Область техники, к которой относится изобретение
Данное изобретение относится к комплексным соединениям трехвалентного железа и 2-оксобутандиамида и содержащим их фармацевтическим композициям для применения в качестве медикаментов, в частности для лечения и/или профилактики симптомов дефицита железа и железодефицитных анемий.
Предпосылки создания изобретения
Почти для всех живых существ железо является жизненно важным микроэлементом, участвуя, в частности, в росте организма и в кроветворении. В этих процессах метаболизм железа регулируется в основном на уровне утилизации железа, содержащегося в гемоглобине стареющих эритроцитов и на уровне поглощения железа, содержащегося в пище, в двенадцатиперстной кишке. Высвободившееся железо поглощается клетками кишечника, в частности, посредством специальных транспортных систем (транспортер двухвалентных металлов DMT-1, ферропортин, трансферрин, рецепторы трансферрина), поступает в кровоток и переносится с кровью в соответствующие органы и ткани.
В организме человека железо играет ключевую роль в переносе кислорода, поглощении кислорода, и в некоторых клеточных функциях, а именно в транспорте электронов в митохондриях; в итоге этот элемент важен для всего обмена энергии в целом.
В организме человека содержится в среднем 4-5 г железа; оно присутствует в некоторых ферментах, в гемоглобине и миоглобине, а также в запасной форме (депо) в виде ферритина и гемосидерина.
Приблизительно половина всего количества железа - около 2 г - представлена гемовым железом, то есть находится в связанном виде в составе гемоглобина эритроцитов. Поскольку время жизни эритроцитов ограничено (75-150 суток), эти клетки постоянно обновляются: образуются новые эритроциты (более 2 миллионов клеток в секунду), а старые ликвидируются. Столь высокая регенеративная способность обеспечивается, помимо прочего, фагоцитозом: макрофаги поглощают стареющие эритроциты и разрушают их, так что содержавшееся в них железо может быть повторно использовано в метаболизме. Этим путем поставляется большая часть из того количества железа (25 мг/сут), которое требуется для эритропоэза.
У взрослого человека постоянная потребность в железе составляет 0,5-1,5 мг/сут, детям и беременным женщинам требуется 2-5 мг/сут. Организм все время теряет железо, например, путем десквамации кожи и эпителиальной выстилки органов, однако количество это невелико; оно может возрастать при кровотечениях, например при менструациях у женщин. Обычно из-за потери крови уровень железа в организме значительно снижается, поскольку с каждыми 2 мл крови уходит 1 мг железа. У здорового взрослого человека нормальные потери железа, составляющие около 1 мг/сут, обычно восполняются за счет поступления его с пищей. Уровень железа в организме регулируется его всасыванием: содержащееся в пище железо обычно поглощается на 6-12%, но при дефиците железа его поглощение усиливается и возрастает до 25%. Поглощение железа регулируется в зависимости от потребности в нем организма и от величины имеющихся запасов железа. У человека утилизуются ионы как двух-, так и трехвалентного железа. Обычно соединения трехвалентного железа растворяются в содержимом желудка, где pH достаточно кислый, и таким образом становятся доступными для всасывания. Всасывание железа осуществляется клетками слизистой верхнего отдела тонкого кишечника. В ходе этого процесса вначале трехвалентное негемовое железо восстанавливается до двухвалентного в мембране кишечных клеток, например при участии ферроредуктазы (дуоденального связанного с мембраной цитохрома b), и тогда может транспортироваться внутрь кишечных клеток посредством белка - транспортера двухвалентных металлов DMT1. Гемовое железо проникает через клеточную мембрану энтероцитов без изменений. Внутри энтероцитов железо либо запасается в виде ферритина, либо выделяется в кровоток посредством транспортного белка ферропортина. В этом процессе центральную роль играет гепсидин - наиболее важный регуляторный фактор поглощения железа. Двухвалентное железо, перенесенное в кровоток при участии ферропортина, превращается в трехвалентное под действием оксидаз (церулоплазмина, гефестина), и далее трехвалентное железо с помощью трансферрина транспортируется в соответствующие места организма (см., например, работу "Balancing acts: molecular control of mammalian iron metabolism". M.W. Hentze, Cell 777, 2004, 285-297).
У млекопитающих отсутствует способность к активному выделению железа из организма. Метаболизм железа регулируется в значительной степени гепсидином - через клеточные механизмы выделения железа макрофагами, гепатоцитами и энтероцитами.
В случаях какой-либо патологии пониженный уровень железа в сыворотке крови ведет к понижению уровня гемоглобина, падению продукции эритроцитов и тем самым к анемии.
Внешние проявления анемии включают утомляемость, бледность кожных покровов, ослабление способности к сосредоточению. Клинические симптомы анемии включают низкий уровень железа в сыворотке крови (гипоферремия), низкий уровень гемоглобина, пониженный гематокрит, а также недостаточное количество эритроцитов и ретикулоцитов, повышенные уровни растворимых рецепторов трансферрина.
Симптомы дефицита железа или железодефицитных анемий купируются потреблением железа. Для этого препараты железа принимают через рот или вводят внутривенно. Также для лечения анемий можно усиливать образование эритроцитов, используя эритропоэтин и другие стимуляторы эритропоэза.
Причиной анемии нередко является плохое питание: недостаточный или несбалансированный рацион, или низкое содержание в нем железа. Также анемия может быть обусловлена недостаточным поглощением железа, например из-за тотального удаления желудка или при некоторых заболеваниях, например при болезни Крона. Дефицит железа возможен вследствие возрастания потерь крови, например в связи с хирургическим вмешательством, травмой, усиленной менструацией или донорством. Известно также, что в определенных обстоятельствах: в период интенсивного роста в детском или подростковом возрасте, а также при беременности - потребность организма в железе возрастает. Поскольку дефицит железа приводит не только к снижению продукции эритроцитов, но вследствие этого и к ухудшению снабжения кислородом всего организма, а отсюда к таким симптомам, как утомляемость, бледность кожных покровов, ослабление способности к сосредоточению и к долговременному негативному влиянию на умственное развитие, что особенно касается подростков, очень большой интерес представляют возможности высокоэффективной и хорошо переносимой терапии дефицита железа и железодефицитных анемий.
Применение комплексных соединений трехвалентного железа по данному изобретению открывает возможность эффективного лечения симптомов дефицита железа и железодефицитных анемий путем перорального приема без необходимости мириться с высокой вероятностью побочных эффектов, вызываемых традиционными препаратами - солями двухвалентного железа, например FeSO4, и обусловленных окислительным стрессом. Благодаря этому можно избежать плохого соблюдения пациентами предписаний врача, что зачастую служит причиной неуспешного купирования железодефицитных состояний.
Уровень техники
На сегодняшний день известно множество комплексных соединений железа для лечения железодефицитных состояний.
Очень многие из этих комплексных соединений состоят из полимерных структур. Такие комплексные соединения в большинстве своем являются железо-сахаридными комплексными соединениями (публикации WO 20081455586, WO 2007062546, WO 20040437865; патент США №2003236224, Европейский патент №150085). В настоящее время в продаже имеются лекарственные препараты именно из этой группы, например мальтофер (Maltofer), венофер (Venofer), феринжект (Ferinject), дексферрум (Dexferrum), ферумокситол (Ferumoxytol).
Также многие полимерные комплексные соединения железа состоят из пептидных комплексных соединений железа (патент Китая №101481404, Европейский патент №939083, патент Японии №02083400).
Описаны также комплексные соединения железа, структурно производные от таких макромолекулярных соединений, как гемоглобин, хлорофилл, куркумин и гепарин (патент США №474670, патент Китая №1687089; BioMetals, 2009, 22, 701-710).
Описаны также низкомолекулярные комплексные соединения железа. Многие из этих соединений содержат в качестве лигандов карбоновые кислоты и аминокислоты. Среди них внимание привлекают такие лиганды, как аспартат (патент США №2009035385) и цитрат (Европейский патент №308362) В этой связи описаны также комплексные соединения железа, содержащие производные фенилаланина в качестве лигандов (патент Европейского союза №2044777).
Также описаны комплексные соединения железа, построенные из мономеров-сахаров или представляющие собой сочетание мономеров и полимеров (патент Франции №19671016).
В заявке на патент США №2005/0192315 описываются фармацевтические композиции, содержащие хинолиновые соединения, которые формально включают 2-(гидроксиметилен)-пропандиамид - пропандиамидный структурный элемент. Соответственно, эти соединения не содержат 2-оксобутандиамидного структурного элемента.
Описаны также комплексные соединения железа с гидроксипироном и гидроксипиридоном (Европейские патенты №№159194, 138420 и 107458). Аналогично этому описаны соответствующие 5-кольцевые системы - комплексные соединения железа с гидроксифураноном (см. публикацию WO 2006037449).
Кроме того, описаны комплексные соединения железа с пиримидин-2-ол-1-оксидными лигандами, используемые для лечения железодефицитной анемии (см. публикацию WO 2012130882). В публикации WO 2012163938 описаны Fe(III)-2,4-диоксо-1-карбонильные комплексные соединения, которые тоже можно использовать для лечения железодефицитной анемии.
Также описаны комплексные соединения железа с β-кетоамидными лигандами, которые предлагалось использовать для лечения железодефицитных состояний (см. публикацию WO 2011117225). Однако в этой публикации нет указаний на комплексные соединения трехвалентного железа с 2-оксобутандиамидным лигандом. Кроме того, комплексные соединения железа с β-кетоамидными лигандами нуждаются в усовершенствовании, в частности в отношении их растворимости в воде и утилизации содержащегося в них железа.
Важной оставляющей в лечении симптомов дефицита железа и железодефицитных анемий являются также соли железа (например, сульфат, фумарат, аспартат и сукцинат двухвалентного железа; хлорид трехвалентного железа).
Однако эти соли проблематично использовать - отчасти из-за того, что они плохо переносятся пациентами, часто (до 50% случаев) вызывая тошноту, рвоту, понос, запор и спастические боли в животе. Кроме того, при использовании солей двухвалентного железа в организме образуются свободные ионы Fe(II), способствующие образованию (путем реакции Фентона) активных форм кислорода (ROS). Эти активные формы кислорода вызывают повреждения ДНК, липидов, белков и углеводов, что имеет далеко идущие последствия для клеток, тканей и органов. Эта группа проблем, известная под названием окислительного стресса, хорошо известна и обычно считается причиной плохой переносимости указанных веществ.
Цель изобретения
Цель данного изобретения лежит в области разработки новых терапевтически эффективных веществ, характеризующихся хорошей активностью, высокой степенью утилизации железа, стабильностью комплексных соединений и достаточной растворимостью, в частности высокой стабильностью и хорошей растворимостью в водной среде с нейтральным pH, которые можно использовать путем перорального приема для действенного лечения симптомов дефицита железа и железодефицитных анемий. Для эффективности лечения пероральными препаратами железа особенно важны высокая стабильность и хорошая растворимость.
Также ожидалось, что эти комплексные соединения железа будут вызывать гораздо меньше побочных эффектов или будут проявлять меньшую токсичность, в особенности по сравнению с традиционно применяемыми солями двухвалентного железа. Кроме того, предполагалось, что эти комплексные соединения железа - в противоположность известным полимерным комплексным соединениям железа - будут обладать фиксированной структурой (стехиометрией) и их можно будет получать путем несложного химического синтеза. Эта цель была достигнута путем разработки новых комплексных соединений трехвалентного железа.
Также предполагалось создать такие новые комплексные соединения железа, чтобы они поглощались клетками кишечника непосредственно через клеточную мембрану и таким образом связанное в них железо высвобождалось в прямой доступности для ферритина или трансферрина или поступало в кровоток в виде интактного комплекса. Предполагалось, что благодаря этим свойствам новые комплексные соединения железа не будут вызывать высокой концентрации свободных ионов железа. А именно свободные ионы железа обусловливают образование активных форм кислорода, которые, в конечном счете, приводят к наблюдаемым побочным эффектам.
Чтобы выполнить все эти требования, авторы данного изобретения разработали новые комплексные соединения трехвалентного железа с не слишком большой молекулярной массой, умеренной липофильностью, очень хорошей активностью или утилизацией железа, высокой растворимостью в воде и оптимальной зависимой от pH стабильностью комплекса.
При разработке новых комплексных соединений железа повышение их стабильности, в частности в водной среде с нейтральным pH, не должно было достигаться в ущерб растворимости, так как при пероральном применении растворимость препарата очень важна. Эта двоякая задача решается комплексными соединениями железа по данному изобретению. Они проявляют высокую стабильность в водной среде при нейтральном pH и в то же время обладают очень хорошей растворимостью в воде. Таким образом, комплексные соединения железа по данному изобретению позволяют добиться гораздо более скорого успеха лечения.
Раскрытие изобретения
Авторы данного изобретения обнаружили, что новые комплексные соединения трехвалентного железа с 2-оксобутандиамидными лигандами удивительным образом подходят к описанным выше требованиям. Удалось показать, что с этими комплексными соединениями железа достигается высокая степень поглощения железа, а значит и быстрый терапевтический эффект при лечении железодефицитной анемии. Особенно в сравнении с солями железа комплексные соединения по данному изобретению отличаются высокой степенью и скоростью утилизации железа. Кроме того, эти новые системы вызывают гораздо более слабые побочные эффекты, чем обычно используемые соли железа благодаря тому, что не дают существенного количества свободных ионов железа. Комплексные соединения по данному изобретению не вызывают практически никакого окислительного стресса, поскольку не образуются свободные радикалы. Таким образом, в случае комплексных соединений по данному изобретению отмечаются значительно более слабые побочные эффекты, чем в случае солей железа, известных в данной области техники. Комплексные соединения по данному изобретению обладают высокой стабильностью как при нейтральных, так и при кислых pH, что является особенным преимуществом при пероральном применении. Комплексные соединения по данному изобретению несложно получать и включать в состав лекарственных препаратов, в частности для перорального применения.
Таким образом, объектом данного изобретения являются комплексные соединения трехвалентного железа и 2-оксобутандиамида или их соли, в частности фармацевтически приемлемые их соли, для применения в качестве медикаментов. Также объектом данного изобретения являются комплексные соединения трехвалентного железа и 2-оксобутандиамида или их фармацевтически приемлемые соли для применения при осуществлении способа терапевтического воздействия на организм человека или животного, соответственно.
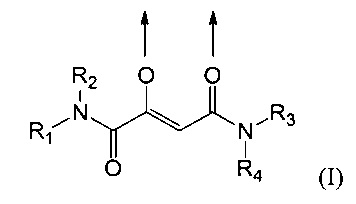
Комплексные соединения трехвалентного железа и 2-оксобутандиамида при использовании по данному изобретению включают, в частности, соединения, содержащие по меньшей мере один лиганд, имеющий формулу (I):
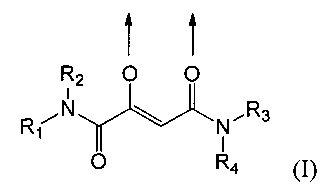
где стрелки соответственно представляют координационную связь, идущую к атому железа,
R1 и R2 одинаковы или же различны и их соответственно выбирают из группы, состоящей из водорода и при необходимости замещенного алкила, или
R1 и R2 вместе с атомом азота, с которым они связаны, образуют при необходимости замещенное 3-6-членное кольцо, которое при необходимости может содержать еще один гетероатом,
R3 и R4 одинаковы или же различны и их соответственно выбирают из группы, состоящей из водорода и при необходимости замещенного алкила, или
R3 и R4 вместе с атомом азота, с которым они связаны, образуют при необходимости замещенное 3-6-членное кольцо, которое при необходимости может содержать еще один гетероатом,
или их фармацевтически приемлемые соли.
Предпочтительными являются в особенности комплексные соединения трехвалентного железа и 2-оксобутандиамида, содержащие по меньшей мере один лиганд, имеющий формулу (I):
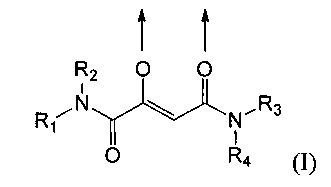
где стрелки соответственно представляют координационные связи, идущие к одному или разным атомам железа,
R1 и R2 одинаковы или же различны и их соответственно выбирают из группы, состоящей из водорода, метила, этила, пропила, изопропила, н-бутила, втор-бутила и трет-бутила, и где указанные алкильные группы могут быть замещенными, причем заместитель выбирают из группы, состоящей из алкокси-группы, алкоксикарбонильной группы, аминокарбонильной (H2NCO-) группы, моноалкиламинокарбонильной группы и диалкиламинокарбонильной группы, или
R1 и R2 вместе с атомом азота, с которым они связаны, образуют при необходимости замещенное 5-6-членное кольцо, которое при необходимости может содержать еще один гетероатом,
R3 и R4 одинаковы или же различны и их соответственно выбирают из группы, состоящей из водорода и при необходимости замещенного алкила, и при необходимости замещенного циклоалкила, где в указанных алкильных группах один или два углеродных атома при необходимости могут быть заменены на кислород, или
R3 и R4 вместе с атомом азота, с которым они связаны, образуют при необходимости замещенное 3-6-членное кольцо, которое при необходимости может содержать еще один гетероатом.
Также предпочтительными являются комплексные соединения трехвалентного железа и 2-оксобутандиамида, которые содержат по меньшей мере один лиганд, имеющий формулу (I):
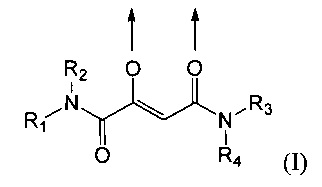
где стрелки соответственно представляют координационные связи, идущие к одному или разным атомам железа,
R1 и R2 одинаковы или же различны и их соответственно выбирают из группы, состоящей из водорода, метила, этила, пропила, изопропила, н-бутила, втор-бутила и трет-бутила, и где указанные алкильные группы могут быть замещенными, причем заместитель выбирают из группы, состоящей из алкокси-группы, алкоксикарбонильной группы, аминокарбонильной (H2NCO-) группы, моноалкиламинокарбонильной группы и диалкиламинокарбонильной группы, или
R1 и R2 вместе с атомом азота, с которым они связаны, образуют пирролидинил,
R3 и R4 одинаковы или же различны и их соответственно выбирают из группы, состоящей из водорода, алкила и циклоалкила, где алкильная и циклоалкильная групп при необходимости могут быть замещенными, причем заместитель выбирают из группы, состоящей из гидроксильной группы, алкокси-группы, алкоксикарбонильной группы, аминокарбонильной (H2NCO-) группы, моноалкиламинокарбонильной группы и диалкиламинокарбонильной группы,
R3 и R4 вместе с атомом азота, с которым они связаны, образуют морфолинил или пирролидинил, где указанные циклические группы могут быть замещенными гидроксильной группой.
В частности предпочтительными являются также комплексные соединения трехвалентного железа и 2-оксобутандиамида, которые содержат по меньшей мере один лиганд, имеющий формулу (I):
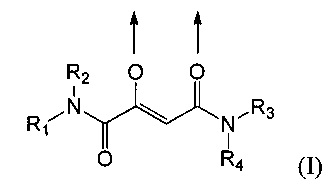
где стрелки соответственно представляют координационные связи, идущие к одному или разным атомам железа,
R1 и R2 одинаковы или же различны и их соответственно выбирают из группы, состоящей из водорода, метила, этила, пропила, изопропила, н-бутила, втор-бутила и трет-бутила,
R3 и R4 одинаковы или же различны и их соответственно выбирают из группы, состоящей из водорода и при необходимости замещенного алкила, или
R3 и R4 вместе с атомом азота, с которым они связаны, образуют при необходимости замещенное 3-6-членное кольцо, которое при необходимости может содержать еще один гетероатом.
Особенно предпочтительные комплексные соединения трехвалентного железа содержат по меньшей мере один лиганд, имеющий формулу (I):
где стрелки соответственно представляют координационные связи, идущие к одному или разным атомам железа,
R1 и R2 одинаковы или же различны и их соответственно выбирают из группы, состоящей из водорода и метила,
R3 и R4 одинаковы или же различны и их соответственно выбирают из группы, состоящей из водорода и при необходимости замещенного алкила.
Особенно предпочтительны комплексные соединения трехвалентного железа, имеющие формулу (II):
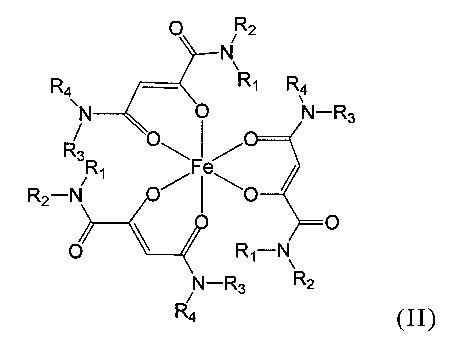
где R1, R2, R3 и R4 такие, как описано выше.
Предпочтительно молекулярная масса комплексных соединений трехвалентного железа и 2-оксобутандиамида по данному изобретению составляет менее 1000 г/моль, более предпочтительно менее 800 г/моль (в каждом случае определяется по структурной формуле).
В общем контексте данного изобретения при необходимости замещенный алкил, в частности в случае заместителей R1-R4, предпочтительно включает: неразветвленный или разветвленный алкил из 1-6 атомов углерода, циклоалкил из 3-6 предпочтительно 5-6 атомов углерода, или алкил из 1-4 атомов углерода, замещенный циклоалкилом, причем указанные алкильные группы при необходимости могут быть замещенными.
Указанные выше алкильные группы при необходимости являются замещенными каждая 1-3 заместителями. Более предпочтительно, что они не являются замещенными.
Заместители алкила предпочтительно выбирают из группы, состоящей из гидроксильной группы, алкокси-группы, алкоксикарбонильной группы, аминокарбонильной (H2NCO-) группы, моноалкиламинокарбонильной группы и диалкиламинокарбонильной группы, в особенности, как описано ниже. В отношении алкильных групп в указанных заместителях - алкокси-группе, алкоксикарбонильной группе, моноалкиламинокарбонильной группе и диалкиламинокарбонильной группе - можно сослаться на определение алкила, приведенное выше и ниже.
Заместители алкила предпочтительно выбирают из группы, состоящей из гидроксильной группы и при необходимости замещенной алкокси-группы, в частности, как описано ниже.
В описанных выше алкильных группах при необходимости один или более атомов углерода могут быть заменены на кислород. Это означает, в частности, что одна или более метиленовых групп (-CH2-) может быть заменена в алкильных группах на кислород (-O-).
Примеры алкильных остатков, содержащих 1-6 атомов углерода, включают: метальную группу, этильную группу, н-пропильную группу, изопропильную группу, н-бутильную группу, изобутильную группу, втор-бутильную группу, трет-бутильную группу, н-пентильную группу, изопентильную группу, втор-пентильную группу, трет-пентильную группу, 2-метилбутильную группу, н-гексильную группу, 1-метилпентильную группу, 2-метилпентильную группу, 3-метилпентильную группу, 4-метилпентильную группу, 1-этилбутильную группу, 2-этилбутильную группу, 3-этилбутильную группу, 1,1-диметилбутильную группу, 2,2-диметилбутильную группу, 3,3-диметилбутильную группу, 1-этил-1-метилпропильную группу и проч. Предпочтительны алкилы, содержащие 1-4 атомов углерода. Наиболее предпочтительны метил, этил, н-пропил и н-бутил.
Циклоалкильные группы с 3-6 атомами углерода предпочтительно включают циклопропильную группу, циклобутильную группу, циклопентильную группу и циклогексильную группу. Циклоалкильные остатки при необходимости могут быть замещенными предпочтительно одним из таких заместителей, как гидроксильная группа, метальная группа или метокси-группа.
Определение алкильных групп, при необходимости замещенных, также включает алкильные группы, замещенные упомянутыми выше циклоалкильными группами, например циклопропилметилом, циклобутилметилом, цилкопентилметилом или циклогексилметилом.
Примеры алкильных остатков, замещенных гидроксильной группой, включают упомянутые выше алкильные остатки, в которых имеются 1-2 гидроксильных остатка, например гидроксиметил, 2-гидроксиэтил, 3-гидроксипропил, 4-гидроксибутил и проч.
В контексте данного изобретения при необходимости замещенные алкокси-группы (RO-) включают, например, неразветвленные или разветвленные алкокси- остатки, предпочтительно содержащие до четырех атомов кислорода, например метокси-группу, этокси-группу, н-пропилокси-группу и изопропилокси-группу. Алкокси-группы при необходимости могут быть замещенными, например упомянутыми выше возможными заместителями алкила, в частности одним-тремя, предпочтительно одним заместителем.
Предпочтительными алкокси-группами являются метокси- и этокси-группа.
В контексте данного изобретения алкоксикарбонил включает упомянутую выше алкокси-группу, с которой связана карбонильная группа (схематически RO(C=O)-, где R представляет упомянутый выше алкил). Примеры алкоксикарбонил а включают метоксикарбонил и этоксикарбонил.
