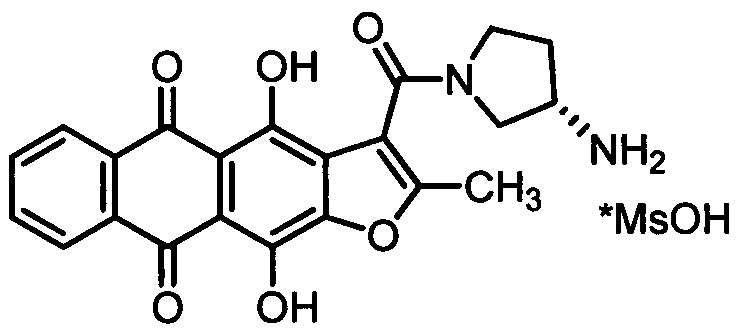
Изобретение относится к медицине, а именно к онкологии, и может быть использовано в качестве средства для лечения онкологических заболеваний, включая случаи, характеризующиеся низкой восприимчивостью к традиционным химиотерапевтическим препаратам. Сущность заявляемого способа заключается в том, что нуждающемуся в лечении пациенту перорально при однократном или многократном режиме применения вводят терапевтически эффективное количество метансульфоната (S)-3-[(3-амино-1-пирролидинил)карбонил]-4,11-дигидрокси-2-метилантра[2,3-b]фуран-5,10-диона (антрафурана), представленного формулой:
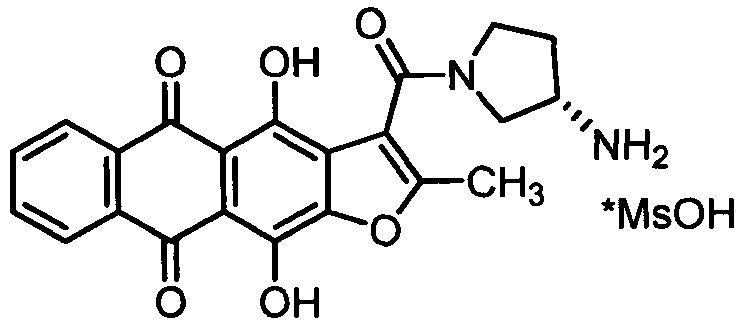
Антрафуран может быть использован самостоятельно или, более предпочтительно, в качестве фармацевтически активного ингредиента жидкой или твердой пероральной фармацевтической композиции, предпочтительно в виде перорального дозированного лекарственного средства (в виде раствора, таблетки или капсулы). Заявленные в настоящем изобретении фармацевтические композиции обеспечивают высокую биофармацевтическую растворимость антрафурана в биорелевантных средах, быстрое высвобождение активного ингредиента из таблеток или капсул (не менее 80% в течение 10 мин), а также стабильны при хранении.
Предпосылки создания настоящего изобретения
Известно, что гетероаренантрацендионы, содержащие в боковой цепи циклические диамины, обладают высокой антипролиферативной активностью [Shchekotikhin А.Е., et al. Eur. J. Med. Chem. 2016, 112, 114; Shchekotikhin A.E., et al. Eur. J. Med. Chem. 2014, 30 (86), 797; Shchekotikhin A.E. et al. Bioorg. Med. Chem., 2005, 13 (6), 2285; RU 2412166]. Так, ранее был описан мультитаргетный (S)-3-[(3-амино-1-пирролидинил)карбонил]-4,11-дигидрокси-2-метилантра[2,3-b]фуран-5,10-дион (далее антрафуран), способный за счет одновременного ингибирования топоизомераз 1, 2 и протеинкиназ индуцировать гибель опухолевых клеток различного гистогенеза, включая резистентные линии [Shchekotikhin А.Е., et al. Eur. J. Med. Chem. 2016, 112, 114; RU №2554939]. Антрафуран и его фармацевтически приемлемые солевые формы, включая метансульфонат, известны в качестве соединений, блокирующих деление опухолевых клеток и имеющих высокую противоопухолевую эффективность при парентеральном применении на моделях гемобластозов (Р388), в том числе и резистентных (P388/DOX), а также солидных опухолей (B16/F10) [Shchekotikhin А.Е., et al. Eur. J. Med. Chem. 2016, 112, 114; RU №2554939].
Хорошо известно, что противоопухолевые средства антрахинонового ряда (например доксорубицин, рубомицин, митоксантрон) вводятся пациентам преимущественно парентерально, зачастую медленной внутривенной инфузией, проводимой медицинским персоналом в условиях стационара [Противоопухолевая химиотерапия. Руководство, Скил Р.Т. (ред.), 2011], поскольку они не могут эффективно вводиться пероральным путем по причине их нестабильности или из-за низкого всасывания из желудочно-кишечного тракта. Поэтому ранее были разработаны фармацевтические противоопухолевые композиции для парентерального применения антрафурана, а также предложен способ лечения опухолевых заболеваний, предусматривающий парентеральное введение нуждающемуся пациенту терапевтически эффективного количества антрафурана или фармацевтических композиций на его основе [RU №2554939].
Неожиданно было обнаружено, что при пероральном способе введения водной композиции антрафуран обладает хорошей биодоступностью и высокой противоопухолевой активностью. Поэтому настоящее изобретение включает новый способ лечения субъекта с онкологическим заболеванием, путем перорального введения такому пациенту антрафурана, представленной выше формулы. Однако исследование кинетики растворения субстанции антрафурана в биорелевантных средах (например, в искусственном желудочном соке при 37°С) показало, что антрафуран относится к препаратам с низкой биофармацевтической растворимостью [Amidon G.L., et al. Pharm. Res., 1995, 12, 413]. Низкая растворимость лимитирует попадание в системный кровоток соединения при пероральном применении, поэтому другой задачей изобретения ставилась разработка фармацевтических композиций, повышающих его биофармацевтическую растворимость и пригодных для создания дозированных лекарственных средств для перорального применения, с высокой скоростью высвобождения активного ингредиента из лекарственной формы.
Авторами изобретения проведены интенсивные исследования, в результате которых разработан ряд жидких и твердых фармацевтических композиций, обеспечивающих высокую растворимость и биодоступность антрафурана при пероральном применении, а также стабильность лекарственного средства при хранении. Разработанные композиции обладают высокой противоопухолевой активностью и могут быть использованы для получения пероральных дозированных лекарственных форм антрафурана. Фармацевтические композиции, описанные в настоящем изобретении, могут находиться в виде разовых дозированных лекарственных форм, предпочтительно в виде капсул (например, мягких или твердых) или таблеток, обеспечивающих простоту введения лекарственного средства. Такие разовые дозированные лекарственные формы содержат заранее определенное терапевтически эффективное количество активного ингредиента в виде порошка или гранул, раствора или суспензии в водной или неводной жидкости.
Одним из аспектов настоящего изобретения является пероральное средство, представляющее собой жидкую фармацевтическую композицию, включающую антрафуран, жидкий фармацевтически приемлемый носитель, и необязательно, вспомогательные эксципиенты. Изобретение включает жидкие композиции, пригодные для получения разовых дозированных форм, например для инкапсулирования в мягкие капсулы.
Жидкая композиция данного изобретения включает метансульфонат антрафурана, а также носитель, содержащий, по меньшей мере, один фармацевтически приемлемый растворитель, такой как полиэтиленгликоль, 1,2-полипропиленгликоль, вода или их смесь.
Жидкая композиция по данному изобретению может предпочтительно содержать любые кристаллические или аморфную формы метансульфоната антрафурана, а также их сольваты, более предпочтительно аморфную форму кристаллогидрата, полученную по методу [RU №2554939], в фармацевтически эффектном количестве, находящемся предпочтительно в диапазоне от приблизительно 1% до приблизительно 40% от общей массы композиции.
В предпочтительных вариантах изобретения в качестве носителя для жидкой композиции может быть использован полиэтиленгликоль, представляющий собой жидкую форму полиэтиленгликоля, имеющего среднюю молекулярную массу, находящуюся в интервале от приблизительно 200 до приблизительно 600 дальтон. В наиболее предпочтительном варианте изобретения применяется полиэтиленгликоль с молекулярной массой 400 дальтон. Полиэтиленгликоль может присутствовать в композиции по данному изобретению в количестве от приблизительно 60% до приблизительно 99% от общей массы композиции.
Помимо активного ингредиента и носителя, жидкая композиция антрафурана может также необязательно содержать сорастворители и вспомогательные эксципиенты, хорошо известные из уровня техники, такие как, без ограничения перечисленным, сорастворители, суспендирующие агенты, поверхностно-активные вещества, солюбилизаторы, эмульгаторы, стабилизаторы, консерванты, антиоксиданты, буферные соединения и другие эксципиенты, хорошо известные из общего уровня техники [Handbook of Pharmaceutical Excipients (Sixth Edition), Rowe R.C., Sheskey P.J. Quinn M.E (eds.), 2009; Modern Pharmaceutics (Fourth Edition), Banker G. et al. (eds.), 2002; Remington's Pharmaceutical Sciences (21th Edition), Troy D.B., Beringer P. (eds.), 2006].
