ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к соединениям, способным модулировать активность GPR40, которые, таким образом, подходят для лечения связанных с GPR40 нарушений. Кроме того, изобретение относится к соединениям, способам их получения, фармацевтическим композициям, содержащим указанные соединения, и к применению указанных соединений для лечения определенных нарушений, связанных с активностью GPR40.
УРОВЕНЬ ТЕХНИКИ
Рецептор, сопряженный с G-белками, GPR40, функционирует в качестве рецептора длинноцепочечных свободных жирных кислот (СЖК) в организме и, соответственно, задействован в большом числе метаболических состояний в организме. Например, существуют доказательства того, что агонисты GPR40 способствуют секреции инсулина, в то же время антагонисты GPR40 ингибируют секрецию инсулина, и, таким образом, в зависимости от конкретного случая, указанные агонисты и антагонисты могут подходящими терапевтическими агентами для ряда связанных с инсулином нарушений, таких как диабет 2 типа, ожирение, нарушение переносимости глюкозы, инсулинорезистентность, нейродегенеративные заболевания и т.д.
Диабет, как правило, представляет собой хроническое заболевание, которое возникает в тех случаях, когда поджелудочная железа не производит достаточного количества инсулина, или когда организм не может эффективно использовать инсулин, который в нем производится, для регуляции уровня глюкозы в крови. Гипергликемия, или повышенное содержание сахара в крови, является общим результатом неконтролируемого диабета и со временем приводит к нежелательным физиологическим изменениям у субъектов, страдающих от указанного заболевания, особенно к изменениям в нервной системе и сердечно-сосудистой системе.
Всемирная организация здравоохранения (ВОЗ) установила, что более 220 миллионов человек по всему миру страдают от диабета. По оценкам в 2005 году от диабета умерло более 1,1 миллиона человек (хотя, вероятно, общее число должно быть еще выше, так как в это количество не включены люди, которые умерли в результате осложнений диабета, таких как заболевания сердца или почечная недостаточность). Из всех случаев смерти от диабета почти 80% происходят в странах с низким и средним уровнем доходов, почти 50% составляют люди, не достигшие 70-летнего возраста, и примерно 55% - женщины. ВОЗ также прогнозирует, что число смертей от диабета удвоится в период с 2005 по 2030 годы, если не предпринимать срочные предупредительные меры для сдерживания или обращения развития этой эпидемий. Несмотря на то, что по меньшей мере часть общего числа заболеваний диабетом может быть связана с генетическими факторами, основной причиной является быстрый эпидемиологический переход, связанный с изменениями режима питания и сниженной физической активностью, что является очевидным с учетом того факта, что диабет наиболее распространен среди городского населения.
Несмотря на то, что правильное питание, регулярная физическая активность, поддержание нормальной массы тела и избегание потребления табака могут предотвращать или задерживать возникновение заболевания, в настоящее время отсутствуют эффективные терапевтические стратегии для профилактики или лечения диабета.
Патогенез диабета 2 типа характеризуется дисфункцией бета-клеток и прогрессирующей инсулинорезистентностью с компенсаторной гиперинсулинемией и с последующим снижением секреции инсулина и повышающейся гипергликемией. Продолжительная адаптация основной массы бета-клеток к повышению концентрации глюкозы происходит, главным образом, за счет увеличения числа бета-клеток в результате гиперплазии и неогенеза (Bonner-Weir S, 2002; Rhodes CJ, 2005).
Диабет 2 типа также характеризуется повышенным уровнем длинноцепочечных СЖК в плазме, что приводит к дополнительному нарушению секреции инсулина в бета-клетках. В норме СЖК являются незаменимым топливом для бета-клеток, но становятся токсичными, когда их содержание хронически находится на повышенном уровне. В эндокринной части поджелудочной железы краткосрочное воздействие бета-клеток на потребляемые жирные кислоты усиливает индуцируемое глюкозой высвобождение инсулина (Haber EP et al., 2003, Yaney GC and Crokey BE, 2003), в то же время продолжительное воздействие нарушает секрецию инсулина и индуцирует недостаточность секреторной функции (липотоксичность; Lee Y et al., 1994, Unger RH, 2002) и апоптоз бета-клеток (липоапоптоз; Shimabukuro М et al., 1998, Lupi R et al., 2002).
Появляется все больше доказательств того, что липиды также могут служить в качестве внеклеточных лигандов для конкретного класса рецепторов и, таким образом, действовать в качестве «датчиков правильного питания» (Nolan CJ et al., 2006). Открытие этих рецепторов позволило сделать предположение о том, что липиды, в частности. свободные жирные кислоты (СЖК) могут регулировать клеточную функцию. Недавно было показано, что свободные жирные кислоты (СЖК) могут действовать в качестве лигандов орфанных рецепторов, сопряженных с G-белками (GPCR), и было выдвинуто предположение, что СЖК играют важную роль в физиологическом гомеостазе глюкозы (Rayasam GV et al., 2007).
GPR40, GPR120, GPR41 и GPR43 являются примерами растущего числа GPRC, для которых подтверждена активация свободными жирными кислотами (Kotarsky К et al., 2003, Brown AJ et al., 2003). GPR40 и GPR120 активируются средне- и длинноцепочечными свободными жирными кислотами, в то же время короткоцепочечные жирные кислоты активируют GPR41 и GPR43 (Kotarsky К et al., 2003, Nilsson NE et al., 2003, Brown AJ et al., 2003).
Каждый GPR имеет характеристическое распределение в тканях. GPR40, главным образом, экспрессируется в бета-клетках поджелудочной железы (Salehi A et al., 2005). Ген, кодирующий GPR40, расположен ниже CD22 на хромосоме 19q13.1 (Sawzdargo M et al., 1997) вблизи области, которая связана с повышенным содержанием триглицеридов в сыворотке у семей с диабетом 2 типа (Elbein SC and Hasstedt SJ, 2002). В гене GPR40 было идентифицировано два типа полиморфизма, замещение Arg211His и редкая мутация Asp175Asn (Haga Н et al., 2002). Позднее экспрессию GPR40 также наблюдали в жировых тканях сальника и в альфа-клетках поджелудочной железы (Fodgren E et al., 2007).
Точно установлено, что жирные кислоты обладают выраженным действием при поддержании секреции инсулина базальными клетками и «подготовке» островковых β-клеток к ответу на глюкозу после длительного голодания (Gravena С et al., 2002). Кроме того, тот факт, что активация рецептора приводила к повышению внутриклеточного содержания Са2+ в результате связывания с Gαq/11, а впоследствии к активации протеинкиназы С (РКС), указал на возможную роль GPR40 в секреции инсулина (Poitout V 2003, Fujiwara К et al., 2005, Schnell S et al., 2007). Понижающая регуляция экспрессии GPR40 в клеточных линиях инсулиномы мышей приводила к снижению способности жирных кислот усиливать секрецию инсулина (Itoh Y et al., 2003, Shapiro H et al., 2005). Было показано, что GPR40 имеет значение не только при модуляции секреции инсулина жирными кислотами, но также при стимулируемой глюкозой секреции инсулина (GSIS) после потребления пищи с высоким содержанием жиров (Kebede M et al., 2008).
Для исследования роли GPR40 в метаболизме несколько групп исследовали фенотип нокаута GPR40 или сверхэкспрессию GPR40 на различных моделях грызунов. Мыши GPR40 -/-, которым давали пищу с высоким содержанием жиров (HFD), страдали от ожирения в той же степени, что и мыши дикого типа (WT), но были защищены от вызванной ожирением гиперинсулинемии, непереносимости глюкозы, гепатического стеатоза, гипертриглицеридемии и повышенного продуцирования глюкозы в печени (Steneberg P et al., 2005). Другая группа при исследовании влияния кормления на мышей GPR40-/- показала недостаточное снижение острой стимулируемой пальмитатом GSIS (50% снижение) в выделенных островках. С другой стороны в этих островках отсутствовало ингибирование GSIS через 72 часа воздействия пальмитата или олеата по сравнению с WT (Latour MG et al., 2007). В другом исследовании, когда GPR40 специфически сверхэкспрессировался в бета-клетках поджелудочной железы, трансгенные мыши обретали непереносимость глюкозы, у них стала отсутствовать первая фаза секреции инсулина, и, наконец, мыши заболевали диабетом. У указанных мышей также изменялась морфология бета-клеток (Steneberg Р et al., 2005).
Несмотря на то, что приведенные выше данные исследований указывают на положительное действие антагонизма GPR40 для контроля диабета, ряд других исследований доказывает обратное. Комплексное исследование с использованием серий высокоактивных и селективных агонистов GPR40 показало, что указанные соединения значительно усиливали GSIS у мышей дикого типа, но не у мышей GPR40-/-. В этом исследовании также было показано снижение содержания глюкозы в крови у мышей, страдающих диабетом, вызванным стрептозотоцином, и у мышей, страдающих ожирением, вызванным приемом пищи с высоким содержанием жиров. Было высказано предположение, что указанные соединения не способствуют хроническому токсическому действию свободных жирных кислот на островковые клетки (Tan CP et al., 2008). В другом недавнем исследовании было показано, что, несмотря на то, что GPR40 необходим для секреции инсулина в ответ на СЖК, мыши GPR40 -/- не были защищены от инсулинорезистентности, вызванной приемом пищи с высоким содержанием жиров, от или гепатического стеатоза (Lan H et al., 2008).
Поскольку жирные кислоты усиливают секрецию инсулина в зависимости от содержания глюкозы, можно предположить, что если действие жирных кислот на секрецию инсулина опосредовано по меньшей мере отчасти GPR40, то агонист GPR40, представляющий собой малую молекулу, может действовать в качестве чувствительного к глюкозе стимулятора секреции (Briscoe CP et al., 2006).
Недавно было показано, что GPR40 экспрессируется в эндокринных клетках желудочно-кишечного тракта, включая клетки, экспрессирующие инкретиновые гормоны: глюкагоноподобный пептид-1 (GLP-1) и глюкозозависимый инсулинотропный полипептид (GIP), а также что GPR40 способствует стимулируемой СЖК секреции инсулина (Edfalk S et al., 2008, Parker HE et al., 2009).
Хорошо известно, что кратковременное воздействие СЖК стимулирует секрецию инсулина, в то же время хроническое воздействие нарушает функцию бета-клеток и индуцирует апоптоз. Было обнаружено, что олеиновая кислота, действие которой по меньшей мере отчасти опосредовано GPR40, может защищать клетки NIT-1 от индуцированного пальмитатами липоапоптоза. Более того, было обнаружено, что олеиновая кислота способствовала активации внеклеточного сигнального пути протеинкиназы-МАРК, главным образом, посредством GPR40, который увеличивал экспрессию гена 1 раннего ответа роста, что приводило к антилипоапоптотическому действию на клетки NIT-1. Было сделано предположение, что GPR40 может быть задействован в контроле пластичности массы бета-клеток (Zhang Y et al., 2007).
Клинические исследования показали, что общее содержание жира в организме связано как с плотностью кости, так и с риском переломов, а также что потребление жира уменьшает обновление костной ткани. Эти явления по меньшей мере отчасти опосредованы эндокринными механизмами, но существует возможность того, что липиды могут действовать непосредственно на кость. Было показано, что рецепторы, связывающие жирные кислоты, экспрессируются в остеобластных (GPR120) и остеокластных (GPR40, 41, 43, 120) клетках. Синтетический агонист GPR40/120 имитировал ингибирующее действие жирных кислот на остеокластогенез (Cornish J et al., 2008).
GPR40 был недавно обнаружен в нейронах мозга. Последние исследования показали, что полиненасыщенные жирные кислоты (ПНЖК) способны улучшать долговременную потенциацию гиппокампа, обучаемость взрослых крыс и когнитивную функцию людей с дефицитом памяти. Возможно, определенные ПНЖК могут действовать в качестве эндогенных лигандов GPR40 на поверхности нейронных клеток (Yamashima Т, 2008).
В другом исследовании, проведенном для взрослых обезьян, было показано, что содержание белка GPR40 значительно повышалось на второй неделе после глобальной ишемии головного мозга по сравнению с контролем. С учетом этих данных полагают, что GPR40 может участвовать в регуляции гиппокампального нейрогенеза у взрослых приматов (Ма D et al., 2007; 2008).
Соответственно, ожидается, что соединения, которые модулируют GPR40, имеют подходящие терапевтические свойства, в частности, в отношении метаболических состояний, таких как диабет, ожирение, гипергликемия, непереносимость глюкозы, инсулинорезистентность, гиперинсулинемия, гиперхолестеринемия, гипертензия, гиперлипопротеинемия, гиперлипидемия, гипертриглицеридемия, дислипидемия, метаболический синдром X, атеросклероз, диабетическая нейропатия, диабетическая ретинопатия и гипогликемия.
Соединения этого типа также могут быть подходящими для лечения когнитивных нарушений, остеопороза, воспалительных нарушений, сердечно-сосудистых заболеваний, заболеваний почек, кетоацидоза, тромботических нарушений, нефропатии, сексуальной дисфункции, дерматопатии, диспепсии, рака и эдемы. Таким образом, существует значительный интерес к разработке соединений с указанным типом действия.
ЗАДАЧИ ИЗОБРЕТЕНИЯ
Основная задача изобретения состоит в том, чтобы предложить соединения, которые представляют собой модуляторы активности рецептора GPR40. Можно ожидать, что указанные соединения подходят для лечения состояний, связанных с GPR40.
Другая задача состоит в том, чтобы предложить фармацевтическую композицию, содержащую соединение, которое представляет собой модулятор активности рецептора GPR40, и фармацевтически приемлемое вспомогательное вещество, разбавитель или носитель.
Другая задача состоит в том, чтобы предложить способ предотвращения или лечения состояния, связанного с функцией рецептора GPR40 у млекопитающего.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В настоящем изобретении предложены соединения формулы (I):
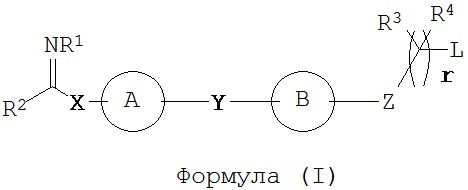
где:
кольцо А выбрано из группы, состоящей из возможно замещенного С3-С12 циклоалкила, возможно замещенного C2-C12 гетероциклоалкила, возможно замещенного С6-С10 арилконденсированного С3-С6 циклоалкила, возможно замещенного C1-C18 гетероарилконденсированного С3-С6 циклоалкила, возможно замещенного С6-С10 арилконденсированного C2-C12 гетероциклоалкила, возможно замещенного C1-C18 гетероарилконденсированного C2-C12 гетероциклоалкила, возможно замещенного С6-C18 арила и возможно замещенного C1-C18 гетероарила;
кольцо B представляет собой возможно замещенную С6-C18 арильную группу, возможно замещенную С6-С10 арилконденсированную C2-C12 гетероциклоалкильную или возможно замещенную C1-C18 гетероарильную группу;
Х представляет собой связь или линкерный фрагмент, содержащий от 1 до 8 атомов в линейной цепи;
Y представляет собой связь или линкерный фрагмент, содержащий от 1 до 8 атомов в линейной цепи;
Z представляет собой связь или выбран из группы, состоящей из возможно; замещенного С6-C18 арила и возможно замещенного C1-C18 гетероарила, -С(=O)-, -C(=NR1)-, -(CR5R6)-, -(CR5R6)O-, -(CR5R6)S-, -(CR5R6)NR'-, или представляет собой гетероатомную группу, выбранную из группы, состоящей из S, О, Р и NR’’, где R’’ выбран из группы, состоящей из Н, возможно замещенного C1-С12 алкила, возможно замещенного С2-С10 гетероалкила, возможно замещенного С3-С12 циклоалкила, возможно замещенного С6-C18 арила и возможно замещенного C1-C18 гетероарила;
L представляет собой группу, способную высвобождать катион, или ее соль;
R1 выбран из группы, состоящей из Н, OR7, возможно замещенного C1-C12 алкила, возможно замещенного C1-C12 галогеналкила, возможно замещенного C2-C12 алкенила, возможно замещенного C2-C12 алкинила, возможно замещенного C1-C12 алкилокси, возможно замещенного C1-C12 галогеналкилокси, возможно замещенного С2-С10 гетероалкила, возможно замещенного С3-С12 циклоалкила, возможно замещенного С3-С12 циклоалкенила, возможно замещенного C2-C12 гетероциклоалкила, возможно замещенного C2-C12 гетероциклоалкенила, возможно замещенного C6-C18 арила и возможно замещенного C1-C18 гетероарила;
R2 представляет собой Н или кольцо, выбранное из группы, состоящей из возможно замещенного С3-С12 циклоалкила, возможно замещенного C2-C12 гетероциклоалкила, возможно замещенного С6-С10 арилконденсированного С3-С6 циклоалкила, возможно замещенного C1-C18 гетероарилконденсированного С3-С6 циклоалкила, возможно замещенного С6-С10 арилконденсированного C2-C12 гетероциклоалкила, возможно замещенного C1-C18 гетероарилконденсированного C2-C12 гетероциклоалкила, возможно замещенного С6-C18 арила и возможно замещенного C1-C18 гетероарила;
каждый R3, R4, R5 и R6 независимо выбран из группы, состоящей из Н, галогена, CN, -NO2, SH, CF3, ОН, CO2H, CONH2, OCF3, возможно замещенного C1-C12 алкила, возможно замещенного C1-C12 галогеналкила, возможно замещенного C2-C12 алкенила, возможно замещенного C2-C12 алкинила, возможно замещенного C1-C12 алкилокси, возможно замещенного C1-C12 галогеналкилокси, возможно замещенного С2-С10 гетероалкила, возможно замещенного С3-С12 циклоалкила, возможно замещенного С3-С12 циклоалкенила, возможно замещенного C2-C12 гетероциклоалкила, возможно замещенного С2-С12 гетероциклоалкенила, возможно замещенного С6-C18 арила и возможно замещенного C1-C18 гетероарила, или
любые два из R3, R4, R5 и R6 совместно с атомами, к которым они присоединены, могут образовывать возможно замещенный циклический фрагмент или двойную или тройную связь между атомами, к которым они присоединены;
R7 выбран из группы, состоящей из Н, возможно замещенного C1-C12 алкила, возможно замещенного C2-C12 алкенила, возможно замещенного С2-С10 гетероалкила, возможно замещенного C2-C10 галогеналкила, возможно замещенного C1-C12 циклоалкила; возможно замещенного С6-C18 арила и возможно замещенного C1-C18 гетероарила;
r представляет собой целое число, выбранное из группы, состоящей из 0, 1 и 2;
или их фармацевтически приемлемые соли, N-оксиды или пролекарства.
Так же, как и в случае любой группы соединений со схожей структурой, которая имеет определенное применение, конкретные варианты значений переменных в соединениях формулы (I) особенно подходят для целевого назначения указанные соединений.
Фрагмент L может представлять собой любой фрагмент или группу, которые способны высвобождать катион, или его соль. Существует множество групп этого типа, что очевидно специалистам в данной области техники. В некоторых вариантах реализации L выбран из группы, состоящей из -СО2Н, -SO3H, -PO3H, -SO2NH2, -CONHSO2CH3 и тетразол-5-ила. В некоторых вариантах реализации L представляет собой СО2Н или его соль.
В соединениях согласно настоящему изобретению r представляет собой целое число, выбранное из группы, состоящей из 0, 1 и 2. В некоторых вариантах реализации r равен 0. В некоторых вариантах реализации r равен 1. В некоторых вариантах реализации r равен 2.
В некоторых вариантах реализации L представляет собой CO2H, а r равен 1, т.е. предложены соединения формулы (II)
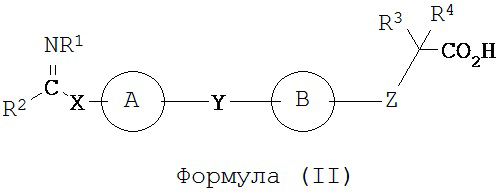
или их фармацевтически приемлемые соли или пролекарства, где R1, R2, R3, R4, А, В, X, Y и Z такие, как определено выше.
В некоторых вариантах реализации каждый R3 и R4 независимо выбран из группы, состоящей из Н, галогена, CN, -NO2, SH, CF3, OCF3, СН3 и СН2СН3. В некоторых вариантах реализации R3 и R4 представляют собой Н.
В некоторых вариантах реализации Z представляет собой -С(=O)-. В некоторых вариантах реализации Z представляет собой -C(=NR1)-. В некоторых вариантах реализации Z представляет собой -(CR5R5)-. В некоторых вариантах реализации Z представляет собой -(CR5R6)-, где R5 представляет собой Н, т.е. Z представляет собой CHR6.
В некоторых вариантах реализации каждый R5 и R6 независимо выбран из группы, состоящей из Н, галогена, CN, -NO2, SH, CF3, OCF3, СН3 и СН2СН3. В некоторых вариантах реализации каждый R представляет собой H, а R6 представляет собой циано.
В некоторых вариантах реализации R5 или R6 совместно с одним из R3 и R4 и атомами, к которым они присоединены, образуют циклический фрагмент. В некоторых вариантах реализации циклический фрагмент представляет собой циклопропильную группу.
В некоторых вариантах реализации L представляет собой CO2H, r равен 1, Z представляет собой -(CR5R6)-, а каждый из R3, R4 и R5 представляет собой Н, т.е. предложены соединения формулы (III)
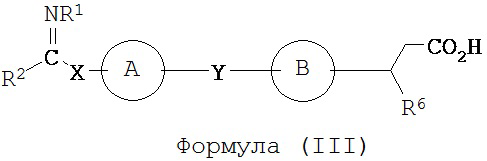
или их фармацевтически приемлемые соли или пролекарства, где R1, R2, R6, А, В, Х и Y такие, как определено выше.
В некоторых вариантах реализации L представляет собой CO2H, r равен 1, Z представляет собой -(CR5R6)-, а каждый из R3, R4, R5 и R6 представляет собой Н, т.е. предложены соединения формулы (IIIa)
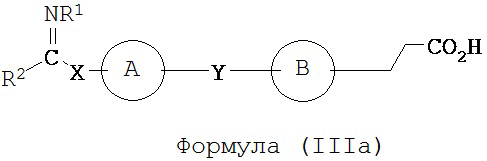
или их фармацевтически приемлемые соли или пролекарства, где R1, R2, А, В, Х и Y такие, как определено выше.
В некоторых вариантах реализации кольцо А и кольцо В независимо выбраны из группы, состоящей из возможно замещенного С6-C18 арила и возможно замещенного C1-C18 гетероарила, и могут представлять собой моноциклические, бициклические или полициклические фрагменты. В конкретных вариантах реализации каждый из А и В представляет собой моноциклический или бициклический фрагмент. В конкретных вариантах реализации каждый из А и В представляет собой моноциклический фрагмент.
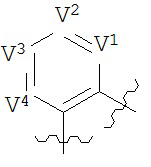
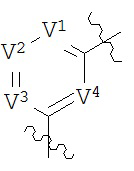
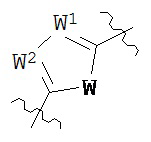
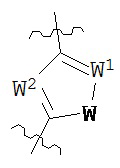
В конкретных вариантах реализации кольцо В выбрано из группы, состоящей из:
 ,
, 

 или
или 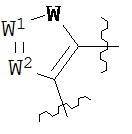
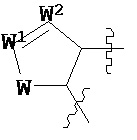
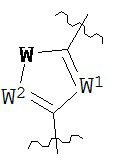
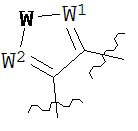
где каждый V1, V2, V3 и V4 независимо выбран из группы, состоящей из N и C(R8);
W выбран из группы, состоящей из О, S и NR8;
каждый W1 и W2 независимо выбран из группы, состоящей из N и CR8;
где каждый R8 независимо выбран из группы, состоящей из Н, галогена, ОН, NO2, CN, SH, NH2, CF3, OCF3, возможно замещенного C1-C12 алкила, возможно замещенного C1-C12 галогеналкила, возможно замещенного C2-C12 алкенила, возможно замещенного С2-C12 алкинила, возможно замещенного C2-C12 гетероалкила, возможно замещенного С3-С12 циклоалкила, возможно замещенного С3-С12 циклоалкенила, возможно замещенного C2-C12 гетероциклоалкила, возможно замещенного C2-C12 гетероциклоалкенила, возможно замещенного C6-C18 арила, возможно замещенного C1-C18 гетероарила, возможно замещенного C1-C12 алкилокси, возможно замещенного C2-C12 алкенилокси, возможно замещенного C2-C12 алкинилокси, возможно замещенного С2-С10 гетероалкилокси, возможно замещенного С3-С12 циклоалкилокси, возможно замещенного С3-С12 циклоалкенилокси, возможно замещенного С2-С12 гетероциклоалкилокси, возможно замещенного С2-С12 гетероциклоалкенилокси, возможно замещенного С6-C18 арилокси, возможно замещенного C1-C18 гетероарилокси, возможно замещенного C1-C12 алкиламино, SR7, SO3H, SO2NR7R7, SO2R10, SONR7R7, SOR7, COR7, COOH, COOR7, CONR7R7, NR7COR7, NR7COOR7, NR7SO2R7, NR7CONR7R7, NR7R7 и ацила.
В некоторых вариантах реализации кольцо В представляет собой возможно замещенную фенильную группу. Указанная группа может быть незамещенной или может быть замещена одним или более возможными заместителями. Широкий ряд возможных заместителей, определенных выше, можно применять. Примеры особенно подходящих возможных заместителей включают, но не ограничиваются ими, OH, F, Br, Cl, метил, CN, трифторметил, этил, 2,2,2-трифторэтил, изопропил, пропил, 2-этилпропил, 3,3-диметилпропил, бутил, изобутил, 3,3-диметилбутил, 2-этилбутил, пентил, 2-метилпентил, пент-4-енил, гексил, гептил, октил, фенил, NH2, фенокси, гидрокси, метокси, этокси, пиррол-1-ил и 3,5-диметилпиразол-1-ил.
В некоторых вариантах реализации кольцо В представляет собой возможно замещенную фенильную группу формулы
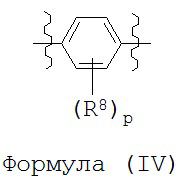
каждый R8 независимо выбран из группы, состоящей из H, галогена, OH, NH2, NO2, CN, C1-C12 алкила, C1-C12 галогеналкила, C1-C12 алкоксила и C1-C12 галогеналкоксила,
где p представляет собой целое число, выбранное из группы, состоящей из 0, 1, 2, 3 и 4.
В соединениях согласно настоящему изобретению p представляет собой целое число, выбранное из группы, состоящей из 0, 1, 2, 3 и 4. В некоторых вариантах реализации p равен 0. В некоторых вариантах реализации p равен 1. В некоторых вариантах реализации p равен 2. В некоторых вариантах реализации р равен 3. В некоторых вариантах реализации p равен 4.
R8 может быть выбран из широкого ряда возможных заместителей, обсуждаемых выше. В некоторых вариантах реализации каждый R8 независимо выбран из группы, состоящей из H, галогена, OH, NO2, CN, C1-C12 алкила, C1-C12 галогеналкила, C1-C12 алкоксила и C1-C12 галогеналкоксила. Типичные заместители R8 включают F, Cl, Br, I, CH3, CH2CH3, OH, OCH3, CF3, OCF3, NO2, NH2 и CN.
В некоторых вариантах реализации L представляет собой СО2Н, r равен 1, Z представляет собой -(CR5R6)-, каждый из R3, R4, R5 представляет собой Н, а кольцо В представляет собой возможно замещенную фенильную группу формулы (IV), т.е. предложены соединения формулы (IVa)
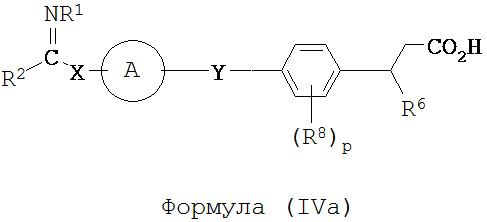
или их фармацевтически приемлемые соли или пролекарства, где R1, R2, R6, R8, А, p, X и Y такие, как определено выше.
В некоторых вариантах реализации L представляет собой CO2H, r равен 1, Z представляет собой -(CR5R6)-, каждый из R3, R4, R5 и R6 представляет собой H, а кольцо B представляет собой возможно замещенную фенильную группу формулы (IV), т.е. предложены соединения формулы (IVb)
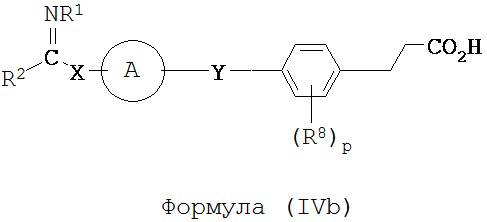
или их фармацевтически приемлемые соли или пролекарства, где R1, R2, R8, A, p, X и Y такие, как определено выше.
В конкретных вариантах реализации кольцо А выбрано из группы, состоящей из:
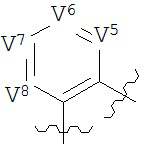 ,
, 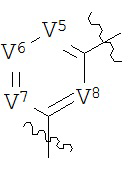
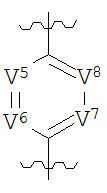
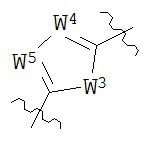
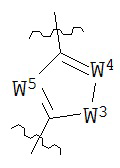
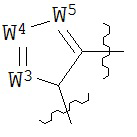
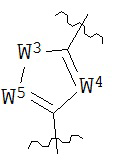
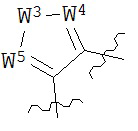 или
или 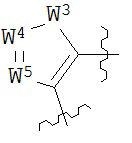
где каждый V5, V6, V7 и V8 независимо выбран из группы, состоящей из N и C(R9);
W3 выбран из группы, состоящей из О, S и NR9;
каждый W4 и W5 независимо выбран из группы, состоящей из N и CR9;
где каждый R9 независимо выбран из группы, состоящей из H, галогена, OH, NO2, CN, SH, NH2, CF3, OCF3, возможно замещенного C1-C12 алкила, возможно замещенного C1-C12 галогеналкила, возможно замещенного C2-C12 алкенила, возможно замещенного С2-C12 алкинила, возможно замещенного C2-C12 гетероалкила, возможно замещенного С3-С12 циклоалкила, возможно замещенного С3-С12 циклоалкенила, возможно замещенного С2-C12 гетероциклоалкила, возможно замещенного С2-С12 гетероциклоалкенила, возможно замещенного С6-C18 арила, возможно замещенного C1-C18 гетероарила, возможно замещенного C1-C12 алкилокси, возможно замещенного C2-C12 алкенилокси, возможно замещенного С2-С12 алкинилокси, возможно замещенного С2-С10 гетероалкилокси, возможно замещенного С3-С12 циклоалкилокси, возможно замещенного С3-С12 циклоалкенилокси, возможно замещенного C2-C12 гетероциклоалкилокси, возможно замещенного C2-C12 гетероциклоалкенилокси, возможно замещенного С6-C18 арилокси, возможно замещенного C1-C18 гетероарилокси, возможно замещенного C1-C12 алкиламино, SR7, SO3H, SO2NR7R7, SO2R7, SONR7R7, SOR7, COR7, COOH, COOR7, CONR7R7, NR7COR7, NR7COOR7, NR7SO2R7, NR7CONR7R7, NR7R7 и ацила.
В некоторых вариантах реализации кольцо А представляет собой возможно замещенную фенильную группу. Указанная группа может быть незамещенной или может быть замещена одним или более возможными заместителями. Широкий ряд возможных заместителей, определенных выше, можно применять. Примеры особенно подходящих возможных заместителей включают, но не ограничиваются ими, ОН, F, Br, Cl, метил, CN, трифторметил, этил, 2,2,2-трифторэтил, изопропил, пропил, 2-этилпропил, 3,3-диметилпропил, бутил, изобутил, 3,3-диметилбутил, 2-этилбутил, пентил, 2-метилпентил, пент-4-енил, гексил, гептил, октил, фенил, NH2, фенокси, гидрокси, метокси, этокси, пиррол-1-ил и 3,5-диметилпиразол-1-ил.
В некоторых вариантах реализации кольцо А представляет собой возможно замещенную фенильную группу. В некоторых вариантах реализации кольцо А представляет собой возможно замещенную фенильную группу, выбранную из группы, состоящей из формулы (Va) и формулы (Vb):
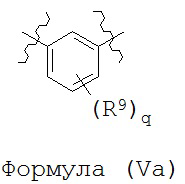 и
и 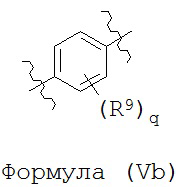
где каждый R9 независимо выбран из группы, состоящей из Н, галогена, OH, NH2, NO2, CN, C1-C12 алкила, C1-C12 галогеналкила, C1-C12 алкоксила и C1-C12 галогеналкоксила;
где q представляет собой целое число, выбранное из группы, состоящей из 0, 1, 2, 3 и 4.
В конкретных вариантах реализации кольцо А представляет собой возможно замещенную фенильную группу формулы:
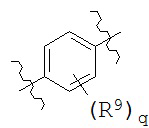
где R9 и q такие, как определено выше.
В соединениях согласно настоящему изобретению q представляет собой целое число, выбранное из группы, состоящей из 0, 1, 2, 3 и 4. В некоторых вариантах реализации q равен 0. В некоторых вариантах реализации q равен 1. В некоторых вариантах реализации q равен 2. В некоторых вариантах реализации q равен 3. В некоторых вариантах реализации q равен 4.
R9 может быть выбран из широкого диапазона возможных заместителей, обсуждаемых выше. В некоторых вариантах реализации R9 независимо выбран из группы, состоящей из Н, галогена, ОН, NO2, CN, C1-C12 алкила, C1-C12 галогеналкила, C1-C12 алкоксила и C1-C12 галогеналкоксила. Типичные заместители R9 включают F, Cl, Br, I, СН3, CH2CH3, OH, OCH3, CF3, OCF3, NO2, NH2 и CN.
В некоторых вариантах реализации L представляет собой CO2H, r равен 1, Z представляет собой -(CR5R6)-, каждый из R3, R4, R5 представляет собой Н, кольцо B представляет собой возможно замещенную фенильную группу формулы (IV), а кольцо A представляет собой возможно замещенную фенильную группу формулы (Va), т.е. предложены соединения формулы (Vc)
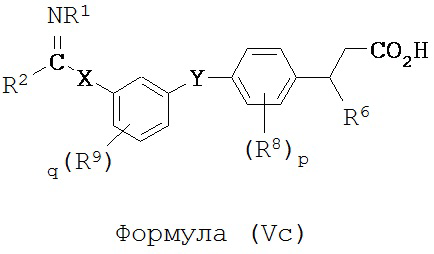
или их фармацевтически приемлемые соли или пролекарства, где R1, R2, R6, R8, R9, q, p, X и Y такие, как определено выше.
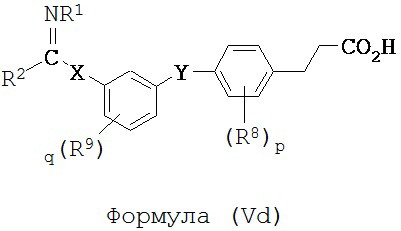
В некоторых вариантах реализации L представляет собой CO2H, r равен 1, Z представляет собой -(CR5R6)-, каждый из R3, R4, R5 и R6 представляет собой H, кольцо B представляет собой возможно замещенную фенильную группу формулы (IV), а кольцо А представляет собой возможно замещенную фенильную группу формулы (Va), т.е. предложены соединения формулы (Vd)
или их фармацевтически приемлемые соли или пролекарства, где R1, R2, R8, R9, q, p, X и Y такие, как определено выше.
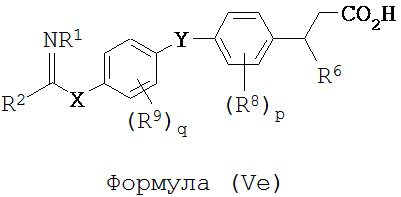
В некоторых вариантах реализации L представляет собой CO2H, r равен 1, Z представляет собой -(CR5R6)-, каждый из R3, R4, R5 представляет собой H, кольцо В представляет собой возможно замещенную фенильную группу формулы (IV), а кольцо А представляет собой возможно замещенную фенильную группу формулы (Vb), т.е. предложены соединения формулы (Ve)
или их фармацевтически приемлемые соли или пролекарства, где R1, R2, R6, R8, R9, q, p, Х и Y такие, как определено выше.
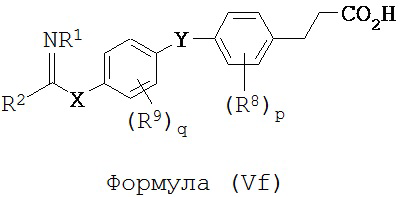
В некоторых вариантах реализации L представляет собой CO2H, r равен 1, Z представляет собой -(CR5R6)-, каждый из R3, R4, R5 и R6 представляет собой H, кольцо В представляет собой возможно замещенную фенильную группу формулы (IV), а кольцо A представляет собой возможно замещенную фенильную группу формулы (Vb), т.е. предложены соединения формулы (Vf)
или их фармацевтически приемлемые соли или пролекарства, где R1, R2, R8, R9, q, p, Х и Y такие, как определено выше.
В соединениях согласно настоящему изобретению Х и Y независимо выбраны таким образом, что они представляют собой связь или содержат от 1 до 8 атомов в линейной цепи.
В некоторых вариантах реализации Y выбран из группы, состоящей из:
(a) связи,
(b) -O-,
(c) -S-,
(d) -S(=O)-,
(e) -S(=O)2-,
(f) -N(R10)-,
(g) -C(R10)2-,
(h) -C(R10)2O-,
(i) -C(R10)2N(R10)-,
(j) -C(=R11),
(k) -OC1-5 алкила-,
(l) -C1-5 алкил-O-,
(m) -С1-5 алкил-OC1-5 алкила,
(n) -N(R10)C1-5 алкила-,
(о) -C1-5 алкил-N(R10)-;
(p) -C1-5 алкил-N(R10)C1-5 алкила-,
(q) -N(R10)CO-,
(r) -N(R10)COC1-5 алкил-,
(s) -C1-5 алкил-N(R10)СО-,
(t) -C1-5 алкил-N(R10)COC1-5 алкил-,
(u) -CON(R10)-,
(v) -C1-5 алкил-CON(R10)-,
(w) -CON(R10)C1-5 алкил-,
(x) -C1-5 алкил-CON(R10)С1-5 алкил-,
(y) -SO2N(R10)-,
(z) -N(R10)SO2-,
(aa) -С1-5 алкил-,
где каждый из алкильных фрагментов может быть дополнительно замещен,
где R10 выбран из группы, состоящей из H, возможно замещенного C1-C12 алкила, возможно замещенного С2-С10 гетероалкила, возможно замещенного С3-С12 циклоалкила, возможно замещенного С6-C18 арила и возможно замещенного C1-C18 гетероарила, или два R10 совместно могут образовывать циклический фрагмент;
где R11 выбран из группы, состоящей из: О, S, NR12 и C(R13R14);
каждый R12 выбран из группы, состоящей из Н, OR15, возможно замещенного C1-C12 алкила, возможно замещенного C1-C12 галогеналкила, возможно замещенного C2-C12 алкенила, возможно замещенного C2-C12 алкинила, возможно замещенного C1-C12 алкилокси, возможно замещенного C1-C12 галогеналкилокси, возможно замещенного С2-С10 гетероалкила, возможно замещенного С3-С12 циклоалкила, возможно замещенного С3-С12 циклоалкенила, возможно замещенного C2-C12 гетероциклоалкила, возможно замещенного C2-C12 гетероциклоалкенила, возможно замещенного С6-C18 арила и возможно замещенного С1-С18 гетероарила,
каждый R13 и R14 независимо выбран из группы, состоящей из Н, галогена, ОН, NO2, CN, SH, NH2, CF3, OCF3, возможно замещенного C1-C12 алкила, возможно замещенного C1-C12 галогеналкила, возможно замещенного C2-C12 алкенила, возможно замещенного C2-C12 алкинила, возможно замещенного C2-C12 гетероалкила, возможно замещенного С3-С12 циклоалкила, возможно замещенного С3-С12 циклоалкенила, возможно замещенного C2-C12 гетероциклоалкила, возможно замещенного C2-C12 гетероциклоалкенила, возможно замещенного С6-C18 арила, возможно замещенного C1-C18 гетероарила, возможно замещенного C1-C12 алкилокси, возможно замещенного C2-C12 алкенилокси, возможно замещенного C2-C12 алкинилокси, возможно замещенного С2-С10 гетероалкилокси, возможно замещенного С3-С12 циклоалкилокси, возможно замещенного С3-С12 циклоалкенилокси, возможно замещенного C2-C12 гетероциклоалкилокси, возможно замещенного C2-C12 гетероциклоалкенилокси, возможно замещенного С6-C18 арилокси, возможно замещенного C1-C18 гетероарилокси, возможно замещенного C1-C12 алкиламино, SR15, SO3H, SO2NR15R16, SO2R15, SONR15R16, SOR15, COR15, COOH, COOR16, CONR15R16, NR15COR16, NR15COOR16, NR15SO2R16, NR15CONR16R17, NR15R16 и ацила;
каждый R15, R16 и R17 независимо выбран из группы, состоящей из Н, возможно замещенного C1-C12 алкила, возможно замещенного С2-С10 гетероалкила, возможно замещенного C1-C12 галогеналкила, возможно замещенного C3-C12 циклоалкила, возможно замещенного C6-C18 арила и возможно замещенного C1-C18 гетероарила.
В некоторых вариантах реализации Y выбран из группы, состоящей из:
(a) -C1-5 алкил-O-,
(b) -С1-5 алкил-N(R10)-;
(c) -N(R10)- и
(d) -CON(R10)-,
где R10 такой, как определено выше.
В некоторых вариантах реализации Y выбран из группы, состоящей из -CH2NH-, -CH2O-, -NH-, -CONH-, -SO2NH- и -CH2CH2NH-. В некоторых вариантах реализации Y представляет собой -CH2NH-. В некоторых вариантах реализации Y представляет собой -CH2O-. В некоторых вариантах реализации Y представляет собой -NH-. В некоторых вариантах реализации Y представляет собой -CONH-. В некоторых вариантах реализации Y представляет собой -SO2NH-. В некоторых вариантах реализации Y представляет собой -CH2CH2NH-.
В некоторых вариантах реализации Y выбран из -CH2NH- и -CH2O-.
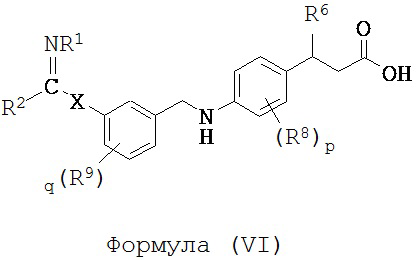
В некоторых вариантах реализации L представляет собой CO2H, r равен 1, Z представляет собой -(CR5R6)-, каждый из R3, R4, R5 представляет собой Н, кольцо В представляет собой возможно замещенную фенильную группу формулы (IV), кольцо А представляет собой возможно замещенную фенильную группу формулы (Va), a Y представляет собой -CH2NH-, т.е. предложены соединения формулы (VI)
или их фармацевтически приемлемые соли или пролекарства, где R1, R2, R6, R8, R9, q, p и X такие, как определено выше.
В некоторых вариантах реализации L представляет собой CO2H, r равен 1, Z представляет собой -(CR5R6)-, каждый из R3, R4, R5 и R6 представляет собой H, кольцо B представляет собой возможно замещенную фенильную группу формулы (IV), кольцо A представляет собой возможно замещенную фенильную группу формулы (Va), a Y представляет собой -CH2NH-, т.е. предложены соединения формулы (VIa)
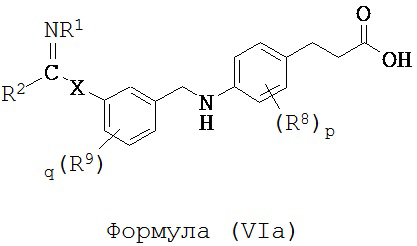
или их фармацевтически приемлемые соли или пролекарства, где R1, R2, R8, R9, q, p и X такие, как определено выше.
В некоторых вариантах реализации L представляет собой CO2H, r равен 1, Z представляет собой -(CR5R6)-, каждый R3, R4, R5 представляет собой H, кольцо B представляет собой возможно замещенную фенильную группу формулы (IV), кольцо A представляет собой возможно замещенную фенильную группу формулы (Vb), a Y представляет собой -CH2NH-, т.е. предложены соединения формулы (VIb)
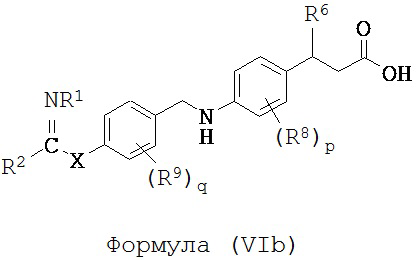
или их фармацевтически приемлемые соли или пролекарства, где R1, R2, R6, R8, R9, q, p и X такие, как определено выше.
В некоторых вариантах реализации L представляет собой CO2H, r равен 1, Z представляет собой -(CR5R6)-, каждый из R3, R4, R5 и R6 представляет собой H, кольцо В представляет собой возможно замещенную фенильную группу формулы (IV), кольцо А представляет собой возможно замещенную фенильную группу формулы (Vb), a Y представляет собой -CH2NH-, т.е. предложены соединения формулы (VIe)
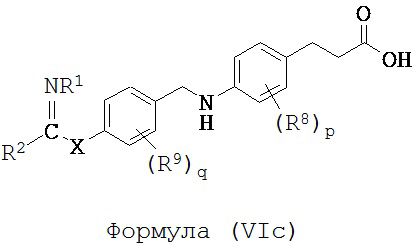
или их фармацевтически приемлемые соли или пролекарства, где R1, R2, R8, R9, q, p и X такие, как определено выше.
В некоторых вариантах реализации L представляет собой CO2H, r равен 1, Z представляет собой -(CR5R6)-, каждый из R3, R4, R5 представляет собой Н, кольцо В представляет собой возможно замещенную фенильную группу формулы (IV), кольцо А представляет собой возможно замещенную фенильную группу формулы (Va), a Y представляет собой -СН2О-, т.е. предложены соединения формулы (VId)
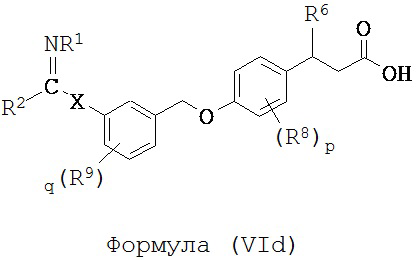
или их фармацевтически приемлемые соли или пролекарства, где R1, R2, R6, R8, R9, q, p и Х такие, как определено выше.
В некоторых вариантах реализации L представляет собой CO2H, r равен 1, Z представляет собой -(CR5R6)-, каждый из R3, R4, R5 и R6 представляет собой Н, кольцо В представляет собой возможно замещенную фенильную группу формулы (IV), кольцо А представляет собой возможно замещенную фенильную группу формулы (Va), a Y представляет собой -CH2O-, т.е. предложены соединения формулы (VIe)
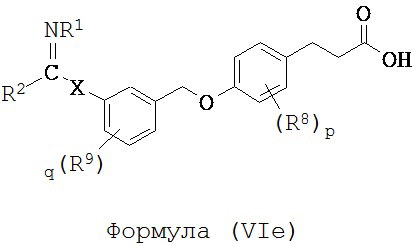
или их фармацевтически приемлемые соли или пролекарства, где R1, R2, R8, R9, q, p и Х такие, как определено выше.
В некоторых вариантах реализации L представляет собой СО2Н, r равен 1, Z представляет собой -(CR5R6)-, каждый из R3, R4, R5 представляет собой Н, кольцо В представляет собой возможно замещенную фенильную группу формулы (IV), кольцо А представляет собой возможно замещенную фенильную группу формулы (Vb), a Y представляет собой -CH2O-, т.е. предложены соединения формулы (VIf)

