Перекрестная ссылка на родственные заявки
Данная заявка на международный патент основана на предварительной заявке США №61/617535 (Attorney Docket # ТРС-11-18), озаглавленной «Способ окислительного дегидрирования с улучшенной регулируемостью для получения бутадиена», зарегистрированной 29 марта 2012 г, содержание которой приведено в описании полностью в качестве ссылки.
Данная заявка на международный патент также основана на предварительной заявке США №61/617506 (Attorney Docket # ТРС-10-25), озаглавленной «Способ окислительного дегидрирования с низкими выбросами для получения бутадиена», зарегистрированной 29 марта 2012 г, содержание которой полностью включено в описание в качестве ссылки.
Предпосылки создания изобретения
Ранее известные способы получения бутадиена из бутенобогащенного углеводородистого питания использовали реакторы, чья форма в значительной степени определялась соображениями падения давления, приводя к реакторам, которые считались неглубокими - глубина слоя (линейный размер в направлении потока) всех четырех слоев слоя часто ограничивалась примерно метром или менее с общей высотой катализатора окислительного дегидрирования только примерно 55-60 см (22-24 дюйм) или менее. В частности, ранее известные способы обычно использовали природный газ для испаривания бутена и тепло смеси углеводородов, предпочтительно, бутенов, кислорода и водяного пара при температуре выше 260°С (500°F), более предпочтительно, выше примерно 315°С (600°F), и, предпочтительно, выше примерно 345°С (650°F), или в некоторых случаях даже выше 370°С (700°F). В традиционном способе реакционная смесь содержит бутены, кислород в количестве от примерно 0,4 моль до примерно 0,8 моль, более обычно, от слегка свыше 0,5 моль до примерно 0,65 моль кислорода на каждый моль бутена в бутенобогащенном углеводородистом питании, и перегретый водяной пар в количествах от примерно 12:1 до примерно 16:1. Нагретую реакционную смесь пропускают через многослойный слой, содержащий четыре слоя: слой распределения инертного потока и удерживания катализатора, который ограничивает течение по каналам реакционной смеси, когда она проходит через каталитический слой, а также служит для удерживания нижних слоев на месте против завихренности, которая может присутствовать выше каталитического слоя; второй слой, содержащий массу слоя, был катализатором окисления/дегидрирования; тогда как третий слой содержит катализатор удаления альдегида и алкина ((УАА) ("AAR")), который превращает алкины и альдегиды в продукте в соединения, которые являются менее вредными для переработки для полимеризации бутадиенов, чем алкины и альдегиды. Самый нижний слой содержит инертный дисперсный твердый материал носителя. Традиционно, общая высота слоя ограничивается примерно метром или менее, тогда как глубина слоя окислительного дегидрирования ограничивается до менее примерно 56 см (22 дюйм).
При прохождении через катализатор окислительного дегидрирования бутены превращаются в бутадиен, что сопровождается высвобождением большого количества тепла, с получением в результате температур около 540°С или 595°С (1000°F или 1100°F). В прошлом, когда глубина каталитического слоя была небольшой, могло быть трудным предотвратить проскок кислорода в катализатор УАА, несмотря на принимаемые меры для обеспечения того, чтобы весь кислород, присутствующий в реакционной смеси, был потреблен перед достижением катализатора УАА. Проскок кислорода может привести как к потере желаемого бутадиенового продукта, так и, даже более серьезно, к разрушению катализатора УАА и/или реакторного сосуда. В результате, во многих случаях указанные соображения приводят к использованию длины довольно консервативного цикла и преждевременных изменений катализатора, так что эффективный срок службы катализатора является короче, чем необходимо, и страдает процент времени на потоке.
После реакции смесь продукта реакции охлаждается, и бутадиен отделяется при контакте с абсорбционным маслом и последующим фракционированием.
Обычно указанные способы дают сырой бутадиен с чистотой в интервале от примерно 50 до примерно 70%, более обычно, от примерно 55 до примерно 65%, который пропускают далее в установку для дальнейшей переработки с использованием известных технологий.
Ссылки, представляющие интерес, рассматриваются ниже.
Lewis, Hydrocarbon Conversion Process Using Novel Metallo Manganese Oxides, патент США №5772898, 30 июня 1998 г, относится к способу конверсии углеводородов, включающему контактирование исходных углеводородов с катализатором, содержащим кристаллическую металломарганцевооксидную композицию, имеющую структуру трехмерной решетки, внутрикристаллическую пористую систему и эмпирический химический состав на безводной основе, выраженный формулой:
AxMn8-xMxO16,
где А представляет собой моделирующий агент, выбранный из щелочных металлов, щелочно-земельных металлов и аммониевого иона, «y» - число молей А и варьируется из группы, состоящей из от примерно 0,5 до примерно 2,0, М представляет собой металл, выбранный из группы, состоящей из хрома, циркония, олова, платины, родия, ниобия, тантала, ванадия, сурьмы, рутения, галлия и германия, «х» - число молей М и варьируется от примерно 0,01 до примерно 4,0 и характеризуется тем, что марганец имеет валентность +3 или +4, М имеет валентность +3, +4 или +5, и композиция имеет голландитную структуру.
Sasaki et al.; Iron-Antimony-Containing Metal Oxide Catalyst Composition And Process For Production The Same; патент США №5139988, 18 августа 1992 г, относится к композиции, которая содержит в качестве неотъемлемых компонентов: кристаллический антимонат железа и, по меньшей мере, один элемент, выбранный из группы, состоящей из ванадия, молибдена и вольфрама, используется в качестве катализатора в реакции окисления органических соединений. Также рассматривается способ получения композиции.
Dejaifve et al., Catalyst For Dehydrogenating Organic Compounds, A Process For Its Preparation And Its Use; патент США №4975407, 4 декабря 1990 г, относится к катализатору, полученному из агентов, обеспечивающих оксиды железа, и агентов, обеспечивающих оксид калия, характеризующемуся тем, что мольное соотношение находится в интервале от 1,5 до 60 и что фаза феррита калия K2Fe12O19 присутствует, нанесенная на октаэдральную матрицу Fe3O4, с показом эпитаксии между гексагональной структурой K2Fe12O19 и (III) - плоскостями шпинельной структуры Fe3O4.
McFarland; Acetylene Removal Process, патент США №4658080 относится к способу удаления ацетилена из потоков органических соединений, в частности, потоков, получаемых в результате окислительного дегидрирования С4-С8-углеводородов с использованием катализатора восстановления ацетилена, содержащего феррит и оксид никеля, оксид щелочноземельного металла, карбонат или гидроксид магния, кальция, стронция или бария и оксидкарбонат щелочного металла или гидроксид на основе лития, калия, натрия или рубидия. Примером использования катализатора является трубчатый реактор, в котором окислительное дегидрирование проводится на С4-С8-углеводородах, и продукт реакции сразу пропускается через слой катализатора восстановления ацетилена (см. также McFarland; Acetylene Removal Process, патенты США №4644088, 17 февраля 1987 г и №4513159, 23 апреля 1985 г).
Patel, Process For Removal α-Acetylenes From Diolefins, патент США №4266086, относится к удалению альфа-ацетиленов, включая винилацетилен и метилацетилен, из исходного сырья, содержащего бутадиен и смешанные моноолефины и алканы, загрязненные альфа-ацетиленами в количестве до примерно 1,0% по массе (масс. %) при контактировании жидкой фазы с металлоксидным (оксид меди, оксид серебра или их смеси) катализатором на носителе при отсутствии водорода при температуре в интервале от примерно 90°С (200°F) до примерно 130°С (260°F).
В Besozzi et al., Purification Of Unsaturated Compounds, патент США №4150063, 17 апреля 1979 г, газообразные потоки, содержащие ненасыщенные углеводороды и карбонильные соединения, контактируют с катализатором, содержащим, по меньшей мере, один металл из групп 8, 1b, 2b, 4b, 6b и, по меньшей мере, один элемент из групп 1а и 2а, с разрушением карбонильных соединений без существенной потери ненасыщенных углеводородов.
Miklas, Method of Activating Zinc-Ferrite Oxidative Dehydrogenation Catalyst, патент США №3953370, 27 апреля 1976 г, относится к использованию водяного пара при температуре 370-700°С (700-1300°F) для активации цинкферритного катализатора окислительного дегидрирования для получения бутадиена из С4-С8-углеводородов.
Tschopp, Diolefin Production And Purification, патент США №3943185, 9 марта 1976 г, относится к способу получения потока окислительно дегидрированных С4-углеводородов, по существу не содержащих кислород, и удаленных инертных неконденсирующихся газов, содержащему абсорбирование С4-углеводородов в масле абсорбера в первой зоне; отгонку кислорода и инертных неконденсирующихся газов из смеси масла адсорбера и С4-углеводородов во второй зоне, которая работает в условиях температуры и давления с поддержанием водной фазы во второй зоне; и выведение (1) определенно водной фазы из второй зоны, (2) головной части определенно каждого из кислорода и инертных неконденсирующихся газов и остатков масла адсорбера и С4-углеводородов, по существу не содержащих кислород, и инертных неконденсирующихся газов.
В Woerner et al., Purification Of Unsaturated Hydrocarbons By Extractive Distillation With Addition Of Liquid Solvent To Stripper Overhead, патенте США №3496070, 17 февраля 1970 г, предусматривается способ сепарации смеси углеводородов, содержащих 4-5 углеродных атомов, включая ненасыщенные углеводороды, который (способ) содержит: экстракционную дистилляцию смеси углеводородов селективным растворителем в экстракционной дистилляционной колонне, в результате чего углеводород селективно экстрагируется в экстракционной дистилляционной колонне с образованием фракции углеводородобогащенного растворителя, которая подается в колонну отгонки растворителя с указанным растворителем, удаляемым как остатки из указанной колонны отгонки растворителя, и выпариваемую углеводородную фракцию отбирают как головную фракцию из указанной колонны отгонки; введение указанного селективного растворителя в жидкой фазе в выпаренную головную часть из отпарного аппарата растворителя со снижением давления в конденсаторе головной части колонны отгонки растворителя и в отпарном аппарате растворителя. То есть продукт способа альтернативно может быть отобран как головная часть из отпарного аппарата растворителя вместо отбора из экстракционной дистилляционной колонны.
Bajars; Dehydrogenation With Magnesium Ferrite; патент США №3284536, 8 ноября 1966 г, относится к дегидрированию углеводородов в паровой фазе при повышенных температурах в присутствии кислорода и катализатора, содержащего феррит магния. Углеводородами, которые подвергаются дегидрированию, согласно способу, являются углеводороды с 4-7 углеродными атомами, предпочтительно, алифатические углеводороды, выбранные из группы, состоящей из насыщенных углеводородов, моноолефинов, диолефинов и их смесей с 4, 5 или 6 углеродными атомами, имеющих неразветвленную цепь с, по меньшей мере, четырьмя углеродными атомами, и циклоалифатические углеводороды. Кислород присутствует в реакционной зоне в количестве в интервале 0,2-2,5 моль кислорода на 1 моль дегидрированного углеводорода. Температура реакции дегидрирования является выше 2 50°С, такая как выше примерно 300°С или 375°С, и максимальная температура в реакторе может быть около 650°С или 750°С, или, возможно, выше в некоторых условиях.
