ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к ингибиторам фермента фосфодиэстераза 4 (PDE4). Более конкретно, изобретение относится к соединениям, которые представляют собой производные 1-фенил-2-пиридинил-алкиловых спиртов, способам получения таких соединений, содержащим их композициям и их терапевтическому применению.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
Обструкция дыхательных путей характеризует ряд тяжелых респираторных заболеваний, включая астму и хроническую обструктивную болезнь легких (COPD). События, приводящие к обструкции дыхательных путей, включают отек стенок дыхательных путей, усиленную продукции слизи и воспаление.
Лекарственные средства для лечения респираторных заболеваний, таких как астма и COPD, в настоящее время вводят посредством ингаляции. Одним из преимуществ ингаляционного пути введения по сравнению с системным путем является возможность доставки лекарственного средства непосредственно в область его действия, снижение системных побочных эффектов и тем самым получение более быстрого клинического ответа и более высокого терапевтического индекса.
Ингалируемые кортикостероиды являются современной поддерживающей терапией выбора для астмы и вместе с бронходилататорными бета2-агонистами для облегчения острых симптомов они образуют основное направление существующей в настоящее время терапии для этого заболевания. Существующий в настоящее время контроль COPD является главным образом симптоматическим посредством бронходилатационной терапии ингалируемыми антихолинэргическими средствами и ингалируемыми агонистами бета2-адренорецепторов. Однако кортикостероиды не снижают воспалительный ответ при COPD, как это имеет место в случае астмы.
Другой класс терапевтических агентов, которые широко исследовались с точки зрении их противовоспалительных эффектов для лечения воспалительных респираторных заболеваний, таких как астма и COPD, представлен ингибиторами ферментов фосфодиэстераз (PDE), в частности фосфодиэстеразы типа 4 (далее в данном описании именуемой как PDE4).
В уровне техники описаны различные соединения, действующие как PDE4 ингибиторы. Однако пригодность некоторых PDE4 ингибиторов первого поколения, таких как ролипрам и пикламиласт, ограничена вследствие их нежелательных побочных эффектов. Указанные эффекты включают тошноту и рвоту из-за их воздействия на PDE4 в центральной нервной системе и секрецию желудочной кислоты из-за их воздействия на PDE4 в париетальных клетках кишечника.
Причина указанных побочных эффектов широко исследовалась.
Было установлено, что PDE4 существует в двух отдельных формах, представляющих разные конформации, которые были обозначены как сайт связывания ролипрама с высокой аффинностью, или HPDE4, главным образом присутствующий в центральной нервной системе и в париетальных клетках, и сайт связывания ролипрама с низкой аффинностью, или LPDE4 (Jacobitz, S et al. Mol. Pharmacol, 1996, 50, 891-899), который обнаруживается в иммунных и воспалительных клетках. Обе формы, по-видимому, проявляют каталитическую активность, однако они различаются в отношении их чувствительности к ингибиторам. В частности, соединения с более высокой аффинностью к LPDE4, по-видимому, в меньшей степени способны индуцировать побочные эффекты, такие как тошнота, рвота и повышенная желудочная секреция.
Попытка таргетирования LPDE4 имела результатом некоторое улучшение в селективности для PDE4 ингибиторов второго поколения, таких как рофлумиласт. Тем не менее, достижение премлемого профиля побочных эффектов для рофлумиласта зависит от дозы.
В уровне техники раскрыты другие классы соединений, действующих как PDE4 ингибиторы.
Например, в EP 1634606 описаны, среди прочих, кетоновые производные, такие как производные бензофурана или 1,3-бензодиоксола.
В WO 9402465 описаны, среди прочих, кетоновые производные общей формулы

где R1 представляет собой низший алкил, и R2 может представлять собой алкил, алкенил, циклоалкил, циклоалкил, циклоалкенил, циклотиоалкил или циклотиоалкенил.
WO 9535281, заявителем в которой является Celltech Therapeutics, касается три-замещенных фенильных производных.
В WO 2009/018909 описаны производные 1-фенил-2-пиридинил-алкиловых спиртов, которые имеют общую формулу, представленную ниже:
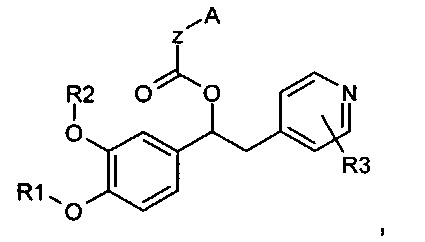
в качестве ингибиторов фермента фосфодиэстераза 4 (PDE4).
В WO 2009/077068 описаны производные 1-фенил-2-пиридинил-алкиловых спиртов, которые имеют общую формулу, представленную ниже:
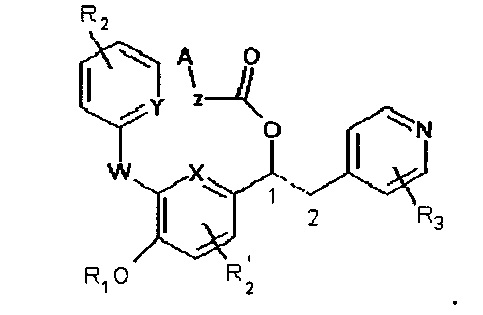
В WO 2010/089107 описаны производные 1-фенил-2-пиридинил-алкиловых спиртов, которые имеют общую формулу, представленную ниже:

в качестве ингибиторов фермента фосфодиэстераза 4 (PDE4).
Хотя некоторые PDE4 ингибиторы были раскрыты в той же мере, что и описанные выше, все еще остается потребность в дополнительных PDE4 ингибиторах. В особенности существует потребность в дополнительных PDE4 ингибиторах с высокой аффинностью в отношении фермента PDE4, которые продемонстрировали бы соответствующий профиль развития в качестве ингаляционного лечения, например, в плане снижения побочных эффектов.
Такое снижение побочных эффектов может быть достигнуто, например, благодаря низкому системному воздействию лекарственного средства; подходящий профиль в плане ряда фармакокинетических характеристик, особенно высокого или умеренного метаболического клиренса, может, таким образом, оказаться ключом к решению этой задачи.
Настоящее изобретение решает указанную выше задачу посредством создания соединений по изобретению.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Изобретение относится к соединениям, действующим как ингибиторы фермента фосфодиэстеразы 4 (PDE4), к способам получения указанных соединений, содержащим их композициям и к их терапевтическому применению.
Изобретение относится к соединениям общей формулы (I)
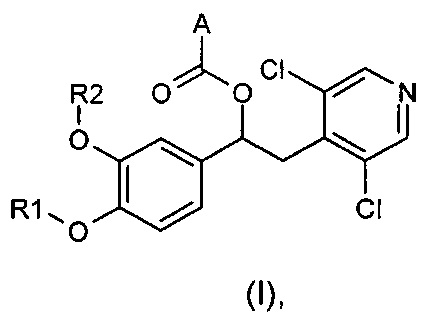
где:
R1 и R2 являются разными или одинаковыми и независимо выбраны из группы, состоящей из:
- Н;
- (C1-C6)алкила, возможно замещенного одним или более заместителями, выбранными из (C3-C7)циклоалкила или (C5-C7)циклоалкенила;
- (C1-C6)галогеналкила;
- (C3-C7)циклоалкила;
- (C5-C7)циклоалкенила;
- (C2-C6)алкенила и
- (C2-C6)алкинила;
А представляет собой моноциклическую гетероарильную кольцевую систему, выбранную из группы, состоящей из радикалов (а), (b), (с) и (d):
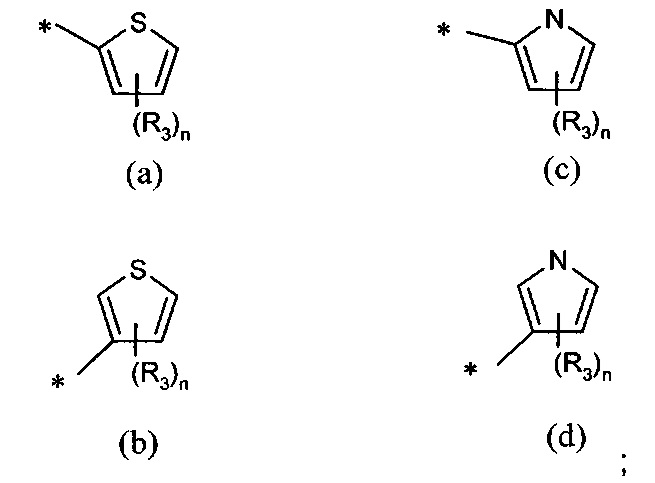
n представляет собой 0, 1 или 2;
R3 представляет собой возможный заместитель, который в каждом случае выбран из группы, состоящей из:
- (C1-C6)алкила, возможно замещенного одним или более (C3-C7)циклоалкилами;
- (C1-C6)галогеналкила;
- OR4, где R4 выбран из группы, состоящей из Н, (C1-C10)алкила, возможно замещенного (C3-C7)циклоалкилом, (C1-C6)галогеналкила, (C3-C7)циклоалкила;
- атомов галогена;
- группы -NR5SO2R6, где
R5 выбран из группы, состоящей из водорода; (C1-C6)алкила; (C1-C6)алкила, который замещен группой -NR7R8, где R7 и R8 независимо представляют собой H, (C1-C6)алкил, или вместе с атомом азота, к которому они присоединены, образуют (C5-C7)гетероциклоалкильную группу; (C1-C6)алкила, который замещен гетероарильной группой; и
R6 представляет собой (C1-C4)алкил, возможно замещенный (C3-C7)циклоалкилом; или фенильное кольцо, возможно замещенное одним или двумя атомами галогена, гидрокси, группами (C1-C4)алкил или (C1-C4)алкокси или группой -C(O)NR9R10, где R9 и R10 независимо представляют собой водород, (C1-C4)алкильные группы, или вместе с атомом азота, к которому они присоединены, образуют (C5-C7)гетероциклоалкильную группу;
- группы -SO2R11, где
R11 выбран из (C1-C6)алкила; фенильного кольца, которое возможно замещено одной или двумя группами, выбранными из списка, состоящего из следующего: атом галогена, гидроксил, (C1-C4)алкил, (C1-C4)алкокси, группа -C(O)NR12R13, где R12 и R13 независимо представляют собой водород, (C1-C4)алкильные группы, или вместе с атомом азота, к которому они присоединены, образуют (C5-C7)гетероциклоалкильную группу, и группа -NR14R15, где R14 и R15 независимо представляют собой водород, (C1-C4)алкильные группы, или вместе с атомом азота, к которому они присоединены, образуют (C5-C7)гетероциклоалкильную группу;
- группы -C(O)R16, где
R16 представляет собой фенильное кольцо, которое возможно замещено одной или двумя группами, выбранными из списка, состоящего из следующего: атом галогена, гидроксил, (C1-C4)алкил, (C1-C4)алкокси, группа -C(O)NR23R24, где R23 и R24 независимо представляют собой водород, (C1-C4)алкильные группы, или вместе с атомом азота, к которому они присоединены, образуют (C5-C7)гетероциклоалкильную группу, и группа -NR25R26, где R25 и R26 независимо представляют собой водород, (C1-C4)алкильные группы, или вместе с атомом азота, к которому они присоединены, образуют (C5-C7)гетероциклоалкильную группу; и
- группы -SO2NR17R18, где
R17 представляет собой Н или (C1-C6)алкил; и
R18 выбран из водорода; (C1-C6)алкила; (C1-C6)алкила, который замещен группой -NR19R20, где R19 и R20 независимо представляют собой H или (C1-C6)алкил, или вместе с атомом азота, к которому они присоединены, образуют (C5-C7)гетероциклоалкильную группу; фенильного кольца, возможно замещенного одним или двумя атомами галогена, гидрокси, группами (C1-C4)алкил или (C1-C4)алкокси, или группой -C(O)NR21R22, где R21 и R22 независимо представляют собой водород, (C1-C4)алкильные группы, или вместе с атомом азота, к которому они присоединены, образуют (C5-C7)гетероциклоалкильную группу;
или их фармацевтически приемлемым солям и N-оксидам по пиридиновому кольцу.
Изобретение также охватывает фармацевтически приемлемые соли и/или сольваты соединений формулы (I).
Изобретение также включает соответствующие N-оксиды по пиридиновому кольцу соединений формулы (I) или их фармацевтически приемлемые соли и/или сольваты.
Далее в данном описании соединения формулы (I), их N-оксиды по пиридиновому кольцу, их воплощения и их фармацевтически приемлемые соли и сольваты, определенные в любом аспекте изобретения (за исключением промежуточных соединений, раскрытых при описании химических способов) именуются как "соединения по изобретению".
Изобретение также включает способ получения соединений по изобретению.
В настоящем изобретении также предложены фармацевтические композиции соединений по изобретению либо самих по себе, либо в комбинации, в смеси с одним или более фармацевтически приемлемыми носителями.
В изобретении также предложено устройство, подходящее для доставки фармацевтических композиций соединений по изобретению.
В дополнительном аспекте настоящего изобретения предложено применение соединений по изобретению в качестве лекарственного средства.
В одном аспекте настоящего изобретения предложено применение соединений по изобретению для изготовления лекарственного средства.
В частности, в настоящем изобретении предложено применение соединений по изобретению для предупреждения и/или лечения любого заболевания, характеризующегося гиперактивностью фосфодиэстеразы 4 (PDE4), и/или при котором желательным является ингибирование активности PDE4.
В частности, соединения по изобретению сами по себе или скомбинированные с другими активными ингредиентами могут быть введены для предупреждения и/или лечения заболевания респираторного тракта, характеризующегося обструкцией дыхательных путей, такого как астма и COPD.
В другом аспекте настоящего изобретения предложено применение соединений по изобретению для изготовления лекарственного средства для предупреждения и/или лечения любого заболевания, характеризующегося гиперактивностью фосфодиэстеразы 4 (PDE4), и/или при котором желательным является ингибирование активности PDE4.
Кроме того, в настоящем изобретении предложен способ предупреждения и/или лечения любого заболевания, при котором желательным является ингибирование активности PDE4, включающий введение нуждающемуся в таком лечении пациенту терапевтически эффективного количества соединения по изобретению.
ОПРЕДЕЛЕНИЯ
Термин "атомы галогена" как он используется в данном описании, включает фтор, хлор, бром и иод, предпочтительно хлор.
Термин "(C1-Cx)алкил", где x представляет собой целое число, большее чем 1, относится к алкильным группам с прямой или разветвленной цепью, где количество составляющих их атомов углерода находится в диапазоне от 1 до x. Конкретные алкильные группы представляют собой метил, этил, н-пропил, изопропил и трет-бутил.
Термин "(C1-Cx)алкоксил", где x представляет собой целое число, большее чем 1, относится к алкокси-группам с прямой или разветвленной цепью, где количество составляющих их атомов углерода находится в диапазоне от 1 до x. Конкретные алкокси-группы представляют собой метоксил, этоксил, н-пропоксил, изопропоксил и трет-бутоксил.
Выражения "(C1-Cx)галогеналкил" относятся к определенным выше группам "(C1-Cx)алкил", где один или более атомов водорода заменены одним или более атомами галогена, которые могут быть одинаковыми или могут отличаться друг от друга.
Примеры указанных (C1-Cx)галогеналкильных групп могут, таким образом, включать галогенированные, поли-галогенированные и полностью галогенированные алкильные группы, где все атомы водорода заменены атомами галогена, например трифторметильные или дифторметильные группы.
Термин "(C3-Cy)циклоалкил", где y представляет собой целое число, большее чем 3, относится к насыщенным циклическим углеводородным группам, содержащим от 3 до y кольцевых атомов углерода. Примеры включают циклопропил, циклобутил, циклопентил, циклогексил и циклогептил.
Производное выражение "(C5-Cz)гетероциклоалкил" относится к насыщенным моноциклическим (C5-Cz)циклоалкильным группам, где z представляет собой целое число, большее чем 5, в которых по меньшей мере один кольцевой атом углерода заменен гетероатом (например N, NH, S или О). Неограничивающие примеры (C5-Cz)гетероциклоалкила представлены следующими группами: пирролидинил, тиазолидинил, пиперазинил, пиперидинил, морфолинил, тиоморфолинил.
Термин "(C2-Cx)алкенил" относится к прямым или разветвленным, конъюгированным или неконъюгированным углеродным цепям с одной или более двойными связями в цис- или транс-конфигурации, где количество атомов находится в диапазоне от 2 до х.
По аналогии, термин "(C5-Cy)циклоалкенил", где y представляет собой целое число, большее чем 5, относится к циклическим углеводородным группам, содержащим от 5 до y кольцевых атомов углерода и одну или две двойные связи.
Термин "(C2-Cx)алкинил" относится к прямым или разветвленным углеродным цепям с одной или более тройными связями, где количество атомов находится в диапазоне от 2 до x.
Выражение "арил" относится к моно- или би-циклическим кольцевым системам, которые содержат от 6 до 10 кольцевых атомов, где по меньшей мере одно кольцо является ароматическим.
Выражение "гетероарил" относится к моно- или би-циклическим кольцевым системам, которые содержат от 5 до 11 кольцевых атомов, в которых по меньшей мере одно кольцо является ароматическим, и в которых по меньшей мере один кольцевой атом представляет собой гетероатом (например N, NH, S или О).
Примеры подходящих моноциклических арильных или гетероарильных систем включают, например, радикалы фенил, тиофен, бензол, пиррол, пиразол, имидазол, изоксазол, оксазол, изотиазол, тиазол, пиридин, имидазолидин, фуран и тому подобное.
Примеры подходящих арильных или гетероарильных бициклических систем включают радикалы нафталин, бифенилен, пурин, птеридин, бензотриазол, хинолин, изохинолин, индол, изоиндол, бензотиофен, дигидробензодиоксин, дигидробензодиоксепин, бензооксазин и тому подобное.
Выражение "гетероарилкарбонил" относится к группам гетероарилCO-, где группа "гетероарил" имеет определенное выше значение.
Как оно используется в данном описании, выражение "кольцевая система" относится к моно- или бициклическим кольцевым системам, которые могут быть насыщенными, частично ненасыщенными или ненасыщенными, такими как арил, (C3-C8)циклоалкил, (C3-C7)гетероциклоалкил или гетероарил.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Изобретение касается класса соединений, действующих как ингибиторы фермента фосфодиэстераза 4 (PDE4).
Указанный класс соединений ингибирует превращение циклических нуклеотидов, в частности циклического аденозинмонофосфата (сАМР), в их неактивные 5'-мононуклеотидные формы.
В дыхательных путях физиологические ответы на повышенные внутриклеточные уровни циклических нуклеотидов, в частности сАМР, приводят к подавлению активности иммунных и провоспалительных клеток, таких как тучные клетки, макрофаги, Т-лимфоциты, эозинофилы и нейтрофилы, результатом чего является снижение высвобождения воспалительных медиаторов, которые включают цитокины, такие как IL (интерлейкин)-1, IL-3 и фактор некроза опухоли-альфа (TNF-α).
Это также ведет к релаксации гладкой маскулатуры дыхательных путей и уменьшению отека.
Настоящее изобретение относится к производным 1-фенил-2-пиридинилалкиловых спиртов общей формулы (I), их фармацевтически приемлемым солям и N-оксидам по пиридиновому кольцу
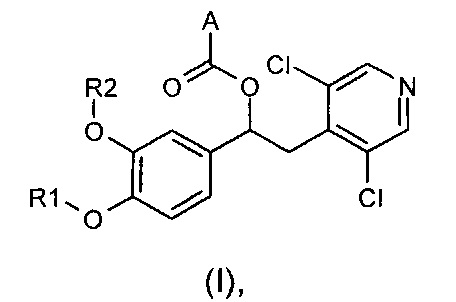
где R1, R2 и A являются такими, как определено выше.
Термин "фармацевтически приемлемые соли", как он используется в данном описании, относится к производным соединений формулы (I), где родительское соединение подходящим образом модифицировано посредством превращения любой свободной кислотной или основной группы, если она присутствует, в соответствующую соль присоединения с любым основанием или кислотой, обычно рассматриваемыми как являющиеся фармацевтически приемлемыми.
Подходящие примеры указанных солей могут, таким образом, включать соли присоединения минеральной или органической кислоты и основных остатков, таких аминогруппы, а также минеральные или органические соли присоединения кислотных остатков, таких как карбоксильные группы.
Катионы неорганических оснований, которые могут удобным образом быть использованы для получения солей согласно изобретению, включают ионы щелочных или щелочно-земельных металлов, таких как калий, натрий, кальций или магний.
Соли, полученные посредством взаимодействия главного соединения, функционирующего как основание, с неорганической или органической кислотой с образованием соли, включают, например соли соляной кислоты, серной кислоты, фосфорной кислоты, метансульфоновой кислоты, камфорсульфоновой кислоты, оксалиновой кислоты, малеиновой кислоты, янтарной кислоты и лимонной кислоты.
Специалисту в данной области очевидно, что соединения общей формулы (I) содержат по меньшей мере один стереогенный центр, а именно, представленный атомом углерода (1), отмеченный звездочкой на структуре, представленной ниже, и поэтому существуют в виде оптических стереоизомеров:

В случае, когда соединения по изобретению имеют по меньшей мере один стереогенный центр, они могут соответственно существовать как энантиомеры. В случае, когда соединения по изобретению имеют два или более стереогенных центра, они могут дополнительно существовать как диастереоизомеры. Следует понимать, что все такие изомеры и их смеси в любых соотношениях входят в объем настоящего изобретения.
В предпочтительном воплощении настоящее изобретение касается соединений формулы (I)', которые представляют собой соединения формулы (I), как они определены выше, где абсолютная конфигурация углерода (1) является такой, как показано ниже:

