ВВЕДЕНИЕ
[0001] Настоящее изобретение относится к фармацевтически активным соединениям. Более конкретно, настоящее изобретение относится к соединениям, которые являются ингибиторами ферментативной активности Aurora киназ. Соединения по изобретению являются также ингибиторами активности FMS-подобной тирозинкиназы-3 (FLT3). Настоящее изобретение также относится к способам получения этих соединений, содержащим их фармацевтическим композициям и к их применению при лечении пролиферативных заболеваний, таких как злокачественное новообразование, а также других заболеваний или состояний, в которых вовлечена активность Aurora киназы и/или FLT3.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
[0002] Пролиферативные заболевания, такие как злокачественное новообразование, характеризуются неконтролируемой и нерегулируемой клеточной пролиферацией. Именно то, что заставляет клетки пролиферировать неконтролируемым и нерегулируемым образом, было объектом интенсивных исследований на протяжении последних десятилетий.
[0003] Aurora киназы, семейство из трех серин-треониновых киназ, обозначаемых как A, B и C, играют ключевую и явную роли на различных стадиях митоза.1,3 На ранних стадиях митоза Aurora-A образует комплекс с белком, связывающимся с моторным белком Xklp2 (TPX2), который регулирует созревание центросом и формирование митотического веретена.4,5 Aurora-B образует комплексы с внутренним центромерным белком (INCENP), сурвивином и бореалином, тем самым регулируя конденсацию хромосом, выравнивание хромосом, контрольную точку митотического цикла и цитокинез.6-9 О сверхэкспрессии Aurora-A и Aurora-B сообщалось для широкого спектра злокачественных новообразований у человека, включая рак молочной железы, рак толстой кишки, рак яичников, глиому, рак щитовидной железы, семиному.10-16 Функция Aurora-C в процессе митоза менее понятна. Однако сообщалось о высокой экспрессии Aurora-C в семенниках.17,18
[0004] В последние годы небольшие молекулы, нацеленные на Aurora киназы, стали общей стратегией разработки новых химиотерапевтических средств при злокачественных новообразованиях, и был описан ряд структурно разнообразных ингибиторов активности Aurora,18-20 включая 1 (VX-680 (МК-0457)),21 2 (AZD1152),22 3 (PHA-739358)23,24 и 4 (AMG 900)25 (смотри далее).
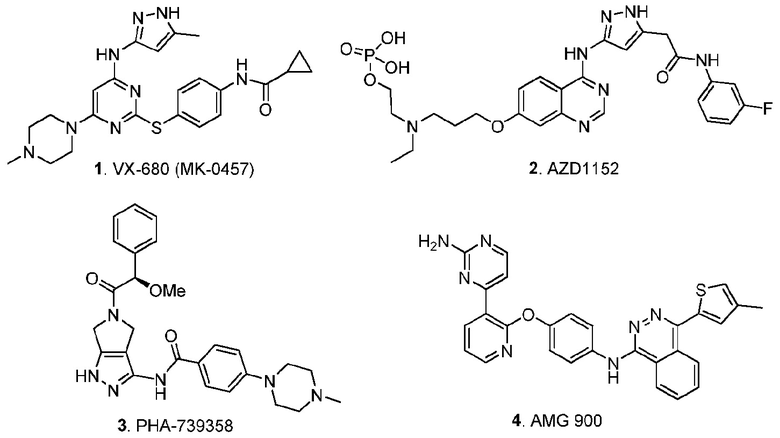
[0005] Однако остается потребность выявления дополнительных терапевтических агентов, способных ингибировать активность Aurora киназ.
[0006] В международных патентных публикациях №№ WO 2007/072017 и WO 2009/001021, в обоих описана серия имидазо[4,5-b]производных пиридина, которые функционируют как ингибиторы активности Aurora киназ, и которые, следовательно, являются потенциально полезными терапевтическими агентами для лечения злокачественного новообразования. Одно конкретное соединение, описанное в WO 2009/001021, показано далее.
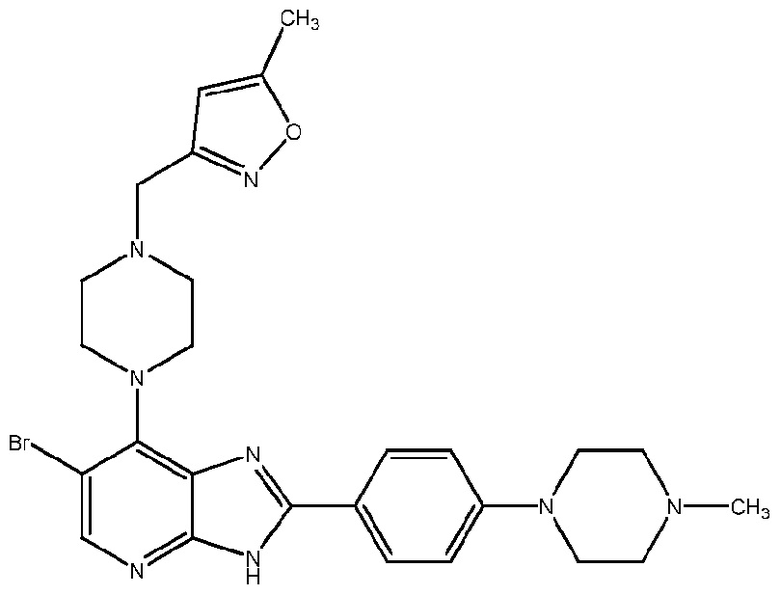
[0007] Конкретное соединение (известное как CCT137690) является мощным и биодоступным при пероральном приеме ингибитором Aurora киназ, которое ингибирует in vivo рост ксенотрансплантата карциномы толстой кишки SW620 человека с сопутствующей модуляцией биомаркера, соответствующей поражению цели.26 Однако доклиническая разработка этого соединения была ограничена ввиду его узкой широты терапевтического диапазона в отношении hERG43 (IC50=3,0 мкМ)26 и его низкой стабильности в микросомах печени человека (86% метаболизируется после 30 мин инкубации, неопубликованные данные).
[0008] Таким образом, объектом настоящего изобретения является обеспечение биодоступности при пероральном приеме ингибиторов ферментативной активности Aurora киназ, пригодных для доклинической и клинической оценки.
[0009] Таким образом, объектом настоящего изобретения является обеспечение биодоступности при пероральном приеме ингибиторов ферментативной активности Aurora киназ, которые обладают приемлемой стабильностью в микросомах человека, пониженным ингибированием активности цитохрома P450 и, в случае некоторых соединений, широким терапевтическим индексом в отношении hERG.
[0010] FLT3 представляет собой транс-мембранную киназу, которая относится к классу III семейства рецепторных тирозинкиназ (RTK). Связывание FLT3-лиганда (FL) с его рецептором приводит к димеризациии, аутофосфорилированию и последующей активации нижележащих сигнальных путей.37 Высокий уровень экспрессии FLT3 был обнаружен в бластных клетках при острой миелоидной лейкемии (AML), и у больных AML были выявлены два основных класса мутаций, т.е. внутренние тандемные дупликации (ITD) и точковые мутации тирозинкиназного домена (TKD).37,38 Внутренние тандемные дупликации были определены у 20-25% больных AML, и точковые мутации тирозинкиназного домена у 5-10% больных AML.37,38 Ряд низкомолекулярных ингибиторов FLT3 были оценены в клинических испытаниях.38,39
[0011] Таким образом, существует потребность в соединениях, которые имеют двойную функцию ингибирования как киназ Aurora, так и FLT3. Такие соединения могли бы быть использованы при лечении заболеваний и/или состояний, в которые вовлечены Aurora и/или FLT3, такие как, например, AML.
[0012] Поэтому следующим объектом настоящего изобретения является разработка соединений, обладающих этой двойной активностью.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
[0013] В одном аспекте настоящее изобретение относится к соединению или его фармацевтически приемлемой соли или сольвату, как определено в настоящем документе.
[0014] В другом аспекте настоящее изобретение относится к фармацевтической композиции, содержащей соединение по изобретению, как определено в настоящем документе, или его фармацевтически приемлемую соль или сольват и один или несколько фармацевтически приемлемых инертных наполнителей.
[0015] В другом аспекте настоящее изобретение относится к соединению по изобретению, как определено в настоящем документе, или его фармацевтически приемлемой соли или сольвату, или фармацевтической композиции, как определено в настоящем документе, для применения в терапии.
[0016] В другом аспекте настоящее изобретение относится к соединению по изобретению, как определено в настоящем документе, или его фармацевтически приемлемой соли или сольвату, или фармацевтической композиции, как определено в настоящем документе, для применения при лечении заболеваний или состояний, в которых вовлечена активность Aurora киназы и/или FLT3.
[0017] В другом аспекте настоящее изобретение относится к применению соединения по изобретению, как определено в настоящем документе, или его фармацевтически приемлемой соли или сольвата, при получении лекарственного средства для применения в лечении заболеваний или состояний, в которых вовлечена активность Aurora киназы и/или FLT3.
[0018] В другом аспекте настоящее изобретение относится к способу лечения заболевания или состояния, в которые вовлечена активность Aurora киназы и/или FLT3, где указанный способ включает введение субъекту при необходимости такого лечения терапевтически эффективного количества соединения по изобретению, как определено в настоящем документе, или его фармацевтически приемлемой соли или сольвата, или фармацевтической композиции, как определено в настоящем документе.
[0019] В другом аспекте настоящее изобретение относится к соединению или его фармацевтически приемлемой соли или сольвату, или фармацевтической композиции, как определено в настоящем документе, для применения в лечении пролиферативных расстройств, таких как злокачественное новообразование. В конкретном варианте злокачественным новообразованием является рак у человека.
[0020] В другом аспекте настоящее изобретение относится к применению соединения или его фармацевтически приемлемой соли или сольвата при получении лекарственного средства для применения в лечении пролиферативных расстройств, таких как злокачественное новообразование. В конкретном варианте злокачественным новообразованием является рак у человека.
[0021] В другом аспекте настоящее изобретение относится к способу лечения пролиферативного расстройства, такого как злокачественное новообразование, где указанный способ включает введение субъекту при необходимости такого лечения терапевтически эффективного количества соединения или его фармацевтически приемлемой соли или сольвата, или фармацевтической композиции, как определено в настоящем документе. В конкретном варианте злокачественным новообразованием является рак у человека.
[0022] В другом аспекте настоящее изобретение относится к соединению или его фармацевтически приемлемой соли или сольвату, или фармацевтической композиции, как определено в настоящем документе, для использования в продуцировании ингибирующего действия Aurora киназы и/или FLT3.
[0023] В другом аспекте настоящее изобретение относится к применению соединения или его фармацевтически приемлемой соли или сольвата при получении лекарственного средства для использования в продуцировании ингибирующего действия Aurora киназы и/или FLT3.
[0024] В другом аспекте настоящее изобретение относится к способу продуцирования in vitro ингибирующего действия Aurora киназы и/или FLT3, где указанный способ включает введение эффективного количества соединения или его фармацевтически приемлемой соли или сольвата.
[0025] В другом аспекте настоящее изобретение относится к способу продуцирования in vivo ингибирующего действия Aurora киназы и/или FLT3, где указанный способ включает введение эффективного количества соединения или его фармацевтически приемлемой соли или сольвата.
[0026] В другом аспекте настоящее изобретение относится к способу ингибирования пролиферации клеток in vitro или in vivo, где указанный способ включает контактирование клетки с эффективным количеством соединения, описанного в настоящем документе, или его фармацевтически приемлемой соли или сольвата.
[0027] Настоящее изобретение далее относится к способу синтеза соединения или его фармацевтически приемлемой соли или сольвата, как определено в настоящем документе.
[0028] В другом аспекте настоящее изобретение относится к соединению или его фармацевтически приемлемой соли или сольвату, которые могут быть получены, либо получены, либо непосредственно получают, описанным в настоящем документе.
[0029] В другом аспекте настоящее изобретение относится к новым промежуточным соединениям, описанным в настоящем документе, которые пригодны для использования в любом из способов синтеза, описанных в настоящем документе.