Levin et al., Process For Removing Aldehydes and/or Ketones From an Olefinic Stream, опубликованная заявка на патент США 2004/0122275, 24 июня 2004 г, относится к удалению кислородсодержщей примеси, выбранной из альдегида и/или кетона, из потока олефинового продукта. Поток продукта контактирует с металлоксидсодержащим катализатором в присутствии С1-С6-спирта в условиях, достаточных для превращения кислородсодержащей примеси в олефин и/или кислородсодержащее соединение с более высоким числом углеродов, чем альдегид и/или кетон.
Металлоксидсодержащий катализатор обычно содержит оксид, по меньшей мере, одного металла, выбранного из группы, состоящей из металлов группы 2, металлов группы 3 (включая металлы лантанидного и актинидного ряда) и металлов группы 4. Катализатор может содержать два или более металлов из одной и той же группы металлов. В одном варианте металлоксидсодержащий катализатор содержит оксид лантана и оксид магния. В другом - катализатор содержит оксид металла, выбранного из группы, состоящей из Ti, Zr и Hf. В еще другом варианте катализатор содержит оксид металла, выбранного из группы, состоящей из Sc, Y, La и Се.
Van Egmond, Distillation Process For Removal Of Methyl Acetylene and/or Propadiene From an Olefin Stream, опубликованная заявка на патент США 2004/0122268, 24 июня 2004 г, относится к способу получения потока пропиленового продукта и/или потока бутиленового продукта из олефинового потока при удалении метилацетилена и/или пропадиена (MAPD) из пропилена и/или бутилена в двухстадийном способе фракционирования.
Welch et al. в работе "Butadiene via Oxidative Dehydrogenation", Hydrocarbon Processing, Nov. 1978, pp. 131-136 рассматривают способ окислительного дегидрирования, в котором водяной пар, воздух или кислород и нормальные бутены нагревают и пропускают через нераскрытый авторегенерируемый гетерогенный катализатор при температуре около 430°С (800°F) с использованием водяного пара в качестве поглотителя тепла с усреднением роста температуры в адиабатической реакторной системе без использования газофазных добавок, таких как галоид и соединения серы. Способ потребляет по существу весь кислород в питании, обычно оставляя уровни кислорода в выходящем потоке ниже 0,3%. Ацетилены и окисленные побочные продукты являются главными побочными продуктами.
Краткое описание изобретения
Настоящее изобретение предусматривает способ получения бутадиена из бутенобогащенного питания, содержащий стадии обеспечения бутенобогащенного углеводородистого питания, испаривания и перегревания указанного бутенобогащенного углеводородистого питания при температуре, по меньшей мере, примерно 205°С (400°F), смешения указанного бутенобогащенного углеводородистого питания с перегретым водяным паром и кислородобогащенным газом с образованием потока реакторного питания, причем моли кислорода в указанном потоке реакторного питания регулируются так, чтобы находиться в интервале, по меньшей мере, примерно 0,4, более предпочтительно, по меньшей мере, примерно 0,5 моль кислорода на моль бутенобогащенного углеводородистого питания, причем поток питания указанного реактора окислительного дегидрирования содержит: главную пропорцию оксида железа, незначительную пропорцию оксида цинка и небольшие количества оксида марганца и фосфорную кислоту вместе с оксидом кальция, производным от неазотистого предшественника кальция, предпочтительно, ацетата кальция, и с образованием в результате потока бутадиенобогащенного продукта. В традиционном варианте нагретая смесь реакционного питания пропускается через многослойный слой, содержащий четыре слоя: слой распределения инертного потока и удерживания катализатора, предпочтительно, содержащий сферы альфа-глинозема, который препятствует протеканию по каналам реакционный смеси, когда она проходит через каталитический слой, а также служит для удержания нижних слоев на месте против завихренности, которая может присутствовать выше каталитического слоя; второй слой, содержащий массу слоя, является катализатором окислительного дегидрирования, который сам имеет глубину более 69 или 70 см (27 дюйм); тогда как третий слой содержит катализатор удаления альдегида и алкина ((УАА) ("AAR")), который превращает алкины и альдегиды в продукте в соединения, которые являются менее вредными для переработки для полимеризации бутадиенов, чем алкины и альдегиды; и самый нижний слой содержит инертный дисперсный твердый материал носителя. Предпочтительно, условия на впуске, в первую очередь, температура, регулируются так, что реакции окислительного дегидрирования вначале имеют место в нижней части каталитического слоя окислительного дегидрирования, так что удается избежать коксования в частях слоя выше начальной реакционной зоны, и, по меньшей мере, 3, предпочтительно, по меньшей мере, 5, более предпочтительно, по меньшей мере, примерно 8 и до примерно 10-75 или более дистанционно считываемых термопар вводятся в часть окислительного дегидрирования слоя для контроля температуры в нем на ряде глубин, а также в местах, отстоящих поперечно по отношению к направлению потока, и температурный профиль контролируется с определением, когда эффективная часть катализатора, ближайшая к УАА-катализатору, становится дезактивированной. Когда это имеет место, температура на впуске слегка увеличивается, так что месторасположение, где имеют место реакции окислительного дегидрирования, может слегка сместиться в каталитическом слое, и новый слой катализатора приводится в эффективное использование. Далее реакционный способ контролируется с определением того, когда вновь используемый слой катализатора становится дезактивированным, и температура на впуске снова увеличивается с перемещением слоя эффективной реакции выше в слое. Таким образом, коксование верхних слоев катализатора может контролироваться так, что слой катализатора, относительно не подвергшийся коксованию, находится всегда в использовании до тех пор, пока самый верхний слой в слое не станет достаточно дезактивированным, так что изменение катализатора подтверждается. Одновременно в данном способе содержание кислорода в УАА-катализаторе, а также в нижних слоях катализатора окислительного дегидрирования может контролироваться как дублирование контроля температурного профиля для дополнительного гарантирования, что удается избежать проскока кислорода в УАА-катализатор и весьма нежелательных последствий этого.
Подходящие ферритные оксидные катализаторы для настоящего изобретения являются обычно до некоторой степени хрупкими и ломкими, так что, когда традиционная технология используется для рецептурирования и загрузки катализатора, имеются значительные трудности в обеспечении того, чтобы каталитический слой не стал частично закупоренным фрагментами частиц. Такие фрагменты могут быть результатом загрузки, относительного движения между частицами в процессе работы или даже просто массы частиц выше, раз достигается полная глубина слоя. Данный вопрос может быть решен как (i) предварительным приведением каталитических частиц перед тем, как они загружаются в реактор, чтобы сделать их более износостойкими, так и (2) загрузкой катализатора с использованием низкоударной технологии загрузки, такой как носок, или даже загрузкой катализатора рукой в противоположность простому сбрасыванию. Идеально как технология предварительного приведения, так и технология низкоударного размещения используются вместе с обеспечением того, что падение давления через слой поддерживается как можно низким.
Краткое описание чертежей
Настоящее изобретение описывается подробно ниже со ссылкой на многочисленные примеры и прилагающиеся чертежи, на которых одинаковые номера обозначают везде подобные части, и где:
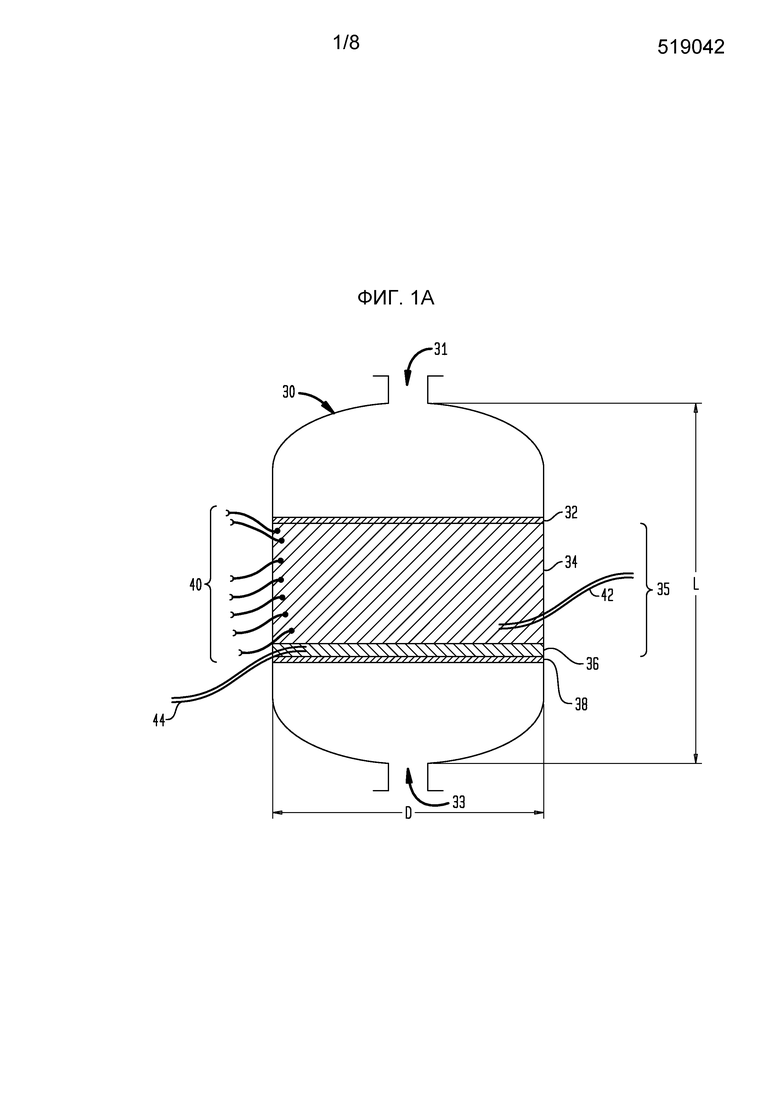
На фигуре 1А схематически представлено сечение реактора, используемого для осуществления настоящего изобретения.
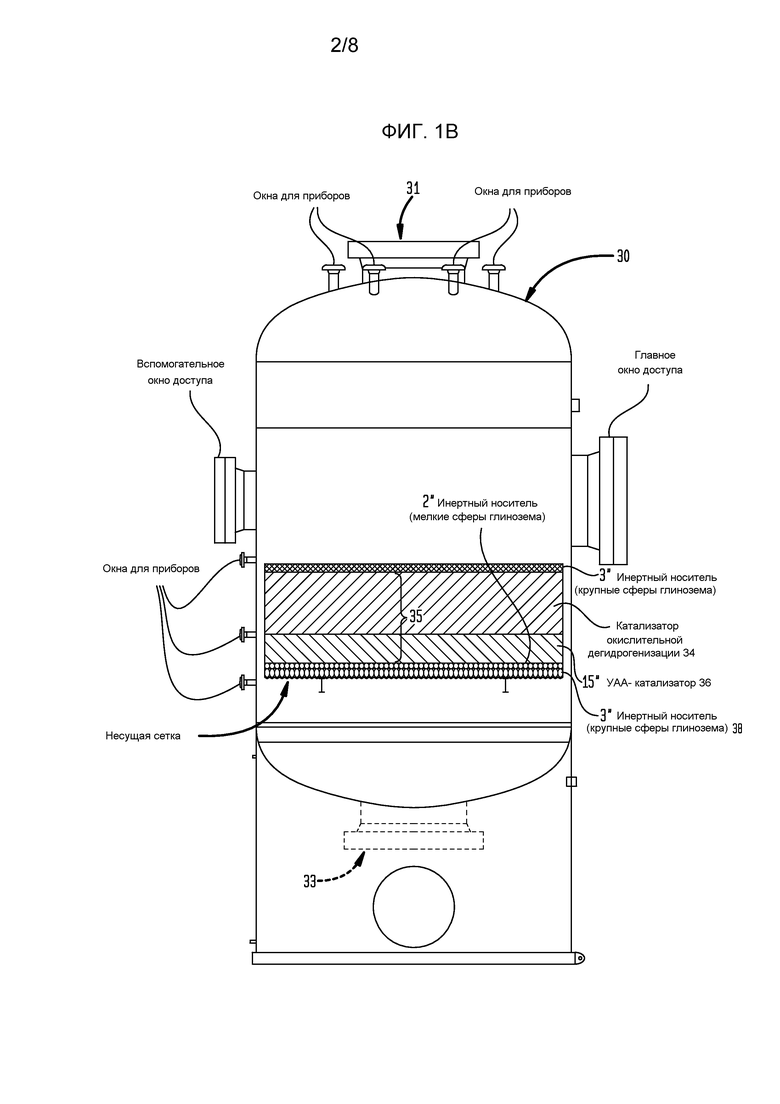
На фигурах 1В и 1С схематически представлено более подробно сечение реактора, показывающее головное пространство в реакторе выше несущей сетки катализатора, а также относительное расположение окон для доступа и приборного оснащения и несущей сетки катализатора.
