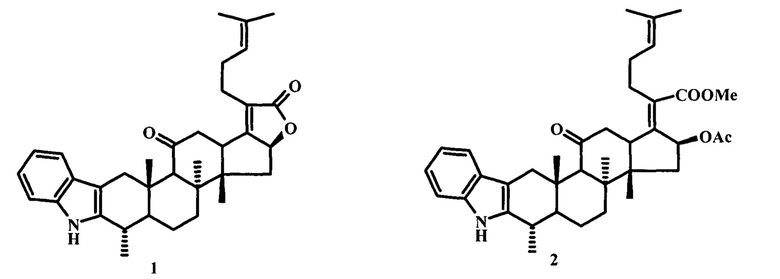
Изобретение относится к области синтеза биологически активных аналогов природных соединений, а именно к получению (3αS,4αS,4βS,7S,13αS)-4α,4β,7,13α-тетраметил-1-(4-метилпент-3-ен-1-ил)-3α,4,4α,4β,5,6,6α,7,8,13,13α,13β,15,15α-тетрадекагидрофуро[3'',2'':3',4']циклопента[1',2':5,6]нафто[2,1-b]карбазол-2,14-диона (1) и метил (2Z)-2-[(2S,3αS,3βS,6S,12αS)-2-(ацетилокси)-3α,3β,6,12α-тетраметил-13-оксо-3,3α,3β,4,5,5α,6,7,12,12α,12β,13,14,14α-тетрадекагидроциклопента[5,6]нафто[2,1-b]карбазол-1(2H)-илиден]-6-метилгепт-5-еноата (2), обладающих высокой активностью по отношению к Staphylococcus aureus (MRSA):
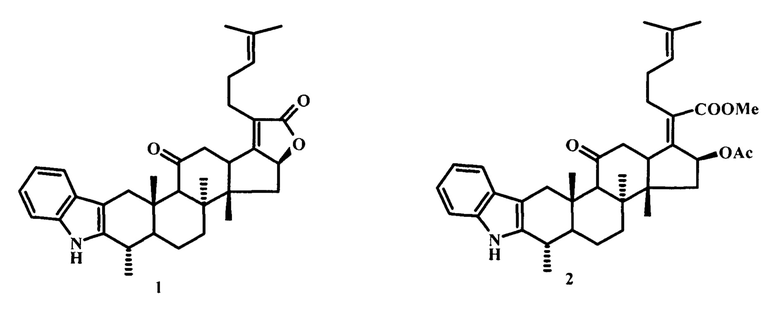
Известны 1Н-дибензо[а, с]карбазолпроизводные дегидроабиетиновой кислоты с различными заместителями в ароматическом ядре (3а-l) [W. Gu, S. Wang. Synthesis and antimicrobial activities of novel lH-dibenzo[a,c]carbazoles from dehydroabietic acid. Eur. J. Med. Chem. 2010, 45, P. 4692-4696] и N-замещенные 1Н-дибензо[а,с]карбазолпроизводные дегидроабиетиновой кислоты, содержащие аминные или гетероциклические группы в боковой алкильной цепи (4a-m) [W. Gu, С. Qiao, S.F. Wang, Y. Нао, Т.Т. Miao. Synthesis and biological evaluation of novel N-substituted lH-dibenzo[a,c]carbazole derivatives of dehydroabietic acid as potential antimicrobial agents. BMC Lett. 2014, 24, P. 328-331] (схема 1).
Схема 1.
Изучение антибактериальной активности синтезированных производных in vitro в отношении штаммов грамположительных (Bacillus subtilis, Staphylococcus aureus) и грамотрицательных (Escherichia coli, Pseudomonas fluorescens) микроорганизмов, а также трех грибковых культур (Candida albicans, Candida tropicalis и Aspergillus niger) показало, что соединения (3d), (3е), (3l), а также производные, содержащие пиперазиновые (4f), (4g) или азольные гетероциклические фрагменты, соединенные гибкими этильными цепями (4j) - (4m), обладали широким спектром антимикробной активностью по отношению ко всем исследованным штаммам при МИК от 0,9 до 15,6 мкг/мл.
