Изобретение относится к медицине, а именно к педиатрии при диагностике наследственных нарушений обмена, и может быть использовано для ранней диагностики наследственной тирозинемии 1 типа (HT1) у детей.
Тирозинемии - нарушения обмена тирозина. Существует 3 вида гипертирозинемии: транзиторная, связанная с недостатком аскорбиновой кислоты; наследственно обусловленная, вызванная, в основном, недостаточностью параоксифенилпировиноградной кислоты (п-ОФП), и тирозинемия вследствие недостаточной способности печени синтезировать тирозинаминотрансферазу.
При генетически детерминированных заболеваниях, обусловленных мутациями в генах, кодирующих образование ферментов, участвующих в метаболизме тирозина, в зависимости от дефекта образования конкретного фермента, различают несколько типов наследственной тирозинемии (НТ): 1 тип, 2 тип и 3 тип. Так, при мутации гена FAH, локализованного на длинном плече 15-й хромосомы 15q25.1 и кодирующего образование фермента фумарилацетоацетатгидролазы, развивается тирозинемия 1 типа. Мутация гена ТАТ, локализованного на длинном плече 16-й хромосомы 16q22.1 и кодирующего образование фермента тирозинаминотрансферазы, обусловливает развитие тирозинемии 2 типа. При мутации гена HPD, локализованного на длинном плече 12-й хромосомы 12q24.31 и кодирующего образование фермента гидроксифенилпируват дегидрогеназы, развивается тирозинемия 3 типа. Недостаток этих ферментов и приводит к тому, что распад тирозина осуществляется по альтернативному патологическому пути с образованием высокотоксичных и канцерогенных продуктов сукцинилацетона, малеилацетоацетата, фумарилацетоацетата, которые приводят к поражению печени, почек, периферических нервов.
Тирозинемия 1 типа (HT1, гепаторенальная тирозинемия) наиболее патогенна в детском возрасте, относится к редким (орфанным) наследственным заболеваниям с аутосомно-рецессивным типом наследования, которое сопровождается распадом тирозина по альтернативному - патологическому - пути, с образованием сукцинилацетона и сукцинилацетоацетата [Tomoeda К., Awata Н., Matsuura Т., Matsuda I., Ploechl Е., Milovac Т., et al; Mutations in the 4-hydroxyphenylpyruvic acid dioxygenase gene are responsible for tyrosinemia type III and hawkinsinuria; Molec. Genet. Metab; 2000; 71: 506-510]. Эти продукты распада тормозят окислительное фосфорилирование в митохондриях и блокируют цикл Кребса, при этом оказывают токсическое действие на клетки печени и проксимальные почечные канальцы, чем и обусловливают клиническую картину заболевания с развитием печеночной недостаточности с тяжелой коагулопатией, цирроза печени (ЦП) и тубулопатии с формированием синдрома Фанкони (гиперфосфатурия, аминоацидурия, метаболический ацидоз, витамин Д-резистентный рахит, гипофосфатемия).
Кроме того, при HT1 происходит поражение нервной системы, проявляющееся острыми кризами перемежающейся порфирии длительностью 1-7 дней, которые характеризуются изменениями в психическом статусе, абдоминальными болями, признаками периферической нейропатии и дыхательными расстройствами.
Выделяют острую (HT1 типа A - НТ1А) и хроническую (HT1 типа Б - НТ1Б) формы наследственной тирозинемии 1 типа, различие которых генетически обусловлено степенью активности фермента фумарилацетоацетазы. НТ1А характеризуется острым течением и ранним началом заболевания, которое проявляется в первые месяцы жизни и характеризуется задержкой физического и психомоторного развития, фебрильной лихорадкой, рвотой, диареей, увеличением живота в объеме за счет как гепатомегалии или гепатоспленомегалии, так и за счет асцита, динамической кишечной непроходимости. На стадии острого гепатита может быть желтуха. В случае острого желудочно-кишечного кровотечения возможно появление мелены и рвоты кофейной гущей. При снижении белково-синтетической функции печени появляются безбелковые отеки, анасарка, кровотечения. НТ1Б отличается более благоприятным течением, хотя возможны обострения процесса. Первые клинические симптомы могут не проявляться вплоть до достижения ребенком шестимесячного возраста. Затем диагностируется прогрессирующее поражение печени с исходом в ЦП и почек в виде тяжелой тубулопатии. Поражение почек определяет различную степень выраженности рахита - от минимальной гипофосфатемии и вальгусной или варусной деформации ног до тяжелой деформации скелета [Holme Е., Lindstedt S. Diagnosis and management of tyrosinemia type I. Curr Opin Pediatr. 1995; 7: 726-321. В некоторых случаях болезнь может начаться с внепеченочных проявлений по типу полинейропатии (боли и слабость в конечностях, абдоминальные боли, параличи конечностей и диафрагмы, сухожильная гипорефлексия, мышечная гипертония), кардиомиопатии и артериальной гипертензии на фоне метаболических процессов и вторично - на фоне поражения почек. При высокой концентрации тирозина в крови (более 600 мкмоль/л) отмечается повышение фоточувствительности глаз, снижение прозрачности роговицы и других оптических сред. Таким образом, имеется полиморфизм клинических проявлений заболевания, а его прогрессирующее течение с поражением различных органов и систем требует ранней диагностики и назначения специфической терапии, направленной на все звенья патогенеза.