или их фармацевтически приемлемые соли или пролекарства, где R1, R2, R6, R8, R9, q, p и Х такие, как определено выше.
В некоторых вариантах реализации L представляет собой CO2H, r равен 1, Z представляет собой -(CR5R6)-, каждый из R3, R4, R5 и R6 представляет собой Н, кольцо В представляет собой возможно замещенную фенильную группу формулы (IV), кольцо А представляет собой возможно замещенную фенильную группу формулы (Vb), a Y представляет собой -CH2O-, т.е. предложены соединения формулы (VIg)
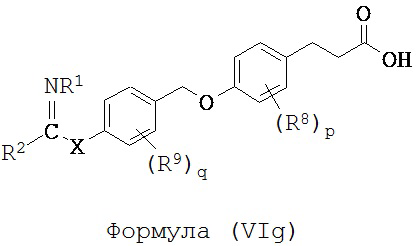
или их фармацевтически приемлемые соли или пролекарства, где R1, R2, R8, R9, q, p и Х такие, как определено выше.
В некоторых вариантах реализации Х выбран из группы, состоящей из:
(а) связи,
(b) -O-,
(c) -S-,
(d) -S(=O)-,
(e) -S(=O)2-,
(f) -N(R18)-,
(g) -C(R18)2-,
(h) -C(=R19)-,
(i) -ОС1-5 алкил-,
(j) -C1-5 алкил-O-,
(k) -С1-5 алкил-ОС1-5 алкил-,
(l) -N(R18)C1-5 алкил-,
(m) -С1-5 алкил-N(R18)-,
(n) -C1-5 алкил-N(R18)С1-5 алкил-,
(o) -N(R18)CO-,
(p) -N(R18)COC1-5 алкил-,
(q) -C1-5 алкил-N(R18)СО-,
(r) -C1-5 алкил-N(R18)COC1-5 алкил-,
(s) -CON(R18)-,
(t) -C1-5 алкил-CON(R18)-,
(u) -CON(R18)C1-5 алкил-,
(v) -C1-5 алкил-CON(R18)C1-5 алкил-,
(w) -SO2N(R18)-,
(x) -N(R18)SO2-,
(y) -С1-5 алкил-,
где каждый из алкильных фрагментов может быть дополнительно замещен,
где R18 выбран из группы, состоящей из H, возможно замещенного C1-C12 алкила, возможно замещенного С2-С10 гетероалкила, возможно замещенного С3-С12 циклоалкила, возможно замещенного С6-C18 арила и возможно замещенного C1-C18 гетероарила;
где R19 выбран из группы, состоящей из: О, S, NR20 и C(R21R22);
где каждый R20 выбран из группы, состоящей из H, OR23, возможно замещенного C1-C12 алкила, возможно замещенного Cl-C12 галогеналкила, возможно замещенного C2-C12 алкенила, возможно замещенного C2-C12 алкинила, возможно замещенного C1-C12 алкилокси, возможно замещенного C1-C12 галогеналкилокси, возможно замещенного С3-С10 гетероалкила, возможно замещенного С3-С12 циклоалкила, возможно замещенного С3-C12 циклоалкенила, возможно замещенного С2-С12 гетероциклоалкила, возможно замещенного C2-C12 гетероциклоалкенила, возможно замещенного С6-C18 арила и возможно замещенного C1-C18 гетероарила;
каждый R21 и R22 независимо выбран из группы, состоящей из Н, галогена, ОН, NO2, CN, SH, NH2, CF3, OCF3, возможно замещенного C1-C12 алкила, возможно замещенного C1-C12 галогеналкила, возможно замещенного C2-C12 алкенила, возможно замещенного C2-C12 алкинила, возможно замещенного C2-C12 гетероалкила, возможно замещенного С3-С12 циклоалкила, возможно замещенного С3-С12 циклоалкенила, возможно замещенного C2-C12 гетероциклоалкила, возможно замещенного C2-C12 гетероциклоалкенила, возможно замещенного С6-C18 арила, возможно замещенного C1-C18 гетероарила, возможно замещенного C1-C12 алкилокси, возможно замещенного C2-C12 алкенилокси, возможно замещенного C2-C12 алкинилокси, возможно замещенного С2-С10 гетероалкилокси, возможно замещенного С3-С12 циклоалкилокси, возможно замещенного С3-С12 циклоалкенилокси, возможно замещенного C2-C12 гетероциклоалкилокси, возможно замещенного C2-C12 гетероциклоалкенилокси, возможно замещенного С6-C18 арилокси, возможно замещенного C1-C18 гетероарилокси, возможно замещенного C1-C12 алкиламино, SR24, SO3H, SO2NR24R25, SO2R24, SONR24R25, SOR24, COR24, COOH, COOR24, CONR24R25, NR24COR25, NR24COOR25, NR24SO2R25, NR24CONR24R25, NR24R25 и ацила;
каждый R23, R24 и R25 независимо выбран из группы, состоящей из Н, возможно замещенного C1-C12 алкила, возможно замещенного С2-С10 гетероалкила, возможно замещенного С3-С12 циклоалкила, возможно замещенного C6-C18 арила и возможно замещенного C1-C18 гетероарила.
В некоторых вариантах реализации Х выбран из группы, состоящей из связи, O и CH2O. В некоторых вариантах реализации Х представляет собой связь. В некоторых вариантах реализации X представляет собой O. В некоторых вариантах реализации X представляет собой CH2O.
В некоторых вариантах реализации L представляет собой CO2H, r равен 1, Z представляет собой -(CR5R6)-, каждый из R3, R4, R5 представляет собой Н, кольцо В представляет собой возможно замещенную фенильную группу формулы (IV), кольцо А представляет собой возможно замещенную фенильную группу формулы (Vb), X представляет собой CH2O и Y представляет собой -CH2O-, т.е. предложены соединения формулы (VIIf)
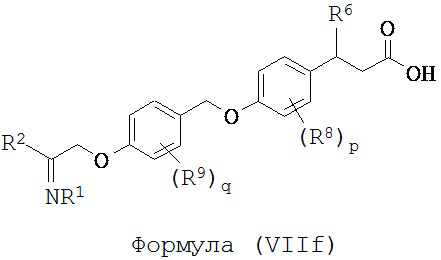
или их фармацевтически приемлемые соли или пролекарства, где R1, R2, R6, R8, R9, q и p такие, как определено выше.
В некоторых вариантах реализации L представляет собой CO2H, r равен 1, Z представляет собой -(CR5R6)-, каждый из R3, R4, R5 и R6 представляет собой Н, кольцо В представляет собой возможно замещенную фенильную группу формулы (IV), кольцо А представляет собой возможно замещенную фенильную группу формулы (Vb), X представляет собой -CH2O- и Y представляет собой -CH2O-, т.е. предложены соединения формулы (VIIg)
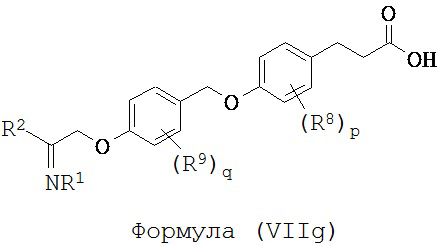
или их фармацевтически приемлемые соли или пролекарства, где R1, R2, R8, R9, q и p такие, как определено выше.
В некоторых вариантах реализации R1 представляет собой OR7. В некоторых вариантах реализации R7 выбран из группы, состоящей из Н, возможно замещенного C1-C12 алкила, возможно замещенного С2-С10 гетероалкила и возможно замещенного C1-C12 галогеналкила.
В некоторых вариантах реализации R7 представляет собой возможно замещенный C1-C12 алкил. В некоторых вариантах реализации R7 выбран из группы, состоящей из метила, этила, изопропила, пропила, 2-этилпропила, 3,3-диметилпропила, бутила, изобутила, 3,3-диметилбутила, 2-этилбутила, пентила, 2-метилпентила, гексила, гептила и октила.
В некоторых вариантах реализации R7 представляет собой возможно замещенную C1-C12 гетероалкильную группу. В некоторых вариантах реализации C2-C12 гетероалкильная группа выбрана из группы, состоящей из гидрокси-С1-С6 алкила, C1-С6 алкилокси-С1-С6 алкила, амино-С1-С6 алкила, C1-С6 алкиламино-С1-С6 алкила и ди(С1-С6 алкил)амино-С1-С6 алкила. Примеры возможных значений R7, представляющего собой C2-C12 гетероалкил, включают гидроксиметил, гидроксиэтил, гидроксипропил, гидроксибутил, гидроксипентил, метоксиметил, 2-метоксиэтил, 3-метоксипропил, 2-этоксиэтил, 3-этоксипропил, аминометил, 2-аминоэтил, 3-аминопропил, 4-аминобутил, 5-аминопентил, метиламинометил, 2-метиламиноэтил, 3-метиламинопропил, 4-метиламинобутил, 5-метиламинопентил, этиламинометил, 2-этиламиноэтил, 3-этиламинопропил, 4-этиламинобутил, 5-этиламинопентил, диметиламинометил, 2-диметиламиноэтил, 3-диметиламинопропил, 4-диметиламинобутил, 5-диметиламинопентил, диэтиламинометил, 2-диэтиламиноэтил, 3-диэтиламинопропил, 4-диэтиламинобутил и 5-диэтиламинопентил.
В некоторых вариантах реализации R1 выбран из группы, состоящей из Н, метокси; дифторметокси; трифторметокси; этокси; 2-циклопропилэтокси; 2,2,2-трифторэтокси; 2-(N,N-диметиламино)этокси; изопропокси; циклопропилокси; и 2-пропинилокси.
В некоторых вариантах реализации R2 представляет собой Н или выбран из группы, состоящей из возможно замещенного С6-C18 арила и возможно замещенного C1-C18 гетероарила.
В некоторых вариантах реализации R2 представляет собой возможно замещенную С1-С18 гетероарильную группу. В некоторых вариантах реализации возможно замещенная С1-С18 гетероарильная группа представляет собой моноциклическую гетероарильную группу. В некоторых вариантах реализации возможно замещенная C1-C18 гетероарильная группа представляет собой бициклическую гетероарильную группу.
В некоторых вариантах реализации R2 представляет собой возможно замещенное 5-членное гетероарильное кольцо. Примеры групп этого типа включают 2-фуран; 3-фуран; 2-тиофен; 3-тиофен; 1-пиррол; 2-пиррол; 3-пиррол; 2-оксазол; 4-оксазол; 5-оксазол; 2-тиазол; 4-тиазол; 5-тиазол; 1-имидазол; 2-имидазол; 4-имидазол; 5-имидазол; 1-пиразол; 3-пиразол; 4-пиразол; 5-пиразол; 3-изоксазол; 4-изоксазол; 5-изоксазол; 3-изотиазол; 4-изотиазол; 5-изотиазол; 4-(1,2,3-оксадиазол); 5-(1,2,3-оксадиазол); 3-(1,2,4-оксадиазол); 5-(1,2,4-оксадиазол); 1-(1,2,3-триазол); 4-(1,2,3-триазол); 5-(1,2,3-триазол); 1-(1,2,4-триазол); 3-(1,2,4-триазол); 5-(1,2,4-триазол); 3-(1,2,4-тиадиазол); 5-(1,2,4-тиадиазол); 2-(1,3,4-тиадиазол), 1-тетразол и 5-тетразол.
В некоторых вариантах реализации R2 представляет собой возможно замещенную C6-C18 арильную группу. В одном из вариантов реализации R2 представляет собой возможно замещенную фенильную группу. Заместители могут быть расположены при любом положении арильного кольца, которое может содержать заместители, что очевидно специалистам в данной области техники. Примеры подходящих возможно замещенных фенилов включают, но не ограничиваются ими, 2-метоксифенил, 3-метоксифенил, 4-метоксифенил, 2-трифторметилфенил, 3-трифторметилфенил, 4-трифторметилфенил, 2-хлорфенил, 3-хлорфенил, 4-хлорфенил, 4-бромфенил, 2-фторфенил, 3-фторфенил, 4-фторфенил, 4-гидроксифенил, 4-фенилфенил, 4-метилфенил, 2,4-дихлорфенил, 3,4-дихлорфенил, 2,5-дихлорфенил, 2,6-дифторфенил, 2-хлор-6-фторфенил, 3-фтор-4-хлорфенил, 3-метил-4-хлорфенил, 3-хлор-4-фторфенил, 3-хлор-4-метилфенил, 2-гидроксифенил, 3-гидроксифенил, 4-гидроксифенил, 4-этоксифенил, 3-феноксифенил, 4-феноксифенил, 2-метилфенил, 3-метилфенил, 4-метилфенил, 4-изопропилфенил, 4-цианофенил, 3,4-диметилфенил, 2,4-диметилфенил, 4-т-бутилфенил, 2,4-диметоксифенил и 3,4-метилендиоксифенил.
В некоторых вариантах реализации R2 представляет собой возможно замещенную фенильную группу формулы (IIX):
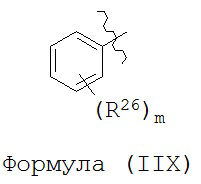
где каждый R26 независимо выбран из группы, состоящей из Н, галогена, ОН, NO2, CN, SH, NH2, CF3, OCF3, возможно замещенного C1-C12 алкила, возможно замещенного C1-C12 галогеналкила, возможно замещенного C2-C12 алкенила, возможно замещенного С2-C12 алкинила, возможно замещенного C2-C12 гетероалкила, возможно замещенного С3-С12 циклоалкила, возможно замещенного С3-С12 циклоалкенила, возможно замещенного C2-C12 гетероциклоалкила, возможно замещенного C2-C12 гетероциклоалкенила, возможно замещенного С6-C18 арила, возможно замещенного C1-C18 гетероарила, возможно замещенного C1-C12 алкилокси, возможно замещенного C2-C12 алкенилокси, возможно замещенного C2-C12 алкинилокси, возможно замещенного С2-С10 гетероалкилокси, возможно замещенного С3-С12 циклоалкилокси, возможно замещенного С3-С12 циклоалкенилокси, возможно замещенного C2-C12 гетероциклоалкилокси, возможно замещенного C2-C12 гетероциклоалкенилокси, возможно замещенного С6-C18 арилокси, возможно замещенного C1-C18 гетероарилокси, возможно замещенного C1-C12 алкиламино, SR7, SO2H, SO2NR7R7, SO2R7, SONR7R7, SOR7, COR7, COOH, COOR7, CONR7R7, NR7COR7, NR7COOR7, NR7SO2R7, NR7ONR7R7, NR7R7 и ацила; и
m представляет собой целое число, выбранное из группы, состоящей из 0, 1, 2, 3, 4 и 5.
В некоторых вариантах реализации L представляет собой СО2Н, r равен 1, Z представляет собой -(CR5R6)-, каждый из R3, R4, R5 представляет собой Н, кольцо В представляет собой возможно замещенную фенильную группу формулы (IV), кольцо А представляет собой возможно замещенную фенильную группу формулы (Vb), Х представляет собой СН2О-, Y представляет собой -СН2О-, а R2 представляет собой возможно замещенную фенильную группу формулы (IIX), т.е. предложены соединения формулы (IIXf)
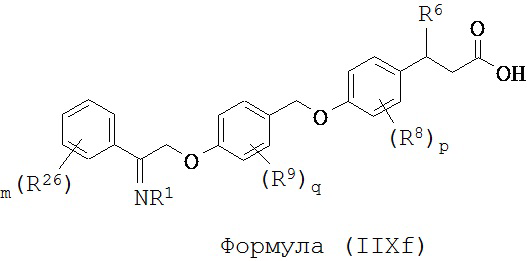
или их фармацевтически приемлемые соли или пролекарства, где R1, R6, R8, R9, R26, m, q и p такие, как определено выше.
В некоторых вариантах реализации L представляет собой СО2Н, r равен 1, Z представляет собой -(CR5R6)-, каждый из R3, R4, R5 и R6 представляет собой Н, кольцо В представляет собой возможно замещенную фенильную группу формулы (IV), кольцо А представляет собой возможно замещенную фенильную группу формулы (Vb), X представляет собой CH2O-, Y представляет собой -СН2О-, а R2 представляет собой возможно замещенную фенильную группу формулы (IIX), т.е. предложены соединения формулы (IIXg)
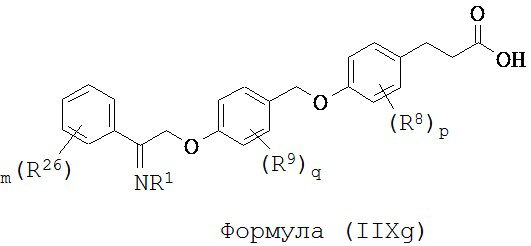
или их фармацевтически приемлемые соли или пролекарства, где R1, R8, R9, R26, m, q и p такие, как определено выше.
В результате наличия оксиминовой двойной связи соединения согласно настоящему изобретению могут существовать в виде Е или Z геометрического изомера. В некоторых вариантах реализации соединения имеют Е геометрию. В некоторых вариантах реализации изомеры имеют Z геометрию.
Большинство, если не все, переменных, обсуждаемых выше, могут быть замещены. Если переменная является возможно замещенной, то в некоторых вариантах реализации каждый возможный заместитель независимо выбран из группы, состоящей из галогена, =O, =S, -CN, -NO2, -CF3, -OCF3, алкила, алкенила, алкинила, галогеналкила, галогеналкенила, галогеналкинила, гетероалкила, циклоалкила, циклоалкенила, гетероциклоалкила, гетероциклоалкенила, арила, гетероарила, циклоалкилалкила, гетероциклоалкилалкила, гетероарилалкила, арилалкила, циклоалкилалкенила, гетероциклоалкилалкенила, арилалкенила, гетероарилалкенила, циклоалкилгетероалкила, гетероциклоалкилгетероалкила, арилгетероалкила, гетероарилгетероалкила, гидрокси, гидроксиалкила, алкилокси, алкилоксиалкила, алкилоксициклоалкила! алкилоксигетероциклоалкила, алкилоксиарила, алкилоксигетероарила, алкилоксикарбонила, алкиламинокарбонила, алкенилокси, алкинилокси, циклоалкилокси, циклоалкенилокси, гетероциклоалкилокси, гетероциклоалкенилокси, арилокси, фенокси, бензилокси, гетероарилокси, арилалкилокси, амино, алкиламино, ациламино, аминоалкила, ариламино, сульфониламино, сульфиниламино, сульфонила, алкилсульфонила, арилсульфонила, аминосульфонила, сульфинила, алкилсульфинила, арилсульфинила, амионосульфиниламиноалкила, -С(=O)ОН, -C(=O)Ra, -C(=O)ORa, C(=O)NRaRb, C(=NOH)Ra, C(=NRa)NRbRc, NRaRb, NRaC(=O)Rb, NRaC(=O)ORb, NRaC(=O)NRbRc, NRaC(=NRb)NRcRd, NRaSO2Rb, -SRa, SO2NRaRb, -ORa, OC(=O)NRaRb, ОС(=O)Ra и ацила,
где каждый Ra, Rb, Rc и Rd независимо выбран из группы, состоящей из Н, C1-C12 алкила, C1-C12 галогеналкила, С2-С12 алкенила, C2-C12 алкинила, C1-С10 гетероалкила, С3-C12 циклоалкила, С3-С12 циклоалкенила, C1-C12 гетероциклоалкила, C1-C12 гетероциклоалкенила, С6-C18 арила, C1-C18 гетероарила и ацила, или любые два или более Ra, Rb, Rc и Rd совместно с атомами, к которым они присоединены, образуют гетероциклическую систему колец, содержащую от 3 до 12 атомов в кольце.
В некоторых вариантах реализации каждый возможный заместитель независимо выбран из группы, состоящей из: F, Cl, Br, =O, =S, -CN, -NO2, алкила, алкенила, гетероалкила, галогеналкила, алкинила, арила, циклоалкила, гетероциклоалкила, гетероарила, гидрокси, гидроксиалкила, алкокси, алкиламино, аминоалкила, ациламино, фенокси, алкоксиалкила, бензилокси, алкилсульфонила, арилсульфонила, аминосульфонила, -C(O)ORa, СООН, SH и ацила.
В некоторых вариантах реализации каждый возможный заместитель независимо выбран из группы, состоящей из: F, Br, Cl, =O, =S, -CN, метила, трифторметила, этила, 2,2,2-трифторэтила, изопропила, пропила, 2-этилпропила, 3,3-диметилпропила, бутила, изобутила, 3,3-диметилбутила, 2-этилбутила, пентила, 2-метилпентила, пент-4-енила, гексила, гептила, октила, фенила, NH2, -NO2, фенокси, гидрокси, метокси, трифторметокси, этокси и метилендиокси.
В качестве альтернативы два возможных заместителя, расположенные при одном фрагменте, могут быть объединены с образованием конденсированного циклического заместителя, присоединенного к фрагменту, который может быть замещенным. Соответственно, термин «возможно замещенный» включает конденсированное кольцо, такое как циклоалкильное кольцо, гетероциклоалкильное кольцо, арильное кольцо или гетероарильное кольцо.
В дополнение к соединениям формулы I описанные варианты реализации также относятся к фармацевтически приемлемым солям, фармацевтически приемлемым N-оксидам, фармацевтически приемлемым пролекарствам и фармацевтически приемлемым метаболитам указанных соединений, а также к фармацевтически приемлемым солям указанных метаболитов.
Изобретение также относится к фармацевтическим композициям, содержащим соединение согласно настоящему изобретению и фармацевтически приемлемый носитель, разбавитель или вспомогательное вещество.
Согласно дополнительному аспекту в настоящем изобретении предложен способ предотвращения или лечения состояния, связанного с функцией рецептора GPR40 у млекопитающего, включающий введение эффективного количества соединения согласно настоящему изобретению.
Согласно еще одному дополнительному аспекту в изобретении предложено применение соединения согласно настоящему изобретению для получения лекарственного средства для лечения состояния, связанного с функцией рецептора GPR40.
Согласно еще одному дополнительному аспекту в изобретении предложено применение соединения согласно настоящему изобретению для лечения состояния, связанного с функцией рецептора GPR40.
В некоторых вариантах реализации состояние выбрано из группы, состоящей из когнитивных нарушений, остеопороза, воспалительных нарушений, диабета, ожирения, гипергликемии, непереносимости глюкозы, инсулинорезистентности, гиперинсулинемии, гиперхолестеринемии, гипертензии, гиперлипопротеинемии, гиперлипидемии, гипертриглицеридемии, дислипидемии, метаболического синдрома, синдрома X, сердечно-сосудистых заболеваний, атеросклероза, заболеваний почек, кетоацидоза, тромботических нарушений, нефропатии, диабетической нейропатии, диабетической ретинопатии, сексуальной дисфункции, дерматопатии, диспепсии, гипогликемии, рака и эдемы.
В некоторых вариантах реализации состояние представляет собой диабет. В некоторых вариантах реализации состояние представляет собой диабет II типа.
Эти и другие идеи изобретения приведены в настоящем описании.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В настоящем изобретении используют ряд терминов, которые хорошо известны специалистам в данной области техники. Тем не менее, для ясности ряду терминов будут даны определения.
Используемый в настоящем описании термин «незамещенный» означает, что заместители отсутствуют или единственными заместителями являются атомы водорода.
Термин «возможно замещенный», используемый в настоящем описании, означает, что группа может быть дополнительно замещена или не замещена или конденсирована или не конденсирована (с образованием конденсированной полициклической системы) с одним или более отличными от водорода заместителями. В конкретных вариантах реализации заместители представляют собой одну или более групп, независимо выбранных из группы, состоящей из галогена, =O, =S, -CN, -NO2, -CF3, -OCF3, алкила, алкенила, алкинила, галогеналкила, галогеналкенила, галогеналкинила, гетероалкила, циклоалкила, циклоалкенила, гетероциклоалкила, гетероциклоалкенила, арила, гетероарила, циклоалкилалкила, гетероциклоалкилалкила, гетероарилалкила, арилалкила; циклоалкилалкенила, гетероциклоалкилалкенила, арилалкенила, гетероарилалкенила, циклоалкилгетероалкила, гетероциклоалкилгетероалкила, арилгетероалкила, гетероарилгетероалкила, гидрокси, гидроксиалкила, алкилокси, алкилоксиалкила, алкилоксициклоалкила, алкилоксигетероциклоалкила, алкилоксиарила, алкилоксигетероарила, алкилоксикарбонила, алкиламинокарбонила, алкенилокси, алкинилокси, циклоалкилокси, циклоалкенилокси, гетероциклоалкилокси; гетероциклоалкенилокси, арилокси, фенокси, бензилокси, гетероарилокси, арилалкилоксй; амино, алкиламино, ациламино, аминоалкила, ариламино, сульфониламино, сульфиниламино, сульфонила, алкилсульфонила, арилсульфонила, аминосульфонила, сульфинила, алкилсульфинила, арилсульфинила, аминосульфиниламиноалкила, -С(=O)ОН, -C(=O)Ra, -C(=O)ORa, C(=O)NRaRb, C(=NOH)Ra, C(=NRa)NRbRc, NRaRb, NRaC(=O)Rb, NRaC(=O)ORb, NRaC(=O)NRbRc, NRaC(=NRb)NRcRd, NRaSO2Rb, -SRa, SO2NRaRb, -ORa, OC(=O)NRaRb, ОС(=O)Ra и ацила,
где каждый Ra, Rb, Rc и Rd независимо выбран из группы, состоящей из H, возможно замещенного С1-С12 алкила, возможно замещенного C1-C12 галогеналкила, возможно замещенного С2-С12 алкенила, возможно замещенного C2-C12 алкинила, возможно замещенного С2-С10 гетероалкила, возможно замещенного С3-С12 циклоалкила, возможно замещенного С3-С12 циклоалкенила, возможно замещенного C2-C12 гетероциклоалкила, C2-C12 гетероциклоалкенила, возможно замещенного С6-C18 арила, возможно замещенного С1-С18 гетероарила и ацила, или любые два или более Ra, Rb, Rc и Rd совместно с атомами, к которым они присоединены, образуют гетероциклическую систему колец, содержащую от 3 до 12 атомов в кольце.
В некоторых вариантах реализации каждый возможный заместитель независимо выбран из группы, состоящей из: галогена, =O, =S, -CN, -NO2, -CF3, -OCF3, алкила, алкенила, алкинила, галогеналкила, галогеналкенила, галогеналкинила, гетероалкила, циклоалкила, циклоалкенила, гетероциклоалкила, гетероциклоалкенила, арила, гетероарила, гидрокси, гидроксиалкила, алкилокси, алкилоксиалкила, алкилоксиарила, алкилоксигетероарила, алкенилокси, алкинилокси, циклоалкилокси, циклоалкенилокси, гетероциклоалкилокси, гетероциклоалкенилокси, арилокси, гетероарилокси, арилалкила, гетероарилалкила, арилалкилокси, амино, алкиламино, ациламино, аминоалкила, ариламино, сульфонила, алкилсульфонила, арилсульфонила, аминосульфонила, аминоалкила, -COOH, -SH и ацила.
Примеры особенно подходящих возможных заместителей включают F, Cl, Br, I, CH3, CH2CH3, OH, OCH3, CF3, OCF3, NO2, NH2 и CN.
В ряде приведенных ниже определений заместителей указано, что «группа может представлять собой терминальную группу или мостиковую группу», это означает, что использование термина охватывает ситуацию, когда группа представляет собой линкер между двумя фрагментами молекулы, а также когда группа представляет собой терминальный фрагмент. Если использовать в качестве примера термин «алкил», в некоторых публикациях используют термин «алкилен» для описания мостиковой группы, и, таким образом, в указанных публикациях существуют различия между терминами «алкил» (терминальная группа) и «алкилен» (мостиковая группа). В настоящей заявке указанные различия отсутствуют, и большинство групп могут представлять собой мостиковую группу или терминальную группу.
«Ацил» означает группу R-C(=O)-, в которой группа R может представлять собой алкил, циклоалкил, гетероциклоалкил, арил или гетероарильную группу, определенные в настоящем описании. Примеры ацилов включают ацетил и бензоил. Указанная группа может представлять собой терминальную группу или мостиковую группу. Если указанная группа представляет собой терминальную группу, то она связана с остатком молекулы через атом углерода в карбониле.
«Ациламино» означает группу R-C(=O)-NH-, в которой группа R может представлять собой алкил, циклоалкил, гетероциклоалкил, арил или гетероарильную группу, определенные в настоящем описании. Указанная группа может представлять собой терминальную группу или мостиковую группу. Если указанная группа представляет собой терминальную группу, то она связана с остатком молекулы через атом азота.
«Алкенил» в качестве группы или части группы означает алифатическую углеводородную группу, содержащую по меньшей мере одну углерод-углеродную двойную связь, которая может быть линейной или разветвленной и предпочтительно содержит 2-12 атомов углерода, более предпочтительно 2-10 атомов углерода, наиболее предпочтительно 2-6 атомов углерода в линейной цепи. Указанная группа может содержать несколько двойных связей в линейной цепи, и ориентация относительно каждой двойной связи может независимо представлять собой Е или Z ориентацию. Алкенильная группа предпочтительно представляет собой 1-алкенильную группу. Типичные алкенильные группы включают, но не ограничиваются ими, этенил, пропенил, бутенил, пентенил, гексенил, гептенил, октенил и ноненил. Указанная группа может представлять собой терминальную группу или мостиковую группу.
«Алкенилокси» относится к группе алкенил-O-, в которой алкенил такой, как определено в настоящем описании. Предпочтительными алкенилоксигруппами являются C1-С6 алкенилоксигруппы. Указанная группа может представлять собой терминальную группу или мостиковую группу. Если указанная группа представляет собой терминальную группу, то она связана с остатком молекулы через атом кислорода.
«Алкил» в качестве группы или части группы относится к линейной или разветвленной алифатической углеводородной группе, предпочтительно к C1-C12 алкилу, более предпочтительно к C1-С10 алкилу, наиболее предпочтительно к C1-С6 алкилу, если не указано иное. Примеры подходящих линейных и разветвленных C1-С6 алкильных заместителей включают метил, этил, н-пропил, 2-пропил, н-бутил, втор-бутил, т-бутил, гексил и т.д. Указанная группа может представлять собой терминальную группу или мостиковую группу.
«Алкиламино» включает моноалкиламино и диалкиламино, если не указано конкретно. «Моноалкиламино» означает группу алкил-NH-, в которой алкил такой, как определено в настоящем описании. «Диалкиламино» означает группу (алкил)2N-, в которой алкильные группы могут быть одинаковыми или различными, но каждая соответствует приведенному в настоящем описании определению алкила. Алкильная группа предпочтительно представляет собой C1-С6 алкильную группу. Указанная группа может представлять собой терминальную группу или мостиковую группу. Если указанная группа представляет собой терминальную группу, то она связана с остатком молекулы через атом азота.
«Алкиламинокарбонил» относится к группе формулы (алкил)x(Н)yNC(=O)-, в которой алкил такой, как определено в настоящем описании, x равен 1 или 2 и сумма x+y=2. Указанная группа может представлять собой терминальную группу или мостиковую группу. Если указанная группа представляет собой терминальную группу, то она связана с остатком молекулы через атом углерода карбонила.
«Алкилокси» относится к группе алкил-O-, в которой алкил такой, как определено в настоящем описании. Предпочтительная алкилоксигруппа представляет собой C1-С6 алкилокси. Примеры включают, но не ограничиваются ими, метокси и этокси. Указанная группа может представлять собой терминальную группу или мостиковую группу.
«Алкилоксиалкил» относится к группе алкилокси-алкил-, в которой алкилокси и алкильный фрагмент такие, как определено в настоящем описании. Указанная группа может представлять собой терминальную группу или мостиковую группу. Если указанная группа представляет собой терминальную группу, то она связана с остатком молекулы через алкильную группу.
«Алкилоксиарил» относится к группе алкилокси-арил-, в которой алкилокси и арильный фрагмент такие, как определено в настоящем описании. Указанная группа может представлять собой терминальную группу или мостиковую группу. Если указанная группа представляет собой терминальную группу, то она связана с остатком молекулы через арильную группу.
«Алкилоксикарбонил» относится к группе алкил-O-С(=O)-, в которой алкил такой, как определено в настоящем описании. Алкильная группа предпочтительно представляет собой C1-С6 алкильную группу. Примеры включают, но не ограничиваются ими, метоксикарбонил и этоксикарбонил. Указанная группа может представлять собой терминальную группу или мостиковую группу. Если указанная группа представляет собой терминальную группу, то она связана с остатком молекулы через атом углерода карбонила.
«Алкилоксициклоалкил» относится к группе алкилокси-циклоалкил-, в которой алкилокси и циклоалкильный фрагмент такие, как определено в настоящем описании. Указанная группа может представлять собой терминальную группу или мостиковую группу. Если указанная группа представляет собой терминальную группу, то она связана с остатком молекулы через циклоалкильную группу.
«Алкилоксигетероарил» относится к группе алкилокси-гетероарил-, в которой алкилокси и гетероарильный фрагмент такие, как определено в настоящем описании. Указанная группа может представлять собой терминальную группу или мостиковую группу. Если указанная группа представляет собой терминальную группу, то она связана с остатком молекулы через гетероарильную группу.
«Алкилоксигетероциклоалкил» относится к группе алкилокси-гетероциклоалкил-, в которой алкилокси и гетероциклоалкильный фрагмент такие, как определено в настоящем описании. Указанная группа может представлять собой терминальную группу или мостиковую группу. Если указанная группа представляет собой терминальную группу, то она связана с остатком молекулы через гетероциклоалкильную группу.
«Алкилсульфинил» означает группу алкил-S(=O)-, в которой алкил такой, как определено в настоящем описании. Алкильная группа предпочтительно представляет собой C1-С6 алкильную группу. Типичные алкилсульфинильные группы включают, но не ограничиваются ими, метилсульфинил и этилсульфинил. Указанная группа может представлять собой терминальную группу или мостиковую группу. Если указанная группа представляет собой терминальную группу, то она связана с остатком молекулы через атом серы.
«Алкилсульфонил» относится к группе алкил-S(=O)2-, в которой алкил такой, как определено в настоящем описании. Алкильная группа предпочтительно представляет собой C1-С6 алкильную группу. Примеры включают, но не ограничиваются ими, метилсульфонил и этилсульфонил. Указанная группа может представлять собой терминальную группу или мостиковую группу. Если указанная группа представляет собой терминальную группу, то она связана с остатком молекулы через атом серы.
«Алкинил» в качестве группы или части группы означает алифатическую углеводородную группу, содержащую углерод-углеродную тройную связь, и которая может быть линейной или разветвленной и предпочтительно содержит 2-12 атомов углерода, более предпочтительно 2-10 атомов углерода, наиболее предпочтительно 2-6 атомов углерода в линейной цепи. Типичные структуры включают, но не ограничиваются ими, этинил и пропинил. Указанная группа может представлять собой терминальную группу или мостиковую группу.
«Алкинилокси» относится к группе алкинил-O-, в которой алкинил такой, как определено в настоящем описании. Предпочтительными алкинилоксигруппами являются C1-С6 алкинилоксигруппы. Указанная группа может представлять собой терминальную группу или мостиковую группу. Если указанная группа представляет собой терминальную группу, то она связана с остатком молекулы через атом кислорода.
«Аминоалкил» означает группу NH2-алкил-, в которой алкильная группа такая, как определено в настоящем описании. Указанная группа может представлять собой терминальную группу или мостиковую группу. Если указанная группа представляет собой терминальную группу, то она связана с остатком молекулы через алкильную группу.
«Аминосульфонил» означает группу NH2-S(=O)2-. Указанная группа может представлять собой терминальную группу или мостиковую группу. Если указанная группа представляет собой терминальную группу, то она связана с остатком молекулы через атом серы.
«Арил» в качестве группы или части группы означает (i) возможно замещенный моноциклический или конденсированный полициклический ароматический карбоцикл (кольцевую структуру, все атомы кольца которой представляют собой атомы углерода), предпочтительно содержащий от 5 до 12 атомов в кольце. Примеры арильных групп включают фенил, нафтил и т.д.; (ii) возможно замещенный частично насыщенный бициклический ароматический карбоциклический фрагмент, в котором фенил и С5-7 циклоалкильная или С5-7 циклоалкенильная группа конденсированы с образованием циклической структуры, такой как тетрагидронафтил, инденил или инданил. Указанная группа может представлять собой терминальную группу или мостиковую группу. Как правило, арильная группа представляет собой С6-C18 арильную группу.
«Арилалкенил» означает группу арил-алкенил-, в которой арил и алкенил такие, как определено в настоящем описании. Типичные арилалкенильные группы включают фенилаллил. Указанная группа может представлять собой терминальную группу или мостиковую группу. Если указанная группа представляет собой терминальную группу, то она связана с остатком молекулы через алкенильную группу.
«Арилалкил» означает группу арил-алкил-, в которой арильный и алкильный фрагменты такие, как определено в настоящем описании. Предпочтительные арилалкильные группы содержат С1-5 алкильный фрагмент. Типичные арилалкильные группы включают бензил, фенэтил, 1-нафталинметил и 2-нафталинметил. Указанная группа может представлять собой терминальную группу или мостиковую группу. Если указанная группа представляет собой терминальную группу, то она связана с остатком молекулы через алкильную группу.
«Арилалкилокси» относится к группе арил-алкил-O-, в которой алкил и арил такие, как определено в настоящем описании. Указанная группа может представлять собой терминальную группу или мостиковую группу. Если указанная группа представляет собой терминальную группу, то она связана с остатком молекулы через атом кислорода.
«Ариламино» включает моноариламино и диариламино, если не указано конкретно. Моноариламино означает группу формулы арил-NH-, в которой арил такой, как определено в настоящем описании. Диариламино означает группу формулы (арил)2NH-, в которой арильные группы могут быть одинаковыми или различными, и каждая группа соответствует приведенному в настоящем описании определению арила. Указанная группа может представлять собой терминальную группу или мостиковую группу. Если указанная группа представляет собой терминальную группу, то она связана с остатком молекулы через атом азота.
«Арилгетероалкил» означает группу арил-гетероалкил-, в которой арильный и гетероалкильный фрагменты такие, как определено в настоящем описании. Указанная группа может представлять собой терминальную группу или мостиковую группу. Если указанная группа представляет собой терминальную группу, то она связана с остатком молекулы через гетероалкильную группу.
«Арилокси» относится к группе арил-O-, в которой арил такой, как определено в настоящем описании. Предпочтительно арилоксигруппа представляет собой C6-C18 арилокси, более предпочтительно С6-С10 арилокси. Указанная группа может представлять собой терминальную группу или мостиковую группу. Если указанная группа представляет собой терминальную группу, то она связана с остатком молекулы через атом кислорода.
«Арилсульфонил» означает группу арил-S(=O)2-, в которой арильная группа такая, как определено в настоящем описании. Указанная группа может представлять собой терминальную группу или мостиковую группу. Если указанная группа представляет собой терминальную группу, то она связана с остатком молекулы через атом серы.
«Связь» представляет собой связь атомов в соединении или молекуле. Связь может представлять собой простую связь, двойную связь или тройную связь.
«Циклоалкенил» означает неароматическую моноциклическую или мультициклическую систему колец, содержащую по меньшей мере одну углерод-углеродную двойную связь, предпочтительно содержащую 5-10 атомов углерода в кольце. Типичные моноциклические циклоалкенильные кольца включают циклопентенил, циклогексенил или циклогептенил. Циклоалкенильная группа может быть замещена одним или более заместителями. Циклоалкенильная группа, как правило, представляет собой С3-С12 циклоалкенильную группу. Указанная группа может представлять собой терминальную группу или мостиковую группу.
«Циклоалкил» относится к насыщенному моноциклическому или конденсированному или спирополициклическому карбоциклу, предпочтительно содержащему от 3 до 9 атомов углерода в кольце, такому как циклопропил, циклобутил, циклопентил, циклогексил и т.д., если не указано иное. Циклоалкил включает моноциклические системы, такие как циклопропил и циклогексил, бициклические системы, такие как декалин, и полициклические системы, такие как адамантан. Циклоалкильная группа, как правило, представляет собой С3-С12 циклоалкильную группу. Указанная группа может представлять собой терминальную группу или мостиковую группу.
«Циклоалкилалкил» означает группу циклоалкил-алкил-, в которой циклоалкильный и алкильный фрагменты такие, как определено в настоящем описании. Типичные моноциклоалкилалкильные группы включают циклопропилметил, циклопентилметил, циклогексилметил и циклогептилметил. Указанная группа может представлять собой терминальную группу или мостиковую группу. Если указанная группа представляет собой терминальную группу, то она связана с остатком молекулы через алкильную группу.
«Циклоалкилалкенил» означает группу циклоалкил-алкенил-, в которой циклоалкильный и алкенильный фрагменты такие, как определено в настоящем описании. Указанная группа может представлять собой терминальную группу или мостиковую группу. Если указанная группа представляет собой терминальную группу, то она связана с остатком молекулы через алкенильную группу.
«Циклоалкилгетероалкил» означает группу циклоалкил-гетероалкил-, в которой циклоалкильный и гетероалкильный фрагменты такие, как определено в настоящем описании. Указанная группа может представлять собой терминальную группу или мостиковую группу. Если указанная группа представляет собой терминальную группу, то она связана с остатком молекулы через гетероалкильную группу.
«Циклоалкилокси» относится к группе циклоалкил-O-, в которой циклоалкил такой, как определено в настоящем описании. Предпочтительно циклоалкилокси представляет собой C1-С6 циклоалкилокси. Примеры включают, но не ограничиваются ими, циклопропанокси и циклобутанокси. Указанная группа может представлять собой терминальную группу или мостиковую группу. Если указанная группа представляет собой терминальную группу, то она связана с остатком молекулы через атом кислорода.
«Циклоалкенилокси» относится к группе циклоалкенил-O-, в которой циклоалкенил такой, как определено в настоящем описании. Предпочтительно Циклоалкенилокси представляет собой C1-С6 Циклоалкенилокси. Указанная группа может представлять собой терминальную группу или мостиковую группу. Если указанная группа представляет собой терминальную группу, то она связана с остатком молекулы через атом кислорода.
«Галогеналкил» относится к алкильной группе, определенной в настоящем описании, в которой один или более атомов водорода заменены на атом галогена, выбранного из группы, состоящей из фтора, хлора, брома и йода. Галогеналкильная группа, как правило, имеет формулу CnH(2n+1-m)Xm, где каждый Х независимо выбран из группы, состоящей из F, Cl, Br и I. В группах этого типа n, как правило, равен от 1 до 10, более предпочтительно от 1 до 6, наиболее предпочтительно от 1 до 3; m, как правило, равен от 1 до 6, более предпочтительно от 1 до 3. Примеры галогеналкилов включают фторметил, дифторметил и трифторметил.
«Галогеналкенил» относится к алкенильной группе, определенной в настоящем описании, в которой один или более атомов водорода заменены на атом галогена, независимо выбранного из группы, состоящей из F, Cl, Br и I.
«Галогеналкинил» относится к алкинильной группе, определенной в настоящем описании, в которой один или более атомов водорода заменены на атом галогена, независимо выбранного из группы, состоящей из F, Cl, Br и I.
«Галоген» представляет собой хлор, фтор, бром или йод.
«Гетероалкил» относится к линейной или разветвленной алкильной группе, предпочтительно содержащей от 2 до 12 атомов углерода, более предпочтительно от 2 до 6 атомов углерода в цепи, в которой один или более атомов углерода (и любых связанных с ними атомов водорода) независимо заменены на гетероатомную группу, выбранную из S, О, P и NR', где R' выбран из группы, состоящей из Н, возможно замещенного С1-С12 алкила, возможно замещенного С3-С12 циклоалкила, возможно замещенного С6-C18 арила и возможно замещенного C1-C18 гетероарила. Типичные гетероалкилы включают простые алкильные эфиры, вторичные и третичные алкиламины, амиды, алкилсульфиды и т.д. Примеры гетероалкилов также включают гидрокси-С1-С6 алкил, C1-С6 алкилокси-С1-С6 алкил, амино-С1-С6 алкил, C1-С6 алкиламино-С1-С6 алкил и ди(С1-С6 алкил)амино-С1-С6 алкил. Указанная группа может представлять собой терминальную группу или мостиковую группу.
«Гетероалкилокси» относится к группе гетероалкил-O-, в которой гетероалкил такой, как определено в настоящем описании. Предпочтительно гетероалкилокси представляет собой С2-С6 гетероалкилокси. Указанная группа может представлять собой терминальную группу или мостиковую группу.
«Гетероарил» отдельно или как часть группы относится к группам, содержащим ароматическое кольцо (предпочтительно 5- или 6-членное ароматическое кольцо), содержащим один или более гетероатомов в качестве атомов кольца в ароматическом кольце, при этом оставшиеся атомы кольца представляют собой атомы углерода. Подходящие гетероатомы включают азот, кислород и серу. Примеры гетероарилов включают тиофен, бензотиофен, бензофуран, бензимидазол, бензоксазол, бензотиазол, бензизотиазол, нафто[2,3-b]тиофен, фуран, изоиндолизин, ксантол, феноксатин, пиррол, имидазол, пиразол, пиридин, пиразин, пиримидин, пиридазин, тетразол, индол, изоиндол, 1H-индазол, пурин, хинолин, изохинолин, фталазин, нафтиридин, хиноксалин, циннолин, карбазол, фенантридин, акридин, феназин, тиазол, изотиазол, фенотиазин, оксазол, изоксазол, фуразан, феноксазин, 2-, 3- или 4-пиридил, 2-, 3-, 4-, 5- или 8-хинолил, 1-, 3-, 4- или 5-изохинолил, 1-, 2- или 3-индолил и 2- или 3-тиенил. Гетероарильная группа, как правило, представляет собой C1-C18 гетероарильную группу. Указанная группа может представлять собой терминальную группу или мостиковую группу.
«Гетероарилалкил» означает группу гетероарил-алкил-, в которой гетероарильный и алкильный фрагменты такие, как определено в настоящем описании. Предпочтительные гетероарилалкильные группы содержат низший алкильный фрагмент. Типичные гетероарилалкильные группы включают пиридилметил. Указанная группа может представлять собой терминальную группу или мостиковую группу. Если указанная группа представляет собой терминальную группу, то она связана с остатком молекулы через алкильную группу.
«Гетероарилалкенил» означает группу гетероарил-алкенил-, в которой гетероарильный и алкенильный фрагменты такие, как определено в настоящем описании. Указанная группа может представлять собой терминальную группу или мостиковую группу. Если указанная группа представляет собой терминальную группу, то она связана с остатком молекулы через алкенильную группу.
«Гетероарилгетероалкил» означает группу гетероарил-гетероалкил-, в который гетероарильный и гетероалкильный фрагменты такие, как определено в настоящем описании. Указанная группа может представлять собой терминальную группу или мостиковую группу. Если указанная группа представляет собой терминальную группу, то она связана с остатком молекулы через гетероалкильную группу.
«Гетероарилокси» относится к группе гетероарил-O-, в которой гетероарил такой, как определено в настоящем описании. Предпочтительно гетероарилокси представляет собой С1-С18 гетероарилокси. Указанная группа может представлять собой терминальную группу или мостиковую группу. Если указанная группа представляет собой терминальную группу, то она связана с остатком молекулы через атом кислорода.
«Гетероцикл» относится к насыщенной, частично ненасыщенной или полностью ненасыщенной моноциклической, бициклической или полициклической системе колец, содержащей по меньшей мере один гетероатом, выбранный из группы, состоящей из азота, серы и кислорода в качестве атома кольца. Примеры гетероциклических фрагментов включают гетероциклоалкил, гетероциклоалкенил и гетероарил.
«Гетероциклоалкенил» относится к гетероциклоалкильной группе, определенной в настоящем описании, содержащей по меньшей мере одну двойную связь. Гетероциклоалкенильная группа, как правило, представляет собой С2-С12 гетероциклоалкенильную группу. Указанная группа может представлять собой терминальную группу или мостиковую группу.
«Гетероциклоалкил» относится к насыщенному моноциклическому, бициклическому или полициклическому кольцу, содержащему по меньшей мере один гетероатом, выбранный из азота, серы, кислорода, предпочтительно от 1 до 3 гетероатомов по меньшей мере в одном кольце. Каждое кольцо предпочтительно является 3-10-членным, более предпочтительно 4-7-членным. Примеры подходящих гетероциклоалкильных заместителей включают пирролидил, тетрагидрофурил, тетрагидротиофуранил, пиперидил, пиперазил, тетрагидропиранил, морфилино, 1,3-диазапан, 1,4-диазапан, 1,4-оксазепан и 1,4-оксатиапан. Гетероциклоалкильная группа, как правило, представляет собой C2-C12 гетероциклоалкильную группу. Указанная группа может представлять собой терминальную группу или мостиковую группу.
«Гетероциклоалкилалкил» относится к группе гетероциклоалкил-алкил-, в которой гетероциклоалкильный и алкильный фрагменты такие, как определено в настоящем описании. Типичные гетероциклоалкилалкильные группы включают (2-тетрагидрофурил)метил, (2-тетрагидротиофуранил)метил. Указанная группа может представлять собой терминальную группу или мостиковую группу. Если указанная группа представляет собой терминальную группу, то она связана с остатком молекулы через алкильную группу.
«Гетероциклоалкилалкенил» относится к группе гетероциклоалкил-алкенил-, в которой гетероциклоалкильный и алкенильный фрагменты такие, как определено в настоящем описании. Указанная группа может представлять собой терминальную группу или мостиковую группу. Если указанная группа представляет собой терминальную группу, то она связана с остатком молекулы через алкенильную группу.
«Гетероциклоалкилгетероалкил» означает группу гетероциклоалкил-гетероалкил-, в которой гетероциклоалкильный и гетероалкильный фрагменты такие, как определено в настоящем описании. Указанная группа может представлять собой терминальную группу или мостиковую группу. Если указанная группа представляет собой терминальную группу, то она связана с остатком молекулы через гетероалкильную группу.
«Гетероциклоалкилокси» относится к группе гетероциклоалкил-O-, в которой гетероциклоалкил такой, как определено в настоящем описании. Предпочтительно гетероциклоалкилокси представляет собой C1-С6 гетероциклоалкилокси. Указанная группа может представлять собой терминальную группу или мостиковую группу. Если указанная группа представляет собой терминальную группу, то она связана с остатком молекулы через атом кислорода.
«Гетероциклоалкенилокси» относится к группе гетероциклоалкенил-O-, в которой гетероциклоалкенил такой, как определено в настоящем описании. Предпочтительно гетероциклоалкенилокси представляет собой C1-С6 гетероциклоалкенилокси. Указанная группа может представлять собой терминальную группу или мостиковую группу. Если указанная группа представляет собой терминальную группу, то она связана с остатком молекулы через атом кислорода.
«Гидроксиалкил» относится к алкильной группе, определенной в настоящем описании, в которой один или более атомов водорода заменены на ОН группу. Гидроксиалкильная группа, как правило, имеет формулу CnH(2n+1-x)(OH)x. В группах этого типа n, как правило, равен от 1 до 10, более предпочтительно от 1 до 6, наиболее предпочтительно от 1 до 3; x, как правило, равен от 1 до 6, более предпочтительно от 1 до 3.
«Сульфинил» означает группу R-S(=O)-, в которой группа R может представлять собой ОН, алкил, циклоалкил, гетероциклоалкил, арил или гетероарильную группу, определенные в настоящем описании. Указанная группа может представлять собой терминальную группу или мостиковую группу. Если указанная группа представляет собой терминальную группу, то она связана с остатком молекулы через атом серы.
«Сульфиниламино» означает группу R-S(=O)-NH-, в которой группа R может представлять собой ОН, алкил, циклоалкил, гетероциклоалкил, арил или гетероарильную группу, определенные в настоящем описании. Указанная группа может представлять собой терминальную группу или мостиковую группу. Если указанная группа представляет собой терминальную группу, то она связана с остатком молекулы через атом азота.
«Сульфонил» означает группу R-S(=O)2-, в которой группа R может представлять собой ОН, алкил, циклоалкил, гетероциклоалкил, арил или гетероарильную группу, определенные в настоящем описании. Указанная группа может представлять собой терминальную группу или мостиковую группу. Если указанная группа представляет собой терминальную группу, то она связана с остатком молекулы через атом серы.
«Сульфониламино» означает группу R-S(=O)2-NH-. Указанная группа может представлять собой терминальную группу или мостиковую группу. Если указанная группа представляет собой терминальную группу, то она связана с остатком молекулы через атом азота.
Понимают, что в число соединений Формулы (I) включены изомерные формы, включая диастереомеры, энантиомеры, таутомеры и геометрические изомеры, «Е» или «Z» конфигурационные изомеры или смеси Е и Z изомеров. Также понимают, что некоторые изомерные формы, такие как диастереомеры, энантиомеры и геометрические изомеры специалисты в данной области техники могут разделять при помощи физических и/или химических способов. Для соединений, в которых возможна геометрическая изомерия, заявитель изобразил предполагаемый изомер соединения, несмотря на это, очевидно, что верную структуру может иметь другой изомер.
Некоторые соединения согласно описанным вариантам реализации могут существовать в виде отдельных стереоизомеров, рацематов и/или смесей энантиомеров и/или диастереомеров. Все указанные отдельные стереоизомеры, рацематы и смеси включены в объем описываемого и заявленного объекта изобретения.
В дополнение, предполагается, что Формула (I) охватывает, в тех случаях, где это возможно, сольватированные, а также несольватированные формы соединений. Таким образом, каждая формула включает соединения, имеющие обозначенную структуру, включая гидратированные, а также негидратированные формы.
Термин «фармацевтически приемлемые соли» относится к солям, которые сохраняют желаемую биологическую активность вышеуказанных соединений, и включает фармацевтически приемлемые соли присоединения кислот и соли присоединения оснований. Подходящие фармацевтически приемлемые соли присоединения кислот к соединениям Формулы (I) можно получать из неорганической кислоты или органической кислоты. Примерами указанных неорганических кислот являются хлороводородная, серная и фосфорная кислоты. Соответствующие органические кислоты могут быть выбраны из алифатических, циклоалифатических, ароматических, гетероциклических карбоновых и сульфоновых органических кислот, примерами которых являются муравьиная, уксусная, пропановая, янтарная, гликолевая, глюконовая, молочная, яблочная, винная, лимонная, фумаровая, малеиновая, алкилсульфокислоты, арилсульфокислоты. Дополнительную информацию о фармацевтически приемлемый солях можно найти в Remington's Pharmaceutical Sciences, 19th Edition, Mack Publishing Co., Easton, PA 1995. В случае твердых агентов специалисты в данной области техники понимают, что соединения согласно настоящему изобретению, агенты и соли могут существовать в различных кристаллических или полиморфных формах, все из которых включены в объем настоящего изобретения и конкретной указанной формулы.
«Пролекарство» означает соединение, которое претерпевает конверсию в соединение формулы (I) в биологической системе, как правило, в результате метаболизма (например, гидролиза, восстановления или окисления). Например, сложноэфирное пролекарство соединения формулы (I), содержащего гидроксильную группу, может превращаться под действием гидролиза in vivo в исходную молекулу. Подходящими сложными эфирами соединений формулы (I), содержащих гидроксильную группу, являются, например, ацетаты, цитраты, лактаты, тартраты, малонаты, оксалаты, салицилаты, пропионаты, сукцинаты, фумараты, малеаты, метилен-бис-β-гидроксинафтоаты, гестизаты, изетионаты, ди-n-толуолтартраты, метансульфонаты, этансульфонаты, бензолсульфонаты, n-толуолсульфонаты, циклогексилсульфаматы и хинаты. В качестве другого примера сложноэфирное пролекарство соединения формулы (I), содержащего карбоксигруппу, может превращаться под действием гидролиза in vivo в исходную молекулу. (Примеры сложноэфирных пролекарств описаны. J. Leinweber, Drug Metab. Res., 18:379, 1987). Аналогично, ацильное пролекарство соединения формулы (I), содержащего аминогруппу, может превращаться под действием гидролиза in vivo в исходную молекулу (Множество примеров пролекарств для указанных и других функциональных групп, включая амины, описаны в Prodrugs: Challenges and Rewards (Parts 1 and 2); Ed V. Stella, R. Borchardt, M. Hageman, R. Oliyai, H. Maag and J Tilley; Springer, 2007).
Термин «терапевтически эффективное количество» или «эффективное количество» представляет собой количество, достаточное для достижения благоприятных или желаемых клинических результатов. Эффективное количество можно вводить за один или более раз. Эффективное количество, как правило, является достаточным для облегчения, ослабления, стабилизации, обращения вспять, замедления или задержки прогрессирования болезненного состояния.
Специфические соединения согласно настоящему изобретению включают:
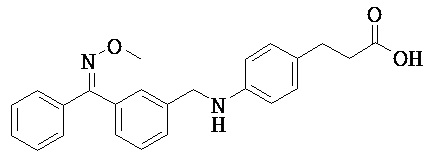
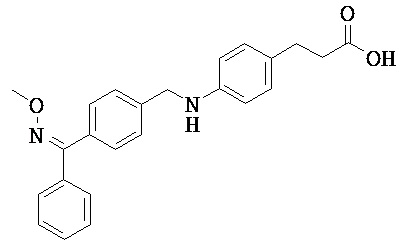
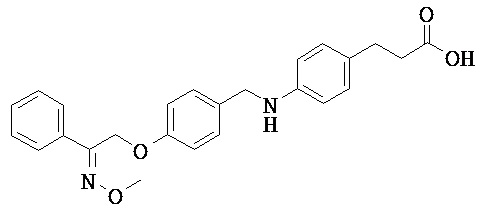
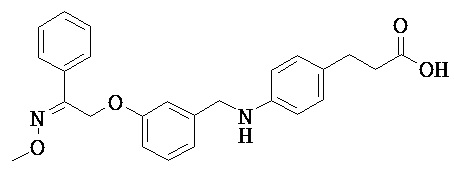
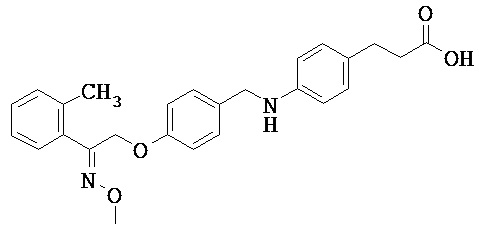
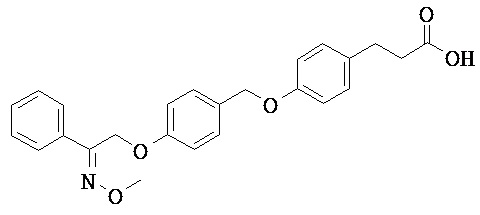
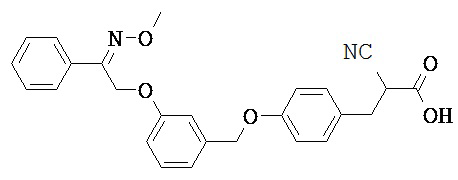
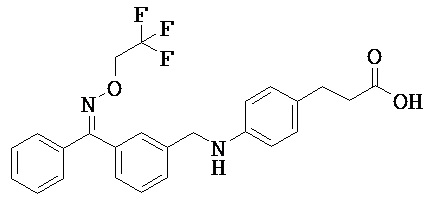
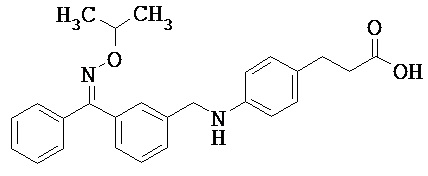
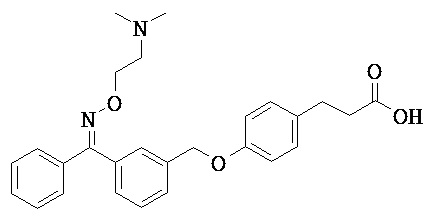
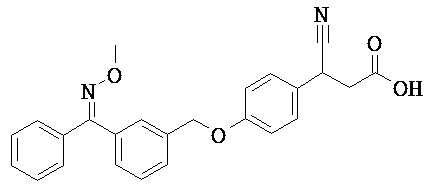
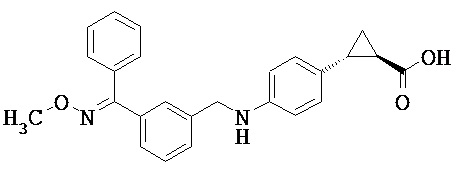
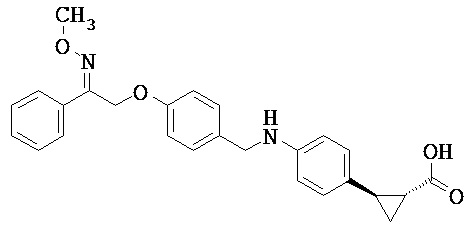
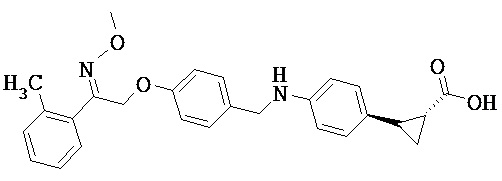
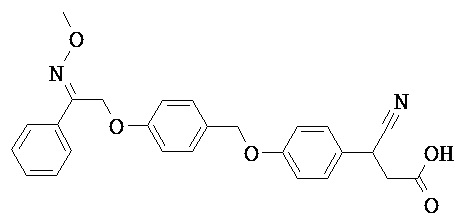
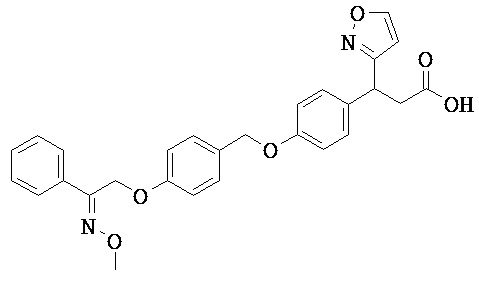
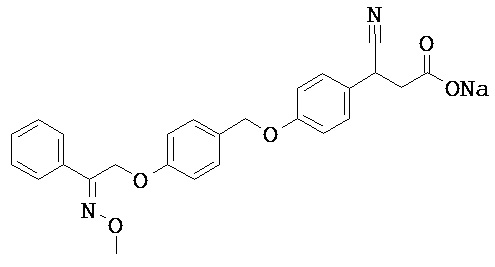
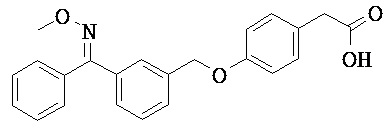
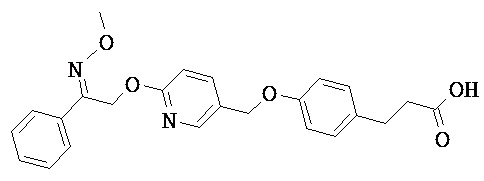
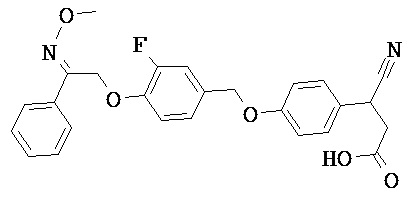
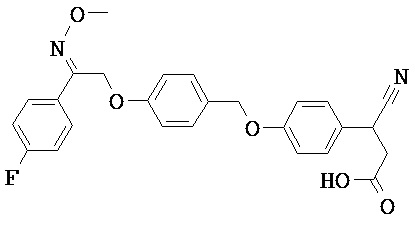
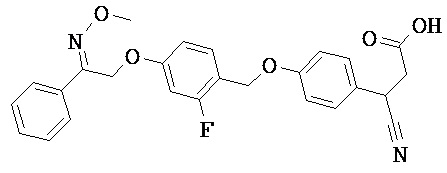
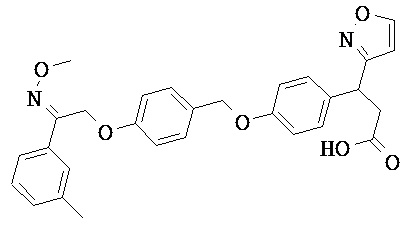
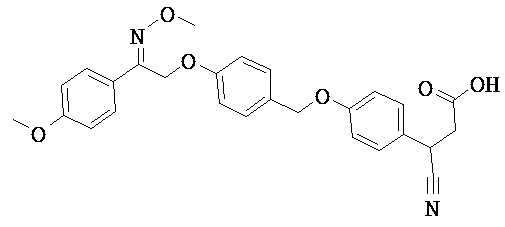
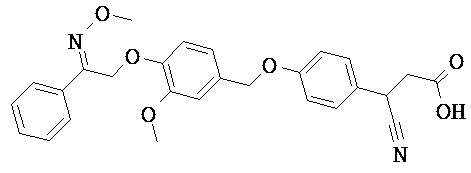
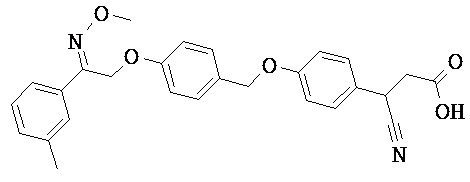
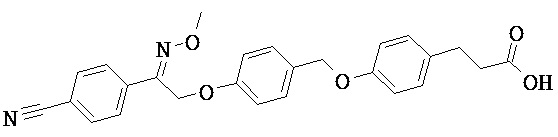
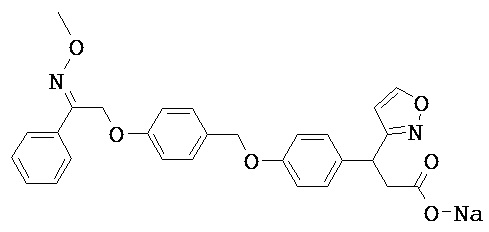
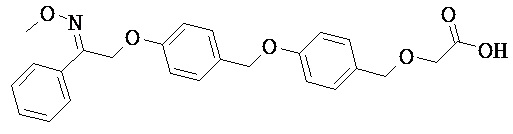
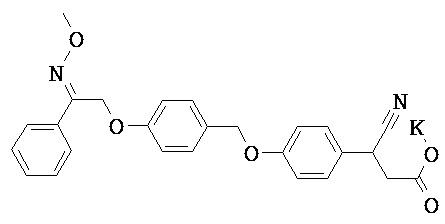
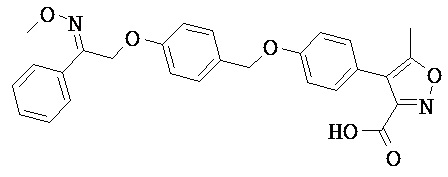
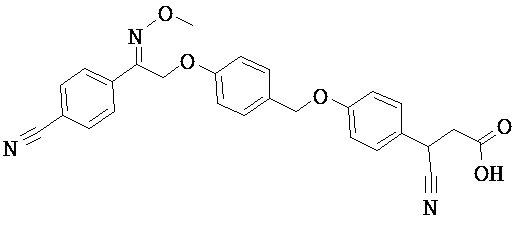
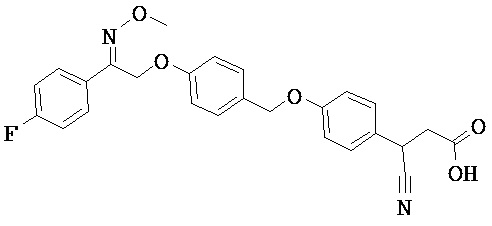
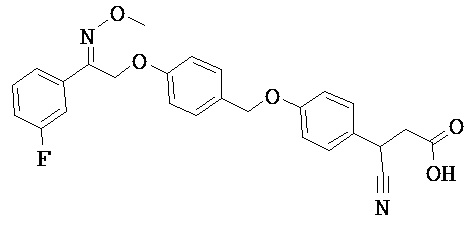
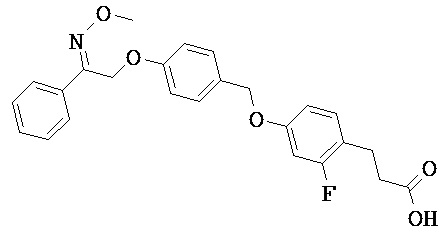
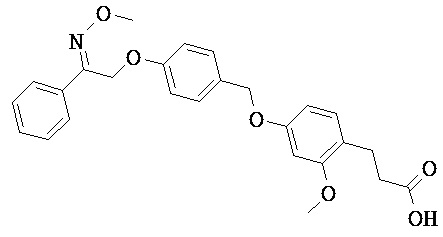
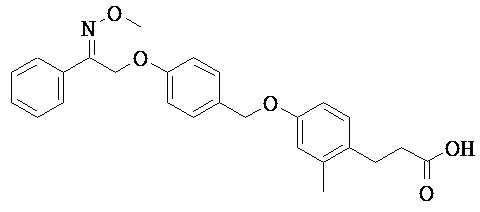
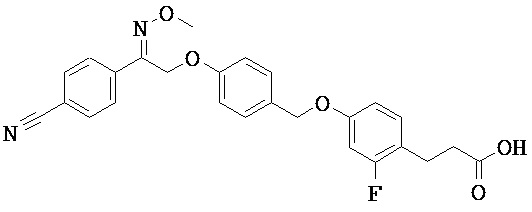
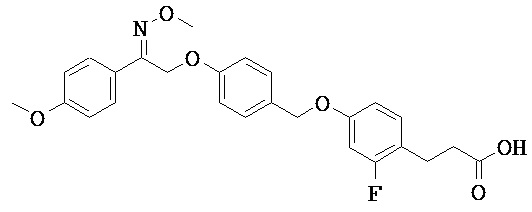
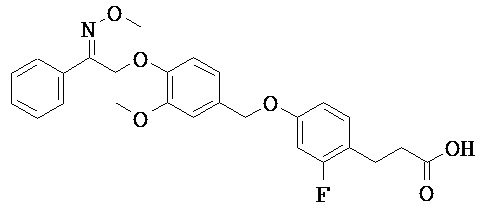
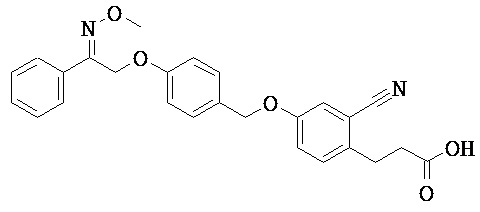
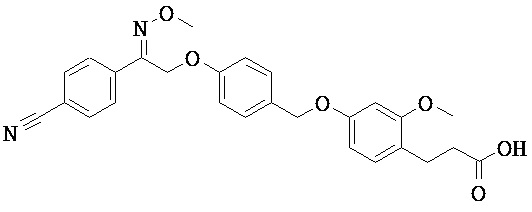
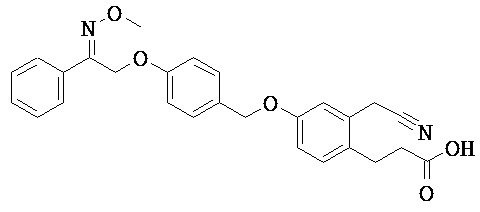
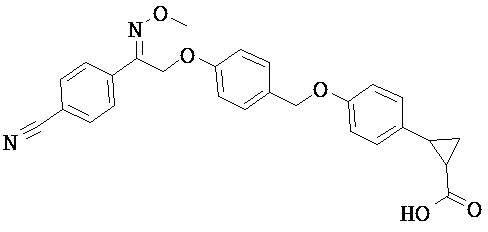
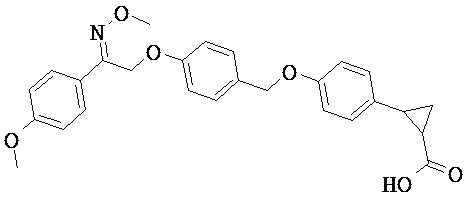
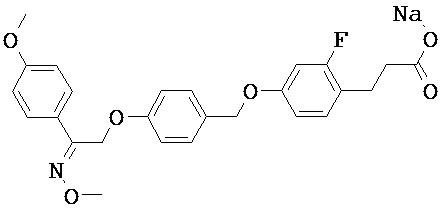
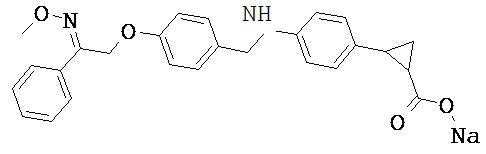
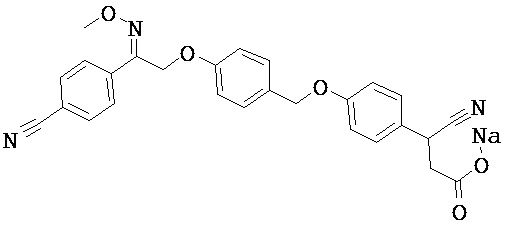
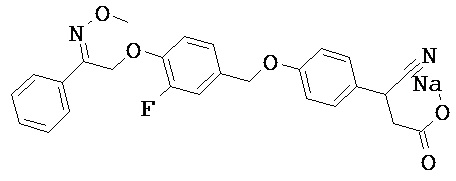
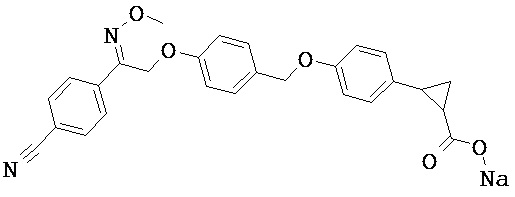
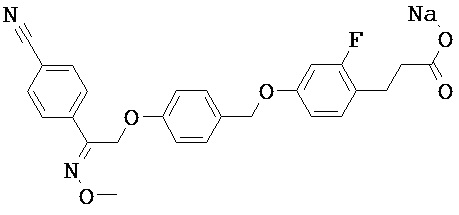
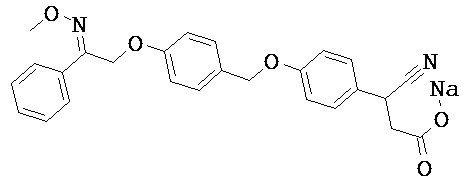

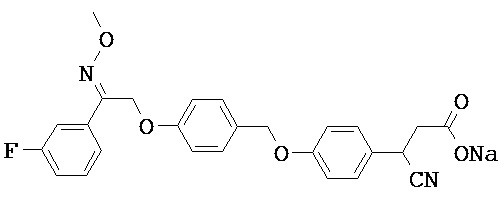
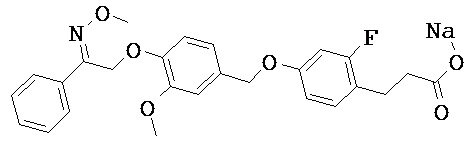
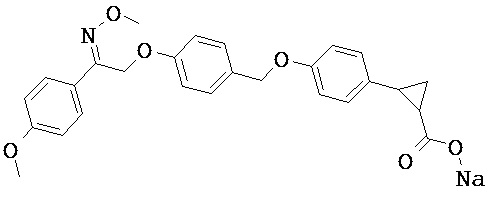
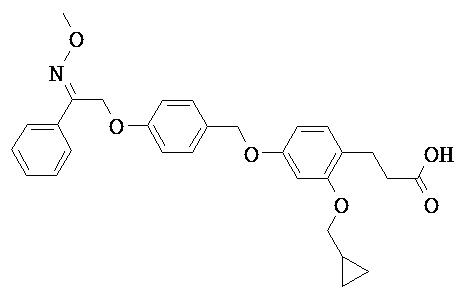
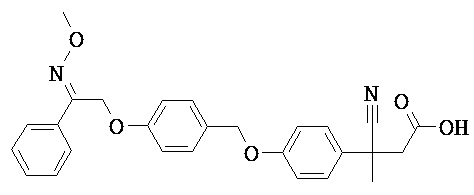
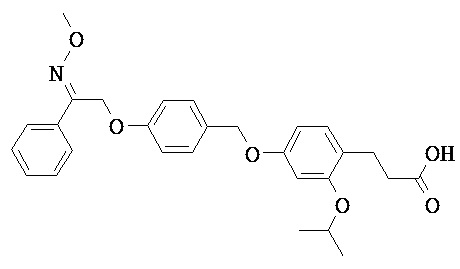
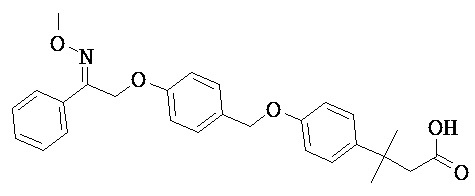
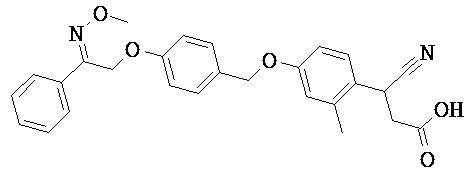
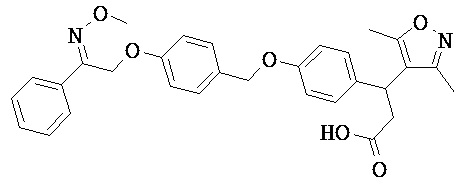
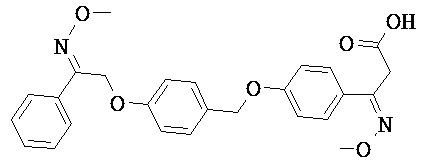
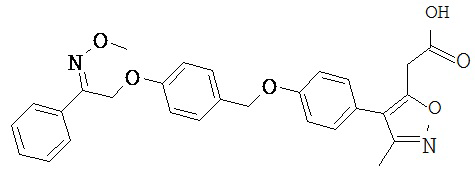
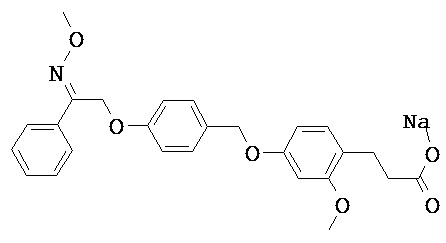
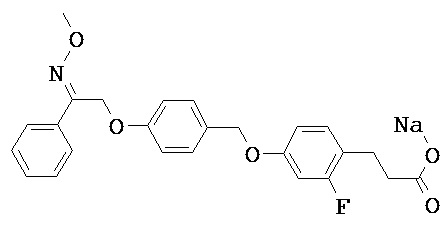
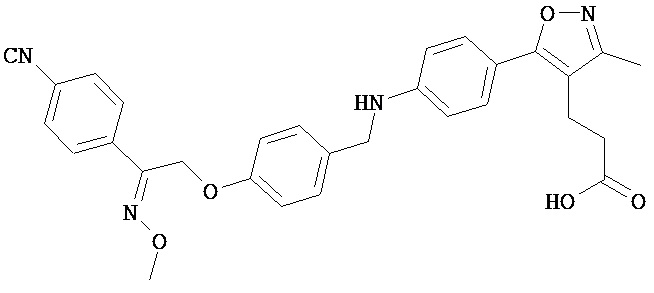
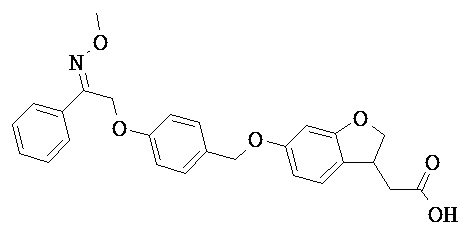
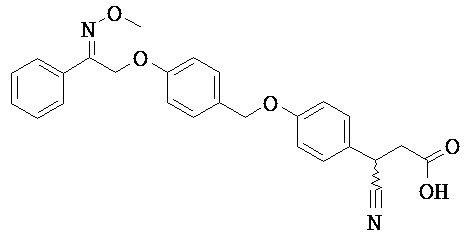
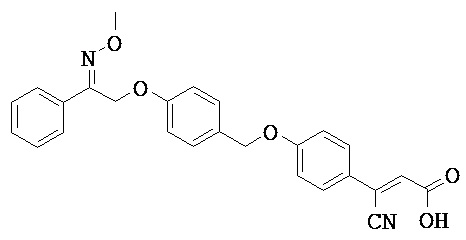
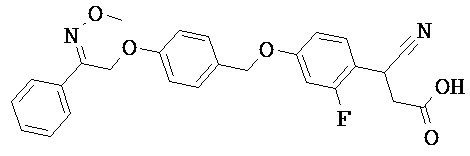

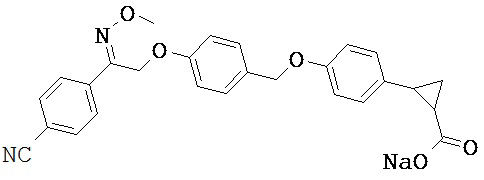
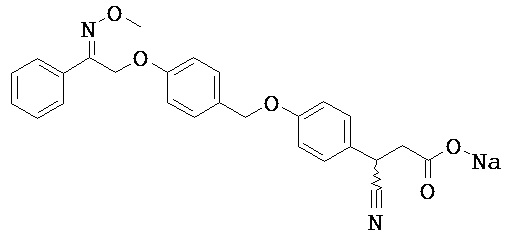
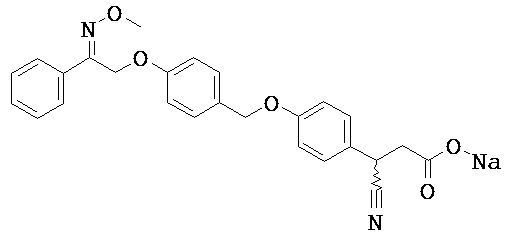
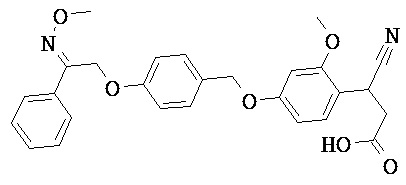
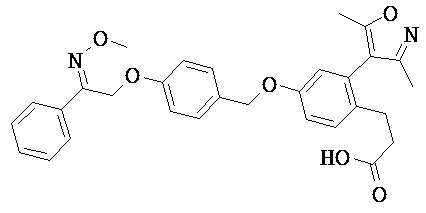
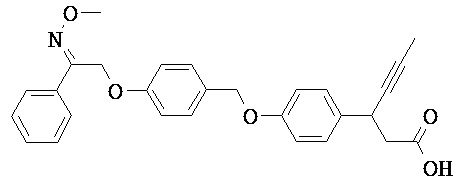
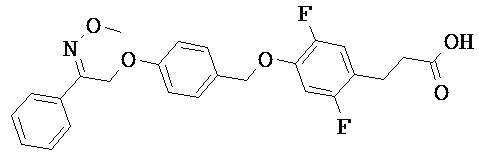
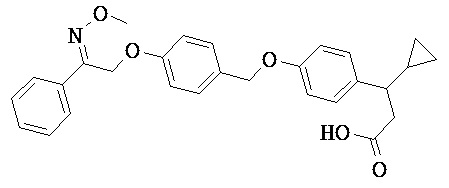
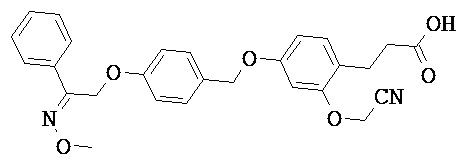
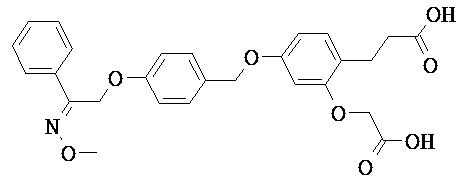
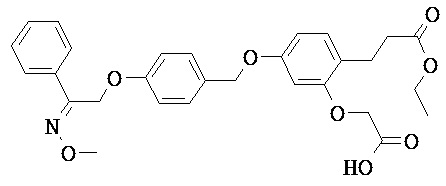
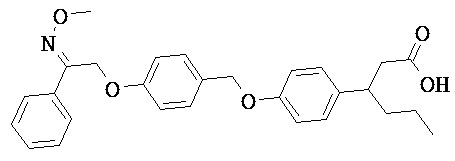
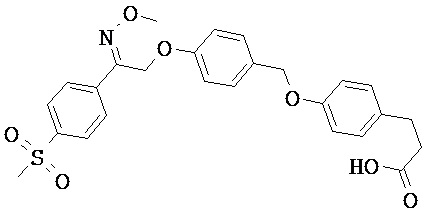
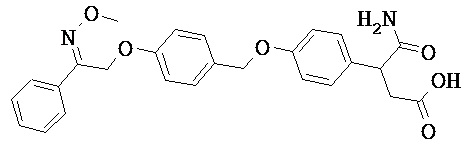
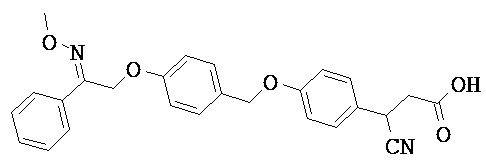

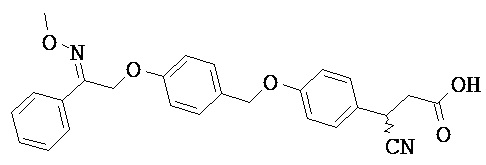
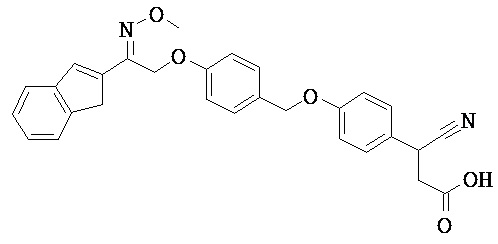
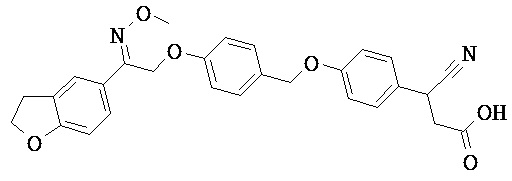
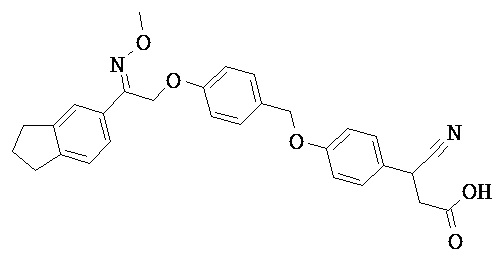
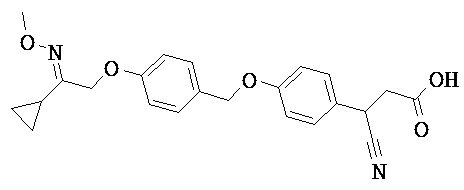
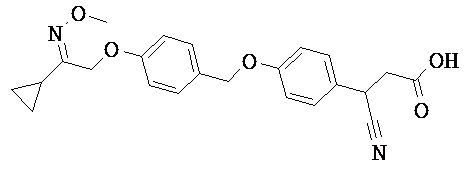
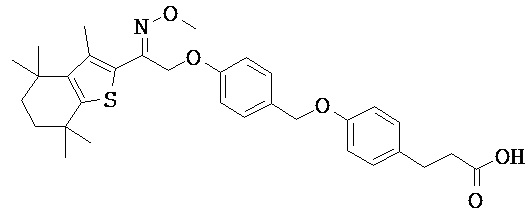
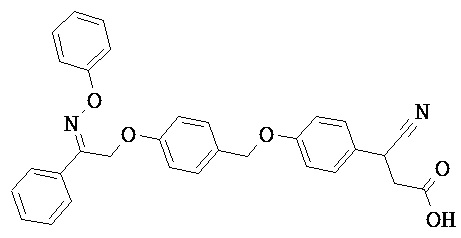
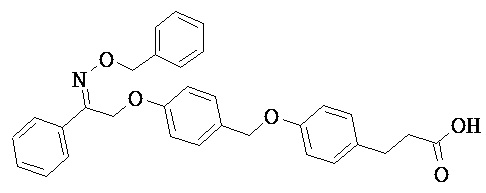

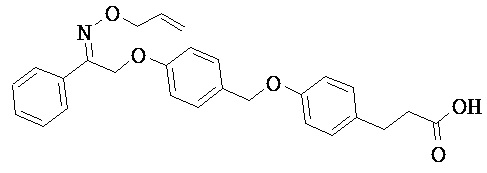
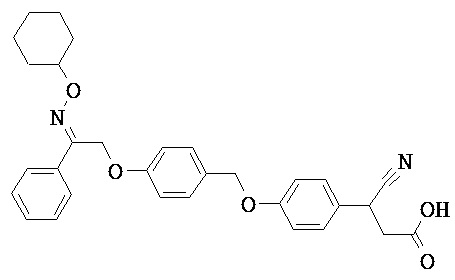
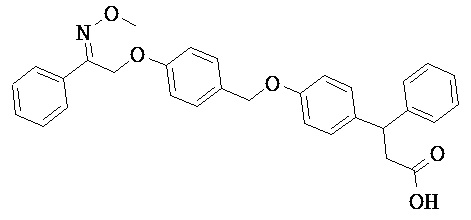