Так, жидкое лекарственное средство может необязательно включать один или более сорастворителей, таких как, без ограничения перечисленным, вода, этанол, 1,2-пропиленгликоль, глицерин, сорбит или их смесь. Общее количество сорастворителей, применяемое в композиции, может находиться в диапазоне от приблизительно 0.1% до приблизительно 30% от общей массы композиции.
Кроме того, жидкая фармацевтическая композиция может необязательно дополнительно включать один или более диспергатор, такой как высокомолекулярный полиэтиленгликоль, представляющий собой твердую форму полиэтиленгликоля, имеющего среднюю молекулярную массу, находящуюся в интервале от приблизительно 1000 до приблизительно 6000 дальтон, полиэтилен/полипропилен/полиэтиленоксидные блок-сополимеры (например, полоксамер или плюроник), твердые эфиры гликоля или твердые неионные компоненты. Общее количество диспергатора в композиции может варьироваться от приблизительно 0.1% до приблизительно 10% от общей массы композиции.
Также, жидкая композиция может включать одно или более ионное или неионное поверхностно-активное вещество, например, без ограничения перечисленным, додецилсульфат натрия, соли желчных кислот, полиоксиэтиленовые эфиры сорбитана и жирной кислоты, такие как твин (Tween®80). Общее количество применяемых поверхностно-активных веществ может находиться в диапазоне от приблизительно 0.1% до приблизительно 1% от общей массы композиции.
Жидкое лекарственное средство, включающее антрафуран, в предпочтительном варианте изобретения может быть инкапсулировано в дозированную форму в виде мягкой капсулы. Оболочка мягкой капсулы может быть изготовлена из любого полимера или полимерной смеси, пригодных для производства фармацевтических дозированных форм в виде мягких капсул. В изобретении могут применятся различные материалы для мягких капсул, которые хорошо известны специалистам в данной области [Modern Pharmaceutics (Fourth Edition), Banker G. et al. (eds.), 2002; Remington's Pharmaceutical Sciences (21th Edition), Troy D.B., Beringer P. (eds.), 2006]. В предпочтительном варианте материал оболочки капсулы может включать желатин, предпочтительно свиной или бычий, или их смеси в массовом отношении от приблизительно 2:1 до 1:2, а также пластификаторы, такие как глицерин, сорбит, пропиленгликоль. Другие добавки, такие как ингибиторы хрупкости, красители, смазывающие средства, могут также использоваться в материале капсулы.
Фармацевтическая композиция по данному изобретению может быть получена с использованием общепринятых методов и оборудования для производства фармацевтических композиций. Мягкие капсулы, содержащие фармацевтическую композицию по настоящему изобретению, могут быть также получены с использованием известных специалистам в области фармации методов производства и оборудования для инкапсуляции. Методы инкапсуляции также хорошо известны специалистам в данной области [Modern Pharmaceutics (Fourth Edition), Banker G. et al. (eds.), 2002; Remington's Pharmaceutical Sciences (21th Edition), Troy D.B., Beringer P. (eds.), 2006]. Полученная дозированная форма в виде мягких капсул по настоящему может содержать антрафурана метансульфанат в терапевтически эффективном количестве, например, приблизительно 50 мг, 100 мг или 200 мг.
Другим аспектом настоящего изобретения является пероральное лекарственное средство, представляющее собой твердую фармацевтическую композицию, являющуюся дисперсией антрафурандиона с одним или более эксципиентами, выбранными из наполнителей, связывающих, разрыхляющих (дезинтегрирующих), смазывающих (глиданты) агентов, поверхностно активных веществ или другими вспомогательными эксципиентами, хорошо известным из общего уровня техники [Handbook of Pharmaceutical Excipients (Sixth Edition), Rowe R.C., Sheskey P.J. Quinn M.E (eds.), 2009; Pharmaceutical Manufacturing Handbook: Production and Processes, Gad S.C. (ed.), 2006; R.K. Khar, S.P. Vyas, F.J. Ahmad, G.K. Jain. The Theory and Practice of Industrial Pharmacy (Forth Edition), 2013; Pharmaceutical Dosage Forms: Tablets. (Second Edition), Volume 1, Lieberman H.A., et al. (eds.), 1989]. Изобретение также включает варианты твердых композиций, пригодных для гранулирования и получения разовых дозированных форм, например в виде твердых капсул или таблеток
Твердая композиция данного изобретения может содержать любые кристаллические или аморфную формы метансульфоната антрафурана и их сольваты, предпочтительно аморфную форму, полученную по методу [RU №2554939], в фармацевтически эффектном количестве, находящемся предпочтительно в диапазоне от приблизительно 10% до приблизительно 80% от общей массы композиции.
В качестве наполнителя (разбавителя) в твердых фармацевтических композициях по настоящему изобретению могут быть использованы один (или более) растворимых или нерастворимых наполнителей, хорошо известных из общего уровня техники [Handbook of Pharmaceutical Excipients (Sixth Edition), Rowe R.C., Sheskey P.J. Quinn M.E (eds.), 2009; R.K. Khar, S.P. Vyas, F.J. Ahmad, G.K. Jain. The Theory and Practice of Industrial Pharmacy (Forth Edition), 2013] или их смесь в любом соотношении. Растворимый наполнитель, который может быть использован в настоящем изобретении, представляет собой наполнитель, растворимый в воде при температуре окружающей среды, например, без ограничения перечисленным, лактоза, глюкоза, маннит, сорбит, мальтодекстрин, мальтоза. Нерастворимый наполнитель, который может быть использован в настоящем изобретении, представляет собой наполнитель, имеющий низкую растворимость в воде при температуре окружающей среды, например, крахмал, различные формы и производные целлюлозы, например микрокристаллическая целлюлоза, двухосновный фосфат кальция, трехосновный фосфат кальция. Количество наполнителя в фармацевтических композициях по настоящему изобретению может составлять от приблизительно 80% до приблизительно 15% от общей массы композиции.
Для обеспечения прочности гранул или таблеток в качестве связывающих агентов в твердых фармацевтических композициях по настоящему изобретению могут быть использованы один или несколько агентов, например, без ограничения перечисленным, поливинилпирролидон (повидон), повидон-винилацетат (коповидон), полиэтиленгликоль, полиэтилен/полипропилен/полиэтиленоксидные блок-сополимеры (например, полоксамер или плюроник), крахмал, гидроксипропилметилцеллюлоза (НРМС), гидроксипропилцеллюлоза, сахароза, желатин, природные камеди (альгинаты, трагаканты, ксантаны, карагенаны, гуараны), и др. Количество связывающих эксципиентов в фармацевтических композициях по настоящему изобретению может составлять от приблизительно 15% до приблизительно 1% от общей массы композиции.
Для увеличения скорости распада таблетированных форм и высвобождения действующего компонента в фармацевтическую композицию по настоящему изобретению может быть включен один или более набухающих разрыхлителей (дезинтегрантов, супердезинтегрантов, суспендирующих агентов), хорошо известных из общего уровня техники [Handbook of Pharmaceutical Excipients (Sixth Edition), Rowe R.C., Sheskey P.J. Quinn M.E. (eds.), 2009; Pharmaceutical Manufacturing Handbook: Production and Processes. Gad S.C. (ed.), 2006; R.K. Khar, S.P. Vyas, F.J. Ahmad, G.K. Jain. The Theory and Practice of Industrial Pharmacy (Forth Edition), 2013; Pharmaceutical Dosage Forms: Tablets., Volume 1. (Second Edition), Lieberman H.A., et al. (eds.), 1989]. Так, композиция может включать такие разрыхлители (дезинтегранты) как, без ограничения перечисленным, целлюлозу и ее различные формы и производные, например метилцелюлозу, гидроксиэтилцелюлозу, карбоксиметилцеллюлозу (кармелозу натрия), кроскармеллозу натрия, микрокристаллическую целлюлозу, силикатизированную микрокристаллическую целлюлозу (просолв), крахмал в том числе химически модифицированный, например крахмалгликолят натрия, мальтоза, кросповидон, коповидон, бентонит, аэросил (коллоидный диоксид кремния). Общее количество разрыхляющих (дезинтегрирующих, супердезинтегрирующих или суспендирующих) агентов, применяемых в композиции, может находиться в диапазоне от приблизительно 1% до приблизительно 50% от общей массы композиции.
