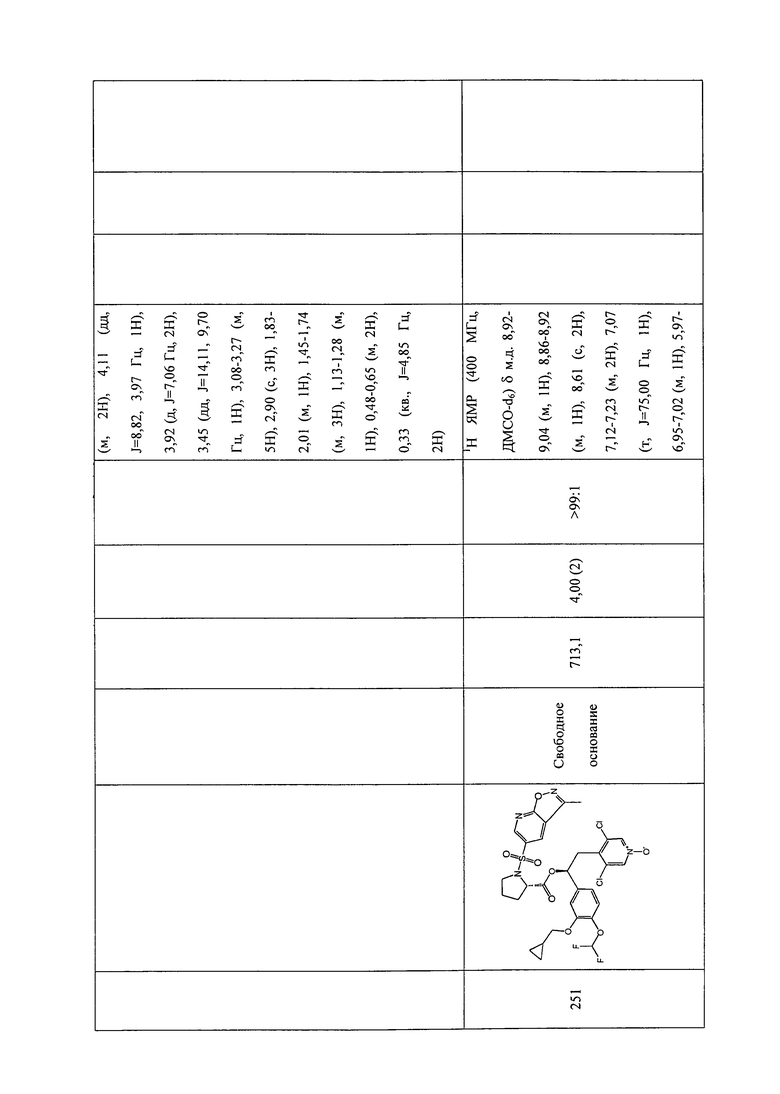
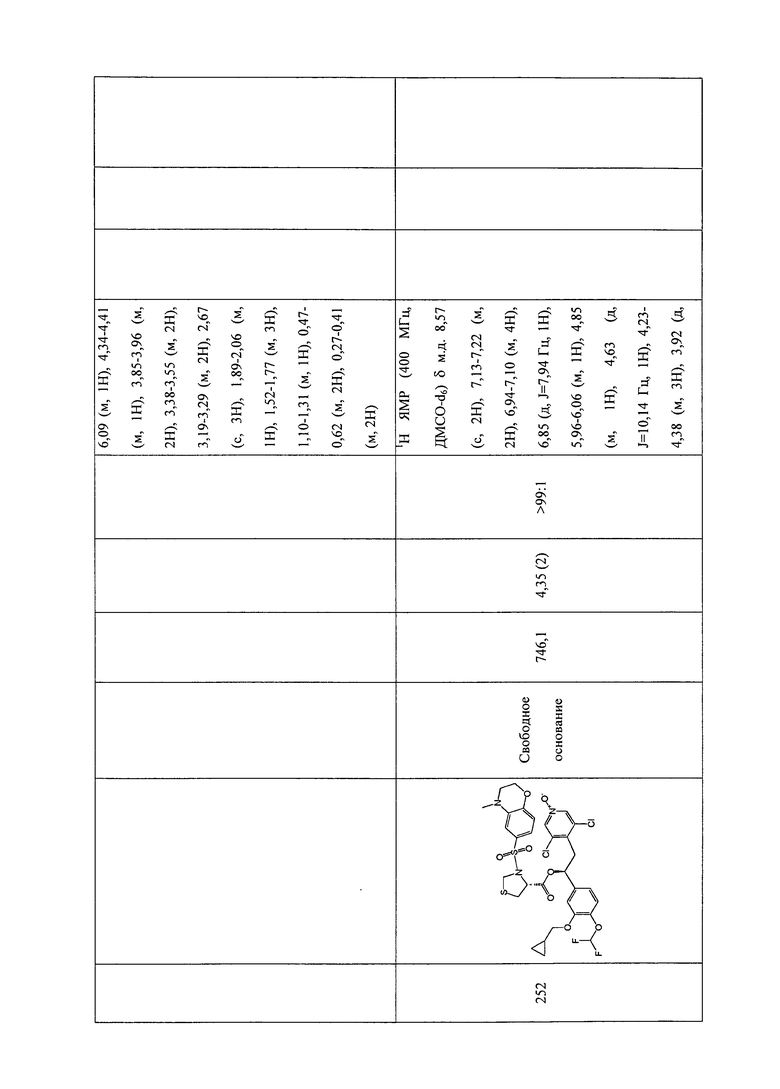
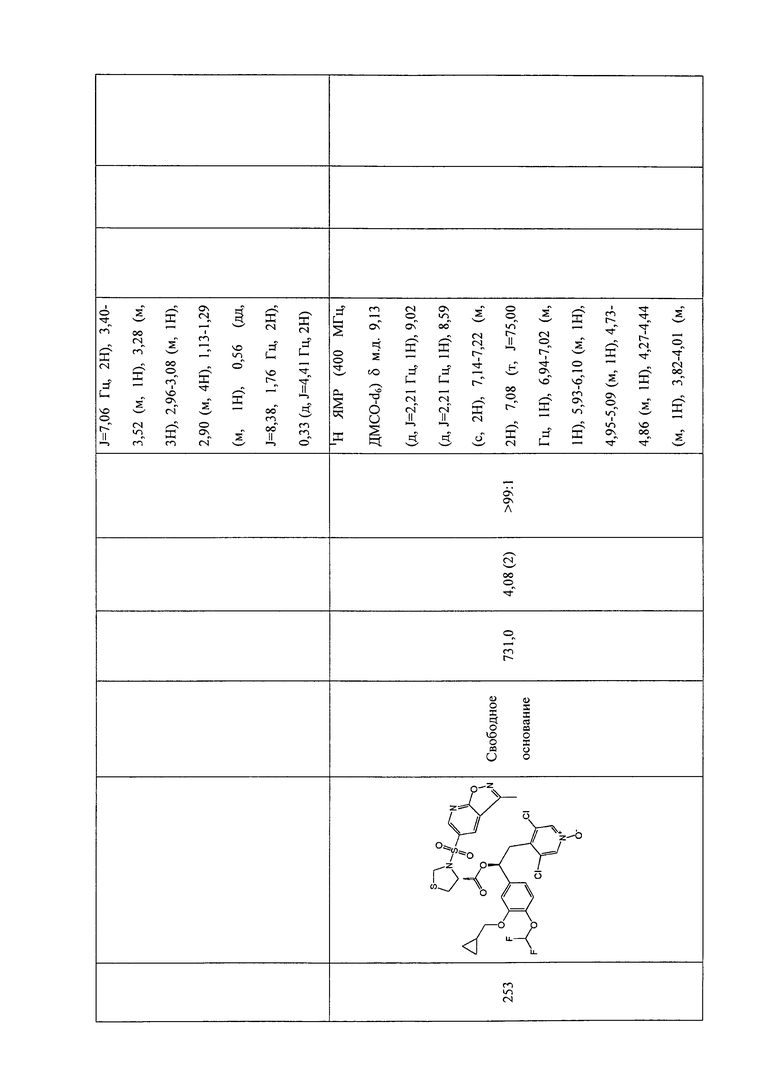
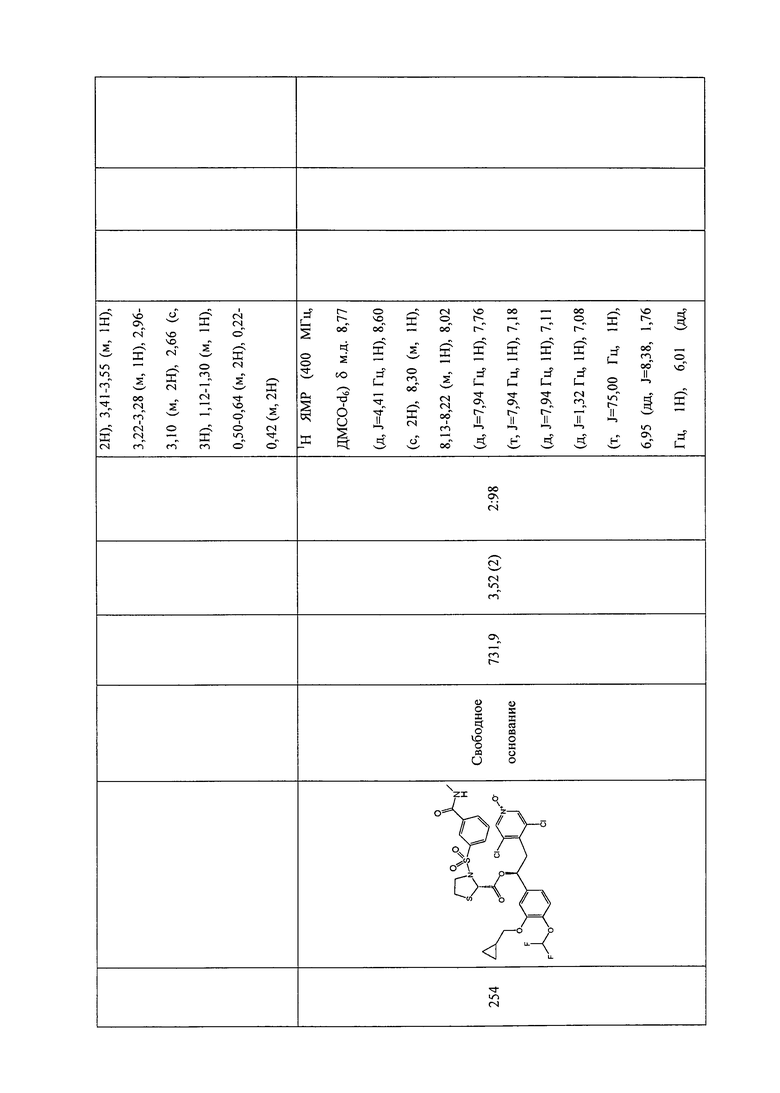

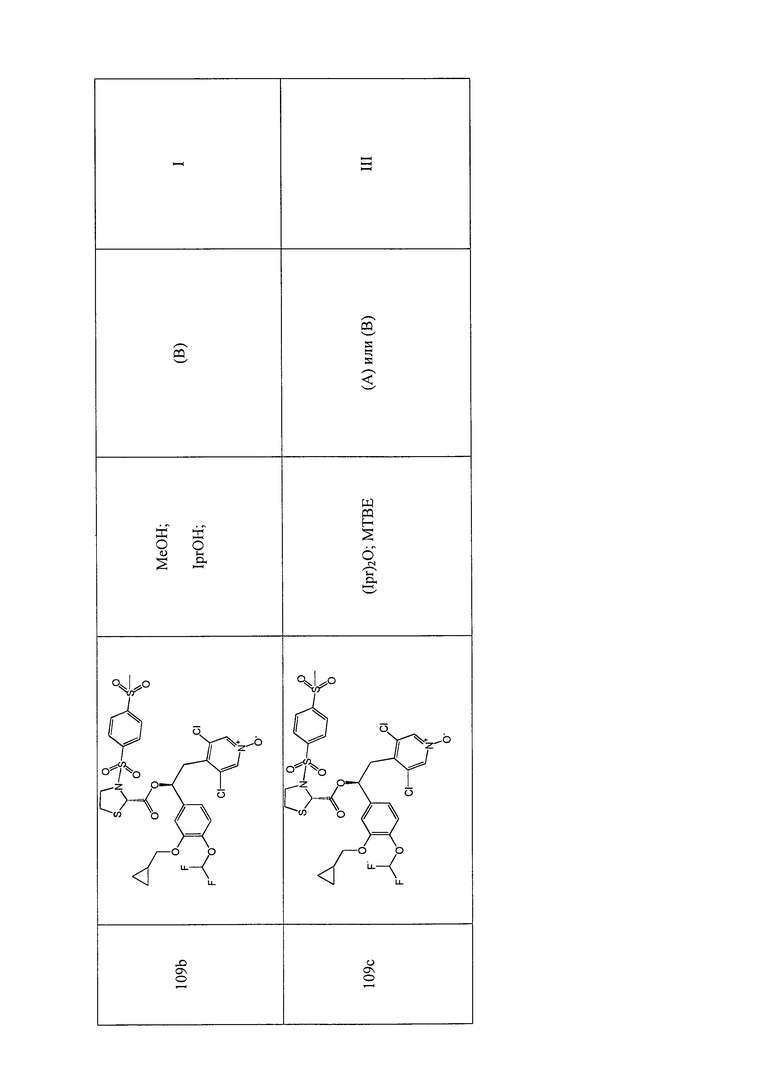
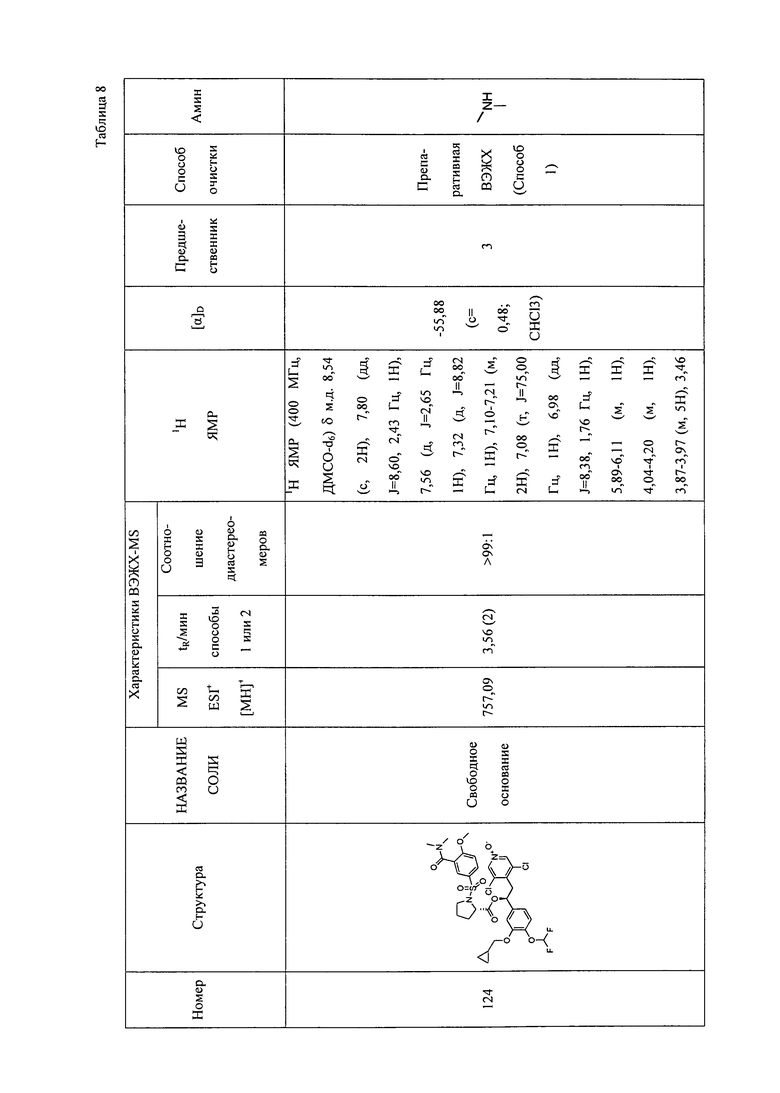
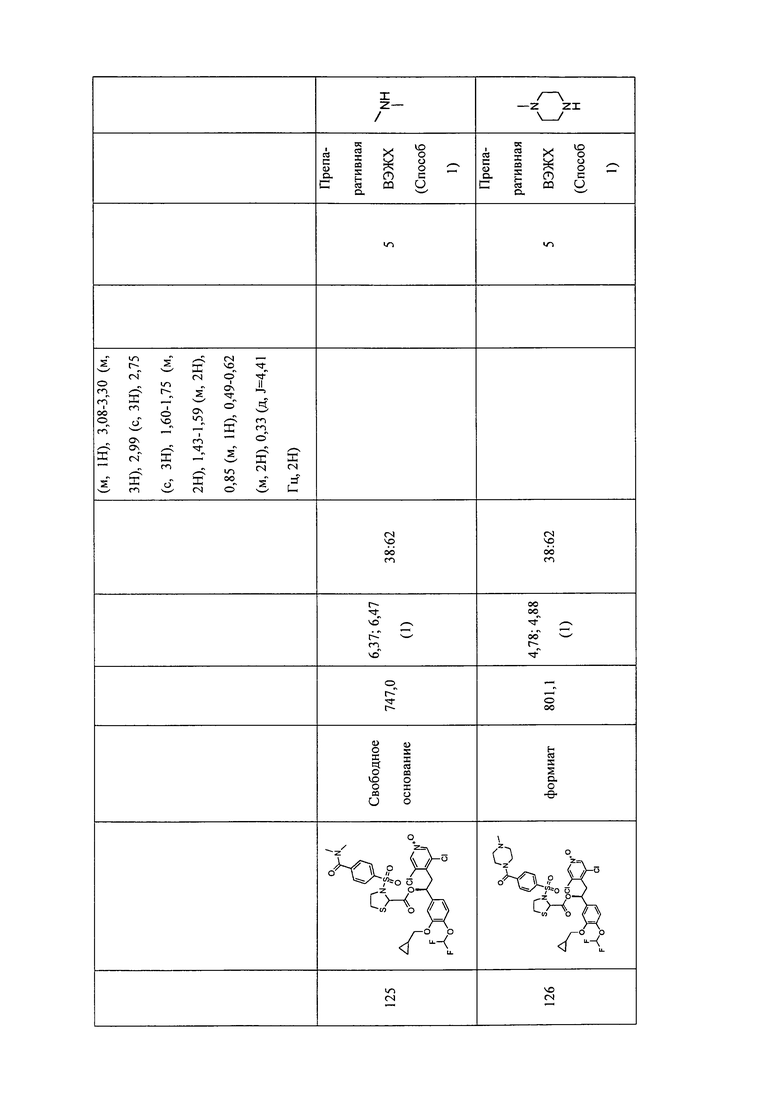
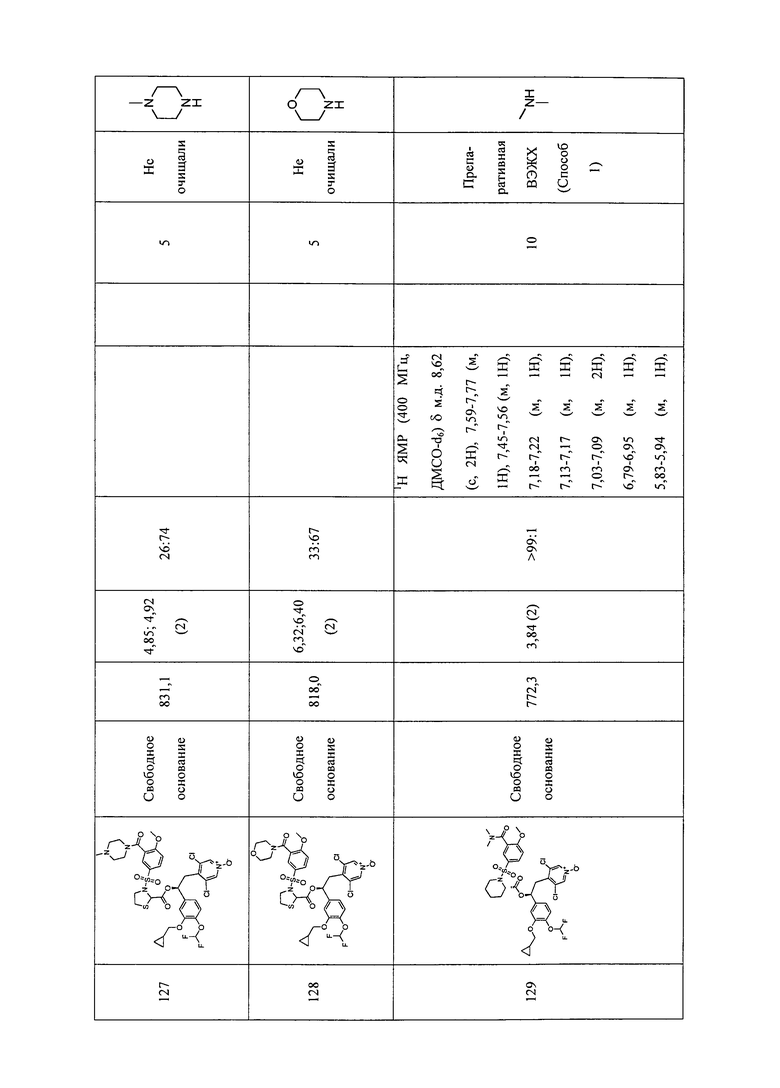
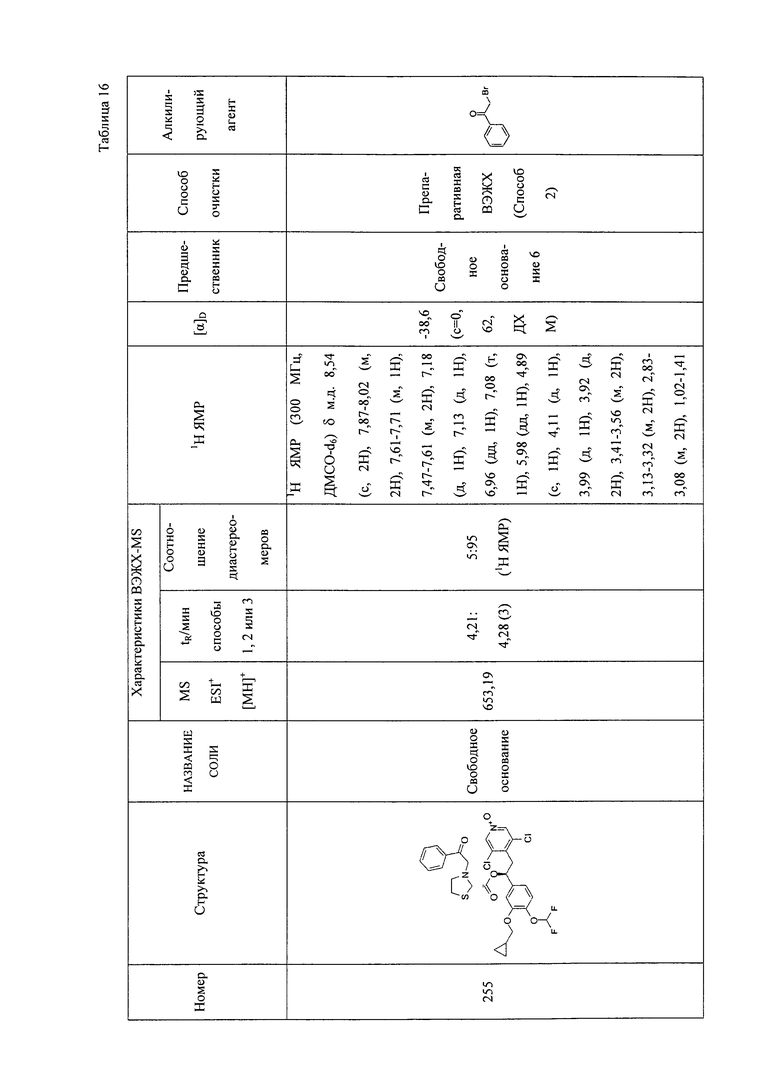
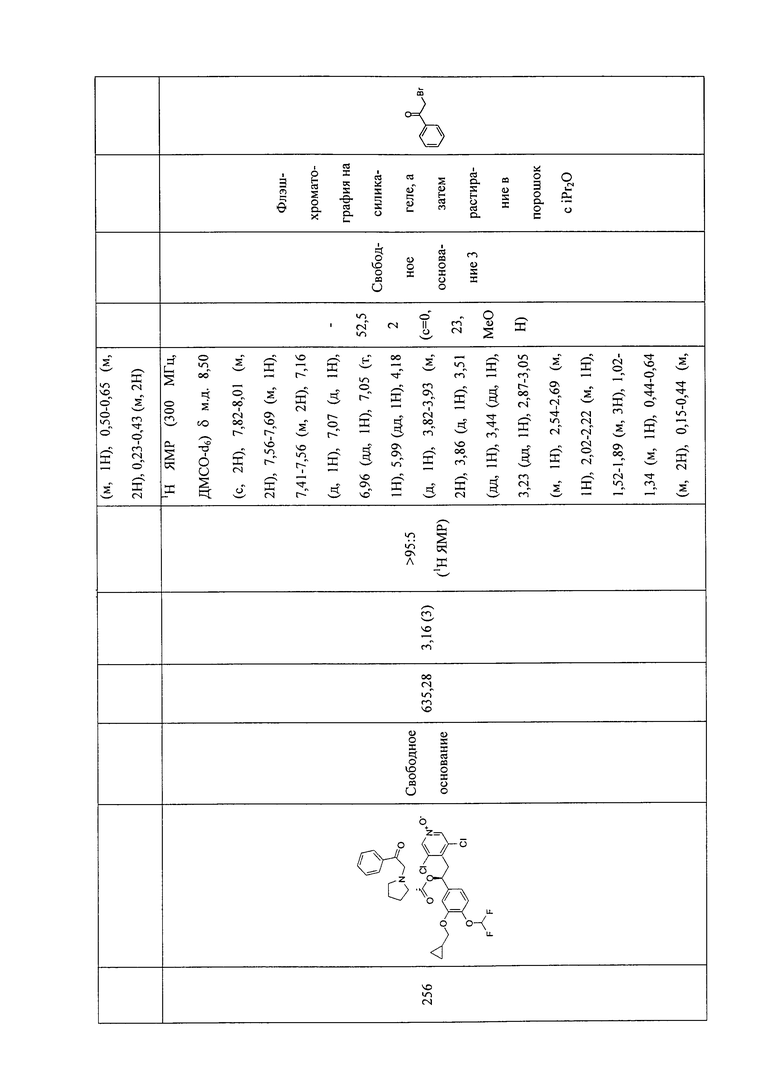
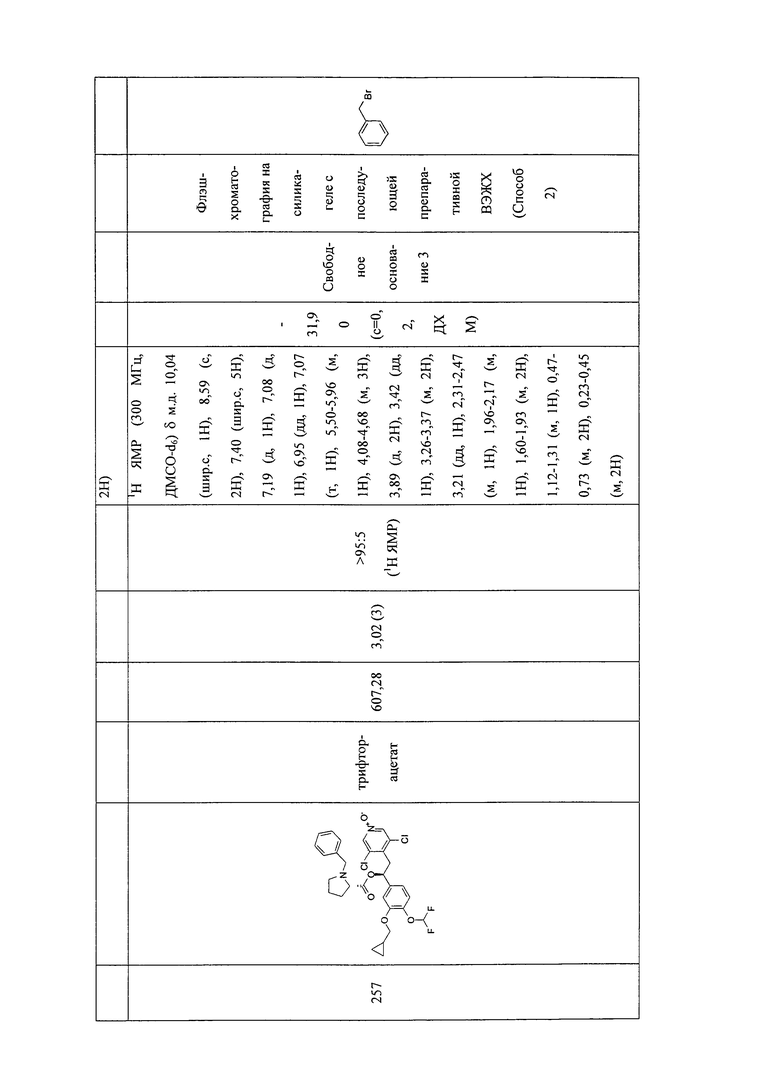
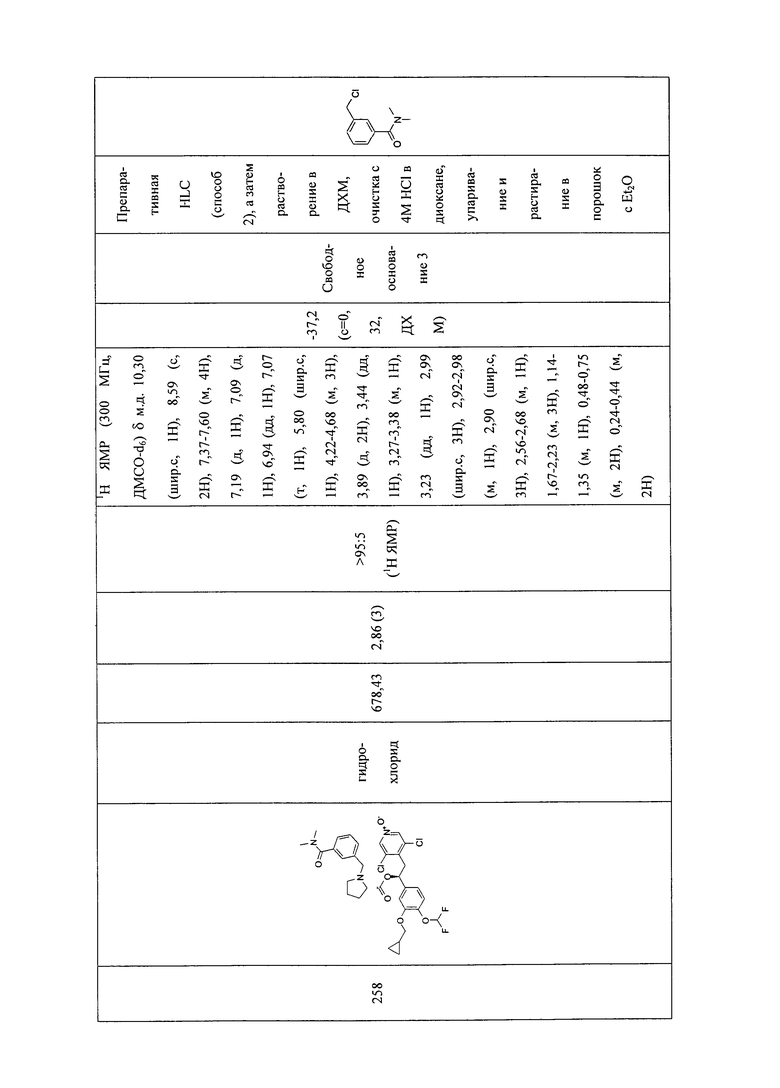

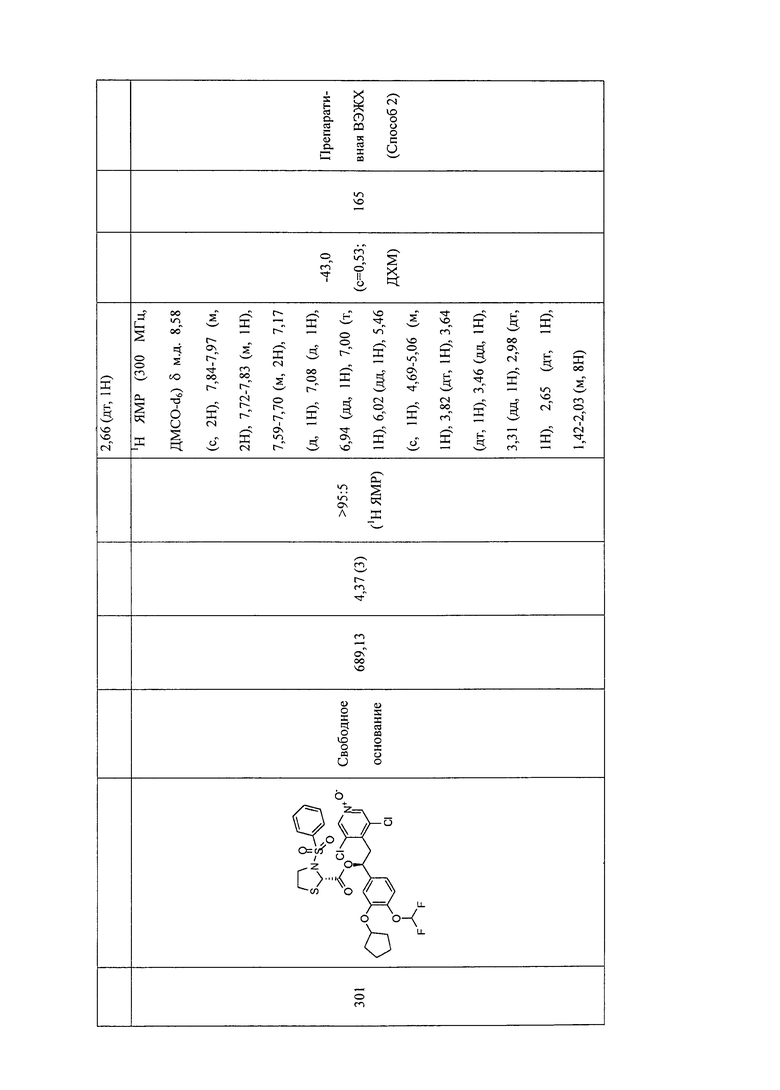
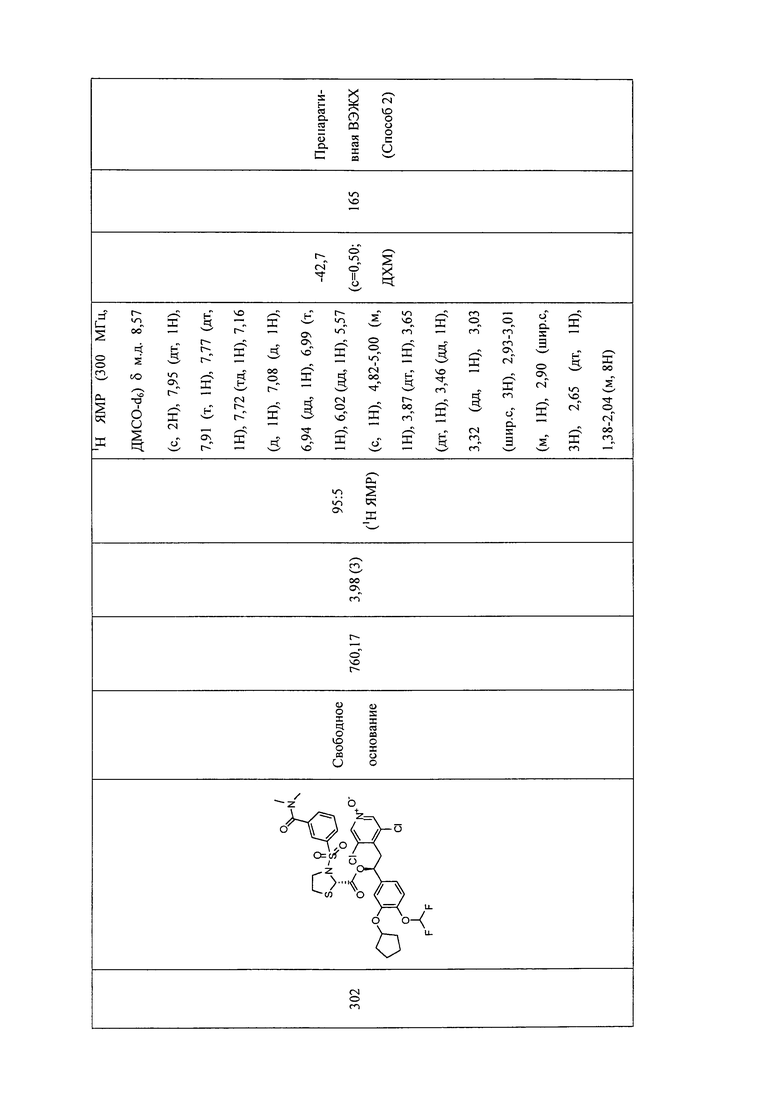
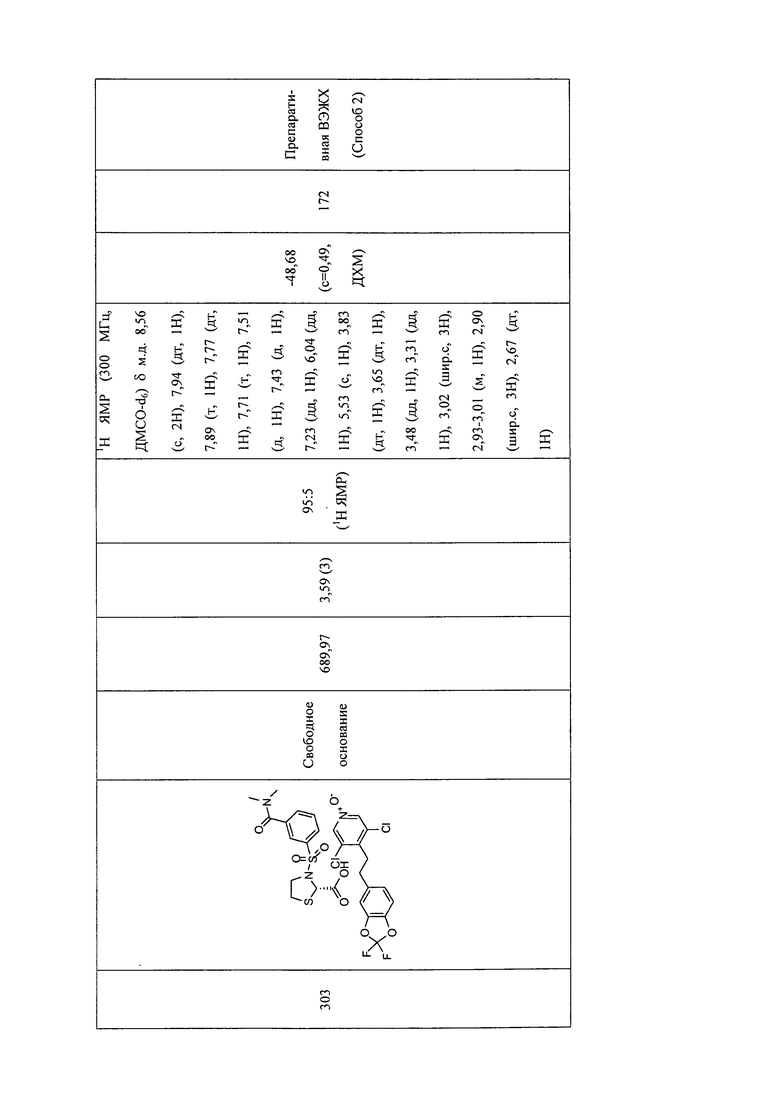
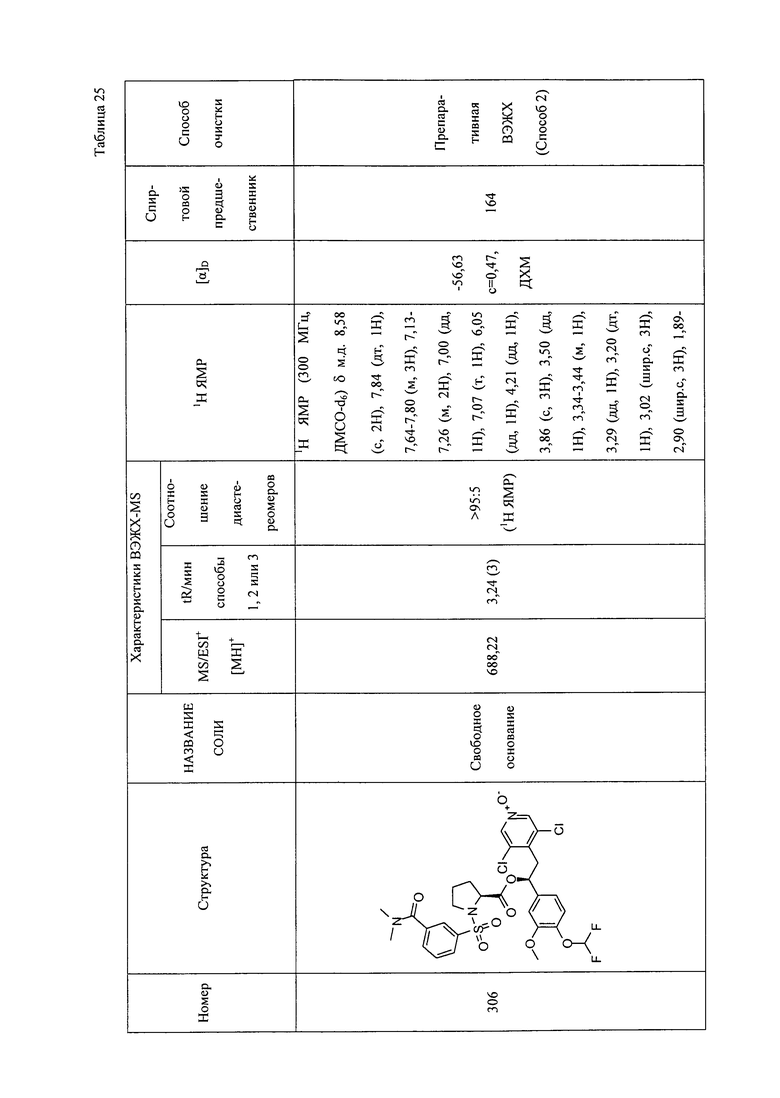
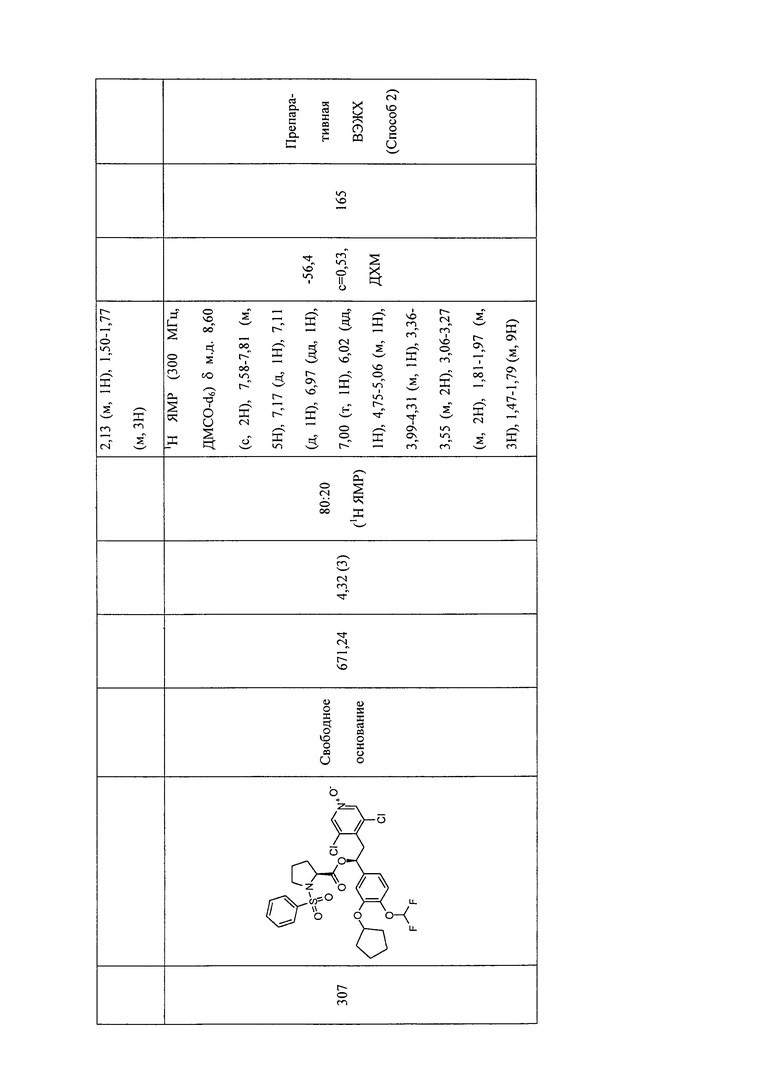
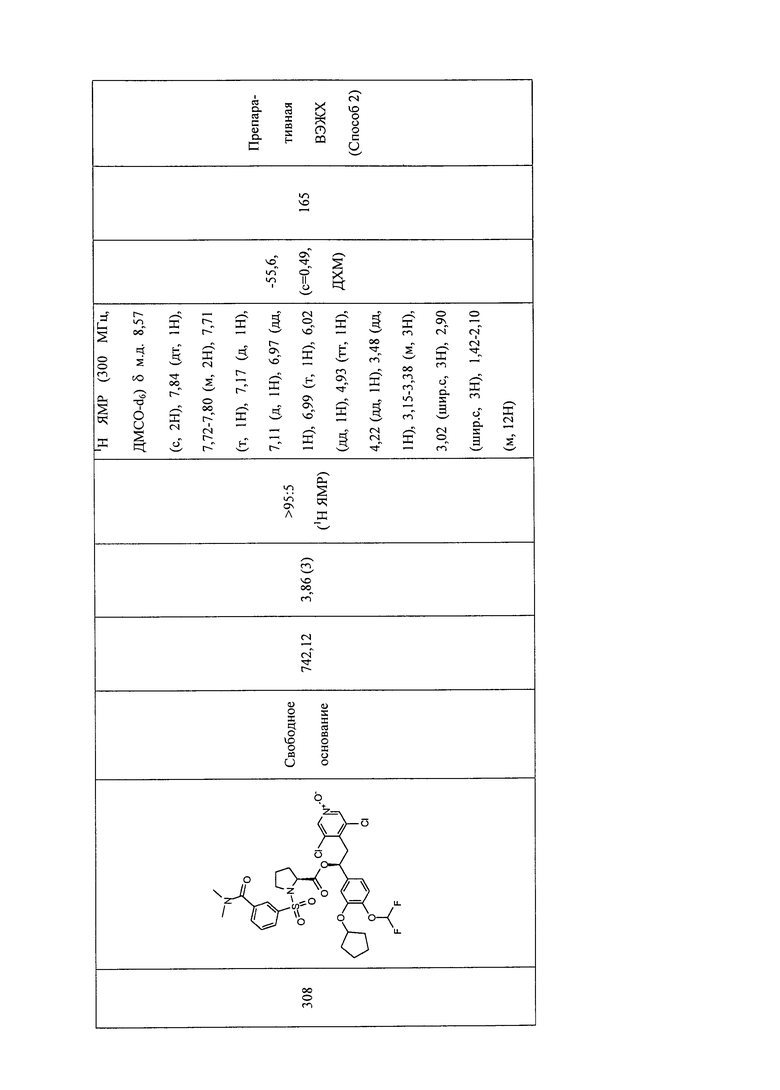
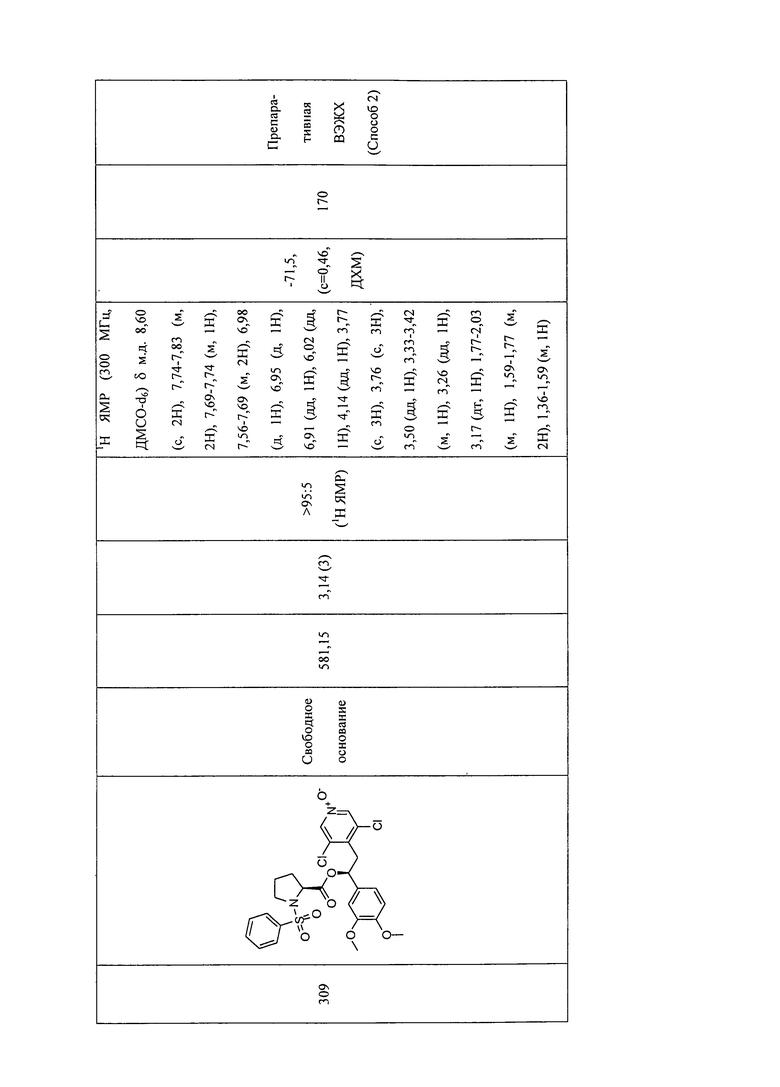
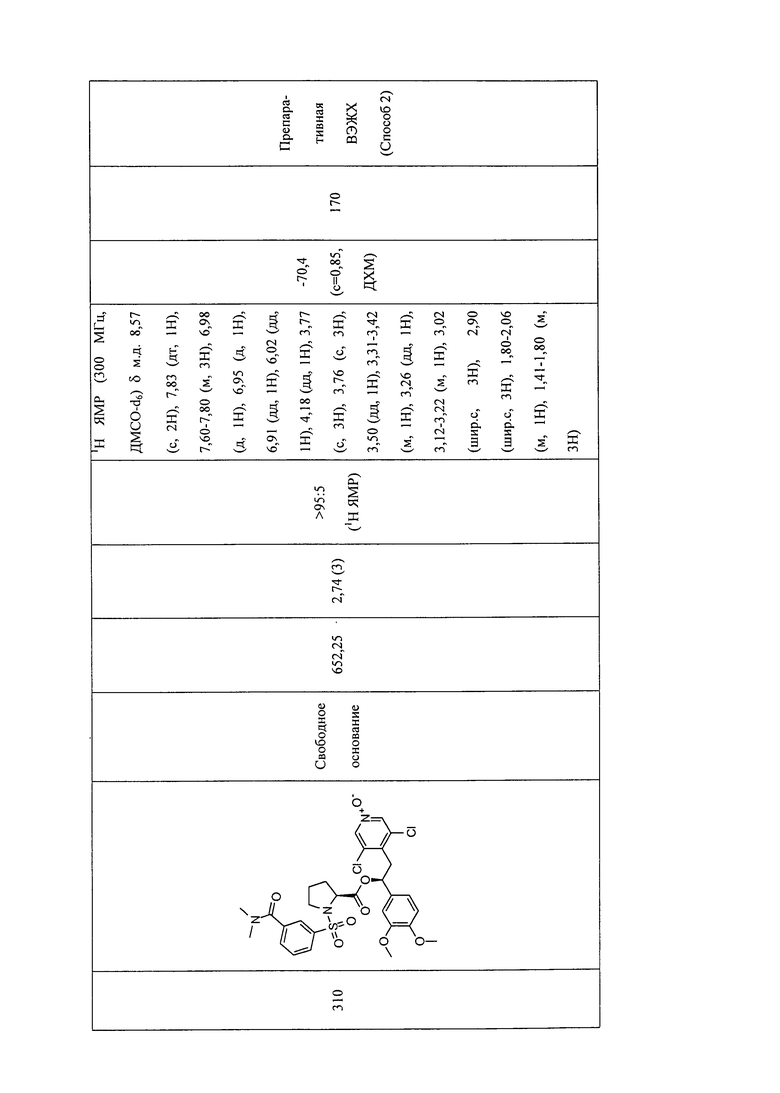
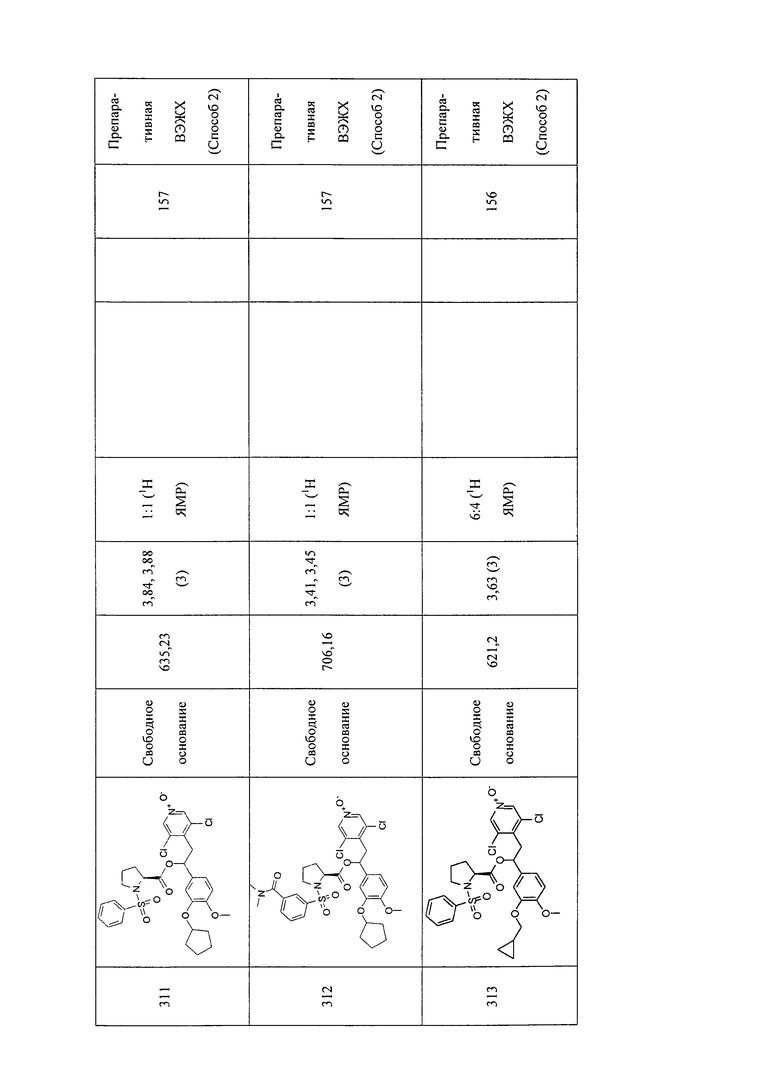
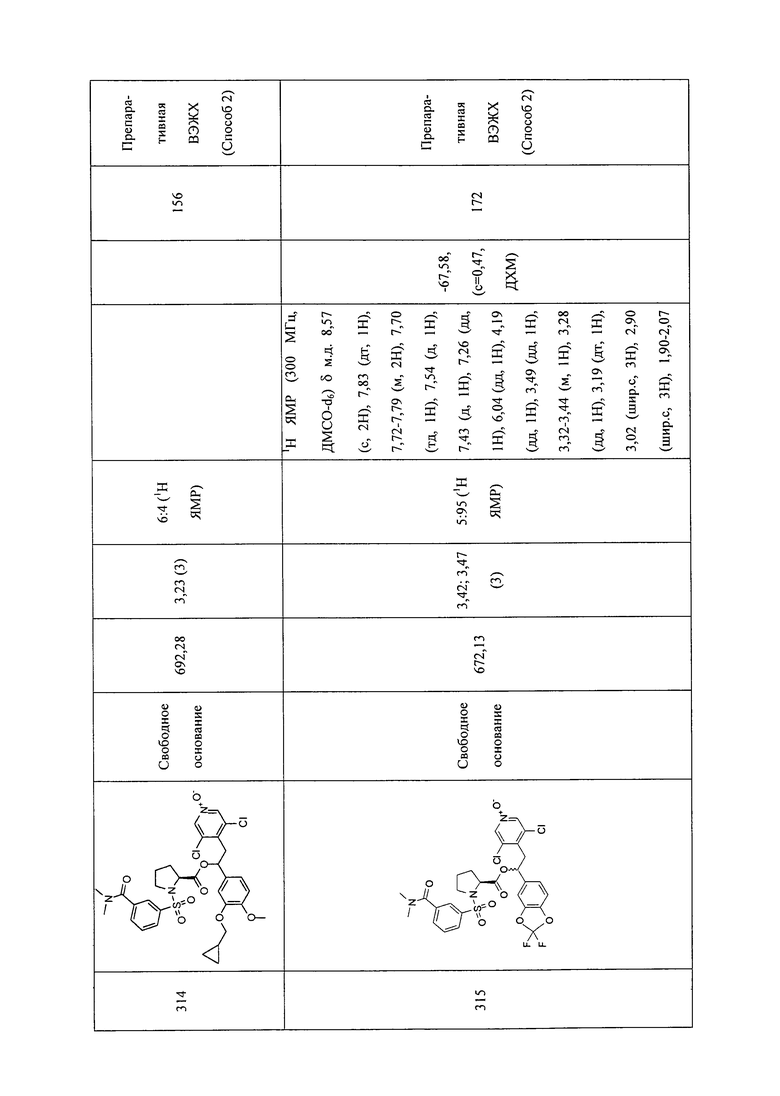
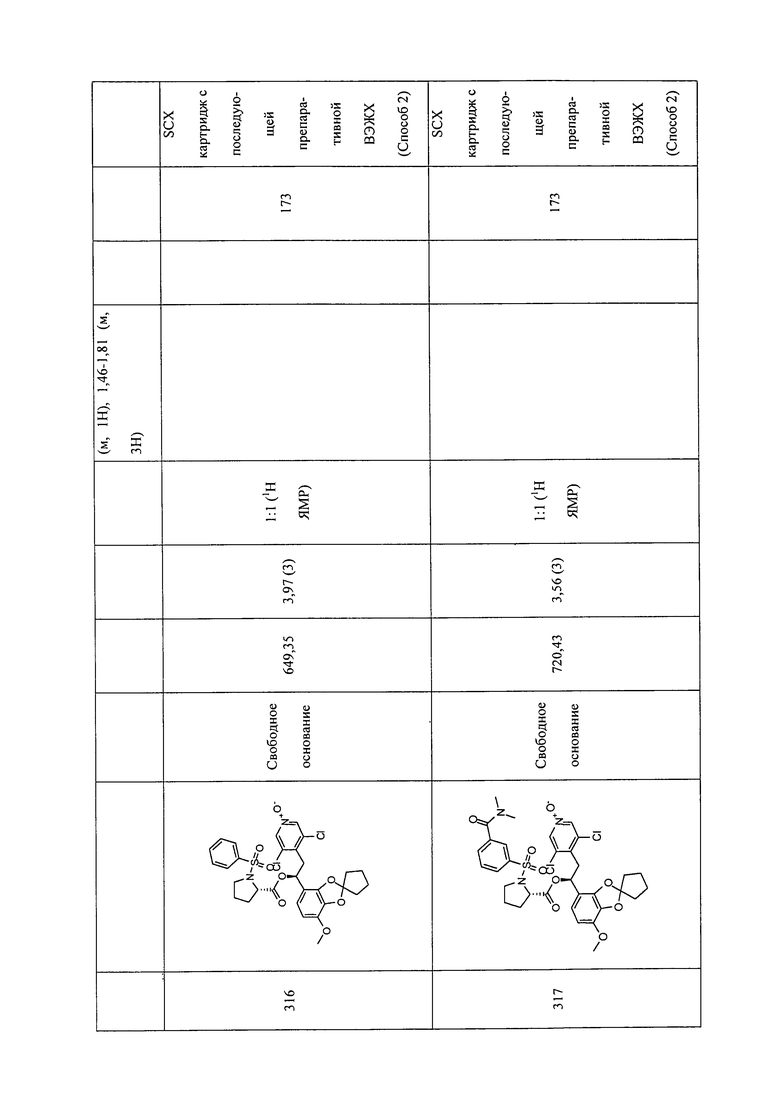
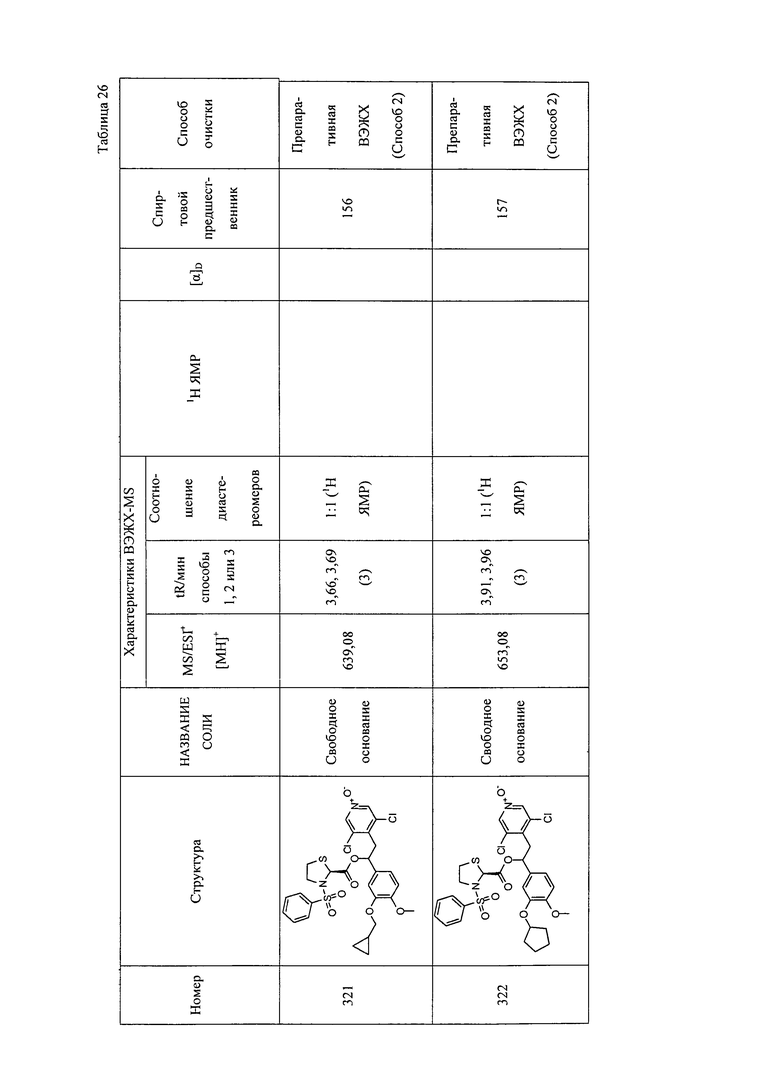
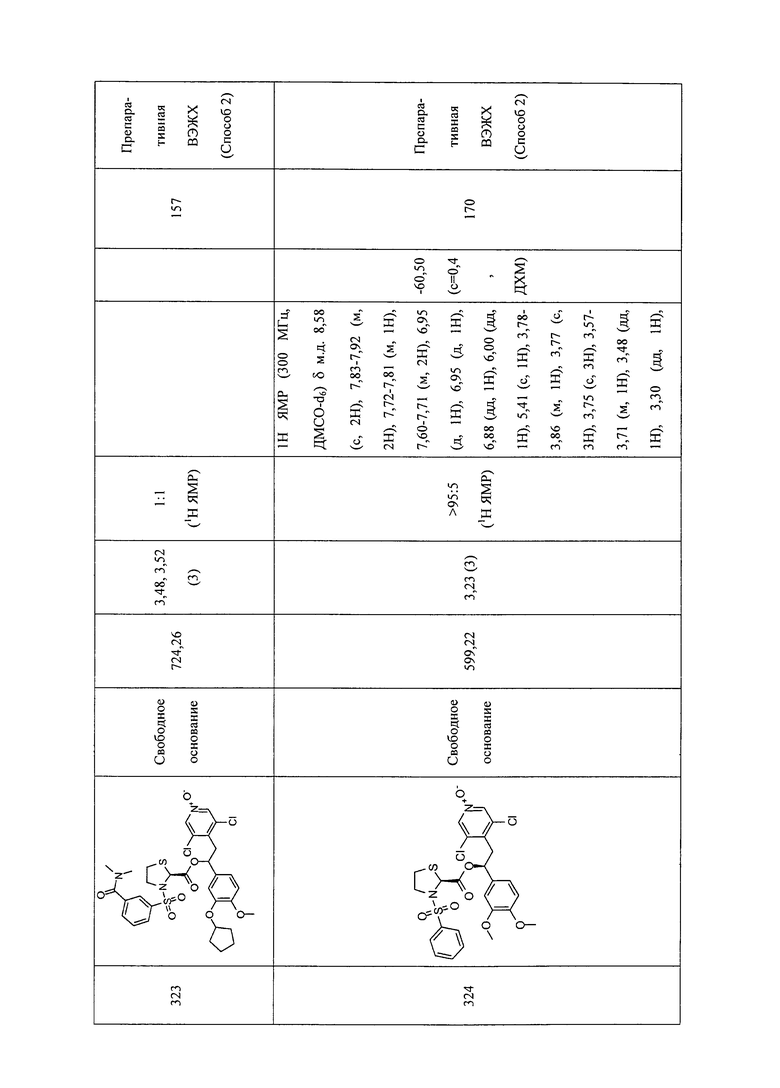
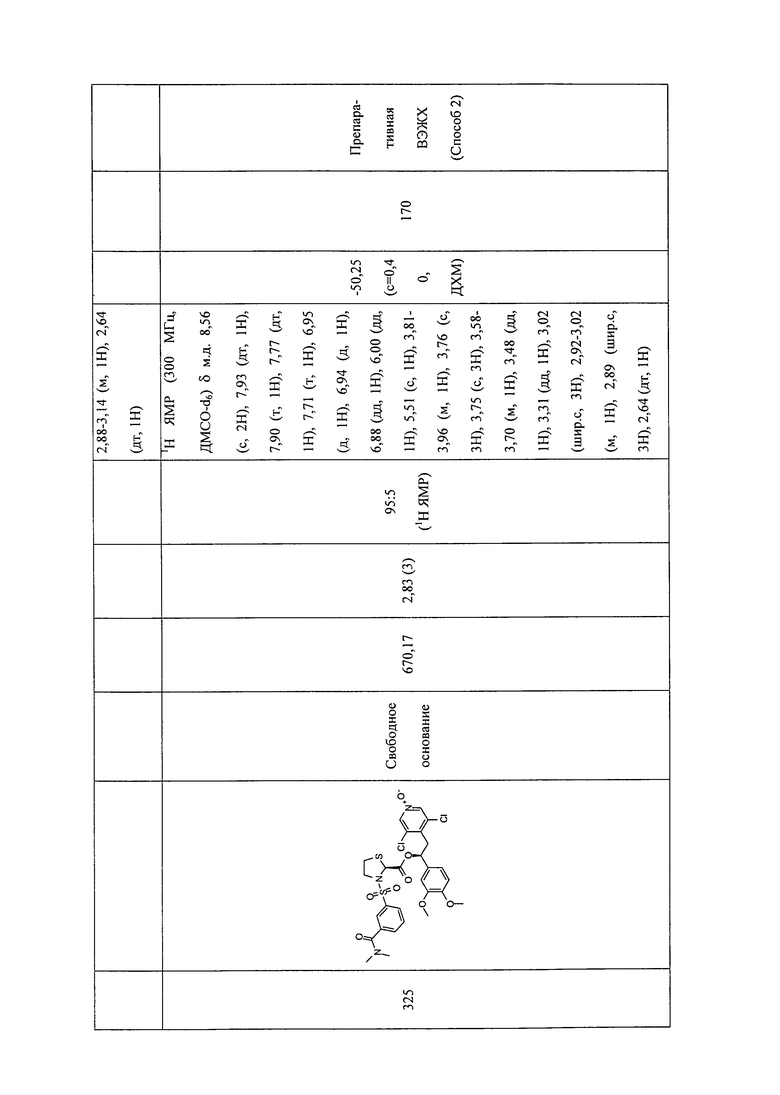
