Область техники
Настоящее изобретение относится к новому гетероциклическому соединению, которое полезно в качестве агента для профилактики или лечения диабета, гиперлипидемии и аналогичных; и аналогичным.
Предпосылки изобретения
Известно, что ретинол-связывающий белок 4 (далее называемый иногда для краткости “RBP4”) является единственным белком, транспортирующим ретинол в крови, продуцируемым главным образом в печени.
В последние годы, кроме того, полагают, что RBP4 является фактором, индуцирующим инсулинорезистентность, как показано ниже.
(1) Поскольку экспрессия RBP4 увеличивается в адипоцитах GLUT4 нокаутированных мышей, показывая стойкость к инсулину, полагают, что RBP4 является потенциальным адипоцитокином, индуцирующим стойкость к инсулину (см. 436, 356-362 (2005) (непатентный документ 1)).
(2) Сверхэкспрессия RBP4 у мышей показывает гипергликемию и гиперинсулинемию, и RBP4 нокаутированные мыши показывают содействие переносимости глюкозы и чувствительности к инсулину в качестве фенотипа (см. 436, 356-362 (2005) (непатентный документ 1)).
(3) Мыши, вскармливаемые кормом с высоким содержанием жира, обнаруживают высокий показатель RBP4 в крови, что соотносится с индуцированием стойкости к инсулину (см. 436, 356-362 (2005) (непатентный документ 1)).
(4) Мыши с моделью заболевания, показывающей диабет и патологию ожирения, такие как ob/ob мыши, мыши с 11β-HCD1 сверхэкспрессией (со специфичной адипозной тканью), MC4R нокаутированные мыши, GLUT4 нокаутированные мыши (со специфичной адипозной тканью и скелетной мышцей) и аналогичные также обнаруживают высокий показатель RBP4 в крови (см. Nature 436, 356-362 (2005) (непатентный документ 1)).
(5) Сообщалось, что концентрация RBP4 в крови и чувствительность к инсулину и скорость удаления сахара у людей соотносятся обратно пропорционально. В испытании с зажимом эугликемической гиперинсулинемической глюкозы скорость вливания глюкозы снижается по мере того, как концентрация RBP4 в крови увеличивается (см. Cell Metab., 6, 79-87 (2007) (непатентный документ 2)).
(6) Хотя известно, что физическая зарядка улучшает чувствительность к инсулину, проявляется крайне высокая корреляция между таким улучшающим эффектом и понижением концентрации RBP4 в крови (см. N. Engl. J. Med., 354, 2552-2563 (2006) (непатентный документ 3)).
(7) WO 2005/059564 (патентная ссылка 1) описывает, что соединение, которое контролирует или регулирует активность RBP4, полезно для лечения стойкости к инсулину.
RBP4 стабильно присутствует в крови в форме комплекса, получающегося в результате связывания ретинола и TTR (транстиретина). Когда RBP4 диссоциируется из TTR и становится свободным, он разлагается и сравнительно быстро выводится из почек. Фенретинид, производное ретинола, ингибирует связывание RBP4 и ретинола, и впоследствии ингибирует образование комплекса с TTR. Известно, что введение фенретинида животным индуцирует понижение RBP4 в крови (см. Biochim. Biophys. Acta, 1294, 48-54 (1996) (непатентный документ 4)).
На основании таких указанных выше находок ожидается, что соединение, которое ингибирует образование комплекса RBP4 и TTR путем ингибирования связывания RBP4 и ретинола, понижает концентрацию RBP4 в крови и впоследствии индуцирует коррекцию гиперглицемии и улучшения стойкости к инсулину.
Как упоминалось выше, соединение способное к понижению концентрации RBP4 в крови, может быть терапевтическим лекарством от диабета.
Кроме того, в последние годы имеется сообщение, свидетельствующее, что показатель RBP4 в крови и TG (триглицерида) в крови или показатель LDL холестерина положительно коррелируют у людей, а показатель RBP4 в крови отрицательно коррелирует с показателем HDL холестерина (см. J. Atheroscler. Thromb., 13, 209-215 (2006) (непатентный документ 5), N. Engl. J. Med., 355, 1392-1395 (2006) (непатентный документ 6), Diabetes, 56 (Supplement 1), A378 (1477-P) (2007) (непатентный документ 7)), предполагая, таким образом, взаимосвязь между RBP4 и метаболизмом липидов.
Ввиду вышеизложенного, лекарственное средство, оказывающее действие по понижению показателя (концентрации) RBP4 в крови (называемое также “RBP4 понижающим действием” в настоящем описании) (называемый в настоящем описании также “RBP4 понижающим агентом”), может быть агентом для профилактики или лечения гиперлипидемии.
Как упоминается выше, лекарственное средство, обладающее действием по понижению показателя (концентрации) RBP4 в крови (называемым также в настоящем описании “RBP4 понижающим действием”) (называемый также в настоящем описании “RBP4 понижающим агентом”), может быть широко применимым от заболеваний, связанных со стилем жизни (диабета, гиперлипидемии и аналогичных).
В качестве соединения, имеющего структуру, сходную со структурой соединения настоящего изобретения, известны следующие соединения.
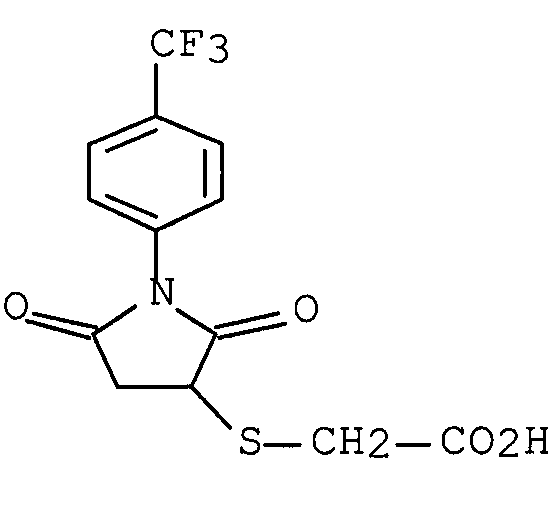
1) WO 03/031984 (патентный документ 2) раскрывает следующее соединение

2) Zhongguo Yaoke Daxue Xuebao (1991), 22(6), 330-3 (непатентный документ 8) раскрывает следующее соединение

CAS регистрационный №: 143247-34-1
3) Следующие соединения зарегистрированы в STN базе данных.

CAS регистрационный №: 736168-64-2
CAS регистрационный №: 452358-00-8

CAS регистрационный №: 1094556-59-8
4) патент США 2004/116417 (патентный документ 3) раскрывает следующее соединение

в которых
R1 представляет собой ароматическую кольцевую группу (ароматическая кольцевая группа необязательно замещена галогеном, C1-4 алкокси, C1-4 алкилом (включая циклический алкил), C1-4 алкилтио, нитро, трифторметилом, трифторметокси, метилендиокси, необязательно замещенным азотсодержащей гетероциклической группой и аналогичными);
R2 представляет водород, C1-3 алкил (необязательно замещенный необязательно сложноэтерифицированной карбоновой кислотой) или аналогичные; и
R3 и R4 представляют собой независимо водород или C1-4 алкил.
5) WO 2006/043064 (патентный документ 4) раскрывает следующие соединения

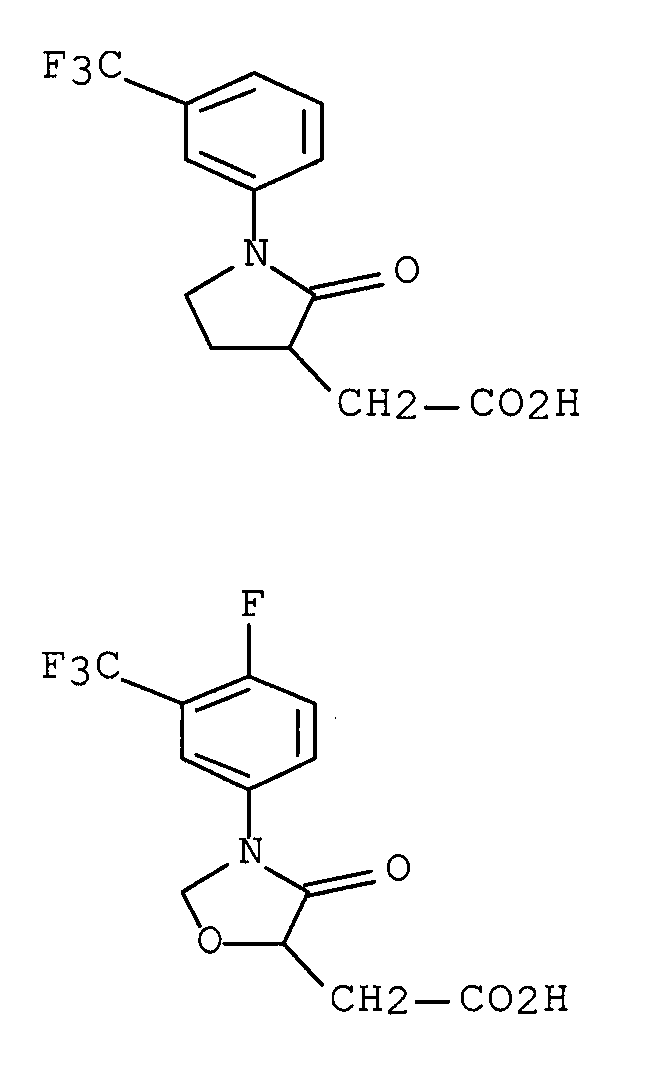
6) WO 95/33719 (патентный документ 5) раскрывает следующие соединения


7) ЕР 200415 А (патентный документ 6) раскрывает следующие соединения

8) JP-A-02-053780 (патентный документ 7) раскрывает следующее соединение

Однако не сообщалось, что вышеупомянутое соединение обладает RBP4 понижающим действием.
Перечень ссылок
Патентные документы
Патентный документ 1: WO 2005/059564
Патентный документ 2: WO 03/31984
Патентный документ 3: US 2004/116417
Патентный документ 4: WO 2006/043064
Патентный документ 5: WO 95/33719
Патентный документ 6: ЕР 200415 А
Патентный документ 7: JP-A-02-053780
Непатентные документы
Непатентный документ 1: 436, 356-362 (2005)
Непатентный документ 2: Cell Metab., 6, 79-87 (2007)
Непатентный документ 3: N. Engl. J. Med., 354, 2552-2563 (2006)
Непатентный документ 4: Biochim. Biophys. Acta, 1294, 48-54 (1996)
Непатентный документ 5: J. Atheroscler. Thromb., 13, 209-215 (2006)
Непатентный документ 6: N. Engl. J. Med., 355, 1392-1395 (2006)
Непатентный документ 7: Diabetes, 56(Supplement 1), A378 (1477-P) (2007)
Непатентный документ 8: Zhongguo Yaoke Daxue Xuebao (1991), 22(6), 330-3
Краткое содержание изобретения
Проблемы, решаемые изобретением
Настоящее изобретение направлено на предоставление соединения, обладающего RBP4 понижающим действием, полезного в качестве лекарственного средства для профилактики или лечения диабета, гиперлипидемии и аналогичных.
Средства решения проблем
В результате интенсивных исследований соединений, обладающих RBP4 понижающим действием, авторы изобретения неожиданно обнаружили соединения, представленные следующей формулой (I), их соль или их пролекарство обладает превосходным RBP4 понижающим действием, что привело в результате к открытию настоящего изобретения.
Соответственно, настоящее изобретение относится к
[1] соединению, представленному формулой

в которой
кольцо А представляет собой 5-членный неароматический гетероцикл, необязательно дополнительно замещенный одним заместителем;
кольцо В представляет собой необязательно дополнительно замещенное бензольное кольцо; и
Х представляет собой связь, O, CH2O, OCH2, CH2, (CH2)2, S, CH2S, SCH2, S(O), CH2S(O), S(O)CH2, S(O)2, CH2S(O)2 или S(O)2CH2,
при условии, что
{(3S,5R)-1-[4-(трифторметил)бензил]-5-[4-(трифторметил)фенил]пирролидин-3-ил}уксусная кислота,
{(3S,5R)-1-[2,5-бис(трифторметил)бензил]-5-[4-(трифторметил)фенил]пирролидин-3-ил}уксусная кислота,
{4-оксо-3-[3-(трифторметил)фенил]-1,3-тиазолидин-5-ил}уксусная кислота,
{2-оксо-1-[3-(трифторметил)фенил]пирролидин-3-ил}уксусная кислота,
{3-[4-фтор-3-(трифторметил)фенил]-4-оксо-1,3-оксазолидин-5-ил}уксусная кислота,
{4-оксо-3-[3-(трифторметил)фенил]-1,3-оксазолидин-5-ил}уксусная кислота,
{3-[2-хлор-5-(трифторметил)фенил]-4-оксо-1,3-тиазолидин-5-ил}уксусная кислота, и
{5-оксо-1-[3-(трифторметил)фенил]-4,5-дигидро-1Н-пиразол-3-ил}уксусная кислота
исключаются,
или к его соли;
[2] соединению упомянутой выше формулы [1], в которой Х представляет собой O, CH2O, OCH2, CH2, S, CH2S, SCH2, S(O) или S(O)2;
[3] соединению упомянутой выше формулы [1], в которой кольцо В представляет бензольное кольцо, необязательно дополнительно замещенное 1-3 заместителями, выбранными из
(а) атома галогена,
(b) C1-6 алкильной группы, необязательно замещенной 1-3 атомами галогена, и
(с) C1-6 алкоксигруппы, необязательно замещенной 1-3 атомами галогена;
[4] соединению упомянутой выше формулы [1], в которой кольцо А представляет собой 5-членный неароматический гетероцикл, необязательно дополнительно замещенный одним заместителем, выбранным из C1-6 алкильной группы и оксогруппы;
[5] соединению упомянутой выше формулы [1], в которой кольцо А представляет собой пирролидиновое кольцо или тетрагидрофурановое кольцо, каждое из которых необязательно дополнительно замещено одной оксогруппой;
[6] соединению упомянутой выше формулы [1], в которой кольцо А представляет собой 5-членный неароматический гетероцикл, необязательно дополнительно замещенный одним заместителем, выбранным из C1-6 алкильной группы и оксогруппы;
кольцо В представляет бензольное кольцо, необязательно дополнительно замещенное 1-3 заместителями, выбранными из
(а) атома галогена,
(b) C1-6 алкильной группы, необязательно замещенной 1-3 атомами галогена, и
(с) C1-6 алкоксигруппы, необязательно замещенной 1-3 атомами галогена; и
Х представляет собой O, CH2O, OCH2, CH2, S, CH2S, SCH2, S(O) или S(O)2;
[7] соединению упомянутой выше формулы [1], в которой кольцо А представляет собой пирролидиновое кольцо или тетрагидрофурановое кольцо, каждое из которых необязательно дополнительно замещено одной оксогруппой;
кольцо В представляет бензольное кольцо, необязательно дополнительно замещенное 1-3 заместителями, выбранными из
(а) атома галогена,
(b) C1-6 алкильной группы, необязательно замещенной 1-3 атомами галогена, и
(с) C1-6 алкоксигруппы, необязательно замещенной 1-3 атомами галогена; и
Х представляет связь;
[8] соединению упомянутой выше формулы [1], в которой кольцо А представляет собой 5-членный неароматический гетероцикл, необязательно дополнительно замещенный одним заместителем, выбранным из C1-6 алкильной группы и оксогруппы;
кольцо В представляет необязательно дополнительно замещенное бензольное кольцо; и
Х представляет собой O, S или CH2;
[9] ({(3S)-1-[3,5-бис(трифторметил)фенил]пирролидин-3-ил}окси)уксусной кислоте или ее соли;
[10] ({1-[4-хлор-3-(трифторметил)фенил]пирролидин-3-ил}сульфанил)уксусной кислоте или ее соли;
[11] 3-{(2R,5S)-5-[3,5-бис(трифторметил)фенил]тетрагидрофуран-2-ил}пропановой кислоте или ее соли;
[12] пролекарству соединения упомянутой выше формулы [1];
[13] лекарственному средству, включающему в себя соединение упомянутой выше формулы [1] или его пролекарство;
[14] лекарственному средству упомянутого выше п.[13], которое является агентом, понижающим ретинол-связывающий белок 4;
[15] лекарственному средству упомянутого выше п.[13], которое является агентом для профилактики или лечения заболевания, связанного с ретинол-связывающим белком 4;
[16] лекарственному средству упомянутого выше п.[13], которое является агентом для профилактики или лечения диабета;
[17] к способу понижения ретинол-связывающего белка 4 у млекопитающих, который включает введение млекопитающему эффективного количества соединения упомянутой выше формулы [1] или его пролекарства;
[18] способу профилактики или лечения диабета у млекопитающих, который включает введение млекопитающему эффективного количества соединения упомянутой выше формулы [1] или его пролекарства;
[19] к применению соединения упомянутой выше формулы [1] или его пролекарства для производства агента, понижающего ретинол-связывающий белок 4;
[20] к применению соединения упомянутой выше формулы [1] или его пролекарства для производства агента для профилактики или лечения диабета;
и аналогичным.
В качестве еще одного воплощения настоящее изобретение относится к
[21] соединению, представленному формулой

в которой
кольцо А' представляет собой необязательно дополнительно замещенный 5-членный неароматический гетероцикл;
кольцо В представляет собой необязательно дополнительно замещенное бензольное кольцо; и
Х представляет собой связь, O, CH2O, OCH2, CH2, (CH2)2, S, CH2S, SCH2, S(O), CH2S(O), S(O)CH2, S(O)2, CH2S(O)2 или S(O)2CH2,
при условии, что
({1-[3,5-бис(трифторметил)фенил]-2,5-диоксоимидазолидин-4-ил}сульфанил)уксусная кислота;
3-{5-оксо-2-тиоксо-1-[3-(трифторметил)фенил]имидазолидин-4-ил}пропановая кислота;
({2,5-диоксо-1-[3-(трифторметил)фенил]пирролидин-3-ил}сульфанил)уксусная кислота; и
({2,5-диоксо-1-[4-(трифторметил)фенил]пирролидин-3-ил}сульфанил)уксусная кислота
исключаются,
или его соли;
[22] соединению упомянутого выше п.[21], в котором кольцо А' представляет собой 5-членный неароматический гетероцикл, необязательно дополнительно замещенный одним заместителем, выбранным из C1-6 алкильной группы и оксогруппы;
[23] пролекарству соединения упомянутого выше п.[21];
[24] лекарственному средству, включающему в себя соединение упомянутого выше п.[21] или его пролекарство;
[25] лекарственному средству упомянутого выше п.[24], которое является агентом, понижающим ретинол-связывающий белок 4;
[26] лекарственному средству упомянутого выше п.[24], которое является агентом для профилактики или лечения диабета;
или аналогичным.
Подробное описание изобретения
Определение каждого символа в формуле (I) и формуле (I') описывается подробно далее.
“Атом галогена” в настоящем описании обозначает, если не указано иное, атом фтора, атом хлора, атом брома или атом йода.
“C1-3 алкилендиоксигруппа” в настоящем описании обозначает, если не указано иное, метилендиокси, этилендиокси или аналогичные.
“C1-6 алкильная группа” в настоящем описании обозначает, если не указано иное, метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил, трет-бутил, пентил, изопентил, неопентил, 1-этилпропил, гексил, изогексил, 1,1-диметилбутил, 2,2-диметилбутил, 3,3-диметилбутил, 2-этилбутил или аналогичные.
“C1-6 алкоксигруппа” в настоящем описании обозначает, если не указано иное, метокси, этокси, пропокси, изопропокси, бутокси, изобутокси, втор-бутокси, трет-бутокси или аналогичные.
“C1-6 алкоксикарбонильная группа” в настоящем описании обозначает, если не указано иное, метоксикарбонил, этоксикарбонил, пропоксикарбонил, трет-бутоксикарбонил или аналогичные.
“C1-6 алкилкарбонильная группа” в настоящем описании обозначает, если не указано иное, ацетил, пропаноил, бутаноил, изобутаноил, пентаноил, изопентаноил, гексаноил или аналогичные.
“C6-14 арилкарбонильная группа” в настоящем описании обозначает, если не указано иное, бензоил, нафтилкарбонил, бифенилкарбонил или аналогичные.
“C1-6 алкилтиогруппа” в настоящем описании обозначает, если не указано иное, метилтио, этилтио, пропилтио, изопропилтио, бутилтио, изобутилтио, втор-бутилтио, трет-бутилтио, пентилтио, изопентилтио, неопентилтио, 1-этилпропилтио, гексилтио, изогексилтио, 1,1-диметилбутилтио, 2,2-диметилбутилтио, 3,3-диметилбутилтио, 2-этилбутилтио или аналогичные.
“C1-6 алкилсульфинильная группа” в настоящем описании обозначает, если не указано иное, метилсульфинил, этилсульфинил, пропилсульфинил, изопропилсульфинил, бутилсульфинил, изобутилсульфинил, втор-бутилсульфинил, трет-бутилсульфинил, пентилсульфинил, изопентилсульфинил, неопентилсульфинил, 1-этилпропилсульфинил, гексилсульфинил, изогексилсульфинил, 1,1-диметилбутилсульфинил, 2,2-диметилбутилсульфинил, 3,3-диметилбутилсульфинил, 2-этилбутилсульфинил или аналогичные.
“C1-6 алкилсульфонильная группа” в настоящем описании обозначает, если не указано иное, метилсульфонил, этилсульфонил, пропилсульфонил, изопропилсульфонил, бутилсульфонил, изобутилсульфонил, втор-бутилсульфонил, трет-бутилсульфонил, пентилсульфонил, изопентилсульфонил, неопентилсульфонил, 1-этилпропилсульфонил, гексилсульфонил, изогексилсульфонил, 1,1-диметилбутилсульфонил, 2,2-диметилбутилсульфонил, 3,3-диметилбутилсульфонил, 2-этилбутилсульфонил или аналогичные.
Кольцо А представляет собой 5-членный неароматический гетероцикл, необязательно замещенный одним заместителем.
Примеры “5-членного неароматического гетероцикла” “5-членного неароматического гетероцикла, необязательно замещенного одним заместителем” для кольца А включают пирролидин, пирролин, имидазолидин, имидазолин, пиразолидин, пиразолин, оксазолидин, оксазолин, тиазолидин, тиазолин, 1,1-диоксидотиазолидин, 1,1-диоксидотиазолин, изоксазолидин, изоксазолин, изотиазолидин, изотиазолин, 1,1-диоксидоизотиазолидин, 1,1-диоксидоизотиазолин, тетрагидрофуран, дигидрофуран, тетрагидротиенил, дигидротиенил, 1,1-диоксидотетрагидротиенил, 1,1-диксидодигидротиенил, диоксолил, диоксоланил и аналогичные. Из них пирролидин, имидазолидин, тетрагидрофуран и 1,1-диоксидоизотиазолидин являются предпочтительными, пирролидин и тетрагидрофуран - более предпочтительными, и пирролидин является особенно предпочтительным.
Кольцо А имеет, помимо кольца В и Х группы, один заместитель в замещаемом положении.
Примеры заместителя включают
(1) C3-10 циклоалкильную группу (например, циклопропил, циклогексил);
(2) C6-14 арильную группу (например, фенил, нафтил), необязательно замещенную 1-3 заместителями, выбранными из
(а) C1-6 алкильной группы, необязательно замещенной 1-3 атомами галогена,
(b) гидроксигруппы,
(с) C1-6 алкоксигруппы, необязательно замещенной 1-3 атомами галогена, и
(d) атома галогена,
(3) ароматическую гетероциклическую группу (например, тиенил, фурил, пиридил, пиразолил, имидазолил, тетразолил, оксазолил, тиазолил, оксадиазолил, тиадиазолил), необязательно замещенную 1-3 заместителями, выбранными из
(а) C1-6 алкильной группы, необязательно замещенной 1-3 атомами галогена,
(b) гидроксигруппы,
(с) C1-6 алкоксигруппы, необязательно замещенной 1-3 атомами галогена, и
(d) атома галогена;
(4) неароматическую гетероциклическую группу (например, тетрагидрофурил, морфолинил, тиоморфолинил, пиперидинил, пирролидинил, пиперазинил), необязательно замещенную 1-3 заместителями, выбранными из
(а) C1-6 алкильной группы, необязательно замещенной 1-3 атомами галогена,
(b) гидроксигруппы,
(с) C1-6 алкоксигруппы, необязательно замещенной 1-3 атомами галогена,
(d) атома галогена, и
(е) оксогруппы;
(5) аминогруппу, необязательно моно- или дизамещенную заместителем(лями), выбранным из
(а) C1-6 алкильной группы, необязательно замещенной 1-3 атомами галогена,
(b) C1-6 алкилкарбонильной группы, необязательно замещенной 1-3 атомами галогена,
(с) C6-14 арилкарбонильной группы, необязательно замещенной 1-3 атомами галогена, и
(d) C1-6 алкоксикарбонильной группы, необязательно замещенной 1-3 атомами галогена,
(е) C1-6 алкилсульфонильной группы (например, метилсульфонила, этилсульфонила, изопропилсульфонила, бутилсульфонила, пентилсульфонила), необязательно замещенной 1-3 атомами галогена,
(f) карбамоильной группы, необязательно моно- или дизамещенной C1-6 алкильной группой (ами), необязательно замещенной 1-3 атомами галогена, и
(g) ароматической гетероциклической группы (например, тиенила, фурила, пиридила, пиразолила, имидазолила, тетразолила, оксазолила, тиазолила, оксадиазолила, тиадиазолила);
[6] C1-6 алкилкарбонильную группу, необязательно замещенную 1-3 атомами галогена,
[7] C1-6 алкоксикарбонильную группу, необязательно замещенную 1-3 заместителями, выбранными из
(а) атома галогена,
(b) C1-6 алкоксигруппы, и
(с) C6-14 арильной группы (например, фенила);
[8] C1-6 алкилсульфонильную группу (например, метилсульфонил, этилсульфонил, изопропилсульфонил), необязательно замещенную 1-3 атомами галогена;
[9] карбамоильную группу, необязательно моно- или дизамещенную заместителем(лями), выбранными из
(а) C1-6 алкильной группы, необязательно замещенной 1-3 атомами галогена,
(b) C1-6 алкилсульфонильной группы (например, метилсульфонила, этилсульфонила, изопропилсульфонила, бутилсульфонила, пентилсульфонила), необязательно замещенной 1-3 атомами галогена,
(с) C6-14 арилсульфонильной группы (например, фенилсульфонила), необязательно замещенной ароматической гетероциклической группой (например, тиенилом, фурилом, пиридилом, пиразолилом, имидазолилом, тетразолилом, оксазолилом, тиазолилом, оксадиазолилом, тиадиазолилом), и
(d) ароматической гетероциклической группы (например, тиенила, фурила, пиридила, пиразолила, имидазолила, тетразолила, оксазолила, тиазолила, оксадиазолила, тиадиазолила);
[10] тиокарбамоильную группу, необязательно моно- или дизамещенную C1-6 алкильной группой(ами), необязательно замещенной 1-3 атомами галогена;
[11] сульфамоильную группу, необязательно моно- или дизамещенную C1-6 алкильной группой(ами), необязательно замещенной 1-3 атомами галогена;
[12] карбоксигруппу;
[13] гидроксигруппу;
[14] C1-6 алкоксигруппу, необязательно замещенную 1-3 заместителями, выбранными из
(а) атома галогена,
(b) карбоксигруппы,
(c) C1-6 алкоксигруппы,
(d) C1-6 алкоксикарбонильной группы, необязательно замещенной 1-3 C6-14 арильными группами (например, фенилом), и
(e) аминогруппы, необязательно моно- или дизамещенной заместителем(лями), выбранными из C1-6 алкильной группы и C1-6 алкоксикарбонильной группы;
[15] C2-6 алкенилоксигруппу (например, этенилокси), необязательно замещенную 1-3 атомами галогена;
[16] C7-13 аралкилоксигруппу (например, бензилокси);
[17] C6-14 арилоксигруппу (например, фенилокси, нафтилокси);
[18] C1-6 алкилкарбонилоксигруппу (например, ацетилокси, трет-бутилкарбонилокси);
[19] C6-14 арилкарбонильную группу, необязательно замещенную 1-3 заместителями, выбранными из
(а) атома галогена, и
(b) C1-6 алкильной группы, необязательно замещенной 1-3 атомами галогена;
[20] неароматическую гетероциклилкарбонильную группу (например, пирролидинилкарбонил, морфолинилкарбонил, тиоморфолинилкарбонил, 1-оксидотиоморфолинилкарбонил), необязательно замещенную 1-3 заместителями, выбранными из C1-6 алкильной группы, необязательно замещенной 1-3 атомами галогена;
[21] меркаптогруппу;
[22] C1-6 алкилтиогруппу (например, метилтио, этилтио), необязательно замещенную 1-3 заместителями, выбранными из
(а) атома галогена, и
(b) C1-6 алкоксикарбонильной группы;
[23] C7-13 аралкилтиогруппу (например, бензилтио);
[24] C6-14 арилтиогруппу (например, фенилтио, нафтилтио);
[25] цианогруппу;
[26] нитрогруппу;
[27] атом галогена;
[28] C1-3 алкилендиоксигруппу;
[29] ароматическую гетероциклилкарбонильную группу (например, пиразолилкарбонил, пиразинилкарбонил, изоксазолилкарбонил, пиридилкарбонил, тиазолилкарбонил), необязательно замещенную 1-3 заместителями, выбранными из C1-6 алкильной группы, необязательно замещенной 1-3 атомами галогена;
[30] формильную группу;
[31] C1-6 алкильную группу, необязательно замещенную 1-3 заместителями, выбранными из
(а) атома галогена,
(b) карбоксигруппы,
(с) гидроксигруппы,
(d) C1-6 алкоксикарбонильной группы,
(е) C1-6 алкоксигруппы, и
(f) аминогруппы, необязательно моно- или дизамещенной C1-6 алкильной группой(ами);
[32] C2-10 алкенильную группу (например, этенил, 1-пропенил), необязательно замещенную 1-3 заместителями, выбранными из
(а) атома галогена,
(b) карбоксигруппы,
(c) гидроксигруппы,
(d) C1-6 алкоксикарбонильной группы,
(e) C1-6 алкоксигруппы,
(f) аминогруппы, необязательно моно- или дизамещенной C1-6 алкильной группой(ами), и
(g) неароматической гетероциклической группы (например, тетрагидрофурила, морфолинила, тиоморфолинила, пиперидинила, пирролидинила, пиперазинила, тиазолидинила), необязательно замещенной 1-3 заместителями, выбранными из
(i) C1-6 алкильной группы, необязательно замещенной 1-3 атомами галогена,
(ii) гидроксигруппы,
(iii) C1-6 алкоксигруппы, необязательно замещенной 1-3 атомами галогена,
(iv) атома галогена, и
(v) оксогруппы;
[33] C7-13 аралкильную группу (например, бензил), необязательно замещенную 1-3 заместителями, выбранными из
(а) C1-6 алкильной группы, необязательно замещенной 1-3 атомами галогена,
(b) гидроксигруппы,
(с) C1-6 алкоксигруппы, и
(d) атома галогена,
[34] оксогруппу,
и аналогичные.
В качестве заместителя предпочтительны C1-6 алкильная группа и оксогруппа, а особенно предпочтительной является оксогруппа.
Кольцо А предпочтительно представляет собой 5-членный неароматический гетероцикл (предпочтительно пирролидин, имидазолидин, тетрагидрофуран, 1,1-диоксидоизотиазолидин), необязательно дополнительно замещенный одним заместителем, выбранным из C1-6 алкильной группы и оксогруппы, более предпочтительно 5-членный неароматический гетероцикл (предпочтительно пирролидин, имидазолидин, тетрагидрофуран, 1,1-диоксидоизотиазолидин), необязательно дополнительно замещенный одной оксогруппой, более предпочтительно пирролидин или тетрагидрофуран, каждый из которых дополнительно замещен одной оксогруппой, особенно предпочтительно пирролидин, не замещенный иным заместителем, чем кольцо В и Х группа.
Кольцо A' представляет собой необязательно дополнительно замещенный 5-членный неароматический гетероцикл.
Примеры “5-членного неароматического гетероцикла” “необязательно дополнительно замещенного 5-членного неароматического гетероцикла” для кольца A' включают в себя гетероциклы, сходные с “5-членным неароматическим гетероциклом” “5-членного неароматического гетероцикла, необязательно дополнительно замещенного одним заместителем” для кольца A. Из них предпочтительными являются пирролидин, имидазолидин, тетрагидрофуран и 1,1-диоксидоизотиазолидин, более предпочтительными являются пирролидин и тетрагидрофуран, и особенно предпочтительным пирролидин.
Кольцо A' необязательно имеет, кроме кольца В и Х группы, 1-3 заместителя в замещаемом положении(ях). Примеры заместителя включают заместители, приведенные для примера в качестве “заместителя”, который необязательно имеет для кольца А “5-членный неароматический гетероцикл” “5-членного неароматического гетероцикла, необязательно дополнительно замещенного одним заместителем”. Когда число заместителей составляет не менее 2, соответствующие заместители могут быть одними и теми же или различными.
В качестве заместителя предпочтительными являются C1-6 алкильная группа и оксогруппа, и особенно предпочтительна оксогруппа.
Кольцом A' предпочтительно является 5-членный неароматический гетероцикл (предпочтительно пирролидин, имидазолидин, тетрагидрофуран, 1,1-диоксидоизотиазолидин), необязательно дополнительно замещенный одним заместителем, выбранным из C1-6 алкильной группы и оксогруппы, более предпочтительно 5-членный неароматический гетероцикл (предпочтительно пирролидин, имидазолидин, тетрагидрофуран, 1,1-диоксидоизотиазолидин), необязательно дополнительно замещенный одной оксогруппой, более предпочтительно пирролидин и тетрагидрофуран, каждый из которых дополнительно замещен одной оксогруппой, особенно предпочтительно пирролидин, не замещенный иным заместителем, чем кольцо В и Х группа.
Кольцо В представляет собой необязательно дополнительно замещенное бензольное кольцо.
Кольцо В имеет необязательно, помимо кольца А и трифторметильной группы, 1-4 заместителя в замещаемом положении. Примеры заместителя включают заместители, приведенные для примера в качестве “заместителя” (исключая оксогруппу), который необязательно имеет для кольца А “5-членный неароматический гетероцикл” “5-членного неароматического гетероцикла, необязательно дополнительно замещенного одним заместителем”. Когда число заместителей составляет не менее 2, соответствующие заместители могут быть одними и теми же или различными.
В качестве заместителя предпочтительными являются
(1) атом галогена (предпочтительно атом фтора, атом хлора, атом брома),
(2) C1-6 алкильная группа, необязательно замещенная 1-3 атомами галогена (предпочтительно атомом фтора),
(3) C1-6 алкоксигруппа, необязательно дополнительно замещенная 1-3 атомами галогена,
(4) цианогруппа,
(5) аминогруппа, необязательно моно- или дизамещенная заместителем(лями), выбранным из
(а) C1-6 алкильной группы, необязательно замещенной 1-3 атомами галогена, и
(b) C1-6 алкилсульфонильной группы, необязательно замещенной 1-3 атомами галогена;
(6) неароматическая гетероциклилкарбонильная группа (например, тиоморфолинилкарбонил, 1-оксидотиоморфолинилкарбонил), необязательно замещенная 1-3 заместителями, выбранными из C1-6 алкильной группы, необязательно замещенной 1-3 атомами галогена,
и аналогичные.
Кольцо В предпочтительно является бензольным кольцом, необязательно дополнительно замещенным 1-4 заместителями, выбранными из
(1) атома галогена (предпочтительно атома фтора, атома хлора, атома брома),
(2) C1-6 алкильной группы, необязательно замещенной 1-3 атомами галогена (предпочтительно атомом фтора), (предпочтительно трифторметил),
(3) C1-6 алкоксигруппы (предпочтительно метокси), необязательно замещенной 1-3 атомами галогена,
(4) цианогруппы,
(5) аминогруппы, необязательно моно- или дизамещенной заместителем(лями), выбранным из
(а) C1-6 алкильной группы (предпочтительно метила), необязательно замещенной 1-3 атомами галогена, и
(b) C1-6 алкилсульфонильной группы (предпочтительно метилсульфонила), необязательно замещенной 1-3 атомами галогена, и
(6) неароматической гетероциклилкарбонильной группы (например, тиоморфолинилкарбонила, 1-оксидотиоморфолинилкарбонила), необязательно замещенной 1-3 заместителями, выбранными из C1-6 алкильной группы, необязательно замещенной 1-3 атомами галогена.
Кольцо В предпочтительно является бензольным кольцом, необязательно дополнительно замещенным 1-4 заместителями, выбранными из атома галогена (предпочтительно атома фтора, атома хлора, атома брома), и C1-6 алкильной группы, необязательно замещенной 1-3 атомами галогена (предпочтительно атомом фтора), (предпочтительно трифторметила).
В качестве еще одного воплощения кольцо В более предпочтительно является бензольным кольцом, необязательно дополнительно замещенным 1-3 заместителями, выбранными из
(1) атома галогена (предпочтительно атома фтора, атома хлора, атома брома),
(2) C1-6 алкильной группы, необязательно замещенной 1-3 атомами галогена (предпочтительно атомом фтора), (предпочтительно трифторметила), и
(3) C1-6 алкоксигруппы (предпочтительно метокси), необязательно замещенной 1-3 атомами галогена.
Согласно еще одному воплощению кольцом В более предпочтительно является бензольное кольцо, дополнительно замещенное 1-3 заместителями, выбранными из
(1) атома галогена (предпочтительно атома фтора, атома хлора, атома брома),
(2) C1-6 алкильной группы, необязательно замещенной 1-3 атомами галогена (предпочтительно атомом фтора), (предпочтительно трифторметила), и
(3) C1-6 алкоксигруппы (предпочтительно метокси), необязательно замещенной 1-3 атомами галогена.
Согласно данному воплощению кольцо В далее представляет собой более предпочтительно бензольное кольцо, дополнительно замещенное одним заместителем, выбранным из
(1) атома галогена (предпочтительно атома фтора, атома хлора, атома брома),
(2) C1-6 алкильной группы, необязательно замещенной 1-3 атомами галогена (предпочтительно атомом фтора), (предпочтительно трифторметила), и
(3) C1-6 алкоксигруппы (предпочтительно метокси), необязательно замещенной 1-3 атомами галогена,
где заместитель присоединен в 3-положении, а трифторметильная группа связана с кольцом В в 5-положении, при этом положением присоединения кольца А является 1-положение.
А именно, далее кольцом В является более предпочтительно бензольное кольцо, представленное формулой:
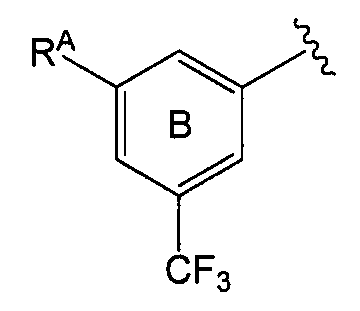
в которой
RA представляет
(1) атом галогена (предпочтительно атом фтора, атом хлора, атом брома),
(2) C1-6 алкильную группу, необязательно замещенную 1-3 атомами галогена (предпочтительно атомом фтора), (предпочтительно трифторметил), и
(3) C1-6 алкоксигруппу (предпочтительно метокси), необязательно замещенную 1-3 атомами галогена.
Особенно предпочтительно кольцом В является бензольное кольцо, представленное формулой:

в которой
RA представляет
(1) атом галогена (предпочтительно атом фтора, атом хлора, атом брома),
(2) C1-6 алкильную группу, необязательно замещенную 1-3 атомами галогена (предпочтительно атомом фтора), (предпочтительно трифторметил),
Х представляет собой связь, O, CH2O, OCH2, CH2, (CH2)2, S, CH2S, SCH2, S(O), CH2S(O), S(O)CH2, S(O)2, CH2S(O)2 или S(O)2CH2,
Х представляет собой предпочтительно связь, O, CH2, S, CH2S, SCH2, S(O) или S(O)2, более предпочтительно O, CH2 или S.
Согласно еще одному воплощению Х представляет собой предпочтительно O, CH2O, OCH2, CH2, S, CH2S, SCH2, S(O) или S(O)2, более предпочтительно O, CH2O, СН2, S, SCH2, S(O) или S(O)2.
Согласно еще одному воплощению Х представляет собой предпочтительно связь.
Предпочтительные примеры соединения (I) включают следующие соединения.
[Соединение I-А]
Соединение, представленное формулой:
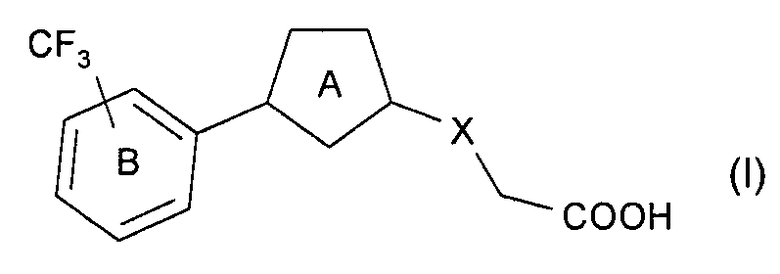
в которой
кольцом A предпочтительно является 5-членный неароматический гетероцикл (предпочтительно пирролидин, имидазолидин, тетрагидрофуран, 1,1-диоксидоизотиазолидин), необязательно дополнительно замещенный одним заместителем, выбранным из C1-6 алкильной группы и оксогруппы;
кольцо В является бензольным кольцом, необязательно дополнительно замещенным 1-4 заместителями, выбранными из атома галогена (предпочтительно атома фтора, атома хлора, атома брома), и C1-6 алкильной группы, необязательно замещенной 1-3 атомами галогена (предпочтительно атомом фтора), (предпочтительно трифторметила); и
Х представляет собой O, S или CH2,
или его соль.
[Соединение I-В]
Соединение, представленное формулой:

в которой
кольцом A является 5-членный неароматический гетероцикл (предпочтительно пирролидин, имидазолидин, тетрагидрофуран, 1,1-диоксидоизотиазолидин), необязательно дополнительно замещенный одним заместителем, выбранным из C1-6 алкильной группы и оксогруппы;
кольцо В является бензольным кольцом, необязательно дополнительно замещенным 1-4 заместителями, выбранными из
(1) атома галогена (предпочтительно атома фтора, атома хлора, атома брома),
(2) C1-6 алкильной группы, необязательно замещенной 1-3 атомами галогена (предпочтительно атомом фтора), (предпочтительно трифторметила),
(3) C1-6 алкоксигруппы (предпочтительно метокси), необязательно замещенной 1-3 атомами галогена,
(4) цианогруппы,
(5) аминогруппы, необязательно моно- или дизамещенной заместителем(лями), выбранным из
(а) C1-6 алкильной группы (предпочтительно метила), необязательно замещенной 1-3 атомами галогена, и
(b) C1-6 алкилсульфонильной группы (предпочтительно метилсульфонила), необязательно замещенной 1-3 атомами галогена, и
(6) неароматической гетероциклилкарбонильной группы (например, тиоморфолинилкарбонила, 1-оксидотиоморфолинилкарбонила), необязательно замещенной 1-3 заместителями, выбранными из C1-6 алкильной группы, необязательно замещенной 1-3 атомами галогена; и
Х представляет собой связь, O, CH2O, OCH2, CH2, (CH2)2, S, CH2S, SCH2, S(O), CH2S(O), S(O)CH2, S(O)2, CH2S(O)2 или S(O)2CH2,
при условии, что
{4-оксо-3-[3-(трифторметил)фенил]-1,3-тиазолидин-5-ил}уксусная кислота,
{2-оксо-1-[3-(трифторметил)фенил]пирролидин-3-ил}уксусная кислота,
{3-[4-фтор-3-(трифторметил)фенил]-4-оксо-1,3-оксазолидин-5-ил}уксусная кислота,
{4-оксо-3-[3-(трифторметил)фенил]-1,3-оксазолидин-5-ил}уксусная кислота,
{3-[2-хлор-5-(трифторметил)фенил]-4-оксо-1,3-тиазолидин-5-ил}уксусная кислота, и
{5-оксо-1-[3-(трифторметил)фенил]-4,5-дигидро-1Н-пиразол-3-ил}уксусная кислота
исключаются,
или его соль.
[Соединение I-С]
Соединение, представленное формулой:

в которой
кольцом A является 5-членный неароматический гетероцикл (предпочтительно пирролидин, имидазолидин, тетрагидрофуран, 1,1-диоксидоизотиазолидин), необязательно дополнительно замещенный одним заместителем, выбранным из C1-6 алкильной группы и оксогруппы;
кольцо В является бензольным кольцом, необязательно дополнительно замещенным 1-3 заместителями, выбранными из
(а) атома галогена (предпочтительно атома фтора, атома хлора, атома брома),
(b) C1-6 алкильной группы, необязательно замещенной 1-3 атомами галогена (предпочтительно атомом фтора), (предпочтительно трифторметила), и
(с) C1-6 алкоксигруппы (предпочтительно метокси), необязательно замещенной 1-3 атомами галогена; и
Х представляет собой O, CH2O, OCH2, CH2, S, CH2S, SCH2, S(O) или S(O)2,
или его соль.
[Соединение I-D]
Соединение, представленное формулой;

в которой
кольцо А представляет собой пирролидиновое или тетрагидрофурановое кольцо, каждое из которых необязательно дополнительно замещено одной оксогруппой;
кольцо В является бензольным кольцом, необязательно дополнительно замещенным 1-3 заместителями, выбранными из
(а) атома галогена (предпочтительно атома фтора, атома хлора, атома брома),
(b) C1-6 алкильной группы, необязательно замещенной 1-3 атомами галогена (предпочтительно атомом фтора), (предпочтительно трифторметила), и
(с) C1-6 алкоксигруппы (предпочтительно метокси), необязательно замещенной 1-3 атомами галогена; и
Х представляет собой связь,
или его соль.
[Соединение I-Е]
Соединение, представленное формулой;

в которой
кольцом A является 5-членный неароматический гетероцикл (предпочтительно пирролидин, имидазолидин, тетрагидрофуран, 1,1-диоксидоизотиазолидин), необязательно дополнительно замещенный одним заместителем, выбранным из C1-6 алкильной группы и оксогруппы;
кольцо В является необязательно дополнительно замещенным бензольным кольцом; и
Х представляет собой O, S или CH2,
или его соль.
[Соединение I-F]
Соединение, представленное формулой;
в которой
кольцо А представляет собой пирролидиновое или тетрагидрофурановое кольцо, каждое из которых необязательно дополнительно замещено одной оксогруппой;
кольцо В является бензольным кольцом, представленным формулой:

