ОБЛАСТЬ ТЕХНИКИ
Настоящая заявка относится к соединениям, которые действуют как ингибиторы циклинзависимой киназы CDK9, к фармацевтическим композициям, содержащим эти соединения, и к способам и применениям для ингибирования активности серинкиназы с применением этих соединений или композиций.
УРОВЕНЬ ТЕХНИКИ
Пролиферация и деление эукариотических клеток является точным и сложным регуляторным процессом. Процесс пролиферации осуществляется посредством клеточного цикла, а упорядоченная прогрессия клеточного цикла происходит с помощью его строгих молекулярных регуляторных механизмов. Было обнаружено, что существует три основных класса молекул, вовлеченных в регуляцию клеточного цикла: циклинзависимые киназы (CDK), циклины и ингибиторы циклинзависимой киназы (CKI), среди которых CDK занимает центральное положение. Было обнаружено 13 членов (CDK1-CDK13) семейства CDK, которые классифицированы на две категории в соответствии с их внутриклеточными функциями: CDK, которые контролируют клеточный цикл, и CDK, которые контролируют клеточную транскрипцию. CDK9 принадлежит к серинкиназе, а его комплекс, образованный с соответствующим циклином, называется положительным фактором элонгации транскрипции b (P-TEFb). Комплекс может фосфорилировать РНК-полимеразу II и некоторые отрицательные факторы элонгации транскрипции (NELF и N-TEF), позволяющие продолжать транскрипцию с сайта инициации, и является основной молекулой для элонгации транскрипции (Sims RJ 3rd et al., Genes Dev, 2004, 18: 2437-68; Yamaguchi Y et al., Mol Cell Biol, 2002, 22: 2918-27). Исследования показали, что аномальные уровни экспрессии CDK9 или (и) аномальная активность киназы будут вызывать аномальную экспрессию различных белков или (и) аномальные уровни мРНК в клетке. Среди прочего, было подтверждено, что антиапоптотические белки, такие как Bcl-2, регуляторные белки, связанные с клеточным циклом, такие как циклин D1, белки, связанные с путем р53, некоторые белки пути NF-κВ и белки, связанные с микроокружением опухоли, такие как VEGF и подобные, тесно связаны с опухолями. Можно сказать, что CDK9 является одной из наиболее важных молекул в развитии опухолей (Shapiro GI. J Clin Oncol, 2006, 24: 1770-83).
РАСКРЫТИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к ингибиторам циклинзависимых киназ. В частности, в настоящем изобретении предложено соединение формулы (I) или его фармацевтически приемлемая соль, сольват, сложный эфир, кислота, метаболит или пролекарство:
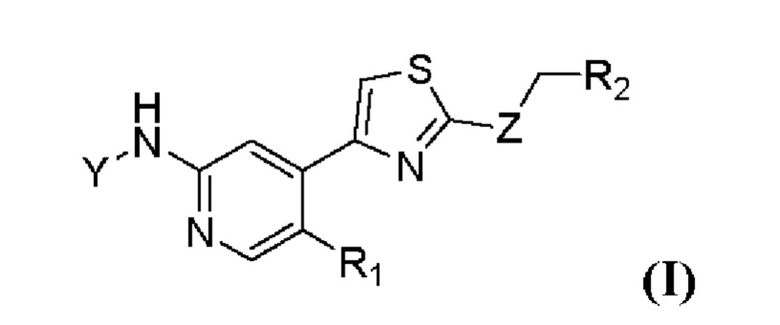
где Y выбран из группы, состоящей из р-фторбензоила, транс-4-аминоциклогексила, в котором N необязательно замещен посредством R3, и транс-4-аминоциклогексилметила, в котором N необязательно замещен посредством R3;
Z выбран из группы, состоящей из NH, S и О;
R1 выбран из группы, состоящей из водорода и галогена;
R2 выбран из группы, состоящей из водорода, С1-С3 алкила, С3-С6 циклоалкила, С3-С6 гетероциклоалкила, необязательно замещенного посредством R4, и фенила, необязательно замещенного посредством R4;
R3 выбран из группы, состоящей из С2-С6 алканоила и С1-С3 алкокси (С1-С3) алкила;
R4 выбран из группы, состоящей из циано и галогена.
В настоящем изобретении также предложена фармацевтическая композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль, сольват, сложный эфир, кислоту, метаболит или пролекарство, и фармацевтически приемлемый носитель или вспомогательное вещество и необязательно другие терапевтические агенты.
Настоящее изобретение также относится к применению соединения формулы (I) или его фармацевтически приемлемой соли, сольвата, сложного эфира, кислоты, метаболита или пролекарства для приготовления лекарственного средства для лечения, предотвращения или облегчения заболевания, расстройства или состояния, регулируемого или обусловленного активностью серинкиназы или связанного с активностью циклинзависимой киназы. Среди них заболевание, расстройство или состояние предпочтительно представляет собой раковое заболевание.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
На фигурах 1a - 1d показаны эффекты, оказываемые соединением 1 на клеточные сигнальные пути в клеточных линиях MV4-11 (фигура 1a), OCI-AML-3 (фигура 1b), HL-60 (фигура 1с) и NB4 (фигура Id);
На фигурах 2а - 2d показаны эффекты, оказываемые соединением 1 на белки, связанные с апоптозом, в клеточных линиях MV4-11 (фигура 2а), OCI-AML-3 (фигура 2b), HL-60 (фигура 2 с) и NB4 (фигура 2d);
На фигурах 3а - 3с показаны эффекты, оказываемые соединением 1 на клеточные циклы в клеточных линиях MV4-11 (фигура За), HL-60 (фигура 3b) и NB4 (фигура 3с).
На фигурах 4а - 4с показаны результаты эксперимента, в котором соединение 1 ингибирует рост опухоли в мышиной модели опухоли, где на фигуре 4а показано изменение относительной массы тела мышей, которым подкожно вводили клетки лейкемии посредством инъекции (рассчитанное на основе массы тела в первый день введения) с течением времени; на фигуре 4b показано изменение размера опухоли, введенной мыши, с течением времени; на фигуре 4с показана окончательная рассчитанная степень ингибирования опухоли (TGI) для каждой группы, и значения для каждой точки данных, показанные на фигурах, отражают среднее значение для каждой экспериментальной группы.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Термины
Если не указано иное, все технические и научные термины, применяемые в настоящем описании, имеют то же значение, которое обычно понимают специалисты в области техники, к которой относится заявленный предмет.
Если не указано иное, в настоящем изобретении используют общепринятые методы, такие как масс-спектрометрия, ЯМР, ВЭЖХ, химия белков, биохимия, методы рекомбинантной ДНК и фармакология в пределах уровня техники. Если конкретное определение не приведено, специалистам в данной области техники известны номенклатура и лабораторные операции и методы, химически связанные с аналитической химией, синтетической органической химией и медицинской и фармацевтической химией, описанные в настоящем документе. В целом, вышеупомянутые методы и процедуры могут быть выполнены традиционными методами, хорошо известными в данной области техники и описанными в различных общих и более конкретных документах, которые процитированы и обсуждаются в настоящем описании.
«Алкил» относится к алифатической углеводородной группе, которая может представлять собой алкил с разветвленной или прямой цепью. В зависимости от структуры алкильная группа может представлять собой одновалентную группу или двухвалентную группу (то есть алкиленовую группу). В настоящем изобретении алкильная группа предпочтительно представляет собой «низшую алкильную группу», содержащую от 1 до 6 атомов углерода, и еще более предпочтительно «низшую алкильную группу», содержащую от 1 до 3 атомов углерода. Типичные алкильные группы включают, но не ограничиваются ими, метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, пентил, гексил и тому подобное.
«Алкокси» относится к -О-алкильной группе, где алкил является таким, как определено в настоящем документе. Типичные алкоксигруппы включают, но не ограничиваются ими, метокси, этокси, пропокси, бутокси, пентилокси, гексилокси и тому подобное.
Термин «арил» обозначает, что плоское кольцо имеет делокализованную систему л-электронов и содержит 4n+2π электронов, где п представляет собой целое число. Арильное кольцо может состоять из пяти, шести, семи, восьми, девяти или более чем девяти атомов. Арильная группа может быть необязательно замещена. Термин «арил» включает карбоциклические арильные группы (такие как фенил) и гетероциклические арильные (или «гетероарильные» или «гетероароматические») группы (такие как пиридин). Термин включает моноциклические или конденсированные полициклические (т.е. кольца, которые имеют смежные пары атомов углерода) группы.
Используемый в настоящем документе термин «арил» обозначает, что каждый из атомов, составляющих кольцо в арильном кольце, представляет собой атом углерода. Арильное кольцо может состоять из пяти, шести, семи, восьми, девяти или более чем девяти атомов. Арильная группа может быть необязательно замещена. Примеры арильных групп включают, но не ограничиваются ими, фенил, нафтил, фенантрил, антрил, флуоренил и флуоренил. В зависимости от структуры арильная группа может представлять собой одновалентную группу или двухвалентную группу (то есть ариленовую группу).
«Алкил (арил)» относится к алкильной группе, как определено в настоящем документе, замещенной арильной группой, как определено в настоящем документе. Неограничивающие алкильные (арильные) группы включают бензил, фенетил и тому подобное.
Термин «циклоалкил» относится к моноциклической или полициклической группе, содержащей только углерод и водород. Циклоалкильная группа включает группу, содержащую от 3 до 10 кольцевых атомов. В зависимости от структуры циклоалкильная группа может представлять собой одновалентную группу или двухвалентную группу (то есть циклоалкиленовую группу). В настоящем изобретении циклоалкильная группа предпочтительно представляет собой циклоалкильную группу, содержащую от 3 до 8 атомов углерода, и еще более предпочтительно «низшую циклоалкильную группу», содержащую от 3 до 6 атомов углерода.
«Алкил (циклоалкил)» относится к алкильной группе, как определено в настоящем документе, замещенной циклоалкильной группой, как определено в настоящем документе. Неограничивающие алкильные (циклоалкильные) группы включают циклопропилметил, циклобутилметил, циклопентилметил, циклогексилметил и тому подобное.
Термин «галоген» («halo») или «галоген» («halogen») относится к фтору, хлору, брому и йоду.
Термины «галоалкил» и «галоалкокси» включают структуры алкила или алкокси, и среди них по меньшей мере один водород замещен атомом галогена. В некоторых вариантах реализации, если два или более атома водорода замещены атомами галогена, атомы галогена либо являются одинаковыми, либо отличаются друг от друга.
Используемый в настоящем описании термин «циано» относится к радикалу формулы -CN.
Термин «карбонил» представляет собой органическую функциональную группу (С=O), образованную связыванием двух атомов углерода и кислорода посредством двойной связи.
Термин «алканоил» или «алкилкарбонил» относится к карбонильной группе, дополнительно замещенной алкильной группой. Типичные алканоильные группы включают, но не ограничиваются ими, ацетил, пропионил, бутирил, пентаноил, гексаноил и тому подобное.
Термин «амино» относится к группе -NH2. Термин «алкиламино» относится к аминозаместителю, дополнительно замещенному одной или двумя алкильными группами, в частности группой -NRR', где R и R' каждый независимо выбран из водорода или низшего алкила, при условии, что -NRR' не является -NH2. Термин «аминоалкил» относится к алкильному заместителю, дополнительно замещенному одной или более аминогруппами. Термин «цианоалкил» относится к алкильному заместителю, дополнительно замещенному одной или более цианогруппами. Используемый в настоящем описании термин «гетероалкил» обозначает, что один или более атомов основных цепей алкильных групп, определенных в настоящем описании, представляют собой гетероатомы, такие как кислород, азот, сера, кремний, фосфор или их комбинации. Гетероатом(ы) может быть расположен в любом месте в гетероалкильной группе или в положении, в котором гетероалкильная группа присоединена к остальной части молекулы.
Термин «гетероарил» относится к арильной группе, содержащей один или более кольцевых гетероатомов, выбранных из группы, состоящей из азота, кислорода и серы. N-содержащий «гетероарильный» фрагмент обозначает, что по меньшей мере один из атомов основной цепи в кольце арильной группы представляет собой азот.В зависимости от структуры гетероарильная группа может представлять собой одновалентную группу или двухвалентную группу (то есть гетероариленовую группу). Примеры гетероарильных групп включают, но не ограничиваются ими, пиридил, имидазолил, пиримидинил, пиразолил, триазолил, пиразинил, тетразолил, фурил, тиенил, изоксазолил, тиазолил, оксазол, изотиазолил, пирролил, хинолил, изохинолил, индолил, бензимидазолил, бензофуранил, индазолил, индолизинил, фталазинил, пиридазинил, изоиндолил, птеридинил, пуринил, оксадиазолил, тиадиазолил, фурил, бензофурил, бензотиенил, бензотиазолил, бензоксазолил, хиназолинил, нафтиридил и фуропиридинил и тому подобные.
Используемый в настоящем описании термин «гетероциклоалкил» обозначает, что один или более атомов, составляющих кольцо в неарильном кольце, представляет собой гетероатом, выбранный из группы, состоящей из азота, кислорода и серы. Гетероциклоалкильное кольцо может состоять из трех, четырех, пяти, шести, семи, восьми, девяти или более чем девяти атомов. Гетероциклоалкильная группа может быть необязательно замещена. Примеры гетероциклоалкильных групп включают, но не ограничиваются ими, лактам, лактон, циклический имин, циклический тиоимин, циклический карбамат, тетрагидротиопиран, 4Н-пиран, тетрагидропиран, пиперидин, 1,3-диоксин, 1,3-диоксан, 1,4-диоксин, 1,4-диоксан, пиперазин, 1,3-оксатиан, 1,4-оксетан, 1,4-оксатиан, тетрагидро-1,4-тиазин, 2Н-1,2-оксазин, малеимид, сукцинимид, барбитуровую кислоту, тиобарбитуровую кислоту, диоксопиперазин, гидантоин, дигидроурацил, морфолин, триоксан, гексагидро-1,3,5-триазин, тетрагидротиофен, тетрагидрофуран, пирролин, пирролидин, имидазолидин, пирролидон, пиразолин, пиразолидин, имидазолин, имидазолидин, 1,3-Диоксол, 1,3-диоксолан, 1,3-дитиолен, 1,3-Дитиолан, изоксазолин, изоксазолидин, оксазолин, оксазолидин, оксазолидинон, тиазолин, тиазолидин и 1,3-оксатиолан. В зависимости от структуры гетероциклоалкильная группа может представлять собой одновалентную группу или двухвалентную группу (то есть гетероциклоалкиленовую группу).
Термин «алкил(гетероарил)» относится к алкильной группе, как определено в настоящем документе, замещенной гетероарильной группой, как определено в настоящем документе.
Термин «алкил(гетероциклоалкил)» относится к алкильной группе, как определено в настоящем документе, замещенной гетероциклоалкильной группой, как определено в настоящем документе.
Термин «необязательно замещенный» или «замещенный» обозначает, что упомянутая группа может быть замещена одной или более дополнительными группами, каждая из которых индивидуально и независимо выбрана из алкильной, циклоалкильной, арильной, гетероарильной, гетероциклильной группы, гидрокси, алкокси, циано, галогена, амида, нитро, галогеналкила, амино, метилсульфонила и тому подобного.
Используемый в настоящем описании термин GI50 относится к концентрации лекарственного средства, необходимой для ингибирования роста 50% клеток, то есть к концентрации лекарственного средства, когда рост 50% клеток (таких как раковые клетки) ингибируется или регулируется.
Используемый в настоящем описании термин IC50 относится к количеству, концентрации или дозе конкретного тестируемого соединения, при котором достигается 50% ингибирование максимального эффекта в анализе, посредством которого измеряют эффект.
Ингибитор киназы CDK9 согласно настоящему изобретению
Настоящее изобретение относится к ингибиторам циклинзависимых киназ CDK9. В частности, в настоящем изобретении предложено соединение формулы (I) или его фармацевтически приемлемая соль, сольват, сложный эфир, кислота, метаболит или пролекарство:
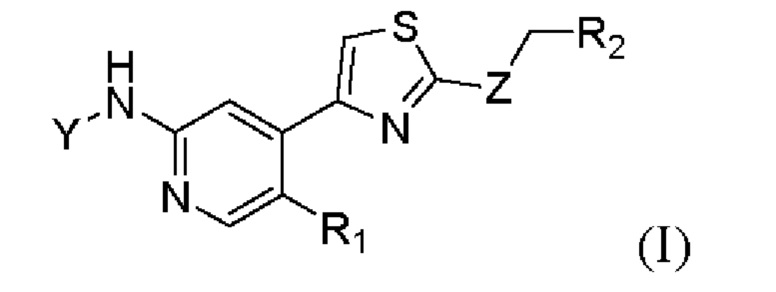
где Y выбран из группы, состоящей из р-фторбензоила, транс-4-аминоциклогексила, в котором N необязательно замещен посредством R3, и транс-4-аминоциклогексилметила, в котором N необязательно замещен посредством R3;
Z выбран из группы, состоящей из NH, S и О;
R1 выбран из группы, состоящей из водорода и галогена;
R2 выбран из группы, состоящей из водорода, С1-С3 алкила, С3-С6 циклоалкила, С3-С6 гетероциклоалкила, необязательно замещенного посредством R4, и фенила, необязательно замещенного посредством R4;
R3 выбран из группы, состоящей из С2-С6 алканоила и С1-С3 алкокси (С1-С3) алкила;
R4 выбран из группы, состоящей из циано и галогена.
В конкретных предпочтительных вариантах реализации Y выбран из следующих структур:
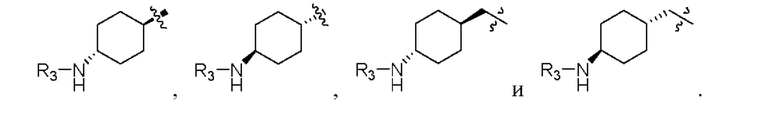
В предпочтительном варианте реализации R1 представляет собой хлор.
В другом предпочтительном варианте реализации R2 выбран из группы, состоящей из водорода, метила, циклопропила, циклогексила, 4-тетрагидропиранила, необязательно замещенного посредством циано, и фенила, необязательно замещенного фтором.
В другом предпочтительном варианте реализации R3 выбран из группы, состоящей из ацетила, 2-метоксиэтила, (R)-1-метил-2-метоксиэтила и (S)-1-метил-2-метоксиэтила.
В настоящем изобретении особенно предпочтительные соединения включают:
4-(((4-(5-хлор-2-(((1R,4r)-4-(((R)-1-метоксипропил-2-ил)амино)циклогексил)амино)пиридин-4-ил)тиазол-2-ил)амино)метил)тетрагидро-2Н-пиран-4-карбонитрил;
(1r,4r)-N1-(5-хлор-4-(2-(((тетрагидро-2Н-пиран-4-ил)метил)амино)тиазол-4-ил)пиридин-2-ил)циклогексан-1,4-диамин;
N-((1r,4r)-4-((5-хлор-4-(2-(((тетрагидро-2H-пиран-4-ил)метил)амино)тиазол-4-ил)пиридин-2-ил)амино)циклогексил)ацетамид;
(1r,4r)-N1-(5-хлор-4-(2-(((тетрагидро-2Н-пиран-4-ил)метил)амино)тиазол-4-ил)пиридин-2-ил)-N4-(2-метоксиэтил)циклогексил-1,4-диамин;
(1S,4r)-N1-(5-хлор-4-(2-(((тетрагидро-2Н-пиран-4-ил)метил)амино)тиазол-4-ил)пиридин-2-ил)-N4-((S)-1-метоксипропан-2-ил)циклогексил-1,4-диамин;
(1R,4r)-N1-(5-хлор-4-(2-(((тетрагидро-2Н-пиран-4-ил)метил)амино)тиазол-4-ил)пиридин-2-ил)-N4-((R)-1-метоксипропан-2-ил)циклогексан-l,4-диамин;
4-(2-((((1r,4r)-4-аминоциклогексил)метил)амино)-5-хлорпиридин-4-ил)-N-((тетрагидро-2Н-пиран-4-ил)метил)тиазол-2-амин;
N-(5-хлор-4-(2-(((тетрагидро-2Н-пиран-4-ил)метил)амино)тиазол-4-ил)пиридин-2-ил)-4-фторбензамид;
(1r,4r)-N1-(5-хлор-4-(2-(метиламино)тиазол-4-ил)пиридин-2-ил)-N4-(2-метоксиэтил)циклогексан-1,4-диамин;
(1r,4r)-N1-(5-хлор-4-(2-((циклогексилметил)амино)тиазол-4-ил)пиридин-2-ил)-N4-(2-метоксиэтил)циклогексан-1,4-диамин;
(1r,4r)-N1-(4-(2-(бензиламино)тиазол-4-ил)-5-хлорпиридин-2-ил)-N4-(2-метоксиэтил)циклогексан-1,4-диамин;
(1r,4r)-N1-(5-хлор-4-(2-((4-фторбензил)амино)тиазол-4-ил)пиридин-2-ил)-N4-(2-метоксиэтил)циклогексан-1,4-диамин;
(1r,4r)-N1-(5-хлор-4-(2-((циклопропилметил)амино)тиазол-4-ил)пиридин-2-ил)-N4-(2-метоксиэтил)циклогексан-1,4-диамин;
4-((4-(5-хлор-2-(((1r,4r)-4-((2-метоксиэтил)амино)циклогексил)амино)пиридин-4-ил)тиазол-2-иламино)метил)тетрагидро-2Н-пиран-4-карбонитрил;
4-(((4-(5-хлор-2-(((1S,4r)-4-(((S)-1-метоксипропил-2-ил)амино)циклогексил)амино)пиридин-4-ил)тиазол-2-ил)амино)метил)тетрагидро-2Н-пиран-4-карбонитрил;
(1r,4r)-N1-(5-хлор-4-(2-((тетрагидро-2Н-пиран-4-ил)метокси)тиазол-4-ил)пиридин-2-ил)-N4-(2-метоксиэтил)циклогексан-1,4-диамин;
(1r,4r)-N1-(5-хлор-4-(2-(((тетрагидро-2Н-пиран-4-ил)метил)меркапто)тиазол-4-ил)пиридин-2-ил)-N4-(2-метоксиэтил)циклогексан-1,4-диамин;
(1r,4r)-N1-(2-метоксиэтил)-N4-(4-(2-(((тетрагидро-2Н-пиран-4-ил)метил)амино)тиазол-4-ил)пиридин-2-ил)циклогексан-1,4-диамин.
