РОДСТВЕННЫЕ ЗАЯВКИ НА ПОЛУЧЕНИЕ ПАТЕНТА
Настоящая заявка испрашивает приоритет согласно предварительным заявкам на патент США №61/494840 и 61/494710, поданным 8 июня 2011 г., которые включены в настоящий документ в полном объеме посредством ссылки.
ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Изобретение относится к применению катионных липидов и соединений жирорастворимых витаминов на нацеливания и усиления активности терапевтических молекул, включая siPHK.
УРОВЕНЬ ТЕХНИКИ
Фиброз печени может быть вызван звездчатыми клетками печени (HSC), приводящими к тому, что множество типов молекул коллагена и фибронектина осаждается на интерстициальной ткани. Это приводит к циррозу печени, печеночной недостаточности и/или гепатоцеллюлярной карциноме. Кроме того, хронический панкреатит развивается в результате панкреатического фиброза по тому же механизму, что и фиброз печени (Madro, et al., 2004; Med Sci Monit. 10: RA166-70; Jaster, 2004, Mol Cancer. 6:26). Кроме того, звездчатые клетки участвуют в нарушениях в голосовых связках и гортани, таких как рубцевание голосовых связок, фиброз слизистой оболочки голосовых связок и ларингеальный фиброз. Для профилактики или лечения фиброза в этих органах и в другом месте в организме необходимо разработать носитель для лекарственного средства и набор носителя для лекарственного средства.
[Звездчатые клетки являются одним из важных целевых кандидатов для лечения фиброза (Fallowfield et al., 2004, Expert Opin Ther Targets. 8:423-35; Pinzani, et al., 2004, Dig Liver Dis. 36: 231-42). В ходе фиброза звездчатые клетки активируются цитокинами из близлежащих клеток. Звездчатые клетки известны как клетки, несущие витамин A и принадлежащие к семейству миофибробластов, и они продуцируют множество факторов, которые вызывают фиброз печени.
С помощью терапевтических способов профилактики или лечения фиброза пытаются контролировать метаболизм коллагена, содействовать системе разрушения коллагена и ингибировать активацию звездчатых клеток. Однако во всех этих случаях низкая специфичность действия и/или низкая специфичность к органу вызывают проблемы, связанные с ограниченной эффективностью и нежелательными побочными эффектами.
Ингибирование синтеза белка коллагена не было установлено как терапевтический способ. Потенциал молекул, направленный на продуцирование коллагена, ограничен возможностью возникновения побочных эффектов. Непосредственное ингибирование продуцирования коллагена обеспечивает другой терапевтический способ профилактики или лечения фиброза. В таком способе требуется контролирование одного или более различных типов коллагена типов I-IV. Этот способ может осуществляться через белок теплового шока (HSP47), специфичный к коллагену молекулярный шаперон, который необходим для внутриклеточного транспорта и молекулярного созревания, необходимого для всех типов коллагена. Поэтому если функция HSP47 может специфически контролироваться в звездчатых клетках, существует возможность ингибирования фиброза печени.
Ряд методик доступен для доставки терапевтического агента, такого как siPHK в клетке, включая применение вирусных трансфекционных систем и невирусных трансфекционных систем. Невирусные трансфекционные системы могут включать, например, полимеры, липиды, липосомы, мицеллы, дендримеры и наноматериалы. Примеры полимеров, которые ранее были изучены для клеточной трансфекции включают катионные полимеры, такие как поли(L-лизин) (PLL), полиэтиленимин (PEI), хитозан и поли(2-диметиламино)этилметакрилат (pDMAEMA).
Система каждого типа имеет свои соответствующие преимущества и недостатки. Например, вирусные системы могут обеспечивать высокую эффективность трансфекции, но не могут быть такими безопасными, как некоторые невирусные системы. Кроме того, вирусные системы могут быть сложными и/или дорогостоящими для получения. Невирусные трансфекционные системы, такие как катионные полимеры, были предложены для трансфекции плазмидной ДНК в клетки. Однако некоторые недостатки катионные полимеров включают их токсичность по отношению к клетке и/или недостаток стабильности.
Таким образом, существует потребность в новых соединениях, композициях и способах применения катионных компонентов для улучшения доставки терапевтических лекарственных средств, включая нуклеиновые кислоты, в клетки, ткани и организмы.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Одним объектом настоящего изобретения являются соединения формулы I
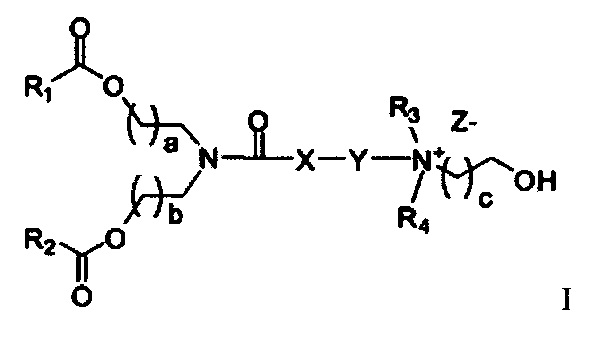
где R1 и R2 независимо выбирается из группы, состоящей из C10-C18 алкильной, C12-C18 алкенильной и олеильной группы; где R3 и R4 независимо выбираются из группы, состоящей из C1-C6алкила и C2-C6алканола; где X выбирается из группы, состоящей из -CH2-, -S-, и -O-, или отсутствует; где Y выбирается из -(CH2)n, -S(CH2)n, -O(CH2)N-, тиофена, -SO2(CH2)n-, и сложного эфира, где n=1-4; где a=1-4; где b=1-4; где c=1-4; и где Z представляет собой противоион.
Одним объектом настоящего изобретения является соединение для облегчения доставки лекарственного средства в целевую клетку, состоящее из структуры (целевая молекула)т-линкер-(целевая молекула)n, где целевой молекулой является ретиноид, имеющий специфический рецептор или сайт активации/связывания на целевой клетке; где тип независимо равны 0, 1, 2 или 3; и где линкер содержит полиэтиленгликоль (ПЭГ) или ПЭГ-подобная молекула.
В одном варианте выполнения настоящего изобретения ретиноид выбирается из группы, состоящей из витамина A, ретиноевой кислоты, третиноина, адапалена, 4-гидрокси(фенил)ретинамида (4-HPR), ретинил пальмитата, ретиналя, насыщенной ретиноевой кислоты и насыщенной деметилированной ретиноевой кислоты.
В другом варианте выполнения настоящего изобретения линкер выбирается из группы, состоящей из бис-амидо-ПЭГ, трис-амидо-ПЭГ, тетра-амидо-ПЭГ, Lys-бис-амидо-ПЭГ Lys, Lys-трис-амидо-ПЭГ-Lys, Lys-тетр-амидо-ПЭГ-Lys, Lys-ПЭГ-Lys, ПЭГ2000, ПЭГ1250, ПЭГ1000, ПЭГ750, ПЭГ550, ПЭГ-Glu, Glu, C6, Gly3, и GluNH.
В другом варианте выполнения настоящего изобретения, соединение выбирается из группы, состоящей из ретиноид-ПЭГ-ретиноид, (ретиноид)2-ПЭГ-(ретиноид)2, VA-ПЭГ2000-VA, (ретиноид)2-бис-амидо-ПЭГ-(ретиноид)2, (ретиноид)2-Lys-бис-амидо-ПЭГ-Lys-(ретиноид)2.
В другом варианте выполнения настоящего изобретения ретиноид выбирается из группы, состоящей из витамина A, ретиноевой кислоты, третиноина, адапалена, 4-гидрокси(фенил)ретинамида (4-HPR), ретинил пальмитата, ретиналя, насыщенной ретиноевой кислоты и насыщенной деметилированной ретиноевой кислоты.
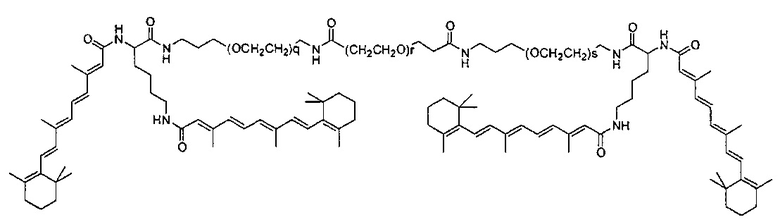
В другом варианте выполнения настоящего изобретения соединение представляет собой композицию формулы
где q, r и s каждый независимо равен 1, 2, 3, 4, 5, 6, 7, 8, 9 или 10.
В другом варианте выполнения настоящего изобретения формула соединения содержит
Другим объектом настоящего изобретения является носитель для лекарственного средства специфичный для звездчатых клеток, содержащий специфичное для звездчатых клеток количество молекулы ретиноида, состоящей из структуры (ретиноид)m-линкер-(ретиноид)n; где m и n независимо равны 0, 1, 2 или 3; и где линкер содержит полиэтиленгликоль (ПЭГ) или ПЭГ-подобную молекулу.
В другом варианте выполнения настоящего изобретения настоящее изобретение обеспечивает композицию, содержащую липосомальную композицию. В других вариантах выполнения настоящего изобретения липосомальная композиция содержит липосому, содержащую бислой липидных молекул.
В определенных вариантах выполнения настоящего изобретения молекула ретиноида по меньшей мере частично выставлена на внешней поверхности носителя для лекарственного средства прежде чем носитель для лекарственного средства достигает звездчатой клетки.
В другом варианте выполнения настоящего изобретения ретиноид составляет от 0.1 мол.% до 20 мол.% липидных молекул.
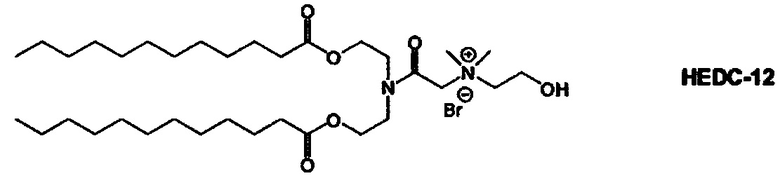
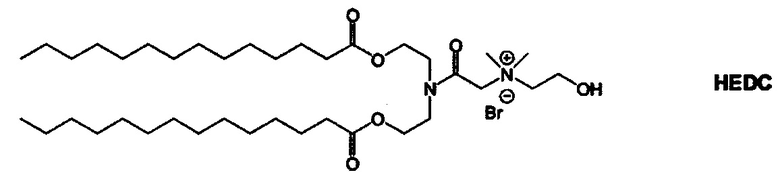
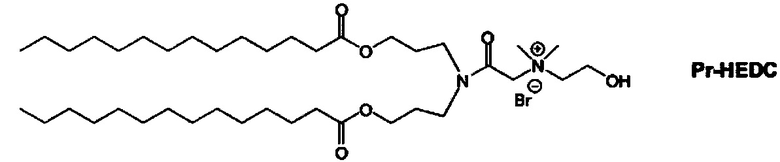
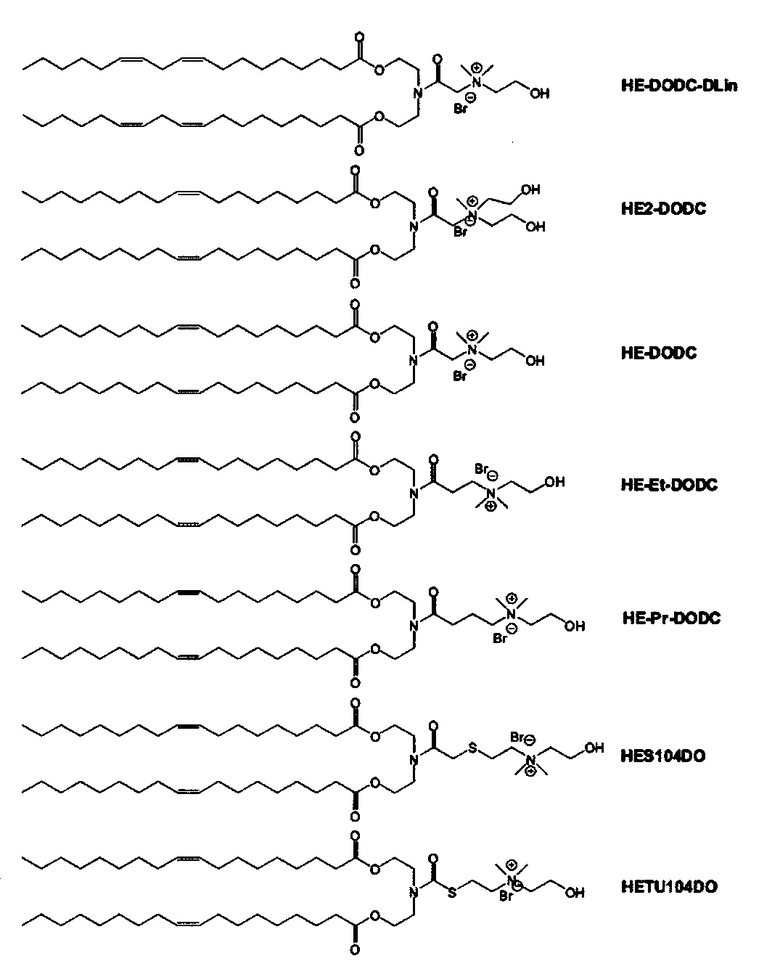
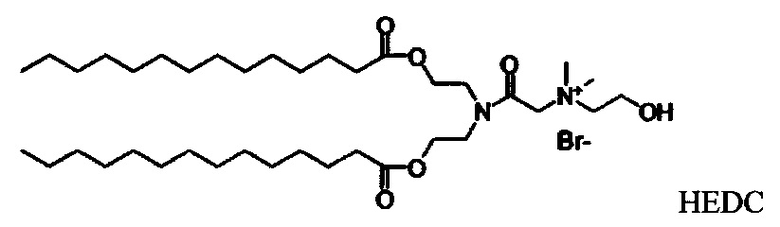
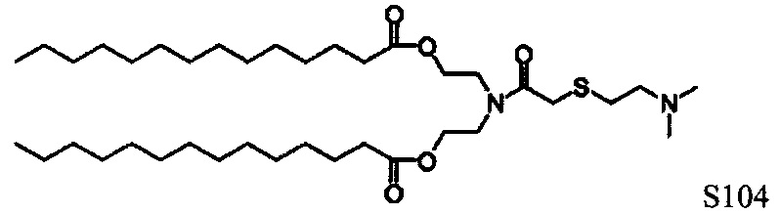
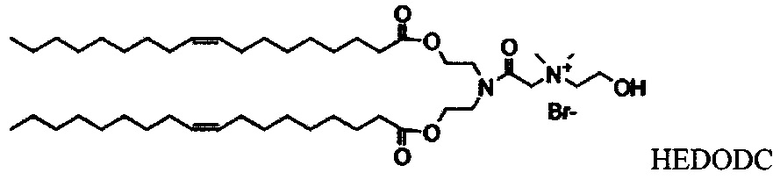
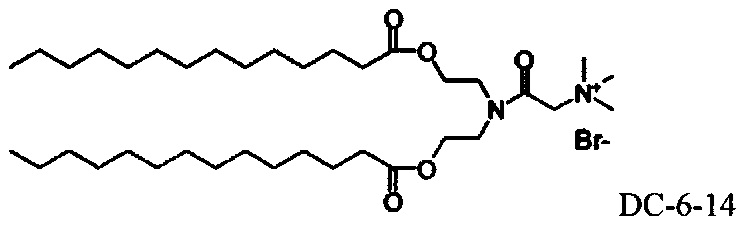
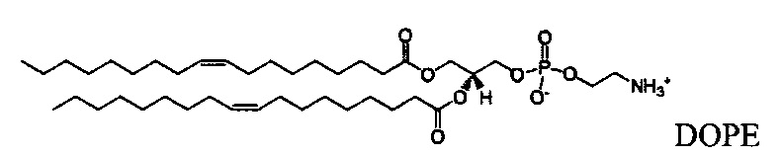
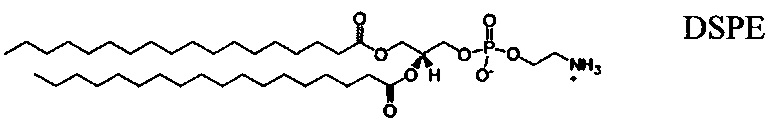
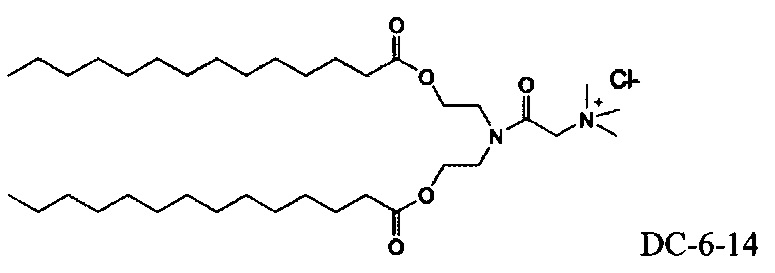
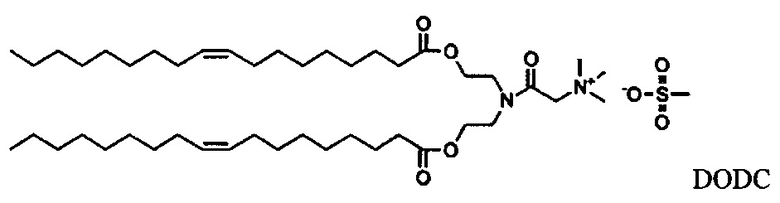
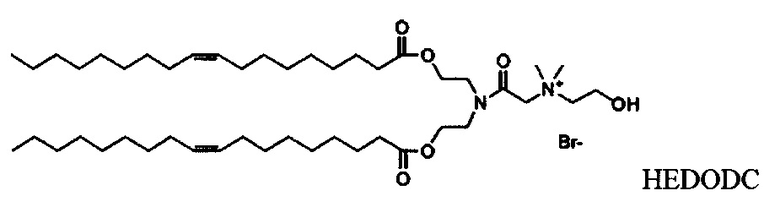
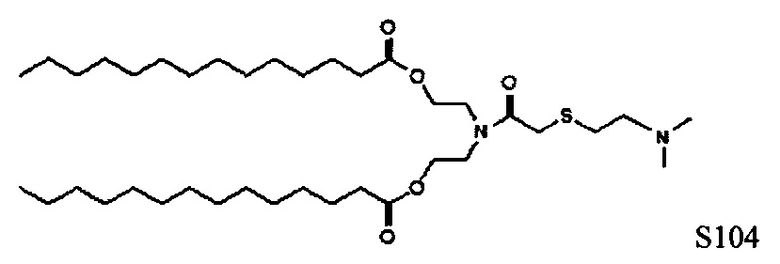
Настоящее изобретение также обеспечивает варианты выполнения настоящего изобретения, где липидные молекулы содержат один или более липидов, выбранных из группы, состоящей из HEDC, DODC, HEDODC, DSPE, DOPE и DC-6-14. В другом варианте выполнения настоящего изобретения липидные молекулы дополнительно содержат S104.
В определенных вариантах выполнения настоящего изобретения носитель для лекарственного средства содержит нуклеиновую кислоту.
В других вариантах выполнения настоящего изобретения нуклеиновой кислотой является siPHK, которая способна подавлять экспрессию hsp47 мРНК в звездчатой клетке.
Другим объектом настоящего изобретения является соединение для облегчения доставки лекарственного средства в целевую клетку, состоящее из структуры (липид)m-линкер-(целевая молекула)n, где целевой молекулой является ретиноид или жирорастворимый витамин, имеющий специфический рецептор или сайт активации/связывания на целевой клетке; где тип независимо равны 0, 1, 2 или 3; и где линкер содержит молекулу полиэтиленгликоля (ПЭГ).
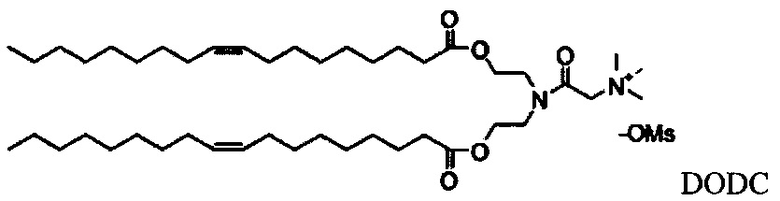
В одном варианте выполнения настоящего изобретения липид выбирается из одного или более члена группы, состоящей из DODC, HEDODC, DSPE, DOPE и DC-6-14.
В другом варианте выполнения настоящего изобретения ретиноид выбирается из группы, состоящей из витамина A, ретиноевой кислоты, третиноина, адапалена, 4-гидрокси(фенил)ретинамида (4-HPR), ретинил пальмитата, ретиналя, насьпценной ретиноевой кислоты и насьпценной деметилированной ретиноевой кислоты.
В другом варианте выполнения настоящего изобретения жирорастворимым витамином является витамин D, витамин E или витамин K.
В другом варианте выполнения настоящего изобретения линкер выбирается из группы, состоящей из бис-амидо-ПЭГ, трис-амидо-ПЭГ, тетра-амидо-ПЭГ, Lys-бис-амидо-ПЭГ Lys, Lys-трис-амидо-ПЭГ-Lys, Lys-тетро-амидо-ПЭГ-Lys, Lys-ПЭГ-Lys, ПЭГ2000, ПЭГ1250, ПЭГ1000, ПЭГ750, ПЭГ550, ПЭГ-Glu, Glu, С6, Gly3 и GluNH.
В другом варианте выполнения настоящего изобретения настоящее изобретение выбирается из группы, состоящей из DSPE-II3r-VA, DSPE-iT3r2000-Glu-VA, DSPE-ror550-VA, DOPE-VA, DOPE-Glu-VA, DOPE-Glu-NH-VA, DOPE-Gly3-VA, DC-VA, DC-6-VA и AR-6-VA.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
На Фиг. 1 показано подавление эффективности некоторых вариантов выполнения настоящего изобретения. Это включает HEDC липосомы по сравнению с DC-6-14 липоплекс контролями.
На Фиг. 2 показано in vitro сравнение подавления гена с применением катионных липидов.
На Фиг. 3 показана оценка генной экспрессии in vivo примерными HEDODC липосомальными композициями согласно настоящему изобретению (* показывает p<0.05).
На Фиг. 4 показана оценка генной экспрессии in vivo примерными HEDC липосомальными композициями согласно Примеру 15. Усы показывают среднеквадратическое отклонение (n=3). Сигмоидальная кривая доза-ответ показана на основе наилучшего соответствия. Значение EC50 было вычислено на основе кривой. Оно соответствует 11.8 нМ.
На Фиг. 5 показаны результаты измерений in vivo с применением крысиной модели DMNQ. После вычитания фоновых уровней gp46 мРНК, вычисленных на основе нативной группы, все значения тестовой группы были нормолизованы по отношению к gp46 мРНК группы со средой (выражается как процент от группы со средой). Средний уровень gp46 мРНК после обработки показал доза-зависимый ответ и кривую, соответствующую сигмоидальной кривой доза-ответ. Вычисленное значение ED50 равно 0.79 мг/кг.
На Фиг. 6 показаны результаты измерений in vivo с применением крысиной модели DMNC. После вычитания фоновых уровней gp46 мРНК, вычисленных на основе нативной группы, все значения тестовой группы были нормализованы по отношению к gp46 мРНК группы со средой (выражается как процент от группы со средой). Уровни мРНК MRPL19 были вычислены на основе анализа количественной ПЦР с обратной транскрипцией (TaqMan®). Уровни мРНК gp46 были нормализованы относительно уровней MRPL19. (*** показывает P<0.02.)
На Фиг. 7 показаны результаты измерений in vivo с применением крысиной модели легочного блеомицина. Столбиковая диаграмма суммирует размеры фиброза (T. Ashcroft) окрашенных азаном частей легкого для каждой группы. Статистический анализ провели с применением One way ANOVA, Bonferroni мульти-сравнительного теста с применением программного обеспечения Prism5.
Фиг. 8 Добавление VA-конъюгата через декорирование усиливает siPHK активность
Фиг. 9 Добавление VA-конъюгата через сосолюбилизацию усиливает siPHK активность
Фиг. 10 Добавление VA-конъюгата через сосолюбилизацию усиливает siPHK активность
Фиг. 11 Добавление VA-конъюгата к липоплексу через сосолюбилизацию усиливает siPHK активность
Фиг. 12 Добавление VA-конъюгата к липоплексу через сосолюбилизацию по сравнению с декорированием.
Фиг. 13 in vivo эффективность на мышах, CCl4 модель
Фиг. 14 in vivo эффективность декорированных по сравнению с солюбилизированными конъюгатами ретиноида
Фиг. 15 in vitro эффективность (pHSC), эффект конъюгатов ретиноида в липосомальных композициях.
На Фиг. 16 показана корреляция содержания конъюгата ретиноида (мол.%) с эффективностью in vivo (крыса DMNQ). Самцам крыс Sprague-Dawley внутривенно вводили либо композиции, содержащие 0, 0.25, 0.5, 1 и 2% DiVA в дозе 0.75 мг/кг siPHK или PBS (среда), один час после последней инъекции DMN.
ПОДРОБНОЕ ОПИСАНИЕ ИЛЛЮСТРАТИВНЫХ ВАРИАНТОВ ВЫПОЛНЕНИЯ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
Согласно настоящему изобретению раскрываются соединения формулы I
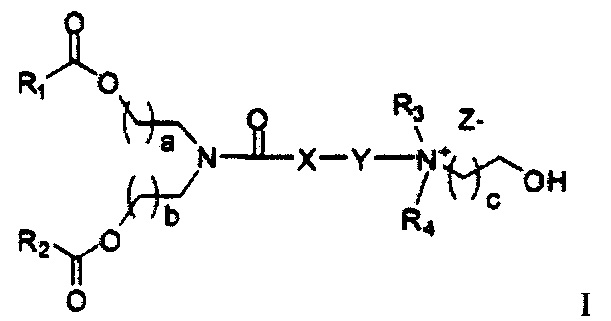
где R1 и R2 независимо выбирается из группы, состоящей из C10-C18 алкильной, C12-C18 алкенильной и олеильной группы; где R3 и R4 независимо выбираются из группы, состоящей из C1-C6элкила и C2-C6алканола; где X выбирается из группы, состоящей из -CH2-, -S- и -O- или отсутствует; где Y выбирается из -(CH2)n, -S(CH2)n, -O(CH2)n-, тиофена, -SO2(CH2)n- и сложного эфира, где n=1-4; где a=1-4; где b=1-4; где c=1-4; и где Z является противоионом.
Соединения согласно настоящему изобретения также упоминаются в настоящей заявке как соединения входящие в класс соединений, известный как "катионные липиды." Катионными липидами являются соединения, которые включают по меньшей мере одну липидную составляющую и положительно заряженный атом азота, связанный с противоионом. "Липиды", как понимается в данной области техники состоят из гидрофобной алкильной или алкенильной составляющей и составляющей карбоновой кислоты или сложного эфира.
Поэтому было обнаружено, что составляющая амино-алкил-гидроксил (-N-алкил-OH) соединений формулы I придает свойства композициям согласно настоящему изобретению, ранее не обнаруженные с другими катионными липидами, ранее описанными. Композиции согласно настоящему изобретению, которые включают соединения формулы I, приводят к превосходному уменьшению экспрессии белка, по сравнению с композициями, которые не включают соединения формулы I. Особенно удивительной является способность композиций согласно настоящему изобретению, которые включают соединения формулы I, уменьшать экспрессию HSP47.
Предпочтительные соединения согласно настоящему изобретению включают те, в которых R1 и R2 каждый независимо представляет собой C10-C30алкил. В более предпочтительных вариантах выполнения настоящего изобретения R1 и R2 каждый независимо представляет собой C10-C20алкил. В еще более предпочтительных вариантах выполнения настоящего изобретения R1 и R2 каждый независимо представляет собой C12-C18алкил. Особенно предпочтительные варианты выполнения настоящего изобретения включают те соединения, в которых R1 и R2 каждый независимо представляет собой C13-C17алкил. Более предпочтительными являются те соединения, в которых R1 и R2 каждый представляет собой С13 алкил.
В других вариантах выполнения настоящего изобретения R1 и R2 каждый независимо представляет собой C10-C30алкенил. В более предпочтительных вариантах выполнения настоящего изобретения R1 и R2 каждый независимо представляет собой C10-C20алкенил. В еще более предпочтительных вариантах выполнения настоящего изобретения R1 и R2 каждый независимо представляет собой C12-C18алкенил. В еще более предпочтительных вариантах выполнения настоящего изобретения R1 и R2 каждый независимо представляет собой C13-C17алкенил. Наиболее предпочтительные соединения согласно настоящему изобретению включают те соединения, в которых R1 и R2 каждый представляет собой C12 алкенил.
Также для соединений формулы I, R3 и R4 независимо выбираются из группы, состоящей из C1-C6алкила. В предпочтительных вариантах выполнения настоящего изобретения R3 и R4 каждый независимо представляет собой C1-C3алкил. Наиболее предпочтительно R3 и R4 каждый представляет собой метил. В других вариантах выполнения настоящего изобретения по меньшей мере один из R3 и R4 представляют собой -CH2CH2OH.
Наиболее предпочтительными являются соединения формулы I, где a=b=c=1.
Z может представлять собой любой противоион азота, так что это термин понятен для специалиста в данной области техники. Предпочтительные противоионы азота включают галогены, причем хлорид и бромид являются особенно предпочтительными, и мезилат (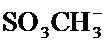
Примерные соединения формулы I включают:
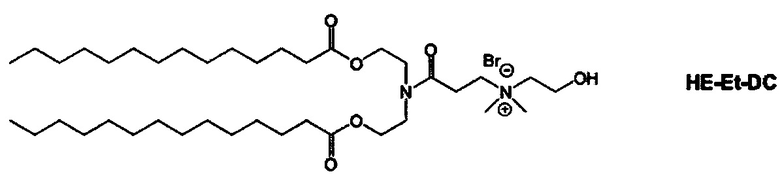
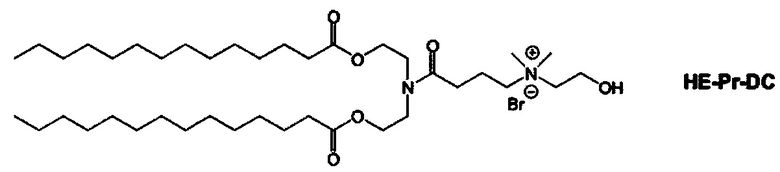
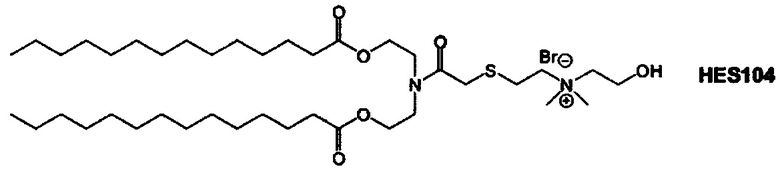
В объем настоящего изобретения входит соединение для облегчения доставки лекарственного средства в целевую клетку, содержащее структуру (нацеливающая молекула)т-линкер-(нацеливающая молекула), где нацеливающая молекула являющаяся ретиноидом или жирорастворимым витамином, имеющим специфический рецептор или [сайт активации/связывания] на целевой клетке; и где тип независимо равны 0, 1, 2 или 3; и где линкер содержит полиэтилегликоль (ПЭГ) или ПЭГ-подобную молекулу и обозначается как "Формула A".
Настоящее изобретение также обеспечивает соединения для облегчения доставки лекарственного средства в целевую клетку, состоящие из структуры (липид)т-линкер-(нацеливающая молекула)n, где нацеливающей молекулой является ретиноид или жирорастворимый витамин, имеющий специфический рецептор на целевой клетке; где тип независимо равны 0, 1, 2 или 3; и где линкер содержит полиэтиленгликоль (ПЭГ) ПЭГ-подобную молекулу и обозначается как "Формула В".
Поэтому было обнаружено, что соединения формулы A или формулы B придают свойства композициям согласно настоящему изобретению, ранее не обнаруженные. Композиции согласно настоящему изобретению, которые включают соединение формулы A или формулы B, приводят к превосходному уменьшению генной экспрессии, по сравнению с композициями, которые не включат эти соединения. Особенно удивительным является способность композиций согласно настоящему изобретению, которые включают соединения Формулы A, уменьшать экспрессию HSP47.
В определенных предпочтительных вариантах выполнения настоящего изобретения ретиноид выбирается из группы, состоящей из витамина A, ретиноевой кислоты, третиноина, адапалена, 4-гидрокси(фенил)ретинамида (4-HPR), ретинил пальмитата, ретиналя, насыщенной ретиноевой кислоты и насыщенной деметилированной ретиноевой кислоты.
Предпочтительные варианты выполнения настоящего изобретения включают соединения, где линкер выбирается из группы, состоящей из бис-амидо-ПЭГ, трис-амидо-ПЭГ, тетра-амидо-ПЭГ, Lys-бис-амидо-ПЭГ Lys, Lys-трис-амидо-ПЭГ-Lys, Lys-TeTpa-aMHflO-ror-Lys, Lys-ror-Lys, ПЭГ2000, ПЭГ1250, ПЭГ1000, ПЭГ750, ПЭГ550, ПЭГ-Glu, Glu, С6, Gly3 и GluNH. В других вариантах выполнения настоящего изобретения ПЭГ представляет собой монодисперсию.
Другой вариант выполнения настоящего изобретения обеспечивает соединение, где формула A выбирается из группы, состоящей из ретиноид-ПЭГ-ретиноид, (ретиноид)2-ПЭГ-(ретиноид)2, VA-ПЭГ2000-УА, (ретиноид)2-бис-амидо-ПЭГ-(ретиноид)2, (ретиноид)2-Lys-бис-амидо-ПЭГ-Lys-(ретиноид)2.
Другим предпочтительным вариантов выполнения настоящего изобретения является соединение формулы
где q, r и s каждый независимо равны 1, 2, 3, 4, 5, 6, 7, 8, 9 или 10.
В других предпочтительных вариантах выполнения настоящего изобретения формула соединения содержит
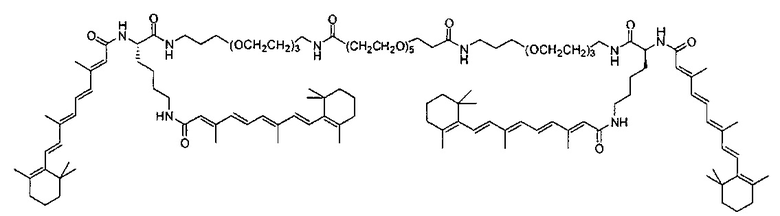
Другие варианты выполнения настоящего изобретения включают структуры, показанные в Таблице 1

Композиции согласно настоящему изобретению, которые включают по меньшей мере одно соединение формул. В предпочтительных вариантах выполнения настоящего изобретения конъюгат ретиноида будет присутствовать в концентрации от около 0.3 до около 30 мас.%, на основе общей массы композиции или состава, что эквивалентно от около 0.1 до около 10 мол.%, что эквивалентно мольному отношению от около 0.1 до около 10. Предпочтительно конъюгатом ретиноида является молекула ретиноид-линкер-липид или молекула ретиноид-линкер-ретиноид.
Примеры конъюгатов ретиноида включают соединения формулы II:
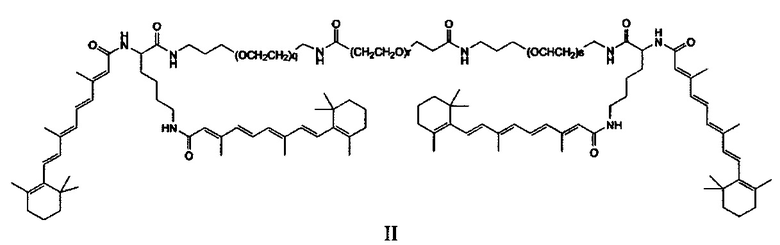
где q, r и s каждый независимо равны 1, 2, 3, 4, 5, 6, 7, 8, 9 или 10 и их энантиомеры и диастереомеры.
Предпочтительные соединения формулы II включают те, где q, r и s каждый независимо равны 1, 2, 3, 4, 5, 6 или 7. Более предпочтительными являются те соединения формулы II, где q, r и s каждый независимо равны 3, 4 или 5. Более предпочтительными являются те соединения формулы II, где q равно 3, r равно 5, и s равно 3. Одним примером соединения формулы II является
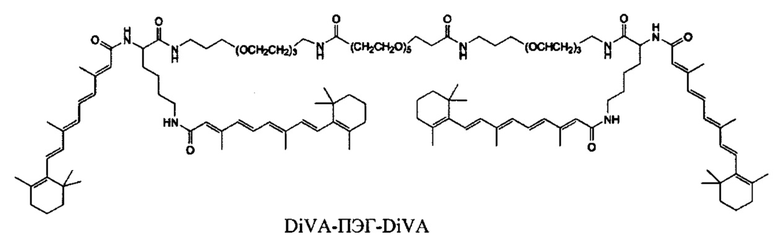
DiVA-ПЭГ-DiVA включают стереоцентры, и все энантиомеры и диастереомеры входят в объем настоящего изобретения.
Концентрация катионных липидов в композициях согласно настоящему изобретению, включающих катионные липиды формулы I, может составлять от 1 до около 80 мас.% от общей массы липидной композиции. Более предпочтительно концентрация составляет от 1 до около 75 мас.%. Даже более предпочтительно концентрация составляет от около 30 до около 75 мас.%. Концентрация от около 30 до около 75 мас.% соответствует до около 30-60 мол.% и мольному отношению около 30-60. Наиболее предпочтительными являются те композиции, которые имеют концентрацию катионных липидов около 50 мас.%. В композициях, которые содержат смесь ионизируемого катионного липида и катионного липида с четвертичным амином формулы I, предпочтительно мол.% составляет от 5% до 45 мол.%, и даже более предпочтительно смесь около 20 мол.% ионизируемого катионного липида и 20 мол.% катионного липида с четвертичным амином формулы I.
Такие композиции могут также включать водную среду. Катионные липиды, включая липиды формулы I, могут быть инкапсулированы внутри липосомы в таких вариантах выполнения настоящего изобретения и могут быть не доступны водной среде. Кроме того, катионные липиды, включая липиды формулы I, могут быть расположены на внешней поверхности липосомы и могут быть доступны водной среде.
Композиции согласно настоящему изобретению, которые включают по меньшей мере одно соединение формулы I и липосому и необязательно конъюгат ретиноида, как например соединение формулы II, может также включать siPHK. Также в объем настоящего изобретения входят композиции, содержащие по меньшей мере одно соединение Формулы A или B и siPHK.
Предполагается, что любая молекула siPHK может применяться в объеме настоящего изобретения. siPHK может включать антисмысловую последовательность к мРНК кодирующей последовательности для человеческого hsp47, примером чего является SEQ ID NO:1, которая показана далее.

