Настоящее изобретение относится к соединениям, которые ингибируют или модулируют активность киназ, а в частности, киназ Aurora и киназ FLT3, к применению соединения при лечении или профилактике болезненных состояний или состояний, опосредованных киназами. Также настоящее изобретение относится к фармацевтическим композициям, содержащим соединения, способам их получения и новым химическим промежуточным соединениям.
ОБЛАСТЬ ТЕХНИКИ
Протеинкиназы составляют большое семейство структурно связанных ферментов, которые ответственны за контроль большого разнообразия процессов сигнальной трансдукции в пределах клетки (Hardie and Hanks (1995) The Protein Kinase Facts Book. I and II, Academic Press, San Diego, CA). Киназы могут быть классифицированы на семейства по субстратам, которые они фосфорилируют (например, протеин-тирозин, протеин-серин/треонин, липиды, и т.д.). Были идентифицированы мотивы последовательности, которые обычно соответствуют каждой из этих семейств киназ (например, Hanks and Hunter, FASEB J., (1995) 9. 576-596; Knighton, и другие, Science, (1991) 253, 407-414; Hiles, и другие, Cell, (1992) 70, 419-429; Kunz, и другие, Cell, (1993) 73, 585-596; Garcia-Bustos, и другие, EMBOJ., (1994) 13, 2352-2361).
Протеинкиназы могут быть характеризированы своими механизмами регулирования. Эти механизмы включают в себя, например, аутофосфорилирование, трансфосфорилирование другими киназами, белок-белок взаимодействия, белок-липид взаимодействия и белок-полинуклеотид взаимодействия. Отдельная протеинкиназа может быть регулируема более чем одним механизмом.
Киназы регулируют многие различные клеточные процессы, включая без ограничения пролиферацию, дифференциацию, апоптоз, подвижность, транскрипцию, трансляцию и другие сигнальные процессы, путем добавления фосфатных групп к целевым белкам. Эти процессы фосфорилирования действуют как молекулярные переключатели «включено-выключено», которые могут модулировать или регулировать биологическую функцию целевого белка. Фосфорилирование целевых белков возникает в ответ на ряд внеклеточных сигналов (гормонов, нейротрансмиттеров, факторов роста и дифференцировки, и т.д.), процессов клеточного цикла, воздействий окружающей среды или воздействий, связанных с питанием, и т.д. Соответствующая протеинкиназа действует в путях передачи сигнала для активации или инактивации (либо прямо, либо косвенно), например, метаболического фермента, регуляторного белка, рецептора, белка цитоскелета, ионного канала или насоса или транскрипционного фактора. Неконтролируемая передача сигнала из-за недостаточного контроля фосфорилирования белка была вовлечена во многие заболевания, включая, например, воспаление, рак, аллергию/астму, заболевание и состояния иммунной системы, заболевание и состояния центральной нервной системы и ангиогенез.
Киназы Aurora
Три члена киназного семейства Aurora до сих пор были обнаружены у млекопитающих (Nigg, Nat. Rev. Mol. Cell Biol. (2001) 2, 21-32). Киназа Aurora А (в литературе также упоминается как Aurora 2) представляет собой серин/треонин киназу, которая вовлечена в G2 и М фазы клеточного цикла, и представляет собой важный регулятор митоза. Полагали, что киназа Aurora А играет роль в митотическом контроле контрольной точки, динамике хромосом и цитокинезе (Adams и другие, Trends Cell Biol, (2001) 11, 49-54). Киназы расположены в центросомах межфазных клеток, на полюсах биполярного веретена и в клеточной пластинке в плоскости экватора веретена митотического аппарата.
Другие две в настоящее время известные киназы Aurora представляют собой Aurora В (в литературе также упоминается как Aurora 1) и Aurora С (в литературе также упоминается как 3). Киназы Aurora обладают чрезвычайно гомологическими каталитическими доменами, но значительно отличаются своими N-конечными частями (Katayama и другие, Cancer Metastasis Rev. (2003) 22(4), 451-64).
Субстраты киназ Aurora А и В идентифицировали как включающие кинезинподобный движущий белок, белки веретена деления, гистон Н3 белок, белок кинетохора и белок-супрессор опухолевого роста р53.
Полагали, что киназы Aurora А были вовлечены в образование веретена и локализировались в центросоме в течение ранней фазы G2, где они фосфорилировали связанные с веретеном белки (Prigent и другие, Cell (2003) 114, 531-535). Hirota и другие, (Cell, (2003) 114, 585-598) обнаружили, что клетки, лишенные протеинкиназы Aurora А, были неспособны попасть в митоз. Более того, обнаружено (Adams, 2001), что мутация или деструкция гена Aurora А у различных видов приводит к митотическим анормальностям, включая отделение центросомы и дефекты при созревании, нарушения веретена и дефекты сегрегации хромосом.
Киназа Aurora А обычно выражена на низком уровне в большинстве нормальных тканей, исключениями являлись ткани с высоким соотношением делящих клеток, например, тимус и семенник. Тем не менее, повышенные уровни киназ Aurora были обнаружены во многих пораженных раком тканях человека (Giet и другие, J. Cell. Sci. (1999) 112, 3591 and Katayama (2003)). Более того, киназа Aurora А наносит на хромосому 20ql3 область, что, как часто находили, была увеличена во многих пораженных раком тканях человека.
Таким образом, например, значительная сверхэкспрессия Aurora А была обнаружена в молочной железе человека, раковых клетках яичника и поджелудочной железы (см. Zhou и другие, Nat. Genet. (1998) 20, 189-193; Tanaka и другие, Cancer Res. (1999) 59, 2041-2044 and Han и другие, Cancer Res. (2002) 62, 2890-2896).
Более того, Isola (American Journal of Pathology (1995) 147, 905-911) сообщал, что амплификация локуса Aurora A (20q13) находится в соотношении с неблагоприятным прогнозом для пациентов с раком молочной железы с негативными узлами.
Амплификацию и/или сверхэкспрессию Aurora-A наблюдали у людей с раком мочевого пузыря, и амплификация Aurora-A связана с анеуплоидией и агрессивным клиническим поведением (см. Sen и другие, J. Natl. Cancer Inst. (2002) 94,1320-1329).
Повышенная экспрессия Aurora-A была определена в более чем 50% с колоректальным раком (см. Bischoff и другие, EMBO J. (1998) 17, 3052-3065 and Takahashi и другие, Jpn. J. Cancer Res. (2000) 91, 1007-1014), раком яичников (см. Gritsko и другие, Clin. Cancer Res. (2003) 9, 1420-1426) и желудочными новообразованиями (см. Sakakura и другие, British Journal of Cancer (2001) 84, 824-831).
Tanaka и другие, (Cancer Research (1999) 59, 2041-2044) обнаружили признаки сверхэкспрессии Aurora А в 94% инвазивных аденокарцином протоков молочной железы.
Высокие уровни киназы Aurora А также были обнаружены в почечных, цервикальных, нейробластомных, меланомных, лимфомных, панкреатических линиях опухолевой клетки и линиях опухолевой клетки предстательной железы (Bischoff и другие, (1998), EMBO J. (1998) 17, 3052-3065; Kimura и другие, J Biol. Chem. (1999) 274, 7334-7340; Zhou и другие, Nature Genetics, 20: 189-193 (1998); Li и другие, Clin Cancer Res. 9(3): 991-7 (2003).
Royce и другие (Cancer. (2004) 100 (1), 12-19) сообщали, что экспрессия гена Aurora 2 (известного как STK15 или ВТАК) была отмечена приблизительно в одной четвертой из первичных опухолей молочной железы.
Reichardt и другие (Oncol Rep. (2003) 10(5), 1275-9) сообщали, что количественный анализ ДНК при помощи PCR для поиска амплификации Aurora в глиомах показал, что 5 из 16 опухолей (31%) различной степени по WHO (1х степень II, 1х степень III, 3х степень IV) показывали амплификацию ДНК гена Aurora 2. Предполагали гипотезу, что амплификация гена Aurora 2 может быть неслучайным генетическим изменением в человеческих глиомах, играя роль в генетических путях онкогенеза.
По результатам Hamada и другие (Br. J. Haematol. (2003) 121(3), 439-47) также предполагали, что Aurora 2 является эффективным кандидатом обозначения не только активности заболевания, но и также онкогенеза неходжкинской лимфомы. Замедление роста опухолевой клетки, полученное в результате ограничения функций этого гена, может быть терапевтическим подходом к неходжкинской лимфоме.
При изучении Gritsko и другие (Clin Cancer Res. (2003) 9(4), 1420-6) киназная активность и уровни белка Aurora А определяли у 92 пациентов с первичными яичниковыми опухолями. In vitro киназные анализы сообщали о повышенной активности киназы Aurora А в 44 случаях (48%). Повышенные уровни белка Aurora А определяли в 52 (57%) случаях. Высокие уровни белка Aurora А хорошо коррелировали с повышенной активностью киназы.
Результаты, полученные Li и другие (Clin. Cancer Res. 2003 Mar; 9(3): 991-7), показали, что ген Aurora А является сверхэкспрессированным в панкреатических опухолях и линиях клетки раковой опухоли, и предполагалось, что сверхэкспрессия Aurora А может играть роль в панкреатическом онкогенезе.
Подобным образом было показано, что амплификация гена Aurora А и связанная повышенная экспрессия митотической киназы, кодирующая его, связаны с анеуплоидией и агрессивным клиническим поведением у людей с раком мочевого пузыря. (J. Natl. Cancer Inst. (2002) 94(17), 1320-9).
При исследовании несколькими группами (Dutertre and Prigent, Mol. Interv. (2003) 3(3), 127-30 and Anand и другие, Cancer Cell. (2003) 3(1), 51-62) полагали, что сверхэкспрессия активности киназы Aurora связана со стойкостью к некоторым современным терапиям рака. Например, сверхэкспрессия Aurora А в фибробластах эмбриона мыши может понижать чувствительность этих клеток к цитотоксичным эффектам производных таксана. Таким образом, ингибиторы киназы Aurora могут найти конкретное применение, касающееся пациентов с развитой стойкостью к существующим терапиям.
На основе работы, выполненной до настоящего времени, предусмотрено, что ингибирование киназы Aurora А докажет эффективное средство подавления развития опухоли.
Также было показано, что присутствует увеличение экспрессии Aurora В в опухолевых клетках по сравнению с нормальными клетками (Adams и другие, Chromasoma. (2001) 110, 65-74). Согласно одному отчету полагали, что сверхэкспрессия Aurora В вызывает анеуплоидию путем усиленного фосфорилирования гистона Н3 при серине 10, и что клетки сверхэкспрессирования Aurora В образуют более агрессивные опухоли и обладают более высокой тенденцией к образованию метастатических опухолей (Ota и другие, Cancer Res. (2002) 62, 5168-5177).
Aurora В необходима как для функции контрольной точки веретена, так и для выравнивания метафазной хромосомы в человеческих клетках (Adams и другие J. Cell Biol. (2001) 153, 865-880; Kallio и другие, Curr. Biol. (2002) 12, 900-905 and Murata-Hori and Wang Curr. Biol. (2002) 12, 894-899). Было показано, что подавление активности киназы Aurora В приводит к компромиссу выравнивания хромосом, функции контрольной точки веретена и цитокинеза (Ditchfield и другие, J.Cell Biol. (2003) 161, 267-280 and Hauf и другие, J. Cell Biol. (2003), 161, 281-294). В результате, после краткой отсрочки клетки покидали митоз без деления и с содержанием 4N ДНК, после чего они быстро теряли свой пролиферативный потенциал.
Harrington и другие (Nat Med. (2004) 10(3), 262-7) показали, что ингибитор киназ Aurora подавляет рост опухоли и вызывает обратное развитие опухоли in vivo. В этом изучении ингибитор киназы Aurora блокирует пролиферацию клетки рака, и также гибель стимулированной клетки в диапазоне линий клеток рака, включая лейкемические, колоректальные линии клеток и линии клеток молочной железы. Кроме того, был показан потенциал для лечения лейкемии путем вызывания апоптоза в лейкозных клетках. VX-680 мощно уничтожали невосприимчивые к лечению первичные клетки острого миелобластного лейкоза (AML) у пациентов (Andrews, Oncogene (2005) 24, 5005-5015).
Manfredi и другие (PNAS (2007) 104, 4106-4111) показали, что низкомолекулярный ингибитор Aurora А подавляет рост опухоли in vivo. В этом изучении дозозависимое ингибирование роста опухоли было показано на НСТ-116 мышах с опухолью и РС-3 мышах с опухолью против мышей, обработанных носителем. Наблюдали ингибирование роста опухоли до 84% против НСТ-116 и 93% против РС-3 клеточных ксенотрансплантатов.
Mortlock и другие (Clin Cancer Res. (2007) 13(12), 3682-3688) показали, что низкомолекулярный ингибитор Aurora В подавляет рост опухоли in vivo. Мышей с иммунодефицитом с установленными SW620, НСТ-116, Colo205, А549, Calu-6 или HL-60 ксенотрансплантатами опухоли дозировали в течение 48 ч подкожным вливанием из мини-насоса низкомолекулярного ингибитора AZD1152. Ингибирование роста опухоли во всех случаях находилось в диапазоне от 55% до 100% с полным обратным развитием опухоли, наблюдаемым у 8 из 11 животных с HL-60 ксенотрансплантатом.
На основе данных, полученных на сегодняшний день, считалось вероятным, что ингибиторы киназы Aurora должны быть особенно полезными при задержке развития опухоли и лечении рака, такого как рак молочной железы, мочевого пузыря, колоректальный рак, рак поджелудочной железы и рак яичников, неходжкинской лимфомы, глиомы, неэндометриоидной карциномы эндометрия, острого миелобластного лейкоза (AML), хронического миелолейкоза (CML), В-клеточной лимфомы (клетка мантийной ткани) и острого лимфобластного лейкоза (ALL).
FLT3
FMS-подобная тирозинкиназа 3 (FLT3) представляет собой тирозинкиназу рецептора, включенную в пролиферацию, дифференциацию и апоптоз гемопоэтических и не гемопоэтических клеток (Scheijen and Griffin, Oncogene (2002) 21, 3314-3333 and Reilly, British Journal of Haematology (2002) 116, 744-757). В результате связывания природного лиганда (FL) рецептор FLT3 димеризуется, что приводит к активации его домена тирозинкиназы, аутофосфорилированию рецептора и восстановлению молекул нисходящего сигнала, таких как р85 подъединица PI3K (фосфатидилинозитол 3 киназа), PLC-гамма (фосфолипаза-С гамма), STAT5a (преобразователь сигнала и активатор транскрипции 5а) и SRC семейства тирозинкиназ (Gilliland and Griffin, Blood (2002) 100(5), 1532-42; Drexler, Leukemia (1996) 10(4), 588-99 and Ravandi и другие, Clin Cancer Res. (2003) 9(2), 535-50).
Активация этих молекул нисходящего сигнала фосфорилированием приводит к пролиферативным и способствующим выживанию эффектам FLT3 (Gilliland and Griffin (2002) and Levis and Small, Leukemia (2003) 17(9), 1738-52).
Соматические мутации FLT3, включающие в себя внутренние тандемные дубликации в околомембранной области рецептора, или посредством точечной мутации D835 в ативационной петле, были показаны приблизительно у 30% пациентов с острым миелолейкозом (AML), раком белых клеток крови, вызванным перепроизводством незрелых миелоидных лейкоцитов (Nakao и другие, Leukemia (1996) 10(12), 1911-8; Thiede и другие, Blood (2002) 99(12), 4326-35; Yamamoto и другие, Blood (2001) 97(8), 2434-9; Abu-Duhier и другие, Br. J. Haematol. (2000) 111(1), 190-5 and Abu-Duhier и другие, Br. J. Haematol. (2001) 113(4), 983-8).
Другие независимые от лиганда активационные мутации FLT3 были описаны совсем недавно, которые способствуют лейкемическому превращению в AML. Присутствие таких мутаций при диагнозе было связано с более плохим прогнозом у некоторых пациентов (Jiang etal, Blood (2004) 104(6), 1855-8 and Kindler и другие, Blood (2005) 105(1), 335-40).
Наша более ранняя международная патентная заявка WO 2008/139161 раскрывает класс замещенных оксазолкарбоксамидов в качестве ингибиторов различных киназ, и в частности, киназы Aurora, киназы FLT3 и киназы FLT4.
В примере М-12 на стр. 132 WO 2008/139161 описано получение соединения амида 2-(1Н-индол-4-ил)-5-(4-пиперазин-1-ил-фенил)-оксазол-4-карбоновой кислоты, структурная формула которого представлена ниже.
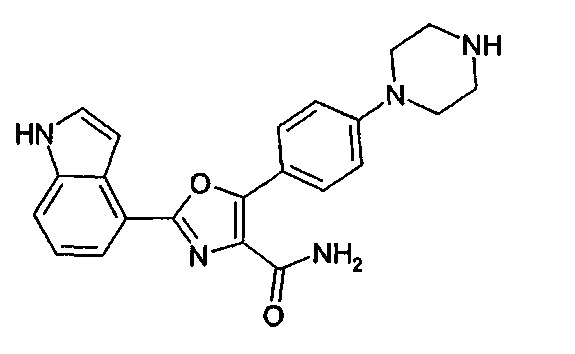
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Было обнаружено, что аналоги представленного выше соединения, но где фрагмент СH2 при 3-положении пиперазинового кольца был замене фрагментом С(СН3)2, значительно усиливали активность относительно одной или нескольких киназ, выбранных из киназ Aurora А и Aurora В и киназы FLT3, и обладали пониженной склонностью к утечке по сравнению с соединением примера М-12 WO. Также было обнаружено, что активность соединений относительно киназ Aurora дополнительно существенно усиливалась с присутствием заместителей при индольной группе.
Соответственно, согласно первому варианту реализации (вариант реализации 1.1), настоящее изобретение относится к соединению формулы (1):
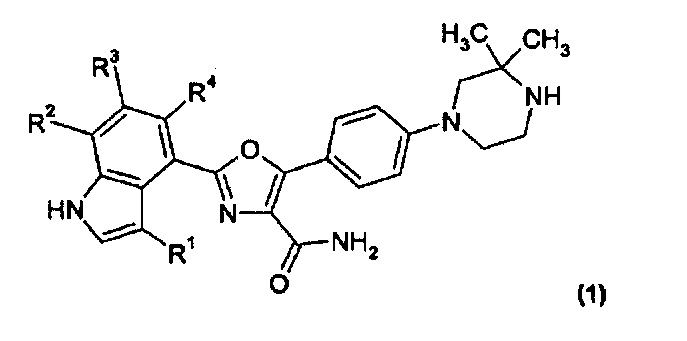
и его солям; где: R1 представляет собой водород или C1-2алкил; и R2, R3 и R4 одинаковые или отличаются, и каждый из них выбран из водорода, С1-2алкила, фтора, хлора, С1-2алкокси и трифторметила, при условии, что не более двух из R2, R3 и R4 отличны от водорода.
Конкретные и предпочтительные соединения по настоящему изобретению таковы, как определено в вариантах реализации 1.2-1.33 ниже.
1.2 Соединение согласно варианту реализации 1.1, где R1 выбран из водорода и метила.
1.3 Соединение согласно варианту реализации 1.2, где R1 представляет собой водород.
1.4 Соединение согласно варианту реализации 1.2, где R1 представляет собой метил.
1.5 Соединение по любому из вариантов реализации 1.1-1.4, где R2 выбран из водорода, фтора, хлора, метила, этила, трифторметила и метокси.
1.6 Соединение по любому из вариантов реализации 1.1-1.4, где R2 выбран из водорода, фтора, хлора, метила, этила и метокси.
1.6А Соединение согласно варианту реализации 1.6, где R2 выбран из водорода, фтора, хлора, метила и метокси.
1.7 Соединение согласно варианту реализации 1.5, где R2 представляет собой водород.
1.8 Соединение согласно варианту реализации 1.5, где R2 представляет собой фтор.
1.9 Соединение согласно варианту реализации 1.5, где R2 представляет собой хлор.
1.10 Соединение согласно варианту реализации 1.5, где R2 представляет собой метил.
1.11 Соединение согласно варианту реализации 1.5, где R2 представляет собой этил.
1.12 Соединение согласно варианту реализации 1.5, где R2 представляет собой метокси.
1.13 Соединение согласно варианту реализации 1.5, где R2 представляет собой трифторметил.
1.14 Соединение по любому из вариантов реализации 1.1-1.13, где R3 выбран из водорода и фтора.
1.15 Соединение согласно варианту реализации 1.14, где R3 представляет собой водород.
1.16 Соединение по любому из вариантов реализации 1.1-1.15, где R4 выбран из водорода, фтора, метила и этила.
1.17 Соединение согласно варианту реализации 1.16, где R4 представляет собой водород.
1.18 Соединение согласно варианту реализации 1.16, где R4 выбран из фтора, метила и этила, и один из R2 и R3 представляет собой водород.
1.19 Соединение согласно варианту реализации 1.18, где R3 представляет собой водород.
1.20 Соединение согласно варианту реализации 1.18 или варианту реализации 1.19, где R4 представляет собой фтор.