[0030] Предпочтительные, подходящие и дополнительные признаки какого-либо конкретного аспекта настоящего изобретения являются также предпочтительными, подходящими и дополнительными признаками любого другого аспекта.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Определения
[0031] Если не указано иное, термины, используемые в описании и в формуле изобретения, имеют следующие значения, указанные далее.
[0032] Должно быть понятно, что ссылки на «лечить» или «лечение» включают в себя профилактику, а также смягчение установленных симптомов заболевания. Термины «лечить» или «лечение» состояния, расстройства или заболевания, следовательно, включают: (1) предупреждение или задержку появления клинических симптомов состояния, расстройства или заболевания, развивающихся у человека, который может страдать или быть предрасположенным к состоянию, расстройству или заболеванию, но еще не ощущает или у которого не проявляются клинические или субклинические симптомы состояния, расстройства или заболевания, (2) подавление состояния, расстройства или заболевания, то есть, остановку, уменьшение или замедление развития заболевания или его рецидива (в случае поддерживающей терапии) или, по крайней мере, одного его клинического или субклинического признака, или (3) снятие или ослабление болезни, то есть, вызывание регресса состояния, расстройства или заболевания, или, по крайней мере, одного из его клинических или субклинических симптомов.
[0033] Термин «терапевтически эффективное количество» означает количество соединения, которое при введении млекопитающему для лечения заболевания, является достаточным, чтобы осуществить такое лечение заболевания. «Терапевтически эффективное количество» будет варьироваться в зависимости от соединения, заболевания и его тяжести, возраста, веса и др. млекопитающего, подвергаемого лечения.
[0034] Фраза «соединение по изобретению» означает соединения, которые описаны в настоящем документе, как в общем, так и в частности.
Соединения по изобретению
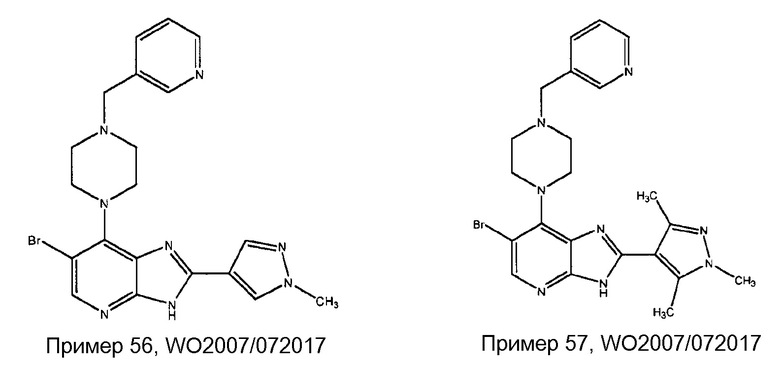
[0035] Как отмечалось ранее, в международной патентной публикации № WO 2007/072017 описана серия производных имидазо[4,5-b]пиридина, которые действуют как ингибиторы активности Aurora киназы. Двумя конкретными соединениями, описанными в WO 2007/072017, являются 6-бром-2-(1-метил-1H-пиразол-4-ил)-7-(4-(пиридин-3-илметил)пиперазин-1-ил)-3H-имидазо[4,5-b]пиридин (пример 56) и 6-бром-7-(4-(пиридин-3-илметил)пиперазин-1-ил)-2-(1,3,5-триметил-1H-пиразол-4-ил)-3H-имидазо[4,5-b]пиридин (пример 57). Структуры этих соединений показаны ниже.
[0036] В первом аспекте настоящее изобретение относится к соединению формулы (I), представленной далее:
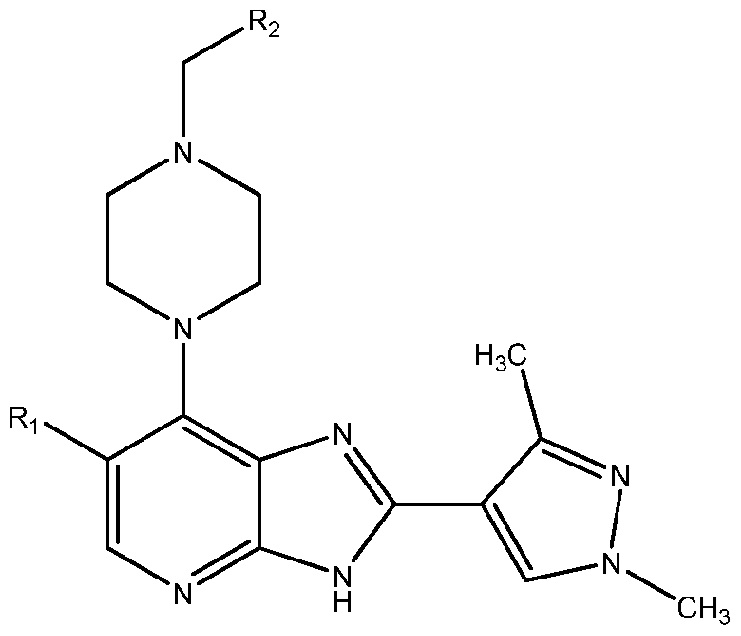
I
где:
R1 представляет собой Br или Cl;
R2 выбран из формулы II или формулы III, показанных далее:

где Ra представляет собой водород или метил;
или его фармацевтически приемлемой соли или сольвату.
[0037] В определении R2 группы выше символ ~~~~ обозначает точку присоединения группы R2 к радикалу -CH2-, имеющемуся в соединениях формулы I.
[0038] Соединения по настоящему изобретению демонстрируют уменьшение ингибирования активности цитохрома P450 по сравнению с соединениями по примерам 56 и 57 в WO 2007/072017. Некоторые соединения по настоящему изобретению также обладают более широким терапевтическим индексом в отношении hERG по сравнению с соединениями по примерам 56 и 57 в WO 2007/072017.
[0039] В частности, соединения по изобретению включают, например, соединения формулы I или их фармацевтически приемлемые соли, где, если не указано иное, каждый из R1 и R2 имеет любое из значений, определенных выше, или в любом из пунктов (1)-(5) здесь далее:
(1) R1 представляет собой Br;
(2) R1 представляет собой Cl;
(3) R2 имеет формулу II;
(4) R2 имеет формулу III, как определено в настоящем документе;
(5) R2 имеет формулу III, как определено в настоящем документе, и Ra представляет собой водород;
(6) R2 имеет формулу III, как определено в настоящем документе, и Ra представляет собой метил;
[0040] Целесообразно, когда R1 представляет собой хлор.
[0041] Целесообразно, когда R2 имеет формулу II (т.е. пара-хлорфенил). В конкретной группе соединений по изобретению, таким образом, соединения имеют структурную формулу Ia, показанную далее:
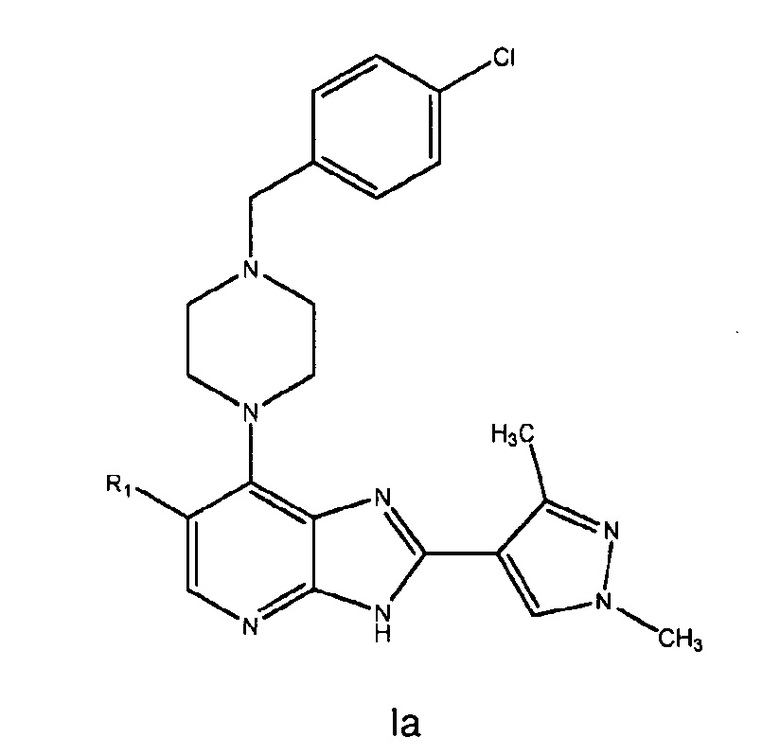
где R1 имеет значения, определенные выше, или их фармацевтически приемлемые соли или сольваты.
[0042] В следующей группе соединений по изобретению R2 имеет формулу III, т.е. соединения имеют структурную формулу Ib, показанную далее:

где R1 и Ra, оба имеют значения, определенные выше, или их фармацевтически приемлемые соли или сольваты.
[0043] Конкретные соединения по настоящему изобретению включают следующие:
6-хлор-7-(4-(4-хлорбензил)пиперазин-1-ил)-2-(1,3-диметил-1H-пиразол-4-ил)-3H-имидазо[4,5-b]пиридин;
3-((4-(6-хлор-2-(1,3-диметил-1H-пиразол-4-ил)-3H-имидазо[4,5-b]пиридин-7-ил)пиперазин-1-ил)метил)-1,2,4-оксадиазол;
3-((4-(6-хлор-2-(1,3-диметил-1H-пиразол-4-ил)-3H-имидазо[4,5-b]пиридин-7-ил)пиперазин-1-ил)метил)-5-метил-1,2,4-оксадиазол;
или их фармацевтически приемлемые соли или сольваты.
[0044] Подходящей фармацевтически приемлемой солью соединения по изобретению является, например, кислотно-аддитивная соль соединения по изобретению, которое является достаточно основным, например, кислотно-аддитивная соль добавления, например, неорганической или органической кислоты, например, хлористоводородной, бромистоводородной, серной, фосфорной, трифторуксусной, муравьиной, лимонной или малеиновой кислоты.
[0045] Настоящее изобретение также охватывает соединения по изобретению, как определено в настоящем документе, которые включают один или несколько изотопных заместителей. Например, H может быть в любой изотопной форме, в том числе 1H, 2H(D) и 3H (T); C может быть в любой изотопной форме, в том числе 12C, 13C и 14C; и тому подобное.
[0046] Следует также учитывать, что некоторые соединения по изобретению могут существовать в сольватированной, а также несольватированной формах, таких как, например, гидратированные формы. Следует также учитывать, что изобретение охватывает все такие сольватированные формы, которые обладают ингибирующей Aurora киназу и/или FLT3 активностью.
[0047] Также следует учитывать, что некоторые соединения по изобретению могут проявлять полиморфизм, и что изобретение охватывает все такие формы, которые обладают ингибирующей Aurora киназу и/или FLT3 активностью.
[0048] Соединения по изобретению могут существовать в виде различных таутомерных форм, и ссылки на соединения по изобретению включают все такие формы. Во избежание сомнений следует учесть, что, когда соединение может существовать в нескольких таутомерных формах, и только одна конкретно описана или показана, все другие, тем не менее, охватываются соединениями по изобретению. Примеры таутомерных форм соединений по настоящему изобретению включают соединения в форме, показанной формулой I выше, а также таутомеры формул (IV) и (V), показанных далее.
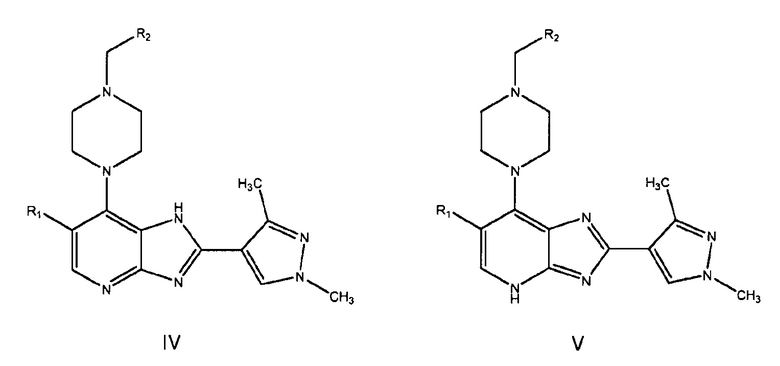
где R1 и R2 имеют значения, определенные выше.