Абсолютная конфигурация углерода (1) присвоена на основе номенклатуры Кана-Ингольда-Прелога (Cahn-Ingold-Prelog), базирующейся на приоритетах групп.
В одном предпочтительном воплощении для соединений формулы (I) абсолютная конфигурация по углероду (1) представляет собой (S).
Следует понимать, что все раскрытые в данном описании предпочтительные группы или воплощения для соединений формулы (I) могут комбинироваться между собой и относятся к соединениям формулы (I)', (IA), (IB), (IC), (ID) и (IE) при внесении необходимых изменений.
В предпочтительном воплощении соединения по изобретению представляют собой N-оксидные производные по пиридиновому кольцу соединений формулы (I) или их фармацевтически приемлемых солей.
В одном предпочтительном воплощении R1 представляет собой (C1-C6)галогеналкил или (C1-C6)алкил.
В другом предпочтительном воплощении R2 представляет собой (C1-C6)алкил, который возможно замещен (C3-C7)циклоалкилом.
В другом предпочтительном воплощении R1 представляет собой (C1-C6)галогеналкил, и R2 представляет собой (C1-C6)алкил, который замещен (C3-C7)циклоалкилом.
В одном предпочтительном воплощении n представляет собой ноль. В другом предпочтительном воплощении n представляет собой 1 или 2. В другом предпочтительном воплощении n представляет собой 1.
В другом предпочтительном воплощении А представляет собой группу (а) или (b). В другом предпочтительном воплощении А представляет собой группу (а).
В одном предпочтительном воплощении А представляет собой группу (с) или (d). В другом предпочтительном воплощении А представляет собой группу (с).
В дополнительном предпочтительном воплощении R1 представляет собой (C1-C6)галогеналкил, R2 представляет собой (C1-C6)алкил, который замещен (C3-C7)циклоалкилом, и А представляет собой группу (а).
В предпочтительном воплощении R3 представляет собой возможный заместитель, который в каждом случае выбран из группы, состоящей из:
- (C1-C6)алкила;
- OR4, где R4 представляет собой водород или (C1-C10)алкил;
- атомов галогена;
- группы -NR5SO2R6, где
R5 выбран из группы, состоящей из водорода; (C1-C6)алкила; (C1-C6)алкила, который замещен группой -NR7R8, где R7 и R8 независимо представляют собой Н, (C1-C6)алкил, или вместе с атомом азота, к которому они присоединены, образуют (C5-C7)гетероциклоалкильную группу; (C1-C6)алкила, который замещен гетероарильной группой; и
R6 представляет собой (C1-C4)алкил, возможно замещенный (C3-C7)циклоалкилом; или фенильное кольцо, возможно замещенное одним или двумя атомами галогена, гидрокси, группами (C1-C4)алкил или (C1-C4)алкокси или группой -C(O)NR9R10, где R9 и R10 независимо представляют собой водород, (C1-C4)алкильные группы, или вместе с атомом азота, к которому они присоединены, образуют (C5-C7)гетероциклоалкильную группу;
- группы -SO2R11, где
R11 выбран из (C1-C6)алкила; фенильного кольца, которое возможно замещено одной или двумя группами, выбранными из списка, состоящего из следующего: атом галогена, гидроксил, (C1-C4)алкил, (C1-C4)алкокси, группа -C(O)NR12R13, где R12 и R13 независимо представляют собой водород, (C1-C4)алкильные группы, или вместе с атомом азота, к которому они присоединены, образуют (C5-C7)гетероциклоалкильную группу, и группа -NR14R15, где R14 и R15 независимо представляют собой водород, (C1-C4)алкильные группы, или вместе с атомом азота, к которому они присоединены, образуют (C5-C7)гетероциклоалкильную группу;
- группы -C(O)R16, где R16 представляет собой фенильное кольцо;
- группы -SO2NR17R18, где
R17 представляет собой Н или (C1-C6)алкил; и
R18 выбран из водорода; (C1-C6)алкила; (C1-C6)алкила, который замещен группой -NR19R20, где R19 и R20 независимо представляют собой Н или (C1-C6)алкил, или вместе с атомом азота, к которому они присоединены, образуют (C5-C7)гетероциклоалкильную группу; фенильного кольца, возможно замещенного одним или двумя атомами галогена, гидрокси, группами (C1-C4)алкил или (C1-C4)алкокси или группой -C(O)NR21R22, где R21 и R22 независимо представляют собой водород, (C1-C4)алкильные группы, или вместе с атомом азота, к которому они присоединены, образуют (C5-C7)гетероциклоалкильную группу.
В предпочтительном воплощении n представляет собой 0, 1 или 2, и R3 выбран из группы, состоящей из:
- (C1-C6)алкила;
- OR4, где R4 представляет собой (C1-C10)алкил;
- группы -NR5SO2R6, где R5 и R6 являются такими, как определено для соединений формулы (I);
- группы -SO2NR17R18, где R17 и R18 являются такими, как определено для соединений формулы (I); и
- группы -SO2R11, где R11 является таким, как определено для соединений формулы (I).
В одном воплощении предложены соединения формулы (IA), где R1, R2, R3 и n имеют значения, как они описаны для формулы (I):

В другом воплощении предложены соединения формулы (IB), где R1, R2, R3 и n имеют значения, как они описаны для формулы (I):
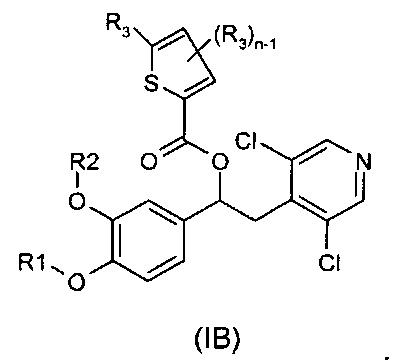
В одном воплощении предложены соединения формулы (IC), где R1, R2, R3 и n имеют значения, как они описаны для формулы (I):

В другом воплощении предложены соединения формулы (ID), где R1, R2, R3 и n имеют значения, как они описаны для формулы (I):
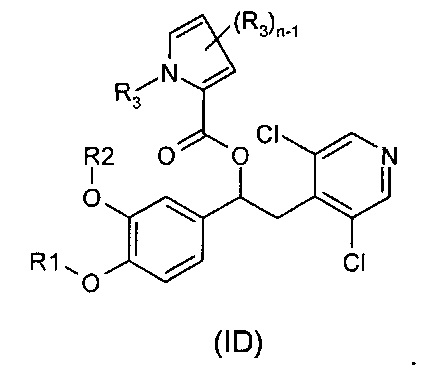
В другом воплощении предложены соединения формулы (IE), где R1, R2, R3 и n имеют значения, как они описаны для формулы (I):
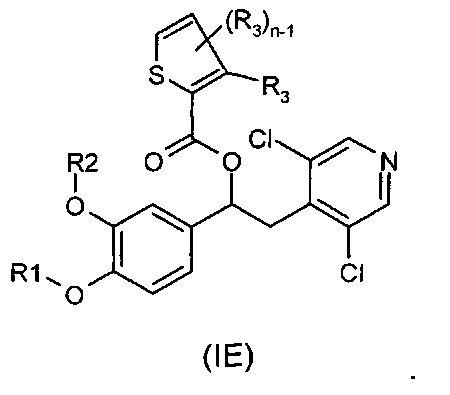
В одном воплощении соединение формулы (I) выбрано из списка, состоящего из:
(S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)-фенил)-2-(тиофен-3-карбонилокси)этил)пиридин-1-оксида;
(S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)-фенил)-2-(тиофен-2-карбонилокси)этил)пиридин-1-оксида;
(S)-4-(2-(1Н-пиррол-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)-3,5-дихлорпиридин-1-оксида;
(S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)-фенил)-2-(5-метилтиофен-2-карбонилокси)этил)пиридин-1-оксида;
(S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)-фенил)-2-(3-метокситиофен-2-карбонилокси)этил)пиридин-1-оксида;
(S)-3,5-дихлор-4-(2-(5-хлортиофен-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)пиридин-1-оксида;
(S)-3,5-дихлор-4-(2-(3-хлор-4-(метилсульфонил)тиофен-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-этил)пиридин-1-оксида;
(S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)-фенил)-2-(5-метокситиофен-2-карбонилокси)этил)пиридин-1-оксида;
(S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)-фенил)-2-(4,5-диметилтиофен-2-карбонилокси)этил)пиридин-1-оксида;
(S)-3,5-дихлор-4-(2-(3-хлортиофен-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)пиридин-1-оксида;
(S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)-фенил)-2-(3-(N-метилсульфамоил)тиофен-2-карбонилокси)этил)пиридин-1-оксида;
(S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)-фенил)-2-(3-(N-метил-N-(2-морфолиноэтил)сульфамоил)тиофен-2-карбонилокси)-этил)пиридин-1-оксида;
(S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)-фенил)-2-(3-(N-(2-морфолиноэтил)метилсульфонамидо)тиофен-2-карбонилокси)-этил)пиридин-1-оксида;
(S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)-фенил)-2-(3-(метилсульфонамидо)тиофен-2-карбонилокси)этил)пиридин-1-оксида;
(S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)-фенил)-2-(5-(N-(2-морфолиноэтил)метилсульфонамидо)тиофен-2-карбонилокси)-этил)пиридин-1-оксида;
(S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)-фенил)-2-(4-(метилсульфонамидо)тиофен-2-карбонилокси)этил)пиридин-1-оксида;
(S)-3,5-дихлор-4-(2-(4-(4-хлорфенилсульфонил)тиофен-3-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)-пиридин-1-оксида;
(S)-4-(2-(3-бензоилтиофен-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)-3,5-дихлорпиридин-1-оксида;
(S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)-фенил)-2-(3-(3-(диметилкарбамоил)фенилсульфонамидо)тиофен-2-карбонилокси)-этил)пиридин-1-оксида;
(S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)-фенил)-2-(5-(N-(3-(диметилкарбамоил)фенил)сульфамоил)тиофен-2-карбонилокси)-этил)пиридин-1-оксида;
(S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)-фенил)-2-(5-(N-(4-(диметилкарбамоил)фенил)сульфамоил)тиофен-2-карбонилокси)-этил)пиридин-1-оксида;
(S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)-фенил)-2-(5-(N-(пиридин-3-илметил)метилсульфонамидо)тиофен-2-карбонилокси)-этил)пиридин-1-оксида;
(S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)-фенил)-2-(5-(N-(2-(пирролидин-1-ил)этил)метилсульфонамидо)тиофен-2-карбонилокси)-этил)пиридин-1-оксида;
(S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)-фенил)-2-(4-метокси-5-(N-(2-морфолиноэтил)метилсульфонамидо)-тиофен-2-карбонилокси)этил)пиридин-1-оксида;
(S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)-фенил)-2-(1-(фенилсульфонил)-1Н-пиррол-2-карбонилокси)этил)пиридин-1-оксида;
(S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)-фенил)-2-(1-(3-(диметилкарбамоил)фенилсульфонил)-1Н-пиррол-2-карбонилокси)-этил)пиридин-1-оксида;
(S)-4-(2-(1-(4-аминофенилсульфонил)-1Н-пиррол-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)-3,5-дихлорпиридин-1-оксида;
(S)-3,5-дихлор-4-(2-(5-(1-циклопропил-N-(2-морфолиноэтил)-метилсульфонамидо)тиофен-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)пиридин-1-оксида;
(S)-3,5-дихлор-4-(2-(5-(1-циклопропил-N-(2-(пирролидин-1-ил)этил)-метилсульфонамидо)тиофен-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)пиридин-1-оксида;
(S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)-фенил)-2-(1-(3-(диметилкарбамоил)-4-гидроксифенилсульфонил)-1Н-пиррол-2-карбонилокси)этил)пиридин-1-оксида;
(S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)-фенил)-2-(1-(3-(диметилкарбамоил)-4-метоксифенилсульфонил)-1Н-пиррол-2-карбонилокси)этил)пиридин-1-оксида;
(S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)-фенил)-2-(1-метил-4-(фенилсульфонамидо)-1Н-пиррол-2-карбонилокси)этил)пиридин-1-оксида;
(S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)-фенил)-2-(4-(3-(диметилкарбамоил)фенилсульфонамидо)-1-метил-1Н-пиррол-2-карбонилокси)этил)пиридин-1-оксида;
(S)-3,5-дихлор-4-(2-(3,4-диметоксифенил)-2-(тиофен-2-карбонилокси)-этил)пиридин-1-оксида;
(S)-3,5-дихлор-4-(2-(4-(дифторметокси)-3-метоксифенил)-2-(тиофен-2-карбонилокси)этил)пиридин-1-оксида;
и их фармацевтически приемлемых солей или сольватов.
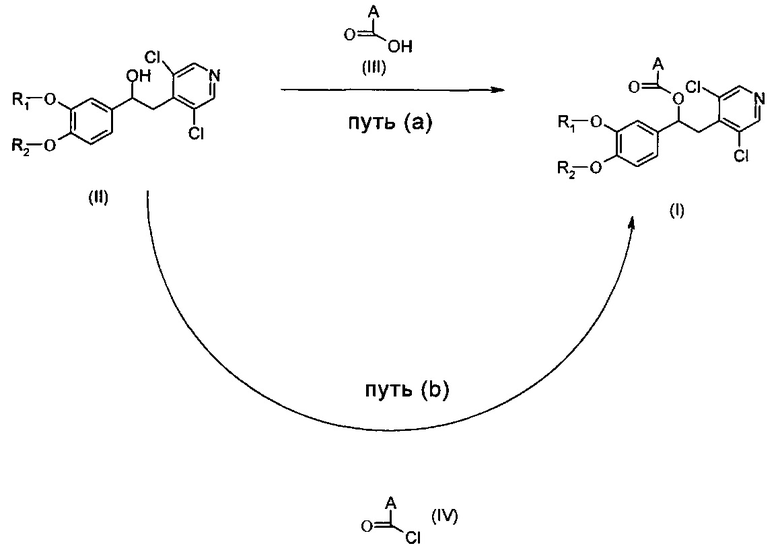
В одном аспекте настоящего изобретения предложен способ получения соединений по изобретению согласно общим путям синтеза (а) или (b), представленным на общей схеме ниже.
Общая схема
Для получения дополнительных соединений по изобретению специалист может осуществить, где это целесообразно, приемлемые изменения в условиях, специфически раскрытых в экспериментальной части, для того, чтобы адаптировать пути синтеза. Такие модификации могут включать, но без ограничения этим, использование подходящих исходных материалов для получения других соединений, изменения в растворителе и температуре реакций, замены реактивов с аналогичной химической функцией, введение или удаление стадий введения защиты/снятия защиты функциональных групп, чувствительных к реакционным условиям и реагентам.
Также предполагается введение или удаление специфических стадий синтеза для дополнительной функционализации химического скелета, и они включены в объем настоящего изобретения.
В Таблице А ниже сделаны ссылки на конкретные схемы синтеза, где пути (а) или (b) и их варианты лучше детализированы, и которые раскрыты в экспериментальной части.

Способы, которые могут быть использованы и которые раскрыты и описаны в Примерах и на Схемах, не следует рассматривать как ограничивающие объем методов синтеза, доступных для получения соединений по изобретению.
N-оксиды по 2-пиридинильному кольцу соединения общей формулы (I) могут быть получены в соответствии с методами, раскрытыми в литературе и хорошо известными специалистам в данной области. Например, они могут быть получены путем растворения соединения общей формулы (I) в CH2Cl2 или CHCl3, затем добавления к полученному раствору окисляющего агента, такого как мета-хлорпербензойная кислота (mCPBA). Другие окисляющие агенты, которые могут быть использованы, представляют собой перекись водорода, пербензойную кислоту и перуксусную кислоту.
Альтернативно, в частности для тех соединений, в которых присутствует функциональная группа, чувствительная к окислению, соответствующие N-оксиды получают посредством проведения стадии окисления перед введением дополнительных функциональных групп, например на соединениях формулы (II), тем самым получая соединения формулы (V).

В предпочтительном воплощении способ получения соединений формулы (I) осуществляют, начиная с N-оксидного соединения формулы (V) по пиридиновому кольцу, тем самым обеспечивая возможность получения соединения формулы (I) в форме N-оксидов по пиридиновому кольцу.
Соединения, используемые в качестве исходных материалов или промежуточных соединений, могут иметься в продаже, их получение может быть специально описано в литературе, либо они могут быть получены в соответствии с методами, раскрытыми в литературе и хорошо известными специалистам в данной области. В ряде случаев процедуры для получения промежуточных соединений также могут быть раскрыты в экспериментальной части, например, как раскрыто на схемах 1, 2, 3, 4, 5, 7, 8, 9, 10, 11, 12, 13, 14, 15.
Соединения формул (II) и (V) также могут быть получены так, как раскрыто в международной заявке на патент WO 2009/018909.
Описанный способ является особенно предпочтительным, поскольку он с легкостью может быть должным образом видоизменен посредством любого приемлемого известного из уровня техники варианта, таким образом, чтобы обеспечивать получение целевых соединений по изобретению. Такие варианты входят в объем настоящего изобретения.
Из всего вышеизложенного специалисту должно быть понятно, что любая из описанных групп может быть представлена как таковая или в соответствующим образом защищенной форме.
В частности, присутствующие в промежуточном соединении и в соединениях функциональные группы, которые могут генерировать нежелательные побочные реакции и побочные продукты, нуждаются в соответствующей защите перед тем, как будет иметь место алкилирование, ацилирование, сочетание или сульфонилирование. Аналогично, последующее снятие защиты с тех же самых защищенных групп может происходить после завершения указанных реакций.
В настоящем изобретении, если не указано иного, термин "защитная группа" означает защитную группу, пригодную для сохранения функции группы, с которой она связана. Обычно защитные группы используются для сохранения амино, гидроксильных или карбоксильных функций. Подходящие защитные группы могут, таким образом, включать, например, бензил, бензилоксикарбонил, трет-бутоксикарбонил, алкильные или бензильные сложные эфиры или тому подобное, которые хорошо известны специалистам в данной области [в качестве общей ссылки смотри T.W. Green; Protective Groups in Organic Synthesis (Wiley, N.Y. 1981)].
Аналогично, селективное введение и снятие защиты с любой из указанных групп, например включающих карбонильные, гидроксильные или аминогруппы, может быть осуществлено согласно очень хорошо известным методам, широко используемым в области химии органического синтеза.
Возможное образование солей соединений формулы (I) или их N-оксидов по пиридиновому кольцу может быть осуществлено посредством соответствующего превращения любой из свободных кислотных или аминогрупп в соответствующие фармацевтически приемлемые соли. В этом случае также условия проведении операций, используемые для возможного получения солей соединений по изобретению, входят в объем знаний специалиста в данной области.
Из всего вышеизложенного специалисту должно быть понятно, что вышеописанный способ, включая любой его вариант, для получения подходящего соединения по изобретению, может быть удобным образом видоизменен таким образом, чтобы адаптировать реакционные условия к специфическим потребностям, например, посредством выбора, в зависимости от конкретного случая, подходящих конденсирующих агентов, растворителей и защитных групп.
На следущих далее Схемах для соединений формул (II)-(XVI), если не указано иного, группы A, n, R1, R2 и R3 имеют такие же значения, как описано для соединений формулы (I) выше.
Реакционный путь (а)
Соединения формулы (I) могут быть получены согласно Схеме А ниже, посредством взаимодействия соединения формулы (II) с соответствующим соединением формулы (III).
Схема А

Типичные реакционные условия включают взаимодействие соединения формулы (II) с соединением формулы (III) в подходящем полярном апротонном растворителе, таком как DMF, хлороформ, ацетонитрил или DCM, в присутствии соответствующего конденсирующего агента, такого как EDC, DCC или CDI, и подходящего катализатора, такого как DMAP, HBTU, НОВТ, 4-пирролидинопиридин (4-PPY) или другой 4-алкиламинопиридин, при комнатной температуре.
Реакционный путь (b)
Соединения формулы (I) могут быть получены согласно Схема В ниже посредством взаимодействия соединения формулы (II) с соответствующим соединением формулы (IV).
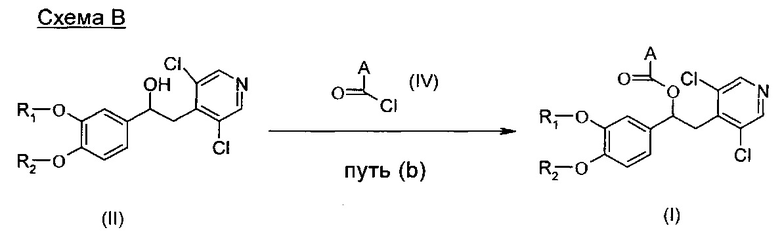
Типичные реакционные условия включают взаимодействие соединения формулы (II) с соединением формулы (IV) в подходящем полярном апротонном растворителе, таком как DCM или хлороформ, в присутствии подходящего основания, такого как DMAP, Py, TEA или DIPEA, при подходящей температуре, такой как например КТ.
Соединения формулы (IIIa), то есть соединения формулы (III), где n представляет собой 1 или 2, R3 представляет собой в каждом случае группу -SO2NR17R18, могут быть получены согласно представленной ниже Схеме С посредством взаимодействия соединения формулы (VI), где PG представляет собой подходящую защитную группу карбоксила, с соответствующим соединением формулы R17R18NH с последующим снятием защиты с карбоксильной группы в подходящих условиях.
Схема С

Типичные реакционные условия включают взаимодействие соединения формулы (VI) с соединением формулы R17R18NH в подходящем полярном апртонном растворителе, таком как DMF, DMSO, ацетонитрил, DCM, THF или хлороформ, в присутствии подходящего основания, такого как DMAP, TEA, метиламин или DIPEA, при подходящей температуре, такой как, например, КТ, или в чистом основании, таком как пиридин, при подходящей температуре, такой как, например, 0 градусов.
Соединения формулы (IIIb), то есть соединения формулы (III), где А представляет собой группу (а) или (b), n представляет собой 1 или 2, R3 представляет собой в каждом случае группу -NR5SO2R6, могут быть получены согласно представленной ниже Схеме D посредством взаимодействия соединения формулы (VII), где PG представляет собой подходящую защитную группу карбоксила, с подходящим сульфонилирующим агентом формулы R6SO2X, где X представляет собой уходящую группу, такую как хлор, с последующим снятием защиты с карбоксильной группы при подходящих условиях.
Схема D

Типичные реакционные условия включают взаимодействие соединения формулы (VII) с соединением формулы R6SO2X в подходящем полярном апротонном растворителе, таком как DMF, DMSO, ацетонитрил, DCM или хлороформ, в присутствии подходящего основания, такого как DMAP, TEA или DIPEA, при подходящей температуре, такой как, например, КТ, или в чистом основании, таком как пиридин, при подходящей температуре, такой как, например, 0 градусов.
Соединения формулы (IIIb1), то есть соединения формулы (IIIb), где R5 представляет собой группу, отличную от водорода, могут быть получены согласно представленной выше Схеме D посредством осуществления промежуточной стадии алкилирования перед тем, как будет иметь место снятие защиты с карбоксильной группы, согласно представленной ниже Схеме Е. Соединения формулы (VIII) [где n представляет собой 1 или 2, А представляет собой группу (а) или (b), и другие группы являются такими, как определено для соединений формулы (I)] подвергают взаимодействию с подходящим алкилирующим агентом формулы R5LG (где LG представляет собой подходящую уходящую группу, такую как галоген, или мезилат, или тозилат, или брозилат), с получением соединений формулы (IX) [где n представляет собой 1 или 2, и другие группы являются такими, как определено для соединений формулы (I)], как представлено ниже на Схеме Е.
Схема Е
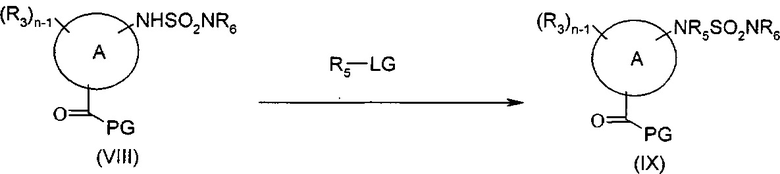
Типичные реакционные условия включают взаимодействие соединения формулы (VIII) с соединением формулы R5LG в подходящем полярном апротонном растворителе, таком как DMF, DMSO, ацетонитрил, DCM или хлороформ, в присутствии подходящего основания, такого как K2CO3, NaH, нBuLi или DMAP, при подходящей температуре, такой как, например, 50 градусов.
Соединения формулы (VII), как они определены выше, могут быть получены согласно представленной ниже Схеме F посредством взаимодействия соединения формулы (X), где PG представляет собой подходящую защитную группу карбоксила, в подходящих восстановительных условиях.
Схема F

Типичные реакционные условия включают взаимодействие соединения формулы (X) в подходящем полярном апротонном растворителе, таком как THF, DMF, ацетонитрил, DCM или хлороформ, с подходящим восстанавливающим агентом, таким как SnCl2 2H2O (дигидрат хлорида олова) или дитионит натрия, при подходящей температуре, такой как, например, КТ. Соединение (X) также может быть восстановлено в подходящем полярном протонном растворителе, таком как МеОН или EtOH, с подходящим катализатором, таким как Pd/C или Ni Рэнея (Raney), в атмосфере водорода при подходящей температуре, такой как, например, КТ.
Соединения формулы (X), как они определены выше, могут быть получены согласно представленной ниже Схеме G посредством взаимодействия соединения формулы (XI), где А представляет собой группу (а) или (b), n представляет собой 1 или 2, и R3 является таким, как определено выше, в подходящих окислительных условиях с последующей защитой карбоксильной группы с помощью защитной группы PG в соответствующих условиях.
Схема G