1.21 Соединение согласно варианту реализации 1.18 варианту реализации 1.19, где R4 представляет собой метил.
1.22 Соединение согласно варианту реализации 1.18 варианту реализации 1.19, где R4 представляет собой этил.
1.22А Соединение согласно варианту реализации 1.1, где R1 представляет собой водород; R2 выбран из водорода, метила, этила, фтора, хлора и метокси; R3 представляет собой водород; и R4 выбран из водорода, фтора и метила.
1.23 Соединение согласно варианту реализации 1.22А, где (i) R1 представляет собой водород; R2 выбран из метила, этила, фтора, хлора и метокси; R3 представляет собой водород; и R4 представляет собой водород; или (ii) R1 представляет собой водород; R2 представляет собой водород; R3 представляет собой водород; и R4 представляет собой метил; или (iii) R1 представляет собой водород; R2 представляет собой фтор; R3 представляет собой водород; и R4 представляет собой метил.
1.24 Соединение согласно варианту реализации 1.1, которое выбрано из соединений прим. 1 - прим. 12 таблицы 1 ниже, и его соли.
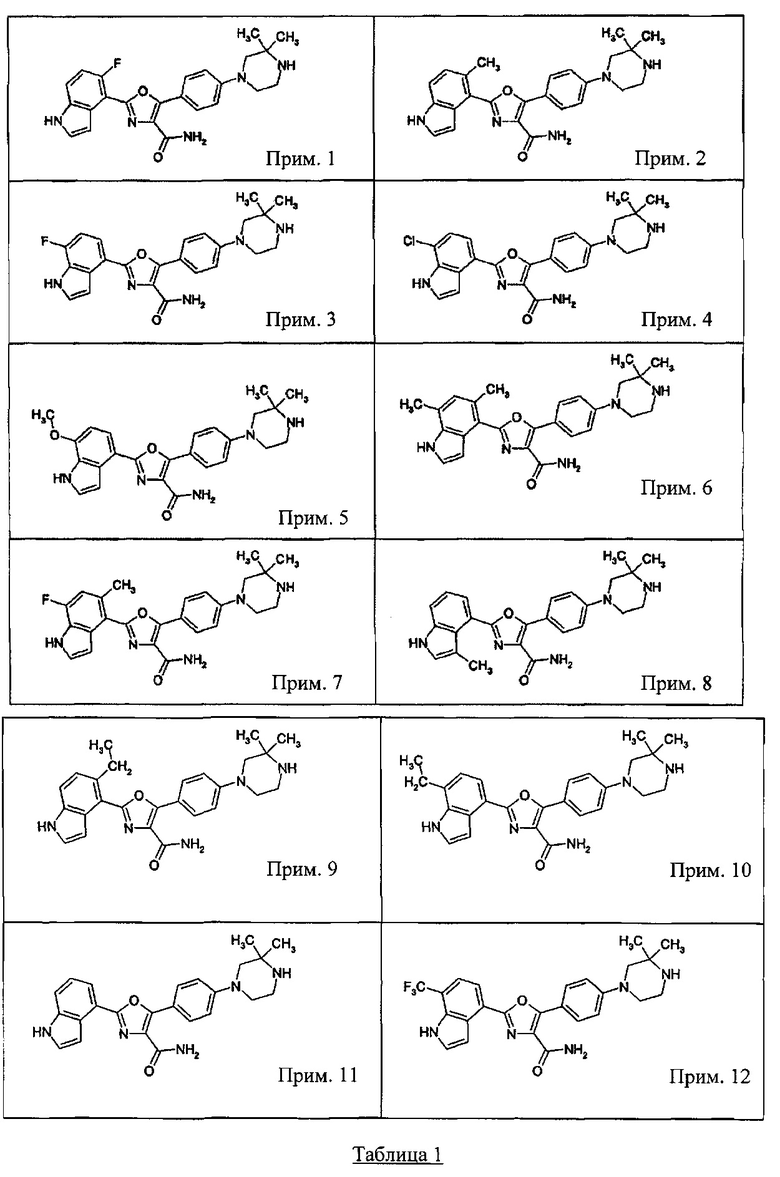
1.25 Соединение согласно варианту реализации 1.24, которое выбрано из соединений прим. 2, прим. 3, прим. 5 и прим. 7 таблицы 1, и его соли.
1.26 Соединение согласно варианту реализации 1.25, которое представляет собой соединение прим. 2, или его соль.
1.27 Соединение согласно варианту реализации 1.25, которое представляет собой соединение прим. 3, или его соль.
1.28 Соединение согласно варианту реализации 1.25, которое представляет собой соединение прим. 5, или его соль.
1.29 Соединение согласно варианту реализации 1.25, которое представляет собой соединение прим. 7, или его соль.
1.30 Соединение по любому из вариантов реализации 1.1-1.29, которое находится в форме свободного основания.
1.31 Соединение по любому из вариантов реализации 1.1-1.29, которое находится в форме соли.
1.32 Соединение согласно варианту реализации 1.31, где соль представляет собой кислотно-аддитивную соль.
1.33 Соединение согласно варианту реализации 1.31 или варианту реализации 1.32, где соль представляет собой фармацевтически приемлемую соль.
Общие предпочтения и определения
Ссылки на киназы в настоящем описании включают в себя не только нормально функционирующую форму рассматриваемой киназы, а также и ее мутантные формы.
Предусматривается, что используемый в настоящем описании термин «модуляция» при использовании относительно активности киназы, определяет изменение уровня биологической активности киназы (киназ). Таким образом, модуляция охватывает физиологические изменения, эффект которых возрастает или понижается при релевантной киназной активности. В последнем случае модуляция может быть описана как «ингибирование».
Используемый в настоящем описании термин «повышенная регуляция» относительно киназы определен как включающий повышенную экспрессию или сверхэкспрессию киназы, включая амплификацию гена (т.е., много копий генов) и повышенную экспрессию путем транскрипционного эффекта, и гиперактивность и активацию киназы, включая активацию мутациями.
Предусматривается, что ссылки в настоящем документе на стадию заболевания или состояние, «опосредованное» конкретной киназой, действуют с ограничением, таким образом, различные стадии заболевания или состояния, к которым применяли этот термин, являлись таковыми, в которых рассматриваемая киназа (или ее мутированная форма) играет биологическую роль. Биологическая роль, которую играет киназа, может быть прямой или косвенной, и может быть необходимой и/или достаточной для проявления симптомов заболевания, стадии или состояния (или его этиологии или прогрессирования).
Соли
Соединения по настоящему изобретению могут быть представлены в форме солей.
Соли (как определено согласно вариантам реализации в 1.31-1.33) типично представляли собой кислотно-аддитивные соли.
Соли могут быть синтезированы из исходного соединения традиционными химическими способами, такими, которые описаны в Pharmaceutical Salts: Properties, Selection, and Use, P. Heinrich Stahl (Editor), Camille G. Wermuth (Editor), ISBN: 3-90639-026-8, Hardcover, 388 pages, August 2002. Как правило, такие соли могут быть получены путем осуществления взаимодействия формы свободного основания соединения с кислотой в воде или органическим растворителе, или в их смеси; как правило, использовали неводную среду, такую как эфир, этилацетат, этанол, изопропанол или ацетонитрил.
Кислотно-аддитивные соли (как определено в варианте реализации 1.32) могут быть образованы с большим разнообразием кислот, как неорганическими, так и органическими. Примеры кислотно-аддитивных солей включают в себя соли, образованные с кислотой, выбранной из группы, состоящей из уксусной, 2,2-дихлоуксусной, адипиновой, альгиновой, аскорбиновой (например, L-аскорбиновой), L-аспарагиновой, бензолсульфоновой, бензойной, 4-ацетамидобензойной, бутановой, (+) камфарной, камфарсульфоновой, (+)-(1S)-камфар-10-сульфоновой, каприновой, капроновой, каприловой, коричной, лимонной, цикламовой, додецилсерной, этан-1,2-дисульфоновой, этансульфоновой, 2-гидроксиэтансульфоновой, муравьиной, фумаровой, галактаровой, гентизиновой, глюкогептоновой, D-глюконовой, глюкуроновой (например, D-глюкуроновой), глутаминовой (например, L-глутаминовой), α-оксоглутаровой, гликолевой, гиппуриновой, бромистоводородной, соляной, йодистоводородной, изэтиновой, (+)-L-молочной, (±)-DL-молочной, лактобионовой, малеиновой, яблочной, (-)-L-яблочной, малоновой, (±)-DL-миндальной, метансульфоновой, нафталин-2-сульфоновой, нафталин-1,5-дисульфоновой, 1-гидрокси-2-нафтойной, никотиновой, азотной, олеиновой, оротовой, щавелевой, пальмитиновой, памовой, фосфорной, пропионовой, L-пироглутаминовой, салициловой, 4-аминосалициловой, себациновой, стеариновой, янтарной, серной, дубильной, (+)-L-виннокаменной, тиоциановой, пара-толуолсульфоновой, ундециленовой и валериановой кислот, а также ацилированных аминокислот и катионообменньгх смол.
Солевые формы соединений по настоящему изобретению типично представляют собой фармацевтически приемлемые соли, и примеры фармацевтически приемлемых солей обсуждали в Berge и другие, 1977, "Pharmaceutically Acceptable Salts," J. Pharm. Sci., Vol. 66, pp. 1-19. Тем не менее, соли, которые не являются фармацевтически приемлемыми, также могут быть получены в форме промежуточных соединений, которые затем могут быть превращены в фармацевтически приемлемые соли. Такие не фармацевтически приемлемые солевые формы, которые могут быть применимы, например, при очистке или разделении соединений по настоящему изобретению, также составляют часть настоящего изобретения.
Изотопы
Соединения по настоящему изобретению, как определено в любом из вариантов реализации 1.1-1.33, могут содержать одно или более изотопных замещений, и ссылка на конкретный элемент включает в себя в пределах своего объема все изотопы элемента. Например, ссылка на водород включает в себя в пределах своего объема 1Н, 2Н (D) и 3Н (Т). Подобным образом, ссылки на углерод и кислород включают в себя в пределах своего объема, соответственно, 12С, 13С и 14С и 16O и 18O.
Аналогичным образом, ссылка на конкретную функциональную группу также включает в себя в пределах своего объема изотопные вариации, если в контексте не отмечено иное.
Например, ссылка на алкильную группу, такую как этильная группа, также охватывает вариации, в которых один или более атомов водорода в группе находятся в форме изотопов дейтерия или трития, например, как в этильной группе, в которой все пять атомов водорода находятся в изотопной форме дейтерия (пердейтероэтильная группа).
Изотопы могут быть радиоактивными или не радиоактивными. Согласно одному варианту реализации настоящего изобретения (вариант реализации 1.34) соединение по любому из вариантов реализации 1.1-1.33 не содержит радиоактивных изотопов. Такие соединения являются предпочтительными для терапевтического применения. Тем не менее, согласно другому варианту реализации (вариант реализации 1.35), соединение по любому из вариантов реализации 1.1-1.33 может содержать один или более радиоактивных изотопов. Соединения, содержащие такие радиоактивные изотопы, могут быть применимы в диагностическом контексте.
Сольваты
Соединения формулы (1), как определено в любом из вариантов реализации 1.1-1.35, могут образовывать сольваты.
Предпочтительными сольватами являются сольваты, образованные включением в твердотельную структуру (например, кристаллическую структуру) соединений по настоящему изобретению молекул не токсичного фармацевтически приемлемого растворителя (ниже называется сольватирующийся растворитель).
Примеры таких растворителей включают в себя воду, спирты (такие как этанол, изопропанол и бутанол) и диметилсульфоксид. Сольваты могут быть получены перекристаллизацией соединений по настоящему изобретению с растворителем или смесью растворителей, содержащей сольватирующийся растворитель. Будет ли образован сольват или нет в любом представленном случае, может быть определено путем подвергания кристаллов соединения анализу с применением хорошо известных и стандартных методик, таких как термогравиметрический анализ (TGE), дифференциальная сканирующая калориметрия (DSC) и рентгеноструктурная кристаллография.
Сольваты могут быть стехиометрическими или не стехиометрическими сольватами.
В частности, предпочтительными сольватами являются гидраты, и примеры гидратов включают в себя гемигидраты, моногидраты и дигидраты.
Соответственно, согласно дополнительным вариантам реализации 1.36 и 1.37, настоящее изобретение обеспечивает:
1.36 Соединение по любому из вариантов реализации 1.1-1.35 в форме сольвата.
1.37 Соединение согласно варианту реализации 1.36, где сольват представляет собой гидрат.
Для более подробного обсуждения сольватов и способов, используемых для их получения и характеристики, см. Bryn и другие, Solid-State Chemistry of Drugs, Second Edition, published by SSCI, Inc of West Lafayette, IN, USA, 1999, ISBN 0-967-06710-3.
Альтернативно, вместо существующего в виде гидрата, соединение по настоящему изобретению может быть безводным. Таким образом, согласно другому варианту реализации (вариант реализации 1.38) настоящее изобретение относится к соединению, как определено в любом из вариантов реализации 1.1-1.35, в безводной форме.
Кристаллические и аморфные формы
Соединения по любому из вариантов реализации 1.1-1.38 могут существовать в кристаллическом или не кристаллическом (например, аморфном) состоянии.
Будет ли соединение существовать в кристаллическом состоянии или нет, легко может быть определено стандартными методиками, такими как рентгеновская порошковая дифрактометрия (XRPD).
Кристаллы и их кристаллические структуры могут быть характеризованы с применением некоторого количества методик, включая рентгеноструктурную кристаллографию монокристаллов, рентгеновскую порошковую дифрактометрию (XRPD), дифференциальную сканирующую калориметрию (DSC) и инфракрасную спектроскопию, например, инфракрасная спектроскопия с Фурье-преобразованием (FTIR). Поведение кристаллов при условиях переменной влажности может быть анализировано изучениями гравиметрической сорбции паров, а также XRPD.
Определение кристаллической структуры соединения может быть выполнено при помощи рентгеноструктурной кристаллографии, которая может быть проведена традиционными способами, такими как те, что описаны в настоящей заявке, и которые описаны в Fundamentals of Crystallography, С Giacovazzo, Н. L. Monaco, D. Viterbo, F. Scordari, G. Gilli, G. Zanotti and M. Catti, (International Union of Crystallography/Oxford University Press, 1992 ISBN 0-19-855578-4 (p/b), 0-19-85579-2 (h/b)). Эта методика включает в себя анализ и интерпретацию дифракции рентгеновских лучей монокристалла.
В аморфном твердом веществе трехмерная структура, которая обычно существует в кристаллической форме, не существует, и положения молекул относительно друг друга в аморфной форме в основном являются беспорядочными, см., например, Hancock и другие J. Pharm. Sci. (1997), 86, 1).
Соответственно, согласно дополнительным вариантам реализации настоящее изобретение обеспечивает:
1.39 Соединение по любому из вариантов реализации 1.1-1.38 в кристаллической форме.
1.40 Соединение по любому из вариантов реализации 1.1-1.38, которое:
(а) от 50% до 100% кристаллическое, и более конкретно, по меньшей мере на 50% кристаллическое, или по меньшей мере на 60% кристаллическое, или по меньшей мере на 70% кристаллическое, или по меньшей мере на 80% кристаллическое, или по меньшей мере на 90% кристаллическое, или по меньшей мере на 95% кристаллическое, или по меньшей мере на 98% кристаллическое, или по меньшей мере на 99% кристаллическое, или по меньшей мере на 99,5% кристаллическое, или по меньшей мере на 99,9% кристаллическое, например, на 100% кристаллическое.
1.41 Соединение по любому из вариантов реализации 1.1-1.38, которое находится в аморфной форме.
Пролекарства
Соединения формулы (1), как определено в любом из вариантов реализации 1.1-1.41, могут находиться в форме пролекарства. Под «пролекарствами» подразумевается, например, любое соединение, которое превращено in vivo в биологически активное соединение формулы (1), как определено в любом из вариантов реализации 1.1-1.41.
Например, некоторые пролекарства представляют собой сложные эфиры активного соединения (например, физиологически приемлемый метаболически лабильный сложный эфир). В течение метаболизма сложноэфирная группа (-C(=O)OR) отщепляется с получением активного лекарственного средства. Такие сложные эфиры могут быть образованы эстерификацией, например, любой из гидроксильных групп, присутствующих в исходном соединении, по необходимости, перед защитой любых других реакционноспособных групп, присутствующих в исходном соединении, со следующим снятием защитных групп, если это потребуется.
Также, некоторые пролекарства активированы ферментативным путем с получением активного соединения, или соединения, которое, при дальнейшей химической реакции, дает в результате активное соединение (например, как в ADEPT, GDEPT, LIDEPT, и т.д.). Например, пролекарство может быть производным сахара или другим гликозидным конъюгатом, или может быть производным аминокислотного сложного эфира.
Соответственно, согласно другому варианту реализации (вариант реализации 1.42) настоящее изобретение относится к пролекарству соединения, как определено в любом из вариантов реализации 1.1-1.41, где соединение содержит функциональную группу, которая является изменяемой при физиологических условиях с образованием гидроксильной группы или аминогруппы.
Комплексы и клатраты
Также формулой (1) в вариантах реализации 1.1-1.42 охватываются комплексы (например, комплексы включения или клатраты с соединениями, такие как циклодекстрины, или комплексы с металлами) соединений согласно вариантам реализации 1.1-1.42.
Соответственно, согласно другому варианту реализации (вариант реализации 1.43) настоящее изобретение относится к соединению по любому из вариантов реализации
1.1-1.42 в форме комплекса или клатрата.
Биологическая активность
Соединения по настоящему изобретению обладают различными терапевтическими применениями.
Соответственно, согласно другому варианту реализации (вариант реализации 2.1) настоящее изобретение относится к соединению формулы (1), как определено в любом из вариантов реализации 1.1-1.43, для применения в медицине.
Более конкретно, соединения по настоящему изобретению являются ингибиторами киназ, например, киназы FLT3 и киназ Aurora, таких как киназа Aurora А и киназа Aurora В.
Таким образом, согласно дополнительным вариантам реализации (2.2-2.14) настоящее изобретение обеспечивает:
2.2 Соединение формулы (1) или его любую подгруппу или примеры, как определено в любом из вариантов реализации 1.1-1.43, для применения при профилактике или лечении стадии заболевания или состояния, опосредованного киназой FLT3 или киназой Aurora (такой как киназа Aurora А или киназа Aurora В).
2.3 Соединение формулы (1) или его любую подгруппу или примеры, как определено в любом из вариантов реализации 1.1-1.43, для применения при профилактике или лечении стадии заболевания или состояния, которое характеризуется анормальной экспрессией (например, сверхэкспрессией) киназы FLT3 или киназы Aurora (такой как киназа Aurora А или киназа Aurora В).
2.4 Применение соединения формулы (1) или его любой подгруппы или примеров, как определено в любом из вариантов реализации 1.1-1.43, для изготовления медикамента для профилактики или лечения стадии заболевания или состояния, опосредованного киназой FLT3 или киназой Aurora (такой как киназа Aurora А или киназа Aurora В).
2.5 Применение соединения формулы (1) или его любой подгруппы или примеров, как определено в любом из вариантов реализации 1.1-1.43, для изготовления медикамента для профилактики или лечения стадии заболевания или состояния, которое характеризуется анормальной экспрессией (например, сверхэкспрессией) киназы FLT3 или киназы Aurora (такой как киназа Aurora А или киназа Aurora В).
2.6 Способ профилактики или лечения стадии заболевания или состояния, опосредованного киназой FLT3 или киназой Aurora (такой как киназа Aurora А или киназа Aurora В), причем способ предусматривает введение субъекту при необходимости соединения формулы (1) или его любой подгруппы или примеров, как определено в любом из вариантов реализации 1.1-1.43.
2.7 Способ профилактики или лечения стадии заболевания или состояния, которое характеризуется анормальной экспрессией (например, сверхэкспрессией) киназы FLT3 или киназы Aurora (такой как киназа Aurora А или киназа Aurora В), причем способ предусматривает введение субъекту при необходимости соединения формулы (1) или его любой подгруппы или примеров, как определено в любом из вариантов реализации 1.1-1.43.
2.8 Способ ослабления или понижения числа случаев стадии заболевания или состояния, опосредованного киназой FLT3 или киназой Aurora (такой как киназа Aurora А или киназа Aurora В), причем способ предусматривает введение субъекту при необходимости соединения формулы (1) или его любой подгруппы или примеров, как определено в любом из вариантов реализации 1.1-1.43.
2.9 Соединение для применения, применение или способ по любому из вариантов реализации 2.1-2.8, где стадия заболевания или состояние является таким, которое опосредовано киназой FLT3 или характеризуется анормальной экспрессией (например, сверхэкспрессией) киназы FLT3.
2.10 Соединение для применения, применение или способ по любому из вариантов реализации 2.1-2.8, где стадия заболевания или состояние является таким, которое опосредовано киназой Aurora (например, киназой Aurora А или Aurora В) или характеризуется анормальной экспрессией (например, сверхэкспрессией) киназы Aurora (например, киназы Aurora А или Aurora В).
2.11 Способ ингибирования киназы FLT3, причем способ предусматривает приведение в контакт киназы с ингибирующим киназу соединением формулы (1) или его любой подгруппой или примерами, как определено в любом из вариантов реализации 1.1-1.43.
2.12 Способ ингибирования киназы Aurora (такой как киназа Aurora А или киназа Aurora В), причем способ предусматривает приведение в контакт киназы с ингибирующим киназу соединением формулы (1) или его любой подгруппой или примерами, как определено в любом из вариантов реализации 1.1-1.43.
2.13 Способ модулирования клеточного процесса (например, клеточного деления) путем ингибирования активности киназы FLT3 с применением соединения формулы (1) или его любой подгруппы или примеров, как определено в любом из вариантов реализации 1.1-1.43.
2.14 Способ модулирования клеточного процесса (например, клеточного деления) путем ингибирования активности киназы Aurora (такой как киназа Aurora А или киназа Aurora В) с применением соединения формулы (1) или его любой подгруппы или примеров, как определено в любом из вариантов реализации 1.1-1.43.
Ожидалось, что вследствие их активности при модулировании и, в частности, ингибировании киназ FLT3 и Aurora, они будут применимы при лечении и предупреждении пролиферативных нарушений, таких как рак.
Соответственно, согласно дополнительным вариантам реализации (варианты реализации 2.15-2.28) настоящее изобретение дополнительно обеспечивает:
2.15 Соединение формулы (1) или его любую подгруппу или примеры, как определено в любом из вариантов реализации 1.1-1.43, для применения при профилактике или лечении пролиферативного заболевания, такого как рак.
2.16 Применение соединения формулы (1) или его любой подгруппы или примеров, как определено в любом из вариантов реализации 1.1-1.43, для изготовления медикамента для применения при профилактике или лечении пролиферативного заболевания, такого как рак.
2.17 Способ лечения пролиферативного заболевания, такого как рак, у субъекта, причем способ предусматривает введение субъекту (например, млекопитающему, такому как человек) соединения формулы (1) или его любой подгруппы или примеров, как определено в любом из вариантов реализации 1.1-1.43.
2.18 Соединение формулы (1) или его любую подгруппу или примеры, как определено в любом из вариантов реализации 1.1-1.43, для применения при профилактике или лечении заболевания или состояния, предусматривающего или возникающего в результате анормального роста клеток.
2.19 Применение соединения формулы (1) или его любой подгруппы или примеров, как определено в любом из вариантов реализации 1.1-1.43, для изготовления медикамента для применения при профилактике или лечении заболевания или состояния, предусматривающего или возникающего в результате анормального роста клеток.
2.20 Способ лечения заболевания или состояния, предусматривающего или возникающего в результате анормального роста клеток, у млекопитающего, причем способ предусматривает введение млекопитающему соединения формулы (1) или его любой подгруппы или примеров, как определено в любом из вариантов реализации 1.1-1.43, в количестве, эффективном при ингибировании анормального роста клеток.
2.21 Способ облегчения или снижения числа случаев заболевания или состояния, предусматривающего или возникающего в результате анормального роста клеток, у млекопитающего, причем способ предусматривает введение млекопитающему соединения формулы (1) или его любой подгруппы или примеров, как определено в любом из вариантов реализации 1.1-1.43, в количестве, эффективном при ингибировании анормального роста клеток.
2.22 Соединение для применения, применение или способ, как определено в любом из вариантов реализации 2.15-2.17, где пролиферативное заболевание представляет собой рак, выбранный из карциномы, например, карциномы мочевого пузыря, молочной железы, толстой кишки (например, колоректальная карцинома, такая как аденокарцинома толстой кишки и аденома толстой кишки), почки, эпидермиса, печени, легких, например аденокарцинома, мелкоклеточный рак легких и не мелкоклеточная карцинома легких, пищевода, желчного пузыря, яичника, поджелудочной железы, например, экзокринная карцинома поджелудочной железы, желудка, шейки матки, щитовидной железы, предстательной железы или кожи, например, плоскоклеточная карцинома; гемопоэтической опухоли лимфоидного происхождения, например, лейкоз, острый лимфоцитарный лейкоз, В-клеточная лимфома, Т-клеточная лимфома, ходжкинская лимфома, неходжкинская лимфома, лимфома ворсистых клеток или лимфома Беркитта; гемопоэтической опухоли миелоидного происхождения, например, острый и хронический миелогенный лейкозы, миелодиспластический синдром или промиелоцитарный лейкоз; фолликулярного рака щитовидной железы; опухоли мезенхимального происхождения, например, фибросаркома или рабдомиосаркома, опухоли центральной или периферической нервной системы, например, астроцитома, нейробластома, глиома или шваннома; меланомы; семиномы; тератокарциномы; остеолитической саркомы; пигментной ксеродермы; кератоакантомы; фолликулярного рака щитовидной железы; или саркомы Капоши.
2.23 Соединение для применения, применение или способ, как определено в варианте реализации 2.22, где рак является таким, который подвержен ингибированию киназой Aurora А, и выбран из рака молочной железы, мочевого пузыря, колоректального, поджелудочной железы и яичников, неходжкинской лимфомы, глиомы, не эндометриоидной карциномы эндометрия, острого миелобластного лейкоза (AML), хронического миелогенного лейкоза (CML), В-клеточной лимфомы (клетка мантийной ткани) и острого лимфобластного лейкоза (ALL).
2.24 Соединение для применения, применение или способ, как определено в варианте реализации 2.22, где рак является таким, который подвержен ингибированию киназой Aurora В, и выбран из колоректального рака, рака легких, острого миелоидного лейкоза, острого лимфобластного лейкоза и острого эозинофильного лейкоза.
2.25 Соединение для применения, применение или способ, как определено в варианте реализации 2.22, где рак является таким, который подвержен ингибированию киназой FLT3, и представляет собой острый миелоидный лейкоз (AML).
2.26 Соединение для применения, применение или способ, как определено в варианте реализации 2.25, где AML связан у пациента с соматической или точечной мутацией FLT3.
2.27 Соединение для применения, применение или способ, как определено в варианте реализации 2.26, где AML связан у пациента с соматической мутацией FLT3, включающей в себя внутренние тандемные дубликации в околомембранной области рецептора FLT3.
2.28 Соединение для применения, применение или способ, как определено в варианте реализации 2.26, где AML связан у пациента с точечной мутацией D835 в активационной петле FLT3.
2.29 Соединение для применения, применение или способ, как определено в варианте реализации 2.22, где пролиферативное заболевание представляет собой гемопоэтическую опухоль.
2.30 Соединение для применения, применение или способ, как определено в варианте реализации 2.29, где гемопоэтическая опухоль выбрана из острого лимфобластного лейкоза (ALL), острого миелоидного лейкоза (AML), хронического миелоидного лейкоза (CML), ходжкинской лимфомы (HL), неходжкинской лимфомы (NHL) и множественной миеломы (ММ).
Независимо от того, является ли конкретный вид рака чувствительным к ингибированию киназой Aurora или киназой FLT3, он может быть определен при помощи анализа роста клеток, например, анализом, который описан в примере ниже или способом, изложенным в разделе под названием «способы диагностики».
Активность соединений по настоящему изобретению в качестве ингибиторов киназ может быть измерена с применением анализов, изложенных в примерах ниже, и уровень активности, показанный данным соединением, может быть определен в значениях IC50. Предпочтительные соединения по настоящему изобретению представляют собой соединения, обладающие значением IC50 менее чем 0,01 мкМ, более предпочтительно, менее чем 0,005 мкМ.
Способы получения соединений по настоящему изобретению
Согласно другому аспекту (вариант реализации 3.1) настоящее изобретение обеспечивает способ получения соединения, как определено в любом из вариантов реализации 1.1-1.35, причем способ предусматривает:
(i) осуществление взаимодействия соединения формулы (10):

где LG1 представляет собой йод или бром (более типично йод), DMB представляет собой 2,4-диметоксибензил защитную группу и boc представляет собой трет-бутилоксикарбонил защитную группу; с соединением формулы (11А) или (11В) или соответствующим боронатным сложным диэфиром:
 ,
,
где PG представляет собой защитную группу, такую как трет-бутилдиметилсилильная группа, при реакционных условиях сочетания Сузуки, и затем удаление защитных групп DMB и boc; или
(ii) осуществление взаимодействия соединения формулы (18):
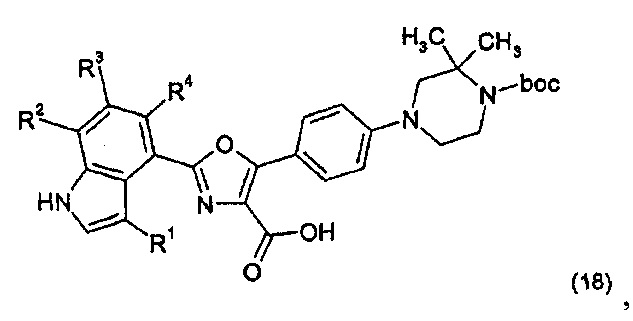
где boc представляет собой трет-бутилоксикарбонил, с аммиаком при амидобразующих условиях, и затем удаление группы boc.
При варианте процесса (i) реакционные условия сочетания Сузуки типично включают в себя присутствие палладиевого катализатора, такого как тетракис(трифенилфосфин)палладий или бис (1,1'-бис(дифенил-фосфино)-ферроцен) палладия дихлорид (Pd(dppf)2Cl2), и основания (например, карбоната, такого как карбонат калия). Реакция может проводиться в полярном растворителе, например, ацетонитриле или диоксане, или их смесях, или в водном растворителе, таком как водный этанол, или в эфире, таком как диметоксиэтан, и реакционную смесь типично нагревали, например, до температуры 80°C или более, например, температуры в диапазоне от 80°C до 100°C.
После прохождения реакции сочетания соединения (10) и или соединения (11А), или соединения (11В), защитные группы могут быть беспрепятственно удалены с применением кислоты, такой как трифторметансульфоновая кислота, в растворителе, таком как дихлорметан, обычно при комнатной температуре или около нее.
Соединение формулы (10), в которой LG1 представляет собой атом йода, может быть получено посерийными взаимодействиями, как показано на схеме 1 ниже.
На схеме 1 йодбензоилхлорид взаимодействовал с этил 2-изоцианоацетатом в полярном апротонном растворителе, таком как тетрагидрофуран, например, при комнатной температуре, с получением сложного эфира йодфенилоксазола (13). Сложный эфир (13) затем взаимодействовал с защищенным пиперазином (14) в присутствии палладиевого соединения, такого как ацетат палладия, и фосфинового лиганда, такого как бифенил-2-илдициклогексилфосфин, с получением замещенного пиперазинилфенил-оксазолового сложного эфира (15). Реакцию типично проводили в апротонном растворителе, таком как толуол, в присутствии основания, такого как карбонат цезия, обычно с умеренным нагревом, например, до температуры приблизительно 70-90°C.
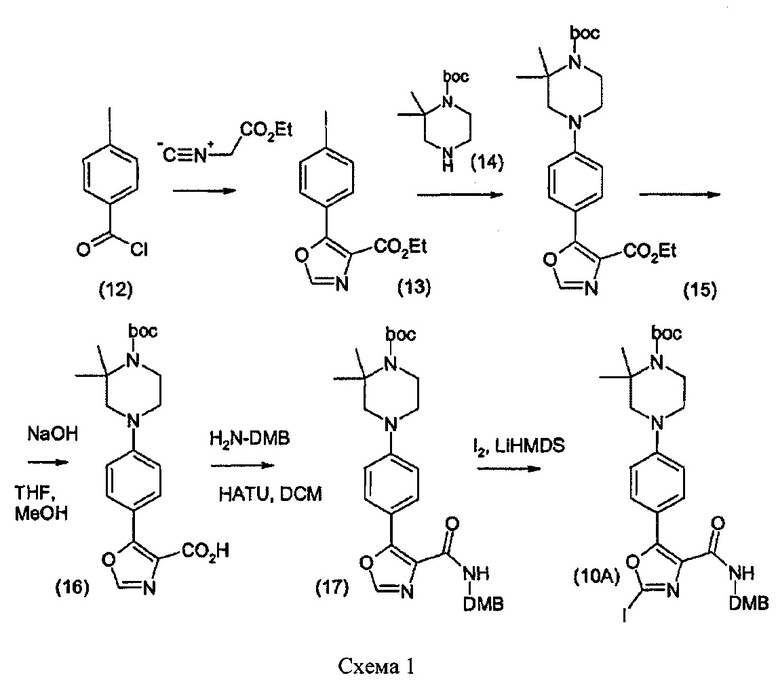
Сложный эфир (15) гидролизовали с применением гидроксида щелочного металла, такого как гидроксид натрия, в полярном растворителе, таком как метанол и/или THF, с получением карбоновой кислоты (16), которую затем превращали в диметоксибензиламид (17) путем осуществления взаимодействия с диметоксибензиламином при условиях образования амида, например, в присутствии реагента такого типа, который обычно используют при образовании амидных связей. Примеры таких реагентов включают в себя агенты реакции сочетания на основе карбодиимида, такие как 1,3-дициклогексилкарбодиимид (DCC) (Sheehan и другие, J. Amer. Chem Soc. 1955, 77, 1067) и 1-этил-3-(3'-диметиламинопропил)-карбодиимид (в настоящем описании называется как EDC или EDCl) (Sheehan и другие, J. Org. Chem., 1961, 26, 2525), которые обычно используют в комбинации с 1-гидрокси-7-азабензотриазолом (НОAt) (L.A. Carpino, J. Amer. Chem. Soc, 1993, 115. 4397) или 1-гидроксибензотраизолом (HOBt) (Konig и другие, Chem. Ber., 103, 708, 2024-2034). Дополнительными примерами таких реагентов являются агенты реакции сочетания на основе урония, такие как O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (HATU). Одним предпочтительным амидным агентом реакции сочетания является HATU.
Реакцию сочетания типично проводят в не водном, не протонном растворителе, таком как диметилформамид, при комнатной температуре в присутствии не взаимодействующего основания, например, третичного амина, такого как триэтиламин или N,N-диизопропилэтиламин.
Литированием диметоксибензиламида (17) с применением лития гексаметилдисилазида (LiHMDS) в THF при пониженной температуре с последующим взаимодействием с йодом получали 2-йодоксазол (10). Соединение (10) затем может быть превращено в соединение формулы (1) описанными выше способами.
При варианте процесса (ii) карбоновая кислота формулы (18) реагировала с аммиаком (или исходной аминогруппой, такой как диметоксибензиламин) при описанном выше типе условий образования амида, например, с применением EDC в комбинации с HOBt.
Соединение формулы (18) может быть получено последовательностью реакций, показанных на схеме 2 ниже.
На схеме 2 сложный эфир (15) (см. схему 1 выше) йодировали путем осуществления взаимодействия с литием гексаметилдисилазидом (LiHMDS) в THF при пониженной температуре с последующим взаимодействием с йодом с получением йодистого соединения (19). Йодистое соединение (19) затем взаимодействовало при условиях реакции сцепления Сузуки (см. выше) с соединением боронатного сложного эфира формулы (11) с получением промежуточного соединения (20), которое гидролизовали с применением гидроксида натрия с получением карбоновой кислоты формулы (18).
После образования многие соединения формулы (1) могут быть превращены в другие соединения формулы (1) с применением стандартных взаимопревращений функциональной группы.
Примеры взаимопревращений функциональной группы, а также реагенты и условия проведения таких превращений, могут быть обнаружены, например, в Advanced Organic Chemistry, by Jerry March, 4th edition, 119, Wiley Interscience, New York, Fiesers' Reagents for Organic Synthesis, Volumes 1-17, John Wiley, edited by Mary Fieser (ISBN: 0-471-58283-2), and Organic Syntheses, Volumes 1-8, John Wiley, edited by Jeremiah P. Freeman (ISBN: 0-471-31192-8).
Во многих описанных выше реакциях может быть необходимым защищать одну или более групп для предотвращения прохождения реакции на нежелательном месте молекулы. Примеры защитных групп, и способы защиты и снятия защиты с функциональных групп могут быть обнаружены в Protective Groups in Organic Synthesis (Т. Green and P. Wuts; 3rd Edition; John Wiley and Sons, 1999).
Соединения по настоящему изобретению могут быть выделены и очищены стандартными методиками, хорошо известными специалисту настоящей области техники. Одной особо применимой методикой очистки соединений является препаративная жидкостная хроматография с применением масс-спектрометрии в качестве средства определения очищенных соединений, выходящих из колонки для хроматографии.
Препаративная LC-MS представляет собой стандартный и эффективный способ, используемый для очистки небольших органических молекул, таких как описанных в настоящем изобретении соединений. Способы жидкостной хроматографии (LC) и масс-спектрометрии (MS) могут меняться с обеспечением лучшего отделения неочищенных материалов и улучшенного определения образцов при помощи MS. Оптимизация градиента препаративного LC способа будет включать в себя переменные колонки, летучие элюенты и модификаторы, и градиенты. Способы являются хорошо известными в области техники для оптимизации препаративных LC-MS способов, и затем их использовали для очистки соединений. Такие способы описаны в Rosentreter U, Huber U.; Optimal fraction collecting in preparative LC/MS; J Comb Chem.; 2004; 6(2), 159-64 and Leister W, Strauss K, Wisnoski D, Zhao Z, Lindsley C, Development of a custom high-throughput preparative liquid chromatography/mass spectrometer platform for the preparative purification and analytical analysis of compound libraries; J Comb Chem.; 2003; 5(3); 322-9.
Промежуточные соединения (10), (10A) и (15)-(20) также составляют часть настоящего изобретения. Соответственно, согласно дополнительному варианту реализации (вариант реализации 3.2), настоящее изобретение относится к соединению, выбранному из промежуточных соединений формулы (10), (10А), (15), (16), (17), (18), (19) и (20), как описано выше.
Фармацевтические составы
Тогда как для активного соединения является возможным его введение отдельно, предпочтительным является представлять его в виде фармацевтической композиции (например, состава), содержащей по меньшей мере одно активное соединение по настоящему изобретению вместе с одним или более фармацевтически приемлемыми носителями, вспомогательными средствами, наполнителями, разбавителями, наполнителями, буферами, стабилизаторами, консервантами, смазывающими веществами или другими материалами, хорошо известными специалистам настоящей области техники и необязательно другими терапевтическими или профилактическими агентами.
Соответственно, согласно другому аспекту (вариант реализации 4.1) настоящее изобретение относится к фармацевтической композиции, содержащей соединение, как определено в любом из вариантов реализации 1.1-1.43, и фармацевтически приемлемый носитель.
Используемый в настоящем описании термин «фармацевтически приемлемый» относится к соединениям, материалам, композициям и/или формам дозировки, которые, в пределах объема медицинской точки зрения, подходят для применения, находясь в контакте с тканями субъекта (например, человека), без избыточной токсичности, раздражения, аллергической реакции или других проблем или осложнений в соответствии с приемлемым соотношением польза/риск. Каждый носитель, наполнитель, и т.д. должен быть «приемлемым» в смысле совместимости с другими ингредиентами состава.
Фармацевтические композиции могут быть в любой форме, подходящей для перорального, парентерального, местного, интраназального, офтальмического, ушного, ректального, внутривагинального или трансдермального введения. Если композиции предусмотрены для парентерального введения, они могут быть сформулированы для внутривенного, внутримышечного, интраперитонеального, подкожного введения или для прямой доставки в целевой орган или ткань инъекцией, инфузией или другими способами доставки.
Согласно одному аспекту (вариант реализации 4.2) фармацевтическая композиция находится в форме, подходящей для i.v. введения, например, инъекцией или инфузией.
Согласно другому варианту реализации (вариант реализации 4.3) фармацевтическая композиция находится в форме, подходящей для подкожного (s.c.) введения.
Согласно дополнительному варианту реализации (вариант реализации 4.4) фармацевтическая композиция находится в форме, подходящей для перорального введения.
Фармацевтические формы дозировки, подходящие для перорального введения, включают в себя таблетки, капсулы, каплеты, пилюли, пастилки, сиропы, растворы, порошки, гранулы, эликсиры и суспензии, подъязычные таблетки, пластинки или пластыри и трансбуккальные пластыри.
Фармацевтические композиции, содержащие соединения формулы (1), могут быть сформулированы в соответствии с известными методиками, см., например, Remington's Pharmaceutical Sciences, Mack Publishing Company, Easton, PA, USA.
Таким образом, таблетированные композиции могут содержать единицу дозировки активного соединения вместе с инертным разбавителем или носителем, таким как сахар или сахарный спирт, например, лактозой, сахарозой, сорбитом или маннитом; и/или разбавителем не сахарного происхождения, таким как карбонат натрия, фосфат кальция, карбонат кальция или целлюлоза или ее производные, такие как метилцеллюлоза, этилцеллюлоза, гидроксипропилметилцеллюлоза, и крахмалы, такие как кукурузный крахмал. Таблетки также могут содержать такие стандартные ингредиенты в качестве связующих и гранулирующих агентов, такие как поливинилпирролидон, агенты, вызывающие дезинтеграцию (например, поддающиеся разбуханию сшитые полимеры, такие как сшитая карбоксиметилцеллюлоза), смазывающие агенты (например, стеараты), консерванты (например, парабены), антиоксиданты (например, ВНТ), буферные агенты (например, фосфатные или цитратные буферы) и шипучие агенты, такие как смеси цитрата/бикарбоната. Такие наполнители являются хорошо известными и нет необходимости в их подробном обсуждении в настоящем описании.
Капсульные составы могут быть разнообразными из твердого желатина или мягкого желатина, и могут содержать активный компонент в твердой, полутвердой или жидкой форме. Желатиновые капсулы могут быть образованы из животного желатина или синтетического, или из его эквивалентов растительного происхождения.
Твердые формы дозировки (например, таблетки, капсулы, и т.д.) могут быть покрытыми или не покрытыми, но обычно содержат покрытия, например, покрытие из защитной плетки (например, воск или лак) или покрытие контролируемого высвобождения. Покрытие (например, полимер типа Eudragitтм) может быть разработано для высвобождения активного компонента на необходимом местоположении в желудочно-кишечном тракте. Таким образом, покрытие может быть выбрано таким образом, чтобы распадаться при определенных условиях pH в желудочно-кишечном тракте, тем самым селективно высвобождая соединение в желудке или в подвздошной кишке или двенадцатиперстной кишке.
Вместо или вдобавок к, покрытию, лекарственное средство может присутствовать в твердой матрице, содержащей агент, контролирующий высвобождение, например, агент, задерживающий высвобождение, который может быть адаптирован к селективному высвобождению соединения при условиях переменной кислотности или щелочности в желудочно-кишечном тракте. Альтернативно, материал матрицы или покрытие, задерживающее высвобождение, может принимать форму разрушаемого полимера (например, полимера малеинового ангидрида), который в основном непрерывно разрушается при прохождении лекарственной формы через желудочно-кишечный тракт. В качестве дополнительной альтернативы, в системе доставки может быть сформулировано активное соединение, которое обеспечивает осмотический контроль высвобождения соединения. Составы осмотического высвобождения и другого длительного высвобождения или замедленного высвобождения могут быть получены способами, хорошо известными специалистам настоящей области техники.
Композиции для местного применения включают в себя мазь, кремы, спрей, пластыри, гели, жидкие капли и вкладки (например, внутриглазные вкладки). Такие композиции могут быть составлены согласно известным способам.
Композиции для парентерального введения типично представлены как стерильные водные или масляные растворы или высокодисперсные суспензии, или могут быть обеспечены в высокодисперсной форме стерильного порошка для составления препарата немедленного приема со стерильной водой для инъекции.
Композиции для парентерального введения могут быть сформулированы для введения в виде отдельных единиц дозировки или могут быть сформулированы для введения инфузией.
Примеры составов для ректального или внутривагинального введения включают в себя пессарии и суппозитории, которые могут быть, например, образованы из пластичного или воскового материала определенной формы, содержащего активное соединение.
Композиции для введения ингаляционным путем могут принимать форму вдыхаемых порошкообразных композиций или жидких или порошкообразных спреев, и могут быть введены в стандартной форме с применением ингаляторных устройств для порошка или устройств, распределяющих аэрозоль. Такие устройства являются хорошо известными. Для введения путем ингаляции порошкообразные составы типично содержат активное соединение вместе с инертным твердым порошкообразным разбавителем, таким как лактоза.
Как правило, соединения по настоящему изобретению будут присутствовать в стандартной лекарственной форме, и, как таковые, типично будут содержать достаточное количество соединения для обеспечения желаемого уровня биологической активности. Например, состав, предназначенный для перорального введения, может содержать от 0,1 миллиграмма до 2 грамм активного ингредиента, более типично, от 10 миллиграмм до 1 грамма, например, от 50 миллиграмм до 500 миллиграмм.
Активное соединение будет вводиться пациенту при необходимости (например, человеку или животному) в количестве, достаточном для достижения желаемого терапевтического эффекта.
Способы лечения
Предусматривается, что соединения формулы (1) и их подгруппы, определенные в настоящем документе, будут применимыми при профилактике или лечении диапазона стадий заболевания или состояний, опосредованных киназой Aurora (например, киназой Aurora А или киназой Aurora В). Примеры таких стадий заболевания или состояний изложены выше.
В частности, предусматривается, что соединения формулы (1) будут применимы при профилактике и лечении пролиферативных заболеваний (таких как рак) и миелопролиферативных нарушений.
Как правило, соединения вводили субъекту при необходимости такого введения, например, человеку или животному, предпочтительно, человеку.
Соединения типично будут вводить в количествах, которые являются терапевтически или профилактически применимыми, и которые, как правило, не токсичны. Тем не менее, в определенных ситуациях (например, в случае опасных для жизни заболеваний) преимущества введения соединения формулы (1) могут перевешивать недостатки любых токсических или побочных эффектов, в этом случае можно считать желательным введение соединений в количествах, которые связаны со степенью токсичности.
Соединения могут быть введены в течение более длительного срока для сохранения преимущественных терапевтических эффектов или могут быть введены только в течение короткого периода. Альтернативно, они могут быть введены пульсирующим или непрерывным способом.
Типичная ежедневная доза соединения может быть в диапазоне от 100 пикограмм до 100 миллиграмм на килограмм массы тела, более типично, от 5 нанограмм до 25 миллиграмм на килограмм массы тела, и более обычно, от 10 нанограмм до 15 миллиграмм на килограмм (например, от 10 нанограмм до 10 миллиграмм) на килограмм массы тела, хотя, если необходимо, могут быть введены более высокие или более низкие дозы. В конечном итоге, количество вводимого соединения и тип используемой композиции будет соответствовать природе заболевания или физиологическому состоянию, которое лечили, и будет на усмотрение врача.
Соединения формулы (1) могут быть введены как единственный терапевтический агент или они могут быть введены в комбинационной терапии с одним или более другими соединениями для лечения конкретной стадии заболевания, например, новообразования, такого как рак, как было определено выше. Примеры других терапевтических агентов, которые могут быть введены вместе (или одновременно, или при разных интервалах времени) с соединениями формулы (1), включают в себя без ограничения ингибиторы топоизомеразы, алкилирующие агенты, антиметаболиты, ДНК связующие и микротрубочковые ингибиторы (направленные на тубулин агенты), такие как цисплатин, циклофосфамид, доксорубицин, иринотекан, флударабин, 5FU, таксаны, митомицин С или радиотерапия. Альтернативно, соединения формулы (1) могут быть введены при комбинационной терапии с моноклональными антителами или ингибиторами сигнальной трансдукции.
Если соединение формулы (1) вводили при комбинационной терапии с одним, двумя, тремя, четырьмя или более другими терапевтическими агентами (предпочтительно, одним или двумя, более предпочтительно, одним), соединения могут быть введены одновременно или последовательно. При последовательном введении они могут быть введены при узких интервалах (например, в течение 5-10 минут) или при более длительных интервалах (например, с интервалом 1, 2, 3, 4 или более часов, или даже при более длительных периодах, если необходимо), точный режим дозировки соответствовал свойствам терапевтического (их) агента (ов).
Соединения по настоящему изобретению также могут быть введены в сочетании с не химиотерапевтическими лечениями, такими как радиотерапия, фотодинамическая терапия, генная терапия; оперативное вмешательство и контролируемая диета.
Для применения в комбинационной терапии с другим химиотерапевтическим агентом соединение формулы (1) с одним, двумя, тремя, четырьмя или более другими терапевтическими агентами может быть, например, сформулировано вместе в лекарственной форме, содержащей два, три, четыре или более терапевтических агентов. В альтернативе отдельные терапевтические агенты могут быть сформулированы отдельно и быть представлены вместе в форме набора, необязательно, с инструкциями их применения.
Способы диагностики
Перед введением соединения формулы (1) пациента осматривали для определения, является ли заболевание или состояние, от которого пациент страдает или может страдать, одним из тех, которое может быть подвержено лечению соединением с активностью против киназы FLT3 или киназы Aurora (например, киназы Aurora А или киназы Aurora В).
Соответственно, согласно дополнительным вариантам реализации (5.1-5.6) настоящее изобретение обеспечивает:
5.1: Соединение, как определено в любом из представленных вариантов реализации 1.1-1.43, или его любую подгруппу или примеры, как определено в настоящем описании, для применения при лечении или профилактике стадии заболевания или состояния у пациента, которого осмотрели и определили как страдающего от, или который находится в зоне риска, заболевания или состояния, которое было подвержено лечению соединением с активностью против киназы Aurora (например, киназы Aurora А или киназы Aurora В).
5.2 Применение соединения, как определено в любом из представленных вариантов реализации 1.1-1.43, или его любую подгруппу или примеры, как определено в настоящем описании, для изготовления медикамента для лечения или профилактики стадии заболевания или состояния у пациента, которого осмотрели и определили как страдающего от, или который находится в зоне риска, заболевания или состояния, которое было подвержено лечению соединением с активностью против киназы Aurora (например, киназы Aurora А или киназы Aurora В).
5.3 Способ диагностики и лечения стадии заболевания или состояния, опосредованного киназой Aurora (например, киназой Aurora А и киназой Aurora В), причем способ предусматривает: (i) осмотр пациента на определение того, является ли заболевание или состояние, от которого пациент страдает или может страдать, одним из тех, которое может быть подвержено лечению соединением с активностью против киназы; и (ii) если отмечено, что это заболевание или состояние, к которому, таким образом, пациент подвержен, соответственно, следует введение пациенту соединения, как определено в любом из представленных вариантов реализации 1.1-1.43, или его любой подгруппы или примеров, как определено в настоящем описании.
5.4 Соединение, как определено в любом из представленных вариантов реализации 1.1-1.43, или его любую подгруппу или примеры, как определено в настоящем описании, для применения при лечении или профилактике стадии заболевания или состояния у пациента, которого осмотрели и определили как страдающего от, или который находится в зоне риска, заболевания или состояния, которое было подвержено лечению соединением с активностью против киназы FLT3.
5.5 Применение соединения, как определено в любом из представленных вариантов реализации 1.1-1.43, или его любой подгруппы или примеров, как определено в настоящем описании, для изготовления медикамента для лечения или профилактики стадии заболевания или состояния у пациента, которого осмотрели и определили как страдающего от, или который находится в зоне риска, заболевания или состояния, которое было подвержено лечению соединением с активностью против киназы FLT3.
5.6 Способ диагностики и лечения стадии заболевания или состояния, опосредованного киназой FLT3, причем способ предусматривает: (i) осмотр пациента на определение того, является ли заболевание или состояние, от которого пациент страдает или может страдать, одним из тех, которое может быть подвержено лечению соединением с активностью против киназы; и (ii) если отмечено, что это заболевание или состояние, к которому, таким образом, пациент подвержен, соответственно, следует введение пациенту соединения, как определено в любом из представленных вариантов реализации 1.1-1.43, или его любой подгруппы или примеров, как определено в настоящем описании.
Биологический образец, взятый у пациента, может быть подвергнут диагностическим испытаниям для определения, является ли состояние или заболевание, такое как рак, от которого пациент страдает или может страдать, одним из тех, которое характеризуется генетической анормальностью (например, мутированной киназой) или анормальной белковой экспрессией, такой как сверхэкспрессия или повышенная регуляция киназы Aurora или киназы FLT3. Пациента могут подвергать диагностическому испытанию для определения проверяющей характеристики повышенной регуляции киназы Aurora или киназы FLT3 или присутствия мутированной киназы Aurora или киназы FLT3. Опухоли с повышенной регуляцией киназы Aurora или киназы FLT3 могут быть особым образом чувствительными к ингибиторам киназы.
Таким образом, опухоли предпочтительно осматривали на повышенную регуляцию кинзы Aurora или киназы FLT3. Диагностические испытания типично проводили на биологическом образце, выбранном из биопсийных образцов опухоли, образцов крови (выделение и обогащение отброшенных опухолевых клеток), биопсийных образцов экскрементов, слюны, анализе хромосом, плевральной жидкости, перитонеальной жидкости или мочи.
Определение особей, несущих мутацию в киназе Aurora или киназе FLT3, может означать, что пациент будет особым образом подходить для лечения ингибитором киназы. Опухоли предпочтительно могут быть осмотрены на присутствие разновидностей перед лечением. Как правило, процесс осмотра включает в себя прямое секвенирование, микроматричный анализ олигонуклеотидов или специфическое антитело мутанта.
Способы определения и анализы мутаций и повышенная регуляция белков известны специалистам настоящей области техники. Способы отбора могут включать в себя без ограничения стандартные способы, такие как полимеразная цепная реакция с обратной траскриптазой (RT-PCR) или in-situ гибридизацию.
При отборе при помощи RT-PCR уровень мРНК в опухоли оценивается путем образования копии кДНК мРНК с последующей амплификацией кДНК путем PCR. Способы амплификации PCR, выбор праймеров и условия амплификации известны специалистам настоящей области техники. Процедуры с нуклеиновой кислотой и PCR проводились стандартными способами, как описано, например, в Ausubel и другие, eds. Current Protocols in Molecular Biology (2004) John Wiley & Sons Inc., or Innis, M.A. и другие, eds. PCR Protocols: a guide to methods and applications (1990) Academic Press, San Diego. Реакции и процедуры, включающие в себя методики с нуклеиновой кислотой, также описаны в Sambrook и другие, 3rd Ed, Molecular Cloning: A Laboratory Manual (2001) Cold Spring Harbor Laboratory Press. Альтернативно, может быть использован коммерчески доступный набор для RT-PCR (например, Roche Molecular Biochemicals), или методология, как изложено в патентах США №№4666828; 4683202; 4801531; 5192659, 5272057, 5882864 и 6218529, и включено в настоящий документ посредством ссылки.
Примером методики гибридизации in-situ для определения мРНК экспрессии будет флуоресцентная гибридизация in-situ (FISH) (см. Angerer, 1987 Meth. Enzymol., 152: 649).
Как правило, гибридизация in-situ включает в себя следующие основные стадии: (1) фиксацию анализируемой ткани; (2) обработку предварительной гибридизацией образца для увеличения доступности мишеневой нуклеиновой кислоты и для снижения неспецифического связывания; (3) гибридизацию смеси нуклеиновых кислот до нуклеиновой кислоты в биологической структуре или ткани; (4) промывания после гибридизации для удаления фрагментов нуклеиновой кислоты, не связанных гибридизацией, и (5) определение гибридизированных фрагментов нуклеиновой кислоты. Пробы, используемые в таких применениях, типично помечали, например, радиоизотопами или флуоресцентными репортерами. Предпочтительные пробы являются довольно длинными, например, от приблизительно 50, 100 или 200 нуклеотидов до приблизительно 1000 или более нуклеотидов, для обеспечения конкретной гибридизации мишеневой(ыми) нуклеиновой(ыми) кислотой(ами) при жестких условиях. Стандартные способы проведения FISH описаны в Ausubel и другие, eds. Current Protocols in Molecular Biology (2004) John Wiley & Sons Inc and Fluorescence In Situ Hybridization: Technical Overview by John M. S. Bartlett in Molecular Diagnosis of Cancer, Methods and Protocols, 2nd ed.; ISBN: 1-59259-760-2; (2004) pps. 077-088; Series: Methods in Molecular Medicine.
Альтернативно, белковые продукты, экспрессированные из мРНК, могут быть анализированы при помощи иммуногистохимии опухолевых образцов, твердофазного иммуноанализа титрационными микропланшетами, вестерн-блоттинга, 2-размерного денатурирующего электрофореза в полиакриламидном геле, ELISA, проточной цитометрии и других способов, известных из области техники, для определения конкретных белков. Способы определения будут включать в себя применение сайт-специфических антител.
КРАТКОЕ ОПИСАНИЕ ФИГУР
На фигуре 1 проиллюстрирован противоопухолевый эффект соединений по примерам 2 и 7 по сравнению с цитарабином в MV4-11 ксенотрансплантате мышиной модели, описанном в примере 17.
На фигуре 2 проиллюстрирован противоопухолевый эффект соединения по примеру 2 по сравнению с цитарабином в MV4-11 ксенотрансплантате мышиной модели.
На фигуре 3 проиллюстрирован противоопухолевый эффект соединения по примеру 7 по сравнению с цитарабином в MV4-11 ксенотрансплантате мышиной модели.
На фигуре 4 проиллюстрированы массы опухолей голой мыши с ксенотрансплантатом MV4-11 на 29 день (конечная точка) после введения носителя, соединений по примерам 2 и 7 и цитарабина.
На фигуре 5 проиллюстрированы изменения массы тела голой мыши с ксенотрансплантатом MV4-11 в течение всего периода изучения, описанного в примере 17, после введения носителя, соединений по примерам 2 и 7 и цитарабина.
ПРИМЕРЫ
Настоящее изобретение будет проиллюстрировано без ограничения ссылкой на конкретные варианты реализации, описанные в следующих примерах.
В примерах использовали следующие аббревиатуры:
Получение промежуточных соединений
А. Получение промежуточного соединения (10A)
Трет-бутиловый сложный эфир 4-{4-[4-(2,4-диметокси-бензилкарбамоил)-2-йод-оксазол-5-ил]-фенил)-2,2-диметил-пиперазин-1-карбоновой кислоты
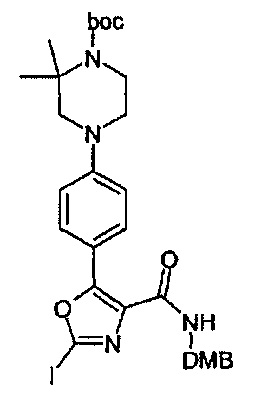
Промежуточное соединение (10А) может быть получено последовательностью реакций, показанных на схеме 1 выше.
Стадия 1
Этиловый сложный эфир 5-(4-йод-фенил)-оксазол-4-карбоновой кислоты (соединение (13) на схеме 1)
К раствору 4-йодбензоил хлорида (14,0 г, 0,052 моль) в 100 мл THF по каплям добавляли TEA (15,6 г, 0,156 моль), и смесь перемешивали в течение 10 минут перед медленным добавлением этил 2-изоцианоацетата (6,5 г, 0,058 моль). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов, растворитель удаляли и остаток обрабатывали EtOAc и водой. Органический слой отделяли, сушили над Na2SO4 и растворитель удаляли. Неочищенный продукт очищали на колонке с получением названного соединения (11,0 г, 61,7%) в виде оранжевого твердого вещества.
1Н ЯМР: CDCl3 400 МГц - δ 7,92 (s, 1Н), 7,80-7,90 (m, 4Н), 4,43 (q, J=7,2 Гц, 2Н), 1,42 (t, J=7,2 Гц, 3Н).
Стадия 2
Трет-бутиловый сложный эфир 4-[4-(4-этоксикарбонил-оксазол-5-ил)-фенил]-2,2-диметил-пиперазин-1-карбоновой кислоты (соединение (15) на схеме 1)
К перемешиваемому раствору этилового сложного эфира 5-(4-йод-фенил)-оксазол-4-карбоновой кислоты (11,0 г, 32 ммоль) в сухом толуоле (300 мл) добавляли трет-бутиловый сложный эфир 2,2-диметил-пиперазин-1-карбоновой кислоты (7,53 г, 35,2 ммоль), Pd(AcO)2 (580 мг, 2,56 ммоль), бифенил-2-илдициклогексилфосфин (0,9 г, 2,56 ммоль) и Cs2CO3 (20,7 г, 64 ммоль) в атмосфере азота при комнатной температуре. Смесь нагревали до 80°C и перемешивали в течение 24 часов. Раствор оставляли охлаждаться до комнатной температуры, а затем разделяли между водой и EtOAc. Органическую фазу сушили над Na2SO4, и растворитель удаляли с получением неочищенного продукта. Его очищали колоночной хроматографией на силикагеле (РЕ/EtOAc=40/1~10/1) с восстановлением непрореагировавшего исходного материала; PE/EtOAc=8/1~5/1 с получением названного соединения (6,0 г, 43%) в виде желтого твердого вещества.
1Н ЯМР: DMSO 400 МГц - δ 8,06 (d, J=8,8 Гц, 2Н), 7,81 (s, 1Н), 6,75 (d, J=9,2 Гц, 2Н), 4,42 (q, 2Н), 3,86 (m, 2Н), 3,50 (m, 2Н), 3,46 (s, 2Н), 1,56 (s, 9Н), 1,43 (s, 6Н).
Стадия 3
Трет-бутиловый сложный эфир 4-{4-[4-{2,4-диметокси-бензилкарбамоил)-оксазол-5-ил]-фенил}-2,2-диметил-пиперазин-1-карбоновой кислоты (соединение (17) на схеме 1)
К раствору трет-бутилового сложного эфира 4-[4-(4-этоксикарбонил-оксазол-5-ил)-фенил]-2,2-диметил-пиперазин-1-карбоновой кислоты (2,4 г, 5,6 ммоль) в 20 мл THF и 20 мл метанола добавляли 2 н водный раствор гидроксида натрия (11,2 мл, 22,4 ммоль). Смесь перемешивали при комнатной температуре в течение 16 часов, и органический растворитель затем удаляли in vacuo. Оставшуюся водную смесь доводили до pH 4-5 добавлением 1 н водного хлорида водорода. Смесь сушили сублимацией с получением 3 г трет-бутилового сложного эфира 4-[4-(4-карбокси-оксазол-5-ил)-фенил]-2,2-диметил-пиперазин-1-карбоновой кислоты в виде порошка желтоватого цвета, который сразу использовали на следующей стадии.
Трет-бутиловый сложный эфир 4-[4-(4-карбокси-оксазол-5-ил)-фенил]-2,2-диметил-пиперазин-1-карбоновой кислоты растворяли в 30 мл DMF. К раствору добавляли 2,4-диметокси-бензиламин (1,26 г, 7,6 ммоль), HATU (2,8 г, 7,4 ммоль) и TEA (1 г, 10 ммоль). Смесь перемешивали при комнатной температуре всю ночь, и растворитель удаляли in vacuo. Остаток очищали колоночной хроматографией на силикагеле (РЕ:EtOAc=5:1-DCM:МеОН=200:1) с получением названного соединения (1,6 г, 3 ммоль, выход: 54% за две стадии).
1Н ЯМР: (400 МГц MeOD) δ 8,10 (d, J=9,2 Гц, 2Н), 8,00 (s, 1Н), 7,20 (d, J=8,0 Гц, 1H), 6,82 (d, J=9,2 Гц, 2H), 6,48-6,58 (m, 2H), 4,49 (s, 2H), 3,84-3,90 (m, 5H), 3,80 (s, 3H), 3,71 (t, 2H), 3,56 (s, 2H), 1,51 (s, 9H), 1,45 (s, 6H).
Стадия 4
Трет-бутиловый сложный эфир 4-{4-[4-(2,4-диметокси-бензилкарбамоил)-2-йод-оксазол-5-ил]-фенил1-2,2-диметил-пиперазин-1-карбоновой кислоты (соединение (10А) на схеме 1)
К раствору трет-бутилового сложного эфира 4-{4-[4-(2,4-диметокси-бензилкарбамоил)-оксазол-5-ил]-фенил}-2,2-диметил-пиперазин-1-карбоновой кислоты (2,6 г, 4,7 ммоль) в 20 мл THF при -78°C добавляли 1 М раствор LiHMDS (44 мл, 44 ммоль) в THF. Смесь перемешивали в течение 0,5 ч, затем частями добавляли I2 (9 г, 35 ммоль). Смесь перемешивали при комнатной температуре в течение 1 ч. Смесь гасили 15% водным Na2S2O3 и экстрагировали EtOAc и промывали водой. Органическую фазу сушили над Na2SO4. Растворитель удаляли in vacuo с получением неочищенного продукта, который очищали колоночной хроматографией на силикагеле (РЕ/EtOAc=2/1-РЕ/EtOAc/DCM=1/1/2) с получением названного соединения (2,4 г, выход: 75%).
1Н ЯМР: CDCl3 400 МГц - δ 8,16 (d, J=9,2 Гц, 2Н), 7,22-7,47 (m, 3Н), 6,70 (d, J=9,2 Гц, 2Н), 6,41-6,46 (m, 3Н), 4,52 (d, J=5,6 Гц, 2Н), 3,82-3,88 (m, 5Н), 3,80 (s, 3Н), 3,46 (t, 2Н), 3,43 (s, 2Н), 1,49 (s, 9Н), 1,41 (s, 6Н).
В. ПОЛУЧЕНИЕ ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ БОРОНОВОЙ КИСЛОТЫ
Способами, которые представлены ниже, получали следующие промежуточные соединения бороновой кислоты/боронатного сложного эфира В-1-В-12.
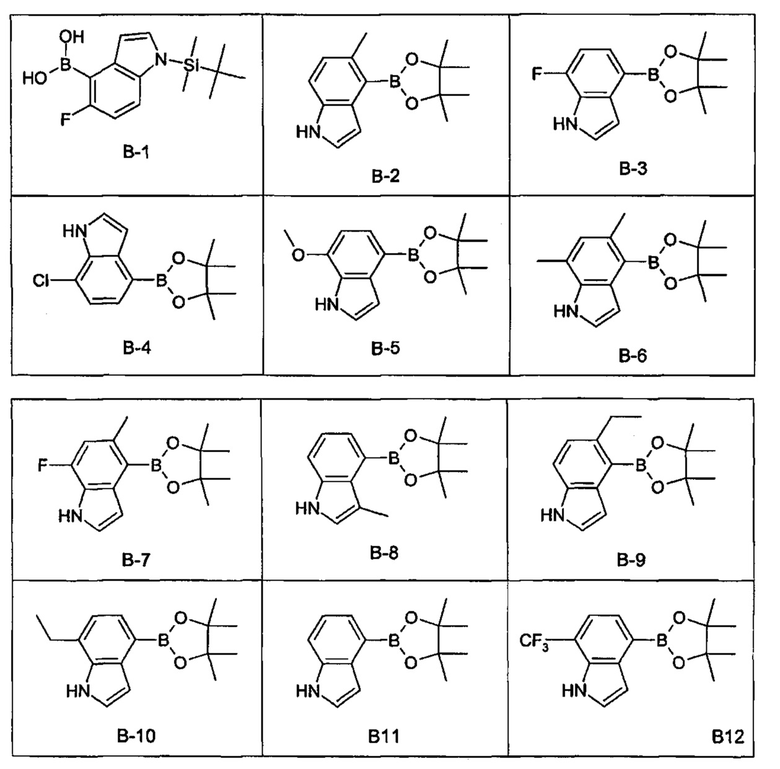
Промежуточное соединение В-1
1-(трет-бутилдиметилсилил)-5-фтор-1Н-индол-4-илбороновая кислота
Стадия 1
Получение 1-(трет-бутилдиметилсилил)-5-фтор-1Н-индола
При 0°C NaH (1,88 г, 46,2 ммоль) добавляли к раствору 5-фтор-1Н-индола (5,2 г, 38,5 ммоль) в 40 мл DMF. Через 10 мин при 0°C добавляли трет-бутилдиметилсилила хлорид (6,96 г, 46,2 ммоль), и смесь оставляли перемешиваться при 0°C в течение дополнительного 1 ч. Смесь нагревали до комнатной температуры и перемешивали всю ночь, затем разбавляли добавлением воды и экстрагировали EtOAc. Органический слой сушили над Na2SO4, концентрировали и очищали колоночной хроматографией на силикагеле с получением названного соединения (6,5 г, 67,7%)
1Н ЯМР CDCl3 400 МГц δ 7,41 (s, 1H), 7,28-7,25 (dd, 1H, J=2,4 и J=8,8), 7,22 (d, 1H, J=3.2), 6,89 (d, 1H, J=2,8), 6,58-6,57 (dd, 1H, J=0,8 и J=3,2), 0,93 (t, 9H), 0,60 (t, 6H)
Стадия 2
Получение 1-(трет-бутилдиметилсилил)-5-фтор-1Н-индол-4-илбороновой кислоты
К смеси продукта стадии 1 (4,98 г, 20 ммоль) и TMEDA (2,32 г, 20 ммоль) в THF при -78°C медленно добавляли 1,3 М раствор втор-BuLi (15,4 мл, 20 ммоль) в циклогексане, и смесь перемешивали в течение 2 ч при -78°C. Триизопропил борат (3,76 г, 20 ммоль) добавляли к смеси при -78°C, и перемешивали в течение дополнительного 1 ч, а затем нагревали до -20°C. Добавляли воду, и смесь экстрагировали EtOAc, и органический слой сушили над Na2SO4 и выпаривали. Остаток перекристаллизовывали (EtOAc и н-гексан) с получением названного соединения (1,2 г, 20,7%).
1H ЯМР DMSO 400 МГц δ 7,48-7,45 (m, 1Н), 7,35 (d, 1H, J=3,2), 6,83 (t, 1Н), 6,50 (s, 2H), 6,62 (dd, 1H, J=0,8 и J-3,2), 0,84 (s, 9H), 0,56 (s, 6H).
Промежуточное соединение B-2
5-Метил-4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-1Н-индол
Стадия 1
3-Бром-2,4-диметил-нитробензол
Дымящую азотную кислоту (32,5 мл) медленно добавляли к раствору 2,6-диметил-бромбензола (10 г, 54 ммоль) в АсОН (75 мл), охлажденному в ледяной бане. Полученную смесь оставляли нагреваться до комнатной температуры, перемешивали в течение 1 ч, и нагревали при 80°C в течение 2 ч. Реакционную смесь охлаждали до комнатной температуры и выливали в ледяную воду при перемешивании. Полученный осадок собирали фильтрованием с отсасыванием с получением названного соединения (10 г), который использовали без дополнительной очистки.
Стадия 2
[2-(2-бром-3-метил-6-нитро-фенил)-винил]-диметил-амин
Смесь 3-бром-2,4-диметил-нитробензола (12 г, 52 ммоль) и пирролидина (2,12 мл) в DMF/DMA (180 мл) нагревали всю ночь при 120°C в запаянной пробирке. Смесь разбавляли водой и экстрагировали EtOAc. Органический слой сушили над Na2SO4 и концентрировали с получением неочищенного названного соединения (10 г).
Стадия 3
4-Бром-5-метилиндол
[2-(2-Бром-3-метил-6-нитро-фенил)-винил]-диметил-амин (10 г) растворяли в АсОН/Н2О (100 мл:25 мл), охлаждали до 0°C и обрабатывали Zn (30 г), медленно добавленным частями. После полного добавления реакционную смесь нагревали при 110°C всю ночь. Смесь разбавляли водой и экстрагировали EtOAc. Органический слой сушили над Na2SO4 и концентрировали с получением неочищенного продукта. Неочищенный продукт очищали колоночной хроматографией на силикагеле с получением названного соединения (1,4 г, 20%)
1Н ЯМР CDCl3 400 МГц δ 2,47 (m, 3Н), 6,50-6,51 (m, 1Н), 6,97-6,99 (m, 1Н), 7,12-7,18 (m, 2Н), 8,12 (s, 1H)
Стадия 4
Получение 5-метил-4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-1Н-индола:
4-Бром-5-метилиндол (0,7 г, 3,35 ммоль), бис(пинаколято)диборон (1,7 г, 6,7 ммоль), КОАс (1 г, 10 ммоль) и Pd(dppf)Cl2 (73 мг, 3 моль. %) суспендировали в сухом DMSO (20 мл) в двух 40 мл стеклянных пробирках, которые плотно запаивали алюминиевым/тефлоновым зажимом. Образцы облучали при 250 Вт, 180°C в течение 40 мин в одномодовом микроволновом реакторе CEM-Discover. После завершения реакций сосуды охлаждали до 60°C, объединяли и неочищенную смесь фильтровали через тонкую целитную пробку. Целитную пробку промывали EtOAc (50 мл), органические фракции объединяли и растворитель удаляли под сниженным давлением. Неочищенный продукт очищали колоночной хроматографией на силикагеле, а затем преп-ВЭЖХ с получением соединения 5 (280 мг, 33%).
1H ЯМР CDCl3 400 МГц δ 1,41 (s, 12Н), 2,63 (s, 3Н), 6,96-6,97 (m, 1Н), 7,01-7,03 (m, 1H), 7,18-7,20 (m, 1Н), 7,32-7,34 (m, 1Н), 8,13 (s, 1Н)
Промежуточное соединение В-3
7-Фтор-4-(4,4,5,5-тетраметил-[1,3,2]дилксаборолан-2-ил)-1Н-индол
Стадия 1
Получение 4-бром-7-фтор-1Н-индола
При -40°C винилмагния бромид (300 мл 1,0 М раствора в THF, 300 ммоль) добавляли по каплям к раствору 3-нитро-4-фтор-бромбензола (22 г, 100 ммоль) в THF (700 мл). Через 1 ч при -40°C смесь гасили водным раствором NH4Cl и экстрагировали EtOAc. Органический слой сушили над Na2SO4 и концентрировали с получением неочищенного продукта, который очищали колоночной хроматографией на силикагеле с получением 4-бром-7-фтор-1Н-индола (5 г, 23,3%).
1Н ЯМР CDCl3 400 МГц δ 6,56-6,58 (m, 1Н), 6,72-6,79 (m, 1H), 7,06-7,17 (m, 1Н), 7,20-7,24 (m, 1H), 8,45 (s, 1H).
Стадия 2
7-Фтор-4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-1Н-индол
4-Бром-7-фтор-1Н-индол (5 г, 23,3 ммоль), бис(пинаколято)диборон (9,5 г, 37,4 ммоль), КОАс (6,85 г, 69,9 ммоль) и Pd(dppf)Cl2 (0,51 г, 3 моль. %) суспендировали в сухом DME (60 мл) в пяти 40 мл стеклянных пробирках, которые плотно запаивали алюминиевым/тефлоновым зажимом. Образцы облучали при 250 Вт, 150°C в течение 25 мин в одномодовом микроволновом реакторе CEM-Discover. После завершения реакций сосуды охлаждали до 60°C, объединяли и неочищенную смесь фильтровали через тонкий слой целита. Целит промывали EtOAc (50 мл), органические фракции объединяли и растворитель удаляли in vacuo. Неочищенный продукт очищали колоночной хроматографией на силикагеле, а затем преп-ВЭЖХ с получением названного соединения (2 г, 32,9%).
1H ЯМР CDCl3 400 МГц δ 1,40 (s, 12Н), 6,86-6,96 (m, 1Н), 7,02-7,14 (m, 1H), 7,20-7,24 (m, 1H), 7,51-7,62 (m, 1H), 8,43 (s, 1H)
Промежуточное соединение B-4
7-Хлор-4-(4,4,5,5-тетраметил-[1.3.2]диоксаборолан-2-ил)-1Н-индол
Стадия 1
Получение 4-бром-1-хлор-2-нитро-бензола:
К раствору 4-хлор-3-нитро-фениламина (17,2 г, 0,1 моль) в 260 мл НВr (48%) при 0°C по каплям добавляли NaNO2 (13,8 г, 0,2 моль) в воде. Реакционную смесь перемешивали в течение 1 ч при 0°C, затем CuBr (24 г, 0,17 моль) частями добавляли к смеси и перемешивали в течение дополнительного 1 ч. Добавляли воду, смесь оставляли нагреваться до комнатной температуры, а затем экстрагировали EtOAc. Неочищенный продукт очищали колоночной хроматографией на силикагеле с получением названного соединения (13 г, 55%).
1Н ЯМР CDCl3 400 МГц δ 8,03-8,02 (m, 1Н), 7,66-7,63 (m, 1H), 7,45-7,42 (m, 1H).
Стадия 2
Получение 4-бром-7-хлориндола:
К раствору 4-бром-1-хлор-2-нитро-бензола (14 г, 0,059 моль) в 400 мл THF при -40°C по каплям добавляли винилмагния бромид (177 мл 1,0 М раствора в THF, 0,177 моль). Реакционную смесь перемешивали в течение 2 ч при -40°C. Добавляли водный NH4Cl, и смесь экстрагировали эфиром. Органическую фазу сушили над Na2SO4, концентрировали и остаток очищали колоночной хроматографией на силикагеле с получением названного соединения (6 г, 44%).
1Н ЯМР CDCl3 400 МГц δ 8,5 (s, 1Н), 7,31-7,29 (m, 1Н), 7,26-7,22 (m, 1Н),7,07-7,05 (m, 1H), 6,65-6,63 (m, 1Н).
Стадия 3
Получение 7-хлор-4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-1Н-индола:
4-Бром-7-хлориндол (0,6 г, 2,6 ммоль), бис(пинаколято)диборон (0,728 г, 2,86 ммоль), КОАс (0,76 г, 7,8 моль) и Pd(dppf)Cl2 (57 мг, 3 моль. %) суспендировали в DME (15 мл) и облучали при 250 Вт, 130°C в течение 35 мин в одномодовом микроволновом реакторе CEM-Discover. Твердое вещество удаляли фильтрацией, добавляли воду и полученную смесь экстрагировали EtOAc. Органическую фазу сушили над Na2SO4, концентрировали и остаток очищали колоночной хроматографией на силикагеле с получением названного соединения (0,3 г, 39%).
1Н ЯМР CDCl3 400 МГц δ 8,5 (s, 1H), 7,57-7,55 (d, 1Н, J=7,6 Гц), 7,3 (s, 1Н), 7,21-7,19 (d, 1H, J=7,6 Гц), 7,08-7,07 (m, 1Н), 1,396-1,385 (m, 12Н).
Промежуточное соединение В-5
7-Метокси-4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-1Н-индол
Стадия 1
Получение 4-бром-3-метил-2-нитро-фенола:
К раствору 3-метил-2-нитро-фенола (20 г, 0,13 моль) в CHCl3 (20 мл) по каплям добавляли раствор Br2 (6,4 мл) в НОАс (15 мл) при 0°C, и смесь перемешивали при 0°C в течение 3 ч. Добавляли лед, и смесь экстрагировали CHCl3. После сушки над Na2SO4 CHCl3 удаляли с получением 4-бром-3-метил-2-нитро-фенола (30 г), который использовали без дополнительной очистки.
Стадия 2
Получение 1-бром-4-метокси-2-метил-3-нитро-бензола:
К раствору 4-бром-3-метил-2-нитро-фенола (30 г, 0,13 моль) в ацетоне (200 мл) добавляли К2СО3 (39 г, 0,28 моль) и йодметан (17,6 мл, 0,28 моль). Смесь перемешивали в течение 18 ч, а затем ее концентрировали, разбавляли водой и экстрагировали CH2Cl2. Органическую фазу сушили над Na2SO4, фильтровали и концентрировали с получением 1-бром-4-метокси-2-метил-3-нитро-бензола (31 г, 96%), который использовали без дополнительной очистки.
Стадия 3
Получение 4-бром-7-метоксииндола:
К раствору 1-бром-4-метокси-2-метил-3-нитро-бензола (31 г, 0,126 моль) в DMF (150 мл) добавляли диметилформамида диметилацеталь (27 мл) и пирролидин (10,5 мл, 0,127 моль). Смесь нагревали при 90°C в течение 18 ч и охлаждали до к. т. Смесь разбавляли водой и экстрагировали CH2Cl2. Органическую фазу сушили над Na2SO4, фильтровали и концентрировали. Остаток растворяли в НОАс (50 мл) и по каплям добавляли к раствору Fe (20,5 г, 0,37 моль) в кипящем НОАс (150 мл). Смесь кипятили с обратным холодильником в течение 1 ч и охлаждали до к. т., добавляли воду и смесь нейтрализовали Na2CO3 и экстрагировали CH2Cl2. Органическую фазу сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали преп-ТСХ (РЕ/EtOAc=5/1) с получением названного соединения (13 г, 44%).
1Н ЯМР CDCl3 400 МГц δ: 8,41 (brs, 1Н), 7,18-7,08 (m, 2Н), 6,49-6,43 (m, 2Н), 3,86 (s, 3Н).
Стадия 4
Получение 7-метокси-4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-1Н-индола:
К раствору 4-бром-7-метоксииндола (5 г, 22,2 ммоль) в 1,4-диоксане (170 мл) добавляли бис(пинаколято)диборон (6,2 г, 24,4 ммоль), КОАс (6,5 г, 66,3 ммоль) и Pd(dppf)Cl2 (1,2 г, 1,7 ммоль) и смесь нагревали до температуры дефлегмации в течение 15 ч. После охлаждения смесь концентрировали и остаток очищали препаративной ТСХ (РЕ/EtOAc=20/1) с получением названного соединения (2,6 г, 43%).
1H ЯМР CDCl3 400 МГц δ: 8,38 (brs, 1Н), 7,60 (d, 1Н), 7,21 (d, 1H), 7,01 (d, 1H), 6,66 (d, 1Н), 3,98 (s, 3H), 1,39 (s, 12Н).
Промежуточное соединение В-6
5.7-Диметил-4-(4,4,5,5-тетраметил-[1.3.2] диоксаборолан-2-ил)-1Н-индол
Стадия 1
Получение 1-бром-2,4-диметил-5-нитро-бензола:
1-Бром-2,4-диметил-бензол (9 г, 48,6 ммоль) добавляли в HNO3 (100 мл, 60%) при к. т. Смесь перемешивали всю ночь при к. т. Смесь выливали в ледяную воду и экстрагировали EtOAc. Органическую фазу затем сушили над Na2SO4 и концентрировали с получением 1-бром-2,4-диметил-5-нитро-бензола (6,5 г), который использовали в последующей стадии без очистки.
Получение 4-бром-5,7-диметилиндола:
К раствору 1-бром-2,4-диметил-5-нитро-бензола (7,5 г, 32,6 ммоль) в THF (100 мл) при -78°C по каплям добавляли винилмагния бромид (110 мл 1,0 М раствора в THF, 1,1 моль). Реакционную смесь оставляли медленно нагреваться до -40°C, затем перемешивали в течение 4 ч. Добавляли воду, и реакционную смесь оставляли медленно нагреваться до к. т. и экстрагировали EtOAc. Органическую фазу затем сушили над Na2SO4 и концентрировали с получением 4-бром-5,7-диметилиндола (2,3 г), который использовали в последующей стадии без очистки.
Получение 5,7-диметил-4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-1Н-индола:
4-Бром-5,7-диметилиндол (2,3 г неочищенного, 7 ммоль), бис(пинаколято)диборон (2,54 г, 10 ммоль), КОАс (2 г, 20 ммоль) и Pd(dppf)Cl2 (150 мг, 3 моль. %) суспендировали в сухом DME (30 мл) в пяти 50 мл стеклянных пробирках, которые плотно запаивали алюминиевым/тефлоновым зажимом. Образцы облучали при 250 Вт, 130°C в течение 50 мин в микроволновом реакторе. После завершения реакций смеси объединяли, разбавляли водой и экстрагировали EtOAc. Органическую фазу сушили над Na2SO4, и растворитель удаляли выпариванием. Препаративной ВЭЖХ получали названное соединение (0,37 г, 15%).
1Н ЯМР: (400 МГц, CDCl3): δ 7,97 (s, 1Н), 7,19 (s, 1H), 7,00 (s, 1Н), 6,85 (s, 1H), 2,61 (s, 3Н), 2,46 (s, 3Н), 1,40 (s, 12Н)
Промежуточное соединение В-7
7-Фтор-5-метил-4-(4,4,5,5-тетраметил-[1.3.2]диоксаборолан-2-ил)-1Н-индол
Стадия 1
Получение 1-бром-4-фтор-2-метил-5-нитро-бензола:
К раствору 1-бром-4-фтор-2-метил-бензола (20 г, 0,11 моль) в 160 мл H2SO4 добавляли одну часть KNO3 (116 г, 0,11 моль) при 0°C. Смесь оставляли нагреваться до к. т. и перемешивали всю ночь. Смесь выливали в ледяную воду, экстрагировали EtOAc, сушили над Na2SO4 и концентрировали с получением 1-бром-4-фтор-2-метил-5-нитробензола (20 г), который использовали в последующей реакции без очистки.
Стадия 2
Получение 4-бром-7-фтор-5-метилиндола:
К раствору 1-бром-4-фтор-2-метил-5-нитро-бензола (20 г, 0,086 моль) в THF (250 мл) при -78°C по каплям добавляли винилмагния бромид (300 мл, 0,3 моль), затем смесь перемешивали при -78°C в течение 2 ч. Добавляли воду, и реакционную смесь медленно оставляли нагреваться до к. т. Смесь экстрагировали EtOAc, сушили над Na2SO4 и концентрировали. Неочищенный продукт очищали колоночной хроматографией на силикагеле с получением 4-бром-7-фтор-5-метилиндола (3,2 г, 16,4%).
Стадия 3
Получение 7-фтор-5-метил-4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-1Н-индола:
4-Бром-7-фтор-5-метилиндол (3,2 г, 0,014 моль), бис(пинаколято)диборон (5,08 г, 0,02 моль), Pd(dppf)Cl2 (0,1 г) и КОАс (4,1 г, 0,042 моль) суспендировали в сухом DME (60 мл) в десяти 50 мл стеклянных пробирках, которые плотно запаивали алюминиевым/тефлоновым зажимом. Образцы облучали при 250 Вт в течение 2 ч при 135°C в микроволновом реакторе. Образцы охлаждали, объединяли, разбавляли водой и экстрагировали EtOAc. Органическую фазу сушили над Na2SO4, и растворитель удаляли выпариванием. Препаративной ВЭЖХ получали названное соединение (0,43 г, 11%).
1Н ЯМР (MDOH 400 МГц)
δ: 7,22-7,21 (d, 1H), 6,87-6,85 (m, 1Н), 6,68-6,65 (d, 1Н), 2,56 (s, 3Н), 1,39 (s, 12Н).
Промежуточное соединение В-8
3-Метил-4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-1Н-индол
Смесь 3-метил-4-броминдола (0,79 г, 3,8 ммоль), бис(пинаколято)диборона (1,1 г, 4,5 ммоль), PdCl2(dppf)2 (83 мг, 0,11 ммоль) и КОАс (1,1 г, 11,3 ммоль) в DMF (10 мл) продували в атмосфере N2 в течение 10 мин, а затем нагревали при 80°C всю ночь. Реакционную смесь разбавляли водой. Водный слой экстрагировали EtOAc и промывали солевым раствором, и сушили над безводным Na2SO4. Неочищенный продукт очищали колоночной хроматографией на силикагеле с получением названного соединения (0,7 г, 72%).
1Н ЯМР (400 МГц, CDCl3): δ 7,93 (brs, 1Н), 7,57 (d, 1Н, J=6,8 Гц), 7,42 (d, 1H, J=8 Гц), 7,17 (t, 1Н, J=7,2 Гц), 7,01 (s, 1H), 2,48 (s, 3Н), 1,40 (s, 12Н).
Промежуточное соединение В-9
5-Этил-4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-1Н-индол
Стадия 1
Получение 6-этил-2-метил-3-нитро-фениламина
2-Этил-6-метил-фениламин (21 г, 156 ммоль) растворяли в ледяной бане при 0°C в 175 мл концентрированной H2SO4. Добавляли 9 мл (171 ммоль, 1,1 экв.) концентрированной HNO3, и реакционную смесь перемешивали в течение 1 ч при 0°C. Реакционную смесь выливали в 1 л льда, и полученный раствор экстрагировали 5 раз EtOAc. Объединенные органические слои сушили над Na2SO4, и растворитель удаляли при пониженном давлении с получением 19 г (выход 68%) смеси 6-этил-2-метил-3-нитро-фениламина и побочного продукта 6-метил-2-этил-3-нитро-фениламина. Смесь использовали в следующей реакции без дополнительной очистки.
Стадия 2
Получение 2-бром-1 -этил-3-метил-4-нитро-бензола
NaNO2 (7,7 г, 110 ммоль) в 15 мл воды медленно добавляли к перемешиваемому раствору продуктов стадии 1 (19 г, 106 ммоль) в 48% водном HBr (35 мл) при 0°C. По каплям добавляли CuBr (1,4 г, 10 ммоль) в 48% водном HBr (10 мл). После перемешивания в течение 15 мин смесь нагревали при 90°C в течение 2 ч. После охлаждения смесь разбавляли водой и экстрагировали 5 раз EtOAc. Объединенные органические фазы концентрировали и очищали колоночной хроматографией на силикагеле (РЕ/EtOAc=50:1) с получением смеси 2-бром-1-этил-3-метила-нитробензола и 2-бром-3-этил-1-метил-4-нитро-бензола (23 г, выход 95%).
Стадия 3
Получение 1-[2-(2-бром-3-этил-6-нитро-фенил)-винил]-пирролидина
Продукт стадии 2 (13 г, 53 ммоль) и пирролидин (2,3 мл) в DMF/DMA (150 мл) нагревали с кипячением с обратным холодильником всю ночь. Смесь охлаждали, а затем концентрировали с получением неочищенного названного соединения, которое использовали сразу на следующей стадии.
Стадия 4
Получение 4-бром-5-этил-1Н-индола
Одну часть раствора неочищенного 1-[2-(2-бром-3-этил-6-нитро-фенил)-винил]-пирролидина стадии 3 в 80 мл АсОН добавляли к нагревающейся с обратным холодильником суспензии железа (13 г) в 50 мл АсОН. Полученную смесь кипятили с обратным холодильником в течение 2 ч. После охлаждения смесь разбавляли водой и нейтрализовали Na2СО3, и экстрагировали 3 раза EtOAc. Объединенные органические фазы концентрировали и очищали колоночной хроматографией на силикагеле и препаративной ВЭЖХ с получением названного соединения (1,5 г, 12% из 6-этил-2-метил-3-нитро-фениламина).
Стадия 5
Получение 5-этил-4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-1Н-индола
4-Бром-5-этил-1Н-индол (1,0 г, 4,48 ммоль), бис(пинаколято)диборон (2,4 г, 9 ммоль), КОАс (1,5 г, 15 ммоль) и Pd(dppf)Cl2 (210 мг, 6 моль. %) суспендировали в сухом диоксане (50 мл) и нагревали при 80°C всю ночь. Смесь охлаждали и фильтровали, и фильтрат концентрировали и очищали препаративной ВЭЖХ с получением названного соединения (300 мг, 25%).
1H ЯМР (CDCl3, 400 МГц): δ 7,90 (brs, 1H), 7,31-7,25 (m, 1Н), 7,15-7,08 (m, 1H), 6,98 (d, 1H, J=8,4 Гц), 6,90-9,85 (m, 1Н), 2,92 (q, 2Н, J=7,6 Гц), 1,34 (s, 12Н), 1,16 (t, 3Н, J=7,6 Гц).
Промежуточное соединение В-10
7-Этил-4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-1Н-индол
Стадия 1
Получение 4-бром-1-этил-2-нитро-бензола
К раствору 1-бром-4-этилбензола (7 г, 37,8 ммоль) в 9 мл концентрированной H2SO4 при -20°C добавляли смесь H2SO4 (3,74 г, 41,9 ммоль) и HNO3 (2,64 г, 41,9 ммоль). Смесь перемешивали при -20°C в течение 2 ч. Добавляли воду, и смесь экстрагировали EtOAc. Органическую фазу промывали водным NaHCO3 и солевым раствором, сушили над Na2SO4 и концентрировали. Остаток очищали колоночной хроматографией на силикагеле с получением названного соединения (3 г, 34%).
1Н ЯМР (CDCl3 400 МГц)
δ: 8,04-8,035 (d, 1H), 7,68-7,65 (m, 1H), 7,29-7,27 (d, 1H), 2,92-2,87 (m, 2Н), 1,31-1,28 (t, 3Н).
Стадия 2
Получение 4-бром-7-этил-1Н-индола
К раствору 4-бром-1-этил-2-нитро-бензола (8,7 г, 0,037 моль) в THF при -40°C по каплям добавляли винилмагния бромид (132 мл, 0,132 моль), и смесь перемешивали при -40°C дополнительные 2 ч. Реакционную смесь разбавляли водой, экстрагировали EtOAc, сушили над Na2SO4 и концентрировали. Неочищенный продукт очищали колоночной хроматографией на силикагеле с получением названного соединения (1,35 г, 16%).
Стадия 3
Получение 7-этил-4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-1Н-индола
4-Бром-7-этил-1Н-индол (1,35 г, 6,02 ммоль), бис(пинаколято)диборон (3,05 г, 12 ммоль), КОАс (1,8 г, 18 ммоль) и Pd(dppf)Cl2 (128 мг) суспендировали в сухом DME (100 мл) и нагревали при 135°C всю ночь. Смесь охлаждали и фильтровали, и фильтрат концентрировали и очищали препаративной ВЭЖХ с получением названного соединения (300 мг, 25%).
1Н ЯМР (CDCl3 400 МГц)
δ: 8,14 (s, 1Н), 7,61-7,60 (d, 1H), 7,28-7,25 (d, 1Н), 7,08-7,05 (m, 2Н), 2,92-2,86 (m, 2Н), 1,38-1,35 (m, 15Н).
Промежуточное соединение В-11
Коммерчески доступно.
Промежуточное соединение В-12
7-Трифторметил-4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-1H-индол
Стадия 1
4-Бром-7-трифторметилиндол
К раствору 4-бром-2-нитро-1-трифторметил-бензола (11,4 г, 0,04 моль) в 200 мл THF добавляли винилмагния бромид (160 мл, 0,16 моль) при -78°C, и смесь перемешивали в течение 1,5 ч перед гашением насыщенным раствором NH4Cl, и экстрагировали EtOAc. Органическую фазу сушили над Na2SO4, и растворитель удаляли in vacuo. Неочищенный продукт очищали колоночной хроматографией на силикагеле с получением названного соединения (2 г, 25%). 1Н ЯМР (CDCl3 400 МГц), δ 8,6 (br, 1H), 7,28-7,38 (m, 3Н), 6,70 (m, 1H).
Стадия 2
7-Трифторметил-4-(4,4,5,5-тетраметил-[1.3.2]диоксаборолан-2-ил)-1Н-индол
Смесь 4-бром-7-трифторметилиндола (2 г, 7,6 ммоль), бис(пинаколято)диборона (2,5 г, 9 ммоль), КОАс (1 г, 0,01 моль) и Pd(dppf)Cl2 (0,2 г) в 30 мл DMSO нагревали при 90°C в N2 всю ночь. Остаток разделяли между водой и EtOAc. Органическую фазу сушили над Na2SO4, и растворитель удаляли in vacuo с получением неочищенного продукта, который очищали колоночной хроматографией на силикагеле с получением названного соединения (1 г, 41%). 1Н ЯМР (CDCl3 400 МГц), δ 8,6 (br, 1H), 7,71 (d, J=7,6 Гц, 1H), 7,47 (d, J=7,6 Гц, 1H), 7,37 (m, 1Н), 7,17 (m, 1Н), 1,42 (s, 12Н)
Получение соединений формулы (1)
ПРИМЕР 1
Амид 2-(5-фтор-1Н-индол-4-ил)-5-[4-(3,3-диметил-пиперазин-1-ил)-фенил]-оксазол-4-карбоновой кислоты

Стадия 1
К смеси промежуточного соединения (10А) (200 мг, 0,3 ммоль) в 4 мл диоксана, 1 мл ацетонитрила и 1 мл воды добавляли промежуточное соединение В1 (161 мг, 0,55 ммоль), К2СО3 (80 мг, 0,6 ммоль) и Pd(dppf)Cl2 (36,5 мг, 0,05 ммоль). Смесь нагревали при 90°C в N2 в течение 16 часов. Растворитель удаляли in vacuo, и остаток очищали препаративной ТСХ (РЕ:EtOAc=1:1) с получением желаемого продукта 13 (120 мг, 60%).
Стадия 2
К раствору соединения 13 (120 мг, 0,18 ммоль) в 3 мл CH2Cl2 добавляли 1 мл трифлатной кислоты, и смесь перемешивали в течение 4 ч при комнатной температуре. Смесь затем медленно добавляли к насыщенному водному NaHCO3 с перемешиванием, дважды экстрагировали EtOAc. Объединенные органически фазы концентрировали в вакууме. Остаток очищали препаративной ВЭЖХ с получением названного соединения (18 мг, 23%) в виде беловатого твердого вещества.
(18 мг, выход от 10: 14%, М+1: 434) 1Н ЯМР (400 МГц DMSO-d6) δ 11,55 (s, 1H), 8,19 (m, 2H), 7,51-7,67 (m, 4H), 7,32 (s, 2H), 7,11 (t, 1H), 6,98 (d, J=8 Гц, 2H), 3,15 (m, 2H), 3,01 (s, 2H), 2,83 (m, 2H), 1,08 (s, 6H).
ПРИМЕР 2
Амид 2-(5-метил-1Н-индол-4-ил)-5-[4-(3,3-диметил-пиперазин-1-ил)-фенил]-оксазол-4-карбоновой кислоты

Стадия 1
К раствору промежуточного соединения (10А) (1,9 г, 2,8 ммоль) в 40 мл диоксана, 10 мл ацетонитрила и 10 мл воды добавляли сложный эфир бороновой кислоты промежуточного соединения В2 (1 г, 3,9 ммоль), К2СО3 (0,7 г, 5,1 ммоль) и Pd(dppf)Cl2 (0,5 г, 0,68 ммоль). Смесь нагревали при 90°C в течение 6 ч в N2. Растворитель удаляли, и остаток очищали колоночной хроматографией на силикагеле с получением соединения 11 (1,32 г, выход: 70%).
Стадия 2
К раствору соединения 11 (1,32 г, 0,2 ммоль) в 20 мл CH2Cl2 добавляли 5 мл трифлатной кислоты. Смесь перемешивали при 25°C в течение 6 ч. Смесь медленно добавляли к водн. NaHCO3 при перемешивании, и дважды экстрагировали EtOAc. Объединенные органические фазы концентрировали in vacuo. Неочищенный продукт очищали препаративной ВЭЖХ с применением системы, описанной ниже, с получением названного соединения (360 мг, 42%) в виде желтоватого твердого вещества.
(22 мг, выход от 10: 17%, М+1: 430) 1Н ЯМР: (400 МГц DMSO-d6) δ 11,3 (s, 1Н), 8,22 (d, J=8,8 Гц, 2H), 7,46-7,60 (m, 4H), 7,1 (d, J=8,0 Гц, 1H), 7,00-7,04 (m, 3H), 3,15 (m, 2H), 3,01 (s, 2H), 2,85 (m, 2H), 2,74 (s, 3H), 1,09 (s, 6H).
Способ разделения преп-ВЭЖХ для SRUM-3:
Прибор: Shimadzu LC-8A для препаративной ВЭЖХ
Колонка: Gemini 250*50 мм в.д. 10u
Подвижная фаза: А: Н2O (0,04% NH3H2O), В: CH3CN
Градиент: 25%-50% (В фаза) за 23 минуты
Скорость потока: 80 мл/мин
Длина волны: 220 и 254 нм
ПРИМЕР 3
Амид 2-(7-фтор-1Н-индол-4-ил)-5-[4-(3,3-диметил-пиперазин-1-ил)-фенил1-оксазол-4-карбоновой кислоты

Получали способом согласно примеру 2 с применением промежуточного соединения В3 сложного эфира бороновой кислоты.
(20 мг, выход от 10: 15%, М+1: 434) 1H ЯМР: (400 МГц DMSO-d6) δ 11,90 (s, 1Н), 8,19 (m, 2H), 7,70-7,78 (m, 2H), 7,57 (s, 1H), 7,45 (s, 1H), 7,37 (s, 1H), 7,06 (q, 1H), 6,96 (d, J=8 Гц, 2H), 3,15 (t, 2H), 3,01 (s, 2H), 2,83 (t, 2H), 1,07 (s, 6H).
ПРИМЕР 4
Амид 2-(7-хлор-1Н-индол-4-ил)-5-[4-(3.3-диметил-пиперазин-1-ил]-фенил]-оксазол-4-карбоновой кислоты
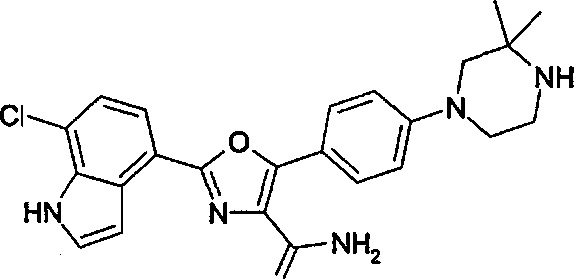
Получали способом согласно примеру 2 с применением промежуточного соединения В4 сложного эфира бороновой кислоты.
(24 мг, выход от 10: 18%, М+1: 450) 1Н ЯМР: (400 МГц DMSO-d6) δ 11,85 (s, 1Н), 8,24 (d, J=9,2 Гц, 2H), 7,82 (d, J=8,0 Гц, 1H), 7,77 (s, 1H), 7,62 (m, 1H), 7,51 (s, 1H), 7,44 (m, 1H), 7,34 (m, 1H) 7,00 (d, J=9,2 Гц, 2H), 3,16 (t, 2H), 3,01 (s, 2H), 2,85 (t, 2H), 1,08 (s, 6H).
ПРИМЕР 5
Амид 2-(7-метокси-1Н-индол-4-ил)-5-[4-(3,3-диметил-пиперазин-1-ил)-фенил]-оксазол-4-карбоновой кислоты
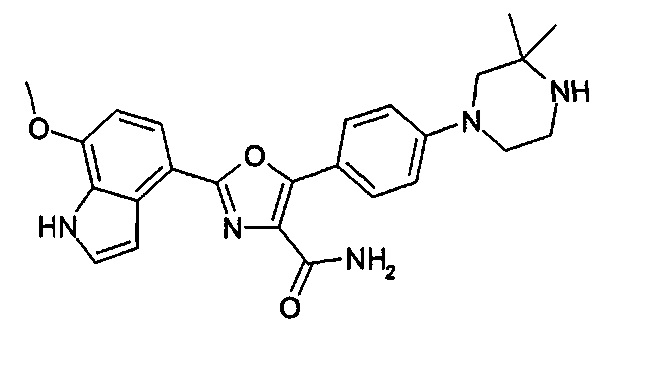
Получали способом согласно примеру 2 с применением промежуточного соединения B5 сложного эфира бороновой кислоты.
(19 мг, выход от 10: 14%, М+1: 446) 1Н ЯМР: (400 МГц DMSO-d6) δ 11,58 (s, 1H), 8,23 (d, J=9,2 Гц, 2H), 7,79 (d, J=8,4 Гц, 2H), 7,68 (s, 1H), 7,47 (s, 1H), 7,42 (t, 1H), 7,28 (t, 1H), 7,00 (d, J=9,2 Гц, 2H), 6,84 (d, J=8,0 Гц, 2H), 3,17-3.14 (m, 2H), 3,01 (s, 2H), 2,87 (t, 2H), 2,65 (s, 3H), 1,09 (s, 6H).
ПРИМЕР 6
Амид 2-(5,7-диметил-1Н-индол-4-ил)-5-[4-(3,3-диметил-пиперазин-1-ил)-фенил]-оксазол-4-карбоновой кислоты

Получали способом согласно примеру 2 с применением промежуточного соединения B6 сложного эфира бороновой кислоты.
(33 мг, выход от 10: 25%, М+1: 444) 1Н ЯМР: (400 МГц DMSO-d6) δ 11,3 (s, 1Н), 8,21 (d, J=8,8 Гц, 2H), 7,44-7,56 (m, 3Н), 6,91-7,05 (m, 4H), 3,15 (m, 2H), 3,01 (s, 2H), 2,85 (m, 2H), 2,70 (s, 3H), 1,08 (s, 6H).