В контексте данного изобретения моноалкилкарбонил и диалкилкарбонил включают остатки, имеющие формулы HRN-C(=O)- и R2N-C(=O)-, где в отношении алкильных групп (R) можно сослаться на приведенное выше определение алкила. Примеры таких групп включают метиламинокарбонил и диметиламинокарбонил.
По данному изобретению 2-оксобутандиамидный лиганд комплексного соединения включает соответствующую основную структуру:
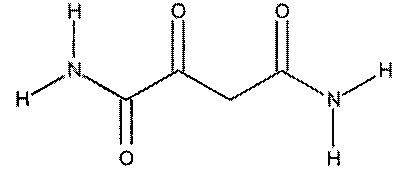
2-оксобутандиамид
а также все соединения, в которых один или более имеющихся атомов водорода замещены другими атомами или группами атомов, соответственно, причем для возникновения описываемой далее кето-енол-таутомерии между двумя группами C=O, образующими координационную связь, должен присутствовать по меньшей мере один атом водорода.
Специалистам в данной области техники ясно, что 2-оксобутандиамидные лиганды комплексных соединений по данному изобретению, в частности лиганды, имеющие формулу (I):
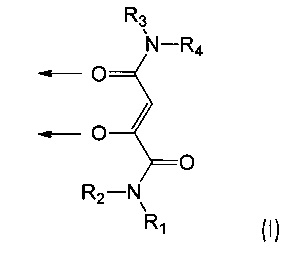
происходят от соответствующих 2-оксобутандиамидных соединений (III):
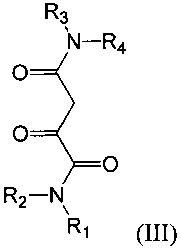 ,
,
в которых имеется кето-енольная таутомерия, а именно:
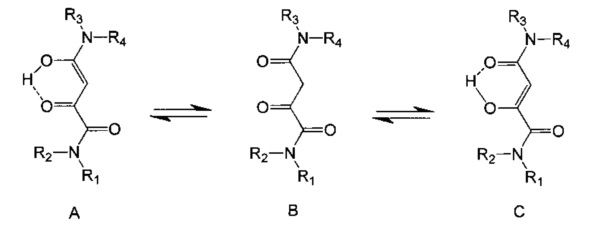 .
.
Мезомерные формы A и C неразличимы аналитическими методами. В контексте данного изобретения в каждом случае включаются все формы, но лиганд, как правило, берется только в кето-форме.
Формально такой лиганд происходит от соответствующей енольной формы A или C путем ухода протона:
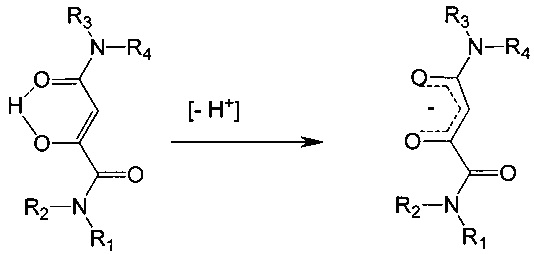 ,
,
и таким образом формально несет один отрицательный заряд. Также в контексте данного изобретения для комплексных соединений железа всегда представляется только одна из локализованных резонансных структур:
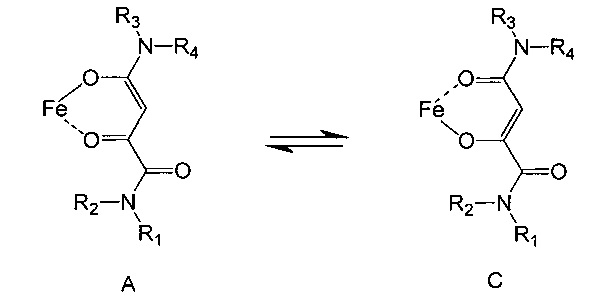
хотя из-за низкой электронной плотности на атоме кислорода амидной группировки в положении 1,3 относительно кето-группы следует ожидать, что превалирует резонансная структура C. Как объяснялось выше, аналитическими методами различить структуры A и C невозможно. Другая амидная группировка, находящаяся в положении 1,2 относительно кето-группы, по данным, полученным аналитическими методами, не участвует, по-видимому, в связывании железа. При исследовании комплексных соединений по настоящему изобретению методом инфракрасной спектроскопии было показано, что для этой амидной группировки между свободным лигандом и комплексом наблюдается лишь очень небольшой сдвиг полос инфракрасного спектра, что свидетельствует против участия данной карбонильной группы в связывании железа при образовании комплексного соединения.
Ниже приведены примеры 2-оксобутандиамидных лигандов, используемых по данному изобретению.
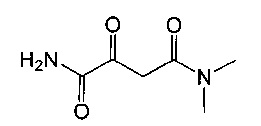
Данное изобретение относится также к способу получения предлагаемых комплексных соединений трехвалентного железа, включающему взаимодействие 2-оксобутандиамидного соединения (III) с солью трехвалентного железа (IV).
2-Оксобутандиамидные соединения включают, в частности, соединения, описываемые формулой (III):
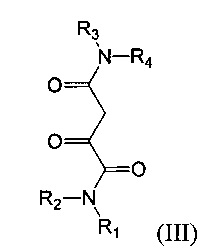
где R1-R4 определены выше.
Примеры солей трехвалентного железа, подходящих для упомянутой выше реакции, включают хлорид, ацетат, сульфат, нитрат и ацетилацетонат, из которых предпочтителен хлорид (FeCl3).
Предпочтительный способ изображен на следующей схеме:
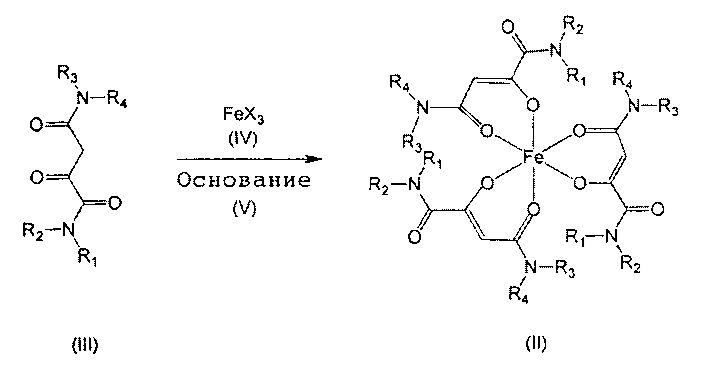
где R1-R4 определены выше, X представляет анион, например галогенид-ион (например, хлорид-ион), карбоксилат-ион (например, ацетат), сульфат-ион, нитрат-ион и ацетилацетонат-ион, а основанием служит обычное органическое или неорганическое основание.
В способе по данному изобретению проводят реакцию предпочтительно между лигандом (III) в количестве 3-5 эквивалентов и подходящей солью трехвалентного железа (IV) (здесь особенно подходят хлорид, ацетат, сульфат и ацетилацетонат) в стандартных условиях образования соответствующих комплексных соединений, имеющих общую формулу (II). При этом синтез осуществляется в условиях pH, оптимальных для образования комплекса. Оптимальный pH достигается путем прибавления по необходимости основания (V), для чего используются предпочтительно ацетат натрия, триэтиламин, карбонат натрия, бикарбонат натрия, метанолят натрия, этанолят натрия, карбонат калия, бикарбонат калия или метанолят калия.
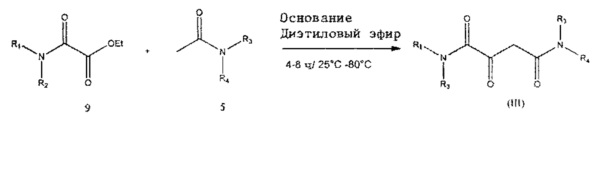
Требующиеся для получения комплексных соединений лиганды (III) имеются в продаже, а также могут быть синтезированы следующим путем, который использовался по данному изобретению.
В случае частично замещенных амидов (III) ((R1=H; R2=Н или R2≠H; R3,R4≠H) вначале путем реакции конденсации получали эфир 6 из диэтилового эфира щавелевой кислоты с общей формулой 4 и диалкилдацетамида общей формулы 5 (R.J. Gobeil et al, Journal of the American Chemical Society, 1945, 67, 511). В качестве конденсирующих агентов подходят различные основания, например бутиллитий, диизопропиламин лития, натрий, гидрид натрия, амид натрия, алкоксиды натрия, калий, гидрид калия, амид калия и алкоксиды калия, причем предпочтителен трет-бутоксид калия.
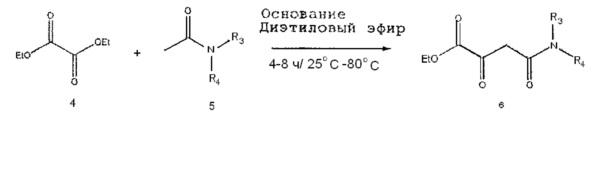
Для получения амида (III) эфир 6 превращают в комплекс с металлом 7. Предпочтительным металлом при этом является медь, хотя подходят и другие переходные металлы. Затем комплекс с металлом 7 взаимодействует с соответствующим амином с образованием амидного комплексного соединения (III). Амид (III) удаляют из комплекса с металлом с помощью разбавленной неорганической кислоты (A. Ichiba et al, Journal of the Scientific Research Institute, Tokyo, 1948, 23, 23-29); в случае меди для этого предпочтительна разбавленная серная кислота. Если амид (III) обладает высокой растворимостью в воде, то удаление амида осуществляют с помощью сероводорода в органическом растворителе, в качестве которого предпочтителен метиловый спирт.
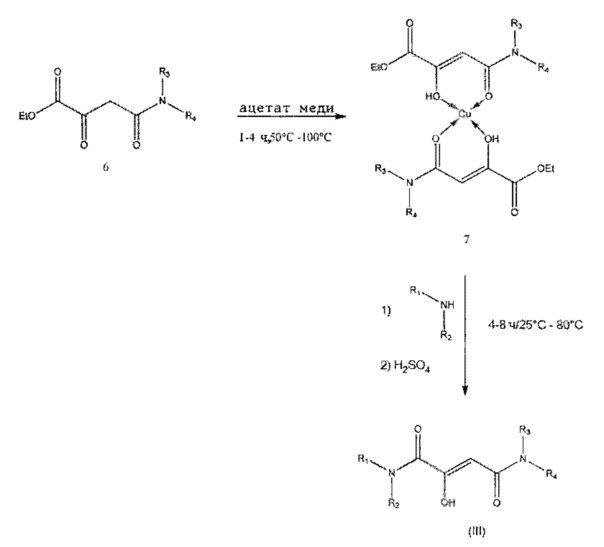
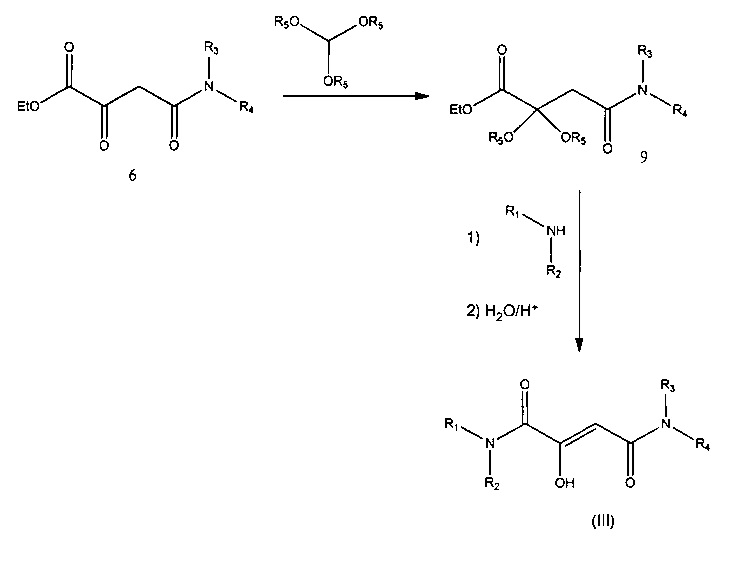
Или же проводят реакцию эфира 6 с алкиловым эфиром ортомуравьиной кислоты с образованием кеталя 8. (J. Perronnet et al, Journal of Heterocyclic Chemistry, 1980, 17, 727-731). Затем последний взаимодействует с соответствующим амином, в результате чего образуется диамидный комплекс, который гидролизуется с образованием амида (III) в кислой водной среде. (W. Kantlehner et al, Liebigs Annalen der Chemie, 1980, 9, 1448-1454).
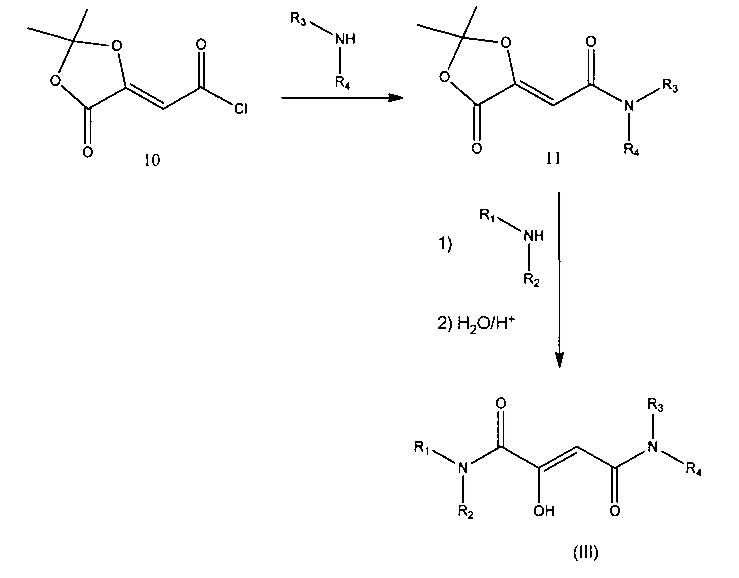
Другим способом получения амида (III) с каким-либо замещением (R1=H или R1≠H; R2=H или R2≠H; R3=H или R3≠H, R4=H или R4≠H) является синтез начиная с (2Z)-(2,2-диметил-5-оксо-1,3-диоксолан-4-илиден)ацетилхлорида. В этом случае хлорид 10 взаимодействует с соответствующим амином с образованием ацетонида 11, который превращается в амид (III) на втором этапе реакции (J. Banville et al, Tetrahedron Letters, 2010, 57, 3170-3173).
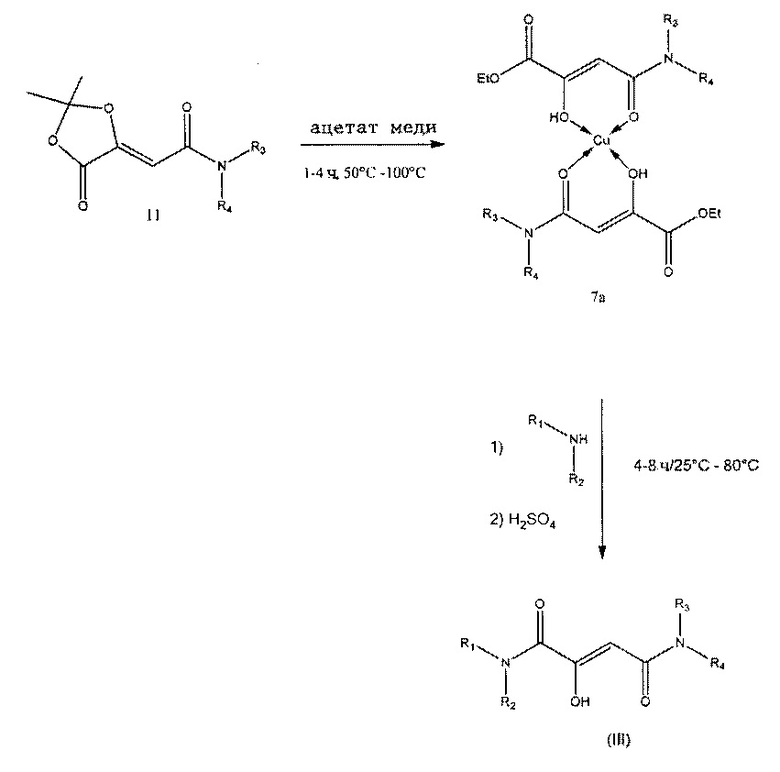
Чтобы улучшить выход на конечной стадии, можно сначала проводить реакцию ацетонида 11 с комплексным соединением металла 7а (с R3=H или R3≠H, R4=H или R4≠H). Предпочтительным металлом при этом является медь, хотя подходят и другие переходные металлы. Затем комплексное соединение металла 7а взаимодействует с соответствующим амином с образованием амидного комплексного соединения металла (III). Амид (III) удаляют из комплекса с металлом с помощью разбавленной неорганической кислоты (А. Ichiba et al, Journal of the Scientific Research Institute, Tokyo, 1948, 23, 23-29); в случае меди для этого предпочтительна разбавленная серная кислота. Если амид (III) обладает высокой растворимостью в воде, то удаление амида осуществляют с помощью сероводорода в органическом растворителе, в качестве которого предпочтителен метиловый спирт.
В случае полного замещения (R1, R2, R3, R4≠H) синтез представляет собой просто классическую реакцию конденсации. При этом имеющийся в продаже алкил N,N-диалкилоксамат 9 и диалкилацетамид 5 непосредственно взаимодействуют с подходящим конденсирующим основанием с образованием амида (III). В качестве конденсирующих агентов подходят различные основания, например бутиллитий, диизопропиламин лития, натрий, гидрид натрия, амид натрия, алкоксиды натрия, калий, гидрид калия, амид калия и алкоксиды калия, причем предпочтителен трет-бутоксид калия.
Здесь остатки R1-R4 такие, как описано и определено, соответственно, выше. Фармацевтически приемлемые соли соединений по данному изобретению, в которых комплекс трехвалентного железа формально несет положительный заряд, включают, например, соли с подходящими анионами, например карбоксилаты, сульфонаты, сульфаты, хлориды, бромиды, иодиды, фосфаты, тартраты, метансульфонаты, гидроксиэтансульфонаты, глицинаты, малеаты, пропионаты, фумараты, толуолсульфонаты, бензолсульфонаты, трифторацетаты, нафталиндисульфонаты-1,5, салицилаты, бензоаты, лактаты, соли яблочной кислоты, соли 3-гидрокси-2-нафтойной-2 кислоты, цитраты и ацетаты.
Фармацевтически приемлемые соли соединений по данному изобретению, в которых комплекс трехвалентного железа формально несет отрицательный заряд, включают, например, соли с фармацевтически приемлемыми основаниями, например соли с гидроксидами щелочных или щелочноземельных металлов, например NaOH, KOH, Ca(ОН)2, Mg(OH)2 и проч.; с аминовыми соединениями, например этиламином, диэтиламином, триэтиламином, этилдиизопропиламином, этаноламином, диэтаноламином, триэтаноламином, метилглюкамином, дициклогексиламином, диметиламиноэтанолом, прокаином, дибензиламином, N-метилморфолином, аргинином, лизином, этилендиамином, N-метилпиперидином, 2-амино-2-метилпропанолом(1), 2-амино-2-метилпропандиолом-(1,3), 2-амино-2-гидроксилметилпропандиолом-(1,3) (TRIS) и др.
На растворимость в воде или в физиологическом солевом растворе - и таким образом также, возможно, на эффективность соединений по данному изобретению - существенно влияет образование соли, в частности выбор противоиона.
Соединения по данному изобретению являются предпочтительно нейтральными комплексными соединениями.
Положительные фармакологические эффекты
Авторы данного изобретения обнаружили, что разработанные ими 2-оксобутандиамидные комплексные соединения трехвалентного железа, составляющие объект изобретения и представленные, в частности, общей структурной формулой (II), являются стабильными биологически доступными комплексами железа и подходят для применения в качестве медикамента с целью лечения и профилактики симптомов дефицита железа и железодефицитных анемий и сопровождающих их симптомов.
Медикаменты, содержащие соединения по данному изобретению, подходят для применения в медицине и ветеринарии.
Соединения по данному изобретению также подходят для получения медикаментов, используемых при лечении индивидов с симптомами железодефицитной анемии, например страдающих от утомляемости, апатии и вялости, неспособности сосредоточиться, низкой эффективности умственной деятельности, трудностей с подбором слов в речи, забывчивости, патологической бледности, раздражительности, ускорения частоты сердечных сокращений (тахикардии), изъязвлений или опухания языка, увеличения селезенки, извращений аппетита, головной боли, отсутствия аппетита, повышенной подверженности инфекциям или депрессивных настроений.
Комплексные соединения трехвалентного железа по данному изобретению также подходят для лечения железодефицитной анемии у беременных женщин; скрытой железодефицитной анемии у детей и подростков; железодефицитной анемии, обусловленной аномалиями желудочно-кишечного тракта; железодефицитной анемии из-за потери крови (например, при кровотечениях в желудочно-кишечном тракте вследствие изъязвлений, карциномы, геморроя, воспалительных расстройств, приема ацетилсалициловой кислоты); железодефицитной анемии, обусловленной менструациями; железодефицитной анемии вследствие повреждений/травм; железодефицитной анемии, связанной с глютеновой энтеропатией; железодефицитной анемии из-за недостаточного потребления железа с пищей, в частности при нарушениях питания и/или пищевого поведения у детей и подростков; иммунодефицита, вызванного железодефицитной анемией; нарушений мозговых функций, вызванных железодефицитной анемией; синдрома беспокойных ног, связанного с железодефицитной анемией; железодефицитных анемий при раковых заболеваниях; железодефицитных анемий, связанных с химиотерапией; железодефицитных анемий, спровоцированных воспалительными процессами; железодефицитных анемий при застойной сердечной недостаточности; железодефицитных анемий при хронической почечной недостаточности на стадиях 3-5; железодефицитных анемий, обусловленных хроническими воспалительными процессами; железодефицитных анемий при ревматоидном артрите; железодефицитных анемий при системной красной волчанке и железодефицитных анемий при воспалительных заболеваниях желудочно-кишечного тракта. Комплексные соединения трехвалентного железа по данному изобретению также полезны для лечения дефицита железа при нормальном уровне гемоглобина. А именно, дефицит железа бывает несмотря на то, что показатели гемоглобина находятся в пределах нормы. Как правило, нормальным считается уровень гемоглобина, наблюдаемый у 96% здоровых людей. По информации, приводимой в Википедии, нормальный уровень гемоглобина у человека следующий:
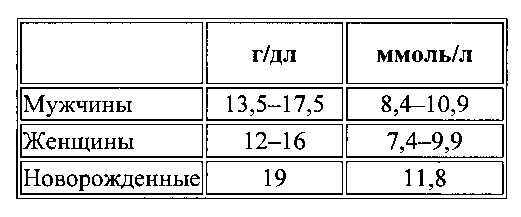
(Содержание гемоглобина можно определять, например, по каталогу «Национальные стандарты Германии» DIN 58931).
Введение препаратов железа может осуществляться на протяжении нескольких месяцев до тех пор, пока статус пациента в отношении уровня железа не улучшится (что отражается, например, в уровне гемоглобина, насыщении трансферрина и уровне ферритина в сыворотке крови) или вплоть до желаемого улучшения состояния здоровья, потерпевшего ущерб из-за железодефицитной анемии.
Препараты по данному изобретению можно принимать детям, подросткам и взрослым.
Соединения по данному изобретению можно вводить в организм перорально либо парентерально. Предпочтителен пероральный прием.
Соединения по данному изобретению и упомянутые выше комбинации соединений по данному изобретению с другими активными веществами или лекарственными препаратами можно использовать, в частности, для получения медикаментов, предназначенных для лечения железодефицитной анемии, например железодефицитной анемии у беременных женщин; скрытой железодефицитной анемии у детей и подростков; железодефицитной анемии, обусловленной аномалиями желудочно-кишечного тракта; железодефицитной анемии из-за потери крови, например, при кровотечениях в желудочно-кишечном тракте (вследствие изъязвлений, карциномы, геморроя, воспалительных расстройств, приема ацетилсалициловой кислоты), при менструациях, повреждениях/травмах; железодефицитной анемии, связанной с глютеновой энтеропатией; железодефицитной анемии из-за недостаточного потребления железа с пищей, в частности при нарушениях питания и/или пищевого поведения у детей и подростков; иммунодефицита, вызванного железодефицитной анемией; нарушений мозговых функций, вызванных железодефицитной анемией; синдрома беспокойных ног, связанного с железодефицитной анемией.
Указанное применение по данному изобретению ведет к улучшению показателей уровня железа, гемоглобина, ферритина и трансферрина, что, в особенности у детей и подростков, но также и у взрослых сопровождается улучшением показателей в тестах по проверке кратковременной памяти (STM) и долговременной памяти (LTM), в тесте Равенна по таблицам с заданиями возрастающей трудности, в тесте по определению уровня интеллектуального развития Векслера (WAIS) и/или в тесте по проверке эмоционального интеллекта (тест Барона-Когана, определение коэффициента эмпатичности, вариант для детей и подростков EQ-i,YV); улучшаются также уровни нейтрофилов и антител и/или функция лимфоцитов.
Также данное изобретение относится к фармацевтическим композициям, содержащим одно или более соединений по данному изобретению, в частности имеющих формулу (II), а также при необходимости один или более других фармацевтически эффективных соединений, а также при необходимости один или более фармакологически приемлемых носителей и/или вспомогательных веществ, и/или растворителей. Указанные фармацевтические композиции содержат, например, до 99% (масса/масса) или до 90% (масса/масса), или до 80% (масса/масса), или до 70% (масса/масса) соединений по данному изобретению, причем в каждом случае остальная часть композиции формируется фармакологически приемлемыми носителями и/или вспомогательными веществами, и/или растворителями. Эти носители, вспомогательные вещества и растворители являются обычно применяемыми в фармацевтической практике.
Указанные выше фармацевтические композиции пригодны, например, для внутривенного, внутрибрюшинного, внутримышечного, интравагинального, интрабуккального, чрескожного, подкожного, через кожу/слизистые, перорального, ректального, чрескожного, местного, интрадермального, интрагастрального или внутрикожного применения и предлагаются в форме, например, таблеток, пилюль, таблеток с кишечно-растворимой оболочкой, таблеток с покрытием из пленки, многослойных таблеток, препаратов с замедленным высвобождением для перорального, подкожного или чрескожного введения (в частности, пластыри), депо-препаратов, драже, суппозиториев, гелей, мазей, сиропов, гранул, суппозиториев, эмульсий, дисперсий, микрокапсул, микропрепаратов, нанопрепаратов, липосомных препаратов, капсул, капсул с кишечно-растворимой оболочкой, порошков, порошков для вдыхания, микрокристаллических препаратов, аэрозолей для вдыхания, порошков для распыления, капель, капель в нос, спреев для носа, аэрозолей, ампул, растворов, экстрактов, суспензий, инфузионных растворов, растворов для инъекций и др.
В одном из предпочтительных воплощений данного изобретения комплексные соединения железа вводят пациенту в форме таблеток или капсул. Они могут быть представлены, например, кислотоустойчивыми или имеющими pH-зависимое покрытие формами.
Комплексные соединения по данному изобретению, а также содержащие их фармацевтические композиции, применяют предпочтительно перорально, хотя возможны и другие пути введения, например парентеральный, в частности внутривенный.
Для введения в организм соединения по данному изобретению предпочтительно включают в состав фармацевтических композиций в форме пилюль, таблеток, таблеток с кишечно-растворимой оболочкой, таблеток, покрытых пленкой, многослойных таблеток, препаратов с замедленным высвобождением для перорального применения, депо-препаратов, драже, гранул, эмульсий, дисперсий, микрокапсул, микропрепаратов, нанопрепаратов, липосомных препаратов, капсул, капсул с кишечно-растворимой оболочкой, порошков, микрокристаллических препаратов, порошков для распыления, капель, ампул, растворов, суспензий, инфузионных растворов или растворов для инъекций.
Соединения по данному изобретению можно вводить в организм в составе фармацевтических композиций, которые могут содержать различные органические или неорганические носители и/или вспомогательные вещества, обычно используемые для фармацевтических целей, в частности в случае твердых лекарственных препаратов, например, такие эксципиенты, как сахароза, крахмал, маннит, сорбит, лактоза, глюкоза, целлюлоза, тальк, фосфат кальция, карбонат кальция; связующие агенты, например целлюлозу, метилцеллюлозу, гидроксипропилцеллюлозу, полипропилпирролидон, желатин, аравийскую камедь, полиэтиленгликоль, сахарозу, крахмал; дезинтегрирующие агенты, например крахмал, гидролизованный крахмал, карбоксиметилцеллюлозу, кальциевую соль карбоксиметилцеллюлозы, гидроксипропил-крахмал, натрия гликоль-крахмал, бикарбонат натрия, фосфат кальция, цитрат кальция; агенты, улучшающие скольжение, например стеарат магния, тальк, лаурилсульфат натрия; ароматизирующие агенты, например лимонную кислоту, ментол, глицин, апельсиновый порошок; консерванты, например бензоат натрия, бисульфит натрия, метилпарабен, пропилпарабен; стабилизирующие агенты, например лимонную кислоту, цитрат натрия, уксусную кислоту и поликарбоновые кислоты типа Titriplex (например, диэтилентриаминпентауксусную кислоту); суспендирующие агенты, например метилцеллюлозу, поливинилпирролидон, стеарат алюминия; диспергирующие агенты; разбавители, например воду, органические растворители; пчелиный воск; масло какао; полиэтиленгликоль; белый вазелин и др.
Жидкие лекарственные составы, например растворы, суспензии и гели, обычно содержат жидкость-носитель, например воду и/или фармацевтически приемлемые органические растворители. Также жидкие лекарственные составы могут содержать агенты для подведения pH, эмульгирующие или диспергирующие агенты, забуферивающие агенты, консервирующие агенты, увлажняющие агенты, желатинизирующие агенты (например, метилцеллюлозу), окрашивающие и/или ароматизирующие агенты. Указанные композиции могут быть изотоничными, то есть обладать таким же осмотическим давлением, как кровь. Изотоничность композиции достигается с помощью хлорида натрия и других фармацевтически приемлемых агентов, например декстрозы, мальтозы, борной кислоты, тартрата натрия, пропиленгликоля и других неорганических или органических растворимых веществ. Нужная вязкость жидких композиций достигается с помощью фармацевтически приемлемых загустителей, например метилцеллюлозы. Другие пригодные загустители включают, например, ксантановую камедь, карбоксиметилцеллюлозу, гидроксипропилцеллюлозу, карбомеры и проч. Предпочтительная концентрация загустителя зависит от его природы. Для того, чтобы увеличить срок хранения жидких препаратов, можно использовать фармацевтически приемлемые консервирующие агенты. Для этого пригоден бензиловый спирт, но можно также использовать многие другие консерванты, включая, например, парабены, тимеросал, хлорбутанол и бензалкония хлорид.
Активное вещество по данному изобретению можно вводит в организм, например, в разовой дозе 0,001-500 мг/кг массы тела, например 1-4 раза в день. Но дозу можно увеличивать или уменьшать в зависимости от возраста, массы тела, состояния пациента, степени тяжести заболевания или способа введения.
Осуществление изобретения
Примеры
Приведенными ниже примерами данное изобретение иллюстрируется более подробно. Эти примеры носят исключительно иллюстративный характер, и специалист в данной области техники может распространить конкретные примеры на другие заявленные соединения. Написание названий соединений определяли с помощью программы ACD/Name (версия 12).
ИСХОДНЫЕ СОЕДИНЕНИЯ
Исходные соединения, использовавшиеся в приведенных ниже примерах, получали следующим образом.
A. N4,N4-диметил-2-оксобутандиамид
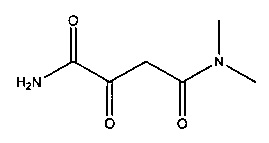
Суспендировали 53,30 г (0,475 моль) трет-бутоксида калия в 300 мл диэтилового эфира. К этой суспензии прибавляли 73,07 г (0,500 моль) диэтилоксалата, охлаждая на ледяной бане. Перемешивали 30 минут с охлаждением, после чего прибавляли 43,57 г (0,500 моль) N,N-диметилапетамида и реакционную смесь перемешивали в течение 2 часов. Для дальнейшей обработки отфильтровывали образовавшееся твердое вещество и дезинтегрировали его в 260 мл 4 н соляной кислоты и 750 мл этилацетата. После разделения фаз водную фазу два раза экстрагировали этилацетатом (по 300 мл). Объединенные органические фазы высушивали над сульфатом натрия и упаривали до сухого состояния с помощью ротационного испарителя. Выход полученного этил-4-(диметиламино)-2,4-диоксобутаноата составлял 54,3 г (58%).
Инфракрасный спектр (в чистом виде, см-1): 2984, 2941, 1738, 1620, 1507, 1467, 1393, 1370, 1355, 1313, 1260, 1170, 1126, 1016, 927, 860, 823, 772, 723, 628.
1H-ЯМР (CDCl3, 400 МГц): δ [ppm] = 1,38 (3H); 3,06 (6Н); 4,33 (2Н); 6,25 (1Н).
Суспендировали 32,56 г (163,1 ммоль) моногидрата ацетата меди в 470 мл этилового спирта и нагревали до температуры 75°C. Затем прибавляли в один прием 61,06 г (326,2 ммоль) этил-4-(диметиламино)-2,4-диоксобутаноата. Полученную смесь перемешивали в течение 40 минут при температуре 75°C. Комплексное соединение меди выпадало в осадок при температуре 4°C. Затем 32,42 г (74,4 ммоль) этого комплексного соединения меди суспендировали в 530 мл раствора аммиака в метаноле (7 н) и перемешивали в течение 4 часов при комнатной температуре, добавляя при этом 10 мл этилового спирта, после чего фильтровали. Для дальнейшей обработки комплексное соединение меди суспендировали в 960 мл хлороформа и перемешивали при комнатной температуре. Затем добавляли 266 мл 10%-ной серной кислоты и перемешивали в течение 30 минут. Разделяли фазы и водную фазу два раза экстрагировали хлороформом (по 960 мл. Объединенные органические фазы высушивали над сульфатом натрия и упаривали до сухого состояния с помощью ротационного испарителя. Полученный остаток кипятили с 156 мл этилацетата и оставляли для кристаллизации на ночь при температуре 4°C. Образовавшийся кристаллический продукт отфильтровывали и высушивали в вакууме. Выход N4,N4-диметил-2-оксобутандиамида составлял 15,5 г (66%),
ИК-спектр (в чистом виде, см-1): 3392, 3154, 2950, 2799, 1699, 1633, 1600, 1576, 1502, 1427, 1378, 1344, 1253, 1170, 1122, 1097, 1054, 990, 925, 908, 811, 761, 730, 665.
1Н-ЯМР (DMSO-d6, 400 МГц): δ [ppm] = 3,0 (6Н), 6,2 (1Н), 7,7 (2Н), 15,3 (1Н).
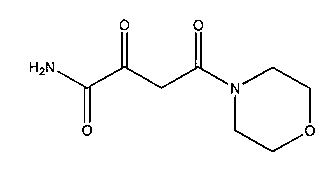
B. 4-(Морфолин-4-ил)-2,4-диоксобутанамид
Помещали 10,2 г (0,1 моль) диизопропиламина в 100 мл безводного тетрагидрофурана (THF) в атмосфере азота и охлаждали в охладительной смеси. Прибавляли медленно по каплям 40 мл н-бутиллития в гексане (2,5М; 0,1 моль). Перемешивали в течение 60 минут, после чего по каплям прибавляли 12,9 г (0,1 моль) ацетилморфолина, Реакционную смесь перемешивали еще 20 минут в охладительной смеси, обеспечивающей температуру ниже 0°C. В другой колбе 43,7 г (0,3 моль) диэтилоксалата охлаждали в 50 мл безводного THF в охладительной смеси, обеспечивающей температуру ниже 0°C и холодную реакционную смесь, перемешивая, разливали небольшими порциями по пробиркам, которые затем оставляли на ночь при перемешивании, так что они согревались до комнатной температуры. Выпаривали THF с помощью ротационного испарителя, остаток собирали в 80 мл 17%-ной соляной кислоты. Водную фазу экстрагировали 5 раз этилацетатом (по 150 мл). Объединенные органические фазы высушивали над сульфатом натрия и выпаривали с помощью ротационного испарителя до сухого состояния. Избыток диэтилоксалата отгоняли (2-3 мбар, 80°C). После кристаллизации из PE получали 5,0 г этил-4-(морфолин-4-ил)-2,4-диоксобутаноата в виде белых кристаллов с температурой плавления 60°C.
1H-ЯМР (CDCl3, 400 МГц): δ [ppm] = 1,35 (3H), 3,50-3,73 (8Н), 4,33 (2Н), 6,20 (1Н), 14,32 (1Н).
Суспендировали 7,68 г (38,5 ммоль) моногидрата ацетата меди в 110 мл этилового спирта и нагревали до температуры 75°C Прибавляли (в один прием) 17,63 г (76,96 ммоль) этил-4-(морфолин-4-ил)-2,4-диоксобутаноата. Полученную смесь перемешивали в течение 15 мин при температуре 72°C. После этого при температуре 4°C выпадало в осадок 19,86 г комплексного соединения меди.
Суспендировали 15,7 г (30,2 ммоль) комплексного соединения меди в 240 мл раствора аммиака в метиловом спирте (7 н) и перемешивали в течение 4 часов при комнатной температуре, добавляя 80 мл этилового спирта, после чего фильтровали.
Для дальнейшей обработки комплексное соединение меди суспендировали в 410 мл хлороформа и перемешивали при комнатной температуре. Прибавляли 122 мл 10%-ной серной кислоты, после чего перемешивали 35 мин. После разделения фаз водную фазу экстрагировали два раза хлороформом (по 400 мл). Объединенные органические фазы высушивали над сульфатом натрия и выпаривали с помощью ротационного испарителя до сухого состояния. Полученный остаток кипятили с 80 мл этилацетата и оставляли на ночь при температуре 4°C для кристаллизации. Кристаллический продукт отфильтровывали и высушивали в вакууме. В результате получали 5,3 г (выход 34%) 4-(морфолин-4-ил)-2,4-диоксобутанамида.
ИК-спектр (в чистом виде, см-1): 3453, 3342, 3285, 3180, 2985, 2932, 2873, 1716, 1635, 1576, 1485, 1463, 1442, 1381, 1330, 1303, 1272, 1243, 1195, 1132, 1103, 1066, 1049, 978, 940, 905, 850, 819, 795, 767, 726, 665.
Енольная форма
1H-ЯМР (DMSO-d6, 400 МГц): δ [ppm] = 3,6 (4Н), 6,2 (1Н), 7,8 (2Н), 15,0 (1Н).
C. N4,N4-диэтил-2-оксобутандиамид
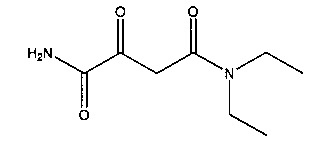
Суспендировали 18,5 г (0,165 моль) трет-бутоксида натрия в 100 мл диэтилового эфира. Прибавляли 25,4 г (0,174 моль) диэтилоксалата, охлаждая на ледяной бане. Перемешивали в течение 30 мин при охлаждении, после чего прибавляли 20,0 г (0,174 моль) N,N-диэтилацетамида и реакционную смесь перемешивали в течение ночи при комнатной температуре. Для дальнейшей обработки прибавляли 260 мл диэтилового эфира и отфильтровывали твердый материал. Этот твердый материал дезинтегрировали в смеси из 35 мл соляной кислоты (6 н), 174 мл этилацетата и 20 мл воды. После разделения фаз водную фазу экстрагировали два раза этилацетатом (по 100 мл). Объединенные органические фазы высушивали над сульфатом натрия и выпаривали с помощью ротационного испарителя до сухого состояния. Неочищенный продукт кристаллизовали из смеси петролейный эфир/диэтиловый эфир при температуре 4°C. Выход 4-(диэтиламино)-2,4-диоксобутаноата составлял 17,6 г (47%).
Енольная форма
1H-ЯМР (CDCl3, 400 МГц): δ [ppm] = 1,2 (6Н), 1,4 (3H), 3,4 (4Н), 4,4 (2Н), 6,2 (1Н).
Суспендировали 8,16 г (40,9 ммоль) моногидрата ацетата меди в 100 мл этилового спирта и нагревали до температуры 73°C. Прибавляли в один прием 17,6 г (81,7 ммоль) этил-4-(диэтиламино)-2,4-диоксобутаноата. Полученную смесь перемешивали в течение 40 мин при температуре 72°C. Затем прибавляли 50 мл толуола и удаляли растворитель при помощи ротационного испарителя. Полученный остаток выпаривали с 50 мл толуола еще два раза. В результате получали 19,9 г неочищенного продукта - комплексного соединения меди, который далее использовали без дополнительной очистки.
Растворяли 9,0 г (18 ммоль) полученного комплексного соединения меди в 25 мл раствора аммиака в метиловом спирте (7 н) и перемешивали в течение 4 часов при комнатной температуре. После этого растворитель полностью удаляли с помощью ротационного испарителя. Для дальнейшей обработки комплексное соединение меди суспендировали в 190 мл дихлорметана и перемешивали при комнатной температуре. Прибавляли 53 мл 10%-ной серной кислоты и перемешивали в течение 5 мин. Фазы разделяли и водную фазу экстрагировали два раза дихлорметаном (по 90 мл). Объединенные органические фазы высушивали над сульфатом натрия и выпаривали с помощью ротационного испарителя до сухого состояния. Полученный остаток очищали путем хроматографии на колонке (силикагель, этилацетат). Выход полученного N4,N4-диэтил-2-оксобутандиамида составлял 6,3 г (94%).
ИК-спектр (в чистом виде, см-1): 3371, 3193, 2974, 2935, 2875, 2324, 2051, 1982, 1787, 1741, 1683, 1635, 1586, 1495, 1459, 1400, 1380, 1362, 1308, 1269, 1239, 1218, 1158, 1130, 1098, 1081, 1048, 960, 896, 827, 782.
Енольная форма
1H-ЯМР (DMSO-d6, 400 МГц): δ [ppm] = 1,1 (6Н), 3,4 (4Н), 6,1 (1H), 7,8 (2Н), 15,4 (1Н).
D. 2,4-Диоксо-4-(пирролидин-1-ил)бутанамид
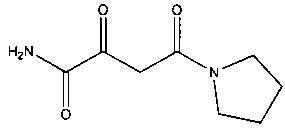
Суспендировали 8,33 г (74,4 ммоль) трет-бутоксида натрия в 47 мл диэтилового эфира. Прибавляли 11,4 г (78,3 ммоль) диэтилоксалата, охлаждая на ледяной бане. Перемешивали в течение 60 мин при охлаждении, после чего прибавляли 8,86 г (78,3 моль) N-ацетилпирролидина и реакционную смесь перемешивали в течение 3 часов при комнатной температуре. Суспензию фильтровали. Полученный твердый материал дезинтегрировали в смеси из 25 мл соляной кислоты (6 н), 120 мл этилацетата и 15 мл воды. После разделения фаз водную фазу экстрагировали два раза этилацетатом (по 50 мл). Объединенные органические фазы высушивали над сульфатом натрия и выпаривали с помощью ротационного испарителя до сухого состояния. Полученный неочищенный продукт очищали путем хроматографии на колонке (силикагель, петролейный эфир, этилацетат 2/1). Выход этил-2,4-диоксо-4-(пирролидин-1-ил)бутаноата составлял 7,7 г (46%).
Енольная форма
1H-ЯМР (DMSO-d6, 400 МГц): δ [ppm] = 1,3 (3H), 1,9 (4Н), 3,4 (2Н), 3,5 (2Н), 4,3 (2Н), 6,1 (1Н), 14,9 (1Н).
Суспендировали 3,59 г (18,0 ммоль) моногидрата ацетата меди в 10 мл этилового спирта и нагревали до температуры 73°C. Прибавляли в один прием 7,66 г (35,9 ммоль) этил-2,4-диоксо-4-(пирролидин-1-ил)бутаноата. Полученную смесь перемешивали в течение 30 мин при температуре 72°C. Затем прибавляли 20 мл этилового спирта и 10 мл воды и перемешивали еще 20 минут. Проводили горячее фильтрование, после чего полученный раствор держали при температуре 4°C. Объединенные органические фазы высушивали над сульфатом натрия и выпаривали с помощью ротационного испарителя до сухого состояния. В результате получалось 4,4 г комплексного соединения меди в кристаллическом виде; этот продукт затем использовали без дальнейшей очистки. Растворяли 4,4 г (9,0 ммоль) полученного комплексного соединения меди в 64 мл раствора аммиака в метиловом спирте (7 н) и перемешивали в течение 4 часов при комнатной температуре. Образовавшуюся суспензию фильтровали и твердый материал высушивали в течение 3 суток при температуре 50°C в глубоком вакууме. Для дальнейшей обработки комплексное соединение меди суспендировали в 100 мл дихлорметана и прибавляли 53 мл 10%-ной серной кислоты. Перемешивали до тех пор, пока не сформировывались две четкие фазы. Эти фазы разделяли и водную фазу экстрагировали два раза дихлорметаном (по 100 мл). Объединенные органические фазы высушивали над сульфатом натрия и выпаривали с помощью ротационного испарителя до сухого состояния. Полученный остаток кристаллизовали из горячего этилацетата. Выход 2,4-диоксо-4-(пирролидин-1-ил) бутанамида составлял 1,7 г (51%),
ИК-спектр (в чистом виде, см-1): 3444, 3293, 3146, 2976, 2890, 1688, 1628, 1586, 1482, 1460, 1399, 1343, 1301, 1230, 1187, 1164, 1128, 1107, 1052, 1019, 970, 913, 852, 819, 748.
Енольная форма
1H-ЯМР (DMSO-d6, 400 МГц): δ [ppm] = 1,9 (4Н), 3,4 (4Н), 6,0 (1Н), 7,8 (2Н), 15,3 (1Н).
Е. N,N`,N`-Тетраметил-2-оксобутандиамид
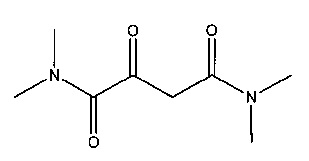
Суспендировали 18,65 г (166,3 ммоль) трет-бутоксида натрия в 170 мл диэтилового эфира. Прибавляли 25,58 г (175,0 ммоль) этил-N,N-диметилоксамата, охлаждая на ледяной бане. Перемешивали 30 мин. Объединенные органические фазы высушивали над сульфатом натрия и выпаривали с помощью ротационного испарителя до сухого состояния. Прибавляли 15,25 г (175,0 ммоль) N,N-диметилацетамида и реакционную смесь перемешивали в течение 2 часов при комнатной температуре. Затем в реакционную смесь добавляли 90 мл соляной кислоты (6 н), 420 мл этилацетата и 15 мл воды и перемешивали 5 мин. После разделения фаз водную фазу экстрагировали этилацетатом (200 мл). Объединенные органические фазы высушивали над сульфатом натрия и выпаривали с помощью ротационного испарителя до сухого состояния. Полученный неочищенный продукт очищали путем хроматографии на колонке (силикагель, дихлорметан/ацетон 5/1). Выход N,N,N`,N`-тетраметил-2-оксобутандиамида составлял 5,3 г (16%).
ИК-спектр (в чистом виде, см-1): 2936, 1720, 1635, 1496, 1456, 1397, 1352, 1259, 1237, 1203, 1130, 1107, 1077, 951, 892, 849, 807, 774, 753, 720.
Кето-форма
1H-ЯМР (DMSO-d6, 400 МГц): δ [ppm] = 2,8-3,0 (12Н), 3,9 (2Н).
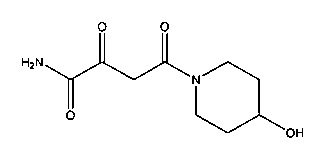
F. 4-(4-Гидроксипиперидин-1-ил)-2,4-диоксобутанамид
Растворяли 120 г (1,19 моль) 4-гидроксипиридина в 1700 мл дихлорметана и прибавляли по каплям 132 г (1,03 моль) триэтиламина. Реакционную смесь охлаждали до -40°C и прибавляли по каплям 93,4 г (1,19 моль) ацетилхлорида. Затем перемешивали в течение 1 часа при комнатной температуре. Образовавшуюся суспензию фильтровали и фильтрат концентрировали с помощью ротационного испарителя до сухого состояния. Полученный остаток собирали в 1700 мл этилацетата и фильтровали еще раз. Фильтрат концентрировали до сухого состояния. В результате было получено 120 г (выход 71%) 1-(4-гидроксипиперидин-1-ил)этанона.
1H-ЯМР (CDCl3, 400 МГц): δ [ppm] = 1,5 (2Н), 1,8 (2Н), 2,1 (3H), 2,7 (1Н), 3,2 (2Н), 3,7 (1Н), 3,9 (1Н), 4,1 (1Н).
Суспендировали 120 г (839 ммоль) 1-(4-гидроксипиперидин-1-ил)этанона в 1,5 л дихлорметана и охлаждали на ледяной бане. Прибавляли по каплям 85,9 г (839 ммоль) триэтиламина и перемешивали в течение 10 мин. Затем прибавляли 101 г (839 ммоль) триметилацетилхлорида и реакционную смесь перемешивали в течение 3 суток при комнатной температуре. После этого реакционную смесь концентрировали до объема 200 мл и фильтровали. Фильтрат концентрировали до сухого состояния. Полученный остаток собирали в 200 мл этилацетата, перемешивали в течение 30 мин и фильтровали. Фильтрат концентрировали с помощью ротационного испарителя до сухого состояния. Для очистки полученный продукт кристаллизовали из н-гептана. В результате было получено 56 г (выход 29%) 1-ацетилпиперидин-4-ил-2,2-диметилпропаноата в виде белых кристаллов.