Типичные реакционные условия включают взаимодействие соединения формулы (XI) в подходящем полярном апротонном растворителе, таком как DMF, ацетонитрил, DCM, диоксан или хлороформ, в присутствии подходящего окисляющего агента, такого как оксон (смесь 2KHSO5⋅KHSO4⋅K2SO4) или PDC (дихромат пиридиния), или в подходящей смеси полярного апротонного растворителя и водного растворителя, такой как диоксан и раствор хлорита натрия в воде, в присутствии подходящего окисляющего агента, такого как сульфаминовая кислота, при подходящей температуре, такой как, например, КТ. Альтернативно окисление может быть осуществлено в подходящем полярном протонном растворителе, таком как ацетон, диоксан или трет-BuOH, и посредством подходящего окисляющего агента, такого как перманганат калия или реагент Джонса (Jones).
Соединения формулы (XI), как они определены выше, могут быть получены согласно представленной ниже Схеме Н посредством взаимодействия соединения формулы (XII), где А представляет собой группу (а) или (b), n представляет собой 1 или 2, и R3 является таким, как определено выше, в подходящих условиях нитрования.
Схема Н

Типичные реакционные условия включают взаимодействие соединения формулы (XII), растворенного в уксусном ангидриде, трифторуксусном ангидриде, водной конц. серной кислоте или трифликовом ангидриде, добавление подходящего источника нитро-группы, такого как азотная кислота в ледяной уксусной кислоте, азотная кислота в серной кислоте, азотная кислота в уксусном ангидриде, водная азотная кислота, нитрат аммония или нитрат калия в водной азотной кислоте, при подходящей температуре, такой как, например, 0 градусов.
Соединения формулы (IIIc), то есть соединения формулы (III), где А представляет собой группу (с) или (d), где азот замещен группой R3, представляющей собой (C1-C6)алкил, n представляет собой 2, R3 представляет собой в каждом случае группу -NR5SO2R6, могут быть получены согласно представленной ниже Схеме J посредством взаимодействия соединения формулы (XIII), где А представляет собой группу (с) или (d), где азот замещен группой R3, представляющей собой (C1-C6)алкил, и n представляет собой 2, с подходящим сульфонилирующим агентом формулы R6SO2X, где X представляет собой уходящую группу, такую как хлор.
Схема J

Типичные реакционные условия включают взаимодействие соединения формулы (XIII) с соединением формулы R6SO2X в подходящем полярном апротонном растворителе, таком как DMF, DMSO, ацетонитрил, DCM или хлороформ, в присутствии подходящего основания, такого как DMAP, TEA или DIPEA, при подходящей температуре, такой как, например, КТ, или в чистом основании, таком как пиридин, при подходящей температуре, такой как, например, 0 градусов.
Соединения формулы (XIII), как они определены выше, могут быть получены согласно представленной ниже Схеме К посредством взаимодействия соединения формулы (XIV), где А представляет собой группу (с) или (d), где азот замещен группой R3, представляющей собой (C1-C6)алкил, в подходящих восстановительных условиях.
Схема К

Типичные реакционные условия включают взаимодействие соединения формулы (XIV) в подходящем полярном апротонном растворителе, таком как THF, ацетонитрил, DCM или хлороформ, в присутствии подходящего восстанавливающего агента, такого как SnCl2 2H2O (дигидрат хлорида олова) или дитионит натрия, или в подходящей смеси полярного апротонного растворителя и водного растворителя, такой как EtOH и вода, в присутствии подходящего восстанавливающего агента, такого как порошок железа и хлорид аммония, при подходящей температуре, такой как, например, КТ. Соединение (X) также может быть восстановлено в подходящем водном растворителе, таком как 1 М карбонат натрия, или полярном протонном растворителе, таком как МеОН или EtOH, с подходящим катализатором, таким как Pd/C или Ni Рэнея, в атмосфере водорода при подходящей температуре, такой как, например, КТ.
Соединения формулы (XIV), как они определены выше, могут быть получены согласно представленной ниже Схеме L посредством взаимодействия соединения формулы (XV), где А представляет собой группу (с) или (d), где азот замещен группой R3, представляющей собой (C1-C6)алкил, в подходящих условиях нитрования.
Схема L

Типичные реакционные условия включают взаимодействие соединения формулы (XV), растворенного в уксусном ангидриде, трифторуксусном ангидриде или трифликовом ангидриде, добавление подходящего источника нитро-группы, такого как азотная кислота в ледяной уксусной кислоте, азотная кислота в серной кислоте, азотная кислота в уксусном ангидриде, нитрат аммония или нитрат меди, при подходящей температуре, такой как, например, 0 градусов.
Соединения формулы (IIId), то есть соединения формулы (III), где А представляет собой группу (с) или (d), n представляет собой 1 или 2, и R3 представляет собой в каждом случае группу -SO2R11, которая связана с атомом азота кольца А, могут быть получены согласно представленной ниже Схеме М посредством взаимодействия соединения формулы (XVI), где PG представляет собой подходящую защитную группу карбоксила, с соответствующим соединением формулы R11SO2Cl с последующим снятием защиты с карбоксильной группы в подходящих условиях.
Схема М

Типичные реакционные условия включают взаимодействие соединения формулы (XVI) с соединением формулы R11SO2Cl в подходящем полярном апротонном растворителе, таком как DMF, ацетонитрил, DCM или хлороформ, в присутствии подходящего основания, такого как DMAP, TEA, NaOHводн. или DIPEA, при подходящей температуре, такой как, например, КТ, в присутствии, если необходимо, подходящего катализатора фазового переноса, такого как гидросульфат тетрабутиламмония, или в чистом основании, таком как пиридин, при подходящей температуре, такой как, например, 0 градусов.
В настоящем изобретении также предложены фармацевтические композиции соединений по изобретению в смеси с одним или более фармацевтически приемлемыми носителями, например описанными в Remington's Pharmaceutical Sciences Handbook, XVII Ed., Mack Pub., N.Y., U.S.A.
Введение соединений по настоящему изобретению можно осуществлять в соответствии с потребностями пациента, например перорально, интраназально, парентерально (подкожно, внутривенно, внутримышечно, интрастернально и посредством инфузии), посредством ингаляции, ректально, вагинально, местно, локально, трансдермально и посредством глазного введения. Для введения соединений по изобретению могут быть использованы различные твердые пероральные лекарственные формы, в том числе такие твердые формы как таблетки, желатиновые капсулы, капсулы, каплеты, гранулы, лепешки и нефасованные порошки. Соединения по настоящему изобретению можно вводить сами по себе или в комбинации с различными фармацевтически приемлемыми носителями, разбавителями (такими как сахароза, маннит, лактоза, крахмалы) и эксципиентами, известными в данной области и включающими, но не ограничивающимися этим, суспендирующие агенты, солюбилизаторы, буферные агенты, связующие агенты, разрыхлители, консерванты, красители, ароматизаторы, смазывающие агенты и тому подобное. Удобными для введения соединений по настоящему изобретению также являются капсулы, таблетки и гели с высвобождением во времени.
Для введения соединений по изобретению также могут быть использованы различные жидкие пероральные лекарственные формы, включающие водные и неводные растворы, эмульсии, суспензии, сиропы и эликсиры. Такие лекарственные формы также могут содержать подходящие инертные разбавители, известные в данной области, такие как вода, и подходящие эксципиенты, известные в данной области, такие как консерванты, увлажняющие агенты, подсластители, ароматизаторы, а также агенты для эмульгирования и/или суспендирования соединений по изобретению. Соединения по настоящему изобретению можно инъецировать, например внутривенно, в форме изотонического стерильного раствора. Также возможными являются другие препараты.
Суппозитории для ректального введения соединений по настоящему изобретению могут быть приготовлены путем смешивания соединения с подходящим эксципиентом, таким как масло какао, салицилаты и полиэтиленгликоли.
Композиции для вагинального введения могут быть в форме крема, геля, пасты, пены или спрея, содержащего, в дополнение к активному ингредиенту, подходящие известные в данной области носители.
Для местного введения фармацевтическая композиция может быть в форме кремов, мазей, линиментов, лосьонов, эмульсий, суспензий, гелей, растворов, паст, порошков, спреев и капель, подходящих для введения на кожу, в глаз, ухо или нос. Местное введение также может включать трансдермальное введение с помощью таких средств, как трансдермальные пластыри.
Для лечения заболеваний респираторного тракта соединения по изобретению предпочтительно вводят посредством ингаляции.
Ингалируемые препараты включают ингалируемые порошки, содержащие пропеллент дозированные аэрозоли или не содержащие пропеллента ингалируемые композиции, все из которых могут быть доставлены с помощью подходящего устройства, известного в данной области.
Для введения в виде сухого порошка могут быть использованы ингаляторы разовой или множественной дозы, известные из уровня техники. В этом случае порошком могут быть заполнены желатиновые, пластиковые или другие капсулы, картриджи или блистерные упаковки или резервуар.
К порошкообразным соединениям по изобретению может быть добавлен разбавитель или носитель, обычно нетоксичный и химически инертный по отношению к соединениям по изобретению, например лактоза или любое другое вспомогательное вещество, подходящее для улучшения вдыхаемой фракции.
Ингалируемые аэрозоли, содержащие газ-пропеллент, такой как гидрофторалканы, могут содержать соединения по изобретению либо в растворе, либо в диспергированной форме. Пропеллентсодержащие композиции могут также содержать другие ингредиенты, такие как сорастворители, стабилизаторы и возможно другие эксципиенты.
Не содержащие пропеллента ингалируемые композиции, содержащие соединения по настоящему изобретению, могут быть в форме растворов или суспензий в водной, спиртовой или водно-спиртовой среде, и их можно доставлять с помощью струйных или ультразвуковых небулайзеров, известных из уровня техники, или с помощью пневматических небулайзеров, таких как Respimat®.
Соединения по изобретению могут быть введены в виде отдельного активного агента, либо в комбинации с другими фармацевтически активными ингредиентами, включая те, которые в настоящее время применяются в лечении респираторных заболеваний, например бета2-агонисты, кортикостероиды и антихолинэргические или антимускариновые агенты.
Дозировки соединений по настоящему изобретению зависят от множества факторов, включая конкретное заболевание, подлежащее лечению, тяжесть симптомов, путь введения, частоту введения доз, конкретное используемое соединение, эффективность, токсикологический профиль и фармакокинетический профиль соединения.
Преимущественно соединения по изобретению можно вводить, например, в дозировке от 0,001 до 1000 мг/сутки, предпочтительно от 0,1 до 500 мг/сутки.
При их введении ингаляционным путем дозировка соединений по изобретению предпочтительно находится между 0,01 и 20 мг/сутки, предпочтительно между 0,1 и 10 мг/сутки.
Предпочтительно соединения по изобретению сами по себе или в комбинации с другими активными ингредиентами могут быть введены для предотвращения и/или лечения любого обструктивного респираторного заболевания, такого как астма, хронический бронхит или хроническая обструктивная болезнь легких (COPD).
Однако соединения по изобретению могут быть введены для предупреждения и/или лечения любого заболевания, где требуется ингибирование PDE4. Указанное заболевание включает аллергические болезненные состояния, такие как атопический дерматит, крапивница, аллергический ринит, аллергический конъюнктивит, весенний конъюнктивит, эозинофильная гранулема, псориаз, воспалительный артрит, ревматоидный артрит, септический шок, язвенный колит, болезнь Крона, реперфузионное поражение миокарда и головного мозга, хронический гломерулонефрит, эндотоксический шок, муковисцидоз, артериальный рестеноз, атеросклероз, кератоз, ревматоидный спондилит, остеоартрит, пирез, сахарный диабет, пневмокониоз, токсическая и аллергическая контактная экзема, атопическая экзема, себорейная экзема, простой лишай, солнечный ожог, зуд в аногенитальной области, очаговая алопеция, гипертрофированные рубцы, дискоидная красная волчанка, системная красная волчанка, фолликулярные и обширные пиодермии, эндогенные и экзогенные акне, розовые акне, болезнь Бехчета, анафилактиодный пурпурный нефрит, воспалительное заболевание кишечника, лейкоз, рассеянный склероз, желудочно-кишечные заболевания, аутоиммунные заболевания и тому подобное.
Они также включают неврологические и психиатрические расстройства, такие как болезнь Альцгеймера, рассеянный склероз, боковой амиотрофический склероз (ALS), множественную системную атрофию (MSA), шизофрению, болезнь Паркинсона, болезнь Гентингтона, болезнь Пика, депрессию, инсульт и поражение спинного мозга.
Настоящее изобретение далее будет дополнительно описано с помощью следующих неограничивающих примеров.
Экспериментальная часть
Химические названия соединений генерировали с помощью программного обеспечения Structure То Name Enterprise 10.0 Cambridge (структура - название).
Сокращения
EDC - 1-этил-3-(3-диметиламинопропил)карбодиимид) гидрохлорид; DMAP - 4-диметиламинопиридин; DMF - диметилформамид; EtOAc или AcOEt - этилацетат; КТ или к.т. - комнатная температура; THF - тетрагидрофуран; DCM - дихлорметан; Et2O - диэтиловый эфир; МеОН - метиловый спирт; EtOH - этиловый спирт; (Ipr)2O - диизопропиловый эфир; TEA - триэтиламин; TFA - трифторуксусная кислота; CH3CN - ацетонитрил; (Вос)2O - ди-трет-бутилдикарбонат; DIPEA - ди-изопропилэтиламин; DMSO - диметилсульфоксид.
Характеристика ЯМР (ядерный магнитный резонанс)
ЯМР спектры снимали одним из следующих способов.
1Н-ЯМР спектры снимали на спектрометре 400 МГц Varian AS400. Химический сдвиг представлен в виде величин δ в м.д. (миллионные доли) относительно триметилсилана (TMS) в качестве внутеннего стандарта. Константы спин-спинового взаимодействия (величины J) приведены в герцах (Гц), а мультиплетности указаны, используя следующие сокращения (s - синглет, d - дублет, t - триплет, q - квартет, m - мультиплет, br - широкий, nd - не определено).
Либо 1Н-ЯМР спектры снимали на спектрометре Bruker ARX300 при 300,13 МГц (1Н), используя дейтерированные растворители, такие как дейтерированный диметилсульфоксид (DMSO-d6) или дейтерированный хлороформ (CDCl3). Прибор был снабжен мультиядерным обратным зондом и температурным контроллером. Химические сдвиги выражены в миллионных долях (м.д.) вниз от тетраметилсилана (d единицы). Мультиплетность указана следующим образом: (s) синглет, (d) дублет, (dd) двойной дублет, (ddd) тройной дублет, (t) триплет, (dt) двойной триплет, (q) квартет, (m) мультиплет, (br s) широкий сигнал. Константы спин-спинового взаимодействия J выражены в единицах герц (Гц).
Препаративная ВЭЖХ - метод 1
Колонка: Waters Symmetry Prep C18 17 мкм 19×300
Поток: 20 мл/мин
Подвижная фаза: 90% H2O, 10% ацетонитрил, 0,05% TFA (А), 10% H2O, 90% ацетонитрил, 0,05% TFA (В)
Градиент:
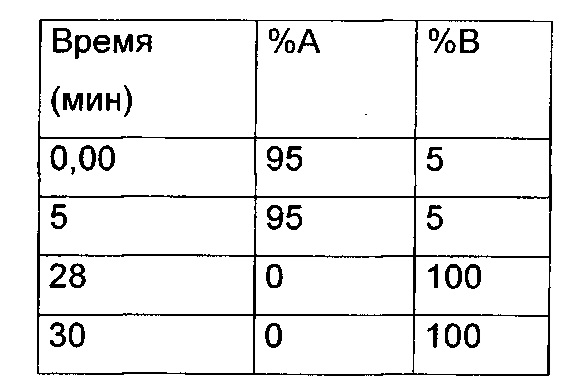
Такой же градиент, но без TFA в подвижной фазе, использовали для препаративной ВЭЖХ в нейтральных условиях.
Препаративная ВЭЖХ - метод 2
Waters Micromass ZQ; устройство управления образцами 2767; детектор с фотодиодной матрицей 2996;
колонка XTerra Prep MS C18 (5 мкм, 19×150 мм, Waters); скорость потока 20 мл/мин, с МС (масс-спектрометрия) детектированием или с UV (ультрафиолетовое облучение) при 254 нм.
Градиент:
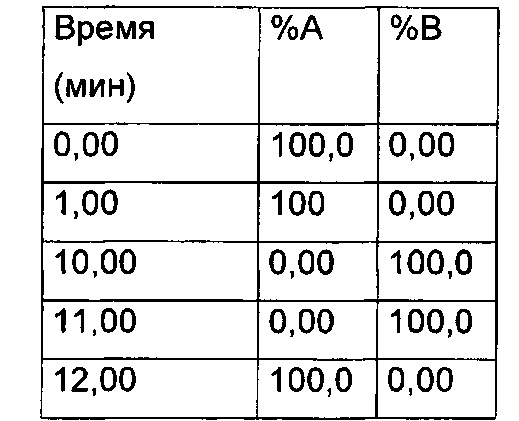
Элюент
Растворитель A (вода:MeCN:HCOOH 95:5:0,05)
Растворитель В (вода:MeCN:НСООН 5:95:0,05)
Препаративная ВЭЖХ - метод 3
Waters Micromass ZQ / устройство управления образцами 2767
Детектор с фотодиодной матрицей: 2996
Колонка: XTERRA Prep MS С18 10 мкм 19×300
Поток: 20 мл/мин
Подвижные фазы: H2O, 0,1% TFA (А); ацетонитрил, 0,1% TFA (В)
Градиент:

Кондиционирование:
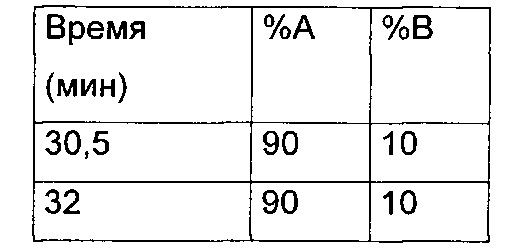
Определение оптического вращения (активности)
Конкретные величины вращения соединений измеряли с помощью поляриметра Perkin Elmer модель 241 или 341.
Температура (°С) 25
Длина пути (дм) 1
Длина волны натриевой D-линии (589 нм)
Система автоматического синтеза с микроволновым облучением
Реакции, стимулируемые микроволновым облучением, проводили с использованием реактора СЕМ Discover.
Мощность выходного микроволнового излучения от 0 до 300 Ватт
Температурный диапазон от 0 до 300 градусов
Диапазон давления от 0 до 300 фунт-сила на кв. дюйм (от 0 до 2068,5 кПа)
Объем сосудов от 5 мл до 125 мл (для реакций, проводимых в атмосферных условиях)
10 мл флаконы с септой (для повышенных температур и давлений)
Величины МС (масс-спектрометрия)/ИЭР+ (ионизация электрораспылением), приведенные в тексте ниже, могут быть получены либо с помощью МС прибора Waters ZQ (или его эквивалента), либо с помощью прибора UPLC Waters.
МС прибор: Waters ZQ (или эквивалент)
ЭР (электрораспыление) в полярном режиме
Напряжение на капилляре (кВ) 3,00
Напряжение на конусе (В) 20,00
Напряжение на экстракторе (В) 3,00
Напряжение на RF линзе (В) 1,0
ЭР в полярном режиме
Напряжение на капилляре (кВ) 3,00
Напряжение на конусе (В) 20,00
Напряжение на экстракторе (В) 3,00
Напряжение на RF линзе (В) 1,0
Температура источника (°C) 110
Температура осушения (°C) 210
Поток газа через конус (л/ч) 150
Скорость потока газа-осушителя (л/ч) 650
Диапазон масс: от 100 до 950
Время сканирования (сек): 0,32
Интервал между сканами (сек): 0,03
LC (жидкостная хроматография) прибор: Acquity Waters UPLC
Прибор: UPLC Waters в сочетании с ZQ micromass и с детектором 2996 PDA
Колонка: Acquity UPLC ВЕН С18 1,7 мкм 50×2,1 мм
Метод: TFA длинный
Условия: ИЭР+, 3,2 КВ, 25 В, 350°C
Длина волны: PBI


Детальные пути синтеза и процедуры для конкретных соединений представлены в Примерах 1-15. Синтез (S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-гидроксиэтил)пиридин-1-оксида может быть осуществлен так, как описано в WO 2010/089107 (соединение 7).
В следующих далее процедурах в некоторых случаях после каждого исходного материала приведена ссылка на номер соединения. Она приводится только для помощи специалисту-химику. Исходный материал не обязательно может быть получен из партии, на которую сделана ссылка.
Когда приводится указание на использование "сходной" или "аналогичной" процедуры, как это понятно специалисту в данной области, такая процедура может включать небольшие модификации, например, в таких параметрах, как температура реакции, количество реагента/растворителя, время реакции, условия обработки или условия хроматографической очистки.
Пример 1
Синтез (S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(1-(фенилсульфонил)-1H-пиррол-2-карбонилокси)этил)пиридин-1-оксида (Соединение 3)
Схема 1
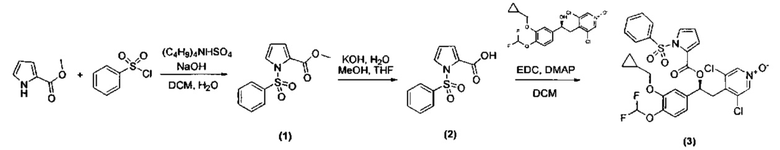
Стадия 1: Получение метил 1-(фенилсульфонил)-1H-пиррол-2-карбоксилата (Промежуточное соединение 1)
К смеси метил-1H-пиррол-2-карбоксилата (1 г, 7,99 ммоль), гидросульфата тетрабутиламмония (0,271 г, 0,799 ммоль) и NaOH (2,88 г, 71,9 ммоль) в воде (5 мл) и DCM (40 мл) при интенсивном перемешивании добавляли по каплям в течение 10 минут при КТ раствор бензолсульфонилхлорида (1,794 мл, 13,99 ммоль) в DCM (5 мл). Реакционную смесь перемешивали при той же температуре в течение 3 ч. Смесь разбавляли водой (50 мл) и DCM (100 мл), органическую фазу отделяли, промывали рассолом (50 мл) и сушили над сульфатом натрия. Растворитель выпаривали и остаток очищали флэш-хроматографией на колонке с силикагелем (петролейный эфир/ацетон 8/2) с получением метил-1-(фенилсульфонил)-1Н-пиррол-2-карбоксилата (Промежуточное соединение 1) (1,448 г, 5,46 ммоль, МС/ИЭР+ 287,9 [MNa]+).
Стадия 2: Получение 1-(фенилсульфонил)-1Н-пиррол-2-карбоновой кислоты (Промежуточное соединение 2)
К раствору метил-1-(фенилсульфонил)-1Н-пиррол-2-карбоксилата (Промежуточное соединение 1) (500 мг, 1,885 ммоль) в МеОН (7 мл) и THF (7 мл) охлаждали при 0°C, добавляли по каплям водный 1 н. КОН (2,827 мл, 2,83 ммоль) и полученную смесь перемешивали при КТ в течение 24 ч. UPLC-MC анализ показал присутствие целевого соединения в смеси с метил-1Н-пиррол-2-карбоксилатом в качестве основного продукта. Смесь вливали в ледяную воду и промывали DCM. Водную фазу подкисляли 37%-ной HCl (pH=2) и экстрагировали DCM. Органический слой сушили над сульфатом натрия, фильтровали и упаривали с получением смеси 1-(фенилсульфонил)-1Н-пиррол-2-карбоновой кислоты (Промежуточное соединение 2) (МС/ИЭР+ 252,0 [МН]+) и метил-1H-пиррол-2-карбоксилата (МС/ИЭР+ 126,0 [МН]+) (234 мг, соотношение примерно 3/7). Эту смесь использовали в таком виде на следующей стадии.
Стадия 3: Получение (S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(1-(фенилсульфонил)-1Н-пиррол-2-карбонилокси)этил)пиридин-1-оксида (Соединение 3)
Смесь неочищенной 1-(фенилсульфонил)-1Н-пиррол-2-карбоновой кислоты (234 мг, соединение получено как указано выше на стадии 2), (S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-гидроксиэтил)пиридин-1-оксида (117 мг, 0,279 ммоль), EDC (161 мг, 0,838 ммоль) и DMAP (34,1 мг, 0,279 ммоль) в DCM (15 мл) перемешивали при КТ в течение 5 ч. Смесь разбавляли DCM и промывали 0,5 н. HCl, насыщ. Na2CO3 и наконец рассолом; органическую фазу сушили над сульфатом натрия и растворитель удаляли под вакуумом. Неочищенный материал очищали препаративной ВЭЖХ (Метод 1) с получением (S)-3,5-дихлор-4-(2-(3- (циклопропилметокси)-4-(дифторметокси)фенил)-2-(1-(фенилсульфонил)-1Н-пиррол-2-карбонилокси)этил)пиридин-1-оксида (Соединение 3) (45 мг, 0,069 ммоль, МС/ИЭР+ 653,07 [МН]+, [αD] равно -25,83, с равно 0,6, DCM). 1Н ЯМР (300 МГц, DMSO-d6) δ м.д. 8.47 (s, 2Н), 7.87 (dd, 1Н), 7.78-7.85 (m, 2Н), 7.68-7.78 (m, 1Н), 7.51-7.64 (m, 2Н), 7.28 (dd, 1Н), 7.16 (d, 1Н), 7.11 (d, 1Н), 6.98 (dd, 1Н), 7.06 (t, 1Н), 6.52 (t, 1Н), 6.01 (dd, 1Н), 3.89 (d, 2Н), 3.44 (dd, 1Н), 3.22 (dd, 1Н), 1.09-1.30 (m, 1Н), 0.47-0.63 (m, 2Н), 0.26-0.41 (m, 2Н).
Пример 2
Синтез (S)-4-(2-(1-(4-аминофенилсульфонил)-1H-пиррол-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)-3,5-дихлорпиридин-1-оксида (Соединение 7)
Схема 2