ПРИМЕР 7
Амид 2-(5-метил-7-фтор-1Н-индол-4-ил)-5-[4-(3,3-диметил-пиперазин-1-ил)-фенил]-оксазол-4-карбоновой кислоты
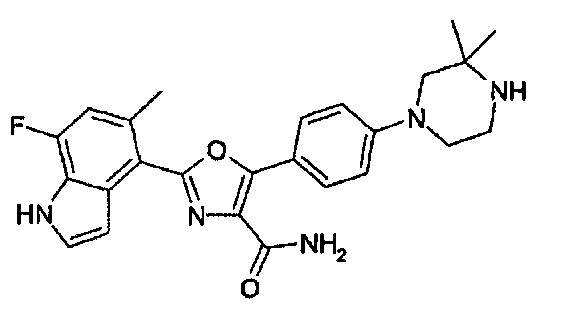
Получали способом согласно примеру 2 с применением промежуточного соединения B7 сложного эфира бороновой кислоты.
(16 мг, выход от 10: 12%, М+1: 448) 1Н ЯМР: (400 МГц DMSO-d6) δ 8,21 (d, J=8,8 Гц, 2Н), 7,46-7,60 (m, 3Н), 6,9-7,1 (m, 3Н), 3,15 (m, 2Н), 3,01 (s, 2Н), 2,85 (m, 2Н), 2,72 (s, 3Н), 1,09 (s, 6Н).
ПРИМЕР 8
Амид 2-(3-метил-1Н-индол-4-ил)-5-[4-(3,3-диметил-пиперазин-1-ил)-фенил]-оксазол-4-карбоновой кислоты
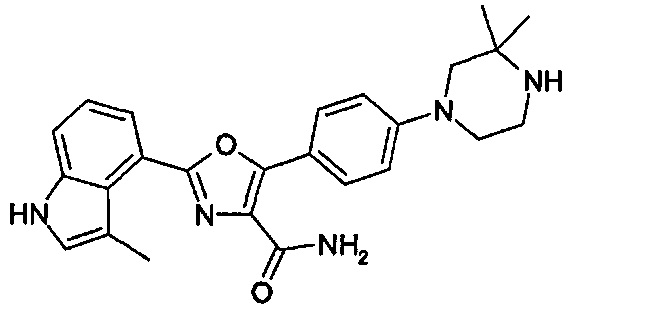
Получали способом согласно примеру 2 с применением промежуточного соединения B8 сложного эфира бороновой кислоты.
(9,4 мг, выход от 10: 7%, М+1: 430) 1Н ЯМР: (400 МГц DMSO-d6) δ 11,17 (br, 1Н), 8,15 (d, J=8,8 Гц, 2Н), 7,46-7,60 (m, 4Н), 7,29 (s, 1Н), 7,16 (t, 1Н), 6,97 (d, J=8,8 Гц, 2Н), 3,15 (m, 2Н), 2,99 (s, 2Н), 2,85 (m, 2Н), 2,25 (s, 3Н), 1,08 (s, 6Н).
ПРИМЕР 9
Амид 2-(5-этил-1Н-индол-4-ил)-5-[4-(3,3-диметил-пиперазин-1-ил)-фенил]-оксазол-4-карбоновой кислоты
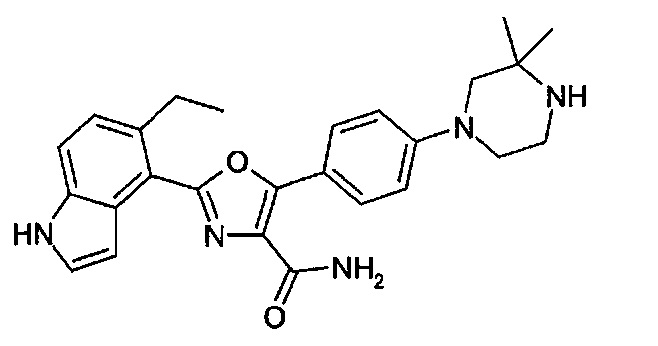
Получали способом согласно примеру 2 с применением промежуточного соединения B9 сложного эфира бороновой кислоты.
(28 мг, выход от 10: 21%, М+1: 444) 1Н ЯМР: (400 МГц DMSO-d6) δ 11,3 (br, 1H), 8,19 (d, J=8,6 Гц, 2H), 7,46-7,51 (m, 4H), 6,92-7,12 (m, 4H), 3,15 (m, 2H), 3,08 (q, 2H), 3,01 (s, 2H), 2,85 (m, 2H), 1,23 (t, 3Н), 1,09 (s, 6H).
ПРИМЕР 10
Амид 2-(7-этил-1Н-индол-4-ил)-5-[4-(3,3-диметил-пиперазин-1-ил)-фенил1-оксазол-4-карбоновой кислоты

Получали способом согласно примеру 2 с применением промежуточного соединения В10 сложного эфира бороновой кислоты.
(32 мг, выход от 10: 24%, М+1: 443) 1Н ЯМР: (400 МГц DMSO-d6) δ 11,4 (br, 1Н), 8,23 (d, J=8,6 Гц, 2H), 6,99-7,79 (m, 8H), 3,15 (m, 2H), 3,01 (s, 2H), 2,94 (q, 2H), 2,85 (m, 2H), 1,29 (t, 3H), 1,09 (s, 6H).
ПРИМЕР 11
Амид 2-(1Н-индол-4-ил)-5-[4-(3,3-диметил-пиперазин-1-ил)-фенил]-оксазол-4-карбоновой кислоты
Получали способом согласно примеру 2 с применением промежуточного соединения B11 сложного эфира бороновой кислоты.
(466 мг, 38% выход от промежуточного соединения 10А, М+1: 416) 1Н ЯМР: (400 МГц DMSO-d6) δ 11,48 (s, 1Н), 8,27 (d, 2H), 7,85 (d, 1H), 7,74 (s, 1H), 7,61-7,53 (m, 3H), 7,33 (s, 1H), 7,26 (t, 1H), 7,02 (d, 2H), 3,19-3,17 (m, 2H), 3,03 (s, 2H), 2,89-2,86 (m, 2H), 1,11 (s, 6H).
ПРИМЕР 12
Амид 2-(7-трифторметил-1Н-индол-4-ил)-5-[4-(3,3-диметил-пиперазин-1-ил)-фенил]-оксазол-4-карбоновой кислоты

Названное соединение получали последовательностью реакций, показанной в общей схеме 2 выше.
Стадия 1
Этил 5-(4-(4-(трет-бутоксикарбонил)-3,3-диметилпиперазин-1-ил)фенил)-2-йодоксазол-4-карбоксилат
Названное соединение получали из промежуточного соединения этилового сложного эфира соединения (15) (см. стадию 2 в получении промежуточного соединения (10А) выше) с применением способа литирования/йодирования, описанного на стадии 4 того же получения.
Стадия 2
Трет-бутиловый сложный эфир 4-{4-[4-этоксикарбонил-2-(7-трифторметил-1Н-индол-4-ил)-оксазол-5-ил]-фенил}-2,2-диметил-пиперазин-1-карбоновой кислоты
Этил 5-(4-(4-(трет-бутоксикарбонил)-3,3-диметилпиперазин-1-ил)фенил)-2-йодоксазол-4-карбоксилат реагировал со промежуточным соединением В-12 сложного эфира бороновой кислоты при условиях реакции сочетания Сузуки, аналогичных описанным в примере 2 выше, с получением названного соединения.
Стадия 3
Трет-бутиловый сложный эфир 4-{4-[4-карбокси-2-(7-трифторметил-1Н-индол-4-ил)-оксазол-5-ил]-фенил}-2,2-диметил-пиперазин-1-карбоновой кислоты
Трет-бутиловый сложный эфир 4-{4-[4-этоксикарбонил-2-(7-трифторметил-1Н-индол-4-ил)-оксазол-5-ил]-фенил}-2,2-диметил-пиперазин-1-карбоновой кислоты (0,2 г, 0,3 ммоль) и NaOH (0,024 г, 0,6 ммоль) в 5 ил метанола, воды и THF перемешивали при 50°C всю ночь. Органический растворитель удаляли in vacuo, оставшийся раствор доводили до pH 5, и полученное названное соединение собирали в виде белого твердого вещества фильтрацией и сушили при пониженном давлении.
Стадия 4
Трет-бутиловый сложный эфир 4-{4-[4-карбамоил-2-(7-трифторметил-1Н-индол-4-ил)-оксазол-5-ил)-фенил}-2,2-диметил-пиперазин-1-карбоновой кислоты
Смесь трет-бутилового сложного эфира 4-{4-[4-карбокси-2-(7-трифторметил-1Н-индол-4-ил)-оксазол-5-ил]-фенил}-2,2-диметил-пиперазин-1-карбоновой кислоты, EDCl HCl (0,36 г, 0,002 моль) и HOBt (0,3 г,0,002 моль) в 15 мл DMF и 15 мл NH3 в диоксане перемешивали в течение 2 ч. Растворители удаляли in vacuo. Остаток разделяли между водой и EtOAc, органическую фазу сушили над Na2SO4 и растворитель удаляли in vacuo с получением неочищенного продукта названного соединения (0,18 г неочищенного).
Стадия 5
Амид 2-(7-трифторметил-1Н-индол-4-ил)-5-[4-(3,3-диметил-пиперазин-1-ил)-фенил]-оксазол-4-карбоновой кислоты
Трет-бутиловый сложный эфир 4-{4-[4-карбамоил-2-(7-трифторметил-1Н-индол-4-ил)-оксазол-5-ил]-фенил}-2,2-диметил-пиперазин-1-карбоновой кислоты (0,18 г неочищенного) растворяли в смеси DCM и TFA (30 мл 2:1), и раствор перемешивали при к. т. в течение 2 ч, если растворитель удаляли in vacuo. Остаток разделяли между водой и EtOAc. Органическую фазу сушили над Na2SO4, и растворитель удаляли in vacuo с получением неочищенного продукта, который очищали преп-ВЭЖХ с получением названного соединения (60 мг, 41% от этил 5-(4-(4-(трет-бутоксикарбонил)-3,3-диметилпиперазин-1-ил)фенил)-2-йодоксазол-4-карбоксилата).
М+1: 484) 1H ЯМР: (400 МГц DMSO-d6) δ 11,85 (s, 1Н), 8,27-8,25 (d, 2H, J=8 Гц), 7,97-7,95 (d, 1H, J=8 Гц), 7,85 (s, 1H), 7,66-7,56 (m, 3H), 7,02-7,00 (d, 2H J=0,8 Гц), 3,19-3,17 (m, 2H), 3,04 (s, 2H), 2,87-2,86 (m, 2H), 1,09 (s, 6H).
БИОЛОГИЧЕСКАЯ АКТИВНОСТЬ
ПРИМЕР 13
Ингибирующая активность FLT3 киназы и Aurora киназы
Способность соединений по настоящему изобретению ингибировать киназу FLT3 и киназы Aurora А и Aurora В определяли с применением описанных ниже способов.
Анализ киназ проводили в Reaction Biology Corp., Malvern, Pennsylvania, USA, с применением следующей общей процедуры:
1) Получали обозначенный субстрат в свежеприготовленном основном буферном растворе (20 мМ Hepes pH 7,5, 10 мМ MgCl2, 1 мМ EGTA, 0,02% Brij35, 0,02 мг/мл BSА, 0,1 мМ Na3VO4, 2 мМ DTT, 1% DMSO).
2) Доставляли кофакторы (1,5 мМ СаСl2, 16 мкг/мл кальмодулина, 2 мМ MnCl2) в вышеописанный субстратный раствор.
3) Доставляли обозначенные киназы в субстратный раствор и осторожно смешивали.
4) Доставляли тестовое соединение различных концентраций в DMSO в киназную реакционную смесь.
5) Доставляли 33Р-АТР (специфическая активность 0,01 □Ci/□л готовой) в реакционную смесь для начала реакции.
6) Инкубировали реакцию киназы в течение 120 мин при комнатной температуре.
7) Реакционные смеси в виде пятен наносили на Р81 бумагу ионообменного фильтра (ватманская бумага №3698-915).
8) Не связанный фосфат удаляли тщательным промыванием фильтров в 0,75% фосфорной кислоте.
9) 33Р сигнал определяли с применением Typhoon phosphorimagers (GE Healthcare). После вычитания фона, полученного из контрольных реакционных смесей, содержащих неактивный фермент, определяли значения IC50 с применением функции нелинейной регрессии с помощью Prism (программное обеспечение Graphpad).
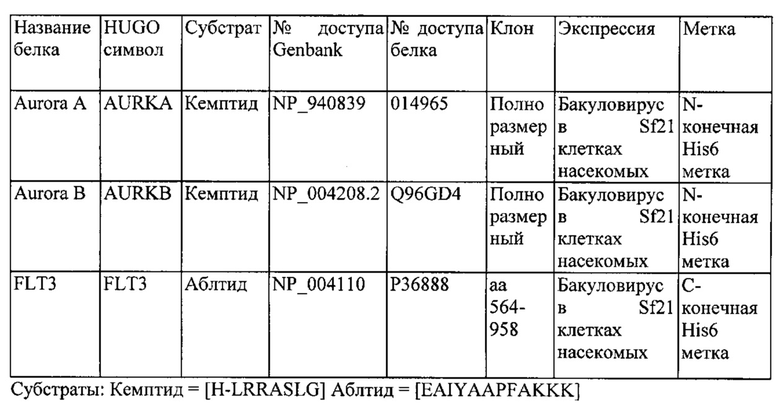
Концентрации тестовых соединений, необходимые для ингибирования 50% ферментативной активности (IC50) каждой из трех киназ, представлены в таблице ниже. Для сравнения также показаны значения IC50 для соединения амид 2-(1Н-индол-4-ил)-5-(4-пиперазин-1-ил-фенил)-оксазол-4-карбоновой кислоты, раскрытого в примере М-12 WO 2008/139161.

ПРИМЕР 14
Антипролиферативная активность
Антипролиферативные активности соединений по настоящему изобретению определяли путем измерения способности соединения ингибировать рост клеточной линии НСТ-116 колоректального рака. Ингибирование клеточного роста измеряли с применением анализа Alamar Blue (Nociari и другие, Journal of Immunological Methods (1998) 213, 157-167). Способ основан на способности жизнеспособных клеток восстанавливать резазурин до его флуоресцентного продукта резофурина. Для каждого пролиферационного анализа клетки высеивали на 96 луночных планшетах и оставляли восстанавливаться в течение 16 часов перед добавлением ингибиторных соединений в течение дополнительных 96 часов. В конце инкубационного периода добавляли 10% (объем/объем) Alamar Blue и инкубировали еще 6 часов перед определением флуоресцентного продукта при возбуждении 535 нм и эмиссии 590 нм. Все клеточные линии получали от ЕСАСС (Европейская коллекция культивируемых клеток).
Активности соединений по настоящему изобретению против клеточной линии НСТ-116 человеческого колоректального рака показаны в таблице ниже. Колонка с заглавием НСТ-116 96 Hrs относится к концентрации соединения, необходимого для понижения пролиферации НСТ-116 клеток на 50% с последующим 96 ч периодом воздействия. Колонка с заглавием [Полиплоидия] относится к самой низкой концентрации соединения, необходимого для получения отдельного полиплоидного фенотипа, как полагалось, вследствие ингибирования Aurora В.
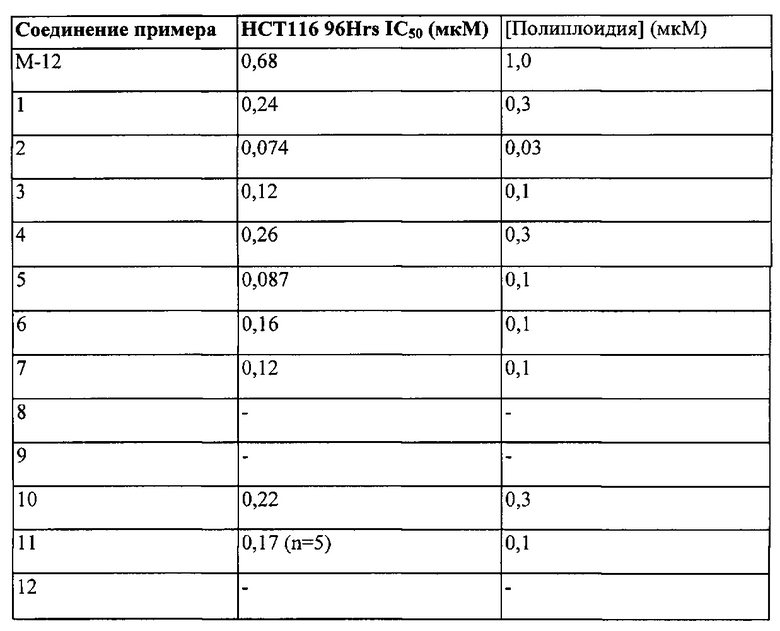
ПРИМЕР 15
Изучения проницаемости с применением клеток Сасо-2
Способность соединения к поглощению и удерживанию клеткой может быть определена проведением изучений проницаемости с применением клеток Сасо-2. Хотя клетки Сасо-2 в основном использовали для предположения биодоступности при пероральном введении соединения, полученные измерения также показывают клеточную способность к проницаемости соединения.
Таким образом, соединения по настоящему изобретению исследовали стандартным анализом Сасо-2, и определяли скорость потока соединений в и из клеток. Исходя из результатов, скорость вытекания рассчитывали как соотношение скорости соединения, выходящего из клетки, к скорости соединения, входящего в клетку.
Результаты представлены в таблице ниже. В таблице колонка с заглавием «Сасо-2 А2В» обозначает скорость, при которой соединение втекает в клетки, а колонка Сасо-2 В2А обозначает скорость, при которой соединение вытекает из клеток. «Соотношение вытекания» представляет собой скорость Сасо-2 В2А: Сасо-2 А2В.
Результаты показывают, что все исследованные соединения обладают более благоприятными соотношениями вытекания, чем соединение примера М-12 WO 2008/139161. В случае соединений примеров 2-11, соотношения вытекания являются более чем в пять раз лучшими, чем соотношение вытекания соединения М-12 известного уровня техники.
Сравнение данных соединения М-12 и соединения примера 11 показывает, что замена группы CH2 в пиперазиновом кольце группой C(CH3)2 существенно улучшает соотношение вытекания.
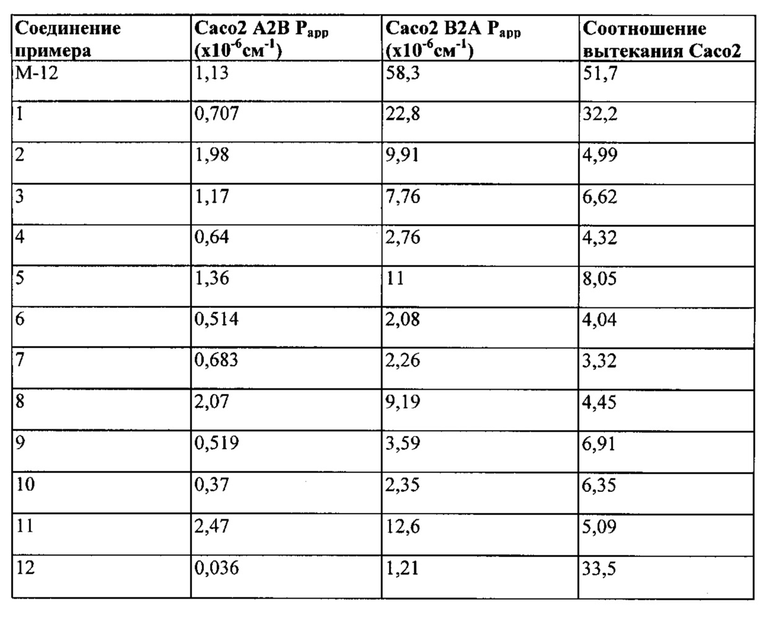
ПРИМЕР 16
Активность относительно человеческих гематологических линий опухолевых клеток
Соединения примеров 2 и 7 исследовали относительно диапазона человеческих гематологических линий клеток с применением анализа CellTiter-Blue® (#G8081, Promega) согласно инструкциям изготовителя. Клетки собирали из культур с экспоненциальной фазой, считали и высеивали в 96 луночных плоскодонных титрационных микропланшетах с плотностью клеток 20000-90000 клеток/лунка. Через 24 ч периода восстановления, чтобы позволить клетками возобновить экспоненциальную фазу роста, добавляли 10 мкл культуральной среды (четыре контроля лунки/планшет) или культуральной среды с тестовым соединением. Соединения использовали в двух экземплярах при десяти концентрациях, и обработку продолжали в течение четырех дней. После обработки клеток добавляли реагент CellTiter-Blue® 10 мкл/лунка. После инкубационного периода до четырех часов измеряли флуоресценцию (FU) с применением многофункционального планшета-ридера EnVision Xcite (возбуждение λ=531 нм, эмиссия λ=615 нм). Соединения исследовали в двух-четырех независимых экспериментах для каждой клеточной линии. Значения IC50 (микромолярные) относительно каждой клеточной линии представлены в таблице ниже.