Структуры предпочтительных соединений согласно настоящему изобретению перечислены ниже.
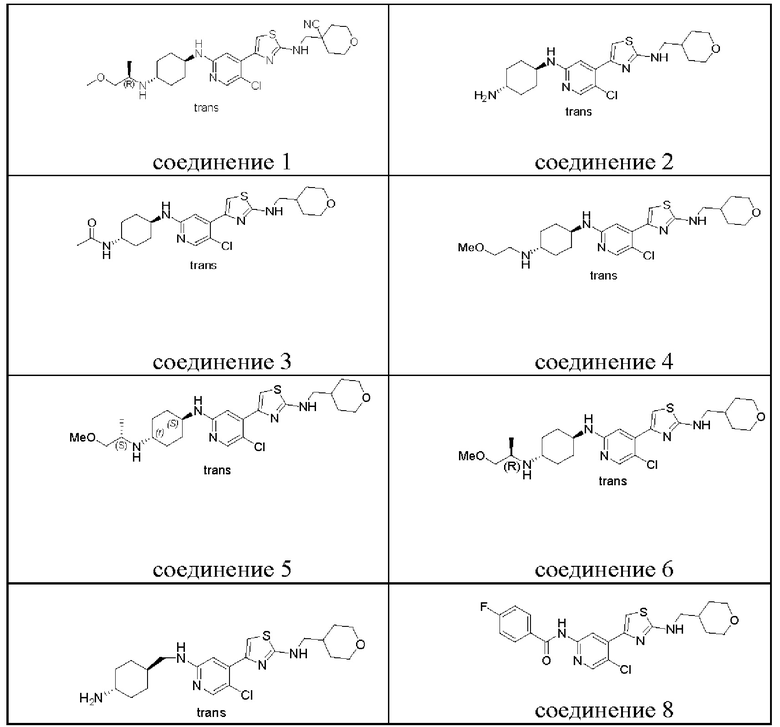
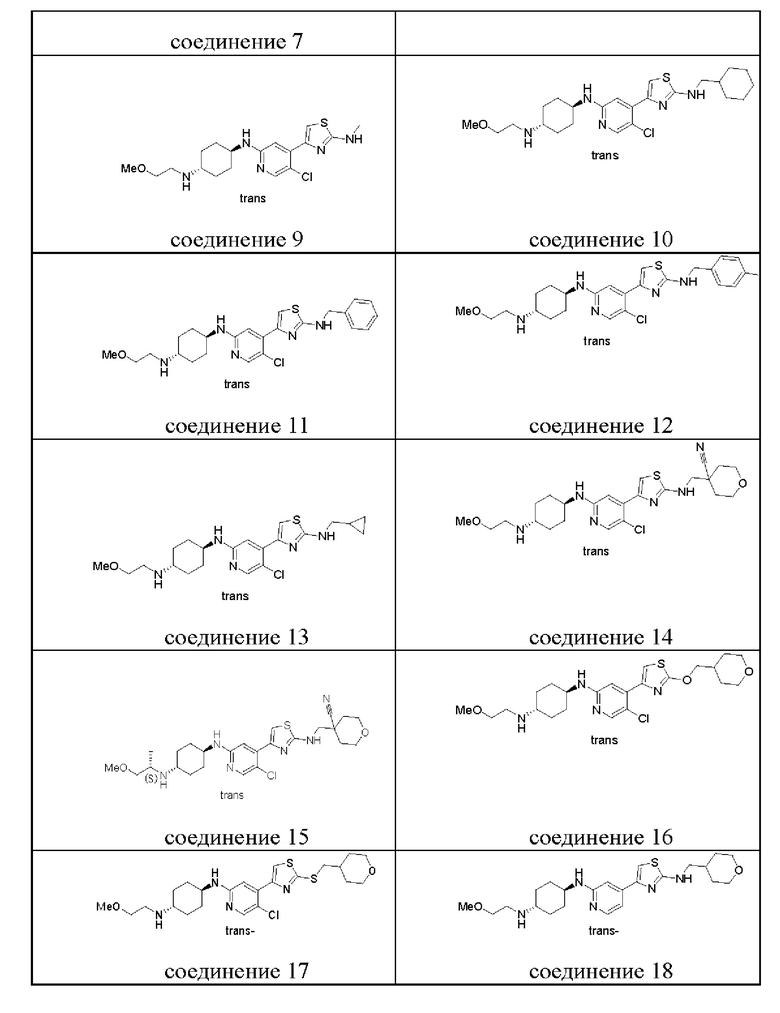
Хотя в приведенной выше таблице перечислены структуры предпочтительных соединений согласно настоящему изобретению, следует понимать, что два атома углерода, соответственно присоединенные к пара-аминогруппе в циклогексильной группе, не являются хиральными центрами, представление химической связи  или
или  является просто обозначением присоединения двух химических связей к пара-аминогруппе и имеют транс-структуру относительно циклогексильной группы, и, таким образом, соединения, представленные сменой этих двух химических связей
является просто обозначением присоединения двух химических связей к пара-аминогруппе и имеют транс-структуру относительно циклогексильной группы, и, таким образом, соединения, представленные сменой этих двух химических связей  и
и  , также входят в объем настоящего изобретения. В настоящем изобретении описаны новые ингибиторы киназы. Фармацевтически приемлемые соли, сольваты, сложные эфиры, кислоты, фармацевтически активные метаболиты и пролекарства этого соединения также описаны в настоящем документе.
, также входят в объем настоящего изобретения. В настоящем изобретении описаны новые ингибиторы киназы. Фармацевтически приемлемые соли, сольваты, сложные эфиры, кислоты, фармацевтически активные метаболиты и пролекарства этого соединения также описаны в настоящем документе.
В дополнительных или других вариантах реализации соединения, описанные в настоящем документе, вводят субъекту, нуждающемуся в этом, для превращения в его организме с образованием метаболитов, которые затем применяют для получения необходимого эффекта, включая необходимый терапевтический эффект.
Описанные в настоящем документе соединения можно превращать и/или применять в форме фармацевтически приемлемых солей. Типы фармацевтически приемлемых солей включают, но не ограничиваются ими: (1) кислотно-аддитивную соль, образованную взаимодействием формы свободного основания соединения с фармацевтически приемлемой неорганической кислотой, такой как хлористоводородная кислота, бромистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота, метафосфорная кислота или тому подобное; или с органической кислотой, такой как уксусная кислота, пропионовая кислота, капроновая кислота, циклопентанпропионовая кислота, гликолевая кислота, пировиноградная кислота, молочная кислота, малоновая кислота, яблочная кислота, лимонная кислота, янтарная кислота, малеиновая кислота, винная кислота, фумаровая кислота, трифторуксусная кислота, бензойная кислота, 3-(4-гидроксибензоил)бензойная кислота, коричная кислота, миндальная кислота, метансульфоновая кислота, этансульфоновая кислота, 1,2-этандисульфоновая кислота, 2-гидроксиэтансульфоновая кислота, бензолсульфоновая кислота, толуолсульфокислота, 4-метилбицикло-[2.2.2]окт-2-ен-1-карбоновая кислота, 2-нафталинсульфоновая кислота, трет-бутилуксусная кислота, глюкогептоновая кислота, 4,4'-метилен-бис-(3-гидрокси-2-ен-1-карбоновая кислота), 3-фенилпропионовая кислота, триметилуксусная кислота, додецилсульфат, глюконовая кислота, глутаминовая кислота, салициловая кислота, гидроксинафтойная кислота, стеариновая кислота, муконовая кислота и т.д.; (2) соль присоединения основания, образованную, когда кислотный протон исходного соединения замещен ионом металла, таким как ион щелочного металла (например, лития, натрия, калия), ион щелочноземельного металла (например, магния или кальция) или ион алюминия; или координирован с органическими основаниями. Приемлемые органические основания включают этаноламин, диэтаноламин, триэтаноламин, триметиламин, N-метилглюкамин и тому подобное. Приемлемые неорганические основания включают гидроксид алюминия, гидроксид кальция, гидроксид калия, карбонат натрия, гидроксид натрия и тому подобное.
Соответствующие противоионы фармацевтически приемлемых солей могут быть проанализированы и охарактеризованы с применением различных методов, включая, но не ограничиваясь ими, ионообменную хроматографию, ионную хроматографию, капиллярный электрофорез, индуктивно связанную плазму, атомно-абсорбционную спектроскопию, масс-спектрометрию или любую их комбинацию.
Соль выделяют с применением по меньшей мере одной из следующих методик: фильтрация, осаждение нерастворителем с последующей фильтрацией, выпаривание растворителя или лиофилизация в случае водного раствора.
Скрининг и характеристика фармацевтически приемлемых солей, полиморфов и/или сольватов можно осуществлять с применением различных методов, включая, но не ограничиваясь этим, термический анализ, рентгеновскую дифракцию, спектроскопию, микроскопию и элементный анализ. Различные применяемые спектральные методы включают, но не ограничиваются ими, рамановский, FTIR, UVIS и ЯМР (жидкое и твердое состояние). Различные методы микроскопии включают, но не ограничиваются ими, ИК-микроскопию и рамановскую микроскопию.
Фармацевтические композиции согласно настоящему изобретению
В настоящем изобретении также предложена фармацевтическая композиция, содержащая по меньшей мере одно соединение формулы (I) или его фармацевтически приемлемую соль, сольват, сложный эфир, кислоту, фармацевтически активный метаболит или пролекарство, и фармацевтически приемлемый носитель или вспомогательное вещество и необязательно другие терапевтические агенты.
Во время лечения его можно применять отдельно или в комбинации с одним или более другими терапевтическими агентами, по необходимости. Лекарственное средство, содержащее соединение согласно настоящему изобретению, можно вводить пациенту посредством по меньшей мере одного способа, выбранного из инъекции, перорального введения, ингаляции, ректального или трансдермального введения.
В варианте реализации настоящего изобретения при лечении пациента в соответствии с настоящим изобретением количество данного лекарственного средства зависит от ряда факторов, таких как конкретный режим дозирования, тип заболевания или расстройства и его тяжесть, а также субъект, нуждающийся в лечении, или уникальность хозяина (например, масса тела), однако, в зависимости от конкретных обстоятельств, включая, например, конкретное применяемое лекарство, способ введения, подлежащее лечению состояние и подлежащего лечению субъекта или хозяина, вводимая доза может быть определена способами, обычно известными в данной области техники. В целом, для применения при лечении взрослого человека вводимая доза обычно составляет от 0,02 до 5000 мг/день, например, от примерно 1 до 1500 мг/день. Необходимая доза может быть удобным образом представлена в виде разовой дозы или в виде одновременных (или в течение короткого периода времени) или отдельных доз с соответствующими интервалами, таких как две, три, четыре или более отдельных доз в день. Специалистам в данной области техники будет понятно, что, хотя приведены вышеупомянутые диапазоны доз, конкретное эффективное количество может быть соответствующим образом скорректировано в зависимости от состояния пациента и в связи с диагнозом врача.
Применение лекарственного средства согласно настоящему изобретению
Соединение формулы (I) или его фармацевтически приемлемую соль, сольват, сложный эфир, кислоту, метаболит или пролекарство или фармацевтическую композицию, содержащую их, можно применять для ингибирования активности циклинзависимых киназ (CDK) и циклинов, особенно активности CDK9. Соединение формулы (I) или его фармацевтически приемлемую соль, сольват, сложный эфир, кислоту, метаболит или пролекарство можно применять для лечения или предотвращения одного или более заболеваний, выбранных из группы, состоящей из немелкоклеточного рака легкого, мелкоклеточного рака легкого, аденокарциномы легкого, плоскоклеточного рака легкого, рака поджелудочной железы, рака предстательной железы, рака мочевого пузыря, рака печени, рака кожи, глиомы, рака молочной железы, меланомы, злокачественной глиомы, рабдомиосаркомы, рака яичников, астроцитомы, саркомы Юинга, ретинобластомы, эпителиальноклеточной карциномы, рака толстой кишки, рака почки, гастроинтестинальной стромальной опухоли, лейкоза, гистиоцитарной лимфомы и назофарингеальной карциномы.
Более предпочтительно, соединение формулы (I), описанное в настоящем документе, или его фармацевтически приемлемую соль, сольват, сложный эфир, кислоту, метаболит или пролекарство, или фармацевтическую композицию, содержащую их, можно применять в качестве ингибитора CDK9, который можно применять для лечения немелкоклеточного рака легкого, мелкоклеточного рака легкого, аденокарциномы легкого, плоскоклеточного рака легкого, рака поджелудочной железы, рака предстательной железы, рака мочевого пузыря, рака печени, рака кожи, глиомы, рака молочной железы, меланомы, злокачественной глиомы, рабдомиосаркомы, рака яичников, астроцитомы, саркомы Юинга, ретинобластомы, эпителиальноклеточной карциномы, рака толстой кишки, рака почки, гастроинтестинальной стромальной опухоли, лейкоза, гистиоцитарной лимфомы и назофарингеальной карциномы при его применении отдельно или в комбинации с другими терапевтическими агентами.
Получение соединения
Соединения формулы (I) можно синтезировать с применением стандартных методик синтеза, известных специалистам в данной области техники, или с применением методов, известных в данной области техники, в комбинации со способами, описанными в настоящем документе. Кроме того, растворители, температуры и другие условия реакции, представленные в настоящем документе, могут варьироваться в зависимости от компетентности в данной области техники. В качестве дополнительного руководства также можно применять следующие методы синтеза.
Реакции можно применять последовательно для получения соединений, описанных в настоящем документе; или их можно применять для синтеза фрагментов, которые впоследствии добавляют способами, описанными в настоящем документе, и/или способами, известными в данной области техники.
В определенных вариантах реализации в настоящем изобретении предложены способы получения соединения-ингибитора серинкиназы, описанные в настоящем документе, и способы его применения. В определенных вариантах реализации соединения, описанные в настоящем документе, можно синтезировать с применением следующих схем синтеза. Соединения можно синтезировать способами, аналогичными описанным ниже, с применением соответствующих исходных материалов.
Исходные материалы для синтеза соединений, описанные в настоящем документе, могут быть синтезированы или могут быть получены из коммерческих источников. Соединения, описанные в настоящем документе, и другие родственные соединения, имеющие различные заместители, могут быть синтезированы с применением методик и исходных материалов, известных специалистам в данной области техники. Общие способы получения соединений, описанных в настоящем изобретении, могут быть получены из реакций, известных в данной области техники, и реакции могут быть модифицированы для введения различных фрагментов в молекулы, представленные в настоящем изобретении, с помощью реагентов и условий, которые специалисты в данной области техники считают подходящими.
При необходимости продукт реакции может быть выделен и очищен с применением традиционных способов, включая, но не ограничиваясь ими, фильтрацию, дистилляцию, кристаллизацию, хроматографию и тому подобное. Эти продукты могут быть охарактеризованы с применением традиционных способов, включая физические константы и спектральные данные.
Неограничивающие примеры схем синтеза для получения соединений формулы (I) описаны ниже.
Примеры
Следующие конкретные неограничивающие примеры следует рассматривать только как иллюстративные и никоим образом не ограничивающие настоящее описание. Хотя никакие дополнительные подробности не описаны, считается, что специалист в данной области техники может в полной мере применять настоящее описание на основании приведенного здесь описания.
Пример 1: Синтез 4-(((4-(5-хлор-2-(((1R,4r)-4-(((R)-1-метоксипропил-2-ил)амино)циклогексил)амино)пиридин-4-ил)тиазол-2-ил)амино)метил)тетрагидро-2Н-пиран-4-карбонитрила
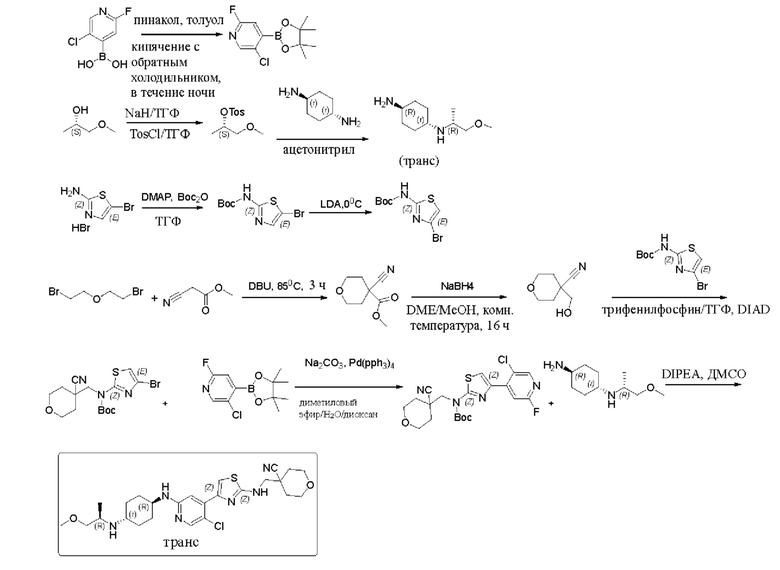
Стадия 1: синтез 5-хлор-2-фтор-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридина
5-хлор-2-фторпиридин-4-бороновую кислоту (0,7 г, 4,46 ммоль) и пинакол (0,63 г, 5,35 ммоль) добавляли к 50 мл толуола, смесь нагревали до 120°С и кипятили с обратным холодильником в течение ночи, и по данным ТСХ оставалось небольшое количество материала. Реакционный раствор охлаждали до комнатной температуры, концентрировали и сушили с помощью масляного насоса с получением 0,92 г соединения 5-хлор-2-фтор-4-(4,4,5,5-тетраметил-1,3,2-д иоксаборолан-2-ил)пиридина в форме белого твердого вещества, выход 80%, MC(ESI): m/z 258,1 (М+Н)+.
Стадия 2: синтез (S)-1-метоксипропан-2-ил 4-метилбензолсульфоната 60%) гидрида натрия NaH (6,52 г, 283 ммоль) добавляли к сухому тетраги дрофу рану ТГФ (200 мл), который охлаждали до 0°С на ледяной бане, и защищали азотом, и затем по каплям добавляли (S)-(+)-1-метокси-2-пропанол (21 г, 233 ммоль). После завершения добавления по каплям смесь перемешивали при комнатной температуре в течение 1,5 часов. Реакционный раствор снова охлаждали до 0°С, и затем по каплям добавляли раствор п-толуолсульфонилхлорида (45,3 г, 283 ммоль) в тетрагидрофуране ТГФ (200 мл). После добавления смесь перемешивали при комнатной температуре в течение ночи. По данным ТСХ исходный материал был полностью израсходован. Реакционную смесь разбавляли этилацетатом (500 мл), гасили посредством добавления воды по каплям (500 мл) при охлаждении льдом, и разделяли. Водную фазу экстрагировали этилацетатом (200 мл) один раз. Органические фазы объединяли, промывали водой (200 мл) и насыщенным солевым раствором (200 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением 43 г бледно-желтого маслянистого неочищенного продукта, который выделяли с применением колонки (петролейный эфир / этилацетат = 5/1) с получением 37 г (8)-1-метоксипропан-2-ил 4-метилбензолсульфоната в форме бледно-желтого масла, выход 65,1%, МС (ESI): m/z 245,1 (М+Н)+.
Стадия 3: синтез (1r,4R)-N1-((R)-1-метоксипропан-2-ил)циклогексан-1,4-диамина
К 50 мл ацетонитрила, который нагревали до 90°С, добавляли (S)-l-метоксипропан-2-ил 4-метилбензолсульфонат (5 г, 20,5 ммоль) и транс-1,4-циклогександиамин (5,84 г, 51,2 ммоль), и реакцию проводили в течение ночи. Реакцию контролировали с помощью ТСХ до ее завершения. Реакционный раствор охлаждали, и затем фильтровали, и фильтрат концентрировали. Остаток растворяли в дихлорметане, смешивали с силикагелем и выделяли с помощью колонки (дихлорметан / метанол = 10/1) с получением 2,5 г соединения (1r,4R)-N1-((R)-1-метоксипропан-2-ил)циклогексан-1,4-диамина в форме бледно-желтой жидкости, выход 65%, МС (ESI): m/z 187,3 (М+Н)+.
Стадия 4: синтез трет-бутил 5-бромтиазол-2-илкарбамата
5-Бромтиазол-2-амина гидробромид (105 г, 403 ммоль) суспендировали в 500 мл тетрагидрофурана, и добавляли диметиламинопиридин (2,41 г, 20 ммоль) для образования белого помутнения. Медленно по каплям добавляли раствор ди-трет-бутил дикарбоната (105,6 г, 484,6 ммоль) в тетрагидрофуране. Проводили реакцию смеси в течение двух дней. Затем реакционный раствор концентрировали, растворяли в дихлорметане (300 мл), перемешивали с силикагелем и выделяли с помощью колонки (элюировали с применением градиента петролейный эфир / этилацетат = 10/1 - 6/1) с получением 45 г трет-бутил 5-бромтиазол-2-илкарбамата в форме почти белого твердого вещества, выход 40%, МС (ESI): m/z 278,98 (М+Н)+.
Стадия 5: синтез трет-бутил 4-бромтиазол-2-илкарбамата
Раствор диизопропиламина (64 мл, 446 ммоль) в 200 мл тетрагидрофурана добавляли в сухую трехгорлую колбу, которую защищали азотом, и охлаждали до 0°С, а затем добавляли н-бутиллитий (2,5 М, 173 мл, 431,7 ммоль). Реакцию проводили в течение 1 часа после завершения добавления. Раствор трет-бутил-5-бромтиазол-2-илкарбамата в 400 мл тетрагидрофурана добавляли по каплям при 0°С, и реакцию проводили в течение 2 часов после завершения добавления. По данным ТСХ реакция была завершена. При 0°С реакцию гасили медленным добавлением ледяной воды (5 мл), перемешивали в течение 30 минут, затем добавляли насыщенный водный раствор хлорида аммония (500 мл), и разделяли. Водный слой экстрагировали дихлорметаном (2×300 мл). Органические фазы объединяли, промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Остаток перекристаллизовывали из смеси петролейный эфир: этилацетат = 30:1 с получением 31 г трет-бутил-4-бромтиазол-2-илкарбамата в форме белого твердого вещества, выход 77,5%, МС (ESI): m/z 278,98 (М+Н)+.