[0049] Соединения по изобретению, содержащие функциональные аминогруппы, также могут образовывать N-оксиды. Отсылка в настоящем документе к соединению формулы I, которое содержит функциональную аминогруппу, также включает N-оксид. Когда соединение содержит несколько функциональных аминогрупп, один или несколько атомов азота могут быть окислены с образованием N-оксида. Конкретные примеры N-оксидов представляют собой N-оксиды по атому азота гетероцикла, содержащего атом азота. N-Оксиды могут быть образованы путем обработки соответствующего амина окисляющим агентом, таким как пероксид водорода или перкислота (например, пероксикарбоновая кислота), см., например, Advanced Organic Chemistry, by Jerry March, 4ой Edition, Wiley Interscience, страницы. Более конкретно, N-оксиды могут быть получены способом, описанным L. W. Deady (Syn. Comm. 1977, 7, 509-514), по которому соединение амина взаимодействует с м-хлорпероксибензойной кислотой (MCPBA), например, в инертном растворителе, таком как дихлорметан.
[0050] Соединения по изобретению могут быть введены в виде пролекарства, которое расщепляется в организме человека или животного, чтобы высвободить соединение по изобретению. Пролекарство может быть использовано для того, чтобы изменить физические свойства и/или фармакокинетические свойства соединения по изобретению. Пролекарство может быть получено, когда соединение по изобретению содержит подходящую группу или заместитель, к которому может быть присоединена модифицирующая свойства группа. Примеры пролекарства in vivo включают расщепляемые амидные производные, которые могут быть образованы по аминогруппе соединения по изобретению.
[0051] Соответственно, настоящее изобретение включает соединения формулы I, как определено выше, которые могут быть получены путем органического синтеза, и которые могут образовываться в организме человека или животного путем расщепления его пролекарства. Соответственно, настоящее изобретение включает соединения формулы I, которые получают путем органического синтеза, а также такие соединения, которые образуются в организме человека или животного путем метаболизма соединения-предшественника, то есть соединение формулы I может быть синтетически полученным соединением или метаболически образованным соединением.
[0052] Подходящее фармацевтически приемлемое пролекарство соединения формулы I представляет собой такое, которое основано на разумном медицинском заключении как подходящее для введения в организм человека или животного без нежелательных фармакологических активностей и без чрезмерной токсичности.
[0053] Различные формы пролекарств были описаны, например, в следующих документах:
a) Methods in Enzymology, Vol. 42, p. 309-396, edited by K. Widder, et al. (Academic Press, 1985);
b) Design of Pro-drugs, edited by H. Bundgaard, (Elsevier, 1985);
c) A Textbook of Drug Design and Development, edited by Krogsgaard-Larsen and H. Bundgaard, Chapter 5 «Design and Application of Pro-drugs», by H. Bundgaard p. 113-191 (1991);
d) H. Bundgaard, Advanced Drug Delivery Reviews, 8, 1-38 (1992);
e) H. Bundgaard, et al., Journal of Pharmaceutical Sciences, 77, 285 (1988);
f) N. Kakeya, et al., Chem. Pharm. Bull.. 32, 692 (1984);
g) T. Higuchi and V. Stella, «Pro-Drugs as Novel Delivery Systems», A.C.S. Symposium Series, Volume 14; и
h) E. Roche (editor), «Bioreversible Carriers in Drug Design», Pergamon Press, 1987.
[0054] Действие in vivo соединения формулы I может проявляться частично одним или более метаболитами, которые образуются в организме человека или животного после введения соединения формулы I. Как указано выше, действие in vivo соединения формулы I также может проявляться при метаболизме соединения-предшественника (пролекарства).
[0055] Следует также иметь в виду, что соединения формулы (I) также могут быть ковалентно присоединены (в любом подходящем положении) к другим группам, таким как, например, солюбилизирующие группы (например, полимеры PEG), группы, которые позволяют им быть связанными с твердой подложкой (такие, как, например, биотин-содержащие группы) и мишеневые лиганды (например, антитела или фрагменты антител).
Синтез
[0056] При рассмотрении описания синтетических способов, представленных далее, и указанных методов синтеза, которые используются для получения исходных продуктов, следует учитывать, что все предлагаемые условия реакции, включая выбор растворителя, атмосферу реакции, температуру реакции, продолжительность эксперимента и способы обработки, могут быть выбраны специалистом в данной области техники.
[0057] Как понятно специалисту в области органического синтеза, функциональные группы, имеющиеся в различных частях молекулы, должны быть совместимы с используемыми реагентами и условиями реакции.
[0058] Необходимые исходные продукты могут быть получены с помощью стандартных способов органической химии. Получение таких исходных продуктов описано в связи со следующими представительными вариантами способа и в прилагаемых примерах. Кроме того, необходимые исходные продукты могут быть получены способами, аналогичными проиллюстрированным, которые находятся в рамках знаний обычного химика-органика.
[0059] Следует иметь в виду, что в ходе синтеза соединений по изобретению в способах, описанных далее, или в процессе синтеза некоторых исходных продуктов может быть желательным защитить некоторые группы заместителей, чтобы предотвратить их от нежелательных взаимодействий. Опытный химик сможет оценить, когда такая защита необходима и как такие защитные группы могут быть введены, а позже удалены.
[0060] В качестве примеров защитных групп смотри один из многих обзоров на эту тему, например, «Protective Groups in Organic Synthesis» by Theodora Green (publisher: John Wiley & Sons). Защитные группы могут быть удалены любым удобным способом, описанным в литературе или известным квалифицированному химику, как необходимые для удаления защитных групп, такие способы выбирают так, чтобы осуществить удаление защитной группы с минимальным затрагиванием других групп в молекуле.
[0061] Таким образом, если реагенты включают, например, такие группы, как амино, карбокси или гидрокси, может быть желательным защитить группу в некоторых реакциях, упомянутых в данном документе.
[0062] В качестве примера подходящая защитная группа для амино или алкиламино группы представляет собой, например, ацильную группу, например, алканоильную группу, такую как ацетил, алкоксикарбонильную группу, например, метоксикарбонильная, этоксикарбонильная или трет-бутоксикарбонильная группы, арилметоксикарбонильную группу, например, бензилоксикарбонил, или ароильную группу, например, бензоил. Условия удаления указанных выше защитных групп обязательно изменяются в зависимости от выбора защитной группы. Так, например, ацильная группа, такая как алканоильная или алкоксикарбонильная группа или ароильная группа, может быть удалена, например, путем гидролиза с помощью подходящего основания, например, гидроксида щелочного металла, например, гидроксида лития или натрия. Альтернативно ацильная группа, такая как трет-бутоксикарбонильная группа, может быть удалена, например, путем обработки подходящей кислотой, такой как соляная, серная или фосфорная кислоты, или в среде трифторуксусной кислоты, и арилметоксикарбонильная группа, такая как бензилоксикарбонильная группа, может быть удалена, например, путем гидрирования над катализатором, таким как палладий-на-углероде, или путем обработки кислотой Льюиса, например BF3⋅OEt2. Соответствующей альтернативной защитной группой для первичной аминогруппы является, например, фталоильная группа, которая может быть удалена путем обработки алкиламином, например диметиламинопропиламином, или гидразином.
[0063] Соединения по настоящему изобретению могут быть получены, используя общие способы синтеза, описанные в WO 2007/072017 и WO 2009/001021, полное содержание которых включено в настоящий документ посредством ссылки.
[0064] В конкретном аспекте настоящее изобретение относится к способу синтеза соединения формулы I или его фармацевтически приемлемой соли или сольвата, где способ включает:
a) взаимодействие соединения формулы A:
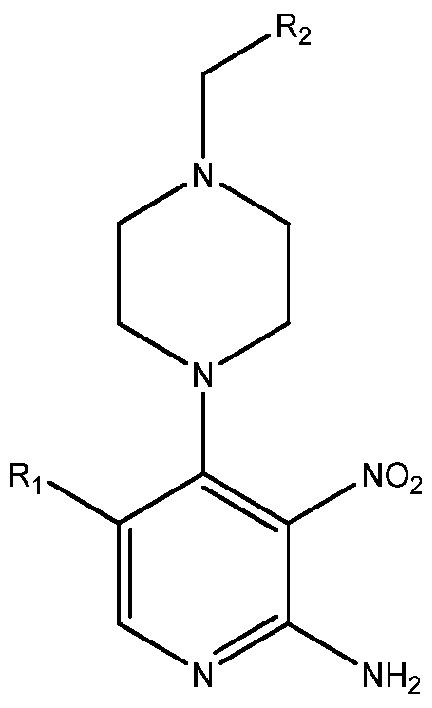
где R1 и R2, каждый, имеют одно из значений, приведенных выше;
с 1,3-диметил-1H-пиразол-4-карбальдегидом в присутствии подходящего восстанавливающего агента; и
b) далее необязательно и если необходимо:
i) удаление любых имеющихся защитных групп;
ii) преобразование соединения формулы I в другое соединение формулы I; и/или
iii) получение его фармацевтически приемлемой соли или сольвата.
[0065] Целесообразно, когда реакция между соединением формулы A и 1,3-диметил-1H-пиразол-4-карбальдегидом имеет место в присутствии подходящего растворителя. В этой реакции могут быть использованы любой подходящий растворитель или смесь растворителей. Примеры подходящих растворителей включают ДМСО, воду, ДМФА и спирты, например, EtOH.
[0066] Целесообразно, когда реакция протекает в присутствии подходящего восстановителя, такого как водный раствор Na2S2O4.26
[0067] Специалист в данной области сможет также подобрать подходящие условия реакции с целью облегчения этого взаимодействия.
[0068] Реакцию также можно проводить при повышенной температуре, например, может использоваться температура в диапазоне от 50 до 190°C (в зависимости от природы растворителя).
[0069] Полученное соединение формулы I может быть выделено и очищено с использованием способов, хорошо известных в данной области техники.
[0070] Способ, описанный в настоящем документе, может дополнительно включать стадию, в которой соединение формулы I подвергают солевому обмену, особенно в ситуациях, когда соединение формулы I образуется в виде смеси различных солевых форм. Солевой обмен соответствующим образом включает иммобилизацию соединения формулы I на подходящей твердой подложке или смоле и элюирование соединений соответствующей кислотой с получением одной соли соединения формулы I.
[0071] Соединения формулы A могут быть получены с помощью способов, известных в данной области техники.
[0072] Пример соответствующего способа получения соединения формулы I через промежуточное соединение формулы A показан на схеме 1 ниже.
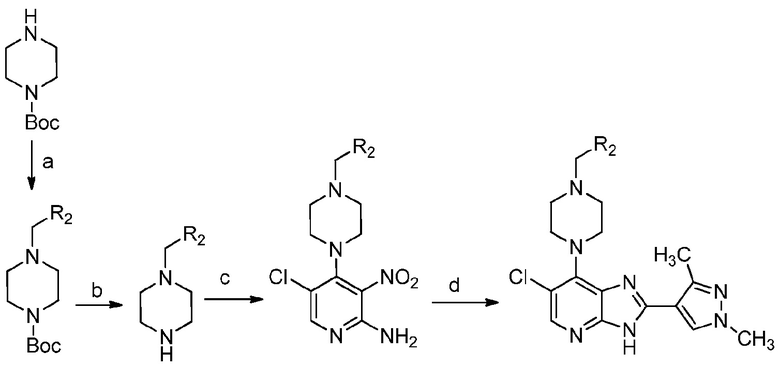
Реагенты и условия: стадии (a) и (b), указанные выше, относятся только к производному 1,2,4-оксадиазола, потому что 1-(4-хлорбензил)пиперазин и 1-((5-метил-1,2,4-оксадиазол-3-ил)метил)пиперазин являются коммерчески доступными: (a) для производного 1,2,4-оксадиазола: CH2Cl2, 3-(хлорметил)-1,2,4-оксадиазол, Et3N, 50°C; (b) для производного 1,2,4-оксадиазола: ТФУ, CH2Cl2, комн. темп.; (c) для производных 4-хлорбензила и 1,2,4-оксадиазола: 2-амино-4,5-дихлор-3-нитропиридин, изо-Pr2NEt, изо-PrOH, нагревание; (d) для производных 4-хлорбензила и 1,2,4-оксадиазола: 1,3-диметил-1H-пиразол-4-карбальдегид, EtOH, 1M водн. Na2S2O4, 80°C.