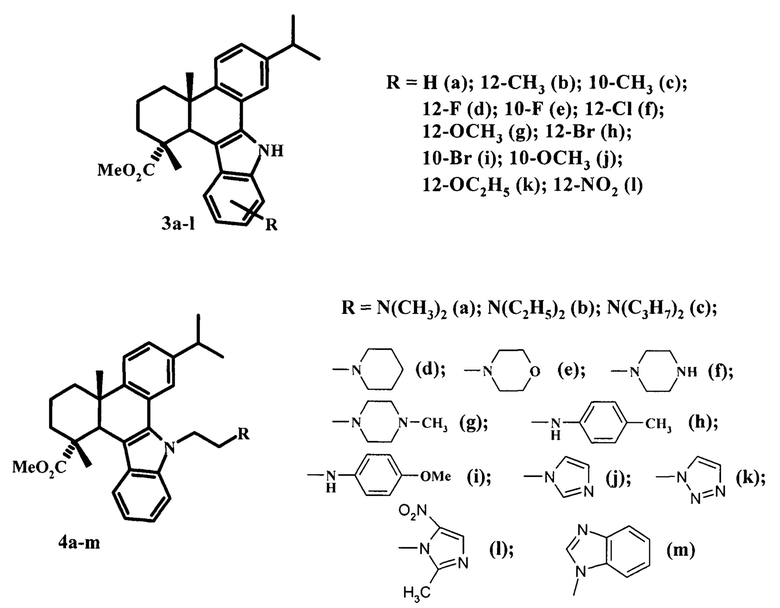
Известны [2,1-b]карбазольные производные урсоловой кислоты (5a-h) (схема 2). Скрининг антибактериальной активности данных соединений в отношении грамположительных (Bacillus subtilis, Staphylococcus aureus) и грамотрицательных (Escherichia coli, Pseudomonas fluorescens) бактерий, а также грибковых культур (Candida albicans, Candida tropicalis и Aspergillus niger) показал, что производное с фториндольным фрагментом и N-(диметиламино)пропиламидной боковой цепью (5b) проявило выраженную антибактериальную активность в отношении всех исследованных бактерий [W. Gu, Y. Нао, G. Zhang, S.F. Wang, Т.Т. Miao, K.Р. Zhang. Synthesis, in vitro antimicrobial and cytotoxic activities of new carbazole derivatives of ursolic acid. BMC Lett. 2015, 25, P. 554-557].
Схема 2.
Таким образом, синтез и антибактериальные свойства нафто[2,1-b]карбазолпроизводных фузидовой кислоты (1) и (2) в литературе не описаны.
Задачей предлагаемого изобретения является синтез и изучение противомикробной активности in vitro (3αS,4αS,4βS,7S,13αS)-4α,4β,7,13α-тетраметил-1-(4-метилпент-3-ен-1-ил)-3α,4,4α,4β,5,6,6α,7,8,13,13α,13β,15,15α-тетрадекагидро-фуро[3'',2'':3',4']циклопента[1',2':5,6]нафто[2,1-b]карбазол-2,14-диона (1) и метил (2Z)-2-[(2S,3αS,3βS,6S,12αS)-2-(ацетилокси)-3α,3β,6,12α-тетраметил-13-оксо-3,3α,3β,4,5,5α,6,7,12,12α,12β,13,14,14α-тетрадекагидро-циклопента[5,6]нафто[2,1-b]карбазол-1(2H)-илиден]-6-метилгепт-5-еноата (2) в отношении 5 различных видов бактерий: Escherichia coli, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa и Staphylococcus aureus, и двух разновидностей грибков: Candida albicans и Cryptococcus neoformans.