Например,
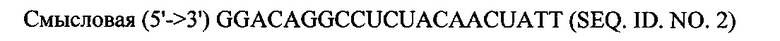
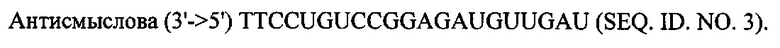
Такие композиции могут также включать водную среду. Предпочтительно такие композиции состоят по существу из одного соединения формулы I в заряженном комплексе с siPHK. Такие композиции, включающие соединение формулы I и siPHK, могут дополнительно содержать жидкую среду. В одном варианте выполнения настоящего изобретения жидкая среда подходит для инъекции живому организму. Жидкие среды в объеме любого их вариантов выполнения настоящего изобретения могут быть водными, то есть полностью состоять из водного растворителя и включать соли, буферы и/или другие фармацевтические эксципиенты. В другом варианте выполнения настоящего изобретения жидкая среда может состоять из водного растворителя в комбинации с другим жидким растворителем, таким как, например, органический растворитель. Жидкие растворители в объеме любого из описанных вариантов выполнения настоящего изобретения могут также включать по меньшей мере один органический растворитель. Органические растворители по существу известны в данной области техники и включают C1-C4 спирты, диметилсульфоксид ("DMSO") и тому подобное. Те липидные среды, которые включают смесь воды и по меньшей мере одного органического растворителя, также входят в объем любого из описанных вариантов выполнения настоящего изобретена.
Также в объем настоящего изобретения входят композиции, которые содержат по меньшей мере одно соединений Формулы I и липосому. Такие варианты выполнения настоящего изобретения могут включать смеси соединений формулы I и липосому. Другие варианты выполнения настоящего изобретения могут включать липосому и одно или более соединений формулы I в дополнение к катионным липидам, которые не охватываются формулой I.
В некоторых вариантах выполнения настоящего изобретения siPHK будет инкапсулироваться липосомой, так что siPHK не доступна внешней среде. В других вариантах выполнения настоящего изобретения siPHK не будет инкапсулироваться липосомой. В таких вариантах выполнения настоящего изобретения siPHK может образовываться на внешней поверхности липосомы. В этих вариантах выполнения настоящего изобретения siPHK доступна водной среде.
Другие варианты выполнения настоящего изобретения включают специфичный к звездчатой клетке носитель для лекарственно средства, содержащий липосомальную композицию. Липосомальная композиция может содержать липосому, содержащую бислой липидных молекул. В определенных вариантах выполнения настоящего изобретения может быть предпочтительным, что молекула ретиноида является по меньшей мере частично выставленной на внешней поверхности носителя для лекарственного средства, прежде чем носитель для лекарственного средства достигает звездчатой клетки.
Определенные варианты выполнения настоящего изобретения обеспечивают липидные молекулы, которые содержат один или более липидов, выбранных из группы, состоящей из HEDC, DODC, HEDODC, DSPE, DOPE и DC-6-14. В других вариантах выполнения настоящего изобретения липидные молекулы могут дополнительно содержать S104.
В некоторых вариантах выполнения настоящего изобретения siPHK будет инкапсулироваться липосомой, так что siPHK является не доступной водной среде. В других вариантах выполнения настоящего изобретения siPHK не будет инкапсулироваться липосомой. В таких вариантах выполнения настоящего изобретения siPHK может образовываться на внешней поверхности липосомы. В этих вариантах выполнения настоящего изобретения siPHK является доступной водной среде.
Другие варианты выполнения настоящего изобретения носитель для лекарственного средства, специфичный к звездчатой клетке, содержащий липосомальную композицию. Липосомальная композиция может включать липосому, содержащую бислой липидных молекул. В других вариантах выполнения настоящего изобретения молекула ретиноида по меньшей мере частично выставлена на внешней поверхности носителя для лекарственного средства до того, как носитель для лекарственного средства достигает звездчатой клетки.
В определенных предпочтительных вариантах выполнения настоящего изобретения ретиноид составляет от 0.1 мол.% до 20 мол.% липидных молекул.
Вышеописанные композиции согласно настоящему изобретению могут также включать ПЭГ-конъюгированные липиды, которые по существу известны в данной области техники, включая ПЭГ-фсфолипиды и ПЭГ-церамиды, включая одну или более молекул, выбранных из следующих: ПЭГ2000-DSPE, ПЭГ2000-DPPE, ПЭГ2000-DMPE, ПЭГ2000-ЭОРЕ, ПЭГ1000-DSPE, ПЭГ1000-DPPE, ПЭГ1000-DMPE, ПЭГ1000-DOPE, ПЭГ550-DSPE, ПЭГ550-ОРРЕ, ПЭГ-550ОМРЕ, ПЭГ-1000DOPE, ПЭГ-холестерин, ПЭГ2000-церамид, ПЭГ1000-церамид, ПЭГ750-церамид и ПЭГ550-церамид.
Вышеописанные композиции согласно настоящему изобретению могут один или более фосфолипидов, таких как, например, 1,2-дистеароил-sn-глицеро-3-фосфолипид ("DSPC"), дипальмитоилфосфатидилхолин ("DPPC"), 1,2-дипальмитоил-sn-глицеро-3-фосфоэтаноламин ("DPPE") и 1,2-диолеоил-sn-глицеро-3-фосфоэтаноламин ("DOPE"). Предпочтительным вспомогательным фосфолипидом является DOPE.
Например, липосомы в объеме настоящего изобретения, были получены с применением различных ПЭГ-липидов, включенных с применением способов солюбилизации, описанных в настоящем изобретении. Эти композиции содержали катионный липид : DOPE : холестерин : DIVA-ПЭГ-DIVA : ПЭГ-липид (50:10:38:5:2 мольное отношение), и каждую композицию протестировали в pHSC in vitro анализе, описанном в настоящей заявке, с применением HSP-47-C siPHK человека/крысы при концентрации 200 нМ. Результаты показаны в следующей таблице:
Вышеописанные композиции согласно настоящему изобретению могут включать один или более фосфолипидов, таких как, например, 1,2-дистеароил- sn -глицеро-3-фосфолипид ("DSPC"), дипальмитоилфосфатидилхолин ("DPPC"), 1,2-дипальмитоил-sn-глицеро-3-фосфоэтаноламин ("DPPE") и 1,2-диолеоил-sn-глицеро-3-фосфоэтаноламин ("DOPE"). Предпочтительным вспомогательным липидом является DOPE.
В дополнение к катионному липиду формулы I, другие липиды могут быть полезны. Они включают ионизируемые катионные липиды, включая S104, показанные ниже.
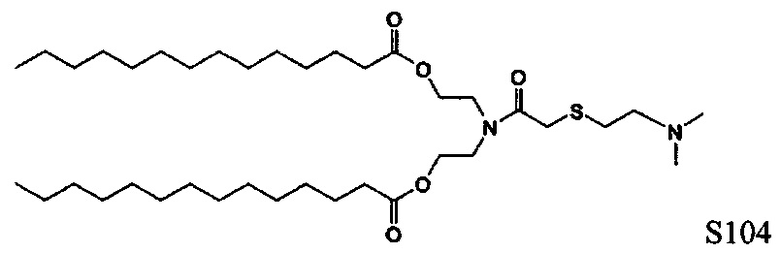
Композиции для доставки могут состоять из катионного липида Формулы I в комбинации с ионизируемым катионным липидом. Ионизируемый катионный липид может включать, например, S104. Ионизируемый катионный липид может присутствовать в концентрации от 0 до 45 мол.%, включая концентрацию, выбранную из 5, 10, 15, 20, 25, 30, 35,40 и 45 мол.%.
В объем настоящего изобретения также входят фармацевтические составы, которые включают любую из вышеупомянутых композиций в дополнение к фармацевтически приемлемому носителю или разбавителю. Фармацевтические составы согласно настоящему изобретению будут включать по меньшей мере один терапевтический агент. Предпочтительно терапевтическим агентом является siPHK. Предполагается, что любая молекула siPHK может применяться в объеме настоящего изобретения.
В объем настоящего изобретения также входят фармацевтические композиции, которые включают любое из вышеупомянутых соединений в дополнение к фармацевтически приемлемому носителю или разбавителю. Фармацевтические композиции согласно настоящему изобретению будут включать по меньшей мере один терапевтический агент. Предпочтительно терапевтическим агентом является siPHK. Предполагается, что любая молекула siPHK может применяться в объеме настоящего изобретения. Как описано ранее, siPHK включает последовательности, обозначенные как SEQ ID NO: 1, SEQ ID NO: 2, SEQ ID NO: 3 и SEQ ID NO: 4.
В предпочтительных композициях согласно настоящему изобретению, включающих siPHK, siPHK инкапсулирована липосомой. В других вариантах выполнения настоящего изобретения siPHK может находиться на внешней стороне липосомы. В этих вариантах выполнения настоящего изобретения siPHK может образовываться на внешней стороне липосомы.
Полезный интервал катионного липида: siPHK (фосфатное отношение азота липида к siPHK, "N:P") составляет от 0.2 до 5.0. Особенно предпочтительным интервалом N:P является 1.5-2.5 для композиций и составов согласно настоящему изобретению.
Предпочтительные композиции согласно настоящему изобретению включают композиции, содержащие HEDC : S104 : DOPE : Холестерин : ПЭГ-ОМРЕ : DIVA-ПЭГ-DiVA (20:20:30:25:5:2 мольное отношение) и HEDC : S104 : DOPE : Холестерин : ПЭГ-DMPE : DiVA-ПЭГ-DiVA (20:20:30:25:5:2 мольное отношение), где DiVA-ПЭГ-DiVA является солюбилизированным. DODC : DOPE : холестерин : ПЭГ-липид:DiVA-ПЭГ-DiVA (50:10:38:2:5 мольное отношение) и DODC : DOPE : холестерин : ПЭГ-липид : DiVA-ПЭГ-DiVA композиции, где DiVA-ПЭГ-DiVA является солюбилизированным.
Другие композиции согласно настоящему изобретению включают композиции, содержащие HEDODC : DOPE : олестерин-ПЭГ-липид : DiBA-ПЭГ-DiVA (50:10:38:2:5 мольное отношение) и HEDODC : DOPE : холестерин-ПЭГ-липид:DiVA-ПЭГ-DiVA композиции, где DiVA-ПЭГ-DiVA является солюбилизированным.
Другие предпочтительные композиции согласно настоящему изобретению включают композиции, содержащие DC-6-14 : DOPE : холестерин : DiVA-ПЭГ-DiVA (40:30:30:5, мольные отношения) и БС-6-14 : DOPE : холестерин : DiVA-TOT-DiVA, где DiVA-ПЭГ-DiVA является солюбилизированным.
Также настоящее изобретение охватывает способы доставки терапевтического агента в организм пациента. Эти способы содержит обеспечение фармацевтической композиции, включая любые вышеприведенные композиции и фармацевтически приемлемый носитель или разбавитель; и введение фармацевтической композиции в организм пациента.
Определения
Согласно настоящему изобретению термин «алкил» относится к полностью насыщенной (без двойных или тройных связей) углеводородной группе с линейной или разветвленной углеводородной цепью, например группе, имеющей общую формулу -CnH2n+1. Алкильная группа может содержать от 1 до 50 атомов углерода (во всех случаях, когда она появляется в настоящем описании, числовой диапазон, такой как «1-50» относится к каждому целому числу в данном диапазоне; например, термин «1-50 атомов углерода» означает, что алкильная группа может состоять из 1 атома углерода, 2 атомов углерода, 3 атомов углерода и т.д., до 50 атомов углерода включительно, несмотря на то что настоящее определение также предусматривает появление термина «алкил», где числовой диапазон не указан). Алкильная группа может также представлять собой алкил среднего размера, содержащий от 1 до 30 атомов углерода. Алкильная группа может также представлять собой низший алкил, содержащий от 1 до 5 атомов углерода. Алкильная группа соединений может быть обозначена «C1-C4алкил» или схожими обозначениями. Только в качестве примера, термин «C1-C4 алкил» означает, что алкильная цепь содержит от одного до четырех атомов углерода, т.е. алкильная цепь выбрана из группы, включающей метил, этил, пропил, изопропил, н-бутил, изобутил, втор-бутил и трет-бутил. Типичные алкильные группы включают, но никоим образом не ограничиваются ими, метил, этил, пропил, изопропил, бутил, изобутил, третичный бутил, пентил, гексил и тому подобное.
Согласно настоящему изобретению термин «алкенил» относится к алкильной группе, содержащей в линейной или разветвленной углеводородной цепи одну или более двойных связей. Алкенильная группа может не содержать заместители или содержать заместители. В случае, когда алкенильная группа содержит заместители, заместитель (заместители) может быть выбран из тех же групп, описанных выше относительно замещения алкильной группы, если не указано иное.
Согласно настоящему изобретению термин «алкинил» относится к алкильной группе, содержащей в линейной или разветвленной углеводородной цепи одну или более тройных связей. Алкинильная группа может не содержать заместители или содержать заместители. В случае, когда алкинильная группа содержит заместители, заместитель (заместители) может быть выбран из тех же групп, описанных выше относительно замещения алкильной группы, если не указано иное.
Согласно настоящему изобретению термин "галоген" относится к F, Cl, Br и I.
Согласно настоящему изобретению термин "мезилат" относится к -OSO2CH3.
Согласно настоящему изобретению Термин «фармацевтическая композиция» относится к смеси композиции согласно настоящему изобретению с одним или более другими химическими компонентами, такими как разбавители или дополнительные фармацевтические носители. Фармацевтическая композиция облегчает введение соединения в организм. В данной области техники существуют многочисленные способы введения фармацевтической композиции, включая, но без ограничения к этому, инъекцию и парентеральное введение.
Согласно настоящему изобретению термин «фармацевтический носитель» относится ко второму химическому соединению, отличающемуся от носителя и используемому в дополнение к нему, облегчающему введение соединения в клетки или ткани. Например, диметилсульфоксид (ДМСО) представляет собой широко используемый носитель, т.к. он облегчает поглощение многих органических соединений клетками или тканями организма.
Согласно настоящему изобретению термин «разбавитель» относится к химическим соединениям, разведенным в воде, которые будут растворять представляющую интерес композицию (например, композиция, которая может содержать соединение, ретиноид, второй липид, стабилизирующий агент и/или терапевтический агент), а также стабилизировать биологически активную форму соединения. Соли, растворенные в буферных растворах, используют в качестве разбавителей в данной области техники. Один из широко используемых буферных растворов представляет собой фосфатный буферный раствор, т.к. он имитирует солевые условия крови человека. Поскольку буферные соли могут контролировать pH раствора в низких концентрациях, буферный разбавитель редко модифицирует биологическую активность соединения. В настоящем описании термин «эксципиент» относится к инертному веществу, которое добавляют в композицию для придания, без ограничения, объема, консистенции, стабильности, связывающей способности, смазывания, способности к распаду и т.д. указанной композиции. «Разбавитель» представляет собой вид экциаиента.
Согласно настоящему изобретению термин «терапевтический агент» представляет собой соединение, которое после введения млекопитающему в терапевтически эффективном количестве обеспечивает полезный терапевтический эффект для указанного млекопитающего. В настоящем описании терапевтический агент может называться лекарственным средством. Для специалиста в данной области техники очевидно, что термин «терапевтический агент» не ограничен лекарственными средствами, которые были одобрены регулирующим органом. «Терапевтический агент» может быть функционально связан с соединением согласно настоящему изобретению, ретиноидом и/или вторым липидом. Например, второй липид согласно настоящему изобретению может формировать липосому, и терапевтический агент может быть функционально связан с липосомой, например, как описано в настоящей заявке.
Согласно настоящему изобретению "композиции липоплекса" относятся к тем композициям, в которых siPHK находится на внешней стороне липосомы. В предпочтительных композициях липоплекса siPHK образуется на внешней стороне липосомы. Другие предпочтительные композиции липоплекса включают те, в которых siPHK связываемый с любой средой, присутствующей на внешней стороне липосомы.
Согласно настоящему изобретению "липосомальные композиции" относятся к тем композициям, в которых siPHK инкапсулируется внутри липосомы. В предпочтительных липосомальных композициях siPHK является не связываемой с любой средой, присутствующей на внешней стороне липосомы.
Согласно настоящему изобретению термин "солюбилизированный" относится к добавлению компонента в смесь катионного липида до формирования пустой везикулы.
Согласно настоящему изобретению термин "декорированный" относится к добавлению компонента после формирования везикулы.
Согласно настоящему изобретению "DC-6-14" относится к следующему катионному липидному соединению:
Согласно настоящему изобретению "DODC" относится к следующему катионному липидному соединению::
Согласно настоящему изобретению "HEDODC" относится к следующему катионному липидному соединению:
Согласно настоящему изобретению «Ретиноид» является представителем класса соединений, состоящих из четырех изопреноидных звеньев, соединенных в конфигурации голова-к-хвосту, см. G.P. Moss, "Biochemical Nomenclature and Related Documents," 2nd Ed. Portland Press, pp. 247-251 (1992). «Витамин A» представляет собой родовой дескриптор для ретиноидов, проявляющих в качественном отношении биологическую активность ретинола. В настоящем описании термин «ретиноид» относится к природным и синтетическим ретиноидам, включая ретиноиды первого поколения, второго поколения и третьего поколения. Неограничивающими примерами встречающихся в природе ретиноидов являются (1) 11-цис-ретиналь, (2) полностью транс-ретинол, (3) ретинилпальмитат, (4) полностью транс-ретиноевую кислоту и (5) 13-цис-ретиноевые кислоты. Кроме того, термин «ретиноид» включает ретинолы, ретинали и ретиноевые кислоты, рексиноиды, деметилированные и/или насыщенные ретиноевае кислоты и их производные.
Согласно настоящему изобретению "конъюгат ретиноида" относится к молекуле, которая включает по меньшей мере одну ретиноидную составляющую.
Согласно настоящему изобретению "молекула ретиноид-линкер-липид" относится к молекуле, которая включает по меньшей мере одну ретиноидную составляющую, прикрепленную к по меньшей мере одной липидной составляющей через по меньшей мере один линкер, такой как, например, ПЭГ составляющая.
Согласно настоящему изобретению "молекула ретиноид-линкер-ретиноид" относится к молекуле, которая включает по меньшей мере одну ретиноидную составляющую, присоединенную к другой ретиноидной составляющей (которая может быть такой же или отличной) через по меньшей мере один линкер, такой как, например, ПЭГ составляющая.
Согласно настоящему изобретению "витамин D" является общим определением для группы витаминов, имеющих противорахитную активность. Группа витамина D включает: витамин D2 (кальциферол), витамин D3 (иррадиированный 22-дигидроэргостерол), витамин D4 (иррадиированный дигидроситостерол) и витамин D5 (иррадиированный дигидроситостерол).
Согласно настоящему изобретению "витамин Е" является общим определением для группы молекул с антиоксидантной активностью. Семейство витамина E включает α-токоферол, β-токоферол, γ-токферол и δ-токоферол, причем α-токоферол является наиболее распространенным. (Brigelius-Flohe and Traber, The FASEB Journal. 1999; 13: 1145-1155).
Согласно настоящему изобретению "витамин K" является общим определением для противогеморрагического фактора и включает витамин K1 (фитонодион), витамин K2 (менахинон), витамин K3, витамин K4 и витамин K5. Витамины K1 и K2 являются природными, тогда как K3-5 являются синтетическими.
Согласно настоящему изобретению "молекула ретиноид-линкер-липид" относится к молекуле, которая включает по меньшей мере одну ретиноидную составляющую, прикрепленную к по меньшей мере одной липидной составляющей через по меньшей мере один линкер, такой как, например, ПЭГ составляющая.
Согласно настоящему изобретению "молекула ретиноид-линкер-ретиноид" относится к молекуле, которая включает по меньшей мере одну ретиноидную составляющую, присоединенную к другой ретиноидной составляющей (которая может быть такой же или отличной) через по меньшей мере один линкер, такой как, например, ПЭГ составляющая.
Согласно настоящему изобретению термины "липид" и "липофильный" применяются в описании настоящего изобретения согласно их общеизвестным значениям, как понимается специалистами в данной области техники. Неограничивающие примеры липидов и липофильных групп включают жирные кислоты, стиролы, C2-C50алкил, C2-C50гетероалкил, C2-C50алкенил, C2-C50гетероалкенил, C5-C50арил, C5-C50гетероарил, C2-C50алкинил, C2-C50гетероалкинил, C2-C50карбоксиалкенил и C2-C50карбксигетероалкенил. Жирной кислотой является насыщенная или ненасыщенная длинноцепочечная монокарбоновая кислота, которая содержит, например, 12-24 атомов углерода Липид характеризуется как являющийся по существу не растворимым в воде, имеющим растворимость в воде менее около 0.01% (массовая основа). Согласно настоящему изобретению термины "липидная составляющая" и "липофильная составляющая" относятся к липиду или его части, которая присоединяется к другой группе. Например, липидная группа может присоединяться к другому соединению (например, мономеру) посредством химической реакции между функциональной группой (такой как группа карбоновой кислоты) липида и соответствующей функциональной группой мономера.
Согласно настоящему изобретению "siPHK" относится к малой интерферирующей РНК, также известной в данной области техники как короткая интерферирующая РНК или сайлесинг РНК. siPHK является классом двухнитиевых молекул РНК, которые оказывают многообразие эффектов, известных в данной области техники, наиболее значимым является влияние на экспрессию специфических генов и экспрессию белка.
Согласно настоящему изобретению признак "инкапсулированный липосомой" относится к компоненту, находящемуся по существу или полностью внутри структуры липосомы.
Согласно настоящему изобретению признак "доступный для водной среды" относится к компоненту, находящемуся в контакте с водной средой.
Согласно настоящему изобретению признак "не доступный для водной среды" относится к компоненту, не находящемуся в контакте с водной средой.
Согласно настоящему изобретению признак "образуемая на внешней поверхности липосомы" относится к компоненту, функционально связанному с внешней поверхностью липосомы.
Согласно настоящему изобретению признак "расположенный на внешней поверхности липосомы" относится к компоненту, находящемуся на или около внешней поверхности липосомы.
Согласно настоящему изобретению термин "заряженное образование" относится к электростатическому связыванию.
Согласно настоящему изобретению термин "функционально связанный" относится к электронному взаимодействию соединения как описано в настоящей заявке, терапевтического агента, ретиноида и/или второго липида. Такое взаимодействие может иметь форму химической связи, включая, но без ограничения к этому, ковалентную связь, полярную ковалентную связь, ионную связь, электростатическое связывание, координированную ковалентную связь, ароматическую связь, водородную связь, дипольное или ван-дер-ваальсовое взаимодействие. Специалисты в данной области техники понимают, что относительные силы таких взаимодействий могут широко варьироваться.
Термин "липосома" применяется в настоящей заявке в его общеизвестном значении, как понимается специалистами в данной области техники, и относится к структуре липидного бислоя, который содержит липиды, присоединенные к полярным, гидрофильным группам, которые образуют по существу закрытую структуру в водной среде. В некоторых вариантах выполнения настоящего изобретения, липосома может быть функционально связана с одним или более соединениями, такими как терапевтический агент и ретиноид или конъюгат ретиноида. Липосома может состоять из одного липидного бислоя (то есть униламеллярная), или она может состоять из двух или более сосредоточненых липидных бислоев (то есть мультиламеллярная). Кроме того, липосома может быть примерно сферической или эллипсоидной по форме.
Термин "облегчение доставки лекарственного средства в целевую клетку" относится к повышению способности соединений ретиноида или жирорастворимого витамина повышать доставку терапевтической молекулы, такой как siPHK, в клетку. Не желая быть связанными конкретной теорией, соединение ретиноида или жирорастворимого витамина взаимодействует со специфическим рецептором или [сайтом активации/связывания] на целевой клетке со специфичностью, которая может быть измерена. Например, связывание является в общем специфическим, когда аффинность связывания (Ka) составляет или более, предпочтительно 107 М-1 или более, более предпочтительно 108 М-1 или более и наиболее предпочтительно 109 М-1 или более. Аффинность связывания антитела может быть легко определена специалистом в данной области техники, например, посредством анализа Scatchard (Scatchard, Ann. NY Acad. Sci. 51: 660, 1949)
Системы для доставки нуклеиновых кислот могут включать, например водные и неводные гели, кремы, многосоставные эмульсии, микроэмульсии, липосомы, мази, водные и неводные растворы, лосьоны, аэрозоли, углеводородные основания и порошки, и могут содержать эксципиенты, такие как солюбилизаторы, усилители проникновения (например, жирные кислоты, сложные эфиры жирных кислот, жирные спирты и аминокислоты) и гидрофильные полимеры (например, поликарбофил и поливинилпиролидон).
В дополнение к катионному липиду Формулы I, другие липиды могут быть полезны. Они включают ионизируемые катионные липиды, включая
Композиции для доставки могут состоять из катионного липида формулы I в комбинации с ионизируемым катионным липидом. Ионизируемый катионный липид может включать, например, S104. Ионизируемый катионный липид может присутствовать в концентрации от 0 до 45 мол.%, включая концентрацию, выбранную из 5, 10, 15, 20, 25, 30, 35, 40 и 45 мол.%.
Липид, конъюгированный с молекулой полиэтиленгликоля (ПЭГ), может присутствовать в липосомных частицах. ПЭГ-липиды включают
- 1,2-димиристолеоил-sn-глицеро-3-фосфоэтаноламин-N-ПЭГ (ПЭГ-DMPE)
- 1,2-дипальмитоил-sn-глицеро-3-фосфоэтаноламин-N-ПЭГ (ПЭГ-DPPE),
- 1,2-дистеароил-sn-глицеро-3-фосфоэтаноламин-N-ПЭГ (ПЭГ-DSPE) или
- 1 Д-диолеоил-sn-глицеро-3-фосфоэтаноламин-N-ПЭГ (ПЭГ-DOPE) и/или
- ПЭГ-церамид.
Композиции доставки могут состоять из катионного липида Формулы I в комбинации с ПЭГ-липидом. ПЭГ-липид может присутствовать в концентрации от 0 до 15 мол.%, предпочтительно от 1 до 10 мол.%, включая концентрацию, выбранную из 1, 2, 3, 4, 5, 6, 7, 8, 9 и 10 мол.%.
Неограничивающие примеры некатионных липидов включают фосфолипиды, такие как лецитин, фосфатидилэтаноламин, лизолецитин, лизофосфатидилэтаноламин, фосфатидилсерин, фосфатидилинозитол, сфингомиелин, яичный сфингомиелин (ESM), цефалин, кардиолипин, фосфатидная кислота, цереброзиды, дицетилфосфат, дистеароилфосфатидилхолин (DSPC), диолеоилфосфатидилхолин (DOPC), дипальмитоилфосфатидилхолин (DPPC), диолеоилфосфатидилглицерол (DOPG), дипальмитоилфосфатидилглицерол (DPPG), диолеоилфосфатидилэтаноламин (DOPE), пальмитоилолеоил-фосфатидилхолин (POPC), пальмитоилолеоил-фосфатидилэтаноламин (POPE), пальмитоилолеоил-фосфатидилглицерол (POPG), диолеоил-фосфатидилэтаноламин 4-(N-малеимидометил)-циклогексан-1-карбоксилат (DOPE-mal), дипальметоил-фосфатидилэтаноламин (DPPE), димиристоил-фосфатидилэтаноламин (DMPE), дистеароил-фосфатидилэтаноламин (DSPE), монометил-фосфатидилэтаноламин, диметил-фосфатидилэтаноламин, диэлаидоил-фосфатидилэтаноламин (DEPE), стеароилолеоил-фосфатидилэтаноламин (SOPE), лизофосфатидилхолин, дилинолеоилфосфатидилхолин и их смеси. Другие диацилфосфатидилхолин и диацилфосфатидилэтаноламин фосфолипиды также могут применяться. Ацильные группы этих липидов предпочтительно представляют собой ацильные группы, полученные из жирных кислот, имеющих C10-C24 углеводородные цепи, например, лауроил, миристоил, пальмитоил, стеароил или олеоил.
В определенных вариантах выполнения настоящего изобретения количество фосфолипида, присутствующего в частицах, составляет от около 0 мол.% до около 55 мол.%, более конкретно при концентрации, выбранной из 5, 10, 15, 20, 25, 30, 35, 40, 45, 50 и 55%. Неограничивающим примером фосфолипида является DOPE.
Дополнительные примеры некатионных липидов включают стеролы, такие как холестерин и его производные, такие как холестанол, холестанол, холестанол, копростанол, холестерил-2'-гидроксиэтиловый эфир, холестерил-4'-гидроксиэтиловый эфир и их смеси.
В определенных вариантах выполнения настоящего изобретения холестерин или производная холестерина, присутствующие в частицах, составляют от около 0 мол.% до около 55 мол.%, более конкретно при концентрации, выбранной из 5, 10, 15, 20, 25, 30, 35, 40, 45, 50 и 55 мол.%. В качестве неограничивающего примера холестерин присутствует в липидных частицах.
В определенных других вариантах выполнения настоящего изобретения, холестерин присутствует в липидных частицах, содержащих смесь фосфолипидов, и холестерин содержит от около 30 мол.% до около 40 мол.%, от около 30 мол.% до около 35 мол.% или от около 35 мол.% до около 40 мол.% от всех присутствующих в частицах липидов. В качестве неограничивающего примера, липидная частица, содержащая смесь фосфолипида и холестерина, может содержать холестерин в количестве около 34 мол.% от всех липидов, присутствующих в частице.
В других вариантах выполнения настоящего изобретения холестерин присутствует в липидных частицах, содержащих смесь фосфолипидов, и холестерин содержит от около 10 мол.% до около 30 мол.%, от около 15 мол.% до около 25 мол.% или от около 17 мол.% до около 23 мол.% от всех присутствующих в частицах липидов. В качестве неограничивающего примера, липидная частица, содержащая смесь фосфолипида и холестерина, может содержать холестерин в количестве около 20 мол.% от всех липидов, присутствующих в частице.
Ретиноид или конъюгат ретиноида, полезный для доставки нуклеиновой кислоты, находится в состоянии, в котором он растворяется в среде или смешивается со средой, которая может растворять или сохранять его.
Любой ретиноид или конъюгат ретиноида может применяться в настоящей заявке до тех пор, пока он активно аккумулируется стволовыми клетками; примеры ретиноида включают, но без ограничения к этому, третиноин, адапален, ретинол пальмитат и, в частности, витамин A, насыщенный витамин A, ретиноевая кислота, примеры конъюгата ретиноида включают ПЭГ-ретиноид конъюгаты. В настоящем изобретении применяется свойство стволовых клеток положительно включать ретиноид и/или конъюгат ретиноида, и посредством ретиноида и/или конъюгата ретиноида в качестве носителя для лекарственного средства или посредством связывания с или включения в другой компонент носителя для лекарственного средства, желательный материал или организм специфически транспортируется в стволовые клетки. Ретиноид является членом класса соединений, имеющих скелет, в котором четыре изопреноидные единицы связываются образом голова к хвосту. See G.P. Moss, "Biochemical Nomenclature и Related Documents," 2nd Ed. Portland Press, pp. 247-251 (1992). Витамин A является общим дескриптором качества ретиноида, показывающий биологическую активность ретинола. Ретиноид согласно настоящему изобретению способствует специфической доставке вещества в раковую клетку и CAF (то есть вещество нацеливается в эти клетки). Такой ретиноид в частности не ограничивается, и его примеры включают ретинол, витамин A, насыщенный витамин A, ретиналь, ретиноевая кислота, сложный эфир ретинола и жирной кислоты, сложный эфир алифатического спирта и ретиноевой кислоты, этретинат, третиноин, изотретиноин, адапален, акитретин, тазаротен и ретинол пальмитат, и аналоги витамина A, такие как фенретинид и бексаротен. Конъюгаты ретиноида включают ПЭГ-конъюгаты, например, diVA-ПЭГ-diVA и VA-ПЭГ-VA.
Носитель для лекарственного средства согласно настоящему изобретению поэтому может содержать компонент носителя для лекарственного средства, отличный от ретиноида и/или конъюгата ретиноида. Такой компонент конкретно не ограничивается, и любой компонент, известный в области медицины и фармацевтики, может применяться, но предпочтительно, чтобы он был способен включать ретиноид и/или конъюгат ретиноида. Кроме того, носитель для лекарственного средства согласно настоящему изобретению может содержать вещество, которое улучшает включение в звездчатые клетки, например, ретинол-связывающий белок (RBP). Связывание ретиноида и/или конъюгата ретиноида с носителем для лекарственного средства согласно настоящему изобретению или включение в него может также осуществляться посредством связывания ретиноида и/или конъюгата ретиноида с другим компонентом носителя для лекарственного средства или включения в него химическим и/или физическим методами. Альтернативно, связывание ретиноида и/или конъюгата ретиноида с носителем для лекарственного средства согласно настоящему изобретению или включение в него может также осуществляться посредством подмешивания ретиноида и/или конъюгата ретиноида, имеющего аффинность к ассоциации, и основных компонентов носителя для лекарственного средства, в компоненты носителя для лекарственного средства в ходе приготовления носителя для лекарственного средства.
Количество ретиноида и/или конъюгата ретиноида, связанного с носителем для лекарственного средства согласно настоящему изобретению, или включенного в него, может составлять от 0.1 мол.% до 20 мол.% в качестве массового отношения относительно компонентов носителя для лекарственного средства, предпочтительно от 0.2% до 10% и более предпочтительно от 0.5% до 5 мол.%, включая концентрацию, выбранную из значений 0.25, 0.5, 1.0, 1.5, 2.0. 2.5, 3.0, 3.5, 4.0, 4.5 и 5.0 мол.%.
В определенных вариантах выполнения настоящего изобретения настоящее изобретение обеспечивает липосому, получаемую смешением в камере. Он включает способ, который включает обеспечение водный раствор, содержащий нуклеиновую кислоту, такую как интерферирующая РНК, в первом резервуаре, обеспечение органического раствора липида во втором резервуаре и смешение водного раствора с органическим раствором липида, так что органический раствор липида смешивается с водным раствором с получением липосомы, инкапсулирующей нуклеиновую кислоту (например, siPHK), при градиенте концентрации органического растворителя.
Липосома, сформированная с применением способа смешивания, как правило, имеет размер от около 40 нм до около 250 нм, от около 50 нм до около 150 нм, от около 60 нм до около 150 нм. Частицы, сформированные таким образом, не агрегируют и при необходимости доводятся до размера для достижения равномерного размера частиц.
Носитель для лекарственного средства согласно настоящему изобретению может быть в любой форме до тех пор, пока желательный материал или организм может быть транспортирован в целевые звездчатые клетки, и примеры формы включают, но без ограничения к этому, полимерную мицеллу, липосому, эмульсию, микросферу и наносферу. Кроме того, носитель для лекарственного средства может включать в его внутренней части вещество, которое должно быть транспортировано, присоединенное к его внешней части вещество, которое должно транспортироваться, или быть смешанным с веществом, которое должно быть транспортировано, до тех пор, пока ретиноид и/или конъюгат ретиноида, включенный в него, по меньшей мере частично выставлен на внешней части препарата пока он не достигнет наконец звездчатых клеток.
Носитель для лекарственного средства согласно настоящему изобретению специфически нацелен на звездчатые клетки и обеспечивает желательный эффект, такой как, например, ингибирование или предупреждение фиброза, показываемый с максимальным эффектом и минимальными побочными эффектами посредством эффективного транспортирования в звездчатые клетки желательного вещества или организма, как например, лекарственное средство для контроля активности или роста звездчатых клеток. Вещество или организм, который доставляет носитель для лекарственного средства согласно настоящему изобретению, в частности не ограничен, но предпочтительно имеет размер, который обеспечивает физическое перемещение в живом организме от места введения до печени, поджелудочной железы и т.д., где присутствуют стволовые клетки. Носитель для лекарственного средства согласно настоящему изобретению поэтому может транспортировать не только вещество, такой как атом, молекула, соединение, белок или нуклеиновая кислота, но также и организм, такой как вирус, частица вируса, клетка, система высвобождения лекарственного средства, составленная из одного или более элементов или микромашины. Вещество или организм предпочтительно имеет свойство оказания некоторого эффекта на звездчатые клетки, и примерами которого является метка звездчатых клеток и контроль активности или роста звездчатых клеток.
Поэтому в одном варианте выполнения настоящего изобретения лекарственное носитель для лекарственного средства доставляет средство контролирует активность и рост звездчатых клеток. Это может быть любое лекарственное средство, которое непосредственно или косвенно ингибирует физико-химические действия звездчатых клеток, которые участвуют в развитии фиброза, и его примеры включают, но без ограничения к этому, ингибиторы активности TGFβ, такие как усеченный рецептор TGFβ типа II и растворимый рецептор TGFβ типа II, препараты фактора роста, такие как HGF и экспрессионные векторы для них, промоторы продукции ММР, как например аденовирусный вектор, содержащий ген ММР, ингибиторы продукции TIMP, такие как антисмысловая TIMP нуклеиновая кислота, PPARγ лиганд, ингибиторы клеточной активности и/или ингибиторы клеточного роста, такие как ингибитор активности ангиотензина, ингибитор активности PDGF, и ингибитор натриевого канала, а также индукторы апоптоза, такие как соединение 861 и глиотоксин, адипонектин, и соединение, имеющее ингибиторную активность в отношении киназы Rho, такое как (+)-транс-4-(1-аминоэтил)-1-(4-пиридилкарбамоил)циклогексан. Кроме того, лекарственное средство для контроля или роста звездчатых клеток согласно настоящему изобретению может быть любым лекарственным средством, которое непосредственно или косвенно вызывает физико-химические действия звездчатых клеток, непосредственно или косвенно участвующих в ингибировании фиброза, и его примеры включают, но без ограничения к этому, лекарственное средство для активации системы разрушения коллагена, например, промоторы продукции ММР, как например ММР экспрессионный вектор, HGF, и лекарственные средства, имеющие HGF-подобную активность, такие как аналоги HGF и их экспрессионные вектора.
Другие примеры лекарственных средств для контроля активности или роста звездчатых клеток согласно настоящему изобретению включают лекарственное средство для контроля метаболизма внеклеточного матрикса, как например коллагена, например, вещество, оказывающее эффект на ингибирование экспрессии целевой молекулы, как например siRNA, рибозим и антисмысловая нуклеиновая кислота (включая РНК, ДНК, PNA и их композит), вещество, имеющее доминантный отрицательный эффект, и вектора, экспрессирующие их, которые нацеливаются, например, на молекулу, составляющую внеклеточный матрикс, продуцируемую звездчатыми клетками, или нацеливаются на одну или более молекулы, которые имеют функцию продуцирования или секретирования молекулы, составляющей внеклеточный матрикс.
Настоящее изобретение также относится к лекарственному средству для лечения нарушения, связанного со звездчатыми клетками, причем лекарственное средство содержит носитель для лекарственного средства и лекарственное средство для контроля активности или роста звездчатых клеток, и относится к применению носителя для лекарственного средства для получения фармацевтической композиции для лечения нарушения, связанного со звездчатыми клетками. Нарушение, связанное со звездчатыми клетками, в контексте настоящего изобретения означает нарушение, в котором звездчатые клетки непосредственно или косвенно участвуют в процессе нарушения, то есть, вспышке, обострении, улучшении, ремиссии, излечении и т.д. нарушения, и его примеры включают нарушения печени, такие как гепатит, в частности хронический гепатит, фиброз печени, цирроз печени и рак печени, и панкреатические нарушения, такие как панкреатит, в частности хронический панкреатит, фиброз поджелудочной железы и рак поджелудочной железы.
В препарате согласно настоящему изобретению носитель для лекарственного средства может включать лекарственное средство в его внутренней части, содержать присоединенное к его внешней части вещество, содержащее лекарственное средство, или быть смешанным с лекарственным средством до тех пор, пока ретиноид и/или конъюгат ретиноида, включенный в носитель для лекарственного средства, по меньшей мере частично выставлен на внешней части препарата до того, как он достигнет, наконец, звездчатых клеток. Поэтому, в зависимости от пути введения или образа, которым лекарственное средство высвобождается, препарат может быть покрыт подходящим веществом, таким как, например, энтеральное покрытие или материал, который разрушается со временем, или может быть включен в соответствующую систему высвобождения лекарственного средства.
Поэтому настоящее изобретение включает носитель для лекарственного средства или набор для получения препарата, содержащий один или более контейнеров, содержащих один или более составляющих носителя для лекарственного средства, ретиноид и/или конъюгат ретиноида, и/или лекарственное средство, а также включает существенный компонент для носителя для лекарственного средства, или препарат, обеспечиваемый в форме такого набора. Набор согласно настоящему изобретению может содержать, в дополнение к описанным выше, описание и т.д., где описывается способ получения или способ введения носителя для лекарственного средства и препарата согласно настоящему изобретению. Кроме того, набор согласно настоящему изобретению может содержать все компоненты для получения носителя для лекарственного средства или препарата согласно настоящему изобретению, но не обязательно содержит все компоненты. Поэтому набор согласно настоящему изобретению необязательно должен содержать реагент или растворитель, который, как правило, доступен на месте лечения, эксперимента и т.д., как например, например, стерильная вода, соляной раствор или раствор глюкозы.
Настоящее изобретение, кроме того, относится к способу лечения нарушения, связанного со звездчатыми клетками, причем способ включает введение эффективного количества препарата субъекту, нуждающемуся в этом. Эффективное количество в контексте настоящего изобретения относится к количеству, которое подавляет вспышку целевого нарушения, уменьшение его симптомов или предотвращение его развития, и предпочтительно представляет собой количество, которое предотвращает вспышку целевого нарушения или излечивает целевое нарушение Оно также представляет собой количество, которое не вызывает побочный эффект, который превышает благоприятный эффект от введения. Такое количество может быть определено подходящим образом посредством in vitro теста с применением культивированных клеток и т.д. или посредством тестирования на животной модели, такой как мышь, крыса, собака или свинья, и такие способы тестирования хорошо известны специалистам в данной области техники.
В способе согласно настоящему изобретению термин `субъект` означает любое живое существо, предпочтительно животное, более предпочтительно млекопитающее и еще более предпочтительно человека. В контексте настоящего изобретения субъект может быть здоровым или подверженным такому нарушению, и в случае лечения рассматриваемого нарушения, субъект, как правило, означает субъект, у которого обнаружено нарушение или имеющий риск развития нарушения.
Кроме того, термин "лечение" включает все типы приемлемого с медицинской точки зрения профилактического и/или терапевтического вмешательства в целях излечения, временного ослабления, профилактики и т.д. нарушения. Например, когда нарушением является фиброз печени, термин "лечение" включает приемлемое с медицинской точки зрения вмешательство в различных целях, включая замедление или остановку развития фиброза, регрессию или исчезновение очагов, профилактику вспышки фиброза или профилактику повторного развития.
Настоящее изобретение также относится к способу доставки лекарственного средства в звездчатые клетки с применением носителя для лекарственного средства. Этот способ включает, но без ограничения к этому, стадию нанесения вещества, подлежащего доставке, на носитель для лекарственного средства, и стадию введения или добавления носителя для лекарственного средства, несущего вещество, подлежащее доставке, в живой организм или среду, содержащую звездчатые клетки, как например, культуральную среду. Эти стадии могут быть достигнуты подходящим способом в соответствии с известным способом, описанным в настоящем изобретении способом и т.д. Этот способ доставки может быть объединен с другим способом доставки, например, другим способом доставки, в котором орган, в котором присутствуют звездчатые клетки, является целевым и т.д.
Молекулы нуклеиновой кислоты могут быть адаптированы для применения для профилактики или лечения фиброза (например, печени, почек, перитонеального и легких), признаков, состояний и/или нарушений, и/или любого другого признака, заболевания, нарушения или состояния, которое связано с или зависит от уровней hsp47 в клетке или ткани, сами по себе или в комбинации с другими терапиями. Молекула нуклеиновой кислоты может включать среду для доставки, включая липосомы, для введения субъекту, носителем и разбавители и их соли, и/или может присутствовать в фармацевтически приемлемых композициях.
Молекулы нуклеиновой кислоты согласно настоящему изобретению могут включать последовательности, показанные в Таблице 3. Примерами таких молекул нуклеиновой кислоты по существу состоят из последовательностей, приведенных в Таблице 3.
Молекулы нуклеиновых кислот можно вводить с помощью введения в легкие, например, посредством вдыхания высушенной лекарственной формы в виде аэрозоля или спрея, вводимой с помощью устройства для ингаляции или распылителя, обеспечивающего быстрое местное поглощение молекул нуклеиновых кислот соответствующими тканями легких. Твердые дисперсные композиции, содержащие подходящие для вдыхания сухие частицы тонко измельченных композиций нуклеиновых кислот, можно изготовить посредством измельчения высушенных или лиофилизированных композиций нуклеиновых кислот и затем пропускания измельченных композиций, например, через сито с размером ячеек 400, чтобы разбить или отделить крупные агломераты. Твердая дисперсная композиция, содержащая композиции нуклеиновых кислот согласно настоящему изобретению, может дополнительно содержать диспергатор, который служит для облегчения образования аэрозоля, а также другие терапевтические соединения. Приемлемым диспергатором является лактоза, которую можно смешивать с соединением нуклеиновой кислоты в любом приемлемом соотношении, например, соотношении 1 к 1 по массе.
Аэрозоли жидких частиц могут включать молекулы нуклеиновых кислот согласно настоящему описанию и могут быть получены любыми приемлемыми способами, например, с помощью распылителя (смотрите, например, патент США №4,501,729). Распылители представляют собой коммерчески доступные устройства, которые превращают растворы или суспензии активного ингредиента в терапевтический аэрозоль, например, с помощью ускорения сжатого газа, обычно воздуха или кислорода, через узкие отверстия трубки Вентури или с помощью ультразвукового перемешивания. Приемлемые композиций для использования в распылителях включают активный ингредиент в жидком носителе в количестве до 40% масс/масс, предпочтительно меньше, чем 20% масс/масс, композиции. Обычно носитель представляет собой воду или разбавленный водно-спиртовой раствор, предпочтительно сделанный изотоничным жидкостям тела посредством добавления, например, хлорида натрия или других приемлемых солей. Возможные добавки включают консерванты, если лекарственная форма изготавливается нестерильной, например, метил гидроксибензоат, антиоксиданты, ароматизаторы, эфирные масла, буферные вещества и эмульгаторы и другие поверхностно активные вещества. Аэрозоли твердых частиц, включая активную композицию и поверхностно активное вещество, также можно изготовить с помощью любого твердодисперсного генератора аэрозолей. Генераторы аэрозолей для введения твердодисперсного лекарственного средства пациенту производят частицы, которые можно вдыхать, как описано выше, и генерируют объем аэрозоля, содержащий предварительно измеренную дозу терапевтической композиции со скоростью, подходящей для введения человеку. Одни примером типа твердодисперсного генератора аэрозолей является инжектор. Приемлемые лекарственные формы для введения посредством вдувания включают тонко измельченные порошки, которые можно вводить с помощью инжектора. В инжекторе порошок, например, его измеренная доза, эффективная для осуществления лечения согласно настоящему описанию, заключена в капсулы или картриджи, обычно сделанные из желатина или пластика, которые прокалывают или открывают in situ, и порошок, доставляемый воздухом, выпускаемым из устройства при вдыхании или с помощью ручной помпы. Порошок, используемый в инжекторе, состоит только из активного ингредиента или из смеси порошков, содержащей активный ингредиент, приемлемый разбавитель порошка, такой как лактоза, и, возможно, поверхностно активное вещество. Активный ингредиент обычно содержит от 0,1 до 100 мас./мас. композиции. Второй тип примеров генераторов аэрозолей включает дозирующий ингалятор. Дозирующие ингаляторы представляют собой аэрозольные распылители под давлением, обычно содержащие суспензию или раствор активного ингредиента в сжиженном пропелленте. В процессе применения указанные устройства выбрасывают лекарственную форму через клапан для доставки измеренного объема для производства тонкодисперсного спрея, содержащего активный ингредиент. Приемлемые пропелленты включают некоторые соединения хлорфторуглеродов, например, дихлордифторметан, трихлорфторметан, дихлортетрафторэтан и их смеси. Дополнительно лекарственная форма может содержать один или несколько сорастворителей, например, этанол, эмульгаторов и других поверхностно активных веществ, таких как масляная кислота или сорбитан триолеат, антиоксидантов и приемлемых ароматизаторов. Другие способы для доставки в легкие описываются, например, в заявке на патент США №20040037780 и патентах США №6592904; 6582728; 6565885. В WO 2008/132723 относится к аэрозольной доставке олигонуклеотидов в общем и в частности siRNA в дыхательную систему.
Молекулы нуклеиновых кислот можно вводить в центральную нервную систему (ЦНС) или периферическую нервную систему (ПНС). Эксперименты показали эффективное поглощение in vivo нуклеиновых кислот нейронами. Смотрите, например, Sommer et al., 1998, Antisense Nuc. Acid Drug Dev., 8: 75; Epa et al., 2000, Antisense Nuc. Acid Drag Dev., 10: 469; Broaddus et al., 1998, J. Neurosurg., 88: 734; Karle et al., 1997, Eur. J. Pharmocol., 340: 153; Bannai et al., 1998, Brain Research, 784: 304; Rajakumar et al., 1997, Synapse, 26: 199; Wu-pong et al., 1999, BioPharm, 12: 32; Bannai et al., 1998, Brain Res. Protoc, 3:83; and Simantov et al., 1996, Neuroscience, 74: 39. Молекулы нуклеиновых кислот, таким образом, подходят для доставки и поглощения клетками в ЦНС и/или ПНС.
Доставка молекул нуклеиновых кислот в ЦНС обеспечивается большим количеством различных стратегий. Традиционные подходы к доставке в ЦНС, которые могут быть использованы, включают, но не ограничиваются, интратекальное и интрацеребровентрикулярное введение, имплантацию катетеров и помп, непосредственную инъекцию или перфузию в место поражения или травмы, инъекцию в систему мозговых артерий или введение посредством химического или осмотического открытия гематоэнцефалического барьера. Другие подходы могут включать использование различных транспортных систем и систем носителей, например, посредством использования конъюгатов и биодеградируемых полимеров. Более того, методы генной терапии, например, согласно описанию Kaplitt et al., патент США №6,180,613 и Davidson, WO 04/013280, могут быть использованы для экспрессии молекул нуклеиновых кислот в ЦНС.
Молекулы нуклеиновой кислоты могут входить в композиции с или быть объединены с полиэтиленимином {например, линейный или разветвленный PEI) и/или производными полиэтиленимина, включая например, привитые PEI, такие как производные, как галактоза PEI, холестерин PEI, полученный из антител PEI, и полиэтиленгликоль PEI (ПЭГ-PEI) (смотрите, например, Ogris et al., 2001, AAPA PharmSci, 3, 1-11; Furgeson et al., 2003, Bioconjugate Hem., 14, 840-847; Kunath et al., 2002, Pharm Res 19:810-17; Choi et al., 2001, Bull. Korean Chem. Soc, 22: 46-52; Bettinger et al., 1999, Bioconjugate Chem., 10: 558-561; Peterson et al., 2002, Bioconjugate Chem. 13: 845-54; Erbacher et al., 1999, J Gene Med 1: 1-18; Godbey et al., 1999., PNAS, 96: 5177-81; Godbey et al., 1999, J Controlled Release, 60: 149-60; Diebold et al., 1999, J Biol Chem, 274: 19087-94; Thomas et al., 2002, PNAS, 99,14640-45; and Sagara, U.S. Pat. No. 6,586,524).
Молекулы нуклеиновых кислот могут включать биоконъюгаты, например, конъюгат нуклеиновой кислоты, как описано в Vargeese et al., патент США №. 10/427160; патент США №. 6528631; патент США №. 6335434; патент США №. 6235886; патент США №. 6153737; патент США №. 5214136; патент США №. 5138045.
Композиции, способы и наборы согласно настоящему описанию могут включать экспрессионный вектор, который включает последовательность нуклеиновой кислоты, кодирующую по меньшей мере одну молекулу нуклеиновой кислоты согласно настоящему изобретению, таким образом, что это способствует экспрессии молекулы нуклеиновой кислоты. Способы введения молекул нуклеиновых кислот или одного или нескольких векторов, способных экспрессировать цепи dsRNA в среде клетки, будут зависеть от типа клетки и состава ее среды. Молекулу нуклеиновой кислоты или векторную конструкцию можно вводить непосредственно в клетку (т.е. внутриклеточно) или вводить внеклеточно в полость, интерстициальное пространство, в кровообращение организма, вводить перорально или можно вводить посредством погружения организма или клетки в раствор, содержащий dsRNA. Клетка предпочтительно представляет собой клетку млекопитающего, более предпочтительно клетку человека. Молекула нуклеиновой кислоты вектора экспрессии может включать смысловую область и антисмысловую область. Антисмысловая область может включать последовательность, комплементарную последовательности РНК или ДНК, кодирующей hsp47, и смысловая область может включать последовательность, комплементарную антисмысловой области. Молекула нуклеиновой кислоты может включать две отдельные цепи, имеющие комплементарные смысловую и антисмысловую области. Молекула нуклеиновой кислоты может включать одну цепь, имеющую комплементарные смысловую и антисмысловую области.
Молекулы нуклеиновых кислот, которые взаимодействуют с целевыми молекулами РНК и подавляют ген, кодирующий целевые молекулы РНК (например, целевые молекулы РНК, обозначаемые в настоящем описании номерами доступа Genbank), могут экспрессироваться из единиц транскрипции, вставленных в векторы ДНК или РНК. Рекомбинантные векторы могут представлять собой ДНК плазмиды и вирусные векторы. Молекула нуклеиновой кислоты, экспрессирующая вирусные векторы, может быть сконструирована на основе, но без ограничения к этому, аденассоциированного вируса, ретровируса, аденовируса или альфавируса. Рекомбинантные векторы, способные к экспрессии молекул нуклеиновых кислот, могут быть доставлены согласно настоящему описанию и сохраняться в клетках-мишенях. В другом случае, можно использовать вирусные векторы, которые способствуют временной экспрессии молекул нуклеиновых кислот. Такие векторы при необходимости можно вводить повторно. После экспрессии молекулы нуклеиновых кислот связываются с и подавляют функцию или экспрессию гена посредством РНК интерференции (RNKi). Доставка молекулы нуклеиновой кислоты, экспрессирующей векторы, может быть системной, например, посредством внутривенного или внутримышечного введения, посредством введения в клетки-мишени эксплантированные из пациента, с последующим обратным введением пациенту, или другими способами, которые позволяют вводить в желательную целевую клетку.
Другим объектом настоящего изобретения является фармацевтическая композиция, содержащая один или более физиологически приемлемых поверхностно-активных агентов, фармацевтических носителей, разбавителей, эксципиентов и суспендирующих агентов или их комбинацию; и к композиции (например, композиция, которая может включать соединение, ретиноид, второй липид, стабилизирующий агент и/или терапевтический агент), как раскрывается в настоящей заявке. Приемлемые дополнительные фармацевтические носители или разбавители для терапевтического использования хорошо известны в фармации и описаны, например, в Remington’s Pharmaceutical Sciences, 18th Ed., Mack Publishing Co., Easton, PA (1990), полностью включенном в настоящее описание посредством ссылки. Консерванты, стабилизаторы, красители и тому подобное, могут быть включены в фармацевтическую композицию. Например, бензоат натрия, аскорбиновая кислота и эфиры п-гидроксибензойной кислоты могут быть добавлены в качестве консервантов. Кроме того, могут быть использованы антиоксиданты и суспендирующие агенты. В различных вариантах выполнения настоящего изобретения спирты, эфиры, сульфатированные алифатические спирты и т.д. могут быть использованы в качестве поверхностно-активных агентов; сахароза, глюкоза, лактоза, крахмал, кристаллическая целлюлоза, маннит, легкий безводный силикат, алюминат магния, метасиликат-алюминат магния, синтетический силикат алюминия, карбонат кальция, кислый карбонат натрия, гидрофосфат кальция, карбоксиметилцеллюлоза кальция и т.д. могут быть использованы в качестве эксципиентов; кокосовое масло, оливковое масло, кунжутное масло", арахисовое масло, соевое масло могут быть использованы в качестве суспендирующих агентов или смазывающих веществ; ацетатфталат целлюлозы в качестве производного углевода, такого как целлюлоза или сахар, или сополимер метилацетат-метакрилат в качестве производного поливинила могут быть использованы в качестве суспендирующих агентов; и пластификаторы, такие как эфир-фталаты и т.д. могут быть использованы в качестве суспендирующих агентов.
Фармацевтические композиции согласно настоящему изобретению могут быть введены пациенту-человеку как таковые или в фармацевтических композициях, в которых их смешивают с другими активными компонентами, как в комбинированной терапии, или подходящими фармацевтическими носителями или эксципиентом (эксципиентами). Способы получения и введения соединений согласно настоящей заявке могут быть найдены в "Remington’s Pharmaceutical Sciences," Mack Publishing Co., Easton, PA, 18th edition, 1990.
Подходящие пути введения могут включать, например, парентеральную доставку, включающую внутримышечные, подкожные, внутривенные, интрамедуллярные инъекции, а также интратекальные, прямые интравентрикулярные, интраперитонеальные, интраназальные или интраокулярные инъекции. Композиция (например, композиция, которая может включать соединение, ретиноид, второй липид, стабилизирующий агент и/или терапевтический агент) также может вводиться в лекарственных формах с замедленным или контролируемым высвобождением, включающих инъекции веществ замедленного всасывания, осмотические помпы и тому подобное для пролонгированного и/или рассчитанного по времени, импульсного введения с заданной скоростью. Кроме того, путь введения может быть местным или системным.
Фармацевтические композиции могут быть изготовлены известным способом, например, способами традиционного смешивания, растворения, гранулирования, получения драже, растирания в порошок, эмульгирования, инкапсулирования, «захвата» или таблетирования
Фармацевтические композиции могут быть изготовлены любым традиционным способом с использованием одного или более физиологически приемлемых фармацевтических носителей, включающих эксципиенты и вспомогательные вещества, облегчающие переработку активных соединений в препараты, которые могут быть фармацевтически использованы. Подходящий состав зависит от выбранного пути введения. Любые из хорошо известных способов, фармацевтических носителей и эксципиентов могут быть использованы в качестве подходящих и понятных в данной области техники; например, в Remington’s Pharmaceutical Sciences, выше.
Лекарственные средства для инъекций могут быть получены в традиционных формах, либо в виде жидких растворов или суспензий, твердых форм, подходящих для растворения или суспендирования в жидкости перед инъекцией, либо в виде эмульсий. Подходящие эксципиенты представляют собой, например, воду, физиологический раствор, декстрозу, маннит, лактозу, лецитин, альбумин, глутамат натрия, гидрохлорид цистеина и т.д. Кроме того, при необходимости фармацевтические композиции для инъекций могут содержать небольшие количества нетоксичных вспомогательных веществ, таких как смачивающие агенты, pH буферные агенты и т.д. Физиологически совместимые буферы включают, но не ограничиваются следующими: раствор Хенкса, раствор Рингера или физиологический буферный раствор. При необходимости, могут быть использованы усиливающие абсорбцию препараты.
Фармацевтические составы для парентерального введения, например, путем болюсной инъекции или непрерывной инфузии, включают водные растворы активных композиций (например, композиция, которая может включать соединение, ретиноид, второй липид, стабилизирующий агент и/или терапевтический агент) в водорастворимой форме. Кроме того, суспензии активных соединений могут быть получены в виде подходящих масляных суспензий для инъекций. Водные суспензии для инъекций могут содержать вещества, повышающие вязкость суспензии, такие как натрийкарбоксиметилцеллюлоза, сорбит или декстран. Возможно, суспензия также может содержать подходящие стабилизаторы или агенты, повышающие растворимость соединений, что позволяет получать высококонцентрированные растворы. Составы для инъекции могут находиться в дозированной лекарственной форме, например в ампулах или в многодозовых контейнерах с добавленным консервантом. Композиции могут находиться в таких формах, как суспензии, растворы или эмульсии в масляных или водных растворителях, и могут содержать вспомогательные вещества, такие как суспендирующие, стабилизирующие и/или диспергирующие агенты. В качестве альтернативы активный компонент может находиться в форме порошка для разбавления подходящим растворителем, например, стерильной апирогенной водой перед применением.
В дополнение к составам, описанным ранее, композиции также могут быть изготовлены в виде депо-препарата. Указанные составы длительного действия могут быть введены путем имплантации (например, подкожно или внутримышечно) или путем внутримышечной инъекции. Таким образом, например, композиции (например, композиция, которая может включать соединение, ретиноид, второй липид, стабилизирующий агент и/или терапевтический агент) могут быть изготовлены совместно с подходящими полимерными или гидрофобными веществами (например, в виде эмульсии в подходящем масле), или ионообменными смолами, или в виде умеренно растворимых производных, например, в виде умеренно растворимой соли.
Некоторые варианты выполнения настоящего изобретения направлены на способ доставки терапевтического агента в клетку. Например, некоторые варианты выполнения настоящего изобретения направлены на способ доставки терапевтического агента, такого как siPHK, в клетку. Подходящие клетки для применения согласно способам, описанным в настоящей заявке, включают прокариоты, дрожи или высшие эукариотические клетки, включая растительные и животные клетки (например, клетки млекопитающих). В некоторых вариантах выполнения настоящего изобретения, клетками могут быть клетки фибросаркомы человека (например, HT1080 клеточная линия). В других вариантах выполнения настоящего изобретения клетками могут быть раковые клетки. Клеточные линии, которые являются модельными системами для рака, могут применяться, включая, но без ограничения к этому, рак молочной железы (MCF-7, MDA-MB-438 клеточные линии), U87 клеточная линия глиобластомы, B16F0 клетки (меланома), HeLa клетки (рак шейки матки), A549 клетки (рак легких), и клеточная линия опухоли крысы GH3 и 9L. В этих вариантах выполнения настоящего изобретения композиции, описанные в настоящем изобретении, могут применяться для трансфекции клетки. Эти варианты выполнения настоящего изобретения могут включать контакт клетки с композицией, описанной в настоящем изобретении, которая включает терапевтический агент, с доставкой таким образом терапевтического агента в клетку.
В настоящем описании указаны способы лечения состояния, характеризующегося аномальным фиброзом, которые могут включать введение терапевтически эффективного количества композиции согласно настоящему изобретению. Состояния, характеризующиеся аномальным фиброзом, могут включать раковое и/или фиброзное заболевание. Виды раковых заболеваний, которые можно лечить или облегчать композицией согласно настоящему изобретению включают, но без ограничения к этому, рак легкого, рак поджелудочной железы, рак молочной железы, рак печени, рак желудка и рак толстой кишки. В одном варианте выполнения настоящего изобретения раковое заболевание, которое можно лечить или облегчать, представляет собой рак поджелудочной железы. В другом варианте выполнения настоящего изобретения раковое заболевание, которое можно лечить или облегчать, представляет собой рак легкого. Виды фиброзного заболевания, которые можно лечить или облегчать терапевтической композицией, указанной в настоящем описании, включают, но не ограничиваются следующими: фиброз печени, цирроз печени, панкреатит, фиброз поджелудочной железы, кистозный фиброз, рубцевание голосовых связок, фиброз слизистой голосовых связок, фиброз гортани, фиброз легких, идиопатический фиброз легких, кистозный фиброз, миелофиброз, ретроперитонеальный фиброз и нефрогенный системный фиброз. В варианте выполнения настоящего изобретения состояние, которое можно лечить или облегчать, представляет собой фиброз печени.
Композиции или фармацевтические композиции согласно настоящему изобретению могут быть введены субъекту любыми подходящими способами. Неограничивающие примеры способов введения включают в том числе: (a) введение путем инъекции, подкожно, интраперитонеально, внутривенно, внутримышечно, внутрикожно, интраорбитально, интракапсулярно, интраспинально, интрастернально или тому подобное, включая доставку посредством инфузионной помпы; (b) локальное введение, например, путем инъекции напрямую в область почки или сердца, например, путем имплантации депо; а также местное введение; по усмотрению специалиста в данной области для приведения активного соединения в контакт с живой тканью.
Фармацевтические композиции, подходящие для введения, включают композиции (например, композиция, которая может включать соединение, ретиноид, второй липид, стабилизирующий агент и/или терапевтический агент), в которых активные компоненты содержатся в количестве, эффективном для достижения намеченной цели. Терапевтически эффективное количество соединений, указанных в настоящем описании, необходимое в качестве дозы, будет зависеть от способа введения, вида животного, включая человека, которого лечат, и физических характеристик конкретного рассматриваемого животного. Доза может быть скорректирована для достижения необходимого эффекта, но будет зависеть от таких факторов, как масса, питание, сопутствующее лечение, и других факторов, очевидных для специалиста в области медицины. Более конкретно, «терапевтически эффективное количество» означает количество соединения, эффективное для предупреждения, уменьшения или облегчения симптомов заболевания или увеличения продолжительности жизни субъекта, которого лечат. Определение терапевтически эффективного количества находится в пределах возможностей специалиста в данной области техники, особенно в свете подробного описания, предложенного в настоящем документе.
Для специалиста в данной области техники очевидно, что подходящая дозировка in vivo для введения и конкретный способ введения будут варьировать в зависимости от возраста, массы и вида млекопитающего, которого лечат, конкретных используемых соединений и конкретного назначения, для которого используют данные соединения. Определение эффективных уровней дозировки, т.е. уровней дозировки, необходимых для достижения необходимого результата, может быть выполнено специалистом в данной области техники с использованием стандартных способов фармакологии. Как правило, клиническое применение продуктов у человека начинают на более низких уровнях дозировки и уровень дозировки увеличивают до достижения необходимого эффекта. В качестве альтернативы подходящие исследования in vitro могут быть использованы для определения подходящих дозировок и способов введения композиций, идентифицированных настоящими способами с использованием известных способов фармакологии.
В исследованиях с участием животных, не относящихся к человеку, применение потенциальных продуктов начинают на более высоких уровнях дозировки и дозировку уменьшают до тех пор пока становится уже невозможным достичь необходимого эффекта или пока не исчезают неблагоприятные побочные эффекты. Дозировка может широко варьировать в зависимости от необходимых эффектов и терапевтического показания. Как правило, дозы могут составлять от примерно 10 микрограмм/кг до примерно 100 мг/кг массы тела, предпочтительно от примерно 100 мк/кг до примерно 10 мг/кг массы тела. В качестве альтернативы дозы могут быть основаны и рассчитаны исходя из площади поверхности тела пациента, как известно специалисту в данной области техники.
Точный состав, способ введения и дозировка фармацевтических композиций могут быть выбраны лечащим врачом с учетом состояния пациента, (смотрите, например, Fingl et al. 1975, in "The Pharmacological Basis of Therapeutics", полностью включенный в настоящее описание посредством ссылки, в частности гл. 1, стр. 1). Как правило, диапазон доз композиции, вводимой пациенту, может составлять от примерно 0.5 до примерно 1000 мг/кг массы тела пациента. Доза может представлять собой однократную дозу или серию двух или более доз, вводимых в течение одного или более дней, в зависимости от того, что необходимо пациенту. В тех случаях, когда человеческие дозы соединений были определены по меньшей мере для какого-либо состояния, дозы будут приблизительно такими же или составлять от примерно 0.1% до примерно 500%, более предпочтительно от примерно 25% до примерно 250% от определенной человеческой дозы. В случае, когда человеческая доза не определена, как будет в случае вновь открываемых фармацевтических композиций, подходящая человеческая доза может быть получена из значений ED50 или ID50 или других подходящих значений, полученных в результате исследований in vitro или in vivo, определенных исследованиями токсичности и исследованиями эффективности у животных.
Следует отметить, что лечащему врачу известно, как и когда прекращать, прерывать или корректировать введение из-за токсичности или дисфункций органов. Напротив, лечащему врачу также известно, как перевести лечение на более высокие уровни, если не был получен достаточный клинический ответ (исключая токсичность). Величина вводимой дозы в лечении представляющего интерес расстройства будет варьировать в зависимости от тяжести состояния, которое лечат, и способа введения. Тяжесть состояния может, например, быть оценена отчасти стандартными прогностическими способами оценки. Кроме того, доза и, возможно, частота введения дозы, также будут варьировать в зависимости от возраста, массы тела и реакции конкретного пациента. Программа, сопоставимая с описанной выше, может быть использована в ветеринарии.
Несмотря на то что точная дозировка будет определена в зависимости от конкретного лекарственного средства, в большинстве случаев можно сделать некоторые обобщения относительно дозировки. Ежедневный режим приема для взрослого пациента-человека может представлять собой, например, пероральную дозу, равную от примерно 0.1 мг до примерно 2000 мг каждого активного компонента, предпочтительно от примерно 1 мг до примерно 500 мг, например, от 5 до 200 мг. В других вариантах реализации используют внутривенную, подкожную или внутримышечную дозу каждого активного компонента, равную от примерно 0.01 мг до примерно 100 мг, предпочтительно от примерно 0.1 мг до примерно 60 мг, например, от примерно 1 до примерно 40 мг. В случаях введения фармацевтически приемлемой соли, дозы могут быть рассчитаны в виде свободного основания. В некоторых вариантах реализации композицию вводят от 1 до 4 раз в сутки. В качестве альтернативы композиции могут быть введены путем непрерывной внутривенной инфузии, предпочтительно в дозировке каждого активного компонента до примерно 1000 мг в сутки. Для специалиста в данной области техники очевидно, что в некоторых ситуациях может быть необходимым введение соединений, указанных в настоящем описании, в количествах, превышающих или даже значительно превышающих вышеприведенный предпочтительный диапазон доз для эффективного и «агрессивного» лечения наиболее агрессивных заболеваний или инфекций. В некоторых вариантах реализации соединения будут вводить на протяжении непрерывной терапии, например, в течение недели или больше, или в течение нескольких месяцев или лет.
Величина дозы и интервал могут быть скорректированы индивидуально для обеспечения уровней активного вещества в плазме, достаточных для поддержания модулирующих эффектов или минимальной эффективной концентрации (МЭК). МЭК будет варьировать для каждого соединения, но может быть определена исходя из данных in vitro. Дозы, необходимые для достижения МЭК, будет зависеть от индивидуальных особенностей и способа введения. Однако анализы ВЭЖХ или биоанализы могут быть использованы для определения концентраций в плазме.
Интервалы между введением лекарственного средства также могут быть определены с использованием значения МЭК. Композиции должны быть введены с использованием режима, при котором поддерживаются уровни в плазме выше МЭК в течение 10-90% времени, предпочтительно 30-90% и наиболее предпочтительно 50-90%.
В случаях местного введения или селективного поглощения эффективная местная концентрация лекарственного средства может быть не связана с концентрацией в плазме.
Количество вводимой композиции может зависеть от подлежащего лечению субъекта, массы субъекта, тяжести заболевания, способа введения и решения лечащего врача.
Композиции согласно настоящему изобретению (например, композиция, которая может включать соединение, ретиноид, второй липид, стабилизирующий агент и/или терапевтический агент) могут быть оценены на предмет эффективности и токсичности с использованием известных способов. Например, токсикология конкретного соединения или подмножества соединений, обладающих некоторыми общими химическими группами, может быть установлена путем определения токсичности in vitro в отношении линии клеток, такой как линия клеток млекопитающего и предпочтительно человека. Результаты указанных исследований часто прогнозируют токсичность у животных, таких как млекопитающие или, в частности, люди. В качестве альтернативы токсичность конкретных соединений в модели у животных, таких как мыши, крысы, кролики или обезьяны, может быть определена с использованием известных способов. Эффективность конкретного соединения может быть определена с использованием некоторых известных способов, таких как способы in vitro, моделей у животных или клинических испытаний с участием человека. Существуют известные модели in vitro почти для каждой группы состояния, включая, но не ограничиваясь следующими: раковое заболевание, сердечнососудистое заболевание и различные виды иммунной дисфункции. Подобным образом подходящие модели у животных могут быть использованы для определения эффективности химических соединений для лечения указанных состояний. При выборе модели для определения эффективности специалист может руководствоваться существующим уровнем техники для выбора подходящей модели, дозы и способа введения, и режима. Конечно, клинические испытания с участием человека также могут быть использованы для определения эффективности соединения у людей.
При необходимости композиции могут быть предложены в упаковке или устройстве для распыления, которое может содержать одну или более дозированных лекарственных форм, содержащих активный компонент. Указанная упаковка может, например, включать металлическую фольгу или пластиковую пленку, как, например, блистерная упаковка. К упаковке или устройству для распыления может прилагаться инструкция по применению. К упаковке или устройству для распыления также может прилагаться памятка, содержащаяся в контейнере, в форме, предусмотренной государственным органом по контролю производства, применения или продажи фармацевтической продукции, отражающая разрешение государственным органом указанной формы лекарственного средства для введения человеку или использования в ветеринарии. Указанная памятка, например, может представлять собой маркировку, разрешенную Управлением по контролю за качеством пищевых продуктов и лекарственных средств США для лекарственных средств, отпускаемых по рецепту, или вкладыш разрешенного продукта. Композиции, содержащие соединение, полученное в совместимом фармацевтическом носителе, также могут быть получены, помещены в подходящий контейнер и помечены как предназначенные для лечения указанного состояния.
Понятно, что в любом соединении, описанном в настоящем изобретении, имеющем один или более стереоцентров, если абсолютная стереохимия точно не указана, тогда каждый центр может независимо иметь R-конфигурацию или S-конфигурацию или их смесь. Таким образом, соединения согласно настоящему изобретению могут быть энантиомерно чистыми или могут представлять собой стереоизомерные смеси. Кроме того, необходимо понимать, что в любом соединении, имеющем одну или более двойных связей, создающих геометрические изомеры, которые могут быть определены как E или Z, каждая двойная связь может независимо быть Е или Z или их смесью. Подобным образом, все таутомерные формы также рассматриваются как включенные.
Настоящее изобретение может быть далее проиллюстрировано посредством следующих примеров. Эти примеры являются только иллюстративными и предназначены для ограничения настоящего изобретения.
ПРИМЕРЫ
Пример 1: Получение 2-(бис(2-(тетрадеканоилокси)этил)амино)-N-(2-гидроксиэтил)-N,N-диметил-2-оксоэтан-аминия бромид (HEDC)