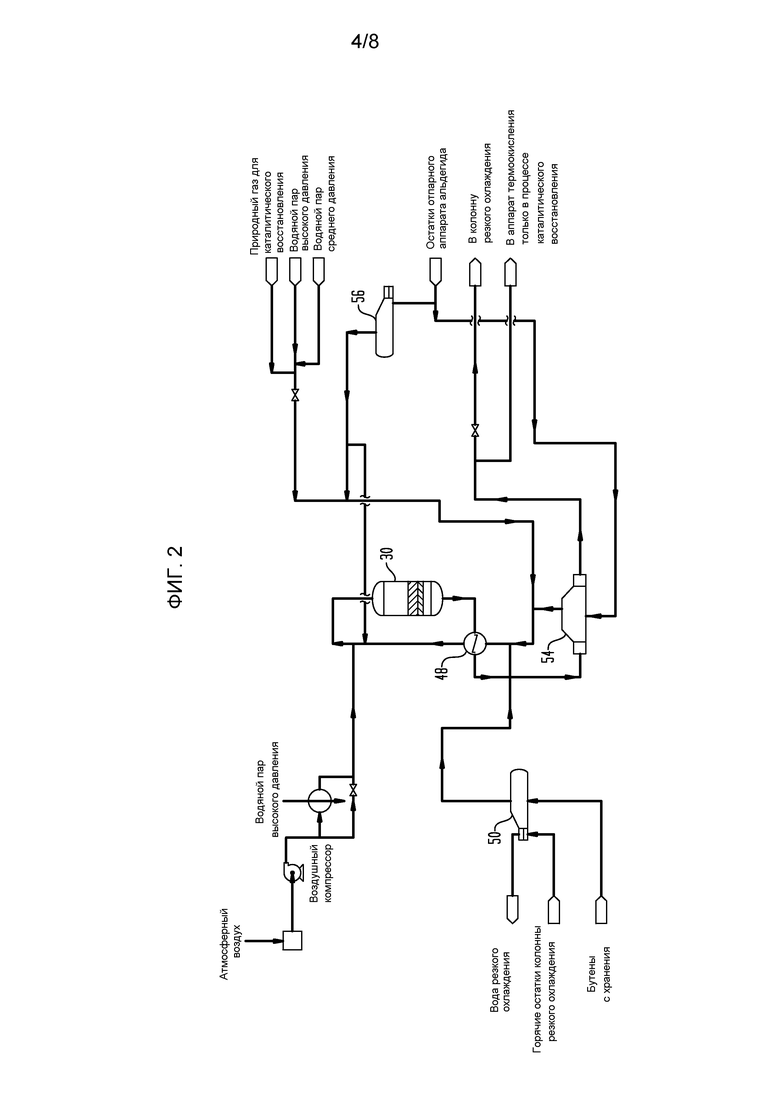
На фигуре 2 представлена блок-схема реакторной секции установки сырого бутадиена, показывающая реактор и оборудование предварительной обработки для приведения бутенобогащенного питания к впускным условиям, требуемым для работы реактора.
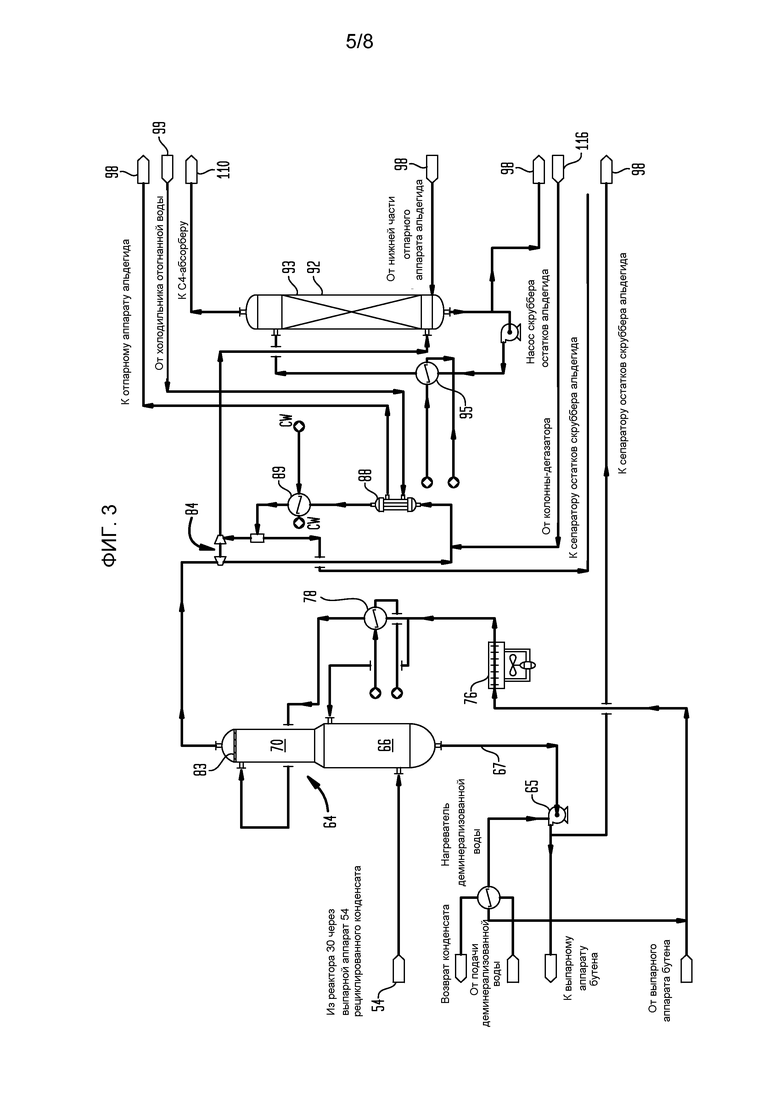
На фигуре 3 представлена блок-схема части установки сырого бутадиена, показывающая газосжимающеее и скрубберное оборудование для начальной переработки потока бутадиенобогащенного продукта, полученного в секции реактора с фигуры 2.
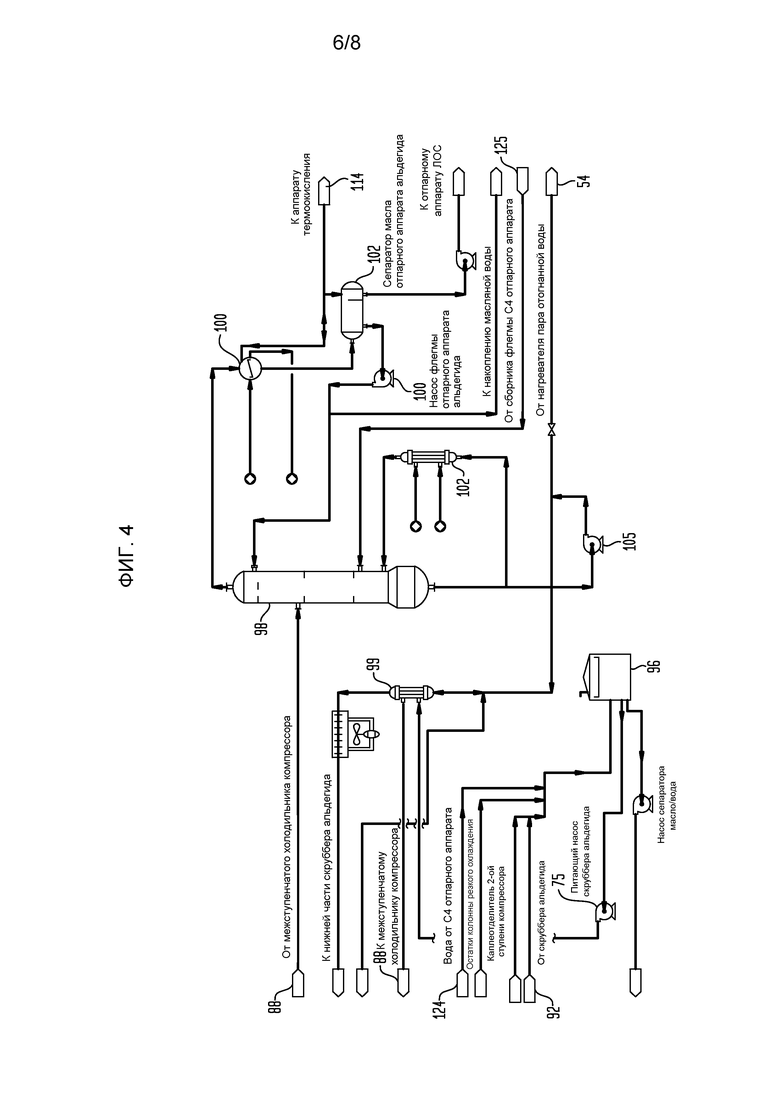
На фигуре 4 представлена блок-схема части установки сырого бутадиена, показывающая отпарной аппарат альдегида и связанное оборудование для переработки потока бутадиенобогащенного продукта после обработки в секции газосжимающего и скрубберного оборудования с фигуры 3.