Стадия 1: Получение метил-1-(4-нитрофенилсульфонил)-1Н-пиррол-2-карбоксилата (Промежуточное соединение 4)
К смеси метил-1Н-пиррол-2-карбоксилата (500 мг, 4,00 ммоль), гидросульфата тетрабутиламмония (136 мг, 0,400 ммоль) и гидроксида натрия (1,438 г, 36,0 ммоль) в воде (2,5 мл) и DCM (40 мл) при интенсивном перемешивании добавляли по каплям в течение 10 минут при КТ 4-нитробензол-1-сульфонилхлорид (1,550 г, 6,99 ммоль). Через 20 ч эту смесь разбавляли водой (50 мл) и DCM (100 мл); органические фазы отделяли, промывали рассолом (80 мл × 2) и сушили над сульфатом натрия. Растворитель выпаривали и неочищенный материал растирали с МеОН (50 мл) в течение ночи с получением после фильтрования метил-1-(4- нитрофенилсульфонил)-1Н-пиррол-2-карбоксилата (Промежут. соед. 4) (555 мг, 1,789 ммоль, МС/ИЭР+ 311,0 [МН]+).
Стадия 2: Получение 1-(4-нитрофенилсульфонил)-1Н-пиррол-2-карбоновой кислоты (Промежуточное соединение 5)
К раствору метил-1-(4-нитрофенилсульфонил)-1H-пиррол-2-карбоксилата (Промежут. соед. 4) (550 мг, 1,773 ммоль) в пиридине (25 мл) добавляли йодид лития (1661 мг, 12,41 ммоль) и полученную смесь кипятили с обратным холодильником в течение 8 ч. Раствор концентрировали под вакуумом и неочищенный материал растворяли в DCM (150 мл) и промывали 1 н. HCl (50 мл × 2) и затем рассолом (50 мл); органический слой сушили над сульфатом натрия и растворитель удаляли с получением 1-(4-нитрофенилсульфонил)-1Н-пиррол-2-карбоновой кислоты (Промежут. соед. 5) (433 мг, 1,462 ммоль).
Стадия 3: Получение (S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(1-(4-нитрофенилсульфонил)-1Н-пиррол-2-карбонилокси)этил)пиридин-1-оксида (Промежуточное соединение 6)
Смесь (S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)-фенил)-2-гидроксиэтил)пиридин-1-оксида (206 мг, 0,490 ммоль), 1-(4-нитрофенилсульфонил)-1Н-пиррол-2-карбоновой кислоты (Промежут. соед. 5) (160 мг, 0,539 ммоль), EDC (282 мг, 1,471 ммоль) и DMAP (59,9 мг, 0,490 ммоль) в DCM (10 мл) перемешивали при КТ в течение 24 ч. Реакционную смесь разбавляли DCM и промывали 1 н. HCl, насыщ. NaHCO3 (40 мл) и наконец рассолом. Органическую фазу сушили над сульфатом натрия и растворитель удаляли под вакуумом; неочищенный материал очищали препаративной ВЭЖХ (Метод 1) с получением (S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(1-(4-нитрофенилсульфонил)-1Н-пиррол-2-карбонилокси)этил)пиридин-1-оксида (Промежут.соед. 6) (95 мг, 0,136 ммоль, МС/ИЭР+ 698,05 [МН]+, [δD] равно -40,97, с равно 0,6, DCM).
1Н ЯМР (300 МГц, DMSO-d6) δ м.д. 8.40 (s, 2Н), 8.36-8.40 (m, 2Н), 8.04-8.18 (m, 2Н), 7.94 (dd, 1Н), 7.36 (dd, 1Н), 7.17 (d, 1Н), 7.13 (d, 1Н), 7.01 (dd, 1Н), 7.05 (t, 1Н), 6.59 (dd, 1Н), 6.03 (dd, 1Н), 3.90 (d, 2Н), 3.45 (dd, 1Н), 3.23 (dd, 1Н), 1.07-1.32 (m, 1Н), 0.46-0.64 (m, 2Н), 0.23-0.43 (m, 2Н).
Стадия 4: Получение (S)-4-(2-(1-(4-аминофенилсульфонил)-1Н-пиррол-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)-фенил)этил)-3,5-дихлорпиридин-1-оксида (Соединение 7)
Смесь (S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)-фенил)-2-(1-(4-нитрофенилсульфонил)-1Н-пиррол-2-карбонилокси)этил)-пиридин-1-оксида (Промежут. соед. 6) (424 мг, 0,607 ммоль) и дигидрата хлорида олова(Н) (548 мг, 2,428 ммоль) в THF (40 мл) нагревали при 45°C в течение 2 ч. Добавляли дополнительное количество дигидрата хлорида олова(II) (1,644 г, 7,284 ммоль) в 3 порциях в течение 3 часов пр 60°C и полученную смесь кипятили с обратным холодильником в течение ночи. Растворитель удаляли и смесь разбавляли этилацетатом; добавляли водный насыщ. NaHCO3 и смесь фильтровали через набивку целита. Отфильтрованные фазы отделяли и органический слой промывали рассолом и сушили над сульфатом натрия; растворитель удаляли и неочищенный материал очищали флэш-хроматографией на колонке с силикагелем (DCM/MeOH в соотношении 98/2) с получением (S)-4-(2-(1-(4-аминофенилсульфонил)-1Н-пиррол-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)-фенил)этил)-3,5-дихлорпиридин-1-оксида (Промежут. соед. 7) (207 мг, 0,310 ммоль, МС/ИЭР+ 667,96 [МН]+, [αD] равно -61,24, с равно 1, DCM).
1Н ЯМР (300 МГц, DMSO-d6) δ м.д. 8.50 (s, 2Н), 7.68 (dd, 1Н), 7.48-7.58 (m, 2Н), 7.11-7.20 (m, 3Н), 6.99 (dd, 1Н), 7.05 (t, 1Н), 6.51-6.63 (m, 2Н), 6.38 (dd, 1Н), 6.32 (s, 2Н), 6.05 (dd, 1Н), 3.90 (m, 2Н), 3.47 (dd, 1Н), 3.29 (dd, 1Н), 1.03-1.37 (m, 1Н), 0.43-0.76 (m, 2Н), 0.22-0.43 (m, 2Н).
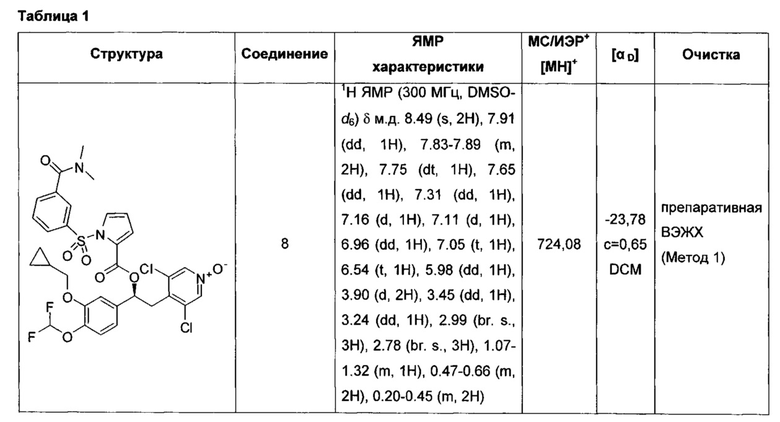
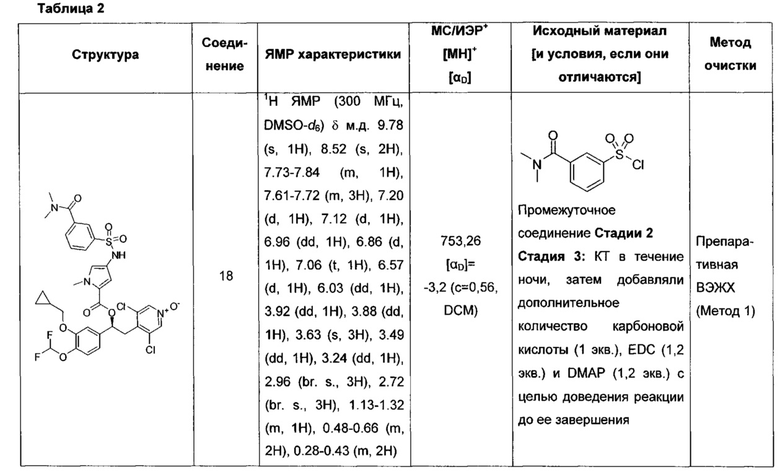
Соединение, указанное в Таблице 1, получали с использованием методики, аналогичной описанной на Схеме 2, Стадии 1, 2 и 3, используя подходящие реагенты.
Пример 3
Синтез (S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(1-(3-(диметилкарбамоил)-4-гидроксифенилсульфонил)-1Н-пиррол-2-карбонилокси)этил)пиридин-1-оксида (Соединение 13) и (5)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(1-(3-(диметилкарбамоил)-4-метоксифенилсульфонил)-1Н-пиррол-2-карбонилокси)этил)пиридин-1-оксида (Соединение 14)
Схема 3

Стадия 1: Получение 3-(диметилкарбамоил)-4-метоксибензол-1-сульфонилхлорида (Промежуточное соединение 9)
К суспензии 5-(хлорсульфонил)-2-метоксибензойной кислоты (1,243 г, 4,96 ммоль) в безводном DCM (20 мл), охлажденной до 0°C, добавляли по каплям (COCl)2 (1,302 мл, 14,88 ммоль) с последующим добавлением нескольких капель безводного DMF и реакционную смесь перемешивали при КТ в течение 1 ч. Смесь концентрировали при пониженном давлении, обрабатывали безводным толуолом (30 мл × 2) и упаривали досуха. Остаток суспендировали в безводном толуоле (20 мл) и полученную суспензию охлаждали до 0°C; добавляли по каплям раствор диметиламина 2,0 М в THF (4,96 мл, 9,92 ммоль) и реакционную смесь перемешивали при КТ в течение ночи. Смесь разбавляли DCM и промывали рассолом; органическую фазу сушили над сульфатом натрия и растворитель удаляли. Неочищенный материал очищали флэш-хроматографией на силикагеле (EtOAc/петролейный эфир в соотношении 60/40) с получением 3-(диметилкарбамоил)-4-метоксибензол-1-сульфонилхлорида (Промежут. соед. 9) (400 мг, 1,440 ммоль, МС/ИЭР+ 278,0 [МН]+).
Стадия 2: Получение метил-1-(3-(диметилкарбамоил)-4-метоксифенилсульфонил)-1H-пиррол-2-карбоксилата (Промежуточное соединение 10)
К смеси метил-1H-пиррол-2-карбоксилата (135 мг, 1,080 ммоль), гидросульфата тетрабутиламмония (36,7 мг, 0,108 ммоль) и гидроксида натрия (389 мг, 9,72 ммоль) в воде (2 мл) и DCM (4 мл) при интенсивном перемешивании добавляли по каплям при КТ в течение 10 минут раствор 3-(диметилкарбамоил)-4-метоксибензол-1-сульфонилхлорида (Промежут. соед. 9) (300 мг, 1,080 ммоль) в DCM (5 мл) и реакционную смесь перемешивали при той же температуре в течение 24 ч. Смесь разбавляли водой и DCM, органическую фазу отделяли, промывали рассолом и сушили над сульфатом натрия. Растворитель выпаривали под вакуумом с получением метил-1-(3-(диметилкарбамоил)-4-метоксифенилсульфонил)-1Н-пиррол-2-карбоксилата (Промежут. соед. 10) (260 мг, 0,710 ммоль, МС/ИЭР+ 367,0 [МН]+), который использовали в таком виде на следующей стадии.
Стадия 3: Получение 1-(3-(диметилкарбамоил)-4-гидроксифенилсульфонил)-1H-пиррол-2-карбоновой кислоты (Промежуточное соединение 12) и 1-(3-(диметилкарбамоил)-4-метоксифенилсульфонил)-1H-пиррол-2-карбоновой кислоты (Промежуточное соединение 11)
К раствору метил-1-(3-(диметилкарбамоил)-4-метоксифенилсульфонил)-1H-пиррол-2-карбоксилата (Промежут. соед. 10) (260 мг, 0,710 ммоль) в пиридине (7 мл) добавляли йодид лития (665 мг, 4,97 ммоль) и смесь кипятили с обратным холодильником в течение 20 ч. Растворитель удаляли под вакуумом и неочищенный материал разбавляли DCM; полученную суспензию промывали 1 н. HCl и затем рассолом. Органическую фазу сушили над сульфатом натрия и растворитель выпаривали; неочищенный материал очищали флэш-хроматографией на колонке с силикагелем (DCM/MeOH в соотношении от 9/1 до 1/1) с получением смеси 1-(3-(диметилкарбамоил)-4-гидроксифенилсульфонил)-1H-пиррол-2-карбоновой кислоты (Промежут. соед. 12) (МС/ИЭР+ 339,0 [МН]+) и 1-(3-(диметилкарбамоил)-4-метоксифенилсульфонил)-1H-пиррол-2-карбоновой кислоты (Промежут. соед. 11) (МС/ИЭР+ 353,0 [МН]+) (240 мг, соотношение примерно 1/1), которую использовали в таком виде на следующей стадии.
Стадия 4: Получение (S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(1-(3-(диметилкарбамоил)-4-гидроксифенилсульфонил)-1Н-пиррол-2-карбонилокси)этил)пиридин-1-оксида (Соединение 13) и (S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(1-(3-(диметилкарбамоил)-4-метоксифенилсульфонил)-1Н-пиррол-2-карбонилокси)этил)пиридин-1-оксида (Соединение 14)
(S)-3,5-Дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)-фенил)-2-гидроксиэтил)-пиридин-1-оксид (298 мг, 0,709 ммоль), EDC (408 мг, 2,128 ммоль), DMAP (95 мг, 0,780 ммоль) и смесь 1-(3-(диметилкарбамоил)-4-гидроксифенилсульфонил)-1H-пиррол-2-карбоновой кислоты (Промежут. соед. 12) и 1-(3-(диметилкарбамоил)-4-метоксифенилсульфонил)-1H-пиррол-2-карбоновой кислоты (Промежут. соед. 11) (245 мг, получена согласно методике, описанной выше на Стадии 3) растворяли в DCM (100 мл) и реакционную смесь перемешивали при КТ в течение 2 суток. Смесь разбавляли водой (80 мл) и осадок собирали фильтрованием и очищали препаративной ВЭЖХ (Метод 1) с получением:
первого элюированного соединения: (S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(1-(3-(диметилкарбамоил)-4-гидроксифенилсульфонил)-1Н-пиррол-2-карбонилокси)этил)пиридин-1-оксид (Соединение 13) (55 мг, 0,074 ммоль, МС/ИЭР+ 740,13 [МН]+, [δD] равно -15,20, с равно 0,8, DCM);
1Н ЯМР (300 МГц, DMSO-d6) δ м.д. 11.23 (s, 1Н), 8.51 (s, 2Н), 7.81 (dd, 1Н), 7.76 (dd, 1Н), 7.67 (d, 1Н), 7.24 (dd, 1Н), 7.15 (d, 1Н), 7.11 (d, 1Н), 7.01 (d, 1Н), 6.97 (dd, 1Н), 7.05 (t, 1Н), 6.47 (t, 1Н), 6.01 (dd, 1Н), 3.90 (d, 2Н), 3.47 (dd, 1Н), 3.29 (dd, 1Н), 2.95 (br. s., 3Н), 2.68 (br. s., 3Н), 1.07-1.33 (m, 1Н), 0.45-0.66 (m, 2Н), 0.21-0.44 (m, 2Н);
второго элюированного соединения: (S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)-фенил)-2-(1-(3-(диметилкарбамоил)-4-метоксифенилсульфонил)-1Н-пиррол-2-карбонилокси)этил)пиридин-1-оксид (Соединение 14) (25 мг, 0,033 ммоль, МС/ИЭР+ 754,15 [МН]+, -28,00, с равно 0,1, DCM);
1Н ЯМР (300 МГц, DMSO-d6) δ м.д. 8.50 (s, 2Н), 7.91 (dd, 1Н), 7.86 (dd, 1Н), 7.71 (d, 1Н), 7.23-7.29 (m, 2Н), 7.16 (d, 1Н), 7.12 (d, 1Н), 6.98 (dd, 1Н), 7.05 (t, 1Н), 6.48 (t, 1Н), 6.02 (dd, 1Н), 3.89 (s, 3Н), 3.90 (d, 2Н), 3.48 (dd, 1Н), 3.27 (dd, 1Н), 2.96 (s, 3Н), 2.63 (br. s., 3Н), 1.05-1.35 (m, 1Н), 0.46-0.65 (m, 2Н), 0.21-0.46 (m, 2Н).
Пример 4
Синтез (S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(1-метил-4-(фенилсульфонамидо)-1Н-пиррол-2-карбонилокси)этил)пиридин-1-оксида (Соединение 17)
Схема 4

Стадия 1: Получение 1-метил-4-нитро-1Н-пиррол-2-карбоновой кислоты (Промежуточное соединение 15)
Уксусный ангидрид (8 мл, 85 ммоль) обрабатывали азотной кислотой (1,020 мл, 15,98 ммоль) (после добавления наблюдалось нагревание); смесь охлаждали до КТ и медленно добавляли к суспензии 1-метил-1H-пиррол-2-карбоновой кислоты (2 г, 15,98 ммоль) в уксусном ангидриде (12 мл, 127 ммоль), охлажденной до -30°C. Реакционную смесь перемешивали при -30°C в течение 0,5 ч, затем при КТ в течение 20 минут. Смесь снова охлаждали до -30°C для осаждения целевого продукта. Твердое вещество быстро отфильтровывали на фриттованной воронке, охлаждаемой сухим льдом; через несколько минут при комнатной температуре твердое вещество становилось гигроскопичным и образовывало густую суспензию/раствор, которую(ый) собирали в отдельную колбу. Суспензию охлаждали при -30°C и промывали дважды смесью гексана (5 мл) и (iPr)2O (1 мл). Полученное твердое вещество сушили под вакуумом, затем обрабатывали 2 н. NaOH до полного растворения (10 мл) и осаждали снова путем добавления 37%-ной HCl (20 мл). Твердое вещество отфильтровывали, промывали водой (10 мл) и наконец сушили под вакуумом в течение 3 суток с получением 1-метил-4-нитро-1Н-пиррол-2-карбоновой кислоты (Промежут. соед. 15) (540 мг, 3,17 ммоль, МС/ИЭР+ 170,9 [МН]+).
Стадия 2: Получение 1-метил-4-(фенилсульфонамидо)-1Н-пиррол-2-карбоновой кислоты (Промежуточное соединение 16)
1-Метил-4-нитро-1Н-пиррол-2-карбоновую кислоту (Промежут. соед. 15) (350 мг, 2,057 ммоль) растворяли в водном 1М карбонате натрия (15 мл, 15,00 ммоль); добавляли 10% Pd/C (219 мг) и смесь гидрировали в аппарате Парра при давлении 40 фунт/кв.дюйм в течение 2 ч. Катализатор отфильтровывали, выдерживая смесь в потоке азота, и фильтрат собирали в охлажденную колбу (ледяную баню). К охлажденному раствору добавляли THF (20 мл) и бензолсульфонилхлорид (0,318 мл, 2,469 ммоль) и полученную смесь перемешивали при 0°C в течение 0,5 ч и затем при КТ в течение 1 ч. Реакционную смесь экстрагировали Et2O и органический слой отбрасывали. Водный слой осторожно подкисляли добавлением твердого KHSO4 до pH 3 и экстрагировали дважды DCM. Объединенные органические слои сушили над Na2SO4 и растворитель удаляли под вакуумом с получением 1-метил-4-(фенилсульфонамидо)-1Н-пиррол-2-карбоновой кислоты (Промежут. соед. 16) (540 мг, 1,927 ммоль, МС/ИЭР+ 280,9 [МН]+), которую использовали без очистки.
Стадия 2: Получение (S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(1-метил-4-(фенилсульфонамидо)-1Н-пиррол-2-карбонилокси)этил)пиридин-1-оксида (Соединение 17)
Раствор 1-метил-4-(фенилсульфонамидо)-1H-пиррол-2-карбоновой кислоты (Промежут. соед. 16) (77 мг, 0,274 ммоль), (S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-гидроксиэтил)пиридин-1-оксида (115 мг, 0,274 ммоль), EDC (79 мг, 0,410 ммоль) и DMAP (50,1 мг, 0,410 ммоль) в DCM (40 мл) перемешивали при КТ в течение 3 суток. Смесь промывали дважды 1 н. HCl и органический слой сушили над Na2SO4; растворитель удаляли под вакуумом и остаток очищали препаративной ВЭЖХ (Метод 1). Полученный продукт дополнительно очищали путем растирания с iPrOH и затем растворяли в CH3CN и воде и упаривали досуха с получением (S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(1-метил-4-(фенилсульфонамидо)-1Н-пиррол-2-карбонилокси)этил)пиридин-1-оксида (Соединение 17) (56 мг, 0,082 ммоль, МС/ИЭР+ 682,09 [МН]+, [αD] равно -1,5, с равно 0,46, DCM).
1Н ЯМР (300 МГц, DMSO-d6) δ м.д. 9.74 (s, 1Н), 8.52 (s, 2Н), 7.69-7.79 (m, 2Н), 7.50-7.69 (m, 3Н), 7.19 (d, 1Н), 7.11 (d, 1Н), 6.95 (dd, 1Н), 6.84 (d, 1Н), 7.06 (t, 1Н), 6.58 (d, 1Н), 6.03 (dd, 1Н), 3.92 (dd, 1Н), 3.89 (dd, 1Н), 3.63 (s, 3Н), 3.49 (dd, 1Н), 3.25 (dd, 1Н), 1.06-1.32 (m, 1Н), 0.45-0.72 (m, 2Н), 0.17-0.45 (m, 2Н).
Соединение, описанное в Таблице 2, получали согласно методикам, аналогичным описанным выше и проиллюстрированным на Схеме 4, Стадия 1-3, путем взаимодействия соответствующих предшественников (имеющихся в продаже или синтезированных специалистом в данной области техники) с подходящими реагентами, и где стадия очистки была осуществлена как указано в Таблице 2.
Пример 5
Синтез (S)-4-(2-(1H-пиррол-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)-3,5-дихлорпиридин-1-оксида (Соединение 20)
Схема 5