Стадия 6: синтез 4-циано-тетрагидро-2Н-пиран-4-метилкарбоната
К 600 мл диметилформамида добавляли метилцианоацетат (39,1 г, 395,3 ммоль) и 2,2-дибромэтиловый эфир (100 г, 434,8 ммоль), и добавляли DBU (90 г, 593 ммоль). Смесь нагревали при 85°С в течение 3 часов. По данным ТСХ исходный материал был полностью израсходован. Смесь фильтровали для удаления твердого вещества, которое промывали этилацетатом (2×300 мл). Фильтрат концентрировали с получением коричневого масла, которое перегоняли при пониженном давлении.
Фракцию, которая представляла собой бесцветную жидкость, получали, когда внутренняя температура составляла 65-70°С, и помещали для кристаллизации с получением 42 г 4-циано-тетрагидро-2Н-пиран-4-метил карбоната в форме белого твердого вещества, выход 62,8%, МС (ESI): m/z 178,2 (М+Н)+.
Стадия 7: синтез 4-(гидроксиметил)-тетрагидро-2Н-пиран-4-карбонитрила
4-циано-тетрагидро-2Н-пиран-4-метил карбонат (42 г, 248,4 ммоль) растворяли в 400 мл диметилового эфира этиленгликоля и 40 мл метанола, который охлаждали до 0°С на ледяной бане, и частями добавляли борогидрид натрия (11,1 г, 149 ммоль). После окончания добавления смесь естественным образом нагревали до комнатной температуры и перемешивали в течение 16 часов. По данным ТСХ реакция была завершена. Затем реакционный раствор концентрировали, затем снова концентрировали после добавления метанола, чтобы погасить избыток борогидрида натрия, и затем концентрировали. Остаток выделяли с применением колонки (петролейный эфир / этилацетат = 5/1) с получением 28 г 4-(гидроксиметил)-тетрагидро-2Н-пиран-4-карбонитрила в форме бледно-желтого масла, выход 79,5%, МС (ESI): m/z 142,1 (М+Н)+.
Стадия 8: синтез трет-бутил (4-бромтиазол-2-ил)((4-цианотетрагидро-2Н-пиран-4-ил)метил)карбамата
4-(гидроксиметил)-тетрагидро-2Н-пиран-4-карбонитрил, трет-бутил-4-бромтиазол-2-илкарбамат и трифенилфосфин добавляли к безводному тетраги дрофу рану ТГФ, который охлаждали до 0°С, и затем по каплям добавляли диизопропилазодикарбоксилат DIAD. Смесь перемешивали при комнатной температуре в течение 10 минут, и затем нагревали до 40°С и перемешивали в течение ночи. Затем реакционный раствор концентрировали. Остаток растворяли в дихлорметане, смешивали с силикагелем и выделяли с применением колонки (петролейный эфир / этилацетат = 50/1, 30/1, 20/1) с получением 365 мг трет-бутил (4-бромтиазол-2-ил)((4-цианотетрагидро-2Н-пиран-4-ил)метил)карбамата в виде белого твердого вещества, выход 50%, МС (ESI): m/z 402,1 (М+Н)+.
Стадия 9: синтез трет-бутил (4-(5-хлор-2-фторпиридин-4-ил)тиазол-2-ил)((4-циано-тетрагидро-2Н-пиран-4-ил)метил)карбамата
5-хлор-2-фтор-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин и карбонат натрия добавляли к смеси диметиловый эфир/Н2О/диоксан, которую дважды продували азотом, и затем добавляли трет-бутил (4-бромтиазол-2-ил) ((4-цианотетрагидро-2Н-пиран-4-ил)метил)карбамат и тетратрифенилфосфин палладий Pd(pph3)4. Систему трижды продували азотом, затем нагревали до 70°С, и реакцию проводили в течение 6 часов. Когда по данным ТСХ оставалась только половина исходного материала, нагревание прекращали, и реакционную смесь обрабатывали. Реакционный раствор охлаждали до комнатной температуры, и затем добавляли этилацетат и метанол. Смесь фильтровали, остаток на фильтре промывали этилацетатом, и фильтрат концентрировали. Затем остаток растворяли в дихлорметане, промывали насыщенным солевым раствором и разделяли. Органическую фазу сушили над безводным сульфатом натрия и фильтровали. Фильтрат смешивали с силикагелем и выделяли с применением колонки (петролейный эфир / этилацетат = 30/1) с получением 3,2 г трет-бутил (4-(5-хлор-2-фторпиридин-4-ил)тиазол-2-ил) ((4-циано-тетрагидро-2Н-пиран-4-ил)метил)карбамата в форме белого пенистого твердого вещества, выход 55%, МС (ESI): m/z 453,1 (М+Н)+.
Стадия 10: синтез 4-(((4-(5-хлор-2-(((1R,4r)-4-(((R)-1-метоксипропил-2-ил)амино)циклогексил)амино)пиридин-4-ил)тиазол-2-ил)амино)метил)тетрагидро-2Н-пиран-4-карбонитрила
Трет-бутил (4-(5-хлор-2-фторпиридин-4-ил)тиазол-2-ил) ((4-циано-тетрагидро-2Н-пиран-4-ил)метил)карбамат (3,2 г, 7,1 ммоль), (1r,4R)-N1-((R)-1- метоксипропан-2-ил)циклогексан-1,4-диамин (3,9 г, 21,2 ммоль) и диизопропилэтиламин DIPEA добавляли к 30 мл диметилсульфоксида, который защищали азотом, и затем нагревали до 100 - 110°С, и реакцию проводили в течение двух дней. Реакцию контролировали посредством ТСХ и LCMS. Когда исходный материал трет-бутил (4-(5-хлор-2-фторпиридин-4-ил)тиазол-2-ил)((4-циано-тетрагидро-2Н-пиран-4-ил) метил)карбамат был полностью израсходован, и оставались некоторые из промежуточных соединений при удалении ВОС, реакцию останавливали. Реакционную смесь охлаждали, и затем разбавляли этилацетатом (60 мл), добавляли воду (150 мл) при охлаждении льдом, и разделяли. Затем водную фазу экстрагировали этилацетатом (2×50 мл). Органические фазы объединяли, промывали насыщенным солевым раствором (100 мл), сушили над безводным сульфатом натрия, фильтровали, и фильтрат концентрировали с получением неочищенного вещества в форме желтовато-коричневого масла. Неочищенное вещество выделяли с применением колонки (ацетонитрил/вода/трифторуксусная кислота = 80/20/0,001) с получением 700 мг 4-(((4-(5-хлор-2-(((1R,4r)-4-(((R)-1-метоксипропил-2-ил)амино)циклогексил)амино)пиридин-4-ил)тиазол-2-ил)амино)метил)тетрагидро-2Н-пиран-4-карбонитрила в форме бледно-желтого твердого вещества, выход 19,1%. 1H ЯМР (400 МГц, CDCl3) δ 8,06 (s, 1Н), 7,38 (s, 1H), 6,97 (s, 1H), 5,92 (brs, 1H), 4,45 (d, J=8,0 Гц, 1H), 4,02 (dd, J1=2,8 Гц, J2=12 Гц, 2H), 3,71-3,74 (m, 4H), 3,54-3,56 (m, 1H), 3,35 (s, 3H), 3,21-3,25 (m, 2H), 3,00-3,05 (m, 1H), 2,50-2,60 (m, 1H), 2,15 (d, J=9,6 Гц, 2H), 2,04-2,07 (m, 1H), 1,95 (d, J=12,8 Гц, 3H), 1,74-1,82 (m, 3H), 1,10-1,30 (m, 4H), 1,00 (d, J=8,4 Гц, 3Н), МС (ESI): m/z 519,3 (M+H)+.
Пример 2: синтез (1r,4r)-N1-(5-хлор-4-(2-(((тетрагидро-2Н-пиран-4-ил)метил)амино)тиазол-4-ил) пиридин-2-ил)циклогексан-1,4-диамина
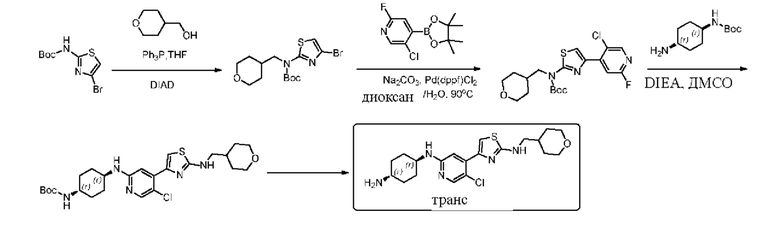
Стадия 1: синтез трет-бутил (4-бромтиазол-2-ил)((тетрагидро-2Н-пиран-4-ил)метил)карбамата
Трет-бутил 4-бромтиазол-2-карбамат (12,53 г, 107,91 ммоль), (тетрагидро-2Н-пиран-4-ил)метанол (20 г, 71,94 ммоль) и трифенилфосфин добавляли к 360 мл безводного ТЕФ (после двойной перегонки), который охлаждали до -10°С, и затем добавляли диизопропил азодикарбоксилат DIAD (21,82 г, 107,91 ммоль). Смесь перемешивали при комнатной температуре в течение 10 минут, и затем нагревали до 50°С, и реакцию проводили в течение 3 часов. По данным ТСХ исходные материалы исчезли. Затем реакционный раствор концентрировали. Остаток растворяли в дихлорметане, смешивали с силикагелем и выделяли с применением колонки (петролейный эфир / этилацетат = 30/1, 20/1) с получением 21 г трет-бутил (4-бромтиазол-2-ил)((тетрагидро-2Н-пиран-4-ил)метил)карбамата в форме белого твердого вещества, выход 87,5%, МС (ESI): m/z 519,3 (М+Н)+.
Стадия 2: синтез трет-бутил (4-(5-хлор-2-фторпиридин-4-ил)тиазол-2-ил)((тетрагидро-2Н-пиран-4-ил)метил)карбамата
Трет-бутил (4-бромтиазол-2-ил)((тетрагидро-2Н-пиран-4-ил)метил)карбамат (21 г, 1,51 ммоль), 5-хлор-2-фтор-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин (30 г, 3,0 ммоль), Pd(dppf)Cl2 (2,04 г, 0,151) и Na2CO3 (15 г, 3,78 ммоль) добавляли к 500 мл диоксана и 100 мл воды, которые защищали азотом, затем нагревали до 90°С, и реакцию проводили в течение ночи. Реакцию контролировали посредством ТСХ и LCMS. Когда трет-бутил (4-бромтиазол-2-ил)((тетрагидро-2Н-пиран-4-ил)метил)карбамат был полностью израсходован, реакцию останавливали. Реакционный раствор охлаждали, и затем добавляли воду (100 мл). Затем водную фазу экстрагировали этилацетатом (3×100 мл). Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали, и концентрировали с получением неочищенного вещества в форме желтовато-коричневого масла. Остаток выделяли с помощью хроматографии (петролейный эфир / этилацетат = 30:1, 25:1) с получением 19,4 г трет-бутил (4-(5-хлор-2-фторпиридин-4-ил)тиазол-2-ил)((тетрагидро-2Н-пиран-4-ил)метил)карбамата в форме белого твердого вещества, выход 81,5%, МС (ESI): m/z 428,1 (М+Н)+.
Стадия 3: синтез трет-бутил ((1r,4r)-4-((5-хлор-4-(2-(((тетрагидро-2Н-пиран-4-ил)метил)амино)тиазол-4-ил)пиридин-2-ил)амино)циклогексил) карбамата
Трет-бутил (4-(5-хлор-2-фторпиридин-4-ил)тиазол-2-ил)((тетрагидро-2Н-пиран-4-ил)метил)карбамат и трет-бутил (1r,4r)-(4-аминоциклогексил) карбамат добавляли к ДМСО, и добавляли диизопропилэтиламин DIEA. Смесь нагревали до 100°С, и реакцию проводили в течение 2 дней. Когда по данным ТСХ исходные материалы исчезли, нагревание прекращали, и реакционную смесь обрабатывали. Реакционный раствор охлаждали до комнатной температуры и выливали в ледяную воду. Водный слой экстрагировали дихлорметаном (3 х 200 мл). Органические фазы промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия и фильтровали. Фильтрат смешивали с силикагелем и выделяли с применением колонки (петролейный эфир / этилацетат = 3/1, 2:1, 1:1) с получением 3,6 г трет-бутил ((1r,4r)-4-((5-хлор-4-(2-(((тетрагидро-2Н-пиран-4-ил) метил)амино)тиазол-4-ил)пиридин-2-ил)амино)циклогексил)карбамата в форме бледно-желтого твердого вещества, выход 40%, МС (ESI): m/z 522,2 (М+Н)+.
Стадия 4: синтез (1r,4r)-N1-(5-хлор-4-(2-(((тетрагидро-2Н-пиран-4-ил) метил)амино)тиазол-4-ил)пиридин-2-ил)циклогексан-1,4-диамина
Трет-бутил ((1r,4r)-4-((5-хлор-4-(2-(((тетрагидро-2Н-пиран-4-ил)метил) амино)тиазол-4-ил)пиридин-2-ил)амино)циклогексил)карбамат (2,9 г, 5,56 ммоль) добавляли к смеси тетрагидрофуран / дихлорметан (20 мл / 20 мл), которую защищали азотом, и охлаждали до 0°С, затем по каплям добавляли 20 мл трифторуксусной кислоты. Проводили реакцию смеси в течение 2 ч при комнатной температуре. Реакцию контролировали посредством ТСХ. Реакционный раствор концентрировали, и затем медленно выливали в ледяную воду. Смесь экстрагировали дихлорметаном (3 × 30 мл). Органическую фазу промывали насыщенным солевым раствором, сушили над сульфатом натрия, фильтровали, и концентрировали с получением неочищенного вещества. Неочищенное вещество взбалтывали со смесью дихлорметан: этилацетат = 2:1, фильтровали, и сушили с получением 1,6 г (1r,4r)-N1-(5-хлор-4-(2-(((тетрагидро-2Н-пиран-4-ил)метил)амино)тиазол-4-ил)пиридин-2-ил)циклогексан-1,4-диамина в форме белого твердого вещества, выход 68%, 1Н ЯМР (400 МГц, CDCl3) δ 8,06 (s, 1Н), 7,33 (s, 1H), 6,96 (s, 1H), 5,21-5,30 (m, 1H), 4,32 (d, J=8,0 Гц, 1H)), 3,99-4,03 (m, 2H), 3,53-3,61 (m, 1H), 3,38-3,44 (m, 2H), 3,23(t, J=6,4 Гц, 2H), 2,68-2,74 (m, 1H), 2,11-2,13 (m, 2H), 1,85-2,13 (m, 3H), 1,70-1,73 (m, 2H), 1,10-1,45 (m, 7H). МС (ESI): m/z 422,2 (M+H)+.
Пример 3: синтез N-((1r,4r)-4-((5-хлор-4-(2-(((тетрагидро-2Н-пиран-4-ил)метил)амино)тиазол-4-ил)пиридин-2-ил)амино)циклогексил)ацетамида
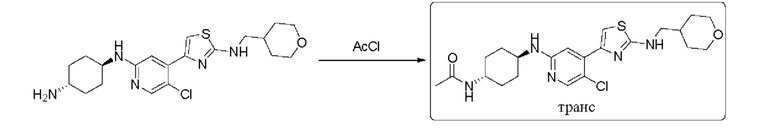
(1r,4r)-N1-(5-хлор-4-(2-(((тетрагидро-2Н-пиран-4-ил)метил)амино)тиазол-4-ил)пиридин-2-ил)циклогексан-1,4-диамин (0,422 г, 1 ммоль) растворяли в 10 мл дихлорметана, который защищали азотом, и добавляли ацетилхлорид. Осаждалось большое количество твердых веществ, и по данным ТСХ исходный материал был полностью израсходован. Смесь фильтровали, взбалтывали с метил трет-бутиловым эфиром и сушили с получением 187 мг N-((1r,4r)-4-((5-хлор-4-(2-(((тетрагидро-2Н-пиран-4-ил)метил)амино)тиазол-4-ил)пиридин-2-ил)амино)циклогексил)ацетамида в форме белого твердого вещества, выход 41%, 1Н ЯМР (400 МГц, CDCl3) δ 8,06 (s, 1H), 7,33 (s, 1H), 6,96 (s, 1H), 5,30-5,34 (m, 1H), 5,20-5,30 (m, 1H), 4,32 (d, J=8,0 Гц, 1H), 3,99-4,03 (m, 2H), 3,78-3,83 (m, 1H), 3,62-3,64 (m, 1H), 3,41 (t, J=12 Гц, 2H), 3,24 (t, J=6,4 Гц, 1H), 2,13-2,15 (m, 2H), 2,00-2,09 (m, 2H), 1,95 (s, 3H), 1,70-1,73 (m, 2H), 1,20-1,49 (m, 7H). MC (ESI): m/z 464,1 (M+H)+.
Пример 4: синтез (1r,4r)-N1-(5-хлор-4-(2-(((тетрагидро-2Н-пиран-4-ил)метил)амино)тиазол-4-ил)пиридин-2-ил)-N4-(2-метоксиэтил)циклогексан-1,4-диамина
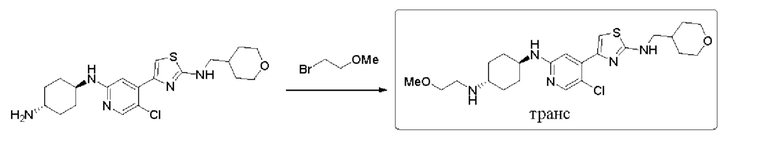
(1r,4r)-N1-(5-хлор-4-(2-(((тетрагидро-2Н-пиран-4-ил)метил)амино)тиазол-4-ил)пиридин-2-ил)циклогексан-1,4-диамин (0,357 г, 0,846 ммоль), 2-бромэтилметиловый эфир (0,118 г, 0,846 ммоль) и карбонат калия (0,116 г, 0,846 ммоль) добавляли к 10 мл ДМФА, который защищали азотом, и затем нагревали до 100°С, и реакцию проводили в течение двух дней. Реакцию контролировали посредством ТСХ и LCMS, и обрабатывали после остановки реакции. Реакционный раствор охлаждали, и затем выливали в ледяную воду (20 мл). Затем водную фазу экстрагировали этилацетатом (3×20 мл). Органическую фазу промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали, и концентрировали с получением неочищенного вещества в форме желтовато-коричневого масла. Остаток выделяли с применением колоночной хроматографии (дихлорметан/метанол = 20:1, 15:1, 10:1) с получением 0,070 г (1r,4r)-N1-(5-хлор-4-(2-(((тетрагидро-2Н-пиран-4-ил)метил)амино)тиазол-4-ил) пиридин-2-ил)-N4-(2-метоксиэтил)циклогексан-1,4-диамина в форме бледно-желтого твердого вещества, выход 17%, 1Н ЯМР (400 МГц, CDCl3) δ 8,06 (s, 1Н), 7,33 (s, 1H), 6,96 (s, 1H), 5,65 (brs, 1H), 4,40 (d, J=8,0 Гц, 1H), 3,95-4,06 (m, 2H), 3,49-3,70 (m, 3H), 3,28-3,45 (m, 5H), 3,18 (t, J=6,4 Гц, 1H), 2,96-3,05 (m, 2H), 2,76-2081 (m, 1H), 2,14-2,28 (m, 6H), 1,85-1,95 (m, 3H), 1,70-1,73 (m, 2H), 1,41-1,60 (m, 2H), 1,13-1,40 (m, 5H). MC (ESI): m/z 480,3 (M+H)+.
Пример 5: синтез (1S,4r)-N1-(5-хлор-4-(2-(((тетрагидро-2Н-пиран-4-ил)метил)амино)тиазол-4-ил)пиридин-2-ил)-N4-((5)-1-метоксипропан-2-ил)циклогексан-1,4-диамина
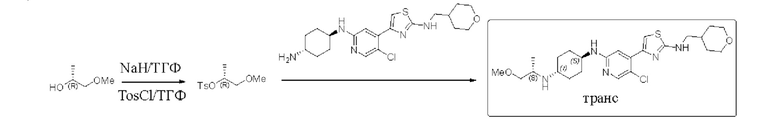
Стадия 1: синтез (R)-1-метоксипропан-2-ол 4-метилбензолсульфоната
Гидрид натрия NaH (1,46 г, 0,037 ммоль) добавляли к сухому тетраги дрофу рану ТГФ (1 л), который охлаждали до 0°С при охлаждении льдом и защищали азотом, и затем по каплям добавляли (R)-(-)-1-метоксипропан-2-ол (3 г, 0,033 ммоль). После окончания добавления по каплям, смесь нагревали до комнатной температуры и перемешивали в течение 1,5 часов. Реакционный раствор снова охлаждали до 0°С, и затем по каплям добавляли раствор п-толуолсульфонил хлорида TosCl (6,47 г, 0,034 ммоль) в тетрагидрофуране ТГФ (80 мл). При добавлении температура составляла ниже 10°С. После добавления смесь перемешивали при комнатной температуре (32°С) в течение ночи. По данным ТСХ исходный материал был полностью израсходован. Реакцию гасили посредством добавления по каплям насыщенного водного раствора хлорида аммония (20 мл) при охлаждении льдом, и разделяли. Водную фазу дважды экстрагировали этилацетатом (30 мл). Органические фазы объединяли, промывали насыщенным солевым раствором (50 мл), сушили над безводным сульфатом натрия, фильтровали, и концентрировали с получением неочищенного вещества в форме бледно-желтого масла. Неочищенное вещество выделяли с применением колонки (петролейный эфир / этилацетат = 5/1) с получением 4,2 г (R)-1-метоксипропан-2-ол 4-метилбензолсульфоната в форме бледно-желтого масла, выход 52%, МС (ESI): m/z 245,1 (М+Н)+.
