Область техники, к которой относится изобретение
Настоящее изобретение относится к антагонистам ванилоидного рецептора, именно к антагонистам TRPV1.
Уровень техники
Ванилоидный рецептор типа 1 (TRPV1 Transient Receptor Potential Vanilloid 1) играет основную роль в развитии поствоспалительной гипералгезии; таким образом, лиганды TRPV1 могут быть клинически пригодным в качестве анальгезирующих и противовоспалительных лекарственных средств.
Известно, что соединения, полученные из природных продуктов и называемые капсаициноидами и резинифероноидами, являются лигандами TRPV1. Среди них ретванил, ваниламид ретиноевой кислоты, является потенциальным агонистом [1].
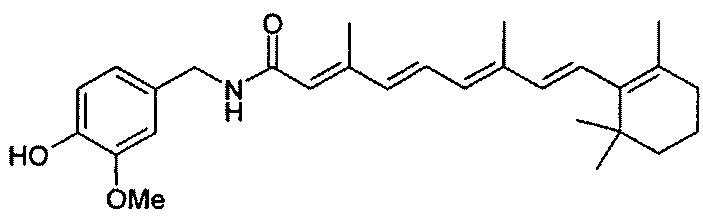
Ber. der Deutschen Chem. Gesellschaft, vol. 70, pp. 1009-1012 раскрывает синтез следующих соединений:
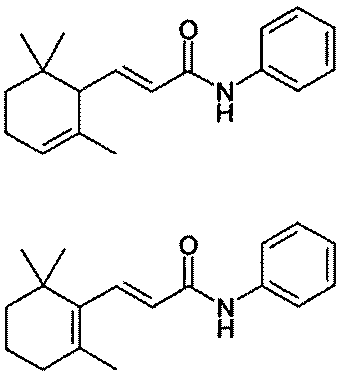
но не упоминает о их биологических свойствах.
WO 03/024920 отмечает использование ретиноидов для лечения артритов и воспалительных дерматологических заболеваний.
Chem. Pharm. Bull. 43(1) 100-107 (1995) раскрывает, в частности, следующие соединения:
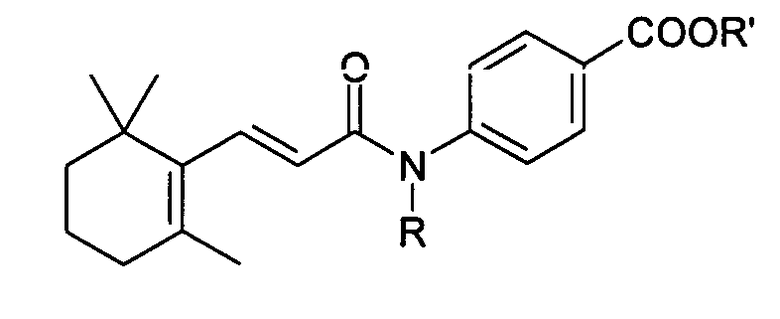
где R представляет собой атом водорода и R' представляет собой атом водорода или метил и их ретиноидную активность.
WO 03/049702, JOC vol. 48, no. 1, 2005, pp. 71-90 и Neuropharmacology, vol. 46, no. 1, 2004, pp. 133-149 раскрывает N-ариламиды коричной кислоты, содержащие группу, которая может быть представлена следующим образом:
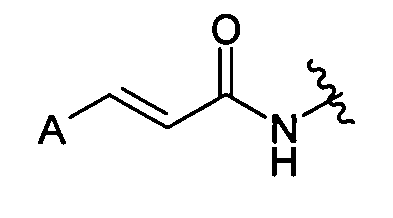
где А представляет замещенный арил. Эти соединения являются антагонистами ванилоидного рецептора и могут быть использованы для лечения ряда воспалительных состояний.
Описание изобретения
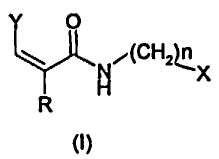
Настоящее изобретение относится к ингибиторам TRPV1 формулы (I)
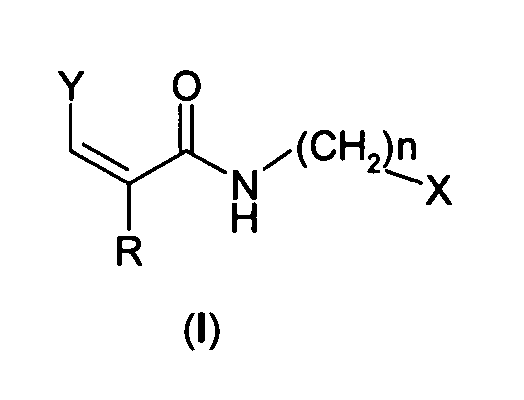
где:
Y представляет собой группу формулы:
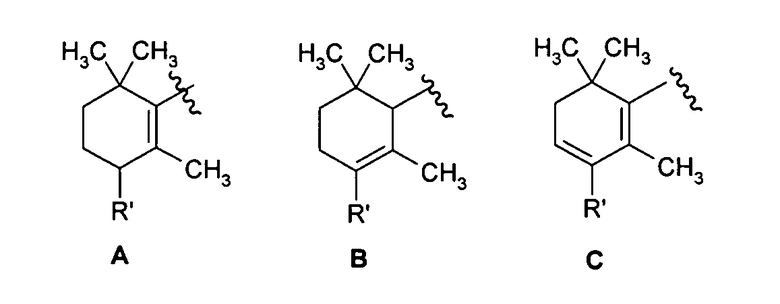
в которой:
R' выбирают из атомов водорода, галогена, гидрокси, (С1-С6)алкила, (С2-С6)алкенила, (С3-С6)алкинила, (С1-С6)алкокси, (С1-С6)алкиламино, фенила, нафтила, фенокси, нафтокси или фениламино, ароматические циклы которых необязательно замещены одним или более атомом галогена, гидрокси, (С1-С4)алкильной, (С1-С4)алкокси и трифторметильной группами;
R представляет собой метил или атом водорода;
n представляет собой 0 или 1;
X выбирают из фенила, пиридинила, нафтила, хинолинила и изохинолинила, необязательно замещенных одним или более группами, выбранными из атома галогена, гидрокси, (С1-С4)алкила, (С1-С4)алкокси и трифторметила;