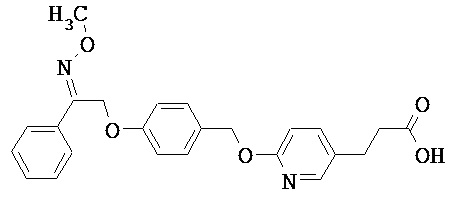
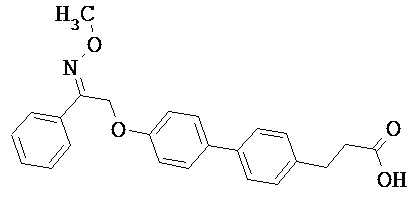
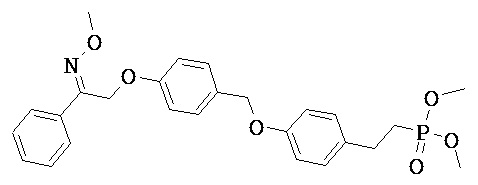
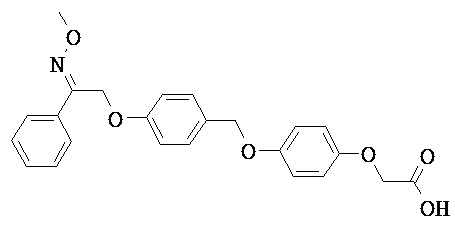
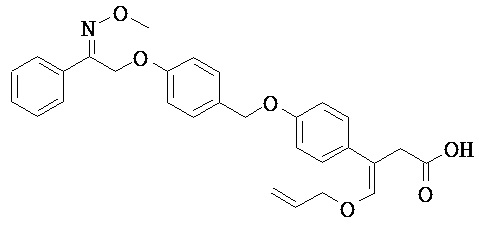
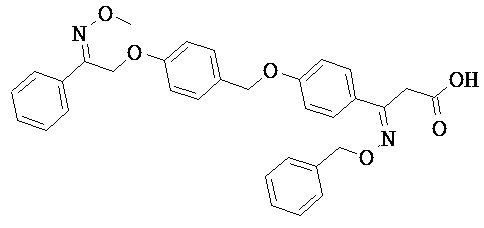
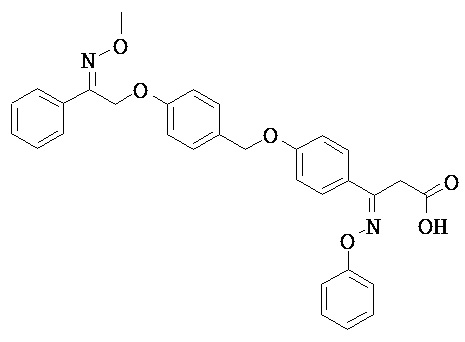
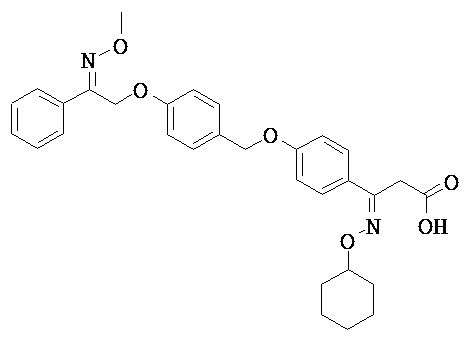 ,
,
или их геометрические изомеры, или фармацевтически приемлемые соли, или пролекарства.
Соединения обладают способностью активации рецептора GPR40. Способность активации рецептора GPR40 может возникать в результате непосредственного действия соединения исключительно на рецептор GPR40 для модуляции/увеличения биологической активности. Тем не менее, понимают, что соединения также могут по меньшей мере отчасти действовать на другие факторы, связанные с активностью рецептора GPR40.
Активацию рецептора GPR40 можно проводить при помощи любого способа из числа хорошо известных в данной области техники. Например, если необходимо проведение активации рецептора GPR40 in vitro, то соответствующее количество соединения можно добавлять в раствор, содержащий рецептор GPR40. В тех случаях, где желательно проведение активации рецептора GPR40 у млекопитающего, активация рецептора GPR40, как правило, включает введение соединения млекопитающему, у которого имеется рецептор GPR40.
Соответственно, соединения могут иметь большое число применений, в которых можно использовать вышеуказанную способность активировать рецептор GPR40.
Соответственно, можно ожидать, что соединения согласно настоящему изобретению имеют подходящие терапевтические свойства, в частности, в отношении метаболических состояний, таких как диабет, ожирение, гипергликемия, непереносимость глюкозы, инсулинорезистентность, гиперинсулинемия, гиперхолестеринемия, гипертензия, гиперлипопротеинемия, гиперлипидемия, гипертриглицеридемия, дислипидемия, метаболический синдром X, атеросклероз, диабетическая нейропатия, диабетическая ретинопатия и гипогликемия.
Соединения согласно настоящему изобретению также могут подходить для лечения когнитивных нарушений, остеопороза, воспалительных нарушений, сердечно-сосудистых заболеваний, заболеваний почек, кетоацидоза, тромботических нарушений, нефропатии, сексуальной дисфункции, дерматопатии, диспепсии, рака и эдемы. Таким образом, существует значительный интерес к разработке соединений с указанным типом действия.
Введение соединений Формулы (I) человеку можно проводить при помощи любого из приемлемых способов энтерального введения, таких как пероральное или ректальное введение, или путем парентерального введения, такого как подкожный, внутримышечный, внутривенный и внутрикожный способы. Инъекцию можно проводить с применением болюса или путем постоянной или периодической инфузии. Активное соединение, как правило, содержится в фармацевтически приемлемом носителе или разбавителе в количестве, достаточном для доставки пациенту терапевтически эффективной дозы. В различных вариантах реализации активное соединение может быть селективно токсичным или обладать более высокой токсичностью в отношении быстро пролиферирующих клеток, например, раковых опухолей, по сравнению с нормальными клетками.
При применении соединений согласно настоящему изобретению их можно вводить в любой форме или при помощи любого способа, которые делают соединение биодоступным. Специалисты в области получения составов могут легко выбрать подходящую форму и способ введения в зависимости от конкретных характеристик выбранного соединения, состояния, требующего лечения, стадии состояния, требующего лечения, и других важных условий. Дополнительную информацию см. в Remingtons Pharmaceutical Sciences, 19th edition. Mack Publishing Co. (1995).
Соединения согласно настоящему изобретению можно вводить отдельно или в форме фармацевтической композиции в комбинации с фармацевтически приемлемым носителем, разбавителем или вспомогательным веществом. Несмотря на то, что соединения согласно настоящему изобретению являются эффективными сами по себе, их, как правило, включают в составы и вводят в форме фармацевтически приемлемых солей, так как эти формы, как правило, являются более стабильными, легко кристаллизующимися и имеют повышенную растворимость.
Соединения, тем не менее, как правило, применяют в форме фармацевтических композиций, которые получают с учетом желаемого способа введения. Таким образом, в некоторых вариантах реализации в настоящем изобретении предложена фармацевтическая композиция, содержащая соединение Формулы (I) и фармацевтически приемлемый носитель, разбавитель или вспомогательное вещество. Композиции получают при помощи способов, хорошо известных в данной области техники.
С других вариантах реализации изобретения предложена фармацевтическая упаковка или набор, содержащая один или более контейнеров, заполненных одним или более ингредиентами фармацевтической композиции согласно настоящему изобретению. В указанной упаковке или наборе может храниться контейнер, содержащий стандартную дозировку агента(ов). Наборы могут включать композицию, содержащую эффективный агент в виде концентрата (включая лиофилизированные композиции), который можно дополнительно разбавлять перед применением, или агент в применяемой концентрации, где пробирки могут содержать одну или более дозировок. Для удобства в наборах стандартные дозировки могут содержаться в стерильных пробирках, и, таким образом, врачи могут напрямую использовать пробирки, где в пробирках содержится(атся) агент(ы) в желаемом количестве и концентрации. В указанном(ых) контейнере(ах) могут содержаться различные письменные материалы, такие как инструкции по применению или уведомления, утвержденные правительственным ведомством, регулирующим производство, применение или реализацию фармацевтических или биологических продуктов, в которых приведено разрешение для введения человеку, одобренное ведомством, регулирующим производство, применение или реализацию фармацевтических или биологических продуктов.
Соединения согласно настоящему изобретению можно применять или вводить в комбинации с одним или более дополнительными лекарственными средствами для лечения указанных нарушений/заболеваний. Компоненты можно вводить в одном составе или в раздельных составах. В случае введения в отдельных составах соединения согласно настоящему изобретению можно вводить последовательно или одновременно с другим(и) лекарственным(и) средством(ами).
В дополнение к возможности введения в комбинации с одним или более дополнительными лекарственными средствами соединения согласно настоящему изобретению можно применять в комбинированной терапии. При проведении комбинированной терапии соединения, как правило, вводят в комбинации друг с другом. Таким образом, одно или более соединений согласно настоящему изобретению можно вводить одновременно (в виде комбинированного препарата) или последовательно для достижения желаемого действия. Комбинированная терапия является особенно желательной в случаях, когда терапевтические профили каждого соединения различаются, и объединенное действие двух лекарственных средств обеспечивает улучшенный терапевтический результат.
Фармацевтические композиции согласно настоящему изобретению для парентеральной инъекции включают фармацевтически приемлемые стерильные водные или неводные растворы, дисперсии, суспензии или эмульсии, а также стерильные порошки, которые необходимо растворять в стерильных инъецируемых растворах или дисперсиях непосредственно перед применением. Примеры подходящих водных и неводных носителей, разбавителей, растворителей или вспомогательных веществ включают воду, этанол, полиолы (такие как глицерин, пропиленгликоль, полиэтиленгликоль и т.д.) и их подходящие смеси, растительные масла (такие как оливковое масло) и инъецируемые органические сложные эфиры, такие как этилолеат. Подходящую текучесть можно поддерживать, например, путем применения оболочечных веществ, таких как лецитин, путем поддержания требуемого размера частиц в случае дисперсий и путем применения поверхностно активных веществ.
Указанные композиции также могут содержать адъюванты, такие как консерванты, увлажнители, эмульгаторы и диспергирующие агенты. Предотвращение действия микроорганизмов можно обеспечивать путем введения различных противобактериальных и противогрибковых агентов, например, парабенов, хлорбутанола, фенолсорбиновой кислоты и т.д. Также может быть желательным включение изотонических агентов, таких как сахара, хлорид натрия и т.д. Продолжительность всасывания инъецируемой фармацевтической формы можно обеспечивать путем введения агентов, которые задерживают всасывание, таких как моностеарат алюминия и желатин.
При желании для более эффективного распределения соединения можно вводить в системы замедленного высвобождения и направленной доставки, такие как полимерные матрицы, липосомы и микросферы.
Инъецируемые составы можно стерилизовать, например, путем фильтрации через удерживающий бактерии фильтр или путем введения стерилизующих агентов в форме стерильных твердых композиций, которые можно растворять или диспергировать в стерильной воде или другой стерильной инъецируемой среде непосредственно перед применением.
Твердые лекарственные формы для перорального введения включают капсулы, таблетки, пилюли, порошки и гранулы. В указанных твердых лекарственных формах активное соединение смешивают по меньшей мере с одним инертным фармацевтически приемлемым вспомогательным веществом или носителем, таким как цитрат натрия или фосфат дикальция и/или a) с наполнителями или добавками, такими как крахмалы, лактоза, сахароза, глюкоза, маннит и кремниевая кислота, b) со связывающими веществами, такими как, например, карбоксиметилцеллюлоза, альгинаты, желатин, поливинилпирролидон, сахароза и аравийская камедь, c) с увлажнителями, такими как глицерин, d) с разрыхлителями, такими как агар-агар, карбонат кальция, картофельный или тапиоковый крахмал, альгиновая кислота, определенные силикаты и карбонат натрия, e) с агентами, затрудняющими растворение, такими как парафин, f) с ускорителями всасывания, такими как четвертичные аммонийные соединения, g) с увлажняющими агентами, такими как, например, цетиловый спирт и глицерина моностеарат, h) с абсорбентами, такими как каолин и бентонитовая глина, и i) со смазками, такими как тальк, стеарат кальция, стеарат магния, твердые полиэтиленгликоли, лаурилсульфат натрия, и их смесями. В случае капсул, таблеток и пилюль лекарственная форма также может содержать буферные агенты.
Твердые композиции аналогичного типа также можно применять в качестве наполнителей твердых и мягких желатиновых капсул с применением таких вспомогательных веществ, как лактоза или молочный сахар, а также высокомолекулярные полиэтиленгликоли и т.д.
Твердые лекарственные формы, такие как таблетки, драже, капсулы, пилюли и гранулы, можно получать с использованием оболочек и покрытий, таких как кишечнорастворимая оболочка и другие оболочки, хорошо известные в области получения фармацевтических составов. Твердые лекарственные формы также могут содержать опалесцирующие агенты, а также могут представлять собой композицию, высвобождающую активный(е) ингредиент(ы) исключительно или преимущественно в определенной части кишечного тракта, возможно замедленным образом. Примеры заливочных композиций, которые можно применять, включают полимерные вещества и воски.
Активные соединения также могут присутствовать в микроинкапсулированной форме, при необходимости совместно с одним или более вышеуказанными вспомогательными веществами.
Жидкие лекарственные формы для перорального введения включают фармацевтически приемлемые эмульсии, растворы, суспензии, сиропы и эликсиры. В дополнение к активным соединениям жидкие лекарственные формы могут содержать инертные разбавители, общепринятые в данной области техники, такие как, например, вода или другие растворители, агенты, повышающие растворимость, и эмульгаторы, такие как этиловый спирт, изопропиловый спирт, этилкарбонат, этилацетат, бензиловый спирт, бензилбензоат, пропиленгликоль, 1,3-бутиленгликоль, диметилформамид, масла (в частности, хлопковое, арахисовое, кукурузное, масло зародышей пшеницы, оливковое, касторовое и кунжутное масла), глицерин, тетрагидрофурфуриловый спирт, полиэтиленгликоли и сложные эфиры жирных кислот и сорбитана, и их смеси.
Помимо инертных разбавителей пероральные композиции также могут содержать адъюванты, такие как увлажнители, эмульгаторы и суспендирующие агенты, подсластители, ароматизаторы и вкусовые добавки.
Суспензии в дополнение к активным соединениям могут содержать суспендирующие агенты, такие как, например, этоксилированный изостеариловый спирт, полиоксиэтиленсорбит и сложные эфиры сорбитана, микрокристаллическая целлюлоза, метагидроксид алюминия, бентонит, агар-агар и трагакантовая камедь, а также их смеси.
Композиции для ректального и вагинального введения предпочтительно представляют собой суппозитории, которые можно получать путем смешения соединений согласно настоящему изобретению и подходящих нераздражающих вспомогательных веществ или носителей, таких как масло какао, полиэтиленгликоль или воск для суппозиториев, которые являются твердыми при комнатной температуре, но жидкими при температуре тела, и, таким образом, плавятся в прямой кишке или влагалищной полости и высвобождают активное соединение.
Лекарственные формы для местного введения, содержащие соединение согласно настоящему изобретению, включают порошки, пластыри, спреи, мази и ингалируемые препараты. Активное соединение смешивают в стерильных условиях с фармацевтически приемлемым носителем и любыми необходимыми консервантами, буферами или газами-вытеснителями.
Соединение предпочтительно вводят в количестве, достаточном для излечения и облегчения или ослабления состояния. Терапевтически эффективное количество могут легко определять назначенные специалисты-диагносты при помощи традиционных способов и путем исследования результатов, полученных в аналогичных случаях. При определении терапевтически эффективного количества необходимо учитывать ряд факторов, которые включают, но не ограничиваются ими, особенности животного, его размер, возраст и общее состояние здоровья, конкретное состояние, тяжесть состояния, ответ пациента на лечение, конкретное вводимое соединение, способ введения, биодоступность вводимого препарата, выбранная схема дозирования, применение других лекарственных средств и другие важные условия.
Предпочтительная дозировка находится в диапазоне примерно от 0,01 до 300 мг на килограмм массы тела в день. Более предпочтительная дозировка находится в диапазоне от 0,1 до 100 мг на килограмм массы тела в день, более предпочтительно от 0,2 до 80 мг на килограмм массы тела в день, еще более предпочтительно от 0,2 до 50 мг на килограмм массы тела в день. Подходящую дозу можно вводить в виде нескольких поддоз в день.
СИНТЕЗ СОЕДИНЕНИЙ СОГЛАСНО НАСТОЯЩЕМУ ИЗОБРЕТЕНИЮ
Агенты согласно различным вариантам реализации можно получать при помощи реакций и схем синтеза, описанных ниже, с использованием техник, доступных в данной области техники, с применением легкодоступных исходных веществ. Получение конкретных соединений согласно вариантам реализации описано подробно в приведенных ниже примерах, но специалистам будет понятным, что описанные химические реакции можно легко адаптировать для получения ряда других агентов согласно различным вариантам реализации. Например, синтез не приведенных в примерах соединений можно успешно проводить с использованием модификаций, очевидных специалистам в данной области техники, например, путем соответствующей защиты мешающих групп, изменения других подходящих реагентов, известных в данной области техники, проведения традиционных модификаций условий проведения реакций. Перечень подходящих защитных групп, используемых в органическом синтезе, можно найти в T.W. Greene's Protective Groups in Organic Synthesis, 3rd Edition, John Wiley & Sons, 1991. В качестве альтернативы, очевидно, что можно применять другие реакции, описанные в настоящей заявке или известные в данной области техники, для получения других соединений согласно различным вариантам реализации.
Реагенты, подходящие для синтеза соединений, можно получать или синтезировать согласно способам, известным в данной области техники.
Символы, сокращения и условные обозначения, используемые при описании способов, схем и примеров, соответствуют используемым в современной научной литературе. В частности, следующие сокращения могут использоваться в примерах и в описании, не ограничивая их.
- г (граммы)
- л (литры)
- Гц (Герц)
- моль (моли)
- КТ (комнатная температура)
- мин. (минуты)
- MeOH (метанол)
- CHCl3 (хлороформ)
- ДХМ (дихлорметан)
- ДМСО (диметилсульфоксид)
- EtOAc (этилацетат)
- мг (миллиграммы)
- мл (миллилитры)
- psi (фунты на квадратный дюйм)
- мМ (миллимолярный)
- МГц (мегагерц)
- ч (часы)
- ТСХ (тонкослойная хроматография)
- EtOH (этанол)
- CDCl3 (дейтерированный хлороформ)
- HCl (хлороводородная кислота)
- ДМФ (N,N-диметилформамид)
- ТГФ (тетрагидрофуран)
- K2CO3 (карбонат калия)
- Na2SO4 (сульфат натрия)
- PC (реакционная смесь)
Если не указано иное, все температуры выражены в °C (градусы Цельсия). Все реакции проводили при комнатной температуре, если не указано иное.
Все применяемые растворители и реагенты являются коммерчески доступными и приобретались в Sigma Aldrich, Fluka, Acros, Spectrochem, Alfa Aesar, Avra, Qualigens, Merck, Rankem и Leonid Chemicals.
1H ЯМР спектры получали на Bruker AV 300. Химические сдвиги выражены в частях на миллион (ppm, единицы δ). Константы спин-спинового взаимодействия выражены в герцах (Гц). Структура расщепления описывает кажущуюся мультиплетность и обозначена s (синглет), d (дублет), t (триплет), q (квартет), m (мультиплет) или br (широкий пик).
Масс-спектры получали на масс-спектрометре с одной квадрупольной линзой Agilent technologies 6120 с применением химической ионизации при атмосферном давлении (ХИАД) или ионизации электронным распылением (ИЭР) или комбинации двух указанных источников.
Все образцы исследовали на системе CHIMADZU с насосом LC-20 AD, диодноматричным детектором SPD-M20A, автодозатором SIL-20A.
СХЕМА СИНТЕЗА 1
Одна из схем получения определенных соединений согласно настоящему изобретению показана ниже на схеме 1, где с использованием традиционных способов бромированный дифенилкетон (промежуточное соединение 1) и 3-(4-гидроксифенил)метилпропионат (промежуточное соединение 2) получают и подвергают взаимодействию с получением промежуточного соединения 3. Метальную группу кислоты затем удаляют, затем подвергают взаимодействию с соответствующим амином с получением соответствующего оксима. Изменяя исходные вещества, применяемые для получения промежуточных соединений 1 и 2, и состав амина, используемого для получения конечного оксима, можно получать широкий ряд соединений согласно настоящему изобретению с применением указанной методики. С использованием указанного способа или его измененных форм в качестве общего правила можно получать все оксимы, содержащие кислород в группе Y.
Схема 1:
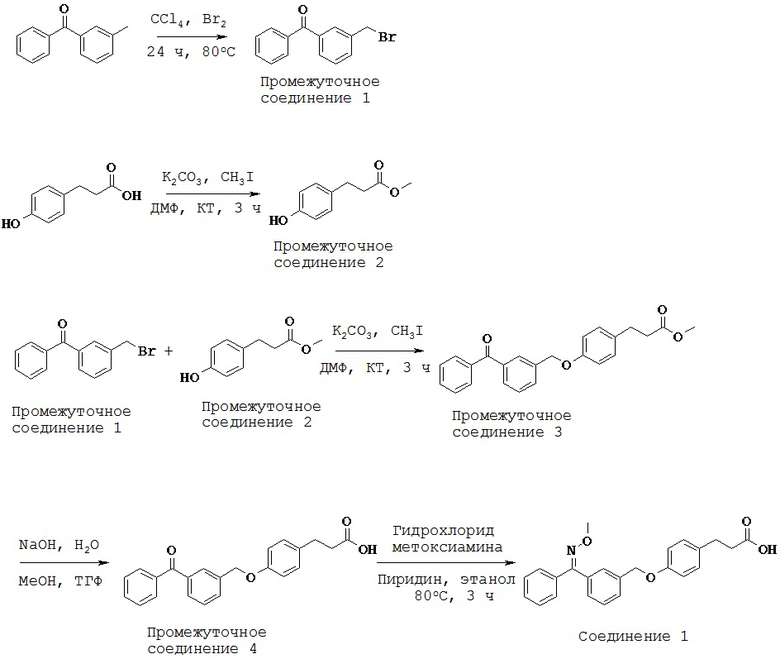
Пример 1
3-[4-({3-[(E,Z)-(метоксиимино)(фенил)метил]бензил}окси)фенил]пропановая кислота (1)
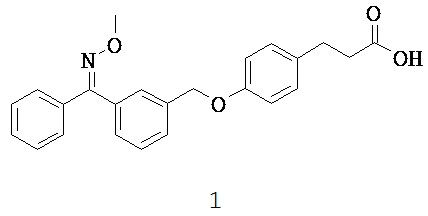
Соединение 1 синтезировали из [3-(бромметил)фенил](фенил)метанона и метил-3-(4-гидроксифенил)пропаноата согласно способу, описанному на Схеме 1.
Промежуточное соединение 1: [3-(бромметил)фенил](фенил)метанон
В 1000 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 375 мл CCl4. Растворитель охлаждали до 0°C и в перемешиваемый растворитель добавляли фенил-м-толуилметанон (15,0 г, 76,45 ммоль), затем добавляли бром (24,43 г, 152,9 ммоль) при 0°C и смесь оставляли перемешиваться при той же температуре в течение 30 минут. Полученный раствор кипятили с обратным холодильником при 80°C в течение 24 часов. После завершения реакции (прохождение отслеживали путем ТСХ) PC охлаждали до 10°C и реакцию гасили путем добавления твердого бикарбоната натрия (15 г, 178,57 ммоль). Органический слой промывали насыщенным водным раствором бикарбоната натрия (300 мл × 2) и насыщенным солевым раствором (300 мл): Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении с получением продукта в виде белого твердого вещества (6,8 г, выход 32,3%): 1Н ЯМР (300 МГц, CDCl3): δ 7,74-7,76 (m, 2H), 7,71-7,72 (t, 2H), 7,51-7,67 (m, 2H), 7,40-7,45 (m, 3H), 4,46 (s, 2H).
Промежуточное соединение 2: Метил-3-(4-гидроксифенил)пропаноат
В 1000 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 250 мл ДМФ, 3-(4-гидроксифенил)пропановую кислоту (25,0 г, 150,43 ммоль) и K2CO3 (41,58 г, 300,87 ммоль). Полученную смесь перемешивали при КТ в течение 30 минут. Метилйодид (25,627 г, 180,51 ммоль) добавляли в полученную смесь, предварительно охлажденную до 0°C. Полученную смесь перемешивали при КТ в течение 3 часов. После завершения реакции (прохождение отслеживали путем ТСХ) растворитель удаляли при пониженном давлении и неочищенную массу растворяли в этилацетате (250 мл). Органический слой промывали водой (250 мл), насыщенным раствором бикарбоната натрия (250 мл × 2) и насыщенным солевым раствором (250 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Продукт получали в виде желтой маслянистой жидкости (26 г, выход 95,9%): МС (ИЭР, 120 эВ): m/z=178,9 (М-Н)+; 1Н ЯМР (300 МГц, CDCl3): δ 6,97-7,00 (d, 2H), 6,66-6,69 (d, 2H), 4,94 (s, 1H), 3,59 (s, 3H), 2,78-2,83 (t, 2H), 2,50-2,55 (t, 2H).
Промежуточное соединение 3: Метил-3-{4-[(3-бензоилбензил)окси]фенил}пропаноат
В 250 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 45 мл ДМФ. В перемешиваемый растворитель добавляли метиловый эфир 3-(4-гидроксифенил)пропановой кислоты (4,5 г, 16,35 ммоль), затем K2CO3 (6,78 г, 49,074 ммоль) и полученную смесь перемешивали при комнатной температуре в течение 30 минут. (3-Бромметилфенил)фенилметанон (2,94 г, 16,35 ммоль) добавляли в полученную выше смесь и перемешивание продолжали при КТ в течение 24 часов. После завершения реакции (прохождение отслеживали путем ТСХ) растворитель удаляли при пониженном давлении. Неочищенное соединение очищали путем колоночной хроматографии на силикагеле (100/200 меш) с применением петролейного эфира (60-80) и этилацетата в качестве элюента с получением продукта в виде желтой маслянистой жидкости (5,1 г, выход 83,3%): МС (ИЭР, 120 эВ): m/z=375,1 (М+Н)+; 1Н ЯМР (300 МГц, CDCl3): δ 7,68 (s, 1Н), 7,62-7,66 (d, 3H), 7,57-7,59 (d, 1H), 7,53 (m, 1H), 7,45-7,50 (m, 3H), 7,11-7,13 (d, 2H), 6,88-6,90 (d, 2H), 5,10 (s, 2H), 3,66 (s, 3H), 2,85-2,92 (m, 2H), 2,56-2,62 (m, 2H).
Промежуточное соединение 4: 3-{4-[(3-бензоилбензил)окси]фенил}пропановая кислота
В 100 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 15 мл ТГФ, 1,0 мл воды и 1,0 мл метанола. В перемешиваемую смесь растворителей добавляли метиловый эфир [4-(3-бензоилбензилокси)фенил]пропановой кислоты (1,5 г, 4,0064 ммоль), затем добавляли гидроксид натрия (0,480 г, 12,02 ммоль). Полученный раствор перемешивали при КТ в течение 15 часов. После завершения реакции (прохождение отслеживали путем ТСХ) PC разбавляли 10 мл воды и промывали 40 мл ДХМ (50 мл × 2). Затем водный слой подкисляли до pH 6 при помощи конц. HCl и экстрагировали ДХМ (75 мл × 3). Слой в ДХМ промывали водой (75 мл), сушили над безводным Na2SO4, затем растворитель удаляли при пониженном давлении. Продукт получали в виде белого твердого вещества (1,4 г, выход 97,2%): МС (ИЭР, 120 эВ): m/z=361,1 (М+Н)+; 1Н ЯМР (300 МГц, CDCl3): δ 12,09 (s, 1H), 7,80 (s, 1H), 7,66-7,73 (m, 5H), 7,56-7,58 (d, 3H), 7,13-7,16 (m, 2H), 6,90-6,93 (d, 2H), 5,18 (s, 2H), 2,72-2,77 (t, 2H), 2,42-2,45 (m, 2H).
Соединение 1: 3-[4-({3-[(Е,Z)-(метоксиимино)(фенил)метил]бензил}окси)фенил]-пропановая кислота
В 100 мл круглодонную колбу, оборудованную магнитной мешалкой и обратным холодильником, помещали 42 мл этанола и 8,4 мл пиридина. В перемешиваемую смесь растворителей добавляли 3-[4-(3-бензоилбензилокси)фенил]пропановую кислоту (1,4 г, 3,884 ммоль), затем гидрохлорид метоксиламина (1,62 г, 19,422 ммоль). Полученный раствор кипятили с обратным холодильником при 80°С в течение 3 часов. После завершения реакции (прохождение отслеживали путем ТСХ) растворитель удаляли при пониженном давлении и полученную неочищенную массу помещали в дихлорметан (100 мл). Органический слой промывали водой (100 мл × 3) и насыщенным солевым раствором (100 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Продукт получали в виде белого твердого вещества (1,1 г, выход 72,8%); чистота 98,3%.
СХЕМА СИНТЕЗА 2
Альтернативная схема получения определенных соединений согласно настоящему изобретению показана ниже на схеме 2. При помощи традиционных способов бромированный дифенилкетон (промежуточное соединение 1) и 3-(4-аминофенил)-пропановую кислоту получали и подвергали взаимодействию с получением промежуточного соединения 5, которое затем вводили в реакцию с соответствующим амином с получением соответствующего оксима. Путем изменения исходных веществ, применяемых для получения промежуточного соединения 1, состава применяемой пропановой кислоты и состава амина, применяемого для получения конечного оксима, можно получать широкий ряд соединений согласно настоящему изобретению при помощи указанной методики. С использованием указанного способа или его измененных форм в качестве общего правила можно получать все оксимы, содержащие кислород в группе Y.
Схема 2:
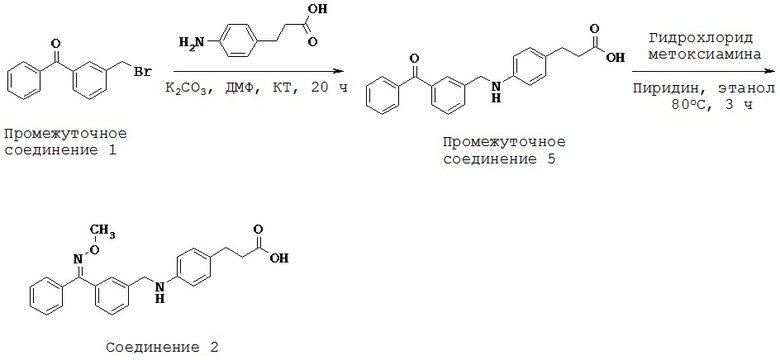
Пример 2
3-[4-({3-[(Е,Z)-(метоксиимино)(фенил)метил]бензил}амино)фенил]пропановая кислота (2)
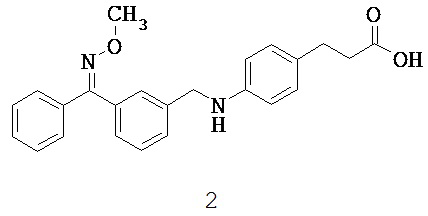
Соединение 2 синтезировали из [3-(бромметил)фенил](фенил)метанона и 3-(4-аминофенил)пропановой кислоты согласно способу, описанному на Схеме 2.
Промежуточное соединение 5: 3-{4-[(3-бензоилбензил)амино]фенил}пропановая кислота
В 250 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 25 мл ДМФ. В перемешиваемый растворитель добавляли 3-(4-аминофенил)пропановую кислоту (0,6 г, 3,635 ммоль), затем K2CO3 (1,507 г, 10,905 ммоль) и смесь перемешивали при КТ в течение 30 минут. В полученную смесь добавляли (3-бромметилфенил)фенилметанон (1,0 г, 3,635 ммоль) и перемешивание продолжали при КТ в течение еще 20 часов. После завершения реакции (прохождение отслеживали путем ТСХ) растворитель удаляли при пониженном давлении и полученное неочищенное соединение растворяли в воде (15 мл). Водный слой подкисляли насыщенным раствором лимонной кислоты в воде (1,5 г в 3 мл воды) до pH 5. Соединение получали после экстракции остатка диэтиловым эфиром (100 мл × 3). Объединенные слои в эфире промывали насыщенным солевым раствором (50 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Полученную неочищенную массу очищали путем колоночной хроматографии на силикагеле (100/200 меш) с применением петролейного эфира (60-80) и этилацетата в качестве элюента. Продукт получали в виде коричневого липкого твердого вещества (0,3 г, выход 22,9%): МС (ИЭР, 120 эВ): m/z=360,1 (М+Н)+; 1Н ЯМР (300 МГц, ДМСО-d6): δ 12,04 (s, 1H), 7,72 (s, 1H), 7,64-7,68 (t, 4H), 7,57-7,60 (d, 1H), 7,48-7,54 (m, 3H), 6,89-6,91 (d, 2H), 6,46-6,49 (d, 2H), 6,18-6,22 (t, 1H), 4,32-4,34 (d, 2H), 2,61-2,66 (d, 2H), 2,38-2,43 (d, 2H).
Соединение 2: 3-[4-({3-[(Е,Z)-(метоксиимино)(фенил)метил]бензил}амино)фенил]-пропановая кислота
В 100 мл круглодонную колбу, оборудованную магнитной мешалкой и обратным холодильником, помещали 9 мл этанола и 1,8 мл пиридина. В перемешиваемый раствор добавляли 3-[4-(3-бензоилбензиламино)фенил]пропановую кислоту (0,3 г, 0,834 ммоль), затем гидрохлорид метоксиламина (0,348 г, 4,173 ммоль). Полученный раствор кипятили с обратным холодильником при 82°C в течение 3 часов. После завершения реакции (прохождение отслеживали путем ТСХ) растворитель удаляли при пониженном давлении и полученную неочищенную массу помещали в дихлорметан (50 мл). Органический слой затем промывали водой (50 мл × 3), насыщенным солевым раствором (50 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Полученное неочищенное вещество очищали путем колоночной хроматографии на силикагеле (100/200 меш) с применением петролейного эфира (60-80) и этилацетата в качестве элюента с получением продукта в виде белого твердого вещества (0,1 г, выход 31,3%); чистота: 97,1%.
Пример 3
3-[4-({4-[(Е,Z)-(метоксиимино)(фенил)метил]бензил}амино)фенил]пропановая кислота (3):
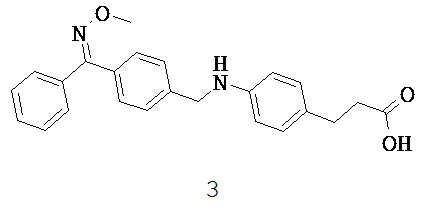
Соединение 3 синтезировали из [4-(бромметил)фенил](фенил)метанона (0,65 г, 2,1 ммоль) и 3-(4-аминофенил)пропановой кислоты (0,25 г, 1,4 ммоль) согласно способу, аналогичному описанному на Схеме 2 (0,006 г, выход 1,5%); чистота 87,98%.
СХЕМА СИНТЕЗА 3
Альтернативная схема синтеза определенных соединений согласно настоящему изобретению показана ниже на схеме 3 и находит применение для синтеза соединений, в которых X отличен от связи. При помощи традиционных способов бромированный оксим (промежуточное соединение 6) и соответствующий замещенный гидроксиальдегид подвергают взаимодействию с получением промежуточного соединения 7, которое затем вводят в реакцию с соответствующим 3-(4-аминофенил)метилпропионатом с получением промежуточного соединения 8, которое можно легко деметилировать с получением соединения 4. Путем изменения исходных веществ, применяемых для получения промежуточного соединения 6, и состава гидроксиальдегида и производного пропановой кислоты можно получать широкий ряд соединений согласно настоящему изобретению при помощи указанной методики.
Схема 3:
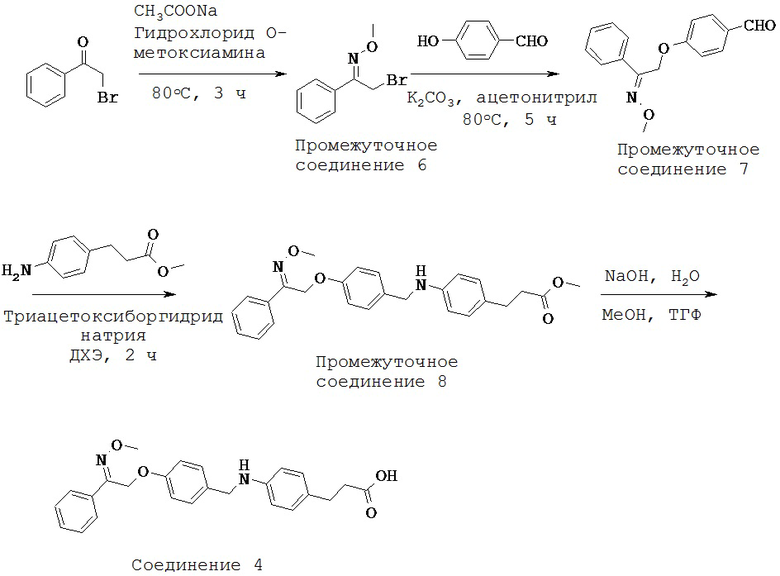
Пример 4
3-{4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)амино]фенил}пропановая кислота (4)
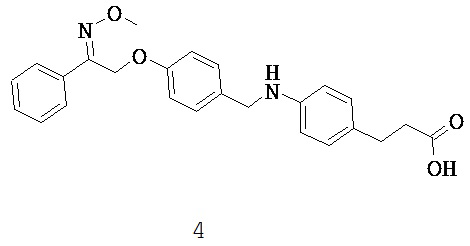
Соединение 4 синтезировали из 4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}-бензальдегида и метил-3-(4-аминофенил)пропаноата согласно способу, описанному на Схеме 3.
Промежуточное соединение 6: (1Z)-2-бром-N-метокси-1-фенилэтанимин
В 100 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 20 мл уксусной кислоты. В перемешиваемый растворитель добавляли 2-бромацетофенон (1 г, 5 ммоль) и ацетат натрия (0,49 г, 5,97 ммоль). Полученную смесь перемешивали в течение 5 минут, гидрохлорид O-метоксиламина (0,4 г, 4,79 ммоль) добавляли в полученную выше смесь и PC нагревали при 80°C и перемешивали при указанной температуре в течение 3 часов. После завершения реакции (прохождение отслеживали путем ТСХ) PC выливали в воду (15 мл) и экстрагировали этилацетатом (100 мл). Органический слой промывали насыщенным солевым раствором (50 мл) и сушили над безводным Na2SO4. Растворитель удаляли при пониженном давлении и остаток очищали путем колоночной хроматографии на силикагеле (100/200 меш) с применением петролейного эфира (60-80) и этилацетата с получением титульного соединения (0,6 г, выход 52,6%): МС (ИЭР, 120 эВ): m/z=228,1 (М+Н)+; 1Н ЯМР (300 МГц, CDCl3): δ 7,62-7,64 (m, 2Н), 7,32-7,34 (m, 3H), 4,28 (s, 2H), 4,02 (s, 3H).
Промежуточное соединение 7: 4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}-бензальдегид
В 100 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 20 мл ацетонитрила. В перемешиваемый растворитель добавляли 4-гидроксибензальдегид (0,21 г, 1,72 ммоль) и K2CO3 (0,46 г, 3,33 ммоль). Затем смесь перемешивали в течение 5 минут. O-метилоксим 2-бром-1-фенилэтанона (0,4 г, 1,7 ммоль) добавляли. Затем PC нагревали до 80°C и перемешивали при указанной температуре в течение 5 часов. После завершения реакции (прохождение отслеживали путем ТСХ) PC концентрировали путем удаления ацетонитрила, воду (10 мл) добавляли и смесь экстрагировали этилацетатом (20 мл × 2). Органический слой промывали насыщенным солевым раствором (20 мл), сушили над безводным Na2SO4 и выпаривали насухо с получением титульного соединения (0,4 г, выход 87,5%): МС (ИЭР, 120 эВ): m/z=270,1 (М+Н)+; 1Н ЯМР (300 МГц, CDCl3): δ 9,80 (s, 1H), 7,72-7,75 (d, 2H), 7,655-7,59 (m, 2H), 7,27-7,29 (m, 3H), 6,92-6,95 (d, 2H), 5,22 (s, 2H), 4,01 (s, 3H).
Промежуточное соединение 8: метил-3-{4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)амино]фенил}пропаноат
В 50 мл кругло донную колбу, оборудованную магнитной мешалкой, помещали 5 мл дихлорэтана. В перемешиваемый растворитель добавляли 4-(2-метоксиимино-2-фенилэтокси)бензальдегид (0,4 г, 1,5 ммоль), метиловый эфир 3-(4-аминофенил)пропановой кислоты (0,27 г, 1,5 ммоль). PC охлаждали до 0°C и триацетоксиборгидрид натрия (0,47 г, 2,25 ммоль) добавляли по частям в течение 15 минут. Реакционную смесь перемешивали при КТ в течение 2 часов. Реакцию гасили раствором NaHCO3 (40 мл 10% раствора в воде) и смесь экстрагировали этилацетатом (20 мл × 2). Органический слой промывали насыщенным солевым раствором (25 мл), сушили над безводным Na2SO4 и выпаривали в вакууме насухо. Полученное неочищенное соединение очищали путем колоночной хроматографии на силикагеле (100/200 меш) с применением петролейного эфира (60-80) и этилацетата в качестве элюента с получением продукта (0,450 г, выход 69,4%): МС (ИЭР, 120 эВ): m/z=433,1 (М+Н)+.
Соединение 4: 3-{4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)амино]-фенил}пропановая кислота
В 25 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 1:2 мл смеси метанол:ТГФ. В перемешиваемый растворитель добавляли метиловый эфир 3-{4-[4-(2-метоксиимино-2-фенилэтокси)бензиламино]фенил}пропановой кислоты (0,4 г, 0,92 ммоль). В перемешиваемый раствор NaOH (0,074 г, 1,85 ммоль) в воде (1 мл) добавляли. Полученный раствор перемешивали при КТ в течение 3 часов. После завершения реакции (прохождение отслеживали путем ТСХ) растворители и воду выпаривали при пониженном давлении. Остаток разбавляли 1 мл воды, охлаждали до 0°C, затем подкисляли 1 н. HCl и экстрагировали этилацетатом (15 мл × 2). Органический слой промывали водой (20 мл) и насыщенным солевым раствором (20 мл) и сушили над безводным Na2SO4. Органический слой выпаривали при пониженном давлении. Продукт получали в виде бесцветного твердого вещества (0,32 г, выход 84,2%); чистота 94,09%.
Пример 5
3-{4-[(3-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)амино]фенил}пропановая кислота (5)
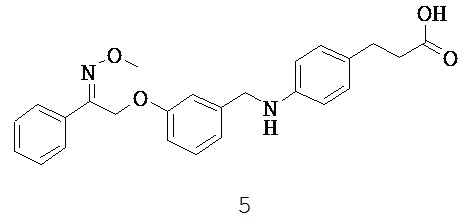
Соединение 5 синтезировали из 3-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}-бензальдегида (0,27 г, 1,0 ммоль) и 3-(4-аминофенил)пропановой кислоты (0,179 г, 1 ммоль) согласно способу, аналогичному описанному на Схеме 3 (0,04 г, выход 9,6%); чистота: 98,54%.
Схема 4:
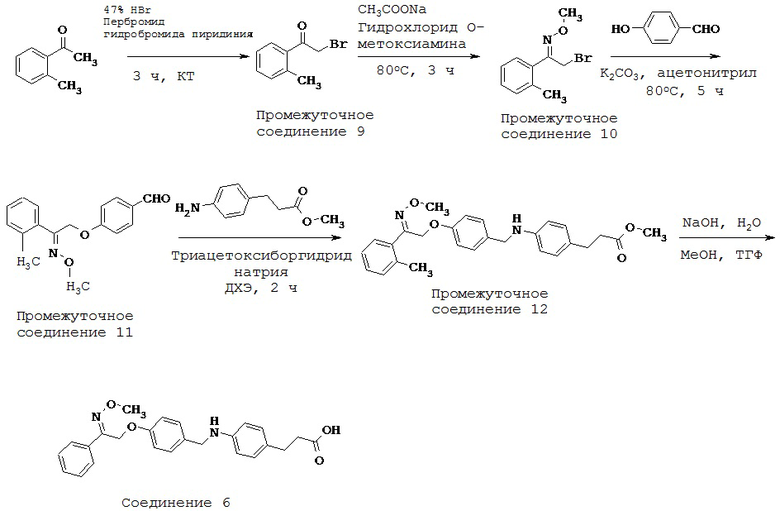
Пример 6
3-{4-[(4-{[(2Z)-2-(метоксиимино)-2-(2-метилфенил)этил]окси}бензил)амино]фенил}пропановая кислота (6)
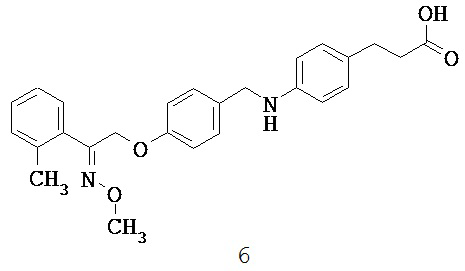
Соединение 6 синтезировали из 4-{[(2Z)-2-(метоксиимино)-2-(2-метилфенил)этил]-окси}бензальдегида и 3-(4-аминофенил)пропановой кислоты согласно способу, описанному на Схеме 4.
Промежуточное соединение 9: 2-бром-1-(2-метилфенил)этанон
В 250 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 60 мл уксусной кислоты. В перемешиваемый растворитель добавляли 1-о-толуилэтанон (3,0 г, 22,35 ммоль). PC охлаждали до 0°C, медленно добавляли 47% бромоводородную кислоту (9,7 мл, 178,63 ммоль), пербромид гидробромида пиридиния (8,6 г, 26,89 ммоль). Полученную смесь перемешивали при КТ в течение 3 часов. Реакцию затем гасили 10% раствором бикарбоната натрия в воде (150 мл) при 0°C и смесь экстрагировали этилацетатом (75 мл × 2). Органический слой промывали насыщенным солевым раствором (50 мл), сушили над безводным Na2SO4 и выпаривали насухо. Полученное неочищенное соединение очищали путем колоночной хроматографии на силикагеле (100/200 меш) с применением петролейного эфира (60-80) и этилацетата в качестве элюента с получением продукта (2,50 г, выход 52,5%).
Промежуточное соединение 10: (1Z)-2-бром-N-метокси-1-(2-метилфенил)этанимин
Способ аналогичен описанному для получения промежуточного соединения 6 на схеме 3.
Промежуточное соединение 11: 4-{[(2Z)-2-(метоксиимино)-2-(2-метилфенил)этил]окси}-бензальдегид
Способ аналогичен описанному для получения промежуточного соединения 7 на схеме 3.
Промежуточное соединение 12: метил-3-{4-[(4-{[(2Z)-2-(метоксиимино)-2-(2-метилфенил)этил]окси}бензил)амино]фенил}пропаноат
Способ аналогичен описанному для получения промежуточного соединения 8 на схеме 3.
Соединение 6: 3-{4-[(4-{[(2Z)-2-(метоксиимино)-2-(2-метилфенил)этил]окси}бензил)амино]фенил}пропановая кислота
Соединение 6 синтезировали из 4-{[(2Z)-2-(метоксиимино)-2-(2-метилфенил)этил]-окси}бензальдегида и 3-(4-аминофенил)пропановой кислоты согласно способу, описанному на Схеме 3 (0,10 г, выход 5,5%); чистота: 97,84%.
Схема 5:
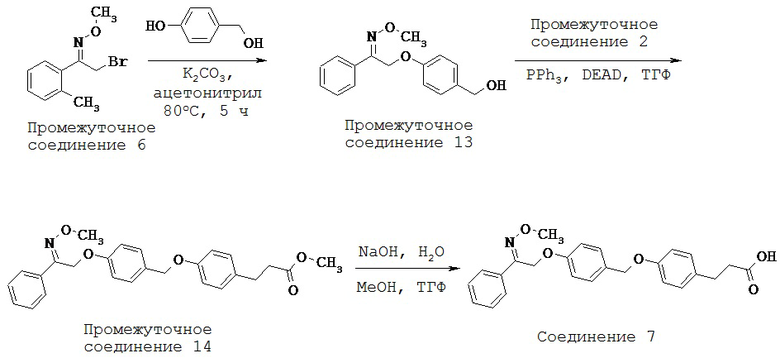
Пример 7
3-{4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропановая кислота (7):
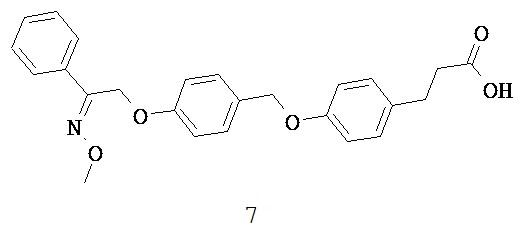
Соединение 7 синтезировали из (4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}фенил)-метанола и метил-3-(4-гидроксифенил)пропаноата согласно способу, описанному на Схеме 5.
Промежуточное соединение 13: (4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}фенил)-метанол
В 100 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 20 мл ацетонитрила. В перемешиваемый растворитель добавляли 4-гидроксибензиловый спирт (0,27 г, 2,175 ммоль) и K2CO3 (0,4 г, 2,89 ммоль). Затем смесь перемешивали в течение 5 минут. O-метилоксим 2-бром-1-фенилэтанона (0,5 г, 2 ммоль) добавляли. Затем PC нагревали при 80°C в течение 5 часов. После завершения реакции (прохождение отслеживали путем ТСХ) PC концентрировали в вакууме для удаления ацетонитрила. К остатку затем добавляли 10 мл воды и экстрагировали этилацетатом (15 мл × 2). Органический слой промывали насыщенным солевым раствором (15 мл), сушили над безводным Na2SO4 и выпаривали насухо с получением титульного соединения (0,5 г, выход 92,2%); 1Н ЯМР (300 МГц, ДМСО-d6): δ 7,61,7,64 (m, 2H), 7,38-7,40 (t, 3H), 7,18-7,21 (d, 2H), 6,83-6,86 (d, 2H), 5.20 (s, 2H), 5,02-5,06 (t, 1H), 4,38-4,39 (d, 2H), 3,99 (s, 3H).
Промежуточное соединение 14: метил-3-{4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропаноат
В 50 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали сухой ТГФ (5,0 мл) и O-метилоксим 2-(4-гидроксиметилфенокси)-1-фенилэтанона (0,25 г, 0,9 ммоль). В полученную выше смесь метиловый эфир 3-(4-гидроксифенил)пропановой кислоты (0,16 г, 0,889 ммоль) добавляли и полученную смесь перемешивали при 0°C в течение 5 минут. Трифенилфосфин (0,3 г, 1,3 ммоль) добавляли в смесь и перемешивали при 0°C в течение 15 минут, затем добавляли диэтилазадикарбоксилат (0,23 г, 1,32 ммоль). После перемешивания полученной смеси при КТ в течение 10 часов PC выпаривали для удаления ТГФ. Остаток разбавляли водой (15 мл) и экстрагировали этилацетатом (15 мл × 2). Органический слой промывали насыщенным солевым раствором (15 мл) и сушили над безводным Na2SO4. Концентрированно растворителя и очистка полученного остатка путем колоночной хроматографии на силикагеле (100/200 меш) с применением петролейного эфира (60-80) и этилацетата в качестве элюента приводили к получению продукта (0,09 г, выход 23,4%): МС (ИЭР, 120 эВ): m/z=434,2 (М+Н)+; 1Н ЯМР (300 МГц, ДМСО-d6): δ 7,62-7,64 (m, 2H), 7,38-7,40 (t, 3H), 7,32-7,34 (d, 2H), 7,09-7,12 (d, 2H), 6,86-6,92 (t, 4H), 5,20-5,22 (d, 2H), 4,93-4,95 (d, 2H), 3,97-3,99 (t, 3H), 3,55-3,56 (d, 3H), 2,74-2,79 (t, 2H), 2,55-2,59 (t, 2H).
Соединение 7: 3-{4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил] окси}бензил)окси] фенил}пропановая кислота
В 25 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 2 мл метанола и 1 мл ТГФ. В перемешиваемый растворитель добавляли метиловый эфир 3-{4-[4-(2-метоксиимино-2-фенилэтокси)бензилокси]фенил}пропановой кислоты (0,085 г, 0,19 ммоль). В перемешиваемый раствор NaOH (0,015 г, 0,375 ммоль) в воде (1 мл) добавляли. Полученный раствор перемешивали при КТ в течение 3 часов. После завершения реакции (прохождение отслеживали путем ТСХ) растворитель выпаривали при пониженном давлении. PC разбавляли 1 мл воды, охлаждали до 0°C, затем подкисляли 1 н. HCl (5 мл) и экстрагировали этилацетатом (10 мл × 2), в органический слой добавляли воду (5 мл) и промывали насыщенным солевым раствором (5 мл), сушили над безводным Na2SO4 и выпаривали насухо с получением бесцветного твердого вещества (0,050 г, выход 63,3%); чистота: 97,65%.
Схема 6:
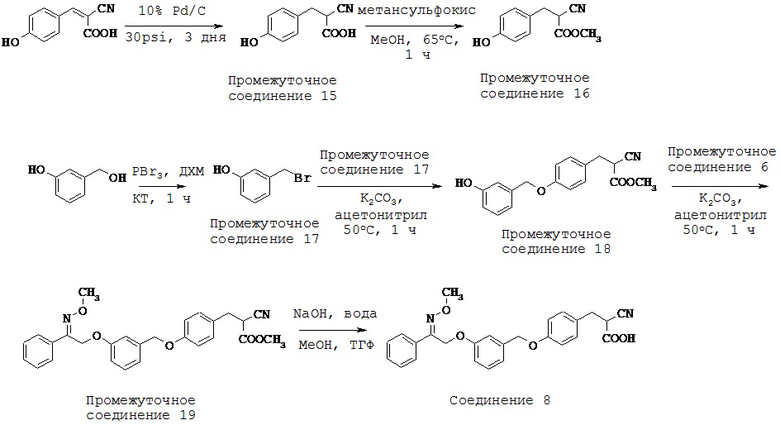
Пример 8
2-циано-3-{4-[(3-{[(2Е,2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропановая кислота (8):
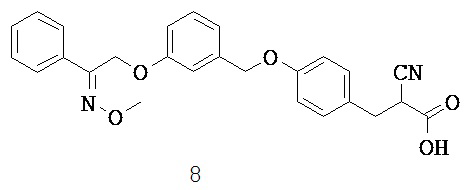
Соединение 8 синтезировали из метил-2-циано-3-(4-гидроксифенил)пропаноата и (3-{[(2Е,2Z)-2-(метоксиимино)-2-фенилэтил]окси}фенил)метанола согласно способу, описанному на Схеме 6.
Промежуточное соединение 15: 2-циано-3-(4-гидроксифенил)пропановая кислота
В 250 мл колбу для встряхивателя Парра помещали 2-циано-3-(4-гидроксифенил)-акриловую кислоту (0,5 г, 2,6 ммоль) в этилацетате (10 мл) и раствор продували азотом в течение 10 минут. Затем 10% палладий на углеродной подложке (0,07 г) добавляли в атмосфере азота. После завершения добавления смесь гидрировали при 30 psi в течение 3 дней. После завершения реакции (прохождение отслеживали путем ТСХ) PC фильтровали через подложку целита и остаток тщательно промывали метанолом (15 мл). После фильтрования фильтрат концентрировали для отгонки растворителя и сушили в вакууме. Продукт получали в виде желтой жидкости (0,5 г, выход 98,9%): МС (ИЭР, 120 эВ): m/z=192,1 (М+Н)+; 1Н ЯМР (300 МГц, ДМСО-d6): δ 7,07-7,10 (d, 2H), 6,70-6,72 (d, 2H), 4,17-4,21 (t, 1H), 2,92-3,09 (m, 2H).
Промежуточное соединение 16; метил-2-циано-3-(4-гидроксифенил)пропаноат
В 25 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 10 мл метанола. В перемешиваемый растворитель добавляли 2-циано-3-(4-гидроксифенил)-пропановую кислоту (0,5 г, 2,62 ммоль), затем метансульфокислоту (0,5 мл, 7,69 ммоль). После завершения добавления PC кипятили с обратным холодильником при 65°С в течение 1 часа. Через 1 час растворитель выпаривали, воду (20 мл) добавляли и органический слой экстрагировали этилацетатом (15 мл × 2). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Продукт получали в виде коричневой жидкости (0,3 г, выход 55,8%): МС (ИЭР, 120 эВ): m/z=204,1 (М+Н)+.
Промежуточное соединение 17: 3-(бромметил)фенол
В 50 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 15 мл дихлорметана. В перемешиваемый растворитель добавляли 3-гидроксиметилфенол (0,5 г, 4,03 ммоль). PC охлаждали до 0°С и трибромид фосфора (1,6 г, 0,56 мл, 6,04 ммоль) добавляли по каплям. После завершения добавления PC перемешивали при КТ в течение 1 часа. Через 1 час PC разбавляли дихлорметаном (25 мл) и органический слой промывали водой (20 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Продукт получали в виде коричневого твердого вещества (0,65 г, выход 86,3%); МС (ИЭР, 120 эВ): m/z=187,0 (М+Н)+; 1Н ЯМР (300 МГц, CDCl3): δ 7,10-7,15 (t, 1Н), 6,86-6,89 (d, 1H), 6,80 (d, 1H), 6,67-6,71 (m, 1H), 4,35-4,36 (d, 2H), 3,80-3,94 (brs, 1H).
Промежуточное соединение 18: метил-2-циано-3-{4-[(3-гидроксибензил)окси]фенил}пропаноат
В 50 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 10 мл ацетонитрила. В перемешиваемый растворитель добавляли метиловый эфир 2-циано-3-(4-гидроксифенил)пропановой кислоты (0,3 г, 1,57 ммоль), затем К2СО3 (0,65 г, 4,71 ммоль) и 3-бромметилфенол (0,294 г, 1,57 ммоль). После завершения добавления PC нагревали при 50°C в течение 1 часа. Через 1 час PC концентрировали для отгонки растворителя, воду (20 мл) добавляли и водный слой экстрагировали этилацетатом (15 мл × 2). Органический слой сушили над безводным Na2SO4. Растворитель удаляли при пониженном давлении. Продукт получали в виде желтой жидкости (0,32 г, выход 65,44%); МС (ИЭР, 120 эВ): m/z=310,0 (М-Н)+.
Промежуточное соединение 19: метил-2-циано-3-{4-[(3-{[(2E,2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропаноат
В 50 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 10 мл ацетонитрила. В перемешиваемый растворитель добавляли метиловый эфир 2-циано-3-[4-(3-гидроксибензилокси)фенил]пропановой кислоты (0,32 г, 1,027 ммоль), затем K2CO3 (0,425 г, 3,081 ммоль). Затем O-метилоксим 1-фенилэтанона (0,234 г, 1,027 ммоль) добавляли. После завершения добавления PC нагревали при 50°C в течение 1 часа. Через 1 час PC концентрировали; воду (20 мл) добавляли и экстрагировали этилацетатом (15 мл × 2). Органический слой сушили над безводным Na2SO4. Растворитель удаляли при пониженном давлении. Продукт получали в виде бледно-желтой жидкости (0,07 г, выход 14,85%); МС (ИЭР, 120 эВ): m/z=459,1 (М+Н)+.
Соединение 8: 2-циано-3-{4-[(3-{[(2Е,2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропановая кислота
В 10 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 3 мл ТГФ. В перемешиваемый растворитель добавляли метиловый эфир 2-циано-3-{4-[3-(2-метоксиимино-2-фенилэтокси)бензилокси]фенил}пропановой кислоты (0,07 г, 0,152 ммоль) и метанол (3 мл). PC охлаждали до 0°C и гидроксид натрия (0,03 г) в воде (1 мл) добавляли по каплям. Затем реакционную смесь перемешивали при КТ в течение 1 часа. Через 1 час растворители выпаривали и неочищенное вещество растворяли в минимальном количестве воды (2 мл) и водный слой промывали диэтиловым эфиром (5 мл × 2). Водный слой подкисляли до рН 3 при помощи 1 н. хлороводородной кислоты. Водный слой экстрагировали этилацетатом (5 мл × 2). Органический слой промывали насыщенным солевым раствором (5 мл) и сушили над безводным Na2SO4. Растворитель удаляли при пониженном давлении, получали бледно-желтое полутвердое вещество (0,015 г, выход 22,1%); чистота: 85,1%.
Схема 7:
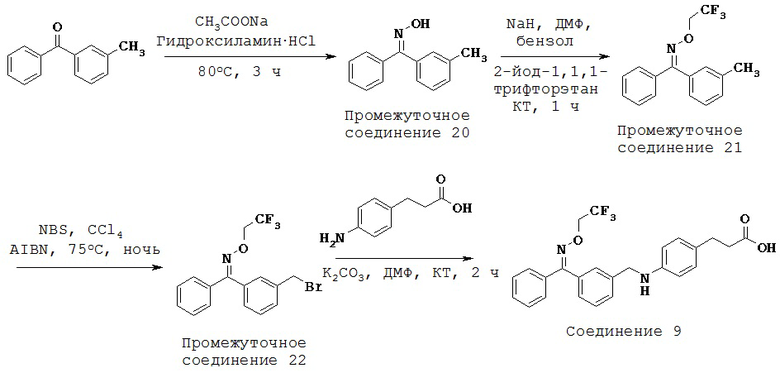
Пример 9
3-{4-[(3-{(E,Z)-фенил[(2,2,2-трифторэтокси)имино]метил}бензил)амино]фенил}пропановая кислота (9):
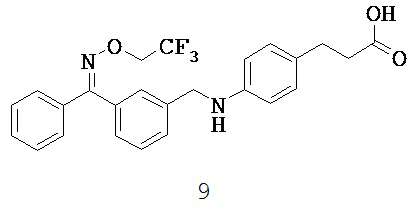
Соединение 9 синтезировали из (Е,Z)-1-[3-(бромметил)фенил]-1-фенил-N-(2,2,2-трифторэтокси)метанамина и 3-(4-аминофенил)пропановой кислоты согласно способу, описанному на Схеме 7.
Промежуточное соединение 20: (Е,Z)-N-гидрокси-1-(3-метилфенил)-1-фенилметанамин
В 100 мл кругло донную колбу, оборудованную магнитной мешалкой, помещали 20 мл уксусной кислоты. В перемешиваемый растворитель добавляли (3-метилфенил)(фенил)-метанон (2 г, 10 ммоль) и ацетат натрия (1,06 г, 1,3 экв.). Затем смесь перемешивали в течение 5 минут, гидрохлорид гидроксиламина (0,89 г, 1,2 экв.) добавляли. PC нагревали при 80°C в течение 3 часов. После завершения реакции (прохождение отслеживали путем ТСХ) PC выливали в воду (75 мл) и экстрагировали этилацетатом (50 мл × 2). Органический слой промывали насыщенным солевым раствором (40 мл), сушили над безводным Na2SO4, выпаривали насухо и очищали на силикагеле (100/200 меш) с применением петролейного эфира (60-80) и этилацетата с получением титульного соединения (1,6 г, выход 74,4%); МС (ИЭР, 120 эВ): m/z=212,1 (М+Н)+; 1Н ЯМР (300 МГц, ДМСО-d6): δ 11,27-11,28 (d, 1H), 7,04-7,48 (m, 9H), 2,27-2,33 (d, 3H).
Промежуточное соединение 21: (E,Z)-1-(3-метилфенил)-1-фенил-N-(2,2,2-трифторэтокси)метанамин
В 50 мл кругло донную колбу, оборудованную магнитной мешалкой, помещали 5 мл ДМФ. В перемешиваемый растворитель добавляли гидрид натрия (60%) при 0°C, затем (Z)-N-гидрокси-1-(3-метилфенил)-1-фенилметанамин (0,6 г, 2,8 ммоль) в смеси 1:1 ДМФ и бензола при 0°C и перемешивали при 0°C в течение 30 минут. 2-Йод-1,1,1-трифторэтан (0,71 г, 3,4 ммоль) добавляли и полученную смесь перемешивали при КТ в течение 1 часа. После завершения реакции (прохождение отслеживали путем ТСХ) PC выливали в воду (5 мл), экстрагировали этилацетатом (15 мл × 2). Органический слой промывали насыщенным солевым раствором (15 мл), сушили над безводным Na2SO4, выпаривали насухо и очищали путем колоночной хроматографии на силикагеле (100/200 меш) с применением петролейного эфира (60-80) и этилацетата с получением титульного соединения (0,23 г, выход 28,1%): МС (ИЭР, 120 эВ): m/z=294,1 (М+Н)+; 1Н ЯМР (300 МГц, CDCl3): δ 7,06-7,39 (m, 9Н), 4,41-4,49 (q, 2H), 2,26-2,30 (d, 3H).
Промежуточное соединение 22: (Е,Z)-1-[3-(бромметил)фенил]-1-фенил-N-(2,2,2-трифторэтокси)метанамин
В 100 мл кругло донную колбу, оборудованную магнитной мешалкой, помещали 50 мл CCl4, добавляли (Е,Z)-1-(3-метилфенил)-1-фенил-Н-(пропан-2-илокси)метанамин (0,22 г, 0,7 ммоль), N-бромсукцинимид (0,14 г, 0,8 ммоль) и AIBN (0,011 г, 0,07 ммоль). Полученную смесь кипятили с обратным холодильником при 75°C в течение 8 часов. После завершения реакции (прохождение отслеживали путем ТСХ) реакцию гасили ледяной водой (5 мл) и смесь экстрагировали хлороформом (10 мл × 2). Органический слой промывали насыщенным солевым раствором (10 мл), сушили над безводным Na2SO4, очищали путем колоночной хроматографии с применением петролейного эфира (60-80) и этилацетата с получением продукта (0,175 г, 67,3%); МС (ИЭР, 120 эВ): m/z=372,0 (М+Н)+; 1Н ЯМР (300 МГц, CDCl3): δ 7,19-7,46 (m, 9Н), 4,39-4,50 (m, 4H).
Соединение 9: 3-{4-[(3-{(Е,Z)-фенил[(2,2,2-трифторэтокси)имино]метил}бензил)амино]фенил}пропановая кислота
В 100 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 5 мл ДМФ. В перемешиваемый растворитель добавляли 3-(4-аминофенил)пропановую кислоту (0,068 г, 0,41 ммоль) и К2СО3 (0,15 г, 1,085 ммоль). Затем смесь перемешивали в течение 15 минут. (Е,Z)-1-[3-(бромметил)фенил]-1-фенил-N-(пропан-2-илокси)метанамин (0,17 г, 0,45 ммоль) добавляли. Затем PC перемешивали при КТ в течение 2 часов. После завершения реакции (прохождение отслеживали путем ТСХ) PC выпаривали для удаления ДМФ, затем добавляли воду (5 мл), экстрагировали этилацетатом (5 мл × 2). Органический слой промывали насыщенным солевым раствором (5 мл), сушили над безводным Na2SO4, выпаривали насухо и очищали путем препаративной ТСХ с применением 30% этилацетата в петролейном эфире (60-80) с получением титульного соединения (0,020 г, выход 10,7%); чистота 95,01%.
Пример 10
3-{4-[(3-{(Е,Z)-фенил[(пропан-2-илокси)имино]метил}бензил)амино]фенил}пропановая кислота (10):
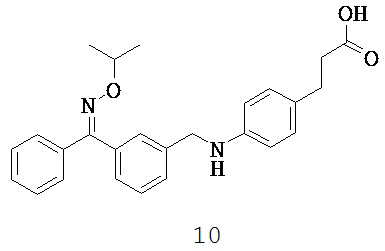
Соединение 10 синтезировали из (Е,Z)-1-[3-(бромметил)фенил]-1-фенил-N-(пропан-2-илокси)метанамина (0,38 г, 1,14 ммоль) и 3-(4-аминофенил)пропановой кислоты (0,17 г, 1,02 ммоль) согласно способу, аналогичному описанному на Схеме 7 (0,02 г, выход 4,2%); чистота 87,98%.
Схема 8:
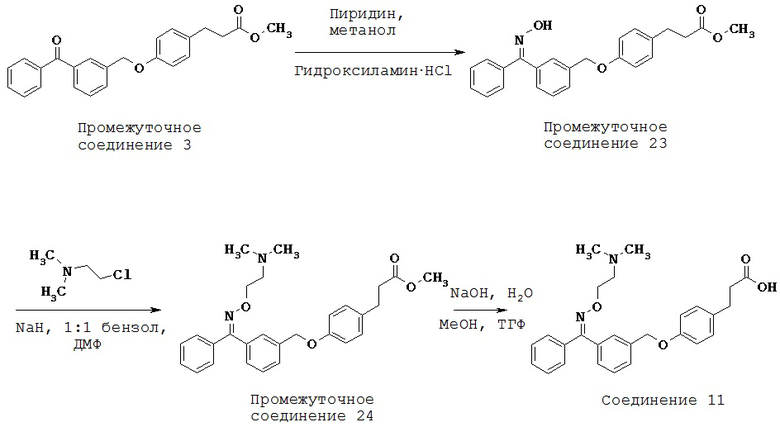
Пример 11
3-[4-({3-[(E,Z)-{[2-(диметиламино)этокси]имино}(фенил)метил]бензил}окси)фенил]-пропановая кислота (11):
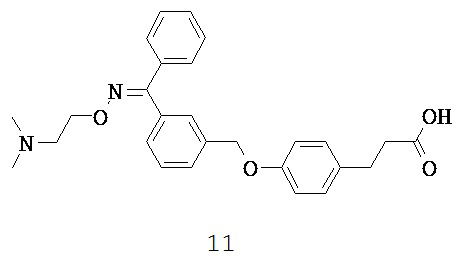
Соединение 11 синтезировали из 2-хлор-N,N-диметилэтанамина и метил-3-[4-({3-[(Z)-(гидроксиимино)(фенил)метил]бензил} окси)фенил]пропаноата согласно способу, описанному на Схеме 8.
Промежуточное соединение 23: метил-3-[4-({3-[(E,Z)-(гидроксиимино)(фенил)метил]бензил}окси)фенил]пропаноат
В 100 мл кругло донную колбу, оборудованную магнитной мешалкой и обратным холодильником, помещали 35 мл этанола и 7,8 мл пиридина. В перемешиваемый раствор добавляли метиловый эфир 3-[4-(3-бензоилбензилокси)фенил]пропановой кислоты (1,31 г, 3,498 ммоль), затем гидрохлорид гидроксиламина (1,2 г, 17,494 ммоль). Полученный раствор кипятили с обратным холодильником при 85°C в течение 3 часов. После завершения реакции (прохождение отслеживали путем ТСХ) растворитель удаляли при пониженном давлении и полученную неочищенную массу помещали в дихлорметан (100 мл). Органический слой промывали водой (100 мл × 2) и насыщенным солевым раствором (100 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Полученное неочищенное соединение очищали путем колоночной хроматографии на силикагеле (100/200 меш) с применением петролейного эфира (60-80) и этилацетата в качестве элюента. Продукт получали в виде желтого вязкого раствора (1,05 г, выход 78,4%): МС (ИЭР, 120 эВ): m/z=390,2 (М+Н)+; 1Н ЯМР (300 МГц, CDCl3): δ 11,36-11,37 (s, 1H), 7,46-7,49 (m, 4H), 7,36-7,43 (m, 2H), 7,25-7,33 (m, 3H), 7,10-7,13 (m, 2H), 6,85-6,92 (m, 2H), 5,05-5,09 (d, 2H), 3,56 (s, 3H), 2,72-2,79 (m, 2H), 2,55-2,60 (m, 2H).
Промежуточное соединение 24: метил-3-[4-({3-[(E,Z)-{[2-(диметиламино)этокси]имино}(фенил)метил]бензил}окси)фенил]пропаноат
В 100 мл круглодонную колбу, оборудованную магнитной мешалкой и обратным холодильником, помещали NaH (0,051 г, 1,28 ммоль) и 6 мл ДМФ в атмосфере азота. PC охлаждали до 0°C и в перемешиваемую массу добавляли метиловый эфир 3-{4-[3-(гидроксииминофенилметил)бензилокси]фенил}пропановой кислоты (0,5 г, 1,28 ммоль) в смеси 1:1 бензола и ДМФ (4 мл). Полученную смесь затем перемешивали в течение 15 минут и добавляли гидрохлорид 2-хлор-НМ-диметилэтиламина (0,20 г, 1,41 ммоль) при 0°C. Полученный раствор нагревали при 45°C в течение 3 часов. После завершения реакции (прохождение отслеживали путем ТСХ) растворитель удаляли при пониженном давлении. Полученную неочищенную массу помещали в диэтиловый эфир (100 мл), промывали водой (50 мл × 2) и насыщенным солевым раствором (50 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Полученное неочищенное соединение очищали путем колоночной хроматографии на силикагеле (100/200 меш) с применением петролейного эфира (60-80) и этилацетата в качестве элюента. Продукт получали в виде желтого вязкого раствора (0,175 г, выход 29,7%); МС (ИЭР, 120 эВ): m/z=461,2 (М+Н)+; 1Н ЯМР (300 МГц, CDCl3): δ 7,39-7,41 (m, 2Н), 7,31-7,36 (m, 4H), 7,24-7,28 (m, 3H), 7,02-7,05 (d, 2H), 6,78-6,84 (m, 2H), 4,93-4,97 (d, 2H), 4,25-4,28 (t, 2H), 3,59 (s, 3H), 2,79-2,85 (t, 2H), 2,66-2,67 (d, 2H), 2,50-2,55 (t, 2H), 2,18-2,28 (m, 6H).
Соединение 11: 3-[4-({3-[(E,Z)-{[2-(диметиламино)этокси]имино}(фенил)метил]бензил}окси)фенил]пропановая кислота
В 50 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 3 мл ТГФ, 0,5 мл воды и 0,5 мл метанола. В перемешиваемый растворитель добавляли метиловый эфир 3-(4-{3-[(2-диметиламиноэтоксиимино)фенилметил]бензилокси}фенил)пропановой кислоты (0,1 г, 0,217 ммоль), затем добавляли гидроксид натрия (0,026 г, 0,651 ммоль). Полученный раствор перемешивали при КТ в течение 15 часов. После завершения реакции (прохождение отслеживали путем ТСХ) PC разбавляли 10 мл воды и промывали дихлорметаном (20 мл × 2). Водный слой подкисляли до pH 3 при помощи концентрированной хлороводородной кислоты и экстрагировали ДХМ (75 мл × 3). Слой в ДХМ сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Продукт получали в виде белого твердого вещества (0,045 г, выход 15,5%); чистота 85,01%.
Схема 9:
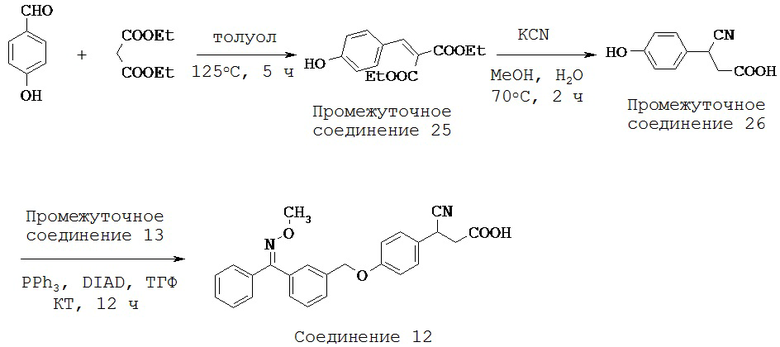
Пример 12
3-Циано-3-[4-({3-[(E,Z)-(метоксиимино)(фенил)метил]бензил}окси)фенил]пропановая кислота (12):
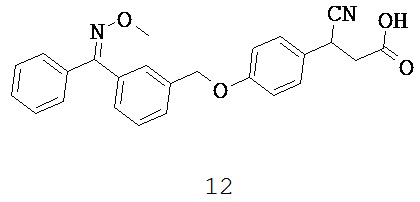
Соединение 12 синтезировали из 3-циано-3-(4-гидроксифенил)пропановой кислоты и (1Е,1Z)-2-бром-N-метокси-1-фенилэтанимина согласно способу, описанному на Схеме 9.
Промежуточное соединение 25: диэтил-(4-гидроксибензилиден)пропандиоат
В 100 мл кругло донную колбу, оборудованную магнитной мешалкой, помещали пиперидин (0,426 г, 0,5 мл, 5 ммоль) и уксусную кислоту (0,3 г, 0,3 мл, 5 ммоль). В полученную смесь добавляли толуол (25 мл), затем 4-гидроксибензальдегид (2 г, 16,4 ммоль) и диэтилмалонат (3,15 г, 19,7 ммоль). PC нагревали при 125°C с использованием насадки Дина-Старка в течение 5 часов. Через 5 часов PC концентрировали; воду (50 мл) добавляли и смесь экстрагировали этилацетатом (50 мл × 2). Органический слой промывали насыщенным солевым раствором (50 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Продукт получали в виде желтого твердого вещества (2 г, выход 46,2%); МС (ИЭР, 120 эВ): m/z=263,1 (М-Н)+; 1Н ЯМР (300 МГц, ДМСО-d6): δ 10,27 (s, 1Н), 7,58 (s, 1H), 7,36-7,38 (d, 2H), 6,81-6,84 (d, 2H), 4,17-4,32 (m, 4H), 4,06-4,14 (q, 1H), 1,21-1,26 (t, 6H).
Промежуточное соединение 26: 3-циано-3-(4-гидроксифенил)пропановая кислота
В 25 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали метанол (6 мл) и воду (1 мл). В перемешиваемый растворитель добавляли диэтиловый эфир 2-(4-гидроксибензилиден)малоновой кислоты (1 г, 3,8 ммоль), затем цианид калия (0,492 г, 7,6 ммоль). PC нагревали при 70°C в течение 2 часов. PC концентрировали, разбавляли насыщенным раствором NaHCO3 (50 мл) и промывали этилацетатом (50 мл × 2). Водный слой подкисляли до рН 3 при помощи 1 н. хлороводородной кислоты и экстрагировали этилацетатом (50 мл × 2). Органический слой промывали водой (50 мл) и насыщенным солевым раствором (50 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Продукт получали в виде коричневой жидкости (0,6 г, выход 72,3%); МС (ИЭР, 120 эВ): m/z=190,0 (М-Н)+; 1Н ЯМР (300 МГц, ДМСО-d6): δ 12,69 (s, 1H), 9,58 (s, 1H), 7,22-7,24 (d, 2H), 6,75-6,78 (d, 2H), 4,28-4,40 (m, 1H), 2,77-3,12 (m, 2H).
Соединение 12: 3-Циано-3-[4-({3-[(E,Z)-(метоксиимино)(фенил)метил]бензил}окси)фенил]пропановая кислота
В 25 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали этанол (3 мл). В перемешиваемый растворитель добавляли 3-циано-3-(4-гидроксифенил)-пропановую кислоту (0,1 г, 0,52 ммоль), O-метилоксим (3-бромметилфенил)-фенилметанона (0,159 г, 0,52 ммоль) и гидроксид натрия (0,03 г, 0,75 ммоль) в воде (1 мл). PC перемешивали при КТ в течение 8 часов. PC концентрировали, воду (10 мл) добавляли и смесь экстрагировали этилацетатом (15 мл × 2). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Продукт получали в виде бледно-желтого твердого вещества (0,017 г, выход 7,8%); чистота: 94,19%.
Схема 10:
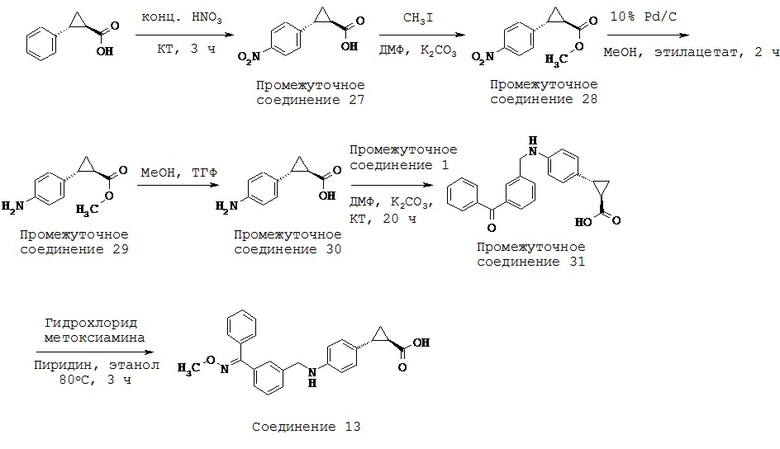
Пример 13
(1R,2R)-2-[4-({3-[(E,Z)-(метоксиимино)(фенил)метил]бензил}амино)фенил]-циклопропанкарбоновая кислота (13):
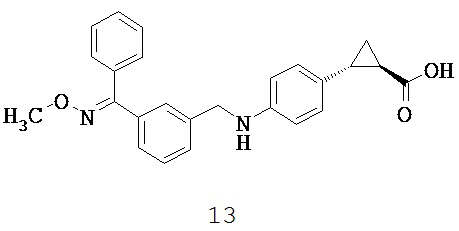
Соединение 13 получали с применением (1R,2R)-2-{4-[(3-бензоилбензил)амино]-фенил}циклопропанкарбоновой кислоты (0,5 г, 1,3 ммоль) и гидрохлорида метоксиламина (0,56 г, 6,7 ммоль) согласно способу, описанному на Схеме 10.
Промежуточное соединение 27: (1R,2R)-2-(4-нитрофенил)циклопропанкарбоновая кислота
В 1000 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 150 мл HNO3, охлажденной до 0°C, и добавляли (1R,2R)-2-фенилциклопропанкарбоновую кислоту (15 г, 92,593 ммоль) по частям в течение получаса, PC нагревали до КТ и перемешивали в течение 3 часов. После завершения реакции PC выливали в ледяную воду, перемешивали в течение 30 минут, отфильтровывали продукт и сушили в вакууме. Полученный продукт представлял собой белое твердое вещество (15 г, выход 78,2%).
Промежуточное соединение 28: метил-(1R,2R)-2-(4-нитрофенил)циклопропанкарбоксилат
В 500 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 120 мл ДМФ. В перемешиваемый растворитель добавляли (1R,2R)-2-(4-нитрофенил)-циклопропанкарбоновую кислоту (15 г, 72 ммоль), K2CO3 (29 г, 217 ммоль) и метилйодид (5,5 мл, 86 ммоль), перемешивали при КТ в течение 8 часов. PC выливали в воду (100 мл) и экстрагировали этилацетатом (150 мл × 2). Органический слой промывали насыщенным раствором NaHCO3 (50 мл), водой (50 мл) и насыщенным солевым раствором (50 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Продукт получали в виде бледно-желтой жидкости (14 г, выход 87,9%). МС (ИЭР, 120 эВ): m/z=206,1 (М-Н)+.
Промежуточное соединение 29: метил-(1R,2R)-2-(4-аминофенил)циклопропанкарбоксилат
В 1000 мл колбу для гидрогенизатора Парра, содержащую метанол (70 мл) и этилацетат (70 мл), добавляли метил-(1R,2R)-2-(4-нитрофенил)циклопропанкарбоксилат (14 г, 63,3 ммоль) и 10% Pd/C (2 г) в атмосфере N2. PC гидрировали при 40 psi в течение 5 часов, PC разбавляли этилацетатом (100 мл), фильтровали через подложку целита для удаления Pd/C и выпаривали насухо при пониженном давлении. Неочищенное вещество очищали путем колоночной флэш-хроматографии на силикагеле (230/400 меш) с получением продукта (4 г, выход 33,1%). МС (ИЭР, 120 эВ): m/z=192,1 (М+Н)+; 1Н ЯМР (300 МГц, CDCl3): δ 6,83-6,86 (d, 2H), 6,58-6,61 (d, 2H), 3,63 (s, 3H), 2,36-2,39 (m, 1H), 1,69-1,75 (m, 1H), 1,42-1,49 (m, 1H), 1,14-1,20 (m, 1H).
Промежуточное соединение 30: (1R,2R)-2-(4-аминофенил)циклопропанкарбоновая кислота
В 25 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 5 мл метанола и 5 мл ТГФ. В перемешиваемый растворитель добавляли метил-(1R,2R)-2-(4-аминофенил)циклопропанкарбоксилат (1 г, 5,23 ммоль) и NaOH (0,627 г, 15,69 ммоль) в воде (3 мл). Полученный раствор перемешивали при КТ в течение 3 часов. После завершения реакции (прохождение отслеживали путем ТСХ) растворитель удаляли при пониженном давлении. PC разбавляли 3 мл воды, охлаждали до 0°C, затем подкисляли до pH 3 при помощи 1 н. HCl и экстрагировали этилацетатом (50 мл × 2). Органический слой промывали водой (50 мл) и насыщенным солевым раствором (30 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Продукт получали в виде белого твердого вещества (0,9 г, выход 97,2%).
Промежуточное соединение 31: (1R,2R)-2-{4-[(3-бензоилбензил)амино]фенил}циклопропанкарбоновая кислота
Получали из (1R,2R)-2-(4-аминофенил)циклопропанкарбоновой кислоты (0,7 г, 3,9 ммоль) и [3-(бромметил)фенил](фенил)метанона (0,9 г, 3,2 ммоль) согласно способу, описанному на Схеме 2 для получения промежуточного соединения 5 (0,5 г, выход 39%). МС (ИЭР, 120 эВ); m/z=372,0 (М+Н)+.
Соединение 13: (1R,2R)-2-[4-({3-[(E,Z)-(метоксиимино)(фенил)метил]бензил}амино)фенил]циклопропанкарбоновая кислота
Получали из (1R,2R)-2-{4-[(3-бензоилбензил)амино]фенил}циклопропанкарбоновой кислоты (0,5 г, 1,3 ммоль) и гидрохлорида метоксиламина (0,56 г, 6,7 ммоль) согласно способу, описанному на Схеме 2 для получения соединения 2 (0,01 г, выход 1,9%); чистота: 94,46% (48,37%: 44,56%).
Пример 14
(1R,2R)-2-{4-[(4-{[(2E,2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)амино]фенил}циклопропанкарбоновая кислота (14):
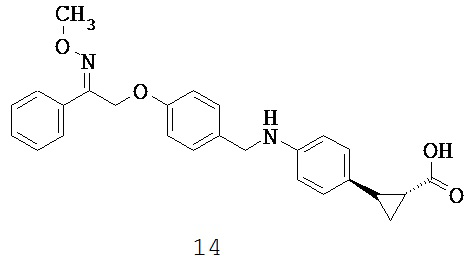
Соединение 14 синтезировали из метил-(1R,2R)-2-(4-аминофенил)-циклопропанкарбоксилата (0,3 г, 1,5 ммоль) и 4-{[(2)-2-(метоксиимино)-2-фенилэтил]окси}-бензальдегида (0,4 г, 1,5 ммоль) согласно способу, описанному на Схеме 10 (0,15 г, выход 23,8%); чистота: 95,4%.
Пример 15
(1R,2R)-2-{4-[(4-{[(2Z)-2-(метоксиимино)-2-(2-метилфенил)этил]окси}бензил)амино]фенил}циклопропанкарбоновая кислота (15):
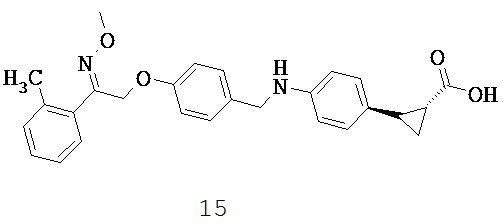
Соединение 15 синтезировали из метил-(1R,2R)-2-(4-аминофенил)-циклопропанкарбоксилата (0,18 г, 0,9 ммоль) и 4-{[(2Z)-2-(метоксиимино)-2-(2-метилфенил)-этил]окси}бензальдегида (0,31 г, 1,1 ммоль) согласно способу, описанному на Схеме 3 (0,12 г, выход 28%); чистота: 91,26%.
Пример 16
3-циано-3-{4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропановая кислота (16):
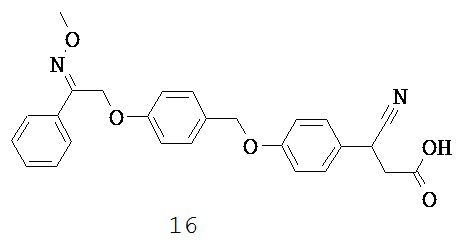
Соединение 16 синтезировали из (4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}-фенил)метанола (0,264 г, 0,98 ммоль) и метил-3-циано-3-(4-гидроксифенил)пропаноата (0,2 г, 0,98 ммоль) согласно способу, описанному на Схеме 5 (0,15 г, выход 34,7%); чистота: 98,2%.
Схема 11:
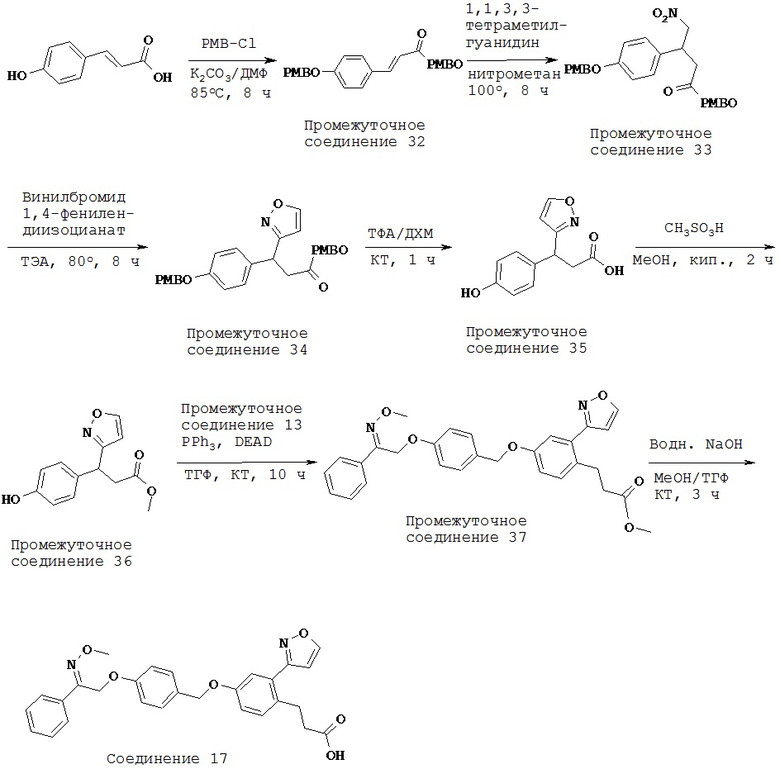
Пример 17
3-{4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}-3-(1,2-оксазол-3-ил)пропановая кислота (17):
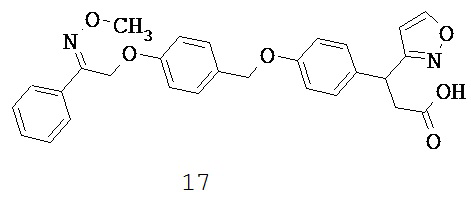
Соединение 17 получали с применением метил-3-(4-гидроксифенил)-3-(1,2-оксазол-3-ил)пропаноата и (4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}фенил)метанола согласно способу, описанному на Схеме 11.
Промежуточное соединение 32: 4-метоксибензил-(2Z)-3-{4-[(4-метоксибензил)окси]фенил}проп-2-еноат
В 250 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 25 мл ДМФ. В перемешиваемый растворитель добавляли (2Z)-3-(4-гидроксифенил)проп-2-еновую кислоту (2 г, 12,18 ммоль), K2CO3 (6,67 г, 48,73 ммоль) и 1-(хлорметил)-4-метоксибензол (3,81 г, 24,36 ммоль), перемешивали при 85°C в течение 8 часов в атмосфере азота. PC концентрировали путем удаления растворителя. Неочищенное вещество растворяли в этилацетате (100 мл). Органический слой промывали водой (50 мл) и насыщенным солевым раствором (50 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Продукт получали в виде белого твердого вещества (4,5 г, выход 91,83%). МС (ИЭР, 120 эВ): m/z=403 (М-Н)+; 1H ЯМР (300 МГц, CDCl3): δ 7,55-7,60 (d, 1H), 7,35-7,38 (d, 2H), 7,25-7,28 (dd, 4H), 6,81-6,88 (m, 5H), 5,09 (s, 2H), 4,92 (s, 2H), 3,73 (s, 6H).
Промежуточное соединение 33: 4-метоксибензил-3-{4-[(4-метоксибензил)окси]фенил}-4-нитробутаноат
В 500 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 10 мл нитрометана. В перемешиваемый растворитель добавляли 4-метоксибензил-(2Е,2Z)-3-{4-[(4-метоксибензил)окси]фенил}проп-2-еноат (1 г, 2,47 ммоль) и 1,1,3,3-тетраметилгуанидин (0,14 г, 1,28 ммоль), перемешивали при КТ в течение 8 часов, затем повышали температуру до 50°C и перемешивали в течение 3 часов, а затем при 100°C в течение 8 часов. PC концентрировали путем удаления растворителя и неочищенное вещество растворяли в этилацетате (100 мл). Органический слой промывали водой (50 мл) и насыщенным солевым раствором (50 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали путем колоночной хроматографии на силикагеле (100/200 меш), продукт получали в виде бледно-желтого твердого вещества (0,4 г, выход 58,82%). 1Н ЯМР (300 МГц, CDCl3): δ 7,26-7,29 (d, 2H), 7,09-7,12 (d, 2H), 7,01-7,04 (d, 2H), 6,76-6,86 (m, 6H), 4,91 (s, 2H), 4,87 (s, 2H), 4,46-4,62 (m, 2H), 3,83-3,88 (t, 1H), 3,74 (s, 3H), 3,72 (s, 3H), 2,66-2,69 (d, 2H).
Промежуточное соединение 34: 4-метоксибензил-3-{4-[(4-метоксибензил)окси]фенил}-3-(1,2-оксазол-3-ил)пропаноат
В 50 мл кругло донную колбу, оборудованную магнитной мешалкой, помещали 3 мл триэтиламина. В колбу добавляли 4-метоксибензил-3-{4-[(4-метоксибензил)окси]фенил}-4-нитробутаноат (0,25 г, 0,537 ммоль), затем добавляли винилбромид (4 мл), 1,4-фенилендиизоцианат (0,3 г, 1,87 ммоль), кипятили с обратным холодильником при 85°C в течение 8 часов. PC охлаждали до КТ, осаждалось твердое вещество. Твердое вещество удаляли путем фильтрования, а фильтрат выпаривали при пониженном давлении. Получали продукт в виде коричневого вязкого вещества (0,1 г, выход 40,2%). 1Н ЯМР (300 МГц, CDCl3): δ 8,17-8,18 (d, 1Н), 7,25-7,28 (d, 2H), 7,05-7,11 (t, 4H), 6,75-6,85 (m, 6H), 5,97-5,98 (d, 1Н), 4,92 (s, 2H), 4,86 (s, 2H), 4,46-4,52 (t, 1Н), 3,73 (s, 3H), 3,72 (s, 3H), 3,18-3,26 (q, 1Н), 2,85-2,93 (q, 1H).
Промежуточное соединение 35: 3-(4-гидроксифенил)-3-(1,2-оксазол-3-ил)пропановая кислота
В 100 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 30 мл ДХМ. В перемешиваемый растворитель добавляли 4-метоксибензил-3-{4-[(4-метоксибензил)окси]фенил}-3-(1,2-оксазол-3-ил)пропаноат (3 г, 6,33 ммоль) и охлаждали до 0°C, в полученную смесь добавляли 30 мл трифторуксусной кислоты по каплям. Полученный раствор перемешивали при КТ в течение 1 часа. После завершения реакции (прохождение отслеживали путем ТСХ) растворитель удаляли при пониженном давлении. PC разбавляли 30 мл метанола, осаждалось твердое вещество. Твердое вещество удаляли путем фильтрования, а фильтрат выпаривали при пониженном давлении. Продукт получали в виде вязкого вещества (1,48 г, выход 99%). МС (ИЭР, 120 эВ): m/z=234,2 (М+Н)+; 1Н ЯМР (300 МГц, CDCl3): δ 8,23 (d, 1Н), 6,98-7,01 (d, 2H), 6,67-6,70 (d, 4H), 6,04-6,05 (d, 1Н), 4,42-4,47 (t, 1Н), 3,07-3,15 (q, 1Н), 2,75-2,83 (q, 1Н).
Промежуточное соединение 36: метил-3-(4-гидроксифенил)-3-(1,2-оксазол-3-ил)пропаноат
В 100 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 20 мл метанола. В перемешиваемый растворитель добавляли 3-(4-гидроксифенил)-3-(1,2-оксазол-3-ил)пропановую кислоту (1,48 г, 6,34 ммоль) и охлаждали до 0°C, добавляли 1 мл метансульфокислоты. Полученную смесь перемешивали при температуре кипения в течение 2 часов. После завершения реакции (прохождение отслеживали путем ТСХ) растворитель удаляли при пониженном давлении и неочищенную массу растворяли в этилацетате (25 мл). Органический слой промывали водой (50 мл), насыщенным солевым раствором (50 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Продукт получали в виде бледно-желтой маслянистой жидкости (0,69 г, выход 44%); МС (ИЭР, 120 эВ): m/z=248,9 (М+Н)+; 1Н ЯМР (300 МГц, CDCl3): δ 8,20 (s, 1Н), 7,01-7,04 (d, 2H), 6,66-6,69 (d, 2H), 6,00 (d, 1H), 5,56 (bro s, 1H), 4,46-4,51 (t, 1H), 3,56 (s, 3H), 3,17-3,25 (q, 1H), 2,83-2,91 (q, 1H).
Промежуточное соединение 37: метил-3-{4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}-3-(1,2-оксазол-3-ил)пропаноат
В 50 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали сухой ТГФ (5,0 мл) и метил-3-(4-гидроксифенил)-3-(1,2-оксазол-3-ил)пропаноат (0,18 г, 0,74 ммоль). В полученную выше смесь (4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}фенил)метанол (0,2 г, 0,74 ммоль) добавляли и полученную смесь перемешивали при 0°C в течение 5 минут. Трифенилфосфин (0,25 г, 0,96 ммоль) добавляли в смесь и перемешивали при 0°C в течение 15 минут, затем добавляли диизопропилазадикарбоксилат (0,19 г, 0,96 ммоль). После перемешивания полученной смеси при КТ в течение 10 часов PC выпаривали для удаления ТГФ. Остаток разбавляли водой (15 мл) и экстрагировали этилацетатом (15 мл × 2). Органический слой промывали насыщенным солевым раствором (15 мл) и сушили над безводным Na2SO4. Концентрирование растворителя и очистка полученного остатка путем колоночной хроматографии на силикагеле (100/200 меш) с применением петролейного эфира (60-80) и этилацетата в качестве элюента приводили к получению продукта (0,1 г, выход 27,47%); МС (ИЭР, 120 эВ): m/z=501,1 (М+Н)+; 1Н ЯМР (300 МГц, CDCl3): δ 8,20 (d, 1H), 7,57-7,60 (t, 2H), 7,22-7,28 (m, 5H), 7,07-7,10 (d, 2H), 6,81-6,85 (m, 4H), 5,99-6,00 (d, 1H), 5,12 (s, 2H), 4,85 (s, 2H), 4,47-4,52 (t, 1H), 3,98 (s, 3H), 3,56 (s, 3H), 3,18-3,26 (q, 1H), 2,82-2,90 (q, 1H).
Соединение 17: 3-{4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}-3-(1,2-оксазол-3-ил)пропановая кислота
В 25 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 2 мл метанола и 2 мл ТГФ. В перемешиваемый растворитель добавляли метил-3-{4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}-3-(1,2-оксазол-3-ил)пропаноат (0,1 г, 0,2 ммоль) и NaOH (0,03 г, 0,6 ммоль) в воде (1 мл). Полученный раствор перемешивали при КТ в течение 3 часов. После завершения реакции (прохождение отслеживали путем ТСХ) растворитель удаляли при пониженном давлении. PC разбавляли 2 мл воды, охлаждали до 0°C, затем подкисляли до рН 3 при помощи 1 н. HCl и экстрагировали этилацетатом (15 мл × 2). Органический слой промывали водой (20 мл) и насыщенным солевым раствором (20 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Продукт получали в виде белого твердого вещества (0,015 г, выход 15,5%); чистота: 97,68%.
Схема 12
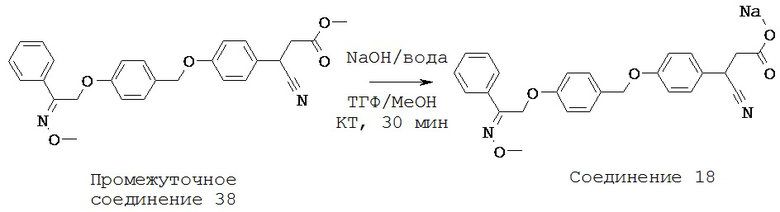
Пример 18
3-циано-3-{4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропаноат натрия (18):