Получение промежуточного соединения 1: 2,2'-(трет-бутоксикарбонилазандиил)бис(этан-2,1-диил)дитетрадеканоат (HEDC-BOC-IN)
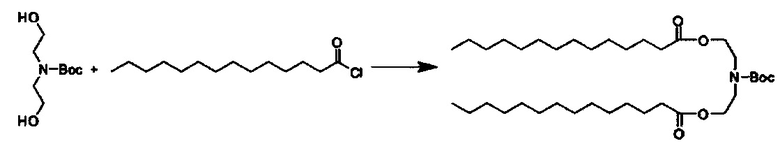
Раствор JV-BOC-диэтаноламина (194 г, 0.946 моль), триэтиламин(201 г, 2.03 моль) и диаминопиридин (23.1 г, 0.19 моль) в DCM (1750 мл) охладили до 0°C. Раствор миристоилхлорида (491 г, 1.99 моль) в DCM (440 мл) добавили за период 50 минут при 0-10°C и смеси дали нагреться до температуры окружающей среды. Полное превращение определили посредством ТСХ через 1.5 часа 20-24°C. Воду (1750 мл) добавили и pH 8.3 измеряли посредством измерителя pH. Органическую фазу отделили, промыли с помощью (1) 6% NaHCO3 (500 мл), (2) 0.3 М HCl (1700 мл), (3) 12.5% хлорида натрия (1700 мл) и высушили с помощью безводного сульфата магния (120 г). Выпаривание фильтрата при 50°C и 50 мБар позволило получить 622 г 2,2,-(трет-бутоксикарбонилазандиил)бис(этан-2,1-диил) дитетрадеканоата (HEDC-BOC-IN). Этот остаток дистилляции, который отвердился при отстаивании, применяли на следующей стадии.
Получение промежуточного соединения 2: 2,2'-(трет-бутоксикарбонилазандиил)бис(этан-2,1-диил)дитетрадеканоата (HEDC-амин-IN) TFA соли
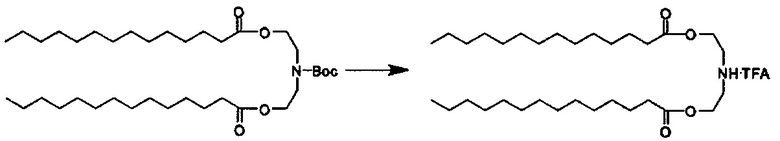
HEDC-BOC-IN (620 г, 0.990 моль) переведи в жидкую форму посредством короткого нагревания на 50°C-бане и затем охладили ниже 25°C. TFA (940 мл, 1.39 кг, 1.18 моль) добавили в жидкость за период 30 минут, с применением умеренного охлаждения для того, чтобы поддерживать температуру не выше 25°C. Добавив две трети TFA, наблюдали значительное выделение газа. Реакционную смесь перемешивали всю ночь при температуре окружающей среды. TCX показала следы HEDC-BOC-IN. Реакционную смесь нагрели для дистилляции TFA при пониженном давлении (125-60 мБар) на водяной бане 50-55°C и дистилляцию проводили до тех пор, пока она не стала очень медленной. TFA пара абсорбировались в скруббере с 10% гидроксидом натрия. Гептан (2000 мл) добавили, перемешали и отогнали при пониженном давлении. Гептан (2000 мл) добавили по частям к отвержденному остатку и смесь нагрели до 45°C, при этой температуре образовался слегка мутный раствор. Раствор охладили, кристаллизовали при 40°C и получили осадок посредством перемешивания в течение 25 минут при 40-36°C. После охлаждения и перемешивания в течение 40 минут при температуре окружающей среды, осадок тяжелых кристаллов отделили посредством фильтрации и осадок на фильтре промыли с помощью гептан (1000 мл). Влажный осадок на фильтре (914 г) высушивали всю ночь при температуре окружающей среды при пониженном давлении (<1 мБар) с получением 635 г (100%) 2,2'-(трет-бутоксикарбонилазандиил)бис(этан-2,1-диил)дитетрадеканоат (HEDC-aMHH-IN) TFA соли в виде белых кристаллов.
Получение промежуточного соединения 3: 2,2'-(2-(диметиламино)ацетилазандиил)бис(этан-2,1-диил)дитетрадеканоата (HEDC-DiMeGly-IN)
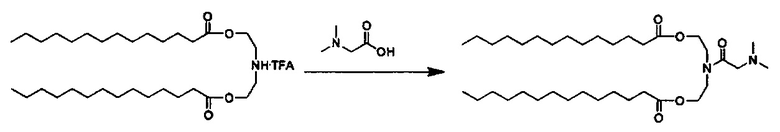
N,N-Диметилглицин (56.6 г, 548 ммоль), HOBt гидрат (83.9 г, 548 ммоль) и EDC гидрохлорид (105 г, 548 ммоль) добавили к DMF (3.5 л) и смесь перемешали при температуре окружающей среды в течение одного часа. Образовался чистый раствор. HEDC-амин-IN TFA соль (270 г, 442 ммоль) смешали с DCM (1.15 л) и 6% бикарбонатом натрия (1.15 л). Отделенную органическую фазу со свободным амином добавили к связывающей смеси в DMF, и наблюдалось осаждение, а также повышение температуры на около 9°C. Триэтиламин (47.0 г, 464 ммоль) добавили и реакционную смесь перемешали при 25-30°C в течение пяти часов. TCX показала неполное превращение и дополнительный EDC гидрохлорид (29.5 г, 154 ммоль) добавили. После перемешивания в течение всей ночи при температуре окружающей среды, получили чистый раствор, и теперь TCX TLC показала полное превращение. Реакционную смесь смешали с DCM (2.3 л) и 2% бикарбонатом натрия (7 л). Органическую фазу дважды промыли с помощью 1.25% хлорида натрия (5 л каждый раз) и высушили с помощью безводного сульфата магния (186 г). Фильтрат выпарили при 50°C и 30 мБар с получением 253 г неочищенного масла. Неочищенное вещество загрузили на колонку, набитую 2.6 кг силикагеля 60 (40-63μ). Продукт элюировали с помощью толуола : этилацетата (8:2) (4 л) и затем с помощью этилацетата : метанола (1:1) (5 л). Содержащий фракции продукт (3.5-8 л) выпарили (50°C/250-20 мБар) с получением 208 г (66%) 2,2'-(2-(диметиламино)ацетилазандиил)бис(этан-2,1-диил)дитетрадеканоата (HEDC-DiMeGly-IN) в виде масла.
Получение HEDC: 2-(бис(2-(тетрадеканоилокси)этил)амино)-N-(2-гидроксиэтил)-N,N-диметил-2-оксетанаминия бромида
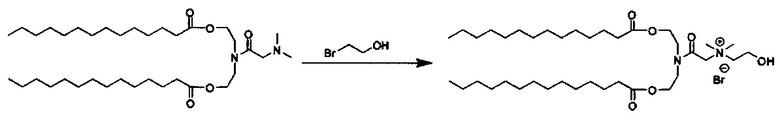
Смесь HEDC-DiMeGly-IN (206 г, 337 ммоль) и 2-бромэтанола (274 г, 2.19 моль) перемешали при 80°C в течение двух часов. ВЭЖХ показала 8.1% непрореагировавшего диметилглицин-промежуточного соединения. После дополнительного периода в 40 минут при 80°C, ВЭЖХ показала 7.8% непрореагировавшего диметилглицин промежуточного соединения. Теплый (65°C) этил ацетат (2 л) добавили. Холостой фильтрат горячего раствора промыли с помощью горячего этил ацетата (0.5 л). Объединенные фильтраты охладили до 0°C и инициировали кристаллизацию. Суспензию продукта охладили медленно и перемешали при от -16 до -18°C в течение 40 минут. Осадок отделили посредством фильтрации и осадок на фильтре промыли с помощью холодного этил ацетата (200 мл). Высушивание в течение всей ночи (20°C/<1 мБар) обеспечило 211 г неочищенного вещества. Вещество затем перекристаллизовали из смеси этил ацетата (2.1 л) и этанола (105 мл) посредством нагревания до 35°C и затравливания при 25°C. Осадок выделили при 10°C, промыли с помощью холодного этил ацетат (300 мл) и высушивали (20°C/<1 мБар) всю ночь с получением 161 г (66%) 2-(бис(2-(тетрадеканоилокси)этил)амино)-N-(2-гидроксиэтил)-N,N-диметил-2-оксоэтанаминия бромида (HEDC). ВЭЖХ показала 99.5% чистоту. QTOF MS ESI+: m/z 655.6 (M+H).
Пример 2: Получение 2-(бис(3-(тетрадеканоилокси)пропил)амино)-N-(2-гидроксиэтил)-N,N-диметил-2-оксо-этанаминия бромида (Pr-HEDC)
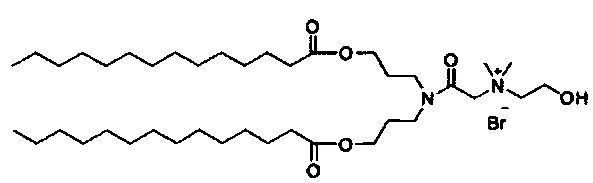
Получение промежуточного соединения 1: 3,3'-азандиилбис(пропан-1-ол)
Смесь 3-амино-1-пропанола (14.5 мл, 19.0 ммоль), 1-хлор-3-гидрокси пропана (8 мл, 95.6 ммоль) и H2O (-50 мл) нагревали с возвратом флегмы в течение 24 часов. Затем добавили гидроксид калия (5.40 г). После растворения, всю H2O выпарили с получением вязкого масла и большого количества хлорида калия. Их отфильтровали и промыли с помощью сухого ацетона и дихлорметана. Органическую фазу высушили над Na2SO4, отфильтровали и концентрировали. Продукт затем очистили с помощью хроматографии на силикагеле с применением DCM/MeOH градиента с получением 12.5 г 3,3'-азандиилбис(пропан-1-ол).
Получение промежуточного соединения 2: трет-бутилбис(3-гидроксипропил)карбамат
3,3'-Азандиилбис(пропан-1-ол) (12.5 г, 95.4 ммоль) разбавили DCM (25 мл). Раствор ди-трет-бутилдикарбоната (26 г, 119.25 ммоль) в DCM (25 мл) медленно добавили при перемешивании под покровом Ar газа. Реакционную смесь перемешивали всю ночь. Реакционную смесь концентрировали. Очищение посредством хроматографии на силикагеле с применением DCM/MeOH градиента дало трет-бутилбис(3-гидроксипропил)карбамат.
Получение промежуточного соединения 3: ((трет-бутоксикарбонил)азандиил)бис(пропан-3,1-диил) дитетрадеканоата
трет-Бутилбис(3-гидроксипропил)карбамат (4.00 г, 17.3 ммоль), триэтиламин (4.80 мл, 34.6 ммоль) и 4-диметиламинопиридин (529 мг, 4.33 ммоль) растворили в хлороформе (50 мл). При перемешивании на ледяной бане, раствор миристоилхлорида добавили за ~15 минут. Добавление осуществляли таким путем, что температура реакции не превысила 30°C. Реакционную смесь перемешивали при комнатной температуре всю ночь. На следующий день MeOH (50 мл) и 0.9% соляной раствор (50 мл) добавили для погашения реакции. Органический слой отделили и промыли с помощью 1М NaHCO3. Растворитель высушили с помощью Na2SO4, отфильтровали и концентрировали in vacuo с получением ((трет-бутоксикарбони л)азан диил)бис(пропан-3,1-диил)дитетрадеканоата в виде масла.
Получение промежуточного соединения 4: азандиилбис(пропан-3,1-диил)дитетрадеканоат TFA соли
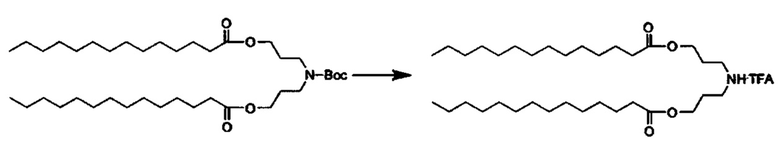
((трет-бутоксикарбонил)азандиил)бис(пропан-3,1-диил)дитетрадеканоат (11.3 г, 17.3 ммоль) растворили в TFA/CHCl3 (1:1, 20 мл) и смесь перемешивали при комнатной температуре в течение 15 минут. Смесь затем концентрировали in vacuo. Это повторили два раза. Осадок затем растворили в DCM и промыли с помощью H2O, высушили с помощью Na2SO4 и концентрировали in vacuo. Очищение посредством хроматографии на силикагеле с применением DCM/MeOH градиента дало азандиилбис(пропан-3,1-диил) дитетрадеканоат в виде TFA соли (750 мг).
Получение промежуточного соединения 5: ((2-(диметиламино)ацетил)азандиил)бис(пропан-3,1-диил)дитетрадеканоата
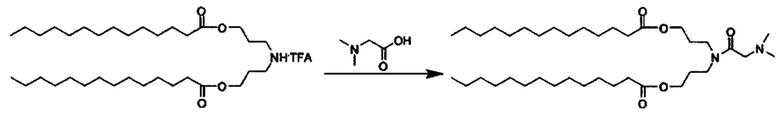
Азандиилбис(пропан-3,1-диил) дитетрадеканоат TFA соль (750 мг, 1.35 ммоль) разбавили с помощью DCM (5 мл) и добавили к предварительно активированной смеси N,N-диметилглицина (154 мг, 1.49 ммоль), HATU (616 мг, 1.62 ммоль) и DIEA (495 мкл, 2.84 ммоль) в DCM (5 мл). Продукт продули аргоном и перемешали при комнатной температуре всю ночь и затем концентрировали. Очищение посредством хроматографии на силикагеле с применением DCM/MeOH градиента дало 465 мг ((2-(диметиламино)ацетил)азандиил)бис(пропан-3,1-диил)дитетрадеканоата.
Получение Pr-HEDC: 2-(бис(3-(тетрадеканоилокси)пропил)амино)-N-(2-гидроксиэтил)-N,N-диметил-2-оксоэтанаминия бромида
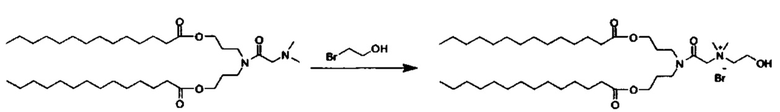
В герметизированной системе, ((2-(диметиламино)ацетил)азандиил)бис(пропан-3,1-диил)дитетрадеканоат (246 мг, 0.385 ммоль) растворили в ACN (10 мл) и 2-бромэтанола (500 мкл) добавили. Реакционный сосуд продули инертным газом и затем запаяли. Смесь нагрели до 80°C, перемешивали всю ночь и затем охладили и концентрировали in vacuo. Очищение посредством хроматографии на силикагеле с применением DCM/MeOH градиента дало 99 мг 2-(бис(3-(тетрадеканоилокси)пропил)амино)-N-(2-гидроксиэтил)-N,N-диметил-2-оксоэтанаминия бромида. QTOF MS ESI+: m/z 683.6 (M+H).
Пример 3: Получение 2-(бис(3-(олеоилокси)пропил)амино)-N-(2-гидроксиэтил)-N,N-диметил-2-оксоэтанаминия бромида (Pr-HE-DODC)
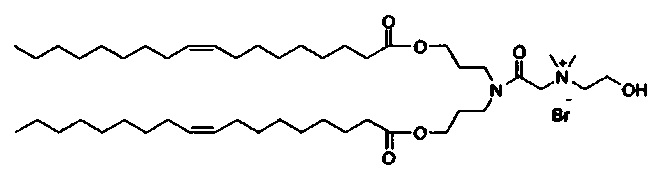
Получение промежуточного соединения 1: (Z)-((трет-бутоксикарбонил)азандиил)бис(пропан-3,1-диил)диолеата
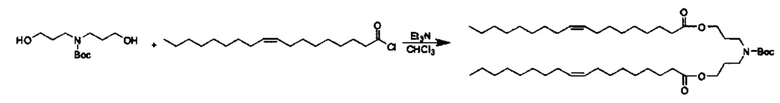
трет-Бутилбис(3-гидроксипропил)карбамат (синтез описан ранее), триэтиламин и DMAP растворили в хлороформе. При перемешивании на ледяной бане, раствор олеилхлорида добавили через 15 минут. Добавление осуществляли таким путем, что температура реакции не превысила 30°C. Реакционную смесь перемешивали при комнатной температуре всю ночь. На следующий день MeOH (50 мл) и 0.9% соляной раствор (50 мл) добавили для погашения реакции. Органический слой отделили и промыли с помощью 1М NaHCO3. Растворитель высушили с помощью Na2SO4, отфильтровали и концентрировали с получением масла. (Z)-((трет-бутоксикарбонил)азандиил)бис(пропан-3,1-диил) диолеат применяли без дальнейшей очистки.
Получение промежуточного соединения 2: (Z)-азандиилбис(пропан-3,1-диил)диолеат TFA соли
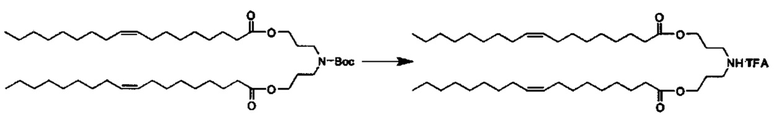
(Z)-((трет-Бутоксикарбонил)азандиил)бис(пропан-3,1-диил)диолеат (13.2 г, 17.3 ммоль) растворили в TFA/CHCl3 (1:1, 20 мл) и смесь перемешивали при комнатной температуре в течение 15 минут. Смесь затем концентрировали in vacuo. Это повторили два раза. Осадок затем растворили в DCM и промыли с помощью H2O, высушили с помощью Na2SO4 и концентрировали. Очищение посредством хроматографии на силикагеле с применением DCM/MeOH градиента дало (Z)-азандиилбис(пропан-3,1-диил) диолеат TFA соль (750 мг).
Получение промежуточного соединения 3: (Z)-((2-(диметиламино)ацетил)азандиил)бис(пропан-3,1-диил)диолеата
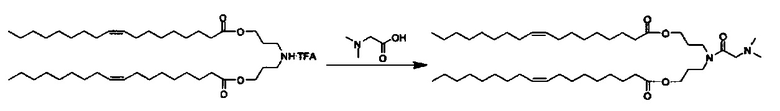
(Z)-азандиилбис(пропан-3,1-диил)диолеат TFA соль (750 мг, 1.13 ммоль) разбавили с помощью DCM (5 мл) и добавили к предварительно активированной смеси N,N-диметилглицин (128 мг, 1.24 ммоль), HATU (517 мг, 1.36 ммоль) и DIEA (413 мкл, 2.37 ммоль) в DCM (5 мл). Колбу продули аргоном и перемешивали при комнатной температуре всю ночь. Реакционную смесь концентрировали и подвергли хроматографии на силикагеле с применением DCM/MeOH градиента с получением (Z)-((2-(диметиламино)ацетил)азандиил)бис(пропан-3,1-диил)диолеата.
Получение Pr-HE-DODC: 2-(бис(3-(олеоилокси)пропил)амино)-N-(2-гидроксиэтил)-N,N-диметил-2-оксоэтанаминия бромида
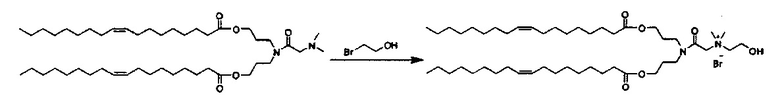
В герметизированной системе, (Z)-((2-(диметиламино)ацетил)азандиил)бис(пропан-3,1-диил) диолеат (269 мг, 0.360 ммоль) растворили в ACN (10 мл) и 2-Бромэтанол (200 мкл) добавили. Реакционный сосуд продули инертным газом и затем запаяли. Реакционную смесь нагрели до 80°C и перемешивали всю ночь. Реакционную смесь охладили и концентрировали. Очищение посредством хроматографии на силикагеле с применением DCM/MeOH градиента дало 2-(бис(3-(олеоилокси)пропил)амино)-N-(2-гидроксиэтил)-N,N-диметил-2-оксоэтанаминия бромид (129 мг). QTOF MS ESI+: m/z 791.7 (M+H).
Пример 4: Получение 3-(бис(2-(тетрадеканоилокси)этил)амино)-N-(2-гидроксиэтил)-N,N-диметил-3-оксорго-пан-1-аминия бромид (HE-Et-DC)
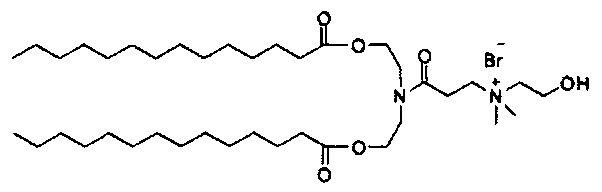
Получение промежуточного соединения 1: ((3-(диметиламино)пропаноил)азандиил)бис(этан-2,1-диил)дитетрадеканоата

Синтез азандиилбис(этан-2,1-диил) дитетрадеканоат TFA соли описан ранее. Азандиилбис(этан-2,1-диил) дитетрадеканоат TFA соль (1.5 г, 2.85 ммоль) разбавили с помощью DCM (10 мл) и добавили к предварительно активированной смеси 3-(диметиламино)пропионовой кислоты HCl соли (482 мг, 3.14 ммоль), HATU (1.30 г, 3.42 ммоль) и DIEA (1.04 мл, 5.98 ммоль) в DCM (10 мл). Круглодонную колбу продули аргоном и реакционную смесь перемешивали при комнатной температуре всю ночь. Реакционную смесь концентрировали. Очищение посредством хроматографии на силикагеле с применением DCM/MeOH градиента дало ((3-(диметиламино)пропаноил)азандиил)бис(этан-2,1-диил)дитетрадеканоат.
Получение HE-Et-DC: 3-(бис(2-(тетрадеканоилокси)этил)амино)-N-(2-гидроксиэтил)-N,N-диметил-3-оксопропан-1-аминия бромид
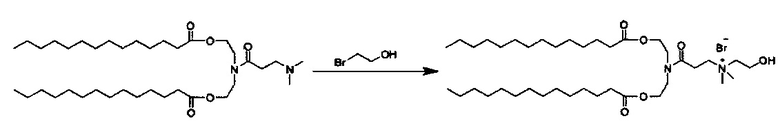
В герметизированной системе, ((3-(диметиламино)пропаноил)азандиил)бис(этан-2,1-диил) дитетрадеканоат (606 мг, 0.970 ммоль) растворили в ACN (10 мл) и 2-Бромэтанола (500 мкл) добавили. Реакционный сосуд продули инертным газом и затем запаяли. Реакционную смесь нагрели до 80°C и перемешивали всю ночь, затем охладили и концентрировали. Очищение посредством хроматографии на силикагеле с применением DCM/MeOH градиента дало 3-(бис(2-(тетрадеканоилокси)этил)амино)-N-(2-гидроксиэтил)-N,N-диметил-3-оксопропан-1-аминия бромид (80 мг). QTOF MS ESI+: m/z 669.6 (M+H).
Пример 5: Получение 3-(бис(2-(олеоилокси)этил)амино)-N-(2-гидроксиэтил)-N,N-диметил-3-оксопропан-1-аминия бромид (HR-Et-DODC)