Схема 1
[0073] 2-Амино-4,5-дихлор-3-нитропиридин (4,5-дихлор-3-нитропиридин-2-амин) и 2-амино-5-бром-4-хлор-3-нитропиридин (5-бром-4-хлор-3-нитропиридин-2-амин), предшественники в синтезе производных A 2-амино-3-нитропиридина, были получены, как описано ранее26 или путем галогенирования 2-амино-4-хлор-3-нитропиридина (4-хлор-3-нитропиридин-2-амин)40.
Фармацевтические композиции
[0074] В соответствии с еще одним аспектом изобретения предложена фармацевтическая композиция, которая содержит соединение по изобретению, описанное выше, или его фармацевтически приемлемую соль или сольват в ассоциации с фармацевтически приемлемым разбавителем или носителем.
[0075] Композиции по изобретению могут быть в виде, подходящем для перорального применения (например, в виде таблеток, лепешек, твердых или мягких капсул, водных или масляных суспензий, эмульсий, диспергируемых порошков или гранул, сиропов или эликсиров), для местного применения (например, в виде кремов, мазей, гелей или водных или масляных растворов или суспензий), для введения путем ингаляции (например, в виде мелкодисперсного порошка или жидкого аэрозоля), для введения путем инсуффляции (например, в виде мелкодисперсного порошка) или для парентерального введения (например, в виде стерильного водного или масляного раствора для внутривенного, подкожного, внутримышечного, внутрибрюшинного или внутримышечного дозирования или в виде суппозиториев для ректального дозирования).
[0076] Композиции по изобретению могут быть получены с помощью обычных способов с использованием обычных фармацевтических инертных вспомогательных веществ, хорошо известных в данной области техники. Таким образом, композиции, предназначенные для перорального применения, могут содержать, например, один или несколько красителей, подсластителей, ароматизаторов и/или консервантов.
[0077] Эффективное количество соединения по настоящему изобретению для применения в терапии пролиферативных заболеваний представляет собой количество, достаточное для симптоматического облегчения симптомов инфекции у теплокровных животных, в частности, у человека, для замедления прогрессирования инфекции или для уменьшения риска ухудшения состояния у пациентов с симптомами инфекции.
[0078] Количество активного ингредиента, которое объединяют с одним или несколькими инертными вспомогательными веществами для получения единой лекарственной формы, будет обязательно меняться в зависимости от субъекта, получающего лечение, и конкретного пути введения. Например, препарат, предназначенный для перорального введения людям, обычно содержит, например, от 0,5 мг до 0,5 г активного агента (более подходяще, от 0,5 до 100 мг, например, от 1 до 30 мг) в сочетании с подходящим и удобным количеством инертного вспомогательного вещества, которое может изменяться от около 5 до около 98% по массе от общей композиции.
[0079] Размер дозы для терапевтических или профилактических целей соединения формулы I будет, естественно, меняться в зависимости от характера и тяжести состояний, возраста и пола животного или пациента и пути введения, в соответствии с хорошо известными принципами медицины.
[0080] При использовании соединения по изобретению в терапевтических или профилактических целях его обычно вводят таким образом, чтобы ежедневная доза была в диапазоне, например, от 0,1 мг/кг до 30 мг/кг массы тела, давая, если необходимо, в виде раздельных доз. Обычно при парентеральном пути введения используются более низкие дозы. Так, например, для внутривенного или внутрибрюшинного введения, обычно используется доза в интервале, например, от 0,1 мг/кг до 30 мг/кг массы тела. Аналогичным образом для введения путем ингаляции используется доза в интервале, например, от 0,05 мг/кг до 25 мг/кг массы тела. Пероральное ведение также может быть подходящим, в частности, в виде таблеток. Как правило, стандартные лекарственные формы будут содержать от около 0,5 мг до 0,5 г соединения по настоящему изобретению.
Терапевтическое использование и применение
[0081] Соединения по изобретению являются ингибиторами активности Aurora киназы и FLT3.
[0082] Таким образом, в другом аспекте настоящее изобретение относится к способу ингибирования активности Aurora киназы и/или FLT3 в клетке, где способ включает введение в указанную клетку соединения формулы I, как определено в настоящем документе, или его фармацевтически приемлемую соль или сольват.
[0083] В следующем аспекте настоящее изобретение относится к способу ингибирования активности Aurora киназы и/или FLT3 in vitro или in vivo, где указанный способ включает контактирование клетки с эффективным количеством соединения или его фармацевтически приемлемой соли или сольвата, как определено в настоящем документе.
[0084] В другом аспекте настоящее изобретение относится к способу ингибирования активности Aurora киназы и/или FLT3 у субъекта человека или животного, при необходимости такого ингибирования, где способ включает введение указанному субъекту эффективного количества соединения формулы I, как определено в настоящем документе, или его фармацевтически приемлемой соли или сольвата.
[0085] Aurora киназа может представлять собой Aurora киназу A, B или C.
[0086] В одном аспекте настоящее изобретение относится к соединению формулы I или его фармацевтически приемлемой соли или сольвату или к фармацевтической композиции, как определено в настоящем документе, для применения в терапии.
[0087] В другом аспекте настоящее изобретение относится к соединению формулы I, как определено в настоящем документе, или его фармацевтически приемлемой соли или сольвату для применения при лечении заболевания или состояния, связанного с активностью Aurora киназы (и/или активностью FLT3).
[0088] В другом аспекте настоящее изобретение относится к применению соединению формулы I, как определено в настоящем документе, или его фармацевтически приемлемой соли или сольвату, при получении лекарственного средства для применения в лечении заболевания или состояния, связанного с активностью Aurora киназы (и/или активностью FLT3).
[0089] В еще одном аспекте настоящее изобретение относится к способу лечения пролиферативного расстройства у субъекта человека или животного, где способ включает введение указанному субъекту терапевтически приемлемого количества соединения формулы I, как определено в настоящем документе, или его фармацевтически приемлемой соли или сольвата.
[0090] В еще одном аспекте настоящее изобретение относится к соединению формулы I, как определено в настоящем документе, или его фармацевтически приемлемой соли или сольвату для применения при лечении пролиферативного расстройства.
[0091] В еще одном аспекте настоящее изобретение относится к применению соединения формулы I, как определено в настоящем документе, или его фармацевтически приемлемой соли или сольвата при получении лекарственного средства для применения в лечении пролиферативного расстройства.
[0092] В другом аспекте настоящее изобретение относится к соединению или его фармацевтически приемлемой соли или сольвату или фармацевтической композиции, как определено в настоящем документе, для применения при лечении злокачественного новообразования.
[0093] В еще одном аспекте настоящее изобретение относится к применению соединения или его фармацевтически приемлемой соли или сольвата, как определено в настоящем документе, при получении лекарственного средства для применения в лечении злокачественного новообразования.
[0094] В еще одном аспекте настоящее изобретение относится к способу лечения злокачественного новообразования у пациента, нуждающегося в таком лечении, где указанный способ включает введение указанному пациенту терапевтически эффективного количества соединения или его фармацевтически приемлемой соли или сольвата или фармацевтической композиции, как определено в настоящем документе.
[0095] Соединения по изобретению могут быть использованы, например, при лечении колоректального рака, рака молочной железы, легких, простаты, поджелудочной железы или мочевого пузыря и почек или лейкоза или лимфомы.
[0096] В частности, соединения по настоящему изобретению могут быть использованы для лечения лейкозов. Высокая экспрессия Aurora киназ была продемонстрирована при лейкозе (клеточные линии и группы пациентов).30-33 Кроме того, внутренние тандемные дупликации гена FLT3 (FLT3-ITD) приводят к активации конститутивной киназы FLT3.34 Существенно, что FLT3-ITD встречается у 20-35% взрослых и 15% детей с неблагоприятным прогнозом AML для обеих возрастных групп.35
[0097] Таким образом, в конкретном варианте осуществления изобретения соединения могут быть использованы для лечения лейкозов, таких как острый миелоидный лейкоз (AML), миелодиспластический синдром (MDS), хронический лимфоцитарный лейкоз (CLL) и множественная миелома. Соединения по настоящему изобретению рассматриваются также как полезные при лечении нейробластомы.
[0098] Соединения по настоящему изобретению, как ожидается, будут представлять конкретную пользу для пациентов, которые не смогли получить лечение с помощью стандартной терапии. Высказывается предположение, что соединения по настоящему изобретению также будут иметь значение при лечении больных пожилого возраста (например, старше 60 лет), страдающих лейкозом (например, AML), потому что у таких пациентов, как ожидается, будет целесообразным ингибирование Aurora киназы.
[0099] Предполагается также, что соединения по настоящему изобретению будут иметь значение при лечении детей с лейкозом (например, диагностированных как AML с мутацией FLT3 и младенческий AML), а также нейробластом.
Способы введения
[00100] Соединения по изобретению или фармацевтическая композиция, содержащая активное соединение, могут быть введены субъекту любым удобным способом введения, либо системно/периферийно, либо местно (т.е. в месте желаемого действия).
[00101] Способы введения включают, но не ограничиваются ими, пероральный (например, при приеме внутрь); буккальный; сублингвальный; трансдермальный (в том числе, например, с помощью накладки, пластыря и др.); трансмукозальный (в том числе, например, с помощью накладки, пластыря и др.); интраназальный (например, назальный спрей); глазной (например, глазные капли); пульмональный (например, с использованием ингаляционной или инсуффляционной терапии, например, с помощью аэрозоля, например, через рот или нос); ректальный (например, в виде свечей или клизм); вагинальный (например, с помощью пессариев); парентеральный, например, путем инъекций, в том числе подкожной, внутрикожной, внутримышечной, внутривенной, внутриартериальной, внутрисердечной, интратекальной, межпозвоночной, внутрикапсульной, субкапсулярной, внутриглазничной, внутрибрюшинной, интратрахеальной, подкожной, внутрисуставной, субарахноидальной и интрастенальной; путем импланта депо или резервуара, например, подкожно или внутримышечно.
Комбинированная терапия
[00102] Соединения по изобретению могут вводиться отдельно в качестве монотерапии или могут вводиться в комбинации с одним или несколькими дополнительными терапевтическими агентами. Выбор одного или более дополнительных терапевтических агентов будет, конечно, различаться в зависимости от заболевания или состояния, подвергаемого лечению, и степени его тяжести.