Соединение 18 синтезировали из (4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}-фенил)метанола (0,264 г, 0,98 ммоль) и метил-3-циано-3-(4-гидркосифенил)пропаноата (0,2 г, 0,98 ммоль) согласно способу, описанному на Схеме 5 (0,15 г, выход 34,7%); чистота: 99,9%.
Пример 19
[4-({3-[(E,Z)-(метоксиимино)(фенил)метил]бензил}окси)фенил]уксусная кислота (19)
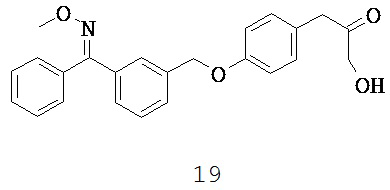
Соединение 19 получали с применением (E,Z)-1-[3-(бромметил)фенил]-N-метокси-1-фенилметанимина (1,0 г, 3,635 ммоль) и (4-гидроксифенил)уксусной кислоты (0,553 г, 3,635 ммоль) согласно способу, описанному на схеме 1. Продукт получали в виде бесцветной жидкости (0,1 г, выход 30,84%); чистота: 94,3%.
Пример 20
3-{4-[(6-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}пиридин-3-ил)метокси]фенил}пропановая кислота (20)
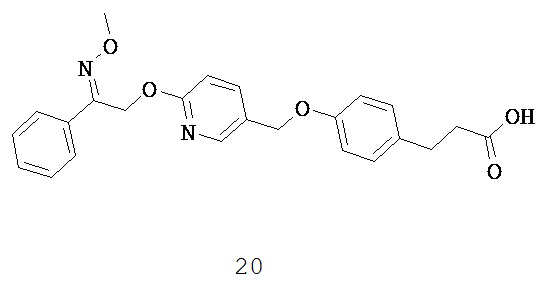
Соединение 20 получали с применением (2Z)-2-(метоксиимино)-2-фенилэтил]окси}пиридин-3-ил)метанола (0,34 г, 1,248 ммоль) и метил-3-(4-гидроксифенил)пропаноата (0,23 г, 1,248 ммоль), а также DIAD вместо DEAD в качестве реагента, согласно способу, описанному на схеме 5. Продукт получали в виде белого твердого вещества (0,08 г, выход 45,93%); чистота 84,8%.
Схема 13
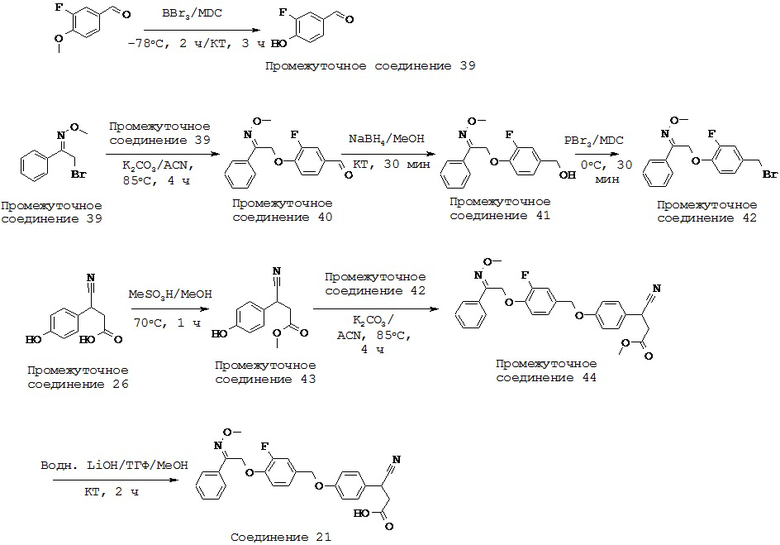
Пример 21
3-циано-3-{4-[(3-фтор-4-{[(2Е,2Z)-2-(метоксиимино)-2-5 фенилэтил]окси}бензил)окси]фенил}пропановая кислота (21)
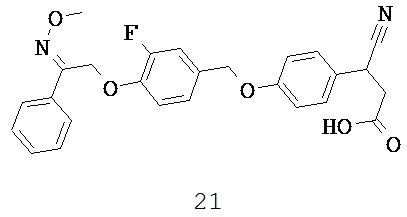
Соединение 21 получали с применением метил-3-циано-3-(4-гидроксифенил)-пропаноата и (1Е,1Z)-2-[4-(бромметил)-2-фторфенокси]-N-метокси-1-фенилэтанимина согласно способу, описанному на схеме 13.
Промежуточное соединение 39: 3-фтор-4-гидроксибензальдегид
В 25 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 20 мл дихлорметана. В перемешиваемый растворитель добавляли 3-фтор-4-метоксибензальдегид (2 г, 12,97 ммоль). Реакционную смесь охлаждали до -78°C и трибромид бора (51 мл) добавляли по каплям, перемешивали в течение 2 часов при указанной температуре, затем при KT в течение примерно 16 часов. Реакцию гасили путем медленного добавления раствора NaHCO3 при 0°C. Реакционную смесь концентрировали для отгонки растворителя; этилацетат (20 мл) добавляли. Органический слой промывали насыщенным раствором NaHCO3 (20 мл), затем солевым раствором (20 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Продукт получали в виде белого твердого вещества (1,0 г, выход 54,94%). 1Н ЯМР (300 МГц, CDCl3): δ 9,77 (s, 1H), 7,54-7,59 (m, 1H), 7,08-7,12 (t, 1H), 6,40 (s, 1H).
Промежуточное соединение 40: 3-фтор-4-{[(2Е,2Z)-2-(метоксиимино)-2-фенилэтил] окси}бензальдегид
В 250 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 75 мл ацетонитрила. В перемешиваемый растворитель добавляли O-метилоксим (1E,1Z)-2-бром-1-фенилэтанона (1,1 г, 5,0 ммоль), карбонат калия (2,3 г, 15,00 ммоль). При 0°C 3-фтор-4-гидроксибензальдегид (0,7 г, 5,0 ммоль) в ацетонитриле (10 мл) добавляли по каплям и перемешивали при 85°C в течение 4 часов в атмосфере азота. PC фильтровали через керамическую воронку и промывали этилацетатом. Фильтрат концентрировали для отгонки растворителя. Воду (30 мл) добавляли и экстрагировали этилацетатом (20 мл × 3). Органический слой промывали водой (30 мл) и насыщенным солевым раствором (30 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали путем колоночной хроматографии на силикагеле с применением этилацетата и петролейного эфира в качестве элюентов. Продукт получали в виде желтой жидкости (1 г, выход 72%). 1Н ЯМР (300 МГц, CDCl3): δ 9,86 (s, 1H), 7,78-7,81 (d, 1H), 7,64-7,68 (m, 3Н), 7,40-7,45 (m, 4H), 5,45 (s, 2H), 4,03 (s, 3Н).
Промежуточное соединение 41: O-метилоксим (1Е,1Z)-2-[2-фтор-4-(гидроксиметил)фенокси]-1-фенилэтанона
В 25 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 4 мл метанола. В перемешиваемый растворитель добавляли 3-фтор-4-{[(2Е,2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензальдегид (0,83 г, 2,89 ммоль). Реакционную смесь охлаждали до 0°C и боргидрид натрия (0,16 г, 4,33 ммоль) добавляли по частям. Реакционную смесь оставляли перемешиваться в течение 30 минут. Затем реакционную смесь концентрировали для отгонки растворителя. Воду (10 мл) добавляли и экстрагировали этилацетатом (10 мл × 3). Органический слой промывали водой (50 мл) и насыщенным солевым раствором (10 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Продукт получали в виде бесцветной жидкости (0,7 г, выход 84,34%). 1Н ЯМР (300 МГц, CDCl3): δ 7,60-7,63 (m, 2H), 7.27-7,29 (m, 3Н), 6,89-6,98 (m, 3Н), 5,19 (s, 2H), 4,49 (s, 2H), 3,97 (s, 3H).
Промежуточное соединение 42: O-метилоксим (1Е,1Z)-2-[4-(бромметил)-2-фторфенокси]-1-фенилэтанона
В 25 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали (3-фтор-4-{[(2Е,2Z)-2-(метоксиимино)-2-фенилэтил]окси}фенил)метанол (0,45 г, 1,55 ммоль) и ДХМ (4 мл). В перемешиваемый раствор трибромид фосфора (0,5 мл, 3 экв.) добавляли при 0°C и перемешивали в течение 30 минут в атмосфере азота. PC выливали в воду (5 мл) и экстрагировали этилацетатом (20 мл). Органический слой промывали водой (10 мл) и насыщенным солевым раствором (10 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Продукт получали в виде бесцветной жидкости (0,24 г, выход 44,44%). 1Н ЯМР (300 МГц, CDCl3): δ 7,60-7,63 (m, 2H), 7,27-7,29 (m, 3Н), 6,98-7,02 (m, 2H), 6,86-6,91 (t, 1H), 5,19 (s, 2H), 4,33 (s, 2H), 3,97 (s, 3Н).
Промежуточное соединение 43: метил-3-циано-3-(4-гидроксифенил)пропаноат
В 50 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали метанол (15 мл). В перемешиваемый растворитель добавляли 3-циано-3-(4-гидроксифенил)-пропановую кислоту (0,6 г, 3,1 ммоль), затем метансульфокислоту (1 мл). Реакционную смесь кипятили с обратным холодильником в течение 1 часа. После завершения реакции реакционную смесь концентрировали для отгонки растворителя. Воду (10 мл) добавляли и экстрагировали этилацетатом (25 мл). Органический слой промывали водой (5 мл) и насыщенным солевым раствором (5 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Продукт получали в виде бледно-желтой жидкости (0,6 г, 93,3%). 1Н ЯМР (300 МГц, CDCl3): δ 7,13-7,15 (d, 2H), 6,75-6,78 (d, 2H), 4,14-4,19 (t, 1H), 3,65 (s, 3Н), 2,89-2,97 (m, 1Н), 2,71-2,79 (m, 1H).
Промежуточное соединение 44: метил-3-циано-3-{4-[(3-фтор-4-{[(2Е,2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропаноат
В 250 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 2 мл ацетонитрила. В перемешиваемый растворитель добавляли (1Е,1Z)-2-[4-(бромметил)-2-фторфенокси]-N-метокси-1-фенилэтанимин (0,24 г, 0,68 ммоль), карбонат калия (0,28 г, 2,0 ммоль) при 0°С, затем добавляли по каплям метил-3-циано-3-(4-гидроксифенил)-пропаноат (0,14 г, 0,68 ммоль) в ацетонитриле (2 мл) и перемешивали при 85°С в течение 4 часов в атмосфере азота. PC фильтровали через керамическую воронку и промывали этилацетатом. Фильтрат концентрировали для отгонки растворителя. Остаток экстрагировали этилацетатом (10 мл). Органический слой промывали водой (5 мл) и насыщенным солевым раствором (5 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали путем колоночной хроматографии на силикагеле (этилацетат:гексан=5:95-30:70). Продукт получали в виде желтой жидкости (0,18 г, выход 56,25%). 1Н ЯМР (300 МГц, CDCl3): δ 7,64-7,67 (m, 2H), 7,35-7,41 (m, 5H), 7,18-7,28 (m, 3H), 6,99-7,02 (d, 2H), 5,30 (s, 2H), 5,01 (s, 2H), 4,42-4,47 (q, 1H), 3,99-4,06 (s, 3H), 3,62 (s, 3H), 3,07-3,16 (q, 1H), 2,90-2,97 (q, 1H).
Соединение 21: 3-циано-3-{4-[(3-фтор-4-{[(2Е,2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропановая кислота
В 10 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 2 мл тетрагидрофурана. В перемешиваемый растворитель добавляли метил-3-циано-3-{4-[(3-фтор-4-{[(2Е,2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропаноат
(0,1 г, 0,21 ммоль) и метанол (2 мл). Реакционную смесь охлаждали до 0°C и гидроксид лития (0,03 г, 1,2 ммоль) в воде (2 мл) добавляли по каплям. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. После завершения реакции реакционную смесь концентрировали для отгонки растворителя. Полученную соль растворяли в воде (1 мл) и экстрагировали диэтиловым эфиром (5 мл). Водный слой подкисляли 1 н. HCl до pH 3 и экстрагировали диэтиловым эфиром (5 мл). Органический слой промывали солевым раствором (5 мл), сушили от растворителя безводным Na2SO4 и концентрировали с получением вязкого твердого вещества (0,09 г, выход 92,73%). МС (ИЭР, 120 эВ): m/z=463,1 (М+Н)+; чистота по данным ВЭЖХ: 89,53%.
Пример 22
3-циано-3-{4-[(4-{[(2Е,2Z)-2-(4-фторфенил)-2-(метоксиимино)этил]окси}бензил)окси]фенил}пропановая кислота (22)
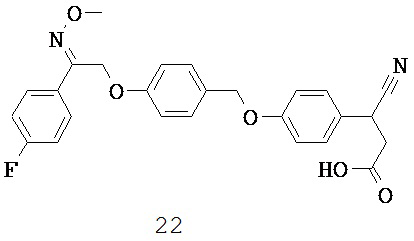
Соединение 22 синтезировали из O-метилоксима (1Е,1Z)-1-(4-фторфенил)-2-[4-(гидроксиметил)фенокси]этанона (0,212 г, 0,731 ммоль) и метил-3-циано-3-(4-гидроксифенил)пропаноата (0,1 г, 0,487 ммоль) согласно способу, описанному на схеме 13 (0,02 г, выход 44%); чистота: 98,58%.
Пример 23
3-циано-3-{4-[(2-фтор-4-{[(2Е,2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропановая кислота (23)
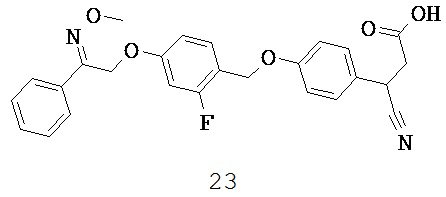
Соединение 23 синтезировали из O-метилоксима (1Е,1Z)-2-[3-фтор-4-(гидроксиметил)-фенокси]-1-фенилэтанона (0,14 г, 0,397 ммоль) и метил-3-циано-3-(4-гидроксифенил)-пропаноата (0,0815 г, 0,397 ммоль) согласно способу, описанному на схеме 13 (0,04 г, выход 41,23%); чистота: 88,0%.
Пример 24
3-{4-[(4-{[(2Е,2Z)-2-(метоксиимино)-2-(3-метилфенил)этил]окси}бензил)окси]фенил}-3-(1,2-оксазол-3-ил)пропановая кислота (24)
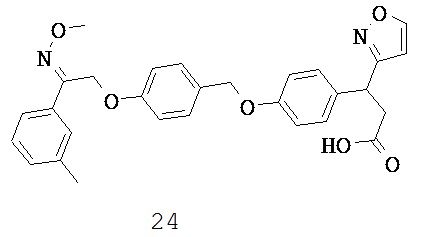
Соединение 24 синтезировали из (1Е,1Z)-2-[4-(бромметил)фенокси]-N-метокси-1-(3-метилфенил)этанимина (0,2 г, 0,5 ммоль) и метил-3-(4-гидроксифенил)-3-(1,2-оксазол-3-ил)пропаноата (0,14 г, 0,56 ммоль) с применением DIAD вместо DEAD в качестве агента сочетания согласно способу, описанному на схемах 11 и 13 (0,04 г, выход 14,0%); чистота: 96,74%.
Пример 25
3-циано-3-{4-[(4-{[(2Е,2Z)-2-(метоксиимино)-2-(4-метоксифенил)этил]окси}бензил)окси]фенил}пропановая кислота (25)
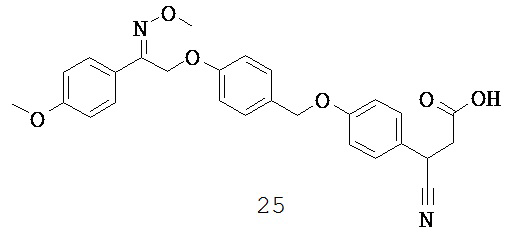
Соединение 25 синтезировали из O-метилоксима (1Е,1Z)-2-[4-(гидроксиметил)-фенокси]-1-(4-метоксифенил)этанона (0,2 г, 0,66 ммоль) и метил-3-циано-3-(4-гидроксифенил)пропаноата (0,136 г, 0,66 ммоль) с применением DIAD вместо DEAD в качестве агента сочетания согласно способу, описанному на схеме 11 (0,0038 г, выход 3,01%); чистота: 89,41%.
Пример 26
3-циано-3-{4-[(3-метокси-4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропановая кислота (26)
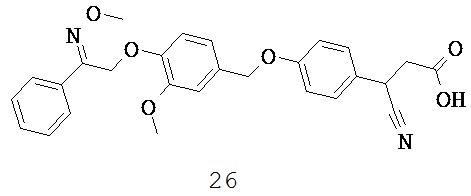
Соединение 26 синтезировали из O-метилоксима (1Z)-2-[4-(гидроксиметил)-2-метоксифенокси]-1-фенилэтанона (0,2 г, 0,66 ммоль) и метил-3-циано-3-(4-гидроксифенил)-пропаноата (0,136 г, 0,66 ммоль) согласно способу, описанному на схеме 11 (0,0032 г, выход 7,77%); чистота: 95,56%.
Пример 27
3-циано-3-{4-[(4-{[(2Е,2Z)-2-(метоксиимино)-2-(3-метилфенил)этил]окси}бензил)окси]фенил}пропановая кислота (27)
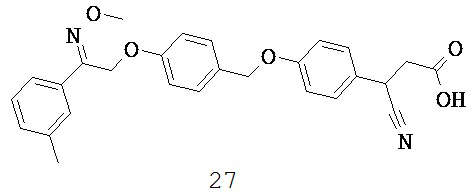
Соединение 27 синтезировали из (1E,1Z)-2-[4-(бромметил)фенокси]-N-метокси-1-(3-метилфенил)этанамина (0,2 г, 0,5 ммоль) и метил-3-(4-гидроксифенил)-3-(1,2-оксазол-3-ил)-пропаноата (0,14 г, 0,56 ммоль) с применением DIAD вместо DEAD в качестве агента сочетания. Затем проводили гидролиз метил-3-{4-[(4-{[(2Е,2Z)-2-(метоксиимино)-2-(3-метилфенил)этил]окси}бензил)окси]фенил}-3-(1,2-оксазол-З -ил)пропаноата (0,150 г, 1,0 экв.) водным раствором NaOH (60 мг, 2,5 экв.) согласно способу, описанному на схемах 11 и 13 (0,04 мг, 14,0%); чистота: 96,2%.
Пример 28
3-{4-[(4-{[(2Е,2Z)-2-(4-цианофенил)-2-(метоксиимино)этил]окси}бензил)окси]фенил}пропановая кислота(28)
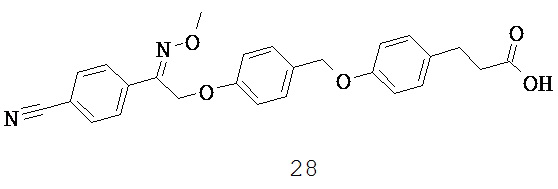
Соединение 28 синтезировали из 4-{(1Е,1Z)-2-[4-(гидроксиметил)фенокси]-N-метоксиэтанимидоил}бензонитрила (0,26 г, 0,668 ммоль) и метил-3-(4-гидроксифенил)-пропаноата (0,144 г, 0,802 ммоль) согласно способу, описанному на схеме 13 (0,098 г, выход 84%); чистота: 93,06%.
Пример 29
3-{4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}-3-(1,2-оксазол-3-ил)пропаноат натрия (29)
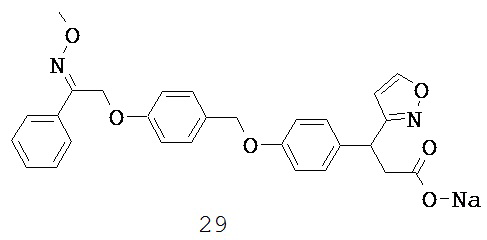
Соединение 29 синтезировали из 3-{4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}-бензил)окси]фенил}-3-(1,2-оксазол-3-ил)пропановой кислоты (0,07 г, 0,14 ммоль) и гидроксида натрия (0,0057 г, 0,14 ммоль) согласно способу, описанному на схеме 12 (0,052 г, выход 71,2%); чистота: 99,00%.
Схема 14

Пример 30
({4-[(4-{[(2E,2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]бензил}окси)уксусная кислота (30)
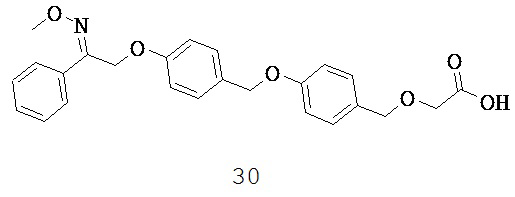
Соединение 30 синтезировали из {4-[(4-{[(2Е,2Z)-2-(метоксиимино)-2-фенилэтил]-окси}бензил)окси]фенил}метанола (0,1 г, 0,26 ммоль) и бромуксусной кислоты (0,022 г, 0,15 ммоль) согласно способу, описанному на схеме 14 (0,01 г, выход: 6,25%); чистота: 59,30%.
Промежуточное соединение 45: 4-[(4-{[(2Е,2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]бензальдегид
В 50 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали сухой ТГФ (5,0 мл) и (4-{[(2Е,2Z)-2-(метоксиимино)-2-фенилэтил]окси}фенил)метанол (1,5 г, 5,5 ммоль), в полученную выше смесь 4-гидроксибензальдегид (0,81 г, 6,6 ммоль) добавляли и полученную смесь перемешивали при 0°C в течение 5 минут. Трифенилфосфин (1,88 г, 7,15 ммоль) добавляли в смесь и перемешивали при 0°C в течение 15 минут, затем добавляли диизопропилазадикарбоксилат (1,44 г, 7,15 ммоль). После перемешивания полученной смеси при КТ в течение 16 часов PC выпаривали для удаления ТГФ. Остаток разбавляли водой (50 мл) и экстрагировали этилацетатом (150 мл × 2). Органический слой промывали насыщенным солевым раствором (50 мл) и сушили над безводным Na2SO4. Концентрирование растворителя при пониженном давлении приводило к получению продукта (0,7 г, выход 35,0%).
Промежуточное соединение 46: {4-[(4-{[(2Е,2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}метанол
В 50 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 10 мл метанола. В перемешиваемый растворитель добавляли 4-[(4-{[(2E,2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]бензальдегид (0,7 г, 1,86 ммоль), PC охлаждали до 0°C и боргидрид натрия (0,105 г, 2,79 ммоль) добавляли по частям в течение 15 минут. Реакционную смесь перемешивали при КТ в течение 1 часа. Реакцию гасили раствором NaHCO3 (40 мл 10% раствора в воде) и смесь экстрагировали этилацетатом (20 мл × 2). Органический слой промывали насыщенным солевым раствором (25 мл), сушили над безводным Na2SO4 и удаляли растворитель при пониженном давлении с получением продукта (0,7 г, выход 100,0%).
Соединение 30: ({4-[(4-{[(2Е,2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]бензил}окси)уксусная кислота
В 25 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 12 мл ТГФ. В перемешиваемый растворитель добавляли гидрид натрия (0,015 г, 0,6 ммоль) по частям при 0°C и перемешивали при указанной температуре в течение 30 минут. Затем {4-[(4-{[(2E,2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}метанол (0,1 г, 0,26 ммоль) в тетрагидрофуране (3 мл) добавляли по каплям. Реакционную смесь перемешивали при КТ в течение 13 часов. Реакцию гасили водой при 0°C, смесь подкисляли и экстрагировали этилацетатом. Органический слой промывали водой и солевым раствором. Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Продукт получали в виде белого твердого вещества (0,01 г, выход 6,25%).
Пример 31
3-Циано-3-{4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропаноат калия (31)
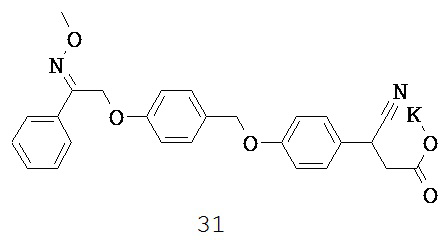
Соединение 31 синтезировали из этил-3-циано-3-{4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропаноата (0,5 г, 1,06 ммоль) и гидроксида калия (0,093 г, 1,66 ммоль) согласно способу, описанному на Схеме 12 (0,4 г, выход 78,34%); чистота: 99,27%.
Схема 15
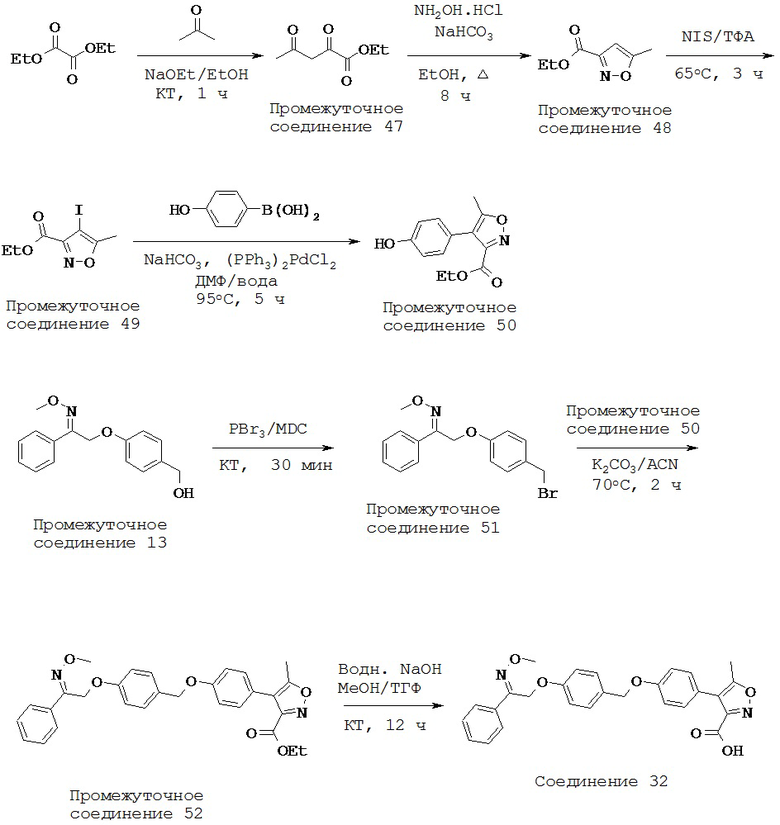
Пример 32
4-{4-[(4-{[(2E,2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси] фенил}-5-метил-1,2-оксазол-3-карбоновая кислота (32)
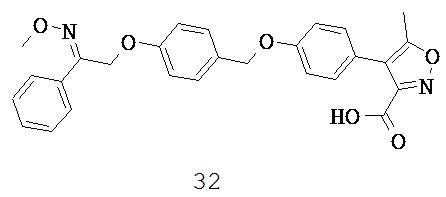
Соединение 32 получали с применением этил-4-(4-гидроксифенил)-5-метил-1,2-оксазол-3-карбоксилата и O-метилоксима (1E,1Z)-2-[3-(бромметил)фенокси]-1-фенилэтанона согласно способу, описанному на схеме 15.
Промежуточное соединение 47: этил-2,4-диоксопентаноат
В 100 мл двухгорлую круглодонную колбу, оборудованную магнитной мешалкой, помещали 25 мл этанола. В перемешиваемый растворитель добавляли кусочки металлического натрия (0,86 г, 37 ммоль) медленно при 0°C в атмосфере азота и перемешивали в течение 30 минут при комнатной температуре. Через 30 минут диэтилоксалат (5 г, 34,2 ммоль) в ацетоне (10 мл) добавляли по каплям. Во время добавления реакционная смесь становилась желтой. После завершения добавления наблюдали получение густой массы, добавляли этанол (10 мл) в реакционную смесь и перемешивали при комнатной температуре в течение 1 часа. Затем реакционную смесь фильтровали; твердые вещества растворяли в ледяной воде (50 мл), подкисляли концентрированной серной кислотой и экстрагировали этилацетатом (100 мл). Органический слой концентрировали при пониженном давлении и следы растворителя удаляли путем растирания с н-гексаном. Продукт получали в виде коричневой жидкости (3,2 г, выход 59,2%). 1Н ЯМР (300 МГц, ДМСО-d6): δ 5,46 (s, 2Н), 4,02-4,09 (q, 2H), 1,89 (s, 3H), 1,18-1,22 (t, 3H).
Промежуточное соединение 48: этил-5-метил-1,2-оксазол-3-карбоксилат
В 100 мл круглодонную колбу, оборудованную холодильником и магнитной мешалкой, помещали 10,5 мл этанола. В перемешиваемый растворитель добавляли этил-2,4-диоксопентаноат (2,5 г, 15 ммоль), гидрохлорид гидроксиламина (1,09 г, 15 ммоль) и бикарбонат натрия (1,32 г, 15 ммоль). После завершения добавления реакционную смесь кипятили с обратным холодильником при 80°C в течение 8 часов в атмосфере азота. После завершения реакции растворитель выпаривали из реакционной смеси. Этилацетат (50 мл) добавляли; органический слой промывали водой (50 мл), затем солевым раствором (25 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Продукт получали в виде бесцветной жидкости (1 г, выход 40,08%). 1Н ЯМР (300 МГц, CDCl3): δ 6,34 (s, 1Н), 4,33-4,40 (q, 2H), 2,43 (s, 3Н), 1,32-1,37 (t, 3Н).
Промежуточное соединение 49: этил-4-йод-5-метил-1,2-оксазол-3-карбоксилат
В 25 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 1 мл ТФА. В перемешиваемый растворитель добавляли этил-5-метил-1,2-оксазол-3-карбоксилат (0,1 г, 0,6 ммоль) и N-йодсукцинимид (0,289 г, 1,2 ммоль). После завершения добавления реакционную смесь нагревали при 65°C в течение 3 часов. После завершения реакции реакционную смесь разбавляли этилацетатом (10 мл), органический слой промывали насыщенным раствором NaHCO3 (10 мл), раствором тиосульфата натрия (10 мл), водой (25 мл) и, наконец, солевым раствором (10 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Продукт получали в виде коричневой жидкости (0,12 г, выход 66,2%). 1Н ЯМР (300 МГц, CDCl3): δ 4,36-4,43 (q, 2Н), 2,49 (s, 3H), 1,34-1,39 (t, 3H).
Промежуточное соединение 50: этил-4-(4-гидроксифенил)-5-метил-1,2-оксазол-3-карбоксилат
В 25 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали ДМФ (2,5 мл). В перемешиваемый растворитель добавляли 4-гидроксифенилбороновую кислоту (0,2 г, 0,7 ммоль), этил-4-йод-5-метил-1,2-оксазол-3-карбоксилат (0,116 г, 0,8 ммоль), бикарбонат натрия (0,18 г, 2,1 ммоль) и воду (0,08 мл) в атмосфере аргона. После завершения добавления реакционную смесь продували аргоном в течение 15 минут. В полученную смесь хлорид бис(трифенилфосфин)палладия (II) (0,05 г, 0,07 ммоль) добавляли, продували аргоном в течение 10 минут. После завершения реакцию гасили водой (10 мл) и смесь экстрагировали этилацетатом (10 мл). Органический слой промывали водой (5 мл) и солевым раствором (5 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали путем колоночной хроматографии на силикагеле с применением этилацетата и петролейного эфира в качестве элюентов. Продукт получали в виде беловатого твердого вещества (0,1 г, выход 57%). 1Н ЯМР (300 МГц, CDCl3): δ 7,10-7,13 (d, 2Н), 6,80-6,82 (d, 2Н), 5,14 (s, 1H), 4,25-4,32 (q, 2Н), 2,36 (s, 3H), 1,23-1,28 (t, 3H).
Промежуточное соединение 51: O-метилоксим (1Е,1Z)-2-[4-(бромметил)фенокси]-1-фенилэтанона
В 50 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 10 мл дихлорметана. В перемешиваемый растворитель добавляли O-метилоксим (1E,1Z)-2-[4-(гидроксиметил)фенокси]-1-фенилэтанона (0,5 г, 1,84 ммоль). При 0°С трибромид фосфора (0,746 г, 2,76 ммоль) добавляли по каплям и перемешивали при комнатной температуре в течение 30 минут. После завершения реакции реакционную смесь выливали в воду (25 мл) и экстрагировали дихлорметаном (50 мл). Органический слой промывали водой (25 мл) и насыщенным солевым раствором (25 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Продукт получали в виде желтоватого вязкого твердого вещества (0,4 г, выход 65%).
Промежуточное соединение 52: этил-4-{4-[(4-{[(2Е,2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}-5-метил-1,2-оксазол-3-карбоксилат
В 25 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 5 мл ацетонитрила. В перемешиваемый растворитель добавляли этил-4-(4-гидроксифенил)-5-метил-1,2-оксазол-3-карбоксилат (0,1 г, 0,4 ммоль), карбонат калия (0,178 г, 1,2 ммоль). Реакционную смесь охлаждали до 0°C, O-метилоксим (1Z)-2-[4-(бромметил)фенокси]-1-фенилэтанона (0,143 г, 0,4 ммоль) в ацетонитриле (2 мл) добавляли по каплям и перемешивали при 70°C в течение 2 часов. Реакционную смесь концентрировали для отгонки растворителя, разбавляли водой (10 мл) и экстрагировали этилацетатом (10 мл). Органический слой промывали водой (5 мл) и насыщенным солевым раствором (5 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали путем колоночной хроматографии на силикагеле с применением этилацетата и петролейного эфира в качестве элюентов. Продукт получали в виде белого твердого вещества (0,11 г, выход 54%). 1Н ЯМР (300 МГц, CDCl3): δ 7,59-7,62 (m, 2H), 7,26-7,34 (m, 5H), 7,14-7,17 (d, 2H), 6,92-6,95 (d, 2H), 6,85-6,88 (d, 2H), 5,14 (s, 2H), 4,93 (s, 2H), 4,24-4,31 (q, 2H), 3,99 (s, 3H), 2,36 (s, 3H), 1,21-1,26 (t, 3H).
Соединение 32: 4-{4-[(4-{[(2E,2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}-5-метил-1,2-оксазол-3-карбоновая кислота
В 10 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 3 мл тетрагидрофурана. В перемешиваемый растворитель добавляли метил-4-{4-[(4-{[(2Е,2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}-5-метил-1,2-оксазол-3-карбоксилат (0,11 г, 0,2 ммоль), этанол (3 мл) и гидроксид натрия (0,035 г, 0,9 ммоль) в воде (0,5 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи. После завершения реакции растворитель удаляли, натриевую соль промывали диэтиловым эфиром (5 мл); водный слой подкисляли 1 н. HCl до рН 2,0 и экстрагировали этилацетатом (5 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали путем колоночной хроматографии на силикагеле с применением метанола и дихлорметана в качестве элюентов. Продукт получали в виде бледно-желтого липкого твердого вещества (0,045 г, выход 38,8%).
Пример 33
3-Циано-3-{4-[(4-{[(2Е,2Z)-2-(4-цианофенил)-2-(метоксиимино)этил]окси}бензил)окси]фенил}пропановая кислота (33)
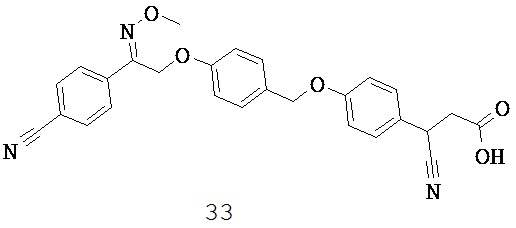
Соединение 33 синтезировали из 4-{(1Е,1Z)-2-[4-(гидроксиметил)фенокси]-N-метоксиэтанимидоил}бензонитрила (0,328 г, 0,9 ммоль) и метил-3-циано-3-(4-гидроксифенил)пропаноата (0,2 г, 0,9 ммоль) согласно способу, описанному на схеме 18 (0,04 г, выход 13,73%); чистота: 78,04%.
Пример 34
3-Циано-3-{4-[(4-{[(2Z)-2-(4-фторфенил)-2-(метоксиимино)этил]окси}бензил)окси]фенил}пропановая кислота (34)
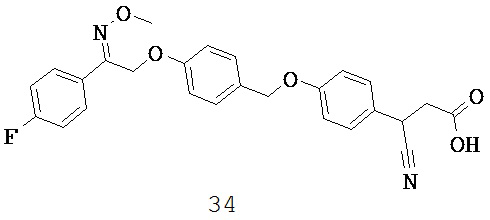
Соединение 34 синтезировали из O-метилоксима (1Z)-1-(4-фторфенил)-2-[4-(гидроксиметил)фенокси]этанона (0,4 г, 1,46 ммоль) и метил-3-циано-3-(4-гидроксифенил)-пропаноата (0,3 г, 1,46 ммоль) согласно способу, описанному на схеме 18 (0,06 г, выход 15,46%); чистота: 95,29%.
Пример 35
3-Циано-3-{4-[(4-{[(2Z)-2-(3-фторфенил)-2-(метоксиимино)этил]окси}бензил)окси]фенил}пропановая кислота (35)
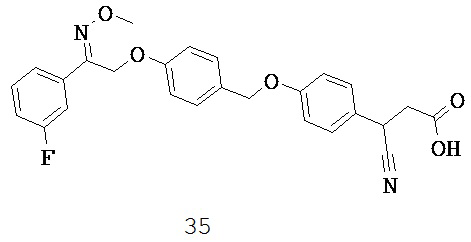
Соединение 35 синтезировали из O-метилоксима (1Z)-1-(3-фторфенил)-2-[4-(гидроксиметил)фенокси]этанона (0,5 г, 1,8 ммоль) и метил-3-циано-3-(4-гидроксифенил)-пропаноата (0,372 г, 1,8 ммоль) согласно способу, описанному на схеме 18 (0,1 г, выход 18,73%); чистота: 95,1%.
Схема 16
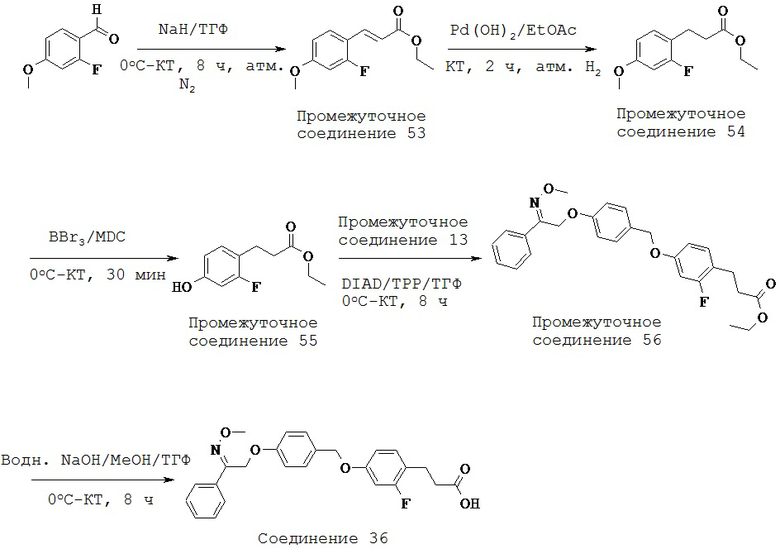
Пример 36
3-{2-Фтор-4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропановая кислота (36)
Соединение 36 получали с применением метил-3-циано-3-(4-гидроксифенил)-пропаноата и O-метилоксима (1Z)-2-[4-(бромметил)-2-фторфенокси]-1-фенилэтанона согласно способу, описанному на схеме 16 (0,186 г, выход 65,97%); чистота: 98,88%.
Промежуточное соединение 53: этил-(2Z)-3-(2-фтор-4-метоксифенил)ацетат
В 100 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 25 мл тетрагидрофурана. В перемешиваемый растворитель добавляли гидрид натрия (0,39 г, 16,2 ммоль) по частям при 0°С, затем триэтилфосфоноацетат (2,9 г, 12,9 ммоль). Реакционную смесь перемешивали при 0°C в течение 30 минут. В перемешиваемый раствор 2-фтор-4-метоксибензальдегид (1 г, 6,4 ммоль) в тетрагидрофуране (2 мл) добавляли по каплям и перемешивали при комнатной температуре в течение ночи. После завершения реакции реакционную смесь выливали в лед и экстрагировали этилацетатом (50 мл). Органический слой промывали водой (25 мл) и насыщенным солевым раствором (25 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали путем колоночной хроматографии на силикагеле с применением этилацетата и петролейного эфира в качестве элюентов. Продукт получали в виде бесцветной маслянистой жидкости (1,2 г, выход 82,49%). 1Н ЯМР (300 МГц, CDCl3): δ 7,65-7,70 (d, 1H), 7,35-7,41 (t, 1H), 6,54-6,66 (m, 2H), 6,31-6,37 (d, 1H), 4,15-4,22 (q, 2H), 3,76 (s, 3H), 1,24-1,29 (t, 3Н).
Промежуточное соединение 54: этил-3-(2-фтор-4-метоксифенил)пропаноат
В 500 мл колбу для встряхивателя Парра помещали этил-(2Е,2Z)-3-(2-фтор-4-метоксифенил)проп-2-еноат (0,5 г, 2,2 ммоль) и этилацетат (15 мл) и продували азотом в течение 10 минут. Гидроксид палладия (20%) добавляли и выдерживали в атмосфере водорода при 50 psi в течение 2 часов. После завершения реакции реакционную смесь фильтровали через целит, промывали тщательно этилацетатом (25 мл) и концентрировали для отгонки растворителя. Продукт получали в виде коричневого твердого вещества (0,462 г, выход 91,58%). 1Н ЯМР (300 МГц, CDCl3): δ 7,00-7,06 (t, 1Н), 6,50-6,56 (m, 2H), 4,01-4,09 (q, 2H), 3,70 (s, 3Н), 2,81-2,86 (t, 2H), 2,49-2,54 (t, 2H), 1,14-1,19 (t, 3Н).
Промежуточное соединение 55: этил-3-(2-фтор-4-гидроксифенил)пропаноат
В 25 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали ТО мл дихлорметана. В перемешиваемый растворитель добавляли этил-3-(2-фтор-4-метоксифенил)-пропаноат (0,45 г, 2 ммоль). Реакционную смесь охлаждали до 0°С и трибромид бора (0,45 мл) добавляли по каплям. После перемешивания в течение 30 минут реакцию гасили путем медленного добавления этанола (1 мл) при 0°С. Реакционную смесь концентрировали для отгонки растворителя; этилацетат (10 мл) добавляли. Органический слой промывали насыщенным раствором NaHCO3 (10 мл), затем солевым раствором (10 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Продукт получали в виде бесцветной маслянистой жидкости (0,421 г, выход 99,75%). 1Н ЯМР (300 МГц, CDCl3): δ 6,93-6,99 (t, 1H), 6,43-6,48 (m, 2H), 5,54 (s, 1H), 4,02-4,09 (q, 2H), 2,79-2,85 (t, 2H), 2,50-2,55 (t, 2H), 1,14-1,19 (t, 3Н).
Промежуточное соединение 56: этил-3-{2-фтор-4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропаноат
В 50 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали тетрагидрофуран (10 мл). В перемешиваемый растворитель добавляли (4-{[(2E,2Z)-2-(метоксиимино)-2-фенилэтил]окси}фенил)метанол (0,524 г, 1,93 ммоль), этил-3-(2-фтор-4-гидроксифенил)пропаноат (0,41 г, 1,93 ммоль) и трифенилфосфин (0,608 г, 2,32 ммоль) в атмосфере аргона. Реакционную смесь охлаждали до 0°C, диизопропилазокарбоксилат (0,43 г, 2,12 ммоль) добавляли по каплям. Реакционную смесь перемешивали при комнатной температуре в течение ночи в атмосфере аргона. После завершения реакции реакционную смесь концентрировали; воду (10 мл) добавляли и экстрагировали этилацетатом (25 мл). Органический слой промывали солевым раствором (10 мл). Неочищенное вещество очищали путем колоночной хроматографии на силикагеле с применением этилацетата и петролейного эфира в качестве элюентов. Продукт получали в виде белого твердого вещества (0,324 г, выход 36,02%). МС (ИЭР, 120 эВ): m/z=466,2 (М+Н)+; 1Н ЯМР (300 МГц, CDCl3): δ 7,58-7,61 (m, 2H), 7,27-7,29 (m, 3H), 7,22-7,25 (m, 2H), 6,99-7,05 (m, 1H), 6,83-6,86 (d, 2H), 6,55-6,61 (m, 2H), 5,13 (s, 2H), 4,85 (s, 2H), 4,01-4,08 (q, 2H), 3,99 (s, 3H), 2,80-2,85 (t, 2H), 2,48-2,54 (t, 2H), 1,14-1,18 (t, 3H).
Соединение 36: 3-{2-Фтор-4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропановая кислота
В 10 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 1 мл тетрагидрофурана. В перемешиваемый растворитель добавляли этил-3-{2-фтор-4-[(4-{[(2Е,2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропаноат (0,3 г, 0,64 ммоль) и метанол (2 мл). Реакционную смесь охлаждали до 0°C и гидроксид натрия (0,13 г, 3,2 ммоль) в воде (1 мл) добавляли по каплям. Реакционную смесь перемешивали в течение ночи. После завершения реакции реакционную смесь концентрировали для отгонки растворителя. Воду (1 мл) добавляли и экстрагировали диэтиловым эфиром (5 мл). Водный слой подкисляли 1 н. HCl до рН 3 и экстрагировали диэтиловым эфиром (5 мл). Органический слой промывали солевым раствором (5 мл), отгоняли растворитель и сушили. Продукт получали в виде беловатого твердого вещества (0,186 г, выход 65,97%). МС (ИЭР, 120 эВ): m/z=438,1 (M+И)+; чистота по данным ВЭЖХ: 98,88%.
Пример 37
3-{2-Метокси-4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропановая кислота (37)
Соединение 37 синтезировали из O-метилоксима (1Z)-2-[4-(гидроксиметил)фенокси]-1-фенилэтанона (0,4 г, 1,197 ммоль) и метил-3-(4-гидрокси-2-метоксифенил)пропаноата (0,268 г, 1,197 ммоль) согласно способу, описанному на схеме 13 (0,2 г, выход 89%); чистота: 99,31%.
Пример 38
3-{4-[(4-{[(2Z)-2-(Метоксиимино)-2-фенилэтил]окси}бензил)окси]-2-метилфенил}пропановая кислота (38)
Соединение 38 синтезировали из O-метилоксима (1Z)-2-[4-(гидроксиметил)фенокси]-1-фенилэтанона (0,45 г, 1,7 ммоль) и метил-3-(4-гидрокси-2-метилфенил)пропаноата (0,34 г, 1,7 ммоль) согласно способу, описанному на схеме 15 (0,003 г, выход 8,0%); чистота: 98,24%.
Пример 39
3-{4-[(4-{[(2Z)-2-(4-Цианофенил)-2-(метоксиимино)этил]окси}бензил)окси]-2-фторфенил}пропановая кислота (39)
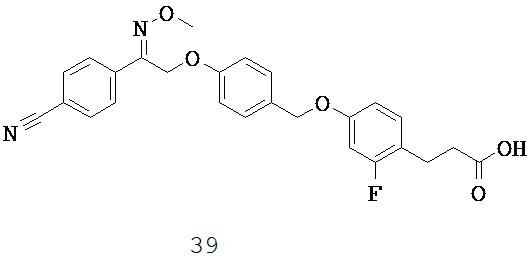
Соединение 39 синтезировали из 4-{(1Z)-2-[4-(гидроксиметил)фенокси]-N-метоксиэтанимидоил}бензонитрила (0,4 г, 1,4 ммоль) и метил-3-(2-фтор-4-гидроксифенил)-пропаноата (0,3 г, 1,4 ммоль) согласно способу, описанному на схеме 13 (0,074 г, выход 50,19%); чистота: 93,47%.
Пример 40
3-{2-Фтор-4-[(4-{[(2Z)-2-(метоксиимино)-2-(4-метоксифенил)этил]окси}бензил)окси]фенил}пропановая кислота (40)
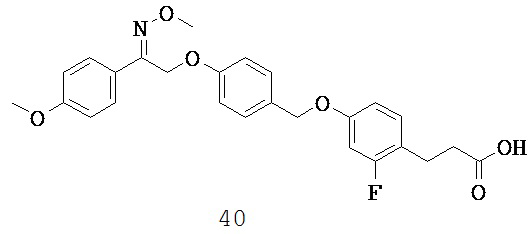
Соединение 40 синтезировали из O-метилоксима (1Z)-2-[4-(гидроксиметил)фенокси]-1-(4-метоксифенил)этанона (0,25 г, 0,84 ммоль) и метил-3-(2-фтор-4-гидроксифенил)-пропаноата (0,2 г, 0,94 ммоль) согласно способу, описанному на схеме 18 (0,1 г, выход 71,00%); чистота: 98,10%.
Пример 41
3-{2-Фтор-4-[(3-метокси-4-{[(2Е,2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропановая кислота (41)
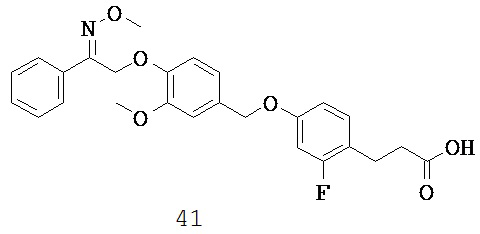
Соединение 41 синтезировали из O-метилоксима (1Е,1Z)-2-[4-(гидроксиметил)-2-метоксифенокси]-1-фенилэтанона (0,464 г, 1,54 ммоль) и метил-3-(2-фтор-4-гидроксифенил)-пропаноата (0,327 г, 1,54 ммоль) согласно способу, описанному на схеме 11 (0,13 г, выход 47,00%); чистота: 89,09%.
Пример 42
3-{2-Циано-4-[(4-{[(2Е,2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропановая кислота (42)
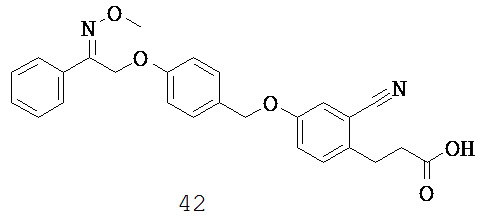
Соединение 42 синтезировали из O-метилоксима (1Е,1Z)-2-[4-(гидроксиметил)-фенокси]-1-фенилэтанона (0,034 г, 0,1 ммоль) и метил-3-(2-циано-4-гидроксифенил)-пропаноата (0,026 г, 0,2 ммоль) согласно способу, описанному на схеме 18 (0,0025 г, выход 12,89%); чистота: 76,00%.
Пример 43
3-{4-[(4-{[(2Z)-2-(4-Цианофенил)-2-(метоксиимино)этил]окси}бензил)окси]-2-метоксифенил}пропановая кислота (43)
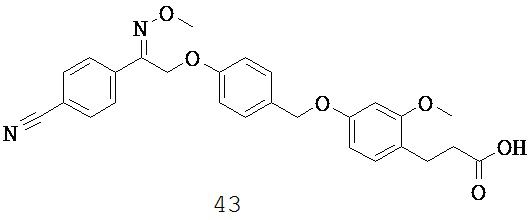
Соединение 43 синтезировали из 4-{(1Z)-2-[4-(гидроксиметил)фенокси]-N-метоксиэтанимидоил}бензонитрила (0,563 г, 1,9 ммоль) и метил-3-(4-гидрокси-2-метоксифенил)пропаноата (0,4 г, 1,9 ммоль) согласно способу, описанному на схеме 18 (0,27 г, выход 71,5%); чистота: 94,96%.
Пример 44
3-{2-(Цианометил)-4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропановая кислота (44)
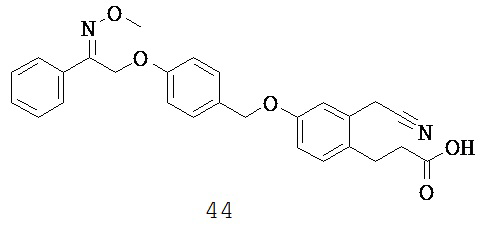
Соединение 44 синтезировали из O-метилоксима (1Z)-2-[4-(гидроксиметил)фенокси]-1-фенилэтанона (0,3 г, 1,28 ммоль) и метил-3-[2-(пианометил)-4-гидроксифенил]пропаноата (0,348 г, 1,28 ммоль) согласно способу, описанному на схеме 18 (0,1 г, выход 71,00%); чистота: 92,72%.
Схема 17
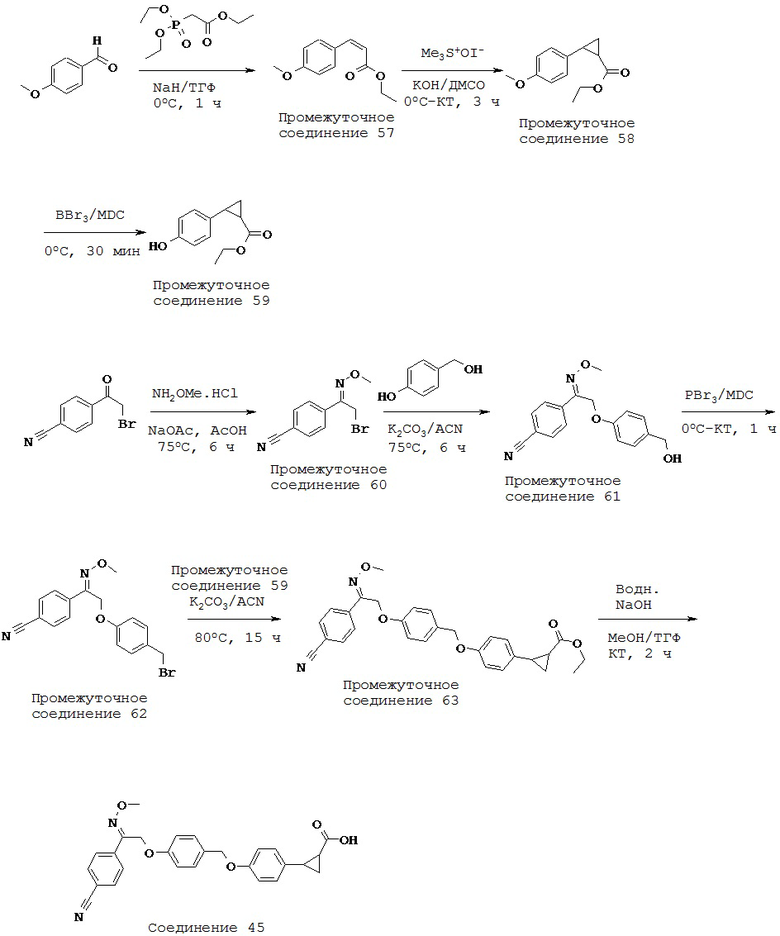
Пример 45
2-{4-[(4-{[(2Z)-2-(4-Цианофенил)-2-(метоксиимино)этил]окси}бензил)окси]фенил}циклопропанкарбоновая кислота (45)
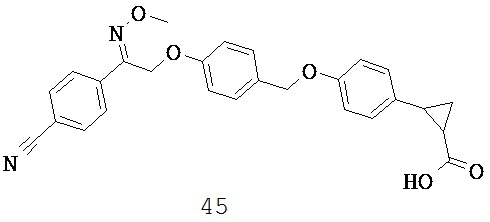
Соединение 45 получали из этил-2-(4-гидроксифенил)циклопропанкарбоксилата (0,1 г, 0,48 ммоль) и 4-{(1Z)-2-[4-(бромметил)фенокси]-N-метоксиэтанимидоил}бензонитрила (0,175 г, 0,48 ммоль) согласно способу, описанному на схеме 17.
Промежуточное соединение 57: этил-(2Z)-3-(4-метоксифенил)проп-2-еноат
В 500 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 160 мл тетрагидрофурана. В перемешиваемый растворитель добавляли этилфосфоноацетат (16,5 г, 73,6 ммоль) в атмосфере аргона. Реакционную смесь охлаждали до 0°C и гидрид натрия (2,65 г, 110 ммоль) добавляли по частям, перемешивали в течение 15 минут при указанной температуре. Затем 4-метоксибензальдегид (5 г, 36,0 ммоль) в тетрагидрофуране (30 мл) добавляли по каплям. Реакционную смесь перемешивали при 0°C в течение 1 часа. Реакцию гасили путем медленного добавления воды при 0°C, смесь перемешивали в течение 10 минут. Слои разделяли; этилацетат (50 мл) добавляли в водный слой и экстрагировали слои. Органический слой промывали солевым раствором (50 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Продукт получали в виде бледно-желтого полутвердого вещества (7,4 г, выход 98,76%). 1Н ЯМР (300 МГц, CDCl3): δ 7,54-7,60 (d, 1H), 7,39-7,42 (d, 2H), 6,82-6,85 (d, 2H), 6,21-6,26 (d, 1H), 4,15-4,22 (q, 2H), 3,76 (s, 3H), 1,24-1,28 (t, 3H).
Промежуточное соединение 58: этил-2-(4-метоксифенил)циклопропанкарбоксилат
В 50 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 14 мл диметилсульфоксида. В перемешиваемый растворитель добавляли йодид триметилсульфоксония (1,53 г, 6,93 ммоль). Реакционную смесь охлаждали до 0°C и порошковый гидроксид калия (0,42 г, 7,49 ммоль) добавляли по частям, перемешивали в течение 10 минут при комнатной температуре. Затем этил-(2Z)-3-(4-метоксифенил)акрилат (1,3 г, 6,31 ммоль) добавляли. Реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Реакцию гасили водой и диэтиловый эфир добавляли. Слои разделяли и органический слой промывали солевым раствором (50 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали путем колоночной хроматографии на силикагеле с применением этилацетата и петролейного эфира в качестве элюентов. Продукт получали в виде бледно-розового твердого вещества (0,6 г, выход 43,22%). 1Н ЯМР (300 МГц, CDCl3): δ 6,95-6,98 (d, 2H), 6,74-6,77 (d, 2H), 4,06-4,13 (q, 2H), 3,72 (s, 3Н), 2,38-2,44 (m, 1H), 1,72-1,78 (m, 1H), 1,43-1,61 (m, 2H), 1,25-1,29 (t, 3H).
Промежуточное соединение 59: этил-2-(4-гидроксифенил)циклопропанкарбоксилат
В 50 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 20 мл дихлорметана. В перемешиваемый растворитель добавляли этил-2-(4-метоксифенил)-циклопропанкарбоксилат (0,9 г, 4 ммоль). Реакционную смесь охлаждали до 0°C и трибромид бора (0,9 мл) добавляли по каплям, перемешивали в течение 30 минут при указанной температуре. Реакцию гасили путем добавления этанола по каплям при 0°C. Реакционную смесь концентрировали для отгонки растворителя; воду добавляли в неочищенное вещество и смесь экстрагировали этилацетатом. Органический слой промывали насыщенным раствором NaHCO3 (25 мл), затем солевым раствором (25 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Продукт получали в виде бледно-желтой жидкости (0,3 г, выход 35,6%). 1Н ЯМР (300 МГц, ДМСО-d6): δ 9,27 (s, 1H), 6,94-6,97 (d, 2H), 6,64-6,67 (d, 2H), 4,05-4,12 (q, 2H), 2,28-2,36 (m, 1H), 1,75-1,80 (m, 1H), 1,35-1,41 (m, 2H), 1,15-1,28 (t, 3Н).
Промежуточное соединение 60: 4-[(1Z)-2-бром-N-метоксиэтанимидоил]бензонитрил
В 1 л круглодонную колбу, оборудованную магнитной мешалкой, помещали 300 мл уксусной кислоты. В перемешиваемый растворитель добавляли 4-(бромацетил)бензонитрил (40 г, 178 ммоль), гидрохлорид O-метоксиламина (22,36 г, 267 ммоль), затем ацетат натрия (21,96 г, 267 ммоль). Реакционную смесь нагревали при 75°C в течение 6 часов. Реакционную смесь подщелачивали до рН 8 насыщенным раствором NaHCO3 (500 мл). Водный слой экстрагировали этилацетатом (250 мл × 3), органический слой промывали водой (200 мл), затем солевым раствором (100 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали путем колоночной хроматографии на силикагеле с применением этилацетата и петролейного эфира в качестве элюентов. Продукт получали в виде бледно-желтой маслянистой жидкости (19 г, выход 42,06%). 1Н ЯМР (300 МГц, CDCl3): δ 7,75-7,78 (d, 2H), 7,61-7,64 (d, 2H), 4,46 (s, 2H), 4,06 (s, 3Н).
Промежуточное соединение 61: 4-{(1Z)-2-[4-(гидроксиметил)фенокси]-N-метоксиэтанимидоил}бензонитрил
В 500 мл круглодонную колбу, оборудованную механической мешалкой, помещали 200 мл ацетонитрила. В перемешиваемый растворитель добавляли 4-[(1E,1Z)-2-бром-N-метоксиэтанимидоил]бензонитрил (19 г, 75 ммоль), 4-(гидроксиметил)фенол (9,31 г, 75 ммоль), затем карбонат калия (31,1 г, 225 ммоль). Реакционную смесь нагревали при 75°C в течение 6 часов. Реакционную смесь фильтровали через керамическую воронку и промывали этилацетатом (100 мл). Фильтрат концентрировали для отгонки растворителя; воду (100 мл) добавляли и экстрагировали этилацетатом (100 мл × 2). Органический слой промывали водой (100 мл), затем солевым раствором (50 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали путем колоночной хроматографии на силикагеле с применением этиацетата и петролейного эфира в качестве элюентов. Продукт получали в виде желтой жидкости (12,2 г, выход 54,83%). 1Н ЯМР (300 МГц, CDCl3): δ 7,71-7,74 (d, 2H), 7,55-7,57 (d, 2H), 7,19-7,21 (d, 2H), 6,79-6,81 (d, 2H), 5,14 (s, 2H), 4,53-4,55 (d, 2H), 4,02 (s, 3Н).
Промежуточное соединение 62: 4-{(1Z)-2-[4-(бромметил)фенокси]-N-метоксиэтанимидоил}бензонитрил
В 25 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 50 мл дихлорметана. В перемешиваемый растворитель добавляли 4-{(1E,1Z)-2-[4-(гидроксиметил)-фенокси]-N-метоксиэтанимидоил}бензонитрил (0,2 г, 6,0 ммоль) при 0°C, трибромид фосфора (0,1 мл, 9,0 ммоль) добавляли по каплям и перемешивали при комнатной температуре в течение 1 часа. После завершения реакции реакционную смесь выливали в воду (10 мл) и экстрагировали дихлорметаном (20 мл). Органический слой промывали водой (25 мл) и насыщенным солевым раствором (20 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Продукт получали в виде желтоватого вязкого твердого вещества (0,2 г, выход 90,9%).
Промежуточное соединение 63: этил-2-{4-[(4-{[(2Z)-2-(4-цианофенил)-2-(метоксиимино)этил]окси}бензил)окси]фенил}циклопропанкарбоксилат
В 25 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 5 мл ацетонитрила. В перемешиваемый растворитель добавляли этил-2-(4-гидроксифенил)-циклопропанкарбоксилат (0,1 г, 0,48 ммоль), карбонат калия (0,2 г, 1,4 ммоль). Реакционную смесь охлаждали до 0°C, 4-{(1Е,1Z)-2-[4-(бромметил)фенокси]-N-метоксиэтанимидоил}-бензонитрил (0,175 г, 0,48 ммоль) в ацетонитриле (5 мл) добавляли по каплям и перемешивали при 70°C в течение 15 часов. Реакционную смесь концентрировали для отгонки растворителя, разбавляли водой (10 мл) и экстрагировали этилацетатом (10 мл). Органический слой промывали водой (5 мл) и насыщенным солевым раствором (5 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали путем колоночной хроматографии на силикагеле с применением этилацетата и петролейного эфира в качестве элюентов. Продукт получали в виде белого твердого вещества (0,19 г, выход 82,6%).
Соединение 45: 2-{4-[(4-{[(2Z)-2-(4-Цианофенил)-2-(метоксиимино)этил]окси}бензил)окси]фенил}циклопропанкарбоновая кислота
В 50 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 30 мл тетрагидрофурана. В перемешиваемый растворитель добавляли этил-2-{4-[(4-{[(2Е,2Z)-2-(4-цианофенил)-2-(метоксиимино)этил]окси}бензил)окси]фенил}циклопропанкарбоксилат (0,07 г, 0,16 ммоль) в метаноле (5 мл). Реакционную смесь охлаждали до 0°C и гидроксид натрия (0,7 г, 0,8 ммоль) в воде (5 мл) добавляли по каплям и перемешивали в течение 4 часов. Реакционную смесь концентрировали для отгонки растворителя; этилацетат (10 мл) добавляли и перемешивали в течение 10 минут. Органический слой концентрировали и неочищенное вещество подкисляли насыщенным раствором лимонной кислоты до рН 6. Полученные твердые вещества фильтровали и сушили. Продукт получали в виде белого твердого вещества (0,012 г, выход 18,5%). МС (ИЭР, 120 эВ): m/z=455,1 (М-Н)+; чистота по данным ВЭЖХ: 97,19%; 1Н ЯМР (300 МГц, ДМСО-d6): δ 7,80-7,89 (m, 4H), 7,32-7,35 (d, 2Н), 7,05-7,08 (d, 2H), 6,87-6,91 (m, 4H), 5,27 (s, 2H), 4,97 (s, 2H), 4,04 (s, 3H), 2,27-2,35 (m, 1H), 1,67-1,73 (m, 1H), 1,23-1,41 (m, 2H).
Пример 46
2-{4-[(4-{[(2Z)-2-(Метоксиимино)-2-(4-метоксифенил)этил]окси}бензил)окси]фенил}циклопропанкарбоновая кислота (46)
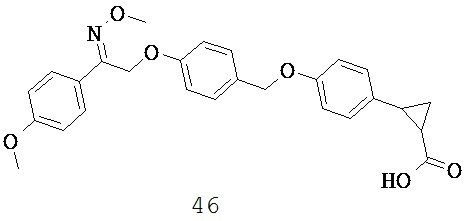
Соединение 46 синтезировали из O-метилоксима (1Z)-2-[4-(гидроксиметил)фенокси]-1-(4-метоксифенил)этанона (0,146 г, 0,48 ммоль) и этил-2-(4-гидроксифенил)-циклопропанкарбоксилата (0,1 г, 0,4 ммоль) согласно способу, описанному на схеме 18 (0,01 г, выход 13,26%); чистота: 92,24%.
Пример 47
3-{2-Фтор-4-[(4-{[(2Z)-2-(метоксиимино)-2-(4-метоксифенил)этил]окси}бензил)окси]фенил}пропаноат натрия (47)
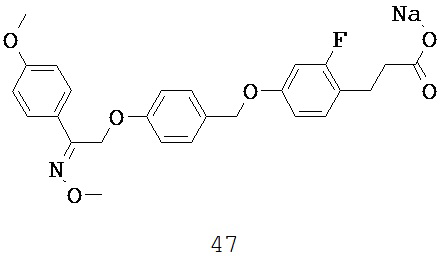
Соединение 47 синтезировали из гидроксида натрия (0,007 г, 0,18 ммоль) и 3-{2-фтор-4-[(4-{[(2Z)-2-(метоксиимино)-2-(4-метоксифенил)этил]окси}бензил)окси]фенил}пропановой кислоты (0,087 г, 0,18 ммоль) согласно способу, описанному на схеме 12 (0,07 г, выход 79,00%); чистота: 99,79%.
Пример 48
2-{4-[(4-{[(2Е,2Z)-2-(Метоксиимино)-2-фенилэтил]окси}бензил)амино]фенил}циклопропанкарбоксилат натрия (48)
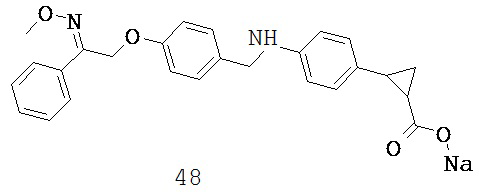
Соединение 48 синтезировали из 2-{4-[(4-{[(2E,2Z)-2-(метоксиимино)-2-фенилэтил]-окси}бензил)амино]фенил}циклопропанкарбоновой кислоты (0,093 г, 0,216 ммоль) и гидроксида натрия (0,0086 г, 0,216 ммоль) согласно способу, описанному на схеме 12 (0,075 г, выход 81,50%); чистота: 91,51%.
Пример 49
3-Циано-3-{4-[(4-{[(2E,2Z)-2-(4-цианофенил)-2-(метоксиимино)этил]окси}бензил)окси]фенил}пропаноат натрия (49)
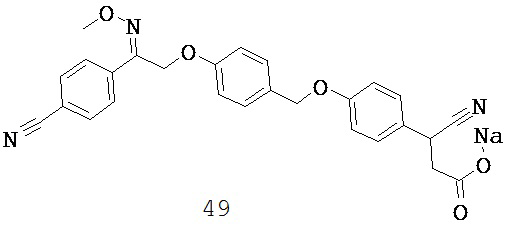
Соединение 49 синтезировали из 3-циано-3-{4-[(4-{[(2E,2Z)-2-(4-цианофенил)-2-(метоксиимино)этил]окси}бензил)окси]фенил}пропановой кислоты (1,5 г, 3 ммоль) и гидроксида натрия (0,3 г, 1М раствор) согласно способу, описанному на схеме 12 (0,51 г, выход 34,40%); чистота: 98,20%.
Пример 50
3-Циано-3-{4-[(3-фтор-4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропаноат натрия (50)
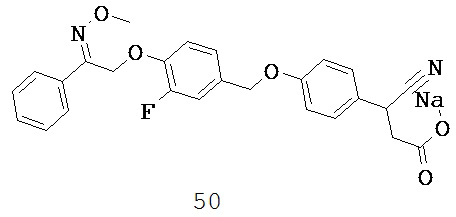
Соединение 50 синтезировали из 3-циано-3-{4-[(3-фтор-4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропановой кислоты (0,08 г, 0,0002 ммоль), гидроксида натрия (0,01 г, 1М раствор) согласно способу, описанному на схеме 12 (0,072 г, выход 82,76%); чистота: 88,00%.
Пример 51
2-{4-[(4-{[(2Z)-2-(4-Цианофенил)-2-(метоксиимино)этил]окси}бензил)окси]фенил}циклопропанкарбоксилат натрия (51)
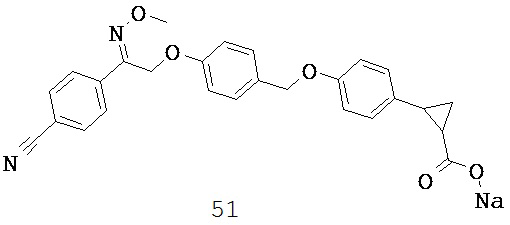
Соединение 51 синтезировали из гидроксида натрия (0,04 г, 0,96 ммоль) и 2-{4-[(4-{[(2Z)-2-(4-цианофенил)-2-(метоксиимино)этил]окси}бензил)окси]фенил}-циклопропанкарбоновой кислоты (0,22 г, 0,48 ммоль) согласно способу, описанному на схеме 12 (0,15 г, выход 72,40%); чистота: 90,60%.
Пример 52
3-{4-[(4-{[(2Z)-2-(4-Цианофенил)-2-(метоксиимино)этил]окси}бензил)окси]-2-фторфенил}пропаноат натрия (52)
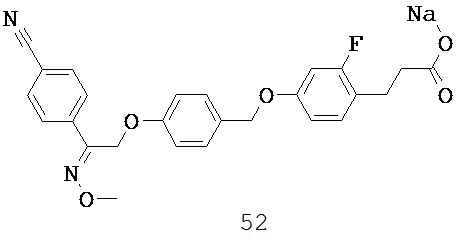
Соединение 52 синтезировали из 3-{4-[(4-{[(2Z)-2-(4-цианофенил)-2-(метоксиимино)-этил]окси}бензил)окси]-2-фторфенил}пропановой кислоты (0,06 г, 0,12 ммоль) и гидроксида натрия (0,0049 г, 0,12 ммоль) согласно способу, описанному на схеме 12 (0,046 г, выход 85,2%); чистота: 88,58%.
Пример 53
3-Циано-3-{4-[(4-{[(2Е)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропаноат натрия (53)
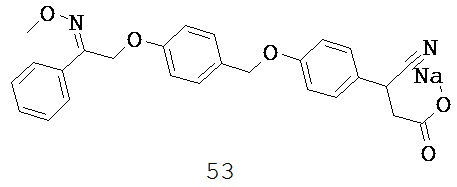
Соединение 53 синтезировали из гидроксида натрия (0,05 г, 1,27 ммоль) и этил-3-циано-3-{4-[(4-{[(2E)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропаноата (0,3 г, 0,64 ммоль) согласно способу, описанному на схеме 12 (0,2 г, выход 67,38%); чистота: 89,99%.
Пример 54
3-Циано-3-{4-[(4-{[(2Z)-2-(4-фторфенил)-2-(метоксиимино)этил]окси}бензил)окси]фенил}пропаноат натрия (54)
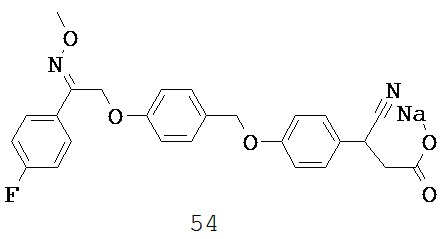
Соединение 54 синтезировали из гидроксида натрия (0,067 г, 1,6 ммоль) и 3-циано-3-{4-[(4-{[(2Z)-2-(4-фторфенил)-2-(метоксиимино)этил]окси}бензил)окси]фенил}пропановой кислоты (0,4 г, 0,8 ммоль) согласно способу, описанному на схеме 12 (0,3 г, выход 73,84%); чистота: 96,50%.
Пример 55
3-Циано-3-{4-[(4-{[(2Z)-2-(3-фторфенил)-2-(метоксиимино)этил]окси}бензил)окси]фенил}пропаноат натрия (55)
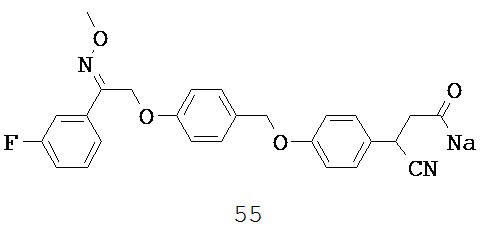
Соединение 55 синтезировали из 3-циано-3-{4-[(4-{[(2Z)-2-(3-фторфенил)-2-(метоксиимино)этил]окси}бензил)окси]фенил}пропановой кислоты (0,55 г, 1,2 ммоль) и гидроксида натрия (0,092 г, 2,3 ммоль) согласно способу, описанному на схеме 12 (0,4 г, выход 71,59%); чистота: 96,74%.
Пример 56
3-{2-Фтор-4-[(3-метокси-4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропаноат натрия (56)
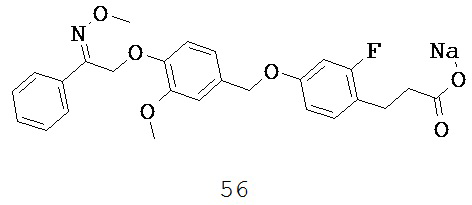
Соединение 56 синтезировали из 3-{2-фтор-4-[(3-метокси-4-{[(2 г)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропановой кислоты (0,3 г, 0,6 ммоль) и гидроксида натрия (0,072 г, 1,8 ммоль) согласно способу, описанному на схеме 12 (0,085 г, выход 30,00%); чистота: 90,03%.
Пример 57
2-{4-[(4-{[(2Z)-2-(Метоксиимино)-2-(4-метоксифенил)этил]окси}бензил)окси]фенил}циклопропанкарбоксилат натрия (57)
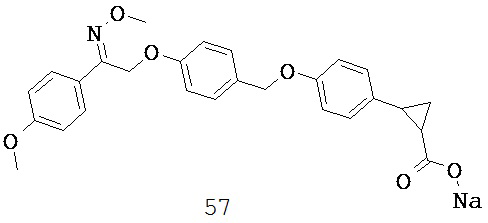
Соединение 57 синтезировали из 2-{4-[(4-{[(2Z)-2-(метоксиимино)-2-(4-метоксифенил)-этил]окси}бензил)окси]фенил}циклопропанкарбоновой кислоты (0,08 г, 0,2 ммоль) и гидроксида натрия (0,013 г, 0,3 ммоль) согласно способу, описанному на схеме 12 (0,022 г, выход 27,84%); чистота: 98,61%.
Пример 58
3-{2-(Циклопропилметокси)-4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропановая кислота (58)
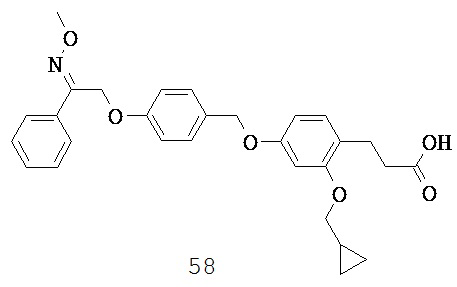
Соединение 58 синтезировали из O-метилоксима (1Z)-2-[4-(гидроксиметил)фенокси]-1-фенилэтанона (0,656 г, 2,4 ммоль) и метил-3-[2-(циклопропилметокси)-4-гидроксифенил]-пропаноата (0,6 г, 2,4 ммоль) согласно способу, описанному на схеме 18 (0,04 г, выход 4,20%); чистота: 98,15%.
Пример 59
3-Циано-3-{4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}бутановая кислота (59)
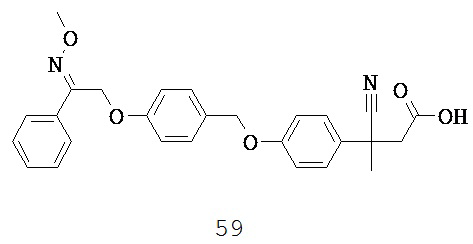
Соединение 59 синтезировали из O-метилоксима (1Z)-2-[4-(гидроксиметил)фенокси]-1-фенилэтанона (2,34 г, 8,66 ммоль) и этил-3-циано-3-(4-гидроксифенил)бутаноата (2,4 г, 9,6 ммоль) согласно способу, описанному на схеме 18 (0,15 г, выход 57,47%); чистота: 98,34%.
Пример 60
3-{4-[(4-{[(2Z)-2-(Метоксиимино)-2-фенилэтил]окси}бензил)окси]-2-(пропан-2-илокси)фенил}пропановая кислота (60)
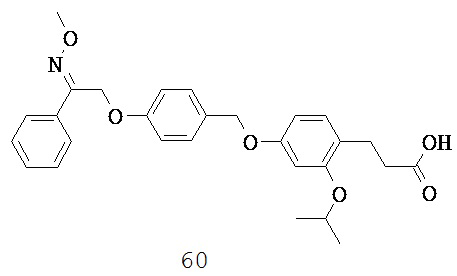
Соединение 60 синтезировали из O-метилоксима (1Z)-2-[4-(гидроксиметил)фенокси]-1-фенилэтанона (0,268 г, 0,99 ммоль) и этил-3-(4-гидрокси-2-изопропоксифенил)пропаноата (0,25 г, 0,99 ммоль) согласно способу, описанному на схеме 15 (0,04 г, выход 13,73%); чистота: 97,21%.
Пример 61
3-{4-[(4-{[(2Е,2Z)-2-(Метоксиимино)-2-фенилэтил]окси}бензил)окси] фенил}-3-метилбутановая кислота (61)

Соединение 61 синтезировали из O-метилоксима (1E,1Z)-2-[4-(гидроксиметил)-фенокси]-1-фенилэтанона (0,128 г, 0,47 ммоль) и метил-3-(4-гидроксифенил)-3-метилбутаноата (0,11 г, 0,52 ммоль) согласно способу, описанному на схеме 18 (0,001 г, выход 1,04%); чистота: 65,13%.
Пример 62
3-Циано-3-{4-((4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]-2-метилфенил}пропановая кислота (62)
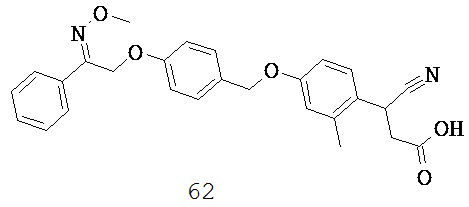
Соединение 62 синтезировали из O-метилоксима (1Z)-2-[4-(гидроксиметил)фенокси]-1-фенилэтанона (0,5 г, 1,85 ммоль) и метил-3-циано-3-(4-гидрокси-2-метилфенил)пропаноата (0,404 г, 1,85 ммоль) согласно способу, описанному на схеме 18 (0,65 г, выход 95,69%); чистота: 93,64%.
Схема 18
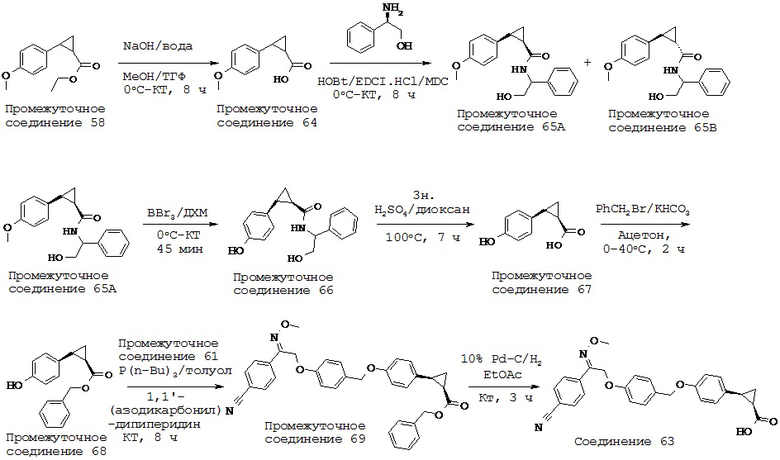
Пример 63
(1R,2S)-2-{4-[(4-{[(2Z)-2-(4-Цианофенил)-2-(метоксиимино)этил]окси}бензил)окси]фенил}циклопропанкарбоновая кислота (63)
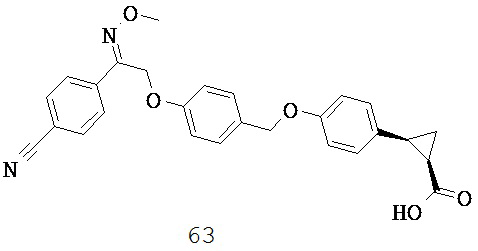
Соединение 63 получали с применением бензил-(1R,2S)-2-(4-гидроксифенил)-циклопропанкарбоксилата и 4-{(1Е,1Z)-2-[4-(гидроксиметил)фенокси]-N-метоксиэтанимидоил}бензонитрила согласно способу, описанному на схеме 17.
Промежуточное соединение 64: 2-(4-метоксифенил)циклопропанкарбоновая кислота
В 100 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 20 мл тетрагидрофурана. В перемешиваемый растворитель добавляли этил-2-(4-метоксифенил)-циклопропанкарбоксилат (8 г, 36 ммоль). Реакционную смесь охлаждали до 0°C и гидроксид натрия (2,1 г, 54 ммоль) в воде (5 мл) добавляли по каплям, затем этанол (10 мл). Реакционную смесь перемешивали в течение 8 часов при комнатной температуре. Реакционную смесь концентрировали для отгонки растворителя; неочищенное вещество подкисляли 1 н. хлороводородной кислотой до кислого pH и экстрагировали этилацетатом (50 мл). Слои разделяли; органический слой промывали солевым раствором (20 мл). Органический слой сушили над Na2SO4 и растворитель удаляли при пониженном давлении. Продукт получали в виде беловатого твердого вещества (6,5 г, выход 94,2%). 1H ЯМР (300 МГц, CDCl3): δ 6,96-6,99 (d, 2H), 6,74-6,77 (d, 2H), 3,71 (s, 3H), 2,46-2,53 (m, 1Н), 1,72-1,78 (m, 1Н), 1,52-1,58 (m, 1Н), 1,27-1,32 (m, 1Н).
Промежуточные соединения 65A и 65B: (1R,2S)-N-(2-гидрокси-1-фенилэтил)-2-(4-метоксифенил)ииклопропанкарбоксамид
В 250 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 70 мл дихлорметана. В перемешиваемый растворитель добавляли 2-(4-метоксифенил)-циклопропанкарбоновую кислоту (4,5 г, 33 ммоль), D-(-)-α-фенилглицинол (3,2 г, 50 ммоль). Реакционную смесь охлаждали до 0°C, затем добавляли гидрохлорид N-(3-диметиламинопропил)-N-этилкарбодиимида (6,7 г, 50 ммоль), N-гидроксибензотриазол (3,1 г, 33 ммоль) и реакционную смесь оставляли перемешиваться при комнатной температуре в течение 8 часов. Реакционную смесь разбавляли дихлорметаном (100 мл) и промывали водой (100 мл). Органический слой промывали солевым раствором (25 мл). Органический слой сушили над Na2SO4 и растворитель удаляли при пониженном давлении. Продукт получали в виде белого твердого вещества (2 г, выход 27,44%).
Промежуточное соединение 66: (1R,2S)-2-(4-гидроксифенил)-N-(2-гидрокси-1-фенилэтил)циклопропанкарбоксамид
В 250 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 100 мл дихлорметана. В перемешиваемый растворитель добавляли (1R,2S)-N-(2-гидрокси-1-фенилэтил)-2-(4-метоксифенил)циклопропанкарбоксамид (1,9 г, 6 ммоль). Реакционную смесь охлаждали до 0°C и трибромид бора (0,86 мл, 9 ммоль) добавляли по каплям. Реакционную смесь оставляли перемешиваться при комнатной температуре в течение 45 минут. Реакцию гасили этанолом (5 мл), реакционную смесь концентрировали для отгонки растворителя; воду (25 мл) добавляли и экстрагировали этилацетатом (25 мл). Органический слой промывали насыщенным раствором NaHCO3 (10 мл) и солевым раствором (10 мл). Органический слой сушили над Na2SO4 и растворитель удаляли при пониженном давлении. Продукт получали в виде белого твердого вещества (0,55 г, выход 58,7%). 1Н ЯМР (300 МГц, ДМСО-d6): δ 9,20 (s, 1Н), 8,47-8,50 (d, 1H), 7,21-7,30 (m, 5H), 6,88-6,91 (d, 2H), 6,63-6,66 (d, 2H), 4,86-4,90 (t, 2H), 3,52-3,56 (t, 2H), 2,05-2,11 (m, 1H), 1,87-1,93 (m, 1H), 1,24-1,29 (m, 1H), 1,04-1,10 (m, 1H).
Промежуточное соединение 67: (1R,2S)-2-(4-гидроксифенил)циклопропанкарбоновая кислота
В 100 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 22 мл диоксана. В перемешиваемый растворитель добавляли (1R,2S)-2-(4-гидроксифенил)-N-(2-гидрокси-1-фенилэтил)циклопропанкарбоксамид (0,75 г, 2,52 ммоль) и 3н. серную кислоту (22 мл). Реакционную смесь нагревали при 100°C в течение 7 часов. Реакционную смесь выливали в лед и экстрагировали этилацетатом (25 мл). Органический слой промывали солевым раствором (25 мл). Органический слой сушили над Na2SO4 и растворитель удаляли при пониженном давлении. Продукт получали в виде бледно-желтой маслянистой жидкости (0,51 г, выход 100%). 1H ЯМР (300 МГц, ДМСО-d6): δ 12,20 (s, 1Н), 9,25 (s, 1H), 6,93-6,96 (d, 2H), 6,64-6,67 (d, 2H), 2,24-2,31 (m, 1H), 1,63-1,68 (m, 1H), 1,31-1,37 (m, 1H), 1,20-1,26 (m, 1H).
Промежуточное соединение 68: бензил-(1R,2S)-2-(4-гидроксифенил)циклопропанкарбоксилат
В 100 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 50 мл ацетона. В перемешиваемый растворитель добавляли (1R,2S)-2-(4-гидроксифенил)-циклопропанкарбоновую кислоту (0,51 г, 2,86 ммоль). Реакционную смесь охлаждали до 0°C, добавляли бикарбонат калия (0,28 г, 2,8 ммоль), перемешивали в течение 10 минут и бензилбромид (0,3 мл, 2,8 ммоль) добавляли по каплям. Реакционную смесь нагревали при 40°C в течение 2 часов. Реакционную смесь концентрировали для отгонки растворителя; воду (25 мл) добавляли и экстрагировали этилацетатом (25 мл). Органический слой промывали солевым раствором (25 мл). Органический слой сушили над Na2SO4 и растворитель удаляли при пониженном давлении. Продукт получали в виде бледно-желтой жидкости (0,44 г, выход 57,2%). 1Н ЯМР (300 МГц, CDCl3): δ 7,30 (s, 4H), 7,19 (s, 1H), 6,89-6,92 (d, 2H), 6,66-6,68 (d, 2H), 5,08 (s, 2H), 4,89 (s, 1H), 2,41-2,48 (m, 1H), 1,78-1,84 (m, 1H), 1,49-1,56 (m, 1H), 1,17-1,24 (m, 1H).
Промежуточное соединение 69: бензил-(1R,2S)-2-{4-[(4-{[(2Е,2Z)-2-(4-цианофенил)-2-(метоксиимино)этил]окси}бензил)окси]фенил}циклопропанкарбоксилат
В 100 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 20 мл толуола. В перемешиваемый растворитель добавляли бензил-(1R,2S)-2-(4-гидроксифенил)-циклопропанкарбоксилат (0,44 г, 1,6 ммоль) и 4-{(1E,1Z)-2-[4-(гидроксиметил)фенокси]-N-метоксиэтанимидоил}бензонитрил (0,43 г, 1,4 ммоль). Реакционную смесь охлаждали до 0°C; трибутилфосфин (0,61 мл, 2,4 ммоль) добавляли и перемешивали в течение 10 минут. В полученную смесь 1,1-(азодикарбонил)дипиперидин (0,62 г, 2,4 ммоль) в толуоле (2 мл) добавляли по каплям. Реакционную смесь перемешивали при комнатной температуре в течение 8 часов. Реакционную смесь разбавляли этилацетатом (25 мл) и промывали водой (25 мл). Органический слой промывали солевым раствором (25 мл). Органический слой сушили над Na2SO4 и растворитель удаляли при пониженном давлении. Продукт получали в виде бледно-желтой маслянистой жидкости (0,64 г, выход 71,4%).
Соединение 63: (1R,2S)-2-{4-[(4-{[(2Z)-2-(4-Цианофенил)-2-(метоксиимино)этил]окси}бензил)окси]фенил}циклопропанкарбоновая кислота
В 250 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 20 мл этилацетата. В перемешиваемый растворитель добавляли бензил-(1R,2S)-2-{4-[(4-{[(2Е,2Z)-2-(4-цианофенил)-2-(метоксиимино)этил]окси}бензил)окси]фенил}циклопропанкарбоксилат (0,64 г, 1,17 ммоль). Реакционную смесь продували азотом в течение 10 минут. Затем 10% палладий на углеродной подложке (0,07 г) добавляли и перемешивали в атмосфере водорода в течение 3 часов. Через 3 часа реакционную смесь фильтровали через целит и промывали этилацетатом (50 мл). Фильтрат концентрировали для отгонки растворителя и сушили в вакууме. Продукт получали в виде белого твердого вещества (0,19 г, выход 37,1%). МС (ИЭР, 120 эВ): m/z=455,1 (М-Н)+; чистота по данным ВЭЖХ: 94,6%; 1Н ЯМР (300 МГц, ДМСО-d6): δ 12,11 (br s, 1H), 7,80-7,89 (m, 4H), 7,32-7,35 (d, 2H), 7,05-7,08 (d, 2H), 6,87-6,91 (m, 4H), 5,27 (s, 2H), 4,97 (s, 2H), 2,27-2,35 (m, 1H), 1,67-1,73 (m, 1H), 1,33-1,39 (m, 1H).
Пример 64
(1R,2S)-2-{4-[(4-{[(2Е,2Z)-2-(4-Цианофенил)-2-(метоксиимино)этил]окси}бензил)окси]фенил}циклопропанкарбоновая кислота (64)
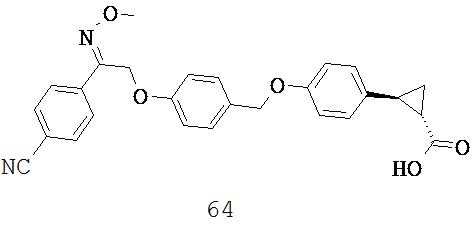
Соединение 64 синтезировали из бензил-(1S,2S)-2-(4-гидроксифенил)-циклопропанкарбоксилата (0,6 г, 2,2 ммоль) и 4-{(1Е,1Z)-2-[4-(гидроксиметил)фенокси]-N-метоксиэтанимидоил}бензонитрила (0,66 г, 2,2 ммоль) согласно способу, описанному на схеме 18 (0,2 г, выход 39,91%); чистота: 89,14%.
Пример 65
3-{4-[(4-{[(2Z)-2-(Метоксиимино)-2-фенилэтил]окси}бензил)окси]-2-(5-метил-1,2-оксазол-3-ил)фенил}пропановая кислота
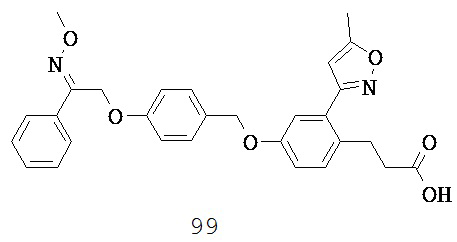
Соединение 65 синтезировали из (4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}-фенил)метанола (0,0012 г, 0,0439 ммоль) и метил-3-[4-гидрокси-2-(5-метил-1,2-оксазол-3-ил)фенил]пропаноата (0,0015 г, 0,0549 ммоль) согласно способу, описанному на схеме 18 (0,006 г, выход 10,20%); чистота: 96,94%.
Пример 66
3-Циано-3-{4-[(4-{[(2Z)-2-(метоксиимино)-2-(4-метоксифенил)этил]окси}бензил)окси]фенил}пропановая кислота
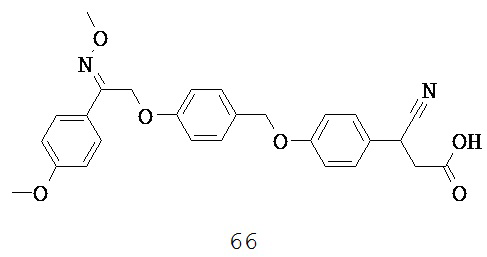
Соединение 66 синтезировали из (4-{[(2Z)-2-(метоксиимино)-2-(4-метоксифенил)этил]-окси}фенил)метанола (0,33 г, 0,9 ммоль) и метил-3-циано-3-(4-гидроксифенил)пропаноата (0,21 г, 0,9 ммоль) согласно способу, описанному на схеме 13 (0,3 г, выход 89,28%); чистота: 85%.
Схема 19
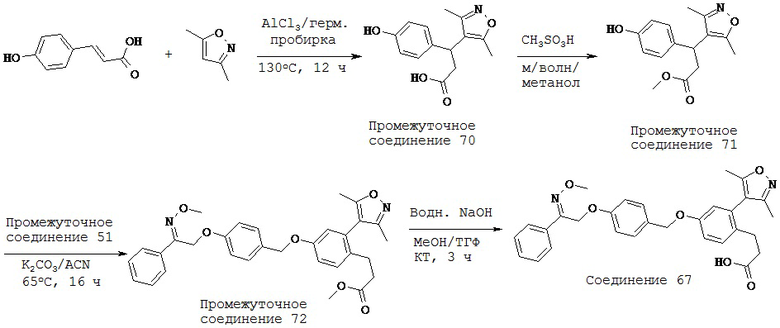
Пример 67
3-(3,5-Диметил-1,2-оксазол-4-ил)-3-{4-[(4-{[(2Е,2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропановая кислота
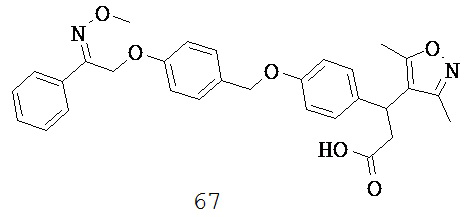
Соединение 67 синтезировали из (1Е,1Z)-2-[4-(бромметил)фенокси]-N-метокси-1-фенидэтанимина (0,121 г, 0,363 ммоль) и метил-3-(3,5-диметил-1,2-оксазол-4-ил)-3-(4-гидроксифенил)пропаноата (0,1 г, 0,363 ммоль) согласно способу, описанному на схеме 18 (0,01 г, выход 5,4%).
Промежуточное соединение 70: 3-(3,5-диметил-1,2-оксазол-4-ил)-3-(4-гидроксифенил)пропановая кислота
В 50 мл герметичную пробирку помещали (2Е)-3-(4-гидроксифенил)проп-2-еновую кислоту (3,0 г, 0,018 ммоль) и 3,5-диметил-1,2-оксазол (3,54 г, 0,054 ммоль) и нагревали при 130°C в течение 16 часов. Реакционную смесь разбавляли водой (10 мл) и экстрагировали ДХМ (20 мл). Объединенные экстракты сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенное вещество применяли на следующей стадии без очистки (1,0 г, выход 20,96%).
Промежуточное соединение 71: метил-3-(3,5-диметил-1,2-оксазол-4-ил)-3-(4-гидроксифенил)пропаноат
В 25 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 20 мл метанола. В перемешиваемый растворитель добавляли 3-(3,5-диметил-1,2-оксазол-4-ил)-3-(4-гидроксифенил)пропановую кислоту (1,16 г, 12,0 ммоль). После завершения добавления PC кипятили с обратным холодильником при 65°C в течение 1 часа. Через 1 час растворитель выпаривали, воду (20 мл) добавляли и органический слой экстрагировали этилацетатом (15 мл × 2). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Продукт получали в виде коричневой жидкости (0,2 г, выход 18,72%).
Промежуточное соединение 72: метил-3-(3,5-диметил-1,2-оксазол-4-ил)-3-{4-[(4-{[(2Е,2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропаноат
В 25 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 5 мл ацетонитрила. В перемешиваемый растворитель добавляли метил-3-(3,5-диметил-1,2-оксазол-4-ил)-3-(4-гидроксифенил)пропаноат (0,1 г, 0,363 ммоль), карбонат калия (0,15 г, 1,09 ммоль). Реакционную смесь охлаждали до 0°C, (1Е,1Z)-2-[4-(бромметил)фенокси]-N-метокси-1-фенилэтанимин (0,121 г, 0,363 ммоль) в ацетонитриле (2 мл) добавляли по каплям и перемешивали при 65°С в течение 14 часов. Реакционную смесь концентрировали для отгонки растворителя; разбавляли водой (10 мл) и экстрагировали этилацетатом (10 мл). Органический слой промывали водой (5 мл) и насыщенным солевым раствором (5 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали путем колоночной хроматографии на силикагеле с применением этилацетата и петролейного эфира в качестве элюентов. Продукт получали в виде белого твердого вещества (0,19 г, выход 100,00%).
Соединение 67: 3-(3,5-Диметил-1,2-оксазол-4-ил)-3-{4-[(4-{[(2E,2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропановая кислота
В 10 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 3 мл тетрагидрофурана. В перемешиваемый растворитель добавляли метил-3-(3,5-диметил-1,2-оксазол-4-ил)-3-{4-[(4-{[(2E,2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}-пропаноат (0,19 г, 0,36 ммоль), метанол (3 мл) и гидроксид натрия (0,030 г, 0,75 ммоль) в воде (1,0 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи. После завершения реакции растворитель удаляли, натриевую соль промывали диэтиловым эфиром (5 мл); водный слой подкисляли 1 н. HCl до pH 2,0 и экстрагировали этилацетатом (5 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали путем колоночной хроматографии на силикагеле с применением метанола и дихлорметана в качестве элюентов. Продукт получали в виде бледно-желтого липкого твердого вещества (0,01 г, выход 5,4%).
Схема 20
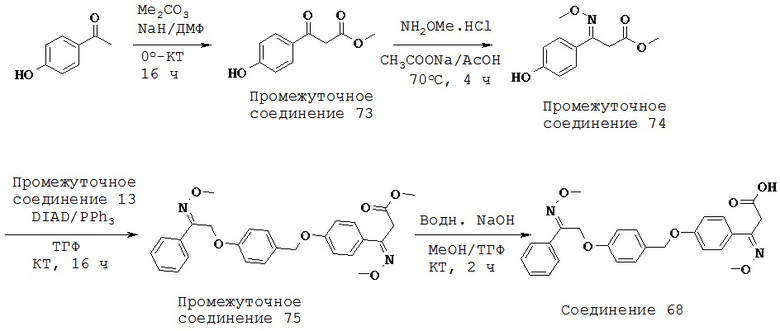
Пример 68
(3E,3Z)-3-(Метоксиимино)-3-{4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропановая кислота
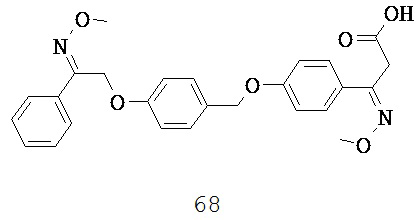
Соединение 68 синтезировали из (4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}-фенил)метанола (0,24 г, 0,89 ммоль) и метил-(3E,3Z)-3-(4-гидроксифенил)-3-(метоксиимино)-пропаноата (0,2 г, 0,89 ммоль) согласно способу, описанному на схеме 20 (0,02 г, выход 25,76%); чистота: 96,32%.
Промежуточное соединение 73: метил-3-(4-гидроксифенил)-3-оксопропаноат
В 250 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали гидрид натрия (3,5 г, 147 ммоль). При 0°C 100 мл ДМФ добавляли медленно при перемешивании. В перемешиваемый растворитель добавляли 1-(4-гидроксифенил)этанон (5 г, 36 ммоль) и диметилкарбонат (15,5 г, 170 ммоль), перемешивали при комнатной температуре в течение ночи. PC выливали в воду (100 мл) и экстрагировали этилацетатом (100 мл). Органический слой промывали водой (50 мл) и насыщенным солевым раствором (50 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали путем колоночной хроматографии на силикагеле с применением этилацетата и петролейного эфира в качестве элюентов. Продукт получали в виде бледно-желтой маслянистой жидкости (3,4 г, выход 47,66%). МС (ИЭР, 120 эВ): m/z=195 (М+Н)+; 1Н ЯМР (300 МГц, CDCl3): δ 7,79-7,82 (d, 2H), 6,81-6,84 (d, 2H), 6,21 (br s, 1H), 3,90 (s, 2H), 3,69 (s, 3H).
Промежуточное соединение 74: метил-(3Е,3Z)-3-(4-гидроксифенил)-3-(метоксиимино)пропаноат
В 50 мл кругло донную колбу, оборудованную магнитной мешалкой, помещали 10 мл уксусной кислоты. В перемешиваемый растворитель добавляли метил-3-(4-гидроксифенил)-3-оксопропаноат (0,5 г, 2,6 ммоль), ацетат натрия (0,31 г, 3,8 ммоль) и гидрохлорид O-метилгидроксиламина (0,32 г, 3,8 ммоль), перемешивали при 70°C в течение 4 часов. PC выливали в воду (50 мл) и экстрагировали этилацетатом (50 мл). Органический слой промывали водой (25 мл) и насыщенным солевым раствором (25 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Продукт получали в виде белого твердого вещества (0,35 г, выход 60,86%). МС (ИЭР, 120 эВ): m/z=224 (М+Н)+; 1Н ЯМР (300 МГц, CDCl3): δ 7,43-7,46 (d, 2H), 6,71-6,74 (d, 2H), 5,45 (s, 1H), 3,91 (s, 3H), 3,68 (s, 2H), 3,63 (s, 3H).
Промежуточное соединение 75: метил-(3E,3Z)-3-(метоксиимино)-3-{4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропаноат
В 50 мл кругло донную колбу, оборудованную магнитной мешалкой, помещали 10 мл тетрагидрофурана. В перемешиваемый растворитель добавляли метил-(3E,3Z)-3-(4-гидроксифенил)-3-(метоксиимино)пропаноат (0,2 г, 0,89 ммоль), (1E,1Z)-2-[4-(гидроксиметил)фенокси]-1-фенилэтанон (0,24 г, 0,89 ммоль) и трифенилфосфин (0,34 г, 1,34 ммоль). Реакционную смесь охлаждали до 0°С и диизопропилазокарбоксилат (0,27 г, 13,4 ммоль) добавляли по каплям и перемешивали при комнатной температуре в течение ночи. PC выливали в воду (25 мл) и экстрагировали этилацетатом (50 мл). Органический слой промывали водой (50 мл) и насыщенным солевым раствором (50 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали путем колоночной хроматографии на силикагеле с применением этилацетата и петролейного эфира в качестве элюентов. Продукт получали в виде бесцветной маслянистой жидкости (0,08 г, выход 18,74%). МС (ИЭР, 120 эВ): m/z=477 (M+H)+; 1Н ЯМР (300 МГц, CDCl3): δ 7,58-7,61 (m, 2H), 7,49-7,52 (d, 2H), 7,26-7,29 (m, 4Н), 7,23 (s, 1H), 6,83-6,89 (t, 4H), 5,13 (s, 2H), 4,92 (s, 2H), 3,99 (s, 3H), 3,91 (s, 3H), 3,68 (s, 2H), 3,62 (s, 3H).
Соединение 68: (3E,3Z)-3-(Метоксиимино)-3-{4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропановая кислота
В 10 мл кругло донную колбу, оборудованную магнитной мешалкой, помещали 4 мл тетрагидрофурана. В перемешиваемый растворитель добавляли метил-(3E,3Z)-3-(метоксиимино)-3-{4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}-пропаноат (0,08 г, 0,16 ммоль) и метанол (2 мл). Затем реакционную смесь охлаждали до 0°C и гидроксид натрия (0,13 г, 0,33 ммоль) в воде (1 мл) добавляли и перемешивали в течение 1 часа. PC концентрировали для отгонки растворителя, неочищенное вещество промывали диэтиловым эфиром, подкисляли 1 н. HCl и экстрагировали этилацетатом (5 мл). Неочищенное вещество очищали путем препаративной ТСХ с применением метанола и хлороформа в качестве элюентов. Продукт получали в виде беловатого твердого вещества (0,02 г, выход 25,76%).
Схема 21
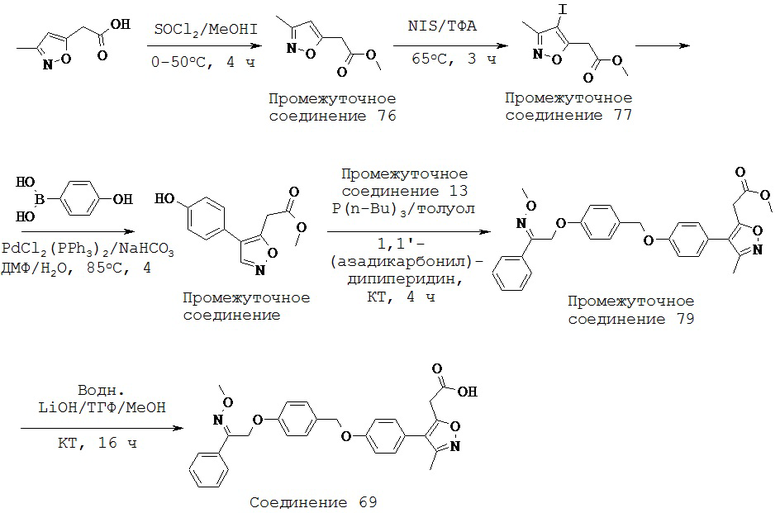
Пример 69
(4-{4-[(4-{[(2Z)-2-(Метоксиимино)-2-фенилэтил] окси}бензил)окси] фенил}-3-метил-1,2-оксазол-5-ил)уксусная кислота
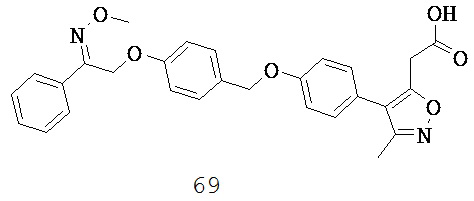
Соединение 69 синтезировали из (4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}-фенил)метанола (0,329 г, 1,2 ммоль) и метил-[4-(4-гидроксифенил)-3-метил-1,2-оксазол-5-ил]ацетата (0,3 г, 1,2 ммоль) согласно способу, описанному на схеме 21 (0,32 г, выход 70,2%); чистота: 96,43%.
Промежуточное соединение 76: метил-(3-метил-1,2-оксазол-5-ил)ацетат
В 50 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 10 мл метанола. В перемешиваемый растворитель добавляли (3-метил-1,2-оксазол-5-ил)уксусную кислоту (1 г, 7 ммоль), затем по каплям добавляли SOCl2 (1,25 г, 10,5 ммоль) в реакционную смесь при 0°C и перемешивали при 50°C в течение 4 часов. После завершения реакции реакционную смесь концентрировали для отгонки растворителя; воду (25 мл) добавляли и экстрагировали этилацетатом (25 мл). Органический слой промывали насыщенным раствором NaHCO3 (10 мл) и солевым раствором (10 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Продукт получали в виде коричневой жидкости (1 г, выход 92%). 1Н ЯМР (300 МГц, CDCl3): δ 6,03 (s, 1H), 3,73 (d, 2H), 3,69 (s, 3H), 2,23 (s, 3H).
Промежуточное соединение 77: метил-(4-йод-3-метил-1,2-оксазол-5-ил)ацетат
В герметичную пробирку, оборудованную магнитной мешалкой, помещали метил-(3-метил-1,2-оксазол-5-ил)ацетат (0,9 г, 5,8 ммоль) и N-йодсукцинимид (2,61 г, 11,6 ммоль) в 10 мл трифторуксусной кислоты. Реакционную смесь нагревали при 65°C в течение 3 часов. Реакцию гасили раствором NaHCO3 (25 мл) и смесь экстрагировали этилацетатом (30 мл). Органический слой промывали водой (30 мл) и насыщенным солевым раствором (30 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Продукт получали в виде коричневого твердого вещества (1,18 г, выход 72,4%).
Промежуточное соединение 78: метил-[4-(4-гидроксифенил)-3-метил-1,2-оксазол-5-ил]ацетат
В 50 мл двухгорлую круглодонную колбу, оборудованную магнитной мешалкой, помещали 5 мл ДМФ. В перемешиваемый растворитель добавляли метил-(4-йод-3-метил-1,2-оксазол-5-ил)ацетат (0,7 г, 2,5 ммоль), (4-гидроксифенил)бороновую кислоту (0,34 г, 2,5 ммоль) и 1 мл водного раствора NaHCO3 (0,62 г, 7,47 ммоль) в атмосфере аргона. После продувания газообразного аргона в течение примерно 10 минут добавляли хлорид бис(трифенилфосфин)палладия (II) (0,21 г, 0,3 ммоль) и нагревали при 85°C в течение 4 часов. PC фильтровали через целит, промывали холодной водой и экстрагировали этилацетатом (25 мл). Органический слой промывали водой (20 мл) и насыщенным солевым раствором (20 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали путем колоночной хроматографии на силикагеле с применением этилацетата и петролейного эфира в качестве элюентов. Продукт получали в виде коричневого твердого вещества (0,4 г, выход 66,6%). 1Н ЯМР (300 МГц, CDCl3): δ 7,07-7,10 (d, 2Н), 6,84-6,86 (d, 2H), 5,82 (s, 1H), 3,69 (s, 2H), 3,66 (s, 3Н), 2,21 (s, 3Н).
Промежуточное соединение 79: метил-(4-{4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}-3-метил-1,2-оксазол-5-ил)ацетат
В 50 мл двухгорлую круглодонную колбу, оборудованную магнитной мешалкой, помещали 15 мл толуола. В перемешиваемый растворитель добавляли метил-[4-(4-гидроксифенил)-3-метил-1,2-оксазол-5-ил]ацетат (0,3 г, 1,21 ммоль), O-метилоксим (1Z)-2-[4-(гидроксиметил)фенокси]-1-фенилэтанона (0,33 г, 1,21 ммоль) и три(н-бутил)фосфин (0,39 г, 1,94 ммоль) в атмосфере азота. Реакционную смесь охлаждали до 0°C и 1,1'-(азодикарбонил)дипиперидин (0,49 г, 1,94 ммоль) добавляли по частям и перемешивали при комнатной температуре в течение 2 часов. После завершения реакции реакционную смесь выливали в воду (20 мл) и экстрагировали этилацетатом (25 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали путем колоночной хроматографии на силикагеле с применением этилацетата и петролейного эфира в качестве элюентов. Продукт получали в виде бесцветной маслянистой жидкости (0,47 г, выход 77%). 1H ЯМР (300 МГц, CDCl3): δ 7,57-7,61 (m, 2Н), 7,26-7,30 (m, 5H), 7,11-7,14 (d, 2H), 6,94-6,97 (d, 2H), 6,85-6,88 (d, 2H), 5,14 (s, 2H), 4,93 (s, 2H), 3,99 (s, 3Н), 3,68 (s, 2H), 3,65 (s, 3H), 2,20 (s, 3H).
Соединение 69: (4-{4-[(4-{[(2Z)-2-(Метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}-3-метил-1,2-оксазол-5-ил)уксусная кислота
В 25 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали тетрагидрофуран (5 мл). В перемешиваемый растворитель добавляли метил-(4-{4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}-3-метил-1,2-оксазол-5-ил)ацетат (0,47 г, 0,94 ммоль), метанол (3 мл) и гидроксид лития (0,07 г, 0,94 ммоль) в воде (3 мл). После завершения добавления реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь концентрировали для отгонки растворителя; неочищенное вещество промывали диэтиловым эфиром (5 мл). Воду (1 мл) добавляли и водный слой подкисляли насыщенным раствором лимонной кислоты до pH 6. Водный слой экстрагировали этилацетатом (5 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Продукт получали в виде белого твердого вещества (0,32 г, выход 70,2%).
Пример 70
3-{2-Метокси-4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропаноат натрия
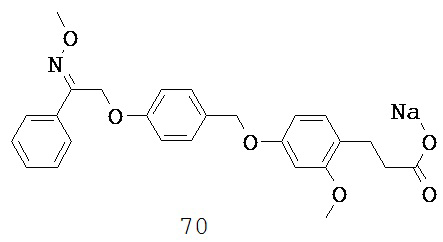
Соединение 70 синтезировали из 3-{2-метокси-4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропановой кислоты (0,204 г, 0,45 ммоль) и гидроксида натрия (0,45 мл, (1М, раствор), 0,45 ммоль) согласно способу, описанному на схеме 12 (0,15 г, выход 71,43%); чистота: 99,11%.
Пример 71
3-{2-Фтор-4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропаноат натрия
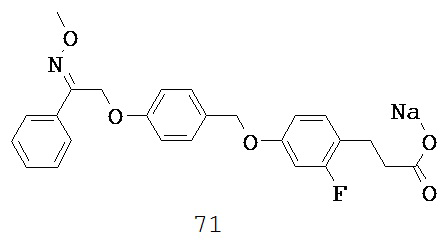
Соединение 71 синтезировали из 3-{2-Фтор-4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропановой кислоты (0,102 г, 0,233 ммоль) и гидроксида натрия (0,233 мл, 1М раствор, 0,233 ммоль) согласно способу, описанному на схеме 12 (0,06 г, выход 56,07%); чистота: 98,81%.
Схема 22
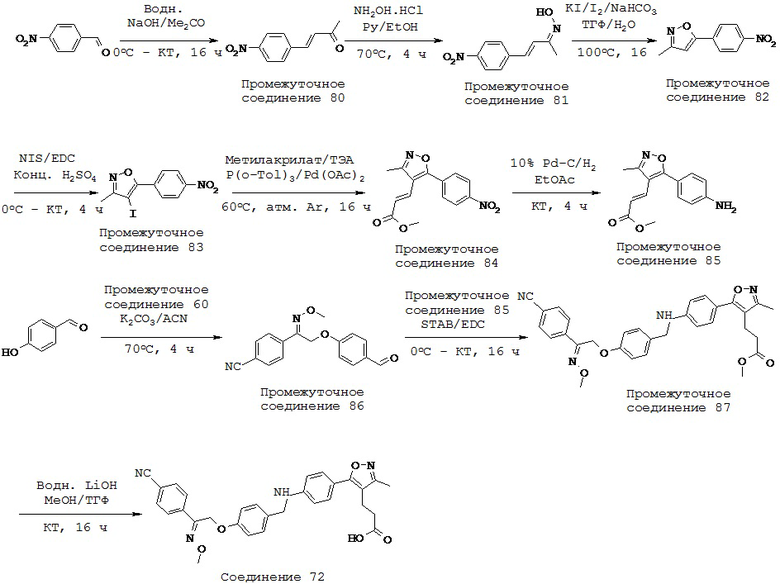
Пример 72
3-(5-{4-[(4-{[(2Е,2Z)-2-(4-Цианофенил)-2-(метоксиимино)этил]окси}бензил)амино]-фенил}-3-метил-1,2-оксазол-4-ил)пропановая кислота (72)
Соединение 72 синтезировали из 4-[(1E,1Z)-2-(4-фтормилфенокси)-N-метоксиэтанимидоил]бензонитрила (0,158 г, 0,54 ммоль) и метил-3-[5-(4-аминофенил)-3-метил-1,2-оксазол-4-ил]пропаноата (0,14 г, 0,54 ммоль) согласно способу, описанному на схеме 22 (0,0056 г, выход 33,84%); чистота: 87,65%.
Промежуточное соединение 80: (3E,3Z)-4-(4-нитрофенил)бут-3-ен-2-он
В 100 мл кругло донную колбу, оборудованную магнитной мешалкой, помещали 100 мл ацетона. В перемешиваемый растворитель добавляли 4-нитробензальдегид (10 г, 66,2 ммоль), затем 4н. раствор NaOH (10 мл) при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь концентрировали для отгонки растворителя. Воду (25 мл) добавляли и экстрагировали этилацетатом (25 мл). Органический слой промывали насыщенным солевым раствором. Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали путем колоночной хроматографии на силикагеле с применением этилацетата и петролейного эфира в качестве элюентов. Продукт получали в виде желтого твердого вещества (2,5 г, выход 19,75%). 1H ЯМР (300 МГц, CDCl3): δ 8,18-8,21 (d, 2H), 7,62-7,65 (d, 2H), 7,44-7,49 (d, 1H), 6,73-6,78 (d, 1H), 2,36 (s, 3H).
Промежуточное соединение 81: оксим (2Z,3E)-4-(4-нитрофенил)бут-3-ен-2-она
В 100 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 10 мл этанола. В перемешиваемый растворитель добавляли (2E,3Z)-4-(4-нитрофенил)бут-3-ен-2-он (0,5 г, 2,62 ммоль), гидрохлорид гидроксиламина (0,27 г, 3,93 ммоль) и пиридин (0,828 г, 10,47 ммоль) и перемешивали при 70°C в течение 4 часов в атмосфере азота. PC выливали в воду (50 мл) и экстрагировали этилацетатом (50 мл). Органический слой промывали 1 н. раствором HCl (25 мл) и насыщенным солевым раствором (25 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Продукт получали в виде желтого твердого вещества (0,516 г, выход 95,7%). МС (ИЭР, 120 эВ): m/z=207,1 (М+Н)+; 1Н ЯМР (300 МГц, ДМСО-d6): δ 11,49 (s, 1Н), 8,19-8,22 (d, 2H), 7,84-7,87 (d, 2H), 7,10 (s, 2H), 2,02 (s, 3H).
Промежуточное соединение 82: 3-метил-5-(4-нитрофенил)-1,2-оксазол
В 100 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 20 мл тетрагидрофурана и воду (10 мл). В перемешиваемый растворитель добавляли (2Z,3E)-N-гидрокси-4-(4-нитрофенил)бут-3-ен-2-имин (0,5 г, 2,42 ммоль), йодид калия (0,81 г, 4,85 ммоль), йод (1,23 г, 4,49 ммоль) и NaHCO3 (0,81 г, 9,7 ммоль). Смесь перемешивали при 100°C в течение ночи в атмосфере азота. Реакционную смесь охлаждали, выливали в воду и экстрагировали этилацетатом. Органический слой промывали водой (25 мл), раствором тиосульфата натрия (30 мл) и насыщенным солевым раствором (30 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали путем колоночной хроматографии на силикагеле с применением этилацетата и петролейного эфира в качестве элюентов. Продукт получали в виде желтого твердого вещества (0,31 г, выход 62,6%). МС (ИЭР, 120 эВ): m/z=205,1 (М+Н)+; 1Н ЯМР (300 МГц, CDCl3): δ 8,24-8,27 (t, 2H), 7,85-7,87 (t, 2H), 6,49 (s, 1Н), 2,33 (s, 3H).
Промежуточное соединение 83: 4-йод-3-метил-5-(4-нитрофенил)-1,2-оксазол
В 100 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 10 мл 1,2-дихлорэтана. В перемешиваемый раствор добавляли 3-метил-5-(4-нитрофенил)-1,2-оксазол (0,27 г, 1,30 ммоль), N-йодсукцинимид (0,28 г, 1,24 ммоль), затем концентрированную H2SO4 (0,2 мл) при 0°C и перемешивали при комнатной температуре в течение 4 часов. Реакционную смесь выливали в воду (30 мл) и экстрагировали этилацетатом (50 мл). Органический слой промывали раствором тиосульфата натрия (30 мл), водой (30 мл) и насыщенным солевым раствором (10 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Продукт получали в виде желтого твердого вещества (0,40 г, выход 93,7%). 1Н ЯМР (300 МГц, CDCl3): δ 8,29-8,32 (dd, 2H), 8,18-8,21 (dd, 2H), 2,33 (s, 3H).
Промежуточное соединение 84: метил-(2Е,2Z)-3-[3-метил-5-(4-нитрофенил)-1,2-оксазол-4-ил]акрилат
В 100 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали триэтиламин (15 мл). В перешиваемый растворитель добавляли 4-йод-3-метил-5-(4-нитрофенил)-1,2-оксазол (0,35 г, 1,06 ммоль) и метилакрилат (0,2 мл, 2,12 ммоль) в атмосфере аргона. После продувания аргона в течение 15 минут три-о-толуилфосфин (0,032 г, 0,11 ммоль) и ацетат палладия (II) (0,024 г, 0,11 ммоль) добавляли в реакционную смесь. Реакционную смесь нагревали при 60°C в течение ночи в атмосфере аргона. Реакционную смесь фильтровали через целит и промывали этилацетатом и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали путем колоночной хроматографии на силикагеле с применением этилацетата и петролейного эфира в качестве элюентов. Продукт получали в виде бледно-желтого твердого вещества (0,175 г, выход 57,4%). 1Н ЯМР (300 МГц, CDCl3): δ 8,25-8,28 (d, 1Н), 7,85-7,88 (d, 1H), 7,80-7,83 (d, 2H), 7,57-7,62 (d, 1H), 6,26-6,32 (d, 1H), 3,76 (s, 3H) 2,44 (s, 3H).
Промежуточное соединение 85: метил-3-[5-(4-аминофенил)-3-метил-1,2-оксазол-4-ил]пропаноат
В колбу для гидрирования помещали этилацетат (15 мл) и метил-(2Е,2Z)-3-[3-метил-5-(4-нитрофенил)-1,2-оксазол-4-ил]проп-2-еноат (0,17 г, 0,59 ммоль), затем 10% Pd/C (0,2 г). Смесь в колбе гидрировали в течение 4 часов при 60 psi. Реакционную смесь фильтровали через целит, промывали этилацетатом и растворитель удаляли при пониженном давлении путем отгонки. Продукт получали в виде желтого твердого вещества (0,148 г, выход 97%). МС (ИЭР, 120 эВ): m/z=261,0 (М+Н)+; 1Н ЯМР (300 МГц, CDCl3): δ 7,41-7,43 (d, 2H), 6,66-6,69 (d, 2H), 3,60 (s, 3H), 2,80-2,85 (t, 2H), 2,44-2,49 (t, 2H), 2,22 (s, 3H).
Промежуточное соединение 86: 4-[(1Е,1Z)-2-(4-формилфенокси)-N-метоксиэтанимидоил]бензонитрил
В 100 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 20 мл ацетонитрила. В перемешиваемый растворитель добавляли 4-гидроксибензальдегид (0,7 г, 5,7 ммоль) и карбонат калия (2,4 г, 17,1 ммоль). Реакционную смесь охлаждали до 0°C и 4-[(1Е,1Z)-2-бром-N-метоксиэтанимидоил]бензонитрил (1,45 г, 5,7 ммоль) в ацетонитриле (5 мл) добавляли по каплям. После завершения добавления реакционную смесь перемешивали при 80°C в течение 3 часов. Реакционную смесь фильтровали через керамическую воронку, промывали этилацетатом (10 мл). Фильтрат промывали водой (20 мл) и солевым раствором (10 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали путем колоночной хроматографии на силикагеле с применением этилацетата и петролейного эфира в качестве элюентов. Продукт получали в виде белого твердого вещества (0,7 г, выход 42%). 1Н ЯМР (300 МГц, CDCl3): δ 9,82 (s, 1H), 7,70-7,77 (m, 4H), 7,56-7,59 (d, 2H), 6,91-6,94 (d, 2H), 5,23 (s, 2H), 4,05 (s, 3H).
Промежуточное соединение 87: метил-3-(5-{4-[(4-{[(2Е,2Z)-2-(4-цианофенил)-2-(метоксиимино)этил]окси}бензил)амино]фенил}-3-метил-1,2-оксазол-4-ил)пропаноат
В 100 мл кругло донную колбу, оборудованную магнитной мешалкой, помещали 10 мл 1,2-дихлорэтана. В перемешиваемый растворитель добавляли 4-[(1E,1Z)-2-(4-формилфенокси)-N-метоксиэтанимидоил]бензонитрил (0,158 г, 0,54 ммоль) и метил-3-[5-(4-аминофенил)-3-метил-1,2-оксазол-4-ил]пропаноат (0,14 г, 0,54 ммоль). Реакционную смесь охлаждали до 0°C и триацетоксиборгидрид натрия (0,29 г, 1,35 ммоль) добавляли по частям. После завершения добавления реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь выливали в воду (10 мл) и экстрагировали дихлорметаном (20 мл). Органический слой промывали водой (20 мл) и солевым раствором (10 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали путем колоночной хроматографии на силикагеле с применением этилацетата и петролейного эфира в качестве элюентов. Продукт получали в виде желтого вязкого твердого вещества (0,017 г, выход 5,9%). 1Н ЯМР (300 МГц, CDCl3): δ 7,71-7,74 (d, 2H), 7,55-7,58 (d, 2H), 7,41-7,44 (d, 2H), 7,18-7,20 (d, 2H), 6,78-6,81 (d, 2H), 6,58-6,61 (d, 2H), 5,13 (s, 2H), 4,23 (s, 2H), 4,02 (s, 3H), 3,59 (s, 3H), 2,79-2,84 (t, 2H), 2,43-2,48 (t, 2H), 2,21 (s, 3H).
Соединение 72: 3-(5-{4-[(4-{[(2E,2Z)-2-(4-Цианофенил)-2-(метоксиимино)этил]окси}-бензил)амино]фенил}-3-метил-1,2-оксазол-4-ил)пропановая кислота
В 25 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 3 мл тетрагидрофурана. В перемешиваемый растворитель добавляли метил-3-(5-{4-[(4-{[(2E,2Z)-2-(4-цианофенил)-2-(метоксиимино)этил]окси}бензил)амино]фенил}-3-метил-1,2-оксазол-4-ил)пропаноат (0,017 г, 0,03 ммоль), метанол (0,5 мл) и гидроксид лития (0,003 г, 0,13 ммоль) в воде (0,5 мл). После завершения добавления реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь концентрировали для отгонки растворителя; неочищенное вещество промывали диэтиловым эфиром (5 мл). Воду (1 мл) добавляли и водный слой подкисляли насыщенным раствором лимонной кислоты до pH 6. Водный слой экстрагировали этилацетатом (5 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали путем препаративной ТСХ с применением метанола и хлороформа в качестве элюентов. Продукт получали в виде желтого твердого вещества (0,0056 г, выход 33,84%). 2Н), 6,74-6,78 (t, 1Н), 6,66-6,69 (d, 2Н), 5,24 (s, 2Н), 4,23-4,25 (d, 2Н), 4,06 (s, 3H), 2,72-2,77 (t, 2Н), 2,40-2,45 (t, 2Н), 2,20 (s, 3H).
Схема 23
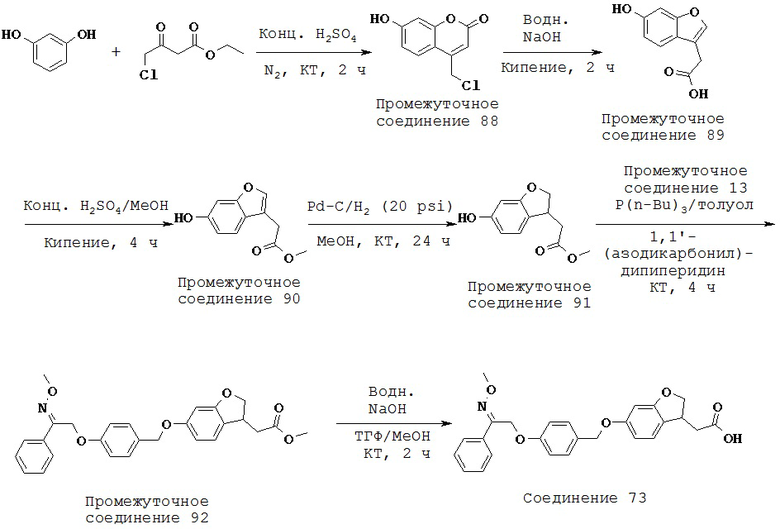
Пример 73
{6-[(4-{[(2Z)-2-(Метоксиимино)-2-фенилэтил]окси}бензил)окси]-2,3-дигидро-1-бензофуран-3-ил}уксусная кислота
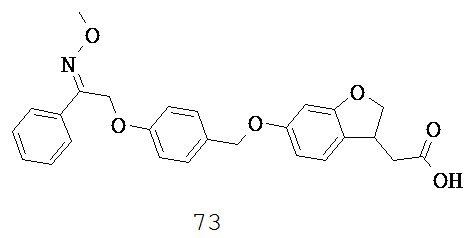
Соединение 73 синтезировали из (4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}-фенил)метанола (0,26 г, 0,96 ммоль) и метил-(6-гидрокси-2,3-дигидро-1-бензофуран-3-ил)ацетата (0,20 г, 0,96 ммоль) согласно способу, описанному на схеме 23 (0,001 г, выход 5,0%); чистота: 97,09%.
Промежуточное соединение 88: 4-(хлорметил)-7-гидрокси-2Н-хромен-2-он
В 250 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали этил-4-хлорацетоацетат (22,12 г, 134,9 ммоль), растворяли в концентрированной серной кислоте (48 мл) при 0°C и резорцин (14,0 г, 127,3 ммоль) добавляли по частям. Смесь перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь выливали в ледяную воду и полученное твердое вещество собирали путем фильтрования, промывали водой и сушили с получением 4-(хлорметил)-7-гидрокси-2Н-хромен-2-она (21,0 г, 75,6%) в виде бежевого твердого вещества.
Промежуточное соединение 89: (6-гидрокси-1-бензофуран-3-ил)уксусная кислота
Смесь полученного 4-(хлорметил)-7-гидрокси-2Н-хромен-2-она (21,0 г, 21,0 ммоль) и 1М водного раствора гидроксида натрия (1 л) перемешивали и кипятили с обратным холодильником в течение 2 часов. Реакционную смесь подкисляли концентрированной серной кислотой и экстрагировали этилацетатом. Экстракт промывали солевым раствором, сушили над безводным сульфатом натрия и концентрировали с получением (6-гидрокси-1-бензофуран-3-ил)уксусной кислоты (18,0 г, 93,75%).
Промежуточное соединение 90: метил-(6-гидрокси-1-бензофуран-3-ил)ацетат
Полученные кристаллы (6-гидрокси-1-бензофуран-3-ил)уксусной кислоты суспендировали в метаноле (100 мл) и в суспензию добавляли концентрированную H2SO4 (10 мл) и смесь перемешивали и кипятили с обратным холодильником в течение 4 часов. После выпаривания растворителя остаток разбавляли диэтиловым эфиром и последовательно промывали водой, насыщенным раствором гидрокарбоната натрия и солевым раствором, сушили над безводным сульфатом натрия и концентрировали с получением метил-(6-гидрокси-1-бензофуран-3-ил)ацетата. Неочищенное вещество очищали путем колоночной хроматографии с получением твердого продукта (11,23 г, 58%).
Промежуточное соединение 91: метил-(6-гидрокси-2,3-дигидро-1-бензофуран-3-ил)ацетат
Полученный метил-(6-гидрокси-1-бензофуран-3-ил)ацетат (6 г, 29,13 ммоль) гидрировали над 10% палладием на углеродной подложке (1 г в 1 мл воды) в метаноле (60 мл) в атмосфере водорода (20 psi) в течение примерно 24 часов. Катализатор удаляли путем фильтрования и фильтрат концентрировали. Остаток очищали путем колоночной хроматографии на силикагеле с получением твердого вещества (2,9 г, 47,93%).
Промежуточное соединение 92: метил-{6-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]-2,3-дигидро-1-бензофуран-3-ил}ацетат
В 50 мл двухгорлую круглодонную колбу, оборудованную магнитной мешалкой, помещали 5 мл толуола. В перемешиваемый растворитель добавляли (4-{[(2E,2Z)-2-(метоксиимино)-2-фенилэтил]окси}фенил)метанол (0,26 г, 0,96 ммоль), метил-(6-гидрокси-2,3-дигидро-1-бензофуран-3-ил)ацетат (0,20 г, 0,96 ммоль) и три(н-бутил)фосфин (0,25 г, 1,2 ммоль) в атмосфере азота. Реакционную смесь охлаждали до 0°С и 1,1'-(азодикарбонил)-дипиперидин (0,30 г, 1,2 ммоль) добавляли по частям и перемешивали при комнатной температуре в течение 2 часов. После завершения реакции реакционную смесь выливали в воду (10 мл) и экстрагировали этилацетатом (15 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали путем колоночной хроматографии на силикагеле с применением этилацетата и петролейного эфира в качестве элюентов. Продукт получали в виде бесцветной маслянистой жидкости (0,21 г, выход 53,1%).
Соединение 73: {6-[(4-{[(2Z)-2-(Метоксиимино)-2-фенилэтил]окси}бензил)окси]-2,3-дигидро-1-бензофуран-3-ил}уксусная кислота
В 25 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 5 мл тетрагидрофурана. В перемешиваемый растворитель добавляли метил-{6-[(4-{[(2E,2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]-2,3-дигидро-1-бензофуран-3-ил}ацетат (0,2 г, 0,43 ммоль) в метаноле (5 мл). Реакционную смесь охлаждали до 0°C и гидроксид натрия (0,035 г, 0,86 ммоль) в воде (5 мл) добавляли по каплям и перемешивали в течение 2 часов. Реакционную смесь концентрировали для отгонки растворителя; этилацетат (10 мл) добавляли и перемешивали в течение 10 минут. Органический слой удаляли и неочищенное вещество подкисляли насыщенным раствором лимонной кислоты до pH 6. Полученное твердое вещество отфильтровывали и сушили. Продукт получали в виде белого твердого вещества (0,01 г, выход 5,0%).
Схема 24
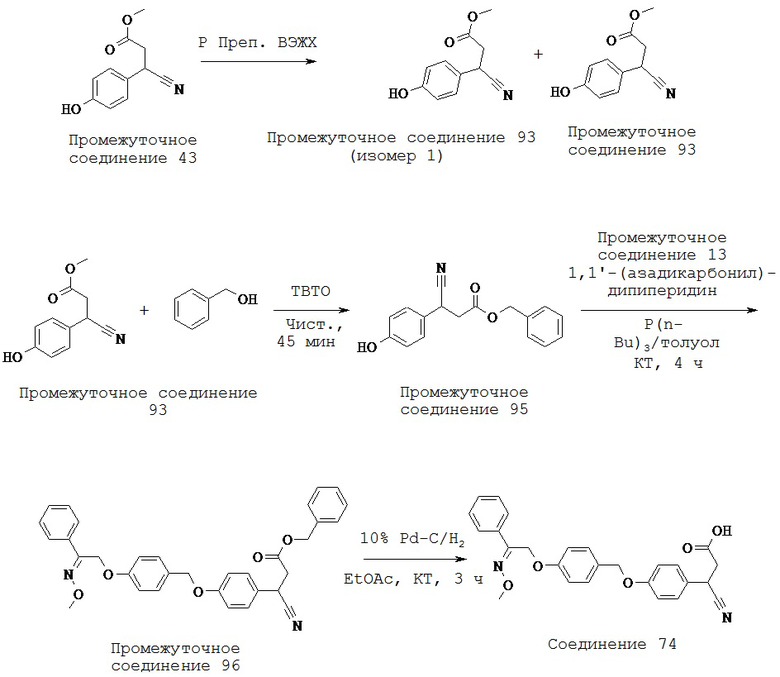
Пример 74
(+)-3-Циано-3-{4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропановая кислота (74)
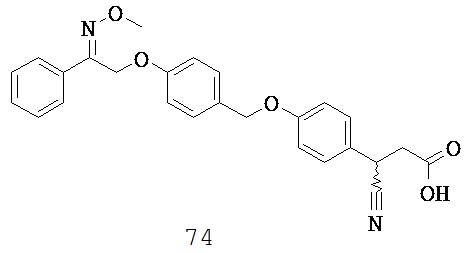
Соединение 74 синтезировали из (4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}-фенил)метанола (0,04 г, 2,2 ммоль) и бензил-3-циано-3-(4-гидроксифенил)пропаноата (0,66 г, 2,2 ммоль) согласно способу, описанному на схеме 24 (0,2 г, выход 39,91%); чистота: 98,51%.
Промежуточное соединение 93 (изомер 1); (+)-метил-3-циано-3-(4-гидроксифенил)пропаноат
Метил-3-циано-3-(4-гидроксифенил)пропаноат разделяли путем нормально-фазовой препаративной ВЭЖХ [CHIRALPAC IC (250*4,6) мм, мобильная фаза: гексан:ИПС:ТФА (80:20:0,1, об./об./об.), расход 1,0 мл/мин; темп. колонки: 25°C]. Изомер, метил-3-циано-3-(4-гидроксифенил)пропаноат (+ve) (время удерживания 10,22 мин) получали таким образом с ЭИ=99,34%.
Промежуточное соединение 94 (изомер 2): (-)-метил-3-циано-3-(4-гидроксифенил)пропаноат
Метил-3-циано-3-(4-гидроксифенил)пропаноат разделяли путем нормально-фазовой препаративной ВЭЖХ [CHIRALPAC IC (250*4,6) мм, мобильная фаза: гексан:ИПС:ТФА (80:20:0,1, об./об./об.), расход 1,0 мл/мин; темп. колонки: 25°C]. Изомер, метил-3-циано-3-(4-гидроксифенил)пропаноат (-ve) (время удерживания 12,75 мин) получали таким образом с ЭИ=98,03%.
Промежуточное соединение 95: бензил-3-циано-3-(4-гидроксифенил)пропаноат
В пробирку для микроволнового реактора помещали метил-3-циано-3-(4-гидроксифенил)пропаноат (+ve) (0,5 г, 2,4 ммоль) и бензиловый спирт (0,5 мл, 4,8 ммоль), добавляли оксид трибутилолова (0,03 г, 5% масс./масс.). Устанавливали температуру, равную 90°C, при мощности 250 Вт в течение 30 минут. После завершения реакции (отслеживали путем ТСХ) PC экстрагировали этилацетатом. Органический слой промывали водой и насыщенным солевым раствором, сушили над безводным Na2SO4 и выпаривали насухо с получением титульного соединения (0,040 г, выход 6,0%).
Промежуточное соединение 96: (+)-бензил-3-циано-3-{4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропаноат
В 50 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали толуол (20 мл). В перемешиваемый растворитель добавляли 0-метилоксим 2-(4-гидроксиметил)-1-фенилэтанона (0,039 г, 0,14 ммоль) и бензил-3-циано-3-(4-гидроксифенил)пропаноат (0,04 г, 0,14 ммоль) при 0°C в течение 5 минут. Затем добавляли трибутилфосфин (0,043 г, 0,2 ммоль), перемешивали при 0°C в течение 15 минут, добавляли 1,1'-аза(дикарбонил)-дипиперидин (0,054 г, 0,2 ммоль), полученную массу перемешивали при КТ в течение 16 часов. Растворитель удаляли, остаток экстрагировали этилацетатом. Органический слой промывали водой и солевым раствором и сушили безводным сульфатом натрия, выпаривали, очищали на колонке с оксидом кремния (100-200 меш) с применением петролейного эфира и этилацетата. Продукт получали в виде бесцветной маслянистой жидкости (0,06 г, 79,00%).
Соединение 74: (+)-3-Циано-3-{4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропановая кислота (+ve)
В 500 мл колбу для гидрогенизатора Парра, содержащую этилацетат (15 мл), добавляли бензил-3-циано-3-{4-[(4-{[(2E,2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}-пропаноат (0,06 г, 0,1 ммоль) и 10% Pd/C (0,006 г) в атмосфере N2. PC гидрировали в течение 16 часов. PC разбавляли этилацетатом (10 мл), фильтровали через подложку целита для удаления Pd/C и выпаривали насухо при пониженном давлении. Неочищенное вещество очищали путем колоночной флэш-хроматографии на силикагеле (100/200 меш) с получением продукта (0,0097 г, выход 18,00%).
Пример 75
(-)-3-Циано-3-{4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропановая кислота
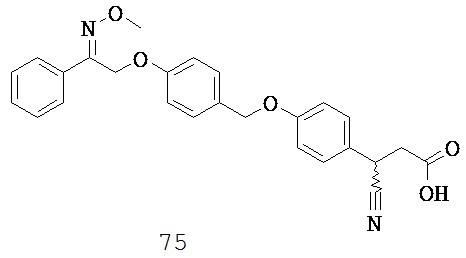
Соединение 75 синтезировали из бензил-3-циано-3-(4-гидроксифенил)пропаноата (0,27 г, 0,95 ммоль), полученного из промежуточного соединения 96, который затем вводили в реакцию сочетания с (4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}фенил)метанолом (0,26 г, 0,95 ммоль) согласно способу, описанному на схеме 24 (0,08 г, выход 88,89%); чистота: 89,58%.
Схема 25
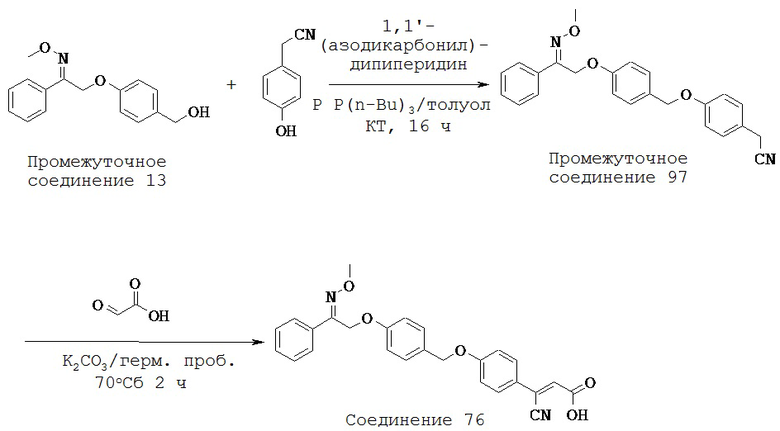
Пример 76
(2Z)-3-Циано-3-{4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}проп-2-еновая кислота
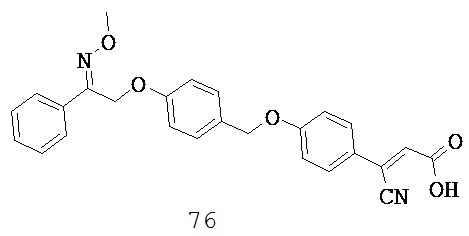
Соединение 76 синтезировали из (4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}-фенил)метанола (0,18 г, 0,47 ммоль) и оксоуксусной кислоты (0,064 г, 0,70 ммоль) согласно способу, описанному на схеме 25 (0,012 г, выход 5,8%); чистота: 96,28%.
Промежуточное соединение 97: {4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}ацетонитрил
В 50 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали сухой толуол. В перемешиваемый растворитель добавляли O-метилоксим 2-(4-гидроксиметилфенокси)-1-фенилэтанона (0,20 г, 0,74 ммоль) и (4-гидроксифенил)-ацетонитрил (0,098 г, 0,74 ммоль) при 0°C в течение 5 минут. Затем добавляли трибутилфосфин (0,024 г, 1,2 ммоль), перемешивали при 0°C в течение 15 минут, добавляли 1,1'-аза(дикарбонил)дипиперидин (0,303 г, 1,2 ммоль), полученную массу перемешивали при КТ в течение 16 часов. Растворитель удаляли, остаток экстрагировали этилацетатом. Органический слой промывали водой и солевым раствором и сушили безводным сульфатом натрия, выпаривали, очищали на колонке с оксидом кремния (100-200 меш) с применением петролейного эфира и этилацетата. Продукт получали в виде бесцветной маслянистой жидкости (0,18 г, 60,00%).
Соединение 76: (2Z)-3-Циано-3-{4-[(4-{[(2Е,2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}проп-2-еновая кислота
В 50 мл герметичную пробирку, оборудованную магнитной мешалкой, помещали метанол (5 мл). В перемешиваемый растворитель добавляли {4-[(4-{[(2E,2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}ацетонитрил (0,18 г, 0,47 ммоль), карбонат калия (0,26 г, 1,9 ммоль) и оксоуксусную кислоту (0,064 г, 0,70 ммоль) в атмосфере азота. Реакционную смесь нагревали при 70°C в течение 2 часов. Затем удаляли растворитель при пониженном давлении, полученный остаток растворяли в воде (10 мл), промывали диэтиловым эфиром (20 мл). Водный слой подкисляли 1 н. HCl, экстрагировали этилацетатом, промывали водой и солевым раствором. Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении с получением продукта (0,012 г, выход 5,80%).
Пример 77
3-Циано-3-{2-фтор-4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропановая кислота
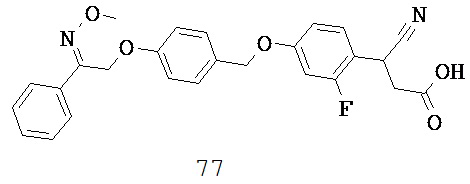
Соединение 77 синтезировали из (4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}-фенил)метанола (0,607 г, 2,24 ммоль) и метил-3-циано-3-(2-фтор-4-гидроксифенил)-пропаноата (0,5 г, 2,24 ммоль) согласно способу, описанному на схеме 18 (0,2 г, выход 95,4%); чистота: 95,68%.
Пример 78
3-Циано-3-{4-[(4-{[(2E,2Z)-2-(гидроксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропановая кислота
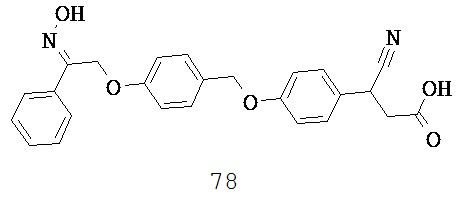
Соединение 78 синтезировали из 2-[4-(гидроксиметил)фенокси]-1-фенилэтанона (0,5 г, 2,06 ммоль) и метил-3-циано-3-(4-гидроксифенил)пропаноата (0,452 г, 2,06 ммоль) согласно способу, описанному на схеме 8 (0,023 г, 15,00%); чистота: 68,90%.
Пример 79
2-{4-[(4-{[(2Z)-2-(4-Цианофенил)-2-(метоксиимино)этил]окси}бензил)окси]фенил}-циклопропанкарбоксилат натрия (энантиомер 1)
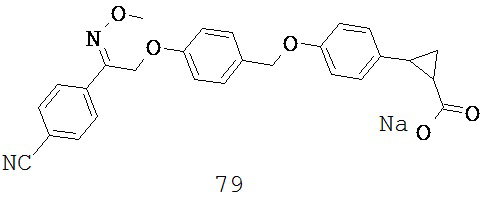
Соединение 79 синтезировали из 2-{4-[(4-{[(2Z)-2-(4-цианофенил)-2-(метоксиимино)-этил]окси}бензил)окси]фенил}циклопропанкарбоновой кислоты (0,1 г, 2,4 ммоль) и бикарбоната натрия (0,25 мл, 2,4 ммоль) согласно способу, описанному на схеме 12 (0,095 г, выход 90,56%); чистота: 89,00%.
Пример 80
2-{4-[(4-{[(2Z)-2-(4-Цианофенил)-2-(метоксиимино)этил]окси}бензил)окси]фенил}-циклопропанкарбоксилат натрия (энантиомер 2)
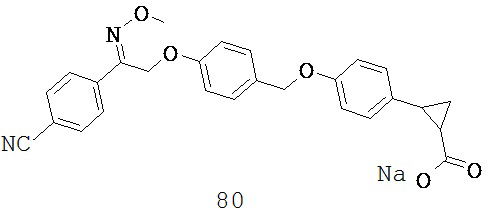
Соединение 80 синтезировали из 2-{4-[(4-{[(2Z)-2-(4-цианофенил)-2-(метоксиимино)-этил]окси}бензил)окси]фенил}циклопропанкарбоновой кислоты (0,026 г, 0,0567 ммоль) и бикарбоната натрия (0,06 мл, 1М раствор) согласно способу, описанному на схеме 12 (0,0051 г, выход 18,89%); чистота: 96,10%.
Пример 81
(+)-3-циано-3-{4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропаноат натрия
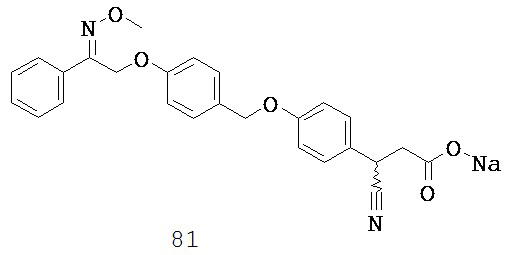
Соединение 81 синтезировали из 3-циано-3-{4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропановой кислоты (0,78 г, 1,75 ммоль) и раствора NaHCO3 (1,75 мл, 1М раствор) согласно способу, описанному на схеме 12 (0,58 г, выход 70,00%); чистота: 89,10%.
Пример 82
(-)-3-Циано-3-{4-[(4-{[(2E,2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропаноат натрия
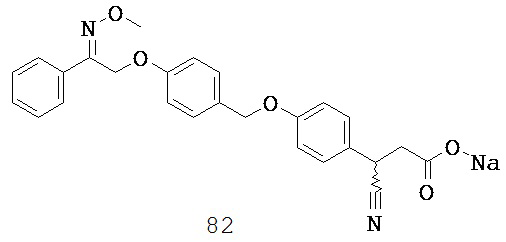
Соединение 82 синтезировали из энантиомерно чистой 3-циано-3-{4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропановой кислоты (0,45 г, 1,0135 ммоль) и бикарбоната натрия (1,0 мл, 1,0135 ммоль) согласно способу, описанному на схеме 12 (0,312 г, выход 95,74%), растворитель удаляли при пониженном давлении при 25°C; чистота: 98,88%. Оптическое вращение: -1,74 (25°C, вода).
Схема 26
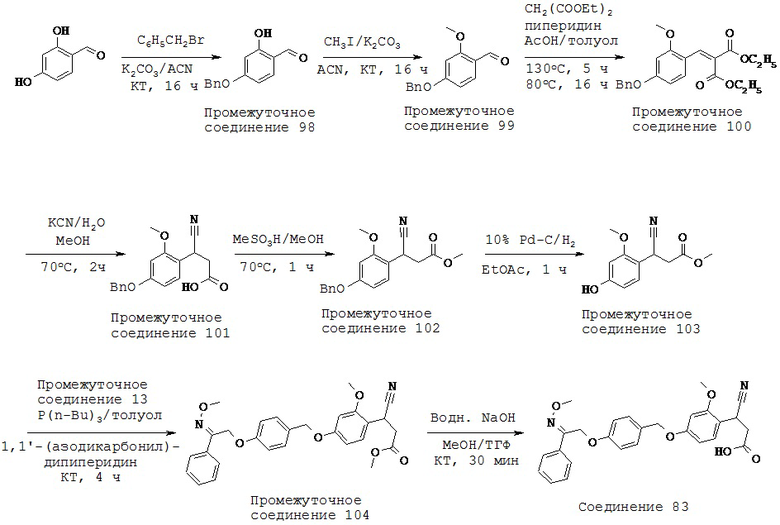
Пример 83
3-Циано-3-{2-метокси-4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропановая кислота
Соединение 83 синтезировали из (4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}-фенил)метанола (0,231 г, 0,85 ммоль) и метил-3-циано-3-(4-гидрокси-2-метоксифенил)-пропаноата (0,2 г, 0,85 ммоль) согласно способу, описанному на схеме 26 (0,001 г, выход 6,86%); чистота: 95,70%.
Промежуточное соединение 98: 4-(бензилокси)-2-гидроксибензальдегид
В 250 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 30 мл ацетонитрила. В перемешиваемый растворитель добавляли 2,4-дигидроксибензальдегид (8,0 г, 57,0 ммоль), карбонат калия (15,77 г, 114,0 ммоль). Реакционную смесь охлаждали до 0°C, добавляли бензилбромид (9,905 г, 57,0 ммоль) в ацетонитриле (100 мл) по каплям и перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь концентрировали для отгонки растворителя. Остаток экстрагировали этилацетатом (80 мл). Органический слой промывали водой (50 мл) и насыщенным солевым раствором (50 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Продукт получали в виде белого твердого вещества (4,0 г, выход 30,77%).
Промежуточное соединение 99: 4-(бензилокси)-2-метоксибензальдегид
В 100 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 10 мл ацетонитрила. В перемешиваемый растворитель добавляли 4-(бензилокси)-2-гидроксибензальдегид (1,0 г, 4,38 ммоль), карбонат калия (1,21 г, 8,76 ммоль). Реакционную смесь охлаждали до 0°C, добавляли метилйодид (1,135 г, 8,76 ммоль) в ацетонитриле (5 мл) по каплям и перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь концентрировали для отгонки растворителя. Остаток экстрагировали этилацетатом (50 мл). Органический слой промывали водой (40 мл) и насыщенным солевым раствором (40 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Продукт получали в виде белого твердого вещества (1,06 г, выход 100,0%).
Промежуточное соединение 100: диэтил-[4-(бензилокси)-2-метоксибензилиден]пропандиоат
В 100 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали пиперидин (0,108 г, 0,13 мл, 1,35 ммоль) и уксусную кислоту (0,081 г, 0,08 мл, 1,35 ммоль). В полученную смесь добавляли толуол (20 мл), затем 4-(бензилокси)-2-метоксибензальдегид (1,1 г, 4,5 ммоль) и диэтилмалонат (0,87 г, 5,5 ммоль). PC нагревали при 125°C с применением насадки Дина-Старка в течение 5 часов. Через 5 часов PC концентрировали; воду (50 мл) добавляли и экстрагировали этилацетатом (50 мл × 2). Органический слой промывали насыщенным солевым раствором (50 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Продукт получали в виде желтого твердого вещества (0,775 г, выход 44,2%).
Промежуточное соединение 101: 3-[4-(бензилокси)-2-метоксифенил]-3-цианопропановая кислота
В 25 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали метанол (20 мл) и воду (5 мл). В перемешиваемый растворитель добавляли диэтил-[4-(бензилокси)-2-метоксибензилиден]пропандиоат (0,775 г, 2,02 ммоль), затем цианид калия (0,262 г, 4,04 ммоль). PC нагревали при 70°C в течение 2 часов. PC концентрировали, разбавляли насыщенным раствором NaHCO3 (50 мл) и промывали этилацетатом (50 мл × 2). Водный слой подкисляли до pH 3 при помощи 1 н. хлороводородной кислоты и экстрагировали этилацетатом (50 мл × 2). Органический слой промывали водой (50 мл) и насыщенным солевым раствором (50 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Продукт получали в виде коричневой жидкости (0,62 г, выход 100,0%).
Промежуточное соединение 102: метил-3-[4-(бензилокси)-2-метоксифенил]-3-цианопропаноат
В 25 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали метанол (10 мл). В перемешиваемый растворитель добавляли 3-[4-(бензилокси)-2-метоксифенил]-3-цианопропановую кислоту (0,62 г, 2,56 ммоль) и метансульфокислоту (1 мл), нагревали при 65°C в течение 2 часов. PC концентрировали, экстрагировали этилацетатом (50 мл × 2). Органический слой промывали водой (50 мл) и насыщенным солевым раствором (50 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Продукт получали в виде коричневой жидкости (0,65 г, выход 100,0%).
Промежуточное соединение 103: метил-3-циано-3-(4-гидрокси-2-метоксифенил)пропаноат
В 500 мл колбу для гидрогенизатора Парра, содержащую этилацетат (15 мл), добавляли метил-3-циано-3-(4-гидрокси-2-метоксифенил)пропаноат (0,65 г, 2,0 ммоль) и 10% Pd/C (0,1 г) в атмосфере N2. PC гидрировали в течение 1 часа. PC разбавляли этилацетатом (10 мл), фильтровали через подложку целита для удаления Pd/C и выпаривали насухо при пониженном давлении. Неочищенное вещество очищали путем колоночной флэш-хроматографии на силикагеле (100/200 меш) с получением продукта (0,2 г, выход 42,50%).
Промежуточное соединение 104: метил-3-циано-3-{2-метокси-4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропаноат
В 50 мл кругло донную колбу, оборудованную магнитной мешалкой, помещали 15 мл толуола. В перемешиваемый растворитель добавляли (4-{[(2E,2Z)-2-(метоксиимино)-2-фенилэтил]окси}фенил)метанол (0,23 г, 0,85 ммоль), метил-3-циано-3-(4-гидрокси-2-метоксифенил)пропаноат (0,2 г, 0,85 ммоль). Реакционную смесь охлаждали до 0°C; трибутилфосфин (0,22 г, 1,1 ммоль) добавляли и перемешивали в течение 10 минут. В перемешиваемый раствор 1,1'-(азодикарбонил)дипиперидин (0,28 г, 1,1 ммоль) в толуоле (2 мл) добавляли по каплям при указанной температуре и перемешивали при комнатной температуре в течение 4 часов. Реакционную смесь разбавляли водой (10 мл) и экстрагировали этилацетатом (15 мл × 2). Органический слой промывали водой (10 мл), затем солевым раствором (10 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали путем колоночной хроматографии на силикагеле с применением этилацетата и петролейного эфира в качестве элюентов. Продукт получали в виде бесцветной маслянистой жидкости (0,15 г, выход 36,60%).
Соединение 83: 3-Циано-3-{2-метокси-4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропановая кислота
В 50 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 5 мл тетрагидрофурана. В перемешиваемый растворитель добавляли метил-3-циано-3-{2-метокси-4-[(4-{[(2E,2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропаноат (0,15 г, 0,31 ммоль) в метаноле (5 мл). Реакционную смесь охлаждали до 0°C и гидроксид натрия (0,037 г, 0,29 ммоль) в воде (5 мл) добавляли по каплям и перемешивали в течение 30 часов. Реакционную смесь концентрировали для отгонки растворителя; этилацетат (10 мл) добавляли и перемешивали в течение 10 минут. Органический слой удаляли и неочищенное вещество подкисляли насыщенным раствором лимонной кислоты до pH 6. Полученное твердое вещество отфильтровывали и сушили. Продукт получали в виде белого твердого вещества (0,01 г, выход 6,86%).
Схема 27
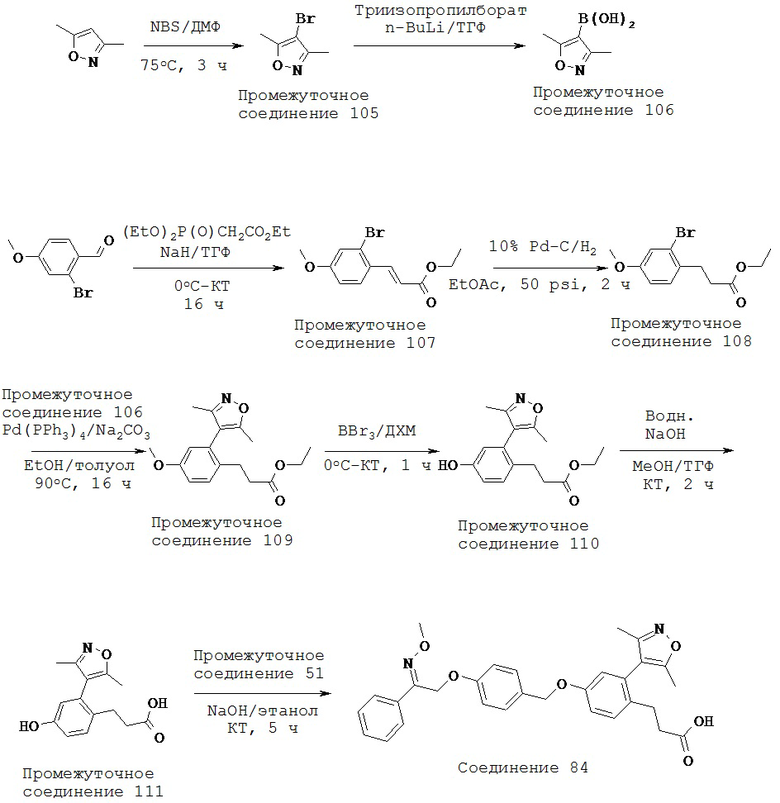
Пример 84
3-{2-(3,5-Диметил-1,2-оксазол-4-ил)-4-[(4-{[(2Е,2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропановая кислота
Соединение 84 синтезировали из 3-[2-(3,5-диметил-1,2-оксазол-4-ил)-4-гидроксифенил]пропановой кислоты (0,04 г, 0,15 ммоль) и (1Е,1Z)-2-[4-(бромметил)-фенокси]-N-метокси-1-фенилэтанимина (0,051 г, 0,15 ммоль) согласно способу, описанному на схеме 27 (0,04 г, выход 6,20%); чистота: 80,10%.
Промежуточное соединение 105: 4-бром-3,5-диметил-1,2-оксазол
В 250 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали ДМФ (53 мл). В перемешиваемый растворитель добавляли 3,5-диметил-1,2-оксазол (5,3 г, 54,57 ммоль) и N-йодсукцинимид (11,659 г, 65,49 ммоль), после завершения добавления реакционную смесь нагревали при 75°C в течение 3 часов. После завершения реакции реакционную смесь разбавляли этилацетатом (100 мл), органический слой промывали насыщенным раствором NaHCO3 (100 мл), раствором тиосульфата натрия (100 мл), водой (200 мл) и, наконец, солевым раствором (100 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении с получением продукта (6,1 г, выход 63,4%).
Промежуточное соединение 106: (3,5-диметил-1,2-оксазол-4-ил)бороновая кислота
В 500 мл трехгорлую круглодонную колбу, оборудованную магнитной мешалкой, помещали 4-бром-3,5-диметил-1,2-оксазол (4,0 г, 22,7 ммоль) в ТГФ (40 мл), охлаждали до -78°C. В перемешиваемый раствор добавляли н-бутиллитий (28,4 мл, 1,6М раствор, 45,0 ммоль) по каплям и перемешивали при -65°C в течение примерно 30 минут. PC охлаждали до -78°C, добавляли триизопропилборат (12,81 г, 68,0 ммоль), после чего температуру повышали до комнатной и перемешивали в течение примерно 16 часов. Затем удаляли растворитель при пониженном давлении и реакцию гасили насыщенным раствором NH4Cl и смесь экстрагировали этилацетатом. Органический слой промывали водой, сушили над безводным Na2SO4 и удаляли растворитель при пониженном давлении. Полученное неочищенное вещество очищали путем колоночной хроматографии на силикагеле с получением белого твердого продукта (0,4 г, выход 12,5%).
Промежуточное соединение 107: этил-(2E,2Z)-3-(2-бром-4-метоксифенил)проп-2-еноат
В 250 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 12 мл этилацетата. В перемешиваемый растворитель добавляли этилфосфоноацетат (2,5 г, 11,0 ммоль) в атмосфере азота. Реакционную смесь охлаждали до 0°C и гидрид натрия (0,401 г, 16,0 ммоль) добавляли по частям, перемешивали в течение 1 часа при указанной температуре. Затем 2-бром-4-метоксибензальдегид (1,2 г, 5,5 ммоль) в тетрагидрофуране (10 мл) добавляли по каплям. Реакционную смесь перемешивали при КТ в течение 16 часов. Реакцию гасили путем медленного добавления воды при 0°C, перемешивали в течение 10 минут. Слои разделяли; этилацетат (50 мл) добавляли в водный слой и экстрагировали слои. Органический слой промывали солевым раствором (50 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Продукт получали в виде бледно-желтого полутвердого вещества (0,29 г, выход 18,50%).
Промежуточное соединение 108: этил-3-(2-бром-4-метоксифенил)пропаноат
В 500 мл колбу для гидрогенизатора Парра, содержащую этилацетат (10 мл), добавляли этил-(2E)-3-(2-бром-4-метоксифенил)проп-2-еноат (0,2 г, 0,7 ммоль) и 10% Pd/C (0,05 г) в атмосфере азота. PC гидрировали при 50 psi в течение 1 часа. Затем PC разбавляли этилацетатом (10 мл), фильтровали через подложку целита для удаления Pd/C и выпаривали насухо при пониженном давлении. Неочищенное вещество очищали путем колоночной флэш-хроматографии на силикагеле (100/200 меш) с получением продукта (0,18 г, выход 90,00%).
Промежуточное соединение 109: этил-3-[2-(3,5-диметил-1,2-оксазол-4-ил)-4-метоксифенил]пропаноат
В 25 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали толуол (1 мл). В перемешиваемый растворитель добавляли (3,5-диметил-1,2-оксазол-4-ил)бороновую кислоту (0,059 г, 0,42 ммоль), этил-(2-бром-4-метоксифенил)пропаноат (0,08 г, 0,28 ммоль), бикарбонат натрия (0,5 мл, 2М раствор) в этаноле (1 мл) в атмосфере аргона. После завершения добавления реакционную смесь продували аргоном в течение 15 минут. В реакционную смесь добавляли тетракис(трифенилфосфин)палладий (0) (0,016 г, 0,01 ммоль) и продували аргоном в течение 10 минут. После завершения добавления реакционную смесь нагревали при 95°C в течение 5 часов в атмосфере аргона. После завершения реакцию гасили водой (10 мл) и смесь экстрагировали этилацетатом (10 мл). Органический слой промывали водой (5 мл) и солевым раствором (5 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенный продукт (0,08 г, выход 94,10%) применяли на следующей стадии без очистки.
Промежуточное соединение 110: этил-3-[2-(3,5-диметил-1,2-оксазол-4-ил)-4-гидроксифенил]пропаноат
В 25 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 10 мл дихлорметана. В перемешиваемый растворитель добавляли этил-3-[2-(3,5-диметил-1,2-оксазол-4-ил)-4-метоксифенил]пропаноат (0,120 г, 0,4 ммоль). Реакционную смесь охлаждали до 0°C и трибромид бора (0,099 г, 0,4 ммоль) добавляли по каплям. После 30-минутного перемешивания реакцию гасили путем медленного добавления этанола (1 мл) при 0°C. Реакционную смесь концентрировали для отгонки растворителя; этилацетат (10 мл) добавляли. Органический слой промывали насыщенным раствором NaHCO3 (10 мл), затем солевым раствором (10 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Продукт получали в виде бесцветной маслянистой жидкости (0,1 г, выход 90,90%).
Промежуточное соединение 111: 3-[2-(3,5-диметил-1,2-оксазол-4-ил)-4-гидроксифенил]пропановая кислота
В 10 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 3 мл тетрагидрофурана. В перемешиваемый растворитель добавляли этил-3-[2-(3,5-диметил-1,2-оксазол-4-ил)-4-гидроксифенил]пропаноат (0,10 г, 0,35 ммоль), этанол (3 мл) и гидроксид натрия (0,015 г, 0,35 ммоль) в воде (0,5 мл). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. После завершения реакции растворитель удаляли, натриевую соль промывали диэтиловым эфиром (5 мл); водный слой подкисляли 1 н. HCl до pH 2,0 и экстрагировали этилацетатом (5 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении с получением продукта (0,04 г, выход 44,4%).
Соединение 84: 3-{2-(3,5-диметил-1,2-оксазол-4-ил)-4-[(4-{[(2E,2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропановая кислота
В 25 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 5 мл ацетонитрила. В перемешиваемый растворитель добавляли 3-[2-(3,5-диметил-1,2-оксазол-4-ил)-4-гидроксифенил]пропановую кислоту (0,04 г, 0,15 ммоль), гидроксид натрия (0,019 г, 0,4 ммоль). Реакционную смесь охлаждали до 0°C, (1E,1Z)-2-[4-(бромметил)фенокси]-N-метокси-1-фенилэтанимин (0,051 г, 0,15 ммоль) в этаноле (2 мл) добавляли по каплям и перемешивали при КТ в течение 5 часов. После завершения реакции растворитель удаляли, натриевую соль промывали диэтиловым эфиром (5 мл); водный слой подкисляли 1 н. HCl до pH 2,0 и экстрагировали этилацетатом (5 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении с получением продукта (0,006 г, выход 6,2%).
Схема 28
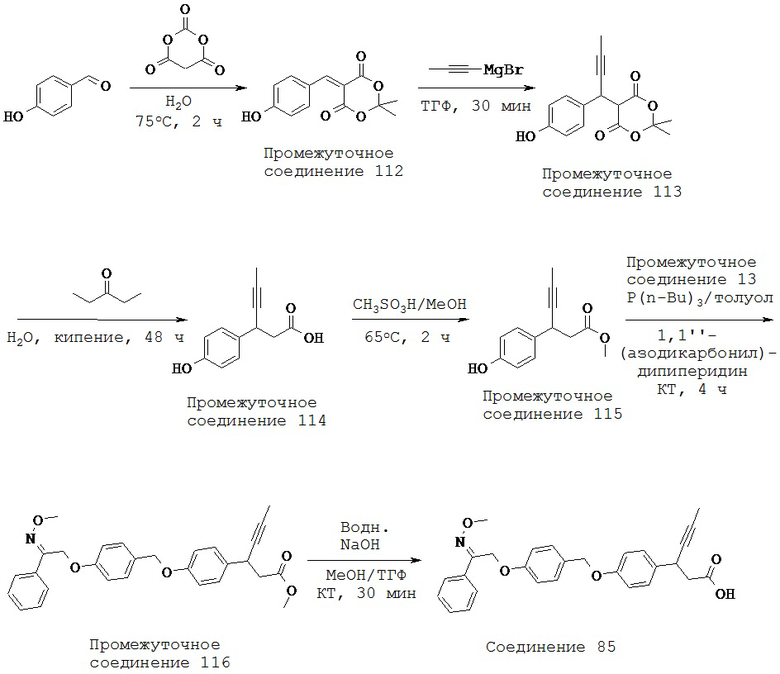
Пример 85
3-{4-[(4-{[(2Z)-2-(Метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}гекс-4-иновая кислота (85)
Соединение 85 синтезировали из (4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}-фенил)метанола (0,2 г, 0,74 ммоль) и метил-3-(4-гидроксифенил)гекс-4-иноата (0,16 г, 0,74 ммоль) согласно способу, описанному на схеме 28 (0,03 г, выход 20,59%); чистота: 98,37%.
Промежуточное соединение 112: 5-(4-гидроксибензилиден)-2,2-диметил-1,3-диоксан-4,6-дион
В 500 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали воду (160 мл). В перемешиваемый растворитель добавляли 4-гидроксибензальдегид (25,0 г, 122,12 ммоль) и 2,2-диметил-1,3-диоксан-4,6-дион (25,0 г). Смесь нагревали при 75°C в течение 2 часов. PC охлаждали с получением твердого вещества, фильтровали и промывали водой, затем сушили с получением продукта (34,0 г, выход 83,74%).
Промежуточное соединение 113: 5-[1-(4-гидроксифенил)бут-2-ин-1-ил]-2,2-диметил-1,3-диоксан-4,6-дион
В 250 мл 3-горлую круглодонную колбу, оборудованную магнитной мешалкой, помещали бромид (проп-1-ин-1-ил)магния (42,5 мл, 211 ммоль) в ТГФ (30 мл). В PC добавляли медленно 5-(4-гидроксибензилиден)-2,2-диметил-1,3-диоксан-4,6-дион (5,0 г, 100,8 ммоль) в ТГФ (20 мл) в атмосфере азота. PC перемешивали при комнатной температуре в течение примерно 30 минут. Реакционную смесь разбавляли раствором NH4Cl и гексаном. Водный слой подкисляли раствором KHSO4 до pH 2 и экстрагировали этилацетатом, промывали водой, затем сушили с получением продукта (5,8 г, выход 100,0%).
Промежуточное соединение 114: 3-(4-гидроксифенил)гекс-4-иновая кислота
В 250 мл 3-горлую круглодонную колбу, оборудованную магнитной мешалкой, помещали 5-[1-(4-гидроксифенил)бут-2-ин-1-ил]-2,2-диметил-1,3-диоксан-4,6-дион (5,8 г, 20,13 ммоль) в диэтилкетоне, добавляли воду (13 мл), реакционную смесь кипятили с обратным холодильником в течение 48 часов. PC подкисляли 1 н. HCl и экстрагировали этилацетатом. Органический слой промывали водой, сушили над безводным сульфатом натрия. Затем удаляли растворитель при пониженном давлении с получением продукта (4,11 г, выход 100,0%).
Промежуточное соединение 115: метил-3-(4-гидроксифенил)гекс-4-иноат
В 250 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали метанол (10 мл). В перемешиваемый растворитель добавляли 3-(4-гидроксифенил)гекс-4-иновую кислоту (0,62 г, 20,14 ммоль) и метансульфокислоту (3 мл), смесь нагревали при 65°C в течение 2 часов. PC концентрировали, экстрагировали этилацетатом (50 мл × 2). Органический слой промывали водой (50 мл) и насыщенным солевым раствором (50 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении с получением продукта (0,4 г, выход 90,9%).
Промежуточное соединение 116: метил-3-{4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил] окси}бензил)окси] фенил}гекс-4-иноат
В 50 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 15 мл толуола. В перемешиваемый растворитель добавляли (4-{[(2E,2Z)-2-(метоксиимино)-2-фенилэтил]окси}фенил)метанол (0,2 г, 0,74 ммоль), метил-3-(4-гидроксифенил)гекс-4-иноат (0,16 г, 0,74 ммоль). Реакционную смесь охлаждали до 0°C; трибутилфосфин (0,195 г, 0,96 ммоль) добавляли и перемешивали в течение 10 минут. В перемешиваемый раствор 1,1’-(азодикарбонил)дипиперидин (0,242 г, 0,96 ммоль) в толуоле (2 мл) добавляли по каплям при указанной температуре и перемешивали при комнатной температуре в течение 4 часов. Реакционную смесь разбавляли водой (10 мл) и экстрагировали этилацетатом (15 мл × 2). Органический слой промывали водой (10 мл), затем солевым раствором (10 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали путем колоночной хроматографии на силикагеле с применением этилацетата и петролейного эфира в качестве элюентов. Продукт получали в виде бесцветной маслянистой жидкости (0,15 г, выход 42,86%).
Соединение 85: 3-{4-[(4-{[(2Z)-2-(Метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}гекс-4-иновая кислота
В 50 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 5 мл тетрагидрофурана. В перемешиваемый растворитель добавляли метил-3-{4-[(4-{[(2E,2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}гекс-4-иноат (0,15 г, 0,32 ммоль) в метаноле (5 мл). Реакционную смесь охлаждали до 0°C и гидроксид натрия (0,025 г, 0,64 ммоль) в воде (5 мл) добавляли по каплям и перемешивали в течение 30 часов. Реакционную смесь концентрировали для отгонки растворителя; этилацетат (10 мл) добавляли и перемешивали в течение 10 минут. Органический слой концентрировали и неочищенное вещество подкисляли насыщенным раствором лимонной кислоты до pH 6. Полученное твердое вещество отфильтровывали и сушили. Продукт получали в виде белого твердого вещества (0,03 г, выход 20,59%).
Схема 29
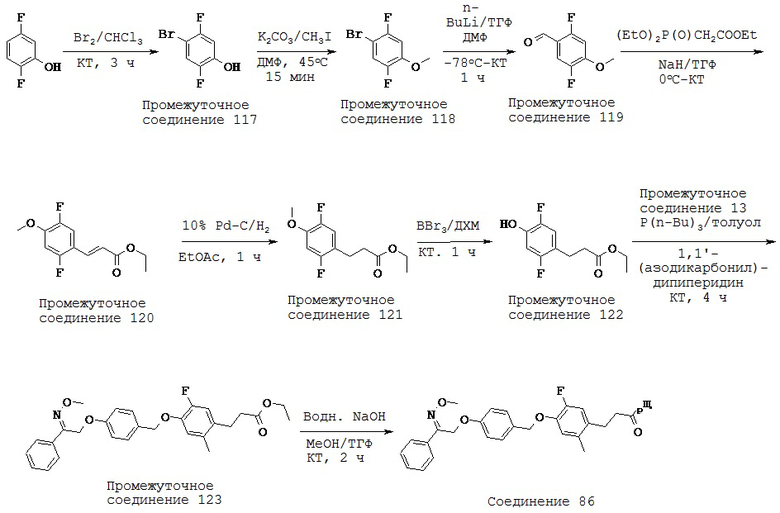
Пример 86
3-{2,5-Дифтор-4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропановая кислота (86)
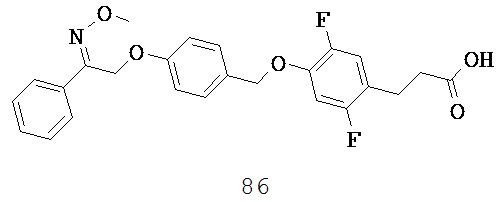
Соединение 86 синтезировали из (4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}-фенил)метанола (0,053 г, 0,109 ммоль) и этил-3-(2,5-дифтор-4-гидроксифенил)пропаноата (0,05 г, 1,25 ммоль) согласно способу, описанному на схеме 29 (0,008 г, выход 16,00%); чистота: 94,64%.
Промежуточное соединение 117: 4-бром-2,5-дифторфенол
В 250 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 100 мл CHCl3. В перемешиваемый растворитель добавляли 2,5-дифторфенол (5,0 г, 38,4 ммоль), бром (6,14 г, 38,4 ммоль) при 0°C и перемешивали при комнатной температуре в течение 3 часов. Реакцию гасили раствором тиосульфата натрия (20 мл) и смесь экстрагировали этилацетатом (15 мл × 2). Органический слой промывали водой (50 мл), затем солевым раствором (20 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении с получением продукта (6,5 г, выход 81,05%).
Промежуточное соединение 118: 1-бром-2,5-дифтор-4-метоксибензол
В 100 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 20 мл ДМФ. В перемешиваемый растворитель добавляли 4-бром-2,5-дифторфенол (2,0 г, 9,6 ммоль), K2CO3 (3,98 г, 28,8 ммоль) и метилйодид (1,63 г, 11,53 ммоль), перемешивали при 45°C в течение 15 минут в атмосфере азота. Реакцию гасили водой, смесь экстрагировали этилацетатом (100 мл). Органический слой промывали водой (50 мл) и насыщенным солевым раствором (50 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Продукт получали в виде белого твердого вещества (1,2 г, выход 56,07%).
Промежуточное соединение 119: 2,5-дифтор-4-метоксибензальдегид
В 250 мл трехгорлую круглодонную колбу, оборудованную магнитной мешалкой, помещали 1-бром-2,5-дифтор-4-метоксибензол (1,0 г, 4,5 ммоль) в 10 мл ТГФ, охлаждали до -78°C. В перемешиваемый раствор добавляли н-бутиллитий (3,09 мл, 1,6М раствор, 4,95 ммоль) по каплям и перемешивали при указанной температуре, добавляли ДМФ (0,375 г, 5,13 ммоль), после чего температуру повышали до комнатной и перемешивали в течение примерно 16 часов. Затем реакцию гасили ледяной водой и смесь экстрагировали этилацетатом. Органический слой промывали водой, сушили над безводным Na2SO4. Удаление растворителя при пониженном давлении приводило к получению жидкого продукта (0,77 г, выход 100,0%).
Промежуточное соединение 120: этил-(2Z)-3-(2,5-дифтор-4-метоксифенил)проп-2-еноат
В 100 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 10 мл тетрагидрофурана. В перемешиваемый растворитель добавляли гидрид натрия (0,23 г, 9,86 ммоль) по частям при 0°C, затем триэтилфосфоноацетат (1,66 г, 7,4 ммоль). Реакционную смесь перемешивали при 0°C в течение 30 минут. В перемешиваемый раствор 2,5-дифтор-4-метоксибензальдегид (2,3 г, 7,2 ммоль) в тетрагидрофуране (2 мл) добавляли по каплям и перемешивали при комнатной температуре в течение ночи. После завершения реакции реакционную смесь выливали в лед и экстрагировали этилацетатом (25 мл). Органический слой промывали водой (25 мл) и насыщенным солевым раствором (25 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали путем колоночной хроматографии на силикагеле с применением этилацетата и петролейного эфира в качестве элюентов. Продукт получали в виде бесцветной маслянистой жидкости (0,1 г, выход 8,4%).
Промежуточное соединение 121: этил-3-(2,5-дифтор-4-метоксифенил)пропаноат
В 500 мл колбу для встряхивателя Парра помещали этил-(2E)-3-(2,5-дифтор-4-метоксифенил)проп-2-еноат (0,1 г, 0,41 ммоль), этилацетат (5 мл). В реакционную смесь добавляли 10% Pd-C (20%) и гидрировали с применением диафрагмы в течение 1 часа. После завершения реакции реакционную смесь фильтровали через целит, промывали тщательно этилацетатом (20 мл) и концентрировали для отгонки растворителя. Продукт получали в виде коричневого твердого вещества (0,080 г, выход 80,0%).
Промежуточное соединение 122: этил-3-(2,5-дифтор-4-гидроксифенил)пропаноат
В 25 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 3 мл дихлорметана. В перемешиваемый растворитель добавляли этил-3-(2,5-дифтор-4-метоксифенил)пропаноат (0,08 г, 0,327 ммоль). Реакционную смесь охлаждали до 0°C и трибромид бора (0,04 мл, 0,425 ммоль) добавляли по каплям. После 30-минутного перемешивания реакцию гасили путем медленного добавления этанола (1 мл) при 0°C. Реакционную смесь концентрировали для отгонки растворителя; этилацетат (10 мл) добавляли. Органический слой промывали насыщенным раствором NaHCO3 (10 мл), затем солевым раствором (10 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Продукт получали в виде бесцветной маслянистой жидкости (0,06 г, выход 80,0%).
Промежуточное соединение 123: этил-3-{2,5-дифтор-4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропаноат
В 100 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 2 мл толуола. В перемешиваемый растворитель добавляли (4-{[(2Е,2Z)-2-(метоксиимино)-2-фенилэтил]окси}фенил)метанол (0,0529 г, 0,195 ммоль) и этил-3-(2,5-дифтор-4-гидроксифенил)пропаноат (1,0 г, 5,0 ммоль). Реакционную смесь охлаждали до 0°C; трибутилфосфин (0,057 г, 0,282 ммоль) добавляли и перемешивали в течение 10 минут. В реакционную смесь добавляли 1,1-(азодикарбонил)дипиперидин (0,71 г, 0,282 ммоль) в толуоле (2 мл) по каплям. Реакционную смесь перемешивали при комнатной температуре в течение 4 часов. Реакционную смесь разбавляли этилацетатом (10 мл) и промывали водой (25 мл). Органический слой промывали солевым раствором (25 мл). Органический слой сушили над Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали путем колоночной хроматографии на силикагеле с применением этилацетата и петролейного эфира в качестве элюентов с получением продукта (0,048 г, 48,0%).
Соединение 86: 3-{2,5-Дифтор-4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропановая кислота
В 10 мл кругло донную колбу, оборудованную магнитной мешалкой, помещали 3 мл тетрагидрофурана. В перемешиваемый растворитель добавляли этил-3-{2,5-дифтор-4-[(4-{[(2E,2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропаноат (0,053 г, 0,109 ммоль) и метанол (3,0 мл). Реакционную смесь охлаждали до 0°C и гидроксид натрия (0,05 г, 1,25 ммоль) в воде (1 мл) добавляли по каплям. Реакционную смесь перемешивали в течение ночи. После завершения реакции реакционную смесь концентрировали для отгонки растворителя. Полученную соль растворяли в воде (1 мл) и экстрагировали диэтиловым эфиром (5 мл). Водный слой подкисляли 1 н. HCl до pH 3 и экстрагировали этилацетатом (15 мл). Органический слой сушили над Na2SO4 и растворитель удаляли при пониженном давлении с получением продукта (0,008 г, 16,00%).
Схема 30
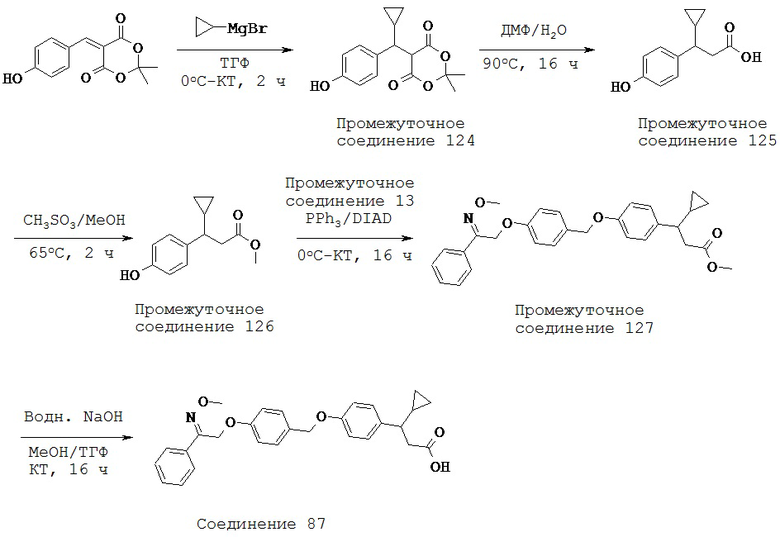
Пример 87
3-Циклопропил-3-{4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропановая кислота (87)
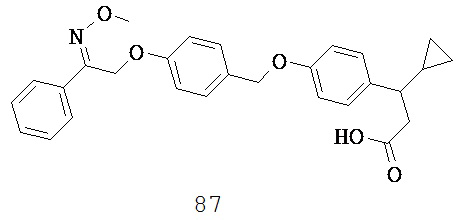
Соединение 87 синтезировали из (4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}-фенил)метанола (0,22 г, 0,8146 ммоль) и метил-3-циклопропил-3-(4-гидроксифенил)-пропаноата (0,2 г, 0,9082 ммоль) согласно способу, описанному на схеме 30 (0,04 г, выход 22,90%); чистота: 98,07%.
Промежуточное соединение 124: 5-[циклопропил(4-гидроксифенил)метилиден]-2,2-диметил-1,3-диоксан-4,6-дион
В 250 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 6 мл сухого ТГФ в атмосфере аргона. В перемешиваемый растворитель добавляли 5-(гидроксибензилиден)-2,2-диметил-1,3-диоксан-4,6-дион (0,6 г, 2,41 ммоль). Полученную смесь охлаждали до 0°C и добавляли бромид циклопропилмагния (25 мл, 0,5М в ТГФ, 7,25 ммоль). Полученную смесь перемешивали при температуре окружающей среды в течение 2 часов. Затем реакцию гасили 20 мл 1 н. HCl, затем этилацетат (50 мл) добавляли, хорошо перемешивали и слои разделяли. Органический слой промывали водой (100 мл × 3), солевым раствором. Затем органический слой сушили над безводным Na2SO4. Растворитель удаляли при пониженном давлении и полученное неочищенное соединение очищали путем колоночной хроматографии на силикагеле (60-120 меш) с применением петролейного эфира (60-80) и этилацетата в качестве элюентов. Продукт получали в виде желтого твердого вещества. Выход: 42,76% (0,3 г). МС (ИЭР, 120 эВ): m/z=291,1 (M+1). 1Н ЯМР (300 МГц, ДМСО-d6): δ 9,16 (s, 1H), 6,95-6,98 (d, 2H), 6,53-6,56 (d, 2H), 4,42-4,43 (d, 1H), 2,54-2,59 (m, 1H), 1,61 (s, 4H), 1,29 (s, 3H), 0,40-0,49 (m, 2H), 0,23-0,28 (m, 1H), 0,03-0,01 (m, 1H).
Промежуточное соединение 125: 3-циклопропил-3-(4-гидроксиметил)пропановая кислота
В 100 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали ДМФ (10 мл) и воду (2 мл). В перемешиваемый растворитель добавляли 5-[циклопропил(4-гидроксифенил)метилиден]-2,2-диметил-1,3-диоксан-4,6-дион (0,3 г). Полученную смесь перемешивали при 90°C в течение 15 часов. После завершения реакции (прохождение отслеживали путем ТСХ) растворитель из реакционной массы удаляли при пониженном давлении и полученную неочищенную массу помещали в 4н. NaOH (50 мл). Водный слой промывали диэтиловым эфиром (100 мл × 3). Затем водный слой подкисляли 3 н. HCl (50 мл) и экстрагировали ДХМ (100 мл × 3). Объединенные слои в ДХМ промывали насыщенным солевым раствором (100 мл). Затем органический слой сушили над безводным Na2SO4. Растворитель удаляли при пониженном давлении. Продукт получали в виде коричневого вязкого раствора. Выход: 75,01% (160 мг). 1Н ЯМР (300 МГц, ДМСО-d6): δ 11,93 (s, 1Н), 9,14 (s, 1Н), 7,01-7,04 (d, 2H), 6,64-6,66 (d, 2H), 2,53-2,60 (m, 2H), 2,14-2,22 (m, 1Н), 0,93-0,95 (m, 1Н), 0,45-0,47 (m, 1Н), 0,27-0,30 (m, 1H), 0,16-0,19 (m, 1Н), 0,05-0,07 (m, 1H).
Промежуточное соединение 126: метил-3-циклопропил-3-(4-гидроксифенил)пропаноат
В 100 мл круглодонную колбу, оборудованную магнитной мешалкой и обратным холодильником, помещали 5 мл метанола. В перемешиваемый растворитель добавляли 3-циклопропил-3-(4-гидроксифенил)пропановую кислоту (160 мг, 0,7759 ммоль), затем метансульфокислоту (74,56 мг, 0,7759 ммоль). Полученную смесь перемешивали при 65°C в течение 2 часов. После завершения реакции (прохождение отслеживали путем ТСХ) растворитель из реакционной массы удаляли при пониженном давлении и полученную неочищенную массу помещали в этилацетат (50 мл) и промывали водой (100 мл × 3), раствором бикарбоната натрия (100 мл × 3), насыщенным солевым раствором (100 мл). Затем органический слой сушили над безводным Na2SO4. Растворитель удаляли при пониженном давлении. Продукт получали в виде коричневого вязкого раствора; выход 98,0% (0,23 г, неочищенный); 1Н ЯМР (300 МГц, ДМСО-d6): δ 9,16 (s, 1H), 7,01-7,04 (d, 2H), 6,64-6,67 (d, 2H), 3,49 (s, 3Н), 2,63-2,68 (m, 2H), 2,14-2,22 (m, 1H), 0,93-0,97 (m, 1H), 0,45-0,49 (m, 1H), 0,26-0,33 (m, 1H), 0,13-0,17 (m, 1H), 0,04-0,07 (m, 1H).
Промежуточное соединение 127: метил-3-циклопропил-3-{4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропаноат
В 100 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 10 мл ТГФ. В перемешиваемый растворитель добавляли (4-{[(2Е,2Z)-2-(метоксиимино)-2-фенилэтил]окси}фенил)метанол (220,7 мг, 0,8146 ммоль), затем метил-3-циклопропил-3-(4-гидроксифенил)пропаноат (0,2 г, 0,9082 ммоль). Полученную смесь охлаждали до 0°C и добавляли трифенилфосфин (0,3 г, 1,18 ммоль), затем DIAD (0,24 г, 1,18 ммоль). Полученную смесь перемешивали при температуре окружающей среды в течение 15 часов. Затем растворитель из реакционной массы удаляли при пониженном давлении и полученную неочищенную массу помещали в этилацетат (50 мл) и промывали водой (100 мл × 3), насыщенным солевым раствором (100 мл). Затем органический слой сушили над 3 г безводного Na2SO4. Растворитель удаляли при пониженном давлении и полученное неочищенное соединение очищали путем колоночной хроматографии на силикагеле (60-120 меш) с применением петролейного эфира (60-80) и этилацетата в качестве элюентов. Продукт получали в виде желтого вязкого раствора. Выход: 46,8% (0,18 г); МС (ИЭР, 120 эВ): m/z=474,1 (М+Н)+; 1Н ЯМР (300 МГц, CDCl3): δ 7,57-7,60 (m, 2H), 7,24-7,27 (m, 5H), 7,04-7,07 (d, 2H), 6,79-6,84 (m, 4H), 5,11 (s, 2H), 4,84 (s, 2H), 3,96 (s, 3H), 3,50 (s, 3Н), 2,60-2,65 (m, 2H), 2,20-2,28 (m, 1H), 1,33-1,35 (m, 2H), 0,45-0,50 (m, 1H), 0,31-0,35 (m, 1H), 0,13-0,18 (m, 1H), 0,04-0,08 (m, 1H).
Соединение 87: 3-Циклопропил-3-{4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропановая кислота
В 100 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 10 мл ТГФ и 1 мл метанола. В перемешиваемый растворитель добавляли метил-3-циклопропил-3-{4-[(4-{[(2E,2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропаноат (180 мг, 0,38 ммоль), в полученную смесь добавляли гидроксид натрия (0,0456 г, 1,1404 ммоль) в воде (1 мл). Полученную смесь перемешивали при температуре окружающей среды в течение 15 часов. После завершения реакции (прохождение отслеживали путем ТСХ) растворитель из реакционной массы удаляли при пониженном давлении и полученную неочищенную массу помещали в воду (50 мл). Водный слой промывали диэтиловым эфиром (100 мл × 3). Затем водный слой подкисляли 3 н. HCl (50 мл) и экстрагировали ДХМ (100 мл × 3). Объединенные слои в ДХМ промывали насыщенным солевым раствором (100 мл). Затем органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Полученную неочищенную массу очищали путем препаративной ТСХ. Продукт получали в виде белого твердого вещества. Выход: 22,9% (40 мг); МС (ИЭР, 120 эВ): m/z=460,1 (M+1).
Схема 31
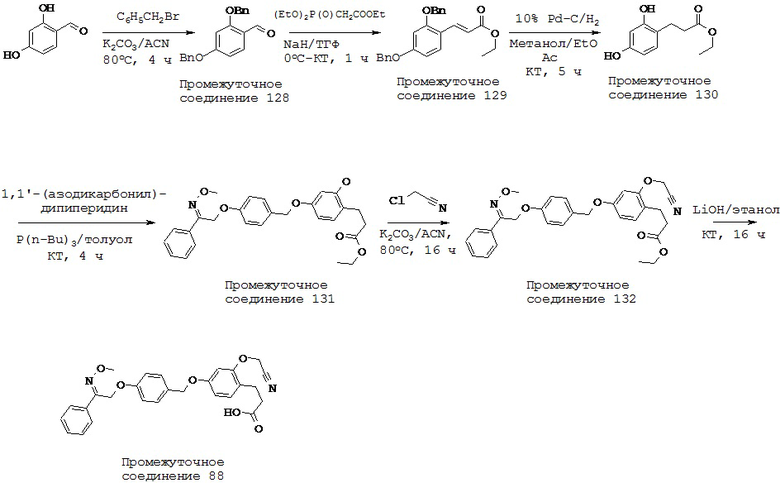
Пример 88
3-{2-(Цианометокси)-4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропановая кислота (88)
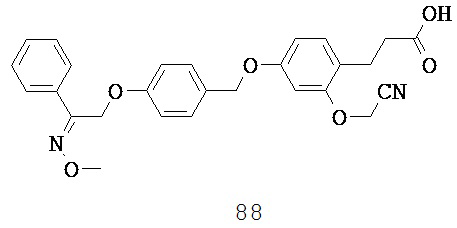
Соединение 88 синтезировали из (4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}-фенил)метанола (1,0 г, 5,0 ммоль) и этил-3-(2,4-дигидроксифенил)пропаноата (1,16 г, 4,0 ммоль) согласно способу, описанному на схеме 31 (0,0031 г, выход 8,16%).
Промежуточное соединение 128: 2,4-бис(бензилокси)бензальдегид
В 250 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 100 мл ацетонитрила. В перемешиваемый растворитель добавляли 2,4-дигидроксибензальдегид (10,0 г, 72,0 ммоль) и K2CO3 (19,87 г, 144,0 ммоль). Затем смесь перемешивали в течение 15 минут, медленно добавляли бензилбромид (12,38 г, 72,0 ммоль), PC нагревали до 80°C в течение 3 часов. Затем растворитель удаляли при пониженном давлении и соединение экстрагировали этилацетатом (50 мл × 3). Органический слой промывали водой и насыщенным солевым раствором, сушили над безводным Na2SO4 и выпаривали насухо с получением неочищенного продукта. Неочищенное вещество очищали путем колоночной хроматографии на силикагеле с применением этилацетата и петролейного эфира в качестве элюентов с получением продукта (2,3 г, выход 10,03%).
Промежуточное соединение 129: этил-(2E)-3-[2,4-бис(бензилокси)фенил]проп-2-еноат
В 100 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 25 мл тетрагидрофурана. В перемешиваемый растворитель добавляли гидрид натрия (0,35 г, 14,0 ммоль) по частям при 0°C, затем триэтилфосфоноацетат (3,23 г, 14,0 ммоль). Реакционную смесь перемешивали при 0°C в течение 30 минут. В перемешиваемый раствор 2,4-бис(бензилокси)бензальдегид (2,3 г, 7,2 ммоль) в тетрагидрофуране (2 мл) добавляли по каплям и перемешивали при комнатной температуре в течение 16 часов. После завершения реакции реакционную смесь выливали в лед и экстрагировали этилацетатом (50 мл). Органический слой промывали водой (25 мл) и насыщенным солевым раствором (25 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали путем колоночной хроматографии на силикагеле с применением этилацетата и петролейного эфира в качестве элюентов. Продукт получали в виде бесцветной маслянистой жидкости (2,3 г, выход 82,14%).
Промежуточное соединение 130: этил-3-(2,4-дигидроксифенил)пропаноат
В 500 мл колбу для встряхивателя Парра помещали этил-(2E)-3-[2,4-бис(бензилокси)-фенил]проп-2-еноат (2,3 г, 5,92 ммоль), этилацетат (10 мл) и метанол (10 мл). В реакционную смесь добавляли гидроксид палладия (20%) и гидрировали при 50 psi в течение 2 часов. После завершения реакции реакционную смесь фильтровали через целит, промывали тщательно этилацетатом (25 мл) и концентрировали для отгонки растворителя. Продукт получали в виде коричневого твердого вещества (1,2 г, выход 96,8%).
Промежуточное соединение 131: этил-3-{2-гидрокси-4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропаноат
В 100 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 20 мл толуола. В перемешиваемый растворитель добавляли (4-{[(2E,2Z)-2-(метоксиимино)-2-фенилэтил]окси}фенил)метанол (1,16 г, 4,0 ммоль) и этил-3-(2,4-дигидроксифенил)-пропаноат (1,0 г, 5,0 ммоль). Реакционную смесь охлаждали до 0°C; трибутилфосфин (1,5 г, 7,0 ммоль) добавляли и перемешивали в течение 10 минут. В реакционную смесь добавляли 1,1-(азодикарбонил)дипиперидин (1,2 г, 5,0 ммоль) в толуоле (2 мл) по каплям. Реакционную смесь перемешивали при комнатной температуре в течение 8 часов. Реакционную смесь разбавляли этилацетатом (25 мл) и промывали водой (25 мл). Органический слой промывали солевым раствором (25 мл). Органический слой сушили над Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали путем колоночной хроматографии на силикагеле с применением этилацетата и петролейного эфира в качестве элюентов. Продукт получали в виде бесцветной маслянистой жидкости (0,15 г, 6,48%).
Промежуточное соединение 132: этил-3-{2-(цианометокси)-4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропаноат
В 25 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 2 мл ацетонитрила. В перемешиваемый растворитель добавляли этил-3-{2-гидрокси-4-[(4-{[(2E,2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропаноат (0,1 г, 0,2 ммоль), карбонат калия (0,055 г, 0,4 ммоль). Реакционную смесь охлаждали до 0°C, добавляли хлорацетонитрил (0,024 г, 3,0 ммоль) в ацетонитриле (20 мл) по каплям и перемешивали при 80°C в течение 16 часов. Реакционную смесь концентрировали для отгонки растворителя. Полученный остаток растворяли в воде и экстрагировали этилацетатом (20 мл). Органический слой промывали водой (10 мл) и насыщенным солевым раствором (10 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали путем колоночной хроматографии на силикагеле с применением этилацетата и петролейного эфира в качестве элюентов с получением продукта (0,05 г, выход 50,0%).
Соединение 88: 3-{2-(Цианометокси)-4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропановая кислота
В 10 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 1 мл тетрагидрофурана. В перемешиваемый растворитель добавляли этил-3-{2-(цианометокси)-4-[(4-{[(2E,2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропаноат (0,04 г, 0,079 ммоль) и этанол (0,5 мл). Реакционную смесь охлаждали до 0°C и гидроксид лития (0,0057 г, 0,23 ммоль) в воде (1 мл) добавляли по каплям. Реакционную смесь перемешивали в течение ночи. После завершения реакции реакционную смесь концентрировали для отгонки растворителя. Соль растворяли в воде (1 мл) и экстрагировали диэтиловым эфиром (5 мл). Водный слой подкисляли 1 н. HCl до pH 3 и экстрагировали диэтиловым эфиром (5 мл). Органический слой промывали солевым раствором (5 мл), отгоняли растворитель и сушили. Неочищенное вещество очищали путем колоночной хроматографии на силикагеле с применением этилацетата и петролейного эфира в качестве элюентов с получением продукта (0,0031 г, выход 8,16%).
Пример 89
3-{2-(Карбоксиметокси)-4-[(4-{[(2E,2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропановая кислота (89)
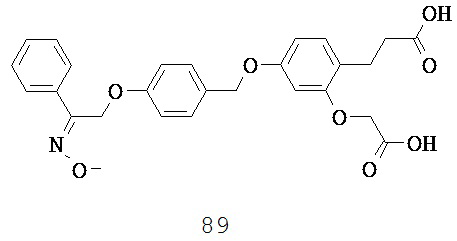
Соединение 89 синтезировали из (4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}-фенил)метанола (1,0 г, 5,0 ммоль) и этил-3-(2,4-дигидроксифенил)пропаноата (1,16 г, 4,0 ммоль) согласно способу, описанному на схеме 31 (0,006 г, выход 15,38%).
Пример 90
{2-(3-Этокси-3-оксопропил)-5-[(4-{[(2E,2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенокси}уксусная кислота (90)
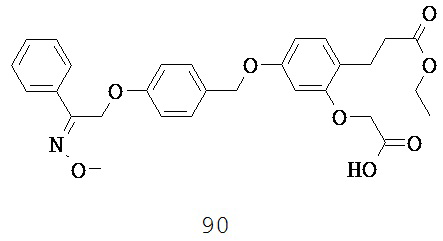
Соединение 90 синтезировали из (4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}-фенил)метанола (1,0 г, 5,0 ммоль) и этил-3-(2,4-дигидроксифенил)пропаноата (1,16 г, 4,0 ммоль) согласно способу, описанному на схеме 31 (0,0008 г, выход 19,28%).
Схема 32
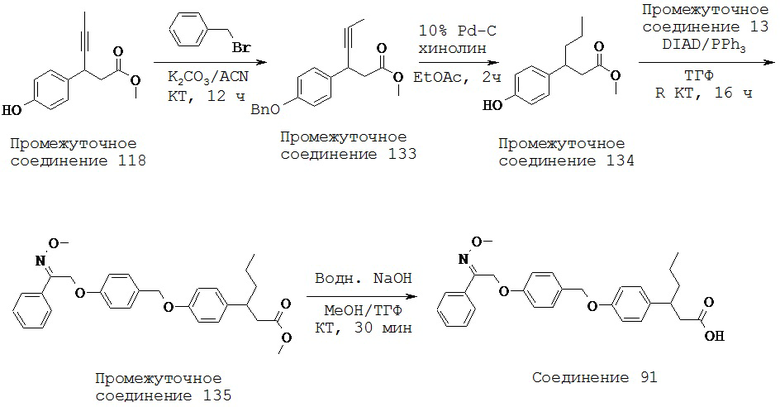
Пример 91
3-{4-[(4-{[(2Z)-2-(Метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}гексановая кислота (91)
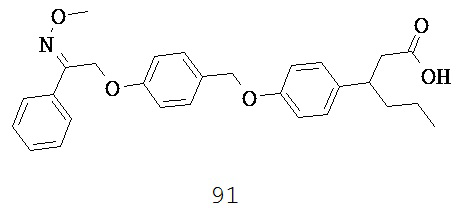
Соединение 91 синтезировали из (4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}-фенил)метанола (0,134 г, 0,5 ммоль) и метил-3-(4-гидроксифенил)гексаноата (0,11 г, 0,5 ммоль) согласно способу, описанному на схеме 32 (0,012 г, выход 24,73%); чистота: 87,06%.
Промежуточное соединение 133: метил-3-[4-(бензилокси)фенил]гекс-4-иноат
В 250 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 30 мл ацетонитрила. В перемешиваемый растворитель добавляли метил-3-(4-гидроксифенил)гекс-4-иноат (3,8 г, 17,4 ммоль), карбонат калия (7,2 г, 52,2 ммоль). Реакционную смесь охлаждали до 0°C, добавляли бензилбромид (4,47 г, 1,5 ммоль) в ацетонитриле (20 мл) по каплям и перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь концентрировали для отгонки растворителя. Воду (20 мл) добавляли и экстрагировали этилацетатом (60 мл). Органический слой промывали водой (20 мл) и насыщенным солевым раствором (20 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали путем колоночной хроматографии на силикагеле с применением этилацетата и петролейного эфира в качестве элюентов с получением продукта (4,2 г, выход 78,35%).
Промежуточное соединение 134: метил-3-(4-гидроксифенил)гексаноат
В 500 мл колбу для гидрогенизатора Парра, содержащую этилацетат (5 мл) и хинолин (0,117 г, 1,4 ммоль) добавляли метил-3-[4-(бензилокси)фенил]гекс-4-иноат (0,2 г, 65,0 ммоль) и 10% Pd/C (0,015 г) в атмосфере N2. PC гидрировали в течение 2 часов. PC разбавляли этилацетатом (10 мл), отфильтровывали катализатор через целит и выпаривали насухо при пониженном давлении. Неочищенное вещество очищали путем колоночной флэш-хроматографии на силикагеле (100/200 меш) с получением продукта (0,11 г, выход 76,38%).
Промежуточное соединение 135; метил-3-{4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}гексаноат
В 50 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали сухой ТГФ (5,0 мл) и (4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}фенил)метанол (0,134 г, 0,5 ммоль). В полученную выше смесь метил-3-(4-гидроксифенил)гексаноат (0,11 г, 0,5 ммоль) добавляли и полученную смесь перемешивали при 0°C в течение 5 минут. Трифенилфосфин (0,17 г, 0,65 ммоль) добавляли в смесь и перемешивали при 0°C в течение 15 минут, затем добавляли диэтилазадикарбоксилат (0,13 г, 0,65 ммоль). После перемешивания полученной смеси при КТ в течение 16 часов PC выпаривали для удаления ТГФ. Остаток разбавляли водой (15 мл) и экстрагировали этилацетатом (15 мл × 2). Органический слой промывали насыщенным солевым раствором (15 мл) и сушили над безводным Na2SO4. Концентрирование растврителя и очистка полученного остатка путем колоночной хроматографии на силикагеле (100/200 меш) с применением петролейного эфира и этилацетата в качестве элюентов приводили к получению продукта (0,05 г, выход 21,74%).
Соединение 91: 3-{4-[(4-{[(2Z)-2-(Метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}гексановая кислота
В 25 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 3 мл тетрагидрофурана. В перемешиваемый растворитель добавляли метил-3-{4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}гексаноат (0,017 г, 0,03 ммоль), метанол (0,5 мл) и гидроксид натрия (0,013 г, 0,32 ммоль) в воде (0,5 мл). После завершения добавления реакционную смесь перемешивали при комнатной температуре в течение 30 минут. Реакционную смесь концентрировали для отгонки растворителя; неочищенное вещество промывали диэтиловым эфиром (5 мл). Соль растворяли в воде (1 мл) и водный слой подкисляли насыщенной лимонной кислотой до pH 6. Водный слой экстрагировали этилацетатом (5 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали путем препаративной ТСХ с применением метанола и хлороформа в качестве элюентов. Продукт получали в виде белого полутвердого вещества (0,012 г, выход 24,73%). МС (ИЭР, 120 эВ): m/z=462,1 (М+Н)+.
Пример 92
3-(4-{[4-({(2Z)-2-(Метоксиимино)-2-[4-(метилсульфонил)фенил]этил}окси)бензил]окси}фенил)пропановая кислота (92)
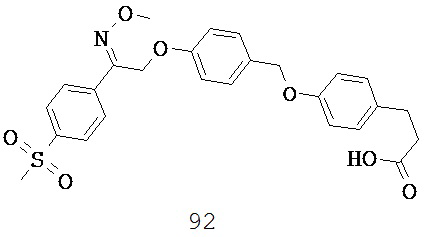
Соединение 92 синтезировали из [4-({(2Z)-2-(метоксиимино)-2-[4-(метилсульфонил)-фенил] этил }окси)фенил] метанола (0,57 г, 1,63 ммоль) и метил-3-(4-гидроксифенил)-пропаноата (0,293 г, 1,63 ммоль) согласно способу, описанному на схеме 5 (0,2 г, выход 39,91%); чистота: 96,52%.
Схема 33
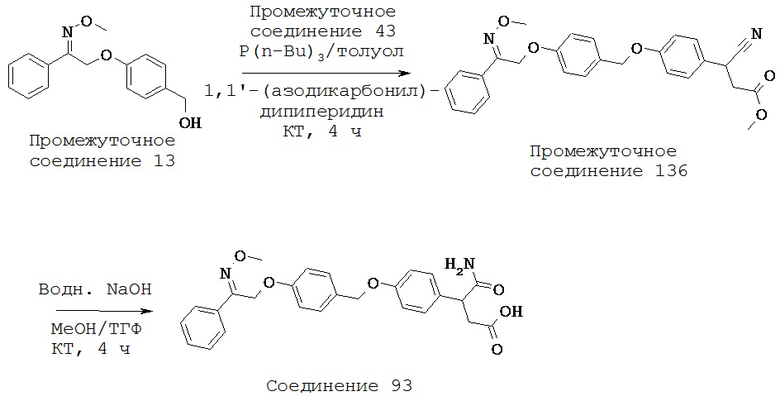
Пример 93
4-Амино-3-{4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}-4-оксобутановая кислота (93)
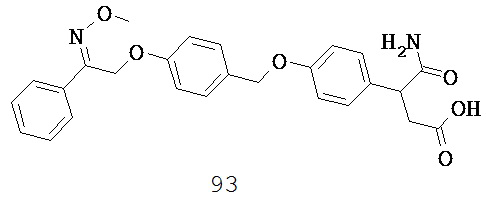
Соединение 93 синтезировали из (4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}-фенил)метанола (30,93 г, 114,0 ммоль) и метил-3-циано-3-(4-гидроксифенил)пропаноата (26,0 г, 127,0 ммоль) согласно способу, описанному на схеме 33 (2,1 г, выход 9,05%); чистота: 96,52%.
Промежуточное соединение 136: метил-3-циано-3-{4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропаноат
В 500 мл 3-горлую круглодонную колбу, оборудованную магнитной мешалкой, помещали 100 мл толуола. В перемешиваемый растворитель добавляли (4-{[(2E,2Z)-2-(метоксиимино)-2-фенилэтил]окси}фенил)метанол (30,93 г, 114,0 ммоль) и метил-3-циано-3-(4-гидроксифенил)пропаноат (26,0 г, 127,0 ммоль). Реакционную смесь охлаждали до 0°C; трибутилфосфин (33,35 г, 4,26 ммоль) добавляли и перемешивали в течение 20 минут. В реакционную смесь добавляли 1,1'-(азодикарбонил)дипиперидин (41,56 г, 5,53 ммоль) в толуоле (50 мл) по каплям. Реакционную смесь перемешивали при комнатной температуре в течение 4 часов. Реакционную смесь разбавляли этилацетатом (250 мл) и промывали водой. Наконец, органический слой промывали солевым раствором и сушили над Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали путем колоночной хроматографии на силикагеле с применением этилацетата и петролейного эфира в качестве элюентов. Продукт получали в виде бесцветной маслянистой жидкости (45,0 г, 86,09%).
Соединение 93: 4-Амино-3-{4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}-4-оксобутановая кислота
В 500 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 115 мл тетрагидрофурана. В перемешиваемый растворитель добавляли метил-3-циано-3-{4-[(4-{[(2E,2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропаноат (23,0 г, 0,502 ммоль), метанол (115 мл) и гидроксид натрия (25,0 мл, 4н. раствор). Затем реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь концентрировали для отгонки растворителя; неочищенное вещество промывали этилацетатом (100 мл). В остаток добавляли воду (50 мл) и водный слой подкисляли насыщенным 1 н. раствором NaOH до pH 6. Водный слой экстрагировали этилацетатом (200 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенное вещество (5,0 г) очищали путем кристаллизации в смеси этилацетата (30,0 мл) и метанола (20,0 мл). Продукт получали в виде белого твердого вещества (2,1 г, выход 9,05%).
Пример 94
3-{3-Метокси-4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропановая кислота (94)
Соединение 94 синтезировали из (4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}-фенил)метанола (0,12 г, 0,446 ммоль) и этил-3-(4-гидрокси-3-метоксифенил)пропаноата (0,1 г, 0,446 ммоль) согласно способу, описанному на схеме 16 (0,06 г, выход 47,62%); чистота: 98,66%.
Схема 34
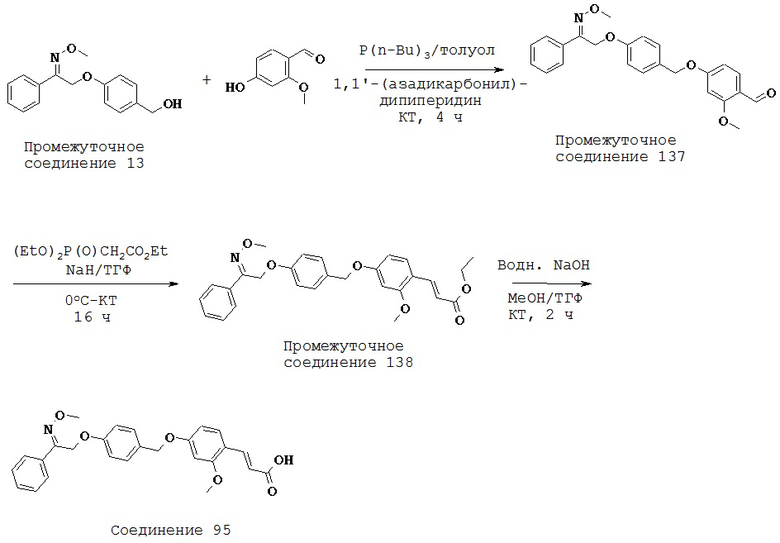
Пример 95
(2E,2Z)-3-{2-Метокси-4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}проп-2-еновая кислота (95)
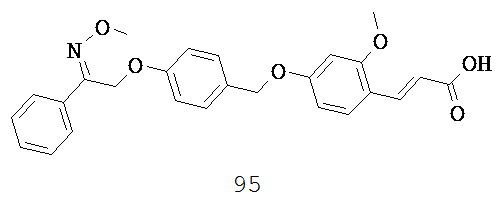
Соединение 95 синтезировали из (4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}-фенил)метанола (0,8 г, 2,95 ммоль) и (2E,2Z)-3-(4-гидрокси-2-метоксифенил)проп-2-еновной кислоты (0,5 г, 3,28 ммоль) согласно способу, описанному на схеме 34 (0,3 г, выход 25,0%); чистота: 89,76%.
Промежуточное соединение 137: 2-метокси-4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]бензальдегид
В 100 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 20 мл толуола. В перемешиваемый растворитель добавляли (4-{[(2E,2Z)-2-(метоксиимино)-2-фенилэтил]окси}фенил)метанол (0,8 г, 2,95 ммоль) и 4-гидрокси-2-метоксибензальдегид (0,5 г, 3,28 ммоль). Реакционную смесь охлаждали до 0°C; трибутилфосфин (0,86 г, 4,26 ммоль) добавляли и перемешивали в течение 10 минут. В реакционную смесь добавляли 1,1-(азодикарбонил)дипиперидин (1,39 г, 5,53 ммоль) в толуоле (2 мл) по каплям. Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь разбавляли этилацетатом (25 мл) и промывали водой (25 мл). Органический слой промывали солевым раствором (25 мл). Органический слой сушили над Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали путем колончоной хроматографии на силикагеле с применением этилацетата и петролейного эфира в качестве элюентов. Продукт получали в виде бесцветной маслянистой жидкости (0,3 г, 25,0%).
Промежуточное соединение 138: этил-(2E,2Z)-3-{2-метокси-4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}проп-2-еноат
В 100 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 10 мл тетрагидрофурана. В перемешиваемый растворитель добавляли гидрид натрия (0,035 г, 1,47 ммоль) по частям при 0°C, затем триэтилфосфоноацетат (0,25 г, 1,1 ммоль). Реакционную смесь перемешивали при 0°C в течение 30 минут. В перемешиваемый раствор добавляли по каплям 2-метокси-4-[(4-{[(2E,2Z)-2-(метоксиимино)-2-фенилэтил]окси}-бензил)окси]бензальдегид (0,3 г, 0,739 ммоль) в тетрагидрофуране (2 мл) и перемешивали при комнатной температуре в течение 16 часов. После завершения реакции реакционную смесь выливали в лед и экстрагировали этилацетатом (50 мл). Органический слой промывали водой (25 мл) и насыщенным солевым раствором (25 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали путем колоночной хроматографии на силикагеле с применением этилацетата и петролейного эфира в качестве элюентов. Продукт получали в виде бесцветной маслянистой жидкости (0,3 г, выход 8,5%).
Соединение 95: (2E,2Z)-3-{2-Метокси-4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}проп-2-еновая кислота
В 25 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 3 мл тетрагидрофурана. В перемешиваемый растворитель добавляли этил-(2E,2Z)-3-{2-метокси-4-[(4-{[(2E,2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}проп-2-еноат (0,3 г, 0,63 ммоль), метанол (3 мл) и гидроксид натрия (5,0 мл, 2н. раствор) в воде (0,5 мл). После завершения добавления реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь концентрировали для отгонки растворителя; неочищенное вещество промывали этилацетатом (25 мл). В остаток добавляли воду (1 мл) и водный слой подкисляли насыщенным раствором лимонной кислоты до pH 6. Водный слой экстрагировали этилацетатом (20 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали путем препаративной ТСХ с применением метанола и хлороформа в качестве элюентов. Продукт получали в виде белого полутвердого вещества (0,2 г, выход 71,42%).
Пример 96
3-{2-Гидрокси-4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропановая кислота (96)
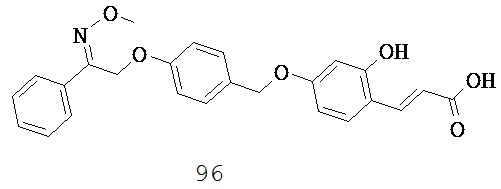
Соединение 96 синтезировали из этил-3-{2-(цианометокси)-4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил] окси} бензил)окси] фенил} пропаноата (промежуточное соединение 134, 0,1 г, 0,199 ммоль) и Pd-C (0,02 г) согласно способу гидролиза, описанному на схеме 16 (0,005 г, выход 36,0%); чистота: 93,97%.
Пример 97
(2E,2Z)-3-{4-[(4-{[(2Z)-2-(Метоксиимино)-2-фенилэтил]окси}бензил)окси]-2-(5-метил-1,2-оксазол-3-ил)фенил}проп-2-еновая кислота
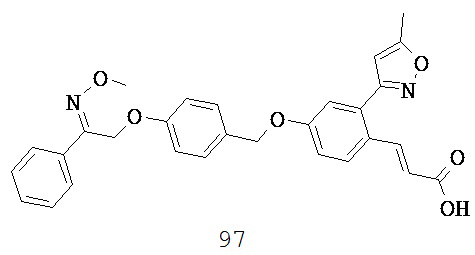
Соединение 97 синтезировали из (4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}-фенил)метанола (0,02 г, 0,0545 ммоль) и метил-(2E)-3-[4-гидрокси-2-(5-метил-1,2-оксазол-3-ил)фенил]проп-2-еноата (0,012 г, 0,0436 ммоль) согласно способу, описанному на схеме 18 (0,008 г, выход 13,70%); чистота: 95,32%.
Пример 98
3-Циано-3-{4-[(4-{[(2E)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропановая кислота (рацемат) (98)
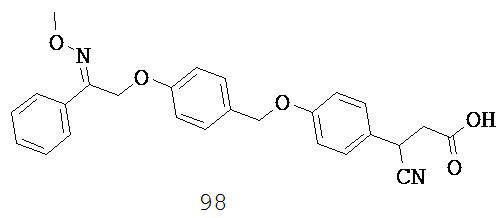
Соединение 98 синтезировали из (4-{[(2E)-2-(метоксиимино)-2-фенилэтил]окси}-фенил)метанола (0,55 г, 2,03 ммоль) и метил-3-циано-3-(4-гидроксифенил)пропаноата (0,46 г, 2,22 ммоль) согласно способу, описанному на схеме 5 (0,008 г, выход 30,1%); чистота: 93,18%.
Пример 99
3-Циано-3-{4-[(4-{[(2E)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропановая кислота (энантиомер-1) (99)
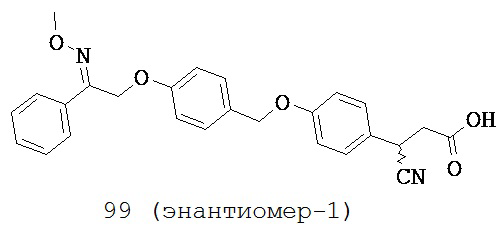
Соединение 99 синтезировали из 3-циано-3-{4-[(4-{[(2E)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропановой кислоты (соединение 98) путем хиральной препаративной ВЭЖХ (0,008 г, выход 15%); чистота: 84,93%; хиральная чистота: 98%.
Пример 100
3-Циано-3-{4-[(4-{[(2E)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропановая кислота (энантиомер-2) (100)
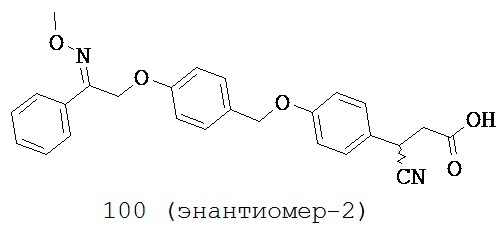
Соединение 100 синтезировали из 3-циано-3-{4-[(4-{[(2E)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропановой кислоты (соединение 98) путем хиральной препаративной ВЭЖХ (0,008 г, выход 15%); чистота: 92,37%; хиральная чистота: 95%.
Пример 101
3-Циано-3-{4-[(4-{[(2Z)-2-(1H-инден-2-ил)-2-(метоксиимино)этил]окси}бензил)окси]фенил}пропановая кислота (101)
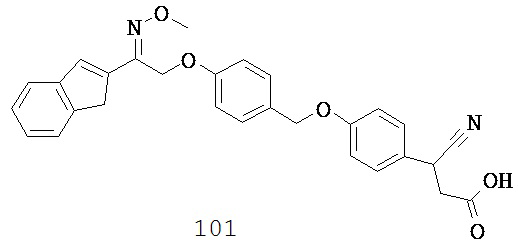
Соединение 101 синтезировали из (4-{[(2Z)-2-(1H-инден-2-ил)-2-(метоксиимино)этил]-окси}фенил)метанола (0,079 г, 0,4 ммоль) и метил-3-циано-3-(4-гидроксифенил)пропаноата (0,11 г, 0,4 ммоль) согласно способу, описанному на схеме 5 (0,008 г, выход 20,5%); чистота: 97,17%.
Пример 102
3-Циано-3-{4-[(4-{[(2Z)-2-(2,3-дигидро-1-бензофуран-5-ил)-2-(метоксиимино)этил]окси}бензил)окси]фенил}пропановая кислота (102)
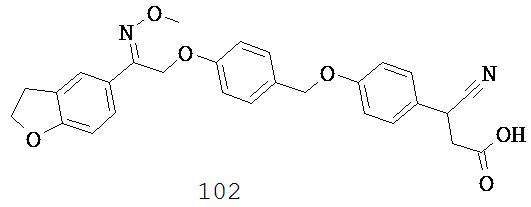
Соединение 102 синтезировали из (4-{[(2Z)-2-(2,3-дигидро-1-бензофуран-5-ил)-2-(метоксиимино)этил]окси}фенил)метанола (0,330 г, 1,0 ммоль) и метил-3-циано-3-(4-гидроксифенил)пропаноата (0,216 г, 1,0 ммоль) согласно способу, описанному на схеме 5 (0,008 г, выход 56,2%); чистота: 94,55%.
Пример 103
3-Циано-3-{4-[(4-{[(2Z)-2-(2,3-дигидро-1H-инден-5-ил)-2-(метоксиимино)этил]окси}бензил)окси]фенил}пропановая кислота (103)
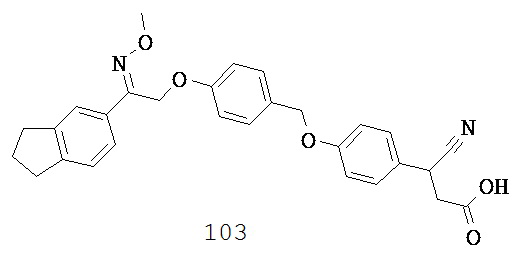
Соединение 103 синтезировали из (4-{[(2Z)-2-(2,3-дигидро-1H-инден-5-ил)-2-(метоксиимино)этил]окси}фенил)метанола (0,25 г, 0,8 ммоль) и метил-3-циано-3-(4-гидроксифенил)пропаноата (0,214 г, 1,04 ммоль) согласно способу, описанному на схеме 5 (0,008 г, выход 38,4%); чистота: 95,42%.
Пример 104
3-Циано-3-{4-[(4-{[(2E)-2-циклопропил-2-(метоксиимино)этил]окси}бензил)окси]фенил}пропановая кислота (104)
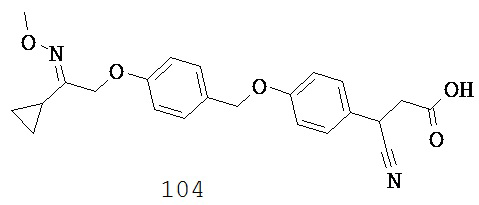
Соединение 104 синтезировали из (4-{[(2E)-2-циклопропил-2-(метоксиимино)этил]-окси}фенил)метанола (0,5 г, 2,12 ммоль) и метил-3-циано-3-(4-гидроксифенил)пропаноата (0,44 г, 2,12 ммоль) согласно способу, описанному на схеме 5 (0,008 г, выход 44,24%); чистота: 85,80%.
Пример 105
3-Циано-3-{4-[(4-{[(2Z)-2-циклопропил-2-(метоксиимино)этил]окси}бензил)окси]фенил}пропановая кислота (105)
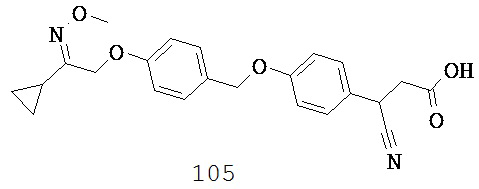
Соединение 105 синтезировали из (4-{[(2Z)-2-циклопропил-2-(метоксиимино)этил]-окси}фенил)метанола (0,5 г, 2,12 ммоль) и метил-3-циано-3-(4-гидроксифенил)пропаноата (0,44 г, 2,12 ммоль) согласно способу, описанному на схеме 5 (0,008 г, выход 56,32%); чистота: 84,26%.
Пример 106
3-{4-[(4-{[(2E)-2-(Метоксиимино)-2-(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)этил]окси}бензил)окси]фенил}пропановая кислота (106)
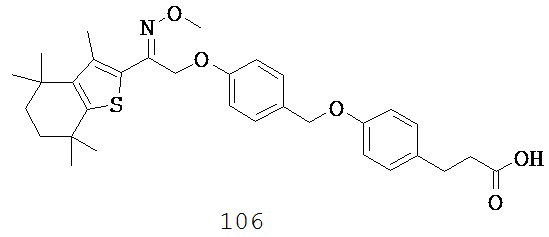
Соединение 106 синтезировали из (4-{[(2E)-2-(метоксиимино)-2-(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)этил]окси}фенил)метанола (0,3 г, 0,7 ммоль) и метил-3-(4-гидроксифенил)пропаноата (0,135 г, 0,7 ммоль) согласно способу, описанному на схеме 5 (0,008 г, выход 2,16%); чистота: 97,59%.
Схема 35:
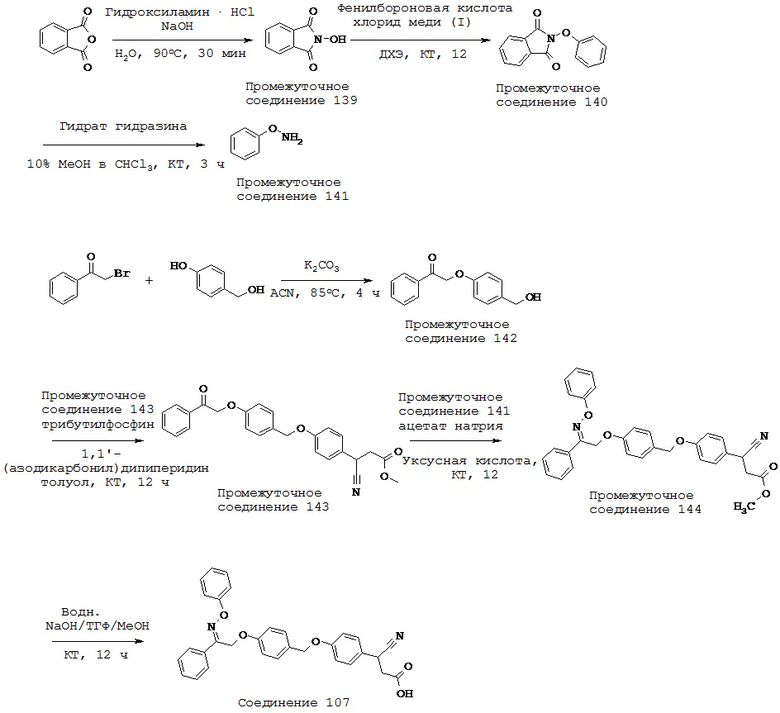
Пример 107
3-Циано-3-{4-[(4-{[(2E,2Z)-2-(феноксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропановая кислота (107)
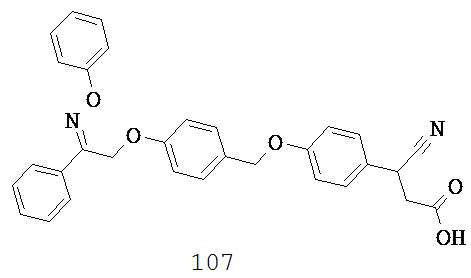
Соединение 107 синтезировали из O-фенилгидроксиламина (аминооксибензола) (0,15 г, 1,4 ммоль) и метил-3-циано-3-(4-{[4-(2-оксо-2-фенилэтокси)бензил]окси}фенил)пропаноата (0,2 г, 0,46 ммоль) согласно способу, описанному на схеме 35 (0,008 г, выход 10%); чистота: 69,92% и 26,24%.
Промежуточное соединение 139: 2-гидрокси-1H-изоиндол-1,3(2H)-дион
В 250 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали гидрохлорид гидроксиламина (5 г, 73 ммоль) в воде, затем медленно добавляли 50% раствор NaOH. В перемешиваемый раствор добавляли фталевый ангидрид (11,26 г, 76 ммоль), перемешивали при 90°C в течение 40 минут. Затем охлаждали до КТ, желтоватое твердое вещество удаляли путем фильтрования. Фильтрат нейтрализовали 30% H2SO4, и осаждалось твердое вещество. Твердое вещество отфильтровывали через воронку Бюхнера и сушили с использованием вакуума с получением титульного соединения (6,5 г, выход 55,34%); 1H ЯМР (300 МГц, ДМСО-d6): δ 10,80 (s, 1H), 7,84 (s, 4H).
Промежуточное соединение 140: 2-фенокси-1H-изоиндол-1,3(2H)-дион
В 50 мл одногорлую круглодонную колбу помещали 2-гидрокси-1Н-изоиндол-1,3(2Н)-дион (1 г, 6,13 ммоль), молекулярные сита 4Å (1 г) и хлорид меди (I) (0,61 г, 0,06 ммоль) в ДХЭ на открытом воздухе. В перемешиваемую реакционную смесь добавляли фенилбороновую кислоту (1,5 г, 12,2 ммоль), затем пиридин (0,53 г, 6,7 ммоль) по каплям. Полученную смесь перемешивали при КТ в течение 12 часов. Реакционную смесь разбавляли ДХМ, а затем фильтровали через подложку целита. Органический слой промывали насыщенным солевым раствором (50 мл) и сушили над безводным Na2SO4. Растворитель удаляли при пониженном давлении с получением беловатого твердого вещества. 1H ЯМР (300 МГц, CDCl3): δ 7,83-7,86 (q, 2H), 7,73-7,77 (m, 2H), 7,28-7,30 (m, 2H), 7,07-7,11 (m, 3H).
Промежуточное соединение 141: O-фенилгидроксиламин (аминооксибензол)
В 50 мл круглодонную колбу помещали 2-фенокси-1H-изоиндол-1,3(2H)-дион (0,6 г, 2,5 ммоль) и 10% МеОН в CHCl3 (15 мл). В перемешиваемый раствор добавляли гидрат гидразина (0,37 мл, 7,53 ммоль) по каплям при КТ и продолжали перемешивание в течение 3 часов. Фталазин (побочный продукт), выпадавший в виде твердого осадка, отфильтровывали и промывали твердое вещество 10% MeOH в CHCl3. Фильтрат сушили над безводным Na2SO4 и концентрировали с получением неочищенного продукта, который немедленно применяли на следующей стадии.
Промежуточное соединение 142: 2-[4-(гидроксиметил)фенокси]-1-фенилэтанон
В 100 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 20 мл ацетонитрила. В перемешиваемый растворитель добавляли 4-гидроксибензиловый спирт (6,74 г, 25,1 ммоль) и K2CO3 (10,4 г, 75,3 ммоль). Затем смесь перемешивали в течение 5 минут. 2-Бром-1-фенилэтанон (5 г, 25,1 ммоль) добавляли. Затем PC нагревали при 80°C в течение 2 часов. После завершения реакции (прохождение отслеживали путем ТСХ) PC концентрировали в вакууме для удаления ацетонитрила. В остаток затем добавляли 10 мл воды и экстрагировали этилацетатом (15 мл × 2). Органический слой промывали насыщенным солевым раствором (15 мл), сушили над безводным Na2SO4 и выпаривали насухо с получением титульного соединения (5 г, выход 82,2%); 1H ЯМР (300 МГц, CDCl3): δ 7,92-7,94 (d, 2H), 7,53-7,58 (t, 1H), 7,41-7,46 (t, 2H), 7,21-7,24 (d, 2H), 6,85-6,86 (d, 2H), 5,22 (s, 2H), 4,55 (s, 2H).
Промежуточное соединение 143: метил-3-циано-3-(4-{[4-(2-оксо-2-фенилэтокси)бензил]окси}фенил)пропаноат
В 100 мл двухгорлую круглодонную колбу помещали 2-[4-(гидроксиметил)фенокси]-1-фенилэтанон (2,5 г, 10,3 ммоль) и метил-3-циано-3-(4-гидроксифенил)пропаноат (2,5 г, 12,4 ммоль) в 50 мл толуола. В полученную смесь добавляли трибутилфосфин (3,33 г, 16,5 ммоль) при 0°C и добавляли 1,1'-(азодикарбонил)дипиперидин (4,165 г, 16,5 ммоль) по частям в течение 15 минут при 0°C. Через 12 часов перемешивания при КТ смесь разбавляли н-гексаном и перемешивали в течение 10 минут. Полученное твердое вещество отфильтровывали и фильтрат концентрировали с получением неочищенного продукта, из которого после растирания с этилацетатом и петролейным эфиром получали чистый продукт (2,5 г, выход 56,1%); 1Н ЯМР (300 МГц, ДМСО-d6): δ 8,01-8,03 (d, 2H), 7,67-7,72 (t, 1H), 7,55-7,60 (t, 2H), 7,34-7,37 (d, 4H), 6,96-7,02 (t, 4H), 5,59 (s, 2H), 5,01 (s, 2H), 4,37-4,46 (m, 1H), 3,61 (s, 3H), 3,07-3,16 (m, 1H), 2,90-2,97 (m, 1H).
Промежуточное соединение 144: метил-3-циано-3-{4-[(4-{[(2E,2Z)-2-(феноксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропаноат
В 50 мл одногорлую круглодонную колбу, содержащую метил-3-циано-3-(4-{[4-(2-оксо-2-фенилэтокси)бензил]окси}фенил)пропаноат (0,2 г, 0,466 ммоль) в уксусной кислоте (2 мл), добавляли O-фенилгидроксиламин (0,15 г, 1,4 ммоль), затем ацетат натрия (0,076 г, 9,32 ммоль). Реакционную смесь перемешивали при КТ в атмосфере азота в течение 3 часов. Затем PC разбавляли этилацетатом (25 мл) и органический слой промывали водой (20 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Продукт получали в виде маслянистой жидкости (0,12 г, выход 42,95%); МС (ИЭР, 120 эВ): m/z=521,2 (М+Н)+; 1H ЯМР (300 МГц, CDCl3): δ 7,70-7,74 (m, 2H), 7,34-7,35 (d, 4H), 7,23-7,29 (m, 8H), 6,86-6,94 (m, 4H), 5,36 (s, 2H), 4,89 (s, 2H), 4,14-4,19 (t, 1H), 3,65 (s, 3H), 2,88-2,96 (dd, 1H), 2,70-2,78 (dd, 1H).
Соединение 107: 3-Циано-3-{4-[(4-{[(2E,2Z)-2-(феноксиимино)-2-фенилэтил] окси}бензил)окси] фенил}пропановая кислота
В 10 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 3 мл ТГФ. В перемешиваемый растворитель добавляли метил-3-циано-3-{4-[(4-{[(2E,2Z)-2-(феноксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропаноат (0,11 г, 0,211 ммоль) и метанол (3 мл). PC охлаждали до 0°C и гидроксид натрия (0,03 г) в воде (1 мл) добавляли по каплям. Затем реакционную смесь перемешивали при КТ в течение 12 часов. Затем растворители выпаривали и неочищенное вещество растворяли в минимальном количестве воды (2 мл) и водный слой промывали диэтиловым эфиром (5 мл × 2). Водный слой подкисляли до pH 3 при помощи 1 н. хлороводородной кислоты. Водный слой экстрагировали этилацетатом (5 мл × 2). Органический слой промывали насыщенным солевым раствором (5 мл) и сушили над безводным Na2SO4. Растворитель удаляли при пониженном давлении, продукт получали в виде полутвердого вещества (0,008 г, выход 8,0%); чистота: 69,92% и 26,24%.
Пример 108
3-(4-{[4-({(2E,2Z)-2-[(Бензилокси)имино]-2-фенилэтил}окси)бензил]окси}фенил)пропановая кислота (108)
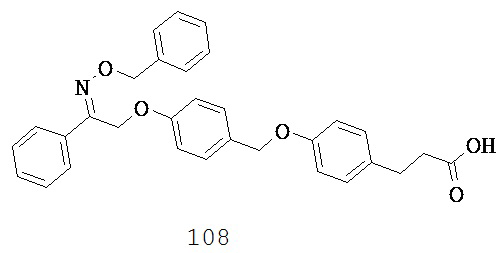
Соединение 108 синтезировали из O-бензилгидроксиламина ([(аминоокси)метил]-бензола) (0,07 г, 0,4 ммоль) и метил-3-(4-{[4-(2-оксо-2-фенилэтокси)бензил]окси}фенил)-пропаноата (0,15 г, 0,4 ммоль) согласно способу, описанному на схеме 35 (0,06 г, выход 32,5%); чистота: 84,22% и 10,16%.
Пример 109
3-Циано-3-{4-[(4-{[(2Z)-2-(гидроксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропановая кислота (109)
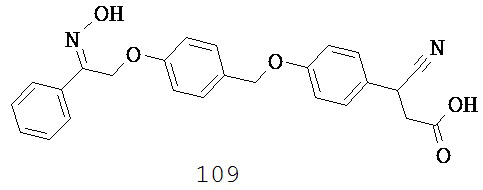
Соединение 109 синтезировали из соли HCl и O-гидроксиламина (0,34 г, 1,15 ммоль) и метил-3-циано-3-(4-{[4-(2-оксо-2-фенилэтокси)бензил]окси}фенил)пропаноата (1,4 г, 3,26 ммоль) согласно способу, описанному на схеме 35 (0,26 г, выход 14,8%); чистота: 92,16%.
Пример 110
3-(4-{[4-({(2E,2Z)-2-Фенил-2-[(проп-2-ен-1-илокси)имино]этил}окси)бензил]окси}фенил)пропановая кислота (110)
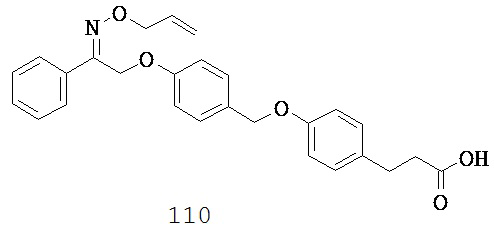
Соединение 110 синтезировали из O-проп-2-ен-1-илгидроксиламина (3-(аминоокси)-проп-1-ена) (0,2 г, 1,8 ммоль), метил-3-(4-{[4-(2-оксо-2-фенилэтокси)бензил]окси}фенил)-пропаноата (0,37 г, 0,09 ммоль) согласно способу, описанному на схеме 35 (0,08 г, выход 12%); чистота: 86,5% и 9,06%.
Пример 111
3-Циано-3-(4-{[4-({(2Z)-2-[(циклогексилокси)имино]-2-фенилэтил}окси)бензил]окси}фенил)пропановая кислота (111)
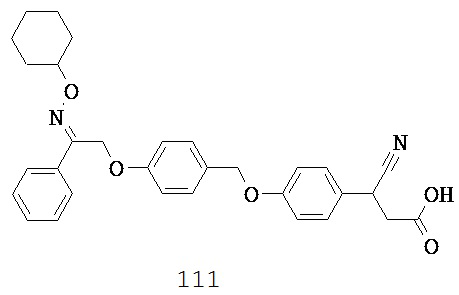
Соединение 111 синтезировали из соли O-гидроксиламина и HCl (0,16 г, 1,4 ммоль) и метил-3-циано-3-(4-{[4-(2-оксо-2-фенилэтокси)бензил]окси}фенил)пропаноата (0,2 г, 0,46 ммоль) согласно способу, описанному на схеме 35 (0,06 г, выход 24%); чистота: 95%.
Схема 36:
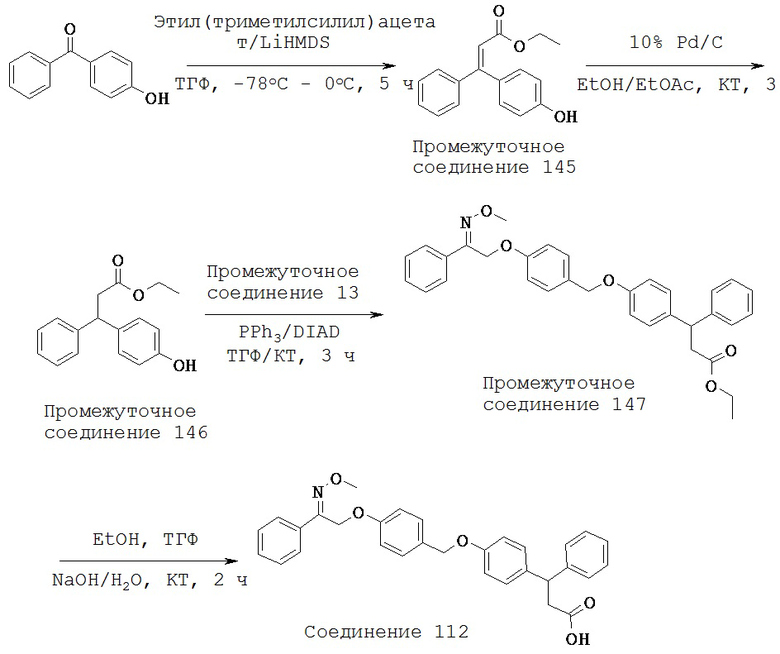
Пример 112
3-{4-[(4-{[(2Z)-2-(Метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}-3-фенилпропановая кислота (112)
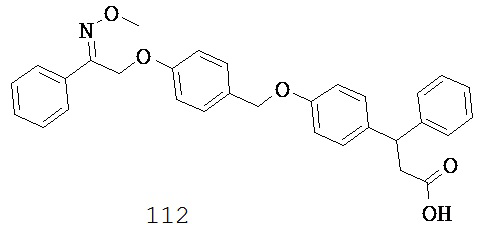
Соединение 112 синтезировали из этил-3-(4-гидроксифенил)-3-фенилпропаноата (0,6 г, 2,2 ммоль) и (4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}фенил)метанола (0,6 г, 2,2 ммоль) согласно способу, описанному на схеме 36 (0,2 г, выход 18,2%); чистота: 98,55%.
Промежуточное соединение 145: этил-(2Z)-3-(4-гидроксифенил)-3-фенилпроп-2-еноат
Раствор ГМДС (21,17 г, 131,1 ммоль) в ТГФ (200 мл) охлаждали до -40°C. Затем добавляли n-BuLi (56,7 мл, 136,22 ммоль) и перемешивали при указанной температуре в течение 45 минут, после чего (4-гидроксифенил)(фенил)метанон (2 г, 10 ммоль) в ТГФ добавляли медленно в течение 10 минут и перемешивали при -40° - -30°C в течение одного часа. Затем этил(триметилсилил)ацетат (2,42 г, 15,1 ммоль) добавляли по каплям и перемешивали при 0°С-15°C до израсходования исходного вещества (4 ч). После завершения реакцию гасили хлоридом аммония, а затем ТГФ выпаривали в вакууме, полученный раствор разделяли в воде и этилацетате. Объединенные органические слои сушили и выпаривали с получением неочищенного вещества, которое очищали путем хроматографии и растирали с н-гексаном с получением красной маслянистой жидкости в качестве продукта (1,5 г, выход 55,9%); 1Н ЯМР (300 МГц, CDCl3): δ 7,31-7,33 (m, 4H), 7,01-7,74 (d, 1H), 6,7-7,16 (m, 4H), 6,25 (s, 1H), 3,94-3,99 (q, 2H), 1,07-1,10 (t, 3H).
Промежуточное соединение 146: этил-3-(4-гидроксифенил)-3-фенилпропаноат
500 мл колбу для встряхивателя Парра, содержащую этил-(2Z)-3-(4-гидроксифенил)-3-фенилпроп-2-еноат (1 г, 3,7 ммоль) в смеси этанол/этилацетат, дегазировали азотом в течение 2 минут. Добавляли 10% Pd/C (0,4 г, 10%) и подводили H2 под давлением 50 psi в течение 3 часов. Реакционную смесь фильтровали через подложку целита и промывали подложку избытком этанола. Органическую фазу сушили над безводным Na2SO4 и концентрировали с получением маслянистой жидкости в качестве продукта (0,6 г, выход 60,1%); 1H ЯМР (300 МГц, ДМСО-d6): δ 9,21 (s, 1H), 7,07-7,29 (m, 7H), 6,63-6,66 (d, 2H), 4,29-4,34 (t, 1H), 3,89-3,96 (q, 2H), 3,02-3,03 (d, 2H), 0,99-1,04 (t, 3H).
Промежуточное соединение 147: этил-3-{4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}-3-фенилпропаноат
В 100 мл двухгорлую круглодонную колбу помещали этил-3-(4-гидкросифенил)-3-фенилпропаноат (0,6 г, 2,2 ммоль) и (4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}фенил)-метанол (0,6 г, 2,2 ммоль) в 20 мл ТГФ. В полученную смесь добавляли трифенилфосфин (0,755 г, 2,88 ммоль) при 0°C и DIAD (0,59 г, 2,88 ммоль) по частям в течение 15 минут при 0°C. Через 12 часов перемешивания при КТ смесь разбавляли этилацетатом, промывали последовательно водой и солевым раствором. Органическую фазу сушили над безводным Na2SO4 и концентрировали с получением желтой маслянистой жидкости в качестве продукта (0,6 г, выход 52,17%); 1Н ЯМР (300 МГц, ДМСО-d6): δ 7,57-7,60 (m, 2H), 7,06-7,31 (m, 12H), 6,84-6,86 (m, 4H), 5,12 (s, 2H), 4,84 (s, 2H), 4,39-4,45 (t, 1H), 3,86-4,01 (m, 5H), 2,92-2,95 (d, 2H), 1,00-1,05 (t, 3H).
Соединение 112: 3-{4-[(4-{[(2Z)-2-(Метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}-3-фенилпропановая кислота
В 50 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 3 мл ТГФ. В перемешиваемый растворитель добавляли этил-3-{4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}-3-фенилпропаноат (0,6 г, 1,1 ммоль) и метанол (3 мл). PC охлаждали до 0°C и гидроксид натрия (0,09 г) в воде (1 мл) добавляли по каплям. Затем реакционную смесь перемешивали при КТ в течение 12 часов. Затем растворители выпаривали и неочищенное вещество растворяли в минимальном количестве воды (2 мл) и водный слой промывали диэтиловым эфиром (5 мл × 2). Водный слой подкисляли до pH 3 при помощи 1 н. хлороводородной кислоты. Водный слой экстрагировали этилацетатом (5 мл × 2). Органический слой промывали насыщенным солевым раствором (5 мл) и сушили над безводным Na2SO4. Растворитель удаляли при пониженном давлении, продукт получали в виде полутвердого вещества (0,2 г, выход 35,71%); чистота: 35,7%.
Схема 37
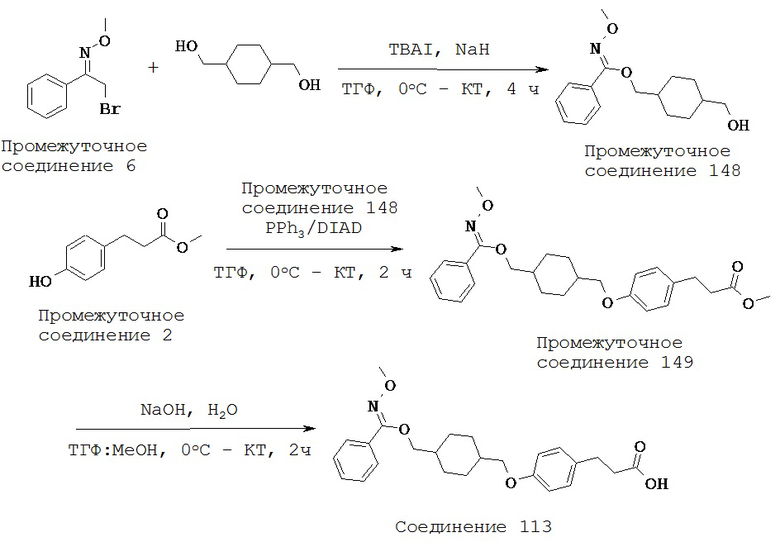
Пример 113
3-(4-{[4-({[(2E,2Z)-2-(Метоксиимино)-2-фенилэтил]окси}метил)циклогексил]метил}фенил)пропановая кислота (113)
Соединение 113 синтезировали из [4-({[(2E,2Z)-2-(метоксиимино)-2-фенилэтил]окси}-метил)циклогексил] метанола (0,7 г, 2,4 ммоль) и метил-3-(4-гидроксифенил)пропаноата (0,43 г, 2,4 ммоль) согласно способу, описанному на схеме 37 (0,4 г, выход 38,5%); чистота: 49,3% и 50,04%.
Промежуточное соединение 148: [4-({[(2E,2Z)-2-(метоксиимино)-2-фенилэтил]окси}метил)циклогексил]метанол
В 100 мл 2-горлую круглодонную колбу, содержащую суспензию NaH (0,33 г, 13,8 ммоль) в ТГФ (5 мл), в атмосфере азота добавляли циклогексан-1,4-диилдиметанол (1 г, 6,93 ммоль) в ТГФ по каплям при 0°C. Полученный раствор перемешивали при указанной температуре в течение 5 минут, затем добавляли (1Z)-2-бром-N-метокси-1-фенилэтанимин (1,72 г, 9,5 ммоль) и TBAI (0,128 г, 0,34 ммоль) медленно. Реакционную смесь перемешивали при КТ в течение 4 часов. Избыток NaH гасили льдом и перемешивали в течение 5 минут. Затем PC разбавляли этилацетатом (50 мл) и органический слой промывали водой (20 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Продукт получали в виде маслянистой жидкости (0,7 г, выход 35%); 1Н ЯМР (300 МГц, CDCl3): δ 7,59-7,93 (m, 2H), 7,27-7,29 (m, 3H), 4,57 (s, 2H), 3,92 (s, 3H), 3,34-3,41 (dd, 2H), 3,13-3,23 (dd, 2H), 1,19-1,40 (m, 8H).
Промежуточное соединение 149: метил-3-(4-{[4-({[2Е,2Z)-2-(метоксиимино)-2-фенилэтил]окси}метил)циклогексил]метокси}фенил)пропаноат
В 100 мл двухгорлую круглодонную колбу помещали [4-({[(2E,2Z)-2-(метоксиимино)-2-фенилэтил]окси}метил)циклогексил]метанол (0,7 г, 2,4 ммоль) и метил-3-(4-гидроксифенил)пропаноат (0,43 г, 2,4 ммоль) в 20 мл ТГФ. В полученную смесь добавляли трифенилфосфин (0,82 г, 3,12 ммоль) при 0°C и DIAD (0,63 г, 3,12 ммоль) по частям в течение 15 минут при 0°C. Через 12 часов перемешивания при КТ смесь разбавляли этилацетатом, промывали последовательно водой и солевым раствором. Органическую фазу сушили над безводным Na2SO4 и концентрировали с получением желтой маслянистой жидкости в качестве продукта (0,6 г, выход 55,17%); 1Н ЯМР (300 МГц, CDCl3): δ 7,60-7,62 (t, 2H), 7,19-7,29 (t, 3H), 7,02-7,04 (m, 2H), 6,70-6,74 (m, 2H), 4,57 (s, 2H), 3,92 (s, 3H), 3,59-3,69 (m, 5H), 3,14-3,25 (m, 2H), 2,81-2,82 (dd, 2H), 2,52-2,55 (dd, 2H), 1,19-1,54 (m, 8H).
Соединение 113: 3-(4-{[4-({[(2H,2Z)-2-(Метоксиимино)-2-фенилэтил]окси}метил)циклогексил]метил}фенил)пропановая кислота
В 50 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 3 мл ТГФ. В перемешиваемый растворитель добавляли метил-3-(4-{[4-({[2E,2Z)-2-(метоксиимино)-2-фенилэтил]окси}метил)циклогексил]метокси} фенил)пропаноат (0,6 г, 1,32 ммоль) и метанол (3 мл). PC охлаждали до 0°C и гидроксид натрия (0,09 г) в воде (1 мл) добавляли по каплям. Затем реакционную смесь перемешивали при КТ в течение 12 часов. Затем растворители выпаривали и неочищенное вещество растворяли в минимальном количестве воды (2 мл) и водный слой промывали диэтиловым эфиром (5 мл × 2). Водный слой подкисляли до pH 3 при помощи 1 н. хлороводородной кислоты. Водный слой экстрагировали этилацетатом (5 мл × 2). Органический слой промывали насыщенным солевым раствором (5 мл) и сушили над безводным Na2SO4. Растворитель удаляли при пониженном давлении, продукт получали в виде маслянистой жидкости (0,2 г, выход 70%); чистота: 49,3% и 50,04%.
Схема 38:
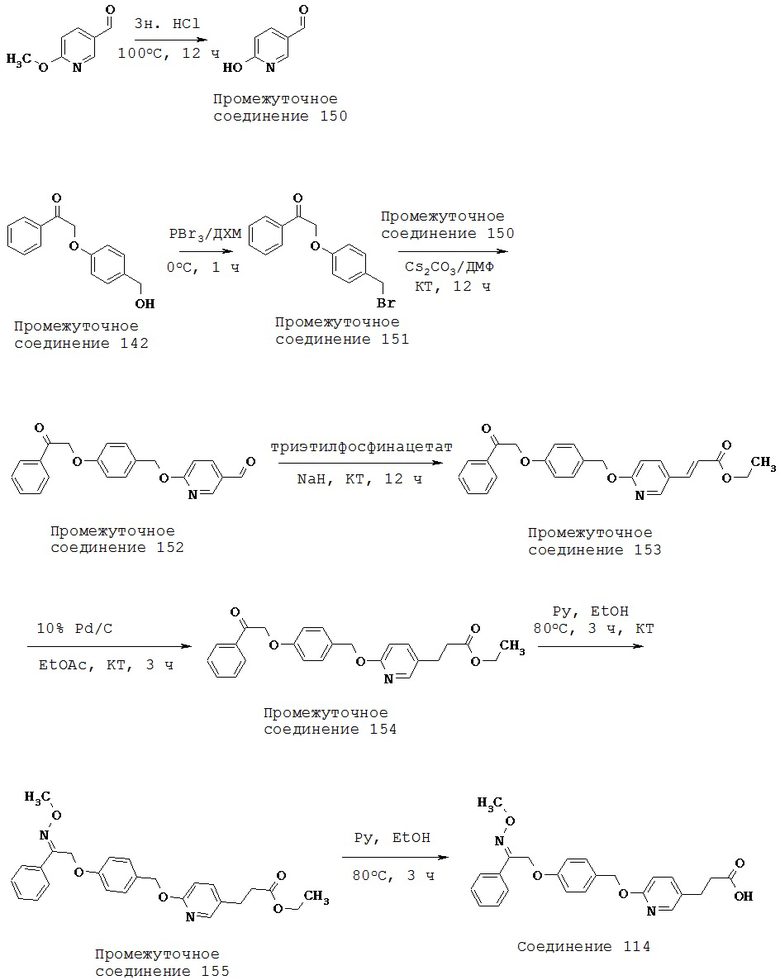
Пример 114:
3-{6-[(4-{[(2Z)-2-(Метоксиимино)-2-фенилэтил]окси}бензил)окси]пиридин-3-ил}пропановая кислота (114)
Соединение 114 синтезировали из 2-[4-(бромметил)фенокси]-1-фенилэтанона (0,61 г, 2 ммоль) и 6-гидроксипиридин-3-карбальдегида (0,25 г, 2 ммоль) согласно способу, описанному на схеме 38 (0,01 г, выход 21,8%); чистота: 85%.
Промежуточное соединение 150: 6-гидроксипиридин-3-карбальдегид
В 100 мл кругло донную колбу помещали 6-метоксипиридин-3-карбальдегид (1 г, 7 ммоль), добавляли 3 н. HCl (20 мл), а затем кипятили с обратным холодильником при 100°C в течение 12 часов. PC охлаждали медленно до КТ. Твердое вещество образовывалось при охлаждении, его отфильтровывали через воронку Бюхнера и сушили с использованием вакуума с получением титульного соединения в виде белых кристаллов (0,3 г, выход 37%).
Промежуточное соединение 151: 2-[4-(бромметил)фенокси]-1-фенилэтанон
В 50 мл кругло донную колбу, содержащую 2-[4-(гидроксиметил)фенокси]-1-фенилэтанон (10 г, 41,2 ммоль) в ДХМ, добавляли трибромид фосфора (16,76 г, 61,9 ммоль) по каплям в течение 5 минут при 0°C. Реакционную смесь перемешивали при КТ в течение 1 часа, затем реакцию гасили ледяной водой. Разбавляли избытком ДХМ, промывали последовательно водой, нас. раствором NaHCO3 и солевым раствором. Органическую фазу сушили над безводным Na2SO4 и концентрировали с получением продукта (9 г, выход 71,4%); 1Н ЯМР (300 МГц, ДМСО-d6): δ 7,90-7,93 (t, 2H), 7,53-7,58 (t, 1H), 7,41-7,46 (t, 2H), 7,23-7,26 (d, 2H), 6,81-6,84 (t, 2H), 5,20 (s, 2H), 4,40 (s, 2H).
Промежуточное соединение 152: 6-{[4-(2-оксо-2-фенилэтокси)бензил]окси}пиридин-3-карбальдегид
В раствор метил-6-гидроксипиридин-3-карбальдегида (0,25 г, 2 ммоль) в ДМФ (10 мл) добавляли карбонат цезия (0,97 г, 3 ммоль) и перемешивали при КТ в течение 30 минут, затем добавляли 2-[4-(бромметил)фенокси]-1-фенилэтанон (0,61 г, 2 ммоль), полученную массу перемешивали при 80°C в течение 12 часов, реакцию гасили водой и смесь экстрагировали этилацетатом. Органический слой промывали водой и солевым раствором, сушили над безводным Na2SO4 и концентрировали с получением неочищенного продукта, который очищали путем колоночной хроматографии с получением бледно-коричневого твердого вещества в качестве продукта (0,4 г, выход 60%); 1Н ЯМР (300 МГц, CDCl3): δ 9,59 (s, 1H) 7,95-8,02 (m, 2H), 7,67-7,78 (m, 3H), 7,54-7,59 (m, 2H), 7,30-7,33 (d, 2H), 6,93-6,96 (d, 2H), 6,49-6,53 (d, 1H), 5,57 (s, 2H), 5,11 (s, 2H).
Промежуточное соединение 153: этил-(2E)-3-(6-{[4-(2-оксо-2-фенилэтокси)бензил]окси}пиридин-3-ил)проп-2-еноат
В перемешиваемую суспензию NaH (0,025 г, 1 ммоль) в сухом ТГФ (10 мл) при 0°C добавляли триэтилфосфоноацетат (0,224 г, 1 ммоль) медленно и перемешивали в течение 30 минут, затем добавляли 6-{[4-(2-оксо-2-фенилэтокси)бензил]окси}пиридин-3-карбальдегид (0,4 г, 1 ммоль), растворенный в ТГФ, и перемешивали при КТ в течение 12 часов. Реакцию гасили льдом и смесь разбавляли этилацетатом, органический слой промывали водой и солевым раствором, сушили над безводным Na2SO4 и концентрировали. Неочищенный продукт очищали путем колоночной хроматографии с получением бледно-желтой маслянистой жидкости в качестве продукта (0,13 г, выход 31%); МС (ИЭР, 120 эВ): m/z=418 (М+Н)+.
Промежуточное соединение 154: этил-3-(6-{[4-(2-оксо-2-фенилэтокси)бензил]окси}пиридин-3-ил)пропаноат
Раствор этил-(2E)-3-(6-{[4-(2-оксо-2-фенилэтокси)бензил]окси}пиридин-3-ил)проп-2-еноата (0,13 г, 0,31 ммоль) в смеси этилацетат/этанол дегазировали, добавляли палладий на углеродной подложке (10 мл) и перемешивали в атмосфере водорода с применением диафрагмы в течение 3 часов. Реакционную массу фильтровали через подложку целита и концентрировали с получением бесцветной маслянистой жидкости (0,12 г, выход 95%); МС (ИЭР, 120 эВ): m/z=420 (М+Н)+.
Промежуточное соединение 155: этил-3-{6-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]пиридин-3-ил}пропаноат
В раствор этил-3-(6-{[4-(2-оксо-2-фенилэтокси)бензил]окси}пиридин-3-ил)пропаноата (0,13 г, 0,3 ммоль) в этаноле добавляли пиридин (0,038 г, 0,45 ммоль) и гидрохлорид O-метоксиламина, полученную массу кипятили с обратным холодильником при 80°C в течение 3 часов. Реакцию гасили водой и смесь экстрагировали этилацетатом, органический слой промывали водой и солевым раствором, сушили над безводным Na2SO4 и концентрировали с получением бесцветной маслянистой жидкости (0,03 г, выход 23%); МС (ИЭР, 120 эВ): m/z=449 (М+Н)+.
Соединение 114: 3-{6-[(4-{[(2Z)-2-(Метоксиимино)-2-фенилэтил]окси}бензил)окси]пиридин-3-ил}пропановая кислота
В перемешиваемый раствор этил-3-{6-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}-бензил)окси]пиридин-3-ил}пропаноата (0,03 г, 0,067 ммоль) в смеси ТГФ и этанола добавляли водный NaOH (0,03 г, 0,067 ммоль) при 0°C, затем перемешивали при КТ в течение 2 часов. После израсходования исходного вещества реакционную массу концентрировали в вакууме при 25°C для удаления растворителей и разбавляли водой. Водную фракцию нейтрализовали лимонной кислотой до нейтрального pH, затем экстрагировали этилацетатом, органический слой промывали водой и солевым раствором, сушили над Na2SO4 и концентрировали. Неочищенный продукт очищали путем препаративной ТСХ с получением бесцветной маслянистой жидкости в качестве продукта (0,01 г, выход 35%); чистота: 85%.
Схема 39:
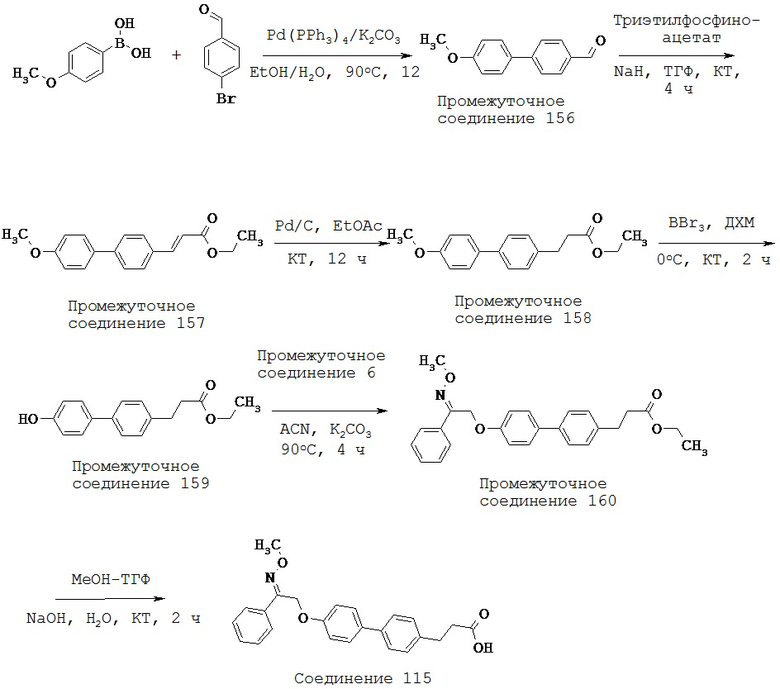
Пример 115
3-(4'-{[(2Z)-2-(Метоксиимино)-2-фенилэтил]окси}бифен-4-ил)пропановая кислота (115)
Соединение 115 синтезировали из этил-3-(4'-гидроксибифен-4-ил)пропаноата (0,27 г, 1 ммоль) и (1Z)-2-бром-N-метокси-1-фенилэтанимина (0,23 г, 1 ммоль) согласно способу, описанному на схеме 39 (0,2 г, 49,8%); чистота: 99,5%.
Промежуточное соединение 156: 4'-метоксибифенил-4-карбальдегид
В раствор (4-метоксифенил)бороновой кислоты (1 г, 6,6 ммоль) и 4-бромбензальдегида (1,2 г, 3,5 ммоль) в смеси этанол/вода добавляли карбонат калия (1,7 г, 12,0 ммоль), дегазировали и добавляли тетракис(трифенилфосфин)палладий (0) (0,7 г, 0,6 ммоль) и перемешивали в атмосфере аргона в течение 12 часов, реакционную массу разбавляли водой и экстрагировали этилацетатом, органический слой промывали водой и солевым раствором, сушили над безводным Na2SO4 и концентрировали с получением коричневого твердого вещества в качестве продукта (1 г, выход 71,4%). 1Н ЯМР (300 МГц, CDCl3): δ 9,96 (s, 1H), 7,84 (d, 2H), 7,63-7,66 (d, 2H), 7,51-7,54 (s, 2H), 6,93-6,95 (d, 2H), 3,80 (s, 3H).
Промежуточное соединение 157: этил-(2Z)-3-(4'-метоксибифен-4-ил)проп-2-еноат
В перемешиваемую суспензию NaH (0,14 г, 5,8 ммоль) в сухом ТГФ (10 мл) при 0°C добавляли триэтилфосфоноацетат (1,3 г, 5,8 ммоль) медленно и перемешивали в течение 30 минут, затем добавляли 4'-метоксибифенил-4-карбальдегид (1 г, 4,7 ммоль), растворенный в ТГФ, и перемешивали при КТ в течение ночи. Реакцию гасили льдом и смесь разбавляли этилацетатом, органический слой промывали водой и солевым раствором, сушили над безводным сульфатом натрия и концентрировали с получением титульного соединения в виде белого твердого вещества (1 г, выход 74,4%); 1Н ЯМР (300 МГц, CDCl3): δ 7,47-7,61 (m, 8H), 6,90-6,93 (d, 2H), 4,17-4,24 (m, 2H), 3,78 (s, 3H), 1,25-1,30 (t, 3H).
Промежуточное соединение 158: этил-3-(4'-метоксибифен-4-ил)пропаноат
В раствор этил-(2E)-3-(4'-метоксибифен-4-ил)проп-2-еноата (1 г, 3,5 ммоль) в этилацетате добавляли уксусную кислоту, дегазировали аргоном и добавляли палладий на углеродной подложке (100 мг), перемешивали в атмосфере водорода в течение ночи, анализ ТСХ показывал образование нового пятна, PC фильтровали через целит, промывали этилацетатом и концентрировали с получением белого твердого вещества (0,7 г, выход 71,4%).
Промежуточное соединение 159: этил-3-(4'-гидроксибифен-4-ил)пропаноат
В раствор этил-3-(4'-метоксибифен-4-ил)пропаноата (0,35 г, 1,2 ммоль) в дихлорметане добавляли трибромид бора (0,4 г, 1,6 ммоль) при 0°C и перемешивали при КТ в течение 30 минут. Реакцию гасили этанолом и разбавляли водой, экстрагировали дихлорметаном, органический слой промывали NaHCO3, водой и солевым раствором и сушили над сульфатом натрия, затем концентрировали с получением белого твердого вещества (0,27 г, выход 83,3%); 1H ЯМР (300 МГц, CDCl3): δ 9,49 (s, 1H), 7,43-7,48 (t, 4H), 7,23-7,26 (d, 2H), 6,81-6,84 (d, 2H), 4,01-4,08 (m, 2H), 2,83-2,88 (t, 2H), 2,59-2,64 (t, 2H), 1,13-1,18 (t, 3H).
Промежуточное соединение 160: этил-3-(4'-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бифен-4-ил)пропаноат
В раствор этил-3-(4'-гидроксибифен-4-ил)пропаноата (0,27 г, 0,001 ммоль) в ацетонитриле добавляли карбонат калия (0,27 г, 0,002 ммоль) и (1Z)-2-бром-N-метокси-1-фенилэтанимин (0,23 г, 0,001 ммоль) и кипятили с обратным холодильником при 80°C в течение 3 часов. PC разбавляли водой и экстрагировали этилацетатом. Органический слой промывали водой и солевым раствором, сушили над безводным Na2SO4 и концентрировали с получением неочищенного продукта, который очищали путем флэш-хроматографии с получением белого вязкого твердого вещества в качестве продукта (0,25 г, выход 60%); МС (ИЭР, 120 эВ): m/z=418 (М+Н)+; 1H ЯМР (300 МГц, CDCl3): δ 7,59-7,63 (m, 2H), 7,37-7,42 (m, 4H), 7,26-7,28 (m, 3H), 7,15 (d, 2H), 6,88-6,90 (d, 2H), 5,15 (s, 2H), 4,03-4,10 (m, 2H), 3,99 (s, 3H), 2,87-2,93 (t, 2H), 2,54-2,60 (t, 2H), 1,17-1,19 (t, 3H).
Соединение 115: 3-(4'-{[(2Z)-2-(Метоксиимино)-2-фенилэтил]окси}бифен-4-ил)пропановая кислота
В раствор этил-3-(4'-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бифен-4-ил)пропаноата (0,25 г, 0,6 ммоль) в ТГФ добавляли этанол и водный раствор NaOH и перемешивали при КТ в течение 2 часов, реакционную массу концентрировали для удаления растворителей и разбавляли водой, нейтрализовали 1 н. HCl и экстрагировали этилацетатом, органический слой промывали водой и солевым раствором, сушили над безводным Na2SO4 и концентрировали с получением белого твердого вещества (0,2 г, 83,3%); чистота: 99,5%.
Схема 40:
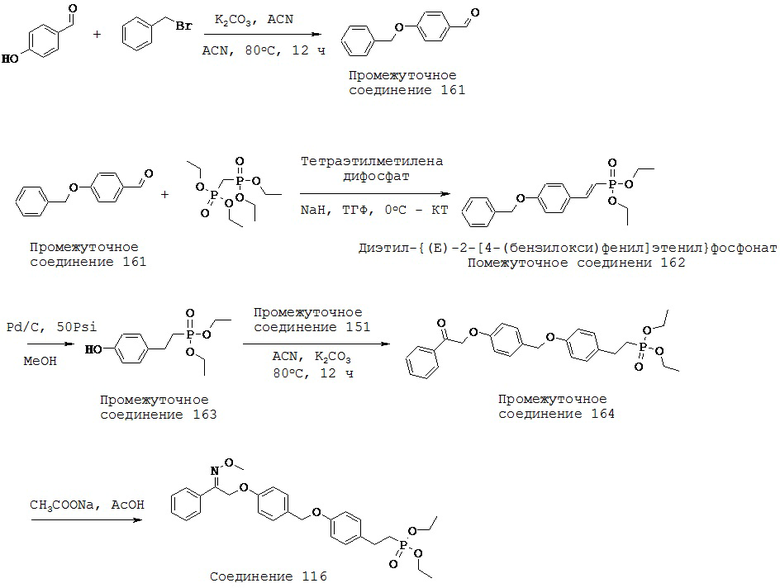
Пример 116:
Диэтил-(2-{4-[(4-{[(2E,2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}этил)фосфонат (116)
Соединение 116 синтезировали из 2-[4-(бромметил)фенокси]-1-фенилэтанона (0,2 г, 0,657 ммоль) и диметил-[2-(4-гидроксифенил)этил]фосфоната (0,17 г, 0,657 ммоль) согласно способу, описанному на схеме 40 (0,006 г, выход 12,6%); чистота: 79,9% и 12,52%.
Промежуточное соединение 161: 4-(бензилокси)бензальдегид
В 100 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 80 мл ацетонитрила. В перемешиваемый растворитель добавляли 4-гидроксибензальдегид (4 г, 33 ммоль) и K2CO3 (9 г, 65 ммоль). Затем смесь перемешивали в течение 5 минут. Бензилбромид (6,7 г, 39 ммоль) добавляли. Затем PC нагревали при 80°C в течение 2 часов. После завершения реакции (прохождение отслеживали путем ТСХ) PC концентрировали в вакууме для удаления ацетонитрила. В остаток затем добавляли 100 мл воды и экстрагировали этилацетатом (150 мл × 2). Органический слой промывали насыщенным солевым раствором (50 мл), сушили над безводным Na2SO4 и выпаривали насухо с получением титульного соединения (4,5 г, 64,75%).
Промежуточное соединение 162: диэтил-{(E)-2-[4-(бензилокси)фенил]этенил}фосфонат
В 100 мл 2-горлую круглодонную колбу, содержащую суспензию NaH (0,14 г, 5,67 ммоль) в ТГФ (50 мл), в атмосфере азота добавляли тетраэтилметандиилбис(фосфонат) (1,63 г, 5,67 ммоль) в ТГФ по каплям при 0°C. Полученный раствор перемешивали при указанной температуре в течение 5 минут, затем добавляли 4-(бензилокси)бензальдегид медленно. Реакционную смесь перемешивали при КТ в течение 4 часов. Избыток NaH гасили льдом и перемешивали в течение 5 минут. Затем PC разбавляли этилацетатом (50 мл) и органический слой промывали водой (20 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Продукт получали в виде маслянистой жидкости (1,6 г, выход 98%). 1H ЯМР (300 МГц, CDCl3): δ 7,28-7,45 (m, 8H), 6,89-7,23 (d, 2H), 5,96-6,08 (t, 1H), 5,03 (s, 2H), 4,0-4,1 (m, 3H), 1,26-1,30 (t, 3H).
Промежуточное соединение 163: диэтил-[2-(4-гидроксифенил)этил]фосфонат
500 мл колбу для встряхивателя Парра, содержащую диэтил-{(E)-2-[4-(бензилокси)-фенил]этенил}фосфонат (1,6 г, 4,62 ммоль) в MeOH, дегазировали азотом в течение 2 минут. Добавляли 10% Pd/C (0,5 г, 10%) и подводили Н2 под давлением 50 psi в течение 3 часов. Реакционную смесь фильтровали через подложку целита и промывали подложку избытком MeOH. Органическую фазу сушили над сульфатом натрия и концентрировали с получением маслянистой жидкости в качестве продукта (1,01 г, выход 84,16%).
Промежуточное соединение 164: диэтил-[2-(4-{[4-(2-оксо-2-фенилэтокси)бензил]окси}фенил)этил]фосфонат
В 50 мл кругло донную колбу, оборудованную магнитной мешалкой, помещали 10 мл ацетонитрила. В перемешиваемый растворитель добавляли диэтил-[2-(4-гидроксифенил)-этил]фосфонат (0,17 г, 0,66 ммоль) и K2CO3 (0,27 г, 1,97 ммоль). Затем смесь перемешивали в течение 5 минут. 2-[4-(Бромметил)фенокси]-1-фенилэтанон (0,2 г, 0,66 ммоль) добавляли. Затем PC нагревали при 80°C в течение 2 часов. После завершения реакции (прохождение отслеживали путем ТСХ) PC концентрировали в вакууме для удаления ацетонитрила. В остаток затем добавляли 100 мл воды и экстрагировали этилацетатом (30 мл × 2). Органический слой промывали насыщенным солевым раствором (20 мл), сушили над безводным Na2SO4 и выпаривали насухо с получением титульного соединения (0,14 г, 45,2%); 1Н ЯМР (300 МГц, ДМСО-d6): δ 8,01-8,04 (d, 2H), 7,68-7,70 (t, 1H), 7,55-7,60 (d, 2H), 7,33-7,36 (d, 2H), 7,14-7,17 (d, 2H), 6,96-6,98 (d, 2H), 6,89-6,92 (d, 2H), 5,59 (s, 2H), 4,98 (s, 2H), 3,92-4,04 (m, 4H), 2,66-2,75 (m, 2H), 1,94-2,05 (m, 2H), 1,18-1,91 (m, 6H).
Соединение 116: Диэтил-(2-{4-[(4-{[(2E,2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}этил)фосфонат
В 50 мл одногорлую круглодонную колбу, содеражщую диэтил-[2-(4-{[4-(2-оксо-2-фенилэтокси)бензил]окси}фенил)этил]фосфонат (0,02 г, 0,041 ммоль) в уксусной кислоте (2 мл) добавляли гидрохлорид O-метоксиламина (0,007 г, 0,062 ммоль), затем ацетат натрия (0,005 г, 0,083 ммоль). Реакционную смесь перемешивали при КТ в атмосфере азота в течение 3 часов. Затем PC разбавляли этилацетатом (15 мл) и органический слой промывали нас. раствором NaHCO3 (5 мл) и водой (5 мл). Органический слой сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Продукт получали в виде маслянистой жидкости (0,006 г, выход 28,57%); МС (ИЭР, 120 эВ): m/z=512 (М+Н)+; чистота: 79,9% и 12,52%.
Схема 41
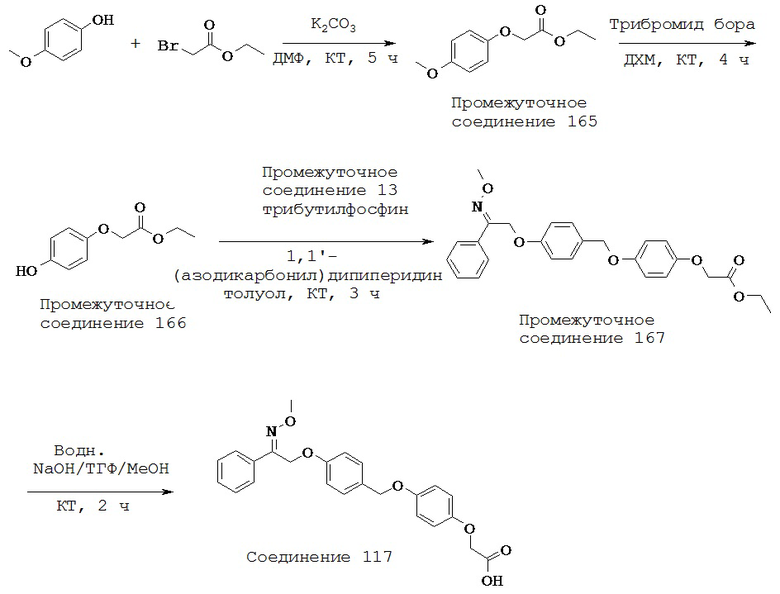
Пример 117
{4-[(4-{[(2Z)-2-(Метоксиимино)-2-фенилэтил]окси}бензил)окси]фенокси}уксусная кислота (117)
Соединение 117 синтезировали из промежуточного соединения 13 (0,22 г, 1,020 ммоль) и этил-(4-гидроксифенокси)ацетата (0,20 г, 1,020 ммоль) согласно способу, описанному на схеме 41 (0,060 г, выход 13,72%); чистота: 97,98%.
Промежуточное соединение 165: этил-(4-метоксифенокси)ацетат
В 100 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 12 мл ДМФ. В перемешиваемый растворитель добавляли K2CO3 (2,2 г, 16 ммоль), 4-метоксифенол (1 г, 8 ммоль) и этилбромацетат (1,1 мл, 9,6 ммоль). Затем реакционную смесь перемешивали при КТ в течение 5 часов. После завершения реакции (прохождение отслеживали путем ТСХ) реакционную смесь концентрировали и экстрагировали этилацетатом. Затем органический слой сушили над безводным Na2SO4. Растворитель удаляли при пониженном давлении с получением соединения (2 г, выход 90%).
Промежуточное соединение 166: этил-(4-гидроксифенокси)ацетат
В 100 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 20 мл ДХМ, в перемешиваемый растворитель добавляли этил-(4-метоксифенокси)ацетат (2 г, 9,5 ммоль) и трибромид бора (1,85 г, 11,0 ммоль) добавляли при 0°C. Затем смесь перемешивали при 0°C в течение 30-45 минут. После завершения (прохождение реакции отслеживали путем ТСХ) реакцию гасили бикарбонатом натрия и концентрировали реакционную смесь, экстрагировали этилацетатом. Органический слой промывали солевым раствором и сушили над безводным Na2SO4 и выпаривали насухо, полученное неочищенное соединение очищали путем колоночной хроматографии на силикагеле с применением петролейного эфира и этилацетата в качестве элюентов с получением продукта (0,2 г, выход 20%); 1Н ЯМР (300 МГц, ДМСО-d6): δ 8,97 (s, 1Н), 6,72-6,75 (d, 2H), 6,64-6,67 (d, 2H), 4,62 (s, 2H) 4,11-4,18 (q, 2H), 1,18-1,22 (t, 3H).
Промежуточное соединение 167: этил-{4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенокси}ацетат
В 50 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 10 мл толуола. В перемешиваемый растворитель добавляли этил-(4-гидроксифенокси)ацетат (0,2 г, 1,030 ммоль) и (4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}фенил)метанол (0,22 г, 0,810 ммоль), затем добавляли трибутилфосфин (0,268 г, 1,32 ммоль) и 1,1'-(азодикарбонил)-дипиперидин (0,33 г, 1,326 ммоль). Затем реакционную смесь перемешивали при КТ в течение 3 часов. После завершения реакции (прохождение отслеживали путем ТСХ) реакционную смесь экстрагировали этилацетатом. Затем органический слой сушили над безводным Na2SO4. Растворитель удаляли при пониженном давлении и полученное неочищенное соединение очищали путем колоночной хроматографии на силикагеле с применением петролейного эфира и этилацетата в качестве элюентов (0,13 г, выход 28,3%); 1Н ЯМР (300 МГц, CDCl3): δ 7,57-7,60 (m, 2H), 7,18-7,28 (m, 5H), 6,82-6,84 (d, 3H), 6,78-6,79 (d, 3H), 5,12 (s, 2H), 4,83 (s, 2H), 4,49 (s, 2H), 4,15-4,22 (m, 2H), 3,98 (s, 3H), 1,19-1,24 (t, 3H).
Соединение 117: {4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенокси}уксусная кислота
В 25 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 3 мл этанола и 3 мл ТГФ. В перемешиваемый растворитель добавляли этил-{4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенокси}ацетат (0,13 г, 1,289 ммоль) и NaOH, после чего добавляли воду (1 мл) при 0°C. Затем смесь перемешивали при КТ в течение 5 часов. После завершения реакции (прохождение отслеживали путем ТСХ) реакционную смесь полностью выпаривали и неочищенное вещество промывали диэтиловым эфиром и разбавляли водой, затем нейтрализовали 1 н. HCl, затем экстрагировали этилацетатом, органический слой промывали водой и солевым раствором, сушили над сульфатом натрия. Органический слой выпаривали на роторном испарителе с получением продукта (0,06 г, выход 49,5%); чистота: 97,98%.
Схема 42
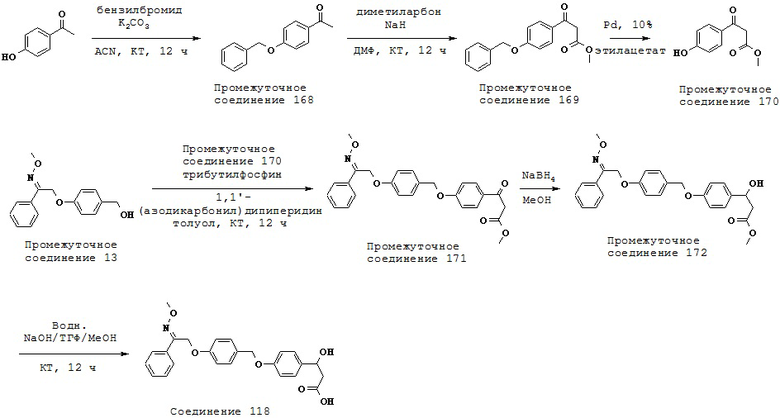
Пример 118
3-Гидрокси-3-{4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропановая кислота (118)
Соединение 118 синтезировали из метил-3-гидрокси-3-{4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропаноата согласно способу, описанному на схеме 42 (0,02 г, выход 26,5%); чистота: 45,3% и 43,6%.
Промежуточное соединение 168: 1-[4-(бензилокси)фенил]этанон
В перемешиваемый раствор 1-(4-гидроксифенил)этанона (5 г, 36 ммоль) в ацетонитриле (50 мл) добавляли карбонат калия (9,9 г, 72 ммоль) и бензилбромид (7,5 г, 44 ммоль) и перемешивали при КТ в течение ночи; реакционную массу разбавляли водой и экстрагировали этилацетатом, органический слой промывали водой и солевым раствором, сушили над Na2SO4, выпаривание при пониженном давлении приводило к получению титульного соединения в виде белого твердого вещества (7 г, выход 84,1%); 1Н ЯМР (300 МГц, CDCl3): δ 7,85-7,88 (d, 2H), 7,25-7,37 (m, 5H), 6,92-6,95 (d, 2H), 5,06 (s, 2H), 2,48 (s, 3H).
Промежуточное соединение 169: метил-3-[4-(бензилокси)фенил]-3-оксопропаноат
В перемешиваемую суспензию NaH (3,7 г, 154,2 ммоль) в ДМФ (25 мл) добавляли 1-[4-(бензилокси)фенил]этанон (7 г, 31 ммоль) при -5°C и перемешивали в течение 30 минут, затем диметилкарбонат (13,5 г, 150 ммоль, растворенный в ДМФ) добавляли при -5°C медленно (выделение тепла) и нагревали медленно до КТ, перемешивали при КТ в течение ночи. Реакцию гасили ледяной водой и экстрагировали этилацетатом, органический слой промывали водой и солевым раствором, сушили над Na2SO4. Органический слой концентрировали с получением продукта (7 г, выход 92%); 1Н ЯМР (300 МГц, CDCl3): δ 7,84-7,87 (d, 2H), 7,28-7,35 (m, 5H), 6,94-6,96 (d, 2H), 5,07 (s, 2H), 3,89 (s, 2h), 3,68 (s, 3H).
Промежуточное соединение 170: метил-3-(4-гидроксифенил)-3-оксопропаноат
В 50 мл кругло донную колбу, оборудованную магнитной мешалкой, помещали этилацетат (15 мл), в перемешиваемый растворитель добавляли метил-3-[4-(бензилокси)-фенил]-3-оксопропаноат (0,3 г, 1,05 ммоль) и 10% палладий на углеродной подложке (80 мг). После завершения добавления баллон водорода соединяли с сосудом с реакционной смесью и перемешивали при комнатной температуре в течение 1 часа. Через 3 часа реакционную смесь фильтровали через целит. Затем органический слой концентрировали и сушили. Продукт получали в виде бесцветной жидкости (0,2 г, выход 98%).
Промежуточное соединение 171: метил-3-{4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}-3-оксопропаноат
В 50 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 10 мл толуола, в перемешиваемый растворитель добавляли метил-3-(4-гидроксифенил)-3-оксопропаноат (0,2 г, 1,036 ммоль) и (4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}фенил)-метанол (0,22 г, 0,82 ммоль), затем трибутилфосфин (0,271 г, 1,324 ммоль) и 1,1'-(азодикарбонил)дипиперидин по частям при 0°C (0,339 г, 1,346 ммоль). Затем реакционную смесь перемешивали при КТ в течение 12 часов. После завершения реакции (прохождение отслеживали путем ТСХ) реакционную смесь концентрировали и экстрагировали этилацетатом. Затем органический слой сушили над безводным Na2SO4. Растворитель удаляли при пониженном давлении и полученное неочищенное соединение очищали путем колоночной хроматографии на силикагеле с применением петролейного эфира и этилацетата в качестве элюентов (0,05 г, выход 14%); МС (ИЭР, 120 эВ): m/z=448,1 (М+H)+.
Промежуточное соединение 172: метил-3-гидрокси-3-{4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропаноат
В 25 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 4 мл МеОН, в перемешиваемый растворитель добавляли метил-3-{4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}-3-оксопропаноат (0,08 г, 0,178 ммоль) и боргидрид натрия (0,008 г, 1,21 ммоль) при 0°C. Затем смесь перемешивали при КТ в течение 6 часов, после завершения (прохождение реакции отслеживали путем ТСХ) реакцию гасили ледяной водой, концентрировали реакционную смесь и экстрагировали этилацетатом. Органический слой промывали солевым раствором, сушили над безводным Na2SO4 и выпаривали насухо, полученное неочищенное соединение очищали путем колоночной хроматографии на силикагеле с применением петролейного эфира и этилацетата в качестве элюентов (0,04 г, выход 50%); 1Н ЯМР (300 МГц, ДМСО-d6): δ 7,62-7,65 (m, 2H), 7,32-7,40 (m, 5H), 7,23-7,26 (d, 2H), 6,90-6,93 (m, 4H), 5,36-5,38 (d, 1H) 5,22 (s, 2H), 4,92 (s, 2H), 4,85-4,89 (t, 1H), 4,04 (s, 3H), 3,57 (s, 3H), 2,57-2,59 (d, 2H).
Соединение 118: 3-Гидрокси-3-{4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропановая кислота
В 25 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 2 мл метанола и 2 мл ТГФ. В перемешиваемый растворитель добавляли метил-3-гидрокси-3-{4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропаноат (0,04 г, 0,08 ммоль) и NaOH, затем воду (0,5 мл). Затем смесь перемешивали при КТ в течение 12 часов, после завершения реакции (прохождение отслеживали путем ТСХ) реакционную смесь полностью выпаривали и неочищенное вещество промывали диэтиловым эфиром и разбавляли водой, а затем нейтрализовали 1 н. HCl, затем экстрагировали этилацетатом, органический слой промывали водой и солевым раствором, сушили над Na2SO4. Органический слой выпаривали на роторном испарителе при пониженном давлении с получением продукта в виде белого твердого вещества (0,02 г, выход 52,6%); чистота: 45,3% и 43,6%.
Схема 43
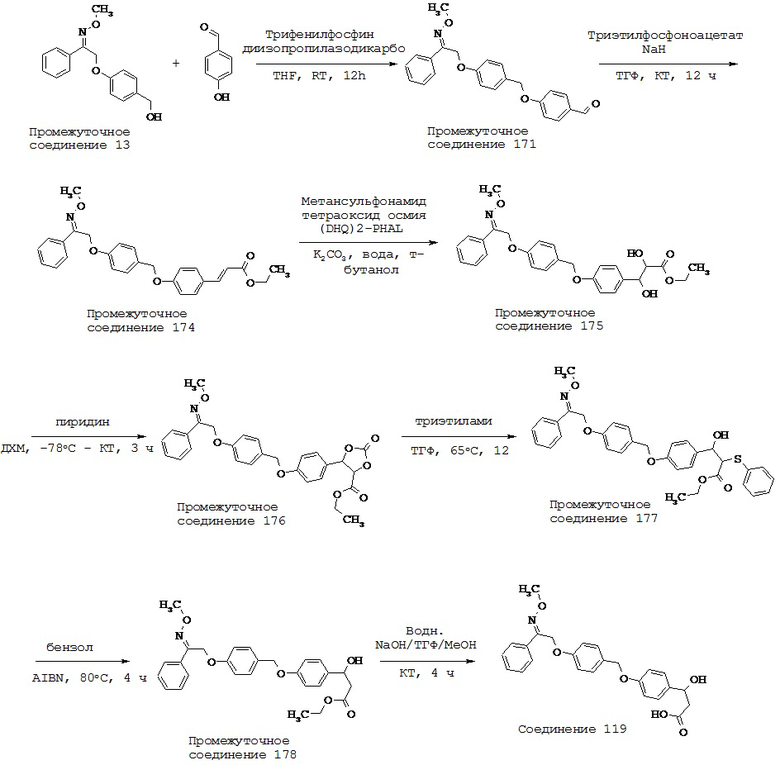
Пример 119
3-Гидрокси-3-{4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропановая кислота (119)
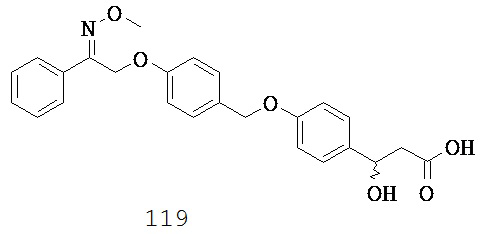
Соединение 119 синтезировали из этил-3-гидрокси-3-{4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропаноата согласно способу, описанному на схеме 43 (0,008 г, выход 5,1%); чистота: 96,8%.
Промежуточное соединение 173: 4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]бензальдегид
В перемешиваемый раствор (4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}фенил)-метанола (10 г, 37 ммоль) и 4-гидроксибензальдегида (4,5 г, 37 ммоль) в ТГФ (100 мл) добавляли трифенилфосфин (12,2 г, 46,5 ммоль) и диизопропилазодикарбоксилат (11,2 г, 55 ммоль) при 0°C, затем перемешивали при КТ в течение 12 часов. PC разбавляли водой и экстрагировали этилацетатом. Органический слой промывали солевым раствором, сушили над Na2SO4 и концентрировали с получением бледно-коричневой маслянистой жидкости. Неочищенное вещество очищали на колонке (оксид кремния 100-200 меш) с применением 8% этилацетата с получением титульного соединения в виде белого твердого вещества (5 г, выход 36,2%). 1Н ЯМР (300 МГц, CDCl3): δ 9,81 (s, 1H), 7,74-7,77 (d, 2H), 7,58-7,61 (m, 2H), 7,24- 7,29 (m, 5H), 6,97-6,99 (d, 2H), 6,85-6,87 (d, 2H), 5,13 (s, 2H), 4,98 (s, 2H), 3,98 (s, 3H).
Промежуточное соединение 174: этил-(2E)-3-{4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}проп-2-еноат
В перемешиваемую суспензию NaH (0,63 г, 26 ммоль) в сухом ТГФ (25 мл) при 0°C добавляли триэтилфосфоноацетат (3,8 г, 16,9 ммоль) медленно и перемешивали в течение 30 минут, затем добавляли 4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]-бензальдегид (5 г, 13 ммоль), растворенный в ТГФ, и перемешивали при КТ в течение 12 часов. Реакцию гасили ледяной 1 н. HCl и смесь разбавляли этилацетатом, органический слой промывали водой и солевым раствором, сушили над сульфатом натрия и концентрировали (Примечание: во время реакции также проходил гидролиз, и полученную кислоту превращали в сложный эфир с применением метанола и метансульфокислоты) (5 г, выход 83,7%). 1H ЯМР (300 МГц, CDCl3): δ 7,54-7,60 (m, 3H), 7,38-7,41 (d, 2H), 7,23-7,29 (m, 4H), 6,83-6,89 (m, 4H), 6,21-6,26 (d, 1H), 5,13 (s, 2H), 4,92 (s, 2H), 3,98 (s, 3H), 3,7 (s, 3H).
Промежуточное соединение 175: этил-2,3-дигидрокси-3-{4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропаноат
В 500 мл круглодонную колбу помещали феррицианид калия (4,7 г, 14 ммоль), карбонат калия (1,98 г, 14 ммоль), (DHQ)2PHAL (0,075 г, 0,09 ммоль) и метансульфонамид (0,45 г, 4,8 ммоль) в 1:1 смеси т-бутанола и воды и перемешивали при КТ в течение 5 минут, затем охлаждали до 0°C, затем добавляли тетраоксид осмия (25 мг, растворенные в 0,5 мл толуола) и этил-(2E)-3-{4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]-фенил}проп-2-еноат (2 г, 4,8 ммоль), после чего перемешивали при КТ в течение 12 часов. Реакцию гасили сульфитом натрия и массу экстрагировали этилацетатом, органический слой промывали водой и солевым раствором, сушили над сульфатом натрия и концентрировали с получением бледно-коричневой маслянистой жидкости, которую очищали на колонке (оксид кремния, 60-120 меш) с применением 40% этилацетата в петролейном эфире с получением титульного соединения в виде белого твердого вещества (1,3 г, выход 60,7%). 1Н ЯМР (300 МГц, CDCl3): δ 7,62-7,64 (m, 2H), 7,32-7,39 (m, 5H), 7,21-7,24 (d, 2H), 6,9-6,91 (m, 4H), 5,38-5,40 (d, 1H), 5,30-5,33 (d, 1H), 5,22 (s, 2H), 4,97 (s, 2H), 4,72-4,75 (t, 1H), 4,05-4,08 (m, 1H), 3,99 (s, 3Н), 3,55 (s, 3H).
Промежуточное соединение 176: этил-5-{4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}-2-оксо-1,3-диоксолан-4-карбоксилат
В перемешиваемый раствор этил-2,3-дигидрокси-3-{4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропаноата (1,3 г, 2,8 ммоль) в дихлорметане (20 мл) добавляли пиридин (1,4 г, 17,7 ммоль) при -78°C, затем раствор трифосгена (0,44 г, 15 ммоль) в дихлорметане и постепенно нагревали до КТ, затем перемешивали при КТ в течение 4 часов. Реакцию гасили 1 н. HCl и смесь экстрагировали дихлорметаном. Органический слой промывали водой, NaHCO3 и солевым раствором, сушили над безводным Na2SO4 и концентрировали с получением бесцветной маслянистой жидкости, которую очищали на колонке (оксид кремния, 60-120 меш) с применением 20% этилацетата в петролейном эфире с получением титульного соединения в виде бесцветной маслянистой жидкости (0,8 г, выход 58%). 1H ЯМР (300 МГц, CDCl3): δ 7,62-7,64 (m, 2H), 7,34-7,45 (m, 8H), 7,06-7,09 (d, 2H), 6,91-6,94 (d, 2H), 5,88-5,91 (d, 1H), 5,41-5,44 (d, 1H), 5,23 (s, 2H), 5,04 (s, 2H), 3,99 (s, 3H), 3,76 (s, 3H).
Промежуточное соединение 177: этил-3-гидрокси-3-{4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси] фенил}-2-(фенилсульфанил)пропаноат
В раствор этил-5-{4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]-фенил}-2-оксо-1,3-диоксолан-4-карбоксилата (0,8 г, 1,6 ммоль) в ТГФ добавляли триэтиламин (0,32 г, 3,2 ммоль) и тиофенол (0,34 г, 3,2 ммоль) при 0°C в атмосфере аргона, затем медленно нагревали до КТ и продолжали перемешивание при 60°C в течение 12 часов. Реакцию гасили водой и массу экстрагировали этилацетатом, органический слой промывали водой и солевым раствором, сушили над сульфатом натрия и концентрировали с получением желтой маслянистой жидкости, которую дополнительно очищали на колонке (оксид кремния, 60-120 меш) с применением 20% этилацетата в петролейном эфире с получением титульного соединения в виде желтой маслянистой жидкости (0,55 г, выход 61%). 1Н ЯМР (300 МГц, ДМСО): δ 7,62-7,65 (m, 2H), 7,27-7,40 (m, 8H), 7,22-7,24 (m, 2H), 7,11-7,14 (m, 2H), 6,90-6,93 (d, 4H), 5,21 (s, 2H), 5,01 (s, 2H), 4,63-4,73 (m, 1H), 4,01 (s, 3H), 3,89-3,91 (d, 1H), 3,59 (s, 3H).
Промежуточное соединение 178: этил-3-гидрокси-3-{4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропаноат
В раствор этил-3-гидрокси-3-{4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}-бензил)окси]фенил}-2-(фенилсульфанил)пропаноата (0,05 г, 0,09 ммоль) в бензоле добавляли AIBN (каталитическое количество) и гидрид трибутилолова, полученную массу кипятили с обратным холодильником при 80°С в течение 4 часов. Реакцию гасили водой, смесь экстрагировали этилацетатом. Органический слой промывали водой и солевым раствором, сушили над безводным Na2SO4 и концентрировали с получением желтой маслянистой жидкости, которую дополнительно очищали путем препаративной ТСХ с применением 10% этилацетата в хлороформе с получением титульного соединения в виде желтой маслянистой жидкости (0,015 г, выход 37,5%). 1Н ЯМР (300 МГц, CDCl3): δ 7,63-7,65 (m, 2H), 7,32-740 (m, 5H), 7,23-7,26 (d, 2H), 6,9-6,93 (m, 4H), 5,37-5,38 (d, 1H), 5,22 (s, 2H), 4,98 (s, 2H), 4,85-4,92 (t, 1H), 3,99 (s, 3H), 3,57 (s, 3H), 3,15-3,17 (d, 1H), 2,57-2,59 (d, 2H).
Соединение 119: 3-Гидрокси-3-{4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропановая кислота
В перемешиваемый раствор этил-3-гидрокси-3-{4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропаноата (0,015 г, 0,0334 ммоль) в смеси ТГФ и этанола добавляли NaOH (0,003 г, 0,075 ммоль) в 0,5 мл воды при 0°С, затем перемешивали при КТ в течение 2 часов. После израсходования исходного вещества реакционную массу концентрировали в вакууме для удаления растворителей и разбавляли водой. Водную фракцию нейтрализовали 1 н. HCl до нейтрального pH, затем экстрагировали этилацетатом, органический слой промывали водой и солевым раствором, сушили над безводным Na2SO4. Выпаривание в вакууме приводило к получению титульного соединения в виде белого твердого вещества (0,008 г, выход 13,70%); чистота: 96,8%.
Схема 44
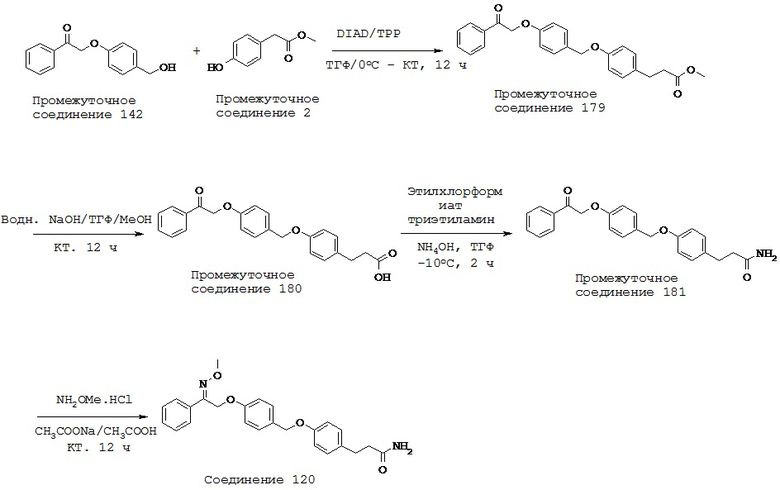
Пример 120
3-{4-[(4-{[(2Z)-2-(Метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропанамид (120)
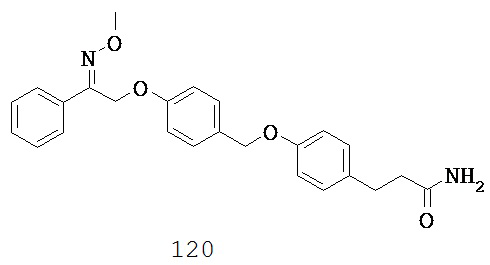
Соединение 120 синтезировали из 3-(4-{[4-(2-оксо-2-фенилэтокси)бензил]окси}фенил)-пропанамида согласно способу, описанному на схеме 44 (0,02 г, выход 19,8%); чистота: 91,0%.
Промежуточное соединение 179: метил-3-(4-{[4-(2-оксо-2-фенилэтокси)бензил]окси}фенил)пропаноат
В 250 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали сухой ТГФ, добавляли 2-[4-(гидроксиметил)фенокси]-1-фенилэтанон (10,5 г, 43 ммоль) и метил-3-(4-гидроксифенил)пропаноат (10,1 г, 56 ммоль), перемешивали при 0°C в течение 5 минут, затем добавляли трифенилфосфин (18,1 г, 39 ммоль), перемешивали при 0°C в течение 15 минут, затем добавляли диэтилазадикарбоксилат (14 г, 69 ммоль), полученную массу перемешивали при КТ в течение ночи, PC выпаривали для удаления ТГФ, затем добавляли воду, экстрагировали этилацетатом. Органический слой промывали солевым раствором и сушили над безводным Na2SO4, выпаривали и очищали на колонке с оксидом кремния (100-200 меш) с применением петролейного эфира и этилацетата (8 г, выход 45%).
Промежуточное соединение 180: 3-(4-{[4-(2-оксо-2-фенилэтокси)бензил]окси}фенил)пропановая кислота
В 100 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 5 мл метанола и 5 мл ТГФ. В перемешиваемый растворитель добавляли 4-{[4-(3-метокси-3-оксопропил)фенокси]метил}фенил-бензоат (1 г, 2,4 ммоль) и NaOH, затем воду (1 мл). Затем смесь перемешивали при КТ в течение 12 часов. После завершения реакции (прохождение отслеживали путем ТСХ) реакционную смесь полностью выпаривали и неочищенное вещество промывали диэтиловым эфиром и разбавляли водой, а затем нейтрализовали 1 н. HCl, экстрагировали этилацетатом, органический слой промывали водой и солевым раствором, сушили над Na2SO4. Органический слой выпаривали на роторном испарителе с получением продукта (1 г, выход 99%). МС (ИЭР, 120 эВ): m/z=389,2 (М-Н)+.
Промежуточное соединение 181: 3-(4-{[4-(2-оксо-2-фенилэтокси)бензил]окси}фенил)пропанамид
В 100 мл кругло донную колбу, оборудованную магнитной мешалкой, помещали 12 мл ТГФ, в перемешиваемый растворитель добавляли 3-(4-{[4-(бензоилокси)бензил]окси}-фенил)пропановую кислоту (1 г, 2,4 ммоль), этилхлорформиат (0,36 мл, 38 ммоль) и триэтиламин (1,4 мл, 10 ммоль) при -10°C. Затем смесь перемешивали при указанной температуре в течение 45 минут, после чего добавляли NH4OH, продолжали перемешивание при указанной температуре в течение 1 часа. После завершения реакции (прохождение отслеживали путем ТСХ) реакционную массу экстрагировали этилацетатом и водой. Органический слой промывали солевым раствором, сушили над Na2SO4 и выпаривали насухо с получением соединения (1 г, выход 99%). МС (ИЭР, 120 эВ): m/z=390,2 (М-H)+.
Соединение 120: 3-{4-[(4-{[(2Z)-2-(Метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропанамид
В 100 мл круглодонную колбу, оборудованную магнитной мешалкой, помещали 15 мл уксусной кислоты, в перемешиваемый растворитель добавляли 3-(4-{[4-(2-оксо-2-фенилэтокси)бензил]окси}фенил)пропанамид (1 г, 2,5 ммоль), гидрохлорид O-метоксиламина (0,32 г, 3,8 ммоль) и ацетат натрия (0,311 г, 3,8 ммоль). Затем реакционную смесь перемешивали при КТ в течение 12 часов. После завершения реакции (прохождение отслеживали путем ТСХ) реакционную смесь подщелачивали при помощи карбоната натрия, затем экстрагировали этилацетатом. Затем органический слой сушили над безводным Na2SO4. Растворитель удаляли при пониженном давлении; полученное неочищенное соединение очищали путем колоночной хроматографии на силикагеле с применением петролейного эфира и этилацетата в качестве элюентов с получением титульного соединения в виде белого твердого вещества (0,02 г, выход 20%).
Схема 45:
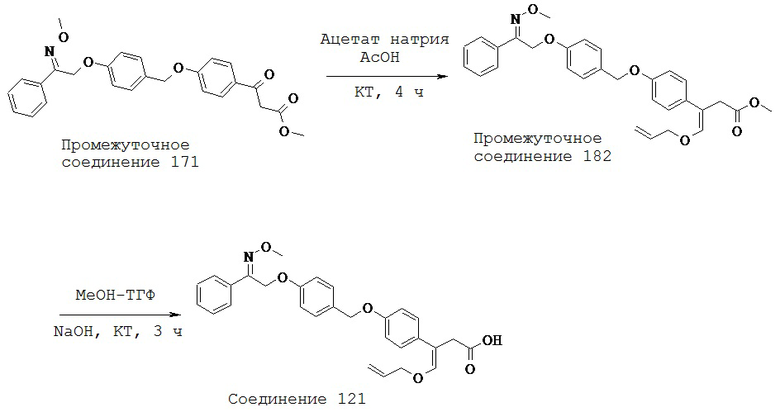
Пример 121
(3Z)-3-{4-[(4-{[(2E,2Z)-2-(Метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}-3-[(проп-2-ен-1-илокси)имино]пропановая кислота (121)
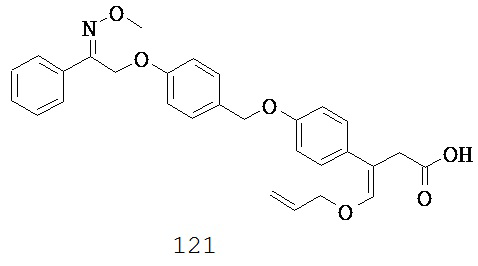
Соединение 121 синтезировали из метил-3-{4-[(4-{[(2E,2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}-3-оксопропаноата (1,2 г, 2,74 ммоль) и гидрохлорида O-аллилгидроксиламина (0,2 г, 1,8 ммоль) при помощи способа, описанного на схеме 46 (0,09 г, выход 11%); чистота: 57,5% и 29,89%.
Промежуточное соединение 182: метил-(3Z)-3-{4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}-3-[(проп-2-ен-1-илокси)имино]пропаноат
В 50 мл одногорлую круглодонную колбу, содержащую метил-3-{4-[(4-{[(2E,2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}-3-оксопропаноат (1,2 г, 2,74 ммоль) в уксусной кислоте (2 мл), добавляли гидрохлорид O-аллилгидроксиламина (0,2 г, 1,8 ммоль), затем ацетат натрия (0,14 г, 1,8 ммоль). Реакционную смесь перемешивали при КТ в атмосфере азота в течение 2-3 часов. Реакционную смесь разбавляли этилацетатом (40 мл), промывали последовательно водой (3×20 мл), нас. NaHCO3 (2×20 мл) и водой (1×20 мл). Органическую фазу, наконец, промывали солевым раствором (1×20 мл), затем сушили над сульфатом натрия и концентрировали с получением бледно-желтого геля (0,22 г, выход 24,32%). 1H ЯМР (300 МГц, CDCl3): δ 7,58-7,61 (m, 4H), 7,23-7,29 (m, 5H), 6,83-6,89 (m, 4H), 5,92-6,03 (m, 1H), 5,13-5,33 (m, 5H), 4,9 (s, 2H), 4,64-4,70 (m, 3H), 3,98 (s, 3H), 3,67 (s, 3H).
Соединение 121: (3Z)-3-{4-[(4-{[(2E,2Z)-2-(Метоксиимино)-2-фенилэтил]окси}бензил)-окси]фенил}-3-[(проп-2-ен-1-илокси)имино]пропановая кислота
2н. раствор NaOH (0,4 мл, 0,796 ммоль) добавляли в охлажденный раствор метил-(3E)-3-{4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}-3-[(проп-2-ен-1-илокси)имино]пропаноата (0,2 г, 0,39 ммоль) в смеси МеОН-ТТФ. Реакционную смесь перемешивали при КТ в течение 12 часов. Избыток растворителей удаляли при пониженном давлении и полученную вязкую маслу разбавляли водой, нейтрализовали 1 н. HCl, а затем экстрагировали этилацетатом. Органическую фазу сушили над сульфатом натрия и концентрировали с получением твердого вещества (0,09 г, выход 46%); чистота: 57,5% и 29,89%.
Пример 122
(3E,3Z)-3-[(Бензилокси)имино]-3-{4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропановая кислота (122)
Соединение 122 синтезировали из соли O-бензилгидроксиламина и HCl (0,2 г, 1,25 ммоль) и метил-3-{4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]-фенил}-3-оксопропаноата (1,12 г, 2,5 ммоль) согласно способу, описанному на схеме 46 (0,26 г, выход 23,1%); чистота: 38%.
Пример 123
(3E,3Z)-3-{4-[(4-{[(2Z)-2-(Метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}-3-(феноксиимино)пропановая кислота (123)
Соединение 123 синтезировали из гидрохлорида O-фенилгидроксиламина (0,12 г, 1,11 ммоль) и метил-3-{4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]-фенил}-3-оксопропаноата (0,25 г, 0,56 ммоль) согласно способу, описанному на схеме 46 (0,03 г, выход 1,05%); чистота: 87,35%.
Пример 124
(3E,3Z)-3-[(Циклогексилокси)имино]-3-{4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)окси]фенил}пропановая кислота (124)
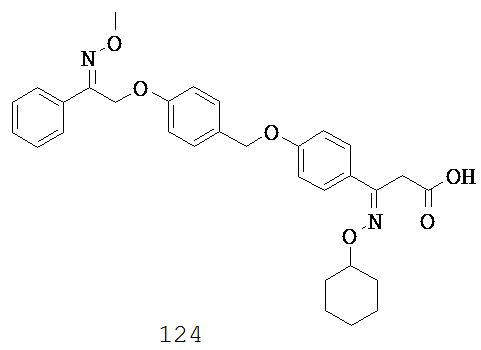
Соединение 124 синтезировали из гидрохлорида O-циклогексилгидроксиламина (0,154 г, 1,34 ммоль) и метил-3-{4-[(4-{[(2Z)-2-(метоксиимино)-2-фенилэтил]окси}бензил)-окси]фенил}-3-оксопропаноата (0,3 г, 0,67 ммоль) согласно способу, описанному на схеме 46 (0,07 г, выход 19,58%); чистота: 86,4% и 9%.
Необходимо отметить, что в вышеизложенных экспериментальных способах во многих случаях продукт был геометрическим чистым, т.е. получали только Е или Z изомер. В этих случаях в тексте содержится указание на экспериментальное определение структуры Е или Z изомера, и изображенная структура является правильной. В других случаях вещество представляло собой смесь Е и Z изомеров, что также определено в экспериментальной части. В этих случаях для удобства читателя показана только одна из двух структур геометрических изомеров (как правило, показан Z изомер), но специалистам в данной области при прочтении экспериментальной части будет понятно, что присутствуют оба изомера.
Соединения, указанные в Таблице 1, или их геометрические изомеры синтезировали согласно вышеизложенным способам или их вариантам.
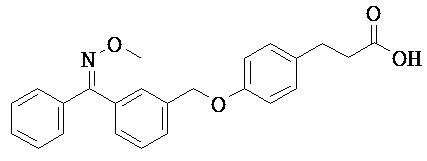
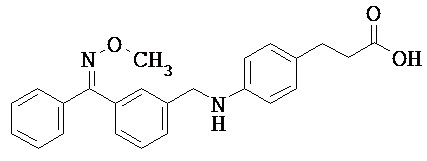
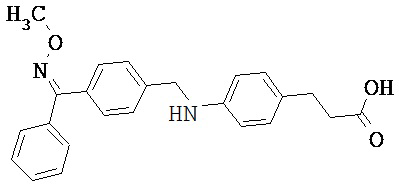
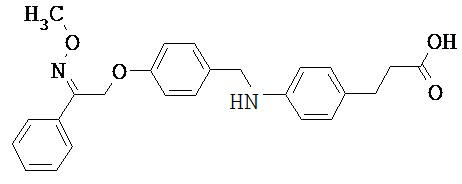
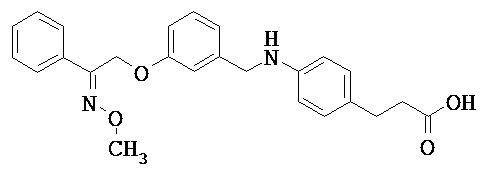
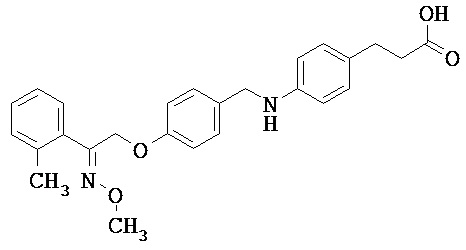
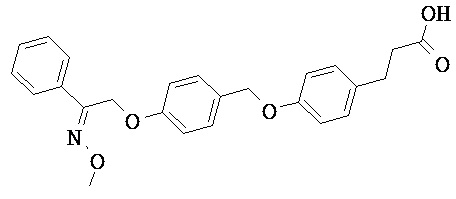
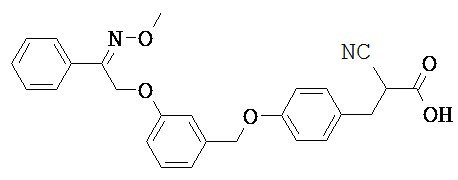
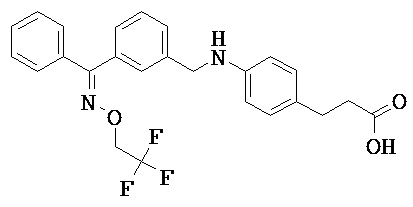
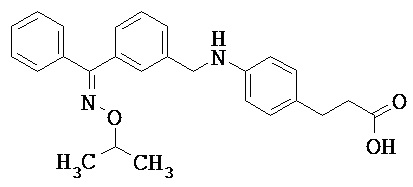
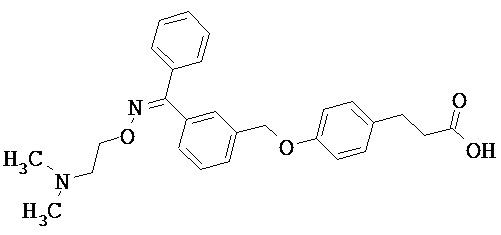
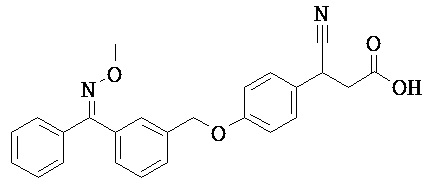
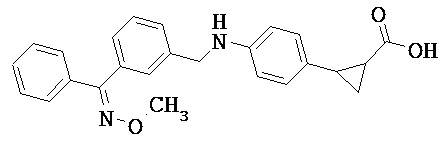
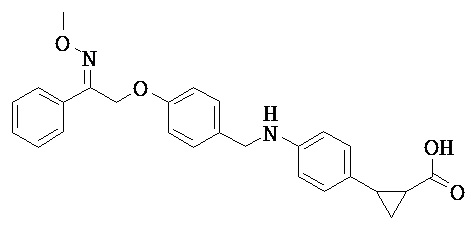
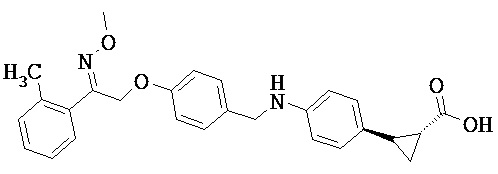
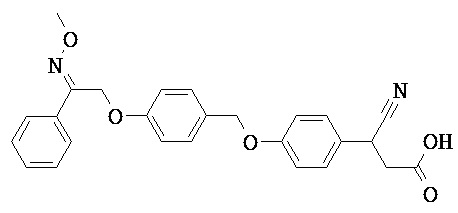
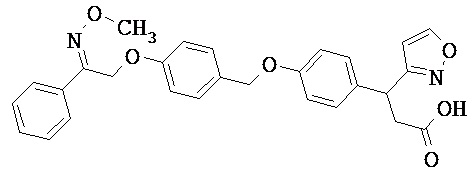
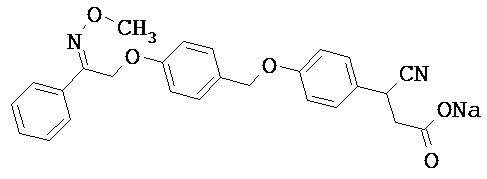