Получение промежуточного соединения 1: (Z)-((трет-бутоксикарбонил)азандиил)бис(этан-2,1-диил)диолеата
N-Boc диэтаноламина (Aldrich 15268, Lot# 0001406013, MW 205.25; 17.81 г, 0.087 моль), триэтиламин(Aldrich, MW 101.19; 24.4 мл, 0.176 моль) и 4-(диметиламино)пиридин (Aldrich, MW 122.17; 2.76 г, 1.3 г, 0.023 моль) растворили в 350 мл хлороформа. При перемешивании, раствор олеилхлорида (MW 300.91; 61.6 г, 0.174 моль) в 100 мл хлороформа добавили через 10 минут (Альтернативно, раствор в хлороформе N-Boc диэтаноламина погрузили на ледяную/водяную баню при добавлении олеилхлорида). Добавление осуществляли таким путем, что температура реакционной смеси не превысила 50°C. Реакционную смесь перемешали при комнатной температуре в течение 2 часов. Смесь 200 мл метанола (EMD МХ0485-5, Lot 50350) и 200 мл 0.9% соляного раствора (Хлорид натрия, BDH, BDH8014, Lot# 92717) добавили для погашения реакции. Органический слой отделили и промыли с помощью 2×100 мл разбавленного водного бикарбонатом натрия (Aldrich S6014, Batch# 095К0143). Растворитель удалили посредством роторного испарителя с получением 59.5 г неочищенного продукта в виде масла бледно желтого цвета (MW 734.14; 59.5 г, 0.081 моль, 100% выход). Это вещество применяли на следующей стадии без дальнейшей очистки. 1H ЯМР (400 МГц, CDCl3) 0.87 (т, 6H, CH3), 1.20-1.40 (м, 40H, CH2), 1.45 (с, 9H, tBu CH3), 1.59 (м, 4H, CH2CH2C(=O)), 2.00(м, 8H, CH2CH=CH), 2.33 (т, 4H, CH2C(=O)), 3.48 (м, 4Н, NCH2CH2O), 4.18 (м, 4H, NCH2CH2O), 5.33 (м, 4H, CH=CH).
Получение промежуточного соединения 2: (2)-азандиилбис(этан-2,1-диил)диолеат TFA соли
(2)-((трет-бутоксикарбонил)азандиил)бис(этан-2,1-диил)диолеат (59.5 г, 0.081 моль) дважды обработали с помощью 100 мл трифторуксусной кислоты (Alfa Aesar Stock# A12198, Lot# D07W005, MW 114.02; 100 мл, 1.35 моль) и 100 мл хлороформа (Aldrich 154733, Lot# KBF5943V). Каждая обработка состояла из перемешивания при комнатной температуре в течение десяти минут и растворитель удаляли на роторном испарителе в конце каждой обработки. После второй обработки реакционную смесь концентрировали посредством роторного испарителя. Осадок растворили в 200 мл метиленхлорида и смесь промыли с помощью 100 мл воды дважды. Осадок очистили с помощью хроматографии на силикагеле с применением смеси метанола (EMD МХ0485-5, Lot 50350) и метиленхлорида (EMD DX0835-5, Lot 51090) в качестве разбавителя с получением 44 г (2)-азандиилбис(этан-2,1-диил) диолеат TFA соли (44.0 г). 1H ЯМР (400 МГц, CDCl3) 0.87 (т, 6H, CH3), 1.20-1.40 (м, 40H, CH2), 1.59 (м, 4H, CH2CH2C(=О)),2.00 (м, 8H, CH2CH=CH), 2.33 (т, 4H, CH2C(=O)), 3.31 (м, 4H, NCH2CH2O), 4.38 (м, 4Н, NCH2CH2O), 5.33 (м, 4H, CH=CH).
Получение промежуточного соединения 3: (((Z)-((3-(диметиламино)пропаноил)азандиил)бис(этан-2,1-диил) диолеата
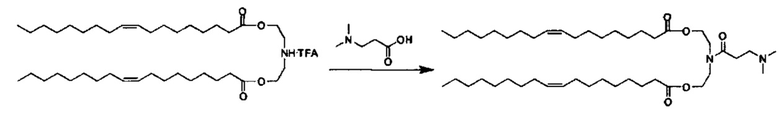
(Z)-азандиилбис(этан-2,1-диил) диолеат TFA соль (1.50 г, 2.37 ммоль) разбавили с помощью DCM (10 мл) и добавили к предварительно активированной смеси 3-(диметиламино)пропионовой кислоты HCl соли (383 мг, 2.49 ммоль), HATU (1034 мг, 2.72 ммоль) и DIEA (831 мкл, 4.77 ммоль) в DCM (10 мл). Круглодонную колбу продули аргоном и реакционную смесь перемешивали при комнатной температуре всю ночь. Реакционную смесь концентрировали. Очищение посредством хроматографии на силикагеле с применением DCM/MeOH градиента дало (Z)-((3-(диметиламино)пропаноил)азандиил)бис(этан-2,1-диил)диолеат.
Получение HE-Et-DODC: 3-(бис(2-(олеоилокси)этил)амино)-N-(2-гидроксиэтил)-N,N-диметил-3-оксо-пропан-1-аминия бромида
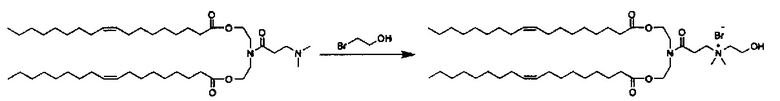
В герметизированной системе, (((Z)-((3-(диметиламино)пропаноил)азандиил)бис(этан-2,1-диил) диолеат (588 мг, 0.802 ммоль) растворили в ACN (10 мл) и 2-бромэтанол (200 мкл) добавили. Реакционный сосуд продули инертным газом и затем запаяли. Реакционную смесь нагрели до 80°C и перемешивали всю ночь, затем охладили и концентрировали in vacuo. Очищение посредством хроматографии на силикагеле с применением DCM/MeOH градиента дало 3-(бис(2-(олеоилокси)этил)амино)-N-(2-гидроксиэтил)-N,N-диметил-3-оксопропан-1-аминия бромид (160 мг). QTOF MS ESI+: m/z 764.3 (M+H).
Пример 6: Получение 4-(бис(2-(тетрадеканоилокси)этил)амино)-N-(2-гидроксиэтил)-N,N-диметил-4-оксобутан-1-аминия бромид (HE-Pr-DC)
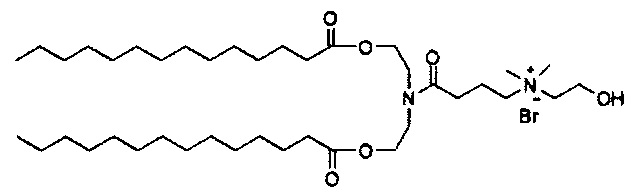
Получение промежуточного соединения 1: ((4-(диметиламино)бутаноил)азандиил)бис(этан-2,1-диил)дитетрадеканоата
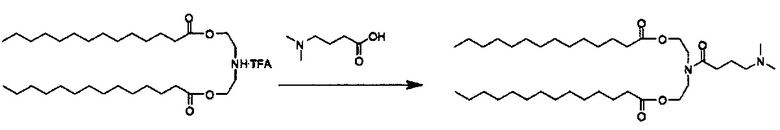
Синтез азандиилбис(этан-2,1-диил)дитетрадеканоат TFA соли описан ранее. Азандиилбис(этан-2,1-диил) дитетрадеканоат TFA соль (1.00 г, 1.90 ммоль) разбавили с помощью DCM (5 мл) и добавили к предварительно активированной смеси 4-(диметиламино) масляной кислоты HCl соли (382 мг, 2.28 ммоль), HATU (867 мг, 2.28 ммоль) и DIEA (728 мкл, 4.18 ммоль) в DCM (5 мл). Колбу продули аргоном и реакционную смесь перемешали при комнатной температуре всю ночь, затем концентрировали. Очищение посредством хроматографии на силикагеле с применением DCM/MeOH градиента дало ((4-(диметиламино)бутаноил)азандиил)бис(этан-2,1-диил) дитетрадеканоат.
Получение HE-Pr-DC: 4-(бис(2-(тетрадеканоилокси)этил)амино)-N-(2-гидроксиэтил)-N,N-диметил-4-оксобутан-1-аминия бромида
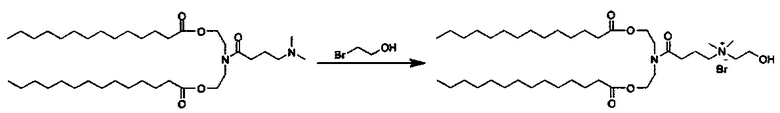
В герметизированной системе, ((4-(диметиламино)бутаноил)азандиил)бис(этан-2,1-диил)дитетрадеканоат (300 мг, 0.469 ммоль) растворили в ACN (5 мл) и 2-бромэтанол (500 мкл) добавили. Реакционный сосуд продули инертным газом и затем запаяли. Реакционную смесь нагрели до 80°C и перемешивали всю ночь, затем концентрировали. Очищение посредством хроматографии на силикагеле с применением DCM/MeOH градиента дало 4-(бис(2-(тетрадеканоилокси)этил)амино)-N-(2-гидроксиэтил)-N,N-диметил-4-оксобутан-1-аминия бромид (140 мг). LCMS ESI+: т/г 684.4 (M+H).
Пример 7: Получение 4-(бис(2-(олеоилокси)этил)амино)-N-(2-гидроксиэтил)-N,N-диметил-4-оксобутан-1-аминия бромида (HE-Pr-DODC)
Получение промежуточного соединения 1: (Z)-((4-(диметиламино)бутаноил)азандиил)бис(этан-2,1-диил)диолеат
Синтез (Z)-азандиилбис(этан-2,1-диил)диолеат TFA соли описан ранее. (Z)-азандиилбис(этан-2,1-диил) диолеат TFA соль (1.00 г, 1.58 ммоль) разбавили с помощью DCM (5 мл) и добавили к предварительно активированной смеси 4-(диметиламино) масляной кислоты HCl соли (317 мг, 1.89 ммоль), HATU (719 мг, 1.89 ммоль) и DIEA (606 мкл, 3.48 ммоль) в DCM (5 мл). Колбу продули аргоном и реакционную смесь перемешали при комнатной температуре всю ночь, затем концентрировали. Очищение посредством хроматографии на силикагеле с применением DCM/MeOH градиента дало (Z)-((4-(диметиламино)бутаноил)азандиил)бис(этан-2,1-диил) диолеат.
Получение HE-Pr-DODC: 4-(бис(2-(олеоилокси)этил)амино)-N-(2-гидроксиэтил)-N,N-диметил-4-оксобутан-1-аминия бромида
В герметизированной системе, ((4-(диметиламино)бутаноил)азандиил)бис(этан-2,1-диил)дитетрадеканоат (400 мг, 0.535 ммоль) растворили в ACN (5 мл) и 2-бромэтанол (500 мкл) добавили. Реакционный сосуд продули инертным газом и затем запаяли. Реакционную смесь нагрели до 80°C и перемешивали всю ночь, затем концентрировали. Очищение посредством хроматографии на силикагеле с помощью DCM/MeOH градиента дало 4-(бис(2-(олеоилокси)этил)амино)-N-(2-гидроксиэтил)-N,N-диметил-4-оксобутан-1-аминия бромид (255 мг). LCMS ESI+: m/z 792.5 (M+H).
Пример 8: Получение 2-(бис(2-(олеоилокси)этил)амино)-N,N-бис(2-гидроксиэтил)-N-метил-2-оксоэтанаминия бромида (HE2DODC)
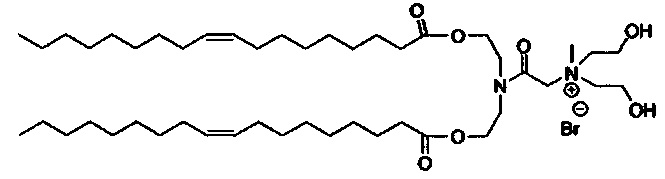
Получение промежуточного соединения 1: (Z)-((2-бромацетил)азандиил)бис(этан-2,1-диил)диолеат
Синтез (Z)-азандиилбис(этан-2,1-диил)диолеат TFA соли описан ранее. (Z)-азандиилбис(этан-2,1-диил) диолеат TFA соль (1.50 г, 2.34 ммоль) растворили в DCM (20 мл) и разместили на ледяной бане. Бромацетилбромид (214 мкл, 2.46 ммоль) добавили с последующим добавлением триэтиламина (685 мкл, 4.91 ммоль). Ледяную баню удалили и реакционную смесь перемешивали всю ночь при комнатной температуре в атмосфере инертного газа, затем разбавили с помощью DCM до 100 мл и промыли с помощью 1М HCl (75 мл), H2O (75 мл), насыщенного NaHCO3 раствора (75 мл) и насыщенного раствора хлористого натрия (75 мл). Все водные промывки обратно экстрагировали с помощью DCM (25 мл). Высушенные с помощью MgSO4 органические фазы отфильтровали и концентрировали in vacuo. Очищение посредством хроматографии на силикагеле с помощью этил ацетата дало (2)-((2-бромацетил)азандиил)бис(этан-2,1-диил)диолеат (1.22 г).
Получение HE2DODC: 2-(бис(2-(олеоилокси)этил)амино)-N,N-бис(2-гидроксиэтал)-N-метил-2-оксоэтанаминия бромид
В герметизированной системе, (2)-((2-бромацетил)азандиил)бис(этан-2,1-диил) диолеат (2.08 г, 2.75 ммоль), объединенный с N-метилдиэтиламином (1.58 мл, 13.8 ммоль), добавили. Реакционный сосуд продули инертным газом и затем запаяли. Реакционную смесь нагрели до 50°C и перемешивали всю ночь и затем концентрировали. Очищение посредством хроматографии на силикагеле с помощью DCM/MeOH градиента дало 2-(бис(2-(олеоилокси)этил)амино)-N,N-бис(2-гидроксиэтил)-N-метил-2-оксоэтанаминия бромид (479 мг). LCMS ESI+: m/z 793.7 (M+H).
Пример 9: Получение 2-(бис(2-((9Z,12Z)-октадека-9,12-диеноилокси)этил)амино)-N-(2-гвдроксиэтш)-N,N-диметил-2-оксоэтанаминия бромид (HEDC-DLin)
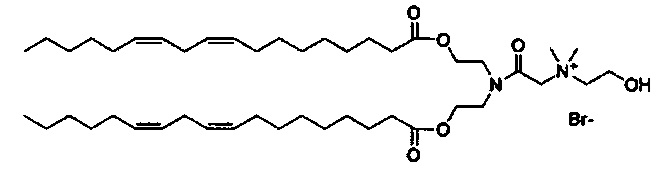
2-(бис(2-((9Z,12Z)-октадека-9,12-диеноилокси)этил)амино)-N-(2-гидроксиэтил)-N,N-диметил-2-оксоэтанаминия бромид получили подобным образом как и HEDC с замещением (9Z,12Z)-октадека-9,12-диеноилхлорида на миристоилхлорид.
Пример 10: Получение 2-(бис(2-(додеканоилокси)этил)амино)-N-(2-гидроксиэтил)-N,N-диметил-2-оксоэтанаминия бромид (HEDC-12)
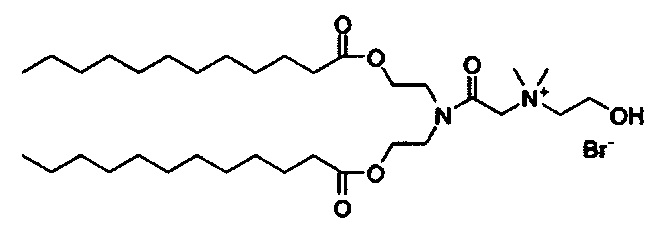
2-(бис(2-(додеканоилокси)этал)амино)-N-(2-гидроксиэтил)-N,N-диметил-2-оксоэтанаминия бромид получили подобным образом как и HEDC с замещением додеканоилхлорида на миристоилхлорид.
Пример 11: Получение 2-((2-(бис(2-(тетрадеканоилокси)этил)амино)-2-оксоэтил)тио)-N-(2-гидроксиэтил)-N,N-диметилэтанаминия бромид (HES104)
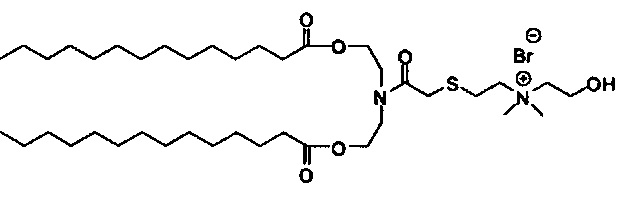
Получение промежуточного соединения 1: ((2-((2-(диметиламино)этил)тио)ацетил)азандиил)бис(этан-2,1-диил)дитетрадеканоат (S104).

Синтез азандиилбис(этан-2,1-диил) дитетрадеканоат TFA соли описан ранее. Азандиилбис(этан-2,1-даил) дитетрадеканоат TFA соль (152 г, 238 ммоль) перемешали с DCM (2.3 л) и 10% бикарбонатом калия (1.15 л) при 0-5°C. Органическую фазу отделили и водную фазу затем экстрагировали с помощью DCM (1.15 л). Объединенные органические фазы перемешали с гидраотм сульфата магния (236 г) в течение 30 минут при 0-5°C, отфильтровали и промыли с помощью DCM (1.15 л). К объединенным фильтратам добавили 2-((2-(диметиламино)этил)тио)уксусной кислоты гидрохлорид (57.0 г, 285 ммоль), EDC гидрохлорид (68.4 г, 357 ммоль) и DMAP (2.91 г, 23.8 ммоль) и суспензию перемешивали всю ночь при температуре окружающей среды, после этого периода времени был получен чистый раствор. MQ-воду (2.3 л) и метанол (460 мл) добавили и после перемешивания в течение 10 минут отделили чистую органическую фазу. Мутную водную фазу (pH 3.0) экстрагировали с помощью DCM (575 мл). Объединенные органические экстракты концентрировали с получением 143 г неочищенного вещества в виде соли гидрохлорида. Неочищенное вещество (142.6 g) перенесли в дистилляционную колбу с DCM (500 мл) и этил ацетат (1 л) добавили. Раствор нагрели о дистилляции при атмосферном давлении и дистилляцию проводили за период 70 минут, для того чтобы получить температуру остатка 76°C. Общий объем 1.4 л был получен посредством добавления этилацетата (800 мл) и этанол (70 мл) добавили. Чистый раствор при 50°C охладили до 37°C и добавили затравочные кристаллы. Наблюдая инициацию значительной кристаллизации за период 10 минут при 37-35°C, суспензию охладили и перемешивали при 0°C всю ночь и осадок отделили посредством фильтрации и промыли с помощью холодного этилацетата (210 мл). Высушивание до постоянной массы при температуре окружающей среды в масляном вакуумном насосе за период 4.5 часов дало 134 г перекристаллизованного вещества в виде соли гидрохлорида, белое кристаллическое твердое вещество. Фосфат трикалия (85 г, 0.40 моль) и гидрофосфат дикалия (226 г, 1.30 моль) добавили в очищенную воду (1.7 л) и образованный раствор с pH 10.9 охладили до 18-20°C. DCM (1.3 л) и перекристаллизованный S104 гидрохлорид (133 г, 0.188 моль) добавили и смесь перемешали в течение 10 минут. Чистую органическую фазу удалили при умеренной скорости (за период 35 минут) и мутную водную фазу далее экстрагировали с помощью DCM (650 мл). Объединенные органические фазы перемешали с безводным сульфатом натрия (65 г) в течение 40 минут и смесь отфильтровали, промывая с помощью DCM (200 мл). Объединенные фильтраты выпарили на 50°C водяной бане при пониженном давлении (понижение до 20 мБар, при этом давлении испарение продолжалось в течение одного часа). Дополнительное испарение на 15-20°C водяной бане с применением у вакуумного масляного насоса, дало 126 г частично отвержденного масла. Охлаждение в -20°C охлаждающей бане дало полное отверждение, и после высушивания при -20°C с помощью вакуумного масляного насоса мы получили 126 г ((2-((2-(диметиламино)этил)тио)ацетил)азандиил)бис(этан-2,1-диил)дитетрадеканоата (S104). ВЭЖХ показала 98.1% чистоту.
Получение HES104: 2-((2-(бис(2-(тетрадеканоилокси)этил)амино)-2-оксоэтил)тио)-N-(2-гидроксиэтил)-N,N-диметилэтанаминия бромида
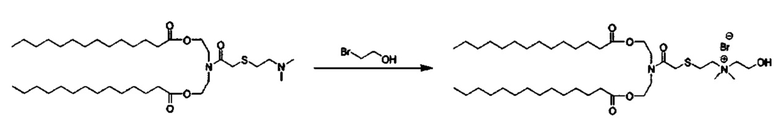
В герметизированной системе, ((2-((2-(диметиламино)этил)тио)ацетил)азандиил)бис(этан-2,1-диил)дитетрадеканоат (1.00 г, 1.49 ммоль), объединенный с 2-бромэтанолом (687 мкл, 9.69 ммоль), добавили. Реакционный сосуд продули инертным газом и затем запаяли. Реакционную смесь нагрели до 75°C и перемешивали всю ночь, затем охладили и концентрировали in vacuo. Очищение посредством хроматографии на силикагеле с помощью DCM/MeOH градиента дало 2-((2-(бис(2-(тетрадеканоилокси)этил)амино)-2-оксоэтил)тио)-N-(2-гидроксиэтил)-N,N-диметилэтанаминия бромид (HES104) (790 мг). LCMS ESI+: m/z 715.7 (M+H).
Пример 12: Получение 2-((2-(бис(2-(олеоилокси)этил)амино)-2-оксоэтил)тио)-N-(2-гтздроксиэтал)-N,N-диметилэтанаминия бромид (HES104-DO)
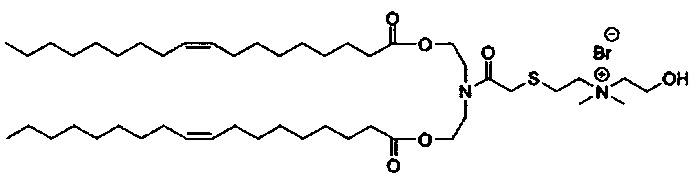
Получение промежуточного соединения 1: (Z)-((2-((2-(диметиламино)этил)тио)ацетил)азандиил)бис(этан-2,1-диил)диолеат
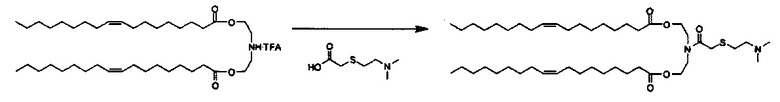
Синтез (Z)-азандиилбис(этан-2,1-диил)диолеат TFA соли описан ранее. (Z)-азандиилбис(этан-2,1-диил)диолеат TFA соль (4.06 г, 6.41 ммоль) перемешали в DCM (60 мл) с 10% K2CO3 (30 мл) при 0-5°C. Через 30 минут, органическую фазу отделили и водную фазу далее экстрагировали с помощью DCM (30 мл). Объединенные органические фазы перемешали с безводным MgSO4 в течение 30 минут при 0-5°C, отфильтровали и промыли с помощью DCM (30 мл). К объединенным фильтратам добавили 2-((2-(диметиламино)этил)тио)уксусную кислоту (1.26 г, 7.70 ммоль), EDC HCl соль (1.84 г, 9.62 ммоль), DMAP (78.3 мг, 0.64 ммоль). Тонкую суспензию перемешивали всю ночь при комнатной температуре; после этого периода времени был получен чистый раствор. На следующий день деионизированную воду (60 мл) и метанол (30 мл) добавили. После перемешивания в течение 10 минут, выделили чистый органический слой. Мутную водную фазу экстрагировали с помощью DCM. Объединенные органические экстракты концентрировали. Неочищенное вещество отфильтровали через слой оксида кремния и растворили в DCM (40 мл) и PBS (pH=11, 50 мл) добавили. Смесь перемешали при комнатной температуре в течение 10 минут. Затем органическую фазу отделили и водную фазу снова экстрагировали с помощью DCM (15 мл). Объединенные органические фазы перемешали с безводным MgSO4 в течение 30 минут. Смесь затем отфильтровали и промыли с помощью DCM. Объединенные фильтраты концентрировали in vacuo с получением (Z)-((2-((2-(диметиламино)этил)тио)ацетил)азандиил)бис(этан-2,1-диил) диолеата (3.44 г).
Получение HES104-DO
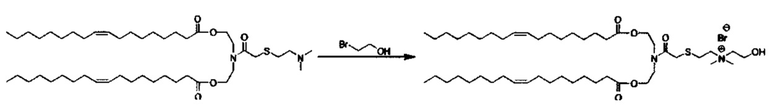
В герметизированной системе, (Z)-((2-((2-(диметиламино)этил)тио)ацетил)азандиил)бис(этан-2,1-диил)диолеат (540 мг, 0.693 ммоль) объединили с 2-бромэтанола (319 мкл, 4.50) добавили. Реакционный сосуд продули инертным газом и затем запаяли. Реакционную смесь нагрели до 75°C и перемешивали всю ночь. На следующий день охладили и концентрировали in vacuo. Очищение посредством хроматографии на силикагеле с помощью DCM/MeOH градиента дало 2-((2-(бис(2-(олеоилокси)этил)амино)-2-оксоэтил)тио)-N-(2-гидроксиэтил)-N,N-диметилэтанаминия бромид (324 мг).
LCMS ESI+: m/z 823.8 (M+H).
Пример 13: Получение 2-((бис(2-(олеоилокси)этил)карбамоил)тио)-N-(2-гидроксиэтил)-N,N-диметилэтан-аминия бромида (HETU104-DO)
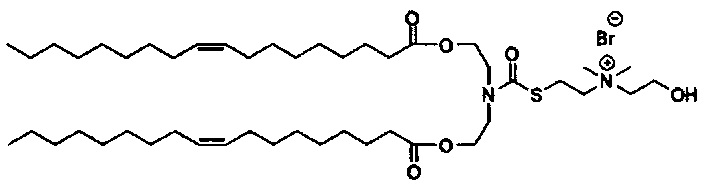
Получение промежуточного соединения 1: (Z)-((((2-(диметиламино)этил)тио)карбонил)азандиил)бис(этан-2,1-диил)диолеата
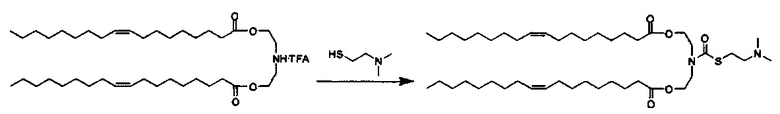
Синтез (Z)-((трет-бутоксикарбонил)азандиил)бис(этан-2,1-диил)диолеат описан ранее. (Z)-((трет-бутоксикарбонил)азандиил)бис(этан-2,1-диил)диолеат (4.2 г, 5.72 ммоль) растворили в DCM (20 мл) и охладили до 0°C на ледяной бане. TFA (20 мл) добавили и смесь перемешивали под покровом инертного газа в течение 20 минут. После этого смесь концентрировали in vacuo. Осадок распределили между 10% K2CO3 (20 мл) и DCM (20 мл). Смесь перемешали на ледяной бане в течение 20 минут. Органическую часть собрали и мутный водный слой экстрагировали с помощью DCM (2×10 мл). К объединенным органическим экстрактам добавили безводный MgSO4 и перемешали при 0°C в течение 20 минут. Суспензию отфильтровали и промыли DCM (10 мл). Дифосген (1.38 мл, 11.4 ммоль) добавили к веществу (2)-азандиилбис(этан-2,1-диил)диолеат в DCM и перемешали под покровом инертного газа при комнатной температуре. На следующий день DCM и избыток дифосгена удалили in vacuo. 2-(диметиламино)этан тиола HCl соль (4.05 г, 28.6 ммоль) растворили в DCM (50 мл) и триэтиламине (5.2 мл, 37.2 ммоль) и добавили к (Z)-((хлоркарбонил)азандиил)бис(этан-2,1-диил)диолеатному остатку. Реакционную смесь перемешивали всю ночь при комнатной температуре. На следующий день смесь разбавили с помощью DCM и промыли с помощью 0.3М HCl (75 мл), воды (75 мл) и 10% K2CO3 (75 мл). Обратно экстрагировали все водные промывки с помощью DCM (25 мл). Органическую фазу высушили над безводным MgSO4, отфильтровали и концентрировали in vacuo. Очищение посредством хроматографии на силикагеле с помощью DCM/MeOH градиента дало (Z)-((((2-(диметиламино)этил)тио)карбонил)азандиил)бис(этан-2,1-диил)диолеат (1.90 г).
Получение HETU104DO
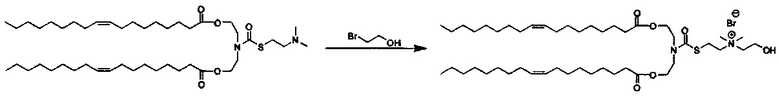
В герметизированной системе, (Z)-((((2-(диметиламино)этил)тио)карбонил)азандиил)бис(этан-2,1-диил)диолеат (615 мг, 0.804 ммоль), объединенный с 2-бромэтанолом (370 мкл, 5.22 ммоль), добавили. Реакционный сосуд продули инертным газом и затем запаяли. Реакционную смесь нагрели до 75°C и перемешивали всю ночь, затем охладили и концентрировали in vacuo. Очищение с помощью хроматографии на силикагеле с применением DCM/MeOH градиента дало 2-((бис(2-(олеоилокси)этил)карбамоил)тио)-N-(2-гидроксиэтил)-N,N-диметилэтанаминия бромид (473 мг). LCMS ESI+: m/z 809.8 (M+H).
Пример 14: Синтез Dope-Glu-VA
Получение (Z)-(2R)-3-(((2-(5-(((2E,4E,6E,8E)-3,7-диметил-9-(2,6,6-триметилциклогекс-1-ен-1-ил)нона-2,4,6,8-тетраен-1-ил)окси)-5-оксопентанамидо)этокси)(гидрокси)фосфорил)окси)пропан-1,2-диилдиолеат (DOPE-Glu-VA)
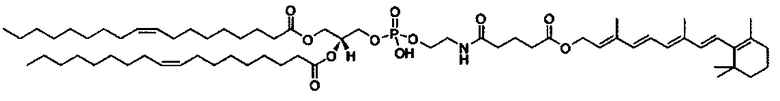
Получение промежуточного соединения 1: 5-(((2E,4E,6E,8E)-3,7-диметил-9-(2,6,6-триметилциклогекс-1-ен-1-ил)нона-2,4,6,8-тетраен-1-ил)окси)-5-оксопентановая кислота
Глутаровый альдегид (220 мг, 1.93 ммоль) и ретинол (500 мг, 1.75 ммоль) растворили в дихлорметане (5 мл) в окрашенном в янтарный цвет флаконе. Триэтиламин (513 мкл, 3.68 ммоль) добавили и флакон продули аргоном. Реакционную смесь перемешивали при комнатной температуре в течение 4 часов. Вещество концентрировали и очищение с помощью хроматографии на силикагеле с применением дихлорметан/метанол градиента. Фракции объединили и концентрировали с получением масла желтоватого цвета (700 мг). Продукт подтвердили с помощью ЯМР.
Получение DOPE-Glu-VA: (Z)-(2R)-3-(((2-(5-(((2E,4E,6E,8E)-3,7-диметил-9-(2,6,6-триметилциклогекс-1-ен-1-ил)нона-2,4,6,8-тетраен-1-ил)окси)-5-оксопентанамидо)этокси)(гидрокси)фосфорил)окси)пропан-1,2-диил диолеата
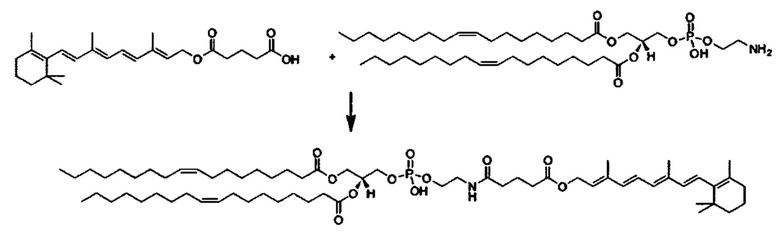
1,2-диолеил-sn-глицеро-3-фосфоэтаноламин (500 мг, 0.672 ммоль), N,N,N',N'-Тетраметил-O-(7-азабензотриазол-1-ил)урония гексафторфосфат (306.5 мг, 0.806 ммоль) и 5-(((2E,4E,6E,8E)-3,7-диметил-9-(2,6,6-триметилциклогекс-1-ен-1-ил)нона-2,4,6,8-тетраен-1-ил)окси)-5-оксопентановую кислоту (269 мг, 0.672 ммоль) растворили в хлороформе/DMF (10 мл, 1:1 смесь) в окрашенном в янтарный цвет флаконе, продутом аргоном и N,N-Диизопропилэтиламин (300 мкл, 1.68 ммоль) добавили. Реакционную смесь перемешивали всю ночь при комнатной температуре. Реакционную смесь концентрировали и затем очищали посредством хроматографии на силикагеле с применением дихлорметан/метанол градиента. Фракции объединили и концентрировали с получением масла желтоватого цвета (460 мг, 61%). Продукт подтвердили посредством ЯМР. 1H ЯМР (400 МГц), δH: 8.6 (д, 1H), 8.27 (д, 1H), 6.57-6.61 (дд, 1H), 6.08-6.25 (м, 4H), 5.57 (т, 1H), 5.30-5.34 (м, 4H), 5.18 (м, 1H), 4.68-4.70 (д, 2H), 4.28-4.35 (м, 1H), 4.05-4.15 (м, 1H), 3.81-3.97 (м, 4H), 3.52-3.62 (м, 1H), 3.35-3.45 (м, 2H), 2.95-3.05 (м, 1H), 2.33-2.35 (т, 3H), 2.2-2.3 (м, 7H), 1.9-2.05 (м, 17H), 1.85 (с, 3H), 1.69 (с, 3H), 1.5-1.65 (м, 6H), 1.4-1.5 (м, 2H), 1.18-1.38 (м, ~40H), 1.01 (с, 3H), 0.84-0.88 (м, 12H).
Пример 15: DOPE-Glu-NH-VA
Получение (Z)-(2R)-3-(((2-(4-((2E,4E,6E,8E)-3,7-диметил-9-(2,6,6-триметилциклогекс-1-ен-1-ил)нона-2,4,6,8-тетраенамидо)бутанамидо)этокси)(гидрокси)фосфорил)окси)пропан-1,2-диил диолеата (DOPE-Glu-NH-VA)
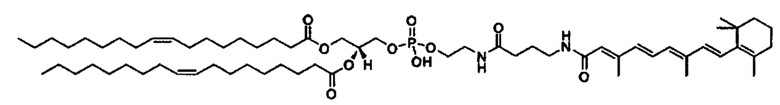
Получение промежуточного соединения h (Z)-(2R)-3-(((2-(4-аминобутанамидо)этокси)(гидрокси)фосфорил)окси)пропан-1,2-диил диолеат
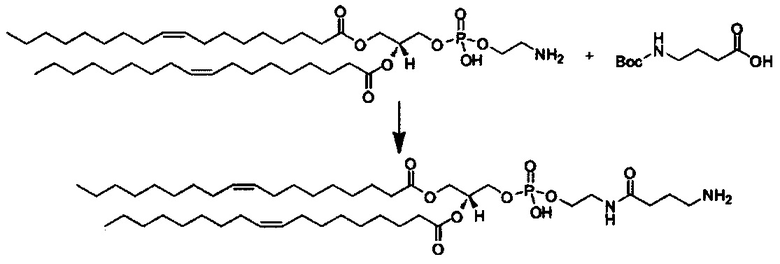
1,2-диолеил-sn-глицеро-3-фосфоэтаноламин (2500 мг, 3.36 ммоль), Boc-GABA-OH (751 мг, 3.70 ммоль) и N,N,N',N'-Тетраметил-O-(7-азабензотриазол-1-ил)урония гексафторфосфат (1531 мг, 4.03 ммоль) растворили в DMF/хлороформ (25 мл, 1:1 смесь). N,N-Диизопропилэтиламин (880 мкл, 5.05 ммоль) добавили и смесь перемешивали при комнатной температуре всю ночь под аргоном. Реакционную смесь разбавили с помощью -200 мл H2O и продукт экстрагировали с помощью дихлорметана (3×100 мл). Продукт промыли с помощью ~75 мл pH 4.0 PBS буфера, высушенные с помощью сульфата натрия органические фракции отфильтровали и концентрировали. Вещество затем очистили с помощью хроматографии на силикагеле с применением дихлорметан/метанол градиента и концентрировали с получением бесцветного масла (2.01 г, 64%). Продукт подтвердили с помощью ЯМР. Вещество затем растворили в 30 мл 2М HCl/диэтиловый простой эфир. Реакционную смесь перемешивали при комнатной температуре на H2O бане. Через 2 часа раствор концентрировали с получением (Z)-(2R)-3-(((2-(4-аминобутанамидо)этокси)(гидрокси)фосфорил)окси)пропан-1,2-диил диолеата.
Получение DOPE-Glu-NH-VA: (Z)-(2R)-3-(((2-(4-((2E,4E,6E,8E)-3,7-диметил-9-(2,6,6-триметилциклогекс-1-ен-1-ил)нона-2,4,6,8-тетраенамидо)бутанамидо)этокси)(гидрокси)фосфорил)окси)пропан-1,2-диил диолеата
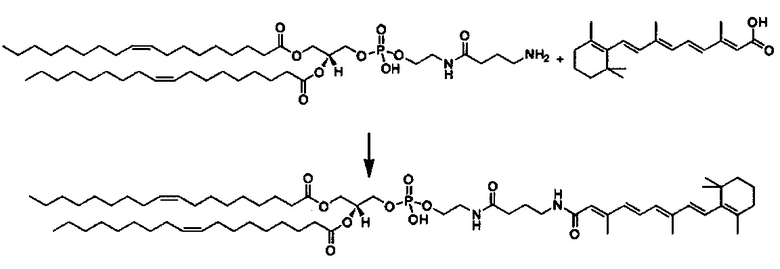
(Z)-(2R)-3-(((2-(4-аминобутанамидо)этокси)(гидрокси)фосфорил)окси)пропан-1,2-диил диолеат (1200 мг, 1.45 ммоль), ретиноевую кислоту (500 мг, 1.66 ммоль) и N,N,N',N'-Тетраметил-O-(7-азабензотриазол-1-ил)урония гексафторфосфат (689 мг, 1.81 ммоль) суспендировали в DMF/хлороформе (10 мл, 1:1 mixture). N,N-Диизопропилэтиламин (758 мкл, 4.35 ммоль) добавили. Круглодонную колбу продули аргоном и накрыли алюминиевой фольгой. Реакционную смесь перемешали при комнатной температуре в течение 4 часов, растворили в дихлорметане (75 мл) и H2O (75 мл), экстрагировали в дихлорметане, высушили (сульфат натрия), отфильтровали и концентрировали. Очищение с помощью хроматографии на силикагеле с применением дихлорметан/метанол градиента дало (Z)-(2R)-3-(((2-(4-((2E,4E,6E,8E)-3,7-диметил-9-(2,6,6-триметилциклогекс-1-ен-1-ил)нона-2,4,6,8-тетраенамидо)бутанамидо)этокси)(гидрокси)фосфорил)окси)пропан-1,2-диил диолеат (292 мг, 18%). Продукт охарактеризовали с помощью LCMS & ЯМР. 1H ЯМР (400 МГц), δН: 8.55 (с, 1H), 8.2 (д, 1H), 7.3 (с, 1H), 6.6 (дд, 1H), 6.10-6.27 (м, 5H), 5.5 (т, 1H), 5.31 (с, 4H), 5.1-5.2 (м, 2H), 4.68 (д, 2H), 4.3 (д, 2H), 4.1 (м, 2H), 3.9 (м, 8H), 3.58 (кв, 4H), 3.4 (с, 4H), 3.0 (кв, 4H), 2.33-2.35 (т, 3H), 2.2-2.3 (м, 7H), 1.9-2.05 (м, 17H), 1.85 (с, 3H), 1.69 (с, 3H), 1.5-1.65 (м, 6H), 1.4-1.5 (м, 2H), 1.18-1.38 (м, ~40H), 1.01 (с, 3H), 0.84-0.88 (м, 12H). MS: m/z 1112.44 (M+H+).
Пример 16: DSPE-II3r550-VA
Получение (2R)-3-(((((45E,47E,49E,51E)-46,50-диметил-4,44-диоксо-52-(2,6,6-триметилциклогекс-1-ен-1-ил)-7,10,13,16,19,22,25,28,31,34,37,40-додекаоксо-3,43-диазадопентаконта-45,47,49,51-тетраен-1-ил)окси)(гидрокси)фосфорил)окси)пропан-1,2-диил дистеарата (DSPE-n3r550-VA)

Получение промежуточного соединения 1: (2R)-3-((((2,2-диметил-4,44-диоксо-3,8,11,14,17,20,23,26,29,32,35,38,41-тридекаокса-5,45-диазагептатетраконтан-47-ил)окси)(гидрокси)фосфорил)окси)пропан-1,2-диил дистеарата
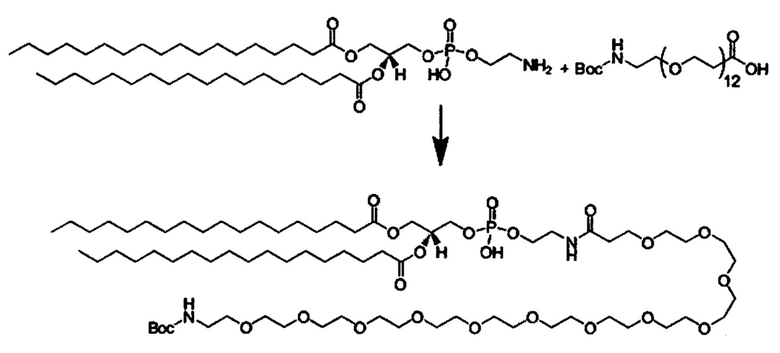
1,2-дистеароил-зп-глицеро-3-фосфоэтаноламин (200 мг, 0.267 ммоль), t-Boc-N-амидо-dПЭГ12-кислоту (211 мг, 0.294 ммоль) и N,N,N',N'-Тетраметил-O-(7-азабензотриазол-1-ил)урония гексафторфосфат (122 мг, 0.320 ммоль) растворили в хлороформ/метанол/H2O (6 мл, 65:35:8) в 20 мл сцинтилляционном флаконе, продутом аргоном. N,N-Диизопропилэтиламин (116 мкл, 0.668 ммоль) добавили. Реакционную смесь перемешивали при 25°C в течение 4 часов и концентрировали. Вещество затем очистили с помощью хроматографии на силикагеле с применением дихлорметан/метанол градиента с получением (2R)-3-((((2,2-диметил-4,44-диоксо-3,8,11,14,17,20,23,26,29,32,35,38,41-тридекаокса-5,45-диазагептатетраконтан-47-ил)окси)(гидрокси)фосфорил)окси)пропан-1,2-диил дистеарата в виде масла (252 мг, 65%).
Получение DSPE-ПЭГ550-VA: (2R)-3-(((((45E,47E,49E,51E)-46,50-диметил-4,44-диоксо-52-(2,6,6-триметилциклогекс-1-ен-1-ил)-7,10,13,16,19,22,25,28,31,34,37,40-додекаоксо-3,43-диазадопентаконта-45,47,49,51-тетраен-1-ил)окси)(гидрокси)фосфорил)окси)пропан-1,2-диил дистеарата
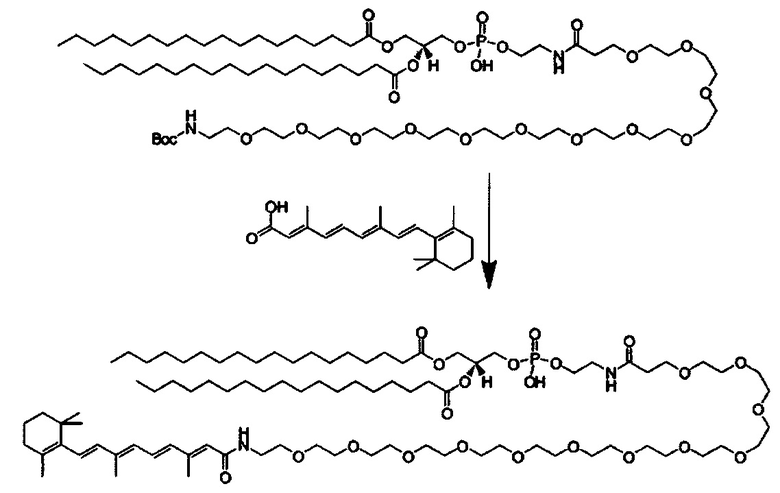
(2R)-3-((((2,2-диметил-4,44-диоксо-3,8,11,14,17,20,23,26,29,32,35,38,41-тридекаокса-5,45-диазагептатетраконтан-47-ил)окси)(гидрокси)фосфорил)окси)пропан-1,2-диил дистеарат (252 мг, 0.174 ммоль) растворили в диэтиловом простом эфире (5 мл), реакционную смесь разместили на H2O бане при комнатной температуре. 2М HCl/простой диэтиловый эфир (2 мл, 4 ммоль) добавили и смесь перемешивали в течение около 1 часа. После этого растворитель и избыток HCl удалили in vacuo. Суспендировали вещество в 2 мл N,N-Диметилформамиде в круглодонной колбе, продутой аргоном. Ретиноевую кислоту (57.5 мг, 0.191 ммоль), N,N,N',N'-Тетраметил-O-(7-азабензотриазол-1-ил)урония гексафторфосфат (79 мг, 0.209 ммоль) и N,N-Диизопропилэтиламин (106 мкл, 0.609 ммоль) добавили. Вещество растворилось не полность, поэтому добавили больше хлороформа/метанола/H2O (1 мл, 65:35:8 об:об:об смесь) для полученная гомогенной реакционной смеси. Через 3.5 часа реакционную смесь концентрировали. Вещество затем очистили с помощью хроматографии на силикагеле с применением дихлорметан/метанол градиента с получением (2R)-3-(((((45E,47E,49E,51E)-46,50-диметил-4,44-диоксо-52-(2,6,6-триметилциклогекс-1-ен-1-ил)-7,10,13,16,19,22,25,28,31,34,37,40-додекаоксо-3,43-диазадопентаконта-45,47,49,51-тетраен-1-ил)окси)(гидрокси)фосфорил)окси)пропан-1,2-диил дистеарата в виде твердого вещества желтовато-коричневого цвета (210 мг, 74%). Продукт подтвердили посредством ЯМР & LCMS. 1H ЯМР (400 МГц), δH: 8.6 (с, 1H), 8.25 (д, 1H), 6.8-6.9 (дд, 1H), 6.3-6.4 (м, 1H), 6.12-6.25 (дд, 5H), 5.71 (с, 1H), 5.18 (м, 2H), 4.33 (дд, 2H), 4.13 (м, 2H), 3.95 (м, 2H), 3.74 (м, 8H), 3.63 (с, ~48H), 3.0 (кв, 2H), 2.5 (т, 3H), 2.35 (с, 3H), 2.25 (т, 8H), 1.97 (м, 7H), 1.7 (3, 3H), 1.5 (м, 2H), 1.36 (м, 12H), 1.23 (м, ~56H), 1.01 (с, 6H), 0.86 (т, 12H). MS: m/z 1630.28 (M+H+).
Пример 17: DSPE-ПЭГ2000-Glu-VA
Получение DSPE-ПЭГ2000-Glu-VA