Стадия 1: Получение 1Н-пиррол-2-карбонилхлорида (Промежуточное соединение 19)
К раствору 1Н-пиррол-2-карбоновой кислоты (200 мг, 1,80 ммоль) в CHCl3 (5 мл) добавляли дихлорметилметиловый эфир (3,8 г, 3 мл, 33,05 ммоль) и смесь кипятили с обратным холодильником в течение 3 часов. Смесь оставляли нагреваться до КТ и растворитель выпаривали при пониженном давлении. Неочищенный материал переносили в хлороформ и упаривали досуха (×3) с получением 1H-пиррол-2-карбонилхлорида (Промежут. соед. 19) (200 мг, 1,54 ммоль). МС/ИЭР+ 129,00 [МН]+.
Стадия 2: Получение (S)-4-(2-(1Н-пиррол-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)-3,5-дихлорпиридин-1-оксида (Соединение 20)
К раствору (S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-гидроксиэтил)пиридин-1-оксида (30 мг, 0,071 ммоль) и 1Н-пиррол-2-карбонилхлорида (Промежут. соед. 19) (200 мг, 1,54 ммоль) в безводном DCM (5 мл) добавляли DMAP (20 мг, 0,164 ммоль). Смесь перемешивали в течение ночи при КТ. Смесь разбавляли DCM и промывали 1 н. HCl. Органическую фазу сушили над Na2SO4 и упаривали досуха; неочищенный материал очищали препаративной ВЭЖХ (Метод 2) с получением (S)-4-(2-(1Н-пиррол-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)-3,5-дихлорпиридин-1-оксида (Соединение 20) (20 мг, 0,039 ммоль, МС/ИЭР+ 513,32 [МН]+, [αD] равно -20,61, с равно 0,59, DCM).
1Н ЯМР (400 МГц, ацетон) δ м.д. 10.92 (br. s., 1Н), 8.26 (s, 2Н), 7.27 (d, J=1,96 Гц, 1Н), 7.11-7.22 (m, 2Н), 7.07 (td, J=2,75, 1.59 Гц, 1Н), 6.98 (ddd, J=3,79, 2,38, 1,53 Гц, 1Н), 6.90 (t, J=75,00 Гц, 1Н), 6.27 (dd, J=9,41, 4,77 Гц, 1Н), 6.22 (dt, J=3,76, 2,40 Гц, 1Н), 3.90-4.04 (m, 2Н), 3.65 (dd, J=14,00, 9,35 Гц, 1Н), 3.39 (dd, J=14,00, 4,83 Гц, 1Н), 1.21-1.32 (m, 1Н), 0.47-0.65 (m, 2Н), 0.31-0.44 (m, 2Н).
Соединение, описанное в Таблице 3, получали согласно методике синтеза, аналогичной описанной выше и проиллюстрированной на Схеме 5, Стадия 1, путем взаимодействия соответствующих предшественников (имеющихся в продаже или синтезированных специалистом в данной области техники) с подходящими реагентами, и где стадию очистки осуществляли как указано в Таблице 3.

Пример 6
Синтез (S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(5-метилтиофен-2-карбонилокси)-этил)пиридин-1-оксида (Соединение 22)
Схема 6

Получение (S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(5-метилтиофен-2-карбонилокси)этил)пиридин-1-оксида (Соединение 22)
Смесь (S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)-фенил)-2-гидроксиэтил)пиридин-1-оксида (100 мг, 0,238 ммоль), 5-метилтиофен-2-карбоновой кислоты (40,6 мг, 0,286 ммоль), EDC (137 мг, 0,714 ммоль) и DMAP (29,1 мг, 0,238 ммоль) в DCM (10 мл) перемешивали в течение ночи при КТ. Реакционную смесь разбавляли DCM, промывали 1 н. HCl, затем водным 5%-ным NaHCO3 и наконец рассолом. Органическую фазу сушили над сульфатом натрия и растворитель выпаривали; неочищенный материал очищали флэш-хроматографией на силикагеле (DCM, затем DCM/EtOAc в соотношении 3/1) и полученную аморфную фазу обрабатывали МеОН и упаривали с получением (S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(5-метилтиофен-2-карбонилокси)этил)-пиридин-1-оксида (Соединение 22) (114 мг, 0,209 ммоль, МС/ИЭР+ 544,17 [МН]+, [αD] равно -68,2, с равно 0,5, CHCl3).
1Н ЯМР (300 МГц, DMSO-d6) δ м.д. 8.56 (s, 2Н), 7.67 (d, 1Н), 7.21 (d, 1Н), 7.18 (d, 1Н), 7.05 (dd, 1Н), 6.94 (dd, 2Н), 6.14 (dd, 1Н), 3.93 (d, 2Н), 3.56 (dd, 1Н), 3.31 (dd, 1Н), 1.03-1.32 (m, 1Н), 0.45-0.69 (m, 2Н), 0.16-0.44 (m, 2Н).
Соединения, перечисленные в Таблице 4, получали согласно методикам, аналогичным описанному выше в Примере 6, Схема 6, используя соответствующие исходные материалы.
Соединение 82, указанное в Таблице 4а, получали согласно методике, аналогичной описанной выше в Примере 6, Схема 6, используя в качестве исходного материала подходящий спирт (S)-3,5-дихлор-4-(2-(3,4-диметоксифенил)-2-гидроксиэтил)пиридин-1-оксид, полученный как описано в WO 2012/168226 (Пример 18, Схема 24, соединение 170).

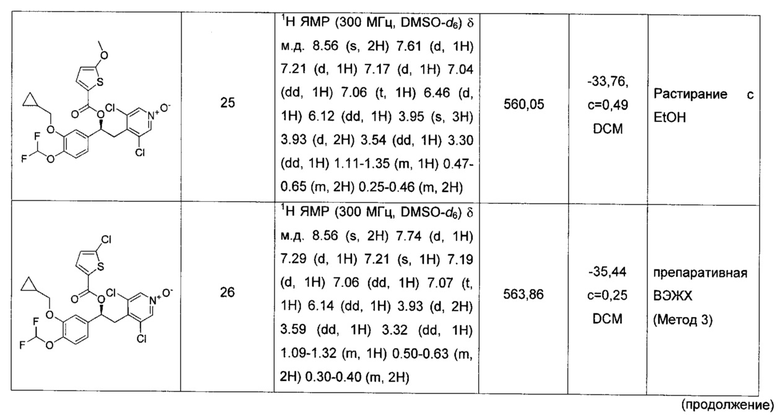
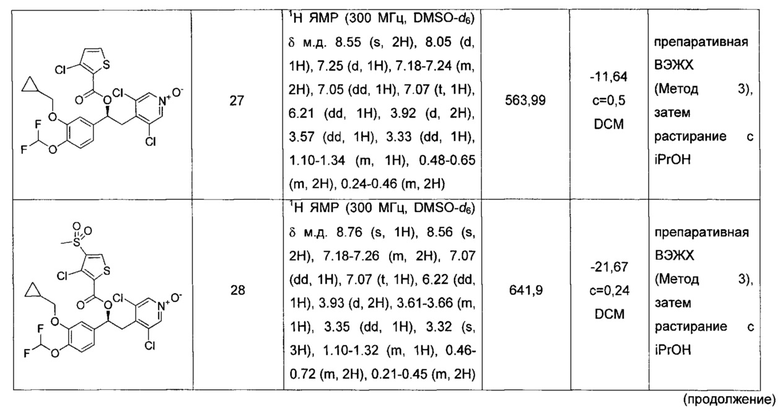



Пример 7
Синтез (S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(3-(N-метилсульфамоил)тиофен-2-карбонилокси)этил)пиридин-1-оксида (Соединение 36)
Схема 7
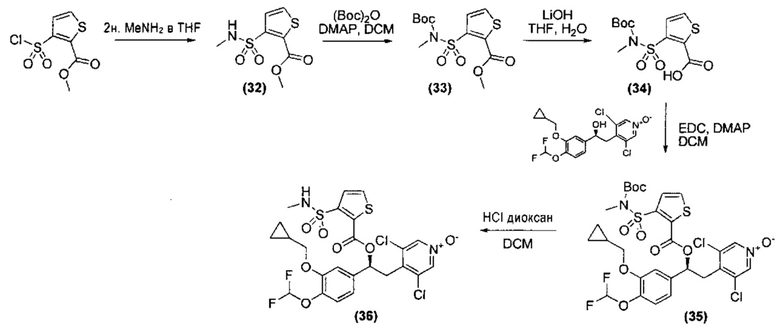
Стадия 1: Получение метил-3-(N-метилсульфамоил)тиофен-2-карбоксилата (Промежуточное соединение 32)
К раствору метил-3-(хлорсульфонил)тиофен-2-карбоксилата (500 мг, 2,077 ммоль) в DCM (20 мл) добавляли 2 н. раствор метанамина в THF (4,155 мл, 8,31 ммоль) и реакционную смесь перемешивали в течение 1 ч при КТ. Смесь распределяли между 2 н. HCl и DCM и водную фазу экстрагировали DCM. Объединенные органические слои сушили над сульфатом натрия и растворитель удаляли с получением метил-3-(N-метилсульфамоил)тиофен-2-карбоксилата (Промежут. соед. 32) (430 мг, 1,828 ммоль, МС/ИЭР+ 235,8 [МН]+).
Стадия 2: Получение метил-3-(N-(/прел7-бутоксикарбонил)-1H-метилсульфамоил)тиофен-2-карбоксилата (Промежут. соед. 33)
К раствору метил-3-(N-метилсульфамоил)тиофен-2-карбоксилата (Промежут. соед. 32) (430 мг, 1,828 ммоль) в DCM (20 мл) добавляли DMAP (246 мг, 2,010 ммоль) и ди-трет-бутилдикарбонат (399 мг, 1,828 ммоль) и реакционную смесь перемешивали в течение ночи при КТ. Смесь распределяли между водой и DCM и водную фазу экстрагировали DCM (×3); объединенные органические слои сушили над сульфатом натрия и растворитель удаляли с получением метил-3-(N-(трет-бутоксикарбонил)-N-метилсульфамоил)тиофен- 2-карбоксилата (Промежут. соед. 33), который использовали без какой-либо дополнительной очистки (500 мг, 1,491 ммоль, МС/ИЭР+ 357,8 [MNa]+).
Стадия 3: Получение 3-(N-(трет-бутоксикарбонил)-N-метилсульфамоил)тиофен-2-карбоновой кислоты (Промежуточное соединение 34)
К раствору метил-3-(N-(трет-бутоксикарбонил)-N-метилсульфамоил)-тиофен-2-карбоксилата (Промежут. соед. 33) (500 мг, 1,491 ммоль) в смеси THF и Н2О 1/1 (100 мл) добавляли гидроксид лития (357 мг, 14,91 ммоль) и реакционную смесь перемешивали при КТ в течение 2 ч. Органический растворитель выпаривали под вакуумом и водный остаток подкисляли 1 н. HCl (рН 4). Полученный осадок собирали фильтрованием с получением 3-(N-(трет-бутоксикарбонил)-N-метилсульфамоил)тиофен-2-карбоновой кислоты (Промежут. соед. 34) (370 мг, 1,151 ммоль, МС/ИЭР+ 343,8 [MNa]+).
Стадия 4: Получение (S)-4-(2-(3-(N-(трет-бутоксикарбонил)-N-метилсульфамоил)тиофен-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)-3,5-дихлорпиридин-1-оксида (Промежуточное соединение 35)
К раствору 3-(N-(трет-бутоксикарбонил)-N-метилсульфамоил)-тиофен-2-карбоновой кислоты (Промежут. соед. 34) (150 мг, 0,467 ммоль) в DCM (5 мл) добавляли (S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)-фенил)-2-гидроксиэтил)пиридин-1-оксид (196 мг, 0,467 ммоль), EDC (268 мг, 1,400 ммоль) и DMAP (28,5 мг, 0,233 ммоль) и смесь подвергали взаимодействию при КТ в течение ночи. Растворитель удаляли и неочищенный материал очищали флэш-хроматографией на силикагеле (DCM/MeOH в соотношении 95/5) с получением (S)-4-(2-(3-(N-(трет-бутоксикарбонил)-N-метилсульфамоил)тиофен-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)-3,5-дихлорпиридин-1-оксида (Промежут. соед. 35) (230 мг, 0,318 ммоль, МС/ИЭР+ 722,8 [МН]+).
Стадия 5: Получение (S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(3-(N-метилсульфамоил)тиофен-2-карбонилокси)этил)пиридин-1-оксида (Соединение 36)
К раствору (S)-4-(2-(3-(N-(трет-бутоксикарбонил)-N-метилсульфамоил)-тиофен-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-этил)-3,5-дихлорпиридин-1-оксида (Промежут. соед. 35) (230 мг, 0,318 ммоль) в DCM (10 мл) добавляли 4М HCl в диоксане (0,795 мл, 3,18 ммоль) и смесь перемешивали при КТ в течение 48 ч. Растворитель выпаривали и полученный неочищенный материал очищали препаративной ВЭЖХ (Метод 3) с получением (S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(3-(N-метилсульфамоил)тиофен-2-карбонилокси)этил)пиридин-1-оксида (Соединение 36) (71,3 мг, 0,114 ммоль, МС/ИЭР+ 623,02 [МН]+, [αD] равно -10,04, с равно 0,5, DCM).
1Н ЯМР (300 МГц, DMSO-d6) δ м.д. 8.54 (s, 2Н), 8.06 (dd, 1Н), 7.43 (dd, 1Н), 7.21 (d, 1Н), 7.21 (d, 1Н), 6.99-7.07 (m, 2Н), 7.08 (t, 1Н), 6.19 (dd, 1Н), 3.93 (d, 2Н), 3.58 (dd, 1Н), 3.37 (dd, 1Н), 2.48 (d, 3Н), 1.14-1.23 (m, 1Н), 0.50-0.63 (m, 2Н), 0.28-0.42 (m, 2Н).
Пример 8
Синтез (S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(3-(метилсульфонамидо)тиофен-2-карбонилокси)этил)пиридин-1-оксида (Соединение 39)
Схема 8
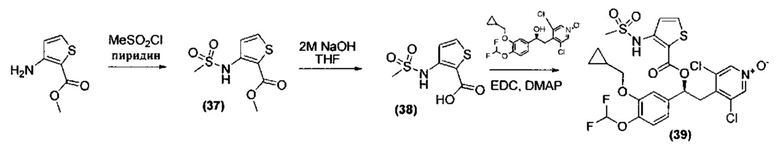
Стадия 1: Получение метил-3-(метилсульфонамидо)тиофен-2-карбоксилата (Промежуточное соединение 37)
К раствору метил-3-аминотиофен-2-карбоксилата (1,013 г, 6,44 ммоль) в пиридине (15 мл), охлажденного до 0°C, добавляли метансульфонилхлорид (0,552 мл, 7,09 ммоль) и реакционную смесь перемешивали при КТ в течение 4 ч. Добавляли дополнительное количество метансульфонилхлорида (0,100 мл, 1,289 ммоль) и перемешивание продолжали в течение ночи при КТ. Растворитель выпаривали и остаток распределяли между EtOAc и 1 н. HCl; органический слой промывали 1 н. HCl, сушили над Na2SO4 и упаривали. Неочищенный материал растирали с Et2O/DCM 95/5 с получением метил-3-(метилсульфонамидо)тиофен-2-карбоксилата (Промежуточное соединение 37) (1,272 г, 5,41 ммоль, МС/ИЭР+ 235,9 [МН]+).
Стадия 2: Получение 3-(метилсульфонамидо)тиофен-2-карбоновой кислоты (Промежуточное соединение 38)
Смесь метил-3-(метилсульфонамидо)тиофен-2-карбоксилата (Промежут. соед. 37) (193 мг, 0,820 ммоль) в водном 2М NaOH (4,10 мл, 8,20 ммоль) и THF (4,1 мл) нагревали в условиях микроволнового облучения при 80°C в течение 30 минут. Добавляли 2 н. HCl (pH 2) и смесь экстрагировали EtOAc (×3). Объединенные органические слои сушили над Na2SO4 и упаривали досуха с получением 3-(метилсульфонамидо)тиофен-2-карбоновой кислоты (Промежут. соед. 38) (180 мг, 0,814 ммоль, МС/ИЭР+ 221,8 [МН]+).
Стадия 3: Получение (S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(3-(метилсульфонамидо)тиофен-2-карбонилокси)этил)пиридин-1-оксида (Соединение 39)
Смесь 3-(метилсульфонамидо)тиофен-2-карбоновой кислоты (Промежут. соед. 38) (110 мг, 0,497 ммоль), (S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-гидроксиэтил)пиридин-1-оксида (209 мг, 0,497 ммоль), EDC (286 мг, 1,491 ммоль) и DMAP (30,4 мг, 0,249 ммоль) в DCM (25 мл) перемешивали при КТ в течение 3 суток. Смесь разбавляли DCM и промывали 1 н. HCl и водным NaHCO3; органический слой сушили над Na2SO4, фильтровали и упаривали. Неочищенный материал очищали флэш-хроматографией на картридже с силикагелем (DCM/MeOH в соотношении от 99,5/0,5 до 99/1) с получением (S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)-фенил)-2-(3-(метилсульфонамидо)-тиофен-2-карбонилокси)этил)пиридин-1-оксида (Промежут. соед. 39) (95 мг, 0,152 ммоль, МС/ИЭР+ 623,05 [МН]+, [αD] равно -28,6, с равно 0,3, DCM). 1H ЯМР (300 МГц, DMSO-d6) δ м.д. 8.57 (s, 3Н) 7.97 (d, 1Н) 7.27 (d, 1Н) 7.23 (d, 1Н) 7.19 (d, 1Н) 7.04 (dd, 1Н) 6.94 (t, 1Н) 6.16 (dd, 1Н) 3.93 (d, 2Н) 3.58 (dd, 1Н) 3.34-3.45 (m, 1Н) 3.16 (s, 3Н) 1.04-1.35 (m, 1Н) 0.49-0.65 (m, 2Н) 0.24-0.42 (m, 2Н).
Пример 9
Синтез (S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(4-(метилсульфонамидо)тиофен-2-карбонилокси)этил)пиридин-1-оксида (Соединение 46)
Схема 9
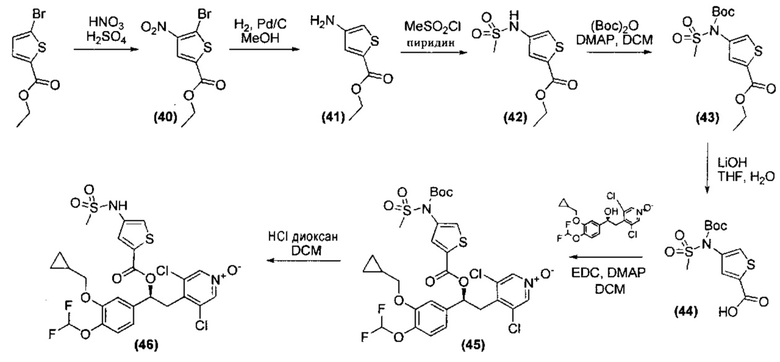
Стадия 1: Получение этил-5-бром-4-нитротиофен-2-карбоксилата (Промежуточное соединение 40)
К раствору этил-5-бромтиофен-2-карбоксилата (1,5 г, 6,38 ммоль) в водной конц. H2SO4 (6 мл), охлажденному до 0°C, добавляли водную 65%-ную HNO3 (2 мл) и реакционную смесь оставляли нагреваться до КТ и перемешивали в течение 1 ч. Смесь вливали в ледяную воду и экстрагировали EtOAc (×3); объединенные органические слои сушили над сульфатом натрия и растворитель удаляли с получением этил-5-бром-4-нитротиофен-2-карбоксилата (Промежут. соед. 40), который использовали без какой-либо дополнительной очистки (550 мг, 1,964 ммоль).
Стадия 2: Получение этил-4-аминотиофен-2-карбоксилата (Промежуточное соединение 41)
Раствор этил-5-бром-4-нитротиофен-2-карбоксилата (Промежут. соед. 40) (500 мг, 1,785 ммоль) в МеОН (35 мл) гидрировали в проточном реакторе (аппарат H-Cube) (скорость потока: 1,0 мл/мин, темп.: 50°C, давление H2: 20 бар). Растворитель выпаривали с получением этил-4-аминотиофен-2-карбоксилата (Промежут. соед. 41), который использовали без какой-либо дополнительной очистки (350 мг, МС/ИЭР+ 172,1 [МН]+).
Стадия 3: Получение этил-4-(метилсульфонамидо)тиофен-2-карбоксилата (Промежуточное соединение 42)
К раствору этил-4-аминотиофен-2-карбоксилата (Промежут. соед. 41) (теоретически 1,785 ммоль) в пиридине (20 мл) добавляли метансульфонилхлорид (0,239 мл, 3,07 ммоль) и смесь перемешивали в течение ночи при КТ. Растворитель выпаривали и остаток распределяли между 1 н. HCl и EtOAC; водную фазу экстрагировали EtOAc и объединенные органические слои сушили над сульфатом натрия и упаривали с получением этил-4-(метилсульфонамидо)тиофен-2-карбоксилата (Промежут. соед. 42), который использовали без какой-либо дополнительной очистки (370 мг, 1,484 ммоль, МС/ИЭР+ 249,8 [МН]+).
Стадия 4: Получение этил-4-(N-(трет-бутоксикарбонил)-метилсульфонамидо)тиофен-2-карбоксилата (Промежуточное соединение 43)
К раствору этил-4-(метилсульфонамидо)тиофен-2-карбоксилата (22) (370 мг, 1,484 ммоль) в DCM (20 мл) добавляли DMAP (272 мг, 2,226 ммоль) и ди-трет-бутилдикарбонат (486 мг, 2,226 ммоль) и смесь перемешивали при КТ в течение 5 ч. Растворитель удаляли и неочищенный материал очищали флэш-хроматографией на силикагеле (гексан/EtOAc в соотношении 1/1) с получением этил-4-(N-(трет-бутоксикарбонил)-метилсульфонамидо)тиофен-2-карбоксилата (Промежут. соед. 43) (300 мг, 0,859 ммоль).
Стадия 5: Получение 4-(N-(трет-бутоксикарбонил)-метилсульфонамидо)тиофен-2-карбоновой кислоты (Промежуточное соединение 44)
К раствору этил-4-(N-(трет-бутоксикарбонил)метилсульфонамидо)-тиофен-2-карбоксилата (Промежут. соед. 43) (300 мг, 0,859 ммоль) в смеси THF/вода 1/1 (20 мл) добавляли гидроксид лития (61,7 мг, 2,58 ммоль) и смесь подвергали взаимодействию в течение ночи при КТ. Органический растворитель выпаривали и водный остаток подкисляли 1 н. HCl (pH 5); полученный осадок собирали фильтрованием с получением 4-(N-(трет-бутоксикарбонил)-метилсульфонамидо)тиофен-2-карбоновой кислоты (Промежут. соед. 44) (220 мг, 0,685 ммоль, МС/ИЭР+ 343,7 [MNa]+).
Стадия 6: Получение (S)-4-(2-(4-(N-(трет-бутоксикарбонил)-метилсульфонамидо)тиофен-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)-3,5-дихлорпиридин-1-оксида (Промежуточное соединение 45)
К раствору 4-(N-(трет-бутоксикарбонил)метилсульфонамидо)-тиофен-2-карбоновой кислоты (Промежут. соед. 44) (100 мг, 0,311 ммоль) в DCM (5 мл) добавляли (S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)- фенил)-2-гидроксиэтил)пиридин-1-оксид (131 мг, 0,311 ммоль), EDC (179 мг, 0,934 ммоль) и DMAP (19,01 мг, 0,156 ммоль) и реакционную смесь перемешивали при КТ в течение ночи. Смесь разбавляли DCM (10 мл) и промывали 1 н. HCl; водную фазу экстрагировали EtOAc и объединенные органические слои сушили над сульфатом натрия. Растворитель удаляли под вакуумом и неочищенный материал очищали флэш-хроматографией на силикагеле (DCM/MeOH в соотношении 98/2) с получением (S)-4-(2-(4-(N-(трет-бутоксикарбонил)-метилсульфонамидо)тиофен-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)-3,5-дихлорпиридин-1-оксида (Промежут. соед. 45) (72 мг, 0,100 ммоль, МС/ИЭР+ 722,9 [МН]+).
Стадия 7: Получение (S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(4-(метилсульфонамидо)тиофен-2-карбонилокси)этил)пиридин-1-оксида (Соединение 46)
К раствору (S)-4-(2-(4-(N-(трет-бутоксикарбонил)-метилсульфонамидо)-тиофен-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-этил)-3,5-дихлорпиридин-1-оксида (Промежут.соед. 5) (72 мг, 0,100 ммоль) в DCM (5 мл) добавляли 4М HCl в диоксане (0,249 мл, 0,995 ммоль) и смесь подвергали взаимодействию в течение ночи при КТ. Растворитель удаляли и неочищенный материал растирали с МеОН; дополнительная очистка препаративной ВЭЖХ (Метод 3) потребовалась для получения (S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(4-(метилсульфонамидо)-тиофен-2-карбонилокси)этил)пиридин-1-оксида (Соединение 46) (30,3 мг, 0,049 ммоль, МС/ИЭР+ 623,05 [МН]+, [αD] равно -29,28, с равно 0,25, DCM).
1Н ЯМР (300 МГц, DMSO-d6) δ м.д. 10.06 (s, 1Н), 8.55 (s, 2Н), 7.60 (d, 1Н), 7.40 (d, 1Н), 7.22 (d, 1Н), 7.19 (d, 1Н), 7.05 (dd, 1Н), 7.07 (t, 1Н), 6.13 (dd, 1Н), 3.93 (d, 2Н), 3.59 (dd, 1Н), 3.32 (dd, 1Н), 3.00 (s, 3Н), 1.12-1.32 (m, 1Н), 0.48-0.62 (m, 2Н), 0.27-0.44 (m, 2Н).
Пример 10
Синтез (S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(3-(N-метил-N-(2-морфолиноэтил)-сульфамоил)-тиофен-2-карбонилокси)этил)пиридин-1-оксида (Соединение 49)
Схема 10

