ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее раскрытие относится к соединениям и их производным, которые могут быть полезны в качестве агонистов STING (стимулятор генов интерферона), активирующих путь STING. Настоящее раскрытие также относится к композициям, содержащим такие соединения, способам синтеза таких соединений и к применению таких соединений, включая введение таких соединений для индукции иммунного ответа, индукции STING-зависимой продукции интерферона типа I и/или для лечения нарушения пролиферации клеток, такого как рак.
ССЫЛКА НА ПЕРЕЧЕНЬ ПОСЛЕДОВАТЕЛЬНОСТЕЙ, ПРЕДСТАВЛЕННЫЙ ЭЛЕКТРОННЫМ ОБРАЗОМ
Перечень последовательностей настоящей заявки представлен в электронном виде через EFS-Web в виде перечня последовательностей в формате ASCII с именем файла «24578_SEQLIST-FEB2019», датой создания 1 марта 2019 г. и размером 25 КБ. Этот перечень последовательностей, представленный через EFS-Web, является частью описания и полностью включен в настоящий документ посредством ссылки.
УРОВЕНЬ ТЕХНИКИ
Иммунная система сформировалась в процессе эволюции, чтобы распознавать и нейтрализовать различные типы угроз в целях поддержания гомеостаза хозяина, и обычно подразделяется на две ветви: адаптивную и врожденную. Адаптивная иммунная система специализируется на распознавании в качестве чужеродных тех антигенов, которые не экспрессируются в природе в организме хозяина, и на создании анти-антигенного ответа посредством скоординированных действий многих подгрупп лейкоцитов. Отличительной чертой адаптивных иммунных ответов является способность обеспечивать «память» или длительный иммунитет против обнаруженного антигена. Хотя этот специфический и продолжительный эффект имеет решающее значение для здоровья и выживания хозяина, адаптивный иммунный ответ требует времени для выработки полноценного ответа.
Врожденная иммунная система компенсирует эту задержку по времени и специализируется на быстром реагировании на различные повреждающие факторы или сигналы опасности. Она обеспечивает первую линию защиты от бактерий, вирусов, паразитов и других инфекционных угроз, но также сильно реагирует на определенные сигналы опасности, связанные с повреждением клеток или тканей. Врожденная иммунная система не обладает антигенной специфичностью, но реагирует на множество эффекторных механизмов. Опсонизация, фагоцитоз, активация системы комплемента и продукция растворимых биоактивных молекул, таких как цитокины или хемокины, все это является механизмами, с помощью которых врожденная иммунная система опосредует свой ответ. Реагируя на эти молекулярные паттерны, ассоциированные с повреждением (DAMP), или патоген-ассоциированные молекулярные паттерны (РАМР), описанные выше, врожденная иммунная система способна обеспечить широкую защиту от широкого спектра угроз для хозяина.
Свободные цитозольные ДНК и РНК входят в число этих РАМР и DAMP. Недавно было продемонстрировано, что основным сенсором цитозольной ДНК является cGAS (циклическая GMP-AMP-синтаза). При распознавании цитозольной ДНК cGAS катализирует образование циклического динуклеотида 2'3'-cGAMP, атипичного второго мессенджера, который прочно связывается с ER-трансмембранным адаптерным белком STING. STING после связывания с cGAMP претерпевает конформационное изменение, он перемещается в перинуклеарный компартмент и индуцирует активацию критических факторов транскрипции IRF-3 и NF-κВ. Это приводит к сильной индукции интерферонов типа I и продукции провоспалительных цитокинов, таких как IL-6, TNF-α и IFN-γ.
Важность интерферонов типа I и провоспалительных цитокинов для различных клеток иммунной системы была установлена очень хорошо. В частности, эти молекулы в значительной степени усиливают активацию Т-клеток, повышая способность дендритных клеток и макрофагов захватывать, процессировать, презентировать и кросс-презентировать антигены Т-клеткам. Стимулирующая способность этих антигенпрезентирующих клеток активировать Т-клетки усиливается за счет активации критических костимулирующих молекул, таких как CD80 или CD86. Наконец, интерфероны типа I могут быстро взаимодействовать со своими когнатными рецепторами и запускать активацию интерферон-чувствительных генов, которые могут вносить значительный вклад в активацию адаптивных иммунных клеток.
С терапевтической точки зрения показано, что интерфероны типа I обладают противовирусной активностью, напрямую подавляя репликацию вируса гепатита В и вируса гепатита С человека, а также стимулируя иммунные ответы на инфицированные вирусом клетки. Соединения, которые могут индуцировать продукцию интерферона I типа, применяются в вакцинах, где они действуют как адъюванты, усиливая специфические иммунные ответы на антигены и минимизируя побочные эффекты за счет снижения дозировки и расширения иммунного ответа.
Кроме того, интерфероны и соединения, которые могут индуцировать продукцию интерферона, потенциально могут применяться при лечении онкологических заболеваний у человека. Такие молекулы являются потенциально полезными в качестве противораковых агентов с разнообразными путями активности. Интерфероны могут напрямую ингибировать пролиферацию опухолевых клеток человека и могут действовать синергетически с различными одобренными химиотерапевтическими агентами. Интерфероны типа I могут значительно усиливать противоопухолевые иммунные ответы, индуцируя активацию клеток как адаптивной, так и врожденной иммунной системы. Наконец, инвазивность опухоли может подавляться интерферонами путем модуляции экспрессии ферментов, связанных с ремоделированием тканей.
Принимая во внимание перспективность интерферонов типа I и соединений, индуцирующих интерферон типа I, в качестве противовирусных и противораковых агентов, остается потребность в новых агентах, которые могут индуцировать эффективную продукцию интерферона типа I. В связи с растущим объемом данных, демонстрирующих, что путь cGAS-STING, сенсор цитозольной ДНК, обладает значительной способностью индуцировать интерфероны типа I, разработка агентов, активирующих STING, быстро занимает важное место в сегодняшнем направлении противоопухолевой терапии.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение включает соединения общей формулы (I), соединения общей формулы (II), соединения общей формулы (III), соединения общей формулы (IV), соединения общей формулы (V), соединения общей формулы (VI) и их фармацевтически приемлемые соли. Эти соединения и их фармацевтически приемлемые соли могут быть полезны в качестве агентов для индукции иммунных ответов, для индукции STING-опосредованной продукции интерферона типа I и/или для лечения нарушения пролиферации клеток.
Настоящее изобретение относится к новым соединениям общей формулы (I). В частности, настоящее изобретение относится к соединениям, имеющим общую структурную формулу (I):
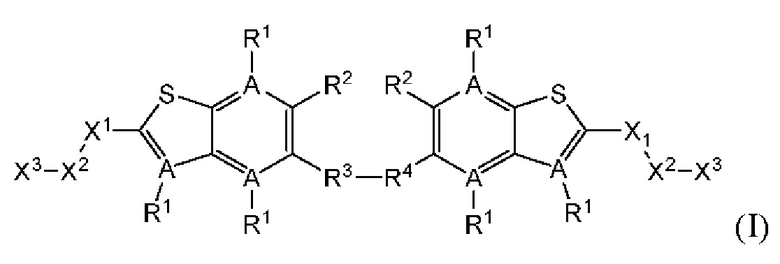
или их фармацевтически приемлемым солям, как описано в настоящем документе. Также раскрыты применения соединений общей формулы (I) и способы получения соединений общей формулы (I).
Настоящее изобретение также относится к новым соединениям общей формулы (II). В частности, настоящее изобретение относится к соединениям, имеющим общую структурную формулу (II):
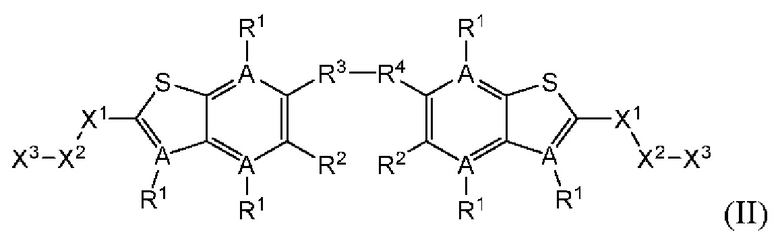
или их фармацевтически приемлемым солям, как описано в настоящем документе. Также раскрыты применения соединений общей формулы (II) и способы получения соединений общей формулы (II).
Настоящее изобретение также относится к новым соединениям общей формулы (III). В частности, настоящее изобретение относится к соединениям, имеющим общую структурную формулу (III):
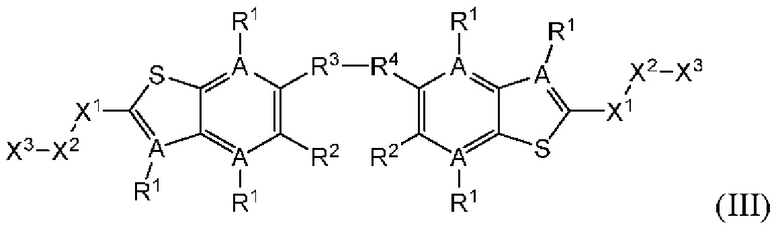
или их фармацевтически приемлемым солям, как описано в настоящем документе. Также раскрыты применения соединений общей формулы (III) и способы получения соединений общей формулы (III).
Настоящее изобретение также относится к новым соединениям общей формулы (IV). В частности, настоящее изобретение относится к соединениям, имеющим общую структурную формулу (IV):
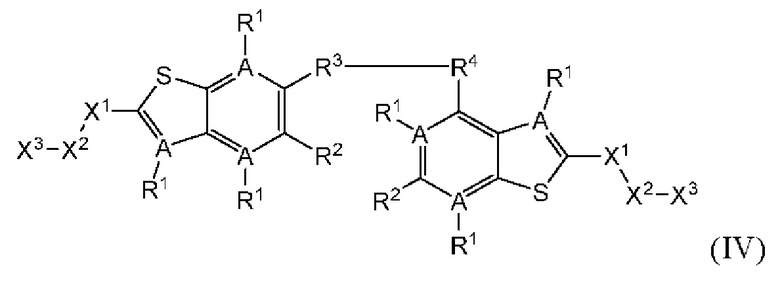
или их фармацевтически приемлемым солям, как описано в настоящем документе. Также раскрыты применения соединений общей формулы (IV) и способы получения соединений общей формулы (IV).
Настоящее изобретение также относится к новым соединениям общей формулы (V). В частности, настоящее изобретение относится к соединениям, имеющим общую структурную формулу (V):
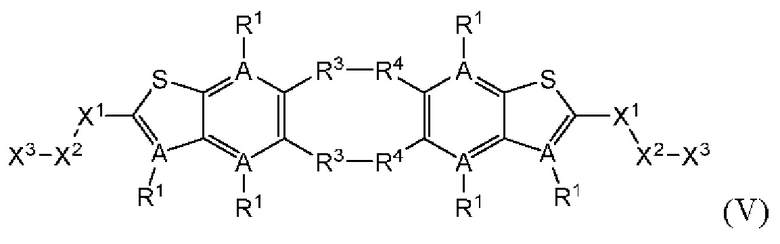
или их фармацевтически приемлемым солям, как описано в настоящем документе. Также раскрыты применения соединений общей формулы (V) и способы получения соединений общей формулы (V).
Настоящее изобретение также относится к новым соединениям общей формулы (VI). В частности, настоящее изобретение относится к соединениям, имеющим общую структурную формулу (VI):
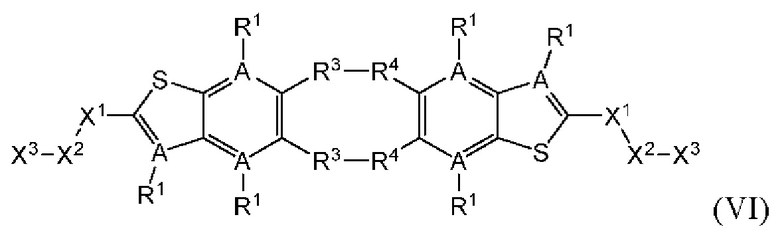
или их фармацевтически приемлемым солям, как описано в настоящем документе. Также раскрыты применения соединений общей формулы (VI) и способы получения соединений общей формулы (VI).
Другие варианты осуществления, аспекты и отличительные признаки настоящего изобретения либо дополнительно описаны, либо будут очевидны из последующего описания, примеров и прилагаемой формулы изобретения.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение включает соединения общей формулы (I), соединения общей формулы (II), соединения общей формулы (III), соединения общей формулы (IV), соединения общей формулы (V), соединения общей формулы (VI) и их фармацевтически приемлемые соли. Эти соединения и их фармацевтически приемлемые соли могут быть полезны в качестве агентов для индукции иммунных ответов, для индукции STING-опосредованной продукции интерферона типа I и/или для лечения нарушения пролиферации клеток.
Первый вариант осуществления относится к соединениям общей формулы (I):
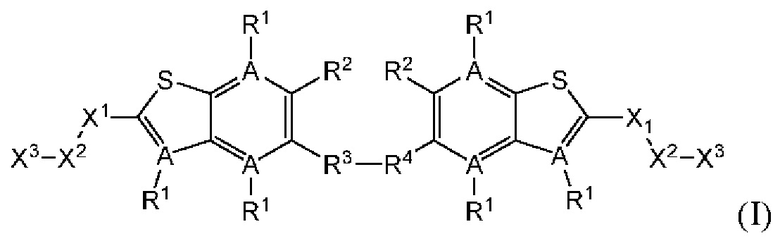
или их фармацевтически приемлемой соли, где каждый A-R1 независимо выбран из группы, состоящей из C-R1 и N; каждый R1 независимо выбран из группы, состоящей из Н, галогена, OR6, N(R6)2, C1-C6 алкила, C1-С6 галоалкила, C1-С6 алкила, замещенного OR6, C1-C6 алкила, замещенного N(R6)2, COOR6, и C(O)N(R6)2; каждый R2 независимо выбран из группы, состоящей из Н, галогена, CN, OR6, N(R6)2, COOR6, C(O)N(R6)2, SO2R6, C1-C6 алкила, C1-C6 галоалкила, C1-C6 алкила, замещенного OR6, C2-C6 алкенила, C2-C6 галоалкенила, C2-C6 алкенила, замещенного OR6, С2-С6 алкинила, С2-С6 галоалкинила, С2-С6 алкинила, замещенного OR6, С3-С6 циклоалкила, и от 3- до 6-членного гетероциклического кольца, включая 1-2 кольцевых члена, выбранных из группы, состоящей из О, S и N(R6); R3 и R4 независимо выбраны из группы, состоящей из O-(C1-C4 алкилен или галоалкилен), С1-С5 алкилена или галоалкилена и N(R6)-(C1-C4 алкилен или галоалкилен); необязательно, R4 может быть взят совместно с соседним C-R1 и атомом, к которому они присоединены, с образованием конденсированного кольца Е, которое выбрано из фенила или 5- или 6-членного гетероциклического кольца, включая 1-2 кольцевых члена, выбранных из группы, состоящей из О, S, N и N(R6), где связь с R3 от указанного кольца Е проходит от атома на указанном кольце Е с открытой валентностью для замещения, и где указанные фенил или гетероциклическое кольцо необязательно замещены одним или более членами группы, состоящей из галогена, С1-С3 алкила, и С1-С3 галоалкила; каждый R6 независимо выбран из группы, состоящей из Н, C1-С6 алкила, и C1-С6 галоалкила; каждый X1 независимо выбран из группы, состоящей из С=O, -СН2-, -CHF-, и -CF2-; каждый X2 независимо выбран из (C(R8)2)(1-3), где каждый R8 независимо выбран из группы, состоящей из Н, галогена, C1-C6 алкила, CN, OR6, N(R6)2, C1-С6 галоалкила, С3-С6 циклоалкила, C1-C6 алкила, замещенного OR6, и C1-C6 алкила, замещенного N(R6)2; необязательно, 2 R8 на разных атомах углерода могут быть взяты вместе совместно с атомами, к которым они присоединены, с образованием от 3- до 6-членного конденсированного кольца; и необязательно, 2 R8 на одном атоме углерода могут быть взяты вместе совместно с атомом, к которому они присоединены, с образованием от 3- до 6-членного спироцикла; каждый X3 независимо выбран из группы, состоящей из COOR6, C(O)SR6, C(S)OR6,  SO2R6, C(O)N(R9)2, и CN; и каждый R9 независимо выбран из группы, состоящей из Н, COOR, и SO2R6.
SO2R6, C(O)N(R9)2, и CN; и каждый R9 независимо выбран из группы, состоящей из Н, COOR, и SO2R6.
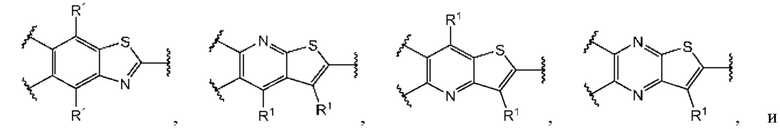
В первом аспекте первого варианта осуществления каждый A-R1 независимо выбран из группы, состоящей из C-R1 и N. В конкретных примерах этого аспекта каждый 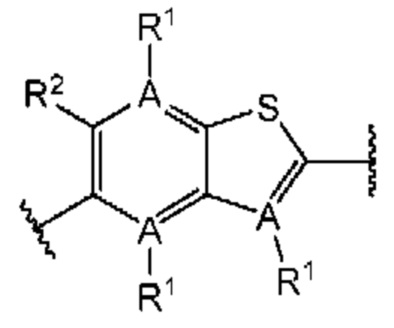 независимо выбран из группы, состоящей из
независимо выбран из группы, состоящей из 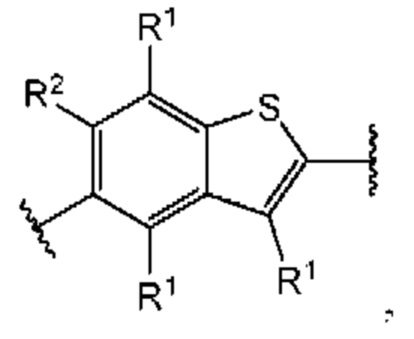
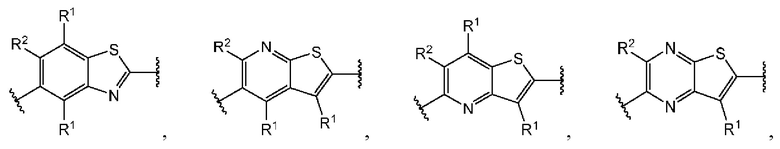 и
и 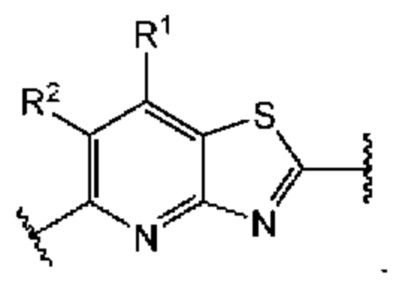 В более конкретных случаях этого аспекта каждый
В более конкретных случаях этого аспекта каждый 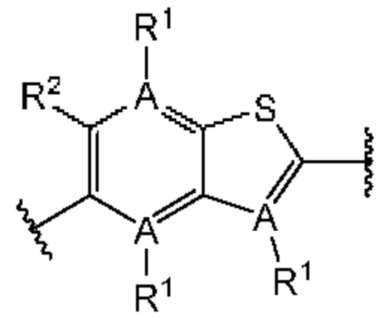 независимо выбран из группы, состоящей из
независимо выбран из группы, состоящей из 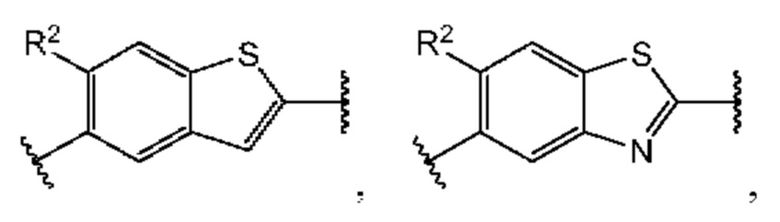
 и
и 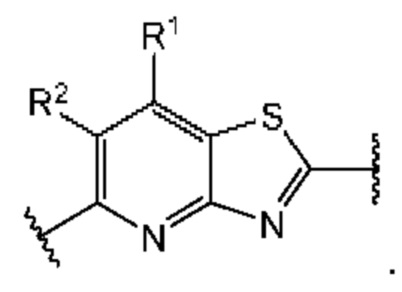 В этом аспекте все другие группы являются такими, как указано выше в общей формуле (I) первого варианта осуществления.
В этом аспекте все другие группы являются такими, как указано выше в общей формуле (I) первого варианта осуществления.
Во втором аспекте первого варианта осуществления каждый R1 независимо выбран из группы, состоящей из Н, галогена, OR6, N(R6)2, C1-C6 алкила, C1-C6 галоалкила, C1-C6 алкила, замещенного OR6, C1-С6 алкила, замещенного N(R6)2, COOR6, и C(O)N(R6)2. В случаях этого аспекта каждый R1 независимо выбран из группы, состоящей из Н, галогена, C1-С3 алкила, и C1-С3 галоалкила. В конкретных примерах этого аспекта каждый R1 независимо выбран из группы, состоящей из Н и галогена. В более конкретных случаях этого аспекта каждый R1 независимо выбран из группы, состоящей из Н и F. В этом аспекте все другие группы являются такими, как указано выше в общей формуле (I) первого варианта осуществления или в первом аспекте выше.
В третьем аспекте первого варианта осуществления каждый R2 независимо выбран из группы, состоящей из Н, галогена, CN, OR6, N(R6)2, COOR6, C(O)N(R6)2, SO2R6, C1-C6 алкила, C1-С6 галоалкила, C1-С6 алкила, замещенного OR6, С2-С6 алкенила, С2-С6 галоалкенила, С2-С6 алкенила, замещенного OR6, С2-С6 алкинила, С2-С6 галоалкинила, С2-С6 алкинила, замещенного OR6, С3-С6 циклоалкила, и от 3- до 6-членного гетероциклического кольца, включая 1-2 кольцевых члена, выбранных из группы, состоящей из О, S и N(R6). В случаях этого аспекта каждый R2 независимо выбран из группы, состоящей из Н, галогена, C1-С3 алкила, С1-С3 галоалкила, ОС1-С3 алкила, С2-С3 алкенила, и N(R6)2. В конкретных примерах этого аспекта каждый R2 независимо выбран из группы, состоящей из Н, Br, Cl, СН3, СН2СН3, СН=СН2, ОСН3, OCFH2, OCF2H, OCF3, и N(R6)2. В более конкретных случаях этого аспекта каждый R2 независимо выбран из группы, состоящей из Н, СН3, ОСН3, и OCF2H. В этом аспекте все другие группы являются такими, как указано в общей формуле (I) первого варианта осуществления или в первом или втором аспектах, описанных выше.
В четвертом аспекте первого варианта осуществления R3 и R4 независимо выбраны из группы, состоящей из O-(C1-C4 алкилен или галоалкилен), С1-С5 алкилена или галоалкилена, и N(R6)-(C1-C4 алкилен или галоалкилен); необязательно, R4 может быть взят совместно с соседним C-R1 и атомом, к которому они присоединены, с образованием конденсированного кольца Е, которое выбрано из фенила или 5- или 6-членного гетероциклического кольца, включая 1-2 кольцевых члена, выбранных из группы, состоящей из О, S, N и N(R6), где связь с R3 от указанного кольца Е проходит от атома на указанном кольце Е с открытой валентностью для замещения, и где указанные фенил или гетероциклическое кольцо необязательно замещены одним или более членами группы, состоящей из галогена, C1-С3 алкила, и C1-С3 галоалкила. В примерах этого четвертого аспекта R3-R4 выбран из группы, состоящей из -(СН2)2-8-, -O(CH2)1-7-, -O(СН2)1-6О-, -NH(CH2)1-7-, и -NH(CH2)1-6O-. В определенных примерах этого четвертого аспекта R3-R4 выбран из группы, состоящей из -(СН2)2-, -(CH2)3-, -(СН2)4-, -O(СН2)2-, -O(СН2)3-, -O(CH2)4-, -O(СН2)2O-, -O(СН2)3О-, -ОСН2СН(СН3)CH2O-, -O(CH2)4O-, -O(CH2)5O-, -NH(CH3)2-, -NH(CH2)3-, и -NH(CH2)3O-. В конкретных примерах этого четвертого аспекта R4 может быть взят совместно с соседним C-R1 и атомом, к которому они присоединены, с образованием конденсированного кольца Е, которое выбрано из фенила или 5- или 6-членного гетероциклического кольца, включая 1-2 кольцевых члена, выбранных из группы, состоящей из О, S, N и N(R6), где связь с R3 от указанного кольца Е проходит от атома на указанном кольце Е с открытой валентностью для замещения, и где указанные фенил или гетероциклическое кольцо необязательно замещены одним или более членами группы, состоящей из галогена, С1-С3 алкила, и С1-С3 галоалкила. В этом случае структура общей формулы (I) представляет собой формулу (Ia):
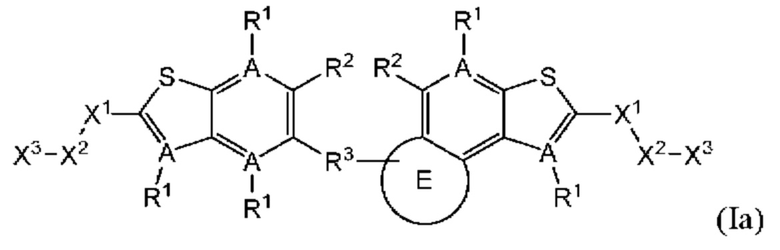
где все группы имеют значения, указанные в общей формуле (I). В этом аспекте все другие группы являются такими, как указано в общей формуле (I) первого варианта осуществления или в первом-третьем аспектах, описанных выше.
В пятом аспекте первого варианта осуществления каждый R6 независимо выбран из группы, состоящей из Н, C1-С6 алкила, и C1-С6 галоалкила. В примерах этого аспекта каждый R6 независимо выбран из группы, состоящей из Н, C1-С3 алкила, и C1-С3 галоалкила. В конкретных примерах этого аспекта каждый R6 независимо выбран из группы, состоящей из Н, СН3, и CHF2. В этом аспекте все другие группы являются такими, как указано в общей формуле (I) первого варианта осуществления или в первом-четвертом аспектах, описанных выше.
В шестом аспекте первого варианта осуществления каждый X1 независимо выбран из группы, состоящей из С=O, -CH2-, -CHF-, и -CF2-. В примерах этого аспекта X1 выбран из группы, состоящей из С=O и -СН2-. В конкретных примерах этого аспекта X1 представляет собой С=O. В этом варианте осуществления все другие группы являются такими, как указано в общей формуле (I) первого варианта осуществления или в первом-пятом аспектах, описанных выше.
В седьмом аспекте первого варианта осуществления каждый X2 независимо выбран из (C(R8)2)(1-3), где каждый R8 независимо выбран из группы, состоящей из Н, галогена, C1-C6 алкила, CN, OR6, N(R6)2, C1-С6 галоалкила, С3-С6 циклоалкила, C1-C6 алкила, замещенного OR6, и C1-C6 алкила, замещенного N(R6)2; необязательно, 2 R8 на разных атомах углерода могут быть взяты вместе совместно с атомами, к которым они присоединены, с образованием от 3- до 6-членного конденсированного кольца; и, необязательно, 2 R8 на одном атоме углерода могут быть взяты вместе, совместно с атомом, к которому они присоединены, с образованием от 3- до 6-членного спироцикла. В первом случае этого аспекта каждый X2 представляет собой CH2CHR8, где R8 выбран из группы, состоящей из Н, С1-С3 алкила, C1-С3 алкила, замещенного ОН, С1-С3 алкила, замещенного ОС1-С3 алкилом, и С3-С6 циклоалкила. В конкретных примерах этого первого случая каждый X2 представляет собой CH2CHR8, где R8 выбран из группы, состоящей из Н, СН3, СН2ОН, СН2СН3, СН2СН2СН3, СН(СН3)2, СН2ОСН3, и циклопропила. Во втором случае этого аспекта каждый X2 представляет собой CHR8CHR8, где каждый R8 независимо выбран из группы, состоящей из Н, С1-С3 алкила, С1-С3 алкила, замещенного ОН, С1-С3 алкила, замещенного ОС1-С3 алкилом, и С3-С6 циклоалкила, и, необязательно, 2 R8 на разных атомах углерода взяты вместе совместно с атомами, к которым они присоединены, с образованием от 3- до 6-членного конденсированного кольца. В конкретных примерах этого второго случая каждый X2 представляет собой CHR8CHR8, где каждый R8 независимо выбран из группы, состоящей из Н и C1-С3 алкила, и, необязательно, 2 R8 на разных атомах углерода взяты вместе совместно с атомами, к которым они присоединены, с образованием от 3- до 6-членного конденсированного кольца. В третьем случае этого аспекта каждый X2 представляет собой CH2C(R8)2, где каждый R8 независимо выбран из группы, состоящей из Н, C1-С3 алкила, C1-С3 алкила, замещенного ОН, C1-С3 алкила, замещенного ОС1-С3 алкилом, и С3-С6 циклоалкила, и, необязательно, 2 R8 на одном атоме углерода взяты вместе совместно с атомами, к которым они присоединены, с образованием от 3- до 6-членного спироцикла. В конкретных примерах этого третьего случая каждый X2 представляет собой CH2C(R8)2, где каждый R8 независимо выбран из группы, состоящей из Н и C1-С3 алкила, и, необязательно, 2 R8 на одном атоме углерода взяты вместе совместно с атомами, к которым они присоединены, с образованием от 3- до 6-членного спироцикла. В этом аспекте все другие группы являются такими, как указано в общей формуле (I) первого варианта осуществления или в первом-шестом аспектах, описанных выше.
В восьмом аспекте первого варианта осуществления каждый X3 независимо выбран из группы, состоящей из COOR6, C(O)SR6, C(S)OR6,  SO2R6, C(O)N(R9)2, и CN. В примерах этого аспекта каждый X3 независимо выбран из группы, состоящей из COOR6,
SO2R6, C(O)N(R9)2, и CN. В примерах этого аспекта каждый X3 независимо выбран из группы, состоящей из COOR6,  SO2R6, C(O)N(R9)2, и CN. В конкретных примерах этого аспекта каждый X3 независимо выбран из группы, состоящей из COOR6, C(O)N(R9)2, и CN. В даже более конкретных случаях этого аспекта каждый X3 независимо выбран из группы, состоящей из СООН, СООСН3, CONH2, и CN. В этом аспекте все другие группы являются такими, как указано в общей формуле (I) первого варианта осуществления или в первом-седьмом аспектах, описанных выше.
SO2R6, C(O)N(R9)2, и CN. В конкретных примерах этого аспекта каждый X3 независимо выбран из группы, состоящей из COOR6, C(O)N(R9)2, и CN. В даже более конкретных случаях этого аспекта каждый X3 независимо выбран из группы, состоящей из СООН, СООСН3, CONH2, и CN. В этом аспекте все другие группы являются такими, как указано в общей формуле (I) первого варианта осуществления или в первом-седьмом аспектах, описанных выше.
В девятом аспекте первого варианта осуществления каждый R9 независимо выбран из группы, состоящей из Н, COOR6, и SO2R6. В примерах этого аспекта каждый R9 независимо представляет собой Н. В этом аспекте все другие группы являются такими, как указано в общей формуле (I) первого варианта осуществления или в первом-восьмом аспектах, описанных выше.
Десятый аспект первого варианта осуществления относится к фармацевтической композиции, где указанная фармацевтическая композиция содержит (а) соединение в соответствии с приведенной выше общей формулой (I) первого варианта осуществления или с первым-девятым аспектами, описанными выше, или его фармацевтически приемлемую соль; и (b) фармацевтически приемлемый носитель.
Одиннадцатый аспект первого варианта осуществления относится к способам индукции иммунного ответа у пациента, нуждающегося в терапии, включающим введение пациенту терапевтически эффективного количества соединения в соответствии с приведенной выше общей формулой (I) первого варианта осуществления или с первым-девятым аспектами, описанными выше, или его фармацевтически приемлемой соли.
Двенадцатый аспект первого варианта осуществления относится к способам индукции иммунного ответа у пациента, нуждающегося в терапии, включающим введение пациенту терапевтически эффективного количества композиции согласно десятому аспекту, описанному выше.
Тринадцатый аспект первого варианта осуществления относится к способам индукции STING-зависимой продукции интерферона типа I у пациента, нуждающегося в терапии, включающим введение пациенту терапевтически эффективного количества соединения согласно приведенной выше общей формуле (I) первого варианта осуществления или с первого по девятый аспектам, описанным выше, или его фармацевтически приемлемой соли.
Четырнадцатый аспект первого варианта осуществления относится к способам индукции STING-зависимой продукции интерферона I типа у пациента, нуждающегося в терапии, включающим введение этому пациенту терапевтически эффективного количества композиции согласно десятому аспекту, описанному выше.
Пятнадцатый аспект первого варианта осуществления относится к способам индукции STING-зависимой продукции цитокинов у пациента, нуждающегося в терапии, включающим введение этому пациенту терапевтически эффективного количества соединения согласно приведенной выше общей формуле (I) первого варианта осуществления или с первого по девятый аспектам, описанным выше, или его фармацевтически приемлемой соли.
Шестнадцатый аспект первого варианта осуществления относится к способам индукции STING-зависимой продукции цитокинов у пациента, нуждающегося в терапии, включающим введение этому пациенту терапевтически эффективного количества композиции согласно десятому аспекту, описанному выше.
Семнадцатый аспект первого варианта осуществления относится к способам лечения нарушения пролиферации клеток у пациента, нуждающегося в терапии, включающим введение этому пациенту терапевтически эффективного количества соединения общей формулы (I) или его фармацевтически приемлемой соли. В случаях этого семнадцатого аспекта первого варианта осуществления нарушение пролиферации клеток представляет собой рак.
Восемнадцатый аспект первого варианта осуществления относится к способам лечения нарушения пролиферации клеток у пациента, нуждающегося в терапии, при этом указанный способ включает введение пациенту терапевтически эффективного количества композиции в соответствии с одиннадцатым аспектом, описанным выше. В случаях этого восемнадцатого аспекта первого варианта осуществления нарушение пролиферации клеток представляет собой рак.
В каждом аспекте первого варианта осуществления, описанного в настоящем документе, переменные R1, R2, R3, R4, R6, R8, R9, А, X1, X2 и X3 общей формулы (I) первого варианта осуществления и его различных аспектов и случаев, каждый независимо выбран из каждого, при условии, что, по меньшей мере, один из R1, R2, R3, R4, R6, R8, и R9 не представляет собой Н.
Второй вариант осуществления относится к соединениям общей формулы (II):
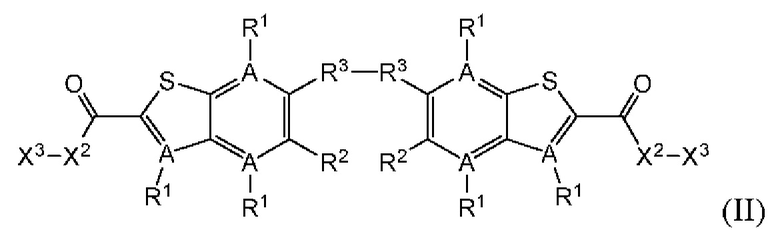
или их фармацевтически приемлемой соли, где каждый A-R1 независимо выбран из группы, состоящей из C-R1 и N; где каждый R1 независимо выбран из группы, состоящей из Н, галогена, OR6, N(R6)2, C1-С6 алкила, C1-C6 галоалкила, C1-С6 алкила, замещенного OR6, C1-С6 алкила, замещенного N(R6)2, COOR6, и C(O)N(R6)2; каждый R2 независимо выбран из группы, состоящей из Н, галогена, CN, OR6, N(R6)2, COOR6, C(O)N(R6)2, SO2R6, C1-C6 алкила, C1-С6 галоалкила, C1-С6 алкила, замещенного OR6, С2-С6 алкенила, С2-С6 галоалкенила, С2-С6 алкенила, замещенного OR6, С2-С6 алкинила, С2-С6 галоалкинила, С2-С6 алкинила, замещенного OR6, С3-С6 циклоалкила, и от 3- до 6-членного гетероциклического кольца, включая 1-2 кольцевых члена, выбранных из группы, состоящей из О, S и N(R6); R3 и R4 независимо выбраны из группы, состоящей из O-(C1-C4 алкилен или галоалкилен), С1-С5 алкилена или галоалкилена, и N(R6)-(C1-C4 алкилен или галоалкилен); необязательно, R4 может быть взят совместно с соседним C-R1 и атомом, к которому они присоединены, с образованием конденсированного кольца Е, которое выбрано из фенила или 5- или 6-членного гетероциклического кольца, включая 1-2 кольцевых члена, выбранных из группы, состоящей из О, S, N и N(R6), где связь с R3 от указанного кольца Е проходит от атома на указанном кольце Е с открытой валентностью для замещения, и где указанные фенил или гетероциклическое кольцо необязательно замещены одним или более членами группы, состоящей из галогена, C1-С3 алкила, и C1-С3 галоалкила; каждый R6 независимо выбран из группы, состоящей из Н, C1-C6 алкила, и C1-С6 галоалкила; каждый X1 независимо выбран из группы, состоящей из С=O, -СН2-, -CHF-, и -CF2-; каждый X2 независимо выбран из (C(R8)2)(1-3), где каждый R8 независимо выбран из группы, состоящей из Н, галогена, C1-С6 алкила, CN, OR6, N(R6)2, C1-С6 галоалкила, С3-С6 циклоалкила, C1-C6 алкила, замещенного OR6, и C1-C6 алкила, замещенного N(R6)2; необязательно, 2 R8 на разных атомах углерода могут быть взяты вместе совместно с атомами, к которым они присоединены, с образованием от 3- до 6-членного конденсированного кольца; и, необязательно, 2 R8 на одном атоме углерода могут быть взяты вместе совместно с атомом, к которому они присоединены, с образованием от 3- до 6-членного спироцикла; каждый X3 независимо выбран из группы, состоящей из COOR6, C(O)SR6, C(S)OR6,  SO2R6, C(O)N(R9)2, и CN; и каждый R9 независимо выбран из группы, состоящей из Н, COOR6, и SO2R6.
SO2R6, C(O)N(R9)2, и CN; и каждый R9 независимо выбран из группы, состоящей из Н, COOR6, и SO2R6.
В первом аспекте второго варианта осуществления каждый A-R1 независимо выбран из группы, состоящей из C-R1 и N. В конкретных примерах этого аспекта каждый 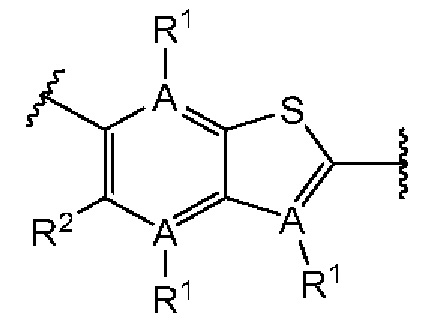 независимо выбран из группы, состоящей из
независимо выбран из группы, состоящей из 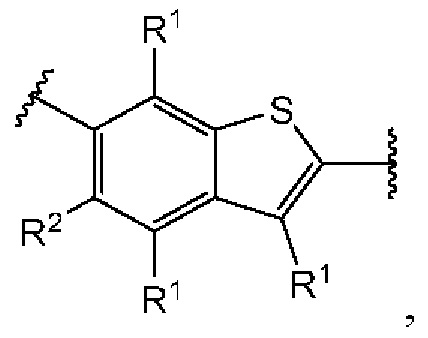
 и
и 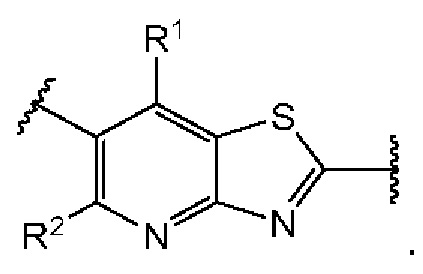 В более конкретных случаях этого аспекта каждый
В более конкретных случаях этого аспекта каждый 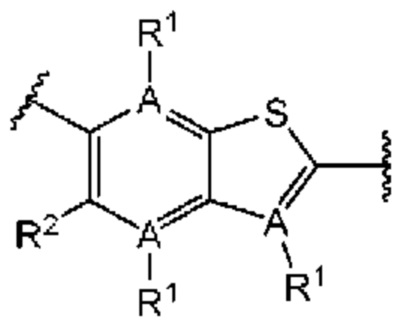 независимо выбран из группы, состоящей из
независимо выбран из группы, состоящей из 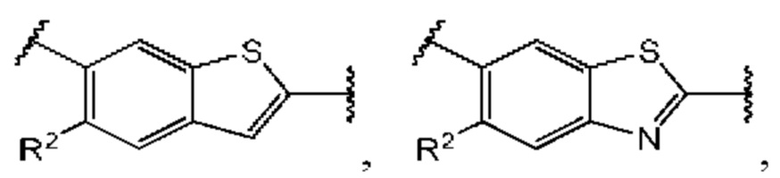
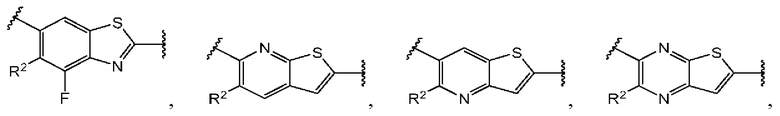 и
и 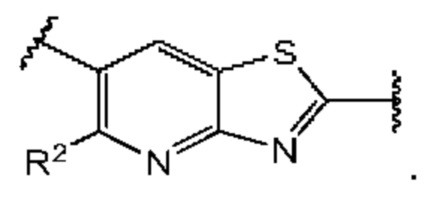 В этом аспекте все другие группы имеют значения, указанные в приведенной выше общей формуле (II) второго варианта осуществления.
В этом аспекте все другие группы имеют значения, указанные в приведенной выше общей формуле (II) второго варианта осуществления.
Во втором аспекте второго варианта осуществления каждый R1 независимо выбран из группы, состоящей из Н, галогена, OR6, N(R6)2, C1-C6 алкила, C1-C6 галоалкила, C1-C6 алкила, замещенного OR6, C1-С6 алкила, замещенного N(R6)2, COOR6, и C(O)N(R6)2. В примерах этого аспекта каждый R1 независимо выбран из группы, состоящей из Н, галогена, C1-С3 алкила и C1-С3 галоалкила. В конкретных примерах этого аспекта каждый R1 независимо выбран из группы, состоящей из Н и галогена. В более конкретных случаях этого аспекта каждый R1 независимо выбран из группы, состоящей из Н и F. В этом аспекте все другие группы имеют значения, указанные в приведенной выше общей формуле (II) второго варианта осуществления или в первом аспекте.
В третьем аспекте второго варианта осуществления каждый R2 независимо выбран из группы, состоящей из Н, галогена, CN, OR6, N(R6)2, COOR6, C(O)N(R6)2, SO2R6, C1-C6 алкила, C1-С6 галоалкила, C1-C6 алкила, замещенного OR6, С2-С6 алкенила, С2-С6 галоалкенила, С2-С6 алкенила, замещенного OR6, С2-С6 алкинила, С2-С6 галоалкинила, С2-С6 алкинила, замещенного OR6, С3-С6 циклоалкила, и от 3- до 6-членного гетероциклического кольца, включая 1-2 кольцевых члена, выбранных из группы, состоящей из О, S и N(R6). В примерах этого аспекта каждый R2 независимо выбран из группы, состоящей из Н, галогена, С1-С3 алкила, С1-С3 галоалкила, ОС1-С3 алкила, С2-С3 алкенила и N(R6)2. В конкретных примерах этого аспекта каждый R2 независимо выбран из группы, состоящей из Н, Br, Cl, СН3, СН2СН3, СН=СН2, ОСН3, OCFH2, OCF2H, OCF3, и N(R6)2. В более конкретных случаях этого аспекта каждый R2 независимо выбран из группы, состоящей из Н, СН3, ОСН3, и OCF2H. В этом аспекте все другие группы имеют значения, указанные в общей формуле (II) второго варианта осуществления или в первом или втором аспектах, описанных выше.
В четвертом аспекте второго варианта осуществления R3 и R4 независимо выбраны из группы, состоящей из O-(C1-C4 алкилен или галоалкилен), С1-С5 алкилена или галоалкилена, и N(R6)-(C1-C4 алкилен или галоалкилен); необязательно, R4 может быть взят совместно с соседним C-R1 и атомом, к которому они присоединены, с образованием конденсированного кольца Е, которое выбрано из фенила или 5- или 6-членного гетероциклического кольца, включая 1-2 кольцевых члена, выбранных из группы, состоящей из О, S, N и N(R6), где связь с R3 от указанного кольца Е проходит от атома на указанном кольце Е с открытой валентностью для замещения, и где указанные фенил или гетероциклическое кольцо необязательно замещены одним или более членами группы, состоящей из галогена, С1-С3 алкила, и С1-С3 галоалкила. В примерах этого четвертого аспекта R3-R4 выбран из группы, состоящей из -(CH2)2-8-, -O(CH2)1-7-, -O(CH2)1-6O-, -NH(CH2)1-7-, и -NH(CH2)1-6O-. В отдельных случаях этого четвертого аспекта R3-R4 выбран из группы, состоящей из -(СН2)2-, -(СН2)3-, -(СН2)4-, -O(СН2)2-, -O(СН2)3-, -O(СН2)4-, -O(СН2)2O-, -O(СН2)3O-, -ОСН2СН(СН3)CH2O-, -O(CH2)4O-, -O(CH2)5O-, -NH(CH2)2-, -NH(CH2)3-, и -NH(CH2)3O-. В конкретных случаях этого четвертого аспекта R4 может быть взят совместно с соседним C-R1 и атомом, к которому они присоединены, с образованием конденсированного кольца Е, которое выбрано из фенила или 5- или 6-членного гетероциклического кольца, включая 1-2 кольцевых члена, выбранных из группы, состоящей из О, S, N и N(R6), где связь с R3 от указанного кольца Е проходит от атома на указанном кольце Е с открытой валентностью для замещения, и где указанные фенил или гетероциклическое кольцо необязательно замещены одним или более членами группы, состоящей из галогена, C1-С3 алкила и C1-С3 галоалкила. В этом случае структура общей формулы (II) представляет собой формулу (IIa):
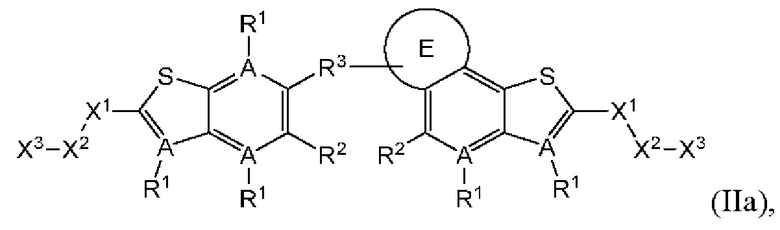
где все группы имеют значения, указанные в общей формуле (II). В этом аспекте все другие группы имеют значения, указанные в общей формуле (II) второго варианта осуществления или в первом-третьем аспектах, описанных выше.
В пятом аспекте второго варианта осуществления каждый R6 независимо выбран из группы, состоящей из Н, C1-С6 алкила и C1-С6 галоалкила. В примерах этого аспекта каждый R6 независимо выбран из группы, состоящей из Н, C1-С3 алкила, и С1-С3 галоалкила. В конкретных примерах этого аспекта каждый R6 независимо выбран из группы, состоящей из Н, СН3, и CHF2. В этом аспекте все другие группы имеют значения, указанные в общей формуле (II) второго варианта осуществления или в первом-четвертом аспектах, описанных выше.
В шестом аспекте второго варианта осуществления каждый X1 независимо выбран из группы, состоящей из С=O, -CH2-, -CHF-, и -CF2-. В примерах этого аспекта X1 выбран из группы, состоящей из С=O и -СН2-. В конкретных примерах этого аспекта X1 представляет собой С=O. В этом варианте осуществления все другие группы имеют значения, указанные в общей формуле (II) второго варианта осуществления или в первом-пятом аспектах, описанных выше.
В седьмом аспекте второго варианта осуществления каждый X2 независимо выбран из (C(R8)2)(1-3), где каждый R8 независимо выбран из группы, состоящей из Н, галогена, C1-C6 алкила, CN, OR6, N(R6)2, C1-С6 галоалкила, С3-С6 циклоалкила, C1-C6 алкила, замещенного OR6, и C1-С6 алкила, замещенного N(R6)2; необязательно, 2 R8 на разных атомах углерода могут быть взяты вместе совместно с атомами, к которым они присоединены, с образованием от 3- до 6-членного конденсированного кольца; и, необязательно, 2 R8 на одном атоме углерода могут быть взяты вместе совместно с атомом, к которому они присоединены, с образованием от 3- до 6-членного спироцикла. В первом случае этого аспекта каждый X2 представляет собой CH2CHR8, где R8 выбран из группы, состоящей из Н, C1-С3 алкила, C1-С3 алкила, замещенного ОН, C1-С3 алкила, замещенного ОС1-С3 алкилом, и С3-С6 циклоалкила. В отдельных примерах этого первого случая каждый X2 представляет собой CH2CHR8, где R8 выбран из группы, состоящей из Н, СН3, СН2ОН, СН2СН3, СН2СН2СН3, СН(СН3)2, СН2ОСН3, и циклопропила. Во втором случае этого аспекта каждый X2 представляет собой CHR8CHR8, где каждый R8 независимо выбран из группы, состоящей из Н, С1-С3 алкила, С1-С3 алкила, замещенного ОН, С1-С3 алкила, замещенного ОС1-С3 алкилом, и С3-С6 циклоалкила, и, необязательно, 2 R8 на разных атомах углерода взяты вместе совместно с атомами, к которым они присоединены, с образованием от 3- до 6-членного конденсированного кольца. В отдельных примерах этого второго случая каждый X2 представляет собой CHR8CHR8, где каждый R8 независимо выбран из группы, состоящей из Н и C1-С3 алкила, и, необязательно, 2 R8 на разных атомах углерода взяты вместе совместно с атомами, к которым они присоединены, с образованием от 3- до 6-членного конденсированного кольца. В третьем случае этого аспекта каждый X2 представляет собой CH2C(R8)2, где каждый R8 независимо выбран из группы, состоящей из Н, C1-С3 алкила, C1-С3 алкила, замещенного ОН, C1-С3 алкила, замещенного ОС1-С3 алкилом, и С3-С6 циклоалкила, и, необязательно, 2 R8 на одном атоме углерода взяты вместе совместно с атомами, к которым они присоединены, с образованием от 3- до 6-членного спироцикла. В отдельных примерах этого третьего случая каждый X2 представляет собой CH2C(R8)2, где каждый R8 независимо выбран из группы, состоящей из Н и C1-С3 алкила, и, необязательно, 2 R8 на одном атоме углерода взяты вместе совместно с атомами, к которым они присоединены, с образованием от 3- до 6-членного спироцикла. В этом аспекте все другие группы имеют значения, указанные в общей формуле (II) второго варианта осуществления или в первом-шестом аспектах, описанных выше.
В восьмом аспекте второго варианта осуществления каждый X3 независимо выбран из группы, состоящей из COOR6, C(O)SR6, C(S)OR6,  SO2R6, C(O)N(R9)2, и CN. В примерах этого аспекта каждый X3 независимо выбран из группы, состоящей из COOR6,
SO2R6, C(O)N(R9)2, и CN. В примерах этого аспекта каждый X3 независимо выбран из группы, состоящей из COOR6,  SO2R6, C(O)N(R9)2, и CN. В конкретных примерах этого аспекта каждый X3 независимо выбран из группы, состоящей из COOR6, C(O)N(R9)2, и CN. В даже более конкретных случаях этого аспекта каждый X3 независимо выбран из группы, состоящей из СООН, СООСН3, CONH2, и CN. В этом аспекте все другие группы имеют значения, указанные в общей формуле (II) второго варианта осуществления или в первом-седьмом аспектах, описанных выше.
SO2R6, C(O)N(R9)2, и CN. В конкретных примерах этого аспекта каждый X3 независимо выбран из группы, состоящей из COOR6, C(O)N(R9)2, и CN. В даже более конкретных случаях этого аспекта каждый X3 независимо выбран из группы, состоящей из СООН, СООСН3, CONH2, и CN. В этом аспекте все другие группы имеют значения, указанные в общей формуле (II) второго варианта осуществления или в первом-седьмом аспектах, описанных выше.
В девятом аспекте второго варианта осуществления каждый R9 независимо выбран из группы, состоящей из Н, COOR6, и SO2R6. В примерах этого аспекта каждый R9 независимо представляет собой Н. В этом аспекте все другие группы имеют значения, указанные в общей формуле (II) второго варианта осуществления или в первом-восьмом аспектах, описанных выше.
Десятый аспект второго варианта осуществления относится к фармацевтической композиции, где указанная фармацевтическая композиция содержит (а) соединение согласно вышеуказанной общей формуле (II) второго варианта осуществления или первому-девятому аспектам, описанным выше, или его фармацевтически приемлемую соль; и (b) фармацевтически приемлемый носитель.
Одиннадцатый аспект второго варианта осуществления относится к способам индукции иммунного ответа у пациента, нуждающегося в терапии, включающим введение этому пациенту терапевтически эффективного количества соединения согласно вышеуказанной общей формуле (II) второго варианта осуществления или первому-девятому аспектам, описанным выше, или его фармацевтически приемлемой соли.
Двенадцатый аспект второго варианта осуществления относится к способам индукции иммунного ответа у пациента, нуждающегося в терапии, включающим введение этому пациенту терапевтически эффективного количества композиции согласно десятому аспекту, описанному выше.
Тринадцатый аспект второго варианта осуществления относится к способам индукции STING-зависимой продукции интерферона I типа у пациента, нуждающегося в терапии, включающим введение этому пациенту терапевтически эффективного количества соединения согласно вышеуказанной общей формуле (II) второго варианта осуществления или первому-девятому аспектам, описанным выше, или его фармацевтически приемлемой соли.
Четырнадцатый аспект второго варианта осуществления относится к способам индукции STING-зависимой продукции интерферона I типа у пациента, нуждающегося в терапии, включающим введение этому пациенту терапевтически эффективного количества композиции согласно десятому аспекту, описанному выше.
Пятнадцатый аспект второго варианта осуществления относится к способам индукции STING-зависимой продукции цитокинов у пациента, нуждающегося в терапии, включающим введение этому пациенту терапевтически эффективного количества соединения согласно вышеуказанной общей формуле (II) второго варианта осуществления или первому-девятому аспектам, описанным выше, или его фармацевтически приемлемой соли.
Шестнадцатый аспект второго варианта осуществления относится к способам индукции STING-зависимой продукции цитокинов у пациента, нуждающегося в терапии, включающим введение этому пациенту терапевтически эффективного количества композиции согласно десятому аспекту, описанному выше.
Семнадцатый аспект второго варианта осуществления относится к способам лечения нарушения пролиферации клеток у пациента, нуждающегося в терапии, включающим введение этому пациенту терапевтически эффективного количества соединения общей формулы (II) или его фармацевтически приемлемой соли. В случаях этого семнадцатого аспекта второго варианта осуществления нарушение пролиферации клеток представляет собой рак.
Восемнадцатый аспект второго варианта осуществления относится к способам лечения нарушения пролиферации клеток у пациента, нуждающегося в терапии, при этом указанный способ включает введение пациенту терапевтически эффективного количества композиции в соответствии с одиннадцатым аспектом, описанным выше. В случаях этого восемнадцатого аспекта второго варианта осуществления нарушение пролиферации клеток представляет собой рак.
В каждом аспекте второго варианта осуществления, описанного в настоящем документе, переменные R1, R2, R3, R4, R6, R8, R9, А, X1, X2, и X3 общей формулы (II) второго варианта осуществления и его различных аспектов и случаев, каждый независимо выбран из каждого, при условии, что, по меньшей мере, один из R1, R2, R3, R4, R6, R8, и R9 не представляет собой Н.
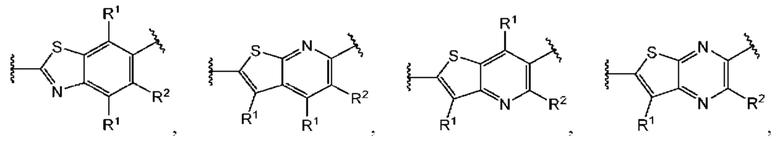
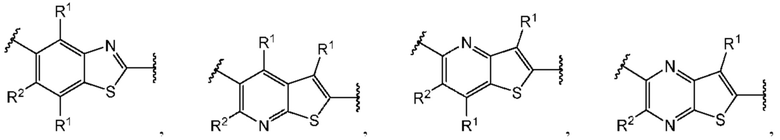
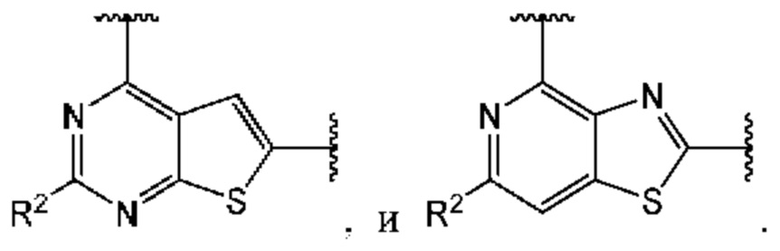
Третий вариант осуществления относится к соединениям общей формулы (III):
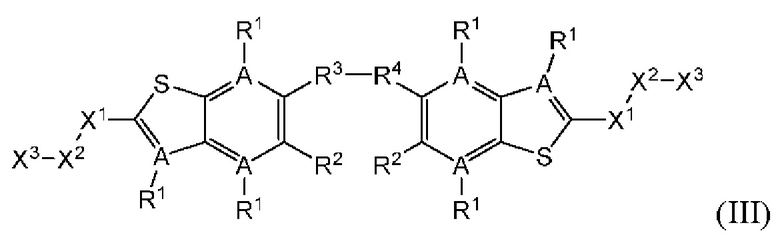
или их фармацевтически приемлемой соли, где каждый A-R1 независимо выбран из группы, состоящей из C-R1 и N; каждый R1 независимо выбран из группы, состоящей из Н, галогена, OR6, N(R6)2, C1-C6 алкила, C1-С6 галоалкила, C1-С6 алкила, замещенного OR6, C1-С6 алкила, замещенного N(R6)2, COOR6, и C(O)N(R6)2; каждый R2 независимо выбран из группы, состоящей из Н, галогена, CN, OR6, N(R6)2, COOR6, C(O)N(R6)2, SO2R6, C1-C6 алкила, C1-С6 галоалкила, C1-C6 алкила, замещенного OR6, С2-С6 алкенила, С2-С6 галоалкенила, С2-С6 алкенила, замещенного OR6, С2-С6 алкинила, С2-С6 галоалкинила, С2-С6 алкинила, замещенного OR6, С3-С6 циклоалкила, и от 3- до 6-членного гетероциклического кольца, включая 1-2 кольцевых члена, выбранных из группы, состоящей из О, S и N(R6); R3 и R4 независимо выбраны из группы, состоящей из O-(C1-C4 алкилен или галоалкилен), С1-С5 алкилена или галоалкилена, и N(R6)-(C1-C4 алкилен или галоалкилен); необязательно, R3 может быть взят совместно с соседним C-R1 и атомом, к которому они присоединены, с образованием конденсированного кольца G, которое выбрано из фенила или 5- или 6-членного гетероциклического кольца, включая 1-2 кольцевых члена, выбранных из группы, состоящей из О, S, N и N(R6), где связь с R3 от указанного кольца G проходит от атома на указанном кольце G с открытой валентностью для замещения, и где указанные фенил или гетероциклическое кольцо необязательно замещены одним или более членами группы, состоящей из галогена, С1-С3 алкила, и С1-С3 галоалкила; необязательно, R4 может быть взят совместно с соседним C-R1 и атомом, к которому они присоединены, с образованием конденсированного кольца Е, которое выбрано из фенила или 5- или 6-членного гетероциклического кольца, включая 1-2 кольцевых члена, выбранных из группы, состоящей из О, S, N и N(R6), где связь с R4 от указанного кольца Е проходит от атома на указанном кольце Е с открытой валентностью для замещения, и где указанные фенил или гетероциклическое кольцо необязательно замещены одним или более членами группы, состоящей из галогена, С1-С3 алкила, и С1-С3 галоалкила; каждый R6 независимо выбран из группы, состоящей из Н, C1-C6 алкила, и C1-C6 галоалкила; каждый X1 независимо выбран из группы, состоящей из С=O, -CH2-, -CHF-, и -CF2-; каждый X2 независимо выбран из (C(R8)2)(1-3), где каждый R8 независимо выбран из группы, состоящей из Н, галогена, C1-C6 алкила, CN, OR6, N(R6)2, C1-C6 галоалкила, С3-С6 циклоалкила, C1-C6 алкила, замещенного OR6, и C1-C6 алкила, замещенного N(R6)2; необязательно, 2 R8 на разных атомах углерода могут быть взяты вместе совместно с атомами, к которым они присоединены, с образованием от 3- до 6-членного конденсированного кольца; и, необязательно, 2 R8 на одном атоме углерода могут быть взяты вместе совместно с атомом, к которому они присоединены, с образованием от 3- до 6-членного спироцикла; каждый X3 независимо выбран из группы, состоящей из COOR6, C(O)SR6, C(S)OR6,  SO2R6, C(O)N(R9)2, и CN; и каждый R9 независимо выбран из группы, состоящей из Н, COOR6, и SO2R6.
SO2R6, C(O)N(R9)2, и CN; и каждый R9 независимо выбран из группы, состоящей из Н, COOR6, и SO2R6.
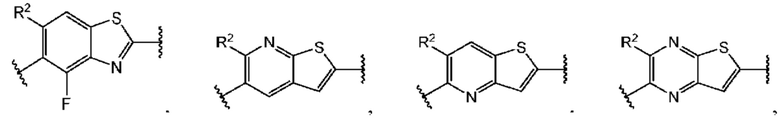
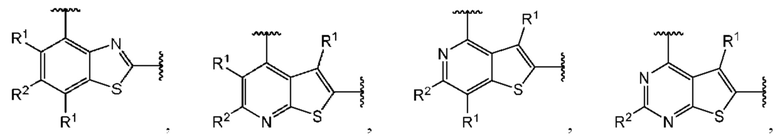
В первом аспекте третьего варианта осуществления каждый A-R1 независимо выбран из группы, состоящей из C-R1 и N. В конкретных примерах этого аспекта каждый  независимо выбран из группы, состоящей из
независимо выбран из группы, состоящей из 
 и
и 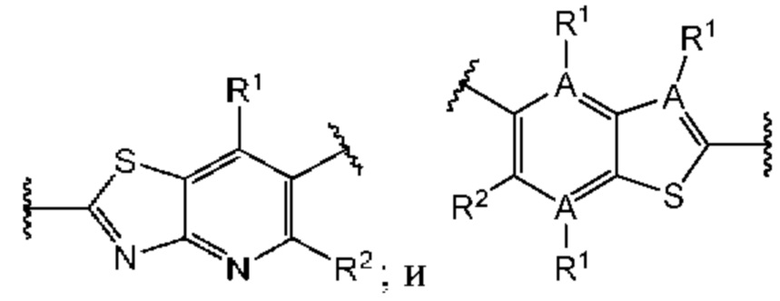 выбран из группы, состоящей из
выбран из группы, состоящей из 
 и
и 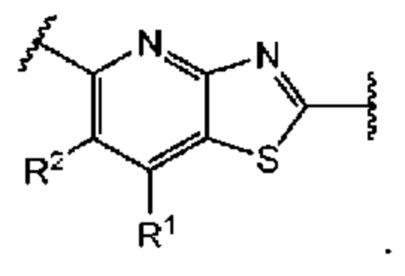 В более конкретных случаях этого аспекта каждый
В более конкретных случаях этого аспекта каждый 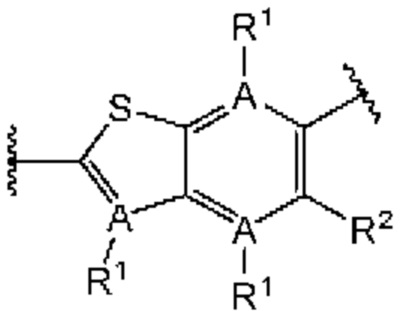 независимо выбран из группы, состоящей из
независимо выбран из группы, состоящей из 
 и
и 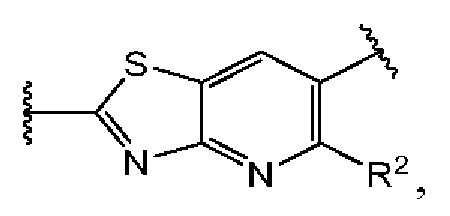 и каждый
и каждый 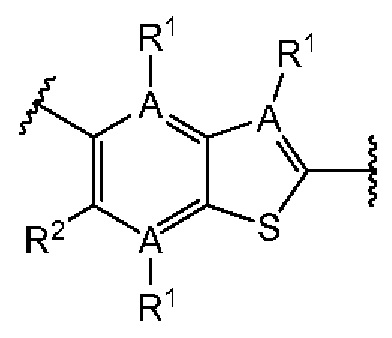 выбран из группы, состоящей из
выбран из группы, состоящей из 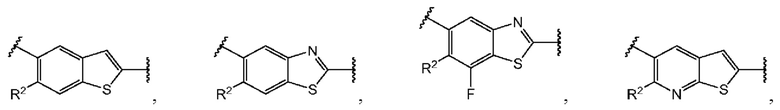
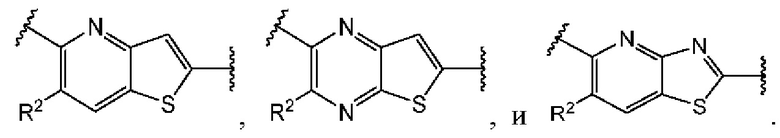 В этом аспекте все остальные группы соответствуют вышеуказанной общей формуле (III) третьего варианта осуществления.
В этом аспекте все остальные группы соответствуют вышеуказанной общей формуле (III) третьего варианта осуществления.
Во втором аспекте третьего варианта осуществления каждый R1 независимо выбран из группы, состоящей из Н, галогена, OR6, N(R6)2, C1-C6 алкила, C1-C6 галоалкила, C1-C6 алкила, замещенного OR6, C1-C6 алкила, замещенного N(R6)2, COOR6, и C(O)N(R6)2. В примерах этого аспекта каждый R1 независимо выбран из группы, состоящей из Н, галогена, C1-С3 алкила, и C1-С3 галоалкила. В конкретных примерах этого аспекта каждый R1 независимо выбран из группы, состоящей из Н и галогена. В более конкретных случаях этого аспекта каждый R1 независимо выбран из группы, состоящей из Н и F. В этом аспекте все остальные группы соответствуют вышеуказанной общей формуле (III) третьего варианта осуществления или первому аспекту выше.
В третьем аспекте третьего варианта осуществления каждый R2 независимо выбран из группы, состоящей из Н, галогена, CN, OR6, N(R6)2, COOR6, C(O)N(R6)2, SO2R6, C1-C6 алкила, C1-С6 галоалкила, C1-C6 алкила, замещенного OR6, С2-С6 алкенила, С2-С6 галоалкенила, С2-С6 алкенила, замещенного OR6, С2-С6 алкинила, С2-С6 галоалкинила, С2-С6 алкинила, замещенного OR6, С3-С6 циклоалкила, и от 3- до 6-членного гетероциклического кольца, включая 1-2 кольцевых члена, выбранных из группы, состоящей из О, S и N(R6). В примерах этого аспекта каждый R2 независимо выбран из группы, состоящей из Н, галогена, C1-С3 алкила, С1-С3 галоалкила, ОС1-С3 алкила, С2-С3 алкенила, и N(R6)2. В конкретных примерах этого аспекта каждый R2 независимо выбран из группы, состоящей из Н, Br, Cl, СН3, СН2СН3, СН=СН2, ОСН3, OCFH2, OCF2H, OCF3, и N(R6)2. В более конкретных случаях этого аспекта каждый R2 независимо выбран из группы, состоящей из Н, СН3, ОСН3, и OCF2H. В этом аспекте все остальные группы являются такими, как указано в общей формуле (III) третьего варианта осуществления или в первом или втором аспектах, описанных выше.
В четвертом аспекте третьего варианта осуществления R3 и R4 независимо выбраны из группы, состоящей из O-(C1-C4 алкилен или галоалкилен), С1-С5 алкилена или галоалкилена, и N(R6)-(C1-C4 алкилен или галоалкилен); необязательно, R3 может быть взят совместно с соседним C-R1 и атомом, к которому они присоединены, с образованием конденсированного кольца G, которое выбрано из фенила или 5- или 6-членного гетероциклического кольца, включая 1-2 кольцевых члена, выбранных из группы, состоящей из О, S, N и N(R6), где связь с R3 от указанного кольца G проходит от атома на указанном кольце G с открытой валентностью для замещения, и где указанные фенил или гетероциклическое кольцо необязательно замещены одним или более членами группы, состоящей из галогена, C1-С3 алкила, и C1-С3 галоалкила; необязательно, R4 может быть взят совместно с соседним C-R1 и атомом, к которому они присоединены, с образованием конденсированного кольца Е, которое выбрано из фенила или 5- или 6-членного гетероциклического кольца, включая 1-2 кольцевых члена, выбранных из группы, состоящей из О, S, N и N(R6), где связь с R4 от указанного кольца Е проходит от атома на указанном кольце Е с открытой валентностью для замещения, и где указанные фенил или гетероциклическое кольцо необязательно замещены одним или более членами группы, состоящей из галогена, C1-С3 алкила, и C1-С3 галоалкила. В примерах этого четвертого аспекта R3-R4 выбран из группы, состоящей из -(CH2)2-8-, -O(CH2)1-7-, -O(CH2)1-6O-, -NH(CH2)1-7-, и -NH(CH2)1-6O-. В отдельных случаях этого четвертого аспекта R3-R4 выбран из группы, состоящей из -(СН2)2-, -(СН2)3-, -(СН2)4-, -O(СН2)2-, -O(СН2)3-, -O(СН2)4-, -O(СН2)2O-, -O(СН2)3О-, -ОСН2СН(СН3)CH2O-, -O(CH2)4O-, -O(CH2)5O-, -NH(CH2)2-, -NH(CH2)3-, и -NH(CH2)3O-. В конкретных случаях этого четвертого аспекта R3 может быть взят совместно с соседним C-R1 и атомом, к которому они присоединены, с образованием конденсированного кольца G, которое выбрано из фенила или 5- или 6-членного гетероциклического кольца, включая 1-2 кольцевых члена, выбранных из группы, состоящей из О, S, N и N(R6), где связь с R4 от указанного кольца G проходит от атома на указанном кольце Е с открытой валентностью для замещения, и где указанные фенил или гетероциклическое кольцо необязательно замещены одним или более членами группы, состоящей из галогена, С1-С3 алкила, и С1-С3 галоалкила. В этом случае структура общей формулы (III) представляет собой формулу (IIIa):
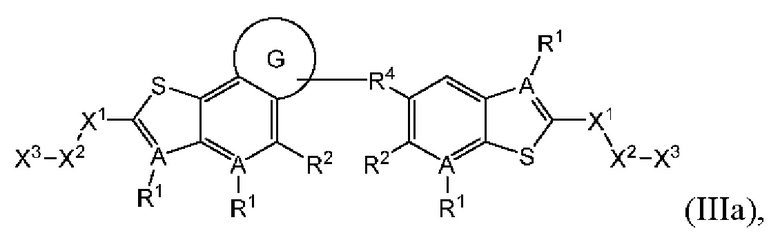
где все группы имеют значения, указанные в общей формуле (III). В дополнительных конкретных случаях этого четвертого аспекта R4 может быть взят совместно с соседним С-R1 и атомом, к которому они присоединены, с образованием конденсированного кольца Е, которое выбрано из фенила или 5- или 6-членного гетероциклического кольца, включая 1-2 кольцевых члена, выбранных из группы, состоящей из О, S, N и N(R6), где связь с R3 от указанного кольца Е проходит от атома на указанном кольце Е с открытой валентностью для замещения, и где указанные фенил или гетероциклическое кольцо необязательно замещены одним или более членами группы, состоящей из галогена, C1-С3 алкила, и C1-С3 галоалкила. В этом случае структура общей формулы (III) представляет собой формулу (IIIb):
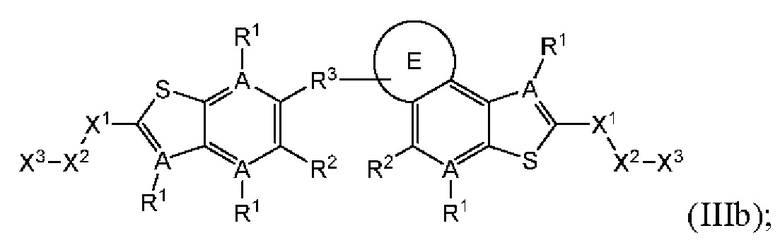
где все группы имеют значения, указанные в общей формуле (III). В этом аспекте все остальные группы являются такими, как указано в общей формуле (III) первого варианта осуществления или в первом-третьем аспектах, описанных выше.
В пятом аспекте третьего варианта осуществления каждый R6 независимо выбран из группы, состоящей из Н, C1-С6 алкила, и C1-C6 галоалкила. В примерах этого аспекта каждый R6 независимо выбран из группы, состоящей из Н, C1-С3 алкила, и C1-С3 галоалкила. В конкретных примерах этого аспекта каждый R6 независимо выбран из группы, состоящей из Н, СН3, и CHF2. В этом аспекте все остальные группы являются такими, как указано в общей формуле (III) третьего варианта осуществления или в первом-четвертом аспектах, описанных выше.
В шестом аспекте третьего варианта осуществления каждый X1 независимо выбран из группы, состоящей из С=O, -CH2-, -CHF-, и -CF2-. В примерах этого аспекта X1 выбран из группы, состоящей из С=O и -СН2-. В конкретных примерах этого аспекта X1 представляет собой С=O. В этом варианте осуществления все остальные группы являются такими, как указано в общей формуле (III) третьего варианта осуществления или в первом-пятом аспектах, описанных выше.
В седьмом аспекте третьего варианта осуществления каждый X2 независимо выбран из (C(R8)2)(1-3), где каждый R8 независимо выбран из группы, состоящей из Н, галогена, C1-C6 алкила, CN, OR6, N(R6)2, C1-С6 галоалкила, С3-С6 циклоалкила, C1-C6 алкила, замещенного OR6, и C1-С6 алкила, замещенного N(R6)2; необязательно, 2 R8 на разных атомах углерода могут быть взяты вместе совместно с атомами, к которым они присоединены, с образованием от 3- до 6-членного конденсированного кольца; и, необязательно, 2 R8 на одном атоме углерода могут быть взяты вместе совместно с атомом, к которому они присоединены, с образованием от 3- до 6-членного спироцикла. В первом случае этого аспекта каждый X2 представляет собой CH2CHR8, где R8 выбран из группы, состоящей из Н, C1-С3 алкила, C1-С3 алкила, замещенного ОН, C1-С3 алкила, замещенного ОС1-С3 алкилом, и С3-С6 циклоалкила. В отдельных примерах этого первого случая каждый X2 представляет собой CH2CHR8, где R8 выбран из группы, состоящей из Н, СН3, СН2ОН, СН2СН3, СН2СН2СН3, СН(СН3)2, СН2ОСН3, и циклопропила. Во втором случае этого аспекта каждый X2 представляет собой CHR8CHR8, где каждый R8 независимо выбран из группы, состоящей из Н, C1-С3 алкила, C1-С3 алкила, замещенного ОН, C1-С3 алкила, замещенного ОС1-С3 алкилом, и С3-С6 циклоалкила, и, необязательно, 2 R8 на разных атомах углерода взяты вместе совместно с атомами, к которым они присоединены, с образованием от 3- до 6-членного конденсированного кольца. В отдельных примерах этого второго случая каждый X2 представляет собой CHR8CHR8, где каждый R8 независимо выбран из группы, состоящей из Н и C1-С3 алкила, и, необязательно, 2 R8 на разных атомах углерода взяты вместе совместно с атомами, к которым они присоединены, с образованием от 3- до 6-членного конденсированного кольца. В третьем случае этого аспекта каждый X2 представляет собой CH2C(R8)2, где каждый R8 независимо выбран из группы, состоящей из Н, C1-С3 алкила, C1-С3 алкила, замещенного ОН, C1-С3 алкила, замещенного ОС1-С3 алкилом, и С3-С6 циклоалкила, и, необязательно, 2 R8 на одном атоме углерода взяты вместе совместно с атомами, к которым они присоединены, с образованием от 3- до 6-членного спироцикла. В отдельных примерах этого третьего случая каждый X2 представляет собой CH2C(R8)2, где каждый R8 независимо выбран из группы, состоящей из Н и C1-С3 алкила, и, необязательно, 2 R8 на одном атоме углерода взяты вместе совместно с атомами, к которым они присоединены, с образованием от 3- до 6-членного спироцикла. В этом аспекте все остальные группы являются такими, как указано в общей формуле (III) третьего варианта осуществления или в первом-шестом аспектах, описанных выше.
В восьмом аспекте третьего варианта осуществления каждый X3 независимо выбран из группы, состоящей из COOR6, C(O)SR6, C(S)OR6,  SO2R6, C(O)N(R9)2, и CN. В примерах этого аспекта каждый X3 независимо выбран из группы, состоящей из COOR6,
SO2R6, C(O)N(R9)2, и CN. В примерах этого аспекта каждый X3 независимо выбран из группы, состоящей из COOR6,  SO2R6, C(O)N(R9)2, и CN. В конкретных примерах этого аспекта каждый X3 независимо выбран из группы, состоящей из COOR6, C(O)N(R9)2, и CN. В даже более конкретных случаях этого аспекта каждый X3 независимо выбран из группы, состоящей из СООН, СООСН3, CONH2, и CN. В этом аспекте все остальные группы являются такими, как указано в общей формуле (III) третьего варианта осуществления или в первом-седьмом аспектах, описанных выше.
SO2R6, C(O)N(R9)2, и CN. В конкретных примерах этого аспекта каждый X3 независимо выбран из группы, состоящей из COOR6, C(O)N(R9)2, и CN. В даже более конкретных случаях этого аспекта каждый X3 независимо выбран из группы, состоящей из СООН, СООСН3, CONH2, и CN. В этом аспекте все остальные группы являются такими, как указано в общей формуле (III) третьего варианта осуществления или в первом-седьмом аспектах, описанных выше.
В девятом аспекте третьего варианта осуществления каждый R9 независимо выбран из группы, состоящей из Н, COOR6, и SO2R6. В примерах этого аспекта каждый R9 независимо представляет собой Н. В этом аспекте все остальные группы являются такими, как указано в общей формуле (III) третьего варианта осуществления или в первом-шестом аспектах, описанных выше.
Десятый аспект третьего варианта осуществления относится к фармацевтической композиции, где указанная фармацевтическая композиция содержит (а) соединение согласно общей формуле (III) третьего варианта осуществления или первому-девятому аспектам, описанным выше, или его фармацевтически приемлемую соль; и (b) фармацевтически приемлемый носитель.
Одиннадцатый аспект третьего варианта осуществления относится к способам индукции иммунного ответа у пациента, нуждающегося в терапии, включающим введение этому пациенту терапевтически эффективного количества соединения согласно общей формуле (III) третьего варианта осуществления или первому-девятому аспектам, описанным выше, или его фармацевтически приемлемой соли.
Двенадцатый аспект третьего варианта осуществления относится к способам индукции иммунного ответа у пациента, нуждающегося в терапии, включающим введение этому пациенту терапевтически эффективного количества композиции согласно десятому аспекту, описанному выше.
Тринадцатый аспект третьего варианта относится к способам индукции STING-зависимой продукции интерферона I типа у пациента, нуждающегося в терапии, включающим введение этому пациенту терапевтически эффективного количества соединения согласно вышеуказанной общей формуле (III) третьего варианта осуществления или первому-девятому аспектам, описанным выше, или его фармацевтически приемлемой соли.
Четырнадцатый аспект третьего варианта осуществления относится к способам индукции STING-зависимой продукции интерферона I типа у пациента, нуждающегося в терапии, включающим введение этому пациенту терапевтически эффективного количества композиции согласно десятому аспекту, описанному выше.
Пятнадцатый аспект третьего варианта осуществления относится к способам индукции STING-зависимой продукции цитокинов у пациента, нуждающегося в терапии, включающим введение этому пациенту терапевтически эффективного количества соединения согласно вышеуказанной общей формуле (III) третьего варианта осуществления или первому-девятому аспектам, описанным выше, или его фармацевтически приемлемой соли.
Шестнадцатый аспект третьего варианта осуществления относится к способам индукции STING-зависимой продукции цитокинов у пациента, нуждающегося в терапии, включающим введение этому пациенту терапевтически эффективного количества композиции согласно десятому аспекту, описанному выше.
Семнадцатый аспект третьего варианта осуществления относится к способам лечения нарушения пролиферации клеток у пациента, нуждающегося в терапии, включающим введение этому пациенту терапевтически эффективного количества соединения общей формулы (III) или его фармацевтически приемлемой соли. В случаях этого семнадцатого аспекта третьего варианта осуществления нарушение пролиферации клеток представляет собой рак.
Восемнадцатый аспект третьего варианта осуществления относится к способам лечения нарушения пролиферации клеток у пациента, нуждающегося в терапии, при этом указанный способ включает введение пациенту терапевтически эффективного количества композиции в соответствии с одиннадцатым аспектом, описанным выше. В случаях этого восемнадцатого аспекта третьего варианта осуществления нарушение пролиферации клеток представляет собой рак.
В каждом аспекте третьего варианта осуществления, описанного в настоящем документе, переменные R1, R2, R3, R4, R6, R8, R9, А, X1, X2, и X3 общей формулы (III) третьего варианта осуществления и его различных аспектов и случаев, каждый независимо выбран из каждого, при условии, что, по меньшей мере, один из R1, R2, R3, R4, R6, R8, и R9 не представляет собой Н.
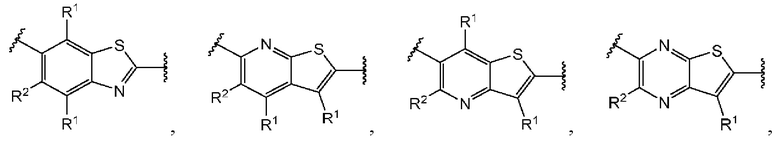
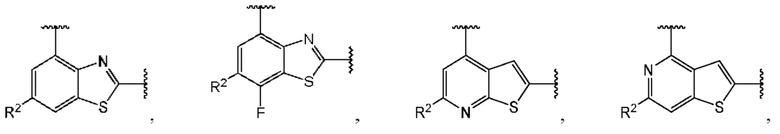
Четвертый вариант осуществления относится к соединениям общей формулы (IV):
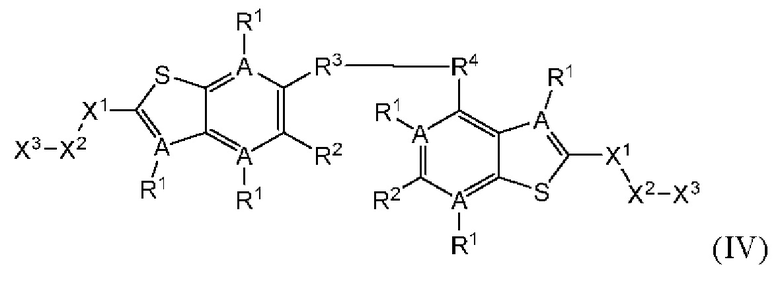
или их фармацевтически приемлемой соли, где каждый A-R1 независимо выбран из группы, состоящей из C-R1 и N; каждый R1 независимо выбран из группы, состоящей из Н, галогена, OR6, N(R6)2, C1-C6 алкила, C1-С6 галоалкила, C1-C6 алкила, замещенного OR6, C1-C6 алкила, замещенного N(R6)2, COOR6, и C(O)N(R6)2; каждый R2 независимо выбран из группы, состоящей из Н, галогена, CN, OR6, N(R6)2, COOR6, C(O)N(R6)2, SO2R6, C1-C6 алкила, C1-С6 галоалкила, C1-C6 алкила, замещенного OR6, С2-С6 алкенила, С2-С6 галоалкенила, С2-С6 алкенила, замещенного OR6, С2-С6 алкинила, С2-С6 галоалкинила, С2-С6 алкинила, замещенного OR6, С3-С6 циклоалкила, и от 3- до 6-членного гетероциклического кольца, включая 1-2 кольцевых члена, выбранных из группы, состоящей из О, S и N(R6); R3 и R4 независимо выбраны из группы, состоящей из O-(C1-C4 алкилен или галоалкилен), С1-С5 алкилена или галоалкилена, и N(R6)-(C1-C4 алкилен или галоалкилен); каждый R6 независимо выбран из группы, состоящей из Н, C1-C6 алкила, и C1-C6 галоалкила; каждый X1 независимо выбран из группы, состоящей из С=O, -СН2-, -CHF-, и -CF2-; каждый X2 независимо выбран из (C(R8)2)(1-3), где каждый R8 независимо выбран из группы, состоящей из Н, галогена, C1-С6 алкила, CN, OR6, N(R6)2, C1-C6 галоалкила, С3-С6 циклоалкила, C1-C6 алкила, замещенного OR6, и C1-C6 алкила, замещенного N(R6)2; необязательно, 2 R8 на разных атомах углерода могут быть взяты вместе совместно с атомами, к которым они присоединены, с образованием от 3- до 6-членного конденсированного кольца; и, необязательно, 2 R8 на одном атоме углерода могут быть взяты вместе, совместно с атомом, к которому они присоединены, с образованием от 3- до 6-членного спироцикла; каждый X3 независимо выбран из группы, состоящей из COOR6, C(O)SR6, C(S)OR6,  SO2R6, C(O)N(R9)2, и CN; и каждый R9 независимо выбран из группы, состоящей из Н, COOR6, и SO2R6.
SO2R6, C(O)N(R9)2, и CN; и каждый R9 независимо выбран из группы, состоящей из Н, COOR6, и SO2R6.
В первом аспекте четвертого варианта осуществления каждый A-R1 независимо выбран из группы, состоящей из C-R1 и N. В конкретных примерах этого аспекта каждый  независимо выбран из группы, состоящей из
независимо выбран из группы, состоящей из 
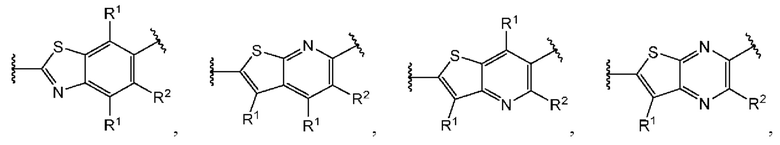 и
и 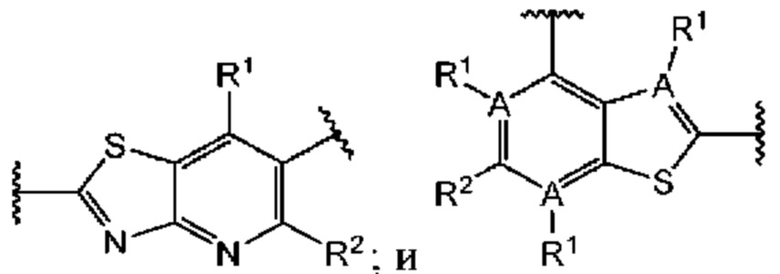 выбран из группы, состоящей из
выбран из группы, состоящей из 
 и
и 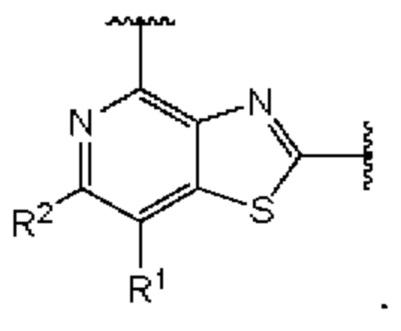 В более конкретных случаях этого аспекта каждый
В более конкретных случаях этого аспекта каждый 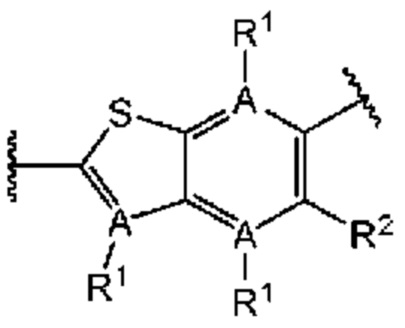 независимо выбран из группы, состоящей из
независимо выбран из группы, состоящей из 
 и
и 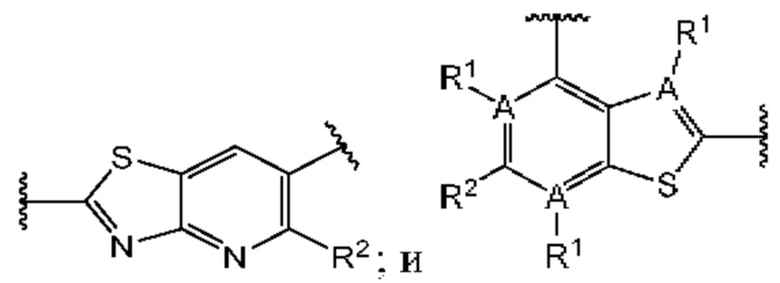 выбран из группы, состоящей из
выбран из группы, состоящей из 
 В этом аспекте все остальные группы являются такими, как указано в общей формуле (IV) четвертого варианта осуществления выше.
В этом аспекте все остальные группы являются такими, как указано в общей формуле (IV) четвертого варианта осуществления выше.
Во втором аспекте четвертого варианта осуществления каждый R1 независимо выбран из группы, состоящей из Н, галогена, OR6, N(R6)2, C1-C6 алкила, C1-C6 галоалкила, C1-C6 алкила, замещенного OR6, C1-С6 алкила, замещенного N(R6)2, COOR6, и C(O)N(R6)2. В примерах этого аспекта каждый R1 независимо выбран из группы, состоящей из Н, галогена, C1-С3 алкила, и C1-С3 галоалкила. В конкретных примерах этого аспекта каждый R1 независимо выбран из группы, состоящей из Н и галогена. В более конкретных случаях этого аспекта каждый R1 независимо выбран из группы, состоящей из Н и F. В этом аспекте все остальные группы являются такими, как указано в вышеприведенной общей формуле (IV) четвертого варианта осуществления или в первом аспекте выше.
В третьем аспекте четвертого варианта осуществления каждый R2 независимо выбран из группы, состоящей из Н, галогена, CN, OR6, N(R6)2, COOR6, C(O)N(R6)2, SO2R6, C1-C6 алкила, C1-С6 галоалкила, C1-C6 алкила, замещенного OR6, С2-С6 алкенила, С2-С6 галоалкенила, С2-С6 алкенила, замещенного OR6, С2-С6 алкинила, С2-С6 галоалкинила, С2-С6 алкинила, замещенного OR6, С3-С6 циклоалкила, и от 3- до 6-членного гетероциклического кольца, включая 1-2 кольцевых члена, выбранных из группы, состоящей из О, S и N(R6). В примерах этого аспекта каждый R2 независимо выбран из группы, состоящей из Н, галогена, C1-С3 алкила, C1-С3 галоалкила, ОС1-С3 алкила, С2-С3 алкенила, и N(R6)2. В конкретных примерах этого аспекта каждый R2 независимо выбран из группы, состоящей из Н, Br, Cl, СН3, СН2СН3, СН=СН2, ОСН3, OCFH2, OCF2H, OCF3, и N(R6)2. В более конкретных случаях этого аспекта каждый R2 независимо выбран из группы, состоящей из Н, СН3, ОСН3, и OCF2H. В этом аспекте все остальные группы являются такими, как указано в общей формуле (IV) четвертого варианта осуществления или в первом или втором аспектах, описанных выше.
В четвертом аспекте четвертого варианта осуществления R3 и R4 независимо выбраны из группы, состоящей из O-(C1-C4 алкилен или галоалкилен), С1-С5 алкилена или галоалкилена, и N(R6)-(C1-C4 алкилен или галоалкилен). В примерах этого четвертого аспекта R3-R4 выбран из группы, состоящей из -(CH2)2-8-, -O(CH2)1-7-, -O(CH2)1-6O-, -NH(CH2)1-7-, и -NH(CH2)1-6O-. В отдельных случаях этого четвертого аспекта R3-R4 выбран из группы, состоящей из -(СН2)2-, -(СН2)3-, -(СН2)4-, -O(СН2)2-, -O(СН2)3-, -O(СН2)4-, -O(СН2)2О-, -O(СН2)3О-, -ОСН2СН(СН3)СН2О-, -O(CH2)4O-, -O(CH2)5O-, -NH(CH2)2-, -NH(CH2)3-, и -NH(CH2)3O-. В этом аспекте все остальные группы являются такими, как указано в общей формуле (IV) четвертого варианта осуществления или в первом-третьем аспектах, описанных выше.
В пятом аспекте четвертого варианта осуществления каждый R6 независимо выбран из группы, состоящей из Н, C1-С6 алкила, и C1-C6 галоалкила. В примерах этого аспекта каждый R6 независимо выбран из группы, состоящей из Н, C1-С3 алкила, и C1-С3 галоалкила. В конкретных примерах этого аспекта каждый R6 независимо выбран из группы, состоящей из Н, СН3, и CHF2. В этом аспекте все остальные группы являются такими, как указано в общей формуле (IV) четвертого варианта осуществления или в первом-четвертом аспектах, описанных выше.
В шестом аспекте четвертого варианта осуществления каждый X1 независимо выбран из группы, состоящей из С=O, -CH2-, -CHF-, и -CF2-. В примерах этого аспекта X1 выбран из группы, состоящей из С=O и -СН2-. В конкретных примерах этого аспекта X1 представляет собой С=O. В этом варианте осуществления все остальные группы являются такими, как указано в общей формуле (IV) четвертого варианта осуществления или в первом-пятом аспектах, описанных выше.
В седьмом аспекте четвертого варианта осуществления каждый X2 независимо выбран из (C(R8)2)(1-3), где каждый R8 независимо выбран из группы, состоящей из Н, галогена, C1-C6 алкила, CN, OR6, N(R6)2, C1-С6 галоалкила, С3-С6 циклоалкила, C1-C6 алкила, замещенного OR6, и C1-С6 алкила, замещенного N(R6)2; необязательно, 2 R8 на разных атомах углерода могут быть взяты вместе совместно с атомами, к которым они присоединены, с образованием от 3- до 6-членного конденсированного кольца; и, необязательно, 2 R8 на одном атоме углерода могут быть взяты вместе совместно с атомом, к которому они присоединены, с образованием от 3- до 6-членного спироцикла. В первом случае этого аспекта каждый X2 представляет собой CH2CHR8, где R8 выбран из группы, состоящей из Н, C1-С3 алкила, C1-С3 алкила, замещенного ОН, C1-С3 алкила, замещенного ОС1-С3 алкилом, и С3-С6 циклоалкила. В отдельных примерах этого первого случая каждый X2 представляет собой CH2CHR8, где R8 выбран из группы, состоящей из Н, СН3, СН2ОН, СН2СН3, СН2СН2СН3, СН(СН3)2, СН2ОСН3, и циклопропила. Во втором случае этого аспекта каждый X2 представляет собой CHR8CHR8, где каждый R8 независимо выбран из группы, состоящей из Н, С1-С3 алкила, С1-С3 алкила, замещенного ОН, С1-С3 алкила, замещенного OC1-С3 алкилом, и С3-С6 циклоалкила, и, необязательно, 2 R8 на разных атомах углерода взяты вместе совместно с атомами, к которым они присоединены, с образованием от 3- до 6-членного конденсированного кольца. В отдельных примерах этого второго случая каждый X2 представляет собой CHR8CHR8, где каждый R8 независимо выбран из группы, состоящей из Н и C1-С3 алкила, и, необязательно, 2 R8 на разных атомах углерода взяты вместе совместно с атомами, к которым они присоединены, с образованием от 3- до 6-членного конденсированного кольца. В третьем случае этого аспекта каждый X2 представляет собой CH2C(R8)2, где каждый R8 независимо выбран из группы, состоящей из Н, C1-С3 алкила, C1-С3 алкила, замещенного ОН, C1-С3 алкила, замещенного ОС1-С3 алкилом, и С3-С6 циклоалкила, и, необязательно, 2 R8 на одном атоме углерода взяты вместе совместно с атомами, к которым они присоединены, с образованием от 3- до 6-членного спироцикла. В отдельных примерах этого третьего случая каждый X2 представляет собой CH2C(R8)2, где каждый R8 независимо выбран из группы, состоящей из Н и C1-С3 алкила, и, необязательно, 2 R8 на одном атоме углерода взяты вместе совместно с атомами, к которым они присоединены, с образованием от 3- до 6-членного спироцикла. В этом аспекте все остальные группы являются такими, как указано в общей формуле (IV) четвертого варианта осуществления или в первом-шестом аспектах, описанных выше.
В восьмом аспекте четвертого варианта осуществления каждый X3 независимо выбран из группы, состоящей из COOR6, C(O)SR6, C(S)OR6, 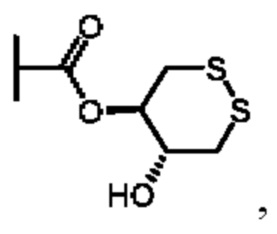 SO2R6, C(O)N(R9)2, и CN. В примерах этого аспекта каждый X3 независимо выбран из группы, состоящей из COOR6,
SO2R6, C(O)N(R9)2, и CN. В примерах этого аспекта каждый X3 независимо выбран из группы, состоящей из COOR6, 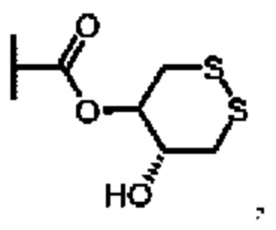 SO2R6, C(O)N(R9)2, и CN. В конкретных примерах этого аспекта каждый X3 независимо выбран из группы, состоящей из COOR6, C(O)N(R9)2, и CN. В даже более конкретных случаях этого аспекта каждый X3 независимо выбран из группы, состоящей из СООН, СООСН3, CONH2, и CN. В этом аспекте все остальные группы являются такими, как указано в общей формуле (IV) четвертого варианта осуществления или в первом-седьмом аспектах, описанных выше.
SO2R6, C(O)N(R9)2, и CN. В конкретных примерах этого аспекта каждый X3 независимо выбран из группы, состоящей из COOR6, C(O)N(R9)2, и CN. В даже более конкретных случаях этого аспекта каждый X3 независимо выбран из группы, состоящей из СООН, СООСН3, CONH2, и CN. В этом аспекте все остальные группы являются такими, как указано в общей формуле (IV) четвертого варианта осуществления или в первом-седьмом аспектах, описанных выше.
В девятом аспекте четвертого варианта осуществления каждый R9 независимо выбран из группы, состоящей из Н, COOR6, и SO2R6. В примерах этого аспекта каждый R9 независимо представляет собой Н. В этом аспекте все остальные группы являются такими, как указано в общей формуле (IV) четвертого варианта осуществления или в первом-восьмом аспектах, описанных выше.
Десятый аспект четвертого варианта осуществления относится к фармацевтической композиции, где указанная фармацевтическая композиция содержит (а) соединение согласно общей формуле (IV) четвертого варианта осуществления или первому-девятому аспектам, описанным выше, или его фармацевтически приемлемую соль; и (b) фармацевтически приемлемый носитель.
Одиннадцатый аспект четвертого варианта осуществления относится к способам индукции иммунного ответа у пациента, нуждающегося в терапии, включающим введение этому пациенту терапевтически эффективного количества соединения согласно общей формуле (IV) четвертого варианта осуществления или первому-девятому аспектам, описанным выше, или его фармацевтически приемлемой соли.
Двенадцатый аспект четвертого варианта осуществления относится к способам индукции иммунного ответа у пациента, нуждающегося в терапии, включающим введение этому пациенту терапевтически эффективного количества композиции согласно десятому аспекту, описанному выше.
Тринадцатый аспект четвертого варианта осуществления относится к способам индукции STING-зависимой продукции интерферона I типа у пациента, нуждающегося в терапии, включающим введение этому пациенту терапевтически эффективного количества соединения согласно вышеуказанной общей формуле (IV) четвертого варианта осуществления или первому-девятому аспектам, описанным выше, или его фармацевтически приемлемой соли.
Четырнадцатый аспект четвертого варианта осуществления относится к способам индукции STING-зависимой продукции интерферона I типа у пациента, нуждающегося в терапии, включающим введение этому пациенту терапевтически эффективного количества композиции согласно десятому аспекту, описанному выше.
Пятнадцатый аспект четвертого варианта осуществления относится к способам индукции STING-зависимой продукции цитокинов у пациента, нуждающегося в терапии, включающим введение этому пациенту терапевтически эффективного количества соединения согласно вышеуказанной общей формуле (IV) четвертого варианта осуществления или первому-девятому аспектам, описанным выше, или его фармацевтически приемлемой соли.
Шестнадцатый аспект четвертого варианта осуществления относится к способам индукции STING-зависимой продукции цитокинов у пациента, нуждающегося в терапии, включающим введение этому пациенту терапевтически эффективного количества композиции согласно десятому аспекту, описанному выше.
Семнадцатый аспект четвертого варианта осуществления относится к способам лечения нарушения пролиферации клеток у пациента, нуждающегося в терапии, включающим введение этому пациенту терапевтически эффективного количества соединения общей формулы (IV) или его фармацевтически приемлемой соли. В случаях этого семнадцатого аспекта четвертого варианта осуществления нарушение пролиферации клеток представляет собой рак.
Восемнадцатый аспект четвертого варианта осуществления относится к способам лечения нарушения пролиферации клеток у пациента, нуждающегося в терапии, при этом указанный способ включает введение пациенту терапевтически эффективного количества композиции в соответствии с одиннадцатым аспектом, описанным выше. В случаях этого восемнадцатого аспекта четвертого варианта осуществления нарушение пролиферации клеток представляет собой рак.
В каждом аспекте четвертого варианта осуществления, описанного в настоящем документе, переменные R1, R2, R3, R4, R6, R8, R9, А, X1, X2, и X3 общей формулы (IV) четвертого варианта осуществления и его различных аспектов и случаев каждый независимо выбран из каждого, при условии, что, по меньшей мере, один из R1 , R2 , R3 , R4, R6, R8, и R9 не представляет собой Н.
Пятый вариант осуществления относится к соединениям общей формулы (V):
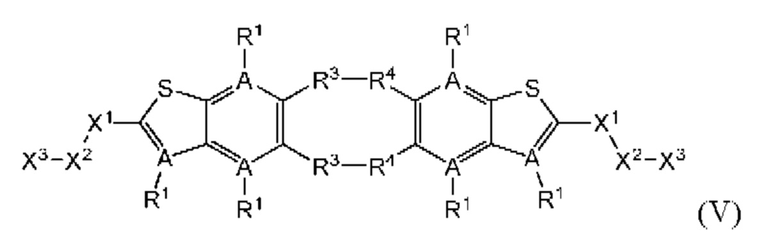
или их фармацевтически приемлемой соли, где каждый A-R1 независимо выбран из группы, состоящей из C-R1 и N; каждый R1 независимо выбран из группы, состоящей из Н, галогена, OR6, N(R6)2, C1-C6 алкила, C1-C6 галоалкила, C1-C6 алкила, замещенного OR6, C1-C6 алкила, замещенного N(R6)2, COOR6, и C(O)N(R6)2; R3 и R4 независимо выбраны из группы, состоящей из O-(C1-C4 алкилен или галоалкилен), С1-С5 алкилена или галоалкилена, и N(R6)-(C1-C4 алкилен или галоалкилен); каждый X1 независимо выбран из группы, состоящей из С=O, -CH2-, -CHF-, и -CF2-; каждый X2 независимо выбран из (C(R8)2)(1-3), где каждый R8 независимо выбран из группы, состоящей из Н, галогена, C1-C6 алкила, CN, OR6, N(R6)2, C1-C6 галоалкила, С3-С6 циклоалкила, C1-C6 алкила, замещенного OR6, и C1-C6 алкила, замещенного N(R6)2; необязательно, 2 R8 на разных атомах углерода могут быть взяты вместе совместно с атомами, к которым они присоединены, с образованием от 3- до 6-членного конденсированного кольца; и, необязательно, 2 R8 на одном атоме углерода могут быть взяты вместе совместно с атомом, к которому они присоединены, с образованием от 3- до 6-членного спироцикла; каждый X3 независимо выбран из группы, состоящей из COOR6, C(O)SR6, C(S)OR6, 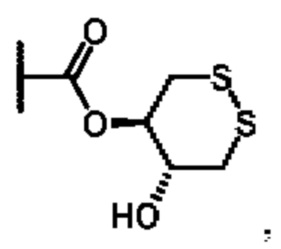 S02R6, C(O)N(R9)2, и CN; и каждый R9 независимо выбран из группы, состоящей из Н, COOR, и SO2R6.
S02R6, C(O)N(R9)2, и CN; и каждый R9 независимо выбран из группы, состоящей из Н, COOR, и SO2R6.
В первом аспекте пятого варианта осуществления каждый A-R1 независимо выбран из группы, состоящей из C-R1 и N. В конкретных примерах этого аспекта каждый 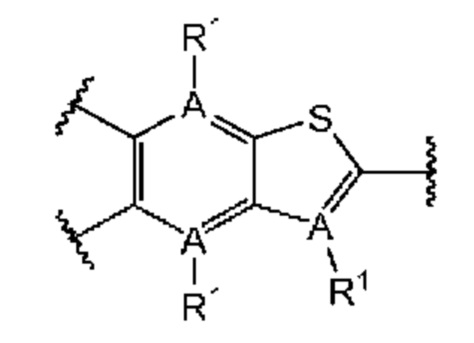 независимо выбран из группы, состоящей из
независимо выбран из группы, состоящей из 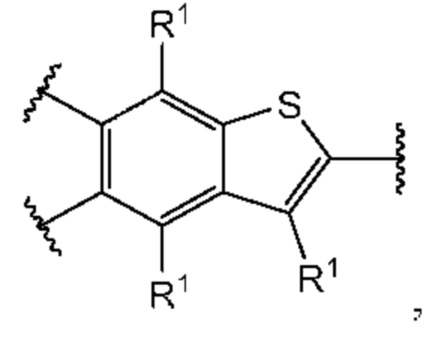
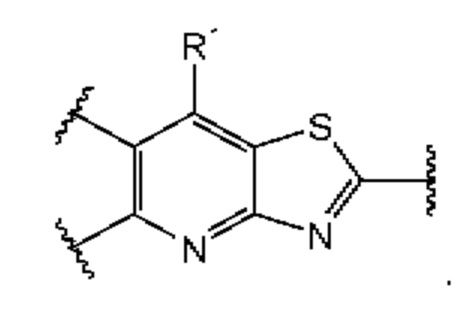 В более конкретных случаях этого аспекта каждый
В более конкретных случаях этого аспекта каждый 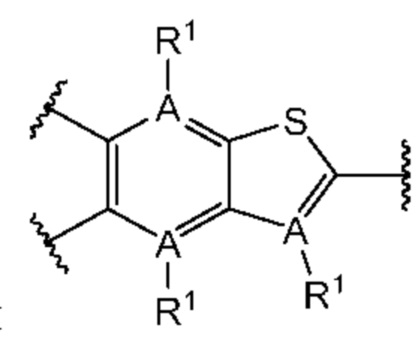 независимо выбран из группы, состоящей из
независимо выбран из группы, состоящей из 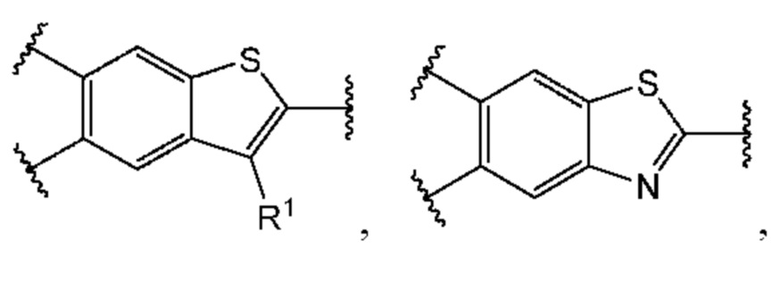
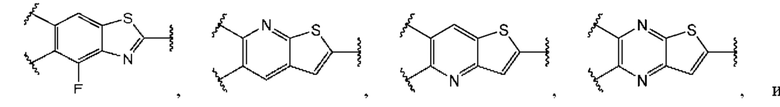
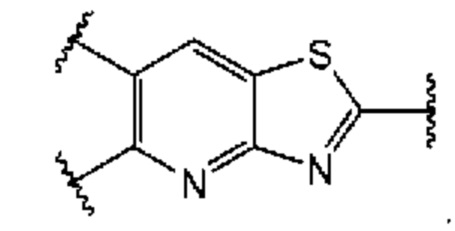 В этом аспекте все остальные группы являются такими, как указано в общей формуле (V) пятого варианта осуществления выше.
В этом аспекте все остальные группы являются такими, как указано в общей формуле (V) пятого варианта осуществления выше.
Во втором аспекте пятого варианта осуществления каждый R1 независимо выбран из группы, состоящей из Н, галогена, OR6, N(R6)2, C1-C6 алкила, C1-C6 галоалкила, C1-C6 алкила, замещенного OR6, C1-C6 алкила, замещенного N(R6)2, COOR6, и C(O)N(R6)2. В примерах этого аспекта каждый R1 независимо выбран из группы, состоящей из Н, галогена, C1-С3 алкила, и C1-С3 галоалкила. В конкретных примерах этого аспекта каждый R1 независимо выбран из группы, состоящей из Н и галогена. В более конкретных случаях этого аспекта каждый R1 независимо выбран из группы, состоящей из Н и F. В этом аспекте все остальные группы являются такими, как указано в вышеприведенной общей формуле (V) пятого варианта осуществления или в первом аспекте выше.
В третьем аспекте пятого варианта осуществления R3 и R4 независимо выбраны из группы, состоящей из O-(C1-C4 алкилен или галоалкилен), С1-С5 алкилена или галоалкилена, и N(R6)-(C1-C4 алкилен или галоалкилен). В примерах этого четвертого аспекта R3-R4 выбран из группы, состоящей из -(CH2)2-8-, -O(СН2)1-7-, -O(СН2)1-6 О-, -NH(CH2)1-7-, и -NH(CH2)1-6O-. В отдельных случаях этого четвертого аспекта R3-R4 выбран из группы, состоящей из -(СН2)2-, -(СН2)3-, -(СН2)4-, -O(СН2)2-, -O(СН2)3-, -O(СН2)4-, -O(СН2)2O-, -O(СН2)3О-, -ОСН2СН(СН3)CH2O-, -O(CH2)4O-, -O(CH2)5O-, -NH(CH2)2-, -NH(CH2)3-, и -NH(CH2)3O-. В этом аспекте все остальные группы являются такими, как указано в общей формуле (V) пятого варианта осуществления или в первом и втором аспектах, описанных выше.
В четвертом аспекте пятого варианта осуществления каждый R6 независимо выбран из группы, состоящей из Н, C1-C6 алкила, и C1-C6 галоалкила. В примерах этого аспекта каждый R6 независимо выбран из группы, состоящей из Н, C1-С3 алкила, и C1-С3 галоалкила. В конкретных примерах этого аспекта каждый R6 независимо выбран из группы, состоящей из Н, СН3, и CHF2. В этом аспекте все остальные группы являются такими, как указано в общей формуле (V) пятого варианта осуществления или в первом-третьем аспектах, описанных выше.
В пятом аспекте пятого варианта осуществления каждый X1 независимо выбран из группы, состоящей из С=O, -СН2-, -CHF-, и -CF2-. В примерах этого аспекта X1 выбран из группы, состоящей из С=O и -СН2-. В конкретных примерах этого аспекта X1 представляет собой С=O. В этом варианте осуществления все остальные группы являются такими, как указано в общей формуле (V) пятого варианта осуществления или в первом-четвертом аспектах, описанных выше.
В шестом аспекте пятого варианта осуществления каждый X2 независимо выбран из (C(R8)2)(1-3), где каждый R8 независимо выбран из группы, состоящей из Н, галогена, C1-C6 алкила, CN, OR6, N(R6)2, C1-C6 галоалкила, С3-С6 циклоалкила, C1-С6 алкила, замещенного OR6, и C1-C6 алкила, замещенного N(R6)2; необязательно, 2 R8 на разных атомах углерода могут быть взяты вместе совместно с атомами, к которым они присоединены, с образованием от 3- до 6-членного конденсированного кольца; и, необязательно, 2 R8 на одном атоме углерода могут быть взяты вместе совместно с атомом, к которому они присоединены, с образованием от 3- до 6-членного спироцикла. В первом случае этого аспекта каждый X2 представляет собой CH2CHR8, где R8 выбран из группы, состоящей из Н, С1-С3 алкила, C1-С3 алкила, замещенного ОН, С1-С3 алкила, замещенного ОС1-С3 алкилом, и С3-С6 циклоалкила. В отдельных примерах этого первого случая каждый X2 представляет собой CH2CHR8, где R8 выбран из группы, состоящей из Н, СН3, СН2ОН, СН2СН3, СН2СН2СН3, СН(СН3)2, СН2ОСН3, и циклопропила. Во втором случае этого аспекта каждый X2 представляет собой CHR8CHR8, где каждый R8 независимо выбран из группы, состоящей из Н, C1-С3 алкила, C1-С3 алкила, замещенного ОН, C1-С3 алкила, замещенного ОС1-С3 алкилом, и С3-С6 циклоалкила, и, необязательно, 2 R8 на разных атомах углерода взяты вместе совместно с атомами, к которым они присоединены, с образованием от 3- до 6-членного конденсированного кольца. В отдельных примерах этого второго случая каждый X2 представляет собой CHR8CHR8, где каждый R8 независимо выбран из группы, состоящей из Н и C1-С3 алкила, и, необязательно, 2 R8 на разных атомах углерода взяты вместе совместно с атомами, к которым они присоединены, с образованием от 3- до 6-членного конденсированного кольца. В третьем случае этого аспекта каждый X2 представляет собой CH2C(R8)2, где каждый R8 независимо выбран из группы, состоящей из Н, C1-С3 алкила, C1-С3 алкила, замещенного ОН, C1-С3 алкила, замещенного ОС1-С3 алкилом, и С3-С6 циклоалкила, и, необязательно, 2 R8 на одном атоме углерода взяты вместе совместно с атомами, к которым они присоединены, с образованием от 3- до 6-членного спироцикла. В отдельных примерах этого третьего случая каждый X2 представляет собой CH2C(R8)2, где каждый R8 независимо выбран из группы, состоящей из Н и C1-С3 алкила, и, необязательно, 2 R8 на одном атоме углерода взяты вместе совместно с атомами, к которым они присоединены, с образованием от 3- до 6-членного спироцикла. В этом аспекте все остальные группы являются такими, как указано в общей формуле (V) пятого варианта осуществления или в первом-пятом аспектах, описанных выше.
В седьмом аспекте пятого варианта осуществления каждый X3 независимо выбран из группы, состоящей из COOR6, C(O)SR6, C(S)OR6,  , SO2R6, C(O)N(R9)2, и CN. В примерах этого аспекта каждый X3 независимо выбран из группы, состоящей из COOR6,
, SO2R6, C(O)N(R9)2, и CN. В примерах этого аспекта каждый X3 независимо выбран из группы, состоящей из COOR6,  SO2R6, C(O)N(R9)2, и CN. В конкретных примерах этого аспекта каждый X3 независимо выбран из группы, состоящей из COOR6, C(O)N(R9)2, и CN. В даже более конкретных случаях этого аспекта каждый X3 независимо выбран из группы, состоящей из СООН, СООСН3, CONH2, и CN. В этом аспекте все остальные группы являются такими, как указано в общей формуле (V) пятого варианта осуществления или в первом-шестом аспектах, описанных выше.
SO2R6, C(O)N(R9)2, и CN. В конкретных примерах этого аспекта каждый X3 независимо выбран из группы, состоящей из COOR6, C(O)N(R9)2, и CN. В даже более конкретных случаях этого аспекта каждый X3 независимо выбран из группы, состоящей из СООН, СООСН3, CONH2, и CN. В этом аспекте все остальные группы являются такими, как указано в общей формуле (V) пятого варианта осуществления или в первом-шестом аспектах, описанных выше.
В восьмом аспекте пятого варианта осуществления каждый R9 независимо выбран из группы, состоящей из Н, COOR6, и SO2R6. В примерах этого аспекта каждый R9 независимо представляет собой Н. В этом аспекте все остальные группы являются такими, как указано в общей формуле (V) пятого варианта осуществления или в первом-седьмом аспектах, описанных выше.
Девятый аспект пятого варианта осуществления относится к фармацевтической композиции, где указанная фармацевтическая композиция содержит (а) соединение согласно общей формуле (V) пятого варианта осуществления или с первого по восьмой аспектам, описанным выше, или его фармацевтически приемлемую соль; и (b) фармацевтически приемлемый носитель.
Десятый аспект пятого варианта осуществления относится к способам индукции иммунного ответа у пациента, нуждающегося в терапии, включающим введение этому пациенту терапевтически эффективного количества соединения согласно общей формуле (V) пятого варианта осуществления или с первого по восьмой аспектам, описанным выше, или его фармацевтически приемлемой соли.
Одиннадцатый аспект пятого варианта осуществления относится к способам индукции иммунного ответа у пациента, нуждающегося в терапии, включающим введение этому пациенту терапевтически эффективного количества композиции согласно девятому аспекту, описанному выше.
Двенадцатый аспект пятого варианта осуществления относится к способам индукции STING-зависимой продукции интерферона I типа у пациента, нуждающегося в терапии, включающим введение этому пациенту терапевтически эффективного количества соединения согласно вышеприведенной общей формуле (V) пятого варианта осуществления или с первого по восьмой аспектам, описанным выше, или его фармацевтически приемлемой соли.
Тринадцатый аспект пятого варианта осуществления относится к способам индукции STING-зависимой продукции интерферона I типа у пациента, нуждающегося в терапии, включающим введение этому пациенту терапевтически эффективного количества композиции согласно девятому аспекту, описанному выше.
Четырнадцатый аспект пятого варианта осуществления относится к способам индукции STING-зависимой продукции цитокинов у пациента, нуждающегося в терапии, включающим введение этому пациенту терапевтически эффективного количества соединения согласно вышеприведенной общей формуле (V) пятого варианта осуществления или с первого по восьмой аспектам, описанным выше, или его фармацевтически приемлемой соли.
Пятнадцатый аспект пятого варианта осуществления относится к способам индукции STING-зависимой продукции цитокинов у пациента, нуждающегося в терапии, включающим введение этому пациенту терапевтически эффективного количества композиции согласно девятому аспекту, описанному выше.
Шестнадцатый аспект пятого варианта осуществления относится к способам лечения нарушения пролиферации клеток у пациента, нуждающегося в терапии, включающим введение этому пациенту терапевтически эффективного количества соединения согласно вышеприведенной общей формуле (V) пятого варианта осуществления или с первого по восьмой аспектам, описанным выше, или его фармацевтически приемлемой соли. В случаях этого шестнадцатого аспекта пятого варианта осуществления нарушение пролиферации клеток представляет собой рак.
Семнадцатый аспект пятого варианта осуществления относится к способам лечения нарушения пролиферации клеток у пациента, нуждающегося в терапии, где указанный способ включает введение этому пациенту терапевтически эффективного количества композиции в соответствии с девятым аспектом, описанным выше. В случаях этого семнадцатого аспекта пятого варианта осуществления нарушение пролиферации клеток представляет собой рак.
В каждом аспекте пятого варианта осуществления, описанного в настоящем документе, переменные R1 , R2 , R3 , R4, R6, R8, R9, А, X1, X2 и X3 общей формулы (V) пятого варианта осуществления и их различных аспектов и случаев, каждый независимо выбран из каждого, при условии, что, по меньшей мере, один из R1 , R2 , R3 , R4, R6, R8, и R9 не представляет собой Н.
Шестой вариант осуществления относится к соединениям общей формулы (VI):
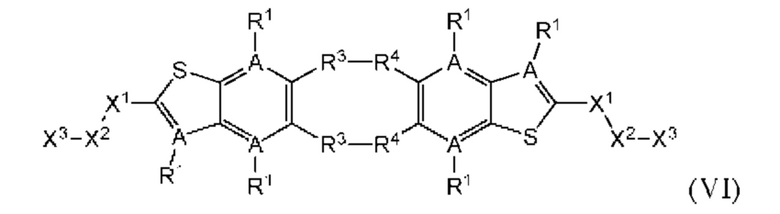
или их фармацевтически приемлемой соли, где каждый A-R1 независимо выбран из группы, состоящей из C-R1 и N; каждый R1 независимо выбран из группы, состоящей из Н, галогена, OR6, N(R6)2, C1-C6 алкила, C1-C6 галоалкила, C1-C6 алкила, замещенного OR6, C1-C6 алкила, замещенного N(R6)2, COOR6, и C(O)N(R6)2; R3 и R4 независимо выбраны из группы, состоящей из O-(C1-C4 алкилен или галоалкилен), С1-С5 алкилена или галоалкилена, и N(R6)-(C1-C4 алкилен или галоалкилен); каждый R6 независимо выбран из группы, состоящей из Н, C1-C6 алкила, и C1-C6 галоалкила; каждый X1 независимо выбран из группы, состоящей из С=O, -СН2-, -CHF-, и -CF2-; каждый X2 независимо выбран из (C(R8)2)(1-3), где каждый R8 независимо выбран из группы, состоящей из Н, галогена, C1-C6 алкила, CN, OR6, N(R6)2, C1-C6 галоалкила, С3-С6 циклоалкила, C1-C6 алкила, замещенного OR6, и C1-C6 алкила, замещенного N(R6)2; необязательно, 2 R8 на разных атомах углерода могут быть взяты вместе совместно с атомами, к которым они присоединены, с образованием от 3- до 6-членного конденсированного кольца; и, необязательно, 2 R8 на одном атоме углерода могут быть взяты вместе совместно с атомом, к которому они присоединены, с образованием от 3- до 6-членного спироцикла; каждый X3 независимо выбран из группы, состоящей из COOR6, C(O)SR6, C(S)OR6,  SO2R6, C(O)N(R9)2, и CN; и каждый R9 независимо выбран из группы, состоящей из Н, COOR6, и SO2R6.
SO2R6, C(O)N(R9)2, и CN; и каждый R9 независимо выбран из группы, состоящей из Н, COOR6, и SO2R6.
В первом аспекте шестого варианта осуществления каждый A-R1 независимо выбран из группы, состоящей из C-R1 и N. В конкретных примерах этого аспекта каждый 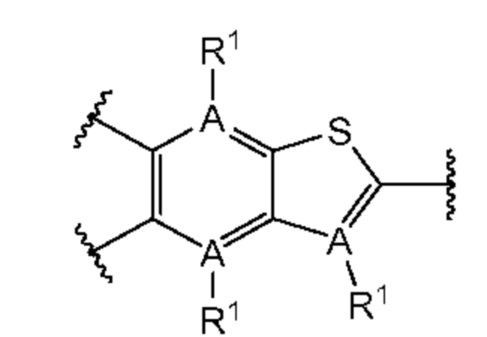 независимо выбран из группы, состоящей из
независимо выбран из группы, состоящей из 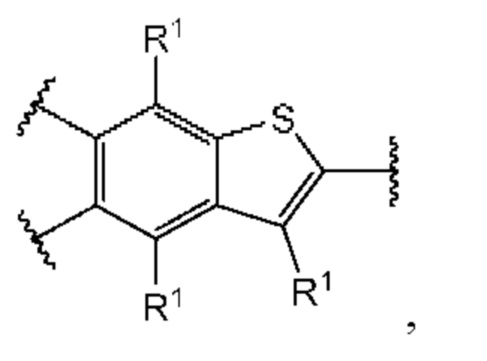
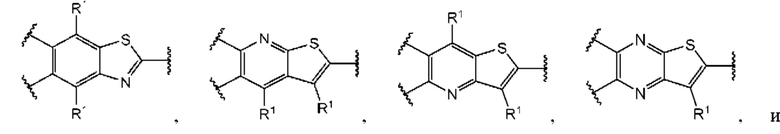
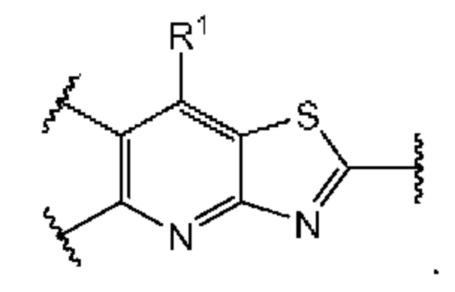 В более конкретных случаях этого аспекта каждый
В более конкретных случаях этого аспекта каждый 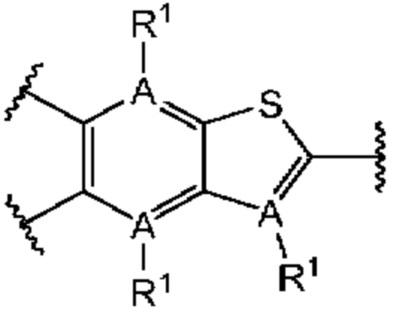 независимо выбран из группы, состоящей из
независимо выбран из группы, состоящей из 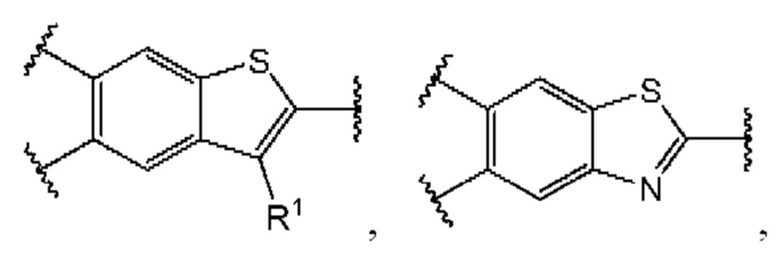
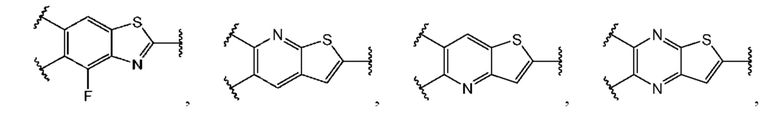 и
и 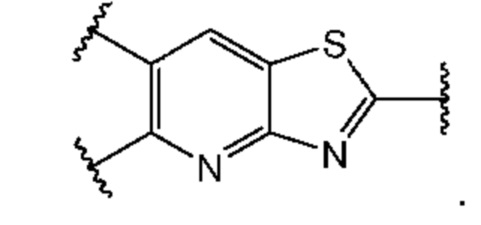 В этом аспекте все остальные группы являются такими, как указано в общей формуле (VI) шестого варианта осуществления выше.
В этом аспекте все остальные группы являются такими, как указано в общей формуле (VI) шестого варианта осуществления выше.
Во втором аспекте шестого варианта осуществления каждый R1 независимо выбран из группы, состоящей из Н, галогена, OR6, N(R6)2, C1-C6 алкила, C1-C6 галоалкила, C1-C6 алкила, замещенного OR6, C1-C6 алкила, замещенного N(R6)2, COOR6, и C(O)N(R6)2. В примерах этого аспекта каждый R1 независимо выбран из группы, состоящей из Н, галогена, С1-С3 алкила, и С1-С3 галоалкила. В конкретных примерах этого аспекта каждый R1 независимо выбран из группы, состоящей из Н и галогена. В более конкретных случаях этого аспекта каждый R1 независимо выбран из группы, состоящей из Н и F. В этом аспекте все остальные группы являются такими, как указано в приведенной выше общей формуле (VI) шестого варианта осуществления или в первом аспекте выше.
В третьем аспекте шестого варианта осуществления R3 и R4 независимо выбраны из группы, состоящей из O-(C1-C4 алкилен или галоалкилен), С1-С5 алкилена или галоалкилена, и N(R6)-(C1-C4 алкилен или галоалкилен). В примерах этого четвертого аспекта R3-R4 выбран из группы, состоящей из -(СР2)2-8-, -O(СН2)1-7-, -O(СН2)1-6О-, -NH(CH2)1-7-, и -NH(CH2)1-6O-. В отдельных случаях этого четвертого аспекта R3-R4 выбран из группы, состоящей из -(СН2)2-, -(СН2)3-, -(СН2)4-, -O(СН2)2-, -O(СН2)3-, -O(СН2)4-, -O(СН2)2O-, -O(СН2)3О-, -ОСН2СН(СН3)CH2O-, -O(CH2)4O-, -O(CH2)5O-, -NH(CH2)2-, -NH(CH2)3-, и -NH(CH2)3O-. В этом аспекте все остальные группы являются такими, как указано в общей формуле (VI) шестого варианта осуществления или в первом и втором аспектах, описанных выше.
В четвертом аспекте шестого варианта осуществления каждый R6 независимо выбран из группы, состоящей из Н, C1-C6 алкила, и C1-C6 галоалкила. В примерах этого аспекта каждый R6 независимо выбран из группы, состоящей из Н, C1-С3 алкила, и С1-С3 галоалкила. В конкретных примерах этого аспекта каждый R6 независимо выбран из группы, состоящей из Н, СН3, и CHF2. В этом аспекте все остальные группы являются такими, как указано в общей формуле (VI) шестого варианта осуществления или в первом-третьем аспектах, описанных выше.
В пятом аспекте шестого варианта осуществления каждый X1 независимо выбран из группы, состоящей из С=O, -СН2-, -CHF-, и -CF2-. В примерах этого аспекта X1 выбран из группы, состоящей из С=O и -СН2-. В конкретных примерах этого аспекта X1 представляет собой С=O. В этом варианте осуществления все остальные группы являются такими, как указано в общей формуле (VI) шестого варианта осуществления или в первом-четвертом аспектах, описанных выше.
В шестом аспекте шестого варианта осуществления каждый X2 независимо выбран из (C(R8)2)(1-3), где каждый R8 независимо выбран из группы, состоящей из Н, галогена, C1-C6 алкила, CN, OR6, N(R6)2, C1-C6 галоалкила, С3-С6 циклоалкила, C1-C6 алкила, замещенного OR6, и C1-C6 алкила, замещенного N(R6)2; необязательно, 2 R8 на разных атомах углерода могут быть взяты вместе совместно с атомами, к которым они присоединены, с образованием от 3- до 6-членного конденсированного кольца; и, необязательно, 2 R8 на одном атоме углерода могут быть взяты вместе совместно с атомом, к которому они присоединены, с образованием от 3- до 6-членного спироцикла. В первом случае этого аспекта каждый X2 представляет собой CH2CHR8, где R8 выбран из группы, состоящей из Н, С1-С3 алкила, С1-С3 алкила, замещенного ОН, С1-С3 алкила, замещенного ОС1-С3 алкилом, и С3-С6 циклоалкила. В отдельных примерах этого первого случая каждый X2 представляет собой CH2CHR8, где R8 выбран из группы, состоящей из Н, СН3, СН2ОН, СН2СН3, СН2СН2СН3, СН(СН3)2, СН2ОСН3, и циклопропила. Во втором случае этого аспекта каждый X2 представляет собой CHR8CHR8, где каждый R8 независимо выбран из группы, состоящей из Н, C1-С3 алкила, О-С3 алкила, замещенного ОН, C1-С3 алкила, замещенного ОС1-С3 алкилом, и С3-С6 циклоалкила, и, необязательно, 2 R8 на разных атомах углерода взяты вместе совместно с атомами, к которым они присоединены, с образованием от 3- до 6-членного конденсированного кольца. В отдельных примерах этого второго случая каждый X2 представляет собой CHR8CHR8, где каждый R8 независимо выбран из группы, состоящей из Н и С1-С3 алкила, и, необязательно, 2 R8 на разных атомах углерода взяты вместе совместно с атомами, к которым они присоединены, с образованием от 3- до 6-членного конденсированного кольца. В третьем случае этого аспекта каждый X2 представляет собой CH2C(R8)2, где каждый R8 независимо выбран из группы, состоящей из Н, С1-С3 алкила, С1-С3 алкила, замещенного ОН, С1-С3 алкила, замещенного ОС1-С3 алкилом, и С3-С6 циклоалкила, и, необязательно, 2 R8 на одном атоме углерода взяты вместе совместно с атомами, к которым они присоединены, с образованием от 3- до 6-членного спироцикла. В отдельных примерах этого третьего случая каждый X2 представляет собой CH2C(R8)2, где каждый R8 независимо выбран из группы, состоящей из Н и С1-С3 алкила, и, необязательно, 2 R8 на одном атоме углерода взяты вместе совместно с атомами, к которым они присоединены, с образованием от 3- до 6-членного спироцикла. В этом аспекте все остальные группы являются такими, как указано в общей формуле (VI) шестого варианта осуществления или в первом-пятом аспектах, описанных выше.
В седьмом аспекте шестого варианта осуществления каждый X3 независимо выбран из группы, состоящей из COOR6, C(O)SR6, C(S)OR6,  , SO2R6, C(O)N(R9)2, и CN. В примерах этого аспекта каждый X3 независимо выбран из группы, состоящей из COOR6,
, SO2R6, C(O)N(R9)2, и CN. В примерах этого аспекта каждый X3 независимо выбран из группы, состоящей из COOR6,  SO2R6, C(O)N(R9)2, и CN. В конкретных примерах этого аспекта каждый X3 независимо выбран из группы, состоящей из COOR6, C(O)N(R9)2, и CN. В даже более конкретных случаях этого аспекта каждый X3 независимо выбран из группы, состоящей из СООН, СООСН3, CONH2, и CN. В этом аспекте все остальные группы являются такими, как указано в общей формуле (VI) шестого варианта осуществления или в первом-шестом аспектах, описанных выше.
SO2R6, C(O)N(R9)2, и CN. В конкретных примерах этого аспекта каждый X3 независимо выбран из группы, состоящей из COOR6, C(O)N(R9)2, и CN. В даже более конкретных случаях этого аспекта каждый X3 независимо выбран из группы, состоящей из СООН, СООСН3, CONH2, и CN. В этом аспекте все остальные группы являются такими, как указано в общей формуле (VI) шестого варианта осуществления или в первом-шестом аспектах, описанных выше.
В восьмом аспекте шестого варианта осуществления каждый R9 независимо выбран из группы, состоящей из Н, COOR6, и SO2R6. В примерах этого аспекта каждый R9 независимо представляет собой Н. В этом аспекте все остальные группы являются такими, как указано в общей формуле (V) пятого варианта осуществления или в первом-седьмом аспектах, описанных выше.
Девятый аспект шестого варианта осуществления относится к фармацевтической композиции, где указанная фармацевтическая композиция содержит (а) соединение согласно общей формуле (VI) шестого варианта осуществления или с первого по восьмой аспектам, описанным выше, или его фармацевтически приемлемую соль; и (b) фармацевтически приемлемый носитель.
Десятый аспект шестого варианта осуществления относится к способам индукции иммунного ответа у пациента, нуждающегося в терапии, включающим введение этому пациенту терапевтически эффективного количества соединения согласно общей формуле (VI) шестого варианта осуществления или с первого по восьмой аспектам, описанным выше, или его фармацевтически приемлемой соли.
Одиннадцатый аспект шестого варианта осуществления относится к способам индукции иммунного ответа у пациента, нуждающегося в терапии, включающим введение этому пациенту терапевтически эффективного количества композиции согласно девятому аспекту, описанному выше.
Двенадцатый аспект шестого варианта осуществления относится к способам индукции STING-зависимой продукции интерферона I типа у пациента, нуждающегося в терапии, включающим введение этому пациенту терапевтически эффективного количества соединения согласно приведенной выше общей формуле (VI) шестого варианта осуществления или с первого по восьмой аспектам, описанным выше или его фармацевтически приемлемой соли.
Тринадцатый аспект шестого варианта осуществления относится к способам индукции STING-зависимой продукции интерферона I типа у пациента, нуждающегося в терапии, включающим введение этому пациенту терапевтически эффективного количества композиции согласно девятому аспекту, описанному выше.
Четырнадцатый аспект шестого варианта осуществления относится к способам индукции STING-зависимой продукции цитокинов у пациента, нуждающегося в терапии, включающим введение этому пациенту терапевтически эффективного количества соединения согласно приведенной выше общей формуле (VI) шестого варианта осуществления или с первого по восьмой аспектам, описанным выше, или его фармацевтически приемлемой соли.
Пятнадцатый аспект шестого варианта осуществления относится к способам индукции STING-зависимой продукции цитокинов у пациента, нуждающегося в терапии, включающим введение этому пациенту терапевтически эффективного количества композиции согласно девятому аспекту, описанному выше.
Шестнадцатый аспект шестого варианта осуществления относится к способам лечения нарушения пролиферации клеток у пациента, нуждающегося в терапии, включающим введение этому пациенту терапевтически эффективного количества соединения согласно приведенной выше общей формуле (VI) шестого варианта осуществления или с первого по восьмой аспектам, описанным выше, или его фармацевтически приемлемой соли. В случаях этого шестнадцатого аспекта шестого варианта осуществления нарушение пролиферации клеток представляет собой рак.
Семнадцатый аспект шестого варианта осуществления относится к способам лечения нарушения пролиферации клеток у пациента, нуждающегося в терапии, где указанный способ включает введение пациенту терапевтически эффективного количества композиции в соответствии с девятым аспектом, описанным выше. В случаях этого семнадцатого аспекта шестого варианта осуществления нарушение пролиферации клеток представляет собой рак.
В каждом аспекте шестого варианта осуществления, описанного в настоящем документе, переменные R1 , R2 , R3 , R4, R6, R8, R9, А, X1, X2, и X3 общей формулы (VI) шестого варианта осуществления и его различных аспектов и случаев, каждый независимо выбран из каждого, при условии, что, по меньшей мере, один из R1 , R2 , R3 , R4, R6, R8, и R9 не представляет собой Н.
Седьмой вариант осуществления относится к соединению, выбранному из группы, состоящей из
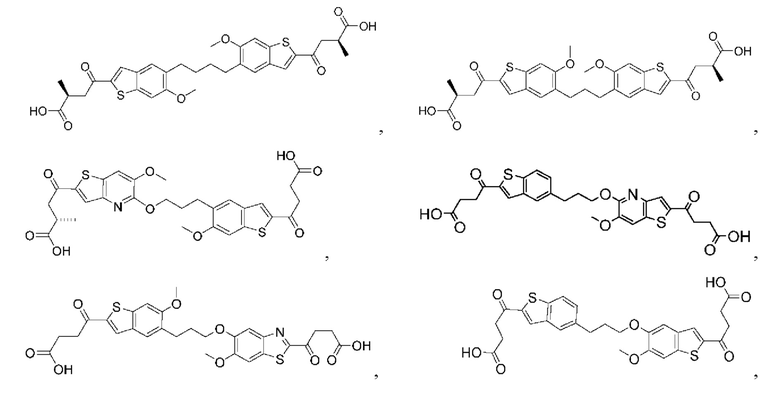
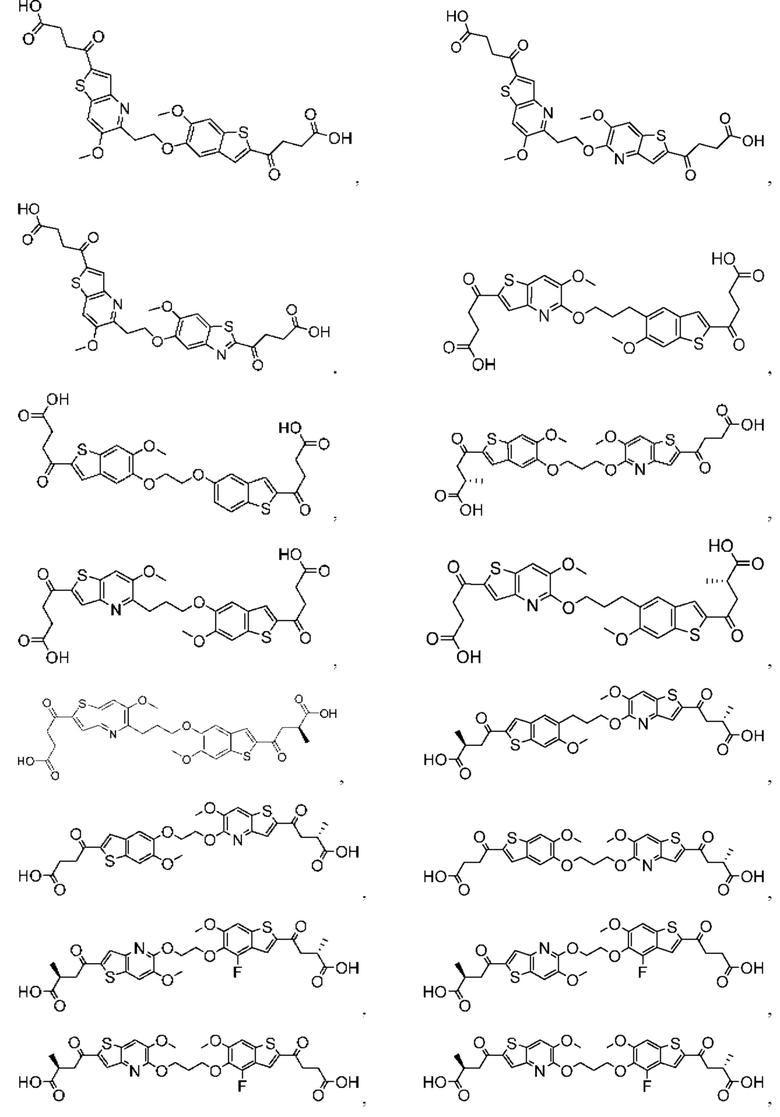
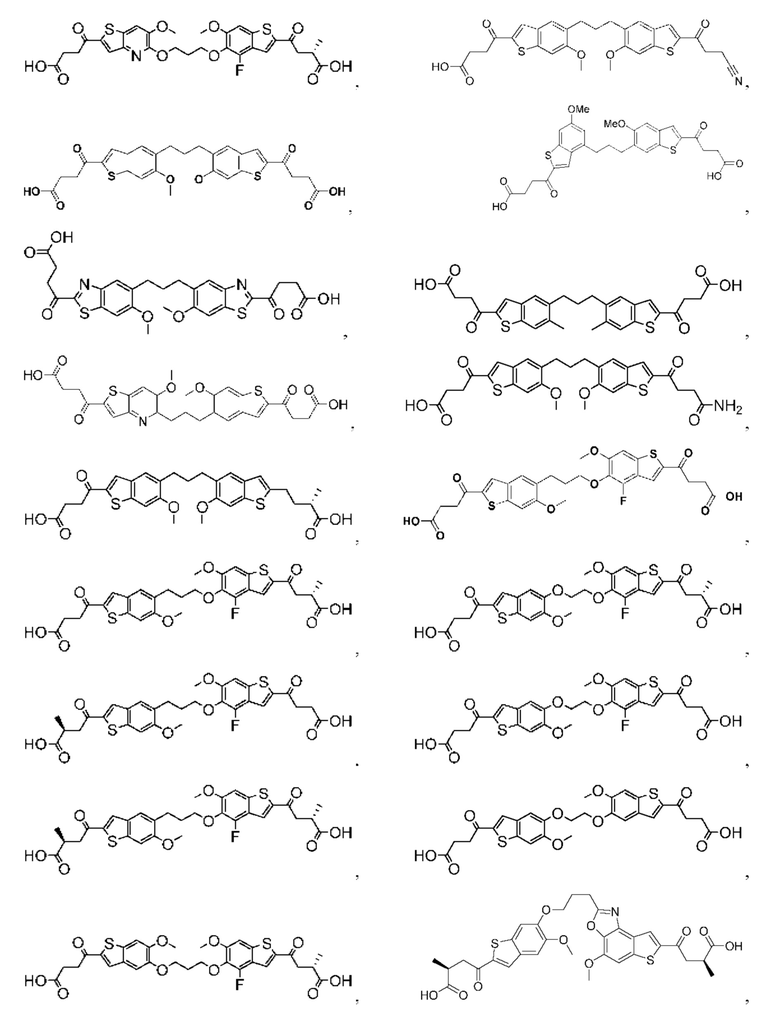
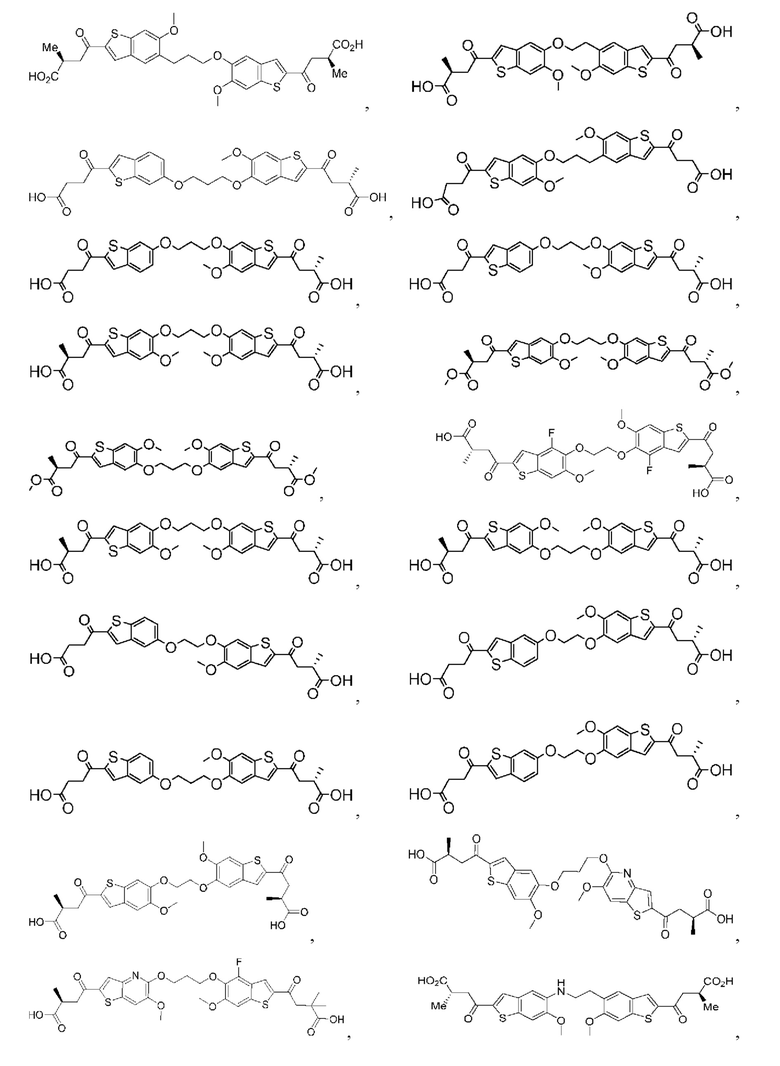
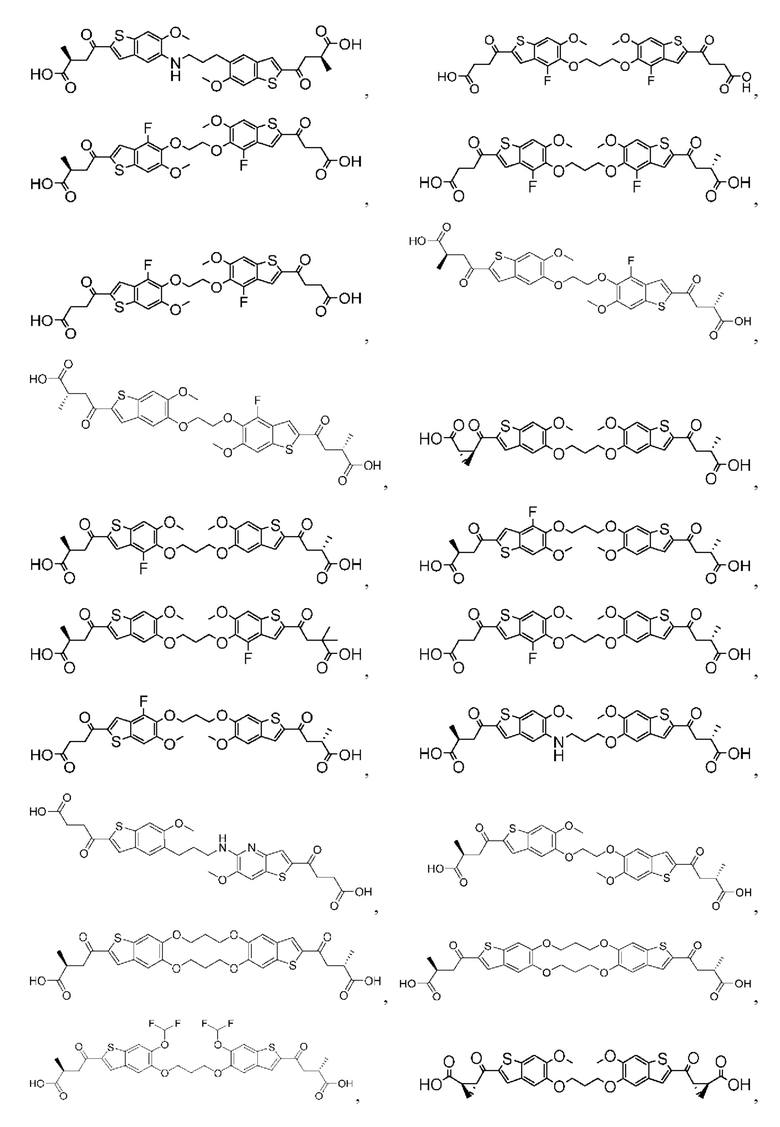
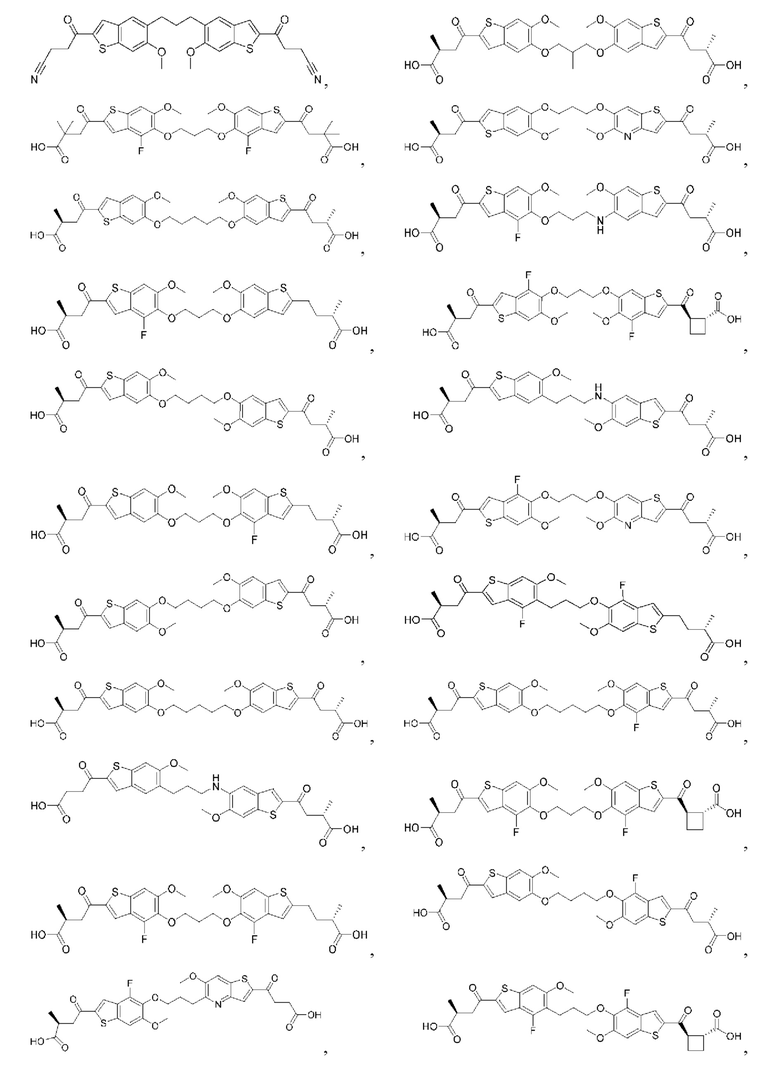
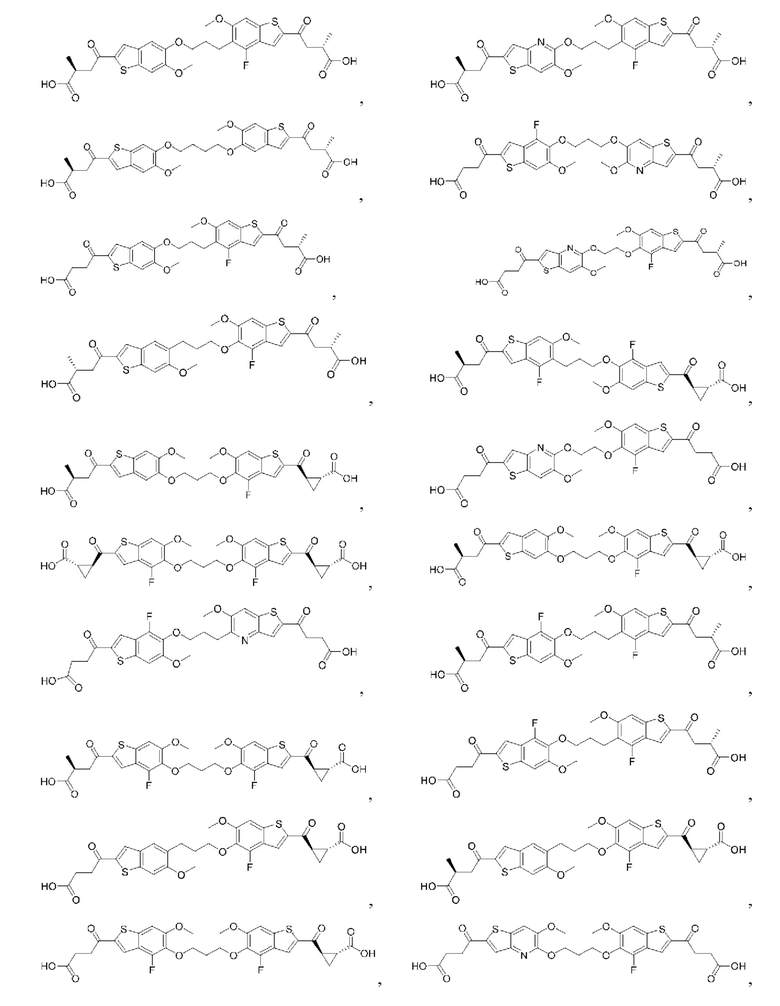
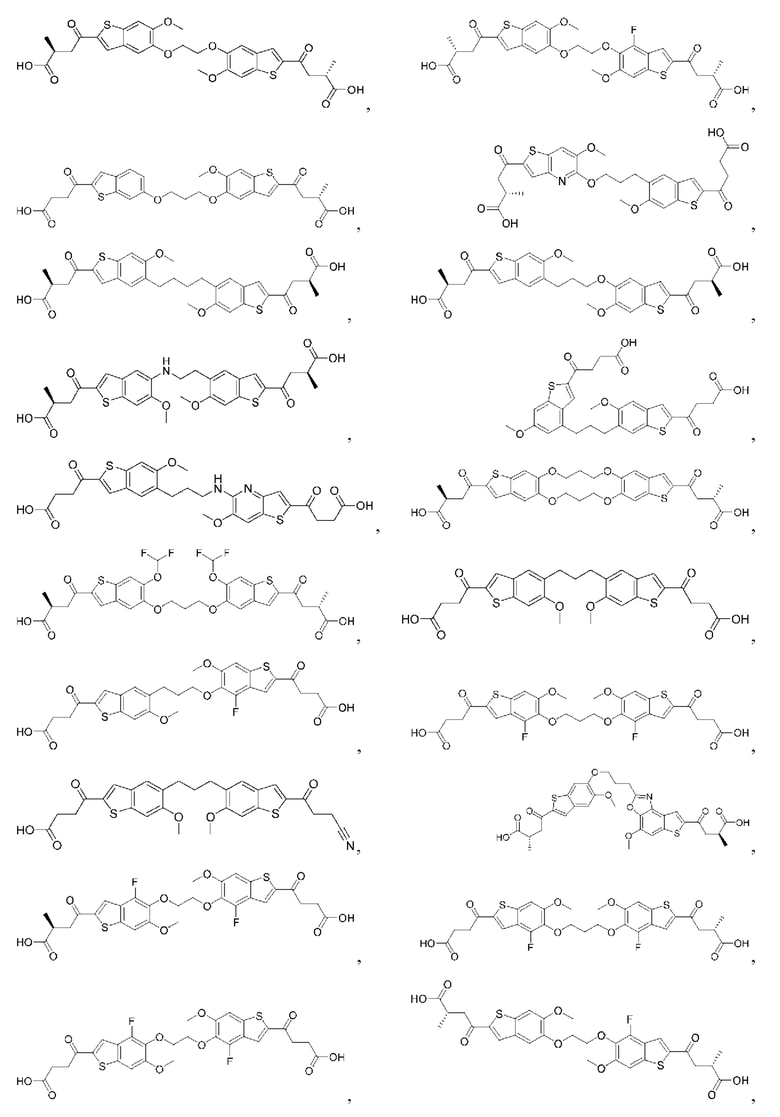
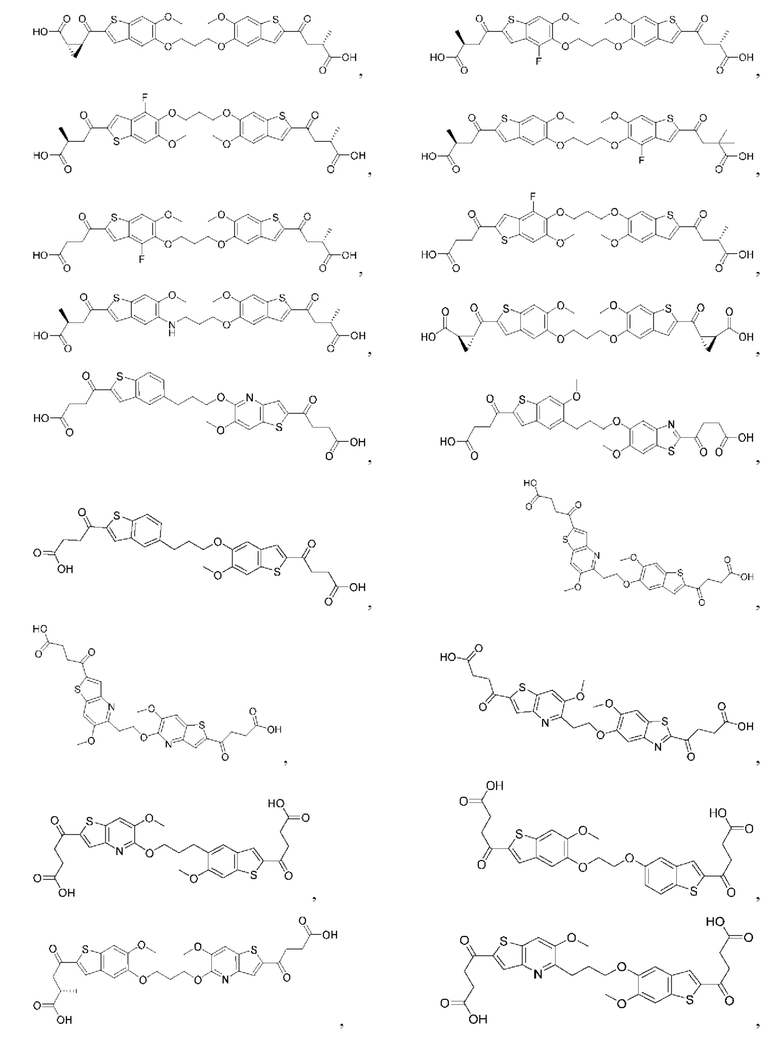
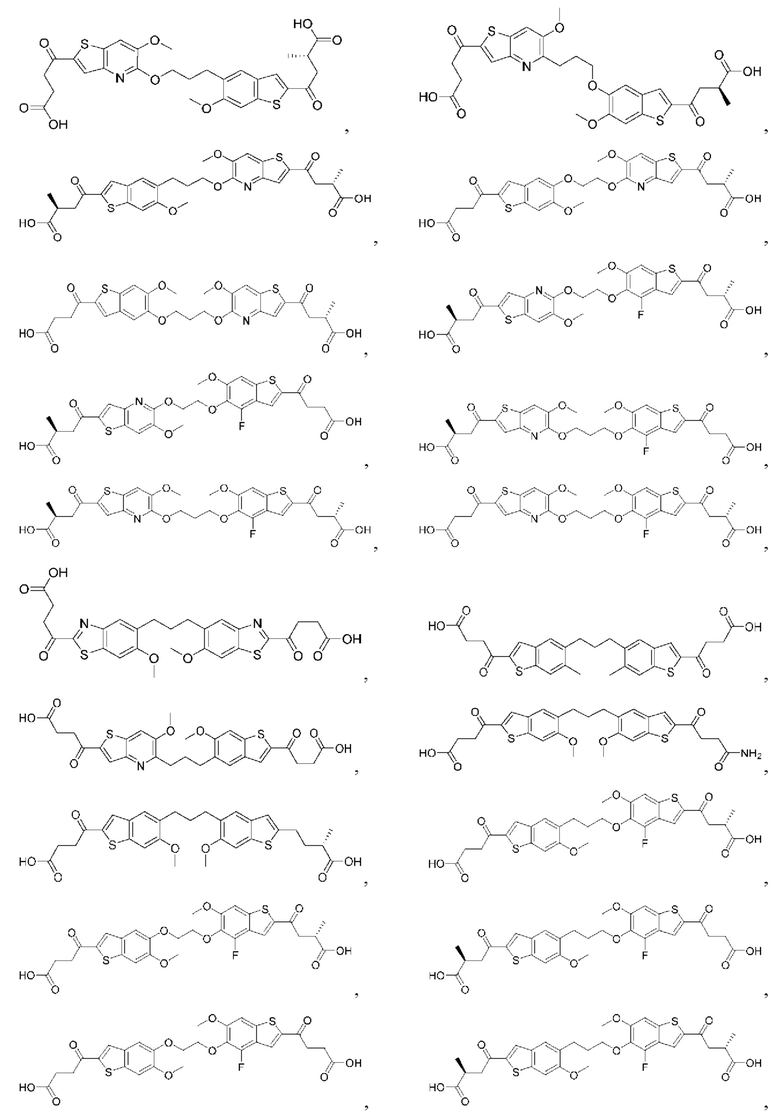
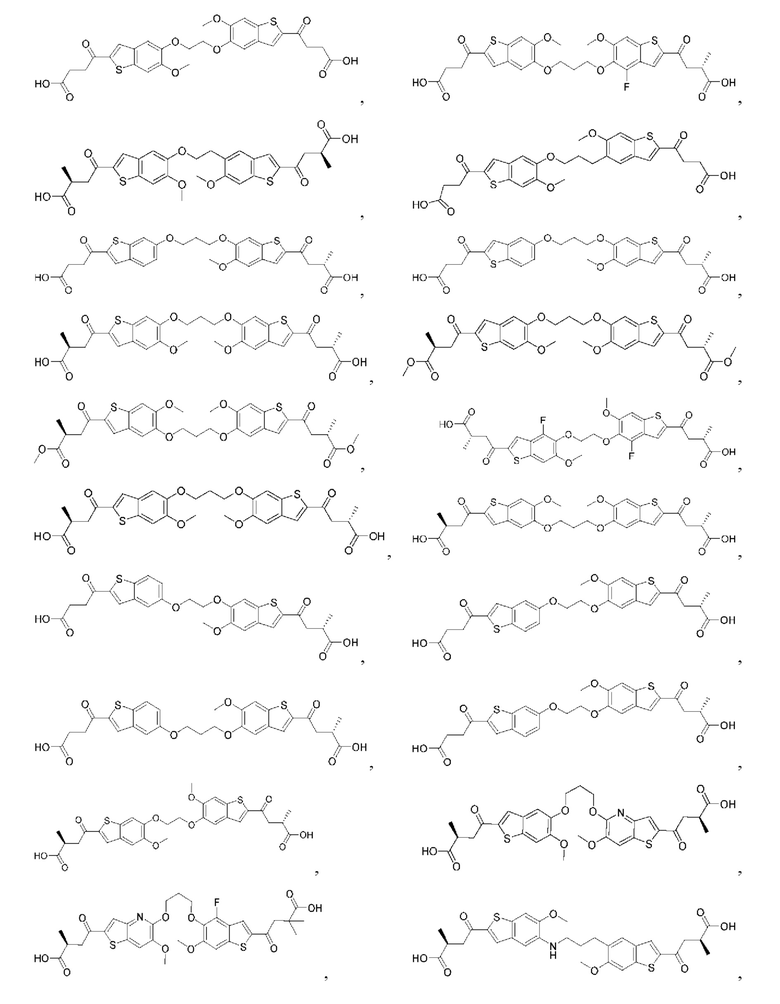
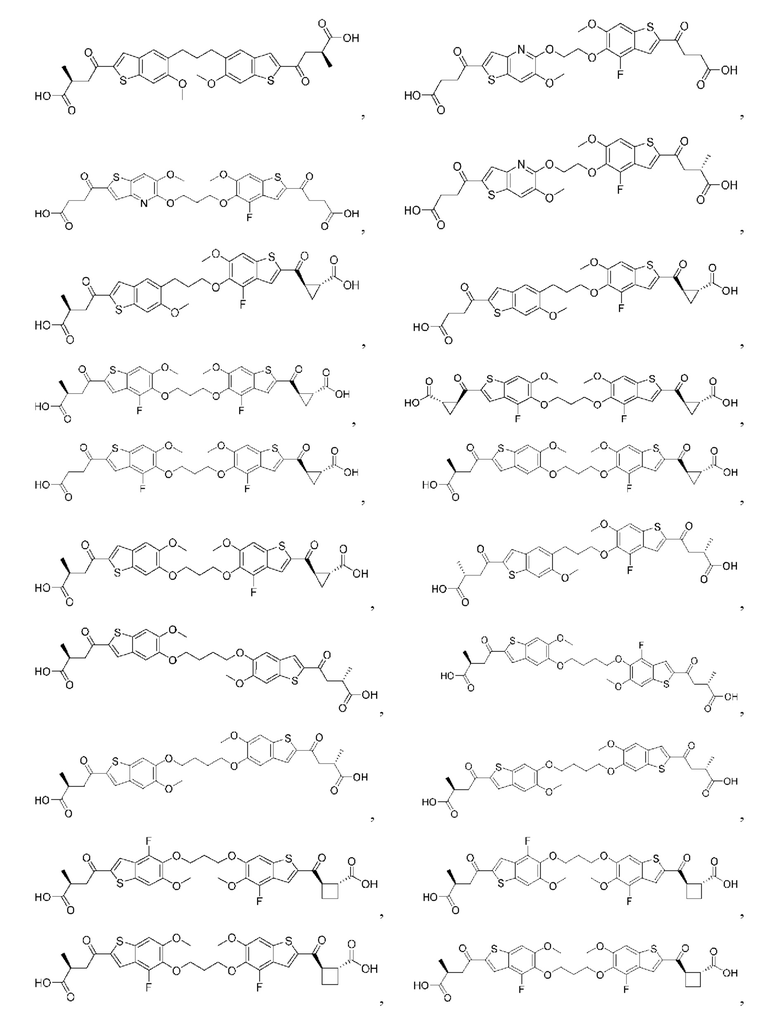
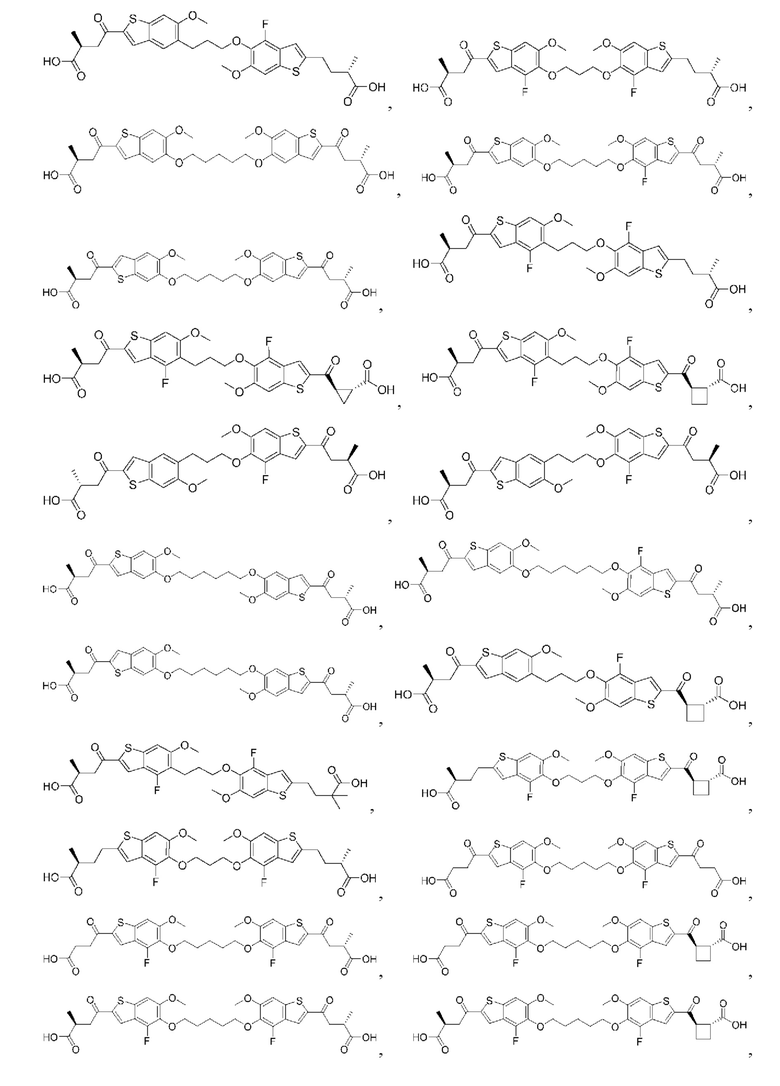
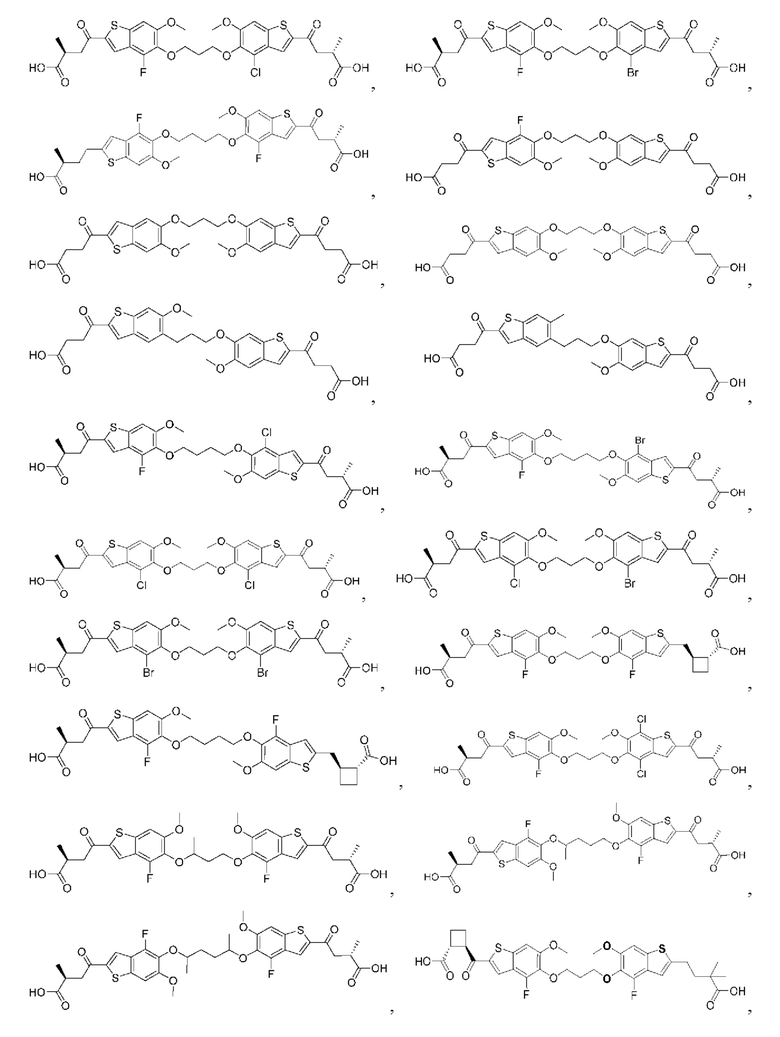
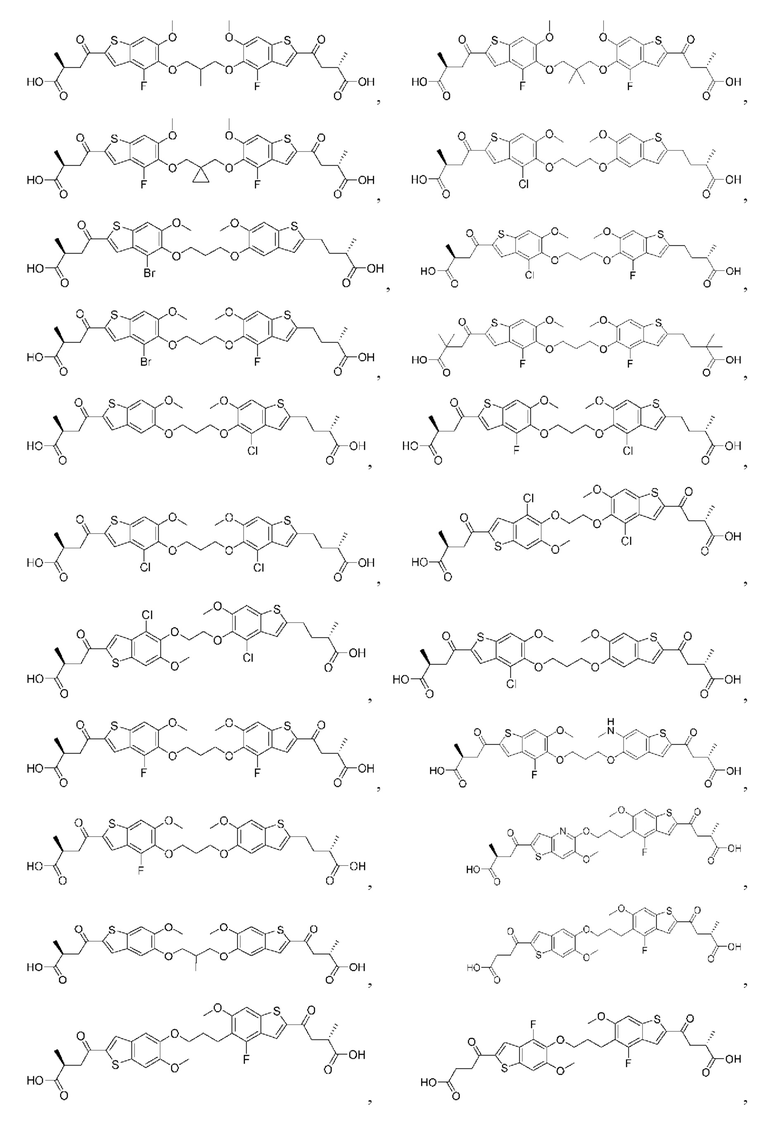
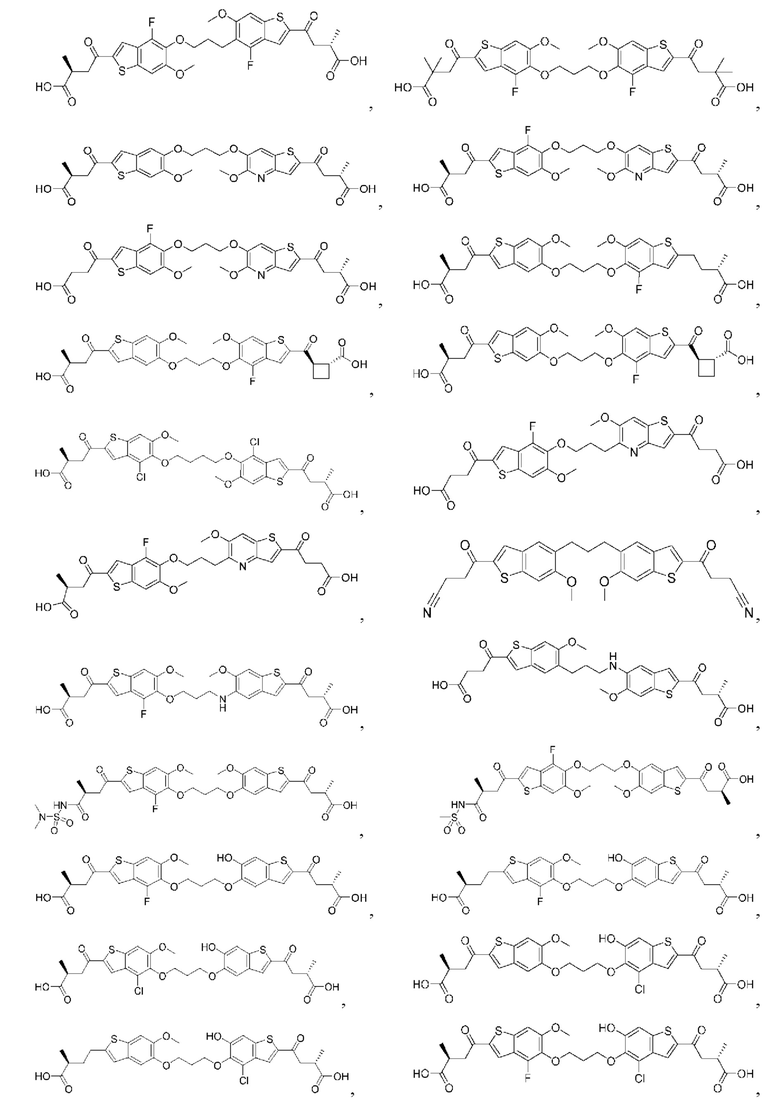
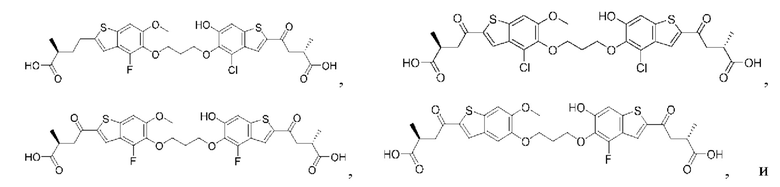
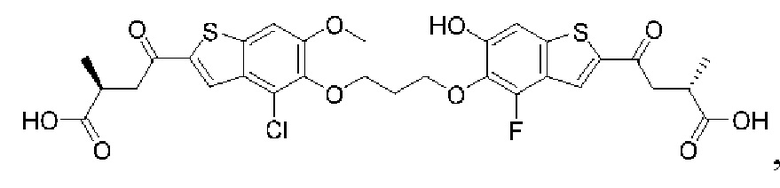 и его фармацевтически приемлемые соли. Конкретные аспекты этого седьмого варианта осуществления относятся к соединению, выбранному из группы, состоящей из
и его фармацевтически приемлемые соли. Конкретные аспекты этого седьмого варианта осуществления относятся к соединению, выбранному из группы, состоящей из
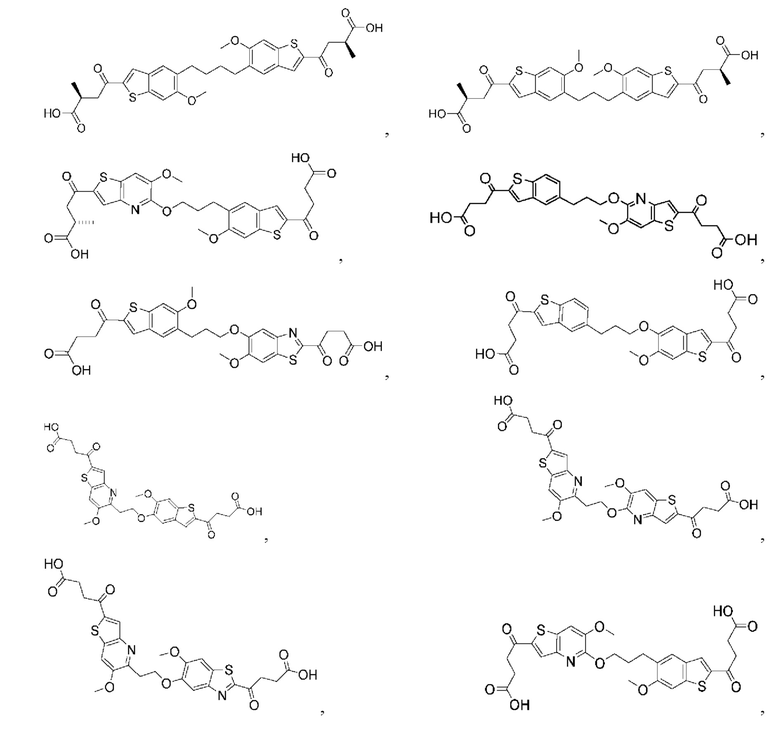
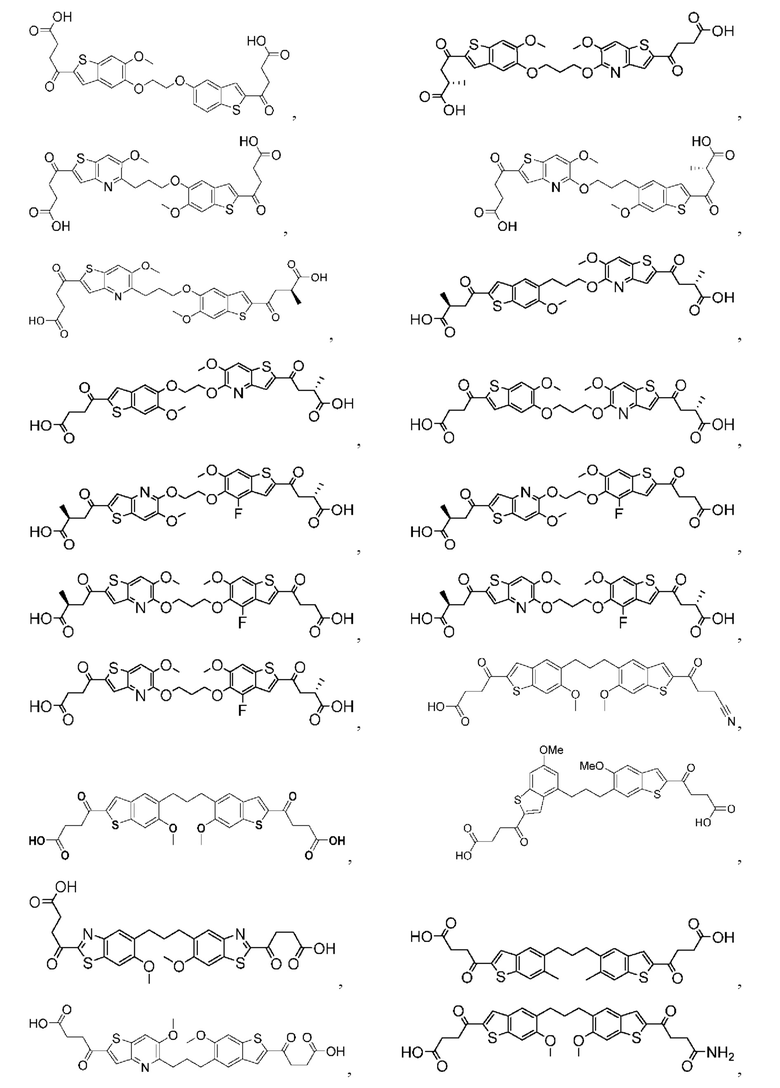
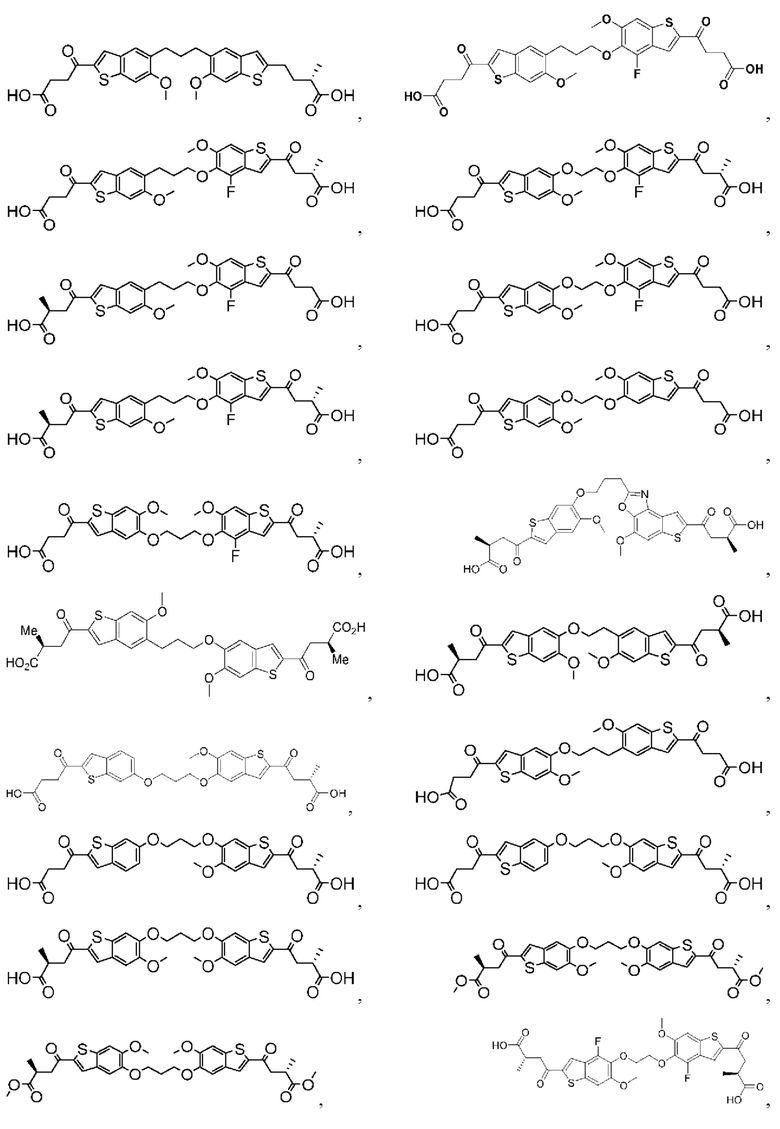
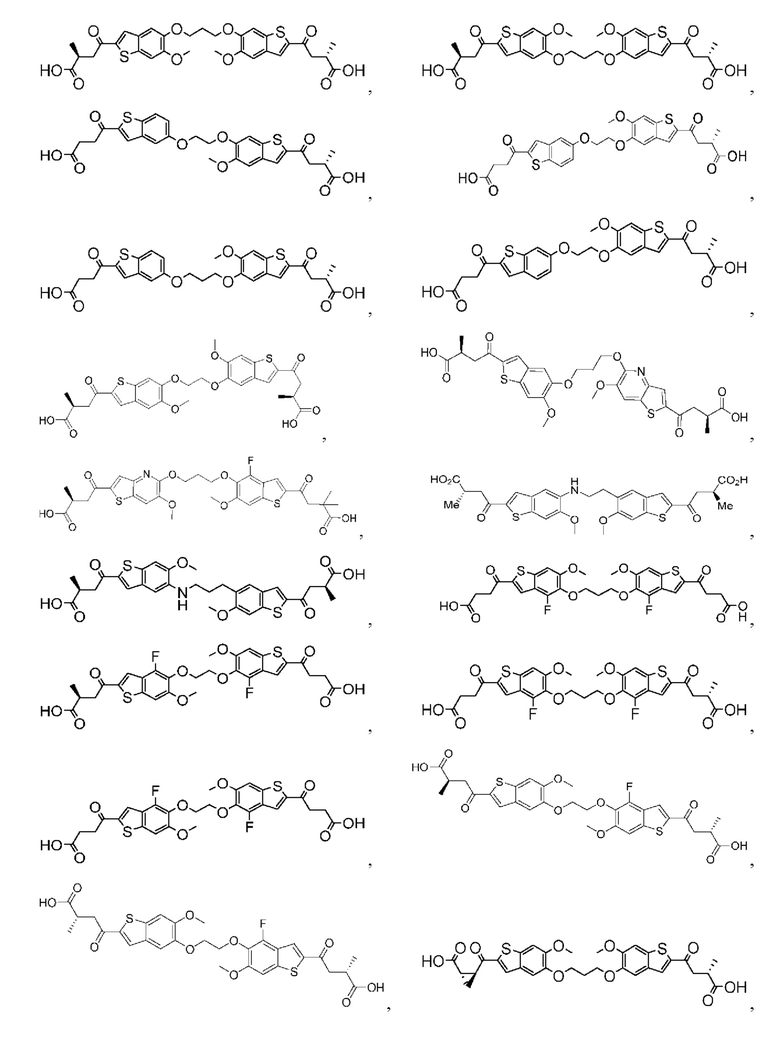
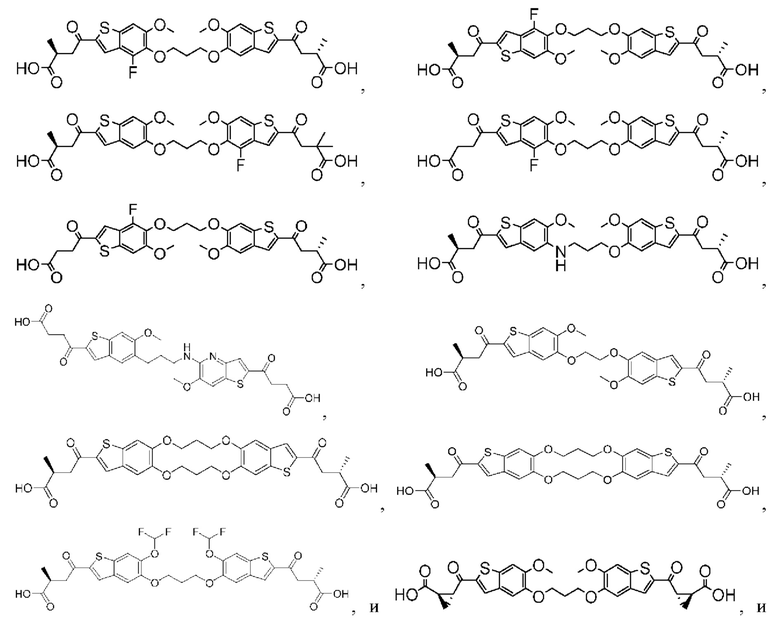
его фармацевтически приемлемым солям.
Первый аспект седьмого варианта осуществления относится к способам индукции иммунного ответа у пациента, нуждающегося в терапии, включающим введение этому пациенту терапевтически эффективного количества соединения согласно приведенному выше седьмому варианту осуществления или его фармацевтически приемлемой соли. Второй аспект седьмого варианта осуществления относится к способам индукции иммунного ответа у пациента, нуждающегося в терапии, включающим введение этому пациенту терапевтически эффективного количества композиции, содержащей соединение согласно приведенному выше седьмому варианту осуществления или его фармацевтически приемлемую соль.
Третий аспект седьмого варианта осуществления относится к способам индукции STING-зависимой продукции интерферона I типа у пациента, нуждающегося в терапии, включающим введение этому пациенту терапевтически эффективного количества соединения согласно приведенному выше седьмому варианту осуществления или его фармацевтически приемлемой соли.
Четвертый аспект седьмого варианта осуществления относится к способам индукции STING-зависимой продукции интерферона I типа у пациента, нуждающегося в терапии, включающим введение этому пациенту терапевтически эффективного количества композиции, содержащей соединение согласно приведенному выше седьмому варианту осуществления или его фармацевтически приемлемую соль.
Пятый аспект седьмого варианта осуществления относится к способам индукции STING-зависимой продукции цитокинов у пациента, нуждающегося в терапии, включающим введение этому пациенту терапевтически эффективного количества соединения согласно приведенному выше седьмому варианту осуществления или его фармацевтически приемлемой соли.
Шестой аспект седьмого варианта осуществления относится к способам индукции STING-зависимой продукции цитокинов у пациента, нуждающегося в терапии, включающим введение этому пациенту терапевтически эффективного количества композиции, содержащей соединение согласно приведенному выше седьмому варианту осуществления или его фармацевтически приемлемую соль.
Седьмой аспект седьмого варианта осуществления относится к способам лечения нарушения пролиферации клеток у пациента, нуждающегося в терапии, включающим введение этому пациенту терапевтически эффективного количества соединения согласно приведенному выше седьмому варианту осуществления или его фармацевтически приемлемой соли. В случаях седьмого аспекта седьмого варианта осуществления нарушение пролиферации клеток представляет собой рак.
Восьмой аспект седьмого варианта осуществления относится к способам лечения нарушения пролиферации клеток у пациента, нуждающегося в терапии, где указанный способ включает введение этому пациенту терапевтически эффективного количества композиции, содержащей соединение согласно седьмому варианту осуществления, приведенному выше. В случаях этого восьмого аспекта седьмого варианта осуществления нарушение пролиферации клеток представляет собой рак.
Восьмой вариант осуществления относится к соединению, выбранному из иллюстративных образцов, изображенных в Примерах с 1 по 190, показанных ниже.
Первый аспект восьмого варианта осуществления относится к способам индукции иммунного ответа у пациента, нуждающегося в терапии, включающим введение этому пациенту терапевтически эффективного количества соединения согласно приведенному выше восьмому варианту осуществления или его фармацевтически приемлемой соли.
Второй аспект восьмого варианта осуществления относится к способам индукции иммунного ответа у пациента, нуждающегося в терапии, включающим введение этому пациенту терапевтически эффективного количества композиции, содержащей соединение согласно приведенному выше восьмому варианту осуществления или его фармацевтически приемлемую соль.
Третий аспект восьмого варианта осуществления относится к способам индукции STING-зависимой продукции интерферона I типа у пациента, нуждающегося в терапии, включающим введение этому пациенту терапевтически эффективного количества соединения согласно приведенному выше восьмому варианту осуществления или его фармацевтически приемлемой соли.
Четвертый аспект восьмого варианта осуществления относится к способам индукции STING-зависимой продукции интерферона I типа у пациента, нуждающегося в терапии, включающим введение этому пациенту терапевтически эффективного количества композиции, содержащей соединение согласно приведенному выше восьмому варианту осуществления или его фармацевтически приемлемую соль.
Пятый аспект восьмого варианта осуществления относится к способам индукции STING-зависимой продукции цитокинов у пациента, нуждающегося в терапии, включающим введение этому пациенту терапевтически эффективного количества соединения согласно приведенному выше восьмому варианту осуществления или его фармацевтически приемлемой соли.
Шестой аспект восьмого варианта осуществления относится к способам индукции STING-зависимой продукции цитокинов у пациента, нуждающегося в терапии, включающим введение этому пациенту терапевтически эффективного количества композиции, содержащей соединение согласно приведенному выше восьмому варианту осуществления или его фармацевтически приемлемую соль.
Седьмой аспект восьмого варианта осуществления относится к способам лечения нарушения пролиферации клеток у пациента, нуждающегося в терапии, включающим введение этому пациенту терапевтически эффективного количества соединения согласно приведенному выше восьмому варианту осуществления или его фармацевтически приемлемой соли. В случаях этого седьмого аспекта восьмого варианта осуществления нарушение пролиферации клеток представляет собой рак.
Восьмой аспект восьмого варианта осуществления относится к способам лечения нарушения пролиферации клеток у пациента, нуждающегося в терапии, где указанный способ включает введение этому пациенту терапевтически эффективного количества композиции, содержащей соединение согласно приведенному выше восьмому варианту осуществления. В случаях этого восьмого аспекта восьмого варианта осуществления нарушение пролиферации клеток представляет собой рак.
Другие варианты осуществления настоящего раскрытия включают следующее:
(a) Фармацевтическую композицию, содержащую эффективное количество соединения общей формулы (I), соединения общей формулы (II), соединения общей формулы (III), соединения общей формулы (IV), соединения общей формулы (V), соединения общей формулы (VI) или их фармацевтически приемлемой соли и фармацевтически приемлемый носитель.
(b) Фармацевтическую композицию по (а), дополнительно содержащую активный агент, выбранный из группы, состоящей из соединений-агонистов STING, противовирусных соединений, антигенов, адъювантов, антагонистов путей CTLA-4 и PD-1 и других иммуномодулирующих агентов, липидов, липосом, пептидов, противораковых и химиотерапевтических агентов.
(c) Фармацевтическую комбинацию, которая представляет собой (i) соединение общей формулы (I), соединение общей формулы (II), соединение общей формулы (III), соединение общей формулы (IV), соединение общей формулы (V), соединение общей формулы (VI), или их фармацевтически приемлемую соль, и (ii) активный агент, выбранный из группы, состоящей из соединений-агонистов STING, противовирусных соединений, антигенов, адъювантов, антагонистов путей CTLA-4 и PD-1 и других иммуномодулирующих агентов, липидов, липосом, пептидов, противораковых и химиотерапевтических агентов; где соединение общей формулы (I), соединение общей формулы (II), соединение общей формулы (III), соединение общей (формулы IV), соединение общей (формулы V), соединение общей формулы (VI) или их фармацевтически приемлемая соль и активный агент каждый применяется в количестве, которое делает комбинацию эффективной для индукции иммунного ответа у пациента.
(d) Способ индукции иммунного ответа у пациента, который включает введение пациенту, нуждающемуся в терапии, терапевтически эффективного количества соединения общей формулы (I), соединения общей формулы (II), соединения общей формулы (III), соединения общей формулы (IV), соединения общей формулы (V), соединения общей формулы (VI) или их фармацевтически приемлемой соли.
(e) Способ индукции иммунного ответа у пациента, который включает введение пациенту, нуждающемуся в терапии, терапевтически эффективного количества композиции по (а), композиции по (b) или комбинации по (с).
(f) Способ индукции STING-зависимой продукции интерферона I типа у пациента, который включает введение пациенту, нуждающемуся в терапии, терапевтически эффективного количества соединения общей формулы (I), соединения общей формулы (II), соединения общей формулы (III), соединения общей формулы (IV), соединения общей формулы (V), соединения общей формулы (VI) или их фармацевтически приемлемой соли. Способ индукции STING-зависимой продукции интерферона I типа у пациента, который включает введение пациенту, нуждающемуся в терапии, терапевтически эффективного количества композиции по (а), композиции по (Ъ) или комбинации по (с).
(h) Способ индукции STING-зависимой продукции цитокинов у пациента, который включает введение пациенту, нуждающемуся в терапии, терапевтически эффективного количества соединения общей формулы (I), соединения общей формулы (II), соединения общей формулы (III), соединения общей формулы (IV), соединения общей формулы (V), соединения общей формулы (VI) или их фармацевтически приемлемой соли.
(i) Способ индукции STING-зависимой продукции цитокинов у пациента, который включает введение пациенту, нуждающемуся в терапии, терапевтически эффективного количества композиции по (а), композиции по (b) или комбинации по (с).
(j) Способ лечения нарушения пролиферации клеток у пациента, нуждающегося в терапии, где указанный способ включает введение пациенту терапевтически эффективного количества соединения общей формулы (I), соединения общей формулы (II), соединения общей формулы (III), соединения общей формулы (IV), соединения общей формулы (V), соединения общей формулы (VI) или их фармацевтически приемлемой соли, (k) Способ по (j), где нарушение пролиферации клеток представляет собой рак. (1) Способ лечения нарушения пролиферации клеток у пациента, нуждающегося в терапии, где указанный способ включает введение пациенту терапевтически эффективного количества композиции по (а), композиции по (b) или комбинации по (с), (m) Способ по (1), где нарушение пролиферации клеток представляет собой рак. Настоящее раскрытие также включает соединение по настоящему изобретению для применения (i) в, (ii) в качестве лекарственного средства для, или (iii) при приготовлении лекарственного средства для: (а) индукции иммунного ответа у пациента, или (b) индукции STING-зависимой продукции цитокинов у пациента. В этих применениях соединения по настоящему изобретению необязательно могут применяться в комбинации с одним или более активными агентами, выбранными из соединений-агонистов STING, противовирусных соединений, антигенов, адъювантов, антагонистов путей CTLA-4 и PD-1 и других иммуномодулирующих агентов, липидов, липосом, пептидов, противораковых агентов и химиотерапевтических агентов.
Дополнительные варианты осуществления настоящего изобретения включают фармацевтические композиции, комбинации и способы, изложенные выше в пунктах (а) -(т) и применения, изложенные в предыдущем абзаце, где соединение по настоящему изобретению, применяемое в них, представляет собой соединение по одному из вариантов осуществления, аспектов, случаев, примеров или отличительных признаков соединений, описанных выше. Во всех этих вариантах осуществления соединение необязательно может применяться в форме фармацевтически приемлемой соли, если это необходимо. В вариантах осуществления соединения, представленных выше, следует понимать, что каждый вариант осуществления может быть объединен с одним или более другими вариантами осуществления в той степени, в которой такая комбинация обеспечивает стабильное соединение и согласуется с описанием вариантов осуществления. Кроме того, следует понимать, что варианты осуществления композиций и способов, представленные как (а) - (m) выше, подразумеваются как включающие все варианты осуществления соединений, включая такие варианты осуществления, которые являются результатом комбинаций вариантов осуществления.
Термин «субъект» (альтернативно, «пациент») в контексте настоящего описания относится к млекопитающему, которое являлось объектом лечения, наблюдения или эксперимента. Млекопитающее может быть самцом или самкой. Млекопитающее может быть одним или в нескольких экземплярах, выбранным из группы, состоящей из людей, крупного рогатого скота (например, коров), представителей подотряда свинообразных (например, свиней), мелких жвачных (например, овец), парнокопытных (например, коз), представителей семейства лошадиных (например, лошадей), представителей семейства псовых (например, домашних собак), представителей семейства кошачьих (например, домашних кошек), зайцеобразных (кроликов), грызунов (например, крыс или мышей), семейства енотовых (например, енотов). В конкретных вариантах осуществления субъектом является человек. В контексте данного документа термин «иммунный ответ» относится к любому одному или более из следующего: специфический иммунный ответ, неспецифический иммунный ответ, как специфический, так и неспецифический иммунный ответ, врожденный иммунный ответ, первичный иммунный ответ, адаптивный иммунитет, вторичный иммунный ответ, иммунологическая память, активация иммунных клеток, пролиферация иммунных клеток, дифференцировка иммунных клеток и экспрессия цитокинов. В некоторых вариантах осуществления соединение общей формулы (I), соединение общей формулы (II), соединение общей формулы (III), соединение общей формулы (IV), соединение общей формулы (V), соединение общей формулы (VI) или фармацевтически приемлемая соль вышеперечисленных соединений вводится в сочетании с одним или более дополнительными терапевтическими агентами, включая противовирусные соединения, вакцины, предназначенные для стимуляции иммунного ответа на один или более из заранее определенных антигенов, адъюванты, антагонисты путей CTLA-4 и PD-1 и другие иммуномодулирующие агенты, липиды, липосомы, пептиды, противораковые агенты, химиотерапевтические агенты и так далее. В некоторых вариантах осуществления соединение общей формулы (I), соединение общей формулы (II), соединение общей формулы (III), соединение общей формулы (IV), соединение общей формулы (V), соединение общей формулы (VI) или фармацевтически приемлемая соль вышеперечисленных соединений вводится в сочетании с одной или более дополнительными композициями, включая противовирусные соединения, вакцины, предназначенные для стимуляции иммунного ответа на один или более заранее определенных антигенов, адъюванты, антагонисты путей CTLA-4 и PD-1 и другие иммуномодулирующие агенты, липиды, липосомы, пептиды, противораковые агенты, химиотерапевтические агенты и так далее.
Соединения
В контексте данного документа термин «алкил» относится к насыщенному алифатическому углеводородному радикалу с одновалентной прямой или разветвленной цепью, содержащему число атомов углерода в указанном диапазоне. Так, например, "C1-6 алкил" (или "C1-C6 алкил") относится к любому из изомеров гексилалкила и пентилалкила, а также к н-, изо-, втор- и трет-бутилу, н- и изопропилу, этилу и метилу. В качестве другого примера, «C1-4 алкил» относится к н-, изо-, втор- и трет-бутилу, н- и изопропилу, этилу и метилу.
В контексте данного документа термин «алкилен» относится к насыщенному алифатическому углеводородному радикалу с двухвалентной прямой цепью, содержащему число атомов углерода в указанном диапазоне.
В контексте данного документа термин «алкенил» относится к одновалентному ненасыщенному алифатическому углеводородному радикалу с прямой или разветвленной цепью, содержащему число атомов углерода в указанном диапазоне и включающему одну или более двойных связей.
В контексте данного документа термин «алкенилен» относится к двухвалентному ненасыщенному алифатическому углеводородному радикалу с прямой цепью, содержащему число атомов углерода в указанном диапазоне и включающему одну или более двойных связей.
В контексте данного документа термин «алкинил» относится к одновалентному ненасыщенному алифатическому углеводородному радикалу с прямой или разветвленной цепью, содержащему число атомов углерода в указанном диапазоне и включающему одну или более тройных связей.
В контексте данного документа термин «алкинилен» относится к двухвалентному ненасыщенному алифатическому углеводородному радикалу с прямой цепью, содержащему число атомов углерода в указанном диапазоне и включающему одну или более тройных связей.
В контексте данного документа термин «галоген» (или «гало») обозначает фтор, хлор, бром и йод (альтернативно, фтор, хлор, бром, и йод или F, О, Br и I).
В контексте данного документа термин «галоалкил» относится к алкильной группе, как определено выше, в которой один или более атомов водорода заменены галогеном. Таким образом, например, "C1-6 галоалкил" (или "C1-С6 галоалкил") относится к линейной или разветвленной от C1 до С6 алкильной группе, как определено выше, с одним или более галогеновыми заместителями. Термин «фторалкил» имеет аналогичное значение, за исключением того, что галогеновые заместители ограничиваются фтором. Подходящие фторалкилы включают ряд (CH2)0-4CF3 (то есть, трифторметил, 2,2,2-трифторэтил, 3,3,3-трифтор-н-пропил и т.д.).
В контексте данного документа термин «галоалкилен» относится к алкиленовой группе, как определено выше, в которой один или более атомов водорода заменены галогеном, как в галоалкильной группе, определенной выше.
В контексте данного документа термин «галоалкенил» относится к алкенильной группе, как определено выше, в которой один или более атомов водорода заменены галогеном.
В контексте данного документа термин «галоалкенилен» относится к алкениленовой группе, как определено выше, в которой один или более атомов водорода заменены галогеном.
В контексте данного документа термин «галоалкинил» относится к алкинильной группе, как определено выше, в которой один или более атомов водорода заменены галогеном.
В контексте данного документа термин «галоалкинилен» относится к алкиниленовой группе, как определено выше, в которой один или более атомов водорода заменены галогеном.
В контексте данного документа термин «алкокси» сам по себе или в комбинации включает алкильную группу, соединенную с атомом, соединяющим кислород. Термин «алкокси» также включает группы алкилового эфира, где термин «алкил» определен выше, и термин «эфир» обозначает две алкильные группы с атомом кислорода между ними. Примеры подходящих алкоксигрупп включают метокси, этокси, н-пропокси, изопропокси, н-бутокси, втор-бутокси, трет-бутокси, метоксиметан (также называемый «диметиловый эфир») и метоксиэтан (также называемый «этил метиловый эфир»).
В контексте данного документа термин «циклоалкил» относится к насыщенному углеводороду, содержащему одно кольцо с определенным числом атомов углерода. Примеры циклоалкила включают циклопропил, циклобутил, циклопентил и циклогексил. В контексте данного документа термин «гетероцикл», «гетероциклил» или «гетероциклический» представляет собой стабильный от 3- до 6-членный моноциклический фрагмент, который является либо насыщенным, либо ненасыщенным, и который состоит из атомов углерода и от одного до двух гетероатомов, выбранных из группы, состоящей из N, О и S. Гетероциклическое кольцо может быть присоединено к любому гетероатому или атому углерода, что приводит к образованию стабильной структуры. Термин включает гетероарильные фрагменты. Примеры таких гетероциклических элементов включают, но не ограничиваются ими, азепинил, бензимидазолил, бензизоксазолил, бензофуразанил, бензопиранил, бензотиопиранил, бензофурил, бензотиазолил, бензотиенил, бензоксазолил, хроманил, циннолинил, дигидробензофурил, дигидробензотиенил, дигидробензотиопиранил, дигидробензотиопиранил сульфон, 1,3-диоксоланил, фурил, имидазолидинил, имидазолинил, имидазолил, индолинил, индолил, изохроманил, изоиндолинил, изохинолинил, изотиазолидинил, изотиазолил, изотиазолидинил, морфолинил, нафтиридинил, оксадиазолил, 2-оксоазепинил, оксазолил, 2-оксопиперазинил, 2-оксопиперидинил, 2-оксопирролидинил, пиперидил, пиперазинил, пиридил, пиразинил, пиразолидинил, пиразолил, пиридазинил, пиримидинил, пирролидинил, пирролил, хиназолинил, хинолинил, хинохалинил, тетрагидрофурил, тетрагидроизохинолинил, тетрагидрохинолинил, тиаморфолинил, тиаморфолинил сульфоксид, тиазолил, тиазолинил, тиенофурил, тиенотиенил, триазолил и тиенил. В контексте данного документа термин «конденсированное кольцо» относится к циклической группе, образованной заместителями на отдельных атомах в прямом или разветвленном алкане, или к циклической группе, образованной заместителями на отдельных атомах в другом кольце.
В контексте данного документа термин «спироцикл» или «спироциклическое кольцо» относится к боковой циклической группе, образованной заместителями на отдельном атоме.
Если прямо не указано иное, все диапазоны, указанные в данном документе, являются охватывающими; то есть, диапазон включает значения для верхнего и нижнего пределов диапазона, а также все значения между ними. В качестве примера, температурные диапазоны, процентное содержание, диапазоны эквивалентов и тому подобное, описанные здесь, включают верхний и нижний пределы диапазона и любое значение в промежутке между ними. Представленные здесь числовые значения и применение термина «около» могут включать вариации ±1%, ±2%, ±3%, ±4%, ±5%, ±10%, ±15% и ±20% и их числовые эквиваленты.
В контексте данного документа термин «один или более» элементов включает один элемент, выбранный из перечня, а также сочетание двух или более элементов, выбранных из перечня.
В соединениях общей формулы (I), соединениях общей формулы (II), соединениях общей формулы (III), соединениях общей (формулы IV), соединениях общей (формулы V), соединениях общей формулы (VI) и фармацевтически приемлемых солях вышеперечисленных соединений, атомы могут иметь свое природное изотопное содержание, или один или более атомов могут быть искусственно обогащены конкретным изотопом, имеющим тот же атомный номер, но атомную массу или массовое число, отличное от атомной массы или массового числа атома, преимущественно присутствующего в природе. Подразумевается, что настоящее описание включает все подходящие изотопные варианты соединений общей формулы (I), соединений общей формулы (II), соединений общей формулы (III), соединений общей формулы (IV), соединений общей формулы (V), соединений общей формулы (VI) и фармацевтически приемлемых солей вышеперечисленных соединений. Например, различные изотопные формы водорода (Н) включают протай (1Н), дейтерий (2Н) и тритий (3Н). Протий представляет собой преобладающий изотоп водорода, встречающийся в природе. Обогащение дейтерием может давать определенные терапевтические преимущества, такие как увеличение периода полувыведения in vivo или снижение требований к дозировке, или может обеспечить соединение, применяемое в качестве стандарта для определения характеристик биологических образцов. Изотопнообогащенные соединения общей формулы (I), соединения общей формулы (II), соединения общей формулы (III), соединения общей формулы (IV), соединения общей формулы (V), соединения общей формулы (VI) и фармацевтически приемлемые соли вышеперечисленных соединений могут быть получены без лишнего экспериментального исследования с помощью обычных методик, хорошо известных специалистам в данной области, или способами, аналогичными тем, которые описаны в Схемах и Примерах в данном документе, с применением подходящих реагентов и/или промежуточных соединений, обогащенных изотопами.
В конкретных вариантах осуществления соединений общей формулы (I), соединений общей формулы (II), соединений общей формулы (III), соединений общей формулы (IV), соединений общей формулы (V), соединений общей формулы (VI) и фармацевтически приемлемых солей вышеперечисленных соединений, соединения изотопно обогащены дейтерием. В аспектах этих вариантов осуществления один или более из R1, R2, R3, R6, R8 и R9 может включать дейтерий.
Как показано в общих структурных формулах и структурах конкретных соединений, представленных в настоящем документе, прямая линия в хиральном центре включает как (R), так и (S) стереоизомеры и их смеси. Кроме того, если не указано иное (например, 100% очищенное соединение), ссылка на конкретную стереохимию в положении обеспечивает соединение, имеющее указанную стереохимию, но не исключает присутствие стереоизомеров, имеющих различную стереохимию в указанном положении. Перечисление или описание конкретного соединения в формуле изобретения (то есть, видового пункта) без обозначения конкретной стереоконфигурации или с таким обозначением менее чем для всех хиральных центров предназначено для охвата таких необозначенных хиральных центров, рацемата, рацемических смесей, каждого индивидуального энантиомера, смеси диастереоизомеров и каждого индивидуального диастереомера соединения, где такие формы возможны благодаря наличию одного или более асимметричных центров. Разделение смеси стереоизомеров может проводиться на промежуточной стадии во время синтеза соединения общей формулы (I), соединения общей формулы (II), соединения общей формулы (III), соединения общей формулы (IV), соединения общей формулы (V), соединения общей формулы (VI) или фармацевтически приемлемой соли вышеперечисленных соединений, или это может быть осуществлено с конечным рацемическим продуктом. Абсолютная стереохимия может быть определена с помощью рентгеновской кристаллографии кристаллических продуктов или кристаллических промежуточных соединений, которые при необходимости дериватизируют с помощью реагента, содержащего стереогенный центр известной конфигурации. В качестве альтернативы, абсолютная стереохимия может быть определена с помощью спектрального анализа вибрационного кругового дихроизма (VCD). Настоящее изобретение включает все такие изомеры, а также соли, сольваты (включая гидраты) и сольватированные соли таких рацематов, энантиомеров, диастереомеров, таутомеров и их смеси.
Изобретение включает все возможные энантиомеры и диастереомеры и смеси двух или более стереоизомеров, например, смеси энантиомеров и/или диастереомеров, во всех соотношениях. Таким образом, энантиомеры являются объектом изобретения в энантиомерно чистой форме, как левовращающие, так и правовращающие антиподы, в форме рацематов и в виде смесей двух энантиомеров во всех соотношениях. В случае цис/транс-изомерии изобретение включает как цис-форму, так и транс-форму, а также смеси этих форм во всех соотношениях. При желании получение отдельных стереоизомеров можно проводить путем разделения смеси общепринятыми методами, например, с помощью хроматографии или кристаллизации, с применением стереохимически однородных исходных материалов для синтеза или путем стереоселективного синтеза. Необязательно, дериватизацию можно проводить до разделения стереоизомеров. Разделение смеси стереоизомеров можно проводить на промежуточной стадии во время синтеза соединения общей формулы (I), соединения общей формулы (II), соединения общей формулы (III), соединения общей формулы (IV), соединения общей формулы (V), соединения общей формулы (VI) или фармацевтически приемлемой соли вышеперечисленных соединений, или это можно сделать с конечным рацемическим продуктом. Абсолютная стереохимия может быть определена с помощью рентгеновской кристаллографии кристаллических продуктов или кристаллических промежуточных соединений, которые при необходимости дериватизируют с помощью реагента, содержащего стереогенный центр известной конфигурации. Если не указан конкретный изомер, соль, сольват (включая гидраты) или сольватированная соль такого рацемата, энантиомера или диастереомера, настоящее изобретение включает все такие изомеры, а также соли, сольваты (включая гидраты) и сольватированные соли таких рацематов, энантиомеров, диастереомеров и их смеси.
Термин «соединение» относится к соединению и, в некоторых вариантах осуществления, к любому его гидрату или сольвату, если они стабильны. Гидрат представляет собой соединение, образующее комплекс с водой, и сольват представляет собой соединение, которое образует комплекс с растворителем, которым может быть органический растворитель или неорганический растворитель.
«Стабильное» соединение представляет собой соединение, которое может быть получено и выделено, и структура и свойства которого остаются или могут оставаться практически неизменными в течение периода времени, достаточного для применения соединения для целей, описанных в данном документе (например, терапевтическое введение пациенту). Соединения по настоящему изобретению ограничены стабильными соединениями, охватываемыми общей формулой (I), общей формулой (II), общей формулой (III), общей формулой (IV), общей формулой (V), общей формулой (VI) или их фармацевтически приемлемыми солями.
Соли
Как указано выше, соединения по настоящему изобретению могут применяться в форме фармацевтически приемлемых солей. Специалисты в данной области определят те случаи, в которых соединения по изобретению могут образовывать соли. Примеры таких соединений описаны здесь со ссылкой на возможные соли. Данная ссылка предназначена только для иллюстрации. Фармацевтически приемлемые соли могут применяться с соединениями для лечения пациентов. Однако нефармацевтические соли могут быть полезны при получении промежуточных соединений.
Термин «фармацевтически приемлемая соль» относится к соли (включая внутреннюю соль, такую как цвиттерион), которая обладает эффективностью, аналогичной исходному соединению, и которая не является биологически или иным образом нежелательной (например, не является ни токсичной, ни иным образом вредной для ее реципиента). Таким образом, вариант осуществления изобретения относится к фармацевтически приемлемым солям соединений по изобретению. Термин «соль(и)» в контексте данного документа означает любое из следующего: кислотные соли, образованные с неорганическими и/или органическими кислотами, а также основные соли, образованные с неорганическими и/или органическими основаниями. Соли соединений по изобретению могут быть образованы способами, известными специалистам в данной области, например, путем взаимодействия соединения по изобретению с некоторым количеством кислоты или основания, таким как эквивалентное количество, в такой среде, в которой соль осаждается, или в водной среде с последующей лиофилизацией.
Примеры солей присоединения кислоты включают ацетаты, аскорбаты, бензоаты, бензолсульфонаты, бисульфаты, бораты, бутираты, цитраты, камфораты, камфорсульфонаты, фумараты, гидрохлориды, гидробромиды, гидроиодиды, лактаты, малеаты, метансульфонаты («мезилаты»), нафталинсульфонаты, нитраты, оксалаты, фосфаты, пропионаты, салицилаты, сукцинаты, сульфаты, тартараты, тиоцианаты, толуолсульфонаты («тозилаты») и тому подобное. Подходящие соли включают соли присоединения кислоты, которые, например, могут быть образованы путем смешивания раствора соединения с раствором фармацевтически приемлемой кислоты, такой как соляная кислота, серная кислота, уксусная кислота, трифторуксусная кислота или бензойная кислота. Кроме того, кислоты, которые обычно считаются подходящими для образования фармацевтически полезных солей из основных фармацевтических соединений, рассматриваются, например, в P. Stahl et al, Camille G. (eds.), Handbook of Pharmaceutical Salts. Properties, Selection and Use. (2002) Zurich: Wiley-VCH; S. Berge et al, Journal of Pharmaceutical Sciences (1977) 66(1) 1-19; P. Gould, International J. of Pharmaceutics (1986) 33 201-217; Anderson et al, The Practice of Medicinal Chemistry (1996), Academic Press, New York; и в The Orange Book (Food & Drug Administration, Washington, D.C. на их веб-сайте). Эти раскрытия включены в настоящий документ посредством ссылки.
Примеры основных солей включают соли аммония, соли щелочных металлов, такие как соли натрия, лития и калия, соли щелочноземельных металлов, такие как соли кальция и магния, соли с органическими основаниями (например, органическими аминами), такими как дициклогексиламин, трет-бутиламин, холин, и соли с аминокислотами, такими как аргинин, лизин и тому подобное. Основные азотсодержащие группы могут быть кватернизованы такими агентами, как низшие алкилгалогениды (например, метил, этил, бутилхлориды, бромиды и йодиды), диалкилсульфаты (например, диметил, диэтил и дибутилсульфаты), галогениды с длинной цепью (например, децил, лаурил и стеарилхлориды, бромиды и иодиды), аралкилгалогениды (например, бензил и фенэтилбромиды) и другие. Соединения, содержащие кислотный фрагмент, могут быть смешаны с подходящими фармацевтически приемлемыми солями, чтобы получить, например, соли щелочных металлов (например, соли натрия или калия), соли щелочноземельных металлов (например, соли кальция или магния) и соли, образованные с подходящими органическими лигандами, такие как соли четвертичного аммония. Также, в случае присутствия кислотной (-СООН) или спиртовой группы, могут применяться фармацевтически приемлемые сложные эфиры для изменения характеристик растворимости или гидролиза соединения.
Подразумевается, что все такие кислые соли и основные соли являются фармацевтически приемлемыми солями в пределах объема изобретения, и все кислые и основные соли считаются эквивалентными свободным формам соответствующих соединений для целей изобретения.
Кроме того, когда соединение по изобретению содержит как основную группу, такую как, но не ограничиваясь ими, алифатический первичный, вторичный, третичный или циклический амин, ароматический или гетероариламин, пиридин или имидазол, так и кислотную группу, такую как, но не ограничиваясь ими, тетразол или карбоновая кислота, могут образовываться цвиттерионы («внутренние соли»), включенные в термины «соль(и)», применяемые в данном документе. Понятно, что определенные соединения по изобретению могут существовать в цвиттерионной форме, обладая как анионными, так и катионными центрами в одном и том же соединении и суммарным нейтральным зарядом. Такие цвиттерионы включены в изобретение.
Способы получения соединений
Несколько способов получения соединений общей формулы (I), соединений общей формулы (II), соединений общей формулы (III), соединений общей формулы (IV), соединений общей формулы (V), соединений общей формулы (VI) и фармацевтически приемлемых солей вышеперечисленных соединений описаны в следующих Схемах и Примерах. Исходные материалы и промежуточные соединения приобретаются из коммерческих источников, получают известными способами или проиллюстрированы другими способами. В некоторых случаях порядок проведения стадий реакционных схем может быть изменен для облегчения реакции или во избежание нежелательных продуктов реакции.
В следующих Способах и Схемах LG представляет собой уходящую группу, которая может быть галогенидной или трифлатной группой. Переменные, включенные в Способы и Схемы, имеют указанные значения; примерные катализаторы определены в сокращениях (ниже).
Способ 1
Димеры бензотиофена 1D и 1Е и их фармацевтически приемлемые соли могут быть получены несколькими способами. Один из способов показан на Схеме 1. Последовательность начинается с аллил-бензотиофена 1А. Путем кросс-метатезиса с катализатором Граббса 2G (второго поколения) получают олефиновые димеры 1В и 1С. Гидрированием и затем гидролизом получают димеры 1D и 1Е с разной длиной связки.
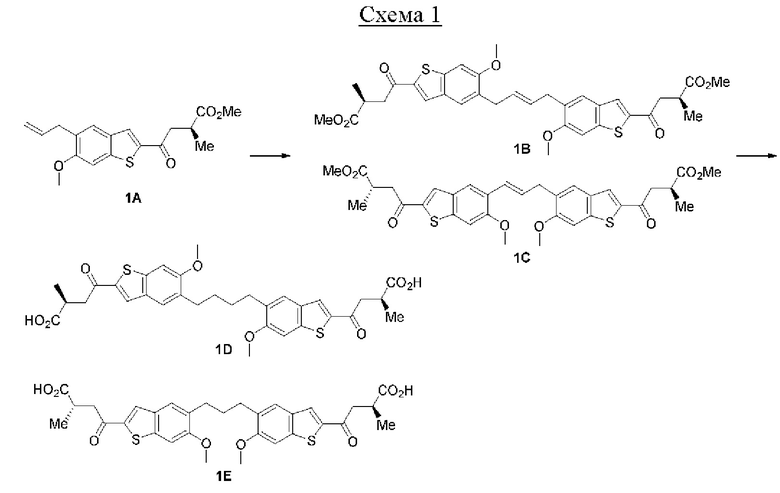
Способ 2
Другой способ получения димеров бензотиофена и их фармацевтических солей подробно показан на Схеме 2. Последовательность начинается с соответствующим образом замещенного арилгалогенида 2А. Путем перекрестного сочетания с первичным спиртом, содержащим бензотиофен (2В), с применением RockPhos Pd G3 (третьего поколения) с последующим гидролизом, получают димер 2С.
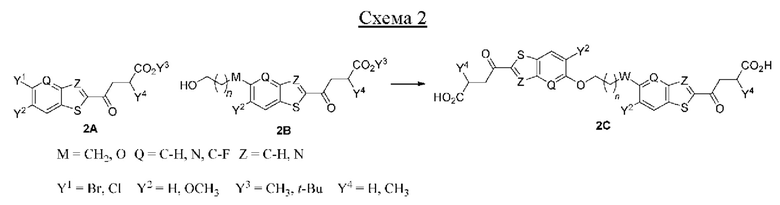
Способ 3
Другой способ получения димеров бензотиофена и их фармацевтически приемлемых солей подробно показан на Схеме 3. Путем перекрестного сочетания алкилбромида бензотиофена 3А и арилбромида 3В получают димер нитрильного эфира 3С. С помощью гидролиза водным гидроксидом натрия образуется димер 3D.
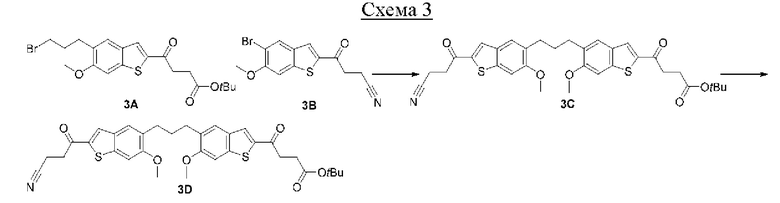
Способ 4
Другой способ получения димеров бензотифена и их фармацевтически приемлемых солей подробно показан на Схеме 4. Путем алкильной реакции Сузуки между арилбромидом 4А и алкилборонатным эфиром 4В в присутствии палладиевого катализатора с последующим гидролизом сложного эфира получают димер 4С.
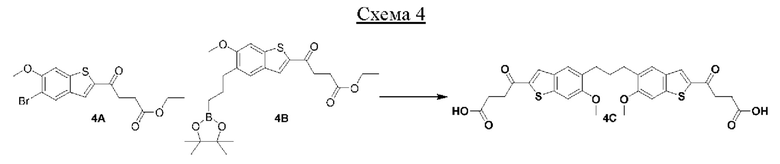
Способ 5
Другой способ получения димеров бензотиофена и их фармацевтически приемлемых солей подробно показан на Схеме 5. Путем перекрестного сочетания алкилбромида бензотиофена 5А и арилбромида 5В получают димер 5С. С помощью гидролиза получают димер 5D. Аналогичным образом арилгалогенид 5Е может быть связан с алкилбромидом 5F с образованием промежуточного соединения 5G. С помощью гидролиза получают кислоту 5Н.
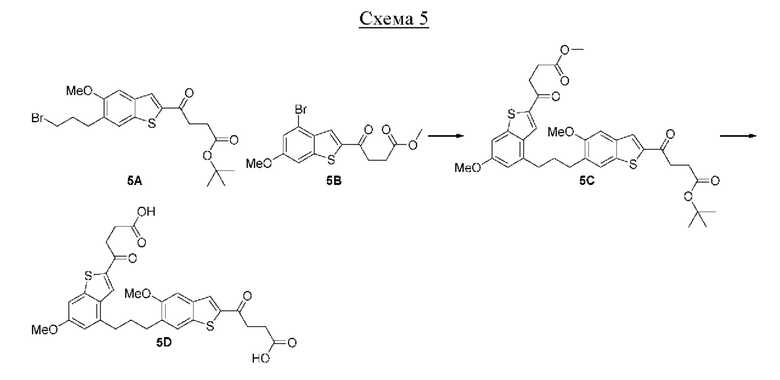
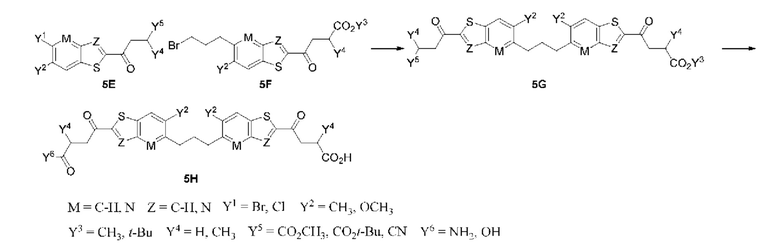
Способ 6
Другой способ получения димеров бензотиофена и их фармацевтически приемлемых солей подробно показан на Схеме 6. Последовательность начинается с алкилгалогенида 6А. Путем замещения галогенида соответствующим образом замещенным фенолом 6В в основных условиях с последующим гидролизом получают димер 6С. Аналогично, алкилхлорид 6D может быть замещен фенолом 6Е в основных условиях с получением 6F после превращения в двухосновную кислоту. Фенол 6G также может применяться для замещения алкилхлорида 6D с получением целевого 6Н после превращения в двухосновную кислоту. Наконец, алкилбромид 61 может быть замещен фенолом 6J с получением 6K после превращения в двухосновную кислоту.
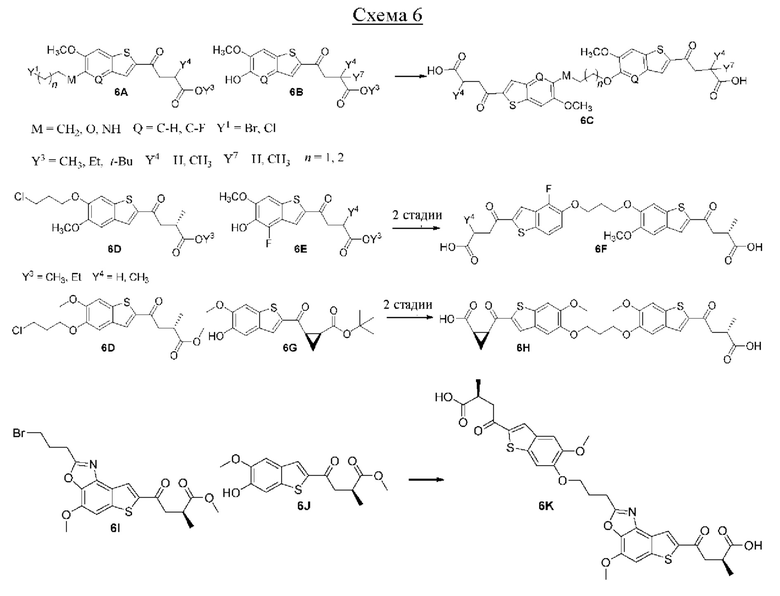
Способ 7
Другой способ получения димеров бензотиофена и их фармацевтически приемлемых солей подробно показан на Схеме 7. Последовательность начинается с реакции Мицунобу между первичным спиртом 7А и фенолом 7В с получением 7С после гидролиза сложного диэфира. Альтернативно, фенол 7D может вступать в реакцию со спиртом 7Е в условиях реакции Мицунобу. Последующим омылением получают двухосновную кислоту 7F. Фенол 7G также может применяться в реакции Мицунобу с 7Н с получением 7I после превращения сложного диэфира в двухосновную кислоту.
Аналогичным образом бис-фенол бензотиофен 7J и соответствующим образом замещенный диол (7K) могут быть подвергнуты условиям реакции Мицунобу с получением смеси 7L и 7М. С помощью гидролиза получают смесь двухосновных кислот 7N и 7O.
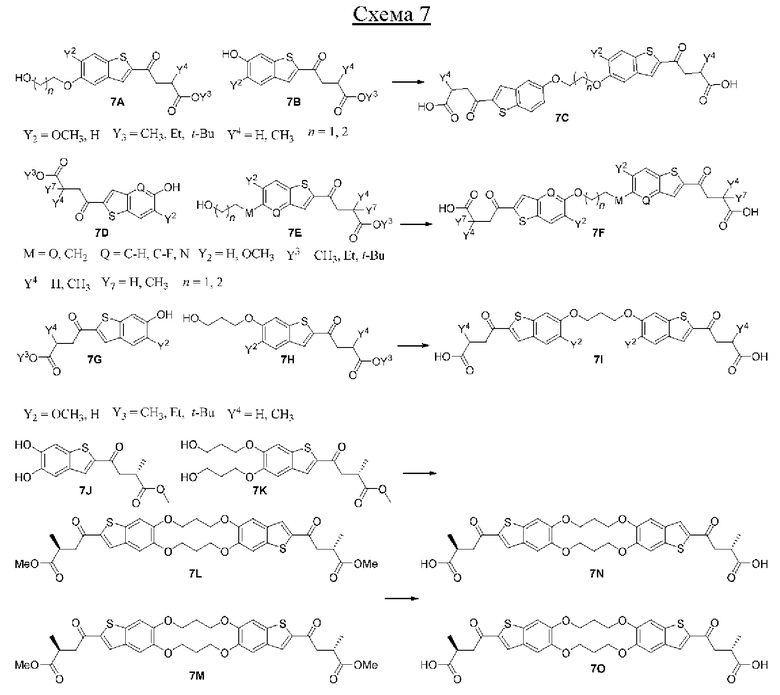
Способ 8
Другой способ получения димеров бензотиофена и их фармацевтически приемлемых солей подробно показан на Схеме 8. Последовательность начинается с соответствующим образом замещенного арилбромида 8А. Путем реакции с алкиламином 8В в присутствии палладиевого катализатора получают сложный диэфир димера алкиламина 8С. С помощью гидролиза получают целевую двухосновную кислоту 8D.
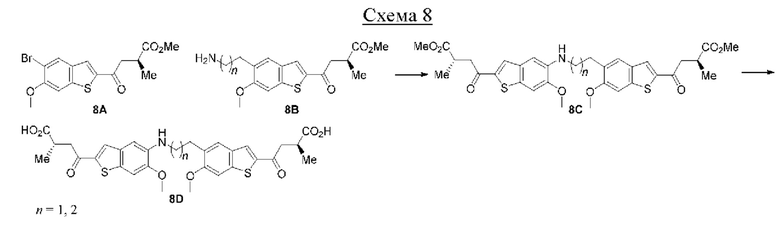
Способ 9
Другой способ получения димеров бензотиофена и их фармацевтически приемлемых солей подробно показан на Схеме 9. Последовательность начинается с восстановительного аминирования между альдегидом 9А и аминоазабензотифеном 9В в присутствии триацетоксиборгидрида натрия. Путем добавления TFA к неочищенной смеси получают целевую двухосновную кислоту 9С.
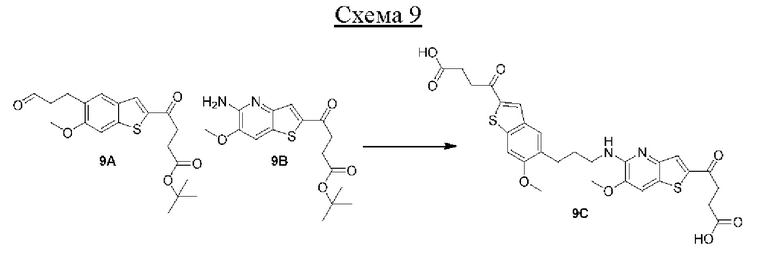
Способ 10
Другой способ получения димеров бензотиофена и их фармацевтически приемлемых солей подробно показан на Схеме 10. Последовательность начинается с бисалкилирования алкилдигалогенида соответствующим образом замещенным фенольным бензотиофеном 10А с получением сложного диэфира димера 10В. С помощью гидролиза получают целевую двухосновную кислоту 10С. Аналогично, дигалогенид может быть бисалкилирован 10D с получением двухосновной кислоты 10Е после превращения сложного диэфира в двухосновную кислоту.
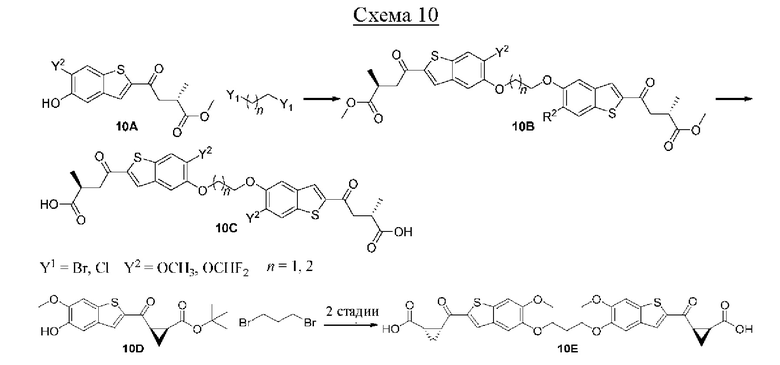
Способ 11
Другой способ получения димеров бензотиофена и их фармацевтически приемлемых солей подробно показан на Схеме 11. Последовательность начинается с алкилирования анилина 11В алкилбромидом 11А с получением 11С. Путем гидролиза сложного диэфира получают 11D.
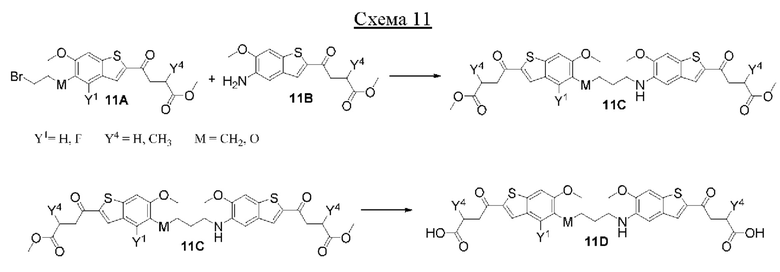
Способ 12
Другой способ получения димеров бензотиофена и их фармацевтически приемлемых солей подробно показан на Схеме 12. Последовательность начинается с алкилирования фенола 12В алкилбромидом 12А с получением 12С. Путем селективного гидролиза, опосредованного основанием, получают монокислоту 12D. Сочетанием 12D либо с сульфонамидом, либо с сульфамидом получают 12Е. С помощью кислотного гидролиза получают 12F.
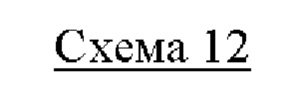
Способ 13
Другой способ получения димеров бензотиофена и их фармацевтически приемлемых солей подробно показан на Схеме 13. Последовательность начинается с алкилирования фенола 13В алкилбромидом 13А с получением 13С. Путем гидролиза сложного диэфира с последующим опосредованным кислотой снятием защиты у фенола, защищенного метоксиметилацеталем (MOM), получают 13D.
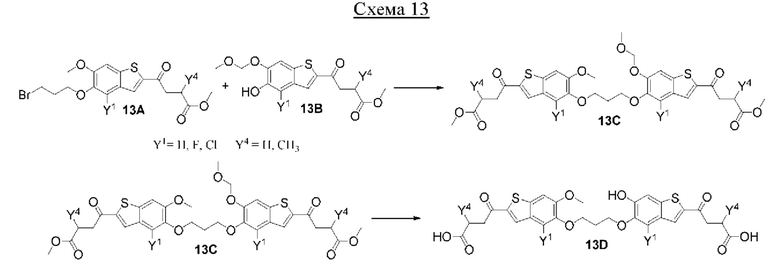
Способы применения
Описанные в настоящем документе соединения, имеющие терапевтическое применение, такие как соединения общей формулы (I), соединения общей формулы (II), соединения общей формулы (III), соединения общей формулы (IV), соединения общей формулы (V), соединения общей формулы (VI), соединения Примеров с 1 по 190 и фармацевтически приемлемые соли вышеперечисленных соединений могут быть введены пациенту с целью индукции иммунного ответа, индукции STING-зависимой продукции цитокинов и/или индукции противоопухолевой активности. Термин «введение» и его варианты (например, «введение» соединения) означает предоставление соединения пациенту, нуждающемуся в лечении. Когда соединение предоставляется в комбинации с одним или более дополнительными активными агентами (например, противовирусными агентами, полезными для лечения инфекции HCV или противоопухолевыми агентами для лечения онкологических заболеваний), «введение» и его варианты каждый понимаются как включающие одновременное и последовательное предоставление соединения или соли и других агентов.
Описанные здесь соединения могут быть агонистами STING. Эти соединения являются потенциально полезными для лечения заболеваний или нарушений, включая, но не ограничиваясь ими, нарушения пролиферации клеток. Нарушения клеточной пролиферации включают, но не ограничиваются ими, онкологические заболевания, доброкачественный папилломатоз, гестационные трофобластические заболевания и доброкачественные неопластические заболевания, такие как папиллома кожи (кондиломы) и папиллома половых органов.
В конкретных вариантах осуществления заболевание или нарушение, которое необходимо лечить, представляет собой нарушение пролиферации клеток. В некоторых вариантах осуществления нарушение пролиферации клеток представляет собой рак. В отдельных вариантах осуществления рак выбран из онкологических заболеваний головного мозга и спинного мозга, онкологических заболеваний головы и шеи, лейкемии и онкологических заболеваний крови, онкологических заболеваний кожи, онкологических заболеваний репродуктивной системы, онкологических заболеваний желудочно-кишечной системы, онкологических заболеваний печени и желчных протоков, онкологических заболеваний почек и мочевого пузыря, онкологических заболеваний костей, онкологических заболеваний легких, злокачественной мезотелиомы, сарком, лимфом, аденокарцином, онкологических заболеваний щитовидной железы, опухолей сердца, герминогенных опухолей, злокачественных нейроэндокринных (карциноидных) опухолей, срединных онкологических заболеваний и онкологических заболеваний без выявленного первичного очага (то есть, онкологических заболеваний, при которых обнаружен метастазирующий рак, но первичная локализация рака неизвестна). В конкретных вариантах осуществления рак присутствует у взрослого пациента; в дополнительных вариантах осуществления рак присутствует у пациента детского возраста. В конкретных вариантах осуществления рак связан со СПИДом.
В конкретных вариантах осуществления рак выбран из онкологических заболеваний головного мозга и спинного мозга. В конкретных вариантах осуществления рак выбран из группы, состоящей из анапластических астроцитом, глиобластом, астроцитом и эстезионейробластом (также известных как ольфакторные нейробластомы). В конкретных вариантах осуществления рак головного мозга выбран из группы, состоящей из астроцитарной опухоли (например, пилоцитарной астроцитомы, субэпендимальной гигантоклеточной астроцитомы, диффузной астроцитомы, плеоморфной ксантоастроцитомы, анапластической астроцитомы, астроцитомы, гигантоклеточной глиобластомы, глиобластомы, вторичной глиобластомы, первичной глиобластомы взрослых и первичной глиобластомы детского возраста), олигодендроглиальной опухоли (например, олигодендроглиомы и анапластической олигодендроглиомы), олигоастроцитарной опухоли (например, олигоастроцитомы и анапластической олигоастроцитомы), эпендимомы (например, миксопапиллярной эпендимомы и анапластической эпендимомы); медуллобластомы, примитивной нейроэктодермальной опухоли, шванномы, менингиомы, атипичной менингиомы, анапластической менингиомы, аденомы гипофиза, глиомы ствола головного мозга, астроцитомы мозжечка, церебральной астроцитомы/злокачественной глиомы, глиомы зрительного пути и гипоталамической глиомы, и первичной лимфомы центральной нервной системы. В конкретных случаях этих вариантов осуществления рак головного мозга выбран из группы, состоящей из глиомы, мультиформной глиобластомы, параганглиомы и супратенториальных примитивных нейроэктодермальных опухолей (sPNET).
В конкретных вариантах осуществления рак выбран из онкологических заболеваний головы и шеи, включая онкологические заболевания носоглотки, полости носа и околоносовых пазух, онкологические заболевания гипофарингеальной области, онкологические заболевания ротовой полости (например, плоскоклеточные карциномы, лимфомы и саркомы), онкологические заболевания губ, онкологические заболевания ротоглотки, опухоли слюнных желез, онкологические заболевания гортани (например, плоскоклеточные карциномы гортани, рабдомиосаркомы) и онкологические заболевания глаза или онкологические заболевания глазной области. В конкретных вариантах осуществления онкологическое заболевание глаза выбрано из группы, состоящей из внутриглазной меланомы и ретинобластомы.
В конкретных вариантах осуществления рак выбран из лейкемии и онкологических заболеваний крови. В конкретных вариантах осуществления рак выбран из группы, состоящей из миелопролиферативных новообразований, миелодиспластических синдромов, миелодиспластических/миелопролиферативных новообразований, острого миелоидного лейкоза (AML), миелодиспластического синдрома (MDS), хронического миелоидного лейкоза (CML), миелопролиферативного новообразования (MPN), пост-MPN АМЛ, пост-MDS АМЛ, del(5q)-ассоциированного MDS или AML высокого риска, хронического миелоидного лейкоза в бластной фазе, ангиоиммунобластной лимфомы, острого лимфобластного лейкоза, гистиоцитоза из клеток Лангерганса, волосатоклеточного лейкоза и новообразований плазматических клеток, включая плазмоцитомы множественные миеломы. Упомянутые здесь лейкозы могут быть острыми или хроническими.
В конкретных вариантах осуществления рак выбран из онкологических заболеваний кожи. В отдельных вариантах осуществления рак кожи выбран из группы, состоящей из меланомы, плоскоклеточного рака и базальноклеточного рака.
В конкретных вариантах осуществления рак выбран из онкологических заболеваний репродуктивной системы. В отдельных вариантах осуществления рак выбран из группы, состоящей из онкологических заболеваний груди, онкологических заболеваний шейки матки, онкологических заболеваний влагалища, онкологических заболеваний яичников, онкологических заболеваний простаты, онкологических заболеваний полового члена и онкологических заболеваний яичек. В конкретных случаях этих вариантов осуществления рак представляет собой рак груди, выбранный из группы, состоящей из протоковых карцином и филлоидных цистосарком. В конкретных случаях этих вариантов осуществления рак груди может представлять собой рак груди у мужчин или рак груди у женщин. В конкретных случаях этих вариантов осуществления рак представляет собой рак шейки матки, выбранный из группы, состоящей из плоскоклеточных карцином и аденокарцином. В конкретных случаях этих вариантов осуществления рак представляет собой рак яичника, выбранный из группы, состоящей из эпителиальных онкологических заболеваний.
В конкретных вариантах осуществления рак выбран из онкологических заболеваний желудочно-кишечной системы. В отдельных вариантах осуществления рак выбран из группы, состоящей из онкологических заболеваний пищевода, онкологических заболеваний желудочно-кишечного тракта (также известных как рак желудка), карциноидных опухолей желудочно-кишечного тракта, онкологических заболеваний поджелудочной железы, онкологических заболеваний желчного пузыря, онкологических заболеваний прямой кишки и рака анального канала. В случаях этих вариантов осуществления рак выбран из группы, состоящей из плоскоклеточных карцином пищевода, аденокарцином пищевода, аденокарцином желудка, карциноидных опухолей желудочно-кишечного тракта, стромальных опухолей желудочно-кишечного тракта, лимфом желудка, лимфом желудочно-кишечного тракта, солидных псевдопапиллярных опухолей поджелудочной железы, панкреатобластомы, инсулином, карцином поджелудочной железы, включая ацинарно-клеточные карциномы и протоковые аденокарциномы, аденокарциномы желчного пузыря, колоректальные аденокарциномы и плоскоклеточные карциномы анального канала.
В конкретных вариантах осуществления рак выбран из онкологических заболеваний печени и желчных протоков. В конкретных вариантах осуществления рак представляет собой рак печени (также известный как гепатоцеллюлярная карцинома). В конкретных вариантах осуществления рак представляет собой рак желчного протока (также известный как холангиокарцинома); в случаях этих вариантов осуществления рак желчного протока выбран из группы, состоящей из внутрипеченочной холангиокарциномы и внепеченочной холангиокарциномы.
В конкретных вариантах осуществления рак выбран из рака почки и мочевого пузыря. В конкретных вариантах осуществления рак представляет собой рак почки, выбранный из группы, состоящей из почечно-клеточного рака, опухолей Вильмса и переходно-клеточных онкологических заболеваний. В конкретных вариантах осуществления рак представляет собой рак мочевого пузыря, выбранный из группы, состоящей из уротелиальной карциномы (переходно-клеточной карциномы), плоскоклеточных карцином и аденокарцином. В конкретных вариантах осуществления рак выбран из онкологических заболеваний костей. В конкретных вариантах осуществления рак кости выбран из группы, состоящей из остеосаркомы, злокачественной фиброзной гистиоцитомы кости, саркомы Юинга, хордомы (рак кости, возникающий из остатков спинной струны вблизи позвоночника). В конкретных вариантах осуществления рак выбран из онкологических заболеваний легких. В конкретных вариантах осуществления рак легкого выбран из группы, состоящей из немелкоклеточного рака легкого, мелкоклеточного рака легкого, бронхиальных опухолей и плевропульмональных бластом.
В конкретных вариантах осуществления рак выбран из злокачественной мезотелиомы. В конкретных вариантах осуществления рак выбран из группы, состоящей из эпителиальной мезотелиомы и саркоматоидных карцином.
В конкретных вариантах осуществления рак выбран из сарком. В отдельных вариантах осуществления саркома выбрана из группы, состоящей из центральной хондросаркомы, центральной и периостальной хондромы, фибросаркомы, светлоклеточной саркомы сухожильных влагалищ и саркомы Капоши.
В конкретных вариантах осуществления рак выбран из лимфом. В отдельных вариантах осуществления рак выбран из группы, состоящей из лимфомы Ходжкина (например, клеток Рида-Штернберга), неходжкинской лимфомы (например, диффузной В-клеточной крупноклеточной лимфомы, фолликулярной лимфомы, грибовидного микоза, синдрома Сезари, первичной лимфомы центральной нервной системы), Т-клеточных лимфом кожи, первичных лимфом центральной нервной системы.
В конкретных вариантах осуществления рак выбран из железистых онкологических заболеваний. В отдельных вариантах осуществления рак выбран из группы, состоящей из адренокортикального рака (также известного как адренокортикальная карцинома или карцинома коры надпочечников), феохромоцитом, параганглиом, опухолей гипофиза, тимомы и карцином тимуса.
В конкретных вариантах осуществления рак выбран из онкологических заболеваний щитовидной железы. В отдельных вариантах осуществления рак щитовидной железы выбран из группы, состоящей из медуллярных карцином щитовидной железы, папиллярных карцином щитовидной железы и фолликулярных карцином щитовидной железы. В отдельных вариантах осуществления рак выбран из группы, состоящей из злокачественных экстракраниальных эмбрионально-клеточных опухолей и злокачественных экстрагонадных эмбрионально-клеточных опухолей. В конкретных случаях этих вариантов осуществления злокачественные экстрагонадные эмбрионально-клеточные опухоли выбраны из группы, состоящей из несемином и семином. В конкретных вариантах осуществления рак выбран из опухолей сердца. В отдельных вариантах осуществления опухоль сердца выбрана из группы, состоящей из злокачественной тератомы, лимфомы, рабдомиосакромы, ангиосаркомы, хондросаркомы, фибросаркомы у детей и синовиальной саркомы.
В конкретных вариантах осуществления нарушение пролиферации клеток выбрано из доброкачественного папилломатоза, доброкачественных неопластических заболеваний и гестационных трофобластических заболеваний. В отдельных вариантах осуществления доброкачественное неопластическое заболевание выбрано из папилломы кожи (кондилом) и папилломы половых органов. В отдельных вариантах осуществления гестационное трофобластическое заболевание выбрано из группы, состоящей из пузырного заноса и гестационной трофобластической неоплазии (например, инвазивного пузырного заноса, хориокарцином, трофобластических опухолей в плацентарной области и эпителиоидных трофобластических опухолей).
В контексте данного документа термины «терапия» и «лечение» относятся ко всем процессам, в которых может иметь место замедление, прерывание, остановка, контроль или остановка прогрессирования заболевания или расстройства, описанного здесь. Эти термины не обязательно указывают на полное устранение всех симптомов заболевания или расстройства.
Термины «назначение» и/или «введение» соединения следует понимать как включающие предоставление пациенту соединения, описанного в данном документе, или его фармацевтически приемлемой соли и композиций из вышеперечисленных. Количество соединения, вводимого пациенту, является количеством, достаточным для индукции иммунного ответа и/или для индукции STING-зависимой продукции интерферона I типа у пациента. В одном варианте осуществления количество соединения может быть «эффективным количеством» или «терапевтически эффективным количеством», таким образом, чтобы рассматриваемое соединение вводилось в количестве, которое вызовет, соответственно, биологическую или медицинскую (то есть, предназначенную для лечения) ответную реакцию ткани, системы, животного или человека, которую добивается получить исследователь, ветеринар, врач или другой клиницист. Эффективное количество не обязательно включает критерии токсичности и безопасности, связанные с введением соединения.
Эффективное количество соединения будет варьироваться в зависимости от конкретного выбранного соединения (например, с учетом активности, эффективности и/или периода полувыведения соединения); выбранного способа введения; состояния, которое подлежит лечению; тяжести состояния, которое подлежит лечению; возраста, роста, веса и физического состояния пациента, проходящего лечение; истории болезни пациента, проходящего лечение; продолжительности лечения; характера сопутствующей терапии; целевого терапевтического эффекта и подобных факторов, и могут быть определены по стандартным методикам специалистом в данной области.
Описанные в настоящем документе соединения могут быть введены любым подходящим путем, включая пероральное и парентеральное введение. Парентеральное введение обычно осуществляется путем инъекции или инфузии и включает внутривенную, внутримышечную и подкожную инъекцию или инфузию.
Соединения, раскрытые в данном документе, могут быть введены однократно или в соответствии со схемой введения, при которой несколько доз вводят с различными интервалами времени в течение заданного периода времени. Например, дозы могут быть введены один, два, три или четыре раза в день. Дозы могут быть введены до достижения желаемого терапевтического эффекта или неограниченно долго для поддержания желаемого терапевтического эффекта. Подходящие режимы дозирования для соединения, описанного здесь, зависят от фармакокинетических свойств этого соединения, таких как абсорбция, распределение и период полувыведения, которые могут быть определены квалифицированным специалистом. Кроме того, подходящие режимы дозирования, включая продолжительность таких режимов, для соединения, описанного в настоящем документе, зависят от заболевания или состояния, подлежащего лечению, тяжести заболевания или состояния, возраста и физического состояния пациента, проходящего лечение, медицинской истории пациента, проходящего лечение, характера сопутствующей терапии, желаемого терапевтического эффекта и подобных факторов в пределах компетенции и профессиональных знаний квалифицированного специалиста. Таким квалифицированным специалистам также будет понятно, что подходящие режимы дозирования могут потребовать корректировки, учитывая индивидуальную ответную реакцию пациента на режим дозирования или с течением времени, когда индивидуальные потребности пациента изменяются. Типичные суточные дозировки могут варьироваться в зависимости от конкретного выбранного пути введения.
Один вариант осуществления настоящего раскрытия относится к способу лечения нарушения пролиферации клеток, включающему введение терапевтически эффективного количества соединения общей формулы (I), соединения общей формулы (II), соединения общей формулы (III), соединения общей формулы (IV), соединения общей формулы (V), соединения общей формулы (VI) и фармацевтически приемлемых солей вышеперечисленных соединений пациенту, нуждающемуся в таком лечении. В вариантах осуществления изобретения заболевание или нарушение, которое необходимо лечить, представляет собой нарушение пролиферации клеток. В аспектах этих вариантов осуществления нарушение пролиферации клеток представляет собой рак. В дополнительных аспектах этих вариантов осуществления рак выбран из онкологических заболеваний головного мозга и спинного мозга, онкологических заболеваний головы и шеи, лейкемии и онкологических заболеваний крови, онкологических заболеваний кожи, онкологических заболеваний репродуктивной системы, онкологических заболеваний желудочно-кишечной системы, онкологических заболеваний печени и желчных протоков, онкологических заболеваний почек и мочевого пузыря, онкологических заболеваний костей, онкологических заболеваний легких, злокачественной мезотелиомы, сарком, лимфом, железистых онкологических заболеваний, онкологических заболеваний щитовидной железы, опухолей сердца, эмбрионально-клеточных опухолей, злокачественных нейроэндокринных (карциноидных) опухолей, онкологических заболеваний срединного тракта и онкологических заболеваний с неизвестной первичной локализацией.
В одном варианте осуществления в настоящем документе раскрыто применение соединения общей формулы (I), соединения общей формулы (II), соединения общей формулы (III), соединения общей формулы (IV), соединения общей формулы (V), соединения общей формулы (VI) или фармацевтически приемлемой соли вышеперечисленных соединений в терапии. Соединение может быть полезным в способе индукции иммунного ответа и/или индукции STING-зависимой продукции интерферона типа I у пациента, такого как млекопитающее, нуждающегося в таком ингибировании, включающем введение эффективного количества соединения пациенту. В одном варианте осуществления в настоящем документе раскрыта фармацевтическая композиция, содержащая, по меньшей мере, одно соединение общей формулы (I), по меньшей мере, одно соединение общей формулы (II), по меньшей мере, одно соединение общей формулы (III), по меньшей мере, одно соединение общей формулы (IV), по меньшей мере, одно соединение общей формулы (V), по меньшей мере, одно соединение общей формулы (VI) или, по меньшей мере, одну фармацевтически приемлемую соль вышеперечисленных соединений, для применения в предполагаемом лечении с целью индукции иммунного ответа и/или индукции STING-зависимой продукции интерферона I типа.
Один из вариантов осуществления, раскрытый в данном документе, представляет собой применение соединения общей формулы (I), соединения общей формулы (II), соединения общей формулы (III), соединения общей формулы (IV), соединения общей формулы (V), соединения общей формулы (VI) или фармацевтически приемлемой соли вышеперечисленных соединений при изготовлении лекарственного средства для индукции иммунного ответа и/или для индукции STING-зависимой продукции интерферона типа I. В вариантах осуществления изобретения заболевание или нарушение, подлежащее лечению, представляет собой нарушение пролиферации клеток. В аспектах этих вариантов осуществления нарушение пролиферации клеток представляет собой рак. В дополнительных аспектах этих вариантов осуществления рак выбран из онкологических заболеваний головного мозга и спинного мозга, онкологических заболеваний головы и шеи, лейкемии и онкологических заболеваний крови, онкологических заболеваний кожи, онкологических заболеваний репродуктивной системы, онкологических заболеваний желудочно-кишечной системы, онкологических заболеваний печени и желчных протоков, онкологических заболеваний почек и мочевого пузыря, онкологических заболеваний костей, онкологических заболеваний легких, злокачественной мезотелиомы, сарком, лимфом, железистых онкологических заболеваний, онкологических заболеваний щитовидной железы, опухолей сердца, эмбрионально-клеточных опухолей, злокачественных нейроэндокринных (карциноидных) опухолей, онкологических заболеваний срединного тракта и онкологических заболеваний с неизвестной первичной локализацией.
Композиции
Термин «композиция» в контексте данного документа предназначен для охвата лекарственной формы, содержащей указанное соединение в указанном количестве, а также любой лекарственной формы, которая является прямым или косвенным результатом комбинации указанного соединения в указанном количестве. Такой термин предназначен для охвата лекарственной формы, содержащей соединение общей формулы (I), соединение общей формулы (II), соединение общей формулы (III), соединение общей формулы (IV), соединение общей формулы (V), соединение общей формулы (VI) или фармацевтически приемлемую соль вышеперечисленных соединений и один или более фармацевтически приемлемых носителей или вспомогательных веществ. Соответственно, композиции по настоящему раскрытию охватывают любую композицию, полученную путем смешивания соединения по настоящему раскрытию и одного или более фармацевтически приемлемых носителей или вспомогательных веществ. Под термином «фармацевтически приемлемый» подразумевается, что носители или вспомогательные вещества совместимы с описанным здесь соединением и с другими ингредиентами композиции.
С целью индукции иммунного ответа и/или индукции STING-зависимой продукции интерферона типа I соединения общей формулы (I), соединения общей формулы (II), соединения общей формулы (III), соединения общей формулы (IV), соединения общей формулы (V), соединения общей формулы (VI) или фармацевтически приемлемые соли вышеперечисленных соединений могут быть введены с помощью средств, обеспечивающих контакт активного агента с местом действия агента. Соединения могут быть введены обычными способами, доступными для применения в сочетании с фармацевтическими препаратами, либо в виде отдельных терапевтических агентов, либо в комбинации терапевтических агентов. Соединения могут быть введены отдельно, но обычно вводятся с фармацевтическим носителем, выбранным на основе выбранного пути введения и стандартной фармацевтической практики.
В одном варианте осуществления в настоящем документе раскрыта композиция, содержащая соединение общей формулы (I), соединение общей формулы (II), соединение общей формулы (III), соединение общей формулы (IV), соединение общей формулы (V), соединение общей формулы (VI) или фармацевтически приемлемую соль вышеперечисленных соединений и один или более фармацевтически приемлемых носителей или вспомогательных веществ. Композиция может быть приготовлена и упакована в нерасфасованной форме, в которой терапевтически эффективное количество соединения по настоящему изобретению может быть извлечено и затем введено пациенту, например, с порошками или сиропами. Альтернативно, композиция может быть приготовлена и упакована в единичную лекарственную форму, в которой каждая физически дискретная единица содержит терапевтически эффективное количество соединения общей формулы (I), соединения общей формулы (II), соединения общей формулы (III), соединения общей формулы (IV), соединения общей формулы (V), соединения общей формулы (VI) или фармацевтически приемлемую соль вышеперечисленных соединений. Раскрытые здесь соединения и фармацевтически приемлемый носитель или вспомогательное вещество (вспомогательные вещества) как правило будут входить в состав дозированной формы, адаптированной для введения пациенту желаемым путем введения. Например, лекарственные формы включают формы, адаптированные для (1) перорального введения, такие как таблетки, капсулы, каплеты, пилюли, пастилки, порошки, сиропы, эликсиры, суспензии, растворы, эмульсии, саше и облатки; и (2) парентерального введения, такого как стерильные растворы, суспензии и порошки для восстановления. Подходящие фармацевтически приемлемые носители или вспомогательные вещества будут варьироваться в зависимости от выбранной конкретной лекарственной формы. Кроме того, подходящие фармацевтически приемлемые носители или вспомогательные вещества могут быть выбраны для конкретной функции, которую они могут выполнять в композиции. Например, некоторые фармацевтически приемлемые носители или вспомогательные вещества могут быть выбраны из-за их способности облегчать производство однородных лекарственных форм. Некоторые фармацевтически приемлемые носители или вспомогательные вещества могут быть выбраны из-за их способности облегчать производство стабильных лекарственных форм. Некоторые фармацевтически приемлемые носители или вспомогательные вещества могут быть выбраны из-за их способности облегчать перенос или транспортировку соединения, раскрытого в данном документе, после введения пациенту, из одного органа или части тела в другой орган или другую часть тела. Некоторые фармацевтически приемлемые носители или вспомогательные вещества могут быть выбраны из-за их способности улучшать соблюдение пациентом режима лечения.
Подходящие фармацевтически приемлемые вспомогательные вещества включают следующие типы вспомогательных веществ: разбавители, смазки, связывающие вещества, разрыхлители, наполнители, скользящие вещества, гранулирующие агенты, покрывающие агенты, смачивающие агенты, растворители, сорастворители, суспендирующие агенты, эмульгаторы, подсластители, ароматизаторы, агенты, маскирующие запахи, красители, агенты, предотвращающие слеживание, гумектанты, хелатирующие агенты, пластификаторы, агенты, увеличивающие вязкость, антиоксиданты, консерванты, стабилизаторы, поверхностно-активные вещества и буферные агенты.
Квалифицированный специалист обладает знаниями и профессиональными навыками в данной области, чтобы выбрать подходящие фармацевтически приемлемые носители и вспомогательные вещества в подходящих количествах для применения в композициях по настоящему раскрытию. Кроме того, специалистам в данной области доступен ряд ресурсов, в которых описаны фармацевтически приемлемые носители и вспомогательные вещества, и они могут быть полезны при выборе подходящих фармацевтически приемлемых носителей и вспомогательных веществ. Примеры включают Remington's Pharmaceutical Sciences (Mack Publishing Company), The Handbook of Pharmaceutical Additives (Gower Publishing Limited) и The Handbook of Pharmaceutical Excipients (the American Pharmaceutical Association and the Pharmaceutical Press).
Композиции по раскрытию получают с применением методик и способов, известных специалистам в данной области. Некоторые способы, обычно используемые в данной области техники, описаны в Remington's Pharmaceutical Sciences (Mack Publishing Company).
В одном варианте осуществления настоящее изобретение относится к твердой пероральной лекарственной форме, такой как таблетка или капсула, содержащей терапевтически эффективное количество соединения общей формулы (I), соединения общей формулы (II), соединения общей формулы (III), соединения общей формулы (IV), соединения общей формулы (V), соединения общей формулы (VI) или фармацевтически приемлемой соли вышеперечисленных соединений и разбавитель или наполнитель. Подходящие разбавители и наполнители включают лактозу, сахарозу, декстрозу, маннит, сорбит, крахмал (например, кукурузный крахмал, картофельный крахмал и предварительно желатинизированный крахмал), целлюлозу и ее производные (например, микрокристаллическую целлюлозу), сульфат кальция и двухосновный фосфат кальция. Твердая пероральная лекарственная форма может дополнительно содержать связывающее вещество. Подходящие связывающие вещества включают крахмал (например, кукурузный крахмал, картофельный крахмал и предварительно желатинизированный крахмал), желатин, камедь, альгинат натрия, альгиновую кислоту, трагакант, гуаровую камедь, повидон и целлюлозу и ее производные (например, микрокристаллическую целлюлозу). Твердая пероральная лекарственная форма может дополнительно содержать разрыхлитель. Подходящие разрыхлители включают кросповидон, натрийгликолят крахмала, кроскармелозу, альгиновую кислоту и натрий карбоксиметилцеллюлозу. Твердая пероральная лекарственная форма может дополнительно содержать смазывающее вещество. Подходящие смазывающие вещества включают стеариновую кислоту, стеарат магния, стеарат кальция и тальк.
Где это целесообразно, лекарственные формы для перорального введения могут быть микрокапсулированы. Композиция также может быть приготовлена для продления или задержки высвобождения, например, путем покрытия или заключения вещества в виде частиц в полимеры, воск или тому подобное.
Соединения, раскрытые в данном документе, также могут быть связаны с растворимыми полимерами в качестве нацеливаемых носителей лекарственных средств. Такие полимеры могут включать поливинилпирролидон, пирансополимер, полигидроксипропилметакриламидфенол, полигидроксиэтиласпартамидфенол или полиэтиленоксидполилизин, замещенный остатками пальмитоила. Кроме того, соединения по настоящему описанию могут быть соединены с классом биоразлагаемых полимеров, полезных для достижения контролируемого высвобождения лекарственного средства, например, полимолочной кислотой, полиэпсилонкапролактоном, полигидроксимасляной кислотой, сложными полиортоэфирами, полиацеталями, полидигидропиранами, полицианакрилатами и поперечно-сшитыми или амфипатическими блок-сополимерами гидрогелей.
В одном варианте осуществления изобретение относится к жидкой пероральной лекарственной форме. Жидкости для перорального применения, такие как растворы, сиропы и эликсиры, могут быть приготовлены в форме единичной дозы таким образом, что данное количество содержит заранее определенное количество соединения или его фармацевтически приемлемой соли, раскрытых в настоящем документе. Сиропы могут быть приготовлены путем растворения соединения по настоящему изобретению в подходящим образом ароматизированном водном растворе; тогда как эликсиры готовят при помощи нетоксичного спиртового носителя. Суспензии могут быть составлены путем диспергирования соединения по настоящему изобретению в нетоксичном носителе. Также могут быть добавлены солюбилизаторы и эмульгаторы, такие как этоксилированные изостеариловые спирты и полиоксиэтиленсорбитоловые эфиры, консерванты, вкусовые добавки, такие как масло перечной мяты или другие натуральные подсластители, или сахарин или другие искусственные подсластители и тому подобное.
В одном варианте осуществления изобретение относится к композициям для парентерального введения. Композиции, адаптированные для парентерального введения, включают водные и неводные стерильные растворы для инъекций, которые могут содержать антиоксиданты, буферы, бактериостаты и растворенные вещества, которые делают композицию изотонической с кровью предполагаемого реципиента; и водные и неводные стерильные суспензии, которые могут включать суспендирующие агенты и загустители. Композиции могут быть представлены в однодозовых или многодозовых контейнерах, например, запаянных ампулах и флаконах, и могут храниться в сублимированном (лиофилизированном) состоянии, требующем только добавления стерильного жидкого носителя, например, воды для инъекций, непосредственно перед применением. Экстемпоральные растворы для инъекций и суспензии могут быть приготовлены из стерильных порошков, гранул и таблеток.
Комбинации
Соединения общей формулы (I), соединения общей формулы (II), соединения общей формулы (III), соединения общей формулы (IV), соединения общей формулы (V), соединения общей формулы (VI) и/или фармацевтически приемлемые соли вышеперечисленных соединений могут быть введены в комбинации с одним или более дополнительными активными агентами. В вариантах осуществления изобретения одно или более соединений общей формулы (I), соединений общей формулы (II), соединений общей формулы (III), соединений общей формулы (IV), соединений общей формулы (V), соединений общей формулы (VI) или одна или более фармацевтически приемлемых солей вышеперечисленных соединений, и один или более дополнительных активных агентов могут быть введены совместно. Дополнительный активный агент (агенты) может быть введен в виде разовой лекарственной формы с соединением общей формулы (I), соединением общей формулы (II), соединением общей формулы (III), соединением общей формулы (IV), соединением общей формулы (V), соединением общей формулы (VI) или фармацевтически приемлемой солью вышеперечисленных соединений, или дополнительный активный агент (агенты) может быть введен в отдельной лекарственной форме (формах) из дозированной формы, содержащей соединение общей формулы (I), соединение общей формулы (II), соединение общей формулы (III), соединение общей формулы (IV), соединение общей формулы (V), соединение формулы формулы (VI) или фармацевтически приемлемую соль вышеперечисленных соединений.
Дополнительный активный агент (агенты) может быть предоставлен в виде фармацевтически приемлемой соли, где это необходимо.
Дополнительный активный агент (агенты) может представлять собой один или более агентов, выбранных из группы, состоящей из соединений-агонистов STING, противовирусных соединений, антигенов, адъювантов, противораковых агентов, антагонистов пути CTLA-4, LAG-3 и PD-1, липидов, липосом, пептидов, цитотоксических агентов, химиотерапевтических агентов, иммуномодулирующих клеточные линии, ингибиторов контрольных точек, ингибиторов рецептора фактора роста эндотелия сосудов (VEGF), ингибиторов топоизомеразы II, агентов алкилирования, противоопухолевых антибиотиков, антиметаболитов, ретиноидов и иммуномодулирующих агентов, включая, но не ограничиваясь ими, противораковые вакцины. Следует понимать, что такой дополнительный активный агент (агенты) может быть предоставлен в виде фармацевтически приемлемой соли. Следует понимать, что описания указанных выше дополнительных активных агентов могут частично совпадать. Также будет понятно, что комбинации для лечения подлежат оптимизации, и понятно, что лучшая комбинация для применения соединений общей формулы (I), соединений общей формулы (II), соединений общей формулы (III), соединений общей формулы (IV), соединений общей формулы (V), соединений общей формулы (VI) или фармацевтически приемлемых солей вышеперечисленных соединений и одного или более дополнительных активных агентов будет определяться на основе индивидуальных потребностей пациента.
Соединение, раскрытое в данном документе, может применяться в комбинации с одним или более другими активными агентами, включая, но не ограничиваясь ими, другие противораковые агенты, которые применяются для профилактики, лечения, контроля, уменьшения интенсивности или снижения риска конкретного заболевания или состояния (например, нарушений пролиферации клеток). В одном варианте осуществления соединение, раскрытое в данном документе, комбинируется с одним или более другими противораковыми агентами для применения в профилактике, лечении, контроле, уменьшении интенсивности или снижении риска конкретного заболевания или состояния, для которого раскрытые в данном документе соединения являются полезными. Такие другие активные агенты могут быть введены способом и в обычно применяемом для них количестве, одновременно или последовательно с соединением по настоящему изобретению.
Когда соединение, раскрытое в данном документе, применяется одновременно с одним или более другими активными агентами, предполагается композиция, содержащая такие другие активные агенты в дополнение к соединению, раскрытому в данном документе. Соответственно, композиции по настоящему раскрытию включают те, которые также содержат один или более другие активные ингредиенты в дополнение к соединению, раскрытому в данном документе. Соединение, описанное в данном документе, может быть введено либо одновременно, либо до или после одного или более другого активного агента (агентов). Соединение, раскрытое в данном документе, может быть введено отдельно, одним и тем же или другим путем введения или вместе в той же фармацевтической композиции, что и другой агент (агенты).
Препараты, представленные в виде комбинаций, могут включать композицию, содержащую соединение общей формулы (I), соединение общей формулы (II), соединение общей формулы (III), соединение общей формулы (IV), соединение общей формулы (V), соединение общей формулы (VI) или фармацевтически приемлемую соль вышеперечисленных соединений, и один или более другой активный агент (агенты) вместе в одной фармацевтической композиции, или могут включать композицию, содержащую соединение общей формулы (I), соединение общей формулы (II), соединение общей формулы (III), соединение общей формулы (IV), соединение общей формулы (V), соединение общей формулы (VI) или фармацевтически приемлемую соль вышеперечисленных соединений, и композицию, содержащую один или более другой активный агент (агенты) в отдельной форме, например, в форме набора или в любой форме, предназначенной для раздельного введения либо одновременно, либо по отдельным схемам дозирования.
Массовое соотношение соединения общей формулы (I), соединения общей формулы (II), соединения общей формулы (III), соединения общей формулы (IV), соединения общей формулы (V), соединения общей формулы (VI) или фармацевтически приемлемой соли вышеперечисленных соединений со вторым активным агентом может варьироваться и будет зависеть от терапевтически эффективной дозы каждого агента. Обычно будет применяться терапевтически эффективная доза каждого. Комбинации соединения, раскрытого в данном документе, и других активных агентов, как правило, также будут находиться в пределах вышеуказанного диапазона, но в каждом случае следует применять терапевтически эффективную дозу каждого активного агента. В таких комбинациях соединение, раскрытое здесь, и другие активные агенты могут быть введены отдельно или совместно. Кроме того, введение одного элемента может происходить до, одновременно или после введения другого агента (агентов).
В одном варианте осуществления это раскрытие относится к композиции, содержащей соединение общей формулы (I), соединение общей формулы (II), соединение общей формулы (III), соединение общей формулы (IV), соединение общей формулы (V), соединение общей формулы (VI) или фармацевтически приемлемую соль вышеперечисленных соединений, и, по меньшей мере, один другой активный агент, в виде комбинированного препарата для одновременного, раздельного или последовательного применения в терапии. В одном варианте осуществления терапия представляет собой лечение нарушения пролиферации клеток, такого как рак.
В одном варианте осуществления раскрытие обеспечивает набор, содержащий две или более отдельных фармацевтических композиций, по меньшей мере, одна из которых содержит соединение общей формулы (I), соединение общей формулы (II), соединение общей формулы (III), соединение общей формулы (IV), соединение общей формулы (V), соединение общей формулы (VI) или фармацевтически приемлемую соль вышеперечисленных соединений. В одном варианте осуществления набор содержит средства для раздельного хранения указанных композиций, такие как контейнер, разделенный флакон или разделенный пакет из фольги. Примером такого набора является блистерная упаковка, которая обычно применяется для упаковки таблеток, капсул и тому подобного.
Набор по настоящему раскрытию может применяться для введения различных лекарственных форм, например, пероральных и парентеральных, для введения отдельных композиций с различными интервалами дозирования или для титрования отдельных композиций относительно друг друга. Чтобы облегчить соблюдение требований, набор по раскрытию обычно включает инструкции по введению.
В настоящем документе раскрыто применение соединения общей формулы (I), соединения общей формулы (II), соединения общей формулы (III), соединения общей формулы (IV), соединения общей формулы (V), соединения общей формулы (VI) или фармацевтически приемлемой соли вышеперечисленных соединений для лечения нарушения пролиферации клеток, где лекарственное средство готовят для введения с другим активным агентом. В раскрытии также предлагается применение другого активного агента для лечения нарушения пролиферации клеток, где лекарственное средство вводят с соединением общей формулы (I), соединением общей формулы (II), соединением общей формулы (III), соединением общей формулы (IV), соединением общей формулы (V), соединением общей формулы (VI) или фармацевтически приемлемой солью вышеперечисленных соединений. В раскрытии также предусмотрено применение соединения общей формулы (I), соединения общей формулы (II), соединения общей формулы (III), соединения общей формулы (IV), соединения общей формулы (V), соединения общей формулы (VI) или фармацевтически приемлемой соли вышеперечисленных соединений для лечения нарушения пролиферации клеток, где пациента ранее (например, в течение 24 часов) лечили другим активным агентом. В раскрытии также предлагается применение другого активного агента для лечения нарушения пролиферации клеток, где пациента ранее (например, в течение 24 ч) лечили соединением общей формулы (I), соединением общей формулы (II), соединением общей формулы (III), соединением общей формулы (IV), соединением общей формулы (V), соединением общей формулы (VI) или фармацевтически приемлемой солью вышеперечисленных соединений. Второй агент может быть введен через неделю, несколько недель, месяц или несколько месяцев после введения соединения, описанного в настоящем дкументе.
Соединения-агонисты STING, которые могут применяться в комбинации с соединениями общей формулы (I), соединениями общей формулы (II), соединениями общей формулы (III), соединениями общей формулы (IV), соединениями общей формулы (V), соединениями общей формулы (VI) или фармацевтически приемлемыми солями вышеперечисленных соединений, раскрытые в настоящем документе, включают, но не ограничиваются ими, циклические динуклеотидные соединения, такие как раскрытые, например, в публикациях Международных заявок на патент по РСТ №№WO2014093936, WO2014189805, WO2014189806, WO2015185565, WO2016120305, WO2016096174, WO2016096577, WO2017027645, WO2017027646, WO2017075477, WO2017093933 и WO2018009466.
Противовирусные соединения, которые могут применяться в комбинации с соединениями общей формулы (I), соединениями общей формулы (II), соединениями общей формулы (III), соединениями общей формулы (IV), соединениями общей формулы (V), соединениями общей формулы (VI) или фармацевтически приемлемыми солями вышеперечисленных соединений, раскрытых в настоящем документе, включают ингибиторы вируса гепатита В (HBV), ингибиторы протеазы вируса гепатита С (HCV), ингибиторы полимеразы HCV, ингибиторы NS4A HCV, ингибиторы NS5A HCV, ингибиторы NS5b HCV и ингибиторы вируса иммунодефицита человека (ВИЧ). Такие противовирусные соединения могут быть предоставлены в виде фармацевтически приемлемой соли, где это целесообразно.
Антигены и адъюванты, которые могут применяться в комбинации с соединениями общей формулы (I), соединениями общей формулы (II), соединениями общей формулы (III), соединениями общей формулы (IV), соединениями общей формулы (V), соединениями общей формулы (VI) или фармацевтически приемлемыми солями вышеперечисленных соединений, включают ко стимулирующую молекулу В7, интерлейкин-2, интерферон-γ GM-CSF, антагонисты CTLA-4, лиганд ОХ-40/0Х-40, лиганд CD40/CD40, сарграмостим, левамизол, вирус осповакцины, бациллы Кальметта-Герена (BCG), липосомы, квасцы, полный или неполный адъювант Фрейнда, детоксифицированные эндотоксины, минеральные масла, поверхностно-активные вещества, такие как липолецитин, плюроновые полиолы, полианионы, пептиды и масляные или углеводородные эмульсии. Адъюванты, такие как гидроксид алюминия или фосфат алюминия, могут быть добавлены для увеличения способности вакцины вызывать, усиливать или продлевать иммунный ответ. Дополнительные соединения, такие как цитокины, хемокины и последовательности бактериальных нуклеиновых кислот, такие как CpG, агонист толл-подобного рецептора (TLR) 9, а также дополнительные агонисты для TLR 2, TLR 4, TLR 5, TLR 7, TLR 8, TLR 9, включая липопротеин, LPS, монофосфориллипид А, липотейхоевую кислоту, имиквимод, резиквимод и, кроме того, агонисты гена I, индуцируемого ретиноевой кислотой (RIG-I), такие как поли 1:С, применяемые отдельно или в комбинации с описанными композициями, также являются потенциальными адъюванты. Такие антигены и адъюванты могут быть предоставлены в виде фармацевтически приемлемой соли, где это целесообразно.
Пути CLTA-4 и PD-1 являются важными негативными регуляторами иммунного ответа. Активированные Т-клетки активируют CTLA-4, который связывается с антиген-презентирующими клетками и ингибирует стимуляцию Т-клеток, экспрессию гена IL-2 и пролиферацию Т-клеток; эти противоопухолевые эффекты наблюдались на мышиных моделях карциномы толстой кишки, метастатического рака простаты и метастатической меланомы. PD-1 связывается с активными Т-клетками и подавляет активацию Т-клеток. Антагонисты PD-1 также продемонстрировали противоопухолевые эффекты. Антагонисты пути CTLA-4 и PD-1, которые могут применяться в комбинации с соединениями общей формулы (I), соединениями общей формулы (II), соединениями общей формулы (III), соединениями общей формулы (IV), соединениями общей формулы (V), соединениями общей формулы (VI) или фармацевтически приемлемыми солями вышеперечисленных соединений, раскрытые в настоящем документе, включают ипилимумаб, тремелимумаб, ниволумаб, пембролизумаб, СТ-011, АМР-224 и MDX-1106.
«Антагонист PD-1» или «антагонист пути PD-1» означает любое химическое соединение или биологическую молекулу, которое блокирует связывание PD-L1, экспрессируемого на раковой клетке, с PD-1, экспрессируемым на иммунной клетке (Т-клетке, В-клетке или NKT-клетке) и предпочтительно также блокирует связывание PD-L2, экспрессируемого на раковой клетке, с иммунной клеткой, экспрессирующей PD-1. Альтернативные названия или синонимы PD-1 и его лигандов включают: PDCD1, PD1, CD279 и SLEB2 для PD-1; PDCD1L1, PDL1, В7Н1, В7-4, CD274 и В7-Н для PD-L1; и PDCD1L2, PDL2, B7-DC, Btdc и CD273 для PD-L2. В любом из способов лечения, лекарственных средств и применений по настоящему раскрытию, в которых лечат пациента-человека, антагонист PD-1 блокирует связывание человеческого PD-L1 с человеческим PD-1 и предпочтительно блокирует связывание и человеческих PD- L1, и PD-L2 к человеческому PD-1. Аминокислотные последовательности PD-1 человека можно найти в NCBI Locus No. NP_005009. Аминокислотные последовательности PD-L1 и PD-L2 человека можно найти в NCBI Locus No. NP_054862 и NP_079515, соответственно.
Антагонисты PD-1, применимые в любом способе лечения, лекарственных средствах и применениях по настоящему раскрытию, включают моноклональное антитело (mAb) или его антигенсвязывающий фрагмент, которое специфически связывается с PD-1 или PD-L1, и предпочтительно специфически связывается с человеческим PD-1 или человеческим PD-L1. mAb может быть человеческим антителом, гуманизированным антителом или химерным антителом и может включать человеческую константную область. В некоторых вариантах осуществления человеческая константная область выбрана из группы, состоящей из константных областей IgG1, IgG2, IgG3 и IgG4, и в предпочтительных вариантах осуществления человеческая константная область представляет собой константную область IgG1 или IgG4. В некоторых вариантах осуществления антигенсвязывающий фрагмент выбран из группы, состоящей из фрагментов Fab, Fab'-SH, F(ab')2, scFv и Fv.
Примеры mAb, которые связываются с человеческим PD-1, и применяемых в способе лечения, лекарственных средствах и применениях по настоящему раскрытию, описаны в патентах США №№US7488802, US7521051, US8008449, US8354509 и US8168757, публикациях международных патентных заявок по РСТ WO 2004/004771, WO 2004/072286 и WO 2004/056875 и публикации заявки на патент США №US2011/0271358.
Примеры mAb, которые связываются с человеческим PD-L1, и применяемых в способе лечения, лекарственных средствах и применениях по настоящему раскрытию, описаны в международных патентных заявках по РСТ №№WO2013/019906 и WO2010/077634 А1 и в патенте США №US8383796. Специфические моноклональные антитела против PD-L1 человека, применяемые в качестве антагониста PD-1 в способе лечения, лекарственных средствах и применениях, описанных в настоящем изобретении, включают MPDL3280A, BMS-936559, MEDI4736, MSB0010718C и антитело, которое содержит вариабельные области тяжелой цепи и легкой цепи SEQ ID NO:24 и SEQ ID NO:21, соответственно, из WO2013/019906.
Другие антагонисты PD-1, применимые в любом способе лечения, лекарственных средствах и применениях по настоящему раскрытию, включают иммунную адгезию, которая специфически связывает с PD-1 или PD-L1, и предпочтительно специфически связывает с PD-1 или PD-L1 человека, например, слитый белок, содержащий внеклеточную или связывающую PD-1 часть PD-L1 или PD-L2, слитую с константной областью, такой как область Fc молекулы иммуноглобулина. Примеры молекул иммунной адгезии, которые специфически связываются с PD-1, описаны в публикациях международных патентных заявок по РСТ WO2010/027827 и WO2011/066342. Специфические слитые белки, применяемые в качестве антагониста PD-1 в способе лечения, лекарственных средствах и применениях по настоящему раскрытию, включают АМР-224 (также известный как В7-DCIg), который представляет собой слитый белок PD-L2-FC и связывается с PD-1 человека. Раскрытие дополнительно относится к способу лечения рака у пациента-человека, включающему введение пациенту соединения, раскрытого в данном документе (то есть, соединения общей формулы (I), соединения общей формулы (II), соединения общей формулы (III), соединения общей формулы (IV), соединения общей формулы (V), соединения общей формулы (VI) или фармацевтически приемлемой соли вышеперечисленных соединений) и антагониста PD-1. Соединение по изобретению и антагонист PD-1 могут быть введены одновременно или последовательно.
В конкретных вариантах осуществления антагонист PD-1 представляет собой антитело против PD-1 или его антигенсвязывающий фрагмент. В альтернативных вариантах осуществления антагонист PD-1 представляет собой антитело против PD-L1 или его антигенсвязывающий фрагмент.В некоторых вариантах осуществления антагонист PD-1 представляет собой пембролизумаб (KEYTRUDA™, Merck & Co., Inc., Kenilworth, NJ, USA), ниволумаб (OPDIVO™, Bristol-Myers Squibb Company, Princeton, NJ, USA), цемиплимаб (LIBTAYO™, Regeneron Pharmaceuticals, Inc., Tarrytown, NY, USA), атезолизумаб (TECENTRIQ™, Genentech, San Francisco, CA, USA), дурвалумаб (IMFINZI™, AstraZeneca Pharmaceuticals LP, Wilmington, DE) или авелумаб (BAVENCIO™, Merck KGaA, Darmstadt, Germany).
В некоторых вариантах осуществления антагонист PD-1 представляет собой пембролизумаб. В конкретных подвариантах осуществления способ включает введение пациенту 200 мг пембролизумаба примерно каждые три недели. В других подвариантах осуществления способ включает введение пациенту 400 мг пембролизумаба примерно каждые шесть недель.
В дополнительных подвариантах осуществления способ включает введение пациенту 2 мг/кг пембролизумаба примерно каждые три недели. В конкретных подвариантах осуществления пациент является педиатрическим пациентом.
В некоторых вариантах осуществления антагонист PD-1 представляет собой ниволумаб. В конкретных подвариантах осуществления способ включает введение пациенту 240 мг ниволумаба примерно каждые две недели. В других подвариантах осуществления способ включает введение пациенту 480 мг ниволумаба примерно каждые четыре недели. В некоторых вариантах осуществления антагонист PD-1 представляет собой цемиплимаб. В конкретных вариантах осуществления способ включает введение пациенту 350 мг цемиплимаба примерно каждые 3 недели.
В некоторых вариантах осуществления антагонист PD-1 представляет собой атезолизумаб. В конкретных подвариантах осуществления способ включает введение пациенту 1200 мг атезолизумаба примерно каждые три недели.
В некоторых вариантах осуществления антагонист PD-1 представляет собой дурвалумаб. В конкретных подвариантах осуществления способ включает введение пациенту 10 мг/кг дурвалумаба примерно каждые две недели.
В некоторых вариантах осуществления антагонист PD-1 представляет собой авелумаб. В конкретных подвариантах осуществления способ включает введение пациенту 800 мг авелумаба примерно каждые две недели.
Примеры цитотоксических агентов, которые могут применяться в комбинации с соединениями общей формулы (I), соединениями общей формулы (II), соединениями общей формулы (III), соединениями общей формулы (IV), соединениями общей формулы (V), соединениями общей формулы (VI) или фармацевтически приемлемыми солями вышеперечисленных соединений, включают, но не ограничиваются ими, триоксид мышьяка (продается под торговым названием TRISENOX®), аспарагиназу (также известную как L-аспарагиназа и L-аспарагиназа Erwinia, продаваемая под торговыми названиями ELSPAR® и KIDROLASE®).
Химиотерапевтические агенты, которые могут применяться в комбинации с соединениями общей формулы (I), соединениями общей формулы (II), соединениями общей формулы (III), соединениями общей формулы (IV), соединениями общей формулы (V), соединениями общей формулы (VI) или фармацевтически приемлемыми солями вышеперечисленных соединений, раскрытыми в настоящем документе, включают абиратерона ацетат, алтретамин, ангидровинбластин, ауристатин, бексаротен, бикалутамид, BMS 184476, 2,3,4,5,6-пентафтор-N-(3-фтор-4-метоксифенил) бензолсульфонамид, блеомицин, N,N-диметил-L-валил-L-валил-N-метил-L-валил-L-пролил-1-L-пролин-трет-бутиламид, кахектин, цемадотин, хлорамбуцил, циклофосфамид, 3',4'-дидегидро-4'-дезокси-8'-норвинкалейкобластин, доцетаксел, доксетаксел, циклофосфамид, карбоплатин, кармустин, цисплатин, криптофицин, цитарабин, дакарбазин (DTIC), дактиномицин, даунорубицин, децитабин, доластатин, доксорубицин (адриамицин), этопозид, 5-фторурацил, финастерид, флутамид, гидроксимочевину и таксаны, ифосфамид, лиарозол, лонидамин, ломустин (CCNU), MDV3100, мехлорэтамин (мустарген), мелфалан, мивобулин изетионат, ризоксин, сертенеф, стрептозоцин, митомицин, метотрексат, таксаны, нилутамид, ниволумаб, онапристон, паклитаксел, пембролизумаб, преднимустин, прокарбазин, RPR109881, эстрамустина фосфат, тамоксифен, тазонермин, таксол, третиноин, винбластин, винкристин, виндезина сульфат и винфлунин. Такие химиотерапевтические агенты могут быть предоставлены в виде фармацевтически приемлемой соли, где это целесообразно.
Примеры ингибиторов рецепторов фактора роста эндотелия сосудов (VEGF) включают, но не ограничиваются ими, бевацизумаб (продается под торговым названием AVASTIN), акситиниб (описан в международной патентной публикации согласно РСТ №WO01/002369), бриваниб аланинат ((S)-((R)-1-(4-(4-фтор-2-метил-1Н-индол-5-илокси)-5-метилпирро[2,1-f][1,2,4]триазин-6-илокси)пропан-2-ил)2-аминопропаноат, также известный как BMS-582664), мотесаниб (N-(2,3-дигидро-3,3-диметил-1Н-индол-6-ил)-2-[(4-пиридинилметил)амино]-3-пиридинкарбоксамид, описанный в публикации международной патентной заявки согласно РСТ №WO02/068470), пасиреотид (также известный как SO 230, и описанный в публикации международного патента согласно РСТ №WO02/010192) и сорафениб (продается под торговым названием NEXAVAR). Такие ингибиторы могут быть предоставлены в виде фармацевтически приемлемой соли, где это целесообразно.
Примеры ингибиторов топоизомеразы II включают, но не ограничиваются ими, этопозид (также известный как VP-16 и этопозид фосфат, продаваемый под торговыми названиями TOPOSAR, VEPESID и ETOPOPHOS) и тенипозид (также известный как VM-26, продаваемый под торговыми названиями VUMON). Такие ингибиторы могут быть предоставлены в виде фармацевтически приемлемой соли, где это целесообразно.
Примеры алкилирующих агентов включают, но не ограничиваются ими, 5-азацитидин (продается под торговым названием VIDAZA), децитабин (продается под торговым названием DECOGEN), темозоломид (продается под торговыми названиями TEMCAD, TEMODAR и TEMODAL), дактиномицин (также известный как актиномицин-D и продаваемый под торговым названием COSMEGEN), мелфалан (также известный как L-РАМ, L-сарколизин и фенилаланиновая горчица, продаваемый под торговым названием ALKERAN), алтретамин (также известный как гексаметилмеламин (НММ), продается под торговым названием TEKCALEN®), кармустин (продается под торговым названием BCNU), бендамустин (продается под торговой маркой TREANDA), бусульфан (продается под торговыми названиями BUSULFEX® и MYLERAN®), карбоплатин (продается под торговым названием PARAPLATIN®), ломустин (также известный как CCNU, продается под торговым названием CeeNU®), цисплатин (также известный как CDDP, продается под торговыми названиями PLATINOL® и PLATINOL®-AQ), хлорамбуцил (продается под торговым названием LEUKERAN®), циклофосфамид (продается под торговыми названиями CYTOKCAN® и NEOSAR®), дакарбазин (также известный как DTIC, DIC и имидазолкарбоксамид, продается под торговым названием DTIC-Dome®), ифосфамид (продается под торговым названием IFEX®), прокарбазин (продается под торговым названием MATULANE®), мехлорэтамин (также известный как мустарген, мустин и гидрохлорид мехлорэтамина, продается под торговым названием MUSTARGEN®), стрептозоцин (продается под торговым названием ZANOSAR®), тиотепа (также известный как тиофосфоамид, TESPA и TSPA, и продается под торговым названием THIOPLEX®. Такие алкилирующие агенты могут быть предоставлены в виде фармацевтически приемлемой соли, где это целесообразно.
Примеры противоопухолевых антибиотиков включают, но не ограничиваются ими, доксорубицин (продается под торговыми названиями ADRIAMYCIN® и RUBEX®), блеомицин (продается под торговым названием LENOKCANE®), даунорубицин (также известный как гидрохлорид даунорубицина, дауномицин и гидрохлорид рубидомицина, продается под торговым названием CERUBIDLNE®), липосомальный даунорубицин (липосома даунорубицина цитрата, продается под торговым названием DAUNOXOME®), митоксантрон (также известный как DHAD, продается под торговым названием NOVANTRONE®), эпирубицин (продается под торговым названием ELLENCE™), идарубицин (продается под торговыми названиями IDAMYCIN®, IDAMYCIN PFS®) и митомицин С (продается под торговым названием MUTAMYCIN®). Такие противоопухолевые антибиотики могут быть предоставлены в виде фармацевтически приемлемой соли, где это целесообразно.
Примеры антиметаболитов включают, но не ограничиваются ими, кларибин (2-хлордезоксиаденозин, продаваемый под торговым названием LEUSTATIN®), 5-фторурацил (продаваемый под торговым названием ADRUCIL®), 6-тиогуанин (продаваемый под торговым названием PURINETHOL®), пеметрексед (продается под торговым названием ALIMTA®), цитарабин (также известный как арабинозилцитозин (Ага-С), продаваемый под торговым названием CYTOSAR-U®), липосомальный цитарабин (также известный как липосомальный Ага-С, продаваемый под торговым названием DEPOCYT™), децитабин (продается под торговым названием DACOGEN®), гидроксимочевина (продается под торговыми названиями HYDREA®, DROXIA™ и MYLOCEL™), флударабин (продается под торговым названием FLUDARA®), флоксуридин (продается под торговым названием FUDR®), кладрибин (также известный как 2-хлордезоксиаденозин (2-CdA), продается под торговым названием LEUSTATIN™), метотрексат (также известный как аметоптерин, метотрексат натрия (МТХ), продается под торговыми названиями RHEUMATREX® и TREXALL™) и пентостатин (продается под торговым названием NIPENT®). Такие антиметаболиты могут быть предоставлены в виде фармацевтически приемлемой соли, где это целесообразно.
Примеры ретиноидов включают, но не ограничиваются ими, алитретиноин (продается под торговым названием PANRETIN®), третиноин (полностью транс-ретиноевая кислота, также известная как ATRA, продаваемая под торговым названием VESANOID®), изотретиноин (13-с/8-ретиноевая кислота, продаваемая под торговыми названиями ACCUTANE®, AMNESTEEM®, CLARAVIS®, CLARUS®, DECUTAN®, IZOTAH®, IZOTECH®, ORATANE®, IZOTRET® и SOTRET®) и бексаротен (продается под торговым названием TARGRETIN®). Такие соединения могут быть предоставлены в виде фармацевтически приемлемой соли, где это целесообразно.
Активность: биохимический конкурентный анализ STING [3H]cGAMP
Отдельные соединения, описанные в приведенных здесь Примерах, определены как агонисты STING путем (i) связывания с белком STING, о чем свидетельствует снижение связывания меченного тритием лиганда cGAMP с белком STING, по меньшей мере, на 20% при 20 мкМ (концентрация тестируемого соединения) в STING биохимическом конкурентном анализе [3H]cGAMP и/или (ii) демонстрации продукции интерферона с 6% или большей индукцией секреции IFN-β при 30 мкМ в клеточном анализе ТНР1 (где индукция, вызванная cGAMP при 30 мкМ, была установлена как 100%).
Способность соединений связывать STING количественно оценивалась по способности конкурировать с меченным тритием лигандом cGAMP за мембрану рецептора STING человека с применением радиоактивного анализа вещества по степени его связывания с мембранным фильтром. В анализе связывания используют рецептор STING, полученный из клеточных мембран Hi-Five, сверхэкспрессирующий полноразмерный STING HAQ, полученный в лаборатории, и меченный тритием лиганд cGAMP, также очищенный в лаборатории.
В нижеследующей экспериментальной части подробно описано получение конкретных примеров по настоящему раскрытию. Соединения примеров представлены в их нейтральных формах в способах и таблицах ниже. В некоторых случаях соединения выделяли в виде солей в зависимости от способа, применяемого для их окончательной очистки, и/или собственных молекулярных свойств. Примеры приведены только для иллюстративных целей и никоим образом не предназначены для ограничения объема настоящего раскрытия.
ПРИМЕРЫ





Получение 1: 3-(трет-бутокси)-3-оксопропаноат магния
Этанолат магния (3.57 г, 31.2 ммоль) добавляли к смеси 3-(трет-бутокси)-3-оксопропановой кислоты (10.0 г, 62.4 ммоль) в THF (100 мл) при 20°С. Реакционную смесь перемешивали при 20°С в течение 18 ч в атмосфере Ar. Реакционную смесь затем концентрировали под пониженным давлением. Остаток высушивали под пониженным давлением с получением 3-(трет-бутокси)-3-оксопропаноата магния. 1Н ЯМР (499 МГц, DMSO-d6) δ 2.96 (s, 4Н), 1.39 (s, 18Н).
Получение 2: трет-Бутил 3-(2,3-диметокситиено[2,3-b]пиразин-6-ил)-3-оксопропаноат
Стадия 1: 3-бром-5,6-диметоксипиразин-2-карбальдегид
К раствору 2,2,6,6-тетраметилпиперидина (5.12 мл, 30.1 ммоль) в THF (40 мл) при -78°С добавляли по каплям раствор n-BuLi (2.5 М в гексанах, 11.5 мл, 28.8 ммоль). Реакционную смесь перемешивали в течение 10 мин при -78°С, затем нагревали до 0°С и перемешивали в течение 20 мин. Реакционную смесь затем снова охлаждали до -78°С и добавляли раствор 5-бром-2,3-диметоксипиразина (3.00 г, 13.7 ммоль) в THF (10 мл) на протяжении 5 мин. Реакционную смесь перемешивали при -78°С в течение 1 ч, и затем реакцию останавливали DMF (1.06 мл, 13.7 ммоль). Реакционную смесь нагревали до 0°С и перемешивали в течение дополнительных 20 мин. АсОН (3.0 мл) добавляли при 0°С, и реакционную смесь нагревали до комнатной температуры и перемешивали на протяжении ночи. Смесь разбавляли EtOAc (300 мл) и затем промывали Н2О (2×150 мл) и насыщенным водным NaCl. Органический слой отделяли, высушивали над Na2SO4, фильтровали и концентрировали под пониженным давлением. Остаток очищали с помощью хроматографии на силикагеле (элюируя EtOAc в гексанах) с получением 3-бром-5,6-диметоксипиразин-2-карбальдегида. ЖХ-МС (C7H8BrN2O3) (ES, m/z): 247, 249 [М+Н]+. 1Н ЯМР (500 МГц, CDCl3) δ 10.19 (s, 1H), 4.17 (s, 3Н), 4.14 (s, 3Н).
Стадия 2: трет-бутил 2,3-диметокситиепо[2,3-b]пиразин-6-карбоксилат
трет-Бутил 2-сульфанилацетат (424 мкл, 2.92 ммоль) и DMF (2.9 мл) добавляли к 3-бром-5,6-диметоксипиразин-2-карбальдегиду (650 мг, 2.63 ммоль) при комнатной температуре. Затем к реакционной смеси добавляли порциями К2СО3 (1090 мг, 7.89 ммоль) при комнатной температуре. Реакционную смесь нагревали до 80°С и перемешивали на протяжении ночи. Реакционную смесь затем охлаждали до комнатной температуры, разбавляли Et2O и останавливали H2O. Реакционную смесь экстрагировали Et2O, и объединенные органические вещества промывали насыщенным водным NaCl, высушивали над Na2SO4, фильтровали и концентрировали под пониженным давлением. Остаток очищали с помощью хроматографии на силикагеле (элюируя EtOAc в гексанах) с получением трет-бутил 2,3-диметокситиено[2,3-b]пиразин-6-карбоксилата. ЖХ-МС (C13H17N2O4S) (ES, m/z): 297 [М+Н]+. 1Н ЯМР (500 МГц, DMSO-d6) δ 7.89 (s, 1Н), 4.02 (s, 3Н), 3.99 (s, 3Н), 1.55 (s, 9H).
Стадия 3: 2,3-диметокситиено[2,3-b]пиразин-6-карбоноеая кислота
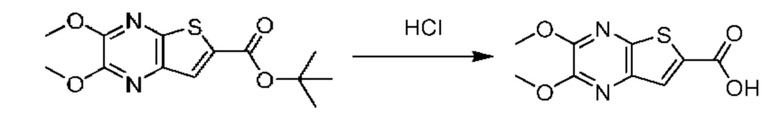
К перемешанному раствору трет-бутил 2,3-диметокситиено[2,3-b]пиразин-6-карбоксилата (400 мг, 1.35 ммоль) в DCM (6.0 мл) добавляли HCl (4.0 М в диоксане, 1.7 мл, 6.8 ммоль) при комнатной температуре. Реакционную смесь перемешивали на протяжении ночи при комнатной температуре, и затем разбавляли путем добавления по каплям гексаном (50 мл) и перемешивали в течение 1 ч при комнатной температуре. Реакционную смесь фильтровали, и собранные вещества промывали гексаном (2x10 мл) и высушивали под пониженным давлением с получением 2,3-диметокситиено[2,3-b]пиразин-6-карбоновой кислоты. ЖХ-МС (C9H9N2O4S) (ES, m/z): 241 [М+Н]+. 1Н ЯМР (500 МГц, DMSO-d6) δ 12.71 (br s, 1Н), 7.90 (s, 1H), 4.03 (s, 3H), 3.99 (s, 3H).
Стадия 4: трет-бутил 3-(2,3-диметокситиено[2,3-b]пиразин-6-ил)-3-оксопропаноат
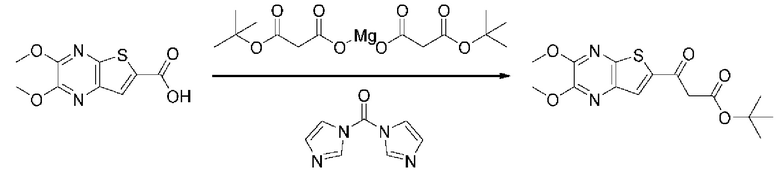
Смесь 2,3-диметокситиено[2,3-b]пиразин-6-карбоновой кислоты (80 мг, 0.33 ммоль) и CDI (324 мг, 2.00 ммоль) в THF (5.5 мл) перемешивали при комнатной температуре в течение 3 ч. К смеси добавляли бис(3-трет-бутокси-3-оксопропаноат) магния (628 мг, 1.83 ммоль) и полученную в результате смесь перемешивали на протяжении ночи при комнатной температуре. Реакционную смесь концентрировали под пониженным давлением. Остаток очищали с помощью хроматографии на силикагеле (элюируя (25% EtOH в EtOAc) в гексанах) с получением трет-бутил 3-(2,3-диметокситиено[2,3-b]пиразин-6-ил)-3-оксопропаноата. ЖХ-МС (C15H19N2O5S) (ES, m/z): 339 [М+Н]+. 1H ЯМР (500 МГц, DMSO-d6) δ 8.32 (s, 1H), 4.13 (s, 2Н), 4.04 (s, 3Н), 4.00 (s, 3Н), 1.41 (s, 9Н).
Получение 3: 5,6-диметокситиено[3,2-b]пиридин-2-карбоновая кислота
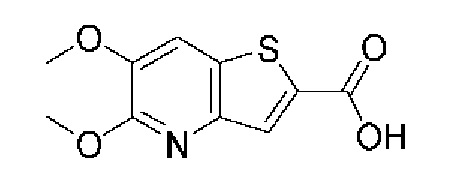
Стадия 1: трет-бутил 5,6-диметокситиено[3,2-b]пиридин-2-карбоксилат
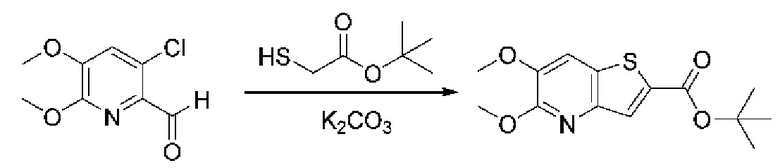
К2СО3 (1180 мг, 8.56 ммоль) добавляли к смеси 3-хлор-5,6-диметоксипиколинальдегида (575 мг, 2.86 ммоль) и трет-бутил 2-сульфанилацетата (0.456 мл, 3.14 ммоль) в DMF (8.3 мл) при комнатной температуре. Реакционную смесь перемешивали и нагревали до 60°С в течение 3 дней. Реакционную смесь охлаждали до комнатной температуры, и затем разбавляли Et2O и Н2О. Органический слой отделяли, промывали насыщенным водным NaCl, высушивали над Na2SO4, фильтровали и концентрировали под пониженным давлением. Остаток очищали с помощью хроматографии на силикагеле (элюируя EtOAc в гексанах) с получением трет-бутил 5,6-диметокситиено[3,2-b]пиридин-2-карбоксилата. 1Н ЯМР (499 МГц, DMSO-d6) δ 7.95 (s, 1H), 7.84 (s, 1H), 3.93 (s, 3Н), 3.87 (s, 3Н), 1.54 (s, 9H).
Стадия 2: 5,6-диметокситиепо[3,2-b]пиридип-2-карбоповая кислота
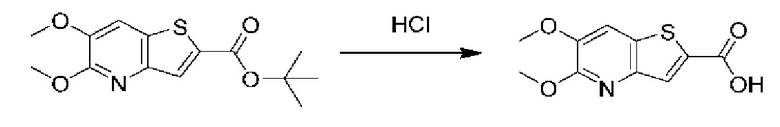
HCl (4.0 M в H2O, 2.1 мл, 8.4 ммоль) добавляли к раствору трет-бутил 5,6-диметокситиено[3,2-b]пиридин-2-карбоксилата (493 мг, 1.67 ммоль) в DCM (7.4 мл) при комнатной температуре. Реакционную смесь перемешивали на протяжении ночи при комнатной температуре, и затем разбавляли путем добавления по каплям гексана (50 мл). Смесь перемешивали в течение 1 ч и затем фильтровали. Собранные вещества промывали гексаном (2x10 мл) и затем высушивали под пониженным давлением с получением 5,6-диметокситиено[3,2-b]пиридин-2-карбоновой кислоты. ЖХ-МС (C10H10NO4S) (ES, m/z): 240 [М+Н]+. 1H ЯМР (499 МГц, DMSO-d6) δ 7.97 (s, 1Н), 7.85 (s, 1H), 3.95 (s, 3H), 3.88 (s, 3H).
Получение 4: трет-Бутил 3-(5,6-диметокситиено[3,2-b]пиридин-2-ил)-3-оксопропаноат
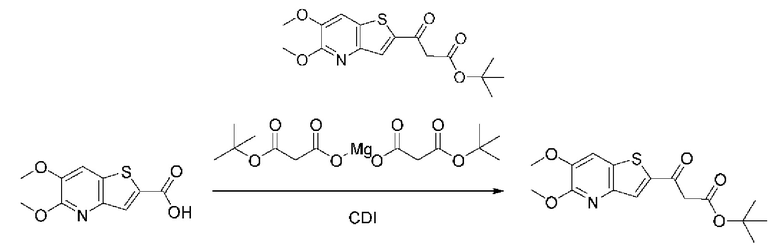
CDI (508 мг, 3.13 ммоль) добавляли к смеси 5,6-диметокситиено[3,2-b]пиридин-2-карбоновой кислоты (500 мг, 2.09 ммоль) в THF (5 мл). Реакционную смесь перемешивали при комнатной температуре в течение 3 ч. Реакционную смесь добавляли в отдельную колбу, содержащую 3-(трет-бутокси)-3-оксопропаноат магния (1220 мг, 3.55 ммоль). Реакционную смесь разбавляли дополнительным THF (4 мл) и перемешивали на протяжении ночи при комнатной температуре. Реакционную смесь затем нагревали до 50°С в течение 1 ч. Реакционную смесь охлаждали до комнатной температуры и разбавляли H2O (20 мл). Добавляли дигидрат трехосновного цитрата натрия (2 г) и EtOAc (50 мл). Органический слой отделяли, и водный слой экстрагировали EtOAc. Органические слои объединяли, промывали насыщенным водным NaCl, высушивали над безводным Na2SO4, фильтровали и концентрировали под пониженным давлением. Остаток очищали с помощью хроматографии на силикагеле (элюируя EtOAc в гексанах) с получением трет-бутил 3-(5,6-диметокситиено[3,2-b]пиридин-2-ил)-3-оксопропаноата. ЖХ-МС (C16H20NO5S) (ES, m/z): 338 [М+Н]+. 1H ЯМР (500 МГц, CDCl3) δ 7.97 (s, 1H), 7.44 (s, 1H), 4.13 (s, 3Н), 3.99 (s, 3H), 3.92 (s, 2Н), 1.48 (s, 9Н).
Получение 5: C-Phos Pd G4
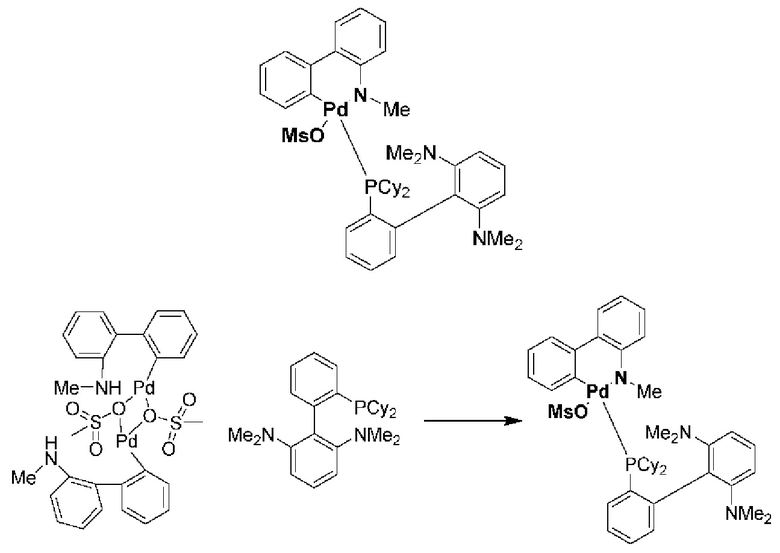
Смесь димера (2'-метиламино-1,1'-бифенил-2-ил)метансульфоната палладия (II) (439 мг, 0.573 ммоль) и 2'-(дициклогексилфосфино)-N2,N2,N6,N6-тетраметил-[1,1'-бифенил]-2,6-диамина (500 мг, 1.15 ммоль) в DCM (6 мл) перемешивали при комнатной температуре в течение 2 ч. Раствор затем разбавляли Et2O (30 мл). Раствор фильтровали и концентрировали под пониженным давлением. Остаток затем суспендировали в пентанах и снова концентрировали под пониженным давлением с получением C-Phos Pd G4. См. Bruno, N. С; Niljianskul, N.; Buchwald, S. L. J. Org. Chem. 2014, 79, 4161.
Промежуточное соединение 1: метил (S)-4-(5-хлол-6-метокситиено[3,2-b]пиримидин-2-ил)-2-метил-4-оксобутаноат
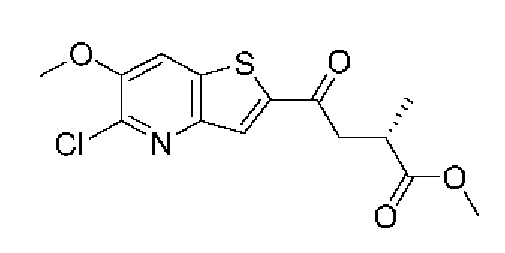
Стадия 1: 5-хлор-6-метокситиено[3,2-b]пиридин-2-карбонилхлорид

POCl3 (1.17 мл, 12.5 ммоль) добавляли по каплям к перемешанной смеси 5,6-диметокситиено[3,2-b]пиридин-2-карбоновой кислоты (1.00 г, 4.1 ммоль) в DMF (10.45 мл) при 0°С в атмосфере N2. Через 10 мин реакционную смесь оставляли нагреваться до комнатной температуры. Реакционную смесь затем нагревали до 100°С и перемешивали в течение 45 мин. Реакционную смесь добавляли в ледяную воду (100 мл) и перемешивали. Смесь фильтровали, и собранные вещества промывали водой (2x30 мл) и гексаном (50 мл). Собранные вещества разбавляли Et2O (50 мл) и фильтровали. Собранные вещества растворяли в CH2Cl2 (60 мл) и смесь высушивали над безводным Na2SO4, фильтровали и концентрировали под пониженным давлением с получением 5-хлор-6-метокситиено[3,2-b]пиридин-2-карбонилхлорида. 1Н ЯМР (500 МГц, CDCl3) δ 8.32 (s, 1H), 7.60 (s, 1H), 4.06 (s, 3H).
Стадия 2: метил (S)-4-(5-хлор-6-метокситиено[3,2-b]пиридин-2-ил)-2-метил-4-оксобутаноат
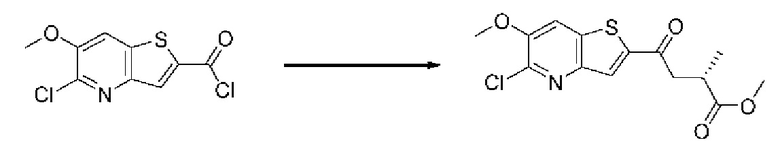
CuI (0.073 г, 0.38 ммоль) помещали под вакуум и нагревали в течение 1 мин тепловым феном. Колбу оставляли охлаждаться до комнатной температуры и затем открывали в атмосфере N2. Еще дважды колбу откачивали и затем снова заполняли N2. В колбе поддерживали положительное давление N2 с помощью резиновой септы и присоединенного входного клапана для N2. THF (2 мл) добавляли в колбу, и реакционную смесь охлаждали на бане с ледяной водой. Раствор (R)-(3-метокси-2-метил-3-оксопропил) цинка (II) бромида в THF (0.50 М, 1.68 мл, 0.84 ммоль) добавляли по каплям к реакционной смеси в течение 5 мин. Реакционную смесь перемешивали в течение 105 мин при 0°С. Смесь 5-хлор-6-метокситиено[3,2-b]пиридин-2-карбонилхлорида (0.200 г, 0.763 ммоль) в NMP (3 мл) затем добавляли по каплям в течение 5 мин. Реакционную смесь затем перемешивали в течение 3 ч при 0°С. Реакционную смесь затем добавляли к перемешанной смеси изопропилацетата (50 мл) и цитрата натрия (20% масс/об. в воде, 50 мл). После перемешивания в течение 20 мин слои разделяли, и водный слой экстрагировали изопропилацетатом (30 мл). Органические слои объединяли, промывали насыщенным водным NaCl (2x50 мл), высушивали над безводным Na2SO4, фильтровали и концентрировали под пониженным давлением. Остаток очищали с помощью хроматографии на силикагеле (элюируя EtOAc в гексанах) с получением (S)-метил 4-(5-хлор-6-метокситиено[3,2-b]пиридин-2-ил)-2-метил-4-оксобутаноата. ЖХ-МС (C14H15CINO4S) (ES, m/z): 328 [М+Н]+. 1Н ЯМР (500 МГц, CDCl3) δ 7.99 (s, 1H), 7.60 (s, 1H), 4.03 (s, 3H), 3.72 (s, 3H), 3.51 (dd, J=17.2, 7.9 Гц, 1Н), 3.21-3.12 (m, 1H), 3.06 (dd, J=17.2, 5.2 Гц, 1H), 1.32 (d, J=7.1 Гц, 3H).
Промежуточное соединение 2: трет-бутил 4-(5-(3-гидуоксипуопил)-6-метоксибензо[b]тиофен-2-ил)-4-оксобутаноат
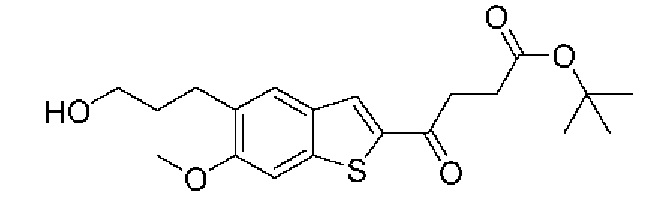
Стадия 1: трет-бутил 4-(5-(3-((трет-бутилдиметилсилил)окси)пропил)-6-метоксибензо[b]тиофен-2-ил)-4-оксобутаноат
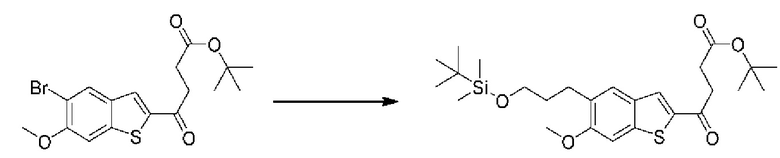
трет-Бутил 4-(5-бром-6-метоксибензо[b]тиофен-2-ил)-4-оксобутаноат (1.75 г, 4.38 ммоль; 81% чистоты) и C-Phos Pd G3 (0.177 г, 0.219 ммоль) добавляли в 40 мл виалу с винтовой крышкой с септой. Виалу три раза откачивали и снова заполняли N2. В виалу добавляли THF (15.7 мл) в атмосфере N2 с перемешиванием. При перемешивании полученной суспензии при комнатной температуре добавляли (3-((трет-бутилдиметилсилил)окси)пропил)цинк (II) бромид (0.50 М в THF, 17.5 мл, 8.75 ммоль) по каплям с перемешиванием. Смесь перемешивали при комнатной температуре в течение 18 ч. Реакционную смесь затем разделяли между EtOAc (75 мл) и 10% водным цитратом натрия (75 мл) и перемешивали энергично в течение 5 мин. Слои разделяли, и водный слой экстрагировали EtOAc (20 мл). Органические слои объединяли, промывали насыщенным водным NaCl (50 мл), высушивали над безводным Na2SO4, фильтровали и концентрировали под пониженным давлением. Остаток очищали с помощью хроматографии на силикагеле (0→40% градиент EtOAc в гексанах) с получением трет-бутил 4-(5-(3-((трет-бутилдиметилсилил)окси)пропил)-6-метоксибензо[b]тиофен-2-ил)-4-оксобутаноата. ЖХ-МС (C26H40NaO5SSi) (ES, m/z): 515 [M+Na]+. 1Н ЯМР (500 МГц, CDCl3) δ 7.89 (s, 1H), 7.62 (s, 1H), 7.24 (s, 1H), 3.92 (s, 3H), 3.67 (t, J=6.3 Гц, 2Н), 3.28 (t, J=6.8 Гц, 2Н), 2.76 (t, J=7.2 Гц, 2Н), 2.72 (t, J=7.2 Гц, 2Н), 1.85 (р, J=6.5 Гц, 2Н), 1.46 (s, 9Н), 0.94 (s, 9Н), 0.08 (s, 6Н).
Стадия 2: трет-бутил 4-(5-(3-гидуоксипуопил)-6-метоксибеизо[b]тиофен-2-ил)-4-оксобутапоат
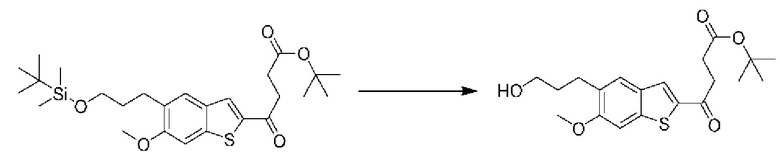
К смеси трет-бутил 4-(5-(3-((трет-бутилдиметилсилил)окси)пропил)-6-метоксибензо[b]тиофен-2-ил)-4-оксобутаноата (1.45 г, 2.94 ммоль) в МеОН (5.0 мл) добавляли воду (5.0 мл) и НОАс (5.0 мл). Полученную в результате суспензию перемешивали при комнатной температуре в течение 18 ч. Реакционную смесь разделяли между EtOAc и водным NaCl. Слои разделяли, и водный слой экстрагировали EtOAc. Органические слои объединяли, дважды промывали насыщенным водным NaCl, высушивали над безводным Na2SO4, фильтровали и концентрировали под пониженным давлением с получением сырого остатка. Полученный в результате остаток очищали с помощью хроматографии на силикагеле (0→100% градиент EtOAc в гексанах) с получением трет-бутил 4-(5-(3-гидроксипропил)-6-метоксибензо[b]тиофен-2-ил)-4-оксобутаноата. ЖХ-МС (C20H26NaO5S) (ES, m/z): 401 [M+Na]+. 1Н ЯМР (500 МГц, CDCl3) δ 7.89 (s, 1H), 7.64 (s, 1H), 7.26 (s, 1H), 3.94 (s, 3H), 3.68 (t, J=5.5 Гц, 2Н), 3.28 (t, J=6.6 Гц, 2Н), 2.82 (t, J=7.3 Гц, 2Н), 2.72 (t, J=6.6 Гц, 2Н), 1.96-1.83 (m, 2Н), 1.46 (s, 9Н).
Промежуточное соединение 3: трет-бутил 4-(5-(3-6ромпропил)-6-метоксибензо[b]тиофен-2-ил)-4-оксобутаноат
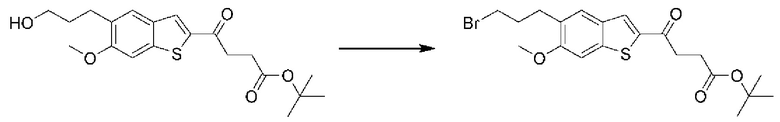
Трифенилфосфин (0.24 г, 0.91 ммоль) добавляли к смеси трет-бутил 4-(5-(3-гидроксипропил)-6-метоксибензо[b]тиофен-2-ил)-4-оксобутаноата (0.22 г, 0.57 ммоль) в THF (2.8 мл). Полученную в результате смесь охлаждали до 0°С, и добавляли NBS (0.15 г, 0.85 ммоль) одной порцией. После перемешивания в течение 30 мин при 0°С, реакцию разбавляли насыщенным водным AlCl3 и EtOAc. Органический слой отделяли, высушивали над Na2SO4, фильтровали и концентрировали под пониженным давлением. Полученное в результате вещество затем очищали с помощью хроматографии на силикагеле с получением трет-бутил 4-(5-(3-бромпропил)-6-метоксибензо[b]тиофен-2-ил)-4-оксобутаноата. ЖХ-МС (C16H18BrO4S) (ES, m/z): 385, 387 [М-С4Н8]+. 1Н ЯМР (500 МГц, DMSO-d6) δ 8.27 (s, 1H), 7.78 (s, 1H), 7.62 (s, 1H), 3.90 (s, 3H), 3.60-3.49 (m, 2Н), 3.30-3.21 (m, 2Н), 2.88-2.75 (m, 2Н), 2.67-2.56 (m, 2Н), 2.18-2.06 (m, 2Н), 1.38 (s, 9Н).
Промежуточное соединение 4: 4-(5-бром-6-метоксибензо[b]тиофен-2-ил)-4-оксобутаннитрил
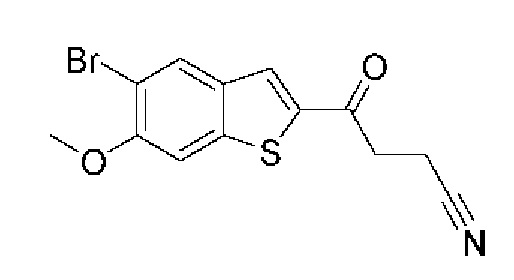
Стадия 1: метил 5-бром-6-метоксибепзо[b]тиофен-2-карбоксилат
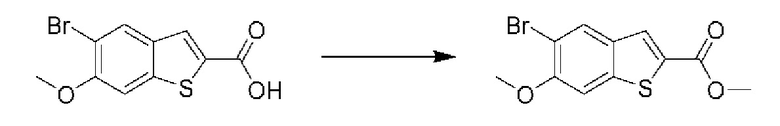
Концентрированную H2SO4 (3.0 мл, 56 ммоль) добавляли к суспензии 5-бром-6-метоксибензо[b]тиофен-2-карбоновой кислоты (5.0 г, 17 ммоль) в МеОН (60 мл). Реакционную смесь нагревали до 70°С в течение 4.5 дней. Смесь затем охлаждали до комнатной температуры и разбавляли водой. К смеси добавляли 30% IPA в CHCl3. Органический слой отделяли, высушивали над Na2SO4, фильтровали и концентрировали под пониженным давлением с получением метил 5-бром-6-метоксибензо[b]тиофен-2-карбоксилата. 1Н ЯМР (500 МГц, DMSO-d6) δ 8.30 (s, 1H), 8.09 (s, 1H), 7.83 (s, 1H), 3.95 (s, 3H), 3.88 (s, 3H).
Стадия 2: (5-бром-6-метоксибензо[b]тиофен-2-ил)метанол
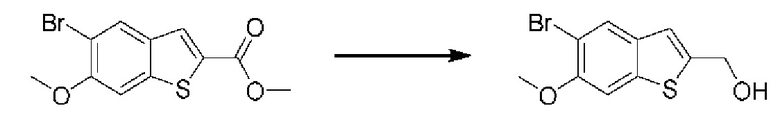
LAH (1.0 М в THF, 2.8 мл, 2.8 ммоль) медленно добавляли к смеси метил 5-бром-6-метоксибензо[b]тиофен-2-карбоксилата (0.78 г, 2.3 ммоль) в THF (9.0 мл) при 0°С. Через 40 мин реакционную смесь разбавляли насыщенным водным AlCl3. EtOAc добавляли, и органический слой отделяли, высушивали над Na2SO4, фильтровали и концентрировали под пониженным давлением. Полученное в результате вещество очищали с помощью хроматографии на силикагеле с получением (5-бром-6-метоксибензо[b]тиофен-2-ил)метанола. ЖХ-МС (C10H8BrOS) (ES, m/z): 255, 257 [М-ОН]+. 1Н ЯМР (500 МГц, DMSO-d6) δ 8.01 (s, 1H), 7.69 (s, 1Н), 7.14 (s, 1H), 5.69-5.59 (m, 1H), 4.73-4.64 (m, 2H), 3.89 (s, 3H).
Стадия 3: 5-буом-6-метоксибензо[b]тиофен-2-каубалъдегид
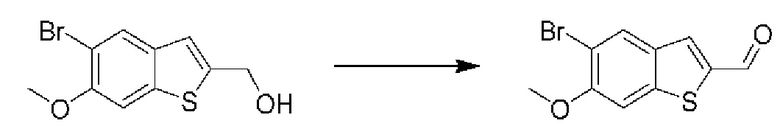
Диоксид марганца (6.3 г, 73 ммоль) добавляли к смеси (5-бром-6-метоксибензо[b]тиофен-2-ил)метанола (4.0 г, 15 ммоль) в DCM (97 мл). Реакционную смесь перемешивали при комнатной температуре в течение 20 ч, затем фильтровали через слой целита. Фильтрат концентрировали под пониженным давлением. Полученное в результате вещество растирали в МеОН, и смесь пропускали через стеклянную фритту с получением 5-бром-6-метоксибензо[b]тиофен-2-карбальдегида. ЖХ-МС (C10H8BrO2S) (ES, m/z): 271, 273 [М+Н]+. 1Н ЯМР (500 МГц, DMSO-d6) δ 10.07 (s, 1Н), 8.40 (s, 1H), 8.29 (s, 1H), 7.87 (s, 1H), 3.97 (s, 3H).
Стадия 4: 4-(5-буом-6-метоксибензо[b]тиофен-2-ил)-4-оксобутаннитуил

ACN (0.15 мл, 2.2 ммоль) добавляли к суспензии 5-бром-6-метоксибензо[b]тиофен-2-карбальдегида (0.30 г, 1.1 ммоль), 2-мезитил-2,5,6,7-тетрагидро-пирроло[2,1-с][1,2,4]триазол-4-ийхлорида (0.029 г, 0.11 ммоль) и К3РО4 (0.24 г, 1.1 ммоль) в толуоле (2.2 мл). Реакционную смесь помещали в атмосферу Ar и перемешивали при комнатной температуре в течение 18 ч. Реакционную смесь затем концентрировали под пониженным давлением, и полученный в результате остаток очищали с помощью хроматографии на силикагеле с получением 4-(5-бром-6-метоксибензо[b]тиофен-2-ил)-4-оксобутаннитрила. ЖХ-МС (C13H11BrNO2S) (ES, m/z): 324, 326 [М+Н]+. 1H ЯМР (500 МГц, DMSO-d6) δ 8.33-8.22 (m, 2Н), 7.84 (s, 1H), 3.96 (s, 3H), 3.59-3.44 (m, 2Н), 2.88-2.73 (m, 2Н).
Промежуточное соединение 5: этил 4-(5-бром-6-метоксибензо[b]тиофен-2-ил)-4-оксобутаноат
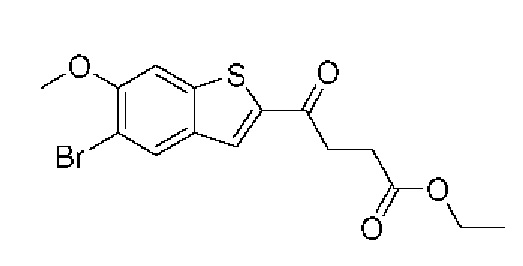
Стадия 1: 5-бром-2-фтор-4-метоксибензальдегид
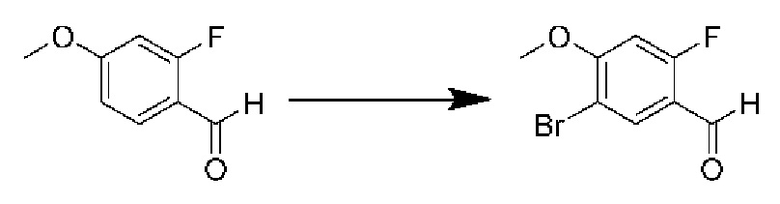
2-Фтор-4-метоксибензальдегид (9.0 г, 58 ммоль) медленно добавляли (порциями) к раствору Br2 (6.0 мл, 120 ммоль) в МеОН (40 мл) при 0°С. Реакционную смесь перемешивали при 0°С в течение 2 ч. К реакционной смеси медленно добавляли раствор NaHSO3 (24.3 г, 234 ммоль) в Н2О (300 мл) при 0°С. Полученную в результате суспензию затем перемешивали в течение 30 мин при 0°С. Реакционную смесь фильтровали, и фильтрат промывали дополнительным объемом Н2О (3x25 мл). Фильтрат затем высушивали под пониженным давлением с получением 5-бром-2-фтор-4-метоксибензальдегида. Продукт применяли без очистки. 1Н ЯМР (500 МГц, DMSO-d6): δ 10.02 (s, 1H), 7.98 (d, J=7.5 Гц, 1H), 7.26 (d, J=13.0 Гц, 1H), 3.97 (s, 3H).
Стадия 2: трет-бутил 5-буом-6-метоксибензо[b]тиофен-2-каубоксилат
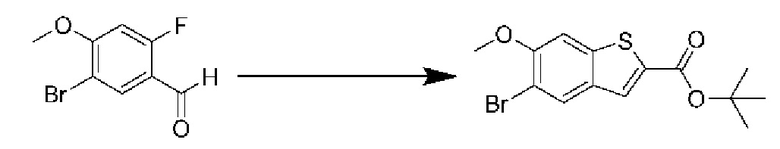
К2СО3 (19.0 г, 137 ммоль) медленно добавляли (порциями) к раствору 5-бром-2-фтор-4-метоксибензальдегида (10.7 г, 45.8 ммоль) и трет-бутил 2-меркаптоацетата (6.65 мл, 45.8 ммоль) в DMF (50 мл) при 20°С в атмосфере Ar. Реакционную смесь перемешивали и нагревали до 100°С в течение 16 ч. Реакционную смесь затем охлаждали до комнатной температуры и разбавляли Et2O (1000 мл). Смесь затем промывали H2O (500 мл, затем 2x250 мл) и объединенные водные слои экстрагировали Et2O (2x200 мл). Органические слои затем объединяли и промывали насыщенным водным NaCl (50 мл). Органический слой отделяли, высушивали над MgSO4, фильтровали и концентрировали под пониженным давлением с получением трет-бутил 5-бром-6-метоксибензо[b]тиофен-2-карбоксилата. Продукт применяли без очистки. 1H ЯМР (500 МГц, DMSO-d6): δ 8.26 (s, 1H), 7.96 (s, 1H), 7.78 (s, 1H), 3.92 (s, 3H), 1.55 (s,9H).
Стадия 3: 5-бром-6-метоксибепзо[b]тиофеп-2-карбоповая кислота
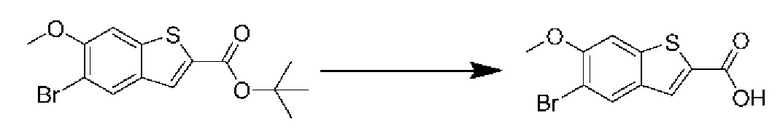
HCl (56 мл, 4.0 М в 1,4-диоксане, 230 ммоль) добавляли к раствору трет-бутил 5-бром-6-метоксибензо[6]тиофен-2-карбоксилата (15.5 г, 45.0 ммоль) в DCM (200 мл) при 20°С. Реакционную смесь перемешивали при 20°С в течение 3 дней. Реакционную смесь затем разбавляли путем добавления по каплям гексана (500 мл). Полученную после добавления суспензию перемешивали в течение дополнительных 2 ч при комнатной температуре. Реакционную смесь фильтровали, и собранное вещество промывали гексаном (2x50 мл) и высушивали под пониженным давлением с получением 5-бром-6-метоксибензо[b]тиофен-2-карбоновой кислоты, которую применяли без очистки. 1Н ЯМР (500 МГц, DMSO-d6): δ 13.42 (s, 1H), 8.26 (s, 1H), 7.98 (s, 1H), 7.80 (s, 1Н), 3.93 (s, 3H).
Стадия 4: 5-буом-6-метоксибензо[b]тиофен-2-каубонилхлоуид
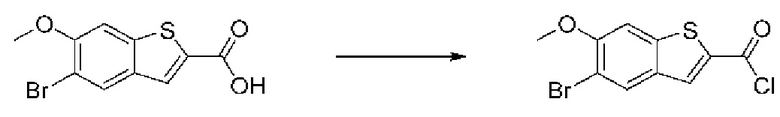
DMF (0.049 мл, 0.63 ммоль) медленно добавляли (по каплям) к раствору 5-бром-6-метоксибензо[b]тиофен-2-карбоновой кислоты (6.0 г, 21 ммоль) и (COCl)2 (5.5 мл, 63 ммоль) в THF (100 мл) при 0°С в атмосфере Ar. Реакционную смесь перемешивали при 0°С в течение 2 ч и затем оставляли нагреваться до комнатной температуры. Реакционную смесь перемешивали в течение 18 ч при комнатной температуре. Реакционную смесь затем концентрировали под пониженным давлением с получением 5-бром-6-метоксибензо[b]тиофен-2-карбонилхлорида. Продукт применяли без очистки.
Стадия 5: этил 4-(5-бром-6-метоксибензо[b]тиофен-2-ил)-4-оксобутаноат

Раствор (3-этокси-3-оксопропил)цинка (II) бромида (13.8 мл, 0.50 М в THF, 6.9 ммоль) добавляли в высушенную в печи колбу, содержащую ((тиофен-2-карбонил)окси)медь (1.31 г, 6.87 ммоль) в атмосфере Ar при 0°С. Реакционную смесь перемешивали в течение 20 мин при 0°С в атмосфере Ar. Дегазированный Ar раствор 5-бром-6-метоксибензо[b]тиофен-2-карбонил хлорид а (1.52 г, 4.98 ммоль) в THF (25.0 мл) затем добавляли через канюлю к реакционной смеси при 0°С; полученную в результате суспензию оставляли нагреваться до комнатной температуры и перемешивали в течение 3 ч. Реакционную смесь охлаждали до 0°С и останавливали насыщенным водным NH4Cl (50 мл). Смесь оставляли нагреваться до комнатной температуры и перемешивали в течение 10 мин. Смесь фильтровали, и фильтрат разбавляли EtOAc (500 мл) и насыщенным водным NaCl (50 мл). Органический слой отделяли, промывали насыщенным водным NaCl (25 мл), высушивали над MgSO4, фильтровали и концентрировали под пониженным давлением. Остаток очищали с помощью колоночной хроматографии на силикагеле (EtOAc в DCM) с получением этил 4-(5-бром-6-метоксибензо[b]тиофен-2-ил)-4-оксобутаноата. ЖХ-МС (C15H16BrO4S) (ES, m/z): 371, 373 [М+Н]+. 1H ЯМР (500 МГц, DMSO-d6): δ 8.27 (s, 1H), 8.26 (s, 1H), 7.81 (s, 1H), 4.07-4.02 (m, 2Н), 3.94 (s, 3H), 3.35-3.25 (m, 2Н), 2.68-2.64 (m, 2Н), 1.20-1.14 (m, 3H).
Промежуточное соединение 6: трет-бутил 4-(5-бром-6-метоксибензо[b]тиофен-2-ил)-4-оксобутаноат
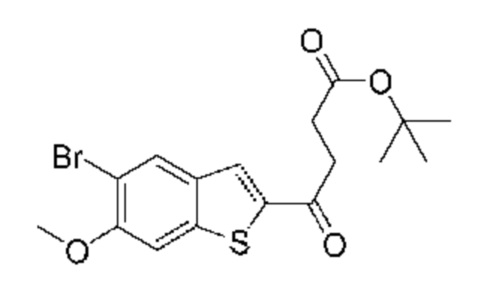
Промежуточное соединение 6 может быть получено в соответствии со способами, аналогичными описанным выше для Промежуточного соединения 5, с применением соответствующих исходных материалов, описанных как приготовленные или полученных из коммерчески доступных источников.
Промежуточное соединение 7: этил 4-(6-метокси-5-(3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пропил)бензо[b]тиофен-2-ил)-4-оксобутаноат
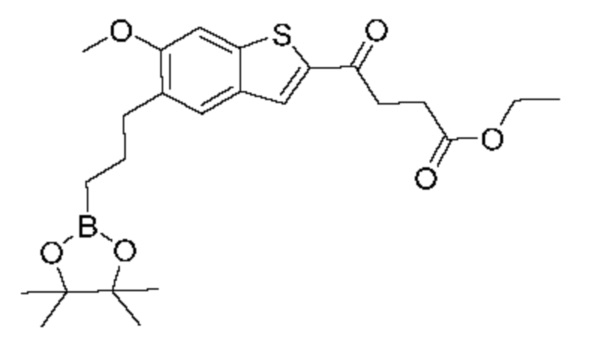
Стадия 1: этил 4-(5-аллил-6-метоксибензо[b]тиофен-2-ил)-4-оксобутаноат
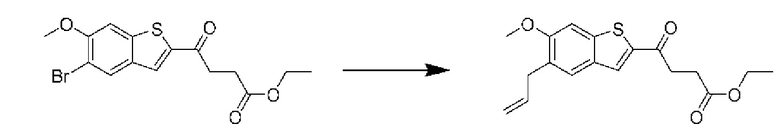
В виалу, содержащую этил 4-(5-бром-6-метоксибензо[b]тиофен-2-ил)-4-оксобутаноат (5.0 г, 13 ммоль), Pd(Ph3P)4 (1.6 г, 1.3 ммоль) и диоксан (15 мл), добавляли аллилтри-н-бутилолово (5.4 мл, 18 ммоль). Реакцию нагревали до 90°С в течение 18 ч. После охлаждения до комнатной температуры смесь разбавляли DCM, фильтровали через целит и добавляли в колбу, содержащую водный KF (0.5 М, 200 мл). Смесь перемешивали, и органический слой затем отделяли, высушивали над Na2SO4, фильтровали и концентрировали под пониженным давлением. Полученный в результате остаток очищали с помощью колоночной хроматографии на силикагеле (0→30% градиент EtOAc в гексанах) с получением этил 4-(5-аллил-6-метоксибензо[b]тиофен-2-ил)-4-оксобутаноата. ЖХ-МС (C18H21O4S) (ES, m/z): 333 [М+Н]+. 1Н ЯМР (600 МГц, DMSO-d6) δ 8.23 (s, 1Н), 7.69 (s, 1Н), 7.58 (s, 1Н), 5.96 (dq, J=15.9, 6.6 Гц, 1Н), 5.04 (d,J=4.5 Гц, 1H), 5.02 (s, 1H), 4.01 (q, J=7.0 Гц, 2Н), 3.85 (s, 3H), 3.37 (d, J=6.3 Гц, 2Н), 3.27 (dd, J=11.0, 4.3 Гц, 2Н), 2.62 (t, J=6.1 Гц, 2Н), 1.13 (t, J=7.1 Гц, 3H).
Стадия 2: этил 4-(6-метокси-5-(3-(4,4,5,5-тетраметил-1,3,2-диоксаборолап-2-ил)пропил)бензо[b]тиофен-2-ил)-4-оксобутаноат
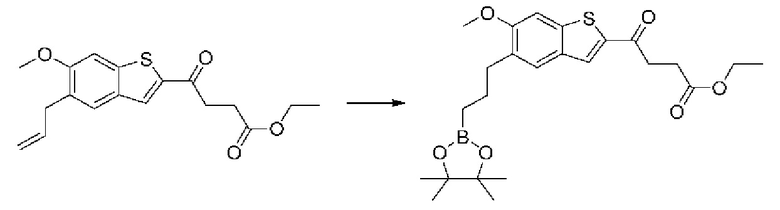
К смеси 1,4-бис(дифенилфосфино)бутана (0.45 г, 1.1 ммоль), димера хлор(1,5-циклооктадиен)иридия (i) (0.35 г, 0.53 ммоль), этил 4-(5-аллил-6-метоксибензо[b]тиофен-2-ил)-4-оксобутаноата (3.5 г, 11 ммоль) и THF (20 мл) добавляли пинаколборан (1.0 М в THF, 15.8 мл, 15.8 ммоль). Реакцию перемешивали при комнатной температуре в течение 4 ч. Растворитель затем удаляли под пониженным давлением, и остаток очищали с помощью колоночной хроматографии на силикагеле (0→20% градиент EtOAc в гексанах) с получением этил 4-(6-метокси-5-(3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пропил)бензо[b]тиофен-2-ил)-4-оксобутаноата. 1Н ЯМР (600 МГц, DMSO-d6) δ 8.21 (s, 1H), 7.66 (s, 1Н), 7.53 (s, 1H), 4.02 (q, J=7.0 Гц, 2H), 3.84 (s, 3H), 3.27 (t, J=6.2 Гц, 2H), 2.62 (t, J=6.1 Гц, 2H), 2.58 (t, J=7.4 Гц, 2H), 1.58 (p, J=7.4 Гц, 2H), 1.16-1.11 (m, 15H), 0.67(t, J=7.6 Гц, 2H).
Промежуточное соединение 8: трет-Бутил 4-(6-(3-бромпропил)-5-метоксибензо[b]тиофен-2-ил)-4-оксобутаноат
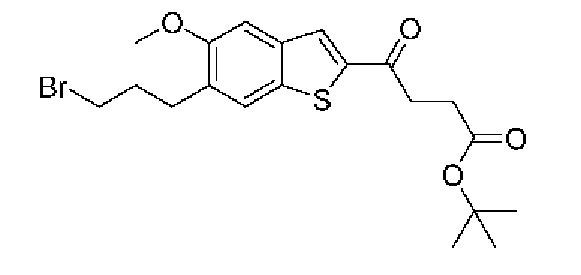
Стадия 1: метил 6-бром-5-метоксибензо[b]тиофен-2-карбоксилат
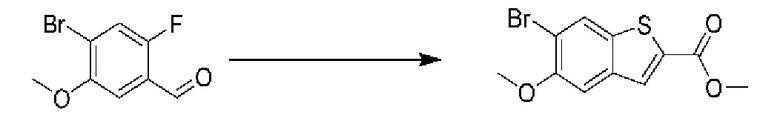
К перемешанному раствору 4-бром-2-фтор-5-метоксибензальдегида (5.00 г, 21.5 ммоль) в DMF (100 мл) добавляли метил 2-меркаптоацетат (2.51 г, 23.6 ммоль) и K2CO3 (8.90 г, 64.4 ммоль). Реакционную смесь 3 раза дегазировали N2. Полученную в результате смесь затем перемешивали при комнатной температуре в течение 15 ч. К реакционной смеси добавляли EtOAc (500 мл) и Н2О (1200 мл). Органический слой отделяли и промывали насыщенным водным NaCl (2×200 мл), высушивали над безводным Na2SO4, фильтровали и концентрировали под пониженным давлением. Остаток очищали с помощью колоночной хроматографии на силикагеле (EtOAc в РЕ) с получением метил 6-бром-5-метоксибензо[b]тиофен-2-карбоксилата. ЖХ-МС (C11H10BrO3S) (ES, m/z): 301, 303 [М+Н]+. 1Н ЯМР (400 МГц, CDCl3): δ=8.01 (s, 1H), 7.93 (s, 1H), 7.26 (s, 1H), 3.96 (s, 3Н), 3.94 (s, 3Н).
Стадия 2: 6-бром-5-метоксибензо[b]тиофен-2-каубоноеая кислота
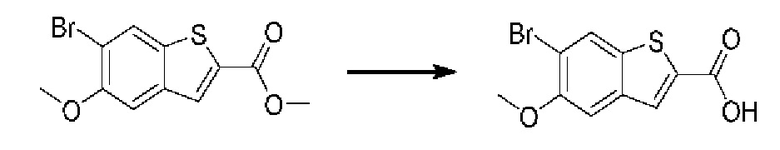
К суспензии метил 6-бром-5-метоксибензо[b]тиофен-2-карбоксилата (1.45 г, 4.81 ммоль) в МеОН (20 мл), THF (20 мл) и H2O (20 мл) добавляли NaOH (1.93 г, 48.1 ммоль). Полученную в результате суспензию нагревали до 50°С в течение 0.5 ч. Реакционную смесь концентрировали под пониженным давлением для удаления растворителя. К остатку добавляли Н2О (200 мл) и добавляли лимонную кислоту, чтобы довести раствор до рН=6. Полученную водную суспензию экстрагировали EtOAc (3×50 мл). Объединенные органические слои промывали насыщенным водным NaCl (100 мл), высушивали над Na2SO4, фильтровали и концентрировали под пониженным давлением с получением 6-бром-5-метоксибензо[b]тиофен-2-карбоновой кислоты, которую применяли без дополнительной очистки. 1H ЯМР (400 МГц, DMSO-d6): δ=13.52 (br s, 1Н), 8.35 (s, 1H), 8.01 (s, 1H), 7.65 (s, 1H), 3.90 (s, 3H).
Стадия 3: 6-бром-5-метоксибензо[b]тиофен-2-каубонилхлоуид
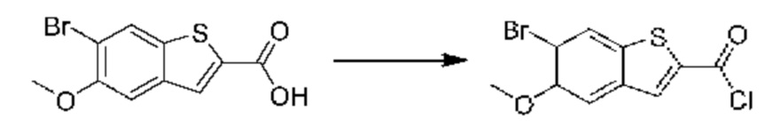
К перемешанному раствору 6-бром-5-метоксибензо[b]тиофен-2-карбоновой кислоты (800 мг, 2.79 ммоль) в безводном THF (6 мл) добавляли (COCl)2 (1.06 г, 8.36 ммоль) по каплям при 0°С. Смесь затем нагревали при 75°С в течение 15 ч и затем охлаждали до комнатной температуры. Растворитель удаляли под пониженным давлением с получением неочищенного 6-бром-5-метоксибензо[b]тиофен-2-карбонилхлорида, который применяли без дополнительной очистки.
Стадия 4: трет-бутил 4-(6-бром-5-метоксибензо[b]тиофен-2-ил)-4-оксобутаноат
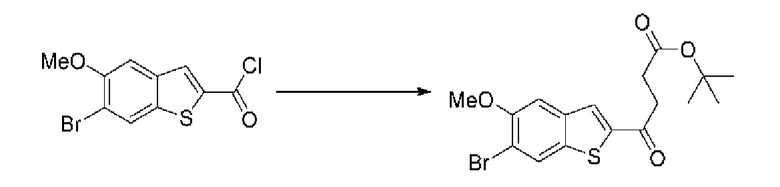
В круглодонную колбу добавляли CuI (0.24 г, 2.4 ммоль). Колбу откачивали и затем заполняли N2. Процедуру повторяли три раза. Добавляли THF (4.0 мл) и смесь охлаждали до 0°С. Смесь (3-(трет-бутокси)-3-оксопропил)цинка (II) бромида (0.50 М в THF, 9.6 мл, 4.8 ммоль) добавляли по каплям при 0°С в течение 10 мин. Полученную в результате смесь оставляли перемешиваться в течение 30 мин. Добавляли 6-бром-5-метоксибензо[b]тиофен-2-карбонилхлорид (0.73 г, 2.4 ммоль). Смесь удаляли из ледяной бани и оставляли нагреваться до комнатной температуры. Смесь перемешивали в течение 2 ч. Смесь затем охлаждали до 0°С, и добавляли концентрированный NH4OH (4.5 мл). К полученной в результате суспензии добавляли воду (240 мл) и МеОН (60 мл). Смесь перемешивали в течение 5 мин и обрабатывали ультразвуком на бане с соникатором. Смесь затем разбавляли EtOAc, и органический слой отделяли, высушивали над MgSO4, фильтровали и концентрировали под пониженным давлением. Остаток сырого вещества очищали с помощью хроматографии на силикагеле с получением трет-бутил 4-(6-бром-5-метоксибензо[b]тиофен-2-ил)-4-оксобутаноата. ЖХ-МС (C17H19BrO4S) (ES, m/z): 421, 423 [M+Na]+.
Стадия 5: трет-бутил 4-(6-(3-((трет-бутилдиметилсилил)окси)пропил)-5-метоксибензо[b]тиофен-2-ил)-4-оксобутаноат
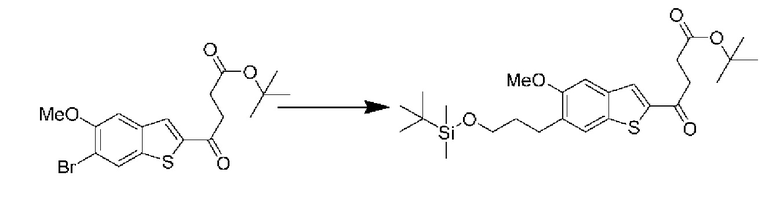
В колбу, содержащую трет-бутил 4-(6-бром-5-метоксибензо[b]тиофен-2-ил)-4-оксобутаноат (0.16 г, 0.40 ммоль) и THF (2.0 мл), добавляли [(2-дициклогексилфосфино-2',6'-бис(N,N-диметиламино)-1,1'-бифенил)-2-(2'-амино-1,1'-бифенил)]палладий (II) метан сульфонат (C-Phos Pd G3, 16 мг, 0.020 ммоль). Колбу 3 раза откачивали и снова заполняли N2. Добавляли (3-((трет-бутилдиметилсилил)окси)пропил)цинк (II) бромид (0.50 М в THF, 2.4 мл, 1.2 ммоль) и смесь оставляли перемешиваться при комнатной температуре в течение 2.5 ч. Затем реакцию останавливали смесью EtOAc и 10% водного цитрата натрия. Органический слой отделяли, промывали насыщенным водным NaCl, высушивали над MgSO4, фильтровали и концентрировали под пониженным давлением. Сырой остаток очищали с помощью хроматографии на силикагеле с получением трет-бутил 4-(6-(3-((трет-бутилдиметилсилил)окси)пропил)-5-метоксибензо[b]тиофен-2-ил)-4-оксобутаноата. ЖХ-МС (C26H41O5SSi-C4H8) (ES, m/z): 437 [М-С4Н8]+.
Стадия 6: трет-бутил 4-(б-(3-гидроксипропил)-5-метоксибензо[b]тиофен-2-ил)-4-оксобутапоат
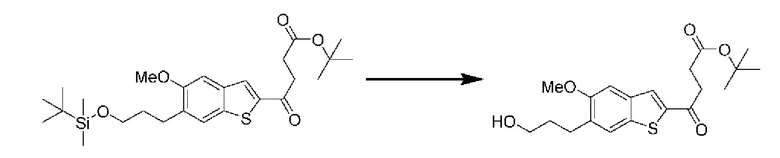
В колбу, содержащую трет-бутил 4-(6-(3-((трет-бутилдиметилсилил)окси)пропил)-5-метоксибензо[b]тиофен-2-ил)-4-оксобутаноат (0.11 г, 0.23 ммоль), добавляли МеОН (1.5 мл), воду (1.5 мл) и НО Ас (1.5 мл). Смесь оставляли перемешиваться в течение 4 ч. Смесь разбавляли EtOAc и затем промывали водой (3×50 мл). Органический слой высушивали над MgSO4, фильтровали и концентрировали под пониженным давлением. Сырой остаток очищали с помощью колоночной хроматографии на силикагеле с получением трет-бутил 4-(6-(3-гидроксипропил)-5-метокси-бензо[b]тиофен-2-ил)-4-оксобутаноата. ЖХ-МС (C20H27O5S-C4H8) (ES, m/z): 323 [М-С4Н8]+.
Стадия 7: трет-бутил 4-(6-(3-бромпуопш)-5-метоксибензо[b]тиофен-2-ил)-4-оксобутапоат

К смеси трет-бутил 4-(6-(3-гидроксипропил)-5-метоксибензо[b]тиофен-2-ил)-4-оксобутаноата (74 мг, 0.20 ммоль) и трифенилфосфина (82 мг, 0.31 ммоль) в THF (1.0 мл) при 0°С добавляли NBS (52 мг, 0.29 ммоль). Через 15 мин при 0°С смесь останавливали насыщенным водным NH4Cl и разбавляли EtOAc. Органический слой отделяли, высушивали над MgSO4, фильтровали и концентрировали под пониженным давлением. Сырой остаток очищали с помощью колоночной хроматографии на силикагеле с получением трет-бутил 4-(6-(3-бромпропил)-5-метоксибензо[b]тиофен-2-ил)-4-оксобутаноата. ЖХ-МС (C20H26BrO4S-C4H8) (ES, m/z): 385, 387 [М-С4Н8].
Промежуточное соединение 9: Метил 4-(4-бром-6-метоксибензо[b]тиофен-2-ил)-4-оксобутаноат
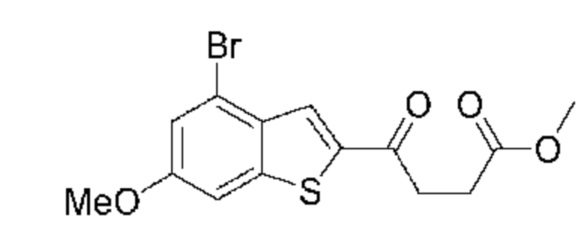
Стадия 1: 2-бром-6-фтор-4-метоксибензальдегид
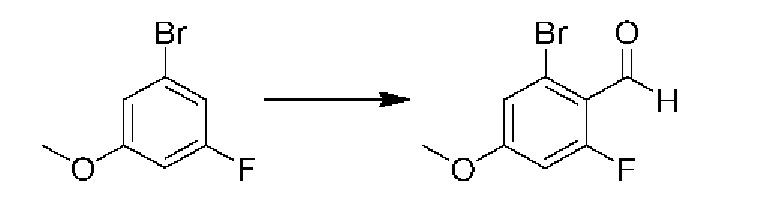
К смеси 1-бром-3-фтор-5-метоксибензола (7.5 г, 37 ммоль) в THF (120 мл) при -78°С добавляли LDA (2.0 М в THF, 22 мл, 44 ммоль) и смесь оставляли перемешиваться в течение 30 мин при -78°С. Через 30 мин добавляли DMF (3.4 мл, 44 ммоль) по каплям, и смесь затем оставляли перемешиваться в течение 30 мин. Затем реакцию останавливали водой, нагревали до комнатной температуры и затем добавляли EtOAc. Слои разделяли, и водный слой экстрагировали EtOAc еще два раза. Объединенные органические слои высушивали над MgSO4, фильтровали и концентрировали под пониженным давлением. Остаток сырого вещества очищали с помощью хроматографии на силикагеле с получением 2-бром-6-фтор-4-метоксибензальдегида. ЖХ-МС (C8H7BrFO2) (ES, m/z): 233, 235 [М+Н]+.
Стадия 2: метил 4-бром-6-метоксибензо[b]тиофен-2-карбоксилат
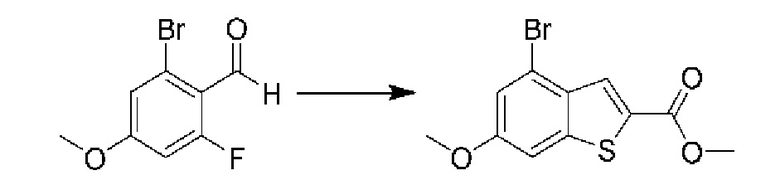
К смеси 2-бром-6-фтор-4-метоксибензальдегида (2.5 г, 11 ммоль) в DMSO (54 мл) добавляли TEA (3.0 мл, 21 ммоль). Через 10 мин добавляли метил тиогликолят (3.1 мл, 32 ммоль) и смесь оставляли перемешиваться в течение 30 мин при комнатной температуре. Через 30 мин смесь нагревали до 60°С в течение 1 ч. После охлаждения до комнатной температуры смесь разбавляли насыщенным водным NaHCO3 и EtOAc. Органический слой отделяли, высушивали над MgSO4, фильтровали и концентрировали под пониженным давлением. Остаток сырого вещества очищали с помощью хроматографии на силикагеле (0→15% градиент EtOAc в гексанах) с получением метил 4-бром-6-метоксибензо[b]тиофен-2-карбоксилата. ЖХ-МС (C11H10BrO3S) (ES, m/z): 301, 303 [М+Н]+. 1H ЯМР (500 МГц, DMSO-d6) δ 7.93 (s, 1H), 7.73 (s, 1H), 7.44-7.37 (m, 1Н), 3.89 (s, 3Н), 3.87 (s, 3Н).
Стадия 3: 4-бром-6-метоксибензо[b]тиофеп-2-карбоновая кислота
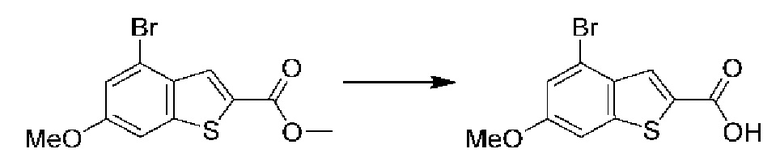
К смеси метил 4-бром-6-метоксибензо[b]тиофен-2-карбоксилата (1.7 г, 5.5 ммоль) в THF (14 мл), МеОН (7.0 мл) и воде (7.0 мл) добавляли LiOH (0.66 г, 28 ммоль) и смесь нагревали до 40°С в течение 2 ч. Через 2 ч смесь оставляли охлаждаться до комнатной температуры. Смесь останавливали водной HCl (2.0 М в воде, 14 мл, 28 ммоль). Смесь фильтровали, и остаток промывали EtOAc. Остаток затем высушивали под вакуумом и применяли без дополнительной очистки. ЖХ-МС (C10H8BrO3S) (ES, m/z): 287, 289 [М+Н]+. 1Н ЯМР (500 МГц, DMSO-d6) δ 13.57 (s, 1H), 7.86 (s, 1H), 7.71 (s, 1H), 7.38 (d, J=1.1 Гц, 1H), 3.86 (s, 3Н).
Промежуточное соединение 10: Этил 4-(5-(3-бромпропил)-6-метоксибензо[b]тиофен-2-ил)-4-оксобутаноат
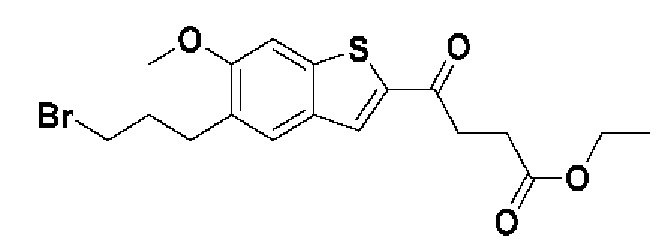
Стадия 1: этил 4-(6-метокси-5-(3-((тетрагидро-2Н-пирап-2-ил)окси)пропил)бензо[b]тиофен-2-ил)-4-оксобутаноат
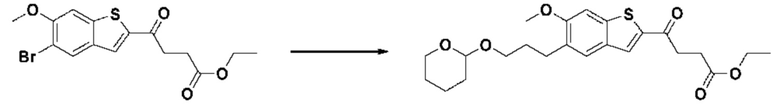
К смеси этил 4-(5-бром-6-метоксибензо[b]тиофен-2-ил)-4-оксобутаноата (13 г, 35 ммоль) и C-Phos Pd G4 (1.4 г, 1.7 ммоль) добавляли однократно (3-((тетрагидро-2Н-пиран-2-ил)окси)пропил)цинк (II) бромид (0.50 М в THF, 100 мл, 50 ммоль). Реакцию нагревали до 40°С в течение 2 ч. Смесь затем оставляли охлаждаться до комнатной температуры и фильтровали через целит. Фильтрат концентрировали под пониженным давлением. Полученный в результате остаток очищали с помощью колоночной хроматографии на силикагеле (0→30% градиент EtOAc в гексанах) с получением этил 4-(6-метокси-5-(3-((тетрагидро-2Н-пиран-2-ил)окси)пропил)бензо[b]тиофен-2-ил)-4-оксобутаноата. ЖХ-МС (C23H31O6S) (ES, m/z): 435 [М+Н]+. 1Н ЯМР (600 МГц, DMSO-d6) δ 8.21 (s, 1H), 7.71 (s, 1H), 7.55 (s, 1H), 4.50 (s, 1H), 4.02 (q, J=7.0 Гц, 2H), 3.85 (s, 3H), 3.70 (t, J=8.1 Гц, 1H), 3.65-3.58 (m, 1H), 3.40-3.34 (m, 1H), 3.33-3.29 (m, 3H), 2.73-2.59 (m, 4H), 1.79 (p, J=6.7 Гц, 2H), 1.69 (d, J=8.7 Гц, 1H), 1.58 (t, J=7.9 Гц, 1H), 1.48-1.34 (m, 4H), 1.14 (t, J=7.1 Гц, 3H).
Стадия 2: этил 4-(5-(3-бромпропил)-6-метоксибензо[b]тиофен-2-ил)-4-оксобутаноат
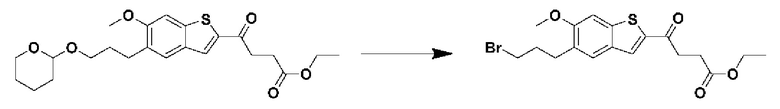
К смеси 4-(6-метокси-5-(3-((тетрагидро-2Н-пиран-2-ил)окси)пропил)бензо[b]тиофен-2-ил)-4-оксобутаноата (6.2 г, 14 ммоль) и DCM (100 мл) при 0°С добавляли трифенилфосфин дибромид (9.03 г, 21.4 ммоль) порциями. Смесь оставляли нагреваться до комнатной температуры и затем перемешивали в течение 1 ч. Затем реакцию останавливали водой и разбавляли DCM. Органический слой отделяли, высушивали над Na2SO4, фильтровали и концентрировали под пониженным давлением. Полученный в результате остаток очищали с помощью колоночной хроматографии на силикагеле (0→30% градиент EtOAc в гексанах) с получением этил 4-(5-(3-бромпропил)-6-метоксибензо[b]тиофен-2-ил)-4-оксобутаноата. ЖХ-МС (C18H22BrO4S) (ES, m/z): 413, 415 [М+Н]+. 1H ЯМР (600 МГц, DMSO-d6) δ 8.23 (s, 1H), 7.73 (s, 1H), 7.58 (s, 1H), 4.02 (q, J=7.0 Гц, 2H), 3.86 (s, 3Н), 3.50 (t, J=6.5 Гц, 2H), 3.27 (d, J=6.4 Гц, 2H), 2.75 (t, J=7.3 Гц, 2H), 2.63 (t, J=6.2 Гц, 2H), 2.07 (p, J=6.7 Гц, 2H), 1.14 (t, J=7.1 Гц, 3Н).
Промежуточное соединение 11: Этил 4-(4-фтор-5-гидрокси-6-метоксибензо[b]тиофен-2-ил)-4-оксобутаноат
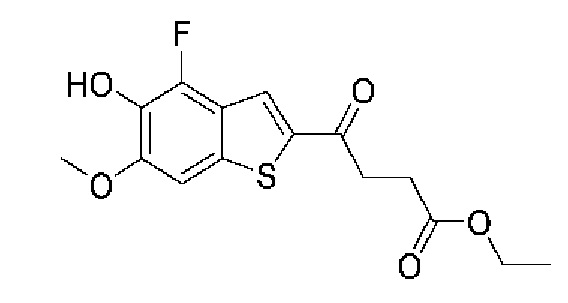
Стадия 1: метил 4-фтор-5,6-диметоксибензо[b]тиофен-2-карбоксилат

1-Хлорметил-4-фтор-1,4-диазониабицикло[2.2.2]октан бис(тетрафторборат) (Selectfluor™, 77 мг, 0.22 ммоль) добавляли к смеси метил 5,6-диметоксибензо[b]тиофен-2-карбоксилата (50 мг, 0.20 ммоль) в ACN (1 мл) при комнатной температуре. Полученную в результате смесь перемешивали при 45°С в течение 15 ч. Смесь охлаждали до комнатной температуры, разбавляли насыщенным водным NaHCO3 (10 мл) и экстрагировали EtOAc (3×10 мл). Объединенные органические слои промывали насыщенным водным NaCl (10 мл), высушивали над безводным Na2SO4, фильтровали и концентрировали под пониженным давлением. Остаток очищали с помощью препаративной TLC (SiO2, EtOAc в РЕ) с получением метил 4-фтор-5,6-диметоксибензо[b]тиофен-2-карбоксилата. ЖХ-МС (C12H12FO4S) (ES, m/z): 293 [М+Н]+. 1H ЯМР (400 МГц, CDCl3): δ 8.05 (s, 1H), 7.08 (s, 1H), 3.99 (s, 3Н), 3.97 (s, 3Н), 3.94 (s, 3Н).
Стадия 2: 4-фтор-5,6-диметоксибензо[b]тиофен-2-карбоновая кислота

LiOH⋅Н2О (71.4 мг, 1.70 ммоль) добавляли порциями к смеси метил 4-фтор-5,6-диметоксибензо[b]тиофен-2-карбоксилата (46 мг, 0.170 ммоль) в THF (3 мл), МеОН (1 мл) и Н2О (1 мл) при комнатной температуре. Затем смесь перемешивали в течение 15 ч. Смесь доводили до рН = 5 с помощью 1 н. HCl и экстрагировали EtOAc (3×10 мл). Объединенные органические слои промывали насыщенным водным NaCl (10 мл), высушивали над безводным Na2SO4, фильтровали и концентрировали под пониженным давлением. Остаток очищали с помощью препаративной ВЭЖХ (ACN/H2O с 0.1% TFA) с получением 4-фтор-5,6-диметоксибензо[b]тиофен-2-карбоновой кислоты. ЖХ-МС (C11H9FO4S) (ES, m/z): 257 [М+Н]+. 1H ЯМР (400 МГц, CDCl3): δ 8.12 (s, 1Н), 7.09 (s, 1Н), 3.99 (s, 3Н), 3.97 (s, 3Н).
Стадия 3: 4-фтор-5,6-диметоксибензо[b]тиофен-2-карбонилхлорид
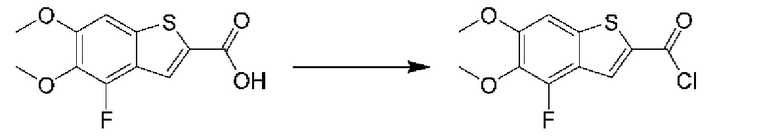
К перемешанному раствору 4-фтор-5,6-диметоксибензо[b]тиофен-2-карбоновой кислоты (153 мг, 0.60 ммоль) в безводном THF (5 мл) добавляли (COCl)2 (0.21 мл, 2.40 ммоль) по каплям при 0°С. Смесь перемешивали при 0°С в течение 1 ч и затем при комнатной температуре в течение 1 ч. Растворитель удаляли под пониженным давлением с получением 4-фтор-5,6-диметоксибензо[b]тиофен-2-карбонилхлорида, который применяли без дополнительной очистки.
Стадия 4: этил 4-(4-фтор-5,6-диметоксибензо[b]тиофен-2-ил)-4-оксобутаноат
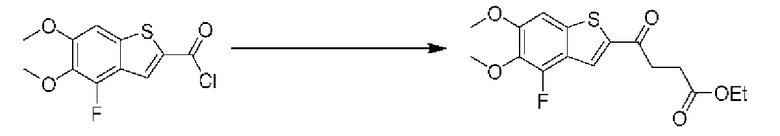
Суспензию тиофен-2-карбоксилата меди (I) (125 мг, 0.65 ммоль) продували N2 в течение 5 мин и затем охлаждали до 0°С. Раствор (3-этокси-3-оксопропил)цинка (II) бромида (17.7 мл, 0.5 М в THF, 8.83 ммоль) добавляли в атмосфере N2 при 0°С, и реакционную смесь перемешивали в течение 20 мин при 0°С.Затем добавляли продутый N2 раствор 4-фтор-5,6-диметоксибензо[6]тиофен-2-карбонилхлорида (130 мг, 0.47 ммоль) в THF (3 мл) при 0°С. Полученную в результате суспензию оставляли нагреваться до комнатной температуры и перемешивали в течение 8 ч. Смесь выливали в насыщенный водный NH4Cl (20 мл) с перемешиванием. Смесь экстрагировали EtOAc (2×20 мл). Объединенные органические слои промывали Н2О и насыщенным водным NaCl, высушивали над MgSO4, фильтровали и концентрировали под пониженным давлением. Остаток очищали с помощью колоночной хроматографии на силикагеле (EtOAc в гексанах) с получением этил 4-(4-фтор-5,6-диметоксибензо[b]тиофен-2-ил)-4-оксобутаноата. ЖХ-МС (C16H18FO5S) (ES, m/z): 341 [М+Н]+. 1Н ЯМР (500 МГц, CDCl3) δ 8.02 (d, J=0.7 Гц, 1H), 7.10 (t, J=1.0 Гц, 1H), 4.19 (q, J=7.2 Гц, 2Н), 4.05-3.97 (m, 6Н), 3.36 (t, J=6.7 Гц, 2Н), 2.81 (t, J=6.7 Гц, 2Н), 1.29 (t, J=7.2 Гц, 3Н).
Стадия 5: этил 4-(4-фтор-5-гидрокси-6-метоксибензо[b]тиофен-2-ил)-4-оксобутаноат
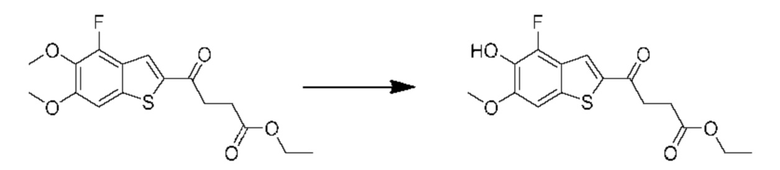
К смеси этил 4-(4-фтор-5,6-диметоксибензо[b]тиофен-2-ил)-4-оксобутаноата (3.6 г, 11 ммоль) и DCM (50 мл) добавляли AlCl3 (5.64 г, 42.3 ммоль). Реакционную смесь оставляли перемешиваться при комнатной температуре в течение 18 ч. К реакционному сосуду затем присоединяли капельную воронку, и к смеси медленно добавляли воду (50 мл) с энергичным перемешиванием с последующим добавлением водной HCl (1 н., 50 мл). Смесь затем разбавляли 20% IPA/DCM. Органический слой отделяли, высушивали над Na2SO4, фильтровали и концентрировали под пониженным давлением. Полученный в результате остаток очищали с помощью колоночной хроматографии на силикагеле (100% DCM) с получением этил 4-(4-фтор-5-гидрокси-6-метоксибензо[b]тиофен-2-ил)-4-оксобутаноата. ЖХ-МС (C15H16FO5S) (ES, m/z): 327 [М+Н]+. 1H ЯМР (600 МГц, DMSO-d6) δ 9.53 (s, 1Н), 8.25 (s, 1H), 7.47 (s, 1H), 4.06 (q, J=7.1 Гц, 2H), 3.92 (s, 3Н), 3.39-3.34 (m, 2Н), 2.68-2.63 (m, 2Н), 1.18 (t, J=7.1 Гц, 3Н).
Промежуточное соединение 12: (S)-метил 4-(5-(3-гидроксипропил)-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноат
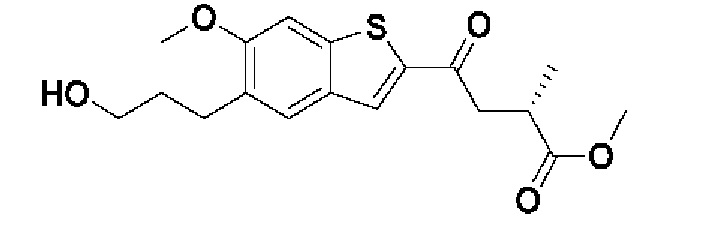
Стадия 1: (2S)-метил-4-(6-метокси-5-(3-((тетрагидро-2Н-пиран-2-ил)окси)пропил)бензо[b]тиофен-2-ил)-2-метил-4-оксобутаноат

К смеси (S)-метил-4-(5-бром-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата (7.0 г, 19 ммоль) и C-Phos Pd G4 (0.76 г, 0.94 ммоль) добавляли (3-((тетрагидро-2Н-пиран-2-ил)окси)пропил)цинк (II) бромид (0.50 М в THF, 100 мл, 50 ммоль). Смесь нагревали до 40°С в течение 2 ч. Смесь затем оставляли охлаждаться до комнатной температуры и фильтровали через целит. Фильтрат концентрировали под пониженным давлением. Остаток очищали с помощью колоночной хроматографии на силикагеле (0→30% градиент EtOAc в гексанах) с получением этил (2S)-метил-4-(6-метокси-5-(3-((тетрагидро-2Н-пиран-2-ил)окси)пропил)бензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата. ЖХ-МС (C23H31O6S) (ES, m/z): 435 [М+Н]+. 1Н ЯМР (600 МГц, DMSO-d6) δ 8.21 (s, 1H), 7.70 (s, 1H), 7.55 (s, 1H), 4.50 (s, 1H), 3.85 (s, 3Н), 3.70 (dd, J=13.2, 5.3 Гц, 1H), 3.64-3.58 (m, 1H), 3.56 (s, 3H), 3.42-3.35 (m, 2H), 3.35-3.29 (m, 1H), 3.15 (dd, J=17.4, 4.9 Гц, 1H), 2.94 (dt, J=12.9, 7.1 Гц, 1H), 2.67 (hept, J=7.6, 7.1 Гц, 2H), 1.79 (p, J=6.7 Гц, 2H), 1.69 (d, J=8.7 Гц, 1H), 1.58 (t, J=7.9 Гц, 1H), 1.48-1.35 (m, 4H), 1.18-1.11 (m, 3H).
Стадия 2: (S)-метил 4-(5-(3-гидроксипропил)-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноат
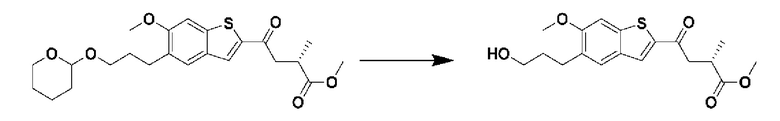
К смеси (2S)-метил-4-(6-метокси-5-(3-((тетрагидро-2Н-пиран-2-ил)окси)пропил)бензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата (2.62 г, 6.03 ммоль) и МеОН (50 мл) добавляли pTsOH (1.72 г, 9.04 ммоль). Смесь оставляли перемешиваться при комнатной температуре в течение 1 ч. Затем реакцию останавливали водой и разбавляли DCM. Органический слой отделяли и затем промывали водным насыщенным NaHCO3. Органический слой затем высушивали над Na2SO4, фильтровали и концентрировали под пониженным давлением с получением (S)-метил 4-(5-(3-гидроксипропил)-6-метоксибензо[b]тиофен-2-ил)-2метил-4-оксобутаноата. ЖХ-МС (C18H23O5S) (ES, m/z): 351 [М+Н]+. 1H ЯМР (600 МГц, DMSO-d6) δ 8.21 (s, 1H), 7.69 (s, 1H), 7.54 (s, 1H), 4.44 (t, J=5.0 Гц, 1H), 3.85 (s, 3H), 3.56 (s, 3H), 3.42-3.35 (m, 3H), 3.15 (dd, J=17.4, 4.9 Гц, 1H), 2.93 (h, J=7.0 Гц, 1H), 2.64 (t, J=7.6 Гц, 2H), 1.68 (p, J=6.6 Гц, 2H), 1.15 (d, J=7.1 Гц, 3Н).
Промежуточное соединение 13: Метил 4-(5-(3-бромпропил)-6-метилбензо[b]тиофен-2-ил)-4-оксобутаноат
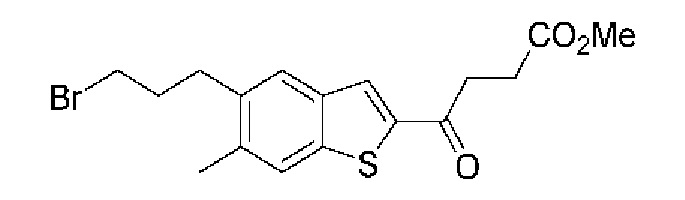
Стадия 1: метил 5-бром-6-метилбензо[b]тиофен-2-карбоксилат
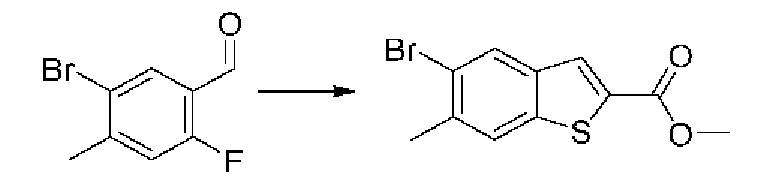
К смеси 5-бром-2-фтор-4-метилбензальдегида (5.0 г, 23 ммоль) в DMSO (120 мл) добавляли TEA (6.4 мл, 46 ммоль). Через 10 мин добавляли метил тиогликолят (6.7 мл, 69 ммоль) и смесь затем нагревали до 60°С в течение 18 ч. Через 18 ч смесь охлаждали до комнатной температуры и разбавляли EtOAc и водой. Органический слой отделяли, высушивали над MgSO4, фильтровали и концентрировали под пониженным давлением. Смесь разбавляли DCM. Смесь фильтровали, и остаток высушивали под вакуумом. К маточному раствору добавляли силикагель (50 г) и смесь концентрировали. Смесь затем очищали с помощью хроматографии на силикагеле с получением метил 5-бром-6-метилбензо[b]тиофен-2-карбоксилата. 1H ЯМР (500 МГц, DMSO-d6) δ 8.31 (s, 1H), 8.13 (s, 1H), 8.08 (s, 1H), 3.89 (s, 3Н), 2.48 (s, 3Н).
Стадия 2: 5-бром-6-метилбензо[b]тиофен-2-каубоноеая кислота
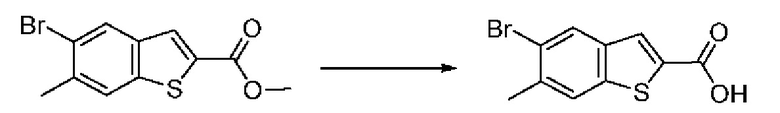
К смеси метил 5-бром-6-метилбензо[b]тиофен-2-карбоксилата (2.76 г, 9.68 ммоль) в THF (24 мл), воде (12 мл) и МеОН (12 мл) добавляли LiOH (1.16 г, 48.4 ммоль) и смесь перемешивали в течение 30 мин при комнатной температуре. Смесь затем подкисляли почти до нейтрального рН с помощью HCl (1.0 М в воде, 48 мл, 48 ммоль). Смесь затем разбавляли EtOAc и водой. Органический слой отделяли, высушивали над MgSO4, фильтровали и концентрировали с получением 5-бром-6-метилбензо[b]тиофен-2-карбоновой кислоты. Продукт применяли без очистки. ЖХ-МС (C10H8BrO2S) (ES, m/z): 271, 273 [М+Н]+. 1Н ЯМР (499 МГц, DMSO-d6) δ 13.56 (s, 1H), 8.29 (s, 1H), 8.05 (s, 1H), 8.04 (s, 1H), 2.47 (s, 3Н).
Стадия 3: 5-бром-6-метилбензо[b]тиофен
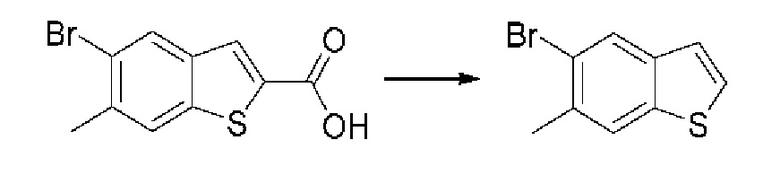
К 5-бром-6-метилбензо[b]тиофен-2-карбоновой кислоте (5.7 г, 21 ммоль) добавляли DMA (100 мл). Смесь затем разделяли поровну в 5 виал. DBU (1.6 мл) добавляли к каждой виале, и каждую виалу затем облучали в микроволновой печи до 200°С в течение 2 ч. После завершения пять виал объединяли и затем разбавляли EtOAc и насыщенным водным NaHCO3. Органический слой отделяли, высушивали над MgSO4, фильтровали и концентрировали под пониженным давлением. Остаток очищали с помощью хроматографии на силикагеле с получением 5-бром-6-метилбензо[b]тиофена. 1Н ЯМР (500 МГц, DMSO-d6) δ 8.15 (s, 1H), 8.00 (s, 1Н), 7.75 (d, J=5A Гц, 1Н), 7.39 (d, J=5.3 Гц, 1Н), 2.45 (s, 3Н).
Стадия 4: 4-(5-бром-6-метилбензо[b]тиофен-2-ил)-4-оксобутановая кислота
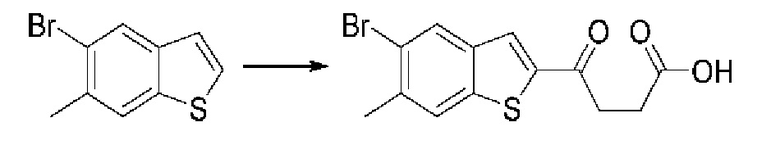
К смеси 5-бром-6-метилбензо[b]тиофена (2.0 г, 8.8 ммоль) в DCM (88 мл) при 0°С добавляли сукциновый ангидрид (1.1 г, 11 ммоль) и затем AlCl3 (2.3 г, 18 ммоль). Смесь нагревали до комнатной температуры и перемешивали в течение 18 ч. Смесь затем разбавляли EtOAc и HCl (1.0 н. в воде). Органический слой отделяли, высушивали над MgSO4 и фильтровали. К фильтрату добавляли силикагель (10 г) и смесь концентрировали под пониженным давлением. Смесь помещали под вакуум в течение 18 ч и затем очищали с помощью хроматографии на силикагеле (0→50% градиент EtOAc в гексанах) с получением 4-(5-бром-6-метилбензо[b]тиофен-2-ил)-4-оксобутановой кислоты. ЖХ-МС (C13H12BrO3S) (ES, m/z): 327, 329 [М+Н]+.
Стадия 5: метил 4-(5-бром-6-метилбензо[b]тиофеп-2-ил)-4-оксобутаноат
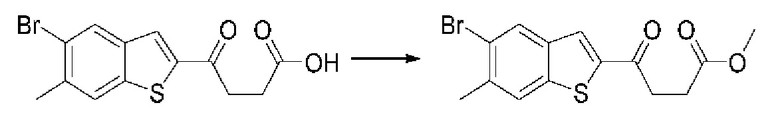
К смеси 4-(5-бром-6-метилбензо[b]тиофен-2-ил)-4-оксобутановой кислоты (0.99 г, 3.0 ммоль) в DMF (20 мл) добавляли K2CO3 (1.0 г, 7.6 ммоль). Через 10 мин добавляли CH3I (0.95 мл, 15 ммоль) и смесь оставляли перемешиваться до завершения реакции по данным ЖХ-МС. Смесь затем разбавляли EtOAc и водой. Органический слой отделяли, высушивали над MgSO4, фильтровали и концентрировали под пониженным давлением. Полученный в результате остаток очищали с помощью хроматографии на силикагеле с получением метил 4-(5-бром-6-метилбензо[b]тиофен-2-ил)-4-оксобутаноата. ЖХ-МС (C14H14BrO3S) (ES, m/z): 341, 343 [М+Н]+. 1Н ЯМР (500 МГц, DMSO-d6) δ 8.32 (s, 1H), 8.30 (s, 1H), 8.07 (s, 1H), 3.61 (s, 3Н), 3.37 (t, J=6.3 Гц, 2Н), 2.70 (t, J=6.3 Гц, 2Н), 2.48 (s, 3Н).
Стадия 6: метил 4-(5-(3-((трет-бутилдиметилсилил)окси)пропил)-6-метилбензо[b]тиофен-2-ил)-4-оксобутаноат
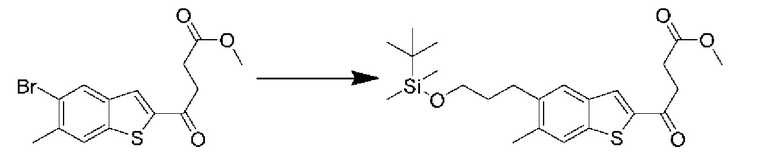
В колбу, содержащую метил 4-(5-бром-6-метилбензо[b]тиофен-2-ил)-4-оксобутаноат (0.40 г, 1.2 ммоль) и THF (5.9 мл), добавляли C-Phos Pd G3 (47 мг, 0.059 ммоль) и смесь три раза откачивали и снова заполняли N2. Добавляли (3-((трет-бутилдиметилсилил)окси)пропил)цинк (II) бромид (0.50 М в THF, 7.0 мл, 3.5 ммоль) и смесь оставляли перемешиваться при комнатной температуре в течение 2.5 ч. Затем реакцию останавливали смесью EtOAc и 10% водного цитрата натрия. Органический слой отделяли, промывали насыщенным водным NaCl, высушивали над MgSO4, фильтровали и концентрировали под пониженным давлением. Остаток очищали с помощью хроматографии на силикагеле с получением метил 4-(5-(3-((трет-бутилдиметилсилил)окси)пропил)-6-метилбензо[b]тиофен-2-ил)-4-оксобутаноата. ЖХ-МС (C23H35O4SSi) (ES, m/z): 435 [М+Н]+.
Стадия 7: метил 4-(5-(3-гидроксипропил)-6-метилбензо[b]тиофеп-2-ил)-4-оксобутапоат
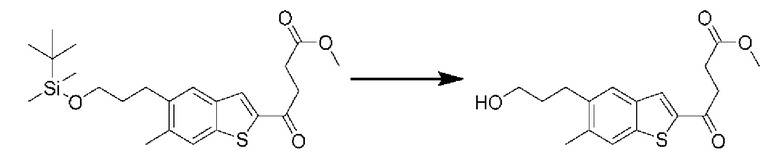
К смеси метил 4-(5-(3-((трет-бутилдиметилсилил)окси)пропил)-6-метил-бензо[b]тиофен-2-ил)-4-оксобутаноата (0.23 г, 0.54 ммоль) в THF (2.7 мл) добавляли TBAF (1.0 М в THF, 1.0 мл, 1.0 ммоль). Через 1.5 ч смесь разбавляли EtOAc и насыщенным водным NH4Cl. Органический слой отделяли, высушивали над MgSO4, фильтровали и концентрировали под пониженным давлением. Остаток очищали с помощью хроматографии на силикагеле (0→70% градиент EtOAc в гексанах) с получением метил 4-(5-(3-гидроксипропил)-6-метилбензо[b]тиофен-2-ил)-4-оксобутаноата. ЖХ-МС (C17H21O4S) (ES, m/z): 321 [М+Н]+. 1Н ЯМР (499 МГц, DMSO-d6) δ 8.30 (s, 1H), 7.81 (s, 1H), 7.78 (s, 1H), 4.55 (t, J=5.1 Гц, 1H), 3.61 (s, 3H), 3.51-3.46 (m, 2H), 3.38-3.34 (m, 2H), 2.77-2.66 (m, 4H), 2.41 (s, 3H), 1.79-1.67 (m, 2H).
Стадия 8: метил 4-(5-(3-бромпропил)-6-метилбензо[b]тиофен-2-ил)-4-оксобутаноат
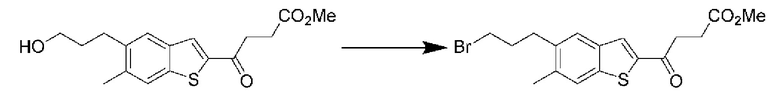
К перемешанной смеси метил 4-(5-(3-гидроксипропил)-6-метилбензо[b]тиофен-2-ил)-4-оксобутаноата (97 мг, 0.30 ммоль) и Ph3P (130 мг, 0.48 ммоль) в THF (1.5 мл) при 0°С добавляли NBS (81 мг, 0.45 ммоль) одной порцией. Через 30 мин реакцию останавливали насыщенным водным NH4Cl и EtOAc. Органический слой отделяли, высушивали над MgSO4, фильтровали и концентрировали под пониженным давлением. Остаток очищали с помощью хроматографии на силикагеле (0→25% градиент EtOAc в гексанах) с получением метил 4-(5-(3-бромпропил)-6-метилбензо[b]тиофен-2-ил)-4-оксобутаноата. ЖХ-МС (C17H20BrO3S) (ES, m/z): 383, 385 [М+Н]+.
Промежуточное соединение 14: трет-бутил 4-(5-(3-гидроксипропил)-6-метокситиено[3,2-b]пиридин-2-ил)-4-оксобутаноат
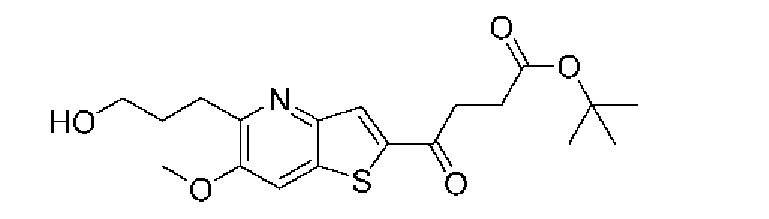
Стадия 1: трет-бутил 4-(5-хлор-6-метокситиено[3,2-b]пиридин-2-ил)-4-оксобутаноат
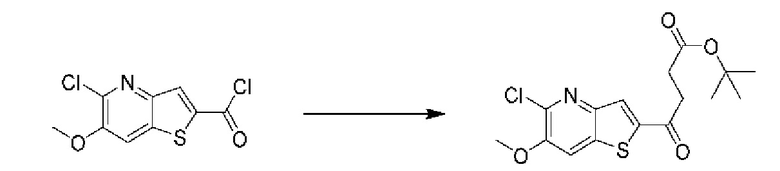
CuCl (0.721 г, 7.29 ммоль) добавляли в 250 мл круглодонную колбу с мешалкой. Колбу три раза откачивали и затем продували N2. В колбу добавляли THF (14.6 мл) и затем перемешивали и охлаждали до 0°С на бане с ледяной водой. Затем добавляли (3-(трет-бутокси)-3-оксопропил)цинк (II) (0.50 М THF, 30 мл, 15 ммоль) по каплям в течение 10 мин, перемешивая при 0°С. Полученную в результате смесь перемешивали при 0°С в течение 35 мин. Добавляли 5-хлор-6-метокси тиено[3,2-b]пиридин-2-карбонилхлорид (1.91 г, 7.29 ммоль) с последующим добавлением NMP (14.6 мл). Полученную в результате смесь перемешивали в течение 7 ч при 0°С. К реакционной смеси добавляли концентрированный NH4OH (4 мл) с быстрым перемешиванием при 0°С.К этой суспензии добавляли воду : МеОН (4:1 140 мл) вместе с ~20 г дигидрата трехосновного цитрата натрия. Смесь перемешивали в течение 20 мин. Полученную в результате суспензию фильтровали, и осадок на фильтре промывали водой. Осадок затем суспендировали в гексане и дважды фильтровали. Осадок вакуумировали с продуванием N2 в течение 72 ч с получением трет-бутил 4-(5-хлор-6-метокситиено[3,2-b]пиридин-2-ил)-4-оксобутаноата. ЖХ-МС (C16H19ClNO4S) (ES, m/z): 356 [М+Н]+. 1Н ЯМР (500 МГц, CDCl3) δ 8.03 (s, 1H), 7.61 (s, 1H), 4.03 (s, 3Н), 3.30 (t, J=6.5 Гц, 2Н), 2.87-2.59 (m, 2Н), 1.46 (s, 9Н).
Стадия 2: трет-бутил 4-(5-(3-((трет-бутилдиметилсилил)окси)пропил)-6-метокситиено[3,2-b]пиридин-2-ил)-4-оксобутаноат
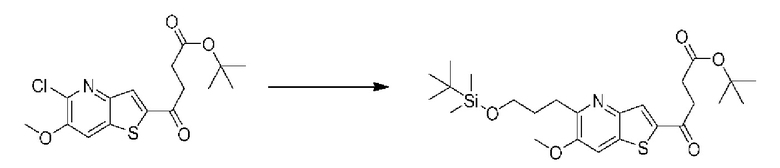
трет-Бутил 4-(5-хлор-6-метокситиено[3,2-b]пиридин-2-ил)-4-оксобутаноат (1.00 г, 2.81 ммоль) и CPhos Pd G3 (0.113 г, 0.141 ммоль) добавляли в 40 мл виалу с винтовой крышкой с септой. Виалу три раза откачивали и снова заполняли N2. В виалу добавляли THF (10.0 мл) в атмосфере N2 с перемешиванием. При перемешивании полученной суспензии при комнатной температуре добавляли по каплям (3-трет-бутилдиметилсилил)окси)пропил)цинк (II) бромид (0.50 М в THF, 11.2 мл, 5.60 ммоль). Полученную в результате смесь перемешивали в течение 3 ч при комнатной температуре. Смесь разделяли между EtOAc (75 мл) и 10% водным цитратом натрия (75 мл) и энергично перемешивали в течение 5 мин. Слои разделяли, и водный слой экстрагировали EtOAc (20 мл). Органические слои объединяли, промывали насыщенным водным NaCl (50 мл), высушивали над безводным Na2SO4, концентрировали под пониженным давлением с получением сырого остатка. Полученный в результате остаток очищали с помощью хроматографии на силикагеле (0→40% градиент EtOAc в гексанах) с получением трет-бутил 4-(5-(3-((трет-бутилдиметилсилил)окси)пропил)-6-метокситиено[3,2-b]пиридин-2-ил)-4-оксобутаноата. ЖХ-МС (C25H40NO5SSi) (ES, m/z): 494 [М+Н]+. 1Н ЯМР (500 МГц, CDCl3) δ 8.08 (s, 1Н), 7.48 (s, 1H), 3.94 (s, 3Н), 3.75 (t, J=6.5 Гц, 2Н), 3.30 (t, J=6.6 Гц, 2Н), 3.02-2.95 (m. 2Н), 2.73 (t, J=6.6 Гц, 2Н), 2.03-1.96 (т, 2Н), 1.46 (s, 9Н), 0.92 (s, 9Н), 0.07 (s, 6Н).
Стадия 3: трет-бутил 4-(5-(3-гидуоксипуопил)-6-метокситиено[3,2-b]пиридин-2-ил)-4-оксобутаноат
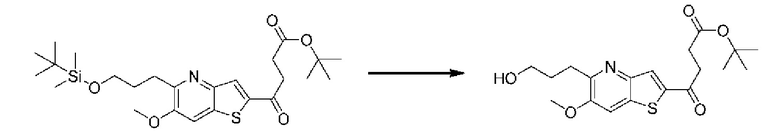
К смеси трет-бутил 4-(5-(3-((трет-бутилдиметилсилил)окси)пропил)-6-метокситиено[3,2-b]пиридин-2-ил)-4-оксобутаноата (0.735 г, 1.49 ммоль) в МеОН (3.0 мл) добавляли воду (3.0 мл) и затем НОАс (3.0 мл). Полученную в результате смесь перемешивали при комнатной температуре в течение 18 ч. Смесь затем разделяли между EtOAc (50 мл), водой (25 мл) и насыщенным водным NaCl (25 мл). Слои разделяли, и водный слой экстрагировали EtOAc (50 мл). Органические слои объединяли, промывали насыщенным водным NaCl (50 мл), высушивали над безводным Na2SO4, фильтровали и концентрировали под пониженным давлением. Полученный в результате остаток очищали с помощью хроматографии на силикагеле (0→100% градиент EtOAc в гексане, затем изократическое элюирование при 100% EtOAc) с получением трет-бутил 4-(5-(3-гидроксипропил)-6-метокситиено[3,2-b]пиридин-2-ил)-4-оксобутаноата. ЖХ-МС (C19H26NO5S) (ES, m/z): 380 [М+Н]+. 1Н ЯМР (500 МГц, CDCl3) δ 8.06 (s, 1Н), 7.51 (s, 1Н), 3.95 (s, 3Н), 3.73 (t, J=5.8 Гц, 2Н), 3.28 (t, J=6.6 Гц, 2Н), 3.10 (t, J=6.9 Гц, 2Н), 2.72 (t, J=6.6 Гц, 2Н), 2.06 (р, J=6.5 Гц, 2Н), 1.45 (s, 9Н).
Промежуточное соединение 15: трет-Бутил 4-(5-бром-6-метоксибензо[b]тиазол-2-ил)-4-оксобутаноат
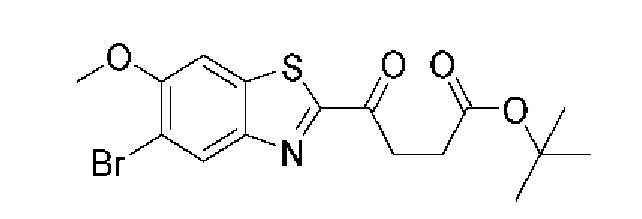
Стадия 1: метил 2-((3-6ром-4-метоксифенил)амино)-2-оксоацетат
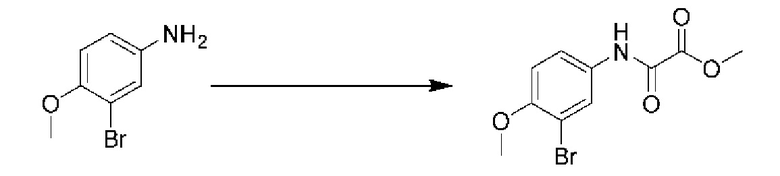
В 5 л 4-хгорлую круглодонную колбу, которую продували и в которой поддерживали инертную атмосферу N2, помещали смесь 3-бром-4-метоксианилина (232 г, 1.15 моль) в DCM (3.0 л), DIPEA (171 г, 1.32 моль) и метил 2-хлор-2-оксоацетата (148 г, 1.21 моль). Полученную в результате смесь перемешивали в течение 1 ч при комнатной температуре. Реакцию затем останавливали добавлением смеси воды/льда (2 л). Полученную в результате смесь экстрагировали DCM (3×1 л). Органические слои объединяли и концентрировали под пониженным давлением с получением метил 2-((3-бром-4-метоксифенил)амино)-2-оксоацетата, который применяли без очистки или определения характеристик.
Стадия 2: О-метил 2-((3-бром-4-метоксифенил)амино)-2-оксоэтантиоат
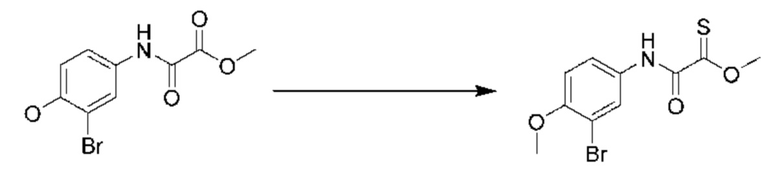
В 3 л 4-хгорлую круглодонную колбу, которую продували и в которой поддерживали инертную атмосферу N2, помещали смесь метил 2-((3-бром-4-метоксифенил)амино)-2-оксоацетата (111 г, 386 ммоль) в толуоле (1.5 л) и реагента Лавессона (86.4 г, 214 ммоль). Полученную в результате смесь нагревали до 85°С в течение 16 ч. Реакционную смесь затем охлаждали до комнатной температуры. Полученные в результате вещества удаляли путем фильтрования и промывали DCM (3×500 мл). Фильтрат концентрировали под пониженным давлением, и остаток очищали с помощью хроматографии на силикагеле (EtOAc/РЕ (1:20)) с получением О-метил 2-((3-бром-4-метоксифенил)амино)-2-оксоэтантиоата, который применяли без определения характеристик.
Стадия 3: 5-бром-6-метоксибензо[d]тиазол-2-карбоновая кислота, калиевая соль и 7-бром-6-метоксибензо[d]тиазол-2-карбоновая кислота, калиевая соль (смесь изомеров 2:1)
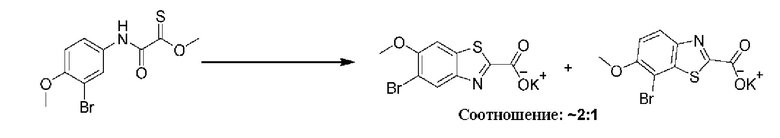
В 2 л 4-хгорлую круглодонную колбу, которую продували и в которой поддерживали инертную атмосферу N2, помещали метил О-метил 2-((3-бром-4-метоксифенил)амино)-2-оксоэтантиоат (84.5 г, 278 ммоль). В реакционную смесь добавляли смесь KOH (50 г, 90 ммоль) в Н2О (500 мл) в течение 10 мин. Затем к реакционной смеси добавляли смесь гидрата ферроцианида калия (III) (242 г, 735 ммоль) в Н2О (2 л) в течение 10 мин. рН полученной в результате смеси доводили до 2 с помощью водной HCl (2.0 М). Затем добавляли воду (500 мл). Полученную в результате смесь оставляли перемешиваться в течение 1 ч при комнатной температуре. Полученное в результате вещество затем собирали путем фильтрования и промывали DCM (1 л). Остаток суспендировали в водном KOH (2.0 М, 500 мл, 1 моль) в течение 0.5 ч. Полученное в результате вещество затем собирали путем фильтрования и промывали Н2О (2×500 мл) с получением 2:1 смеси 5-бром-6-метоксибензо[d]тиазол-2-карбоновой кислоты, калиевой соли и 7-бром-6-метоксибензо[d]тиазол-2-карбоновой кислоты, калиевой соли. Характеристики для 5-бром-6-метоксибензо[d]тиазол-2-карбоновой кислоты, калиевой соли (главный изомер): 1Н ЯМР (400 МГц, DMSO-d6, ppm) δ 8.13 (s, 1H), 7.70 (s, 1H), 3.80 (s, 3Н). Характеристики для 7-бром-6-метоксибензо[d]тиазол-2-карбоновой кислоты, калиевой соли (минорный изомер): 1Н ЯМР (400 МГц, DMSO-d6, ррь) 7.90 (d, J=8.9 Гц, 1H), 7.26 (d, J=8.9 Гц, 1H), 3.91 (s, 3Н).
Стадия 4: 5-бром-6-метоксибензо[d]тиазол-2-карбоновая кислота и 7-бром-6-метоксибензо[d]тиазол-2-карбоновая кислота (смесь изомеров 2:1)
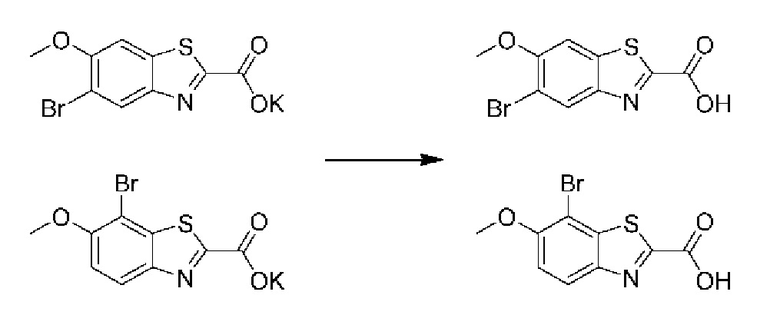
В 1 л колбу добавляли 5-бром-6-метоксибензо[d]тиазол-2-карбоновую кислоту, калиевую соль и 7-бром-6-метоксибензо[d]тиазол-2-карбоновую кислоту, калиевую соль (смесь изомеров 2:1) (22.4 г, 49.4 ммоль), воду (300 мл), ACN (180 мл), МеОН (120 мл) и TFA (11.4 мл, 148 ммоль). Смесь энергично перемешивали в течение 15 мин при комнатной температуре. Полученное в результате вещество собирали путем фильтрования и промывали водой (2×20 мл), МеОН (2×5 мл) и Et2O (2×10 мл) с получением смеси 2:1 5-бром-6-метоксибензо[d]тиазол-2-карбоновой кислоты и 7-бром-6-метоксибензо[d]тиазол-2-карбоновой кислоты. Характеристики для 5-бром-6-метоксибензо[d]тиазол-2-карбоновой кислоты (главный изомер): 1Н ЯМР (500 МГц, DMSO-d6) δ 8.45 (s, 1H), 7.97 (s, 1H), 3.96 (s, 3Н). Характеристики для 7-бром-6-метоксибензо[d]тиазол-2-карбоновой кислоты (минорный изомер): 1H ЯМР (500 МГц, DMSO-d6) δ 8.21 (d, J=9.0 Гц, 1H), 7.51 (d, J=9.0 Гц, 1H), 4.00 (s, 3Н).
Стадия 5: метил 5-бром-6-метоксибензо[d]тиазол-2-каубоксилат
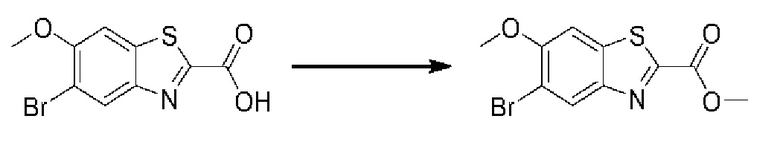
В 250 мл колбу добавляли 5-бром-6-метоксибензо[d]тиазол-2-карбоновую кислоту и 7-бром-6-метоксибензо[d]тиазол-2-карбоновую кислоту (смесь изомеров 2:1) (17.2 г, 41.8 ммоль) и МеОН (150 мл). Смесь энергично перемешивали и охлаждали до 0°С. К перемешиваемой смеси добавляли SOCl2 (6.1 мл, 84 ммоль) по каплям в течение 10 мин. Смесь затем нагревали до кипения с обратным холодильником в течение 18 ч. После охлаждения до комнатной температуры полученное в результате вещество собирали путем фильтрования и промывали МеОН (2×20 мл). Вещество очищали с помощью хроматографии на силикагеле (0→30% градиент EtOAc в DCM) с получением метил 5-бром-6-метоксибензо[d]тиазол-2-карбоксилата в виде единственного изомера. ЖХ-МС (C10H9BrNO3S) (ES, m/z): 302, 304 (М+Н)+. 1Н ЯМР (500 МГц, CDCl3) δ 8.42 (s, 1Н), 7.40 (s, 1H), 4.09 (s, 3Н), 4.02 (s, 3Н).
Стадия б: 5-бром-6-метоксибензо[d]тиазол-2-каубоновая кислота
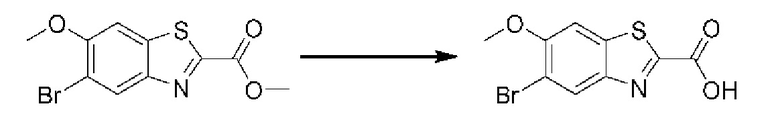
В 500 мл круглодонную колбу добавляли метил 5-бром-6-метоксибензо[d]тиазол-2-карбоксилат (7.3 г, 24 ммоль) и МеОН (150 мл). К энергично перемешиваемой смеси добавляли водный NaOH (2.0 М, 37 мл, 74 ммоль). Смесь нагревали до кипения с обратным холодильником в течение 30 мин. После охлаждения до комнатной температуры добавляли по каплям водный HCl (2.0 М, 37 мл, 74 ммоль). Смесь энергично перемешивали при комнатной температуре в течение 18 ч. Полученное в результате вещество собирали путем фильтрования и промывали водой (2×50 мл) и МеОН (2×20 мл) с получением 5-бром-6-метоксибензо[d]тиазол-2-карбоновой кислоты. ЖХ-МС (C9H7BrNO3S) (ES, m/z): 288, 290 (М+Н). 1H ЯМР (500 МГц, DMSO-d6) δ 8.45 (s, 1H), 7.97 (s, 1H), 3.96 (s, 3Н).
Стадия 7: 5-бром-6-метоксибензо[d]тиазол-2-карбонилхлорид
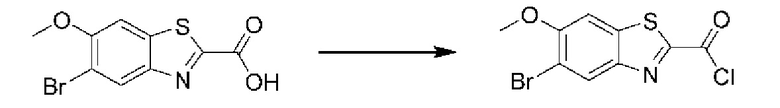
В 100 мл круглодонную колбу добавляли 5-бром-6-метоксибензо[с1]тиазол-2-карбоновую кислоту (1.46 г, 5.07 ммоль), DCM (25 мл) и DMF (0.080 мл, 1.0 ммоль). К смеси добавляли (СОС1)г (5.32 мл, 10.6 ммоль) по каплям в течение 1 мин, и смесь энергично перемешивали при комнатной температуре в течение 15 мин. Смесь затем фильтровали через целит. Фильтрат концентрировали под пониженным давлением с получением 5-бром-6-метоксибензо[d]тиазол-2-карбонилхлорида, который применяли без дополнительной очистки или определения характеристик.
Стадия 8: трет-бутил 4-(5-бром-6-метоксибензо[с1]тиазол-2-ил)-4-оксобутаноат
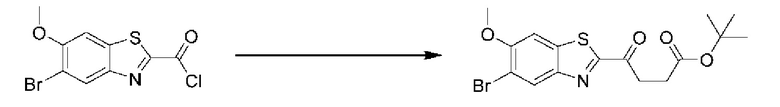
В 250 мл круглодонную колбу добавляли CuCl (0.45 г, 4.6 ммоль). Колбу три раза откачивали и заполняли N2. Добавляли THF (10 мл) и смесь перемешивали и охлаждали до 0°С. К смеси добавляли (3-(трет-бутокси)-3-оксопропил)цинк (II) бромид (0.50 М в THF, 18 мл, 9.0 ммоль). Через 10 мин добавляли по каплям смесь 5-бром-6-метокси-бензо[d]тиазол-2-карбонилхлорида (1.4 г, 4.6 ммоль) в NMP (30 мл) в течение 5 мин. Через 5 мин смесь оставляли нагреваться до комнатной температуры и затем перемешивали в течение 1 ч. К смеси добавляли воду (30 мл) и концентрированный водный NH4OH (15 мл). Смесь экстрагировали EtOAc (125 мл) и органический слой промывали водой (2×75 мл). Органический слой высушивали над Na2SO4, фильтровали и концентрировали под пониженным давлением. Остаток очищали с помощью хроматографии на силикагеле (0→75% градиент EtOAc в гексанах) с получением трет-бутил 4-(5-бром-6-метоксибензо[d]тиазол-2-ил)-4-оксобутаноата. ЖХ-МС (C16H18BrNO4S + Na) (ES, m/z): 422, 424 [M+Na]+. 1H ЯМР (500 МГц, CDCl3) δ 8.38 (s, 1H), 7.40 (s, 1H), 4.02 (s, 3H), 3.51 (t, J=6.6 Гц, 2H), 2.75 (t, J=6.6 Гц, 2H), 1.46 (s, 9H).
Промежуточное соединение 16: трет-бутил 4-(5-(3-6ромпропил)-6-метоксибензо[d]тиазол-2-ил)-4-оксобутаноат
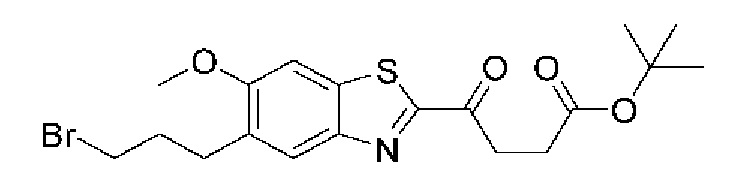
Стадия 1: трет-бутил 4-(5-(3-((трет-бутилдиметилсилил)окси)пропил)-6-метоксибензо[d]тиазол-2-ил)-4-оксо6утаиоат
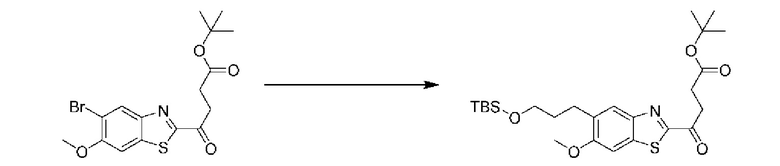
В 4 мл виалу добавляли трет-бутил 4-(5-бром-6-метоксибензо[d]тиазол-2-ил)-4-оксобутаноат (233 мг, 0.581 ммоль) и C-Phos Pd G3 (14 мг, 0.018 ммоль). Виалу три раза откачивали и заполняли N2. В виалу добавляли THF (0.60 мл) с последующим добавлением (3-((трет-бутилдиметилсилил)окси)пропил)цинка(II) бромида (0.50 М в THF, 2.90 мл, 1.45 ммоль). Смесь перемешивали при комнатной температуре в течение 20 мин. К смеси добавляли дополнительный (3-((трет-бутилдиметилсилил)окси)-пропил)цинк (II) бромид (0.50 М в THF, 1.4 мл, 0.73 ммоль). Через 30 мин смесь разбавляли EtOAc (30 мл) и промывали 10% водным трехосновным цитратом натрия (30 мл). Органический слой отделяли, высушивали над Na2SO4, фильтровали и концентрировали под пониженным давлением. Полученный в результате остаток очищали с помощью хроматографии на силикагеле (0→40% градиент EtOAc в гексанах) с получением трет-бутил 4-(5-(3-((трет-бутилдиметилсилил)окси)пропил)-6-метоксибензо[d]тиазол-2-ил)-4-оксобутаноата. ЖХ-МС (C25H40NO5SSi) (ES, m/z): 494 [М+Н]+. 1H ЯМР (500 МГц, CDCl3) δ 7.93 (s, 1Н), 7.31 (s, 1H), 3.94 (s, 3Н), 3.70 (t, J=6.3 Гц, 2Н), 3.53 (t, J=6.6 Гц, 2Н), 2.86-2.78 (m, 2Н), 2.74 (t, J=6.6 Гц, 2Н), 1.94-1.84 (т, 2Н), 1.45 (s, 9Н), 0.93 (s, 9Н).
Стадия 2: трет-бутил 4-(5-(3-гидроксипропил)-6-метоксибензо[d]тиазол-2-ил)-4-оксобутапоат
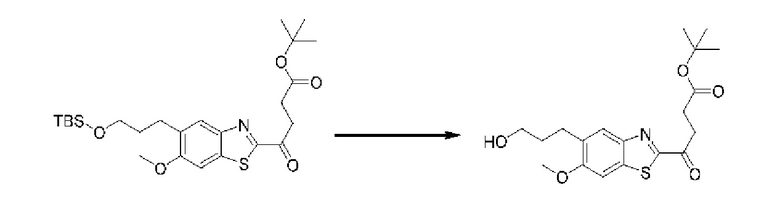
В 100 мл колбу добавляли трет-бутил 4-(5-(3-((трет-бутилдиметилсилил)окси)пропил)-6-метоксибензо[d]тиазол-2-ил)-4-оксобутаноат (229 мг, 0.464 ммоль), МеОН (5.0 мл), воду (5.0 мл) и АсОН (5.0 мл). Смесь оставляли перемешиваться при комнатной температуре в течение 4 ч. Смесь затем разбавляли EtOAc (50 мл) и промывали водой (3×50 мл). Органический слой отделяли, высушивали над Na2SO4, фильтровали и концентрировали под пониженным давлением с получением трет-бутил 4-(5-(3-гидроксипропил)-6-метоксибензо[d]тиазол-2-ил)-4-оксобутаноата. ЖХ-МС (C19H26NO5S) (ES, m/z): 380 [М+Н]+. 1H ЯМР (500 МГц, CDCl3) δ 7.95 (s, 1H), 7.34 (s, 1H), 3.96 (s, 3Н), 3.70 (t, J=6.3 Гц, 2Н), 3.52 (t, J=6.1 Гц, 2Н), 2.87 (t, J=7.5 Гц, 2Н), 2.75 (t, J=6.6 Гц, 2Н), 2.00-1.90 (m, 2Н), 1.46 (s, 9Н).
Стадия 3: трет-бутил 4-(5-(3-бромпропил)-6-метоксибензо[d]тиазол-2-ил)-4-оксобутаноат
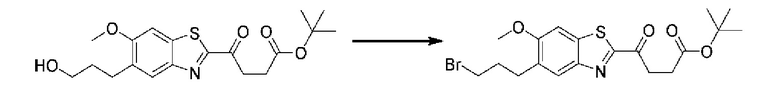
В 4 мл виалу добавляли CBr4 (72 мг, 0.22 ммоль), трифенилфосфин (62 мг, 0.24 ммоль) и трет-бутил 4-(5-(3-гидроксипропил)-6-метоксибензо[d]тиазол-2-ил)-4-оксобутаноат (69 мг, 0.18 ммоль). Виалу охлаждали до 0°С, и добавляли DCM (1.0 мл). Смесь оставляли нагреваться до комнатной температуры в течение 90 мин. Смесь затем сразу очищали с помощью хроматографии на силикагеле (0→30% градиент EtOAc в гексанах) с получением трет-бутил 4-(5-(3-бромпропил)-6-метоксибензо[d]тиазол-2-ил)-4-оксобутаноата. ЖХ-МС (C19H25BrNO4S) (ES, m/z): 442, 444 [М+Н]+. 1Н ЯМР (499 МГц, CDCl3) δ 7.96 (s, 1H), 7.33 (s, 1H), 3.52 (t, J=6.6 Гц, 2Н), 3.95 (s, 3Н), 3.45 (t, J=6.6 Гц, 2Н), 2.93 (t, J=7.2 Гц, 2Н), 2.75 (t, J=6.6 Гц, 2Н), 2.30-2.19 (m, 2Н), 1.46 (s, 9Н).
Промежуточное соединение 17: трет-бутил 4-(5-гидрокси-6-метоксибензо[b]тиофен-2-ил)-4-оксобутаноат
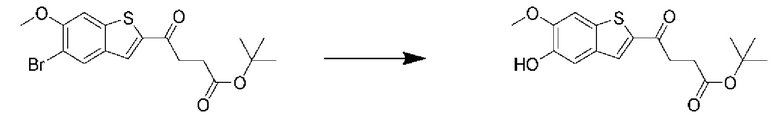
К смеси RockPhos Pd G3 (0.105 г, 0.125 ммоль), бензальдоксима (3.03 г, 25.0 ммоль), трет-бутил 4-(5-бром-6-метоксибензо[b]тиофен-2-ил)-4-оксобутаноата (5.0 г, 13 ммоль) и Cs2CO3 (12.2 г, 37.6 ммоль) добавляли DMF (40 мл). Реакцию нагревали до 80°С в течение 18 ч. Реакционную смесь затем оставляли охлаждаться до комнатной температуры и выливали в колбу, содержащую водную HCl (0.5 М, 100 мл). Полученную в результате смесь экстрагировали DCM. Органический слой затем высушивали над Na2SO4, фильтровали и концентрировали под пониженным давлением. Полученный в результате остаток очищали с помощью колоночной хроматографии на силикагеле (0→50% градиент EtOAc в гексанах) с получением трет-бутил 4-(5-гидрокси-6-метоксибензо[b]тиофен-2-ил)-4-оксобутаноата. ЖХ-МС (C17H20O5SNa) (ES, m/z): 359 [M+Na]+. 1H ЯМР (600 МГц, DMSO-d6) δ 9.35 (s, 1H), 8.12 (s, 1Н), 7.49 (s, 1H), 7.27 (s, 1H), 3.83 (s, 3H), 3.18 (t, J=6.2 Гц, 2H), 2.52 (t, J=6.2 Гц, 2H), 1.33 (s, 9H).
Промежуточное соединение 18: Этил 4-(5-(3-гидроксипропил)-6-метоксибензо[b]тиофен-2-ил)-4-оксобутаноат
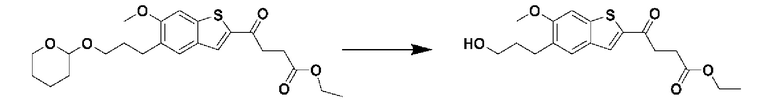
К смеси этил 4-(6-метокси-5-(3-((тетрагидро-2Н-пиран-2-ил)окси)пропил)бензо[b]тиофен-2-ил)-4-оксобутаноата (5.0 г, 12 ммоль) и EtOH (100 мл) добавляли pTsOH (4.4 г, 23 ммоль). Реакцию оставляли перемешиваться при комнатной температуре в течение 1 ч. Реакцию затем останавливали водой и разбавляли DCM. Органический слой отделяли и промывали водным насыщенным NaHCO3. Органический слой высушивали над Na2SO4, фильтровали и концентрировали под пониженным давлением с получением этил 4-(5-(3-гидроксипропил)-6-метоксибензо[b]тиофен-2-ил)-4-оксобутаноата. ЖХ-МС (C18H23O5S) (ES, m/z): 351 [М+Н]+. 1Н ЯМР (600 МГц, DMSO-d6) δ 8.21 (s, 1H), 7.70 (s, 1H), 7.55 (s, 1H), 4.44 (t, J=5.0 Гц, 1H), 4.02 (q, J=7.0 Гц, 2H), 3.85 (s, 3H), 3.40 (q, J=6.0 Гц, 2H), 3.27 (d, J=6.4 Гц, 2H), 2.63 (q, J=7.0, 5.9 Гц, 4H), 1.68 (p, J=6.6 Гц, 2H), 1.14 (t, J=7.1 Гц, 3Н).
Промежуточное соединение 19: метил (2S)-4-[5-(2-аминоэтил)-6-метокси-1-бензотиофен-2-ил]-2-метил-4-оксобутаноат
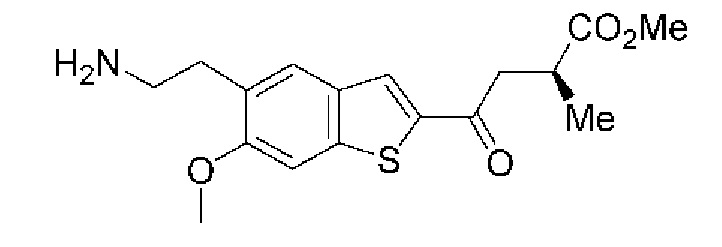
Стадия 1: метил (2S)-4-(5-{2-[(трет-бутоксикарбонил)амино]этил}-6-метокси-1-бензотиофен-2-ил)-2-метил-4-оксобутаноат
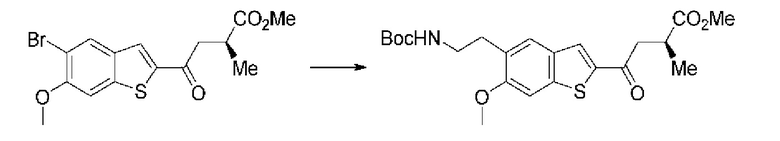
К перемешиваемой смеси метил (2S)-4-(5-бром-6-метокси-1-бензотиофен-2-ил)-2-метил-4-оксобутаноата (124 мг, 0.334 ммоль), трис(триметилсилил)силана (103 мкл, 0.334 ммоль) и безводного Na2CO3 (71 мг, 0.67 ммоль) в дегазированном DME (1.7 мл) в атмосфере N2, добавляли смесь Ir(2-(2,4-дифторфенил)-5-(трифторметил)пиридин)2 (4,4'-ди-трет-бутил-2,2'-бипиридин)PF6 (3.8 мг, 3.3 мкмоль) в дегазированном DME (1.2 мл). Добавляли суспензию комплекса никель (II) хлорид этиленгликоль диметиловый эфир (0.37 мг, 1.7 мкмоль) и 4,4'-ди-трет-бутил-2,2'-бипиридин (0.45 мг, 1.7 мкмоль) в дегазированном DME (445 мкл) и полученную в результате смесь перемешивали в атмосфере N2 в течение 15 мин при комнатной температуре. Добавляли трет-бутил N-(2-бромэтил)карбамат (150 мг, 0.67 ммоль) одной порцией в атмосфере N2, и реакционную смесь перемешивали и облучали двумя 34 W синими светодиодными лампами (на расстоянии 7 см с каждой стороны) в течение 18 ч при комнатной температуре. Смесь затем сразу очищали с помощью колоночной флэш-хроматографии на силикагеле (EtOAc в гексанах) с получением метил (2S)-4-(5-{2-[(трет-бутоксикарбонил)амино]этил}-6-метокси-1-бензотиофен-2-ил)-2-метил-4-оксобутаноата. ЖХ-МС (C22H30NO6S) (ES, m/z): 436 [М+Н]+. 1Н ЯМР (500 МГц, DMSO-d6): δ 8.25 (s, 1H), 7.70 (s, 1H), 7.60 (s, 1H), 6.86 (br, 1H), 3.88 (s, 3Н), 3.59 (s, 3Н), 3.43 (dd, J=17.5, 8.6 Гц, 1H), 3.19 (dd, J=17.5, 5.0 Гц, 1H), 3.15 (t, J=7.0 Гц, 2H), 3.01-2.93 (m, 1H), 2.77 (t, J=7.0 Гц, 2H), 1.34 (s, 9H), 1.19 (d, J=7.1 Гц, 3Н).
Стадия 2: метил (2S)-4-[5-(2-аминоэтил)-6-метокси-1-бензотиофен-2-ил]-2-метил-4-оксобутаноат
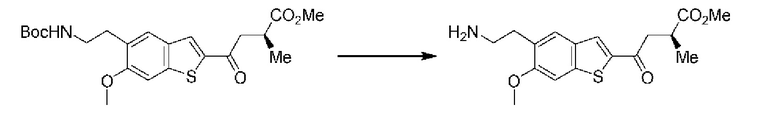
К перемешиваемому раствору метил (2S)-4-(5-{2-[(трет-бутоксикарбонил)амино]этил}-6-метокси-1-бензотиофен-2-ил)-2-метил-4-оксобутаноата (54 мг, 0.12 ммоль) в CH2Cl2 (2.8 мл) добавляли TFA (476 мкл, 6.18 ммоль) одной порцией при комнатной температуре, и реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Смесь концентрировали, и остаток растворяли в CH3CN и воде и сушили в вакууме на протяжении ночи с получением метил (2S)-4-[5-(2-аминоэтил)-6-метокси-1-бензотиофен-2-ил]-2-метил-4-оксобутаноата. ЖХ-МС (C17H22NO4S) (ES, m/z): 336 [М+Н]+. 1H ЯМР (500 МГц, DMSO-d6): δ 8.29 (s, 1H), 7.80 (br, 2Н), 7.79 (s, 1H), 7.68 (s, 1H), 3.91 (s, 3H), 3.60 (s, 3H), 3.45-3.39 (m, 1H), 3.20 (dd, J=17.5, 5.0 Гц, 1H), 3.10-3.01 (m, 2H), 3.01-2.93 (m, 1H), 2.95 (t, 7=7.0 Гц, 2H), 1.20 (d, J=7.1 Гц, 3Н).
Промежуточное соединение 20: Этил 4-(5-(3-бромпропил)-6-метоксибензо[b]тиофен-2-ил)-4-оксобутаноат
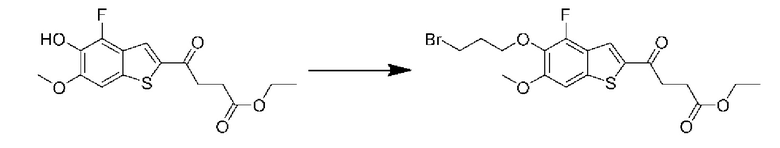
К смеси этил 4-(4-фтор-5-гидрокси-6-метоксибензо[b]тиофен-2-ил)-4-оксобутаноата (0.065 г, 0.2 ммоль), Cs2CO3 (0.326 г, 1.00 ммоль) и ACN (2 мл) добавляли 1,3-дибромпропан (1.0 мл, 9.9 ммоль). Смесь нагревали до 65°С в течение 2 ч. После охлаждения до комнатной температуры смесь фильтровали, и отфильтрованное вещество промывали THF. Фильтрат разбавляли гексаном, и смесь затем концентрировали под пониженным давлением с получением этил 4-(6-метокси-5-(3-((тетрагидро-2Н-пиран-2-ил)окси)пропил)бензо[b]тиофен-2-ил)-4-оксобутаноата. ЖХ-МС (C18H21BrFO5S) (ES, m/z): 447, 449 [М+Н]+.
Промежуточные соединения с 21 по 23 и с 62 по 86, как показано в Таблице 1 ниже, или могут быть получены в соответствии со способами, аналогичными описанным в Промежуточном соединении 20 выше, с применением соответствующих исходных материалов, описанных в способах получения, или получены из коммерческих источников.
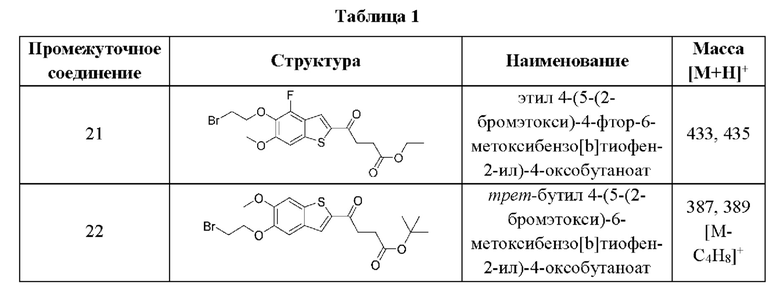
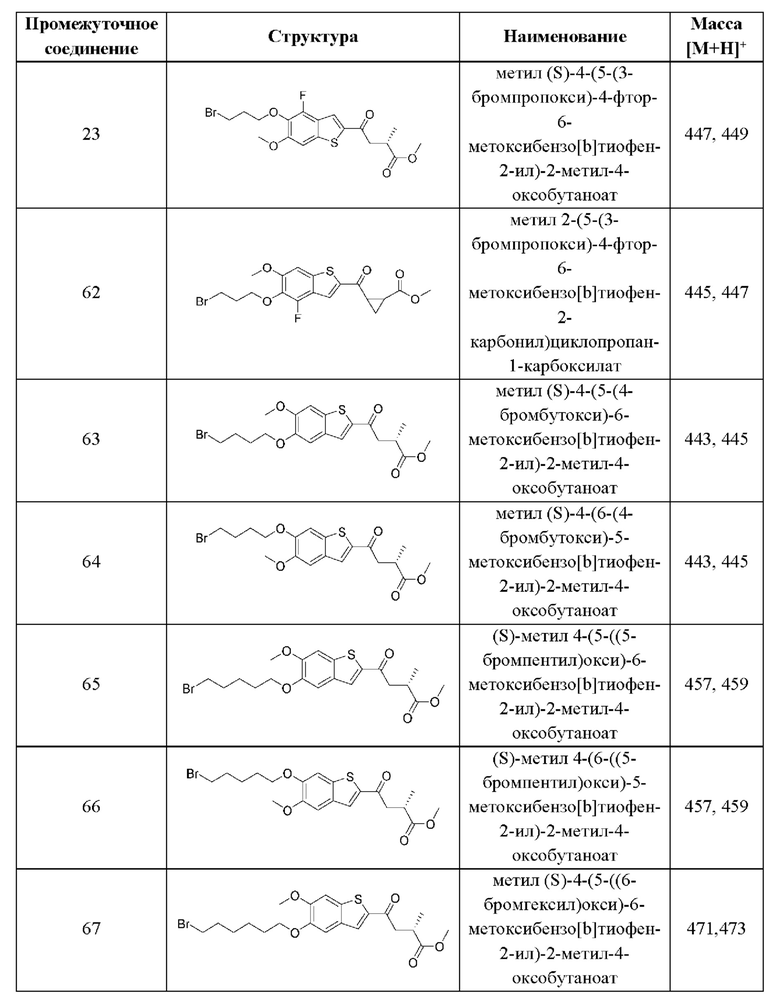
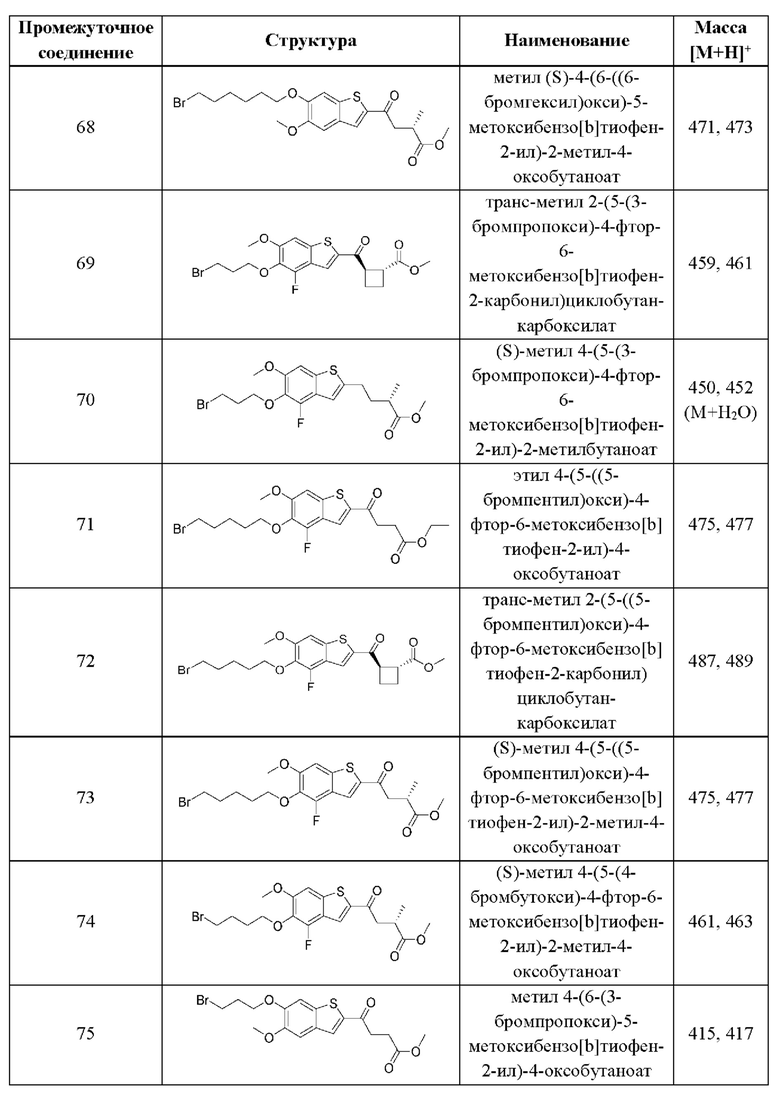
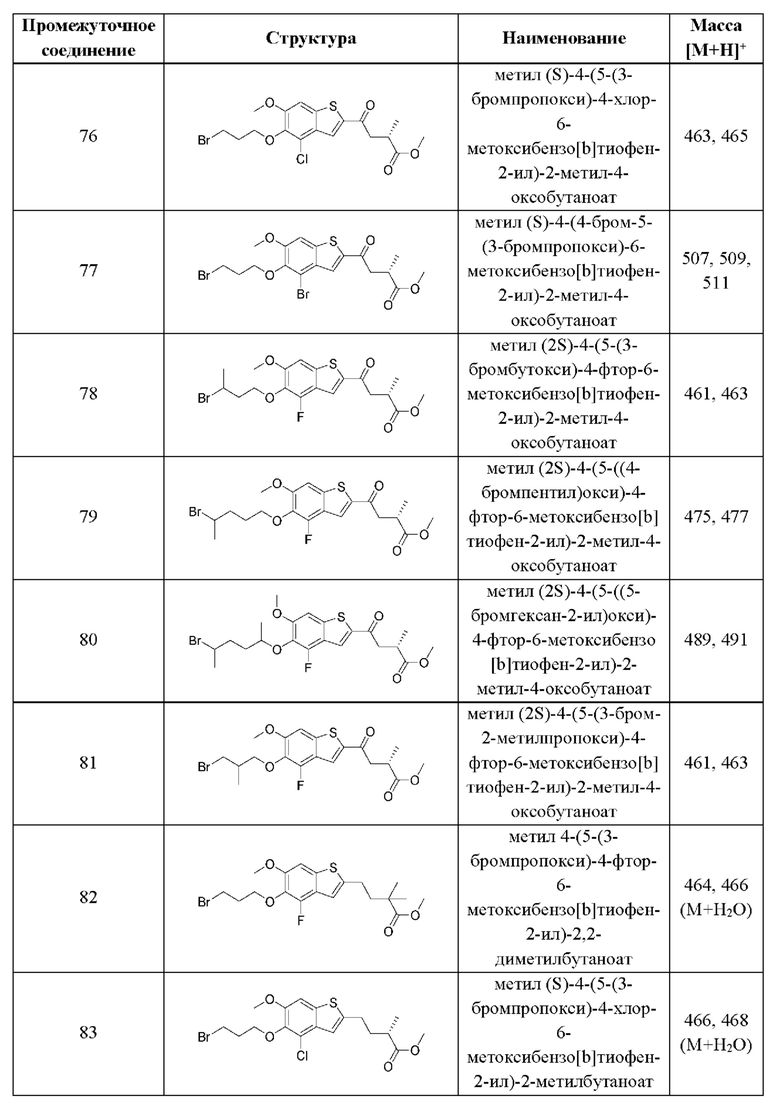
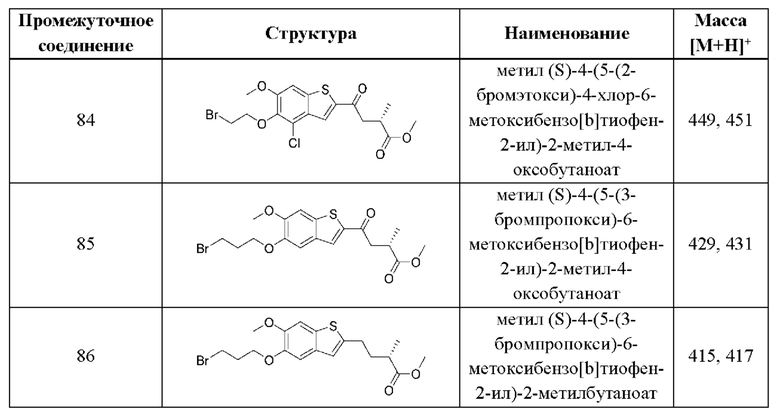
Промежуточное соединение 24: (S)-метил 4-(4-фтор-5-гидрокси-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноат
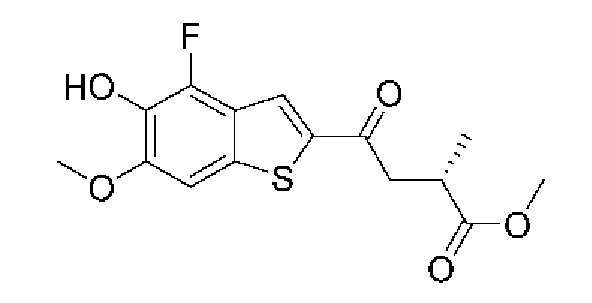
Стадия 1: (S)-метил 4-(4-фтор-5,6-диметоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноат
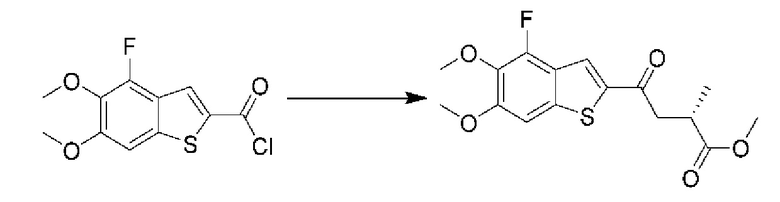
К смеси 4-фтор-5,6-диметоксибензо[b]тиофен-2-карбонилхлорида (4.3 г, 16 ммоль), C-Phos Pd G4 (0.26 г, 0.31 ммоль) и THF (25 мл) добавляли (R)-(3-метокси-2-метил-3-оксопропил)цинк (II) бромид (0.50 М в THF, 25.0 мл, 12.5 ммоль). Реакцию оставляли перемешиваться в течение 2 ч при комнатной температуре. Реакцию затем останавливали водным насыщенным NH4Cl и разбавляли DCM. Органический слой отделяли, высушивали над Na2SO4, фильтровали и концентрировали под пониженным давлением. Полученный в результате остаток очищали с помощью колоночной хроматографии на силикагеле (0→50% градиент EtOAc в гексанах) с получением (S)-метил 4-(4-фтор-5,6-диметоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата. ЖХ-МС (C16H18FO5S) (ES, m/z): 341 [M+H]+. 1H ЯМР (600 МГц, DMSO-d6) δ 8.33 (s, 1H), 7.58 (s, 1H), 3.92 (s, 3H), 3.86 (s, 3H), 3.60 (s, 3H), 3.50 (dd, J=17.7, 8.7 Гц, 1H), 3.25 (dd, J=17.7, 5.0 Гц, 1H), 3.00-2.92 (m, 1H), 1.20 (d, J=7.3 Гц, 3Н).
Стадия 2: (S)-метил 4-(4-фтор-5-гидрокси-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноат
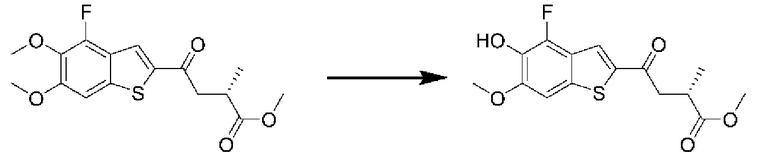
К смеси (S)-метил 4-(4-фтор-5,6-диметоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата (4.0 г, 12 ммоль) и DCM (50 мл) добавляли AlCl3 (6.27 г, 47.0 ммоль). Реакционную смесь оставляли перемешиваться при комнатной температуре в течение 18 ч. Затем к смеси медленно добавляли воду (50 мл) с энергичным перемешиванием с последующим добавлением водной HCl (1 н., 50 мл). Смесь затем выливали в делительную воронку и добавляли 20% IPA/DCM. Органический слой отделяли, высушивали над Na2SO4, фильтровали, и фильтрат концентрировали под пониженным давлением. Полученный в результате остаток очищали с помощью колоночной хроматографии на силикагеле (100% DCM) с получением (S)-метил 4-(4-фтор-5-гидрокси-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата. ЖХ-МС (C15H16FO5S) (ES, m/z): 327 [М+Н]+. 1H ЯМР (600 МГц, DMSO-d6) δ 9.53 (s, 1H), 8.25 (s, 1H), 7.47 (s, 1H), 3.92 (s, 3H), 3.60 (s, 3H), 3.48 (dd, J=17.7, 8.7 Гц, 1H), 3.24 (dd, J=17.7, 5.0 Гц, 1H), 3.01-2.89 (m, 1H), 1.19 (d, J=7.2 Гц, 3H).
Промежуточное соединение 25: метил (R)-4-(5-гидрокси-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноат
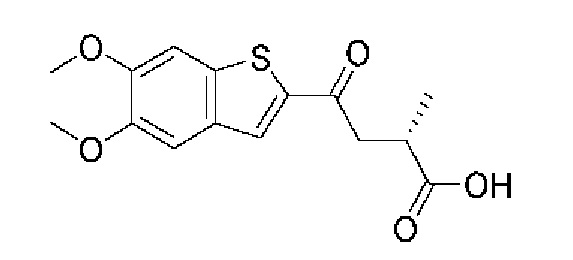
Стадия 1: 5,6-диметоксибензо[b]тиофен-2-каубонилхлоуид
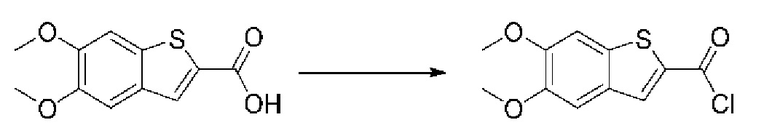
К перемешиваемому раствору 5,6-диметоксибензо[b]тиофен-2-карбоновой кислоты (5.0 г, 21 ммоль) в THF (200 мл) при 0°С в атмосфере Ar добавляли (COCl)2 (5.5 мл, 63 ммоль) с последующим добавлением DMF (0.1 мл, 1.3 ммоль). Реакционную смесь перемешивали при 0°С в течение 1 ч, затем оставляли нагреваться до комнатной температуры и перемешивали на протяжении ночи. Реакционную смесь концентрировали под пониженным давлением, и полученный в результате 5,6-диметоксибензо[b]тиофен-2-карбонилхлорид применяли без очистки. 1Н ЯМР (600 МГц, CH3CN-d3): δ 8.25 (s, 1Н), 7.46 (s, 1H), 7.45 (s, 1H), 3.92 (s, 3Н), 3.88 (s, 3Н).
Стадия 2: метил (S)-4-(5,6-диметоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноат

В высушенную в печи продутую Ar круглодонную колбу, содержащую медь (I) тиофен-2-карбоксилат (797 мг, 4.2 ммоль), при 0°С добавляли по каплям (R)-(3-метокси-2-метил-3-оксопропил)цинк (II) бромид (7.8 мл, 0.5 М в THF, 3.9 ммоль). Реакционную смесь перемешивали при 0°С в течение 20 мин. К реакционной смеси добавляли суспензию 5,6-диметоксибензо[b]тиофен-2-карбонилхлорида (777 мг, 3.0 ммоль) в THF (15 мл) по каплям. Реакционную смесь оставляли нагреваться до комнатной температуры и перемешивали в течение 6 ч. Реакционную смесь разбавляли насыщенным водным раствором NH4Cl (15 мл) с последующим добавлением DCM (30 мл). Выпавшие в осадок вещества удаляли путем фильтрования перед экстракцией. Слои разделяли, и водный слой экстрагировали DCM (3×30 мл). Объединенные органические слои концентрировали под пониженным давлением. Остаток очищали с помощью колоночной хроматографии на силикагеле ((25% EtOH в EtOAc) в гексанах) с получением метил (S)-4-(5,6-диметоксибензо[6]тиофен-2-ил)-2-метил-4-оксобутаноата. ЖХ-МС (C16H19O5S) (ES, m/z): 323 [М+Н]+. 1Н ЯМР (500 МГц, CDCl3): δ 7.89 (s, 1Н), 7.26 (s, 2Н), 4.00 (s, 3Н), 3.97 (s, 3Н), 3.72 (s, 3Н), 3.48 (dd, J=16.9, 7.6 Гц, 1H), 3.22-3.16 (m, 1H), 3.05 (dd, J=16.9, 6.0 Гц, 1H), 1.31 (d, J=7.2 Гц, 3Н).
Стадия 3: метил (R)-4-(5-гидрокси-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноат
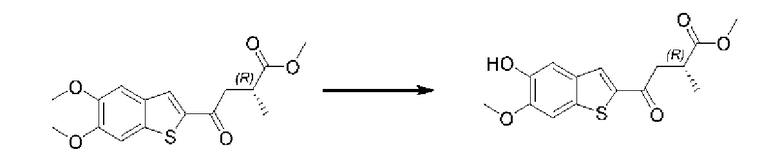
AlCl3 (1.0 г, 7.5 ммоль) добавляли к (R)-метил 4-(5,6-диметоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноату (1.0 г, 3.0 ммоль) в CH2Cl2 (40 мл) при 0°С. Реакционную смесь оставляли нагреваться до комнатной температуры в течение 18 ч. Реакционную смесь затем охлаждали до 0°С, и добавляли МеОН (85 мл). Смесь оставляли перемешиваться при 0°С в течение 30 мин. Смесь затем оставляли нагреваться до комнатной температуры и концентрировали под пониженным давлением. Полученный в результате остаток очищали с помощью колоночной хроматографии на силикагеле (градиент EtOAc в гексанах) с получением неочищенного метил (R)-4-(5-гидрокси-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата. Смесь затем очищали с помощью хиральной SFC (колонка AD-H (21×250 мм), 30% МеОН с 0.25% DMEA в CO2) с получением метил (R)-4-(5-гидрокси-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата, время удерживания 4.7 мин. ЖХ-МС (C15H17O5S) (ES, m/z): 309 [М+Н]+. 1Н ЯМР (DMSO-d6) δ: 9.41 (s, 1H), 8.17 (s, 1H), 7.54 (s, 1H), 7.31 (s, 1H), 3.87 (s, 3H), 3.60 (s, 3H), 3.40 (dd, J=17.4, 8.6 Гц, 1H), 3.17 (dd, J=17.5, 5.1 Гц, 1H), 3.02-2.91 (m, 1H), 1.19 (d, J=7.1 Гц, 3Н).
Промежуточные соединения 26-27, как показано в Таблице 2 ниже, или могут быть получены в соответствии со способами, аналогичными описанным в Промежуточном соединении 25 выше, с применением соответствующих исходных материалов, описанных в способах получения, или получены из коммерческих источников.
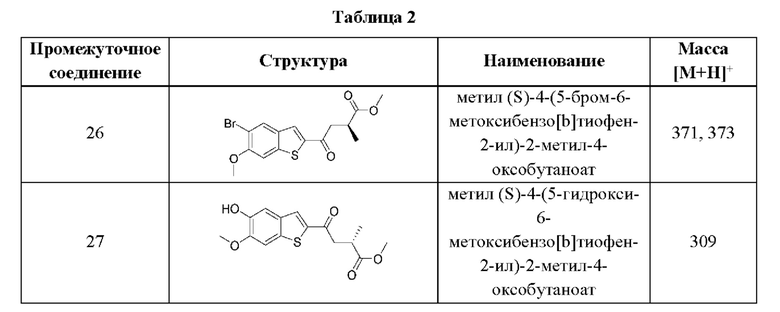
Промежуточное соединение 28: метил (S)-4-(5-(2-хлорэтокси)-4-фтор-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноат
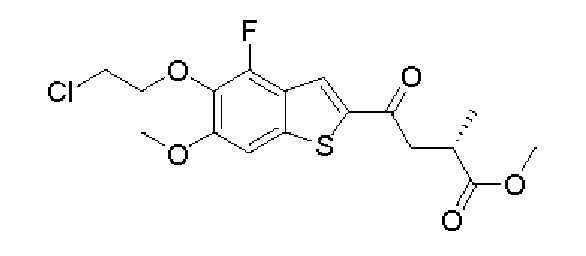
Стадия 1: метил (S)-4-(4-фтор-5,6-диметоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноат
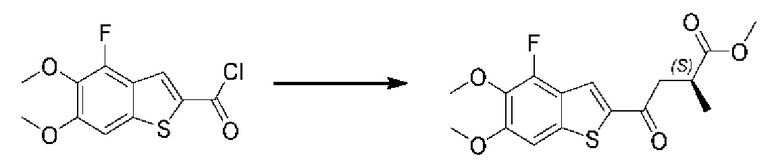
Колбу, содержащую CuCl (2.5 г, 25 ммоль), продували Ar и затем охлаждали до 0°С. Добавляли по каплям 3-метокси-(2R)-(+)-метил-3-оксопропилцинка бромид (0.50 М в THF, 50 мл, 25 ммоль). К перемешиваемой реакционной смеси добавляли по каплям смесь 4-фтор-5,6-диметоксибензо[b]тиофен-2-карбонилхлорида (5.0 г, 20 ммоль) в THF (25 мл) и NMP (25 мл). Реакционную смесь затем оставляли нагреваться до комнатной температуры в течение 18 ч. Затем реакцию останавливали насыщенным водным NH4Cl (100 мл) и экстрагировали EtOAc (3×100 мл). Объединенные органические вещества высушивали над MgSO4, фильтровали и концентрировали под пониженным давлением. Полученный в результате остаток очищали с помощью колоночной хроматографии на силиагеле с нормальными фазами (EtOAc в гексанах) с получением (S)-метил 4-(4-фтор-5,6-диметоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата. ЖХ-МС (C16H18FO5S) (ES, m/z): 341 [М+Н]+. 1Н ЯМР (CDCl3) δ: 7.99 (s, 1H), 7.09 (s, 1H), 4.00 (s, 3Н), 3.98 (s, 3Н), 3.73 (s, 3Н), 3.51 (dd, J=17.0, 7.9 Гц, 1Н), 3.24-3.12 (m, 1H), 3.06 (dd, J=17.0, 5.6 Гц, 1H), 1.32 (d, J=7.1 Гц, 3Н).
Стадия 2: (S)-метил 4-(4-фтор-5-гидрокси-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноат
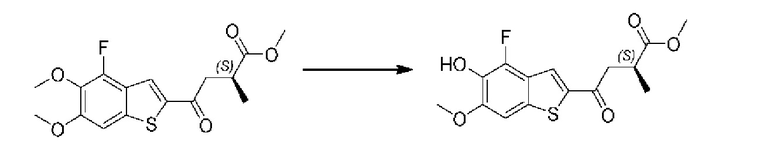
К смеси метил (S)-4-(4-фтор-5,6-диметоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата (1.0 г, 2.9 ммоль) и СН=CCl2 (40 мл) добавляли AlCl3 (1.0 г, 7.5 ммоль). Реакционную смесь оставляли перемешиваться при комнатной температуре в течение 18 ч. Реакционную смесь затем охлаждали до 0°С и разбавляли МеОН (40 мл). Смесь затем оставляли нагреваться до комнатной температуры и концентрировали под пониженным давлением. Полученный в результате остаток очищали с помощью колоночной хроматографии на силикагеле (EtOAc в гексанах) с получением (S)-метил 4-(4-фтор-5-гидрокси-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата. ЖХ-МС (C15H16FO5S) (ES, m/z): 327 [М+Н]+. 1Н ЯМР (метанол-d4) δ: 8.11 (s, 1H), 7.29 (s, 1H), 3.98 (s, 3Н), 3.69 (s, 3Н), 3.50 (dd, J=17.5, 8.6 Гц, 1H), 3.23-3.16 (m, 1H), 3.11-3.02 (m, 1H), 1.28 (d, J=7.2 Гц, 3Н).
Стадия 3: метил (S)-4-(5-(2-хлорэтокси)-4-фтор-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноат
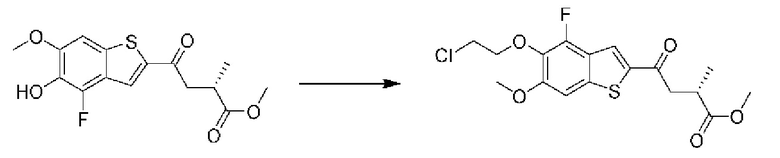
K2CO3 (170 мг, 1.2 ммоль) добавляли к перемешиваемой смеси (S)-метил 4-(4-фтор-5-гидрокси-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата (200 мг, 0.61 ммоль) и DMF (2.7 мл). К перемешиваемой реакционной смеси добавляли 1-бром-2-хлорпентан (50 мкл, 0.6 ммоль) и реакционную смесь затем нагревали до 80°С в течение 18 ч. После охлаждения до комнатной температуры реакционную смесь концентрировали под пониженным давлением. Полученный в результате остаток очищали с помощью колоночной хроматографии на силикагеле (EtOAc в гексанах) с получением метил (S)-4-(5-(2-хлорэтокси)-4-фтор-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата. ЖХ-МС (C17H19ClFO5S) (ES, m/z): 389 [М+Н]+.
Промежуточные соединения с 29 по 31 и 87, как показано в Таблице 3 ниже, или могут быть получены в соответствии со способами, аналогичными описанным в Промежуточном соединении 28 выше, с применением соответствующих исходных материалов, описанных в способах получения, или получены из коммерческих источников.
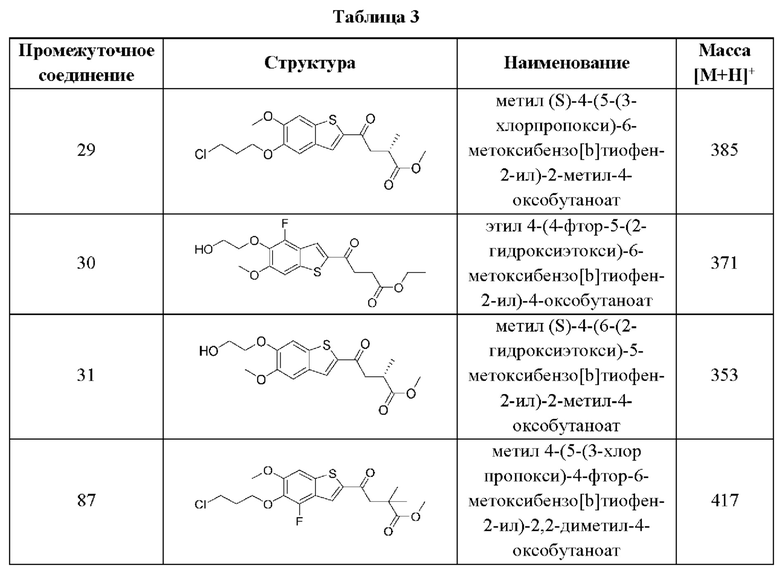
Промежуточное соединение 32: трет-бутил 4-(5-(2-гидроксиэтил)-6-метокситиено[3,2-b]пиридин-2-ил)-4-оксобутаноат, соль уксусной кислоты
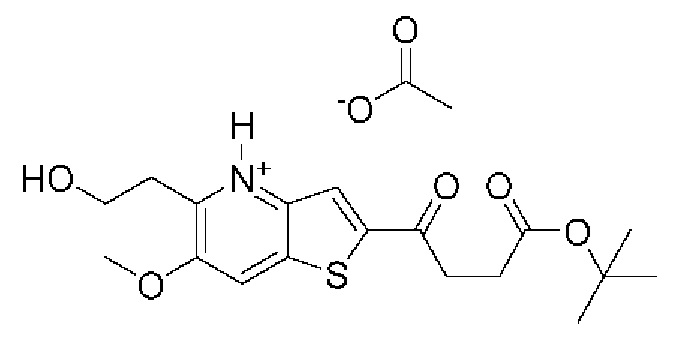
Стадия 1: трет-бутил 4-(6-метокси-5-(2-((тетрагидро-2Н-пиран-2-ил)окси)этил)тиено[3,2-b]пиридин-2-ил)-4-оксобутаноат
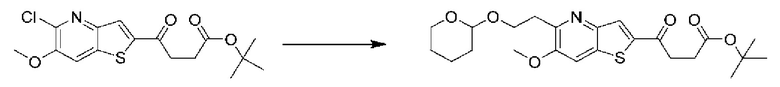
В 4 мл виалу добавляли бис(ди-трет-бутил(4-диметиламинофенил)фосфин)дихлор-палладий (II) (5.0 мг, 7.0 мкмоль), Cs2CO3 (137 мг, 0.422 ммоль), трет-бутил 4-(5-хлор-6-метокситиено[3,2-b]пиридин-2-ил)-4-оксобутаноат (50.0 мг, 0.141 ммоль) и калий трифтор(2-((тетрагидро-2Н-пиран-2-ил)окси)этил)борат (39.8 мг, 0.169 ммоль). В виалу добавляли толуол (0.50 мл) и воду (0.10 мл). Виалу дегазировали N2 в течение 5 мин. Смесь нагревали до 100°С в течение 18 ч. После охлаждения до комнатной температуры смесь фильтровали через целит, и целит промывали EtOAc. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали с помощью хроматографии на силикагеле (0→75% градиент EtOAc в гексанах) с получением трет-бутил 4-(6-метокси-5-(2-((тетрагидро-2Н-пиран-2-ил)окси)этил)тиено[3,2-b]пиридин-2-ил)-4-оксобутаноата. ЖХ-МС (C23H32NO6S) (ES, m/z): 450 [М+Н]+. 1Н ЯМР (500 МГц, CDCl3) δ 8.45 (s, 1Н), 7.80 (s, 1H), 4.71 (s, 1H), 4.26-4.15 (m, 1Н), 4.04 (s, 3Н), 3.78 (t, J=8.3 Гц, 1H), 3.65-3.42 (m, 4Н), 3.33 (t, J=6.3 Гц, 2Н), 2.73 (t, J=6.3 Гц, 2Н), 1.64-1.48 (m, 6Н), 1.46 (s, 9Н).
Стадия 2: трет-бутил 4-(5-(2-гидроксиэтил)-6-метокситиено[3,2-b]пиридин-2-ил)-4-оксобутаноат, соль уксусной кислоты
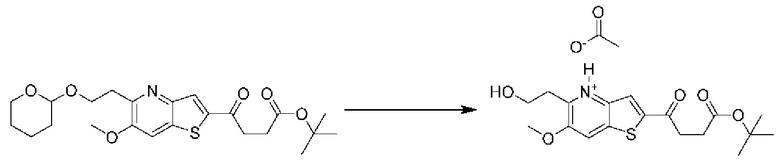
В 4 мл виалу добавляли трет-бутил 4-(6-метокси-5-(2-((тетрагидро-2Н-пиран-2-ил)окси)этил)-тиено[3,2-b]пиридин-2-ил)-4-оксобутаноат (28 мг, 0.062 ммоль), НОАс (0.50 мл), МеОН (0.50 мл) и воду (0.5 мл). Смесь нагревали до 50°С в течение 75 мин. После охлаждения до комнатной температуры смесь разбавляли EtOAc (30 мл) и промывали водой (2×30 мл). Органический слой высушивали над Na2SO4, фильтровали и концентрировали под пониженным давлением с получением трет-бутил 4-(5-(2-гидроксиэтил)-6-метокситиено[3,2-b]пиридин-2-ил)-4-оксобутаноата, соли уксусной кислоты. ЖХ-МС (C18H24NO5S) (ES, m/z): 366 [М+Н]+. 1H ЯМР (500 МГц, CDCl3) δ 8.22 (s, 1Н), 7.68 (s, 1Н), 4.13 (t, J=5.4 Гц, 2Н), 4.01 (s, 3Н), 3.39-3.22 (m, 4Н), 2.74 (t, J=6.5 Гц, 2Н), 1.46 (s, 9Н).
Промежуточное соединение 33: трет-бутил 4-(5-(3-гидроксипропил)бензо[b]тиофен-2-ил)-4-оксобутаноат
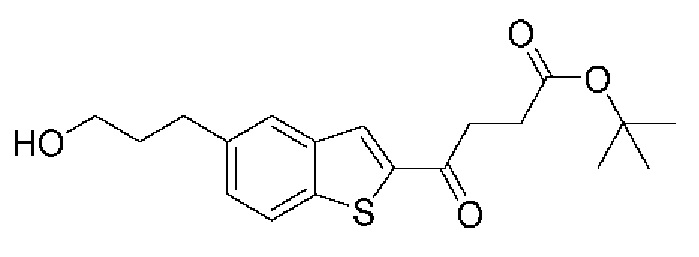
Стадия 1: трет-бутил 4-(5-(3-((трет-бутилдиметилсилил)окси)пропил)бензо[b]тиофен-2-ил)-4-оксобутаноат
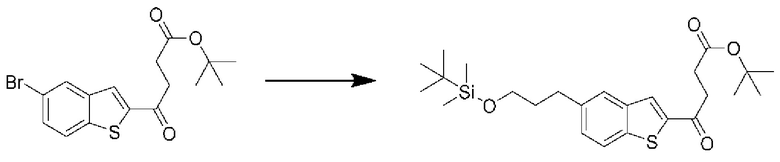
трет-Бутил 4-(5-бромбензо[b]тиофен-2-ил)-4-оксобутаноат (0.207 г, 0.561 ммоль) и хлор(2-дициклогексилфосфино-2',4,'6'-триизопропил-1,1'-бифенил)[2-(2'-амино-1,1'-бифенил)]палладий (II) (0.022 г, 0.028 ммоль) помещали в 20 мл виалу с завинчивающейся крышкой с магнитной мешалкой. Виалу 3 раза откачивали и снова заполняли N2. Виалу закрывали впускным клапаном для N2 и затем добавляли THF (2.0 мл). Добавляли по каплям (3-((трет-бутилдиметилсилил)окси)пропил)цинк (II) бромид (0.50 М в THF, 3.4 мл, 1.7 ммоль) с перемешиванием. После завершения добавления реакционную смесь перемешивали при комнатной температуре в атмосфере N2 в течение 1.5 ч. Реакционную смесь затем разделяли между EtOAc (50 мл) и 10% водным цитратом натрия (10 мл) и перемешивали в течение 30 мин. Слои затем разделяли, и органический слой промывали насыщенным водным NaCl, высушивали над безводным MgSO4, фильтровали и концентрировали под пониженным давлением. Полученный в результате остаток очищали с помощью хроматографии на силикагеле (0→55% градиент EtOAc в гексанах) с получением трет-бутил 4-(5-(3-((трет-бутилдиметилсилил)окси)пропил)бензо[b]тиофен-2-ил)-4-оксобутаноата. ЖХ-МС (C25H38NaO4SSi) (ES, m/z): 485 [M+Na]+. 1H ЯМР (500 МГц, CDCl3) δ 7.96 (s, 1H), 7.78 (d, Г=8.2 Гц, 1H), 7.70 (s, 1H), 7.33 (d, J=8.2 Гц, 1H), 3.66 (t, J=5.8 Гц, 2Н), 3.31 (t, J=6.5 Гц, 2Н), 2.82 (t, J=7.5 Гц, 2Н), 2.73 (t, J=6.5 Гц, 2Н), 1.93-1.86 (m, 2Н), 1.47 (s, 9Н), 0.94 (s, 9Н), 0.08 (s, 6Н).
Стадия 2: трет-бутил 4-(5-(3-гидроксипропил)бензо[b]тиофен-2-ил)-4-оксобутаноат
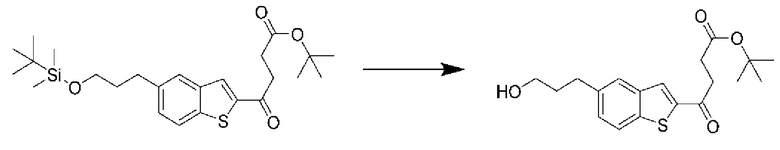
TBAF (1.0 M в THF, 0.44 мл, 0.44 ммоль) добавляли к перемешиваемой смеси трет-бутил 4-(5-(3-((трет-бутилдиметилсилил)окси)пропил)бензо[b]тиофен-2-ил)-4-оксобутаноата (102 мг, 0.220 ммоль) в THF (1.3 мл) при комнатной температуре в атмосфере N2. Полученную в результате смесь перемешивали при комнатной температуре в течение 4.5 ч. Реакционную смесь затем разделяли между Et2O и насыщенным водным NH4Cl и перемешивали при комнатной температуре в течение 1 ч. Слои разделяли, и органический слой промывали насыщенным водным NaCl, высушивали над безводным MgSO4, фильтровали и концентрировали под пониженным давлением с получением сырого остатка. Полученный в результате остаток очищали с помощью хроматографии на силикагеле (0→60% градиент EtOAc в гексанах) с получением трет-бутил 4-(5-(3-гидроксипропил)бензо[b]тиофен-2-ил)-4-оксобутаноата. ЖХ-МС (C19H24NaO4S) (ES, m/z): 371 [M+Na]+. 1Н ЯМР (500 МГц, CDCl3) δ 7.95 (s, 1H), 7.77 (d, J=8.3 Гц, 1H), 7.70 (s, 1H), 7.33 (d, J=8.3 Гц, 1Н), 3.71 (t, J=6.3 Гц, 2H), 3.30 (t, J=6.7 Гц, 2H), 2.84 (t, J=7.7 Гц, 2H), 2.72 (t, J=6.7 Гц, 2H), 1.98-1.91 (m, 2H), 1.46 (s, 9H).
Промежуточное соединение 34: (S)-метил 4-(4-фтор-5-(3-гидроксипропокси)-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноат
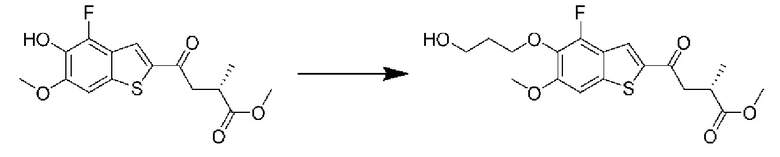
К смеси (S)-метил 4-(4-фтор-5-гидрокси-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата (0.065 г, 0.2 ммоль) и Cs2CO3 (0.33 г, 1.0 ммоль) в ACN (2.0 мл) добавляли (3-бромпропокси)(трет-бутил)диметилсилан (0.10 мл, 0.20 ммоль). Реакцию затем нагревали до 65°С в течение 2 ч. После охлаждения до комнатной температуры смесь затем фильтровали и промывали THF (5 мл). К полученному в результате фильтрату добавляли воду (2 мл) с последующим добавлением MP-TsOH (загрузка 4.38 ммоль/г, 1.00 г, 4.38 ммоль). Смесь затем нагревали до 60°С в течение 30 мин. После охлаждения до комнатной температуры смесь фильтровали и промывали THF. Фильтрат концентрировали под пониженным давлением с получением (S)-метил 4-(4-фтор-5-(3-гидроксипропокси)-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата, который применяли без дополнительной очистки. ЖХ-МС (C18H22FO6S) (ES, m/z): 385 [М+Н]+.
Промежуточные соединения 35-36, как показано в Таблице 4 ниже, или могут быть получены в соответствии со способами, аналогичными описанным в Промежуточном соединении 34 выше, с применением соответствующих исходных материалов, описанных в способах получения, или получены из коммерческих источников.
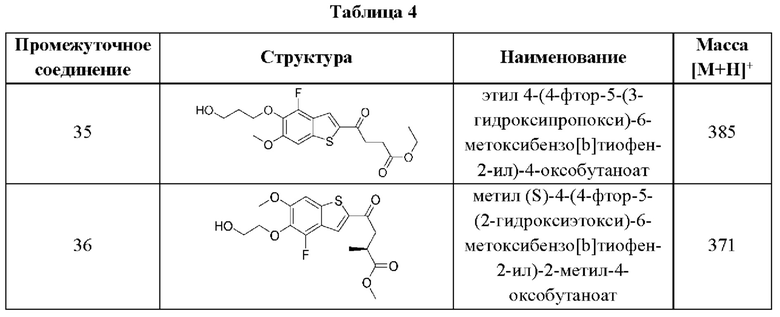
Промежуточное соединение 37: метил (2S)-4-[5-(3-бромпропил)-6-метокси-1-бензотиофен-2-ил]-2-метил-4-оксобутаноат
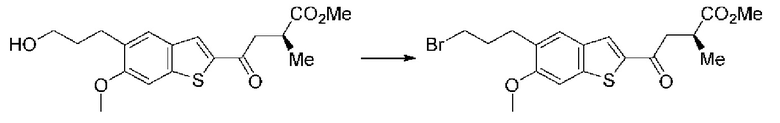
К перемешиваемой смеси метил (2S)-4-[5-(3-гидроксипропил)-6-метокси-1-бензотиофен-2-ил]-2-метил-4-оксобутаноата (187 мг, 0.534 ммоль) и Ph3P (224 мг, 0.854 ммоль) в THF (2.7 мл) при 0°С добавляли NBS (142 мг, 0.800 ммоль) одной порцией в атмосфере N2. Реакционную смесь перемешивали в течение 20 мин при 0°С. Смесь сразу очищали с помощью колоночной флэш-хроматографии на силикагеле (EtOAc в гексанах) с получением метил (2S)-4-[5-(3-бромпропил)-6-метокси-1-бензотиофен-2-ил]-2-метил-4-оксобутаноата. ЖХ-МС (C18H22BrO4S) (ES, m/z): 413, 415 [М+Н]+. 1Н ЯМР (500 МГц, DMSO-d6): δ 8.27 (s, 1Н), 7.76 (s, 1H), 7.62 (s, 1H), 3.89 (s, 3H), 3.59 (s, 3H), 3.54 (t, J=6.8 Гц, 2H),3.43 (dd, J=17.5, 8.6 Гц, 1H), 3.19 (dd, J=17.5, 5.0 Гц, 1H), 3.02-2.92 (m, 1H), 2.79(t, J=7.0 Гц, 2H), 2.10 (pentet, J=6.9 Гц, 2H), 1.19 (d, J=7.1 Гц, 3H).
Промежуточное соединение 38: трет-бутил 4-(5-(3-гидроксипропокси)-6-метокситиено[3,2-b]пиридин-2-ил)-4-оксобутаноат
Стадия 1: трет-бутил 4-(5-(3-((трет-бутилдиметилсилил)окси)пропокси)-6-метокситиено[3,2-b]пиридин-2-ил)-4-оксобутаноат
трет-Бутил 4-(5-хлор-6-метокситиено[3,2-b]пиридин-2-ил)-4-оксобутаноат (265 мг, 0.745 ммоль), 3-((трет-бутилдиметилсилил)окси)пропан-1-ол (156 мг, 0.819 ммоль), RockPhos Pd G3 (31 мг, 0.037 ммоль) и Cs2CO3 (364 мг, 1.12 ммоль) добавляли в 20 мл виалу с завинчивающейся крышкой с магнитной мешалкой. Виалу закрывали, и вставляли впускную иглу для N2. Через эту иглу виалу три раза откачивали и снова заполняли N2. В атмосфере N2 добавляли толуол (2.5 мл), впускную иглу для N2 удаляли, и герметично закрытую виалу нагревали до 110°С в течение 18 ч. Реакцию оставляли охлаждаться до комнатной температуры, и добавляли МеОН (3.0 мл), воду (3.0 мл) и НОАс (3.0 мл). Реакционную смесь перемешивали в течение 7 ч при комнатной температуре, затем разделяли между EtOAc и насыщенным водным NaCl. Слои разделяли, и водный слой экстрагировали EtOAc.Органические слои объединяли, промывали водным NaCl, высушивали над безводным Na2SO4, фильтровали и концентрировали под пониженным давлением с получением сырого остатка. Остаток очищали с помощью хроматографии на силикагеле (0→100% 3:1 EtOAc : EtOH градиент в гексанах) с получением трет-бутил 4-(5-(3-((трет-бутилдиметилсилил)окси)пропокси)-6-метокситиено[3,2-b]пиридин-2-ил)-4-оксобутаноата. ЖХ-МС (C25H40NO6SSi) (ES, m/z): 510 [М+Н]+. 1H ЯМР (500 МГц, CDCl3) δ 7.95 (s, 1H), 7.39 (s, 1H), 4.56 (t, J=6.5 Гц, 2H), 3.94 (s, 3Н), 3.83 (t, J=6.0 Гц, 2H), 3.26 (t, J=6.1 Гц, 2H), 2.70 (t, J=6.7 Гц, 2H), 2.12-2.05 (m, 2H), 1.44 (s, 9H), 0.89 (s, 9H), 0.07 (s, 6H).
Стадия 2: трет-бутил 4-(5-(3-гидроксипропокси)-6-метокситиено[3,2-b]пиридин-2-ил)-4-оксобутаноат
В виалу, содержащую трет-бутил 4-(5-(3-((трет-бутилдиметилсилил)окси)пропокси)-6-метокситиено[3,2-b]пиридин-2-ил)-4-оксобутаноат (83 мг, 0.16 ммоль), добавляли МеОН (1.0 мл), воду (1.0 мл) и затем НОАс (1.0 мл). Полученную в результате смесь перемешивали в течение 30 мин при комнатной температуре. Добавляли МеОН (1.0 мл) и продолжали перемешивание. Через 30 мин добавляли THF (1.0 мл) и смесь оставляли перемешиваться в течение 18 ч при комнатной температуре. Смесь разделяли между EtOAc (25 мл) и водным NaCl (25 мл) и перемешивали. Слои разделяли, и водный слой экстрагировали EtOAc.Органические слои объединяли, дважды промывали насыщенным водным NaCl, высушивали над безводным Na2SO4, фильтровали и концентрировали под пониженным давлением с получением сырого остатка. Полученный в результате остаток очищали с помощью хроматографии на силикагеле (0→100% градиент EtOAc в гексанах, затем изократическое элюирование при 100% EtOAc) с получением трет-бутил 4-(5-(3-гидроксипропокси)-6-метокситиено[3,2-b]пиридин-2-ил)-4-оксобутаноата. ЖХ-МС (C19H26NO6S) (ES, m/z): 396 [М+Н]+. 1Н ЯМР (500 МГц, CDCl3) δ 7.93 (s, 1H), 7.44 (s, 1H), 4.69 (t, J=5.6 Гц, 2H), 3.97 (s, 3H), 3.80-3.76 (m, 2H), 3.31-3.20 (m, 2H), 2.71 (t, J=6.5 Гц, 2H), 2.14-2.07 (m, 2H), 1.46 (s, 9H).
Промежуточное соединение 39, как показано в Таблице 5 ниже, или может быть получено в соответствии со способами, аналогичными описанным в Промежуточном соединении 38 выше, с применением соответствующих исходных материалов, описанных в способах получения, или получено из коммерческих источников.
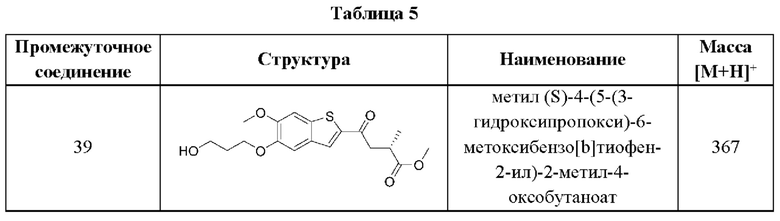
Промежуточное соединение 40: Метил 4-(6-гидроксибензо[b]тиофен-2-ил)-4-оксобутаноат
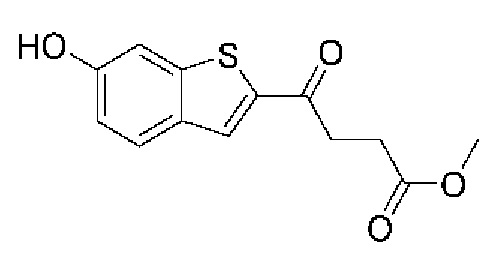
Стадия 1: 6-(бензилокси)бензо[b]тиофен
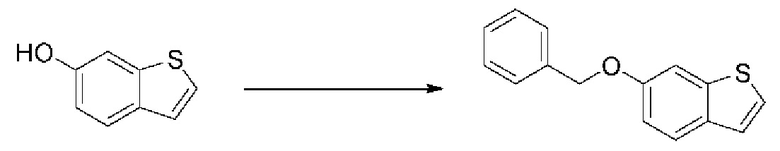
K2CO3 (2.62 г, 19.0 ммоль) добавляли к смеси бензо[b]тиофен-6-ола (1.9 г, 13 ммоль) и бензилбромида (1.51 мл, 12.7 ммоль) в DMF (10.0 мл) при 20°С в атмосфере Ar. Реакционную смесь перемешивали и нагревали до 50°С в течение 18 ч. После охлаждения до комнатной температуры реакционную смесь затем разбавляли EtOAc (500 мл) и водой (100 мл). Органический слой отделяли, промывали водой (50 мл) и затем насыщенным водным NaCl (50 мл), высушивали над MgSO4, фильтровали и концентрировали под пониженным давлением с получением неочищенного остатка. Остаток сырого вещества очищали с помощью хроматографии на силикагеле (0→100% градиент EtOAc в гексанах) с получением 6-(бензилокси)бензо[b]тиофена. ЖХ-МС (C15H13OS) (ES, m/z): 241 [М+Н]+. 1H ЯМР (500 МГц, DMSO-d6) δ 7.77 (d, J=8.6 Гц, 1H), 7.67 (s, 1H), 7.54 (d, J=5.2 Гц, 1H), 7.49 (d, J=7.2 Гц, 2Н), 7.44-7.38 (m, 2H), 7.37-7.32 (m, 2Н), 7.08 (d, J=8.5 Гц, 1H), 5.17 (s, 2Н).
Стадия 2: 6-(бензилокси)бензо[b]тиофен-2-карбальдегид

LDA (2.0 М в THF, 7.3 мл, 15 ммоль) добавляли к смеси 6-(бензилокси)бензо[b]тиофена (2.92 г, 12.2 ммоль) в THF (10.0 мл) при -78°С в атмосфере Ar. Реакционную смесь перемешивали при -78°С в течение 20 мин. К реакционной смеси добавляли DMF (2.4 мл, 30 ммоль) при -78°С, и реакционную смесь затем оставляли медленно нагреваться до комнатной температуры. Реакционную смесь перемешивали в течение 15 мин при комнатной температуре. Реакционную смесь останавливали лимонной кислотой (1.0 М в воде, 24 мл, 24 ммоль) при 0°С и затем разбавляли EtOAc (200 мл). Суспензию перемешивали в течение 15 мин. Органический слой отделяли, высушивали над MgSO4, фильтровали и концентрировали под пониженным давлением с получением неочищенного остатка. Сырой остаток очищали с помощью хроматографии на силикагеле (0→100% градиент EtOAc в гексанах) с получением 6-(бензилокси)бензо[b]тиофен-2-карбальдегида. ЖХ-МС (C16H13O2S) (ES, m/z): 269 [М+Н]+. 1H ЯМР (500 МГц, DMSO-d6) δ 10.05 (s, 1Н), 8.34 (s, 1H), 8.01 (d, J=8.8 Гц, 1H), 7.78 (s, 1H), 7.50 (d, J=7.3 Гц, 2H), 7.42 (t, J=7.3 Гц, 2H), 7.40-7.33 (m, 1H), 7.20 (d, J=8.4 Гц, 1H), 5.23 (s, 2H).
Стадия 3: трет-бутил 4-(6-(бепзилокси)бензо[b]тиофен-2-ил)-4-оксобутаноат
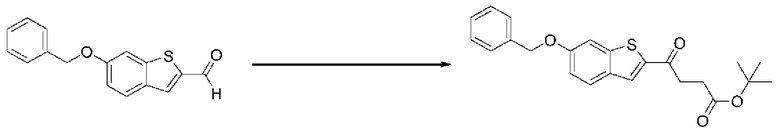
Смесь 6-(бензилокси)бензо[b]тиофен-2-карбальдегида (2.02 г, 7.53 ммоль), 2-мезитил-6,7-дигидро-5Н-пирроло[2,1-с][1,2,4]триазол-2-ий хлорида (0.099 г, 0.38 ммоль) и трехосновного фосфата калия (1.6 г, 7.5 ммоль) продували Ar в течение 5 мин при 20°С. Затем добавляли толуол (15 мл) и трет-бутилакрилат (2.2 мл, 15 ммоль) при 20°С. Реакционную смесь оставляли перемешиваться в течение 18 ч при 20°С. Реакционную смесь затем разбавляли EtOAc (200 мл) и фильтровали для удаления неорганических солей. Фильтрат концентрировали под пониженным давлением с получением неочищенного остатка. Остаток сырого вещества очищали с помощью хроматографии на силикагеле (0→100% градиент EtOAc в гексанах) с получением трет-бутил 4-(6-(бензилокси)бензо[b]тиофен-2-ил)-4-оксобутаноата. ЖХ-МС (C23H25O4S) (ES, m/z): 397 [М+Н]+. 1Н ЯМР (500 МГц, DMSO-d6) δ 8.35 (s, 1H), 7.96 (d, J=7.0 Гц, 1H), 7.75 (s, 1H), 7.56-7.39 (m, 2Н), 7.47-7.33 (m, 3Н), 7.25-7.15 (m, 1Н), 5.24 (s, 2H), 3.34-3.23 (m, 2H), 2.65-2.55 (m, 2H), 1.40 (s, 9H).
Стадия 4: 4-(6-гидуоксибензо[b]тиофен-2-ил)-4-оксобутановая кислота
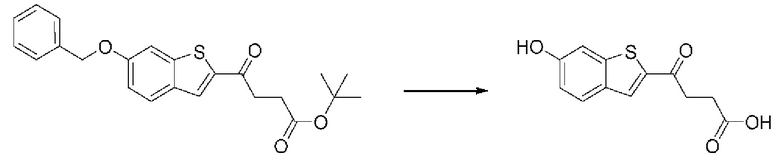
HCl (37% в воде, 19.6 мл, 238 ммоль) добавляли к смеси трет-бутил 4-(6-(бензилокси)бензо[b]тиофен-2-ил)-4-оксобутаноата (2.36 г, 5.96 ммоль) в диоксане (100 мл). Реакционную смесь нагревали до 90°С в течение 2 дней. Реакционную смесь охлаждали до комнатной температуры и разбавляли EtOAc (500 мл). Органический слой отделяли, промывали водой (3×100 мл) и затем насыщенным водным NaCl (50 мл), высушивали над MgSO4, фильтровали и концентрировали под пониженным давлением с получением 4-(6-гидроксибензо[b]тиофен-2-ил)-4-оксобутановой кислоты, которую применяли без дополнительной очистки. ЖХ-МС (C12H11O4S) (ES, m/z): 251 [М+Н]+.
Стадия 5: метил 4-(б-гидроксибензо[b]тиофен-2-ил)-4-оксобутаноат
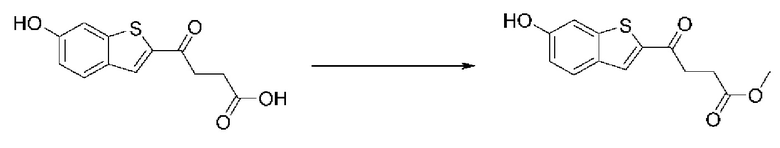
TMS-диазометан (2.0 М в гексанах, 3.0 мл, 6.0 ммоль) добавляли к смеси 4-(6-гидроксибензо[b]тиофен-2-ил)-4-оксобутановой кислоты (1.5 г, 6.0 ммоль) в DCM (25 мл) и МеОН (25 мл) при 0°С. Реакционную смесь перемешивали при 0°С в течение 15 мин (пока не прекратилось выделение газа). Добавляли НОАс (несколько капель), чтобы погасить весь оставшийся TMS-диазометан. Реакционную смесь затем концентрировали под пониженным давлением с получением неочищенного остатка. Остаток сырого вещества очищали с помощью хроматографии на силикагеле (0→100% градиент EtOAc в гексанах) с получением метил 4-(6-гидроксибензо[b]тиофен-2-ил)-4-оксобутаноата. ЖХ-МС (C13H13O4S) (ES, m/z): 265 [М+Н]+. 1Н ЯМР (500 МГц, DMSO-d6) δ 10.17 (s, 1H), 8.26 (s, 1H), 7.84 (d, J=8.7 Гц, 1H), 7.30 (s, 1H), 6.97 (d, J=8.2 Гц, 1H), 3.76-3.65 (m, 1H), 3.60 (s, 3Н), 3.54-3.44 (m, 1H), 2.72-2.67 (m, 2H).
Промежуточное соединение 41, как показано в Таблице 6 ниже, или может быть получено в соответствии со способами, аналогичными описанным в Промежуточном соединении 40 выше, с применением соответствующих исходных материалов, описанных в способах получения, или получено из коммерческих источников.
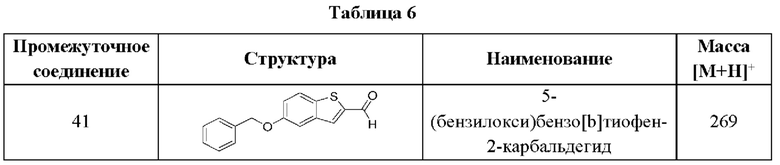
Промежуточное соединение 42: Метил (S)-4-(6-(3-хлорпропокси)-5-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноат
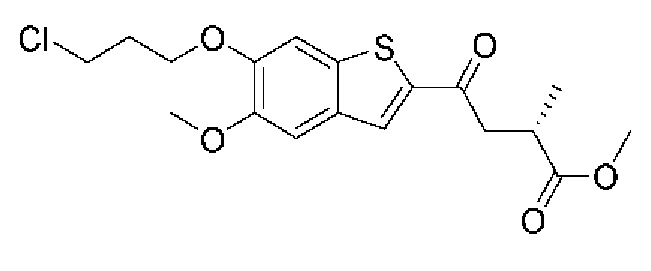
Стадия 1: 6-(бензилокси)-5-метоксибензо[b]тиофен-2-карбоновая кислота
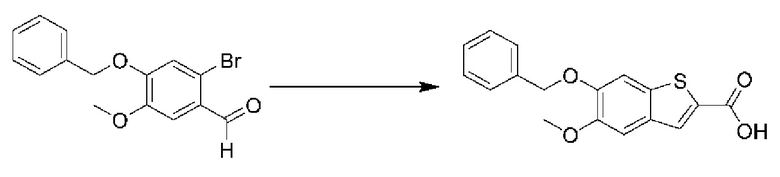
Смесь 4-(бензилокси)-2-бром-5-метоксибензальдегида (13 г, 41 ммоль), K2CO3 (11 г, 81 ммоль), 18-Crown-6 (2.1 г, 8.1 ммоль) и метил 2-меркаптоацетата (6.0 мл, 67 ммоль) в DMF (150 мл) нагревали до 90°С в атмосфере Ar в течение 14 ч. После охлаждения до комнатной температуры реакционную смесь останавливали водой (400 мл), подкисляли с помощью 1 н. HCl до рН~5 и экстрагировали EtOAc (3×250 мл). Объединенные органические слои промывали 10% водным LiCl (3×50 мл), высушивали над безводным Na2SO4, фильтровали и концентрировали под пониженным давлением. Остаток разбавляли EtOAc/МеОН (50 мл/50 мл) и обрабатывали водным LiOH (2.0 М, 80 мл, 160 ммоль) при 20°С.Реакционную смесь нагревали до 60°С в течение 2 ч. После охлаждения до комнатной температуры смесь концентрировали под пониженным давлением. Остаток разбавляли водным NaOH (1.0 М, 50 мл, 50 ммоль), водой (300 мл) и EtOAc (300 мл). Слои разделяли, и водный слой промывали EtOAc (3×200 мл). Водный слой затем подкисляли 6 н. HCl до рН~5, и экстрагировали EtOAc (3×300 мл). Органические вещества объединяли, промывали насыщенным водным NaCl (50 мл), высушивали над безводным Na2SO4, фильтровали и концентрировали под пониженным давлением. Остаток растирали с 1:1 EtOAc/PE (3×30 мл) и 1:1 DCM/PE (3×30 мл) с получением 6-(бензилокси)-5-метоксибензо[b]тиофен-2-карбоновой кислоты. ЖХ-МС (C17H15O4S) (ES, m/z): 315 [М+Н]+. 1Н ЯМР (400 МГц, DMSO-d6) δ 13.16 (br s, 1H), 7.95 (s, 1H), 7.69 (s, 1H), 7.51 (s, 1H), 7.50-7.46 (m, 2H), 7.44-7.32 (m, 3H), 5.17 (s, 2H), 3.83 (s, 3H).
Стадия 2: 6-(бензилокси)-5-метоксибензо[b]тиофен-2-карбонилхлорид

DMF (0.023 мл, 0.29 ммоль) добавляли к смеси 6-(бензилокси)-5-метоксибензо[b]тиофен-2-карбоновой кислоты (3.05 г, 9.70 ммоль) и (COCl)2 (2.55 мл, 29.1 ммоль) в THF (50 мл) при 0°С в атмосфере Ar. Смесь перемешивали при 0°С в течение 30 мин и затем нагревали до комнатной температуры. Реакционную смесь затем перемешивали в течение 2 ч при комнатной температуре. Реакционную смесь концентрировали под пониженным давлением с получением 6-(бензилокси)-5-метоксибензо[b]тиофен-2-карбонилхлорида, который применяли без очистки.
Стадия 3: метил (8)-4-(6-(бензилокси)-5-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноат
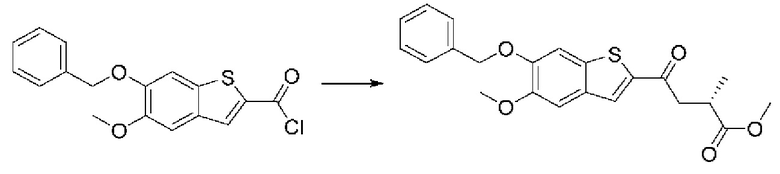
Смесь (R)-(3-метокси-2-метил-3-оксопропил)цинка (II) бромида (0.50 М в THF, 25 мл, 13 ммоль) медленно добавляли в колбу, содержащую ((тиофен-2-карбонил)окси)медь (2.38 г, 12.5 ммоль) в атмосфере Ar при 0°С. Реакционную смесь перемешивали в течение 20 мин при 0°С в атмосфере Ar. Дегазированную Ar смесь 6-(бензилокси)-5-метоксибензо[b]тиофен-2-карбонилхлорида (3.2 г, 9.6 ммоль) в THF (50 мл) затем медленно добавляли к смеси через канюлю при 0°С. Смесь оставляли нагреваться до комнатной температуры, и затем перемешивали в течение 16 ч при комнатной температуре. Реакционную смесь охлаждали до 0°С и останавливали добавлением насыщенного водного NH4Cl (50 мл), воды (100 мл) и EtOAc (500 мл). Полученную в результате двухфазную смесь нагревали до комнатной температуры и перемешивали в течение 1 ч при комнатной температуре. Смесь затем фильтровали через фритту Сбыте, и фильтрат разделяли с использованием делительной воронки. Органический слой отделяли, промывали насыщенным водным NaCl (50 мл), высушивали над MgSO4, фильтровали и концентрировали под пониженным давлением. Остаток очищали с помощью хроматографии на силикагеле (0→50% градиент EtOAc в гексанах) с получением целевого продукта, который повторно очищали с помощью хроматографии на силикагеле (элюируя 0-50% EtOAc в DCM) с получением (S)-метил 4-(6-(бензилокси)-5-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата. ЖХ-МС (C22H23O5S) (ES, m/z): 399 [М+Н]+. 1Н ЯМР (499 МГц, DMSO-d6) δ 8.22 (s, 1H), 7.72 (s, 1H), 7.52-7.47 (m, 3Н), 7.42 (t, J=7.3 Гц, 2Н), 7.38-7.34 (m, 1H), 5.19 (s, 2Н), 3.85 (s, 3Н), 3.60 (s, 3Н), 3.42 (dd, J=17.5, 8.6 Гц, 1H), 3.19 (dd, J=17.4, 4.9 Гц, 1H), 3.04-2.91 (m, 1H), 1.20 (d, J=7.1 Гц, 3Н).
Стадия 4: метил (3)-4-(6-гидрокси-5-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноат
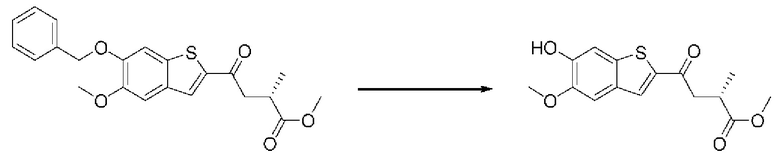
Смесь (S)-метил 4-(6-(бензилокси)-5-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата (1.60 г, 4.02 ммоль) и Pd/C (1.07 г, 1.00 ммоль) дегазировали Ar. К смеси медленно добавляли EtOAc (50 мл), МеОН (50 мл) и HCl (37% в воде, 0.66 мл, 8.0 ммоль) в атмосфере потока Ar. Полученную реакционную смесь дегазировали в вакууме и снова заполняли Н2. Реакционную смесь перемешивали в атмосфере Н2 в течение 3 ч. Реакционную смесь фильтровали через целит, промывая EtOAc.Фильтрат концентрировали под пониженным давлением. Остаток очищали с помощью хроматографии на силикагеле (элюируя 0-100% EtOAc в гексанах) с получением (S)-метил 4-(6-гидрокси-5-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата. ЖХ-МС (C15H17O5S) (ES, m/z): 309 [М+Н]+. 1H ЯМР (499 МГц, DMSO-d6) δ 9.93 (s, 1Н), 8.17 (s, 1Н), 7.44 (s, 1H), 7.30 (s, 1H), 3.85 (s, 3Н), 3.60 (s, 3Н), 3.44-3.38 (m, 1H), 3.16 (dd, J=17.3, 4.7 Гц, 1H), 3.01-2.92 (m, 1H), 1.19 (d, J=7.0 Гц, 3Н).
Стадия 5: метил (S)-4-(6-(3-хлорпропокси)-5-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноат
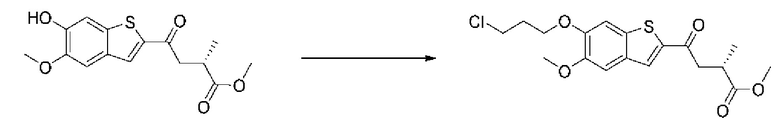
Смесь (S)-метил 4-(6-гидрокси-5-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата (141 мг, 0.457 ммоль), 1-бром-3-хлорпропана (360 мг, 2.3 ммоль) и K2CO3 (379 мг, 2.74 ммоль) дегазировали Ar. К смеси добавляли ACN (4.0 мл) и реакционную смесь нагревали до 50°С в течение 18 ч. После охлаждения до комнатной температуры реакционную смесь затем разбавляли DCM (25 мл) и фильтровали. Фильтрат концентрировали под пониженным давлением и затем очищали с помощью хроматографии на силикагеле (0→100% градиент EtOAc в DCM) с получением (S)-метил 4-(6-(3-хлорпропокси)-5-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата. ЖХ-МС (C18H22ClO5S) (ES, m/z): 385 [М+Н]+.
Промежуточное соединение 43, как показано в Таблице 7 ниже, или может быть получено в соответствии со способами, аналогичными описанным в Промежуточном соединении 42 выше, с применением соответствующих исходных материалов, описанных в способах получения, или получено из коммерческих источников.

Промежуточное соединение 44: метил (2S)-4-[5-(2-гидроксиэтил)-6-метокси-1-бензотиофен-2-ил]-2-метил-4-оксобутаноат
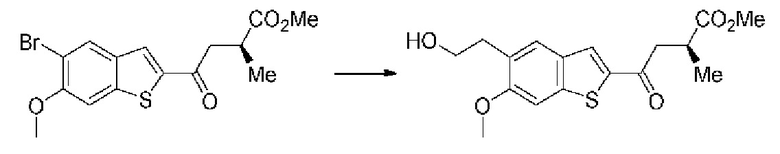
К перемешиваемой смеси метил (2S)-4-(5-бром-6-метокси-1-бензотиофен-2-ил)-2-метил-4-оксобутаноата (1.24 г, 3.34 ммоль), трис(триметилсилил)силана (1.0 мл, 3.3 ммоль) и безводного Na2CO3 (708 мг, 6.68 ммоль) в дегазированном DME (16.5 мл) в атмосфере N2 добавляли смесь Ir(2-(2,4-дифторфенил)-5-(трифторметил)пиридин)2 (4,4'-ди-трет-бутил-2,2'-бипиридин)PF6 (38 мг, 33 мкмоль) в дегазированном DME (12 мл). Добавляли суспензию комплекса никель (II) хлорид этиленгликоль диметиловый эфир (3.7 мг, 17 мкмоль) и 4,4'-ди-трет-бутил-2,2'-бипиридина (4.5 мг, 17 мкмоль) в дегазированном DME (4.5 мл) и полученную в результате смесь перемешивали в атмосфере N2 в течение 15 мин при комнатной температуре. 2-Бромэтанол (835 мг, 6.68 ммоль) добавляли одной порцией в атмосфере N2, и реакционную смесь перемешивали и облучали в фотореакторе с интенсивностью света 20% в течение 24 ч при комнатной температуре. Смесь затем фильтровали, и фильтрат концентрировали под пониженным давлением. Остаток очищали с помощью колоночной флэш-хроматографии на силикагеле (EtOAc в гексанах) с получением метил (2S)-4-[5-(2-гидроксиэтил)-6-метокси-1-бензотиофен-2-ил]-2-метил-4-оксобутаноата. ЖХ-МС (C17H21O5S) (ES, m/z): 337 [М+Н]+. 1H ЯМР (500 МГц, DMSO-d6): δ 8.26 (s, 1H), 7.76 (s, 1H), 7.58 (s, 1H), 4.64 (t, J=5.2 Гц, 1H), 3.88 (s, 3H), 3.64-3.55 (m, 2H), 3.59 (s, 3H), 3.42 (dd, J=17.5, 8.6 Гц, 1H), 3.19 (dd, J=17.5, 5.0 Гц, 1H), 3.01-2.92 (m, 1H), 2.81 (t, J=6.9 Гц, 2H), 1.19 (d, J=7.1 Гц, 3Н).
Промежуточное соединение 45: трет-бутил 4-(6-метокси-5-(3-оксопропил)бензо[b]тиофен-2-ил)-4-оксобутаноат
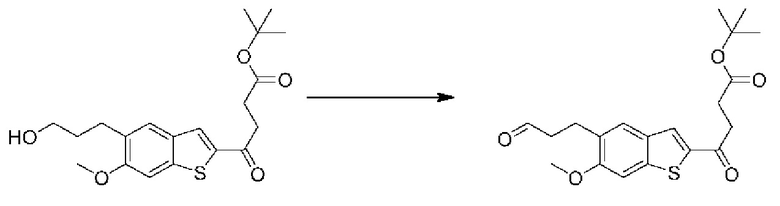
В 4 мл виалу добавляли трет-бутил 4-(5-(3-гидроксипропил)-6-метоксибензо[b]тиофен-2-ил)-4-оксобутаноат (30 мг, 0.079 ммоль), периодинан Десса-Мартина (1,1,1-триацетокси-1,1-дигидро-1,2-бензодиоксол-3(1Н)-он; 50 мг, 0.1 ммоль) и DCM (1.0 мл). Смесь перемешивали при комнатной температуре в течение 1 ч. Смесь очищали с помощью хроматографии на силикагеле (0→55% градиент EtOAc в гексанах) с получением трет-бутил 4-(6-метокси-5-(3-оксопропил)бензо[b]тиофен-2-ил)-4-оксобутаноата. ЖХ-МС (C20H24O5SNa) (ES, m/z): 399 [M+Na]+. 1H ЯМР (500 МГц, CDCl3) δ 9.85 (s, 1H), 7.89 (s, 1H), 7.64 (s, 1H), 3.27 (t, J=6.8 Гц, 2H), 3.05 (t, J=7.4 Гц, 2H), 2.80 (t, J=7.2 Гц, 2H), 2.72 (t, J=6.7 Гц, 2H), 1.46 (s, 9H).
Промежуточное соединение 46: трет-бутил 4-(5-амино-6-метокситиено[3,2-b]пиридин-2-ил)-4-оксобутаноат
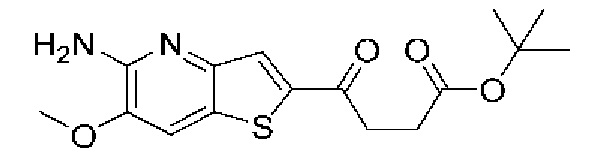
Стадия 1: трет-бутил 4-(5-((дифенилметилен)амино)-6-метокситиено[3,2-b]пиридин-2-ил)-4-оксобутаноат

В 4 мл виалу добавляли Cs2CO3 (252 мг, 0.773 ммоль), дифенилметанимин (129 мкл, 0.773 ммоль), трет-бутил 4-(5-хлор-6-метокситиено[3,2-b]пиридин-2-ил)-4-оксобутаноат (138 мг, 0.387 ммоль), Rac BINAP Pd G3 (19 мг, 0.019 ммоль) и толуол (2.0 мл). Смесь нагревали до 120°С в течение 3.5 ч. После охлаждения до комнатной температуры смесь сразу очищали с помощью хроматографии на силикагеле (0→50% градиент EtOAc в гексанах) с получением трет-бутил 4-(5-((дифенилметилен)амино)-6-метокситиено[3,2-b]пиридин-2-ил)-4-оксобутаноата. ЖХ-МС (C29H29N2O4S) (ES, m/z): 501 [М+Н]+.
Стадия 2: трет-бутил 4-(5-амино-6-метокситиено[3,2-b]пиридин-2-ил)-4-оксобутаноат

В 20 мл виалу добавляли трет-бутил 4-(5-((дифенилметилен)амино)-6-метокситиено[3,2-b]пиридин-2-ил)-4-оксобутаноат (146 мг, 0.291 ммоль), АсОН (2.0 мл), МеОН (2.0 мл), воду (2.0 мл) и THF (6.0 мл). Смесь нагревали до 50°С в течение 1 ч. После охлаждения до комнатной температуры смесь затем оставляли перемешиваться при комнатной температуре в течение 24 ч. Смесь затем разбавляли EtOAc (50 мл) и промывали водным насыщенным NaHCO3 (50 мл) и водой (50 мл). Органический слой высушивали над Na2SO4, фильтровали и концентрировали под пониженным давлением. Полученный в результате остаток очищали с помощью хроматографии на силикагеле (0→50% градиент EtOAc в гексанах) с получением трет-бутил 4-(5-амино-6-метокситиено[3,2-b]пиридин-2-ил)-4-оксобутаноата. ЖХ-МС (C16H21N2O4S) (ES, m/z): 337 [М+Н]+. 1H ЯМР (500 МГц, CDCl3) δ 8.02 (s, 1H), 7.53 (s, 1H), 4.11 (s, 3Н), 3.24 (t, J=6.4 Гц, 2Н), 2.74-2.70 (m, 2Н), 1.46 (s, 9Н).
Промежуточное соединение 47: Метил (S)-4-(6-(3-гидроксипропокси)-5-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноат
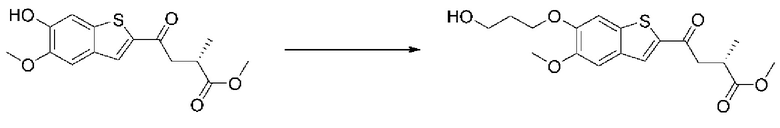
Смесь (S)-метил 4-(6-гидрокси-5-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата (25 мг, 0.081 ммоль), 3-хлорпропан-1-ола (10 мг, 0.1 ммоль) и K2CO3 (22 мг, 0.16 ммоль) дегазировали Ar. К смеси добавляли DMF (0.50 мл), и реакционную смесь облучали в микроволновом реакторе до 100°С в течение 1 ч. Реакционную смесь оставляли охлаждаться до комнатной температуры и затем разбавляли EtOAc (25 мл) и водой (5 мл). Органический слой отделяли, промывали дополнительным объемом воды (5 мл) и затем насыщенным водным NaCl (5 мл), высушивали над MgSO4, фильтровали и концентрировали под пониженным давлением с получением неочищенного остатка. Остаток сырого вещества очищали с помощью хроматографии на силикагеле (элюируя 0-100% [5% МеОН в EtOAc] в DCM) с получением (S)-метил 4-(6-(3-гидроксипропокси)-5-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата. ЖХ-МС (C18H23O6S) (ES, m/z): 367 [М+Н]+.
Промежуточное соединение 48, как показано в Таблице 8 ниже, или может быть получено в соответствии со способами, аналогичными описанным в Промежуточном соединении 47 выше, с применением соответствующих исходных материалов, описанных в способах получения, или получено из коммерческих источников.
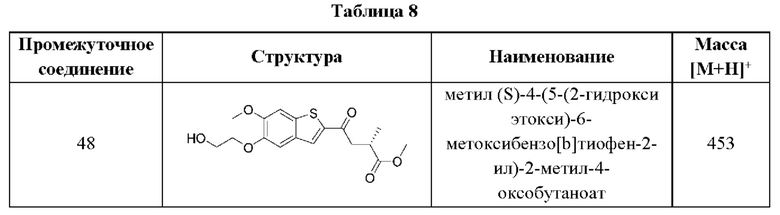
Промежуточное соединение 49: трет-бутил 4-(5-гидрокси-6-метоксибензо[b]тиофен-2-ил)-4-оксобутаноат
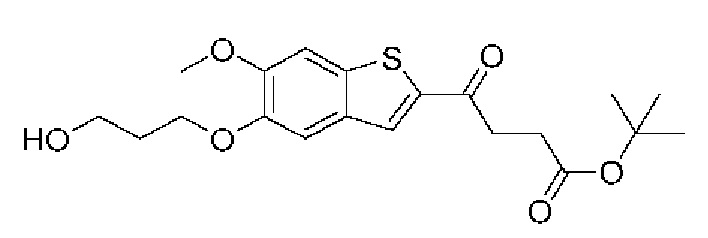
Стадия 1: трет-бутил 4-(6-метокси-5-(3-((тетрагидро-2Н-пиран-2-ил)окси)пропокси)бензо[b]тиофен-2-ил)-4-оксобутаноат
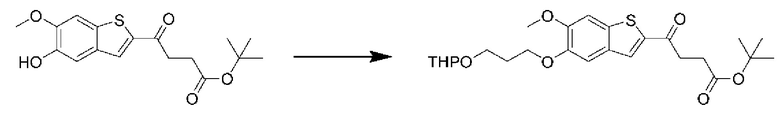
К смеси трет-бутил 4-(5-гидрокси-6-метоксибензо[b]тиофен-2-ил)-4-оксобутаноата (3.0 г, 8.9 ммоль), K2CO3 (7.4 г, 54 ммоль) и ACN (20 мл) добавляли 2-(3-бромпропокси)тетрагидро-2Н-пиран (3.8 мл, 22 ммоль). Реакцию нагревали до 65°С в течение 4 ч. После охлаждения до комнатной температуры смесь фильтровали, промывали ACN, и затем растворитель удаляли под пониженным давлением. Полученный в результате остаток растворяли в DCM (10 мл) и медленно добавляли гексан (100 мл). Полученный в результате осадок отфильтровывали, промывали гексаном и высушивали на воздухе с получением трет-бутил 4-(6-метокси-5-(3-((тетрагидро-2Н-пиран-2-ил)окси)пропокси)бензо[b]тиофен-2-ил)-4-оксобутаноата. ЖХ-МС (C25H34O7SNa) (ES, m/z): 501 [M+Na]+. 1Н ЯМР (600 МГц, DMSO-d6) δ 8.16 (s, 1H), 7.56 (s, 1H), 7.46 (s, 1H), 4.54 (s, 1H), 4.06 (t, J=6.2 Гц, 2H), 3.83 (s, 3H), 3.78 (dt, J=9.6, 6.4 Гц, 1H), 3.69 (dd, J=13.5, 5.5 Гц, 1H), 3.52-3.45 (m, 1H), 3.41-3.35 (m, 1H), 3.20 (t, J=6.2 Гц, 2H), 2.54 (t, J=6.2 Гц, 2H), 2.02-1.95 (m, 2H), 1.71-1.62 (m, 1H), 1.58 (t, J=7.8 Гц, 1H), 1.47-1.39 (m, 3H), 1.36-1.33 (m, 10H).
Стадия 2: трет-бутил 4-(5-гидрокси-6-метоксибензо[b]тиофен-2-ил)-4-оксобутаноат
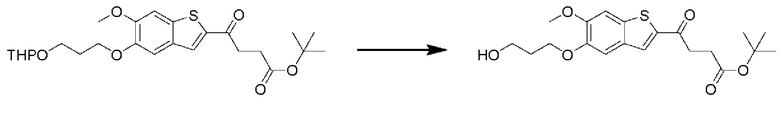
К смеси трет-бутил 4-(6-метокси-5-(3-((тетрагидро-2Н-пиран-2-ил)окси)пропокси)бензо[b]тиофен-2-ил)-4-оксобутаноата (3.78 г, 7.90 ммоль) и EtOH (50 мл) добавляли pTsOH (2.2 г, 12 ммоль), и смесь оставляли перемешиваться при комнатной температуре в течение 2 ч. Смесь затем разбавляли DCM и останавливали водным насыщенным ИаНСОз. Органический слой затем отделяли, высушивали над Na2SO4, фильтровали и концентрировали под пониженным давлением с получением трет-бутил 4-(5-гидрокси-6-метоксибензо[b]тиофен-2-ил)-4-оксобутаноата. ЖХ-МС (C20H26O6SNa) (ES, m/z): 417 [M+Na]+. 1H ЯМР (600 МГц, DMSO-d6) δ 8.15 (s, 1H), 7.55 (s, 1H), 7.44 (s, 1H), 4.53 (t, J=4.7 Гц, 1H), 4.05 (t, J=6.2 Гц, 2H), 3.83 (s, 3H), 3.55 (q, J=5.4 Гц, 2H), 3.20 (t, J=6.l Гц, 2H), 2.54 (t, J=6.1 Гц, 2H), 1.88 (p, J=5.8 Гц, 2H), 1.33 (s, 9H).
Промежуточное соединение 50, как показано в Таблице 9 ниже, или может быть получено в соответствии со способами, аналогичными описанным в Промежуточном соединении 49 выше, с применением соответствующих исходных материалов, описанных в способах получения, или получено из коммерческих источников.
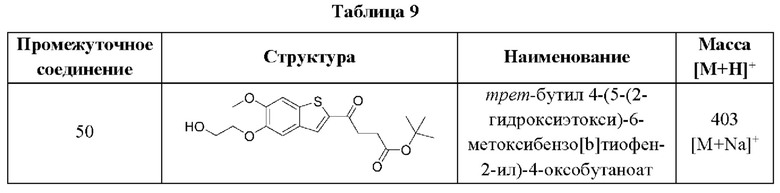
Промежуточное соединение 51: Метил 4-(5-гидроксибензо[b]тиофен-2-ил)-4-оксобутаноат
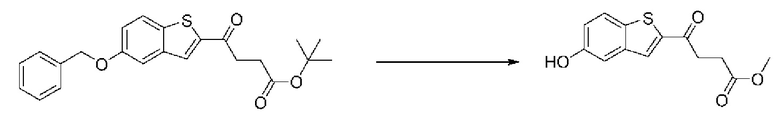
Смесь трет-бутил 4-(5-(бензилокси)бензо[b]тиофен-2-ил)-4-оксобутаноата (1.43 г, 3.60 ммоль) и Pd/C (10% масс/масс, 1.5 г, 1.4 ммоль) дегазировали Ar. К смеси медленно добавляли МеОН (25 мл), EtOAc (25 мл) и HCl (37% в воде, 0.59 мл, 7.2 ммоль) в атмосфере потока Ar. Полученную реакционную смесь дегазировали с помощью вакуума и снова заполняли H2. Полученную в результате смесь перемешивали в атмосфере H2 в течение 24 ч. Реакционную смесь затем фильтровали через целит, и целит промывали EtOAc. Фильтрат концентрировали под пониженным давлением с получением неочищенного остатка. Остаток сырого вещества очищали с помощью хроматографии на силикагеле (0→100% градиент EtOAc в гексанах) с получением метил 4-(5-гидроксибензо[b]тиофен-2-ил)-4-оксобутаноата. ЖХ-МС (C13H13O4S) (ES, m/z): 265 [М+Н]+. 1Н ЯМР (500 МГц, DMSO-d6) δ 9.74 (s, 1Н), 8.28 (s, 1H), 7.85 (d, J=8.5 Гц, 1H), 7.36 (s, 1H), 7.07 (d, J=8.0 Гц, 1H), 3.63 (s, 3H), 2.75-2.67 (m, 2H); 2 алифатических протона закрыты пиком растворителя и не видны.
Промежуточное соединение 52: метил (2S)-4-(5,6-дигидрокси-1-бензотиофен-2-ил)-2-метил-4-оксобутаноат
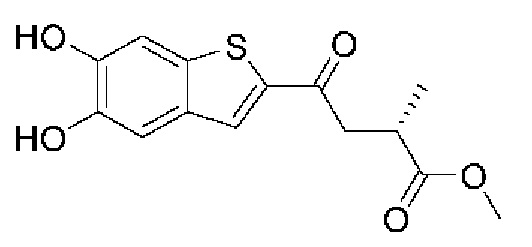
Стадия 1: (2S)-4-(5,6-дигидрокси-1-бензотиофен-2-ил)-2-метил-4-оксобутановая кислота
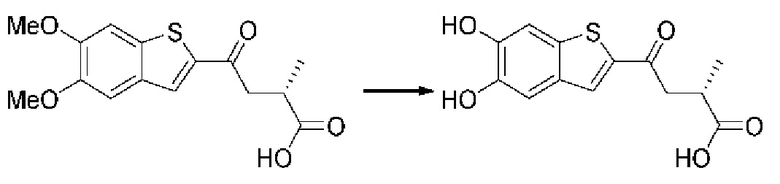
К перемешанному раствору (2S)-4-(5,6-диметокси-1-бензотиофен-2-ил)-2-метил-4-оксобутановой кислоты (2.0 г, 6.5 ммоль) в DCM (65 мл) добавляли BBr3 (1 M в DCM, 19.5 мл, 19.5 ммоль) при 0°С. Реакционную смесь оставляли нагреваться до комнатной температуры в течение 2.5 ч. Смесь затем охлаждали до 0°С, обрабатывали водой и концентрировали под пониженным давлением. Остаток фильтровали, промывали водой и высушивали под высоким вакуумом с получением (2S)-4-(5,6-дигидрокси-1-бензотиофен-2-ил)-2-метил-4-оксобутановой кислоты, которую применяли без дополнительной очистки. ЖХ-МС (C13H13O5S) ES, m/z): 281 [М+Н]+. 1Н ЯМР (500 МГц, DMSO-d6) δ 12.18 (s, 1H), 9.78 (brs, 1H), 9.49 (brs, 1H), 8.12 (s, 1H), 7.28 (s, 1H), 7.24 (s, 1H), 3.39-3.33 (m, 1H), 3.03 (dd, J=17.2, 5.2 Гц, 1H), 2.91-2.84 (m, 1H), 1.16 (d, J=7.1 Гц, 3Н).
Стадия 2: метил (2S)-4-(5,6-дигидрокси-1-бензотиофен-2-ил)-2-метил-4-оксобутаноат

К перемешанному раствору (2S)-4-(5,6-дигидрокси-1-бензотиофен-2-ил)-2-метил-4-оксобутановой кислоты (1.35 г, 4.82 ммоль) в DCM (24 мл) и МеОН (24 мл) добавляли TMS-диазометан (2 M в гексанах, 3.6 мл, 7.2 ммоль). Смесь оставляли перемешиваться в течение 30 мин, обрабатывали НОАс (0.28 мл, 4.8 ммоль) и концентрировали под пониженным давлением. Остаток очищали с помощью хроматографии на силикагеле (0→10% градиент МеОН в DCM) с получением метил (2S)-4-(5,6-дигидрокси-1-бензотиофен-2-ил)-2-метил-4-оксобутаноата. ЖХ-МС (C14H15O5S) (ES, m/z): 295 [М+Н]+. 1H ЯМР (500 МГц, DMSO-d6) δ 9.85 (s, 1H), 9.45 (s, 1H), 8.13 (s, 1H), 7.28 (s, 1H), 7.25 (s, 1H), 3.59 (s, 3H), 3.41-3.34 (m, 1H), 3.17-3.11 (m, 1H), 2.98-2.91 (m, 1H), 1.17 (d, J=7.1 Гц, 3Н).
Промежуточное соединение 53: Метил (2S)-4-(5,6-бис(3-гидроксипропокси)-1-бензотиофен-2-ил]-2-метил-4-оксобутаноат
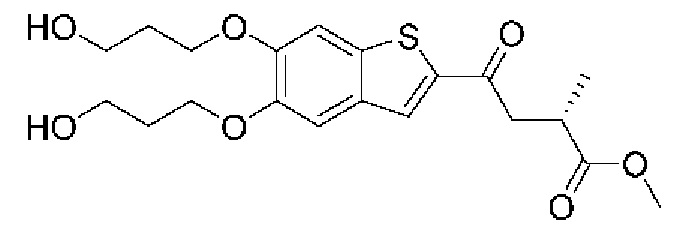
Смесь метил (2S)-4-(5,6-дигидрокси-1-бензотиофен-2-ил)-2-метил-4-оксобутаноата (230 мг, 0.78 ммоль), 3-хлор-1-пропанола (196 мкл, 2.34 ммоль) и K2CO3 (432 мг, 3.13 ммоль) в DMF (7.8 мл) облучали в микроволновом реакторе до 100°С в течение 1 ч. После охлаждения до комнатной температуры смесь разбавляли EtOAc и насыщенным водным NaCl. Органический слой отделяли, высушивали над Na2SO4, фильтровали и затем концентрировали под пониженным давлением. Полученный в результате остаток очищали с помощью флэш-хроматографии (0→10% градиент МеОН в DCM) с получением метил (2S)-4-[5,6-бис(3-гидроксипропокси)-1-бензотиофен-2-ил]-2-метил-4-оксобутаноата. ЖХ-МС (C20H27O7S) (ES, m/z): 411 [М+Н]+
Промежуточное соединение 54: метил (2S)-4-[6-(дифторметокси)-5-гидрокси-1-бензотиофен-2-ил]-2-метил-4-оксобутаноат
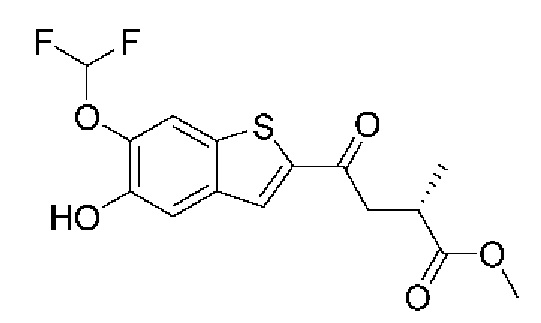
К замороженной смеси метил (2S)-4-(5,6-дигидрокси-1-бензотиофен-2-ил)-2-метил-4-оксобутаноата (177 мг, 0.601 ммоль) и KOH (1.0 М в воде, 120 мкл, 1.2 ммоль) в ACN (5.5 мл) и воде (0.55 мл) при -78°С добавляли диэтил(бромдифторметил)фосфонат(170 мкл, 0.96 ммоль). Охлаждающую баню удаляли, и реакционную смесь оставляли перемешиваться в течение 4 ч. Смесь разбавляли EtOAc и водой. Слои разделяли, и водный слой повторно экстрагировали EtOAc (х3). Объединенные органические вещества высушивали над Na2SO4, фильтровали и затем концентрировали под пониженным давлением. Полученную в результате смесь очищали с помощью флэш-хроматографии (0-50% EtOAc в DCM) с получением неочищенного метил (2S)-4-[6-(дифторметокси)-5-гидрокси-1-бензотиофен-2-ил]-2-метил-4-оксобутаноата. Целевые фракции очищали с помощью SFC (15% МеОН (+0.25% DMEA) в СО2) с получением метил (2S)-4-[6-(дифторметокси)-5-гидрокси-1-бензотиофен-2-ил]-2-метил-4-оксобутаноата, время удерживания 4.4 мин. ЖХ-МС (C15H15F2O5S) (ES, m/z): 345 [М+Н]+. 1H ЯМР (500 МГц, DMSO-d6) δ 10.25 (s, 1H), 8.28 (s, 1H), 7.83 (s, 1H), 7.50 (s, 1H), 7.18 (t, J=74.4 Гц, 1H), 3.59 (s, 3H), 3.44 (dd, J=17.6, 8.7 Гц, 1H), 3.20 (dd, J=17.6, 4.9 Гц, 1H), 3.01-2.94 (m, 1H), 1.19(d, J=7.1 Гц, 3Н).
Промежуточное соединение 55: Метил 4-(4-фтор-5-гидрокси-6-метоксибензо[b]тиофен-2-ил)-2,2-диметил-4-оксобутаноат
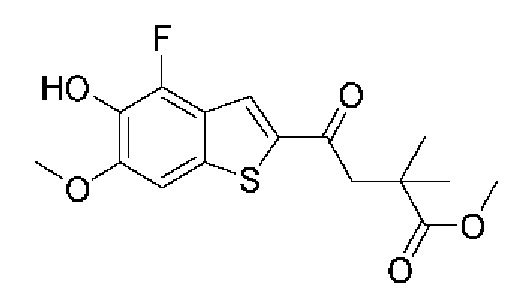
Стадия 1: 4-(4-фтор-5,6-диметоксибензо[b]тиофен-2-ил)-2,2-диметил-4-оксобутановая кислота
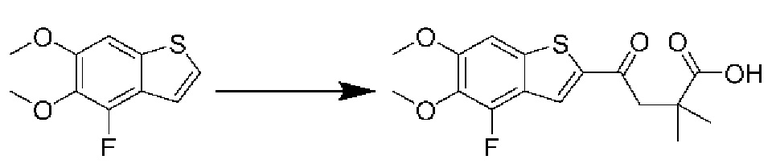
3,3-Диметилдигидрофуран-2,5-дион (4.6 г, 36 ммоль) добавляли к смеси 4-фтор-5,6-диметоксибензо[b]тиофена (3.8 г, 18 ммоль) и AlCh (3.1 г, 23 ммоль) в DCM (100 мл) при 0°С в атмосфере Ar. Реакционную смесь перемешивали при 0°С в течение 2 ч и затем нагревали до комнатной температуры и перемешивали в течение 16 ч. Реакцию затем останавливали медленным добавлением реакционной смеси к смеси воды (200 мл) и EtOAc (500 мл) при 0°С. Полученную в результате смесь перемешивали в течение 1 ч при 20°С и затем разбавляли HCl (2.0 М в воде, 36 мл, 72 ммоль). Органический слой отделяли, промывали насыщенным водным NaCl (50 мл), высушивали над MgSO4, фильтровали и концентрировали под пониженным давлением. Смесь затем очищали с помощью хроматографии на силикагеле с получением 4-(4-фтор-5,6-диметоксибензо[b]тиофен-2-ил)-2,2-диметил-4-оксобутановой кислоты. ЖХ-МС (C16H18FO5S) (ES, m/z): 341 [М+Н]+.
Стадия 2: метил 4-(4-фтор-5,6-диметоксибензо[b]тиофен-2-ил)-2,2-диметил-4-оксобутаноат
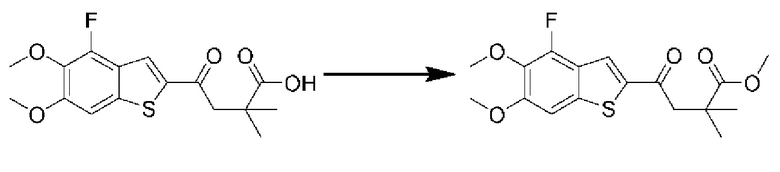
К смеси 4-(4-фтор-5,6-диметоксибензо[b]тиофен-2-ил)-2,2-диметил-4-оксобутановой кислоты (2.3 г, 6.8 ммоль) в DMF (45 мл) добавляли K2CO3 (2.3 г, 17 ммоль). Через 10 мин добавляли CH3I (2.1 мл, 34 ммоль), и смесь перемешивали в течение 18 ч при комнатной температуре. Смесь затем разбавляли водой и Et2O. Органический слой отделяли, высушивали над MgSO4, фильтровали и концентрировали под пониженным давлением. Смесь затем очищали с помощью хроматографии на силикагеле с получением метил 4-(4-фтор-5,6-диметоксибензо[b]тиофен-2-ил)-2,2-диметил-4-оксобутаноата. ЖХ-МС (C17H20FO5S) (ES, m/z): 355 [М+Н]+. 1H ЯМР (500 МГц, DMSO-d6) δ 8.32 (s, 1H), 7.59 (s, 1Н), 3.92 (s, 3Н), 3.85 (s, 3Н), 3.57 (s, 3Н), 3.44 (s, 2H), 1.23 (s, 6H).
Стадия 3: метил 4-(4-фтор-5-гидрокси-6-метоксибензо[b]тиофен-2-ил)-2,2-диметил-4-оксобутаноат
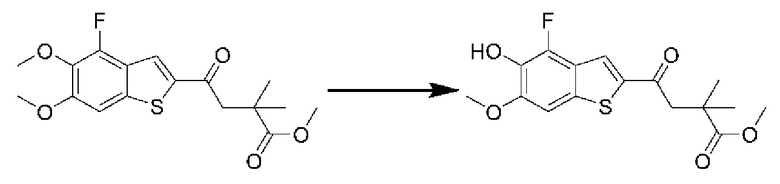
К смеси метил 4-(4-фтор-5,6-диметоксибензо[b]тиофен-2-ил)-2,2-диметил-4-оксобутаноата (1.1 г, 3.1 ммоль) и DCM (20 мл) добавляли AlCl3 (1.7 г, 12 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 18 ч. Реакционную смесь выливали в колбу, содержащую лед и 1 н. HCl, и перемешивали в течение 5 мин. Затем добавляли EtOAc. Органический слой отделяли, высушивали над MgSO4, фильтровали и концентрировали под пониженным давлением. Смесь затем очищали с помощью хроматографии на силикагеле с получением метил 4-(4-фтор-5-гидрокси-6-метоксибензо[b]тиофен-2-ил)-2,2-диметил-4-оксобутаноата. ЖХ-МС (C16H18FO5S) (ES, m/z): 341 [М+Н]+. 1H ЯМР (500 МГц, DMSO-d6) δ 9.55 (s, 1H), 8.24 (s, 1H), 7.47 (s, 1H), 3.91 (s, 3Н), 3.57 (s, 3Н), 3.43 (s, 2H), 1.23 (s, 6H).
Промежуточное соединение 56: трет-Бутил (1S,2R и 1R,2S)-2-(5-гидрокси-6-метоксибензо[b]тиофен-2-карбонил)циклопропан-1-карбоксилат
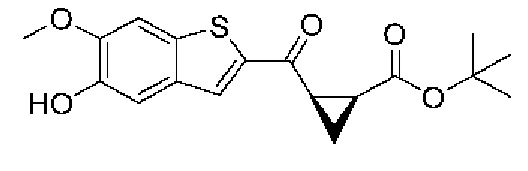
Стадия 1: (1S,2R и 1R,2S)-2-(5-буом-6-метоксибензо[b]тиофен-2-карбонил)циклопропан-1-карбоновая кислота
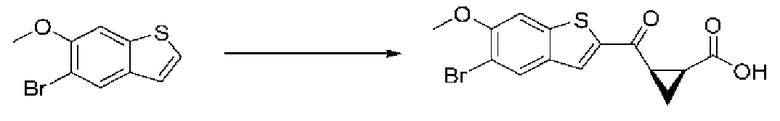
3-Оксабицикло[3.1.0]гексан-2,4-дион (3.78 г, 33.7 ммоль) добавляли к смеси 5-бром-6-метоксибензо[b]тиофена (4.1 г, 17 ммоль) и AlCl3 (2.92 г, 21.9 ммоль) в DCM (100 мл) при 0°С в атмосфере Ar. Реакционную смесь перемешивали при 0°С в течение 2 ч и затем оставляли нагреваться до комнатной температуры. Реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Реакцию затем останавливали медленным добавлением реакционной смеси к смеси воды (200 мл) и EtOAc (500 мл) при 0°С. Полученную в результате смесь оставляли нагреваться до комнатной температуры и перемешивали в течение 1 ч. Смесь затем разбавляли HCl (2.0 М в воде, 34 мл, 68 ммоль). Органический слой отделяли, промывали насыщенным водным NaCl (50 мл), высушивали над MgSO4, фильтровали и концентрировали под пониженным давлением с получением неочищенного остатка. Остаток сырого вещества очищали с помощью хроматографии на силикагеле (элюируя 0-100% [5% МеОН в EtOAc] в DCM) с получением (1S,2R и 1R,2S)-2-(5-бром-6-метоксибензо[b]тиофен-2-карбонил)циклопропанкарбоновой кислоты. ЖХ-МС (C14H12BrO4S) (ES, m/z): 355, 357 [М+Н]+.
Стадия 2: трет-бутил (1S,2R и 1R,2S)-2-(5-бром-6-метоксибензо[b]тиофен-2-каубонил)циклопропан-1-карбоксилат

трет-Бутанол (25.8 мл, 270 ммоль) добавляли к смеси ВОС-ангидрида (6.3 мл, 27 ммоль), DMAP (0.33 г, 2.7 ммоль) и (1S,2R и 1R,2S)-2-(5-бром-6-метоксибензо[b]тиофен-2-карбонил)циклопропанкарбоновой кислоты (4.8 г, 14 ммоль) в DCE (30 мл) при комнатной температуре в атмосфере Ar. Реакционную смесь перемешивали и нагревали до 50°С в течение 2 ч. Реакционную смесь затем охлаждали до комнатной температуры и концентрировали под пониженным давлением с получением неочищенного остатка. Остаток сырого вещества очищали с помощью хроматографии на силикагеле (0→100% градиент EtOAc в гексанах) с получением (1S,2R и 1R,2S)-трет-бутил 2-(5-бром-6-метоксибензо[b]тиофен-2-карбонил)циклопропанкарбоксилата. ЖХ-МС (C18H20BrO4S-С4Н8) (ES, m/z): 355, 357 [M+H-tBu]+. 1Н ЯМР (500 МГц, DMSO-d6) δ 8.33 (s, 1H), 8.26 (s, 1H), 7.81 (s, 1H), 3.95 (s, 3Н), 3.10-3.03 (m, 1H), 2.32-2.25 (m, 1H), 1.57-1.52 (m, 1H), 1.32-1.28 (m, 1H), 1.14 (s, 9H).
Стадия 3: трет-бутил (1S,2R и 1R,2S)-2-(5-гидрокси-6-метоксибензо[b]тиофен-2-карбопил)циклопропан-1-карбоксилат

Смесь (1S,2R и 1R,2S)-трет-бутил 2-(5-бром-6-метоксибензо[b]тиофен-2-карбонил)циклопропанкарбоксилата (1.64 г, 3.99 ммоль), RockPhos Pd G3 (0.100 г, 0.120 ммоль), Cs2CO3 (3.90 г, 12.0 ммоль) и (E)-бензальдегидоксима (0.725 г, 5.98 ммоль) дегазировали Ar в течение 5 мин. Добавляли DMF (10.0 мл) в атмосфере Ar, и смесь затем дегазировали Ar в течение 5 мин. Реакционную смесь перемешивали и нагревали до 80°С в течение 18 ч. Реакционную смесь охлаждали до комнатной температуры и разбавляли водой (100 мл) и EtOAc (250 мл). Органический слой отделяли, промывали водой (50 мл) и затем насыщенным водным NaCl (50 мл). Органический слой отделяли, высушивали над MgSO4, фильтровали и концентрировали под пониженным давлением с получением неочищенного остатка. Остаток сырого вещества очищали с помощью хроматографии на силикагеле (0→75% градиент EtOAc в гексанах) с получением (1S,2R и 1R,2S)-трет-бутил 2-(5-гидрокси-6-метоксибензо[b]тиофен-2-карбонил)циклопропанкарбоксилата. ЖХ-МС (C18H20O5SNa) (ES, m/z): 371 [M+Na]+.
Промежуточное соединение 57: (S)-Метил-4-(5-амино-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноатоксилат
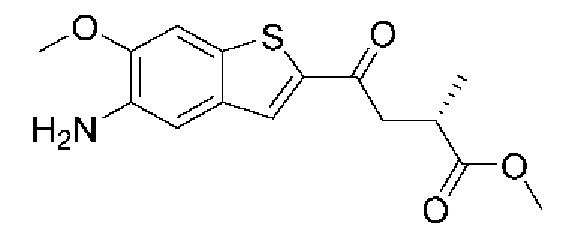
Стадия 1: метил (S)-4-(5-((дифенилметилен)амино)-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноат
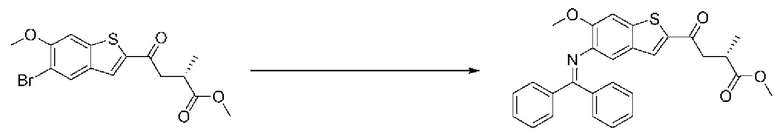
Смесь (S)-метил 4-(5-бром-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата (0.50 г, 1.4 ммоль), бензофенонимина (0.45 мл, 2.7 ммоль), Rac BINAP Pd G3 (0.067 г, 0.067 ммоль) и Cs2CO3 (0.878 г, 2.69 ммоль) дегазировали Ar в течение 5 мин. Добавляли толуол (10.0 мл) при 20°С в атмосфере Ar, и смесь затем дегазировали Ar в течение 5 мин. Реакционную смесь перемешивали и нагревали до 110°С в течение 18 ч в атмосфере Ar. Реакционную смесь затем охлаждали до комнатной температуры и разбавляли DCM (25 мл). Смесь фильтровали, и фильтрат концентрировали под пониженным давлением с получением метил (S)-4-(5-((дифенилметилен)амино)-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата, которую применяли без очистки. ЖХ-МС (C28H26NO4S) (ES, m/z): 472 [М+Н]+.
Стадия 2: (S)-метил 4-(5-амино-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноат
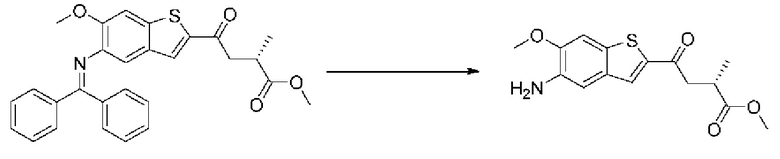
TFA (2.1 мл, 27 ммоль) добавляли к смеси (S)-метил 4-(5-((дифенилметилен)амино)-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата (635 мг, 1.35 ммоль) в DCM (5.0 мл) при 20°С. Полученную в результате смесь перемешивали при 20°С в течение 30 мин. Реакционную смесь затем концентрировали под пониженным давлением с получением неочищенного остатка. Остаток сырого вещества очищали с помощью хроматографии на силикагеле (0→100% градиент EtOAc в DCM) с получением (S)-метил 4-(5-амино-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата. ЖХ-МС (C15H18NO4S) (ES, m/z): 308 [М+Н]+.
Промежуточное соединение 58: трет-Бутил 4-(6-гидроксибензо[b]тиофен-2-ил)-4-оксобутаноат
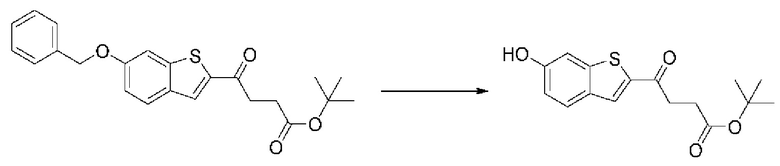
Смесь трет-бутил 4-(6-(бензилокси)бензо[b]тиофен-2-ил)-4-оксобутаноата (256 мг, 0.646 ммоль) и Pd/C (172 мг, 0.161 ммоль) дегазировали Ar. К смеси медленно добавляли EtOAc (5.0 мл), МеОН (5.0 мл) и HCl (37% в воде, 0.053 мл, 0.65 ммоль) в атмосфере Ar. Полученную выше реакционную смесь дегазировали с помощью вакуума и снова заполняли Н2. Полученную в результате смесь перемешивали в атмосфере Н2 в течение 24 ч. Реакционную смесь затем фильтровали через целит, промывая EtOAc. Фильтрат концентрировали под пониженным давлением с получением неочищенного остатка. Остаток сырого вещества очищали с помощью хроматографии на силикагеле (элюируя 0-100% EtOAc в гексанах) с получением трет-бутил 4-(6-гидроксибензо[b]тиофен-2-ил)-4-оксобутаноата. ЖХ-МС (C16H19O4S-C4H8) (ES, m/z): 251 [M+H-tBu]+.
Промежуточное соединение 59: трет-бутил 4-(5-бромбензо[b]тиофен-2-ил)-4-оксобутаноат
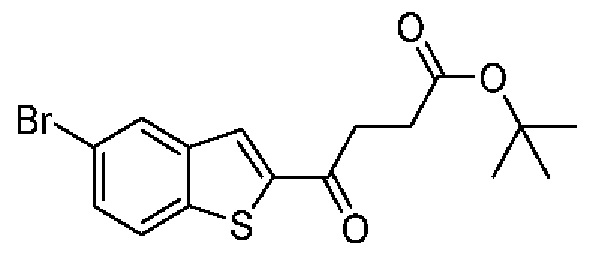
Стадия 1: 5-бромбензо[b]тиофен-2-карбопилхлорид
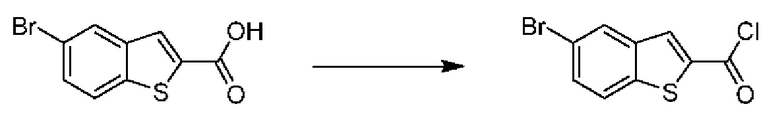
(COCl)2 (0.30 мл, 3.4 ммоль) добавляли по каплям с перемешиванием к смеси 5-бромбензо[b]тиофен-2-карбоновой кислоты (0.750 г, 2.92 ммоль) в CH2Cl2 (15 мл) при 0°С в течение 3 мин. К реакционной смеси добавляли DMF (0.023 мл, 0.29 ммоль) с последующим добавлением дополнительного (COCl)2 (0.20 мл, 2.3 ммоль). Реакционную смесь затем перемешивали при 0°С в течение 1 ч. Реакционную колбу удаляли из бани с ледяной водой, и продолжали перемешивание при комнатной температуре в течение 1.5 ч. Реакционную смесь концентрировали с получением 5-бромбензо[b]тиофен-2-карбонилхлорида, который применяли без дополнительной очистки или определения характеристик.
Стадия 2: трет-бутил 4-(5-бромбензо[b]тиофен-2-ил)-4-оксобутаноат
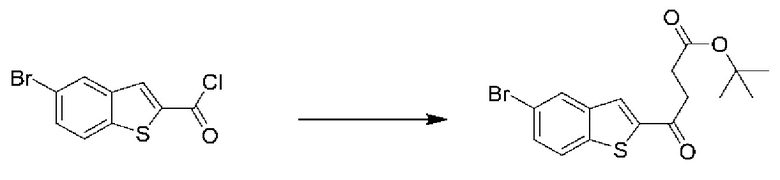
CuCl (0.269 г, 2.72 ммоль) добавляли в 100 мл круглодонную колбу с мешалкой. Колбу три раза откачивали и затем снова заполняли N2. THF (6.0 мл) добавляли в колбу, затем перемешивали и охлаждали до 0°С на бане с ледяной водой. Добавляли по каплям (3-(трет-бутокси)-3-оксопропил)цинк (II) бромид (0.50 М в THF, 11 мл, 5.5 ммоль) с перемешиванием при 0°С в течение 3 мин до смеси с CuCl. Полученную в результате смесь перемешивали при 0°С в течение 30 мин. Добавляли по каплям смесь 5-бромбензо[b]тиофен-2-карбонилхлорида (0.75 г, 2.7 ммоль) в NMP (24 мл) в течение 7 мин при 0°С с перемешиванием. Полученную в результате смесь перемешивали при 0°С в течение 2 ч. Реакционную смесь затем разделяли между изопропилацетатом (300 мл) и 10% водным цитратом натрия (300 мл). Полученную в результате смесь перемешивали в течение 20 мин. Слои разделяли, и водный слой экстрагировали изопропилацетатом (150 мл). Органические слои объединяли, промывали водным NaCl, затем насыщенным водным NaCl, высушивали над безводным MgSO4 и фильтровали, и фильтрат оставляли стоять на протяжении ночи. Фильтрат затем фильтровали и концентрировали с получением сырого остатка. Сырой остаток загружали на колонку с силикагелем и ацетоном. Колонку высушивали продуванием через нее находящегося под давлением N2. Высушенную колонку затем подвергали элюированию с градиентом 0→30% EtOAc с гексаном. Все содержащие продукт фракции собирали, концентрировали и очищали с помощью хроматографии на силикагеле (0→25% градиент EtOAc в гексанах). Содержащие продукт фракции концентрировали и очищали с помощью ахиральной SFC (колонка Phenomenex Biphenyl, 21 мм × 250 мм, 90:10 CO2 : МеОН масс./0.25% DMEA, 70 мл/мин, давление на выходе 100 бар, концентрация загрузки 18.5 мг/мл в MeOH/MeCN, объем вводимой пробы 1.6 мл, детектирование при 215 нм) с получением трет-бутил 4-(5-бромбензо[b]тиофен-2-ил)-4-оксобутаноата. ЖХ-МС (C16H17BrNaO3S) (ES, m/z): 391, 393 [M+Na]+. 1H ЯМР (500 МГц, CDCl3) δ 8.05 (s, 1H), 7.94 (s, 1H), 7.75 (d, J=8.6 Гц, 1H), 7.57 (d, J=8.6 Гц, 1H), 3.31 (t, J=6.6 Гц, 2H), 2.74 (t, J=6.6 Гц, 2H), 1.46 (s, 9H).
Промежуточное соединение 60: трет-Бутил 4-(5-хлор-6-метокситиено[3,2-b]пиридин-2-ил)-4-оксобутаноат
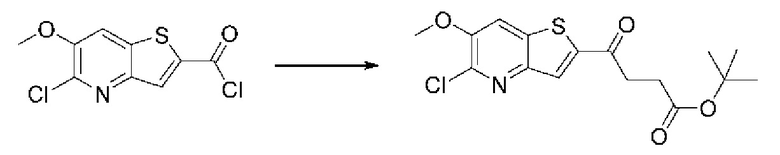
Колбу, содержащую CuCl (0.76 г, 7.6 ммоль) откачивали и затем три раза продували N2. Добавляли THF (15 мл), и смесь охлаждали до 0°С. Добавляли по каплям (3-(трет-бутокси)-3-оксопропил)цинк (II) бромид (0.50 М в THF, 31 мл, 16 ммоль) в течение 10 мин. Через 30 мин добавляли 5-хлор-6-метокситиено[3,2-b]пиридин-2-карбонилхлорид (2.0 г, 7.6 ммоль) с последующим добавлением NMP (15 мл). Смесь затем оставляли нагреваться до комнатной температуры. Через 1 ч смесь охлаждали до 0°С и добавляли концентрированный водный NH4OH (4.5 мл). К этой смеси добавляли воду (240 мл) и МеОН (60 мл). Смесь перемешивали в течение 5 мин и обрабатывали ультразвуком на водяной бане с соникатором. Полученную в результате смесь фильтровали, и выпавшие в осадок вещества промывали водой и затем гексаном. Выпавшие в осадок вещества выделяли и высушивали под вакуумом. Осадок затем разделяли между EtOAc и 10% водным цитратом натрия. Слои разделяли, и водный слой промывали EtOAc. Объединенные органические слои высушивали над Na2SO4, фильтровали и концентрировали под пониженным давлением. Во время концентрирования выпадал осадок, который собирали фильтрацией, промывали EtOAc и высушивали под вакуумом с получением трет-бутил 4-(5-хлор-6-метокситиено[3,2-b]пиридин-2-ил)-4-оксобутаноата. ЖХ-МС (C16H19ClNO4S) (ES, m/z): 356 [М+Н]+. 1Н ЯМР (500 МГц, CDCl3) δ 8.03 (s, 1H), 7.62 (s, 1Н), 4.04 (s, 3Н), 3.30 (t, J=6.4 Гц, 2Н), 2.74 (t, J=6.4 Гц, 2Н), 1.46 (s, 9Н).
Промежуточное соединение 61: метил (S)-4-(2-(3-бромпропил)-4-метокситиено[2',3':5,6]бензо[1,2-d]оксазол-7-ил)-2-метил-4-оксобутаноат
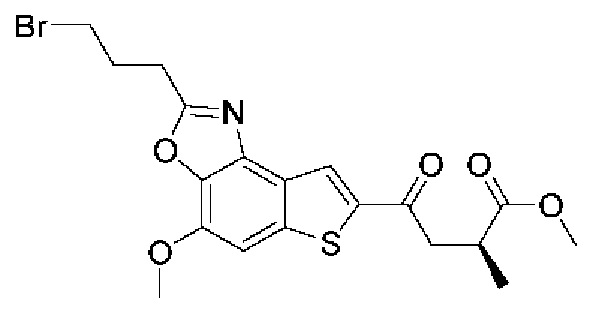
Стадия 1: метил (S)-4-(5-гидрокси-6-метокси-4-нитробензо[b]тиофен-2-ил)-2-метил-4-оксобутаноат

В 20 мл виалу добавляли (S)-метил 4-(5-гидрокси-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноат (100 мг, 0.21 ммоль) и EtOAc (3.0 мл). HNO3 (0.017 мл, 0.27 ммоль) добавляли, и смесь оставляли перемешиваться в течение 1 ч. Через 1 ч смесь концентрировали под пониженным давлением и затем растирали с Et2O (3.0 мл). Полученные в результате вещества собирали путем фильтрования с получением (S)-метил 4-(5-гидрокси-6-метокси-4-нитробензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата. 1Н ЯМР (500 МГц, CDCl3) δ 12.08 (s, 1H), 8.75 (s, 1H), 7.53 (s, 1H), 4.07 (s, 3Н), 3.74 (s, 3Н), 3.59 (dd, J=17.1, 7.9 Гц, 1H), 3.23-3.16 (m, 1H), 3.16-3.09 (m, 1H), 1.35 (d, J=7.1 Гц, 3Н).
Стадия 2: метил (S)-4-(2-(3-бромпропил)-4-метокситиено[2',3':5,6]бензо[1,2-d]оксазол-7-ил)-2-метил-4-оксобутаноат
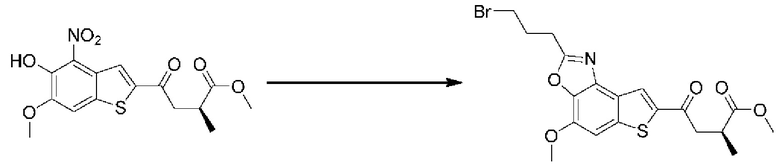
В 4 мл виалу добавляли (S)-метил 4-(5-гидрокси-6-метокси-4-нитробензо[b]тиофен-2-ил)-2-метил-4-оксобутаноат (45 мг, 0.13 ммоль), Pd (0.040 г, 0.038 ммоль) и МеОН (0.60 мл). К смеси добавляли 4-бром-1,1,1-триметоксибутан (0.61 мл, 3.8 ммоль). Смесь перемешивали в атмосфере Н2 в течение 1 ч. Смесь затем фильтровали, и фильтрат концентрировали под пониженным давлением. Полученный в результате остаток очищали с помощью хроматографии на силикагеле (0→70% градиент EtOAc в гексанах) с получением (S)-метил 4-(2-(3-бромпропил)-4-метокситиено[2',3':5,6]бензо[1,2-d]оксазол-7-ил)-2-метил-4-оксобутаноата. ЖХ-МС (C19H21BrNO5S) (ES, m/z): 454, 456 [М+Н]+. 1Н ЯМР (500 МГц, CDCl3) δ 8.36 (s, 1H), 7.25 (s, 1H), 4.11 (s, 3Н), 3.73 (s, 3Н), 3.62 (t, J=6.4 Гц, 2H), 3.55 (dd, J=16.7, 7.5 Гц, 1H), 3.24 (t, J=7.3 Гц, 2H), 3.18 (dd, J=13.5, 6.8 Гц, 1H), 3.12 (dd, J=16.7, 5.8 Гц, 1H), 2.53 (р, J=6.8 Гц, 2Н), 1.32 (d, J=7.0 Гц, 3Н).
Промежуточное соединение 88: Метил (S)-4-(5-бром-6-метоксибензо[b]тиофен-2-ил)-2-метилбутаноат
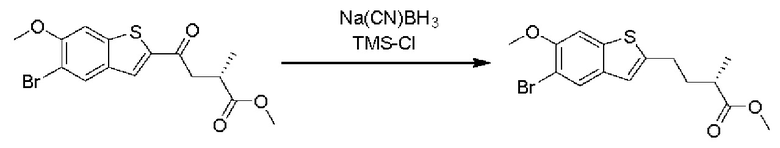
К охлажденному раствору метил (S)-4-(5-бром-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата (0.637 г, 1.72 ммоль) в ACN (15 мл) добавляли TMS-Cl (1.32 мл, 10.3 ммоль). Добавляли цианоборогидрид натрия (0.65 г, 10 ммоль), и реакционную смесь перемешивали при 0°С в течение 2 ч. Реакционную смесь останавливали насыщенным водным NH4Cl и экстрагировали DCM. Органический слой отделяли, высушивали над Na2SO4, фильтровали и концентрировали под пониженным давлением. Остаток очищали с помощью хроматографии на силикагеле (EtOAc/гексаны) с получением метил (S)-4-(бром-6-метоксибензо[b]тиофен-2-ил)-2-метилбутаноата. 1Н ЯМР (600 МГц, DMSO-d6) δ 7.96 (s, 1H), 7.66 (s, 1Н), 7.04 (s, 1H), 3.88 (s, 3Н), 3.61 (s, 3Н), 2.87-2.83 (m, 2Н), 2.57-2.52 (m, 1Н), 2.00-1.94 (m, 1H), 1.82-1.73 (m, 1H), 1.14 (d, J=7.0 Гц, 3Н).
Промежуточные соединения с 89 по 93, как показано в Таблице 10 ниже, или могут быть получены в соответствии со способами, аналогичными описанным в Промежуточном соединении 88 выше, с применением соответствующих исходных материалов, описанных в способах получения, или получены из коммерческих источников.
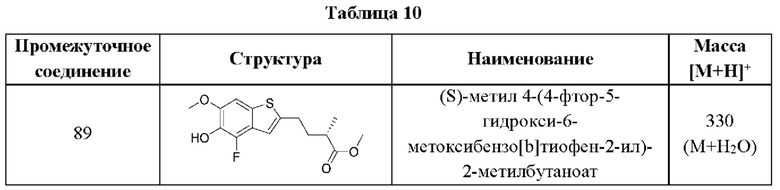

Промежуточное соединение 94: Метил (2S)-4-(5-(3-гидрокси-2-метилпропокси)-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноат
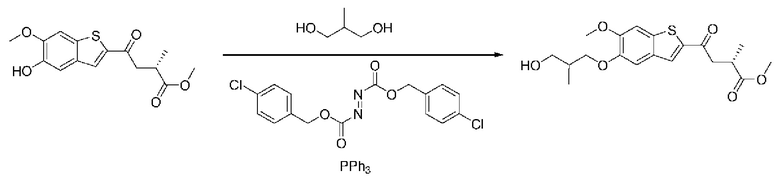
Смесь 2-метилпропан-1,3-диола (146 мг, 1.62 ммоль), (S)-метил 4-(5-гидрокси-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата (50 мг, 0.16 ммоль), (Е)-бис(4-хлорбензил)диазин-1,2-дикарбоксилата (60 мг, 0.16 ммоль) и трифенилфосфина (43 мг, 0.16 ммоль) в NMP (0.30 мл) дегазировали Ar и затем перемешивали и нагревали до 100°С в течение 3 ч. Реакционную смесь охлаждали до комнатной температуры и сразу очищали с помощью ВЭЖХ с обратными фазами (ACN/вода с 0.1% TFA) с получением (2S)-метил 4-(5-(3-гидрокси-2-метилпропокси)-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата. ЖХ-МС (C19H25O6S) (ES, m/z): 381 [М+Н]+.
Промежуточное соединение 95: 4-(5-(3-Бромпропил)-6-метоксибензо[b]тиофен-2-ил)-4-оксобутаннитрил
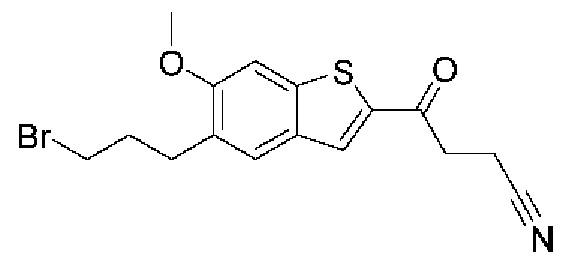
Стадия 1: 4-(5-(3-((трет-бутилдиметилсилил)окси)пропил)-6-метоксибензо[b]тиофен-2-ил)-4-оксобутаннитуил
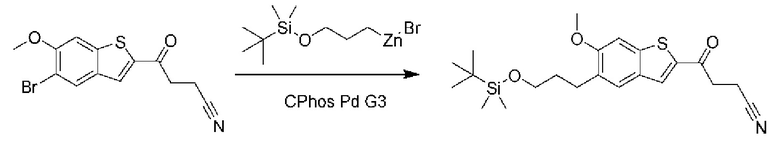
4-(5-Бром-6-метоксибензо[b]тиофен-2-ил)-4-оксобутаннитрил (161 мг, 0.496 ммоль) и CPhos Pd G3 (20 мг, 0.025 ммоль) добавляли к THF (2.5 мл). Реакционную смесь герметично закрывали, и вышеуказанную реакционную смесь откачивали и снова заполняли азотом (3х). К реакционной смеси добавляли 3-(трет-бутилдиметилсилокси)пропилцинк бромид (0.50 М в THF, 3.0 мл, 1.5 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 96 ч. Реакционную смесь разбавляли EtOAc и затем экстрагировали водным NaHCO3. Водный слой отделяли и экстрагировали EtOAc (3х). Органические слои объединяли, промывали рассолом, высушивали над Na2SO4, фильтровали и концентрировали под пониженным давлением с получением 4-(5-(3-((трет-бутилдиметилсилил)окси)пропил)-6-метокси-бензо[b]тиофен-2-ил)-4-оксобутаннитрила. ЖХ-МС (C22H32NO3SSi) (ES, m/z): 418 [М+Н]+.
Стадия 2: 4-(5-(3-гидроксипропил)-6-метоксибензо[b]тиофен-2-ил)-4-оксобутаннитрил
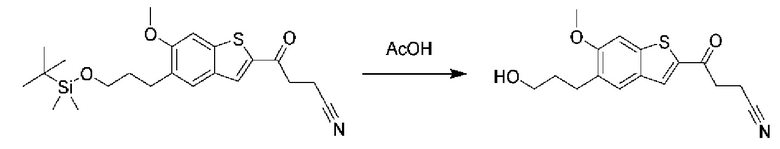
4-(5-(3-((трет-Бутилдиметилсилил)окси)пропил)-6-метоксибензо[b]тиофен-2-ил)-4-оксобутаннитрил (165 мг, 0.395 ммоль) суспендировали в смеси МеОН (2.0 мл), воды (2.0 мл) и НОАс (2.0 мл). Полученную в результате суспензию перемешивали при комнатной температуре в течение 19 ч. Реакционную смесь разбавляли водным NaHCO3 и затем экстрагировали EtOAc (3х). Органические слои объединяли, промывали рассолом, высушивали над Na2SO4, фильтровали и концентрировали под пониженным давлением с получением 4-(5-(3-гидроксипропил)-6-метоксибензо[b]тиофен-2-ил)-4-оксобутаннитрила. ЖХ-МС (C16H18NO3S) (ES, m/z): 304 [М+Н]+. 1Н ЯМР (499 МГц, DMSO-d6) δ 8.27 (s, 1H), 7.75 (s, 1H), 7.61 (s, 1H), 4.50-4.47 (m, 1H), 3.90 (s, 3Н), 3.53-3.40 (m, 4Н), 2.79 (t, J=6.5 Гц, 2Н), 2.68 (t, J=7.4 Гц, 2Н), 1.79-1.66 (m, 2Н).
Стадия 3: 4-(5-(3-буомпропил)-6-метоксибензо[b]тиофен-2-ил)-4-оксобутаннитуил
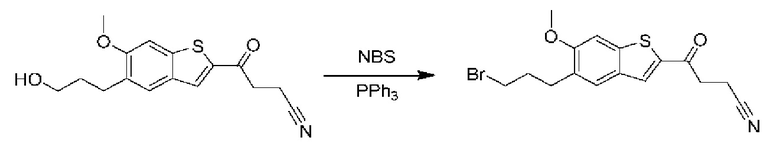
Трифенилфосфин (39 мг, 0.15 ммоль) добавляли к раствору 4-(5-(3-гидроксипропил)-6-метоксибензо[b]тиофен-2-ил)-4-оксобутаннитрила (45 мг, 0.15 ммоль) в THF (1.0 мл). Смесь охлаждали до 0°С, и затем добавляли NBS (26 мг, 0.15 ммоль). Через 1 ч к реакционной смеси добавляли дополнительные трифенилфосфин (23 мг, 0.089 ммоль) и NBS (13 мг, 0.074 ммоль). Смесь перемешивали в течение дополнительных 10 мин при 0°С.Реакционную смесь останавливали водным насыщенным NH4Cl и затем разбавляли EtOAc. Органический слой отделяли, промывали рассолом, высушивали над Na2SO4, фильтровали и концентрировали под пониженным давлением. Остаток очищали с помощью хроматографии на силикагеле (EtOAc/гексан) с получением 4-(5-(3-бромпропил)-6-метокси-бензо[b]тиофен-2-ил)-4-оксобутаннитрила. ЖХ-МС (C16H17BrNO2SNa) (ES, m/z): 388, 390 [M+Na]+.
Промежуточное соединение 96: Метил (S)-4-(6-(3-хлорпропокси)-5-метокситиено[3,2-b]пиридин-2-ил)-2-метил-4-оксобутаноат
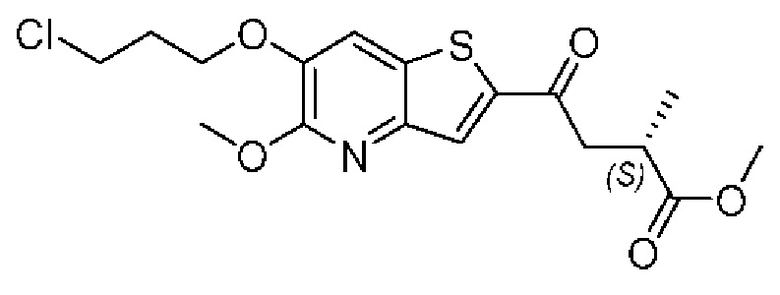
Стадия 1: 6-бромтиено[3,2-b]пиридин
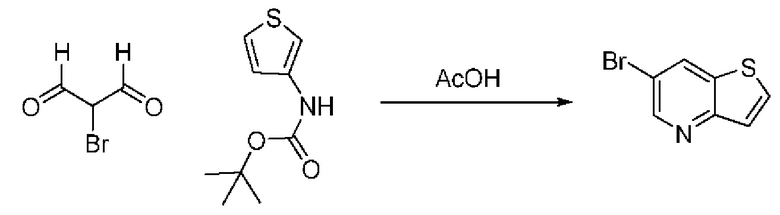
АсОН (50 мл) добавляли к смеси 2-броммалональдегида (3.56 г, 23.6 ммоль) и трет-бутил тиофен-3-илкарбамата (4.70 г, 23.6 ммоль) при комнатной температуре в воздушной атмосфере. Реакционную смесь перемешивали и нагревали до 100°С в течение 24 ч. Реакционную смесь затем охлаждали до комнатной температуры и разбавляли EtOAc (300 мл). Добавляли насыщенный водный NaHCO3, пока не прекратилось выделение газа. Органический слой отделяли, промывали рассолом (50 мл), высушивали над MgSO4, фильтровали и концентрировали под пониженным давлением. Остаток очищали с помощью хроматографии на силикагеле (элюируя EtOAc в гексанах) с получением 6-бромтиено[3,2-b]пиридина. ЖХ-МС (C7H5BrNS) (ES, m/z): 214, 216 [M+H]+. 1H ЯМР (499 МГц, DMSO-d6) δ 8.86 (s, 1H), 8.74 (s, 1H), 8.18 (d, J=5.4 Гц, 1H), 7.58 (d, J=5.4 Гц, 1H).
Стадия 2: 6-бромтиено[3,2-b]пиридин 4-оксид
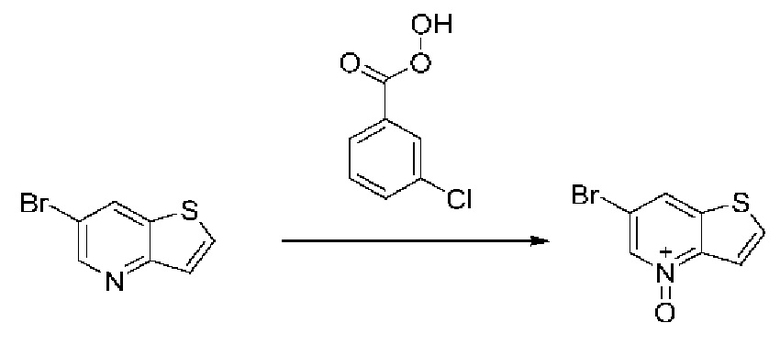
mCPBA (1.49 г, 6.63 ммоль) добавляли к смеси 6-бромтиено[3,2-b]пиридина (1.42 г, 6.63 ммоль) в DCM (50 мл) при 0°С в атмосфере Ar. Реакционную смесь затем оставляли нагреваться до комнатной температуры и перемешивали в течение дополнительных 24 ч. Реакционную смесь затем концентрировали под пониженным давлением. Остаток очищали с помощью хроматографии на силикагеле (элюируя [5% МеОН в EtOAc] в DCM) с получением 6-бромтиено[3,2-b]пиридин 4-оксида. ЖХ-МС (C7H5BrNOS) (ES, m/z): 230, 232 [М+Н]+.
Стадия 3: 6-бромтиено[3,2-b]пиридин-5-илацетат
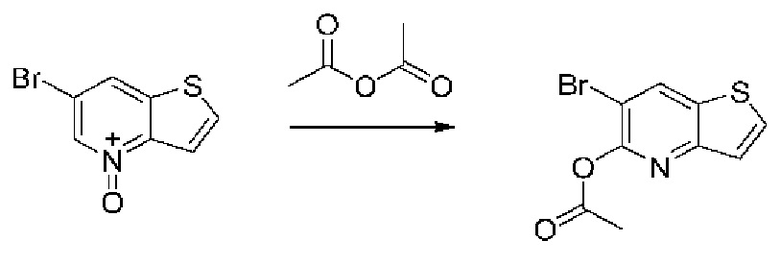
Уксусный ангидрид (20 мл, 210 ммоль) добавляли к 6-бромтиено[3,2-b]пиридин 4-оксиду (1.38 г, 6.00 ммоль) при комнатной температуре в атмосфере N2 (газообразный). Реакционную смесь затем нагревали до 140°С и перемешивали в течение 4 ч. Реакционную смесь затем охлаждали до комнатной температуры и разбавляли EtOAc (200 мл) и Н2О (200 мл). NaHCO3 медленно добавляли порциями к реакционной смеси, пока не прекратилось выделение газа. Органический слой отделяли, промывали рассолом (25 мл), высушивали над MgSO4, фильтровали и концентрировали под пониженным давлением. Остаток очищали с помощью хроматографии на силикагеле (элюируя [5% МеОН в EtOAc] в DCM) с получением 6-бромтиено[3,2-b]пиридин-5-ил ацетата. ЖХ-МС (C9H7BrNO2S-С2Н2О) (ES, m/z): 230, 232 [М+Н-ацетат]+. 1Н ЯМР (499 МГц, DMSO-d6) δ 9.04 (s, 1H), 8.29 (d, J=5.2 Гц, 1H), 7.54 (d, J=5.4 Гц, 1H), 2.41 (s, 3Н).
Стадия 4: 6-бромтиено[3,2-b]пиридин-5-ол
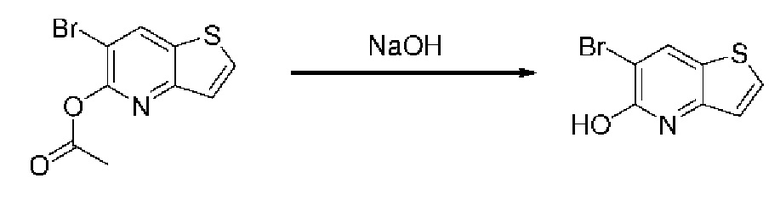
NaOH (2.0 M в H2O, 4.0 мл, 8.0 ммоль) добавляли к раствору 6-бромтиено[3,2-b]пиридин-5-ил ацетата (431 мг, 1.58 ммоль) в МеОН (5.0 мл) при 20°С в атмосфере N2 (газообразный). Реакционную смесь затем перемешивали в течение 1 ч при 20°С. Реакцию останавливали HCl (1.0 М в H2O, 8.0 мл, 8.0 ммоль) и затем разбавляли добавлением Н2О (10 мл). Реакционную смесь перемешивали в течение 30 мин и затем фильтровали. Собранное вещество промывали дополнительным объемом H2O (10 мл) и затем высушивали под пониженным давлением с получением 6-бромтиено[3,2-b]пиридин-5-ола. ЖХ-МС (C7H5BrNOS) (ES, m/z): 230, 232 [М+Н]+. 1H ЯМР (499 МГц, DMSO-d6) δ 12.65 (s, 1H), 8.59 (s, 1H), 7.91 (d, J=5.3 Гц, 1H), 6.99 (d, J=5.1 Гц, 1H).
Стадия 5: 6-буом-5-хлортиено[3,2-b]пиридин
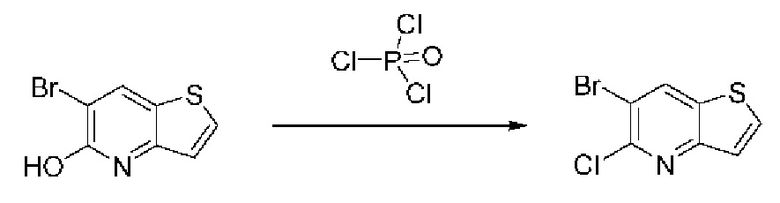
Оксихлорид фосфора (15.2 мл, 163 ммоль) добавляли к 6-бромтиено[3,2-b]пиридин-5-олу (375 мг, 1.63 ммоль) при 20°С в атмосфере N2 (газ). Реакционную смесь затем перемешивали и нагревали до 100°С в течение 2 дней. Реакционную смесь охлаждали до комнатной температуры, и реакцию останавливали путем добавления по каплям к раствору водного насыщенного раствора NaHCO3. Реакционную смесь разбавляли EtOAc (250 мл) и перемешивали. Органический слой отделяли, промывали рассолом (25 мл), высушивали над MgSO4, фильтровали и концентрировали под пониженным давлением с получением 6-бром-5-хлортиено[3,2-b]пиридина, который применяли без очистки. ЖХ-МС (C7H4BrClNS) (ES, m/z): 248, 250 [М+Н]+.
Стадия 6: 6-бром-5-метокситиено[3,2-b]пиридин
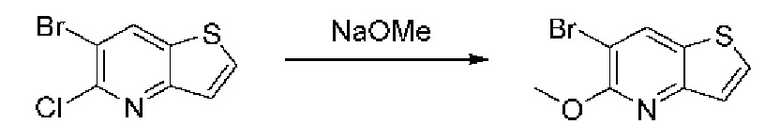
NaOMe (25% в МеОН, 3.24 мл, 14 ммоль) добавляли к смеси 6-бром-5-хлортиено[3,2-b]пиридина (352 мг, 1.42 ммоль) в МеОН (10 мл) при 20°С в атмосфере N2 (газ). Реакционную смесь перемешивали и нагревали до 100°С в течение 1 ч в микроволновом реакторе. Реакционную смесь останавливали лимонной кислотой (1.0 М в H2O, 28 мл, 28 ммоль) и разбавляли EtOAc (250 мл). Органический слой отделяли, промывали рассолом (25 мл), высушивали над MgSO4, фильтровали и концентрировали под пониженным давлением. Остаток очищали с помощью хроматографии на силикагеле (элюируя [5% МеОН в EtOAc] в DCM) с получением 6-бром-5-метокситиено[3,2-b]пиридина. ЖХ-МС (C8H7BrNOS) (ES, m/z): 244, 246 [M+H]+. 1H ЯМР (499 МГц, DMSO-d6) δ 8.78 (s, 1H), 8.09 (d, J=5.3 Гц, 1H), 7.44 (d, J=5.2 Гц, 1H), 3.99 (s, 3H).
Стадия 7: 6-бром-5-метокситиено[3,2-b]пиридин-2-карбоновая кислота
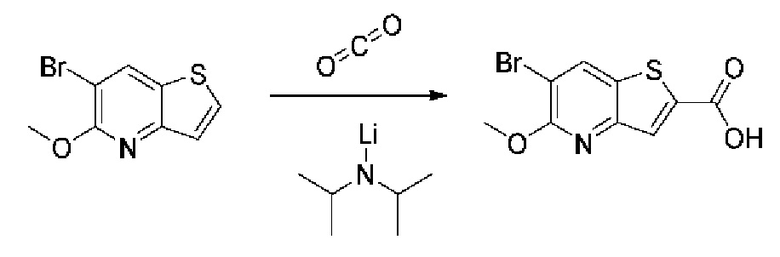
LDA (2.0 M в THF, 12.3 мл, 24.6 ммоль) добавляли к раствору 6-бром-5-метокситиено[3,2-b]пиридина (5.0 г, 20 ммоль) в THF (100 мл) при -78°С. Смесь выдерживали в течение 15 минут, и затем реакцию останавливали путем добавления CO2(газ) при -78°С. Реакционную смесь затем нагревали до 0°С в течение 10 мин. Реакцию останавливали HCl (2.0 М в воде, 12.3 мл, 24.6 ммоль) при 0°С и затем разбавляли EtOAc (500 мл). Органический слой отделяли, промывали рассолом (50 мл), высушивали над MgSO4, фильтровали и концентрировали под пониженным давлением. Остаток суспендировали в DCM (90 мл) и перемешивали в течение 1 ч. Затем добавляли по каплям гексан (250 мл) через капельную воронку в течение ~4 ч при комнатной температуре. Полученную в результате суспензию перемешивали в течение дополнительных 16 ч при комнатной температуре. Суспензию затем фильтровали, и остаток промывали смесью 4:1 гексан/DCM (50 мл). Остаток высушивали под вакуумом с получением 6-бром-5-метокситиено[3,2-b]пиридин-2-карбоновой кислоты. ЖХ-МС (C9H7BrNO3S) (ES, m/z): 288, 290 [М+Н]+. 1ЯМР (499 МГц, DMSO-d6) δ 13.79 (s, 1H), 8.87 (s, 1H), 7.93 (s, 1H), 4.01 (s, 3Н).
Стадия 8: 6-бром-5-метокситиено[3,2-b]пиридин-2-карбопилхлорид
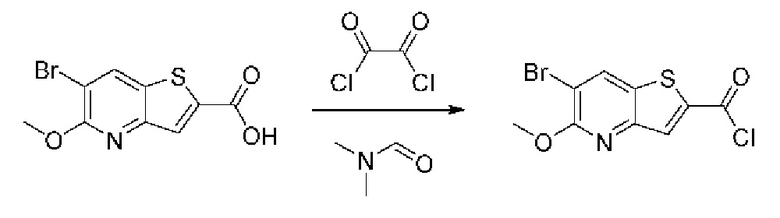
DMF (8 мкл, 0.1 ммоль) добавляли к раствору 6-бром-5-метокситиено[3,2-b]пиридин-2-карбоновой кислоты (1.05 г, 3.64 ммоль) и оксалилхлорида (0.64 мл, 7.3 ммоль) в THF (40 мл) при 0°С в атмосфере Ar. Реакционную смесь перемешивали при 0°С в течение 1 ч. Реакционную смесь концентрировали под пониженным давлением с получением 6-бром-5-метокситиено[3,2-b]пиридин-2-карбонилхлорида, который сразу применяли на следующей стадии.
Стадия 9: метил (S)-4-(6-бром-5-метокситиено[3,2-b]пиридин-2-ил)-2-метил-4-оксобутаноат
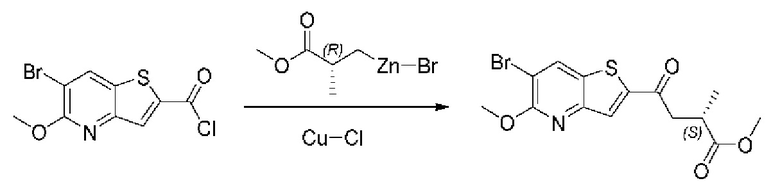
Раствор (R)-(3-метокси-2-метил-3-оксопропил)цинка (II) бромида (0.5 М в THF, 14.6 мл, 7.29 ммоль) медленно добавляли в высушенную в печи колбу, содержащую хлорид меди (I) (0.361 г, 3.64 ммоль) в атмосфере Ar при 0°С. Реакционную смесь перемешивали в течение 20 мин при 0°С в атмосфере Ar. Дегазированный Ar раствор 6-бром-5-метокситиено[3,2-b]пиридин-2-карбонилхлорида (1.12 г, 3.64 ммоль) в THF (10 мл) и NMP (5.0 мл) затем медленно добавляли через канюлю к реакционной смеси при 0°С; полученный в результате раствор нагревали до комнатной температуры и перемешивали в течение дополнительных 18 ч при комнатной температуре. Реакционную смесь охлаждали до 0°С, и реакцию останавливали добавлением насыщенного водного раствора NH4Cl (50 мл) и EtOAc (100 мл). Полученную в результате двухфазную смесь нагревали до комнатной температуры и перемешивали в течение дополнительного 1 ч. Смесь затем фильтровали, и фильтрат разбавляли дополнительным EtOAc (250 мл) и рассолом (50 мл). Органический слой отделяли, промывали дополнительным рассолом (50 мл), высушивали над MgSO4, фильтровали и концентрировали под пониженным давлением. Остаток очищали с помощью хроматографии на силикагеле (EtOAc/гексан) с получением (S)-метил 4-(6-бром-5-метокситиено[3,2-b]пиридин-2-ил)-2-метил-4-оксобутаноата. ЖХ-МС (C14H15BrNO4S) (ES, m/z): 372, 374 [М+Н]+. 1H ЯМР (499 МГц, DMSO-d6) δ 8.87 (s, 1Н), 8.37 (s, 1H), 4.02 (s, 3H), 3.59 (s, 3H), 3.52 (dd, J=17.9, 8.7 Гц, 1H), 3.27 (dd, J=18.0, 4.5 Гц, 1H), 3.03-2.91 (m, 1H), 1.20 (d, J=7.1 Гц, 3H).
Стадия 10: (S)-(5-метокси-2-(4-метокси-3-метил-4-оксобутаноил)тиено[3,2-b]пиридин-6-ил)6ороновая кислота
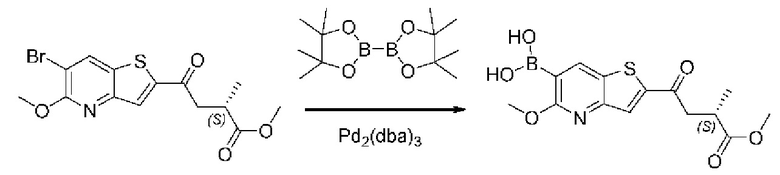
Смесь (S)-метил 4-(6-бром-5-метокситиено[3,2-b]пиридин-2-ил)-2-метил-4-оксобутаноата (700 мг, 1.88 ммоль), 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолан) (597 мг, 2.351 ммоль), Pd2(dba)3 (43 мг, 0.047 ммоль), трициклогексилфосфина (53 мг, 0.19 ммоль) и ацетата калия (высушивали в печи) (295 мг, 3.01 ммоль) дегазировали Ar в течение 5 мин. Добавляли диоксан (15 мл) при комнатной температуре, и полученную в результате смесь дегазировали Ar в течение 5 мин. Реакционную смесь затем нагревали до 80°С и перемешивали в течение 18 ч в атмосфере Ar. Реакционную смесь охлаждали до комнатной температуры и затем разбавляли EtOAc (20 мл). Суспензию перемешивали при комнатной температуре в течение 10 мин и затем фильтровали через целит, промывая EtOAc (20 мл). Фильтрат концентрировали под пониженным давлением с получением (S)-(5-метокси-2-(4-метокси-3-метил-4-оксобутаноил)тиено[3,2-b]пиридин-6-ил)бороновой кислоты, которую применяли без очистки на следующей стадии. ЖХ-МС (C14H17BNO6S) (ES, m/z): 338 [М+Н]+.
Стадия 11: метил (S)-4-(6-гидуокси-5-метокситиено[3,2-b]пиридин-2-ил)-2-метил-4-оксобутаноат
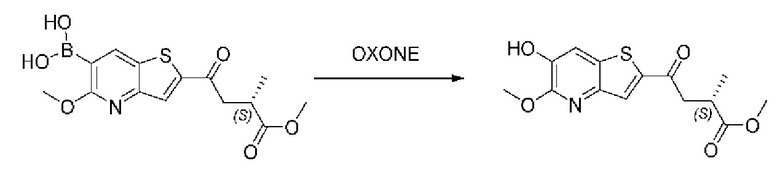
Раствор OXONE® ( ; 0.2 М в воде, 14.10 мл, 2.82 ммоль) добавляли к смеси (S)-(5-метокси-2-(4-метокси-3-метил-4-оксобутаноил)тиено[3,2-b]пиридин-6-ил)бороновой кислоты (634 мг, 1.88 ммоль) в ацетоне (20 мл) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 30 мин. Реакцию останавливали добавлением раствора бисульфата натрия (587 мг, 5.64 ммоль) в воде (5 мл) и затем перемешивали в течение 5 мин. Реакционную смесь разбавляли DCM (200 мл). Органический слой отделяли, и водный слой промывали дополнительным DCM (2×50 мл). Органические слои объединяли, высушивали над MgSO4, фильтровали и концентрировали под пониженным давлением. Остаток очищали с помощью хроматографии на силикагеле (EtOAc/DCM) с получением (S)-метил 4-(6-гидрокси-5-метокситиено [3,2-b]пиридин-2-ил)-2-метил-4-оксобутаноата. ЖХ-МС (C14H16NO5S) (ES, m/z): 310 [М+Н]+. 1H ЯМР (600 МГц, DMSO-d6) δ 10.46 (s, 1H), 8.23 (s, 1Н), 7.66 (s, 1H), 3.97 (s, 3Н), 3.60 (s, 3Н), 3.44 (dd, J=17.5, 8.6 Гц, 1H), 3.20 (dd, J=17.5, 5.0 Гц, 1Н), 3.00-2.92 (m, 1H), 1.19 (d, J=7.2 Гц, 3Н).
; 0.2 М в воде, 14.10 мл, 2.82 ммоль) добавляли к смеси (S)-(5-метокси-2-(4-метокси-3-метил-4-оксобутаноил)тиено[3,2-b]пиридин-6-ил)бороновой кислоты (634 мг, 1.88 ммоль) в ацетоне (20 мл) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 30 мин. Реакцию останавливали добавлением раствора бисульфата натрия (587 мг, 5.64 ммоль) в воде (5 мл) и затем перемешивали в течение 5 мин. Реакционную смесь разбавляли DCM (200 мл). Органический слой отделяли, и водный слой промывали дополнительным DCM (2×50 мл). Органические слои объединяли, высушивали над MgSO4, фильтровали и концентрировали под пониженным давлением. Остаток очищали с помощью хроматографии на силикагеле (EtOAc/DCM) с получением (S)-метил 4-(6-гидрокси-5-метокситиено [3,2-b]пиридин-2-ил)-2-метил-4-оксобутаноата. ЖХ-МС (C14H16NO5S) (ES, m/z): 310 [М+Н]+. 1H ЯМР (600 МГц, DMSO-d6) δ 10.46 (s, 1H), 8.23 (s, 1Н), 7.66 (s, 1H), 3.97 (s, 3Н), 3.60 (s, 3Н), 3.44 (dd, J=17.5, 8.6 Гц, 1H), 3.20 (dd, J=17.5, 5.0 Гц, 1Н), 3.00-2.92 (m, 1H), 1.19 (d, J=7.2 Гц, 3Н).
Стадия 12: метил (S)-4-(6-(3-хлоупуопокси)-5-метокситиено[3,2-b]пиридин-2-ил)-2-метил-4-оксобутаноат
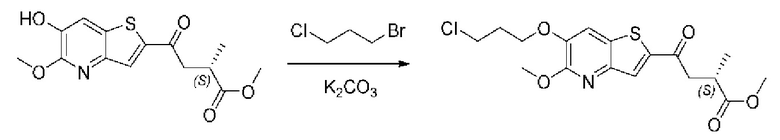
1-Бром-3-хлорпропан (244 мг, 1.55 ммоль) добавляли к смеси (S)-метил 4-(6-гидрокси-5-метокситиено[3,2-b]пиридин-2-ил)-2-метил-4-оксобутаноата (96 мг, 0.31 ммоль) и карбоната калия (257 мг, 1.86 ммоль) в DMF (1.0 мл) при комнатной температуре. Реакционную смесь перемешивали и нагревали до 50°С в течение 4 ч. Реакционную смесь охлаждали до комнатной температуры и фильтровали, и затем сразу очищали с помощью хроматографии на силикагеле (EtOAc/DCM) с получением (S)-метил 4-(6-(3-хлорпропокси)-5-метокситиено[3,2-b]пиридин-2-ил)-2-метил-4-оксобутаноата. ЖХ-МС (C17H21ClNO5S) (ES, m/z): 386 [М+Н]+.
Промежуточное соединение 97: Метил (S)-4-(5-(3-бромпропил)-4-фтор-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноат
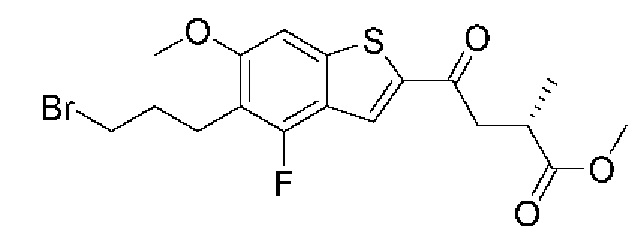
Стадия 1: метил (S)-4-(4-фтор-6-метокси-5-(((трифторметил)сульфонил)окси)бензо[b]тиофен-2-ил)-2-метил-4-оксобутаноат
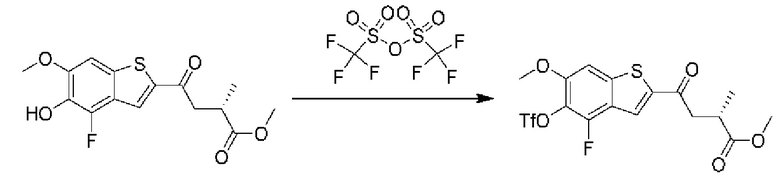
Метил (S)-4-(4-фтор-5-гидрокси-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноат (750 мг, 2.30 ммоль), DCM (10 мл), основание Хунига (2.0 мл, 11 ммоль) и трифторметансульфоновый ангидрид (1.0 М в DCM, 3.5 мл, 3.5 ммоль) объединяли. Реакционную смесь перемешивали при комнатной температуре в течение 20 мин. Затем реакцию останавливали водой и разбавляли DCM. Органический слой отделяли, промывали насыщенным NaHCO3, высушивали над Na2SO4, фильтровали и концентрировали под пониженным давлением. Остаток очищали с помощью хроматографии на силикагеле (EtOAc/гексан) с получением (S)-метил 4-(4-фтор-6-метокси-5-(((трифторметил)сульфонил)окси)бензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата. ЖХ-МС (C16H15F4O7S2) (ES, m/z): 459 [М+Н]+. 1H ЯМР (600 МГц, DMSO-d6) δ 8.52 (s, 1H), 7.93 (s, 1H), 4.03 (s, 3Н), 3.61 (s, 3Н), 3.53 (dd, J=17.8, 8.7 Гц, 1H), 3.27 (dd, J=17.8, 5.0 Гц, 1H), 3.05-2.92 (m, 1H), 1.21 (d, J=7.2 Гц, 3Н).
Стадия 2: метил (2S)-4-(4-фтор-6-метокси-5-(3-((тетрагидро-2Н-пирап-2-ил)окси)пропил)бензо[b]тиофен-2-ил)-2-метил-4-оксобутаноат
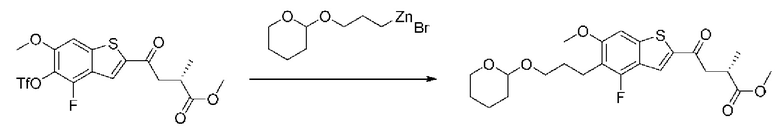
(S)-Метил 4-(4-фтор-6-метокси-5-(((трифторметил)сульфонил)окси)бензо-[b]тиофен-2-ил)-2-метил-4-оксобутаноат (1.00 г, 2.18 ммоль), CPhos Pd G4 (0.088 г, 0.11 ммоль) и (3-((тетрагидро-2Н-пиран-2-ил)окси)пропил)цинк (II) бромид (0.50 М в THF, 8.7 мл, 4.4 ммоль) объединяли в виале. Реакционную смесь нагревали при 40°С в течение 2 ч. Реакционную смесь фильтровали через целит и затем концентрировали под пониженным давлением. Остаток очищали с помощью хроматографии на силикагеле (EtOAc/гексан) с получением метил (28)-4-(4-фтор-6-метокси-5-(3-((тетрагидро-2Н-пиран-2-ил)окси)пропил)бензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата. ЖХ-МС (C23H30FO6S) (ES, m/z): 453 [М+Н]+. 1H ЯМР (600 МГц, DMSO-d6) δ 8.31 (s, 1H), 7.52 (s, 1H), 4.54 (t, J=3.4 Гц, 1H), 3.92 (s, 3Н), 3.75-3.70 (m, 1Н), 3.69-3.62 (m, 1H), 3.60 (s, 3Н), 3.49 (dd, J=17.7, 8.7 Гц, 1H), 3.44-3.39 (m, 1H), 3.38-3.28 (m, 1H), 3.25 (dd, J=17.6, 5.0 Гц, 1H), 3.02-2.92 (m, 1H), 2.80-2.70 (m, 2Н), 1.84-1.76 (m, 2Н), 1.76-1.67 (m, 1H), 1.63-1.57 (m, 1H), 1.50-1.40 (m, 4Н), 1.20-1.17 (m, 3Н).
Стадия 3: метил (Б)-4-(5-(3-бромпропил)-4-фтор-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноат
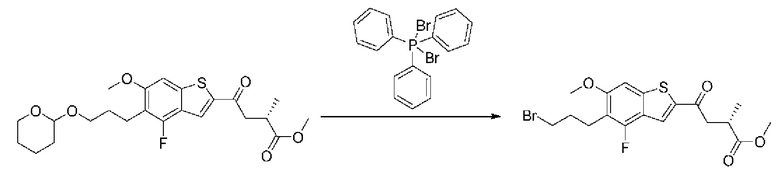
Метил (28)-4-(4-фтор-6-метокси-5-(3-((тетрагидро-2Н-пиран-2-ил)окси)пропил)бензо[b]тиофен-2-ил)-2-метил-4-оксобутаноат (350 мг, 0.773 ммоль) и DCM (5 мл) объединяли в виале. К реакционной смеси добавляли трифенилфосфин дибромид (653 мг, 1.55 ммоль) и перемешивали в течение 30 мин. Реакционную смесь останавливали водой. Органический слой отделяли, высушивали над Na2SO4, фильтровали и концентрировали под пониженным давлением. Остаток очищали с помощью хроматографии на силикагеле (EtOAc/гексан) с получением метил (S)-4-(5-(3-бромпропил)-4-фтор-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата. ЖХ-МС (C18H21BrFO4S) (ES, m/z): 431, 433 [М+Н]+.
Промежуточное соединение 98: Метил (S)-4-(4-фтор-5-(3-гидроксипропил)-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноат
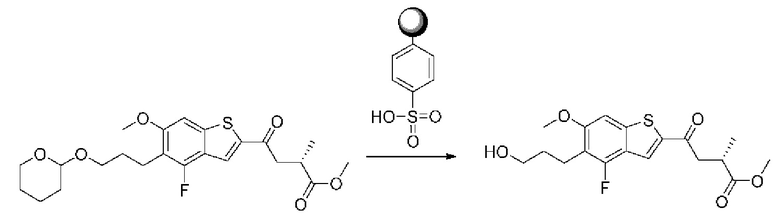
Метил (2S)-4-(4-фтор-6-метокси-5-(3-((тетрагидро-2Н-пиран-2-ил)окси)пропил)бензо[b]тиофен-2-ил)-2-метил-4-оксобутаноат (350 мг, 0.773 ммоль), МеОН (5 мл) и Mp-TsOH (1.00 г, 4.33 ммоль) объединяли в виале. Реакционную смесь встряхивали в течение 2 ч. Реакционную смесь затем фильтровали, промывали МеОН и концентрировали под пониженным давлением с получением метил (S)-4-(4-фтор-5-(3-гидроксипропил)-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата. ЖХ-МС (C18H22FO5S) (ES, m/z): 369 [М+Н]+. 1Н ЯМР (600 МГц, DMSO-d6) δ 8.31 (s, 1H), 7.51 (s, 1Н), 3.92 (s, 3Н), 3.60 (s, 3Н), 3.50 (dd, J=17.7, 8.7 Гц, 1H), 3.44 (t, J=6.5 Гц, 2H), 3.25 (dd, J=17.4, 4.7 Гц, 1H), 3.02-2.93 (m, 1H), 2.71 (t, J=7.4 Гц, 2H), 1.69-1.63 (m, 2H), 1.20 (d, J=7.0 Гц, 3Н).
Промежуточное_соединение 99: Метил 2-(4-фтор-5-гидуокси-6-метоксибензо[b]тиофен-2-карбонил)циклопропан-1-карбоксилат
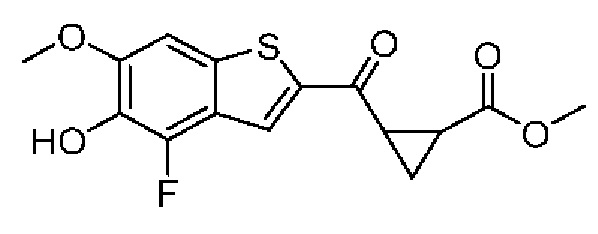
Стадия 1: 2-(4-фтор-5,6-диметоксибензо[b]тиофен-2-карбонил)циклопропан-1-карбоновая кислота
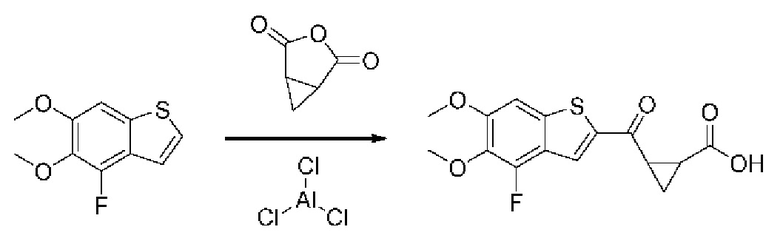
Хлорид алюминия (0.923 г, 6.92 ммоль) добавляли к смеси 4-фтор-5,6-диметоксибензо[b]тиофена (1.13 г, 5.32 ммоль) и 3-оксабицикло[3.1.0]гексан-2,4-диона (1.19 г, 10.7 ммоль) в DCM (20.0 мл) при 0°С. Реакционную смесь нагревали до комнатной температуры и затем перемешивали в течение 18 ч. Реакционную смесь охлаждали до 0°С и останавливали водой (50 мл). Смесь затем разбавляли EtOAc (500 мл). Органический слой отделяли, промывали рассолом, высушивали над MgSO4, фильтровали и концентрировали под пониженным давлением с получением 2-(4-фтор-5,6-диметоксибензо[b]тиофен-2-карбонил)циклопропан-1-карбоновой кислоты, которую применяли без очистки на следующей стадии. ЖХ-МС (C15H14FO5S) (ES, m/z): 325 [М+Н]+.
Стадия 2: метил 2-(4-фтор-5,6-диметоксибензо[b]тиофен-2-карбонил)циклопропан-1-карбоксилат
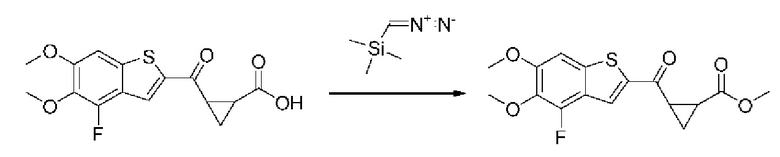
TMS-диазометан (2.0 M в диэтиловом эфире, 4.3 мл, 8.6 ммоль) добавляли по каплям к смеси 2-(4-фтор-5,6-диметоксибензо[b]тиофен-2-карбонил)циклопропанкарбоновой кислоты (1.40 г, 4.32 ммоль) в DCM (20 мл) и МеОН (20 мл) при 0°С. Реакционную смесь перемешивали при 0°С в течение 15 мин. Реакционную смесь останавливали НОАс. Реакционную смесь затем концентрировали под пониженным давлением. Остаток очищали с помощью хроматографии на силикагеле (EtOAc/DCM) с получением метил 2-(4-фтор-5,6-диметоксибензо[b]тиофен-2-карбонил)циклопропанкарбоксилата. ЖХ-МС (C16H16FO5S) (ES, m/z): 339 [М+Н]+. 1H ЯМР (499 МГц, DMSO-d6) δ 8.37 (s, 1Н), 7.59 (s, 1H), 3.93 (s, 3H), 3.87 (s, 3H), 3.48 (s, 3H), 3.31-3.25 (m, 1H), 2.47-2.41 (m, 1H), 1.62-1.58 (m, 1H), 1.44-1.40 (m, 1H).
Стадия 3: метил 2-(4-фтор-5-гидрокси-6-метоксибензо[b]тиофен-2-карбонил)циклопропан-1-карбоксилат
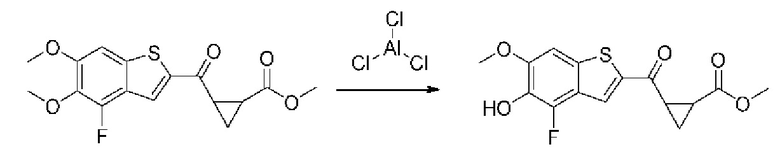
Хлорид алюминия (2.66 г, 20.0 ммоль) добавляли порциями к смеси метил 2-(4-фтор-5,6-диметоксибензо[b]тиофен-2-карбонил)циклопропанкарбоксилата (1.50 г, 4.43 ммоль) в DCM (40 мл) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 6 ч в атмосфере Ar. Реакцию охлаждали до 0°С и затем медленно останавливали водой (50 мл). Реакционную смесь разбавляли дополнительным DCM (250 мл). Органический слой отделяли, промывали рассолом (25 мл), высушивали над MgSO4, фильтровали и концентрировали под пониженным давлением. Остаток очищали с помощью хроматографии на силикагеле (EtOAc/DCM) с получением метил 2-(4-фтор-5-гидрокси-6-метоксибензо[b]тиофен-2-карбонил)циклопропанкарбоксилата. ЖХ-МС (C15H14FO5S) (ES, m/z): 325 [М+Н]+. 1Н ЯМР (499 МГц, DMSO-d6) δ 9.55 (s, 1H), 8.29 (s, 1H), 7.48 (s, 1H), 3.92 (s, 3Н), 3.48 (s, 3Н), 3.30-3.23 (m, 1H), 2.45-2.39 (m, 1H), 1.61-1.57 (m, 1H), 1.43-1.38 (m, 1H).
Промежуточное соединение 100: Метил (R)-4-(5-(3-бромпропил)-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноат
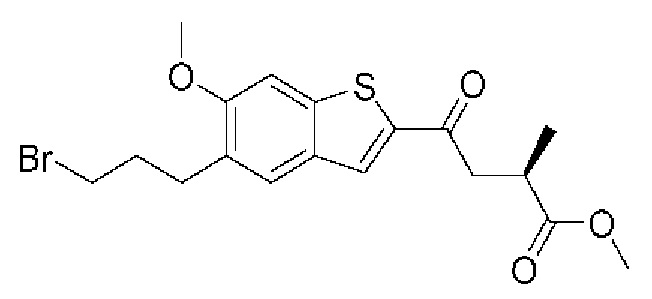
Стадия 1: метил (R)-4-(5-бром-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноат
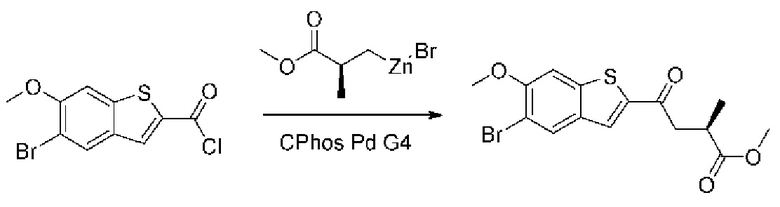
5-Бром-6-метоксибензо[b]тиофен-2-карбонилхлорид (3.00 г, 9.82 ммоль) и THF (98 мл) объединяли. Реакционную смесь затем дегазировали Ar в течение 10 мин. К реакционной смеси добавляли CPhos Pd G4 (0.081 г, 0.098 ммоль), и смесь охлаждали до 0°С. Затем к реакционной смеси добавляли (S)-(3-метокси-2-метил-3-оксопропил)цинк (II) бромид (0.50 М в THF, 21 мл, 11 ммоль) с помощью капельной воронки. Смесь перемешивали при 0°С в течение 1 ч. Реакционную смесь останавливали насыщенным водным NH4Cl и разбавляли EtOAc. Органический слой отделяли, высушивали над MgSO4, фильтровали и концентрировали под пониженным давлением. Остаток очищали с помощью хроматографии на силикагеле (EtOAc/гексан) с получением метил (R)-4-(5-бром-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата. ЖХ-МС (C15H16BrO4S) (ES, m/z): 371, 373 [М+Н]+. 1H ЯМР (499 МГц, DMSO-d6) δ 8.28-8.26 (m, 2Н), 7.82 (s, 1H), 3.95 (s, 3Н), 3.60 (s, 3Н), 3.44 (dd, J=17.6, 8.6 Гц, 1H), 3.21 (dd, J=17.6, 5.1 Гц, 1H), 3.03-2.93 (m, 1H), 1.20 (d, J=7.2 Гц, 3Н).
Стадия 2: метил (R)-4-(5-(3-((трет-бутилдиметилсилил)окси)пропил)-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноат
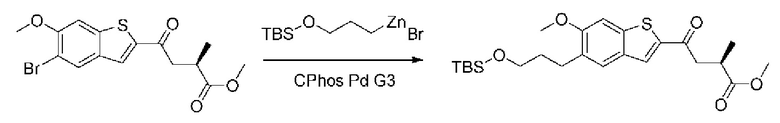
CPhos Pd G3 (174 мг, 0.215 ммоль) и (3-((трет-бутилдиметилсилил)окси)пропил)цинк (II) бромид (0.50 М в THF, 9.7 мл, 4.9 ммоль) добавляли к смеси (R)-метил 4-(5-бром-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата (1.00 г, 2.69 ммоль) в THF (13.5 мл). Реакционную смесь нагревали при 40°С в течение 3 ч. Реакционную смесь охлаждали до комнатной температуры, останавливали водой и разбавляли EtOAc. Органический слой отделяли, высушивали над MgSO4, фильтровали и концентрировали под пониженным давлением. Остаток очищали с помощью хроматографии на силикагеле (EtOAc/гексан) с получением метил (R)-4-(5-(3-((трет-бутилдиметилсилил)окси)пропил)-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата. ЖХ-МС (C24H37O5SSi) (ES, m/z): 465 [М+Н]+.
Стадия 3: метил (R)-4-(5-(3-гидроксипропил)-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноат
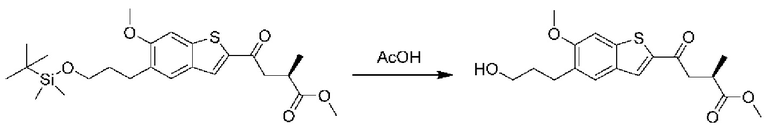
(R)-Метил 4-(5-(3-((трет-бутилдиметилсилил)окси)пропил)-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноат (1.04 г, 2.24 ммоль) суспендировали в смеси МеОН (4 мл), воды (4 мл) и НОАс (4 мл). Реакционную смесь перемешивали при комнатной температуре в течение 3.5 ч. Реакционную смесь разбавляли EtOAc и водой. Водный слой отделяли и экстрагировали дополнительным EtOAc (3х). Органические слои объединяли, промывали рассолом, высушивали над Na2SO4, фильтровали и концентрировали под пониженным давлением. Остаток очищали с помощью хроматографии на силикагеле (EtOAc/гексан) с получением метил (R)-4-(5-(3-гидроксипропил)-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата. ЖХ-МС (C18H23O5S) (ES, m/z): 351 [М+Н]+.
Стадия 4: метил (R)-4-(5-(3-бромпропил)-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноат
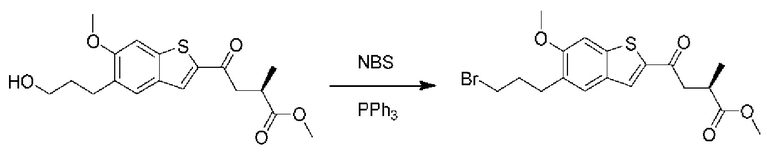
NBS (518 мг, 2.91 ммоль) добавляли к смеси (R)-метил 4-(5-(3-гидроксипропил)-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата (680 мг, 1.94 ммоль) и трифенилфосфина (814 мг, 3.10 ммоль) в THF (9.7 мл) при 0°С. Через 1.5 ч реакционную смесь останавливали насыщенным водным NH4Cl и затем разбавляли EtOAc. Органический слой отделяли, промывали рассолом, высушивали над MgSO4, фильтровали и концентрировали под пониженным давлением. Остаток очищали с помощью хроматографии на силикагеле (EtOAc/гексан) с получением метил (R)-4-(5-(3-бромпропил)-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата. ЖХ-МС (C18H22BrO4S) (ES, m/z): 413, 415 [М+Н]+. 1Н ЯМР (499 МГц, DMSO-d6) δ 8.27 (s, 1H), 7.77 (s, 1H), 7.62 (s, 1H), 3.90 (s, 3Н), 3.60 (s, 3Н), 3.55 (t, J=6.6 Гц, 2Н), 3.43 (dd, J=17.5, 8.6 Гц, 1H), 3.20 (dd, J=17.5, 5.1 Гц, 1H), 3.04-2.94 (m, 1H), 2.83-2.77 (m, 2Н), 2.14-2.08 (m, 2Н), 1.20 (d, J=7.2 Гц, 3Н).
Промежуточные соединения 101, 102, 103 и 104: Метил (1R,2R или 1S,2S)-2-(4-фтор-6-гидрокси-5-метоксибензо[b]тиофен-2-карбонил)циклобутан-1-карбоксилат, метил (1R,2R или 1S,2S)-2-(4-фтор-6-гидрокси-5-метоксибензо[b]тиофен-2-карбонил)циклобутан-1-карбоксилат, метил (1R,2R или 1S,2S)-2-(4-фтор-5-гидрокси-6-метоксибензо[b]тиофен-2-карбонил)циклобутан-1-карбоксилат, метил (1R,2R или 1S,2S)-2-(4-фтор-5-гидрокси-6-метоксибензо[b]тиофен-2-карбонил)циклобутан-1-карбоксилат
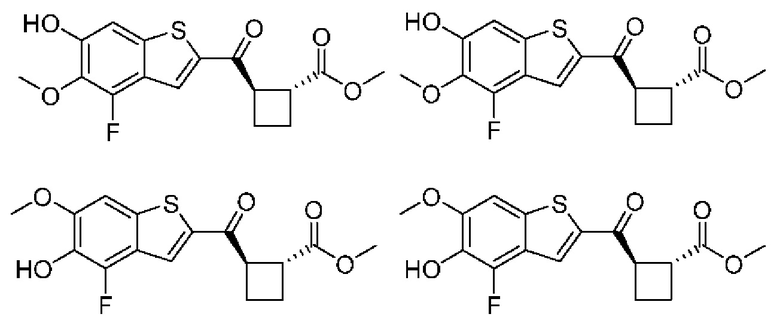
Стадия 1: (цис)-2-(4-фтор-5,6-диметоксибензо[b]тиофен-2-карбонил)циклобутан-1-карбоновая кислота
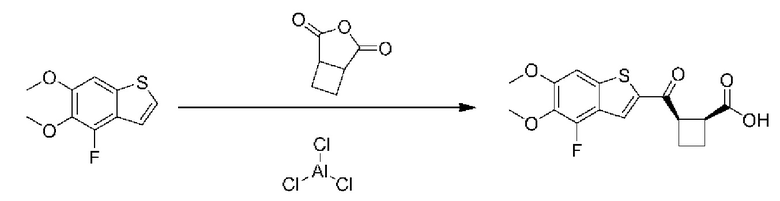
Хлорид алюминия (776 мг, 5.82 ммоль) добавляли к смеси 4-фтор-5,6-диметоксибензо[b]тиофена (950 мг, 4.48 ммоль) и 3-оксабицикло[3.2.0]гептан-2,4-диона (1130 мг, 8.95 ммоль) в DCM (20.0 мл) при 0°С. Реакционную смесь нагревали до комнатной температуры и затем перемешивали в течение 18 ч. Реакционную смесь охлаждали до 0°С и затем останавливали путем добавления по каплям воды (50 мл). Смесь затем разбавляли дополнительным DCM (200 мл). Органический слой отделяли, промывали рассолом, высушивали над MgSO4, фильтровали и концентрировали под пониженным давлением с получением (цис)-2-(4-фтор-5,6-диметоксибензо[b]тиофен-2-карбонил)циклобутан-1-карбоновой кислоты, которую применяли без очистки на следующей стадии. ЖХ-МС (C16H16FO5S) (ES, m/z): 339 [М+Н]+.
Стадия 2: метил (цис)-2-(4-фтор-5,6-диметоксибензо[b]тиофен-2-карбонил)циклобутан-1-карбоксилат
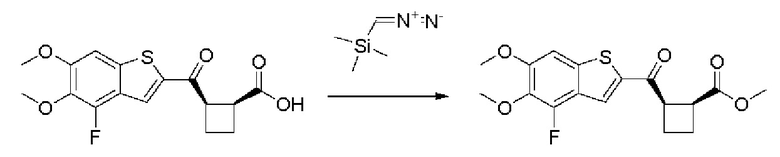
TMS-диазометан (2.0 M в диэтиловом эфире, 4.0 мл, 8.0 ммоль) добавляли по каплям к смеси (цис)-2-(4-фтор-5,6-диметоксибензо[b]тиофен-2-карбонил)циклобутанкарбоновой кислоты (1.35 г, 3.99 ммоль) в DCM (20 мл) и МеОН (20 мл) при 0°С. Реакционную смесь перемешивали при 0°С в течение 15 мин. Реакционную смесь останавливали НОАс. Реакционную смесь затем концентрировали под пониженным давлением. Остаток очищали с помощью хроматографии на силикагеле (EtOAc/DCM) с получением (цис)-метил 2-(4-фтор-5,6-диметоксибензо[b]тиофен-2-карбонил)циклобутанкарбоксилата. ЖХ-МС (C17H18FO5S) (ES, m/z): 353 [М+Н]+. 1H ЯМР (499 МГц, DMSO-d6) δ 8.15 (s, 1H), 7.58 (s, 1H), 4.59-4.50 (m, 1H), 3.93 (s, 3H), 3.86 (s, 3H), 3.68 (q, J=8.9, 8.5 Гц, 1H), 3.41 (s, 3H), 2.34-2.10 (m, 4H).
Стадия 3: (транс)-2-(4-фтор-5,6-диметоксибензо[b]тиофен-2-карбонил)циклобутан-1-карбоновая кислота
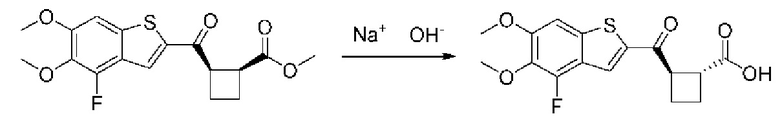
NaOH (5.0 M, 1.8 мл, 9.0 ммоль) добавляли к смеси (цис)-метил 2-(4-фтор-5,6-диметоксибензо[b]тиофен-2-карбонил)циклобутанкарбоксилата (650 мг, 1.85 ммоль) в МеОН (25 мл) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 4 ч. Реакционную смесь останавливали TFA (0.85 мл, 11 ммоль) и затем разбавляли DCM (250 мл). Органический слой отделяли, промывали рассолом (50 мл), высушивали над MgSO4, фильтровали и концентрировали под пониженным давлением. Остаток очищали с помощью хроматографии на силикагеле (MeOH/DCM) с получением (транс)-2-(4-фтор-5,6-диметоксибензо[b]тиофен-2-карбонил)циклобутанкарбоновой кислоты. ЖХ-МС (C16H16FO5S) (ES, m/z): 339 [М+Н]+.
Стадия 4: метил (транс)-2-(4-фтор-5,6-диметоксибензо[b]тиофен-2-карбонил)циклобутан-1-карбоксилат
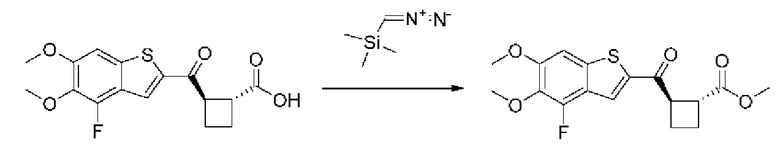
TMS-диазометан (2.0 М в диэтиловом эфире, 1.7 мл, 3.4 ммоль) добавляли по каплям к смеси (транс)-2-(4-фтор-5,6-диметоксибензо[b]тиофен-2-карбонил)циклобутанкарбоновой кислоты (570 мг, 1.69 ммоль) в DCM (20 мл) и МеОН (20 мл) при 0°С. Реакционную смесь перемешивали при 0°С в течение 15 мин. Реакционную смесь останавливали НОАс. Реакционную смесь затем концентрировали под пониженным давлением. Остаток очищали с помощью хроматографии на силикагеле (EtOAc/DCM) с получением (транс)-метил 2-(4-фтор-5,6-диметоксибензо[b]тиофен-2-карбонил)циклобутанкарбоксилата. ЖХ-МС (C17H18FO5S) (ES, m/z): 353 [М+Н]+. 1Н ЯМР (499 МГц, DMSO-d6) δ 8.22 (s, 1H), 7.60 (s, 1Н), 4.42 (q, J=9.0 Гц, 1H), 3.93 (s, 3Н), 3.86 (s, 3Н), 3.63 (s, 3Н), 3.58-3.48 (m, 1H), 2.38-2.30 (m, 1H), 2.26-2.18 (m, 1H), 2.17-2.10 (m, 2H).
Стадия 5: метил (1R,2R или 1S,2S)-2-(4-фтор-6-гидрокси-5-метоксибензо[b]тиофен-2-карбонил)циклобутан-1-карбоксилат, метил (1R,2R или 1S,2S)-2-(4-фтор-6-гидрокси-5-метоксибензо[b]тиофен-2-карбонил)циклобутан-1-карбоксилат, метил (1R,2R или 1S,2S)-2-(4-фтор-5-гидрокси-6-метоксибензо[b] тиофен-2-карбонил)циклобутан-1-карбоксилат, метил (1R,2R или 1S,2S)-2-(4-фтор-5-гидрокси-6-метоксибензо[b]тиофен-2-карбонил)циклобутан-1-карбоксилат
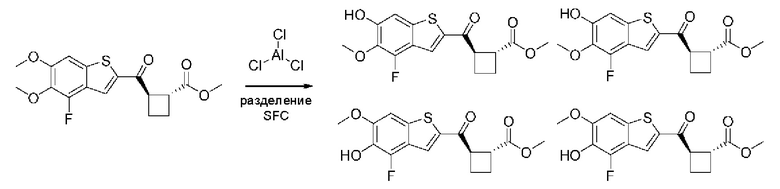
AlCl3 (1.10 г, 8.27 ммоль) добавляли к смеси (транс)-метил 2-(4-фтор-5,6-диметоксибензо[b]тиофен-2-карбонил)циклобутанкарбоксилата (530 мг, 1.50 ммоль) в DCM (40 мл) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 6 ч в атмосфере Ar. Реакционную смесь охлаждали до 0°С и затем медленно останавливали водой (50 мл). Реакционную смесь затем разбавляли дополнительным DCM (250 мл). Органический слой отделяли, промывали рассолом (25 мл), высушивали над MgSO4, фильтровали и концентрировали под пониженным давлением. Остаток очищали с помощью хроматографии на силикагеле (EtOAc/DCM) с получением смеси (+/- транс)-метил-2-(4-фтор-5-гидрокси-6-метоксибензо[b]тиофен-2-карбонил)циклобутан-1-карбоксилата и (+/- транс)-метил-2-(4-фтор-6-гидрокси-5-метоксибензо[b]тиофен-2-карбонил)циклобутан-1-карбоксилата. Смесь рацемических региоизомеров очищали с помощью хиральной SFC (колонка ССА, 20% [МеОН с 0.25% DMEA] в СО2) с получением:
Пик 1 (3.0 мин): метил (1R,2R или 1S,2S)-2-(4-фтор-6-гидрокси-5-метоксибензо[b]тиофен-2-карбонил)циклобутан-1-карбоксилат. ЖХ-МС (C16H16FO5S) (ES, m/z): 339 [М+Н]+. 1Н ЯМР (499 МГц, DMSO-d6) δ 10.58 (br s, 1H), 8.15 (s, 1H), 7.24 (s, 1H), 4.45-4.34 (m, 1H), 3.86 (s, 3H), 3.62 (s, 3H), 3.56-3.47 (m, 1H), 2.36-2.24 (m, 1H), 2.25-2.16 (m, 1H), 2.16-2.07 (m, 2H). Пик 2 (3.5 мин): метил (1R,2R или 1S,2S)-2-(4-фтор-6-гидрокси-5-метоксибензо[b]тиофен-2-карбонил)циклобутан-1-карбоксилат. ЖХ-МС (C16H16FO5S) (ES, m/z): 339 [М+Н]+. Пик 3 (3.9 мин): метил (1R,2R или 1S,2S)-2-(4-фтор-5-гидрокси-6-метоксибензо[b]тиофен-2-карбонил)циклобутан-1-карбоксилат. ЖХ-МС (C16H16FO5S) (ES, m/z): 339 [М+Н]+. 1H ЯМР (499 МГц, DMSO-d6) δ 9.55 (s, 1H), 8.14 (s, 1H), 7.48 (s, 1H), 4.45-4.38 (m, 1H), 3.92 (s, 3H), 3.62 (s, 3H), 3.58-3.49 (m, 1H), 2.38-2.27 (m, 1H), 2.25-2.19 (m, 1H), 2.17-2.09 (m, 2H). Пик 4 (5.0 мин): метил (1R,2R или 1S,2S)-2-(4-фтор-5-гидрокси-6-метоксибензо[b]тиофен-2-карбонил)циклобутан-1-карбоксилат. ЖХ-МС (C16H16FO5S) (ES, m/z): 339 [M+H]+. 1H ЯМР (499 МГц, DMSO-d6) δ 9.54 (s, 1H), 8.14 (s, 1Н), 7.48 (s, 1H), 4.44-4.38 (m, 1H), 3.92 (s, 3H), 3.62 (s, 3H), 3.55-3.49 (m, 1H), 2.38-2.26 (m, 1H), 2.26-2.17 (m, 1H), 2.17-2.10 (m, 2H).
Промежуточное соединение 105: Метил (R)-4-(4-фтор-5-гидрокси-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноат
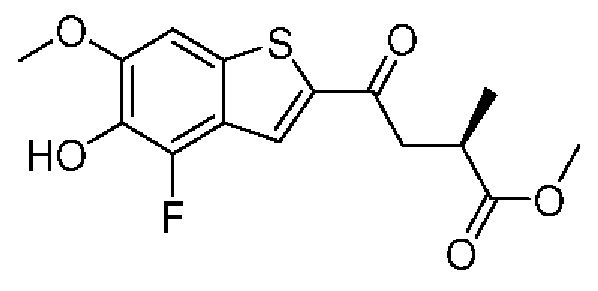
Стадия 1: метил (R)-4-(4-фтор-5,6-диметоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноат
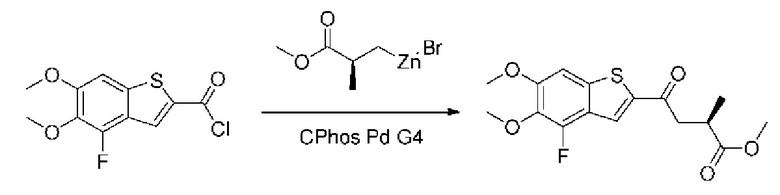
Смесь 4-фтор-5,6-диметоксибензо[b]тиофен-2-карбонилхлорида (1.00 г, 3.64 ммоль) и THF (36.4 мл) дегазировали Ar в течение 10 мин. CPhos Pd G4 (0.030 г, 0.036 ммоль) добавляли к смеси, и реакционную смесь охлаждали до 0°С. (S)-(3-метокси-2-метил-3-оксопропил)цинк (II) бромид (0.50 М в THF, 8.0 мл, 4.0 ммоль) затем добавляли к реакционной смеси через капельную воронку. Смесь перемешивали при 0°С в течение 1 ч. Реакционную смесь останавливали насыщенным водным NH4Cl и затем разбавляли EtOAc. Органический слой отделяли, высушивали над MgSO4, фильтровали и концентрировали под пониженным давлением. Остаток очищали с помощью хроматографии на силикагеле (EtOAc/гексан) с получением метил (R)-4-(4-фтор-5,6-диметоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата. ЖХ-МС (C16H18FO5S) (ES, m/z): 341 [М+Н]+.
Стадия 2: метил (R)-4-(4-фтор-5-гидрокси-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноат
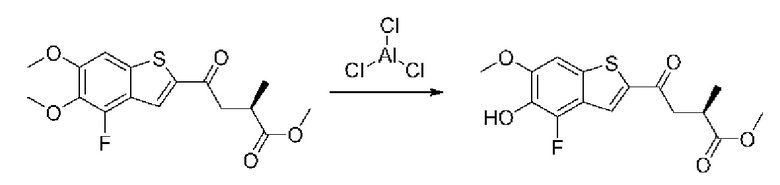
AlCl3 (1.16 г, 8.72 ммоль) добавляли к смеси (R)-метил 4-(4-фтор-5,6-диметоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата (0.742 г, 2.18 ммоль) в DCM (22 мл) при комнатной температуре. Реакционную смесь перемешивали в течение 18 ч. Реакционную смесь затем охлаждали до 0°С и останавливали добавлением воды (50 мл) и HCl (1.0 М в воде, 50 мл, 50 ммоль). Реакционную смесь затем разбавляли EtOAc. Органический слой отделяли, высушивали над MgSO4, фильтровали и концентрировали под пониженным давлением с получением метил (R)-4-(4-фтор-5-гидрокси-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата, который применяли без очистки. ЖХ-МС (C15H16FO5S) (ES, m/z): 327 [М+Н]+.
Промежуточное соединение 106: Метил (S)-4-(4-хлор-5-гидрокси-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноат
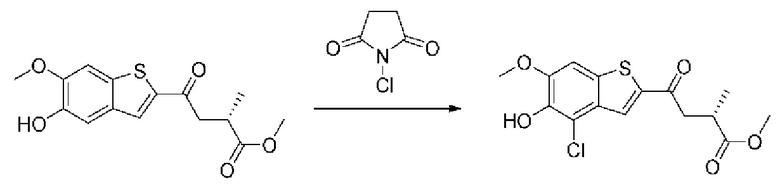
NCS (11 мг, 0.081 ммоль) добавляли к смеси (S)-метил 4-(5-гидрокси-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата (25 мг, 0.081 ммоль) в DMF (0.25 мл) при комнатной температуре. Реакционную смесь затем нагревали до 40°С и перемешивали в течение 2 ч. Реакционную смесь охлаждали до комнатной температуры и сразу очищали с помощью хроматографии на силикагеле (EtOAc/гексан) с получением (S)-метил 4-(4-хлор-5-гидрокси-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата. ЖХ-МС (C15H16ClO5S) (ES, m/z): 343 [М+Н]+. 1H ЯМР (499 МГц, DMSO-d6) δ 9.78 (s, 1H), 8.16 (s, 1H), 7.63 (s, 1H), 3.94 (s, 3H), 3.60 (s, 3H), 3.50 (dd, J=17.8, 8.7 Гц, 1H), 3.31-3.23 (m, 1H), 3.02-2.92 (m, 1H), 1.20 (d, J=7.2 Гц, 3Н).
Промежуточное соединение 107: Метил (S)-4-(4-бром-5-гидрокси-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксо6утаноат
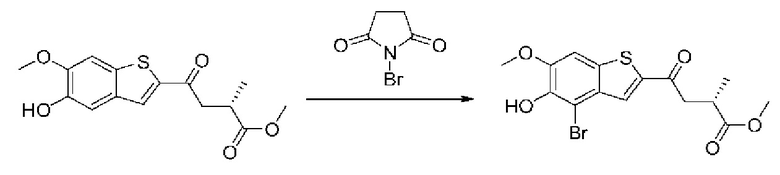
NBS (14 мг, 0.081 ммоль) добавляли к смеси (S)-метил 4-(5-гидрокси-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата (25 мг, 0.081 ммоль) в DMF (0.25 мл) при комнатной температуре. Реакционную смесь затем нагревали до 40°С и перемешивали в течение 2 ч. Реакционную смесь охлаждали до комнатной температуры и сразу очищали с помощью хроматографии на силикагеле (EtOAc/DCM) с получением (S)-метил 4-(4-бром-5-гидрокси-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата. ЖХ-МС (C15H16BrO5S) (ES, m/z): 387, 389 [М+Н]+. 1H ЯМР (499 МГц, DMSO-d6) δ 9.82 (s, 1H), 8.08 (s, 1Н), 7.66 (s, 1H), 3.94 (s, 3Н), 3.60 (s, 3Н), 3.50 (dd, J=17.8, 8.7 Гц, 1H), 3.31-3.24 (m, 1H), 3.03-2.92 (m, 1H), 1.21 (d, J=7.2 Гц, 3Н).
Промежуточное соединение 108: Метил 4-(6-гидрокси-5-метоксибензо[b]тиофен-2-ил)-4-оксобутаноат
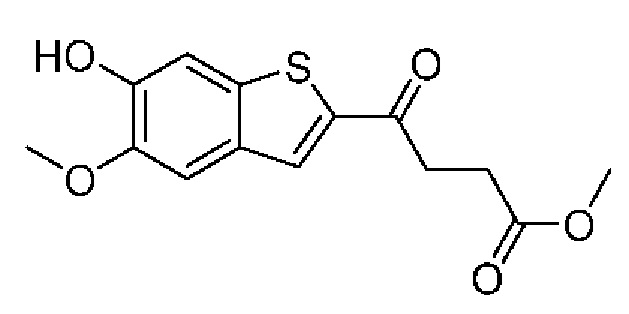
Стадия 1: метил 4-(5,6-диметоксибензо[b]тиофен-2-ил)-4-оксобутаноат
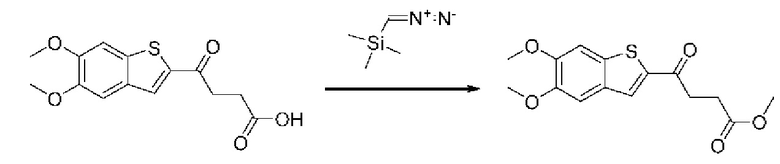
TMS-диазометан (2.0 М в диэтиловом эфире, 5.5 мл, 11 ммоль) добавляли по каплям к смеси 4-(5,6-диметоксибензо[b]тиофен-2-ил)-4-оксобутановой кислоты (2.15 г, 7.30 ммоль) в DCM (50 мл) и МеОН (50 мл) при 0°С. Реакционную смесь перемешивали при 0°С в течение 1 ч. Реакционную смесь останавливали НОАс. Реакционную смесь концентрировали под пониженным давлением с получением метил 4-(5,6-диметоксибензо[b]тиофен-2-ил)-4-оксобутаноата, который применяли без очистки на следующей стадии. ЖХ-МС (C15H17O5S) (ES, m/z): 309 [М+Н]+. 1Н ЯМР (499 МГц, DMSO-d6) δ 8.22 (s, 1H), 7.61 (s, 1H), 7.49 (s, 1H), 3.87 (s, 3H), 3.84 (s, 3H), 3.61 (s, 3H), 3.35-3.30 (m, 2H), 2.71-2.66 (m, 2H).
Стадия 2: метил 4-(6-гидрокси-5-метоксибензо[b]тиофен-2-ил)-4-оксобутаноат и метил 4-(5-гидрокси-6-метоксибензо[b]тиофен-2-ил)-4-оксобутаноат
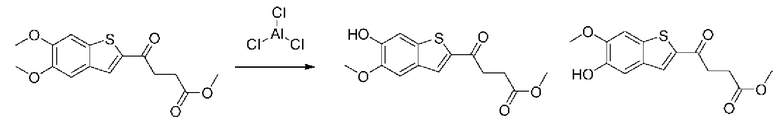
AlCl3 (5.71 г, 42.8 ммоль) добавляли к смеси метил 4-(5,6-диметоксибензо[b]тиофен-2-ил)-4-оксобутаноата (2.20 г, 7.13 ммоль) в DCM (250 мл) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 24 ч. Реакционную смесь охлаждали до 0°С и останавливали водой (50 мл, добавляли по каплям с помощью капельной воронки). Реакционную смесь затем нагревали до комнатной температуры и разбавляли дополнительным DCM (250 мл). Органический слой отделяли, высушивали над MgSO4, фильтровали и концентрировали под пониженным давлением. Остаток очищали с помощью хроматографии на силикагеле (EtOAc/DCM) с получением неразделенной смеси метил 4-(6-гидрокси-5-метоксибензо[b]тиофен-2-ил)-4-оксобутаноата (77%) и метил 4-(5-гидрокси-6-метоксибензо[b]тиофен-2-ил)-4-оксобутаноата (23%). ЖХ-МС (C14H15O5S) (ES, m/z): 295 [М+Н]+.
Стадия 3: метил 4-(5-(((бензилокси)карбонил)окси)-6-метоксибензо[b]тиофен-2-ил)-4-оксобутаноат и метил 4-(6-(((бензилокси)карбонил)окси)-5-метоксибензо[b]тиофен-2-ил)-4-оксобутаноат
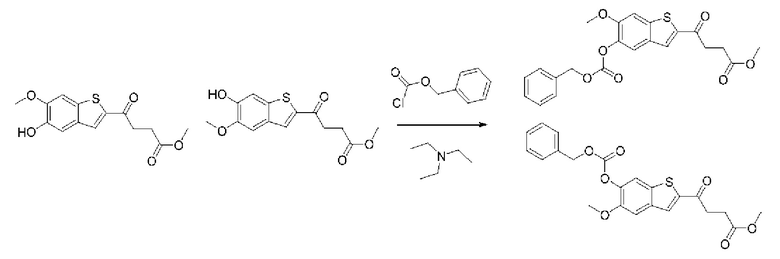
CBZ-C1 (1.06 мл, 7.42 ммоль) добавляли к смеси метил 4-(6-гидрокси-5-метоксибензо[b]тиофен-2-ил)-4-оксобутаноата (77%) и метил 4-(5-гидрокси-6-метоксибензо[b]тиофен-2-ил)-4-оксобутаноата (23%) (1.82 г, 6.18 ммоль) и TEA (1.29 мл, 9.28 ммоль) в DCM (30 мл) при 0°С. Реакционную смесь затем нагревали до комнатной температуры и перемешивали в течение дополнительных 2 ч. Реакционную смесь концентрировали под пониженным давлением, и остаток очищали с помощью хроматографии на силикагеле (EtOAc/гексан) с получением:
Пик 1: метил 4-(5-(((бензилокси)карбонил)окси)-6-метоксибензо[b]тиофен-2-ил)-4-оксобутаноат. ЖХ-МС (C22H21O7S) (ES, m/z): 429 [М+Н]+. 1Н ЯМР (499 МГц, DMSO-d6) δ 8.30 (s, 1Н), 7.89 (s, 1H), 7.84 (s, 1H), 7.48-7.38 (m, 5H), 5.30 (s, 2H), 3.87 (s, 3H), 3.61 (s, 3H), 3.40-3.33 (m, 2H), 2.69 (t, J=6.4 Гц, 2H).
Пик 2: метил 4-(6-(((бензилокси)карбонил)окси)-5-метоксибензо[b]тиофен-2-ил)-4-оксобутаноат. ЖХ-МС (C22H21O7S) (ES, m/z): 429 [М+Н]+. 1Н ЯМР (499 МГц, DMSO-d6) δ 8.32 (s, 1Н), 8.01 (s, 1H), 7.71 (s, 1H), 7.47-7.37 (m, 5H), 5.31 (s, 2H), 3.85 (s, 3H), 3.62 (s, 3H), 3.41-3.36 (m, 2H), 2.74-2.68 (m, 2H).
Стадия 4: метил 4-(6-гидрокси-5-метоксибензо[b]тиофен-2-ил)-4-оксобутаноат
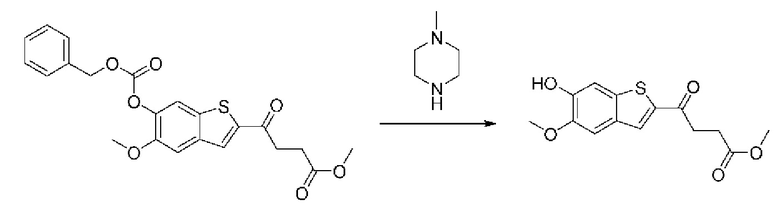
1-Метилпиперазин (1.4 мл, 13 ммоль) добавляли к смеси метил 4-(6-(((бензилокси)карбонил)окси)-5-метоксибензо[b]тиофен-2-ил)-4-оксобутаноата (1.84 г, 4.29 ммоль) в DMF (5 мл) и МеОН (5 мл) при комнатной температуре. Реакционную смесь затем нагревали до 50°С и перемешивали в течение дополнительных 30 мин. Реакционную смесь охлаждали до комнатной температуры и частично концентрировали под пониженным давлением. Сырую смесь очищали с помощью хроматографии на силикагеле (EtOAc/DCM) с получением метил 4-(6-гидрокси-5-метоксибензо[b]тиофен-2-ил)-4-оксобутаноата. ЖХ-МС (C14H15O5S) (ES, m/z): 295 [М+Н]+. 1Н ЯМР (499 МГц, DMSO-d6) δ 9.88 (s, 1H), 8.18 (s, 1H), 7.47 (s, 1H), 7.32 (s, 1H), 3.86 (s, 3Н), 3.61 (s, 3Н), 3.33-3.29 (m, 2H), 2.71-2.66 (m, 2H).
Промежуточное соединение 109: трет-Бутил (S)-4-(5-гидрокси-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноат
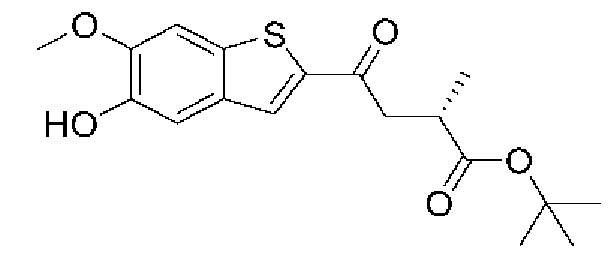
Стадия 1: (S)-4-(5-гидрокси-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутановая кислота
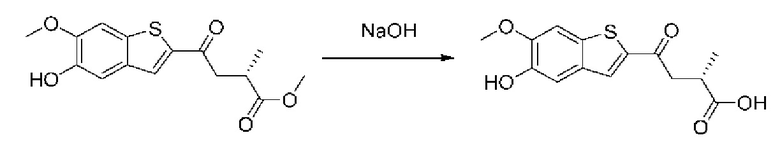
К смеси (S)-метил 4-(5-гидрокси-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата (3.00 г, 9.73 ммоль) в МеОН (97 мл) и THF (97 мл) добавляли NaOH (5.0 М в воде, 39 мл, 200 ммоль). Смесь нагревали до 50°С в течение 1.5 ч. Реакционную смесь охлаждали до комнатной температуры и подкисляли до рН~3 с помощью HCl (2.0 М в воде, 100 мл, 200 ммоль). Смесь разбавляли EtOAc и водой. Органический слой отделяли, высушивали над MgSO4, фильтровали и концентрировали под пониженным давлением с получением (S)-4-(5-гидрокси-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутановой кислоты. ЖХ-МС (C14H15O5S) (ES, m/z): 295 [М+Н]+. 1H ЯМР (499 МГц, DMSO-d6) δ 12.18 (s, 1H), 9.39 (s, 1Н), 8.17 (s, 1H), 7.54 (s, 1H), 7.32 (s, 1H), 3.88 (s, 3Н), 3.38 (dd, J=17.3, 8.5 Гц, 1H), 3.07 (dd, J=17.3, 5.3 Гц, 1H), 2.94-2.83 (m, 1H), 1.18 (d, J=7.2 Гц, 3Н).
Стадия 2: трет-бутил (3)-4-(5-гидрокси-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноат
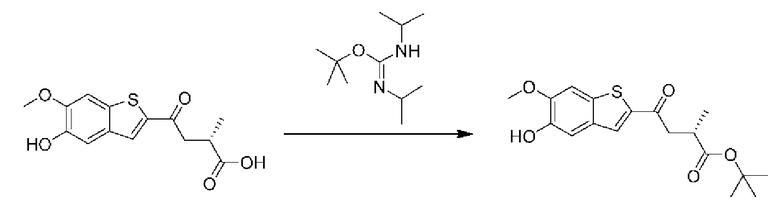
2-трет-Бутил-1,3-диизопропилизомочевину (2.3 мл, 10 ммоль) добавляли к смеси (S)-4-(5-гидрокси-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутановой кислоты (1.00 г, 3.40 ммоль) в DMF (6.8 мл) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Смесь разбавляли EtOAc и водой. Органический слой отделяли, высушивали над MgSO4, фильтровали и концентрировали под пониженным давлением. Остаток очищали с помощью хроматографии на силикагеле (EtOAc/гексан) с получением трет-бутил (S)-4-(5-гидрокси-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата. ЖХ-МС (C18H23O5S+Na) (ES, m/z): 373 [M+Na]+. 1H ЯМР (499 МГц, DMSO-d6) δ 9.40 (s, 1Н), 8.17 (s, 1H), 7.54 (s, 1H), 7.31 (s, 1H), 3.88 (s, 3H), 3.36-3.30 (m, 1H), 3.05 (dd, J=17.1, 5.0 Гц, 1H), 2.90-2.79 (m, 1H), 1.35 (s, 9H), 1.16 (d, J=7.2 Гц, 3Н).
Промежуточное соединение 110: Метил (S)-4-(4,7-дихлор-5-гидрокси-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноат
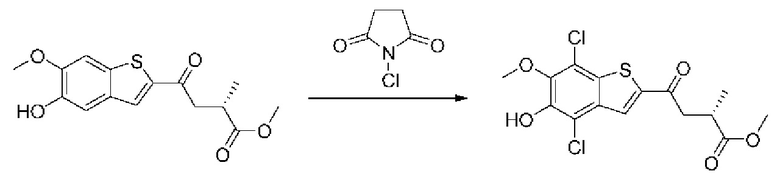
NCS (433 мг, 3.24 ммоль) добавляли к смеси (S)-метил 4-(5-гидрокси-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата (500 мг, 1.62 ммоль) в DMF (4.0 мл) при 20°С. Реакционную смесь затем нагревали до 40°С и перемешивали в течение 2 ч. Реакционную смесь охлаждали до комнатной температуры и разбавляли EtOAc (100 мл). Смесь промывали водой (3×25 мл) и рассолом (1×25 мл). Органический слой отделяли, высушивали над MgSO4, фильтровали и концентрировали под пониженным давлением. Остаток очищали с помощью хроматографии на силикагеле (EtOAc/гексан) с последующей дополнительной очисткой с помощью хроматографии на силикагеле (EtOAc/DCM) с получением метил (S)-4-(4,7-дихлор-5-гидрокси-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата. ЖХ-МС (C15H15Cl2O5S) (ES, m/z): 377, 379 [М+Н]+.
Промежуточное соединение 111: Метил (S)-4-(5-(3-бром-2,2-диметилпуопокси)-4-фтор-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноат
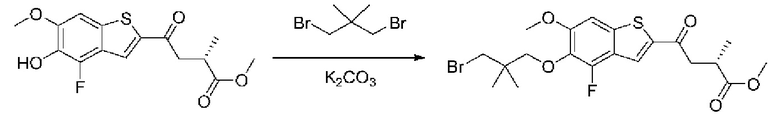
1,3-Дибром-2,2-диметилпропан (176 мг, 0.766 ммоль) добавляли к смеси (S)-метил 4-(4-фтор-5-гидрокси-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата (25 мг, 0.077 ммоль) и карбоната калия (42 мг, 0.31 ммоль) в DMF (0.5 мл) при комнатной температуре. Реакционную смесь затем перемешивали и нагревали до 100°С в течение 24 ч. Реакционную смесь охлаждали до комнатной температуры, разбавляли DCM (2 мл) и фильтровали. Фильтрат сразу очищали с помощью хроматографии на силикагеле (EtOAc/гексан) с получением метил (S)-4-(5-(3-бром-2,2-диметилпропокси)-4-фтор-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата. ЖХ-МС (C20H25BrFO5S) (ES, m/z): 475, 477 [М+Н]+.
Промежуточное соединение 112: Метил (S)-4-(5-((1-(бромметил)циклопропил)метокси)-4-фтор-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноат
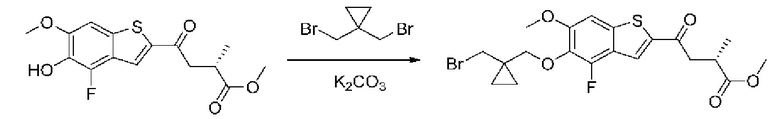
1,1-Бис(бромметил)циклопропан (175 мг, 0.766 ммоль) добавляли к смеси (S)-метил 4-(4-фтор-5-гидрокси-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата (25 мг, 0.077 ммоль) и карбоната калия (42 мг, 0.31 ммоль) в DMF (0.5 мл) при комнатной температуре. Реакционную смесь затем перемешивали и нагревали до 40°С в течение 4 ч. Реакционную смесь охлаждали до комнатной температуры, разбавляли DCM (2 мл) и фильтровали. Фильтрат сразу очищали с помощью хроматографии на силикагеле (EtOAc/гексан) с получением метил (S)-4-(5-((1-(бромметил)циклопропил)метокси)-4-фтор-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата. ЖХ-МС (C20H23BrFO5S) (ES, m/z): 473, 475 [М+Н]+.
Промежуточное соединение 113: Метил (S)-4-(5-гидрокси-6-(метоксиметокси)бензо[b]тиофен-2-ил)-2-метил-4-оксобутаноат
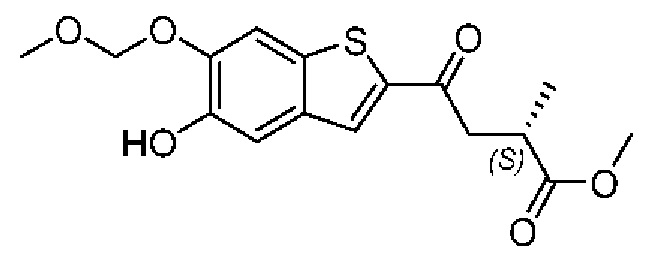
Стадия 1: метил (S)-4-(5-бром-6-гидроксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноат
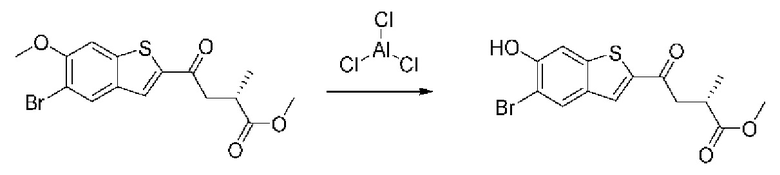
AlCl3 (3.23 г, 24.2 ммоль) добавляли к смеси метил (S)-4-(5-бром-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата (1.50 г, 4.04 ммоль) в DCM (35 мл) при комнатной температуре. Реакционную смесь перемешивали и нагревали до 35°С в течение 24 ч. Реакционную смесь охлаждали до 0°С и затем останавливали медленным добавлением воды (10 мл). Смесь затем нагревали до комнатной температуры и перемешивали в течение дополнительных 15 мин. Реакционную смесь разбавляли DCM (250 мл), и органический слой отделяли. Органический слой промывали рассолом (50 мл). Органический слой отделяли, высушивали над MgSO4, фильтровали и концентрировали под пониженным давлением. Остаток очищали с помощью хроматографии на силикагеле (EtOAc/гексан) с получением метил (S)-4-(5-бром-6-гидроксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата. ЖХ-МС (C14H14BrO4S) (ES, m/z): 357, 359 [М+Н]+.
Стадия 2: метил (S)-4-(5-бром-6-(метоксиметокси)бензо[b]тиофен-2-ил)-2-метил-4-оксобутаноат
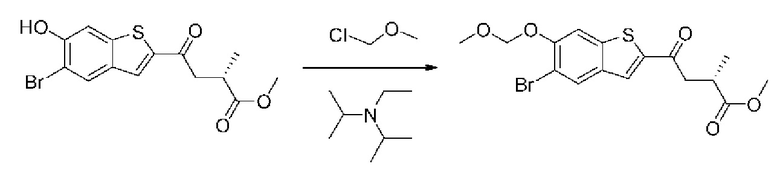
МОМ-Cl (0.91 мл, 12 ммоль) добавляли к смеси метил (S)-4-(5-бром-6-гидроксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата (1.42 г, 3.98 ммоль) и основания Хунига (4.2 мл, 24 ммоль) в DCM (25 мл) при 0°С. Реакционную смесь перемешивали при 0°С в течение 1 ч и затем нагревали до комнатной температуры и перемешивали в течение дополнительных 24 ч. Реакционную смесь останавливали добавлением насыщенного водного NaHCO3 (10 мл) и затем разбавляли EtOAc (250 мл) и водой (50 мл). Органический слой отделяли, промывали рассолом (25 мл), высушивали над MgSO4, фильтровали и концентрировали под пониженным давлением. Остаток очищали с помощью хроматографии на силикагеле (EtOAc/гексан) с получением метил (S)-4-(5-бром-6-(метоксиметокси)бензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата. ЖХ-МС (C16H18BrO5S) (ES, m/z): 401, 403 [М+Н]+. 1Н ЯМР (499 МГц, DMSO-d6) δ 8.30 (s, 1Н), 8.28 (s, 1H), 7.87 (s, 1H), 5.41 (s, 2H), 3.60 (s, 3H), 3.48-3.42 (m, 1H), 3.45 (s, 3H), 3.22 (dd, J=17.6, 5.1 Гц, 1H), 3.04-2.93 (m, 1H), 1.20 (d, J=7.4 Гц, 3Н).
Стадия 3: метил (S)-4-(6-(метоксиметокси)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензо[b]тиофен-2-ил)-2-метил-4-оксобутаноат
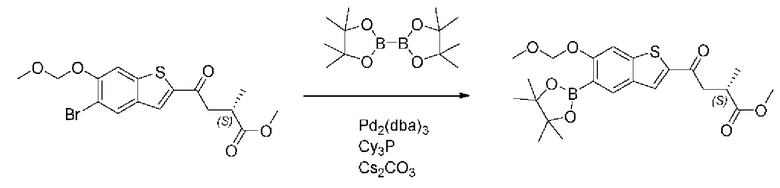
Смесь метил (S)-4-(5-бром-6-(метоксиметокси)бензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата (1.57 г, 3.91 ммоль), 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолан) (1.19 г, 4.70 ммоль), Pd2(dba)3 (0.179 г, 0.196 ммоль), трициклогексилфосфина (0.219 г, 0.783 ммоль) и ацетата калия (0.614 г, 6.26 ммоль) дегазировали Ar в течение 5 мин. Диоксан (20 мл) добавляли при комнатной температуре, и полученную в результате смесь дегазировали Ar в течение 5 мин. Реакционную смесь затем нагревали до 90°С и перемешивали в течение 6 ч в атмосфере Ar. Реакционную смесь охлаждали до комнатной температуры и затем разбавляли EtOAc (50 мл). Суспензию перемешивали при комнатной температуре в течение 10 мин, и затем фильтровали через целит, промывая EtOAc (50 мл). Фильтрат концентрировали под пониженным давлением с получением метил (S)-4-(6-(метоксиметокси)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата, который применяли без очистки на следующей стадии. ЖХ-МС (C22H30BO7S) (ES, m/z): 449 [М+Н]+.
Стадия 4: метил (S)-4-(5-гидрокси-6-(метоксиметокси)бензо[b]тиофен-2-ил)-2-метил-4-оксобутаноат
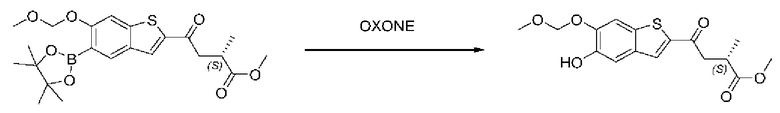
Раствор OXONE® (0.20 М в воде, 21 мл, 4.3 ммоль) добавляли к раствору метил (S)-4-(6-(метоксиметокси)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата (1.75 г, 3.90 ммоль) в ацетоне (50 мл) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 60 мин. Реакционную смесь останавливали добавлением раствора бисульфита натрия (0.81 г, 7.8 ммоль) в воде (5 мл) и затем перемешивали в течение 5 мин. Реакционную смесь разбавляли EtOAc (250 мл). Органический слой отделяли, и водный слой промывали дополнительным объемом EtOAc (2×100 мл). Органические слои объединяли, промывали рассолом (25 мл), высушивали над MgSO4, фильтровали и концентрировали под пониженным давлением. Остаток очищали с помощью хроматографии на силикагеле (EtOAc/гексан) с получением метил (S)-4-(5-гидрокси-6-(метоксиметокси)бензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата, загрязненного побочным продуктом пинакола. Выделенное вещество повторно очищали с помощью хроматографии на силикагеле (EtOAc/DCM) с получением метил (S)-4-(5-гидрокси-6-(метоксиметокси)-бензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата. ЖХ-МС (C16H19O6S) (ES, m/z): 339 [М+Н]+. 1Н ЯМР (499 МГц, DMSO-d6) δ 9.52 (s, 1Н), 8.20 (s, 1H), 7.61 (s, 1H), 7.37 (s, 1H), 5.29 (s, 2H), 3.60 (s, 3H), 3.44 (s, 3H), 3.43-3.38 (m, 1H), 3.18 (dd, J=17.5, 5.1 Гц, 1H), 3.03-2.91 (m, 1H), 1.19 (d, J=7.2 Гц, 3Н).
Промежуточное соединение 114: Метил (S)-4-(4-хлор-5-гидрокси-6-(метоксиметокси)бензо[b]тиофен-2-ил)-2-метил-4-оксобутаноат
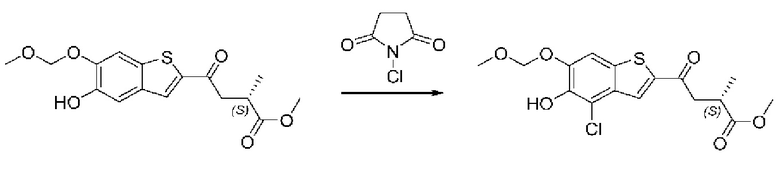
NCS (99 мг, 0.74 ммоль) добавляли к смеси метил (S)-4-(5-гидрокси-6-(метоксиметокси)бензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата (250 мг, 0.74 ммоль) в DMF (2.0 мл) при комнатной температуре. Реакционную смесь перемешивали и нагревали до 40°С в течение 2 ч. Реакционную смесь охлаждали до комнатной температуры и затем сразу очищали с помощью хроматографии на силикагеле (EtOAc/гексан) с получением метил (S)-4-(4-хлор-5-гидрокси-6-(метоксиметокси)бензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата. ЖХ-МС (C16H18ClO6S) (ES, m/z): 373 [M+H]+. 1H ЯМР (499 МГц, DMSO-d6) δ 9.83 (s, 1H), 8.17 (s, 1H), 7.67 (s, 1H), 5.35 (s, 2Н), 3.60 (s, 3Н), 3.51 (dd, J=17.8, 8.7 Гц, 1H), 3.46 (s, 3Н), 3.27 (dd, J=17.8, 5.0 Гц, 1H), 3.02-2.91 (m, 1H), 1.20 (d, J=7.2 Гц, 3Н).
Промежуточное соединение 115: Метил (S)-4-(5-(3-бромпропокси)-4-фтор-6-(метоксиметокси)бензо[b]тиофен-2-ил)-2-метил-4-оксобутаноат
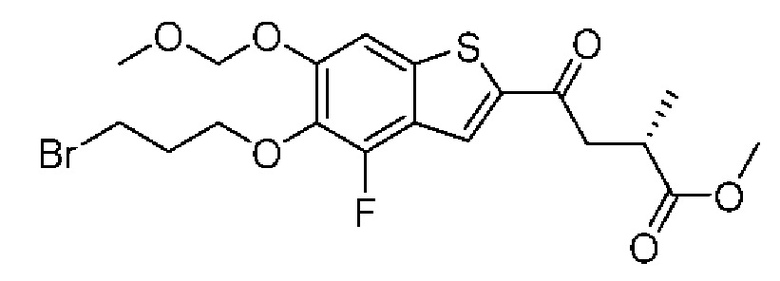
Стадия 1: (S)-4-(4-фтор-5,6-дигидроксибензо[b]тиофен-2-ил)-2-метил-4-оксобутановая кислота
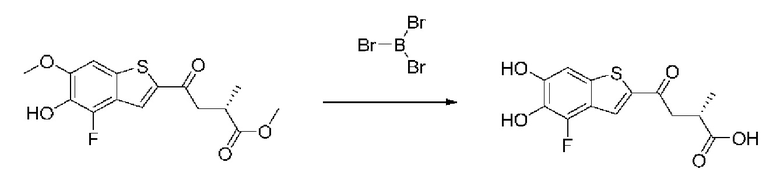
Трибромид бора (1.0 M в DCM, 31 мл, 31 ммоль) добавляли к смеси метил (S)-4-(4-фтор-5-гидрокси-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата (2.0 г, 6.1 ммоль) в DCM (100 мл) при 0°С. Реакционную смесь перемешивали и нагревали до 30°С в течение 2 ч. Реакционную смесь охлаждали до 0°С, и реакцию затем останавливали путем добавления по каплям МеОН (10 мл). Реакционную смесь перемешивали в течение дополнительных 15 мин. Реакционную смесь затем разбавляли водой (100 мл) и дополнительным DCM (500 мл). Органический слой отделяли, промывали рассолом (50 мл), высушивали над MgSO4, фильтровали и концентрировали под пониженным давлением с получением (S)-4-(4-фтор-5,6-дигидроксибензо[b]тиофен-2-ил)-2-метил-4-оксобутановой кислоты, которую применяли без очистки на следующей стадии. ЖХ-МС (C13H12FO5S) (ES, m/z): 299 [М+Н]+.
Стадия 2: метил (S)-4-(4-фтор-5,6-дигидроксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноат
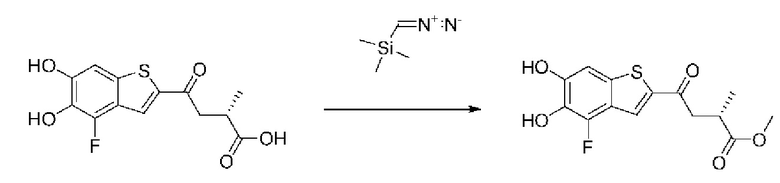
TMS-диазометан (2.0 М в диэтиловом эфире, 3.1 мл, 6.2 ммоль) добавляли к смеси (S)-4-(4-фтор-5,6-дигидроксибензо[b]тиофен-2-ил)-2-метил-4-оксобутановой кислоты (1.83 г, 6.14 ммоль) в DCM (10 мл) и МеОН (10 мл) при 0°С. Реакционную смесь затем перемешивали при 0°С в течение 15 мин. Реакционную смесь останавливали НОАс и затем концентрировали под пониженным давлением. Остаток очищали с помощью хроматографии на силикагеле (EtOAc/гексан) с получением целевого продукта. Выделенное вещество очищали с помощью хроматографии на силикагеле ([25% EtOH в EtOAc]/гексан) с получением метил (S)-4-(4-фтор-5,6-дигидроксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата. ЖХ-МС (C14H14FO5S) (ES, m/z): 313 [М+Н]+.
Стадия 3: метил (S)-4-(4-фтор-5-гидрокси-6-(метоксиметокси)бензо[b]тиофен-2-ил)-2-метил-4-оксобутаноат и метил (S)-4-(4-фтор-6-гидрокси-5-(метоксиметокси)бензо[b]тиофен-2-ил)-2-метил-4-оксобутаноат
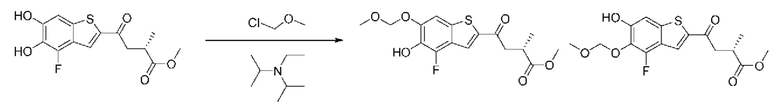
MOM-Cl (0.12 мл, 1.6 ммоль) добавляли к смеси метил (S)-4-(4-фтор-5,6-дигидроксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата (460 мг, 1.47 ммоль) и основания Хунига (0.52 мл, 3.0 ммоль) в DCM (25 мл) при 0°С. Реакционную смесь перемешивали при 0°С в течение 1 ч и затем нагревали до комнатной температуры и перемешивали в течение дополнительных 24 ч. Реакционную смесь останавливали добавлением насыщенного водного NaHCO3 (10 мл) и затем разбавляли EtOAc (250 мл) и водой (50 мл). Органический слой отделяли, промывали рассолом (25 мл), высушивали над MgSO4, фильтровали и концентрировали под пониженным давлением. Остаток очищали с помощью хроматографии на силикагеле (EtOAc/гексан) с получением неразделенной смеси метил (S)-4-(4-фтор-5-гидрокси-6-(метоксиметокси)бензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата и метил (S)-4-(4-фтор-6-гидрокси-5-(метоксиметокси)бензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата. Смесь применяли в следующей реакции без дополнительной очистки. ЖХ-МС (C18H18FO6S) (ES, m/z): 357 [М+Н]+.
Стадия 4: метил (S)-4-(6-(3-бромпропокси)-4-фтор-5-(метоксиметокси)бензо[b]тиофен-2-ил)-2-метил-4-оксобутаноат и метил (S)-4-(5-(3-бромпропокси)-4-фтор-6-(метоксиметокси)бензо[b]тиофен-2-ил)-2-метил-4-оксобутаноат
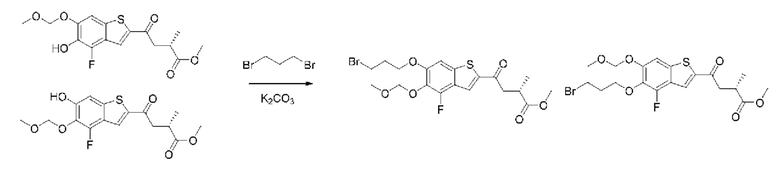
1,3-Дибромпропан (0.58 мл, 5.6 ммоль) добавляли к смеси метил (S)-4-(4-фтор-5-гидрокси-6-(метоксиметокси)бензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата и метил (S)-4-(4-фтор-6-гидрокси-5-(метоксиметокси)бензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата (270 мг, 0.75 ммоль) и карбоната калия (260 мг, 1.9 ммоль) в DMF (2.0 мл) при комнатной температуре. Смесь перемешивали при комнатной температуре в течение 18 ч. Реакционную смесь разбавляли EtOAc (250 мл) и водой (50 мл). Органический слой отделяли, промывали рассолом (50 мл), высушивали над MgSO4, фильтровали и концентрировали под пониженным давлением. Остаток очищали с помощью хроматографии на силикагеле (EtOAc/гексаны) с получением смеси региоизомеров. Смесь региоизомеров очищали с помощьюВЭЖХ с обратными фазами (ACN/вода с 0.1% TFA) с получением:
Пик, элюируемый первым, ВЭЖХ: метил (S)-4-(6-(3-бромпропокси)-4-фтор-5-(метоксиметокси)бензо[b]тиофен-2-ил)-2-метил-4-оксобутаноат. ЖХ-МС (C19H23BrFO6S) (ES, m/z): 477, 479 [М+Н]+.
Пик, элюируемый вторым, ВЭЖХ: метил (S)-4-(5-(3-бромпропокси)-4-фтор-6-(метоксиметокси)бензо[b]тиофен-2-ил)-2-метил-4-оксобутаноат. ЖХ-МС (C19H23BrFO6S) (ES, m/z): 477, 479 [М+Н]+.
Промежуточное_соединение 116: Метил (S)-4-(5-гидрокси-6-(метиламино)бензо[b]тиофен-2-ил)-2-метил-4-оксобутаноат
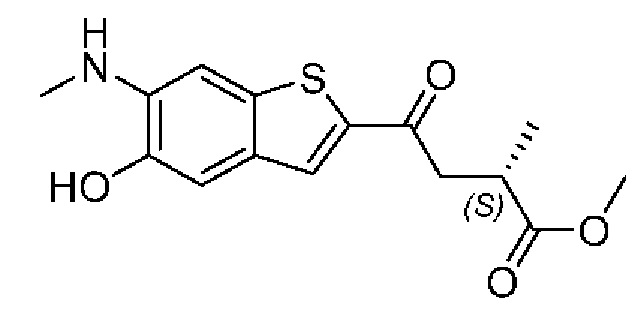
Стадия 1: метил (S)-4-(6-бром-5-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноат
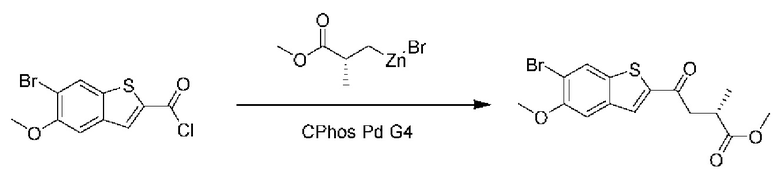
6-Бром-5-метоксибензо[b]тиофен-2-карбонилхлорид (5.96 г, 19.5 ммоль) и CPhos Pd G4 (0.160 г, 0.195 ммоль) объединяли в колбе и дегазировали Ar в течение 5 мин. THF (75 мл) добавляли в атмосфере Ar, и смесь охлаждали до 0°С. Затем добавляли по каплям (R)-(3-метокси-2-метил-3-оксопропил)цинк (II) бромид (0.50 М в THF, 40 мл, 20 ммоль), и полученную в результате смесь перемешивали при 0°С в течение 2 ч и затем нагревали до комнатной температуры. Смесь перемешивали при комнатной температуре в течение 3 дней. Реакционную смесь останавливали насыщенным водным NH4Cl (25 мл) и затем разбавляли EtOAc (500 мл). Органический слой отделяли, промывали рассолом (50 мл), высушивали над MgSO4, фильтровали и концентрировали под пониженным давлением. Остаток очищали с помощью хроматографии на силикагеле (EtOAc/гексаны) с получением метил (S)-4-(6-бром-5-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата. ЖХ-МС (C15H16BrO4S) (ES, m/z): 371, 373 [М+Н]+. 1Н ЯМР (499 МГц, DMSO-d6) δ 8.39 (s, 1H), 8.30 (s, 1H), 7.65 (s, 1Н), 3.94 (s, 3Н), 3.61 (s, 3Н), 3.52-3.43 (m, 1H), 3.28-3.18 (m, 1H), 3.05-2.95 (m, 1H), 1.23-1.18 (m, 3H).
Стадия 2: метил (S)-4-(6-бром-5-гидроксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноат
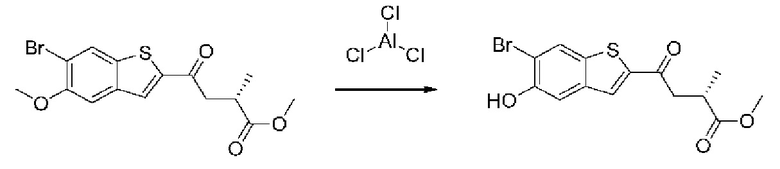
AlCl3 (366 мг, 2.75 ммоль) добавляли к смеси метил (S)-4-(6-бром-5-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата (170 мг, 0.458 ммоль) в DCM (35 мл) при комнатной температуре. Реакционную смесь перемешивали и нагревали до 45°С в течение 3 дней. Реакционную смесь охлаждали до 0°С, и реакцию затем останавливали медленным добавлением воды (10 мл). Смесь затем нагревали до комнатной температуры и перемешивали в течение дополнительных 15 мин. Реакционную смесь разбавляли DCM (250 мл), и органический слой отделяли. Органический слой промывали рассолом (50 мл). Органический слой отделяли, высушивали над MgSO4, фильтровали и концентрировали под пониженным давлением. Остаток очищали с помощью хроматографии на силикагеле (EtOAc/гексан) с получением метил (S)-4-(6-бром-5-гидроксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата. ЖХ-МС (C14H14BrO4S) (ES, m/z): 357, 359 [М+Н]+.
Стадия 3: метил (3)-4-(6-бром-5-(метоксиметокси)бензо[b]тиофен-2-ил)-2-метил-4-оксобутаноат
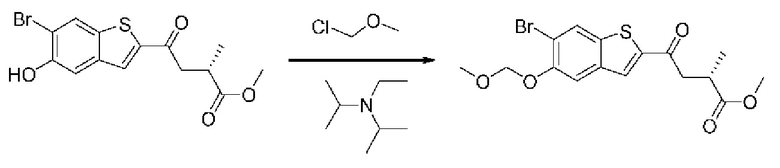
Основание Хунига (0.44 мл, 2.5 ммоль) добавляли к смеси метил (S)-4-(6-бром-5-гидроксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата (150 мг, 0.42 ммоль) и МОМ-Cl (0.096 мл, 1.3 ммоль) в DCM (5.0 мл) при 0°С. Реакционную смесь перемешивали при 0°С в течение 1 ч. Реакционную смесь сразу очищали с помощью хроматографии на силикагеле (EtOAc/гексан) с получением метил (S)-4-(6-бром-5-(метоксиметокси)бензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата. ЖХ-МС (C16H18BrO5S) (ES, m/z): 401, 403 [M+H]+. 1H ЯМР (499 МГц, DMSO-d6) δ 8.42 (s, 1H), 8.33 (s, 1H), 7.77 (s, 1H), 5.38 (s, 2H), 3.61 (s, 3H), 3.53-3.42 (m, 4H), 3.23 (dd, J=17.7, 3.0 Гц, 1H), 3.07-2.95 (m, 1H), 1.21 (d, J=5.0 Гц, 3Н).
Стадия 4: метил (S)-4-(6-((трет-бутоксикарбонил)(метил)амино)-5-(метоксиметокси)бензо[b]тиофен-2-ил)-2-метил-4-оксобутаноат
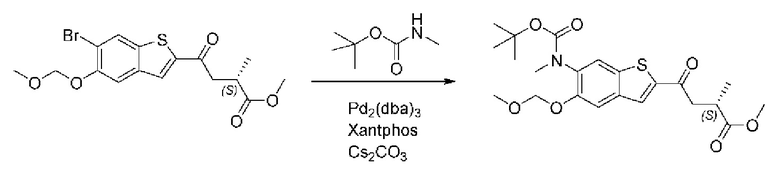
К дегазированной Ar смеси метил (S)-4-(6-бром-5-(метоксиметокси)бензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата (144 мг, 0.359 ммоль), трет-бутилметилкарбамата (71 мг, 0.54 ммоль), Pd2(dba)3 (16 мг, 0.018 ммоль), Xantphos (31 мг, 0.054 ммоль) и карбоната цезия (234 мг, 0.718 ммоль) добавляли диоксан (4.0 мл) при комнатной температуре, дегазируя Ar. Смесь перемешивали в течение 5 мин, дегазируя Ar, затем смесь нагревали до 90°С и перемешивали в атмосфере Ar в течение 18 ч. Реакционную смесь охлаждали до комнатной температуры и разбавляли EtOAc (20 мл). Полученную в результате суспензию фильтровали через целит. Фильтрат концентрировали под пониженным давлением. Остаток очищали с помощью хроматографии на силикагеле (EtOAc/гексан) с получением указанного в заголовке соединения. ЖХ-МС (C22H30NO7S) (ES, m/z): 452 [М+Н]+.
Стадия 5: метил (S)-4-(5-гидрокси-6-(метиламино)бензо[b]тиофен-2-ил)-2-метил-4-оксобутаноат
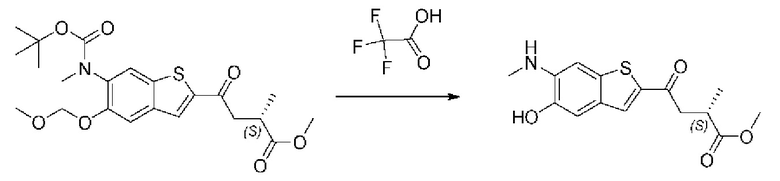
TFA (1.0 мл, 13 ммоль) добавляли к смеси метил (S)-4-(6-((tpet-бутоксикарбонил)(метил)амино)-5-(метоксиметокси)бензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата (18 мг, 0.040 ммоль) в DCM (2.0 мл) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Реакционную смесь разбавляли EtOAc (10 мл), и реакцию медленно останавливали добавлением насыщенного водного раствора NaHCO3 (10 мл). Полученную в результате смесь перемешивали в течение 10 мин. Органический слой отделяли, промывали рассолом (5 мл), высушивали над MgSO4, фильтровали и концентрировали под пониженным давлением. Остаток очищали с помощью хроматографии на силикагеле (EtOAc/гексан) с получением метил (S)-4-(5-гидрокси-6-(метиламино)бензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата. ЖХ-МС (C15H18NO4S) (ES, m/z): 308 [М+Н]+.
Пример 1 (2S,2'S)-4,4'-[бутан-1,4-диилбис(6-метокси-1-бензотиен-5,2-диил)]бис(2-метил-4-оксобутановая кислота) и
Пример 2: (2S,2'S)-4,4'-[пропан-1,3-диилбис(6-метокси-1-бензотиен-5,2-диил)]бис(2-метил-4-оксобутановая кислота)
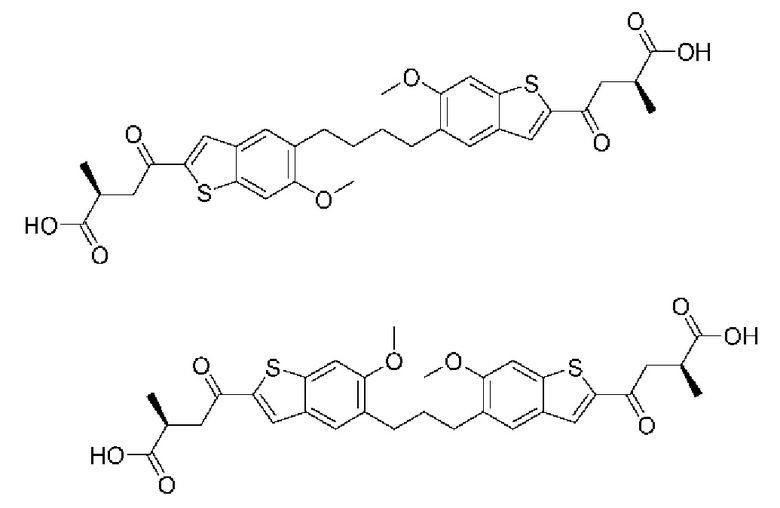
Стадия 1: метил (2S)-4-[6-метокси-5-(проп-2-еп-1-ил)-1-бензотиофен-2-ил]-2-метил-4-оксобутаноат
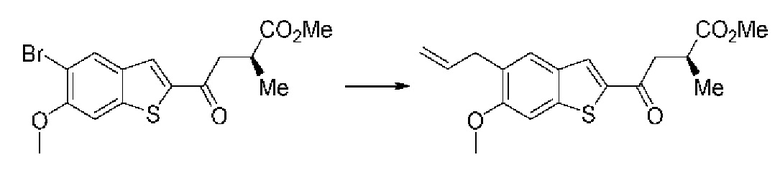
К перемешиваемой смеси метил (2S)-4-(5-бром-6-метокси-1-бензотиофен-2-ил)-2-метил-4-оксобутаноата (120 мг, 0.32 ммоль), бис(дибензилиденацетон)палладия (7.1 мг, 12 мкмоль) и аллилтрибутилолова (120 мкл, 0.39 ммоль) в толуоле (0.49 мл) добавляли три-трет-бутилфосфин (1.0 М в толуоле, 26 мкл, 26 мкмоль) в атмосфере N2. Реакционную смесь нагревали до 65°С в течение 18 ч. После охлаждения до комнатной температуры смесь разбавляли Et2O, и затем добавляли CsF. Неочищенную смесь перемешивали в течение 5 мин при комнатной температуре. Смесь затем фильтровали, и фильтрат сразу очищали с помощью колоночной хроматографии на силикагеле (EtOAc в гексанах) с получением метил (2S)-4-[6-метокси-5-(проп-2-ен-1-ил)-1-бензотиофен-2-ил]-2-метил-4-оксобутаноата. ЖХ-МС (C18H21O4S) (ES, m/z): 333 [М+Н]+. 1Н ЯМР (500 МГц, DMSO-d6): δ 8.27 (s, 1H), 7.72 (s, 1H), 7.62 (s, 1H), 6.06-5.94 (m, 1H), 5.07 (d, J=12.5 Гц, 2Н), 3.89 (s, 3Н), 3.59 (s, 3Н), 3.46-3.38 (т, 3Н), 3.19 (dd, J=17.5, 5.0 Гц, 1H), 3.02-2.92 (m, 1H), 1.19 (d, J=7.1 Гц, 3Н).
Стадия 2: диметил (2S,2'S)-4,4'-[бут-2-ен-1,4-диилбис(6-метокси-1-бензотиен-5,2-диил)]бис(2-метил-4-оксобутаноат) и диметил (2S,2'S)-4,4'-[проп-1'-ен-1,3-диилбис(6-метокси-1-бензотиеп-5,2-диил)]бис(2-метил-4-оксобутаноат)
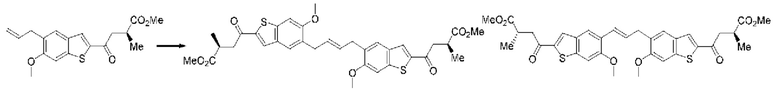
К смеси метил (2S)-4-[6-метокси-5-(проп-2-ен-1-ил)-1-бензотиофен-2-ил]-2-метил-4-оксобутаноата (44 мг, 0.13 ммоль) в CH2Cl2 (1.3 мл) добавляли катализатор Граббса 2G (11 мг, 0.013 ммоль) в CH2Cl2 (1.3 мл) одной порцией в атмосфере N2. Реакционную смесь нагревали до 65°С в течение 18 ч. После охлаждения до комнатной температуры смесь сразу очищали с помощью колоночной хроматографии на силикагеле (EtOAc в гексанах) с получением смеси диметил (2S,2'S)-4,4'-[бут-2-ен-1,4-диилбис(6-метокси-1-бензотиен-5,2-диил)]бис(2-метил-4-оксобутаноат) и диметил (28,2'8)-4,4'-[проп-1-ен-1,3-диилбис(6-метокси-1-бензотиен-5,2-диил)]бис(2-метил-4-оксобутаноат). Характеристики диметил (2S,2'S)-4,4'-[бут-2-ен-1,4-диилбис(6-метокси-1-бензотиен-5,2-диил)]бис(2-метил-4-оксобутаноат): ЖХ-МС (C34H37O8S2) (ES, m/z): 637 [М+Н]+. Характеристики диметил (2S,2'S)-4,4'-[проп-1-ен-1,3-диилбис(6-метокси-1-бензотиен-5,2-диил)]бис(2-метил-4-оксобутаноат): ЖХ-МС (C33H35O8S2) (ES, m/z): 623 [М+Н]+.
Стадия 3: диметил ('2S,2'S)-4,4'-[бутан-1,4-диилбис(6-метокси-1-бензотиен-5,2-диил)]бис(2-метил-4-оксобутаноат) и диметил (2S,2'S)-4,4'-[пропан-1,3-диилбис(6-метокси-1-бензотиен-5,2-диил)]бис(2-метил-4-оксобутаноат)
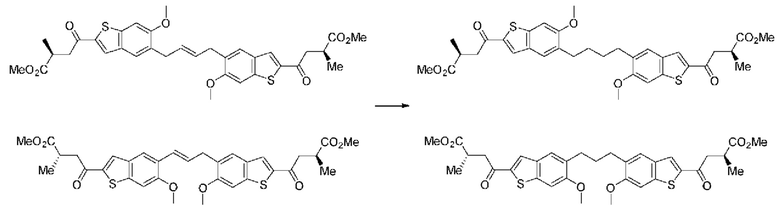
К смеси диметил (2S,2'S)-4,4'-[бут-2-ен-1,4-диилбис(6-метокси-1-бензотиен-5,2-диил)]бис(2-метил-4-оксобутаноат) и диметил (2S,2'S)-4,4'-[проп-1-ен-1,3-диилбис(6-метокси-1-бензотиен-5,2-диил)]бис(2-метил-4-оксобутаноат) (57.0 мг, смесь ~ 3/2 по данным 1H-ЯМР) в EtOAc (0.5 мл) в атмосфере N2 добавляли 10% Pd/C (6.0 мг, 5.4 мкмоль) одной порцией при комнатной температуре. Реакционную смесь дегазировали и снова заполняли H2 (три раза) и перемешивали в атмосфере H2 (баллонный) в течение 18 ч при комнатной температуре. Смесь затем концентрировали под пониженным давлением с получением неочищенной смеси диметил (2S,2'S)-4,4'-[бутан-1,4-диилбис(6-метокси-1-бензотиен-5,2-диил)]бис(2-метил-4-оксобутаноат) и диметил (2S,2'S)-4,4'-[пропан-1,3-диилбис(6-метокси-1-бензотиен-5,2-диил)]бис(2-метил-4-оксобутаноат). Смесь применяли без дополнительной очистки или определения характеристик.
Стадия 4: (2S,2'S)-4,4'-[бутан-1,4-диилбис(6-метокси-1-бензотиен-5,2-диил))'бис(2-метил-4-оксобутановая кислота) и (2S,2'S)-4,4'-[пуопаи-1,3-диилбис(6-метокси-1-бензотиен-5,2-диил)]бис(2-метил-4-оксобутановая кислота) I
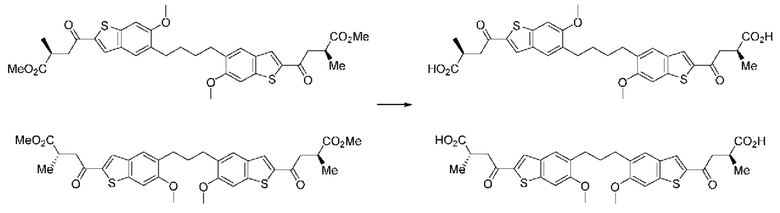
К смеси диметил (28,2'8)-4,4'-[бутан-1,4-диилбис(6-метокси-1-бензотиен-5,2-диил)]бис(2-метил-4-оксобутаноат) и диметил (2S,2'S)-4,4'-[пропан-1,3-диилбис(6-метокси-1-бензотиен-5,2-диил)]бис(2-метил-4-оксобутаноат) (57 мг) в THF (1.0 мл), МеОН (1.0 мл) и воде (0.20 мл) добавляли LiOH (26 мг, 1.1 ммоль) одной порцией при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 1 ч и затем реакцию останавливали водной HCl (2 н., 0.52 мл). Смесь разбавляли DMSO, и полученную в результате смесь фильтровали. Фильтрат очищали с помощью RP-HPLC [колонка С18, вода (0.1% TFA)-CH3CN] с получением (2S,2'S)-4,4'-[бутан-1,4-диилбис(6-метокси-1-бензотиен-5,2-диил)]бис(2-метил-4-оксобутановая кислота) и (2S,2'S)-4,4'-[пропан-1,3-диилбис(6-метокси-1-бензотиен-5,2-диил)]бис(2-метил-4-оксобутановая кислота).
Данные характеристики для (2S,2'S)-4,4'-[бутан-1,4-диилбис(6-метокси-1-бензотиен-5,2-диил)]бис(2-метил-4-оксобутановая кислота): ЖХ-МС (C32H34O8S2Na) (ES, m/z): 633 [M+Na]+. 1H ЯМР (500 МГц, DMSO-A): δ 12.20 (br, 2Н), 8.21 (s, 2Н), 7.72 (s, 2Н), 7.57 (s, 2Н), 3.87 (s, 6Н), 3.39 (dd, J=17.5, 8.6 Гц, 2Н), 3.07 (dd, J=17.5, 5.0 Гц, 2Н), 2.94-2.84 (m, 2Н), 2.69 (br, 4Н), 1.62 (br, 4Н), 1.18 (d, J=7.1 Гц, 6Н).
Данные характеристики для (2S,2'S)-4,4'-[пропан-1,3-диилбис(6-метокси-1-бензотиен-5,2-диил)]бис(2-метил-4-оксобутановая кислота): ЖХ-МС (C31H32O8S2Na) (ES, m/z): 619 [M+Na]+. 1H ЯМР (500 МГц, DMSO-J6): δ 12.21 (br, 2H), 8.23 (s, 2H), 7.76 (s, 2H), 7.59 (s, 2H), 3.88 (s, 6H), 3.40 (dd, J=17.5, 8.6 Гц, 2H), 3.08 (dd, J=17.5, 5.0 Гц, 2H), 2.94-2.85 (m, 2H), 2.71 (t, J=7.1 Гц, 4H), 1.91 (pentet, J=7.1 Гц, 2H), 1.18 (d, J=7.1 Гц, 6H).
Пример 3: (S)-4-(5-(3-(2-(3-карбоксипропаноил)-6-метоксибензо[b]тиофен-5-ил)пропокси)-6-метокситиено[3,2-b]пиридин-2-ил)-2-метил-4-оксобутановая кислота
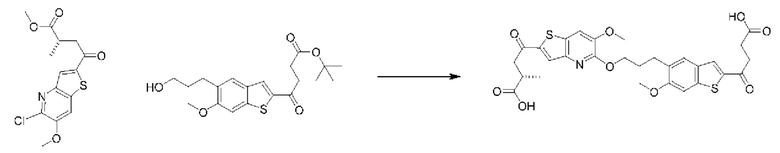
В виалу 1 драм с завинчивающейся крышкой с магнитной мешалкой загружали (S)-метил 4-(5-хлор-6-метокситиено[3,2-b]пиридин-2-ил)-2-метил-4-оксобутаноат (60 мг, 0.18 ммоль), трет-бутил 4-(5-(3-гидроксипропил)-6-метоксибензо[b]тиофен-2-ил)-4-оксобутаноат (76 мг, 0.20 ммоль), RockPhos Pd G3 (7.7 мг, 9.2 мкмоль) и CS2CO3 (89 мг, 0.28 ммоль) и закрывали крышкой с септой. Виалу 3 раза откачивали и снова заполняли N2. Добавляли толуол (0.61 мл), и суспензию перемешивали на вортексе, обрабатывали ультразвуком и затем нагревали до 110°С с перемешиванием в течение 2.25 ч. Реакцию затем оставляли охлаждаться до комнатной температуры. Добавляли TFA (0.60 мл, 7.8 ммоль), и смесь перемешивали при комнатной температуре в течение 1.5 ч. Смесь затем концентрировали под пониженным давлением. К полученному в результате остатку добавляли THF (1.0 мл) и МеОН (0.50 мл). Добавляли водный NaOH (2.0 М, 0.50 мл, 1.0 ммоль), и полученную в результате смесь нагревали до 40°С в течение 5 ч. После охлаждения до комнатной температуры добавляли дополнительный водный NaOH (2.0 М, 0.50 мл, 1.0 ммоль), и смесь нагревали до 40°С в течение дополнительных 2.5 ч. Реакцию затем оставляли охлаждаться до комнатной температуры и концентрировали под пониженным давлением. Добавляли DMSO (1.0 мл), и полученную в результате смесь фильтровали через шприцевой фильтр. Фильтрат затем очищали с помощью RP-HPLC (С 18, градиент MeCN/вода, с NH4OH). Фракцию, содержащую продукт, концентрировали под пониженным давлением и затем дополнительно очищали с помощью RP-HPLC (С 18, градиент MeCN/вода, с TFA) с получением (S)-4-(5-(3-(2-(3-карбоксипропаноил)-6-метоксибензо[b]тиофен-5-ил)пропокси)-6-метокситиено[3,2-b]пиридин-2-ил)-2-метил-4-оксобутановой кислоты. ЖХ-МС (C29H30NO9S2) (ES, m/z): 600 [М+Н]+. 1H ЯМР (500 МГц, DMSO-d6) δ 12.18 (s, 2Н), 8.22 (s, 1H), 8.20 (s, 1H), 7.97 (s, 1H), 7.78 (s, 1H), 7.60 (s, 1H), 4.39 (t, J=6.3 Гц, 2H), 3.90 (s, 3H), 3.87 (s, 3H), 3.43 (dd, J=17.5, 8.6 Гц, 1H), 3.29-3.22 (m, 2H), 3.15-3.06 (m, 1H), 2.91-2.80 (m, 3H), 2.63-2.56 (m, 2H), 2.18-2.06 (m, 2H), 1.17 (d, J=7.2 Гц, 3Н).
Примеры с 4 по 23 и 81-83, как показано в Таблице 11 ниже, или могут быть получены в соответствии со способами, аналогичными описанным в Примере 3 выше, с применением подходящих исходных материалов, описанных выше в Способах получения или Промежуточных соединениях, или получены из коммерческих источников.
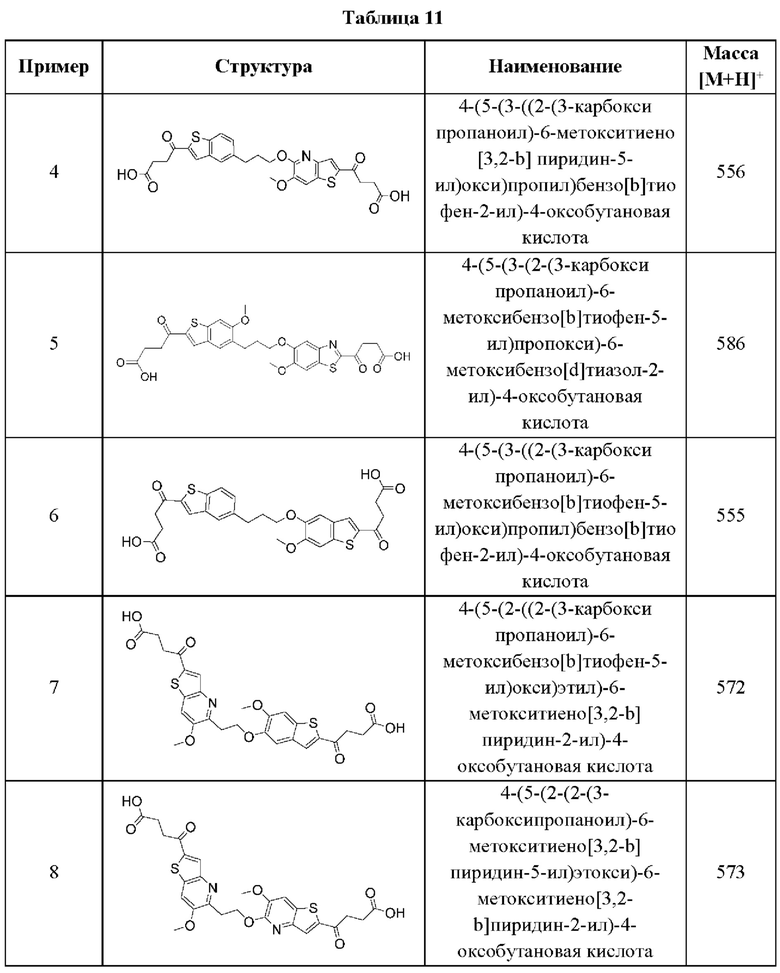
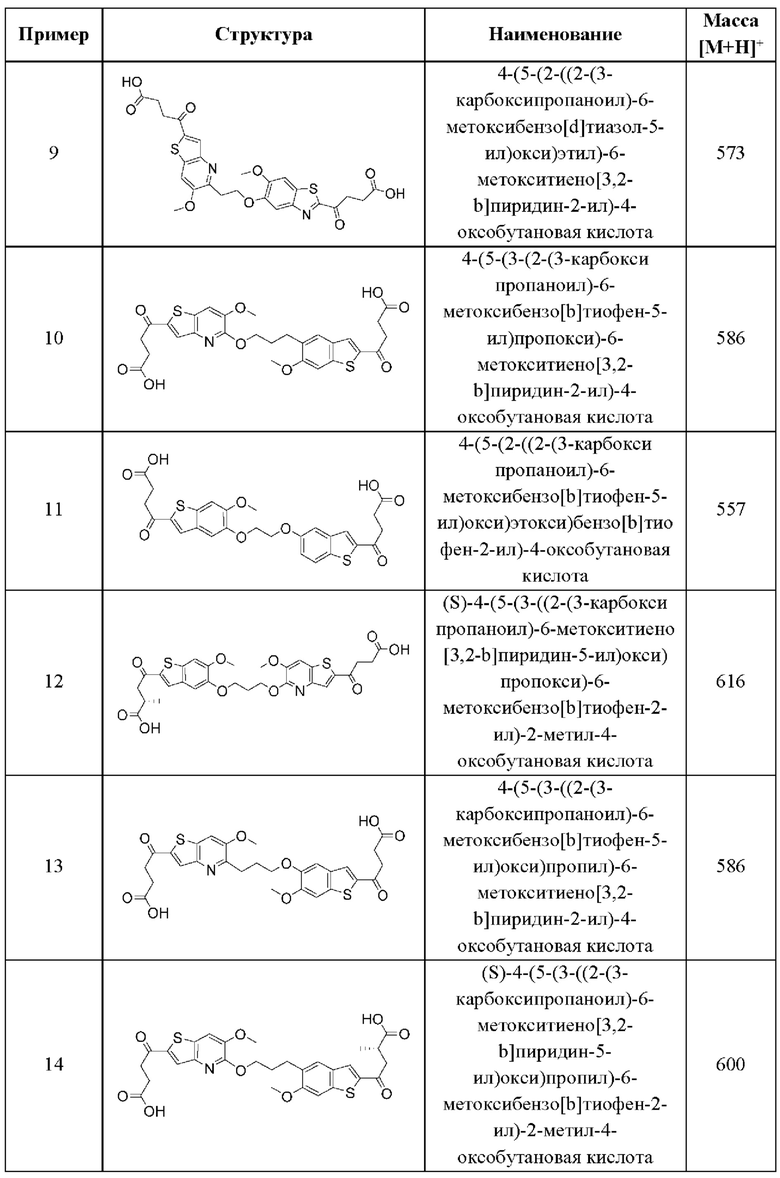
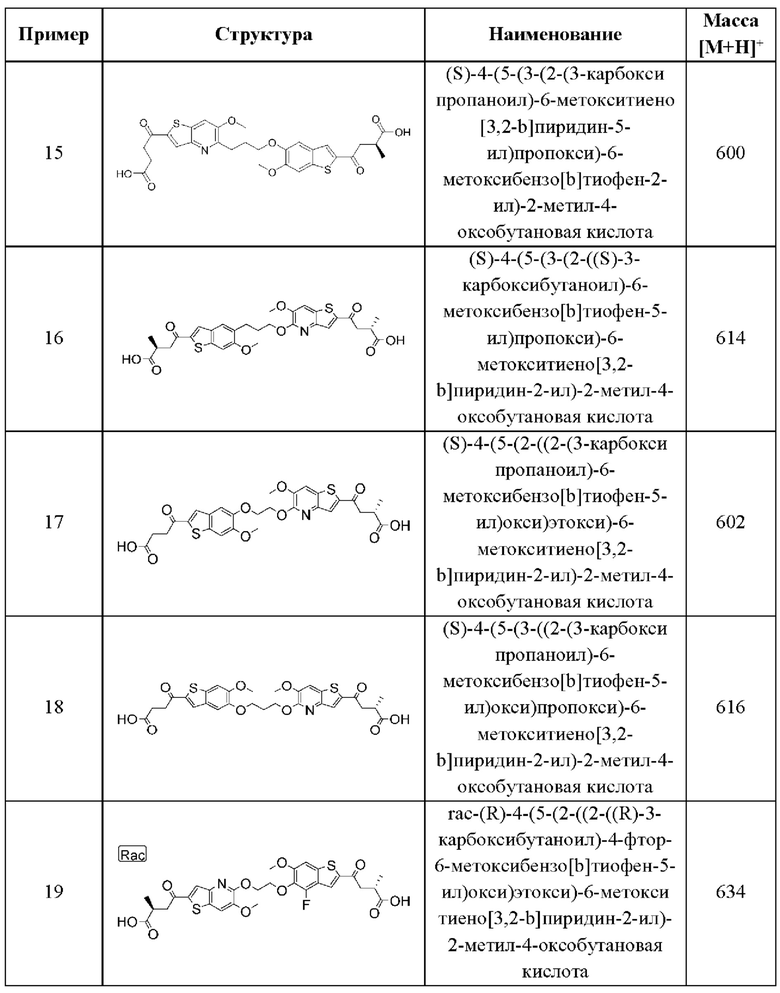
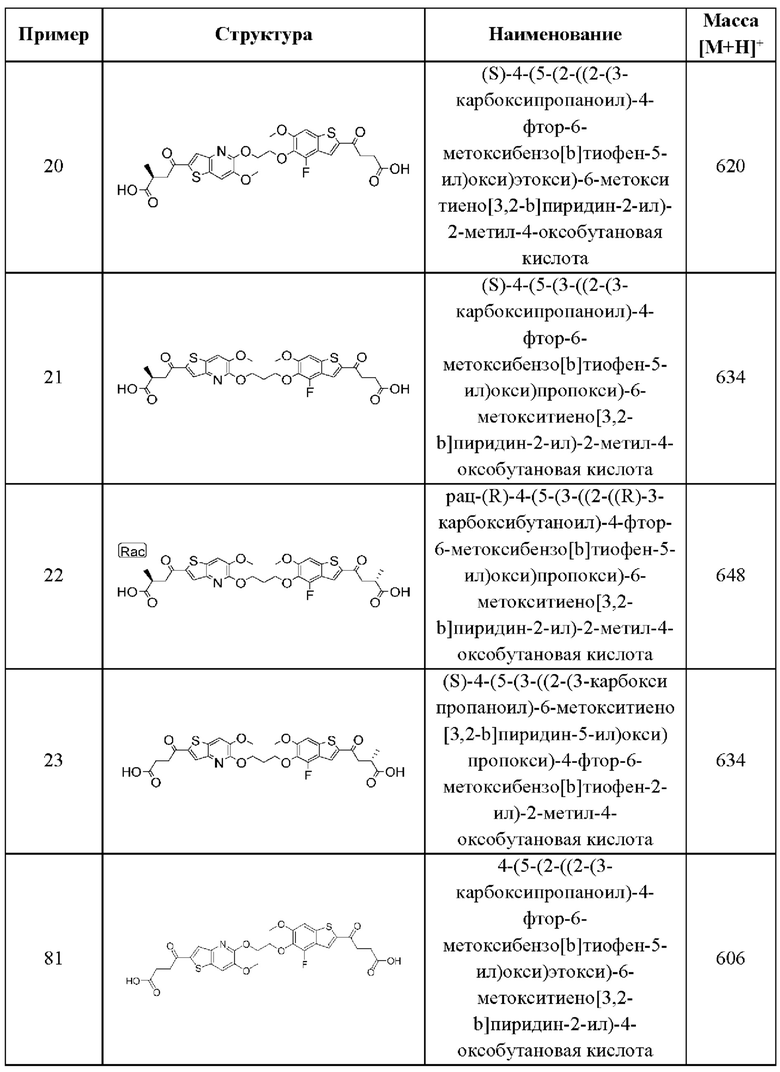
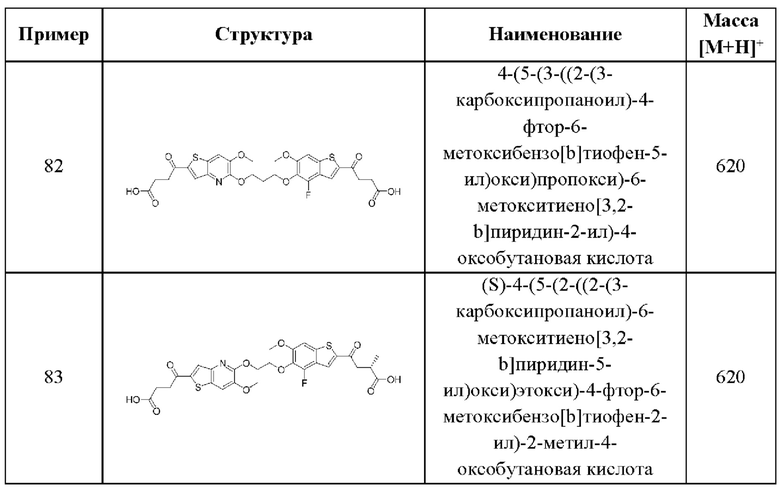
Пример 24: 4-(5-(3-(2-(3-цианопропаноил)-6-метоксибензо[b]тиофен-5-ил)пропил)-6-метоксибензо[b]тиофен-2-ил)-4-оксобутановая кислота
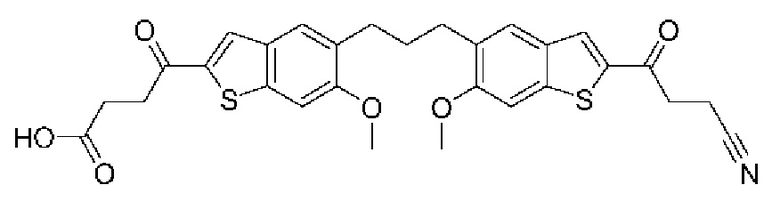
Стадия 1: трет-бутил 4-(5-(3-(2-(3-цианопропаноил)-6-метоксибензо[b]тиофен-5-ил)пропил)-6-метоксибензо[b]тиофен-2-ил)-4-оксобутапоат
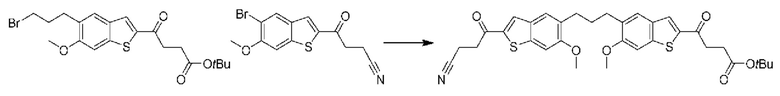
В виалу добавляли трет-бутил 4-(5-(3-бромпропил)-6-метоксибензо[Ь]тиофен-2-ил)-4-оксобутаноат (45 мг, 0.10 ммоль), 4-(5-бром-6-метоксибензо[b]тиофен-2-ил)-4-оксобутаннитрил (33 мг, 0.10 ммоль), NaI (7.6 мг, 0.051 ммоль), комплекс никель (II) бромид этиленгликоль диметиловый эфир (9.4 мг, 0.031 ммоль), Mn (22 мг, 0.41 ммоль) и 4,4'-диметокси-2,2-бипиридин (6.6 мг, 0.031 ммоль). В виалу добавляли DMPU (1.0 мл) с последующим добавлением 5% об./об. растворов DMPU Ру (82 мкл, 0.051 ммоль) и TMS-Cl (78 мкл, 0.031 ммоль). Виалу дегазировали Ar в течение 5 мин. Смесь нагревали до 90°С в течение 2 ч. Через 2 ч смесь оставляли охлаждаться до комнатной температуры и сразу очищали с помощью препаративной ВЭЖХ (ACN/H2O с 0.1% TFA) с получением трет-бутил 4-(5-(3-(2-(3-цианопропаноил)-6-метоксибензо[b]тиофен-5-ил)пропил)-6-метоксибензо[b]тиофен-2-ил)-4-оксобутаноата. ЖХ-МС (C33H35NO6S2Na) (ES, m/z): 628 [M+Na]+.
Стадия 2: 4-(5-(3-(2-(3-цианопропапоил)-6-метоксибензо[b]тиофен-5-ил)пропил)-6-метоксибензо[b]тиофен-2-ил)-4-оксобутановая кислота

К смеси трет-бутил 4-(5-(3-(2-(3-цианопропаноил)-6-метоксибензо[b]тиофен-5-ил)пропил)-6-метоксибензо[b]тиофен-2-ил)-4-оксобутаноата (8.5 мг, 0.014 ммоль), МеОН (500 мкл) и ACN (500 мкл) добавляли NaOH (5.0 М в воде, 0.56 мкл, 0.28 ммоль). Смесь нагревали до 40°С в течение 4 ч. После охлаждения до комнатной температуры смесь очищали с помощью препаративной ВЭЖХ (ACN/H2O с 0.1% TFA) с получением 4-(5-(3-(2-(3-цианопропаноил)-6-метоксибензо[b]тиофен-5-ил)пропил)-6-метоксибензо[b]тиофен-2-ил)-4-оксобутановой кислоты. ЖХ-МС (C29H28NO6S2) (ES, m/z): 550 [М+Н]+. 1Н ЯМР (500 МГц, DMSO-d6) δ 12.20 (s, 1H), 8.30-8.20 (m, 2Н), 7.82-7.75 (m, 2Н), 7.65-7.56 (m, 2Н), 3.89 (s, 6Н), 3.54-3.46 (m, 2Н), 2.82-2.76 (m, 2Н), 2.76-2.58 (m, 8Н), 1.99-1.86 (m, 2Н).
Пример 25: 4,4-(5,5-(пропан-1,3-Диил)бис(6-метоксибензо[b]тиофен-5,2-диил))бис(4-оксобутановая кислота)
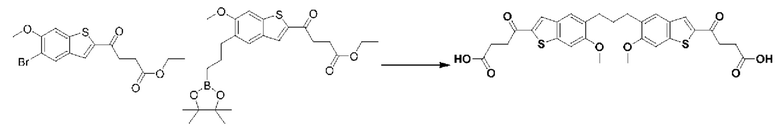
К смеси этил 4-(6-метокси-5-(3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пропил)бензо[b]тиофен-2-ил)-4-оксобутаноата (69 мг, 0.15 ммоль), этил 4-(5-бром-6-метоксибензо[b]тиофен-2-ил)-4-оксобутаноата (84 мг, 0.23 ммоль), PdCl2(dppf)-CH2Cl2 (25 мг, 0.030 ммоль) и CS2CO3 (195 мг, 0.600 ммоль) добавляли диоксан (0.80 мл) и воду (0.2 мл). Реакцию нагревали до 100°С в течение 18 ч. После охлаждения до комнатной температуры смесь затем фильтровали, и остаточные вещества промывали диоксаном. Фильтрат концентрировали под пониженным давлением. Полученный в результате остаток затем очищали с помощью препаративной ВЭЖХ (ACN/H2O с 0.1% NH4OH) с получением 4,4-(5,5-(пропан-1,3-диил)бис(6-метоксибензо[b]тиофен-5,2-диил))бис(4-оксобутановая кислота). ЖХ-МС (C29H27O8S2) (ES, m/z): 566 [М-Н]-. 1H ЯМР (600 МГц, DMSO-d6) δ 8.11 (s, 2Н), 7.68 (s, 2Н), 7.50 (s, 2Н), 3.82 (s, 6Н), 3.16-3.09 (m, 4Н), 2.66 (t, J=6.6 Гц, 4Н), 2.40 (s, 4Н), 1.93-1.83 (m, 2Н).
Пример 26: 4-(4-(3-(2-(3-карбоксипропаноил)-5-метоксибензо[b]тиофен-6-ил)пропил)-6-метоксибензо[b]тиофен-2-ил)-4-оксобутановая кислота
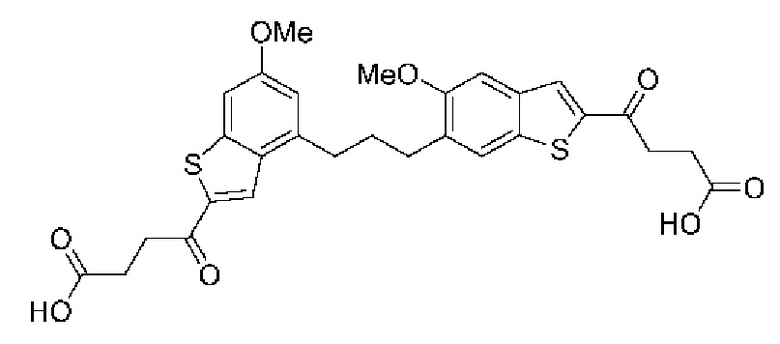
Стадия 1: трет-бутил 4-(5-метокси-6-(3-(6-метокси-2-(4-метокси-4-оксобутаноил)бензо[b]тиофен-4-ил)пропил)бензо[b]тиофен-2-ил)-4-оксобутаноат
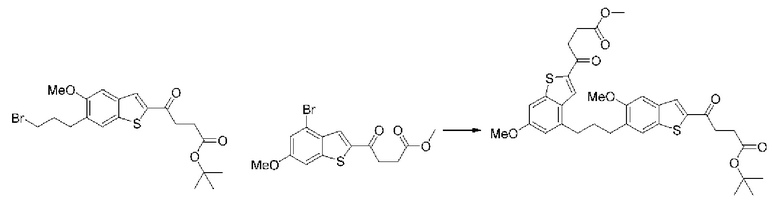
В виалу добавляли трет-бутил 4-(6-(3-бромпропил)-5-метоксибензо[b]тиофен-2-ил)-4-оксобутаноат (72 мг, 0.16 ммоль), метил 4-(4-бром-6-метоксибензо[b]тиофен-2-ил)-4-оксобутаноат (58 мг, 0.16 ммоль), NaI (12 мг, 0.082 ммоль), комплекс никель (II) бромид этиленгликоль диметиловый эфир (15 мг, 0.049 ммоль), Mn (36 мг, 0.65 ммоль) и 4,4-диметокси-2,2-бипиридин (11 мг, 0.049 ммоль). В виалу добавляли DMPU (1.6 мл) с последующим добавлением 5% об./об. растворов DMPU Ру (130 мкл, 0.082 ммоль) и TMS-Cl (130 мкл, 0.049 ммоль). Виалу дегазировали Ar в течение 5 мин. Смесь нагревали до 90°С в течение 1 ч. Через 1 ч смесь оставляли охлаждаться до комнатной температуры и затем разбавляли EtOAc и водой. Органический слой отделяли, высушивали над MgSO4, фильтровали и концентрировали под пониженным давлением. Полученную в результате смесь применяли без дополнительной очистки или определения характеристик.
Стадия 2: 4-(4-(3-(2-(3-карбоксипропаноил)-5-метоксибензо[b]тиофен-6-ил)пропил)-6-метоксибензо[b]тиофен-2-ил)-4-оксобутановая кислота
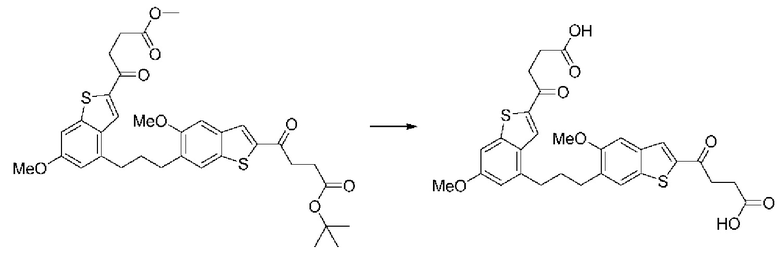
К смеси трет-бутил 4-(5-метокси-6-(3-(6-метокси-2-(4-метокси-4-оксобутаноил)бензо[b]тиофен-4-ил)пропил)бензо[b]тиофен-2-ил)-4-оксобутаноата (100 мг, 0.16 ммоль) и МеОН (1.6 мл) добавляли NaOH (5 М в воде, 0.65 мл, 3.3 ммоль), и смесь нагревали до 50°С в течение 1 ч. После охлаждения до комнатной температуры смесь очищали с помощью препаративной ВЭЖХ (ACN/H2O с 0.1% TFA) с получением 4-(4-(3-(2-(3-карбоксипропаноил)-5-метоксибензо[b]тиофен-6-ил)пропил)-6-метоксибензо[b]тиофен-2-ил)-4-оксобутановой кислоты. ЖХ-МС (C29H29O8S2) (ES, m/z): 569 [М+Н]+. 1H ЯМР (500 МГц, DMSO-d6) δ 12.23 (s, 2Н), 8.27 (s, 1H), 8.25 (s, 1Н), 7.82 (s, 1H), 7.50 (s, 1H), 7.44 (s, 1H), 6.93 (s, 1H), 3.89-3.79 (m, 6H), 3.34-3.19 (m, 4H), 3.03 (t, J=7.3 Гц, 2H), 2.82-2.74 (m, 2H), 2.66-2.55 (m, 4H), 2.06-1.96 (m, 2H).
Примеры с 27 по 31, как показано в Таблице 12 ниже, или могут быть получены в соответствии со способами, аналогичными описанным в Примере 26 выше, с применением подходящих исходных материалов, описанных выше в Способах получения или Промежуточных соединениях, или получены из коммерческих источников.
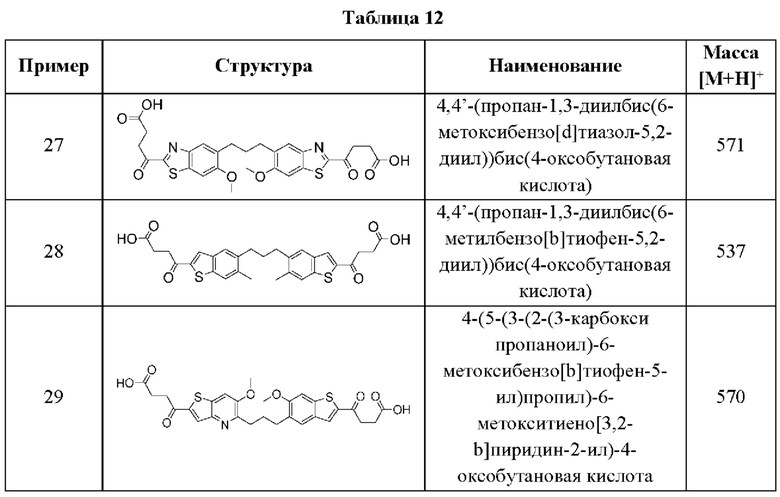
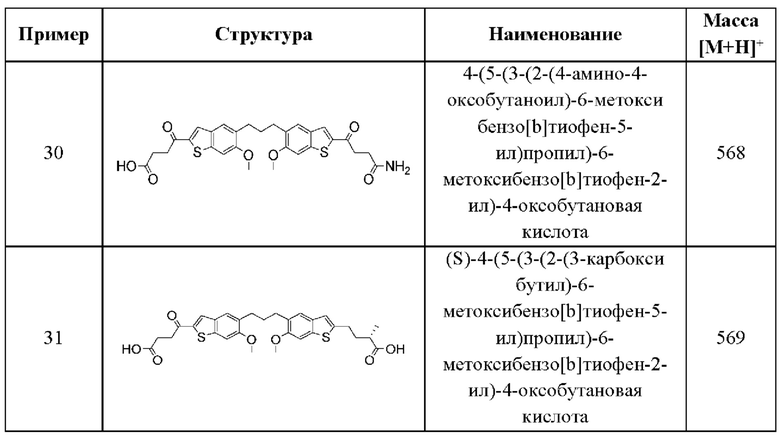
Пример 32: 4-(5-(3-((2-(3-карбоксипропаноил)-4-фтор-6-метоксибензо[b]тиофен-5-ил)окси)пропил)-6-метоксибензо[b]тиофен-2-ил)-4-оксобутановая кислота
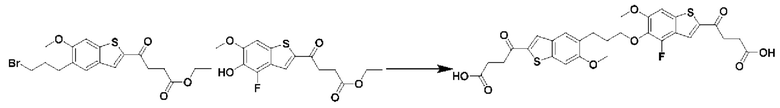
К смеси этил 4-(5-(3-бромпропил)-6-метоксибензо[b]тиофен-2-ил)-4-оксобутаноата (83 мг, 0.20 ммоль), этил 4-(4-фтор-5-гидрокси-6-метоксибензо[b]тиофен-2-ил)-4-оксобутаноата (78 мг, 0.24 ммоль) и K2CO3 (28 мг, 0.20 ммоль) добавляли ACN (1 мл). Реакцию нагревали до 65°С в течение 18 ч. После охлаждения до комнатной температуры смесь разбавляли ACN (4 мл) и фильтровали. Фильтрат затем концентрировали под пониженным давлением. Затем добавляли THF (2.0 мл), МеОН (0.50 мл), воду (1.0 мл) и LiOH (48 мг, 2.0 ммоль), и смесь оставляли перемешиваться при комнатной температуре в течение 2 ч. Затем реакцию останавливали АсОН (0.40 мл), и смесь концентрировали под пониженным давлением. Полученный в результате остаток затем очищали с помощью препаративной ВЭЖХ (ACN/H2O с 0.1% TFA) с получением 4-(5-(3-((2-(3-карбоксипропаноил)-4-фтор-6-метоксибензо[b]тиофен-5-ил)окси)пропил)-6-метоксибензо[b]тиофен-2-ил)-4-оксобутановой кислоты. ЖХ-МС (C29H28FO9S2) (ES, m/z): 603 [М+Н]+. 1Н ЯМР (600 МГц, DMSO-d6) δ 8.28 (s, 1Н), 8.20 (s, 1H), 7.73 (d, J=13.7 Гц, 1H), 7.55 (d,.J=11.4 Гц, 2Н), 4.06-4.00 (m, 2Н), 3.90-3.81 (m, 6Н), 3.28 (t, J=6.1 Гц, 2Н), 3.23 (t, J=6.2 Гц, 2Н), 2.82 (s, 2Н), 2.58-2.56 (m, 4Н), 1.95-193 (m, 2Н).
Примеры с 33 по 40 и с 84 по 157, как показано в Таблице 13 ниже, или могут быть получены в соответствии со способами, аналогичными описанным в Примере 32 выше, с применением подходящих исходных материалов, описанных выше в Способах получения или Промежуточных соединениях, или получены из коммерческих источников.
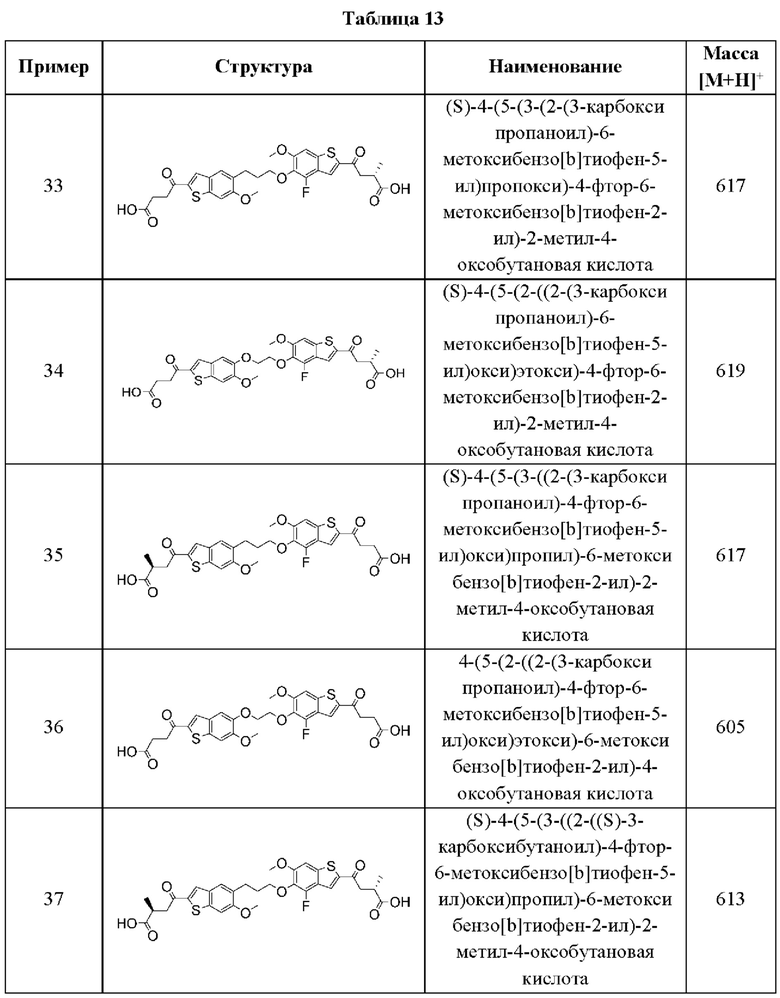
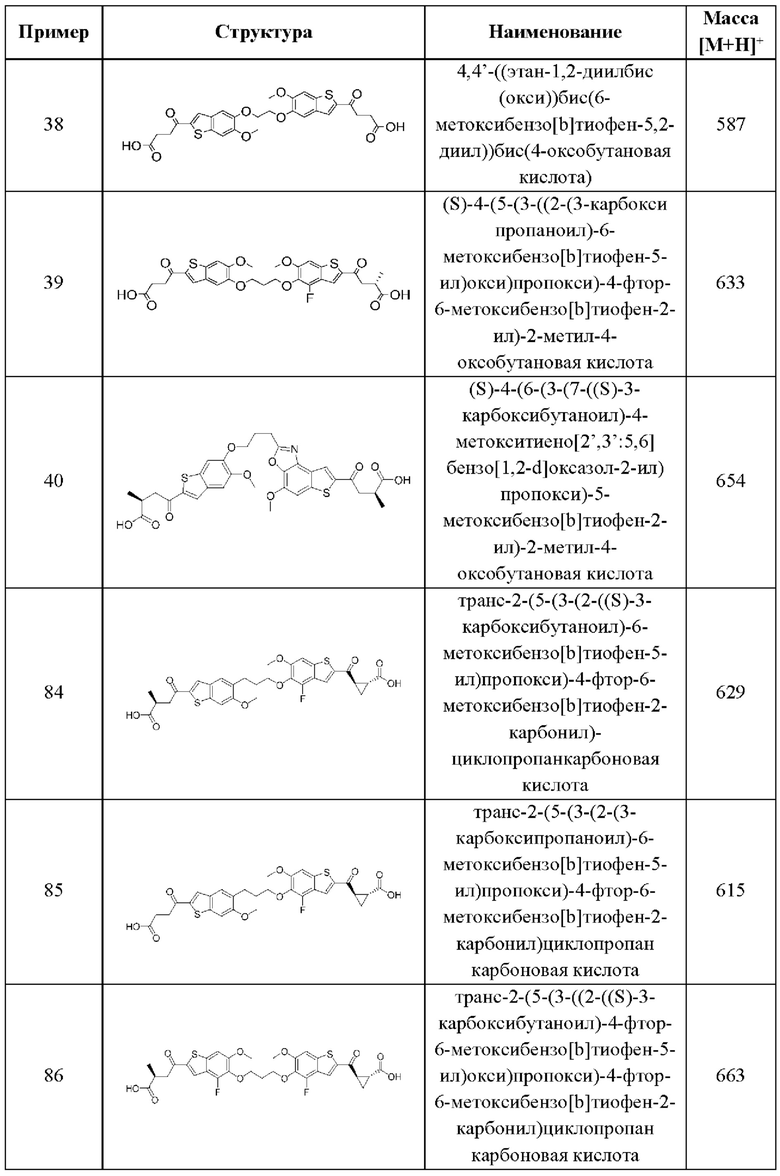
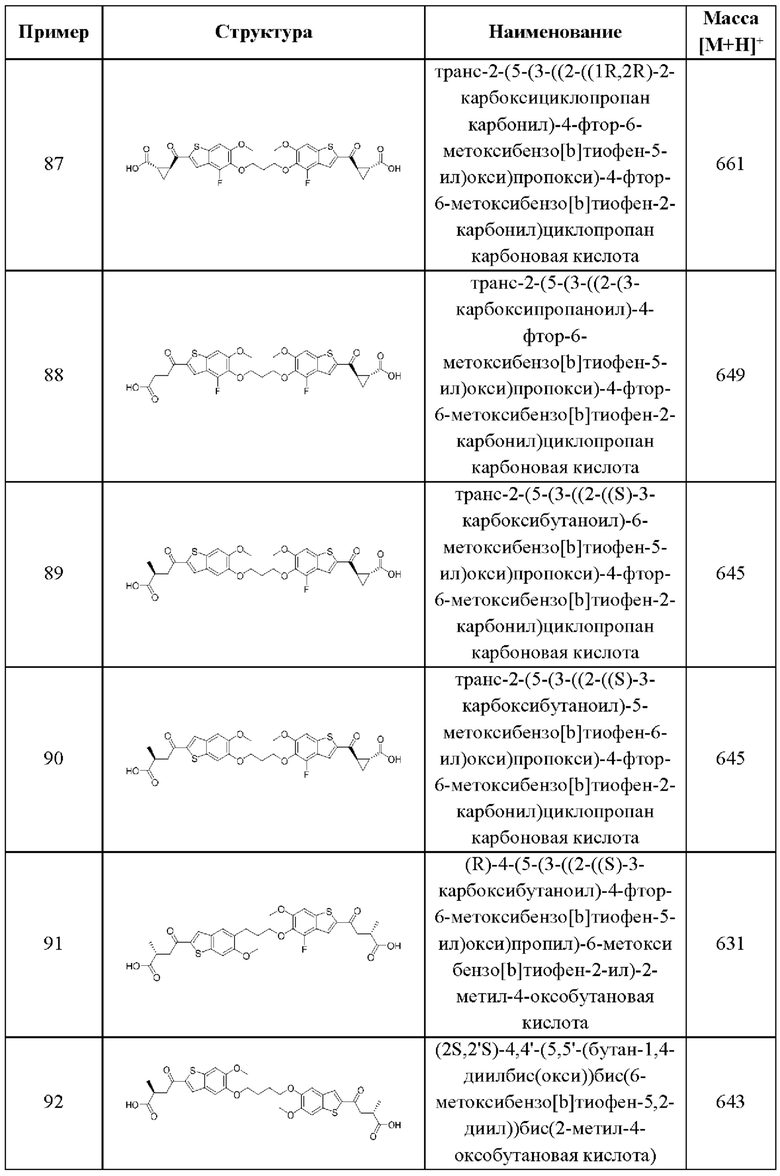
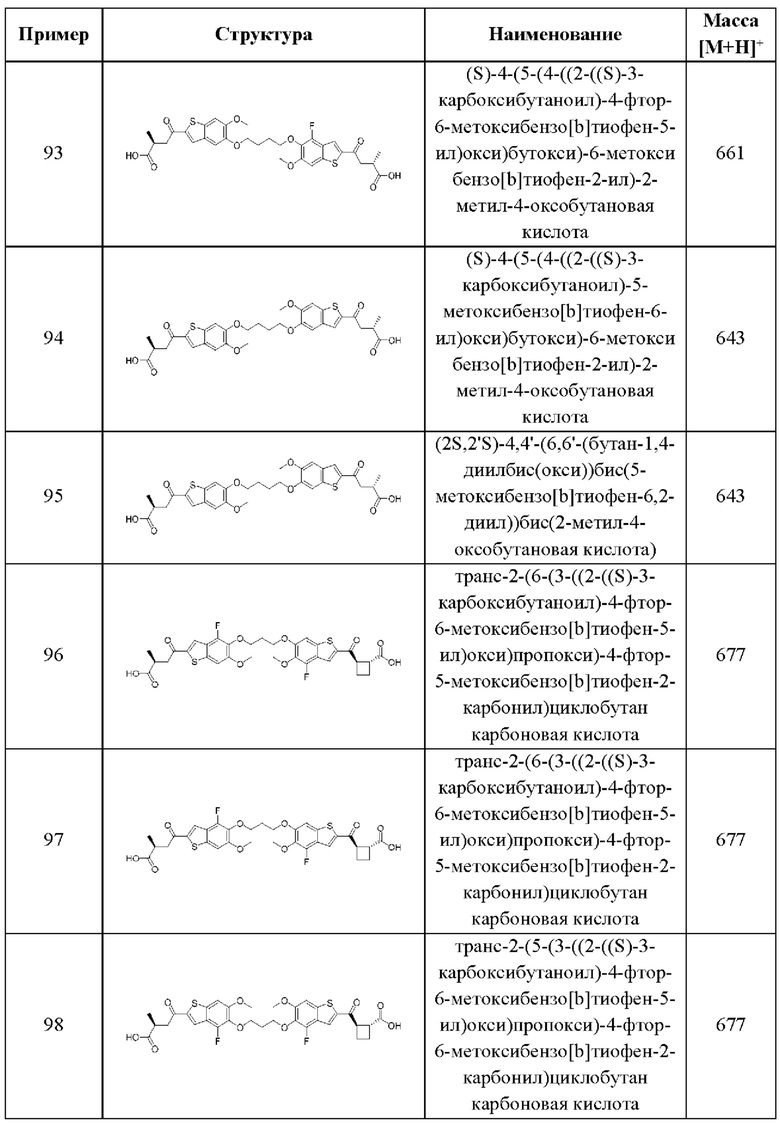
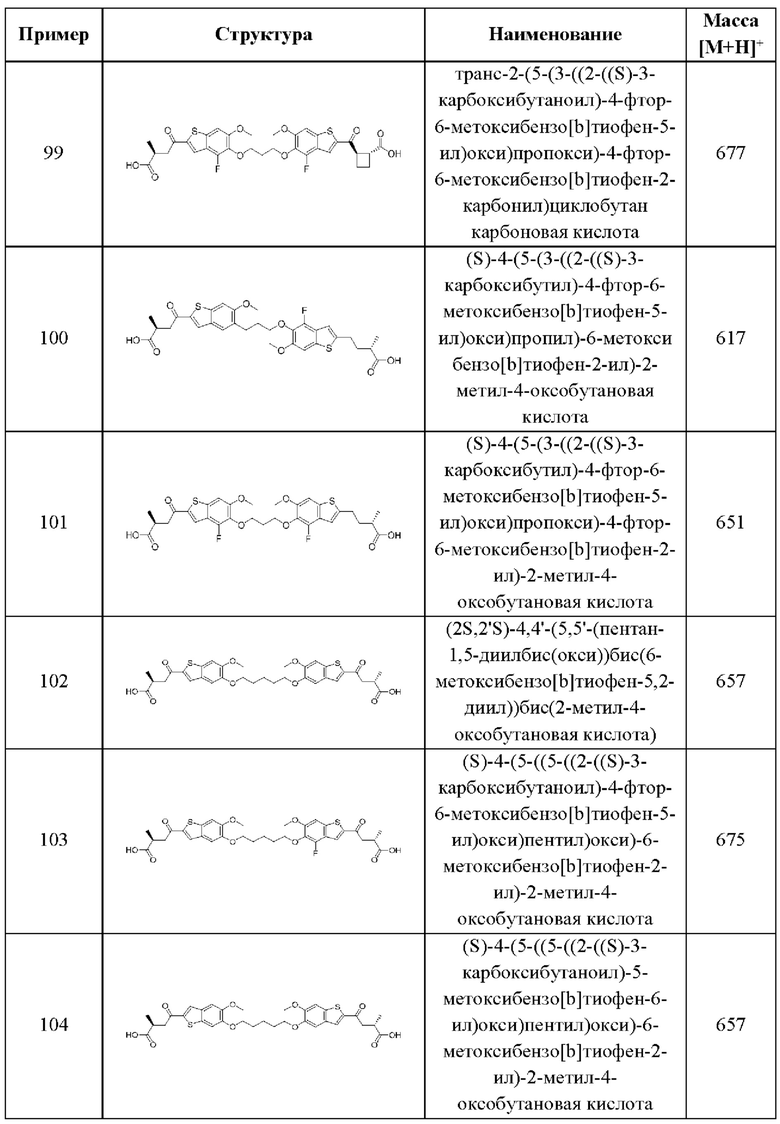
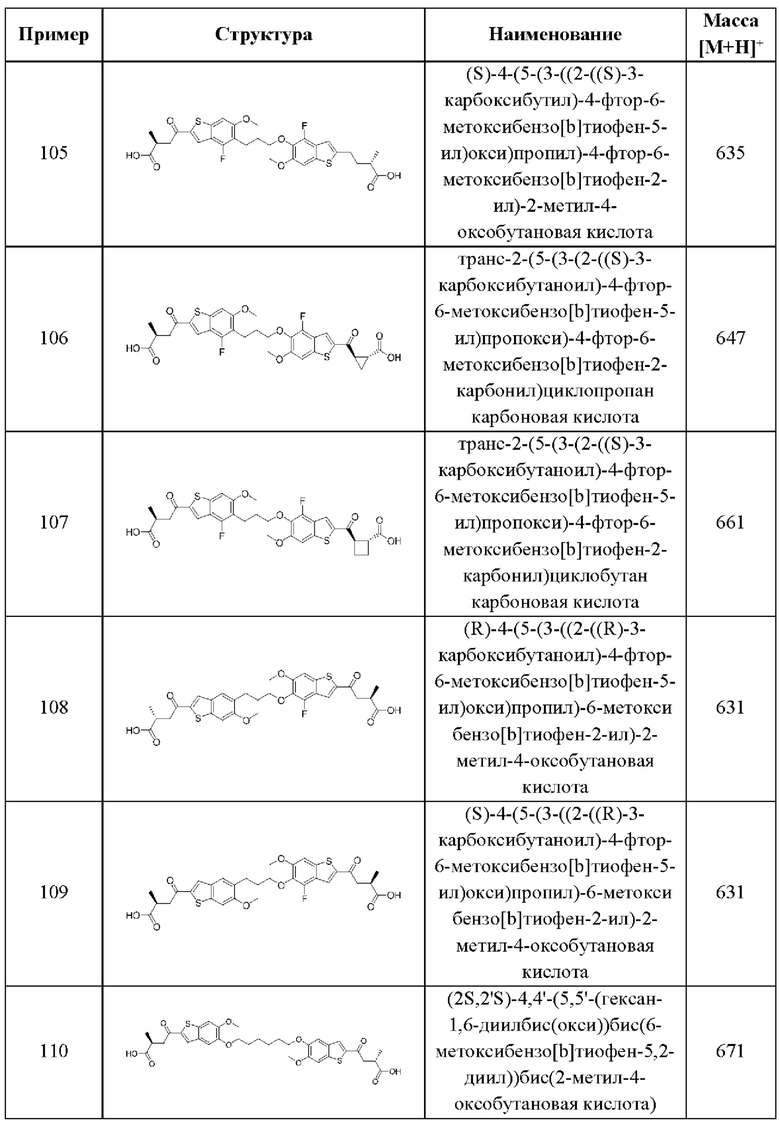
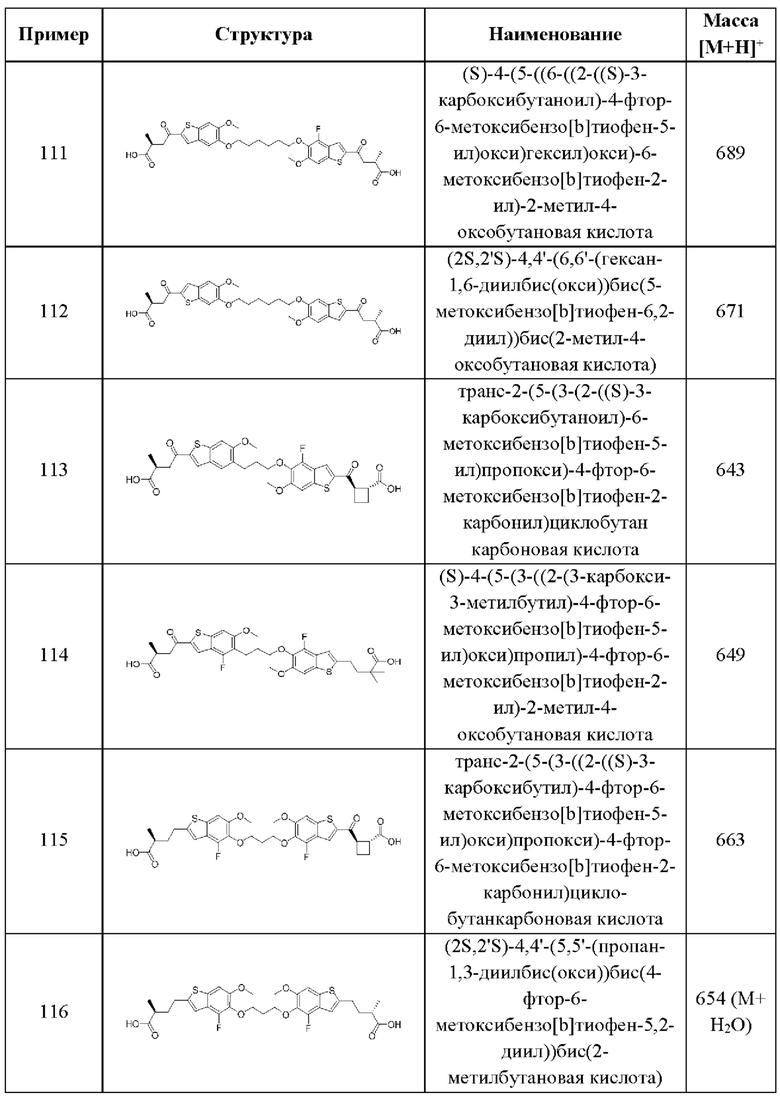
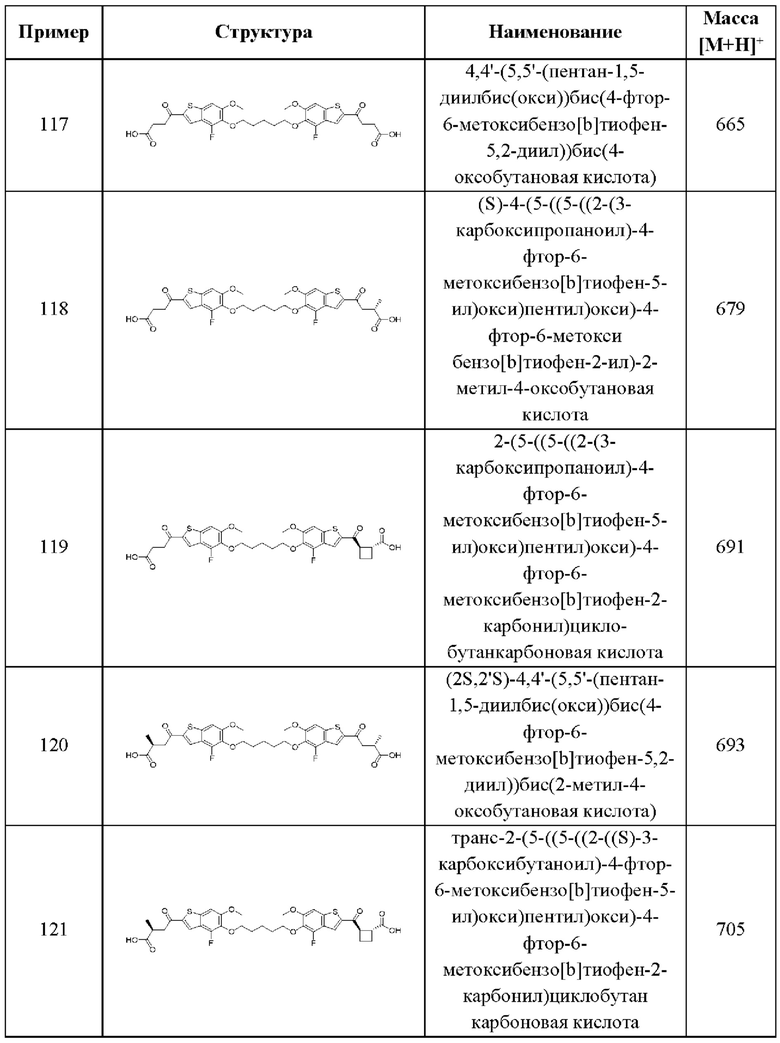
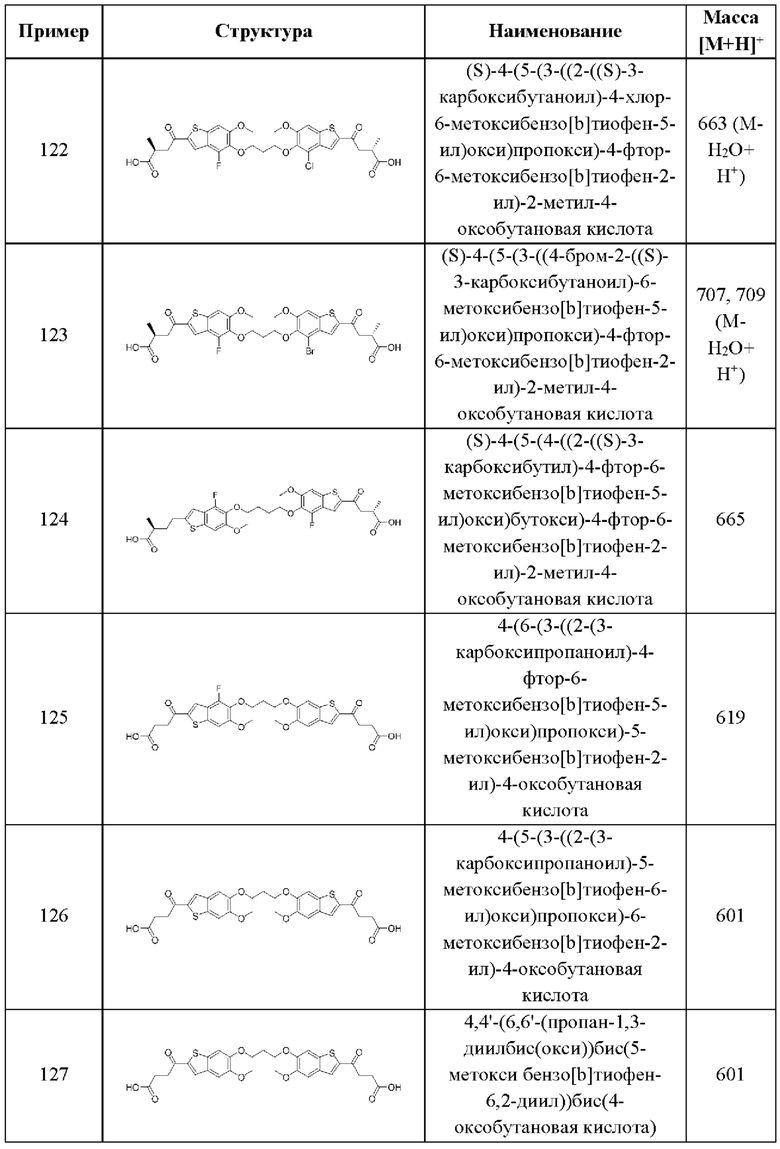
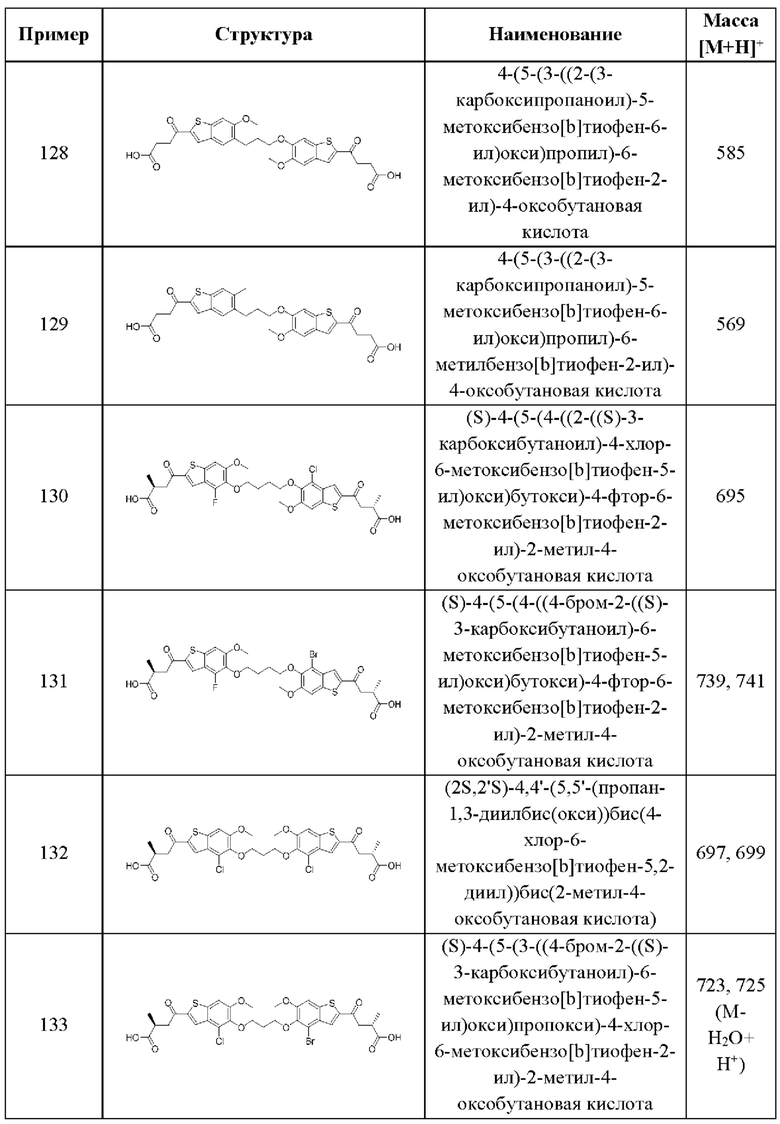
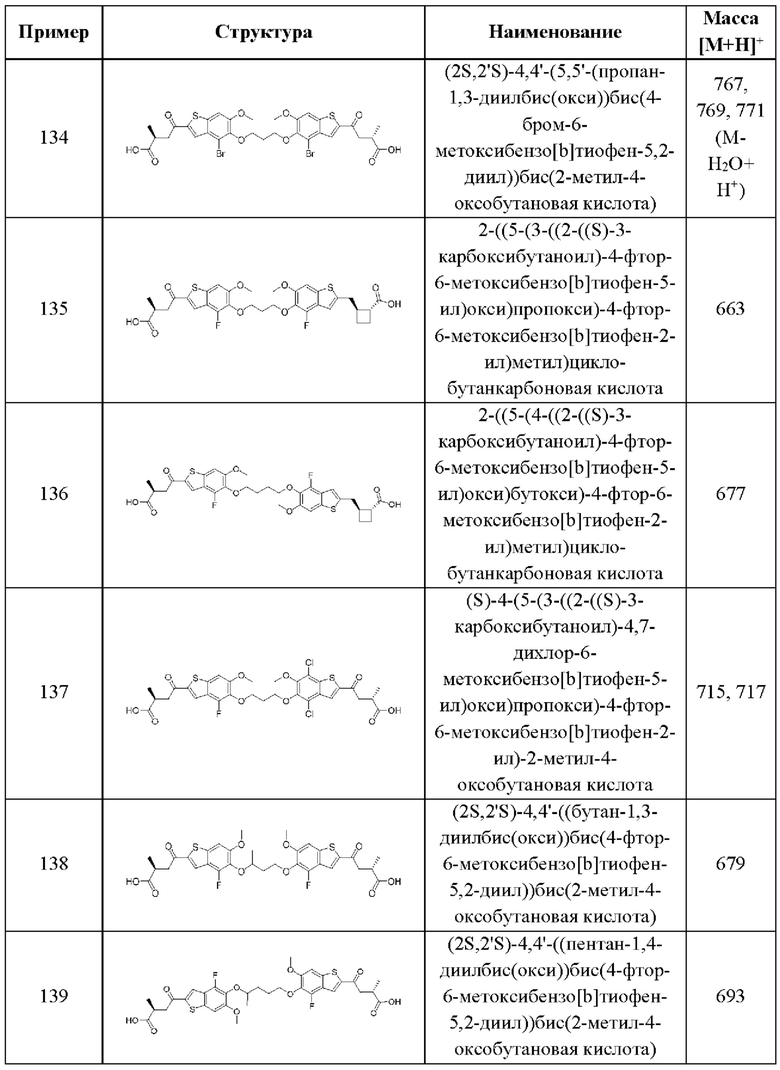
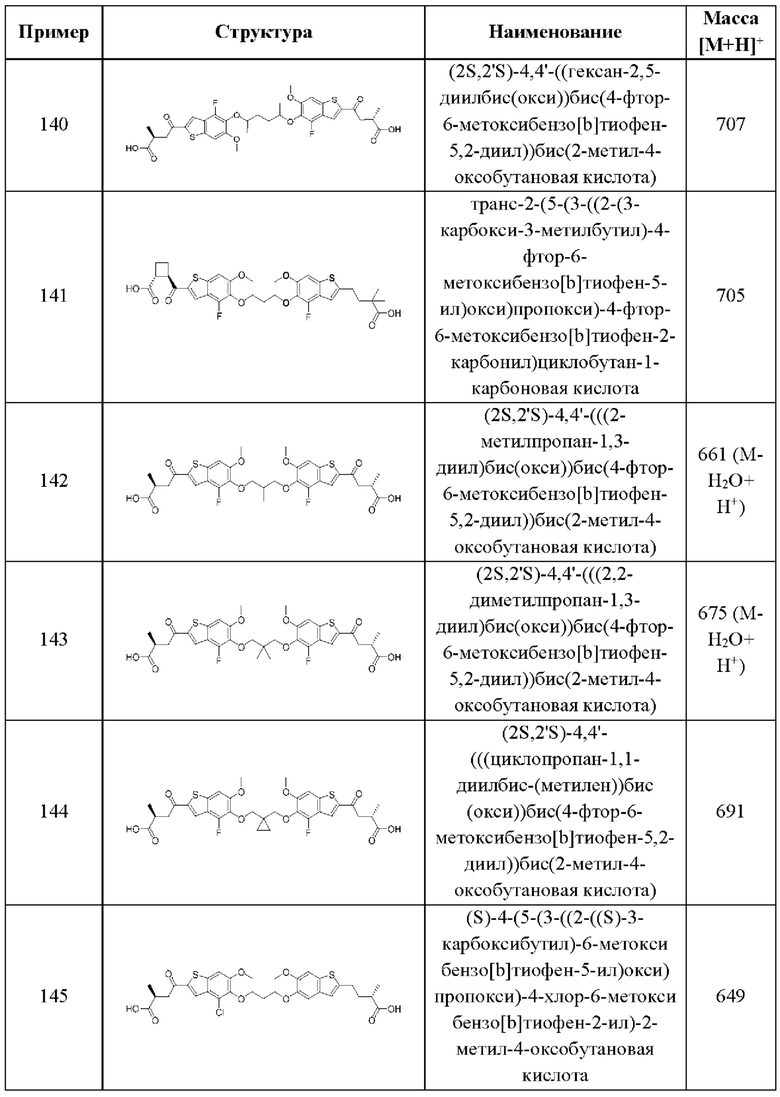
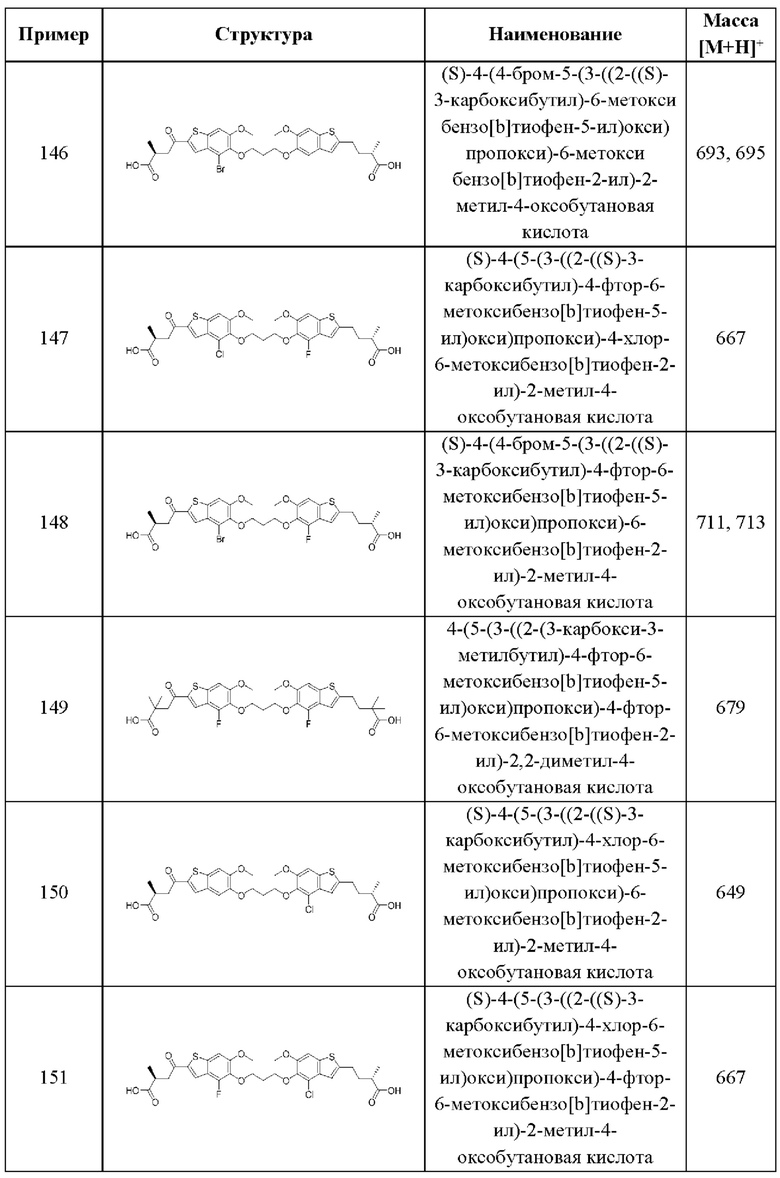
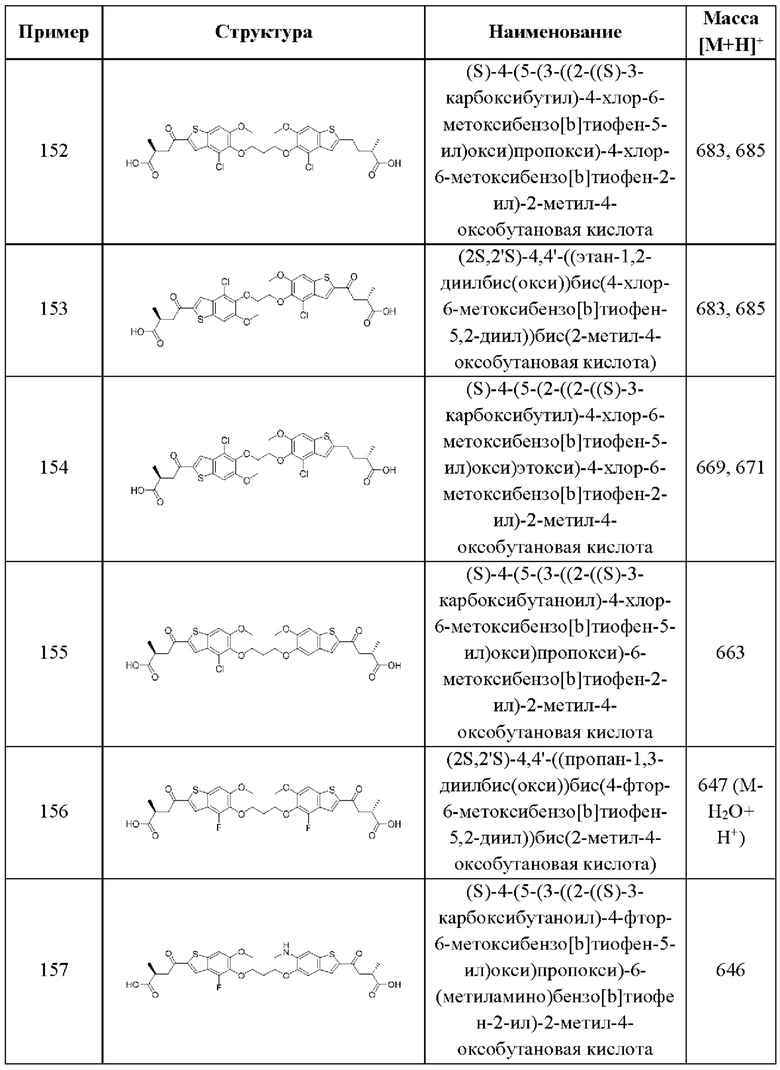
Пример 41: (2S)-4-(5-[3-({2-[(3S)-3-карбоксибутаноил]-6-метокси-1-бензотиофен-5-ил}окси)пропил]-6-метокси-1-бензотиофен-2-ил}-2-метил-4-оксобутановая кислота
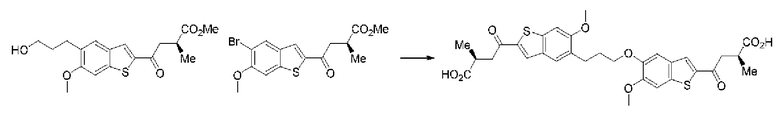
К смеси метил (2S)-4-(5-бром-6-метокси-1-бензотиофен-2-ил)-2-метил-4-оксобутаноата (107 мг, 0.287 ммоль), RockPhos Pd G3 (11 мг, 0.013 ммоль) и Cs2CO3 (128 мг, 0.392 ммоль) в атмосфере N2 добавляли смесь метил (2S)-4-[5-(3-гидрокси-пропил)-6-метокси-1-бензотиофен-2-ил]-2-метил-4-оксобутаноата (92 мг, 0.26 ммоль) в толуоле (870 мкл). Реакционную смесь продували N2 в течение 5 мин и затем нагревали до 110°С в течение 2 ч. После охлаждения до комнатной температуры добавляли THF (1.0 мл), МеОН (1.0 мл) и воду (0.25 мл) с последующим добавлением LiOH⋅Н2О (157 мг, 3.75 ммоль) одной порцией при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 4 ч, затем реакцию останавливали водной HCl (2.0 н., 1.35 мл). Добавляли DMSO, и полученную в результате смесь фильтровали. Фильтрат очищали с помощью ВЭЖХ с обратными фазами [колонка С18, вода (0.1% TFA)-CH3CN] с получением (2S)-4-{5-[3-({2-[(3S)-3-карбоксибутаноил]-6-метокси-1-бензотиофен-5-ил}окси)пропил]-6-метокси-1-бензотиофен-2-ил}-2-метил-4-оксобутановой кислоты. ЖХ-МС (C31H32O9S2Na) (ES, m/z): 635 [M+Na]+. 1Н ЯМР (500 МГц, DMSO-d6): δ 12.19 (br, 2Н), 8.23 (s, 1H), 8.16 (s, 1Н), 7.77 (s, 1H), 7.61 (s, 1H), 7.60 (s, 1H), 7.44 (s, 1H), 4.05 (d, J=6.2 Гц, 2H), 3.88 (s, 3H), 3.88 (s, 3H), 3.06 (dt, J=17.5, 4.3 Гц, 2H), 2.94-2.80 (m, 4H), 2.10 (pentet, J=6.9 Гц, 2H), 1.17 (d, J=7.1 Гц, 6H).
Примеры 42, 158 и 159, как показано в Таблице 14 ниже, или могут быть получены в соответствии со способами, аналогичными описанным в Примере 41 выше, с применением подходящих исходных материалов, описанных выше в Способах получения или Промежуточных соединениях, или получены из коммерческих источников.
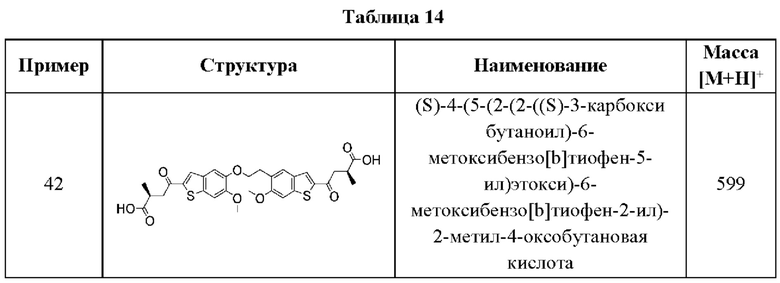
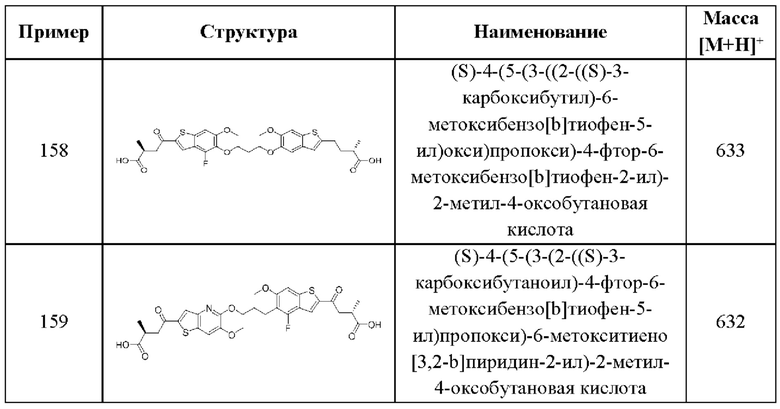
Пример 43: (S)-4-(5-(3-((2-(3-карбоксипропаноил)бензо[b]тиофен-6-ил)окси)пропокси)-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутановая кислота
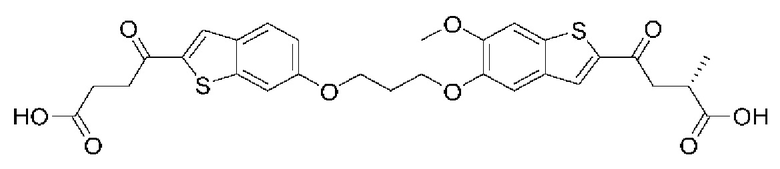
Стадия 1: метил (S)-4-(6-метокси-5-(3-((2-(4-метокси-4-оксобутаноил)бензо[b]тиофен-6-ил)окси)пропокси)бензо[b]тиофен-2-ил)-2-метил-4-оксобутаноат
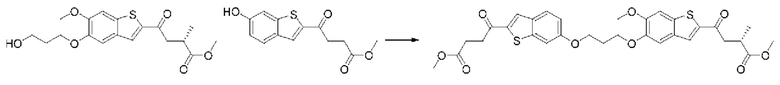
Смесь (S)-метил 4-(5-(3-гидроксипропокси)-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата (16 мг, 0.044 ммоль), метил 4-(6-гидроксибензо[b]тиофен-2-ил)-4-оксобутаноата (14 мг, 0.052 ммоль), (Е)-диазин-1,2-диилбис(пиперидин-1-илметанон) (28 мг, 0.11 ммоль) и Ph3P (29 мг, 0.11 ммоль) в THF (1.0 мл) дегазировали Ar и затем перемешивали при 20°С в течение 18 ч. Реакционную смесь затем разбавляли DMSO (1.0 мл) и фильтровали. Смесь затем сразу очищали с помощью ВЭЖХ с обратными фазами (ACN в воде, 0.1% TFA, С-18 неподвижная фаза) с получением метил (S)-4-(6-метокси-5-(3-((2-(4-метокси-4-оксобутаноил)бензо[b]тиофен-6-ил)окси)пропокси)бензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата. ЖХ-МС (C31H33O9S2) (ES, m/z): 613 [М+Н]+.
Стадия 2: (S)-4-(5-(3-((2-(3-карбоксипропапоил)бензо[b]тиофен-6-ил)окси)пропокси)-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутановая кислота
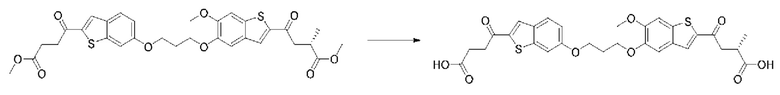
NaOH (5.0 M в воде, 21 мкл, 0.11 ммоль) добавляли к смеси метил (S)-4-(6-метокси-5-(3-((2-(4-метокси-4-оксобутаноил)бензо[b]тиофен-6-ил)окси)пропокси)бензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата (6.3 мг, 10 мкмоль) в МеОН (1.0 мл) при комнатной температуре. Реакционную смесь перемешивали и нагревали до 50°С в течение 2 ч. Неочищенную реакционную смесь охлаждали до комнатной температуры, останавливали TFA (16 мкл, 0.21 ммоль), разбавляли DMSO (1.0 мл) и затем фильтровали. Фильтрат затем сразу очищали с помощью ВЭЖХ с обратными фазами (ACN в воде, 0.1% TFA, С-18 неподвижная фаза) с получением (S)-4-(5-(3-((2-(3-карбоксипропаноил)бензо[b]тиофен-6-ил)окси)пропокси)-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутановой кислоты. ЖХ-МС (C29H29O9S2) (ES, m/z): 585 [М+Н]+. 1Н ЯМР (500 МГц, DMSO-d6) δ 12.22 (s, 2Н), 8.33 (s, 1Н), 8.19 (s, 1H), 7.94 (d, J=7.8 Гц, 1H), 7.69 (s, 1H), 7.63 (s, 1H), 7.55 (s, 1H), 7.15 (d, J=7.6 Гц, 1H), 4.34-4.18 (m, 4H), 3.89 (s, 3H), 3.44-3.25 (m, 3H), 3.13-3.05 (m, 1H), 2.95-2.87 (m, 1H), 2.66-2.60 (m, 2H), 2.34-2.27 (m, 2H), 1.25-1.14 (m, 3H).
Примеры с 44 по 59 и 160, как показано в Таблице 15 ниже, или могут быть получены в соответствии со способами, аналогичными описанным в Примере 43 выше, с применением подходящих исходных материалов, описанных выше в Способах получения или Промежуточных соединениях, или получены из коммерческих источников.
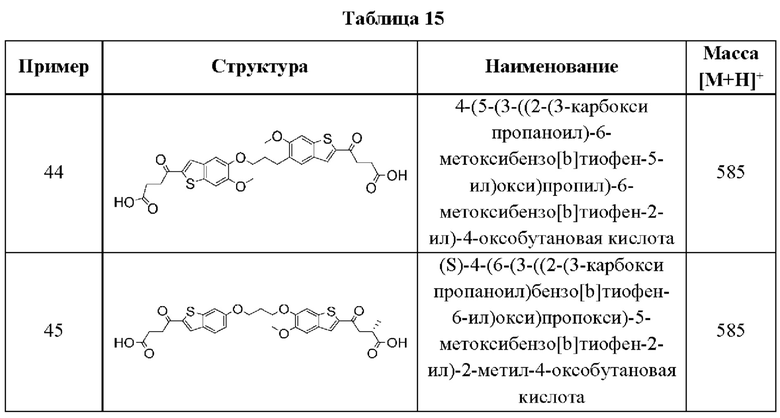
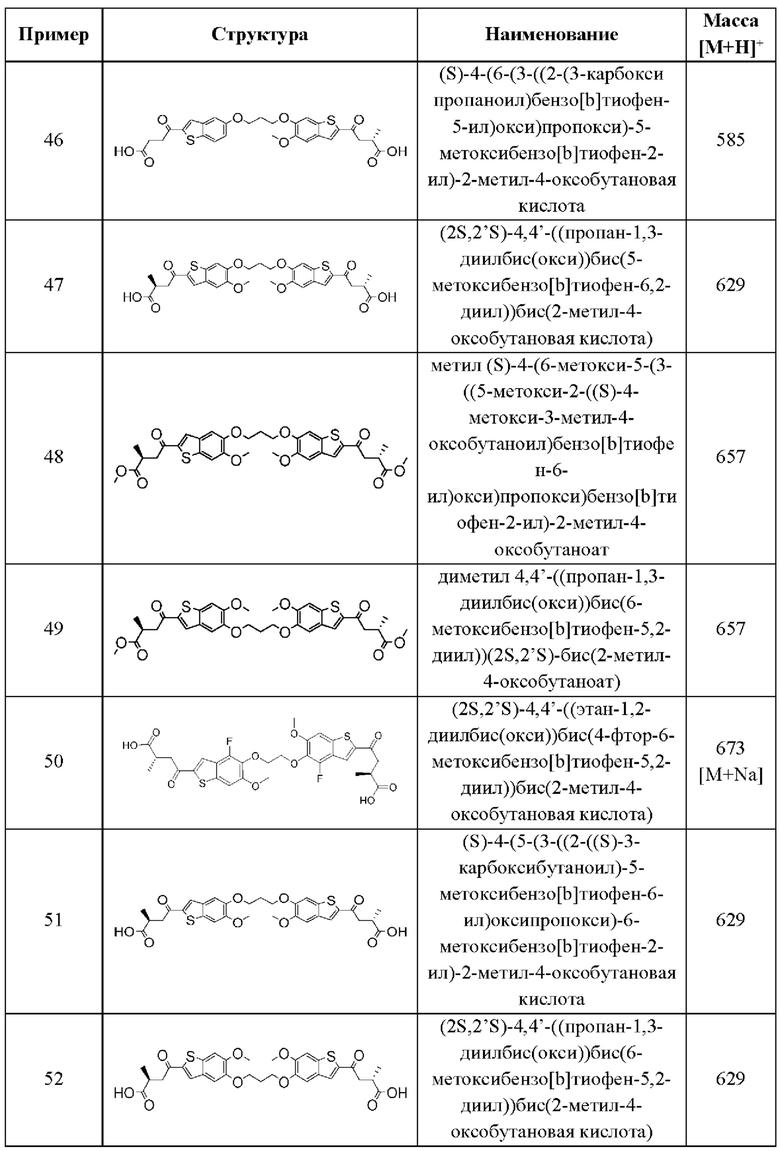
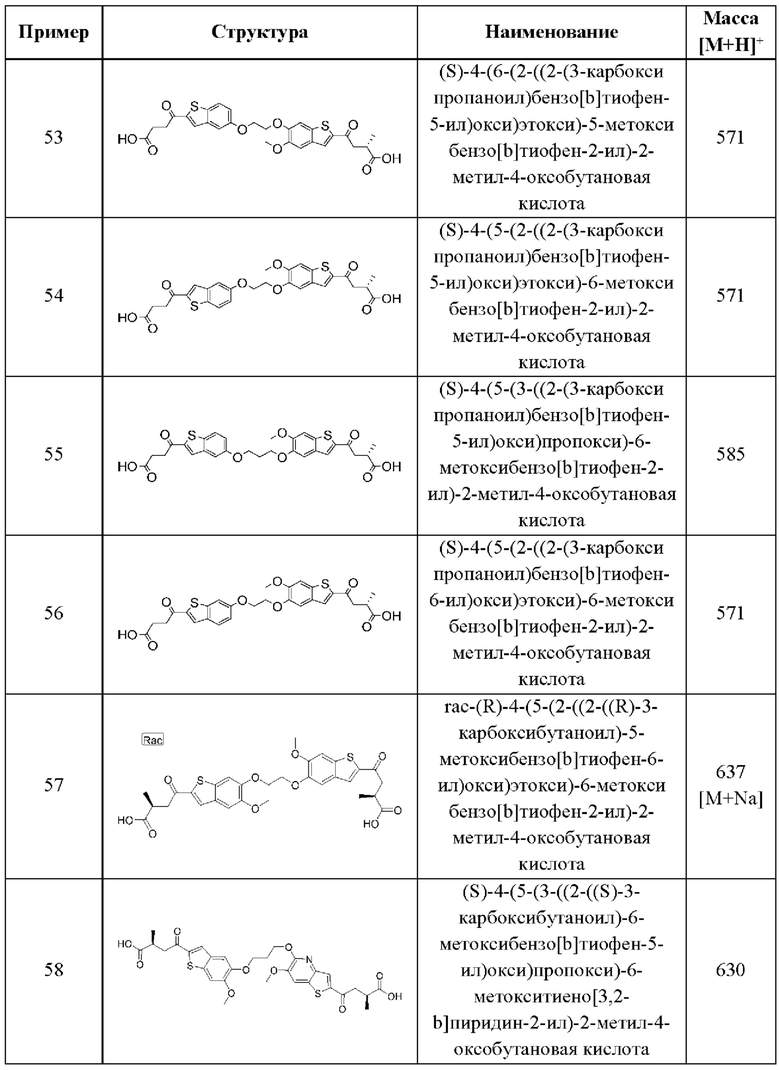
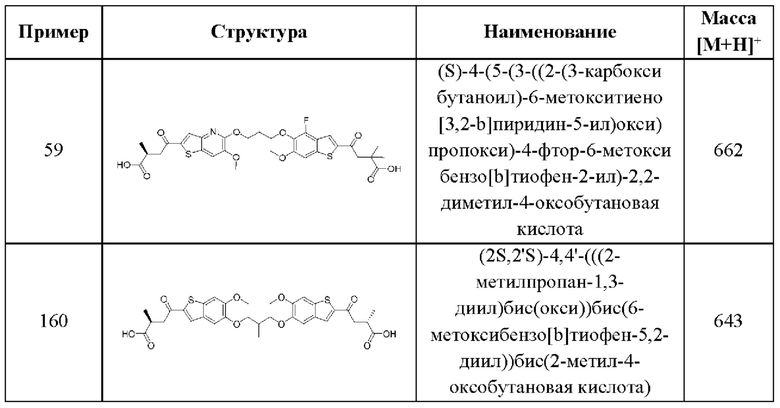
Пример 60: (2S)-4-{5-[2-({2-[(3S)-3-карбоксибутаноил]-6-метокси-1-бензотиофен-5-ил}амино)этил]-6-метокси-1-бензотиофен-2-ил}-2-метил-4-оксобутановая кислота
Стадия 1: метил (2S)-4-{6-метокси-5-[2-({6-метокси-2-[(3S)-4-метокси-3-метил-4-оксобутаноил]-1-бензотиофен-5-ил}амино)этил]-1-бензотиофен-2-ил}-2-метил-4-оксобутапоат
К смеси метил (2S)-4-(5-бром-6-метокси-1-бензотиофен-2-ил)-2-метил-4-оксобутаноата (54 мг, 0.14 ммоль), рацемического BINAP Pd G3 (12 мг, 0.012 ммоль) и Cs2CO3 (117 мг, 0.360 ммоль) в атмосфере N2 добавляли суспензию метил (2S)-4-[5-(2-аминоэтил)-6-метокси-1-бензотиофен-2-ил]-2-метил-4-оксобутаноата (54 мг, 0.120 ммоль) в толуоле (1.6 мл). Реакционную смесь продували N2 в течение 45 мин, затем нагревали до 110°С в течение 12 ч. После охлаждения до комнатной температуры смесь очищали с помощью колоночной хроматографии на силикагеле (EtOAc в гексанах) с получением метил (2S)-4-{6-метокси-5-[2-({6-метокси-2-[(3S)-4-метокси-3-метил-4-оксобутаноил]-1-бензотиофен-5-ил}амино)этил]-1-бензотиофен-2-ил}-2-метил-4-оксобутаноата. ЖХ-МС (C32H35NO8S2Na) (ES, m/z): 648 [M+Na]+.
Стадия 2: (2S)-4-{5-[2-({2-[(3S)-3-карбоксибутаноил]-6-метокси-1-бензотиофен-5-ил}амино)этил]-6-метокси-1-бензотиофен-2-ил}-2-метил-4-оксобутановая кислота
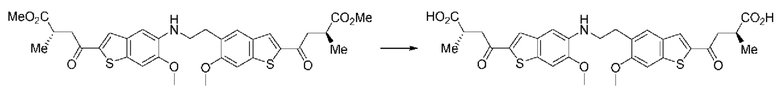
К смеси метил (2S)-4-{6-метокси-5-[2-({6-метокси-2-[(3S)-4-метокси-3-метил-4-оксобутаноил]-1-бензотиофен-5-ил}амино)этил]-1-бензотиофен-2-ил}-2-метил-4-оксобутаноата (20 мг, 0.03 ммоль) в THF (280 мкл), МеОН (280 мкл) и воде (70 мкл) добавляли LiOH⋅Н2О (13 мг, 0.32 ммоль) одной порцией при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 2 ч, затем реакцию останавливали водной HCl (2 н., 170 мкл). Смесь разбавляли DMSO, и полученную в результате смесь фильтровали. Фильтрат очищали с помощью ВЭЖХ с обратными фазами [колонка С18, вода (0.1% TFA)-CH3CN] с получением (2S)-4-{5-[2-({2-[(3S)-3-карбоксибутаноил]-6-метокси-1-бензотиофен-5-ил}амино)этил]-6-метокси-1-бензотиофен-2-ил}-2-метил-4-оксобутановой кислоты. ЖХ-МС (C30H32NO8S2) (ES, m/z): 598 [М+Н]+. 1H ЯМР (500 МГц, DMSO-d6): δ 12.12 (br, 2Н), 8.27 (s, 1H), 8.11 (s, 1H), 7.82 (s, 1H), 7.66 (s, 1H), 7.44 (s, 1H), 7.07 (s, 1H), 3.98 (s, 3Н), 3.90 (s, 3Н), 3.41 (dd, J=17.5, 8.6 Гц, 2Н), 3.36 (t, J=6.5 Гц, 2Н), 3.14-2.96 (m, 4Н), 2.94-2.84 (m, 2Н), 1.18 (d, J=7.1 Гц, 6Н).
Пример 61, как показано в Таблице 16 ниже, или может быть получен в соответствии со способами, аналогичными описанным в Примере 60 выше, с применением подходящих исходных материалов, описанных выше в Способах получения или Промежуточных соединениях, или получен из коммерческих источников.
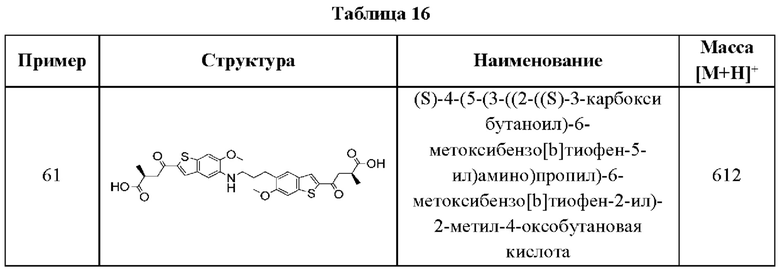
Пример 62: 4,4-((пропан-1,3-диилбис(окси))бис(4-фтор-6-метоксибензо[b]тиофен-5,2-диил))бис(4-оксобутановая кислота)
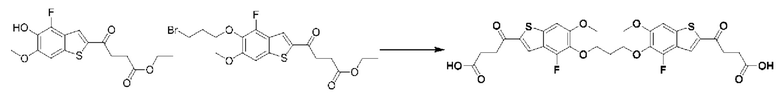
К смеси этил 4-(4-фтор-5-гидрокси-6-метоксибензо[b]тиофен-2-ил)-4-оксобутаноата (0.024 г, 0.073 ммоль), этил 4-(5-(3-бромпропокси)-4-фтор-6-метоксибензо[b]тиофен-2-ил)-4-оксобутаноата (0.025 г, 0.056 ммоль) и CS2CO3 (0.091 г, 0.28 ммоль) добавляли ACN (1.0 мл). Реакционную смесь затем нагревали до 65°С в течение 1 ч. После охлаждения до комнатной температуры смесь разбавляли THF, фильтровали, и фильтрат концентрировали под пониженным давлением. К полученному в результате остатку добавляли THF (1.0 мл), МеОН (0.20 мл), воду (0.5 мл) и LiOH (0.013 г, 0.56 ммоль). Смесь оставляли перемешиваться при комнатной температуре в течение 2 ч. Затем реакцию останавливали АсОН, и смесь концентрировали под пониженным давлением. Полученный в результате остаток очищали с помощью препаративной ВЭЖХ (ACN/H2O с 0.1% TFA) с получением 4,4-((пропан-1,3-диилбис(окси))бис(4-фтор-6-метоксибензо[b]тиофен-5,2-диил))бис(4-оксобутановая кислота). ЖХ-МС (C29H27F2O10S2) (ES, m/z): 637 [М+Н]+. 1Н ЯМР (600 МГц, DMSO-d6) δ 8.26 (s, 2Н), 7.55 (s, 2Н), 4.31-4.24 (m, 4Н), 3.99-3.80 (m, 6Н), 3.34-3.32 (m, 4Н), 2.60 (q, J=6.4 Гц, 4Н), 2.09 (dt, J=11.5, 5.8 Гц, 2Н).
Примеры с 63 по 65 и с 161 по 164, как показано в Таблице 17 ниже, или могут быть получены в соответствии со способами, аналогичными описанным в Примере 62 выше, с применением подходящих исходных материалов, описанных выше в Способах получения или Промежуточных соединениях, или получены из коммерческих источников.
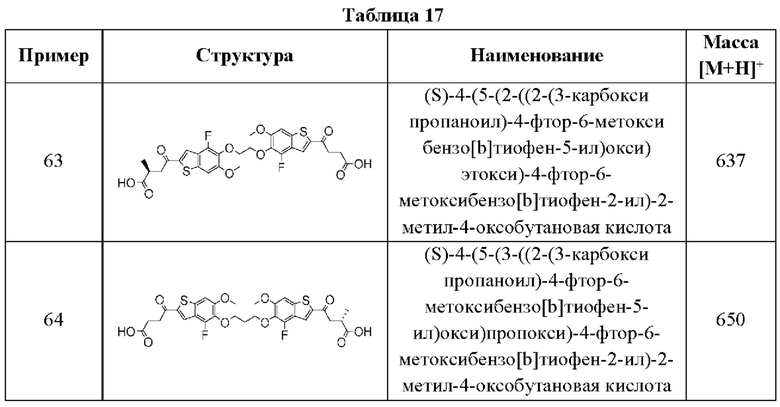
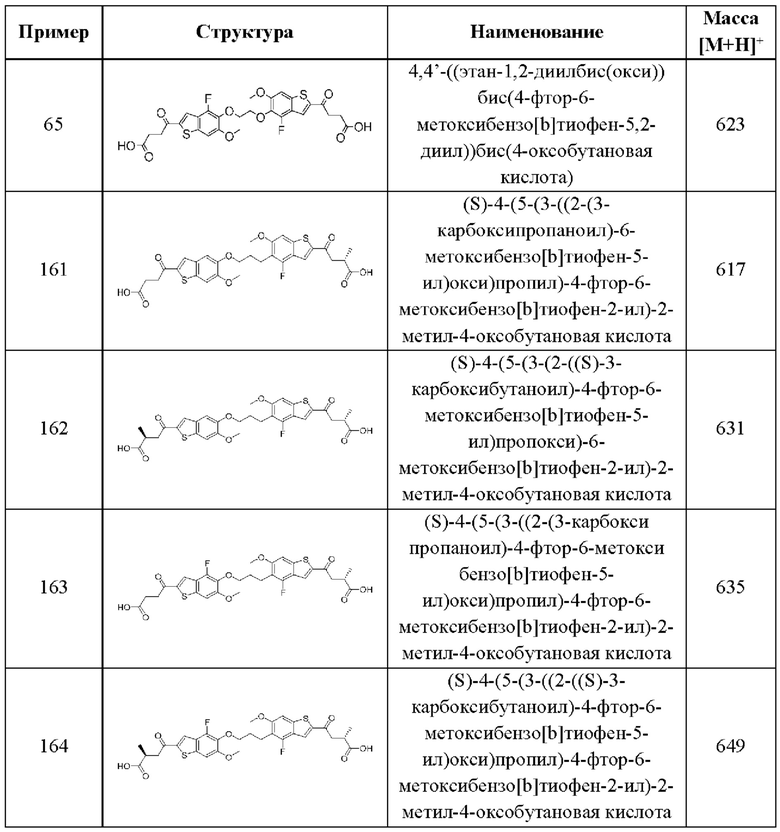
Пример 66: (R)-4-(5-(2-((2-((S)-3-карбоксибутаноил)-4-фтор-6-метоксибензо[b]тиофен-5-ил)окси)этокси)-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутановая кислота
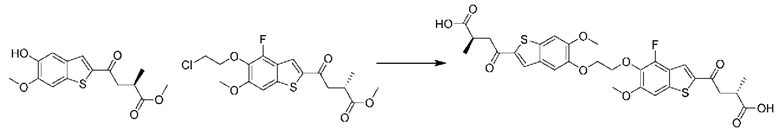
(R)-метил 4-(5-гидрокси-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноат (0.20 М в DMF, 500 мкл, 0.11 ммоль) добавляли к перемешиваемой суспензии метил (S)-4-(5-(2-хлорэтокси)-4-фтор-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата (0.20 М в DMF, 500 мкл, 0.11 ммоль) и K2CO3 (30 мг, 0.2 ммоль). Реакционную смесь нагревали до 100°С в течение 18 ч. После охлаждения до комнатной температуры добавляли DMSO (500 мкл), и реакционную смесь фильтровали. Добавляли воду (500 мкл) с последующим добавлением LiOH⋅H2O (30 мг, 0.7 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 18 ч. Смесь фильтровали, и вещество очищали с помощью колоночной хроматографии с обращенными фазами, колонка С-18 (ACN/вода + 0.1% TFA) с получением (R)-4-(5-(2-((2-((S)-3-карбоксибутаноил)-4-фтор-6-метоксибензо[b]тиофен-5-ил)окси)этокси)-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутановой кислоты. ЖХ-МС (C30H29FO10S2Na) (ES, m/z): 655 [M+Na]+. 1H ЯМР (DMSO-d6) δ: 12.18 (s, 2H), 8.31 (s, 1H), 8.18 (s, 1H), 7.58 (s, 2H), 7.50 (s, 1H), 4.50-4.41 (m, 2H), 4.36-4.30 (m, 2H), 3.88 (s, 3H), 3.75 (s, 3H), 3.46 (dd, J=18, 9 Гц, 1H), 3.40 (dd, J=17, 9 Гц, 1H), 3.10 (ddd, J=22, 18, 5 Гц, 2H), 2.95-2.81 (m, 2H), 1.24-1.14 (m, 6H).
Примеры с 67 по 74 и с 165 по 171, как показано в Таблице 18 ниже, или могут быть получены в соответствии со способами, аналогичными описанным в Примере 66 выше, с применением подходящих исходных материалов, описанных выше в Способах получения или Промежуточных соединениях, или получены из коммерческих источников.
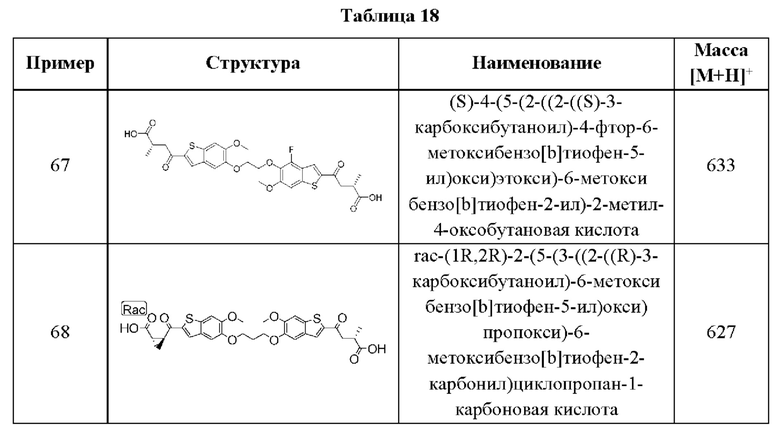
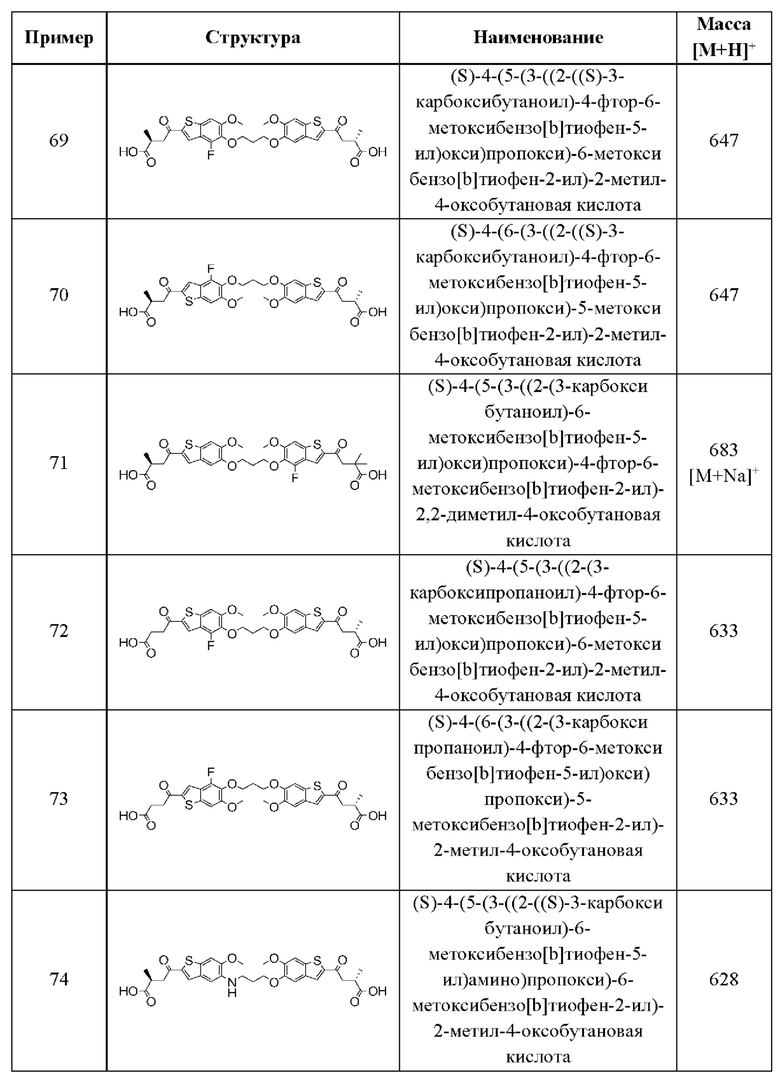
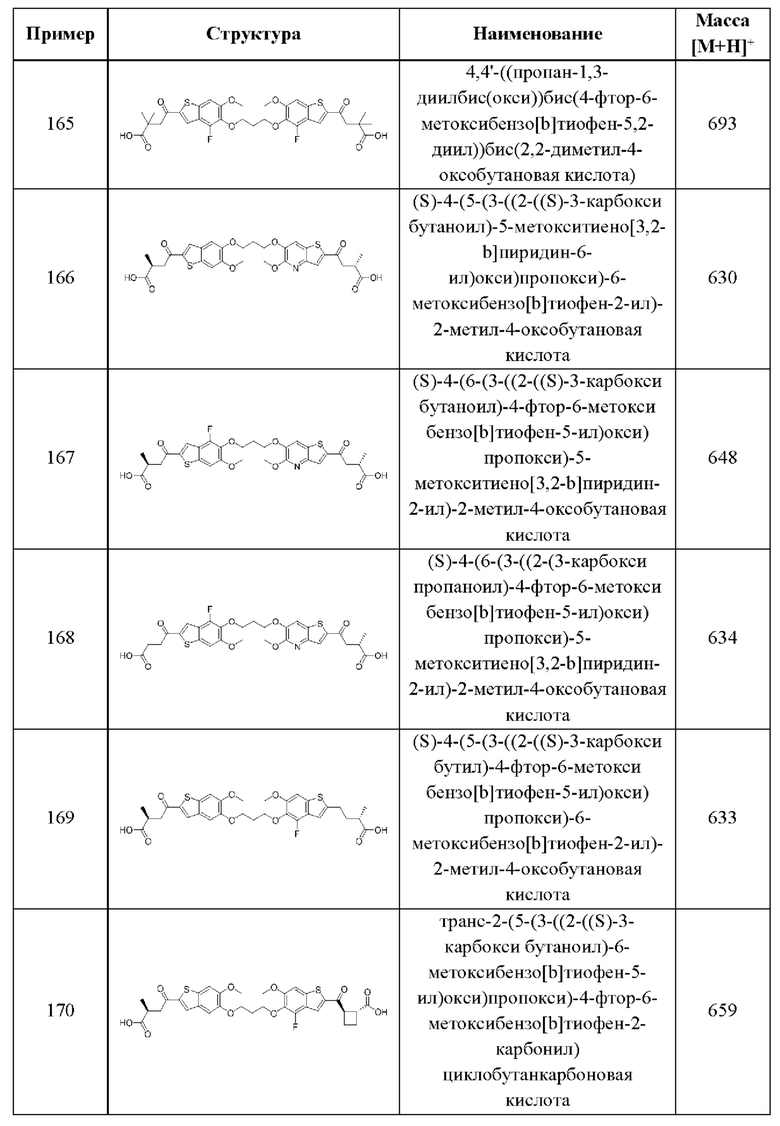
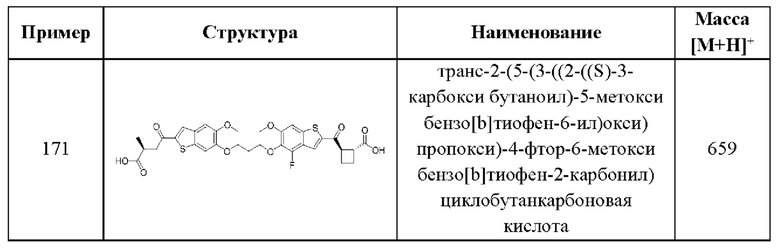
Пример 75: 4-(5-((3-(2-(3-карбоксипропаноил)-6-метоксибензо[b]тиофен-5-ил)пропил)амино)-6-метокситиено[3,2-b]пиридин-2-ил)-4-оксобутановая кислота
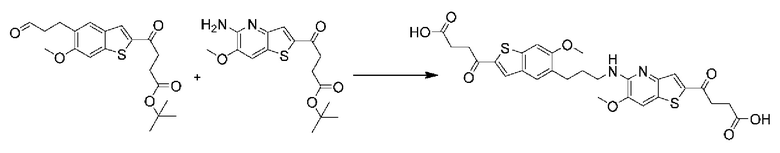
В 4 мл виалу добавляли трет-бутил 4-(6-метокси-5-(3-оксопропил)бензо[b]тиофен-2-ил)-4-оксобутаноат (10 мг, 0.03 ммоль), mpem-бутил 4-(5-амино-6-метокситиено[3,2-b]пиридин-2-ил)-4-оксобутаноат (8.9 мг, 0.027 ммоль) и THF (0.50 мл). К суспензии добавляли АсОН (0.015 мл; 0.27 ммоль). Смесь нагревали до 55°С в течение 10 мин. После охлаждения до комнатной температуры добавляли триацетоксиборогидрид натрия (17 мг, 0.080 ммоль). Смесь затем нагревали до 55°С в течение 3 ч. После охлаждения до комнатной температуры добавляли TFA (1 мл), и смесь оставляли перемешиваться при комнатной температуре в течение 1 ч. Смесь затем концентрировали под пониженным давлением и затем разбавляли DMSO (1 мл). Смесь очищали с помощью препаративной ВЭЖХ (ACN/H2O масс./0.1% TFA) с получением 4-(5-((3-(2-(3-карбоксипропаноил)-6-метоксибензо[b]тиофен-5-ил)пропил)амино)-6-метокситиено[3,2-b]пиридин-2-ил)-4-оксобутановой кислоты. ЖХ-МС (C28H29N2O8S2) (ES, m/z): 585 [M+H]+. 1H ЯМР (500 МГц, DMSO-d6) δ 12.18 (br, 2H), 8.22 (s, 1H), 8.03 (s, 1H), 7.79 (s, 1H), 7.63 (s, 1H), 7.59 (s, 1H), 6.55 (br s, 1H), 3.90 (s, 3H), 3.88 (s, 3H), 3.48-3.43 (m, 2H), 3.27-3.24 (m, 4H), 2.73 (t, J=7.3 Гц, 2H), 2.61-2.56 (m, 4H), 1.98-1.91 (m, 2H).
Пример 76: (2S,2'S)-4,4'-((этан-1,2-диилбис(окси))бис(6-метоксибензо[b]тиофен-5,2-диил))бис(2-метил-4-оксобутановая кислота)
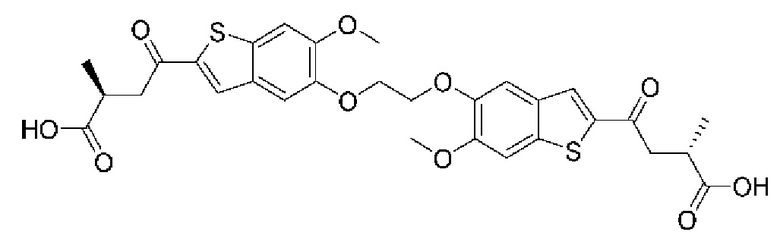
Стадия 1: диметил 4,4'-((этан-1,2-диилбис(окси))бис(6-метоксибензо[b]тиофен-5,2-диил))(2S,2'S)-бис(2-метил-4-оксобутаноат)
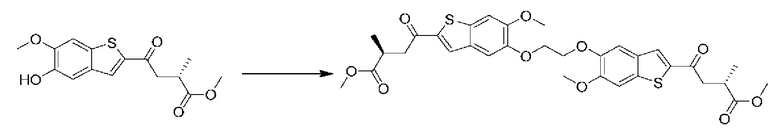
Смесь метил (S)-4-(5-гидрокси-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата (50 мг, 0.17 ммоль) и K2CO3 (47 мг, 0.34 ммоль) разбавляли DMF (750 мкл). Добавляли 1-бром-2-хлорпентан (18 мкл, 0.22 ммоль), и реакционную смесь нагревали до 100°С в течение 18 ч. Реакционную смесь концентрировали под пониженным давлением и применяли без дополнительной очистки или определения характеристик.
Стадия 2: (2S,2'S)-4,4'-((этан-1,2-диилбис(окси))бис(6-метоксибензо[b]тиофен-5,2-диил))бис(2-метил-4-оксобутановая кислота)
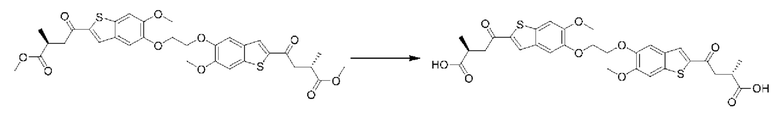
LiOH (16 мг, 0.67 ммоль) добавляли к перемешиваемому раствору диметил 4,4'-((этан-1,2-диилбис(окси))бис(6-метоксибензо[b]тиофен-5,2-диил))(2S,2'S)-бис(2-метил-4-оксобутаноат) (50 мг, 0.08 ммоль) в THF (340 мкл)/воде (80 мкл). Реакционную смесь оставляли перемешиваться при комнатной температуре в течение 4 ч. Реакционную смесь фильтровали и концентрировали под пониженным давлением. Остаток растворяли в DMSO (4 мл) и фильтровали. Вещество очищали с помощью колоночной хроматографии с обращенными фазами, колонка С18 (ACN/вода с 0.1% TFA) с получением (2S,2'S)-4,4'-(5,5'-(этан-1,2-диилбис(окси))бис(6-метоксибензо[b]тиофен-5,2-диил))бис(2-метил-4-оксобутановая кислота). ЖХ-МС (C30H30O10S2Na) (ES, m/z): 637 [M+Na]+. 1H ЯМР (500 МГц, DMSO-d6) δ: 12.21 (br s, 2H), 8.20 (s, 2H), 7.64 (s, 2H), 7.59 (s, 2H), 4.44 (s, 4H), 3.87 (s, 6H), 3.41 (dd, J=17, 9 Гц, 2H), 3.09 (dd, J=17, 5 Гц, 2H), 3.05-2.58 (m, 2H), 1.19 (d, J=7 Гц, 6Н).
Пример 77: (2S,2'S)-4,4'-(7,8,17,18-тетрагидро-6H,16H-бис[1]бензотиено[5,6-b:6',5'-i][1,4,8,11]тетраоксациклотетрадецин-2,12-диил)бис(2-метил-4-оксобутановая кислота) и
Пример 78: (2S,2'S)-4,4'-(7,8,17,18-тетрагидро-6H,16H-бис[1]бензотиено[5,6-b:5',6'-i][1,4,8,11]тетраоксациклотетрадецин-2,12-диил)бис(2-метил-4-оксобутановая кислота)
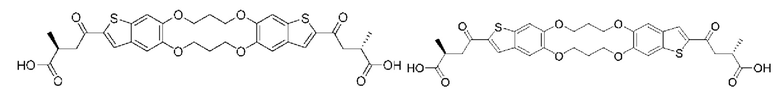
Стадия 1: диметил (2S,2'S)-4,4'-(7,8,17,18-тетрагидро-6Н,16Н-бис[1]бензотиено[5,6-b:5',6'-i][1,4,8,11]тетраоксациклотетрадецин-2,12-диил)бис(2-метил-4-оксобутаноат) и диметил (2S,2'S)-4,4'-(7,8,17,18-тетрагидро-6Н,16Н-бис[1]бензотиено[5,6-b:6',5'-i][1,4,8,11] тетраоксациклотетрадецин-2,12-диил)бис(2-метил-4-оксобутаноат)
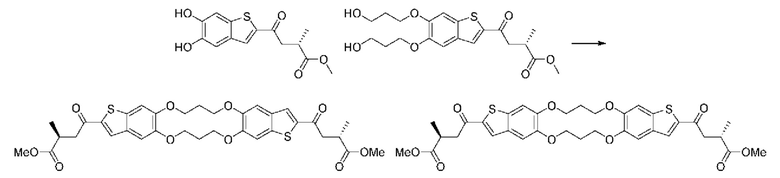
Смесь метил (2S)-4-(5,6-дигидрокси-1-бензотиофен-2-ил)-2-метил-4-оксобутаноата (60 мг, 0.20 ммоль), метил (2S)-4-[5,6-бис(3-гидроксипропокси)-1-бензотиофен-2-ил]-2-метил-4-оксобутаноата (120 мг, 0.20 ммоль), Ph3P (193 мг, 0.737 ммоль) и 1,1'-(азодикарбонил)дипиперидина (186 мг, 0.737 ммоль) в THF (2.0 мл) оставляли перемешиваться при комнатной температуре в течение 18 ч. Смесь затем фильтровали и очищали с помощью ВЭЖХ с обратными фазами (градиент MeCN/вода с 0.1% TFA) с получением смеси 1:1 диметил (2S,2'S)-4,4'-(7,8,17,18-тетрагидро-6H,16H-бис[1]бензотиено[5,6-b:5',6'-i][1,4,8,11]тетраоксациклотетрадецин-2,12-диил)бис(2-метил-4-оксобутаноат) и диметил (2S,2'S)-4,4'-(7,8,17,18-тетрагидро-6H,16H-бис[1]бензотиено[5,6-b:6',5'-i][1,4,8,11]тетраоксациклотетрадецин-2,12-диил)бис(2-метил-4-оксобутаноат). ЖХ-МС (C34H36O10S2Na) (ES, m/z): 691 [M+Na]+.
Стадия 2: (2S,2'S)-4,4'-(7,8,17,18-тетрагидро-6Н,16Н-бис[1]бензотиено[5,6-b:5',6'-i][1,4,8,11]тетраоксациклотетрадецин-2,12-диил)бис(2-метил-4-оксобутановая кислота) и (2S,2'S)-4,4'-(7,8,17,18-тетрагидро-6Н,16Н-бис[1]бензотиено[5,6-b:6',5'-i][1,4,8,11]тетраоксациклотетрадецин-2,12-диил)бис(2-метил-4-оксобутановая кислота)
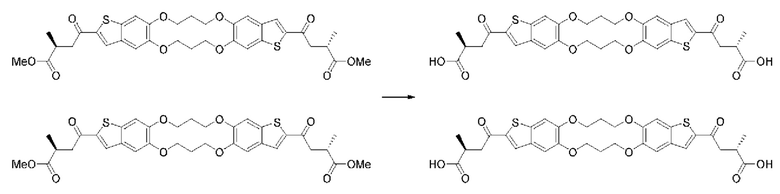
К перемешанному раствору смеси 1:1 диметил (2S,2'S)-4,4'-(7,8,17,18-тетрагидро-6H,16H-бис[1]бензотиено[5,6-b:5',6'-i][1,4,8,11]тетраоксациклотетрадецин-2,12-диил)бис(2-метил-4-оксобутаноат) и диметил (2S,2'S)-4,4'-(7,8,17,18-тетрагидро-6H,16H-бис[1]бензотиено[5,6-b:6',5'-i][1,4,8,11]тетраоксациклотетрадецин-2,12-диил)бис(2-метил-4-оксобутаноат) (67 мг, 0.10 ммоль) в МеОН (1.0 мл), THF (1.0 мл) и воде (0.2 мл) добавляли LiOH (24 мг, 1.0 ммоль). Реакционную смесь оставляли перемешиваться в течение 4 ч и затем очищали с помощью ВЭЖХ с обратными фазами (градиент MeCN/вода с 0.1% TFA) с получением смеси 1:1 (2S,2'S)-4,4'-(7,8,17,18-тетрагидро-6H,16H-бис[1]бензотиено[5,6-b:5',6'-i][1,4,8,11]тетраоксациклотетрадецин-2,12-диил)бис(2-метил-4-оксобутановая кислота) и (2S,2'S)-4,4'-(7,8,17,18-тетрагидро-6H,16H-бис[1]бензотиено[5,6-b:6',5'-i][1,4,8,11]тетраоксацикло-тетрадецин-2,12-диил)бис(2-метил-4-оксобутановая кислота). ЖХ-МС (C32H33O10S2) (ES, m/z): 641 [M+H]+. 1H ЯМР (500 МГц, DMSO-d6) δ 12.20 (brs, 2H), 8.18 (s, 2H), 7.52 (s, 2H), 7.40 (s, 2H), 4.27-4.18 (m, 8H), 3.42-3.34 (m, 2H), 3.10-3.03 (m, 2H), 2.92-2.85 (m, 2H), 2.29 (brs, 4H), 1.17 (d, J=6.8 Гц, 6Н).
Пример 79: (2S,2'S)-4,4'-(1,3-пропандиилбис{окси[6-(дифторметокси)-1-бензотиен-5,2-диил]})бис(2-метил-4-оксобутановая кислота)
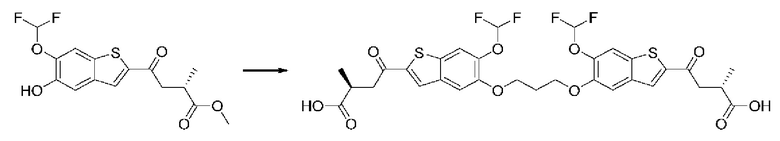
К перемешанному раствору метил (2S)-4-[6-(дифторметокси)-5-гидрокси-1-бензотиофен-2-ил]-2-метил-4-оксобутаноата (54 мг, 0.16 ммоль) и K2CO3 (109 мг, 0.788 ммоль) в DMF (0.8 мл) добавляли 1,3-дибромпропан (8 мкл, 0.08 ммоль). Смесь нагревали до 50°С в течение 4 ч. После охлаждения до комнатной температуры смесь обрабатывали LiOH (2М в воде, 790 мкл, 1.58 ммоль) и оставляли перемешиваться при комнатной температуре в течение 4 ч. Смесь затем концентрировали под пониженным давлением и очищали с помощью ВЭЖХ с обратными фазами (градиент MeCN/вода с 0.1% TFA) с получением (2S,2'S)-4,4'-(1,3-пропандиилбис{окси[6-(дифторметокси)-1-бензотиен-5,2-диил]})бис(2-метил-4-оксобутановая кислота). ЖХ-МС (C31H28F4O10S2Na) (ES, m/z): 723 [M+23]+. 1H ЯМР (500 МГц, DMSO-d6) δ 12.25 (s, 2H), 8.27 (s, 2H), 7.94 (s, 2H), 7.73 (s, 2H), 7.20 (t, J=74.0 Гц, 2Н), 4.33-4.29 (m, 4Н), 3.46-3.40 (m, 2H), 3.12 (dd, J=17.5, 4.9 Гц, 2Н), 2.94-2.87 (m, 2H), 2.37-2.32 (m, 2H), 1.19 (d, J=7.1 Гц, 6Н).
Примеры 80 и 172, как показано в Таблице 19 ниже, или могут быть получены в соответствии со способами, аналогичными описанным в Примере 79 выше, с применением подходящих исходных материалов, описанных выше в Способах получения или Промежуточных соединениях, или получены из коммерческих источников.
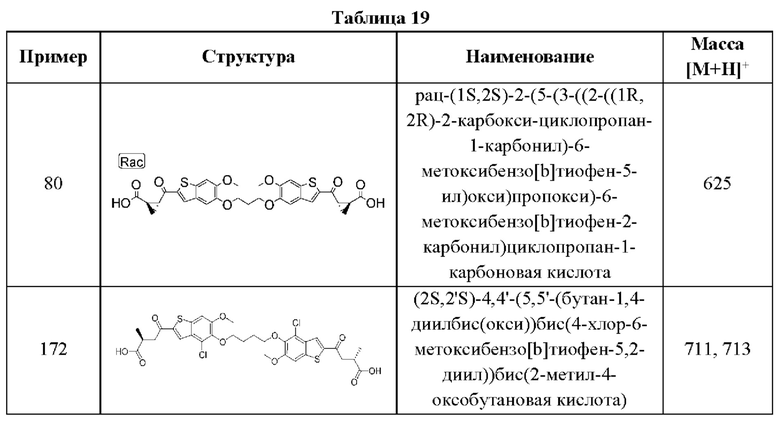
Пример 173: (4-(5-(3-((2-(3-Карбоксипропаноил)-4-фтор-6-метоксибензо[b]тиофен-5-ил)окси)пропил)-6-метокситиено[3,2-b]пиридин-2-ил)-4-оксобутановая кислота
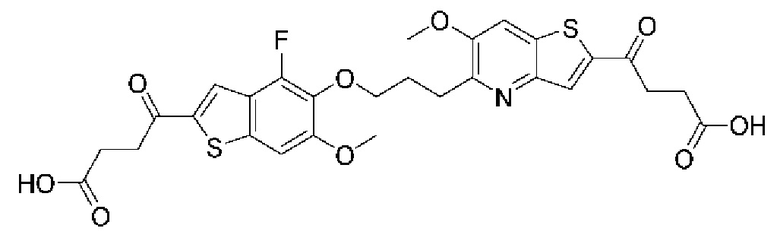
трет-Бутил 4-(5-(3-гидроксипропил)-6-метокситиено[3,2-b]пиридин-2-ил)-4-оксобутаноат (76 мг, 0.20 ммоль) добавляли в виалу, содержащую этил 4-(4-фтор-5-гидрокси-6-метоксибензо[b]тиофен-2-ил)-4-оксобутаноат (250 мг, 0.77 ммоль), PS-TPP (319 мг, 0.600 ммоль), DIAD (0.117 мл, 0.600 ммоль) и THF (3 мл). Реакционную смесь перемешивали в течение 3 ч при комнатной температуре. Реакционную смесь фильтровали, промывали THF (5 мл). Добавляли МеОН (2 мл), воду (3 мл) и LiOH (48 мг, 2.0 ммоль), и реакционную смесь перемешивали в течение дополнительных 2 ч. Затем реакцию останавливали НОАс и концентрировали под пониженным давлением. Остаток суспендировали в DCM (5 мл) и добавляли TFA (1 мл). Реакционную смесь перемешивали в течение 1 ч. Реакционную смесь затем концентрировали под пониженным давлением, и остаток очищали с помощью ВЭЖХ с обратными фазами (градиент ацетонитрил/вода с 0.1% TFA) с получением (4-(5-(3-((2-(3-карбоксипропаноил)-4-фтор-6-метоксибензо[b]тиофен-5-ил)окси)пропил)-6-метокситиено[3,2-b]пиридин-2-ил)-4-оксобутановой кислоты. ЖХ-МС (C28H27FNO9S2) (ES, m/z): 604 [M+H]+. 1H ЯМР (600 МГц, DMSO-d6) δ 8.34 (s, 1H), 8.31 (s, 1H), 8.07 (s, 1H), 7.57 (s, 1H), 4.17 (t, J=6.3 Гц, 2H), 3.93 (s, 3H), 3.90 (s, 3H), 3.35-3.29 (m, 4H), 3.10-3.04 (m, 2H), 2.63-2.58 (m, 4H), 2.18-2.13 (m, 2H).
Пример 174, как показано в Таблице 20 ниже, или может быть получен в соответствии со способами, аналогичными описанным в Примере 173 выше, с применением подходящих исходных материалов, описанных выше в Способах получения или Промежуточных соединениях, или получен из коммерческих источников.
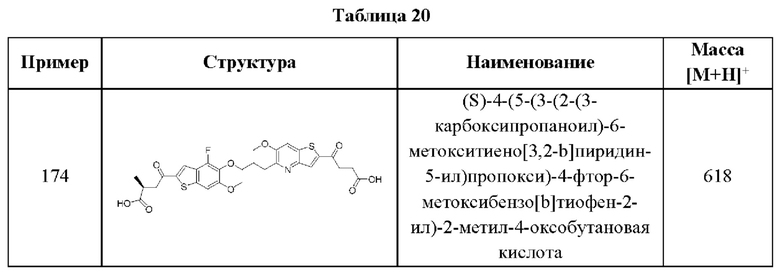
Пример 175: 4,4'-(Пропан-1,3-диилбис(6-метоксибензо[b]тиофен-5,2-диил))бис(4-оксобутаннитрил)
4-(5-Бром-6-метоксибензо[b]тиофен-2-ил)-4-оксобутаннитрил (35 мг, 0.11 ммоль), комплекс никель (II) бромид этиленгликоль диметиловый эфир (10 мг, 0.033 ммоль), марганец (24 мг, 0.44 ммоль), NaI (8 мг, 0.06 ммоль) и 4,4'-диметокси-2,2'-бипиридин (7 мг, 0.03 ммоль) объединяли в виале. В виалу добавляли раствор 4-(5-(3-бромпропил)-6-метоксибензо[b]тиофен-2-ил)-4-оксобутаннитрила (40 мг, 0.11 ммоль) в DMPU (1.1 мл). К реакционной смеси добавляли 5% об./об. растворы в DMPU Ру (88 мкл, 0.055 ммоль) и TMS-C1 (84 мкл, 0.033 ммоль). Реакционную смесь дегазировали Ar в течение 5 мин, и затем перемешивали и нагревали при 90°С в течение 2 ч. Реакционную смесь охлаждали до комнатной температуры, разбавляли DMSO (2 мл) и фильтровали. Фильтрат очищали с помощью ВЭЖХ с обратными фазами (ACN/вода с модификатором TFA) с получением 4,4'-(пропан-1,3-диилбис(6-метоксибензо[b]тиофен-5,2-диил))бис(4-оксобутаннитрил). ЖХ-МС (C29H27N2O4S2) (ES, m/z): 531 [М+Н]+. 1H ЯМР (499 МГц, DMSO-d6) δ 8.26 (s, 2H), 7.78 (s, 2H), 7.61 (s, 2H), 3.89 (s, 6H), 3.55-3.45 (m, 4H), 2.81-2.76 (m, 4H), 2.74-2.69 (m, 4H), 1.95-1.90 (m, 2H).
Пример 176: (S)-4-(5-((3-((2-((S)-3-Карбоксибутаноил)-4-фтор-6-метоксибензо[b]тиофен-5-ил)окси)пропил)амино)-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутановая кислота
Смесь (S)-метил 4-(5-амино-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата (37 мг, 0.12 ммоль), (S)-метил 4-(5-(3-бромпропокси)-4-фтор-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата (54 мг, 0.12 ммоль) и карбоната калия (67 мг, 0.48 ммоль) дегазировали Ar. К смеси добавляли DMF (1.0 мл), и реакционную смесь перемешивали и нагревали при 60°С в течение 2 дней. Реакционную смесь охлаждали до комнатной температуры и затем добавляли NaOH (1.0 M в воде, 0.48 мл, 0.48 ммоль). Смесь дополнительно разбавляли DMSO (1.5 мл) и затем перемешивали при комнатной температуре в течение 1 ч. Реакцию останавливали TFA (0.056 мл, 0.72 ммоль) и фильтровали. Фильтрат очищали с помощью ВЭЖХ с обратными фазами (ACN/вода с 0.1% TFA) с получением (S)-4-(5-((3-((2-((S)-3-карбокси бутаноил)-4-фтор-6-метоксибензо[b]тиофен-5-ил)окси)пропил)амино)-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутановой кислоты. ЖХ-МС (C31H33FNO9S2) (ES, m/z): 646 [М+Н]+. 1H ЯМР (499 МГц, DMSO-d6) δ 8.32 (s, 1Н), 8.11 (s, 1H), 7.60 (s, 1H), 7.45 (s, 1H), 7.04 (s, 1H), 4.17 (t, J=5.9 Гц, 2H), 3.92 (s, 3H), 3.90 (s, 3H), 3.47 (dd, J=17.6, 8.6 Гц, 1H), 3.43-3.34 (m, 3H), 3.17-3.05 (m, 2H), 2.94-2.95 (m, 2H), 2.09-2.02 (m, 2H), 1.20-1.15 (m, 6H).
Пример 177, как показано в Таблице 21 ниже, или может быть получен в соответствии со способами, аналогичными описанным в Примере 176 выше, с применением подходящих исходных материалов, описанных выше в Способах получения или Промежуточных соединениях, или получен из коммерческих источников.
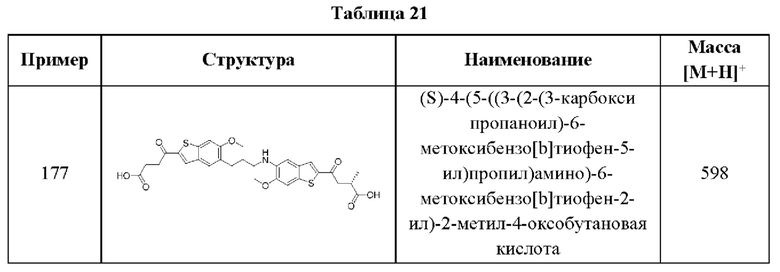
Пример 178: (S)-4-(5-(3-((2-((S)-4-((N,N-диметилсульфамоил)амино)-3-метил-4-оксобутаноил)-4-фтор-6-метоксибензо[b]тиофен-5-ил)окси)пропокси)-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутановая кислота
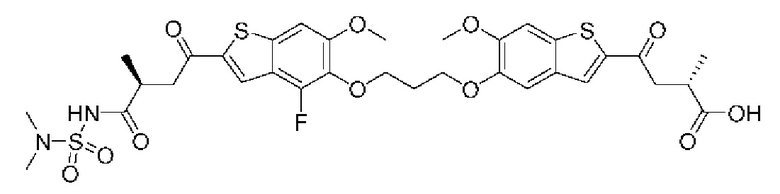
Стадия 1: трет-бутил (S)-4-(5-(3-((4-фтор-6-метокси-2-((S)-4-метокси-3-метил-4-оксобутаноил)бензо[b]тиофен-5-ил)окси)пропокси)-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноат
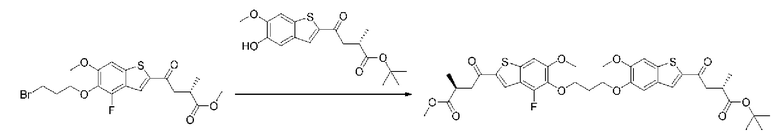
Смесь (S)-метил 4-(5-(3-бромпропокси)-4-фтор-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата (111 мг, 0.248 ммоль), (S)-трет-бутил 4-(5-гидрокси-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата (87 мг, 0.25 ммоль) и карбоната калия (137 мг, 0.993 ммоль) дегазировали Ar. К смеси добавляли DMF (1.0 мл), и реакционную смесь перемешивали и нагревали при 40°С в течение 18 ч. Реакционную смесь охлаждали до комнатной температуры и затем разбавляли EtOAc и водой. Органический слой отделяли, высушивали над MgSO4, фильтровали и концентрировали под пониженным давлением. Остаток очищали с помощью хроматографии на силикагеле (EtOAc/гексан) с получением трет-бутил (S)-4-(5-(3-((4-фтор-6-метокси-2-((S)-4-метокси-3-метил-4-оксобутаноил)бензо[b]тиофен-5-ил)окси)пропокси)-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата. ЖХ-МС (C36H42FO10S2) (ES, m/z): 739 [M+Na]+. 1H ЯМР (499 МГц, DMSO-d6) δ 8.30 (s, 1H), 8.21 (s, 1H), 7.59 (s, 1H), 7.57 (s, 1H), 7.51 (s, 1H), 4.26 (q, J=5.9 Гц, 4H), 3.86 (s, 3Н), 3.85 (s, 3Н), 3.60 (s, 3Н), 3.48 (dd, J=17.7, 8.7 Гц, 1H), 3.39-3.33 (m, 1H), 3.23 (dd, J=17.7, 5.0 Гц, IH), 3.07 (dd, J=17.1, 5.0 Гц, 1H), 3.02-2.93 (m, 1H), 2.91-2.81 (m, 1H), 2.23-2.18 (m, 2H), 1.35 (s, 9H), 1.20-1.15 (m, 6H).
Стадия 2: (S)-4-(5-(3-((2-((S)-4-(трет-бутокси)-3-метил-4-оксобутаноил)-6-метоксибензо[b]тиофен-5-ил)окси)пропокси)-4-фтор-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутановая кислота
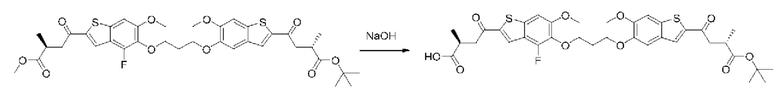
NaOH (1.0 M в воде, 0.94 мл, 0.94 ммоль) добавляли к смеси (S)-трет-бутил 4-(5-(3-((4-фтор-6-метокси-2-((S)-4-метокси-3-метил-4-оксобутаноил)-бензо[b]тиофен-5-ил)окси)пропокси)-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата (135 мг, 0.188 ммоль) в THF (0.94 мл) и МеОН (0.94 мл). Смесь перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь останавливали HCl (2.0 М в воде, 0.47 мл, 0.94 ммоль) и разбавляли EtOAc и водой. Органический слой отделяли, высушивали над MgSO4, фильтровали и концентрировали под пониженным давлением с получением (S)-4-(5-(3-((2-((S)-(трет-бутокси)-3-метил-4-оксобутаноил)-6-метоксибензо[b]тиофен-5-ил)окси)пропокси)-4-фтор-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутановой кислоты. ЖХ-МС (C35H40FO10S2) (ES, m/z): 725 [M+Na]+.
Стадия 3: (S)-4-(5-(3-((2-((S)-4-((N,N-диметилсульфамоил)амино)-3-метил-4-оксобутаноил)-4-фтор-6-метоксибензо[b]тиофен-5-ил)окси)пропокси)-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутановая кислота
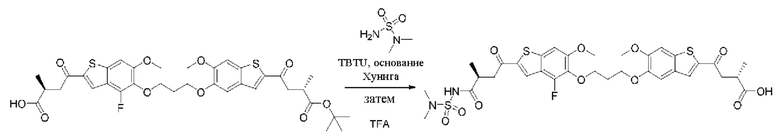
Основание Хунига (0.062 мл, 0.36 ммоль) и TBTU (23 мг, 0.071 ммоль) добавляли к смеси (S)-4-(5-(3-((2-((S)-4-(трет-бутокси)-3-метил-4-оксобутаноил)-6-метокси-бензо[b]тиофен-5-ил)окси)пропокси)-4-фтор-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутановой кислоты (50 мг, 0.071 ммоль) в DCM (0.7 мл). Смесь перемешивали при комнатной температуре в течение 30 мин. Добавляли N,N-диметилсульфамид (11 мг, 0.085 ммоль), и смесь перемешивали в течение дополнительных 4 ч. Затем добавляли TFA (0.055 мл, 0.71 ммоль); и смесь перемешивали и нагревали до 45°С в течение 2 ч. Смесь охлаждали до комнатной температуры и концентрировали под пониженным давлением. Остаток очищали с помощью ВЭЖХ с обратными фазами (ACN/вода с 0.1% TFA) с получением (S)-4-(5-(3-((2-S)-4-((N,N-диметилсульфамоил)амино)-3-метил-4-оксобутаноил)-4-фтор-6-метоксибензо[b]тиофен-5-ил)окси)пропокси)-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутановой кислоты. ЖХ-МС (C33H38FN2O11S3) (ES, m/z): 775 [M+Na]+. 1H ЯМР (499 МГц, DMSO-d6) δ 11.50 (s, 1Н), 8.30 (s, 1Н), 8.22 (s, 1Н), 7.59 (s, 1Н), 7.56 (s, 1H), 7.52 (s, 1H), 4.26 (q, J=5.9 Гц, 4H), 3.87 (s, 3H), 3.85 (s, 3H), 3.48 (dd, J=17.8, 9.8 Гц, 1Н), 3.40 (dd, J=17.3, 8.4 Гц, 1Н), 3.16 (dd, J=17.8, 4.4 Гц, 1Н), 3.09 (dd, J=17.3, 5.3 Гц, 1Н), 3.00-2.94 (m, 1Н), 2.94-2.87 (m, 1Н), 2.79 (s, 6H), 2.23-2.17 (m, 2H), 1.21-1.15 (m, 6H).
Пример 179, как показано в Таблице 22 ниже, или может быть получен в соответствии со способами, аналогичными описанным в Примере 178 выше, с применением подходящих исходных материалов, описанных выше в Способах получения или Промежуточных соединениях, или получен из коммерческих источников.
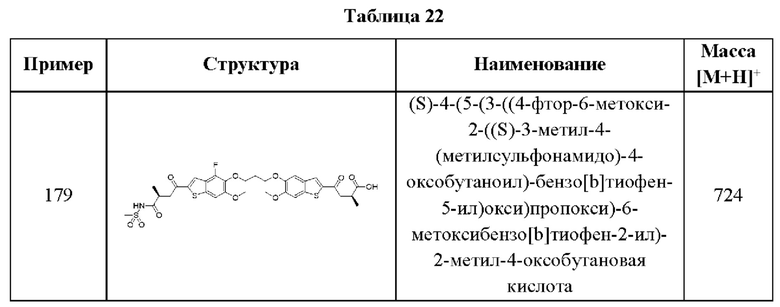
Пример 180: (S)-4-(5-(3-((2-((S)-3-Карбоксибутаноил)-4-фтор-6-метоксибензо[b]тиофен-5-ил)окси)пропокси)-6-гидроксибензо[b]тиофен-2-ил)-2-метил-4-оксобутановая кислота
Стадия 1: метил (S)-4-(5-(3-((4-фтор-6-метокси-2-((S)-4-метокси-3-метил-4-оксобутаноил)бензо[b]тиофен-5-ил)окси)пропокси)-6-(метоксиметокси)бензо[b]тиофен-2-ил)-2-метил-4-оксобутаноат
Смесь метил (S)-4-(5-(3-бромпропокси)-4-фтор-6-метоксибензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата (30 мг, 0.068 ммоль), метил (S)-4-(5-гидрокси-6-(метоксиметокси)бензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата (23 мг, 0.068 ммоль) и карбоната калия (38 мг, 0.27 ммоль) дегазировали Ar. К смеси добавляли DMF (0.5 мл), и реакционную смесь перемешивали и нагревали при 40°С в течение 24 ч. Реакционную смесь охлаждали до комнатной температуры и затем применяли без обработки или очистки в следующей реакции. ЖХ-МС (C34H38FO11S2) (ES, m/z): 705 [M+H]+.
Стадия 2: (S)-4-(5-(3-((2-((S)-3-карбоксибутаноил)-4-фтор-6-метоксибензо[b]тиофен-5-ил)окси)пропокси)-6-гидроксибензо[b]тиофен-2-ил)-2-метил-4-оксобутановая кислота
NaOH (0.55 мл, 1.0 М в воде, 0.55 ммоль) добавляли к раствору метил (S)-4-(5-(3-((4-фтор-6-метокси-2-((S)-4-метокси-3-метил-4-оксобутаноил)бензо[b]тиофен-5-ил)окси)пропокси)-6-(метоксиметокси)бензо[b]тиофен-2-ил)-2-метил-4-оксобутаноата (48 мг, 0.068 ммоль) в DMSO (2.5 мл) при 20°С. Реакционную смесь затем перемешивали при 20°С в течение 15 мин. Реакционную смесь останавливали HCl (0.280 мл, 37% в воде, 3.41 ммоль). Реакционную смесь перемешивали при 20°С в течение 20 ч. Реакционную смесь фильтровали. Остаток очищали с помощью ВЭЖХ с обратными фазами (ACN/вода с 0.1% TFA) с получением (S)-4-(5-(3-((2-((S)-3-карбоксибутаноил)-4-фтор-6-метоксибензо[b]тиофен-5-ил)окси)пропокси)-6-гидроксибензо[b]тиофен-2-ил)-2-метил-4-оксобутановой кислоты. ЖХ-МС (C30H30FO10S2) (ES, m/z): 633 [M+H]+. 1H ЯМР (499 МГц, DMSO-d6) δ 12.17 (s, 2H), 9.70 (s, 1Н), 8.30 (s, 1H), 8.17 (s, 1H), 7.57 (s, 1H), 7.48 (s, 1H), 7.33 (s, 1H), 4.31-4.26 (m, 4H), 3.87 (s, 3H), 3.46 (dd, J=17.6, 8.6 Гц, 1H), 3.38 (dd, J=17.2, 8.4 Гц, 1H), 3.13 (dd, J=17.6, 5.1 Гц, 1H), 3.06 (dd, J=17.2, 5.3 Гц, 1H), 2.91-2.85 (m, 2H), 2.24-2.18 (m, 2H), 1.19 (d, J=7.2 Гц, 6Н).
Примеры с 181 по 190, как показано в Таблице 23 ниже, или могут быть получены в соответствии со способами, аналогичными описанным в Примере 180 выше, с применением подходящих исходных материалов; описанных выше в Способах получения или Промежуточных соединениях, или получены из коммерческих источников.
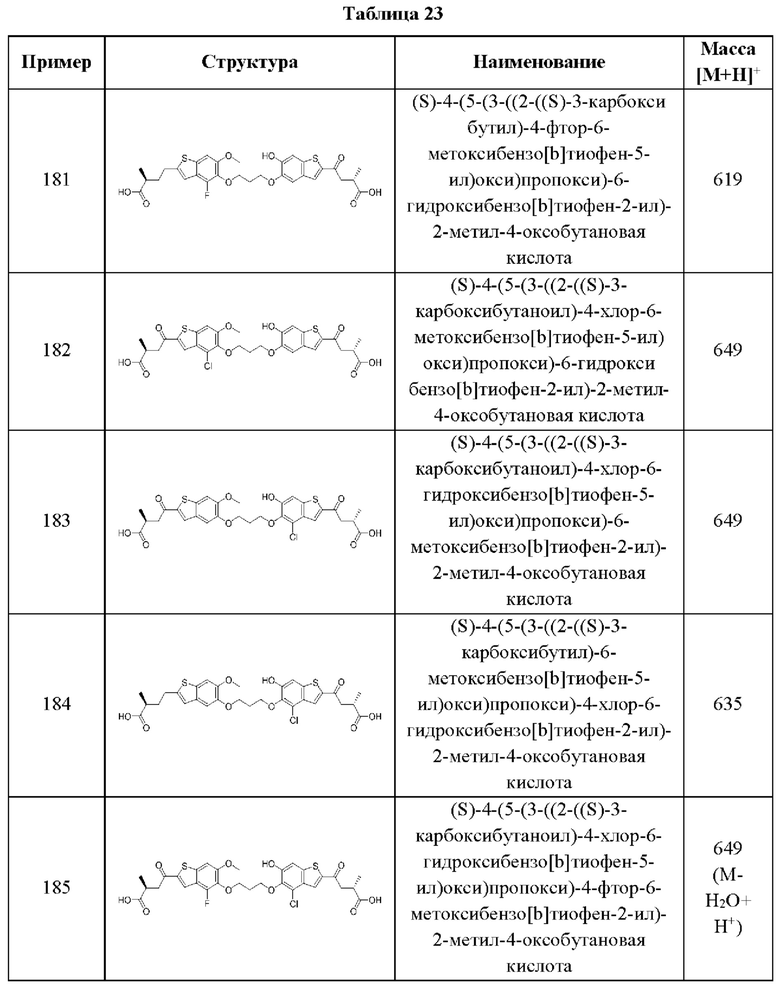
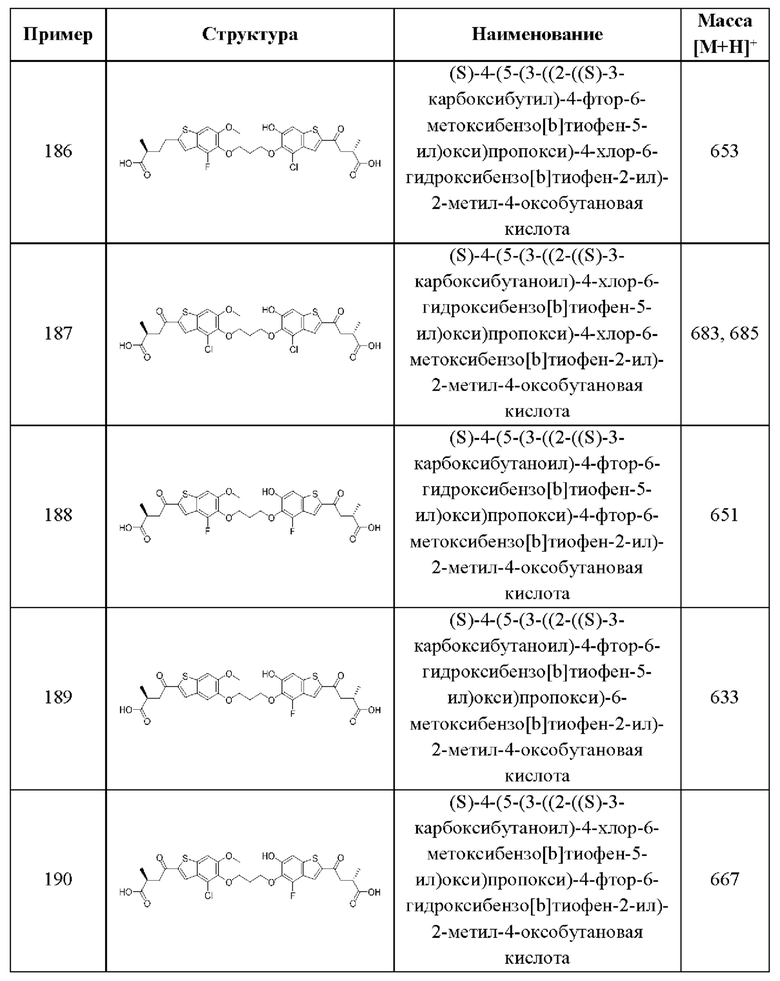
ОЦЕНКА БИОЛОГИЧЕСКОГО ДЕЙСТВИЯ
Отдельные соединения, описанные в приведенных здесь Примерах, определяются как агонисты STING путем (i) связывания с белком STING, о чем свидетельствует снижение связывания меченного тритием лиганда cGAMP с белком STING, по меньшей мере, на 20% при 20 мкМ (концентрация тестируемого соединения) в биохимическом анализе конкурентного связывания [3H]cGAMP с STING и (ii) определения продукции интерферона с 6% или большей индукцией секреции IFN-β при 30 мкМ в клеточном анализе ТНР1 (где индукция, вызванная cGAMP при 30 мкМ, была установлена как 100%).
Синтез [3H]-cGAMP
В пластиковую пробирку AMICON объемом 50 мл добавляли 2.3 мл буферного раствора, содержащего 80 мМ TrisCl, 200 мМ MgCl2 и 20 мМ NaCl с последующим добавлением 0.32 мл 10 мМ водного раствора GTP. Затем добавляли раствор [3H]ATP (21 Ки/ммоль, 45 мКи) в 0.5 мл H2O с последующим добавлением 1 мл раствора 1 мг/мл ДНК (активатора ДНК из семенников сельди, Sigma, #D6898) и 53 мкл 47 мМ раствора фермента cGAS. Добавляли дополнительную H2O, чтобы довести общий объем до 10 мл.
Реакционную смесь перемешивали в течение 2 часов при 37°С, и затем добавляли непосредственно в центрифужную пробирку Amicon Ultra-15 10K и центрифугировали в течение 1 часа при 4,000 g. Собранный раствор затем очищали на полупрепаративной колонке Mono Q, используя следующие подвижные фазы:
А: 0.05 М TrisCl, pH 8.5, доведенный 1 М NaOH.
В: 0.05 М TrisCl, 0.5 М NaCl, pH 8.5, доведенный 1 М NaOH.
Градиент: 100% А в течение 5 минут с последующим линейным градиентом до 50:50 (А:В) в течение 25 минут, 3 мл/мин, 254 нм.
Собранные фракции продукта объединяли и доводили до общего объема 30 мл с помощью буфера А. Суммарный выход 15.5 мКи [3H]cGAMP был выделен с радиохимической чистотой 98.0% при удельной радиоактивности 21.5 Ки/ммоль.
Фермент cGAS
Рекомбинантный ДНК-вектор был химически синтезирован для экспрессии усеченного фермента cGAS человека (остатки 161-522). Чтобы способствовать экспрессии и очистке, амино-конец содержит гексагистидиновую метку, метку SUMO и сайт расщепления TEV. Рекомбинантный фермент был сверхэкспрессирован в отдельных компетентных клетках ROSETTA™ 2(DE3) (Novagen). Аффинную очистку проводили с использованием HIS-Select HF Nickel Affinity Gel (Sigma) с последующей эксклюзионной хроматографией с использованием колонки Hi-Load 26/60 SUPERDEX200 (GE Healthcare) препаративной степени очистки. Фракции объединяли, концентрировали, быстро замораживали в жидком азоте и хранили при -80°С до возникновения необходимости.
Анализ связывания с фильтром 3H-cGAMP (HAQ STING)
Способность соединений связывать STING количественно оценивается по их способности конкурировать с меченным тритием лигандом cGAMP за мембрану человеческого рецептора STING с использованием радиоактивного анализа связывания с фильтром. В анализе связывания используется рецептор STING, полученный из клеточных мембран Trichoplusia ni (Т.ni; Expression Systems, номер по каталогу 94-002F, www.expressionsystems.com), сверхэкспрессирующий полноразмерный HAQ STING и меченный тритием лиганд cGAMP.
Базовый протокол анализа с фильтром HAQ STING представляет собой следующее:
Соединения последовательно титровали с помощью Hamilton STARPlus CORE в 96-луночном планшете (Greiner, #651201) с использованием 1:3 десятиточечного формата дозовой зависимости. После приготовления соединения рабочая концентрация мембраны STING (SEQ. ID. No. 1) 2.2 мкг/мл была приготовлена путем разбавления концентрированной мембраны в буфере для анализа (1x PBS; Invitrogen #SH30028.02) и 7-х гомогенизации в гомогенизаторе Даунса с использованием ручного гомогенизатора тканей (Wheaton, #357546). 148 мкл приготовленной мембраны затем вручную добавляли в каждую лунку 96-луночного полипропиленового планшета с глубокими лунками (Fisher Scientific, #12-566-121). После добавления мембраны в соответствующие лунки добавляли либо 2 мкл титрованного тестируемого соединения, контроля DMSO (Sigma #276855), либо холодного контроля cGAMP с помощью BIOMEK FX. Затем соединение и мембрану предварительно инкубировали в течение 60 минут при комнатной температуре, чтобы дать возможность связываемому соединению прийти в равновесие. После уравновешивания готовили 8 нМ лиганда [3Н] c-GAMP путем разбавления в буфере для анализа, а затем вручную добавляли 50 мкл этого рабочего раствора в каждую лунку планшета для анализа. Затем планшеты инкубировали при комнатной температуре в течение 60 минут, и затем содержимое каждого аналитического планшета фильтровали через 96-луночный фильтровальный планшет GF/B (PerkinElmer, #6005250) с использованием харвестера клеток TomTec Mach III, снабженного 20 мМ буфера HEPES (Fisher Scientific, #BP299500). Затем фильтровальные планшеты высушивали при 55°С в течение 30 минут, используя сушильный шкаф под давлением, после чего в каждую лунку добавляли 30 мкл сцинтиллята Ultima Gold F. Затем измеряли уровни трития для каждой реакционной лунки, используя планшет-ридер PerkinElmer TopCount. После нормализации до контролей рассчитывали процент активности для каждой концентрации соединения путем измерения количества оставшейся радиоактивности. График процентной активности в зависимости от логарифма концентрации соединения строили с помощью 4-параметрического уравнения доза - эффект для расчета значений ЕС50.
Конечные условия реакции представляли собой следующие:
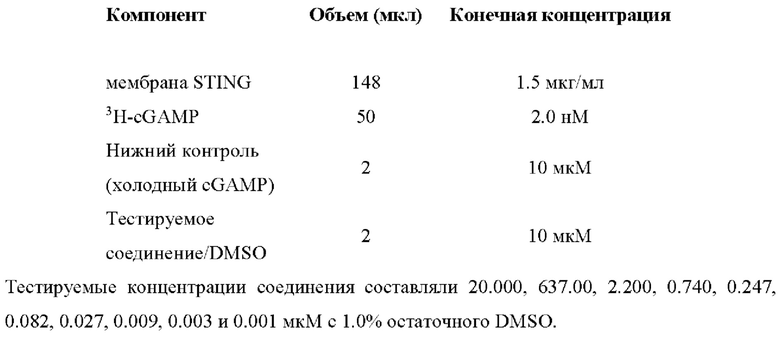
Генерация полноразмерного вируса STING (HAQ)
Вирус STING был создан с использованием бакуловирусной системы на основе клеток насекомых. Клетки Sƒ21 Spodoptera frugiperda (Kempbio, Inc.) разводили до 5е5 клеток/мл в среде Sf-900II SFM (Life Technologies #10902088) без антибиотиков. Суспензию клеток добавляли в каждую лунку обработанного 6-луночного планшета (2 мл на лунку, всего 1е6 клеток), и клетки оставляли прикрепляться в течение, по меньшей мере, 30 минут. В это время 1 мл смеси для котрансфекции собирали путем соединения 500 нг ДНК HAQ STING [STING(l-379)R71H,G230A,H232R,R293Q-GG-AviTag-GS-HRV3C-HIS8/pBAC1] (заказной синтез Genewiz) с 1 мл среды Sf-900II SFM, содержащей 10 мкл реагента Cellfectin® II (Invitrogen #10362100) и 100 нг вирусного остова BestBac 2.0, линеаризованная ДНК бакуловируса с удаленными v-cath/chiA (Expression Systems #91-002). Смеси для трансфекции инкубировали в течение 30 мин. После инкубации осторожно удаляли среду из прикрепившихся клеток в 6-луночном планшете, добавляли 1 мл трансфекционных смесей (1 мл на лунку), и планшет помещали в увлажненный инкубатор при 27°С. На следующий день в каждую лунку 6-луночного планшета добавляли 1 мл среды Sf-900II SFM (без антибиотиков). После добавления среды клетки оставляли инкубироваться с ДНК (SEQ. ID. No. 2) при 27°С в течение 5-7 дней, чтобы получить запас вируса Р0. Для создания запасов вируса P1 0.5 мл супернатанта вируса Р0 добавляли к 50 мл неинфицированных клеток Sƒ21 (высеянных за день до инфицирования с плотностью 5×105 клеток/мл, чтобы обеспечить удвоение в течение одной ночи) в среде Sf-900II SFM, содержащей 5 мкг/мл гентамицина (Invitrogen #15710072). Затем инфицированные клетки инкубировали при 27°С в течение 3 дней, встряхивая при 110 об/мин (ATR Biotech Multitron Infors HT #AJ118). На 3 день подсчитывали культуры P1 с использованием ViCell XR (Beckman Coulter Life Sciences #383556), чтобы подтвердить, что произошло инфицирование (размер клеток на ≥3 мкм больше, чем у неинфицированных клеток, и жизнеспособность приблизительно 85-95%). Культуры собирали в конические пробирки на 50 мл и центрифугировали при 2000xg в течение 10 минут при 4°С. Супернатанты вируса Р1 сливали в чистые центрифужные пробирки объемом 50 мл, и оставшиеся клеточные осадки Р1 использовали для получения клеток насекомых, инфицированных бакуловирусом (BIIC). Среда для криоконсервации, содержащая среду Sf-900II SFM с 10% инактивированной нагреванием FBS, 10% DMSO (Sigma #D2650) и 5 мкг/мл гентамицина, была приготовлена и стерилизована через фильтр 0.22 мкМ непосредственно перед использованием. Клеточные осадки Р1 ресуспендировали до плотности 2е7 клеток/мл и аликвотировали в криопробирки (1 мл на пробирку). Криопробирки помещали в контейнеры для замораживания клеток  на ночь при -80°С и на следующий день переносили в жидкий азот для длительного хранения. Для создания запаса вируса Р2 0.5 мл супернатанта вируса Р1 добавляли к 50 мл неинфицированных клеток Sƒ21 (высеянных за день до инфицирования с плотностью 5×105 клеток/мл, чтобы обеспечить удвоение в течение одной ночи) в среде Sf-900II SFM, содержащей 5 мкг/мл гентамицина. Эти клетки инкубировали при 27°С в течение 3 дней при встряхивании при 110 об/мин перед сбором запаса Р2 с центрифугированием при 2000xg в течение 10 минут при 4°С. Вирусные супернатанты Р2 сливали и отбрасывали, тогда как клеточные осадки Р2 использовали для генерации Р2 BIIC по тому же протоколу, который описан выше. Протокол генерации бакуловирусов был валидирован для последовательного получения BIIC Р1/Р2 с титрами 2е9 БОЕ/мл (2е7 клеток/мл × 100 БОЕ/клетку).
на ночь при -80°С и на следующий день переносили в жидкий азот для длительного хранения. Для создания запаса вируса Р2 0.5 мл супернатанта вируса Р1 добавляли к 50 мл неинфицированных клеток Sƒ21 (высеянных за день до инфицирования с плотностью 5×105 клеток/мл, чтобы обеспечить удвоение в течение одной ночи) в среде Sf-900II SFM, содержащей 5 мкг/мл гентамицина. Эти клетки инкубировали при 27°С в течение 3 дней при встряхивании при 110 об/мин перед сбором запаса Р2 с центрифугированием при 2000xg в течение 10 минут при 4°С. Вирусные супернатанты Р2 сливали и отбрасывали, тогда как клеточные осадки Р2 использовали для генерации Р2 BIIC по тому же протоколу, который описан выше. Протокол генерации бакуловирусов был валидирован для последовательного получения BIIC Р1/Р2 с титрами 2е9 БОЕ/мл (2е7 клеток/мл × 100 БОЕ/клетку).
Экспрессия полноразмерного STING (HAQ)
Для образования мембран STING, BIIC Р1/Р2 амплифицировали в течение ночи путем добавления размороженных BIIC к клеткам Sƒ21, высеянным с плотностью 1.0×106 клеток/мл. Объем BIIC, использованный для заражения культуры, рассчитывали с использованием предполагаемого титра BIIC 2е9 БОЕ/мл для достижения MOI 10 при амплификации в течение ночи. После культивирования в течение ночи клетки подсчитывали на ViCell XR, чтобы подтвердить, что произошло инфицирование (размер клеток на ≥3 мкм больше; чем у неинфицированных клеток, и жизнеспособность примерно 80-90%). Объем инфицированных клеток Sƒ21 после амплификации на протяжении ночи, использованный для инфицирования крупномасштабной экспрессии Trichoplusia ni (T.ni; Expression Systems, номер по каталогу 94-002F, www.expressionsystems.com), высеянных с плотностью 1.0×106 в среде для культивирования клеток (ESF921 SFM, содержащей 5 мкг/мл гентамицина) при MOI - 2.0, рассчитывали на основе (100 БОЕ/инфицированная клетка Sƒ21). Клетки оставляли экспрессироваться в течение 48 часов при 27°С перед сбором клеточного осадка путем центрифугирования при 3,400xg в течение 10 минут при 4°С. Клетки Т. ni подсчитывали на ViCell XR для подтверждения наличия инфекции (размер клеток на ≥3 мкм больше, чем неинфицированных клеток, и жизнеспособность приблизительно 80-90%) до сбора клеток.
Генерация полноразмерных мембран STING (HAQ)
Исходные буферные реагенты:
1) 1 М HEPES рН 7.5, Teknova, номер по каталогу Н1035;
2) 5 М NaCl, Sigma Aldrich, номер по каталогу S5150-1L;
3) KCl, Sigma Aldrich, номер по каталогу 319309-500ML;
4) Таблетки ингибитора протеаз cOmplete без EDTA, Roche Diagnostics, номер по каталогу 11873580001;
5) Бензоназа, универсальная нуклеаза. Pierce, номер по каталогу 88702.
Буфер для лизиса [25 мМ HEPES рН 7.5, 10 мМ MgCl2, 20 мМ KCl, (бензоназа 1:5000, таблетка ингибитора протеаз cOmplete/50 мл)] добавляли к осадку клеток, экспрессирующих полноразмерный STING (HAQ), полученный выше, в 5 мл буфера для лизиса на грамм клеточного осадка. Осадок ресуспендировали и гомогенизировали двадцать раз с использованием гомогенизатора Wheaton Dounce для разрушения клеточной мембраны. Затем гомогенизированный лизат пропускали через микрофлюидизатор EMULSIFLEX-C5 при давлении, близком к 5000 PSI. Ресуспендированный осадок центрифугировали при 36,000 об/мин (100,000xg) в сверхвысокоскоростной центрифуге с ротором 45Ti в течение 45 минут при 4°С. Супернатант удаляли. Затем осадок ресуспендировали в промывочном буфере [(25 мМ HEPES рН 7.5, 1 мМ MgCl2, 20 мМ KCl, 1 М NaCl (таблетка ингибитора протеаз cOmplete/50 мл)] в объеме 50 мл осадка/центрифужную пробирку. Смесь осадка/промывочного буфера затем гомогенизировали, используя стеклянный гомогенизатор на льду (20 ходов), с последующим центрифугированием при 36,000 об/мин в течение 45 минут при 4°С. Супернатант удаляли. Стадию промывки повторяли еще раз. Полученную мембрану ресуспендировали в 20 мМ HEPES рН 7.5, 500 мМ NaCl, 10% глицерина, ингибиторах протеаз, не содержащих EDTA (1 таблетка/50 мл). Концентрацию белка измеряли с помощью анализа Брэдфорда (Bio-Rad Protein Assay, номер по каталогу 500-0006), и обогащение белками определяли с помощью SDS-PAGE и подтверждали анализом Вестерн-блоттинг. Ресуспендированные мембраны хранили при -80°С.
Аминокислотная последовательность полноразмерного HAQ STING [STING(1-379)R71H,G230A,H232R,R293Q-GG-AviTag-GS-HRV3C-HIS8]:

Последовательность плазмидной ДНК полноразмерного HAQ [STING(1-379)R71H,G230A,H232R,R293Q-GG-AviTag-GS-HRV3C-HIS8/pBAC1]:




Некоторые соединения по настоящему описанию оценивали в анализе связывания HAQ STING in vitro, как описано выше. В следующей таблице приведены биологические данные для этих соединений в виде значений ЕС50.
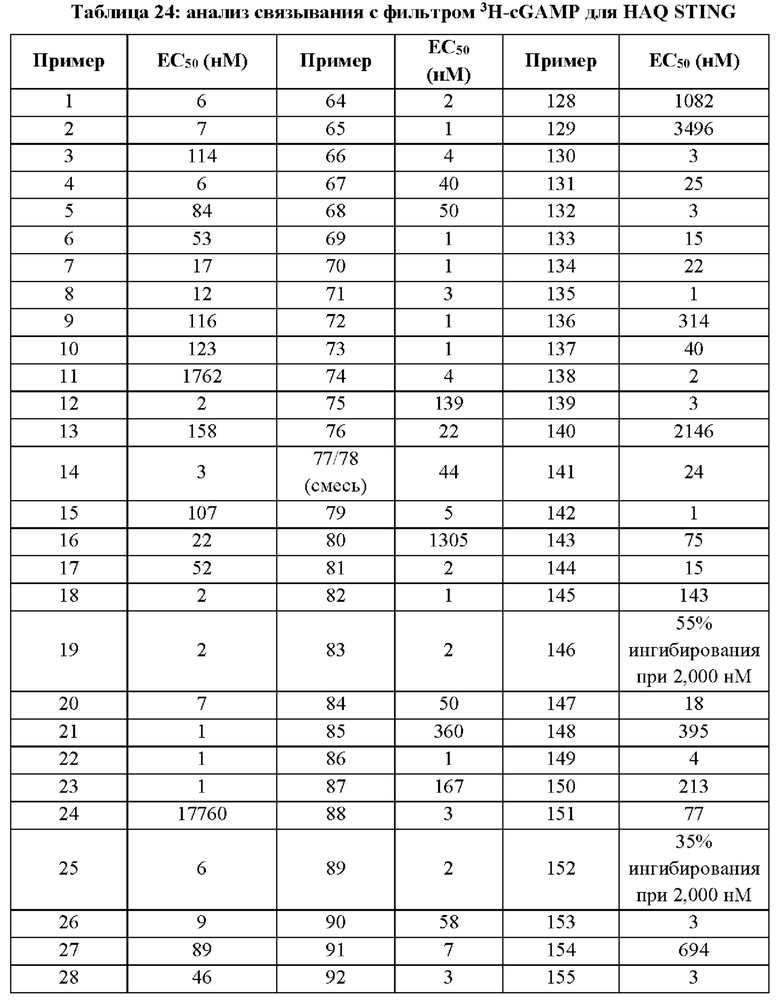
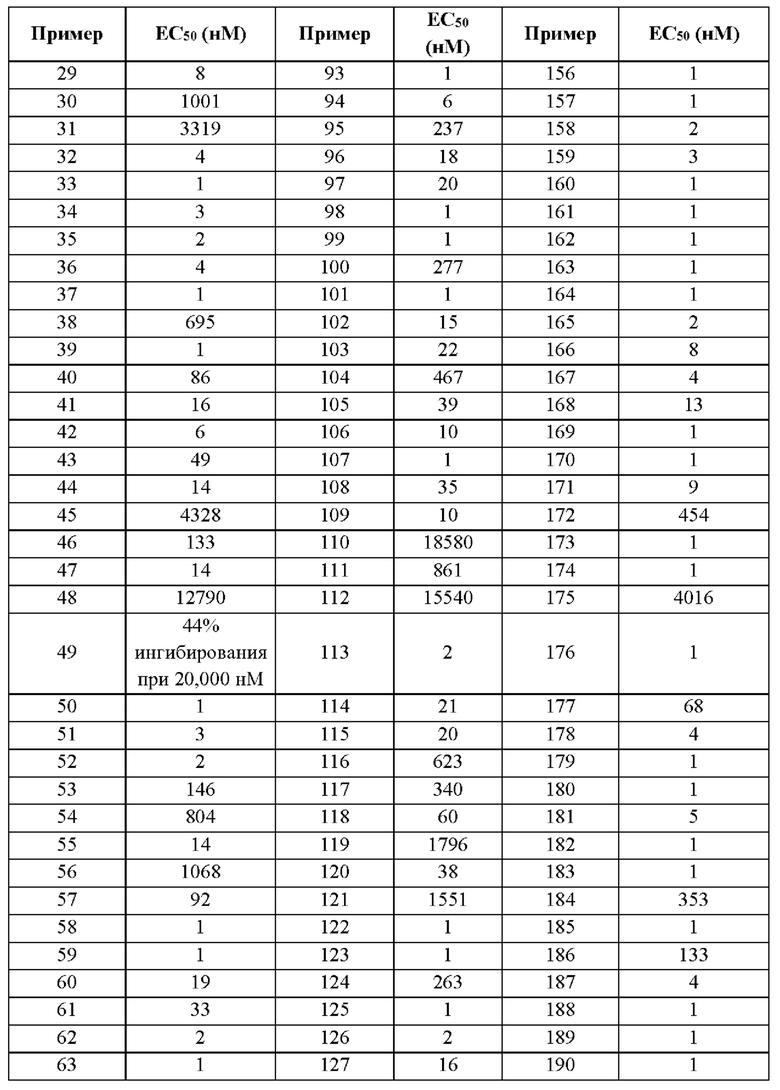
Анализ связывания с фильтром 3H-cGAMP (WT STING)
Способность соединений связывать STING количественно оценивается по их способности конкурировать с меченным тритием лигандом cGAMP за мембрану человеческого рецептора STING с использованием радиоактивного анализа связывания с фильтром. В анализе связывания используется рецептор STING, полученный из клеточных мембран Trichoplusia ni (T.ni; Expression Systems, номер по каталогу 94-002F, www.expressionsystems.com). сверхэкспрессирующий полноразмерный WT STING и меченный тритием лиганд cGAMP.
Базовый протокол анализа с фильтром WT STING представляет собой следующее: 16 нМ лиганда [3Н] c-GAMP получали разбавлением в буфере для анализа, и 50 мкл этого рабочего раствора добавляли вручную в каждую лунку планшета для анализа. После добавления лиганда в соответствующие лунки с помощью BIOMEK FX добавляли 2 мкл либо титрованного тестируемого соединения, контроля DMSO (Sigma #276855), либо холодного контроля cGAMP. Серийно титрованное соединение получали на приборе Hamilton STARPlus CORE в 96-луночном планшете (Greiner, #651201) с использованием 1:3 десятиточечного формата дозовой зависимости. После добавления соединения рабочая концентрация 2.2 мкг/мл мембраны STING (SEQ. ID. No. 3) была приготовлена путем разбавления концентрированной мембраны в буфере для анализа (1x PBS; Invitrogen #SH30028.02) и 7-х гомогенизации в гомогенизаторе Даунса с использованием ручного гомогенизатора тканей (Wheaton, #357546). 148 мкл этой приготовленной мембраны затем вручную добавляли в каждую лунку 96-луночного полипропиленового планшета с глубокими лунками (Fisher Scientific, #12-566-121). Затем соединение, лиганд и мембрану инкубировали в течение 60 минут при комнатной температуре перед тем, как содержимое каждого аналитического планшета фильтровали через 96-луночный фильтровальный планшет GF/B (PerkinElmer, #6005250) с использованием харвестера клеток TomTec Mach III, снабженного 20 мМ буфера HEPES (Fisher Scientific, #BP299500). Затем фильтровальные планшеты высушивали при 55°С в течение 30 минут, используя сушильный шкаф под давлением VWR, после чего в каждую лунку добавляли 30 мкл сцинтиллята Ultima Gold F. Затем измеряли уровни трития для каждой реакционной лунки, используя планшет-ридер PerkinElmer TopCount.
После нормализации до контролей рассчитывали процент активности для каждой концентрации соединения путем измерения количества оставшейся радиоактивности. График процентной активности в зависимости от логарифма концентрации соединения строили с помощью 4-параметрического уравнения доза - эффект для расчета значений ЕС50.
Конечные условия реакции представляли собой следующие:

Генерация полноразмерного вируса STING (WT)
Вирус STING был создан с использованием бакуловирусной системы на основе клеток насекомых. Клетки Sƒ21 Spodoptera frugiperda (Kempbio, Inc.) разводили до 5е5 клеток/мл в среде Sf-900II SFM (Life Technologies #10902088) без антибиотиков. Суспензию клеток добавляли в каждую лунку обработанного 6-луночного планшета (2 мл на лунку, всего 1е6 клеток), и клетки оставляли прикрепляться в течение, по меньшей мере, 30 минут. В это время 1 мл смеси для котрансфекции собирали путем соединения 500 нг WT STING[STING(1-379)H232R-gg-AviTag-gs-HRV3C-HIS8/pBAC1] (заказной синтез Genewiz) с 1 мл среды Sf-900II SFM, содержащей 10 мкл реагента Cellfectin® II (Invitrogen #10362100) и 100 нг вирусного остова BestBac 2.0, линеаризованная ДНК бакуловируса с удаленными v-cath/chiA (Expression Systems #91-002). Смеси для трансфекции инкубировали в течение 30 мин. После инкубации осторожно удаляли среду из прикрепившихся клеток в 6-луночном планшете, добавляли 1 мл трансфекционных смесей (1 мл на лунку), и планшет помещали в увлажненный инкубатор при 27°С. На следующий день в каждую лунку 6-луночного планшета добавляли 1 мл среды Sf-900II SFM (без антибиотиков). После добавления среды клетки оставляли инкубироваться с ДНК [(SEQ. ID. No. 4) и линеаризованный вирусный остов BestBac 2.0] при 27°С в течение 5-7 дней, чтобы получить запас вируса Р0. Для создания запасов вируса Р1 0.5 мл супернатанта вируса Р0 добавляли к 50 мл не инфицированных клеток Sƒ21 (высеянных за день до инфицирования с плотностью 5×105 клеток/мл, чтобы обеспечить удвоение в течение одной ночи) в среде Sf-900II SFM, содержащей 5 мкг/мл гентамицина (Invitrogen #15710072). Затем инфицированные клетки инкубировали при 27°С в течение 3 дней, встряхивая при 110 об/мин (ATR Biotech Multitron Infors HT #AJ118). На 3 день подсчитывали культуры Р1 с использованием ViCell XR (Beckman Coulter Life Sciences #383556), чтобы подтвердить, что произошло инфицирование (размер клеток на ≥3 мкм больше, чем у неинфицированных клеток, и жизнеспособность приблизительно 85-95%). Культуры собирали в конические пробирки на 50 мл и центрифугировали при 2000xg в течение 10 минут при 4°С. Супернатанты вируса Р1 сливали в чистые центрифужные пробирки объемом 50 мл, и оставшиеся клеточные осадки Р1 использовали для получения клеток насекомых, инфицированных бакуловирусом (BIIC). Среда для криоконсервации; содержащая среду Sf-900II SFM с 10% инактивированной нагреванием FBS; 10% DMSO (Sigma #D2650) и 5 мкг/мл гентамицина, была приготовлена и стерилизована через фильтр 0.22 мкМ непосредственно перед использованием. Клеточные осадки Р1 ресуспендировали до плотности 2е7 клеток/мл и аликвотировали в криопробирки (1 мл на пробирку). Криопробирки помещали в контейнеры для замораживания клеток на ночь при -80°С и на следующий день переносили в жидкий азот для длительного хранения. Для создания запаса вируса Р2 0.5 мл супернатанта вируса Р1 добавляли к 50 мл неинфицированных клеток Sƒ21 (высеянных за день до инфицирования с плотностью 5×105 клеток/мл, чтобы обеспечить удвоение в течение одной ночи) в среде Sf-900II SFM, содержащей 5 мкг/мл гентамицина. Эти клетки инкубировали при 27°С в течение 3 дней при встряхивании при 110 об/мин перед сбором запаса Р2 с центрифугированием при 2000xg в течение 10 минут при 4°С. Вирусные супернатанты Р2 сливали и отбрасывали, тогда как клеточные осадки Р2 использовали для генерации Р2 BIIC по тому же протоколу, который описан выше. Протокол генерации бакуловирусов был валидирован для последовательного получения BIIC Р1/Р2 с титрами 2е9 БОЕ/мл (2е7 клеток/мл × 100 БОЕ/клетку).
Экспрессия полноразмерного STING (WT)
Для образования мембран STING, BIIC Р1/Р2 амплифицировали в течение ночи путем добавления размороженных BIIC к клеткам Sƒ21, высеянным с плотностью 1.0×106 клеток/мл. Объем BIIC, использованный для заражения культуры, рассчитывали с использованием предполагаемого титра BIIC 2е9 БОЕ/мл для достижения MOI 10 при амплификации в течение ночи. После культивирования в течение ночи клетки подсчитывали на ViCell XR, чтобы подтвердить; что произошло инфицирование (размер клеток на ≥3 мкм больше; чем у неинфицированных клеток; и жизнеспособность примерно 80-90%). Объем инфицированных клеток Sƒ21 после амплификации на протяжении ночи, использованный для инфицирования крупномасштабной экспрессии Trichoplusia ni (T.ni; Expression Systems, номер по каталогу 94-002F, www.expressionsystems.com), высеянных с плотностью 1.0×106 в среде для культивирования клеток (ESF921 SFM, содержащей 5 мкг/мл гентамицина) при MOI=2.0, рассчитывали на основе (100 БОЕ/инфицированная клетка Sƒ21). Клетки оставляли экспрессироваться в течение 48 часов при 27°С перед сбором клеточного осадка путем центрифугирования при 3,400xg в течение 10 минут при 4°С. Клетки Т. ni подсчитывали на ViCell XR для подтверждения наличия инфекции (размер клеток на ≥3 мкм больше, чем неинфицированных клеток, и жизнеспособность приблизительно 80-90%) до сбора клеток.
Генерация полноразмерной мембраны STING (WT)
Исходные буферные реагенты:
1) 1 М HEPES рН 7.5, Teknova, номер по каталогу Н1035;
2) 5 М NaCl, Sigma Aldrich, номер по каталогу S5150-1L;
3) KCl, Sigma Aldrich, номер по каталогу 319309-500ML;
4) Таблетки ингибитора протеаз cOmplete без EDTA, Roche Diagnostics, номер по каталогу 11873580001;
5) Бензоназа, универсальная нуклеаза. Pierce, номер по каталогу 88702.
Буфер для лизиса [25 мМ HEPES рН 7.5, 10 мМ MgCl2, 20 мМ KCl, (бензоназа 1:5000, таблетка ингибитора протеаз cOmplete/50 мл)] добавляли к осадку клеток, экспрессирующих полноразмерный STING (WT), полученный выше, в 5 мл буфера для лизиса на грамм клеточного осадка. Осадок ресуспендировали и гомогенизировали двадцать раз с использованием гомогенизатора Wheaton Dounce для разрушения клеточной мембраны. Затем гомогенизированный лизат пропускали через микрофлюидизатор Emulsiflex-C5 при давлении, близком к 5000 PSI. Ресуспендированный осадок центрифугировали при 36,000 об/мин (100.000xg) в сверхвысокоскоростной центрифуге с ротором 45Ti в течение 45 минут при 4°С. Супернатант удаляли. Затем осадок ресуспендировали в промывочном буфере [(25 мМ HEPES рН 7.5, 1 мМ MgCl2, 20 мМ KCl, 1 М NaCl (таблетка ингибитора протеаз cOmplete/50 мл)] в объеме 50 мл осадка/центрифужную пробирку. Смесь осадка/промывочного буфера затем гомогенизировали, используя стеклянный гомогенизатор на льду (20 ходов), с последующим центрифугированием при 36,000 об/мин в течение 45 минут при 4°С. Супернатант удаляли. Стадию промывки повторяли еще раз. Полученную мембрану ресуспендировали в 20 мМ HEPES рН 7.5, 500 мМ NaCl, 10% глицерина, ингибиторах протеаз, не содержащих EDTA (1 таблетка/50 мл). Концентрацию белка измеряли с помощью анализа Брэдфорда (Bio-Rad Protein Assay, номер по каталогу 500-0006), и обогащение белками определяли с помощью SDS-PAGE и подтверждали анализом Вестерн-блоттинг. Ресуспендированные мембраны хранили при -80°С.
Аминокислотная последовательность полноразмерного STING WT[STING(1-379)H232R-gg-AviTag-gs-HRV3C-HIS8]:

Последовательность плазмидной ДНК полноразмерного WT STING [STING(1-379)H232R-gg-AviTag-gs-HRV3C-HIS8/pBAC1]:




Некоторые соединения по настоящему описанию оценивали в анализе связывания WT STING in vitro, как описано выше. В следующей таблице приведены биологические данные для этих соединений в виде значений ЕС50.
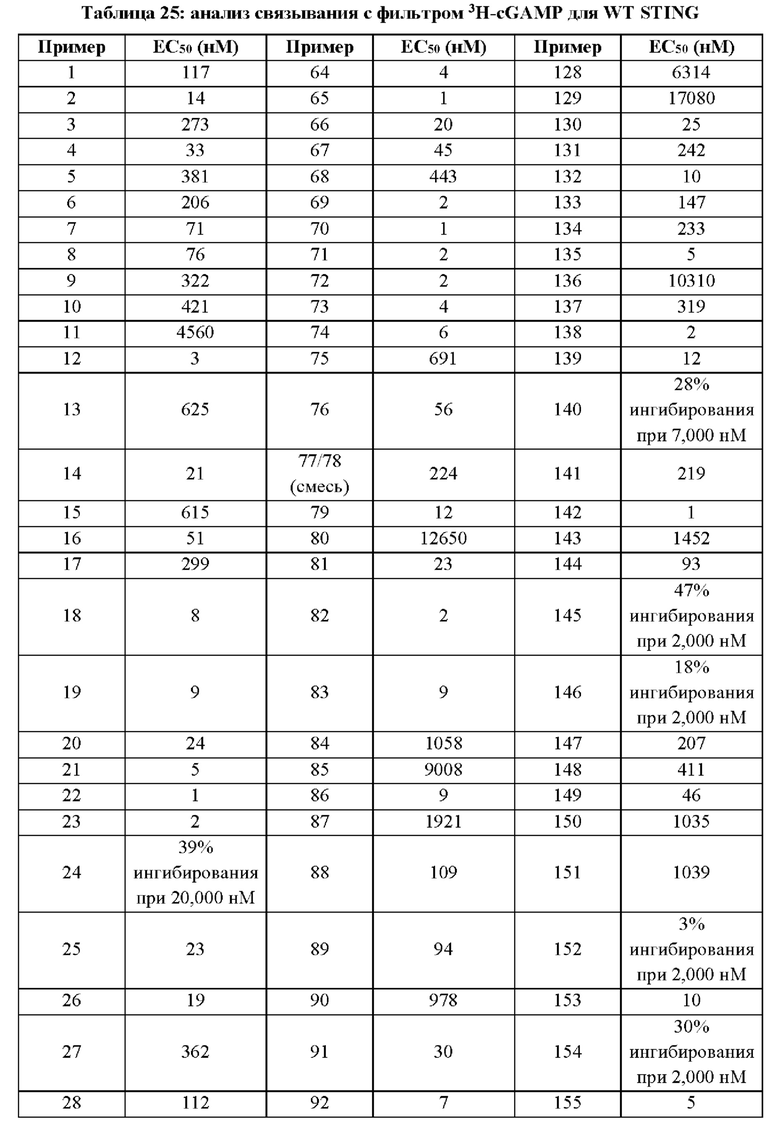
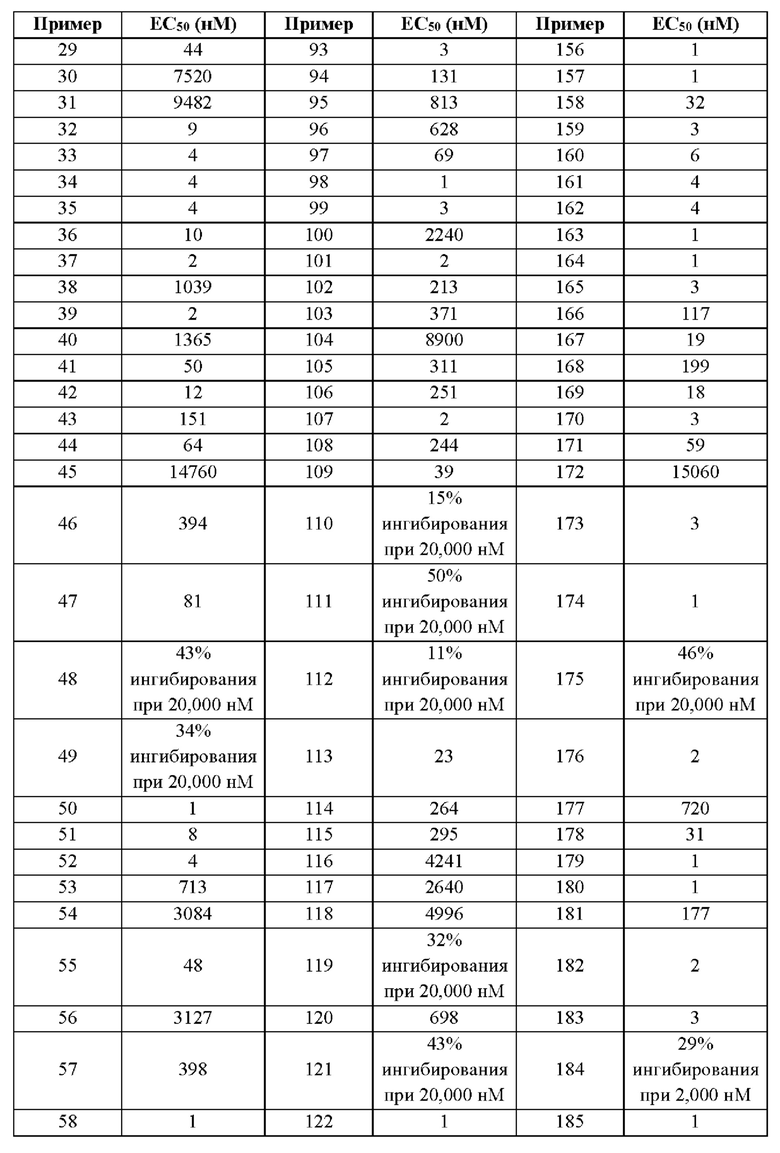
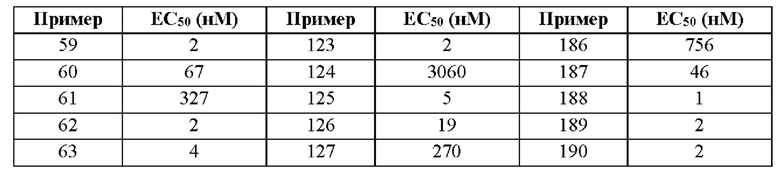
Секреция IFN-β в культуре клеток ТНР1 (5 ч)
Способность соединений стимулировать секрецию интерферона-бета из клеток ТНР1 измеряли с использованием набора IFN-β AlphaLISA человека (Perkin Elmer, номер по каталогу AL265F). Базовый протокол представляет собой следующее: Акустический дозатор Labcyte Echo 550 использовали для переноса 120 нл соединения, растворенного в DMSO, в лунки пустого стерильного 384-луночного микропланшета (Corning, номер по каталогу 3712). Клетки ТНР1 (American Type Culture Collection (Американская коллекция типовых культур), номер по каталогу TIB202), предварительно замороженные в среде для восстановления (Life Technologies, номер по каталогу 12648-010), размораживали и сразу же разводили 10-кратно в среде для анализа при 37°С (RPMI 1640+L-глутамин & феноловый красный, Life Technologies, номер по каталогу 11875-085; 0.5% инактивированной нагреванием фетальной бычьей сыворотки, Sigma Aldrich, номер по каталогу F4135; 1 мМ пирувата натрия, Life Technologies, номер по каталогу 11360-070; 1х неотносящихся к незаменимым аминокислот; Life Technologies, номер по каталогу 11140-050). Жизнеспособность и количество клеток определяли с использованием счетчика клеток Beckman Coulter V-Cell XR. Суспензию клеток центрифугировали при 200×g в течение 5 мин при комнатной температуре. Клетки ресуспендировали до плотности 0.8×106 /мл в аналитической среде при 37°С. Последующие переносы жидкости выполнялись либо с помощью электронной многоканальной пипетки Matrix, либо с помощью автоматизированной платформы для работы с жидкостями Agilent Bravo. Анализ начинали с внесения 40 мкл предварительно приготовленной клеточной суспензии в лунки планшета, содержащего соединения. После 5 часов инкубации при 37°С и 5% СО2 в увлажненной атмосфере планшет с клетками и соединениями центрифугировали при 200×g в течение 5 минут при комнатной температуре. Из каждой лунки 5 мкл супернатанта переносили в соответствующие лунки белого 384-луночного планшета (Perkin Elmer, номер по каталогу №6005620). В эти лунки, содержащие супернатант, добавляли 10 мкл 5× акцепторных гранул с анти-аналитом (50 мкг/мл буфера AlphaLISA HiBlock) и инкубировали в течение 30 минут при комнатной температуре, встряхивая на орбитальном шейкере для планшетов. В каждую лунку добавляли 10 мкл 5× биотинилированного антитела против анализируемого вещества (5 нМ в буфере AlphaLISA HiBlock) и инкубировали на орбитальном шейкере для планшетов в течение 60 минут при комнатной температуре или на протяжении ночи при 4°С. В каждую лунку добавляли 25 мкл 2× донорных гранул SA (80 мкг/мл в буфере AlphaLISA HiBlock) и инкубировали в течение 30-45 мин при комнатной температуре в темноте, встряхивая на орбитальном шейкере для планшетов. Затем планшет считывали на Perkin Elmer Envision (λех=680 нм, λem=570 нм). Эффект в процентах сигнала AlphaLISA при каждой концентрации соединения рассчитывали на основе положительных контролей 30 мкМ cGAMP и отрицательных контролей 0.3% DMSO. График зависимости эффекта в процентах от логарифма концентрации соединения соответствовал 4-параметрическому уравнению концентрация-эффект для расчета значений ЕС50. Исследуемые соединения тестировали при концентрациях 30000, 10000, 3333, 1111, 370.4, 123.4, 41.2, 13.7, 4.6 и 1.5 нМ с 0.3% остаточного DMSO. Контрольное соединение, cGAMP, тестировали при концентрациях 100000, 33333, 11111, 3704, 1235, 412, 137, 46 и 15 нМ с 0.3% остаточного DMSO. Соединения по настоящему описанию оценивали на секрецию IFN-β в культуре клеток ТНР1, как описано выше. В следующей таблице представлены биологические данные для этих соединений в виде процента активации по отношению к 2'3'-cGAMP при концентрации 30 мкМ.
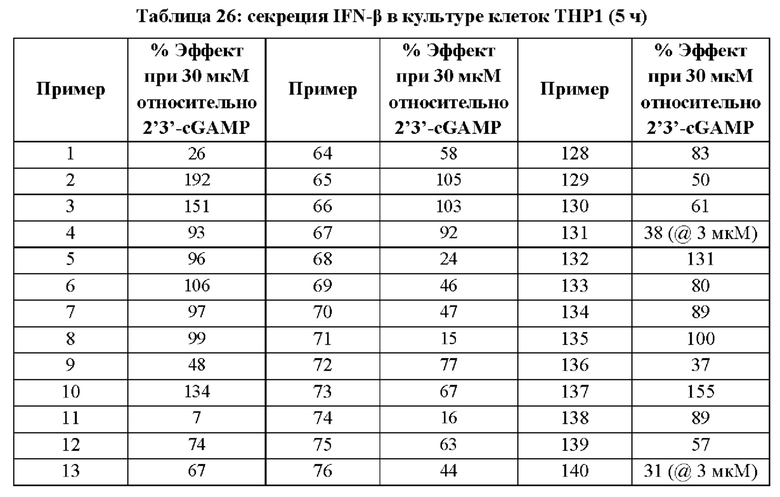
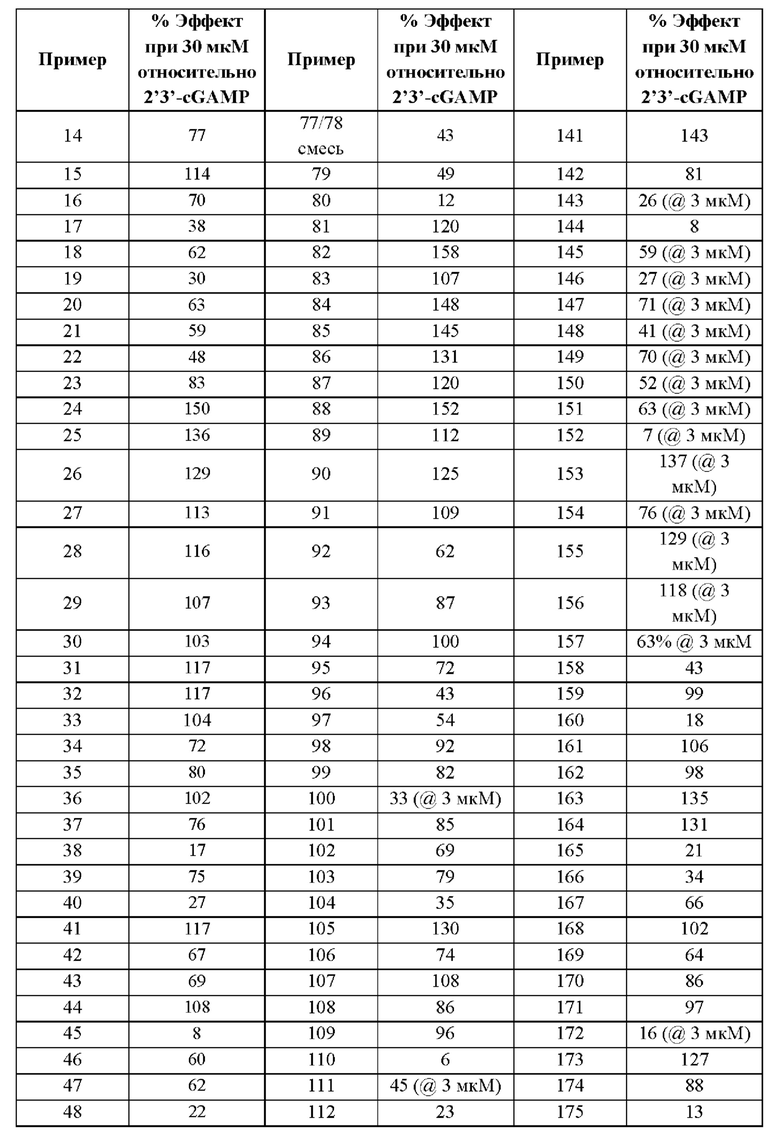
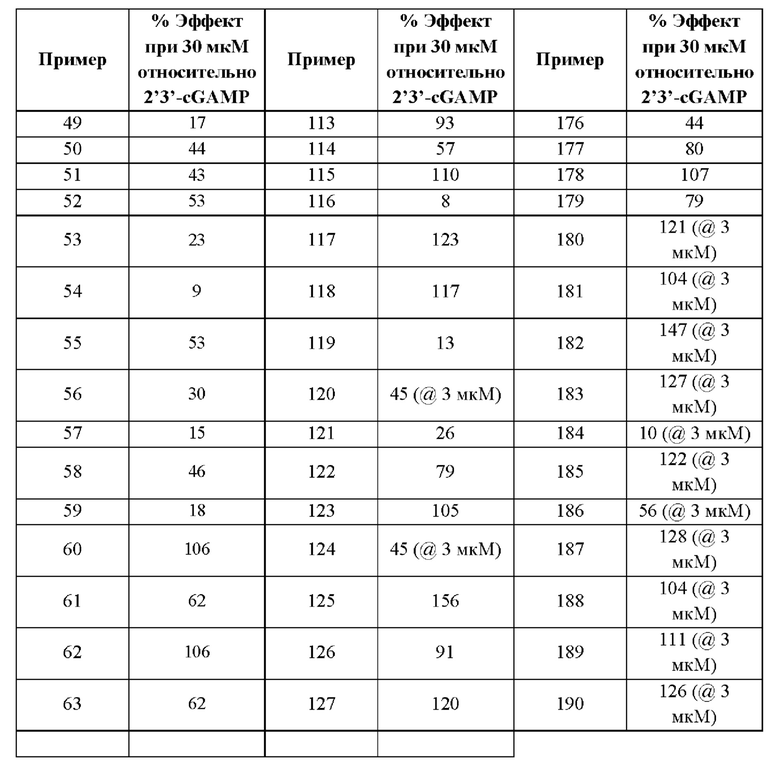
Следует понимать, что целый ряд из рассмотренных выше и других отличительных признаков и функций или их альтернативы желательно могут быть объединены во многие другие системы или заявки. Также будет понятно, что целый ряд в настоящее время непредвиденных или непредусмотренных альтернатив, модификаций, вариаций или улучшений в них может быть впоследствии сделан специалистами в данной области техники, и также предполагается, что они охватываются следующей формулой изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЗАМЕЩЕННЫЕ БЕНЗОИЛАМИНОИНДАН-2-КАРБОНОВЫЕ КИСЛОТЫ И РОДСТВЕННЫЕ СОЕДИНЕНИЯ | 2008 |
|
RU2477279C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ | 2015 |
|
RU2711502C2 |
| 6, 6-БИЦИКЛИЧЕСКИЕ КОЛЬЦЕВЫЕ ЗАМЕЩЕННЫЕ ГЕТЕРОБИЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ ПРОТЕИНКИНАЗ | 2005 |
|
RU2379308C2 |
| СОЛИ ТИАЗОЛИДИНДИОНА СО СНИЖЕННЫМ СРОДСТВОМ К PPAR ДЛЯ ЛЕЧЕНИЯ НАРУШЕНИЙ ОБМЕНА ВЕЩЕСТВ | 2010 |
|
RU2564661C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ | 2015 |
|
RU2734390C2 |
| ЗАМЕЩЕННЫЕ ИМИДАЗОПИРИДИНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ HDM2 | 2013 |
|
RU2690663C2 |
| ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛА, ПОЛЕЗНЫЕ ПРИ ЛЕЧЕНИИ РЕСПИРАТОРНО-СИНЦИТИАЛЬНОЙ ВИРУСНОЙ ИНФЕКЦИИ | 2012 |
|
RU2612530C2 |
| БИЦИКЛИЧЕСКИЕ АГОНИСТЫ СТИМУЛЯТОРА ГЕНОВ ИНТЕРФЕРОНА STING | 2020 |
|
RU2800072C1 |
| ПРОИЗВОДНЫЕ КОНДЕНСИРОВАННОГО 1,4-ДИГИДРОДИОКСИНА В КАЧЕСТВЕ ИНГИБИТОРОВ ФАКТОРА ТРАНСКРИПЦИИ 1 БЕЛКОВ ТЕПЛОВОГО ШОКА | 2014 |
|
RU2671979C2 |
| СОЕДИНЕНИЕ НА ОСНОВЕ ДИГИДРОНАФТИРИДИНОНА, И СПОСОБ ЕГО ПОЛУЧЕНИЯ, И ЕГО ПРИМЕНЕНИЕ В МЕДИЦИНЕ | 2021 |
|
RU2809869C1 |
Изобретение относится к соединениям общей формулы (I), общей формулы (II), общей формулы (III), общей формулы (IV), общей формулы (V), общей формулы (VI) и их фармацевтически приемлемым солям, где все переменные определены в настоящем документе, которые могут быть полезны в качестве индукторов продукции интерферона I типа, конкретно в качестве активных агентов STING. Также предложены фармацевтическая композиция, содержащая такие соединения, и способы применения таких соединений, включая введение таких соединений для индукции иммунного ответа, индукции STING-зависимой продукции интерферона I типа и/или лечения нарушения пролиферации клеток, такого как рак. 17 н. и 16 з.п. ф-лы, 26 табл., 190 пр.
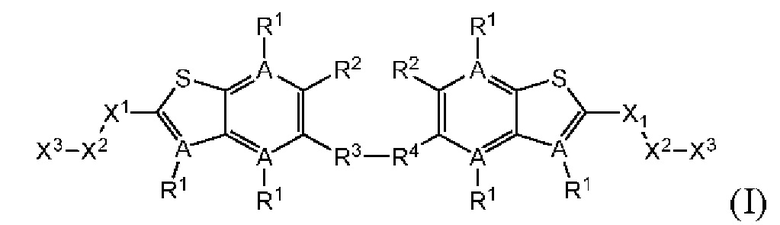
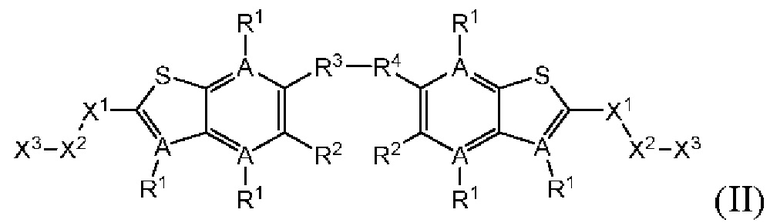
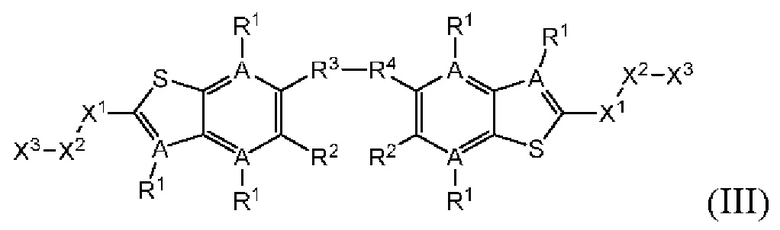
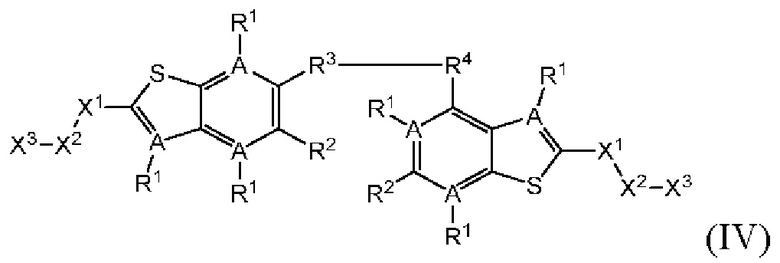
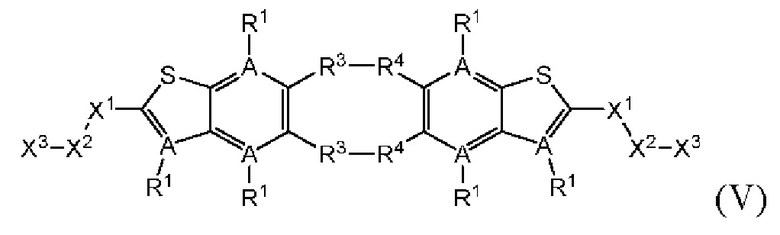
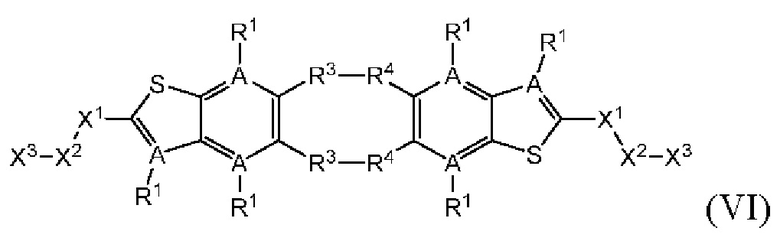
1. Соединение общей формулы (I):
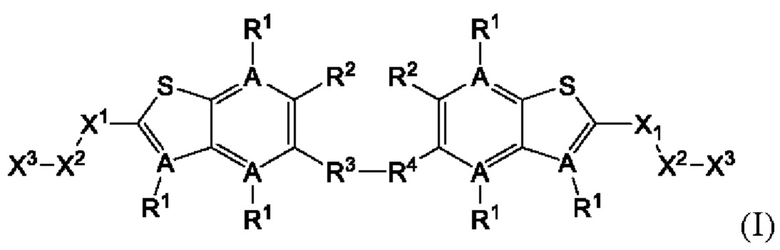
или его фармацевтически приемлемая соль, где
каждый 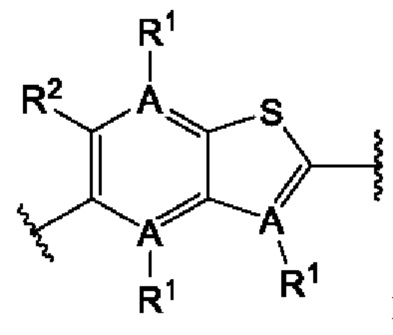 независимо выбран из группы, состоящей из
независимо выбран из группы, состоящей из
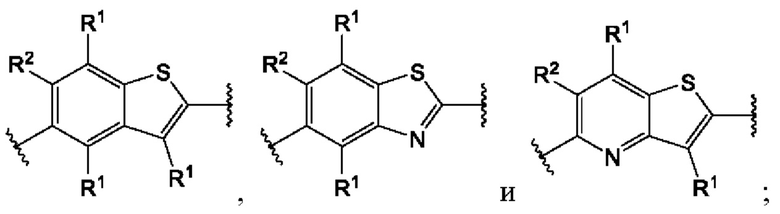
где каждый R1 независимо выбран из группы, состоящей из Н и галогена; каждый R2 независимо выбран из группы, состоящей из Н, OR6, N(R6)2, и C1-C6 алкила;
R3 и R4 независимо выбраны из группы, состоящей из O-(C1-C4 алкилен), С1-С5 алкилена, и N(R6)-(C1-C4 алкилен);
каждый R6 независимо выбран из группы, состоящей из Н, C1-C6 алкила и C1-C6 галоалкила;
каждый X1 представляет собой С=O;
каждый X2 независимо выбран из (C(R8)2)(1-3), где каждый R8 независимо выбран из группы, состоящей из Н и C1-C6 алкила;
необязательно, 2 R8 на разных атомах углерода могут быть взяты вместе совместно с атомами, к которым они присоединены, с образованием от 3- до 6-членного конденсированного кольца; и
каждый X3 независимо выбран из группы, состоящей из COOR6, C(O)N(R9)2 и CN; и
каждый R9 независимо выбран из группы, состоящей из Н и SO2R6.
2. Соединение общей формулы (II):
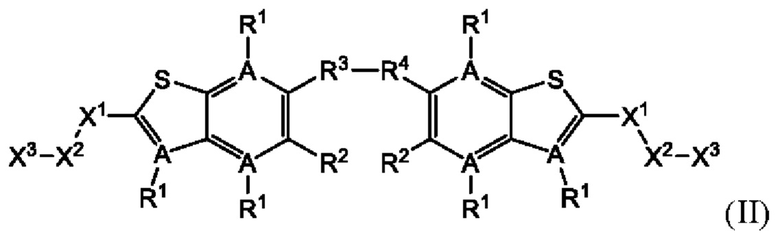
или его фармацевтически приемлемая соль, где
каждый 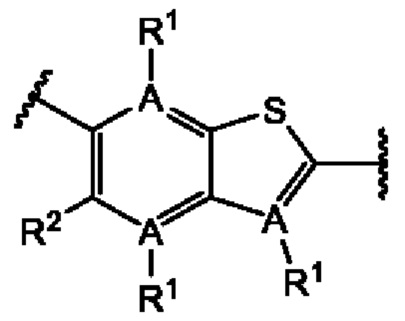 независимо выбран из группы, состоящей из
независимо выбран из группы, состоящей из
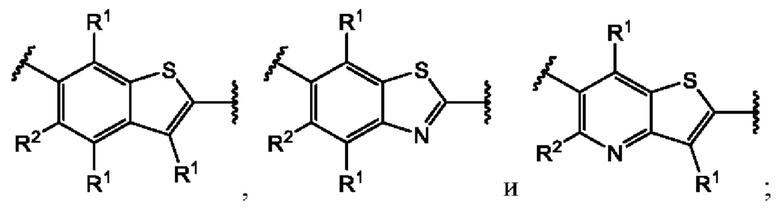
где каждый R1 независимо выбран из группы, состоящей из Н и галогена;
каждый R2 независимо выбран из группы, состоящей из Н, OR6, N(R6)2, и C1-C6 алкила;
R3 и R4 независимо выбраны из группы, состоящей из O-(C1-C4 алкилен), С1-С5 алкилена, и N(R6)-(C1-C4 алкилен);
каждый R6 независимо выбран из группы, состоящей из Н, C1-C6 алкила и C1-C6 галоалкила;
каждый X1 представляет собой С=O;
каждый X2 независимо выбран из (C(R8)2)(1-3), где каждый R8 независимо выбран из группы, состоящей из Н и C1-C6 алкила;
необязательно, 2 R8 на разных атомах углерода могут быть взяты вместе совместно с атомами, к которым они присоединены, с образованием от 3- до 6-членного конденсированного кольца; и
каждый X3 независимо выбран из группы, состоящей из COOR6, C(O)N(R9)2 и CN; и
каждый R9 независимо выбран из группы, состоящей из Н и SO2R6.
3. Соединение общей формулы (III):
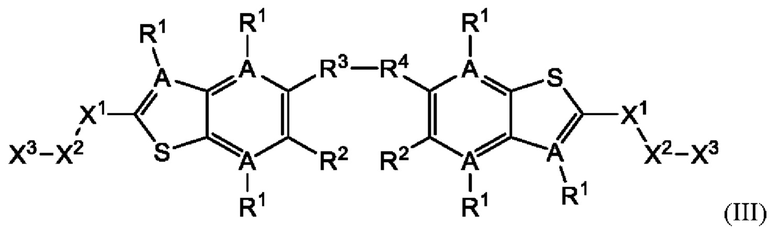
или его фармацевтически приемлемая соль, где
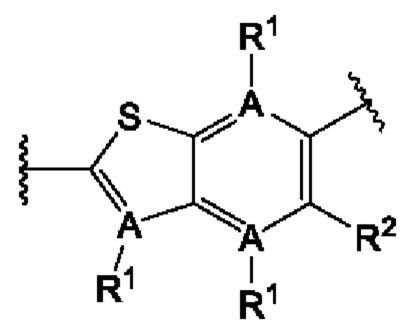 независимо выбран из группы, состоящей из
независимо выбран из группы, состоящей из 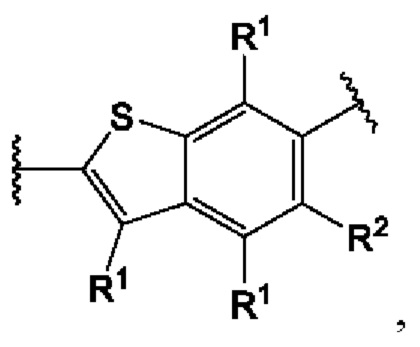
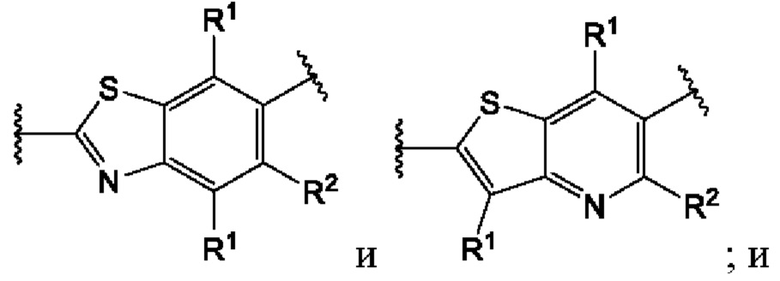
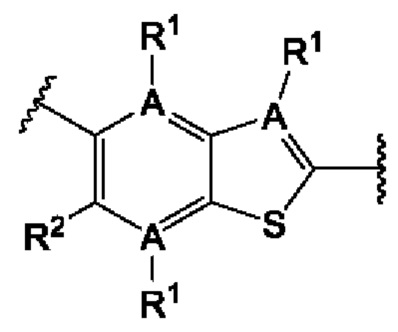 выбран из группы, состоящей из
выбран из группы, состоящей из 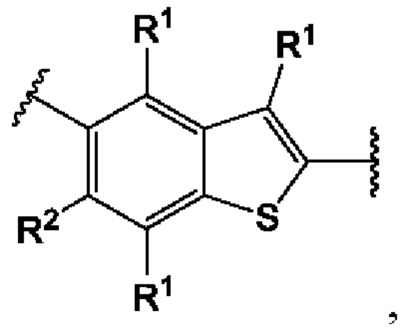
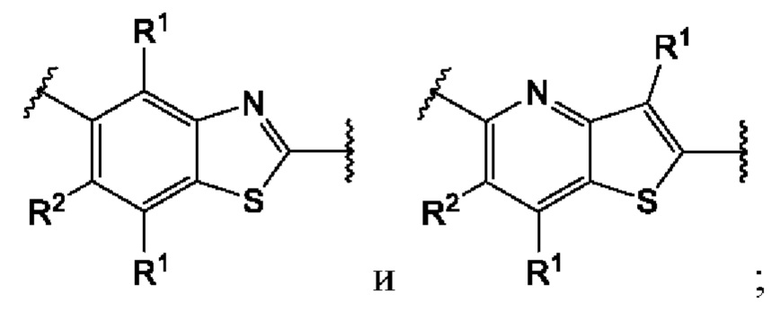
где каждый R1 независимо выбран из группы, состоящей из Н и галогена; каждый R2 независимо выбран из группы, состоящей из Н, OR6, N(R6)2, и C1-C6 алкила;
R3 и R4 независимо выбраны из группы, состоящей из O-(C1-C4 алкилен), С1-С5 алкилена, и N(R6)-(C1-C4 алкилен);
каждый R6 независимо выбран из группы, состоящей из Н, С1-С6 алкила и С1-С6 галоалкила;
каждый X1 представляет собой С=O;
каждый X2 независимо выбран из (C(R8)2)(1-3), где каждый R8 независимо выбран из группы, состоящей из Н и C1-C6 алкила;
необязательно, 2 R8 на разных атомах углерода могут быть взяты вместе совместно с атомами, к которым они присоединены, с образованием от 3- до 6-членного конденсированного кольца; и
каждый X3 независимо выбран из группы, состоящей из COOR6, C(O)N(R9)2 и CN; и
каждый R9 независимо выбран из группы, состоящей из Н и SO2R6.
4. Соединение общей формулы (IV):
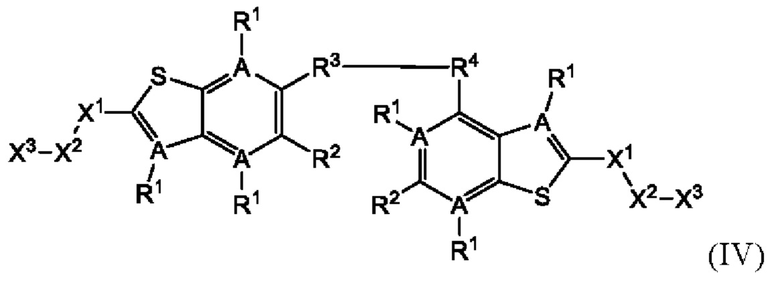
или его фармацевтически приемлемая соль, где
 независимо выбран из группы, состоящей из
независимо выбран из группы, состоящей из 
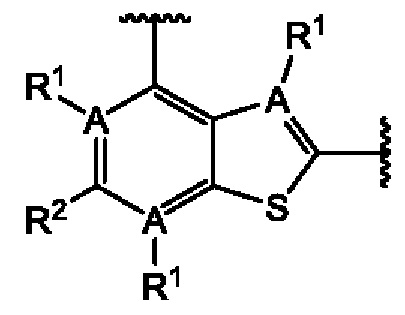 выбран из группы, состоящей из
выбран из группы, состоящей из 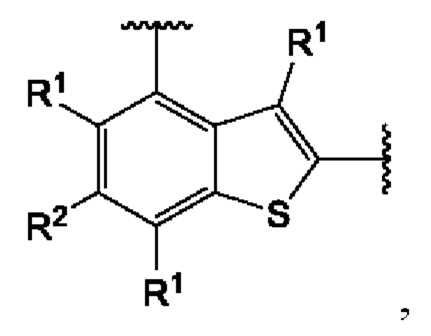
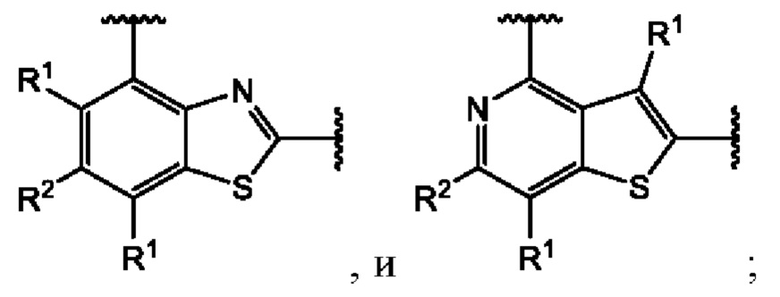
где каждый R1 независимо выбран из группы, состоящей из Н и галогена;
каждый R2 независимо выбран из группы, состоящей из Н, OR6, N(R6)2, и C1-C6 алкила;
R3 и R4 независимо выбраны из группы, состоящей из O-(C1-C4 алкилен), С1-С5 алкилена, и N(R6)-(C1-C4 алкилен);
каждый R6 независимо выбран из группы, состоящей из Н, C1-C6 алкила и C1-C6 галоалкила;
каждый X1 представляет собой С=O;
каждый X2 независимо выбран из (C(R8)2)(1-3), где каждый R8 независимо выбран из группы, состоящей из Н и C1-C6 алкила;
необязательно, 2 R8 на разных атомах углерода могут быть взяты вместе совместно с атомами, к которым они присоединены, с образованием от 3- до 6-членного конденсированного кольца; и
каждый X3 независимо выбран из группы, состоящей из COOR6, C(O)N(R9)2 и CN; и
каждый R9 независимо выбран из группы, состоящей из Н и SO2R6.
5. Соединение общей формулы (V):
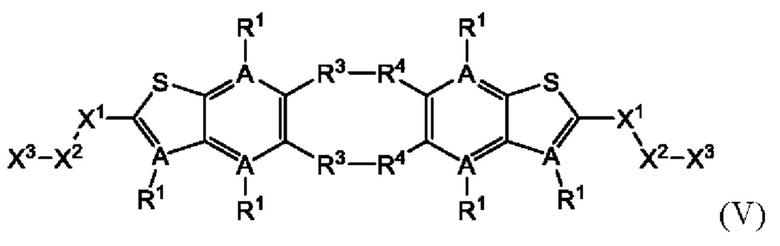
или его фармацевтически приемлемая соль, где
каждый 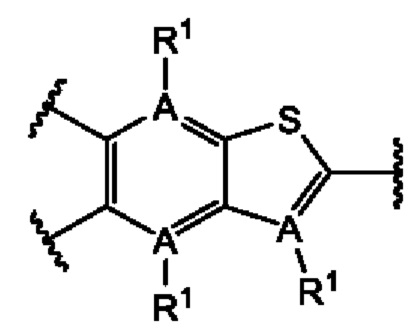 независимо выбран из группы, состоящей из
независимо выбран из группы, состоящей из 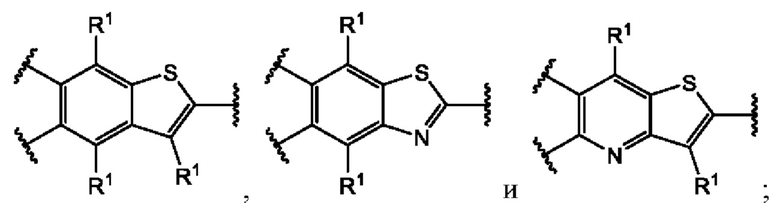
где каждый R1 независимо выбран из группы, состоящей из Н и галогена;
R3 и R4 независимо выбраны из группы, состоящей из O-(C1-C4 алкилен), С1-С5 алкилена, и N(R6)-(C1-C4 алкилен);
каждый R6 независимо выбран из группы, состоящей из Н, C1-C6 алкила и C1-C6 галоалкила;
каждый X1 представляет собой С=O;
каждый X2 независимо выбран из (C(R8)2)(1-3), где каждый R8 независимо выбран из группы, состоящей из Н и C1-C6 алкила;
необязательно, 2 R8 на разных атомах углерода могут быть взяты вместе совместно с атомами, к которым они присоединены, с образованием от 3- до 6-членного конденсированного кольца; и
каждый X3 независимо выбран из группы, состоящей из COOR6, C(O)N(R9)2, и CN; и
каждый R9 независимо выбран из группы, состоящей из Н и SO2R6.
6. Соединение общей формулы (VI):
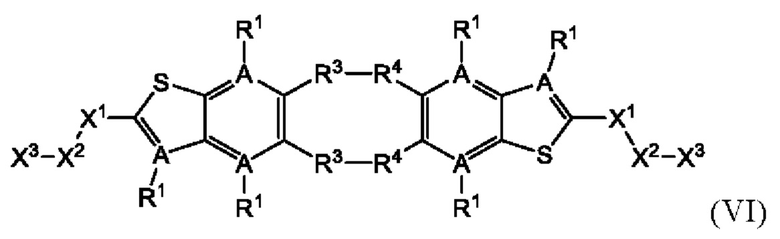
или его фармацевтически приемлемая соль, где
каждый 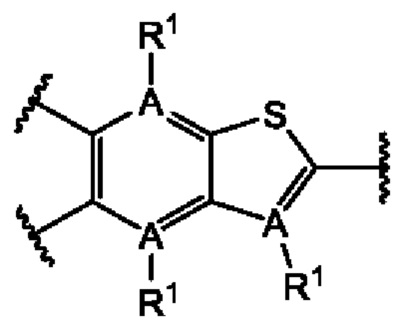 независимо выбран из группы, состоящей из
независимо выбран из группы, состоящей из 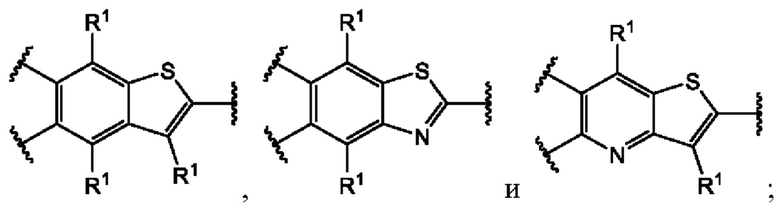
где каждый R1 независимо выбран из группы, состоящей из Н и галогена;
R3 и R4 независимо выбраны из группы, состоящей из O-(C1-C4 алкилен), С1-С5 алкилена, и N(R6)-(C1-C4 алкилен);
каждый R6 независимо выбран из группы, состоящей из Н, C1-C6 алкила и C1-C6 галоалкила;
каждый X1 представляет собой С=O;
каждый X2 независимо выбран из (C(R8)2)(1-3), где каждый R8 независимо выбран из группы, состоящей из Н и C1-C6 алкила;
необязательно, 2 R8 на разных атомах углерода могут быть взяты вместе совместно с атомами, к которым они присоединены, с образованием от 3- до 6-членного конденсированного кольца; и
каждый X3 независимо выбран из группы, состоящей из COOR6, C(O)N(R9)2, и CN; и
каждый R9 независимо выбран из группы, состоящей из Н и SO2R6.
7. Соединение, выбранное из группы, состоящей из:



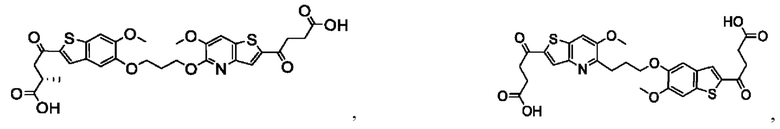

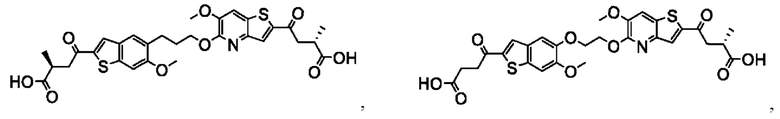
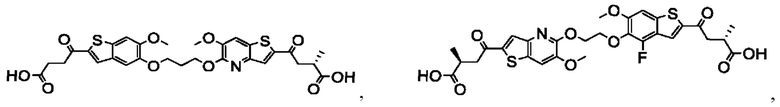
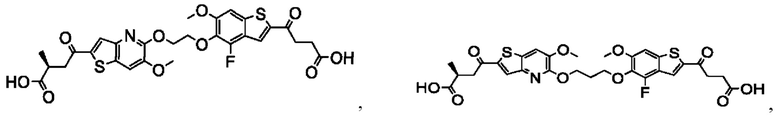
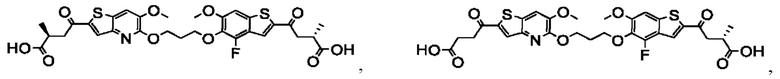

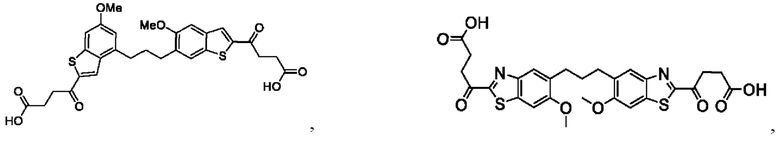
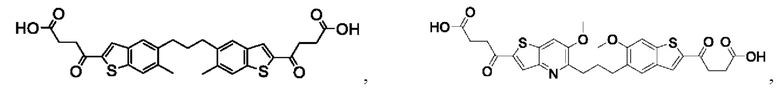
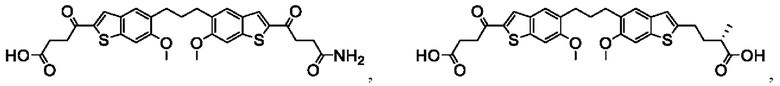
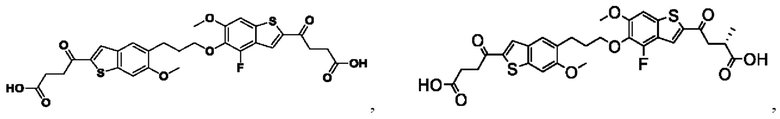
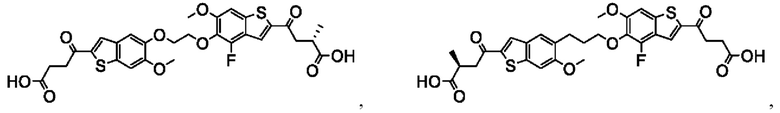
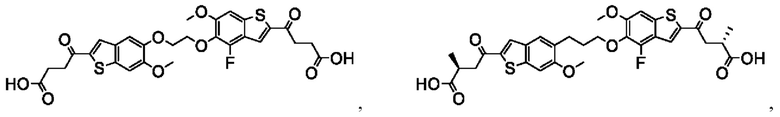
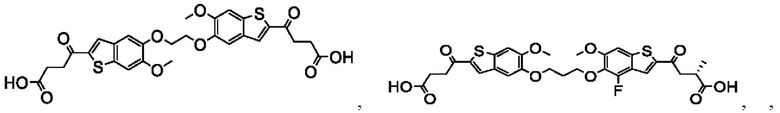
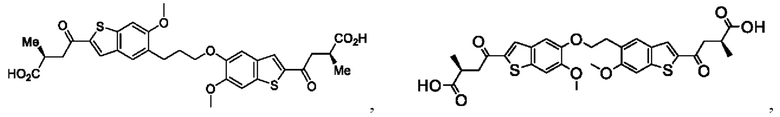
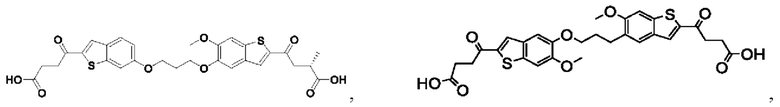
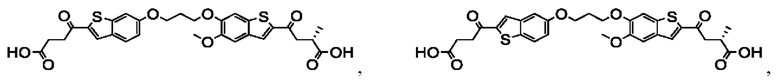


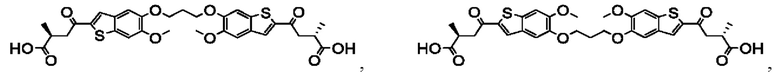

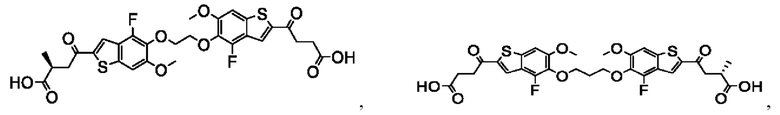
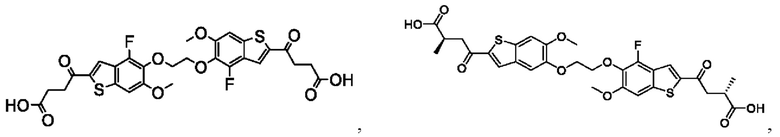
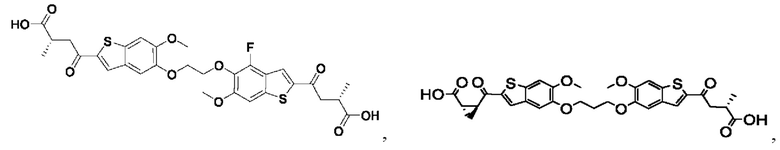
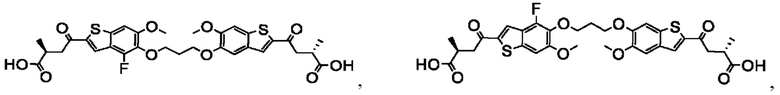
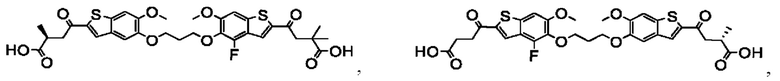
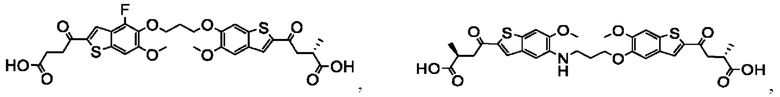
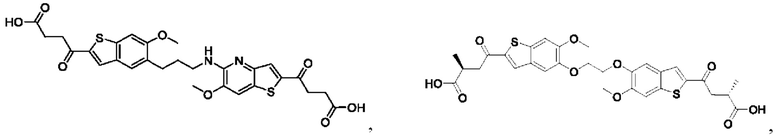
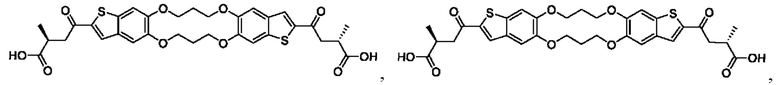
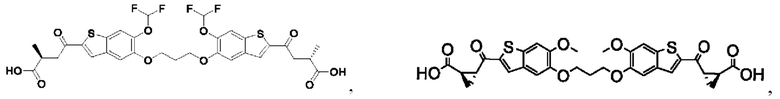
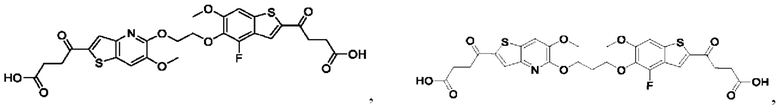
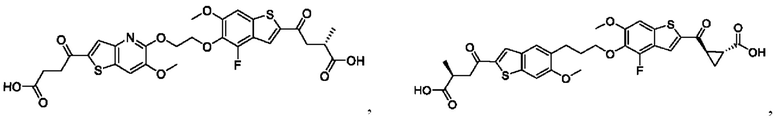
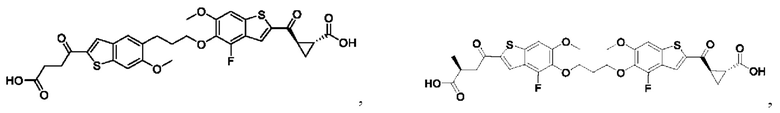
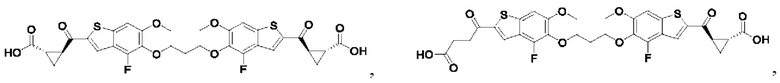
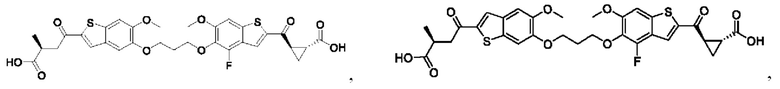
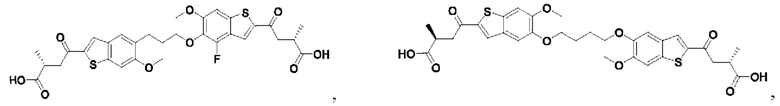
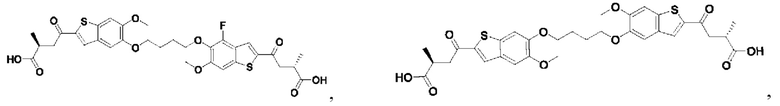
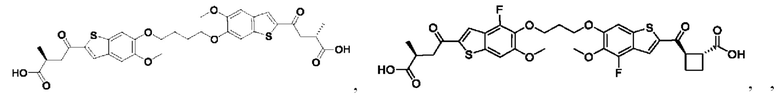
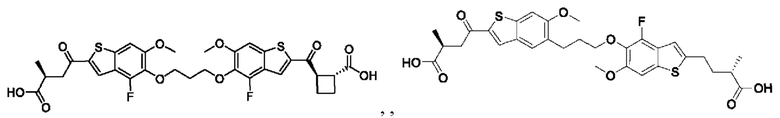
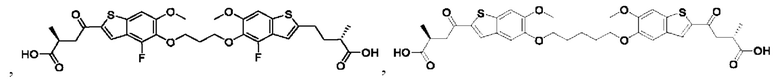
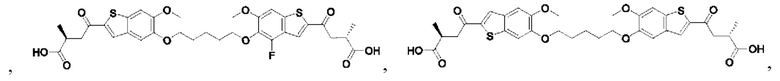
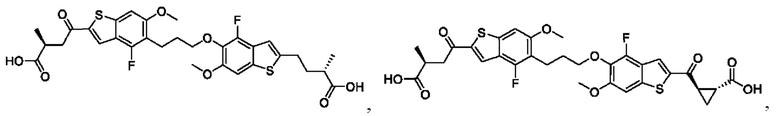
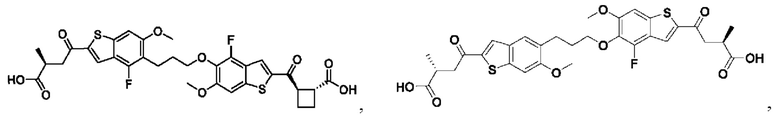
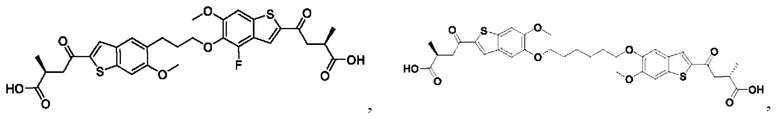
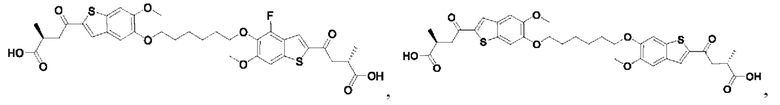
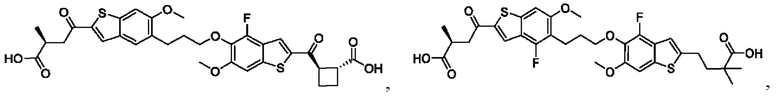
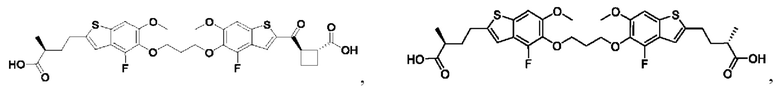
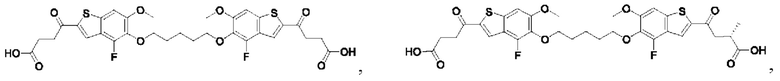

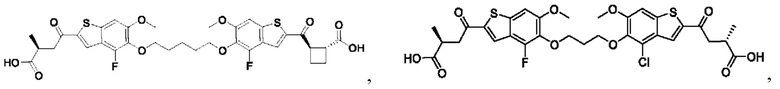
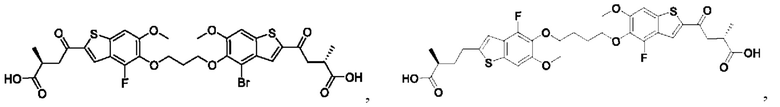
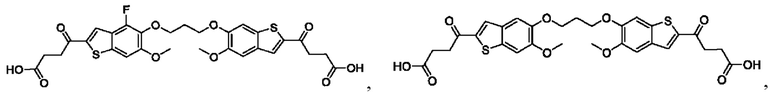
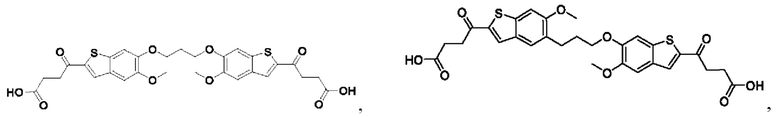
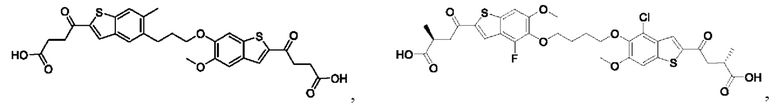
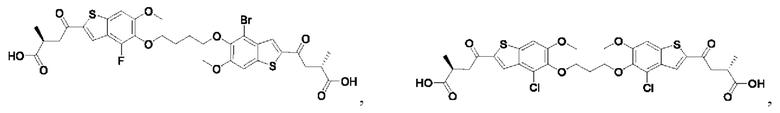
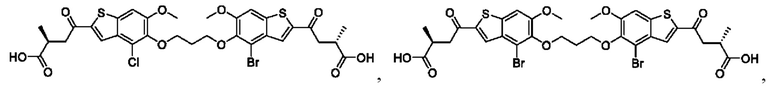
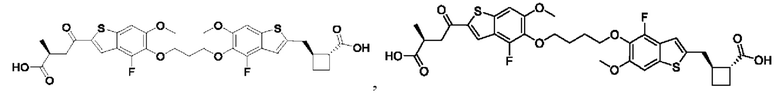
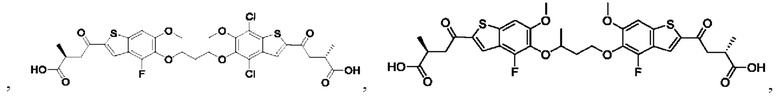
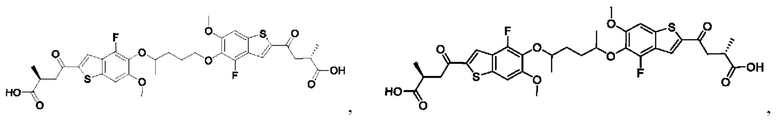
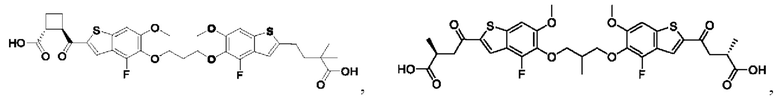
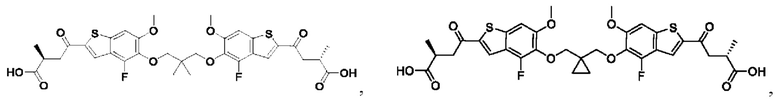
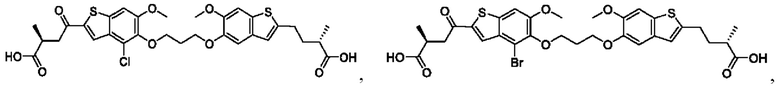
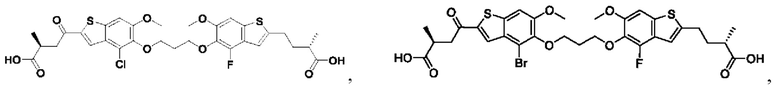
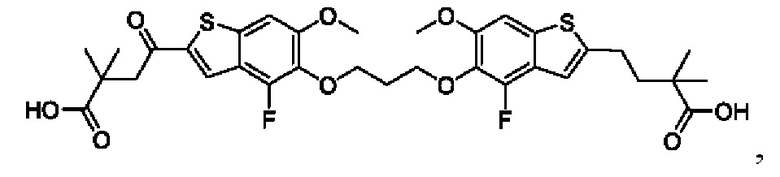
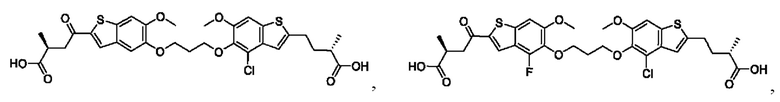
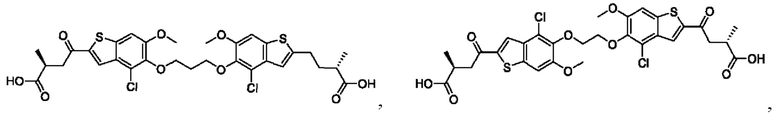
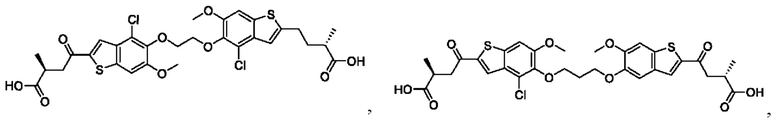
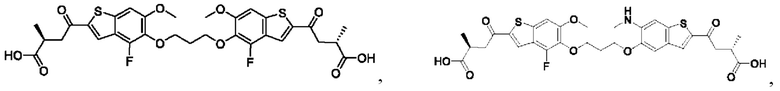
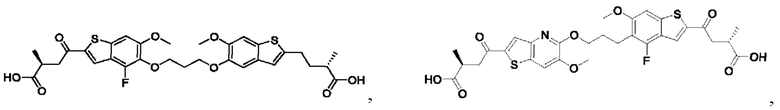
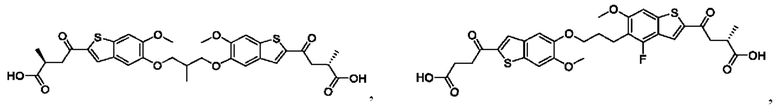
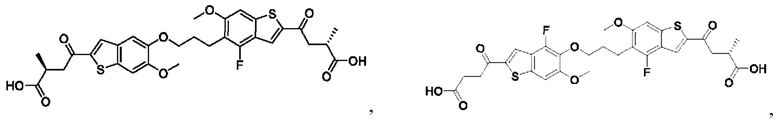
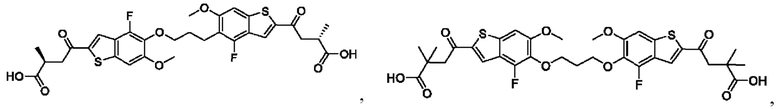
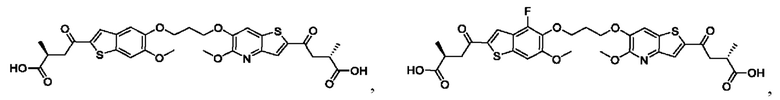
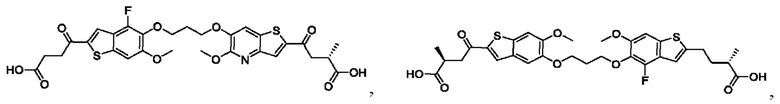
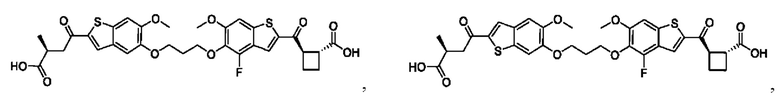
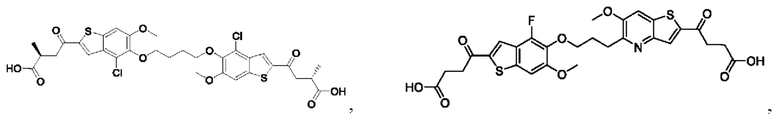
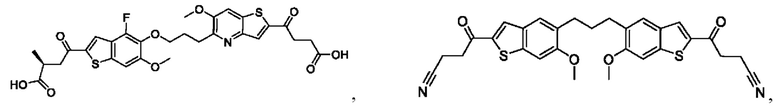
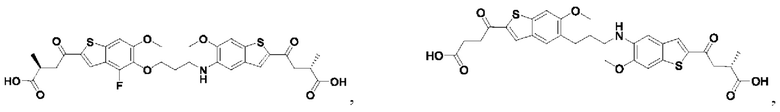
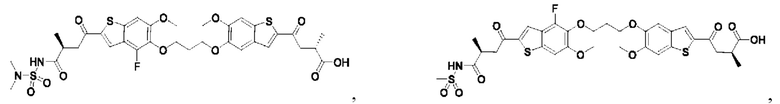
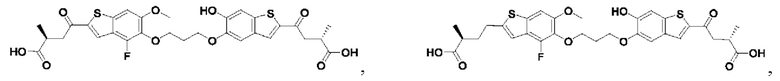
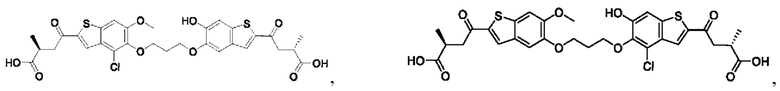
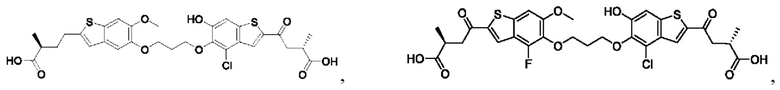
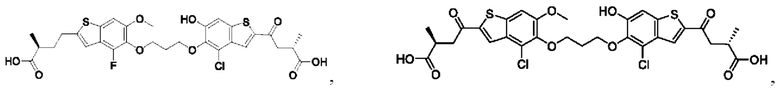
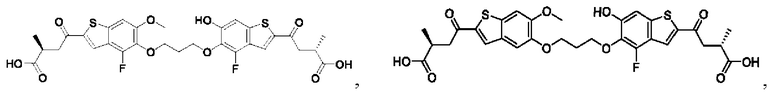 и
и 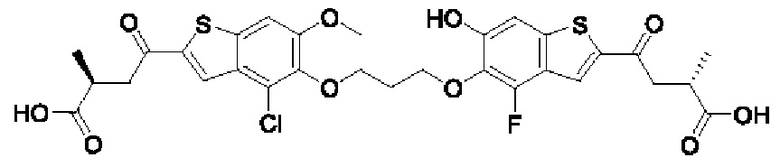
и его фармацевтически приемлемые соли.
8. Фармацевтическая композиция для индукции STING-зависимой продукции интерферона I типа, содержащая соединение по любому из пп. 1-7 или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель.
9. Способ индукции иммунного ответа у субъекта, где указанный способ включает: введение субъекту терапевтически эффективного количества соединения по любому из пп. 1-7 или его фармацевтически приемлемой соли.
10. Способ индукции иммунного ответа у субъекта, где указанный способ включает:
введение субъекту терапевтически эффективного количества фармацевтической композиции по п. 8.
11. Способ индукции STING-зависимой продукции интерферона типа I у субъекта, где указанный способ включает введение субъекту терапевтически эффективного количества соединения по любому из пп. 1-7 или его фармацевтически приемлемой соли.
12. Способ индукции STING-зависимой продукции интерферона I типа у субъекта, где указанный способ включает введение субъекту терапевтически эффективного количества фармацевтической композиции по п. 8.
13. Способ лечения нарушения пролиферации клеток, поддающегося лечению путем индукции STING-зависимой продукции интерферона типа I у субъекта, где указанный способ включает введение субъекту терапевтически эффективного количества соединения по любому из пп. 1-7 или его фармацевтически приемлемой соли.
14. Способ по п. 13, где нарушение пролиферации клеток представляет собой онкологическое заболевание.
15. Способ лечения нарушения пролиферации клеток, поддающегося лечению путем индукции STING-зависимой продукции интерферона типа I у субъекта, где указанный способ включает введение субъекту терапевтически эффективного количества фармацевтической композиции по п. 8.
16. Способ по п. 15, где нарушение пролиферации клеток представляет собой онкологическое заболевание.
17. Применение соединения по любому из пп. 1-7 или его фармацевтически приемлемой соли для индукции STING-зависимой продукции интерферона I типа в терапии.
18. Применение соединения по любому из пп. 1-7 или его фармацевтически приемлемой соли для лечения нарушения пролиферации клеток, поддающегося лечению путем индукции STING-зависимой продукции интерферона типа I у субъекта.
19. Применение по п. 18, где нарушение пролиферации клеток представляет собой онкологическое заболевание.
20. Применение фармацевтической композиции по п. 8 для лечения нарушения пролиферации клеток, поддающегося лечению путем индукции STING-зависимой продукции интерферона типа I у субъекта.
21. Применение по п. 20, где нарушение пролиферации клеток представляет собой онкологическое заболевание.
22. Соединение по п. 7, выбранное из группы, состоящей из 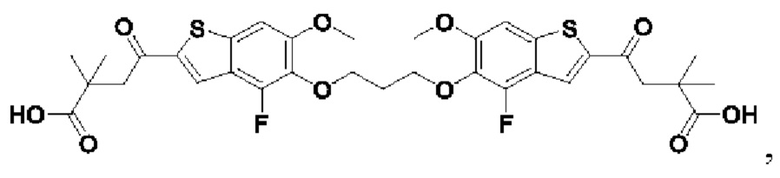 и его фармацевтически приемлемые соли.
и его фармацевтически приемлемые соли.
23. Соединение по п. 22, где соединение представляет собой 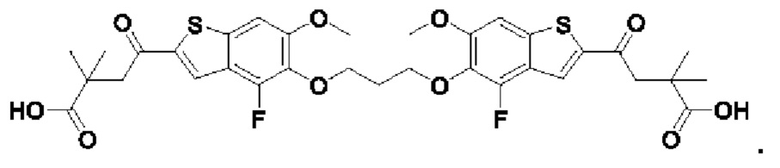
24. Соединение по п. 7, выбранное из группы, состоящей из 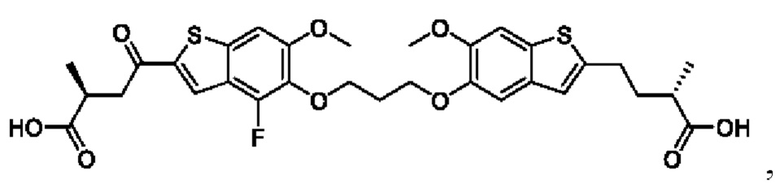 и его фармацевтически приемлемые соли.
и его фармацевтически приемлемые соли.
25. Соединение по п. 24, где соединение представляет собой 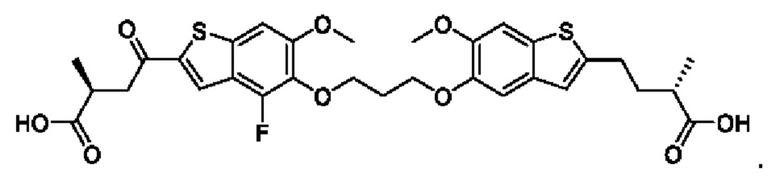
26. Соединение по п. 7, выбранное из группы, состоящей из 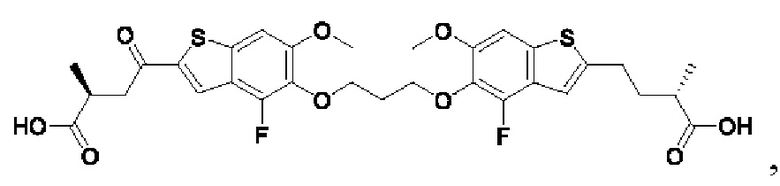 и его фармацевтически приемлемые соли.
и его фармацевтически приемлемые соли.
27. Соединение по п. 26, где соединение представляет собой 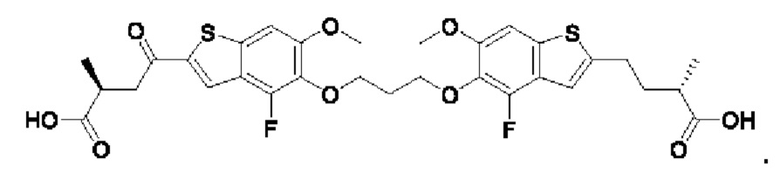
28. Соединение по п. 7, выбранное из группы, состоящей из 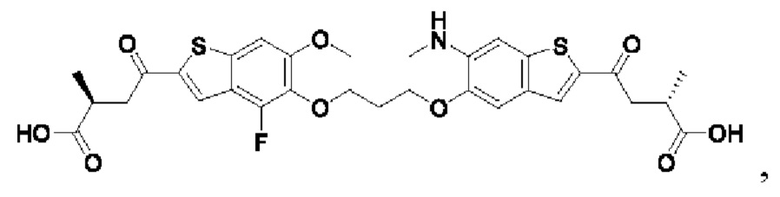 и его фармацевтически приемлемые соли.
и его фармацевтически приемлемые соли.
29. Соединение по п. 28, где соединение представляет собой 
30. Соединение по п. 7, выбранное из группы, состоящей из 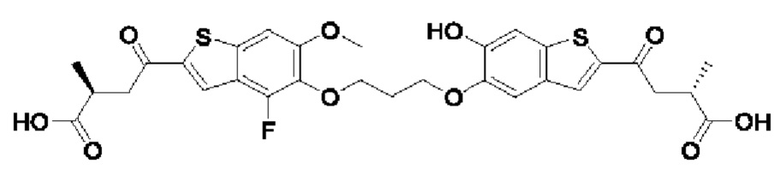 , и его фармацевтически приемлемые соли.
, и его фармацевтически приемлемые соли.
31. Соединение по п. 30, где соединение представляет собой 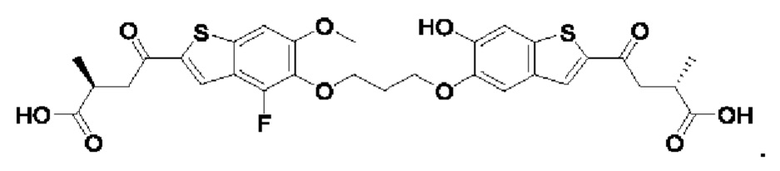
32. Соединение по п. 7, выбранное из группы, состоящей из 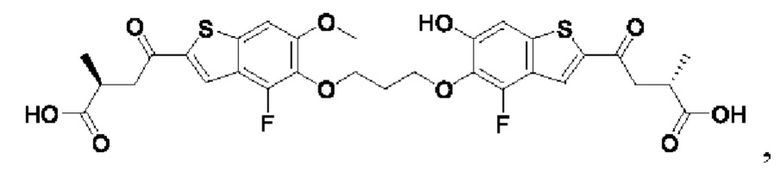 и его фармацевтически приемлемые соли.
и его фармацевтически приемлемые соли.
33. Соединение по п. 32, где соединение представляет собой 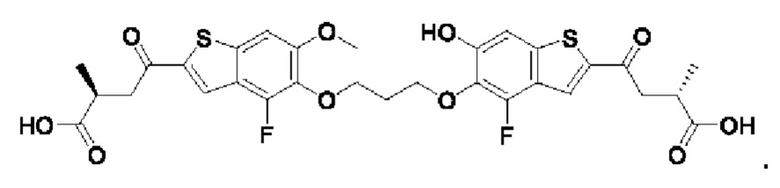
| WO 2015077354 A1, 28.05.2015 | |||
| ГИДРАВЛИЧЕСКИЙ УСИЛИТЕЛЬ | 0 |
|
SU350990A1 |
| GOPINATH P | |||
| et al., As many as six tandem reactions in one step! Unprecedented formation of highly functionalized benzothiophenes, Chem | |||
| Commun., 2009, v | |||
| Способ изготовления звездочек для французской бороны-катка | 1922 |
|
SU46A1 |
| ДВЕРЦЫ ДЛЯ СТЕННЫХ МУСОРНЫХ КАНАЛОВ | 1927 |
|
SU7131A1 |
| ИСАКОВ Д.В | |||
| и др., Циклоферон: механизмы действия и новые перспективы применения в клинической практике, | |||