При отсутствии специфического лечения пациенты умирают в детском возрасте [Sniderman King L, Trahms C, Scott CR.; Tyrosinemia Type I; GeneReviews® [Internet]; Eds. Pagon R.A, Adam M.P, Ardinger H.H, et al.; 2006 Jul 24; 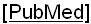 , Доказано, что при раннем начале специфической терапии снижается необходимость ранней трансплантации органов, выживаемость увеличивается более чем в 90% случаев, улучшаются функции печени, предупреждается развитие ЦП и гепатоцеллюлярной карциномы (ГЦК), происходит коррекция почечного канальцевого ацидоза. Лечение эффективно как на стадии ЦП (при НТ1Б), так и на стадии острой печеночной недостаточности (при НТ1А). Чем раньше начата терапия, тем лучше ее результат. Верификация HT1 проводится на основании повышенного уровня тирозина в сыворотке крови и повышении уровня сукцинилацетона в крови и моче. Диагноз подтверждается при проведении генетического исследования на мутации в гене FAH.
, Доказано, что при раннем начале специфической терапии снижается необходимость ранней трансплантации органов, выживаемость увеличивается более чем в 90% случаев, улучшаются функции печени, предупреждается развитие ЦП и гепатоцеллюлярной карциномы (ГЦК), происходит коррекция почечного канальцевого ацидоза. Лечение эффективно как на стадии ЦП (при НТ1Б), так и на стадии острой печеночной недостаточности (при НТ1А). Чем раньше начата терапия, тем лучше ее результат. Верификация HT1 проводится на основании повышенного уровня тирозина в сыворотке крови и повышении уровня сукцинилацетона в крови и моче. Диагноз подтверждается при проведении генетического исследования на мутации в гене FAH.
Известен способ диагностики наследственных нарушений обмена, приводящих к развитию неонатальной гипербилирубинемии. Предлагается тест-система, представленная набором олигонуклеотидов, позволяющих выявлять точковые мутации в генах FAN SERPINA1, которые вызывают соответственно тирозинемию 1 типа и недостаточность альфа-1-антитрипсина. Описан способ детекции указанных точковых мутаций, предусматривающий проведение двухраундной мультиплексной ПЦР с использованием соответствующих наборов специфических праймеров и гибридизацию полученных ПЦР-продуктов с биочипом, содержащим тест-систему (Патент РФ №2458131).
Как правило, исследования, проводимые с целью установления диагноза, проходят в несколько этапов - пошаговая диагностика.
Известен способ пошаговой диагностики и дифференциальной диагностики инфекционных заболеваний у детей [Бегайдарова Р.Х., Стариков Ю.Г., Алшынбекова Г.К., Балтынова Р.З., Дюсембаева А.Е. Диагностика и дифференциальная диагностика инфекционных заболеваний у детей. Учебное пособие. - 2014. - Гэотар-Медиа. - 140 с.]. Недостатком способа является применение его при инфекционных заболеваниях у детей, а не с целью диагностики генетически детерминированных заболеваний у детей.
Известен способ пошаговой диагностики вторичных лицевых болей (заявка №2012106346 от 22.02.2012 г.) на патент на изобретение Российской Федерации), заключающийся в детальном стоматологическом и неврологическом клиническом обследовании пациента, изучении биоэлектрической активности жевательных мышц, функционального состояния височно-нижнечелюстного сустава, окклюзионных взаимоотношений зубов, биоэлектрической активности сердца с последующим анализом вариабельности сердечного ритма, что позволяет выявить этиологический фактор возникновения вторичных лицевых болей, провести дифференциальную диагностику и выявить сочетанные патологии.
Недостатком способа является использование его при лицевых болях у взрослых пациентов, а не при генетически детерминированных заболеваниях у детей.
Известна пошаговая диагностика анемий, помогающая врачу быстро определиться с направлением обследования пациента и тактикой его ведения на основании оценки параметров, получаемых с помощью современных гематологических анализаторов. Шаг первый - взятие крови, с определением среднего объема эритроцитов в кубических микрометрах и среднего содержания гемоглобина в одном эритроците, шаг второй - оценка гематокрита, шаг третий - оценка количества гемоглобина, эритроцитов, лейкоцитов, тромбоцитов, шаг четвертый: оценка эритроцитарных индексов (Селиванов Е.В., Звягинцев Е.Н. Диагностика анемий по результатам анализов, выполненных на современных гематологических анализаторах // Вестник «Лаборатории ДНК-диагностики» - 2010 - №2 - С. 9-12).