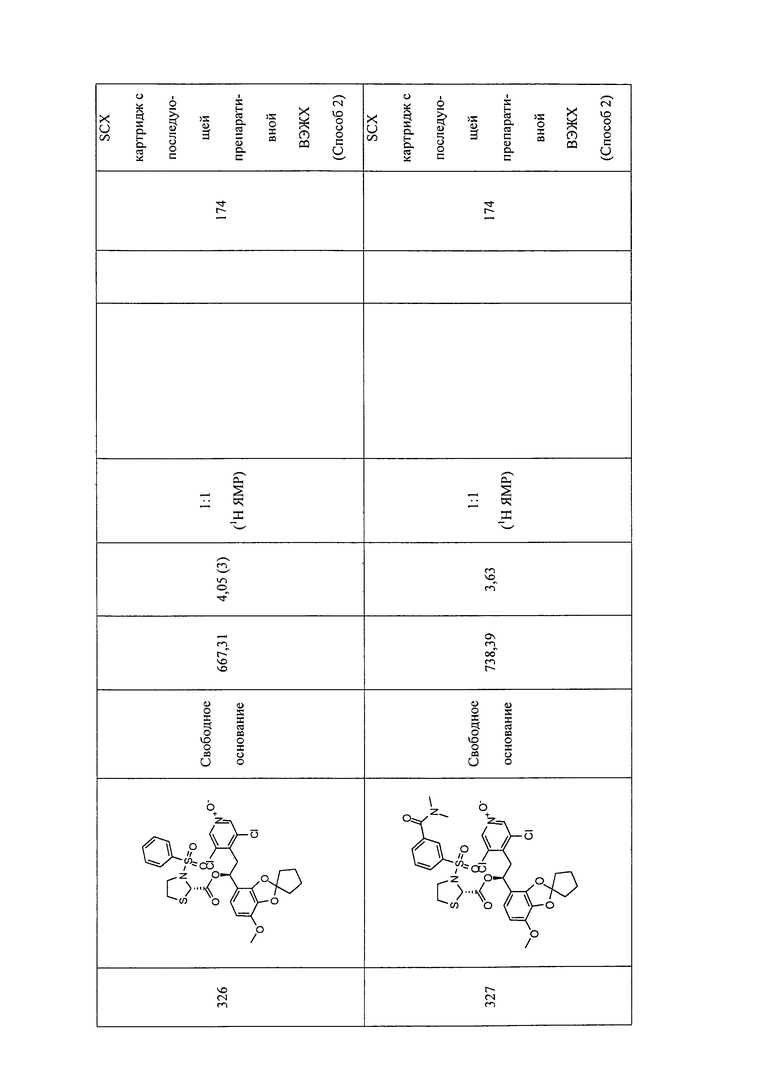
Изобретение относится к соединению общей формулы (I)
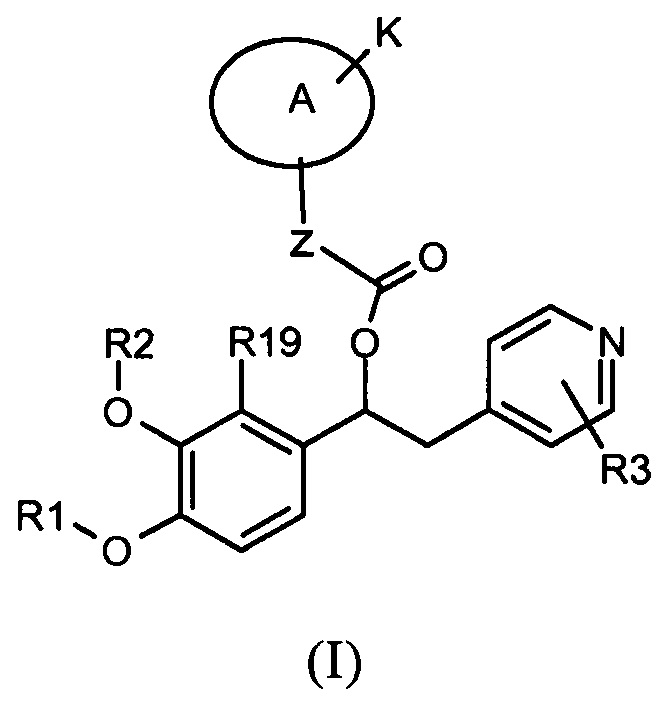
где: R1 выбран из группы, состоящей из: метила; трифторметила; R2 выбран из группы, состоящей из: метила, необязательно замещенного циклопропилом; циклопентила; или R1 и R2, вместе с соединяющими их атомами, образуют 2,2-дифтор-1,3-диоксолановое кольцо формулы (q), конденсированное с фенильной группой, которая несет группы -OR1 и -OR2, где звездочки указывают на атомы углерода, совместно используемые таким фенильным кольцом:
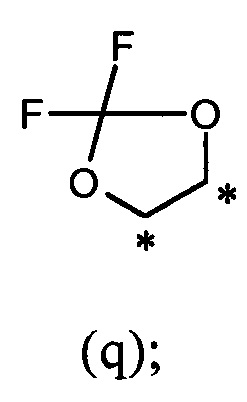
R19 представляет собой водород; R3 является одним или несколькими заместителями, независимо выбранными из атомов галогена; Z представляет собой группу -(СН2)n-, где n равно 0 или 1; А представляет собой насыщенную и моноциклическую (С3-С7) гетероциклоалкиленовую группу, выбранную из представленного ниже списка ди-радикалов: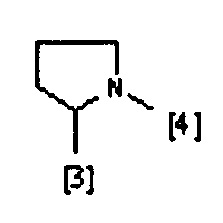 ;
; 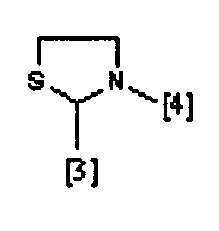 ;
; 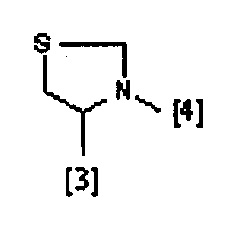 ;
;
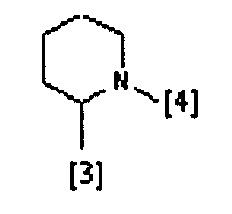 ;