Также, твердая композиция может включать одно или более ионное или неионное поверхностно-активное вещество или их смесь, например, без ограничения перечисленным, додецилсульфат натрия (ДСН), соли желчных кислот, полиоксиэтиленовые эфиры сорбитана и жирной кислоты, такие как Твин 80. Общее количество поверхностно-активных веществ, применяемых в композиции, может находиться в диапазоне от приблизительно 0.1% до приблизительно 5% от массы композиции.
Другие общепринятые эксципиенты, которые могут быть введены в композицию по настоящему изобретению, включают консерванты, стабилизаторы, антиоксиданты, модификаторы текучести на основе диоксида кремния, агенты против прилипания (скользящие агенты или лубриканты), смазывающие агенты (глиданты), например стеариновая кислота и стеарат магния, тальк и другие эксципиенты, хорошо известные из общего уровня техники [Pharmaceutical Manufacturing Handbook: Production and Processes. Gad S.C. (ed.), 2006; Pharmaceutical Dosage Forms: Tablets. (Second Edition), Volume 1. Lieberman H.A., et al. (eds.), 1989; Handbook of Pharmaceutical Excipients (Sixth Edition). Rowe R.C., Sheskey P.J. Quinn M.E (eds.), 2009; Modern Pharmaceutics (Fourth Edition). Banker G. et al. (eds.), 2002; Remington's Pharmaceutical Sciences (21th Edition). Troy D.B., Beringer P. (eds.), 2006].
Твердую фармацевтическую композицию, раскрытую в настоящем изобретении, можно приготовить любыми способами, обычно применяемыми в фармацевтике [Pharmaceutical Dosage Forms: Tablets. (Second Edition), Volume 1. Lieberman H.A., et al. (eds.), 1989; Modern Pharmaceutics (Fourth Edition). Banker G. et al. (eds.), 2002; Remington's Pharmaceutical Sciences (21th Edition). Troy D.B., Beringer P. (eds.), 2006], например путем диспергирования активного ингредиента с наполнителем (разбавителем) и другими необходимыми эксципиентами до получения однородной твердой дисперсии, прямым смешиванием в смесителе или совместным измельчением компонентов смеси. Технология приготовления фармацевтической композиции может включать скрининг активного ингредиента, эксципиентов или их смесей путем просеивания через сито, например имеющего отверстия размером примерно от 100 до 400 мкм.
Полученная таким образом твердая дисперсия антафурана и одного или более эксципиентов может быть использована в производстве дозированных пероральных лекарственных форм, технологии получения которых также хорошо известны из общего уровня техники [Pharmaceutical Manufacturing Handbook: Production and Processes. Gad S.C. (ed.), 2006; Pharmaceutical Dosage Forms: Tablets. (Second Edition), Volume 1. Lieberman H.A., et al. (eds.), 1989; Modern Pharmaceutics (Fourth Edition). Banker G. et al. (eds.), 2002; Pharmaceutics. The Science of Dosage Form Design (2nd Edition). Aulton M.E. (ed.), 2002; Remington's Pharmaceutical Sciences (21th Edition). Troy D.B., Beringer P. (eds.), 2006]. В одном из вариантов изобретения полученная твердая композиция может быть сразу использована для инкапсулирования например, в твердые желатиновые капсулы или для непосредственного прессования в таблетки.
Для приготовления дозированных пероральных лекарственных форм, в предпочтительном варианте, дисперсия активного вещества и наполнителя подвергается гранулированию для достижения более однородного распределения ингредиентов по всему объему смеси и придания лекарственной композиции оптимальных характеристик. Примеры таких характеристик включают, не ограничиваясь перечисленным, приемлемую сыпучесть (текучесть) композиции, плотность, снижение риска сегрегации и повышение гомогенности [Pharmaceutics. The Science of Dosage Form Design (2nd Edition). Aulton M.E. (ed.), 2002; Remington's Pharmaceutical Sciences (21th Edition). Troy D.B., Beringer P. (eds.), 2006].
Гранулирование фармацевтической композиции может быть выполнено методами, хорошо известными из общего уровня технологии приготовления лекарственных форм, например методами сухого гранулирования (без использования жидкости) или влажного гранулирования (с применением жидкости для образования гранул) [Remington's Pharmaceutical Sciences (21th Edition). Troy D.B., Beringer P. (eds.), 2006; Modern Pharmaceutics (Fourth Edition) Banker G. et al. (eds.), 2002; Pharmaceutical Dosage Forms: Tablets. (Second Edition), Volume 1. Lieberman H.A., et al. (eds.), 1989; Pharmaceutics. The Science of Dosage Form Design (2nd Edition). Aulton M.E. (ed.), 2002]. При сухом гранулировании исходную сухую дисперсию агрегируют под давлением (или прессованием), после чего спрессованный материал размалывают с использованием подходящей техники размола с получением гранулированного материала. Влажная грануляция включает получение гранулята с помощью гранулирующей жидкости, содержащей нетоксичный растворитель (обычно воду или этанол) или смесь растворителей, которые можно удалить сушкой. Гранулирующая жидкость может содержать связывающие или другие вспомогательные эксципиенты для обеспечения получения гранул композиции с требуемыми характеристиками. При необходимости перед приготовлением конечной дозированной лекарственной формы полученный гранулят может подвергаться опудриванию или смешению со вспомогательными эксципиентами, такими как, без ограничения перечисленным, связующими, скользящими и/или разрыхляющими агентами, или их смесью.
Гранулы композиции могут быть затем использованы для получения разовых дозированных форм путем инкапсулирования в твердые капсулы или формирования таблеток путем прессования гранулята. Поэтому в одном аспекте настоящего изобретения описана фармацевтическая композиция для получения разовой дозированной лекарственной формы, такой как твердая желатиновая капсула. Инкапсулирование лекарственных форм, являющихся предметом настоящего изобретения, можно провести заключением композиции в оболочку капсул желаемого размера, изготовленную из желатина, метилцеллюлозы или альгината натрия с использованием подходящего аппарата для заполнения капсул. Дозированное средство в виде твердой капсулы, по настоящему изобретению, может содержать метансульфонат антрафурана в терапевтически эффективном количестве, например, приблизительно 50 мг, 100 мг, 200 мг или 400 мг.
Другим аспектом изобретения является твердая композиция для получения разовой дозированной лекарственной формы, такой как таблетка. Таблетки можно приготовить с использованием подходящего аппарата прессованием твердой композиции в виде порошка или гранул, необязательно в смеси с наполнителем, или другими эксципиентами, указанными выше, такими как диспергирующие агенты или поверхностно-активные вещества. Таблетки могут быть покрыты оболочкой с использованием стандартных способов нанесения пленочной оболочки в установке (коаторе) с применением водных или неводных жидкостей. Для нанесения покрытия на ядра таблеток могут быть использованы коммерчески доступные покрывающее смеси, например Opadry, Sepifilm, Acryl-EZE и т.д. или другие пленочные композиции, хорошо известные из общего уровня техники [Pharmaceutical Manufacturing Handbook: Production and Processes. Gad S.C. (ed.), 2006].
Дозированное лекарственное средство в виде таблетки, по настоящему изобретению, может содержать метансульфонат антрафурана в количестве, например, приблизительно 50 мг, 100 мг, 200 мг или 400 мг.
Состав фармацевтических композиций, являющихся предметом настоящего изобретения, должен обеспечивать оптимальные характеристики дозированных лекарственных форм, прежде всего показателей времени распадаемости, скорости растворения и твердости таблеток. Низкие скорости растворения композиции или распадаемости таблеток могут снижать биологическую доступность действующего вещества. Предложенные фармацевтические композиции для перорального применения лекарственного средства на основе антрафурандиона, являющиеся предметом настоящего изобретения, в предпочтительных вариантах, должны обеспечивать высокую скорость и степень растворения действующего вещества (более 80% за 10 мин). Кроме того, твердая композиция для таблетированных дозированных форм должна обеспечивать высокую скорость распада таблеток (менее 10 мин) при сохранении приемлемой твердости и показателя истирания таблеток.
Кроме того, желательно, чтобы композиции были химически стабильными, поскольку разложение посредством окисления, гидролиза, изомеризации, фотолиза, полимеризации или в результате смешивания с эксципиентами, или из-за других причин может приводить к снижению биологической доступности активного ингредиента лекарственного средства. В связи с этим, композиции по этому изобретению, представляющие особый интерес, включают в себя, например, конкретные воплощения, обеспечивающие стабильность антрафурандиона при хранении.
Настоящее изобретение также включает способ лечения нуждающихся пациентов, страдающих от онкологического заболевания, связанного с неконтролируемой тканевой или клеточной пролиферацией (т.е. гиперпролиферацией), предусматривающий пероральное введение терапевтически эффективного количества метансульфоната (S)-3-[(3-амино-1-пирролидинил)карбонил]-4,11-дигидрокси-2-метилантра[2,3-b]фуран-5,10-диона (антрафурана) или лекарственного средства на его основе в соответствии с изобретением, обеспечивающего после его перорального применения создание терапевтической концентрации антрафурана в крови, биологических средах и тканях организма пациента. Антрафуран, а также заявленные в настоящем изобретении лекарственные средства могут быть использованы и для лечения онкологических заболеваний, осложненных резистентностью к другим противоопухолевым препаратам.
Способ, раскрытый в настоящем изобретении, можно использовать для лечения первичных, в том числе, метастатических, а также устойчивых к химиотерапии опухолей. Примеры злокачественных заболеваний включают, без ограничения перечисленным, опухоли молочной железы, легкого, кожи, желудочно-кишечного канала, печени, почек, мозга, мочевого пузыря, яичников, яичка, матки, предстательной железы, костей и мягких тканей, саркомы Вилмса, Ирвинга и Капоши, нейробластомы, гемобластозы, лимфолейкоз, лимфогранулематоз, неходжкинскую лимфому.
Термин "антрафуран", использованный в настоящем описании, означает, (S)-3-[(3-амино-1-пирролидинил)карбонил]-4,11-дигидрокси-2-метилантра[2,3-b]фуран-5,10-дион.
Термин "стабильная" по отношению к композиции или лекарственной форме, означает, что активный ингредиент в композиции или лекарственной форме существенно не деградирует в течение определенного периода времени или становится модифицированным иным образом (например, по данным ВЭЖХ). В некоторых вариантах осуществления примерно 99% соединения (или выше) остается стабильным после определенного периода времени. Кроме того, термин "Стабильная" по отношению к композиции или лекарственной форме может означать стабильность функциональных характеристик композиции или дозированных форм, например растворимости, распадаемости, прочности, истираемости.
Термин "фармацевтическая композиция", использованный в настоящем описании, означает, например, смесь, содержащую определенное количество терапевтического агента, например, терапевтически эффективное количество лекарственного соединения, по меньшей мере, с одним фармацевтически приемлемым носителем или другим эксципиентом, предназначенную для введения человеку.
Термин "фармацевтически приемлемый", использованный в настоящем описании, относится к соединениям, композициям и/или лекарственным формам, которые при контакте с тканями млекопитающих, прежде всего, тканями человека не вызывают аллергических реакций, раздражения, осложнений или других токсических проявлений, и указанные соединения характеризуются достаточно высоким соотношением польза/риск.
"Фармацевтически приемлемая соль" в настоящем изобретении означает обычные соли, получаемые прибавлением кислоты к основанию, которые сохраняют терапевтическую эффективность и свойства антрафурана и образуются из его свободного основания и подходящих органических или неорганических кислот. Примеры фармацевтически приемлемых солей, образующихся при прибавлении кислоты, включают соли, полученные из органических кислот, например, метансульфоновой, этансульфоновой, 2-гидроксиэтансульфоновой, п-толуолсульфоновой или неорганических кислот, например, хлористоводородной, бромистоводородной, йодистоводородной, серной, сульфаминовой и т.п. Кроме того, термин "фармацевтически приемлемая соль" также включает фармацевтически приемлемые сольваты, предпочтительно гидраты.
Термин "фармацевтически приемлемый эксципиент" или "фармацевтически приемлемый носитель" в настоящем изобретении означает твердый или жидкий разбавитель, наполнитель, разрыхлитель или другой эксципиент, который подходит для введения млекопитающему, предпочтительно человеку. В качестве фармацевтически приемлемых носителей или эксципиентов в композициях по настоящему изобретению могут использоваться любые ингредиенты, хорошо известные из общего уровня техники получения лекарственных средств [Handbook of Pharmaceutical Excipients (Sixth Edition). Rowe R.C., Sheskey P.J. Quinn M.E (eds.), 2009; Modern Pharmaceutics (Fourth Edition). Banker G. et al. (eds.), 2002; Remington's Pharmaceutical Sciences (21th Edition). Troy D.B., Beringer P. (eds.), 2006].
Концентрация терапевтического агента в фармацевтической композиции составляет определенное значение, обеспечивающее введение терапевтически эффективного количества лекарственного средства, которое зависит от скорости абсорбции, инактивации и выведения из организма препарата, а также от тяжести состояния пациента или от других факторов, известных специалистам в данной области техники.
Термин "эксципиент", использованный в настоящем описании, означает вспомогательный компонент фармацевтической композиции, необходимый например, для обеспечения требуемых функциональных характеристик лекарственного средства.
"Эффективное количество" или "терапевтически эффективное количество" соединения в настоящем изобретении означает количество антрафурана или его фармацевтически приемлемой соли, например метансульфоната, которое значительно подавляет пролиферацию опухолевых клеток, рост опухли и эффективно для предупреждения, ослабления или смягчения симптомов заболевания или увеличения продолжительности жизни субъекта, подвергающегося лечению. Определение терапевтически эффективного количества вещества относится к компетенции специалиста в данной области техники. Терапевтически эффективное количество или дозировка соединения, предлагаемого в настоящем изобретении, может меняться в широких пределах и может определяться способом, известным специалистам в данной области техники.
Для конкретного реципиента соответствующий курс лечения подбирается с учетом индивидуальной потребности и мнения врача, который вводит лекарственное средство пациенту или назначает его введение. Суточную дозу терапевтического агента можно вводить однократно в виде разовой дозы или многократно в виде разделенных доз, которые вводят через определенные периоды времени. Таким образом, необходимое количество лекарственного средства, например необходимое терапевтически эффективное количество, определяет специалист в данной области медицины. Например, доза терапевтического агента может варьироваться в зависимости от возраста, веса тела или условий в интервале от приблизительно 1 мг до приблизительно 100 мг в расчете на антрафуран или его фармацевтически приемлемую соль на килограмм массы тела реципиента в сутки.
В соответствии с принципами, хорошо известными из данной области фармации, можно осуществлять модификации способов получения лекарственных средств по настоящему изобретению, изменив, например, состав, порядок добавления компонентов фармацевтических композиций, их просеивание, гранулирование и методы приготовления конечных дозированных лекарственных форм. Такие модификации также входят в объем прилагаемой формулы изобретения, а нижеследующие примеры приводятся лишь для иллюстрации настоящего изобретения, а не для ограничения объема притязаний.
Краткое описание чертежей
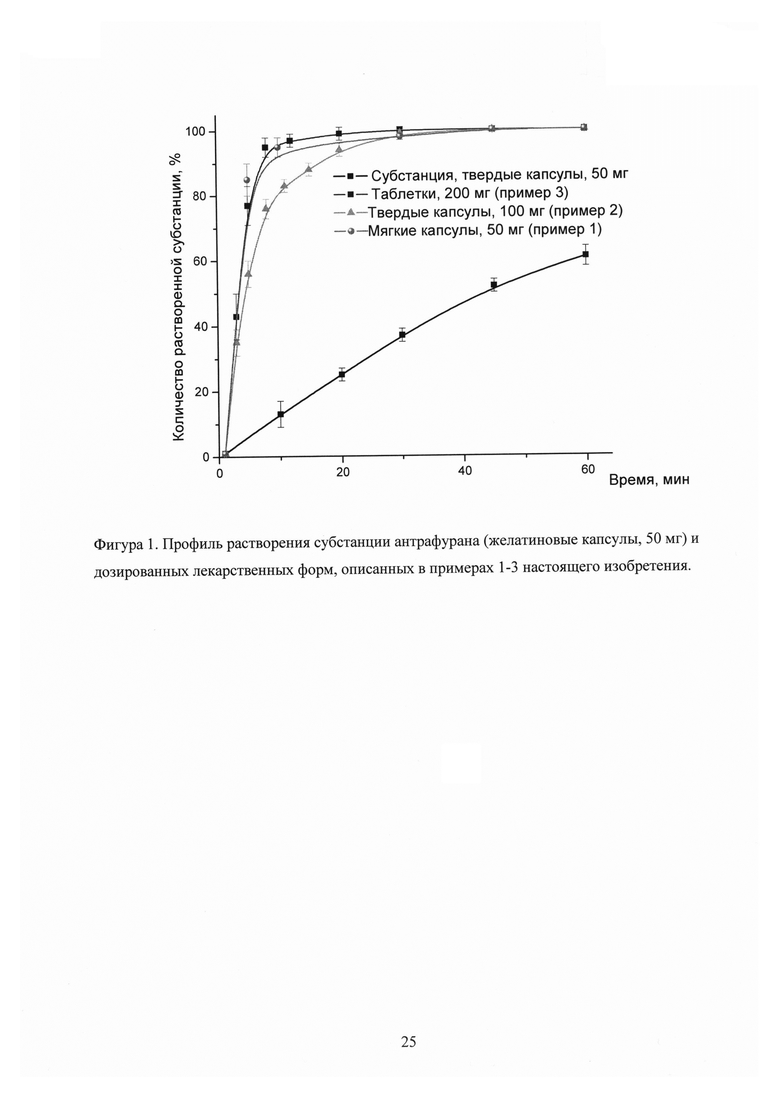
Фигура 1. Профиль растворения субстанции антрафурана (желатиновые капсулы, 50 мг) и дозированных лекарственных форм, описанных в примерах 1-3 настоящего изобретения.
Пример 1. Жидкая композиция для мягких капсул
Компоненты берут в соответствии с рецептурой жидкой композиции по настоящему изобретению, представленной в Таблице 1. К смеси метансульфоната антрафурана и полиэтиленгликоля 400 прибавляют раствор плюроника в смеси этанола и воды и гомомогенизируют в смесителе.
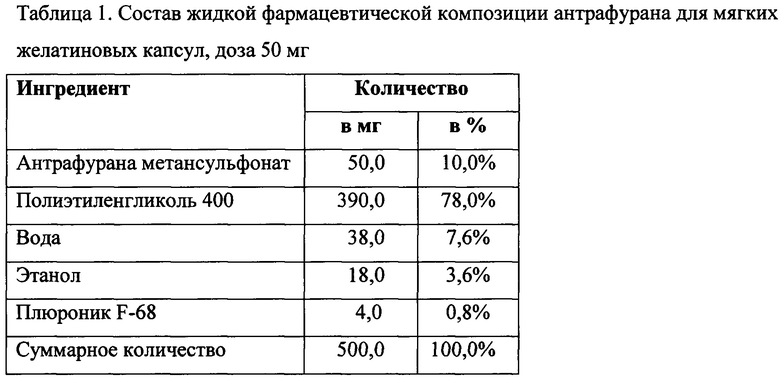
Полученную жидкую композицию инкапсулируют по 500 мг (что соответствует 50 мг антрафурана метансульфоната в капсуле) в мягкие капсулы с оболочкой, изготовленной из свиного и бычьего желатина в соотношении 2:1.
Пример 2. Твердая композиция для твердых капсул
Компоненты взвешивают в соответствии с рецептурой композиции, представленной в Таблице 2. Метансульфонат антрафурана гомогенизируют в смесителе в течение 10 мин, прибавляют просеянную смесь лактозы, крахмала, микрокристаллической целлюлозы и поливинилпирролидона, повидона после чего гомогенизируют в смесителе в течение 15 мин. Смесь просеивают через сито 100 мкм и повторно гомогенизируют в смесителе в течение 15 мин. Навеску додецилсульфата натрия и твина 80 растворяют в смеси воды и этанола, и полученным связующим раствором увлажняют твердую дисперсию при тщательном перемешивании. Влажную смесь пропускают через сито 250 мкм и сушат 8 ч при 40°С, пока остаточная влага в грануляте составит не более 3%. Гранулят пропускают через сито 100 мкм, опудривают аэросилом и инкапсулируют по 400 мг гранулята в твердые желатиновые капсулы размером 1.
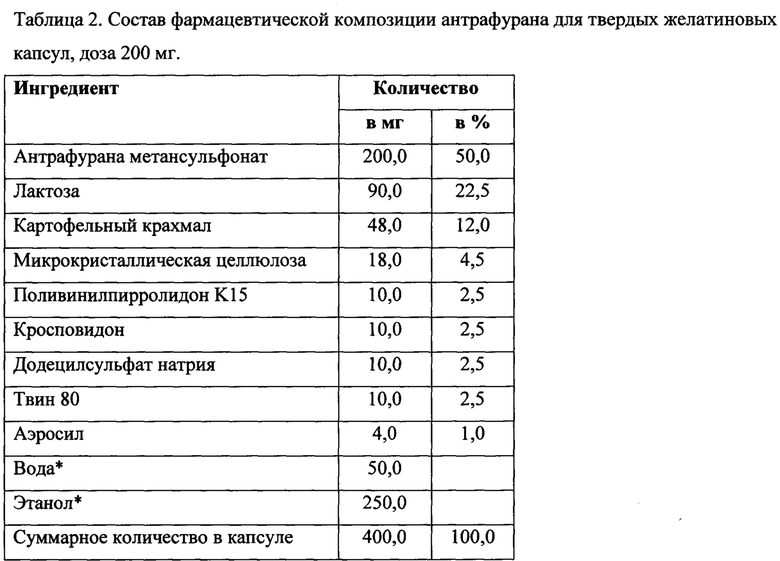
* Этанол (96%) и очищенная вода, используемые в качестве растворителей, испаряются в процессе сушки гранулята, поэтому они отсутствуют в конечном продукте.
Пример 3. Твердая композиция для таблеток в соответствии с изобретением
Компоненты внутренней и внешней фаз взвешивают в соответствии с рецептурой, представленной в Таблице 3. Компоненты внутренней фазы смешивают и гранулируют по методике влажной грануляции, описанной в примере 2. Компоненты внешней фазы (эксплосол, просолв, кросповидон, крахмал, поливинилпирролидон) просеивают через сито 100 мкм, смешивают в смесителе в течение 10 мин. Полученную смесь просеивают через сито 100 мкм и увлажняют дисперсию смесью воды и этанола. Влажную композицию тщательно перемешивают и пропускают через сито 250 мкм. Полученный гранулят сушат 8 ч при 40°С до содержания остаточной влаги 1-2% и протирают через сито 100 мкм. Стеарат магния, стеариновую кислоту и тальк смешивают в смесителе в течение 10 мин, просеивают через сито 100 мкм, и полученной смесью опудривают гранулят внешней фазы.

* Этанол (96%) и очищенная вода, используемые в качестве растворителей, испаряются в процессе сушки гранулята, поэтому они отсутствуют в конечном продукте.
Внешнюю и внутреннюю фазы смешивают в смесителе до получения гомогенной смеси, которую пропускают через сито 100 мкм. Полученный продукт прессуют в таблетки диаметром 9 мм с помощью таблеточного пресса (давление прессования - 3,0 кН).
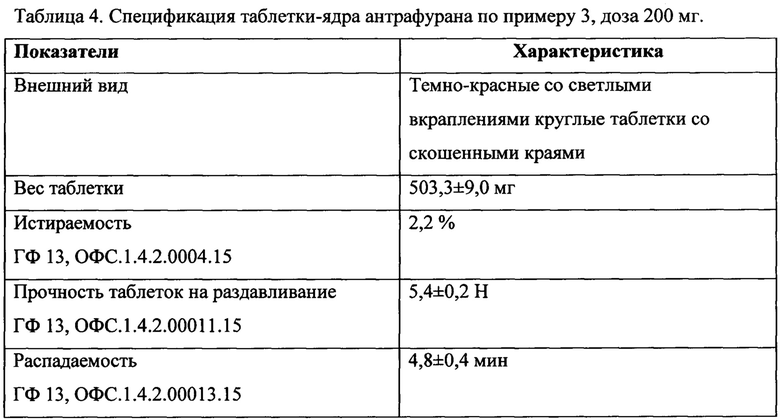
Полученные таблетки-ядра обладают приемлемой истираемостью и прочностью, а также высокой скоростью распадаемости (таблица 4).
Таблетки-ядра покрывают пленочной оболочкой в установке для нанесения покрытия (коаторе) с применением коммерчески доступной покрывающей смеси на основе гипромелозы Opadry® в количестве 12-14 мг на таблетку.
Пример 4. Профиль растворения дозированных лекарственных форм по настоящему изобретению
Для исследования профиля растворения использованы образцы лекарственных средств, описанных в примерах 1-3. Профиль растворения дозированных лекарственных форм исследовали в соответствии с общей фармакопейной статьей (ОФС) ГФ 13, ОФС.1.4.2.00014.15 «Растворение», с использованием аппарата «Лопастная мешалка», при температуре среды растворения +37,0±0,5°С и при скорости вращения лопастей мешалки 50 об/мин. В качестве среды растворения использовали 1000 мл раствора 0,1 н. соляной кислоты (среда, имитирующая желудочный сок человека); в качестве препарата сравнения использована субстанция, инкапсулированная в твердые желатиновые капсулы (доза 50 мг).
Для каждого типа дозированных форм исследовали по 6 образцов из разных серий. В начале эксперимента (0 минут), а также через 3, 5, 8, 11, 15, 20, 30, 45 и 60 мин отбирали пробу раствора, фильтровали через мембранный фильтр, отбрасывали первые порции фильтрата и определяли (методом УФ-спектроскопии) процент высвободившегося в среду антрафурана. Результаты исследования представлены на Фигуре 1 (в координатах: количество растворенного антрафурана, % - время, мин).
Фигура 1.
Фигура 1. Профиль растворения субстанции антрафурана (желатиновые капсулы, 50 мг) и дозированных лекарственных форм, описанных в примерах 1-3 настоящего изобретения.
Представленные экспериментальные данные убедительно показывают, что описанные в примерах фармацевтические композиции, являющиеся предметом настоящего изобретения, обеспечивают высокую скорость растворения антрафурана, а полученные разовые дозированные формы способны быстро (в течение 10 мин) высвобождать не менее 80% активного ингредиента. В противоположность лекарственным композициям, субстанция антрафурана (капсулы, 50 мг) обладает низкой скоростью растворения (менее 60% за 60 мин), что может являться причиной его низкой биодоступности.
Пример 5. Стабильность лекарственных композиций по настоящему изобретению
Стабильность лекарственных форм, описанных в примерах 1-3, оценивали методом ускоренного старения (хранение при 60°), и определяли содержание примесей в течение 0; 1; 2 месяцев. Для определения содержания действующего вещества и примесей использовали градиентную ВЭЖХ в следующих условиях:

Результаты опытов (таблица 5) свидетельствуют о том, что изменения содержания действующего вещества в фармацевтических композициях, не превышают 0,5% в течение всего периода испытаний (2 месяца). Функциональные характеристики лекарственных форм также достоверно не изменялись в течение всего периода испытаний. Таким образом, композиции, раскрытые в настоящем изобретении, обладают адекватной стабильностью для клинических и других применений.
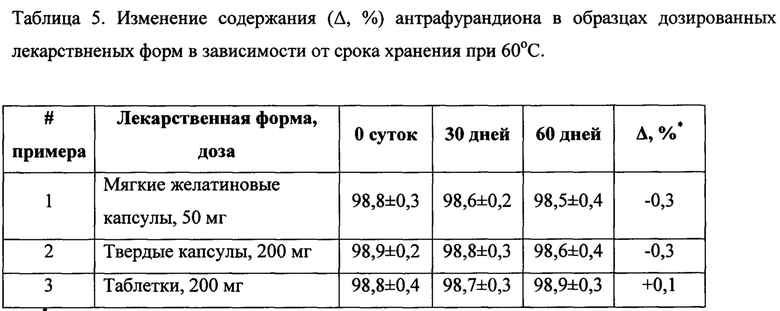
*Δ% - изменение процентного содержания антрафурандиона через 60 дней хранения.
Пример 6. Эффективность противоопухолевых средств на основе антрафурана на мышах с внутрибрюшинно перевитым лимфолейкозом Р388
В опытах использованы мыши-самки гибриды BDF1, массой тела 20-21 г. Перед опытом мышей ранжировали по группам, в каждой группе 6 особей. Одну группу мышей с опухолью (n=6) оставляли без специфического воздействия и считали контрольной. Основные группы получали лечение одним из вариантов лекарственных средств, описанных в примерах 1, 2.
Исследование выполнено in vivo на перевиваемом лимфолейкозе Р388, прогностически значимом для гемобластозов человека [Anticancer Drug Development Guide. Preclinical screening, clinical trials, and approval. Second edt. .- Humana Press. - Totowa. - New Jersey. - // Teicher B.A. and Andrews P.A. (eds.), 2004; Руководство no проведению доклинических исследований лекарственных средств. Часть первая // Трещалина Е.М., и соавт. 2012. - гл. 39. стр. 642-657]. Инокулят лейкоза готовили ex tempore в питательной среде 199 и трансплантировали мышам в брюшную полость по 1 млн. клеток на мышь.
Основные группы животных получали перорально жидкую композицию (пример 1) или твердую композицию (пример 2) в виде свежеприготовленных водного раствора или водной суспензии, содержащих 10 мг/мл метансульфоната антрафурана в дозах 60 или 80 мг/кг. Мыши контрольных групп получали перорально растворитель в адекватном объеме и режиме лечения. Введение растворов животным осуществляли в индивидуальных дозах, рассчитанных на массу тела, один раз в сутки ежедневно в течение 5 дней, начиная со вторых суток после перевивки опухоли.
Оценка эффективности выполнена по увеличению продолжительности жизни мышей в рамках критерия (УПЖ≥25%) в леченых группах по сравнению с контрольной в соответствии с действующими с РФ рекомендациями [Руководство по проведению доклинических исследований лекарственных средств. Часть первая // Трещалина Е.М., и соавт. 2012. - гл. 39. стр. 642-657]. Для этого определяли продолжительность жизни мышей индивидуальную (ПЖ) и среднюю в группе (СПЖ), последняя использована для расчета УПЖ. Полученные данные поступали прямо в статистическую программу Exel Windows 7 для подсчета индивидуальных и средних величин с определением стандартного отклонения (m), доверительного интервала (d) и величины Т-критерия Стьюдента. Достоверными считали различия при р<0,05.
Результаты исследования противоопухолевой активности показывают, что описанные в примерах 1, 2 фармацевтические композиции антрафурана (таблица 6) при пятикратном ежедневном пероральном введении в разовых дозах 60 мг/кг или 80 мг/кг (суммарно 300 или 400 мг/кг) проявляют значимый (более минимального критерия) высокий достоверный противоопухолевый эффект: УПЖ=58-109% и УПЖ=75-102% (р<0,05) независимо от величины примененной дозы (достоверных различий между эффективностью этих доз нет). Переносимость лекарственных форм хорошая, их применение не вызывает гибели мышей от токсичности.
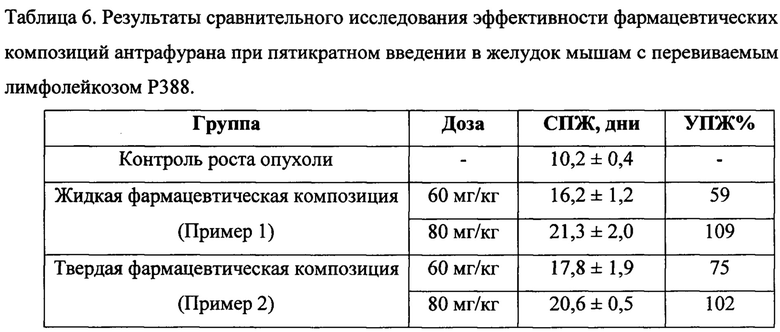
Как видно из результатов эксперимента на мышах с перевиваемым лимфолейкозом Р388, приведенные примеры пероральных средств, являющиеся предметом настоящего изобретения, обладают высокой достоверной противоопухолевой активностью, позволяющей прогнозировать эффективность фармацевтических композиций антрафурана для лечения гемобластозов, например, без ограничения перечисленным, лимфолейкоза, лимфогранулематоза, неходжкинской лимфомы.
Пример 7. Эффективность противоопухолевых средств на основе антрафурана на мышах с подкожно трансплантированной солидной карциномой легкого LLC
Исследование выполнено in vivo на перевиваемой эпидермоидной карциноме легкого Льюис (LLC), прогностически значимом для солидных опухолей человека различного гистогенеза [Anticancer Drug Development Guide. Preclinical screening, clinical trials, and approval. Second edt. .- Humana Press. - Totowa. - New Jersey. - // Teicher B.A. and Andrews P.A. (eds.), 2004; Руководство no проведению доклинических исследований лекарственных средств. Часть первая // Трещалина Е.М., и соавт. 2012. - гл. 39. стр. 642-657].
В опытах использованы мыши-самцы гибриды BDF1 массой тела 18-22 г. Перед опытом мышей ранжировали по группам, в каждой группе 12-13 особей. Одну группу мышей с опухолью (n=10) оставляли без специфического воздействия и считали контрольной. Основные группы (n=13) получали лечение одним из вариантов фармацевтических композиций антрафурана, описанных в примерах 1, 2.
Штамм LLC поддерживали in vivo на половозрелых мышах-самцах линии C57Bl6j. В опытах использован опухолевый инокулят, полученный из 2 или 3 пассажей in vivo. Инокулят готовили ex tempore в виде 10%-ной взвеси в питательной среде 199 и трансплантировали мышам-гибридам BDF1 под кожу бока по 50 мг опухолевой ткани на мышь. Оценка эффективности противоопухолевых средств выполнена в соответствии с рекомендованной методикой [Руководство по проведению доклинических исследований лекарственных средств. Часть первая, Трещалина Е.М., и соавт. 2012. - гл. 39. стр. 642-657].
Основные группы животных получали перорально жидкую композицию (пример 1) или твердую композицию (пример 2) в виде свежеприготовленных водного раствора или водной суспензии, содержащих 10 мг/мл антрафурана метансульфоната. Введение фармацевтических композиций животным осуществляли в соответствующих индивидуальным дозам объемах один раз в сутки в дозе 80 мг/кг в течение пяти дней, начиная с 3-го дня после трансплантации опухоли. Мыши контрольной группы получали перорально 5% раствор глюкозы в адекватном объеме и режиме лечения.