Этот способ пошаговой диагностики анемии выбран нами в качестве прототипа.
Задачей изобретения является разработка способа пошаговой диагностики наследственной тирозинемии 1 типа у детей с помощью многофакторного статистического анализа клинико-диагностических показателей.
Техническим решением поставленной задачи является алгоритм ранней диагностики наследственной тирозинемии 1 типа у детей для своевременного назначения специфической терапии с целью предупреждения развития цирроза печени и гепатоцеллюлярной карциномы, профилактики инвалидизации и смертности вследствие наследственной тирозинемии 1 типа.
Сущность изобретения заключается в том, что у детей первых 3-х месяцев жизни, имеющих симптомокомплекс, состоящий из лихорадки неясного генеза, отеков, желтухи и диспепсического синдрома, а у детей в возрасте 4-х месяцев и старше - гепато- или гепатоспленомегалию и клинические проявления острого рахита, не купирующиеся применением стандартных противорахитических средств, которые можно считать 1-м шагом в ранней диагностике НТ1, проводят 2-й шаг в диагностике HT1: клинический анализ крови с оценкой уровня гемоглобина и эритроцитов (для выявления анемического синдрома), а также количества тромбоцитов и биохимический анализ крови (показатели, отражающие выраженность цитолиза - аланинамино- и аспарагинаминотрансферазы (АЛТ, ACT); уровни общего билирубина и его фракций; показатели, отражающие лабораторные признаки острого рахита - повышенный уровень щелочной фосфатазы (ЩФ) в сыворотке крови, гипокальциемия и гипофосфатемия, гиперфосфатурия); коагулограмма. Целесообразно определить уровень альфа-фетопротеина (АФП), который бывает повышенным. Детям с выявленной анемией, тромбоцитопений, повышенным уровнем АЛТ, ACT, билирубина, АФП и лабораторными признаками острого рахита, - проводят исследование уровня тирозина (и фенилаланина) в крови методом тандемной масс-спектрометрии и исследование на сукцинилацетон в крови и моче - 3-й шаг в диагностике HT1. При повышенном уровне тирозина (и, возможно, фенилаланина), а также повышении сукцинилацетона в крови выше 2 ммоль/л и более 2 ммоль/моль креатинина в моче проводится молекулярно-генетического исследования на мутации в гене FAH, что можно рассматривать как 4-й шаг в алгоритме ранней диагностики HT1.
В гастроэнтерологическом отделении с гепатологической группой ФГАУ «Научный центр здоровья детей» Минздрава России за период с 2006 года по 2015 гг. под наблюдением находились 17 детей (8 мальчиков и 9 девочек) с тирозинемией 1 типа. Из них 5 пациентов с тирозинемией 1A типа и 12 пациентов с тирозинемией 1Б типа. Диагноз был установлен на основании клинико-лабораторных и инструментальных данных и подтвержден результатами молекулярно-генетического исследования.
Проводилось изучение анамнеза жизни и заболевания пациентов, оценивались клинико-лабораторные данные в дебюте заболевания по месту жительства, при первичной госпитализации в ФГАУ НЦЗД МЗ и в динамике через 6 месяцев от начала проводимой терапии. Исследование свертывающей системы крови (коагулограмма) в дебюте заболевания по месту жительства проведено у 8 детей из 17 (47%).
Статистическая обработка данных была проведена в операционной среде Windows XP с использованием компьютерных программ Microsoft Excel 2010 и пакета статистического анализа данных SPSS Statistic (version 20) и StatSoftStatistica (версия 6,0). Количественные переменные описывались числом пациентов (n), средним арифметическим значением (М), m - стандартная ошибка среднего. Использовались непараметрические методы статистики. При анализе выборок, не подчиняющихся закону нормального распределения, использовали непараметрический метод - критерий Манна-Уитни. Различия между величинами считали статистически значимыми при p<0,05.
Анализ диагностической значимости клинико-лабораторных показателей в дебюте заболевания в зависимости от возраста при оптимальном сочетании чувствительности и специфичности осуществляли методом многофакторного статистического анализа и построения ROC-кривых. Количественная интерпретация ROC-анализа оценивалась по показателю AUC (area under curve) - численное значение клинической значимости диагностического теста. По экспертной шкале для значений AUC показатель в пределах 0,5-0,6 свидетельствует о неудовлетворительном качестве диагностического теста, в пределах 0,6-0,7 - о среднем качестве диагностического теста, в пределах 0,7-0,8 - о хорошем качестве диагностического теста, в пределах 0,8-0,9 - очень хорошем, 0,9-1,0 - отличном качестве диагностического теста. Информативность показателя оценивалась по величине площади под кривой и считалась достоверной при AUC>0,6.
Возраст детей при первичной госпитализации в НЦЗД колебался в диапазоне от 5 месяцев до 12 лет 3 месяцев, при этом у 10 детей, госпитализированных в возрасте до 3 лет, средний возраст был 12,3±5,8 месяцев, а у 7 детей, госпитализированных в возрасте старше 3 лет - 75,3±40,8 мес.