Стадия 2: синтез (1S,4r)-N1-(5-хлор-4-(2-(((тетрагидро-2Н-пиран-4-ил)метил)амино)тиазол-4-ил)пиридин-2-ил)-N4-((8)-1-метоксипропан-2-ил)циклогексан-1,4-диамина
(1r,4r)-N1-(5-хлор-4-(2-(((тетрагидро-2Н-пиран-4-ил)метил)амино)тиазол-4-ил)пиридин-2-ил)циклогексан-1,4-диамин (600 мг, 1,2 ммоль), (R)-1-метокси пропан-2-ол 4-метилбензолсульфонат (293 мг, 1,42 ммоль) и карбонат калия (327 мг, 2,4 ммоль) добавляли к 20 мл ацетонитрила, который защищали азотом, и нагревали до 90°С и перемешивали в течение ночи. Реакцию контролировали с помощью LC-MS. Реакционный раствор охлаждали до комнатной температуры, фильтровали, и концентрировали с получением неочищенного вещества в форме бледно-желтого масла. Неочищенное вещество выделяли с помощью пластины с толстым слоем, используемой в препаративных целях (дихлорметан/метанол = 8/1) с получением 30 мг (1S,4r)-N1-(5-хлор-4-(2-(((тетрагидро-2Н-пиран-4-ил)метил)амино)тиазол-4-ил)пиридин-2-ил)-N4-((8)-1-метоксипропан-2-ил)циклогексан-1,4-диамина в форме белого твердого вещества, выход 4,3%, 1Н ЯМР (600 МГц, CDCl3) δ 8,06 (s, 1H), 7,29 (s, 1H), 6,96 (s, 1H), 5,59 (brs, 1H), 4,36 (d, J=8,0 Гц, 1H), 3,95-4,06 (m, 2H), 3,49-3,65 (m, 2H), 3,40-3,49 (m, 1H), 3,22-3,39 (m, 6H), 3,11-3,20 (m, 2H), 2,95-3,10 (m, 1H), 2,08-2,30 (m, 4H), 1,79-1,96 (m, 2H), 1,62-1,71 (m, 2H), 1,09-1,40 (m, 12H), 0,72-0,98 (m, 2H). MC (ESI): m/z 494,3 (M+H)+.
Пример 6: синтез (1R,4r)-N1-(5-хлор-4-(2-(((тетрагидро-2Н-пиран-4-ил)метил)амино)тиазол-4-ил)пиридин-2-ил)-N4-((R)-1-метоксипропан-2-ил)циклогексан-1,4-диамина
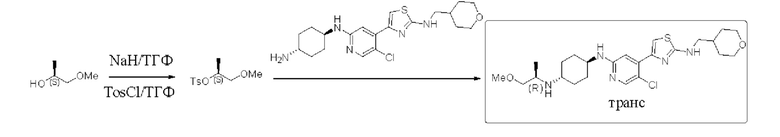
Стадия 1: синтез (8)-1-метоксипропан-2-ол 4-метилбензолсульфоната Гидрид натрия NaH (60%, 1,46 г, 0,037 моль) добавляли к сухому тетраги дрофу рану ТГФ (1 L), который охлаждали до 0°С при охлаждении льдом и защищали азотом, и затем по каплям добавляли (S)-(+)-1-метоксипропан-2-ол (3 г, 0,033 моль). После окончания добавления по каплям, смесь нагревали до комнатной температуры и перемешивали в течение 1,5 часов. Реакционный раствор снова охлаждали до 0°С, и затем по каплям добавляли раствор п-толуолсульфонил хлорида TosCl в тетрагидрофуране ТГФ. При добавлении температура составляла ниже 10°С. После добавления смесь перемешивали при комнатной температуре (32°С) в течение ночи. По данным ТСХ исходный материал был полностью израсходован. Реакцию гасили посредством добавления по каплям насыщенного водного раствора хлорида аммония (20 мл) при охлаждении льдом, и разделяли. Водную фазу дважды экстрагировали этилацетатом (30 мл). Органические фазы объединяли, промывали насыщенным солевым раствором (50 мл), сушили над безводным сульфатом натрия, фильтровали, и концентрировали с получением неочищенного вещества в форме бледно-желтого масла. Неочищенное вещество выделяли с применением колонки (петролейный эфир / этилацетат = 5/1) с получением 4,5 г (S)-1-метоксипропан-2-ол 4-метилбензолсульфоната в форме бледно-желтого масла, выход 55%, МС (ESI): m/z 245,1 (М+Н)+.
Стадия 2: синтез (1R,4r)-N1-(5-хлор-4-(2-(((тетрагидро-2Н-пиран-4-ил)метил) амино)тиазол-4-ил)пиридин-2-ил)-N4-((R)-1-метоксипропан-2-ил)циклогексан-1,4-диамина
(1r,4r)-N1-(5-хлор-4-(2-(((тетрагидро-2Н-пиран-4-ил)метил)амино)тиазол-4-ил)пиридин-2-ил)циклогексан-1,4-диамин (422 мг, 1 ммоль), (S)-1-метоксипропан-2-ол 4-метилбензолсульфонат (122 мг, 0,5 ммоль) и карбонат калия (276 мг, 2 ммоль) добавляли к 15 мл ацетонитрила, который защищали азотом и нагревали до 90°С и перемешивали в течение ночи. Реакцию контролировали с помощью LC-MS до ее осуществления на 25%. Реакционный раствор охлаждали до комнатной температуры, фильтровали, и концентрировали с получением неочищенного вещества в форме бледно-желтого масла. Неочищенное вещество выделяли с помощью пластины с толстым слоем, используемой в препаративных целях (дихлорметан/метанол = 8/1) с получением 83 мг (1R,4r)-N1-(5-хлор-4-(2-(((тетрагидро-2Н-пиран-4-ил)метил)амино)тиазол-4-ил)пиридин-2-ил)-N4-((R)-1-метоксипропан-2-ил)циклогексан-1,4-диамина в форме белого твердого вещества, выход 17%), 1Н ЯМР (400 МГц, CDCl3) 5 8,06 (s, 1Н), 7,33 (s, 1H), 6,96 (s, 1H), 5,30 (brs, 1H), 4,37 (d, J=8,0 Гц, 1H), 3,99-4,03 (m, 2H), 3,52-3,59 (m, 1H), 3,25-3,49 (m, 4H), 3,36 (s, 3H), 3,16-3,25 (m, 2H), 3,06-3,10 (m, 1H), 2,60-2,65 (m, 1H), 2,16 (d, J=10,8 Гц, 2H), 2,00-2,08 (m, 2H), 1,89-1,95 (m, 2H), 1,33-1,45 (m, 4H), 1,12-1,29 (m, 4H), 1,07 (d, J=6,4 Гц, 3Н). МС (ESI): m/z 494,2 (M+H)+.
Пример 7: синтез 4-(2-((((1r,4r)-4-аминоциклогексил)метил)амино)-5-хлорпиридин-4-ил)-N-((тетрагидро-2Н-пиран-4-ил)метил)тиазол-2-амина
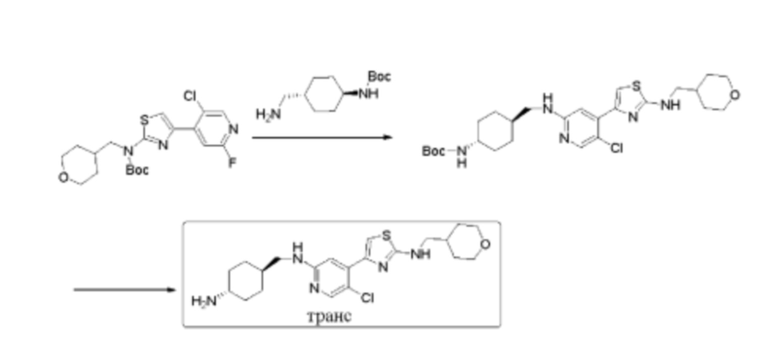
Стадия 1: синтез трет-бутил ((1r,4r)-4-(((5-хлор-4-(2-(((тетрагидро-2Н-пиран-4-ил)метил)амино)тиазол-4-ил)пиридин-2-ил)амино)метил)циклогексил)карбамата
Трет-бутил (4-(5-хлор-2-фторпиридин-4-ил)тиазол-2-ил) ((тетрагидро-2Н-пиран-4-ил)метил)карбамат (0,7 г, 1,6 ммоль), трет-бутил (1r,4r)-4-(аминометил) циклогексилкарбамат (0,748 г, 3,2 ммоль) и триэтиламин (0,458 г, 4,8 ммоль) добавляли к 10 мл диметилсульфоксида. Смесь нагревали до 110°С и перемешивали в течение 48 часов. По данным ТСХ исходный материал был полностью израсходован. После охлаждения до комнатной температуры реакционный раствор выливали в ледяную воду. Смесь экстрагировали этилацетатом (3×20 мл). Органические фазы объединяли, промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Остаток выделяли с применением колоночной хроматографии (петролейный эфир / этилацетат = 10:1, 2:1) с получением трет-бутил ((1r,4r)-4-(((5-хлор-4-(2-(((тетрагидро-2Н-пиран-4-ил)метил)амино) тиазол-4-ил)пиридин-2-ил)амино)метил)циклогексил)карбамата в форме желтого твердого вещества, выход 26%, МС (ESI): m/z 536,2 (М+Н)+.
Стадия 2: синтез 4-(2-((((1r,4r)-4-аминоциклогексил)метил)амино)-5-хлорпиридин-4-ил)-N-((тетрагидро-2Н-пиран-4-ил)метил)тиазол-2-амина
Трет-бутил ((1r,4r)-4-(((5-хлор-4-(2-(((тетрагидро-2Н-пиран-4-ил)метил)амино) тиазол-4-ил)пиридин-2-ил)амино)метил)циклогексил)карбамат (230 мг, 0,43 ммоль) добавляли к дихлорметану (10 мл), который защищали азотом, и охлаждали до 0°С, и затем по каплям добавляли трифторуксусную кислоту. Реакция в смеси проходила в течение 1 часа при комнатной температуре. Реакцию контролировали посредством ТСХ. Реакционный раствор концентрировали, и затем медленно выливали в ледяную воду. Смесь экстрагировали дихлорметаном (3×30 мл). Органическую фазу промывали насыщенным солевым раствором, сушили над сульфатом натрия, фильтровали, и концентрировали с получением неочищенного вещества. Неочищенное вещество выделяли с помощью пластины с толстым слоем, используемой в препаративных целях (дихлорметан/метанол = 5/1) с получением 0,065 г 4-(2-((((1r,4r)-4-аминоциклогексил)метил)амино)-5-хлорпиридин-4-ил)-N-((тетрагидро-2Н-пиран-4-ил)метил)тиазол-2-амина в форме бледно-желтого масла, выход 34,8%, 1Н ЯМР (400 МГц, MeOD) δ 7,82 (s, 1Н), 7,11 (s, 1H), 6,95 (s, 1H), 3,84-3,88 (m, 2H), 3,32 (t, J=11,2 Гц, 2H), 3,16-3,17 (m, 2H), 3,16 (d, J=6,8 Гц, 2H), 3,04 (d, J=6,8 Гц, 2H), 2,75-2,80 (m, 1H), 1,81-1,92 (m, 5H), 1,61-1,64 (m, 2H), 1,49-1,51 (m, 1H), 1,12-1,29 (m, 5H), 0,92-1,05 (m, 2H). MC (ESI): m/z 436,3 (M+H)+.
Пример 8: синтез N-(5-хлор-4-(2-(((тетрагидро-2Н-пиран-4-ил)метил)амино)тиазол-4-ил)пиридин-2-ил)-4-фторбензамида
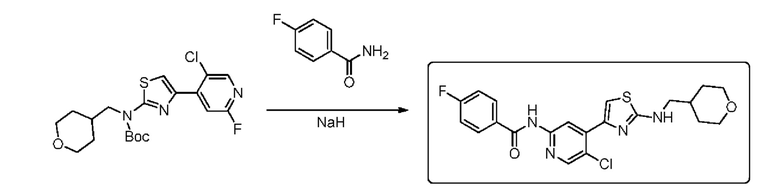
4-фторбензамид (0,65 г, 4,68 ммоль) растворяли в N,N-диметилформамиде ДМФА (15 мл), и добавляли NaH (0,19 г, 4,68 ммоль) при комнатной температуре. Реакционный раствор перемешивали при комнатной температуре в течение 10 мин, и затем добавляли трет-бутил (4-(5-хлор-2-фторпиридин-4-ил)тиазол-2-ил ((тетрагидро-2Н-пиран-4-ил)метил)карбамат (1 г, 2,34 ммоль). Реакционный раствор нагревали до 55°С, и реакцию проводили в течение 4 ч. Реакцию контролировали посредством ТСХ. Реакцию останавливали, и затем реакционный раствор выливали в воду и экстрагировали посредством ЕА (3×20 мл). Органическую фазу промывали насыщенным солевым раствором, сушили над сульфатом натрия, фильтровали, и концентрировали с получением неочищенного вещества. Неочищенное вещество выделяли с помощью пластины с толстым слоем, используемой в препаративных целях (РЕ:ЕА=1:1) с получением 0,032 г N-(5-хлор-4-(2-(((тетрагидро-2Н-пиран-4-ил)метил)амино)тиазол-4-ил)пиридин-2-ил)-4-фторбензамида в форме белого твердого вещества, выход 3,1%, 1Н ЯМР (400 МГц, CDCl3) δ 8,96 (s, 1Н), 8,52 (s, 1H), 8,30 (s, 1H), 7,93-7,96 (m, 2H), 7,41 (s, 1H), 7,19 (t, J=8,4 Гц, 2H), 5,35-5,38 (m, 1H), 4,00-4,04 (m, 2H), 3,40-3,50 (m, 2H), 3,24 (t, J=6,4 Гц, 2H), 1,95-2,01 (m, 1H), 1,72-1,76 (m, 2H), 1,36-1,45 (m, 2H). (ESI+): m/z 447,1 [M+H]+.
Пример 9: синтез (1r,4r)-N1-(5-хлор-4-(2-(метиламино)тиазол-4-ил)пиридин-2-ил)-N4-(2-метоксиэтил)циклогексан-1,4-диамина
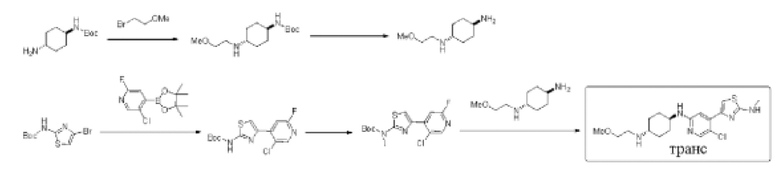
Стадия 1: синтез трет-бутил ((1r,4r)-4-((2-метоксиэтил)амино)циклогексил) карбамата
Трет-бутил (1r,4r)-(4-аминоциклогексил)карбамат (10,0 г, 46,7 ммоль), 2-бромэтилметиловый эфир (5,2 г, 37,4 ммоль) и карбонат калия (12,9 г, 93,4 ммоль) добавляли к ацетонитрилу (150 мл). Реакционную смесь перемешивали при 80°С в течение 16 ч. Реакцию контролировали посредством ТСХ. Когда оставалось небольшое количество исходного материала, реакцию останавливали. Реакционный раствор охлаждали до комнатной температуры и фильтровали. Фильтрат сушили с помощью роторного испарителя, смешивали с силикагелем и выделяли с помощью колоночной хроматографии на силикагеле (дихлорметан/метанол = 20:1) с получением 6,3 г трет-бутил ((1r,4r)-4-((2-метоксиэтил)амино)циклогексил) карбамата в форме желтовато-белого твердого вещества. Выход 50%, МС (ESI): m/z 273,2 (М+Н)+.
Стадия 2: синтез (1r,4r)-N1-(2-метоксиэтил)циклогексан-1,4-диамина Трет-бутил ((1r,4r)-4-((2-метоксиэтил)амино)циклогексил)карбамат (6 г, 22,0 ммоль) растворяли в смеси разбавленная хлористоводородная кислота-тетрагидрофуран (80 мл). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч, и осаждалось большое количество твердого вещества. Реакционный раствор фильтровали. Остаток сушили с получением 5,1 г (1r,4r)-N1-(2-метоксиэтил)циклогексан-1,4-диамина (дигидрохлорида) в форме белого твердого вещества, выход 94,8%, МС (ESI): m/z 173,2 (М+Н)+.
Стадия 3: синтез трет-бутил (4-(5-хлор-2-фторпиридин-4-ил)тиазол-2-ил) карбамата
Трет-бутил 4-бромтиазол-2-ил карбамат (20,0 г, 71,7 ммоль), 5-хлор-2-фтор-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин (37,0 г, 143,4 ммоль), Pd(dppf)Cl2 (2,6 г, 0,151) и Na2CO3 (22,8 г, 245 ммоль) растворяли в смеси 1,4-диоксан/H2O (350 мл/40 мл), которую трижды продували азотом, и затем перемешивали при 90°С в течение 16 ч. Реакцию контролировали с помощью LCMS. После того, как исходные материалы оставались в незначительном количестве, добавляли 5-хлор-2-фтор-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин (18,5 г, 71,7 ммоль). Реакционную смесь трижды продували азотом и перемешивали при 85°С в течение дополнительных 18 ч. Реакцию контролировали с помощью LCMS. Примерно 95% исходных материалов превращались в продукты. Реакционный раствор охлаждали до комнатной температуры и фильтровали. Фильтрат сушили с помощью роторного испарителя, смешивали с силикагелем и выделяли с помощью колоночной хроматографии на силикагеле (петролейный эфир / этилацетат = 10:1) с получением 11,0 г трет-бутил (4-(5-хлор-2-фторпиридин-4-ил)тиазол-2-ил)карбамата в форме белого твердого вещества, выход 47%, и других 10 г неочищенного продукта. МС (ESI): m/z 330,0 (М+Н)+.
Стадия 4: синтез трет-бутил (4-(5-хлор-2-фторпиридин-4-ил)тиазол-2-ил) (метил)карбамата
Трет-бутил (4-(5-хлор-2-фторпиридин-4-ил)тиазол-2-ил)карбамат (200 мг, 0,61 ммоль) и трифенилфосфин (239 мг, 0,91 ммоль) растворяли в ТГФ (4 мл), который трижды продували азотом, и добавляли метанол МеОН (78 мг, 2,43 ммоль). Смесь перемешивали при комнатной температуре в течение 1 минуты, и затем добавляли диизопропил азодикарбоксилат DIAD (184 мг, 0,91 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч. По данным ТСХ исходный материал был полностью израсходован. Реакционный раствор выделяли с помощью препаративной тонкослойной хроматографии с применением в качестве проявляющего растворителя смеси петролейный эфир / этилацетат = 10:1 с получением 205 мг трет-бутил (4-(5-хлор-2-фторпиридин-4-ил)тиазол-2-ил)(метил)карбамата в форме белого твердого вещества, выход 98%, МС (ESI): m/z 344,1 (М+Н)+.
Стадия 5: синтез (1r,4r)-N1-(5-хлор-4-(2-(метиламино)тиазол-4-ил)пиридин-2-ил)-N4-(2-метоксиэтил)циклогексан-1,4-диамина
Трет-бутил (4-(5-хлор-2-фторпиридин-4-ил)тиазол-2-ил)(метил)карбамат (200 мг, 0,58 ммоль), (1r,4r)-N1-(2-метоксиэтил)циклогексан-1,4-диамин (150 мг, 0,64 ммоль), диизопропилэтиламин DIEA (375 мг, 2,9 моль), и фторид цезия (265 мг, 1,74 ммоль) растворяли в диметилсульфоксиде (3 мл). Реакционную смесь перемешивали при 120°С в течение 2 дней. Реакцию контролировали с помощью LCMS. После образования продукта к реакционному раствору добавляли воду (40 мл). Смесь экстрагировали этилацетатом (2×30 мл). Экстракт сушили над безводным сульфатом натрия, концентрировали с применением роторного испарителя, и затем выделяли с помощью препаративной тонкослойной хроматографии с применением в качестве проявляющего растворителя смеси дихлорметан/метанол = 6:1 с получением 80 мг (1r,4r)-N1-(5-хлор-4-(2-(метиламино)тиазол-4-ил)пиридин-2-ил)-N4-(2-метоксиэтил) циклогексан-1,4-диамина в форме бледно-желтого твердого вещества, выход 35%, 1Н ЯМР (400 МГц, ДМСО) δ 7,97 (s, 1H), 7,61-7,62 (m, 1Н), 7,29 (s, 1Н), 7,04 (s, 1H), 6,70 (d, J=7,6 Гц, 1Н), 3,59-3,61 (m, 2Н), 3,37-3,42 (m, 3Н), 3,25 (s, 3Н), 2,87 (d, J=4,8 Гц, 2Н), 2,74-2,77 (m, 2Н), 1,90-1,96 (m, 4Н), 1,12-1,23 (m, 4Н). (ESI+): m/z 396,2 [М+Н]+.
Пример 10: синтез (1r,4r)-N1-(5-хлор-4-(2-((циклогексилметил)амино)тиазол-4-ил)пиридин-2-ил)-N4-(2-метоксиэтил)циклогексан-1,4-диамина
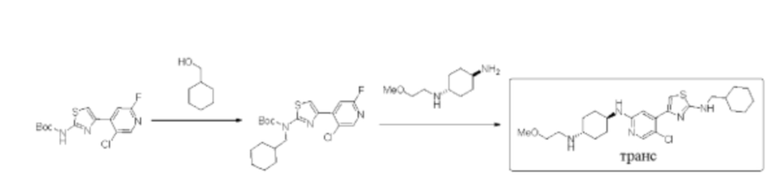
Стадия 1: синтез трет-бутил (4-(5-хлор-2-фторпиридин-4-ил)тиазол-2-ил) (циклогексилметил)карбамата
Трет-бутил (4-(5-хлор-2-фторпиридин-4-ил)тиазол-2-ил)карбамат (200 мг, 0,61 ммоль) и трифенилфосфин (239 мг, 0,91 ммоль) растворяли в ТГФ (5 мл), который трижды продували азотом, и добавляли циклогексилметанол (207 мг, 1,82 ммоль). Смесь перемешивали при комнатной температуре в течение 5 минут, и затем добавляли диизопропил азодикарбоксилат DIAD (184 мг, 0,91 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч. По данным ТСХ исходный материал был полностью израсходован. Реакционный раствор выделяли с помощью препаративной тонкослойной хроматографии с применением в качестве проявляющего растворителя смеси петролейный эфир / этилацетат = 10:1 с получением 255 мг трет-бутил (4-(5-хлор-2-фторпиридин-4-ил)тиазол-2-ил)(циклогексилметил) карбамата в форме белого твердого вещества, выход 99%, (ESI+): m/z 426,1 [М+Н]+.