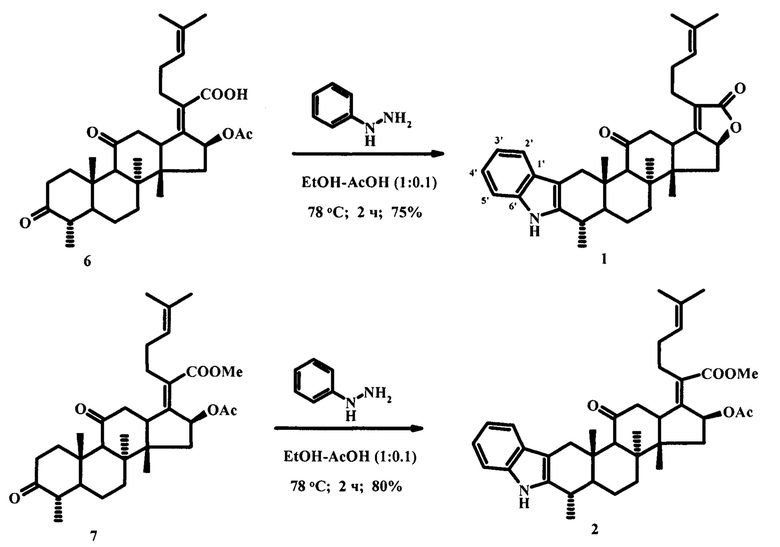
Синтез заявленных индолов осуществляли по реакции Фишера, используя в качестве исходных соединений 3,11-диоксопроизводное фузидовой кислоты (6) или ее метилового эфира (7). Дикетоны (6) или (7) вовлекали во взаимодействие с 3 экв. фенилгидразина в смеси сухого этанола и ледяной уксусной кислоты. Реакционную массу нагревали в течение 2 часов и обрабатывали ледяной водой (схема 3). В результате реакции выделяли производные (1) и (2) с выходами 75 и 80%, соответственно.
Схема 3.
Сущность изобретения поясняется следующими примерами.
Пример 1. Дикетон (6) (0.2 г, 0.32 ммоль) растворили в 5 мл сухого этанола, добавили фенилгидразин (0.1 г, 0.96 ммоль) и 0.5 мл ледяной уксусной кислоты. Смесь нагревали при 78°С в течение 2 часов. Реакционную массу охладили и добавили 30 мл ледяной воды. Продукт реакции экстрагировали хлороформом (3×30 мл), объединенные органические экстракты промывали раствором соли и сушили над CaCl2. Растворитель упаривали при пониженном давлении, получая сырой продукт, который очищали колоночной хроматографией на силикагеле, элюируя смесью петролейный эфир / этилацетат, 1:1.
(3αS,4αS,4βS,7S,13αS)-4α,4β,7,13α-Тетраметил-1-(4-метилпент-3-ен-1-ил)-3α,4,4α,4β,5,6,6α,7,8,13,13α,13β,15,15α-тетрадекагидрофуро[3'',2'':3',4']циклопента[1',2':5,6]нафто[2,1-b]карбазол-2,14-дион (1). Порошок коричневого цвета; выход 75%; т.пл. 194-196°С, [α] 20D +31.2° (с 0.855, CHCl3). Спектр ЯМР 1Н (CDCl3, δ, м.д.): 1.03 (3Н, с, Н-19), 1.14 (3Н, с, Н-18), 1.19-1.22 (1Н, м, Н-6а), 1.28-1.36 (1Н, м, Н-7а), 1.33 (3Н, д, J = 6.5 Гц, Н-28), 1.38 (3Н, с, Н-30), 1.49 (1Н, д, J = 7.1 Гц, Н-15а), 1.63 (3Н, с, Н-27), 1.72 (3Н, с, Н-26), 1.74-1.91 (1Н, м, Н-5), 1.75-1.87 (1H, м, Н-6b), 2.04-2.14 (1H, м, Н-7b), 2.21-2.31 (2Н, м, Н-23), 2.25-2.39 (1Н, м, Н-15b), 2.37-2.48 (2Н, м, Н-22), 2.61 (1H, с, Н-9), 2.64-2.73 (1Н, м, Н-4), 2.74-2.88 (2Н, м, Н-12), 2.75-2.82 (1Н, м, Н-1а), 3.37 (1Н, дд, J = 5.0, 13.0 Гц, Н-13), 3.77 (1Н, д, J = 16.0 Гц, Н-1b), 5.02 (1Н, дд, J = 4.5, 10.5 Гц, Н-16), 5.13 (1Н, т, J = 6.0 Гц, Н-24), 7.09 (1Н, ддд, J3'-2' = 7.5 Гц, J3'-4' = 7.5 Гц, J3'-5' = 1.4 Гц, Н-4'), 7.15 (1Н, ддд, J4'-2' = 1.4 Гц, J4'-3' = 7.5 Гц, J4'-5' = 7.1 Гц, Н-3'), 7.32 (1Н, ддд, J5'-2' = 0.1 Гц, J5'-4' = 7.1 Гц, J5'-3' = 1.4 Гц, Н-5'), 7.55 (1Н, ддд, J2'-5' = 0.1 Гц, J2'-3' = 7.5 Гц, J2'-4' = 1.4 Гц, Н-2').