Получение промежуточного соединения 1: 5-(((2E,4E,6E,8E)-3,7-диметил-9-(2,6,6-триметилциклогекс-1-ен-1-ил)нона-2,4,6,8-тетраен-1-ил)окси)-5-оксопентановая кислота
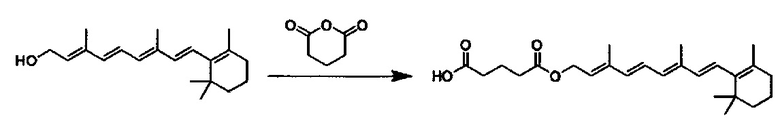
Глутаровый альдегид (115 мг, 1.01 ммоль) и ретинол (240 мг, 0.838 ммоль) растворили в дихлорметане (3 мл) в окрашенном в янтарный цвет флаконе. Триэтиламин (257 мкл, 1.84 ммоль) добавили и флакон продули аргоном. Реакционную перемешивали при комнатной температуре всю ночь. Реакционную смесь концентрировали и затем очистили с помощью хроматографии на силикагеоле с применением дихлорметан/метанол градиента с получением 5-(((2E,4E,6E,8E)-3,7-диметил-9-(2,6,6-триметилциклогекс-1-ен-1-ил)нона-2,4,6,8-тетраен-1-ил)окси)-5-оксопентановой кислоты в виде масла желтоватого цвета (700 мг, 78%). Вещество охарактеризовали посредством ЯМР.
Получение DSPE-ПЭГ2000-Glu-VA
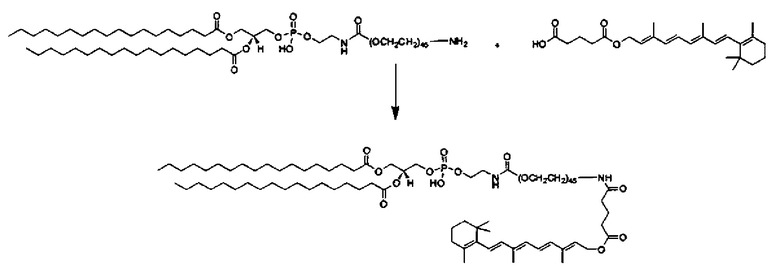
5-(((2E,4E,6E,8E)-3,7-диметил-9-(2,6,6-триметилциклогекс-1-ен-1-ил)нона-2,4,6,8-тетраен-1-ил)окси)-5-оксопентановую кислоту (43 мг, 0.108 ммоль), DSPE-ПЭГ2000-NH2 (250 мг, 0.090 ммоль) и N,N,N',N'-тетраметил-О-(7-азабензотриазол-1-ил)урония гексафторфосфат (45 мг, 0.117 ммоль) растворили в N,N-диметилформамиде (2 мл) окрашенном в янтарный цвет сцинтилляционном флаконе, продутом газом аргоном. N,N-диизопропилэтиламин (47 мкл, 0.270 ммоль) добавили и реакционную смесь перемешивали всю ночь при комнатной температуре, затем очистили с помощью хроматографии на силикагеле с применением дихлорметан/метанол градиента с получением масла желтоватого цвета (59 мг, 20.7%). Продукт подтвердили посредством ЯМР. 1H ЯМР (400 МГц), δH: 706 (м, 1H), 6.59-6.66 (дд, 1H), 6.06-6.30 (m 5H), 5.56-5.60 (т, 1H), 5.17-5.23 (м, 2H), 4.35-4.42 (дд, 2H), 4.12-4.25 (м, 5H), 3.96-3.97 (м, 6H), 3.79-3.81 (т, 1H), 3.66 (м, ~180H), 3.51-3.58 (м, 2H), 3.4-3.48 (м, 4H), 3.3-3.38 (м, 2H), 2.25-2.45 (м, 14H), 1.5-2.0 (м, 15H), 1.23-1.32 (м, ~56H), 1.01 (с, 3H), 0.85-0.88 (т, 12H).
Пример 18: DOPE-Gly3-VA
Получение (Z)-(2R)-3-(((((14E,16E,18E,20E)-15,19-диметил-4,7,10,13-тетраоксо-21-(2,6,6-триметилциклогекс-1-ен-1-ил)-3,6,9,12-тетраазагеникоза-14,16,18,20-тетраен-1-ил)окси)(гидрокси)фосфорил)окси)пропан-1,2-диил диолеата (DOPE-Gly3-VA)
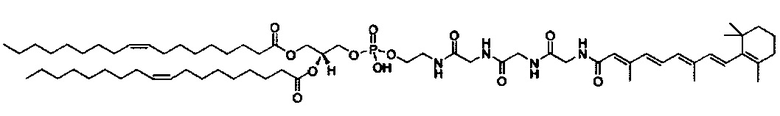
Получение промежуточного соединения 1: (Z)-(2R)-3-(((2-(2-(2-(2-аминоацетамидо)ацетамидо)ацетамидо)этокси)(гидрокси)фосфорил)окси)пропан-1,2-диил диолеат
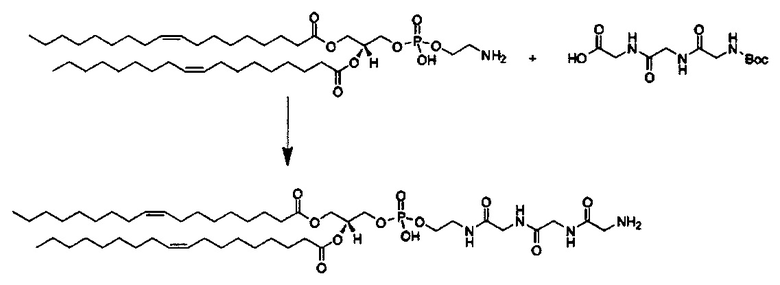
Boc-Gly-Gly-Gly-OH (382 мг, 1.34 ммоль) и N,N,N',N'-Тетраметил-O-(7-азабензотриазол-1-ил)урония гексафторфосфат (532 мг, 1.4 ммоль) растворили в DMF (5 мл). N,N-Диизопропилэтиламин (488 мкл, 2.8 ммоль) добавили и смесь перемешивали при комнатной температуре в течение 10-15 минут. После этого раствор 1,2-диолеил-sn-глицеро-3-фосфоэтаноламина (833 мг, 1.12 ммоль) в хлороформе (5 мл) добавили и реакционный сосуд продули аргоном. Через 16 часов при комнатной температуре, реакционную смесь концентрировали и распределили между дихлорметаном (50 мл) и H2O (50 мл), экстрагировали с помощью дихлорметана (3×50 мл), высушили с помощью сульфата натрия, отфильтровали и концентрировали. Вещество очистили с помощью хроматографии на силикагеле с применением дихлорметан/метанол градиента с получением бесцветного масляного остатка. К этому 2 М HCl/простой диэтиловый эфир (5 мл) добавили и реакционную смесь перемешивали на H2O бане в течение около 2 часов. Реакционную смесь концентрировали и осадок растворили в дихлорметане (75 мл), промыли с помощью насыщенного раствора бикарбонатом натрия (75 мл), экстрагировали продукт с помощью дихлорметана (3×75 мл), высушили с помощью сульфата натрия, отфильтровали и концентрировали с получением (Z)-(2R)-3-(((2-(2-(2-(2-аминоацетамидо)ацетамидо)ацетамидо)этокси)(гидрокси)фосфорил)окси)пропан-1,2-диил диолеата в виде полутвердого вещества (765 мг, 90%). Подтвердили с помощью ЯМР.
Получение DOPE-GlvrVA: (Z)-(2R)-3-(((((14E,16E,18E,20E)-15,19-диметил-4,7,10,13-тетраоксо-21-(2,6,6-триметилциклогекс-1-ен-1-ил)-3,6,9,12-тетраазагеникоза-14,16,18,20-тетраен-1-ил)окси)(гидрокси)фосфорил)окси)пропан-1,2-диил диолеат
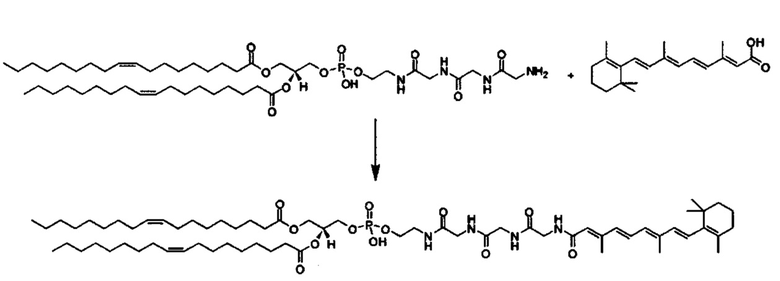
(Z)-(2R)-3-(((2-(2-(2-(2-аминоацетамидо)ацетамидо)ацетамидо)этокси)(гидрокси)фосфорил)окси)пропан-1,2-диил диолеат (765 мг, 0.836 ммоль), ретиноевую кислоту (301 мг, 1.00 ммоль) и N,N,N',N'-тетраметил-O-(7-азабензотриазол-1-ил)урония гексафторфосфат (413 мг, 1.09 ммоль) суспендировали в N,N-диметилформамиде (5 мл). N,N-Диизопропилэтиламин (437 мкл, 2.51 ммоль) добавили и реакционный сосуд продули газом аргоном. Добавили хлороформ (5 мл) с целью растворения веществ. Реакционную смесь перемешивали в течение ~4 часов при комнатной температуре в кругл о донной колбе, покрытой алюминиевой фольгой. Вещество распределили между H2O (100 мл) и дихлорметаном (100 мл). Экстрагировали с помощью дихлорметана (3×100 мл), высушили с помощью сульфата натрия, отфильтровали и концентрировали. Вещество затем очистили с помощью хроматографии на силикагеле с применением дихлорметан/метанол градиента с получением (Z)-(2R)-3-(((((14E,16E,18E,20E)-15,19-диметил-4,7,10,13-тетраоксо-21-(2,6,6-триметилциклогекс-1-ен-1-ил)-3,6,9,12-тетраазагеникоза-14,16,18,20-тетраен-1-ил)окси)(гидрокси)фосфорил)окси)пропан-1,2-диил диолеата в виде масла оранжевого цвета (704 мг, 70%). Продукт очистили с помощью LCMS & ЯМР. 1H ЯМР (400 МГц), δH: 6.90 (т, 1H), 6.21 (кв, 2H), 6.08-6.12 (д, 2H), 5.83 (с, 1H), 5.31 (с, 4H), 5.30 (с, 2H), 4.37 (д, 1H), 4.15 (м, 1H), 3.91 (м, 8H), 3.59 (м, 2H), 3.29 (м, 2H), 3.01 (м, 2H), 2.28 (м, 6H), 1.95-1.98 (м, 12H), 1.44 (с, 3H), 1.5-1.6 (м, 2H), 1.44 (м, 6H), 1.24 (м, ~48H), 1.00 (с, 6H), 0.86 (т, 3H). MS: m/z 1198.42 (M+H+).
Пример 19: VA-ПЭГ-VA
Получение N1,N19-бис((16E,18E,20E,22E)-17,21-диметил-15-оксо-23-(2,6,6-триметилциклогекс-1-ен-1-ил)-4,7,10-триокса-14-азатрикоза-16,18,20,22-тетраен-1-ил)-4,7,10,13,16-пентаоксанонадекан-1,19-диамид (VA-ПЭГ-VA)

Получение VA-ПЭГ-VA: N1,N19-бис((16E,18E,20E,22E)-17,21-диметил-15-оксо-23-(2,6,6-триметилциклогекс-1-ен-1-ил)-4,7,10-триокса-14-азатрикоза-16,18,20,22-тетраен-1-ил)-4,7,10,13,16-пентаоксанонадекан-1,19-диамид
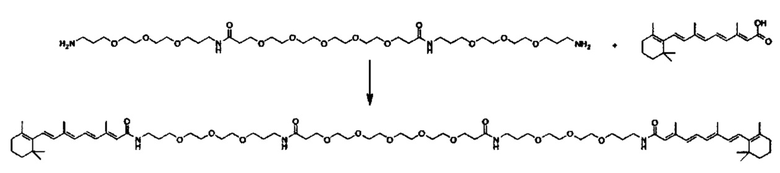
Ретиноевую кислоту (2913 мг, 9.70 ммоль), N,N,N',N'-тетраметил-O-(7-азабензотриазол-1-ил)урония гексафторфосфат (3992 мг, 10.50 ммоль) и диамидо-dПЭГ11-диамин (3000 мг, 4.04 ммоль) суспендировали в N,N-диметилформамиде (10 мл). N,N-Диизопропилэтиламин (4222 μL, 24.24 ммоль) добавили и сосуд продули аргоном. Реакционную смесь перемешивали при комнатной температуре всю ночь в кругло донной колбе, покрытой алюминиевой фольгой. На следующий день вещество распределили между этил ацетатом (125 мл) и водой (125 мл). Экстрагировали этилацетатом (3×125 мл), высушили с помощью сульфата натрия, отфильтровали и концентрировали. Вещество затем очистили с помощью хроматографии на силикагеле с применением дихлорметан/метанол градиента. Объединили фракции и концентрировали с получением масла желтого цвета (2900 мг, 54.9%). Продукт подтвердили с помощью LCMS & ЯМР. 1H ЯМР (400 МГц), δH: 7.1 (с, 2H), 6.87 (т, 2H), 6.51 (т, 2H), 6.12-6.20 (дд, 8H), 5.66 (с, 2H), 3.6-3.8 (м, ~44H), 3.4 (кв, 4H), 3.3 (кв, 4H), 2.46 (т, 4H), 2.32 (с, 6H), 1.9-2.05 (м, 10H), 1.7-1.85 (м, 15H), 1.6 (м, 4H), 1.3-1,5 (м, 6H), 1.01 (с, 12H). QTOF MS: m/z 1306 (M+H+).
Пример 20: VA-ПЭГ2000-VA
Получение (2E,2'E,4E,4'E,6E,6'E,8E,8'E)-N,N-(3,6,9,12,15,18,21,24,27,30,33,36,39,42,45,48,51,54,57,60,63,66,69,72,75,78,81,84,
87,90,93,96,99,102,105,108,111,114,117,120,123,126,129,132,135,138-гексатетраконтаоксатетраконтагектана-1,140-диил)бис(3,7-диметил-9-(2,6,6-триметилциклогекс-1-ен-1-ил)нона-2,4,6,8-тетраенамида) (VA-ПЭГ2000-VA)
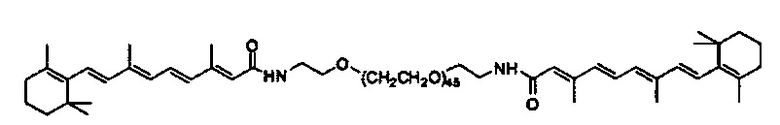
Получение VA-ПЭГ2000-VA: (2E,2'E.4E,4'E,6E,6'E.8E,8'E)-N,N'-(3,6,9,12,15,18,21,24,27,30,33,36,39,42,45,48,51,54,57,60,63,66,69,72,75,78,81,84,
87,90,93,96,99,102,105,108,111,114,117,120,123,126,129,132,135,138-гексатетраконтаоксатетраконтагектана-1,140-диил)бис(3,7-диметил-9-(2,6,6-триметилциклогекс-1-ен-1-ил)нона-2,4,6,8-тетраенамида)
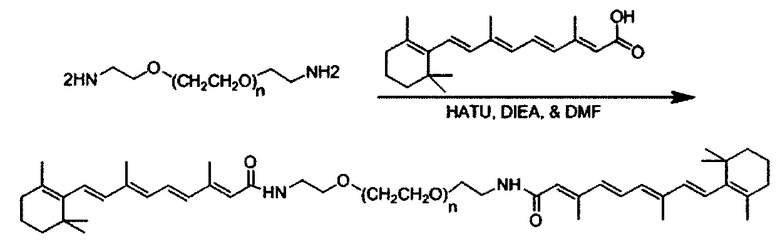
Ретиноевую кислоту (109 мг, 0.362 ммоль), N,N,N',N'-тетраметил-O-(7-азабензотриазол-1-ил)урония гексафторфосфат (149 мг, 0.392 ммоль) и амин-ПЭГ2K-амин (333 мг, 0.151 ммоль) суспендировали в N,N-диметилформамиде (3 мл). N,N-диизопропилэтиламин (158 μL, 0.906 ммоль) добавили и сосуд продули аргоном. Реакционную смесь перемешивали при комнатной температуре всю ночь в круглодонной колбе, покрытой алюминиевой фольгой. На следующий день вещество распределили между этил ацетат (30 мл) и воду (30 мл). Экстрагировали этилацетатом (3×30 мл), высушили с помощью сульфата натрия, отфильтровали и концентрировали. Вещество затем очистили с помощью хроматографии на силикагеле с применением дихлорметан/метанол градиента. Объединили фракции и концентрировали с получением (2E,2'E,4E,4'E,6E,6'E,8E,8'E)-N,N'-(3,6,9,12,15,18,21,24,27,30,33,36,39,42,45,48,51,54,57,60,63,66,69,72,75,78,81,84,
87,90,93,96,99,102,105,108,111,114,117,120,123,126,129,132,135,138-гексатетраконтаоксатетраконтагектана-1,140-диил)бис(3,7-диметил-9-(2,6,6-триметилциклогекс-1-ен-1-ил)нона-2,4,6,8-тетраенамида) в виде масла желтого цвета (97 мг, 23%). Продукт подтвердили с помощью LCMS & ЯМР. 1H ЯМР (400 МГц), 6H: 6.85-6.92 (т, 2h), 6.20-6.32 (м, 6H), 6.08-6.12 (д, 4H), 5.72 (с, 2H), 3.55-3.70 (м, ~180H), 3.4-3.5 (м, 4H), 2.79 (м, 4H), 2.78 (с, 6H), 2.33 (с, 6H), 2.05 (м, 4H), 1.97 (с, 6H), 1.80 (м, 2H), 1.79 (с, 6H), 1.69 (с, 6H), 1.60 (м, 4H), 1.45 (м, 4H), 1.01 (с, 12H). QTOF MS: m/z 2651 (M+H+).
Пример 21: DSPE-ПЭГ2000-VA
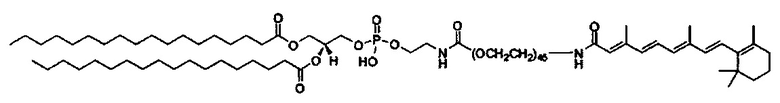
Получение DSPE-ПЭГ2000-VA
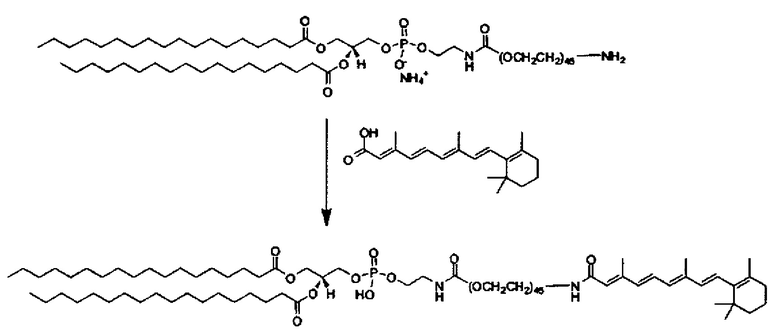
DSPE-ПЭГ2000-NH2 (250 мг, 0.090 ммоль), ретиноевую кислоту (33 мг, 0.108 ммоль) и N,N,N',N'-тетраметил-O-(7-азабензотриазол-1-ил)урония гексафторфосфат (45 мг, 0.117 ммоль) растворили в N,N-Диметилформамиде. N,N-Диизопропилэтиламин (47 мкл, 0.270 ммоль) добавили к смеси. Окрашенный в янтарный цвет сцинтилляционный флакон продули аргоном и перемешивали 3 дня при комнатной температуре. Вещество затем очистили с помощью хроматографии на силикагеле с применением дихлорметан/метанол градиента. Объединили фракции и концентрировали с получением DSPE-ПЭГ2000-VA в виде масла желтого цвета (245 мг, 89%). Продукт подтвердили посредством ЯМР. 1H ЯМР (400 МГц), δH: 6.86 (дд, 1H), 6.25 (м, 1H), 6.09-6.21 (дд, 4H), 5.71 (с, 1H), 5.1-5.2 (м, 1H), 4.3-4.4 (д, 1H), 4.1-4.2 (м, 3H), 3.85-4.0 (м, 4H), 3.8 (т, 1H), 3.5-3.75 (м, ~180H), 3.4-3.5 (м, 8H), 3.3 (м, 2H), 2.35 (с, 3H), 2.26 (м, 4H), 1.70 (с, 3H), 1.55-1.65 (м, 6H), 1.47 (м, 2H), 1.23 (с, ~60H), 1.01 (с, 6H), 0.85 (т, 6H).
Пример 22: diVA-ПЭГ-diVA, также известный как "DiVA"
Получение N1,N19-бис((c,23E,25E,27E,29E)-16-((2E,4E,6E,8E)-3,7-диметил-9-(2,6,6-триметилцикло-гекс-1-ен-1-ил)нона-2,4,6,8-тетраенамидо)-24,28-диметил-15,22-диоксо-30-(2,6,6-триметилциклогекс-1-ен-1-ил)-4,7,10-триокса-14,21-диазатриаконта-23,25,27,29-тетраен-1-ил)-4,7,10,13,16-пентаоксанонадекан-1,19-диамид (diVA)
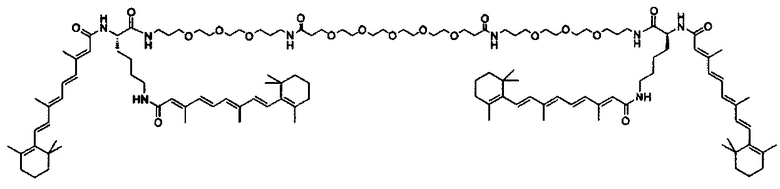
Получение промежуточного соединения 1: тетрабензил((5S,57S)-6,22,40,56-тетраоксо-11,14,17,25,28,31,34,37,45,48,51-ундекаокса-7,21,41,55-тетраазагенгексаконтан-1,5,57,61-тетраил)тетракарбамат, также известный как Z-DiVA-ПЭГ-DiVA-IN
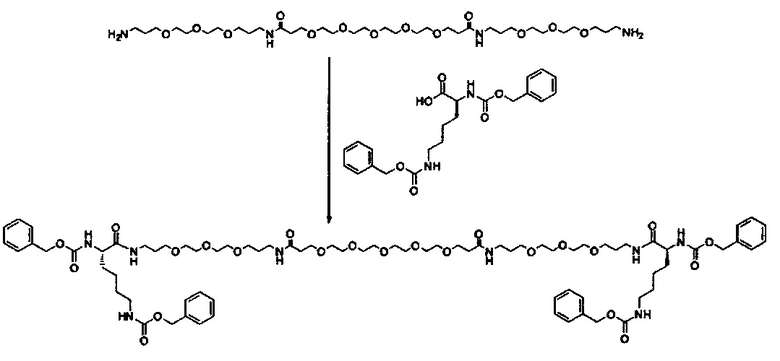
Колбу объемом один литр охладили до 5-10°C и продули азотом и загрузили дихлорметаном (300 мл), d-ПЭГ-11-диамином (Quanta lot EK1-A-1100-010, 50.0 г, 0.067 моль), Z-(L)-Lys(Z)-OH (61.5 г, 0.15 моль) и HOBt гидратом (22.5 г, 0.15 моль). 4-Метилморфолин (4-ММР) (15.0 г, 0.15 моль) добавили к суспензии, и легкая экзотермическая реакция наблюдалась. Суспензию EDC гидрохлорид (43.5 г, 0.23 моль) и 4-ММР (20.0 г, 0.20 моль) в дихлорметане (150 мл) добавили за период 30 минут, и потребовалось умеренное охлаждение, чтобы поддерживать температуру 20-23°C. Слегка мутный раствор перемешивали всю ночь при температуре окружающей среды, и ВЭЖХ показала завершение реакции. Деоинизированную воду (300 мл) добавили и после перемешивания в течение 10 минут наблюдалось быстрое разделение фаз. Водную фазу экстрагировали с помощью дихлорметана (150 мл) - со слегка более медленным разделением фаз. Объединенные органические экстракты промыли с помощью 6% бикарбонатом натрия (300 мл) и высушили с помощью сульфата магния (24 г). Выпаривание на 40-45°C водяной бане при пониженном давлении дало 132 г неочищенного продукта. Раствор неочищенного продукта (131 г) в 8% метаноле в этилацетате в наполняющем колонку силикагеле 60 (40-63 μ), наполненной 8% метанолом в этилацетате. Колонку элюировали 8% метанолом в этилацетате (7.5 л). Фракции, содержащие по существу чистый продукт (5.00-7.25 л), выпарили на 45°C водяной бане при пониженном давлении и 83.6 г очищенного продукта. Раствор очищенного продукта (83.6 г) в дихлорметане (200 мл) загрузили в колонку Dowex 650 С (H+) (200 г), которую промывали дихлорметаном (250 мл). Колонку элюировали дихлорметаном (200 мл). Объединенный продукт, содержащий фракции (300-400 мл), высушили с помощью сульфата магния (14 г) и выпаривали на 45°C водяной бане при пониженном давлении с получением тетрабензил((5S,57S)-6,22,40,56-тетраоксо-11,14,17,25,28,31,34,37,45,48,51-ундекаокса-7,21,41,55-тетраазагенгексаконтан-1,5,57,61-тетраил)тетракарбамата, также известного как Z-DiVA-ПЭГ-DiVA-IN (77.9 г, ВЭЖХ чистота 94.1%).
Получение промежуточного соединения 2: N1,N19-бис((S)-16,20-диамино-15-оксо-4,7,10-триокса-14-азаикозил)-4,7,10,13,16-пентаоксанонадекан-1,19-диамида, также известного как DiVA-ПЭГ-DiVA-IN
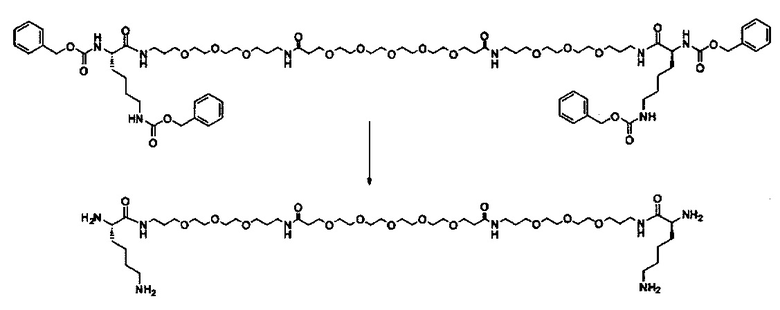
Колбу объемом один литр и продули азотом и загрузили метанол (600 мл) и Z-DiVA-ПЭГ-DiVA-IN (92.9, 60.5 ммоль). Смесь перемешали в атмосфере аргона до тех пор, пока не был получен раствор. Катализатор, 10% РЬ7С/50%воду (Aldrich, 10 г), добавили. Смесь удалили и затем давление выравнили засчет азота. Обеспечили равномерный низкий поток водорода через реакционную смесь, начали перемешивание. Гидрирование продолжали в потоке водорода в течение одного часа. Систему затем закрылм и гидрирование продолжали при ~0.1 бар в течение одного часа. Смесь удалили и затем повторно создали давление ~0.1 бар с помощью водорода. Через еще один час гидрирования смесь удалили и затем повторно создали давление ~0.1 бар с помощью водорода. Перемешивание в атмосфере водорода продолжали в течение 15 часов, после этого времени исходные вещества уже нельзя было определить с помощью ВЭЖХ. Смесь удалили и затем давление уравновесили с помощью азота. Смесь удалили и затем давление уравновесили с помощью азота. Реакционную смесь затем отфильтровали на подушке из целита 545. Осадок на фильтре промыли с помощью метанола (100 мл). Объединенный фильтрат концентрировали, окончательно при 45°C и при давлении менее 50 мБар. Толуол (100 мл) добавили и полученную смесь окончательно концентрировали окончательно при 45°C и при давлении менее 40 мБар с получением N1,N19-бис((S)-16,20-диамино-15-оксо-4,7,10-триокса-14-азаикозил)-4,7,10,13,16-пентаоксанонадекан-1,19-диамида, также известного как DiVA-ПЭГ-DiVA-IN (63.4 г), в виде масла, которое отверждается при отстаивании.
Получение DiVA-ПЭГ-DiVA: N1,N19-бис((c,23E,25E,27E,29E)-16-((2E,4E,6E,8E)-3,7-диметил-9-(2,6,6-триметилциклогекс-1-ен-1-ил)нона-2,4,6,8-тетраенамидо)-24,28-диметил-15,22-диоксо-30-(2,6,6-три-метилциклогекс-1-ен-1-ил)-4,7,10-триокса-14,21-диазатриаконта-23,25,27,29-тетраен-1-ил)-4,7,10,13,16-пентаоксанонадекан-1,19-диамида
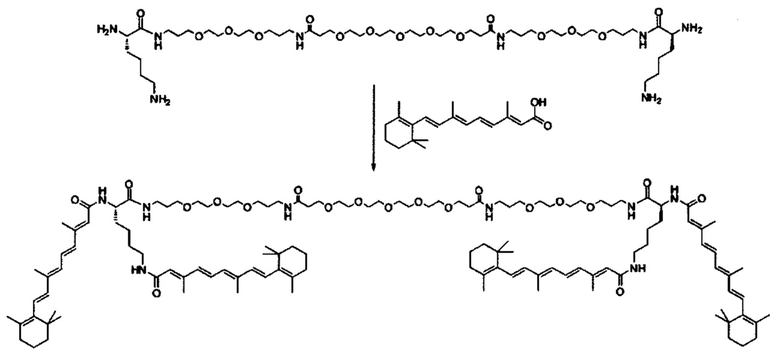
Реактор объемом два литра заполнили аргоном и загрузили дихлорметане (500 мл), DiVA-ПЭГ-DiVA-IN (52.3 г, 52.3 ммоль), ретиноевую кислоту (70.6 г, 235 ммоль) и 4-N,N-диметиламинопиридин (2.6 г, 21.3 ммоль). Смесь перемешали в атмосфере аргона до растворения (~20 минут). Поддерживая температуру реактора при 10-20°C, 1-этил-3-(3-диметиламинопропил) карбодиимид) (EDCI) (70.6 г, 369 ммоль) добавили по частям за период 10-15 минут (реакция была слегка экзотермической в течение первых 30-60 минут). Реактор накрыли алюминиевой фольгой и смесь перемешали при 18-21°C в течение 15-20 часов. Бутилированный гидрокситолуол (ВНТ) (25 мг) добавили и реакционную смесь затем перелили на безводный 6% гидрокарбонат натрия (500 мл) при поддержании атмосферы аргона над смесью. Органическую фазу отделили. Водную фазу промыли с помощью дихлорметане (50 мл). Объединенную органическую фазу высушили с помощью сульфата магния (150 г) в инертной атмосфере и защищали от света. Высушивающий агент отфильтровали (предпочтительна фильтрация под давлением) и осадок на фильтре промыли с помощью дихлорметане (500 мл). Фильтрат концентрировали посредством испарения при пониженном давлении с применением водяной бани 35-40°C. К масляному остатку добавили толуол (150 мл) и снова выпарили с получением полутвердого остатка 210 г. Этот остаток растворили в дихлорметане (250 мл) и нанесли на колонку с силикагелем 60 (1.6 кг) и 0.5% метанолом в дихлорметане) (4 л). Колонку элюировали дихлорметаном (7.2 л), 2), 3% метанол в дихлорметане (13 л), 5% метанол в дихлорметане (13 л), 10% метанол в дихлорметане (18 л). Одну фракцию объемом 10 л взяли и затем взяли фракции объемом 2.5 л. Взяли пробы фракций, защищенных от света, продули аргоном и герметизировали, фракции подвергли анализу посредством ТСХ (10% метанол в дихлорметане, УФ). Фракции, содержащие DiVA-ПЭГ-DiVA, затем проанализировали с помощью ВЭЖХ. 5 фракций<85% чистоты (дали 32 г остатка выпаривания) повторно очистили таким же образом, применяя только 25% от изначального количества силикагеля и растворителей. Фракции>85% чистоты по ВЭЖХ объединили и выпарили при пониженном давлении, применяя водяную баню 35-40°C. Осадок выпаривания (120 г) повторно растворили в дихлорметане (1.5 л) и медленно пропустили (около 1 часа) через колонку, наполненную ионообменником Dowex 650C, H+ форма (107 г). Колонку затем промыли с помощью дихлорметана (1 л). Объединенный элюат (3277.4 г) хорошо смешали и образец (25 мл, 33.33 г) выпарили полностью при комнатной температуре и давлении<0.1 мБар с получением 0.83 г пены. Из этого было вычислено общее количество твердого материала с получением 80.8 г (72.5%). Оставшиеся 3.24 кг раствора концентрировали до 423 г. 266 г этого раствора концентрировали далее с получением сиропа и затем снова растворили в абсолютном этаноле (200 мл). Продукты выпаривания при пониженном давлении, с применением водяной бани 35-40°C, объединили с получением конечного раствора этанола 94.8 г, содержащего 50.8 г (53.6% мас/мас) N1,N19-бис((c,23E,25E,27E,29E)-16-((2E,4E,6E,8E)-3,7-диметил-9-(2,6,6-триметилциклогекс-1-ен-1-ил)нона-2,4,6,8-тетраенамидо)-24,28-диметил-15,22-диоксо-30-(2,6,6-триметилциклогекс-1-ен-1-ил)-4,7,10-триокса-14,21-диазатриаконта-23,25,27,29-тетраен-1-ил)-4,7,10,13,16-пентаоксанонадекан-1,19-диамида, также известного как DiVA-ПЭГ-DiVA, также известного как "DiVA". Охарактеризовали с помощью ЯМР & QTOF. 1H ЯМР (400 МГц), δH: 7.07 (т, 2H), 7.01 (т, 2H), 6.87-6.91 (м, 4.0H), 6.20-6.24 (м, 10H), 6.10-6.13 (м, 8H), 5.79 (с, 2H), 5.71 (с, 2H), 4.4 (кв, 2H), 3.70 (т, 6H), 3.55-3.65 (м, ~34H), 3.59 (т, 6H), 3.4 (м, 2H), 3.25-3.33 (м, 10H), 3.16 (м, 2H), 2.44 (т, 4H), 2.33 (с, 12H), 1.97-2.01 (м, 12H), 1.96 (с, 6H), 1.7-1.9 (м, 12H), 1.69 (с, 12H), 1.5-1.65 (м, 12H), 1.35-1.5(м, 24H), 1.01 (с, 24H). QTOF MS ESI+: m/z 2128 (M+H+).
Пример 23: DOPE-VA
Получение (Z)-(2R)-3-(((2-((2E,4E,6E,8E)-3,7-диметил-9-(2,6,6-триметилциклогекс-1-ен-1-ил)нона-2,4,6,8-тетраенамидо)этокси)(гидрокси)фосфорил)окси)пропан-1,2-диил диолеата (DOPE-VA)
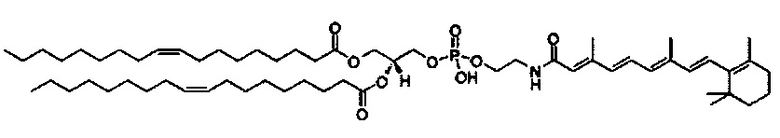
Получение DOPE-VA: (Z)(2R)-3-(((2-((2E,4E,6E,8E)-3,7-диметил-9-(2,6,6-триметилциклогекс-1-ен-1-ил)нона-2,4,6,8-тетраенамидо)этокси)(гидрокси)фосфорил)окси)пропан-1,2-диил диолеата
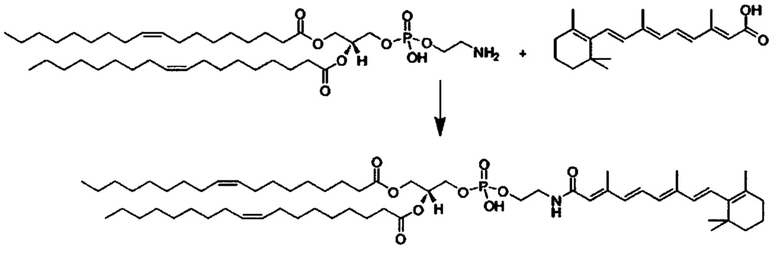
К раствору ретиноевой кислоты (250 мг, 0.83 ммоль) в простом диэтиловом эфире, перемешиваемом (20 мл) при -78°C, раствор (диэтиламино)серы трифторида (130 мкл, 0.90 ммоль) в холодном простом эфире (20 мл) добавили с помощью шприца. Реакционную смесь сняли с ледяной бани и перемешивание продолжали при комнатной температуре в течение еще 2 часов. В конце растворитель удаляли на роторном испарителе. Остаток повторно растворили хлороформом (50 мл) в присутствии твердого Na2CO3(50 мг). К этому раствору добавили 1,2-диолеил-sn-глицеро-3-фосфоэтаноламин (600 мг, 0.81 ммоль) и реакционную смесь перемешали при комнатной температуре в течение еще 24 часов. Растворитель удаляли на роторном испарителе. Остаток очистили с помощью хроматографии на силикагеле с применением дихлорметан/метанол градиента с получением Z)-(2R)-3-(((2-((2E,4E,6E,8E)-3,7-диметил-9-(2,6,6-триметилциклогекс-1-ен-1-ил)нона-2,4,6,8-тетраенамидо)этокси)(гидрокси)фосфорил)окси)пропан-1,2-диил диолеата (240 мг, 28%). 1H ЯМР (400 МГц, CDCl3) ™ 0.87 (т, 6H, CH3), 1.01 (с, 6H, CH3) 1.20-1.40 (м, 40H, CH2), 1.40-1.60 (м, 8H, CH2), 1.70 (с, 3H, CH3-C=С), 1.80-2.10 (м, 8H), 2.32 (м, 4H, CH2C(=O)), 3.50 (м, 2H), 3.92-4.18 (м, 5H), 4.35 (м, 2H), 5.20 (м, 1H, NHC(=O)), 5.31 (м, 4H, CH=CH), 5.80-6.90 (м, 6H, CH=CH).
Пример 24: DC-VA
Получение (((2E,4E,6E,8E)-3,7-диметил-9-(2,6,6-триметилциклогекс-1-ен-1-ил)нона-2,4,6,8-тетраеноил)азандиил)бис(этан-2,1-диил) дитетрадеканоата (DC-VA)
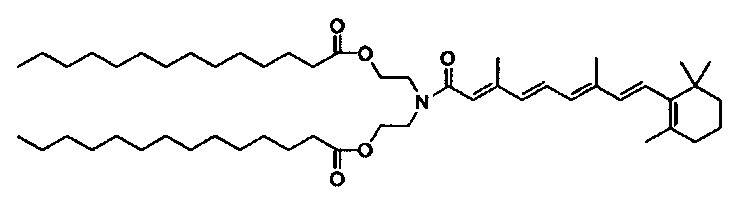
Получение DC-VA: (((2E,4E,6E,8E)-3,7-диметил-9-(2,6,6-триметилциклогекс-1-ен-1-ил)нона-2,4,6,8-тетраеноил)азандиил)бис(этан-2,1-диил)дитетрадеканоата
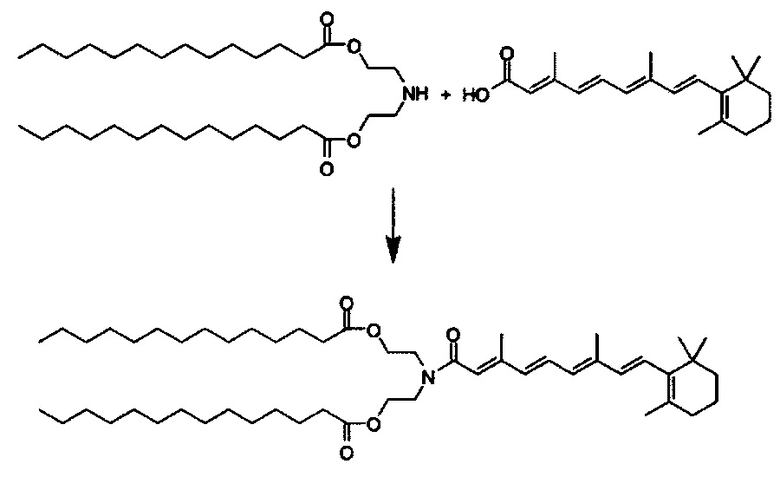
К раствору ретиноевой кислоты (600 мг, 2.0 ммоль) в простом диэтиловом эфире (25 мл), перемешиваемом при -78°C, раствор (диэтиламино)серы трифторид (0.3 мл, 2.1 ммоль) в 5 мл холодного простого эфира добавили посредством шприца. Реакционную смесь сняли с ледяной бани и перемешивание продолжали при комнатной температуре в течение еще 1 часа. После удаления растворителя на роторном испарителе, остаток повторно растворили в дихлорметане (20 мл) в присутствии твердого Na2CO3 (25 мг). К этому раствору добавили азандиилбис(этан-2,1-диил) дитетрадеканоат (1.05 г, 2.0 ммоль) и реакционную смесь перемешали при комнатной температуре в течение еще 24 часов. Реакционную смесь разбавили с помощью дихлорметане (50 мл) и высушили над MgSO4. После удаления растворителя на роторном испарителе, остаток очищение с помощью хроматографии на силикагеле с применением дихлорметан/метанол градиента с получением (((2E,4E,6E,8E)-3,7-диметил-9-(2,6,6-триметилциклогекс-1-ен-1-ил)нона-2,4,6,8-тетраеноил)азандиил)бис(этан-2,1-диил)дитетрадеканоата (800 мг, 50%). 1H ЯМР (400 МГц, CDCl3) ™ 0.87 (т, 6H, CH3), 1.02 (с, 6H, CH3) 1.20-1.40 (м, 40H, CH2), 1.40-1.60 (м, 8H, CH2), 1.70 (с, 3H, CH3-C=C), 1.97(с, 3H, CH3-C=C), 2.05 (м, 2H, CH2), 2.15(с, 3H, CH3-C=C), 2.32 (м, 4H, CH2C(=O)), 3.67 (м, 4H, NCH2CH2O), 4.15-4.30 (м, 4Н, NCH2CH2O), 5.80-6.90 (м, 6H, CH=CH).
Пример 25: DC-6-VA
Получение ((6-((2E,4E,6E,8E)-3,7-диметил-9-(2,6,6-триметилциклогекс-1-ен-1-ил)нона-2,4,6,8-тетраенамидо)гексаноил)азандиил)бис(этан-2,1-диил)дитетрадеканоата (DC-6-VA)
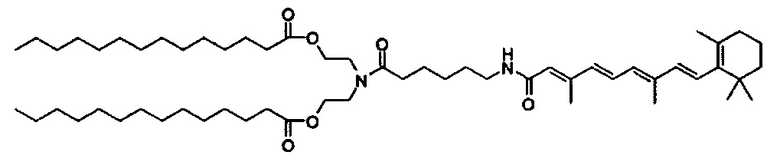
Получение промежуточного соединения 1: ((6-аминогексаноил)азандиил)бис(этан-2,1-диил)дитетрадеканоат TFA соли
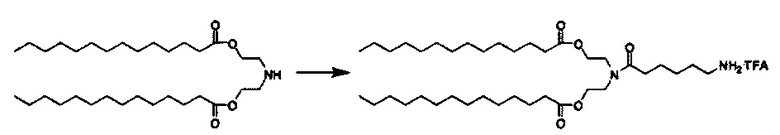
Смесь азандиилбис(этан-2,1-диил)дитетрадеканоат (2.5 г, 4.8 ммоль), Boc-амино капроновой кислоты (1.3 г, 5.6 ммоль), N,N'-дициклогексилкарбодиимида (1.3 г, 6.3 ммоль) и N,N-диизопропилэтиламина (2.6 мл, 0.015 ммоль) растворили в пиридине (40 мл). Раствор перемешивали при 60°C всю ночь. Смесь разбавили с помощью дихлорметана (50 мл) и промыли с помощью соляного раствора(3×50 мл). После концентрирования посредством роторного испарителя, остаток обработали трифторуксусной кислоты/дихлорметана (100 мл, 1:1). Смесь концентрировали и растворили в дихлорметане (50 мл) и промыли с помощью соляного раствора (3×50 мл). Органический слой отделили и концентрировали с получением ((6-аминогексаноил)азандиил)бис(этан-2,1-диил)дитетрадеканоат TFA соли (1.5 г, 33%).
Получение DC-6-VA: ((6-((2E,4E,6E,8E)-3,7-диметил-9-(2,6,6-триметилциклогекс-1-ен-1-ил)нона-2,4,6,8-тетраенамидо)гексаноил)азандиил)бис(этан-2,1-диила)
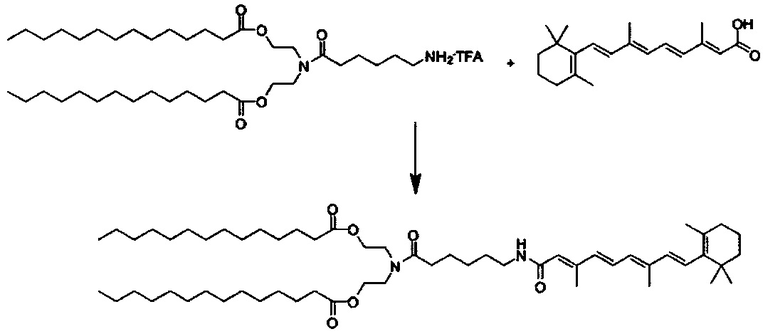
К раствору ретиноевой кислоты (800 мг, 2.67 ммоль) в простом диэтиловом эфире (40 мл), перемешиваемом при -78°C, раствор (диэтиламино)серы трифторида (0.4 мл, 22.80 ммоль) в холодном простом эфире (7 мл) добавили посредством шприца. Реакционную смесь сняли с ледяной бани и перемешивание продолжали при комнатной температуре в течение еще одного часа. После удаления растворителя на роторном испарителе, остаток повторно растворили в дихлорметане (25 мл) в присутствии твердого Na2CO3 (40 мг). К этому раствору добавили ((6-аминогексаноил)азандиил)бис(этан-2,1-диил)дитетрадеканоат TFA соль (1.5 г, 1.6 ммоль) и реакционную смесь перемешали при комнатной температуре еще 24 часов. Реакционную смесь разбавили с помощью дихлорметана (50 мл) и высушили над MgSO4. После удаления растворителя на роторном испарителе, остаток очистили с помощью колоночной хроматографии с применением 5% метанол/дихлорметана в качестве элюента с получением ((6-((2E,4E,6E,8E)-3,7-диметил-9-(2,6,6-триметилциклогекс-1-ен-1-ил)нона-2,4,6,8-тетраенамидо)гексаноил)азандиил)бис(этан-2,1-диила) (360 мг, 24%). 1H ЯМР (400 МГц, CDCl3) ™ 0.87 (т, 6H, CH3), 1.02 (с, 6H, CH3) 1.20-1.40 (м, 42H, CH2), 1.40-1.60 (м, 12H, CH2), 1.70 (с, 3H, CH3-C=C), 1.97(с, 3H, CH3-C=C), 2.05 (м, 2H, CH2), 2.15(с, 3H, CH3-C=C), 2.32 (м, 6H, CH2C(=O)), 3.20 (м, 2H, CH2NHC(=O)), 3.56 (м, 4H, NCH2CH2O), 4.15-4.30 (м, 4H, NCH2CH2O), 5.10 (м, 1H), 5.80-6.90 (м, 6H, CH=CH).
Пример 26: СИНТЕЗ satDiVA
Получение N1,N19-бис((16S)-16-(3,7-диметил-9-(2,6,6-триметилциклогекс-1-ен-1-ил)нонанамидо)-24,28-диметил-15,22-диоксо-30-(2,6,6-триметилциклогекс-1-ен-1-ил)-4,7,10-триокса-14,21-диазатриаконтил)-4,7,10,13,16-пентаоксанонадекан-1,19-диамида (насыщенный DIVA).
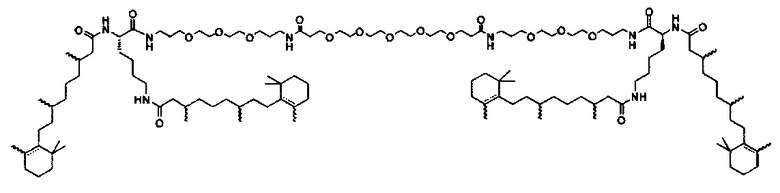
Получение промежуточного соединения 1: 3,7-диметил-9-(2,6,6-триметилциклогекс-1-ен-1-ил)нонановой кислоты
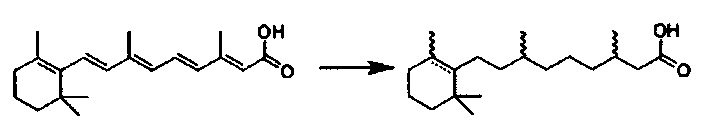
Полностью-транс ретиноевую кислоту (2000 мг, 6.66 ммоль) растворили в гексанах/IPA (3:1, 40 мл) с помощью обработки ультразвуком. Вещество разместили в сосуде шейкере Парра и продули инертным газом. 10% Pd/C (200 мг) добавили и сосуд снова продули инертным газом. Вещество разместили в шейкер Парра на всю ночь с >70 пси газа водорода. Реакционную смесь затем отфильтровали через слой целита и концентрировали с получением 3,7-диметил-9-(2,6,6-триметилциклогекс-1-ен-1-ил)нонановой кислоты (2 г).
Получение насыщенного DIVA: N1,N 19-бис((16S)-16-(3,7-диметил-9-(2,6,6-триметилциклогекс-1-ен-1-ил)нонанамидо)-24,28-диметил-15,22-диоксо-30-(2,6,6-триметилциклогекс-1-ен-1-ил)-4,7,10-триокса-14,21-диазатриаконтил)-4,7,10,13,16-пентаоксанонадекан-1,19-диамида
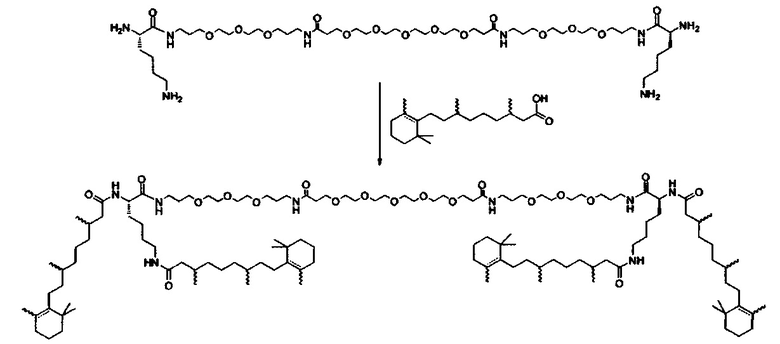
N1,N19-бис((16S)-16-(3,7-диметил-9-(2,6,6-триметилциклогекс-1-ен-1-ил)нонанамидо)-24,28-диметил-15,22-диоксо-30-(2,6,6-триметилциклогекс-1-ен-1-ил)-4,7,10-триокса-14,21-диазатриаконтил)-4,7,10,13,16-пентаоксанонадекан-1,19-диамида, также известный как насыщенный DIVA, получили подобным образом как и diva-ПЭГ-diVA из описанного ранее N1,N19-бис((S)-16,20-диамино-15-оксо-4,7,10-триокса-14-азаикозил)-4,7,10,13,16-пентаоксанонадекан-1,19-диамида с замещением 3,7-диметил-9-(2,6,6-триметилциклогекс-1-ен-1-ил)нонановой кислоты на полностью-транс ретиноевую кислоту. QTOF MS ESI+: m/z 2161, 2163, 2165 & 2167 (M+H+)
Пример 27: СИНТЕЗ simDiVA
Получение N1,N19-бис((S)-15,22-диоксо-30-(2,6,6-триметилциклогекс-1-ен-1-ил)-16-(9-(2,6,6-триметилциклогекс-1-ен-1-ил)нонанамидо)-4,7,10-триокса-14,21-диазатриаконтил)-4,7,10,13,16-пентаоксанонадекан-1,19-диамида (simDiVA).
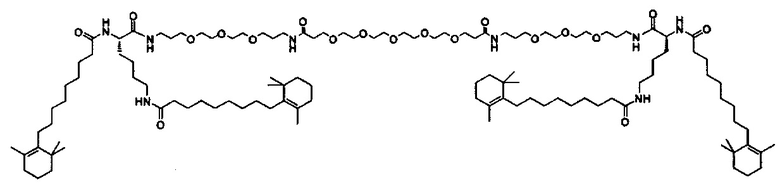
Получение промежуточного соединения 1: 2,6,6-триметилциклогекс-1-ен-1-ил трифторметансульфонат
К раствору 2,2,6-триметилциклогексанона в сухом THF при -78°C в атмосфере азота добавили по каплям 2 М раствор лития диизопропиламида. Смесь перемешали при -78°C в течение 3 часов. Раствор N-фенил-бис(трифторметансульфонимид) в THF затем добавили по каплям (при -78°C). Реакционную колбу заполнили сухим льдом и перемешивали всю ночь. Перемешивание продолжали при комнатной температуре в течение 3 часов, за это время все вещества растворились. Реакционную смесь концентрировали и остаток добавили медленно к гексану (350 мл) при интенсивном перемешивании. Твердое вещество удалил при фильтрации и промыли с помощью гексана (2×50 мл). Фильтрат концентрировали и добавили еще гексана (150 мл). Твердое вещество удалили посредством фильтрации и фильтрат концентрировали. Осаждение повторили еще раз, после чего остаток очистили посредством флеш-хроматографии (оксид кремния, гексан) с получением 2,6,6-триметилциклогекс-1-ен-1-ил трифторметансульфоната в виде бесцветного масла (23.2 г, 60% выход).
Получение промежуточного соединения 2: этил 9-(бромцинцио)нонаноата