; 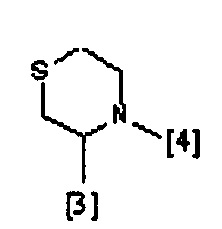 ;
; 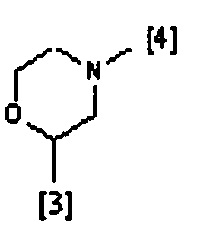
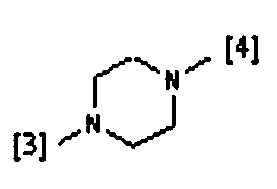 ;
; 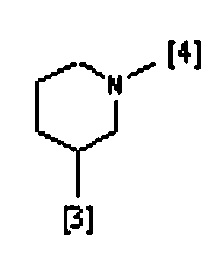 ;
;
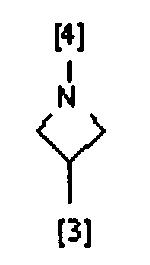 ;
; 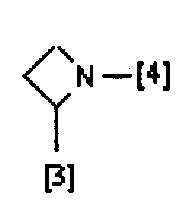 ,
,
где символы [3] и [4] указывают на точки присоединения группы А с группами Z и К соответственно; K выбран из группы, состоящей из: -(СН2)mC(О)R4, где m может быть равно 0 или 1; -С(О)(CH2)jR4, где j может быть равно 1 или 2; -SO2(СН2)pR4, где p может быть равно 0, 1 или 2; -(СН2)ySO2R4, где y может быть равно 1 или 2; -(CH2)zR4, где z может быть равно 1; и -С(O)(CH2)2SO2R4; R4 представляет собой кольцевую систему, которая представляет собой моно- или бициклическое кольцо, которое может быть насыщенным, частично ненасыщенным или полностью ненасыщенным, выбранную из фенила, (С3-С8) циклоалкила, (С3-С7) гетероциклоалкила, в котором по меньшей мере один кольцевой атом углерода заменен гетероатомом, выбранным из N, NH и О, или гетероарила, такое кольцо необязательно замещено одной или