[00103] Является обычным использовать комбинированные терапии для лечения пролиферативных расстройств, таких как злокачественное новообразование. Таким образом, антипролиферативное лечение, определенное выше, может применяться в качестве монотерапии или может включать, в дополнение к соединению по изобретению, обычную хирургическую операцию или лучевую терапию или химиотерапию. Такая химиотерапия может включать один или несколько следующих категорий противоопухолевых агентов:-
(i) другие антипролиферативные/противоопухолевые препараты и их комбинации, используемые в медицинской онкологии, такие как алкилирующие агенты (например, цисплатин, оксалиплатин, карбоплатин, циклофосфамид, нитроген мустард, мелфалан, хлорамбуцил, бусульфан, темозоламид и нитрозомочевины); антиметаболиты (например, гемцитабин и антифолаты, такие как фторпиримидины, такие как 5-фторурацил и тегафур, ралтитрексид, метотрексат, цитозин-арабинозид и гидроксимочевину); противоопухолевые антибиотики (например, антрациклины, такие как адриамицин, блеомицин, доксорубицин, дауномицин, эпирубицин, идаруцибин, митомицин-C, дактиномицин и митрамицин); антимитотические агенты (например, алкалоиды барвинка, такие как винкристин, винбластин, виндезин и винорелбин, и таксоиды, такие как таксол и таксотер, и ингибиторы polo-подобной киназы); и ингибиторы топоизомеразы (например, эпиподофиллотоксины, такие как этопозид и тенипозид, амсакрин, топотекан и камптотецин);
(ii) цитостатические агенты, такие как антиэстрогены (например, тамоксифен, фулвестрант, торемифен, ралоксифен, дролоксифен и йодоксифен), антиандрогены (например, бикалютамид, флутамид, нилутамид и ципротерон ацетат), антагонисты LHRH или агонисты LHRH (например, гозерелин, лейпрорелин и бусерелин), прогестагены (например, мегестрол (мегестрол ацетат), ингибиторы ароматазы (например, анастрозол, летрозол, воразол и экземестан) и ингибиторы 5α-редуктазы, такие как финастерид;
(iii) противоинвазивные агенты [например, ингибиторы семейства c-Src киназ, такие как 4-(6-хлор-2,3-метилендиоксианилино)-7-[2-(4-метилпиперазин-1-ил)этокси]-5-тетрагидропиран-4-илоксихиназолин (AZD0530; международная патентная заявка WO 01/94341), N-(2-хлор-6-метилфенил)-2-{6-[4-(2-гидроксиэтил)пиперазин-1-ил]-2-метилпиримидин-4-иламино}тиазол-5-карбоксамид (дазатиниб, BMS-354825; J. Med. Chem., 2004, 47, 6658-6661) и босутиниб (SKI-606), и ингибиторы металлопротеиназы, такие как маримастат, ингибиторы функции активатора плазминогена типа рецептора урокиназы или антител к гепараназе];
(iv) ингибиторы действия фактора роста: например, такие ингибиторы включают антитела к факторам роста и антитела к рецептору фактора роста (например, анти-erbB2 антитело трастузумаб [Herceptin™], анти-EGFR антитело панитумумаб [Вектибикс®], анти-erbB1 антитело цетуксимаб [Erbitux®, C225] и антитела к фактору роста или к рецептору фактора роста, описанные Stern et al. Critical reviews in oncology/haematology, 2005, Vol. 54, pp 11-29); такие ингибиторы включают также ингибиторы тирозинкиназы, например, ингибиторы семейства эпидермального фактора роста (например, ингибиторы семейства EGFR тирозинкиназы, такие как N-(3-хлор-4-фторфенил)-7-метокси-6-(3-морфолинопропокси)хиназолин-4-амин (гефитиниб, ZD1839), N-(3-этинилфенил)-6,7-бис(2-метоксиэтокси)хиназолин-4-амин (эрлотиниб, OSI-774) и 6-акриламидо-N-(3-хлор-4-фторфенил)-7-(3- морфолинопропокси)хиназолин-4-амин (Cl 1033), ингибиторы erbB2 тирозинкиназы, такие как лапатиниб); ингибиторы семейства фактора роста гепатоцитов; ингибиторы семейства инсулиноподобных факторов роста; ингибиторы семейства тромбоцитарного фактора роста, такие как иматиниб и/или нилотиниб (AMN107); ингибиторы серин/треонин киназ (например, ингибиторы сигнального пути Ras/Raf, такие как ингибиторы фарнезил трансферазы, например, сорафениб (BAY 43-9006), типифарниб (R1 15777) и лонафарниб (SCH66336)), ингибиторы клеточных сигнальных путей через MEK и/или AKT киназы, ингибиторы c-kit, ингибиторы abl киназы, ингибиторы киназы PI3, ингибиторы киназы Plt3, ингибиторы CSF-1R киназы, ингибиторы рецептора IGF киназы (инсулиноподобный фактор роста); ингибиторы Aurora киназы (например AZD1152, PH739358, VX-680, MLN8054, R763, MP235, MP529, VX-528 и AX39459) и ингибиторы циклин-зависимых киназ, такие как CDK2 и/или CDK4 ингибиторов;
(v) антиангиогенные агенты, такие как те, которые ингибируют действие фактора роста эндотелия сосудов [например, антитело к фактору роста анти-сосудистых эндотелиальных клеток бевацизумаб (Avastin™) и, например, ингибитор VEGF рецептора тирозин-киназы, такой как вандетаниб (ZD6474), ваталаниб (PTK787), сунитиниб (SU11248), акситиниб (AG-013736), пазопаниб (GW 786034) и 4-(4-фтор-2-метилиндол-5-илокси)-6-метокси-7-(3-пирролидин-1-илпропокси)хиназолин (AZD2171; пример 240 в WO 00/47212), соединения, такие как те, которые описаны в международных патентных заявках WO 97/22596, WO 97/30035, WO 97/32856 и WO 98/13354, и соединения, которые действуют по другим механизмам (например, линомид, ингибиторы действия интегрина ανβ3 и ангиостатин)];
(vi) вещества, которые повреждают сосуды, такие как комбретастатин А4 и соединения, описанные в международных патентных заявках WO 99/02166, WO 00/40529, WO 00/41669, WO 01 /92224, WO 02/04434 и WO 02/08213;
(vii) антагонист рецептора эндотелина, например, зиботентан (ZD4054) или атразентан;
(viii) антисмысловая терапия, например, такая, которая направлена на мишени, перечисленные выше, такие как ISIS 2503, антисмысловая терапия на основе гена ras;
(ix) способы генной терапии, включая, например, способы замены аберрантных генов с использованием соединений по изобретению в комбинации с онколитическими аденовирусами, такие как способы аберрации р53 или аберрации BRCA1 или BRCA2, GDEPT (пролекарственная терапия, направленная на ген фермента), такие как способы с использованием деаминазы цитозина, тимидинкиназы или бактериальной нитроредуктазы и способы повышения устойчивости пациента к химиотерапии или радиотерапии, такие как генная терапия резистентности ко многим лекарственным средствам; и
(x) способы иммунотерапии, включая, например, способы повышения иммуногенности опухолевых клеток пациента в условиях ex vivo и in vivo, такие как трансфекция цитокинами, такими как интерлейкин 2, интерлейкин 4, или фактор стимуляции колоний гранулоцитов-макрофагов, способы снижения Т-клеточной анергии, способы с использованием трансфектированных иммунных клеток, таких как цитокин-трансфектированные дендритные клетки, способы с использованием цитокин-трансфектированных линий опухолевых клеток и способы с использованием анти-идиотипичных антител.
[00104] Такое совместное/комбинированное лечение может быть достигнуто путем одновременного, последовательного или раздельного дозирования отдельных компонентов лечения. В такой комбинации продуктов используются соединения по настоящему изобретению в интервалах дозирования, описанных выше, и другой фармацевтически активный агент в пределах утвержденного диапазона доз.
[00105] В соответствии с конкретным аспектом изобретение относится к комбинации, подходящей для применения при лечении заболевания или состояния, в которые вовлечена активность протеинкиназы, как определено в настоящем документе (например, злокачественное новообразование), содержащей соединение по изобретению, как определено ранее, или его фармацевтически приемлемую соль или сольват и другой терапевтический агент (например, в качестве противоопухолевого агента).
[00106] В соответствии с этим аспектом изобретение относится к комбинации, подходящей для применения при лечении злокачественного новообразования (например, злокачественного новообразования, включая солидную опухоль), содержащей соединение по изобретению, как определено ранее, или его фармацевтически приемлемую соль или сольват и любой анти-опухолевый агент, перечисленный в пунктах (i)-(ix) выше.
[00107] В соответствии со следующим аспектом изобретение относится к соединению по изобретению или его фармацевтически приемлемой соли или сольвату в комбинации с противоопухолевым агентом, выбранным из перечисленных здесь в пунктах (i)-(ix) выше.
[00108] В настоящем документе термин «комбинация» используется, как следует понимать, в отношении одновременного, раздельного или последовательного введения. В одном аспекте изобретения «комбинация» означает одновременное введение. В другом аспекте изобретения «комбинация» относится к раздельному введению. В следующем аспекте изобретения термин «комбинация» относится к последовательному введению. Когда введение является последовательным или раздельным, задержка введения второго компонента не должна быть такой, чтобы был утрачен положительный эффект комбинации.
[00109] В соответствии с еще одним аспектом изобретение относится к фармацевтической композиции, которая содержит соединение по изобретению или его фармацевтически приемлемую соль или сольват в комбинации с одним или несколькими дополнительными терапевтическими агентами (например, противоопухолевый агент, выбранный из перечисленных в пунктах (i)-(ix), приведенных выше), в ассоциации с фармацевтически приемлемым разбавителем или носителем.
[00110] Соединения по настоящему изобретению, как ожидается, будут особенно полезны в качестве части комбинированной терапии с существующим стандартом лечения при лечении пожилых больных (т.е. пациентов старше 60 лет), поскольку для таких больных может быть успешным ингибирование Aurora киназы (независимо от их FLT3 статуса).
[00111] Соединения по настоящему изобретению также, как ожидается, будут особенно полезны в качестве части комбинированной терапии с существующим стандартом лечения для лечения детей, страдающих лейкемией (например, AML) или нейробластомой.
ПРИМЕРЫ
КРАТКОЕ ОПИСАНИЕ ФИГУР
[00112] На фигуре 1 показано действие соединения по примеру 1 на ксенотрансплантаты опухоли MV4-11 молочной железы человека у мышей без вилочковой железы: (A) относительные объемы опухоли ± SEM. (B) Масса тела мыши.
[00113] На фигуре 2 показано: (A) Общие концентрации препарата в плазме и опухоли и концентрации препарата в свободной плазме по результатам дозирования 50 и 100 мг/кг, образцы отбирали через 2 часа после конечного дозирования. (B) Соединение по примеру 1 ингибирует фосфорилирование гистона H-3 по S10 и фосфорилирование STAT5 по Y694 в трансплантатах опухолей MV4-11 человека (4-дневное исследование). Образцы опухолей получали через 2 часа после последнего дозирования. В качестве внутреннего контроля использовали суммарный гистон H3, суммарный Stat5 и GAPDH.
СИНТЕЗ СОЕДИНЕНИЙ
Примеры 1-3
Общие материалы и способы
[00114] Коммерчески доступные исходные продукты, реагенты и сухие растворители были использованы в том виде, как получены. Флэш колоночную хроматографию осуществляли с использованием Merck силикагеля 60 (0,025-0,04 мм). Колоночную хроматографию осуществляли на FlashMaster personal unit с использованием силикагелевых колонок isolute Flash или системы очищения Biotage SP1 с использованием силикагелевых картриджей Biotage Flash. Препаративную ТСХ проводили на пластинах Analtech или Merck. Ионообменную хроматографию проводили с использованием кислотных картриджей Isolute Flash SCX-II. 1H ЯМР спектры регистрировали на Bruker Avance-500. Образцы готовили в виде растворов в дейтерированном растворителе и соотносили к соответствующему внутреннему пику не-дейтерированного растворителя или к тетраметилсилану. Химические сдвиги регистрировали в м.д. (δ) относительно тетраметилсилана. ЖХ-МС анализ проводили на Waters LCT с разделительным модулем Waters Alliance 2795 и двухволновым детектором поглощения Waters 2487, соединенным с Waters/Micromass LCT времяпролетным масс спектрометром с источником ESI. Аналитическое разделение проводили при 30°C либо на Merck Chromolith SpeedROD колонке (RP-18e, 50×4,6 мм) с использованием скорости потока 2 мл/мин при 3,5 минутном градиентном элюировании с определением при 254 нм, либо на Merck Purospher STAR колонке (RP-18e, 30×4 мм) с использованием скорости потока 1,5 мл/мин при 3,5 минутном градиентном элюировании с определением при 254 нм. Подвижная фаза состояла из смеси метанола (растворитель A) и воды (растворитель B), оба содержащие муравьиную кислоту по 0,1%. Градиентное элюирование было следующим: от 1:9 (A/B) до 9:1 (A B) в течение 2,25 мин, 9:1 (A/B) в течение 0,75 мин, а затем возвращение обратно 1:9 (A/B) в течение более 0,3 мин, наконец, 1:9 (B) в течение 0,2 мин.
