Перекрестная ссылка на родственные заявки
Настоящая заявка испрашивает приоритет согласно заявке на патент Китая No. 201810943005.6, под названием “КОМБИНАЦИЯ ИНГИБИТОРА ГИСТОНДЕАЦЕТИЛАЗЫ И ИНГИБИТОРА ПРОТЕИНКИНАЗЫ И ЕЕ ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ”, поданной 17 августа 2018 г. в Национальное управление интеллектуальной собственности Китая, содержание которой полностью включено в настоящую заявку посредством ссылки.
Область техники
Настоящее раскрытие относится к области техники низкомолекулярного противоопухолевого лекарственного средства направленного действия и, в частности, относится к применению комбинации ингибитора гистондеацетилазы и ингибитора киназы в получении лекарственного средства для лечения или профилактики опухоли, и фармацевтической композиции, включающей ингибитор гистондеацетилазы и ингибитор киназы в качестве активных фармацевтических ингредиентов. Настоящее раскрытие, в частности, относится к фармацевтической композиции, включающей чиаураниб или его фармацевтически приемлемую соль и чидамид или его фармацевтически приемлемую соль в качестве активных ингредиентов, и способу лечения или профилактики рака с помощью комбинации чиаураниба или его фармацевтически приемлемой соли и чидамида или его фармацевтически приемлемой соли.
Уровень техники
Рак представляет собой серьезное заболевание, угрожающее здоровью человека. Лечение рака привлекло внимание всего мира. Обычные химиотерапевтические лекарственные средства неспецифически блокируют деление клеток, вызывая гибель клеток, но они также повреждают нормальные клетки человека, убивая опухолевые клетки. Кроме того, многие цитотоксические лекарственные средства имеют ограниченный диапазон лечения и склонны вызывать побочные реакции, которые могут повышать лекарственную устойчивость после длительного приема.
В предшествующем уровне техники были разработаны различные типы противоопухолевых лекарственных средств, среди которых ингибиторы гистондеацетилазы (HDAC) являются важным классом противоопухолевых соединений.
Гистондеацетилаза (HDAC) представляет собой класс протеаз, играющих важную роль в структурной модификации хромосом и регуляции экспрессии генов. В общем, ацетилирование гистонов способствует диссоциации ДНК и октамеров гистонов и релаксации структуры нуклеосом, так что различные факторы транскрипции и кооперативные факторы транскрипции могут специфически связываться с участками связывания ДНК и активировать транскрипцию генов. В ядре ацетилирование и деацетилирование гистонов находятся в динамическом равновесии и регулируются гистонацетилтрансферазами (HAT) и HDAC. HAT переносят ацетильную группу от Acetyl-CoA к определенным остаткам лизина на аминоконцах гистонов. HDAC деацетилируют гистоны и прочно связываются с отрицательно заряженной ДНК, в результате чего хроматин становится более компактным и изогнутым, что приводит к транскрипционной репрессии. В раковых клетках сверхэкспрессия HDAC приводит к увеличению деацетилирования. За счет восстановления положительного заряда гистонов взаимодействие между ДНК и гистонами усиливается, и свободные нуклеосомы становятся компактными, что неблагоприятно для экспрессии определенных генов, включая некоторые гены-супрессоры опухолей. Ингибиторы HDAC, как новый класс противоопухолевых препаратов, могут увеличивать ацетилирование гистонов в определенных областях хроматина, тем самым регулируя экспрессию и стабильность белков, связанных с апоптозом и дифференцировкой клеток, и индуцируя апоптоз и дифференцировку клеток. Ингибиторы HDAC не только обладают хорошим терапевтическим действием на различные гематологические опухоли и солидные опухоли, но также обладают преимуществами относительно высокой селективности к опухолевым клеткам и низкой токсичности.
Литература показала, что ингибирование активности HDAC может эффективно ингибировать рост, метастазирование и инвазию опухолевых клеток. Ингибиторы HDAC стали важными противоопухолевыми кандидатами. Исследования показали, что ингибиторы HDAC могут эффективно ингибировать пролиферацию опухолевых клеток, индуцировать апоптоз и дифференцировку опухолевых клеток, а также антиангиогенез и оказывать ингибирующее действие на миграцию, инвазию и метастазирование опухолевых клеток.
Ингибиторы HDAC активируют гены-супрессоры опухоли, регулируя ацетилирование и деацетилирование остатков лизина на N-конце гистонов, тем самым ингибируя рост опухолевых клеток и вызывая апоптоз опухолевых клеток (Apoptosis on hepatoma cells but not on primary hepatocytes by histone deacetylase inhibitors valproate and ITF2357. J Hepatol, 2005, 42: 210-217). Yindong Kang et al., в 2009 году, опубликовали обзор исследований иммуномодулирующего действия ингибиторов гистондеацетилазы через дендритные клетки. (Yindong Kang et al., Studies on the Immunomodulation Effect of Histone Deacetylase Inhibitors through Dendritic Cells, Chinese Journal of Transplantation, Volume 3, Issue 2, 2009). В этой обзорной статье описана взаимосвязь между ингибиторами гистондеацетилазы и дендритными клетками (DC), а также иммунная регуляция. Ингибиторы HDAC проявляют сильную противоопухолевую активность при высоких концентрациях и различные иммуномодулирующие эффекты при низких концентрациях. Когда ингибиторы HDAC непосредственно действуют на DC, они ослабляют способность DC мигрировать к хемокину CCL19 во время созревания, тем самым ограничивая DC от достижения и проникновения в лимфоидные органы для осуществления своих антигенпрезентирующих функций. Ингибиторы HDAC влияют на дифференцировку, созревание и миграцию DC и мешают процессу презентации антигена DC, как наиболее важных антигенпрезентирующих клеток, которые играют важную роль в активации антигенспецифических Т-лимфоцитов. Как правило, ингибиторы HDAC напрямую действуют на DC, подавляя иммунный ответ.
В последние годы исследования функций HDAC быстро прогрессировали. Разработка селективного ингибитора HDAC постепенно становится горячей точкой исследований в этой области. В настоящее время различные ингибиторы HDAC уже коммерчески доступны как в стране, так и за рубежом, например, вориностат, чидамид. Однако существующие ингибиторы HDAC все еще нуждаются в улучшении в аспектах активности, эффективности, метаболизма лекарств и побочных эффектов. Противоопухолевое лекарственное средство с более высокой эффективностью и лучшим эффектом срочно требуется в уровне техники, для удовлетворения растущих потребностей людей в области здравоохранения.
Сущность изобретения
В исследовании авторы настоящего изобретения неожиданно обнаружили, что комбинация ингибитора гистондеацетилазы и ингибитора протеинкиназы может обеспечивать синергические противоопухолевые эффекты.
В одном аспекте настоящее изобретение предоставляет применение комбинации ингибитора гистондеацетилазы и ингибитора протеинкиназы в получении лекарственного средства для лечения или профилактики опухолей.
В другом аспекте настоящее изобретение предоставляет способ лечения или профилактики опухоли, включающий введение ингибитора гистондеацетилазы и ингибитора протеинкиназы субъекту, нуждающемуся в этом.
Протеинкиназы представляют собой семейство ферментов, которые катализируют фосфорилирование определенных остатков в белках. Они широко классифицируются на тирозиновые и серин/треониновые протеинкиназы, которые представляют собой большое семейство белков, которые играют важную роль в регулировании различных клеточных процессов и поддержании функций клеток. Протеинкиназы представляют собой ферменты, связанные с путями передачи сигнала, которые катализируют перенос концевого фосфата АТФ на гидроксильную группу остатков тирозина, серина и/или треонина белков. Сверхэкспрессия или неправильная экспрессия нормальных или мутантных протеинкиназ у млекопитающих широко изучалась и, как было показано, играет важную роль в развитии многих заболеваний, включая рак. Эти киназы частично включают, но не ограничиваются ими, нерецепторные тирозинкиназы, например, семейство Янус-киназы (Jak1, Jak2, Jak3 и Tyk2); рецепторные тирозинкиназы, например, рецептор тромбоцитарного фактора роста (PDGFR); и серин/треониновые протеинкиназы, например b-RAF. Аномальная активность киназы наблюдается при многих заболеваниях, включая доброкачественные и злокачественные пролиферативные нарушения, а также заболевания, вызванные неадекватной активацией иммунной и нервной систем.
Как большое семейство структурно связанных ферментов, протеинкиназы отвечают за регулирование различными процессами передачи сигналов в клетках. (См., например, Hardie and Hanks, The Protein Kinase Facts Book, I and II, Academic Press, San Diego, Calif., 1995). Считается, что протеинкиназы произошли от общего предкового гена благодаря сохранению их структуры и каталитической функции. Почти все киназы содержат аналогичный каталитический домен из 250-300 аминокислот. Киназы можно разделить на различные семейства по их рецепторам фосфорилирования (например, белок-тирозин, белок-серин/треонин, липиды и т.п.). Были определены мотивы последовательности, которые в целом соответствуют каждому из этих семейств. (см., например, Hanks and Hunter, (1995), FASEB J.9:576-596; Knighton etc., Science, (1991) 253:407-414; Hiles etc., Cell, (1992), 70:419-429; Kunz etc., Cell (1993), 73:585-596; Garcia-Bustos etc., EMBO J., (1994), 13:2352-2361).