несколькими группами R5, которые могут быть одинаковыми или различными, и которые независимо выбраны из группы, состоящей из: (C1-С6) алкила, необязательно замещенного одной или несколькими группами, независимо выбранными из списка, состоящего из: -ОН; (С3-С7) гетероциклоалкил(С1-С4) алкила, в котором по меньшей мере один кольцевой атом углерода заменен гетероатомом, выбранным из N, NH, О; 5-6-членного гетероарила, в котором по меньшей мере один кольцевой атом углерода заменен гетероатомом, выбранным из N и О; группы -OR6, где R6 выбран из группы, состоящей из (C1-С6) галогеналкила; группы -SO2R7, где R7 представляет собой (С1-С4) алкил; и метила, необязательно замещенного одним или несколькими (С3-С7) циклоалкилами; атомов галогена; CN; NR8R9, где R8 и R9 являются различными или одинаковыми и независимо выбраны из группы, состоящей из: Н; (С1-С4) алкилен-NR13R14, где R13 и R14 являются различными или одинаковыми и независимо выбраны из группы, состоящей из: (C1-С6) алкила, или они образуют с атомом азота, к которому они присоединены насыщенное (С3-С7)гетероциклическое кольцо; группы -SO2R15, где R15 выбран из группы, состоящей из: (C1-С4) алкила; группы -C(O)OR17, где R17 выбран из группы, состоящей из: (C1-С6) алкила; или они образуют с атомом азота, к которому они присоединены, насыщенное или частично насыщенное гетероциклическое кольцо, которое необязательно замещено одним или несколькими (C1-С6) алкилом; (С1-С2) алкилен-NR8R9, как указано выше; COR10, где R10 представляет собой (C1-С6) алкил; оксо; -SO2R11, где R11 представляет собой (С1-С4) алкил или NR8R9, где R8 и R9 такие, как указано выше; -COOR12, где R12 представляет собой Н, (С1-С4) алкил или (С1-С4) алкилен-NR8R9, где R8 и R9 такие, как указано выше; и -CONR8R9, где R8 и R9 такие, как указано выше; где группы R6, R8, R9, R10, R11, R12, R13, R14, R15, R17 и R19 в каждом случае могут иметь одинаковое или разное значение, если присутствуют в количестве более одной группы; и их N-оксидные производные на пиридиновом кольце или их фармацевтически приемлемым солям. Соединение являются ингибиторами фермента фосфодиэстеразы 4 (PDE4). 7 н. и 12 з.п. ф-лы, 26 табл., 36 пр.
