Настоящее изобретение относится к области органической химии, а именно, к получению новых терпеновых β-гидроксиаминов пинановой структуры
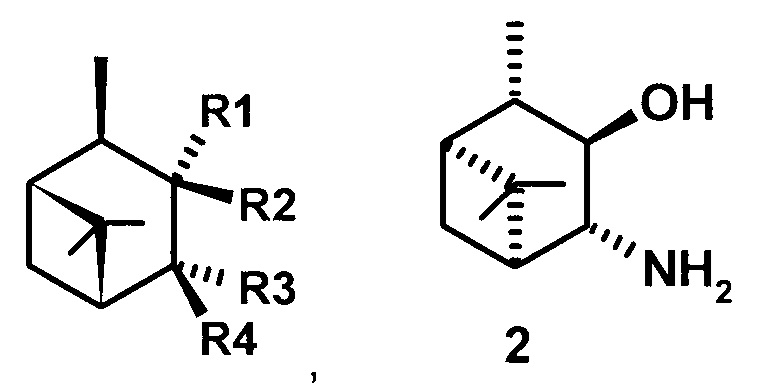
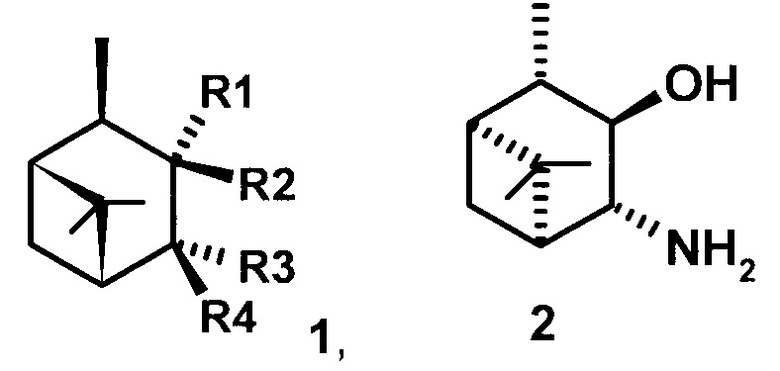 или
или 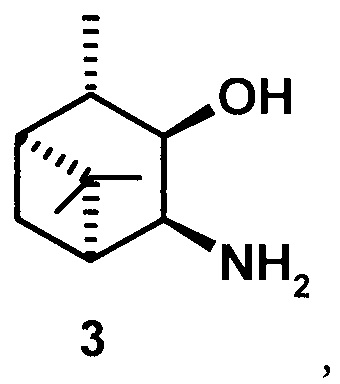
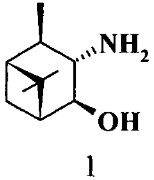
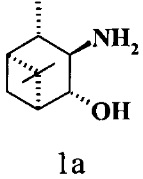
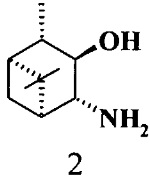
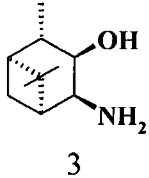
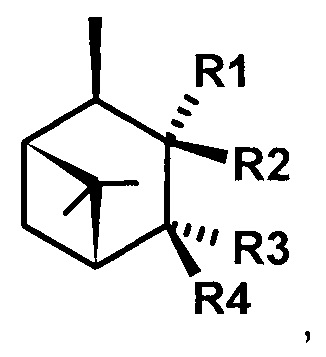
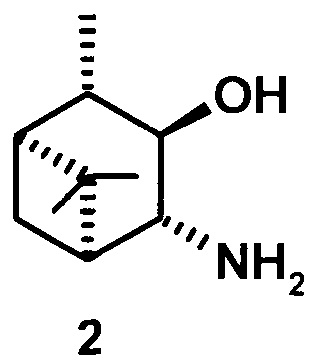
где R1 - NH2, R2 - Н, R3 - Н, R4 - ОН и соединение (1) представляет собой (+) или (-) 3α-амино-4β-гидрокси-10β-пинан; соединение (2) представляет собой 3α-гидрокси-4β-амино-10β-пинан и соединение (3) представляет собой 3α-гидрокси-4α-амино-10β-пинан.
Терпеновые β-аминоспирты являются важнейшими строительными блоками в синтезе энантиомерно обогащенных природных соединений, фармакологически активных веществ, хиральных катализаторов, в том числе и органокатализаторов.
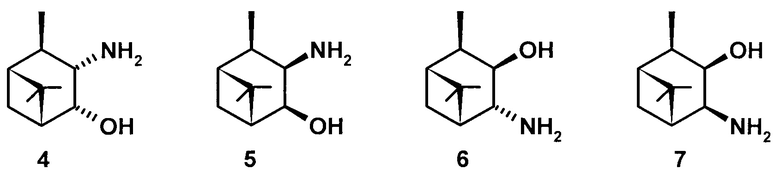
В литературе известны следующие изомерные 3,4-гидроксиамины пинановой структуры 4-7 - 3α-амино-4α-гидрокси-10β-пинан (4), 3β-амино-4β-гидрокси-10β-пинан (5), 3β-гидрокси-4α-амино-10β-пинан (6), 3β-гидрокси-4β-амино-10β-пинан (7).
Соединения 4 и 5 были получены на основе цис-вербанона 8 (схема 1) [Burak K., Chabudzinski Z. Amino alcohols in pinane series. Part III. Synthesis and stereochemistry of 3-amino-4-hydroxy-cis-pinanes and their deuterated analogs. // Polish Journal of Chemistry (Formerly Roczniki Chemii), 1981, V. 55(10), p. 2015-2023]. Нитрозирование соединения 8 проводят следующим образом: раствор цис-вербанона 8 в диэтиловом эфире охлаждают до 0°С и при перемешивании добавляют Na, затем добавляют изоамилнитрит (мольное соотношение 8: Na : изоамилнитрит - 1:1:1) с такой скоростью, чтобы температура реакционной смеси не поднималась выше +10°С и помещают в холодильник на 48 часов.Затем реакционную смесь разбавляют водой и экстрагируют диэтиловым эфиром.