Оценка противоопухолевого эффекта выполнена с использованием стандартного показателя торможения роста опухоли (ТРО%), рассчитанного по разнице средних объемов (Vcp) опухоли в леченой и контрольной группах, выраженной в процентах [Руководство по проведению доклинических исследований лекарственных средств. Часть первая, Трещалина Е.М., и соавт. 2012. - гл. 39. стр. 642-657]. Индивидуальный объем опухоли определяли с помощью электронного измерителя, как произведение двух взаимно перпендикулярных диаметров. Достоверными считали различия при р≤0,05. Отсутствие опухоли у леченых мышей в течение всего периода наблюдения (22 дня) считали полной ремиссией (ПР).
Полученные результаты свидетельствуют, что фармацевтические композиции антрафурандиона, описанные в примерах 1, 2, являющиеся предметом настоящего изобретения, обладают достоверной высокой противоопухолевой активностью при пятикратном применении в разовой дозе 80 мг/кг (суммарно 400 мг/кг). Лучший противоопухолевый эффект на уровне ТРО=85% (n=13, р<0,05) и ТРО=72% (n=12, р<0,05) получен при указанных дозах на 12 сутки после трансплантации опухоли в период формирования опухолевых узлов при большом числе мышей без опухоли. При этом соответственно дозе почти у половины мышей в группах до 22 суток наблюдения рост опухоли был ингибирован полностью, ПР=7/13-5/12 (таблица 7). Достоверный, хотя и незначимый противоопухолевый эффект (ТРО=48 и 37%) с одним случаем полной ремиссии сохранялся до 22 суток после трансплантации опухоли. Переносимость фармацевтических композиций хорошая и их применение не вызывает гибели мышей от токсичности.
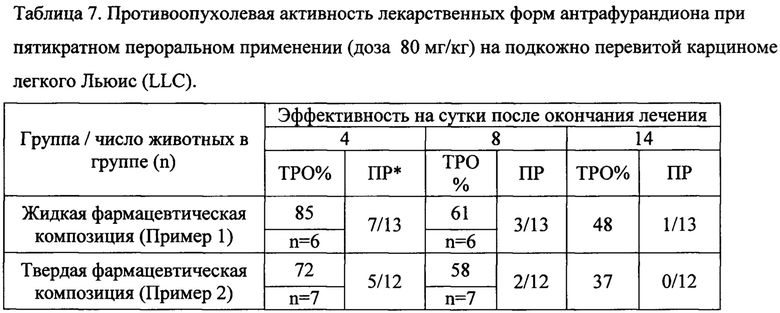
*ПР - число мышей без опухоли при завершении наблюдения (22 сутки)
Как видно из результатов эксперимента на мышах с перевиваемым карциномой легкого Льюис (LLC), пероральные лекарственные средства, являющиеся предметом настоящего изобретения, обладают высокой противоопухолевой активностью. Они вызывают значимое достоверное ингибирование роста опухоли с достижением полной ремиссии почти у половины животных. Данные о высокой достоверной противоопухолевой активности, полученные на этой модели, позволяют прогнозировать эффективность фармацевтических композиций антрафурана для лечения солидных опухолей, например, без ограничения перечисленным, рака молочной и щитовидной желез, яичников, яичка, желудка, нейробластомы, саркомы Вилмса, Ирвинга и остеогенной, трофобластической опухоли.
Пример 8. Эффективность противоопухолевых средств на основе антрафурана на мышах с подкожными ксенографтами рака молочной железы человека T47D
В опытах использованы 8-9 недельные иммунодефицитные мыши-самки Balb/c nude массой тела 20-22 г собственного разведения ФГБУ НМИЦ онкологии имени Н.Н. Блохина Минздрава России. Мышей содержали в специализированном однокоридорном кондиционированном отсеке при нормированном температурно-влажностном режиме на брикетированном стерильном корме, бумажной подстилке и воде с соблюдением требований, предъявляемым к конвенциональным животным [Иммунодефицитные мыши Balb/c nude и моделирование различных вариантов опухолевого роста для доклинических исследований. Трещалина Е.М. // РБЖ, 2017, №3. - стр. 4-13].
Для трансплантации взята адоптированная к росту in vivo культура клеток рака молочной железы человека T47D (АТСС № НТВ 133b-у). Опухоль представляет собой протоковый рак («ductal carcinoma»), клетки которого получены из плеврального выпота больной раком молочной железы с метастатическим плевритом и содержат положительную экспрессию рецепторов: кальцитонин, андрогены, эстрогены, прогестероны, глюкокортикоиды, пролактины. Имплантация 1-го пассажа выполнена 3,0×106 клеток на мышь. При окраске трипановым синим популяция жизнеспособных клеток составила 98%. В опытах использованы 2-й и 3-й пассажи, пассирование in vivo выполнено взвесью опухолевой ткани из предшествующего пассажа по 50 мг на мышь в питательной среде 199. Опухоль трансплантировали билатерально под кожу правого и левого бока.
Мышей перед опытом ранжировали по группам: группу с опухолью без специфического воздействия считали контрольной, в экспериментальной группе животные получали лечение фармацевтической композицией антрафурана, описанной в примере 2, в виде свежеприготовленной водной суспензии в стерильной дистиллированной воде (10 мг/мл). Введение фармацевтических композиций животным осуществляли в соответствующих индивидуальным дозам объемах один раз в сутки в дозе 80 мг/кг в течение пяти дней (суммарно 400 мг/кг), начиная с 3-го дня после трансплантации опухоли. Мыши контрольной группы получали перорально растворитель (стерильную дистиллированную воду) в адекватном объеме и режиме лечения.
Оценка противоопухолевого эффекта выполнена в соответствии с рекомендованной методикой исследованием динамики роста опухоли с использованием в качестве показателя торможение роста для опухоли человека (Т/С<42%). Определение объемов опухолей выполняли, как описано в примере 7. Показатель Т/С% определяли, как соотношение с [Vсропыта/Vсрконтроля]×100% [Рациональная фармакотерапия в онкологии, (ред.) Давыдов М.И., Горбунова В.А. М. Изд. «Литтерра», Трещалина Е.М., и соавт., 2015, с. 844].
Полученные результаты свидетельствуют, что фармацевтическая композиция антрафурандиона, описанная в примере 2, являющаяся предметом настоящего изобретения, обладает достоверной высокой противоопухолевой активностью при пятикратном применении в разовой дозе 80 мг/кг (суммарно 400 мг/кг).
Показано (таблица 8), что в группе контроля после трансплантации опухолевые узлы T47D растут достаточно быстро (латентный период 3-5 дней) и увеличиваются в течение недели (от 11 до 17 суток) почти в 4 раза, что свидетельствует об экспоненциальном росте. Размеры опухолей на 11, 14 и 17 сутки после трансплантации (сроки оценки эффективности в группе лечения), соответственно Vcp=1156±613 мм3, Vcp=2525±1034 мм3 и Vcp=3656±2301 мм3. В группе получавшей лечение пероральной фармацевтической композицией антрафурандиона на 7 сутки после окончания лечения размеры опухолей были достоверно и значимо меньше контроля, соответственно: Vcp=957±442 мм3 против Vcp=2525±1034 мм3, Т/С=38% (T-test=0,002, р<0,05). На 4 и 10 сутки наблюдения эффект был практически на пороговом уровне, Т/С=44-42%. Переносимость фармацевтической композиции хорошая, применение не вызывает гибели мышей от токсичности.
Как видно из результатов эксперимента на мышах с подкожными ксенографтами рака молочной железы человека T47D, пероральное лекарственное средство, являющееся предметом настоящего изобретения, обладает высокой противоопухолевой активностью, позволяющей прогнозировать эффективность фармацевтической композиции антрафурана в клинику солидных опухолей, в частности, рака молочной железы человека.