в которой
RA представляет
(1) атом галогена (предпочтительно атом фтора, атом хлора, атом брома),
(2) C1-6 алкильную группу, необязательно замещенную 1-3 атомами галогена (предпочтительно атомом фтора), (предпочтительно трифторметил), или
(3) C1-6 алкоксигруппу (предпочтительно метокси), необязательно замещенную 1-3 атомами галогена; и
Х представляет собой связь, O, CH2O, OCH2, CH2, (CH2)2, S, CH2S, SCH2, S(O), CH2S(O), S(O)CH2, S(O)2, CH2S(O)2 или S(O)2CH2,
или его соль.
[Соединение I-G]
({(3S)-1-[3,5-бис(трифторметил)фенил]пирролидин-3-ил}окси)уксусная кислота или ее соль.
({1-[4-хлор-3-(трифторметил)фенил]пирролидин-3-ил}сульфанил)уксусная кислота или ее соль.
3-{(2R,5S)-5-[3,5-бис(трифторметил)фенил]тетрагидрофуран-2-ил}пропановая кислота или ее соль.
В качестве соли соединения, представленного формулой (I) или формулой (I'), предпочтительной является фармакологически приемлемая соль. Примеры такой соли включают соли с неорганическим основанием, соли с органическим основанием, соли с неорганической кислотой, соли с органической кислотой, соли с основной или кислой аминокислотой и аналогичные.
Предпочтительные примеры соли с неорганическим основанием включают соли щелочных металлов, такие как соль натрия, калия и аналогичные; соли щелочно-земельных металлов, такие как соль кальция, магния и аналогичные; соль алюминия; аммониевую соль и аналогичные.
Предпочтительные примеры соли с органическим основанием включают соли с триметиламином, триэтиламином, пиридином, пиколином, этаноламином, диэтаноламином, триэтаноламином, трометамин[трис(гидроксиметил)метиламином], трет-бутиламином, циклогексиламином, бензиламином, дициклогексиламином, N,N-дибензилэтилендиамином и аналогичные.
Предпочтительные примеры соли с неорганической кислотой включают соли с соляной кислотой, бромистоводородной кислотой, азотной кислотой, серной кислотой, фосфорной кислотой и аналогичные.
Предпочтительные примеры соли с органической кислотой включают соли с муравьиной, уксусной, трифторуксусной, фталевой, фумаровой, щавелевой, винной, малеиновой, лимонной, янтарной, яблочной, метансульфоновой, бензолсульфоновой, п-толуолсульфоновой кислотами и аналогичные.
Предпочтительные примеры соли с основными аминокислотами включают соли с аргинином, лизином, орнитином и аналогичные.
Предпочтительные примеры соли с кислыми аминокислотами включают соли с аспарагиновой кислотой, глутаминовой кислотой и аналогичные.
Пролекарство соединения, представленного формулой (I) или формулой (I') (называемого далее также совместно соединением (I), обозначает соединение, которое превращается в соединение (I) с помощью реакции, проходящей благодаря ферменту, желудочной кислоте и пр. в физиологических условиях в живом организме, а именно, соединение, которое превращается в соединение (I) окислением, восстановлением, гидролизом и др., обусловленными активностью фермента; соединение, которое превращается в соединение (I) гидролизом и др. благодаря желудочной кислоте, и др.
Примеры пролекарства соединения (I) включают соединение, в котором аминогруппа соединения (I) подвергается ацилированию, алкилированию или фосфорилированию (например, соединение, в котором аминогруппа соединения (I) подвергается эйкозаноилированию, аланилированию, пентиламинокарбонилированию, (5-метил-2-оксо-1,3-диоксолен-4-ил)метоксикарбонилированию, тетрагидрофуранилированию, пирролидилметилированию, пивалоилоксиметилированию и трет-бутилированию);
соединение, в котором гидроксигруппа соединения (I) подвергается ацилированию, алкилированию, фосфорилированию или борированию (напримр, соединение, в котором гидроксигруппа соединения (I) подвергается ацилированию, пальмитоилированию, пропаноилированию, пивалоилированию, сукцинилированию, фумарилированию, аланилированию или диметиламинометилкарбонилированию);
соединение, в котором карбоксильная группа соединения (I) подвергается сложной этерификации или амидированию (например, соединение, в котором карбоксильная группа соединения (I) подвергается этерификации этилом, фенилом, карбоксиметилом, диметиламинометилом, пивалоилоксиметилом, этоксикарбонилоксиэтилом, фталидилом, (5-метил-2-оксо-1,3-диоксолен-4-ил)метилом, подвергается этерификации циклогексилоксикарбонилэтилом или метиламидированию) и аналогичные. Данные соединения могут получаться из соединения (I) с помощью способа, известного самого по себе.
Пролекарством для соединения (I) может быть также соединение, которое превращается в соединение (I) в физиологических условиях, таких как описаны в IYAKUHIN no KAIHATSU, Development of Pharmaceuticals, Vol. 7, Design of Molecules, стр. 163-198, Published by HIROKAWA SHOTEN, 1990.
Соединением (I) может быть сольват (например, гидрат) или не сольват (например, ангидрид).
В дополнение, соединение (I) может быть мечено изотопом (например, 3Н, 14C, 35S, 125I) и аналогичными.
В дополнение, соединением (I) охватывается также дейтериевый конвертер, в котором 1H превращен в 2H(D).
Соединение (I) или его пролекарство (называемое здесь далее иногда для простоты как соединение настоящего изобретения) обладает низкой токсичностью и может использоваться в качестве агента для профилактики или лечения разнообразных заболеваний, упоминаемых ниже, у млекопитающих (например, людей, мышей, крыс, кроликов, собак, кошек, крупного рогатого скота, лошадей, свиней, обезьян) непосредственно или в форме фармацевтической композиции при смешении с фармакологически приемлемым носителем и аналогичными.
Примеры фармакологически приемлемого носителя включают различные органические или неорганические вещества-носители, обычно используемые в качестве материалов препарата, которые добавляются в качестве эксиципиента, смазки, связующего или дезинтегранта для твердых дозированных форм; в качестве растворителя, солюбилизирующего агента, суспендирующего агента, агента изотоничности, буферного или успокоительного агента для жидкого препарата, и аналогичные. Когда необходимо, могут также использоваться добавки к препарату, такие как консерванты, антиоксиданты, окрашивающие агенты, подсластители и аналогичные.
Предпочтительные примеры эксципиента включают лактозу, сахарозу, D-маннит, D-сорбит, крахмал, преджелатинированный крахмал, декстрин, кристаллическую целлюлозу, низкозамещенную гидроксипропилцеллюлозу, натриевую карбоксиметилцеллюлозу, аравийскую камедь, пуллулан, легкую безводную кремниевую кислоту, синтетический силикат алюминия и алюминометасиликат магния.
Предпочтительные примеры смазки включают стеарат магния, стеарат кальция, тальк и коллоидную двуокись кремния.
Предпочтительные примеры связующего включают преджелатинированный крахмал, сахарозу, желатин, аравийскую камедь, метилцеллюлозу, карбоксиметилцеллюлозу, натриевую карбоксиметилцеллюлозу, кристаллическую целлюлозу, сахарозу, D-маннит, трегалозу, декстрин, пуллулан, гидроксипропилцеллюлозу, гидроксипропилметилцеллюлозу и поливинилпирролидон.
Предпочтительные примеры дезинтегрирующего агента включают лактозу, сахарозу, крахмал, карбоксиметилцеллюлозу, кальциевую карбоксиметилцеллюлозу, натриевую кроскармелозу, натриевый карбоксиметилкрахмал, легкую безводную кремниевую кислоту и низкозамещенную гидроксипропилцеллюлозу.
Предпочтительные примеры растворителя включают воду для инъекции, физиологический раствор, раствор Рингера, спирт, пропиленгликоль, полиэтиленгликоль, кунжутное масло, кукурузное масло, оливковое масло и хлопковое масло.
Предпочтительные примеры солюбилизирующего агента включают полиэтиленгликоль, пропиленгликоль, D-маннит, трегалозу, бензилбензоат, этанол, трисаминометан, холестерин, триэтаноламин, карбонат натрия, цитрат натрия, салицилат натрия и ацетат натрия.
Предпочтительные примеры суспендирующего агента включают поверхностно-активные вещества, такие как стеарилтриэтаноламин, лаурилсульфат натрия, лауриламинопропионовую кислоту, лецитин, хлорид бензалкония, бензетонийхлорид, глицерилмоностеарат и аналогичные; гидрофильные полимеры, такие как поливиниловый спирт, поливинилпирролидон, натриевую карбоксиметилцеллюлозу, метилцеллюлозу, гидроксиметилцеллюлозу, гидроксиэтилцеллюлозу, гидроксипропилцеллюлозу и аналогичные; полисорбаты и полиоксиэтилен-гидрированное касторовое масло.
Предпочтительные примеры агента изотоничности включают хлорид натрия, глицерин, D-маннит, D-сорбит и глюкозу.
Предпочтительные примеры буферного агента включают такие буферные агенты, как фосфат, ацетат, карбонат, цитрат и аналогичные.
Предпочтительные примеры успокоительного агента включают бензиловый спирт.
Предпочтительные примеры консерванта включают параоксибензоаты, хлорбутанол, бензиловый спирт, фенэтиловый спирт, дегидроуксусную кислоту и сорбиновую кислоту.
Предпочтительные примеры антиоксиданта включают сульфит, аскорбат и аналогичные.
Предпочтительные примеры окрашивающего агента включают водные пищевые гудроновые красители (например, пищевые красители, такие как пищевой красный № 2 и № 3, пищевой желтый № 4 и № 5, пищевой синий № 1 и № 2, и пр.), не растворимый в воде краплак (например, алюминиевая соль упомянутого выше водного пищевого гудронового окрашивающего агента) и натуральный краситель (например, β-каротен, хлорофилл, оксид железа красный).
Предпочтительные примеры подслащивающего агента включают натриевый сахарин, дикалийглицирризинат, аспартам и стевия.
Примеры дозированных форм вышеупомянутой фармацевтической композиции включают в себя пероральные препараты, такие как таблетки (включая покрытые сахаром таблетки, покрытые пленкой таблетки, подъязычные таблетки, измельчающиеся во рту таблетки), капсулы (включая мягкие капсулы, микрокапсулы), гранулы, порошки, пастилки, сиропы, эмульсии, суспензии, пленки (например, измельчаемые во рту пленки) и аналогичные; и парентеральные агенты, такие как инъекции (например, подкожные инъекции, внутривенные, внутримышечные, интраперитонеальные инъекции, капельные вливания), наружные препараты (например, дермальные препараты, мази), свечи (например, ректальные свечи, вагинальные свечи), пилюли, назальные препараты, легочные препараты (ингаляционные), глазные капли и аналогичные. Их можно безопасно вводить перорально или парентерально (например, местно, ректально, внутривенно).
Данными препаратами могут быть препараты регулируемого высвобождения (например, микрокапсулы длительного высвобождения), такие как препарат немедленного высвобождения, препарат длительного высвобождения и аналогичные.
Фармацевтическую композицию можно получить с помощью способа, используемого обычно в области фармацевтических препаратов, например, по методу, описанному в японской фармакопее, и аналогичным.
Содержание соединения настоящего изобретения в фармацевтической композиции составляет около 0,01-100% масс., предпочтительно около 2-85% масс., от всей композиции.
Хотя доза соединения настоящего изобретения варьирует в зависимости от субъекта, которому она вводится, способа введения, заболевания и аналогичных факторов, она составляет, например, около 1-1000 мг, предпочтительно около 3-300 мг, более предпочтительно около 10-200 мг, в количестве соединения настоящего изобретения в качестве активного ингредиента перорального препарата для введения взрослому (с весом тела около 60 кг) в качестве профилактического или терапевтического лекарственного средства от диабета, и данная доза может вводиться в виде одной или нескольких порций в день.
Во время получения перорального препарата можно применять покрытие, необходимое с целью маскировки вкуса, энтерических свойств или продолжительности или длительности введения.
Примеры покрывающей основы, используемой для покрытия, включают сахарную основу покрытия, водную пленочную основу покрытия, энтерическую пленочную основу покрытия и пленочную основу покрытия длительного высвобождения.
В качестве сахарной основы покрытия используют сахарозу. Кроме того, один или более видов, выбираемых из талька, осажденного карбоната кальция, желатина, аравийской камеди, пуллулана, карнаубского воска и аналогичных, могут использоваться в сочетании.
Примеры водной пленочной основы покрытия включают полимеры целлюлозы, такие как гидроксипропилцеллюлоза, гидроксипропилметилцеллюлоза, гидроксиэтилцеллюлоза, метилгидроксиэтилцеллюлоза и др.; синтетические полимеры, такие как диэтиламиноацетат поливинилацеталя, аминоалкилметакрилатный сополимер Е [Eudragit E (торговая марка)], поливинилпирролидон и др.; и полисахариды, такие как пуллулан и др.
Примеры энтерической пленочной основы покрытий включают полимеры целлюлозы, такие как фталат гидроксипропилметилцеллюлозы, сукцинат ацетата гидроксипропилметилцеллюлозы, карбоксиметилэтилцеллюлоза, фталат ацетата целлюлозы и др.; акриловые полимеры, такие как сополимер L метакриловой кислоты [Eudragit L (торговая марка)], сополимер LD метакриловой кислоты [Eudragit L-30D55 (торговая марка)], сополимер S метакриловой кислоты [Eudragit S (торговая марка)] и др.; и встречающиеся в природе вещества, такие как шеллак и др.
Примеры пленочной основы покрытия длительного высвобождения включают полимеры целлюлозы, такие как этилцеллюлоза и др.; и акриловые полимеры, такие как сополимер RS аминоалкилметакрилата [Eudragit RS (торговая марка)], суспензия этилакрилат-метилметарилатного сополимера [Eudragit NE (торговая марка)], и др.
Упомянутые выше основы покрытий могут использоваться после смешения с двумя или более их видами в соответствующих соотношениях. Для покрытия может использоваться, например, экранирующий свет агент, такой как оксид титана, красный оксид железа(3) и аналогичные.
Соединение настоящего изобретения показывает низкую токсичность (например, острую токсичность, хроническую токсичность, генетическую токсичность, репродуктивную токсичность, кардиотоксичность, карциногенность и др.) и мало побочных эффектов. Поэтому оно может использоваться в качестве агента для профилактики или лечения, или диагностики различных заболеваний млекопитающих (например, человека, крупного рогатого скота, лошадей, собак, кошек, обезьян, мышей, крыс).
Соединение настоящего изобретения обладает превосходным RBP4 (ретинол-связывающий белок 4) понижающим действием. Соответственно, соединение настоящего изобретения полезно в качестве агента для профилактики или лечения заболеваний и состояний, связанных с увеличением RBP4.
Соединение настоящего изобретения, в частности, может использоваться в качестве агента для профилактики или лечения ожирения, диабета (например, диабета типа 1, диабета типа 2, диабета беременных, диабета тучных), гиперлипидемии (например, гипертриглицеридемии, гиперхолестеринемии, высокой LDL-холестеринемии, низкой HDL-холестеринемии, происходящей после приема пищи гиперлипемии), гипертензии, сердечной недостаточности, диабетических осложнений [например, нейропатии, нефропатии, ретинопатии, диабетической кардиомиопатии, катаракты, макроангиопатии, остеопении, гиперосмолярной диабетической комы, инфекций (например, респираторной инфекции, инфекции мочевого тракта, желудочно-кишечной инфекции, инфекции дермальных мягких тканей, инфекции нижних конечностей), диабетической гангрены, ксеростомии, гипакузии (патологически повышенного восприятия звуков), сосудисто-мозговых расстройств, нарушения периферической циркуляции крови], метаболического синдрома (патологии, имеющей три или более выбранных из гипертриглицеридемии (TG), низкого HDL холестерина (HDL-C), гипертензии, тучности живота и ухудшенной переносимости глюкозы), саркопении и аналогичных.
В отношении критериев диагностики диабета Японское диабетическое общество (Japan Diabetes Society) в 1999 г. сообщило новые критерии диагностики.
Согласно данному изобретению диабетом является состояние, при котором уровень глюкозы в крови при любом воздержании от еды (концентрация глюкозы внутривенной плазмы) не менее 126 мг/дл; уровень глюкозы через 2 часа при испытании на пероральную переносимость глюкозы (75 г OGTT) (концентрация глюкозы внутривенной плазмы) не менее 200 мг/дл; и уровень глюкозы в крови при приеме пищи (концентрация глюкозы внутривенной плазмы) не менее 200 мг/дл. Состояние, не подпадающее под упомянутый выше диабет и отличающееся от “состояния, показывающего уровень глюкозы в крови при приеме пищи (концентрацию глюкозы внутривенной плазмы) менее 110 мг/дл или через 2 часа при испытании на пероральную переносимость глюкозы (75 г OGTT) (концентрация глюкозы внутривенной плазмы) менее 140 мг/дл” (нормальный тип) называется “пограничный тип”.
В дополнение, ADA (Американская диабетическая ассоциация (American Diabetes Association)) в 1997 г. и WHO в 1998 г. сообщили новые критерии диагностики диабета.
Согласно данному изобретению диабетом является состояние, показывающее любой уровень глюкозы в крови при воздержании от еды (концентрацию глюкозы внутривенной плазмы) не менее 126 мг/дл; и уровень глюкозы через 2 часа (75 г) при испытании на пероральную переносимость глюкозы (концентрация глюкозы внутривенной плазмы) не менее 200 мг/дл.
Согласно изобретению ухудшенной переносимостью глюкозы является состояние, показывающее уровень сахара в крови при воздержании от еды (концентрация глюкозы внутривенной плазмы) не менее 126 мг/дл, и уровень глюкозы через 2 часа (75 г) при испытании на пероральную переносимость глюкозы (концентрацию глюкозы внутривенной плазмы) не менее 140 мг/дл и менее 200 мг/дл. Согласно изобретению ADA состояние, при котором уровень глюкозы в крови при воздержании от еды (концентрация глюкозы внутривенной плазмы) не менее 110 мг/дл и менее 126 мг/дл, называется IFG (ухудшенная глюкоза при воздержании от еды). Согласно сообщению WHO, среди IFG (ухудшенной глюкозы при воздержании от еды), состояние, при котором уровень глюкозы через 2 часа (75 г) при испытании на пероральную переносимость глюкозы (концентрацию глюкозы внутривенной плазмы) менее 140 мг/дл, называется IFG (ухудшенная глицемия при воздержании от еды).
Соединение настоящего изобретения может также использоваться в качестве агента для профилактики или лечения диабета, пограничного типа, ухудшенной переносимости глюкозы, IFG (ухудшенной глюкозы при воздержании от еды) и IFG (ухудшенной глицемии при воздержании от еды), по определению согласно упомянутым выше новым критериям диагностики. Кроме того, соединение настоящего изобретения может предотвращать прогрессирование пограничного типа, ухудшенной переносимости глюкозы, IFG (ухудшенной глюкозы при воздержании от еды) или IFG (ухудшенной глицемии при воздержании от еды) в диабет.
Соединение настоящего изобретения может также использоваться в качестве агента для профилактики или лечения остеопороза, кахексии (например, карциноматозной кахексии, туберкулезной кахексии, диабетической кахексии, гемопатической кахексии, эндокринопатической кахексии, инфекционной кахексии или кахексии, вызываемой синдромом приобретенного иммунодефицита), жировой дистрофии печени, синдрома поликистозного яичника, почечных заболеваний (например, диабетической нефропатии, гломерулонефрита, гломерулосклероза, синдрома нефроза, гипертензивного нефросклероза, терминального почечного расстройства), мышечной дистрофии, инфаркта миокарда, стенокардии, расстройства сосудов головного мозга (например, инфаркта головного мозга, апоплексии головного мозга), болезни Альцгеймера, болезни Паркинсона, тревоги, деменции, синдрома инсулинорезистентности, синдрома Х, гиперинсулинемии, сенсорной аномалии при гиперинсулинемии, опухоли (например, лейкемии, рака молочной железы, рака предстательной железы, рака кожи), синдрома раздраженного кишечника, острой или хронической диареи, воспалительных заболеваний (например, ревматоидного артрита, спондилитной деформации, остеоартрита, люмбаго, подагры, постоперативных или посттравматических воспалений, набухания, невралгии, фаринголарингита, цистита, гепатита (включая неалкогольный стеатогепатит), пневмонии, панкреатита, энтерита, воспалительной болезни кишечника (включая воспалительный колит), язвенного колита, мукозного повреждения желудка (включая мукозное повреждение желудка, вызываемое аспирином)), мукозного повреждения тонкой кишки, мальабсорбции, дисфункции семенников, синдрома висцерального ожирения, саркопении или возрастной макулярной дегенерации.
Соединение настоящего изобретения может далее использоваться для вторичного предотвращения или подавления прогрессирования упомянутых выше различных заболеваний (например, сердечно-сосудистых событий, таких как инфаркт миокарда и аналогичные).
С целью усиления действия соединения настоящего изобретения или снижения дозы соединения и аналогичных, соединение может использоваться в сочетании с другими лекарственными средствами, такими как терапевтические агенты от диабета, терапевтические агенты от диабетических осложнений, терапевтические агенты от гиперлипидемии, антигипертензивные агенты, агенты против ожирения, диуретики, антитромботические агенты и аналогичные (называемые далее для краткости как сопутствующие лекарственные средства). Время введения соединения настоящего изобретения и введения сопутствующего лекарственного средства не ограничивается, и они могут вводиться субъекту, подвергаемому введению, одновременно или ступенчатым способом. В дополнение, соединения настоящего изобретения и сопутствующее лекарственное средство могут вводиться в виде двух видов препаратов, содержащих соответствующие активные ингредиенты, или в виде одного препарата, содержащего оба активных ингредиента.
Доза сопутствующего лекарственного средства может надлежащим образом определяться на основе клинически принимаемой дозы. В дополнение, соотношение смешения соединения настоящего изобретения и сопутствующего лекарственного средства может определяться соответствующим образом согласно субъекту введения, способу введения, болезни, состоянию, комбинации и аналогичным. Например, когда субъектом введения является человек, сопутствующее лекарственное средство может использоваться в количестве 0,01-100 частей по весу на 1 часть по весу соединения настоящего изобретения.
Примеры терапевтических агентов от диабета включают в себя препараты инсулина (например, препараты животного инсулина, экстрагируемые из поджелудочной железы крупного рогатого скота или свиней; инсулиновые препараты человека, генетически синтезируемые с использованием Escherichia coli или дрожжей; цинковый инсулин; протаминцинковый инсулин; фрагмент или производное инсулина (например, INS-1), пероральный инсулиновый препарат), стимуляторы инсулина (например, пиоглитазон или его соль (предпочтительно гидрохлорид), росиглитазон или его соль (предпочтительно малеат), Tesaglitazar, Ragaglitazar, Muraglitazar, Edaglitazone, Metaglidasen, Naveglitazar, AMG-131, THR-0921, TAK-379), ингибиторы α-глюкозидазы (например, воглибоза, акарбоза, миглитол, эмиглитат), бигуаниды (например, метформин, буформин или его соль (например, гидрохлорид, фумарат, сукцинат), средство, стимулирующее секрецию инсулина [например, сульфонилмочевину (например, толбутамид, глибенкламид, гликлазид, хлорпропамид, толазамид, ацетогексамид, гликлопирамид, глимепирид, глипизид, глибузол), репаглинид, натеглинид, митиглинид или его гидрат кальциевой соли], ингибиторы дипептидилпептидазы IV (например, алоглиптин, вилдаглиптин, ситаглиптин, саксаглиптин, Т-6666, ТС-021), β3 агонисты (например, AJ-9677), GPR40 агонисты, агонисты GLP-1 рецептора [например, GLP-1, GLP-1MR агент, NN-2211, AC-2993 (эксендин-4), BIM-51077, Aib(8,35)hGLP-1(7,37)NH2, CJC-1131], агонисты амилина (например, прамлинтид), ингибиторы фосфотирозинфосфатазы (например, ванадат натрия), ингибиторы глюконеогенеза (например, ингибиторы гликогенфосфорилазы, ингибиторы глюкозо-6-фосфатазы, антагонисты глюкагона), ингибиторы CGLUT (сопереносчик натрий-глюкозы (например, Т-1095), ингибиторы 11β-гидроксистероиддегидрогеназы (например, BVT-3498), адипонектин или его агонисты, ингибиторы IKK (например, AS-2868), лекарственные средства, улучшающие стойкость к лептину, агонисты соматостатинового рецептора, активаторы глюкокиназы (например, Ro-28-1675), GIP (глюкоза-зависимый инсулинотропный пептид) и аналогичные.
Примеры терапевтического агента от диабетических осложнений включают ингибиторы альдозоредуктазы (например, толрестат, эпалрестат, зенарестат, зополрестат, миналрестат, фидарестат, CT-112), нейротрофические факторы и их усиливающие лекарственные средства (например, NGF, NT-3, BDNF, агенты, промотирующие продуцирование/секрецию нейтротрофина (например, 4-(4-хлорфенил)-2-(2-метил-1-имидазолил)-5-[3-(2-метилфенокси)-пропил]оксазол), описанные в WO01/14372, TAK-583), промоторы нервной регенерации (например, Y-128), ингибиторы PKC (например, мезилат рубоксистаурина), ингибиторы AGE (например, ALT946, пимагедин, пиратоксантин, бромид N-фенацилтриазолия (ALT766), ALT-711, EXO-226, пиридорин, пиридоксамин), акцепторы активного кислорода (например, тиоктовую кислоту), церебральные сосудорасширяющие средства (например, тиапурид, мексилетин), агонисты рецептора соматостатина (например, BIM23190), ингибитор сигнал-регулирующей киназы 1 апаптоза (ASK-1) и аналогичные.
Примеры терапевтического агента от гиперлипидемии включают статиновые соединения (например, церивастатин, правастатин, симвастатин, ловастатин, аторвастатин, флувастатин, итавастатин, розувастатин, питавастатин или его соль (например, соль натрия, соль кальция), ингибиторы скваленсинтазы (например, ацетат лапаквистата), фибратные соединения (например, безафибрат, клофибрат, симфибрат, клинофибрат), ингибиторы ACAT (например, Avasimibe, Eflucimibe), анионообменные смолы (например, колестирамин), пробукол, никотиновокислотные лекарственные средства (например, никомол, ницеритрол), этиликозапентат, фитостерины (например, сойстерин, γ-оризанол) и аналогичные.
Примеры антигипертензивного агента включают ингибиторы ангиотензин-превращающего фермента (например, каптоприл, эналаприл, делаприл), антагонисты ангиотензина II (например, кандесартан цилексетил, лосартан, эпросартан, валсартан, телмисартан, ирбесартан, тасосартан, 1-[[2'-(2,5-дигидро-5-оксо-4H-1,2,4-оксадиазол-3-ил)бифенил-4-ил]метил]-2-этокси-1H-бензимидазол-7-карбоновую кислоту, TAK-491), кальциевые антагонисты (например, манидипин, нифедипин, амлодипин, эфонидипин, никардипин), агенты, открывающие калиевые каналы (например, левкромакалим, L-27152, AL 0671, NIP-121), клонидин и аналогичные.
Примеры агента против ожирения включают лекарственные средства против ожирения центральной нервной системы (например, дексфенфлурамин, фенфлурамин, фентермин, сибутрамин, амфепрамон, дексамфетамин, мазиндол, фенилпропаноламин, клобензорекс; антагонисты МСН рецептора (например, SB-568849; SNAP-7941; соединения, описанные в WO01/82925 и WO01/87834); антагонисты нейропептида Y (например, CP-422935); антагонисты рецептора каннабиноида (например, SR-141716, SR-147778); антагонист грелина; ингибиторы 11β-гидроксистероиддегидрогеназы (например, BVT-3498)), ингибиторы панкреатической липазы (например, орлистат, цетилистат, β3 агонисты (например, AJ-9677, AZ40140), аноректические пептиды (например, лептин, CNTF (цилиарный нейротрофический фактор)), агонисты холецистокинина (например, линтитрипт, FPL-15849), анорексигенные агенты (например, P-57) и аналогичные.
Примеры диуретиков включают производные ксантина (например, салицилат теобромин-натрия, салицилат теобромин-кальция), тиазидные препараты (например, этиазид, циклопентиазид, трихлорметиазид, гидрохлортиазид, гидрофлуметиазид, бензилгидрохлортиазид, пенфлутизид, политиазид, метиклотиазид), препараты антиальдостерона (например, спиронолактон, триамтерен), ингибиторы карбоновой ангидразы (например, ацетазоламид), хлорбензолсульфонамидные агенты (например, хлорталидон, мефрусид, индапамид), азосемид, изосорбит, этакриновую кислоту, пиретанид, буметанид, фуросемид и аналогичные.
Примеры антитромботического агента включают гепарин (например, гепариннатрий, гепаринкальций, дальтепариннатрий), варфарин (например, варфаринкалий), антитромбиновые лекарства (например, арагатробан, дабигатран), тромболитические агенты (например, ирокиназу, тизокиназу, альтеплазу, натеплазу, монтеплазу, памитеплазу), ингибиторы агрегации тромбоцитов (например, гидрохлорид тиклопидина, цилостазол, этиликозапентат, берапростнатрий, гидрохлорид сарпогрелата), празугрел, Е5555, SHC530348), FXa ингибиторы (например, TAK-442, ривароксабан, апиксабан, DU-156, YM150) и аналогичные.
В следующем тексте поясняются способы получения соединения (I) настоящего изобретения.
Соединение (I) может быть получено в соответствии со способом, известным самим по себе, например, по способу, описанному подробно ниже, или по аналогичному ему способу.
Каждый символ в реакционных схемах, R1, R2, X1, X2, L1, L2, L3 и кольцо С имеют значения, определенные ниже. Если не указано конкретно, другие символы имеют значения, определенные выше.
R1 представляет атом водорода или карбоксил-защищающую (или -защитную) группу. Конкретные примеры карбоксил-защищающей группы включают группы, упомянутые ниже. Из них предпочтительными являются C1-6 алкильная группа, необязательно замещенная 1-3 атомами галогена; C7-20 аралкильная группа (например, бензил, тритил), необязательно замещенная 1-5 заместителями, выбранными из атома галогена, C1-6 алкоксигруппы, нитрогруппы и аналогичных; и аналогичные.
R2 представляет защитную группу для амина. Конкретные примеры защитной группы амина включают группы, аналогичные амино-защитной группе, упомянутой ниже.
X1 представляет О или S.
X2 представляет CH2, О или S.
L1, L2 и L3 представляют собой независимо уходящую группу.
Предпочтительные конкретные примеры L1 включают в себя атом галогена (предпочтительно хлора, брома, йода), C1-6 алкилсульфонилокси группу, необязательно замещенную 1-3 атомами галогена (например, метансульфонилокси, этансульфонилокси, трифторметансульфонилокси, арилсульфонилокси, необязательно имеющую заместитель(тели) (например, бензолсульфонилокси, п-толуолсульфонилокси) и аналогичные.
Предпочтительные конкретные примеры L2 включают диалкилфосфоногруппу (предпочтительно диметилфосфоногруппу, диэтилфосфоногруппу), трифенилфосфониевую группу и аналогичные.
Предпочтительные примеры L3 включают дигидроксиборанильную группу, диалкоксиборанильную группу (предпочтительно 4,4,5,5-тетраметил-1,3,2-диоксаборан-2-ил), триалкилстаннильную группу (предпочтительно триметилстаннильную группу, н-трибутилстаннильную группу) и аналогичные.
Кольцо С представляет 5-членный ароматический гетероцикл или ненасыщенный гетероцикл. Примеры 5-членного ароматического гетероцикла включают тиофен, фуран, пиррол, имидазол, пиразол, триазол, тетразол, тиазол, изотиазол, тиадиазол, оксазол, изоксазол, оксадиазол и аналогичные. Примеры 5-членного ненасыщенного гетероцикла включают кольцо, имеющее по крайней мере одну двойную связь, среди них кольца, приведенные для примера в качестве “5-членного неароматического гетероцикла” “необязательно дополнительно замещенного 5-членного неароматического гетероцикла” для кольца А.
Конкретные примеры кольца С включают фуран, пиррол и аналогичные.
В следующих способах получения “простые эфирные растворители”, “галоидированные углеводородные растворители”, “ароматические растворители”, “нитрильные растворители”, “сложные эфирные растворители”, “амидные растворители”, “кетоновые растворители”, “сульфоксидные растворители”, “спиртовые растворители”, “органические кислотные растворители” означают следующее.
Примеры “простых эфирных растворителей” включают диэтиловый эфир, тетрагидрофуран (ТГФ), 1,4-диоксан, 1,2-диметоксиэтан и аналогичные.
Примеры “галоидированных углеводородных растворителей” включают дихлорметан, хлороформ, 1,2-дихлорэтан, четыреххлористый углерод, 1,1,2,2-тетрахлорэтан и аналогичные.
Примеры “ароматических растворителей” включают бензол, толуол, ксилол, пиридин, мезитилен и аналогичные.
Примеры “нитрильных растворителей” включают ацетонитрил, пропионитрил и аналогичные.
Примеры “сложноэфирных растворителей” включают этилацетат, метилацетат и аналогичные.
Примеры “амидных растворителей” включают N,N-диметилформамид (ДМФ), N,N-диметилацетамид, N-метилпирролидон и аналогичные.
Примеры “кетоновых растворителей” включают ацетон, метилэтилкетон и аналогичные.
Примеры “сульфоксидных растворителей” включают диметилсульфоксид (ДМСО) и аналогичные.
Примеры “спиртовых растворителей” включают метанол, этанол, изопропанол, трет-бутанол и аналогичные.
Примеры “органических кислотных растворителей” включают муравьиную, уксусную, трифторуксусную, метансульфоновую кислоту и аналогичные.
Если не указано иное, материал исходного соединения в следующих способах получения является промышленно доступным, или может быть получен в соответствии с известным самим по себе способом или способом, аналогичным ему.
Соединение в качестве исходного материала может использоваться в форме соли. Примеры соли включают соли, аналогичные вышеупомянутой соли соединения, представленного формулой (I).
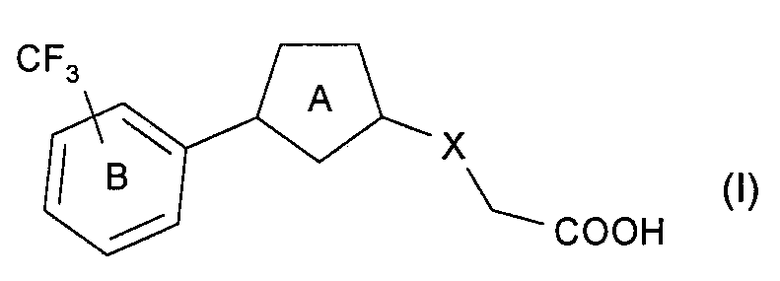
(Способ получения А)
Из соединений (I) настоящего изобретения, соединение, представленное следующей формулой (Ia) или (Ib) (соединение (Ia) или соединение (Ib)) может быть получено, например, согласно следующей реакционной схеме 1.
(Реакционная схема 1)

В данном способе получения соединение (Ia) или соединение (Ib) может быть получено из соединения (IIa) с помощью следующих стадий.
Стадия 1А: стадия получения соединения (IVa), в которой соединение (IIa) подвергают реакции алкилирования соединением (IIIa);
Стадия 2А: стадия получения соединения (Ia) путем удаления R1, которой является карбоксилзащитная группа соединения (IVa);
Стадия 3А: стадия получения соединения (Va), в которой гидроксигруппу соединения (IIa) подвергают реакции галогенирования или сульфоновой сложной этерификации;
Стадия 4А: стадия получения соединения (VIIa), в которой соединение (Va) подвергают реакции алкилирования соединением (VIa);
Стадия 2В: стадия получения соединения (Ib) путем удаления R1, которая является карбоксилзащитной группой соединения (VIIa).
Каждая стадия поясняется подробно далее.
(Стадия 1А)
Соединение (IVa) может быть получено с помощью реакции соединения (IIa) с соединением (IIIa) в присутствии основания.
Примеры основания включают амины (например, триэтиламин, N,N-диизопропилэтиламин, пиридин, 4-диметиламинопиридин, 1,8-диазабицикло[5.4.0]-7-ундецен); карбонаты щелочных металлов (например, карбонат калия, карбонат натрия, карбонат цезия, гидрокарбонат натрия, гидрокарбонат калия); фосфаты щелочных металлов (например, трикалийфосфат, тринатрийфосфат); ацетаты щелочных металлов (ацетат натрия, ацетат калия); гидриды щелочных металлов (например, гидрид натрия, гидрид калия); гидроксиды щелочных металлов (например, гидроксид натрия, гидроксид калия); C1-6 алкоксиды щелочных металлов (например, метоксид натрия, трет-бутоксид натрия, трет-бутоксид калия) и аналогичные. Из них предпочтительными являются гидрид натрия, карбонат натрия, трет-бутоксид калия и аналогичные.
Количество используемого основания составляет обычно 0,1-100 эквивалентов, предпочтительно 1-10 эквивалентов, на 1 эквивалент соединения (IIa).
Конкретные примеры соединения (IIIa) включают метилбромацетат, трет-бутилбромацетат, хлорацетат натрия и аналогичные.
Соединением (IIIa) может быть промышленно доступный продукт, или оно может быть получено согласно известному самому по себе способу или согласно аналогичному ему способу.
Используемое количество соединения (IIIa) составляет обычно 1-100 эквивалентов, предпочтительно 1-5 эквивалентов, на 1 эквивалент соединения (IIa).
Данная реакция осуществляется без растворителя или в инертном растворителе. Примеры инертного растворителя включают простые эфирные растворители, галоидированные углеводородные растворители, нитрильные растворители, ароматические растворители, сложные эфирные растворители, амидные растворители, воду и аналогичные. Данные растворители могут использоваться в виде смеси двух или более видов их в соответствующем соотношении. Из них предпочтительными являются тетрагидрофуран, ацетонитрил, N,N-диметилформамид, N,N-диметилацетамид, диметилсульфоксид и аналогичные.
Когда необходимо, для данной реакции может использоваться катализатор фазового переноса (например, бромид тетрабутиламмония, гидросульфат тетрабутиламмония и др.).
Количество используемого катализатора фазового переноса составляет обычно 0,01-0,5 эквивалентов, предпочтительно 0,01-0,1 эквивалента, на 1 эквивалент соединения (IIa).
Температура данной реакции обычно составляет примерно от 30°С до 200°С, предпочтительно 50°С-120°С.
Время данной реакции составляет обычно от 0,5 часа до 24 часов.
Соединение (IIa) может быть получено согласно известному способу (например, способу USP 5670656; WO 2006/21401; WO 2004/110994) или аналогичному ему способу.
(Стадия 2А)
Удаление защитной группы R1 соединения (IVa) осуществляется в соответствии с известным самим по себе способом, например, способом, описанным в Protective Groups in Organic Synthesis, John Wiley and Sons (1980), и аналогичным. Примеры удаления защитной группы включают способ с использованием кислоты, основания и аналогичных, и аналогичные способы.
(Стадия 3А)
Соединение (Va) может быть получено с помощью превращения гидроксигруппы соединения (IIa) в атом галогена с использованием галогенирующего реагента.
Примеры галогенирующего реагента включают тионилхлорид, тионилбромид, треххлористый фосфор, пентахлорид фосфора, оксихлорид фосфора, тетрабромид углерода и аналогичные.
Количество используемого галогенирующего реагента составляет обычно 1-100 эквивалентов, предпочтительно 1-10 эквивалентов, на 1 эквивалент соединения (IIa).
Данная реакция обычно осуществляется в инертном растворителе (например, простых эфирных растворителях, галоидированных углеводородных растворителях, ароматических растворителях и др.) или без растворителя. Данные растворители могут быть использованы в виде смеси двух или более видов их в соответствующем соотношении. Из них предпочтительными являются тетрагидрофуран, толуол, тетрахлорид углерода и аналогичные.
Температура данной реакции обычно составляет от -20°С до 200°С, предпочтительно 0°С-100°С.
Время данной реакции составляет обычно от 0,5 часа до 24 часов.
Соединение (Va) может быть получено также с помощью превращения гидроксигруппы соединения (IIa) в необязательно галоидированную C1-6 алкилсульфонилокси группу, арилсульфонилокси группу, необязательно имеющую заместитель(тели) (например, бензолсульфонилокси, п-толуолсульфонилокси) и аналогичные с использованием необязательно галоидированного C1-6 алкилсульфонилхлорида (например, метансульфонилхлорида, этансульфонилхлорида, трифторметансульфонилхлорида), арилсульфонилхлорида, необязательно имеющего заместитель(тели) (например, бензолсульфонилхлорида, п-толуолсульфонилхлорида) и аналогичных.
Количество используемого необязательно галоидированного C1-6 алкилсульфонилхлорида или арилсульфонилхлорида, необязательно имеющего заместитель(тели), составляет обычно 1-10 эквивалентов, предпочтительно 1-5 эквивалентов, на 1 эквивалент соединения (IIa).
Данная реакция обычно осуществляется в инертном растворителе (например, простых эфирных растворителях, галоидированных углеводородных растворителях, ароматических растворителях, амидных растворителях и др.) или без растворителя. Данные растворители могут быть использованы в виде смеси двух или более видов их в соответствующем соотношении. Из них предпочтительными являются тетрагидрофуран, толуол, тетрахлорид углерода, N,N-диметилформамид и аналогичные.
Когда необходимо, для данной реакции может быть использовано основание.
Примеры основания включают амины (например, триэтиламин, N,N-диизопропилэтиламин, пиридин, 4-диметиламинопиридин); карбонаты щелочных металлов (например, карбонат калия, карбонат натрия, карбонат цезия); фосфаты щелочных металлов (например, трикалийфосфат, тринатрийфосфат); гидриды щелочных металлов (например, гидрид натрия, гидрид калия); гидроксиды щелочных металлов (например, гидроксид натрия, гидроксид калия) и аналогичные. Из них предпочтительными являются пиридин, триэтиламин, карбонат калия, трикалийфосфат, гидрид натрия и аналогичные.
Количество используемого основания составляет обычно 1-100 эквивалентов, предпочтительно 1-10 эквивалентов, на 1 эквивалент соединения (IIa).
Температура данной реакции обычно составляет от -20°С до 200°С, предпочтительно 0°С-100°С.
Время данной реакции составляет обычно от 0,5 часа до 48 часов.
(Стадия 4А)
Соединение (VIIa) может быть получено с помощью реакции соединения (Va) с соединением (VIa) в присутствии основания.
Примеры основания включают амины (например, триэтиламин, N,N-диизопропилэтиламин, пиридин, 4-диметиламинопиридин, 1,8-диазабицикло[5.4.0]-7-ундецен); карбонаты щелочных металлов (например, карбонат калия, карбонат натрия, карбонат цезия, гидрокарбонат натрия, гидрокарбонат калия); фосфаты щелочных металлов (например, трикалийфосфат, тринатрийфосфат); ацетаты щелочных металлов (ацетат натрия, ацетат калия); гидриды щелочных металлов (например, гидрид натрия, гидрид калия); гидроксиды щелочных металлов (например, гидроксид натрия, гидроксид калия); C1-6 алкоксиды щелочных металлов (например, метоксид натрия, трет-бутоксид натрия, трет-бутоксид калия) и аналогичные. Из них предпочтительными являются гидрид натрия, карбонат натрия, трет-бутоксид калия и аналогичные.
Количество используемого основания составляет обычно 1-100 эквивалентов, предпочтительно 1-10 эквивалентов, на 1 эквивалент соединения (Va).
Конкретные примеры соединения (VIa) включают этилтиогликолят, этилгликолят и аналогичные.
Соединение (VIa) может быть промышленно доступным продуктом, или может быть получено в соответствии с известным самим по себе способом или аналогичным ему способом.
Количество используемого соединения (Va) составляет обычно 1-10 эквивалентов, предпочтительно 1-5 эквивалентов, на 1 эквивалент соединения (Va).
Данная реакция осуществляется без растворителя или в инертном растворителе. Примеры инертного растворителя включают простые эфирные растворители, галогенированные углеводородные растворители, нитрильные растворители, ароматические растворители, сложные эфирные растворители, амидные растворители, воду и аналогичные. Данные растворители могут использоваться в виде смеси двух или более видов их в соответствующем соотношении. Из них предпочтительными являются тетрагидрофуран, ацетонитрил, N,N-диметилформамид, N,N-диметилацетамид, диметилсульфоксид и аналогичные.
Когда необходимо, для данной реакции может быть использован катализатор фазового переноса (например, бромид тетрабутиламмония, кислый сульфат тетрабутиламмония и др.).
Количество используемого катализатора фазового переноса составляет обычно 0,01-0,5 эквивалентов, предпочтительно 0,01-0,1 эквивалента, на 1 эквивалент соединения (Va).
Температура данной реакции обычно составляет от около 30°С до 200°С, предпочтительно 50°С-120°С.
Время данной реакции составляет обычно от 0,5 часа до 24 часов.
(Стадия 2В)
Соединение (Ib) может быть получено из соединения (VIIa) в условиях и по способу, аналогичным тем, что приведены для примера на стадии 2А.
(Способ получения В)
Из соединения (I) настоящего изобретения соединение, представленное следующей формулой (Ic) (соединение (Ic)) может быть получено, например, в соответствии со следующей реакционной схемой 2.
(Реакционная схема 2)
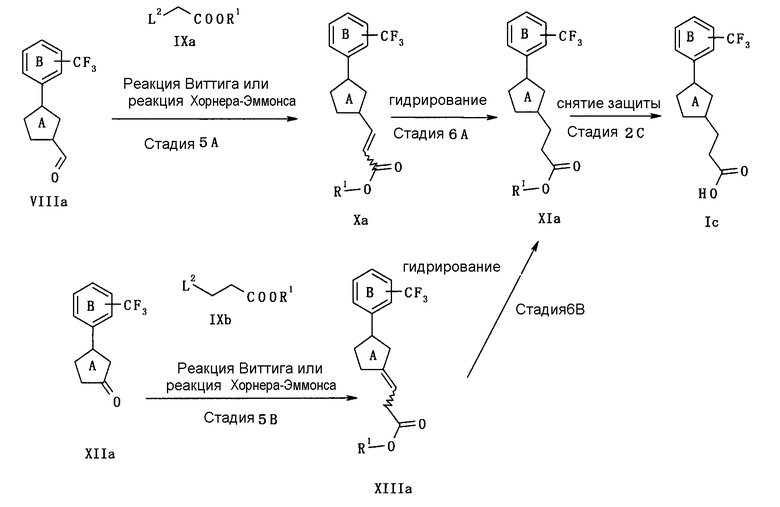
В данном способе получения соединение (Ic) может быть получено из соединения (VIIIa) или соединения (XIIa) с помощью следующих стадий.
Стадия 5А: стадия получения соединения (Ха), в которой соединение (VIIIa) подвергают реакции Виттига или реакции Хорнера-Эммонса с соединением (IXa);
Стадия 6А: стадия получения соединения (XIa), в которой соединение (Xa) подвергают реакции гидрирования;
Стадия 5В: стадия получения соединения (XIIIa), в которой соединение (XIIa) подвергают реакции Виттига или реакции Хорнера-Эммонса с соединением (IXb);
Стадия 6В: стадия получения соединения (XIa), в которой соединение (XIIIa) подвергают реакции гидрирования;
Стадия 2С: стадия получения соединения (Ic) удалением R1, которым является карбоксилзащитная группа соединения (XIa).
Каждая стадия поясняется подробно далее.
(Стадия 5А)
Соединение (Xa) может быть получено в E-форме, Z-форме или в виде смеси E-формы и Z-формы подверганием соединения (VIIIa) реакции Виттига или реакции Хорнера-Эммонса с соединением (IXa).
Реакция Виттига или реакция Хорнера-Эммонса обычно осуществляется с использованием основания в соответствии с известным способом (например, J. Chem. Soc. Perkin Trans. 1, 2895 (1996), 5th edition Jikken Kagaku Koza, 13 vol., стр. 118-139 (2005), Maruzen).
Конкретные примеры соединения (IXa) включают диэфир алкилфосфоновой кислоты (например, этилдиэтилфосфоноацетат, трет-бутилдиэтилфосфоноацетат) или галогениды трифенилфосфинилидов (например, (этоксикарбонилметил)трифенилфосфонийбромид, (трет-бутоксикарбонилметил)трифенилфосфонийхлорид) и аналогичные.
Соединение (IXa) может быть промышленно доступным продуктом или может быть получено с помощью известного способа или способа, аналогичного ему.
Количество используемого соединения (IXa) составляет обычно 0,8-10 эквивалентов, предпочтительно 0,8-3 эквивалента, на 1 эквивалент соединения (VIIIa).
Примеры основания включают амины (например, триэтиламин, N,N-диизопропилэтиламин, пиридин, 4-диметиламинопиридин, 1,8-диазабицикло[5.4.0]-7-ундецен); карбонаты щелочных металлов (например, карбонат калия, карбонат натрия, карбонат цезия, гидрокарбонат натрия, гидрокарбонат калия); фосфаты щелочных металлов (например, трикалийфосфат, тринатрийфосфат); ацетаты щелочных металлов (ацетат натрия, ацетат калия); гидриды щелочных металлов (например, гидрид натрия, гидрид калия); гидроксиды щелочных металлов (например, гидроксид натрия, гидроксид калия); C1-6 алкоксиды щелочных металлов (например, метоксид натрия, трет-бутоксид натрия, трет-бутоксид калия); органические соединения лития (например, н-бутиллитий, втор-бутиллитий, трет-бутиллитий); амиды металлов (например, литийдиизопропиламид, калийгексаметилдисилазид) и аналогичные. Из них предпочтительными являются гидрид натрия, карбонат натрия, трет-бутоксид калия, н-бутиллитий и аналогичные.
Количество используемого основания составляет 1-5 эквивалентов, более предпочтительно 1-2 эквивалента, на 1 эквивалент соединения (VIIIa).
Данная реакция осуществляется в инертном растворителе (например, растворителях, приведенных в качестве примера на стадии 1А). Данные растворители могут использоваться в виде смеси двух или более видов их в соответствующем соотношении. Из них предпочтительными являются диэтиловый эфир, тетрагидрофуран, ацетонитрил, N,N-диметилформамид, этанол и аналогичные.
Данная реакция предпочтительно осуществляется в инертном газе, таком как сухой аргон, сухой азот и аналогичные.
Температура данной реакции обычно составляет от около -78°С до 150°С, предпочтительно -78°С-100°С.
Время данной реакции составляет обычно от 0,5 часа до 24 часов.
Соединение (VIIIa) может быть получено согласно известному способу (например, Synth. Commun. 16, 1343 (1986); J. Am. Chem. Soc. 99, 7020 (1977); WO 2005/92099) или по аналогичному ему способу.
(Стадия 6А)
Соединение (XIa) может быть получено путем подвергания соединения (Xa) реакции гидрирования.
Реакция гидрирования обычно осуществляется с использованием катализатора по известному самому по себе способу (например, Jikken Kagaku Koza (Courses in Experimental Chemistry), 15 vol., oxidation and reduction (II), стр. 333-448 (1977), Maruzen).
Примеры катализатора включают в себя палладий на угле, палладий на угле-этилендиаминовый комплекс, черный палладий, диоксид платины, никель Ренея, кобальт Ренея и аналогичные. Из них предпочтительными являются палладий на угле, комплекс палладий на угле-этилендиамин, диоксид платины и аналогичные.
Количество используемого катализатора составляет обычно 5-1000% масс., предпочтительно 5-300% масс., относительно соединения (Xa).
В реакции гидрирования вместо водородного газа могут использоваться различные источники водорода (например, муравьиная кислота, формиат аммония, формиат триэтиламмония, фосфинат натрия, гидразин).
Количество используемого источника водорода составляет обычно 1-10 эквивалентов, предпочтительно 1-5 эквивалентов, на 1 эквивалент соединения (Ха).
Данная реакция осуществляется в инертном растворителе (например, растворителях, приведенных в качестве примера на стадии 1А), органических кислотных растворителях и аналогичных. Данные растворители могут быть использованы в виде смеси двух или более видов их в соответствующем соотношении. Из них предпочтительными являются метанол, этанол, уксусная кислота, тетрагидрофуран, этилацетат и аналогичные.
Когда необходимо, данная реакция может быть осуществлена под давлением. При проведении под давлением оно составляет обычно 2-10 атм., предпочтительно 2-5 атм.
Температура данной реакции обычно составляет примерно от -20°С до 100°С, предпочтительно 0°С-80°С.
Время данной реакции составляет обычно от 0,5 часа до 100 часов, предпочтительно 0,5-50 часов.
(Стадия 5В)
Соединение (XIIIa) может быть получено из соединения (XIIa) и соединения (IXb) в условиях и по способу, аналогичных тем, что приведены в качестве примера на стадии 5А.
Соединение (XIIa) может быть получено по известному способу (например, Bioorg. Med. Chem. 11, 145 (2003); J. Org. Chem. 54, 220 (1989); J. Org. Chem. 49, 2500 (1984)) или по аналогичному ему способу.
Соединение (IXb) может быть промышленно доступным продуктом (например, 2-(этоксикарбонил)этилтрифенилфосфонийбромид), или может также синтезироваться по известному способу (например, 4th edition, Jikken Kagaku Koza, 19 vol., стр. 57-61 (1992), Maruzen) или по аналогичному ему способу.
(Стадия 6В)
Соединение (XIa) может быть также получено из соединения (XIIIa) в условиях и по способу, аналогичных тем, что приведены в качестве примера на стадии 6А.
(Стадия 2С)
Соединение (Ic) может быть получено из соединения (XIa) в условиях и по способу, аналогичных тем, что приведены в качестве примера на стадии 2А.
(Способ получения С)
Из соединения (I) настоящего изобретения соединение, представленное следующей формулой (Ic) (соединение (Ic)), может быть получено, например, согласно следующей реакционной схеме 3.
(Реакционная схема 3)
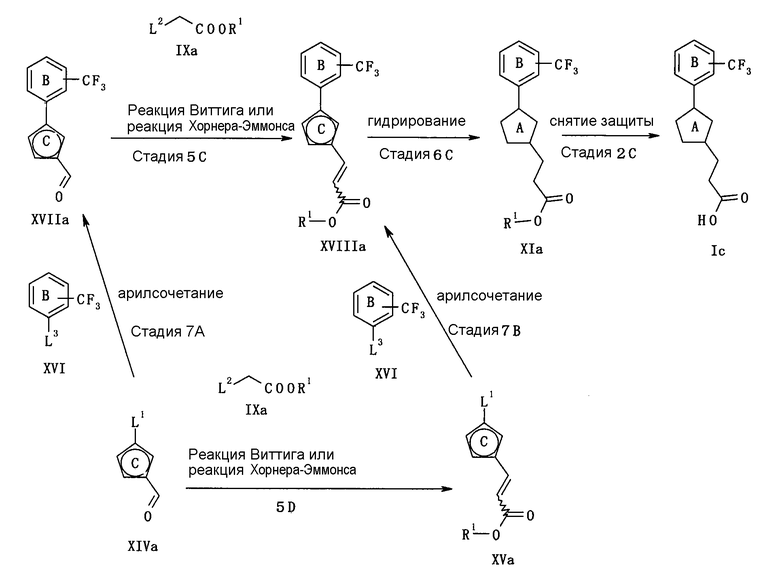
В данном способе получения соединение (Ic) может быть получено из соединения (XIVa) с помощью следующих стадий.
Стадия 7А: стадия получения соединения (XVIIa), в которой соединение (XIVa) подвергают реакции арилсочетания с производным арилбороновой кислоты (XVI);
Стадия 5С: стадия получения соединения (XVIIIa), в которой соединение (XVIIa) подвергают реакции Виттига или реакции Хорнера-Эммонса с соединением (IXa);
Стадия 5D: стадия получения соединения (XVa), в которой соединение (XIVa) подвергают реакции Виттига или реакции Хорнера-Эммонса с соединением (IXa);
Стадия 7В: стадия получения соединения (XVIIIa), в которой соединение (XVa) подвергают реакции арилсочетания с производным арилбороновой кислоты (XVI);
Стадия 6С: стадия получения соединения (XIa), в которой соединение (XVIIIa) подвергают реакции гидрирования;
Стадия 2С: стадия получения соединения (Ic) удалением R1, которым является карбоксилзащитная группа соединения (XIa).
Каждая стадия поясняется подробно далее.
(Стадия 7А)
Соединение (XVIIa) может быть получено подверганием соединения (XIVa) реакции арилсочетания с соединением (XVI).
Количество соединения (XVI) составляет обычно 1-10 эквивалентов, предпочтительно 1-5 эквивалентов, на 1 эквивалент соединения (XIVa).
Реакция арилсочетания обычно осуществляется с использованием катализатора на основе переходного металла в присутствии основания согласно известному способу (например, 5th edition Jikken Kagaku Koza, 18 vol., стр. 327-351 (2005), Maruzen; Chem. Rev., 102, 1359 (2002)).
Примеры катализатора на основе переходного металла включают палладиевые комплексы (например, тетракис(трифенилфосфин)палладий(0), дихлорбис(трифенилфосфин)палладий(II), бис(дибензилиденацетон)палладий(0), ацетат палладия(II), хлорид палладия(II)), комплексы никеля (например, дихлор[1,2-бис(дифенилфосфино)этан]никель(II), бис(1,5-циклооктадиен)никель(0)) и аналогичные. Из них предпочтительным является тетракис(трифенилфосфин)палладий(0).
Количество используемого катализатора на основе переходного металла составляет обычно 0,00001-5 эквивалентов, предпочтительно 0,0001-1 эквивалент, на 1 эквивалент соединения (XIVa).
Для успешного прохождения данной реакции может совместно использоваться фосфиновый лиганд к переходно-металлическому катализатору.
Примеры фосфинового лиганда включают трифенилфосфин, трис(2-метилфенил)фосфин, 1,2-бис(дифенилфосфино)этан, 1,3-бис(дифенилфосфино)пропан, 1,1'-бис(дифенилфосфино)ферроцен и аналогичные.
Количество используемого фосфинового лиганда составляет обычно 1-50 эквивалентов, предпочтительно 2-20 эквивалентов, на 1 эквивалент катализатора на основе переходного металла.
Примеры основания включают основания, приведенные в качестве примера на стадии 5А. Из них предпочтительными являются карбонат натрия, карбонат цезия, трет-бутоксид калия и аналогичные.
Количество используемого основания составляет обычно 1-20 эквивалентов, предпочтительно 1-10 эквивалентов, на 1 эквивалент соединения (XIVa).
Данная реакция осуществляется в инертном растворителе (например, растворителях, приведенных в качестве примера на стадии 1А). Данные растворители могут использоваться в виде смеси двух или более видов их в соответствующем соотношении. Из них предпочтительными являются диметоксиэтан, тетрагидрофуран, бензол, толуол, N,N-диметилформамид, вода и аналогичные.
Данная реакция предпочтительно осуществляется в инертном газе, таком как аргон, азот и аналогичные.
Температура данной реакции обычно составляет примерно от 10°С до 200°С, предпочтительно 50°С-150°С.
Время данной реакции составляет обычно от 0,5 часа до 100 часов, предпочтительно 5-80 часов.
Соединение (XIVa) может быть промышленно доступным продуктом или может быть получено по известному способу (например, Can. J. Chem. 68, 1305 (1990)) или по аналогичному ему способу.
Соединение (XVI) может быть промышленно доступным продуктом или может быть получено по известному способу (например, 5th edition, Jikken Kagaku Koza, 18 vol., стр. 95-102, 183-188 (2005), Maruzen; US 2003/0225106) или по аналогичному ему способу.
(Стадия 5С)
Соединение (XVIIIa) может быть получено из соединения (XVIIa) и соединения (IXa) в условиях и по способу аналогичных тем, что приведены в качестве примера на стадии 5А.
(Стадия 5D)
Соединение (XVa) может быть получено из соединения (XIVa) и соединения (IXa) в условиях и по способу, аналогичных тем, что приведены в качестве примера на стадии 5А.
(Стадия 7В)
Соединение (XVIIIa) может быть также получено из соединения (XVa) и соединения (XVI) в условиях и по способу, аналогичных тем, что приведены в качестве примера на стадии 7А.
(Стадия 6С)
Соединение (XIa) может быть получено из соединения (XVIIIa) в условиях и по способу, аналогичных тем, что приведены в качестве примера на стадии 6А.
(Способ получения D)
Из соединения (I) настоящего изобретения, соединение, представленное следующей формулой (Id) (соединение (Id)) может быть получено, например, согласно следующей реакционной схеме 4.
(Реакционная схема 4)