1. Соединение общей формулы (I)
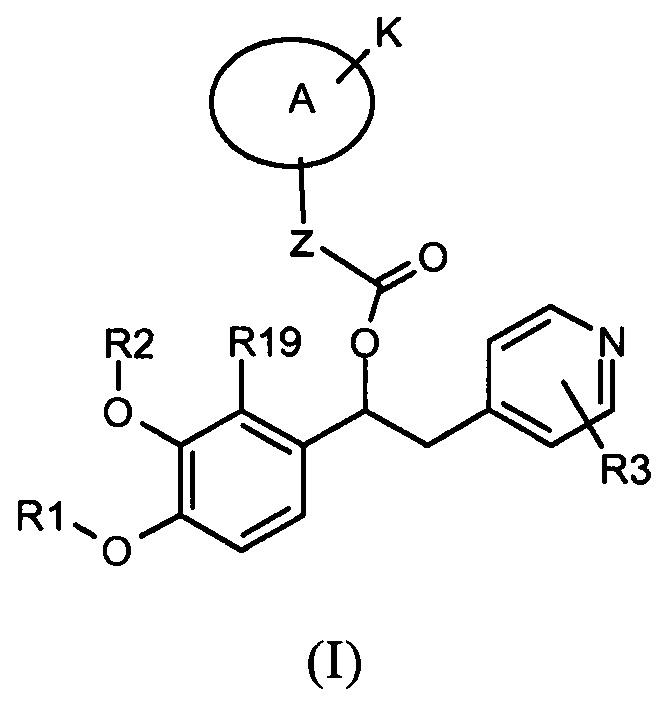
где:
R1 выбран из группы, состоящей из:
- метила;
- трифторметила;
R2 выбран из группы, состоящей из:
- метила, необязательно замещенного циклопропилом;
- циклопентила;
или R1 и R2, вместе с соединяющими их атомами, образуют 2,2-дифтор-1,3-диоксолановое кольцо формулы (q), конденсированное с фенильной группой, которая несет группы -OR1 и -OR2, где звездочки указывают на атомы углерода, совместно используемые таким фенильным кольцом:
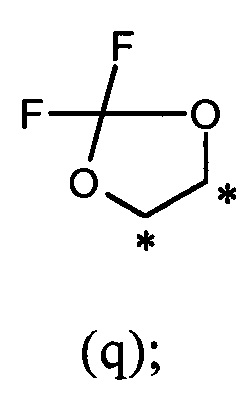
R19 представляет собой водород;
R3 является одним или несколькими заместителями, независимо выбранными из атомов галогена;
Z представляет собой группу -(СН2)n-, где n равно 0 или 1;
А представляет собой насыщенную и моноциклическую (С3-С7) гетероциклоалкиленовую группу, выбранную из представленного ниже списка ди-радикалов:
 ;
; 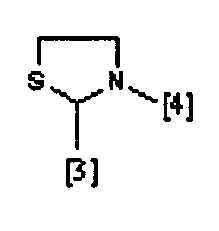 ;
; 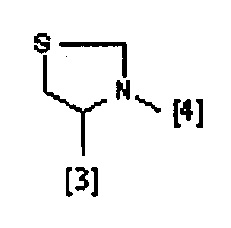 ;
;
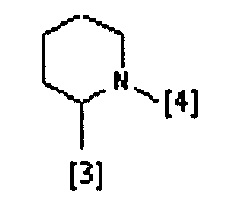 ;
; 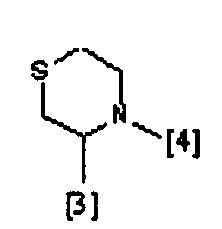 ;
; 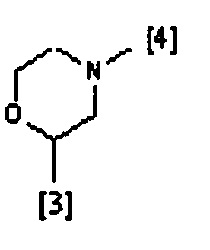
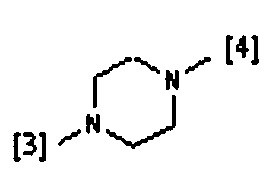 ;
; 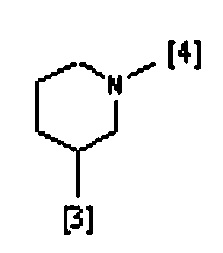 ;
;
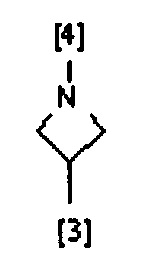 ;
; 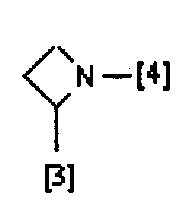 ,
,
где символы [3] и [4] указывают на точки присоединения группы А с группами Z и К соответственно;
K выбран из группы, состоящей из:
- -(СН2)mC(О)R4, где m может быть равно 0 или 1;
- -С(О)(CH2)jR4, где j может быть равно 1 или 2;
- -SO2(СН2)pR4, где p может быть равно 0, 1 или 2;
- -(СН2)ySO2R4, где y может быть равно 1 или 2;
- -(CH2)zR4, где z может быть равно 1; и
- -С(O)(CH2)2SO2R4;
R4 представляет собой кольцевую систему, которая представляет собой моно- или бициклическое кольцо, которое может быть насыщенным, частично ненасыщенным или полностью ненасыщенным, выбранную из фенила, (С3-С8) циклоалкила, (С3-С7) гетероциклоалкила, в котором по меньшей мере один кольцевой атом углерода заменен гетероатомом, выбранным из N, NH и О, или гетероарила, такое кольцо необязательно замещено одной или нескольими группами R5, которые могут быть одинаковыми или различными и которые независимо выбраны из группы, состоящей из:
- (C1-С6) алкила, необязательно замещенного одной или несколькими группами, независимо выбранными из списка, состоящего из: -ОН;
- (С3-С7) гетероциклоалкил(С1-С4) алкила, в котором по меньшей мере один кольцевой атом углерода заменен гетероатомом, выбранным из N, NH, О;
- 5-6-членного гетероарила, в котором по меньшей мере один кольцевой атом углерода заменен гетероатомом, выбранным из N и О;
- группы -OR6, где R6 выбран из группы, состоящей из
- (C1-С6) галогеналкила;
- группы -SO2R7, где R7 представляет собой (С1-С4) алкил; и
- метила, необязательно замещенного одним или несколькими (С3-С7) циклоалкилами;
- атомов галогена;
- CN;
- NR8R9, где R8 и R9 являются различными или одинаковыми и независимо выбраны из группы, состоящей из:
- Н;
(С1-С4) алкилен-NR13R14, где R13 и R14 являются различными или одинаковыми и независимо выбраны из группы, состоящей из:
(C1-С6) алкила, или они образуют с атомом азота, к которому они присоединены насыщенное (С3-С7)гетероциклическое кольцо;
- группы -SO2R15, где R15 выбран из группы, состоящей из: (C1-С4) алкила;
- группы -C(O)OR17, где R17 выбран из группы, состоящей из: (C1-С6) алкила;
или они образуют с атомом азота, к которому они присоединены, насыщенное или частично насыщенное гетероциклическое кольцо, которое необязательно замещено одним или несколькими (C1-С6) алкилом;
- (С1-С2) алкилен-NR8R9, как указано выше;
- COR10, где R10 представляет собой (C1-С6) алкил;
- оксо;
- -SO2R11, где R11 представляет собой (С1-С4) алкил или NR8R9, где R8 и R9 такие, как указано выше;
- -COOR12, где R12 представляет собой Н, (С1-С4) алкил или (С1-С4) алкилен-NR8R9, где R8 и R9 такие, как указано выше; и
- -CONR8R9, где R8 и R9 такие, как указано выше;
где группы R6, R8, R9, R10, R11, R12, R13, R14, R15, R17 и R19 в каждом случае могут иметь одинаковое или разное значение, если присутствуют в количестве более одной группы; и
их N-оксидные производные на пиридиновом кольце или их фармацевтически приемлемые соли.
2. Соединение формулы (IC) по п. 1,
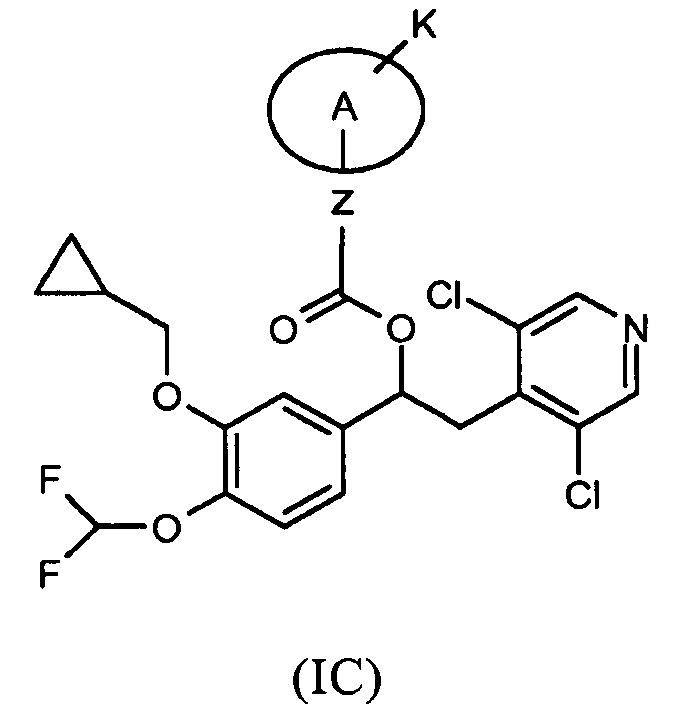
где K, Z и А определены для соединения формулы (I); N-оксидное производное на пиридиновом кольце или их фармацевтически приемлемые соли.
3. Соединение формулы (IF) по п. 1 или 2:
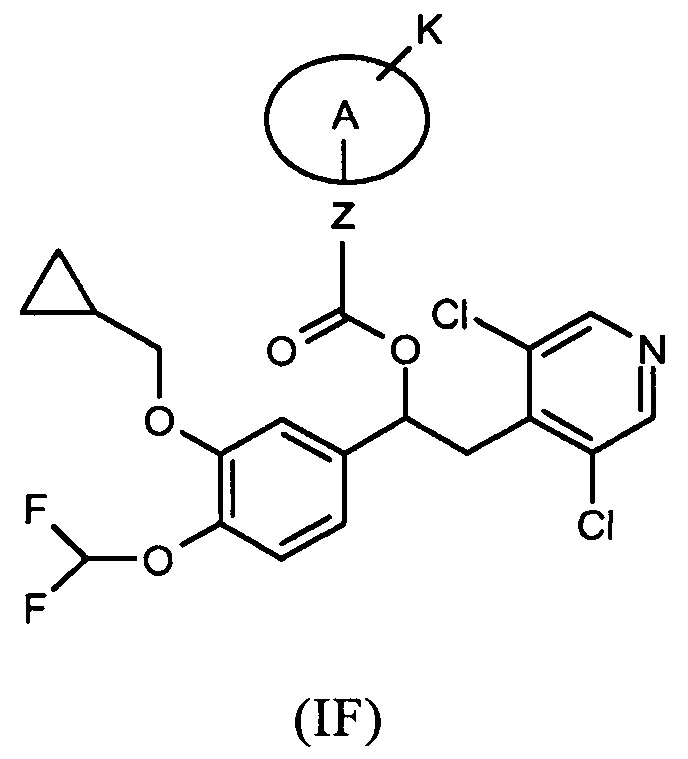
где K выбран из перечня групп, состоящего из:
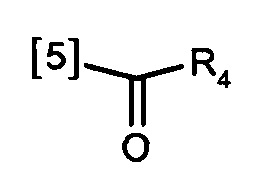 ;
; 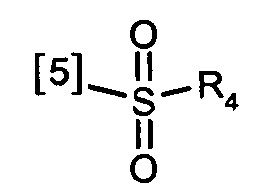 ;
; 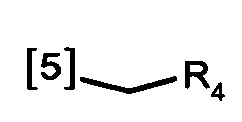 ;
; 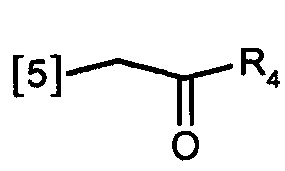
R4 представляет собой фенильную группу или 5,6-членную гетероарильную группу, каждая из которых необязательно замещена одной или несколькими группами R5; и N-оксидное производное на пиридиновом кольце или их фармацевтически приемлемые соли.
4. Соединение формулы (IH) по п. 1 или 2, которое представляет собой N-оксид на пиридиновом кольце:
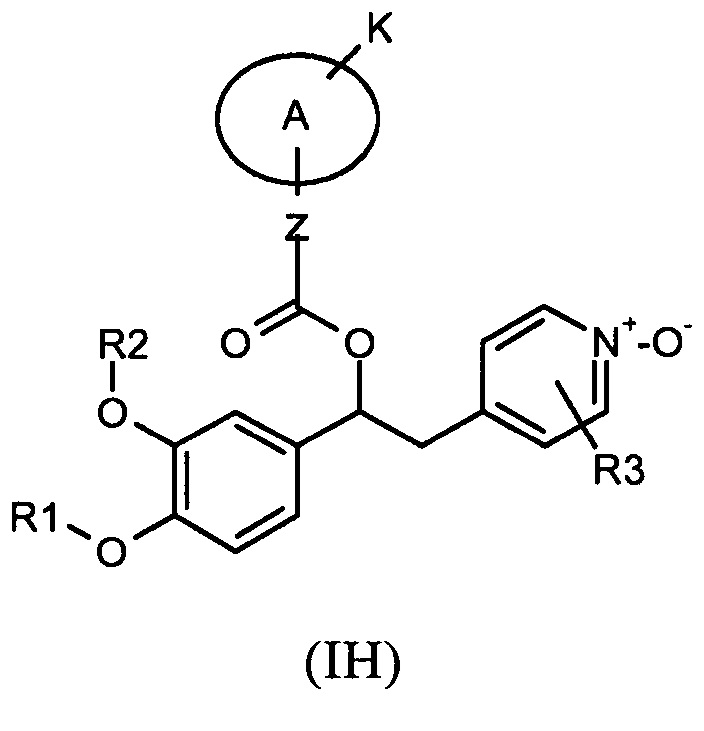
где R1, R2, R3, Z, А и K определены для соединений формулы (I); или его фармацевтически приемлемые соли.