1H-ЯМР (CDCl3, 400 МГц): δ [ppm] = 1,2 (9Н), 1,6 (2Н), 1,8 (2Н), 2,1 (3H), 3,4 (1Н), 3,6 (2Н), 3,7 (1Н), 5,0 (1Н).
Суспендировали 9,88 г (88,0 ммоль) трет-бутоксида натрия в 80 мл диэтилового эфира и прибавляли 12,9 г (88,0 ммоль) диэтилоксалата. Растворяли 20,0 г (88,0 ммоль) 1-ацетилпиперидин-4-ил-2,2-диметилпропаноата в 80 мл диэтилового эфира и прибавляли по каплям в указанную реакционную смесь. Перемешивали в течение 20 мин, после чего оставляли реакционную смесь на ночь без перемешивания. Затем прибавляли 160 мл петролейного эфира и полученную суспензию фильтровали. Оставшийся на фильтре материал собирали в 60 мл соляной кислоты (1 н) и доводили pH до 7 раствором гидроксида натрия. Затем три раза экстрагировали этилацетатом (по 200 мл), высушивали над сульфатом натрия и концентрировали до сухого состояния с помощью ротационного испарителя. В результате получалось 15,3 г (выход 53%) этил-4-{4-[(2,2-диметилпропаноил)окси]пиперидин-1-ил}-2,4-диоксобутаноата. Этот продукт использовали без дальнейшей очистки.
Енольная форма
1H-ЯМР (DMSO-d6, 400 МГц): δ [ppm] = 1,2 (9Н), 1,4 (3H), 1,7 (2Н), 1,9 (2Н), 3,5-3,8 (4Н), 4,3 (2Н), 5,0 (1Н), 6,3 (1Н), 14,5 (1Н).
Растворяли 189 г (577 ммоль) этил-4-{4-[(2,2-диметилпропаноил)окси]пиперидин-1-ил}-2,4-диоксобутаноата в 900 мл безводного этилового спирта и нагревали до температуры 50°C, после чего прибавляли по каплям 393 г 21%-ного раствора этоксида натрия (1,21 моль) и оставляли на ночь при перемешивании и температуре 50°C. Реакционную смесь концентрировали с помощью ротационного испарителя до сухого состояния, и полученный остаток собирали в 2 л соляной кислоты (1 н). Экстрагировали 3 раза этилацетатом (по 2 л), высушивали над сульфатом натрия и концентрировали до сухого состояния. Полученный неочищенный продукт очищали путем хроматографии на колонке (силикагель, ацетон/дихлорметан 1/1). В результате получали 74,9 г (выход 53%) этил-4-(4-гидроксипиперидин-1-ил)-2,4-диоксобутаноата.
Енольная форма
1H-ЯМР (DMSO-d6, 400 МГц): δ [ppm] = 1,3 (3H), 1,4 (2Н), 1,8 (2Н), 3,3 (2Н), 3,7 (1Н), 3,8 (2Н), 4,2 (2Н), 4,8 (1H), 6,3 (1Н), 14,9 (1H).
Суспендировали 8,78 г (44,0 ммоль) моногидрата ацетата меди в 125 мл этилового спирта и нагревали до кипения. Прибавляли порциями 21,3 г (88,0 ммоль этил-4-(4-гидроксипиперидин-1-ил)-2,4-диоксобутаноата. Полученную смесь кипятили с обратным холодильником в течение 40 мин. Затем фильтровали и полученный раствор оставляли на ночь при температуре 2,2°C. В результате получалось 13,2 г комплексного соединения меди в кристаллическом виде; этот продукт использовали без дальнейшей очистки.
Растворяли 7,18 г (13,8 ммоль) полученного комплексного соединения меди в 98 мл раствора аммиака в метиловом спирте (7 н) и перемешивали в течение 4 часов при комнатной температуре. Образовавшуюся суспензию фильтровали и оставшийся на фильтре материал высушивали в вакууме при температуре 50°C. Для дальнейшей обработки комплексное соединение меди суспендировали в 125 мл метилового спирта и через эту суспензию в течение 30 минут пропускали сероводород (H2S). Перемешивали до тех пор, пока не сформировались две четкие фазы. Фильтровали два раза через диатомовую землю (Celite®), после чего фильтрат концентрировали с помощью ротационного испарителя. Полученный остаток перекристаллизовывали из этилацетата. В результате получалось 0,5 г (выход 8%) 4-(4-гидроксипиперидин-1-ил)-2,4-диоксобутанамида.
ИК-спектр (в чистом виде, см-1): 3282, 3118, 2981, 2324, 2164, 2051, 1981, 1797, 1546, 1413, 1349, 1264, 1161, 1138, 1086, 1043, 962, 925, 899, 821, 786, 728, 676.
Енольная форма
1H-ЯМР (DMSO-d6, 400 МГц): δ [ppm] = 1,3 (2Н), 1,7 (2Н), 3,3 (2Н), 3,7-3,9 (3H), 4,8 (1H), 6,2 (1Н), 7,7 (2Н), 15,3 (1Н).
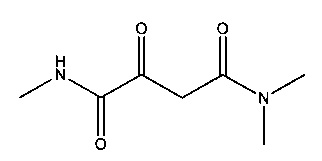
G. N1,N4,N4-Триметил-2-оксобутандиамид
Суспендировали 53,30 г (0,475 моль) трет-бутоксида натрия в 300 мл диэтилового эфира. Прибавляли 73,07 г (0,500 моль) диэтилоксалата, охлаждая на ледяной бане. Перемешивали в течение 30 мин при охлаждении, после чего прибавляли 43,57 г (0,500 моль) N,N-диметилацетамида и реакционную смесь перемешивали в течение 2 часов. Для дальнейшей обработки эту смесь фильтровали и полученное твердое вещество дезинтегрировали в 260 мл соляной кислоты (4 н) и 750 мл этилацетата. После разделения фаз водную фазу два раза экстрагировали этилацетатом (по 300 мл). Объединенные органические фазы высушивали над сульфатом натрия и выпаривали с помощью ротационного испарителя до сухого состояния. В результате получалось 54,3 г (выход 58%) 4-(диметиламино)-2,4-диоксобутаноата.
1H-ЯМР (CDCl3, 400 МГц): δ [ppm] = 1,38 (3H), 3,06 (6Н), 4,33 (2Н), 6,25 (1Н).
Суспендировали 32,56 г (163,1 ммоль) моногидрата ацетата меди в 470 мл этилового спирта и нагревали до температуры 75°C. Прибавляли в один прием 61,06 г (326,2 ммоль) этил-4-(диметиламино)-2,4-диоксобутаноата. Полученную смесь перемешивали в течение 40 мин при температуре 75°C. Образующееся комплексное соединение меди при температуре 4°C выпадало в осадок. Полученное комплексное соединение меди в количестве 10,0 г (23,0 ммоль) суспендировали в 57 мл раствора метиламина в этиловом спирте (33%) и перемешивали в течение 2 часов при комнатной температуре. Реакционную смесь концентрировали до сухого состояния. Для дальнейшей обработки комплексное соединение меди растворяли в 100 мл хлороформа. В полученный раствор прибавляли 170 мл 10%-ной серной кислоты и тщательно перемешивали фазы. После разделения фаз водную фазу экстрагировали два раза хлороформом (по 50 мл). Объединенные органические фазы высушивали над сульфатом натрия и концентрировали с помощью ротационного испарителя до сухого состояния. В результате получалось 4,1 г (выход 52%) N1,N4,N4-триметил-2-оксобутандиамида.
ИК-спектр (в чистом виде, см-1): 3398, 3315, 3105, 2931, 2324, 1783, 1671, 1632, 1599, 1526, 1500, 1405, 1361, 1347, 1252, 1160, 1063, 1018, 935, 907, 838, 776, 726.
Енольная форма:
1H-ЯМР (DMSO-d6, 400 МГц): δ [ppm] = 2,7 (3H), 3,0 (6Н), 6,1 (1H), 8,4 (1Н), 15,5 (1H).
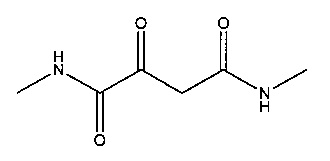
H. N,N'-Диметил-2-оксобутандиамид
Суспендировали 38,2 г (0,556 моль) метиламина гидрохлорида в 70 мл дихлорметана и прибавляли 57,3 г (0,556 моль) триэтиламина. Реакционную смесь охлаждали до температуры -60°C и прибавляли по каплям 108 г (0,556 моль) (2Z)-(2,2-диметил-5-оксо-1,3-диоксолан-4-илиден)ацетилхлорида (полученного, как описано в работе J. Banville et al. Tetrahedron Letters, 2010, 51, 3170-3173), растворенного в 70 мл дихлорметана. Оставляли на ночь при перемешивании при комнатной температуре, после чего кипятили с обратным холодильником в течение 4 часов. Реакционную смесь концентрировали до сухого состояния, и полученный остаток собирали в 400 мл этилацетата. Эту суспензию фильтровали, фильтрат концентрировали до сухого состояния. Полученный остаток собирали в 600 мл диэтилового эфира. Образовавшуюся суспензию фильтровали и фильтрат концентрировали до сухого состояния. В результате получалось 49,1 г (выход 47%) (2Z)-(2,2-диметил-5-оксо-1,3-диоксолан-4-илиден)-N-метилацетамида.
1H-ЯМР (DMSO-d6, 400 МГц): δ [ppm] = 1,7 (6Н), 2,6 (3H), 5,8 (1Н), 8,0 (1H).
Суспендировали 13,1 г (66,0 ммоль) моногидрата ацетата меди в 600 мл этилового спирта и прибавляли 25,7 г (132 ммоль) (2Z)-(2,2-диметил-5-оксо-1,3-диоксолан-4-илиден)-N-метилацетамида. Эту смесь кипятили с обратным холодильником в течение 4 часов. Образовавшуюся суспензию концентрировали до сухого состояния. Полученный остаток три раза десорбировали толуолом, чтобы удалить уксусную кислоту. Суспендировали 10,6 г (25,9 ммоль) полученного комплексного соединения меди в 31 мл раствора метиламина в этиловом спирте (33%) и перемешивали в течение 2 часов при комнатной температуре. После этого реакционную смесь концентрировали до сухого состояния. Для дальнейшей обработки комплексное соединение меди растворяли в 200 мл дихлорметана. Прибавляли 60 мл 10%-ной серной кислоты и 30 мл воды и тщательно перемешивали фазы. После разделения фаз водную фазу два раза экстрагировали дихлорметаном (по 100 мл). Объединенные органические фазы высушивали над сульфатом натрия и концентрировали с помощью ротационного испарителя до сухого состояния. Полученный продукт кристаллизовали из горячего этилацетата. В результате получалось 0,4 г (выход 5%) N,N'-диметил-2-оксобутандиамида.
ИК-спектр (в чистом виде, см-1): 3367, 3310, 3133, 2942, 2051, 1624, 1584, 1532, 1396, 1275, 1247, 1133, 1078, 946, 838, 776, 752, 674.
Енольная форма:
1H-ЯМР (DMSO-d6, 400 МГц): δ [ppm] = 2,7 (6Н), 5,9 (1H), 8,4 (2Н), 14,3 (1Н).
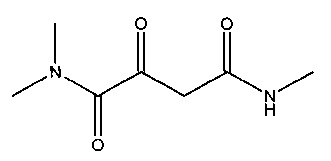
I. N1,N1,N4-Триметил-2-оксобутандиамид
Суспендировали 38,2 г (0,556 моль) метиламина гидрохлорида в 70 мл дихлорметана и прибавляли 57,3 г (0,556 моль) триэтиламина. Реакционную смесь охлаждали до температуры -60°C и прибавляли по каплям 108 г (0,556 моль) (2Z)-(2,2-диметил-5-оксо-1,3-диоксолан-4-илиден)ацетилхлорида (полученного, как описано в работе J. Banville et al. Tetrahedron Letters, 2010, 51, 3170-3173), растворенного в 70 мл дихлорметана. Оставляли на ночь при перемешивании при комнатной температуре, после чего кипятили с обратным холодильником в течение 4 часов. Реакционную смесь концентрировали до сухого состояния, и полученный остаток собирали в 400 мл этилацетата. Эту суспензию фильтровали, фильтрат концентрировали до сухого состояния. Полученный остаток собирали в 600 мл диэтилового эфира. Эту суспензию фильтровали и фильтрат концентрировали до сухого состояния. В результате получалось 49,1 г (выход 47%) (2Z)-(2,2-диметил-5-оксо-1,3-диоксолан-4-илиден)-N-метилацетамида.
1H-ЯМР (DMSO-d6, 400 МГц): δ [ppm] = 1,7 (6Н), 2,6 (3H), 5,8 (1Н), 8,0 (1H).
Растворяли 1,0 г (5,4 ммоль) (2Z)-(2,2-диметил-5-оксо-1,3-диоксолан-4-илиден)-N-метилацетамида в 9,6 мл раствора метиламина в этиловом спирте (33%) и перемешивали в течение 5 мин при комнатной температуре. Прибавляли 6 мл соляной кислоты (6 н) и перемешивали еще 5 мин. Реакционную смесь концентрировали до сухого состояния, и полученный остаток собирали в 15 мл воды. Водную фазу экстрагировали два раза этилацетатом (по 50 мл). Объединенные органические фазы высушивали над сульфатом натрия и концентрировали с помощью ротационного испарителя до сухого состояния. Полученный неочищенный продукт очищали путем хроматографии на колонке (силикагель, этилацетат). В результате получалось 280 мг (выход 30%) N1,N1,N4-триметил-2-оксобутандиамида.
ИК-спектр (в чистом виде, см-1): 3303, 3099, 2941, 1766, 1719, 1624, 1561, 1502, 1449, 1405, 1338, 1255, 1227, 1160, 1079, 1041, 947, 910, 829, 773, 657.
Кето-форма:
1H-ЯМР (DMSO-d6, 400 МГц): δ [ppm] = 2,6 (3H), 2,9 (6Н), 3,6 (2Н), 8,1 (1Н).
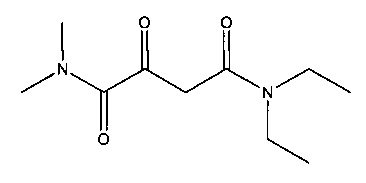
J. N4,N4-диэтил-N1,N1-диметил-2-оксобутандиамид
Суспендировали 8,0 г (71 ммоль) трет-бутоксида натрия в 60 мл диэтилового эфира. Прибавляли 10,9 г (75,0 ммоль) этил-N,N-диметилоксамата, охлаждая на ледяной бане. Перемешивали в течение 30 мин при комнатной температуре. Затем прибавляли 8,6 г (75 ммоль) N,N-диэтилацетамида и реакционную смесь перемешивали в течение 2 часов при комнатной температуре. После этого в реакционную смесь добавляли 45 мл соляной кислоты (6 н) и 200 мл этилацетата. После разделения фаз водную фазу экстрагировали два раза этилацетатом (по 50 мл). Объединенные органические фазы высушивали над сульфатом натрия и концентрировали с помощью ротационного испарителя до сухого состояния. Полученный неочищенный продукт очищали путем хроматографии на колонке (силикагель, петролейный эфир/этилацетат). В результате получалось 3,5 г (выход 22%) N4,N4-диэтил-N1,N1-диметил-2-оксобутандиамида.
ИК-спектр (в чистом виде, см-1): 3496, 2975, 2936, 1721, 1626, 1487, 1448, 1400, 1380, 1362, 1308, 1271, 1217, 1200, 1141, 1100, 1077, 954, 927, 789, 771, 734.
Кето-форма
1H-ЯМР (DMSO-d6, 400 МГц): δ [ppm] = 1,1 (6Н), 3,0 (6Н), 3,3 (4Н), 3,9 (2Н).
K. N,N-Диметил-3,4-диоксо-4-(пирролидин-1-ил)бутанамид
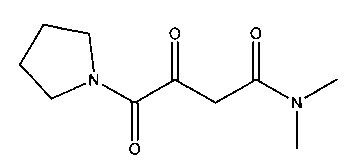
Растворяли 19,2 г (270 ммоль) пирролидина и 27,3 г (270 ммоль) триэтиламина в 400 мл диэтилового эфира и охлаждали на ледяной бане. Прибавляли по каплям 36,9 г (270 ммоль) этилхлороксоацетата, после чего реакционную смесь нагревали до комнатной температуры. Полученную суспензию фильтровали и фильтрат концентрировали с помощью ротационного испарителя. В результате получалось 46,6 г этил-оксо(пирролидин-1-ил)ацетата в виде желтого масла, который использовали без дальнейшей очистки.
Суспендировали 27,2 г (242 ммоль) трет-бутоксида натрия в 200 мл диэтилового эфира. Прибавляли 43,6 г (255 ммоль) этил-оксо(пирролидин-1-ил)ацетата, охлаждая на ледяной бане. Перемешивали в течение 30 мин при комнатной температуре. Затем прибавляли 22,2 г (255 ммоль) N,N-диметилацетамида и реакционную смесь перемешивали в течение 2 часов при комнатной температуре. После этого в реакционную смесь добавляли 136 мл соляной кислоты (6 н), 170 мл этилацетата и 70 мл воды. После разделения фаз водную фазу экстрагировали два раза этилацетатом (по 100 мл). Объединенные органические фазы высушивали над сульфатом натрия и концентрировали с помощью ротационного испарителя до сухого состояния. Полученный неочищенный продукт очищали путем хроматографии на колонке (силикагель, петролейный эфир/этилацетат). В результате получалось 37,1 г (выход 69%) N,N-диметил-3,4-диоксо-4-(пирролидин-1-ил)бутанамида.
ИК-спектр (в чистом виде, см-1): 2952, 2880, 1722, 1632, 1602, 1503, 1445, 1415, 1354, 1338, 1253, 1224, 1163, 1141, 1060, 1033, 975, 952, 915, 890, 870, 848, 812, 765, 728, 695.
Енольная форма
1H-МР (DMSO-d6, 400 МГц): δ [ppm] = 1,8 (4Н), 3,0 (6Н), 3,4 (2Н), 3,6 (2Н), 5,9 (1Н), 15,6 (1Н).
L. N4-[2-(Диметиламино)-2-оксоэтил]-N1,N1,N4-триметил-2-оксобутандиамид
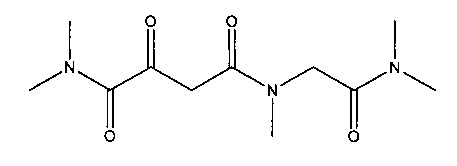
Суспендировали 17,1 г (0,112 моль) N,N-диметил-2-(метиламино)ацетамида гидрохлорида (Sigma-Aldrich: CDS007544) в 300 мл дихлорметана и прибавляли 22,6 г (0,224 моль) триэтиламина. В полученную смесь порциями добавляли 21,3 г (0,112 моль) (2Z)-(2,2-диметил-5-оксо-1,3-диоксолан-4-илиден)ацетилхлорида (полученного, как описано в работе J. Banville et al, Tetrahedron Letters, 2010, 51, 3170-3173). После этого перемешивали в течение 30 мин при комнатной температуре и затем кипятили с обратным холодильником в течение 2 часов. Образовавшуюся суспензию концентрировали до сухого состояния, и сухой остаток собирали в 300 мл этилацетата. Эту суспензию фильтровали и фильтрат концентрировали до сухого состояния. В результате получалось 9,8 г (выход 32%) N2-[(2Z)-2-(2,2-диметил-5-оксо-1,3-диоксолан-4-илиден)ацетил]-N,N,N2-триметилглицинамида.
1Н-ЯМР (DMSO-d6, 400 МГц): δ [ppm] = 1,7 (6Н), 2,8-3,0 (9Н), 4,2-4,3 (2Н), 5,8-6,1 (1H).
Растворяли 8,8 г (32 ммоль) N2-[(2Z)-2-(2,2-диметил-5-оксо-1,3-диоксолан-4-илиден)ацетил]-N,N,N2-триметилглицинамида в 58 мл раствора диметиламина (33%) в этиловом спирте и перемешивали в течение 30 минут при комнатной температуре. Эту реакционную смесь концентрировали до сухого состояния. Полученный неочищенный продукт очищали путем хроматографии на колонке (силикагель, дихлорметиан:метанол 20:1). В результате получалось 3,0 г (выход 36%) N4-[2-(диметиламино)-2-оксоэтил]-N1,N1,N4-триметил-2-оксобутандиамида.
ИК-спектр (в чистом виде, см-1): 3492, 2936, 1720, 1631, 1492, 1398, 1364, 1332, 1261, 1239, 1217, 1139, 1106, 1078, 951, 898, 860, 812, 762, 720, 675.
Кето-форма:
1H-ЯМР (DMSO-d6, 400 МГц): δ [ppm] = 2,7-3,0 (15Н), 3,7-4,0 (2Н), 4,1-4,3 (2Н).
M. N4-(2-Метоксиэтил)-N1,N1,N4-триметил-2-оксобутандиамид
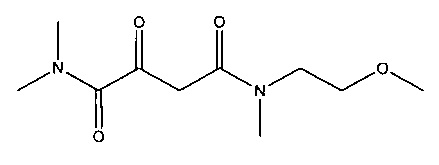
Растворяли 9,09 г (102 ммоль) (2-метоксиэтил)метиламина и 10,3 г (102 ммоль) триэтиламина в ПО мл диэтилового эфира и охлаждали на ледяной бане. В этот раствор прибавляли по каплям 8,01 г (102 ммоль) ацетилхлорида и смесь нагревали до комнатной температуры. Полученную суспензию фильтровали и фильтрат концентрировали с помощью ротационного испарителя. В результате получалось 10 г N-(2-метоксиэтил)-N-метилацетамида в виде бесцветного масла, который использовали без дальнейшей очистки.
Суспендировали 8,08 г (72,0 ммоль) трет-бутоксида натрия в 100 мл диэтилового эфира. В эту суспензию прибавляли 10,5 г (72,0 ммоль) этил-N,N-диметилоксамата, охлаждая на ледяной бане, и перемешивали в течение 15 мин при комнатной температуре. Прибавляли 9,44 г (72,0 ммоль) N-(2-метоксиэтил)-N-метилацетамида и реакционную смесь перемешивали в течение 2 часов при комнатной температуре. Затем в реакционную смесь добавляли 140 мл соляной кислоты (6 н), 180 мл этилацетата и 45 мл воды. После разделения фаз водную фазу экстрагировали 3 раза этилацетатом (по 80 мл). Объединенные органические фазы высушивали над сульфатом натрия и концентрировали с помощью ротационного испарителя до сухого состояния. Полученный неочищенный продукт очищали путем хроматографии на колонке (силикагель, петролейный эфир/этилацетат). В результате получалось 2,98 г (выход 18%) N4-(2-метоксиэтил)-N1,N1,N4-триметил-2-оксобутандиамида.
ИК-спектр (в чистом виде, см-1): 2935, 1612, 1498, 1458, 1427, 1362, 1260, 1193, 1153, 1114, 1062, 1022, 950, 842, 814, 787, 724, 694, 674.
Кето-форма:
1H-ЯМР (DMSO-d6, 400 МГц): δ [ppm] = 2,8-3,0 (9Н), 3,2-3,3 (3H), 3,4-3,6 (4Н), 3,9-4,0 (2Н).
N. N4-(2-Метоксиэтил)-N1,N1-диметил-2-оксобутандиамид
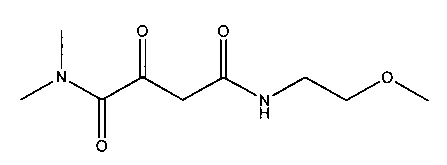
Суспендировали 7,1 г (94 ммоль) 2-метоксиэтиламина в 300 мл дихлорметана и прибавляли 19,0 г (188 ммоль) триэтиламина. Растворяли 18,0 г (94 ммоль) (2Z)-(2,2-диметил-5-оксо-1,3-диоксолан-4-илиден)ацетилхлорида (полученного, как описано в работе J. Banville et al, Tetrahedron Letters, 2010, 51, 3170-3173) в 70 мл дихлорметана. Перемешивали в течение 30 мин при комнатной температуре, после чего кипятили с обратным холодильником в течение 4 часов. Образовавшуюся суспензию концентрировали до сухого состояния, и полученный остаток собирали в 300 мл этилацетата. Эту суспензию фильтровали, фильтрат концентрировали до сухого состояния. Полученный остаток собирали в 350 мл диэтилового эфира и перемешивали. Образовавшуюся суспензию фильтровали и фильтрат концентрировали до сухого состояния. В результате получалось 15,4 г (выход 71%) (2Z)-2-(2,2-диметил-5-оксо-1,3-диоксолан-4-илиден)-N-(2-метоксиэтил)ацетамида.
1H-ЯМР (DMSO-d6, 400 МГц): δ [ppm] = 1,7 (6Н), 3,3 (3H), 3,3-3,4 (4Н), 5,9 (1H), 8,2 (1H).