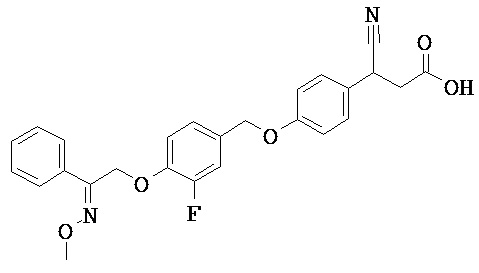
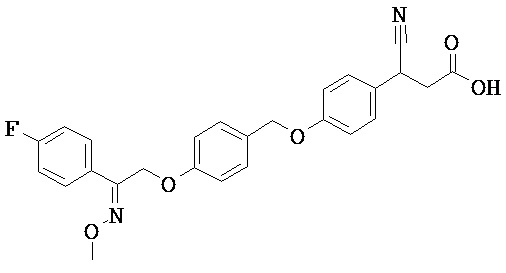
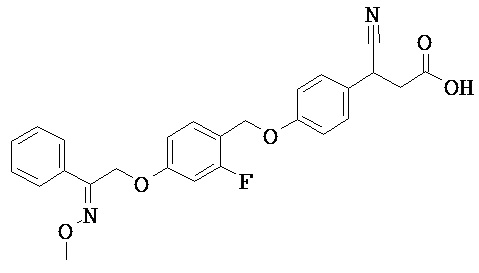
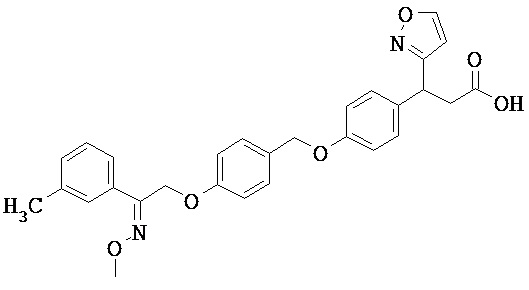
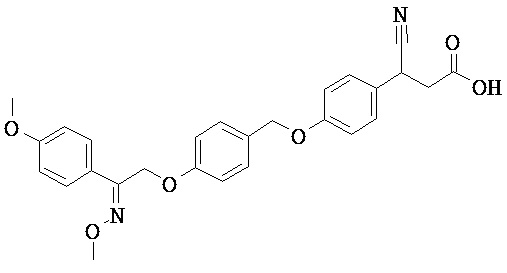
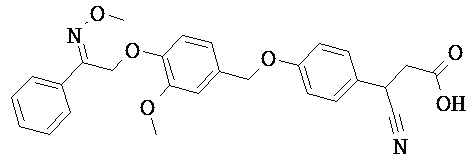
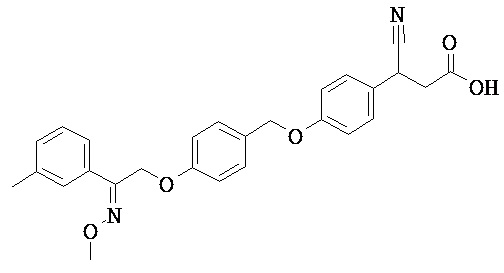
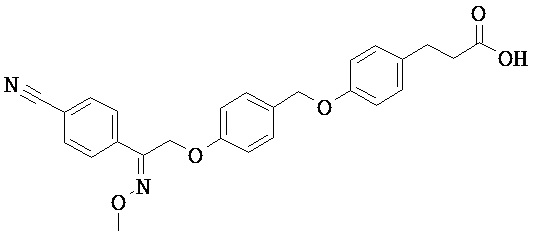
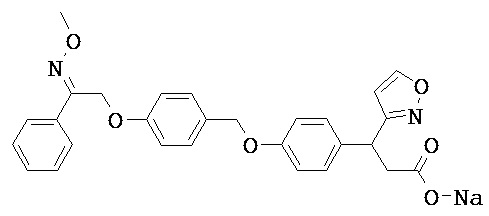
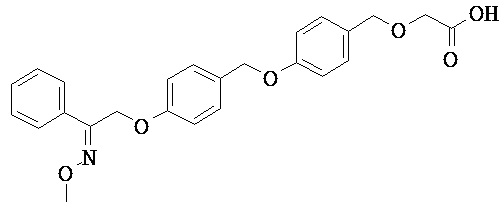
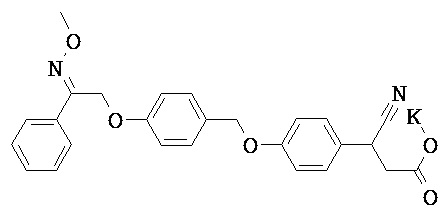
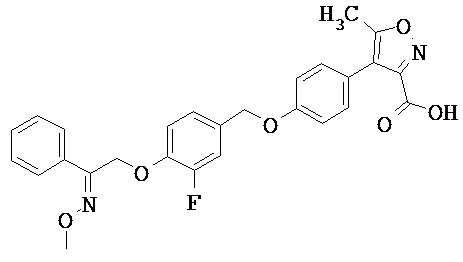
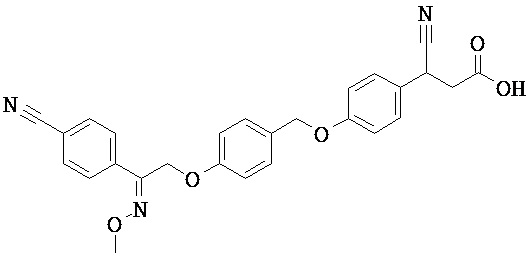
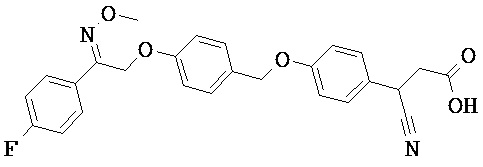
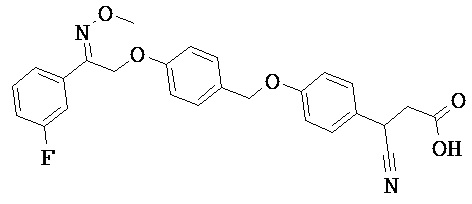
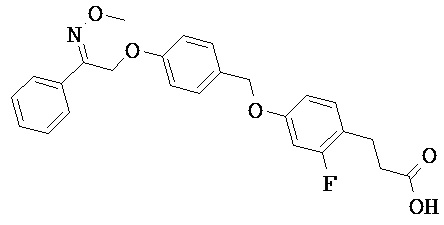
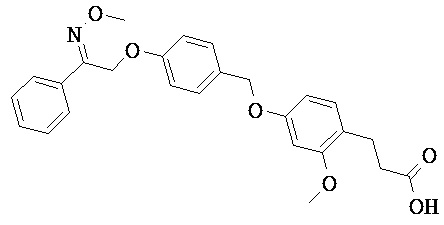
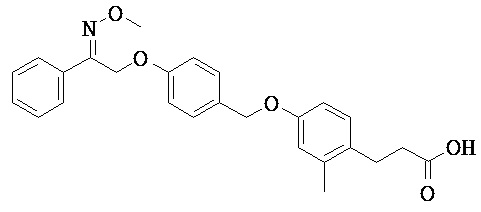
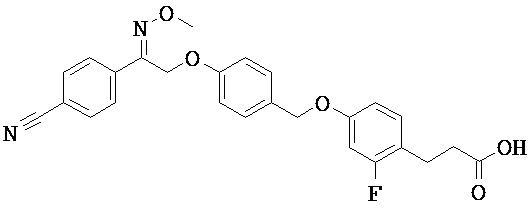
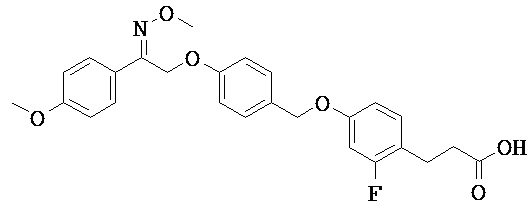
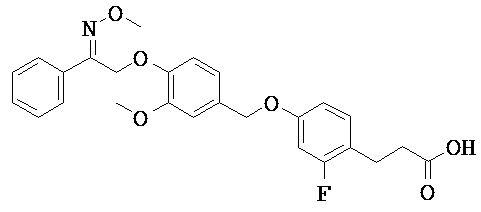
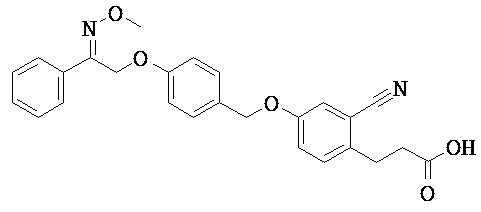
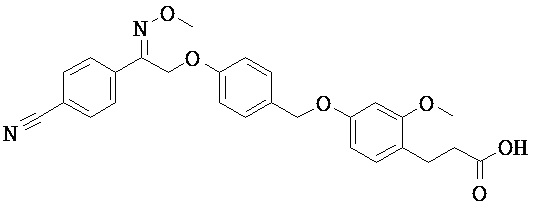