Многие заболевания связаны с неадекватной активностью киназы, вызванной мутацией, сверхэкспрессией или неадекватной регуляцией, аномальной регуляцией или дисрегуляцией, а также с суперпродукцией или недостаточной продукцией факторов роста или цитокинов, включая, помимо прочего, рак и другие заболевания. Протеинкиназы стали классом важных ферментов в качестве мишеней для терапевтического вмешательства. В частности, сверхактивация тирозинкиназы cKit связана с гематологическими злокачественными новообразованиями и является мишенью для лечения рака (Heinrich, Griffith etc., Blood 2000, 96 (3): 925-32). Точно так же передача сигналов JAK3 участвует в лейкемии и лимфоме, которые в настоящее время используются в качестве потенциальной терапевтической мишени (Heinrich, Griffith etc., 2000). Протеинкиназы также играют ключевую роль в регуляции клеточного цикла. Было обнаружено, что различные заболевания, в том числе многие типы рака, могут быть вызваны дефектами различных компонентов путей передачи сигнала (Gaestel et al., Current Medicinal Chemistry, (2007) 14: 2214-2234). В последние годы протеинкиназы, относящиеся к путям онкогенной передачи сигналов, стали важными мишенями для лекарственного средства при лечении различных заболеваний, включая многие типы рака. В качестве противоопухолевых лекарственных средств также использовали различные ингибиторы протеинкиназы.
В настоящем раскрытии, предпочтительно, ингибитор протеинкиназы выбирают из ингибитора серинкиназы, ингибитора треонинкиназы и ингибитора тирозинкиназы. Предпочтительно ингибитор протеинкиназы представляет собой чиаураниб или его фармацевтически приемлемую соль.
В настоящем раскрытии предпочтительно ингибитор гистондеацетилазы представляет собой чидамид или его фармацевтически приемлемую соль.
В одном особенно предпочтительном аспекте настоящего раскрытия ингибитор гистондеацетилазы представляет собой чидамид или его фармацевтически приемлемую соль, и ингибитор киназы представляет собой чиаураниб или его фармацевтически приемлемую соль. Более предпочтительно, ингибитор гистондеацетилазы представляет собой чидамид и ингибитор киназы представляет собой чиаураниб.
В другом аспекте настоящее раскрытие предоставляет фармацевтическую композицию, включающую ингибитор гистондеацетилазы и ингибитор протеинкиназы в качестве активных фармацевтических ингредиентов и необязательно включающую фармацевтически приемлемые эксципиенты и/или носители.
В фармацевтической композиции по настоящему изобретению предпочтительно ингибитор протеинкиназы выбран из ингибиторов серинкиназы, ингибиторов треонинкиназы и ингибиторов тирозинкиназы. Особенно предпочтительно ингибитор протеинкиназы представляет собой чиаураниб или его фармацевтически приемлемую соль. Особенно предпочтительно активные фармацевтические ингредиенты состоят из чиаураниба или его фармацевтически приемлемой соли и чидамида или его фармацевтически приемлемой соли. В конкретном варианте осуществления активные фармацевтические ингредиенты состоят из чиаураниба и чидамида.
В особенно предпочтительном варианте осуществления настоящего раскрытия фармацевтическая композиция представлена в стандартной лекарственной форме, содержащей 5-100 мг чидамида и 5-100 мг чиаураниба. Более предпочтительно количество чидамида и чиаураниба составляет 5-60 мг и 10-100 мг, соответственно.
В некоторых вариантах осуществления настоящего раскрытия в фармацевтической композиции ее фармацевтически приемлемые эксципиенты и/или носители включают повидон, коповидон, гидроксипропилметилцеллюлозы и привитые сополимеры поливинилкапралактам-поливинилацетат-полиэтиленгликоль (например, Soluplus®). В некоторых других вариантах осуществления фармацевтически приемлемые эксципиенты и/или носители включают микрокристаллическую целлюлозу, повидон, коповидон, лактозу, маннит, кросповидон и карбоксиметилцеллюлозу натрия.
Фармацевтическая композиция по настоящему изобретению предпочтительно находится в форме гранул, твердых дисперсий, капсул или таблеток.
В другом аспекте настоящее раскрытие предоставляет набор для лечения или профилактики рака или опухоли, содержащий ингибитор гистондеацетилазы и ингибитор протеинкиназы в качестве активных ингредиентов, где ингибитор гистондеацетилазы и ингибитор протеинкиназы можно вводить одновременно или последовательно.