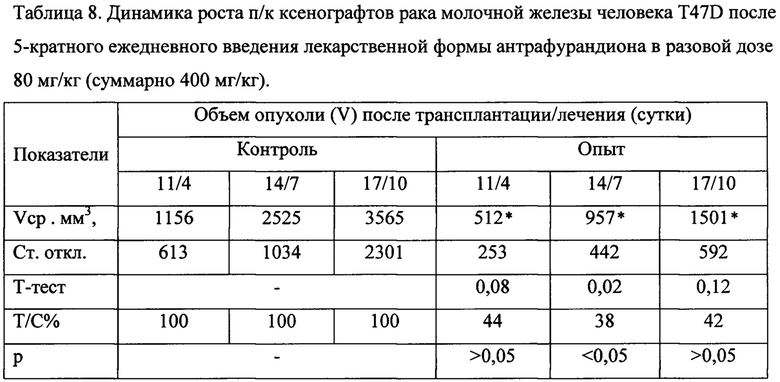
Примечание: *Одна мышь не имела опухоли на 6 сутки после окончания курса
Группа изобретений относится к химико-фармацевтической промышленности и представляет собой пероральное средство для лечения онкологических заболеваний, включающее: терапевтически эффективное количество метансульфоната (S)-3-[(3-амино-1-пирролидинил)карбонил]-4,11-дигидрокси-2-метилантра[2,3-b]фуран-5,10-диона, представленного формулой:
 и жидкий фармацевтически приемлемый носитель, включающий один или более растворителей, выбранных из воды, полиэтиленгликоля, 1,2-полипропиленгликоля, этанола, глицерина. Пероральное средство может представлять собой твердую дисперсию. Группа изобретений включает в себя способ лечения онкологического заболевания, включающий пероральное введение нуждающемуся пациенту терапевтически эффективного количества средства, описанного ранее. Заявленные в настоящем изобретении фармацевтические средства обеспечивают высокую растворимость антрафурана, быстрое высвобождение активного ингредиента из таблеток или капсул, а также стабильны при хранении. 3 н. и 10 з.п. ф-лы, 1 ил., 8 табл., 7 пр.
и жидкий фармацевтически приемлемый носитель, включающий один или более растворителей, выбранных из воды, полиэтиленгликоля, 1,2-полипропиленгликоля, этанола, глицерина. Пероральное средство может представлять собой твердую дисперсию. Группа изобретений включает в себя способ лечения онкологического заболевания, включающий пероральное введение нуждающемуся пациенту терапевтически эффективного количества средства, описанного ранее. Заявленные в настоящем изобретении фармацевтические средства обеспечивают высокую растворимость антрафурана, быстрое высвобождение активного ингредиента из таблеток или капсул, а также стабильны при хранении. 3 н. и 10 з.п. ф-лы, 1 ил., 8 табл., 7 пр.
1. Пероральное средство для лечения онкологических заболеваний, включающее:
a) терапевтически эффективное количество метансульфоната (S)-3-[(3-амино-1-пирролидинил)карбонил]-4,11-дигидрокси-2-метилантра[2,3-b]фуран-5,10-диона, представленного формулой:
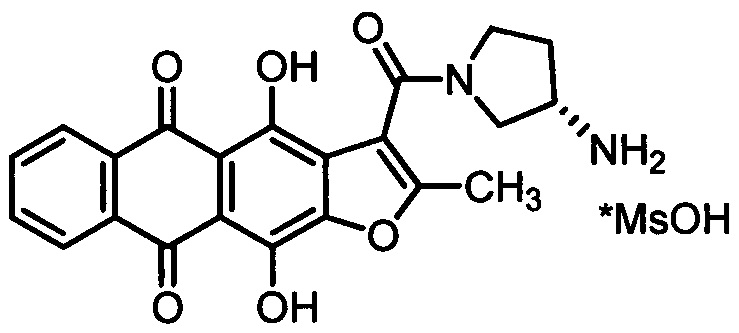
b) жидкий фармацевтически приемлемый носитель, включающий один или более растворителей, выбранных из воды, полиэтиленгликоля, 1,2-полипропиленгликоля, этанола, глицерина.
2. Средство по п. 1, отличающееся тем, что носитель дополнительно содержит один или более эксципиентов, выбранных из сорастворителей, диспергаторов, поверхностно-активных веществ, солюбилизаторов, эмульгаторов, стабилизаторов, консервантов, антиоксидантов, буферных соединений.
3. Средство по п. 1, отличающееся тем, что указанный полиэтиленгликоль имеет среднюю молекулярную массу 400 дальтон и присутствует в количестве от 40 мас. % до приблизительно 95% от общей массы композиции.
4. Средство по п. 1, отличающееся тем, что оно дополнительно содержит оболочку капсулы, в которую указанное средство инкапсулировано.
5. Пероральное средство, представляющее собой твердую дисперсию для лечения онкологических заболеваний, включающее:
a) терапевтически эффективное количество метансульфоната (S)-3-[(3-амино-1-пирролидинил)карбонил]-4,11-дигидрокси-2-метилантра[2,3-b]фуран-5,10-диона, представленного формулой:
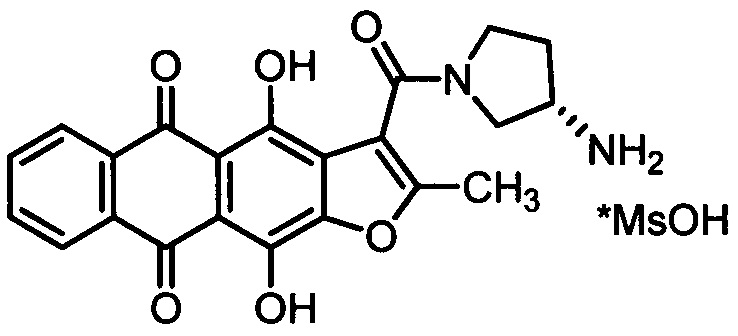
b) по меньшей мере, один или более фармацевтически приемлемых эксципиентов, выбранных из наполнителей (разбавителей, носителей), связывающих, разрыхляющих (дезинтегрирующих), скользящих, смазывающих (глиданты) агентов, поверхностно активных веществ.
6. Средство по п. 5, отличающееся тем, что один или более эксципиентов для твердой дисперсии антрафурана выбраны из целлюлозы и ее любых форм, микрокристаллической целлюлозы, силикатизированной микрокристаллической целлюлозы, производных целлюлозы, метилцеллюлозы, гидроксиэтилцеллюлозы, гидроксипропилцеллюлозы, гидроксипропилметилцеллюлозы, кармелозы натрия, карбоксиметилцеллюлозы, кроскармелозы натрия, крахмала, химически модифицированного крахмала, крахмалгликолята натрия, глюкозы, маннита, сорбита, лактозы, мальтозы, сахарозы, мальтодекстрина, поливинилпирролидона, повидона, повидон-винилацетата, коповидона, кросповидона, полиэтиленгликоля, полиэтилен/полипропилен/полиэтиленоксидных блок-сополимеров, двухосновного фосфата кальция, трехосновного фосфата кальция, желатина, альгината, трагаканта, ксантана, карагенана, гуарана, бентонита, диоксида кремния, коллоидного диоксида кремния, додецилсульфата натрия, солей желчных кислот, полиоксиэтиленовых эфиров сорбитана и жирных кислот, твина 80, стериновой кислоты и стеарата магния, талька.
7. Средство по п. 5, отличающееся тем, что метансульфонат (S)-3-[(3-амино-1-пирролидинил)карбонил]-4,11-дигидрокси-2-метилантра[2,3-b]фуран-5,10-диона присутствует в количестве от 10% до 80% по массе.
8. Средство по п. 5, отличающееся тем, что оно получено совместным измельчением метансульфоната (S)-3-[(3-амино-1-пирролидинил)карбонил]-4,11-дигидрокси-2-метилантра[2,3-b]фуран-5,10-диона с одним или более эксципиентами.
9. Средство по п. 6, отличающееся тем, что оно получено способом сухого или влажного гранулирования.
10. Средство по п. 5, отличающееся тем, что оно дополнительно включает оболочку капсулы, в которую указанное средство инкапсулировано в количестве, содержащем от 50 мг до 400 мг метансульфоната (S)-3-[(3-амино-1-пирролидинил)карбонил]-4,11-дигидрокси-2-метилантра[2,3-b]фуран-5,10-диона.
11. Средство по п. 5, отличающееся тем, что оно получено прессованием в форме таблетки, содержащей от 50 мг до 400 мг метансульфоната (S)-3-[(3-амино-1-пирролидинил)карбонил]-4,11-дигидрокси-2-метилантра[2,3-b]фуран-5,10-диона.
12. Способ лечения онкологического заболевания, включающий пероральное введение нуждающемуся пациенту терапевтически эффективного количества средства по одному из пп. 1-11.
13. Способ лечения онкологического заболевания по п. 12, в котором заболевание выбрано из опухоли молочной железы, легкого, кожи, желудочно-кишечного канала, печени, почек, мозга, мочевого пузыря, яичников, яичка, матки, предстательной железы, костей и мягких тканей, саркомы Вилмса, Ирвинга и Капоши, нейробластомы, гемобластозов, лимфолейкоз, лимфогранулематоз, неходжкинской лимфомы.
| ПРОТИВООПУХОЛЕВЫЙ АНТРАФУРАНДИОН И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ НА ЕГО ОСНОВЕ | 2014 |
|
RU2554939C1 |
| СПОСОБ ПОЛУЧЕНИЯ АНТРА[2,3-b]ФУРАН-3-КАРБОНОВОЙ КИСЛОТЫ | 2014 |
|
RU2554937C1 |
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |