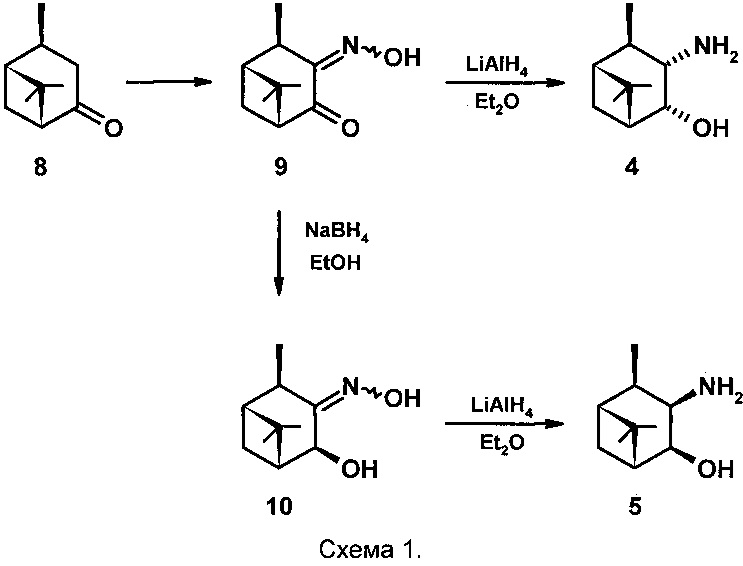
Водный слой отделяют и титруют 10% водным раствором уксусной кислоты до выпадения желтого осадка (рН 5-5,5) - 3-гидроксиимино,цис-пинанона-4 (9) выход которого составляет 47% с соотношением syn : anti изомеров 70:30. Кетооксим 9 восстанавливают 4 эквивалентами LiAlH4 в Et2O при кипячении в течение 16 часов, охлаждают реакционную смесь, отфильтровывают осадок Al(ОН)3, промывают его Et2O, эфирные экстракты сушат MgSO4. После удаления растворителя выход продукта составляет 51% (выход на 2 стадии 24%) и содержит по данным ТСХ два вещества. Смесь этих веществ обрабатывают эфирным раствором HCl, полученный гидрохлорид отфильтровывают, перекристаллизовывают из смеси EtOH и Et2O, получают хроматографически индивидуальный гидрохлорид, который обрабатывают 10% водным раствором KОН и получают гидроксиаминоспирт 4. Выход чистого соединения 4 не указан.
Синтез соединения 5 проводят в две стадии: кетооксим 9 растворяют в EtOH, охлаждают до 0°С, при перемешивании добавляют NaBH4 (мольное соотношение 9 : NaBH4 - 1:1,8), поддерживая температуру не выше +5°С. После окончания реакции (~1,5 час.) удаляют растворитель, к остатку добавляют воду и экстрагируют диэтиловым эфиром, сушат над MgSO4, фильтруют. После удаления растворителя выход кристаллического 3-гидроксиимино,цис-пинан-4β-ола 10 составляет 81%, его пререкристаллизовывают из гептана, получают хроматографически индивидуальное соединение 10, которое восстанавливают LiAlH4 также как это описано для соединения 4. Выход маслянистого продукта составляет 89% (выход на 3 стадии 34%). Его обрабатывают раствором HCl в Et2O, получают кристаллический гидрохлорид, который перекристаллизовывают из смеси EtOH и Et2O, получают хроматографически индивидуальный гидрохлорид, который обрабатывают 10% водным раствором KОН и получают гидроксиаминоспирт 5. Выход чистого соединения 5 не указан.
Соединения 6 и 7 получены основе 2α-гидроксипинанона-3 11 [Burak K., Chabudzinski Z. Amino alcohols in pinane series. Part II. Synthesis and stereochemistry of epimeric 3β-hydroxy-4-amino-cis-pinanes. // Polish Journal of Chemistry (Formerly Roczniki Chemii), 1981, V. 55 (2), pp. 387-392] (схема 2). Раствор 2α-гидроксипинанона-3 в Et2O охлаждают до 0°С и при перемешивании прибавляют Na, затем i-AmONO (мольное соотношение 11 : Na : i-AmONO - 1:1:1), поддерживая температуру не выше +5°С, перемешивают еще 15 мин и помещают в холодильник на 20 ч. Далее в реакционную смесь добавляют воду и лед, отделяют органический слой, водный дважды экстрагируют диэтиловым эфиром.
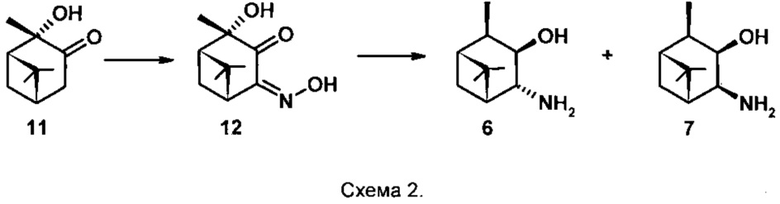
Водный слой, содержащий натриевую соль гидроксииминокетола 12, титруют 10% водным раствором уксусной кислоты до выпадения желтого осадка (рН 3,8-4,2), фильтруют. Выход соединения 12 составил 54,5%, который кристаллизуют из воды, затем из этилацетата и получают хроматографически индивидуальное соединение 12. Далее соединение 12 растворяют в n-пентаноле, добавляют Na (мольное соотношение 12 : Na - 1:18) и доводят до кипения, затем добавляют еще столько же Na и кипятят еще 1 час, пока реакционная смесь не станет бесцветной. После охлаждения до комнатной температуры добавляют воду, отделяют органический слой, водный еще трижды экстрагируют Et2O. Объединяют органические экстракты, сушат над Na2SO4, фильтруют, удаляют растворитель и перегоняют под вакуумом, отбирая фракцию с температурой кипения 92-120°С / 4 торр. (выход 62%), которая по данным ГЖХ содержит два вещества в соотношении 68:32 (общий выход на две стадии 34%). Эту смесь обрабатывают раствором HCl в Et2O, получают смесь двух гидрохлоридов, которые разделяют колоночной хроматографией на SiO2, используя систему растворителей CHCl3:МеОН - 4:1. Далее каждый индивидуальный гидрохлорид обрабатывают 10% водным раствором KОН и получают соединения 6 и 7, окончательный выход которых не указан.
Недостатком является низкий выход гидроксиаминов 4-7.
Задачей настоящего изобретения является разработка селективных способов получения соединений 1, 2, 3, которые в литературе не описаны. Технический результат состоит в расширении арсенала терпеновых β-аминоспиртов, являющихся важнейшими строительными блоками в синтезе энантиомерно обогащенных природных соединений, фармакологически активных веществ, хиральных катализаторов, в том числе и органокатализаторов.
Технический результат достигается тем, что 3,4-Гидроксиамины структурных формул (1, 2, 3)
 или
или 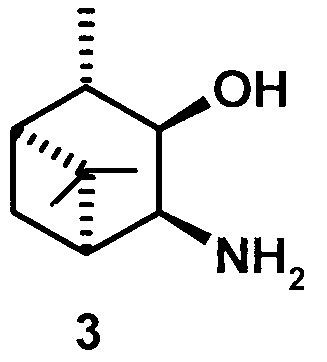
где R1 - NH2, R2 - Н, R3 - Н, R4 - ОН и соединение (1) представляет собой (+) или (-) 3α-амино-4β-гидрокси-10β-пинан; соединение (2) представляет собой 3α-гидрокси-4β-амино-10β-пинан и соединение (3) представляет собой 3α-гидрокси-4α-амино-10β-пинан.
Синтез соединение 1 обеспечивается тем, что исходным соединением является цис-вербанон, легко получаемый восстановлением (гидрированием) вербенона. В качестве оксимирующего реагента используется система t-BuOK -
i-AmONO-ТГФ. В качестве восстановителя оксимной группы используется Zn в растворе NaOH, в качестве восстановителя кетогруппы - система LiBHEt3-ТГФ.
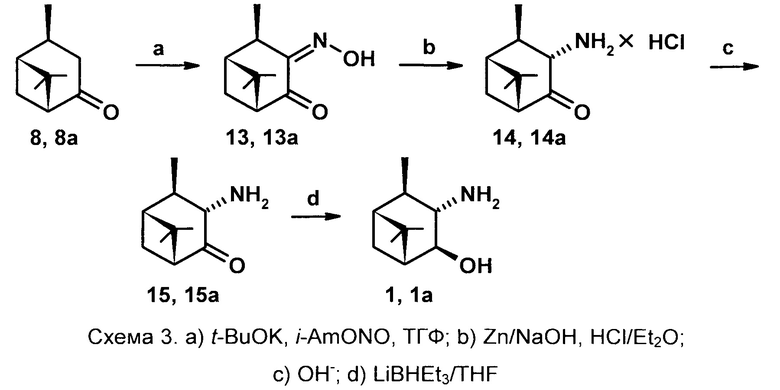
Способ получения новых соединений осуществляется следующим образом:
Раствор цис-вербанона 8 в сухом ТГФ прибавляют при перемешивании к охлажденной до -70-80°С суспензии трет-бутоксида калия в ТГФ, пермешивают 10 мин, прибавляют изо-амилнитрит (мольное соотношение 8: t-BuOK : i-AmONO - 1:1,2:1,2), медленно доводят температуру смеси до комнатной и оставляют на ночь. Далее удаляют ТГФ, к остатку добавляют воду, экстрагируют Et2O. Водную фракцию разбавляют водой и добавляют уксусную кислоту до рН 6. Образовавшийся осадок отфильтровывают, промывают водой, сушат на воздухе, затем в эксикаторе над безводным CaCl2. Выход единственного Z-оксима 13 составляет 70-75%. К суспензии оксима 13 в 30% водном растворе NaOH прибавляют цинковую пыль (мольное соотношение 13: Zn 1:3) и через 5 минут прибавляют диэтиловый эфир, перемешивают еще 5 мин, экстрагируют Et2O, экстракт промывают водой, насыщенным раствором NaCl, сушат Na2SO4 5 мин, фильтруют, к фильтрату добавляют раствор HCl в Et2O. Образующийся осадок отделяют фильтрованием, промывают диэтиловым эфиром, сушат под вакуумом. Выход гидрохлорида 14 87-93%. Далее в раствор гидрохлорида 14 в воде прибавляют водный раствор Na2CO3, экстрагируют эфиром, сушат 5 мин над Na2SO4, фильтруют, отгоняют растворитель. Остаток растворяют в сухом ТГФ, охлаждают до -70-80°С и при перемешивании в инертной атмосфере по каплям прибавляют раствор LiBHEt3 в ТГФ (мольное соотношение 15: LiBHEt3 1:1.1). Перемешивают еще 3-5 часов, постепенно поднимая температуру реакционной смеси до -20°С, добавляют 10% водный раствор NaOH, доводят температуру до комнатной, добавляют воду, экстрагируют этилацетатом. Органические экстракты промывают водой, насыщенным раствором NaCl, сушат Na2SO4, фильтруют, отгоняют растворитель, остаток кристаллизуют из смеси Et2O-гексан. Выход гидроксиамина 1 составляет 60-65%. Общий выход на три стадии индивидуального 3α-амино-4β-гидрокси-10β-пинана 1 37-45%.
Технический результат синтеза соединений 2 и 3 достигается тем, что исходным соединением является 3α-гидрокси-10β-пинанон-4 16, который легко получают окислением транс-3,4-диола диоксидом хлора в ДМФА [патент RU 2509073 C1 Фролова Л.Л., Кучин А.В., Попов А.В. «Способ получения 3α-гидрокси-10β-пинанона-4». 10.03.2014/ Бюл. №7]. В качестве оксимирующего реагента используется NH2OH×HCl (солянокислый гидроксиламин), в качестве восстановителя кетогруппы - система LiAlH4-Et2O.
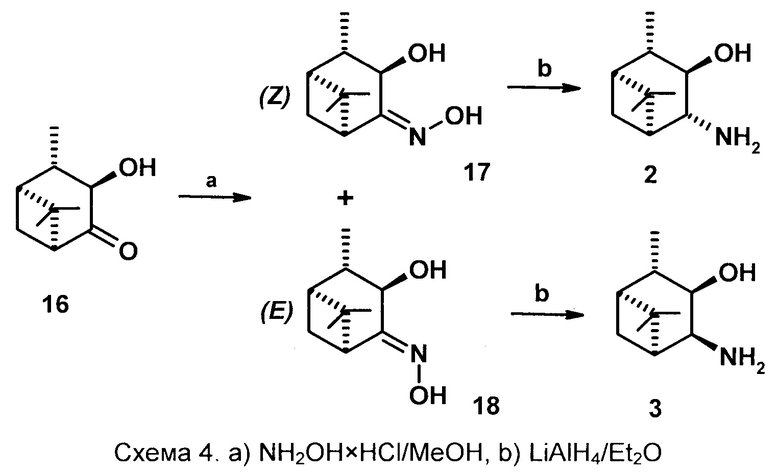
Способ осуществляется следующим образом. Смесь гидроксикетона 16, NaHCO3, гидрохлорида гидроксиламина в метаноле нагревают при температуре 65-67°С в течении 4-7 часов, реакцию контролируют по ТСХ. Далее реакционную смесь разбавляют водой, экстрагируют диэтиловым эфиром, экстракты промывают 5% раствором NaHCO3, насыщенным раствором NaCl, сушат над MgSO4. После удаления растворителя выход смеси оксимов количественный с соотношением Z:E - 70:30-40:60 за 4 и 7 часов
соответственно. Далее смесь оксимов разделяют колоночной хроматографией на SiO2 с использованием системы растворителей петролейный эфир (или гексан) - диэтиловый эфир. Далее гидроксиоксимы подвергают восстановлению 3 эквивалентами LiAlH4 в диэтиловом эфире в течении 5-6 часов при температуре 32-34°С. Далее в реакционную смесь осторожно по каплям прибавляют 3-5% NaOH до разделения слоев, экстрагируют Et2O, промывают насыщенным NaCl, сушат над MgSO4, удаляют растворитель. Соединение 3 дважды перекристаллизовывают из смеси EtOH и Et2O. Выход 41-45%.
Соединение 2 выделяют через гидрохлорид: реакционную смесь после восстановления обрабатывают раствором конц. HCl в диэтиловом эфире, отделяют выпавший осадок гидрохлорида, перекристаллизовывают из смеси EtOH и Et2O, обрабатывают 10% раствором KОН (1:2), экстрагируют Et2O, промывают насыщенным NaCl, сушат над MgSO4. После удаления растворителя выход 32-35%.
Пример 1. К суспензии трет-бутоксида калия (6.73 г, 60 ммоль) в 20 мл сухого ТГФ при -80°С медленно прибавляли раствор (+)-цис-вербанона (8) или (-)-цис-вербанона (8а) (7.61 г., 50 ммоль) в 10 мл сухого ТГФ. Смесь перемешивали 10 мин. Затем в течение 20 мин прибавляли изоамилнитрит (7.03 г, 60 ммоль), смесь перемешивали, медленно нагревали до комнатной температуры и оставляли на ночь. ТГФ отгоняли под вакуумом, к остатку прибавляли 50 мл воды и экстрагировали диэтиловым эфиром (3×15 мл). Водную фракцию разбавляли 150 мл воды и доводили рН до 6 уксусной кислотой. Выпавший осадок отфильтровывали, промывали водой, сушили на воздухе, затем в эксикаторе над безводным CaCl2. Выход единственного Z-оксима (13) или (13а) 70-75%. Белый порошок, т.пл. 135°С, [α]23=-12.0 (с=1.0, CHCl3) для (13), [α]23=+11.9 (с=1.0, CHCl3) для (13а). 1Н ЯМР-спектр (DMSO-d6): δ 0.95 (с, 3Н, Ме-8), 1.31 (д, 3Н, J=7.8 Hz, Ме-10), 1.34 (д, 1H, J=9.6 Hz, Н-7ах), 1.36 (с, 3Н, Ме-9), 2.15 (тд, 1Н, J=5.9, 2.9 Hz, Н-1), 2.53 (т, 1Н, J=5.4 Hz, Н-5), 2.65 (дт, 1Н, J=11.6, 5.8 Hz, H-7eq), 3.09 (кд, 1Н, J=7.1, 2.9 Hz, Н-2), 12.33 (уш.с, 1Н, N-OH). 13С ЯМР-спектр (DMSO-d6): 16.4 (С-10), 24.1 (С-8), 27.4 (С-9), 28.0 (С-7), 36.2 (С-2), 42.1 (С-6), 45.6 (С-1), 56.5 (С-5), 155.9 (С-3), 198.8 (С-4), ИК-спектр (KBr) 1719, 3235 см-1.
К суспензии оксима 13 (5.44 г, 30 ммоль) в 25 мл 30% водного раствора NaOH, прибавляли цинковую пыль (10 мкм, 5.89 г., 90 ммоль). Смесь
разогревается и после завершения реакции (5 мин) прибавляли 70 мл Et2O и перемешивали еще 5 мин. Органический слой отделяли, остаток экстрагировали Et2O (2×30 мл). Объединенные органические слои промывали 20 мл воды, насыщенным раствором NaCl, сушили 5 мин над Na2SO4, отфильтровывали, к фильтрату прибавляли 4.5 М раствор HCl (7.34 мл, 33 ммоль) в Et2O. Осадок отфильтровывали и промывали Et2O (2×30 мл), сушили под вакуумом. Выход гидрохлорида транс-аминокетона (14) или (14а) 87-93%. Розовый порошок, т.пл. 225-230°С с разлож., [α]23 -35.3 (с 0.9, CHCl3) для (14), [α]23 +31.7 (с 1.2, CHCl3) для (14а). ИК-спектр (KBr, v, см-1): 3429, 1724. ЯМР 1Н (300 МГц, DMSO-d6, δ/м.д., J/Гц): 1.04 (с, 3Н, Ме-8), 1.30 (д, 3Н, J=7.6, Ме-10), 1.34 (с, 3Н, Ме-9), 1.78 (д, 1H, J=11.2, Н-7ах), 2.11 (т, 1Н, J=5.0, Н-1), 2.25 (квинтет, 1Н, J=6.8, Н-2), 2.66 (т, 1Н, J=5.6, Н-5), 2.65 (ддд, 1Н, J=11.5, 5.6, 5.0, H-7eq), 3.93 (уш.с, 1Н, Н-3), 8.74 (уш.с, 3Н, NH3+).). ЯМР 13С (75 МГц, DMSO-d6, δ, м.д.): 207.0 (С-4), 57.9 (С-5), 57.6 (С-3), 47.5 (С-1), 41.3 (С-6), 38.1 (С-2), 29.7 (С-7), 27.2 (С-9), 24.8 (С-8), 19.2 (С-10).
К раствору гидрохлорида транс-аминокетона 14 (0.611 г, 3 ммоль) в 15 мл воды прибавляли раствор Na2CO3 (1 г., 9 ммоль) в 5 мл воды, экстрагировали Et2O (3×20 мл), объединенные органические слои сушили 5 мин над Na2SO4, отфильтровывали. Растворитель отгоняли под вакуумом. Остаток растворяли в 5 мл сухого ТГФ, охлаждали до -80°С и в атмосфере аргона медленно прибавляли 1М раствор LiBHEt3 в ТГФ (3.3 мл, 3.3 ммоль). Перемешивание продолжали 3-5 часов с нагревом системы до -20°С. Добавляли 2 мл 10% раствора NaOH. Нагревали до комнатной температуры, прибавляли еще 20 мл воды, экстрагировали EtOAc (3×20 мл), объединенные органические слои промывали 20 мл воды, насыщенным раствором NaCl, сушили над Na2SO4, отфильтровывали, отгоняли растворитель под вакуумом. Остаток перекристаллизовывали в системе Et2O-гексан при -20°С. Выход гидроксиаминопинана (1) или (1а) 60-65%.
3α-амино-4β-гидрокси-10β-пинан (1 или 1а). Белый порошок, т.пл. 148°С, [α]23 +7.2 (с 0.9, CHCl3) для (1), [α]23 -6.7 (с 1.1, CHCl3) для (1а). ИК-спектр (KBr, v, см-1): 3445, 3260, 1605, 1344. ЯМР 1Н (300 МГц, CDCl3, δ/м.д., J/Гц): 0.73 (д, 1H, J=10.1, Н-7ах), 1.11 (с, 3Н, Ме-8), 1.12 (д, 3Н, J=7.4, Ме-10), 1.24 (с, 3Н, Ме-9), 3.09 (квинтет, 1Н, J=7.0, 2.9, Н-2), 1.81 (т, 1Н, J=5.4, Н-1), 2.06 (м, 1Н, Н-5), 2.35 (дт, 1Н, J=10.1, 6.3, H-7eq), 2.64 (уш.с, 3Н, NH2, ОН), 3.03 (дд, 1Н, J=6.9, 5.7, Н-3),
3.78 (дд, 1Н, J=5.2, 2.1, Н-4). ЯМР 13С (75 МГц, CDCl3, δ, м.д.): 82.2 (С-4), 61.0 (С-3), 56.5 (С-5), 48.5 (С-1), 46.5 (С-2), 32.4 (С-7), 32.4 (С-7), 28.8 (С-9), 24.0 (С-8), 19.8 (С-10).
Пример 2. Смесь 2.75 г (16 ммоль) гидроксикетона 16, 2.51 г (23 ммоль) NaHCO3, 1.59 г (23 ммоль) NH2OH*HCl в 20 мл метанола, содержащего 1.5 мл воды, нагревали при перемешивании 7 час при температуре 65-67°С. После охлаждения в реакционную смесь добавляли 20 мл воды до полного растворения осадка, экстрагировали эфиром (3×20 мл), эфирные экстракты промывали 5% водным раствором NaHCO3, насыщенным раствором NaCl и сушили над MgSO4, после удаления растворителя выход продукта количественный с соотношением Z:E изомеров 42:58 по данным ЯМР-спектроскопии. Колоночной хроматографией на SiO2 с использованием системы петролейный эфир - диэтиловый эфир, было выделено 1.18 г (39%) Z-изомера (17) в виде масла и 1.64 г (54%) Е-изомера (18) в виде кристаллического продукта.
Z-оксим (17). Густое масло, Rf 0.64 (Et2O), [α]D +9.1° (с 0.5, EtOH). ИК-спектр (v, см-1): 3259, 2955, 1653, 1467, 1384, 1333, 1248, 1037, 1005, 947, 893, 794, 494. ЯМР 1Н (300 МГц, DMSO-d6, δ/м.д., J/Гц): 0.77 с (3Н, СН3-8), 1.14 д (3Н, СН3-10, J=7.4), 1.28 с (3Н, СН3-9), 1.32 д (1Н, СН-7, J=9.1), 1.93 м (1Н, СН-1), 1.99 м (1Н, СН-2), 2.49 м (1Н, СН-5), 2.52 м (1Н, СН-7), 4.45 дд (1Н, СН-3, J=3.1+3.2), 4.58 д (-ОН, J=2.9), 10.62 с (=N-OH). ЯМР 13С (75 МГц, DMSO-d6, δ, м.д.): 163.1 (С-4), 68.4 (С-3), 48.0 (С-5), 46.9 (С-1), 43.2 (С-2), 40 (С-6), 29.5 (С-7), 26.8 (С-9), 25.0 (С-8), 20.3 (С-10).
Е-оксим (18). Кристаллическое вещество, Rf 0.41 (Et2O), [α]D -58.8° (с. 0.9 EtOH). ИК-спектр (v, см-1): 3296, 2906, 1653, 1468, 1442, 1384, 1342, 974, 908, 740, 654. ЯМР 1Н (300 МГц, DMSO-d6, δ/м.д., J/Гц): 0.79 с (3Н, СН3-8), 1.30 д (3Н, СН3-10, J=7.5), 1.30 с (3Н, СН3-9), 1.42 д (1Н, СН-7, J=9.7), 1.90 ддд (1Н, СН 1), 1.97 ддк (1Н, СН-2, J=2.8+5.9+), 2.38 ддд (1Н, СН-7, J=5.9+5.6+9.7), 3.38 дд (1Н, СН-5, J=5.5+5.6), 4.01 дд (1Н, СН-3, J=3.1), 5.07 д (-ОН), 10.2 с (=N-OH). ЯМР 13С (75 МГц, DMSO-d6, δ, м.д.): 162.9 (С-4), 71.7 (С-3), 47.1 (С-1), 44.6 (С-2), 41.6 (С-5), 40.7 (С-6), 28.2 (С-7), 27.2 (С-9), 24.8 (С-8), 20.2 (С-10).
К суспензии 0.672 г (17.6 ммоль) LiAlH4 в 50 мл абс. Et2O при перемешивании в инертной атмосфере по каплям прибавляли раствор 1.08 г (5.9 ммоль) соединения 17 в 15 мл абс. Et2O. Реакционную смесь перемешивали 6 часов при температуре 32-34°С, охлаждали и оставляли на ночь. Далее в реакционную смесь по каплям добавляли 5% водный раствор NaOH до разделения слоев, отделяли эфирный слой, водный еще три раза экстрагировали эфиром (3×25 мл), экстракты объединяли и промывали насыщенным раствором NaCl и сушили над MgSO4, после удаления растворителя выход маслянистого продукта 0.858 г (87%), который по данным ЯМР-спектроскопии содержит два вещества 2 и 3 в соотношении 7:2. Далее продукт растворяли в 5 мл Et2O, добавляли 4,5 М раствор HCl (5.6 ммоль, 1.25 мл) в Et2O, выпавший осадок отфильтровывали под вакуумом и перекристаллизовывали из смеси Et2O-EtOH. Выход индивидуального по данным ЯМР-спектроскопии гидрохлорида составил 0.576 г (68%).
2×HCl. т.пл. 220°С (с разлож.), [α]D +7.9° (с 0.5 EtOH). ИК-спектр (v, см-1): 3232, 2964, 1525 (NH3+), 1473, 1427, 1261, 1145, 1053, 1006, 921. ЯМР 1Н (300 МГц, DMSO-d6, δ/м.д., J/Гц): 0.94 д (СН2-7, J=9.8), 1.05 с (3Н, СН3-8), 1.09 д (3Н, СН3-10, J=7.2), 1.22 с (3Н, СН3-8), 1.78 дд (1Н, С Н-1, J=5.3, 5.6), 1.85 м (1Н, СН-2, J=7.2), 1.83 дд (Н, СН-5, J=5.3, 6.7), 2.45 ддд (СН2-7, J=6.3, 6.7, 9.8), 3.28 дд (1Н, СН-4, J=4.9, 5.4), 3.91 дд (1Н, СН-3, J=6.2, 6.3), 8.3 уш. (3Н, NH3). ЯМР 13С (75 МГц, DMSO-d6, δ, м.д.): 74.16 (С 3), 62.25 (С-4), 47.73 (С-1), 45.51 (С-5), 44.67 (С-2), 38.20 (С-6), 33.75 (С-7), 28.48 (С-9), 24.97 (С-8), 19.87 (С-10).
Гидрохлорид растворяли в 5 мл Н2О, добавляли 3 мл 10% раствора KОН, перемешивали, экстрагировали эфиром (3×25 мл), эфирные экстракты промывали насыщенным раствором NaCl, сушили MgSO4, удаляли растворитель. Выход соединения 2 составил 0.32 г (32% на исходный гидроксиоксим 17).
3α-гидрокси-4β-амино-10β-пинан (2), [α]D +4.6° (с 0.4 CHCl3). т.пл. 92-93°С. ИК-спектр (v, см-1): 3323, 3251, 3163, 2991, 2900, 1593 (NH2), 1460, 1367, 1319, 1259, 1147, 1120, 1072, 1031, 985, 816, 881, 837, 765. ЯМР 1Н (300 МГц, DMSO-d6, δ/м.д., J/Гц): 0.82 д (СН2-7, J=9.6), 1.05 с (3Н, СН3-8), 1.06 д (3Н, СН3-10, J=7), 1.21 с (3Н, СН3-9), 1.70-1.80 м (2Н, СН-1 и СН-2), 1.83 ддд (Н, СН-5, J=1.8, 6.3, 6.8), 2.29 ддд (СН2-7, J=6.3, 6.5, 9.6), 2.95 дд (1Н, СН-4, J=1.8, 5.3), 3.57 дд (1Н, СН-3, J=5.3, 6.3). ЯМР 13С (75 МГц, DMSO-d6, δ, м.д.): 80.45 (С 3), 63.83 (С-4), 49.52 (С-5), 48.57 (С-1), 45.33 (С-2), 38.75 (С-6), 33.96 (С-7), 29.35 (С-9), 25.05 (С-8), 20.46 (С-10).
К суспензии 0.766.г (20 ммоль) LiAlH4 в 50 мл абс. Et2O при перемешивании в инертной атмосфере по каплям прибавляли раствор 1.23 г (6.7 ммоль) соединения 18 в 15 мл абс. Et2O, кипятили 6 ч, охлаждали и оставляли на ночь. Далее в реакционную смесь по каплям добавляли 5% водный раствор NaOH до разделения слоев, отделяли эфирный слой, водный еще три раза экстрагировали эфиром (3×25 мл), экстракты объединяли и промывали насыщенным раствором NaCl и сушили над MgSO4. После удаления растворителя выход кристаллического продукта 1.024 г (90%), который по данным ЯМР-спектроскопии содержит два вещества 2 и 3 в соотношении 1:5. Продукт дважды перекристаллизовывали из смеси Et2O-EtOH. Получили 0.511 индивидуального по данным ЯМР гидроксиамина 3 (45% на исходный гидроксиоксим 18).
3α-гидрокси-4α-амино-10β-пинан (3), [α]D +41.3° (с 0.5 CHCl3). т.пл. 78-80°С. ИК-спектр (v, см-1): 3354, 3280, 2904, 2723, 1577, 1469, 1375, 1053, 991, 964. ЯМР 1Н (300 МГц, DMSO-d6, δ/м.д., J/Гц): 0.94 с (3Н, СН3-8), 1.06 д (3Н, СН3-10, J=7), 1.04 д (СН2-7), 1.22 с (3Н, СН3-9), 1.67-1.75 м (2Н, СН-1 и СН-2), 1.97 м (Н, СН-5), 2.08 м (СН2-7), 3.3 ддд (1Н, СН-4, J=8.2+3.5+4.8), 3.63 дд (1Н, СН-3, J=5.3+8.2). ЯМР 13С (75 МГц, DMSO-d6, δ, м.д.): 70.5 (С 3), 51.19 (С-4), 49.45 (С-5), 47.55 (С-1), 46.76 (С-2), 39.4 (С-6), 28.67 (С-7), 28.29 (С-9), 23.52 (С-8), 21.37 (С-10).
Полученные 3,4-пинановые аминоспирты были протестированы в качестве органокатализаторов в кросс-альдольной реакции изатина с ацетоном.
Изатин является ценным прекурсором в синтезе большого числа физиологически активных соединений, в частности - такрин, дибукаин, метисазон. Продуктом конденсации 4,6-дибромпроизводного изатина с ацетоном в присутствии хирального вицинального аминоспирта является R-конволутамидин, который оказался мощным ингибитором дифференциации клеток лейкемии HL-60 [A. Malkov, M. Kabeshov, M. Bella, O. Kysilka, D.Malyshev,  ,
, 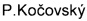 , Org. Lett., 9, 5473 (2007)].
, Org. Lett., 9, 5473 (2007)].
Альдольная реакция изатина с ацетоном, катализируемая аминоспиртами 1-3: 20 mol% cat, 30 экв. ацетона, 2 экв. Воды
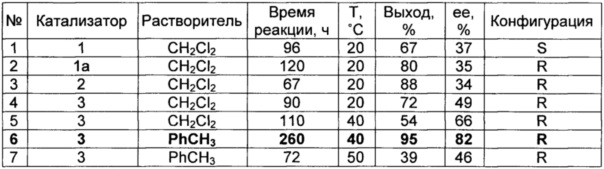
Наилучшие результаты получены при использовании катализатора 3 при проведении реакции в толуоле - выход продукта достигает 95%, энантиоселективность образования R-изомера 82%.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ТЕРПЕНОВЫХ α-ХЛОРКЕТОНОВ ИЛИ ХЛОРГИДРОКСИКЕТОНОВ | 2015 |
|
RU2569896C1 |
| СПОСОБ ПОЛУЧЕНИЯ 3α-ГИДРОКСИ-10β-ПИНАНОНА-4 | 2012 |
|
RU2509073C1 |
| ФТОРСОДЕРЖАЩИЕ ПРОИЗВОДНЫЕ ТЕВИНОЛА И ОРВИНОЛА И СПОСОБЫ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ) | 2012 |
|
RU2506265C1 |
| 7α-(1-ГИДРОКСИ-2,2,2-ТРИФТОРЭТИЛ)-17-МЕТИЛ-3,6-ДИМЕТОКСИ-4,5α-ЭПОКСИ-6α,14αЭТЕНОИЗОМОРФИНАН И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2012 |
|
RU2503678C1 |
| 3,6-ДИМЕТОКСИ-17-МЕТИЛ-7АЛЬФА-(ТРИФТОРАЦЕТИЛ)-4,5АЛЬФА-ЭПОКСИ-6АЛЬФА,14АЛЬФА-ЭТЕНОИЗОМОРФИНАН И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2012 |
|
RU2503677C1 |
| ПРОИЗВОДНЫЕ АРИЛЦИКЛОАЛКИЛАМИНОВ, НЕЙРОПРОТЕКТОР (ВАРИАНТЫ), ВЕЩЕСТВО, ОБЛАДАЮЩЕЕ СОЧЕТАННЫМ НЕЙРОПРОТЕКТОРНЫМ, АНАЛЬГЕТИЧЕСКИМ И АНТИДЕПРЕССИВНЫМ ДЕЙСТВИЕМ, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ НА ЕГО ОСНОВЕ | 2016 |
|
RU2637928C2 |
| СЕЛЕКТИВНЫЕ ЛИГАНДЫ G-КВАДРУПЛЕКСНЫХ СТРУКТУР НУКЛЕИНОВЫХ КИСЛОТ | 2015 |
|
RU2588131C1 |
| Способ получения гидроксамовых кислот, производных 2-арил-2,3-дигидрохиназолин-4(1Н)-онов | 2020 |
|
RU2744750C1 |
| НОВЫЕ НАФТО[2,1-b]КАРБАЗОЛПРОИЗВОДНЫЕ ФУЗИДОВОЙ КИСЛОТЫ, СПОСОБ ПОЛУЧЕНИЯ И АНТИМИКРОБНЫЕ СВОЙСТВА | 2019 |
|
RU2746947C2 |
| 5-АМИНО-3-(2-АМИНОПРОПИЛ)-[1,2,4]ТИАДИАЗОЛЫ | 2011 |
|
RU2449997C1 |
Изобретение относится к новым изомерным формам терпеновых β-гидроксиаминов пинановой структуры-(+)3α-амино-4β-гидрокси-10β-пинану или (-)3α-амино-4β-гидрокси-10β-пинану соответствующих общей формуле (1), 3α-гидрокси-4β-амино-10β-пинану формулы (2) или 3α-гидрокси-4α-амино-10β-пинану формулы (3). Терпеновые аминоспирты настоящего изобретения могут использоваться для получения прекурсоров физиологически активных соединений в качестве хиральных органокатализаторов. Например, в кросс-альдольной реакции изатина с ацетоном в среде толуола выход продукта достигает 95% при энантиоселективности образования R-изомера 82%. Соединения также могут использоваться как строительные блоки в синтезе энантиомерно обогащенных природных соединений, в качестве фармакологически активных веществ.
 1,
1,
 или
или 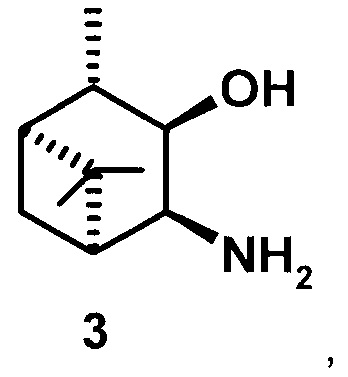
В формуле (1) R1 - NH2, R2 - Н, R3 - Н, R4 – ОН. 2 н.п. ф-лы, 1 табл., 1 пр.
1. 3,4-Гидроксиамины структурной формулы (1)
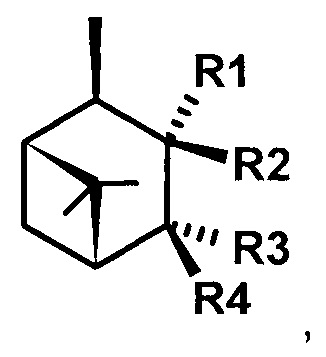
где R1 - NH2, R2 - Н, R3 - Н, R4 - ОН и соединение (1) представляет собой (+)3α-амино-4β-гидрокси-10β-пинан или (-) 3α-амино-4β-гидрокси-10β-пинан.
2. 3,4-Гидроксиамины структурной формулы (2) или (3)
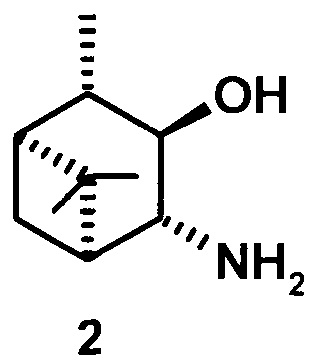 или
или 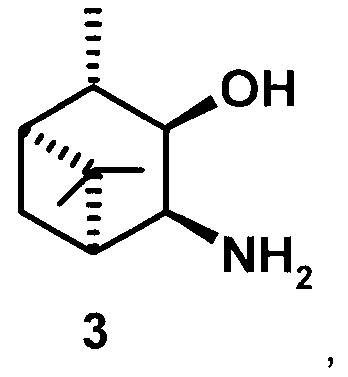
где соединение (2) представляет собой 3α-гидрокси-4β-амино-10β-пинан и соединение (3) представляет собой 3α-гидрокси-4α-амино-10β-пинан.
| Burak, Krzysztof; Chabudzinski, Zenon, Amino alcohols in pinane series | |||
| Part III | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Burak, Krzysztof; Chabudzinski,Zenon, Amino alcohols in pinnae series | |||
| Part II | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Polish Journal of Chemistry, 1981, 55(2), 387-92 (English) (Найдено ACS on STN, реферат, 96:20272, см | |||
| CAS Registry, соединение с RN 80138-55-2P 80185-63-3P) | |||
| Burak, Krzysztof; Chabudzinski, Zenon, Amino alcohols in cis-pinane series | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Symp.Pap.IUPAC Int | |||
| Symp.Chem | |||
| Nat | |||
| Prod., 1978, 11th, Volume 2, 75-8 | |||
| Editor(s): Marekov, N.; Ognyanov, I.; Orahovats, A | |||
| Izd | |||
| BAN: Sofia, Bulg | |||
| (English) (Найдено ACS on STN, 92:76675 реферат, 96:20272, CAS Registry, соединение с RN 72596-09-9P) | |||
| WO 2009057827 A1, 07.05.2009 | |||
| Способ получения производных пинана в виде оптически активных изомеров | 1975 |
|
SU604479A3 |
| DATABASE REGISTRY, CHEMICAL ABSTRACTS SERVICE, COLUMBIO OHIO, US: abstracts, (online!) 26.05.2016, RN-1933776-42-1 (Retrieved on STN). | |||