Предпочтительно, набор по настоящему раскрытию включает чидамид или его фармацевтически приемлемую соль и чиаураниб или его фармацевтически приемлемую соль в качестве активных ингредиентов, которые можно вводить одновременно или последовательно. В особенно предпочтительном аспекте набор по настоящему раскрытию включает чидамид или его фармацевтически приемлемую соль, вводимые первым, и чиаураниб или его фармацевтически приемлемую соль, вводимые вторыми, в качестве активных ингредиентов.
В другом аспекте настоящее раскрытие предоставляет способ лечения или профилактики опухоли, включающий одновременное или последовательное введение ингибитора гистондеацетилазы и ингибитора протеинкиназы субъекту, нуждающемуся в этом.
Авторы изобретения неожиданно обнаружили, что комбинация ингибитора гистондеацетилазы и ингибитора протеинкиназы обладает значительным синергическим противоопухолевым действием, что было подтверждено в экспериментах на “голых” мышах, показавших, что комбинированное введение двух ингредиентов может синергически индуцировать апоптоз раковых клетки и синергически ингибируют образование колоний опухолевых клеток. Авторы изобретения также неожиданно обнаружили, что предварительная обработка ингибитором гистондеацетилазы может повысить чувствительность клеток к ингибитору протеинкиназы и более эффективно усилить противоопухолевые эффекты ингибитора протеинкиназы.
В анализах MTS, колониеобразования, анализе клеточного цикла в проточной цитометрии, каспазы 3/7 и других экспериментах по настоящему раскрытию было обнаружено, что комбинированное введение ингибитора гистондеацетилазы и ингибитора протеинкиназы может синергически индуцировать ингибирование клеточного цикла и апоптоз клеточных линий гепатоцеллюлярной карциномы, например, Bel-7404 и Bel-7402. Такая синергическая противоопухолевая эффективность за счет комбинации ингибитора гистондеацетилазы и ингибитора протеинкиназы также была доказана на ксенотрансплантатной модели клеток Bel-7404 на “голых” мышах.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
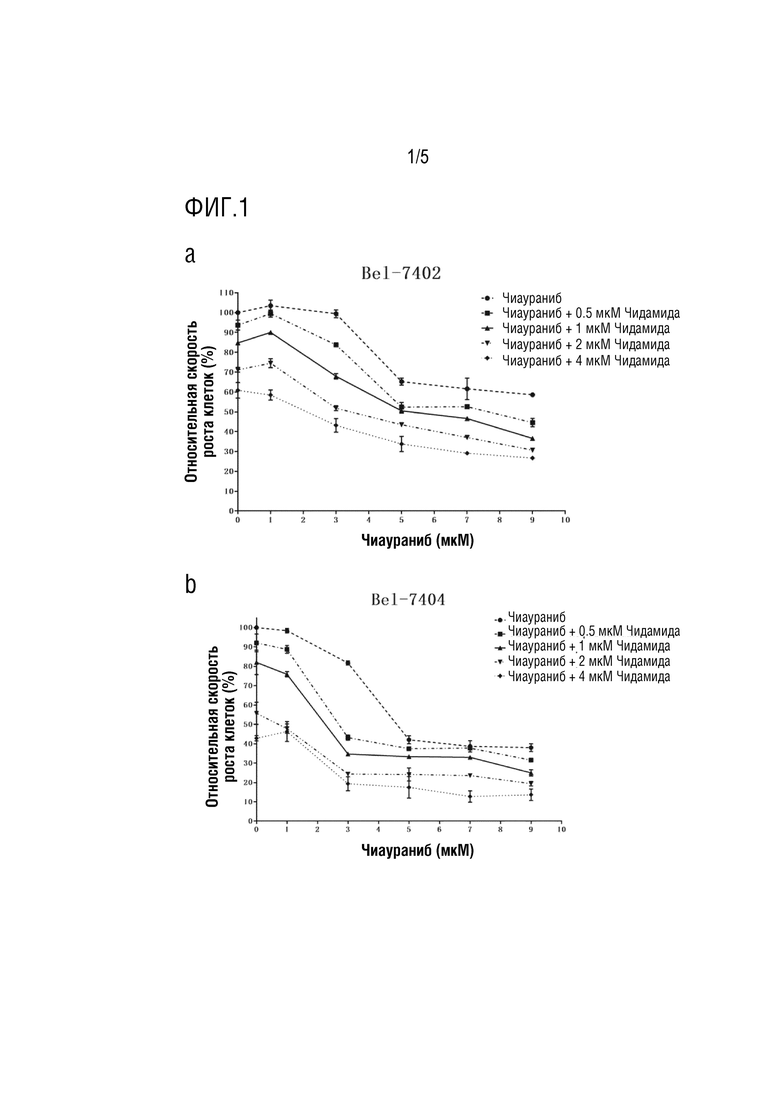
На фиг. 1 показана противоопухолевая эффективность ингибитора протеинкиназы (чиаураниб), синергетически сенсибилизируемого ингибитором гистондеацетилазы (чидамид);
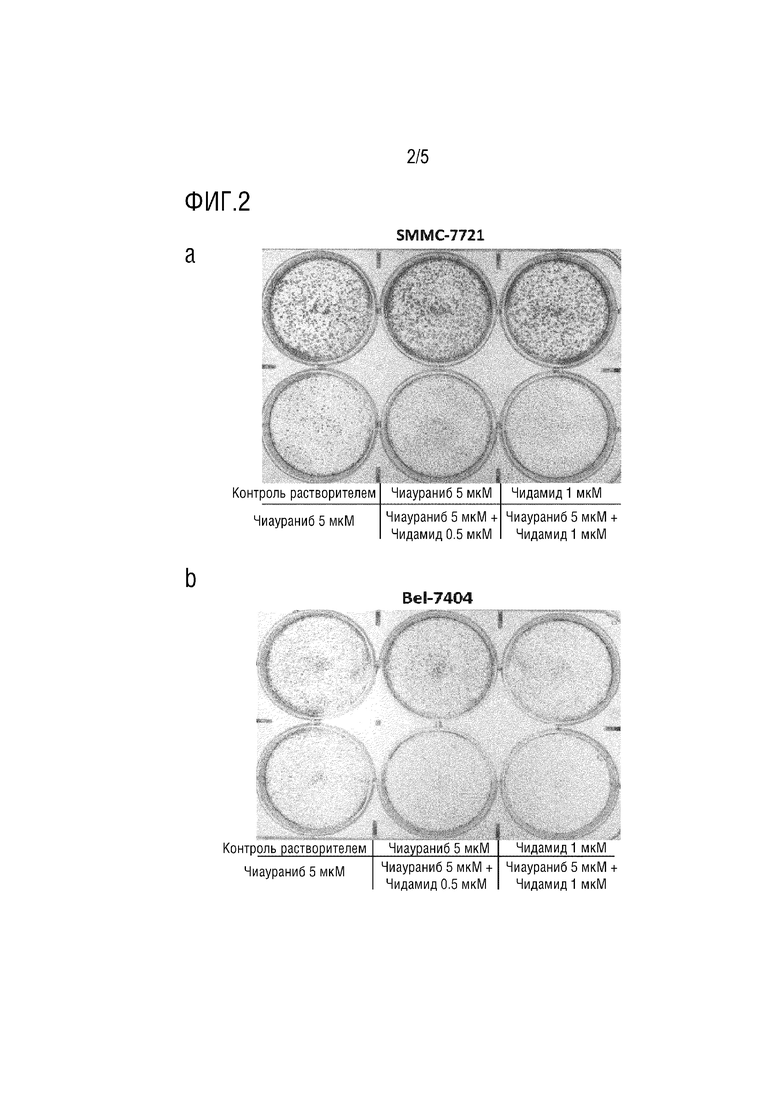
На фиг.2 показано, что комбинация ингибитора гистондеацетилазы и ингибитора протеинкиназы может синергически ингибировать клоногенность опухолевых клеток, как показано в анализе кристаллического фиолетового.
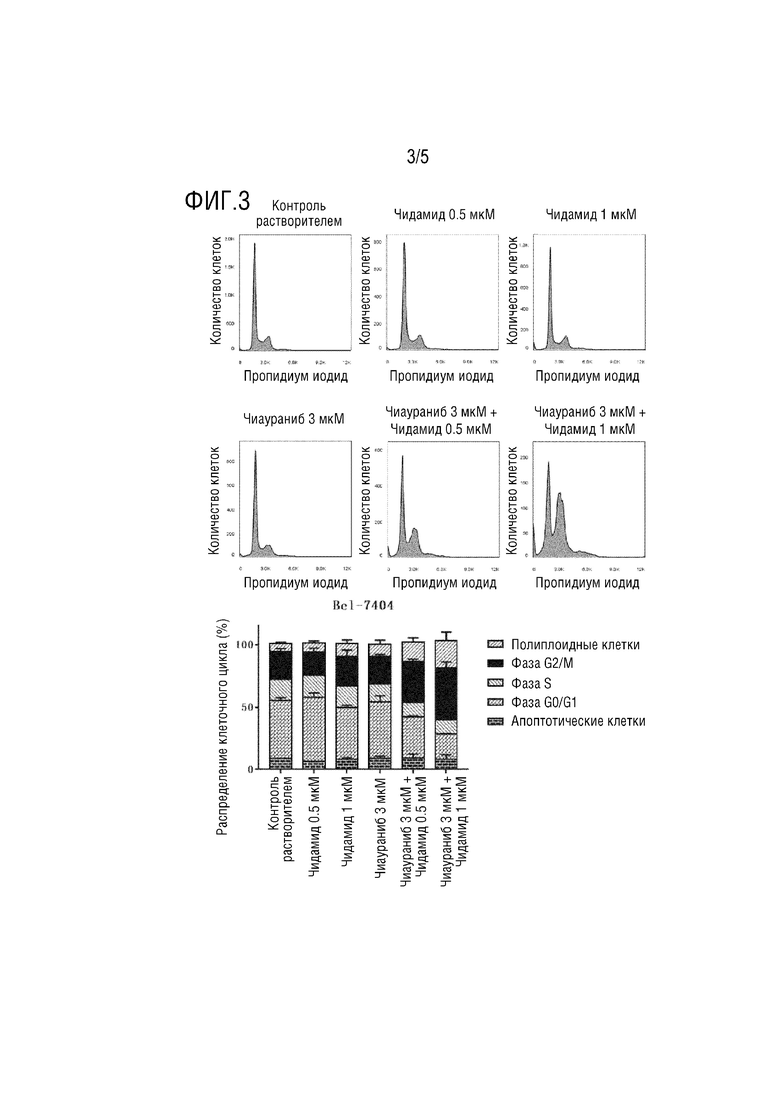
На фиг.3 показано, что комбинация ингибитора гистондеацетилазы и ингибитора протеинкиназы может усиливать ингибирование цикла деления опухлевых клеток, тестируемого методом проточной цитометрии с пропидиум иодидом (PI);
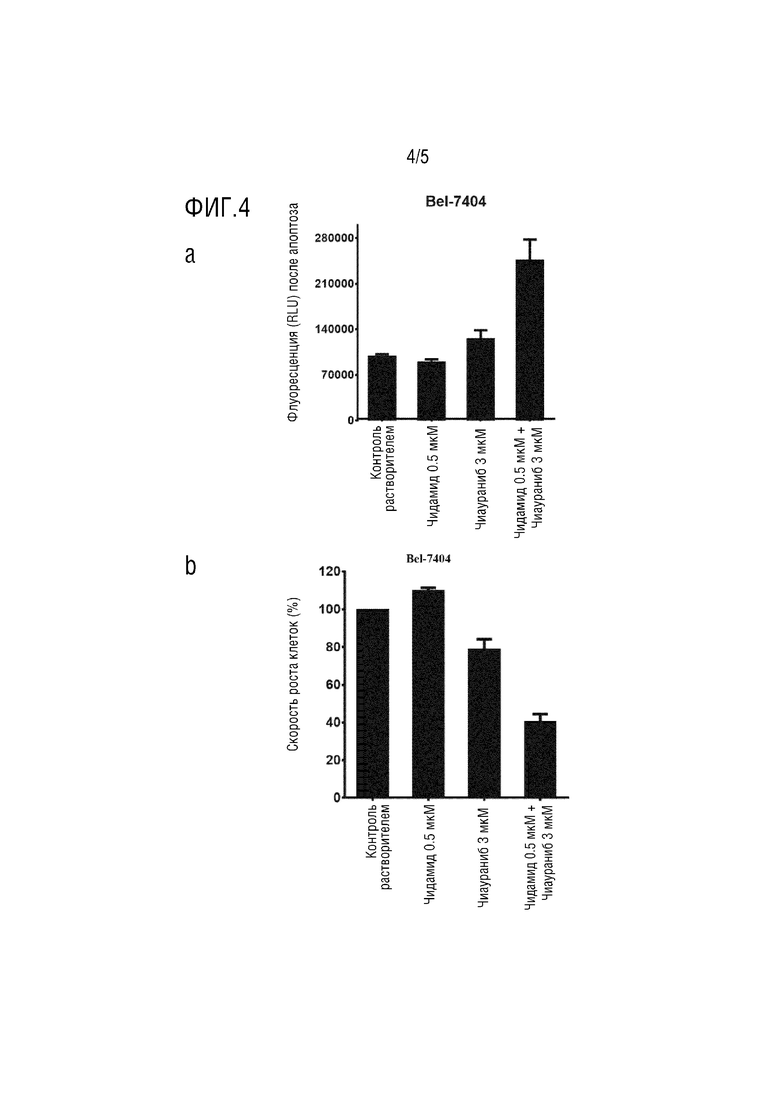
На фиг. 4 показано, что комбинация ингибитора гистондеацетилазы и ингибитора протеинкиназы может усиливать индукцию апоптоза опухоли, протестированную с помощью анализа каспазы 3/7;
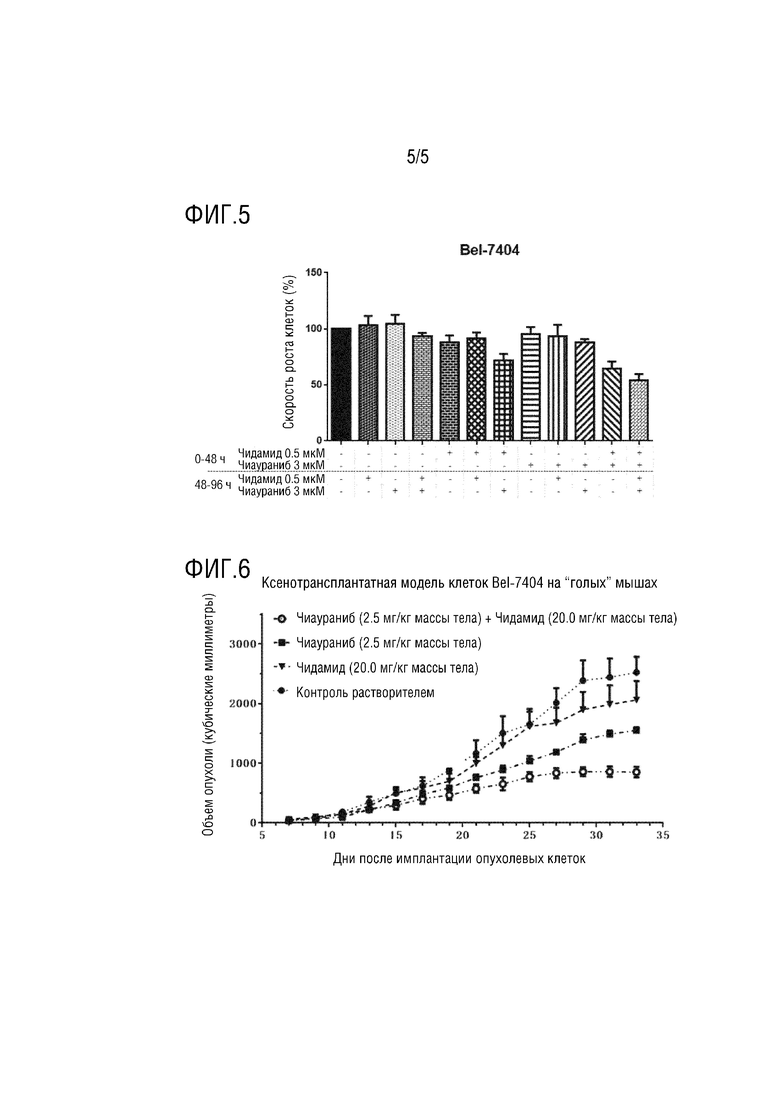
На фиг. 5 показано, что предварительное лечение ингибитором гистондеацетилазы (чидамид) может усиливать противоопухолевый эффект ингибитора протеинкиназы (чиаураниб) в экспериментах с последовательным введением;
На фиг. 6 показана синергическая противоопухолевая эффективность комбинации ингибитора гистондеацетилазы (чидамид) и ингибитора протеинкиназы (чиаураниб) на моделях «голых» мышей.
ПОДРОБНОЕ ОПИСАНИЕ
Используемые в настоящем описании термины используются для описания конкретных вариантов осуществления, но не предназначены для ограничения настоящего раскрытия. Используемые в настоящем описании формы единственного числа также предназначены для включения форм множественного числа, если нет четкого указания в контексте. Кроме того, термины «включать», «содержать», «заключать» или их варианты в описании и/или формуле настоящего раскрытия предназначены для использования аналогично термину «содержать».
Используемые в настоящем описании термины “лечить”, “облегчать” и “улучшать” могут использоваться взаимозаменяемо. Эти термины относятся к способам, связанным с благоприятными или ожидаемыми эффектами (включая, но не ограничиваясь, терапевтическую пользу и/или профилактическую пользу).