У 8 из 17 пациентов на момент первичной госпитализации диагноз тирозинемии был установлен в других медицинских учреждениях, из них трое уже получали специфическую патогенетическую терапию нитизиноном, а пять детей начали специфическое лечение лишь спустя определенное время после верификации диагноза (от 2 месяцев до 2 лет 11 месяцев). У троих детей в терапии нитизиноном отмечались неоднократные перерывы длительностью от 3 месяцев до 1 года.
Анализ историй развития детей показал, что при рождении антропометрические данные (масса и длина тела) у всех пациентов были в пределах референсных значений. У 9 детей на первом году жизни отмечалась задержка психомоторного развития и отставание в физическом развитии. При изучении анамнеза заболевания выявлено, что у 7 детей из 17 дебют заболевания отмечался в возрасте первых трех месяцев жизни (из них у 5 пациентов диагностирована HT1A). У 4 пациентов заболевание дебютировало в возрасте 4-6 месяцев, у 4 - в период от 7 месяцев до 1 года, у двоих детей - в возрасте старше года. У 8 из 17 детей (из них у 6 пациентов диагностирована НТ1А в дебюте заболевания) отмечалось повышение температуры тела до фебрильных и субфебрильных значений, что расценивалось как проявления острого респираторного заболевания. У 4 детей первыми проявлениями заболевания были диарея и рвота, что расценивалось как острая кишечная инфекция. Двое детей находились на лечении в хирургическом стационаре с диагнозом «инвагинация кишечника» и «динамическая непроходимость кишечника». У одного ребенка (НТ1Б) выявлялись клинические признаки полинейропатии. У трех пациентов отмечались повторные носовые кровотечения. В 4 случаях заболевание дебютировало с желтухи.
При осмотре у 14 детей выявляли признаки гепато- и гепатоспленомегалии (у 9 - гепатомегалия, у 5 - гепатоспленомегалия), в 7-ми случаях сопровождающиеся асцитом, верифицированным с помощью УЗИ.
В возрасте от 7 месяцев до 2 лет у 7 детей с НТ1Б в дебюте заболевания отмечались выраженные клинические признаки рахита, характеризующиеся не только рахитическими «четками», «браслетками», но и значительной деформацией костей черепа (лобных бугров), Х-образной деформацией ног, вальгусной установкой стоп, которые не купировались при длительном использовании противорахитических средств. К 1-5 годам у пациентов формировались стойкие изменения скелета: у одного ребенка к возрасту 1 год 2 месяца отмечалась выраженная деформация голеней; у одного ребенка к 2 годам развилась выраженная деформация нижних конечностей, вплоть до потери опорной функции, вывиха коленных чашечек и тазобедренных суставов; у одного ребенка к 5 годам сформировалась деформация позвоночника и грудной клетки с развитием дыхательной недостаточности 2б степени.
Анемический синдром диагностирован у 12 пациентов, из них пятерым, в связи со снижением уровня гемоглобина до критических цифр (19-25 г/л), неоднократно проводились инфузии эритроцитарной массы, а у 7 пациентов выявлена тромбоцитопения. 8 детям была выполнена коагулограмма, по результатам которой у всех выявлены признаки коагулопатии, характеризующиеся удлинением протромбинового времени и АЧТВ, снижением ПТИ и уровня фибриногена. У 11 детей определялось повышение уровня АЛТ и ACT, у 4 - гипербилирубинемия. У 4-х детей выявлялась гипогликемия. Снижение уровня альбуминов отмечалось у 8 детей, что клинически проявлялось безбелковыми отеками и асцитом. У одного пациента верифицирован гидроторакс.
По данным анамнеза, дебют заболевания на возраст первых трех месяцев жизни приходился у всех пяти детей с НТ1А и у двоих - с НТ1Б. Учитывая ранний дебют заболевания, внешних признаков рахита у этих 7 пациентов выявлено не было, не у всех, соответственно, была проведена оценка лабораторных признаков вторичной нефропатии. При комплексном обследовании у 10 детей (у 7 пациентов с различными деформациями скелета и у 3 пациентов - методом случайного поиска) выявлены лабораторные признаки острого рахита, характеризующиеся повышением ЩФ и снижением уровня фосфора в крови, что обусловливало необходимость проведения более углубленного обследования и последующей верификации заболевания. Следует отметить, что 5 детей из 17 длительное время наблюдались в различных клиниках с диагнозами: злокачественная опухоль печени (в течение одного и двух месяцев), гликогеновая болезнь 4 типа (в течение 7-ми месяцев и 3-х лет), мукополисахаридоз 4 типа (в течение 4 лет).