Стадия 2: синтез (1r,4r)-N1-(5-хлор-4-(2-((циклогексилметил)амино)тиазол-4-ил)пиридин-2-ил)-N4-(2-метоксиэтил)циклогексан-1,4-диамина
Трет-бутил (4-(5-хлор-2-фторпиридин-4-ил)тиазол-2-ил)(циклогексилметил) карбамат (250 мг, 0,59 ммоль), (1r,4r)-N1-(2-метоксиэтил) циклогексан-1,4-диамин (288 мг, 1,17 ммоль), диизопропилэтиламин DIEA (379 мг, 2,93 моль), и фторид цезия (268 мг, 1,76 ммоль) растворяли в диметилсульфоксиде (8 мл). Реакционную смесь перемешивали при 120°С в течение 2 дней. Реакцию контролировали с помощью LCMS. После образования продукта к реакционному раствору добавляли воду (30 мл). Смесь экстрагировали смесью дихлорметан/изопропанол = 3:1 (2×30 мл). Экстракт промывали солевым раствором, сушили над безводным сульфатом натрия, концентрировали с применением роторного испарителя, затем смешивали с силикагелем и выделяли с помощью колоночной хроматографии на силикагеле (дихлорметан/метанол = 50:1→20:1) с получением неочищенного вещества в форме желтого масла. Неочищенное вещество выделяли с помощью препаративной тонкослойной хроматографии с применением в качестве проявляющего растворителя смеси дихлорметан/метанол = 8:1 с получением 100 мг (1r,4r)-N1-(5-хлор-4-(2-(циклогексилметил)амино)тиазол-4-ил)пиридин-2-ил)-N4-(2-метоксиэтил)циклогексан-1,4-диамина в форме бледно-желтого твердого вещества, выход 30%, 1Н ЯМР (400 МГц, ДМСО) δ 7,97 (s, 1H), 7,67-7,69 (m, 1Н), 7,25 (s, 1Н), 7,01 (s, 1Н), 6,71 (d, J=7,6 Гц, 1Н), 3,50-3,53 (m, 1Н), 3,36-3,47 (m, 2Н), 3,13 (t, J=6,0 Гц, 2Н), 2,94-2,97 (m, 2Н), 2,72-2,81 (m, 1H), 1,99-2,02 (m, 4Н), 1,61-1,77 (m, 5Н), 1,19-1,33 (m, 7Н), 0,91-1,01 (m, 2Н). (ESI+): m/z 478,3 [М+Н]+.
Пример 11: синтез (1r,4r)-N1-(4-(2-(бензиламино)тиазол-4-ил)-5-хлорпиридин-2-ил)-N4-(2-метоксиэтил)циклогексан-1,4-диамина
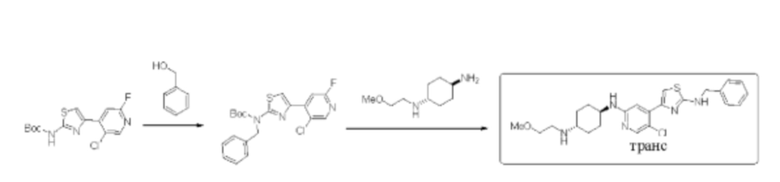
Стадия 1: синтез трет-бутил бензил (4-(5-хлор-2-фторпиридин-4-ил)тиазол-2-ил)карбамата
Трет-бутил (4-(5-хлор-2-фторпиридин-4-ил)тиазол-2-ил)карбамат (200 мг, 0,61 ммоль) и трифенилфосфин (239 мг, 0,91 ммоль) растворяли в ТГФ (5 мл), который трижды продували азотом, и затем добавляли бензиловый спирт (131 мг, 1,21 ммоль). Смесь перемешивали при комнатной температуре в течение 5 минут, и затем добавляли диизопропилэтиламин DIEA (184 мг, 0,91 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч. По данным ТСХ исходный материал был полностью израсходован. Реакционный раствор выделяли с помощью препаративной тонкослойной хроматографии с применением в качестве проявляющего растворителя смеси петролейный эфир / этилацетат = 8:1 с получением 246 мг трет-бутил бензил (4-(5-хлор-2-фторпиридин-4-ил)тиазол-2-ил)карбамата в форме белого твердого вещества, выход 97%, (ESI+): m/z 420,1 [М+Н]+.
Стадия 2: синтез (1r,4r)-N1-(4-(2-(бензиламино)тиазол-4-ил)-5-хлорпиридин-2-ил)-N4-(2-метоксиэтил)циклогексан-1,4-диамина
Трет-бутил бензил (4-(5-хлор-2-фторпиридин-4-ил)тиазол-2-ил)карбамат (240 мг, 0,57 ммоль), (1r,4r)-N1-(2-метоксиэтил)циклогексан-1,4-диамин (280 мг, 1,14 ммоль), диизопропилэтиламин DIEA(369 мг, 2,86 моль) и фторид цезия (268 мг, 1,71 ммоль) растворяли в диметилсульфоксиде (8 мл). Реакционную смесь перемешивали при 120°С в течение 3 дней. Реакцию контролировали с помощью LCMS. После образования продукта к реакционному раствору добавляли воду (30 мл). Смесь экстрагировали смесью дихлорметан/изопропанол = 3:1 (2×35 мл). Экстракт промывали солевым раствором, сушили над безводным сульфатом натрия, концентрировали с применением роторного испарителя, затем смешивали с силикагелем и выделяли с помощью колоночной хроматографии на силикагеле (дихлорметан/метанол = 20:1) с получением неочищенного вещества в форме желтого масла. Неочищенное вещество выделяли с помощью препаративной тонкослойной хроматографии с применением в качестве проявляющего растворителя смеси дихлорметан/метанол=6:1 с получением 100 мг (1r,4r)-N1-(4-(2-(бензиламино)тиазол-4-ил)-5-хлорпиридин-2-ил)-N4-(2-метоксиэтил)циклогексан-1,4-диамина в форме бледно-желтого твердого вещества, выход 30%, 1Н ЯМР (400 МГц, ДМСО) δ 8,21 (t, J=6,0 Гц, 1Н), 7,98 (s, 1Н), 7,26-7,40 (m, 6Н), 7,05 (s, 1Н), 6,72 (d, J=7,6 Гц, 1Н), 4,52 (d, J=5,6 Гц, 2Н), 3,46-3,53 (m, 4Н), 2,97 (brs, 2Н), 2,81 (brs, 1H), 1,99-2,01 (m, 4Н), 1,18-1,34 (m, 4Н). (ESI+): m/z 472,1 [М+Н]+.
Пример 12: синтез (1r,4r)-N1-(5-хлор-4-(2-((4-фторбензил)амино) тиазол-4-ил)пиридин-2-ил)-N4-(2-метоксиэтил)циклогексан-1,4-диамина
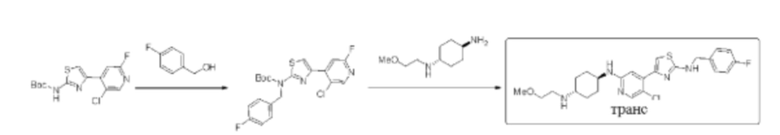
Стадия 1: синтез трет-бутил (4-(5-хлор-2-фторпиридин-4-ил)тиазол-2-ил) (4-фторбензил)карбамата
Трет-бутил (4-(5-хлор-2-фторпиридин-4-ил)тиазол-2-ил)карбамат (200 мг, 0,61 ммоль) и трифенилфосфин (239 мг, 0,91 ммоль) растворяли в ТГФ (5 мл), который трижды продували азотом, и затем добавляли 4-фторбензиловый спирт (153 мг, 1,21 ммоль). Смесь перемешивали при комнатной температуре в течение 5 минут, и затем добавляли DIAD (184 мг, 0,91 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч. По данным ТСХ исходный материал был полностью израсходован. Реакционный раствор выделяли с помощью препаративной тонкослойной хроматографии с применением в качестве проявляющего раствора смеси РЕ/ЕА = 10:1 с получением 248 мг трет-бутил (4-(5-хлор-2-фторпиридин-4-ил)тиазол-2-ил) (4-фторбензил)карбамата в форме бледно-желтого твердого вещества, выход 93%, (ESI+): m/z 438,1 [М+Н]+.
Стадия 2: синтез (1r,4r)-N1-(5-хлор-4-(2-((4-фторбензил)амино) тиазол-4-ил)пиридин-2-ил)-N4-(2-метоксиэтил)циклогексан-1,4-диамина
Трет-бутил (4-(5-хлор-2-фторпиридин-4-ил)тиазол-2-ил) (4-фторбензил)карбамат (240 мг, 0,55 ммоль), (1r,4r)-N1-(2-метоксиэтил) циклогексан-1,4-диамин (268 мг, 1,10 ммоль), диизопропилэтиламин DIEA (353 мг, 2,74 моль), и фторид цезия (251 мг, 1,65 ммоль) растворяли в смеси диметилсульфоксид/N,N-диметилацетамид (3 мл/3 мл). Реакционную смесь перемешивали при 120°С в течение 3 дней. Реакцию контролировали с помощью LCMS. После образования продукта к реакционному раствору добавляли воду (35 мл). Смесь экстрагировали смесью дихлорметан/изопропанол = 3:1 (2×30 мл). Экстракт промывали солевым раствором, сушили над безводным сульфатом натрия, концентрировали с применением роторного испарителя, затем смешивали с силикагелем и выделяли с помощью колоночной хроматографии на силикагеле (дихлорметан/метанол = 20:1) с получением неочищенного вещества в форме желтого масла. Неочищенное вещество выделяли с помощью препаративной тонкослойной хроматографии с применением в качестве проявляющего растворителя смеси дихлорметан/метанол = 8:1 с получением 100 мг (1r,4r)-N1-(5-хлор-4-(2-((4-фторбензил))амино)тиазол-4-ил)пиридин-2-ил)-N4-(2-метоксиэтил)циклогексан-1,4-диамина в форме бледно-желтого твердого вещества, выход 30%, 1Н ЯМР (400 МГц, ДМСО) δ 8,22-8,24 (m, 1H), 7,98 (s, 1H), 7,41-7,44 (m, 2Н), 7,32 (s, 1H), 7,18 (t, J=8,8 Гц, 2Н), 7,04 (s, 1H), 6,73 (d, J=7,6 Гц, 1Н), 4,50 (d, J=5,6 Гц, 2Н), 3,52-3,55 (m, 3Н), 3,29 (s, 3Н), 2,96 (brs, 2Н), 2,97 (brs, 1H), 1,98-2,01 (m, 4Н), 1,21-1,23 (m, 4Н). (ESI+): m/z 490,2 [М+Н]+.
Пример 13: синтез (1r,4r)-N1-(5-хлор-4-(2-((циклопропилметил)амино)тиазол-4-ил)-пиридин-2-ил)-N4-(2-метоксиэтил)-циклогексан-1,4-диамина
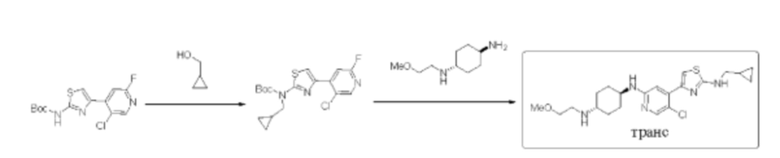
Стадия 1: синтез трет-бутил (4-(5-хлор-2-фторпиридин-4-ил)тиазол-2-ил)(циклопропилметил)карбамата
Трет-бутил (4-(5-хлор-2-фторпиридин-4-ил)тиазол-2-ил)карбамат (200 мг, 0,61 ммоль) и трифенилфосфин (239 мг, 0,91 ммоль) растворяли в тетраги дрофу ране (5 мл), который трижды продували азотом, и затем добавляли циклопропилметанол (131 мг, 1,82 ммоль). Смесь перемешивали при комнатной температуре в течение 5 минут, и затем добавляли диизопропил азодикарбоксилат DIAD (184 мг, 0,91 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч. По данным ТСХ исходный материал был полностью израсходован. Реакционный раствор выделяли с помощью препаративной тонкослойной хроматографии с применением в качестве проявляющего растворителя смеси петролейный эфир / этилацетат = 10:1 с получением 230 мг трет-бутил (4-(5-хлор-2-фторпиридин-4-ил)тиазол-2-ил)(циклопропилметил)карбамата в форме желтовато-белого твердого вещества, выход 98%, (ESI+): m/z 384,1 [М+Н]+.
Стадия 2: синтез (1r,4r)-N1-(5-хлор-4-(2-((циклопропилметил)амино)тиазол-4-ил)пиридин-2-ил)-N4-(2-метоксиэтил)циклогексан-1,4-диамина
Трет-бутил(4-(5-хлор-2-фторпиридин-4-ил)тиазол-2-ил)(циклопропил метил)карбамат (220 мг, 0,57 ммоль), (1r,4r)-N1-(2-метоксиэтил) цикло гексан-1,4-диамин (280 мг, 1,15 ммоль), диизопропилэтиламин DIEA (370 мг, 2,86 моль), и фторид цезия (262 мг, 1,72 ммоль) растворяли в смеси диметилсульфоксид/N,N-диметилацетамид (3 мл/3 мл). Реакционную смесь перемешивали при 120°С в течение 2 дней. Реакцию контролировали с помощью LCMS. После образования продукта к реакционному раствору добавляли воду (35 мл). Смесь экстрагировали смесью дихлорметан/изопропанол = 3:1 (2×30 мл). Экстракт промывали солевым раствором, сушили над безводным сульфатом натрия, концентрировали с применением роторного испарителя, затем смешивали с силикагелем и выделяли с помощью колоночной хроматографии на силикагеле (дихлорметан/метанол = 20:1) с получением неочищенного вещества в форме желтого масла. Неочищенное вещество выделяли с помощью препаративной тонкослойной хроматографии с применением в качестве проявляющего растворителя смеси дихлорметан/метанол = 7:1 с получением 100 мг (1r,4r)-N1-(5-хлор-4-(2-((циклопропилметил)амино)тиазол-4-ил)пиридин-2-ил)-N4-(2-метоксиэтил)циклогексан-1,4-диамина в форме бледно-желтого твердого вещества, выход 35%, 1Н ЯМР (400 МГц, ДМСО) δ 7,97 (s, 1Н), 7,84 (t, J=5,6 Гц, 1H), 7,28 (s, 1Н), 7,04 (s, 1H), 7,74 (d, J=8,0 Гц, 2Н), 3,52-3,55 (m, 3Н), 3,29 (s, 3Н), 3,17 (t, J=6,4 Гц, 2Н), 2,94 (brs, 1H), 2,70-2,85 (m, 1H), 1,97-2,01 (m, 4H), 1,18-1,23 (m, 5H), 0,46-0,49 (m, 2H), 0,23-0,24 (m, 2H). (ESI+): m/z 436,3 [M+H]+.
Пример 14: синтез 4-((4-(5-хлор-2-(((1r,4r)-4-((2-метоксиэтил)амино)циклогексил)амино)пиридин-4-ил)тиазол-2-иламино)метил)тетрагидро-2Н-пиран-4-карбонитрила
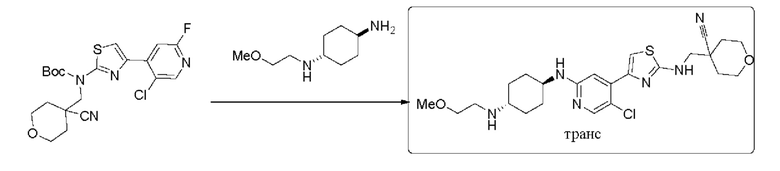
Трет-бутил (4-(5-хлор-2-фторпиридин-4-ил)тиазол-2-ил)((4-циано-тетрагидро-2Н-пиран-4-ил)метил)карбамат (250 мг, 0,55 ммоль), (1r,4r)-N1-(2-метоксиэтил) циклогексан-1,4-диамин (270 мг, 1,10 ммоль), диизопропилэтиламин DIEA (355 мг, 2,75 моль), и фторид цезия (251 мг, 1,65 ммоль) растворяли в смеси диметилсульфоксид/N,N-диметилацетамид (3 мл/3 мл). Реакционную смесь перемешивали при 120°С в течение 3 дней. Реакцию контролировали с помощью LCMS. После образования продукта к реакционному раствору добавляли воду (30 мл). Смесь экстрагировали смесью дихлорметан/изопропанол = 3:1 (3×30 мл). Экстракт сушили над безводным сульфатом натрия, концентрировали с применением роторного испарителя, затем смешивали с силикагелем и выделяли с помощью колоночной хроматографии на силикагеле (дихлорметан/метанол = 10:1) с получением неочищенного вещества в форме желтого твердого вещества. Неочищенное вещество выделяли с помощью препаративной тонкослойной хроматографии с применением в качестве проявляющего растворителя смеси дихлорметан/метанол = 5:1 с получением 80 мг 4-((4-(5-хлор-2-(((1r,4r)-4-((2-метоксиэтил)амино)циклогексил)амино)пиридин-4-ил) тиазол-2-иламино)метил)тетрагидро-2Н-пиран-4-карбонитрила в форме бледно-желтого твердого вещества, выход 28%, 1Н ЯМР (400 МГц, ДМСО) δ 8,13 (m, J=6,0 Гц, 1H), 7,99 (s, 1H), 7,35 (s, 1Н), 7,03 (s, 1H), 6,71 (d, J=7,6 Гц, 1Н), 3,91-3,95 (m, 2H), 3,67 (d, J=6,4 Гц, 2H), 3,45-3,54 (m, 6Н), 3,30 (s, 3Н), 2,98 (brs, 2Н), 2,81 (brs, 1H), 2,00-2,02 (m, 4H), 1,86-1,89 (m, 2H), 1,69-1,72 (m, 2H), 1,19-1,32 (m, 5H). (ESI+): m/z 505,3 [M+H]+.
Пример 15: синтез 4-(((4-(5-хлор-2-(((1S,4r)-4-(((5)-1-метокси пропил-2-ил)амино)циклогексил)амино)пиридин-4-ил)тиазол-2-ил)амино)метил) тетрагидро-2Н-пиран-4-карбонитрила
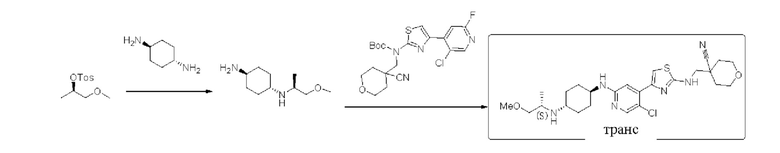
Стадия 1: синтез (1r,4S)-N1-(S)-1-метоксипропан-2-ил)циклогексан-1,4-диамина
(R)-1-метоксипропан-2-ол 4-метилбензолсульфонат (2,0 г, 8,2 ммоль) растворяли в ацетонитриле (20 мл), и добавляли шраяо1,4-циклогександиамин (2,34 г, 20,5 ммоль). Реакционную смесь перемешивали и кипятили с обратным холодильником при 85°С в течение 16 ч. По данным ТСХ исходный материал был полностью израсходован. Реакционный раствор охлаждали до комнатной температуры и фильтровали. Фильтрат сушили с помощью роторного испарителя, затем смешивали с силикагелем и выделяли с помощью колоночной хроматографии на силикагеле (дихлорметан/метанол (0,1% содержание 28% водного раствора аммиака) = 10:1) с получением 600 мг (1r,4S)-N1-((S)-1-метокси пропан-2-ил)циклогексан-1,4-диамина в форме желтого масла, выход 40%, (ESI+): m/z 187,2 [М+Н]+.
Стадия 2: синтез 4-(((4-(5-хлор-2-(((1S,4r)-4-(((S)-1-метоксипропил-2-ил)амино)циклогексил)амино)пиридин-4-ил)тиазол-2-ил)амино)метил)тетрагидро-2Н-пиран-4-карбонитрила
Трет-бутил (4-(5-хлор-2-фторпиридин-4-ил)тиазол-2-ил)((4-циано-тетра гидро-2Н-пиран-4-ил)метил)карбамат (200 мг, 0,44 ммоль), (1r,4S)-N1-((S)-1-метоксипропан-2-ил)циклогексан-1,4-диамин (200 мг, 1,08 ммоль) и диизопропилэтиламин DIPEA (284 мг, 2,9 моль) растворяли в диметилсульфоксиде (2 мл). Реакционную смесь перемешивали при 130°С в течение 2,5 дней. Реакцию контролировали с помощью LCMS. После образования продукта к реакционному раствору добавляли воду (30 мл). Смесь экстрагировали смесью дихлорметан/изопропанол = 3:1 (3×30 мл). Экстракт сушили над безводным сульфатом натрия, концентрировали с применением роторного испарителя, затем смешивали с силикагелем и выделяли с помощью колоночной хроматографии на силикагеле (дихлорметан/метанол = 10:1) с получением неочищенного вещества в форме коричневого масла. Неочищенное вещество выделяли с помощью препаративной тонкослойной хроматографии с применением в качестве проявляющего растворителя смеси дихлорметан/метанол = 8:1 с получением 50 мг 4-(((4-(5-хлор-2-(((1S,4r)-4-(((S)-1-метоксипропил-2-ил)амино)циклогексил)амино) пиридин-4-ил)тиазол-2-ил)амино)метил)тетрагидро-2Н-пиран-4-карбонитрила в форме бледно-желтого твердого вещества, выход 22%, 1Н ЯМР (400 МГц, ДМСО) δ 8,12 (m, J=6,0 Гц, 1Н), 7,98 (s, 1Н), 7,35 (s, 1H), 7,03 (s, 1Н), 6,69 (d, J=8,0 Гц, 1Н), 3,91-3,95 (m, 2Н), 3,66 (d, J=6,4 Гц, 2Н), 3,55-3,65 (m, 1H), 3,47-3,51 (m, 3Н), 3,29 (s, 3Н), 3,17 (d, J=4,8 Гц, 1H), 1,86-1,99 (m, 6Н), 1,66-1,74 (m, 2Н), 0,99-1,26 (m, 8Н). (ESI+): m/z 519,3 [М+Н]+.