за исключением следующих соединений:
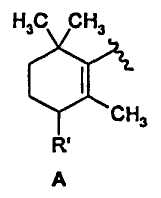
Согласно первому предпочтительному варианту осуществления изобретение относится к соединениям формулы (I), где n представляет собой 0 и X представляет собой 5-изохинолинил. Среди них особенно предпочтительными являются соединения, где R представляет собой атом водорода и Y представляет собой группу формулы:
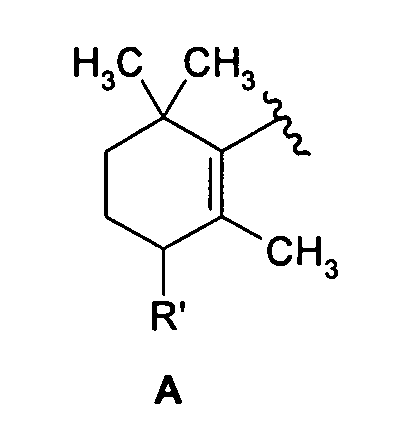
где R' имеет вышеуказанные значения, более предпочтительно означает атом водорода, метокси или фенокси, необязательно замещенный, как указано выше.
Примеры соединений формулы (I) представляют собой следующие:
(2Е)-N-(4-хлорфенил)-3-(2,6,6-триметилциклогекс-1-енил)акриламид;
(2Е)-N-(4-хлорбензил)-3-(2,6,6-триметилциклогекс-1-енил)акриламид;
(2Е)-N-(изохинолин-5-ил)-3-(2,6,6-триметилциклогекс-1-енил)акриламид;
(2Е)-N-(4-хлорфенил)-3-(2,6,6-триметилциклогекс-2-енил)акриламид;
(2Е)-3-(2,6,6-триметилциклогекс-1-енил)-N-(нафталин-1-ил)акриламид;
(2Е)-N-(4-хлорфенил)-3-(2,6,6-триметил-3-феноксициклогекс-1-енил)акриламид;
(2Е)-N-(3-метоксифенил)-3-(2,6,6-триметилциклогекс-1-енил)акриламид;
(2Е)-N-(5-хлорпиридин-2-ил)-3-(2,6,6-триметилциклогекс-1-енил)акриламид;
(2Е)-N-(4-хлорфенил)-3-(2,6,6-триметилциклогекса-1,3-диенил)акриламид;
(2Е)-N-(4-(трифторметил)фенил)-3-(2,6,6-триметилциклогекс-1-енил)акриламид;
(2Е)-3-(2,6,6-триметилциклогекс-1-енил)-N-(хинолин-3-ил)акриламид;
(2Е)-3-(2,6,6-триметилциклогекс-1-енил)-N-(хинолин-5-ил)акриламид;
(2Е)-N-(изохинолин-5-ил)-3-(3-метокси-2,6,6-триметилциклогекс-1-енил)акриламид;
(2Е)-N-(изохинолин-5-ил)-3-(2,6,6-триметил-3-феноксициклогекс-1-енил)акриламид;
(2Е)-N-(изохинолин-5-ил)-3-(3-(3-метоксифенил)-2,6,6-триметилциклогекс-1-енил)акриламид;
(2Е)-3-(3-(4-хлорфенокси)-2,6,6-триметилциклогекс-1-енил)-N-(изохинолин-5-ил)акриламид;
(2Е)-3-(3-(4-фторфенокси)-2,6,6-триметилциклогекс-1-енил)-N-(изохинолин-5-ил)акриламид;
(2Е)-3-(3-(3-фторфенокси)-2,6,6-триметилциклогекс-1-енил)-N-(изохинолин-5-ил)акриламид;
(2Е)-3-(3-(3,4-дифторфенокси)-2,6,6-триметилциклогекс-1-енил)-N-(изохинолин-5-ил)акриламид.
Соединения формулы (I) могут быть получены стандартными методами, такими как взаимодействие соединения формулы (II)
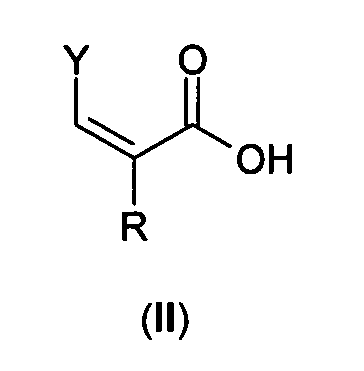
где Y и R соответствуют вышеуказанному определению, и карбоксильная группа активирована подходящим способом для реакции амидирования
с коммерчески доступным соединением формулы (III)
X(CH2)nNH2 (III)
где X соответствует вышеуказанному определению.