Стадия 1: Получение метил-3-(N-метил-N-(2-морфолиноэтил)-сульфамоил)тиофен-2-карбоксилата (Промежуточное соединение 47)
К раствору метил-3-(N-метилсульфамоил)тиофен-2-карбоксилата (полученного способом, аналогичным описанному в Примере 10, Схема 10, Стадия 1, 530 мг, 2,253 ммоль) в безводном DMF (20 мл) в атмосфере азота добавляли К2СО3 (623 мг, 4,51 ммоль) и гидрохлорид 4-(2-хлорэтил)морфолина (838 мг, 4,51 ммоль) и эту смесь подвергали взаимодействию при 60°C в течение ночи и затем при 80°C в течение 5 ч. Нерастворимые неорганические соли отфильтровывали, фильтрат упаривали и остаток очищали флэш-хроматографией на силикагеле (DCM/MeOH в соотношении 9/1) с получением метил-3-(N-метил-N-(2-морфолиноэтил)сульфамоил)тиофен-2-карбоксилата (Промежут. соед. 47) (510 мг, 1,464 ммоль, МС/ИЭР+ 348,8 [МН]+).
Стадия 2: Получение 3-(N-метил-N-(2-морфолиноэтил)-сульфамоил)-тиофен-2-карбоновой кислоты (Промежут. соед. 48)
К раствору метил-3-(N-метил-N-(2-морфолиноэтил)сульфамоил)-тиофен-2-карбоксилата (Промежут. соед. 47) (510 мг, 1,464 ммоль) в смеси 1:1 THF и H2O добавляли гидроксид лития (351 мг, 14,64 ммоль) и смесь перемешивали при КТ в течение ночи. Органический растворитель выпаривали и водный остаток подкисляли 2 н. HCl (pH 7); осадок собирали фильтрованием с получением 3-(N-метил-N-(2-морфолиноэтил)-сульфамоил)тиофен-2-карбоновой кислоты (Промежут. соед. 48) (410 мг, 1,226 ммоль, МС/ИЭР+ 334,9 [МН]+).
Стадия 3: Получение (S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(3-(N-метил-N-(2-морфолиноэтил)-сульфамоил)-тиофен-2-карбонилокси)этил)пиридин-1-оксида (Соединение 49)
К раствору 3-(N-метил-N-(2-морфолиноэтил)сульфамоил)-тиофен-2-карбоновой кислоты (Промежут. соед. 48) (95 мг, 0,284 ммоль) в DCM (5 мл) добавляли (S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)- фенил)-2-гидроксиэтил)пиридин-1-оксид (119 мг, 0,284 ммоль), DMAP (52,1 мг, 0,426 ммоль) и EDC (163 мг, 0,852 ммоль) и смесь перемешивали в течение ночи при КТ. Растворитель выпаривали и остаток очищали фильтрованием через SCX картридж (МеОН, затем МеОН/конц. NH4OH водн. в соотношении 9/1) с получением (S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)-фенил)-2-(3-(N-метил-N-(2-морфолиноэтил)сульфамоил)тиофен-2-карбонилокси)этил)пиридин-1-оксида (Соединение 49) (70,1 мг, 0,095 ммоль, МС/ИЭР+ 736,17 [МН]+, [αD] равно +8,96, с равно 0,25, DCM).
1Н ЯМР (300 МГц, DMSO-d6) δ м.д. 8.54 (s, 2Н) 8.03 (d, 1Н) 7.47 (d, 1Н) 7.00-7.27 (m, 3Н) 7.07 (t, 1Н) 6.18 (dd, 1Н) 3.92 (d, 2Н) 3.54-3.65 (m, 1Н) 3.46-3.54 (m, 4Н) 3.36 (dd, 1Н) 3.21-3.25 (m, 2Н) 2.82 (s, 3Н) 2.39 (t, 2Н) 2.26-2.35 (m, 4Н) 1.19-1.31 (m, 1Н) 0.50-0.64 (m, 2Н) 0.27-0.40 (m, 2Н).
Пример 11
Синтез (S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(3-(N-(2-морфолиноэтил)-метилсульфонамидо)-тиофен-2-карбонилокси)этил)пиридин-1-оксида (Соединение 52)
Схема 11

Стадия 1: Получение метил-3-(N-(2-морфолиноэтил)-метилсульфонамидо)тиофен-2-карбоксилата (Промежуточное соединение 50)
Смесь метил-3-(метилсульфонамидо)тиофен-2-карбоксилата (полученного способом, аналогичным описанному в Примере 8, Схема 8, Стадия 1, 500 мг, 2,125 ммоль), гидрохлорида 4-(2-хлорэтил)морфолина (435 мг, 2,338 ммоль) и карбоната калия (646 мг, 4,68 ммоль) в безводном DMF (13 мл) нагревали при 80°C в течение 5 ч и перемешивали при КТ в течение 3 суток. Растворитель выпаривали, остаток разбавляли EtOAc и промывали H2O и рассолом; органический слой сушили над Na2SO4, фильтровали и упаривали. Неочищенный материал очищали флэш-хроматографией на силикагеле (DCM/EtOAc в соотношении от 8/2 до 7/3 и затем DCM/MeOH в соотношении 95/5) с получением метил-3-(N-(2-морфолиноэтил)метилсульфонамидо)-тиофен-2-карбоксилата (Промежут. соед. 50) (655 мг, 1,880 ммоль, МС/ИЭР+ 348,9 [МН]+).
Стадия 2: Получение 3-(N-(2-морфолиноэтил)-метилсульфонамидо)-тиофен-2-карбоновой кислоты (Промежуточное соединение 51)
Смесь метил-3-(N-(2-морфолиноэтил)метилсульфонамидо)-тиофен-2-карбоксилата (Промежуточное соединение 50) (348 мг, 0,999 ммоль) в водном 2М гидроксиде натрия (4,994 мл, 9,99 ммоль) и THF (4,9 мл) перемешивали при 60°C в течение 6 часов и при КТ в течение ночи. Смесь нейтрализовали 2 н. HCl и растворитель удаляли. Неочищенный материал очищали флэш-хроматографией на силикагеле (DCM/MeOH в соотношении от 8/2 до 75/25) с получением 3-(N-(2-морфолиноэтил)метилсульфонамидо)тиофен-2-карбоновой кислоты (Промежуточное соединение 51) (312 мг, 0,933 ммоль, МС/ИЭР+ 334,9 [МН]+).
Стадия 3: Получение (S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(3-(N(2-морфолиноэтил)-метилсульфонамидо)-тиофен-2-карбонилокси)этил)пиридин-1-оксида (Соединение 52)
Смесь 3-(N-(2-морфолинозтил)метилсульфонамидо)тиофен-2-карбоновой кислоты (Промежут. соед. 51) (159 мг, 0,476 ммоль), (S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-гидроксиэтил)-пиридин-1-оксида (200 мг, 0,476 ммоль), EDC (274 мг, 1,428 ммоль) и DMAP (87 мг, 0,714 ммоль) в DCM (25 мл) перемешивали при КТ в течение ночи. Добавляли 0,5 н. HCl, фазы разделяли и органический слой промывали водным NaHCO3 и сушили над Na2SO4. Растворитель удаляли и неочищенный материал очищали фильтрованием через SCX картридж (DCM/MeOH в соотношении 1/1, затем МеОН/конц. NH4OH водн. в соотношении 95/5); основные фракции разбавляли H2O и полученный водный раствор экстрагировали EtOAc (×3). Объединенные органические слои сушили над Na2SO4, фильтровали и упаривали. Выделенный продукт дополнительно очищали флэш-хроматографией на картридже с силикагелем (DCM/MeOH в соотношении от 99,5/0,5 до 97/3) с получением (S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(3-(N-(2-морфолиноэтил)-метилсульфонамидо)тиофен-2-карбонилокси)этил)пиридин-1-оксида (Соединение 52) (50,2 мг, 0,068 ммоль, МС/ИЭР+ 736,21 [MH]+, [αD] равно -9,88, с равно 0,48, DCM).
1Н ЯМР (300 МГц, DMSO-d6) δ м.д. 8.40 (s, 2Н), 7.89 (d, 1Н), 7.24 (d, 1Н), 7.19 (d, 1Н), 7.19 (d, 1Н), 7.05 (dd, 1Н), 6.98 (t, 1Н), 6.25 (dd, 1Н), 3.95 (d, 2Н), 3.72 (br. s., 2Н), 3.59 (dd, 1Н), 3.51 (br. s., 4Н), 3.42 (dd, 1Н), 2.95 (s, 3Н), 2.31-2.46 (m, 6Н), 1.12-1.33 (m, 1Н), 0.51-0.66 (m, 2Н), 0.23-0.45 (m, 2Н).
Пример 12
Синтез (S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(5-(N-(2-морфолиноэтил)-метилсульфонамидо)-тиофен-2-карбонилокси)этил)пиридин-1-оксида (Соединение 60)
Схема 12

Стадия 1: Получение 5-нитротиофен-2-карбоновой кислоты (Промежуточное соединение 53)
Смесь 5-нитротиофен-2-карбальдегида (1,685 г, 10,72 ммоль) и сульфаминовой кислоты (1,249 г, 12,87 ммоль) в диоксане (30 мл) охлаждали при 0°C и добавляли по каплям раствор хлорита натрия (1,940 г, 21,44 ммоль) в воде (14 мл). Смесь оставляли нагреваться до КТ и перемешивали в течение 2 ч. Вторую партию 5-нитротиофен-2-карбальдегида (1,966 г, 12,1 ммоль) подвергали взаимодействию в тех же условиях с использованием молярных соотношений, описанных выше. Две реакционные смеси объединяли и распределяли между этилацетатом и водой. Органический слой экстрагировали дважды 5%-ным NaHCO3 и органическую фазу отбрасывали. Основную водную фазу подкисляли 2 н. HCl (pH 2) и экстрагировали дважды этилацетатом. Объединенные органические слои промывали рассолом и сушили над сульфатом натрия; растворитель удаляли с получением 5-нитротиофен-2-карбоновой кислоты (Промежут. соед. 53) (3,6 г, 20,79 ммоль, МС/ИЭР+ 173,9 [МН]+).
Стадия 2: Получение трет-бутил-5-нитротиофен-2-карбоксилата (Промежуточное соединение 54)
Раствор 5-нитротиофен-2-карбоновой кислоты (Промежут. соед. 53) (3,6 г, 20,79 ммоль) в безводном толуоле (260 мл) кипятили с обратным холодильником и добавляли по каплям 1,1-ди-трет-бутокси-N,N-диметилметанамин (9,97 мл, 41,6 ммоль). Реакционную смесь кипятили с обратным холодильником в течение 20 ч. Смесь промывали насыщенным раствором NaHCO3, затем рассолом, сушили над Na2SO4 и упаривали досуха с получением трет-бутил-5-нитротиофен-2-карбоксилата (Промежут. соед. 54) (2,23 г, 9,73 ммоль, МС/ИЭР+ недетектируемый [МН]+).
Стадия 3: Получение трет-бутил-5-аминотиофен-2-карбоксилата (Промежуточное соединение 55)
К суспензии трет-бутил-5-нитротиофен-2-карбоксилата (Промежут. соед. 54) (2,230 г, 9,73 ммоль) в смеси EtOH (60 мл) и воды (30 мл) добавляли порошок железа (3,26 г, 58,4 ммоль) и хлорид аммония (0,364 г, 6,81 ммоль). Реакционную смесь перемешивали при 80°C в течение 3 ч и затем при КТ в течение ночи. Смесь фильтровали через набивку целита и фильтрат упаривали досуха. Неочищенный материал затем растворяли в EtOAc и промывали рассолом; органическую фазу сушили над сульфатом натрия и растворитель выпаривали под вакуумом. Остаток очищали фильтрованием через картридж с силикагелем (петролейный эфир/EtOAC в соотношении от 9/1 до 6/4) с получением трет-бутил-5-аминотиофен-2-карбоксилата (Промежут. соед. 55) (1,481 г, 7,43 ммоль, МС/ИЭР+ 200,0 [МН]+).
Стадия 4: Получение трет-бутил-5-(метилсульфонамидо)-тиофен-2-карбоксилата (Промежуточное соединение 56) и трет-бутил-5-(N- (метилсульфонил)-метилсульфонамидо)тиофен-2-карбоксилата (Промежуточное соединение 57)
К раствору трет-бутил-5-аминотиофен-2-карбоксилата (Промежут. соед. 55) (1,481 г, 7,43 ммоль) в пиридине (50 мл), охлажденному до 0°C, добавляли по каплям метансульфонилхлорид (0,695 мл, 8,92 ммоль). Реакционную смесь нагревали до КТ и перемешивали в течение 3 суток. Растворитель удаляли под вакуумом и остаток распределяли между EtOAc и 1 н. HCl; органическую фазу промывали рассолом, сушили над сульфатом натрия, фильтровали и упаривали досуха. Неочищенный материал очищали флэш-хроматографией на картридже с силикагелем (петролейный эфир/EtOAC в соотношении 8/2) с получением трет-бутил-5-(метилсульфонамидо)тиофен-2-карбоксилата (Промежут. соед. 56) (773 мг, 2,79 ммоль, МС/ИЭР+ 299,9 [MNa]+) и трет-бутил-5-(N-(метилсульфонил)-метилсульфонамидо)тиофен-2-карбоксилата (Промежут. соед. 57) (420 мг, 1,182 ммоль, МС/ИЭР+ недетектируемый [МН]+).
Стадия 5а-b: Получение трет-бутил-5-(N-(2-морфолиноэтил)-метилсульфонамидо)тиофен-2-карбоксилата (Промежуточное соединение 58)
Смесь трет-бутил-5-(метилсульфонамидо)тиофен-2-карбоксилата (Промежут. соед. 56) (773 мг, 2,79 ммоль), гидрохлорида 4-(2-хлорэтил)-морфолина (778 мг, 4,18 ммоль) и К2СО3 (963 мг, 6,97 ммоль) в DMF (30 мл) нагревали до 80°C в течение 6 ч. Параллельно смесь трет-бутил-5-(N-(метилсульфонил)метилсульфонамидо)тиофен-2-карбоксилата (Промежут. соед. 57) (420 мг, 1,182 ммоль), гидрохлорида 4-(2-хлорэтил)-морфолина (330 мг, 1,772 ммоль) и К2СО3 (408 мг, 2,95 ммоль) в DMF (15 мл) нагревали до 80°C в течение 4 ч, перемешивали при КТ в течение ночи и нагревали до 80°C в течение дополнительных 2 ч. Две реакционные смеси объединяли и распределяли между EtOAc и водой. Водную фазу экстрагировали EtOAc и объединенные органические слои промывали несколько раз рассолом, сушили над сульфатом натрия, фильтровали и концентрировали досуха. Остаток очищали флэш-хроматографией на силикагеле (от смеси петролейный эфир/EtOAc в соотношении 6/4 до EtOAc 100%) с получением трет-бутил-5-(N-(2-морфолиноэтил)- метилсульфонамидо)тиофен-2-карбоксилата (Промежут. соед. 58) (1,3 г, 3,33 ммоль, МС/ИЭР+ 391,0 [МН]+).
Стадия 6: Получение соли 5-(N-(2-морфолиноэтил)-метилсульфонамидо)-тиофен-2-карбоновой кислоты с 2,2,2-трифторуксусной кислотой (Промежуточное соединение 59)
К раствору трет-бутил-5-(N-(2-морфолиноэтил)-метилсульфонамидо)-тиофен-2-карбоксилата (Промежут. соед. 58) (1,3 г, 3,33 ммоль) в DCM (50 мл) добавляли по каплям при 0°C TFA (2,56 мл, 33,3 ммоль) и затем коричневый раствор перемешивали при КТ в течение ночи. Добавляли дополнительное количество TFA (7,69 мл, 99,9 ммоль) в двух порциях в течение 4 часов, охлаждали до 0°C и перемешивали при КТ. Летучие вещества удаляли под вакуумом с получением соли 5-(N-(2-морфолиноэтил)метилсульфонамидо)-тиофен-2-карбоновой кислоты с 2,2,2-трифторуксусной кислотой (Промежут. соед. 59) (1,436 г, 3,21 ммоль, МС/ИЭР+ 335,0 [МН]+).
Стадия 7: Получение (S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(5-(N-(2-морфолиноэтил)-метилсульфонамидо)-тиофен-2-карбонилокси)этил)-пиридин-1-оксида (Соединение 60)
(S)-3,5-Дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)-фенил)-2-гидроксиэтил)пиридин-1-оксид (1,387 г, 3,3 ммоль), соль 5-(N-(2-морфолиноэтил)метилсульфонамидо)тиофен-2-карбоновой кислоты с 2,2,2-трифторуксусной кислотой (Промежут. соед. 59) (1,436 г, 3,21 ммоль), EDC (1,914 г, 9,99 ммоль) и DMAP (0,813 г, 6,66 ммоль) растворяли в DCM (100 мл) и полученную смесь перемешивали при КТ в течение 7 часов. Добавляли вторую порцию EDC (1,914 г, 9,99 ммоль) и DMAP (0,813 г, 6,66 ммоль) и смесь перемешивали при КТ в течение 3 суток. Осуществляли добавление дополнительного количества EDC (0,447 г, 2,330 ммоль) и DMAP (0,285 г, 2,330 ммоль) и смесь нагревали при 40°C в течение 5 часов; после нового добавления EDC (0,319 г, 1,664 ммоль) и DMAP (0,203 г, 1,664 ммоль) смесь перемешивали при 40°C в течение ночи. Смесь разбавляли DCM (100 мл) и промывали 0,5 н. HCl (3x80 мл), 5%-ным МаНСО3 (80 мл) и наконец рассолом (80 мл); органическую фазу сушили над сульфатом натрия, фильтровали и концентрировали под вакуумом. Неочищенный материал очищали хроматографией на картридже с силикагелем (DCM/EtOAc 1/1, затем промывка колонки DCM, затем смесью DCM/MeOH в соотношении 99/2) с получением не совсем белого твердого вещества (1,6 г), которое растирали с МеОН. Полученное твердое вещество растворяли в смеси 1/1 DCM/MeOH и растворители удаляли под вакуумом; эту операцию повторяли дважды. Остаток сушили с получением (S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(5-(N-(2-морфолиноэтил)метилсульфонамидо)-тиофен-2-карбонилокси)этил)пиридин-1-оксида (Соединение 60) (1,230 г, 1,670 ммоль, МС/ИЭР+ 736,05 [МН]+, [αD] равно -38,54, с равно 1, МеОН).
1Н ЯМР (300 МГц, DMSO-d6) 5 м.д. 8.55 (s, 2Н), 7.71 (d, 1Н), 7.21 (d, 1Н), 7.19 (d, 1Н), 7.11 (d, 1Н), 7.05 (dd, 1Н), 7.07 (t, 1Н), 6.13 (dd, 1Н), 3.93 (d, 2Н), 3.81 (t, 2Н), 3.58 (dd, 1Н), 3.48-3.54 (m, 4Н), 3.32 (dd, 1Н), 3.19 (s, 3Н), 2.47 (t, 2Н), 2.34-2.41 (m, 4Н), 1.04-1.34 (m, 1Н), 0.46-0.64 (m, 2Н), 0.25-0.45 (m, 2Н).
Соединения, перечисленные в Таблице 5, получали согласно методикам синтеза, аналогичным описанным выше и проиллюстрированным на Схеме 12, Стадия 1-7 (Стадия 5а), путем взаимодействия соответствующих предшественников (имеющихся в продаже или синтезированных специалистом в данной области техники) с подходящими реагентами, и где стадию очистки осуществляли как указано в Таблице 5.