Спектр ЯМР 13С (CDCl3, δ, м.д.): 17.18 (СН3, С-28), 17.83 (СН3, С-27), 19.73 (СН3, С-18), 20.34 (СН2, С-6), 20.60 (СН3, С-19), 24.12 (СН2, С-22), 25.68 (СН3, С-30), 25.72 (СН3, С-26), 27.16 (СН2, С-23), 29.80 (СН, С-4), 30.96 (СН2, С-1), 33.64 (СН2, С-7), 33.73 (СН2, С-15), 37.47 (С, С-10), 40.50 (СН2, С-12), 40.79 (СН, С-13), 41.60 (С, С-8), 47.51 (СН, С-5), 54.52 (С, С-14), 58.60 (СН, С-9), 81.65 (СН, С-16), 110.19 (С, С-2), 110.44 (СН, С-5'), 118.18 (СН, С-2'), 119.19 (СН, С-3'), 121.18 (СН, С-4'), 122.86 (СН, С-24), 125.42 (С, С-20), 128.42 (С, С-1'), 133.32 (С, С-25), 136.06 (С, С-6'), 137.36 (СН, С-3), 166.89 (С, С-17), 175.85 (С, С-21), 208.54 (С, С-11). Масс-спектр (MALDI TOF/TOF), m/z (Iотн., %): 525.416 [М] (100), 548.903 [M+Na] (49). Вычислено для C35H43NO3: С, 79.96; Н, 8.24; N, 2.66%. Найдено: С, 79.79; Н, 8.21; N, 2.68%.
Пример 2. Дикетон (7) (0.1 г, 0. 21 ммоль) растворили в 5 мл сухого этанола, добавили фенилгидразин (0.07 г, 0.63 ммоль) и 0.5 мл ледяной уксусной кислоты. Смесь нагревали при 78°С в течение 2 часов. Реакционную массу охладили и добавили 30 мл ледяной воды. Продукт реакции экстрагировали хлороформом (3×30 мл), объединенные органические экстракты промывали раствором соли и сушили над CaCl2. Растворитель упаривали при пониженном давлении, получая сырой продукт, который очищали колоночной хроматографией на силикагеле, элюируя смесью петролейный эфир / этилацетат, 2:1.
Метил (2Z)-2-[(2S,3αS,3βS,6S,12αS)-2-(ацетилокси)-3α,3β,6,12α-тетраметил-13-оксо-3,3α,3β,4,5,5α,6,7,12,12α,12β,13,14,14α-тетрадекагидроциклопента[5,6]нафто[2,1-b]карбазол-1(2H)-илиден]-6-метилгепт-5-еноат (2).