Растворяли 3,0 г (13 ммоль) (2Z)-2-(2,2-диметил-5-оксо-1,3-диоксолан-4-илиден)-N-(2-метоксиэтил)ацетамида в 23 мл раствора диметиламина (33%) в этиловом спирте и перемешивали в течение 5 мин при комнатной температуре. Прибавляли 18 мл раствора HCl (6 н), перемешивали в течение 5 мин и концентрировали с помощью ротационного испарителя до сухого состояния. В результате получалось 2,3 г (выход 82%) N4-(2-метоксиэтил-N1,N1-диметил-2-оксобутандиамида.
ИК-спектр (в чистом виде, см-1): 3307, 3086, 2932, 2884, 1720, 1634, 1548, 1503, 1452, 1401, 1324, 1266, 1195, 1121, 1084, 1041, 948, 819, 772, 718.
Кето-форма:
1H-NMR (DMSO-d6, 400 МГц): δ [ppm] = 2,9 (6Н), 3,3 (3H), 3,2-3,4 (4Н), 3,6 (2Н), 8,2 (1Н).
O. Этил-N-[4-(диметиламино)-3,4-диоксобутаноил]-N-метилглицинат
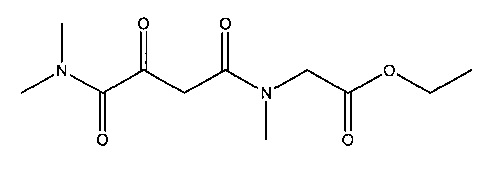
Суспендировали 15,0 г (97,7 ммоль) гидрохлорида этилового эфира саркозина в 300 мл дихлорметана и прибавляли 19,8 г (195 ммоль) триэтиламина. В эту смесь добавляли 18,8 г (98,7 ммоль) (2Z)-(2,2-диметил-5-оксо-1,3-диоксолан-4-илиден)ацетилхлорида (полученного, как описано в работе J. Banville et al, Tetrahedron Letters, 2010, 51, 3170-3173), растворенного в 70 мл дихлорметана. Перемешивали в течение 30 мин при комнатной температуре, после чего кипятили с обратным холодильником в течение 4 часов. Полученную суспензию концентрировали до сухого состояния, и остаток собирали в 300 мл этилацетата. Эту суспензию фильтровали, фильтрат концентрировали до сухого состояния. В результате получалось 12 г (выход 45%) этил-N-[(2Z)-2-(2,2-диметил-5-оксо-1,3-диоксолан-4-илиден)ацетил]-N-метилглицината.
1H-ЯМР (DMSO-d6, 400 МГц): δ [ppm] = 1,2 (3H), 1,8 (6Н), 3,1 (3H), 4,1 (2Н), 4,1-4,3 (2Н).
Растворяли 3,0 г (11 ммоль) этил-N-[(2Z)-2-(2,2-диметил-5-оксо-1,3-диоксолан-4-илиден)ацетил]-N-метилглицината в 20 мл 33%-ного раствора диметиламина в этиловом спирте и перемешивали в течение 5 минут при комнатной температуре. Прибавляли 18 мл раствора HCl (6 н), перемешивали в течение 5 мин и реакционную смесь концентрировали до сухого состояния. Сухой остаток собирали в 50 мл воды и экстрагировали три раза этилацетатом (по 150 мл). Объединенные органические фазы высушивали над сульфатом натрия. В результате получалось 2,3 г (выход 81%) этил-1N-[4-(диметиламино)-3,4-диоксобутаноил]-N-метилглицината.
ИК-спектр (в чистом виде, см-1): 2938, 1743, 1638, 1489, 1399, 1374, 1354, 1197, 1107, 1077, 1031, 950, 900, 851, 811, 765, 717.
Кето-форма:
1H-ЯМР (DMSO-d6, 400 МГц): δ [ppm] = 1,2 (3H), 2,8-3,1 (9Н), 3,8-4,0 (2Н), 4,1 (2Н), 4,1-4,3 (2Н).
P. N1-(2-Метоксиэтил)-N4,N4-диметил-2-оксобутанамид
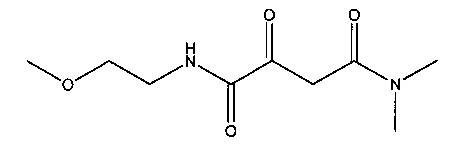
Суспендировали 53,30 г (0,475 моль) трет-бутоксида натрия в 300 мл диэтилового эфира. Прибавляли 73,07 г (0,500 моль) диэтилоксалата, охлаждая на ледяной бане. Перемешивали в течение 30 мин, прибавляли 43,57 г (0,500 моль) N,N-диметилацетамида и реакционную смесь перемешивали в течение 2 часов. Для дальнейшей обработки эту смесь фильтровали и оставшееся твердое вещество дезинтегрировали в 260 мл соляной кислоты (4 н) и 750 мл этилацетата. После разделения фаз водную фазу экстрагировали два раза этилацетатом (по 300 мл). Объединенные органические фазы высушивали над сульфатом натрия и концентрировали с помощью ротационного испарителя до сухого состояния. В результате получалось 54,3 г (выход 58%) 4-(диметиламино)-2,4-диоксобутаноата.
1H-ЯМР (CDCl3, 400 МГц): δ [ppm] = 1,38 (3H), 3,06 (6Н), 4,33 (2Н), 6,25 (1Н).
Суспендировали 32,56 г (163,1 ммоль) моногидрата ацетата меди в 470 мл этилового спирта и нагревали до температуры 75°C. Прибавляли в один прием 61,06 г (326,2 ммоль) этил-4-(диметиламино)-2,4-диоксобутаноата. Полученную смесь перемешивали в течение 40 мин при температуре 75°C. Образовавшееся комплексное соединение меди выпадало в осадок при температуре 4°C.
Растворяли 10,0 г (22,9 ммоль) комплексного соединения меди в 40 мл этилового спирта, прибавляли 17,2 г (229 ммоль) 2-метоксиэтиламина и перемешивали в течение 2 часов при комнатной температуре. После этого реакционную смесь концентрировали до сухого состояния. Для дальнейшей обработки полученное комплексное соединение меди суспендировали в 80 мл дихлорметана и прибавляли 70 мл 10%-ной серной кислоты. После энергичного перемешивания фазы разделяли и водную фазу экстрагировали два раза дихлорметаном (по 50 мл). Объединенные органические фазы высушивали над сульфатом натрия и концентрировали с помощью ротационного испарителя до сухого состояния. Остаток перекристаллизовывали в смеси диэтиловый эфир/петролейный эфир. Кристаллический продукт отфильтровывали и высушивали в вакууме. В результате получалось 5,96 г (выход 60%) N1-(2-метоксиэтил)-N4,N4-диметил-2-оксобутанамида.
ИК-спектр (в чистом виде, см-1): 3372, 3311, 3202, 2974, 2934, 2879, 2830, 1787, 1741, 1674, 1634, 1530, 1495, 1473, 1439, 1400, 1358, 1267, 1193, 1176, 1158, 1124, 1107, 1064, 1016, 960, 901, 825, 815, 767, 715.
Енольная форма
1H-ЯМР (DMSO-d6, 400 МГц): δ [ppm] = 3,0 (6Н), 3,2 (3H), 3,3-3,4 (4Н), 6,1 (1Н), 8,2 (1Н), 15,5 (1Н).
Q. Этил-N-[4-(диметиламино)-2,4-диоксобутаноил]-N-метилглицинат
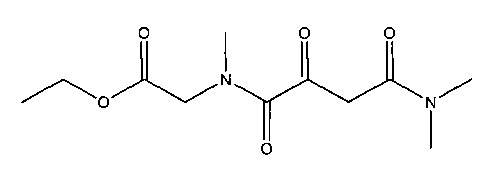
Суспендировали 4,73 г (58,0 ммоль) диметиламина гидрохлорида в 100 мл дихлорметана и прибавляли по каплям 11,1 г (58,0 ммоль) (2Z)-(2,2-диметил-5-оксо-1,3-диоксолан-4-илиден)ацетилхлорида (полученного, как описано в работе J. Banville et al, Tetrahedron Letters, 2010, 51, 3170-3173), растворенного в 100 мл дихлорметана. Оставляли на ночь при перемешивании при комнатной температуре, после чего прибавляли 11,7 г (116 ммоль) триэтиламина и затем кипятили с обратным холодильником в течение 2 часов. Полученную суспензию фильтровали и фильтрат концентрировали до сухого состояния. Остаток собирали в 170 мл этилацетата, фильтровали и еще раз концентрировали до сухого состояния. В результате получалось 11 г (выход 55%) (2Z)-2-(2,2-диметил-5-оксо-1,3-диоксолан-4-илиден)-N,N-диметилацетамида.
1H-ЯМР (DMSO-d6, 400 МГц): δ [ppm] = 1,7 (6Н), 2,9 (3H), 3,0 (3H), 6,0 (1H).
Суспендировали 15,4 г (100 ммоль) гидрохлорида этилового эфира саркозина и 10,1 г (100 ммоль) триэтиламина в 100 мл дихлорметана. Прибавляли 5,0 г (25 ммоль) (2Z)-2-(2,2-диметил-5-оксо-1,3-диоксолан-4-илиден)-N,N-диметилацетамида, после чего кипятили с обратным холодильником в течение 1 часа. Реакционную смесь концентрировали до сухого состояния, и остаток собирали в 20 мл этилацетата. После фильтрования фильтрат концентрировали до сухого состояния. Полученный неочищенный продукт очищали путем хроматографии на колонке (селикагель, этилацетат). В результате получалось 2,7 г (выход 42%) этил-N-[4-(диметиламино)-2,4-диоксобутаноил]-N-метилглицината.
ИК-спектр (в чистом виде, см-1): 3351, 2982, 2939, 1729, 1682, 1634, 1508, 1397, 1375, 1353, 1257, 1198, 1096, 1021, 982, 916, 864, 822, 771.
Кето-форма:
1H-ЯМР (DMSO-d6, 400 МГц): δ [ppm] = 1,2 (3H), 2,8-3,1 (9Н), 3,8-4,0 (2Н), 4,1 (2Н), 4,1-4,3 (2Н).
R. N1-[2-(Диметиламино)-2-оксоэтил]-N1,N4,N4-триметил-2-оксобутандиамид
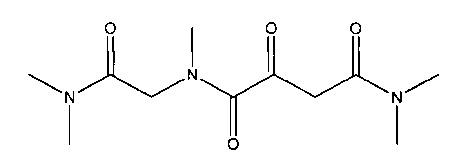
Суспендировали 4,73 г (58,0 ммоль) диметиламина гидрохлорида в 100 мл дихлорметана и прибавляли по каплям 11,1 г (58,0 ммоль) (2Z)-(2,2-диметил-5-оксо-1,3-диоксолан-4-илиден)ацетилхлорида (полученного, как описано в работе J. Banville et al, Tetrahedron Letters, 2010, 51, 3170-3173), растворенного в 100 мл дихлорметана. Оставляли на ночь при перемешивании при комнатной температуре, после чего прибавляли 11,7 г (116 ммоль) триэтиламина и затем кипятили с обратным холодильником в течение 2 часов. Полученную суспензию фильтровали и фильтрат концентрировали до сухого состояния. Остаток собирали в 170 мл этилацетата, фильтровали и еще раз концентрировали до сухого состояния. В результате получалось 11 г (выход 55%) (2Z)-2-(2,2-диметил-5-оксо-1,3-диоксолан-4-илиден)-N,N-диметилацетамида.
1H-ЯМР (DMSO-d6, 400 МГц): δ [ppm] = 1,7 (6Н), 2,9 (3H), 3,0 (3H), 6,0 (1Н),
Суспендировали 3,83 г (25,1 ммоль) N,N-диметил-2-(метиламино)ацетамида гидрохлорида (Sigma-Aldrich: CDS007544) и 2,79 г (27,6 ммоль) триэтиламина в 8 мл этилового спирта. Прибавляли 1,00 г (5,02 ммоль) (2Z)-2-(2,2-диметил-5-оксо-1,3-диоксолан-4-илиден)-N,N-диметилацетамида и перемешивали в течение 30 мин при комнатной температуре. Прибавляли 5 мл раствора HCl (6 н) и реакционную смесь концентрировали до сухого состояния. Остаток собирали в 10 мл воды и 20 мл этилацетата. Фазы разделяли и водную фазу экстрагировали два раза этилацетатом. Органические фазы высушивали над сульфатом натрия и концентрировали до сухого состояния. Полученный неочищенный продукт очищали путем хроматографии на колонке (силикагель, дихлорметан/этиловый спирт). В результате получалось 0,94 г (выход 77%) N1-[2-(диметиламино)-2-оксоэтил]-N1,N4,N4-триметил-2-оксобутандиамида.
ИК-спектр (в чистом виде, см-1): 3487, 2935, 1780, 1721, 1632, 1492, 1397, 1354, 1335, 1305, 1259, 1144, 1095, 1054, 981, 942, 857, 813, 760.
Кето-форма:
1H-ЯМР (DMSO-d6, 400 МГц): δ [ppm] = 2,9-3,1 (15Н), 3,9 (2Н), 4,1-4,3 (2Н).
S. Этил-N-[4-(диметиламино)-2,4-диоксобутаноил] глицинат
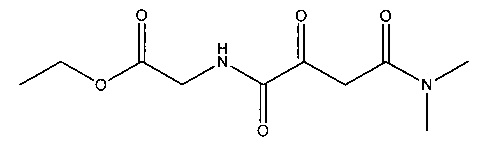
Суспендировали 53,30 г (0,475 моль) трет-бутоксида натрия в 300 мл диэтилового эфира. К этой суспензии прибавляли 73,07 г (0,500 моль) диэтилоксалата, охлаждая на ледяной бане. Перемешивали 30 минут с охлаждением, после чего прибавляли 43,57 г (0,500 моль) N,N-диметилацетамида и реакционную смесь перемешивали в течение 2 часов. Для дальнейшей обработки отфильтровывали образовавшееся твердое вещество и дезинтегрировали его в 260 мл раствора соляной кислоты (4 н) и 750 мл этилацетата. После разделения фаз водную фазу два раза экстрагировали этилацетатом (по 300 мл). Объединенные органические фазы высушивали над сульфатом натрия и концентрировали до сухого состояния с помощью ротационного испарителя. Выход полученного этил-4-(диметиламино)-2,4-диоксобутаноата составлял 54,3 г (58%).
1H-ЯМР (CDCl3, 400 МГц): δ [ppm] = 1,38 (3H), 3,06 (6Н), 4,33 (2Н), 6,25 (1Н).
Суспендировали 32,56 г (163,1 ммоль) моногидрата ацетата меди в 470 мл этилового спирта и нагревали до температуры 75°C. Прибавляли в один прием 61,06 г (326,2 ммоль) этил-4-(диметиламино)-2,4-диоксобутаноата. Полученную смесь перемешивали в течение 40 мин при температуре 75°C. Комплексное соединение меди выпадало в осадок при температуре 4°C.
Растворяли 7,0 г (68 ммоль) этилового эфира глицина (полученного по работе Е. Fischer, Chemische Berichte, 1906, vol, 39, p, 541) в 13 мл этилового спирта, прибавляли 1,0 г (2,3 ммоль) полученного комплексного соединения меди и перемешивали в течение 1 часа при комнатной температуре. После этого реакционную смесь концентрировали до сухого состояния. Для дальнейшей обработки комплексное соединение меди суспендировали в 80 мл дихлорметана и прибавляли 50 мл 10%-ной серной кислоты. После энергичного перемешивания разделяли фазы и водную фазу экстрагировали два раза дихлорметаном (по 50 мл). Объединенные органические фазы высушивали над сульфатом натрия и концентрировали с помощью ротационного испарителя до сухого состояния. Полученный неочищенный продукт очищали путем хроматографии на колонке (силикагель, этилацетат/петролейный эфир). В результате получалось 0,5 г (выход 45%) этил-N-[4-(диметиламино)-2,4-диоксобутаноил]глицината.
ИК-спектр (в чистом виде, см-1): 3389, 2983, 2942, 2324, 2083, 1982, 1739, 1678, 1631, 1608, 1520, 1404, 1354, 1278, 1255, 1179, 1144, 1097, 1084, 1065, 1023, 973, 915, 860, 819, 773, 717.
Енольная форма:
1H-ЯМР (DMSO-d6, 400 МГц): δ [ppm] = 1,2 (3H), 3,0 (6Н), 3,9 (2Н), 4,1 (2Н), 6,2 (1H), 8,7 (1Н), 15,5 (1Н).
T. N1-(2-Метоксиэтил)-N1,N4,N4-триметил-2-оксобутандиамид
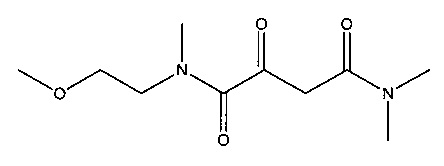
Растворяли 19,5 г (219 ммоль) N-(2-метоксиэтил)метиламина и 22,2 г (219 ммоль) триэтиламина в 400 мл диэтилового эфира и охлаждали на ледяной бане. Прибавляли по каплям 30,0 г (219 ммоль) этилхлороксоацетата и реакционную смесь оставляли согреваться до комнатной температуры. Полученную суспензию фильтровали и фильтрат концентрировали до сухого состояния. В результате получали 36,9 г этил-[(2-метоксиэтил)(метил)амино](оксо)ацетата в виде желтого масла, который, не очищая, использовали для дальнейшей обработки.
Суспендировали 20,8 г (185 ммоль) трет-бутоксида натрия в 200 мл диэтилового эфира. Прибавляли 36,9 г (195 ммоль) этил-[(2-метоксиэтил)(метил)амино](оксо)ацетата, охлаждая на ледяной бане. Перемешивали в течение 30 мин при комнатной температуре. Затем прибавляли 17,0 г (195 ммоль) N,N-диметилацетамида и реакционную смесь перемешивали в течение 2 часов при комнатной температуре. В реакционную смесь добавляли 123 мл раствора соляной кислоты (6 н), 130 мл этилацетата и 30 мл воды. После разделения фаз водную фазу экстрагировали три раза этилацетатом (по 100 мл). Объединенные органические фазы высушивали над сульфатом натрия и концентрировали с помощью ротационного испарителя до сухого состояния. Полученный неочищенный продукт очищали путем хроматографии на колонке (силикагель, дихлорметан/ацетон). В результате получали 4,5 г (выход 10%) N1-(2-метоксиэтил)-N1,N4,N4-триметил-2-оксобутанамида.
ИК-спектр (в чистом виде, см-1): 3482, 2935, 1720, 1631, 1492, 1454, 1401, 1355, 1293, 1260, 1198, 1115, 1069, 1014, 972, 828, 772721.
Кето-форма:
1H-ЯМР (DMSO-d6, 400 МГц): δ [ppm] = 2,8-3,0 (9Н), 3,3 (3H), 3,4-3,5 (4Н), 3,9 (2Н).
U. N4-(2-Гидроксиэтил)-N1,N1,N4-триметил-2-оксобутандиамид
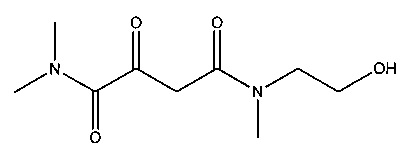
Растворяли 1,18 г (15,7 ммоль) 2-(метиламино)этанола в 50 мл дихлорметана и прибавляли 2,39 г (23,6 ммоль) триэтиламина. В эту смесь добавляли 3,00 г (15,7 ммоль) (2Z)-(2,2-диметил-5-оксо-1,3-диоксолан-4-илиден)ацетилхлорида (полученного, как описано в работе J. Banville et al, Tetrahedron Letters, 2010, 51, 3170-3173) и перемешивали в течение 30 мин при комнатной температуре. Затем кипятили с обратным холодильником в течение 4 часов. Реакционную смесь концентрировали до сухого состояния, и остаток собирали в 300 мл этилацетата. Полученную суспензию фильтровали и фильтрат концентрировали до сухого состояния. Остаток очищали путем хроматографии на колонке (силикагель, хлорметан/этиловый спирт). В результате получали 1,5 г (выход 42%) (2Z)-2-(2,2-диметил-5-оксо-1,3-диоксолан-4-илиден)-N-(2-гидроксиэтил)-N-метилацетамида.
1H-ЯМР (DMSO-d6, 400 МГц): δ [ppm] = 1,7 (6Н), 3,0 (3H), 3,4-3,6 (4Н), 4,7-4,9 (1Н), 6,0-6,2 (1H).
Растворяли 1,0 г (4,4 ммоль) (2Z)-2-(2,2-диметил-5-оксо-1,3-диоксолан-4-илиден)-N-(2-гидроксиэтил)-N-метилацетамида в 8 мл раствора диметиламина в этиловом спирте (33%) и перемешивали в течение 15 мин при комнатной температуре. Эту смесь концентрировали до сухого состояния, в результате чего получали 0,95 г (выход 99%) N4-(2-гидроксиэтил)-N1,N1,N4-триметил-2-оксобутандиамида.
ИК-спектр (в чистом виде, см-1): 3401, 2935, 1720, 1626, 1490, 1448, 1401, 1359, 1297, 1261, 1208, 1110, 1074, 1047, 950, 880, 863, 806, 773, 719.
Кето-форма:
1H-ЯМР (DMSO-d6, 400 МГц): δ [ppm] = 2,8-3,1 (9Н), 3,3-3,5 (4Н), 3,9-4,0 (2Н), 4,9 (1Н).
Способ анализа
Утилизацию железа, происходящую при использовании комплексных соединений железа по данному изобретению, оценивали с помощью модели на мышах.
(В каждом случае брали 6 особей на одно вещество. Для отрицательного контроля брали 6 особей, получавших воду, и для положительного контроля брали 6 особей, получавших FeSO4.)
Использовали мышей самцов линии NMRI (SPF) возрастом приблизительно 3 недели, которые получали корм с низким содержанием железа (приблизительно 5 ppm) в течение приблизительно 3 недель. Затем животным вводили (с помощью желудочного зонда) комплексные соединения железа в дозировке 2 мг железа/кг массы тела/сутки на протяжении двух периодов продолжительностью по 5 сут с интервалом 2 сут (дни 1-5 и 8-12). На 15-й день определяли утилизацию железа по возрастанию уровня гемоглобина (Hb) и увеличению массы тела (BW) по формуле

0,07 = Коэффициент для 70 мл крови на 1 кг массы тела (BW)
0,0034 = Коэффициент для 0,0034 г Fe/г Hb
Hb1 = Уровень гемоглобина (г/л) в День 1.
Hb2(3) = Уровень гемоглобина (г/л) в День 8 (или 15)
BW4 = Масса тела (г) в День 1
BW9(14) = Масса тела (г) в День 8 (или 15)
Hb1 Контроль = Средний уровень гемоглобина (г/л) в День 1 в контрольной группе
Hb2(3) Контроль = Средний уровень гемоглобина (г/л) в День 8 (или 15) в контрольной группе
BW4 Контроль = Средняя масса тела (г) в День 1 в контрольной группе BW9(14) Контроль = Средняя масса тела (г) в День 8 (или 15) в контрольной группе
Доза Fe = Все введенное количество Fe (мг) за 5 или 10 суток
Fe ут. = (Hb2(3) * BW9(14) - Hb1 * BW4) * 0,07 * 0,0034 (mg Fe)
Δ Утилизации = Fe суммарное утилизованное (опытная группа) - Fe утилизованное контрольная группа, утилизованное из пищи (мг Fe).
Приведенная ниже таблица 1 демонстрирует утилизацию соединений по примеру 1 и сравнение соответственных величин, полученных для соединения по примеру 32 публикации WO 11117225 A1:
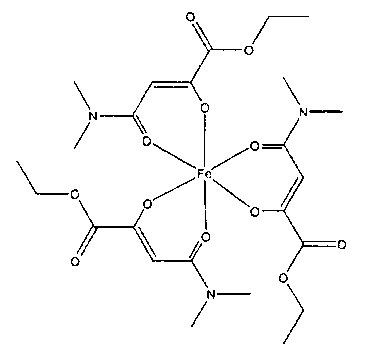

Приведенная ниже таблица демонстрирует, что структурные изменения соединений по сравнению с таковыми из WO 11117225 A1, как правило, улучшают утилизацию.