Изобретение далее иллюстрируется посредством следующих примеров и схем.
ПРИМЕРЫ
Все коммерчески доступные соединения были куплены в Aldrich и использовались без дополнительной очистки. За ходом реакций следили методом тонкослойной хроматографии на силикагеле (покрытые пластины F254 Merck), пятна рассматривали под УФ-излучением и проявляли водным раствором KMnO4. Флэш-хроматографию проводили с использованием силикагеля Merck (230-240 меш). Спектры 1H-ЯМР регистрировали на спектрометре Varian 400 МГц, используя ТМС в качестве внутреннего стандарта. Масс-спектры были получены на спектрометре Waters-Micromass ZMD. Температуры плавления определяли в аппарате Buchi-Tottoli и не исправляли.
Пример 1 - (2Е)-N-(изохинолин-5-ил)-3-(2,6,6-триметилциклогекс-1-енил)акриламид Ia (схема 1)
Кислота 1 была получена из коммерчески доступного β-ионона галоформной реакцией, как описано в литературе [2]. 1,0 ммоль (194 мг) кислоты 1 растворили в 8 мл безводного ДМФА. EDCI (1-этил-3-(3-диметиламинопропил)карбодиимид) (1,2 экв., 1,2 ммоль, 230 мг), HOBt (N-гидроксибензотриазол) (1,2 экв., 1,2 ммоль, 162 мг) и 5-аминоизохинолин (1,2 экв., 1,2 ммоль, 173 мг) последовательно добавили при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 20 ч. Растворитель упарили при пониженном давлении и остаток растворили в 50 мл этилацетата. Органическую фазу промыли водой (2 х 20 мл), насыщенным раствором хлорида натрия (1 х 10 мл), сушили над сульфатом натрия и концентрировали в вакууме. Неочищенный остаток очищали методом колоночной хроматографии (силикагель, 3/7 этилацетат/гексан, за которым следовал этилацетат) и в заключении перекристаллизовали из диэтилового эфира с образование 150 мг бежевого твердого вещества. Выход = 47%. Тпл: (диэтиловый эфир) 131-133°С. 1H ЯМР (CDCl3, 400 МГц) δ 1,10 (6H, с), 1,49 (2H, м), 1,62 (2H, м), 1,81 (3H, с), 2,05 (2H, м), 6,18 (1H, д), 7,62 (2H, м), 7,70 (2H, м), 7,81 (1H, д), 8,38 (1H, уш. с), 8,53 (1H, д, J=5,6 Гц), 9,25 (1H, с); [М+1] 321,7 (C21H24N2O вычислено 320,43).
Пример 2 - (2Е)-N-(изохинолин-5-ил)-3-(3-метокси-2,6,6-триметилциклогекс-1-енил)акриламид Ib (схема 2)
Синтез 1
Синтез (2Е)-метил 3-(3-метокси-2,6,6-триметилциклогекс-1-енил)акрилат 3 [3]
Суспензию сложного эфира 2 (8 ммоль, 1,66 г) и N-бромсукцинимида (NBS) (1,1 экв., 8,8 ммоль, 1,56 г) в CCl4 (30 мл) кипятили в течение 1 ч. После фильтрования через слой целлита растворитель упарили. Остаток растворили в MeOH (20 мл) и реакцию кипятили всю ночь. Растворитель упарили и неочищенный остаток растворили в диэтиловом эфире (30 мл) и промыли водой (1 х 20 мл). Органическую фазу сушили над сульфатом натрия и сконцентрировали в вакууме. Очистку неочищенного остатка проводили методом колоночной хроматографии с использованием в качестве элюента смеси 1/9 этилацетат/петролейный эфир и получили 715 мг бесцветного масла. Выход = 37,5% (две стадии). 1Н ЯМР (CDCl3, 400 МГц) δ 1,02 (3H, с), 1,04 (3H, с), 1,38 (2H, м), 1,62 (2H, м), 1,79 (3H, с), 3,37 (3H, с), 3,51 (1H, м), 3,75 (3H, с), 5,84 (1H, д, J=16 Гц), 7,33 (1H, д, J=16 Гц); [М+1] 239,1 (С14Н22O3 вычислено 238,32).
Синтез (2Е)-3-(3-метокси-2,6,6-триметилциклогекс-1-енил) акриловой кислоты 4
LiOH (5 экв., 630 мг) добавили при 0°С к раствору сложного эфира 3 (3 ммоль, 715 мг) в смеси 3:1:1 ТГФ/МеОН/вода (15 мл) и смесь перемешивали при комнатной температуре всю ночь. Растворители удалили при пониженном давлении и остаток разбавили водой (20 мл). Кислоту высадили добавлением 10% HCl и затем экстрагировали AcOEt (3 × 15 мл). Объединенные органические слои сушили над Na2SO4 и упарили в вакууме и получили 600 мг маслообразного продукта. Выход = 89%. 1H ЯМР (CDCl3, 400 МГц) δ 1,03 (3H, с), 1,05 (3H, с), 1,39 (2H, м), 1,62 (2H, м), 1,80 (3H, с), 3,38 (3H, с), 3,52 (1H, м), 5,86 (1H, д, J=16 Гц), 7,45 (1H, д, J=16 Гц); [М+1] 225,5 (С13Н20O3 вычислено 224,3).