[00115] LC-HRMS анализ осуществляли на HPLC серии Agilent 1200 и диодно-матричном детекторе в сочетании с 6520 квадрупольным времяпролетным масс спектрометром с двойной фокусировкой источника APCl/ESI. Аналитическое разделение осуществляли при 30°C на Merck Purospher STAR колонке (RP-18e, 30×4 мм) с использованием скорости потока 1,5 мл/мин с 4-минутным градиентным элюированием с определением при 254 нм. Подвижная фаза состояла из смеси метанола (растворитель A) и воды (растворитель B), оба содержащие муравьиную кислоту по 0,1%. Градиентное элюирование было следующим: от 1:9 (A/B) до 9:1 (A/B) в течение 2,5 мин, 9:1 (A/B) в течение 1 мин, а затем возвращение обратно 1:9 (A/B) в течение более 0,3 мин, наконец, 1:9 (A/B) в течение 0,2 мин. Следующие ссылочные массы были использованы для HRMS анализа: кофеин [M+H]+ 195.087652; (гексакис(1H,1H,3H-тетрафторпентокси)фосфазен [M+H]+ 922.009798) и гексакис (2,2-дифторэтокси)фосфазен [M+H]+ 622.02896 или резерпин [M+H]+ 609.280657.
Пример 1 - Получение 6-хлор-7-(4-(4-хлорбензил)пиперазин-1-ил)-2-(1,3-диметил-1H-пиразол-4-ил)-3H-имидазо[4,5-b]пиридина
4-Хлор-3-нитропиридин-2-амин40

[00116] В 100 миллиметровую круглодонную колбу, содержащую 2-амино-4-хлорпиридин (0,480 г, 3,75 ммоль), охлаждаемую на ледяной бане, добавляли концентрированную серную кислоту (5,4 г). Реакционную смесь перемешивали в течение 5 мин и затем добавляли по каплям азотную кислоту (70%; 0,36 г). Реакционную смесь перемешивали при 0°C в течение 10 мин, затем нагревали до 55°C и перемешивали при этой температуре в течение 1 часа. Охлаждали до комнатной температуры и разбавляли ледяной водой. Аккуратно доводили pH до ~7,5 с помощью 10%-ного водного раствора NaOH, после чего образовывался желтый осадок. Его отфильтровывали, промывали водой и сушили в вакууме над P2O5. Продукт очищали с помощью колоночной хроматографии на силикагеле (элюирование дихлорметаном) с целью получения при элюировании 4-хлор-3-нитропиридин-2-амина в виде твердого вещества желтого цвета (0,210 г, 32%), 1H-ЯМР (500 МГц, ДМСО-d6) 6,87 (д, J=5,2 Гц, 1H, пиридин C-H), 7,21 (с, 2H, NH2), 8,11 (д, J=5,2 Гц, 1H, пиридин C-H).
[00117] 4-Хлор-5-нитропиридин-2-амин (0,080 г, 12%): 1H-ЯМР (500 МГц, ДМСО-d6) 6,58 (с, 1H, пиридин C-H), 7,58 (с, 2H, NH2), 8,79 (с, 1H, пиридин C-H).
4,5-Дихлор-3-нитропиридин-2-амин

[00118] 4-Хлор-3-нитропиридин-2-амин (0,10 г, 0,58 ммоль) растворяли в сухом ацетонитриле (20 мл). К перемешиваемому раствору затем добавляли N-хлорсукцинимид (0,094 г, 0,70 ммоль) и реакционную смесь нагревали при 80°C в течение 1 часа. Летучие компоненты удаляли в вакууме и остаток очищали путем колоночной хроматографии на силикагеле (элюирование дихлорметаном) с получением указанного в заголовке соединения в виде порошка бледно-коричневого цвета (0,125 г, 85%). 1H-ЯМР (500 МГц, ДМСО-d6) 7,35 (с, 2H, NH2), 8,36 (с, 1H, 6-H).
5-Хлор-4-(4-(4-хлорбензил)пиперазин-1-ил)-3-нитропиридин-2-амин

[00119] К смеси 2-амино-4,5-дихлор-3-нитропиридина (0,152 г, 0,73 ммоль) и изопропанола (22 мл) добавляли 1-(4-хлорбензил)пиперазин (0,165 г, 0,78 ммоль), затем диизопропилэтиламин (0,17 мл, 0,97 ммоль). Реакционную смесь нагревали при 45°C в течение 18 ч, затем дали охладиться до комнатной температуры и разбавляли изопропанолом (5 мл). Осадок собирали фильтрацией, промывали изопропанолом и диэтиловым эфиром. Указанное в заголовке соединение было получено таким образом в виде твердого вещества желтого цвета (0,215 г, 77%); 1H-ЯМР (500 МГц, ДМСО-d6) 2,48 (ушир. с, перекрываемый пиком ДМСО, 4Н, пиперазин C-H), 3,06 (ушир. т, J=4,3 Гц, 4Н, пиперазин C-H), 3,52 (с, 2H, NCH2C6H4Cl), 6,95 (с, 2H, NH2), 7,35 (д, J=8,5 Гц, 2Н) и 7,38 (д, J=8,5 Гц, 2Н) (3,5-ArH и 2,6-ArH), 8,06 (с, 1H, 6-H); LC-MS (ESI, m/z): Rt=1,70 мин - 382, 384, 386 [(M+H)+, Cl2 изотопное распределение].
6-Хлор-7-(4-(4-хлорбензил)пиперазин-1-ил)-2-(1,3-диметил-1H-пиразол-4-ил)-3H-имидазо[4,5-b]пиридин
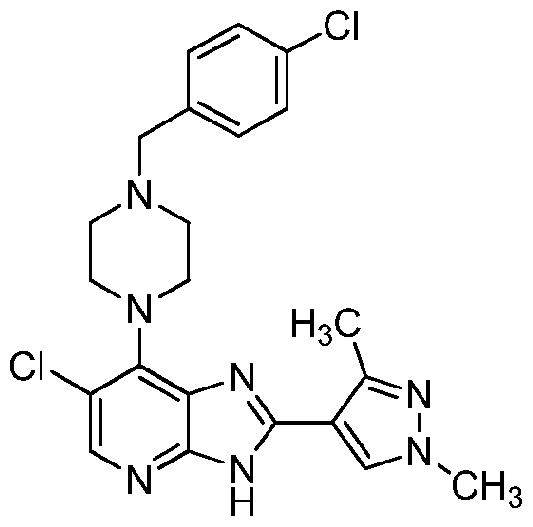
[00120] К смеси 5-хлор-4-(4-(4-хлорбензил)пиперазин-1-ил)-3-нитропиридин-2-амина (0,076 г, 0,20 ммоль) и EtOH (4,0 мл) добавляли 1,3-диметил-1H-пиразол-4-карбальдегид (0,027 г, 0,22 ммоль), затем свежеприготовленный водный раствор Na2S2O4 (1М; 0,85 мл, 0,85 ммоль). Реакционную смесь перемешивали при 80°C в течение 24 ч, затем давали охладиться до комнатной температуры, концентрировали в вакууме и остаток абсорбировали на силикагеле и помещали на 10 г колонку isolute с силикагелем. Элюирование смесью этилацетат/дихлорметан (об./об.; 1:1), а затем смесью 4% метанол в этилацетате/дихлорметан (об./об.; 1:1) давало после растирания в диэтиловом эфире указанное в заголовке соединение в виде твердого вещества белого цвета (0,023 г, 25%).
[00121] 1H-ЯМР (500 МГц, ДМСО-d6) 2,51 (с, перекрываемый пиком растворителя, пиразол 3-CH3), 2,57 (ушир. с, 4H, пиперазин C-H), 3,54 (с, 2H, N-CH2C6H4Cl), 3,68 (ушир. с, 4H, пиперазин C-H), 3,84 (с, 3Н, пиразол N-Me), 7,37 (д, J=8,5 Гц, 2Н) и 7,40 (д, J=8,5 Гц, 2Н) (C6H4Cl), 8,02 (с, 1H), 8,18 (с, 1H) (пиразол 5-H и имидазо[4,5-b]пиридин-5-H), 12,95 (ушир. с, 1H, имидазо[4,5-b]пиридин N-H); LC-MS (ESI, m/z): Rt=1,97 мин - 456, 458, 460 [(M+H)+, Cl2 изотопное распределение].
[00122] HRMS: найдено: 456,1457, рассчитано для C22H24Cl2N7 (М+H)+: 456,1465.
[00123] Это соединение было также получено в больших количествах в диапазоне от 0,80 г до 1,80 г и с выходами в пределах от 54% до 70%. Был использован тот же способ, как описано выше, но в процессе обработки реакционную смесь распределяли между водой и хлороформом. Водный слой экстрагировали хлороформом и этилацетатом и объединенные органические фракции сушили и концентрировали в вакууме. Также вместо EtOH в качестве растворителя был использован ДМСО и в этом случае реакционную смесь перемешивали при 120°C в течение 3 ч.
Пример 2. Получение 3-((4-(6-хлор-2-(1,3-диметил-1H-пиразол-4-ил)-3H-имидазо[4,5-b]пиридин-7-илпиперазин-1-ил)метил)-1,2,4-оксадиазола
трет-Бутил 4-((1,2,4-оксадиазол-3-ил)метил)пиперазин-1-карбоксилат
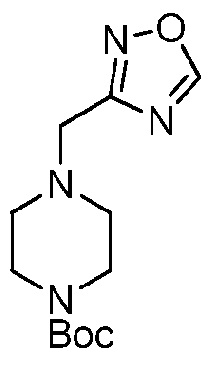
[00124] К раствору Boc-пиперазина (571 мг, 3,07 ммоль) и 3-(хлорметил)-1,2,4-оксадиазола (400 мг, 3,37 ммоль) в CH2Cl2 (30 мл) добавляли триэтиламин (1,70 мл, 12,3 ммоль). Реакционную смесь перемешивали в течение 22 ч при 50°C, затем концентрировали в вакууме с получением сырого маслянистого твердого вещества белого цвета. Очистку осуществляли путем флэш-хроматографии на силикагеле (4×12), элюируя смесью MeOH/CH2Cl2 (5%) с получением указанного в заголовке соединения (555 мг, 67%) в виде твердого вещества белого цвета. 1H-ЯМР (500 МГц, CDCl3) 1,43 (с, 9H, C(CH3)3), 2,52 (приблизительно т, J=4,9 Гц, 4H, CH2), 3,45 (приблизительно т, J=4,9 Гц, 4H, CH2), 3,78 (с, 2H, CH2C-), 8,71 (с, 1H, CHar); LC-MS (ESI, m/z): Rt=1,67 мин - 213 (М - трет-Bu)+, 169 (М - Boc)+.
4-(4-((1,2,4-Оксадиазол-3-ил)метил)пиперазин-1-ил)-5-хлор-3-нитропиридин-2-амин

[00125] К раствору трет-бутил 4-((1,2,4-оксадиазол-3-ил)метил)пиперазин-1-карбоксилата (213 мг, 0,790 ммоль) в CH2Cl2 (18 мл) добавляли ТФУ (1,8 мл, на 23,8 ммоль) и раствор перемешивали при комнатной температуре в течение 1½ ч. Реакционную смесь концентрировали в вакууме, подвергали азеотропной отгонке с толуолом (×2) и сушили в вакуум-эксикаторе (содержащем KOH) в течение ночи с получением масла желтого цвета. Сырое масло растворяли в изо-PrOH (4,4 мл) и добавляли 2-амино-3-нитро-4,5-дихлорпиридин (190 мг, 0,752 ммоль) и DIPEA (520 мкл, 3,00 ммоль). Раствор перемешивали при 50°C в течение 4 ч. После охлаждения выпадал осадок желтого цвета, который отфильтровывали, промывали Et2O, сушили в вакууме с получением указанного в заголовке соединение в виде твердого вещества желтого цвета (165 мг, 0,486, 65%). После этого фильтровали, концентрировали в вакууме с получением 715 мг маслянистого твердого вещества желтого цвета. Очистку осуществляли путем флэш-хроматографии на силикагеле (4×11), элюируя смесью EtOAc/гексан (40-50%) с получением указанного в заголовке соединения (42 мг, 16%) в виде твердого вещества желтого цвета.