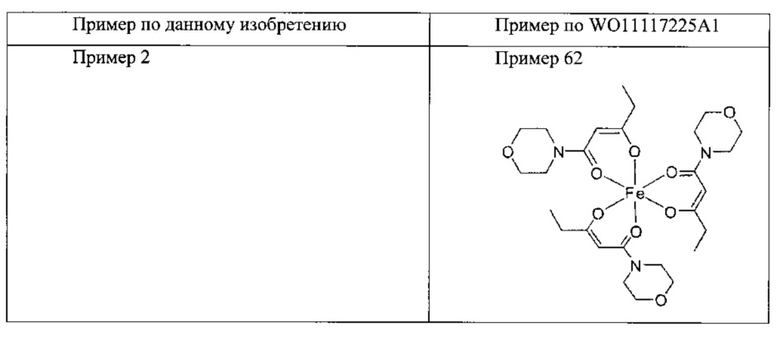
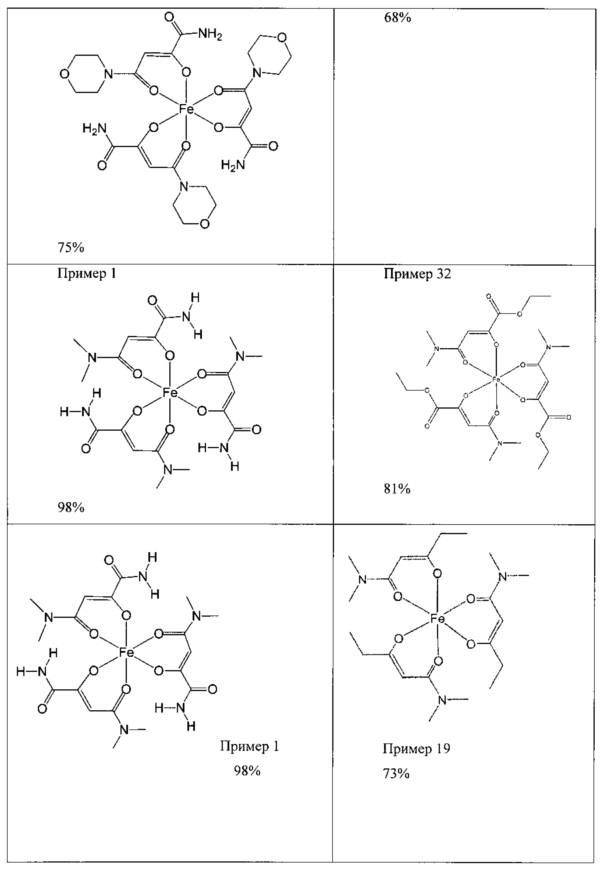
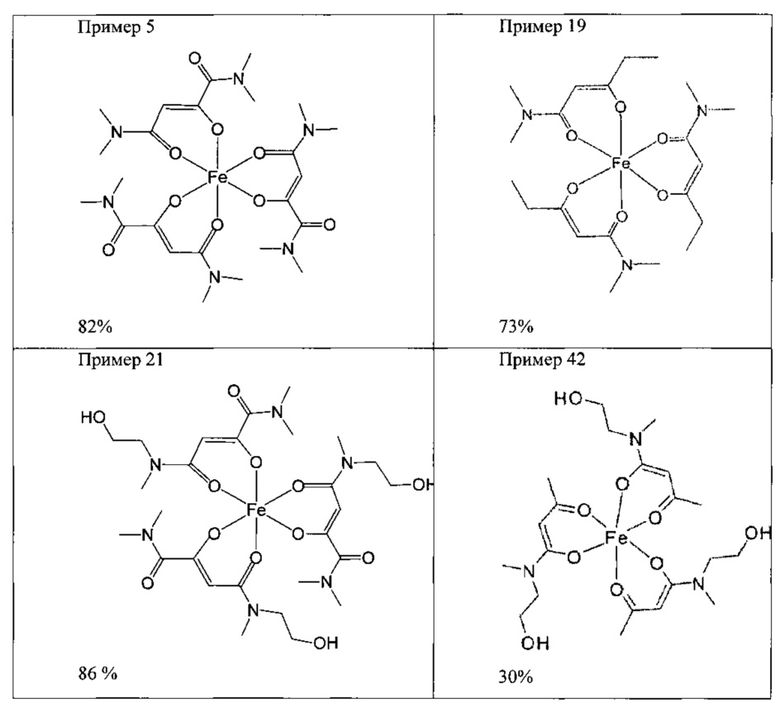
Количественное определение утилизации железа дает очень важный показатель с точки зрения назначения лечения дефицита железа и железодефицитной анемии, потому что этот показатель отражает не только поглощение железа, но и взаимосвязь между массой тела и потреблением железа, что особенно существенно в случае использования в животной модели растущих особей. Если рассматривать только величины по гемоглобину, представляющие реально поглощенное и использованное железо, то та часть, которая обусловлена ростом животного, остается неучтенной. Таким образом, величина утилизации железа является более точной мерой, хотя эта величина и уровень гемоглобина обычно коррелируют друг с другом. Рассмотрение только уровня железа в сыворотке крови, который тоже подлежит количественному определению, менее ценно, так как, хотя эта величина указывает на количество железа, поступившее в организм, но не дает указания на количество железа, используемое организмом.
Результаты проделанного анализа демонстрируют, что комплексные соединения железа по данному изобретению обеспечивают превосходную утилизацию железа, так что они полезны в качестве агентов для лечения железодефицитной анемии и связанных с ней симптомов.
Результаты проделанного анализа также демонстрируют, что в случае комплексного соединения железа по примеру 1 настоящей заявки наблюдается значительно лучшая утилизация железа по сравнению с примером 32 публикации WO 11117225 A1.
Сравнение растворимости, зависимой от pH
Для этой цели определенное количество комплексного соединения трехвалентного железа (навеску), подлежащего исследованию, обычно в концентрации 0,4-0,8 мг/мл (исходя из количества железа в комплексном соединении) помещали в водную среду (воду) с доведенным pH (pH 6,5) и перемешивали указанное время (4 часа) при температуре 25°C. Доведение pH до 6,5 осуществляли с помощью фталатного буферного раствора (0,1 М). Затем определяли содержание железа в растворе (мг железа/мл) путем фотометрического анализа (кюветы: одноразовые кюветы 1 см Plastibrand PS 2,5 мл стандарты ISO 9001 14001; прибор: спектрофотометр ультрафиолетового диапазона SPECORD 205 производства Jena Analytik). Получаемая величина представляет растворимость комплексного соединения железа. Если отвешенное количество комплексного соединения железа растворялось полностью, измеренное значение указывалось значком 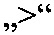 . Результаты этого определения показаны в таблице 2 ниже. Видно, что соединения по данному изобретению обладают значительно большей растворимостью, чем соединении по примеру 32 из WO 11117225 A1.
. Результаты этого определения показаны в таблице 2 ниже. Видно, что соединения по данному изобретению обладают значительно большей растворимостью, чем соединении по примеру 32 из WO 11117225 A1.
Комплексные соединения трехвалентного железа по данному изобретению предпочтительно обладают растворимостью в воде при температуре 25°C и pH 6,5 по меньшей мере 0,3 мг железа/мл (определение проводили фотометрически, как описано выше). Предпочтительные комплексные соединения трехвалентного железа имеют растворимость в воде при температуре 25°C и pH 6,5 по меньшей мере 0,4 мг железа/мл. Особенно предпочтительные комплексные соединения трехвалентного железа имеют растворимость в воде при температуре 25°C и pH 6,5 по меньшей мере 0,5 мг железа/мл.
Хорошая растворимость в воде комплексных соединений трехвалентного железа - необходимое предварительное условие для улучшенного поглощения железа при пероральном приеме. Чем лучше водорастворимость перорального препарата, тем лучше организм поглощает железо. Как правило, не растворимые в воде соединения нельзя вводить в организм перорально, так как они практически не поглощаются.
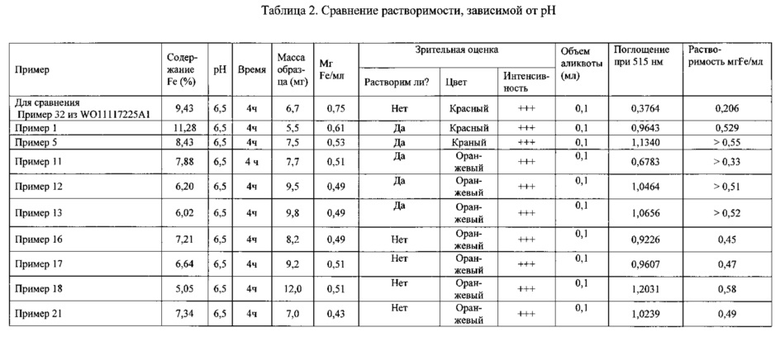
Примеры получения
Пример 1. Комплексное соединение трис-(N4,N4-диметил-2-оксобутандиамид)-Fe(III)
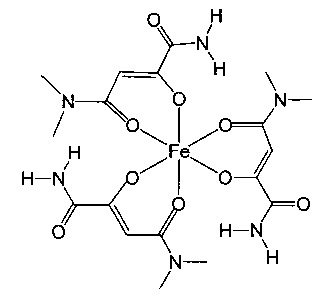
Растворяли 1,75 г (8,78 ммоль) ацетата трехвалентного железа в 160 мл 94%-ного этилового спирта и прибавляли 5,01 г (31,6 ммоль) N4,N4-диметил 2-оксобутандиамида. Смесь кипятили с обратным холодильником в течение 1 часа. Образовавшееся комплексное соединение железа выпадало в осадок при температуре 4°C. Осадок комплексного соединения железа отфильтровывали и высушивали в вакууме при температуре 50°C в течение суток. В результате получалось 3,74 г (выход 81%) искомого продукта в виде твердого вещества красного цвета.
ИК-спектр (в чистом виде, см-1): 3437, 3405, 3153, 2931, 2323, 1680, 1594, 1567, 1498, 1403, 1352, 1258, 1171, 1142, 1058, 1001, 936, 814, 766, 735, 686.
Элементный состав: С 41,17%, H 5,2%, N 15,9%.
Содержание железа: 10,3% [масса/масса].
Пример 2. Комплексное соединение трис-(4-(морфолин-4-ил)-2,4-диоксобутанамид)-Fe(III)
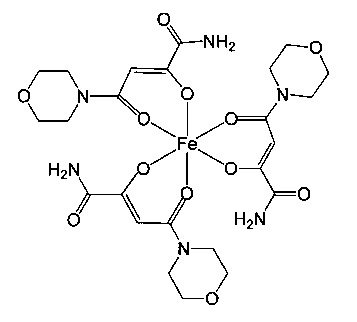
Растворяли 0,60 г (3,1 ммоль) ацетата трехвалентного железа в 150 мл 94%-ного этилового спирта и прибавляли 2,00 г (10,0 ммоль) 4-(морфолин-4-ил)-2,4-диоксобутандиамида. Смесь кипятили с обратным холодильником. После добавления затравочных кристаллов комплексное соединение железа выпадало в осадок при температуре 4°C и его отфильтровывали. Полученный продукт высушивали в вакууме в течение суток при температуре 50°C. В результате получалось 1,1 г (выход 54%) искомого продукта в виде твердого вещества красного цвета.
ИК-спектр (в чистом виде, см-1): 3608, 3352, 3135, 2960, 2918, 2850, 2324, 2050, 1981, 1685, 1570, 1498, 1460, 1443, 1373, 1301, 1273, 1245, 1145, 1112, 1050, 1017, 986, 936, 850, 817, 764, 737, 676.
Элементный состав: С 42,71%, Н 5,7%, N 11,94%.
Содержание железа: 8,3% [масса/масса].
Пример 3. Комплексное соединение трис-(N4,N4-диэтил-2-оксобутандиамид)-Fe(III)
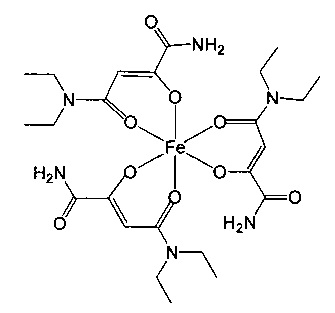
Растворяли 0,13 г (0,83 ммоль) хлорида трехвалентного железа в 10 мл воды и прибавляли 0,50 г (2,5 ммоль) N4,N4-диэтил-2-оксобутандиамида. Перемешивали в течение 15 минут при комнатной температуре, после чего смесь охлаждали на ледяной бане. Прибавляли 0,45 г (3,3 ммоль) тригидрата ацетата натрия, затем перемешивали еще 15 минут на ледяной бане. Выпавшее в осадок комплексное соединение железа отфильтровывали и высушивали в высоком вакууме в течение ночи при температуре 50°C. В результате получали 0,5 г (выход 92%) искомого продукта в виде твердого вещества оранжевого цвета.
ИК-спектр (в чистом виде, см-1): 3458, 3301, 2975, 2935, 1681, 1618, 1566, 1496, 1453, 1437, 1376, 1355, 1307, 1270, 1215, 1160, 1078, 1035, 964, 925, 818, 771, 743, 659.
Элементный состав: С 46,31%, Н 6,5%, N 13,56%.
Содержание железа: 9,1% [масса/масса].
Пример 4. Комплексное соединение трис-(2,4-диоксо-4-(пирролидин-1-ил)бутанамид)-Fe(III)
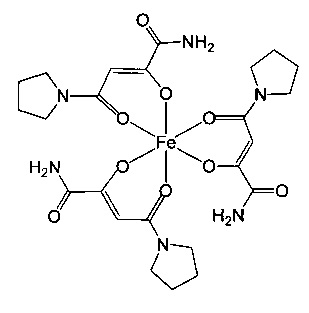
Суспендировали 0,28 г (1,4 ммоль) ацетата трехвалентного железа в 10 мл этилацетата и прибавляли 0,80 г (4,3 ммоль) 2,4-диоксо-4-(пирролидин-1-ил)бутанамида. Реакционную смесь нагревали до температуры 50°C. Затем прибавляли 30 мл этилацетата и 20 мл этилового спирта и полученную суспензию нагревали до температуры 90°C. После этого реакционную смесь охлаждали на ледяной бане. Выпавшее в осадок комплексное соединение железа отфильтровывали и высушивали в высоком вакууме в течение 3 суток при температуре 50°C. В результате получали, 0,67 г (выход 62%) искомого продукта в виде твердого вещества красного цвета.
ИК-спектр (в чистом виде, см-1): 3454, 3367, 3178, 2968, 2877, 1685, 1627, 1560, 1493, 1474, 1457, 1367, 1254, 1225, 1182, 1134, 1116, 1059, 1026, 972, 938, 916, 856, 814, 769, 717, 659.
Элементный состав: С 47,45%, Н 5,6%, N 13,32%.
Содержание железа: 8,8% [масса/масса].
Пример 5. Комплексное соединение трис-(N,N,N`,N`-тетраметил-2-оксобутандиамид)-Fe(III)
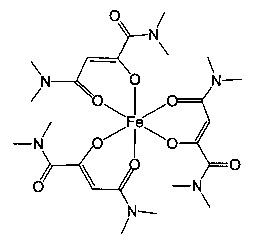
Растворяли 0,54 г (3,3 ммоль) хлорида трехвалентного железа в 20 мл воды и прибавляли 2,0 г (10 ммоль) N,N,N`,N`-тетраметил-2-оксобутандиамида, Перемешивали в течение 15 минут при комнатной температуре, после чего реакционную смесь охлаждали на ледяной бане. Прибавляли 1,8 г (13 ммоль) тригидрата ацетата натрия и перемешивали еще 10 минут на ледяной бане. Эту водную реакционную смесь экстрагировали хлороформом три раза по 50 мл. Объединенные органические фазы концентрировали с помощью ротационного испарителя до сухого состояния и еще два раза выпаривали с 50 мл толуола. Остаток высушивали в высоком вакууме в течение ночи при температуре 50°C. В результате получали 1,6 г (выход 73%) искомого продукта в виде твердого вещества красного цвета.
ИК-спектр (в чистом виде, см-1): 3496, 2929, 1634, 1568, 1486, 1392, 1354, 1259, 1203, 1174, 1118, 1059, 1006, 962, 900, 811, 771, 710, 665. Элементный состав: С 45,95%, Н 6,4%, N 13,19%. Содержание железа: 9,1% [масса/масса].
Пример 6. Комплексное соединение трис-(4-(4-гидроксипиперидин-1-ил)-2,4-диоксобутанамид)-Fe(III)
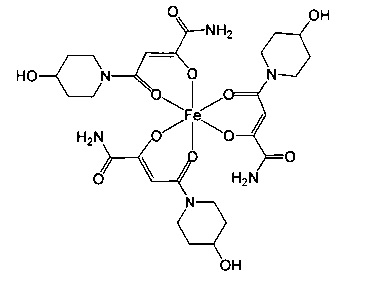
Суспендировали 144 мг (0,734 ммоль) ацетата трехвалентного железа в 50 мл этилового спирта и прибавляли 471 мг (2,20 ммоль) 4-(4-гидроксипиперидин-1-ил)-2,4-диоксобутанамида. Реакционную смесь кипятили с обратным холодильником в течение 1 часа. Затем прибавляли 60 толуола и концентрировали до сухого состояния. Остаток дважды собирали смесью этиловый спирт/толуол (40 мл/60 мл) и концентрировали до сухого состояния. Полученный материал высушивали в высоком вакууме при температуре 50°C. В результате получали 0,5 г (выход 98%) искомого продукта в виде твердого вещества красного цвета.
ИК-спектр (в чистом виде, см-1): 3445, 3298, 2946, 2924, 2324, 2051, 1981, 1687, 1666, 1598, 1561, 1493, 1448, 1370, 1262, 1227, 1138, 1075, 1050, 1023, 985, 928, 834, 816, 767, 664.
Элементный состав: С 46,26%, Н 5,8%, N 11,60%.
Содержание железа: 7,7% [масса/масса].
Пример 7. Комплексное соединение трис-(N1,N4,N4-триметил-2-оксобутандиамид)-Fe(III)
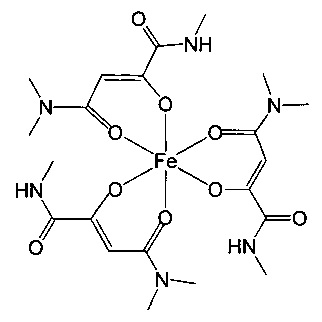
Растворяли 0,784 г (4,83 ммоль) хлорида трехвалентного железа в 20 мл воды и прибавляли 2,55 г (14,5 ммоль) N1,N4,N4-триметил-2-оксобутандиамида. Перемешивали в течение 15 минут при комнатной температуре, после чего реакционную смесь охлаждали на ледяной бане. Прибавляли 2,16 г (19,2 ммоль) тригидрата ацетата натрия и перемешивали в течение 15 минут на ледяной бане. Выпавшее в осадок комплексное соединение железа отфильтровывали и высушивали в высоком вакууме в течение ночи при температуре 50°C. В результате получали 1,5 г (выход 53%) искомого продукта в виде твердого вещества оранжевого цвета.
ИК-спектр (в чистом виде, см-1): 3352, 2934, 2877, 2324, 1674, 1598, 1566, 1504, 1405, 1356, 1253, 1176, 1152, 1063, 992, 980, 961, 930, 896, 847, 813, 769, 748, 736, 706.
Элементный состав: С 43,74%, Н 5,9%, N 14,53%.
Содержание железа: 9,5% [масса/масса].
Пример 8. Комплексное соединение трис-(N,N`-диметил-2-оксобутандиамид)-Fe(III)
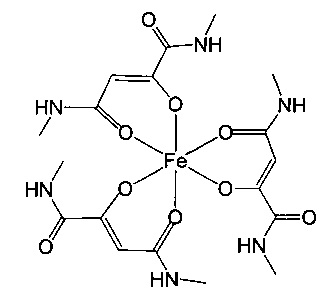
Растворяли 242 мг (1,49 ммоль) хлорида трехвалентного железа в 10 мл воды и прибавляли 707 мг (4,47 ммоль) N,N`-диметил-2-оксобутандиамида. Перемешивали в течение 15 минут при комнатной температуре, после чего прибавляли 812 мг (5,97 ммоль) тригидрата ацетата натрия и перемешивали еще 30 минут. Выпавшее в осадок комплексное соединение железа отфильтровывали и высушивали в высоком вакууме в течение 4 суток при температуре 50°C. В результате получали 587 мг (выход 74%) искомого продукта в виде твердого вещества оранжевого цвета.
ИК-спектр (в чистом виде, см-1): 3334, 3256, 3129, 2940, 1673, 1603, 1546, 1507, 1407, 1278, 1239, 1160, 1139, 1087, 1028, 963, 897, 817, 800, 777, 724, 691.
Элементный состав: С 40,04%, Н 5,1%, N 15,49%.
Содержание железа: 10,2% [масса/масса].
Пример 9. Комплексное соединение трис-(N1,N1,N4-триметил-2-оксобутандиамид)-Fe(III)
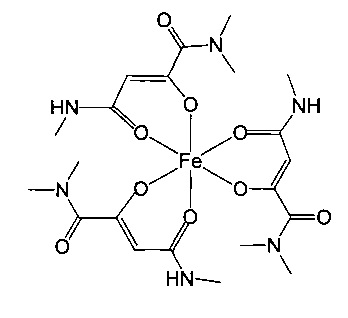
Растворяли 75 мг (0,46 ммоль) хлорида трехвалентного железа в 3 мл воды и прибавляли 239 мг (1,39 ммоль) N1,N1,N4-триметил-2-оксобутандиамида. Перемешивали в течение 15 минут при комнатной температуре, после чего прибавляли 252 мг (1,85 ммоль) тригидрата ацетата натрия и перемешивали еще 30 минут. Реакционную смесь концентрировали до сухого состояния, и остаток собирали в 20 мл хлороформа. Не растворившийся материал отфильтровывали, а фильтрат концентрировали до сухого состояния. Высушивали в высоком вакууме в течение ночи при температуре 50°C. В результате получали 230 мг искомого продукта чистотой 78%.
ИК-спектр (в чистом виде, см-1): 3270, 3118, 2932, 2051, 1578, 1491, 1433, 1395, 1278, 1192, 1157, 1123, 1076, 966, 902, 778.
Элементный состав: С 41,89%, Н 6,1%, N 11,20%.
Содержание железа: 7,7% [масса/масса].
Пример 10. Комплексное соединение трис-(N4,N4-диэтил-N1,N1-диметил-2-оксобутандиамид)-Fe(III)
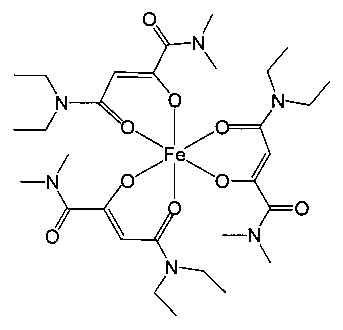
Растворяли 0,49 г (3,0 ммоль) хлорида трехвалентного железа в 20 мл воды и прибавляли 2,0 г (9,0 ммоль) N4,N4-диэтил-N1,N1-диметил-2-оксобутандиамида. Перемешивали в течение 15 минут при комнатной температуре, после чего смесь охлаждали на ледяной бане. Прибавляли 1,63 г (11,8 ммоль) тригидрата ацетата натрия и перемешивали в течение 30 минут. Затем реакционную смесь экстрагировали три раза хлороформом (по 50 мл). Полученный материал высушивали над сульфатом натрия и концентрировали до сухого состояния. Остаток трижды десорбировали толуолом (по 50 мл), чтобы удалить уксусную кислоту. Остаток высушивали в высоком вакууме в течение ночи при температуре 50°C. В результате получали 1,8 г (выход 83%) искомого продукта в виде твердого вещества вино-красного цвета.
ИК-спектр (в чистом виде, см-1): 3481, 2973, 2933, 2050, 1636, 1602, 1563, 1511, 1487, 1435, 1392, 1376, 1357, 1308, 1274, 1218, 1198, 1160, 1120, 1080, 1036, 964, 929, 887, 808, 789, 767, 707.
Элементный состав: С 51,12%, Н 7,2%, N 11,68%.
Содержание железа: 8,0% [масса/масса].
Пример 11. Комплексное соединение трис-(N,N-диметил-3,4-диоксо-4-(пирролидин-1-ил)бутанамид)-Fe(III)
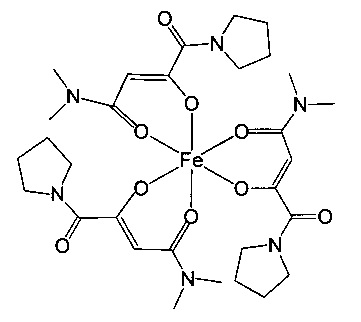
Растворяли 0,48 г (3,0 ммоль) хлорида трехвалентного железа в 20 мл воды и прибавляли 2,0 г (8,9 ммоль) N,N-диметил-3,4-диоксо-4-(пирролидин-1-ил)бутанамида. Перемешивали в течение 15 минут при комнатной температуре, после чего смесь охлаждали на ледяной бане. Прибавляли 1,61 г (11,8 ммоль) тригидрата ацетата натрия и перемешивали еще 30 минут. Реакционную смесь экстрагировали хлороформом три раза по 50 мл, затем высушивали над сульфатом натрия и концентрировали до сухого состояния. Остаток десорбировали толуолом три раза по 50 мл. Остаток высушивали в высоком вакууме в течение ночи при температуре 50°C. В результате получали 2,1 г (выход 97%) искомого продукта в виде твердого вещества винно-красного цвета.
ИК-спектр (в чистом виде, см-1): 3465, 2949, 2875, 2324, 2051, 1694, 1605, 1563, 1504, 1402, 1357, 1294, 1255, 1225, 1162, 1061, 1012, 980, 960, 920, 896, 850, 809, 762, 705.