Используемый в настоящем описании термин «противоопухолевый» относится к лечению, облегчению или улучшению «опухолевого состояния». Термин «опухолевое состояние» относится к существованию клеток с аномальными характеристиками роста, такими как неконтролируемая пролиферация, бесконечная пролиферация, метастатический потенциал, быстрый рост и высокая скорость пролиферации, неупорядоченная онкогенная передача сигналов и определенная уникальная морфология. Оно включает, но не ограничивается ими, рост следующих клеток: (1) доброкачественные или злокачественные клетки (например, опухолевые клетки), связанные со сверхэкспрессией гистондеацетилаз, тирозиновых или серин/треониновых протеинкиназ; (2) доброкачественные или злокачественные клетки (например, опухолевые клетки), связанные с аномально высокой активностью гистондеацетилаз, тирозиновых или серин/треониновых протеинкиназ.
Специалисты в данной области будет понятно, что в комбинации лекарственных средств или фармацевтической композиции согласно настоящему раскрытию активные фармацевтические ингредиенты используются в эффективном количестве или терапевтически эффективном количестве. Термины «эффективное количество» или «терапевтически эффективное количество» относятся к количеству описанного в настощем документе ингибитора, которое достаточно для достижения желаемого результата (включая, но не ограничиваясь этим, применение при лечении заболеваний), как определено ниже. Терапевтически эффективное количество может варьироваться в зависимости от предлагаемого применения (in vitro или in vivo) или субъекта, подлежащего лечению, и состояния заболевания, такого как масса тела и возраст субъектов, тяжесть заболевания, способ введения и т.п., которое может быть легко определено специалистом в данной области. Этот термин также применяется к дозировке, которая может вызвать специфический ответ (например, снижение пролиферации или понижающая регуляция активности целевого белка) в клетке-мишени. Конкретная дозировка может варьироваться в зависимости от конкретного выбранного соединения, режима дозирования, которого необходимо придерживаться, от того, вводится ли оно в комбинации с другими соединениями, времени введения, ткани, подлежащей введению, и физической системы доставки для его переноса.
«Синергизм» или «синергический эффект» означает, что, когда один активный фармацевтический ингредиент объединен с эффективным количеством другого активного фармацевтического ингредиента или другой терапией, он дает лучший эффект, чем использование каждого из них по отдельности. В некоторых вариантах осуществления синергическое использование активных фармацевтических ингредиентов в эффективных терапевтических количествах или средств для лечения дает лучший эффект, чем аддитивный эффект каждого из двух активных фармацевтических ингредиентов или терапевтических средств при использовании по отдельности.
Термин «ингибитор» относится к соединению, способному ингибировать биологическую функцию целевого белка путем ингибирования активности или экспрессии целевого белка. Биологическая активность ингибитора связана с развитием, ростом или распространением опухоли или нежелательными иммунными ответами при аутоиммунном заболевании.
Термины «комбинированное введение», «совместное введение» и их грамматические эквиваленты включают применение двух или более фармацевтически активных ингредиентов у субъекта таким образом, чтобы фармацевтически активные ингредиенты и/или их метаболиты могли одновременно присутствовать в организме животного. Комбинированное введение включает введение активных ингредиентов одновременно или в разное время в виде отдельных композиций или введение композиции, включающей два ее активных ингредиента. Совместно вводимые активные фармацевтические ингредиенты могут находиться в одном и том же препарате, или в разных препаратах, или в одном и том же продукте, например, в виде набора.
Используемый в настоящем описании термин «терапевтический эффект» включает терапевтические и/или профилактические преимущества, как описано выше. Профилактический эффект включает отсрочку или устранение появления заболевания или состояния, отсрочку или устранение начала заболевания или состояния, замедление, остановку или обращение прогрессирования заболевания или состояния, или любую их комбинацию.