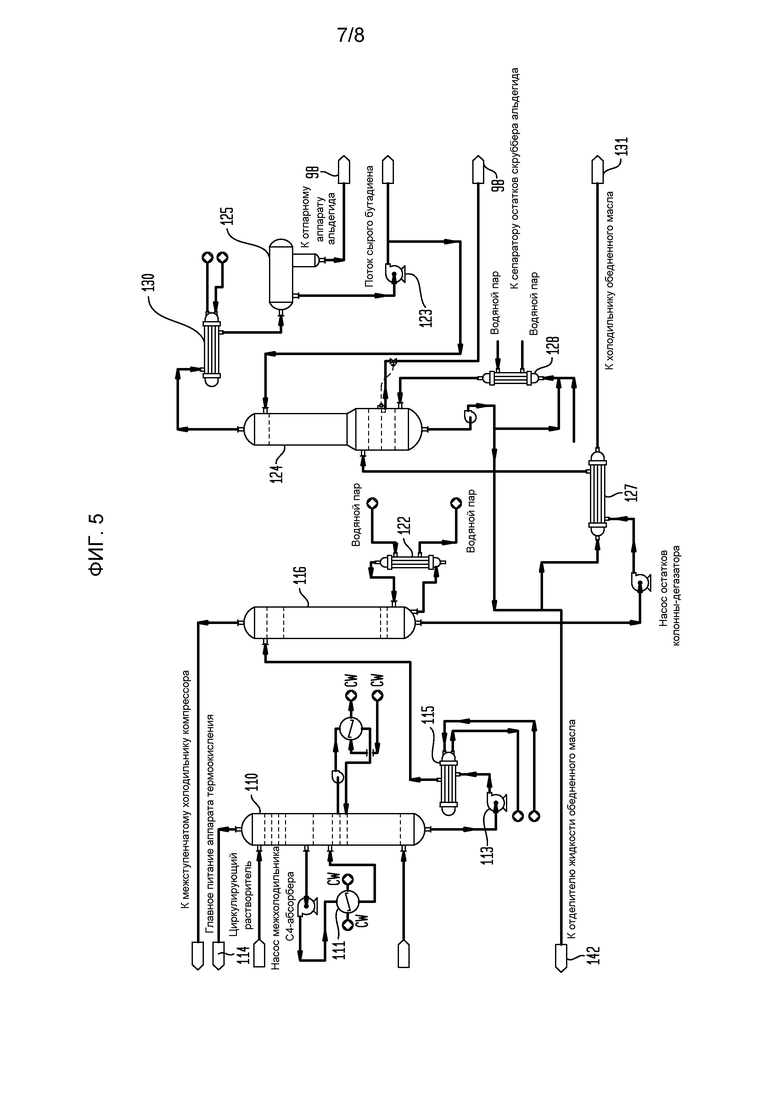
На фигуре 5 представлена блок-схема части установки сырого бутадиена, показывающая оборудование абсорбции и отпаривания C4-соединений для получения сырого потока примерно 50% бутадиена при переработке потока бутадиенобогащенного продукта, полученного из секции отпарного аппарата альдегида с фигуры 4.
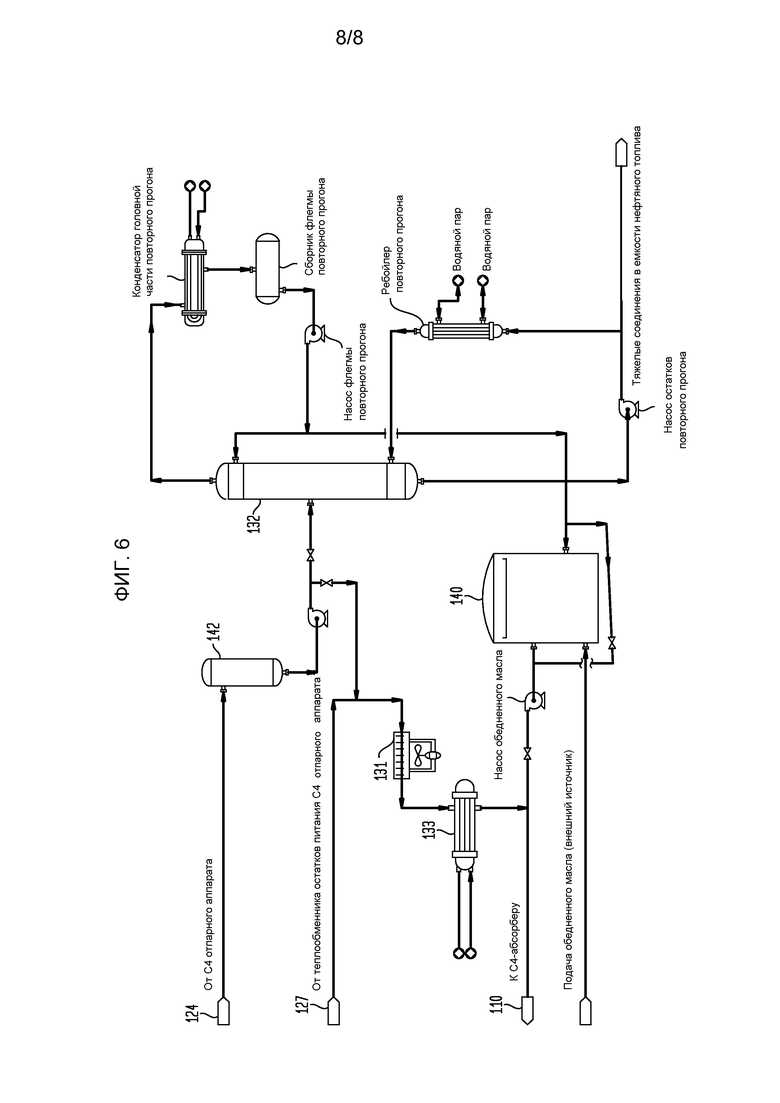
На фигуре 6 представлена блок-схема части установки сырого бутадиена, показывающая части системы, используемой для обработки обедненного масла после отпаривания из него C4-соединений.
Подробное описание предпочтительных вариантов
Настоящее изобретение описывается подробно с использованием фигур только в целях иллюстрации. Изобретение определяется в прилагаемой формуле изобретения. Терминология, используемая в описании и в формуле изобретения, дает обычные значения, например, термин «косвенный теплоперенос» относится к теплопереносу от одного носителя к другому носителю через стенку теплообменника, и давление относится к манометрическому давлению, если не указано иное. При осуществлении способа изобретения, предпочтительно, тепло переносится через одну стенку теплообменника от высокотемпературного потока к низкотемпературному потоку так, как тепло переносится от выходящего потока реактора к питанию реактора в перегревателе питания, как описано далее. Косвенный теплоперенос может быть выполнен в соответствии с изобретением с использованием подходящего оборудования, такого как теплообменники типа труба-в-трубе или теплообменники типа пластина-и-рамка.
Если не указано иное, термин «бутадиен», или «БД», относится к 1,3-бутадиену или к смесям, содержащим 1,3-бутадиен.
Переднее завершение системы получения бутадиена по настоящему изобретению содержит множественные в значительной степени идентичные последовательности способа, причем каждая последовательность способа имеет один реактор 30, дающий поток бутадиенобогащенного продукта, из которого используемое тепло извлекается непрямым теплообменом перед поступлением в колонну 64 резкого охлаждения, в точке которой все потоки способа объединяются. Будет показана только одна последовательность, чтобы избежать излишней сверхсложности.
Что касается фигур 1А-1С, смесь реакционного питания, содержащая бутенобогащенный углеводородистый газ, кислород в соотношении примерно 0,55 моль кислорода на моль углеводорода и водяной пар в соотношении примерно 15 моль на моль углеводорода, поступает в реактор 30 через верхнее впускное окно 31 реактора 30 и течет вниз до столкновения со слоем 32 инертных гранул глинозема, имеющих средний размер частиц около 3-10 мм. Обычно указанные инертные частицы являются глиноземом с низкой площадью поверхности, такие как, возможно, альфа-глинозем в большей степени, чем формы с высокой площадью поверхности, обычно в общих чертах относящиеся к гамма-глинозему, хотя имеются несколько промежуточных форм глинозема, показывающих более высокую площадь поверхности, чем альфа-глинозем. Сферы альфа-глинозема, имеющие следующие характеристики, являются довольно подходящими как для самого верхнего, так и самого низкого слоев в каталитическом слое:
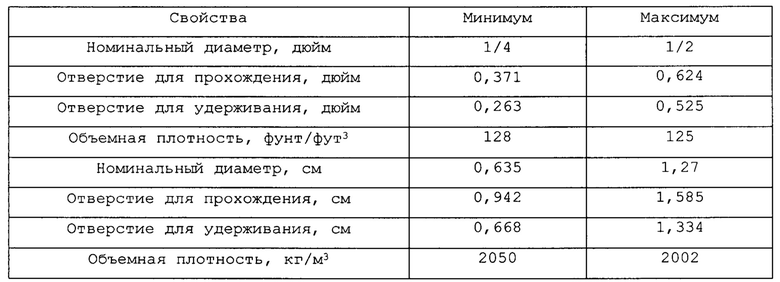
Верхний слой 32 может быть глубиной от примерно 50 мм до примерно 100 мм, такой как от примерно 65 до 85 мм, и в некоторых случаях от примерно 70 до 80 мм. Каждый слой в каталитическом слое, включая верхний слой 32, устанавливают с использованием низкоударного размещения, такого как технология загрузки носком, чтобы избежать разрушения слоев катализатора ниже, тогда как глубина верхнего слоя 32 ограничивается для того, чтобы избежать раздавливания катализатора окислительного дегидрирования ниже. Когда частицы катализатора загружаются носком, важно избежать воздействия на них больших усилий, таких как усилия в результате падения частиц с большой высоты. Если высота носка выше слоя регулируется так, что частицам не позволяют падать с высоты более примерно 91 см (36 дюйм), имеется небольшая опасность значительного разрушения частиц, хотя падение с высоты 185 см (70-75 дюйм) иногда может быть допустимо, в частности, если частицы предварительно приводятся или иным образом термообрабатываются для улучшения их прочности. Аналогично, когда катализатор помещается в бункер на верхний конец носка, точно также принимаются меры, чтобы частицы катализатора не падали с излишней высоты. Также может использоваться загрузка рукой.
В других конфигурациях секции слоя 35, которые включают слои 34, 36, могут быть размещены как кольцевые секции с реагентами, текущими радиально через слой. Теоретически инертные гранулы с распределением потока имеют размеры и конфигурированы для предотвращения нарушения слоя 34 окислительного дегидрирования благодаря какой-либо турбулентности, или завихренности, в потоке смеси реакционного питания при приближении к каталитическому слою. Частицы катализатора окислительного дегидрирования могут быть любого физического размещения, которое обеспечивает эффективный контакт между каталитическими активными частицами и реагентами, включая диспергированные на инертном носителе, но, скорее, массивными частицами, чем активными, диспергированными на носителе катализатора с высокой площадью поверхности. Предпочтительно, частицы катализатора имеют размер от примерно 1 до примерно 25-30 мм, часто принимая форму экструдатов или сфер диаметром от примерно 1 мм до примерно 5 мм. В частности, частицы катализатора, предпочтительно, используемые в связи с настоящим изобретением, должны быть слегка крупней, чем обычно используемые в предшествующей практике, для ограничения падения давления через каталитический слой, то есть авторы изобретения предпочитают использовать каталитический слой, который является глубже, чем обычно используемый ранее. Более высокое падение давления требует более высокого давления в системе, что снижает селективность. Авторы изобретения предпочитают использовать частицы катализатора, имеющие два ключевых отличия от предшествующей практики: (i) частицы «предварительно приводятся» или иным образом термообрабатываются перед загрузкой для придания им прочности на раздавливание, необходимой, чтобы быть используемыми в слое, имеющем глубину от примерно 50 см до примерно 150 см (от примерно 20 дюйм до примерно 60 дюйм), подходяще глубину от примерно 65 см до примерно 130 см (от примерно 25 дюйм до примерно 50 дюйм) или от примерно 75 см до примерно 100 см (от примерно 30 дюйм до примерно 40 дюйм), тогда как объемная плотность частиц составляет не более примерно 1100 кг/м3 (примерно 70 фунт/фут3), подходяще от примерно 880 кг/м3 до примерно 1050 кг/м3 (примерно 55-65 фунт/фут3) или от примерно 920 кг/м3 до примерно 1010 кг/м3 (примерно 58-63 фунт/фут3); и (2) предпочтительно, удается избегать использовать нитраты, которые традиционно используются в качестве предшественников соединений кальция, часто вводимых в указанные катализаторы. Ацетат кальция является подходящим предшественником в данном отношении и имеет преимущество снижения выбросов NOx, хотя хлорид кальция и карбонат кальция являются также подходящими.
Частицы катализатора окислительного дегидрирования, имеющие состав, как представлено здесь в сопутствующей заявке «Способ окислительного дегидрирования с низкими выбросами для получения бутадиена», указанной выше, размещаются в слое или слое 34, имеющем глубину от свыше 69 см (27 дюйм) до примерно 152 см (60 дюйм), предпочтительно, в интервале от примерно 71 см (28 дюйм) до 127 см (50 дюйм), более предпочтительно, в интервале от примерно 76 см (30 дюйм) до 102 см (40 дюйм), бутенобогащенное углеводородистое питание превращается в поток бутадиенобогащенного продукта, который проходит ниже слоя или слоя 34 окислительного дегидрирования через слой или слой 36 УАА-катализатора.
В слое 36 частиц УАА-катализатора алкины и альдегиды в потоке бутадиенобогащенного продукта превращаются в более безвредные частицы, которые не являются такими вредными для использования бутадиена в обычных последующих реакциях полимеризации. Предпочтительно, слой 36 УАА-катализатора присутствует на глубине от примерно 40% до примерно 60% глубины катализатора окислительного дегидрирования, более предпочтительно, примерно 50%. Альтернативно, глубина может составлять от примерно 30 см (12 дюйм) до примерно 51 см (20 дюйм), более предпочтительно, от примерно 33 см (13 дюйм) до примерно 48 см (19 дюйм), и, наиболее предпочтительно, от примерно 36 см (14 дюйм) до примерно 46 см (18 дюйм). Ниже слоя 36 УАА-катализатора лежит слой 38 инертного носителя, состоящий из сфер глинозема, имеющих диаметр в интервале от примерно 1,0 см (0,4 дюйм) до 2,54 см (1 дюйм), причем слой 38 инертного носителя имеет глубину, предпочтительно, от примерно 2,54 см (1 дюйм) до примерно 20 см (8 дюйм), предпочтительно, от примерно 5,08 см (2 дюйм) до примерно 10 см (4 дюйм), более предпочтительно, от примерно 6,4 см до 8,9 см (2,5-3,5 дюйм), и, даже более предпочтительно, от примерно 6,99 см до 7,62 см (2,75-3 дюйм). В других случаях слой более крупных шариков может быть отделен от УАА-катализатора слоем более мелких шариков, как показано на фигуре 1А. После выхода из слоя 38 инертного носителя поток бутадиенобогащенного продукта выходит из реактора 30 через нижнее выпускное окно 33 для последующего извлечения теплотворной способности, содержащейся в нем, и концентрации содержания бутадиена в потоке сырого бутадиена, имеющего концентрацию приблизительно 50-60% бутадиена.
Обычно каталитический способ инициируется повышением температуры каталитического слоя до примерно 425°С (800°F); введением реагентов до тех пор, пока не наблюдается конверсии, затем снижением температуры на впуске с регулированием температуры каталитического слоя. В большинстве случаев природный газ используется для доведения потоков до нужной температуры; затем использование природного газа резко сокращается или отключается полностью, как только наблюдается конверсия. При установившейся работе, когда бутенобогащенное питание вначале воздействует на каталитический слой, условия на впуске тщательно контролируются, так что большая часть конверсии бутенов в бутадиен имеет место в последних нескольких сантиметрах слоя 34 катализатора окислительного дегидрирования выше УАА-катализатора, что вначале регистрируется как значительное изменение стадии в температуре, регистрируемое только самой нижней из термопар 40, распределенных в слое 34 катализатора окислительного дегидрирования, термопар в слое катализатора окислительного дегидрирования, в котором имеет место реакция. Чрезвычайно важно, что реакция по существу завершается до того, как реагенты достигают УАА-катализатора, что, главным образом, контролируется внимательным наблюдением за изменением температуры в реакторе с обеспечением того, что реакционная зона располагается выше УАА-катализатора и перемещается вверх, когда нижние слои катализатора окислительного дегидрирования начинают терять каталитическую активность. Дополнительно, обеспечение месторасположение реакционной зоны может быть подтверждено измерением содержания кислорода чуть выше самого нижнего слоя катализатора окислительного дегидрирования, а также в самом УАА-катализаторе: присутствие любого количества кислорода считается весьма вредным, даже хотя такое высокое содержание кислорода, как 0,3-0,5%, может быть допустимым в течение коротких периодов времени. Когда реакция прогрессирует, катализатор окислительного дегидрирования в самой нижней части слоя 34 катализатора окислительного дегидрирования становится дезактивированным, на что указывает снижение регистрируемой температуры, и что может быть отражено также в измерениях селективности или выхода. Когда самые нижние термопары в массиве начинают регистрировать снижение температуры так, что имеется какой-либо значительный риск проскока кислорода в УАА-катализатор, температура на впуске слегка увеличивается с перемещением реакционной зоны вверх в катализаторе окислительного дегидрирования. Таким образом, удается избежать коксования катализатора в слоях катализатора окислительного дегидрирования выше слоя использования. Во всем способе содержание кислорода или, более точно, отсутствие значительного содержания кислорода в УАА-катализаторе тщательно контролируется для подтверждения того, что кислород не проскакивает в слой УАА-катализатора. Когда самый верхний слой катализатора окислительного дегидрирования становится дезактивириованным в такой степени, что требуется замена катализатора, способ прерывается, и подается новый слой катализатора. Во многих случаях может быть достигнут срок службы катализатора свыше 80 суток (1920 ч) до примерно 1 года (8760 ч), хотя нет необходимости предпринимать попытки устанавливать рекорды без достаточной степени уверенности, что удастся избежать проскока кислорода. Обычно температура на впуске должна увеличиваться постепенно в ходе прогона, так что экономика способа иногда испытывает трудности к концу прогона, дополнительно отбивая охоту пытаться устанавливать рекорды долговечности. В отсутствие некоторого сбоя авторы изобретения ожидают срок службы катализатора минимум 180 суток (4320 ч), если строго соблюдаются вышеуказанные меры предосторожности.
На фигурах 1В и 1С показана конструкция реактора 30 настоящего изобретения, в которой окно (окна) доступа имеют размеры, которые обеспечивают введение в боковой стенке реактора 30 несущей сетки 38 катализатора, расположенной ниже, обеспечивая головное пространство подходяще, по меньшей мере, примерно 1,8 м (6 фут) зазора между полным уровнем каталитического слоя и верхней поверхностью реакторной камеры.
Распределение потока является также важным для того, чтобы избежать течения по каналам и горячих точек в каталитическом слое. Предпочтительный режим течения является полностью турбулентным и улучшается наличием впускного распределителя. В частности, впускной распределитель преимущественно предусматривается для обеспечения однородного распределения потока через каталитический слой и предотвращения течения по каналам и возможного создания горячих точек, что, вероятно, сокращает срок службы катализатора. Одна предпочтительная конструкция указанного впускного распределительного устройства находится в форме перегородок и колец, которые устанавливаются в паровом пространстве выше каталитического слоя, способствуя достаточному распределению потока и минимизируя потери давления на впуске.
Что касается фигуры 2, бутенобогащенное питание испаривается в выпарном аппарате 50 бутена, в котором тепло, необходимое для испаривания, подается при отводе тепла от остатков колонны 64 резкого охлаждения, которые, как будет рассмотрено далее, нагреваются при контакте с горячим продуктом реакции сразу, как достигается установившаяся работа в настоящем способе. После прохождения через выпарной аппарат 50 бутена выпаренное бутеновое питание смешивается с водяным паром, причем водяной пар генерируется в двух выпарных аппаратах 54 и 56 рециклированного конденсата. Водяной пар, генерированный в выпарном аппарате 54 рециклированного конденсата, получается при косвенном теплообмене с потоком бутадиенобогащенного продукта, выходящим из перегревателя 48 реакторного питания. Тепло, необходимое для генерирования водяного пара в выпарном аппарате 56 рециклированного конденсата, предпочтительно, подается водяным паром либо из сети установки, либо, предпочтительно, из аппарата термоокисления или другого удобно доступного источника. Предпочтительно, водяной пар полностью испаривается в выпарном аппарате 56 рециклированного конденсата перед смешением с выпаренным бутеном перед пропусканием через перегреватель 48 реакторного питания, в котором реакторное питание предварительно нагревается при косвенном теплообмене с потоком бутадиенобогащенного продукта, выходящим из реактора 30, с получаемым объединенным входящим потоком, имеющим температуру, по меньшей мере, примерно 345°С (примерно 650°F), предпочтительно в интервале от примерно 345°С до 400°С (от примерно 650°F до 750°F). Таким образом, питание реактора 30 нагревается до требуемой температуры при косвенном теплообмене с выходящим потоком, который, как будет рассмотрено далее, находится обычно при температуре выше 535°С (1000°F), более предпочтительно около 595°С (1100°F). Существенно, что извлеченное тепло проходит только через одну стенку трубы в противоположность схемам, в которых используется промежуточная текучая среда. Предварительно нагретое реакторное питание, выходящее из перегревателя 48 реакторного питания, смешивается со сжатым кислороднесущим газом, обычно воздухом, с тщательно регулируемым количеством воздушного питания, так что приблизительно 0,5-0,6 моль кислорода подается на каждый моль углеводорода в питании, проходящем в реактор. В некоторых случаях бывает удобно предварительно нагревать кислороднесущий газ от примерно 205 до примерно 235°С (от примерно 400 до примерно 450°F) с использованием водяного пара высокого давления. После смешения поток реакционной смеси пропускается в адиабатический реактор 30 с огнеупорной обкладкой, показанный на фигуре 1, где питание бутен/водяной пар/воздух внутри реактора 30 проходит сначала через слой 33 распределения инертного потока, затем в каталитический слой 34 окислительного дегидрирования, имеющий глубину 83,8 см (33 дюйм) или около этого, каталитический слой 34 удаления альдегида и ацетилена (УАА) и слой 38 инертного носителя (сферы глинозема).
Месторасположение интенсивной экзотермической реакции, имеющей место в каждом реакторе, контролируется рядом дистанционно считываемых термопар 40, пространственно расположенных по высоте каталитического слоя 34 окислительного дегидрирования, так что месторасположение в нем реакционной зоны может быть определено. Количество кислорода, остающегося в потоке продукта, контролируется анализатором 42 кислорода, расположеннным вблизи нижней части слоя 34, так что удается избежать проскока кислорода в УАА-слой 36, как рассмотрено далее более подробно. Также предусматривается нижнее окно 44 для отбора проб для анализатора затухания в слое 36, так что состав может контролироваться на нижнем экстремуме реактора. Термопары 40 также необязательно расположены в слое 36 для контроля температуры в УАА-зоне. Вместо термопар может быть использовано любое подходящее температуровоспринимающее устройство, такое как детекторы сопротивления температуры или неконтактные датчики в подходящей конструкции реактора.
Для того, чтобы регулировать систему, заданная температура для реакционной зоны предварительно выбирается и поддерживается в реакционной зоне. Реакционная зона в слое 34 вначале располагается вблизи нижней части слоя 34. Реакционная зона или «активный» слой каталитического слоя 34 окислительного дегидрирования характеризуется относительно резким подъемом температуры в относительно небольшой глубине слоя до предварительно выбранной заданной температуры. Обычно реакционная зона характеризуется ростом температуры от 100°F до 300°F (от 55°С до 167°С) через изменение глубины слоя от 1 до 5 дюйм (от 2,5 см до 13 см) при заданной температуре. Более предпочтительно, активный слой характеризуется ростом температуры от 150°F до 250°F (от 83°С до 139°С) через глубину слоя от 2 до 4 дюйм (от 5 см до 10 см). Ниже реакционной зоны в слое 34, предпочтительно, отсутствует дополнительный рост температуры, если система регулируется соответствующим образом, поскольку кислород полностью или почти полностью истощается в реакционной зоне и больше не присутствует в системе.
Подходящие рабочие заданные температуры для реакционной зоны окислительного дегидрирования составляют от 1000°F до 1200°F (от 340°С до 650°С). Когда заданная температура реакционной зоны начинает падать, температура на впуске реактора повышается, и активная зона смещается вверх в слое 34. Можно вычислить время проскока кислорода на основе скорости изменения температур в слое, которая проявляется в скорости смещения вверх реакционной зоны и оставшейся глубиной слоя выше реакционной зоны. Расчет времени проскока основан на показаниях температуры в слоях выше реакционной зоны (которые являются ниже заданной температуры для реакционной зоны) больше, чем на температурах в или ниже реакционной зоны, т.к. температура выше реакционной зоны является показателем относительно свежего катализатора, доступного для катализа реакции. Таким образом, если изменение температуры во времени показывает, что реакционная зона смещается вверх со скоростью 0,5 см/сутки (24 ч), и самая верхняя термопара (термопары) показывает слой свежего катализатора 5 см, тогда только 10 суток (240 ч) работы остается до проскока кислорода при условии, что скорость истощения катализатора окислительного дегидрирования остается относительно постоянной.
При регулировании смещения реакционной зоны описанным здесь образом катализатор окислительного дегидрирования дает наилучшие характеристики в течение длительного времени.
Как указано ранее, поток горячего продукта реакции из реактора 30 проходит через перегреватель 48 реакторного питания (фигура 2), который подает часть тепла, используемого для доведения питания реактора 30 до требуемой рабочей температуры, и затем продукт реакции, выходящий из перегревателя 48 реакторного питания, проходит через парогенератор 54, в котором часть значительного тепла, содержащегося в нем, используется для испаривания и/или перегревания водяного пара, проходящего в реактор 30.
Позже бутадиенобогащенный реакционный продукт, выходящий из парогенератора 54, проходит в колонну 64 резкого охлаждения (фигура 3), входя на высоте слегка выше максимального уровня жидкости, ожидаемого в процессе нормальной работы. Как указано, в предпочтительном варианте изобретения поток бутадиенобогащенного продукта из реактора 30 объединяется с другими потоками бутадиенобогащенного продукта из других реакторов (не показано) перед поступлением в колонну 64 резкого охлаждения. В одном варианте нижняя секция 66 колонны 64 резкого охлаждения оборудована тарелками клапана, тогда как верхняя секция 70 снабжена гофрированной металлической структурной набивкой, такой как Koch Flexipac, подобной описанной в Lantz et al., патент США 6874769, Stuctured Packing Plate and Element and Method of Fabricating Same or Rucovena, патент США 4740334. Альтернативно, распылительные форсунки могут быть использованы для всей колонны. Ожидается, что во многих случаях будет возможно подавать выходящую смесь парообразного и жидкого продукта реакции прямо в колонну 64 резкого охлаждения без какой-либо предварительной фазовой сепарации, но такая предварительная фазовая сепарация может быть легко осуществлена, если целесообразно, при введении испарительной емкости или подобного устройства фазовой сепарации. Жидкая фаза конденсата, собранная на нижнем выпуске 67 колонны 64 резкого охлаждения, содержащая, главным образом, конденсированный водяной пар и воду резкого охлаждения, подается обратно через горячую сторону выпарного аппарата 50 бутена с возвратом охлажденной жидкости, пропускаемой обратно через воздушный холодильник 76 быстроохлажденного кондесата и затем в холодильник 78 с циркуляцией колонны быстрого охлаждения перед подачей в колонну 64 резкого охлаждения в месте расположения намного выше верха секции 70 с набивкой колонны 64 резкого охлаждения, но ниже подушки 83 туманоуловителя. Предпочтительно, воздушный холодильник 76 быстроохлажденного кондесата оборудован модульными блоками труб, отдельно регулируемыми вентиляторами и лопастями вентиляторов с различным шагом для облегчения регулирования температуры в ряду окружающих условий. Во многих случаях можно извлекать дополнительное тепло из нижнего потока колонны 64 резкого охлаждения для использования где-либо еще в связанной установке, снижая размер и стоимость холодильников 76 и 78 колонны резкого охлаждения.
Пары сырого бутадиена выходят из верхней секции 70 колонны 64 резкого охлаждения (фигура 3), проходя через подушку 83 туманоуловителя, который включен, главным образом, для защиты газового компрессора 84 от попадания любых уловленных капель жидкости и поступают на сторону отсасывания двухступенчатого центробежного газового компрессора 84. Косвенное межступенчатое охлаждение обеспечивается межступенчатыми холодильниками 88 и 89 компрессора с охлаждением в межступенчатом холодильнике 88 компрессора, питаемом потоком способа, выходящим из холодильника 99 выпаренной воды, и нагретый поток от стороны оболочки межступенчатого холодильника 88 газового компрессора подается в отпарной аппарат 98 альдегида (фигура 4). Охлаждение в межступенчатом холодильнике 88 компрессора удобно питается водой охлаждающей колонны установки.
Захваченные капли жидкости, коалесцированные на подушке 83 туманоуловителя, текут обратно через колонну 64 резкого охлаждения, тогда как сжатый бутадиенобогащенный продукт, сжатый до 1140 кПа абс. (около 150 фунт/кв. дюйм), выходит со второй ступени, и он пропускается в скруббер 92 альдегида, верхняя часть 93 которого, предпочтительно, набита структурной набивкой, которая может быть подобна структурной набивке Norton Intallox или набивкам, описанным выше. Часть остатков из скруббера 92 альдегида рециклируется через структурную набивку через холодильник 95 остатков скруббера альдегида, тогда как остальная часть пропускается в отпарной аппарат 98 через сепаратор 96 остатков скруббера альдегида (фигура 4), который получает жидкость из остатков колонны 64 резкого охлаждения с помощью насоса 65 остатков колонны резкого охлаждения, а также из каплеотделителя второй ступени газового компрессора 84. Содержание воды сепаратора 96 остатков скруббера альдегида может быть возвращено в колонну 64 резкого охлаждения в месте ниже подушки 83 туманоуловителя. Важным аспектом данного изобретения является то, что в тех случаях, когда значительные количества углеводородов легче С4 или других малоценных летучих могут быть удалены из различных потоков здесь, такие отходящие газы подаются в аппарат термоокисления, где они сжигаются с получением водяного пара, который может использоваться для подачи тепла, когда необходимо, для различных частей общего способа, поэтому значительно снижая необходимость сжигания природного газа при установившейся работе и поэтому также снижая загрязняющее выделение монооксида углерода и диоксида углерода.
Отпарной аппарат альдегида (фигура 4) получает водную фазу из остатков скруббера альдегида после отгонки масляной фазы. Указанный поток подают насосом сначала на оболочечную сторону холодильника 99 выпаренной воды, откуда она достигает оболочечной стороны межступенчатого холодильника 88 компрессора, что способствует увеличению ее температуры путем интеграции тепла перед подачей в отпарной аппарат 98 альдегида, часть указанных паров головной части из отпарного аппарата 98 альдегида идет в конденсатор 100 головной части отпарного аппарата альдегида и затем возвращается в отпарной аппарат 98 альдегида как флегма с поддержанием равновесия пар/жидкость в колонне и отводом альдегидов головной части, содержащихся в питании данной колонны 98. Баланс потока водяного пара головной части из отпарного аппарата 98 альдегида, обходящего конденсатор 100 головной части, объединяется с другими малоценными горючими соединениями и направляется в аппарат термоокисления (не показано) для получения перегретого водяного пара. Тяжелые углеводороды, унесенные в конденсированном потоке головной части из конденсатора 100 головной части, собираются коагулятором остатков, а также удаляются обработкой на традиционном устройстве маслянистой воды (не показано). Ребойлер 102 отпарного аппарата альдегида использует водяной пар, преимущественно водяной пар среднего давления, для испаривания части остатков отпарного аппарата альдегида из отпарного аппарата 98 альдегида и повторно вводит водяной пар ниже нижней тарелки отпарного аппарата 98 альдегида, тогда как остальная часть прокачивается с использованием насоса 105 остатков отпарного аппарата альдегида в два местоположения: (1) обратно в нижнюю часть скруббера 92 альдегида ниже набивки через два холодильника выпаренной воды (не показано), и (2) в выпарные аппараты рециклированного конденсата, где он генерирует очень большой, если не весь, объем водяного пара, используемого для реакции окислительного дегидрирования.
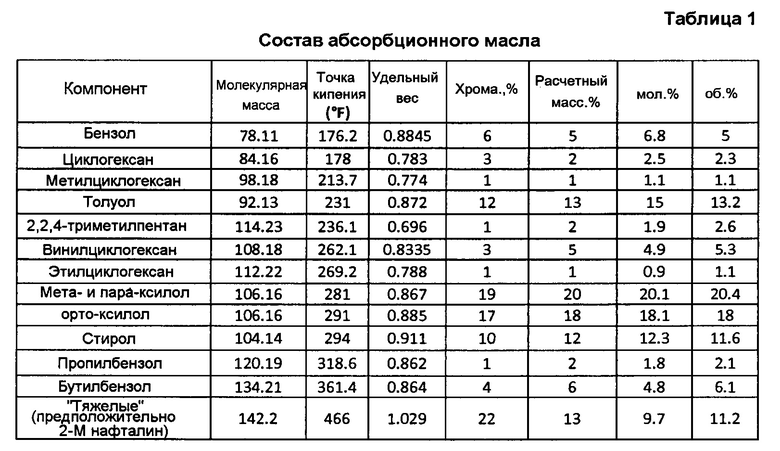
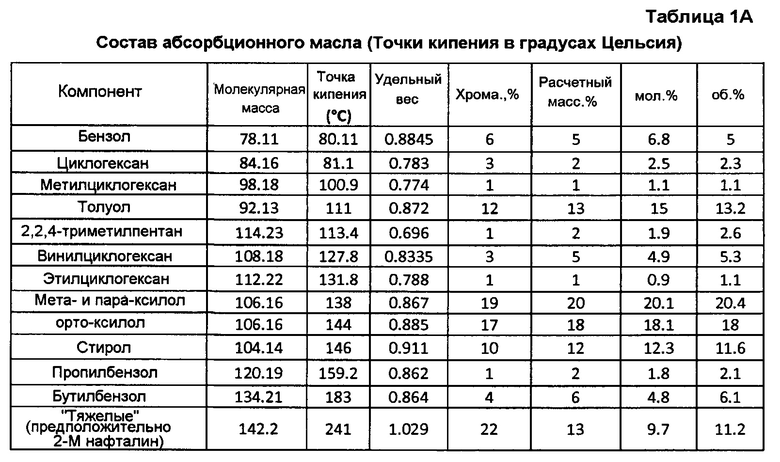
Продукт реакции из головной части скруббера 92 альдегида (фигура 3) проходит в нижнюю часть С4-абсорбера 110 (фигура 5), содержащего многочисленные тарелки или другие известные устройства для способствования контактированию газ/жидкость и оборудованного, по меньшей мере, одним взаимным холодильником 111. Абсорбционное масло (также иногда называемое обедненным маслом), используемое в абсорбере 110, может быть подходяще парафиновым, или смесью парафиновых соединений и ароматических соединений, хотя, по-видимому, подобные лучшие результаты получаются с использованием масел, которые являются богаче, частично или даже полностью, винилциклогексаном (бутадиеновым димером). Хорошие коммерческие результаты получаются, когда свежим абсорбционным маслом является, главным образом, Espersol 250, ароматический нафта-продукт с точкой кипения 90-150°С (200-300°F), имеющий состав, показанный в таблице 1 (точки кипения в градусах Цельсия представлены в таблице 1А). Альтернативно может использоваться нафта-продукт с подобными точками кипения.
Бутадиен в потоке продукта абсорбируется в абсорбционном масле, введенном в верхнюю часть С4-абсорбера 110, остатки из которого прокачиваются в верхнюю часть колонны-дегазатора 116 насосом 113 С4-абсорбера и в холодильник 115 питания дегазатора. Колонна-дегазатор 116 работает при низком давлении для облегчения удаления остаточных газов, в частности, диоксида углерода, азота и водорода, которые пропускают через межступенчатый холодильник 88 двухступенчатого газового компрессора 84 в поток бутадиенобогащенного продукта перед прохождением через скруббер 92 альдегида. Дегазированный газ головной части из дегазатора 116 рециклируется обратно на вторую ступень компрессора 84 и затем в скруббер 92 и абсорбер 110, откуда он, в конечном итоге, находит свой путь в аппарат 114 термоокисления. Ребойлер 122 дегазатора поддерживает температуру в жидкой фазе колонны-дегазатора 116 достаточно высокой, чтобы позволить остаточным газам быть отогнанными при прохождении через аппарат 114 термоокисления, как описано выше. Остатки из колонны-дегазатора 116, содержащие в значительной степени сырой бутадиен и смешанные С4-соединения в абсорбционном масле, пропускаются в отпарной аппарат 124 С4-соединений через теплообменник 127 остатков питания отпарного аппарата 124 С4-соединений, где указанный поток остатков нагревается при прохождении горячего абсорбционного масла из отпарного аппарата 124 остатков С4-соединений через трубы теплообменника 127 остатков/питания отпарного аппарата С4-соединений. Нагретые остатки дегазатора вводятся в отпарной аппарат 124 С4-соединений на промежуточной высоте. Сырой бутадиен и С4-соединения отпариваются из нагретого абсорбционного масла в отпарном аппарате 124 С4-соединений, проходя как поток головной части в конденсатор 130 потока головной части отпарного аппарата С4-соединений, тогда как отработанное абсорбционное масло, собранное в остатках из отпарного аппарата 124 С4-соединений, повторно нагревается в ребойлере 128 отпарного аппарата С4-соединений, и водяной пар головной части из отпарного аппарата 124 С4-соединений конденсируется в конденсаторе 130 потока головной части отпарного аппарата С4-соединений с частью конденсированной жидкости, накопленной в сборнике 125 орошающей фракции отпарного аппарата С4-соединений, где остаточная вода может быть отделена от углеводородной фазы и направлена обратно в отпарную колонну 98 альдегида, тогда как сырой бутадиеновый продукт прокачивается с помощью насоса 123 флегмы отпарного аппарата С4-соединений на дополнительную переработку, хотя достаточно сырого бутадиена рециклируется как обратный поток с обеспечением того, что достаточная сепарация достигается в отпарном аппарате 124 С4-соединений.
Остатки, выходящие из отпарного аппарата 124 С4-соединений, содержат абсорбционное масло, имеющее бутадиен и другие отпаренные С4-соединения, которые разделены на три части, одна из которых рециклируется в отпарной аппарат 124 С4-соединений через ребойлер 128 отпарного аппарата С4-соединений, вторая часть пропускается в отделитель 142 абсорбционного масла (фигура 6), остальная часть используется, как указано ранее, для нагревания смеси бутадиен/абсорбционное масло при прохождении через теплообменник 127 питания/остатков отпарного аппарата С4-соединений, где она и масло, рециклированное из отделителя 142 абсорбционного масла, пропускаются в воздушный холодильник 131 абсорбционного масла и холодильник 133 абсорбционного масла перед возвращением в С4-абсорбер 110 для повторного использования. Так как абсорбционное масло разрушается с образованием тяжелых молекул, свежее полученное масло вводится в систему, тогда как баланс направляется в колонну повторного прогона для очистки тяжелых молекул. При достаточном накоплении тяжелых молекул в абсорбционном масле для выравнивания или по необходимости работы колонны 132 повторного прогона адсорбционного масла часть масла, рециркулированного из отделителя 142 абсорбционного масла, перегоняется с удалением тяжелых компонентов в остатки колонны 132 повторного прогона адсорбционного масла с потоком головной части, подаваемым насосом обратно в контур рециркуляции абсорбционного масла. Необязательно извлеченное масло может подаваться насосом в емкость 140 для хранения, где хранится свежее абсорбционное масло.
В таблицах 2 и 2А представлен энергетический баланс для трех возможных конфигураций установки производительностью 23000 кг/ч (50600 фунт/ч) бутадиена: одна, не имеющая аппарата термоокисления; одна, имеющая небольшой аппарат термоокисления, имеющий размеры, главным образом, для малоценных горючих веществ, получаемых в способе конверсии бутена в бутадиен; и одна, имеющая размеры для малоценных горючих веществ, получаемых в способе конверсии бутена в бутадиен, а также получаемых в способе очистки сырого бутадиена до пользующегося спросом сорта. Можно заметить, что требования к энергии для испаривания и перегревания различных потоков, подаваемых в реактор в процессе установившейся работы способа для конверсии бутенов в бутадиен, являются удивительно небольшими, когда значительное тепло в потоке продукта реакции объединяется с энергией, происходящей от термического окисления малоценных горючих веществ от как получения, так и очистки бутадиена.

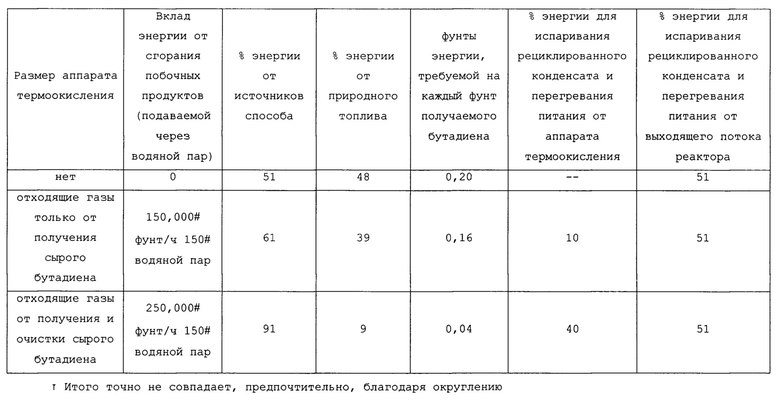

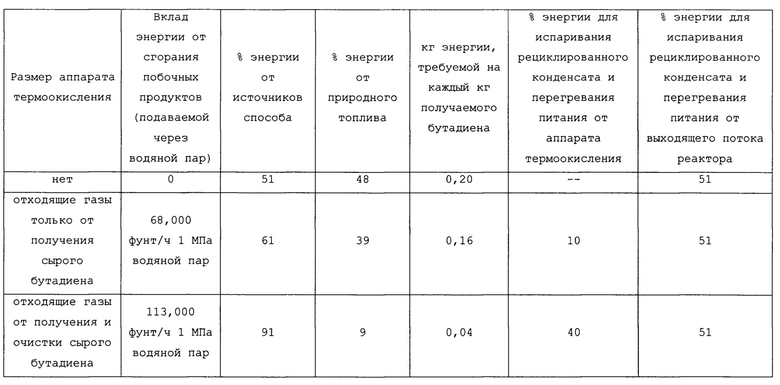
Требования к энергии для реакционной секции также могут быть выражены в кДж/кг (БТЕ/фунт) БД (бутадиена), полученного, как представлено в таблицах 3 и 3А ниже.


Вся энергия для перегревателя 48, свыше 4400 кДж/кг (1900 БТЕ/фунт) бутадиена, может быть подана при косвенном теплообмене значительного тепла от выходящего продукта реактора при высокой температуре с выходящим потоком продукта значительно выше 370°С (700°F). Точно так же вся энергия для выпарного аппарата 54 может быть аналогично подана при косвенном теплообмене при до некоторой степени более низкой температуре выходящего потока продукта. Извлечение тепла из потока способа улучшается при извлечении тепла из выходящего продукта, когда поток находится при относительно высокой температуре в целях перегревания питания, и затем извлечении тепла из выходящего продукта реактора при относительно низкой температуре в целях испаривания питания. Энергия для выпарного аппарата 56 может быть подана из сети водяного пара установки, которая отводит тепло от термического окисления летучих органических соединений, образованных в связи со способом окислительного дегидрирования, как описано здесь.