Пример 13
Синтез 2,2,2-трифторацетата (S)-3,5-дихлор-4-(2-(3-
(циклопропилметокси)-4-(дифторметокси)фенил)-2-(4-метокси-5-(N-(2-морфолиноэтил)-метилсульфонамидо)тиофен-2-карбонилокси)этил)-пиридин-1-оксида (Соединение 72)
Схема 13
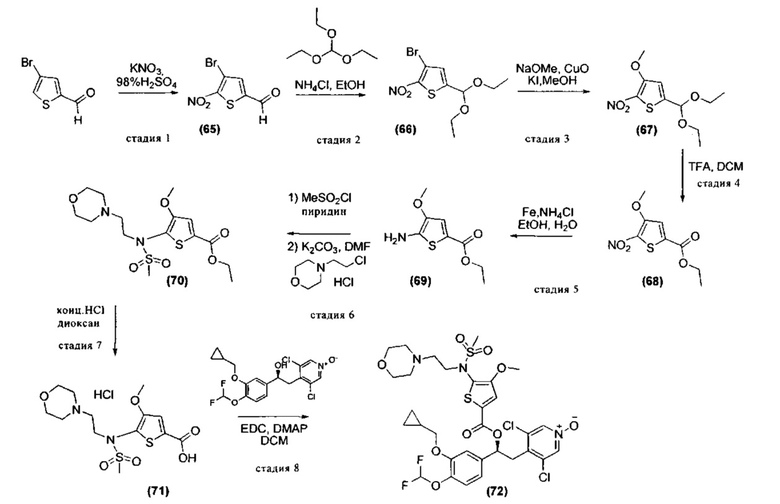
Стадия 1: Получение 4-бром-5-нитротиофен-2-карбальдегида (Промежуточное соединение 65)
Раствор KNO3 (3,38 г, 33,4 ммоль) в 96%-ной водной H2SO4 (30 мл) добавляли к раствору 4-бромтиофен-2-карбальдегида (5,8 г, 30,4 ммоль) в 96%-ной водной H2SO4 (60 мл) в течение 45 минут, поддерживая температуру ниже 4°C. Полученную смесь перемешивали при 0°C в течение 2 часов и затем при КТ в течение ночи. Реакционную смесь вливали в лед (300 мл) и полученный осадок отфильтровывали и промывали водой и гексаном с получением 4-бром-5-нитротиофен-2-карбальдегида (Промежут. соед. 65) (6,987 г, 29,6 ммоль).
Стадия 2: Получение 3-бром-5-(диэтоксиметил)-2-нитротиофена (Промежуточное соединение 66)
К раствору 4-бром-5-нитротиофен-2-карбальдегида (Промежут.соед. 65) (6,981 г, 29,6 ммоль) в EtOH (50 мл) добавляли хлорид аммония (0,475 г, 8,87 ммоль) и триэтилортоформиат (7,39 мл, 44,4 ммоль) и полученный раствор перемешивали при 60°C в течение 14 часов. Добавляли дополнительное количество триэтилортоформиата (7,39 мл, 44,4 ммоль) и смесь подвергали интенсивному кипячению с обратным холодильником в течение дополнительных 2 суток. Растворитель выпаривали под вакуумом и неочищенный материал распределяли между водой и EtOAc. Органическую фазу промывали рассолом,, сушили над сульфатом натрия, фильтровали и упаривали. Неочищенный материал очищали хроматографией на картридже с силикагелем (петролейный эфир/EtOAc в соотношении 9/1) с получением 3-бром-5-(диэтоксиметил)-2-нитротиофена (Промежут. соед. 66) (7,456 г, 24,04 ммоль).
Стадия 3: Получение 5-(диэтоксиметил)-3-метокси-2-нитротиофена (Промежуточное соединение 67)
К раствору 3-бром-5-(диэтоксиметил)-2-нитротиофена (Промежут. соед. 66) (1 г, 3,22 ммоль) в МеОН (10 мл) добавляли оксид меди(М) (0,051 г, 0,645 ммоль), KI (10,7 мг, 0,064 ммоль) и метанолат натрия (0,662 г, 12,25 ммоль) и смесь нагревали в условиях микроволнового (MW) излучения при 120°C в течение 2 ч. Реакционную смесь фильтровали через набивку целита, фильтрат концентрировали под вакуумом и затем распределяли между 0,5 н. HCl и EtOAc. Органическую фазу сушили над сульфатом натрия, фильтровали и упаривали с получением 5-(диэтоксиметил)-3-метокси-2-нитротиофена (Промежут. соед. 67) (680 мг, 2,60 ммоль, МС/ИЭР+ 261,0 [МН]+). Этот неочищенный материал использовали без какой-либо дополнительной очистки.
Стадия 4: Получение этил-4-метокси-5-нитротиофен-2-карбоксилата (Промежуточное соединение 68)
К раствору 5-(диэтоксиметил)-3-метокси-2-нитротиофена (Промежут. соед. 67) (680 мг, 2,60 ммоль) в DCM (15 мл), охлажденному до 0°C, добавляли TFA (5 мл, 64,9 ммоль) и полученную смесь перемешивали при КТ в течение 2 часов и затем концентрировали досуха. Неочищенный материал очищали флэш-хроматографией на силикагеле (от смеси петролейный эфир/EtOAc в соотношении 99/1 до смеси петролейный эфир/EtOAc в соотношении 9/1) с получением этил-4-метокси-5-нитротиофен-2-карбоксилата (Промежут. соед. 68) (300 мг, 1,297 ммоль, содержащий примерно 30% соответствующего метилового сложного эфира, МС/ИЭР+ 231,9 [МН]+). Это промежуточное соединение использовали в таком виде на следующих стадиях, считая что это был единственный продукт.
Стадия 5: Получение этил-5-амино-4-метокситиофен-2-карбоксилата (Промежуточное соединение 69)
Смесь этил-4-метокси-5-нитротиофен-2-карбоксилата (Промежут. соед. 68) (300 мг, 1,97 ммоль) (полученного как описано на Стадии 4, описанной выше) и хлорида аммония (139 мг, 2,59 ммоль) в смеси EtOH/вода в соотношении 2/1 (18 мл) нагревали при 50°C до полного растворения. Добавляли порошок железа (435 мг, 7,78 ммоль) и реакционную смесь нагревали при 80°C в течение 2 часов. Смесь фильтровали и фильтрат концентрировали под вакуумом; остаток распределяли между рассолом и EtOAc и органическую фазу сушили над сульфатом натрия. Растворитель удаляли с получением этил-5-амино-4-метокситиофен-2-карбоксилата (Промежут. соед. 69) в смеси с приблизительно 30% соответствующего метилового сложного эфира (261 мг, 1,297 ммоль, МС/ИЭР+ 201,9 [МН]+). Это промежуточное соединение использовали в таком виде на следующих стадиях.
Стадия 6: Получение этил-4-метокси-5-(N-(2-морфолиноэтил)-метилсульфонамидо)тиофен-2-карбоксилата (Промежуточное соединение 70)
К раствору этил-5-амино-4-метокситиофен-2-карбоксилата (Промежут. соед. 69) (261 мг, 1,297 ммоль) (полученного как описано на Стадии 5, описанной выше) в пиридине (10 мл), охлажденному до 0°C, добавляли метансульфонилхлорид (0,131 мл, 1,686 ммоль) и реакционную смесь оставляли нагреваться до КТ и перемешивали в течение ночи. Растворитель удаляли под вакуумом и остаток распределяли между EtOAc и 10%-ной HCl; органическую фазу промывали рассолом и сушили над сульфатом натрия. Растворитель удаляли под вакуумом и остаток растворяли в безводном DMF (10 мл); добавляли одной порцией К2СО3 (448 мг, 3,24 ммоль) и гидрохлорид 4-(2-хлорэтил)морфолина (362 мг, 1,946 ммоль) и полученную смесь нагревали до 80°C и перемешивали при этой температуре в течение 3 часов. Растворитель удаляли под вакуумом и остаток растворяли в EtOAc и промывали рассолом. Органическую фазу сушили над сульфатом натрия, фильтровали и упаривали. Остаток очищали фильтрованием на картридже с силикагелем (от смеси DCM/EtOAc в соотношении 8/2 до EtOAc) с получением этил-4-метокси-5-(N-(2-морфолиноэтил)метилсульфонамидо)тиофен-2-карбоксилата (Промежут. соед. 70) в смеси с приблизительно 30% соответствующего метилового сложного эфира (375 мг, 0,955 ммоль, МС/ИЭР+ 392,8 [МН]+). Это промежуточное соединение использовали в таком виде на следующих стадиях, считая его единственным продуктом.
Стадия 7: Получение гидрохлорида 4-метокси-5-(N-(2-морфолиноэтил)-метилсульфонамидо)тиофен-2-карбоновой кислоты (Промежуточное соединение 71)
К раствору этил-4-метокси-5-(N-(2-морфолиноэтил)-метилсульфонамидо)тиофен-2-карбоксилата (Промежут. соед. 70, 375 мг, 0,955 ммоль) (полученного как описано на Стадии 6, описанной выше) в диоксане (8 мл) добавляли по каплям водную 37%-ную HCl (3 мл) и полученную смесь нагревали в условиях MW излучения при 100°C в течение 3,5 часов. Летучие вещества удаляли под вакуумом с получением гидрохлорида 4-метокси-5-(N-(2-морфолиноэтил)метилсульфонамидо)-тиофен-2-карбоновой кислоты (Промежут. соед. 71) (383 мг, 0,955 ммоль, МС/ИЭР+ 364,9 [МН]+).
Стадия 8: Получение 2,2,2-трифторацетата (S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(4-метокси-5-(N-(2-морфолиноэтил)-метилсульфонамидо)тиофен-2-карбонилокси)этил)-пиридин-1-оксида (Соединение 72)
(S)-3,5-Дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)-фенил)-2-гидроксиэтил)пиридин-1-оксид (401 мг, 0,955 ммоль), гидрохлорид 4-метокси-5-(N-(2-морфолиноэтил)метилсульфонамидо)тиофен-2-карбоновой кислоты (Промежут. соед. 71) (383 мг, 0,955 ммоль), EDC (549 мг, 2,87 ммоль) и DMAP (233 мг, 1,911 ммоль) растворяли в DCM (30 мл) и раствор перемешивали при КТ в течение 4 суток. Смесь разбавляли DCM и промывали 0,5 н. HCl, насыщ. Na2CO3 и наконец рассолом. Органическую фазу сушили над сульфатом натрия и растворитель удаляли под вакуумом. Неочищенный материал очищали флэш-хроматографией на силикагеле (DCM/MeOH/конц.NH4OH в соотношении 98/2/0,2) и затем дополнительной флэш-хроматографией на силикагеле (DCM/MeOH/конц.NH4OH в соотношении 99/1/0,1). Дополнительная очистка препаративной ВЭЖХ (Метод 1) потребовалась для получения 2,2,2- трифторацетата (S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(4-метокси-5-(N-(2-морфолиноэтил)-метилсульфонамидо)тиофен-2-карбонилокси)этил)пиридин-1-оксида (Соединение 72) (150 мг, 0,170 ммоль, МС/ИЭР+ 766,05 [МН]+, [αD] равно -27,72, с равно 0,5, DCM).
1Н ЯМР (300 МГц, DMSO-d6) δ м.д. 9.65 (s, 1Н), 8.58 (s, 2Н), 7.76 (s, 1Н), 7.15-7.28 (m, 2Н), 6.94-7.15 (m, 1Н), 7.07 (t, 1Н), 6.14 (dd, 1Н), 3.92 (s, 3Н), 3.74-4.06 (m, 10Н), 3.60 (dd, 1Н), 3.35 (dd, 1Н), 3.14 (s, 3Н), 2.99-3.28 (m, 4Н), 1.00-1.33 (m, 1Н), 0.46-0.73 (m, 2Н), 0.32-0.39 (m, 2Н).
Пример 14
Синтез (S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(5-(N-(4-(диметилкарбамоил)фенил)-сульфамоил)тиофен-2-карбонилокси)этил)пиридин-1-оксида (Соединение 77)
Схема 14

Стадия 1: Синтез трет-бутил-4-(диметилкарбамоил)-фенилкарбамата (Промежуточное соединение 73)
К раствору 4-(трет-бутоксикарбониламино)бензойной кислоты (1 г, 4,21 ммоль) в DMF (10 мл) добавляли CDI (1,025 г, 6,32 ммоль). Смесь перемешивали при КТ в течение 2 часов. Добавляли диметиламин 2М в THF (3,16 мл, 6,32 ммоль) и смесь перемешивали при КТ в течение 3 часов. Смесь вливали в воду и экстрагировали AcOEt (2×). Органическую фазу сушили над Na2SO4 и упаривали под вакуумом с получением 1,25 г Промежут. соед. 73, которое кристаллизуется при стоянии.
Стадия 2: Синтез 4-амино-N,N-диметилбензамида (Промежуточное соединение 74)
К раствору трет-бутил-4-(диметилкарбамоил)фенилкарбамата (Промежут. соед. 73) (1,11 г, 4,21 ммоль) в AcOEt (20 мл) добавляли 4М HCl в AcOEt (18 мл, 76 ммоль) и смесь перемешивали в течение 5 часов при КТ. Значение рН доводили до 6/7 путем добавления насыщенного раствора МаНСО3 и водную фазу экстрагировали AcOEt (×3). Органическую фазу сушили над Na2SO4 и упаривали при пониженном давлении с получением 615 мг указанного в заголовке продукта (Промежут. соед. 74).
Стадия 3: Синтез метил-5-(N-(4-(диметилкарбамоил)фенил)-сульфамоил)тиофен-2-карбоксилата (Промежуточное соединение 75)
К раствору 4-амино-N,N-диметилбензамида (Промежут. соед. 74) (57 г, 347 ммоль) в пиридине (570 мкл, 7,06 ммоль), поддерживаемому при 0 градусов в бане лед/вода, добавляли метил-5-(хлорсульфонил)тиофен-2-карбоксилат (100 г, 417 ммоль) и смесь перемешивали при 0 градусов в течение 3 часов. Реакционную смесь разбавляли 1 н. HCl и экстрагировали AcOEt (×2). Органическую фазу сушили над Na2SO4 и концентрировали под вакуумом с получением 100 мг 5-(N-(4-(диметилкарбамоил)фенил)сульфамоил)тиофен-2-карбоксилата (Промежут. соед. 75).
Стадия 4: Синтез 5-(N-(4-(диметилкарбамоил)фенил)-сульфамоил)-тиофен-2-карбоновой кислоты (Промежуточное соединение 76)
К раствору метил-5-(N-(4-(диметилкарбамоил)фенил)-сульфамоил)-тиофен-2-карбоксилата (Промежут. соед. 75) (100 мг, 0,271 ммоль) в метаноле (10 мл) добавляли 1М гидроксид натрия (1 мл) и смесь перемешивали при КТ в течение 4 суток, контролируя развитие реакции и добавляя дополнительные количества 1М NaOH до завершения. МеОН выпаривали при пониженном давлении и неочищенный материал переносили в 1 н. HCl и AcOEt. Органическую фазу сушили над Na2SO4 и концентрировали под вакуумом с получением 54 мг 5-(N-(4-(диметилкарбамоил)фенил)сульфамоил)тиофен-2-карбоновой кислоты (Промежут. соед. 76).
Стадия 5: Синтез (S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(5-(N-(4-(диметилкарбамоил)фенил)- сульфамоил)тиофен-2-карбонилокси)этил)пиридин-1-оксида (Соединение 77)
5-(N-(4-(Диметилкарбамоил)фенил)сульфамоил)тиофен-2-карбоновую кислоту (Промежут. соед. 76) (54 мг, 0,152 ммоль), DMAP (6,20 мг, 0,051 ммоль) и EDC (38,9 мг, 0,203 ммоль) растворяли в DMF. Добавляли (S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)-фенил)-2-гидроксиэтил)пиридин-1-оксид (42,7 мг, 0,102 ммоль). Реакционную смесь перемешивали при КТ в течение 4 часов. Реакционную смесь разбавляли водой и экстрагировали AcOEt. Органическую фазу сушили над Na2SO4 и концентрировали под вакуумом с получением неочищенного материала, который очищали препаративной ВЭЖХ (Метод 2) с получением 10 мг целевого продукта (Соединение 77).
Соединение, описанное в Таблице 6, получали согласно методике, аналогичной описанной на Схеме 14, начиная с имеющихся в продаже подходящих реагентов.
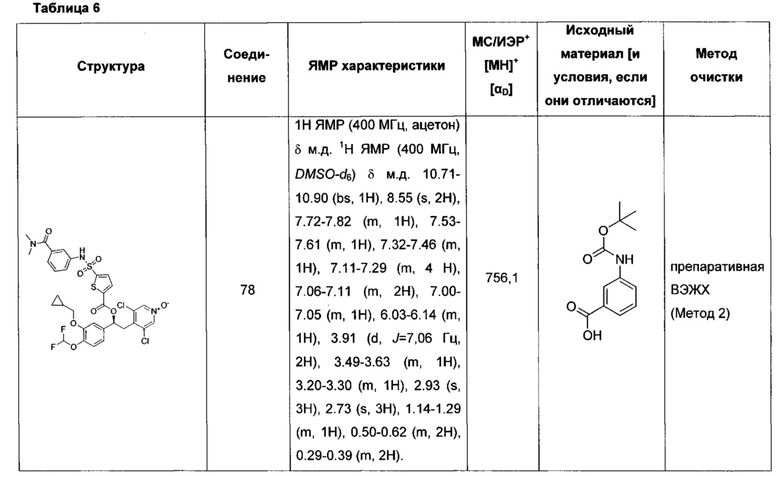
Пример 15
Синтез (S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(3-(3-(диметилкарбамоил)-фенилсульфонамидо)тиофен-2-карбонилокси)этил)пиридин-1-оксида (Соединение 81)
Схема 15

Стадия 1: Синтез метил-3-(3-(диметилкарбамоил)фенил-сульфонамидо)тиофен-2-карбоксилата (Промежуточное соединение 79)
Метил-3-аминотиофен-2-карбоксилат (93 мг, 0,592 ммоль) растворяли в пиридине (Py) (1,5 мл). Добавляли 3-(диметилкарбамоил)бензол-1-сульфонилхлорид (220 мг, 0,887 ммоль) и реакционную смесь перемешивали при КТ в течение 3 часов до завершения реакции. Реакционную смесь разбавляли 1 н. HCl и экстрагировали AcOEt. Органическую фазу промывали 1 н. HCl и рассолом, сушили над Na2SO4 и концентрировали под вакуумом с получением метил-3-(3-(диметилкарбамоил)фенилсульфонамидо)тиофен-2-карбоксилата (Промежут. соед. 79, 200 мг, 0,543 ммоль).
Стадия 2: Синтез 3-(3-(диметилкарбамоил)-фенилсульфонамидо)-тиофен-2-карбоновой кислоты (Промежуточное соединение 80)
Метил-3-(3-(диметилкарбамоил)фенилсульфонамидо)тиофен-2-карбоксилат (Промежут. соед. 79, 200 мг, 0,543 ммоль) растворяли в МеОН (4 мл, 99 ммоль). Добавляли 1 М NaOH (2 мл) и реакционную смесь перемешивали при КТ в течение 8 часов до завершения реакции.
Реакционную смесь разбавляли 1 н. HCl и экстрагировали AcOEt. Органическую фазу промывали 1 н. HCl и рассолом, сушили над Na2SO4 и концентрировали под вакуумом с получением 3-(3-(диметилкарбамоил)-фенилсульфонамидо)тиофен-2-карбоновой кислоты (Промежут. соед. 80, 150 мг, 0,423 ммоль).
Стадия 3: Синтез (S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(3-(3-(диметилкарбамоил)-фенилсульфонамидо)тиофен-2-карбонилокси)этил)пиридин-1-оксида (Соединение 81)
(S)-3,5-Дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)-фенил)-2-гидроксиэтил)пиридин-1-оксид (50 мг, 0,119 ммоль), 3-(3-(диметилкарбамоил)-фенилсульфонамидо)тиофен-2-карбоновую кислоту (Промежут. соед. 80, 127 мг, 0,357 ммоль), DMAP (17,44 мг, 0,143 ммоль) и EDC (228 мг, 1,190 ммоль) растворяли в DMF. Реакционную смесь перемешивали при КТ в течение 8 часов до завершения реакции.
Реакционную смесь разбавляли водой и осадок промывали водой, растворяли в AcOEt и экстрагировали 1 н. HCl, насыщенным раствором Na2CO3 и рассолом. Органическую фазу сушили над Na2SO4 и концентрировали под вакуумом. Неочищенный продукт очищали препаративной ВЭЖХ (Метод 1) с получением (S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(3-(3-(диметилкарбамоил)фенилсульфонамидо)-тиофен-2-карбонилокси)этил)пиридин-1-оксида (Соединение 81, 40 мг, 0,053 ммоль).
Пример 16
Синтез (S)-3,5-дихлор-4-(2-(4-(дифторметокси)-3-метоксифенил)-2-(тиофен-2-карбонилокси)этил)пиридин-1-оксида (Соединение 84)
Схема 16
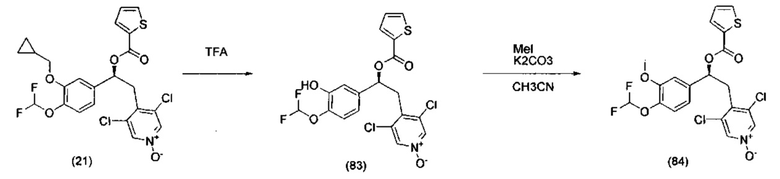
Стадия 1: Синтез (S)-3,5-дихлор-4-(2-(4-(дифторметокси)-3-гидроксифенил)-2-(тиофен-2-карбонилокси)этил)пиридин-1-оксида (Промежуточное соединение 83)
(S)-3,5-Дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-2-(тиофен-2-карбонилокси)этил)пиридин-1-оксид (180 мг, 0,339 ммоль) (Соединение 21), полученный как описано ранее в Примере 5, Схема 5, Таблица 3, обрабатывали трифторуксусной кислотой (TFA) (4 мл, 51,9 ммоль) и полученный раствор перемешивали при комнатной температуре в течение 2,5 часов. Реакционную смесь разбавляли CH2Cl2 (40 мл) и промывали водой (3×40 мл). Органический слой сушили над Na2SO4 и упаривали досуха. Остаток очищали хроматографией на SiO2 (CH2Cl2:AcOEt в соотношении 3:2) с получением (S)-3,5-дихлор-4-(2-(4-(дифторметокси)-3-гидроксифенил)-2-(тиофен-2-карбонилокси)этил)-пиридин-1-оксида (83) (67 мг, 0,141 ммоль, выход 41,4%, МС/ИЭР+ 474,99 [МН]+).
Стадия 2: Синтез (S)-3,5-дихлор-4-(2-(4-(дифторметокси)-3-метоксифенил)-2-(тиофен-2-карбонилокси)этил)пиридин-1-оксида (Соединение 84)
Суспензию (S)-3,5-дихлор-4-(2-(4-(дифторметокси)-3-гидроксифенил)-2-(тиофен-2-карбонилокси)этил)пиридин-1-оксида (67 мг, 0,141 ммоль) (83), карбоната калия (38,9 мг, 0,281 ммоль) и метилйодида (0,018 мл, 0,281 ммоль) в ацетонитриле (10 мл) интенсивно перемешивали при комнатной температуре в течение 24 часов. Добавляли дополнительное количество карбоната калия (38,9 мг, 0,281 ммоль) и метилйодида (0,018 мл, 0,281 ммоль) и смесь перемешивали при комнатной температуре в течение дополнительных 24 часов. Твердый К2СО3 отфильтровывали из реакционной смеси, остаток концентрировали в вакууме и очищали препаративной ВЭЖХ (Метод 3). Собранную фракцию упаривали досуха с получением (S)-3,5-дихлор-4-(2-(4-(дифторметокси)-3-метоксифенил)-2-(тиофен-2-карбонилокси)этил)пиридин-1-оксида (соединение 84) (37 мг, 0,075 ммоль, выход 54,1%, МС/ИЭР+ 489,97 [МН]+, [αD] равно -23,00, с равно 0,49, DCM). 1Н ЯМР (300 МГц, DMSO-d6) δ м.д. 8.56 (s, 2Н), 7.99 (dd, 1Н), 7.86 (dd, 1Н), 7.19-7.26 (m, 3Н), 7.07 (dd, 1Н), 7.06 (t, 1Н), 6.19 (dd, 1Н), 3.86 (s, 3Н), 3,60 (dd, 1Н), 3.35 (dd, 1Н).
ФАРМАКОЛОГИЧЕСКАЯ АКТИВНОСТЬ СОЕДИНЕНИЙ ПО ИЗОБРЕТЕНИЮ
Пример 17
Определение активности ингибирования фосфодиэстеразы 4 (PDE4) в бесклеточном in vitro анализе
Активность PDE4 определяли на супернатантах лизата клеток моноцитов U937 человека. Клетки культивировали, собирали и фракцию супернатантов получали по существу как описано в Torphy TJ et al. J. Pharmacol. Exp. Ther. 1992; 263:1195-1205.
Клетки U937 (банк клеток, коллекция клеточных линий Interlab, ICLC HTL94002) выращивали при 37° и 5% CO2 в среде RPMI 1640 (Roswell Park Memorial Institute) с GlutaMAX™-I, дополненной 10%-ной фетальной бычьей сывороткой и 100 мкг/мл Pen-strep (Gibco).
Клетки собирали и промывали дважды центрифугированием (150×г, 8 мин) в охлажденном, забуференном фосфатом физиологическом растворе (PBS). Промытые клетки ресуспендировали в охлажденном буфере Кребса-Рингера-Хенселайта при конечной концентрации 20×106 клеток/мл и обрабатывали ультразвуком. После центрифугирования при 1500×g в течение 20 мин супернатанты объединяли, разделяли на аликвоты и хранили при -80°C.
Активность PDE4 определяли в клеточных супернатантах посредством анализа исчезновения сАМР из смесей инкубирования.
Концентрация тестируемых соединений находилась в интервале от 10-12 М до 10-6 М. Реакции останавливали тепловой инактивацией фермента (2,5 минуты при 100°C) и содержание остаточного сАМР определяли с использованием ‛LANCE сАМР Assay’ от PerkinElmer, следуя инструкциям производителя.
Процент ингибирования активности PDE4 рассчитывали, принимая исчезновение сАМР в отсутствие ингибиторов за 100% и исчезновение сАМР в образцах с тепловой инактивацией за 0%.
Результаты для тестированных иллюстративных соединений по изобретению, выраженные в виде среднего значения ± стандартное отклонение нМ концентрации тестируемого соединения, которая вызывает 50%-ное ингибирование исчезновения сАМР (IC50), составили менее 2,5 нМ. Для группы предпочтительных соединений (27 из 30 тестированных) результаты составили менее 0,5 нМ. Для группы более предпочтительных соединений (6 из 30 тестированных) результаты составили менее 0,05 нМ.
Пример 18
Определение активности ингибирования PDE4 в in vitro анализе мононуклеарных клеток периферической крови (РВМС)
Анализ, основанный на известной ингибирующей активности под действием ингибиторов PDE4 в отношении индуцируемого липополисахаридом (LPS) фактора некроза опухоли альфа (высвобождение TNF-α в мононуклеарных клетках периферической крови (РВМС)), осуществляли в соответствии с описанной ранее методикой (Hatzelmann A et al. J. Pharmacol. Exp. Ther. 2001; 297:267-279; Draheim R et al. J. Pharmacol. Exp. Ther. 2004; 308:555-563).
Криоконсервированные человеческие РВМС (100 мкл/лунка) инкубировали в 96-луночных планшетах (105 клеток/лунка) в течение 30 мин в присутствии или в отсутствие (50 мкл) тестируемых соединений в интервале концентраций от 10-12 М до 10-6 М или от 10-13 М до 10-7 М. Затем добавляли LPS (3 нг/мл).
Через 18 ч инкубации при 37°C в увлажняемом инкубаторе в атмосфере 95% воздуха и 5% CO2 культуральные среды собирали и измеряли TNF-α посредством твердофазного иммуноферментного анализа (ELISA).
Эффекты тестированных соединений рассчитывали в виде процента ингибирования высвобождения TNF-α, принимая LPS-индуцированную продукцию TNF-α в отсутствие ингибиторного соединения за 100% и фоновую продукцию TNF-α клетками РВМС в отсутствие LPS за 0%.
Результаты для тестированных иллюстративных соединений по изобретению, выраженные в виде среднего значения ±95%-ный доверительный интервал молярной концентрации тестируемого соединения, которая вызывает 50%-ное ингибирование LPS-индуцированной продукции TNF-α (IC50), составили менее 33 нМ. Для группы предпочтительных соединений результаты составили менее 1 нМ. Для группы более предпочтительных соединений результаты составили менее 0,1 нМ.
Пример 19
Определение in vitro собственного клиренса в человеческих микросомах печени
Методика (а)
Тестируемые соединения растворяли в DMSO в двух повторностях при конечной концентрации 1 мкМ (конечная концентрация DMSO 0,5% об./об.) и предварительно инкубировали в течение 10 мин при 37°C в буфере фосфата калия с рН 7,4, 3 мМ MgCl2, с микросомами печени при конечной концентрации 0,5 мг/мл.
По окончании периода предварительной инкубации инициировали начало реакций путем добавления смеси кофакторов (NADP, Glc6P, Glc6P-DH); образцы отбирали в моменты времени 0, 5, 10, 15, 20 и 30 мин, добавляли к ацетонитрилу для остановки реакции и центрифугировали. Супернатанты анализировали и подвергали количественному анализу посредством ЖХ-МС/МС.
Контрольный образец без кофакторов всегда добавляли для проверки стабильности тестируемых соединений в матриксе.
7-Этоксикумарин добавляли в качестве эталонного стандарта.
Фиксированную концентрацию верапамила добавляли в каждый образец в качестве внутреннего стандарта для ЖХ-МС/МС.
Нулевое время инкубации использовали в качестве 100%-ного значения. Потерю субстрата в процентах при инкубации определяли посредством оценки in-vitro периода полувыведения и in-vitro собственного клиренса соединений.
Константу скорости k (мин-1), выведенную из уравнения экспоненциального затухания (площадь пика против времени), использовали для расчета скорости собственного клиренса (CLi) соединений, используя следующее уравнение:
CLi (мл/мин/г печени)=k×V×y,
где:
k рассчитывают из аппроксимации экспоненциального затухания значений площади;
V равно объему инкубации (мл)/мг белка;
y означает выход микросомального белка равный 52,5 мг/г печени.
Соединения по изобретению проявляют высокий или умеренный собственный метаболический клиренс.
Некоторые из иллюстративных соединений по изобретению при тестировании согласно этому протоколу показали собственный клиренс менее 15 мл/мин/г.
Методика (б)
Тестируемые соединения инкубировали в двух повторностях при концентрации 1 мкМ с микросомами печени (0,8 мг белка/мл) в буфере Дульбекко (рН 7,4) при 37°C в присутствии 1 мМ NADPH. В разные моменты времени (0, 5, 10, 20, 30 и 60 мин) отбирали аликвоты инкубатов по 50 мкл, добавляли 80 мкл охлажденного на льду ацетонитрила и 20 мкл 1 мкМ варфарина в ацетонитриле (инъекционная проверка) для остановки реакции и образцы центрифугировали. Супернатант анализировали посредством ЖХ-МС/МС на предмет неизмененных соединений.
Тестируемые соединения инкубировали с микросомами печени в буфере Дульбекко в отсутствие NADPH в течение 0 и 60 минут в качестве контроля. Мидазолам при концентрации 1 мкМ инкубировали с микросомами в качестве положительного контроля для фазы I активности микросом. Контрольные образцы обрабатывали так же, как и образцы тестируемых соединений.
Собственный клиренс определяли с использованием подхода полувыведения.
Период полувыведения рассчитывали из зависимости:

Угол наклона (SLOPE) относится к кривой, построенной по значениям натуральных логарифмов (LN) площади пика остающегося соединения против времени, и рассчитывается анализом линейной регрессии.
Результаты записывали в виде значений периода полувыведения в минутах и в виде значений собственного клиренса in vitro, выраженных в мкл/мин/мг белка (для инкубации с микросомами).
Соединения по изобретению проявляют высокий или умеренный собственный метаболический клиренс.
Некоторые из иллюстративных соединений по изобретению при тестировании согласно этому протоколу показали собственный клиренс менее 300 мкл/мин/мг и период полувыведения менее 3 минут.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ 1-ФЕНИЛ-2-ПИРИДИНИЛАЛКИЛЬНЫХ СПИРТОВ В КАЧЕСТВЕ ИНГИБИТОРОВ ФОСФОДИЭСТЕРАЗЫ | 2012 |
|
RU2626956C2 |
| ПРОИЗВОДНЫЕ 1-ФЕНИЛ 2-ПИРИДИНИЛАЛКИЛОВЫХ СПИРТОВ В КАЧЕСТВЕ ИНГИБИТОРОВ ФОСФОДИЭСТЕРАЗЫ | 2012 |
|
RU2617401C2 |
| Производные 1-фенил-2-пиридинилалкиловых спиртов в качестве ингибиторов фосфодиэстеразы | 2013 |
|
RU2655170C2 |
| ИНГИБИТОРЫ РЕПЛИКАЦИИ ВИРУСА ГРИППА, СПОСОБЫ ИХ ПРИМЕНЕНИЯ И ИСПОЛЬЗОВАНИЕ | 2016 |
|
RU2737190C2 |
| КОНДЕНСИРОВАННЫЕ ТРИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛОВ В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2689777C1 |
| НОВЫЕ 1,2-БИС-СУЛЬФОНАМИДНЫЕ ПРОИЗВОДНЫЕ КАК МОДУЛЯТОРЫ ХЕМОКИНОВОГО РЕЦЕПТОРА | 2011 |
|
RU2654213C9 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ | 2014 |
|
RU2681849C2 |
| КОНДЕНСИРОВАННЫЕ ТРИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ИМИДАЗОЛА В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2679914C9 |
| НОВЫЕ ГИДРОКСИСЛОЖНОЭФИРНЫЕ ПРОИЗВОДНЫЕ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2016 |
|
RU2734418C2 |
| ФЕНИКОЛОВЫЕ ПРОТИВОБАКТЕРИАЛЬНЫЕ СРЕДСТВА | 2013 |
|
RU2593204C2 |
Изобретение относится к cоединениям общей формулы (I)

где R1 и R2 являются разными или одинаковыми и независимо выбраны из группы, состоящей из: - Н; - (С1-С6)галогеналкила; - (С1-С6)алкила, возможно замещенного одним или более заместителями, выбранными из (С3-С7)циклоалкила; и - (С3-С7)циклоалкила; А представляет собой моноциклическую гетероарильную кольцевую систему, выбранную из группы, состоящей из радикалов (а), (b), (с) и (d):



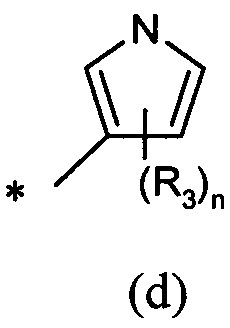 ;
;
n представляет собой 0, 1 или 2; R3 представляет собой возможный заместитель, который в каждом случае выбран из группы, состоящей из: - (С1-С6)алкила, возможно замещенного (С3-С7)циклоалкилом; - OR4, (значение остальных радикалов представлено в п.1 формулы), его соответствующему N-оксиду по пиридиновому кольцу и фармацевтически приемлемым солям или сольватам. Полученные соединения являются ингибиторами фермента фосфодиэстеразы 4 (PDE4). 5 н. и 9 з.п. ф-лы, 8 табл., 19 пр.
1. Соединение общей формулы (I)

где R1 и R2 являются разными или одинаковыми и независимо выбраны из группы, состоящей из:
- Н;
- (С1-С6)галогеналкила;
- (С1-С6)алкила, возможно замещенного одним или более заместителями, выбранными из (С3-С7)циклоалкила; и
- (С3-С7)циклоалкила; А представляет собой моноциклическую гетероарильную кольцевую систему, выбранную из группы, состоящей из радикалов (а), (b), (с) и (d):


 ;
;
n представляет собой 0, 1 или 2;
R3 представляет собой возможный заместитель, который в каждом случае выбран из группы, состоящей из:
- (С1-С6)алкила, возможно замещенного (С3-С7)циклоалкилом;
- OR4, где R4 выбран из группы, состоящей из Н, (С1-С10)алкила, возможно замещенного (С3-С7)циклоалкилом, и (С3-С7)циклоалкила;
- атомов галогена;
- группы -NR5SO2R6,
где R5 выбран из группы, состоящей из водорода; (С1-С6)алкила, который замещен группой -NR7R8, где R7 и R8 вместе с атомом азота, к которому они присоединены, образуют (С5-С7)гетероциклоалкильную группу; (С1-С6)алкила, который замещен гетероарильной группой; и
R6 представляет собой (С1-С4)алкил, возможно замещенный (С3-С7)циклоалкилом; или фенильное кольцо, возможно замещенное группой -C(O)NR9R10, где R9 и R10 независимо представляют собой (С1-С4)алкильные группы;
- группы -SO2R11,
где R11 выбран из (С1-С6)алкила; фенильного кольца, которое возможно замещено одной или двумя группами, выбранными из списка, состоящего из следующего: атом галогена, гидроксил, (С1-С4)алкокси, группа -C(O)NR12R13, где R12 и R13 независимо представляют собой (С1-С4)алкильные группы, и группа -NR14R15, где R14 и R15 независимо представляют собой водород, (C1-С4)алкильные группы, или вместе с атомом азота, к которому они присоединены, образуют (С5-С7)гетероциклоалкильную группу;
- группы -C(O)R16,
где R16 представляет собой фенильное кольцо, которое возможно замещено одной или двумя группами, выбранными из атома галогена и группы -NR25R26, где R25 и R26 независимо представляют собой водород; и
- группы -SO2NR17R18,
где R17 представляет собой Н или (С1-С6)алкил; и
R18 выбран из водорода; фенильного кольца, возможно замещенного группой -C(O)NR21R22, где R21 и R22 независимо представляют собой водород или (С1-С4)алкильные группы;
его соответствующий N-оксид по пиридиновому кольцу и фармацевтически приемлемые соль или сольват.
2. Соединение формулы (I) по п. 1, где R1 представляет собой (C1-С6)галогеналкил, и R2 представляет собой (С1-С6)алкил, который замещен (С3-С7)циклоалкилом.
3. Соединение формулы (I) по п. 1, которое представляет собой N-оксид по пиридиновому кольцу.
4. Соединение формулы (IA) по п. 1
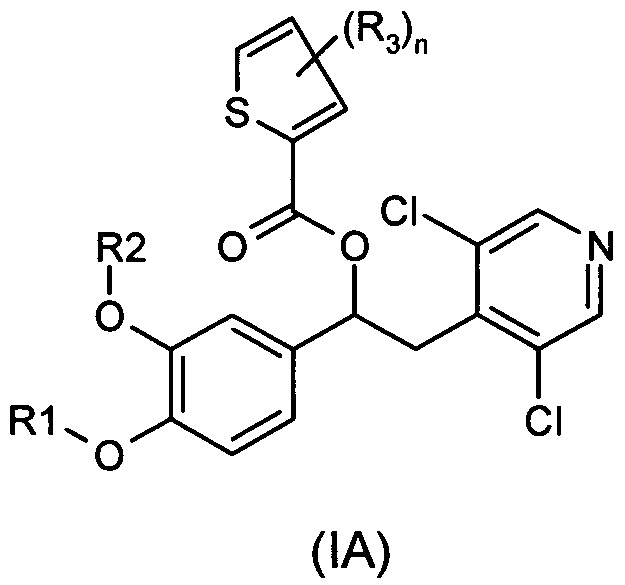 ,
,
где R1, R2, R3 и n имеют значения, как они раскрыты выше.
5. Соединение формулы (I)' по п. 1, которое представляет собой соединение формулы (I), где абсолютная конфигурация углерода (1) является такой, как показано ниже:

6. Соединение формулы (I) по п. 1, которое выбрано из группы, состоящей из:
(S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)-фенил)-2-(тиофен-3-карбонилокси)этил)пиридин-1-оксида;
(S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)-фенил)-2-(тиофен-2-карбонилокси)этил)пиридин-1-оксида;
(S)-4-(2-(1Н-пиррол-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)-3,5-дихлорпиридин-1-оксида;
(S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)-фенил)-2-(5-метилтиофен-2-карбонилокси)этил)пиридин-1-оксида;
(S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)-фенил)-2-(3-метокситиофен-2-карбонилокси)этил)пиридин-1-оксида;
(S)-3,5-дихлор-4-(2-(5-хлортиофен-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)пиридин-1-оксида;
(S)-3,5-дихлор-4-(2-(3-хлор-4-(метилсульфонил)тиофен-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)-этил)пиридин-1-оксида;
(S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)-фенил)-2-(5-метокситиофен-2-карбонилокси)этил)пиридин-1-оксида;
(S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)-фенил)-2-(4,5-диметилтиофен-2-карбонилокси)этил)пиридин-1-оксида;
(S)-3,5-дихлор-4-(2-(3-хлортиофен-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)пиридин-1-оксида;
(S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)-фенил)-2-(3-(N-метилсульфамоил)тиофен-2-карбонилокси)этил)пиридин-1-оксида;
(S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)-фенил)-2-(3-(N-метил-N-(2-морфолиноэтил)сульфамоил)тиофен-2-карбонилокси)-этил)пиридин-1-оксида;
(S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)-фенил)-2-(3-(N-(2-морфолиноэтил)метилсульфонамидо)тиофен-2-карбонилокси)-этил)пиридин-1-оксида;
(S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)-фенил)-2-(3-(метилсульфонамидо)тиофен-2-карбонилокси)этил)пиридин-1-оксида;
(S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)-фенил)-2-(5-(N-(2-морфолиноэтил)метилсульфонамидо)тиофен-2-карбонилокси)-этил)пиридин-1-оксида;
(S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)-фенил)-2-(4-(метилсульфонамидо)тиофен-2-карбонилокси)этил)пиридин-1-оксида;
(S)-3,5-дихлор-4-(2-(4-(4-хлорфенилсульфонил)тиофен-3-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)-пиридин-1-оксида;
(S)-4-(2-(3-бензоилтиофен-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)-3,5-дихлорпиридин-1-оксида;
(S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)-фенил)-2-(3-(3-(диметилкарбамоил)фенилсульфонамидо)тиофен-2-карбонилокси)-этил)пиридин-1-оксида;
(S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)-фенил)-2-(5-(N-(3-(диметилкарбамоил)фенил)сульфамоил)тиофен-2-карбонилокси)-этил)пиридин-1-оксида;
(S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)-фенил)-2-(5-(N-(4-(диметилкарбамоил)фенил)сульфамоил)тиофен-2-карбонилокси)-этил)пиридин-1-оксида;
(S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)-фенил)-2-(5-(N-(пиридин-3-илметил)метилсульфонамидо)тиофен-2-карбонилокси)-этил)пиридин-1-оксида;
(S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)-фенил)-2-(5-(N-(2-(пирролидин-1-ил)этил)метилсульфонамидо)тиофен-2-карбонилокси)-этил)пиридин-1-оксида;
(S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)-фенил)-2-(4-метокси-5-(N-(2-морфолиноэтил)метилсульфонамидо)-тиофен-2-карбонилокси)этил)пиридин-1-оксида;
(S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)-фенил)-2-(1-(фенилсульфонил)-1Н-пиррол-2-карбонилокси)этил)пиридин-1-оксида;
(S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)-фенил)-2-(1-(3-(диметилкарбамоил)фенилсульфонил)-1Н-пиррол-2-карбонилокси)-этил)пиридин-1-оксида;
(S)-4-(2-(1-(4-аминофенилсульфонил)-1Н-пиррол-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)-3,5-дихлорпиридин-1-оксида;
(S)-3,5-дихлор-4-(2-(5-(1-циклопропил-N-(2-морфолиноэтил)-метилсульфонамидо)тиофен-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)пиридин-1-оксида;
(S)-3,5-дихлор-4-(2-(5-(1-циклопропил-N-(2-(пирролидин-1-ил)этил)-метилсульфонамидо)тиофен-2-карбонилокси)-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)этил)пиридин-1-оксида;
(S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)-фенил)-2-(1-(3-(диметилкарбамоил)-4-гидроксифенилсульфонил)-1Н-пиррол-2-карбонилокси)этил)пиридин-1-оксида;
(S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)-фенил)-2-(1-(3-(диметилкарбамоил)-4-метоксифенилсульфонил)-1Н-пиррол-2-карбонилокси)этил)пиридин-1-оксида;
(S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)-фенил)-2-(1-метил-4-(фенилсульфонамидо)-1Н-пиррол-2-карбонилокси)этил)пиридин-1-оксида;
(S)-3,5-дихлор-4-(2-(3-(циклопропилметокси)-4-(дифторметокси)-фенил)-2-(4-(3-(диметилкарбамоил)фенилсульфонамидо)-1-метил-1Н-пиррол-2-карбонилокси)этил)пиридин-1-оксида;
(S)-3,5-дихлор-4-(2-(3,4-диметоксифенил)-2-(тиофен-2-карбонилокси)-этил)пиридин-1-оксида;
(S)-3,5-дихлор-4-(2-(4-(дифторметокси)-3-метоксифенил)-2-(тиофен-2-карбонилокси)этил)пиридин-1-оксида;
и его фармацевтически приемлемые соли или сольваты.
7. Комбинация для ингибирования фосфодиэстеразы 4 (PDE4), содержащая соединение формулы (I) по любому из пп. 1-6 со вторым фармацевтически активным компонентом, выбранным из классов бета2-агонистов, кортикостроидов и антимускариновых агентов.
8. Фармацевтическая композиция для ингибирования фосфодиэстеразы 4 (PDE4), содержащая соединение формулы (I) по любому из пп. 1-6 или комбинацию по п. 7 и один или более фармацевтически приемлемых носителей и/или эксципиентов.
9. Соединение формулы (I) по любому из пп. 1-6 для применения в качестве лекарственного средства для ингибирования фосфодиэстеразы 4 (PDE4).
10. Соединение формулы (I) по любому из пп. 1-6 для применения в предупреждении и/или лечении заболевания респираторного тракта, характеризующегося обструкцией дыхательных путей, такого как астма или COPD (хроническая обструктивная болезнь легких).
11. Соединение формулы (I) по любому из пп. 1-6 для применения в предупреждении и/или лечении аллергического ринита.
12. Соединение формулы (I) по любому из пп. 1-6 для применения в предупреждении и/или лечении атопического дерматита.
13. Устройство для предупреждения и/или лечения заболевания респираторного тракта, характеризующегося обструкцией дыхательных путей, такого как астма или COPD, содержащее фармацевтическую композицию по п. 8.
14. Набор, содержащий фармацевтическую композицию по п. 8 и устройство, которое может представлять собой однодозовый или многодозовый ингалятор сухого порошка, дозирующий ингалятор или небулайзер.
| WO 2009018909, A3, 12.02.2009 | |||
| Видоизменение фрикционной передачи к ватерным машинам | 1929 |
|
SU19113A1 |
| US 20080015226, A1, 17.01.2008 | |||
| RU 2009135810, A, 10.04.2011. | |||