Элементный состав: С 51,51%. Н 6,4%. N 11,73%.
Содержание железа: 8,1% [масса/масса].
Пример 12. Комплексное соединение трис-(N4-[2-(диметиламино)-2-оксоэтил]-N1,N1,N4-триметил-2-оксобутандиамид)-Fe(III)
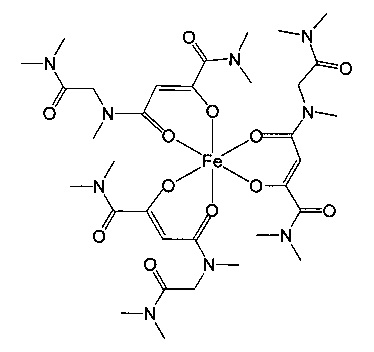
Растворяли 0,40 г (2,5 ммоль) хлорида трехвалентного железа в 20 мл воды и прибавляли 2,0 г (7,5 ммоль) N4-[2-(диметиламино)-2-оксоэтил]-N1,N1,N4-триметил-2-оксобутандиамида, растворенного в 3 мл этилового спирта. Перемешивали в течение 15 минут при комнатной температуре, после чего прибавляли 1,4 г (9,9 ммоль) тригидрата ацетата натрия и перемешивали еще 30 минут. Реакционную смесь экстрагировали хлороформом три раза по 50 мл, затем высушивали над сульфатом натрия и концентрировали до сухого состояния. Остаток десорбировали толуолом два раза по 50 мл. Высушивали в высоком вакууме в течение ночи при температуре 50°C. В результате получали 2,1 г (выход 98%) искомого продукта в виде твердого вещества красного цвета.
ИК-спектр (в чистом виде, см-1): 3453, 2933, 2324, 2164, 2051, 1981, 1628, 1566, 1510, 1485, 1395, 1364, 1335, 1298, 1254, 1218, 1143, 1112, 1060, 1039, 964, 905, 825, 808, 763, 713, 668.
Элементный состав: С 46,06%. Н 6,8%. N 14,46%.
Содержание железа: 6,2% [масса/масса].
Пример 13. Комплексное соединение трис-(N4-(2-метоксиэтил)-N1,N1,N4-триметил-2-оксобутандиамид)-Fe(III)
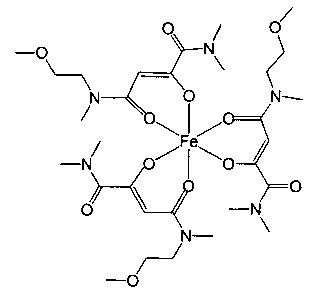
Растворяли 0,44 г (2,7 ммоль) хлорида трехвалентного железа в 20 мл воды и прибавляли 1,9 г (8,1 ммоль) N4-(2-метоксиэтил)-N1,N1,N4-триметил-2-оксобутандиамида. Перемешивали в течение 15 минут при комнатной температуре, после чего прибавляли 1,47 г (10,8 ммоль) тригидрата ацетата натрия и перемешивали еще 20 минут. Реакционную смесь экстрагировали хлороформом три раза по 50 мл, затем высушивали над сульфатом натрия и концентрировали до сухого состояния. Остаток десорбировали толуолом два раза по 50 мл. Высушивали в высоком вакууме в течение 3 суток при температуре 50°C. В результате получали 1,8 г искомого продукта чистотой 80%. ИК-спектр (в чистом виде, см-1): 3487, 2929, 1719, 1635, 1602, 1564, 1514, 1486, 1391, 1359, 1302, 1261, 1197, 1164, 1112, 1064, 1033, 1004, 964, 906, 889, 827, 807, 770, 713, 664. Элементный состав: С 48,17%. Н 6,9%. N 10,98%. Содержание железа: 6,0% [масса/масса].
Пример 14. Комплексное соединение трис-(N4-(2-метоксиэтил)-N1,N1-диметил-2-оксобутандиамид)-Fe(III)
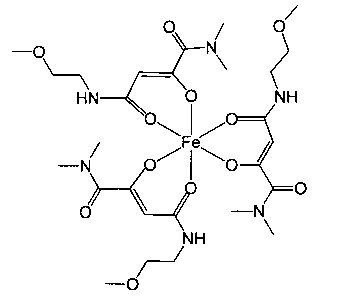
Растворяли 358 мг (2,20 ммоль) хлорида трехвалентного железа в 16 мл воды и прибавляли 1,43 г (6,61 ммоль) N4-(2-метоксиэтил)-N1,N1-диметил-2-оксобутандиамида. Перемешивали в течение 15 минут при комнатной температуре, после чего прибавляли 1,2 г (8,82 ммоль) тригидрата ацетата натрия и перемешивали еще 30 минут. Реакционную смесь экстрагировали хлороформом пять раз по 50 мл, затем высушивали над сульфатом натрия и концентрировали до сухого состояния. Остаток десорбировали толуолом два раза по 50 мл. Высушивали в высоком вакууме в течение трех суток при температуре 50°C. В результате получали 1,39 г искомого продукта чистотой 83%.
ИК-спектр (в чистом виде, см-1): 3274, 3119, 2930, 1720, 1622, 1569, 1523, 1491, 1437, 1393, 1273, 1193, 1118, 1085, 1025, 962, 861, 777, 721, 691.
Элементный состав: С 45,27%. Н 6,5%. N 11,64%.
Содержание железа: 6,6% [масса/масса].
Пример 15. Комплексное соединение трис-(этил-N-[4-(диметиламино)-3,4-диоксобутаноил]-N-метилглицинат)-Fe(III)
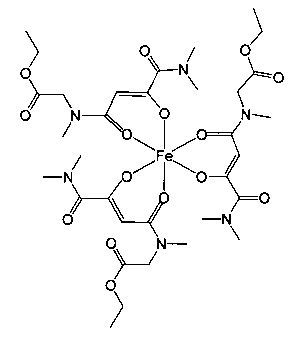
Растворяли 237 мг (1,46 ммоль) хлорида трехвалентного железа в 16 мл воды и прибавляли 1,13 г (4,38 ммоль) этил-N-[4-(диметиламино)-3,4-диоксобутаноил]-N-метилглицината. Перемешивали в течение 15 минут при комнатной температуре, после чего прибавляли 794 мг (5,84 ммоль) тригидрата ацетата натрия и перемешивали еще 30 минут. Реакционную смесь экстрагировали хлороформом пять раз, высушивали над сульфатом натрия и концентрировали до сухого состояния. Остаток десорбировали толуолом два раза по 50 мл. Высушивали в высоком вакууме в течение трех суток при температуре 50°C. В результате получали 860 мг (выход 71%) искомого продукта.
ИК-спектр (в чистом виде, см-1): 3468, 2936, 2324, 2051, 1981, 1739, 1634, 1603, 1563, 1515, 1487, 1444, 1392, 1362, 1294, 1256, 1195, 1149, 1113, 1044, 1022, 964, 905, 861, 817, 764, 707, 667. Элементный состав: С 46,67%. Н 6,1%. N 9,80%. Содержание железа: 6,6% [масса/масса].
Пример 16. Комплексное соединение трис-(N1-(2-метоксиэтил)-N4,N4-диметил-2-оксобутанамид)-Fe(III)
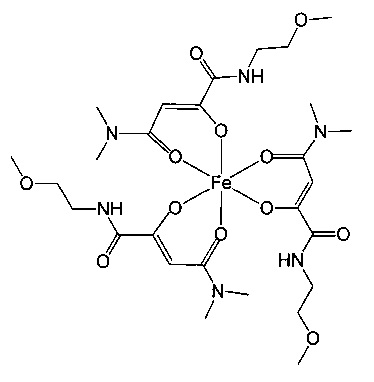
Растворяли 0,49 г (3,0 ммоль) хлорида трехвалентного железа в 10 мл воды и прибавляли 2,0 г (9,1 ммоль) N1-(2-метоксиэтил)-N4,N4-диметил-2-оксобутанамида. Перемешивали в течение 15 минут при комнатной температуре, после чего реакционную смесь охлаждали на ледяной бане. Прибавляли 1,66 г (12,2 ммоль) тригидрата ацетата натрия и перемешивали еще 15 минут. Реакционную смесь экстрагировали хлороформом три раза, высушивали над сульфатом натрия и концентрировали до сухого состояния. Остаток десорбировали толуолом два раза по 50 мл. Высушивали в высоком вакууме в течение ночи при температуре 50°C. В результате получали 1,5 г (выход 70%) искомого продукта в виде твердого вещества красного цвета.
ИК-спектр (в чистом виде, см-1): 3397, 2929, 2885, 1671, 1619, 1580, 1516, 1480, 1403, 1353, 1259, 1173, 1149, 1117, 1088, 1026, 1001, 941, 901, 881, 815, 769, 734. Элементный состав: С 44,99%. Н 6,6%. N 11,43%. Содержание железа: 7,7% [масса/масса].
Пример 17. Комплексное соединение трис-(этил-N-[4-(диметиламино)-2,4-диоксобутаноил]-N-метилглицинат)Fe(III)
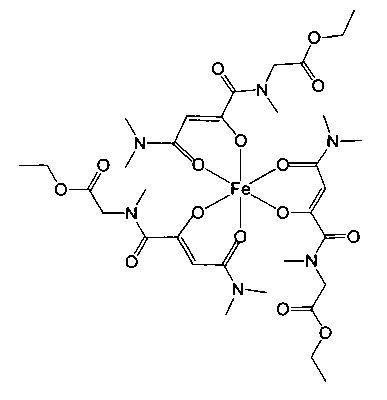
Растворяли 0,21 г (1,3 ммоль) хлорида трехвалентного железа в 10 мл воды и прибавляли 1,02 г (3,8 ммоль) этил-N-[4-(диметиламино)-2,4-диоксобутаноил]-N-метилглицината. Перемешивали в течение 15 минут при комнатной температуре, после чего смесь охлаждали на ледяной бане. Прибавляли 0,71 г (5,2 ммоль) тригидрата ацетата натрия и перемешивали еще 15 минут. Реакционную смесь экстрагировали хлороформом три раза по 50 мл, высушивали над сульфатом натрия и концентрировали до сухого состояния. Остаток десорбировали толуолом два раза по 50 мл. Высушивали в высоком вакууме в течение ночи при температуре 50°C. В результате получали 0,9 г (выход 82%) искомого продукта в виде масла красного цвета.
ИК-спектр (в чистом виде, см-1): 3485, 2981, 2936, 1738, 1641, 1601, 1571, 1508, 1477, 1444, 1397, 1357, 1297, 1258, 1199, 1176, 1105, 1027, 989, 951, 905, 865, 814, 766, 723, 662.
Элементный состав: С 47,10%. Н 6,7%. N 9,11%.
Содержание железа: 6,6% [масса/масса].
Пример 18. Комплексное соединение трис-(N1-[2-(диметиламино)-2-оксоэтил]-N1,N4,N4-триметил-2-оксобутандиамид)-Fe(III)
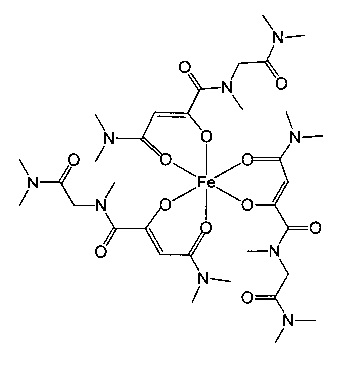
Растворяли 0,11 г (0,70 ммоль) хлорида трехвалентного железа в 10 мл воды и прибавляли 0,54 г (2,1 ммоль) N1-[2-(диметиламино)-2-оксоэтил]-N1,N4,N4-триметил-2-оксобутандиамида в 0,1 мл этилового спирта. Перемешивали в течение 15 минут при комнатной температуре, после чего прибавляли 0,38 г (2,8 ммоль) тригидрата ацетата натрия и перемешивали еще 15 минут. Реакционную смесь экстрагировали хлороформом пять раз по 50 мл, затем высушивали над сульфатом натрия и концентрировали до сухого состояния. Остаток десорбировали толуолом два раза по 50 мл. Высушивали в высоком вакууме в течение ночи при температуре 50°C. В результате получали 0,52 г искомого продукта чистотой 85%.
ИК-спектр (в чистом виде, см-1): 3455, 2934, 1779, 1630, 1572, 1508, 1479, 1398, 1357, 1303, 1259, 1178, 1152, 1106, 1060, 1027, 989, 945, 905, 823, 804, 765, 720.
Элементный состав: С 43,02%. Н 6,9%. N 13,05%.
Содержание железа: 5,8% [масса/масса].
Пример 19. Комплексное соединение трис-(этил-N-[4-(диметиламино)-2,4-диоксобутаноил]глицинат)-Fe(III)
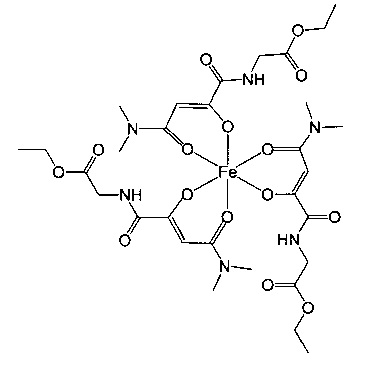
Растворяли 97 мг (0,60 ммоль) хлорида трехвалентного железа в 10 мл воды и прибавляли 459 мг (1,80 ммоль) этил-N-[4-(диметиламино)-2,4-диоксобутаноил]глицината. Перемешивали в течение 15 минут при комнатной температуре, после чего смесь охлаждали на ледяной бане и прибавляли 327 мг (2,40 ммоль) тригидрата ацетата натрия и перемешивали еще 20 минут. Выпавший в осадок твердый материал отфильтровывали и высушивали в высоком вакууме в течение ночи при температуре 50°C. В результате получали 0,35 г (выход 71%) искомого продукта в виде твердого вещества оранжевого цвета.
ИК-спектр (в чистом виде, см-1): 3396, 3317, 2983, 2938, 2289, 2051, 1760, 1743, 1723, 1668, 1625, 1588, 1483, 1428, 1406, 1361, 1280, 1256, 1201, 1178, 1095, 1061, 1022, 994, 931, 873, 853, 822, 770, 751.
Элементный состав: С 44,88%. Н 5,9%. N 10,4%.
Содержание железа: 6,8% [масса/масса].
Пример 20. Комплексное соединение трис-(N1-(2-метоксиэтил)-N1,N4,N4-триметил-2-оксобутандиамид)-Fe(III)
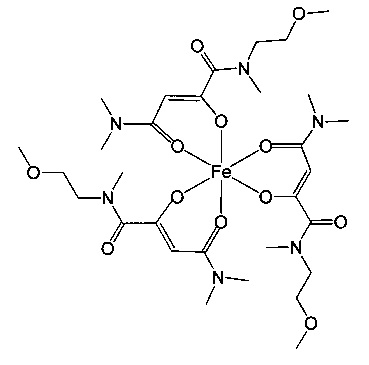
Растворяли 0,46 г (2,8 ммоль) хлорида трехвалентного железа в 20 мл воды и прибавляли 2,0 г (8,5 ммоль) N1-(2-метоксиэтил)-N1,N4,N4-триметил-2-оксобутандиамида. Перемешивали в течение 15 минут при комнатной температуре, после чего реакционную смесь охлаждали на ледяной бане. Прибавляли 1,5 г (11 ммоль) тригидрата ацетата натрия и перемешивали еще 20 минут. Реакционную смесь экстрагировали хлороформом три раза по 50 мл, затем высушивали над сульфатом натрия и концентрировали до сухого состояния. Остаток десорбировали толуолом два раза по 50 мл. Высушивали в высоком вакууме в течение ночи при температуре 50°C. В результате получали 2,0 г (выход 93%) искомого продукта в виде твердого вещества красного цвета.
ИК-спектр (в чистом виде, см-1): 3492, 2929, 1633, 1605, 1568, 1510, 1479, 1449, 1427, 1398, 1354, 1287, 1260, 1200, 1175, 1108, 1067, 1006, 977, 938, 904, 892, 827, 809, 769, 712, 662.
Элементный состав: С 48,06%. Н 7,0%. N 11,0%.
Содержание железа: 6,9% [масса/масса].
Пример 21. Комплексное соединение трис-(N4-(2-гидроксиэтил)-N1,N1,N4-триметил-2-оксобутандиамид)-Fe(III)
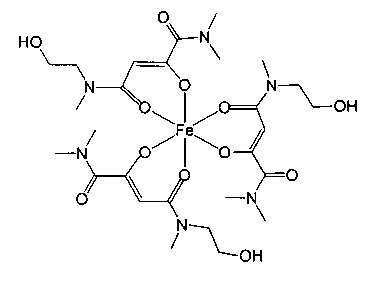
Суспендировали 0,20 г (1,0 ммоль) ацетата трехвалентного железа в 10 мл этилацетата и прибавляли 0,65 г (3,0 ммоль) N4-(2-гидроксиэтил)-N1,N1,N4-триметил-2-оксобутандиамида. Реакционную смесь нагревали до температуры 50°C и прибавляли 20 мл этилового спирта. Перемешивали в течение 20 минут и реакционную смесь концентрировали до сухого состояния. Остаток собирали смесью этиловый спирт/толуол два раза по 5 мл/50 мл и концентрировали до сухого состояния. Остаток высушивали в высоком вакууме при температуре 50°C. В результате получали 0,7 г (выход 99%) искомого продукта в виде твердого вещества красного цвета.
ИК-спектр (в чистом виде, см-1): 3380, 2931, 1621, 1604, 1567, 1516, 1485, 1441, 1396, 1358, 1298, 1260, 1207, 1117, 1051, 1013, 964, 940, 906, 889, 860, 805, 770, 713, 663.
Элементный состав: С 44,50%. Н 6,7%. N 10,85%.
Содержание железа: 8,3% [масса/масса].
| название | год | авторы | номер документа |
|---|---|---|---|
| КОМПЛЕКСНЫЕ СОЕДИНЕНИЯ ЖЕЛЕЗА(III) ДЛЯ ЛЕЧЕНИЯ И ПРОФИЛАКТИКИ СИМПТОМОВ ЖЕЛЕЗОДЕФИЦИТА И ЖЕЛЕЗОДЕФИЦИТНОЙ АНЕМИИ | 2011 |
|
RU2589710C2 |
| ЗАМЕЩЕННЫЕ ЦИКЛОГЕКСИЛДИАМИНЫ | 2009 |
|
RU2526251C2 |
| НОВЫЙ ИНГИБИТОР ЦИКЛИНЗАВИСИМОЙ КИНАЗЫ CDK9 | 2018 |
|
RU2738654C1 |
| СПОСОБ ПОЛУЧЕНИЯ ИЗОБРАЖЕНИЯ КРОВОСНАБЖЕНИЯ МИОКАРДА | 2012 |
|
RU2648358C2 |
| ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ 4-АМИНОЦИКЛОГЕКСАНА | 2009 |
|
RU2503660C2 |
| ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ 4-АМИНОЦИКЛОГЕКСАНА | 2009 |
|
RU2532545C2 |
| АНТИБАКТЕРИАЛЬНЫЕ АГЕНТЫ | 2000 |
|
RU2269525C2 |
| ПРОИЗВОДНЫЕ ПИРИДИНСУЛЬФОНАМИДА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И ПРИМЕНЕНИЕ | 2002 |
|
RU2309148C2 |
| ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ 4-АМИНОЦИКЛОГЕКСАНА | 2009 |
|
RU2525236C2 |
| ПРОИЗВОДНЫЕ 6-АМИНОХИНАЗОЛИНА ИЛИ 3-ЦИАНОХИНОЛИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРА РЕЦЕПТОРНЫХ ТИРОЗИНКИНАЗ EGFR ИЛИ HER-2 | 2010 |
|
RU2536102C2 |
Группа изобретений относится к медицине, в частности к комплексному соединению трехвалентного железа или его фармацевтически приемлемой соли, содержащему, по меньшей мере, один лиганд формулы (I):
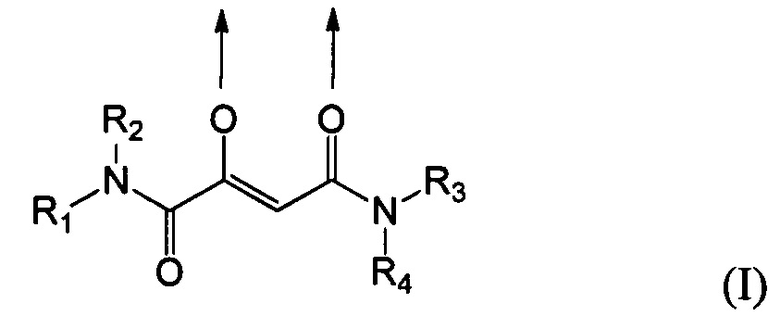
медикаментам и композиции, содержащим комплексное соединение трехвалентного железа. Осуществление изобретения позволит получить терапевтически активное вещество с хорошей активностью, высокой степенью утилизации железа, стабильностью и достаточной растворимостью. 3 н. и 13 з.п. ф-лы, 4 табл., 21 пр.
1. Комплексное соединение трехвалентного железа или его фармацевтически приемлемые соли для применения при лечении и профилактике симптомов дефицита железа и железодефицитных анемий, содержащее, по меньшей мере, один лиганд, имеющий формулу (I):
где стрелки соответственно представляют координационные связи, идущие к одному или разным атомам железа,
R1 и R2 одинаковы или же различны, и их соответственно выбирают из группы, состоящей из водорода, алкильной группы, выбираемой из метила, этила, пропила, изопропила, н-бутила, втор-бутила и трет-бутила, и где указанные алкильные группы могут быть замещенными, причем заместитель выбирают из группы, состоящей из алкокси-группы, алкоксикарбонильной группы, аминокарбонильной (H2NCO-) группы, моноалкиламинокарбонильной группы и диалкиламинокарбонильной группы, или
R1 и R2 вместе с атомом азота, с которым они связаны, образуют 5-6-членное кольцо, которое при необходимости может содержать еще один гетероатом,
R3 и R4 одинаковы или же различны, и их соответственно выбирают из группы, состоящей из водорода, при необходимости замещенного С1-С6-алкила и при необходимости замещенного С3-С6-циклоалкила, где указанные алкил и циклоалкил могут быть замещенными, причем заместитель выбирают из группы, состоящей из гидроксильной группы, алкокси-группы, алкоксикарбонильной группы, аминокарбонильной (H2NCO-) группы, моноалкиламинокарбонильной группы и диалкиламинокарбонильной группы, или
R3 и R4 вместе с атомом азота, с которым они связаны, образуют при необходимости замещенное 3-6-членное кольцо, которое при необходимости может содержать еще один гетероатом, где указанные циклические группы могут быть замещенными гидроксильной группой.
2. Комплексное соединение трехвалентного железа или его фармацевтически приемлемые соли для применения при лечении и профилактике симптомов дефицита железа и железодефицитных анемий по п.1, содержащее, по меньшей мере, один лиганд, имеющий формулу (I):
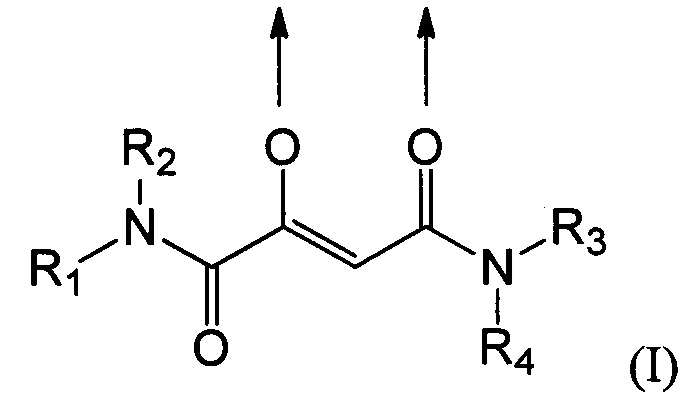
где стрелки соответственно представляют координационные связи, идущие к одному или разным атомам железа,
R1 и R2 одинаковы или же различны, и их соответственно выбирают из группы, состоящей из водорода, алкильной группы, выбираемой из метила и этила, и где указанные алкильные группы могут быть замещенными, причем заместитель выбирают из группы, состоящей из алкокси-группы, алкоксикарбонильной группы, аминокарбонильной (H2NCO-) группы, моноалкиламинокарбонильной группы и диалкиламинокарбонильной группы, или
R1 и R2 вместе с атомом азота, с которым они связаны, образуют пирролидинил,
R3 и R4 одинаковы или же различны, и их соответственно выбирают из группы, состоящей из водорода, алкильной группы, выбираемой из метила и этила, где указанные алкильные группы при необходимости могут быть замещенными, причем заместитель выбирают из группы, состоящей из гидроксильной группы, алкокси-группы, алкоксикарбонильной группы, аминокарбонильной (H2NCO-) группы, моноалкиламинокарбонильной группы и диалкиламинокарбонильной группы,
R3 и R4 вместе с атомом азота, с которым они связаны, образуют морфолинил, пиперидинил или пирролидинил, где указанные циклические группы могут быть замещенными гидроксильной группой.