В сухой пробирке в атмосфере азота находились заряженная цинковая пыль (3.70 г, 56.6 ммоль), иодин (479 мг, 1.89 ммоль) и сухой DMA (20 мл). Смесь перемешали при комнатной температуре пока не исчезла окраска иодина. Этил 9-бромнонаноат добавили и смесь перемешали при 80°C в течение 4 часов и затем при комнатной температуре всю ночь. (Завершение реакции введения цинка проверили с помощью GCMS анализа гидролизованной реакционной смеси.) Реакционную смесь применяли без дальнейшей обработки на следующей стадии. GCMS m/z 186 [M]+(этил нонаноат).
Получение промежуточного соединения 3: этил 9-(2,6,6-триметилциклогекс-1-ен-1-ил)нонаноата
К свежеприготовленному этил 9-(бромцинцио)нонаноату (37.7 ммоль) в диметилацетамиде в атмосфере аргона в реакционной пробирке добавили 2,6,6-триметилциклогекс-1-ен-1-ил трифторметансульфонат (10.8 г, 39.6 ммоль) и затем тетракис(трифенилфосфин)палладий(0) (872 мг, 0.754 ммоль). Пробирку запаяли и смесь перемешали при 95°C в течение 2 часов. Реакционной смеси дали охладиться и затем перелили в простой диэтиловый эфир (100 мл). Верхний слой декантировали и нижний слой дважды промыли простым диэтиловым эфиром (2×25 мл). Объединенные эфирные слои промыли с помощью насыщенного NH4Cl и соляного раствора, высушили с помощью (MgSO4) и концентрировали с получением неочищенного вещества (~12 г). Вещество очистили с помощью флеш-хроматографии (оксид кремния, 0-1.5% EtAc в гексане). Полученное вещество перемешивали под вакуумом в течение 8 часов для того чтобы удалить большую часть побочного продукта, этил нонаноата и затем очистили посредством второй флеш-хроматографии (оксид кремния, 0-15% толуол в гексане). Фракции проанализировали посредством LCMS и GCMS. Самые чистые фракции собрали и концентрировали при температуре ниже 25°C с получением этил 9-(2,6,6-триметилциклогекс-1-ен-1-ил)нонаноата в виде бесцветного масла (6.16 г, 53% выход за две стадии). LCMS ESI+m/z 309 [M+H]+; GCMS m/z 308 [M]+.
Получение промежуточного соединения 4: 9-(2,6,6-триметилциклогекс-1-ен-1-ил)нонановая кислота

К этил 9-(2,6,6-триметилциклогекс-1-ен-1-ил)нонаноату (13.2 г, 42.9 ммоль) в этаноле (80 мл) добавили 4 М КОН (43 мл). Смесь перемешали при комнатной температуре в течение 1.5 часов. Воду (350 мл) добавили и раствор промыли с помощью трет-бутилметилового простого эфира (2×100 мл). SimVA водную фазу охладили, подкислили с помощью 4М HCl (~45 мл) и экстрагировали с помощью пентана (3×100 мл). Объединенные пентановые экстракты промыли с помощью воды (200 мл), высушили (MgSO4), отфильтровали, концентрировали и высушили под высоким вакуумом. Вещество повторно растворили в пентане (100 мл), концентрировали и высушили под высоким вакуумом еще раз с получением 9-(2,6,6-триметилциклогекс-1-ен-1-ил)нонановой кислоты в виде бесцветного масла (11.1 г, 92% выход). MS ESI- m/z 279 [M-H]-.
Получение simdiVA: N1,N19-бис((S)-15,22-диоксо-30-(2,6,6-триметилциклогекс-1-ен-1-ил)-16-(9-(2,6,6-триметилциклогекс-1-ен-1-ил)нонанмидо)-4,7,10-триокса-14,21-диазатриаконтил)-4,7,10,13,16-пентаоксанонадекан-1,19-диамида.
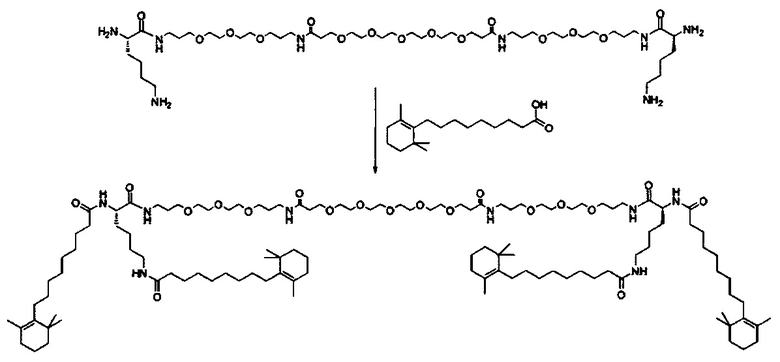
simDIVA получили подобным образом как и diVA как описано ранее для N1,N19-бис((S)-16,20-диамино-15-оксо-4,7,10-триокса-14-азаикозил)-4,7,10,13,16-пентаоксанонадекан-1,19-диамида с замещением 9-(2,6,6-триметилциклогекс-1-ен-1-ил)нонановой кислоты на полностью-транс ретиноевую кислоту. QTOF MS ESI+: m/z 2050 (M+H+)
Пример 28: СИНТЕЗ DiVA-ПЭГ18
Получение (2E,2'E,2ʺE,4E,4'E,4ʺE,6E,6'E,6ʺE,8E,8'E,8ʺE)-N,N',Nʺ-((5R,69R,76E,78E,80E,82E)-77,81-диметил-6,68,75-триоксо-83-(2,6,6-триметилциклогекс-1-ен-1-ил)-10,13,16,19,22,25,28,31,34,37,40,43,46,49,52,55,58,61,64-нонадекаокса-7,67,74-триазатриоктаконта-76,78,80,82-тетраен-1,5,69-триил)трис(3,7-диметил-9-(2,6,6-триметилциклогекс-1-ен-1-ил)нона-2,4,6,8-тетраенамида) (DIVA-ПЭГ 18).

(2E,2'E,2ʺE,4E,4'E,4ʺE,6E,6'E,6ʺE,8E,8'E,8ʺE)-N,N',Nʺ-((5R,69R,76E,78E,80E,82E)-77,81-диметил-6,68,75-триоксо-83-(2,6,6-триметилциклогекс-1-ен-1-ил)-10,13,16,19,22,25,28,31,34,37,40,43,46,49,52,55,58,61,64-нонадекаокса-7,67,74-триазатриоктаконта-76,78,80,82-тетраен-1,5,69-триил)трис(3,7-диметил-9-(2,6,6-триметилциклогекс-1-ен-1-ил)нона-2,4,6,8-тетраенамид), также известный как DIVA-ПЭГ18, получили подобным образом как и diVA с замещением ПЭП18 диамина на диамидо -dПЭГ11-диамин. LCMS ESI+: m/z 2305 (M+Na).
Пример 29: СИНТЕЗ TriVA
Получение TriVA
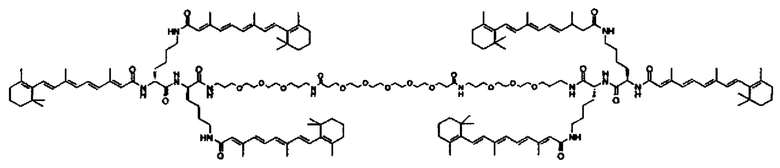
Получение промежуточного соединения 1: (S)-метил 6-(((бензилокси)карбонил)амино)-2-((8)-2,6-бис(((бензилокси)карбонил)амино)гексанамидо) гексаноата
Колбу продули инертным газом и H-Lys(Z)-OMe HCl соль (4 г, 12.1 ммоль), HOBt гидрат (1.84 г, 13.6 ммоль), Z-Lys(Z)-OH (5.64 г, 13.6 ммоль) суспендировали в дихлорметане (50 мл). NMM (1.5 мл, 13.6 ммоль) добавили к суспензии, и раствор стал прозрачным. Суспензию EDC HCl соли (4.01 г, 20.9 ммоль) и NMM (2.0 мл, 18.2 ммоль) в дихлорметане (50 мл) добавили за период 10 минут. Реакционную смесь перемешивали всю ночь при комнатной температуре, затем промыли с помощью 1М HCl (100 мл), H2O (100 мл), насыщенным раствором бикарбоната (100 мл) и насыщенным соляным раствором (100 мл). Все водные промывки обратно экстрагировали с помощью дихлорметана (50 мл). Высушили органические фазы с помощью Na2SO4, отфильтровали и концентрировали. Вещество очистили с помощью хроматографии на силикагеле с применением дихлорметан/метанол градиента с получением (8)-метил 6-(((бензилокси)карбонил)амино)-2-((8)-2,6-бис(((бензилокси)карбонил)амино)гексанамидо)гексаноата (6.91 г).
Получение промежуточного соединения 2: (S)-6-(((бензилокси)карбонил)амино)-2-((8)-2,6-бис(((бензилокси)карбонил)амино)гексанамидо)гексановой кислоты
6-(((бензилокси)карбонил)амино)-2-((S)-2,6-бис(((бензилокси)карбонил)амино)гексанамидо) гексаноат (6.91 г, 10 ммоль) растворили с помощью метанола (50 мл). Добавили КОН (2.24 г, 40 ммоль) и смесь перемешали при 35°C. Через 2 часа реакцию загасили посредством добавления H2O (200 мл) и промыли смесь простым диэтиловым эфиром (50 мл). После этого установили pH ~2 с помощью 1М HCl кислоты. Экстрагировали продукт с помощью дихлорметана (3×100 мл), высушили с помощью Na2SO4, отфильтровали и концентрировали с получением (S)-6-(((бензилокси)карбонил)амино)-2-((S)-2,6-бис(((бензилокси)карбонил)амино)гексанамидо)гексановой кислоты (4 г).
Получение промежуточного соединения 3: (Cbz)6-защищенны N1,N19-бис((16S,19S)-19,23-диамино-16-(4-аминобуитил)-15,18-диоксо-4,7,10-триокса-14,17-диазатрикозил)-4,7,10,13,16-пентаоксанонадекан-1,19-диамид
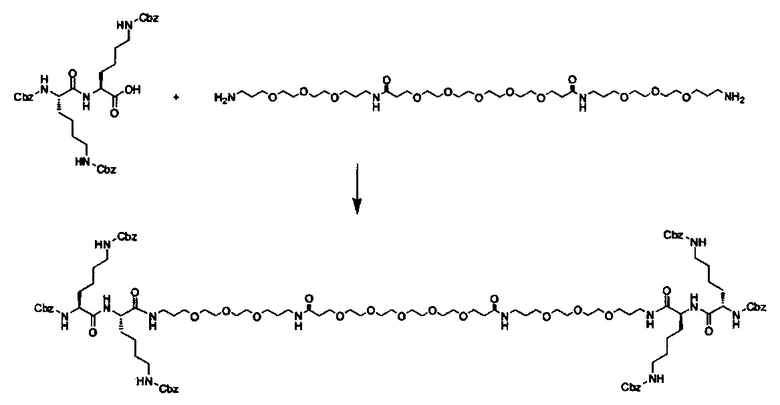
Круглодонную колбу продули инертным газом и диамидо -dПЭГ11-диамин (1 , 1.35 ммоль), (S)-6-(((бензилокси)карбонил)амино)-2-((S)-2,6-бис(((бензилокси)карбонил)амино)гексанамидо)гексановую кислоту (2.05 г, 3.03 ммоль), HOBt гидрат (409 мг, 3.03 ммоль) суспендировали в дихлорметане (25 мл). NMM (333 мкл, 3.03 ммоль) добавили к суспензии, и раствор стал прозрачным. Суспензию EDC HCl соли (893 мг, 4.66 ммоль) и NMM (445 мкл, 4.05 ммоль) в дихлорметане (25 мл) добавили за период 10 минут. Реакционную смесь перемешивали всю ночь при комнатной температуре, затем промыли с помощью 1М HCl (100 мл), H2O (100 мл), насыщенным раствором бикарбоната (100 мл) и насыщенным соляным раствором (100 мл). Все водные промывки обратно экстрагировали с помощью дихлорметане (50 мл). Высушили органические фазы с помощью Na2SO4, отфильтровали и концентрировали. Вещество очистили с помощью хроматографии на силикагеле с применением дихлорметан/метанол градиента с получением (Cbz)6-protected N1,N9-бис((168,198)-19,23-диамино-16-(4-аминобуитил)-15,18-диоксо-4,7,10-триокса-14,17-диазатрикозил)-4,7,10,13,16-пентаоксанонадекан-1,19-диамида (480 мг).
Получение промежуточного соединения 4: N1,N 19-бис((16S, 19S)-19,23-диамино-16-(4-аминобуитил)-15,18-диоксо-4,7,10-триокса-14,17-диазатрикозил)-4,7,10,13,16-пентаоксанонадекан-1,19-диамида
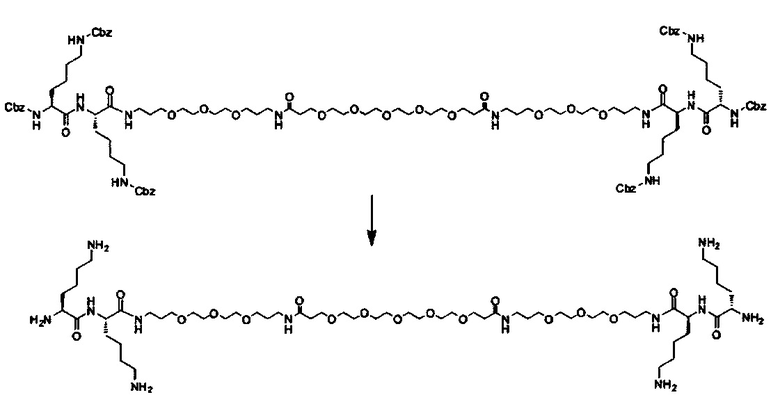
(Cbz)6-защищенный N1,N19-бис((16S,19S)-19,23-диамино-16-(4-аминобуитил)-15,18-диоксо-4,7,10-триокса-14,17-диазатрикозил)-4,7,10,13,16-пентаоксанонадекан-1,19-диамид растворили в метаноле (30 мл) в кругл о донной колбе и продули инертным газом. 10% Pd/C (135 мг) добавили и колбу снова продули инертным газом и затем весь воздух удалили с помощью вакуумного насоса. 8ʺ H2 баллон добавили и реакционную смесь перемешивали при комнатной температуре. Через 2 часа, Pd/C удалили посредством фильтрации через подушку целита, промыли метанолом и концентрировали с получением N1,N19-бис((16S,19S)-19,23-диамино-16-(4-аминобуитил)-15,18-диоксо-4,7,10-триокса-14,17-диазатрикозил)-4,7,10,13,16-пентаоксанонадекан-1,19-диамида (823 мг).
Получение TriVA
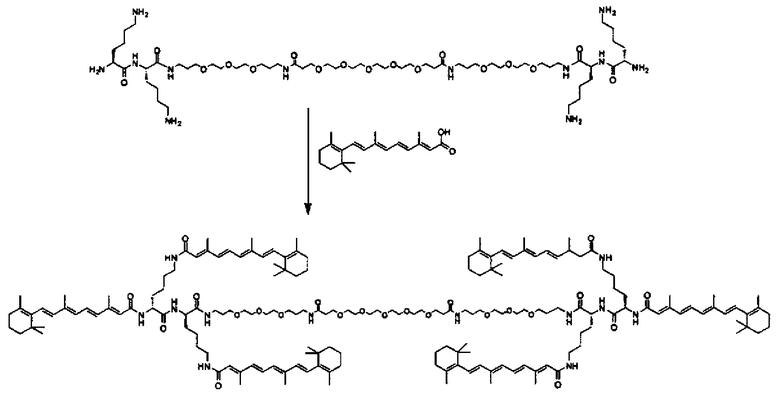
N1,N19-бис((16S,19S)-19,23-диамино-16-(4-аминобуитил)-15,18-диоксо-4,7,10-триокса-14,17-диазатрикозил)-4,7,10,13,16-пентаоксанонадекан-1,19-диамид перемешали в дихлорметане и DMAP и ретиноевую кислоту добавили. NMM добавили и раствор перемешали в накрытой алюминиевой фольгой круглодонной колбе, продутой инертным газом, при комнатной температуре. Суспензию EDC HCl соли & NMM в дихлорметане (20 мл) медленно добавили к реакционной смеси за период 10 минут. Реакционную смесь перемешивали всю ночь при комнатной температуре. На следующий день разбавили с помощью дихлорметана до 100 мл. Промыли с помощью H2O (100 мл), насыщенным раствором бикарбоната (100 мл) и насыщенным соляным раствором (100 мл). Все водные промывки обратно экстрагировали с помощью дихлорметане (50 мл). Высушили органические фазы с помощью Na2SO4, отфильтровали и концентрировали. Вещество очистили с помощью хроматографии на алюминиевой подложке, элюируя дихлорметан/этанол градиентом с получением TriVA (780 мг). LCMS ESI+: m/z 2972 (M+Na).
Пример 30: СИНТЕЗ 4TTNPB
Получение N1,N19-бис((R)-1,8-диоксо-7-(4-((E)-2-(5,5,8,8-тетраметил-5,6,7,8-тетрагидро-нафталин-2-ил)проп-1-ен-1-ил)бензамидо)-1-(4-((Е)-2-(5,5,8,8-тетраметил-5,6,7,8-тетратетрагидронафталин-2-ил)проп-1-ен-1-ил)фенил)-13,16,19-триокса-2,9-диазадокозан-22-ил)-4,7,10,13,16-пентаоксанонадекан-1,19-диамида (4TTNPB).
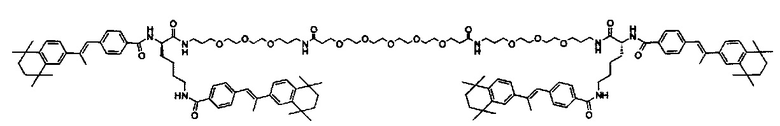
N1,N19-бис((R)-1,8-диоксо-7-(4-((E)-2-(5,5,8,8-тетраметил-5,6,7,8-тетрагидро-нафталин-2-ил)проп-1-ен-1-ил)бензамидо)-1-(4-((E)-2-(5,5,8,8-тетраметил-5,6,7,8-тетратетрагидронафталин-2-ил)проп-1-ен-1-ил)фенил)-13,16,19-триокса-2,9-диазадокозан-22-ил)-4,7,10,13,16-пентаоксанонадекан-1,19-диамида, также известный как 4TTNPB, получили подобным образом как и N1,N19-бис((c,23E,25E,27E,29E)-16-((2E,4E,6E,8E)-3,7-диметил-9-(2,6,6-триметилцикло-гекс-1-ен-1-ил)нона-2,4,6,8-тетраенамидо)-24,28-диметил-15,22-диоксо-30-(2,6,6-триметилциклогекс-1-ен-1-ил)-4,7,10-триокса-14,21-диазатриаконта-23,25,27,29-тетраен-1-ил)-4,7,10,13,16-пентаоксанонадекан-1,19-диамид, также известный как diVA, из N1,N19-бис((S)-16,20-диамино-15-оксо-4,7,10-триокса-14-азаикозил)-4,7,10,13,16-пентаоксанонадекан-1,19-диамида с замещением TTNPB на полностью-транс ретиноевую кислоту. LCMS ESI+: m/z 2343 (M+Na).
Пример 31: СИНТЕЗ 4Myr
Получение N1,N19-бис((R)-15,22-диоксо-16-тетрадеканамидо-4,7,10-триокса-14,21-диазапента-триаконтил)-4,7,10,13,16-пентаоксанонадекан-1,19-диамид (4Myr).
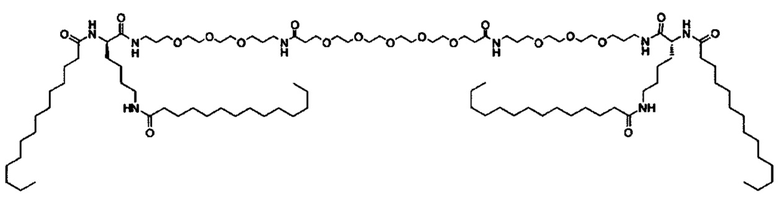
Получение 4Myr: N1,N19-бис((R)-15,22-диоксо-16-тетрадеканамидо-4,7,10-триокса-14,21-диаза-пента-триаконтил)-4,7,10,13,16-пентаоксанонадекан-1,19-диамида
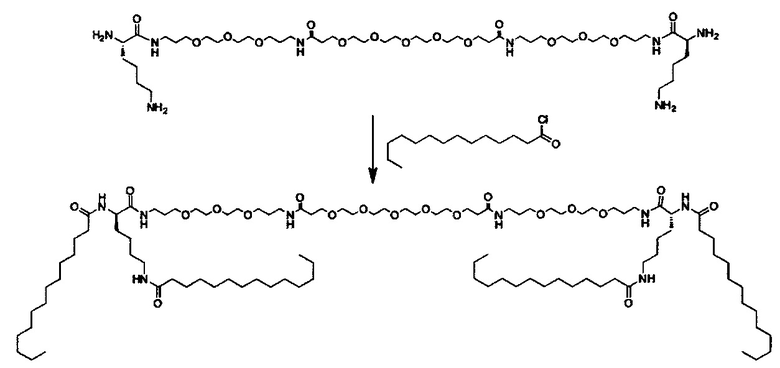
N1,N19-бис((S)-16,20-диамино-15-оксо-4,7,10-триокса-14-азаикозил)-4,7,10,13,16-пентаоксанонадекан-1,19-диамид (синтез описан ранее) растворили в дихлорметане и разместили на ледяной бане. Миристоилхлорид добавили и затем добавили триэтиламин. Ледяную баню удалили и реакционную смесь перемешивали всю ночь при комнатной температуре в атмосфере инертного газа. На следующий день разбавили с помощью дихлорметана до 100 мл и промыли с помощью 1М HCl (75 мл), H2O (75 мл), насыщенным раствором бикарбоната (75 мл) и насыщенным соляным раствором (75 мл). Обратно экстрагировали все водные промывки с помощью дихлорметана (25 мл). Высушили органические фазы с помощью MgSO4, отфильтровали и концентрировали. Очищение посредством хроматографии на силикагеле с применением дихлорметан/метанол градиента дало N1,N19-бис((R)-15,22-диоксо-16-тетрадеканамидо-4,7,10-триокса-14,21-диаза-пента-триаконтил)-4,7,10,13,16-пентаоксанонадекан-1,19-диамид (410 мг). LCMS ESI+: m/z 1841 (M+H).
Пример 32: СИНТЕЗ DiVA-242
Получение N1,N16-бис((R,18E,20E,22E,24E)-11-((2E,4E,6E,8E)-3,7-диметил-9-(2,6,6-триметил-циклогекс-1-ен-1-ил)нона-2,4,6,8-тетраенамидо)-19,23-диметил-10,17-диоксо-25-(2,6,6-триметилциклогекс-1-ен-1-ил)-3,6-диокса-9,16-диазапентакоза-18,20,22,24-тетраен-1-ил)-4,7,10,13-тетраоксагексадекан-1,16-диамида, также известного как DIVA-242
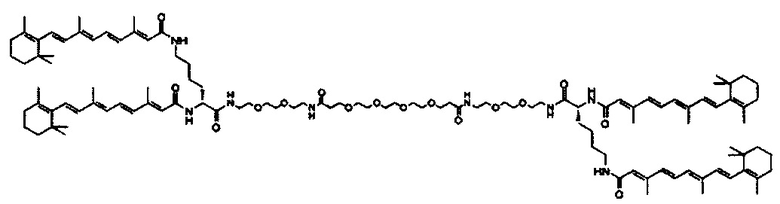
Получение промежуточного соединения 1: ди-трет-бутил(10,25-диоксо-3,6,13,16,19,22,29,32-октаокса-9,26-диазатетратриаконтан-1,34-диил)дикарбамата

Круглодонную колбу, содержащую дихлорметан (25 мл) продули инертным газом и бис-dПЭГ4 кислоту (1000 мг, 3.40 ммоль), N-Boc-3,6-dioкca-1,8-октан диамин (1816 мкл, 7.65 ммоль) и HOBt гидрат (1034 мг, 7.65 ммоль) добавили. NMM (841 мкл, 7.65 ммоль) добавили к суспензии, и раствор стал прозрачным. Суспензию EDC HCl соли (2249 мг, 11.7 ммоль) & NMM (1121 мкл, 10.2 ммоль) в дихлорметане (25 мл) добавили и затем добавили DMAP (62 мг, 0.51 ммоль). Реакционную смесь перемешивали всю ночь при комнатной температуре. Затем разбавили с помощью дихлорметана до 100 мл и промыли с помощью H2O (100 мл), 10% K2CO3 (100 мл) и насыщенным соляным раствором (100 мл), обратно экстрагировали все водные промывки с помощью дихлорметана (30 мл), высушили с помощью MgSO4, отфильтровали и концентрировали. Очищение посредством хроматографии на силикагеле с применением дихлорметан/метанол градиента дало ди-трет-бутил(10,25-диоксо-3,6,13,16,19,22,29,32-октаокса-9,26-диазатетратриаконтан-1,34-диил)дикарбамат (2.57 г).
Получение промежуточного соединения 2: N1,N16-бис(2-(2-(2-аминоэтокси)этокси)этил)-4,7,10,13-тетраоксагекса-декан-1,16-диамид TFA соли

ди-трет-бутил(10,25-диоксо-3,6,13,16,19,22,29,32-октаокса-9,26-диазатетратриаконтан-1,34-диил)дикарбамат растворили в дихлорметане (15 мл) и разместили на ледяной бане. Круглодонную колбу продули инертным газом и TFA (15 мл) добавили. Смесь перемешивали в течение 20 минут. После этого реакционную смесь концентрировали с получением N1,N16-бис(2-(2-(2-аминоэтокси)этокси)этил)-4,7,10,13-тетраоксагексадекан-1,16-диамид TFA соли (1885 мг).
Пример 33: Образование частиц нуклеиновой кислоты и молекул
siPHK упоминается в протоколах композиций как последовательность двухнитиевой siPHK с 21-мерным целевым HSP47/gp46, где HSP47 (мышиный) и gp46 (крысиный) являются гомологами - один и тот же ген в различных последовательностях:
крысиного HSP47-C двухнитиевая siPHK применялась для in vitro анализа (крысиный pHSCs)
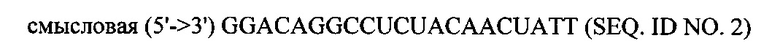
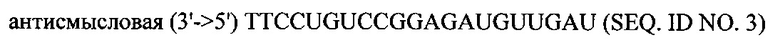
мышиного HSP47-C двухнитиевая siPHK применялась в композициях для in vivo анализа (мышиная модель CCl4)
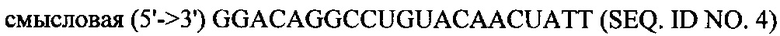
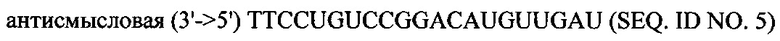
Получение сток-раствора катионного липида. Сток-растворы катионных липидов получали посредством объединения катионного липида с DOPE, холестерином и diVA-ПЭГ-diVA в этаноле при концентрациях 6.0, 5.1 и 2.7 и 2.4 мг/мл соответственно. При необходимости растворы нагревали до около 50°C для облегчения растворения катионных липидов в растворе.
Получение пустых липосом. Сток-раствор катионного липида вводили в быстро перемешиваемую водную смесь при 35-40°C через инъекционную иглу (иглы) при скорости 1.5 мл/мин на инъекционный порт. Отношение сток-раствора катионного липида к водному раствору (об./об.) фиксировалось при 35:65. При смешивании самопроизвольно образовывались пустые везикулы. Пустые везикулы затем уравновешивались при 35-40°C в течение 10 минут прежде чем содержание этанола было уменьшено до ~12%. Пустые везикулы затем диафильтровали с 10× объемами водного буфера для удаления этанола.
Получение липоплекаса. Пустые везикулы, полученные согласно вышеописанному способу, разбавляли до конечного объема 1 мМ при концентрации катионного липида при 9% сахарозе. К перемешиваемому раствору добавили 100 мкл 5% глюкозы в свободной от рибонуклеазы воде на каждый миллилитр разбавленной пустой везикулы ("EV") и тщательно перемешивали. 150 мкл 10 мг/мл siPHK раствора в свободной от рибонуклеазы воде затем добавили за один раз и тщательно смешали. Смесь затем разбавили 5% раствором глюкозы с 1.750 мл на каждый миллилитр EV. Смесь перемешивали при около 200 оборотах в минуту при комнатной температуре в течение 10 минут. Применяя полупроницаемую мембрану с ~100000 MWCO системе перекрестно ультрафильтрации, с применением соответственно выбранного перистальтического насоса (например, Midgee Hoop, UFP-100-H24LA), смесь концентрировали до около 1/3 от изначального объема (или желательного объема) и затем диафильтровали 5 раз объем образца, применяя водный раствор, содержащий 3% сахарозы и 2.9% глюкозы. Продукт затем отфильтровали через комбинированный фильтр с размером пор 0.8/0.2 в асептических условиях перед применением.
Образование липосом, содержащих siPHK без-diVA. Катионный липид, DOPE, холестерин и ПЭГ-конъюгированный липид солюбилизировали в абсолютном этаноле (200 градусов крепости) при молярном отношении 50:10:38:2. SiPHK солюбилизировали в 50 мМ цитратном буфере и температуру довели до 35-40°C. Смесь этанол/липид затем добавили к siPHK-содержащему буферу при перемешивании до самопроизвольного образования загруженных siPHK липосом. Липиды объединили с siPHK с получением конечного общего отношение липид: siPHK равного 15:1 (мас.:мас). Интервал может составлять от 5:1 до 15:1, предпочтительно от 7:1 до 15:1. SiPHK загруженные липосомы подвергли диафильтрации с 0X объемами PBS (pH 7.2) для удаления этанола и замены буфера. Конечный продукт отфильтровали через 0.22 мкм, стерильной марки, PES фильтр для предварительной фильтрации. Этот процесс позволил получить липосомы со средним диаметром частиц 50-100 нм, PDI<0.2 и>85% эффективность включения.
Образование липосом, содержащих siPHK, солюбилизируемые с diVA._siPHK-diVA-липосомальные композиции были получены с применением вышеописанного способа. DiVA-ПЭГ-diVA были солюбилизированы в абсолютном этаноле с другими липидами (катионный липид, DOPE, холестерин и ПЭГ-конъюгированные липиды при отношении 50:10:38:2) до добавления к siPHK содержащему буферу. Молярное отношение diVA-ПЭГ-diVA составляло от 0.1 до 5 (50:10:38:2:0.1-50:10:38:2:5). Этот процесс позволил получить липосомы со средним диаметром частиц 50-100 нм, PDI<0.2 и >85% эффективность включения.
Образование siPHK содержащих липосом с катионными липидами._sWUK-dSM A-липосомальные композиции и siPHK-липосомальные композиции получили с применением вышеописанного способа. Катионным липидом может быть, например, HEDC, HEDODC, DC-6-14 или любая комбинация этих катионных липидов.
Образование siPHK содержащих липосом декорированных diVA. siPHK-липосомальные композиции получили с применением вышеописанного способа и разбавили до концентрации siPHK 0.5 мг/мл в PBS. Катионным липидом может быть HEDC, HEDODC, DC-6-14 или любая комбинация этих катионных липидов. diVA-ПЭГ-diVA разбавили в абсолютном этаноле (200 градусов крепости) до конечной концентрации в интервале от 10 до 50 мг/мл. Подходящее количество этанольного раствора добавили в siPHK-липосомальный раствор с получением конечного мольного содержания от 2 до 10 мол.%. Раствор подняли и опустили с повторениями пипеткой для смешивания. diVA-ПЭГ-diVA концентрацию и добавляемый объем этанола установили для сохранения добавочного объема >1.0 мкл и конечной концентрации этанола <3% (об./об.). Декорированные липосомы затем равномерно перемешали при температуре окружающей среды в течение 1 часа на обычном шейкере до in vitro или in vivo оценки.
В следующих таблицах приведены предпочтительные варианты выполнения настоящего изобретения, выраженные на основании мольных отношений, а также в виде эквивалента мол.% и мас.%.