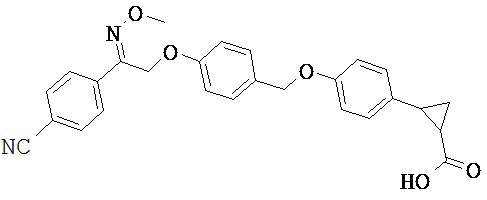
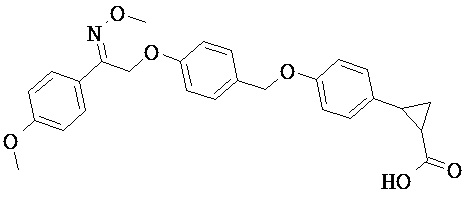
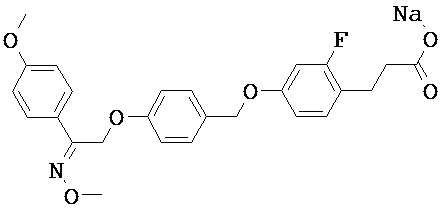
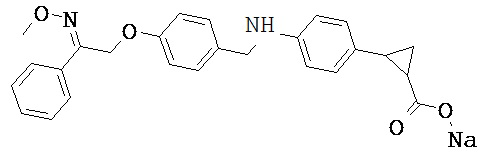
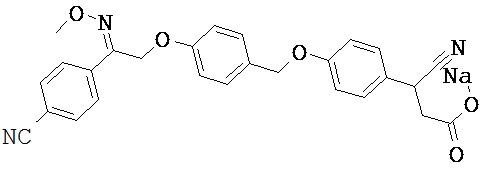
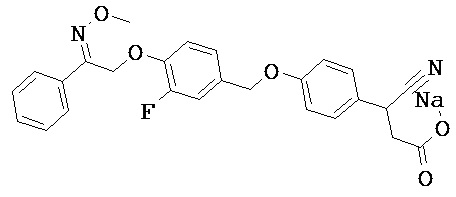
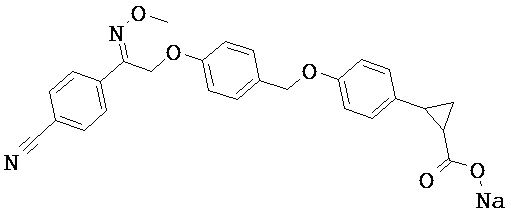
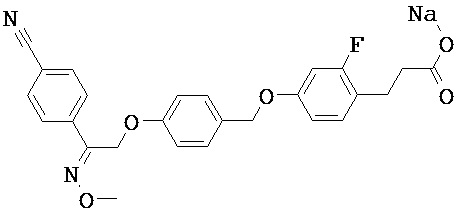
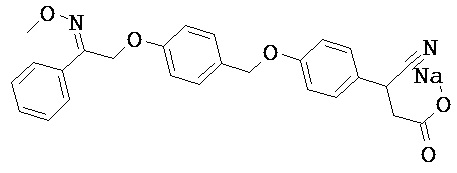
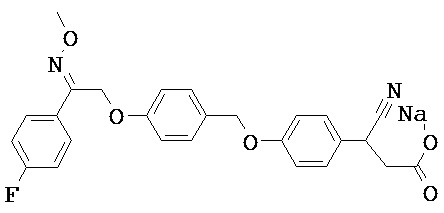
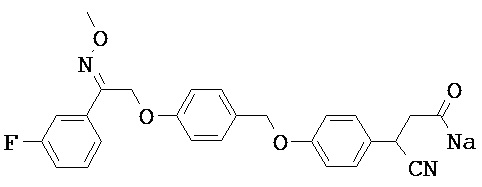
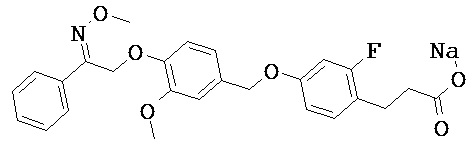
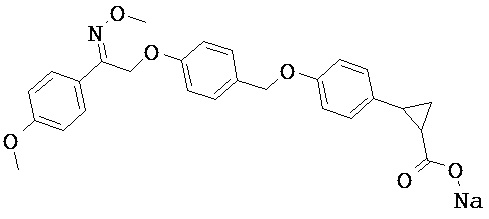
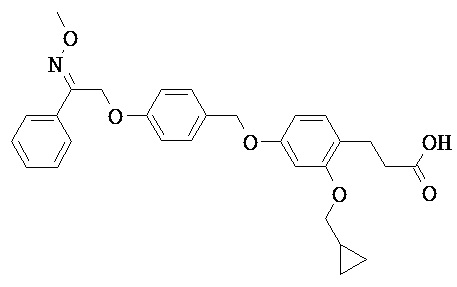
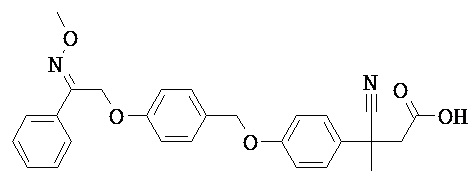
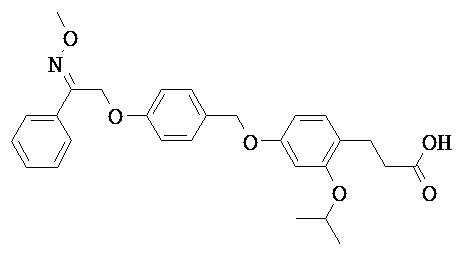
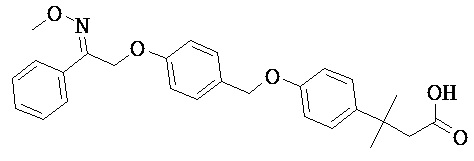
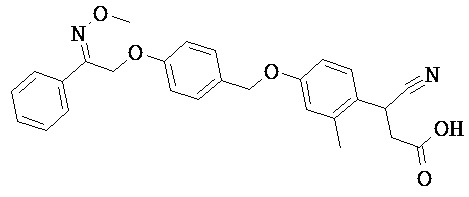
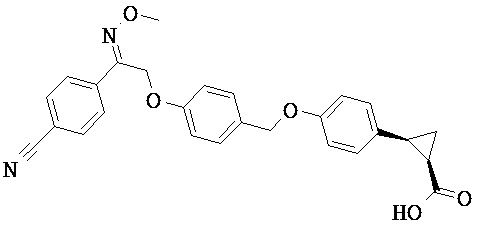
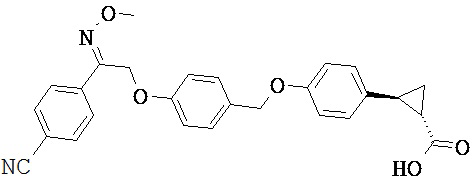
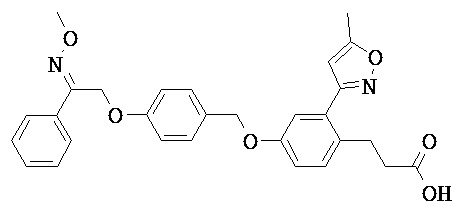
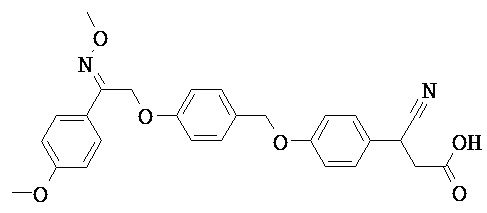
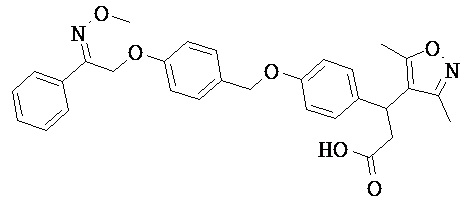
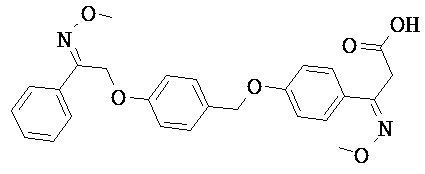
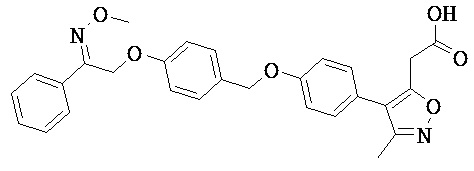
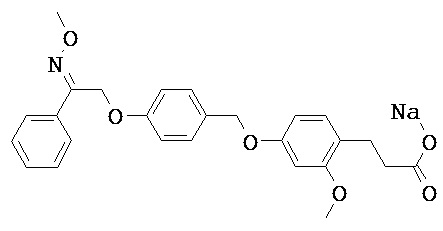