3. Комплексное соединение трехвалентного железа или его фармацевтически приемлемые соли для применения при лечении и профилактике симптомов дефицита железа и железодефицитных анемий по п. 1 или 2, содержащее, по меньшей мере, один лиганд, имеющий формулу (I):
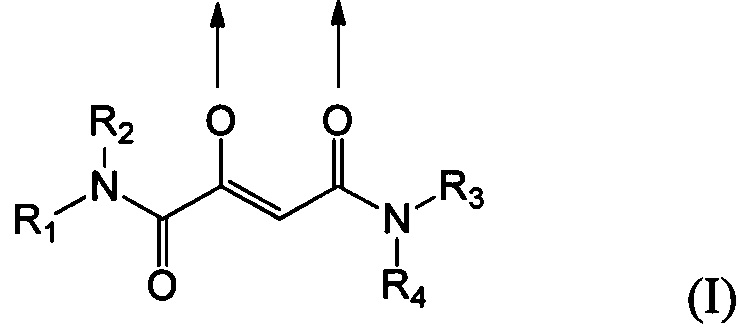
где стрелки соответственно представляют координационные связи, идущие к одному или разным атомам железа,
R1 и R2 одинаковы или же различны, и их соответственно выбирают из группы, состоящей из водорода, метила и этила, и где указанные алкильные группы могут быть замещенными, причем заместитель выбирают из группы, состоящей из алкокси-группы, алкоксикарбонильной группы, или
R1 и R2 вместе с атомом азота, с которым они связаны, образуют пирролидинил,
R3 и R4 одинаковы или же различны, и их соответственно выбирают из группы, состоящей из водорода, алкильной группы, выбираемой из метила и этила, где указанные алкильные группы при необходимости могут быть замещенными, причем заместитель выбирают из группы, состоящей из гидроксильной группы, алкокси-группы, алкоксикарбонильной группы и диалкиламинокарбонильной группы, или
R3 и R4 вместе с атомом азота, с которым они связаны, образуют морфолинил, пиперидинил или пирролидинил, где указанные циклические группы могут быть замещенными гидроксильной группой.
4. Комплексное соединение трехвалентного железа или его фармацевтически приемлемые соли для применения при лечении и профилактике симптомов дефицита железа и железодефицитных анемий по п. 1 или 2, содержащее, по меньшей мере, один лиганд, имеющий формулу (I):
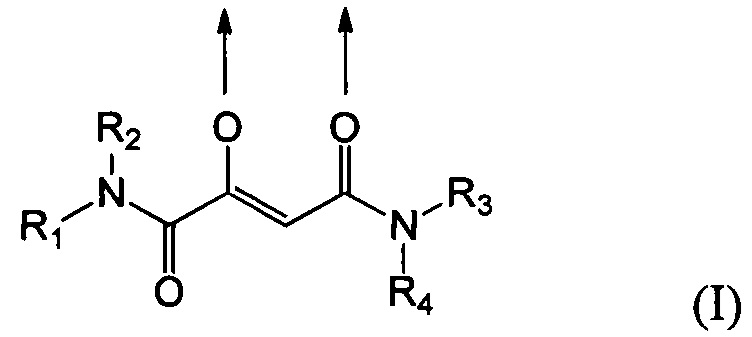
где стрелки соответственно представляют координационные связи, идущие к одному или разным атомам железа,
R1 и R2 одинаковы или же различны, и их соответственно выбирают из группы, состоящей из водорода и метила,
R3 и R4 одинаковы или же различны, и их соответственно выбирают из группы, состоящей из водорода, алкильной группы, выбираемой из метила и этила, где указанные алкильные группы при необходимости могут быть замещенными, причем заместитель выбирают из группы, состоящей из гидроксильной группы, алкокси-группы, алкоксикарбонильной группы и диалкиламинокарбонильной группы.
5. Комплексное соединение трехвалентного железа или его фармацевтически приемлемые соли для применения при лечении и профилактике симптомов дефицита железа и железодефицитных анемий по п. 1 или 2, имеющее формулу (II):
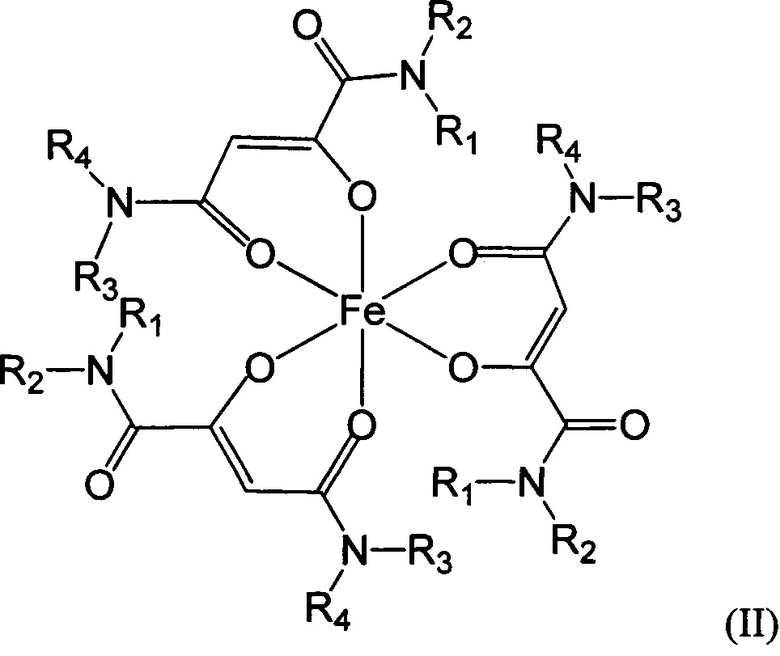
где R1, R2, R3 и R4 определены выше.
6. Комплексное соединение трехвалентного железа или его фармацевтически приемлемые соли для применения при лечении и профилактике симптомов дефицита железа и железодефицитных анемий по п. 1 или 2,
где каждый из R1 и R2 является водородом, а
R3 и R4 одинаковы или же различны, и их выбирают из группы, состоящей из метила и этила.
7. Комплексное соединение трехвалентного железа или его фармацевтически приемлемые соли для применения при лечении и профилактике симптомов дефицита железа и железодефицитных анемий по п. 1 или 2, где комплексное соединение трехвалентного железа выбирают из группы, состоящей из:
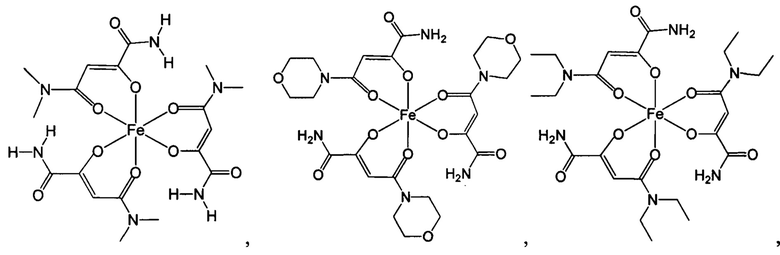
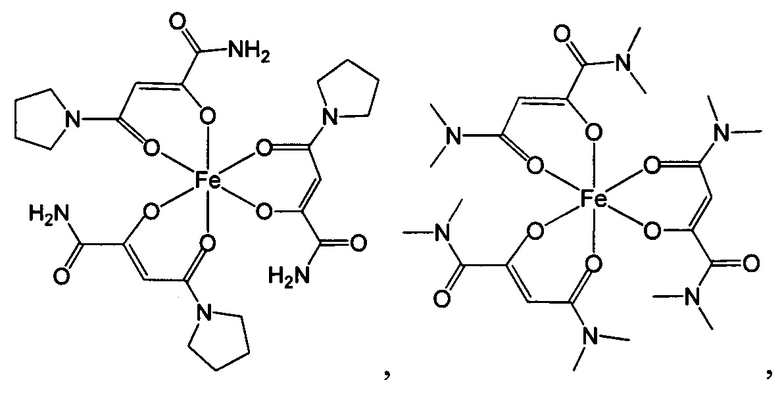
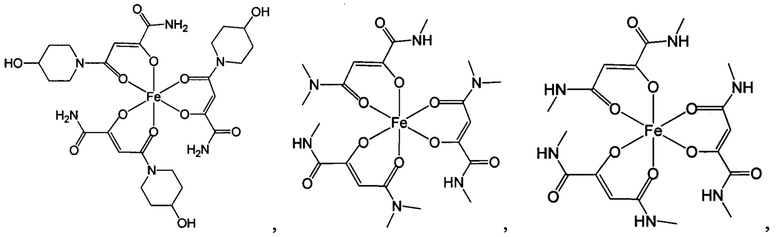


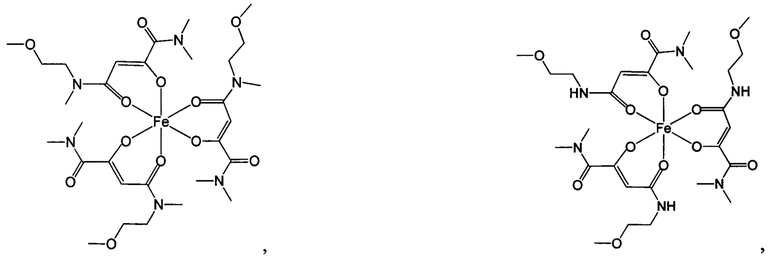
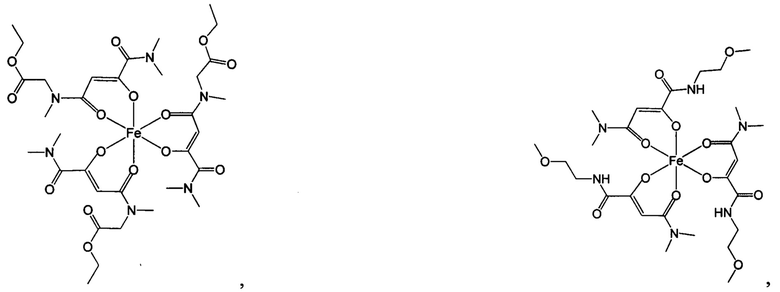
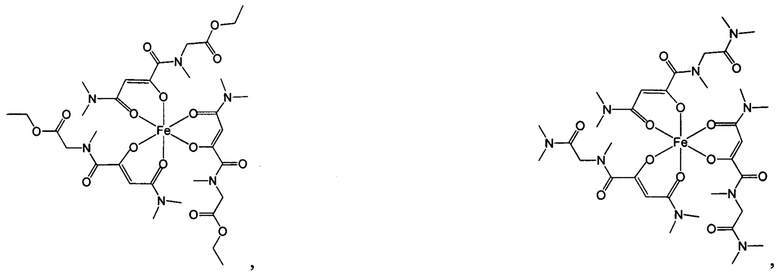
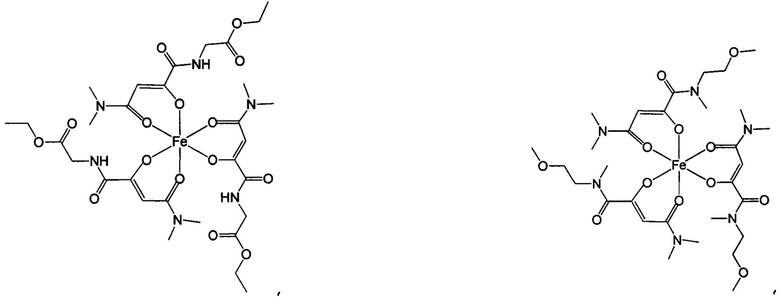 и
и
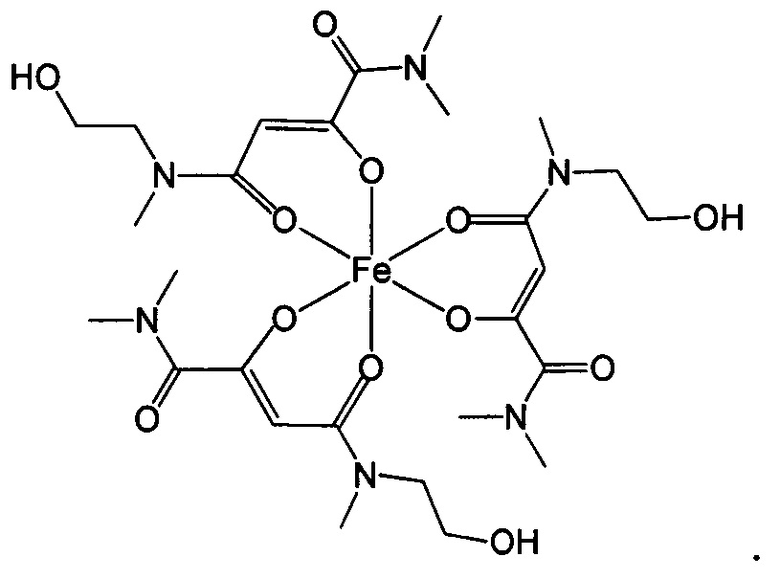
8. Комплексное соединение трехвалентного железа или его фармацевтически приемлемые соли для применения при лечении и профилактике симптомов дефицита железа и железодефицитных анемий по п. 1 или 2, где комплексное соединение трехвалентного железа выбирают из группы, состоящей из:
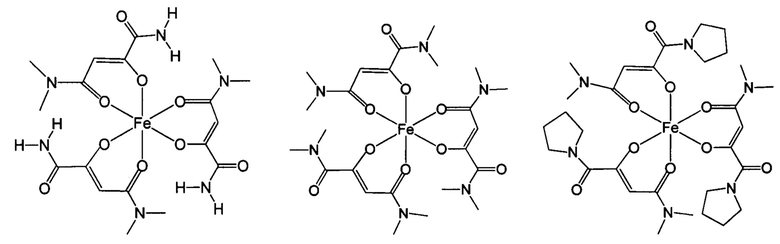
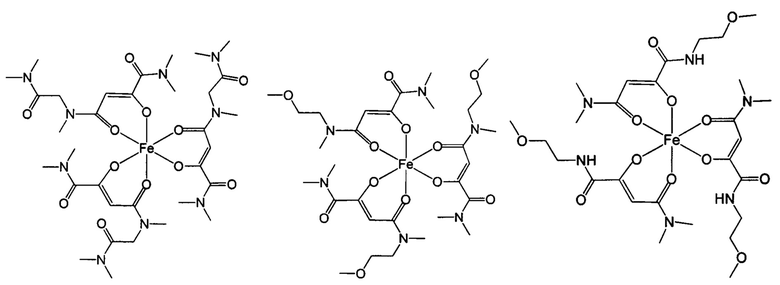
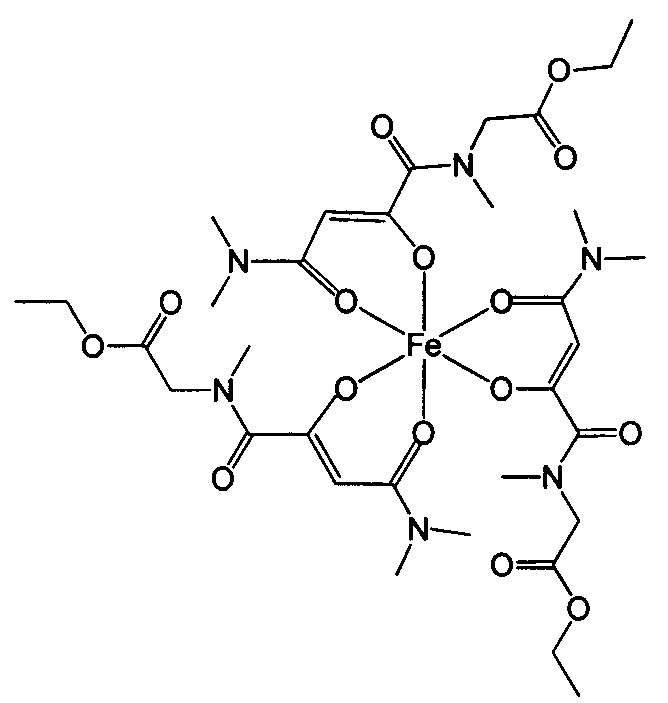
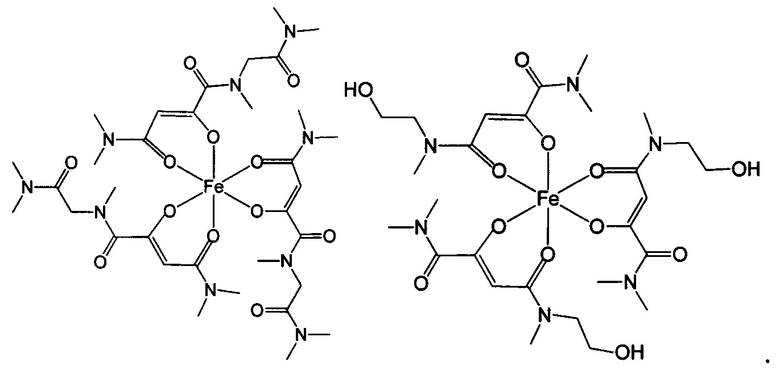
9. Комплексное соединение трехвалентного железа или его фармацевтически приемлемые соли для применения при лечении и профилактике симптомов дефицита железа и железодефицитных анемий по п. 1 или 2, где комплексное соединение трехвалентного железа выбирают из группы, состоящей из:
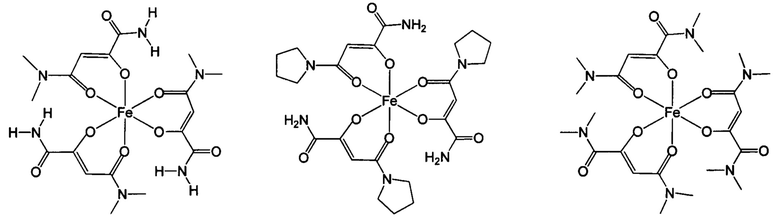
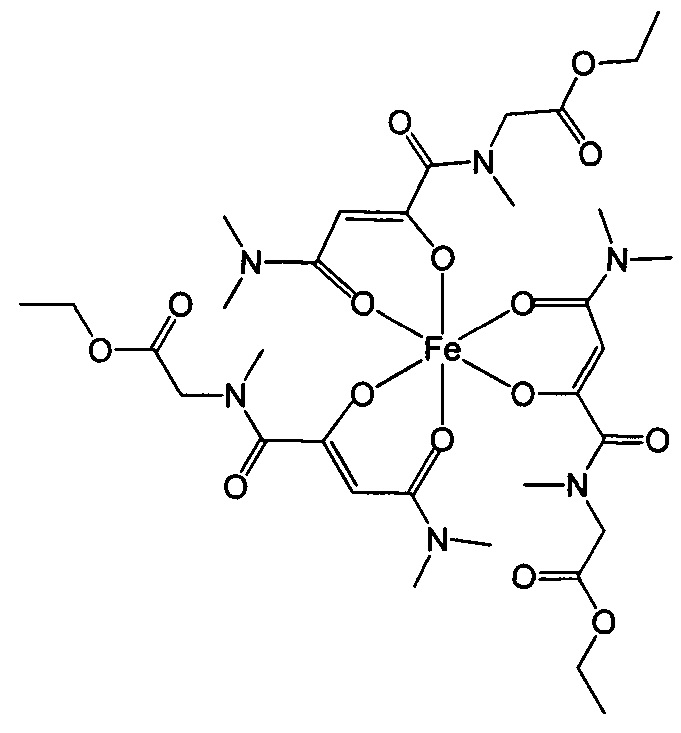 и
и 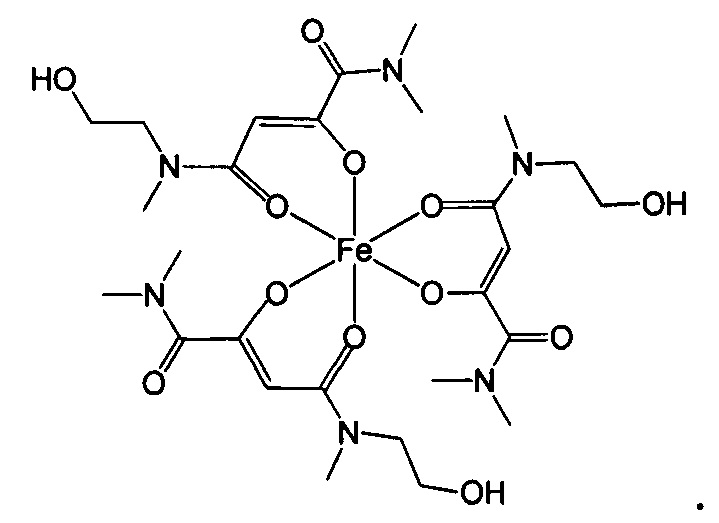
10. Комплексное соединение трехвалентного железа или его фармацевтически приемлемые соли для применения при лечении и профилактике симптомов дефицита железа и железодефицитных анемий по п. 1 или 2, обладающее растворимостью в воде при температуре 25°С и при рН 6,5 по меньшей мере 0,3 мг железа/мл.
11. Комплексное соединение трехвалентного железа или его фармацевтически приемлемые соли для применения при лечении и профилактике симптомов дефицита железа и железодефицитных анемий по п. 1 или 2, где симптомы включают: утомляемость, апатию и вялость, неспособность сосредоточиться, низкую эффективность умственной деятельности, трудности с подбором слов в речи, забывчивость, патологическую бледность, раздражительность, ускорение частоты сердечных сокращений (тахикардию), изъязвления или опухание языка, увеличение селезенки, извращения аппетита, головную боль, отсутствие аппетита, повышенную подверженность инфекциям, депрессивные настроения.
12. Комплексное соединение трехвалентного железа или его фармацевтически приемлемые соли для применения при лечении и профилактике симптомов дефицита железа и железодефицитных анемий по п. 1 или 2 для лечения и профилактики железодефицитной анемии у беременных женщин; скрытой железодефицитной анемии у детей и подростков; железодефицитной анемии, связанной с желудочно-кишечными патологическими состояниями; железодефицитной анемии, вызванной потерей крови, например при желудочно-кишечном кровотечении, обусловленном изъязвлениями, карциномой, геморроем, воспалительными расстройствами, а также приемом ацетилсалициловой кислоты; железодефицитной анемии, обусловленной менструациями; железодефицитной анемии, обусловленной травмой; железодефицитной анемии, обусловленной глютеновой энтеропатией; железодефицитной анемии, обусловленной недостаточным потреблением железа с пищей, в частности при несбалансированном питании у детей и подростков; иммунодефицита, вызванного железодефицитной анемией; нарушении функций головного мозга, обусловленных железодефицитной анемией; синдрома беспокойных ног, обусловленного железодефицитными анемиями; железодефицитных анемий при раке; железодефицитных анемий, обусловленных химиотерапией; железодефицитных анемий, спровоцированных воспалением; железодефицитных анемий при застойной сердечной недостаточности (CHF); железодефицитных анемий при стадиях 3-5 хронической почечной недостаточности (CKD 3-5); железодефицитных анемий, вызванных хроническими воспалительными процессами (ACD); железодефицитных анемий при ревматоидном артрите (RA); железодефицитных анемий при системной красной волчанке (SLE); железодефицитных анемий при воспалительных заболеваниях кишечника (IBD); дефицита железа при нормальном уровне гемоглобина.
13. Комплексное соединение трехвалентного железа или его фармацевтически приемлемые соли для применения при лечении и профилактике симптомов дефицита железа и железодефицитных анемий по п. 1 или 2 для перорального введения.
14. Комплексное соединение трехвалентного железа или его фармацевтически приемлемые соли для применения при лечении и профилактике симптомов дефицита железа и железодефицитных анемий по п. 13 для введения в виде таблеток или капсул.
15. Медикамент в форме пилюль, таблеток, таблеток с кишечно-растворимой оболочкой, таблеток с покрытием из пленки, многослойных таблеток, препаратов с замедленным высвобождением для перорального введения, депо-препаратов, драже, сиропов, гранул, микрокапсул, капсул, капсул с кишечно-растворимой оболочкой, порошков, микрокристаллических препаратов, капель, экстрактов, суспензий, инфузионных растворов или растворов для инъекций, для применения при лечении и профилактике симптомов дефицита железа и железодефицитных анемий, содержащий комплексное соединение трехвалентного железа или его фармацевтически приемлемые соли, как определено в любом из пп. 1-10, и, по меньшей мере, один физиологически приемлемый носитель или иной эксципиент.
16. Композиция, содержащая комплексное соединение трехвалентного железа или его фармацевтически приемлемые соли, как определено в любом из пп. 1-10, для применения при лечении и профилактике симптомов дефицита железа и железодефицитных анемий в сочетании с, по меньшей мере, одним другим медикаментом, влияющим на обмен железа.
| WO 2011117225 A1, 29.09.2011 | |||
| WO 2006037449 A2, 13.04.2006 | |||
| КОМПЛЕКСНЫЕ СОЕДИНЕНИЯ ЖЕЛЕЗА С УГЛЕВОДАМИ | 2008 |
|
RU2423122C2 |