5. Соединение формулы (I)' по п. 1 или 2, которое представляет собой соединение формулы (I), где абсолютная конфигурация углерода (1) такая, как показано ниже:
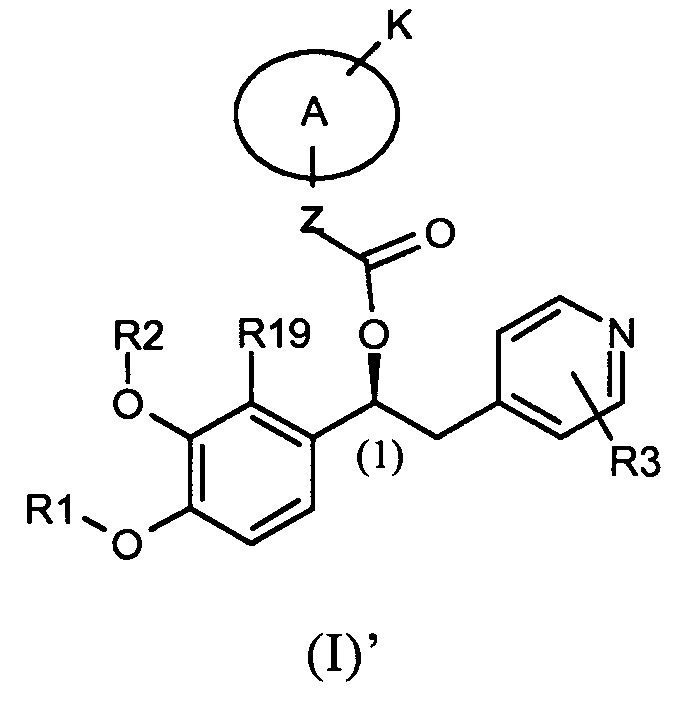
где R1, R2, R3, R19, Z, А и K определены для соединений формулы (I); N-оксидное производное на пиридиновом кольце или его фармацевтически приемлемые соли.
6. Соединение формулы (IDa) по п. 1 или 2:
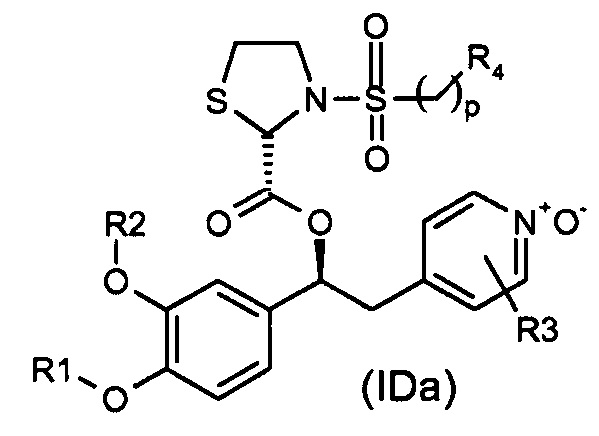
где R1, R2, R3, р, R4 определены для соединений формулы (I); или его фармацевтически приемлемые соли или сольваты.
7. Соединение по п. 1, которое выбрано из группы, состоящей из:
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-1-(3-(циклопропилметокси)-4-(метилсульфонамидо)бензоил)пирролидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-1-(4-метокси-3-(метилсульфонилокси)бензоил)пирролидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-1-(3,4-диметоксифенилсульфонил)пирролидин-2-карбонилокси)этил)пиридина;
1-оксида 4-((2S)-2-(3-(4-аминофенилсульфонил)тиазолидин-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)-3,5-дихлорпиридина;
1-оксида 3,5-дихлор-4-((2S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(3-(4-(метилсульфонамидо)фенилсульфонил)тиазолидин-2-
карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((2S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(3-(4-(N-(2-морфолиноэтил)метилсульфонамидо)фенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 4-((S)-2-((R)-3-(4-аминофенилсульфонил)тиазолидин-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)-3,5-дихлорпиридина;
1-оксида 4-((S)-2-((S)-3-(4-аминофенилсульфонил)тиазолидин-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)-3,5-дихлорпиридина;
1-оксида 4-((S)-2-((S)-1-(4-аминофенилсульфонил)пирролидин-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)-3,5-дихлорпиридина;
1-оксида 3,5-дихлор-4-((2S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(1-((4-(метоксикарбонил)-5-метилфуран-2-ил)метил)пирролидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-1-(3-(диметилкарбамоил)-4-метоксифенилсульфонил)пирролидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((R или S)-4-(3-сульфамоилфенилсульфонил)морфолин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((2S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(3-(2-оксо-2-(тиофен-2-ил)этил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S или R)-3-(4-(диметилкарбамоил)бензил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S или R)-3-(4-(диметилкарбамоил)бензил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 4-((2S)-2-(3-(4-аминобензоил)тиазолидин-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)-3,5-дихлорпиридина;
1-оксида 3,5-дихлор-4-((2S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(3-(3-(диметилкарбамоил)фенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((2S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(4-(3-(диметилкарбамоил)фенилсульфонил)тиоморфолин-3-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((2S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(4-(4-(N-метилсульфамоил)фенилсульфонил)тиоморфолин-3-карбонилокси)этил)пиридина;
1-оксида 4-((2S)-2-(3-(3-амино-4-метоксифенилсульфонил)тиазолидин-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)-3,5-дихлорпиридина;
1-оксида 3,5-дихлор-4-((2S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(3-(фенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 4-((S)-2-((R)-3-(4-аминофенилсульфонил)тиазолидин-4-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)-3,5-дихлорпиридина;
1-оксида 4-((2S)-2-(4-(4-аминофенилсульфонил)морфолин-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)-3,5-дихлорпиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-1-(3-(диметилкарбамоил)фенилсульфонил)пирролидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((2S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(4-(3-(диметилкарбамоил)фенилсульфонил)морфолин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((R)-3-(3-(диметилкарбамоил)фенилсульфонил)тиазолидин-4-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((2S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(3-(1,3-диоксоизоиндолин-5-илсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((2S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(3-(4-(диметилкарбамоил)фенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((R)-1-(3-(диметилкарбамоил)фенилсульфонил)пирролидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((2S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(3-(3-сульфамоилфенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 4-((2S)-2-(3-(3-карбокси-4-метоксифенилсульфонил)тиазолидин-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)-3,5-дихлорпиридина;
1-оксида 3,5-дихлор-4-((2S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(3-(3-фторфенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((2S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(3-(2,4-диметилфенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((2S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(3-(тиофен-2-илсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((2S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(3-(3-(диметилкарбамоил)-4-метоксифенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида (S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(2-(4-(3-(диметилкарбамоил)фенилсульфонил)пиперазин-1-ил)ацетокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((2S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(3-(4-(4-метилпиперазин-1-карбонил)фенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((2S)-2-(3-(3-хлорфенилсульфонил)тиазолидин-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)пиридина;
1-оксида 3,5-дихлор-4-((2S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(3-(1-метил-1Н-имидазол-2-илсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((2S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(3-(циклопропилметилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((2S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(3-(пиридин-3-илсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((2S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(3-(2,4-дифторфенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((2S)-2-(3-(2-хлор-4-фторфенилсульфонил)тиазолидин-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)пиридина;
1-оксида 3,5-дихлор-4-((2S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(3-(4-фтор-2-метилфенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((2S)-2-(3-(2-хлорфенилсульфонил)тиазолидин-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)пиридина;
1-оксида 3,5-дихлор-4-((2S)-2-(3-(циклогексилсульфонил)тиазолидин-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)пиридина;
1-оксида 3,5-дихлор-4-((2S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(3-(2,4-диоксо-1,2,3,4-тетрагидрохиназолин-6-илсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((2S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(3-(тиофен-3-илсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(3-(диметилкарбамоил)фенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-1-(3-(диметилкарбамоил)фенилсульфонил)пиперидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((R)-1-(3-(диметилкарбамоил)фенилсульфонил)пиперидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((2S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(3-(3-(циклопропилметокси)-5-(N-(2-морфолиноэтил)метилсульфонамидо)бензоил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((2S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(3-(3,4-диметоксифенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида (S)-4-(2-(2-(4-(4-аминофенилсульфонил)пиперазин-1-ил)ацетокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)-3,5-дихлорпиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-1-(3-(диметилкарбамоил)-4-метоксифенилсульфонил)пиперидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((2S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(3-(6-морфолинопиридин-3-илсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((2S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(3-(4-метокси-3-(4-метилпиперазин-1-карбонил)фенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((2S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(3-(4-метокси-3-(морфолин-4-карбонил)фенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S или R)-3-(4-метокси-3-(морфолинометил)бензоил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((2S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(3-(3-(N,N-диметилсульфамоил)-4-метоксибензоил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 4-((S)-2-((S или R)-3-(3-карбоксифенилсульфонил)тиазолидин-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)-3,5-дихлорпиридина;
1-оксида 3,5-дихлор-4-((2S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(3-(4-(морфолинометил)бензоил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((2S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(4-(фенилсульфонил)морфолин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((2S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(3-(3,5-диметилизоксазол-4-илсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((2S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(3-(тиазол-5-карбонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((2S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(3-(3-((диметиламино)метил)бензоил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((2S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(3-(оксазол-5-карбонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((2S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(3-(3-(4-метилпиперазин-1-карбонил)фенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-1-(1-метил-1Н-имидазол-2-илсульфонил)пирролидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-1-(фенилсульфонил)пирролидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((2S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(3-(5-(метоксикарбонил)тиофен-2-илсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(пиридин-3-илсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(3-(диметилкарбамоил)-4-метоксифенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(фенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(1-метил-1Н-имидазол-2-илсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(3-сульфамоилфенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((2S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(3-(4-(метилсульфонил)фенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(3,4-диметоксифенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((2S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(3-(5-((диметиламино)метил)тиофен-2-карбонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 4-((2S)-2-(3-(4-(2-аминоэтил)бензоил)тиазолидин-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)-3,5-дихлорпиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(4-(N-метилсульфамоил)фенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(фуран-2-илсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(фуран-3-илсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(3-(N,N-диметилсульфамоил)фенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-1-(3,4-диметоксифенилсульфонил)пиперидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-1-(пиридин-3-илсульфонил)пиперидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(4-(метоксикарбонил)фенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(2-(метоксикарбонил)фенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(3-(диметилкарбамоил)фенилсульфонил)тиазолидин-4-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(3-метоксифенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(3-(трифторметокси)фенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(4-(1,1-диоксотиоморфолинобензоил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 4-((S)-2-((S)-3-(4-карбамоилбензоил)тиазолидин-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)-3,5-дихлорпиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(6-морфолинопиридин-3-илсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 4-((S)-2-((S или R)-3-(4-(аминометил)пиколиноил)тиазолидин-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)-3,5-дихлорпиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(2-метокси-4-метилфенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(2,4-диметилтиазол-5-илсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(4-метил-3,4-дигидро-2Н-бензо[b][1,4]оксазин-6-илсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((2S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(3-пиколиноилтиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S или R)-3-(3-((2-морфолиноэтокси)карбонил)фенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(4-метоксифенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-1-(6-морфолинопиридин-3-илсульфонил)пиперидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-1-(4-нитрофенилсульфонил)пиперидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-1-(3-(N,N-диметилсульфамоил)фенилсульфонил)пиперидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-1-(фенилсульфонил)пиперидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(2,5-диметоксифенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-1-(1-метил-1Н-имидазол-2-илсульфонил)пиперидин-2-карбонилокси)этил)пиридина;
1-оксида 4-((S)-2-((S)-3-(3-ацетилфенилсульфонил)тиазолидин-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)-3,5-дихлорпиридина;
1-оксид 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(4-(морфолинометил)бензоил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(4-(1,1-диоксотиоморфолинометил)бензоил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 4-((S)-2-((S)-3-(3-(аминометил)бензоил)тиазолидин-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)-3,5-дихлорпиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(3-(оксазол-5-ил)бензоил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 4-((S)-2-((S)-3-(3-аминофенилсульфонил)тиазолидин-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)-3,5-дихлорпиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(4-(метилсульфонил)фенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(3-(4-метилпиперазин-1-карбонил)фенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((2S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(3-(3-(N-метилсульфамоил)фенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(1-метил-1Н-имидазол-4-илсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(2-фенилацетил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-((S)-3-(2-циклопропилацетил)тиазолидин-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(3-(фенилсульфонил)пропаноил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(3-морфолинопропаноил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(3-(4-метилпиперазин-1-ил)пропаноил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-1-(3-(диметилкарбамоил)бензоил)пирролидин-2-карбонилокси)этил)пиридина;
1-оксида 4-((S)-2-(2-((S)-1-бензоилпирролидин-2-ил)ацетокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)-3,5-дихлорпиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(2-((S)-1-(3-(диметилкарбамоил)бензоил)пирролидин-2-ил)ацетокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(2-(3-(диметилкарбамоил)фенил)ацетил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-1-(2-(3-(диметилкарбамоил)фенил)ацетил)пирролидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-((S)-3-(2-цианофенилсульфонил)тиазолидин-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(2,3-дигидробензо[b][1,4]диоксин-6-илсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-((S)-3-(2-циано-5-метилфенилсульфонил)тиазолидин-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(2,5-диметилтиофен-3-илсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 4-((S)-2-((S)-3-(4-бром-2-фтор-5-метилфенилсульфонил)тиазолидин-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)-3,5-дихлорпиридина;
1-оксида 4-((S)-2-((S)-3-(3-бром-4-метилфенилсульфонил)тиазолидин-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)-3,5-дихлорпиридина;
1-оксида 3,5-дихлор-4-((S)-2-((S)-3-(4-цианофенилсульфонил)тиазолидин-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-((S)-3-(3-цианофенилсульфонил)тиазолидин-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)пиридина;
1-оксида 4-((S)-2-((S)-3-(4-(1Н-пиразол-1-ил)фенилсульфонил)тиазолидин-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)-3,5-дихлорпиридина;
1-оксида 3,5-дихлор-4-((S)-2-((S)-3-(3-циано-4-фторфенилсульфонил)тиазолидин-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(1-метил-2-оксоиндолин-5-илсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-((S)-3-(2-хлор-5-цианофенилсульфонил)тиазолидин-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(5-метилбензо[b]тиофен-2-илсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(4-(1-метил-1Н-пиразол-3 -ил)фенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(4-(дифторметокси)фенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-((S)-3-(4-хлор-2-(трифторметил)фенилсульфонил)тиазолидин-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(5-фтор-2-метоксифенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 4-((S)-2-((S)-3-(бензо[b]тиофен-2-илсульфонил)тиазолидин-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)-3,5-дихлорпиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(2-оксо-2,3-дигидробензо[d]оксазол-6-илсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(4-(2-оксопирролидин-1-ил)фенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 4-((S)-2-((S)-3-(1-ацетил-1,2,3,4-тетрагидрохинолин-6-илсульфонил)тиазолидин-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)-3,5-дихлорпиридина;
1-оксида 4-((S)-2-((S)-3-(4-(2-ацетамидоэтил)фенилсульфонил)тиазолидин-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)-3,5-дихлорпиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(4-(2,2,2-трифторэтокси)фенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 4-(2-((S)-3-(бензилсульфонил)тиазолидин-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)-3,5-дихлорпиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(фенэтилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 4-((S)-2-((S)-1-(бензилсульфонил)пирролидин-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)-3,5-дихлорпиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((R)-3-(1-метил-2-оксоиндолин-5-илсульфонил)тиазолидин-4-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((R)-1-(фенилсульфонил)пиперидин-3-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(R)-1-(3-(диметилкарбамоил)фенилсульфонил)пиперидин-3-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-1-(фенилсульфонил)пиперидин-3-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-1-(3-(диметилкарбамоил)фенилсульфонил)пиперидин-3-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(R)-4-(фенилсульфонил)морфолин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(R)-4-(3-(диметилкарбамоил)фенилсульфонил)морфолин-2-карбонилокси)этил)пиридина;
3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-4-(фенилсульфонил)морфолин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-4-(3-(диметилкарбамоил)фенилсульфонил)морфолин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(2-((S)-1-(фенилсульфонил)пирролидин-2-ил)ацетокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(2-((S)-1-(3-(диметилкарбамоил)фенилсульфонил)пирролидин-2-ил)ацетокси)этил)пиридина;
1-оксида 4-((S)-2-(2-((S)-1-(бензилсульфонил)пирролидин-2-ил)ацетокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)-3,5-дихлорпиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(2-оксо-2-фенилэтил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-1-(2-оксо-2-фенилэтил)пирролидин-2-карбонилокси)этил)пиридина;
1-оксида 4-((S)-2-((S)-1-бензилпирролидин-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)-3,5-дихлорпиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-1-(3-(диметилкарбамоил)бензил)пирролидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(2-(3-(диметилкарбамоил)фенил)-2-оксоэтил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-1-(2-(3-(диметилкарбамоил)фенил)-2-оксоэтил)пирролидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(циклопропилметил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 4-((S)-2-((S)-3-бензилтиазолидин-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)-3,5-дихлорпиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(3-(диметилкарбамоил)бензил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-фенэтилтиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(2-((S)-1-(3-(диметилкарбамоил)бензил)пирролидин-2-ил)ацетокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(3-(гидроксиметил)фенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 4-((2S)-2-(2-(3-бензоилтиазолидин-2-ил)ацетокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)-3,5-дихлорпиридина;
1-оксида 3,5-дихлор-4-((2S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(2-(3-(3-(диметилкарбамоил)бензоил)тиазолидин-2-ил)ацетокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((2S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(2-(3-(фенилсульфонил)тиазолидин-2-ил)ацетокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((2S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(2-(3-(3-(диметилкарбамоил)фенилсульфонил)тиазолидин-2-ил)ацетокси)этил)пиридина;
1-оксида (S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(1-(3-(диметилкарбамоил)фенилсульфонил)азетидин-3-карбонилокси)этил)пиридина;
1-оксида (S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(1-(фенилсульфонил)азетидин-3-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-1-(3-(диметилкарбамоил)фенилсульфонил)азетидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-1-(фенилсульфонил)азетидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-1-(1-метил-2-оксоиндолин-5-илсульфонил)пирролидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(2-морфолиноэтилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(2-(4-метилпиперазин-1-ил)этилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-1-(2-(фенилсульфонил)этил)пирролидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-1-(2-фенилацетил)пирролидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(3-(диметилкарбамоил)бензилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(4-(дифторметокси)-3-метоксифенил)-2-((S)-3-(3-(диметилкарбамоил)фенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(4-(дифторметокси)-3-метоксифенил)-2-((S)-3-(фенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопентилокси)-4-(дифторметокси)фенил)-2-((S)-3-(фенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопентилокси)-4-(дифторметокси)фенил)-2-((S)-3-(3-(диметилкарбамоил)фенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S или R)-2-(2,2-дифторбензо[d][1,3]диоксол-5-ил)-2-((S)-3-(3-(диметилкарбамоил)фенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(4-(дифторметокси)-3-метоксифенил)-2-((S)-1-(фенилсульфонил)пирролидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(4-(дифторметокси)-3-метоксифенил)-2-((S)-1-(3-(диметилкарбамоил)фенилсульфонил)пирролидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопентилокси)-4-(дифторметокси)фенил)-2-((S)-1-(фенилсульфонил)пирролидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопентилокси)-4-(дифторметокси)фенил)-2-((S)-1-(3-(диметилкарбамоил)фенилсульфонил)пирролидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3,4-диметоксифенил)-2-((S)-1-(фенилсульфонил)пирролидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3,4-диметоксифенил)-2-((S)-1-(3-(диметилкарбамоил)фенилсульфонил)пирролидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-(2-(3-(циклопентилокси)-4-метоксифенил)-2-((S)-1-(фенилсульфонил)пирролидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-(2-(3-(циклопентилокси)-4-метоксифенил)-2-((S)-1-(3-(диметилкарбамоил)фенилсульфонил)пирролидин-2-карбонилокси)этил)пиридина;
1-оксида (S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-метоксифенил)-2-(1-(фенилсульфонил)пирролидин-2-карбонилокси)этил)пиридина;
1-оксида (S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-метоксифенил)-2-(1-(3-(диметилкарбамоил)фенилсульфонил)пирролидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S или R)-2-(2,2-дифторбензо[d][1,3]диоксол-5-ил)-2-((S)-1-(3-(диметилкарбамоил)фенилсульфонил)пирролидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-(2-(4-метоксиспиро[бензо[d][1,3]диоксол-2,1'-циклопентан]-7-ил)-2-((S)-1-(фенилсульфонил)пирролидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-(2-((S)-1-(3-(диметилкарбамоил)фенилсульфонил)пирролидин-2-карбонилокси)-2-(4-метоксиспиро[бензо[d][1,3]диоксол-2,1'-циклопентан]-7-ил)этил)пиридина;
1-оксида 3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-метоксифенил)-2-((S)-3-(3-(диметилкарбамоил)фенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-метоксифенил)-2-((S)-3-(фенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-(2-(3-(циклопентилокси)-4-метоксифенил)-2-((S)-3-(фенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-(2-(3-(циклопентилокси)-4-метоксифенил)-2-((S)-3-(3-(диметилкарбамоил)фенилсульфонил)тиазолидин-2-карбонилокси)этил)-пиридин;
1-оксида 3,5-дихлор-4-((S)-2-(3,4-диметоксифенил)-2-((S)-3-(фенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3,4-диметоксифенил)-2-((S)-3-(3-(диметилкарбамоил)фенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-(2-(4-метоксиспиро[бензо[d][1,3]диоксол-2,1'-циклопентан]-7-ил)-2-((S)-3-(фенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-(2-((S)-3-(3-(диметилкарбамоил)фенилсульфонил)тиазолидин-2-карбонилокси)-2-(4-метоксиспиро[бензо[d][1,3]диоксол-2,1'-циклопентан]-7-ил)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-1-(3-(N,N-диметилсульфамоил)фенилсульфонил)пирролидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(3-метилизоксазоло[5,4-b]пиридин-5-илсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(1,3-диметил-1Н-пиразоло[3,4-b]пиридин-5-илсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(1-метил-5-(метилкарбамоил)-1Н-пиррол-3-илсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(5-(пирролидин-1-карбонил)-1Н-пиррол-3-илсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
(S)-((S)-1-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(3,5-дихлорпиридин-4-ил)этил)-3-(1-метил-1Н-имидазол-2-илсульфонил)тиазолидин-2-карбоксилата;
1-оксида 4-((S)-2-((S)-3-(1Н-1,2,4-триазол-5-илсульфонил)тиазолидин-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)-3,5-дихлорпиридина;
1-оксида 4-((S)-2-((S)-3-бензоилтиазолидин-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)-3,5-дихлорпиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(3-(диметилкарбамоил)бензоил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((R)-1-(фенилсульфонил)пирролидин-2-карбонилокси)этил)пиридина;
1-оксида 4-((S)-2-((R)-1-бензоилпирролидин-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)-3,5-дихлорпиридина;
3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((R)-1-(3-(диметилкарбамоил)бензоил)пирролидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((R)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((R)-1-(3-(диметилкарбамоил)фенилсульфонил)пирролидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((R)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-1-(3-(диметилкарбамоил)фенилсульфонил)пирролидин-2-карбонилокси)этил)пиридина;
(S)-((S)-1-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(3,5-дихлорпиридин-4-ил)этил)-3-(3-(диметилкарбамоил)фенилсульфонил)тиазолидин-2-карбоксилат;
1-оксида 3,5-дихлор-4-((R)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((R)-3-(3-(диметилкарбамоил)фенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((R)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(3-(диметилкарбамоил)фенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-1-(4-метил-3,4-дигидро-2Н-бензо[b][1,4]оксазин-6-илсульфонил)пирролидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-1-(3-метилизоксазоло[5,4-b]пиридин-5-илсульфонил)пирролидин-2-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((R)-3-(4-метил-3,4-дигидро-2Н-бензо[b][1,4]оксазин-6-илсульфонил)тиазолидин-4-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((R)-3-(3-метилизоксазоло[5,4-b]пиридин-5-илсульфонил)тиазолидин-4-карбонилокси)этил)пиридина;
1-оксида 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(3-(метилкарбамоил)фенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина;
или его фармацевтически приемлемая соль.
8. Соединение, представляющее собой 1-оксид 3,5-дихлор-4-((S)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-((S)-3-(3-уреидофенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина или его фармацевтически приемлемую соль.
9. Соединение, представляющее собой 1-оксид 3,5-дихлор-4-((S)-2-(4-(дифторметокси)-3-гидроксифенил)-2-((S)-3-(3-(диметилкарбамоил)фенилсульфонил)тиазолидин-2-карбонилокси)этил)пиридина или его фармацевтически приемлемую соль.
10. Способ получения соединения формулы (IDa) по п. 6, включающий:
- проведение стадии 1):
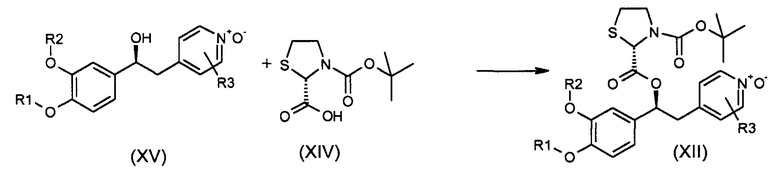
такая стадия 1 включает: а) добавление соединения формулы (XIV), 4-диметиламинопиридина и гидрохлорида 1-этил-3-(3-диметиламинопропил)карбодиимида) к раствору соединения формулы (XV) в диметилформамиде; b) перемешивание смеси; с) выливание смеси в холодную воду; d) фильтрацию остатка;
- а затем проведение стадии 2):
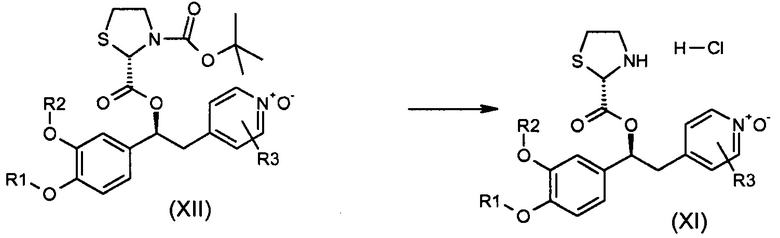
такая стадия 2 включает а) добавление с перемешиванием концентрированного раствора HCl в сухом этилацетате (9 об.) к раствору соединения формулы (XII) в этилацетате при комнатной температуре; b) перемешивание; с) фильтрацию остатка твердого вещества; необязательно, d) промывание полученного твердого вещества этилацетатом;
- а затем проведение стадии 3):
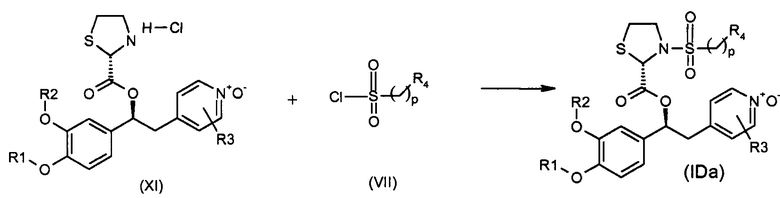
такая стадия 3 включает: а) добавление раствора соединения формулы (VII) в пиридине к охлажденному раствору соединения формулы (XI), перемешивание в пиридине полученного раствора при комнатной температуре; с) заливку раствора в избытке в водную HCl; d) фильтрование осажденного вещества и промывку его водой или d') экстрагирование водной фазы этилацетатом, промывку HCl 1 М, насыщенным солевым раствором и упаривание полученной органической фазы; f) растворение соединения в этаноле (8 об.); g) энергичное перемешивание в течение ночи при комнатной температуре; h) фильтрацию твердого вещества;
где R1, R2, R3, R4 и p в соединениях формул (XV), (XIV), (XII), (XI), (VII) и (IDa) имеют значения в соответствии со значениями соединений формулы (I).
11. Комбинация соединения по любому из пп. 1-9, со вторым фармацевтически активным компонентом, выбранным из бета2-агонистов, выбранных из группы, состоящей из: кармотерола, GSK-642444, индакатерола, милветерола, арформотерола, формотерола, сальбутамола, левалбутерола, тербуталина, AZD-3199, BI-1744-CL, LAS-100977, бамбутерола, изопротеренола, прокатерола, кленбутерола, репротерола, фенотерола и ASF-1020 и их соли, кортикостероидов, выбранных из группы, состоящей из: флутиказона пропионата, флутиказона фуроата, мометазона фуроата, беклометазона дипропионата, циклесонида, будесонида, GSK 685698, GSK 870086 и антимускариновых средств, выбранных из группы, состоящей из: солей аклидиния, тиотропия, ипратропия, троспия, гликопиррония и окситропия.
12. Фармацевтическая композиция, обладающая способностью ингибировать фермент фосфодиэстеразу 4 (PDE4), содержащая терапевтически эффективное количество соединения по любому из пп. 1-9, или комбинацию по п. 11, и один или более фармацевтически приемлемых носителей и/или эксципиентов.
13. Соединение по любому из пп. 1, 7, 8 или 9 для применения в качестве лекарственного средства для профилактики и/или лечения заболевания, характеризующегося сверхактивностью фосфодиэстеразы 4 (PDE4), и/или при котором желательно ингибировать активность PDE4.
14. Соединение по любому из пп. 1, 7, 8 или 9 для применения для профилактики и/или лечения заболевания респираторного тракта, характеризующегося обструкцией дыхательных путей.
15. Соединение для применения по п. 14, в котором заболевание дыхательных путей, характеризующееся обструкцией дыхательных путей, выбирают из астмы и ХОБЛ.
16. Соединение по любому из пп. 1, 7, 8 или 9 для применения в профилактике и/или лечении аллергических ринитов.
17. Соединение по любому из пп. 1, 7, 8 или 9 для применения в профилактике и/или лечении атопического дерматита.
18. Применение соединений по любому из пп. 1-9 для получения лекарственного средства для профилактики и/или лечения заболевания респираторного тракта, характеризующегося обструкцией дыхательных путей.
19. Применение соединений по п. 18, в котором заболевание дыхательных путей, характеризующееся обструкцией дыхательных путей, выбирают из астмы и ХОБЛ.
| СПОСОБ ЛЕЧЕНИЯ И ПРОФИЛАКТИКИ САРКОПТОИДОЗОВ И ДЕМОДЕКОЗОВ ЖИВОТНЫХ | 2000 |
|
RU2216327C2 |
| ШАРНИР МАНИПУЛЯТОРА | 1992 |
|
RU2022783C1 |
| RU 2007117913, A, 20.11.2008. | |||