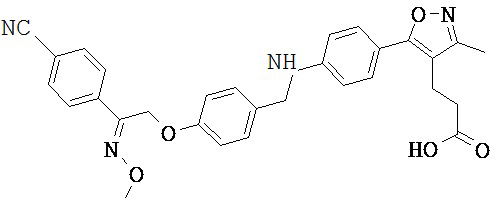
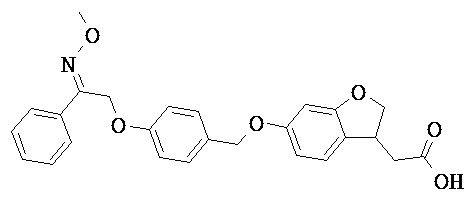
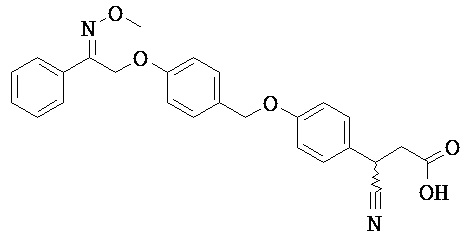
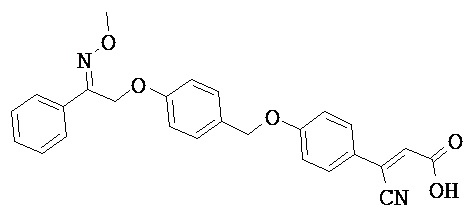
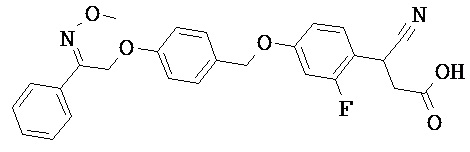
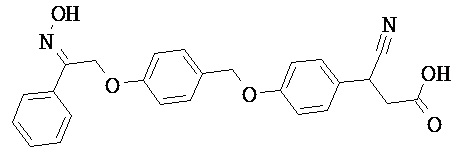
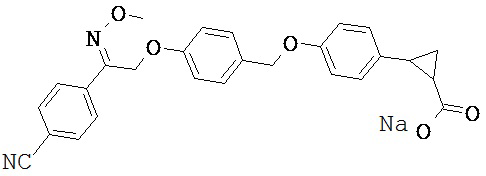
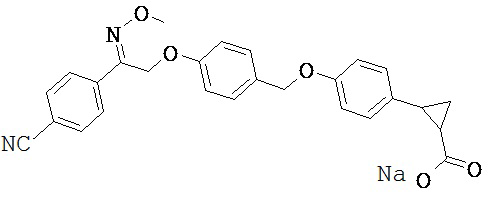
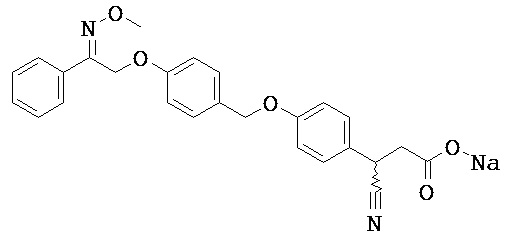
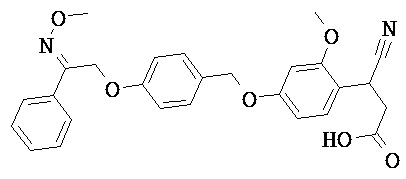
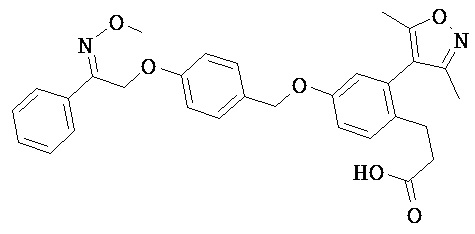
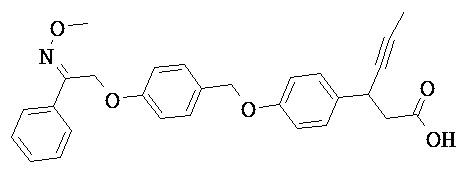
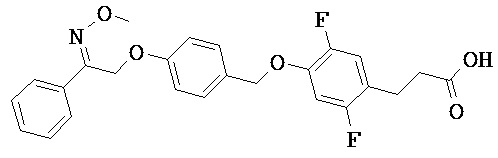
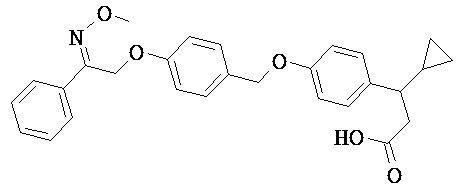
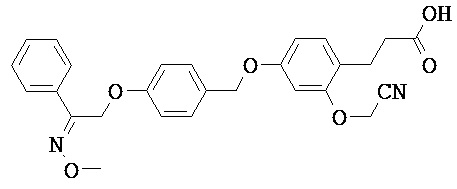
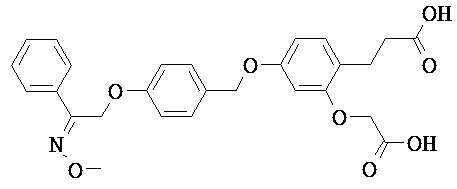
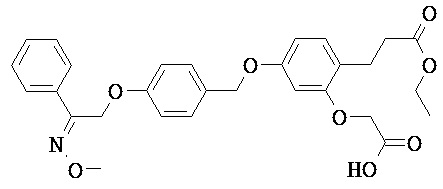
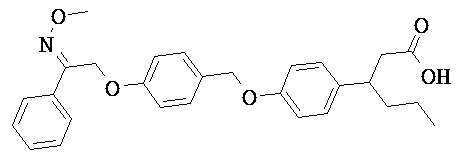
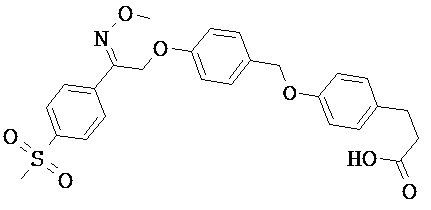
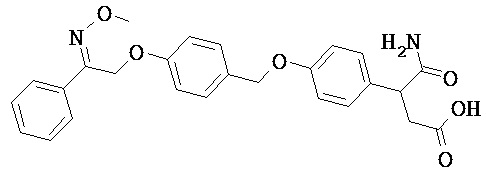
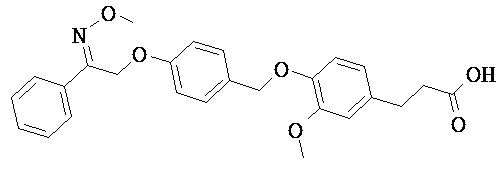
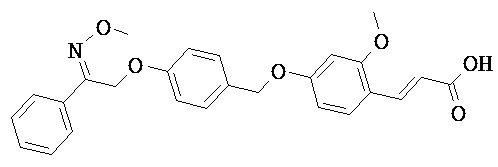
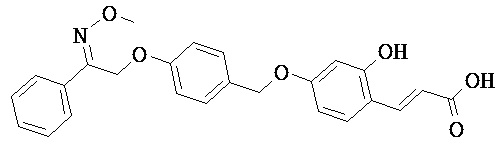
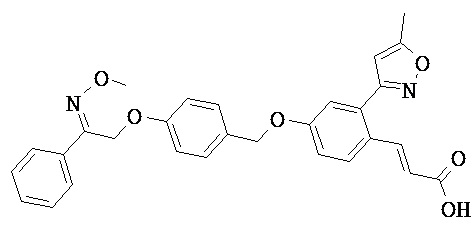
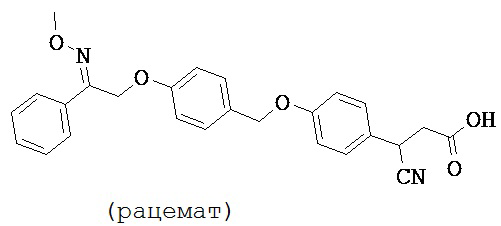
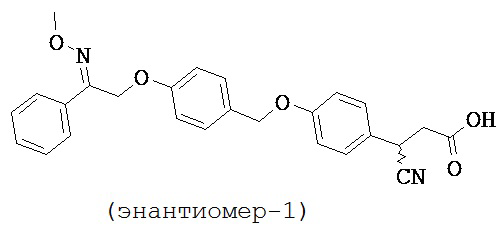
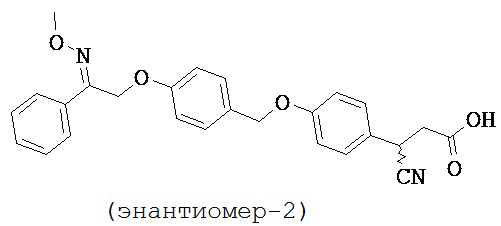
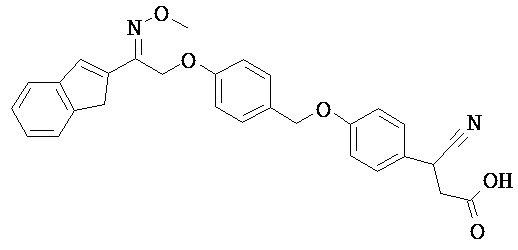
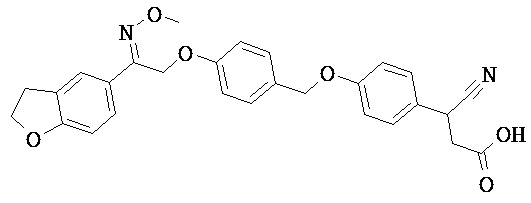
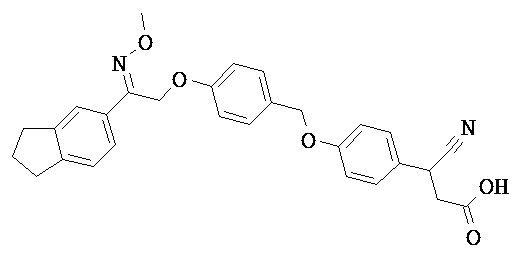
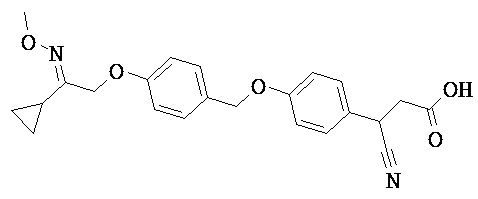
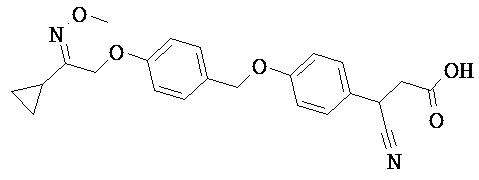
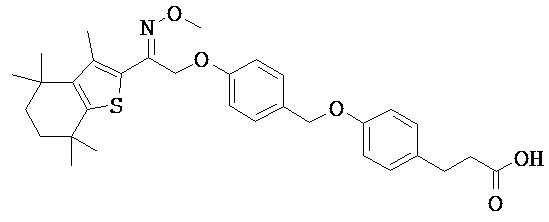
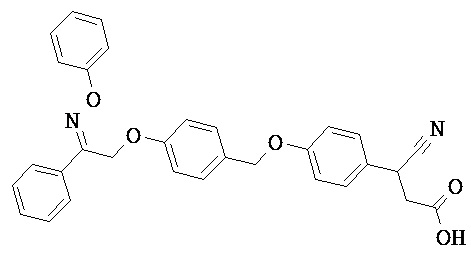
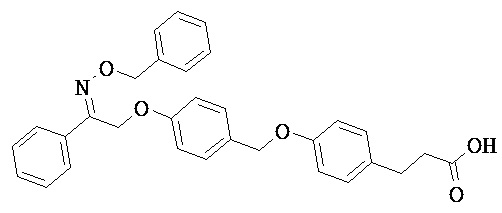
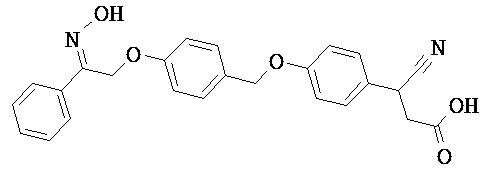
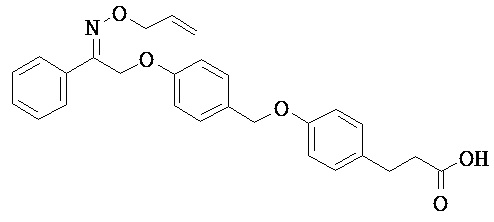
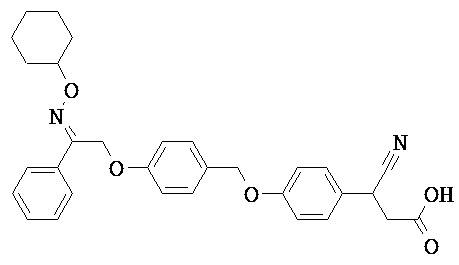
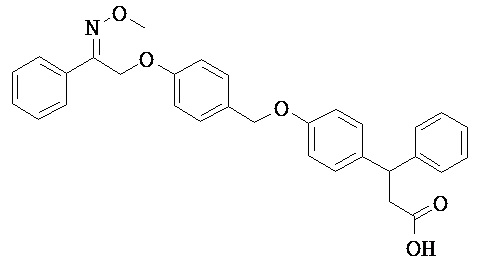
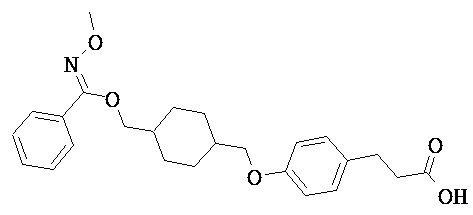
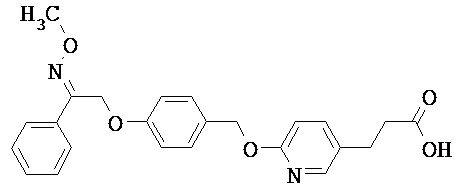
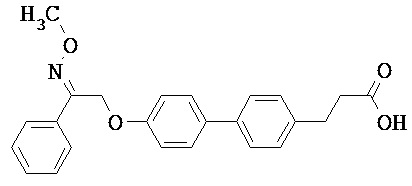
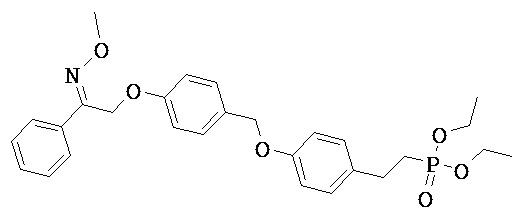
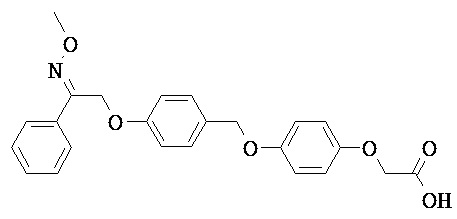
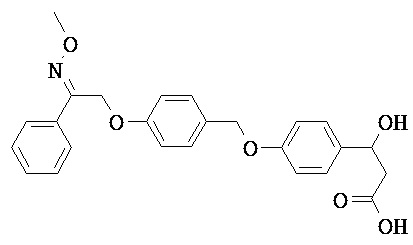
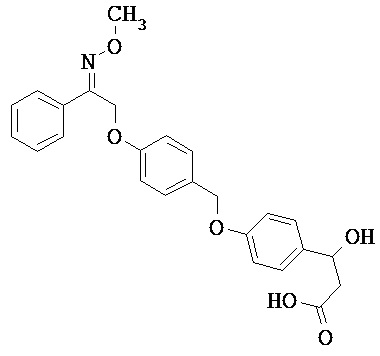
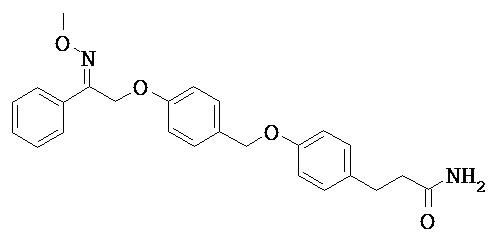
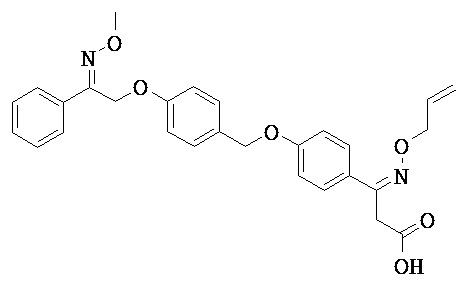

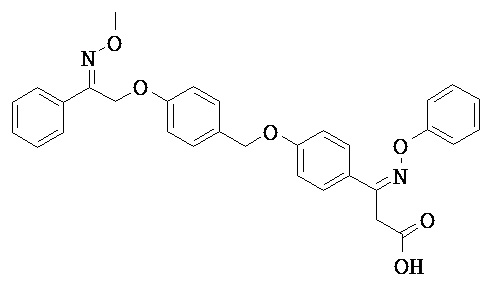
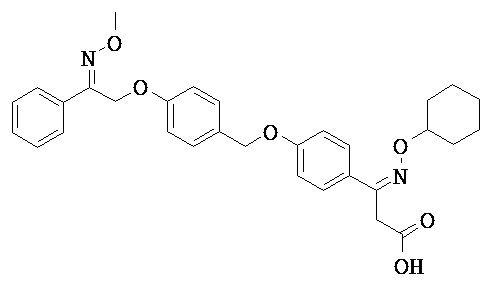
При помощи модифицированных способов, применяемых для получения приведенных выше соединений, также можно получать следующие соединения и их геометрические изомеры.
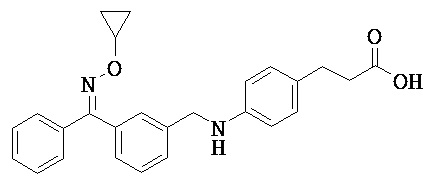
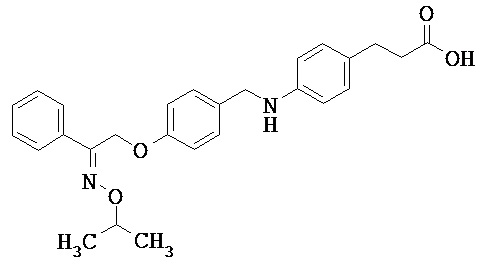
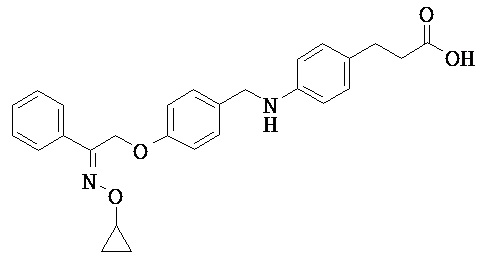
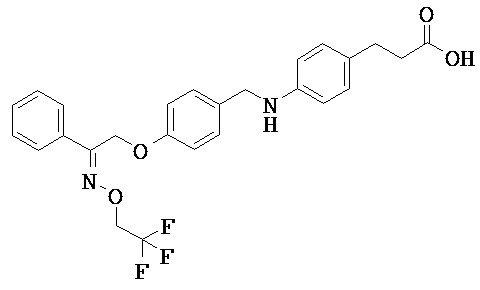
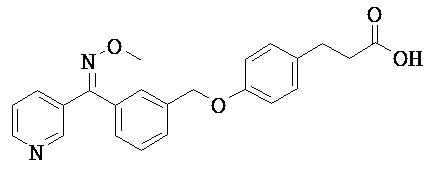

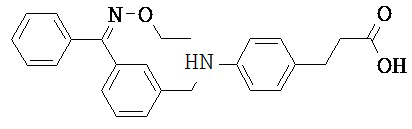
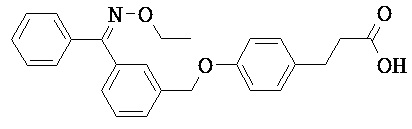
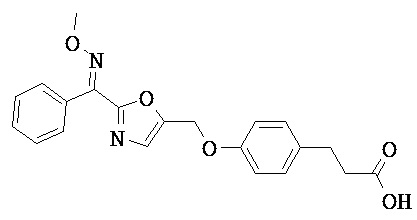
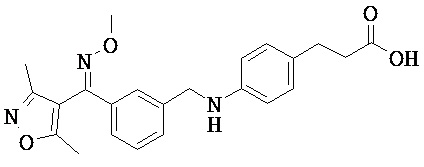
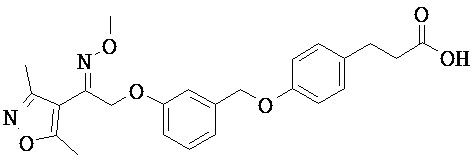

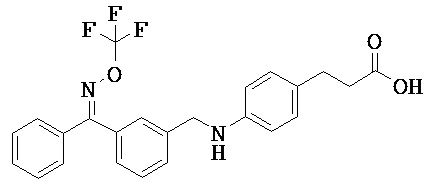
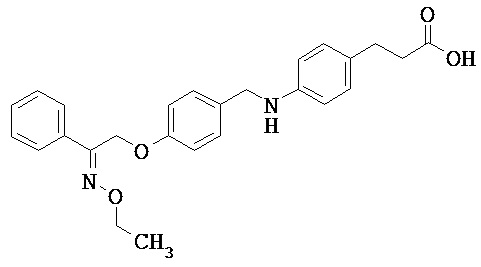
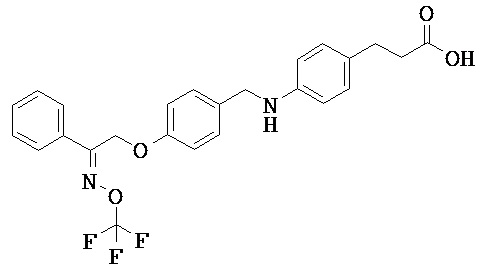
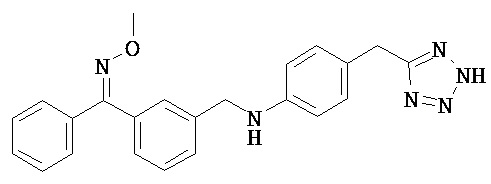
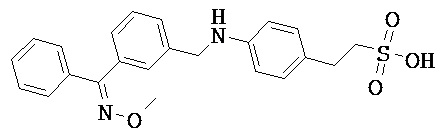
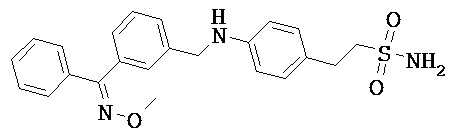
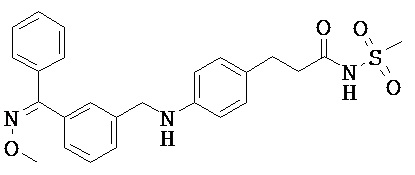
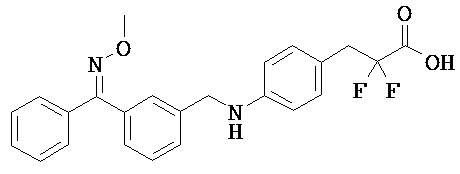
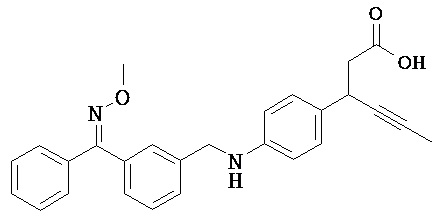
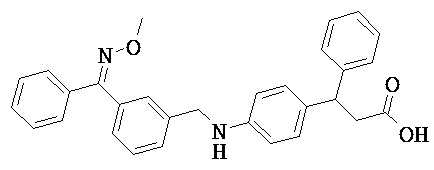
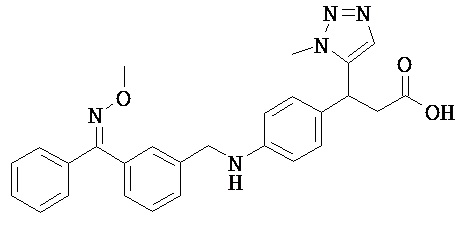
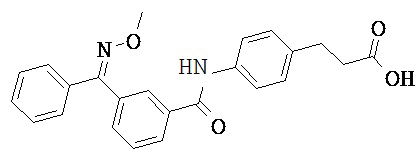
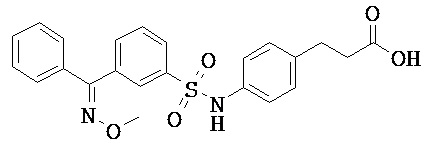
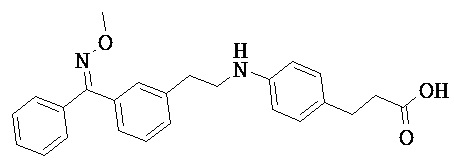
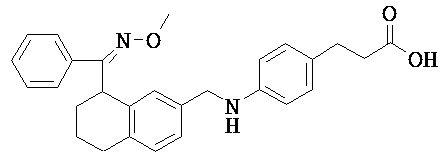
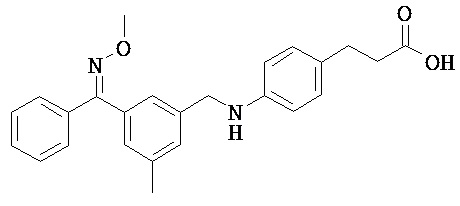
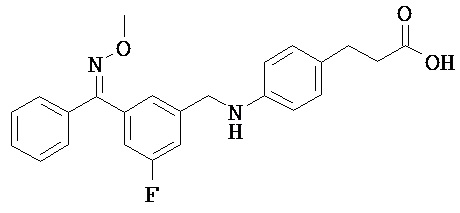
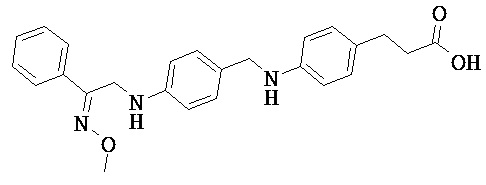
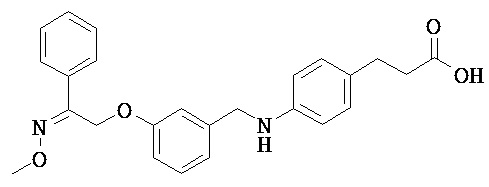
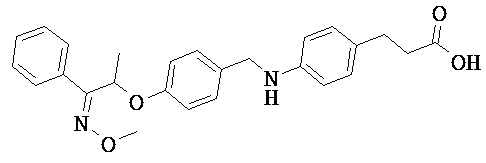
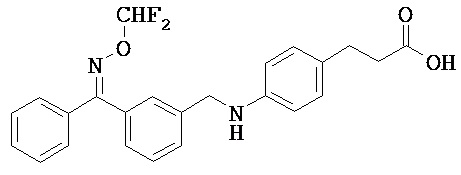
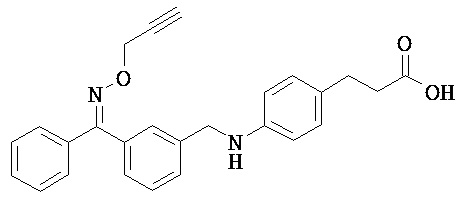
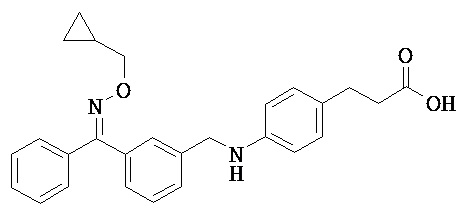
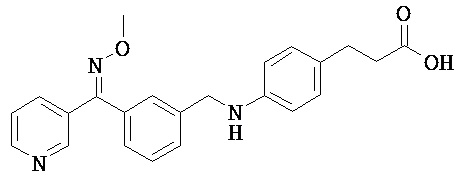
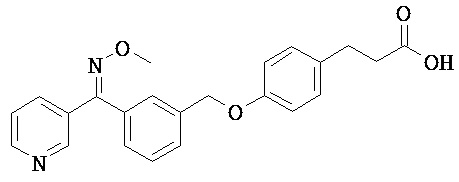
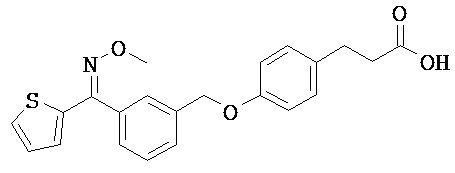
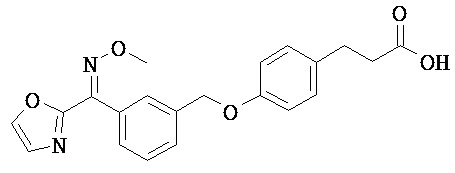
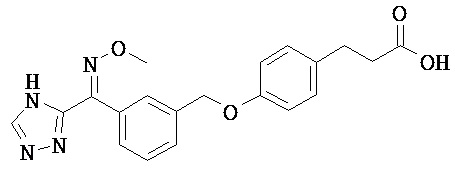
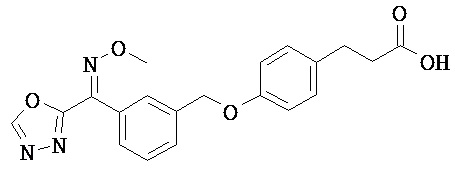
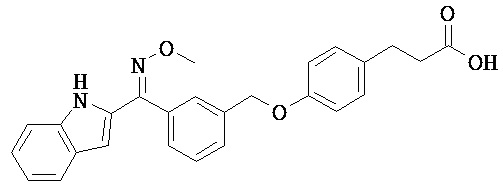
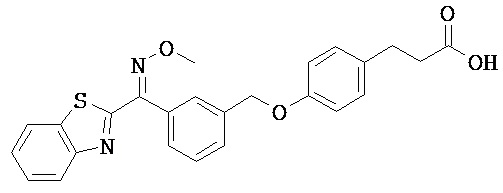
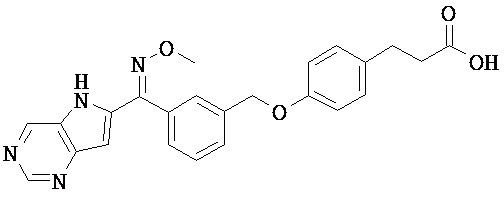
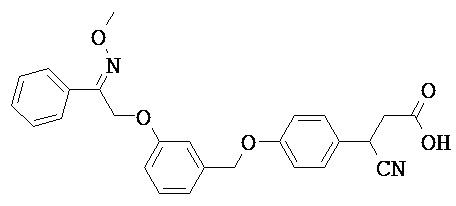
Биологическая активность
Исследование тока кальция для определения активации GPR40:
Клетки СНО-К, стабильно экспрессирующие hGPR40, отбирали в среде, содержащей неомицин/G418. Указанные клетки помещали в планшеты в концентрации 20000 клеток на лунку черного 96-луночного планшета для тканевых культур с прозрачным дном. Проводили культивирование клеток в течение 24 часов при 37°C в увлажненной воздушной среде, содержащей 5% CO2, для обеспечения экспрессии белка.
На следующий день, после удаления среды из лунок добавляли Fluo-4NW (Invitrogen) в концентрации 100 мкл/лунку и инкубировали клетки в течение 30 минут при 37°C и дополнительно в течение 30 минут при КТ. Все исследуемые соединения разбавляли до подходящих концентраций в буфере HEPES. Соединения добавляли к клеткам и проводили анализ содержимого лунок на анализаторе BioTEK Synergy, показания снимали в течение 4 минут с интервалами 5 секунд. Результаты представлены в таблице 3.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ СОЕДИНЕНИЯ, КОТОРЫЕ МОДУЛИРУЮТ РЕЦЕПТОРЫ ТИПА PPARγ, И ИХ ПРИМЕНЕНИЕ В КОСМЕТИЧЕСКИХ И ФАРМАЦЕВТИЧЕСКИХ КОМПОЗИЦИЯХ | 2004 |
|
RU2401836C2 |
| Композиции для лечения гипертензии и/или фиброза | 2017 |
|
RU2752088C1 |
| ИНГИБИТОРЫ URAT1 | 2015 |
|
RU2721844C2 |
| НОВЫЕ БИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2016 |
|
RU2760554C1 |
| ЦИКЛИЧЕСКИЕ АМИДНЫЕ ПРОИЗВОДНЫЕ КАК ИНГИБИТОРЫ 11-БЕТА-ГИДРОКСИСТЕРОИД-ДЕГИДРОГЕНАЗЫ И ИХ ПРИМЕНЕНИЕ | 2012 |
|
RU2669695C2 |
| НОВЫЕ ТИЕНОПИРИМИДИНОВЫЕ ПРОИЗВОДНЫЕ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2014 |
|
RU2605403C2 |
| СПИРОЦИКЛИЧЕСКИЕ АМИНОВЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ S1P | 2011 |
|
RU2602800C2 |
| ГЕТЕРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ | 2009 |
|
RU2490257C2 |
| ПРОИЗВОДНОЕ 1,2,3,4-ТЕТРАГИДРОХИНОКСАЛИНА, МЕТОД ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2019 |
|
RU2804127C2 |
| ПРОИЗВОДНЫЕ 5,6-ДИГИДРОПИРОНА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1994 |
|
RU2140917C1 |
Изобретение относится к соединению формулы (I), где кольцо А представляет собой возможно замещенную фенильную группу, где возможный заместитель представляет собой фтор или метокси; кольцо В представляет собой возможно замещенную фенильную группу, где возможный заместитель выбран из метокси, 1 или 2 атомов фтора, -CH2CN, -О-СН2-С3циклоалкила, изопропокси; изоксазола (который может быть замещен 1 или 2 метильными группами), -О-CH2-CN и -O-СН2-С(O)ОН; X представляет собой связь или -СН2О-; Y представляет собой -CH2O-; Z представляет собой связь или -(CR5R6)-; L представляет собой -СО2Н; R1 представляет собой OR7; R2 представляет собой кольцо, выбранное из группы, состоящей из С3-С12 циклоалкила, С6арилконденсированногоС3-С6 циклоалкила, и возможно замещенного С6 арила, причем каждый возможный заместитель выбран из метила, фтора, метокси, циано и метансульфонила; каждый R3, R4, R5 и R6 независимо выбран из группы, состоящей из Н, CN, ОН, CONH2, С1-С12 алкила, С2-С12 алкинила, С6 арила и возможно замещенного C1-C18 гетероарила, выбранного из изоксазола, причем изоксазол может быть замещен 1 или 2 метильными группами, или любые два из R3, R4, R5 и R6 совместно с атомами, к которым они присоединены, могут образовывать возможно замещенный С3-циклоалкил или двойную связь между атомами, к которым они присоединены; R7 выбран из группы, состоящей из Н, возможно замещенного С1-С12 алкила, причем возможные заместители выбраны из 3 атомов фтора или -N(СН3)2 или фенила, С2-С12 алкенила, С3-С12 циклоалкила и С6 арила; r равен 1; или его фармацевтически приемлемой соли. Соединения формулы (I) по изобретению предназначены для изготовления фармацевтической композиции или лекарственного средства для лечения диабета. Технический результат – соединения, активирующие GPR40. 7 н. и 23 з.п. ф-лы, 3 табл., 124 пр.
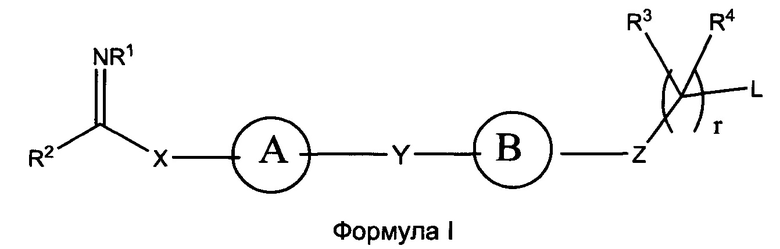
1. Соединение формулы (I):
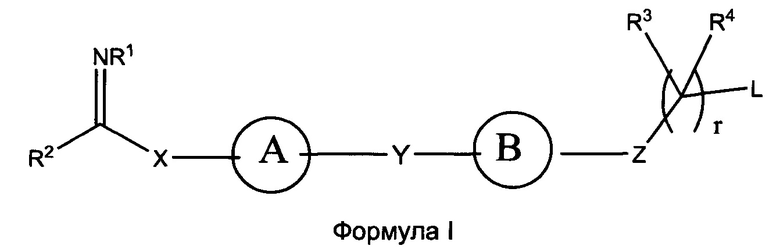
где
кольцо А представляет собой возможно замещенную фенильную группу, где возможный заместитель представляет собой фтор или метокси;
кольцо В представляет собой возможно замещенную фенильную группу, где возможный заместитель выбран из метокси, 1 или 2 атомов фтора, -CH2CN, -О-СН2-С3циклоалкила, изопропокси; изоксазола (который может быть замещен 1 или 2 метильными группами), -О-CH2-CN и -O-СН2-С(O)ОН;
X представляет собой связь или -СН2О-;
Y представляет собой -CH2O-;
Z представляет собой связь или -(CR5R6)-;
L представляет собой -СО2Н;
R1 представляет собой OR7;
R2 представляет собой кольцо, выбранное из группы, состоящей из С3-С12 циклоалкила, С6арилконденсированногоС3-С6 циклоалкила и возможно замещенного С6 арила, причем каждый возможный заместитель выбран из метила, фтора, метокси, циано и метансульфонила;
каждый R3, R4, R5 и R6 независимо выбран из группы, состоящей из Н, CN, ОН, CONH2, С1-С12 алкила, С2-С12 алкинила, С6 арила и возможно замещенного C1-C18 гетероарила, выбранного из изоксазола, причем изоксазол может быть замещен 1 или 2 метильными группами, или
любые два из R3, R4, R5 и R6 совместно с атомами, к которым они присоединены, могут образовывать возможно замещенный С3-циклоалкил, или двойную связь между атомами, к которым они присоединены;
R7 выбран из группы, состоящей из Н, возможно замещенного С1-С12 алкила, причем возможные заместители выбраны из 3 атомов фтора или -N(СН3)2 или фенила, С2-С12 алкенила, С3-С12 циклоалкила и С6 арила;
r равен 1;
или его фармацевтически приемлемая соль.
2. Соединение по п. 1, отличающееся тем, что каждый R3 и R4 представляет собой Н.
3. Соединение по п. 1, отличающееся тем, что Z представляет собой CR5R6.
4. Соединение по п. 3, отличающееся тем, что один из R5 или R6 совместно с одним из R3 и R4 и атомами, к которым они присоединены, образуют циклический фрагмент, представляющий собой циклоалкил.
5. Соединение по п. 4, отличающееся тем, что циклический фрагмент представляет собой циклопропильную группу.
6. Соединение по п. 3, отличающееся тем, что R5 представляет собой Н.
7. Соединение по п. 3, отличающееся тем, что R6 выбран из группы, состоящей из Н, циано и изоксазол-3-ила.
8. Соединение по п. 6, отличающееся тем, что R6 выбран из группы, состоящей из Н, циано и изоксазол-3-ила.
9. Соединение по п. 1, отличающееся тем, что кольцо В представляет собой возможно замещенную фенильную группу формулы:
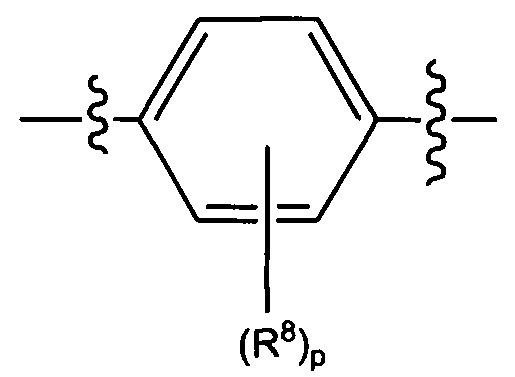
где каждый R8 независимо выбран из группы, состоящей из Н, -CH2CN, -О-СН2-С3циклоалкила, изопропокси; изоксазола (который может быть замещен 1 или 2 метильными группами), -O-CH2-CN и -O-СН2-С(O)ОН,
где р представляет собой целое число, выбранное из группы, состоящей из 0 и 1.
10. Соединение по п. 1, отличающееся тем, что кольцо А представляет собой возможно замещенную фенильную группу, выбранную из группы, состоящей из:
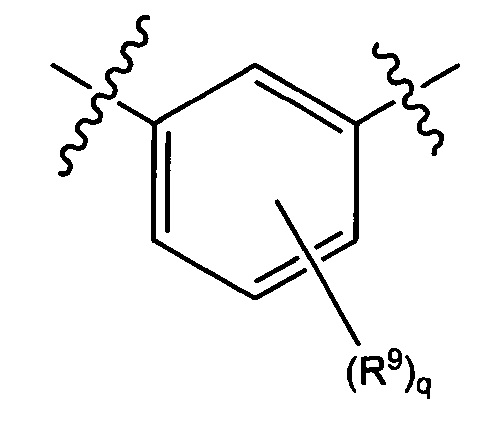 и
и 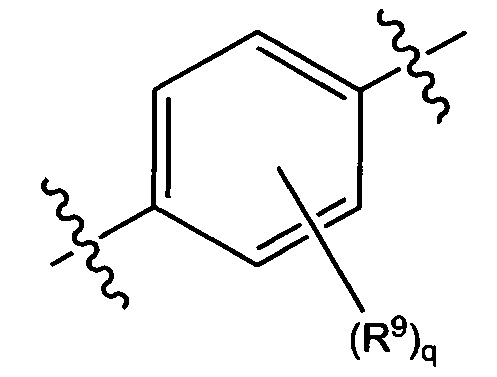
где каждый R9 независимо выбран из группы, состоящей из Н, фтора и метокси; и
где q представляет собой целое число, выбранное из группы, состоящей из 0 и 1.
11. Соединение по п. 10, отличающееся тем, что кольцо А представляет собой возможно замещенную фенильную группу формулы:
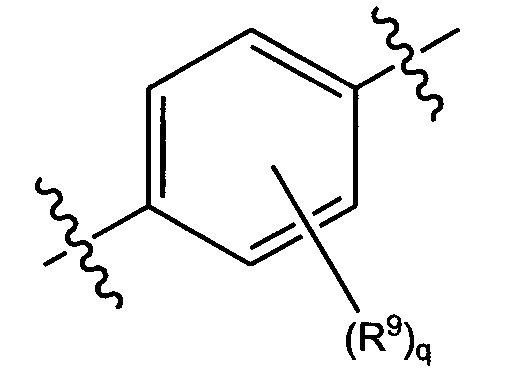
где R9 и q такие, как определено в п. 10.
12. Соединение по п. 1, отличающееся тем, что X представляет собой -СН2О-.
13. Соединение по п. 1, отличающееся тем, что R7 выбран из группы, состоящей из Н и C1-С12 алкила.
14. Соединение по п. 13, отличающееся тем, что R7 представляет собой метил.
15. Соединение по п. 1, отличающееся тем, что R2 представляет собой возможно замещенный фенил.
16. Соединение по п. 15, отличающееся тем, что R2 представляет собой возможно замещенный фенил формулы:
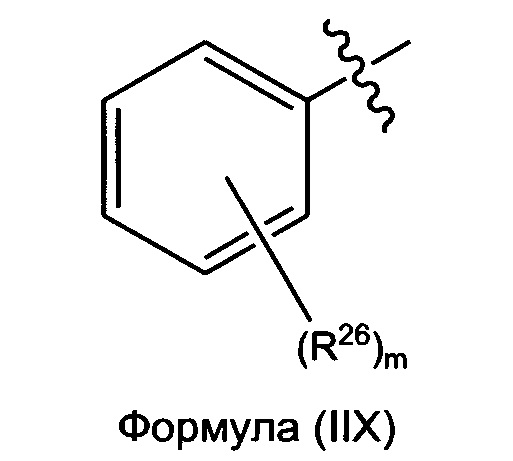
где каждый R26 независимо выбран из группы, состоящей из Н, метила, фтора, метокси, циано и метансульфонила; и
m представляет собой целое число, выбранное из группы, состоящей из 0 и 1.
17. Соединение по п. 1, выбранное из группы, состоящей из:
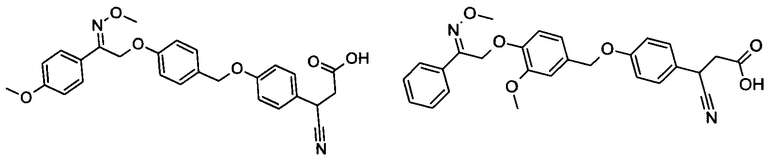
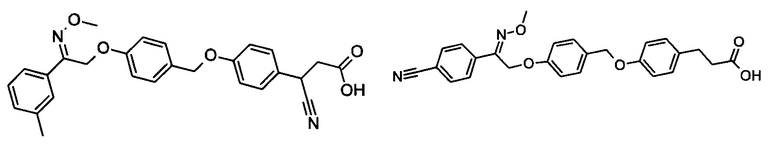
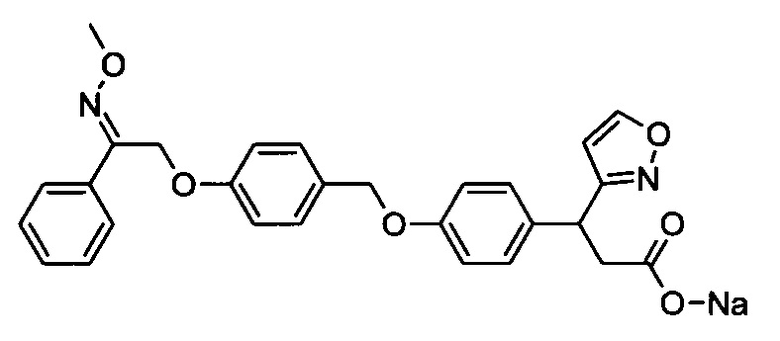
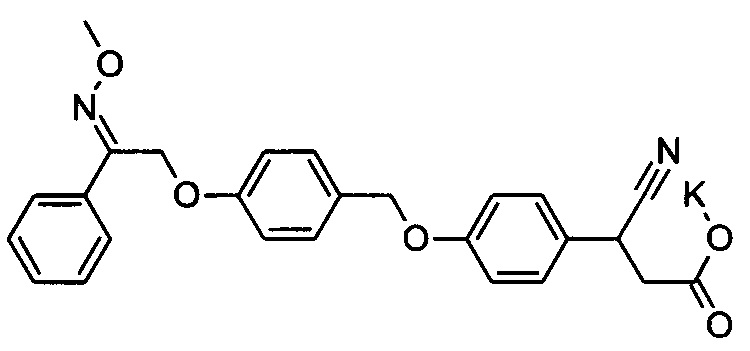
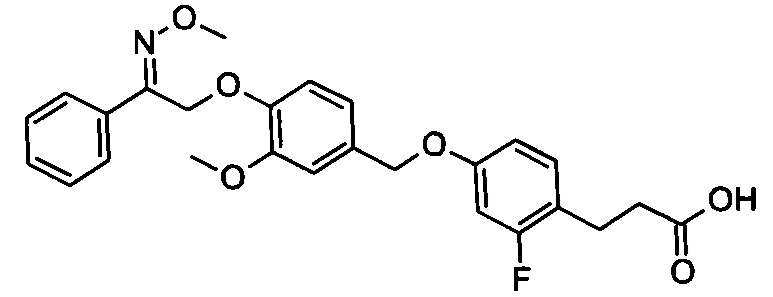

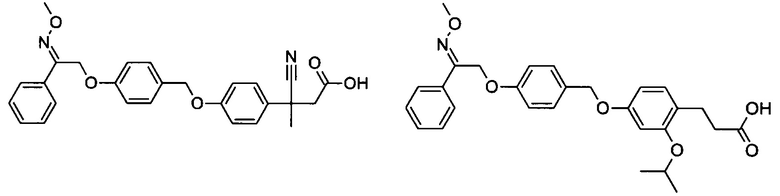
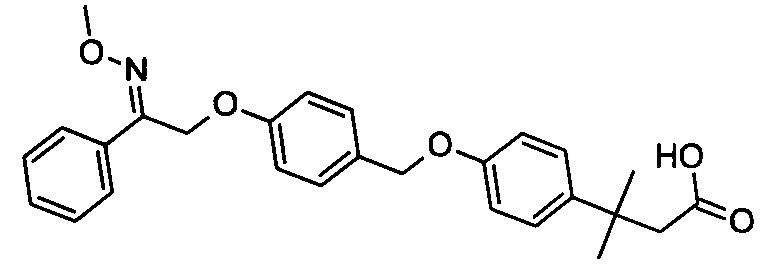
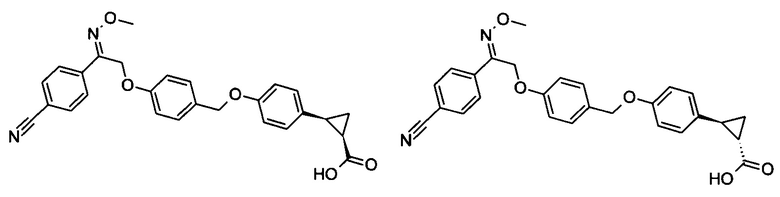
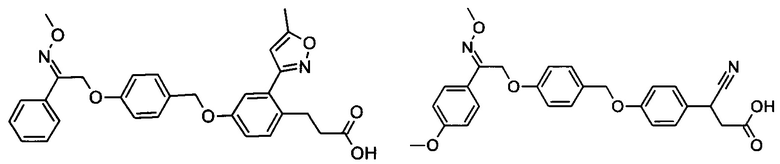
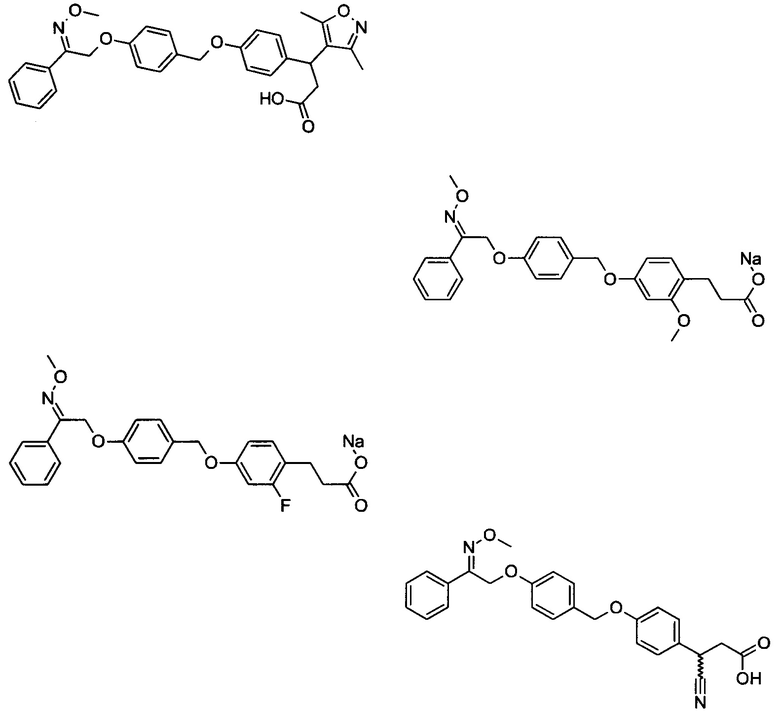
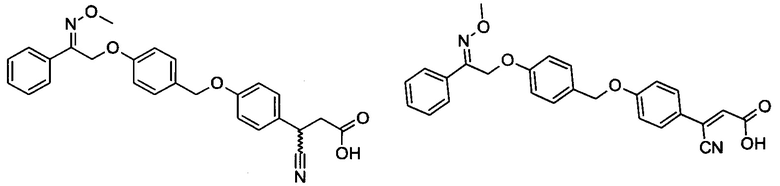
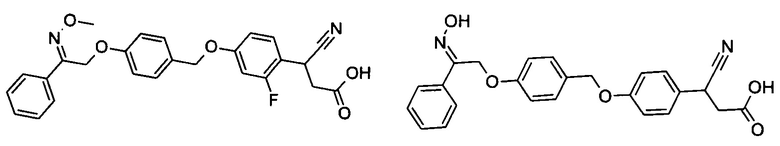
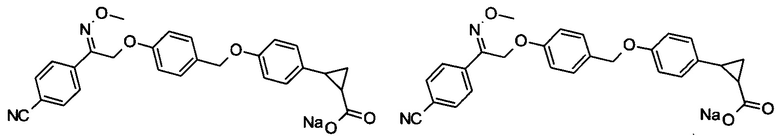
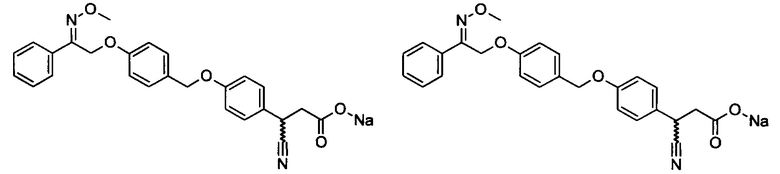
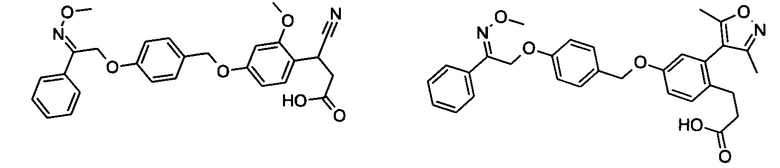
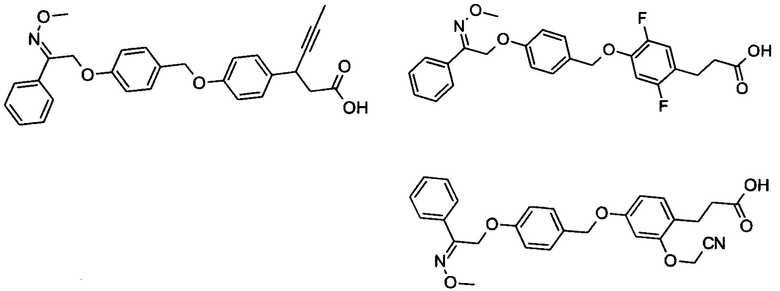
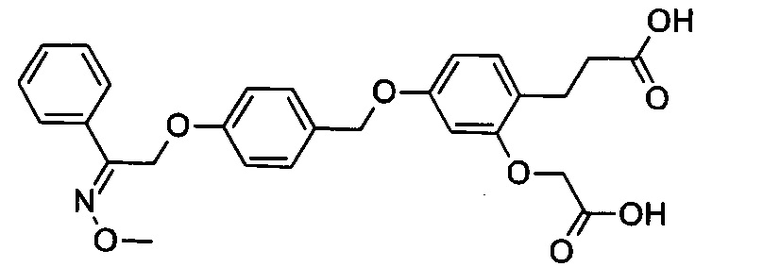
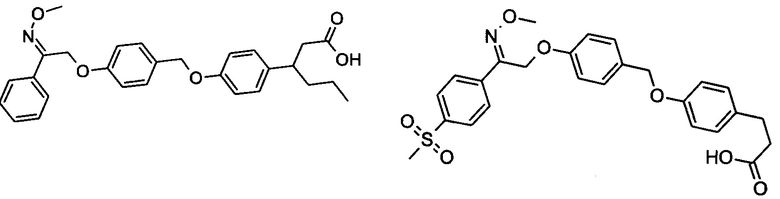
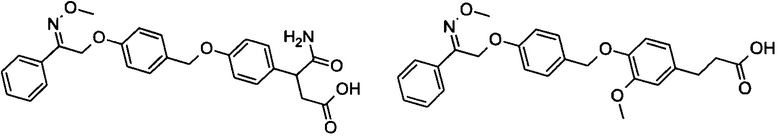

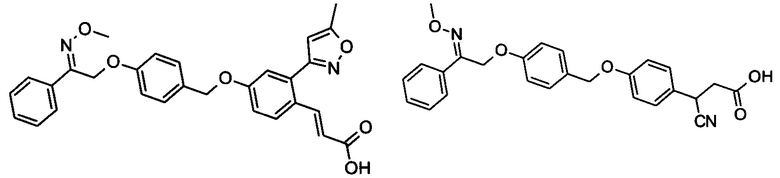
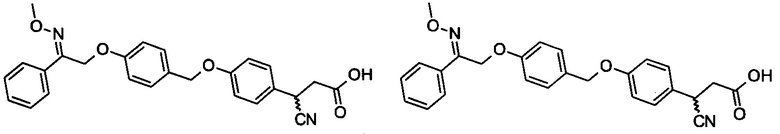
или его геометрический изомер, или фармацевтически приемлемая соль.
18. Фармацевтическая композиция для лечения диабета, содержащая соединение по п. 1 и фармацевтически приемлемый разбавитель, вспомогательное вещество или носитель.
19. Фармацевтическая композиция для лечения диабета, содержащая соединение по п. 17 и фармацевтически приемлемый разбавитель, вспомогательное вещество или носитель.
20. Способ предотвращения или лечения диабета у млекопитающего, включающий введение эффективного количества соединения по п. 1.
21. Способ предотвращения или лечения диабета у млекопитающего, включающий введение эффективного количества соединения по п. 17.
22. Способ по п. 20, отличающийся тем, что диабет представляет собой диабет II типа.
23. Способ по п. 21, отличающийся тем, что диабет представляет собой диабет II типа.
24. Применение соединения по п. 1 для получения лекарственного средства для предотвращения или лечения диабета у млекопитающего.
25. Применение соединения по п. 17 для получения лекарственного средства для предотвращения или лечения диабета у млекопитающего.
26. Применение по п. 24, отличающееся тем, что диабет представляет собой диабет II типа.
27. Применение по п. 25, отличающееся тем, что диабет представляет собой диабет II типа.
28. Соединение по п. 1 для применения для лечения диабета у млекопитающего.
29. Соединение по п. 17 для применения для лечения диабета у млекопитающего.
30. Соединение по п. 28, отличающееся тем, что диабет представляет собой диабет II типа.
| Топчак-трактор для канатной вспашки | 1923 |
|
SU2002A1 |
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| RU 2013103538 A, дата приоритета 12.07.2010 | |||
| US 5484786 A, 16.01.1996 | |||
| US 5516796 A, 14.05.1996 | |||
| СПОСОБ ПОЛУЧЕНИЯ ЧИСТОГО ГЛИНОЗЕМА И ЕГО СОЛЕЙ ИЗ СИЛИКАТОВ ГЛИНОЗЕМА, ПРОСТЫХ ГЛИН И. Т.П. | 1915 |
|
SU280A1 |
| Способ обнаружения утечки в участке напорной сети | 1983 |
|
SU1176139A1 |
| WO 00/01704 A2, 13.01.2000 | |||
| Колосоуборка | 1923 |
|
SU2009A1 |