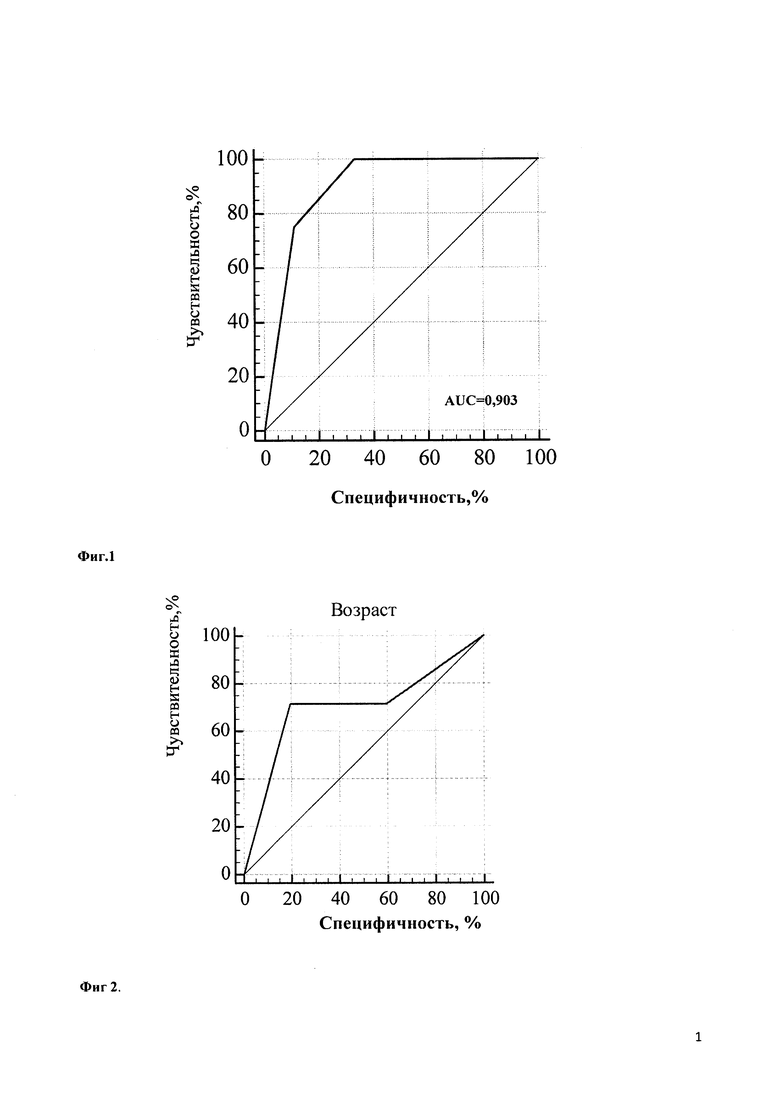
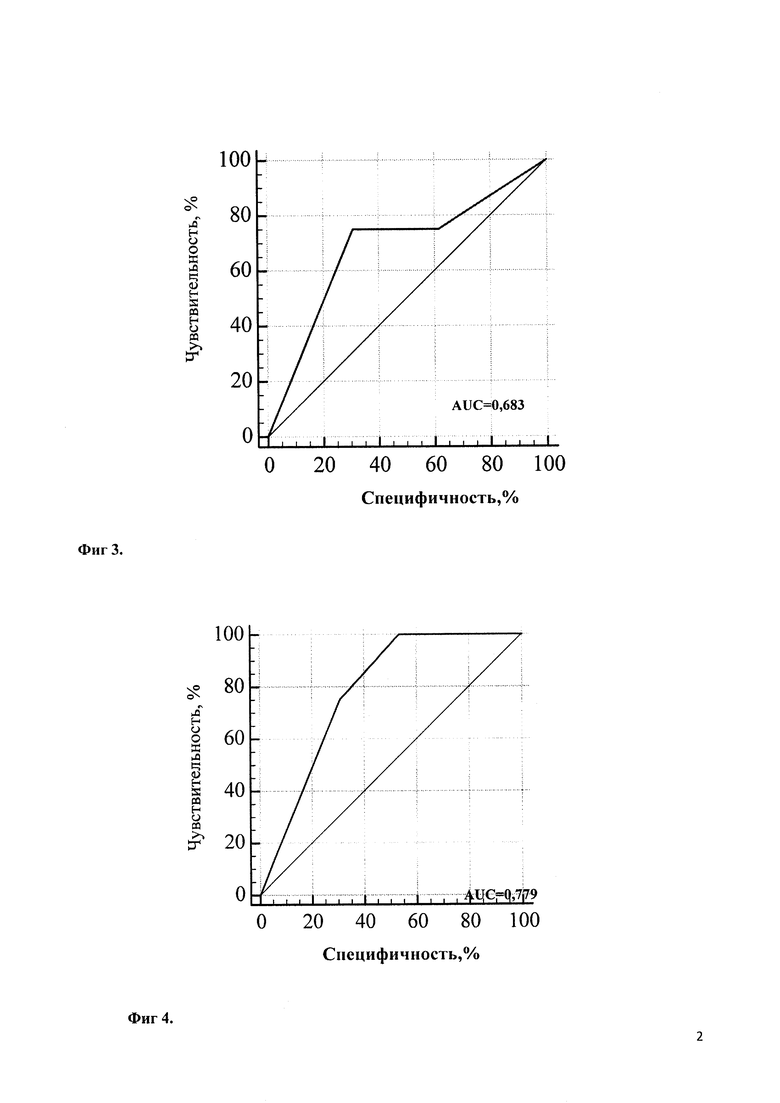
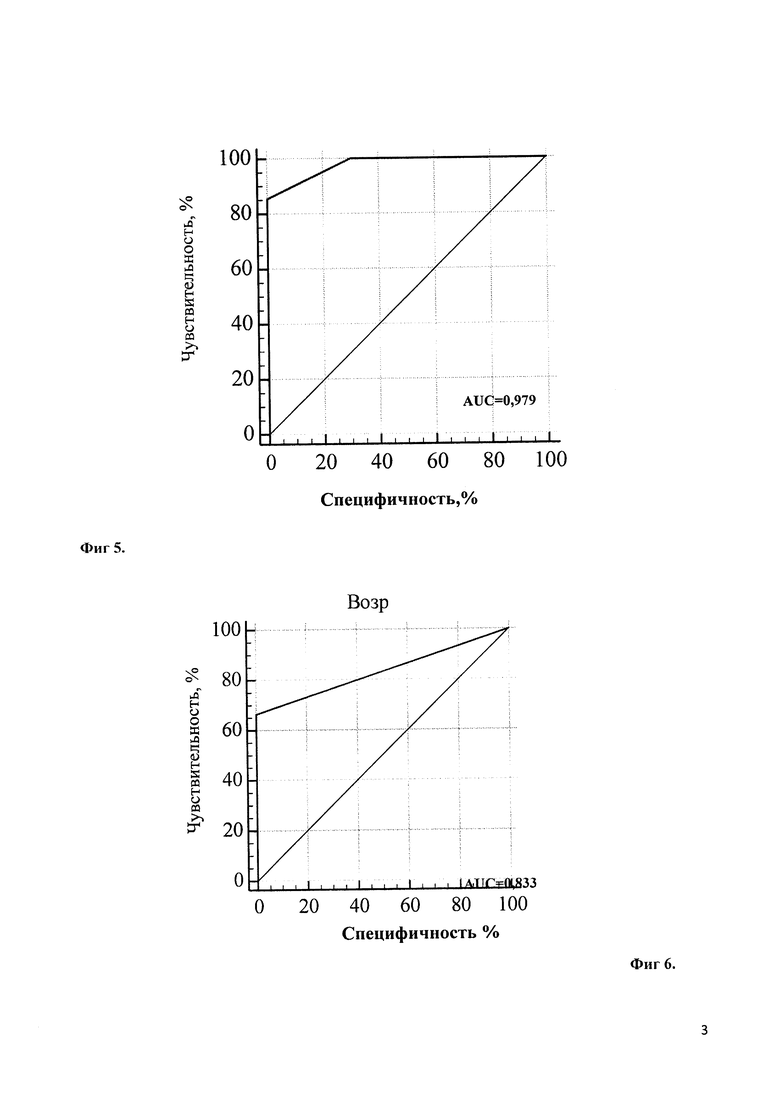
Учитывая выраженный полиморфизм клинических проявлений НТ1 и разнообразие результатов диагностических методов исследования, для определения наиболее значимых показателей был проведен многофакторный статистический анализ. При этом определено, что при дебюте в возрасте первых 3 месяцев жизни наиболее диагностически значимым является симптомокомлекс, состоящий из лихорадки (100,0% чувствительность, 66,7% специфичность, диагностическая значимость которой представлена на Фиг. 1), отеков (71,4% чувствительность, 80,0% специфичность; диагностическая значимость которых представлена на Фиг. 2), желтухи (чувствительность 75,0%, специфичность 69,2%; диагностическая значимость которой представлена на Фиг. 3), диспепсия (чувствительность 100,0%, специфичность 46,1%; диагностическая значимость которой представлена на Фиг. 4). При дебюте в возрасте более 4-х месяцев жизни и старше наиболее диагностически значимыми является острый рахит (чувствительность 85,7%, специфичность 100,0%; диагностическая значимость которого представлена на Фиг. 5) и гепато- или гепатоспленомегалия (чувствительность 66,7%, специфичность 100,0%; диагностическая значимость которых представлена на Фиг. 6).
Способ осуществляется следующим образом: 1-м шагом в алгоритме диагностики HT1 у детей первых 3-х месяцев жизни является сочетание симптомокомплекса лихорадки неясного генеза, отеков, желтухи и диспепсического синдрома. У детей в возрасте 4-х месяцев и старше: гепато- или гепатоспленомегалии и клинические проявления острого рахита, не купирующиеся применением стандартных противорахитических средств.