Хотя настоящее изобретение описано подробно, модификации в духе и объеме изобретения будут легко видны для специалистов в данной области техники. С точки зрения приведенного выше рассмотрения уровня техники и ссылок, включая одновременно рассматриваемые заявки, упомянутые выше в разделах «Предпосылки создания изобретения» и «Подробное описание изобретения», содержание которых полностью приводится здесь в качестве ссылки, дополнительное описание предполагается излишним. Кроме того, должно быть понятно, что аспекты изобретения и части различных вариантов могут быть объединены или взаимно заменены либо полностью, либо частично. Кроме того, специалистам в данной области техники будет ясно, что приведенное выше описание дается только путем примера и не предназначено ограничивать изобретение.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ОКИСЛИТЕЛЬНОГО ДЕГИДРИРОВАНИЯ С НИЗКИМИ ВЫБРОСАМИ ДЛЯ ПОЛУЧЕНИЯ БУТАДИЕНА | 2013 |
|
RU2619114C2 |
| УТИЛИЗАЦИЯ ТЕПЛА В СПОСОБЕ ПРОИЗВОДСТВА БУТАДИЕНА | 2016 |
|
RU2697666C1 |
| СПОСОБ ПОЛУЧЕНИЯ СТИРОЛЬНОГО МОНОМЕРА ОКИСЛИТЕЛЬНЫМ ДЕГИДРИРОВАНИЕМ ЭТИЛБЕНЗОЛА С ИСПОЛЬЗОВАНИЕМ CO В КАЧЕСТВЕ МЯГКОГО ОКИСЛИТЕЛЯ | 2009 |
|
RU2446137C1 |
| ИСПОЛЬЗОВАНИЕ АБСОРБЕРА С4 ДЛЯ ОТПАРИВАНИЯ АЛЬДЕГИДОВ | 2016 |
|
RU2693490C1 |
| ПОЛУЧЕНИЕ 4-ВИНИЛЦИКЛОГЕКСЕНА, ЭТИЛБЕНЗОЛА И СТИРОЛА | 2003 |
|
RU2350593C2 |
| СПОСОБ ОКИСЛИТЕЛЬНОГО ДЕГИДРИРОВАНИЯ ИЗОАМИЛЕНОВ | 1990 |
|
RU2027693C1 |
| СПОСОБ ПОЛУЧЕНИЯ БУТАДИЕНА ПУТЕМ ОКИСЛИТЕЛЬНОГО ДЕГИДРИРОВАНИЯ С ПОСЛЕДУЮЩИМ ПРЯМЫМ ДЕГИДРИРОВАНИЕМ | 2017 |
|
RU2696137C1 |
| СПОСОБЫ И УСТАНОВКИ ДЛЯ ПРОИЗВОДСТВА БУТАДИЕНА | 2016 |
|
RU2674664C1 |
| СПОСОБ И УСТРОЙСТВО ДЛЯ ПРОИЗВОДСТВА ПРОПЕНА И C4 УГЛЕВОДОРОДА | 2016 |
|
RU2726483C1 |
| СПОСОБ ПОЛУЧЕНИЯ, ПО МЕНЬШЕЙ МЕРЕ, ОДНОГО ПРОДУКТА ЧАСТИЧНОГО ОКИСЛЕНИЯ И/ИЛИ АММОКИСЛЕНИЯ ПРОПИЛЕНА | 2003 |
|
RU2346928C9 |
Изобретение относится к двум вариантам способа получения бутадиена из бутенобогащенного питания. Один из вариантов способа содержит следующие стадии: обеспечение бутенобогащенного углеводородистого питания, испаривания и перегревания указанного бутенобогащенного углеводородистого питания при температуре по меньшей мере примерно 345°С (650°F), смешение указанного бутенобогащенного углеводородистого питания с перегретым водяным паром и кислородобогащенным газом с образованием потока реакторного питания; обеспечение каталитического слоя гранул катализатора окислительного дегидрирования, прохождение указанного потока реакторного питания из впуска через указанный каталитический слой и образование в результате потока бутадиенобогащенного продукта; обеспечение указанного каталитического слоя катализатора окислительного дегидрирования связанным с ним множеством температуровоспринимающих устройств, предназначенных для измерения температуры в слое по направлению потока; регулирование условий на впуске указанного реактора, так что реакции окислительного дегидрирования первоначально имеют место в слоях указанного катализатора окислительного дегидрирования, наиболее отдаленных от указанного впуска, включая в реакционной зоне взаимодействие указанного потока реакторного питания с помощью указанного катализатора и образование в результате потока бутадиенобогащенного продукта; контроль температуры по длине слоя и время от времени увеличение температуры на впуске, так что реакционная зона мигрирует относительно указанного впуска в указанном каталитическом слое окислительного дегидрирования. Использование предлагаемого изобретения позволяет избежать последствий проскока кислорода в катализатор. 2 н. и 18 з.п. ф-лы, 6 табл., 8 ил.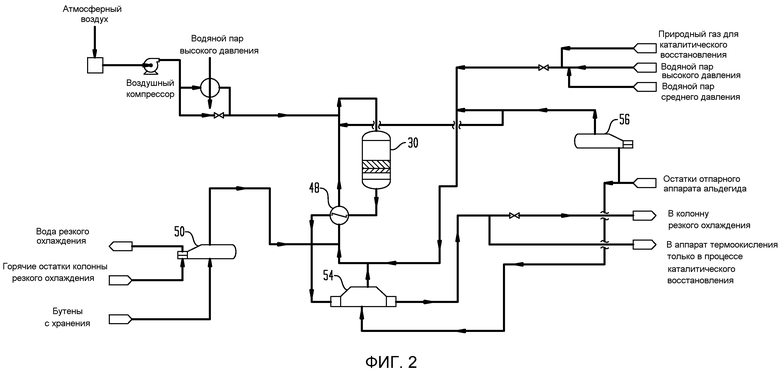
1. Способ получения бутадиена из бутенобогащенного питания, содержащий следующие стадии:
обеспечение бутенобогащенного углеводородистого питания, испаривания и перегревания указанного бутенобогащенного углеводородистого питания при температуре по меньшей мере примерно 345°С (650°F), смешение указанного бутенобогащенного углеводородистого питания с перегретым водяным паром и кислородобогащенным газом с образованием потока реакторного питания;
обеспечение каталитического слоя гранул катализатора окислительного дегидрирования, прохождение указанного потока реакторного питания из впуска через указанный каталитический слой и образование в результате потока бутадиенобогащенного продукта;
обеспечение указанного каталитического слоя катализатора окислительного дегидрирования связанным с ним множеством температуровоспринимающих устройств, предназначенных для измерения температуры в слое по направлению потока;
регулирование условий на впуске указанного реактора, так что реакции окислительного дегидрирования первоначально имеют место в слоях указанного катализатора окислительного дегидрирования, наиболее отдаленных от указанного впуска, включая в реакционной зоне взаимодействие указанного потока реакторного питания с помощью указанного катализатора и образование в результате потока бутадиенобогащенного продукта;
контроль температуры по длине слоя и время от времени увеличение температуры на впуске, так что реакционная зона мигрирует относительно указанного впуска в указанном каталитическом слое окислительного дегидрирования.
2. Способ получения бутадиена из бутенобогащенного питания по п. 1, который дополнительно содержит пропускание потока бутадиенобогащенного продукта через слой катализатора удаления альдегида и алкина, предпочтительно включающий железо, никель, щелочной металл и кислород, причем упомянутый катализатор обеспечивает эффективное удаление ацетиленовых примесей.
3. Способ получения бутадиена из бутенобогащенного питания по п. 1, в котором гранулы катализатора окислительного дегидрирования имеют диаметр в интервале от примерно 1 мм до примерно 30 мм.
4. Способ получения бутадиена из бутенобогащенного питания по п. 1, в котором гранулы катализатора окислительного дегидрирования имеют диаметр в интервале от примерно 1 мм до примерно 5 мм.
5. Способ получения бутадиена из бутенобогащенного питания по п. 1, в котором катализатором окислительного дегидрирования является ферритный катализатор окислительного дегидрирования.
6. Способ получения бутадиена из бутенобогащенного питания по п. 1, в котором ферритный катализатор окислительного дегидрирования по существу не содержит нитрат.
7. Способ получения бутадиена из бутенобогащенного питания по п. 6, в котором катализатор окислительного дегидрирования содержит кислород, главную пропорцию железа, незначительную пропорцию цинка и небольшие количества марганца, фосфора и остаток предшественника кальция, не содержащего нитрат.
8. Способ получения бутадиена из бутенобогащенного питания по п. 1, в котором количество молей кислорода в указанном потоке реакторного питания регулируется так, что обеспечивается по меньшей мере 0,5 моль кислорода на моль бутенобогащенного углеводородистого питания.
9. Способ получения бутадиена из бутенобогащенного питания по п. 1, в котором слой ферритного катализатора окислительного дегидрирования имеет глубину свыше 70 см (27 дюйм).
10. Способ получения бутадиена из бутенобогащенного питания по п. 1, в котором температуровоспринимающие устройства содержат термопары.
11. Способ получения бутадиена из бутенобогащенного питания, содержащий следующие стадии:
обеспечение бутенобогащенного углеводородистого питания, испаривания и перегревания указанного бутенобогащенного углеводородистого питания при температуре по меньшей мере примерно 345°С (650°F), смешение указанного бутенобогащенного углеводородистого питания с перегретым водяным паром и кислородобогащенным газом с образованием потока реакторного питания;
обеспечение каталитического слоя гранул ферритного катализатора окислительного дегидрирования, пропускание указанного потока реакторного питания через указанный каталитический слой и образование в результате потока бутадиенобогащенного продукта;
обеспечение слоя частиц катализатора удаления альдегида и алкина, предпочтительно включающий железо, никель, щелочной металл и кислород, причем упомянутый катализатор удаления альдегида и алкина расположен ниже каталитического слоя гранул ферритного катализатора окислительного дегидрирования;
обеспечение указанного каталитического слоя ферритного катализатора окислительного дегидрирования множеством температуровоспринимающих устройств, заделанных в него через его глубину, включая по меньшей мере одно температуровоспринимающее устройство, отстоящее примерно на 5-10 см выше слоя катализатора удаления альдегида и алкина, и другое, расположенное примерно на 15-25 см выше слоя катализатора удаления альдегида и алкина;
регулирование условий на впуске указанного реактора, так что реакции окислительного дегидрирования первоначально имеют место в самых нижних слоях указанного катализатора окислительного дегидрирования, включая в реакционной зоне взаимодействие указанного потока реакторного питания с помощью указанного катализатора и образование в результате потока бутадиенобогащенного продукта;
контроль температуры во всем слое и время от времени в ответ на падение температуры в реакционной зоне увеличение температуры на впуске, когда активный слой катализатора окислительного дегидрирования в указанной реакционной зоне начинает становиться дезактивированным, так что реакционная зона перемещается вверх в слое окислительного дегидрирования, и прерывание подачи бутенобогащенного углеводородистого питания после того, как температура, указанная температуровоспринимающим устройством, расположенным в самых верхних частях каталитического слоя окислительного дегидрирования, начинает падать.
12. Способ получения бутадиена из бутенобогащенного питания по п. 11, в котором гранулы катализатора окислительного дегидрирования имеют диаметр в интервале от примерно 1 мм до примерно 30 мм.
13. Способ получения бутадиена из бутенобогащенного питания по п. 11, в котором гранулы катализатора окислительного дегидрирования имеют диаметр в интервале от примерно 1 мм до примерно 5 мм.
14. Способ получения бутадиена из бутенобогащенного питания по п. 11, в котором гранулы предварительно приводятся или иным образом термообрабатываются перед загрузкой в степени, достаточной для придания им прочности на раздавливание, необходимой, чтобы быть используемыми в слое, имеющем глубину от примерно 60 см (27 дюйм) до примерно 150 см (60 дюйм), причем объемная плотность предварительно приведенных частиц составляет не более примерно 1121 кг/м3 (70 фунт/фут3).
15. Способ получения бутадиена из бутенобогащенного питания по п. 14, в котором объемная плотность предварительно приведенных гранул находится в интервале от примерно 930 кг/м3 (58 фунт/фут3) до примерно 1010 кг/м3 (63 фунт/фут3).
16. Способ получения бутадиена из бутенобогащенного питания по п. 11, в котором ферритный катализатор окислительного дегидрирования, по существу, не содержит нитрат.
17. Способ получения бутадиена из бутенобогащенного питания по п. 16, в котором ферритный катализатор окислительного дегидрирования содержит кислород, главную пропорцию железа, незначительную пропорцию цинка и небольшие количества марганца, фосфора и остаток предшественника кальция, не содержащего нитрат.
18. Способ получения бутадиена из бутенобогащенного питания по п. 11, в котором количество молей кислорода в указанном потоке реакторного питания регулируется таким образом, что обеспечивается по меньшей мере 0,5 моль кислорода на 1 моль бутенобогащенного углеводородистого питания.
19. Способ получения бутадиена из бутенобогащенного питания по п. 11, в котором ферритный катализатор окислительного дегидрирования имеет глубину свыше 70 см (27 дюйм).
20. Способ получения бутадиена из бутенобогащенного питания по п. 11, в котором температуровоспринимающие устройства содержат термопары.
| US 2011245568 A1, 06.10.2011 | |||
| МАГНИТНЫЙ КОМПАС | 1995 |
|
RU2093792C1 |
| SU 1216938 A1, 20.10.1999 | |||
| US 4658080 A, 14.04.1987. | |||