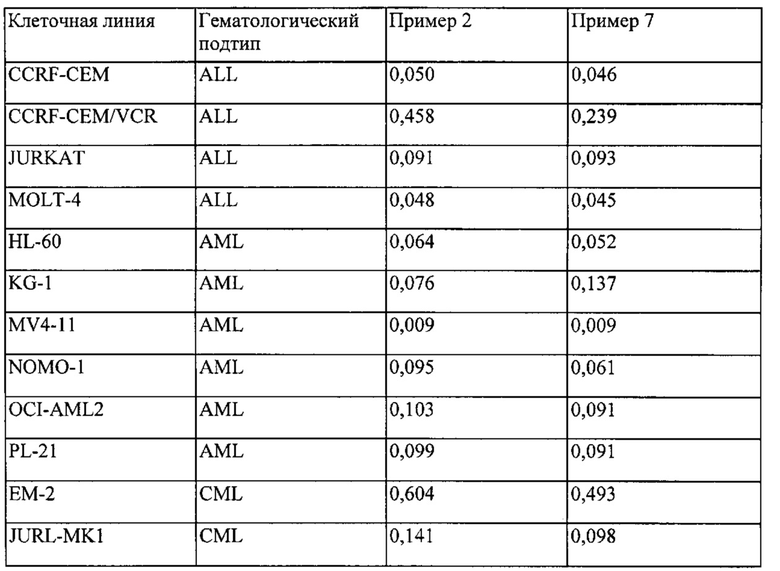
ПРИМЕР 17 Изучения ксенотрансплантата
In vivo антиопухолевая активность соединений по настоящему изобретению исследовали изучением эффекта соединений примеров 2 и 7 на рост опухоли в ксенотрансплантатной модели голой мыши клеточной линии рака MV4-11.
Материалы и способы
2.1. Животные и реагенты
Клеточная линия MV4-11 (АТСС, USA); среда RPMI 1640 (Invitrogen, USA); FBS (Invitrogen, Australia); Balb/c голой мыши (Slac Laboratory Animal Co., Ltd., Shanghai, China): самка, 18-22 г; HP-β-CD (Sigma, USA).
2.2. Процедура
Использовали всего 52 самки голых мышей. Мышей оставляли на 3 дня для периода акклиматизации перед началом эксперимента. В начале изучения в правую боковую поверхность мышей подкожно (s.c.) имплантировали 200 мкл 1×107 MV4-11 опухолевых клеток в 50% Матригеле. Когда опухли достигали среднего объема 100-150 мм3, отбирали 32 мыши из 52 на основе объема опухоли, и произвольно разделяли на 4 группы перед дозированием. Каждая группа для обработки составляла 8 животных с опухолью (n=8/группа).
Мышей с опухолью обрабатывали соединением примера 2, соединением примера 7 или носителем дважды в день в течение 5 рабочих дней, 2 выходных, на протяжении 4 циклов. Группу сравнения мышей обрабатывали цитарабином один раз в день в течение 5 рабочих дней, 2 выходных, на протяжении 4 циклов.
Мышей взвешивали при каждом дозировании, и размеры опухолей регистрировали дважды каждую неделю. За мышами тщательно наблюдали на наличие любых явных нежелательных признаков, связанных с обработкой побочных эффектов, и при наблюдении это регистрировали.
Размер опухоли измеряли дважды каждую неделю в двух размерах с применением кронциркулей, и объем выражали в мм с применением формулы: V=0,5а×b2, где а и b были длинным и коротким диаметрами опухоли, соответственно.
Тестовые составы готовили раз в неделю путем взвешивания соответствующего количества тестового соединения в сосуде, добавлением подходящего объема 20% НР-β-CD для достижения конечной концентрации 2 мг/мл для примера 2 и 3 мг/мл для примера 7 (свободное основание), доводя значение pH до pH 5, встряхиванием сосуда в течение 5 мин, а затем помещением сосуда в баню ультразвукового аппарата для обеспечения прозрачности тестового состава.
Эксперимент подытожен в таблице ниже.
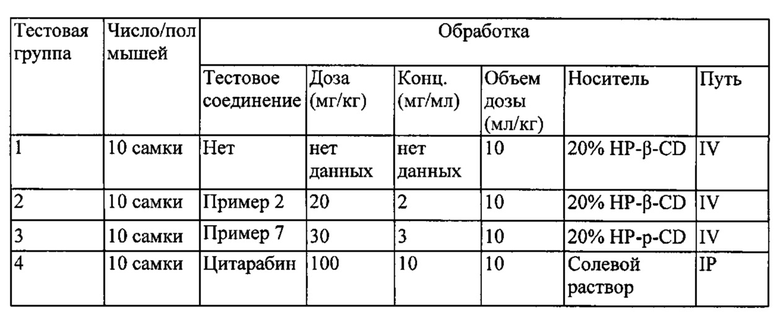

2.3. Результат эксперимента:
Мышей эвтаназировали воздействием CO2 после 4-циклической дозы. Объем опухоли, массу разрезанных опухолей и массу тела мышей измеряли и регистрировали. Ингибиторную скорость (IR) опухоли рассчитывали по формуле IR=(Wv-WT)/Wv×100%.
Различия размеров опухоли между группам анализировали для значения с использованием непарного двустороннего критерия Стьюдента. Полагали, что P<0,05 было статистически значимым.
3. Результаты и пояснения
3.1. Объемы опухоли голых мышей с MV4-11
На фигурах 1-3 показано ингибирование роста соединений примеров 2 и 7 относительно MV4-11 ксенотрансплантатных опухолей у голых мышей. Существенное различие в объеме опухоли наблюдали между опухолями у мышей, получающих тестовые соединения, и у мышей, получающих только носитель, начиная от 4 дня (первое измерение) после первого введения соединений и до конца. Существенное различие в объеме опухоли также наблюдали между опухолями у мышей, получающих тестовые соединения (примеры 2 и 7), и группой с обработкой цитарабином, начиная от 11 дня после первого введения и с дальнейшим продолжением. На фигуре 1 также показано, что и соединение примера 2, и примера 7, полностью ингибировало рост опухоли, тогда как цитарабин замедлял, но полностью не блокировал рост опухоли.
3.2. Массы опухоли голых мышей с MV4-11
Эффект ингибирования опухоли соединений примеров 2 и 7 относительно роста опухоли MV4-11 ксенотрансплантата дополнительно подтверждали существенным отличием масс опухоли между группами обработки и носителя (фигура 4), полученным в конце умерщвлением голых мышей с опухолями.
3.3. Изменение массы тела (%) голых мышей с MV4-11
На фигуре 5 показано изменение массы тела (%) в течение всего периода изучения. Все обработанные мыши потеряли приблизительно 5% веса своего тела в течение первых нескольких дней, а затем восстановили, это означает, что тестовые соединения были хорошо переносимыми при текущих дозировках и путях дозировки голых мышей.
3.4. Расчет ингибиторной скорости (IR) опухоли
Группа соединения примера 2-20 мг/кг: IR=98,13%
Группа соединения примера 7-30 мг/кг: IR=98,24%
Группа цитарабина - 100 мг/кг: IR=66,72%
4. Заключения
При этом изучении исследовали эффекты соединений примера 2 и примера 7 на рост опухоли в ксенотрансплантатных моделях голой мыши от клеточной линии рака MV4-11. Массы тела голых мышей с MV4-11 измеряли как индекс для отражения токсичности in vivo. Различия в объемах опухоли между группами обработки тестовым соединением (примеры 2 и 7) и контрольной группой носителя, а также группой сравнения цитарабина были существенными. При этом изучении, и соединение примера 2, и примера 7 полностью ингибировали рост опухоли, тогда как цитарабин только замедлял, но полностью не блокировал рост опухоли. Значительные различия в массе опухоли между группами обработки и носителя дополнительно подтверждали противоопухолевый эффект. Значения IR для группы соединения примера 2, группы соединения примера 7 и группы цитарабина составляли 98,13%, 98,24% и 66,72%, соответственно.
Профили флуктуации массы тела показали, что мыши, получающие тестовые соединения, теряют массу тела в течение первых нескольких дней, но затем в дальнейшем их масса восстанавливалась, тем самым указывая на то, что тестовые соединения хорошо переносились при текущих дозировках и путях дозирования у голых мышей. В заключение, и соединение примера 2 при дозе 20 мг/кг, и соединение примера 7 при дозе 30 мг/кг, i. v., дважды в день являлись эффективными ингибиторами роста опухоли в ксенотранспланте MV4-11 без очевидной токсичности.
Фармацевтические составы
ПРИМЕР 18 (i) Таблетированный состав
Таблетированную композицию, содержащую соединение формулы (1), получали смешиванием 50 мг соединения с 197 мг лактозы (BP) в качестве разбавителя и 3 мг стеарата магния в качестве смазывающего вещества, и сжатием с образованием таблетки известным способом.
ii) Капсульный состав
Капсульный состав получали смешиванием 100 мг соединения формулы (1) со 100 мг лактозы, и наполнением полученной смесью стандартных непрозрачных твердых желатиновых капсул.
iii) Инъецируемый состав I
Парентеральная композиция для введения путем инъекции может быть получена растворением соединения формулы (1) (например, в солевой форме) в воде, содержащей 10% пропиленгликоля, с получением концентрации активного соединения 1,5% по массе. Затем раствор стерилизовали фильтрацией, наполняли им ампулы и запаивали.
(iv) Инъецируемый состав II
Парентеральную композицию для введения путем инъекции готовили растворением в воде соединения формулы (1) (например, в солевой форме) (2 мг/мл) и маннита (50 мг/мл), стерилизацией фильтрацией раствора и его заполнением в запаиваемые 1 мл пузырьки или ампулы.
(iv) Состав подкожной инъекции
Композицию для подкожного введения получали смешиванием соединения формулы (1) с кукурузным маслом фармацевтической категории с получением концентрации 5 мг/мл Композицию стерилизовали и заполняли в подходящий контейнер.
Эквиваленты
Вышеуказанные примеры представлены с целью иллюстрации настоящего изобретения, и их не следует рассматривать как диктующие ограничение объема настоящего изобретения. Будет легко очевидно, что многочисленные модификации и изменения могут быть сделаны относительно конкретных вариантов реализации настоящего изобретения, описанного выше и иллюстрированного в примерах без отклонения от основ, лежащих в основе настоящего изобретения. Предусматривается, что все такие модификации и изменения охватываются в настоящей заявке.
| название | год | авторы | номер документа |
|---|---|---|---|
| ФАРМАЦЕВТИЧЕСКИ АКТИВНЫЕ СОЕДИНЕНИЯ | 2013 |
|
RU2654942C2 |
| СОЕДИНЕНИЯ И КОМПОЗИЦИИ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕИНКИНАЗ | 2006 |
|
RU2387653C2 |
| ЗАМЕЩЕННЫЕ СОЕДИНЕНИЯ ХИНОЛИНА И СПОСОБЫ ИХ ИСПОЛЬЗОВАНИЯ | 2012 |
|
RU2568258C2 |
| СОЕДИНЕНИЯ ПИРИМИДИНА И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ ДЛЯ ПРЕДУПРЕЖДЕНИЯ ИЛИ ЛЕЧЕНИЯ РАКА | 2019 |
|
RU2807277C2 |
| ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ ИНДОЛ-5-ОЛА И ИХ ТЕРАПЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2013 |
|
RU2674249C2 |
| Замещенные N2-(4-амино-2-метоксифенил)-N4-[2-(диметилфосфорил)-фенил]-5-хлор-пиримидин-2,4-диамины в качестве модуляторов ALK и EGFR, предназначенные для лечения рака | 2015 |
|
RU2607371C1 |
| Бензотиофены и родственные соединения в качестве агонистов STING | 2019 |
|
RU2806274C2 |
| ИНГИБИТОРЫ АКТИВНОСТИ ПРОТЕИНТИРОЗИНКИНАЗЫ | 2009 |
|
RU2533827C2 |
| ХИМИЧЕСКИЕ СОЕДИНЕНИЯ - 759 | 2008 |
|
RU2481348C2 |
| МОДУЛЯТОРЫ АКТИВНОСТИ НЕС1 И СПОСОБЫ ДЛЯ НИХ | 2011 |
|
RU2576036C2 |

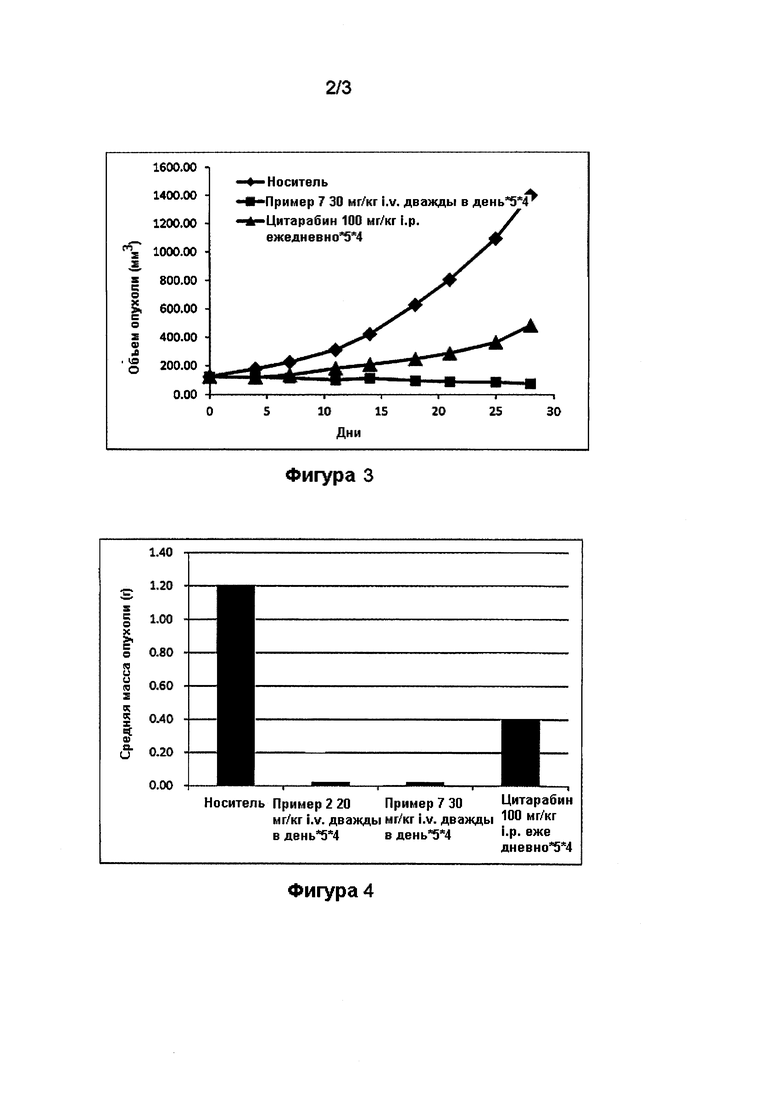

Изобретение относится к соединению формулы (1) и его фармацевтически приемлемым солям; где R1 представляет собой водород или С1-2 алкил; и R2, R3 и R4 одинаковые или отличаются и каждый из них выбран из водорода, С1-2 алкила, фтора, хлора, С1-2 алкокси и трифторметила при условии, что не более двух из R2, R3 и R4 отличны от водорода. Изобретение относится к фармацевтической композиции для профилактики или лечения заболевания, опосредованного киназой Aurora A, Aurora В или киназой FLT3, содержащей терапевтическое количество соединения формулы (1), и фармацевтически приемлемый наполнитель. Соединения предназначены для применения в лечении пролиферативного заболевания, представляющего собой гемопоэтическую опухоль, выбранную из острого лимфобластного лейкоза (ALL), острого миелоидного лейкоза (AML), хронического миелоидного лейкоза (CML), ходжкинской лимфомы (HL), неходжкинской лимфомы (NHL) и множественной миеломы (ММ). Технический результат – соединения для лечения заболеваний, опосредованных киназой Aurora A, Aurora В или киназой FLT3. 3 н. и 13 з.п. ф-лы, 6 табл., 18 пр.

1. Соединение формулы (1):

и его фармацевтически приемлемые соли; где
R1 представляет собой водород или С1-2 алкил; и
R2, R3 и R4 одинаковые или отличаются и каждый из них выбран из водорода, С1-2 алкила, фтора, хлора, С1-2 алкокси и трифторметила при условии, что не более двух из R2, R3 и R4 отличны от водорода.
2. Соединение по п. 1, отличающееся тем, что R1 выбран из водорода и метила.
3. Соединение по п. 1, отличающееся тем, что R2 выбран из водорода, фтора, хлора, метила, этила и метокси.
4. Соединение по п. 1, отличающееся тем, что R3 представляет собой водород.
5. Соединение по п. 1, отличающееся тем, что R4 выбран из водорода, фтора, метила и этила.
6. Соединение по п. 1, отличающееся тем, что (i) R1 представляет собой водород; R2 выбран из метила, этила, фтора, хлора и метокси; R3 представляет собой водород; и R4 представляет собой водород; или (ii) R1 представляет собой водород; R2 представляет собой водород; R3 представляет собой водород; и R4 представляет собой метил; или (iii) R1 представляет собой водород; R2 представляет собой фтор; R3 представляет собой водород; и R4 представляет собой метил.
7. Соединение по п. 1, отличающееся тем, что R1 представляет собой водород; R2 выбран из водорода и фтора; R3 представляет собой водород; и R4 представляет собой метил.
8. Соединение по п.7, отличающееся тем, что R2 представляет собой фтор.
9. Соединение по п.1, отличающееся тем, что указанное соединение выбрано из соединений прим. 1-прим. 12 в таблице ниже и их фармацевтически приемлемых солей:

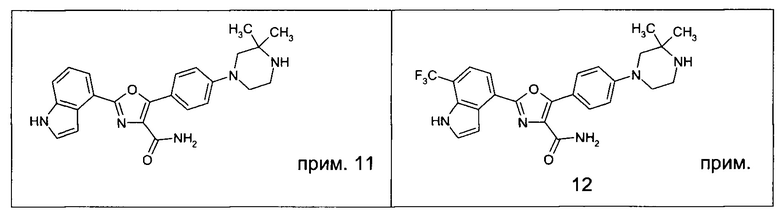
10. Соединение по п. 9, выбранное из соединений прим. 2, прим. 3, прим. 5 и прим. 7 и их фармацевтически приемлемых солей.
11. Соединение по п. 9, представляющее собой:
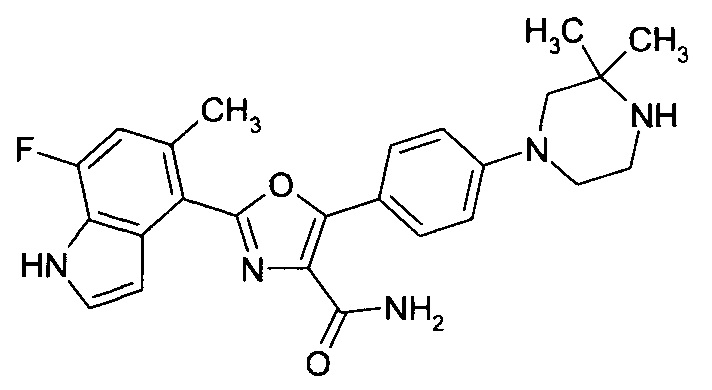
или его фармацевтически приемлемая соль.
12. Фармацевтическая композиция для профилактики или лечения пролиферативного заболевания, причем пролиферативное заболевание представляет собой гемопоэтическую опухоль, выбранную из острого лимфобластного лейкоза (ALL), острого миелоидного лейкоза (AML), хронического миелоидного лейкоза (CML), ходжкинской лимфомы (HL), неходжкинской лимфомы (NHL) и множественной миеломы (ММ), и при этом фармацевтическая композиция содержит терапевтическое количество соединения по любому из пп. 1-11 и фармацевтически приемлемый наполнитель.
13. Фармацевтическая композиция для профилактики или лечения заболевания, опосредованного киназой Aurora A, Aurora В или киназой FLT3, содержащая терапевтическое количество соединения по любому из пп. 1-11 и фармацевтически приемлемый наполнитель.
14. Соединение по любому из пп. 1-11 для применения при профилактике или лечении пролиферативного заболевания, опосредованного киназой Aurora A, Aurora В или киназой FLT3.
15. Соединение по любому из пп. 1-11 для применения в лечении пролиферативного заболевания, отличающееся тем, что указанное пролиферативное заболевание представляет собой гемопоэтическую опухоль, выбранную из острого лимфобластного лейкоза (ALL), острого миелоидного лейкоза (AML), хронического миелоидного лейкоза (CML), ходжкинской лимфомы (HL), неходжкинской лимфомы (NHL) и множественной миеломы (ММ).
16. Соединение для применения по п. 15, отличающееся тем, что пролиферативное заболевание представляет собой рак, чувствительный к ингибированию FLT3 киназой, и представляет собой острый миелоидный лейкоз (AML).
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |
| Приспособление для суммирования отрезков прямых линий | 1923 |
|
SU2010A1 |
| ЕА 200970612 А1, 30.10.2009 | |||
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |

