Настоящее изобретение относится к области органической химии, а именно к способу получения терпеновых α-хлоркетонов или хлоргидроксикетонов, которые широко используются в качестве интермедиатов для получения гетероциклических соединений, эпоксидов конденсаций Дарзана, α-алкил(арил)-тиокарбонильных соединений, β-кетоэфиров и т.д.
Существует значительное количество методов и реагентов для синтеза α-хлоркетонов. Большинство этих методов основано на α-хлорировании кетонов Сl2 [Aston, J.С, Newkirk, J.D., Jenkins, D.M. and Dorsky, J., "Organic Syntheses," Collect. Vol. 111, Wiley, New York, 1955, p. 538], N-хлорсукцинимидом [Buu-Hoi, N.P. and Demerseman, P. // J. Org. Chem., 1953, 18, 649], сульфурилхлоридом [Wyman, D. P. and Kaufman, P.R., // J. Org. Chem., 1964, 29, 1956], оксихлоридом селена [Schaefer, J.P. and Sonnenberg, F . , // J. Org. Chem., 1963, 28, 1128], хлоридом меди (II) [Kosower, E. M., Cole, W. J., Wu, G . S., Cardy, D.E. and Meisters, G., J. Org. Chem., 1963, 28, 630], трихлоризоциануровой кислотой [Hiegel, G. A.; Peyton, К. B. Chlorination of Ketones with Trichloroisocyanuric Acid. Synthetic Communications 1985, 15, 385-392], системой NaClO2- Mn(acac)3 в присутствии влажного Al2O3 [Yakabe, S.; Hirano, M.; Morimoto, T. α-Chlorination of Ketones with Sodium Chlorite, Mn(acac)3 and Alumina in Dichloromethane // Synthetic Communications 1998, 28, 131-138], полимерным аналогом N,N-дихлор-п-толуолсульфонамида в присутствии кислотных катализаторов [Kawasoe, S.; Kobayashi, K.; Ikeda, K.; Ito, Т.; Seok Kwon, Т.; Kondo, S.; Kunisada, H.; Yuki, Y. Preparation of Polymeric Analogs of N,N-Dichloro-p-Toluenesulfonamide and Their Use for Oxidation of Alcohols, Oxidative Lactonization of Diols, and Chlorination of Carbonyl Compounds. // Journal of Macromolecular Science, Part A, 1997, 34, 1429-1438].
И только несколько примеров прямого превращения вторичных спиртов в α-хлоркетоны приведены в литературе. Так, вторичный фенилэтиловый спирт при обработке Сl2 в СН2Сl2 при -50°C образует 2-хлорацетофенон с выходом 16% [Yamauchi, Т., Hattori, K., Mizutaki, S., Tamaki, K., & Uemura, S. (1986). Selenium and tellurium tetrachlorides as reagents for the conversion of alcohols to alkyl chlorides and tellurium tetrachloride as a Lewis acid catalyst for aromatic alkylation. // Bulletin of the Chemical Society of Japan, 59(11), 3617-3620. doi:10.1246/bcsj.59.3617]. С выходом около 50% получены 2-хлор-3-кетостероиды окислительным хлорированием соответствующих спиртов трет-бутилгипохлоритом в уксусной кислоте при Т=70°C [Beereboom, J. J., Djerassi, C, Ginsburg, D., & Fieser, L.F. (1953). Synthesis and Reactions of Chlorinated 3-Ketosteroids // Journal of the American Chemical Society, 75(14), 3500-3505. doi:10.1021/ja01110а057]. При использовании N-хлорсукцинимида или t-BuOCl в трет-бутаноле с небольшим содержанием Н2O выход α-хлор-3-кетостероидов возрастает до 57-84% [Hanze, A. R., Fonken, G.S., Mcintosh, А.V., Searcy, А.М., & Levin, R.Н. (1954). Chemical Studies with 11-Oxygenated Steroids. V. A One-Step Oxidation-Halogenation of 3-Hydroxysteroids // Journal of the American Chemical Society, 76(12), 3179-3181. doi:10.1021/ja01641a020].
Авторами работы [Kim, H.J., Kim, H.R., & Ryu, E.K. (1990). One-Pot Synthesis of α-Chloroketones from Secondary Benzylic Alcohols Using m-Chloroperbenzoic Acid/HCI/DMF System // Synthetic Communications, 20(11), 1625-1629. doi:10.1080/00397919008053082] предложен одностадийный метод синтеза α-хлоркетонов из вторичных бензильных спиртов с использованием системы m-СРВА-HCl-DMF при комнатной температуре в течение 6 часов. Селективность образования целевых продуктов составляет 80-98%, препаративный выход 80-84%. Для окислительного хлорирования этих же субстратов использован N,N-дихлор-п-толуолсульфонамид в ацетонитриле, выход продуктов составляет 92-94% [Kim, Y.Н., Lee, I.S., & Lim, S.С. (1990). One-pot synthesis of α-chloro-ketones from secondary alcohols using N,N-dichloro-p-toluenesulfonamide // Chemistry Letters. Japan Science and Technology Agency (JST), V19, 7, 1125-1128, (1990)]. Для прямого превращения 1-тетралола в α-хлор-1-тетралон использован N-хлорсукцинимид в СН2Сl2 в присутствии сложного катализатора [Tripathi, С.В., & Mukherjee, S. (2012). Lewis Base Catalysis by Thiourea: N-Bromosuccinimide-Mediated Oxidation of Alcohols // The Journal of Organic Chemistry, 77(3), 1592-1598. doi:10.1021/jo202269p]. Известен способ прямого получения α-хлоркетонов с выходом 38-79% окислением ди- и тризамещенных олефинов хромилхлоридом (СrO2Сl2) в ацетоне при температуре минус 70°C [Sharpless, К.В.; Teranishi, A.Y. Chromyl chloride in acetone. α-Chloro ketones and ketones directly from olefins // J. Org. Chem. 1973, 38, 185-186].
α-хлоркетоны с выходом около 50% образуются в результате окисления вторичных спиртов по Сверну при обязательном использовании избытка реагента - оксалилхлорид-ДМСО [Smith, А. В., III; Leenay, Т.L.; Liu, H.-J.; Lloyd A.K.Nelson; Ball, R.G. A Cavaet on the Swern Oxidation // Tetrahedron Letters 1988, 29, 49-52].
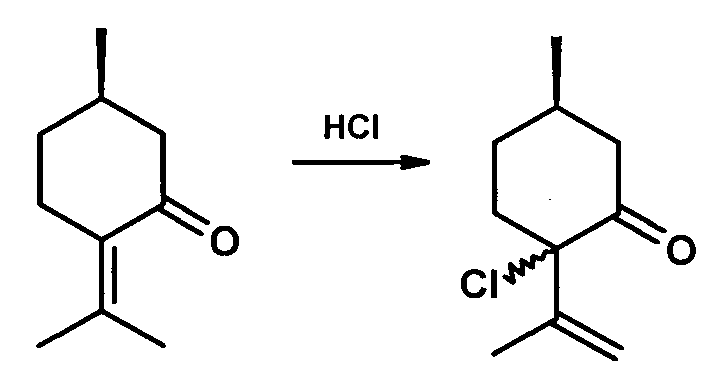
Описан способ получения α-хлоризопулегона в виде смеси стереоизомеров в соотношении 3:2 с выходом 75% при обработке пулегона 1 экв. НОСl в СН2Сl2 при температуре -60°C [Hegde, S. G.; Beckwith, D.; Doti, R.; Wolinsky, J. Synthesis with hypochlorous acid. Conversion to pulegone and isopulegol to menthofuran. Preparation of 3,6-dimethyl-2,6-cycloheptadien-1-one from phorone // J. Org. Chem. 1985, 50, 894-896].
Известен способ получения терпенового 2-хлорпинанона-3 в две стадии: а) взаимодействие пинокарвеола с НСl в абсолютном эфире в течение 24 час приводит к образованию гидрохлорида пинокарвеола (выход продукта не указан); б) окисление последнего CrO3 с выходом 55% дает целевой α-хлоркетон, Тпл. 33-35 [α]D+12 (чистое вещество) [Treibs, W.; Mühlstädt, М.; Megges, R.; Klotz-Herdmann, I. Über Pinocarveol. // Justus Liebigs Ann. Chem. 1960, 634, 118-124].