Порошок коричневого цвета; выход 80%; т.пл. 185-187°С, [α] 20D +78.5° (с 0.923, CHCl3). Спектр ЯМР 1Н (CDCl3, δ, м.д.): 1.03 (3Н, с, Н-19), 1.15-1.23 (1Н, м, Н-6а), 1.18-1.28 (1Н, м, Н-7а), 1.22 (3Н, с, Н-30), 1.25 (3Н, с, Н-18), 1.32 (3Н, д, J = 6.7 Гц, Н-28), 1.47 (1Н, д, J = 14.3 Гц, Н-15а), 1.64 (3Н, с, Н-27), 1.71 (3Н, с, Н-26), 1.72-1.79 (1Н, м, Н-6b), 1.72-1.82 (1H, м, Н-5), 2.04-2.21 (2Н, м, Н-23), 2.05 (3Н, с, O-С(O)СН3), 2.06-2.13 (1H, м, Н-7b), 2.40-2.47 (2Н, м, Н-22), 2.60-2.69 (1Н, м, Н-4), 2.71 (1Н, с, Н-9), 2.72-2.81 (1Н, м, Н-12а), 2.81 (1Н, д, J = 15.9 Гц, Н-1а), 2.93-3.01 (1Н, м, Н-13), 2.96-3.05 (1Н, м, Н-12b), 3.70 (3Н, с, С(О)ОСН3), 3.77 (1Н, д, J = 15.9 Гц, Н-1b), 5.12 (1Н, т, J = 6.5 Гц, Н-24), 5.95 (1Н, д, J = 8.3 Гц, Н-16), 7.14 (1Н, ддд, J4'-2' = 1.5 Гц, J4'-3' = 7.5 Гц, J4'-5' = 7.0 Гц, Н-4'), 7.10 (1Н, ддд, J3'-2' = 7.4 Гц, J3'-4' = 7.5 Гц, J3'-5' = 1.5 Гц, Н-3'), 7.31 (1Н, ддд, J5'-2' = 0.1 Гц, J5'-4' = 7.0 Гц, J5'-3' = 1.5 Гц, Н-5'), 7.55 (1Н, ддд, J2'-5' = 0.1 Гц, J2'-3' = 7.4 Гц, J2'-4' = 1.5 Гц, Н-2').
Спектр ЯМР 13С (CDCl3, δ, м.д.): 16.95 (СН3, С-18), 17.17 (СН3, С-28), 17.78 (СН3, С-27), 20.64 (СН2, С-6), 20.87 (СН3, С-19), 20.97 (СН3, O-С(O)СН3), 24.22 (СН3, С-30), 25.76 (СН3, С-26), 27.89 (СН2, С-23), 29.03 (СН2, С-22), 29.80 (СН, С-4), 30.96 (СН2, С-1), 33.90 (СН2, С-7), 37.27 (С, С-10), 38.20 (СН2, С-15), 40.56 (С, С-8), 44.60 (СН2, С-12), 46.53 (СН, С-13), 47.44 (СН, С-5), 48.74 (С, С-14), 51.57 (СН3, С(O)ОСН3), 57.64 (СН, С-9), 74.28 (СН, С-16), 110.30 (С, С-2), 110.41 (СН, С-5'), 118.20 (СН, С-2'), 119.12 (СН, С-3'), 121.08 (СН, С-4'), 122.60 (СН, С-24), 128.49 (С, С-1'), 131.80 (С, С-20), 133.10 (С, С-25), 136.08 (С, С-6'), 137.47 (СН, С-3), 145.68 (С, С-17), 170.06 (С, С-21), 170.28 (С, O-С(O)СН3), 210.12 (С, С-11). Масс-спектр (MALDI TOF/TOF), m/z (Iотн., %): 599.354 [М] (100), 638.297 [М+К] (63). Вычислено для C38H49NO5: С, 76.09; Н, 8.23; N, 2.34%. Найдено: С, 76.13; Н, 8.20; N, 2.32%.
Контроль реакции осуществляли методом ТСХ на пластинах Sorbfil (Сорбполимер, Краснодар, Россия), проявляли 10% раствором серной кислоты. Для колоночной хроматографии использовали силикагель L (50-160 мкм) марки КСКГ. Температура плавления определена на приборе РНМК 80/2617. Спектры ЯМР 1D (1Н, 13С) и 2D (COSY, NOESY, HSQC, НМВС) сняты на спектрометре Bruker Avance 500 (125.78 МГц для 13С и 500.17 МГц для 1Н) с использованием стандартных импульсных последовательностей фирмы Bruker, внутренний стандарт Me4Si, растворитель - CDCl3. Оптические углы измерены на поляриметре Perkin-Elmer 341. Масс-спектры MALDI TOF/TOF получены на спектрометре Bruker Autoflex ТМ III Smartbeam с использованием матрицы 3-(4-гидрокси-3,5-диметоксифенил)проп-2-еновой кислоты (синапиновая кислота).
Противомикробный скрининг соединений (1) и (2) проводили в CO-ADD (The Community for Antimicrobial Drug Discovery), финансируемым Wellcome Trust (Великобритания) и Университетом Квинсленда (Австралия), на пяти бактериальных штаммах: Escherichia coli (Е. coli) АТСС 25922, Klebsiella pneumoniae (K. pneumoniae) ATCC 700603, Acinetobacter baumannii (A. baumannii) ATCC 19606, Pseudomonas aeruginosa (P. aeruginosa) ATCC 27853 и Staphylococcus aureus (S. aureus) ATCC 43300. Противогрибковую активность определяли на двух грибковых штаммах: Candida albicans (С. albicans) ATCC 90028 и Cryptococcus neoformans (С. neoformans) ATCC 208821.
Первичный скрининг противомикробной активности проводился путем тестов на ингибирование размножения клеток, используя образцы в одной (32 мкг/мл) концентрации. Аликвоту каждого образца в ДМСО помещали в 384-луночный планшет и обрабатывали соответствующей бактериальной культурой. Ингибирование роста бактерий определяли спектрофотометрически при 600 нм на монохромном микропланшетном ридере Tecan M1000 Pro. Процент ингибирования роста рассчитывали для каждой лунки с использованием отрицательного контроля (только для среды) и положительного контроля (бактерии без ингибиторов) на той же пластинке. В случае если один или оба раза наблюдалось ингибирование роста ≥80%, соединение считалось активным (Таблица 1).
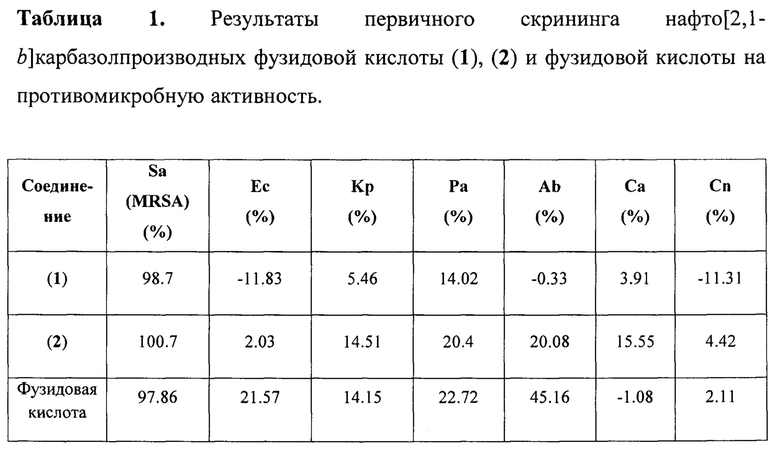
При первичном скрининге было выявлено наличие противомикробной активности у нафто[2,1-b]карбазолпроизводных фузидовой кислоты (1) и (2) в отношении культуры бактерий Staphylococcus aureus. Для соединений (1) и (2) была определена минимальная ингибирующая концентрация в отношении вышеуказанной культуры, а также изучена цитотоксическая и гемолитическая активности.
Минимальную ингибирующую концентрацию (MIC; мкг/мл) устанавливали в соответствии с рекомендациями Института клинических и лабораторных стандартов (Clinical and Laboratory Standards Institute (CLSI), США), определяя наименьшую концентрацию, при которой было обнаружено полное ингибирование бактерий или грибов. Скрининг проводился методом серийных разведений. Образцы готовились в ДМСО в тестовой концентрации 32 мкг/мл. Все бактерии культивировались на агаре Мюллера Хинтона при 37°С в течение ночи. Образец каждой культуры затем разбавлялся в 40 раз и инкубировался при 37°С в течение 1,5-3 ч. Полученные культуры добавлялись в каждую лунку 384-луночного планшета, содержащую исследуемый образец (плотность клеток 5×105 КОЕ/мл и общий объем 50 мкл). Все планшеты накрывались и инкубировались при 37°С в течение 18 ч без встряхивания. Ингибирование роста бактерий определялось измерением поглощения при 600 нм с использованием монохромного микропланшетного ридера Tecan M1000 Pro. Процент ингибирования роста рассчитывался для каждой лунки с использованием отрицательного контроля (только для среды) и положительного контроля (бактерии без ингибиторов) на той же пластине. Значения ингибирования определялись с помощью модифицированных Z-показателей, рассчитанных с использованием медианы и медианного абсолютного отклонения (MAD) образцов (без контроля) на той же пластине. Образцы с величиной ингибирования выше 80% и Z-оценкой выше 2,5 для обеих реплик классифицировались как активные вещества. Образцы с показателями ингибирования от 50 до 80% и Z-оценкой выше 2,5 для обеих реплик классифицировались как частично активные.
Тесты проводились в двойном повторе. Максимальный процент ингибирования роста обозначался как DMax. Хиты были классифицированы при MIC≤16 мкг/мл или MIC≤10 мкМ в любой реплике (n=2 на разных пластинах).
Цитотоксическое действие определяли на клеточной линии эмбриональных почек человека HEK293 путем определения концентрации, вызывающей гибель 50% клеток (Hk СС50). Ингибирование роста клеток HEK293 определяли, измеряя флуоресценцию после добавления 5 мкл 25 мкг/мл резазурина (конечная концентрация 2.3 мкг/мл) и после инкубации в течение еще 3 ч при 37°С в 5% CO2. Интенсивность флуоресценции измеряли с использованием монохромного микропланшетного ридера Tecan M1000 Pro с использованием автоматического вычисления коэффициента усиления. Максимальный процент цитотоксичности обозначали как DMax. Соединение считалось токсичным при СС50≤32 мкг/мл или СС50≤10 мкМ. Кроме того, образцы были отмечены как частичные цитотоксические, если DMax≥50%, даже при СС50 выше максимальной тестируемой концентрации.
Гемолитическую активность (Hm НС10 и НС50 - концентрация при 10% и 50% гемолизе, соответственно) определяли путем измерения поглощения при 405 мм супернатанта - надосадочной жидкости, образованной после инкубации в течение 1 ч при 37°С планшетов, содержащих образцы соединений с добавленными к ним промытыми клетками крови человека, и последующего центрифугирования при 1000 об/мин в течение 10 мин. Абсорбцию измеряли с использованием монохромного микропланшетного ридера Tecan M1000 Pro.
Максимальный процент гемолиза представлен как DMax. Низкое значение DMax при НС10>32 мкг/мл (максимально испытанная концентрация) указывает на образцы без гемолитической активности. Образцы, обладающие гемолитической активностью, были охарактеризованы при НС10≤32 мкг/мл. Кроме того, образцы были помечены как частично гемолитические, если DMax≥50%, даже при НС10>максимальной тестируемой концентрации.
«Колистин» и «Ванкомицин» были использованы в качестве положительных стандартов бактериального ингибирования для грамотрицательных и грамположительных бактерий, соответственно. «Флуконазол» использовали в качестве стандартного ингибитора гриба для С. albicans и С. neoformans. «Тамоксифен» использовали в качестве положительного стандарта цитотоксичности. «Мелиттин» использовали в качестве положительного гемолитического стандарта.
Методика тестирования противомикробной, фунгицидной, цитотоксической и гемолитической активности in vitro соединений приведена также на сайте http://www.co-add.org.