Пример 16: синтез (1r,4r)-N1-(5-хлор-4-(2-((тетрагидро-2Н-пиран-4-ил)метокси)тиазол-4-ил)пиридин-2-ил)-N4-(2-метоксиэтил)циклогексан-1,4-диамина
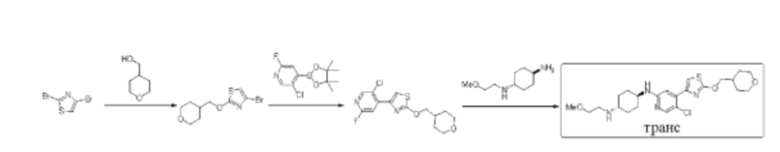
Стадия 1: синтез 4-бром-2-((тетрагидро-2Н-пиран-4-ил)метокси) тиазола
(Тетрагидро-2Н-пиран-4-ил)метанол (5,0 г, 20,8 ммоль) растворяли в 50 мл тетрагидрофурана (50 мл), и затем добавляли NaH (996 мг, 24,9 ммоль). Смесь перемешивали в течение 10 минут при комнатной температуре, и затем добавляли 2,4-дибромтиазол (5,0 г, 20,8 ммоль). Затем смесь перемешивали при комнатной температуре в течение ночи. После добавления к реакционному раствору 100 мл насыщенного раствора хлорида аммония, смесь дважды экстрагировали этилацетатом, по 50 мл каждый раз. Затем органические фазы объединяли, сушили над безводным сульфатом натрия, и затем концентрировали с применением роторного испарителя. Остаток выделяли с применением колоночной хроматографии (петролейный эфир: этилацетат = 100:1) с получением 4,2 г 4-бром-2-((тетрагидро-2Н-пиран-4-ил)метокси)тиазола в форме белого твердого вещества, выход 73%. (ESI+): m/z 278,0 [М+Н]+.
Стадия 2: синтез 4-(5-хлор-2-фторпиридин-4-ил)-2-((тетрагидро-2Н-пиран-4-ил)метокси)тиазола
4-бром-2-((тетрагидро-2Н-пиран-4-ил)метокси)тиазол (2,0 г, 7,22 ммоль) и 5-хлор-2-фтор-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин (3,71 г, 14,44 ммоль) добавляли к смешанному растворителю из 20 мл диоксана и 4 мл воды, и дополнительно добавляли Pd(dppf)Cl2 (161 мг, 0,22 ммоль) и Na2CO3 (2,3 г, 21,66 ммоль). Под защитой азотом смесь нагревали до 80°С и перемешивали в течение ночи. Затем к реакционному раствору добавляли 50 мл воды, смесь дважды экстрагировали этилацетатом, по 50 мл каждый раз. Затем органические фазы объединяли, сушили над безводным сульфатом натрия, и затем концентрировали с применением роторного испарителя. Остаток выделяли с применением колоночной хроматографии (петролейный эфир: этилацетат = 30:1) с получением 1,45 г 4-(5-хлор-2-фторпиридин-4-ил)-2-((тетрагидро-2Н-пиран-4-ил)метокси)тиазола в форме белого твердого вещества. Выход 61,2%. (ESI+): m/z 329,1 [М+Н]+.
Стадия 3: синтез (1r,4r)-N1-(5-хлор-4-(2-((тетрагидро-2Н-пиран-4-ил) метокси)тиазол-4-ил)пиридин-2-ил)-N4-(2-метоксиэтил)циклогексан-1,4-диамина
4-(5-хлор-2-фторпиридин-4-ил)-2-((тетрагидро-2Н-пиран-4-ил)метокси)тиазол (300 мг, 0,915 ммоль), (1r,4r-N1-(2-метоксиэтил)циклогексан-1,4-диамин (245 мг, 1,006 ммоль) и K2CO3 (104 мг, 2,745 ммоль) добавляли к 5 мл ДМСО. Затем смесь при перемешивании нагревали до 100°С, и реакцию проводили в течение 48 часов. Реакцию контролировали с помощью LCMS, и большая часть исходных материалов полностью прореагировала. Реакционный раствор охлаждали до комнатной температуры, и затем добавляли 50 мл воды. Затем смесь дважды экстрагировали этилацетатом, по 10 мл каждый раз. Органические фазы объединяли, сушили над безводным сульфатом натрия, и затем концентрировали с применением роторного испарителя. Полученный неочищенный продукт выделяли с помощью колоночной хроматографии (дихлорметан : метанол = 20:1). В итоге получали 111,0 мг (1r,4r)-N1-(5-хлор-4-(2-((тетрагидро-2Н-пиран-4-ил)-метокси)тиазол-4-ил)пиридин-2-ил)-N4-(2-метоксиэтил)циклогексан-1,4-диамина в форме бледно-коричневого твердого вещества, выход 25,3%. 1Н ЯМР (400 МГц, CDCl3) δ 8,07 (s, 1H), 7,50 (s, 1H), 6,95 (s, 1H), 4,39 (d, J=8,0 Гц, 1H), 4,31 (d, J=6,4 Гц, 2H), 4,03 (dd, J=3,2 Гц, J=11,2 Гц, 2H), 3,59-3,61 (m, 1H), 3,54 (t, J=4,8 Гц, 2H), 3,44-3,47 (m, 2H), 3,37 (s, 3H), 2,85 (t, J=5,2 Гц, 2H), 2,53-2,54 (m, 1H), 2,15-2,18 (m, 3H), 2,00-2,03 (m, 2H), 1,73-1,76 (m, 2H), 1,46-1,52 (m, 2H), 1,15-1,37 (m, 5H). (ESI+): m/z 481,2 [M+H]+.
Пример 17: синтез (1r,4r)-N1-(5-хлор-4-(2-(((тетрагидро-2Н-пиран-4-ил)метил)меркапто)тиазол-4-ил)пиридин-2-ил)-N4-(2-метоксиэтил)циклогексан-1,4-диамина
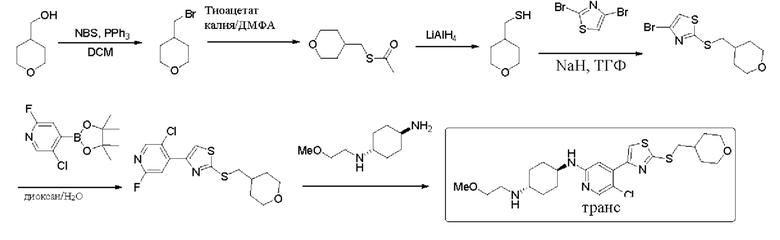
Стадия 1: синтез 4-(бромметил)-тетрагидро-2Н-пирана
(Тетрагидро-2Н-пиран-4-ил)метанол (8,12 г, 10 ммоль) и N-бромсукцинимид NBS (13,71 г, 2448 ммоль) добавляли к 400 мл дихлорметана, который охлаждали до 0°С, и затем медленно частями добавляли трифенилфосфор. Реакционную смесь перемешивали при комнатной температуре в течение 1-2 часов, и по данным ТСХ исходные материалы исчезли. Реакционный раствор выливали в воду (100 мл). Смесь экстрагировали дихлорметаном. Органическую фазу промывали насыщенным солевым раствором, сушили над сульфатом натрия, и выделяли с применением колоночной хроматографии (петролейный эфир / этилацетат = 20/1) с получением 6,2 г 4-(бромметил)-тетрагидро-2Н-пирана в форме бесцветной жидкости, выход 49%, (ESI+): m/z 179,0 [М+Н]+.
Стадия 2: синтез метил 8-(тетрагидро-2Н-пиран-4-ил) тиоацетата
4-(бромметил)-тетрагидро-2Н-пиран и тиоацетат калия добавляли к 60 мл ДМФА. Смесь нагревали до 90°С, и реакцию проводили в течение 2 часов. Когда по данным ТСХ исходные материалы исчезли, нагревание прекращали, и реакционную смесь обрабатывали. Реакционный раствор охлаждали до комнатной температуры, и выливали в ледяную воду. Смесь экстрагировали этилацетатом (3×30 мл). Органическую фазу промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, и фильтровали. Фильтрат смешивали с силикагелем и выделяли с применением колонки (петролейный эфир / этилацетат = 30/1, 20:1, 10:1) с получением 1,8 г метил 8-(тетрагидро-2Н-пиран-4-ил) тиоацетата в форме желтого масла, выход 69%. (ESI+): m/z 175,1 [М+Н]+.
Стадия 3: синтез (тетрагидро-2Н-пиран-4-ил)метил меркаптана
Метил S-(тетрагидро-2Н-пиран-4-ил) тиоацетат добавляли к ТГФ, который защищали азотом и охлаждали до 0°С, медленно партиями добавляли алюмогидрид лития, и реакцию проводили в течение ночи. Реакцию контролировали посредством ТСХ. Реакционный раствор разбавляли тетрагидрофураном (50 мл), и медленно партиями добавляли подходящее количество декагидрата сульфата натрия. Смесь перемешивали в течение 10 мин, фильтровали, и концентрировали с получением 0,68 г неочищенного продукта (тетрагидро-2Н-пиран-4-ил)метил меркаптана в форме желтого масла, выход 100%. (ESI+): m/z 133,1 [М+Н]+.
Стадия 4: синтез 4-бром-2-(((тетрагидро-2Н-пиран-4-ил)метил)сульфидрил) тиазола
(Тетрагидро-2Н-пиран-4-ил)метил меркаптан (0,632 г, 4,8 ммоль) растворяли в тетраги дрофу ране ТГФ, который защищали азотом и охлаждали до 0°С, и затем медленно партиями добавляли гидрид натрия NaH (0,2 г, 4,8 ммоль), и реакцию проводили при комнатной температуре в течение 10 мин. По каплям добавляли раствор 2,4-дибромтиазола в 30 мл тетрагидрофурана ТГФ, и реакцию проводили в течение ночи. Когда по данным ТСХ реакция была почти завершена, реакцию останавливали. Реакционный раствор выливали в насыщенный хлорид аммония и гасили. Смесь экстрагировали этилацетатом (3×30 мл). Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали, и концентрировали с получением неочищенного вещества в форме желтовато-коричневого масла. Неочищенное вещество выделяли с применением колоночной хроматографии (петролейный эфир / этилацетат = 25:1, 20:1) с получением 0,7 г 4-бром-2-(((тетрагидро-2Н-пиран-4-ил)метил) сульфидрил)тиазола в форме почти белого твердого вещества, выход 60%. (ESI+): m/z 294,0 [М+Н]+.
Стадия 5: синтез 4-(5-хлор-2-фторпиридин-4-ил)-2-(((тетрагидро-2Н-пиран-4-ил)метил)сульфидрил)тиазола
4-бром-2-(((тетрагидро-2Н-пиран-4-ил)метил)сульфидрил)тиазол (0,45 г, 1,512 ммоль), 5-хлор-2-фтор-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил) пиридин (0,788 г, 3,0 ммоль), тетракистрифенилфосфин палладий Pd(PPh3)4 (0,18 г, 0,151 ммоль) и карбонат натрия (0,405 г, 3,78 ммоль) добавляли к 20 мл диоксана и 4 мл воды, которые защищали азотом, затем нагревали до 90°С, и реакцию проводили в течение ночи. Реакцию контролировали посредством ТСХ и LCMS. При полном исчезновении исходных материалов реакцию останавливали. Реакционный раствор охлаждали, и затем добавляли воду (80 мл). Смесь экстрагировали этилацетатом (3×30 мл). Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали, и концентрировали с получением неочищенного вещества в форме желтовато-коричневого масла. Неочищенное вещество выделяли с помощью хроматографии (петролейный эфир / этилацетат = 30:1, 25:1) с получением 0,27 г 4-(5-хлор-2-фторпиридин-4-ил)-2-(((тетрагидро-2Н-пиран-4-ил)метил)сульфидрил)тиазола в форме желтого масла, выход 42%. (ESI+): m/z 345,0 [М+Н]+.
Стадия 6: синтез (1r,4r)-N1-(5-хлор-4-(2-(((тетрагидро-2Н-пиран-4-ил)метил)меркапто)тиазол-4-ил)пиридин-2-ил)-N4-(2-метоксиэтил)циклогексан-1,4-диамина
4-(5-Хлор-2-фторпиридин-4-ил)-2-(((тетрагидро-2Н-пиран-4-ил)метил)сульфидрил)тиазол (0,27 г, 0,756 ммоль), (1r,4r)-N1-(2-метоксиэтил)циклогексан-1,4-диамин (0,203 г, 0,831 ммоль) и K2CO3 (0,313 г, 2,268 ммоль) добавляли к ДМСО, который защищали азотом, и затем нагревали до 100°С, и реакцию проводили в течение двух дней. Реакцию контролировали посредством ТСХ и LCMS. Исходный материал 4-(5-хлор-2-фторпиридин-4-ил)-2-(((тетрагидро-2Н-пиран-4-ил)метил)сульфидрил)тиазол оставался, и реакцию останавливали. Реакционную смесь охлаждали и разбавляли этилацетатом (20 мл), воду (800 мл) добавляли на ледяной бане, и разделяли. Затем водную фазу экстрагировали этилацетатом (2×20 мл). Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали, и концентрировали с получением неочищенного вещества в форме желтовато-коричневого масла. Неочищенное вещество выделяли с помощью хроматографии (дихлорметан/метанол = 15:1) с получением 0,135 г (1r,4r)-N1-(5-хлор-4-(2-(((тетрагидро-2Н-пиран-4-ил)метил)сульфидрил)тиазол-4-ил) пиридин-2-ил)-N4-(2-метоксиэтил)циклогексан-1,4-диамина в форме желтого твердого вещества, выход 34,5%. 1Н ЯМР (400 МГц, CDCl3) δ 8,07 (s, 1Н), 7,98 (s, 1H), 6,99 (s, 1H), 4,42 (brs, 1H), 3,91-4,10 (m, 2H), 3,55-3,71 (m, 3H), 2,83-3,52 (m, 12H), 2,13-2,17 (m, 4H), 1,95-2,05 (m, 1H), 1,69-1,87 (m, 2H), 1,31-1,56 (m, 5H), 1,02-1,35 (m, 4H), 0,79-0,95 (m, 1H). (ESI+): m/z 497,2 [M+H]+.
Пример 18: синтез (1r,4r)-N1-(2-метоксиэтил)-N4-(4-(2-(((тетрагидро-2Н-пиран-4-ил)метил)амино)тиазол-4-ил)пиридин-2-ил)циклогексан-1.4-диамина
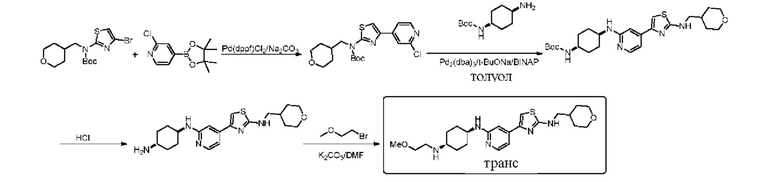
Стадия 1: синтез трет-бутил (4-(2-хлорпиридин-4-ил)тиазол-2-ил)((тетрагидро-2Н-пиран-4-ил)метил)карбамата
Трет-бутил (4-бромтиазол-2-ил)((тетрагидро-2Н-пиран-4-ил)метил) карбамат (1 г, 1,512 ммоль), 2-хлор-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин (0,95 г, 3,0 ммоль), Pd(dppf)Cl2 (0,22 г, 0,151 ммоль) и Na2CO3 (0,703 г, 3,78 ммоль) добавляли к 15 мл диоксана и 30 мл воды, которые защищали азотом, затем нагревали до 80°С, и реакцию проводили в течение ночи. Реакцию контролировали посредством ТСХ и LCMS. При полном исчезновении исходных материалов, реакцию останавливали. Реакционный раствор охлаждали, и затем добавляли воду (50 мл). Смесь экстрагировали этилацетатом (3×30 мл). Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали, и концентрировали с получением неочищенного вещества в форме желтовато-коричневого масла. Неочищенное вещество выделяли с помощью хроматографии (петролейный эфир / этилацетат = 30:1) с получением 0,27 г трет-бутил (4-(2-хлорпиридин-4-ил)тиазол-2-ил) ((тетрагидро-2Н-пиран-4-ил)метил)карбамата в форме желтого масла, выход 42%. (ESI+): m/z 410,1 [М+Н]+.
Стадия 2: синтез ((1r,4r)-4-((4-(2-(((тетрагидро-2Н-пиран-4-ил)метил)амино)тиазол-4-ил)пиридин-2-ил)амино)циклогексил)карбамата
Трет-бутил (4-(2-хлорпиридин-4-ил)тиазол-2-ил)((тетрагидро-2Н-пиран-4-ил) метил)карбамат (0,388 г, 0,95 ммоль), трет-бутил ((lr, 4 г)-4-аминоциклогексил) карбамат (0,244 г, 1,14 ммоль), Pd(dba)3 (0,026 г, 3,8 ммоль), натрия трет-бутоксид, (±)-2,2'-бис-(дифенилфосфино)-1,1'-бинафтил BINAP (0,035 г, 0,0285 ммоль) добавляли к толуолу, который защищали азотом, и затем нагревали до 120°С, и реакцию проводили в течение ночи. Реакцию контролировали посредством ТСХ. При полном исчезновении исходных материалов, реакцию останавливали. Реакционный раствор охлаждали и выливали в насыщенный водный раствор хлорида аммония (20 мл) и разделяли. Затем водную фазу экстрагировали этилацетатом (2 X 20 мл). Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали, и концентрировали с получением 600 мг ((1r,4r)-4-((4-(2-(((тетрагидро-2Н-пиран-4-ил)метил)амино)тиазол-4-ил)пиридин-2-ил) амино)циклогексил)карбамата в форме желтовато-коричневого масла. Неочищенный продукт непосредственно применяли на следующей стадии, и выход рассчитывали на следующей стадии. (ESI+): m/z 488,3 [М+Н]+.
Стадия 3: синтез (1r,4r)-N1-(4-(2-(((тетрагидро-2Н-пиран-4-ил)метил) амино)тиазол-4-ил)пиридин-2-ил)циклогексан-1,4-диамина
К метанолу добавляли трет-бутил ((1r,4r)-4-((4-(2-(((тетрагидро-2Н-пиран-4-ил)метил)амино)тиазол-4-ил)пиридин-2-ил)амино)циклогексил)карбамат (0,6 г, 0,95 ммоль), и добавляли 5 мл хлористоводородной кислоты (6 н.), и реакцию проводили в течение ночи. Затем реакционный раствор концентрировали, и добавляли насыщенный раствор бикарбоната натрия до рН=7. Затем водную фазу экстрагировали этилацетатом (2 X 20 мл) и концентрировали. Остаток замачивали в течение ночи в смеси дихлорметан: метанол=10:1, и фильтровали. Фильтрат концентрировали с получением 0,12 г (1r,4r)-N1-(4-(2-(((тетрагидро-2Н-пиран-4-ил)метил)амино) тиазол-4-ил)пиридин-2-ил)циклогексан-1,4-диамина в форме бледно-желтого масла, выход: 31% (2 стадии). (ESI+): m/z 388,2 [М+Н]+.
Стадия 4: синтез (1r,4r)-N1-(2-метоксиэтил)-N4-(4-(2-(((тетрагидро-2Н-пиран-4-ил)метил)амино)тиазол-4-ил)пиридин-2-ил)циклогексан-1,4-диамина
(1r,4r)-N1-(4-(2-(((тетрагидро-2Н-пиран-4-ил)метил)амино)тиазол-4-ил)пиридин-2-ил)циклогексан-1,4-диамин (0,66 г, 1,7 ммоль), 2-бромэтилметиловый эфир (0,240 г, 1,7 ммоль) и карбонат калия (0,235 г, 1,7 ммоль) добавляли к N,N диметилформамиду, который защищали азотом, и затем нагревали до 100°С, и реакцию проводили в течение двух дней. Реакцию контролировали посредством ТСХ и LCMS. Несмотря на то, что оставалось небольшое количество исходных материалов, реакцию останавливали. Реакционный раствор охлаждали, и затем добавляли воду (30 мл). Смесь экстрагировали этилацетатом (3×20 мл). Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали, и концентрировали с получением неочищенного вещества в форме желтовато-коричневого масла. Неочищенное вещество выделяли с помощью пластины с толстым слоем, используемой в препаративных целях (дихлорметан/метанол = 8:1) с получением 0,057 г (1r,4r)-N1-(2-метоксиэтил)-N4-(4-(2-(((тетрагидро-2Н-пиран-4-ил)метил)амино)тиазол-4-ил)пиридин-2-ил)циклогексан-1,4-диамина в форме желтого масла, выход 15%. 1Н ЯМР (400 МГц, CDCl3) δ 7,95-8,02 (m, 1Н), 6,79-6,89 (m, 3Н), 5,48 (brs, 1H), 4,65-4,85 (m, 1H), 3,98-4,01 (m, 2H), 3,57-3,61 (m, 3H), 3,35-3,42 (m, 5H), 3,18-3,20 (m, 2H), 2,89-2,92 (m, 2H), 2,61-2,68 (m, 1H), 2,17-2,27 (m, 3H), 2,04-2,08 (m, 3H), 1,87-1,96 (m, 1H), 1,69-1,73 (m, 2H), 1,20-1,50 (m, 10H), 0,79-0,95 (m, 2H). (ESI+): m/z 446,3 [M+H]+.
Пример 19: Эффект ингибиторов CDK9 на рост раковых клеток
Посредством тестирования эффекта ингибиторов CDK9 на рост раковых клеток, авторы оценили селективность соединений в отношении ингибирования пролиферации раковых клеток.
В примерах авторы применяли клеточную линию острого миелоцитарного лейкоза (AML) OCI-AML-3, клеточную линию острого промиелоцитарного лейкоза NB-4, клеточную линию MDS-RAEB (тип миелодиспластического синдрома с избытком бластов) SKM-1, клетки лейкоза человека Nomo-1, клеточную линию острого миелоидного лейкоза MOLM14, клеточную линию острого миелоидного лейкоза MOLM13, клеточную линию острого миелоидного лейкоза MV4-11, клеточную линию острого миелоидного лейкоза HL-60, клеточную линию острого миелоидного лейкоза OCI-AML-2, гистиоцитарную лимфому U-937, клеточную линию острого В-клеточного лейкоза МЕС-1, клеточную линию острого В-клеточного лейкоза МЕС-2, острый мегакариобластный лейкоз CMK, клетки легких хомячка CHL, клетки яичника хомяка СНО, клетки немелкоклеточного рака легких человека Н1975, клетки немелкоклеточного рака легкого человека Н358, клетки мелкоклеточного рака легкого человека Н209, клетки аденокарциномы легкого человека Н1395, клетки немелкоклеточного рака легких человека РС-9, клетки рака легких человека Н3122, клетки немелкоклеточного рака легких человека Н2122, клетки немелкоклеточного рака легких человека Н1915, клетки аденокарциномы легких человека Н1355, клетки немелкоклеточного рака легких человека НСС827, клетки рака молочной железы человека MDA-MB-231, клетки рака молочной железы человека MDA-MB-468, клетки рака молочной железы человека MCF-7, клетки рака молочной железы человека T47D, клетки рака молочной железы человека SK-Br-3, вышеуказанные клетки были приобретены у АТСС. Кроме того, в качестве контрольных соединений применяли палбоциклиб (селективный ингибитор CDK4/6, приобретенный у Shanghai Haoyuan Chemical), HY-16462 (CDK9-IN-2, приобретенный у Shanghai Haoyuan) и динациклиб (ингибитор CDK 1/2/5/9, приобретенный у Shanghai Haoyuan Chemical).
В примерах соединения согласно настоящему изобретению с различными концентрациями (0,000508 мкМ, 0,00152 мкМ, 0,00457 мкМ, 0,0137 мкМ, 0,0411 мкМ, 0,123 мкМ, 0,370 мкМ, 1,11 мкМ, 3,33 мкМ, 10 мкМ) и контрольные соединения отдельно добавляли к вышеуказанным клеткам, которые инкубировали в течение 72 часов. Набор для определения жизнеспособности клеток на основе химической люминесценции Cell Titer-Glo® (Promega, США) применяли для определения количества жизнеспособных клеток путем количественного измерения АТФ в живых клетках, согласно которому рассчитывали GI50 и IC50. Результаты показаны в таблицах 1-3: в таблицах 1 и 2 показан GI50 соединений согласно настоящему изобретению относительно протестированных клеточных линий заболевания системы крови; в таблице 3 показаны IC50 соединения 1 относительно типа раковых клеток, отличного от гематологического рака.
На основании результатов таблиц 1 и 2 было обнаружено, что протестированные соединения согласно настоящему изобретению оказывают сильный ингибирующий эффект на тестируемые раковые клетки, такие как клетки лейкоза и клетки лимфомы, и соединения 1 и 14 также показали хорошую селективность: ингибирующее действие на нормальные клетки CHL и СНО отсутствовало, в то время как контрольные лекарственные средства динациклиб и HY-16462 оказывали определенные ингибирующие эффекты на CHL и СНО. Результаты в таблице 3 также показали, что соединение 1 согласно настоящему изобретению также проявляло значительные ингибирующие эффекты на клетки немелкоклеточного рака легких человека, клетки мелкоклеточного рака легкого человека, клетки аденокарциномы легкого и клетки рака молочной железы, тогда как палбоциклиб не демонстрировал очевидного ингибирования на протестированные раковые клетки. Эти результаты послужили важной теоретической основой для применения соединения 1 в качестве менее токсичного селективного ингибитора киназы CDK9 для лечения этих видов рака.
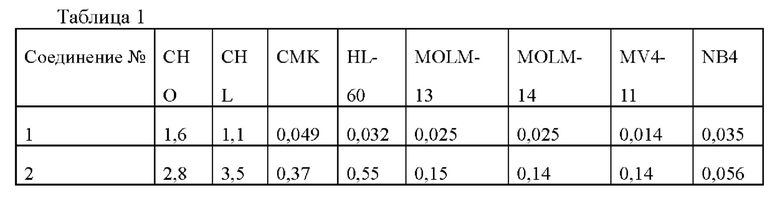
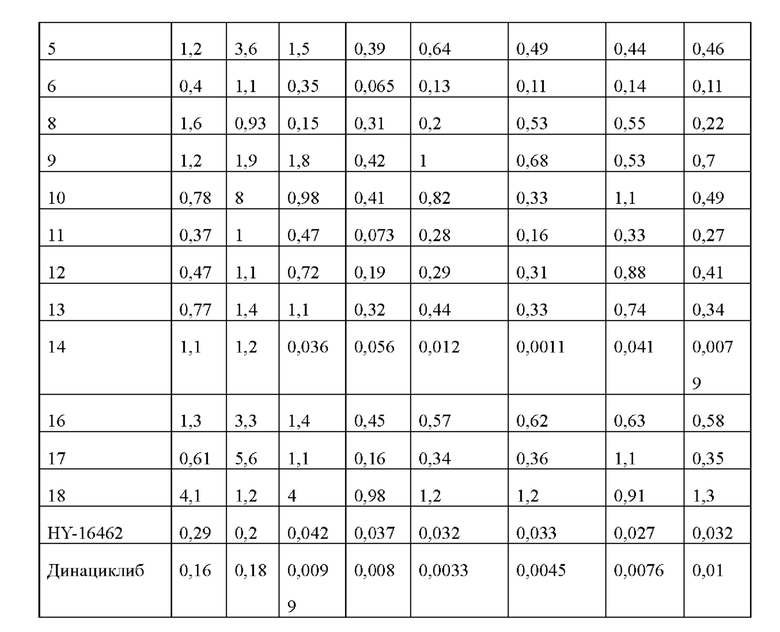
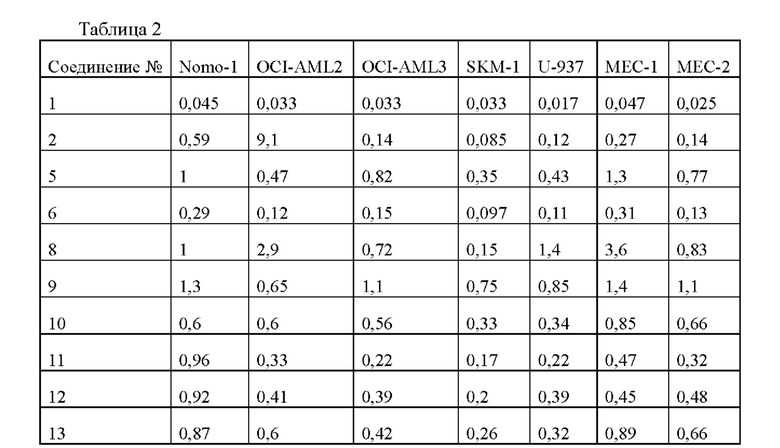
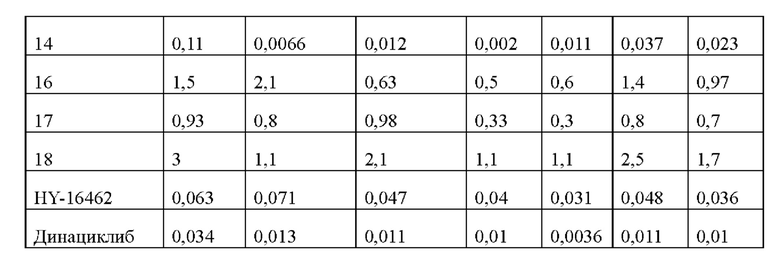
Пример 20: ферментативный анализ ингибирования белка CDK in vitro
Соединения 1 и 14, разведенные в ДМСО, смешивали с детектируемым белком CDK (Invitrogen, США) соответственно, инкубировали при комнатной температуре в течение 30 минут; и затем смешивали со смесью киназа / пептидный субстрат Z-LYTE™ (Invitrogen, США) и 4 × АТФ. Смешанную систему переносили на 384-луночный непрозрачный белый планшет для проведения реакции в течение 1 часа при комнатной температуре; добавляли 5 мкл проявляющего раствора (Invitrogen, США) для проведения реакции при комнатной температуре в течение 1 часа и, наконец, к реакционной смеси добавляли стоп-реагент (Invitrogen, США) для окончания реакции, и для считывания значения флуоресценции применяли считывающее устройство микропланшетов MD SpectraMax I3X (Molecular Devices, США). Значения IC50 для соединений 1 и 14 относительно тестируемого белка CDK рассчитывали на основе считанных значений флуоресценции с применением Prism 5,0 (GraphPad Software, Сан-Диего, Калифорния), и они показаны в таблице 4 ниже.
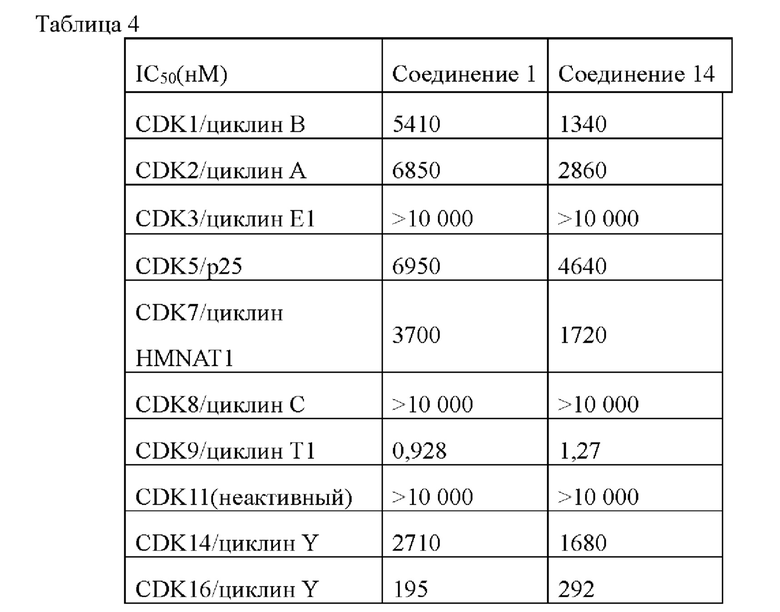
Пример 21: Эффект ингибиторов CDK9 на сигнальный путь
Эффект соединения 1 в клетках оценивали для CDK9 и других протеинкиназ, связанных с его сигнальным путем, например, RNAPII, XIAP, MCL-1, c-MYC, BCL-2 и подобными, относительно четырех типов клеток, клеточной линии острого миелоцитарного лейкоза (AML) OCI-AML-3, клеточной линии острого промиелоцитарного лейкоза NB-4, клеточной линии острого миелоцитарного лейкоза (AML) HL-60 и клеточной линии острого миелоцитарного лейкоза (AML) MV4-11 (все приобретены у АТСС), путем измерения биохимических конечных точек и функциональных конечных точек множества клеток. Соединение 1 (в ДМСО) в различных концентрациях 0 мкМ, 0,03 мкМ, 0,1 мкМ, 0,3 мкМ, 1 мкМ и 3 мкМ, а также лекарственные средства для сравнения динациклиб и HY-16462 (CDK9-IN-2) (приобретено в Shanghai Haoyuan) (в ДМСО) в 1 мкМ применяли для обработки этих клеточных линий в течение 2 часов, и затем производили отбор образцов. Был установлен эффект соединения 1 на фосфорилирование CDK9, RNAPII, XIAP, MCL-1, c-MYC, BCL-2 в этих клеточных линиях (фигуры 1a-d).
В четырех клеточных линиях: клеточной линии острого миелоцитарного лейкоза (AML) OCI-AML-3, клеточной линии острого промиелоцитарного лейкоза NB-4, клеточной линии острого миелоцитарного лейкоза (AML) HL-60 и клеточной линии острого миелоцитарного лейкоза (AML) MV4-11, было обнаружено, что соединение 1 оказывает значительный ингибирующий эффект на фосфорилирование RNAPII, MCL-1 и c-MYC непосредственно после белка CDK9.
Пример 22: Эффект новых ингибиторов киназы на апоптоз
Чтобы доказать, происходит ли гибель клеток в результате апоптоза или некроза, в четырех клеточных линиях, клеточной линии острого миелоцитарного лейкоза (AML) OCI-AML-3, клеточной линии острого промиелоцитарного лейкоза NB-4, клеточной линии острого миелоцитарного лейкоза (AML) HL-60 и клеточной линии острого миелоцитарного лейкоза (AML) MV4-11 (все приобретены у АТСС), в клетках обнаруживали эффект, который соединение 1 оказывает на фермент репарации ДНК полиаденозиндифосфат-рибоза-полимеразу PARP, тесно связанную с апоптозом, и расщепление белка цистеинзависимым аспартатспецифичным протеолитическим ферментом каспазой 3. Соединение 1 (в ДМСО) в различных концентрациях 0 мкМ, 0,01 мкМ, 0,03 мкМ и 0,1 мкМ, динациклиб (в ДМСО) в 0,01 мкМ и HY-16462 (в ДМСО) в 0,1 мкМ применяли для обработки различных клеток, и затем клетки собирали через 24 часа. Для выявления эффекта, оказываемого лекарственными средствами в различных концентрациях на фермент репарации ДНК полиаденозиндифосфат-рибоза-полимеразу PARP и расщепление белка цистеинзависимым аспартатспецифичным протеолитическим ферментом каспазой 3 в разные промежутки времени применяли вестерн-блоттинг.
Результаты эксперимента показаны на фигурах 2a-d: в четырех клеточных линиях: клеточной линии острого миелоцитарного лейкоза (AML) OCI-AML-3, клеточной линии острого промиелоцитарного лейкоза NB-4, клеточной линии острого миелоцитарного лейкоза (AML) HL-60 и клеточной линии острого миелоцитарного лейкоза (AML) MV4-11, очевидно, было обнаружено, что произошло расщепление фермента частичной репарации ДНК полиаденозиндифосфат-рибоза-полимеразы PARP или расположенной далее каспазы 3 PARP. Это продемонстрировало, что соединение 1 может вызывать апоптоз в четырех типах клеток, клеточной линии острого миелоцитарного лейкоза (AML) OCI-AML-3, клеточной линии острого промиелоцитарного лейкоза NB-4, клеточной линии острого миелоцитарного лейкоза (AML) HL-60 и клеточной линии острого миелоцитарного лейкоза (AML) MV4-11.
Пример 23: эффект новых ингибиторов киназы на клеточный цикл
Для изучения того, в каком цикле после введения предотвращалось развитие клеток в трех клеточных линиях: клеточной линии острого промиелоцитарного лейкоза NB-4, клеточной линии острого миелоцитарного лейкоза (AML) HL-60 и клеточной линии острого миелоцитарного лейкоза (AML) MV4-11, тестировали эффект соединения 1 на степень развития клеточного цикла в этих клеточных линиях. Соединение 1 в различных концентрациях 0 мкМ, 0,01 мкМ, 0,03 мкМ и 0,1 мкМ (в ДМСО), ингибитор киназы CDK9 динациклиб в 0,01 мкМ (в ДМСО) и HY-16462 в 0,1 мкМ (в ДМСО) применяли для обработки клеточных линий HL-60, MV4-11 или NB-4, в течение 12 часов, 24 часов или 48 часов, и затем клетки собирали, дважды промывали 1 × PBS-буфером, фиксировали 75% этанолом при -20°С в течение 24 часов, и затем дважды промывали 1 × PBS-буфером. К клеткам добавляли 0,5 мл 1 × буфера PBS и 0,5 мл окрашивающего раствора PI (приобретенного у BD Bioscience, США), и клетки помещали в темноту при 37°С на 15 минут для окрашивания. Степень развития клеточного цикла определяли с помощью проточной цитометрии (BD FACS Calibur). Результаты показаны на фигурах 3а-с.
Результаты показаны на фигурах 3а-с: после обработки трех клеточных линий, клеточной линии острого миелоцитарного лейкоза (AML) HL-60, клеточной линии острого миелоцитарного лейкоза (AML) MV4-11 и клеточной линии острого промиелоцитарного лейкоза NB-4, в течение 12 часов, 24 часов или 48 часов, соответственно, было обнаружено, что соединение 1 оказывало влияние на клеточный цикл этих трех клеток, то есть соединение 1 блокировало клетки в фазе G0-G1.
Пример 24: Экспериментальные результаты соединения 1 в мышиной модели острого гранулоцитарного лейкоза MV4-11
24 самки мышей Bal b/c, 4-6 недель, были приобретены у Shanghai Slack Laboratory Animals Co., Ltd. и содержались в лаборатории SPF. Питьевую воду и подстилку асептически обрабатывали в автоклаве. Все операции проводили в асептических условиях. В день 0,5×106 клеток острого гранулоцитарного лейкоза MV4-11 (приобретенных у АТСС) вводили посредством подкожной инъекции в левую часть спины всех мышей. Начиная с 15 дня всех мышей разделили на четыре группы (по 6 мышей в группе), первой группе мышей перорально вводили растворитель на основе метилцеллюлозы (HKI) ежедневно; второй группе мышей перорально вводили соединение 1 в дозе 10 мг/кг массы тела мыши в день; третьей группе мышей перорально вводили соединение 1 в дозе 20 мг/кг массы тела мыши в день; четвертой группе мышей перорально вводили соединение 1 в дозе 30 мг/кг массы тела мыши в день. С начала введения каждый день измеряли длину/ширину подкожной опухоли с помощью штангенциркуля, и каждый день регистрировали массу тела мыши, и наблюдали эффект, который соединение 1 оказывало на массу тела мыши. На 43-й день мышей умерщвляли, извлекали подкожные опухоли, взвешивали и сравнивали опухоли, а затем из ткани образца опухоли готовили образец белкового лизата для применения. Тенденцию подкожного роста опухоли подсчитывали с 16 дня до 43 дня, и объем опухоли рассчитывали как: длина × ширина × ширина / 2 мм3.
Результаты эксперимента приведены на фигуре. Результаты показали, что для соединения-ингибитора 1, описанного в настоящем изобретении, группа, получавшая высокую дозу (20, 30 мг/кг) влияла на массу тела мышей Bal b/c, но в группе, получавшей низкую дозу (10 мг/кг) масса подкожной опухоли была значительно снижена, и существенного эффекта на массу тела мышей не наблюдалось; ингибирование роста опухоли (TGI) в группе, получавшей высокую дозу (20, 30 мг/кг) может достигать 98,7%. Это указывало на то, что соединение 1 эффективно ингибировало рост подкожных опухолей (фиг. 4а-с).
Промышленная применимость
Настоящее изобретение относится к ингибитору циклинзависимой киназы CDK9, который можно применять для лечения, предотвращения или облегчения заболевания, расстройства или состояния, регулируемого или обусловленного активностью серинкиназы или связанного с активностью циклинзависимой киназы. Таким образом, можно получить соответствующее лекарственное средство, пригодное для промышленного применения.
Хотя настоящее изобретение было подробно описано в настоящем документе, настоящее изобретение не ограничивается этим, и специалисты в данной области техники могут вносить модификации в соответствии с принципами настоящего изобретения. Следовательно, различные модификации в соответствии с принципами настоящего изобретения следует понимать как попадающие в объем настоящего изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПИРАЗОЛПИРИМИДИНОВОЕ ПРОИЗВОДНОЕ И ЕГО ПРИМЕНЕНИЕ | 2016 |
|
RU2735522C2 |
| ГЕТЕРОАРИЛЬНЫЕ СОЕДИНЕНИЯ, СОДЕРЖАЩИЕ ИХ КОМПОЗИЦИИ И СПОСОБЫ ЛЕЧЕНИЯ С ПРИМЕНЕНИЕМ ЭТИХ СОЕДИНЕНИЙ | 2007 |
|
RU2478635C2 |
| ПИРАЗОЛО[3,4-С]ПИРИДИНЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2012 |
|
RU2638116C2 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛАМИНОПИРИМИДИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ ОБОГАЩЕННОЙ ЛЕЙЦИНОВЫМИ ПОВТОРАМИ КИНАЗЫ 2 | 2013 |
|
RU2637947C2 |
| ПРОИЗВОДНЫЕ ДИАМИНОВ | 2002 |
|
RU2319699C2 |
| БИЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ И ЕГО ПРИМЕНЕНИЕ ДЛЯ ИНГИБИРОВАНИЯ SUV39H2 | 2016 |
|
RU2729187C1 |
| N-(2-ЦИАНОГЕТЕРОЦИКЛИЛ)ПИРАЗОЛОПИРИДОНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ЯНУС-КИНАЗЫ | 2014 |
|
RU2669922C2 |
| СОЕДИНЕНИЯ ТИЕНОПИРИМИДИНА | 2013 |
|
RU2637925C2 |
| ПРОИЗВОДНЫЕ ДИАМИНА | 2002 |
|
RU2314303C2 |
| НОВОЕ ПРОИЗВОДНОЕ НИКОТИНАМИДА ИЛИ ЕГО СОЛЬ | 2011 |
|
RU2576623C2 |


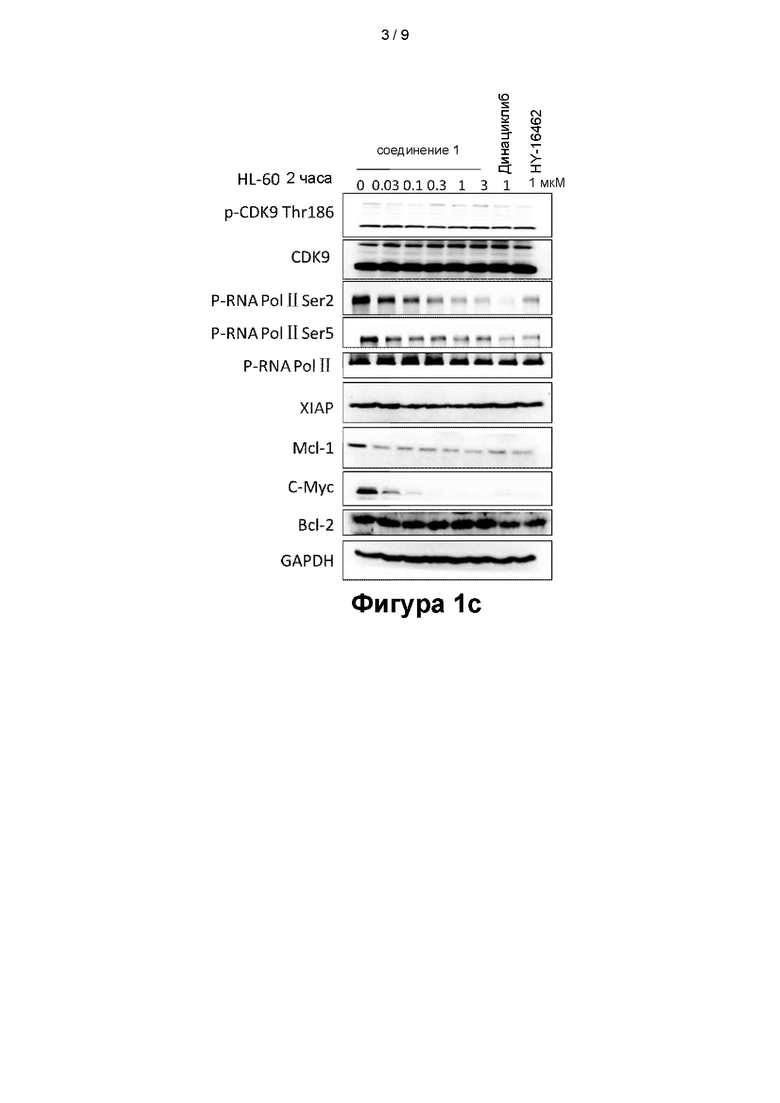
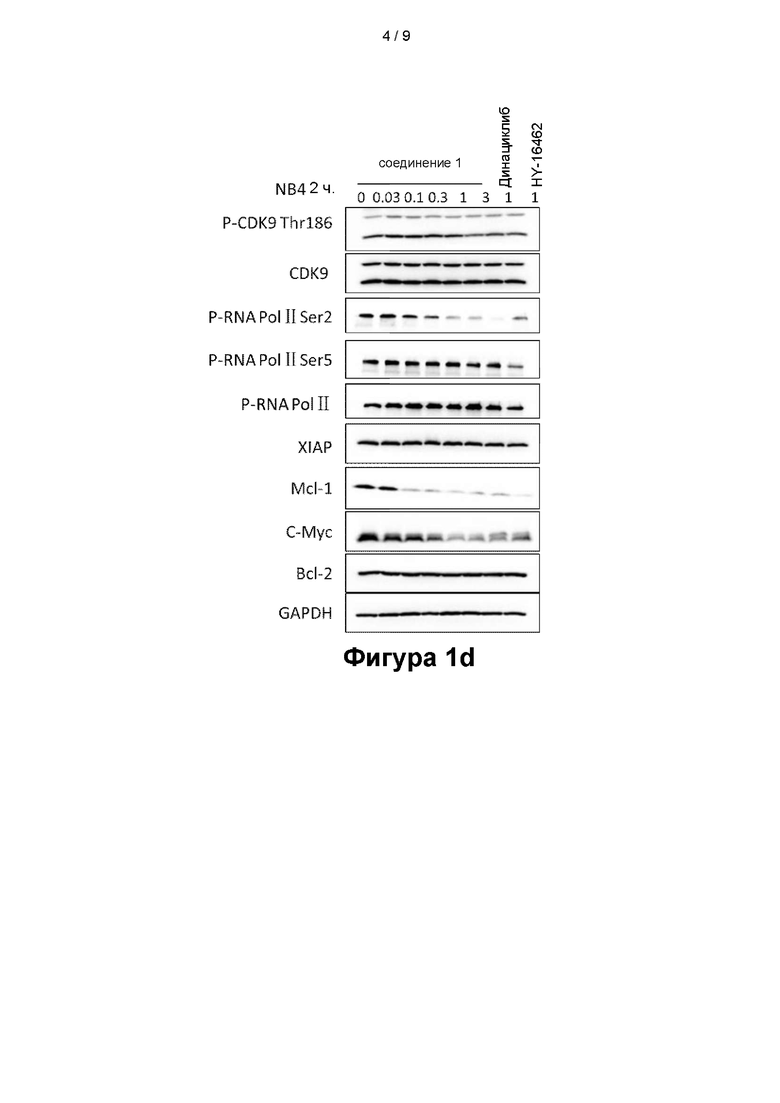
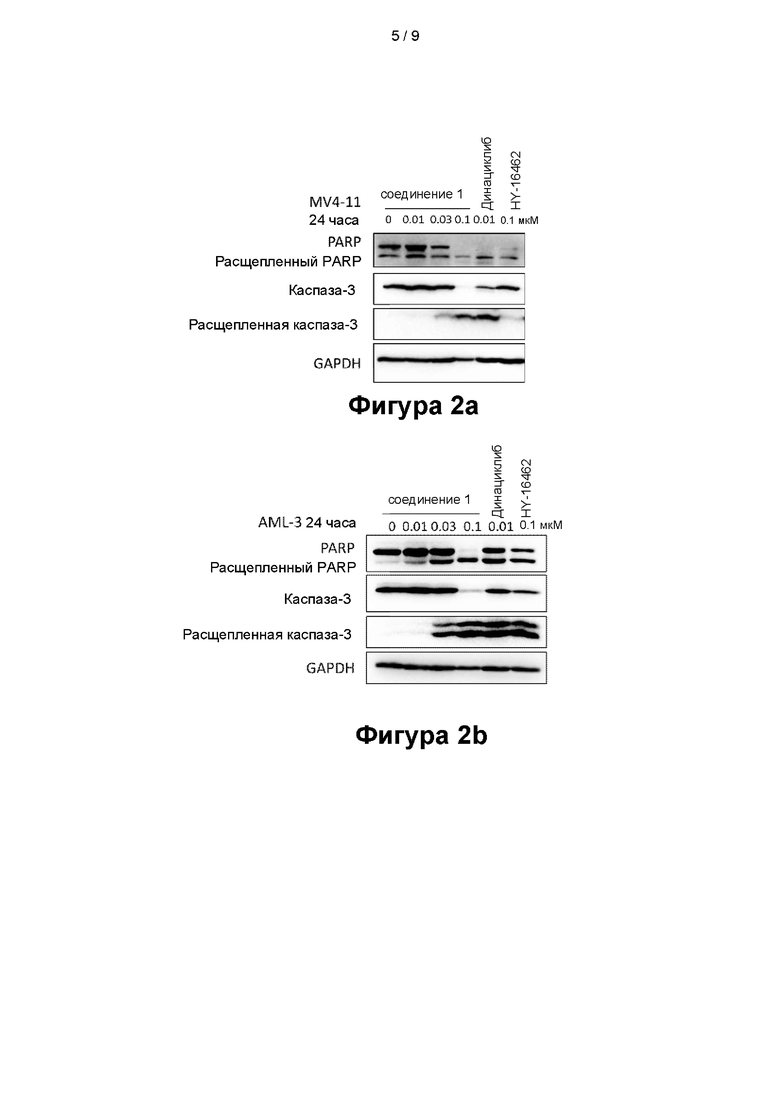
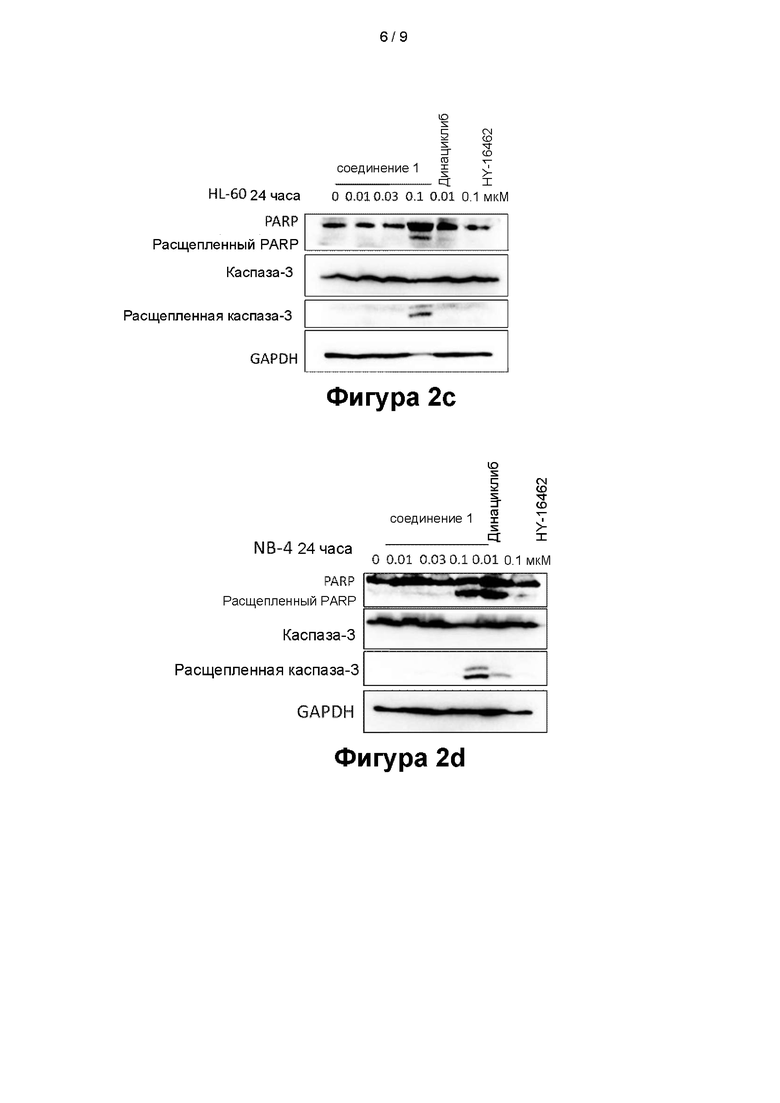


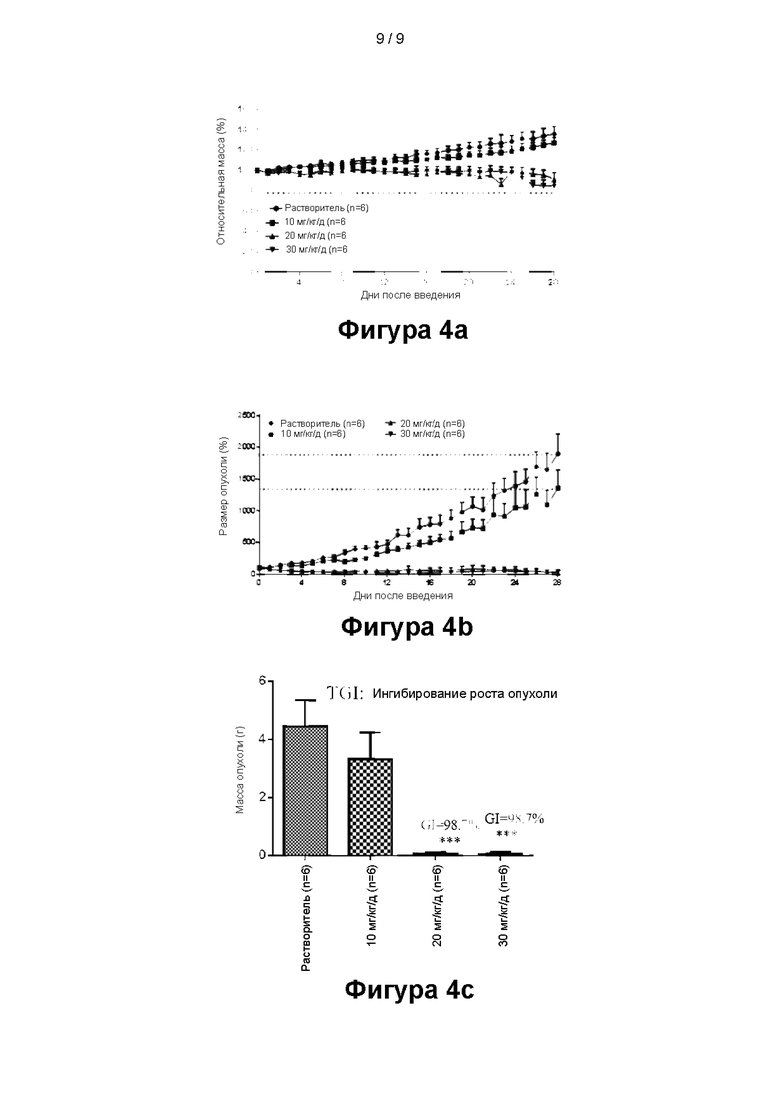
Изобретение относится к соединению формулы (I) или его фармацевтически приемлемой соли, гдe Y выбран из группы, состоящей из p-фторбензоила, транс-4-аминоциклогексила, в котором N необязательно замещен посредством R3, и транс-4-аминоциклогексилметила; Z выбран из группы, состоящей из NH, S и O; R1 выбран из группы, состоящей из водорода и галогена; R2 выбран из группы, состоящей из водорода, C3-C6 циклоалкила, 4-тетрагидропиранила, необязательно замещенного посредством R4, и фенила, необязательно замещенного посредством R4; R3 выбран из группы, состоящей из C2-C6 алканоила и C1-C3 алкокси (C1-C3) алкила; R4 выбран из группы, состоящей из циано и галогена. Изобретение также относится к фармацевтической композиции для ингибирования циклинзависимой киназы CDK9 на основе указанного соединения. Технический результат - получены новые соединения и фармацевтическая композиция на их основе, которые могут найти применение в медицине для лечения раковых заболеваний. 3 н. и 11 з.п. ф-лы, 5 табл., 14 ил., 24 пр.
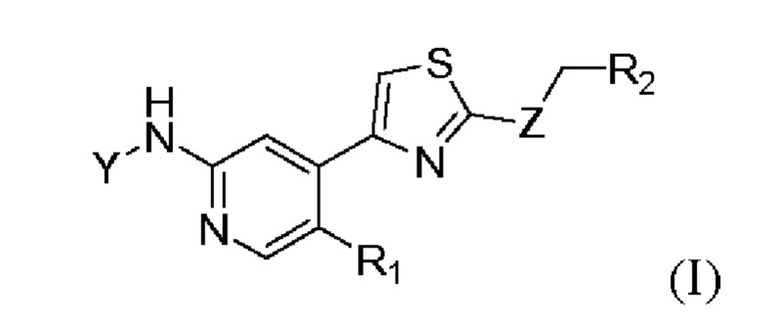
1. Соединение формулы (I):
 (I)
(I)
или его фармацевтически приемлемая соль,
гдe Y выбран из группы, состоящей из p-фторбензоила, транс-4-аминоциклогексила, в котором N необязательно замещен посредством R3, и транс-4-аминоциклогексилметила;
Z выбран из группы, состоящей из NH, S и O;
R1 выбран из группы, состоящей из водорода и галогена;
R2 выбран из группы, состоящей из водорода, C3-C6 циклоалкила, 4-тетрагидропиранила, необязательно замещенного посредством R4, и фенила, необязательно замещенного посредством R4;
R3 выбран из группы, состоящей из C2-C6 алканоила и C1-C3 алкокси (C1-C3) алкила;
R4 выбран из группы, состоящей из циано и галогена.
2. Соединение или его фармацевтически приемлемая соль по п. 1, отличающееся тем, что R1 представляет собой хлор.
3. Соединение или его фармацевтически приемлемая соль по п. 1, отличающееся тем, что R2 выбран из группы, состоящей из водорода, циклопропила, циклогексила, 4-тетрагидропиранила, необязательно замещенного посредством циано, и фенила, необязательно замещенного фтором.
4. Соединение или его фармацевтически приемлемая соль по п. 1, отличающееся тем, что R3 выбран из группы, состоящей из ацетила, 2-метоксиэтила, (R)-1-метил-2-метоксиэтила и (S)-1-метил-2-метоксиэтила.
5. Соединение или его фармацевтически приемлемая соль по любому из пп. 1-4, отличающееся тем, что соединение выбрано из:
4-(((4-(5-хлор-2-(((1R,4r)-4-(((R)-1-метоксипроп-2-ил)амино)циклогексил)амино)пиридин-4-ил)тиазол-2-ил)амино)метил)тетрагидро-2H-пиран-4-карбонитрила;
(1r,4r)-N1-(5-хлор-4-(2-(((тетрагидро-2H-пиран-4-ил)метил)амино)тиазол-4-ил)пиридин-2-ил)циклогексан-1,4-диамина;
N-((1r,4r)-4-((5-хлор-4-(2-(((тетрагидро-2H-пиран-4-ил)метил)амино)тиазол-4-ил)пиридин-2-ил)амино)циклогексил)ацетамида;
(1r,4r)-N1-(5-хлор-4-(2-(((тетрагидро-2H-пиран-4-ил)метил)амино)тиазол-4-ил)пиридин-2-ил)-N4-(2-метоксиэтил)циклогексил-1,4-диамина;
(1S,4r)-N1-(5-хлор-4-(2-(((тетрагидро-2H-пиран-4-ил)метил)амино)тиазол-4-ил)пиридин-2-ил)-N4-((S)-1-метоксипропан-2-ил)циклогексил-1,4-диамина;
(1R,4r)-N1-(5-хлор-4-(2-(((тетрагидро-2H-пиран-4-ил)метил)амино)тиазол-4-ил)пиридин-2-ил)-N4-((R)-1-метоксипропан-2-ил)циклогексан-1,4-диамина;
4-(2-((((1r,4r)-4-аминоциклогексил)метил)амино)-5-хлорпиридин-4-ил)-N-((тетрагидро-2H-пиран-4-ил)метил)тиазол-2-амина;
N-(5-хлор-4-(2-(((тетрагидро-2H-пиран-4-ил)метил)амино)тиазол-4-ил)пиридин-2-ил)-4-фторбензамида;
(1r,4r)-N1-(5-хлор-4-(2-(метиламино)тиазол-4-ил)пиридин-2-ил)-N4-(2-метоксиэтил)циклогексан-1,4-диамина;
(1r,4r)-N1-(5-хлор-4-(2-((циклогексилметил)амино)тиазол-4-ил)пиридин-2-ил)-N4-(2-метоксиэтил)циклогексан-1,4-диамина;
(1r,4r)-N1-(4-(2-(бензиламино)тиазол-4-ил)-5-хлорпиридин-2-ил)-N4-(2-метоксиэтил)циклогексан-1,4-диамина;
(1r,4r)-N1-(5-хлор-4-(2-((4-фторбензил)амино)тиазол-4-ил)пиридин-2-ил)-N4-(2-метоксиэтил)циклогексан-1,4-диамина;
(1r,4r)-N1-(5-хлор-4-(2-((циклопропилметил)амино)тиазол-4-ил)пиридин-2-ил)-N4-(2-метоксиэтил)циклогексан-1,4-диамина;
4-((4-(5-хлор-2-(((1r,4r)-4-((2-метоксиэтил)амино)циклогексил)амино)пиридин-4-ил)тиазол-2-иламино)метил)тетрагидро-2H-пиран-4-карбонитрила;
4-(((4-(5-хлор-2-(((1S,4r)-4-(((S)-1-метоксипроп-2-ил)амино)циклогексил)амино)пиридин-4-ил)тиазол-2-ил)амино)метил)тетрагидро-2H-пиран-4-карбонитрила;
(1r,4r)-N1-(5-хлор-4-(2-((тетрагидро-2H-пиран-4-ил)метокси)тиазол-4-ил)пиридин-2-ил)-N4-(2-метоксиэтил)циклогексан-1,4-диамина;
(1r,4r)-N1-(5-хлор-4-(2-(((тетрагидро-2H-пиран-4-ил)метил)меркапто)тиазол-4-ил)пиридин-2-ил)-N4-(2-метоксиэтил)циклогексан-1,4-диамина; и
(1r,4r)-N1-(2-метоксиэтил)-N4-(4-(2-(((тетрагидро-2H-пиран-4-ил)метил)амино)тиазол-4-ил)пиридин-2-ил)циклогексан-1,4-диамина.
6. Фармацевтическая композиция для ингибирования циклинзависимой киназы CDK9, содержащая эффективное количество соединения или его фармацевтически приемлемой соли по любому из пп. 1-5 и фармацевтически приемлемый носитель или вспомогательное вещество.
7. Применение соединения или его фармацевтически приемлемой соли по любому из пп. 1-5, для получения лекарственного средства для лечения, предотвращения или облегчения заболевания, расстройства или состояния, регулируемого или обусловленного активностью серинкиназы или связанного с активностью циклинзависимой киназы CDK9.
8. Применение по п. 7, отличающееся тем, что заболевание, расстройство или состояние представляет собой раковое заболевание.
9. Применение по п. 8, где раковое заболевание выбрано из группы, состоящей из немелкоклеточного рака легкого, мелкоклеточного рака легкого, аденокарциномы легкого, плоскоклеточного рака легкого, рака молочной железы, рака яичников, лейкоза и гистиоцитарной лимфомы.
10. Применение по п. 7, где заболевание, расстройство или состояние выбрано из группы, состоящей из MDS-RAEB (типа миелодиспластического синдрома с избытком бластов), гистиоцитарной лимфомы, острого В-клеточного лейкоза, острого мегакариобластного лейкоза, острого миелоидного лейкоза и острого промиелоцитарного лейкоза.
11. Соединение или его фармацевтически приемлемая соль по любому из пп. 1-5 для применения для лечения, предотвращения или облегчения заболевания, расстройства или состояния, регулируемого или обусловленного активностью серинкиназы или связанного с активностью циклинзависимой киназы CDK9.
12. Соединение или его фармацевтически приемлемая соль по п. 11, где заболевание, расстройство или состояние представляет собой раковое заболевание.
13. Соединение или его фармацевтически приемлемая соль по п. 12, где заболевание, расстройство или состояние выбрано из группы, состоящей из немелкоклеточного рака легкого, мелкоклеточного рака легкого, аденокарциномы легкого, плоскоклеточного рака легкого, рака молочной железы, рака яичников, лейкоза и гистиоцитарной лимфомы.
14. Соединение или его фармацевтически приемлемая соль по п. 11, где заболевание, расстройство или состояние выбрано из группы, состоящей из MDS-RAEB (типа миелодиспластического синдрома с избытком бластов), гистиоцитарной лимфомы, острого В-клеточного лейкоза, острого мегакариобластного лейкоза, острого миелоидного лейкоза и острого промиелоцитарного лейкоза.
| WO 2007133622 A2, 22.11.2007 | |||
| WO 2013156780 A1, 24.10.2013 | |||
| Приспособление для резки чепраков | 1928 |
|
SU22188A1 |
| Захватный прибор для мешков и тюков | 1927 |
|
SU7717A1 |
| Деревянный щит, склеиваемый из отдельных досок | 1928 |
|
SU13249A1 |