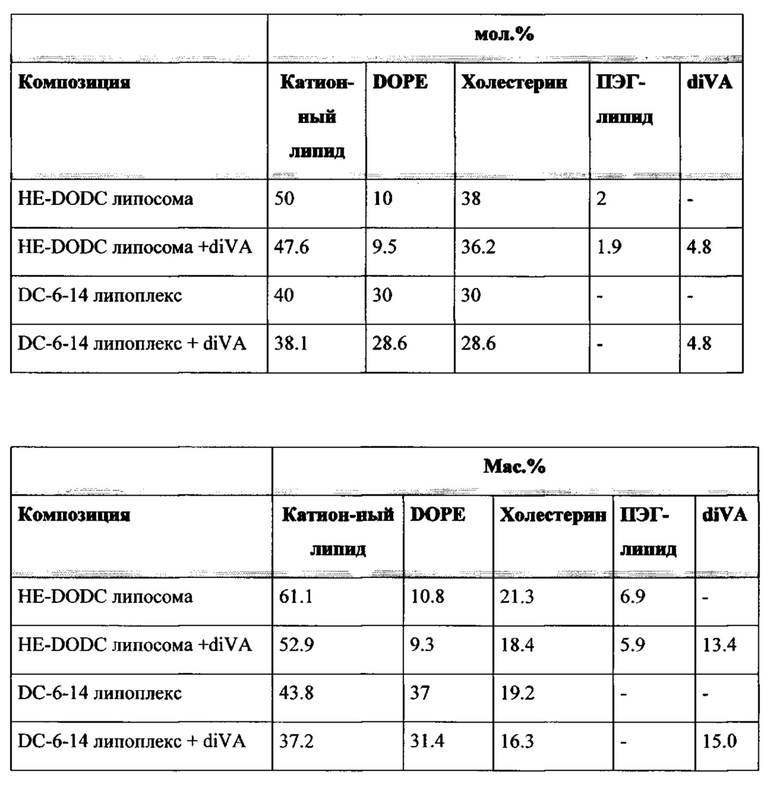
Пример 34: Трансфекция с липосомальными композициями
Способ трансфекции является одинаковым для LX-2 и pHSCs. Липосомальные композиции или композиции липоплекса согласно настоящему изобретения смешали со средой для роста при желательных концентрациях. 100 мкл смеси добавили в клетки в 96-луночном планшете и клетки инкубировали в течение 30 минут при 37°C в инкубаторе с 5% CO2. Через 30 минут среду заменили на свежую среду для роста. Через 48 часов трансфекции клетки обработали с применением Cell-to-Ct® лизисных реагентов (Applied Biosystems) согласно инструкциям производителей.
Количественная ПЦР с обратной транскрипцией для измерения HSP47 мРНК экспрессии (qRT-PCR)
HSP47 и GAPDH TaqMan® анализы и одностадийную ПЦР с обратной транскрипцией master mix получили у Applied Biosystems. Каждая ПЦР реакция включала следующую композицию: одностадийная ПЦР с обратной транскрипцией mix 5 мкл, TaqMan® RT фермент mix 0.25 мкл, зонд анализа экспрессии гена TaqMan® (HSP47) 0.25 мкл, TaqMan® зонд анализа экспрессии гена (GAPDH) 0.5 мкл, свободная от рибонуклеазы вода 3.25 мкл, клеточный лизат 0.75 мкл, общий объем 10 мкл. GAPDH применяли в качестве эндогенного контроля для относительной количественной оценки уровней HSP47 мРНК. Количественную ПЦР с обратной транскрипцией осуществляли в системе ПЦР в реальном времени ViiA 7 (Applied Biosciences) с применением внутреннего способа относительной количественной оценки. Все значения были нормализованы до средней экспрессии HSP47 трансфектированных клеток mock и выражали как процент HSP47 экспрессии по сравнению с mock.
In vivo эксперименты: Самок мышей C57B1/6(Charles River) с диапазоном массы 24-30 грамм применяли для этого исследования. Животных случайным образом распределили по массе в 10 групп по 10 животных в каждой. Все процедуры над животными были одобрены Bio-Quant’s IACUC и/или Attending Veterinarian как необходимо, и условия содержания всех животных были отмечены и задокументированы. Мышей анестезировали с помощью изофлурана и обескровили через полую нижнюю вену.
Повышение экспрессии белка теплового шока 47 (HSP47) было индуцировано посредством интраперитонеальной инъекции CCl4 (CCl4 в оливковом масле, 1:7 (об./об.), 1 л на грамма массы тела добавляя каждый день в течение 7 дней (день 0, 2, 4, 6). С третьего дня мышей обрабатывали в течение четырех последовательных дней (день 3, 4, 5, 6) с композициями липосомы или липоплекса согласно настоящему изобретению или PBS посредством внутривенной инъекции в хвостовую вену. Одну группу из десяти мышей (не подвергавшихся экспериментам) оставили без CCl4 обработки и без внутривенной инъекции, и она служила в качестве контрольной группы для нормальной экспрессии гена HSP47.
Режим эксперимента
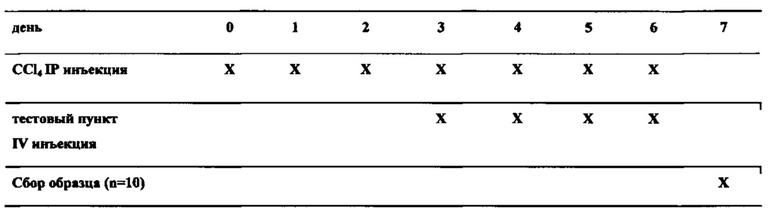
На седьмой день и около 24 часа после последней внутривенной инъекции, всех оставшихся мышей умертвили, и печень опрыскивали PBS прежде чем взять образцы печени для ПЦР анализа. Из каждой мышиной печени взяли образец около 150 мг и поместили в 1.5 мл RNAlater стабилизирующего реагента (Qiagen) и сохраняли при 2-8°C до анализа. Образцы печени собирали из областей поврежденной и/или некрозной чистой и помеченной печени.
Общие уровни РНК мышей экстрагировали с применением RNeasy графы (Qiagen) согласно инструкциям производителя. 20 нг общей РНК применяли для количественной ПЦР с обратной транскрипцией для измерения экспрессии HSP47. HSP47 и GAPDH TaqMan® анализы и одностадийную ПЦР с обратной транскрипцией master mix приобрели у Applied Biosystems. Каждая ПЦР реакция включала следующую композицию: одностадийная ПЦР с обратной транскрипцией mix 5 мкл, TaqMan® RT фермент mix 0.25 мкл, зонд анализа экспрессии гена TaqMan® (HSP47) 0.25 мкл, TaqMan® зонд анализа экспрессии гена (GAPDH) 0.5 мкл, свободная от рибонуклеазы вода 3.25 мкл, клеточный лизат 0.75 мкл, общий объем 10 мкл. GAPDH применяли в качестве эндогенного контроля для относительной количественной оценки уровней HSP47 мРНК. Количественную ПЦР с обратной транскрипцией осуществляли в системе ПЦР в реальном времени ViiA 7 (Applied Biosciences) с применением внутреннего способа относительной количественной оценки. Все значения были нормализованы до средней экспрессии HSP47 группы не участвовавших в экспериментах животных и выражали в виде процента HSP47 экспрессии по сравнению с группой не участвовавших в эксперименте животных.
Композиции, описанные на Фиг. 1, являются следующими:
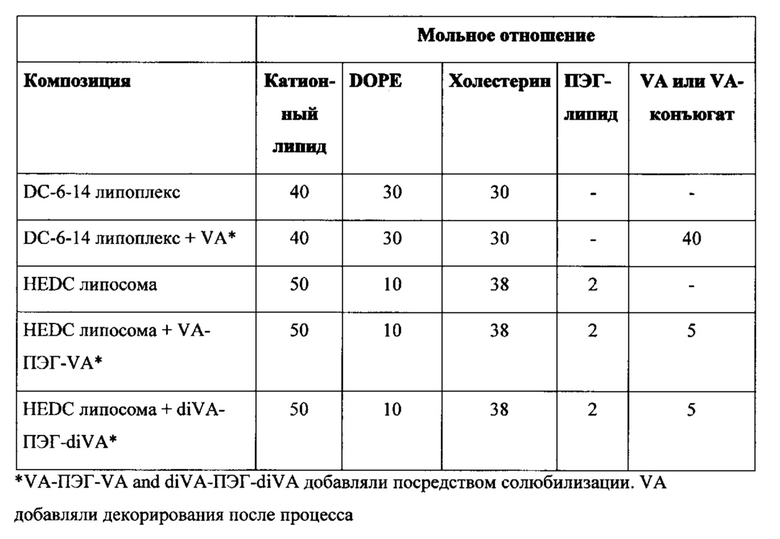
Композиции, описанные на Фиг. 2, являются следующими
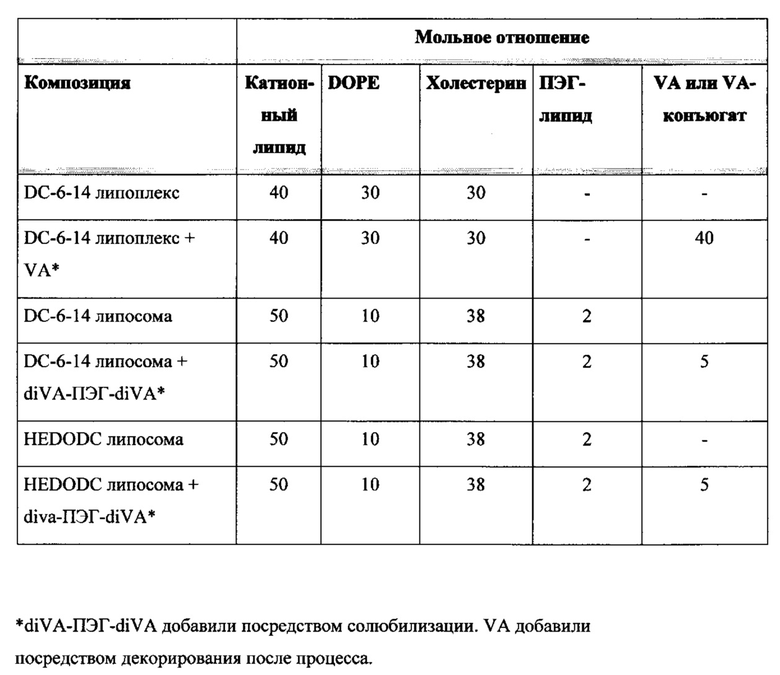
Композиции, описанные на Фиг. 1, являются следующими:
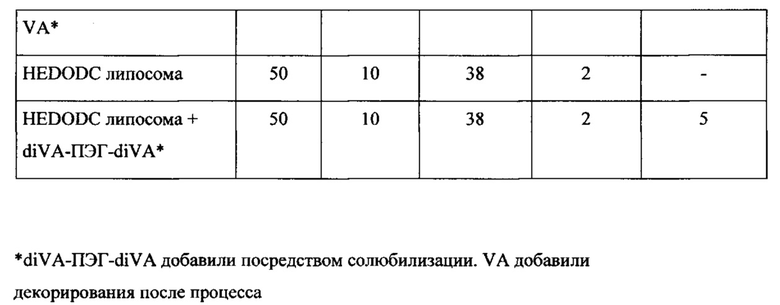
Пример 35: in vitro эффективность (pHSC) - ответ на дозу Описание pHSC in vitro анализа:
Первичные звездчатые клетки печени (pHSCs) в 96-луночном планшете инкубировали с композициями (HEDC : S104 : DOPE : Chol : ПЭГ-DMPE : DiVA, 20:20:30:25:5:2) повышения siPHK концентрации. Через 30 минут клетки промыли и обработали свежей средой для роста и инкубировали при 37°C в течение 48 часов. В это время клетки лизировали и уровни мРНК gp46 и GAPDH измерили посредством анализа количественной ПЦР с обратной транскрипцией (TaqMan®). Уровни мРНК gp46 нормализировали к уровням GAPDH. Нормализированные уровни gp46 выражались как процент от необработанных контрольных клеток. На Фиг. 4 показаны результаты. Усы показывают стандартные отклонения (n=3). Подгонка данных к сигмоидальной кривой доза-ответ с применением Graphpad дала EC50, равную 11.8 нМ.
Пример 36: Токсичность
Описание анализа HepG2 цитотоксичности:
HepG2 клетки, линии адгезивных клеток, полученную из гепатоцеллюлярной карциномы человека, культивировали в MEM/EBSS (Hyclone, Logan, Utah, Cat# SH30024.01) с добавкой 10% FBS (Hyclone, Logan, Utah Cat # SH30910). HepG2 клетки высеивали в 96-луночных черных планшетах Optilux (BD Falcon, Cat # BD353220) при плотности 5000 клеток/лунка всю ночь. Композиции добавили в каждую луну до конечной указанной концентрации siPHK (n=3). Через 48 часов после добавления композиции, жизнеспособность клеток определили с применением CellTiter-Glo Luminescent Cell Viability Assay Kit (Promega, Cat # G7572) согласно инструкциям производителя. Хемилюминесцентный сигнал измерили на Clarity Luminescence Microplate Reader (502-Biotek, Winooski, Vermont). Жизнеспособность вычислили на основе % хемилюминесцентного сигнала в лунке, обработанной композицией, нормализованного к лункам с имитацией обработки.
После исследования композиций с нашими наиболее эффективными катионными липидами с четвертичным амином формулы I были оценены комбинации катионных липидов с четвертичным амином формулы I с их соответствующими ионизируемыми синтетическими предшественниками (как показано в приведенных ниже примерах, i-DC и HEDC, INT4 и DODC, S104 и HES104).

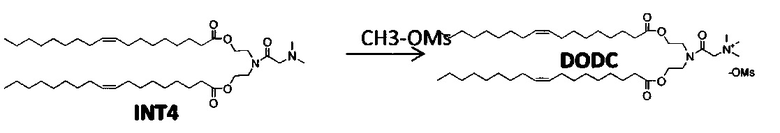
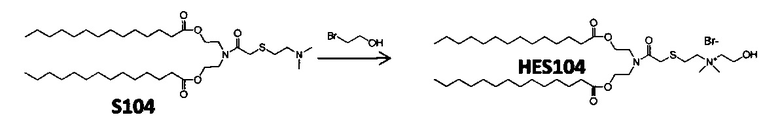
В следующей таблице приводятся примерные результаты для различных композиций. Комбинации катионных липидов с четвертичным амином с ионизируемыми катионными липидами были чрезвычайно и неожиданно менее токсичными, чем липосомы, содержащие один катионный липид (смотрите примеры HEDC по сравнению с HEDC + iDC; и DODC по сравнению с DODC + INT4 в приведенной ниже таблице). Комбинация HEDC + S104 была идентифицирована как другая предпочтительная композиция.
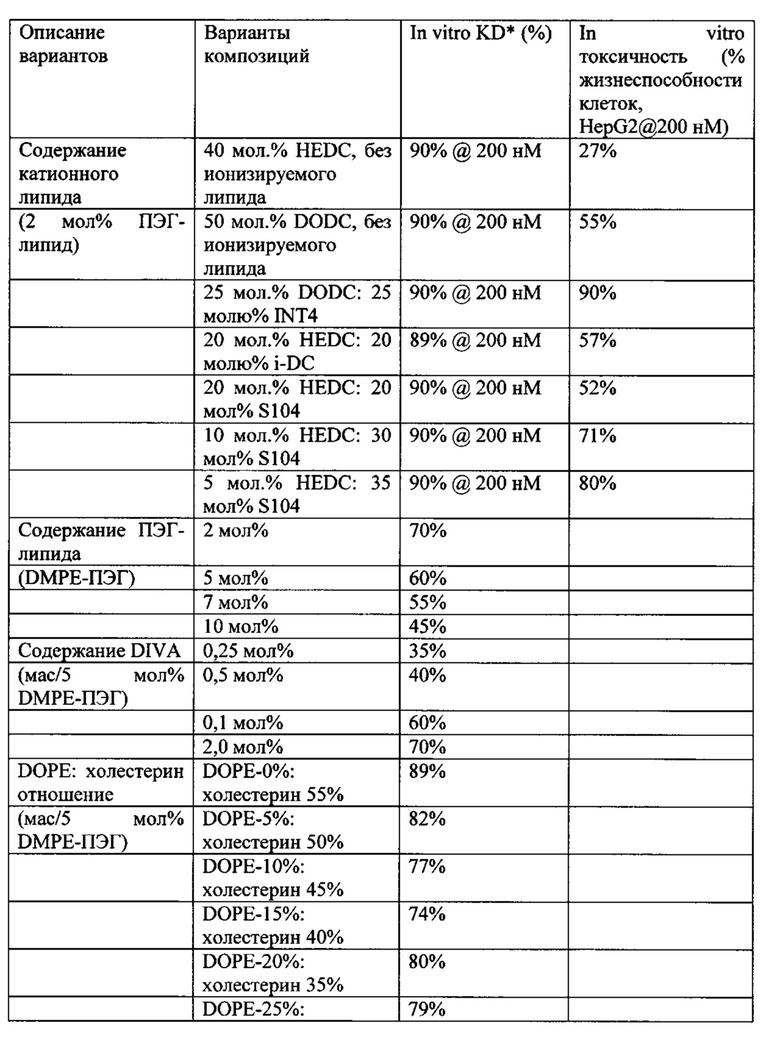
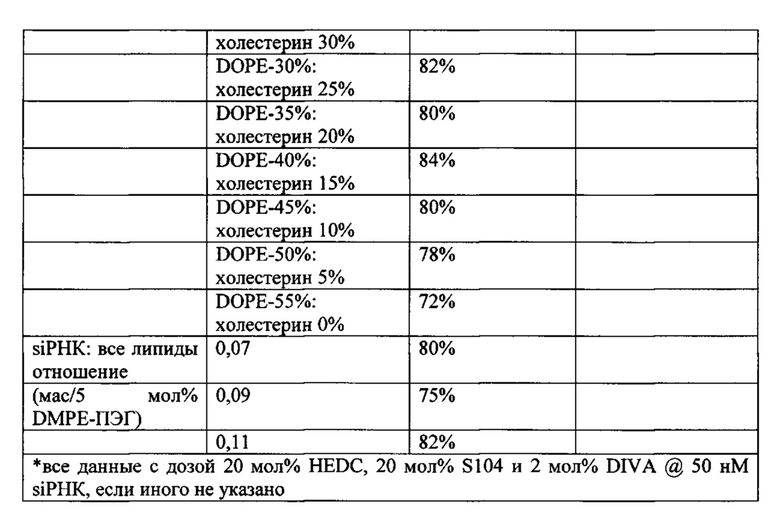
in vivo токсичность
HEDC:S104 (20:20) композиция крайне хорошо переносилась в предварительном in vivo изучении токсичности. Никакой токсичности не наблюдалось, когда композиция вводилась внутривенно при дозах до 25 мг/кг (крыса) и 12 мг/кг (обезьяна). В данной области техники это считается превосходным. Например, Alnylam Pharmaceuticals 1 марта 2012 заявили на конференции AsiaTIDES, что их наименьшая токсичность липидных наночастиц третьего поколения имела NOAEL, равную 10 мг/кг (одна доза, крыса).
Пример 37: in vivo эффективность (DMNQ крысы)
In vivo активность целевой композиции была оценена на модели непродолжительного повреждения печени (упоминается как Quick Model, DMNQ). В этой модели непродолжительное повреждение печени индуцируется обработкой гепатотоксическим агентом, таким как диметилнитрозамин (DMN) и сопровождается оценкой gp46 мРНК уровней. Чтобы индуцировать эти изменения самцов крыс Sprague-Dawley интраперитонеально инъецировали в течение 6 последовательных дней. В конце периода обработки DMN животных случайным образом распредели по группам на основе индивидуальной массы тела животного. Композиции вводили в виде единичной внутривенной дозы и давали через час после последней инъекции DMN. Через двадцать два часа доли печени разрезали и уровни мРНК gp46 и MRPL19 определили посредством анализа количественной ПЦР с обратной транскрипцией (TaqMan®). уровни мРНК для gp46 были нормализованы к уровням MRPL19.
Самцов крыс Sprague-Dawley обрабатывали с DMN при 10 мг/кг на день 1, 2, 3 и 5 мг/кг на день 4, 5, 6 посредством интраперитонеального введения дозы, чтобы индуцировать повреждение печени. Животным (n=8/группа) вводили внутривенные инъекции либо с композициями при дозе 0.5, 0.75, 1.0, 2 мг/кг siPHK в композиции, состоящей из HEDC : S104 : DOPE : Chol : ПЭГ-DMPE : DiVA (20:20:30:25:5:2), или PBS (среда), через один час после последней инъекции DMN. Через двадцать четыре часа вся siPHK была очищена от секции правой доли печени каждого животного и хранилась при 4°C до выделения РНК. Контрольные группы включали группу со средой PBS (DMN-обработанные) и группу не подвергавшихся тестированию животных (необработанные; нет DMN). На Фиг. 5 показаны результаты измерений. После вычитания фоновых значений уровни мРНК gp46 определяли на основе группы не подвергавшихся тестированию животных, все значения для тестовых групп были нормализованы к средней мРНК gp46 группы со средой (выражается как процент от группы со средой). Средний уровень мРНК gp46 после обработки показал доза-зависимый ответ и кривую подогнал к сигмоидальной кривой доза-ответ с получением EC50, равной 0.79 мг/кг.
Пример 38: in vivo эффективность (DMNC крысы)
Самцов крыс Sprague Dawley (130-160 г) обработали DMN посредством интраперитонеального дозирования чтобы индуцировать фиброз печени. Режим обработки DMN составлял 3 раза каждую неделю (понедельник, среда и пятница) 10 мг/кг (то есть, 5.0 мг/мл DMN при дозе 2.0 мл/кг массы тела) в течение первых 3 недель и половина дозы 5 мг/кг (то есть 5 мг/мл DMN при дозе 1.0 мл/кг) с 22 по 57 день. Животным группы плацебо инъецировали PBS (растворитель для DMN) с применением такого же режима. На день 22, 24 часа после последней обработки DMN, образцы крови собрали и проанализировали с помощью биомаркеров заболеваний печени, чтобы подтвердить эффективность DMN обработки. Животных, обработанных DMN, распределили по группам с различной обработкой на основе массы тела и подтвердили, что средние массы тела и диапазоны масс тела животных в каждой группе имеют незначительные различия. Животных из группы предварительной обработки умертвили на день 25, чтобы определить стадию прогрессии заболевания до начала обработки. Обработки композициями, содержащими gp46 siPHK, начали на день 25 с 2 обработками/неделя при конкретной дозе siPHK, в общем 10 раз. На день 59, через 48 часов после последней обработки композицией и 72 часа после последней обработки DMN, животных умертвили посредством ингаляции CO2. Доли печени разрезали и уровни мРНК gp46 и MRPL19 определили посредством анализа количественной ПЦР с обратной транскрипцией (TaqMan®). Уровни мРНК для gp46 были нормализованы к уровням MRPL19.
Самцов крыс Sprague Dawley обрабатывали DMN при 10 мг/кг в течение трех недель (3 раза в неделю) и затем 5.0 мг/мл с 22 по 57 день (три раза в неделю) посредством интраперитонеального введения, чтобы индуцировать фиброз печени. Животным (n = десять/группа) внутривенно инъецировали либо композиции, содержащие HEDC : S104 : DOPE : Chol : ПЭГ-DMPE : DiVA (20:20:30:25:5:2) при 1.5, 1.0, 0.75 и 0.5 мг/кг siPHK, либо PBS (среду) в течение 10 раз (2 раз/неделя), через один час после инъекции DMN. На день 59 вся siPHK была очищена от секции правой доли печени каждого животного и хранилась при 4°C до анализа. Контрольные группы включали группу со средой PBS (DMN-обработанные, PBS обработанные, n=7) и группу с плацебо (обработка PBS вместо DMN и композиции, n=10). На Фиг. 6 показаны результаты измерений. После вычитания фоновых значений уровни мРНК gp46 определяли на основе группы не подвергавшихся тестированию животных, все значения для тестовых групп были нормализованы к средней мРНК gp46 группы со средой (выражается как процент от группы со средой). Животные группы предварительной обработки (n=7) были умерщвлены на день 25 да оценки уровня прогрессии заболевания до начала обработки. One-way Anova анализ с последующим Dunnett’s тестом показал значительное подавление гена gp46 во всех группах с обработкой по сравнению с группой со средой (***, P<0.001).
В следующей таблице суммированы соединения, описанные в настоящей заявке, и результаты, полученные посредством тестирования этих соединений in vivo и in vitro.
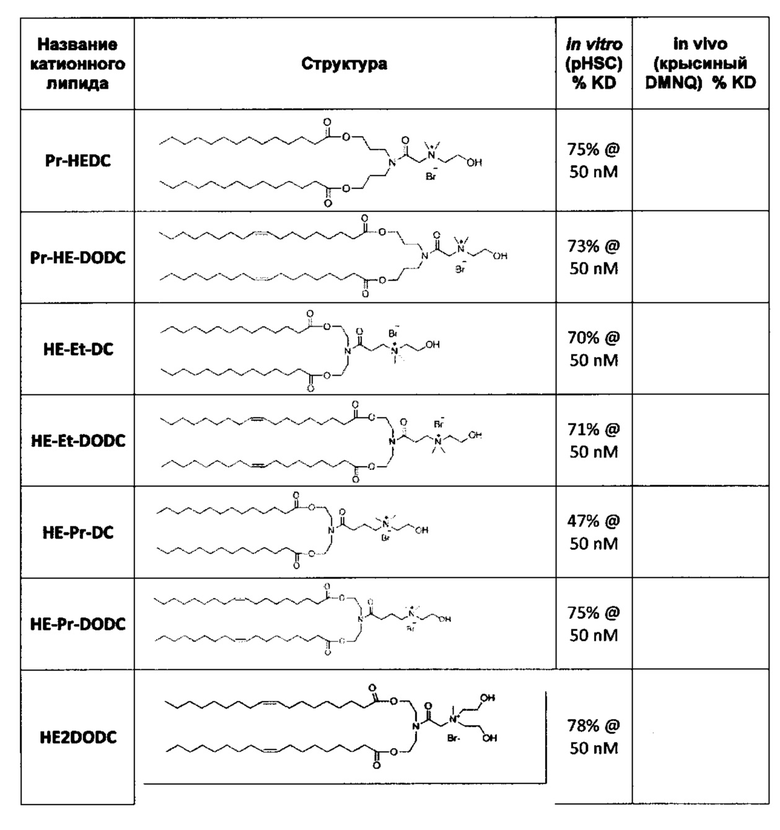
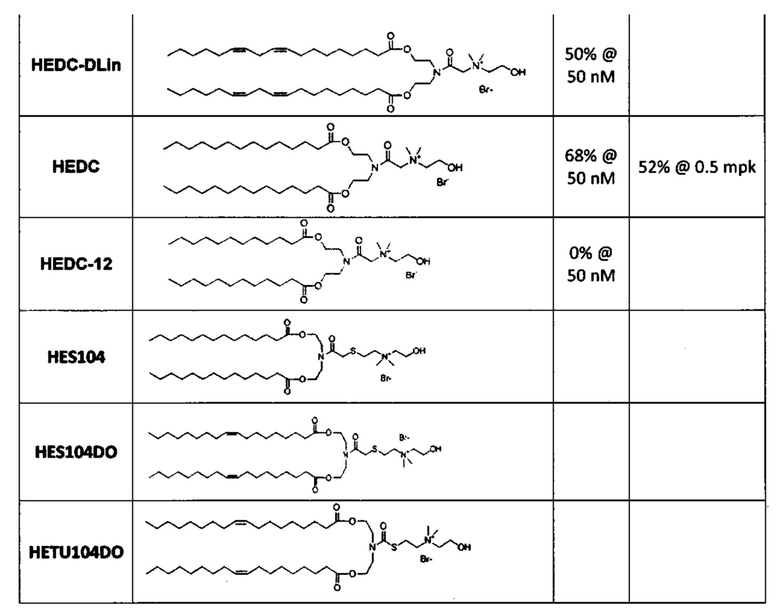
Пример 39: In vivo антилегочный фиброз
S-D самцы крыс (8 крыс/группа, возраст восемь недель, Charles R1ver Laboratories Japan, Inc.) один раз вводили 0.45 мг блеомицина (BLM), растворенного в 0.1 ил соляного раствора, в легкие посредством интратрахеальной канюляции (MicroSprayer, Penn-Century, Inc.) под анестезией с получением модели фиброза легкого, вызванного блеомицином. В этом способе значительный фиброз в общем встречается в легких после 2 недель. Липосомальную композицию (1.5 мг/кг в качестве количества siRNA, 1 мл/кг в объеме, то есть 200 мкл для крысы 200 г) или PBS (1 мл/кг в объеме) вводили крысам через хвостовую вену, начиная со второй недели после введения блеомицина, в общем за 10 раз (на каждый последующий день). Крыс умертвили через два дня после последней обработки, провели гистологическое исследование ткани легких (смотрите Фиг. 30). One way ANOVA и Bonferroni multi сравнительные тесты применяли для оценки статистически значимого различия.
Часть удаленного легкого была зафиксирована формалином обычным способом и подвергли окрашиванию азаном (азокармин, анилиновый сине-оранжевый G раствор).
Как показано по результатам гистологической оценки (T. Ashcroft score) на Фиг. 7, в группе с введением композиции (обработка), размер фиброза был существенно уменьшен.
Пример 40: In vitro оценка VA-siPHK-липосомальных композиций на пониженную эффективность в LX-2 клеточной линии и крысиных первичных звездчатых клетках печени (pHSCs)
LX2 клетки (Dr. S.L. Friedman, Mount Sinai School of Medicine, NY) выращивали в DMEM (Invitrogen) с добавкой 10% фетальной бычьей сыворотки (Invitrogen) при 37°C в инкубаторе с 5% CO2. Клетки трипсинизировали с применением TryPLExpress раствора (Invitrogen) в течение 3 минут при 37°C в инкубаторе. Концентрацию клеток определили посредством подсчета клеток в гемоцитометре и 3000 клеток/лунка высеяли в 96-луночных планшетах. Клетки выращивали в течение 24 часов перед трансфекцией.
Крысиные первичные звездчатые клетки печени (pHSCs) были выделены из крыс Sprague-Dawley согласно ранее опубликованному способу (Nat. Biotechnol. 2008, 26(4): 431-42). pHSCs выращивали в DMEM с добавкой 10% фетальной бычьей сыворотки. Клетки выращивали до двух пассажей после выделения перед их применением для in vitro скрининга. Клетки высеивали при плотности клеток 1000 клеток/лунка в 96-луночных планшетах и выращивали в течение 48 часов перед их применением для трансфекции.
Трансфекция с VA-siPHK-Липосомальные композиции. Способ трансфекции является одинаковым для LX-2 и pHSC клеток. VA-siPHK-липосома или VA siPHK-липоплекс композиции смешали со средой для роста при желательных концентрациях. 100 мкл смеси добавили в клетки в 96-луночном планшете и клетки инкубировали в течение 30 минут при 37°C в инкубаторе с 5% CO2. Через 30 минут среду заменили на свежую среду для роста. Через 48 часов трансфекции клетки обработали с применением Cell-to-Ct® лизисных реагентов (Applied Biosystems) согласно инструкциям производителей.
Количественная (q) ПЦР с обратной транскрипцией для измерения экспрессии HSP47 мРНК. HSP47 и GAPDH TaqMan® анализы и одностадийную ПЦР с обратной транскрипцией master mix получили у Applied Biosystems. Каждая ПЦР реакция включала следующую композицию: одностадийная ПЦР с обратной транскрипцией mix 5 мкл, TaqMan® RT фермент mix 0.25 мкл, зонд анализа экспрессии гена TaqMan® (HSP47) 0.25 мкл, TaqMan® зонд анализа экспрессии гена (GAPDH) 0.5 мкл, свободная от рибонуклеазы вода 3.25 мкл, клеточный лизат 0.75 мкл, общий объем 10 мкл. GAPDH применяли в качестве эндогенного контроля для относительной количественной оценки уровней HSP47 мРНК. Количественную ПЦР с обратной транскрипцией осуществляли в системе ПЦР в реальном времени ViiA 7 (Applied Biosciences) с применением внутреннего способа относительной количественной оценки. Все значения были нормализованы до средней экспрессии HSP47 трансфектированных клеток mock и выражали как процент HSP47 экспрессии по сравнению с mock.
SiPHK упоминаются в протоколах композиций как последовательности двухнитиевых siPHK с 21-мерной нацеливающей HSP47/gp46, где HSP47 (мышиный) и gp46 (крысиный) являются гомологами - один и тот же ген в различных последовательностях:
двухнитиевая siPHK крысиного HSP47-C применялась для in vitro анализа (крысиные pHSC)
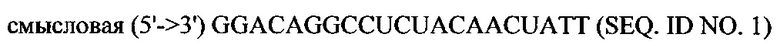
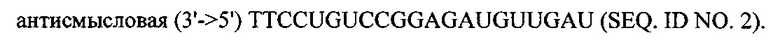
Получение сток-раствора катионного липида. Сток-растворы катионных липидов получали посредством объединения катионного липида с DOPE, холестерином и diVA-ПЭГ-diVA в этаноле при концентрациях 6.0, 5.1 и 2.7 и 2.4 мг/мл соответственно. При необходимости растворы нагревали до около 50°C для облегчения растворения катионных липидов в растворе.
Получение пустых липосом. Сток-раствор катионного липида вводили в быстро перемешиваемую водную смесь 9% сахарозы при 40±1°C через инъекционную иглу (иглы) при скорости 1.5 мл/мин на инъекционный порт. Отношение сток-раствора катионного липида к водному раствору (об./об.) фиксировалось при 35:65. При смешивании самопроизвольно образовывались пустые везикулы. Пустые везикулы затем уравновешивались при 40°C в течение 10 минут прежде чем содержание этанола было уменьшено до ~12%.
Получение липоплекса. Пустые везикулы, полученные согласно вышеописанному способу, разбавляли до конечного объема 1 мМ при концентрации катионного липида при 9% сахарозе. К перемешиваемому раствору добавили 100 мкл 5% глюкозы в свободной от рибонуклеазы воде на каждый миллилитр разбавленной пустой везикулы ("EV") и тщательно перемешивали. 150 мкл 10 мг/мл siPHK раствора в свободной от рибонуклеазы воде затем добавили за один раз и тщательно смешали. Смесь затем разбавили 5% раствором глюкозы с 1.750 мл на каждый миллилитр EV. Смесь перемешивали при около 200 оборотах в минуту при комнатной температуре в течение 10 минут. Применяя полупроницаемую мембрану с ~100000 MWCO системе перекрестно ультрофилтрации, с применением соответственно выбранного перистальтического насоса (например, Midgee Hoop, UFP-100-H24LA), смесь концентрировали до около 1/3 от изначального объема (или желательного объема) и затем диафильтровали 5 раз объем образца, применяя водный раствор, содержащий 3% сахарозы и 2.9% глюкозы. Продукт затем отфильтровали через комбинированный фильтр с размером пор 0.8/0.2 в асептических условиях перед применением.
Образование липосом, содержащих siPHK без-diVA. Катионный липид, DOPE, холестерин и ПЭГ-конъюгированный липид (например, ПЭГ-липид) солюбилизировали в абсолютном этаноле (200 градусов крепости) при молярном отношении 50:10:38:2. SiPHK солюбилизировали в 50 мМ нитратном буфере и температуру довели до 35-40°C. Смесь этанол/липид затем добавили к siPHK-содержащему буферу при перемешивании до самопроизвольного образования загруженных siPHK липосом. Липиды объединили с siPHK с получением конечного общего отношение липид: siPHK равного 15:1 (мас.:мас). Интервал может составлять от 5:1 до 15:1, предпочтительно от 7:1 до 15:1. SiPHK загруженные липосомы подвергли диафильтрации с 0X объемами PBS (pH 7.2) для удаления этанола и замены буфера. Конечный продукт отфильтровали через 0.22 мкм, стерильной марки, PES фильтр для предварительной фильтрации. Этот процесс позволил получить липосомы со средним диаметром частиц 50-100 нм, PDI<0.2 и >85% эффективность включения.
Образование липосом, содержащих siPHK, солюбилизируемые с diVA._siVHK-diVA-липосомальные композиции были получены с применением вышеописанного способа. DiVA-ПЭГ-diVA были солюбилизированы в абсолютном этаноле с другими липидами (катионный липид, DOPE, холестерин и ПЭГ-конъюгированные липиды при отношении 50:10:38:2) до добавления к siPHK содержащему буферу. Молярное отношение diVA-ПЭГ-diVA составляло от 0.1 до 5 (50:10:38:2:0.1-50:10:38:2:5). Этот процесс позволил получить липосомы со средним диаметром частиц 50-100 нм, PDI <0.2, >85% эффективность включения.
Образование siPHK содержащих липосом с катионными липидами._siPHK-diVA-липосомальные композиции и siPHK-липосомальные композиции получили с применением вышеописанного способа. Катионным липидом может быть, например, HEDC, HEDODC, DC-6-14 или любая комбинация этих катионных липидов.
Образование siPHK содержащих липосом декорированных diVA. siPHK-липосомальные композиции получили с применением вышеописанного способа и разбавили до концентрации siPHK 0.5 мг/мл в PBS. Катионным липидом может быть HEDC, HEDODC, DC-6-14 или любая комбинация этих катионных липидов. diVA-ПЭГ-diVA разбавили в абсолютном этаноле (200 градусов крепости) до конечной концентрации в интервале от 10 до 50 мг/мл. Подходящее количество этанольного раствора добавили в siPHK-липосомальный раствор с получением конечного мольного содержания от 2 до 10 мол.%. Раствор подняли и опустили с повторениями пипеткой для смешивания. diVA-ПЭГ-diVA концентрацию и добавляемый объем этанола установили для сохранения добавочного объема >1.0 мкл и конечной концентрации этанола <3% (об./об.). Декорированные липосомы затем равномерно перемешали при температуре окружающей среды в течение 1 часа на обычном шейкере до in vitro или in vivo оценки.
Результаты
На Фиг. 8 показано, что добавление VA-конъюгата к липосомам посредством декорирования улучшило пониженную эффективность siPHK, усилило siPHK активность. ПЭГ-липид. Доза для всех образцов составляла 867 нМ siPHK HSP47-C. Результаты показали, что в каждом случае, где VA-конъюгат добавили к липосомам, siPHK активность усилилась по сравнению с лилпосомами без ретиноида и по сравнению с липосомами, декорированными свободным (неконъюгированным) ретинолом. RNAiMAX был коммерческим реагентом трансфекции.
На Фиг. 9 показано, что добавление VA-конъюгата к липосомам посредством сосолюбилизации улучшило пониженную эффективность siPHK. Это были DODC, содержащие липосомы с VA-конъюгатами, добавленными посредством сосолюбилизации. Композиция представляла собой 50:10:38:2:X, где X=1-10 (DODC : DOPE : холестерин : ПЭГ-липид-конъюгат, мольное отношение). Концентрация в каждом случае составляла 100 нМ siPHK HSP47-C. Результаты показали, что добавление VA-конъюгатов к липосомам посредством сосолюбилизации усиливает siPHK активность.
На Фиг. 10 показано, что добавление VA-конъюгата к липосомам посредством сосолюбилизации значительно улучшило пониженную эффективность siPHK. Результаты включают три различные липосомы, DC-6-14, DODC, HEDODC, с VA конъюгатами, добавленными посредством сосолюбилизации. Композиция была одинаковой для всех, 50:10:38:2, катионный липид:DOPE:холестерин:ПЭГ-липид, с варьированием только катионного липида. Концентрация siPHK составляет 200 нМ siPHK HSP47-C и оинакова для всех случаев. Результаты показали, что добавление VA-конъюгата к липосомам, имеющим различные катионные липиды, значительно усилило siPHK активность, при получении посредством сосолюбилизации.
На Фиг. 11 показано, что добавление VA-конъюгатов к липоплексам, имеющим DC-6-14 катионный липид, посредством сосолюбилизации и siPHK покрытие внешней поверхности липосомы усиливает siPHK активность. Композицией является 40% композиция липоплекса, 40:30:30, BC-6-14 : DOPE : холестерин. Концентрация для всех образцов составляла 867 нМ siPHK HSP47-C. Результаты показали, что добавление VA-конъюгата к липоплексам посредством сосолюбилизации усиливает активность siPHK.
На Фиг. 12 показано добавление VA-конъюгатов к липоплексам, сформированным посредством сосолюбилизации, по сравнению с липоплексами с VA-конъюгатом, добавленным посредством декорирования. Это результаты для DC-6-14 и DODC липоплексов. Композиция состоит из 40:30:30, DC-6-14 : DOPE : холестерин. Концентрация для каждого образца составляет 867 нМ siPHK HSP47-C. Добавление VA-конъюгата посредством сосолюбилизации значительно улучшило пониженную эффективность in vitro, относительно VA-конъюгатов, добавленных посредством декорирования.
Пример 41: In vivo эксперименты
Самок мышей C57B1/6(Charles River) с диапазоном массы 24-30 грамм применяли для этого исследования. Животных случайным образом распределили по массе в 10 групп по 10 животных в каждой. Все процедуры над животными были одобрены Bio-Quant’s IACUC и/или Attending Veterinarian, как необходимо, и условия содержания всех животных были отмечены и документированы. Мышей анестезировали с помощью изофлурана и обескровили через полую нижнюю вену.
Двухнитиевая siPHK мышиного HSP47-C применялась в композициях для in vivo анализа (мышиная модель CCl4)
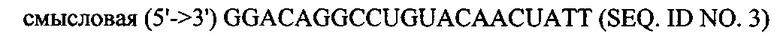
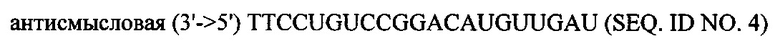
Повышение экспрессии белка теплового шока 47 (HSP47) было индуцировано посредством интраперитонеальной инъекции CCl4 (CCl4 в оливковом масле, 1:7 (об./об.), 1 л на грамма массы тела добавляя каждый день в течение 7 дней (день 0, 2, 4, 6). С третьего дня мышей обрабатывали в течение четырех последовательных дней (день 3, 4, 5, 6) с композициями липосомы или липоплекса согласно настоящему изобретению или PBS посредством внутривенной инъекции в хвостовую вену. Одну группу из десяти мышей (не подвергавшихся экспериментам) оставили без CCl4 обработки и без внутривенной инъекции, и она служила в качестве контрольной группы для нормальной экспрессии гена HSP47.
Режим эксперимента
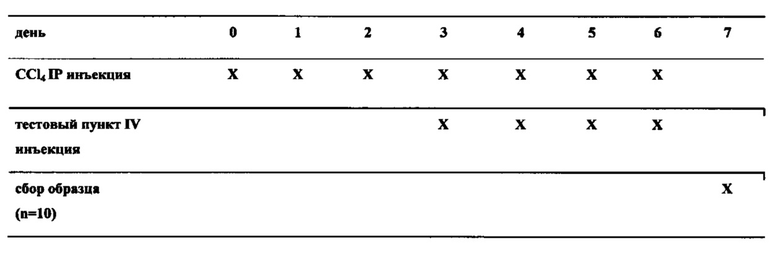
На седьмой день и около 24 часа после последней внутривенной инъекции, всех оставшихся мышей умертвили и печень опрыскивали PBS, прежде чем взять образцы печени для ПЦР анализа. Из каждой мышиной печени взяли образец около 150 мг и поместили в 1.5 мл RNAlater стабилизирующего реагента (Qiagen) и сохраняли при 2-8°C до анализа. Образцы печени собирали из областей поврежденной и/или некрозной чистой и помеченной печени.
Общие уровни РНК мышей экстрагировали с применением RNeasy графы (Qiagen) согласно инструкциям производителя. 20 нг общей РНК применяли для количественной ПЦР с обратной транскрипцией для измерения экспрессии HSP47. HSP47 и GAPDH TaqMan® анализы и одностадийную ПЦР с обратной транскрипцией master mix приобрели у Applied Biosystems. Каждая ПЦР реакция включала следующую композицию: одностадийная ПЦР с обратной транскрипцией mix 5 мкл, TaqMan® RT фермент mix 0.25 мкл, зонд анализа экспрессии гена TaqMan® (HSP47) 0.25 мкл, TaqMan® зонд анализа экспрессии гена (GAPDH) 0.5 мкл, свободная от рибонуклеазы вода 3.25 мкл, клеточный лизат 0.75 мкл, общий объем 10 мкл. GAPDH применяли в качестве эндогенного контроля для относительной количественной оценки уровней HSP47 мРНК. Количественную ПЦР с обратной транскрипцией осуществляли в системе ПЦР в реальном времени ViiA 7 (Applied Biosciences) с применением внутреннего способа относительной количественной оценки. Все значения были нормализованы до средней экспрессии HSP47 группы не участвовавших в экспериментах животных и экспрессировали в виде процента HSP47 экспрессии по сравнению с группой не участвовавших в эксперименте животных.
Пример 42: IN VITRO ЭФФЕКТИВНОСТЬ НАЦЕЛИВАЮЩЕГО КОНЪЮГАТА ЖИРОРАСТВОРИМОГО ВИТАМИНА HEDC : S104 : DOPE : Chol : ПЭГ-DMPE : DiVA
Липосомальные композиции с 50 нМ siPHK были протестированы. Липосомами были либо: HEDC : S104 : DOPE : Chol : ПЭГ-DMPE : DiVA (+DiVA), либо контроли без составляющих витамина A (-DiVA) и их инкубировали в 96-луночных культуральных планшетах, содержащих крысиные звездчатые клетки печени, в течение 30 минут. Через 30 минут среду заменили на свежую среду для роста. Через сорок восемь часов клетки лизировали и уровни мРНК gp46 и GAPDH измерили посредством анализа количественной ПЦР с обратной транскрипцией (TaqMan®) и уровни gp46 были нормализованы до GAPDH уровней.
In vitro эффективность (pHSC), эффект 2% DiVA siPHK была эффективна с 2% diVA и имела EC50 14 нМ. PHSCs в 96-луночном планшете инкубировали композицией без составляющих витамина A для нацеливания (-DiVA) или композиции, которые составляют составляющие витамина A (+DiVA) при 50 нМ siPHK. Через 30 минут, среду заменили на свежую среду для роста. Через сорок восемь часов клетки лизировали и уровни мРНК gp46 и GAPDH измерили посредством анализа количественной ПЦР с обратной транскрипцией (TaqMan®) и уровни gp46 были нормализованы до GAPDH уровней. Нормализованные уровни gp46 выражались как процент от контрольных клеток mock. Усы показывают стандартные отклонения (n=3). средний уровень gp46 после обработки, содержащей DiVA, значительно отличается от фиктивной контрольной обработки (mock) (P<0.001) на основе одностороннего t-теста.
СРАВНЕНИЕ DiVA И satDiVA
Липосомальные композиции протестировали в крысиных pHSC в течение 30 минут в 96-луночных планшетах. Через 48 часов клетки обработали с применением Cells-to-Ct® лизисных реагентов (Applied Biosystems) и мРНК HSP47 количественно определили посредством qRT-PCR. HSP47 экспрессия была нормализована к необработанному контролю. EC50 определили посредством измерения пониженной экспрессии HSP47 (KD) на шести полулогарифмических дозах siPHK и подгонки данных к "Classic sigmoidal dose response function" в Graphpad Prism® 5.04.
Результаты показали, что и DiVA и Sat DiVA повысили KD эффективность (смотрите приведенную ниже таблицу, а также Фиг. 15). EC50 составляет 12 нМ для DiVA, и EC50 составляет 14 нМ для Sat DiVA.

Конъюгат на основе ретиноида по сравнению с конъюгатом без ретиноида
Конъюгаты на основе ретиноида, как было обнаружено, являются более эффективными in vitro по сравнению с эквивалентами без ретиноида (смотрите 4TTNBB и 4Муг по сравнению с эквивалентами конъюгатами с ретиноидом satDiVA и DiVA

Пример 43: IN VITRO ЭФФЕКТИВНОСТЬ НАЦЕЛИВАЮЩЕГО КОНЪЮГАТА ЖИРОРАСТВОРИМОГО ВИТАМИНА HEDC : S104 : DOPE : Chol : ПЭГ-DMPE : DiVA
In vivo активность целевой композиции была оценена на модели непродолжительного повреждения печени (упоминается как Quick Model, DMNQ). В этой модели непродолжительное повреждение печени индуцируется обработкой гепатотоксическим агентом, таким как диметилнитрозамин (DMN) и сопровождается оценкой gp46 мРНК уровней. Чтобы индуцировать эти изменения самцов крыс Sprague-Dawley интраперитонеально инъецировали в течение 6 последовательных дней. В конце периода обработки DMN животных случайным образом распредели по группам на основе индивидуальной массы тела животного. Композиции вводили в виде единичной внутривенной дозы и давали через час после последней инъекции DMN. Через двадцать два часа доли печени разрезали и уровни мРНК gp46 и MRPL19 определили посредством анализа количественной ПЦР с обратной транскрипцией (TaqMan®®). уровни мРНК для gp46 были нормализованы к уровням MRPL19.
Результаты (Фиг. 16) показывают, что корреляция между количеством конъюгата ретиноида и эффективностью очевидна. Только 0.25 мол.% требуется, чтобы увидеть значительный эффект на крысиной модели DMNQ. С 2 мол.% DiVA наблюдается сильное понижение экспрессии gp46. На Фиг. 16 показано, что у самцов крыс Sprague-Dawley, которые были обработаны с DMN при 10 мг/кг на день 1, 2, 3 и 5 мг/кг на день 4, 5, 6 посредством интраперитонеального введения дозы, индуцировалось повреждение печени. Животным (n=8/группа) вводили внутривенные инъекции либо с композициями, содержащими 0, 0.25, 0.5, 1 и 2% DiVA при дозе 0.75 мг/кг siPHK или PBS (среда), через один час после последней инъекции DMN. Через двадцать четыре часа вся siPHK была очищена от секции правой доли печени каждого животного и хранилась при 4°C до выделения РНК. Контрольные группы включали группу со средой PBS (DMN-обработанные) и группу не подвергавшихся тестированию животных (необработанные; нет DMN). После вычитания фоновых значений уровни мРНК gp46 определяли на основе группы не подвергавшихся тестированию животных, все значения для тестовых групп были нормализованы к средней мРНК gp46 группы со средой (выражается как процент от группы со средой).
Самцов крыс Sprague Dawley (130-160 г) обработали DMN посредством интраперитонеального дозирования чтобы индуцировать фиброз печени. Режим обработки DMN составлял 3 раза каждую неделю (понедельник, среда и пятница) 10 мг/кг (то есть, 5.0 мг/мл DMN при дозе 2.0 мл/кг массы тела) в течение первых 3 недель и половина дозы 5 мг/кг (то есть 5 мг/мл DMN при дозе 1.0 мл/кг) с 22 по 57 день. Животным группы плацебо инъецировали PBS (растворитель для DMN) с применением такого же режима. На день 22, 24 часа после последней обработки DMN, образцы крови собрали и проанализировали с помощью биомаркеров заболеваний печени, чтобы подтвердить эффективность DMN обработки. Животных, обработанных DMN, распределили по группам с различной обработкой на основе массы тела и подтвердили, что средние массы тела и диапазоны масс тела животных в каждой группе имеют незначительные различия.
Животных из группы предварительной обработки умертвили на день 25, чтобы определить стадию прогрессии заболевания до начала обработки. Обработки композициями, содержащими gp46 siPHK, начали на день 25 с 2 обработками/неделя при конкретной дозе siPHK, в общем 10 раз. На день 59, через 48 часов после последней обработки композицией и 72 часа после последней обработки DMN, животных умертвили посредством ингаляции С02. Доли печени разрезали и уровни мРНК gp46 и MRPL19 определили посредством анализа количественной ПЦР с обратной транскрипцией (TaqMan®). Уровни мРНК для gp46 были нормализованы к уровням MRPL19.
Содержание статей, патентов и заявок на патент и все другие документы и сведения в электронном виде, которые указаны или цитируются в настоящем документе, приведены в качестве ссылки во всей их полноте в той же степени, как если было бы отдельно указано, что каждая публикация должна быть включена посредством ссылки.
Заявители оставляют за собой право физически включить в публикацию все без исключения материалы и информацию из любых таких статей, патентов, заявок на патент или других физических и электронных документов.
Специалисту в данной области будет очевидно, что различные замены и модификации могут быть внесены в изобретение согласно настоящему описанию без отклонения от объема и сущности изобретения. Таким образом, такие дополнительные варианты реализации находятся в рамках настоящего изобретения и формулы изобретения. Настоящее изобретение учит специалистов в данной области проверять различные сочетания и/или замены химических модификаций согласно настоящему описанию для получения конструкций нуклеиновых кислот с улучшенной активностью для опосредования активности RNAi. Такая улучшенная активность может включать в себя улучшенную стабильность, улучшенную биодоступность и/или улучшенную активацию клеточных ответов, опосредующих RNAi. Таким образом, конкретные варианты согласно настоящему описанию не являются ограничивающими, и специалисты в данной области с легкостью поймут, что можно исследовать определенные комбинации изменений согласно настоящему описанию без неоправданных экспериментов для идентификации молекул нуклеиновых кислот с улучшенной активностью RNAi.
Изобретения наглядно согласно настоящей заявке можно осуществлять на практике в отсутствие любого элемента или элементов, ограничения или ограничений, не конкретно согласно настоящему описанию. Таким образом, например, неопределенный артикль и определенный артикль и похожие определения в контексте изобретения (особенно в контексте следующих пунктов формулы) следует толковать как единственное и множественное число, если не указано иначе или из контекста не следует противоположное. Термины «содержащий», «имеющий», «включающий» и т.п. следует понимать в широком смысле и без ограничения (например, означающими «включая, но не ограничиваясь ими»). Перечисление пределов значений в настоящем описании направлено только на то, чтобы служить сокращенным способом обозначения отдельного значения, попадающего в предел, если не указано иное, и каждое отдельное значение включено в описание, как если бы было отдельно перечислено. Все способы согласно настоящему описанию можно осуществлять в любом порядке, если не указано иное, или не следует иначе из контекста. Использование любых примеров (например, «такие как») согласно настоящему описанию предназначено только для лучшего освещения изобретения и не создает ограничений объема изобретения, если не указано иначе. Ни одно выражение в описании не должно быть истолковано как указание, что любой незаявленный элемент является важным для практики изобретения. Кроме того, термины и выражения, используемые в настоящем описании, были использованы 10 в качестве условия описания, а не ограничения, и указанные термины и выражения не нацелены на исключение любых эквивалентов показанных и описанных функций или их частей, наоборот, признается, что в рамках заявленного изобретения возможны различные модификации. Таким образом, следует понимать, что хотя изобретение было конкретно раскрыто в предпочтительных вариантах и дополнительных функциях, специалисты в данной области могут прибегать к модификациям и изменениям изобретений, изложенных в настоящей публикации, и считается, что такие изменения и вариации находятся в рамках данного изобретения.
Изобретение было описано широко и в общем в настоящей заявке. Каждый узкий вид и подродовая группа, входящие в общее раскрытие информации, также являются частью изобретения. Это включает обобщенное описание изобретения с оговоркой или отрицательным ограничением, удаляющим любой предмет из рода, вне зависимости от того, изложен ли вырезанный материал в настоящем описании. Другие варианты находятся в пределах следующей формулы. Кроме того, если функции и аспекты изобретения, описанного в терминах групп Маркуша, специалистам в данной области будет понятно, что изобретение также описывается в терминах отдельных членов или подгруппы членов группы Маркуша.
| название | год | авторы | номер документа |
|---|---|---|---|
| СОЕДИНЕНИЯ ДЛЯ НАЦЕЛЕННОЙ ДОСТАВКИ ЛЕКАРСТВЕННОГО СРЕДСТВА И УСИЛЕНИЯ АКТИВНОСТИ siPHK | 2012 |
|
RU2769872C2 |
| ЛИПИДЫ ДЛЯ КОМПОЗИЦИЙ ДЛЯ ДОСТАВКИ ТЕРАПЕВТИЧЕСКИХ АГЕНТОВ | 2013 |
|
RU2658007C2 |
| СПОСОБ ПОЛУЧЕНИЯ ЛИПИДНЫХ НАНОЧАСТИЦ ДЛЯ ДОСТАВКИ ЛЕКАРСТВЕННОГО СРЕДСТВА | 2012 |
|
RU2647476C2 |
| ОДНОРАЗОВАЯ СИСТЕМА ДЛЯ СТЕРИЛЬНОГО ПОЛУЧЕНИЯ ЧАСТИЦ ИЗ ЛИПИДОВ И НУКЛЕИНОВЫХ КИСЛОТ | 2012 |
|
RU2642640C2 |
| ФУЗОГЕННЫЕ СОЕДИНЕНИЯ ДЛЯ ДОСТАВКИ БИОЛОГИЧЕСКИ АКТИВНЫХ МОЛЕКУЛ | 2018 |
|
RU2808990C2 |
| СРЕДСТВО ДЛЯ ЛЕЧЕНИЯ ФИБРОЗА КИШЕЧНИКА | 2012 |
|
RU2637372C2 |
| ЛИПОСОМЫ С РЕТИНОИДОМ ДЛЯ УСИЛЕНИЯ МОДУЛЯЦИИ ЭКСПРЕССИИ HSP47 | 2012 |
|
RU2628694C2 |
| ДИСУЛЬФИДНЫЙ ПОЛИКАТИОННЫЙ АМФИФИЛ, КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ С НЕЙТРАЛЬНЫМ ФОСФОЛИПИДОМ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2016 |
|
RU2610271C1 |
| КОМПОЗИЦИЯ ДЛЯ ВОССТАНОВЛЕНИЯ НОРМАЛЬНОЙ ТКАНИ ИЗ ФИБРОЗНОЙ ТКАНИ | 2011 |
|
RU2650796C2 |
| АНТАГОНИСТЫ VRI ВАНИЛОИДНОГО РЕЦЕПТОРА НА ОСНОВЕ ИОНОНА | 2007 |
|
RU2447064C2 |



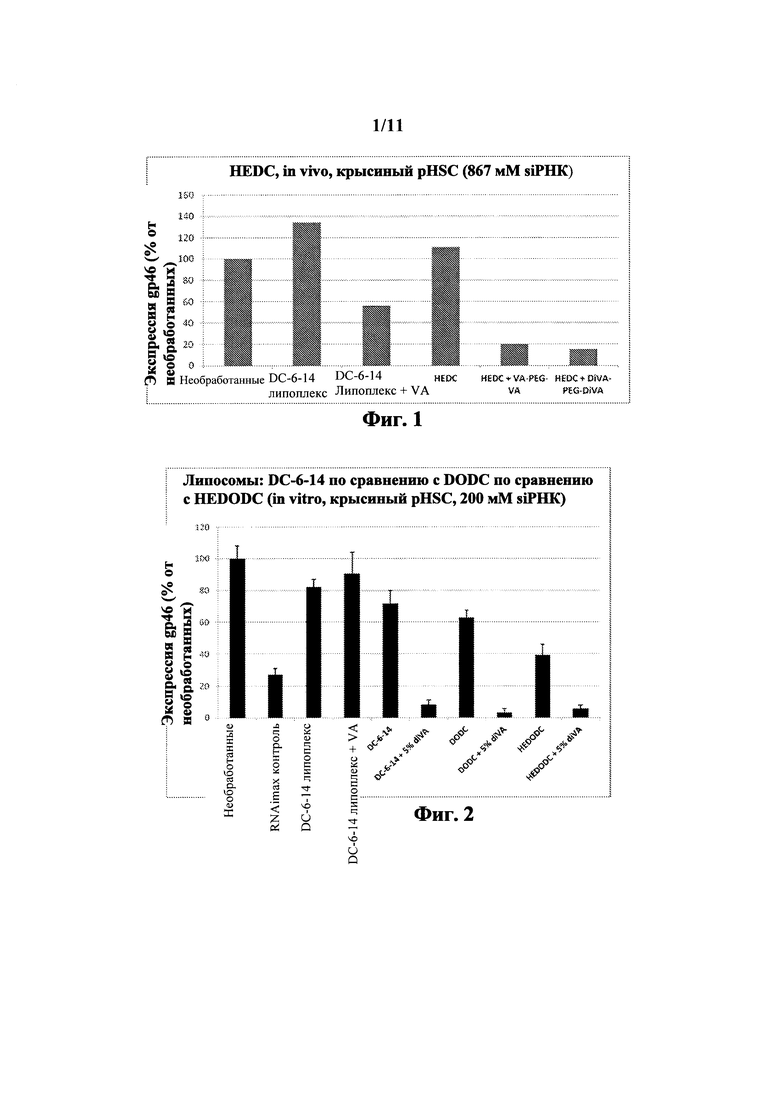
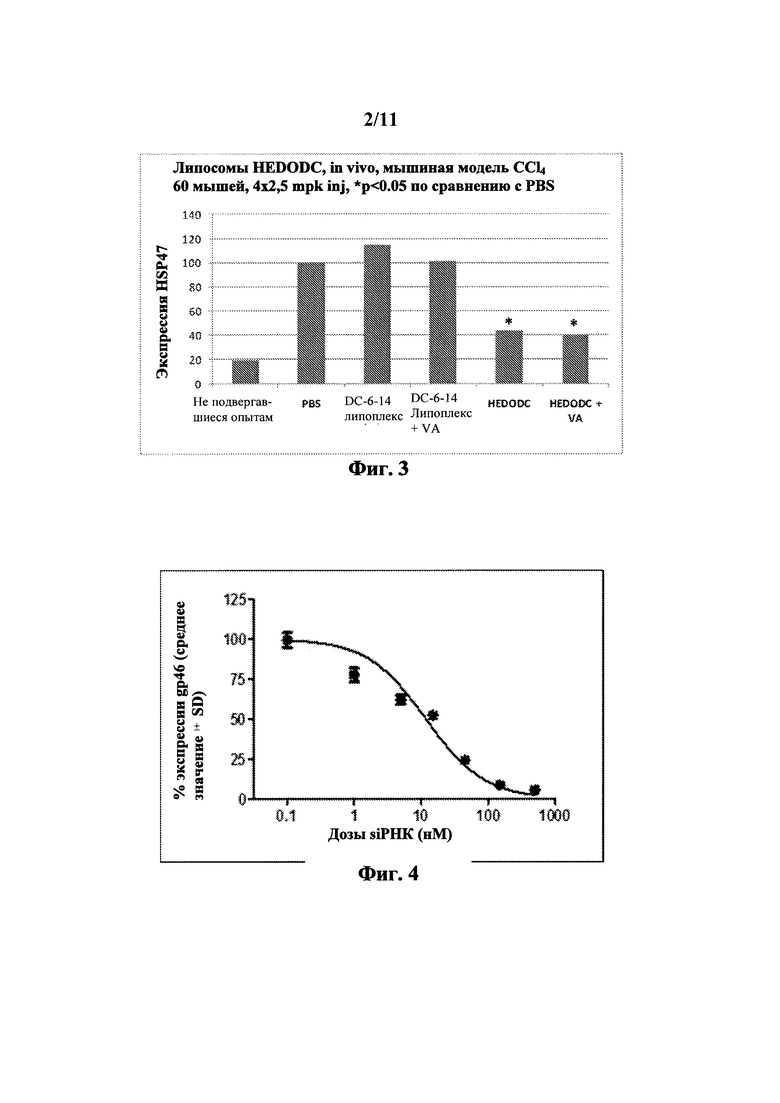
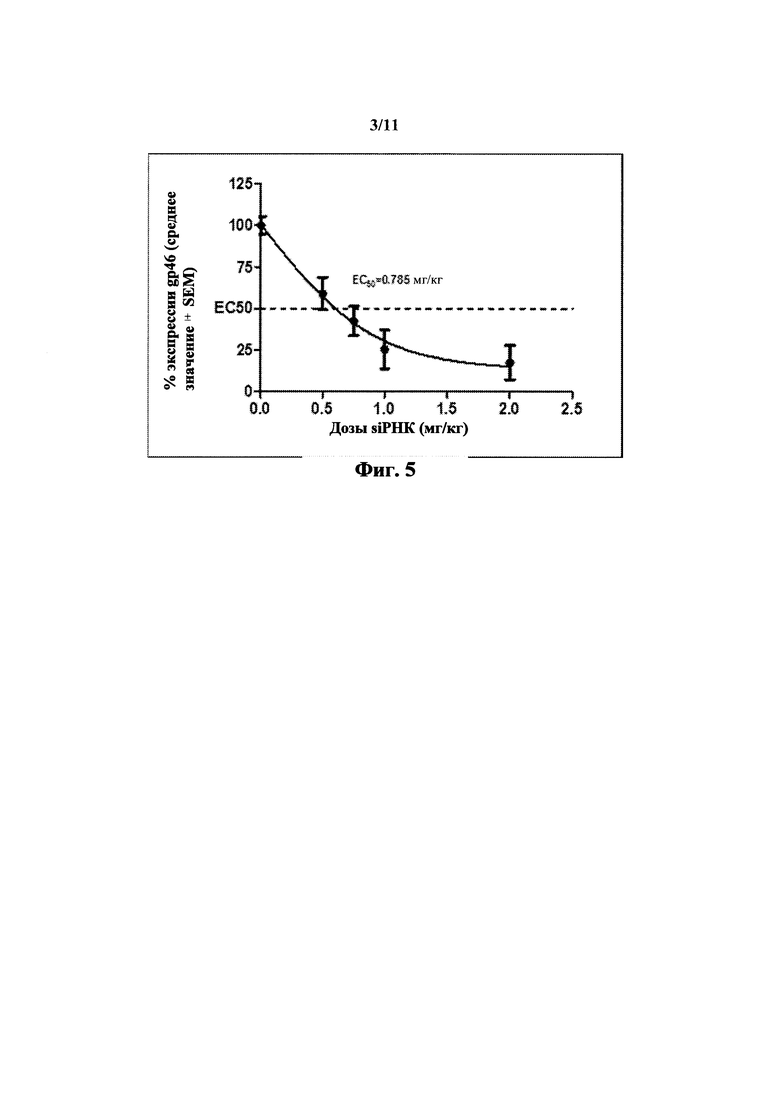
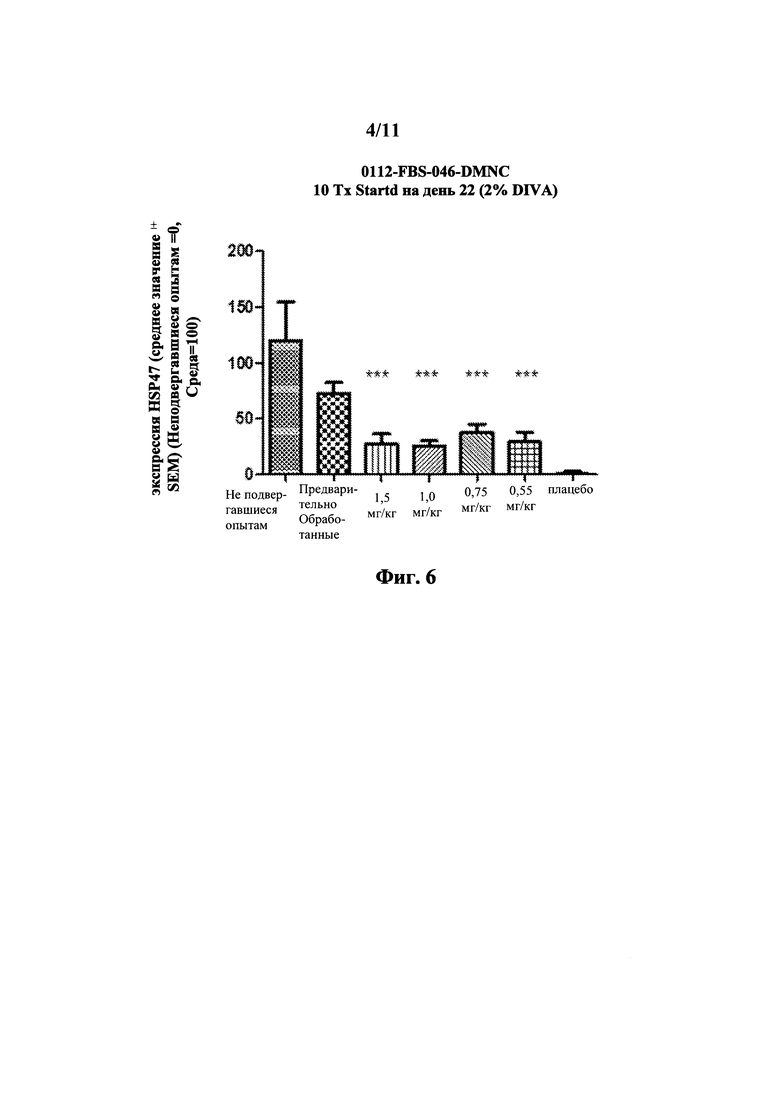
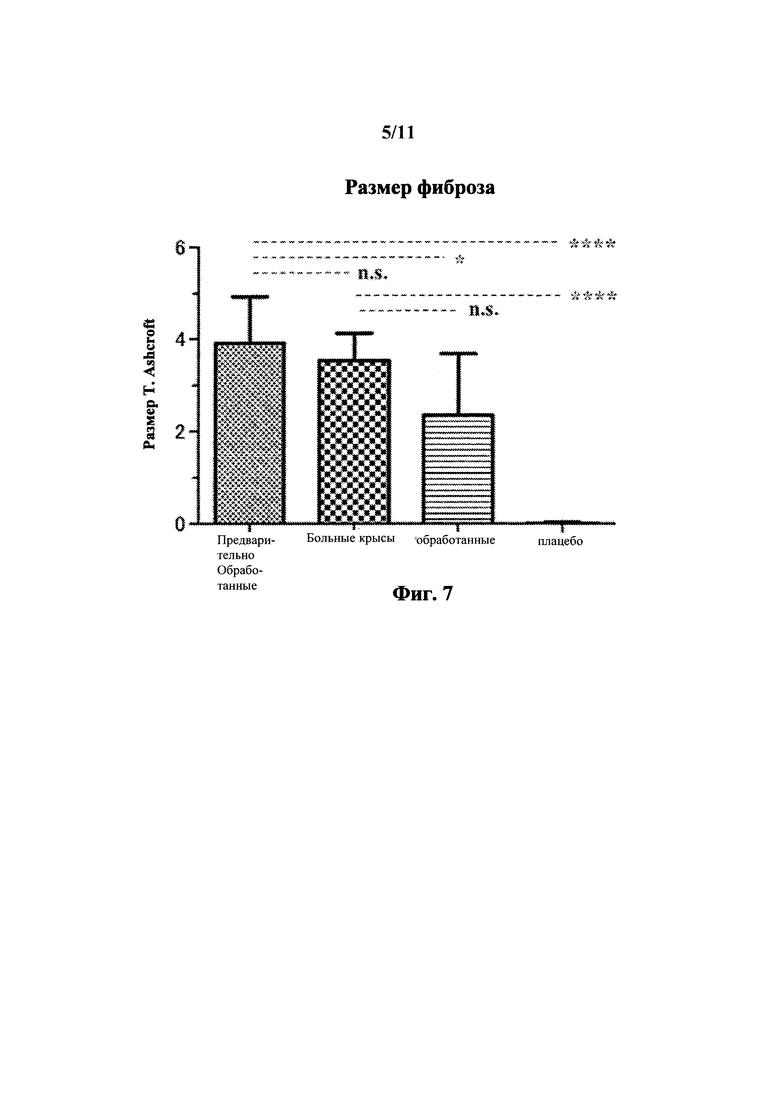
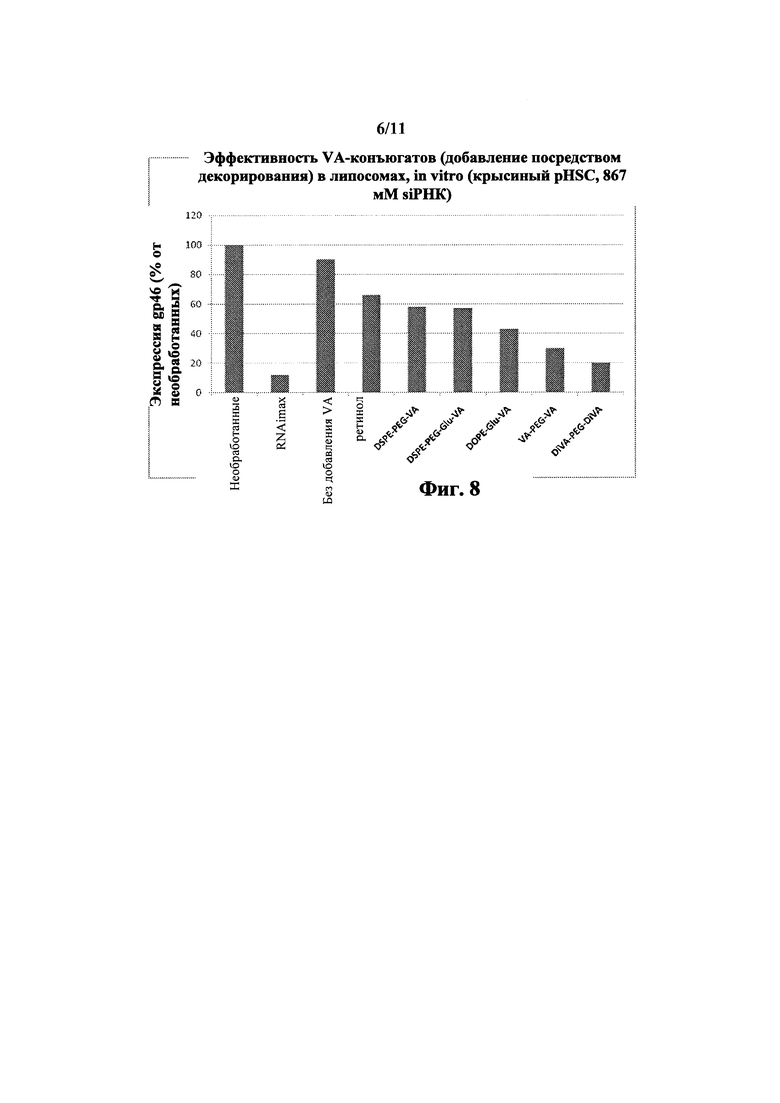

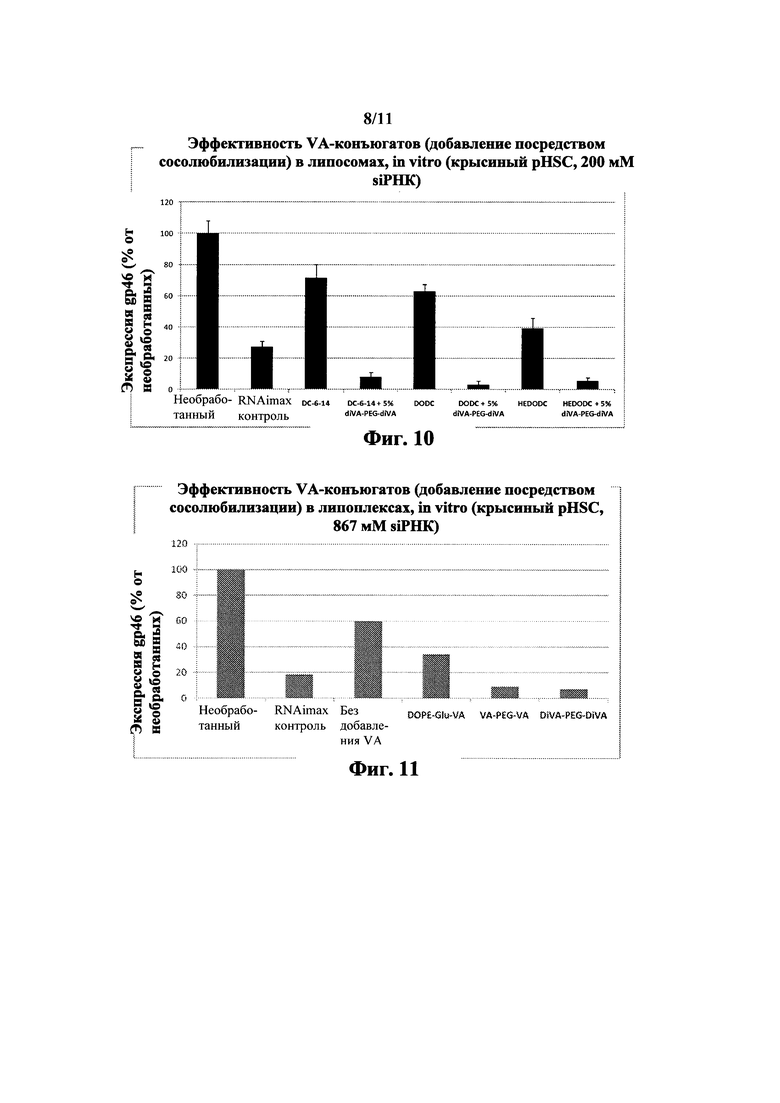
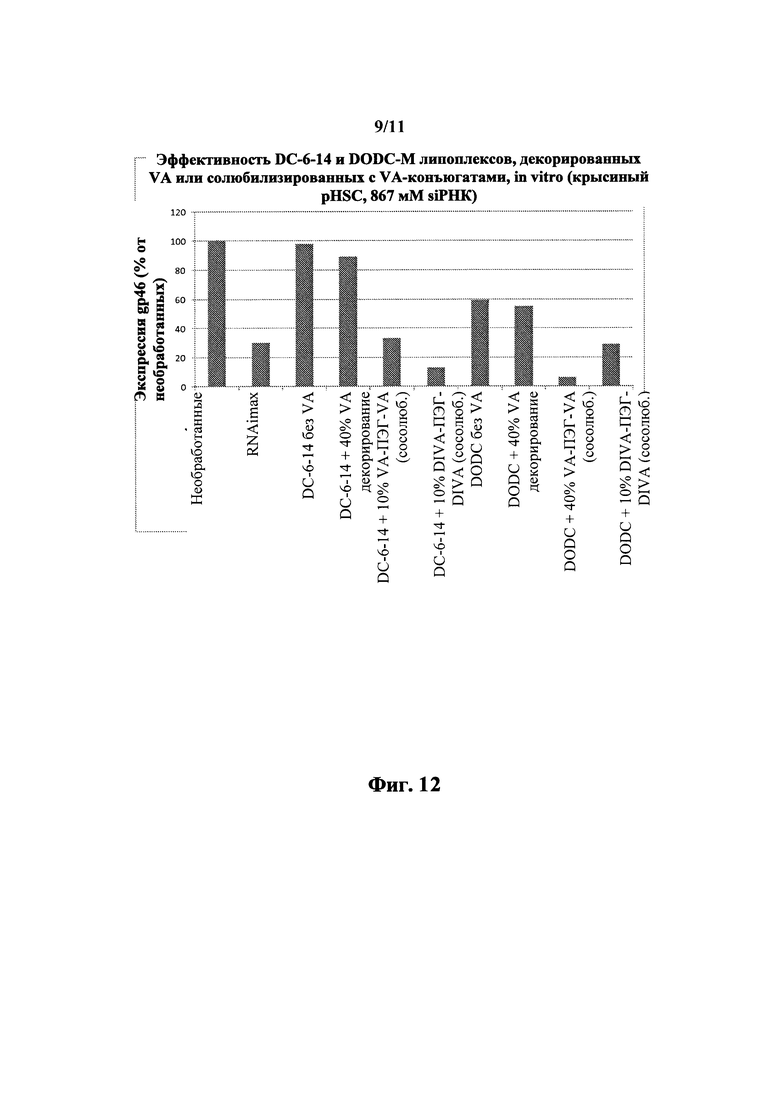
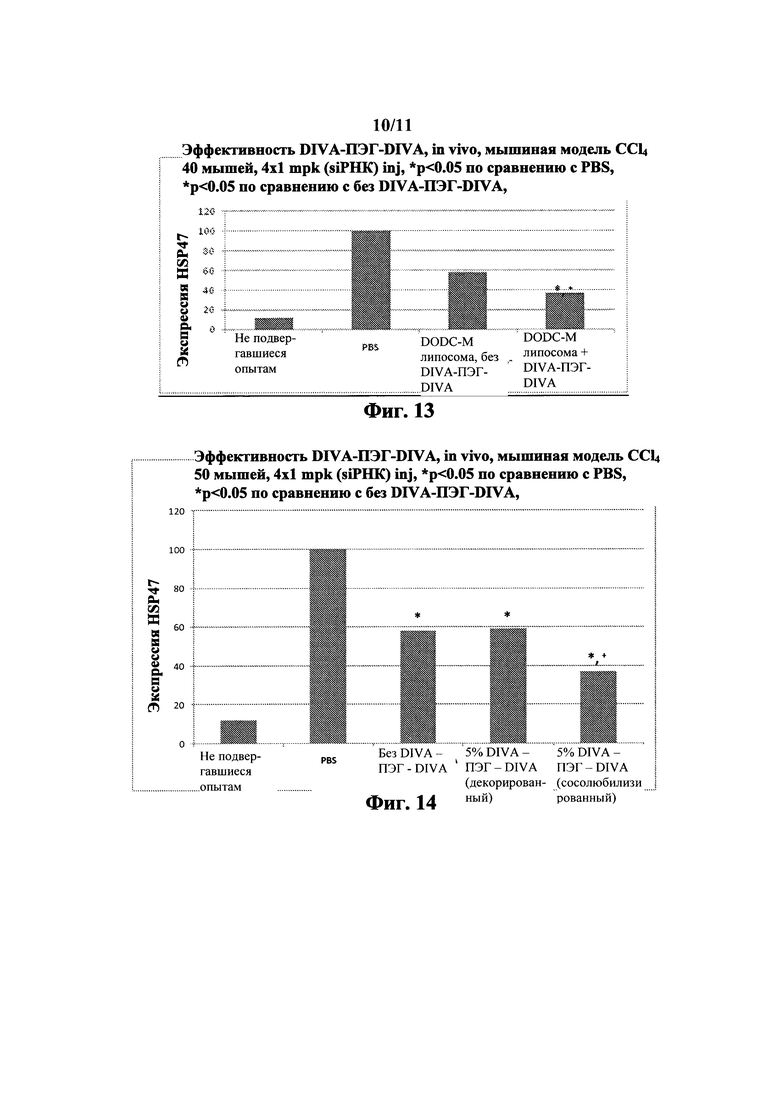
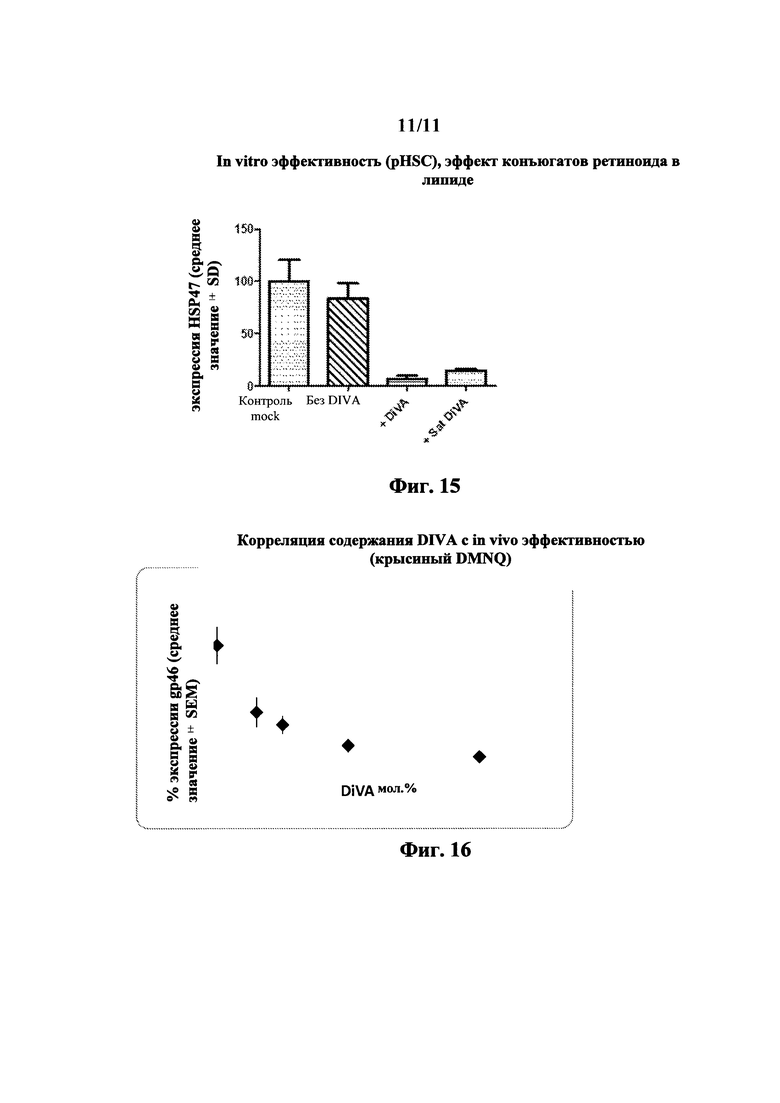
Изобретение относится к соединениям формулы I, которые могут найти применение для улучшения доставки терапевтических лекарственных средств и усиления их активности. В формуле I R1 и R2 независимо выбираются из группы, состоящей из С12-С18 алкильной, С12-С18 алкенильной и олеильной группы; R3 и R4 независимо выбираются из группы, состоящей из С1-С6 алкила и С2-С6 алканола; X выбирается из группы, состоящей из -СН2- и -S-, или отсутствует; Y выбирается из -(CH2)n и -S(CH2)n, где n=1-4; а=1-4; b=1-4; с=1-4 и Z является противоионом. Изобретение относится также к специфичному к звездчатым клеткам носителю для лекарственного средства, содержащему специфичное к звездчатым клеткам количество молекулы ретиноида и катионного липида, состоящего из соединения формулы I. 2 н. и 29 з.п. ф-лы, 16 ил., 14 табл., 43 пр.
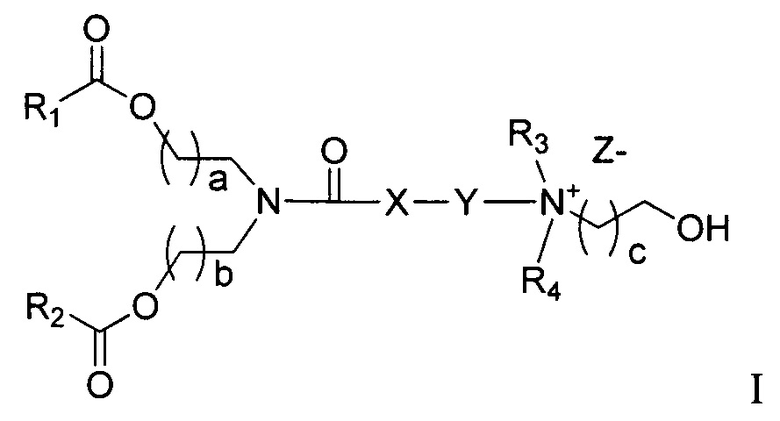
1. Соединение формулы I
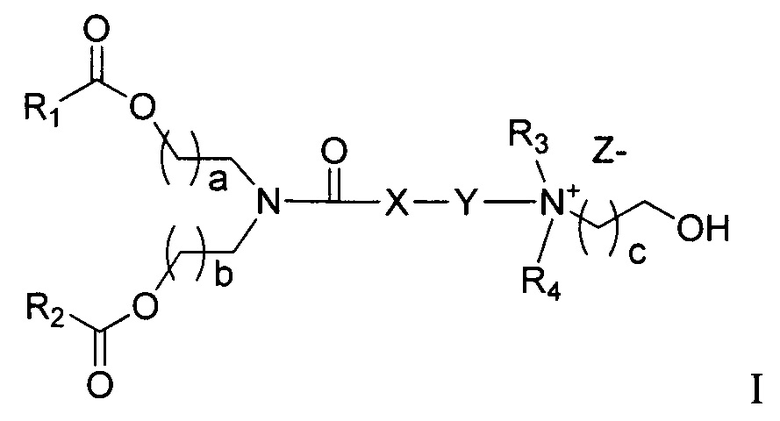 ,
,
где R1 и R2 независимо выбираются из группы, состоящей из С12-С18 алкильной, С12-С18 алкенильной и олеильной группы; где R3 и R4 независимо выбираются из группы, состоящей из С1-С6 алкила и С2-С6 алканола; где X выбирается из группы, состоящей из -СН2- и -S-, или отсутствует; где Y выбирается из -(CH2)n и -S(CH2)n, где n=1-4; где а=1-4; где b=1-4; где с=1-4; и где Z является противоионом.
2. Соединение по п.1, где противоионом является галоген или мезилат.
3. Соединение по п.1, которое выбирается из группы, состоящей из
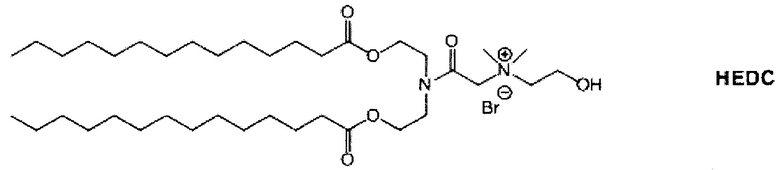
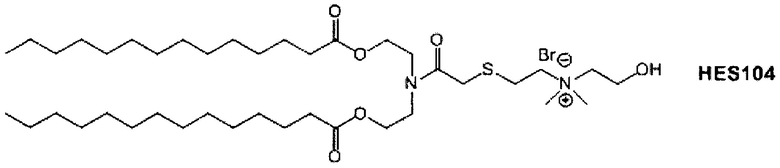
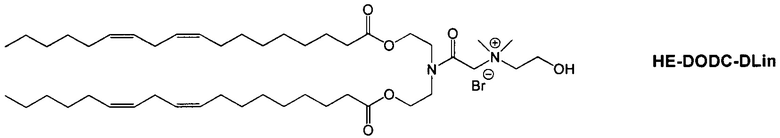
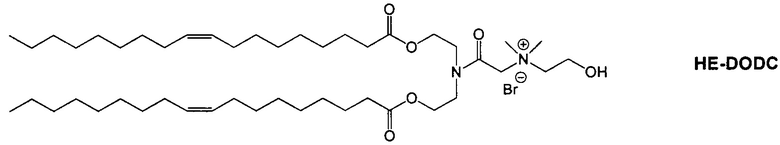
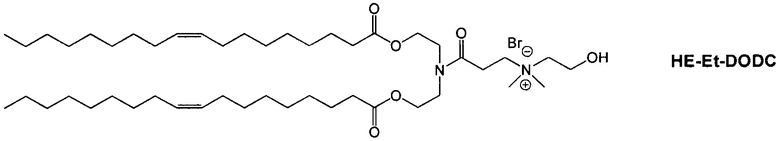
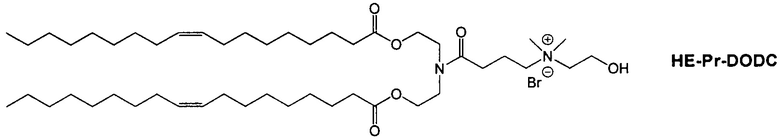
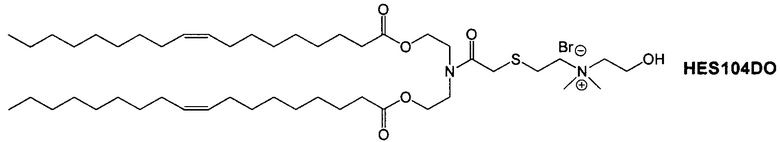
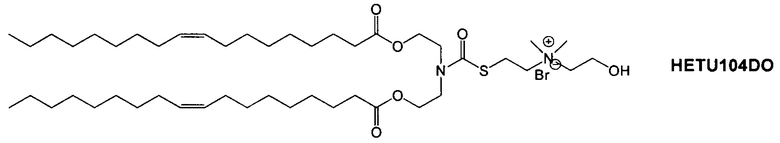 .
.
4. Специфичный к звездчатым клеткам носитель для лекарственного средства, содержащий специфичное к звездчатым клеткам количество молекулы ретиноида и катионного липида, состоящего из соединения по п.3.
5. Носитель для лекарственного средства по п.4 в форме липосомы, содержащей липидные молекулы.
6. Носитель для лекарственного средства по п.4, где молекулой ретиноида является соединение формулы II:
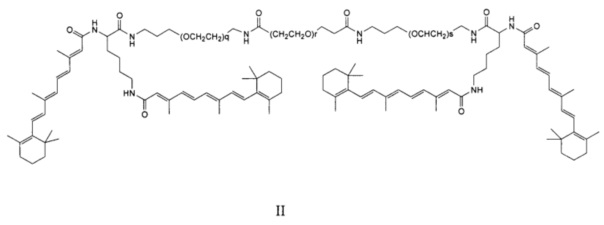 ,
,
где q, r и s каждый независимо равен 1, 2, 3, 4, 5, 6, 7, 8, 9 или 10; или энантиомер, диастереомер или смесь их стереоизомеров.
7. Носитель для лекарственного средства по п.6, где соединением формулы II является
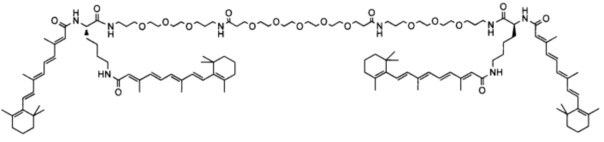 .
.
8. Носитель для лекарственного средства по п.5, где молекула ретиноида, по меньшей мере, частично выставлена на внешней поверхности носителя для лекарственного средства, прежде чем носитель для лекарственного средства достигает звездчатой клетки.
9. Носитель для лекарственного средства по п.4, где ретиноид составляет от 0,2 до 20 мол.% от липидных молекул.
10. Носитель для лекарственного средства по п.5, где катионный липид присутствует в концентрации 5-50 мол.%.
11. Носитель для лекарственного средства по п.5, где катионным липидом является HEDC:
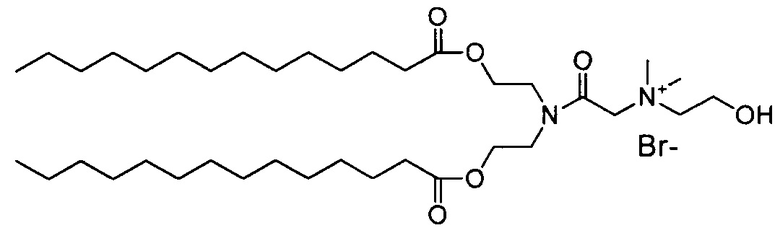 .
.
12. Носитель для лекарственного средства по п.5, где липидные молекулы дополнительно включают липид, конъюгированный с молекулой полиэтиленгликоля (ПЭГ-липид).
13. Носитель для лекарственного средства по п.12, где ПЭГ-липид присутствует в концентрации от 1 до 10 мол.%.
14. Носитель для лекарственного средства по п.12, где ПЭГ-липидом является ПЭГ-димиристоилфосфатидилэтаноламин.
15. Носитель для лекарственного средства по п.5, где липидные молекулы дополнительно содержат ионизируемый катионный липид.
16. Носитель для лекарственного средства по п.15, где ионизируемым катионным липидом является S104:
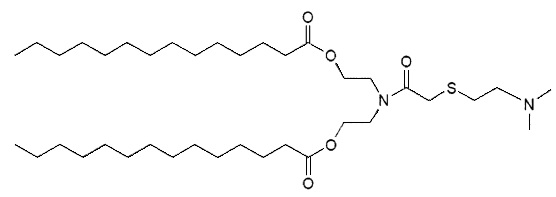 .
.
17. Носитель для лекарственного средства по п.15, где ионизируемый катионный липид присутствует в концентрации от 5 до 45 мол.%.
18. Носитель для лекарственного средства по п.5, где липидные молекулы дополнительно содержат комбинацию катионного липида с четвертичным амином и ионизируемого катионного липида.
19. Носитель для лекарственного средства по п.18, где ионизируемым катионным липидом является непосредственный синтетический предшественник катионного липида с четвертичным амином.
20. Носитель для лекарственного средства по п.18, где катионным липидом с четвертичным амином является HEDC и ионизируемым катионным липидом является S104.
21. Носитель для лекарственного средства по п.5, где липидные молекулы дополнительно содержат некатионный липид.
22. Носитель для лекарственного средства по п.21, где некатионным липидом является фосфолипид.
23. Носитель для лекарственного средства по п.21, где фосфолипидом является диолеоилфосфатидилэтаноламин.
24. Носитель для лекарственного средства по п.21, где фосфолипид присутствует в концентрации от 0 до 55 мол.%.
25. Носитель для лекарственного средства по п.21, где некатионным липидом является холестерин.
26. Носитель для лекарственного средства по п.21, где холестерин присутствует в концентрации от 0 до 55 мол.%.
27. Носитель для лекарственного средства по п.4, дополнительно содержащий нуклеиновую кислоту.
28. Носитель для лекарственного средства по п.27, где нуклеиновой кислотой является siРНК, которая способна подавлять экспрессию hsp47 мРНК в звездчатой клетке.
29. Носитель для лекарственного средства по п.27, дополнительно содержащий siРНК.
30. Носитель для лекарственного средства по п.29, где siРНК инкапсулируется липосомой и не доступна для водной среды.
31. Носитель для лекарственного средства по п.29, где siРНК образуется на внешней поверхности липосомы и доступна для водной среды.
| Устройство для сортировки каменного угля | 1921 |
|
SU61A1 |
| Устройство для сортировки каменного угля | 1921 |
|
SU61A1 |
| Приспособление для суммирования отрезков прямых линий | 1923 |
|
SU2010A1 |
| МОДИФИЦИРОВАННЫЕ АМИНОКИСЛОТЫ, КОМПОЗИЦИИ ДЛЯ ДОСТАВКИ БИОЛОГИЧЕСКИ АКТИВНЫХ АГЕНТОВ В ВЫБРАННЫЕ БИОЛОГИЧЕСКИЕ СИСТЕМЫ, СПОСОБ ПОЛУЧЕНИЯ ТАКИХ КОМПОЗИЦИЙ И ФАРМАКОЛОГИЧЕСКАЯ КОМПОЗИЦИЯ | 1996 |
|
RU2203268C2 |