Термин «фармацевтически приемлемая соль» относится к солям, полученным из множества органических и неорганических противоионов, хорошо известных в данной области. Фармацевтически приемлемые соли включают фармацевтически приемлемые соли присоединения кислот и соли присоединения оснований. Соли присоединения кислоты могут быть образованы с неорганическими и органическими кислотами. Неорганические кислоты, способные к образованию солей, включают, например, хлористоводродную кислоту, бромистоводородную кислоту, серную кислоту, азотную кислоту, фосфорную кислоту ,и тому подобное. Органические кислоты, способные образовывать соли, включают, например, уксусную кислоту, пропионовую кислоту, гликолевую кислоту, пировиноградную кислоту, щавелевую кислоту, малеиновую кислоту, малоновую кислоту, янтарную кислоту, фумаровую кислоту, винную кислоту, лимонную кислоту, бензойную кислоту, коричную кислоту, миндальную кислоту, метансульфоновую кислоту, этансульфоновую кислоту, п-толуолсульфоновую кислоту, салициловую кислоту и тому подобное. Фармацевтически приемлемые соли присоединения оснований могут быть образованы с неорганическими и органическими основаниями. Неорганические основания, способные к образованию солей, включают, например, натрий, калий, литий, аммоний, кальций, магний, железо, цинк, медь, марганец, алюминий и тому подобное. Органические основания, способные образовывать из них соли, включают, например, первичные амины, вторичные амины и третичные амины, замещенные амины, включая природные замещенные амины, циклические амины, основные ионообменные смолы и тому подобное, особенно такие, как изопропиламин, триметиламин, диэтиламин, триэтиламин, трипропиламин и этаноламин. В некоторых вариантах осуществления фармацевтически приемлемые соли присоединения оснований выбирают из солей аммония, калия, натрия, кальция и магния.
«Фармацевтически приемлемый эксципиент и/или носитель» включает любые и все растворители, дисперсионные среды, покрытия, антибактериальные и противогрибковые агенты, изотонические регуляторы и агенты, замедляющие абсорбцию, и тому подобное. Эти среды и агенты, используемые в фармацевтически активных веществах, хорошо известны в данной области. В терапевтических композициях по настоящему изобретению можно использовать любые обычные среды или агенты, за исключением тех, которые несовместимы с активными ингредиентами. В их композиции также могут быть включены дополнительные активные ингредиенты.
В настоящем описании опухоли включают следующие заболевания или состояния: рак груди; рак яичников; рак матки; рак шейки матки; рак простаты; рак мочевого пузыря; лейкоз, включая острый миелоидный лейкоз (AML), острый лимфолейкоз, хронический лимфолейкоз, хронический миелоидный лейкоз, волосатоклеточный лейкоз, остеомиелодисплазию, миелопролиферативные заболевания, острый миелоидный лейкоз (AML), хронический миелолейкоз (CML), мастоцитоз, хронический лимфолейкоз (CLL), множественную миелому (MM) и миелодиспластические синдромы (MDS); рак кости; рак легких; рак кожи, включая базальноклеточную карциному, меланому и плоскоклеточный рак; ретинобластому; кожную или внутриглазную (глазную) меланому; первичную карциному печени; карциному почек; рак щитовидной железы; лимфому, связанную со СПИДом, такую как диффузная крупноклеточная B-клеточная лимфома, иммунобластная B-клеточная лимфома и мелкоклеточная лимфома с нерассеченными ядрами; капосиссаркому; рак центральной нервной системы, такой как первичная опухоль головного мозга, включая нейроглиому; рак периферической нервной системы, включая нейрофиброматоз и неврилеммому, злокачественную фиброзно-клеточную опухоль, злокачественную фиброзную гистиоцитому, злокачественную менингиому, злокачественную мезотелиому и смешанная мезодермальная опухоль; рак ротовой полости и ротоглотки; рак желудка; рак яичек; рак вилочковой железы; рак прямой кишки и рак толстой кишки.
Комбинированное лечение согласно настоящему раскрытию эффективно в широком диапазоне доз. Например, при использовании для лечения взрослых суточная доза ингибитора гистондеацетилазы и ингибитора протеинкиназы может составлять от 1 до 100 мг, предпочтительно от 5 до 100 мг, соответственно. В конкретном варианте осуществления используется комбинация чидамида и чиаураниба, где суточная доза чидамида составляет 5-60 мг, а суточная доза чиаураниба составляет 10-100 мг, соответственно. Точная доза может быть легко определена специалистами в данной области в соответствии с выбранным активным фармацевтическим ингредиентом, путем введения, формой вводимого соединения, субъектом, подлежащим лечению, массой субъекта, подлежащего лечению, а также предпочтениями и опытом врача.
Фармацевтическая комбинация настоящего раскрытия также может использоваться вместе с другими активными фармацевтическими ингредиентами, обладающими противоопухолевой активностью. Соответственно, фармацевтическая композиция или набор по настоящему раскрытию может дополнительно содержать другой активный фармацевтический ингредиент(ы) с противоопухолевой активностью.
В некоторых вариантах осуществления фармацевтическая композиция может быть подходящей для перорального введения, например, в форме гранул, капсул или таблеток. Фармацевтическая композиция по настоящему изобретению, подходящая для перорального введения, может быть в виде диспергируемых лекарственных форм, таких как капсулы или таблетки, жидкости или спреи, каждая из которых включает заранее определенное количество активных ингредиентов, в виде порошков или гранул, растворов или суспензий в водной или неводной жидкости, эмульсий масло-в-воде или жидких эмульсий вода-в-масле, включая жидкие лекарственные формы (такие как суспензии или жидкий раствор) и пероральные твердые лекарственные формы (такие как таблетки или сыпучие порошки). Используемый в настоящем описании термин «таблетка» обычно относится к таблеткам, каплетам, капсулам (включая мягкие желатиновые капсулы) и пастилкам. Пероральные лекарственные формы могут быть представлены в виде таблеток, пилюль, драже, капсул, эмульсий, липофильных и гидрофильных суспензий, жидкостей, гелей, сиропов, жидких растворов, суспензий, принимаемых перорально индивидуумом или пациентом, подлежащим лечению. Такие лекарственные формы можно получить любым фармацевтическим методом. Подходящими эксципиентами могут быть наполнители, такие как сахара, включая лактозу, сахарозу, маннит или сорбитол; целлюлозные наполнители, такие как кукурузный крахмал, пшеничный крахмал, рисовый крахмал, картофельный крахмал, желатин, трагакант, метилцеллюлоза, гидроксипропилметилцеллюлоза, карбоксиметилцеллюлоза натрия и/или поливинилпирролидон (ПВП). Вообще говоря, композицию получают путем равномерного и плотного смешивания активного ингредиента с жидкими носителями или мелко измельченными твердыми носителями или с обоими, а затем при необходимости придания продукту желаемой формы. Например, из нее можно получить таблетку путем прессования или формования, необязательно, с одним или несколькими дополнительными ингредиентами. Она также может быть получена в виде таблетки путем прессования активного ингредиента в сыпучей форме, такой как порошок или гранулы, с использованием подходящей машины. Ее активный ингредиент необязательно смешивают с эксципиентами, такими как, но не ограничиваясь ими, связующие, смазывающие вещества, инертные разбавители и/или поверхностно-активные вещества или диспергирующие вещества. Формованные таблетки могут быть получены формованием смеси порошкообразного соединения, увлажненного инертным жидким разбавителем, в подходящей машине. Носитель может принимать различные формы в зависимости от формы препарата, необходимой для введения. При получении композиции для лекарственных форм для перорального введения в качестве носителя можно использовать любую обычную фармацевтическую среду. В случае жидких лекарственных препаратов для перорального применения (таких как суспензии, растворы и эликсиры) или аэрозолей, таких как вода, гликоли, масла, спирты, ароматизаторы, консерванты, красители и т.п. могут использоваться в качестве носителей. В случае твердых лекарственных препаратов для перорального применения, в некоторых вариантах осуществления, где лактоза не используется, например, в качестве носителей могут использоваться крахмалы, сахара, микрокристаллическая целлюлоза, разбавители, гранулирующие средства, скользящие вещества, связующие и средства для улучшения распадаемости. Например, подходящие носители для твердых лекарственных препаратов для перорального применения включают порошки, капсулы и таблетки. При необходимости на таблетки можно нанести покрытие стандартным водным или неводным способом.
Композиция может дополнительно включать одну или несколько фармацевтически приемлемых добавок и эксципиентов. Такие добавки и эксципиенты включают, но не ограничиваются ими, средства для предотвращения адгезии, противовспенивающие агенты, буферы, полимеры, антиоксиданты, консерванты, хелатирующие агенты, модификаторы вязкости, регуляторы тоничности, ароматизаторы, красители, усилители вкуса, замутнители, суспендирующие агенты, связующие, наполнители, пластификаторы, скользящие вещества и их смеси.
Эксперименты
Экспериментальные материалы
Клеточные линии гепатомы человека Bel-7402 и Bel-7404 приобретали в Cell Resource Center of Shanghai Institutes for Biological Sciences, CAS, и обычно культивировали при 37°C и 5% CO2 в культуральной среде RPMI-1640 (Gibco), содержащей 10% фетальной бычьей сыворотки (FBS; Gibco) и 1% пенициллин-стрептомицин (HyClone). Трипсин приобретали у Gibco. Кристаллический фиолетовый, раствор РНКазы A (10 мг/мл), пропидиум иодид (PI), Triton X-100 приобретали у Biotech (Shanghai) Co., Ltd. Наборы для анализа жизнеспособности клеток MTS, наборы каспаза-Glo 3/7 приобретали у Promega. “Голых” мышей приобретали в Guangdong Medical Laboratory Animal Center.