Пациентам, имеющим указанный симптомокомплекс, необходимо провести 2-й шаг в диагностике HT1: клинический анализ крови с оценкой уровня гемоглобина и эритроцитов (для выявления анемического синдрома), а также количества тромбоцитов и биохимический анализ крови (показатели, отражающие выраженность цитолиза - аланинамино- и аспарагинаминотрансферазы (АЛТ, ACT); уровни общего билирубина и его фракций; показатели, отражающие лабораторные признаки острого рахита - повышенный уровень щелочной фосфатазы (ЩФ) в сыворотке крови, гипокальциемия и гипофосфатемия, гиперфосфатурия); коагулограмма. Целесообразно определить уровень альфа-фетопротеина (АФП), который бывает повышенным.
Детям с выявленной анемией, тромбоцитопений, повышенным уровнем АЛТ, ACT, билирубина, АФП и лабораторными признаками острого рахита, - необходимо проводить исследование уровня тирозина, в крови методом тандемной масс-спектрометрии, и исследование на сукцинилацетон в крови и моче - 3-й шаг в диагностике HT1.
При повышенном уровне тирозина, а также повышении сукцинилацетона в крови выше 2 ммоль/л и более 2 ммоль/моль креатинина в моче, детям показано проведение генетического исследования на мутации в гене FAH, что можно рассматривать как 4-й шаг в алгоритме ранней диагностики HT1. Схема пошаговой диагностики наследственной тирозинемии 1 типа у детей представлена на Фиг. 7.
Пример клинического осуществления способа.
Пример 1. Девочка Ю., 2 года 11 месяцев. В возрасте 2,5 месяца, в дебюте заболевания, отмечали повышение температуры тела до фебрильных цифр, диспепсические проявления. Ребенку в возрасте первых трех месяцев жизни, имеющему указанный симптомокомплекс - 1-й шаг в пошаговой диагностике, назначено проведение обследования, относящегося ко 2-му шагу. При обследовании установлено снижение уровня гемоглобина до 25 г/л, тромбоцитопения до 113×10*9/л, несколько повышенная цитолитическая активность (повышение уровня АЛТ и ACT до 1,5 верхних границ нормы), гипокоагуляция (снижение аналитов: АЧТВ 44,6 с, ПТВ 19,1 с, ПТИ 51%, MHO 1,59), ЩФ 1560 Ед./л. Был определен уровень АФП, который оказался повышенным до 6159 МЕд./л.
Учитывая выявленные клинико-лабораторные изменения, ребенку были проведены исследования, относящиеся к 3-му шагу алгоритма диагностики. Ребенку было проведено измерение уровня ароматических аминокислот и сукцинилацетона в крови (уровень тирозина и фенилаланина не повышен, сукцинилацетон крови повышен до 15 мкмоль/л при норме <2 мкмоль/л).
Учитывая повышенный уровень сукцинилацетона, ребенку было проведено исследование, относящееся к 4-му шагу алгоритма ранней диагностики HT1. Проведено полное секвенирование гена FAH, при этом выявлена мутация c.554-1G>T (гомозигота), которая приводит к развитию наследственной тирозинемии 1 типа.
Был установлен диагноз: наследственная тирозинемия 1 типа. Назначено специфическое лечение в раннем возрасте.
Способ позволяет начать диагностическое обследование детей на ранний стадиях заболевания, уже на 1 уровне оказания медицинской помощи в условиях первичного звена здравоохранения. Своевременная диагностика HT1 и раннее начало специфической терапии позволяют профилактировать инвалидность и смертность вследствие HT1 у детей.
Отбор пациентов для дорогостоящих молекулярно-генетических исследований позволяет максимально уменьшить материальные затраты при проведении диагностики, что с учетом редкой частоты встречаемости заболевания позволит не проводить эти исследования пациентам, не нуждающимся в них.