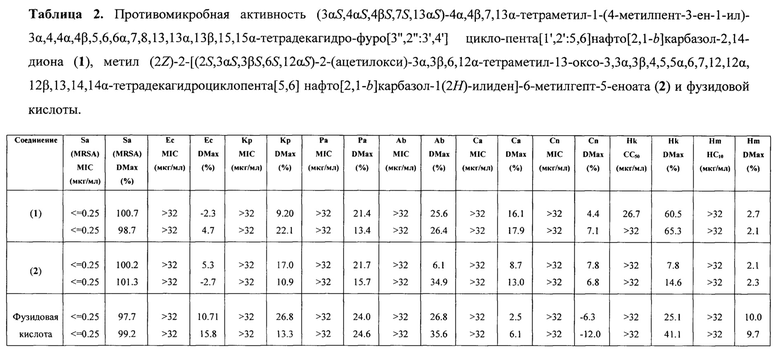
Образцы (3αS,4αS,4βS,7S,13αS)-4α,4β,7,13α-тетраметил-1-(4-метилпент-3-ен-1-ил)-3α,4,4α,4β,5,6,6α,7,8,13,13α,13β,15,15α-тетрадекагидрофуро[3'',2'':3',4']цикло-пента[1',2':5,6]нафто[2,1-b]карбазол-2,14-диона (1) и метил (2Z)-2-[(2S,3αS,3βS,6S,12αS)-2-(ацетилокси)-3α,3β,6,12α-тетраметил-13-оксо-3,3α,3β,4,5,5α,6,7,12,12α,12β,13,14,14α-тетрадекагидроциклопента[5,6]нафто[2,1-b]карбазол-1(2H)-илиден]-6-метилгепт-5-еноата (2) в концентрации <=0.25 мкг/мл показали противомикробную активность, ингибируя рост и размножение >100% грамположительных бактерий Staphylococcus aureus. Гемолитическая активность производных (1) и (2) не превышает 2.7% даже при максимально тестируемой концентрации >32 мкг/мл, что ниже таковой у фузидовой кислоты в 3.5 раза. Цитотоксичность соединения (1) сравнима с фузидовой кислотой, а соединение (2) в 2.8 раз менее токсично по сравнению с нативным антибиотиком (Таблица 2).
Таким образом, нафто[2,1-b]карбазолпроизводные фузидовой кислоты (1) и (2) проявляют антибактериальную активность в отношении патогенных микроорганизмов Staphylococcus aureus, сравнимую с активностью нативного антибиотика, при низкой токсичности и обладая минимальным гемолитическим действием при максимально тестируемой концентрации.
Изобретение относится к нафто[2,1-b]карбазолпроизводному фузидовой кислоты, выбранному из соединения формулы (1) или соединения формулы (2), способу его получения и его применению в медицине:
 .
.
Предложенный способ заключается во взаимодействии соответствующего 3,11-диоксопроизводного фузидовой кислоты с 3 экв. фенилгидразина в среде сухого этанола при кипячении в течение 2 часов в условиях реакции Фишера. Предложено новое нафто[2,1-b]карбазолпроизводное фузидовой кислоты, эффективное в качестве средства с антибактериальной активностью для борьбы с заболеваниями человека и животных, вызванными грамположительными бактериями Staphylococcus aureus, а также новый эффективный способ его получения. 3 н.п. ф-лы, 2 табл., 2 пр.
1. Нафто[2,1-b]карбазолпроизводное фузидовой кислоты формулы (1) или (2)
 .
.
2. Способ получения нафто[2,1-b]карбазолпроизводного фузидовой кислоты по п. 1, выбранного из соединения (1) или соединения (2), заключающийся во взаимодействии соответствующего 3,11-диоксопроизводного фузидовой кислоты с 3 экв. фенилгидразина в среде сухого этанола при кипячении в течение 2 часов в условиях реакции Фишера.
3. Применение нафто[2,1-b]карбазолпроизводного фузидовой кислоты по п. 1, выбранного из соединения (1) или соединения (2), в качестве средства с антибактериальной активностью для борьбы с заболеваниями человека и животных, вызванными грамположительными бактериями Staphylococcus aureus.
| W | |||
| Gu, et al, Bioorganic Med | |||
| Chem | |||
| Lett., 2014, 24, стр | |||
| Способ переработки сплавов меди и цинка (латуни) | 1922 |
|
SU328A1 |
| W | |||
| Gu et al, Bioorganic Med | |||
| Chem | |||
| Lett., 2015, 25, стр | |||
| Ветряный двигатель | 1922 |
|
SU554A1 |
| W | |||
| Gu et al, Eur | |||
| J | |||
| Med | |||
| Chem | |||
| Приспособление для суммирования отрезков прямых линий | 1923 |
|
SU2010A1 |
| ЦИРКУЛЬ ДЛЯ ЧЕРЧЕНИЯ СПИРАЛЕЙ | 1926 |
|
SU4692A1 |
| НОВЫЕ ПРОИЗВОДНЫЕ ФУЗИДОВОЙ КИСЛОТЫ | 2004 |
|
RU2353622C2 |