Пример 1. Анализ MTS
Экспериментный метод
Клеточные линии Bel-7402, Bel-7404 расщепляли трипсином, собирали и подсчитывали, а затем высевали в 96-луночный планшет для культивирования клеток при плотности 3000 клеток/180 мкл в каждой лунке и культивировали при 37°C в условиях 5% СО2. После культивирования в течение ночи клетки вводили в соответствии с группировкой и конечными концентрациями, показанными на фиг. 1 (конечный объем каждой лунки после введения составлял 200 мкл). Дозу для каждой группы помещали в 3 параллельных лунках. После обработки клеток лекарственными средствами в течение 120 часов культуральную среду в 96-луночном планшете удаляли и 100 мкл реагентов MTS для анализа жизнеспособности клеток, содержащих 89,5 мкл RPMI-1640 без фенолового красного, 10 мкл MTS и 0,5 мкл PMS, добавляли в каждую лунку, в то время как такой же объем реагентов MTS для определения жизнеспособности клеток добавляли в лунки без посева клеток в качестве фона обнаружения (OD490-BLK). После инкубации при 37°C в течение примерно 1 часа оптическую плотность каждой лунки считывали при длине волны 490 нм с помощью микропланшетного ридера. Значение OD490-T для каждой лунки с введением и значение OD490-T0 для лунки отрицательного контроля после вычета фона были получены путем вычитания OD490-BLK из значения оптической плотности каждой лунки.
Показатель относительной выживаемости клеток в каждой лунке с введением рассчитывали по следующей формуле:
Показатель выживаемости=OD490-T/OD490-T0 × 100%
Результат эксперимента
Как показано на фиг. 1, в клеточных линиях Bel-7402 и Bel-7404 как монотерапия чиауранибом, так и монотерапия чидамидом показали определенный ингибирующий эффект на пролиферацию опухолевых клеток, который был дозозависимым. Следует отметить, что в Bel-7402, когда эффективная доза чиаураниба достигала 3 мкМ, и эффективная доза чидамида достигала 0,5, 1 или 2 мкМ, соответственно, оба лекарственных средства проявляли значительный синергический эффект, то есть при одной и той же дозе эффективность комбинации двух лекарственных средств была лучше, чем сумма эффективности двух одиночных лекарственных средств, используемых по отдельности. В Bel-7404, когда эффективная доза чиаураниба достигала 3 мкМ, а эффективная доза чидамид достигала 0,5 или 1 мкМ, два лекарственных средства проявляли более значительный синергический эффект, чем эффект в Bel-7402.
Пример 2. Анализы образования колоний
Экспериментный метод
Клеточные линии SMMC-7721, Bel-7404 на стадии логарифмической фазы расщепляли трипсином, собирали и подсчитывали, затем высевали в 6-луночный планшет для культивирования клеток при плотности 500 клеток/1,8 мл в каждой лунке и культивировали при 37°C под 5% CO2. После культивирования в течение ночи клетки вводили в соответствии с группировкой и конечными дозами концентрации, показанными на фиг. 2 (конечный объем каждой лунки после введения составлял 2 мл). Культуральную среду и лекарственные средства заменялись каждые 3 дня для поддержания стабильной систему культивирования и дозировку для каждой лунки.
После культивирования в течение 2-3 недель, когда в культуральном планшете были видны колонии клеток, культивирование прекращали. Супернатанты отбрасывали, и планшет дважды тщательно промывали PBS. 1 мл 75% раствора этанола добавляли в каждую лунку для фиксации клеток, и фиксирующий раствор удаляли через 15 мин. Затем клетки окрашивали в течение 15 мин, добавляя по 1 мл раствора кристаллического фиолетового в каждую лунку, и окрашивающий раствор медленно промывали проточной водой и сушили на воздухе. Окрашенные положительные колонии клеток в каждой лунке культурального планшета фотографировали для сравнения их плотности.
Результат эксперимента
Как показано на фиг. 2, в SMMC-7721, при монообработке чидамидом клеток в дозе 0,5 или 1 мкМ, он не оказывал значительного эффекта на образование колоний клеток. Плотность положительных колоний, окрашенных кристаллическим фиолетовым, в двух лунках была эквивалентна плотности в контрольной лунке, куда добавляли равный объем растворителя DMSO. Однако монообработка чиауранибом в дозе 5 мкМ оказывала относительно значительный эффект на образование колоний клеток. При добавлении 0,5 или 1 мкм дозы чидамида с чиауранибом в такой дозе, образование колоний клеток ингибировалось более значительно, и ингибирование положительно коррелировало с дозой чидамида. В Bel-7404, при монообработке чидамидом клеток в дозировке 0,5 или 1 мкМ, или монообработке чиауранибом клеток в дозе 3 мкМ, все они в некоторой степени влияли на образование колоний клеток, в то время как образование колоний клеток ингибировалось более значительно, когда два препарата объединялись на клетках. Таким образом, комбинация чидамида и чиаураниба оказывала синергический чувствительный эффект на ингибирование колоний SMMC-7721 и Bel-7404.
Пример 3. Клеточный цикл анализировали методом проточной цитометрии с окрашиванием PI
Экспериментный метод
Клетки Bel-7404 на стадии логарифмической фазы расщепляли трипсином, собирали и подсчитывали. Клетки высевали в 6-луночный планшет при плотности 106 клеток на лунку, всего 18 лунок. После нормального культивирования в течение ночи высеянные клетки разделяли на 6 групп, а именно: контрольная группа с растворителем, группа 3 мкМ чиаураниба, группа 0,5 мкМ чидамида, группа 1 мкМ чидамида, группа комбинации 3 мкМ чиаураниба и 0,5 мкМ группа чидамида и группа комбинации 3 мкМ чиаураниба и 1 мкМ чидамида. Каждую группу размещали в 3 параллельных лунках, и соответствующие соединения добавляли в соответствии с указанными выше конечными концентрациями. После культивирования в течение 48 часов образцы клеток собирали трипсинизацией и центрифугировали при 800 об/мин в течение 10 мин. Каждый образец полностью ресуспендировали в 300 мкл фосфатно-солевого буфера (PBS), по каплям добавляли в 700 мкл предварительно охлажденного абсолютного этанола, и затем равномерно перемешивали путем осторожного переворачивания несколько раз для диспергирования и фиксации клеток. Зафиксированные образцы выдерживали при 4°C более 12 часов и анализировали методом проточной цитометрии в течение 1 недели.
PBS, 10 мг/мл маточного раствора PI, раствор 10 мг/мл РНКазы A и Triton X-100 смешивали в соотношении 1000:5:2:1 с образованием рабочего раствора красителя. Зафиксированные образцы клеток центрифугировали при 4°C, 1000 об/мин в течение 10 мин, супернатанты полностью отбрасывали и дважды промывали PBS. Каждый образец ресуспендировали в 300 мкл указанного выше рабочего раствора красителя. После инкубирования при 37°C в течение 30 минут в темноте клетки фильтровали через сито из нержавеющей стали 200 меш. Фильтрат анализировали на клеточные циклы методом проточной цитометрии (каждый образец насчитывал 20000 клеток).
Результат эксперимента
Как показано на фиг. 3, в Bel-7404, по сравнению с контрольной группой с растворителем, клеточный цикл группы 0,5 мкМ чидамида практически не подвергся влиянию. Количество клеток в фазе G2/M и полиплоидных клеток немного увеличилось в группе 1 мкМ чидамида и в группе 3 мкМ чиаураниба. Когда 3 мкМ чиаураниба объединяли с 0,5 мкМ или 1 мкМ чидамида, клетки имели более значительную остановку фазы G2/M и увеличение доли полиплоидных клеток, что указывает на то, что комбинация чидамид и чиаураниба оказывала противоопухолевый эффект, синергически усиливающим ингибирование клеточного цикла и синергически способствующим полиплоидизации клеток.
Пример 4. Апоптоз клеток анализировали с помощью анализа каспаза 3/7
Экспериментный метод
Клетки Bel-7404 на стадии логарифмической фазы расщепляли трипсином, собирали и подсчитывали. Клетки высевали в 96-луночный планшет при плотности 2000 клеток на лунку, всего 18 лунок. После нормального культивирования в течение ночи высеянные клетки разделяли на 6 групп, а именно, холостая группа, контрольная группа с растворителем, группа 3 мкM чиаураниба, группа 0,5 мкM чидамида, группа комбинации 3 мкM чиаураниба и 0,5 мкM чидамида и группа положительного контроля. Каждую группу размещали в 3 параллельных лунках, и соответствующие соединения добавляли в соответствии с указанными выше конечными концентрациями и со-культивировали в течение 48 часов.
Буфер Caspase-Glo3/7 и лиофилизированный порошок Caspase-Glo3/7 в качестве субстрата уравновешивали до 18-22°C. Затем буфер Caspase-Glo3/7 наливали в коричневую бутыль, содержащую субстрат Caspase-Glo3/7, и смешивали их встряхиванием или переворачиванием до полного растворения субстрата с образованием реагента Caspase-Glo.