В данном способе получения соединение (Id) может быть получено из соединения (XIXa) или соединения (XXVa) с помощью следующих стадий.
Стадия 5Е: стадия получения соединения (XXa), в которой соединение (XIXa) подвергают реакции Виттига или реакция Хорнера-Эммонса с соединением (IXa);
Стадия 6D: стадия получения соединения (XXIa), в которой соединение (ХХа) подвергают реакции гидрирования;
Стадия 1В: стадия получения соединения (XXIa), в которой соединение (XXVa) подвергают реакции алкилирования соединением (IIIa);
Стадия 3В: стадия получения соединения (XVIa), в которой гидроксигруппу соединения (XXVa) подвергают реакции галогенирования или сульфоновой сложной этерификации;
Стадия 4В: стадия получения соединения (XXIa), в которой соединение (XVIa) подвергают реакции алкилирования соединением (VIa);
Стадия 8А: стадия получения соединения (XXIIa) удалением R2, которым является защитная группа для амина соединения (XXIa);
Стадия 9А: стадия получения соединения (XXIVa), в которой соединение (XXIIa) подвергают реакции Бухвальда с соединением (XXIIIa);
Стадия 2D: стадия получения соединения (Id) удалением R1, которым является карбоксилзащитная группа соединения (XXIVa).
Каждая стадия поясняется подробно далее.
(Стадия 5Е)
Соединение (XXa) может быть получено из соединения (XIXa) и соединения (IXa) в условиях и по способу, аналогичных тем, что приведены в качестве примера на стадии 5А.
Соединение (XIXa) может быть промышленно доступным продуктом. Альтернативно, соединение (XIXa) может быть также получено в соответствии со способом, известным самим по себе (например, WO 2004/5255; WO 2005/49602 или аналогичным ему способом.
(Стадия 6D)
Соединение (XXIa) может быть получено из соединения (XXa) в условиях и по способу, аналогичных тем, что приведены в качестве примера на стадии 6А.
(Стадия 1В)
Соединение (XXIa) может быть также получено из соединения (XXVa) и соединения (IIIa) в условиях и по способу, аналогичных тем, что приведены в качестве примера на стадии 1А.
(Стадия 3В)
Соединение (XVIa) может быть получено из соединения (XXVa) в условиях и по способу, аналогичных тем, что приведены в качестве примера на стадии 3А.
Соединение (XXVa) может быть промышленно доступным продуктом. Альтернативно, соединение (XXVa) может быть также получено в соответствии с известным способом или аналогичным ему способом.
(Стадия 4В)
Соединение (XXIa) может быть также получено из соединения (XVIa) и соединения (VIa) в условиях и по способу, аналогичных тем, что приведены в качестве примера на стадии 4А.
(Стадия 8А)
Удаление защитной группы R2 соединения (XXIa) может осуществляться в соответствии с известным способом, например, способом, описанным в Protective Groups in Organic Synthesis, John Wiley and Sons (1980), и аналогичным. Примеры удаления защитной группы R2 включают способ с использованием кислоты, основания и аналогичных, гидрирование и аналогичные.
(Стадия 9А)
Соединение (XXIVa) может быть получено подверганием соединения (XXIIa) реакции Бухвальда с соединением (XXIIIa).
Количество используемого соединения (XXIIIa) составляет обычно 1-10 эквивалентов, предпочтительно 1-5 эквивалентов, на 1 эквивалент соединения (XXIIa).
Реакция Buchwald обычно осуществляется с использованием катализатора на основе переходного металла в присутствии основания согласно известному способу (например, Org. Synth. 78, 23 (2000); Org. Lett. 5, 2413 (2003)).
Примеры катализатора на основе переходного металла включают палладиевые комплексы (например, трис(дибензилиденацетон)дипалладий(0), ацетат палладия(II), 1,1'-бис(дифенилфосфино)ферроцендихлорпалладий(II)), бис(дибензилиденацетон)палладий(0)) и аналогичные. Из них предпочтительными является трис(дибензилиденацетон)дипалладий(0) и ацетат палладия (II).
Количество используемого катализатора на основе переходного металла составляет обычно 0,00001-5 эквивалентов, предпочтительно 0,0001-1 эквивалент, на 1 эквивалент соединения (XXIIa).
Для успешного прохождения данной реакции может совместно использоваться фосфиновый лиганд к переходно-металлическому катализатору.
Примеры фосфинового лиганда включают 2,2'-бис(дифенилфосфино)-1,1'-бинафтил, 9,9-диметил-4,5-бис(дифенилфосфино)ксантен и аналогичные.
Количество используемого фосфинового лиганда составляет обычно 1-50 эквивалентов, предпочтительно 2-20 эквивалентов, на 1 эквивалент катализатора на основе переходного металла.
Примеры основания включают амины (например, триэтиламин, N,N-диизопропилэтиламин, пиридин, 4-диметиламинопиридин, 1,8-диазабицикло[5.4.0]-7-ундецен, 1,3,4,6,7,8-гексагидро-1-метил-2Н-пиримидо[1,2-a]пиримидин); карбонаты щелочных металлов (например, карбонат калия, карбонат натрия, карбонат цезия, гидрокарбонат натрия, гидрокарбонат калия); фосфаты щелочных металлов (например, трикалийфосфат, тринатрийфосфат); ацетаты щелочных металлов (ацетат натрия, ацетат калия); гидриды щелочных металлов (например, гидрид натрия, гидрид калия); гидроксиды щелочных металлов (например, гидроксид натрия, гидроксид калия); C1-6 алкоксиды щелочных металлов (например, метоксид натрия, трет-бутоксид натрия, трет-бутоксид калия); органические соединения лития (например, н-бутиллитий, втор-бутиллитий, трет-бутиллитий); амиды металлов (например, литийдиизопропиламид, калийгексаметилдисилазид) и аналогичные. Из них предпочтительными являются карбонат цезия, метоксид натрия, трет-бутоксид натрия, трет-бутоксид калия, трикалийфосфат, 1,3,4,6,7,8-гексагидро-1-метил-2Н-пиримидо[1,2-a]пиримидин) и аналогичные.
Количество используемого основания составляет 1-20 эквивалентов, предпочтительно 1-10 эквивалентов, на 1 эквивалент соединения (XXIIa).
Данная реакция осуществляется в инертном растворителе (например, растворителях, приведенных в качестве примера на стадии 1А). Данные растворители могут быть использованы в виде смеси двух или более видов их в соответствующем соотношении. Из них предпочтительными являются диметоксиэтан, диоксан, тетрагидрофуран, бензол, толуол, N,N-диметилформамид и аналогичные.
Данная реакция предпочтительно осуществляется в инертном газе, таком как аргон, азот и аналогичный инертный газ.
Температура данной реакции обычно составляет примерно от 30°С до 200°С, предпочтительно 50°С-150°С.
Время данной реакции составляет от 0,5 часа до 100 часов, предпочтительно 5-80 часов.
Соединение (XXIIIa) может быть промышленно доступным продуктом или может быть получено по известному способу или аналогичному ему способу.
(Стадия 2D)
Соединение (Id) может быть получено из соединения (XXIVa) в условиях и по способу, аналогичных тем, что приведены в качестве примера на стадии 2А.
(Способ получения Е)
Из соединения (I) настоящего изобретения, соединение, представленное следующей формулой (Ie) (соединение (Ie)), может быть получено, например, согласно следующей реакционной схеме 5.
(Реакционная схема 5)

В данном способе получения соединение (Ie) может быть получено из соединения (XXVIIa) с помощью следующих стадий.
Стадия 10А: стадия получения соединения (XXVIIIa), в которой соединение (XXVIIa) подвергают реакции алкилирования соединением (IIIb);
Стадия 2Е: стадия получения соединения (Ie) удалением R1, которой является карбоксилзащитная группа соединения (XXVIIIa).
Каждая стадия поясняется подробно далее.
(Стадия 10А)
Соединение (XXVIIIa) может быть получено по реакции соединения (XXVIIa) с соединением (IIIb).
Когда необходимо, для данной реакции может использоваться основание.
Примеры основания включают основания, приведенные в качестве примера на стадии 1А. Из них предпочтительными являются гидрид натрия, карбонат натрия, карбонат калия, трет-бутоксид калия, гексаметилдисилазид калия и аналогичные.
Количество используемого основания составляет обычно 0,1-100 эквивалентов, предпочтительно 1-10 эквивалентов, относительно соединения (XXVIIa).
Конкретные примеры соединения (IIIb) включают метил-3-бромпропионат, трет-бутил-3-хлорпропионат, метил-3-хлорпропионат и аналогичные.
Соединение (IIIb) может быть промышленно доступным продуктом или может быть получено по известному способу или по аналогичному ему способу.
Количество используемого соединения (IIIb) составляет обычно 1-100 эквивалентов, предпочтительно 1-5 эквивалентов, на 1 эквивалент соединения (XXVIIa).
Данная реакция осуществляется без растворителя или в инертном растворителе. Примеры инертного растворителя включают растворители, приведенные в качестве примера на стадии 1А. Из них предпочтительными являются тетрагидрофуран, ацетонитрил, N,N-диметилформамид, N,N-диметилацетамид, диметилсульфоксид и аналогичные.
Когда необходимо, для данной реакции может использоваться катализатор фазового переноса (например, бромид тетрабутиламмония, кислый сульфат тетрабутиламмония и др.).
Количество используемого катализатора фазового переноса составляет обычно 0,01-0,5 эквивалентов, предпочтительно 0,01-0,1 эквивалента, на 1 эквивалент соединения (XXVIIa).
Температура данной реакции обычно составляет от -78°С до 200°С, предпочтительно -78°С-120°С.
Время данной реакции составляет обычно от 0,5 часа до 24 часов.
Соединение (XXVIIa) может быть получено по известному способу (например, Tetrahedron Lett. 29, 2525 (1988); USP6, 211, 199; Synthesis 11, 1023 (1991)) или по аналогичному ему способу.
(Стадия 2Е)
Соединение (Ie) может быть получено из соединения (XXVIIIa) в условиях и по способу, аналогичных тем, что приведены в качестве примера на стадии 2А.
(Способ получения F)
Из соединения (I) настоящего изобретения соединение, представленное следующей формулой (If) (соединение (If)), и соединение, представленное следующей формулой (Ig) (соединение (Ig)), может быть получено, например, согласно следующей реакционной схеме 6.
(Реакционная схема 6)
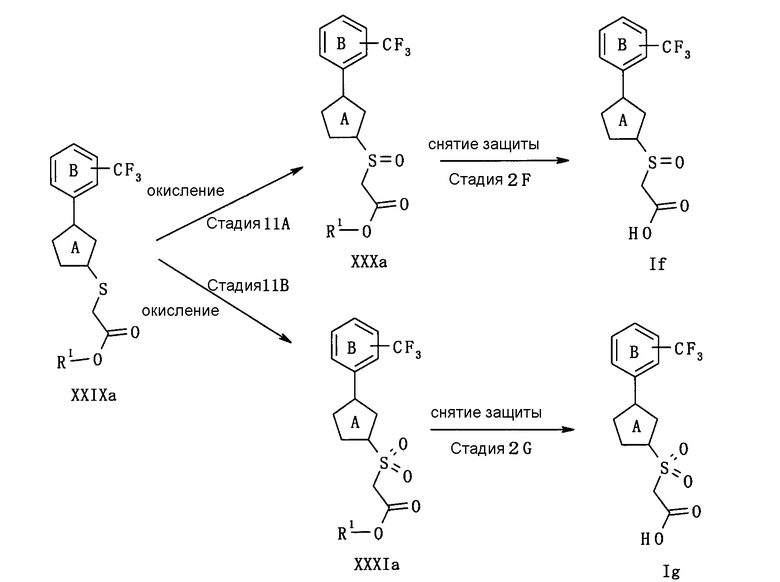
В данном способе получения соединение (If) или соединение (Ig) может быть получено из соединения (XXIXa) с помощью следующих стадий.
Стадия 11А: стадия получения соединения (XXXa), в которой соединение (XXIXa) подвергают реакции окисления;
Стадия 11В: стадия получения соединения (XXXIa), в которой соединение (XXIXa) подвергают реакции окисления;
Стадия 2F: стадия получения соединения (If) удалением R1, которой является карбоксилзащитная группа соединения (XXXa);
Стадия 2G: стадия получения соединения (Ig) удалением R1, которой является карбоксилзащитная группа соединения (XXXIa).
Каждая стадия поясняется подробно далее.
(Стадия 11А)
Соединение (XXXa) может быть получено подверганием соединения (XXIXa) реакции окисления.
Реакция окисления может обычно осуществляться с использованием окислителя в соответствии с известным способом (например, 5th edition, Jikken Kagaku Koza, vol. 17, стр. 205 (2005), Maruzen) или по аналогичному ему способу.
Примеры окислителя включают м-хлорпербензойную кислоту, оксон-персульфатное соединение, бензоилпероксид, бис(триметилсилил)пероксид, диметилдиоксиран, перекись водорода и аналогичные.
Количество используемого окислителя составляет обычно около 1-10 эквивалентов, предпочтительно 1-1,2 эквивалентов, на 1 эквивалент соединения (XXIXa).
Данная реакция может осуществляться, например, в присутствии каталитического количества тетраизопропоксида титана, тартрата натрия, вольфрамата натрия, фенилфосфоновой кислоты, четвертичной аммониевой соли и аналогичных.
Количество используемого катализатора составляет обычно 0,001-0,1 эквивалента, на 1 эквивалент соединения (XXIXa).
Данная реакция обычно осуществляется в инертном растворителе (например, галогенированных углеводородных растворителях, сложно-эфирных растворителях, нитрильных растворителях, простых эфирных растворителях и др.) или без растворителя. Данные растворители могут использоваться в виде смеси двух или более их видов в соответствующем соотношении. Из них предпочтительными являются дихлорметан, этилацетат, ацетонитрил и аналогичные.
Температура данной реакции обычно составляет примерно от 0°С до 100°С, предпочтительно 0°С-80°С.
Время данной реакции составляет, например, от 0,5 часа до 1 дня.
Соединение (XXIXa) может быть получено в соответствии с упомянутой выше стадией 4А, стадией 9А и аналогичными.
(Стадия 11В)
Соединение (XXXIa) может быть получено подверганием соединения (XXIXa) реакции окисления таким же образом, как на стадии 11А.
(Стадия 2F)
Соединение (If) может быть получено из соединения (XXXa) в условиях и по способу, аналогичных тем, что приведены в качестве примера на стадии 2А.
(Стадия 2G)
Соединение (Ig) может быть получено из соединения (XXXIa) в условиях и по способу, аналогичных тем, что приведены в качестве примера на стадии 2А.
(Способ получения G)
Из соединения (I) настоящего изобретения соединение, представленное следующей формулой (Ih) (соединение (Ih)), может быть получено, например, в соответствии со следующей реакционной схемой 7.
(Реакционная схема 7)

В данном способе получения соединение (Ih) может быть получено из соединения (XXVIIa) с помощью следующих стадий.
Стадия 10В: стадия получения соединения (XXXIIa), в которой соединение (XXVIIa) подвергают реакции алкилирования соединением (IIIa);
Стадия 2Н: стадия получения соединения (Ih) удалением R1, которой является карбоксилзащитная группа соединения (XXXIIa).
Каждая стадия поясняется подробно далее.
(Стадия 10В)
Соединение (XXXIIa) может быть получено из соединения (XXVIIa) в условиях и по способу, аналогичных тем, что приведены в качестве примера на стадии 10А.
(Стадия 2Н)
Соединение (Ih) может быть получено из соединения (XXXIIa) в условиях и по способу, аналогичных тем, что приведены в качестве примера на стадии 2А.
(Способ получения Н)
Из соединения (I) настоящего изобретения соединение, представленное следующей формулой (Ii) (соединение (Ii)), может быть получено, например, согласно следующей реакционной схеме 8.
(Реакционная схема 8)
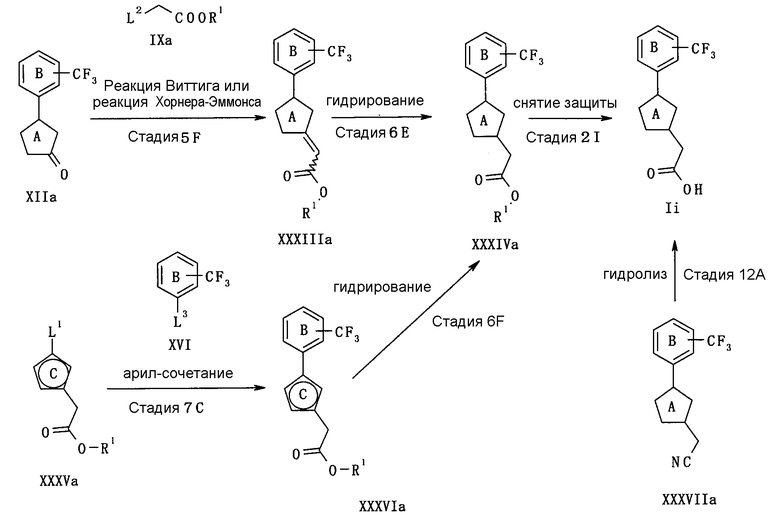
В данном способе получения соединение (Ii) может быть получено из соединения (XIIa), соединения (XXXVa) или соединения (XXXVIIa) с помощью следующих стадий.
Стадия 5F: стадия получения соединения (XXXIIIa), в которой соединение (XIIa) подвергают реакции Виттига или реакции Хорнера-Эммонса с соединением (IXa);
Стадия 6Е: стадия получения соединения (XXXIVa), соединение (XXXIIIa) подвергают реакции гидрирования;
Стадия 7C: стадия получения соединения (XXXVIa), соединение (XXXVa) подвергают реакции арилсочетания с соединением (XVI);
Стадия 6F: стадия получения соединения (XXXIVa), в которой соединение (XXXVIa) подвергают реакции гидрирования;
Стадия 2I: стадия получения соединения (Ii) удалением R1, которой является карбоксилзащитная группа соединения (XXXIVa);
Стадия 12A: стадия получения соединения (Ii), в которой цианогруппу соединения (XXXVIIa) подвергают гидролизу.
Каждая стадия поясняется подробно далее.
(Стадия 5F)
Соединение (XXXIIIa) может быть получено из соединения (XIIa) в условиях и по способу, аналогичных тем, что приведены в качестве примера на стадии 5В.
(Стадия 6Е)
Соединение (XXXIVa) может быть получено из соединения (XXXIIIa) в условиях и по способу, аналогичных тем, что приведены в качестве примера на стадии 6А.
(Стадия 7С)
Соединение (XXXVIa) может быть получено из соединения (XXXa) в условиях и по способу, аналогичных тем, что приведены в качестве примера на стадии 7А.
Соединение (XXXVa) может быть промышленно доступным продуктом. Альтернативно, соединение (XXXVa) может быть также получено согласно известному способу (например, US 2007/244094 или аналогичному ему способу.
(Стадия 6F)
Соединение (XXXIVa) может быть также получено из соединения (XXXVIa) в условиях и по способу, аналогичных тем, что приведены в качестве примера на стадии 6А.
(Стадия 2I)
Соединение (Ii) может быть получено из соединения (XXXIVa) в условиях и по способу, аналогичных тем, что приведены в качестве примера на стадии 2А.
(Стадия 12А)
Гидролиз цианогруппы соединения (XXXVIIa) может осуществляться согласно известному способу (например, 4th edition, Jikken Kagaku Koza, vol. 22, стр. 12-13, 5th edition, Jikken Kagaku Koza, vol. 16, стр. 15-16) и аналогичным. Примеры гидролиза цианогруппы включают способ с использованием кислоты, основания и аналогичных.
Соединение (XXXVIIa) может быть получено согласно известному способу (например, J. Heterocycl. Chem. 22, 129 (1985); USP 5145865) или способу, аналогичному ему.
(Способ получения I)
Из соединения (I) настоящего изобретения соединение, представленное следующей формулой (Ij) (соединение (Ij)), может быть получено, например, согласно следующей реакционной схеме 9.
(Реакционная схема 9)
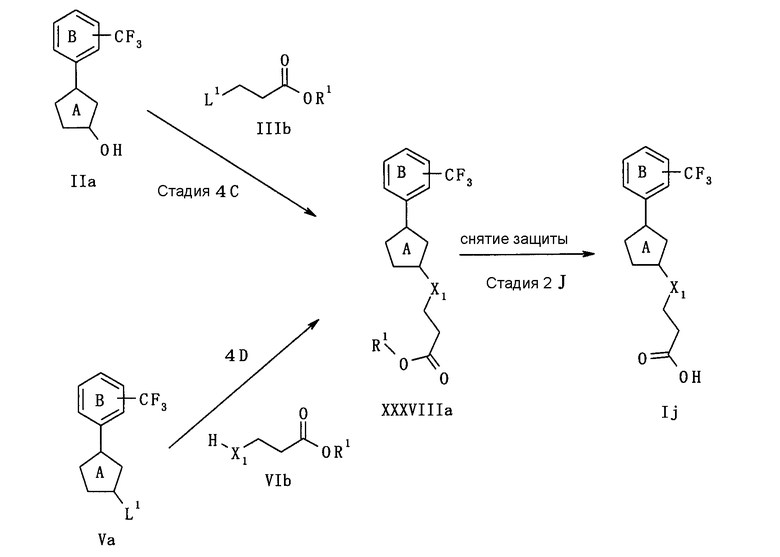
В данном способе получения соединение (Ij) может быть получено из соединения (IIa) или соединения (Va) согласно следующим стадиям.
Стадия 4С: стадия получения соединения (XXXVIIIa), в которой соединение (IIa) подвергают реакции алкилирования соединением (IIIb);
Стадия 4D: стадия получения соединения (XXXVIIIa), в которой соединение (Va) подвергают реакции алкилирования соединением (VIb);
Стадия 2J: стадия получения соединения (Ij) удалением R1, которой является карбоксилзащитная группа соединения (XXXVIIIa).
Каждая стадия поясняется подробно далее.
(Стадия 4С)
Соединение (XXXVIIIa) может быть получено из соединения (IIa) и соединения (IIIb) в условиях и по способу, аналогичных тем, что приведены в качестве примера на стадии 1А.
(Стадия 4D)
Соединение (XXXVIIIa) может быть также получено из соединения (Va) и соединения (VIb) в условиях и по способу, аналогичных тем, что приведены в качестве примера на стадии 4А.
Конкретные примеры соединения (VIb) включают этил-3-меркаптопропионат, трет-бутил-3-гидроксипропионат и аналогичные.
Соединение (VIb) может быть промышленно доступным продуктом или может быть получено по известному способу или аналогичному ему способу.
(Стадия 2J)
Соединение (Ij) может быть получено из соединения (XXXVIIIa) в условиях и по способу, аналогичных тем, что приведены в качестве примера на стадии 2А.
(Способ получения J)
Из соединения (I) настоящего изобретения, соединение, представленное следующей формулой (Ik) (соединение (Ik)), может быть получено, например, согласно следующей реакционной схеме 10.
(Реакционная схема 10)

В данном способе получения соединение (Ik) может быть получено из соединения (XXXIXa) или соединения (XLIa) с помощью следующих стадий.
Стадия 4Е: стадия получения соединения (XLa), в которой соединение (XXXIXa) подвергают реакции алкилирования соединением (IIIa);
Стадия 4F: стадия получения соединения (XLa), в которой соединение (XLIa) подвергают реакции алкилирования соединением (VIa);
Стадия 2К: стадия получения соединения (Ik) удалением R1, которой является карбоксилзащитная группа соединения (XLa).
Каждая стадия поясняется подробно далее.
(Стадия 4Е)
Соединение (XLa) может быть получено из соединения (XXXIXa) и соединения (IIIa) в условиях и по способу, аналогичных тем, что приведены в качестве примера на стадии 1А.
Соединение (XXXIXa) может быть получено согласно известному способу (например, Tetrahedron 63, 3049 (2007); Chem. Pharm. Bull. 47, 1549 (1999) или аналогичному ему способу.
(Стадия 4F)
Соединение (XLa) может быть также получено из соединения (XLIa) и соединения (VIa) в условиях и по способу, аналогичных тем, что приведены в качестве примера на стадии 4А.
Соединение (XLIa) может быть получено по известному способу (например, WO 2005/61470) или по аналогичному ему способу.
(Стадия 2K)
Соединение (Ik) может быть получено из соединения (XLa) в условиях и по способу, аналогичных тем, что приведены в качестве примера на стадии 2А.
(Способ получения K)
Из соединения (I) настоящего изобретения, соединение, представленное следующей формулой (Il) (соединение (Il)), может быть получено, например, согласно следующей реакционной схеме (11).
(Реакционная схема 11)

В данном способе получения соединение (Il) может быть получено из соединения (XIIa), соединения (VIIIa) или (XLVa) с помощью следующих стадий.
Стадия 5G: стадия получения соединения (XLIIa), в которой соединение (XIIa) подвергают реакции Виттига или реакции Хорнера-Эммонса с соединением (IXc);
Стадия 6G: стадия получения соединения (XLIIIa), в которой соединение (XLIIa) подвергают реакции гидрирования;
Стадия 5Н: стадия получения соединения (XLIVa), в которой соединение (VIIIa) подвергают реакции Виттига или реакции Хорнера-Эммонса с соединением (IXb);
Стадия 6Н: стадия получения соединения (XLIIIa), в которой соединение (XLIVa) подвергают реакции гидрирования;
Стадия 5I: стадия получения соединения (XLVIa), в которой соединение (XLVa) подвергают реакции Виттига или реакции Хорнера-Эммонса с соединением (IXa);
Стадия 6I: стадия получения соединения (XLIIIa), в которой соединение (XLVIa) подвергают реакции гидрирования;
Стадия 2L: стадия получения соединения (Il) удалением R1, которой является карбоксилзащитная группа соединения (XLIIIa).
Каждая стадия поясняется подробно далее.
(Стадия 5G)
Соединение (XLIIa) может быть получено из соединения (XIIa) и соединения (IXc) в условиях и по способу, аналогичных тем, что приведены в качестве примера на стадии 5В.
Конкретные примеры соединения (IXc) включают промышленно доступный [3-(этоксикарбонил)пропил]трифенилфосфонийбромид и аналогичные.
Соединение (IXc) может быть промышленно доступным продуктом или может быть получено по известному способу или аналогичному ему способу.
(Стадия 6G)
Соединение (XLIIIa) может быть получено из соединения (XLIIa) в условиях и по способу, аналогичных тем, что приведены в качестве примера на стадии 6В.
(Стадия 5Н)
Соединение (XLIVa) может быть получено из соединения (VIIIa) и соединения (IXb) в условиях и по способу, аналогичных тем, что приведены в качестве примера на стадии 5А.
(Стадия 6Н)
Соединение (XLIIIa) может быть получено из соединения (XLIVa) в условиях и по способу, аналогичных тем, что приведены в качестве примера на стадии 6А.
(Стадия 5I)
Соединение (XLVIa) может быть получено из соединения (XLVa) и соединения (IXa) в условиях и по способу, аналогичных тем, что приведены в качестве примера на стадии 5А.
Соединение (XLVa) может быть получено по известному способу (например, Tetrahedron Lett. 38, 603 (1997); WO 2003/76424; WO 2005/85232) или аналогичному ему способу.
(Стадия 6I)
Соединение (XLIIIa) может быть также получено из соединения (XLVIa) в условиях и по способу, аналогичных тем, что приведены в качестве примера на стадии 6А.
(Стадия 2L)
Соединение (Il) может быть получено из соединения (XLIIIa) в условиях и по способу, аналогичных тем, что приведены в качестве примера на стадии 2А.
(Способ получения L)
Из соединения (I) настоящего изобретения, соединение, представленное следующей формулой (Im) (соединение (Im)), может быть получено, например, согласно следующей реакционной схеме 12.
(Реакционная схема 12)
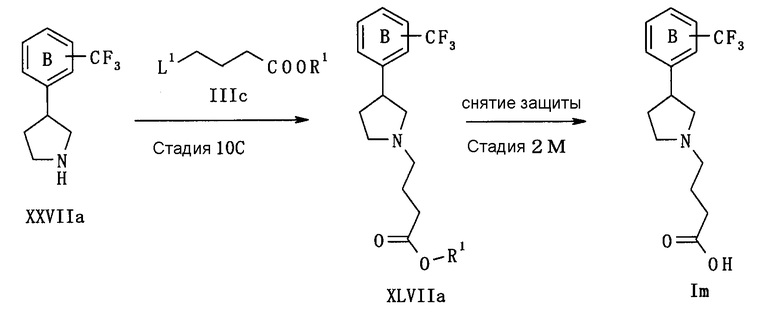
В данном способе получения соединение (Im) может быть получено из соединения (XXVIIa) с помощью следующих стадий.
Стадия 10C: стадия получения соединения (XLVIIa), в которой соединение (XXVIIa) подвергают реакции алкилирования соединением (IIIc);
Стадия 2М: стадия получения соединения (Im) удалением R1, которой является карбоксилзащитная группа соединения (XLVIIa).
Каждая стадия поясняется подробно далее.
(Стадия 10С)
Соединение (XLVIIa) может быть получено из соединения (XXVIIa) и соединения (IIIc) в условиях и по способу, аналогичных тем, что приведены в качестве примера на стадии 10А.
(Стадия 2М)
Соединение (Im) может быть получено из соединения (XLVIIa) в условиях и по способу, аналогичных тем, что приведены в качестве примера на стадии 2А.
(Способ получения М)
Из соединения (I) настоящего изобретения соединение, представленное следующей формулой (In) (соединение (In)), и соединение, представленное следующей формулой (Io) (соединение (Io)), может быть получено, например, в соответствии со следующей реакционной схемой 13.
(Реакционная схема 13)

В данном способе получения соединение (In) или соединение (Io) может быть получено из соединения (XLVIIIa) с помощью следующих стадий.
Стадия 11C: стадия получения соединения (XLIXa), в которой соединение (XLVIIIa) подвергают реакции окисления;
Стадия 11D: стадия получения соединения (La), в которой соединение (XLVIIIa) подвергают реакции окисления;
Стадия 2N: стадия получения соединения (In) удалением R1, которой является карбоксилзащитная группа соединения (XLIXa).
Стадия 2O: стадия получения соединения (Io) удалением R1, которой является карбоксилзащитная группа соединения (La).
Каждая стадия поясняется подробно далее.
(Стадия 11С)
Соединение (XLIXa) может быть получено подверганием соединения (XLVIIIa) реакции окисления с помощью того же способа, что и на стадии 11А.
Соединение (XLVIIIa) может быть получено согласно стадии 4С, стадии 4D и аналогичным.
(Стадия 11D)
Соединение (La) может быть получено подверганием соединения (XLVIIIa) реакции окисления тем же образом, как на стадии 11А.
(Стадия 2N)
Соединение (In) может быть получено из соединения (XLIXa) в условиях и по способу, аналогичных тем, что приведены в качестве примера на стадии 2А.
(Стадия 2O)
Соединение (Io) может быть получено из соединения (La) в условиях и по способу, аналогичных тем, что приведены в качестве примера на стадии 2А.
(Способ получения N)
Из соединения (I) настоящего изобретения соединение, представленное следующей формулой (Ip) (соединение (Ip)), и соединение, представленное следующей формулой (Iq) (соединение (Iq)), может быть получено, например, в соответствии со следующей реакционной схемой 14.
(Реакционная схема 14)

В данном способе получения соединение (Ip) или соединение (Iq) может быть получено из соединения (LIa) с помощью следующих стадий.
Стадия 11E: стадия получения соединения (LIIa), в которой соединение (LIa) подвергают реакции окисления;
Стадия 11F: стадия получения соединения (LIIIa), в которой соединение (LIa) подвергают реакции окисления;
Стадия 2Р: стадия получения соединения (Ip) удалением R1, которой является карбоксилзащитная группа соединения (LIIa);
Стадия 2Q: стадия получения соединения (Iq) удалением R1, которой является карбоксилзащитная группа соединения (LIIIa).
Каждая стадия поясняется подробно далее.
(Стадия 11E)
Соединение (LIIa) может быть получено подверганием соединения (LIa) реакции окисления с помощью того же способа, что и на стадии 11А.
Соединение (LIa) может быть получено согласно стадии 4Е, стадии 4F и аналогичным.
(Стадия 11F)
Соединение (LIIIa) может быть получено подверганием соединения (LIa) реакции окисления с помощью того же способа, что и на стадии 11А.
(Стадия 2P)
Соединение (Ip) может быть получено из соединения (LIIa) в условиях и по способу, аналогичных тем, что приведены в качестве примера на стадии 2А.
(Стадия 2Q)
Соединение (Iq) может быть получено из соединения (LIIIa) в условиях и по способу, аналогичных тем, что приведены в качестве примера на стадии 2А.
(Способ получения О)
Из соединения (I) настоящего изобретения соединение, представленное следующей формулой (Ir) (соединение (Ir)), и соединение, представленное следующей формулой (Ic) (соединение (Ic)), может быть получено, например, в соответствии со следующей реакционной схемой 15.
(Реакционная схема 15)

В данном способе получения соединение (IIb) может быть получено из соединения (XXIII) или соединения (LV), и соединение (Ir) или соединение (Ic) может быть получено из соединения (IIb) с помощью следующих стадий.
Стадия 9B: стадия получения соединения (IIb), в которой соединение (XXIII) подвергают реакции Бухвальда с соединением (LIV);
Стадия 13A: стадия получения соединения (IIb) реакцией соединения (LV) с соединением (LVI);
Стадия 1C: стадия получения соединения (IVb), в которой соединение (IIb) подвергают реакции алкилирования соединением (IIIa);
Стадия 2R: стадия получения соединения (Ir) удалением R1, которой является карбоксилзащитная группа соединения (IVb);
Стадия 3C: стадия получения соединения (Vb), в которой гидроксигруппу соединения (IIb) подвергают галогенированию или сульфоновой сложной этерификации;
Стадия 4G: стадия получения соединения (VIIb), в которой соединение (Vb) подвергают реакции алкилирования соединением (VIa);
Стадия 2S: стадия получения соединения (Is) удалением R1, которой является карбоксилзащитная группа соединения (VIIb).
Каждая стадия поясняется подробно далее.
(Стадия 9B)
Соединение (IIb) может быть получено из соединения (XXIII) и соединения (LIV) в условиях и по способу, аналогичных тем, что приведены в качестве примера на стадии 9А.
Соединение (XXIII) может быть промышленно доступным продуктом или может быть синтезировано согласно известному способу или аналогичному ему способу.
Рацемат или оптически активная форма соединения (LIV) может быть промышленно доступным продуктом или может быть синтезирована согласно известному способу или аналогичному ему способу.
(Стадия 13A)
Соединение (IIb) может быть также получено из соединения (LV) и соединения (LVI) согласно известному способу (например, EP 757051; Org. Lett. 7, 2409 (2005)).
Соединение (LV) и соединение (LVI) могут быть промышленно доступными продуктами или могут быть синтезированы согласно известному способу или аналогичному ему способу.
(Стадия 1C)
Соединение (IVb) может быть получено из соединения (IIb) и соедиения (IIIa) в условиях и по способу, аналогичных тем, что приведены в качестве примера на стадии 1А.
(Стадия 2R)
Соединение (Ir) может быть получено из соединения (IVb) в условиях и по способу, аналогичных тем, что приведены в качестве примера на стадии 2А.
(Стадия 3C)
Соединение (Vb) может быть получено из соединения (IIb) в условиях и по способу, аналогичных тем, что приведены в качестве примера на стадии 3А.
(Стадия 4G)
Соединение (VIIb) может быть получено из соединения (Vb) и соединения (VIa) в условиях и по способу, аналогичных тем, что приведены в качестве примера на стадии 4А.
(Стадия 2S)
Соединение (Is) может быть получено из соединения (VIIb) в условиях и по способу, аналогичных тем, что приведены в качестве примера на стадии 2А.
(Способ получения P)
Из соединения (I) настоящего изобретения, соединение, представленное следующей формулой (It) (соединение (It)), может быть получено, например, в соответствии со следующей реакционной схемой 16.
(Реакционная схема 16)
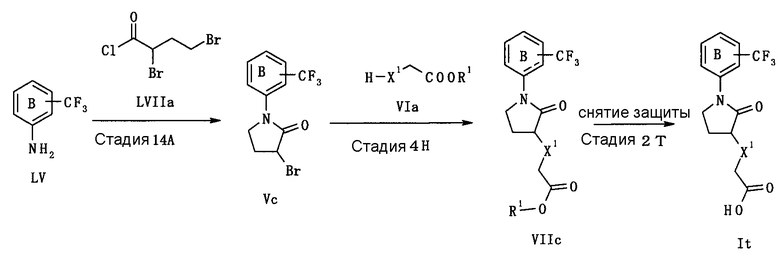
В данном способе получения соединение (It) может быть получено из соединения (LV) с помощью следующих стадий.
Стадия 14A: стадия получения соединения (Vc), в которой соединение (LV) подвергают амидированию соединением (LVIIa), и затем подверганием получающегося в результате соединения реакции внутримолекулярного замыкания кольца;
Стадия 4H: стадия получения соединения (VIIc), в которой соединение (Vc) подвергают реакции алкилирования соединением (VIa);
Стадия 2Т: стадия получения соединения (It) удалением R1, которой является карбоксилзащитная группа соединения (VIIc).
Каждая стадия поясняется подробно далее.
(Стадия 14A)
Соединение (Vc) может быть получено из соединения (LV) и соединения (LVIIa) в соответствии с известным способом (например, US 2003/87909).
Соединение (LVIIa) может быть промышленно доступным продуктом или может быть получено согласно известному способу или аналогичному ему способу.
(Стадия 4H)
Соединение (VIIc) может быть получено из соединения (Vc) и соединения (VIa) в условиях и по способу, аналогичных тем, что приведены в качестве примера на стадии 4A.
(Стадия 2Т)
Соединение (It) может быть получено из соединения (VIIc) в условиях и по способу, аналогичных тем, что приведены в качестве примера на стадии 2А.
(Способ получения Q)
Из соединения (I) настоящего изобретения, соединение, представленное следующей формулой (Iu) (соединение (Iu)), может быть получено, например, в соответствии со следующей реакционной схемой 17.
(Реакционная схема 17)

В данном способе получения соединение (Iu) может быть получено из соединения (LV) с помощью следующих стадий.
Стадия 14B: стадия получения соединения (LVIII), в которой соединение (LV) подвергают сульфонамидированию соединением (LVIIb), и затем подверганием получающегося соединения реакции внутримолекулярного замыкания кольца;
Стадия 15A: стадия получения соединения (VIIIb), в которой соединение (LVIII) подвергают формилированию;
Стадия 5J: стадия получения соединения (Xb), в которой соединение (VIIIb) подвергают реакции Виттига или реакции Хорнера-Эммонса с соединением (IXa);
Стадия 6J: стадия получения соединения (XIb), в которой соединение (Xb) подвергают реакции гидрирования;
Стадия 2U: стадия получения соединения (Iu) удалением R1, которой является карбоксилзащитная группа соединения (XIb).
Каждая стадия поясняется подробно далее.
(Стадия 14B)
Соединение (LVIII) может быть получено из соединения (LV) и соединения (LVIIb) согласно известному способу (например, WO 2003/106405).
Соединение (LVIIb) может быть промышленно доступным продуктом или может быть получено согласно известному способу или аналогичному ему способу.
(Стадия 15A)
Соединение (VIIIb) может быть получено подверганием соединения (LVIII) формилированию.
Формилирование осуществляется по реакции соединения (LVIII) с формилирующим агентом, который является электрофильным, в присутствии металлорганического реагента, которым является основание, согласно известному способу (например, Tetrahedron Lett. 24, 1647 (1983); 5th edition, Jikken Kagaku Koza, vol. 15, стр. 78-87 (2003), Maruzen) или аналогичному ему способу.
Предпочтительные примеры металлорганического реагента включают литийорганические реагенты (н-бутиллитий, втор-бутиллитий, трет-бутиллитий, диизопропиламид лития, гексаметилдисилазид лития).
Количество используемого металлорганического реагента составляет обычно 1-10 эквивалентов, предпочтительно 1-5 эквивалентов, на 1 эквивалент соединения (LVIII).
Конкретные примеры формилирующего агента включают формальдегид, формиат (этилформиат и аналогичные), N,N-диметилформамид, N-формилпиперидин и аналогичные. Они могут быть промышленно доступными продуктами или могут быть получены согласно известному способу или аналогичному ему способу.
Количество используемого формилирующего агента составляет обычно 1-10 эквивалентов, предпочтительно 1-3 эквивалента, на 1 эквивалент соединения (LVIII).
Данная реакция осуществляется в инертном растворителе (например, растворителях, приведенных для примера на стадии 1А). Данные растворители могут использоваться в смеси двух или более видов в соответствующем соотношении. Из них предпочтительными являются диэтиловый эфир, тетрагидрофуран и аналогичные.
Данная реакция предпочтительно осуществляется в инертном газе, таком как сухой аргон, сухой азот и аналогичные.
Температура данной реакции обычно составляет примерно от -78°С до 80°С, предпочтительно -78°С-40°С.
Время данной реакции составляет обычно от 0,5 часа до 16 часов.
Стадия 5J)
Соединение (Xb) может быть получено из соединения (VIIIb) и соединения (IXa) в условиях и по способу, аналогичных тем, что приведены для примера на стадии 5А.
(Стадия 6J)
Соединение (XIb) может быть получено из соединения (Xb) в условиях и по способу, аналогичных тем, что приведены для примера на стадии 6А.
(Стадия 2U)
Соединение (Iu) может быть получено из соединения (XIb) в условиях и по способу, аналогичных тем, что приведены для примера на стадии 2А.
(Способ получения R)
Из соединения (I) настоящего изобретения соединение, представленное следующей формулой (Iv) (соединение (Iv)), может быть получено, например, в соответствии со следующей реакционной схемой 18.
(Реакционная схема 18)

В данном способе получения соединение (Iv) может быть получено из соединения (LV) с помощью следующих стадий.
Стадия 16A: стадия получения соединения (XXVIb), в которой соединение (LV) подвергают обработке соединением (LIX) с образованием производного мочевины, а затем подвергают получающееся в результате соединение реакции внутримолекулярного замыкания кольца;
Стадия 10D: стадия получения соединения (XXVIIa), в которой соединение (XXVIb) подвергают реакции алкилирования соединением (IIIb);
Стадия 2V: стадия получения соединения (Iv) удалением R1, которой является карбоксилзащитная группа соединения (XXVIIa).
Каждая стадия поясняется подробно далее.
(Стадия 16A)
Соединение (XXVIb) может быть получено из соединения (LV) и соединения (LIX) согласно известному способу (например, WO 2004/9558).
Соединение (LIX) может быть промышленно доступным продуктом или может быть получено согласно известному способу или аналогичному ему способу.
(Стадия 10D)
Соединение (XXVIIa) может быть получено из соединения (XXVIb) и соединения (IIIb) в условиях и по способу, аналогичных тем, что приведены в качестве примера на стадии 10А.
(Стадия 2V)
Соединение (Iv) может быть получено из соединения (XXVIIa) в условиях и по способу, аналогичных тем, что приведены в качестве примера на стадии 2А.
(Способ получения S)
Из соединения (I) настоящего изобретения, соединение, представленное следующей формулой (Iw) (соединение (Iw)), может быть получено, например, в соответствии со следующей реакционной схемой 19.
(Реакционная схема 19)

В данном способе получения соединение (Iw) может быть получено из соединения (LV) с помощью следующих стадий.
Стадия 17A:: стадия получения соединения (XVIIb) c помощью реакции соединения ((LV) с соединением (LX);
Стадия 5K: стадия получения соединения (XVIIIb), в которой соединение (XVIIb) подвергают реакции Виттига или реакции Хорнера-Эммонса с соединением (IXa);
Стадия 6K: стадия получения соединения (XIc), в которой соединение (XVIIIb) подвергают реакции гидрирования;
Стадия 2W: стадия получения соединения (Iw) удалением R1, которой является карбоксилзащитная группа соединения (XIc).
Каждая стадия поясняется подробно далее.
(Стадия 17A)
Соединение (XVIIb) может быть получено из соединения (LV) и соединения (LX) согласно известному способу (например, J. Med. Chem. 38, 4950 (1995)).
Соединение (LX) может быть промышленно доступным продуктом или может получено согласно известному способу или аналогичному ему способу.
(Стадия 5К)
Соединение (XVIIIb) может быть получено из соединения (XVIIb) и соединения (IXa) в условиях и по способу, аналогичных тем, что приведены для примера на стадии 5А.
(Стадия 6K)
Соединение (XIc) может быть получено из соединения (XVIIIb) в условиях и по способу, аналогичных тем, что приведены для примера на стадии 6А.
(Стадия 2W)
Соединение (Iw) может быть получено из соединения (XIc) в условиях и по способу, аналогичных тем, что приведены для примера на стадии 2А.
(Способ получения Т)
Из соединения (I) настоящего изобретения, соединение, представленное следующей формулой (Ix) (соединение (Ix)), может быть получено, например, в соответствии со следующей реакционной схемой 20.
(Реакционная схема 20)
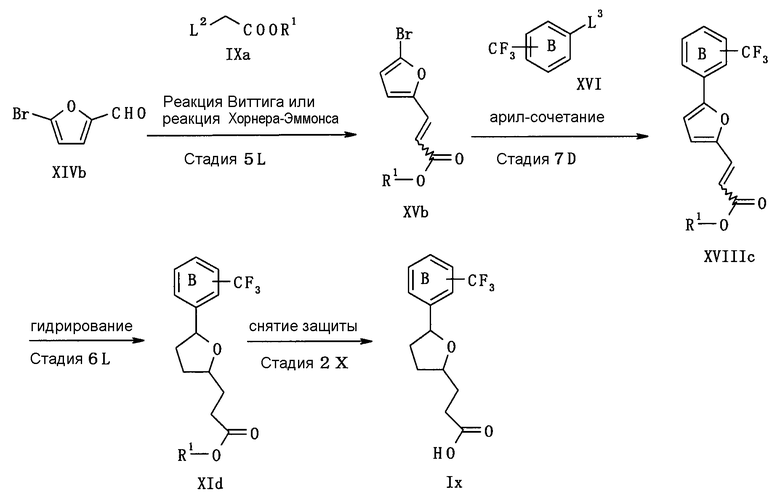
В данном способе получения соединение (Ix) может быть получено из соединения (XIVb) с помощью следующих стадий.
Стадия 5L: стадия получения соединения (XVb), в которой соединение (XIVb) подвергают реакции Виттига или реакции Хорнера-Эммонса с соединением (IXa);
Стадия 7D: стадия получения соединения (XVIIIc), в которой соединение (XVb) подвергают реакции арильного сочетания с соединением (XVI);
Стадия 6L: стадия получения соединения (XId), в которой соединение (XVIIIc) подвергают реакции гидрирования;
Стадия 2X: стадия получения соединения (Ix) удалением R1, которой является карбоксилзащитная группа соединения (XId).
Каждая стадия поясняется подробно далее.
(Стадия 5L)
Соединение (XVb) может быть получено из соединения (XIVb) и соединения (IXa) в условиях и по способу, аналогичных тем, что приведены для примера на стадии 5А.
Соединение (XIVb) может быть промышленно доступным продуктом или может быть получено согласно известному способу или аналогичному ему способу.
(Стадия 7D)
Соединение (XVIIIc) может быть получено из соединения (XVb) и соединения (XVI) в условиях и по способу, аналогичных тем, что приведены для примера на стадии 7А.
(Стадия 6L)
Соединение (XId) может быть получено из соединения (XVIIIc) в условиях и по способу, аналогичных тем, что приведены для примера на стадии 6А.
(Стадия 2X)
Соединение (Ix) может быть получено из соединения (XId) в условиях и по способу, аналогичных тем, что приведены для примера на стадии 2А.
(Способ получения U)
Из соединения (I) настоящего изобретения, соединение, представленное следующей формулой (Iy) (соединение (Iy)), может быть получено, например, в соответствии со следующей реакционной схемой 21.
(Реакционная схема 21)

В данном способе получения соединение (Iy) может быть получено из соединения (IIb) с помощью следующих стадий.
Стадия 18A: стадия получения соединения (XIIb), в которой соединение (IIb) подвергают реакции окисления;
Стадия 5М: стадия получения соединения (XXXIIIb), соединение (XIIb) подвергают реакции Виттига или реакции Хорнера-Эммонса с соединением (IXa);
Стадия 6М: стадия получения соединения (XXXIVb), соединение (XXXIIIb) подвергают реакции гидрирования;
Стадия 2Y: стадия получения соединения (Iy) удалением R1, которой является карбоксилзащитная группа соединения (XXXIVb).
Каждая стадия поясняется подробно далее.
(Стадия 18A)
Соединение (XIIb) может быть получено подверганием соединения (IIb) реакции окисления согласно известному способу (например, Bioorg. Med. Chem. 11, 145 (2003)).
(Стадия 5М)
Соединение (XXXIIIb) может быть получено из соединения (XIIb) и соединения (IXa) в условиях и по способу, аналогичных тем, что приведены для примера на стадии 5А.
(Стадия 6М)
Соединение (XXXIVb) может быть получено из соединения (XXXIIIb) в условиях и по способу, аналогичных тем, что приведены для примера на стадии 6А.
(Стадия 2Y)
Соединение (Iy) может быть получено из соединения (XXXIVb) в условиях и по способу, аналогичных тем, что приведены для примера на стадии 2А.
(Способ получения V)
Из соединения (I) настоящего изобретения, соединение, представленное следующей формулой (Iz) (соединение (Iz)), может быть получено, например, в соответствии со следующей реакционной схемой 22.
(Реакционная схема 22)

В данном способе получения соединение (Iz) может быть получено из соединения (LXI) с помощью следующих стадий.
Стадия 19A: стадия получения соединения (LXII) с помощью реакции соединения (LXI) с соединением (IIIa);
Стадия 20A: стадия получения соединения (XXVIc), в которой цианогруппу соединения (LXII) подвергают реакции внутримолекулярного замыкания кольца вследствие восстановительной реакции;
Стадия 10E: стадия получения соединения (XXXIb), в которой соединение (XXVIc) подвергают реакции алкилирования соединением (IIIa);
Стадия 2Z: стадия получения соединения (Iz) удалением R1, которой является карбоксилзащитная группа соединения (XXXIb).
Каждая стадия поясняется подробно далее.
(Стадия 19А)
Соединение (LXII) может быть получено подверганием соединения (LXI) реакции алкилирования соединением (IIIa) согласно известному способу (например, Tetrahedron 53, 5501 (1997); WO 2004/55016).
Соединение (LXI) может быть промышленно доступным продуктом или может быть получено согласно известному способу или аналогичному ему способу.
(Стадия 20A)
Соединение (XXVIc) может быть получено подверганием соединения (LXII) реакции внутримолекулярного замыкания кольца вследствие реакции восстановления согласно известному способу (например, USP 6211199).
(Стадия 10E)
Соединение (XXXIb) может быть получено из соединения (XXVIc) и соединения (IIIA) в условиях и по способу, аналогичных тем, что приведены для примера на стадии 10А.
(Стадия 2Z)
Соединение (Iz) может быть получено из соединения (XXXIb) в условиях и по способу, аналогичных тем, что приведены для примера на стадии 2А.
В соединениях, получаемых с помощью каждой реакции, упомянутой выше, функциональная группа в молекуле может быть также превращена в целевую функциональную группу с помощью комбинирования известных химических реакций. В данном описании примеры химических реакций включают в себя реакцию окисления, реакцию восстановления, реакцию алкилирования, реакцию ацилирования, реакцию уреирования, гидролиз, реакцию аминирования, реакцию сложной этерификации, реакцию арилсочетания, реакцию снятия защиты и аналогичные.
B упомянутых выше способах получения, когда исходное соединение имеет аминогруппу, карбоксильную группу, гидроксигруппу или карбонильную группу в качестве заместителя, в данные группы может вводиться защитная группа, обычно используемая в пептидной химии, и аналогичные. Указанное в заголовке соединение может быть получено путем удаления защитной группы после реакции, как необходимо.
Примеры амино-защитной группы включают формильную, C1-6 алкилкарбонильную (например, ацетил, пропаноил), C1-6 алкоксикарбонильную (например, метоксикарбонил, этоксикарбонил), бензоильную, C7-13 аралкилкарбонильную (например, бензилкарбонил), С7-13 аралкилокси-карбонильную (например, бензилоксикарбонил, 9-флуоренилметоксикарбонил), С7-13 аралкильную (например, бензил, бензгидрил), тритильную, фталоильную, N,N-диметиламинометиленовую группу, тризамещенную силильную (например, триметилсилил, триэтилсилил, диметилфенилсилил, трет-бутилдиметилсилил, трет-бутилдиэтилсилил), С2-6 алкенильную (например, 1-аллил) и аналогичные. Данные группы являются необязательно замещенными 1-3 заместителями, выбранными из атома галогена, C1-6 алкоксигруппы, нитрогруппы и аналогичных.
Примеры карбоксил-защитной группы включают C1-6 алкильную группу, С7-20 аралкильную (например, бензил, тритил), фенильную, тризамещенную силильную (например, триметилсилил, триэтилсилил, диметилфенилсилил, трет-бутилдиметилсилил, трет-бутилдиэтилсилил), C2-6 алкенильную группу (например, 1-аллил) и аналогичные. Данные группы необязательно замещены 1-5 заместителями, выбранными из атома галогена, C1-6 алкоксигруппы, нитрогруппы и аналогичных.
Примеры гидроксил-защитной группы включают C1-6 алкильную группу, фенильную, тритильную, С7-13 аралкильную (например, бензил), формильную, C1-6 алкилкарбонильную (например, ацетил, пропаноил), бензоильную, С7-13 аралкилкарбонильную (например, бензилкарбонил), 2-тетрагидропиранильную, 2-тетрагидрофуранильную, тризамещенную силильную (например, триметилсилил, триэтилсилил, диметилфенилсилил, трет-бутилдиметилсилил, трет-бутилдиэтилсилил), C2-6 алкенильную группу (например, 1-аллил) и аналогичные. Данные группы необязательно замещены 1-3 заместителями, выбранными из атома галогена, C1-6 алкильной группы, C1-6 алкоксигруппы, нитрогруппы и аналогичных.
Примеры карбонил-защитной группы включают циклический ацеталь (например, 1,3-диоксан), нециклический ацеталь (например, ди-C1-6 алкилацеталь) и аналогичные.
В дополнение, данные защитные группы могут быть введены или удалены согласно известному способу, например, способу, описанному в Protective Groups in Organic Synthesis, John Wiley and Sons (1980) и аналогичным. Примеры способа включают способ с использованием кислоты, основания, ультрафиолетовых лучей, гидразина, фенилгидразина, N-метилдитиокарбамата натрия, тетрабутиламмонийфторида, ацетата палладия, триалкилсилилгалогенида (например, триметилсилилиодида, триметилсилилбромида и др.) и аналогичных, способ восстановления и аналогичные.
Соединение настоящего изобретения, получаемого с помощью каждого из упомянутых выше способов получения, может быть выделено и очищено в соответствии c известными приемами, такими как экстракция растворителем, жидкостная конверсия, перенос растворителем, кристаллизация, перекристаллизация, хроматография и аналогичные. С другой стороны, исходное соединение может непосредственно использоваться в качестве исходного материала следующей стадии в форме реакционной смеси без выделения.
Когда соединение настоящего изобретения содержит оптический изомер, стереоизомер, региоизомер или ротамер, они также охватываются соединением настоящего изобретения и могут быть получены в виде одного продукта в соответствии с известными способами синтеза и разделения. Например, когда соединение настоящего изобретения содержит оптический изомер, оптический изомер, выделяемый из данного соединения, также охватывается соединением настоящего изобретения.
Оптический изомер может быть получен по способу, известному самому по себе.
Соединение настоящего изобретения может быть кристаллом.
Кристаллы соединения настоящего изобретения (далее называемые здесь для краткости иногда кристаллами настоящего изобретения) могут быть получены с помощью кристаллизации в соответствии с известными методами кристаллизации.
В настоящем описании точка плавления обозначает точку, измеряемую с использованием, например, устройства точки микроплавления (Yanako, MP-500D или Buchi, B-545), устройства DSC (дифференциальной сканирующей калориметрии) (SEIKO, EXSTAR6000) или аналогичных.
Обычно точки плавления варьируют в зависимости от измерительных устройств, условий измерения и аналогичных. Кристалл в настоящем описании может показывать величины, отличные от точки плавления, описанной в настоящем описании, пока они находятся в пределах обычной погрешности.
Кристаллы настоящего изобретения является превосходными по физикохимическим свойствам (например, точке плавления, растворимости, стабильности) и биологическим свойствам (например, фармакокинетическим характеристикам (абсорбции, распределению, метаболизму, экскреции), действенности), и, таким образом, они являются чрезвычайно полезными в качестве лекарственного средства.
Настоящее изобретение поясняется более подробно в следующих ссылочных примерах, примерах, экспериментальных примерах и примерах рецептурных форм, которые не должны рассматриваться как ограничительные.
LC-MS анализ в ссылочных примерах и примерах выполняли в следующих условиях.
Прибор для измерения: Waters LC-MS система
ВЭЖХ часть: Agilent HP 1100
МС часть: Micromass ZMD
Колонка: CAPCELL PAK C18 UG120, S-3 мкм, 1,5×35 мм (Shiseido Co., Ltd.)
растворитель: РАСТВОР А; 0,05% содержащая трифторуксусную кислоту вода, РАСТВОР В; 0,04% содержащий трифторуксусную кислоту ацетонитрил
градиентный цикл: 0,00 мин (РАСТВОР А/РАСТВОР В=90/10), 2,00 мин (РАСТВОР А/РАСТВОР В=5/95), 2,75 мин (РАСТВОР А/РАСТВОР В=5/95), 2,76 мин (РАСТВОР А/РАСТВОР В=90/10), 3,60 мин (РАСТВОР А/РАСТВОР В=90/10)
инжекционный объем: 2 мкл, скорость потока: 0,5 мл/мин, способ детекции: УФ220 нм
метод ионизации в МС условиях: ESI
Ссылочный пример 1 1-[3,5-бис(трифторметил)фенил]пирролидин-3-ол
Раствор бромида 3,5-бис(трифторметил)фенила (21,1 г), 3-гидроксипирролидина (6,53 г), трис(дибензилиденацетон)дипалладия(0) (1,47 г), (±)-2,2'-бис(дифенилфосфино)-1,1'-бинафтила (1,99 г) и трет-бутоксида натрия (10,6 г) в толуоле (140 мл) перемешивали в атмосфере газообразного аргона при 100°С в течение 18 часов. После охлаждения до комнатной температуры к реакционной смеси добавляли воду, и смесь экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали, и остаток очищали с помощью колоночной хроматографии на силикагеле (гексан/этилацетат 95:5-70:30), получая указанное в заголовке соединение (13,2 г, выход 61%) в виде бесцветных кристаллов.
1H-ЯМР (300 МГц, CDCl3) δ: 1,82 (уш., 1H), 2,08-2,25 (м, 2H), 3,30-3,34 (м, 1H), 3,39-3,46 (м, 1H), 3,52-3,60 (м, 2H), 4,64-4,68 (м, 1H), 6,85 (с, 2H), 7,11 (с, 1H).
Ссылочный пример 2 Этил ({1-[3,5-бис(трифторметил)фенил]пирролидин-3-ил}сульфанил)ацетат
К раствору 1-[3,5-бис(трифторметил)фенил]пирролидин-3-ола (13,2 г), полученного в ссылочном примере 1, в пиридине (70 мл) добавляли п-толуолсульфонилхлорид (9,7 г), и смесь перемешивали при комнатной температуре в течение 18 часов. К реакционной смеси добавляли воду, и смесь экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали. К полученному остатку добавляли этил тиогликолят (6,13 г), карбонат калия (13,0 г) и N,N-диметилформамид (180 мл), и смесь перемешивали при 120°С в течение 3 часов. После охлаждения до комнатной температуры к реакционной смеси добавляли воду, и смесь экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали, и остаток очищали с помощью колоночной хроматографии на силикагеле (гексан/этилацетат 100:0-80:20), получая указанное в заголовке соединение (6,83 г, выход 39%) в виде бесцветного масла.
1H-ЯМР (300 МГц, CDCl3) δ: 1,31 (т, J=7,2 Гц, 3H), 2,09-2,13 (м, 1H), 2,44-2,50 (м, 1H), 3,29-3,34 (м, 1H), 3,32 (с, 2H), 3,37-3,45 (м, 1H), 3,51-3,56 (м, 1H), 3,69-3,80 (м, 2H), 4,21 (кв, J=7,2 Гц, 2H), 6,84 (с, 2H), 7,12 (с, 1H).
Ссылочный пример 3 Метил ({1-[3,5-бис(трифторметил)фенил]пирролидин-3-ил}сульфонил)ацетат
Раствор ({1-[3,5-бис(трифторметил)фенил]пирролидин-3-ил}сульфанил)уксусной кислоты (500 мг), полученной в Примере 1, метилйодида (93 мкл) и карбоната калия (500 мг) в N,N-диметилформамиде (5 мл) перемешивали при комнатной температуре в течение 18 часов. К реакционной смеси добавляли воду, и смесь экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали. К раствору полученного остатка в дихлорметане (25 мл) добавляли м-хлорпербензойную кислоту (642 мг), и смесь перемешивали при комнатной температуре в течение 3 часов. К реакционной смеси добавляли водный раствор карбоната калия, и смесь экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали, и остаток очищали с помощью NH-силикагельной колоночной хроматографии (гексан/этилацетат 90:10-80:20), получая указанное в заголовке соединение (400 мг, выход 71%) в виде бесцветных кристаллов.
1H-ЯМР (300 МГц, CDCl3) δ: 2,52-2,61 (м, 1H), 2,64-2,71 (м, 1H), 3,48-3,56 (м, 1H), 3,62-3,69 (м, 1H), 3,85 (с, 3H), 3,77-3,90 (м, 2H), 4,07-4,08 (м, 2H), 4,35-4,39 (м, 1H), 6,90 (с, 2H), 7,20 (с, 1H).
Ссылочный пример 4 Этил ({1-[3,5-бис(трифторметил)фенил]-2-оксопирролидин-3-ил}сульфанил)ацетат
К раствору 3,5-бис(трифторметил)анилина (17,0 г) и триэтиламина (13,9 мл) в тетрагидрофуране (500 мл) добавляли 2,4-дибромбутаноилхлорид (10,0 мл), и смесь перемешивали при комнатной температуре в течение 3 часов. К реакционной смеси добавляли воду, и смесь экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали. Полученный остаток растворяли в N,N-диметилформамиде (400 мл), к раствору при 0°С добавляли гидрид натрия (3,2 г), и смесь перемешивали при комнатной температуре в течение 3 часов. К реакционной смеси добавляли воду, и смесь экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали. К полученному остатку добавляли этил тиогликолят (9,13 г), карбонат калия (13,8 г) и N,N-диметилформамид (500 мл), и смесь перемешивали при 60°С в течение 3 часов. После охлаждения до комнатной температуры к реакционной смеси добавляли воду, и смесь экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали, и остаток очищали с помощью колоночной хроматографии на силикагеле (гексан/этилацетат 95:5-80:20), получая указанное в заголовке соединение (19,5 г, выход 64%) в виде бесцветного масла.
1H-ЯМР (300 МГц, CDCl3) δ: 1,29 (т, J=7,2 Гц, 3H), 2,04-2,16 (м, 1H), 2,63-2,70 (м, 1H), 3,36-3,41 (м, 1H), 3,84-4,08 (м, 4H), 4,21 (кв, J=7,2 Гц, 2H), 7,65 (с, 1H), 8,14 (с, 2H).
Ссылочный пример 5 Этил ({1-[3,5-бис(трифторметил)фенил]-2-оксопирролидин-3-ил}сульфинил)ацетат
Раствор этил ({1-[3,5-бис(трифторметил)фенил]-2-оксопирролидин-3-ил}сульфанил)ацетата (3,0 г), полученного в ссылочном примере 4, и м-хлорпербензойной кислоты (1,7 г) в дихлорметане (25 мл) перемешивали при комнатной температуре в течение 18 часов. К реакционной смеси добавляли водный раствор гидрогенсульфита натрия, и смесь экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали, и остаток очищали с помощью колоночной хроматографии на силикагеле (гексан/этилацетат 90:10-70:30), получая указанное в заголовке соединение (1,8 г, выход 58%) в виде бесцветного масла.
1H-ЯМР (300 МГц, CDCl3) δ: 1,26-1,35 (м, 3H), 2,58-2,94 (м, 2H), 3,88-4,79 (м, 7H), 7,64-7,69 (м, 1H), 8,13-8,15 (м, 2H).
Ссылочный пример 6 Этил ({1-[3,5-бис(трифторметил)фенил]-2-оксопирролидин-3-ил}сульфонил)ацетат
Раствор этил ({1-[3,5-бис(трифторметил)фенил]-2-оксопирролидин-3-ил}сульфанил)ацетата (3,0 г), полученного в ссылочном примере 4, и м-хлорпербензойной кислоты (3,48 г) в дихлорметане (50 мл) перемешивали при комнатной температуре в течение 18 часов. К реакционной смеси добавляли водный раствор гидрогенсульфита натрия, и смесь экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали, и остаток очищали с помощью колоночной хроматографии на силикагеле (гексан/этилацетат 90:10-80:20), получая указанное в заголовке соединение (1,6 г, выход 50%) в виде бесцветного масла.
1H-ЯМР (300 МГц, CDCl3) δ: 1,34 (т, J=7,2 Гц, 3H), 2,61-2,75 (м, 1H), 2,88-2,99 (м, 1H), 3,95-4,19 (м, 3H), 4,31 (кв, J=7,2 Гц, 2H), 4,89-5,01 (с, 2H), 7,71 (с, 1H), 8,12 (с, 2H).
Ссылочный пример 7 1-[3-фтор-2-(трифторметил)фенил]пирролидин-3-ол
Указанное в заголовке соединение (6,2 г, выход 24%) получали из 1-бром-3-фтор-2-(трифторметил)бензола и 3-гидроксипирролидина по способу, аналогичному тому, как описано в ссылочном примере 1.
1H-ЯМР (300 МГц, CDCl3) δ: 1,83 (уш., 1H), 1,93-2,01 (м, 1H), 2,10-2,21 (м, 1H), 3,15-3,23 (м, 2H), 3,51-3,63 (м, 2H), 4,50 (уш., 1H), 6,60-6,66 (м, 1H), 6,76-6,79 (м, 1H), 7,25-7,32 (м, 1H).
Ссылочный пример 8 Этил ({1-[3-фтор-2-(трифторметил)фенил]пирролидин-3-ил}сульфанил)ацетат
Указанное в заголовке соединение (1,43 г, выход 16%) получали из 1-[3-фтор-2-(трифторметил)фенил]пирролидин-3-ола, полученного в ссылочном примере 7, по способу, аналогичному тому, как описано в ссылочном примере 2.
1H-ЯМР (300 МГц, CDCl3) δ: 1,28 (т, J=7,2 Гц, 3H), 1,90-1,97 (м, 1H), 2,33-2,39 (м, 1H), 3,19-3,25 (м, 1H), 3,28 (с, 2H), 3,35-3,43 (м, 2H), 3,54-3,59 (м, 1H), 3,65-3,70 (м, 1H), 4,21 (кв, J=7,2 Гц, 2H), 6,61-6,67 (м, 1H), 6,74-6,77 (м, 1H), 7,26-7,32 (м, 1H).
Ссылочный пример 9 1-[2,4-бис(трифторметил)фенил]пирролидин-3-ол
Указанное в заголовке соединение (6,84 г, выход 45%) получали из 1-бром-2,4-бис(трифторметил)бензола и 3-гидроксипирролидина по способу, аналогичному тому, как описано в ссылочном примере 1.
1H-ЯМР (300 МГц, CDCl3) δ: 1,78 (уш., 1H), 2,05-2,17 (м, 2H), 3,32-3,45 (м, 2H), 3,69-3,77 (м, 2H), 4,58-4,59 (м, 1H), 6,93 (д, J=8,7 Гц, 1H), 7,55 (д, J=8,7 Гц, 1H), 7,81 (с, 1H).
Ссылочный пример 10 Этил ({1-[2,4-бис(трифторметил)фенил]пирролидин-3-ил}сульфанил)ацетат
Указанное в заголовке соединение (5,81 г, выход 64%) получали из 1-[2,4-бис(трифторметил)фенил]пирролидин-3-ола, полученного в ссылочном примере 9, по способу, аналогичному тому, как описано в ссылочном примере 2.
1H-ЯМР (300 МГц, CDCl3) δ: 1,29 (т, J=7,2 Гц, 3H), 1,98-2,04 (м, 1H), 2,35-2,42 (м, 1H), 3,30 (с, 2H), 3,37-3,43 (м, 1H), 3,50-3,65 (м, 3H), 3,80-3,86 (м, 1H), 4,20 (кв, J=7,2 Гц, 2H), 6,91 (д, J=9,0 Гц, 1H), 7,56 (дд, J=9,0, 1,5 Гц, 1H), 7,80 (д, J=1,5 Гц, 1H).
Ссылочный пример 11 Этил ({1-[2,4-бис(трифторметил)фенил]пирролидин-3-ил}сульфонил)ацетат
Указанное в заголовке соединение (1,2 г, выход 74%) получали из этил ({1-[2,4-бис(трифторметил)фенил]пирролидин-3-ил}сульфанил)ацетата, полученного в ссылочном примере 10, по способу, аналогичному тому, как описано в ссылочном примере 6.
1H-ЯМР (300 МГц, CDCl3) δ: 1,34 (т, J=7,2 Гц, 3H), 2,42-2,61 (м, 2H), 3,47-3,62 (м, 2H), 3,82 (д, J=7,2 Гц, 2H), 4,02 (д, J=7,2 Гц, 2H), 4,19-4,32 (м, 3H), 7,10-7,13 (м, 1H), 7,63-7,66 (м, 1H), 7,84 (с, 1H).
Ссылочный пример 12 Этил 3-(пирролидин-3-ил)пропаноат
К раствору этил диэтилфосфоноацетата (15,9 г) в тетрагидрофуране (200 мл) при комнатной температуре добавляли гидрид натрия (60% в масле, 2,88 г), и смесь перемешивали в течение 30 минут. К реакционной смеси добавляли бензил 3-формилпирролидин-1-карбоксилат (15,0 г), и смесь перемешивали в течение 4 часов. К реакционной смеси добавляли воду, и смесь экстрагировали этилацетатом. Органический слой промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали, и остаток растворяли в метаноле (500 мл), добавляли гидроксид палладия на угле (2,0 г), и смесь перемешивали в атмосфере водорода (3 атм) при 40°С в течение 4 часов. Реакционному раствору давали охладиться до комнатной температуры, и реакционную систему замещали азотом, фильтровали, и растворитель выпаривали, получая указанное в заголовке соединение (8,0 г, выход 73%) в виде бесцветного масла.
1H-ЯМР (300 МГц, CDCl3) δ: 1,26 (т, J=7,2 Гц, 3H), 1,45-1,57 (м, 1H), 1,69-1,82 (м, 2H), 2,04-2,26 (м, 2H), 2,31-2,36 (м, 2H), 2,65-2,72 (м, 1H), 3,06-3,73 (м, 3H), 4,12 (д, J=7,2 Гц, 2H), 6,57 (уш., 1H).
Ссылочный пример 13 Этил 3-{1-[4-хлор-3-(трифторметил)фенил]пирролидин-3-ил}пропаноат
Указанное в заголовке соединение (5,0 г, выход 61%) получали из 4-бром-1-хлор-2-(трифторметил)бензола и этил 3-(пирролидин-3-ил)пропаноата, полученного в ссылочном примере 12, по способу, аналогичному тому, как описано в ссылочном примере 1.
1H-ЯМР (300 МГц, CDCl3) δ: 1,53 (т, J=7,2 Гц, 3H), 1,65-1,75 (м, 1H), 1,77-1,85 (м, 2H), 2,14-2,24 (м, 1H), 2,27-2,42 (м, 3H), 2,91 (т, J=8,4 Гц, 1H), 3,23-3,46 (м, 3H), 4,14 (кв, J=7,2 Гц, 2H), 6,51-6,55 (м, 1H), 6,73-6,74 (м, 1H), 7,23-7,25 (м, 1H).
Ссылочный пример 14 Этил 3-{1-[2-хлор-3-(трифторметил)фенил]пирролидин-3-ил}пропаноат
Указанное в заголовке соединение (895 мг, выход 13%) получали из 1-бром-2-хлор-3-(трифторметил)бензола и этил 3-(пирролидин-3-ил)пропаноата, полученного в ссылочном примере 12, по способу аналогичному тому, как описано в ссылочном примере 1.
1H-ЯМР (300 МГц, CDCl3) δ: 1,26 (т, J=7,2 Гц, 3H), 1,57-1,64 (м, 1H), 1,78-1,85 (м, 2H), 2,10-2,27 (м, 2H), 2,35-2,40 (м, 2H), 3,18-3,23 (м, 1H), 3,27-3,34 (м, 1H), 3,37-3,43 (м, 1H), 3,53-3,61 (м, 1H), 4,14 (кв, J=7,2 Гц, 2H), 7,02-7,06 (м, 1H), 7,17-7,20 (м, 2H).
Ссылочный пример 15 трет-бутил ({1-[3,5-бис(трифторметил)фенил]пирролидин-3-ил}окси)ацетат
Суспензию гидрида натрия (масло 60%, 1,34 г) в тетрагидрофуране (50 мл) охлаждали льдом. По каплям добавляли раствор 1-[3,5-бис(трифторметил)фенил]пирролидин-3-ола (5,0 г), полученного в ссылочном примере 1, в тетрагидрофуране (100 мл), и смесь перемешивали в течение 30 минут. К смеси добавляли трет-бутилбромацетат (0,59 г), и смесь перемешивали при 55°С в течение 16 часов. Реакционной смеси давали охладиться до комнатной температуры, добавляли насыщенный водный раствор хлорида аммония (10 мл), и смесь распределяли между этилацетатом (200 мл) и водой (200 мл). Органический слой промывали насыщенным солевым раствором, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (гексан/этилацетат 100:0-70:30), получая указанное в заголовке соединение (3,35 г, выход 49%) в виде масла бледно-желтого цвета.
1H-ЯМР (300 МГц, CDCl3) δ: 1,48 (с, 9H), 2,02-2,37 (м, 2H), 3,35-3,61 (м, 4H), 4,03 (с, 2H), 4,28-4,40 (м, 1H), 6,85 (с, 2H), 7,11 (с, 1H).
Ссылочный пример 16 1-[3,5-бис(трифторметил)фенил]имидазолидин-2-он
К раствору 3,5-бис(трифторметил)анилина (21,1 г) и триэтиламина (13,9 г) в толуоле (200 мл) при 0°С медленно добавляли 2-хлорэтилизоцианат (10,0 г), и смесь перемешивали при 60°С в течение 4 часов. К реакционному раствору добавляли этилацетат, и смесь промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток растворяли в N,N-диметилформамиде (250 мл), при 0°С медленно добавляли гидрид натрия (4,0 г), и смесь перемешивали при комнатной температуре в течение 3 часов. К реакционному раствору добавляли этилацетат, и смесь промывали водой и насыщенным солевым раствором и сушили над сульфатом натрия. Растворитель концентрировали при пониженном давлении, получая указанное в заголовке соединение в виде твердого вещества белого цвета (7,0 г, выход 26%).
1H-ЯМР (300 МГц, CDCl3) δ:3,46 (т, J=7,8 Гц, 2H), 3,99 (т, J=7,8 Гц, 2H), 7,45 (с, 1H), 7,61 (с, 1H), 8,19 (с, 2H).
Ссылочный пример 17 Этил 3-{3-[3,5-бис(трифторметил)фенил]-2-оксоимидазолидин-1-ил}пропаноат
К раствору 1-[3,5-бис(трифторметил)фенил]имидазолидин-2-она (0,30 г), полученного в ссылочном примере 16, и этил 3-бромпропионата (214 мг) в N,N-диметилформамиде (5 мл) при комнатной температуре добавляли гидрид натрия (60% в масле, 48 мг), и смесь перемешивали при 100°С в течение 16 часов. К реакционной смеси добавляли этил 3-бромпропионат (100 мг) и гидрид натрия (60% в масле, 15 мг), и смесь перемешивали при 100°С в течение 4 часов. Реакционной смеси давали охладиться до комнатной температуры, добавляли насыщенный водный раствор хлорида аммония (10 мл), и реакционную смесь концентрировали при пониженном давлении. К полученному остатку добавляли воду, и смесь экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (гексан/этилацетат 60:40-20:80), получая указанное в заголовке соединение (181 мг, выход 45%) в виде бесцветного масла.
1H-ЯМР (300 МГц, CDCl3) δ: 1,27 (т, J=7,19 Гц, 3H), 2,64 (т, J=6,63 Гц, 2H), 3,52-3,72 (м, 4H), 3,80-4,02 (м, 2H), 4,17 (кв, J=7,19 Гц, 2H), 7,51 (с, 1H), 8,04 (с, 2H).
Ссылочный пример 18 (3S)-1-[3,5-бис(трифторметил)фенил]пирролидин-3-ол
Раствор 3,5-бис(трифторметил)фенилбромида (44,0 г), гидрохлорида (S)-3-гидроксипирролидина (17,8 г), трис(дибензилиденацетон)дипалладия(0) (5,88 г), (±)-2,2'-бис(дифенилфосфино)-1,1'-бинафтила (8,00 г) и трет-бутоксида натрия (36,0 г) в толуоле (280 мл) нагревали с обратным холодильником при перемешивании в атмосфере газообразного аргона в течение 16 часов. После охлаждения до комнатной температуры к реакционной смеси добавляли воду, и смесь экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали, и остаток очищали с помощью колоночной хроматографии на силикагеле (петролейный эфир/этилацетат 20:1-2:1), получая указанное в заголовке соединение (12,0 г, выход 28%) в виде твердого вещества белого цвета.
1H-ЯМР (300 МГц, CDCl3) δ: 1,67 (уш., 1H), 2,08-2,36 (м, 2H), 3,28-3,38 (м, 1H), 3,38-3,50 (м, 1H), 3,50-3,65 (м, 2H), 4,55-4,81 (м, 1H), 6,87 (с, 2H), 7,13 (с, 1H).
Ссылочный пример 19 (3R)-1-[3,5-бис(трифторметил)фенил]пирролидин-3-ол
Смешанный раствор 3,5-бис(трифторметил)фенилбромида (22,0 г), гидрохлорида (R)-3-гидроксипирролидина (9,23 г), ацетата палладия(II) (0,84 г), (±)-2,2'-бис(дифенилфосфино)-1,1'-бинафтила (4,68 г) и карбоната цезия (73,4 г) в толуоле (300 мл) - 1,4-диоксане (100 мл) перемешивали в атмосфере газообразного аргона при 80°С в течение 16 часов. Твердое вещество отфильтровывали, и фильтрат промывали насыщенным солевым раствором и водой, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали, остаток очищали с помощью колоночной хроматографии на силикагеле (петролейный эфир/этилацетат 10:1-3:1), получая указанное в заголовке соединение (13,7 г, выход 61%) в виде твердого вещества белого цвета.
1H-ЯМР (300 МГц, CDCl3) δ: 1,74 (уш., 1H), 2,06-2,31 (м, 2H), 3,28-3,38 (м, 1H), 3,39-3,50 (м, 1H), 3,50-3,66 (м, 2H), 4,61-4,73 (м, 1H), 6,87 (с, 2H), 7,12 (с, 1H).
Ссылочный пример 20 4-[3,5-бис(трифторметил)фенил]фуран-2-карбальдегид
Раствор 3,5-бис(трифторметил)фенилбороновой кислоты (8,84 г), 4-бром-2-фуральдегида (5,0 г), 2М водного раствора карбоната натрия (71,4 мл), тетракис(трифенилфосфин)палладия(0) (1,65 г) в 1,2-диметоксиэтане (300 мл) перемешивали в атмосфере газообразного аргона при 90°С в течение 16 часов. После охлаждения до комнатной температуры реакционную смесь концентрировали, добавляли воду, и смесь экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали, и остаток очищали с помощью колоночной хроматографии на силикагеле (гексан/этилацетат 98:2-90:10), получая указанное в заголовке соединение (7,26 г, выход 82%) в виде твердого вещества белого цвета.
1H-ЯМР (300 МГц, CDCl3) δ: 7,58 (с, 1H), 7,86 (с, 1H), 7,93 (с, 2H), 8,09 (с, 1H), 9,76 (с, 1H).
Ссылочный пример 21 Этил (2Е)-3-{4-[3,5-бис(трифторметил)фенил]фуран-2-ил}проп-2-еноат
К раствору 4-[3,5-бис(трифторметил)фенил]фуран-2-карбальдегида (2,70 г), полученного в ссылочном примере 20, и этил диэтилфосфоноацетата (2,16 г) в тетрагидрофуране (50 мл) при комнатной температуре добавляли гидрид натрия (60% в масле, 456 мг), и смесь перемешивали в течение 1 часа. К реакционной смеси добавляли насыщенный водный раствор хлорида аммония (10 мл), и смесь концентрировали при пониженном давлении. К полученному остатку добавляли воду, и смесь экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали, получая твердое вещество бледно-желтого цвета. Его перекристаллизовывали из гексана-этилацетата, получая указанное в заголовке соединение (2,51 г, выход 76%) в виде твердого вещества белого цвета.
1H-ЯМР (300 МГц, CDCl3) δ: 1,34 (т, J=7,16 Гц, 3H), 4,27 (кв, J=7,10 Гц, 2H), 6,42 (д, J=15,8 Гц, 1H), 6,93 (с, 1H), 7,46 (д, J=15,8 Гц, 1H), 7,80 (с, 1H), 7,88 (с, 3 H).
Ссылочный пример 22 этил 3-{4-[3,5-бис(трифторметил)фенил]тетрагидрофуран-2-ил}пропаноат
К раствору этил (2Е)-3-{4-[3,5-бис(трифторметил)фенил]фуран-2-ил}проп-2-еноата (500 мг), полученного в ссылочном примере 21, в смешанном растворе из этанола (5 мл) и этилацетата (10 мл) добавляли 10% палладий на угле (содержащий воду (50%), 50 мг), и смесь перемешивали в атмосфере водорода (1 атм) при комнатной температуре в течение 3 часов. Реакционную систему замещали азотом и фильтровали. Растворитель выпаривали, получая стереоизомерную смесь целевого соединения (510 мг, выход >99%) в виде бесцветного масла.
LC-МС ESI(+) m/z:385 (М+H)+, время удерживания 2,68 мин.
Ссылочный пример 23 1-[4-хлор-3-(трифторметил)фенил]пирролидин-3-ол
Указанное в заголовке соединение (8,56 г, выход 90%) получали из бром-4-хлор-3-(трифторметил)бензола и 3-гидроксипирролидина по способу, аналогичному тому, как описано в ссылочном примере 1.
1H-ЯМР (300 МГц, CDCl3) δ: 1,67 (уш., 1H), 2,02-2,29 (м, 2H), 3,21-3,30 (м, 1H), 3,31-3,42 (м, 1H), 3,44-3,58 (м, 2H), 4,58-4,70 (м, 1H), 6,51-6,63 (м, 1H), 6,77-6,82 (м, 1H), 7,26-7,31 (м, 1H).
Ссылочный пример 24 4-метилбензолсульфонат 1-[4-хлор-3-(трифторметил)фенил]пирролидин-3-ила
К раствору 1-[4-хлор-3-(трифторметил)фенил]пирролидин-3-ола (3,0 г), полученного в ссылочном примере 23, в пиридине (20 мл) добавляли п-толуолсульфонилхлорид (2,8 г), и смесь перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь концентрировали, к остатку добавляли воду, и смесь экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали, и полученный остаток очищали с помощью колоночной хроматографии на силикагеле (гексан/этилацетат 90:10-75:25), получая указанное в заголовке соединение (3,66 г, выход 77%) в виде кристаллов белого цвета.
1H-ЯМР (300 МГц, CDCl3) δ: 2,10-2,38 (м, 2H), 2,45 (с, 3H), 3,28-3,61 (м, 4H), 5,13-5,34 (м, 1H), 6,47-6,56 (м, 1H), 6,66-6,71 (м, 1H), 7,23-7,30 (м, 1H), 7,35 (м, 2H), 7,79 (м, 2H).
Ссылочный пример 25 Этил ({1-[4-хлор-3-(трифторметил)фенил]пирролидин-3-ил}сульфанил)ацетат
К раствору 4-метилбензолсульфоната 1-[4-хлор-3-(трифторметил)фенил]пирролидин-3-ила (3,65 г), полученного в ссылочном примере 24, в N,N-диметилформамиде (20 мл) добавляли этил тиогликолят (1,26 г), и карбонат калия (5,92 г), и смесь перемешивали при 120°С в течение 3 часов. После охлаждения до комнатной температуры реакционную смесь концентрировали, добавляли воду, и смесь экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали, и остаток очищали с помощью колоночной хроматографии на силикагеле (гексан/этилацетат 95:5-80:20), получая указанное в заголовке соединение (2,95 г, выход 92%) в виде масла желтого цвета.
1H-ЯМР (300 МГц, CDCl3) δ: 1,31 (т, J=7,1 Гц, 3H), 1,97-2,16 (м, 1H), 2,35-2,51 (м, 1H), 3,16-3,41 (м, 4H), 3,41-3,54 (м, 1H), 3,62-3,76 (м, 2H), 4,22 (кв, J=7,1 Гц, 2H), 6,53-6,61 (м, 1H), 6,74-6,80 (м, 1H), 7,23-7,34 (м, 1H).
Ссылочный пример 26 1-[2-хлор-3-(трифторметил)фенил]пирролидин-3-ол и 1-[3-(трифторметил)фенил]пирролидин-3-ол
Смесь 1-[2-хлор-3-(трифторметил)фенил]пирролидин-3-ола и 1-[3-(трифторметил)фенил]пирролидин-3-ола получали в виде масла коричневого цвета (8,08 г, выход 90%) из 1-бром-2-хлор-3-(трифторметил)бензола и 3-гидроксипирролидина по способу, аналогичному тому, как описано в ссылочном примере 1. Ее использовали для следующей реакции без осуществления дальнейшей очистки.
Ссылочный пример 27 4-метилбензолсульфонат 1-[2-хлор-3-(трифторметил)фенил]пирролидин-3-ила и 4-метилбензолсульфонат 1-[3-(трифторметил)фенил]пирролидин-3-ила
Смесь 1-[2-хлор-3-(трифторметил)фенил]пирролидин-3-ил 4-метилбензолсульфоната и 1-[3-(трифторметил)фенил]пирролидин-3-ил 4-метилбензолсульфоната получали в виде кристаллов белого цвета (3,35 г, выход 69%) из смеси 1-[2-хлор-3-(трифторметил)фенил]пирролидин-3-ола и 1-[3-(трифторметил)фенил]пирролидин-3-ола, полученной в ссылочном примере 26, по способу, аналогичному тому, как описано в ссылочном примере 24. Ее использовали для следующей реакции без осуществления дальнейшей очистки.
LC-МС ESI(+) m/z: 386 (М+H)+, время удерживания 2,74 мин.; 420 (М+H)+, время удерживания 2,74 мин.
Ссылочный пример 28 Этил ({1-[2-хлор-3-трифторметил)фенил]пирролидин-3-ил}сульфанил)ацетат
Указанное в заголовке соединение (1,08 г, выход 61%) получали в виде масла желтого цвета из смеси 4-метилбензолсульфоната 1-[2-хлор-3-(трифторметил)фенил]пирролидин-3-ила и 4-метилбензолсульфоната 1-[3-(трифторметил)фенил]пирролидин-3-ила, полученной в ссылочном примере 27, по способу, аналогичному тому, как описано в ссылочном примере 25.
1H-ЯМР (300 МГц, CDCl3) δ: 1,24-1,34 (м, 3H), 1,89-2,04 (м, 1H), 2,33-2,47 (м, 1H), 3,27-3,41 (м, 3H), 3,42-3,55 (м, 2H), 3,55-3,68 (м, 1H), 3,76-3,86 (м, 1H), 4,15-4,25 (м, 2H), 7,04-7,13 (м, 1H), 7,19-7,29 (м, 2H).
Ссылочный пример 29 Этил ({1-[3-(трифторметил)фенил]пирролидин-3-ил}сульфанил)ацетат
Указанное в заголовке соединение (267 мг, выход 17%) получали в виде масла желтого цвета из смеси 1-[2-хлор-3-(трифторметил)фенил]пирролидин-3-ил 4-метилбензолсульфоната и 1-[3-(трифторметил)фенил]пирролидин-3-ил 4-метилбензолсульфоната, полученной в ссылочном примере 27, по способу, аналогичному тому, как описано в ссылочном примере 25.
1H-ЯМР (300 МГц, CDCl3) δ: 1,31 (т, J=7,2 Гц, 3H), 1,99-2,14 (м, 1H), 2,36-2,52 (м, 1H), 3,21-3,44 (м, 4H), 3,44-3,57 (м, 1H), 3,61-3,79 (м, 2H), 4,22 (кв, J=7,2 Гц, 2H), 6,60-6,74 (м, 2H), 6,87-6,97 (м, 1H), 7,26-7,35 (м, 1H).
Ссылочный пример 30 1-[2,5-бис(трифторметил)фенил]пирролидин-3-ол
Раствор 2,5-бис(трифторметил)фенилбромида (4,45 г), 3-гидроксипирролидина (1,2 г), ацетата палладия(II) (0,16 г), (±)-2,2'-бис(дифенилфосфино)-1,1'-бинафтила (0,87 г) и карбоната цезия (13,6 г) в толуоле (74 мл) перемешивали в атмосфере газообразного аргона при 80°С в течение 16 часов. Реакционной смеси давали охладиться до комнатной температуры, добавляли воду, и смесь экстрагировали этилацетатом. Экстракт промывали насыщенным солевым раствором, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали, и остаток очищали с помощью колоночной хроматографии на силикагеле (гексан/этилацетат 90:10-75:25), получая указанное в заголовке соединение (3,79 г, выход 91%) в виде масла оранжевого цвета.
1H-ЯМР (300 МГц, CDCl3) δ: 1,72 (д, J=4,9 Гц, 1H), 1,95-2,10 (м, 1H), 2,10-2,24 (м, 1H), 3,21-3,43 (м, 2H), 3,60-3,78 (м, 2H), 4,48-4,65 (м, 1H), 7,01-7,16 (м, 1H), 7,16-7,23 (м, 1H), 7,63-7,74 (м, 1H).
Ссылочный пример 31 Этил (2Е)-3-(5-бромфуран-2-ил)проп-2-еноат
К раствору (200 мл) этил диэтилфосфоноацетата (10,5 г) в N,N-диметилформамиде добавляли гидрид натрия (60% в масле, 1,87 г), и смесь перемешивали в атмосфере азота при комнатной температуре в течение 15 минут. К данному раствору добавляли раствор 5-бром-2-фуральдегида (7,45 г) в N,N-диметилформамиде (40 мл), и смесь перемешивали в атмосфере азота при комнатной температуре в течение 1 часа. Реакцию гасили насыщенным раствором хлорида аммония (50 мл), и реакционный раствор распределяли между этилацетатом (900 мл) и водой (900 мл). Органический слой промывали насыщенным солевым раствором, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении, и полученный остаток очищали с помощью колоночной хроматографии на силикагеле (гексан/этилацетат 90:10-85:15), получая указанное в заголовке соединение (10,0 г, выход 96%) в виде твердого вещества бледно-желтого цвета.
1H-ЯМР (300 МГц, CDCl3) δ: 1,32 (т, J=7,2 Гц, 3H), 4,24 (кв, J=7,2 Гц, 2H), 6,31 (д, J=15,6 Гц, 1H), 6,40 (д, J=3,4 Гц, 1H), 6,54 (д, J=3,6 Гц, 1H), 7,31 (д, J=15,8 Гц, 1H).
Ссылочный пример 32 Этил (2Е)-3-{5-[3,5-бис(трифторметил)фенил]фуран-2-ил}проп-2-еноат
К смеси этил (2Е)-3-(5-бромфуран-2-ил)проп-2-еноата (10,0 г), полученного в ссылочном примере 31, 3,5-бис(трифторметил)фенилбороновой кислоты (11,0 г) и 2М карбоната натрия (102 мл) в диметоксиэтане (500 мл) добавляли тетракис(трифенилфосфин)палладий(0) (2,00 г) в атмосфере газообразного аргона, и смесь перемешивали при 110°С в течение 9 часов, и затем при 95°С в течение 15 часов. Реакционному раствору давали охладиться до комнатной температуры, концентрировали, и распределяли между этилацетатом (500 мл) и водой (500 мл). Этилацетатный слой промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении, и полученный остаток очищали с помощью колоночной хроматографии на силикагеле (гексан/этилацетат 90:10-85:15), получая указанное в заголовке соединение (13,2 г, выход 85%) в виде твердого вещества бледно-желтого цвета.
1H-ЯМР (300 МГц, CDCl3) δ: 1,35 (т, J=7,0 Гц, 3H), 4,29 (кв, J=7,2 Гц, 2H), 6,49 (д, J=15,9 Гц, 1H), 6,74 (д, J=3,8 Гц, 1H), 6,92 (д, J=3,4 Гц, 1H), 7,46 (д, J=15,9 Гц, 1H), 7,79 (с, 1H), 8,10 (с, 2H).
Ссылочный пример 33 Этил 3-{5-[3,5-бис(трифторметил)фенил]тетрагидрофуран-2-ил}пропаноат
Раствор этил (2Е)-3-{5-[3,5-бис(трифторметил)фенил]фуран-2-ил}проп-2-еноата (13,1 г), полученного в ссылочном примере 32, и 10% палладия на угле (содержащего воду (50%), 3,48 г) в смеси этанол-тетрагидрофуран (3:1, 320 мл) перемешивали в атмосфере водорода (1 атм) при комнатной температуре в течение 2 дней. Реакционный раствор разбавляли этилацетатом и фильтровали через силикагель. Силикагель промывали этилацетатом (500 мл), фильтрат и смывки объединяли, и растворитель удаляли при пониженном давлении, получая указанное в заголовке соединение (13,2 г, выход 99%) в виде бесцветного масла.
1H-ЯМР (300 МГц, CDCl3) δ: 7,78 (с, 3H), 4,96 (т, J=7,3 Гц, 1H), 4,15 (кв, J=7,2 Гц, 2H), 4,07 (т, J=7,3 Гц, 1H), 2,63-2,32 (м, 3H), 2,20-2,09 (м, 1H), 2,05-1,96 (м, 2H), 1,88-1,60 (м, 2H), 1,26 (т, J=7,1 Гц, 3H).
Ссылочный пример 34 1-[3,5-бис(трифторметил)фенил]-1Н-пиррол-3-карбальдегид
Раствор 3,5-бис(трифторметил)анилина (3,58 г) и 2,5-диметокси-3-тетрагидрофуранкарбальдегида (2,50 г) в уксусной кислоте (16 мл) перемешивали при 90°С в течение 30 минут. После охлаждения до комнатной температуры реакционный раствор концентрировали при пониженном давлении, и остаток распределяли между этилацетатом и насыщенным раствором бикарбоната натрия. Этилацетатный слой промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении, и полученный остаток очищали с помощью колоночной хроматографии на силикагеле (гексан/этилацетат 70:30), получая указанное в заголовке соединение (2,44 г, выход 51%) в виде твердого вещества бледно-желтого цвета.
1H-ЯМР (300 МГц, CDCl3) δ: 6,90 (дд, J=3,1, 1,6 Гц, 1H), 7,18 (т, J=2,4 Гц, 1H), 7,75 (д, J=2,3 Гц, 1H), 7,88 (с, 3H), 9,91 (с, 1H).
Ссылочный пример 35 Этил (2Е)-3-{1-[3,5-бис(трифторметил)фенил]-1Н-пиррол-3-ил}проп-2-еноат
К раствору (35 мл) этил диэтилфосфоноацетата (1,77 г) в N,N-диметилформамиде добавляли гидрид натрия (60% в масле, 0,32 г), и смесь перемешивали в атмосфере азота при комнатной температуре в течение 15 минут. К данному раствору добавляли раствор 1-[3,5-бис(трифторметил)фенил]-1Н-пиррол-3-карбальдегида (2,20 г), полученного в ссылочном примере 34, в N,N-диметилформамиде (10 мл), и смесь перемешивали в атмосфере азота при комнатной температуре в течение 1 часа. Реакцию гасили насыщенным раствором хлорида аммония (10 мл), и реакционный раствор распределяли между этилацетатом (120 мл) и водой (120 мл). Органический слой промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении. Полученное твердое вещество растирали с гексаном, получая указанное в заголовке соединение (2,37 г, выход 88%) в виде твердого вещества белого цвета.
1H-ЯМР (300 МГц, CDCl3) δ: 1,33 (т, J=7,2 Гц, 3H), 4,25 (кв, J=7,2 Гц, 2H), 6,22 (д, J=15,8 Гц, 1H), 6,65 (дд, J=2,9, 1,4 Гц, 1H), 7,14 (т, J=2,4 Гц, 1H), 7,34 (т, J=1,9 Гц, 1H), 7,64 (д, J=15,8 Гц, 1H), 7,75-7,85 (м, 3 H).
Ссылочный пример 36 Этил 3-{1-[3,5-бис(трифторметил)фенил]пирролидин-3-ил}пропаноат
Раствор этил (2Е)-3-{1-[3,5-бис(трифторметил)фенил]-1Н-пиррол-3-ил}проп-2-еноата (0,60 г), полученного в ссылочном примере 35, и 10% палладия на угле (содержащего воду (50%), 0,17 г) в смеси этанол-тетрагидрофуран (4:1, 20 мл) перемешивали в атмосфере водорода (1 атм) при комнатной температуре в течение 16 часов. Реакционный раствор фильтровали через целит, и фильтрат концентрировали. Остаток очищали с помощью колоночной хроматографии на силикагеле (гексан/этилацетат 90:10), получая указанное в заголовке соединение (0,47 г, выход 77%) в виде твердого вещества белого цвета.
1H-ЯМР (300 МГц, CDCl3) δ: 1,28 (т, J=7,2 Гц, 3H), 1,68-1,75 (м, 1H), 1,78-1,87 (м, 2H), 2,20-2,27 (м, 1H), 2,30-2,50 (м, 3H), 2,98 (т, J=8,6 Гц, 1H), 3,23-3,63 (м, 3H), 4,16 (кв, J=7,2 Гц, 2H), 6,83 (с, 2H), 7,09 (с, 1H).
Ссылочный пример 37 N-[3,5-бис(трифторметил)фенил]-3-хлорпропансульфонамид
Раствор 3-хлорпропансульфонилхлорида (5,0 г) и 3,5-бис(трифторметил)анилина (6,47 г) в пиридине (50 мл) перемешивали при комнатной температуре в течение 16 часов. Реакционный раствор концентрировали при пониженном давлении, и остаток распределяли между этилацетатом и 1н соляной кислотой. Органический слой промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении, и полученный остаток очищали с помощью колоночной хроматографии на силикагеле (гексан/этилацетат 95:5-60:40), получая указанное в заголовке соединение (7,20 г, выход 69%) в виде твердого вещества бледно-коричневого цвета.
1H-ЯМР (300 МГц, CDCl3) δ: 2,29-2,42 (м, 2H), 3,37 (т, J=6,1 Гц, 2H), 3,68 (т, J=7,5 Гц, 2H), 7,66 (с, 2H), 7,69 (с, 1H).
Ссылочный пример 38 1,1-диоксид 2-[3,5-бис(трифторметил)фенил]изотиазолидина
К раствору N-[3,5-бис(трифторметил)фенил]-3-хлорпропансульфонамида (7,10 г), полученного в ссылочном примере 37, в N,N-диметилформамиде (100 мл) добавляли гидрид натрия (60% в масле, 0,84 г), и смесь перемешивали при комнатной температуре в течение 3 часов. К реакционной смеси добавляли насыщенный раствор хлорида аммония, и раствор распределяли между этилацетатом и водой. Органический слой промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении, и полученный остаток очищали с помощью колоночной хроматографии на силикагеле (гексан/этилацетат 90:10-40:60), получая указанное в заголовке соединение (2,44 г, выход 38%) в виде твердого вещества бледно-коричневого цвета.
1H-ЯМР (300 МГц, CDCl3) δ: 2,62 (т, J=7,3 Гц, 2H), 3,45 (т, J=7,3 Гц, 2H), 3,86 (т, J=6,6 Гц, 2H), 7,62 (с, 1H), 7,64 (с, 2H).
Ссылочный пример 39 1,1-диоксид 2-[3,5-бис(трифторметил)фенил]изотиазолидин-5-карбальдегида
Раствор 1,1-диоксида 2-[3,5-бис(трифторметил)фенил]изотиазолидина (1,77 г), полученного в ссылочном примере 38, в тетрагидрофуране (60 мл) охлаждали до -78°С в атмосфере аргона, и по каплям добавляли раствор (1,1 моль/л, 14,5 мл) гексаметилдисилазида лития в тетрагидрофуране. Все перемешивали в течение 30 минут, добавляли этилформиат (0,59 г), и смесь перемешивали при -78°С в течение 1 часа и при комнатной температуре в течение 16 часов. К реакционной смеси добавляли насыщенный раствор хлорида аммония, и смесь концентрировали при пониженном давлении, и остаток распределяли между этилацетатом и водой. Этилацетатный слой промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении, и полученный остаток очищали с помощью колоночной хроматографии на силикагеле (гексан/этилацетат 90:10-30:70), получая указанное в заголовке соединение (1,63 г, выход 85%) в виде аморфной пены бледно-желтого цвета.
Данное вещество использовали в следующей реакции без осуществления дальнейшей очистки и идентификации.
Ссылочный пример 40 Этил (2Е)-3-{2-[3,5-бис(трифторметил)фенил]-1,1-диоксидоизотиазолидин-5-ил}проп-2-еноат
К раствору (10 мл) этил диэтилфосфоноацетата (1,01 г) в N,N-диметилформамиде добавляли гидрид натрия (60% в масле, 0,18 г), и смесь перемешивали в атмосфере азота при комнатной температуре в течение 15 минут. К данному раствору добавляли раствор 1,1-диоксида 2-[3,5-бис(трифторметил)фенил]изотиазолидин-5-карбальдегида (1,63 г), полученного в ссылочном примере 39, в N,N-диметилформамиде (6 мл), и смесь перемешивали в атмосфере азота при комнатной температуре в течение 1 часа. Реакцию гасили насыщенным раствором хлорида аммония, и реакционный раствор распределяли между этилацетатом и водой. Органический слой промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении, и полученный остаток очищали с помощью колоночной хроматографии на силикагеле (гексан/этилацетат 90:10-40:60), и полученное твердое вещество желтого цвета перекристаллизовывали из гексана-этилацетата, получая указанное в заголовке соединение (0,43 г, выход 22%) в виде твердого вещества бледно-желтого цвета.
1H-ЯМР (300 МГц, CDCl3) δ: 1,32 (т, J=7,2 Гц, 3H), 2,48-2,68 (м, 1H), 2,67-2,85 (м, 1H), 3,79-3,92 (м, 2H), 4,06-4,22 (м, 1H), 4,26 (кв, J=7,2 Гц, 2H), 6,23 (д, J=15,5 Гц, 1H), 6,90 (дд, J=15,5, 8,5 Гц, 1H), 7,65 (с, 3 H).
Ссылочный пример 41 Этил 3-{2-[3,5-бис(трифторметил)фенил]-1,1-диоксидоизотиазолидин-5-ил}пропаноат
Раствор этил (2Е)-3-{2-[3,5-бис(трифторметил)фенил]-1,1-диоксидоизотиазолидин-5-ил}проп-2-еноата (0,32 г), полученного в ссылочном примере 40, и 20% палладия на гидроксиде углерода (содержащего воду (50%), 0,10 г) в этаноле-тетрагидрофуране (3:1, 20 мл) перемешивали в атмосфере водорода (4 атм) при 50°С в течение 8 часов. Реакционный раствор фильтровали с помощью мембранного фильтра (Advantec, 0,5 мкм), и фильтрат концентрировали, получая указанное в заголовке соединение (0,29 г, выход 91%) в виде твердого вещества белого цвета.
1H-ЯМР (300 МГц, CDCl3) δ: 1,28 (т, J=7,2 Гц, 3H), 2,08-2,43 (м, 3H), 2,58-2,75 (м, 3H), 3,44-3,65 (м, 1H), 3,72-3,85 (м, 2H), 4,17 (кв, J=7,2 Гц, 2H), 7,61 (с, 1H), 7,63 (с, 2H).
Ссылочный пример 42 1-[2-(трифторметил)фенил]пирролидин-3-ол
Указанное в заголовке соединение (3,72 г, выход 67%) получали из 2-(трифторметил)фенилбромида и 3-гидроксипирролидина по способу, аналогичному тому, как описано в ссылочном примере 1.
1H-ЯМР (300 МГц, CDCl3) δ: 1,87 (д, J=6,1 Гц, 1H), 1,92-2,07 (м, 1H), 2,10-2,28 (м, 1H), 3,04-3,31 (м, 2H), 3,39-3,67 (м, 2H), 4,41-4,59 (м, 1H), 6,96 (т, J=7,6 Гц, 1H), 7,08 (д, J=8,3 Гц, 1H), 7,35-7,46 (м, 1H), 7,59 (м, 1H).
Ссылочный пример 43 1-[3-(трифторметил)фенил]пирролидин-3-ол
Указанное в заголовке соединение (2,67 г, выход 54%) получали из 3-(трифторметил)фенилбромида и 3-гидроксипирролидина по способу, аналогичному тому, как описано в ссылочном примере 1.
1H-ЯМР (300 МГц, CDCl3) δ: 1,67 (уш., с, 1H), 2,06-2,32 (м, 2H), 3,24-3,33 (м, 1H), 3,33-3,46 (м, 1H), 3,47-3,65 (м, 2H), 4,52-4,70 (м, 1H), 6,61-6,81 (м, 2H), 6,91 (д, J=7,6 Гц, 1H), 7,30 (т, J=8,0 Гц, 1H).
Ссылочный пример 44 1-[4-(трифторметил)фенил]пирролидин-3-ол
Указанное в заголовке соединение (2,71 г, выход 55%) получали из 4-(трифторметил)фенилбромида и 3-гидроксипирролидина по способу аналогичному тому, как описано в ссылочном примере 1.
1H-ЯМР (300 МГц, CDCl3) δ: 1,67 (д, J=3,8 Гц, 1H), 2,02-2,30 (м, 2H), 3,25-3,34 (м, 1H), 3,36-3,45 (м, 1H), 3,47-3,63 (м, 2H), 4,64 (уш.с, 1H), 6,55 (д, J=8,7 Гц, 2H), 7,44 (д, J=8,7 Гц, 2H).
Ссылочный пример 45 Этил 3-[3,5-бис(трифторметил)фенил]-3-цианопропаноат
Раствор 3,5-бис(трифторметил)фенилацетонитрила (8,30 г) в тетрагидрофуране (80 мл) охлаждали до -78°С в атмосфере газообразного аргона, и по каплям добавляли раствор (1,9 М, 17,3 мл) гексаметилдисилазана натрия в тетрагидрофуране. После завершения капельного добавления раствор перемешивали при 10°С в течение 15 минут и снова охлаждали до -78°С. К раствору добавляли этилбромацетат (5,48 г), и смесь перемешивали при комнатной температуре в течение 16 часов. Реакционный раствор распределяли между этилацетатом и водой, и этилацетатный слой промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении, и полученный остаток очищали с помощью колоночной хроматографии на силикагеле (гексан/этилацетат 100:0-70:30), получая указанное в заголовке соединение (7,50 г, выход 67%) в виде масла коричневого цвета.
1H-ЯМР (300 МГц, CDCl3) δ: 1,25 (т, J=7,2 Гц, 3H), 2,81-2,99 (м, 1H), 3,01-3,19 (м, 1H), 4,19 (кв, 2H), 4,47 (т, J=7,2 Гц, 1H), 7,87 (с, 2H), 7,90 (с, 1H).
Ссылочный пример 46 4-[3,5-бис(трифторметил)фенил]пирролидин-2-он
К раствору этил 3-[3,5-бис(трифторметил)фенил]-3-цианопропаноата (7,40 г), полученного в ссылочном примере 45, и гексагидрата двухлористого кобальта (10,4 г) в метаноле (300 мл) добавляли боргидрид натрия (12,4 г), в то время как температуру реакционного раствора поддерживали не более чем 30°С, и смесь перемешивали при комнатной температуре в течение 18 часов. Реакционный раствор концентрировали при пониженном давлении, и остаток распределяли между этилацетатом и водой. Этилацетатный слой промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении, и полученный остаток очищали с помощью колоночной хроматографии на силикагеле (гексан/этилацетат 50:50-0:100), получая указанное в заголовке соединение (2,67 г, выход 41%) в виде твердого вещества белого цвета.
1H-ЯМР (300 МГц, CDCl3) δ: 2,43-2,56 (м, 1Н), 2,75-2,89 (м, 1Н), 3,38-3,54 (м, 1H), 3,74-3,96 (м, 2H), 6,23 (уш.с, 1H), 7,71 (с, 2H), 7,81 (с, 1H).
Ссылочный пример 47 Метил {4-[3,5-бис(трифторметил)фенил]-2-оксопирролидин-1-ил}ацетат
К раствору 4-[3,5-бис(трифторметил)фенил]пирролидин-2-она (0,70 г), полученного в ссылочном примере 46, в N,N-диметилформамиде (10 мл), при охлаждении льдом добавляли гидрид натрия (60% в масле, 0,10 г), и смесь перемешивали в течение 30 минут. К раствору добавляли раствор метилбромацетата (0,54 г) в N,N-диметилформамиде (5 мл), и смесь перемешивали при комнатной температуре в течение 4 часов. Реакцию гасили насыщенным раствором хлорида аммония, и раствор распределяли между этилацетатом и водой. Органический слой промывали насыщенным солевым раствором, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении, и полученный остаток очищали с помощью колоночной хроматографии на силикагеле (гексан/этилацетат 90:10-50:50), получая указанное в заголовке соединение (0,65 г, выход 75%) в виде масла бледно-коричневого цвета.
1H-ЯМР (300 МГц, CDCl3) δ: 2,48-2,67 (м, 1H), 2,84-3,04 (м, 1H), 3,56-3,66 (м, 1H), 3,72-3,83 (м, 1H), 3,77 (с, 3H), 3,86-3,97 (м, 1H), 3,99-4,10 (м, 1H), 4,21-4,37 (м, 1H), 7,76 (с, 2H), 7,81 (с, 1H).
Ссылочный пример 48 1-[3,5-бис(трифторметил)фенил]пирролидин-3-он
К раствору 1-[3,5-бис(трифторметил)фенил]пирролидин-3-ола (5,0 г), полученного в ссылочном примере 1, и триэтиламина (16,9 г) в диметилсульфоксиде (50 мл), при охлаждении льдом добавляли комплекс пиридина-трехокиси серы (7,98 г), и смесь перемешивали при 0°С в течение 30 минут, а затем при комнатной температуре в течение 20 часов. Реакционный раствор распределяли между этилацетатом и водой, и органический слой промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении, и полученный остаток очищали с помощью колоночной хроматографии на силикагеле (гексан/этилацетат 90:10-40:60), получая указанное в заголовке соединение (3,51 г, выход 71%) в виде масла коричневого цвета.
1H-ЯМР (300 МГц, CDCl3) δ: 2,82 (т, J=7,6 Гц, 2H), 3,80 (т, J=7,5 Гц, 4H), 6,98 (с, 2H), 7,28 (с, 1H).
Ссылочный пример 49 Этил (2Е/Z)-{1-[3,5-бис(трифторметил)фенил]пирролидин-3-илиден}этаноат
К раствору (15 мл) этил диэтилфосфоноацетата (1,24 г) в N,N-диметилформамиде добавляли гидрид натрия (60% в масле, 0,22 г), и смесь перемешивали при комнатной температуре в атмосфере азота в течение 15 минут. К раствору добавляли 1-[3,5-бис(трифторметил)фенил]пирролидин-3-он (1,50 г), полученный в ссылочном примере 48, в N,N-диметилформамиде (10 мл), и смесь перемешивали при комнатной температуре в атмосфере азота в течение 1 часа. Реакцию гасили насыщенным раствором хлорида аммония (10 мл), и реакционный раствор распределяли между этилацетатом и водой. Органический слой промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении, и полученный остаток очищали с помощью колоночной хроматографии на силикагеле (гексан/этилацетат 90:10-60:40), получая указанное в заголовке соединение (1,13 г, выход 61%, смесь Е формы, Z формы) в виде масла бледно-желтого цвета.
LC-МС ESI(+) m/z: 386 (М+H)+, время удерживания 2,11 мин; 386 (М+H)+, время удерживания 2,29 мин.
Ссылочный пример 50 Этил {1-[3,5-бис(трифторметил)фенил]пирролидин-3-ил}ацетат
Раствор этил (2Е/Z)-{1-[3,5-бис(трифторметил)фенил]пирролидин-3-илиден}этаноата (0,70 г), полученного в ссылочном примере 49, и 10% палладия на угле (содержащего воду (50%), 0,20 г) в смеси этанол-тетрагидрофуран (3:1, 320 мл) перемешивали в атмосфере водорода (1 атм) при комнатной температуре в течение 16 часов. Реакционный раствор разбавляли этилацетатом и фильтровали через силикагель. Силикагель промывали этилацетатом (500 мл), и фильтрат и смывки объединяли. Растворитель выпаривали при пониженном давлении, получая указанное в заголовке соединение (13,2 г, выход 99%) в виде бесцветного масла.
1H-ЯМР (300 МГц, CDCl3) δ: 1,29 (т, J=7,2 Гц, 3H), 1,67-1,90 (м, 1H), 2,19-2,38 (м, 1H), 2,41-2,59 (м, 2H), 2,68-2,88 (м, 1H), 2,97-3,09 (м, 1H), 3,29-3,50 (м, 2H), 3,55-3,68 (м, 1H), 4,18 (кв, J=7,2 Гц, 2H), 6,84 (с, 2H), 7,10 (с, 1H).
Ссылочный пример 51 Метил 3-({1-[3,5-бис(трифторметил)фенил]пирролидин-3-ил}сульфанил)пропаноат
Указанное в заголовке соединение (0,46 г, выход 18%) получали из 1-[3,5-бис(трифторметил)фенил]пирролидин-3-ола, полученного в ссылочном примере 1, и метил 3-меркаптопропионата по способу, аналогичному тому, как описано в ссылочном примере 2.
1H-ЯМР (300 МГц, CDCl3) δ: 1,98-2,17 (м, 1H), 2,34-2,53 (м, 1H), 2,60-2,71 (м, 2H), 2,84-2,95 (м, 2H), 3,23-3,35 (м, 1H), 3,36-3,46 (м, 1H), 3,47-3,64 (м, 2H), 3,68-3,80 (м, 4H), 6,84 (с, 2H), 7,13 (с, 1H).
Ссылочный пример 52 1-[4-фтор-2-(трифторметил)фенил]пирролидин-3-ол
Раствор 1-бром-4-фтор-2-(трифторметил)бензола (4,6 г), 3-гидроксипирролидина (1,5 г), ацетата палладия(II) (193 мг), (±)-2,2'-бис(дифенилфосфино)-1,1'-бинафтила (1,07 г) и карбоната цезия (16,8 г) в толуоле (90 мл) перемешивали в атмосфере аргона при 85°C в течение 16 часов. После охлаждения до комнатной температуры к реакционной смеси добавляли воду, и смесь экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали, и остаток очищали с помощью колоночной хроматографии на силикагеле (гексан/этилацетат 90:10-65:35), получая указанное в заголовке соединение (3,58 г, выход 84%) в виде масла желтого цвета.
1H-ЯМР (300 МГц, CDCl3) δ: 1,88-2,03 (м, 2H), 2,14-2,30 (м, 1H), 3,00-3,17 (м, 2H), 3,33-3,41 (м, 1H), 3,41-3,52 (м, 1H), 4,48 (уш.с, 1H), 7,08-7,23 (м, 2H), 7,28-7,37 (м, 1H).
Ссылочный пример 53 1-[4-фтор-3-(трифторметил)фенил]пирролидин-3-ол
Раствор 4-бром-1-фтор-2-(трифторметил)бензола (10 г), 3-гидроксипирролидина (3,56 г), ацетата палладия(II) (462 мг), (±)-2,2'-бис(дифенилфосфино)-1,1'-бинафтила (2,57 г) и карбоната цезия (26,8 г) в толуоле (220 мл) перемешивали в атмосфере аргона при 90°C в течение 16 часов. После охлаждения до комнатной температуры реакционную смесь фильтровали через целит, и целит промывали этилацетатом. Фильтрат и смывки объединяли, и раствор промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали, и остаток очищали с помощью колоночной хроматографии на силикагеле (гексан/этилацетат 80:20-25:75), получая указанное в заголовке соединение (5,91 г, выход 58%) в виде масла бледно-желтого цвета.
1H-ЯМР (300 МГц, CDCl3) δ: 1,72 (д, J=3,4 Гц, 1H), 2,00-2,29 (м, 2H), 3,25 (д, J=10,2 Гц, 1H), 3,33 (тд, J=8,8, 3,6 Гц, 1H), 3,40-3,59 (м, 2H), 4,63 (уш.с, 1H), 6,54-6,70 (м, 2H), 7,05 (т, J=9,4 Гц, 1H).
Ссылочный пример 54 1-[2-фтор-4-(трифторметил)фенил]пирролидин-3-ол
Раствор 1-бром-2-фтор-4-(трифторметил)бензола (4,6 г), 3-гидроксипирролидина (1,5 г), ацетата палладия(II) (137 мг), (±)-2,2'-бис(дифенилфосфино)-1,1'-бинафтила (1,07 г) и карбоната цезия (16,8 г) в толуоле (90 мл) перемешивали в атмосфере аргона при 85°C в течение 16 часов. После охлаждения до комнатной температуры к реакционной смеси добавляли воду, и смесь экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали, и остаток очищали с помощью колоночной хроматографии на силикагеле (гексан/этилацетат 90:10-65:35), получая указанное в заголовке соединение (2,82 г, выход 66%) в виде твердого вещества серого цвета.
1H-ЯМР (300 МГц, CDCl3) δ: 1,63 (д, J=4,5 Гц, 1H), 1,96-2,22 (м, 2H), 3,40-3,57 (м, 2H), 3,61-3,81 (м, 2H), 4,51-4,66 (м, 1H), 6,64 (т, J=9,1 Гц, 1H), 7,15-7,25 (м, 2H).
Ссылочный пример 55 1-[2-фтор-5-(трифторметил)фенил]пирролидин-3-ол
Раствор 2-бром-1-фтор-4-(трифторметил)бензола (4,6 г), 3-гидроксипирролидина (1,5 г), ацетата палладия(II) (193 мг), (±)-2,2'-бис(дифенилфосфино)-1,1'-бинафтила (1,07 г) и карбоната цезия (16,8 г) в толуоле (90 мл) перемешивали в атмосфере аргона при 85°C в течение 16 часов. После охлаждения до комнатной температуры к реакционной смеси добавляли воду, и смесь экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали, и полученный остаток очищали с помощью колоночной хроматографии на силикагеле (гексан/этилацетат 90:10-65:35), получая указанное в заголовке соединение (2,12 г, выход 49%) в виде масла коричневого цвета.
1H-ЯМР (300 МГц, CDCl3) δ: 1,67 (д, J=4,3 Гц, 1H), 1,96-2,23 (м, 2H), 3,35-3,49 (м, 2H), 3,56-3,76 (м, 2H), 4,49-4,65 (м, 1H), 6,79-6,88 (м, 1H), 6,88-6,96 (м, 1H), 6,97-7,10 (м, 1H).
Ссылочный пример 56 1-[2-фтор-3-(трифторметил)фенил]пирролидин-3-ол
2-Фтор-3-(трифторметил)анилин (5,0 г) и 1,4-дибромбутан-2-ол (6,7 г) перемешивали при 100°C в течение 3 часов. После охлаждения до комнатной температуры к реакционной смеси добавляли насыщенный водный раствор карбоната натрия, и смесь экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали, и полученный остаток очищали с помощью колоночной хроматографии на силикагеле (гексан/этилацетат 90:10-50:50), получая указанное в заголовке соединение (1,9 г, выход 27%) в виде твердого вещества бледно-желтого цвета.
1H-ЯМР (300 МГц, CDCl3) δ: 1,70 (д, J=3,8 Гц, 1H), 1,94-2,24 (м, 2H), 3,34-3,52 (м, 2H), 3,60-3,77 (м, 2H), 4,58 (уш.с, 1H), 6,83 (м, J=8,3, 8,3 Гц, 1H), 6,90 (м, J=7,6, 6,1 Гц, 1H), 7,04 (т, J=8,0 Гц, 1H).
Ссылочный пример 57 1-[2,4-бис(трифторметил)фенил]пирролидин-3-ол
Раствор 1-бром-2,4-бис(трифторметил)бензола (4,45 г), 3-гидроксипирролидина (1,2 г), ацетата палладия(II) (156 мг), (±)-2,2'-бис(дифенилфосфино)-1,1'-бинафтила (846 мг) и карбоната цезия (13,6 г) в толуоле (74 мл) перемешивали в атмосфере аргона при 85°C в течение 16 часов. После охлаждения до комнатной температуры к реакционной смеси добавляли воду, и смесь экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали, и полученный остаток очищали с помощью колоночной хроматографии на силикагеле (гексан/этилацетат 90:10-65:35), получая указанное в заголовке соединение (3,28 г, выход 79%) в виде масла коричневого цвета.
1H-ЯМР (300 МГц, CDCl3) δ: 1,66 (д, J=4,2 Гц, 1H), 1,97-2,25 (м, 2H), 3,24-3,54 (м, 2H), 3,64-3,88 (м, 2H), 4,49-4,70 (м, 1H), 6,94 (д, J=9,1 Гц, 1H), 7,56 (дд, J=8,7, 1,9 Гц, 1H), 7,82 (с, 1H).
Ссылочный пример 58 N-[3-бром-5-(трифторметил)фенил]ацетамид
К раствору 3-бром-5-(трифторметил)анилина (10 г) в пиридине (50 мл) добавляли уксусный ангидрид (5,6 г) при 0°C, и смесь перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь концентрировали, добавляли 1М соляную кислоту, и смесь экстрагировали этилацетатом. Органический слой промывали 1М соляной кислотой, насыщенным водным раствором бикарбоната натрия и насыщенным солевым раствором, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали, получая указанное в заголовке соединение (12,9 г, выход четвертичный) в виде твердого вещества белого цвета.
1H-ЯМР (300 МГц, CDCl3) δ: 2,21 (с, 3H), 7,36 (уш.с, 1H), 7,49 (с, 1H), 7,68 (с, 1H), 7,99 (с, 1H).
Ссылочный пример 59 N-[3-бром-5-(трифторметил)фенил]-N-метилацетамид
К раствору N-[3-бром-5-(трифторметил)фенил]ацетамида (6,6 г), полученного в ссылочном примере 58 в ДМФА (диметилформамиде) (71 мл), добавляли гидрид натрия (60% в масле, 1,22 г) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 10 минут, добавляли метилйодид (4,98 г), и смесь перемешивали при комнатной температуре в течение 16 часов. К реакционной смеси добавляли насыщенный водный раствор хлорида аммония, и смесь концентрировали и распределяли между водой и этилацетатом. Органический слой промывали насыщенным солевым раствором, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали, получая указанное в заголовке соединение (6,37 г, выход 92%) в виде масла желтого цвета.
1H-ЯМР (300 МГц, CDCl3) δ: 1,96 (уш.с, 3H), 3,30 (с, 3H), 7,43 (с, 1H), 7,58 (с, 1H), 7,74 (с, 1H).
Ссылочный пример 60 N-[3-(3-гидроксипирролидин-1-ил)-5-(трифторметил)фенил]-N-метилацетамид
Раствор N-[3-бром-5-(трифторметил)фенил]-N-метилацетамида (6,37 г), полученного в ссылочном примере 59, 3-гидроксипирролидина (1,7 г), ацетата палладия(II) (219 мг), (±)-2,2'-бис(дифенилфосфино)-1,1'-бинафтила (1,2 г) и карбоната цезия (19 г) в толуоле (100 мл) перемешивали в атмосфере аргона при 85°C в течение 16 часов. После охлаждения до комнатной температуры к реакционной смеси добавляли воду, и смесь экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали, и остаток очищали с помощью колоночной хроматографии на силикагеле (гексан/этилацетат 34:66-0:100), получая указанное в заголовке соединение (5,02 г, выход 85%) в виде твердого вещества светло-желтого цвета.
1H-ЯМР (300 МГц, CDCl3) δ: 1,92 (с, 3H), 2,01-2,31 (м, 3H), 3,25 (с, 3H), 3,27-3,34 (м, 1H), 3,34-3,46 (м, 1H), 3,46-3,61 (м, 2H), 4,66 (уш.с, 1H), 6,46 (с, 1H), 6,70-6,74 (м, 2H).
Ссылочный пример 61 4-{[3-бром-5-(трифторметил)фенил]карбонил}тиоморфолин
Раствор 3-бром-5-(трифторметил)бензойной кислоты (10 г), тиоморфолина (5,0 г), HOBt (7,4 г) и EDCI (9,26 г) в ацетонитриле (113 мл) перемешивали при комнатной температуре в течение 16 часов. К реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия, и смесь экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали, получая указанное в заголовке соединение (12,3 г, выход 94%) в виде масла желтого цвета.
1H-ЯМР (300 МГц, CDCl3) δ: 2,61 (уш.с, 2H), 2,73 (уш.с, 2H), 3,66 (уш.с, 2H), 4,11 (уш.с, 2H), 7,55-7,59 (м, 1H), 7,69-7,72 (м, 1H), 7,81-7,84 (м, 1H).
Ссылочный пример 62 1-[3-(тиоморфолин-4-илкарбонил)-5-(трифторметил)фенил]пирролидин-3-ол
Раствор 4-{[3-бром-5-(трифторметил)фенил]карбонил}тиоморфолина (5,0 г) полученного в ссылочном примере 61, 3-гидроксипирролидина (1,35 г), ацетата палладия(II) (158,5 мг), (±)-2,2'-бис(дифенилфосфино)-1,1'-бинафтила (884 мг) и карбоната цезия (13,8 г) в толуоле (71 мл) перемешивали в атмосфере аргона при 90°С в течение 16 часов. После охлаждения до комнатной температуры к реакционной смеси добавляли воду, и смесь экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали, и полученный остаток очищали с помощью колоночной хроматографии на силикагеле (гексан/этилацетат 50:50-0:100), получая указанное в заголовке соединение (5,56 г, выход четвертичный) в виде твердого вещества бледно-желтого цвета.
1H-ЯМР (300 МГц, CDCl3) δ: 1,94 (д, J=4,2 Гц, 1H), 2,01-2,27 (м, 2H), 2,57 (уш.с, 2H), 2,73 (уш.с, 2H), 3,22-3,33 (м, 1H), 3,33-3,46 (м, 1H), 3,46-3,58 (м, 2H), 3,66 (уш.с, 2H), 4,01 (уш.с, 2H), 4,57-4,72 (м, 1H), 6,65 (с, 1H), 6,75 (с, 1H), 6,84 (с, 1H).
Ссылочный пример 63 1-оксид 4-{[3-бром-5-(трифторметил)фенил]карбонил}тиоморфолина
К раствору 4-{[3-бром-5-(трифторметил)фенил]карбонил}тиоморфолина (4,5 г), полученного в ссылочном примере 61, в ацетоне (170 мл) добавляли водный раствор (170 мл) Oxone (зарегистрированный товарный знак, 7,8 г) при 0°С, и смесь сразу же перемешивали при 0°С в течение 1 часа. Реакционную смесь экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали, и остаток очищали с помощью колоночной хроматографии на силикагеле (гексан/этилацетат 60:40-0:100), получая указанное в заголовке соединение (2,99 г, выход 84%) в виде аморфного вещества бледно-желтого цвета.
1H-ЯМР (300 МГц, CDCl3) δ: 2,88 (уш.с, 4H), 3,70 (уш.с, 1H), 4,12 (уш.с, 2H), 4,59 (уш.с, 1H), 7,60-7,64 (м, 1H), 7,73-7,77 (м, 1H), 7,85-7,89 (м, 1H).
Ссылочный пример 64 1-{3-[(1-оксидотиоморфолин-4-ил)карбонил]-5-(трифторметил)фенил}пирролидин-3-ол
Раствор 1-оксида 4-{[3-бром-5-(трифторметил)фенил]карбонил}тиоморфолина (2,99 г), полученного в ссылочном примере 63, 3-гидроксипирролидина (774 мг), ацетата палладия(II) (91 мг), (±)-2,2'-бис(дифенилфосфино)-1,1'-бинафтила (504 мг) и карбоната цезия (7,9 г) в смешанном растворителе из толуола (40 мл) и ДМФА (15 мл) перемешивали в атмосфере аргона при 80°С в течение 16 часов. После охлаждения до комнатной температуры реакционную смесь концентрировали, к остатку добавляли воду, и смесь экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали, и остаток очищали с помощью колоночной хроматографии на силикагеле (этилацетат/метанол 100:0-80:20), получая указанное в заголовке соединение (2,10 г, выход 69%) в виде твердого вещества белого цвета.
1H-ЯМР (300 МГц, CDCl3) δ: 1,87 (д, 1H), 2,06-2,27 (м, 2H), 2,96 (уш.с, 4H), 3,30 (д, J=10,6 Гц, 1H), 3,41 (тд, J=8,7, 3,4 Гц, 1H), 3,47-3,62 (м, 2H), 3,77 (уш.с, 1H), 4,12 (уш.с, 2H), 4,57 (уш.с, 1H), 4,62-4,74 (м, 1H), 6,68 (с, 1H), 6,79 (с, 1H), 6,86 (с, 1H).
Ссылочный пример 65 N-[3-бром-5-(трифторметил)фенил]метансульфонамид
К раствору 3-бром-5-(трифторметил)анилина (10 г) в пиридине (50 мл) добавляли метансульфонилхлорид (5,73 г) при 0°С, и смесь перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь концентрировали, добавляли насыщенный водный раствор бикарбоната натрия, и смесь экстрагировали этилацетатом. Органический слой промывали 1М соляной кислотой и насыщенным солевым раствором, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали, получая указанное в заголовке соединение (13,4 г, выход четвертичный) в виде твердого вещества бледно-желтого цвета.
1H-ЯМР (300 МГц, CDCl3) δ: 3,10 (с, 3H), 6,71-7,05 (м, 1H), 7,39 (с, 1H), 7,53-7,62 (м, 2H).
Ссылочный пример 66 N-[3-бром-5-(трифторметил)фенил]-N-метилметансульфонамид
К раствору N-[3-бром-5-(трифторметил)фенил]метансульфонамида (6,77 г), полученного в ссылочном примере 65, в ДМФА (65 мл), добавляли гидрид натрия (60% в масле, 1,1 г) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 10 минут, добавляли метилйодид (4,53 г), и смесь перемешивали при комнатной температуре в течение 16 часов. К реакционной смеси добавляли насыщенный водный раствор хлорида аммония, и смесь концентрировали и распределяли между водой и этилацетатом. Органический слой промывали насыщенным солевым раствором, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали, получая указанное в заголовке соединение (7,19 г, выход четвертичный) в виде масла желтого цвета.
1H-ЯМР (300 МГц, CDCl3) δ: 2,90 (с, 3H), 3,36 (с, 3H), 7,58 (с, 1H), 7,68 (с, 1H), 7,74 (с, 1H).
Ссылочный пример 67 N-[3-(3-гидроксипирролидин-1-ил)-5-(трифторметил)фенил]-N-метилметансульфонамид
Раствор N-[3-бром-5-(трифторметил)фенил]-N-метилметансульфонамида (7,19 г), полученного в ссылочном примере 66, 3-гидроксипирролидина (1,71 г), ацетата палладия(II) (221 мг), (±)-2,2'-бис(дифенилфосфино)-1,1'-бинафтила (1,23 г) и карбоната цезия (19,3 г) в толуоле (100 мл) перемешивали в атмосфере аргона при 85°С в течение 16 часов. После охлаждения до комнатной температуры к реакционной смеси добавляли воду, и смесь экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали, и остаток очищали с помощью колоночной хроматографии на силикагеле (гексан/этилацетат 33:67-0:100), получая указанное в заголовке соединение (5,45 г, выход 82%) в виде аморфного вещества коричневого цвета.
1H-ЯМР (300 МГц, CDCl3) δ: 1,91 (уш.с, 1H), 2,06-2,30 (м, 2H), 2,86 (с, 3H), 3,26-3,34 (м, 1H), 3,32 (с, 3H), 3,34-3,62 (м, 3H), 4,59-4,70 (м, 1H), 6,66 (с, 1H), 6,75 (с, 1H), 6,80 (с, 1H).
Ссылочный пример 68 1-[3-бром-5-(трифторметил)фенил]пирролидин-3-ол
3-Бром-5-(трифторметил)анилин (5,0 г) и 1,4-дибромбутан-2-ол (4,83 г) перемешивали при 100°C в течение 3 часов. После охлаждения до комнатной температуры к реакционной смеси добавляли насыщенный водный раствор карбоната натрия, и смесь экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали, и полученный остаток очищали с помощью колоночной хроматографии на силикагеле (гексан/этилацетат 90:10-50:50), получая указанное в заголовке соединение (5,6 г, выход 22%) в виде масла коричневого цвета.
1H-ЯМР (300 МГц, CDCl3) δ: 1,66 (уш.с, 1H), 2,08-2,27 (м, 2H), 3,23-3,32 (м, 1H), 3,38 (тд, J=8,8, 3,4 Гц, 1H), 3,44-3,59 (м, 2H), 4,65 (уш.с, 1H), 6,65 (с, 1H), 6,80 (т, J=1,9 Гц, 1H), 7,02 (с, 1H).
Ссылочный пример 69 1-[3-метокси-5-(трифторметил)фенил]пирролидин-3-ол
3-Метокси-5-(трифторметил)анилин (15,0 г) и 1,4-дибромбутан-2-ол (18,3 г) перемешивали при 100°C в течение 3 часов. После охлаждения до комнатной температуры к реакционной смеси добавляли насыщенный водный раствор карбоната натрия, и смесь экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали, и остаток очищали с помощью колоночной хроматографии на силикагеле (гексан/этилацетат 90:10-50:50), получая указанное в заголовке соединение (9,57 г, выход 47%) в виде масла оранжевого цвета.
1H-ЯМР (300 МГц, CDCl3) δ: 1,64 (д, J=4,7 Гц, 1H), 2,06-2,27 (м, 2H), 3,23-3,32 (м, 1H), 3,32-3,43 (м, 1H), 3,46-3,59 (м, 2H), 3,82 (с, 3H), 4,55-4,69 (м, 1H), 6,20 (т, J=2,3 Гц, 1H), 6,40 (с, 1H), 6,47 (с, 1H).
Ссылочный пример 70 1-[3-хлор-5-(трифторметил)фенил]пирролидин-3-ол
Раствор 1-бром-3-хлор-5-(трифторметил)бензола (5,0 г), 3-гидроксипирролидина (1,85 г), ацетата палладия(II) (217 мг), (±)-2,2'-бис(дифенилфосфино)-1,1'-бинафтила (1,2 г) и карбоната цезия (18,8 г) в толуоле (96 мл) перемешивали в атмосфере аргона при 85°С в течение 16 часов. После охлаждения до комнатной температуры добавляли воду, и смесь экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали, и остаток очищали с помощью колоночной хроматографии на силикагеле (гексан/этилацетат 80:20-10:90), получая указанное в заголовке соединение (4,11 г, выход 80%) в виде масла коричневого цвета.
1H-ЯМР (300 МГц, CDCl3) δ: 1,65 (д, J=3,8 Гц, 1H), 2,00-2,29 (м, 2H), 3,19-3,32 (м, 1H), 3,38 (тд, J=8,7, 3,4 Гц, 1H), 3,45-3,60 (м, 2H), 4,58-4,72 (м, 1H), 6,61 (с, 1H), 6,63-6,68 (м, 1H), 6,88 (с, 1H).
Ссылочный пример 71 1-[3-фтор-5-(трифторметил)фенил]пирролидин-3-ол
Раствор 1-бром-3-фтор-5-(трифторметил)бензола (10,0 г), 3-гидроксипирролидина (3,58 г), ацетата палладия(II) (461 мг), (±)-2,2'-бис(дифенилфосфино)-1,1'-бинафтила (2,56 г) и карбоната цезия (26,7 г) в толуоле (222 мл) перемешивали в атмосфере газообразного аргона при 90°С в течение 16 часов. После охлаждения до комнатной температуры реакционную смесь фильтровали через целит, и целит промывали этилацетатом. Фильтрат и смывки объединяли, и раствор промывали водой и насыщенным солевым раствором. Все сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали, и остаток очищали с помощью колоночной хроматографии на силикагеле (гексан/этилацетат 98:2-20:80), получая указанное в заголовке соединение (10,65 г, выход четвертичный) в виде масла коричневого цвета.
1H-ЯМР (300 МГц, CDCl3) δ: 1,73 (уш.с, 1H), 2,05-2,28 (м, 2H), 3,27 (д, J=10,6 Гц, 1H), 3,38 (тд, J=8,9, 3,4 Гц, 1H), 3,45-3,59 (м, 2H), 4,55-4,70 (м, 1H), 6,35 (дт, J=11,7, 2,3 Гц, 1H), 6,52 (с, 1H), 6,60 (д, J=8,7 Гц, 1H).
Ссылочный пример 72 (3S)-1-[4-хлор-2-(трифторметил)фенил]пирролидин-3-ол
Раствор 1-бром-4-хлор-2-(трифторметил)бензола (15,0 г), (3S)-пирролидин-3-ола (5,0 г), ацетата палладия(II) (561 мг), (±)-2,2'-бис(дифенилфосфино)-1,1'-бинафтила (3,11 г) и карбоната цезия (37,8 г) в толуоле (280 мл) перемешивали в атмосфере газообразного аргона при 100°С в течение 18 часов. После охлаждения до комнатной температуры к реакционной смеси добавляли воду, и смесь экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали, и остаток очищали с помощью колоночной хроматографии на силикагеле (гексан/этилацетат 95:5-80:20), получая указанное в заголовке соединение (12,4 г, выход 81%) в виде бесцветного масла.
1H-ЯМР (300 МГц, CDCl3) δ: 1,92-2,04 (м, 2H), 2,10-2,22 (м, 1H), 3,17-3,24 (м, 2H), 3,50-3,61 (м, 2H), 4,51 (уш.с, 1H), 6,96 (д, J=9,0 Гц, 1H), 7,31-7,35 (м, 1H), 7,54-7,55 (м, 1H).
Ссылочный пример 73 [2-метокси-3-(трифторметил)фенил]бороновая кислота и [3-метокси-2-(трифторметил)фенил]бороновая кислота
К раствору 1-метокси-2-(трифторметил)бензола (6,30 г, 35,8 ммоль) в ТГФ (тетрагидрофуране) (150 мл) добавляли n-BuLi (21,0 мл, 2,50М раствор гексана, 53,7 ммоль), и смесь перемешивали при комнатной температуре в течение 1 часа. Раствор охлаждали до -78°С, добавляли трис(1-метилэтил)борат (8,08 г, 43,0 ммоль), и смесь перемешивали при -78°С в течение 0,5 часа. Реакционному раствору давали охладиться до комнатной температуры и перемешивали в течение 16 часов. Смесь подкисляли 1М соляной кислотой и экстрагировали этилацетатом (300 мл). Экстракт сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении, получая смесь (6,52 г, выход 83%) [2-метокси-3-(трифторметил)фенил]бороновой кислоты и [3-метокси-2-(трифторметил)фенил]бороновой кислоты.
Ссылочный пример 75 [2-циано-3-(трифторметил)фенил]бороновая кислота
К раствору 2,2,6,6-тетраметилпиперидина (0,99 г, 7,02 ммоль) в ТГФ (25 мл) при -10°С добавляли n-BuLi (2,80 мл, 2,5М раствор гексана, 7,02 ммоль). После перемешивания в течение 10 минут данный раствор охлаждали до -78°С, добавляли трис(1-метилэтил)борат (1,58 г, 8,4 ммоль), и смесь перемешивали в течение 5 минут. К данному раствору добавляли раствор 2-(трифторметил)бензонитрила (1,00 г, 5,85 ммоль) в ТГФ (10 мл), и смесь перемешивали при -78°С в течение 2 часов. Реакционному раствору давали нагреться до комнатной температуры, реакцию гасили уксусной кислотой, и растворитель выпаривали при пониженном давлении. К остатку добавляли этилацетат, осажденное твердое вещество отфильтровывали, и фильтрат концентрировали, получая указанное в заголовке соединение (3,14 г) в виде смеси. Ее использовали для следующей реакции без осуществления дальнейшей очистки и идентификации.
Ссылочный пример 76 Этил (2E)-3-{5-[3-фтор-5-(трифторметил)фенил]фуран-2-ил}проп-2-еноат
Раствор этил (2Е)-3-(5-бромфуран-2-ил)проп-2-еноата (0,64 г, 2,62 ммоль), полученного в ссылочном примере 31, [3-фтор-5-(трифторметил)фенил]бороновой кислоты (0,60 г, 2,88 ммоль), тетракис(трифенилфосфин)палладия (0,15 г, 0,13 ммоль) и 2М раствор карбоната натрия (6,56 мл, 13,1 ммоль) в N,N-диметилацетамиде (30 мл) перемешивали в атмосфере аргона в течение 16 часов. После охлаждения реакционного раствора до комнатной температуры твердое вещество отфильтровывали, и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (петролейный эфир/этилацетат 100:0-95:5), получая указанное в заголовке соединение (0,62 г, выход 72%) в виде твердого вещества желтого цвета.
1H-NMR (300 МГц, CDCl3) δ: 1,35 (т, J=6,9 Гц, 3H), 1,27 (кв, J=7,2 Гц, 2H), 6,45 (д, J=15,6 Гц, 1H), 6,71 (д, J=3,9 Гц, 1H), 6,84 (д, J=3,6 Гц, 1H), 7,26 (м, 1H), 7,45 (д, J=15,9 Гц, 1H), 7,58 (м, 1H), 7,72 (с, 1H).
Ссылочный пример 77 Этил 3-{5-[3-фтор-5-(трифторметил)фенил]тетрагидрофуран-2-ил}пропаноат
Раствор этил (2E)-3-{5-[3-фтор-5-(трифторметил)фенил]фуран-2-ил}проп-2-еноата (0,62 г, 1,89 ммоль), полученного в ссылочном примере 76, и гидроксида палладия (10% на угле, 30 мг) в метаноле (100 мл) перемешивали в атмосфере водорода при комнатной температуре в течение 16 часов. После подтверждения завершения реакции с помощью ТСХ (тонкослойной хроматографии) реакционный раствор фильтровали и концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле (петролейный эфир/этилацетат 95:5), получая указанное в заголовке соединение (0,55 г, выход 88%) в виде масла желтого цвета.
1H-ЯМР (300 МГц, CDCl3) δ: 1,26 (т, J=7,2 Гц, 3H), 1,61-1,85 (м, 2H), 1,97-2,04 (м, 2H), 2,12-2,20 (м, 1H), 2,36-2,47 (м, 1H), 2,50-2,60 (м, 2H), 4,05-4,19 (м, 3H), 4,96 (т, J=7,4 Гц, 1H), 7,78 (с, 3H).
Ссылочный пример 78 Этил (2E)-3-{5-[3-метокси-5-(трифторметил)фенил]фуран-2-ил}проп-2-еноат
Раствор этил (2E)-3-(5-бромфуран-2-ил)проп-2-еноата (0,61 г, 2,50 ммоль), полученного в ссылочном примере 31, [3-метокси-5-(трифторметил)фенил]бороновой кислоты (0,50 г, 2,27 ммоль), тетракис(трифенилфосфин)палладия (0,26 г, 0,23 ммоль) и 2М раствор карбоната натрия (5,68 мл, 11,4 ммоль) в N,N-диметилацетамиде (25 мл) перемешивали в атмосфере аргона в течение 16 часов. После охлаждения реакционного раствора до комнатной температуры твердое вещество отфильтровывали, и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (петролейный эфир/этилацетат 98:2-95:5), получая указанное в заголовке соединение (0,53 г, выход 68%) в виде твердого вещества желтого цвета.
1H-ЯМР (300 МГц, CDCl3) δ: 1,34 (т, J=7,2 Гц, 3H), 3,91 (с, 3H), 4,27 (кв, J=7,2 Гц, 2H), 6,44 (д, J=15,6 Гц, 1H), 6,70 (д, J=3,6 Гц, 1H), 6,80 (д, J=3,6 Гц, 1H), 7,06 (с, 1H), 7,38 (с, 1H), 7,45 (д, J=15,6 Гц, 1H), 7,52 (с, 1H).
Ссылочный пример 79 Этил 3-{5-[3-метокси-5-(трифторметил)фенил]тетрагидрофуран-2-ил}пропаноат
Раствор этил (2E)-3-{5-[3-метокси-5-(трифторметил)фенил]фуран-2-ил}проп-2-еноата (0,48 г, 1,41 ммоль), полученного в ссылочном примере 78, и палладия (10% на угле, 50 мг) в метаноле (50 мл) перемешивали в атмосфере водорода при комнатной температуре в течение 16 часов. После подтверждения завершения реакции с помощью ТСХ, реакционный раствор фильтровали и концентрировали при пониженном давлении, получая указанное в заголовке соединение (0,49 г, выход >99%) в виде бесцветного масла.
1H-ЯМР (300 МГц, CDCl3) δ: 1,26 (т, J=7,2 Гц, 3H), 1,64-1,84 (м, 2H), 1,94-2,01 (кв, J=7,5 Гц, 2H), 2,06-2,13 (м, 1H), 2,27-2,35 (м, 1H), 2,42-2,56 (м, 2H), 3,84 (с, 3H), 4,03-4,07 (м, 1H), 4,13 (кв, J=7,2 Гц, 2H), 4,87 (т, J=7,5 Гц, 1H), 6,70 (с, 1H), 7,07 (с, 1H), 7,15 (с, 1H).
Ссылочный пример 80 Этил (2E)-3-{5-[4-метокси-3-(трифторметил)фенил]фуран-2-ил}проп-2-еноат
Раствор этил (2Е)-3-(5-бромфуран-2-ил)проп-2-еноата (0,61 г, 2,50 ммоль), полученного в ссылочном примере 31, [4-метокси-3-(трифторметил)фенил]бороновой кислоты (0,50 г, 2,27 ммоль), тетракис(трифенилфосфин)палладия (0,26 г, 0,23 ммоль) и 2М раствор карбоната натрия (5,68 мл, 11,4 ммоль) в N,N-диметилацетамиде (25 мл) перемешивали в атмосфере аргона в течение 16 часов. После охлаждения реакционного раствора до комнатной температуры твердое вещество отфильтровывали, и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (петролейный эфир/этилацетат 98:2-95:5), получая указанное в заголовке соединение (0,46 г, выход 60%) в виде твердого вещества желтого цвета.
1H-ЯМР (300 МГц, CDCl3) δ: 1,34 (т, J=7,2 Гц, 3H), 3,95 (с, 3H), 4,27 (кв, J=7,2 Гц, 2H), 6,39 (д, J=15,6 Гц, 1H), 6,65-6,68 (м, 2H), 7,04 (д, J=8,7 Гц, 1H), 7,43 (д, J=15,6 Гц, 1H), 7,84 (дд, J=8,7, 2,1 Гц, 1H), 7,89 (д, J=2,1 Гц, 1H).
Ссылочный пример 81 Этил 3-{5-[4-метокси-3-(трифторметил)фенил]тетрагидрофуран-2-ил}пропаноат
Раствор этил (2E)-3-{5-[4-метокси-3-(трифторметил)фенил]фуран-2-ил}проп-2-еноата (0,39 г, 1,15 ммоль), полученного в ссылочном примере 80, и палладия (10% на угле, 40 мг) в метаноле (25 мл) перемешивали в атмосфере водорода при комнатной температуре в течение 16 часов. После подтверждения завершения реакции с помощью ТСХ реакционный раствор фильтровали и концентрировали при пониженном давлении, получая указанное в заголовке соединение (0,40 г, выход >99%) в виде бесцветного масла.
1H-ЯМР (300 МГц, CDCl3) δ: 1,25 (т, J=7,2 Гц, 3H), 1,59-1,78 (м, 2H), 1,93-2,00 (м, 2H), 2,06-2,14 (м, 1H), 2,23-2,33 (м, 1H), 2,44-2,52 (м, 2H), 3,88 (с, 3H), 4,01-4,05 (м, 1H), 4,14 (кв, J=7,2 Гц, 2H), 4,80 (т, J=7,2 Гц, 1H), 6,96 (д, J=8,4 Гц, 1H), 7,46 (дд, J=8,4, 1,8 Гц, 1H), 7,51 (д, J=1,8 Гц, 1H).
Ссылочный пример 82 Этил (2E)-3-{5-[2-фтор-3-(трифторметил)фенил]фуран-2-ил}проп-2-еноат
Раствор этил (2Е)-3-(5-бромфуран-2-ил)проп-2-еноата (0,65 г, 2,64 ммоль), полученного в ссылочном примере 31, [2-фтор-3-(трифторметил)фенил]бороновой кислоты (0,50 г, 2,40 ммоль), тетракис(трифенилфосфин)палладия (0,28 г, 0,24 ммоль) и 2М раствор карбоната натрия (6,00 мл, 12,0 ммоль) в N,N-диметилацетамиде (25 мл) перемешивали в атмосфере аргона в течение 16 часов. После охлаждения реакционного раствора до комнатной температуры твердое вещество отфильтровывали, и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (петролейный эфир/этилацетат 98:2-95:5), получая указанное в заголовке соединение (0,36 г, выход 46%) в виде твердого вещества желтого цвета.
1H-ЯМР (300 МГц, CDCl3) δ: 1,34 (т, J=7,2 Гц, 3H), 4,27 (кв, J=7,2 Гц, 2H), 6,44 (д, J=15,9 Гц, 1H), 6,74 (д, J=3,6 Гц, 1H), 6,99 (т, J=3,6 Гц, 1H), 7,32 (т, J=7,8 Гц, 1H), 7,45 (д, J=15,9 Гц, 1H), 7,54 (т, J=6,9 Гц, 1H), 8,08 (т, J=6,9 Гц, 1H).
Ссылочный пример 83 Этил 3-{5-[2-фтор-3-(трифторметил)фенил]тетрагидрофуран-2-ил}пропаноат
Раствор этил (2E)-3-{5-[2-фтор-3-(трифторметил)фенил]фуран-2-ил}проп-2-еноата (0,36 г, 1,10 ммоль), полученного в ссылочном примере 82, и палладия (10% на угле, 40 мг) в метаноле (25 мл) перемешивали в атмосфере водорода при комнатной температуре в течение 26 часов. После подтверждения завершения реакции с помощью ТСХ реакционный раствор фильтровали и концентрировали при пониженном давлении, получая указанное в заголовке соединение (0,28 г, выход 76%) в виде бесцветного масла.
1H-ЯМР (300 МГц, CDCl3) δ: 1,25 (т, J=7,2 Гц, 3H), 1,59-1,68 (м, 1H), 1,71-1,81 (м, 1H), 1,96-2,03 (м, 2H), 2,05-2,15 (м, 1H), 2,42-2,60 (м, 3H), 4,02-4,11 (м, 1H), 4,14 (кв, J=7,2 Гц, 2H), 5,14 (т, J=6,9 Гц, 1H), 7,22 (т, J=7,8 Гц, 1H), 7,48 (т, J=6,6 Гц, 1H), 7,72 (т, J=7,8 Гц, 1H).
Ссылочный пример 84 Этил (2E)-3-{5-[3-(трифторметил)фенил]фуран-2-ил}проп-2-еноат
Раствор этил (2Е)-3-(5-бромфуран-2-ил)проп-2-еноата (1,42 г, 5,79 ммоль), полученного в ссылочном примере 31, [3-(трифторметил)фенил]бороновой кислоты (1,00 г, 5,26 ммоль), тетракис(трифенилфосфин)палладия (0,61 г, 0,53 ммоль) и 2М раствор карбоната натрия (13,2 мл, 26,4 ммоль) в N,N-диметилацетамиде (50 мл) перемешивали в атмосфере аргона в течение 16 часов. После охлаждения реакционного раствора до комнатной температуры твердое вещество отфильтровывали, и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (петролейный эфир/этилацетат 98:2-95:5), получая указанное в заголовке соединение (0,96 г, выход 59%) в виде твердого вещества желтого цвета.
Ссылочный пример 85 Этил 3-{5-[3-(трифторметил)фенил]фуран-2-ил}пропаноат
Раствор этил (2E)-3-{5-[3-(трифторметил)фенил]фуран-2-ил}проп-2-еноата (0,36 г, 1,10 ммоль), полученного в ссылочном примере 84, и палладия (10% на угле, 40 мг) в метаноле (25 мл) перемешивали в атмосфере водорода при комнатной температуре в течение 2 часов. После подтверждения завершения реакции с помощью ТСХ, реакционный раствор фильтровали и концентрировали при пониженном давлении, получая указанное в заголовке соединение (0,73 г, выход 75%) в виде бесцветного масла.
1H-ЯМР (300 МГц, CDCl3) δ: 1,25 (т, J=7,2 Гц, 3H), 1,65-1,82 (м, 2H), 1,95-2,08 (м, 2H), 2,16-2,30 (м, 1H), 2,40-2,46 (м, 1H), 2,48-2,54 (м, 2H), 4,04-4,09 (м, 1H), 4,14 (кв, J=7,2 Гц, 2H), 4,90 (т, J=6,9 Гц, 1H), 7,43-7,58 (м, 4H).
Ссылочный пример 86 Этил (2E)-3-{5-[2-циано-3-(трифторметил)фенил]фуран-2-ил}проп-2-еноат
Раствор этил (2Е)-3-(5-бромфуран-2-ил)проп-2-еноата (1,30 г, 5,32 ммоль), полученного в ссылочном примере 31, [2-циано-3-(трифторметил)фенил]бороновой кислоты (3,14 г, 5,85 ммоль), полученной в ссылочном примере 75, тетракис(трифенилфосфин)палладия (0,62 г, 0,53 ммоль) и 2М раствор карбоната натрия (13,3 мл, 26,6 ммоль) в N,N-диметилацетамиде (50 мл) перемешивали в атмосфере аргона в течение 16 часов. После охлаждения реакционного раствора до комнатной температуры твердое вещество отфильтровывали, и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (петролейный эфир/этилацетат 98:2), получая указанное в заголовке соединение (0,70 г, выход 39%) в виде твердого вещества желтого цвета.
Ссылочный пример 87 Этил 3-{5-[2-циано-3-(трифторметил)фенил]тетрагидрофуран-2-ил}пропаноат
Раствор этил (2E)-3-{5-[2-циано-3-(трифторметил)фенил]фуран-2-ил}проп-2-еноата (0,70 г, 2,08 ммоль), полученного в ссылочном примере 86, и палладия (10% на угле, 70 мг) в метаноле (50 мл) перемешивали в атмосфере водорода при комнатной температуре в течение 16 часов. После подтверждения завершения реакции с помощью ТСХ реакционный раствор фильтровали и концентрировали при пониженном давлении, получая указанное в заголовке соединение (0,34 г, выход 48%) в виде бесцветного масла.
Ссылочный пример 88 Этил (2E)-3-{5-[4-фтор-3-(трифторметил)фенил]фуран-2-ил}проп-2-еноат
Раствор этил (2Е)-3-(5-бромфуран-2-ил)проп-2-еноата (0,65 г, 2,64 ммоль), полученного в ссылочном примере 31, [4-фтор-3-(трифторметил)фенил]бороновой кислоты (0,50 г, 2,40 ммоль), тетракис(трифенилфосфин)палладия (0,28 г, 0,24 ммоль) и 2М раствор карбоната натрия (6,56 мл, 13,1 ммоль) в N,N-диметилацетамиде (25 мл) перемешивали в атмосфере аргона в течение 16 часов. После охлаждения реакционного раствора до комнатной температуры твердое вещество отфильтровывали, и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (петролейный эфир/этилацетат 98:2-95:5), получая указанное в заголовке соединение (0,60 г, выход 76%) в виде твердого вещества желтого цвета.
1H-ЯМР (300 МГц, CDCl3) δ: 1,34 (т, J=7,2 Гц, 3H), 4,27 (кв, J=7,2 Гц, 2H), 6,42 (д, J=15,9 Гц, 1H), 6,69 (д, J=3,6 Гц, 1H), 6,74 (д, J=3,6 Гц, 1H), 7,24 (т, J=9,3 Гц, 1H), 7,43 (д, J=15,9 Гц, 1H), 7,83-7,92 (м, 2H).
Ссылочный пример 89 Этил 3-{5-[4-фтор-3-(трифторметил)фенил]тетрагидрофуран-2-ил}пропаноат
Раствор этил (2E)-3-{5-[4-фтор-3-(трифторметил)фенил]фуран-2-ил}проп-2-еноата (0,60 г, 1,83 ммоль), полученного в ссылочном примере 88, и палладия (10% на угле, 60 мг) в метаноле (25 мл) перемешивали в атмосфере водорода при комнатной температуре в течение 20 часов. После подтверждения завершения реакции с помощью ТСХ реакционный раствор фильтровали и концентрировали при пониженном давлении, получая указанное в заголовке соединение (0,54 г, выход 88%) в виде бесцветного твердого вещества.
1H-ЯМР (300 МГц, CDCl3) δ: 1,27 (т, J=7,2 Гц, 3H), 1,62-1,71 (м, 3H), 1,97 (кв, J=12,9 Гц, 2H), 2,08-2,17 (м, 1H), 2,29-2,36 (м, 2H), 4,04-4,08 (м, 1H), 4,15 (кв, J=7,2 Гц, 2H), 4,87 (т, J=6,9 Гц, 1H), 7,16 (т, J=9,6 Гц, 1H), 7,51-7,57 (м, 2H).
Ссылочный пример 90 Этил (2E)-3-{5-[2-метокси-3-(трифторметил)фенил]фуран-2-ил}проп-2-еноат
Ссылочный пример 91 Этил (2E)-3-{5-[3-метокси-2-(трифторметил)фенил]фуран-2-ил}проп-2-еноат
Раствор этил (2Е)-3-(5-бромфуран-2-ил)проп-2-еноата (4,20 г, 17,1 ммоль), полученного в ссылочном примере 31, смесь (6,52 г, 29,6 ммоль) [2-метокси-3-(трифторметил)фенил]бороновой кислоты и [3-метокси-2-(трифторметил)фенил]бороновой кислоты, полученных в ссылочном примере 73, тетракис(трифенилфосфин)палладия (0,60 г, 0,52 ммоль) и 2М раствор карбоната натрия (35 мл, 70 ммоль) в N,N-диметилацетамиде (100 мл) перемешивали в атмосфере аргона в течение 16 часов. После охлаждения реакционного раствора до комнатной температуры твердое вещество отфильтровывали, и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (петролейный эфир/этилацетат 98:2-95:5), получая целевые соединения, оба в виде твердых веществ желтого цвета.
Этил (2E)-3-{5-[2-метокси-3-(трифторметил)фенил]фуран-2-ил}проп-2-еноат (3,08 г, выход 53%):
1H-ЯМР (300 МГц, CDCl3) δ: 1,35 (т, J=7,2 Гц, 3H), 3,81 (с, 3H), 4,27 (кв, J=7,2 Гц, 2H), 6,43 (д, J=15,6 Гц, 1H), 6,75 (д, J=3,6 Гц, 1H), 7,07 (д, J=3,6 Гц, 1H), 7,29 (т, J=7,8 Гц, 1H), 7,47 (д, J=15,6 Гц, 1H), 7,57 (дд, J=7,8, 1,2 Гц, 1H), 8,04 (дд, J=7,8, 1,2 Гц, 1H).
Этил (2E)-3-{5-[3-метокси-2-(трифторметил)фенил]фуран-2-ил}проп-2-еноат (0,32 г, выход 5,5%):
1H-ЯМР (300 МГц, CDCl3) δ: 1,32 (т, J=7,2 Гц, 3H), 3,94 (с, 3H), 4,25 (кв, J=7,2 Гц, 2H), 6,33 (д, J=15,2 Гц, 1H), 6,53 (д, J=3,2 Гц, 1H), 6,66 (д, J=3,2 Гц, 1H), 7,10 (т, J=8,8 Гц, 2H), 7,44 (д, J=15,2 Гц, 1H), 7,50 (т, J=8,0 Гц, 1H).
Ссылочный пример 92 Этил 3-{5-[2-метокси-3-(трифторметил)фенил]тетрагидрофуран-2-ил}пропаноат
Раствор этил (2E)-3-{5-[2-метокси-3-(трифторметил)фенил]фуран-2-ил}проп-2-еноата (0,80 г, 2,35 ммоль), полученного в ссылочном примере 90, и палладия (10% на угле, 80 мг) в метаноле (50 мл) перемешивали в атмосфере водорода при комнатной температуре в течение 5,5 часов. После подтверждения завершения реакции с помощью ТСХ реакционный раствор фильтровали и концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле (петролейный эфир/этилацетат 30:1-10:1), получая указанное в заголовке соединение (0,34 г, выход 42%) в виде бесцветного масла.
1H-ЯМР (300 МГц, CDCl3) δ: 1,26 (т, J=7,2 Гц, 3H), 1,64-1,79 (м, 2H), 1,96-2,04 (м, 2H), 2,07-2,17 (м, 1H), 2,35-2,42 (м, 1H), 2,45-2,54 (м, 2H), 3,85 (с, 3H), 4,01-4,05 (м, 1H), 4,14 (кв, J=7,2 Гц, 2H), 5,17 (т, J=6,9 Гц, 1H), 7,24 (т, J=7,2 Гц, 1H), 7,50 (дд, J=7,8, 1,5 Гц, 1H), 7,70 (дд, J=7,8, 1,5 Гц, 1H).
Ссылочный пример 93 Этил 3-{5-[3-метокси-2-(трифторметил)фенил]тетрагидрофуран-2-ил}пропаноат
Раствор этил (2E)-3-{5-[3-метокси-2-(трифторметил)фенил]фуран-2-ил}проп-2-еноата (0,30 г, 0,88 ммоль), полученного в ссылочном примере 91, и палладия (10% на угле, 120 мг) в метаноле (30 мл) перемешивали в атмосфере водорода при комнатной температуре в течение 16 часов. После подтверждения завершения реакции с помощью ТСХ реакционный раствор фильтровали и концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле (петролейный эфир/этилацетат 30:1-10:1), получая указанное в заголовке соединение (0,17 г, выход 56%) в виде бесцветного масла.
1H-ЯМР (300 МГц, CDCl3) δ: 1,27 (т, J=7,2 Гц, 3H), 1,51-1,58 (м, 1H), 1,67-1,72 (м, 1H), 1,99-2,08 (м, 3H), 2,40-2,59 (м, 3H), 3,88 (с, 3H), 3,98-4,02 (м, 1H), 4,15 (кв, J=7,2 Гц, 2H), 5,29-5,33 (м, 1H), 6,92 (д, J=7,6 Гц, 1H), 7,40-7,47 (м, 2H).
Ссылочный пример 94 5-[3,5-бис(трифторметил)фенил]фуран-2-карбальдегид
Раствор 5-бромфуран-2-карбальдегида (2,54 г, 14,5 ммоль), [3,5-бис(трифторметил)фенил]бороновой кислоты (3,93 г, 15,2 ммоль), тетракис(трифенилфосфин)палладия (0,59 г, 0,51 ммоль) и 2М раствор карбоната натрия (36 мл, 72 ммоль) в ТГФ (150 мл) перемешивали в атмосфере аргона в течение 16 часов. После охлаждения реакционного раствора до комнатной температуры и концентрации при пониженном давлении остаток распределяли между этилацетатом и водой. Органический слой промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом магния, фильтровали и концентрировали. Остаток очищали с помощью колоночной хроматографии на силикагеле (этилацетат), и полученное твердое вещество деструктурировали в гексане, получая указанное в заголовке соединение (4,46 г, выход >99%) в виде твердого вещества бледно-желтого цвета. LC/МС ESI(+) m/z:309 (М+Н)+.
Ссылочный пример 95 {5-[3,5-бис(трифторметил)фенил]фуран-2-ил}метанол
К раствору 5-[3,5-бис(трифторметил)фенил]фуран-2-карбальдегида (5,69 г, 18,5 ммоль), полученного в ссылочном примере 94, в этаноле (170 мл) добавляли боргидрид натрия (1,40 г, 36,9 ммоль), и смесь перемешивали при комнатной температуре в течение 1 часа. Реакционный раствор концентрировали при пониженном давлении, и полученный остаток распределяли между этилацетатом и водой. Органический слой промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом магния, фильтровали и концентрировали. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (гексан/этилацетат 90:10-50:50), получая указанное в заголовке соединение (5,02 г, выход 87%) в виде твердого вещества белого цвета.
1H-ЯМР (300 МГц, CDCl3) δ: 1,81 (т, J=6,1 Гц, 1H), 4,72 (д, J=6,1 Гц, 2H), 6,46 (д, J=3,4 Гц, 1H), 6,80 (д, J=3,4 Гц, 1H), 7,74 (с, 1H), 8,07 (с, 2H).
Ссылочный пример 96 Этил ({5-[3,5-бис(трифторметил)фенил]фуран-2-ил}метокси)ацетат
К раствору {5-[3,5-бис(трифторметил)фенил]фуран-2-ил}метанола (1,00 г, 3,22 ммоль), полученному в ссылочном примере 95, в N,N-диметилформамиде (30 мл) добавляли гидрид натрия (60% в масле, 0,14 г, 3,55 ммоль), и смесь перемешивали при комнатной температуре в течение 15 минут. К раствору добавляли раствор этил бромацетата (0,59 г, 3,55 ммоль) в N,N-диметилформамиде (5 мл), и смесь перемешивали при комнатной температуре в течение 30 минут и при 80°С в течение 5 часов. Реакционному раствору давали охладиться до комнатной температуры, вливали в насыщенный раствор хлорида аммония (100 мл), и смесь экстрагировали этилацетатом (100 мл). Этилацетатный слой промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле (гексан/этилацетат 90:10-30:70), получая указанное в заголовке соединение (0,42 г, выход 33%) в виде масла желтого цвета.
1H-ЯМР (300 МГц, CDCl3) δ: 1,29 (т, J=7,2 Гц, 3H), 4,16 (с, 2H), 4,24 (кв, J=7,1 Гц, 2H), 4,68 (с, 2H), 6,52 (д, J=3,4 Гц, 1H), 6,81 (д, J=3,0 Гц, 1H), 7,74 (с, 1H), 8,07 (с, 2H).
Ссылочный пример 97 Этил ({5-[3,5-бис(трифторметил)фенил]тетрагидрофуран-2-ил}метокси)ацетат
Раствор этил ({5-[3,5-бис(трифторметил)фенил]фуран-2-ил}метокси)ацетата (0,40 г, 1,01 ммоль), полученного в ссылочном примере 96, и палладия (10% на угле, содержащего воду (50%), 100 мг) в этаноле (20 мл) перемешивали в атмосфере водорода при комнатной температуре в течение 16 часов. После подтверждения завершения реакции с помощью ТСХ реакционный раствор фильтровали и концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле (гексан/этилацетат 90:10-40:60), получая указанное в заголовке соединение (0,29 г, выход 72%) в виде бесцветного масла.
1H-ЯМР (300 МГц, CDCl3) δ: 1,28 (т, J=7,2 Гц, 3H), 1,75-2,21 (м, 3H), 2,29-2,55 (м, 1H), 3,65-3,85 (м, 2H), 4,19 (с, 2H), 4,20-4,28 (м, 2H), 4,29-4,43 (м, 1H), 5,03 (т, J=7,3 Гц, 1H), 7,76 (с, 1H), 7,86 (с, 2H).
Ссылочный пример 98 2-[3,5-бис(трифторметил)фенил]-5-(хлорметил)фуран
К раствору {5-[3,5-бис(трифторметил)фенил]фуран-2-ил}метанола (8,26 г, 26,6 ммоль), полученному в ссылочном примере 95, в ТГФ (130 мл) добавляли тионилхлорид (4,76 г, 39,9 ммол) при 0°С, и смесь перемешивали в течение 30 минут. Реакционную смесь затем перемешивали при комнатной температуре в течение 3 часов и концентрировали при пониженном давлении, и остаток распределяли между этилацетатом и водой. Органический слой промывали насыщенным раствором бикарбоната натрия и насыщенным солевым раствором, сушили над безводным сульфатом магния, фильтровали и концентрировали, получая указанное в заголовке соединение (8,43 г, выход 96%) в виде твердого вещества белого цвета.
1Н-ЯМР (300 МГц, CDCl3) δ: 4,67 (с, 2H), 6,53 (д, J=3,4 Гц, 1H), 6,80 (д, J=3,4 Гц, 1H), 7,76 (с, 1H), 8,07 (с, 2H).
Ссылочный пример 99 {5-[3,5-бис(трифторметил)фенил]фуран-2-ил}ацетонитрил
Раствор 2-[3,5-бис(трифторметил)фенил]-5-(хлорметил)фурана (8,40 г, 25,6 ммоль), полученного в ссылочном примере 98, цианида калия (3,33 г, 51,1 ммоль) и 18-краун-6 (6,76 г, 25,6 ммоль) в ацетонитриле (250 мл) перемешивали при 0°C в течение 1 часа и при комнатной температуре в течение 8 часов. Реакционный раствор концентрировали при пониженном давлении, и остаток распределяли между этилацетатом и водой. Органический слой промывали насыщенным солевым раствором, сушили над безводным сульфатом магния и фильтровали, и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле (гексан/этилацетат 90:10-30:70), и полученное желтое твердое вещество промывали гексаном, получая указанное в заголовке соединение (3,92 г, выход 48%) в виде твердого вещества желтого цвета.
1Н-ЯМР (300 МГц, CDCl3) δ: 3,89 (с, 2H), 6,51 (д, J=3,8 Гц, 1H), 6,82 (д, J=3,8 Гц, 1H), 7,76 (с, 1H), 8,04 (с, 2H).
Ссылочный пример 100 {5-[3,5-бис(трифторметил)фенил]фуран-2-ил}уксусная кислота
Раствор {5-[3,5-бис(трифторметил)фенил]фуран-2-ил}ацетонитрила (1,90 г, 5,95 ммоль), полученного в ссылочном примере 99, и 8М раствор гидроксида натрия (5 мл, 40 ммоль) в этаноле (20 мл) нагревали с обратным холодильником в течение 40 минут. Реакционному раствору давали охладиться до комнатной температуры и концентрировали при пониженном давлении. Остаток доводили до значения рН 2 6М соляной кислотой при охлаждении льдом, и распределяли между этилацетатом и водой. Органический слой промывали насыщенным солевым раствором и сушили над безводным сульфатом магния, фильтровали и фильтрат концентрировали при пониженном давлении. Полученное твердое вещество темно-коричневого цвета перекристаллизовывали из толуола-гексана, получая указанное в заголовке соединение (1,43 г, выход 71%) в виде кристаллов бледно-коричневого цвета.
1H-ЯМР (300 МГц, DMCO-d6) δ: 3,82 (с, 2H), 6,49 (д, J=3,4 Гц, 1H), 7,37 (д, J=3,4 Гц, 1H), 7,96 (с, 1H), 8,25 (с, 2H), 12,67 (уш.с, 1H).
Пример 1 ({1-[3,5-бис(трифторметил)фенил]пирролидин-3-ил}сульфанил)уксусная кислота

Раствор этил ({1-[3,5-бис(трифторметил)фенил]пирролидин-3-ил}сульфанил)ацетата (6,80 г), полученного в ссылочном примере 2, и моногидрат гидроксида лития (1,38 г) в тетрагидрофуране (100 мл)-воде (100 мл) перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь доводили до значения рН 5 6н водным раствором соляной кислоты и экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали, получая указанное в заголовке соединение (4,27 г, выход 68%) в виде бесцветных кристаллов.
1H-ЯМР (300 МГц, CDCl3) δ: 2,05-2,17 (м, 1H), 2,43-2,54 (м, 1H), 3,30-3,35 (м, 1H), 3,37 (с, 2H), 3,41-3,46 (м, 1H), 3,52-3,59 (м, 1H), 3,70-3,81 (м, 2H), 6,84 (с, 2H), 7,13 (с, 1H), 11,82 (уш., 1H).
Пример 2 ({1-[3,5-бис(трифторметил)фенил]пирролидин-3-ил}сульфинил)уксусная кислота

Раствор ({1-[3,5-бис(трифторметил)фенил]пирролидин-3-ил}сульфанил)уксусной кислоты (500 мг), полученной в примере 1, и м-хлорпербензойной кислоты (321 мг) в дихлорметане (25 мл) перемешивали при комнатной температуре в течение 2 часов. Растворитель выпаривали, и остаток очищали с помощью препаративной ВЭЖХ (оборудование: Высокопроизводительная очистительная система Gilson Inc.,; колонка: YMC Combiprep ODS-A, S-5 мкм, 50×20 мм; растворитель: РАСТВОР А; 0,1% трифторуксусная кислота, содержащая воду, РАСТВОР В; 0,1% трифторуксусная кислота, содержащая ацетонитрил; градиентный цикл: 0,00 мин (РАСТВОР А/РАСТВОР В=90/10), 1,00 мин (РАСТВОР А/РАСТВОР В=90/10), 4,20 мин (РАСТВОР А/РАСТВОР В=10/90), 5,40 мин (РАСТВОР А/РАСТВОР В=10/90), 5,50 мин (РАСТВОР А/РАСТВОР В=90/10) и 5,60 мин (РАСТВОР А/РАСТВОР В=90/10); скорость потока: 25 мл/мин; метод детекции: UV 220 нм), получая указанное в заголовке соединение (471 мг, выход 90%).
1H-ЯМР (300 МГц, DMCO-d6) δ: 2,25-2,19 (м, 1H), 2,40-2,48 (м, 1H), 3,36-3,52 (м, 3H), 3,66-3,72 (м, 1H), 3,78-3,82 (м, 2H), 4,02-4,09 (м, 1H), 7,08-7,17 (м, 3H), 13,24 (уш., 1H).
Пример 3 ({1-[3,5-бис(трифторметил)фенил]пирролидин-3-ил}сульфонил)уксусная кислота

Указанное в заголовке соединение (245 мг, выход 66%) получали из метил ({1-[3,5-бис(трифторметил)фенил]пирролидин-3-ил}сульфонил)ацетата, полученного в ссылочном примере 3, по способу, аналогичному тому, как описано в примере 1.
1H-ЯМР (300 МГц, DMCO-d6) δ: 2,44-2,47 (м, 2H), 3,46-3,56 (с, 2H), 3,72-3,81 (м, 2H), 4,37-4,53 (м, 3H), 7,10 (с, 2H), 7,19 (с, 1H), 13,56 (уш., 1H).
Пример 4 ({1-[3,5-бис(трифторметил)фенил]-2-оксопирролидин-3-ил}сульфанил)уксусная кислота
Указанное в заголовке соединение (1,0 г, выход 77%) получали из этил ({1-[3,5-бис(трифторметил)фенил]-2-оксопирролидин-3-ил}сульфанил)ацетата, полученного в ссылочном примере 4, по способу, аналогичному тому, как описано в примере 1.
1H-ЯМР (300 МГц, DMCO-d6) δ: 1,97-2,06 (м, 1H), 2,54-2,66 (с, 1H), 3,45-3,68 (м, 2H), 3,95-4,05 (м, 3H), 7,88 (с, 1H), 8,32 (с, 2H), 12,69 (уш., 1H).
Пример 5 ({1-[3,5-бис(трифторметил)фенил]-2-оксопирролидин-3-ил}сульфинил)уксусная кислота

Указанное в заголовке соединение (120 мг, выход 41%) получали из этил ({1-[3,5-бис(трифторметил)фенил]-2-оксопирролидин-3-ил}сульфинил)ацетата, полученного в ссылочном примере 5, по способу, аналогичному тому, как описано в примере 1.
1H-ЯМР (300 МГц, DMCO-d6) δ: 2,43-2,68 (м, 2H), 3,67-3,72 (м, 1H), 4,00-4,34 (м, 4H), 7,92 (с, 1H), 8,34 (с, 2H), 13,35 (уш., 1H).
Пример 6 ({1-[3,5-бис(трифторметил)фенил]-2-оксопирролидин-3-ил}сульфонил)уксусная кислота

Указанное в заголовке соединение (520 мг, выход 69%) получали из этил ({1-[3,5-бис(трифторметил)фенил]-2-оксопирролидин-3-ил}сульфонил)ацетата, полученного в ссылочном примере 6, по способу, аналогичному тому, как описано в примере 1.
1H-ЯМР (300 МГц, DMCO-d6) δ: 2,54-2,62 (м, 2H), 4,04-4,14 (м, 2H), 4,44-4,49 (м, 1H), 4,69-4,74 (м, 1H), 4,94 (т, J= 7,8 Гц, 1H), 7,96 (с, 1H), 8,33 (с, 2H), 13,63 (уш., 1H).
Пример 7 ({1-[3-фтор-2-(трифторметил)фенил]пирролидин-3-ил}сульфанил)уксусная кислота

Указанное в заголовке соединение (101 мг, выход 31%) получали из этил ({1-[3-фтор-2-(трифторметил)фенил]пирролидин-3-ил}сульфанил)ацетата, полученного в ссылочном примере 8, по способу аналогичному тому, как описано в примере 1.
1H-ЯМР (300 МГц, CDCl3) δ: 1,89-2,05 (м, 1H), 2,35-2,43 (м, 1H), 3,17-3,27 (м, 1H), 3,33 (с, 2H), 3,38-3,44 (м, 2H), 3,52-3,71 (м, 1H), 6,61-6,68 (м, 1H), 6,74-6,77 (м, 1H), 7,19-7,33 (м, 1H), 9,15 (уш., 1H).
Пример 8 ({1-[2,4-бис(трифторметил)фенил]пирролидин-3-ил}сульфонил)уксусная кислота
Указанное в заголовке соединение (535 мг, выход 81%) получали из этил ({1-[2,4-бис(трифторметил)фенил]пирролидин-3-ил}сульфонил)ацетата, полученного в ссылочном примере 11, по способу, аналогичному тому, как описано в примере 1.
1H-ЯМР (300 МГц, CDCl3) δ: 2,45-2,62 (м, 2H), 3,45-3,64 (м, 2H), 3,82 (д, J=7,2 Гц, 2H), 4,15-4,03 (м, 2H), 4,17-4,26 (м, 1H), 7,14 (д, J=8,7 Гц, 1H), 7,28 (уш., 1H), 7,66 (д, J=8,7 Гц, 1H), 7,86 (с, 1H).
Пример 9 ({1-[3,5-бис(трифторметил)фенил]пирролидин-3-ил}окси)уксусная кислота

трет-Бутил ({1-[3,5-бис(трифторметил)фенил]пирролидин-3-ил}окси)ацетат (3,35 г), полученный в ссылочном примере 15, растворяли в трифторуксусной кислоте (6,5 мл), и раствор перемешивали при комнатной температуре в течение 5 часов. К реакционной смеси добавляли толуол (10 мл), и смесь концентрировали при пониженном давлении. К полученному остатку добавляли воду (30 мл), и смесь доводили до значения рН 4 насыщенным раствором бикарбоната натрия и экстрагировали этилацетатом (30 мл ×2). Экстракт промывали насыщенным солевым раствором, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении, получая твердое вещество коричневого цвета, которое перекристаллизовывали из гексана-этилацетата, получая указанное в заголовке соединение (1,85 г, выход 64%) в виде твердого вещества белого цвета.
1H-ЯМР (300 МГц, CDCl3) δ: 2,09-2,42 (м, 2H), 3,38-3,65 (м, 4H), 4,21 (с, 2H), 4,33-4,45 (м, 1H), 6,86 (с, 2H), 7,14 (с, 1H).
Пример 10 ({1-[3,5-бис(трифторметил)фенил]пирролидин-3-ил}сульфанил)уксусная кислота
({1-[3,5-Бис(трифторметил)фенил]пирролидин-3-ил}сульфанил)уксусную кислоту (210 мг, два вида рацематов), полученную в примере 1, подвергали хиральной препаративной ВЭЖХ (колонка: CHIRALPAK AS(BF001) 50 мм ID×500 мм L; растворитель: гексан/2-пропанол/муравьиная кислота=970/30/1 (об./об./об.); скорость потока: 80 мл/мин; метод детекции: UV 220 нм; температура: 25°С), получая соединение (tR1), имеющее более короткое время удерживания, которое перекристаллизовывали из этилацетат-гексана, получая указанное в заголовке соединение (68,5 мг, выход 33%).
1H-ЯМР (300 МГц, DMCO-d6) δ: 1,94-2,03 (м, 1H), 2,35-2,51 (м, 1H), 3,30-3,53 (м, 5H), 3,65-3,80 (м, 2H), 7,01 (с, 2H), 7,12 (с, 1H), 12,67 (уш., 1H).
Пример 11 ({1-[3,5-бис(трифторметил)фенил]пирролидин-3-ил}сульфанил)уксусная кислота
({1-[3,5-Бис(трифторметил)фенил]пирролидин-3-ил}сульфанил)уксусную кислоту (210 мг, два вида рацематов), полученную в примере 1, подвергали хиральной препаративной ВЭЖХ (колонка: CHIRALPAK AS(BF001) 50 мм ID×500 мм L; растворитель: гексан/2-пропанол/муравьиная кислота=970/30/1 (об./об./об.); скорость потока: 80 мл/мин; метод детекции: UV 220 нм; температура: 25°С), получая соединение (tR2), имеющее более продолжительное время удерживания, которое перекристаллизовывали из этилацетат-гексана, получая указанное в заголовке соединение (65,6 мг, выход 31%).
1H-ЯМР (300 МГц, DMCO-d6) δ: 1,94-2,03 (м, 1H), 2,35-2,51 (м, 1H), 3,30-3,53 (м, 5H), 3,65-3,80 (м, 2H), 7,01 (с, 2H), 7,12 (с, 1H), 12,67 (уш., 1H).
Пример 12 ({1-[2,4-бис(трифторметил)фенил]пирролидин-3-ил}сульфанил)ацетат кальция
Раствор этил ({1-[2,4-бис(трифторметил)фенил]пирролидин-3-ил}сульфанил)ацетата (1,30 г), полученного в ссылочном примере 10, и моногидрата гидроксида лития (419 мг) в смеси тетрагидрофуран-вода (1:1, 400 мл), перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь доводили до значения рН 5 с помощью 1н водного раствора соляной кислоты, и смесь экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, сушили над безводным сульфатом магния и фильтровали, и фильтрат концентрировали, получая бесцветное масло (1,11 г). Бесцветное масло (1,11 г) растворяли в метаноле (50 мл), добавляли водный раствор (10 мл) бикарбоната калия (297 мг), и смесь перемешивали при комнатной температуре в течение 1 часа. Растворитель выпаривали, и остаток растворяли в метаноле (50 мл). Добавляли водный раствор (10 мл) хлорида кальция (161 мг), и смесь перемешивали при комнатной температуре в течение 1 часа. Растворитель выпаривали, и остаток растворяли в тетрагидрофуране (20 мл). Нерастворимое вещество отфильтровывали, и фильтрат концентрировали, получая указанное в заголовке соединение (1,00 г, выход 86%) в виде бесцветных кристаллов.
1H-ЯМР (300 МГц, DMCO-d6) δ: 1,03-1,05 (м, 1H), 1,88-1,94 (м, 1H), 3,17-3,80 (м, 6H), 7,10 (д, J=9,0 Гц, 1H), 7,69 (д, J=9,0 Гц, 1H), 7,75 (с, 1H).
Пример 13 3-{1-[4-хлор-3-(трифторметил)фенил]пирролидин-3-ил}пропановая кислота
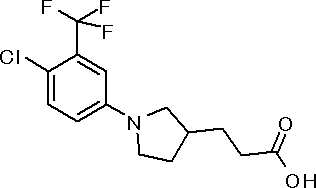
Указанное в заголовке соединение (2,66 г, выход 60%) получали из этил 3-{1-[4-хлор-3-(трифторметил)фенил]пирролидин-3-ил}пропаноата, полученного в ссылочном примере 13, по способу, аналогичному тому, как описано в примере 1.
1H-ЯМР (300 МГц, CDCl3) δ: 1,58-1,67 (м, 3H), 2,10-2,12 (м, 1H), 2,23-2,34 (м, 3H), 2,89 (т, J=8,4 Гц, 1H), 3,19-3,46 (м, 3H), 6,72-6,77 (м, 2H), 7,37-7,40 (м, 1H), 12,08 (с, 1H).
Пример 14 3-{1-[2-хлор-3-(трифторметил)фенил]пирролидин-3-ил}пропановая кислота

Указанное в заголовке соединение (532 мг, выход 66%) получали из этил 3-{1-[2-хлор-3-(трифторметил)фенил]пирролидин-3-ил}пропаноата, полученного в ссылочном примере 14, по способу, аналогичному тому, как описано в примере 1.
1H-ЯМР (300 МГц, DMCO-d6) δ: 1,48-1,54 (м, 1H), 1,56-1,73 (м, 2H), 2,01-2,23 (м, 2H), 2,27-2,32 (м, 2H), 3,15 (т, J=9,6 Гц, 1H), 3,24-3,38 (м, 2H), 3,48-3,56 (м, 1H), 7,24-7,26 (м, 2H), 7,33-7,38 (м, 1H), 12,07 (с, 1H).
Пример 15 3-{3-[3,5-бис(трифторметил)фенил]-2-оксоимидазолидин-1-ил}пропановая кислота

Указанное в заголовке соединение (108 мг, выход 79%) получали из этил 3-{3-[3,5-бис(трифторметил)фенил]-2-оксоимидазолидин-1-ил}пропаноата, полученного в ссылочном примере 17, по способу, аналогичному тому, как описано в примере 1.
1H-ЯМР (300 МГц, CDCl3) δ: 2,61-2,78 (м, 2H), 3,56-3,72 (м, 4H), 3,81-3,95 (м, 2H), 7,52 (с, 1H), 8,03 (с, 2H).
Пример 16 ({(3S)-1-[3,5-бис(трифторметил)фенил]пирролидин-3-ил}окси)уксусная кислота
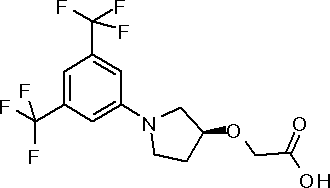
Раствор (3S)-1-[3,5-бис(трифторметил)фенил]пирролидин-3-ола (9,60 г), полученного в ссылочном примере 18, в N,N-диметилформамиде (20 мл) при 60°С добавляли к суспензии гидрида натрия (60% в масле, 1,80 г) в N,N-диметилформамиде (200 мл). После перемешивания в течение 30 минут добавляли хлорацетат натрия (7,50 г) и бромид тетрабутиламмония (1,03 г), и смесь перемешивали при 60°С в течение 16 часов. Реакционной смеси давали охладиться до комнатной температуры, и добавляли воду. Смесь доводили до значения рН 2 с помощью концентрированной соляной кислоты, и смесь экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали, и остаток очищали с помощью колоночной хроматографии на силикагеле (петролейный эфир/этилацетат 1:10), и перекристаллизовывали из смеси гексан-ацетон, получая указанное в заголовке соединение (5,82 г, выход 51%) в виде твердого вещества белого цвета.
1H-ЯМР (300 МГц, DMCO-d6) δ: 2,06-2,16 (м, 2H), 3,33-3,54 (м, 4H), 4,10 (с, 2H), 4,31-4,36 (м, 1H), 7,01 (с, 2H), 7,11 (с, 1H), 12,64 (с, 1H), 97,2% ee, [α]D=+5,9°(c=0,54, MeOH, 22°C).
Пример 17 ({(3R)-1-[3,5-бис(трифторметил)фенил]пирролидин-3-ил}окси)уксусная кислота

Раствор (3R)-1-[3,5-бис(трифторметил)фенил]пирролидин-3-ола (7,45 г), полученного в ссылочном примере 19, в N,N-диметилформамиде (20 мл) при 60°С добавляли к суспензии гидрида натрия (60% в масле, 1,50 г) в N,N-диметилформамиде (200 мл). После перемешивания в течение 30 минут добавляли хлорацетат натрия (3,48 г) и бромид тетрабутиламмония (0,80 г), и смесь перемешивали при 60°С в течение 1 часа. Реакционной смеси давали охладиться до комнатной температуры, и реакцию гасили холодной водой. Реакционную смесь разбавляли водой и доводили до значения рН 2 с помощью концентрированной соляной кислоты, и смесь экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали, и остаток очищали с помощью колоночной хроматографии на силикагеле (этилацетат), и перекристаллизовывали из смеси гексан-ацетон, получая указанное в заголовке соединение (5,82 г, выход 64%) в виде твердого вещества белого цвета.
1H-ЯМР (300 МГц, DMCO-d6) δ: 2,07-2,16 (м, 2H), 3,34-3,54 (м, 4H), 4,10 (с, 2H), 4,33-4,35 (м, 1H), 7,01 (с, 2H), 7,11 (с, 1H), 12,65 (с, 1H). 99,1% ee, [α]D=-5,9°(c=0,56, MeOH, 22°C).
Пример 18 ({1-[3,5-бис(трифторметил)фенил]пирролидин-3-ил}сульфинил)уксусная кислота
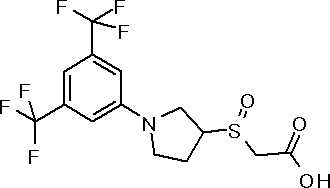
К раствору оптически разделенной tR1 (AS, 300 мг) ({1-[3,5-бис(трифторметил)фенил]пирролидин-3-ил}сульфанил)уксусной кислоты, полученной в примере 10, в ацетоне (10 мл) добавляли водный раствор (5 мл) соединения Oxone-персульфата (500 мг) при 0°С, и смесь перемешивали при 0°С в течение 1 часа. Реакционную смесь экстрагировали этилацетатом, и органический слой сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали, получая указанное в заголовке соединение (291 мг, выход 93%) в виде твердого вещества белого цвета.
1H-ЯМР (300 МГц, DMCO-d6) δ: 2,26-2,53 (м, 2H), 3,37-3,60 (м, 3H), 3,61-3,92 (м, 3H), 3,97-4,10 (м, 1H), 7,08 (с, 1H), 7,14 (с, 1H), 7,18 (с, 1H), 13,26 (уш., 1H).
Пример 19 ({1-[3,5-бис(трифторметил)фенил]пирролидин-3-ил}сульфинил)уксусная кислота
К раствору оптически разделенной tR2 (AS, 300 мг) ({1-[3,5-бис(трифторметил)фенил]пирролидин-3-ил}сульфанил)уксусной кислоты, полученной в примере 11, в ацетоне (10 мл), добавляли водный раствор (5 мл) соединения Oxone-персульфата (500 мг) при 0°С, и смесь перемешивали при 0°С в течение 1 часа. Ацетон выпаривали при пониженном давлении, и осадок собирали фильтрованием. Осадок промывали водой и гексаном, получая указанное в заголовке соединение (311 мг, выход 99%) в виде твердого вещества белого цвета.
1H-ЯМР (300 МГц, DMCO-d6) δ: 2,09-2,55 (м, 2H), 3,39-3,60 (м, 3H), 3,60-3,90 (м, 3H), 3,98-4,11 (м, 1H), 7,08 (с, 1H), 7,14 (с, 1H), 7,18 (с, 1H), 13,24 (уш., 1H).
Пример 20 ({1-[3,5-бис(трифторметил)фенил]пирролидин-3-ил}сульфонил)уксусная кислота

({1-[3,5-Бис(трифторметил)фенил]пирролидин-3-ил}сульфонил)уксусную кислоту (390 мг, два вида рацематов), полученную в примере 3, подвергали хиральной препаративной ВЭЖХ (колонка: CHIRALCEL OJ 50 мм ID×500 мм L; растворитель: гексан/этанол/трифторуксусная кислота=800/200/1 (об./об./об.); скорость потока: 60 мл/мин; метод детекции: UV 220 нм; температура: 35°С), получая соединение (tR1), имеющее более короткое время удерживания, которое перекристаллизовывали из этилацетат-гексана, получая указанное в заголовке соединение (143,7 мг, выход 37%).
1H-ЯМР (300 МГц, DMCO-d6) δ: 2,36-2,48 (м, 2H), 3,40-3,61 (м, 2H), 3,66-3,92 (м, 2H), 4,27-4,54 (м, 3H), 7,11 (с, 2H), 7,20 (с, 1H), 13,60 (уш., 1H).
Пример 21 ({1-[3,5-бис(трифторметил)фенил]пирролидин-3-ил}сульфонил)уксусная кислота
({1-[3,5-Бис(трифторметил)фенил]пирролидин-3-ил}сульфонил)уксусную кислоту (390 мг, два вида рацематов), полученную в примере 3, подвергали хиральной препаративной ВЭЖХ (колонка: CHIRALCEL OJ 50 мм ID×500 мм L; растворитель: гексан/этанол/трифторуксусная кислота=800/200/1 (об./об./об.); скорость потока: 60 мл/мин; метод детекции: UV 220 нм; температура: 35°С), получая соединение (tR2), имеющее более продолжительное время удерживания, которое перекристаллизовывали из этилацетат-гексана, получая указанное в заголовке соединение (136,6 мг, выход 35%).
1H-ЯМР (300 МГц, DMCO-d6) δ: 2,34-2,57 (м, 2H), 3,40-3,63 (м, 2H), 3,67-3,89 (м, 2H), 4,28-4,60 (м, 3H), 7,11 (с, 2H), 7,20 (с, 1H), 13,57 (уш., 1H).
Пример 22 3-{4-[3,5-бис(трифторметил)фенил]тетрагидрофуран-2-ил}пропановая кислота
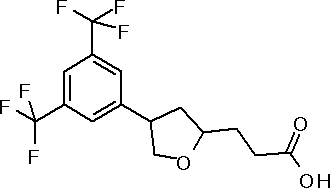
Указанное в заголовке соединение (239 мг, выход 79%) получали из этил 3-{4-[3,5-бис(трифторметил)фенил]тетрагидрофуран-2-ил}пропаноата, полученного в ссылочном примере 22, по способу, аналогичному тому, как описано в примере 1, в виде стереоизомерной смеси.
LC/МС ESI(+) m/z: 357 (М+H)+, время удерживания 2,34 мин.
Пример 23 ({1-[3,5-бис(трифторметил)фенил]пирролидин-3-ил}сульфинил)уксусная кислота
({1-[3,5-Бис(трифторметил)фенил]пирролидин-3-ил}сульфинил)уксусную кислоту (234 мг, смесь двух видов диастереомеров), полученную в примере 19, подвергали хиральной препаративной ВЭЖХ (колонка: CHIRALPAK AD-H 20 мм ID×250 мм L; растворитель: двуокись углерода/метанол/трифторуксусная кислота=800/200/0,2 (объем/объем/объем); скорость потока: 50 мл/мин; метод детекции: UV 254 нм; температура: 35°С), получая соединение (tR1), имеющее более короткое время удерживания, в качестве целевого соединения (128,4 мг, выход 55%).
1H-ЯМР (300 МГц, DMCO-d6) δ: 2,36-2,48 (м, 2H), 3,40-3,62 (м, 3H), 3,63-3,75 (м, 2H), 3,75-3,92 (м, 1H), 4,05 (д, J=14,7 Гц, 1H), 7,08 (с, 2H), 7,18 (с, 1H).
Пример 24 ({1-[3,5-бис(трифторметил)фенил]пирролидин-3-ил}сульфинил)уксусная кислота
({1-[3,5-Бис(трифторметил)фенил]пирролидин-3-ил}сульфинил)уксусную кислоту (234 мг, смесь двух видов диастереомеров), полученную в примере 19, подвергали хиральной препаративной ВЭЖХ (колонка: CHIRALPAK AD-H 20 мм ID×250 мм L; растворитель: двуокись углерода/метанол/трифторуксусная кислота=800/200/0,2 (объем/объем/объем); скорость потока: 50 мл/мин; метод детекции: UV 254 нм; температура: 35°С), получая соединение (tR2), имеющее более продолжительное время удерживания, в качестве целевого соединения (101,2 мг, выход 43%).
1H-ЯМР (300 МГц, DMCO-d6) δ: 2,07-2,26 (м, 1H), 2,31-2,47 (м, 1H), 3,37-3,58 (м, 2H), 3,58-3,72 (м, 1H), 3,72-3,83 (м, 3H), 4,03 (д, J=14,7 Гц, 1H), 7,14 (с, 2H), 7,17 (с, 1H), 13,25 (уш., 1H).
Пример 25 ({1-[3,5-бис(трифторметил)фенил]пирролидин-3-ил}сульфинил)уксусная кислота
({1-[3,5-Бис(трифторметил)фенил]пирролидин-3-ил}сульфинил)уксусную кислоту (197 мг, смесь двух видов диастереомеров), полученную в примере 18, подвергали хиральной препаративной ВЭЖХ (колонка: CHIRALPAK AD-H 20 мм ID×250 мм L; растворитель: двуокись углерода/метанол/трифторуксусная кислота=850/150/0,15 (объем/объем/объем); скорость потока: 50 мл/мин; метод детекции: UV 254 нм; температура: 35°С), получая соединение (tR1), имеющее более короткое время удерживания, в качестве целевого соединения (83,5 мг, выход 42%).
1H-ЯМР (300 МГц, DMCO-d6) δ: 2,08-2,25 (м, 1H), 2,31-2,46 (м, 1H), 3,37-3,58 (м, 2H), 3,58-3,72 (м, 1H), 3,73-3,85 (м, 3H), 4,04 (д, J=14,4 Гц, 1H), 7,14 (с, 2H), 7,17 (с, 1H), 13,23 (уш., 1H).
Пример 26 ({1-[3,5-бис(трифторметил)фенил]пирролидин-3-ил}сульфинил)уксусная кислота
({1-[3,5-Бис(трифторметил)фенил]пирролидин-3-ил}сульфинил)уксусную кислоту (197 мг, смесь двух видов диастереомеров), полученную в примере 18, подвергали хиральной препаративной ВЭЖХ (колонка: CHIRALPAK AD-H 20 мм ID×250 мм L; растворитель: двуокись углерода/метанол/трифторуксусная кислота=850/150/0,15 (объем/объем/объем); скорость потока: 50 мл/мин; метод детекции: UV 254 нм; температура: 35°С), получая соединение (tR2), имеющее более продолжительное время удерживания, в качестве целевого соединения (102,9 мг, выход 52%).
1H-ЯМР (300 МГц, DMCO-d6) δ: 2,30-2,47 (м, 2H), 3,38-3,61 (м, 3H), 3,61-3,75 (м, 2H), 3,75-3,90 (м, 1H), 4,04 (д, J=14,8 Гц, 1H), 7,08 (с, 2H), 7,18 (с, 1H), 13,24 (уш., 1H).
Пример 27 ({1-[4-хлор-3-(трифторметил)фенил]пирролидин-3-ил}окси)ацетат кальция

Желтое масло (0,42 г) получали из 1-[4-хлор-3-(трифторметил)фенил]пирролидин-3-ола, полученного в ссылочном примере 23, по способу, аналогичному тому, как описано в примере 17. Желтое масло (0,18 г) растворяли в метаноле (5 мл), добавляли водный раствор (5 мл) бикарбоната калия (56,9 мг), и смесь перемешивали при комнатной температуре в течение 1 часа. Растворитель выпаривали, и остаток растворяли в метаноле (5 мл). Добавляли водный раствор (5 мл) хлорида кальция (31,5 мг), и смесь перемешивали при комнатной температуре в течение 1 часа. Растворитель выпаривали, и остаток растворяли в этилацетате (20 мл). Нерастворимое вещество отфильтровывали, и фильтрат концентрировали, получая указанное в заголовке соединение (155 мг, выход 68%) в виде кристаллов желтого цвета.
1H-ЯМР (300 МГц, DMCO-d6) δ: 1,93-2,16 (м, 2H), 3,21-3,35 (м, 2H), 3,35-3,46 (м, 2H), 3,70 (с, 2H), 4,25-4,48 (м, 1H), 6,69-6,82 (м, 2H), 7,29-7,47 (м, 1H).
Пример 28 ({1-[4-хлор-3-(трифторметил)фенил]пирролидин-3-ил}сульфанил)уксусная кислота

Указанное в заголовке соединение (2,30 г, выход 89%) получали из этил ({1-[4-хлор-3-(трифторметил)фенил]пирролидин-3-ил}сульфанил)ацетата, полученного в ссылочном примере 25, по способу, аналогичному тому, как описано в примере 1.
1H-ЯМР (300 МГц, CDCl3) δ: 2,00-2,17 (м, 1H), 2,37-2,54 (м, 1H), 3,22-3,55 (м, 5H), 3,63-3,78 (м, 2H), 6,52-6,62 (м, 1H), 6,74-6,81 (м, 1H), 7,27-7,32 (м, 1H).
Пример 29 ({1-[4-хлор-3-(трифторметил)фенил]пирролидин-3-ил}сульфинил)уксусная кислота

Указанное в заголовке соединение (32,0 мг, выход 10%) получали из ({1-[4-хлор-3-(трифторметил)фенил]пирролидин-3-ил}сульфанил)уксусной кислоты, полученной в примере 28, по способу, аналогичному тому, как описано в примере 18.
1H-ЯМР (300 МГц, DMCO-d6) δ: 2,01-2,22 (м, 1H), 2,22-2,45 (м, 1H), 3,17-3,50 (м, 3H), 3,52-3,85 (м, 3H), 3,94-4,10 (м, 1H), 6,78-6,97 (м, 2H), 7,40-7,50 (м, 1H).
Пример 30 ({1-[4-хлор-3-(трифторметил)фенил]пирролидин-3-ил}сульфанил)ацетат кальция
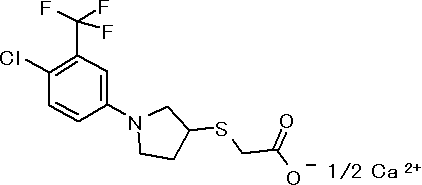
({1-[4-Хлор-3-(трифторметил)фенил]пирролидин-3-ил}сульфанил)уксусную кислоту (246 мг), полученную в примере 28, растворяли в метаноле (10 мл), добавляли водный раствор (10 мл) бикарбоната калия (72,4 мг), и смесь перемешивали при комнатной температуре в течение 1 часа. Растворитель выпаривали, и остаток растворяли в метаноле (10 мл). Добавляли водный раствор (10 мл) хлорида кальция (40,1 мг), и смесь перемешивали при комнатной температуре в течение 1 часа. Растворитель выпаривали, и остаток растворяли в этилацетате (20 мл). Нерастворимые вещества отфильтровывали, и фильтрат концентрировали, получая указанное в заголовке соединение (172 мг, выход 66%) в виде кристаллов желтого цвета.
1H-ЯМР (300 МГц, DMCO-d6) δ: 1,85-2,01 (м, 1H), 2,22-2,39 (м, 1H), 3,08 (с, 2H), 3,11-3,21 (м, 1H), 3,22-3,35 (м, 2H), 3,59-3,72 (м, 2H), 6,70-6,82 (м, 2H), 7,33-7,46 (м, 1H).
Пример 31 ({1-[2-хлор-3-(трифторметил)фенил]пирролидин-3-ил}сульфанил)ацетат калия

Раствор этил ({1-[2-хлор-3-(трифторметил)фенил]пирролидин-3-ил}сульфанил)ацетата (1,02 г), полученного в ссылочном примере 28, и 1н водный раствор гидроксида лития (20 мл) в тетрагидрофуране (20 мл) перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь доводили до величины рН 5 с помощью 1н водного раствора соляной кислоты, и смесь экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, сушили над сульфатом магния и фильтровали, и фильтрат концентрировали, получая желтое масло (893 мг). Желтое масло (160 мг) растворяли в метаноле (5 мл), добавляли водный раствор (10 мл) бикарбоната калия (49,5 мг), и смесь перемешивали при комнатной температуре в течение 1 часа. Растворитель выпаривали, получая указанное в заголовке соединение (136 мг, выход 68%) в виде кристаллов желтого цвета.
1H-ЯМР (300 МГц, DMCO-d6) δ: 1,73-1,89 (м, 1H), 2,15-2,32 (м, 1H), 2,97 (с, 2H), 3,21-3,29 (м, 1H), 3,38-3,46 (м, 2H), 3,47-3,58 (м, 1H), 3,66-3,74 (м, 1H), 7,23-7,28 (м, 1H), 7,28-7,32 (м, 1H), 7,32-7,42 (м, 1H).
Пример 32 ({1-[3-(трифторметил)фенил]пирролидин-3-ил}сульфанил)уксусная кислота

Указанное в заголовке соединение (104 мг, выход 54%) получали из этил ({1-[3-(трифторметил)фенил]пирролидин-3-ил}сульфанил)ацетата, полученного в ссылочном примере 29, по способу, аналогичному тому, как описано в примере 1.
1H-ЯМР (300 МГц, CDCl3) δ: 2,01-2,15 (м, 1H), 2,39-2,54 (м, 1H), 3,21-3,33 (м, 1H), 3,33-3,45 (м, 1H), 3,38 (с, 2H), 3,45-3,59 (м, 1H), 3,61-3,83 (м, 2H), 6,60-6,71 (м, 1H), 6,72 (с, 1H), 6,87-6,98 (м, 1H), 7,27-7,36 (м, 1H).
Пример 33 ({1-[2,5-бис(трифторметил)фенил]пирролидин-3-ил}окси)уксусная кислота
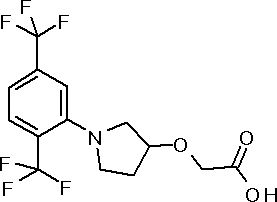
Указанное в заголовке соединение (110 мг, выход 28%) получали из 1-[2,5-бис(трифторметил)фенил]пирролидин-3-ола, полученного в ссылочном примере 30, по способу, аналогичному тому, как описано в примере 17.
1H-ЯМР (300 МГц, CDCl3) δ: 2,16-2,23 (м, 2H), 3,29-3,48 (м, 2H), 3,58-3,76 (м, 2H), 4,08-4,25 (м, 2H), 4,29-4,38 (м, 1H), 7,07-7,15 (м, 1H), 7,18 (с, 1H), 7,63-7,78 (м, 1H).
Пример 34 3-{5-[3,5-бис(трифторметил)фенил]тетрагидрофуран-2-ил}пропановая кислота

Раствор этил 3-{5-[3,5-бис(трифторметил)фенил]тетрагидрофуран-2-ил}пропаноата (13,4 г), полученного в ссылочном примере 33, и 1М раствор гидроксида лития (103 мл) в этаноле (340 мл) перемешивали при комнатной температуре в течение 4 часов. Реакционную смесь доводили до величины рН 3 с помощью 1н раствора соляной кислоты, и концентрировали при пониженном давлении, и остаток распределяли между этилацетатом и водой. Органический слой промывали насыщенным солевым раствором, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали, и полученное бесцветное твердое вещество перекристаллизовывали из гексана, получая указанное в заголовке соединение (10,56 г, выход 86%, рацемат цис-формы) в виде бесцветных кристаллов.
1H-ЯМР (300 МГц, CDCl3) δ: 1,55-1,90 (м, 2H), 1,94-2,08 (м, 2H), 2,08-2,24 (м, 1H), 2,30-2,50 (м, 1H), 2,50-2,70 (м, 2H), 4,02-4,19 (м, 1H), 4,97 (т, J=7,3 Гц, 1H), 7,78 (с, 3H).
Пример 35 3-{(2S,5R)-5-[3,5-бис(трифторметил)фенил]тетрагидрофуран-2-ил}пропановая кислота

3-{5-[3,5-бис(трифторметил)фенил]тетрагидрофуран-2-ил}пропановую кислоту (25,0 г, рацемат цис-формы), полученную в примере 34, подвергали хиральной препаративной ВЭЖХ (колонка: CHIRALPAK AS 50 мм ID×500 мм L; растворитель: гексан/этанол/уксусная кислота=990/10/1 (об./об./об.); скорость потока: 80 мл/мин; метод детекции: UV 220 нм; температура: 30°С), получая соединение (tR1), имеющее более короткое время удерживания, которое перекристаллизовывали из гексана, получая указанное в заголовке соединение (12,25 г, степень восстановления 98%).
1H-ЯМР (300 МГц, CDCl3) δ: 1,55-1,90 (м, 2H), 1,94-2,08 (м, 2H), 2,08-2,24 (м, 1H), 2,30-2,50 (м, 1H), 2,50-2,70 (м, 2H), 4,02-4,19 (м, 1H), 4,97 (т, J=7,3 Гц, 1H), 7,78 (с, 3 H).
Пример 36 3-{(2R,5S)-5-[3,5-бис(трифторметил)фенил]тетрагидрофуран-2-ил}пропановая кислота
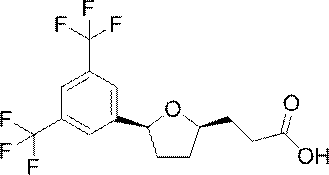
3-{5-[3,5-бис(трифторметил)фенил]тетрагидрофуран-2-ил}пропановую кислоту (25,0 г, рацемат цис-формы), полученную в примере 34, подвергали хиральной препаративной ВЭЖХ (колонка: CHIRALPAK AC 50 мм ID×500 мм L; растворитель: гексан/этанол/уксусная кислота=990/10/1 (об./об./об.); скорость потока: 80 мл/мин; метод детекции: UV 220 нм; температура: 30°С), получая соединение (tR2), имеющее более продолжительное время удерживания, которое перекристаллизовывали из гексана, получая указанное в заголовке соединение (12,25 г, степень восстановления 98%).
1H-ЯМР (300 МГц, CDCl3) δ: 1,55-1,90 (м, 2H), 1,94-2,08 (м, 2H), 2,08-2,24 (м, 1H), 2,30-2,50 (м, 1H), 2,50-2,70 (м, 2H), 4,02-4,19 (м, 1H), 4,97 (т, J=7,3 Гц, 1H), 7,78 (с, 3 H).
Пример 37 3-{1-[3,5-бис(трифторметил)фенил]пирролидин-3-ил}пропановая кислота

Указанное в заголовке соединение (0,13 г, выход 47%) получали из этил 3-{1-[3,5-бис(трифторметил)фенил]пирролидин-3-ил}пропаноата, полученного в ссылочном примере 36, по способу аналогичному тому, как описано в примере 34.
1H-ЯМР (300 МГц, DMCO-d6) δ: 1,54-1,80 (м, 3H), 1,99-2,22 (м, 1H), 2,20-2,41 (м, 3H), 2,87-3,04 (м, 1H), 3,36-3,60 (м, 3H), 6,98 (с, 2H), 7,08 (с, 1H), 12,13 (уш.с, 1H).
Пример 38 3-{2-[3,5-бис(трифторметил)фенил]-1,1-диоксидоизотиазолидин-5-ил}пропановая кислота
Указанное в заголовке соединение (0,23 г, выход 92%) получали из этил 3-{2-[3,5-бис(трифторметил)фенил]-1,1-диоксидоизотиазолидин-5-ил}пропаноата, полученного в ссылочном примере 41, по способу, аналогичному тому, как описано в примере 34.
1H-ЯМР (300 МГц, DMCO-d6) δ: 1,82-2,24 (м, 3H), 2,43-2,53 (м, 2H), 2,54-2,70 (м, 1H), 3,60-3,78 (м, 1H), 3,82-3,96 (м, 2H), 7,73 (с, 2H), 7,82 (с, 1H), 12,35 (с, 1H).
Пример 39 ({1-[2-(трифторметил)фенил]пирролидин-3-ил}окси)ацетат кальция

Коричневое масло (1,06 г, выход 60%) получали из 1-[2-(трифторметил)фенил]пирролидин-3-ола, полученного в ссылочном примере 42, по способу, аналогичному тому, как описано в примере 17. Указанное в заголовке соединение (0,90 г, выход 90%) получали в виде твердого вещества бледно-коричневого цвета из масла коричневого цвета (0,94 г), по способу, аналогичному тому, как описано в примере 27.
1H-ЯМР (300 МГц, DMCO-d6) δ: 1,78-2,11 (м, 2H), 3,19 (м, 2H), 3,29-3,61 (м, 2H), 3,72 (с, 2H), 4,30 (уш.с, 1H), 6,76-6,96 (м, 1H), 6,98-7,16 (м, 1H), 7,32-7,49 (м, 1H), 7,47-7,68 (м, 1H).
Пример 40 ({1-[3-(трифторметил)фенил]пирролидин-3-ил}окси)ацетат кальция

Масло коричневого цвета (1,15 г, выход 66%) получали из 1-[3-(трифторметил)фенил]пирролидин-3-ола, полученного в ссылочном примере 43, по способу, аналогичному тому, как описано в примере 17. Указанное в заголовке соединение (0,90 г, выход 90%) получали в виде твердого вещества бледно-коричневого цвета из масла коричневого цвета, по способу, аналогичному тому, как описано в примере 27.
1H-ЯМР (300 МГц, DMCO-d6) δ: 1,86-2,16 (м, 2H), 3,04-3,52 (м, 4H), 3,73 (с, 2H), 4,36 (уш.с, 1H), 6,67 (с, 1H), 6,75 (д, J=8,7 Гц, 1H), 6,84 (д, J=7,6 Гц, 1H), 7,33 (т, J=7,8 Гц, 1H).
Пример 41 ({1-[4-(трифторметил)фенил]пирролидин-3-ил}окси)уксусная кислота

Указанное в заголовке соединение (1,10 г, выход 63%) в виде твердого вещества бледно-желтого цвета получали из 1-[4-(трифторметил)фенил]пирролидин-3-ола, полученного в ссылочном примере 44, по способу, аналогичному тому, как описано в примере 17.
1H-ЯМР (300 МГц, CDCl3) δ: 2,07-2,39 (м, 2H), 3,31-3,66 (м, 4H), 4,19 (с, 2H), 4,30-4,47 (м, 1H), 6,55 (д, J=8,7 Гц, 2H), 7,45 (д, J=8,7 Гц, 2H).
Пример 42 3-{1-[4-(трифторметил)фенил]пирролидин-3-ил}пропановая кислота

В контейнер микроволнового реактора добавляли этил 3-(пирролидин-3-ил)пропаноат (0,15М диметоксиэтановый раствор, 800 мкл; 120 мкмоль), полученный в ссылочном примере 12, 1-бром-4-(трифторметил)бензол (0,36М диметоксиэтановый раствор, 800 мкл; 288 мкмоль), трет-бутоксид натрия (16,9 мг; 168 мкмоль), (±)-2,2'-бис(дифенилфосфино)-1,1'-бинафтил (4,0 мг; 6,0 мкмоль) и трис(дибензилиденацетон)дипалладий(0) (2,8 мг; 3,0 мкмоль) при комнатной температуре в указанном порядке, контейнер заполняли аргоном и закупоривали, и подвергали облучению в микроволновом реакционном аппарате при 120°С в течение 6 минут. После завершения реакции к реакционной смеси добавляли воду (2 мл), и смесь экстрагировали этилацетатом (3 мл) для отделения органического слоя. Этилацетат выпаривали при пониженном давлении, и остаток растворяли в диметилсульфоксиде (1 мл) и очищали с помощью препаративной ВЭЖХ, получая фракцию высокой чистоты, содержащую этилово-эфирную форму целевого соединения. Растворитель выпаривали, и полученный остаток растворяли в этаноле (400 мкл). При комнатной температуре добавляли 1М водный раствор гидроксида натрия (400 мкл: 400 мкмоль), и смесь перемешивали в течение 16 часов. Смесь нейтрализовали с помощью 1М соляной кислоты (400 мкл: 400 мкмоль) и очищали с помощью препаративной ВЭЖХ (оборудование: Высокопроизводительная очистительная система Gilson Inc.,; колонка: Combiprep Hydrosphere C18, 19×50 мм (YMC); растворитель: РАСТВОР А; 0,1% трифторуксусная кислота, содержащая воду, РАСТВОР В; 0,1% трифторуксусная кислота, содержащая ацетонитрил; градиентный цикл: 0,00 мин (РАСТВОР А/РАСТВОР В=98/2), 1,00 мин (РАСТВОР А/РАСТВОР В=98/2), 5,20 мин (РАСТВОР А/РАСТВОР В=60/40), 5,40 мин (РАСТВОР А/РАСТВОР В=5/95), 6,40 мин (РАСТВОР А/РАСТВОР В=5/95), 6,50 мин (РАСТВОР А/РАСТВОР В=98/2) и 6,60 мин (РАСТВОР А/РАСТВОР В=98/2); скорость потока: 20 мл/мин; метод детекции: UV 220 нм), получая указанное в заголовке соединение.
выход: 2,0 мг
LC-МС анализ: чистота >99,9%
LC/МС ESI(+) m/z: 288 (М+H)+
Пример 43 3-{1-[3-(трифторметил)фенил]пирролидин-3-ил}пропановая кислота
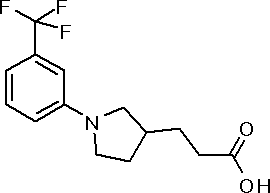
Указанное в заголовке соединение получали из этил 3-(пирролидин-3-ил}пропаноата, полученного в ссылочном примере 12, и 1-бром-3-(трифторметил)бензола, по способу, аналогичному тому, как описано в примере 42.
выход: 0,6 мг
LC-МС анализ: чистота >99,9%
LC/МС ESI(+) m/z: 288 (М+H)+
Пример 44 3-{1-[4-фтор-3-(трифторметил)фенил]пирролидин-3-ил}пропановая кислота
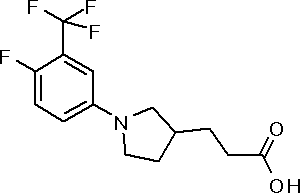
Указанное в заголовке соединение получали из этил 3-(пирролидин-3-ил}пропаноата, полученного в ссылочном примере 12, и 1-бром-4-фтор-3-(трифторметил)бензола, по способу, аналогичному тому, как описано в примере 42.
выход: 0,9 мг
LC-МС анализ: чистота >99,9%
LC/МС ESI(+) m/z: 306 (М+H)+
Пример 45 3-{1-[2-фтор-4-(трифторметил)фенил]пирролидин-3-ил}пропановая кислота

Указанное в заголовке соединение получали из этил 3-(пирролидин-3-ил}пропаноата, полученного в ссылочном примере 12, и 1-бром-2-фтор-4-(трифторметил)бензола, по способу, аналогичному тому, как описано в примере 42.
выход: 0,8 мг
LC-МС анализ: чистота >99,9%
LC/МС ESI(+) m/z: 306 (М+H)+
Пример 46 3-{1-[3-фтор-4-(трифторметил)фенил]пирролидин-3-ил}пропановая кислота

Указанное в заголовке соединение получали из этил 3-(пирролидин-3-ил)пропаноата, полученного в ссылочном примере 12, и 1-бром-3-фтор-4-(трифторметил)бензола, по способу, аналогичному тому, как описано в примере 42.
выход: 1,2 мг
LC-МС анализ: чистота >99,9%
LC/МС ESI(+) m/z: 306 (М+H)+
Пример 47 3-{1-[3-фтор-2-(трифторметил)фенил]пирролидин-3-ил}пропановая кислота

Указанное в заголовке соединение получали из этил 3-(пирролидин-3-ил)пропаноата, полученного в ссылочном примере 12, и 1-бром-3-фтор-2-(трифторметил)бензола, по способу, аналогичному тому, как описано в примере 42.
выход: 0,7 мг
LC-МС анализ: чистота >99,9%
LC/МС ESI(+) m/z: 306 (М+H)+
Пример 48 {4-[3,5-бис(трифторметил)фенил]-2-оксопирролидин-1-ил}уксусная кислота
Указанное в заголовке соединение (0,41 г, выход 79%) получали в виде твердого вещества белого цвета из метил {4-[3,5-бис(трифторметил)фенил]-2-оксопирролидин-1-ил}ацетата, полученного в ссылочном примере 47, по способу, аналогичному тому, как описано в примере 34.
1H-ЯМР (300 МГц, DMCO-d6) δ: 2,39-2,49 (м, 1H), 2,75-2,93 (м, 1H), 3,43-3,57 (м, 1H), 3,75-3,90 (м, 2H), 3,90-4,13 (м, 2H), 7,98 (с, 1H), 8,12 (с, 2H), 12,90 (уш.с, 1H).
Пример 49 {1-[3,5-бис(трифторметил)фенил]пирролидин-3-ил}уксусная кислота

Указанное в заголовке соединение (0,33 г, выход 74%) получали в виде твердого вещества бледно-красного цвета из этил {1-[3,5-бис(трифторметил)фенил]пирролидин-3-ил}ацетата, полученного в ссылочном примере 50, по способу, аналогичному тому, как описано в примере 34.
1H-ЯМР (300 МГц, DMCO-d6) δ: 1,57-1,81 (м, 1H), 2,04-2,28 (м, 1H), 2,38-2,46 (м, 2H), 2,54-2,74 (м, 1H), 2,91-3,12 (м, 1H), 3,26-3,39 (м, 1H), 3,39-3,51 (м, 1H), 3,51-3,60 (м, 1H), 6,96 (с, 2H), 7,10 (с, 1H), 12,23 (с, 1H).
Пример 50 3-({1-[3,5-бис(трифторметил)фенил]пирролидин-3-ил}сульфанил)пропановая кислота

Указанное в заголовке соединение (0,32 г, выход 77%) получали в виде твердого вещества бледно-желтого цвета из метил 3-({1-[3,5-бис(трифторметил)фенил]пирролидин-3-ил}сульфанил)пропаноата, полученного в ссылочном примере 51, по способу, аналогичному тому, как описано в примере 34.
1H-ЯМР (300 МГц, DMCO-d6) δ: 1,83-2,01 (м, 1H), 2,23-2,45 (м, 1H), 2,56 (т, подобно 2H), 2,75-2,84 (м, 2H), 3,17-3,30 (м, 1H), 3,34-3,54 (м, 2H), 3,55-3,67 (м, 1H), 3,73-3,84 (м, 1H), 7,03 (с, 2H), 7,12 (с, 1H), 12,30 (с, 1H).
Пример 51 ({1-[4-фтор-2-(трифторметил)фенил]пирролидин-3-ил}окси)уксусная кислота

К раствору 1-[4-фтор-2-(трифторметил)фенил]пирролидин-3-ола (500 мг), полученного в ссылочном примере 52, в ДМФА (20 мл) добавляли гидрид натрия (60% в масле, 120 мг) при 60°С. После перемешивания в течение 30 минут, к реакционной смеси добавляли хлорацетат натрия (350 мг) и бромид тетрабутиламмония (97 мг), и смесь затем перемешивали при 60°С в течение 1 часа. Реакционной смеси давали охладиться до комнатной температуры, добавляли воду, и смесь концентрировали. Остаток разбавляли простым эфиром, и смесь экстрагировали 1М раствором гидроксида натрия. Водный слой промывали простым эфиром и подкисляли с помощью 6М водного раствора соляной кислоты, и смесь экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали, и остаток очищали с помощью колоночной хроматографии на силикагеле (гексан/этилацетат 80:20-0:100), получая указанное в заголовке соединение (85 мг, выход 14%) в виде твердого вещества серо-белого цвета.
1H-ЯМР (300 МГц, CDCl3) δ: 2,06-2,26 (м, 2H), 3,37-3,50 (м, 1H), 3,50-3,66 (м, 2H), 3,66-3,78 (м, 1H), 4,18 (с, 2H), 4,28-4,38 (м, 1H), 6,72-6,89 (м, 1H), 6,89-6,99 (м, 1H), 6,99-7,11 (м, 1H).
Пример 52 ({1-[4-фтор-3-(трифторметил)фенил]пирролидин-3-ил}окси)ацетат калия

Раствор 1-[4-фтор-3-(трифторметил)фенил]пирролидин-3-ола (6,0 г), полученного в ссылочном примере 53, в ТГФ (45 мл) добавляли к суспензии гидрида натрия (60% в масле, 2,9 г) в ТГФ (300 мл) при 60°С. После перемешивания в течение 30 минут к реакционной смеси добавляли хлорацетат натрия (4,21 г) и бромид тетрабутиламмония (776 мг), и смесь затем перемешивали при 60°С в течение 9 часов. Реакционной смеси давали охладиться до комнатной температуры, добавляли воду, и смесь концентрировали. Смесь подкисляли с помощью 1М водного раствора соляной кислоты, и смесь экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали, и остаток очищали с помощью колоночной хроматографии на силикагеле (гексан/этилацетат 90:10-0:100), получая коричневое масло (6,27 г). Полученное масло (4,81 г) разбавляли метанолом (100 мл), добавляли водный раствор (100 мл) бикарбоната калия (1,65 г), и смесь концентрировали. Остаток разбавляли метанолом (100 мл), добавляли водный раствор (100 мл) хлорида кальция (956 мг), и смесь концентрировали. Остаток разбавляли этилацетатом и фильтровали. Фильтрат концентрировали, получая указанное в заголовке соединение (4,95 г, выход 77%) в виде аморфного вещества желтого цвета.
1H-ЯМР (300 МГц, DMCO-d6) δ: 1,93-2,16 (м, 2H), 2,99-3,33 (м, 3H), 3,33-3,44 (м, 1H), 3,76 (с, 2H), 4,22-4,42 (м, 1H), 6,62 (дд, J=5,7, 3,0 Гц, 1H), 6,74 (дт, J=9,0, 3,4 Гц, 1H), 7,24 (т, J=10,0 Гц, 1H).
Пример 53 ({1-[2-фтор-4-(трифторметил)фенил]пирролидин-3-ил}окси)ацетат калия
К раствору 1-[2-фтор-4-(трифторметил)фенил]пирролидин-3-ола (500 мг), полученного в ссылочном примере 54, в ДМФА (20 мл) добавляли гидрид натрия (60% в масле, 120 мг) при 60°С. После перемешивания в течение 30 минут к реакционной смеси добавляли хлорацетат натрия (351 мг) и бромид тетрабутиламмония (97 мг), и смесь далее перемешивали при 60°С в течение 1 часа. Реакционной смеси давали охладиться до комнатной температуры, добавляли воду, и смесь концентрировали. Остаток разбавляли простым эфиром, и смесь экстрагировали 1М раствором гидроксида натрия. Водный слой промывали простым эфиром и подкисляли с помощью 6М водного раствора соляной кислоты, и смесь экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали, и остаток очищали с помощью колоночной хроматографии на силикагеле (гексан/этилацетат 80:20-0:100), получая желтое масло (311 мг). Полученное масло (310 мг) разбавляли метанолом (20 мл), добавляли водный раствор (20 мл) бикарбоната калия (101 мг) и смесь концентрировали, получая указанное в заголовке соединение (352 мг, выход 51%) в виде аморфного вещества желтого цвета.
1H-ЯМР (300 МГц, DMCO-d6) δ: 1,82-2,13 (м, 2H), 3,40-3,46 (м, 2H), 3,49-3,81 (м, 4H), 4,20-4,48 (м, 1H), 6,60-6,93 (м, 1H), 7,20-7,53 (м, 2H).
Пример 54 ({1-[2-фтор-5-(трифторметил)фенил]пирролидин-3-ил}окси)ацетат кальция

К раствору 1-[2-фтор-5-(трифторметил)фенил]пирролидин-3-ола (500 мг), полученного в ссылочном примере 55, в ДМФА (45 мл) добавляли гидрид натрия (60% в масле, 120 мг) при 60°С. После перемешивания в течение 30 минут к реакционной смеси добавляли хлорацетат натрия (351 мг) и бромид тетрабутиламмония (97 мг), и смесь далее перемешивали при 60°С в течение 1 часа. Реакционной смеси давали охладиться до комнатной температуры, добавляли воду, и смесь концентрировали. Остаток разбавляли эфиром, и смесь экстрагировали 1М раствором гидроксида натрия. Водный слой промывали простым эфиром и подкисляли с помощью 6М водного раствора соляной кислоты, и смесь экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали, и остаток очищали с помощью колоночной хроматографии на силикагеле (гексан/этилацетат 80:20-0:100), получая желтое масло (105 мг). Полученное масло (100 мг) разбавляли метанолом (10 мл), добавляли водный раствор (10 мл) бикарбоната калия (33 мг), и смесь концентрировали. Остаток разбавляли метанолом (10 мл), добавляли водный раствор (10 мл) хлорида кальция (18 мг), и смесь концентрировали. Остаток разбавляли этилацетатом и фильтровали. Фильтрат концентрировали, получая указанное в заголовке соединение (69 мг, выход 11%) в виде аморфного вещества желтого цвета.
1H-ЯМР (300 МГц, DMCO-d6) δ: 1,81-2,11 (м, 2H), 3,06-3,19 (м, 2H), 3,25-3,37 (м, 1H), 3,37-3,50 (м, 1H), 3,66 (с, 2H), 4,23-4,37 (м, 1H), 7,00-7,31 (м, 1H), 7,31-7,54 (м, 2H).
Пример 55 ({1-[2-фтор-3-(трифторметил)фенил]пирролидин-3-ил}окси)ацетат калия
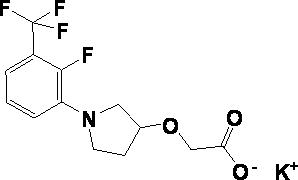
К раствору 1-[2-фтор-3-(трифторметил)фенил]пирролидин-3-ола (500 мг), полученного в ссылочном примере 56, в ДМФА (20 мл) добавляли гидрид натрия (60% в масле, 120 мг) при 60°С. После перемешивания в течение 30 минут, к реакционной смеси добавляли хлорацетат натрия (351 мг) и бромид тетрабутиламмония (97 мг), и смесь далее перемешивали при 60°С в течение 1 часа. Реакционной смеси давали охладиться до комнатной температуры, добавляли воду, и смесь концентрировали. Остаток разбавляли эфиром, и смесь экстрагировали 1М раствором гидроксида натрия. Водный слой промывали простым эфиром и подкисляли с помощью 6М водного раствора соляной кислоты, и смесь экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали, и остаток очищали с помощью колоночной хроматографии на силикагеле (гексан/этилацетат 80:20-0:100), получая желтое масло (130 мг). Полученное масло (80 мг) разбавляли метанолом (10 мл), добавляли водный раствор (10 мл) бикарбоната калия (26 мг) и смесь концентрировали, получая указанное в заголовке соединение (100 мг, выход 14%) в виде аморфного вещества желтого цвета.
1H-ЯМР (300 МГц, DMCO-d6) δ: 1,80-2,08 (м, 2H), 3,35-3,39 (м, 2H), 3,40-3,62 (м, 4H), 4,24-4,38 (м, 1H), 6,79-6,95 (м, 1H), 6,95-7,05 (м, 1H), 7,06-7,23 (м, 1H).
Пример 56 ({1-[2,4-бис(трифторметил)фенил]пирролидин-3-ил}окси)ацетат кальция

К раствору 1-[2,4-бис(трифторметил)фенил]пирролидин-3-ола (500 мг), полученного в ссылочном примере 57, в ДМФА (20 мл) добавляли гидрид натрия (60% в масле, 100 мг) при 60°С. После перемешивания в течение 30 минут к реакционной смеси добавляли хлорацетат натрия (292 мг) и бромид тетрабутиламмония (81 мг), и смесь далее перемешивали при 60°С в течение 1 часа. Реакционной смеси давали охладиться до комнатной температуры, добавляли воду, и смесь концентрировали. Остаток разбавляли простым эфиром, и смесь экстрагировали 1М раствором гидроксида натрия. Водный слой промывали простым эфиром и подкисляли с помощью 6М водного раствора соляной кислоты, и смесь экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали, и остаток очищали с помощью колоночной хроматографии на силикагеле (гексан/этилацетат 80:20-0:100), получая желтое масло (162 мг). Полученное масло (160 мг) разбавляли метанолом (10 мл), добавляли водный раствор (10 мл) бикарбоната калия (45 мг) и смесь концентрировали. Остаток разбавляли метанолом (10 мл), добавляли водный раствор (10 мл) хлорида кальция (25 мг), и смесь концентрировали. Остаток разбавляли этилацетатом и фильтровали. Фильтрат концентрировали, получая указанное в заголовке соединение (188 мг, выход 30%) в виде аморфного вещества желтого цвета.
1H-ЯМР (300 МГц, DMCO-d6) δ: 1,95-2,04 (м, 1H), 2,04-2,17 (м, 1H), 3,15-3,45 (м, 4H), 3,45-3,69 (м, 2H), 4,34 (уш.с, 1H), 7,09 (д, J=9,0 Гц, 1H), 7,70 (д, J=8,7 Гц, 1H), 7,77 (с, 1H).
Пример 57 ({1-[3-(метиламино)-5-(трифторметил)фенил]пирролидин-3-ил}окси)уксусная кислота

К раствору N-[3-(3-гидроксипирролидин-1-ил)-5-(трифторметил)фенил]-N-метилацетамида (1,0 г), полученного в ссылочном примере 60, в ДМФА (33 мл) добавляли гидрид натрия (60% в масле, 198 мг) при 60°С. После перемешивания в течение 30 минут к реакционной смеси добавляли хлорацетат натрия (578 мг) и бромид тетрабутиламмония (160 мг), и смесь далее перемешивали при 60°С в течение 1 часа. Реакционной смеси давали охладиться до комнатной температуры, добавляли воду, и смесь концентрировали. Остаток разбавляли простым эфиром, и смесь экстрагировали 1М раствором гидроксида натрия. Водный слой промывали простым эфиром и подкисляли с помощью 6М водного раствора соляной кислоты, и смесь экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали, и остаток очищали с помощью колоночной хроматографии на силикагеле (гексан/этилацетат 80:20-0:100), получая желтое масло (17 мг). Полученное масло разбавляли ТГФ (тетргидрофураном) (1 мл), добавляли водный раствор (10 мл) гидроксида лития (1 мл), и смесь перемешивали при комнатной температуре в течение 5 часов. Реакционную смесь подкисляли с помощью 1М раствора соляной кислоты, и смесь экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали, получая указанное в заголовке соединение (7,7 мг, выход 0,7%) в виде масла бледно-желтого цвета.
1H-ЯМР (300 МГц, CDCl3) δ: 2,13-2,27 (м, 2H), 2,85 (с, 3H), 3,31-3,62 (м, 5H), 4,16 (с, 2H), 4,31-4,41 (м, 1H), 5,87 (с, 1H), 6,16 (с, 1H), 6,22 (с, 1H).
Пример 58 [(1-{3-[(1-оксидотиоморфолин-4-ил)карбонил]-5-(трифторметил)фенил}пирролидин-3-ил)окси]уксусная кислота
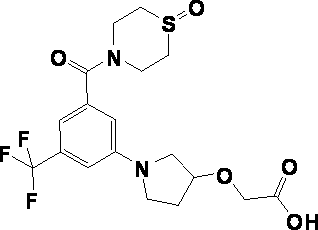
К раствору 1-{3-[(1-оксидотиоморфолин-4-ил)карбонил]-5-(трифторметил)фенил}пирролидин-3-ола (700 мг), полученного в ссылочном примере 64, в ДМФА (35 мл) добавляли гидрид натрия (60% в масле, 112 мг) при 60°С. После перемешивания в течение 30 минут, к реакционной смеси добавляли хлорацетат натрия (325 мг) и бромид тетрабутиламмония (90 мг), и смесь далее перемешивали при 60°С в течение 1 часа. Реакционной смеси давали охладиться до комнатной температуры, добавляли воду, и смесь концентрировали. Остаток разбавляли простым эфиром, и смесь экстрагировали 1М раствором гидроксида натрия. Водный слой промывали простым эфиром и подкисляли с помощью 6М водного раствора соляной кислоты, и смесь экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали, и остаток очищали с помощью колоночной хроматографии на силикагеле (этилацетат/метанол 90:10-50:50), получая указанное в заголовке соединение (67 мг, выход 8%) в виде аморфного вещества желтого цвета.
1H-ЯМР (300 МГц, CDCl3) δ: 2,08-2,22 (м, 1H), 2,22-2,37 (м, 1H), 2,97 (уш., с, 4H), 3,32-3,61 (м, 4H), 3,78 (уш.с, 1H), 4,11 (уш.с, 2H), 4,16 (д, J=1,9 Гц, 2H), 4,32-4,40 (м, 1H), 4,56 (уш.с, 1H), 6,68 (с, 1H), 6,78 (с, 1H), 6,87 (с, 1H).
Пример 59 [(1-{3-[метил(метилсульфонил)амино]-5-(трифторметил)фенил}пирролидин-3-ил)окси]ацетат кальция

К раствору N-[3-(3-гидроксипирролидин-1-ил)-5-(трифторметил)фенил]-N-метилметансульфонамида (1,0 г), полученного в ссылочном примере 67, в ДМФА (30 мл) добавляли гидрид натрия (60% в масле, 177 мг) при 50°С. После перемешивания в течение 30 минут к реакционной смеси добавляли хлорацетат натрия (516 мг) и бромид тетрабутиламмония (143 мг), и смесь далее перемешивали при 50°С в течение 3 часов. Реакционной смеси давали охладиться до комнатной температуры, добавляли воду, и смесь концентрировали. Остаток разбавляли простым эфиром, и смесь экстрагировали 1М раствором гидроксида натрия. Водный слой промывали простым эфиром и подкисляли с помощью 6М водного раствора соляной кислоты, и смесь экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали, и остаток очищали с помощью колоночной хроматографии на силикагеле (гексан/этилацетат 80:20-0:100). Остаток очищали с помощью препаративной ВЭЖХ (оборудование: Высокопроизводительная очистительная система Gilson Inc.,; колонка: YMC Combiprep ODS-A, S-5 мкм, 50×20 мм; растворитель: РАСТВОР А; 0,1% трифторуксусная кислота, содержащая воду, РАСТВОР В; 0,1% трифторуксусная кислота, содержащая ацетонитрил; градиентный цикл: 0,00 мин (РАСТВОР А/РАСТВОР В=90/10), 1,00 мин (РАСТВОР А/РАСТВОР В=90/10), 4,20 мин (РАСТВОР А/РАСТВОР В=10/90), 5,40 мин (РАСТВОР А/РАСТВОР В=10/90), 5,50 мин (РАСТВОР А/РАСТВОР В=90/10), 5,60 мин (РАСТВОР А/РАСТВОР В=90/10); скорость потока: 25 мл/мин; метод детекции: UV 220 нм), получая желтое масло (108 мг). Полученное масло (61 мг) разбавляли метанолом (5 мл), добавляли водный раствор (5 мл) бикарбоната калия (16 мг), и смесь концентрировали. Остаток разбавляли метанолом (5 мл), добавляли водный раствор (5 мл) хлорида кальция (9 мг), и смесь концентрировали. Остаток разбавляли этилацетатом и фильтровали. Фильтрат концентрировали, получая указанное в заголовке соединение (71 мг, выход 6%) в виде аморфного вещества желтого цвета.
1H-ЯМР (300 МГц, DMCO-d6) δ: 1,99-2,15 (м, 2H), 2,97 (с, 3H), 3,25 (с, 3H), 3,27-3,37 (м, 3H), 3,37-3,49 (м, 1H), 3,68 (с, 2H), 4,33-4,43 (м, 1H), 6,62-6,69 (м, 1H), 6,74 (с, 1H), 6,89 (с, 1H).
Пример 60 ({1-[3-бром-5-(трифторметил)фенил]пирролидин-3-ил}окси)уксусная кислота

К раствору 1-[3-бром-5-(трифторметил)фенил]пирролидин-3-ола (500 мг), полученного в ссылочном примере 68, в ДМФА (16 мл), добавляли гидрид натрия (60% в масле, 97 мг) при 60°С. После перемешивания в течение 30 минут к реакционной смеси добавляли хлорацетат натрия (282 мг) и бромид тетрабутиламмония (78 мг), и смесь перемешивали при 60°С в течение 1 часа. Реакционной смеси давали охладиться до комнатной температуры, добавляли воду, и смесь концентрировали. Остаток разбавляли эфиром, и смесь экстрагировали 1М раствором гидроксида натрия. Водный слой промывали простым эфиром и подкисляли с помощью 6М водного раствора соляной кислоты, и смесь экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали, и остаток очищали с помощью колоночной хроматографии на силикагеле (гексан/этилацетат 80:20-0:100). Остаток очищали с помощью препаративной ВЭЖХ (оборудование: Высокопроизводительная очистительная система Gilson Inc.,; колонка: YMC Combiprep ODS-A, S-5 мкм, 50×20 мм; растворитель: РАСТВОР А; 0,1% трифторуксусная кислота, содержащая воду, РАСТВОР В; 0,1% трифторуксусная кислота, содержащая ацетонитрил; градиентный цикл: 0,00 мин (РАСТВОР А/РАСТВОР В=90/10), 1,00 мин (РАСТВОР А/РАСТВОР В=90/10), 4,20 мин (РАСТВОР А/РАСТВОР В=10/90), 5,40 мин (РАСТВОР А/РАСТВОР В=10/90), 5,50 мин (РАСТВОР А/РАСТВОР В=90/10) и 5,60 мин (РАСТВОР А/РАСТВОР В=90/10); скорость потока: 25 мл/мин; метод детекции: UV 220 нм), получая указанное в заголовке соединение (27,6 мг, выход 5%) в виде масла желтого цвета.
1H-ЯМР (300 МГц, CDCl3) δ: 2,08-2,33 (м, 2H), 3,28-3,62 (м, 4H), 4,18 (с, 2H), 4,32-4,45 (м, 1H), 6,64 (с, 1H), 6,80 (с, 1H), 7,03 (с, 1H).
Пример 61 ({1-[3-метокси-5-(трифторметил)фенил]пирролидин-3-ил}окси)уксусная кислота
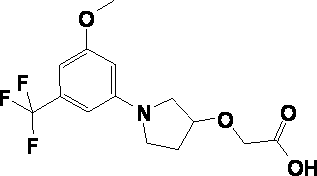
К раствору 1-[3-метокси-5-(трифторметил)фенил]пирролидин-3-ола (1,68 г), полученного в ссылочном примере 69, в ДМФА (129 мл) добавляли гидрид натрия (60% в масле, 386 мг) при 60°С. После перемешивания в течение 30 минут к реакционной смеси добавляли хлорацетат натрия (1,12 г) и бромид тетрабутиламмония (311 мг), и смесь перемешивали при 60°С в течение 3 часов. Реакционной смеси давали охладиться до комнатной температуры, добавляли воду, и смесь концентрировали. Остаток разбавляли простым эфиром, и смесь экстрагировали 1М раствором гидроксида натрия. Водный слой промывали простым эфиром и подкисляли с помощью 6М водного раствора соляной кислоты, и смесь экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали, и остаток очищали с помощью колоночной хроматографии на силикагеле (гексан/этилацетат 50:50-0:100), получая указанное в заголовке соединение (1,86 г, выход 91%) в виде твердого вещества белого цвета.
1H-ЯМР (300 МГц, CDCl3) δ: 2,08-2,35 (м, 2H), 3,32-3,59 (м, 4H), 3,82 (с, 3H), 4,19 (с, 2H), 4,32-4,41 (м, 1H), 6,16-6,24 (м, 1H), 6,39 (с, 1H), 6,48 (с, 1H).
Пример 62 ({1-[3-(тиоморфолин-4-илкарбонил)-5-(трифторметил)фенил]пирролидин-3-ил}окси)уксусная кислота

К раствору 1-[3-(тиоморфолин-4-илкарбонил)-5-(трифторметил)фенил]пирролидин-3-ола (1,0 г), полученного в ссылочном примере 62, в ДМФА (37 мл) добавляли гидрид натрия (60% в масле, 166 мг) при 60°С. После перемешивания в течение 30 минут к реакционной смеси добавляли хлорацетат натрия (484 мг) и бромид тетрабутиламмония (134 мг), и смесь перемешивали при 60°С в течение 2 часов, а затем при 80°С в течение 1 часа. Реакционной смеси давали охладиться до комнатной температуры, добавляли воду, и смесь концентрировали. Смесь подкисляли с помощью 1М водного раствора соляной кислоты, и смесь экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали, и остаток очищали с помощью колоночной хроматографии на силикагеле (гексан/этилацетат 50:50-0:100), получая указанное в заголовке соединение (519 мг, выход 45%) в виде аморфного вещества желтого цвета.
1H-ЯМР (300 МГц, CDCl3) δ: 2,08-2,39 (м, 2H), 2,58 (уш.с, 2H), 2,74 (уш.с, 2H), 3,34-3,59 (м, 4H), 3,70 (уш.с, 1H), 4,09 (уш.с, 2H), 4,16 (с, 2H), 4,30-4,41 (м, 1H), 4,66 (уш.с, 1H), 6,68 (с, 1H), 6,75 (с, 1H), 6,84 (с, 1H).
Пример 63 ({1-[3-хлор-5-(трифторметил)фенил]пирролидин-3-ил}окси)уксусная кислота
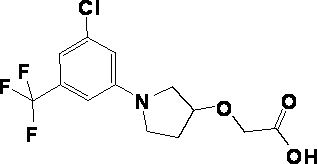
К раствору 1-[3-хлор-5-(трифторметил)фенил]пирролидин-3-ола (1,0 г), полученного в ссылочном примере 70, в ДМФА (38 мл) добавляли гидрид натрия (60% в масле, 226 мг) при 60°С. После перемешивания в течение 30 минут к реакционной смеси добавляли хлорацетат натрия (657 мг) и бромид тетрабутиламмония (123 мг), и смесь перемешивали при 60°С в течение 2 часов, а затем при 80°С в течение 2 часов. Реакционной смеси давали охладиться до комнатной температуры, добавляли воду, и смесь концентрировали. Остаток разбавляли простым эфиром, и смесь экстрагировали 1М раствором гидроксида натрия. Водный слой промывали простым эфиром и подкисляли с помощью 6М водного раствора соляной кислоты, и смесь экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали, и остаток очищали с помощью колоночной хроматографии на силикагеле (гексан/этилацетат 80:20-0:100), получая указанное в заголовке соединение (77,2 мг, выход 6%) в виде масла коричневого цвета.
1H-ЯМР (300 МГц, CDCl3) δ: 2,06-2,38 (м, 2H), 3,26-3,64 (м, 4H), 4,18 (с, 2H), 4,30-4,42 (м, 1H), 6,60 (с, 1H), 6,64 (с, 1H), 6,89 (с, 1H).
Пример 64 ({1-[4-фтор-3-(трифторметил)фенил]пирролидин-3-ил}окси)уксусная кислота

Раствор 1-[4-фтор-3-(трифторметил)фенил]пирролидин-3-ола (3,29 г), полученного в ссылочном примере 53, в ТГФ (30 мл) при 60°С добавляли к суспензии гидрида натрия (60% в масле, 1,58 г) в ТГФ (160 мл). После перемешивания в течение 30 минут к реакционной смеси добавляли хлорацетат натрия (2,31 г) и бромид тетрабутиламмония (427 мг), и смесь затем перемешивали при 60°С в течение 2 часов. Реакционной смеси давали охладиться до комнатной температуры, добавляли воду, и смесь концентрировали. Смесь подкисляли с помощью 1М водного раствора соляной кислоты, и смесь экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали, и остаток очищали с помощью колоночной хроматографии на силикагеле (гексан/этилацетат 90:10-0:100), получая указанное в заголовке соединение (3,18 г, выход 78%) в виде твердого вещества серо-белого цвета.
1H-ЯМР (300 МГц, CDCl3) δ: 2,08-2,33 (м, 2H), 3,26-3,60 (м, 4H), 4,19 (с, 2H), 4,32-4,43 (м, 1H), 6,55-6,71 (м, 2H), 7,05 (т, J=9,4 Гц, 1H).
Пример 65 ({1-[3-фтор-5-(трифторметил)фенил]пирролидин-3-ил}окси)уксусная кислота

Раствор 1-[3-фтор-5-(трифторметил)фенил]пирролидин-3-ола (2,46 г), полученного в ссылочном примере 71, в ТГФ (30 мл) при 60°С добавляли к суспензии гидрида натрия (60% в масле, 1,18 г) в ТГФ (110 мл). После перемешивания в течение 30 минут к реакционной смеси добавляли хлорацетат натрия (1,72 г) и бромид тетрабутиламмония (320 мг), и смесь затем перемешивали при 60°С в течение 2 часов. Реакционной смеси давали охладиться до комнатной температуры, добавляли воду, и смесь концентрировали. Смесь подкисляли с помощью 1М водного раствора соляной кислоты, и смесь экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали, и остаток очищали с помощью колоночной хроматографии на силикагеле (гексан/этилацетат 80:20-0:100), получая указанное в заголовке соединение (2,83 г, выход 93%) в виде твердого вещества белого цвета.
1H-ЯМР (300 МГц, CDCl3) δ: 2,08-2,36 (м, 2H), 3,33-3,60 (м, 4H), 4,19 (с, 2H), 4,38 (тт, J=4,8, 2,4 Гц, 1H), 6,36 (дт, J=11,3, 2,3 Гц, 1H), 6,52 (с, 1H), 6,62 (д, J=8,7 Гц, 1H).
Пример 66 ({(3S)-1-[4-хлор-2-(трифторметил)фенил]пирролидин-3-ил}окси)уксусная кислота

Раствор (3S)-1-[4-хлор-2-(трифторметил)фенил]пирролидин-3-ола (2,66 г), полученного в ссылочном примере 72, в ТГФ (23 мл) при 60°С добавляли к суспензии гидрида натрия (60% в масле, 1,2 г) в ТГФ (120 мл). После перемешивания в течение 30 минут к реакционной смеси добавляли хлорацетат натрия (1,75 г) и бромид тетрабутиламмония (323 мг), и смесь затем перемешивали при 60°С в течение 7 часов. Реакционной смеси давали охладиться до комнатной температуры, добавляли воду, и смесь концентрировали. Смесь подкисляли с помощью 1М водного раствора соляной кислоты, и смесь экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали, и остаток очищали с помощью колоночной хроматографии на силикагеле (гексан/этилацетат 80:20-0:100), получая указанное в заголовке соединение (1,38 г, выход 43%) в виде твердого вещества желтого цвета.
1H-ЯМР (300 МГц, CDCl3) δ: 2,05-2,24 (м, 2H), 3,18-3,40 (м, 2H), 3,44-3,65 (м, 2H), 4,05-4,25 (м, 2H), 4,27-4,37 (м, 1H), 6,97 (д, J=8,7 Гц, 1H), 7,35 (дд, J=8,9, 2,4 Гц, 1H), 7,56 (д, J=2,6 Гц, 1H).
Пример 67 3-{5-[3-фтор-5-(трифторметил)фенил]тетрагидрофуран-2-ил}пропановая кислота

Раствор этил 3-{5-[3-фтор-5-(трифторметил)фенил]тетрагидрофуран-2-ил}пропаноата (0,55 г, 1,65 ммоль), полученного в ссылочном примере 77, и гидроксида лития (0,20 г, 8,23 ммоль) в смеси этанол/вода (20 мл/5 мл) перемешивали при комнатной температуре в течение 4 часов. Реакционный раствор доводили до величины рН 2 с помощью 1М раствора соляной кислоты и концентрировали при пониженном давлении, и остаток распределяли между этилацетатом и водой. Органический слой промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали, и фильтрат концентрировали при пониженном давлении. Полученное твердое вещество белого цвета перекристаллизовывали из гексана, получая указанное в заголовке соединение (0,32 г, выход 63%) в виде твердого вещества белого цвета.
1H-ЯМР (300 МГц, CDCl3) δ: 1,66-1,84 (м, 2H), 1,96-2,03 (м, 2H), 2,07-2,53 (м, 2H), 2,57 (кв, J=7,2 Гц, 2H), 4,04-4,13 (м, 1H), 4,91 (т, J=7,2 Гц, 1H), 7,19-7,26 (м, 2H), 7,35 (с, 1H).
Пример 68 3-{5-[3-метокси-5-(трифторметил)фенил]тетрагидрофуран-2-ил}пропановая кислота

Раствор этил 3-{5-[3-метокси-5-(трифторметил)фенил]тетрагидрофуран-2-ил}пропаноата (0,47 г, 1,36 ммоль), полученного в ссылочном примере 79, и гидроксида лития (0,17 г, 4,08 ммоль) в смеси этанол/вода (20 мл/5 мл) перемешивали при комнатной температуре в течение 2 часов. Реакционный раствор концентрировали, и остаток распределяли между этилацетатом и водой. Водный слой доводили до величины рН 2 с помощью 2М раствора соляной кислоты, и смесь экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом натрия, фильтровали, и фильтрат концентрировали при пониженном давлении, получая указанное в заголовке соединение (0,34 г, выход 79%) в виде твердого вещества белого цвета.
1H-ЯМР (300 МГц, CDCl3) δ: 1,62-1,69 (м, 1H), 1,75-1,86 (м, 1H), 1,98 (т, J=8,7 Гц, 2H), 2,07-2,17 (м, 1H), 2,31-2,38 (м, 1H), 2,53-2,61 (м, 2H), 3,85 (с, 3H), 4,06-4,10 (м, 1H), 4,90 (т, J=7,2 Гц, 1H), 7,01 (с, 1H), 7,08 (с, 1H), 7,15 (с, 1H).
Пример 69 3-{5-[4-метокси-3-(трифторметил)фенил]тетрагидрофуран-2-ил}пропановая кислота

Раствор этил 3-{5-[4-метокси-3-(трифторметил)фенил]тетрагидрофуран-2-ил}пропаноата (0,40 г, 1,16 ммоль), полученного в ссылочном примере 81, и гидроксида лития (0,15 г, 3,47 ммоль) в смеси этанол/вода (20 мл/5 мл) перемешивали при комнатной температуре в течение 2 часов. Реакционный раствор концентрировали, и остаток распределяли между этилацетатом и водой. Водный слой доводили до величины рН 2 с помощью 2М раствора соляной кислоты, и смесь экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом натрия, фильтровали, и фильтрат концентрировали при пониженном давлении, получая указанное в заголовке соединение (0,25 г, выход 88%) в виде твердого вещества белого цвета.
1H-ЯМР (300 МГц, CDCl3) δ: 1,64-1,81 (м, 2H), 1,98 (кв, J=7,2 Гц, 2H), 2,11-2,17 (м, 1H), 2,24-2,33 (м, 1H), 2,51-2,59 (м, 2H), 3,89 (с, 3H), 4,04-4,14 (м, 1H), 4,84 (т, J=7,2 Гц, 1H), 6,97 (д, J=8,4 Гц, 1H), 7,46-7,52 (м, 2H).
Пример 70 3-{5-[2-фтор-3-(трифторметил)фенил]тетрагидрофуран-2-ил}пропановая кислота

Раствор этил 3-{5-[2-фтор-3-(трифторметил)фенил]тетрагидрофуран-2-ил}пропаноата (0,55 г, 1,64 ммоль), полученного в ссылочном примере 83, и гидроксида лития (0,21 г, 4,93 ммоль) в смеси этанол/вода (20 мл/5 мл) перемешивали при комнатной температуре в течение 2 часов. Реакционный раствор концентрировали, и остаток распределяли между этилацетатом и водой. Водный слой доводили до величины рН 2 с помощью 2М раствора соляной кислоты, и смесь экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом натрия, фильтровали, и фильтрат концентрировали при пониженном давлении, получая указанное в заголовке соединение (0,40 г, выход 79%) в виде твердого вещества белого цвета.
1H-ЯМР (300 МГц, CDCl3) δ: 1,61-1,68 (м, 1H), 1,70-1,82 (м, 1H), 1,98-2,05 (м, 2H), 2,09-2,18 (м, 1H), 2,40-2,55 (м, 1H), 2,57-2,68 (м, 2H), 4,04-4,11 (м, 1H), 5,16 (т, J=7,8 Гц, 1H), 7,20-7,25 (м, 1H), 7,48-7,53 (м, 1H), 7,69-7,74 (м, 1H).
Пример 71 3-{5-[3-(трифторметил)фенил]тетрагидрофуран-2-ил}пропановая кислота

Раствор этил 3-{5-[3-(трифторметил)фенил]тетрагидрофуран-2-ил}пропаноата (0,73 г, 2,33 ммоль), полученного в ссылочном примере 85, и гидроксида лития (0,29 г, 7,00 ммоль) в смеси этанол/вода (20 мл/5 мл) перемешивали при комнатной температуре в течение 2 часов. Реакционный раствор концентрировали, и остаток распределяли между этилацетатом и водой. Водный слой доводили до величины рН 2 с помощью 2М раствора соляной кислоты, и смесь экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом натрия, фильтровали, и фильтрат концентрировали при пониженном давлении, получая указанное в заголовке соединение (0,66 г, выход 98%) в виде твердого вещества белого цвета.
1H-ЯМР (300 МГц, CDCl3) δ: 1,62-1,74 (м, 1H), 1,80-1,86 (м, 1H), 1,96-2,03 (м, 2H), 2,13-2,19 (м, 1H), 2,30-2,41 (м, 1H), 2,53-2,61 (м, 2H), 4,07-4,14 (м, 1H), 4,92 (т, J=7,2 Гц, 1H), 7,44-7,54 (м, 3H), 7,58 (с, 1H).
Пример 72 3-{5-[2-циано-3-(трифторметил)фенил]тетрагидрофуран-2-ил}пропановая кислота

Раствор этил 3-{5-[2-циано-3-(трифторметил)фенил]тетрагидрофуран-2-ил}пропаноата (0,34 г, 1,00 ммоль), полученного в ссылочном примере 87, и гидроксида лития (0,13 г, 3,00 ммоль) в смеси этанол/вода (15 мл/4 мл) перемешивали при комнатной температуре в течение 2 часов. Реакционный раствор концентрировали, и остаток распределяли между этилацетатом и водой. Водный слой доводили до величины рН 2 с помощью 2М раствора соляной кислоты, и смесь экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом натрия, фильтровали, и фильтрат концентрировали при пониженном давлении, получая указанное в заголовке соединение (0,29 г, выход 91%) в виде твердого вещества белого цвета.
1H-ЯМР (300 МГц, CDCl3) δ: 1,64-1,81 (м, 2H), 2,02-2,09 (м, 2H), 2,16-2,24 (м, 1H), 2,50-2,70 (м, 3H), 4,10-4,15 (м, 1H), 5,29 (т, J=7,2 Гц, 1H), 7,69-7,71 (м, 2H), 7,88-7,91 (м, 1H).
Пример 73 3-{5-[4-фтор-3-(трифторметил)фенил]тетрагидрофуран-2-ил}пропановая кислота
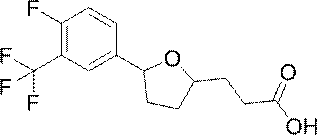
Раствор этил 3-{5-[4-фтор-3-(трифторметил)фенил]тетрагидрофуран-2-ил}пропаноата (0,52 г, 1,58 ммоль), полученного в ссылочном примере 89, и гидроксида лития (0,20 г, 4,76 ммоль) в смеси этанол/вода (20 мл/5 мл) перемешивали при комнатной температуре в течение 2 часов. Реакционный раствор концентрировали, и остаток распределяли между этилацетатом и водой. Водный слой доводили до величины рН 2 с помощью 2М раствора соляной кислоты, и смесь экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом натрия, фильтровали, и фильтрат концентрировали при пониженном давлении, получая указанное в заголовке соединение (0,42 г, выход 88%) в виде масла желтого цвета.
1H-ЯМР (300 МГц, CDCl3) δ: 1,60-1,80 (м, 2H), 1,95-2,02 (м, 2H), 2,09-2,18 (м, 1H), 2,29-2,38 (м, 1H), 2,46-2,59 (м, 2H), 4,04-4,14 (м, 1H), 4,87 (т, J=7,2 Гц, 1H), 7,12-7,18 (м, 1H), 7,50-7,56 (м, 2H).
Пример 74 3-{5-[2-метокси-3-(трифторметил)фенил]тетрагидрофуран-2-ил}пропановая кислота

Раствор этил 3-{5-[2-метокси-3-(трифторметил)фенил]тетрагидрофуран-2-ил}пропаноата (0,34 г, 0,98 ммоль), полученного в ссылочном примере 92, и гидроксида лития (0,12 г, 2,94 ммоль) в смеси этанол/вода (20 мл/5 мл) перемешивали при комнатной температуре в течение 2 часов. Реакционный раствор концентрировали, и остаток распределяли между этилацетатом и водой. Водный слой доводили до величины рН 2 с помощью 2М раствора соляной кислоты, и смесь экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом натрия, фильтровали, и фильтрат концентрировали при пониженном давлении, получая указанное в заголовке соединение (0,42 г, выход 88%) в виде масла желтого цвета.
1H-ЯМР (300 МГц, CDCl3) δ: 1,64-1,83 (м, 2H), 1,99-2,06 (м, 2H), 2,10-2,17 (м, 1H), 2,37-2,48 (м, 1H), 2,54-2,65 (м, 2H), 3,87 (с, 3H), 4,03-4,11 (м, 1H), 5,20 (т, J=7,2 Гц, 1H), 7,22 (т, J=7,8 Гц, 1H), 7,51 (дд, J=7,8, 1,5 Гц, 1H), 7,70 (дд, J=7,8, 1,5 Гц, 1H).
Пример 75 3-{5-[3-метокси-2-(трифторметил)фенил]тетрагидрофуран-2-ил}пропановая кислота

Раствор этил 3-{5-[3-метокси-2-(трифторметил)фенил]тетрагидрофуран-2-ил}пропаноата (0,21 г, 0,61 ммоль), полученного в ссылочном примере 93, и гидроксида лития (76 мг, 1,82 ммоль) в смеси этанол/вода (15 мл/5 мл) перемешивали при комнатной температуре в течение 2 часов. Реакционный раствор концентрировали, и остаток распределяли между этилацетатом и водой. Водный слой доводили до величины рН 2 с помощью 2М раствора соляной кислоты, и смесь экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом натрия, фильтровали, и фильтрат концентрировали при пониженном давлении, получая указанное в заголовке соединение (0,19 г, выход 97%) в виде твердого вещества желтого цвета.
1Н ЯМР (300 МГц, CDCl3): δ 1,52-1,60 (м, 1H), 1,66-1,77 (м, 1H), 2,02-2,11 (м, 3H), 2,41-2,53 (м, 1H), 2,56-2,65 (м, 2H), 3,88 (с, 3H), 3,98-4,07 (м, 1Н), 5,30-5,36 (м, 1Н), 6,92 (д, J=7,8 Гц, 1Н), 7,39-7,49 (м, 2Н).
Пример 76 трометаминовая соль 3-{(2R,5S)-5-[3,5-бис(трифторметил)фенил]тетрагидрофуран-2-ил}пропаноата

К раствору 3-{(2R,5S)-5-[3,5-бис(трифторметил)фенил]тетрагидрофуран-2-ил}пропионовой кислоты (3,00 г, 8,42 ммоль), полученного в ссылочном примере 36, в метаноле (60 мл) добавляли раствор трометамина (1,02 г, 8,42 ммоль) в воде (5 мл), и смесь перемешивали при комнатной температуре в течение 2 часов. Раствор концентрировали при пониженном давлении, и полученное твердое вещество растворяли в смеси этилацетат/толуол (10:1, 50 мл) и концентрировали при пониженном давлении. Полученное твердое вещество деструктурировали в диизопропиловом эфире, получая указанное в заголовке соединение (3,79 г, выход 94%) в виде твердого вещества белого цвета. Т.пл. 110°C. Анализ. Вычислено для C19H25NO6F6: C, 47,80; H, 5,28; N 2,93. Найдено: C, 47,61; H, 5, 47; N 2,99.
Пример 77 ({5-[3,5-бис(трифторметил)фенил]тетрагидрофуран-2-ил}метокси)уксусная кислота

Раствор этил ({5-[3,5-бис(трифторметил)фенил]тетрагидрофуран-2-ил}метокси)ацетата (0,28 г, 0,70 ммоль), полученного в ссылочном примере 97, и 1М раствор гидроксида лития (2,1 мл, 2,1 ммоль) в этаноле (10 мл) перемешивали при комнатной температуре в течение 3 часов. Реакционный раствор доводили до величины рН 3 с помощью 1М раствора соляной кислоты и концентрировали при пониженном давлении. Остаток распределяли между этилацетатом и водой, и органический слой промывали насыщенным солевым раствором, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении, получая указанное в заголовке соединение (0,23 г, выход 88%) в виде твердого вещества белого цвета.
1H-ЯМР (300 МГц, CDCl3) δ: 1,77-2,01 (м, 2H), 2,08-2,22 (м, 1H), 2,31-2,57 (м, 1H), 3,64-3,78 (м, 1H), 3,78-3,89 (м, 1H), 4,24 (с, 2H), 4,28-4,43 (м, 1H), 5,04 (т, J=7,0 Гц, 1H), 7,78 (с, 1H), 7,84 (с, 2H).
Пример 78 {5-[3,5-бис(трифторметил)фенил]тетрагидрофуран-2-ил}уксусная кислота

Раствор {5-[3,5-бис(трифторметил)фенил]фуран-2-ил}уксусной кислоты (1,23 г, 3,64 ммоль), полученного в ссылочном примере 100, и палладия (10% на угле, содержащего воду (50%), 0,36 г) в этаноле (30 мл) перемешивали в атмосфере водорода при комнатной температуре в течение 16 часов. После подтверждения завершения реакции с помощью ТСХ, реакционный раствор фильтровали и концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле (гексан/этилацетат 50:50), и полученное твердое вещество промывали гексаном, получая указанное в заголовке соединение (1,01 г, выход 81%) в виде твердого вещества бледно-желтого цвета.
1H-ЯМР (300 МГц, DMCO-d6) δ: 1,60-1,85 (м, 2H), 2,05-2,25 (м, 1H), 2,34-2,48 (м, 1H), 2,53-2,63 (м, 2H), 4,30-4,48 (м, 1H), 5,04 (т, J=7,0 Гц, 1H), 7,99 (с, 1H), 8,03 (с, 2H), 12,27 (уш.с, 1H).
Экспериментальный пример 1
Действие соединения настоящего изобретения по ингибированию связывания между RBP4 и ретинолом и TTR оценивали с использованием системы Ретинол-RBP4-TTR ELISA (ELISA человеческий тип), показанной ниже.
1A: Клонирование человеческого RBP4 гена и человеческого TTR гена
Человеческий RBP4 ген клонировали с помощью ПЦР реакции с использованием человеческой Universal cDNA (кДНК) (Clontech, QUICK-Clone cDNA) в качестве матрицы и следующего комплекта праймера.
RBPU:
5'-ATATGGATCCACCATGAAGTGGGTGTGGGCGCTC-3' (SEQ ID NO: 1)
RBPL:
5'-ATATGCGGCCGCCTACAAAAGGTTTCTTTCTGATCTGC-3' (SEQ ID NO: 2)
ПЦР реакцию проводили в соответствии с протоколом, прилагаемым к Pyrobest полимеразе (TAKARA SHUZO CO., LTD.). Получаемый ПЦР продукт подвергали электрофорезу с агарозным гелем (1%), и около 0,6 т.п.н. ДНК фрагмента, содержащего RBP4 ген, выделяли из геля и дигерировали с рестрикционными ферментами BamHI и NotI. ДНК, обработанную рестрикционными ферментами, подвергали электрофорезу с агарозным гелем (1%), и около 0,6 т.п.н. ДНК фрагмента выделяли и лигировали с плазмидой pcDNA3.1(+) (Invitrogen), взаимодействующей с рестрикционными ферментами BamHI и NotI, получая экспрессионную плазмиду pcDNA3.1(+)/hRBP4. Подтверждалось, что основная последовательность фрагмента, вставленного в данную плазмиду, соответствует целевой последовательности.
Человеческий TTR ген клонировали с помощью ПЦР реакции с использованием кДНК тонкой кишки человека (Clontech, QUICK-Clone cDNA) в качестве матрицы и следующего комплекта праймера.
TTRU:
5'-ATATGGATCCACCATGGCTTCTCATCGTCTGCTCC-3' (SEQ ID NO: 3)
TTRL:
5'-ATATGCGGCCGCTCATTCCTTGGGATTGGTGACGA-3' (SEQ ID NO: 4)
ПЦР реакцию проводили в соответствии с протоколом, прилагаемым к Pyrobest полимеразе (TAKARA SHUZO CO., LTD.). Получаемый ПЦР продукт подвергали электрофорезу с агарозным гелем (1%), 0,5 т.п.н. ДНК фрагмента, содержащего TTR ген, выделяли из геля и дигерировали с рестрикционными ферментами BamHI и NotI. ДНК, обработанную рестрикционными ферментами, подвергали электрофорезу с агарозным гелем (1%), и около 0,5 т.п.н. ДНК фрагмента выделяли и лигировали с плазмидой pcDNA3.1(+) (Invitrogen), взаимодействующей с рестрикционными ферментами BamHI и NotI, получая экспрессионную плазмиду pcDNA3.1(+)/hTTR. Подтверждалось, что основная последовательность фрагмента, вставленного в данную плазмиду, соответствует целевой последовательности.
1В: Конструирование человеческой RBP4-His экспрессионной плазмиды
EcoRI участок вводили в 3' конец hRBP4 гена с помощью ПЦР реакции с использованием экспрессионной плазмиды pcDNA3.1(+)/hRBP4, полученной в вышеупомянутом п.1А в качестве матрицы и следующего комплекта праймера.
CMVP:
5'-TGGGAGGTCTATATAAGCAGAGCTCG-3' (SEQ ID NO: 5)
RBPECO:
5'-ATATGAATTCTTCCTTGGGATTGGTGAC-3' (SEQ ID NO: 6)
ПЦР реакцию проводили в соответствии с протоколом, прилагаемым к Z-Taq полимеразе (TAKARA SHUZO CO., LTD.). Получаемый ПЦР продукт очищали с использованием QIAquick ПЦР очистительного комплекта (QIAGEN), и дигерировали с рестрикционными ферментами BamHI и EcoRI. ДНК, обработанную рестрикционными ферментами, подвергали электрофорезу с агарозным гелем (1%), полученные примерно 0,6 т.п.н. ДНК выделяли и сшивали с плазмидой pcDNA3.1(+) (Invitrogen), взаимодействующей с рестрикционными ферментами BamHI и EcoRI, получая pcDNA3.1(+)/hRBP4-Есо, имеющую EcoRI сайт на 3' конце hRBP4 гена.
EcoRI сайт вводили в hTTR ген на 3' конце с помощью ПЦР реакции с использованием экспрессионной плазмиды pcDNA3.1(+)/hTTR, полученной в упомянутом выше п.1А, в качестве матрицы и CMVP и TTRECO комплекта праймера.
TTRECO:
5'-ATATGAATTCCAAAAGGTTTCTTTCTGATC-3' (SEQ ID NO: 7)
ПЦР реакцию проводили в соответствии с протоколом, прилагаемым к Z-Taq полимеразе (TAKARA SHUZO CO., LTD.). Получаемый ПЦР продукт очищали с использованием QIAquick ПЦР очистительного комплекта (QIAGEN) и дигерировали с рестрикционными ферментами BamHI и EcoRI. ДНК, обработанную рестрикционными ферментами, подвергали электрофорезу с агарозным гелем (1%), получаемые около 0,6 т.п.н. ДНК фрагмента выделяли и лигировали с плазмидой pcDNA3,1(+) (Invitrogen), взаимодействующей с рестрикционными ферментами BamHI и EcoRI, получая pcDNK3.1(+)/hRBP4-Eco сайт на 3' конце hTTR гена.
TTR-His экспрессионную плазмиду pcDNA3.1(+)/hTTR-His, имеющую His tag, добавляемую к С-концу человеческой TTR, получали с помощью вставки фрагмента синтетического гена, содержащего His tag последовательность, с помощью гибридизации следующей олиго ДНК с EcoRI и NotI сайтами упомянутой выше pcDNA3,1(+)hTTR-Eco.
HISENU:5'-AATTCCATCATCATCATCATCACTAGGC-3' (SEQ ID NO: 8)
HISENL:5'-GGCCGCCTAGTGATGATGATGATGATGG-3' (SEQ ID NO: 9)
HISENU и HISENL каждый растворяли в TE буфере (50 мкл) в концентрации 25 пмоль/мкл, нагревали при 94°С в течение 5 мин и охлаждали постепенно до комнатной температуры, получая фрагмент синтетического гена, содержащего His tag последовательность. pcDNA3.1(+)/hTTR-Eco переваривали с EcoRI и NotI, ДНК, обработанную рестрикционными ферментами, подвергали электрофорезу с агарозным гелем (1%), полученные около 5,9 т.п.н. ДНК выделяли, сегмент синтетического гена, содержащий His последовательность, сшивали с ними, получая TTR-His экспрессионную плазмиду pcDNA3.1(+)/hTTR-His, имеющую His , добавленную к С-концу человеческой TTR.
RBP4-His экспрессионную плазмиду pcDNA3,1(+)/hRBP4-His, имеющую His , добавленную к С-концу человеческой RBP4, получали в соответствии со следующими стадиями.
pcDNA3.1(+)/hRBP4-Eco переваривали с рестрикционными ферментами EcoRI и DraIII, подвергали электрофорезу с агарозным гелем (1%), и полученные около 6,0 т.п.н. ДНК выделяли. pcDNA3.1(+)/hTTR-hTTR-His переваривали с рестрикционными ферментами EcoRI и DraIII, подвергали электрофорезу с агарозным гелем (1%), и полученные около 6,0 т.п.н. ДНК выделяли. Оба ДНК фрагмента сшивали, получая RBP4-His экспрессионную плазмиду pcDNA3.1(+)/hRBP4-His, имеющую His tag, добавленную к С-концу человеческой RBP4.
1С: Получение RBP4-His человека
Человеческую RBP4-His экспрессировали с использованием FreeStyle293 экспрессионной системы (Invitrogen) и экспрессионной плазмиды pcDNA3,1(+)/hRBP4-His, полученной в упомянутом выше п.1В. В соответствии с протоколом, прилагаемым к FreeStyle293 экспрессионной системе, для экспрессии использовали 600 мл культуральной среды. После трансфекции и культивирования в течение 3 дней культуральную надосадочную жидкость, содержащую секретированную/экспрессированную RBP4-His, выделяли. Культуральную надосадочную жидкость повторно концентрировали с использованием VIVACELL250 (молекулярный вес ограничен 10К, VIVASCIENCE) и разбавляли 20 мМ Трис (рН 8), посредством чего буфер заменяли. Жидкость пропускали через TOYOPEARL DEAE-650М колонку (1 см ID×10 см, Tosoh Corporation), уравновешенную 20 мМ Трис буфера (рН 8) при скорости потока 2,5 мл/мин, допуская адсорбцию, и человеческую RBP4-His фракцию получали элюированием 0-0,35М градиентом NaCl. Фракции концентрировали примерно до 5 мл с использованием Vivaspin 20 (молекулярный вес ограничен 10К, VIVASCIENCE). Концентрированный раствор пропускали через HiLoad 26/60 Superdex 200 pg колонку (2,6 см ID×60 см, GE Healthcare), уравновешенную TBS (рН 7,4) и элюируемую TBS (рН 7,4). Фракции, содержащие RBP4-His человека, собирали и концентрировали до около 8 мл с использованием Vivaspin 20 (молекулярный вес ограничен 10К, VIVASCIENCE). Около 8 мг RBP4-His человека получали из 600 мл культуральной среды.
1D: Получение TTR человека
TTR человека экспрессировали с использованием FreeCtyle293 экспрессионной системы (Invitrogen) и экспрессионной плазмиды pcDNA3.1(+)/hTTR, полученной в упомянутом выше п.1А. В соответствии с протоколом, прилагаемым к FreeStyle293 экспрессионной системе, для экспрессии использовали 600 мл культуральной среды. После трансфекции и культивирования в течение 3 дней культуральную надосадочную жидкость, содержащую секретированную/экспрессированную TTR человека выделяли. Культуральную надосадочную жидкость повторно концентрировали с использованием VIVACELL250 (молекулярный вес ограничен 10К, VIVASCIENCE) и разбавляли 20 мМ Трис (рН 8), посредством чего буфер заменяли. Жидкость пропускали через TOYOPEARL DEAE-650М колонку (1 см ID×10 см, Tosoh Corporation), уравновешенную 20 мМ Трис буфера (рН 8) при скорости потока 2,5 мл/мин, допуская адсорбцию, и человеческую TTR фракцию получали элюированием 0-0,55М градиентом NaCl. Данную фракцию повторно концентрировали с использованием Vivaspin 20 (молекулярный вес ограничен 10К, VIVASCIENCE) и разбавляли 20 мМ Трис (рН 8), посредством чего заменяли буфер. Жидкость пропускали через HiLoad Q Sepharose HP колонку (1,6 см ID×10 см, GE Healthcare), уравновешенную 20 мМ Трис буфером (рН 8) при скорости потока 1,0 мл/мин, допуская адсорбцию, и получали TTR человека элюированием 0-0,4М градиентом NaCl. Фракции концентрировали примерно до 5 мл с использованием Vivaspin 20 (молекулярный вес ограничен 10К, VIVASCIENCE). Концентрированный раствор пропускали через HiLoad 26/60 Superdex 75 pg колонку (2,6 см ID×60 см, GE Healthcare), уравновешенную PBS (рН 7,4) и элюируемую PBS (рН 7,4). Фракции, содержащие TTR человека, собирали и концентрировали примерно до 5 мл с использованием Vivaspin 20 (молекулярный вес ограничен 10К, VIVASCIENCE). Около 6 мг TTR человека получали из 600 мл культуральной среды.
1Е: Получение TTR-биотина человека
TTR человека, полученную в упомянутом выше п.1D, метили биотином с использованием комплекта для биотинилирования (Sulfo-Osu) (DOJINDO LABORATORIES) согласно протоколу, прилагаемому к комплекту, посредством чего получали TTR-биотин человека. TTR человека 5,0 мг повторно концентрировали с использованием Vivaspin 6 (молекулярный вес ограничен 10К, VIVASCIENSE) и разбавляли 50 мМ NaHCO3, посредством чего заменяли буфер. Жидкость разбавляли 50 мМ NaHCO3 до концентрации TTR человека 2,0 мг/мл, затем добавляли водный Биотин-(AC5)2 Culfo-Osu раствор (10 мг/мл) (9,9 мкл), полученный в процессе его использования, и смесь подвергали реакции при 25°С в течение 2 часов. После реакции раствор пропускали через NAP-25 колонку (GE Healthcare), уравновешиваемую PBS (рН 7,4), элюировали PBS (рН 7,4), и элюат (3,5 мл), содержащий TTR-биотин человека, собирали.
Экспериментальный пример 2
Анализ связывания с помощью Ретинол-RBP4-TTR ELISA
Анализ связывания выполняли с помощью ELISA системы (Ретинол-RBP4-TTR ELISA) для детектирования ретинол-RBP4-TTR конъюгата с помощью реакции стрептавидин-биотин.
Используемую His-меченую RBP4 человека получали в упомянутом выше п.1С.
Используемую биотинилированную TTR человека получали в упомянутом выше п.1Е.
Стрептавидин (20 мкл) (10 мкг/мл стрептавидина типа II (Wako Pure Chemical Industries, Ltd.), 10 мМ Трис-HCl (рН 7,5), 10мМ NaCl) добавляли на 384 луночную черную планшету maxisorp (Nunc), и планшету подвергали центрифугированию (1000 об/мин, 1 мин) и покрывали на ночь при 4°С. Планшету промывали дважды PBST (PBS, 0,05% Tween 20, 100 мкл/лунку) и блокировали 25% Block Ace (Snow Brand Milk Products Co., Ltd., PBS, 100 мкл/лунку). Планшету подвергали центрифугированию (1000 об/мин, 1 мин) и инкубировали при комнатной температуре в течение 4 часов или на протяжении ночи при 4°С. Планшету промывали дважды PBST (PBS, 0,05% Tween 20, 100 мкл/лунку), и добавляли биотинилированный TTR человека (концентрация исходного раствора 1,3 мг/мл), 1000-кратно разбавленного PBST 20 мкл/лунку. Планшету подвергали центрифугированию (1000 об/мин, 1 мин) и оставляли все еще стоять при комнатной температуре в течение 1,5 часов или на протяжении ночи при 4°С. Планшету промывали 3 раза PBST (100 мкл/лунку), и добавляли His-меченую RBP4 человека (концентрация исходного раствора 0,96 мг/мл), разбавленную 4000-кратно реакционным буфером (50 мМ Трис-HCl, 150 мМ NaCl, 0,005% Tween 20, 1 мМ DTT, 0,1% BSA) по 10 мкл/лунку. Приготавливали ряд разбавлений (8 доз из 10 мМ, 200-кратная концентрация) соединения в ДМСО, и 1 мкл каждого добавляли к реакционному буферу (200 мкл), содержащему ретинол (0,5 мкМ) (Sigma Aldrich Co.). В качестве положительного контроля использовали реакционный буфер (200 мкл), содержащий ретинол с добавлением ДМСО, а в качестве отрицательного контроля использовали реакционный буфер (200 мкл), свободный от ретинола с добавлением ДМСО. Смешанные растворы ретинола и соединения добавляли на планшету 15 мкл/лунку. Смесь перемешивали в планшетном миксере, подвергали центрифугированию (1000 об/мин, 1 мин) и подвергали реакции при комнатной температуре в течение 2 часов. Добавляли 35% Block Ace раствор, разбавленный реакционным буфером 10 мкл/лунку, центрифунировали (1000 об/мин, 1 мин) и подвергали реакции при комнатной температуре в течение 30 минут. Планшету промывали 3 раза PBST (100 мкл/лунку) и добавляли реагент субстрата максимальной чувствительности Super Signal ELISA Femto (PIERCE) по 30 мкл/лунку, и с помощью считывателя с планшеты (Wallac) измеряли люминесценцию.
Степень активности связывания соединения определяли по формуле 100×(величина испытуемого соединения-величина отрицательного контроля)/(величина положительного контроля-величина отрицательного контроля). Активность по ингибированию связывания ИК50 (IC50) вычисляли на основании степени активности связывания при каждой концентрации соединения с использованием программного обеспечения с вычерчиванием графика, Prism (GraphPad Software Inc.). Результаты показаны ниже.
Из приведенных выше результатов ясно, что соединение настоящего изобретения ингибирует связывание RBP4 с ретинолом и TTR.
Экспериментальный пример 3
Действие соединения настоящего изобретения по понижению RBP4 в крови с использованием C57BL/6J мышей.
Самцов мышей C57BL/6J (Charles River Laboratories Japan Inc.) в возрасте 7-15 недель содержали индивидуально для акклиматизации в течение 4-6 дней в условиях свободного доступа к твердому корму CE-2 (CLEA Japan, Inc.), и на основе веса тела группировали (5 на группу). На следующий день после группировки брали образцы или пробы крови из орбитального венозного сплетения и отделяли плазму (величина 0 час.). После этого испытуемые соединения (примеров 1, 13, 15, 16, 17, 27 и 34) вводили перорально в дозе 50 мг/кг (растворитель: 0,5% раствор метилцеллюлозы (10 мл/кг)). Через 4, 7 и 24 часа после введения соединений брали пробы крови из орбитального венозного сплетения и отделяли плазму. Контрольной группе вводили перорально 0,5% раствор метилцеллюлозы (10 мл/кг).
Уровень RBP4 собранной плазмы измеряли с помощью ELISA. RBP4 количественно определяли с помощью следующих стадий с использованием кроличьих анти-мышиных RBP4 поликлональных антител (Hokudo Co, Ltd). На 96-луночную планшету ELISA наносили 50 мкг/мл антител (100 мкл) и оставляли стоять в течение ночи при 4°С. После блокирования с помощью BlockAce (DAINIPPON PHARMACEUTICAL CO., LTD.), добавляли мышиную RBP4 или образец (100 мкл) и планшету оставляли стоять при комнатной температуре в течение 2 часов. После промывки с помощью PBC-0,5% Tween 20, добавляли HRP-меченые анти-RBP4 антитела (полученные мечением RBP4 поликлональные антитела (Hokudo Co., Ltd) с HRP (DOJINDO LABORATORIES)) 100 мкл, и планшету оставляли стоять при комнатной температуре в течение 1 часа. После промывки добавляли TMB (Dako Citomations), и смесь оставляли стоять при комнатной температуре в течение 20 минут, чтобы позволить проявиться окраске, и реакцию гасили 2н серной кислотой. После этого с помощью считывателя с планшет измеряли абсорбцию при А450 нм. Количество изменений от начальной величины каждого животного определяли как относительную величину от контрольной группы (% начальной/контроль) в каждый момент времени. Результаты показаны ниже в виде среднего±стандартное отклонение (n=5).
№
часа
Все из упомянутых выше соединений показали более низкие уровни, чем контрольная группа, через 4 часа и наиболее низкий уровень через 7 часов после введения при одном пероральном введении. Данные результаты показывают, что соединение настоящего изобретения проявляет действие, понижающее RBP4 в крови.
Экспериментальный пример 4
Гипогликемическое действие соединения настоящего изобретения оценивали с использованием крыс Zucker fa/fa.
Экспериментальный пример 4А
Самцов крыс Zucker fa/fa 19-недельного возраста (Takeda Company Limited) содержали в группе на твердом корме CE-2 (CLEA Japan Inc.) до 12 недельного возраста, после этого держали на корме с высоким содержанием жира D06110702 (LSG Corporation) для акклиматизации в условиях свободного доступа к корму. В 19-недельном возрасте крыс распределяли по группам на основе веса тела, уровня глюкозы в крови, уровня гликированного гемоглобина и величины уровня RBP4 в крови (6 на группу). После этого перорально вводили испытуемое соединение (пример 34) в дозе 30 мг/кг (растворитель: 0,5% раствор метилцеллюлозы (5 мл/кг)) в течение 2 недель. Через 24 часа после окончательного введения соединения брали пробы крови из хвостовой вены и отделяли плазму. С использованием собранной плазмы измеряли (1) концентрацию RBP4, (2) уровень гликированного гемоглобина и (3) уровень глюкозы в крови. (1) определяли количественно в соответствии с протоколом, описанным в экспериментальном примере 3. (2) измеряли с использованием автоматического анализатора гликогемоглобина фирмы Tosoh Corporation (HLC-723GHbV Alc2.2 или HLC-723G7GHbV Alc2.2), и (3) измеряли с использованием полностью автоматического анализатора фирмы Hitachi (7070 или 7080), соответственно.
Контрольной группе перорально вводили 0,5% раствор метилцеллюлозы (5 мл/кг). В результате концентрация RBP4 в крови, изменение гликированного гемоглобина и уровень глюкозы в крови показали значительное снижение при пероральном введении (30 мг/кг) соединения (пример 34) в течение 2 недель. Результаты показаны ниже в виде среднее±стандартное отклонение (n=6). В следующей таблице изменение гликированного гемоглобина получают вычитанием величины до введения из величины после введения.
(мкг/мл)
Соединение настоящего изобретения показало значительное снижение гликированного гемоглобина и уровня глюкозы в крови в связи со снижением концентрации RBP4 в крови.
Экспериментальный пример 4В
Самцов крыс Zucker fa/fa 25-недельного возраста (Takeda Pharmaceutical Company Limited) содержали для акклиматизации в группах на твердом корме CE-2 (CLEA Japan Inc.) до 12 недельного возраста, после этого на корме с высоким содержанием жира D06110702 (LCG Corporation) в условиях свободного доступа к корму. В 25-недельном возрасте крыс распределяли по группам на основе веса тела, уровня глюкозы в крови, уровня гликированного гемоглобина и уровня RBP4 в крови (5 на группу). Затем перорально вводили испытуемые соединения (примеры 16 и 17) в дозе 10 мг/кг (растворитель: 0,5% раствор метилцеллюлозы (3 мл/кг)) в течение 2 недель. Через 24 часа после окончательного введения соединений брали пробы крови из хвостовой вены и отделяли плазму. С использованием собранной плазмы измеряли (1) концентрацию RBP4, (2) уровень гликированного гемоглобина и (3) уровень глюкозы в крови. (1) определяли количественно в соответствии с протоколом, описанным в экспериментальном примере 3. (2) измеряли с использованием автоматического анализатора гликогемоглобина фирмы Tosoh Corporation (HLC-723GHbV Alc2.2 или HLC-723G7GHbV Alc2.2), и (3) измеряли с использованием полностью автоматического анализатора фирмы Hitachi (7070 или 7080).
Контрольной группе перорально вводили 0,5% раствор метилцеллюлозы (3 мл/кг). Результаты показаны ниже в виде среднее±стандартное отклонение (n=5). В следующей таблице изменение гликированного гемоглобина получают вычитанием величины до введения из величины после введения.
Соединение настоящего изобретения показало значительное снижение гликированного гемоглобина и уровня глюкозы в крови в связи со снижением концентрации RBP4 в крови.
Пример рецептурной формы 1 (получение капсул)
1), 2), 3) и 4) смешивают и заполняют в желатиновые капсулы.
Пример 2 рецептурной формы (получение таблеток)
Все количество 1), 2) и 3) и 4) (30 г) смешивают с водой, сушат в вакууме и просеивают. Просеянный продукт смешивают с 4) (14 г) и 5) (1 г) и штампуют с помощью таблетирующего устройства, получая тем самым 1000 таблеток, содержащих 30 мг соединения примера 1 на таблетку.
Промышленная применимость
Соединение настоящего изобретения обладает превосходным RBP4-понижающим действием, и является полезным в качестве лекарственного средства для профилактики или лечения заболеваний и состояний, опосредуемых повышенным RBP4, таких как диабет, гиперлипидемия и аналогичные.
Данная заявка основана на патентной заявке № 61/129032, поданной в США, содержание которой целиком охватывается настоящей заявкой.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ 2-ПИРИДОНА | 2010 |
|
RU2551847C2 |
| ЗАМЕЩЕННЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СУЛЬФОНАМИДНЫЕ СОЕДИНЕНИЯ, ПОЛЕЗНЫЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ TRPA 1 | 2014 |
|
RU2675792C2 |
| ИНГИБИТОРЫ TrkA КИНАЗЫ, ОСНОВАННЫЕ НА НИХ КОМПОЗИЦИИ И СПОСОБЫ | 2015 |
|
RU2672583C2 |
| НОВЫЕ СОЕДИНЕНИЯ - АНТАГОНИСТЫ РЕЦЕПТОРА НЕЙРОКИНИНА 1 | 2013 |
|
RU2631319C2 |
| ЧАСТИЧНО НАСЫЩЕННОЕ АЗОТСОДЕРЖАЩЕЕ ГЕТЕРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ | 2013 |
|
RU2641291C2 |
| ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ МОРФОЛИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ, СПОСОБ СНИЖЕНИЯ КОЛИЧЕСТВА ТАХИКИНИНОВ ПРИ ЛЕЧЕНИИ ИЛИ ПРОФИЛАКТИКЕ ФИЗИОЛОГИЧЕСКИХ СОСТОЯНИЙ, АССОЦИИРУЕМЫХ С ИЗБЫТКОМ ТАХИКИНИНОВ | 1994 |
|
RU2131426C1 |
| ФЕНИЛОКСАЗОЛИДИНОНЫ, ИМЕЮЩИЕ С-С-СВЯЗЬ С 4-8-ЧЛЕННЫМИ ГЕТЕРОЦИКЛИЧЕСКИМИ КОЛЬЦАМИ | 1996 |
|
RU2175324C2 |
| ПРОИЗВОДНОЕ ЦИКЛОГЕКСАНА И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2009 |
|
RU2478621C2 |
| ФЕНИКОЛОВЫЕ ПРОТИВОБАКТЕРИАЛЬНЫЕ СРЕДСТВА | 2013 |
|
RU2593204C2 |
| ГЕТЕРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ | 2017 |
|
RU2748693C2 |


Изобретение относится к гетероциклическим соединениям формулы:

или их фармацевтически приемлемой соли, где: кольцо A означает 5-членный неароматический гетероцикл с одним гетероатомом, выбранным из N, O, или с двумя атомами N, необязательно дополнительно замещенный одним заместителем =O; кольцо B означает бензольное кольцо, необязательно дополнительно замещенное 1-3 заместителями, выбранными из: С1-6алкокси, необязательно замещенной 1-3 галогенами, галогена, С1-6алкила, необязательно замещенного 1-3 галогенами; X означает связь, O, CH2O, OCH2, CH2, (CH2)2, S, CH2S, SCH2, S(O), CH2S(O), S(O)CH2, S(O)2, CH2S(O)2 или S(O)2CH2, при условии что
{2-оксо-1-[3-(трифторметил)фенил]-пирролидин-3-ил}уксусная кислота и
{5-оксо-1-[3-(трифторметил)фенил]-4,5-дигидро-1H-пиразол-3-ил}уксусная кислота исключаются. Соединения используют для получения лекарственного средства, понижающего ретинол-связывающий белок 4, что позволяет использовать их для лечения диабета. 7 н. и 7 з.п. ф-лы, 22 схемы, 4 табл., 78 пр.
1. Соединение, представленное формулой
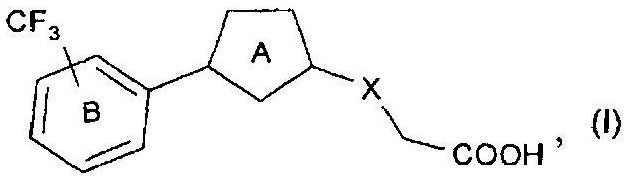
в которой кольцо A представляет собой 5-членный неароматический гетероцикл, содержащий один гетероатом, выбранный из азота, кислорода, или содержащий два азота и необязательно дополнительно замещенный одним заместителем оксо;
кольцо B означает бензольное кольцо, необязательно дополнительно замещенное 1-3 заместителями, выбранными из:
(a) атома галогена;
(b) C1-6алкильной группы, необязательно замещенной 1-3 атомами галогена; и
(c) C1-6алкокси группы, необязательно замещенной 1-3 атомами галогена; и
X представляет собой связь, O, CH2O, OCH2, CH2, (CH2)2, S, CH2S, SCH2, S(O), CH2S(O), S(O)CH2, S(O)2, CH2S(O)2 или S(O)2CH2,
при условии, что
{2-оксо-1-[3-(трифторметил)фенил]-пирролидин-3-ил}уксусная кислота, и
{5-оксо-1-[3-(трифторметил)фенил]-4,5-дигидро-1H-пиразол-3-ил} уксусная кислота исключаются, или его фармацевтически приемлемая соль.
2. Соединение или его фармацевтически приемлемая соль по п.1, в котором X представляет собой O, CH2O, OCH2, CH2, S, CH2S, SCH2, S(O) или S(O)2.
3. Соединение или его фармацевтически приемлемая соль по п.1, в котором кольцо A представляет собой 5-членный неароматический гетероцикл, содержащий один или два атома азота и необязательно дополнительно замещенный одной оксогруппой.
4. Соединение или его фармацевтически приемлемая соль по п.1, в котором кольцо A представляет собой пирролидиновое кольцо, необязательно дополнительно замещенное одной оксогруппой, или тетрагидрофурановое кольцо.
5. Соединение или его фармацевтически приемлемая соль по п.1, в котором кольцо A представляет собой 5-членный неароматический гетероцикл, содержащий один или два атома азота и необязательно дополнительно замещенный одним заместителем, выбранным из оксогруппы; и X представляет собой O, CH2O, OCH2, CH2, S, CH2S, SCH2, S(O) или S(O)2.
6. Соединение или его фармацевтически приемлемая соль по п.1, в котором кольцо A представляет собой пирролидиновое кольцо, замещенное одной оксогруппой, или тетрагидрофурановое кольцо; и X представляет связь.
7. Соединение или его фармацевтически приемлемая соль по п.1, в котором кольцо А представляет собой 5-членный неароматический гетероцикл, содержащий один или два атома азота и необязательно дополнительно замещенный одним заместителем, выбранным из оксогруппы; и X представляет собой O, S или CH2.
8. ({(3S)-1-[3,5-бис(трифторметил)фенил]пирролидин-3-ил} окси)уксусная кислота или ее фармацевтически приемлемая соль.
9. Лекарственное средство, понижающее ретинол-связывающий белок 4, включающее соединение по п.1 или его фармацевтически приемлемую соль.
10. Лекарственное средство по п.9, которое является агентом, для профилактики или лечения диабета.
11. Способ понижения ретинол-связывающего белка 4 у млекопитающих, который включает введение эффективного количества соединения по п.1 или его фармацевтически приемлемой соли млекопитающему.
12. Способ профилактики или лечения диабета у млекопитающего, который включает введение эффективного количества соединения по п.1 или его фармацевтически приемлемой соли млекопитающему.
13. Применение соединения по п.1 или его фармацевтически приемлемой соли для производства агента, понижающего ретинол-связывающий белок 4.
14. Применение соединения по п.1 или его фармацевтически приемлемой соли для производства агента для профилактики или лечения диабета.
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| Прибор для очистки паром от сажи дымогарных трубок в паровозных котлах | 1913 |
|
SU95A1 |
| СМЕСИТЕЛЬ КОРМОВ | 0 |
|
SU200415A1 |
| J | |||
| of Pharmacology and Experimental Therapeutics, 2007, v.320, №3, p.1038-1049 | |||
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| Перекатываемый затвор для водоемов | 1922 |
|
SU2001A1 |
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| СРЕДСТВО ДЛЯ ЛЕЧЕНИЯ САХАРНОГО ДИАБЕТА | 1996 |
|
RU2139718C1 |