Синтез 2
1,0 ммоль (224 мг) кислоты 4 растворили в 10 мл безводного ДМФА. EDCI (1-этил-3-(3-диметиламинопропил)карбодиимид) (1,2 экв., 1,2 ммоль, 230 мг), HOBt (N-гидроксибензотриазол) (1,2 экв., 1,2 ммоль, 162 мг) и 5-аминоизохинолин (1,2 экв., 1,2 ммоль, 173 мг) были добавлены последовательно при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 20 ч. Растворитель упарили при пониженном давлении и остаток растворили в 50 мл этилацетата. Органическую фазу промыли водой (3 × 20 мл) и насыщенным раствором хлорида натрия (1 × 10 мл), сушили над сульфатом натрия и концентрировали в вакууме. Неочищенный остаток очищали методом колоночной хроматографии (силикагель, этилацетат) и в заключение перекристаллизовали из диэтилового эфира с образование 160 мг желтого аморфного твердого вещества. Выход = 45%. 1H ЯМР (CDCl3, 400 МГц) δ 1,07 (6H, с), 1,42 (2H, м), 1,66 (2H, м), 1,86 (3H, c), 3,40 (3H, c), 3,48 (1H, м), 6,18 (1Н, д), 7,51 (1Н, м), 7,65 (3H, м), 7,84 (1H, д), 8,38 (1H, уш.с), 8,55 (1H, д, J=6 Гц), 9,26 (1H, с); [М+1] 351,2 (С22Н26N2O2 вычислено 350,45).
Пример 3 - (2Е)-N-(изохинолин-5-ил)-3-(2,6,6-триметил-3-феноксициклогекс-1-енил)акриламид Iс (схема 3)
Синтез 1
Синтез (2Е)-метил 3-(2,6,6-триметил-3-феноксициклогекс-1-енил)акрилата 5с [4]
Суспензию сложного эфира 2 (3,12 ммоль, 650 мг) и N-бромсукцинимида (NBS) (1,1 экв., 3,43 ммоль, 611 мг) в CCl4 (15 мл) кипятили в течение 1 ч. После фильтрования через слой целлита растворитель упарили. Остаток растворили в MeOH (5 мл) и по каплям добавили к раствору феноксида натрия (6,24 ммоль) в метаноле (10 мл). Полученную смесь перемешивали при комнатной температуре всю ночь. Реакцию вылили в холодный 5% водный раствор гидроксида натрия (15 мл) и продукт экстрагировали эфиром (2 × 20 мл). Органический слой промыли водой (1 × 10 мл), насыщенным раствором хлорида натрия (1 × 5 мл), сушили над безводным сульфатом натрия и сконцентрировали в вакууме. Очистку неочищенного продукта проводили методом колоночной хроматографии с использованием в качестве элюента смеси 1/9 этилацетат/петролейный эфир и получили 350 мг бесцветного масла. Выход = 37,5% (две стадии). 1H ЯМР (CDCl3, 400 МГц) δ 1,07 (3H, с), 1,12 (3H, с), 1,42 (2H, м), 1,78 (2H, м), 1,86 (3H, с), 3,78 (3H, с), 4,57 (1H, м), 5,92 (1H, д, J=16,4 Гц), 6,95 (3H, м), 7,29 (2H, м), 7,44 (1H, д, J=16,4 Гц); [М+1] 301,2 (С19Н24O3 вычислено 300,39).
Синтез (2Е)-3-(2,6,6-триметил-3-феноксициклогекс-1-енил) акриловой кислоты 6с
LiOH (5 экв., 243 мг) добавили при 0°С к раствору сложного эфира 5 (1,16 ммоль, 350 мг) в смеси 3:1:1 ТГФ/МеОН/вода (12,5 мл) и смесь перемешивали при комнатной температуре всю ночь. Растворители удалили при пониженном давлении и остаток разбавили водой (20 мл). Кислоту высадили добавлением 10% HCl и затем экстрагировали AcOEt (3 × 15 мл). Объединенные органические слои сушили над Na2SO4 и упарили в вакууме с получением 300 мг белого твердого вещества. Выход = 90%. 1H ЯМР (CDCl3, 400 МГц) δ 1,08 (3H, с), 1,13 (3H, с), 1,44 (2H, м), 1,78 (2H, м), 1,87 (3H, с), 4,58 (1H, м), 5,94 (1H, д, J=16,4 Гц), 6,96 (3H, м), 7,29 (2H, м), 7,52 (1H, д, J=16,4 Гц); [М+1] 287,5 (С18Н22O3 вычислено 286,37).
Синтез 2
0,5 ммоль (143 мг) кислоты 6с растворили в 5 мл безводного ДМФА. EDCI (1,2 экв., 0,6 ммоль, 115,2 мг), HOBt (1,2 экв., 0,6 ммоль, 81 мг) и 5-аминоизохинолин (1,2 экв., 0,6 ммоль, 86,51 мг) добавили последовательно при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 20 ч. Растворитель упарили при пониженном давлении и остаток растворили в 30 мл этилацетата. Органическую фазу промыли водой (2 х 10 мл) и насыщенным раствором хлорида натрия (1 × 10 мл), сушили над сульфатом натрия и концентрировали в вакууме. Неочищенный твердый продукт очищали методом колоночной хроматографии (силикагель, этилацетат/петролейный эфир 8:2) и в заключении перекристаллизовали из диэтилового эфира с образование 100 мг белого твердого вещества. Выход = 48,5%. Тпл.: (диэтиловый эфир) 141-143°С. 1H ЯМР (CDCl3, 400 МГц) δ 1,14 (3H, с), 1,17 (3H, с), 1,47 (2H, м), 1,78 (2H, м), 1,95 (3H, с), 4,60 (1H, м), 6,41 (1H, д), 6,97 (2H, д, J=7,2 Гц), 7,26 (4H, м), 7,59 (1H, д, J=16 Гц), 7,80 (1H, т, J=8 Гц), 7,92 (1H, д, J=8 Гц), 8,17 (1H, м), 8,37 (1H, м), 8,52 (1H, уш.с), 9,26 (1H, с); [М+1] 413,6 (С27Н28N2O2 вычислено 412,52).
Пример 4 - (2Е)-3-(3-(4-хлорфенокси)-2,6,6-триметилциклогекс-1-енил)-N-(изохинолин-5-ил)акриламид Id (схема 3)
Согласно синтезу 2, исходя из 0,5 ммоль кислоты 6d, получили 150 мг соединения Id в виде белого твердого вещества. Выход = 67%. Тпл.: (диэтиловый эфир) 168°С. 1H ЯМР (CDCl3, 400 МГц) δ 1,11 (3H, с), 1,14 (3H, с), 1,45 (2H, м), 1,66 (2H, м), 1,86 (3H, с), 4,76 (1H, м), 6,61 (1H, д), 7,06 (2H, д, J=8,8 Гц), 7,31 (2H, м), 7,34 (2H, д, J=8,8 Гц), 7,69 (1H, т, J=8 Гц), 7,95 (1H, д, J=8 Гц), 8,04 (1H, м), 8,27 (1H, уш.с), 8,58 (1H, д, J=6 Гц), 9,33 (1H, с); [М+1] 448,4 (С27Н27СlN2O2 вычислено 446,97).
Пример 5 - (2Е)-3-(3-(4-фторфенокси)-2,6,6-триметилциклогекс-1-енил)-N-(изохинолин-5-ил)акриламид Iе (схема 3)
Согласно синтезу 2, исходя из 0,5 ммоль кислоты 6е, получили 100 мг соединения Iе в виде белого твердого вещества. Выход = 46%. Тпл.: (диэтиловый эфир) 135-137°С. 1H ЯМР (CDCl3, 400 МГц) δ 1,11 (3H, с), 1,15 (3H, с), 1,44 (2H, м), 1,71 (2H, м), 1,92 (3H, с), 4,50 (1H, м), 6,21 (1H, д), 6,91 (2H, м), 6,98 (2H, м), 7,54 (1H, д, J=15,6 Гц), 7,72 (3H, м), 7,87 (1H, д), 8,41 (1H, уш.с), 8,55 (1H, д, J=6,4 Гц), 9,28 (1H, с); [М+1] 431,6 (С27Н27FN2O2 вычислено 430,51).
Пример 6 (2Е)-3-(3-(3-фторфенокси)-2,6,6-триметилциклогекс-1-енил)-N-(изохинолин-5-ил)акриламид If (схема 3)
Согласно синтезу 2, исходя из 0,5 ммоль кислоты 6f, получили 90 мг соединения If в виде белого твердого вещества. Выход = 42%. Тпл.: (диэтиловый эфир) 147°С. 1H ЯМР (CDCl3, 200 МГц) δ 1,11 (3H, с), 1,14 (3H, с), 1,57 (2H, м), 1,71 (2H, м), 1,88 (3H, с), 4,56 (1H, м), 6,20 (1H, д), 6,70 (4H, м), 7,58 (1H, д, J=15,6 Гц), 7,65 (3H, м), 7,85 (1H, д), 8,38 (1H, уш.с), 8,59 (1H, д, J=5,8 Гц), 9,29 (1H, с); [М+1] 431,5 (С27Н27FN2O2 вычислено 430,51).
(2Е)-3-(3-(3,4-дифторфенокси)-2,6,6-триметилциклогекс-1-енил)-N-(изохинолин-5-ил)акриламид Ig (схема 3)
Согласно синтезу 2, исходя из 0,5 ммоль кислоты 6g, получили 90 мг соединения Ig в виде белого твердого вещества. Выход = 40%. Тпл.: (диэтиловый эфир) 155°С. 1H ЯМР (CDCl3, 200 МГц) δ 1,11 (3H, с), 1,14 (3H, с), 1,53 (2H, м), 1,75 (2H, м), 1,89 (3H, с), 4,55 (1H, м), 6,20 (1H, д), 6,68 (3H, м), 7,50 (1H, д, J=15,6 Гц), 7,65 (3H, м), 7,85 (1H, д), 8,40 (1H, уш.с), 8,60 (1H, д, J=5,8 Гц), 9,29 (1H, с); [М+1] 449,7 (С27Н26F2N2O2 вычислено 448,51).
Схема 1
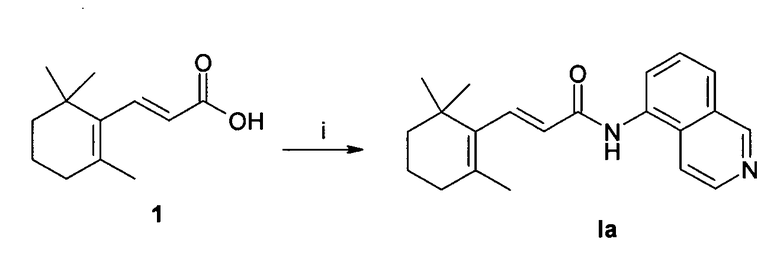
Реагенты: i) EDCI, HOBt, 5-аминоизохинолин, ДМФА, Ткомн.
Схема 2
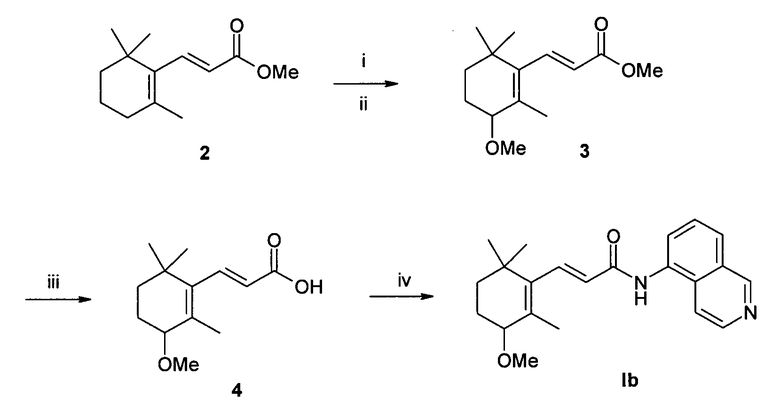
Реагенты: i) NBS/CCl4; ii) MeOH, кипячение; iii) LiOH, ТГФ/MeOH/вода, Ткомн.; iv) EDCI, HOBt, 5-аминоизохинолин, ДМФА, Ткомн.
Схема 3
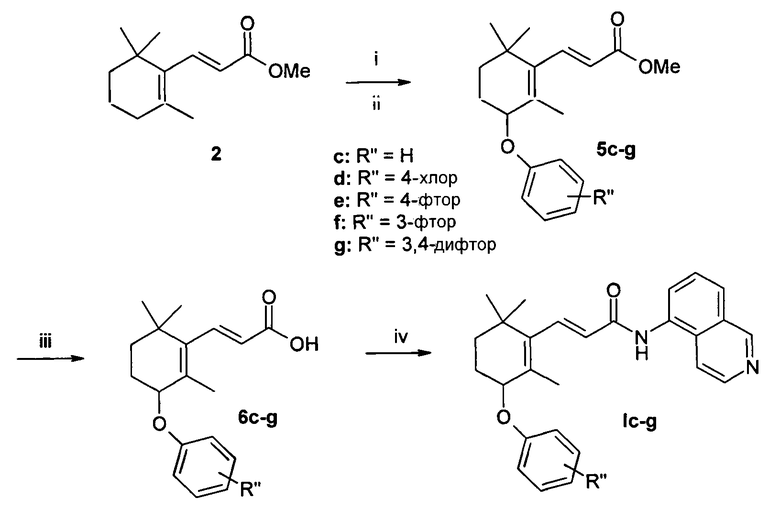
Реагенты: i) NBS/CCl4; ii) R”PhONa, EtOH, Ткомн.; iii) LiOH, ТГФ/MeOH/вода, Ткомн.; iv) EDCI, HOBt, 5-аминоизохинолин, ДМФА, Ткомн.
Количественное определение биологической активности
Использовались новорожденные и зрелые крысы линии Sprague-Dawley (~250 г) (Harlam, Италия). Все эксперименты соответствовали национальным протоколам и были одобрены региональным комитетом по этическим нормам.
Анализ радиолигандного связывания
Для испытаний использовались самцы крыс линии Sprague-Dawley с массой тела между 250 и 350 г к моменту исследования. Для анализа связывания крыс декапитировали под анестезией, извлекали спинной мозг и разрушали с использованием тканевого гомогенизатора Polytron в ледяном буфере, содержащем 5 мМ KCl, 5,8 мМ NaCl, 0,75 мМ CaCl2, 2мМ MgCl2, 320 мМ сахарозы, 10 мМ Hepes (4-(2-гидроксиэтил)-1-пиперазинэтансульфоновая кислота), pH 8,6 [5]. Гомогенизированную ткань центрифугировали при 1000 × g в течение 10 мин при 4°С и супернатант снова центрифугировали в течение 30 мин при 35000 × g при 4°С (Beckman Avanti J25). Осадок ресуспендировали в том же буфере, описанном выше, и использовали в экспериментах по связыванию. В экспериментах по насыщению 150 мкг белок/образец из суспензий мембран инкубировали с [3Н]-резинифератоксином ([3H]RTX) (0,003-3 нМ) в исследуемом буфере, содержащем 0,25 мг/мл бычьего сывороточного альбумина (BSA) без жирных кислот, при 37°С в течение 60 мин. В экспериментах по конкурирующему связыванию мембраны инкубировали при 37°С в течение 60 мин с [3H]RTX (0,4 нМ) и повышая концентрации изучаемых соединений в интервале от 0,1 нМ до 3 мкМ. Неспецифическое связывание определяли в присутствии 1 мкМ RTX. После инкубации реакционную смесь охладили до 0°С и в течение 15 мин инкубировали с бычьим α1-кислым гликопротеином (200 мкг на пробирку) для уменьшения неспецифического связывания RTX. Мембраносвязанный RTX отделили от свободного RTX центрифугированием образцов при 18500 × g в течение 15 мин. Конец микроцентрифужной пробирки, содержащий осадок, отрезали и радиоактивность измеряли сцинтилляционным методом (Packard 2500 TR). Концентрацию белка определяли согласно методу Bio-Rad c бычьим сывороточным альбумином в качестве стандарта (Bradford, 1976). Результаты исследования по насыщению и конкурентному связыванию анализировали программой Ligand [6].
Измерение Са2+ методом флуоресцентного анализа в культивированных ганглиях тройничного нерва крысы
Двухдневных и новорожденных крыс необратимо анестезировали и декапитировали. Ганглии тройничного нерва были выделены и быстро помещены в холодный раствор фосфатного буфера (PBS) перед перенесением в раствор коллагеназы/диспазы (1 мг/мл растворенный в свободном от Ca2+ и Mg2+ PBS) на 35 мин при 37°С [7]. После ферментативной обработки ганглии промыли три раза свободным от Ca2+ и Mg2+ PBS и затем поместили в 2 мл холодной среды DMEM (модифицированная по способу Дульбекко среда Игла) с добавлением 10% сыворотки эмбрионов коров (FBS, инактивированная нагреванием), 2 мМ L-глутамина, 100 μ/мл пенициллина и 100 мкг/мл стрептомицина. Ганглии диссоциировали на одиночные клетки несколькими пропусканиями через группу инъекционных игл (23G вплоть до 25G). В заключение среду и клетки ганглиев фильтровали через 40 мкм фильтр для удаления дебриса и добавили 8 мл среды DMEM и центрифугировали (200 × g 5 мин). Целевой осадок клеток ресуспендировали в среде DMEM [с добавлением 100 нг/мл фактора роста нервов мыши (мыши NGF-7S) и свободного основания цитозин-β-D-арабинофуранозида (ARA-C) 2,5 мкМ]. Клетки высеивали на покрытые поли-L-лизином (8,3 мкМ) и ламинином (5 мкМ) 25 мм покровные стекла и выдерживали в течение от 5 до 8 дней при 37°С во влажной камере, заполненной 5% СО2 и воздухом. К высеянным нейронам добавили эфир Fura-2-АМ (3 мкМ) в растворе Са2+ буфера со следующим составом (мМ): CaCl2 1,4, KCl 5,4, MgSO4 0,4, NaCl 135, D-глюкоза 5, НЕРЕS (4-(2-гидроксиэтил)-1-пиперазинэтансульфоновая кислота) 10 с BSA (бычий сывороточный альбумин) (0,1%), при pH 7,4 на 40 мин при 37°С. Высеянные нейроны затем дважды промыли раствором Са2+ буфера и перенесли в ячейку на предметном столике микроскопа Nikon eclipse TE300. Эфир Fura-2-АМ возбуждали при 340 нм и 380 нм, чтобы измерить относительные изменения концентрации [Са2+]i с помощью отношения F340/F380, которое фиксировалось системой анализа динамического изображения (Laboratory Automation 2.0, RCS, Флоренция, Италия). После перенесения высеянных нейронов в ячейку их оставили (по крайней мере, на 10 мин) для достижения стабильной флуоресценции перед началом эксперимента. Калибровочная кривая была получена с использованием буфера, содержащего эфир Fura-2-АМ и заданную концентрацию свободного Са2+. Затем эту кривую использовали для преобразования данных, полученных из отношения F340/F380 в [Ca2+]i (нМ) [8]. Было изучено влияние премедикации капсазепином (CPZ), SB366791 и соединениями формулы (I) на повышение [Ca2+]i, обусловленное 0,1 мкМ капсаицина.
Вызванная капсаицином вторичная аллодиния у крыс
Капсаицин (20 нмоль/50 мкл/лапа) был введен в лишенный волос участок кожи подошвенной поверхности правой лапы крысы, под анестезией диэтиловым эфиром (Chaplan et al., 1994). Соединение Id было введено перорально (10 мг/кг) за 2 часа до инъекции капсаицина. Тактильная аллодиния была оценена через 90 мин после введения капсаицина.
Лекарственные вещества и реагенты
Лекарственные вещества и реагенты были получены у указанных компаний: [3Н]-Резинифератоксин (Perkin Elmer, Бостон, MA), SB-366791 (Tocris, Великобритания), капсаицин, капсазепин, иономицин, ламинин, поли-L-лизин, вещество P (Sigma, Италия); мыши NGF-7S и коллагеназа/диспаза (Roche Diagnostics, Италия); модифицированная по способу Дульбекко среда Игла (DMEM), сыворотка эмбрионов коров (FBS), инактивированная нагреванием, L-глутамин (200 мМ), пенициллин/стрептомицин (10,000 МЕ/мл ± 10,000 мкг/мл), (Gibco, Италия); эфир Fura-2-AM (Società Italiana Chimici, Италия). Исходные концентрации капсаицина (10 мМ), капсазепина (10 мМ), SB-366791 (1 мМ) и соединений формулы (I) были приготовлены в 50% DMSO и 50% Твина 80 (Tween 80). Эфир Fura-2-AM и иономицин растворяли в 100% DMSO. Все другие лекарственные вещества растворяли в дистиллированной воде. Растворы с подходящими степенями разбавления затем были приготовлены в буферном растворе Кребса.
Результаты
Количественное определение биологической активности
Кривая насыщения [3H]-RTX на рецепторах TRPV1, выделенных из спинного мозга крыс, показывает значение KD, равное 0,21 (0,16-0,27), и значение Bmax, равное 57 (53-62) фмоль/мг белка. График Скэтчарда фактически линейный, и компьютерный анализ данных показывает, что присутствует только один тип сайтов связывания с высокой аффинностью. Эксперименты по конкурентному связыванию [3H]-RTX показали, что соединения Ia, Ib, Ic, Id, Ie, If, Ig и эталонного соединения (Е)-3-(4-хлорфенил)-N-(3-метоксифенил)-акриламид (SB-366791) обладают значениями Ki, равными 66 (56-78) нМ, 26,2 (21,1-32,6) нМ, 4,93 (3,40-7,16) нМ, 27 (23-32) нМ, 14,8 (10,2-21,5) нМ, 8,14 (6,87-9,65) нМ, 10,3 (7,9-13,4) нМ и 36 (30-43) нМ соответственно.
Ca2+ флуоресценция
Капсаицин (0,1 мкМ) вызвал увеличение [Ca2+] в подавляющем большинстве (95%) нейронов тройничного нерва, которые по этой причине были определены как TRPV1 экспрессирующие нейроны. Значения IC50 соединений Ia, Ib, Ic, Id, Ie, If и Ig, ингибирующие вызываемую капсаицином активацию [Ca2+]i, составили 44 (11-184) нМ, 28,4 (25,2-31,9) нМ, 2,12 (1,44-2,82) нМ, 18,2 (4-98) нМ, 5,25 (4,11-6,70) нМ, 0,38 (0,36-0,40) нМ и 0,65 (0,62-0,68) нМ соответственно. Эталонные антагонисты TRPV1, капсазепин и SB-366791 ингибировали отклик капсаицина со значениями IC50, равными 948 (676-1330) нМ и 8,7 (3,4-17,3) нМ соответственно. Результаты выражали в виде среднего значения и границ 95% доверительного интервала.
Вызванная капсаицином вторичная аллодиния у крыс
Через 90 мин после введения капсаицина соединение Id вызвало превентивный эффект (54%) против проаллодинического эффекта капсаицина.
ССЫЛКИ
1. Appendino, G. and Szallasi A. Progress in Medicinal Chemistry 2006, 44, 145-180.
2. Shimasaki, H.; Kagechika, H.; Fukasawa, H. Kawachi, E.; Shudo, K. Chemical & Pharmaceutical Bulletin 1995, 43, 100-7.
3. a) Baasov, T. and Sheves, M. Angew. Chem. 1984, 23, 803-804. b) Baraldi, P. G.; Pollini, G. P.; Simoni, D.; Zanirato, V.; Barco, A.; Benetti, S. Synthesis 1986, 9, 781-2.
4. Nanasawa, M. and Kamogawa, H. Bull. Chem. Soc. Jpn. 1982, 55, 3655-3656.
5. a) Szallasi A. and Blunberg P. M. Neurosciences 1992, 8, 368. b) Szallasi A. and Blunberg P.M. Naunyn Schmiedeberg's Arch Pharmacol. 1993, 347, 84-91.
6. Munson, P.J.; Rodbard, D. Anal. Biochem. 1980, 107, 220-239.
7. Rigoni, M.; Trevisani, M.; Gazzieri, D.; Nadaletto, R.; Tognetto, M.; Creminon, C; Davis, J.B.; Campi, B.; Amatesi, S.; Geppetti, P.; Harrison, S. Br. J. Pharmacol. 2003, 138, 977-985.
8. Kudo, Y.; Ozaki, K.; Miyakawa, A.; Amano, T.; Ogura, A. Jap. J. Pharmacol. 1986, 41, 345-351.
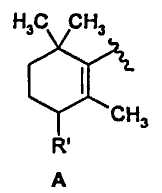
Настоящее изобретение относится с новым соединениям формулы (I), где Y представляет собой группу формулы А, в которой: R′ выбирают из атомов водорода, (С1-С6)алкокси или фенокси, ароматический цикл которого необязательно замещен одним или более атомом галогена; R представляет собой атом водорода; n представляет собой 0; X представляет собой изохинолинил. Также изобретение относится к применению соединения формулы (I) и к фармацевтической композиции на его основе. Технический результат: получены новые соединения, обладающие свойствами антагониста ванилоидного рецептора типа 1. 3 н. и 2 з.п. ф-лы, 6 пр.
1. Соединение формулы (I)
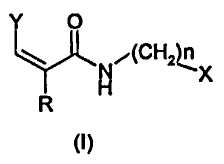
где Y представляет собой группу формулы
в которой
R' выбирают из атомов водорода, (C1-C6)алкокси или фенокси, ароматический цикл которого необязательно замещен одним или более атомом галогена;
R' представляет собой атом водорода;
n представляет собой 0;
X представляет собой изохинолинил.
2. Соединение по п.1, где X представляет собой 5-изохинолинил.
3. Соединения по п.1 или 2 для применения в качестве лекарственного средства, обладающего свойствами антагониста ванилоидного рецептора типа 1.
4. Фармацевтическая композиция, обладающая свойствами антагониста ванилоидного рецептора типа 1, включающая соединение по любому из пп.1-4 в сочетании с одним или более носителем и/или эксципиентом.
5. Применение соединений по п.1 или 2 для приготовления анальгезирующих и/или противовоспалительных лекарственных средств.
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Doherty Elizabeth M; et al: "Discovery of potent, orally available vanilloid receptor-1 antagonists | |||
| Structure-activity relationship of N-aryl cinnamides", JOURNAL OF MEDICINAL CHEMISTRY, 48(1), pp.71-90, 2005 | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Способ получения амидов или их кислотноаддитивных солей | 1986 |
|
SU1440342A3 |