96-луночные планшеты, содержащие обработанные клетки, вынимали из инкубатора и уравновешивали до комнатной температуры. 70 мкл реагента Caspase-Glo (культуральная среда: реагент Caspase-Glo =1:1) добавляли в каждую лунку 96-луночного планшета, содержащего 70 мкл холостого контроля, контрольной группы с растворителем, групп, обработанных лекарственным средством, и положительной контрольной группы. Содержимое каждой лунки осторожно перемешивали, используя планшетный шейкер при 300-500 об/мин в течение 30 с. Клетки инкубировали при комнатной температуре (18-22°C) в течение от 30 мин до 3 ч. Значение флуоресценции каждого образца измеряли люминометром. Значение флуоресценции может напрямую отражать долю апоптотических клеток в каждой лунке и может использоваться для расчета и сравнения жизнеспособности клеток различных групп.
Результат эксперимента
Как показано на фиг. 4, в Bel-7404, после обработки клеток только 0,5 мкМ чидамида, доля апоптотических клеток не имела значительного отличия от контрольной группы растворителя с равным объемом DMSO. Монообработка 3 мкМ чиауранибом в определенной степени увеличивала долю апоптотических клеток. Когда два препарата в вышеуказанных дозах объединяли для обработки клеток, доля апоптотических клеток была значительно увеличена, что было значительно выше, чем сумма доли апоптоза, увеличенной двумя отдельными лекарственными средства по отдельности. Два лекарственных средства синергетически способствовали апоптозу опухолевых клеток. Соответственно, комбинация вызвала ингибирование активности опухолевых клеток, которое было значительно больше, чем сумма ингибирования, вызванного применением каждого из двух лекарственных средств по отдельности.
Пример 5. Эксперимент по последовательному введению
Экспериментный метод
Клетки Bel-7404 на стадии логарифмической фазы расщепляли трипсином, собирали и подсчитывали, высевали в 96-луночный планшет для культивирования клеток при плотности 5000 клеток/180 мкл в каждой лунке и культивировали в атмосфере 5% CO2 при 37°C. После культивирования клеток в течение ночи добавляли лекарственные средства, соответствующие группировке и конечным концентрациям в течение 0-48 часов, как показано на фиг. 5, и конечный объем в каждой лунке достигал 200 мкл. После культивирования клеток в течение 48 часов культуральную среду осторожно отсасывали из 96-луночного планшета. В каждую лунку добавляли 180 мкл свежей культуральной среды и лекарственные средства, соответствующие группировке и конечным концентрациям в течение 48-96 часов, как показано на фиг. 5 (так, чтобы конечный объем в каждой лунке достигал 200 мкл). После того, как клетки культивировали еще 48 часов, то есть общее время обработки лекарственным средством достигло 96 часов, культуральную среду отбрасывали в 96-луночном планшете и добавляли реагенты MTS для анализа жизнеспособности клеток для обнаружения и рассчета выживаемости клеток в каждой лунке.
Результат эксперимента
Как показано на фиг. 5, в Bel-7404, будь то первые 48 часов или следующие 48 часов, ингибирующий эффект на клетки, вызванный добавлением двух лекарств одновременно, был больше, чем эффект при добавлении каждого из двух лекарств по отдельности. Следует отметить, что предварительная обработка клеток монообработкой чидамидом в первые 48 часов и затем обработка клеток монообработкой чиауранибом в следующие 48 часов после отмены чидамида сильно отличалась от предварительной обработки клеток монообработкой чиауранибом в первые 48 часов и затем обработки клеток монообработкой чидамидом в следующие 48 часов после отмены чиаураниба. Предварительная обработка чидамидом сделала клетки более чувствительными к чиауранибу, но предварительная обработка чиауранибом не сделала клетки чувствительными к последующему эффекту монообработки чидамидом, что указывает на то, что механизм синергической сенсибилизации, возникающий в результате комбинации двух лекарственных средств, заключался в том, что эпигенетическая модификация клеток с помощью чидамида изменяла чувствительность клеток к фармакологическим эффектам чиаураниба.
Пример 6. Эксперименты на модели с ксенотрансплантатом опухолевых клеток у “голых” мышах
Экспериментный метод
Клетки Bel-7404 культивировали для использования большого количества и поддерживали в состоянии логарифмической фазы. После того как количество клеток достигло необходимого количества, их расщепляли трипсином, собирали, дважды промывали большим количеством PBS для удаления трипсина и сыворотки и центрифугировали при 800 об/мин в течение 10 мин при комнатной температуре, а супернатанты клеток отбрасывали. Клетки ресуспендировали в культуральной среде RPIM-1640 без FBS и доводили плотность клеток до 107/300 мкл.
В асептических условиях каждой “голой” мыши вводили подкожно в спину 300 мкл/инъекцию, по одной инъекции на “голую” мышь. Для инъекции использовался одноразовый медицинский шприц объемом 1 мл, и место инъекции и направление были примерно одинаковыми для каждой “голой” мыши.
Через день после имплантации клеток “голых” мышей случайным образом разделяли на четыре группы (а именно, контрольная группа с растворителем, группа 2,5 мг/кг чиаураниба, группа 20 мг/кг чидамида и группа с комбинированным введением), помещали в отдельные клетки после маркирования, вводили группами и опухолевое образование каждой мыши наблюдали каждый день. После измерения массы тела каждой “голой” мыши перед введением, “голым” мышам вводили внутрижелудочно в дозе, рассчитанной на килограмм массы тела, то есть 10 мкл раствора CMC-Na на грамм массы тела для контрольной группы с растворителем, 10 мкл 0,25 мг/мл суспензии чиаураниб-CMC-Na на грамм массы тела для группы 2,5 мг/кг чиаураниба, 10 мкл 2 мг/мл суспензии чидамид-CMC-Na на грамм массы тела для группы 20 мг/кг чидамида, и 10 мкл суспензии CMC-Na, содержащей 0,25 мг чиаураниба и 2 мг чидамида на миллилитр, на грамм массы тела для группы комбинированного введения. Каждые 2 дня измеряли самый длинный диаметр опухоли (длину) и самый широкий диаметр (ширину), перпендикулярный самому длинному диаметру, с помощью штангенциркуля. Объем опухоли рассчитывали по формуле TS=длина×(ширина)2/2 и записывали. Каждой “голой” мыши вводили внутрижелудочно регулярно один раз в день. Эксперимент завершали после последнего введения на 33-е сутки.
Результат эксперимента
Как показано на фиг. 6, по сравнению с контрольной группой растворителем, в двух группах, которым вводили одно лекарственное средство 2,5 мг/кг чиаураниба или 20 мг/кг чидамида, объем опухоли у “голых” мышей с опухолями был в определенной степени подавлен, в то время как конечная степень ингибирования опухоли в группе комбинированного введения была выше, чем сумма степеней ингибирования опухоли в двух группах, которым вводили одно лекарственное средство. Это продемонстрировало, что два лекарственных средства имели синергетически сенсибилизированную противоопухолевую активность у “голых мышей”, несущих опухоль.
Пример 7. Получение (в 1000 единицах) фармацевтической композиции соединения 1 (гранулы или капсулы)
Композиция чидамида в форме твердой дисперсии
Композиция соединения в форме гранул
Способ получения
(1) На основании количеств компонентов в композиции чидамида в форме твердой дисперсии, чидамид, повидон и медицинский этанол взвешивали, смешивали и перемешивали до полного растворения всех компонентов при 80°C для получения прозрачного раствора. Раствор подвергали ротационному выпариванию или распылительной сушке с получением белого порошка, то есть твердой дисперсии чидамида;
(2) На основании количеств компонентов в композиции соединения в форме гранул, взвешивали твердую дисперсию чидамида, чиаураниб, повидон, микрокристаллическую целлюлозу и лактозу. Во влажном грануляторе добавляли эксципиенты и равномерно перемешивали, а затем добавляли медицинский этанол для получения гранул при влажном гранулировании. Полученные гранулы сушили в печи или сушилке с псевдоожиженным слоем и просеивали через сито 20 меш, чтобы получить гранулы, содержащие фармацевтические вспомогательные вещества.
(3) Гранулы, полученные на стадии (2), смешивали со стеаратом магния в пропорциях в течение 2 мин для получения общего количества смешанных гранул.
(4) Общее количество смешанных гранул, полученное на стадии (3), упаковывали для получения гранул или заполняли полые капсулы для получения капсул.
Пример 8. Получение (в 1000 единицах) фармацевтической композиции соединения 2 (гранулы, капсулы или таблетки)
Композиция чидамида в форме твердой дисперсии
Композиция соединения в форме гранул
Способ получения
(1) На основании количеств компонентов в композиции чидамида в форме твердой дисперсии, чидамид, коповидон и медицинский этанол взвешивали, смешивали и перемешивали до полного растворения всех компонентов при 80°C для получения прозрачного раствора. Раствор подвергали ротационному выпариванию или распылительной сушке с получением примерно белого порошка, то есть твердой дисперсии чидамида;
(2) На основании количеств компонентов в композиции соединения в форме гранул, взвешивали твердую дисперсию чидамида, чиаураниб, кросповидон, микрокристаллическую целлюлозу и маннит. Эксципиенты добавляли в трехмерный смеситель и однородно перемешивали для получения гранул при сухой грануляции. Полученные гранулы просеивали через сито 20 меш, для получения гранул, содержащие фармацевтические вспомогательные вещества.
(3) Гранулы, полученные на стадии (2), смешивали со стеаратом магния в пропорциях в течение 2 мин для получения общего количества смешанных гранул.
(4) Общее количество смешанных гранул, полученное на стадии (3), оформляли для получения гранул с быстрым высвобождением, или заполняли полые капсулы для получения капсул, или прессовали в получаемые таблетки.
Пример 9. Получение (в 1000 единицах) фармацевтической композиции соединения 3 (гранулы или таблетки)
Композиция чидамида в форме твердой дисперсии
Композиция соединения в форме гранул
Способ получения
(1) На основании количеств компонентов в композиции чидамида в форме твердой дисперсии, гидроксипропилметилцеллюлозу взвешивали и растворяли в воде в количестве, указанном в композиции, куда затем добавляли чидамид, перемешивали до его растворения при 80°C для получения прозрачного раствора. Раствор подвергали ротационному выпариванию или распылительной сушке с получением не совсем белого порошка, то есть твердой дисперсии чидамида.
(2) На основании количеств компонентов в композиции соединения в форме гранул, взвешивали твердую дисперсию чидамида, чиаураниб, кросповидон, микрокристаллическую целлюлозу и лактозу. Эксципиенты добавляли в трехмерный смеситель и однородно перемешивали, для получения гранул при сухой грануляции. Полученные гранулы просеивали через сито 20 меш, для получения гранул, содержащие фармацевтические вспомогательные вещества.
(3) Гранулы, полученные на стадии (2), смешивали со стеаратом магния в пропорциях в течение 2 мин для получения общего количества смешанных гранул.
(4) Общее количество смешанных гранул, полученное на стадии (3), упаковывали для получения гранул с быстрым высвобождением или прессовали в получаемые таблетки.
Пример 10. Получение (в 1000 единицах) фармацевтической композиции соединения 4 (капсулы)
Композиция раствора для нанесения лекарственного средства для чидамида
Композиция раствора для нанесения лекарственного средства для чиаураниба
Композиция ядра пилюли
(микрокристаллическая целлюлоза)
Способ получения
(1) На основании количеств компонентов в композиции раствора для нанесения лекарственного средства для чидамида, чидамид, повидон и медицинский этанол взвешивали, смешивали и перемешивали до полного растворения всех компонентов при 80°C, для получения прозрачного раствора, то есть раствора для нанесения для чиаураниба.
(2) На основании количеств компонентов в композиции раствора для нанесения лекарственного средства для чиаураниба, повидон и медицинский этанол взвешивали. Повидон растворяли в этаноле. Чиаураниб равномерно диспергировали в растворе повидона, для получения раствора для нанесения лекарственного средства для чиаураниба.
(3) Покрытие: пустое ядро пилюли точно взвешивали и наносили лекарственные средства путем распыления раствора для нанесения лекарственного средства для чидамид и раствора для нанесения лекарственного средства для чиаураниба отдельно в псевдоожиженном слое; сердцевину пиллюли после распыления взвешивали для расчета количества нанесенного лекарственного средства, таким образом получали пеллеты комбинированного лекарственного средства, содержащие чидамид и чиаураниб.
(4) Пеллетами, полученными на стадии (2) путем покрытия, заполняли полые капсулы с получением капсул.
Пример 11. Получение (в 1000 единицах) фармацевтической композиции соединения 5 (капсулы)
Композиция раствора для нанесения комбинированного лекарственного средства для чидамида и чиаураниба
Композиция ядра пилюли:
(микрокристаллическая целлюлоза)
Способ получения
(1) На основании количеств компонентов в композиции раствора для нанесения комбинированного лекарственного средства для чидамида и чиаураниба, чидамид, повидон и медицинский этанол взвешивали, смешивали и перемешивали до полного растворения при 80°C, для получения прозрачного раствора. Чиаураниб был равномерно диспергирован в прозрачном растворе чидамида, чтобы получить раствор для нанесения комбинированного лекарственного средства для чидамида и чиаураниба.
(2) Покрытие: пустое ядро пилюли точно взвешивали и наносили лекарственные средства путем распыления жидкости для нанесения комбинации чидамида и чиаураниба в псевдоожиженном слое; сердцевину пиллюли после распыления взвешивали для расчета количества нанесенного лекарственного средства, таким образом получали пеллеты комбинированного лекарственного средства, содержащие чидамид и чиаураниб.
(3) Пеллетами, полученными на стадии (2) путем покрытия, заполняли полые капсулы с получением капсул.
Полученные выше лекарственные формы тестировали на растворение лекарственного средства in vitro в среде хлористоводродной кислоты 0,1 моль/л в соответствии с Pharmacopoeia of the People’s Republic of China 2015 edition. В результате более 85% лекарственных средств растворились в течение 15 минут, что соответствовало требованиям к высвобождению.
Изобретение относится к применению комбинации ингибитора гистондеацетилазы и ингибитора протеинкиназы в получении лекарственного средства для лечения или профилактики опухоли, где ингибитором гистондеацетилазы является чидамид или его фармацевтически приемлемая соль, ингибитором протеинкиназы является чиаураниб или его фармацевтически приемлемая соль, а опухоль представляет собой карциному печени, где массовое соотношение ингибитора гистондеацетилазы и ингибитора протеинкиназы составляет 5-60 мг в расчете на чидамид: 10-100 мг в расчете на чиаураниб, а также фармацевтической композиции и набору для лечения карциномы печени, включающим такую комбинацию. Технический результат: получение лекарственного средства для лечения карциномы печени комбинацией ингибитора гистондеацетилазы чидамида и ингибитора протеинкиназы чиаураниба, обладающей синергическим противоопухолевым эффектом. 3 н. и 5 з.п. ф-лы, 6 ил., 11 пр.
1. Применение комбинации ингибитора гистондеацетилазы и ингибитора протеинкиназы в получении лекарственного средства для лечения или профилактики опухоли, где ингибитором гистондеацетилазы является чидамид или его фармацевтически приемлемая соль, ингибитором протеинкиназы является чиаураниб или его фармацевтически приемлемая соль, а опухоль представляет собой карциному печени, где массовое соотношение ингибитора гистондеацетилазы и ингибитора протеинкиназы составляет 5-60 мг в расчете на чидамид: 10-100 мг в расчете на чиаураниб.
2. Применение по п. 1, где ингибитор гистондеацетилазы представляет собой чидамид, и ингибитор протеинкиназы представляет собой чиаураниб.
3. Фармацевтическая композиция для лечения карциномы печени, включающая ингибитор гистондеацетилазы и ингибитор протеинкиназы в качестве активных фармацевтических ингредиентов и, необязательно, фармацевтически приемлемый эксципиент и/или носитель, где активные фармацевтические ингредиенты состоят из чиаураниба и чидамида, и при этом в стандартной дозировке количество чидамида составляет 5-60 мг, и количество чиаураниба составляет 10-100 мг.
4. Фармацевтическая композиция по п. 3, где фармацевтически приемлемый эксципиент и/или носитель включает повидон, коповидон, гидроксипропилметилцеллюлозу и привитые сополимеры поливинилкапралактам-поливинилацетат-полиэтиленгликоль.
5. Фармацевтическая композиция по любому из пп. 3, 4, где фармацевтическая композиция находится в форме гранул, твердых дисперсий, капсул или таблеток.
6. Фармацевтическая композиция по п. 5, включающая фармацевтически приемлемые эксципиенты и/или носители, выбранные из группы, состоящей из микрокристаллической целлюлозы, повидона, коповидона, лактозы, маннита, кросповидона и карбоксиметилцеллюлозы натрия.
7. Набор для лечения или профилактики карциномы печени, включающий ингибитор гистондеацетилазы и ингибитор протеинкиназы в качестве активных ингредиентов, где ингибитор гистондеацетилазы и ингибитор протеинкиназы вводятся одновременно или последовательно, где ингибитором гистондеацетилазы является чидамид или его фармацевтически приемлемая соль, ингибитором протеинкиназы является чиаураниб или его фармацевтически приемлемая соль, а опухоль представляет собой гепатому, где массовое соотношение ингибитора гистондеацетилазы и ингибитора протеинкиназы составляет 5-60 мг в расчете на чидамид: 10-100 мг в расчете на чиаураниб.
8. Набор по п. 7, включающий чидамид или его фармацевтически приемлемую соль, вводимый первым, и чиаураниб или его фармацевтически приемлемую соль, вводимый вторым, в качестве активных ингредиентов.
| Rasheed, W | |||
| K., Johnstone, R | |||
| W., & Prince, H | |||
| M | |||
| "Histone deacetylase inhibitors in cancer therapy", Expert Opinion on Investigational Drugs, 2007, 16(5), 659-678 | |||
| CN 103432077 A, 11.12.2013 | |||
| CN 102441167 A, 09.05.2012 | |||
| CN 101837129 В, 12.12.2012 | |||
| RU 2010128239 A, 20.01.2012. |