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ обследования детей с подозрением на синдром Алажилля | 2016 |
|
RU2629828C1 |
| Способ обследования детей с подозрением на прогрессирующий семейный внутрипеченочный холестаз (ПСВХ) | 2016 |
|
RU2627615C1 |
| СПОСОБ ДИФФЕРЕНЦИАЛЬНОЙ ДИАГНОСТИКИ ВРОЖДЁННЫХ ХОЛЕСТАТИЧЕСКИХ БОЛЕЗНЕЙ У ДЕТЕЙ РАННЕГО ВОЗРАСТА | 2017 |
|
RU2670619C9 |
| ТЕСТ-СИСТЕМА ДЛЯ ОПРЕДЕЛЕНИЯ МУТАЦИЙ В ГЕНАХ ФУМАРИЛАЦЕТОАЦЕТАТ ГИДРОЛАЗЫ И АЛЬФА-1-АНТИТРИПСИНА ЧЕЛОВЕКА | 2010 |
|
RU2458131C1 |
| СПОСОБ ДИАГНОСТИКИ ЗАТЯЖНОГО ВАРИАНТА ТЕЧЕНИЯ ЖЕЛТУХИ У НОВОРОЖДЕННЫХ И ДЕТЕЙ ПЕРВОГО ГОДА ЖИЗНИ | 2009 |
|
RU2440579C2 |
| СПОСОБ ЛЕЧЕНИЯ КОНЪЮГАЦИОННЫХ ГИПЕРБИЛИРУБИНЕМИЙ У ДЕТЕЙ ПЕРВОГО ГОДА ЖИЗНИ | 2014 |
|
RU2570479C1 |
| Способ одновременной диагностики наследственных заболеваний | 2015 |
|
RU2627115C2 |
| СПОСОБ ОПРЕДЕЛЕНИЯ РИСКА РАЗВИТИЯ ГЕПАТОЦЕЛЛЮЛЯРНОЙ КАРЦИНОМЫ У БОЛЬНЫХ ХРОНИЧЕСКИМ ГЕПАТИТОМ С | 2019 |
|
RU2723891C1 |
| СПОСОБ ПРОГНОЗИРОВАНИЯ ТЕЧЕНИЯ НЕОНАТАЛЬНЫХ ГЕПАТИТОВ У ДЕТЕЙ ПЕРВОГО ГОДА ЖИЗНИ | 2011 |
|
RU2488829C2 |
| СПОСОБ ОЦЕНКИ СТЕПЕНИ ЦИТОЛИЗА КАРДИОМИОЦИТОВ ПРИ ИНФЕКЦИОННЫХ ПОРАЖЕНИЯХ МИОКАРДА | 2011 |
|
RU2487361C1 |

Изобретение относится к медицине, а именно к педиатрии, и касается способа ранней диагностики наследственной тирозинемии 1 типа (HT1). Сущность способа заключается в том, что детям первых 3-х месяцев жизни, у которых имеет место сочетание симптомокомплекса, состоящего из лихорадки неясного генеза, отеков, желтухи и диспепсического синдрома, а у детей в возрасте 4 месяцев и старше - гепато- или гепатоспленомегалии и клинических проявлений острого рахита, проводят исследование крови с оценкой уровня гемоглобина и количества эритроцитов, количества тромбоцитов, уровня АЛТ, ACT, билирубина и его фракций, уровня щелочной фосфатазы, кальция, фосфора, АФП, коагулограммы. В случае выявления анемии, тромбоцитопении, при повышенном уровне АЛТ, ACT, билирубина, АФП и лабораторных признаков острого рахита проводят исследование уровня тирозина в крови методом тандемной масс-спектрометрии и исследование на сукцинилацетон в крови и моче. В случае выявления повышенного уровня тирозина, а также повышении уровня сукцинилацетона в крови выше 2 ммоль/л и более 2 ммоль/л креатинина в моче проводят генетическое исследование на мутации в гене FAH. Использование способа позволяет с высокой точностью диагностировать HT1 на ранних стадиях. 7 ил., 1 пр.
Способ обследования детей раннего возраста для выявления наследственной тирозинемии 1 типа (TH1), включающий следующий алгоритм:
- детям первых 3-х месяцев жизни, у которых имеет место сочетание симптомокомплекса, состоящего из лихорадки неясного генеза, отеков, желтухи и диспепсического синдрома, а у детей в возрасте 4 месяцев и старше - гепато- или гепатоспленомегалии и клинических проявлений острого рахита, проводят исследование крови с оценкой уровня гемоглобина и количества эритроцитов, количества тромбоцитов, уровня АЛТ, ACT, билирубина и его фракций, уровня щелочной фосфатазы, кальция, фосфора, АФП, коагулограммы;
- в случае выявления анемии, тромбоцитопении, при повышенном уровне АЛТ, ACT, билирубина, АФП и лабораторных признаков острого рахита проводят исследование уровня тирозина в крови методом тандемной масс-спектрометрии и исследование на сукцинилацетон в крови и моче;
- в случае выявления повышенного уровня тирозина, а также повышении уровня сукцинилацетона в крови выше 2 ммоль/л и более 2 ммоль/л креатинина в моче проводят генетическое исследование на мутации в гене FAH.
| ПОЛЯКОВА С.И | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| ТЕСТ-СИСТЕМА ДЛЯ ОПРЕДЕЛЕНИЯ МУТАЦИЙ В ГЕНАХ ФУМАРИЛАЦЕТОАЦЕТАТ ГИДРОЛАЗЫ И АЛЬФА-1-АНТИТРИПСИНА ЧЕЛОВЕКА | 2010 |
|
RU2458131C1 |
| Holme E | |||
| Lindstedt S | |||
| Diagnosis and management of tyrosinemia type I // Curr Opin Pediatr | |||
| Топка с качающимися колосниковыми элементами | 1921 |
|
SU1995A1 |
| Найдно в БД PubMed PMID: 8776026 | |||
| S | |||
| Bijarnia, et al., Tyrosinemia type I - diagnostic issues and prenatal diagnosis // Indian Journal of Pediatrics, February, 2006, V | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| Деревянное стыковое устройство | 1920 |
|
SU163A1 |