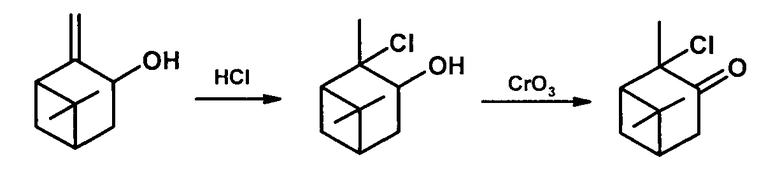
Недостатком этого метода является проведение реакции в две стадии, применение абсолютного эфира, длительность обеих стадий (24 и 12 часов соответственно), средний выход целевого продукта (~55%).
Прототипом предлагаемого изобретения взят способ окисления диоксидом хлора в пиридине вицинальных терпеновых диолов карановой и пинановой структуры [Л.Л. Фролова, А.В. Попов, Л.В. Безуглая, И.Н. Алексеев, П.А. Слепухин, А.В. Кучин. Окисление терпеновых диолов диоксидом хлора: синтез кетолов и α-хлоргидроксикетонов карановой и пинановой структуры, ЖОХ, 2013, т.83(145), вып.8, 1311-1317]. Способ заключается в окислении 3α,4α-карандиола диоксидом хлора в течение 10 часов в присутствии 5% мол ZrOCl2, при этом образуется, кроме основного гидроксикетона, смесь (5:4) диастереомерных 5-хлор, 3α-гидроксикаранонов-4, препаративный выход которой составил 3%. Недостатком данного способа является 1) образование смеси хлоргидроксикетонов, которые не разделяются хроматографией или кристаллизацией; 2) низкий выход этой смеси.
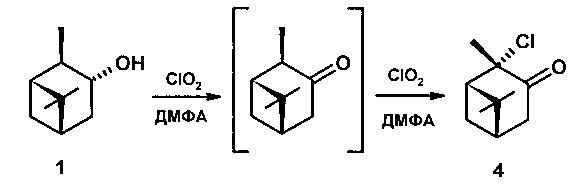
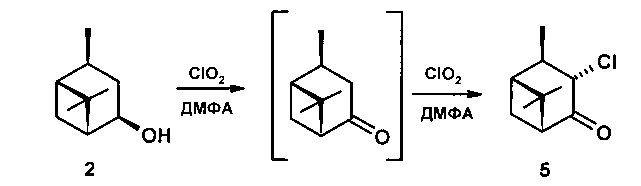
Задачей настоящего изобретения является разработка одностадийного способа получения терпеновых α-хлоркетонов или хлоргидроксикетонов, позволяющего получать новые соединения, а именно 2α-хлорпинанон-3,3α-хлор-10β-пинанон-4,5β-хлор-3β-гидроксикаранон-4 окислением диоксидом хлора. В этом и состоит технический результат.
Технический результат достигается тем, что способ получения терпеновых α-хлоркетонов или хлоргидроксикетонов включает пропускание через раствор исходного соединения в растворителе, без или в присутствии катализаторов, тока газообразного диоксида хлора в течение определенного времени, удаление растворителя, экстракцию диэтиловым или метил-трет-бутиловым эфиром, промывание и сушку реакционной смеси над безводным MgSO4, выделение конечного продукта хроматографией на SiO2 или кристаллизацией, согласно изобретению, в качестве исходного соединения взяты изопинокамфеол, либо неоизовербанол, либо 3β,4β-карандиол.
Способ осуществляется следующим образом.
Исходными соединениями являются изопинокамфеол (1), неоизовербанол (2), 3β,4β-карандиол (3). В качестве окислительно-хлорирующего реагента применяется диоксид хлора (промышленный продукт, используемый для отбелки целлюлозы и очистки питьевой и сточных вод), в качестве растворителя - диметилформамид, в качестве катализаторов - оксихлорид циркония или ацетилацетонат ванадила или хлорид молибдена (V).
Через раствор исходного соединения 1-3 в диметилформамиде без или в присутствии катализаторов хлорида молибдена (МоСl5) или оксихлорида циркония ZrOCl2 или ацетилацетоната ванадила (VO(acac)2) в течение определенного времени пропускают ток газообразного диоксида хлора. Реакцию контролируют по ТСХ и ГЖХ, после удаления растворителя реакционную смесь разбавляют водой, продукты реакции экстрагируют диэтиловым или метил-трет-бутиловым эфиром, эфирные вытяжки промывают насыщенным водным раствором NaCl и сушат над безводным MgSO4. После удаления растворителя продукты анализируют методом ГЖХ, выделяют колоночной хроматографией на силикагеле или кристаллизацией.
Пример 1. Через раствор 0,5 г изопинокамфеола [[α]D-35.8 (с 0,7 EtOH), т.пл. 51-52°C] и 0,01 г ZrOCl2 в 10 мл ДМФА при перемешивании при комнатной температуре пропускали ток газообразного СlO2. Реакцию контролировали по ТСХ и ГЖХ. Через 3 часа из реакционной смеси отогнали ДМФА на роторном испарителе, остаток разбавили 15 мл Н2O, продукты экстрагировали метилтретбутиловым эфиром (3*15), эфирные вытяжки промывали насыщенным раствором NaCl, сушили над MgSO4. После удаления растворителя продукт выделяли колоночной хроматографией на SiO2 (элюент - петролейный эфир:диэтиловый эфир). Выход 0,355 г (71.0%). Результаты экспериментов приведены в таблице 1.
2α-хлорпинанон-3 (4). Rf 0.72 (гексан:Еt2O 2:1), [α]D+96.4 (с 1.1 ЕtOН). ИК спектр (ν, см-1) : 2978, 2935, 2875, 1728 (С=O), 1471, 1448, 1409, 1373, 1321, 1267, 1240, 1209, 1145, 1099, 1047, 945, 914, 866, 831, 729 (С-С1), 628, 551. Спектр ЯМР 13С (75 МГц, CDCl3, δ, м.д.) : 206.02 (С3), 73.98 (С2), 52.84 (С1), 43.06 (С4), 40.26 (С6), 38.27 (С5), 31.55 (С7), 27.47 (С9), 27.50 (С10), 22.79 (С8). ЯМР 1H (300 МГц, CDCl3, δ/м.д., J/Гц): 0.93 с (3Н, СН3-8), 1.41 с (3Н, СН3-9), 1.79 с (3Н, СН3-10), 1.91 д (1Н, Н-7` J=11), 2.17 м (1Н, Н-5, J=2.4, 6.1), 2.41 дд (1H, Н-1, J=6.1, 6.2), 2.59 м (1H, Н-7``, J=11), 2.70 ддд (1H, Н-4` J=3.0, 6.2, 19), 2.77 дд (1H, Н-4``, J=2.4, 19).
Пример 2. Через раствор 0,26 г неоизовербанола [[α]D-1.5 (с 1,0 EtOH), т.пл. 65-66°C] в 5 мл ДМФА при перемешивании при комнатной температуре пропускали ток газообразного СlO2. Реакцию контролировали по ТСХ и ГЖХ. Через 2 часа из реакционной смеси отогнали ДМФА на роторном испарителе, остаток разбавили 10 мл Н2O, продукты экстрагировали диэтиловым эфиром (3*15), эфирные вытяжки промывали насыщенным раствором NaCl, сушили над MgSO4. После удаления растворителя продукт выделяли колоночной хроматографией на SiO2 (элюент - петролейный эфир:диэтиловый эфир). Выход 0,164 г (63.0%). Результаты экспериментов приведены в таблице 2.
3α-хлор, 10β-пинанон-4 (5). Rf 0.58 (гексан:Еt2O 1:1), [α]D+1.8 (с 0.4 ЕtOН). ИК спектр (ν, см-1): 2958, 2939, 2879, 1728 (С=O), 1467, 1382, 1294, 1249, 1188, 983, 881, 773 (С-С1), 650, 626, 586, 503. Спектр ЯМР 13С (75 МГц, DMSO-d6, δ, м.д.): 206.08 (С4), 62.25 (С3), 58.49 (С5), 47.80 (С1), 45.38 (С2), 39.62 (С6), 29.37 (С7), 26.96 (С9), 24.93 (С8), 19.11 (С10). Спектр ЯМР 1Н (300 МГц, DMSO-d6, δ/м.д., J/Гц): 0.98 с (3Н, СН3-8), 1.24 д (3Н, СН3-10, J=7.4), 1.32 с (3Н, СН3-9), 1.39 д (1H, Н-7`, J=11), 2.16 ддд (1Н, Н-1, J=2.3, 5.7, 5.8), 2.42 ддк (1H, Н-2, J=2.3, 5.7, 7.4), 2.68 дд (1H, Н-5, J=5.5, 5.8), 2.77 ддд (1H, Н-7``, J=4.8, 6.0, 11), 4.59 д (1H, Н-3, J=5.7).
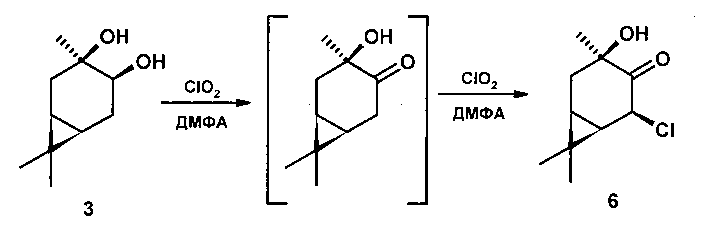
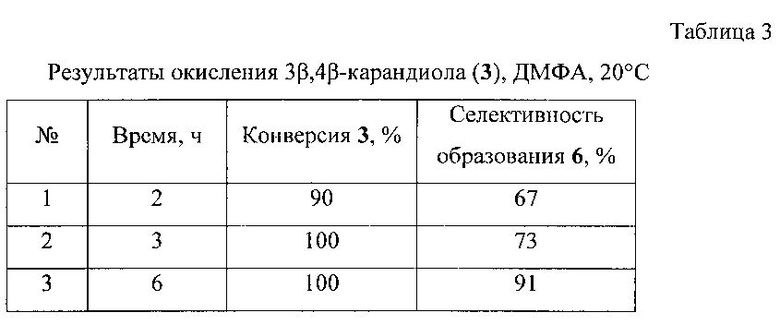
Пример 3. Через раствор 0,21 г 3β,4β-карандиола [[α]D+42.7 (с 10 СНСl3), т.пл. 43-44°C] в 5 мл ДМФА при перемешивании при комнатной температуре пропускали ток газообразного СlO2. Реакцию контролировали по ТСХ и ГЖХ. Через 6 часов из реакционной смеси отогнали ДМФА на роторном испарителе, остаток разбавили 10 мл Н2O, продукты экстрагировали диэтиловым эфиром (3*15 мл), эфирный экстракт промывали насыщенным раствором NaCl, сушили над MgSO4. После удаления растворителя продукт выделяли колоночной хроматографией на SiO2 (элюент - петролейный эфир:диэтиловый эфир). Выход 0,161 г (77%). Результаты экспериментов приведены в таблице 3.
5β-хлор-3β-гидроксикаранон-4 (6). Rf 0.38 (Et2O : гексан 1:1), [α]D+179.1 (с. 0.3 EtOH), т.пл. 85-87°C. ИК спектр (ν, см-1): 3495(ОН), 3012, 2983, 2954, 2933, 2912, 1724(С=O), 1450, 1382, 1361, 1282, 1226, 1186, 1145, 1089, 1033, 970, 939, 812, 790, 759(С-С1), 734. Спектр ЯМР 1Н (300 МГц, DMSO-d6, δ/м.д., J/Гц): 0.84 (3Н, с, СН3-8), 1.04 (3Н, с, СН3-9), 1.16 ддд (Н, Н-1, J 3.9, 9.0, 13), 1.39 с (3Н, с, СН3-10), 1.72 дд (1Н, Н-2, J 3.8, 14.6), 1.76 дд (1H, Н-6, J 8.4, J 9.0), 2.23 дд (1H, Н-2, J 9.8, 14.6), 5.73 д (1Н, Н-5, J 8.4). Спектр ЯМР 13С (75 МГц, DMSO-d6, δ, м.д.): 207.8 (С-4), 76.7 (С-3), 64.8 (С-5), 37.4 (С-2), 33.0 (С-6), 28.2 (С-9), 25.7 (С-10), 22.3 (С-7), 22.2 (С-1), 16.0 (С-8).


| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ 3α-ГИДРОКСИ-10β-ПИНАНОНА-4 | 2012 |
|
RU2509073C1 |
| Новые 3,4-гидроксиамины пинановой структуры | 2016 |
|
RU2631871C1 |
| 3 β -ХЛОР-4 b -ОКСИ-24S-ЭТИЛ-5 b -ХОЛЕСТАН-6-ОН, ОБЛАДАЮЩИЙ ФИТОРОСТОСТИМУЛИРУЮЩЕЙ АКТИВНОСТЬЮ, И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 1986 |
|
SU1371003A1 |
| НОВЫЕ НАФТО[2,1-b]КАРБАЗОЛПРОИЗВОДНЫЕ ФУЗИДОВОЙ КИСЛОТЫ, СПОСОБ ПОЛУЧЕНИЯ И АНТИМИКРОБНЫЕ СВОЙСТВА | 2019 |
|
RU2746947C2 |
| Сульфопроизводные α-пинена | 2017 |
|
RU2657730C1 |
| СПОСОБ ПОЛУЧЕНИЯ 3-АЛКИЛ(ФЕНИЛ)-2,5-ДИФЕНИЛ-1Н-ПИРРОЛОВ | 2017 |
|
RU2677470C2 |
| Способ получения хиральных S-монотерпенилцистеинов | 2018 |
|
RU2675238C1 |
| Сесквитерпеновый тиоацетат (варианты) | 2016 |
|
RU2608944C1 |
| СПОСОБ ГИДРИРОВАНИЯ АЛЬФА, БЕТА-НЕНАСЫЩЕННЫХ КЕТОНОВ | 2013 |
|
RU2529032C1 |
| Способ получения монотерпеновых сульфокислот | 2016 |
|
RU2651791C2 |
Настоящее изобретение относится к способу получения новых терпеновых α-хлоркетонов или хлоргидроксикетонов, которые широко используются в качестве интермедиатов для получения гетероциклических соединений, эпоксидов конденсаций Дарзана, α-алкил(арил)-тиокарбонильных соединений, β-кетоэфиров. Способ включает пропускание через раствор исходного соединения в растворителе, без или в присутствии катализаторов, тока газообразного диоксида хлора в течение определенного времени, удаление растворителя, экстракцию диэтиловым или метил-трет-бутиловым эфиром, промывание и сушку реакционной смеси над безводным MgSO4, выделение конечного продукта хроматографией на SiO2 или кристаллизацией. При этом в качестве исходного соединения используют изопинокамфеол, либо неоизовербанол, либо 3β,4β-карандиол. 3 табл., 3 пр.
Способ получения терпеновых α-хлоркетонов или хлоргидроксикетонов, включающий пропускание через раствор исходного соединения в растворителе, без или в присутствии катализаторов, тока газообразного диоксида хлора в течение определенного времени, удаление растворителя, экстракцию диэтиловым или метил-трет-бутиловым эфиром, промывание и сушку реакционной смеси над безводным MgSO4, выделение конечного продукта хроматографией на SiO2 или кристаллизацией, отличающийся тем, что в качестве исходного соединения взяты изопинокамфеол, либо неоизовербанол, либо 3β,4β-карандиол.
| Фролова Л.Л | |||
| и др | |||
| Окисление терпеновых диолов диоксидом хлора: синтез кетолов и α-хлоргидроксикетонов карановой и пинановой структуры | |||
| ЖОХ, 2013, т | |||
| Пуговица | 0 |
|
SU83A1 |
| Регенератор | 1925 |
|
SU1412A1 |
| US 3661998A1, 09.05.1972 | |||
| СПОСОБ ПОЛУЧЕНИЯ 3α-ГИДРОКСИ-10β-ПИНАНОНА-4 | 2012 |
|
RU2509073C1 |