[00126] 1H-ЯМР (500 МГц, CDCl3) 2,74 (приблизительно т, J=4,1 Гц, 4H, CH2-), 3,25 (т, J=4,8 Гц, 4H, CH2-), 3,85 (с, 2H, -CH2C-), 5,77 (с, 2H, NH2), 7,99 (с, 1H, CHar), 8,72 (с, 1H, -C(Cl)CH-).
[00127] LC-MS (ESI, m/z): Rt=1,56 мин - 340, 342 [(M+H)+, Cl изотопное распределение].
3-((4-(6-хлор-2-(1,3-диметил-1H-пиразол-4-ил)-3H-имидазо[4,5-b]пиридин-7-ил)пиперазин-1-ил)метил)-1,2,4-оксадиазол

[00128] К раствору 4-(4-((1,2,4-оксадиазол-3-ил)метил)пиперазин-1-ил)-5-хлор-3-нитропиридин-2-амина (50,0 мг, 0,147 ммоль) и 1,3-диметил-1H-пиразол-4-карбальдегида (19,2 мг, 0,155 ммоль) в EtOH (3,4 мл) добавляли 1M Na2S2O4 (0,588 мл, 0,588 ммоль, свежеприготовленный) и раствор нагревали при 80°C и перемешивали в течение 15 ч, при этом оставляя открытым для воздуха. После охлаждения реакционную смесь упаривали в вакууме и сухой остаток помещали на силикагель. Очистку осуществляли путем флэш-хроматографии на силикагеле (2×14), элюируя смесью MeOH/CH2Cl2 (5-7,5%) с получением указанного в заголовке соединения (26 мг, 43%) в виде твердого вещества бледно-желтого цвета.
[00129] 1H-ЯМР (500 МГц, CDCl3) 2,58 (с, 3H, CH3), 2,81 (приблизительно т, J=4,4 Гц, 4H, CH2), 3,82 (приблизительно с, 4H, CH2), 3,85 (с, 3H, NCH3), 3,88 (с, 2H, -CH2-), 7,62 (ушир. с, 1H, CHar), 7,87 (ушир. с, 1H, CHar), 8,74 (с, 1H, CHar), 13,04 (с, 1H, NH);
[00130] LC-MS (ESI, m/z): Rt=1,91 мин - 414, 416 [(M+H)+, Cl изотопное распределение];
[00131] HRMS: найдено: 436,1374, рассчитано для C18H20N9OClNa (M+Na)+: 436,1372.
Пример 3 - Получение 3-((4-(6-хлор-2-(1,3-диметил-1H-пиразол-4-ил)-3H-имидазо[4,5-b]пиридин-7-ил)пиперазин-1-ил)метил)-5-метил-1,2,4-оксадиазола
2-Амино-5-хлор-4-(4-(5-метил-1,2,4-оксадиазол-3-ил)метилпиперазин-1-ил)-3-нитропиридин
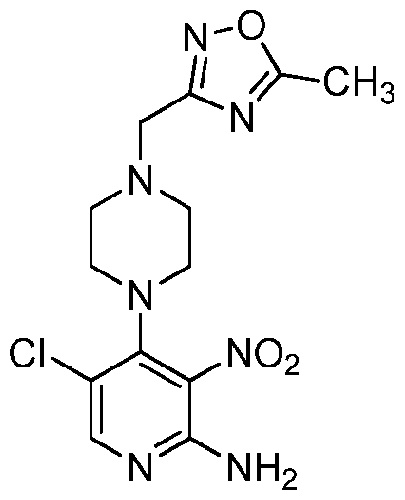
[00132] Гидрохлорид 1-[(5-метил-1,2,4-оксадиазол-3-ил)метил]пиперазина (217 мг, 0,99 ммоль) и 2-амино-4,5-дихлор-3-нитропиридина (208 мг, 1,0 ммоль) перемешивали в 2-пропаноле (5 мл) и добавляли диизопропилэтиламин (523 мкл, 387 мг, 3,0 ммоль). Смесь перемешивали и нагревали при 45°С в течение 23 ч. Реакцию охлаждали и продукт отфильтровывали и промывали 2-пропанолом. Сушка в вакууме давала продукт (246 мг, 69%). 1H-ЯМР (500 МГц, CDCl3,) 2,63 (с, 3H, CH3), 2,77 (ушир. м, 4Н, пиперазин C-H), 3,29 (м, 4Н, пиперазин C-H), 3,76 (с, 2H, CH2), 5,27 (с, 2H, NH2), 8,02 (с, 1H, пиридин 6-H).
[00133] LC-MS (ESI, m/z): Rt=1,66 мин - 354 (M+H)+, изотоп 35Cl.
3-((4-(6-Хлор-2-(1,3-диметил-1H-пиразол-4-ил)-3H-имидазо[4,5-b]пиридин)-7-ил)пиперазин-1-ил)метил)-5-метил-1,2,4-оксадиазол
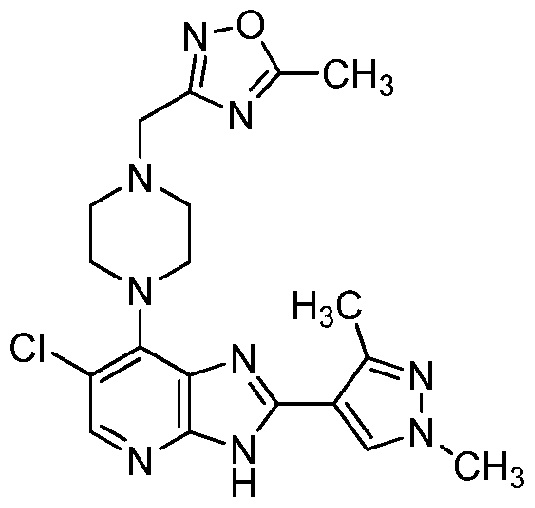
[00134] К раствору 5-хлор-4-(4-((5-метил-1,2,4-оксадиазол-3-ил)метил)пиперазин-1-ил)-3-нитропиридин-2-амина (60,0 мг, 0,170 ммоль) и 1,3-диметил-1H-пиразол-4-карбальдегида (22,2 мг, 0,179 ммоль) в EtOH (3,8 мл) добавляли 1М Na2S2O4 (0,678 мл, 0,678 ммоль, свежеприготовленный) и раствор нагревали при 80°C и перемешивали в течение 16 ч, при этом оставляя открытым для воздуха. После охлаждения реакционную смесь упаривали в вакууме и сухой остаток помещали на силикагель. Очистку осуществляли путем флэш-хроматографии на силикагеле (3×14), элюируя смесью MeOH/CH2Cl2 (5-7,5%), получая указанное в заголовке соединение в виде твердого вещества бледно-желтого цвета. Перекристаллизация в смеси EtOAc/Et2O давала указанное в заголовке соединение (20 мг, 27%) в виде твердого вещества белого цвета. Фильтрат концентрировали в вакууме, получая дополнительное количество указанного в заголовке соединения (12 мг, 16%) в виде твердого вещества бледно-желтого цвета. 1H-ЯМР (500 МГц, CDCl3) 2,60 (с, 3H, CH3), 2,62 (с, 3H, CH3), 2,81 (приблизительно т, J=4,5 Гц, 4H, CH2), 3,76 (с, 2H, -CH2-), 3,87 (приблизительно с, 4H, CH2), 3,90 (с, 3H, NCH3), 7,77 (ушир. с, 1H, CHar), 7,96 (ушир. с, 1H, CHar), 12,18 (с, 1H, NH);
[00135] LC-MS (ESI, m/z): Rt = 1,95 мин - 428, 430 [(M+H)+Cl изотопное распределение];
[00136] HRMS: найдено: 450,1527, рассчитано для C19H22N9OClNa (M+Na)+: 450,1528.
ОЦЕНКА СОЕДИНЕНИЙ ПО ПРИМЕРАМ 1-3
Общие материалы и способы
[00137] Анализ Aurora киназы: значения IC50 Aurora киназы были определены, как описано ранее.26,36
[00138] Анализ жизнеспособности клеток: значения GI50 (концентрация, на 50% ингибирующая рост клеток) определяли, как описано ранее.26,36
Определение клеточных значений IC50 соединения по примеру 1 для ингибирования Aurora A и Aurora B:
[00139] Myc-тегированную Aurora трансфицировали в клетки Hela с использованием Lipofectamine LTX в 24-луночных планшетах и через 24 часа после трансфекции клетки обрабатывали различными концентрациями соединения по примеру 1 в течение 2 часов. Клетки затем лизировали в буфере 2X LDS sample buffer. Белки из разных образцов разделяли с помощью 4-12% Bis-Tris NuPage (Invitrogen) гелей и анализировали с помощью вестерн-блоттинга с использованием антител к P-гистонам H3 (S10) и P-Aurora A (T288). Полосы P-гистона H3 и P-Aurora A были количественно оценены с использованием программного обеспечения Image J software и значения IC50 были рассчитаны с использованием Graphpad Prism.
Микросомальная стабильность мышиной печени:
[00140] Соединения (10 мкМ) инкубировали с белком микросом печени самцов мышей CD1 (1 мг⋅мл-1) в присутствии NADPH (1 мм), UDPGA (2,5 мМ) и MgCl2 (3 мМ) в фосфатно-солевом буфере (10 мМ) при 37°C. Инкубирование осуществляли в течение 0 и 30 минут. Контрольное инкубирование проводили при отсутствии в инкубационной среде NADPH и UDPGA. Процентный состав оставшихся соединений определяли после анализа с помощью LCMS.
Микросомальная стабильность печени человека:
[00141] Соединения (10 мкМ) инкубировали с белком микросом печени человека различного пола (1 мг⋅мл-1) в присутствии NADPH (1 мм), UDPGA (2,5 мМ) и MgCl2 (3 мМ) в фосфатно-солевом буфере (10 мМ) при 37°C. Инкубирование осуществляли в течение 0 и 30 минут. Контрольное инкубирование проводили при отсутствии в инкубационной среде NADPH и UDPGA. Процентный состав оставшихся соединений определяли после анализа с помощью LCMS.
[00142] Ингибирование hERG: Все проценты ингибирования hERG при концентрации соединения в 10 мкМ определяли с помощью Millipore путем клеточного электрофизиологического анализа высокой пропускной способности по ингибированию хвостового тока hERG,41 и значения представляли как среднее из нескольких определений. Отрицательный контроль с наполнителем 0,3% водным ДМСО давал ингибирование в 7-16%. Положительный контроль с цисапридом (1 мкМ) давал ингибирование в 96-104%. Все значения IC50 hERG определялись с помощью Millipore,41 и IC50 hERG для примера 1 также определяли с помощью Cyprotex pic измерения хвостового тока hERG путем клеточной фиксации потенциала.42
[00143] Физико-химические свойства: Измерения LogD и pKa проводили с помощью Pharmorphix® Solid State Services, Member of the Sigma-Aldrich Group, Cambridge, UK.
[00144] Анализ селективности киназ: Анализ киназ с использованием метода KINOMEScan™ и определение Kd осуществляли с помощью KINOMEscan, a Division of DiscoveRx Corporation, San Diego, California, USA; www.kinomescan.com.
[00145] in vivo полная PK (соединение по примеру 1): Мышам (самки Balb/C) вводили перорально или внутривенно соединение по примеру 1 (5 мг/кг-1) в 10% ДМСО, 5% Tween 20 в физиологическом растворе. После введения мышей умерщвляли на 5, 15, 30 минутах и 1, 2, 4, 6 и 24 ч. Кровь удаляли путем пункции сердца и центрифугировали для получения образцов плазмы. Образцы плазмы (100 мкл) добавляли к аналитическому внутреннему стандарту (Olomoucine; IS), затем высаживали белок с помощью 300 мкл метанола. После центрифугирования (1200×g, 30 мин, 4°C) полученный супернатант анализировали на содержание соединения по примеру 1 с помощью LCMS, используя обращенно-фазовую аналитическую колонку Acquity UPLC C18 (Waters, 50×2,1 мм) и режим регистрации положительных ионов ESI MRM методом жидкостной хроматографии системы Agilent 1200 в сочетании с масс-спектрометром 6410 (Agilent Ltd.) с тройным квадруполем.
[00146] Исследования действия на ксенотрансплантат опухоли человека: Способы с участием животных были проведены в рамках руководящих принципов, установленных институтом The Institute of Cancer Research's Animal Ethics Committee и в соответствии с национальными рекомендациями: Workman P, Aboagye EO, Balkwill F, Balmain A, Bruder G, Chaplin DJ, Double JA, Everitt J, Farningham D, Glennie MJ, Kelland LR, Robinson V, Stratford IJ, Tozer GM, Watson S, Wedge SR, Eccles SA. Guidelines for the welfare and use of animals in cancer research. Brit J Cancer 102: 1555-1577, 2010.
[00147] Самкам бестимусных мышей CrTacNCr-Fox1 (nu) имплантировали подкожно по 107 лейкозных клеток FL73-ITD-MV4-11. Когда ксенотрансплантаты хорошо приживались (что означало, что через 10 дней после имплантации объемы опухолей были не менее 100 мм3), животных обрабатывали либо наполнителем (10% ДМСО, 20% ПЭГ-400, 5% Tween 80 и 65% воды), либо соединением по примеру 1 путем введения перорально в два приема, 50 мг/кг и 100 мг/кг (n=5 в каждой группе). Дозирование осуществляли два раза в день в течение 7 дней и один раз в день в течение еще 4 дней.
[00148] Исследование PK/PD: 4-дневное исследование PK/PD осуществляли путем перорального введения носителя, как описано выше, или 50 мг/кг и 100 мг/кг соединения по примеру 1 два раза в день бестимусным мышам с хорошо приживленными ксенотрансплантатами MV4-11 (1-7 дней после имплантации). Образцы плазмы и опухолей отбирали через 2 ч и 6 ч после последнего дозирования.
Результаты
Активность Aurora киназы, клеточная активность, микросомальная стабильность, ингибирование hERG и физикохимические свойства
[00149] Активность соединений по примерам в отношении Aurora A (биохимический анализ) на основе клеток GI50 в SW620 клетках и hERG показана в таблице 1 вместе с данными, касающихся микросомальной стабильности этих соединений и их соответствующих значений clogP.
GI50 (мкМ)
a Результаты представляют собой средние значения для образцов при трехкратном повторении.
b MLM/HLM: Процент метаболизированного исходного соединения после 30 мин инкубации.
c LogD7,4=3,84 (экспериментально определенное значение).
н.о. = не определено.
[00150] Соединение по примеру 1 показало меньшую ингибирующую активность в отношении hERG по сравнению с соединениями по примерам 56 и 57 в WO 2007/072017.
[00151] Основываясь на этих результатах соединение по примеру 1 было выбрано для дальнейшего исследования in vitro и in vivo характеристик.
Селективность киназы
[00152] Селективность киназы оценивали путем анализа соединения по примеру 1 на панели 442-киназы (386 не-мутантных киназ) при концентрации 1 мкM с использованием методики KINOMEScan™.28 Оценка селективности S(10), которую рассчитывали путем деления количества не-мутантных киназ, для которых в анализе наблюдалась конкуренция >90% (это рассчитывалось как <10% от контроля) с помощью общего количества не-мутантных киназ, была определена как 0,057, т.е. 22 хита из 386 не-мутантных исследуемых киназ. Aurora-A, -B и -C были сильно ингибированы с % контрольными значениями, определяемыми как 3,4%, 1% и 16% соответственно. Этот первичный скрининг также выявил более 94% конкуренцию для FLT3 киназы и FLT3 мутантов, включая FLT3-ITD, FLT3(D835Y) и FLT3(D835H).
[00153] Ингибирующая FLT3 и Aurora активность соединения по примеру 1 были впоследствии подтверждены путем определения значения Kd (метод KINOMEScan™), как показано в таблице 2.
Значения Kd для соединения по примеру 1
[00154] Соединения по примерам 2 и 3 также были сильнодействующими ингибиторами FLT3 и FLT3-ITD. Значения Kd соединения по примеру 2 против FLT3 и FLT3-ITD были определены как 4,4 нм и 14 нм соответственно. Аналогично значения Kd соединения по примеру 3 против FLT3 и FLT3-ITD были определены как 5,6 нм и 26 нм соответственно.
[00155] Взятые вместе, эти данные показывают, что соединение по примеру 1 является мощным двойной ингибитором FLT3 и Aurora киназы с небольшим отклонением в активности киназ среди кином.
Оценка клеточного анализа
[00156] В соответствии с двойной ингибирующей FLT3/Aurora активностью соединение по примеру 1 показало антипролиферативное действие относительно клеточных линий опухолей человека, включая HCT116 карциному толстой кишки человека (GI50=0,300 мкМ), и клеточные линии клеток AML человека, экспрессирующие FLT3-ITD MOLM-13 (GI50=0,104 мкМ) и MV4-11 (GI50=0,291 мкМ). В клетках Hela соединение по примеру 1 ингибировало как клеточные Aurora-A, так и Aurora-B при значениях IC50 0,030 мкМ и 0,148 мкМ соответственно. В этих клетках на основе анализов снижение фосфорилирования H3 по S10 было использовано в качестве биомаркера для ингибирования Aurora-B и аутофосфорилирование Aurora-A по T288 в качестве биомаркера для ингибирования Aurora-A.29
In Vivo PK
[00157] Результаты in vivo PK для соединения по примеру 1 на мышах приведены в таблице 3. Оно является перорально высоко биодоступным соединением (F=100%) с коэффициентом очищения, определенным как 0,058 л/ч (~48 мл/мин/кг) и Vd как 0,066 л (~3,3 л/кг).
Соединение по примеру 1: Связывание с белками плазмы мышей и параметры PK
(внутривенное дозирование: 5 мг/кг, пероральное дозирование:
5 мг/кг)
Модель ксенотрансплантата AML
[00158] Активность соединения по примеру 1 на модели ксенотрансплантата человека показана на фигуре 1.
[00159] Обращаясь к фиг. 1, можно видеть, что соединение по примеру 1 сильно ингибирует рост трансплантатов опухоли MV4-11 человека в зависимой от дозы образом с не наблюдаемой токсичностью, что определено по потере массы тела. Когда терапия прекращалась через 11 дней, опухоли были неопределяемыми у мышей, получавших 100 мг/кг режим дозирования соединения по примеру 1 и снижалась до 42% от первоначального объема у мышей, получавших 50 мг/кг режим дозирования. Контрольных мышей забивали на 18 день от начала терапии, когда средний объем опухоли увеличивался более чем на 500%. Напротив, отдельные мыши было забиты, когда опухоли достигали этой стадии, следующим образом: дни 28 и 31 по 50 мг/кг и дни 46 дней и 56 по 100 мг/кг. У трех из 5 мышей в каждой группе обработки (60%) не развивались прогрессивно растущие опухоли, в этот момент исследование прекращали на день 60.
[00160] В результате этого мощного ингибирующего действия in vivo опухоли, подвергаемые обработке, были слишком малы, чтобы обеспечить материал для фармакокинетического/фармакодинамического анализа. Таким образом, было осуществлено повторное 4-дневное PK/PD исследование путем перорального введения 50 мг/кг и 100 мг/кг соединения по примеру 1 два раза в день. Фармакодинамический анализ показал четкое ингибирование фосфорилирования гистона H-3 и ингибирование фосфорилирования STAT5, который является нижестоящей мишенью FLT3 (фигура 2). Кроме того, концентрации препарата в свободной плазме в образцах, полученных 2 ч спустя после последнего дозирования, были определены при 222 нм и 488 нм для 50 мг/кг и 100 мг/кг режимов дозирования соответственно (фигура 2). Концентрация препарата в свободной плазме явно выше значения Kd соединения по примеру 1 против релевантных киназ, т.е. Aurora-A (Kd=7,5 нм), Aurora-B (Kd=48 нм), FLT3 (Kd=6,2 нм), FLT3-ITD (Kd=38 нм). Данные выводы показывают, что соединение по примеру 1 значительно ингибирует рост FLT3-ITD в позитивной модели ксенотрансплантата человека in vivo, с модуляцией биомаркера и воздействием свободного лекарственного средства в соответствии с двойным поражением цели FLT3 и Aurora киназы.
Ингибирование изоформ цитохрома Р450
Материалы и способы
[00161] В данном исследовании были использованы два сравнительных соединения, а именно 6-бром-2-(1-метил-1H-пиразол-4-ил)-7-(4-(пиридин-3-илметил)пиперазин-1-ил)-3H-имидазо[4,5-b]пиридин (сравнительный препарат 1, пример 56, WO 2007/072017) и 6-бром-7-(4-(пиридин-3-илметил)пиперазин-1-ил)-2-(1,3,5-триметил-1H-пиразол-4-ил)-3H-имидазо[4,5-b]пиридин (сравнительный препарат 2, пример 57, WO 2007/072017).
[00162] Сравнительные соединения и соединения по примерам 1-3 выше инкубировали с микросомами печени человека (0,5 мг⋅мл-1) при 10 мкМ и 50 мкМ.
[00163] Ингибирование изоферментов CYP определяли с использованием смеси маркерных субстратов (таблица 4). Образцы инкубировали в течение 10 минут, затем белки высаживали метанолом. Метаболиты субстратов в каждом образце определяли с помощью LC/MS/MS с использованием обращенно-фазовой жидкостной хроматографии и режима регистрации положительных ионов ESI с мониторингом множественных реакций (MRM).
Концентрации маркерных субстратов изоферментов CYP и определенные метаболиты
Результаты
Оцененные значения IC50 по ингибированию изоферментов человека CYP с помощью исследуемых соединений
[00164] Примеры 1-3 не показали какого-либо существенного ингибирования изоферментов CYP (таблица 5), оцененные значения IC50 были выше, чем 10 мкМ.
[00165] Никакие соединения не показали значительного ингибирования CYP1A2, CYP2A6, CYP2C9 или CYP2C19. Оба сравнительных соединения показали значительное ингибирование CYP3A4 с приблизительными значениями IC50, составляющими меньше 1 мкМ, и препарат сравнения 1 также ингибировал CYP2D6. Соединения по настоящему изобретению, таким образом, обладали значительно уменьшенным ингибированием CYP3A4 по сравнению с обоими соединениями сравнения.
Ссылки
1. Carmena, M et al.; Nat. Rev. Mol. Cell Biol. 2003, 4, 842-854.
2. Ducat D. et al.; Exp. Cell Res. 2004, 301, 60-67.
3. Marumoto, T. et al.; Nat. Rev. Cancer 2005, 5, 42-50.
4. Barr, A. R. et al., J. Cell Sci. 2007, 120, 2987-2996.
5. Bayliss, R et al.; Mol. Cell. 2003, 12, 851-862.
6. Giet, R. et al.; J. Cell Biol. 2001, 152, 669-681.
7. Gassmann, R. et al.; J. Cell Biol. 2004, 166, 179-191.
8. Sessa, F. et al.; Mol. Cell, 2005, 18, 379-391.
9. Bishop, J. D. et al.; J. Biol. Chem. 2002, 277, 27577-27580.
10. Tanaka, T. et al.; Cancer Res. 1999, 59, 2041-2044.
11. Bischoff, J. R. et al.; EMBO J. 1998, 17, 3052-3065.
12. Gritsko, T. M. et al.; Clin. Cancer. Res. 2003, 9, 1420-1426.
13. Reichardt, W. et al.; Oncol Rep. 2003, 10, 1275-1279.
14. Chieffi, P. et al.; J. Endocrinol. 2004, 181, 263-270.
15. Araki, K. et al.; J Neurooncol 2004, 67, 53-64.
16. Sorrentino, R. et al.; J Clin Endocrinol Metab. 2005, 90, 928-935.
17. Kimura, M. et al.; J. Biol. Chem. 1999, 274, 7334-7340.
18. Pollard, J. R. et al.; J. Med. Chem. 2009, 52, 2629-2651.
19. Green, M. R. et al.; Expert Opin. Drug Discov. 2011, 6, 291-307.
20. Cheung, C. H. A. et al.; Expert Opin. Ther. Patents 2011, 21, 857-884.
21. Harrington, E. A. et al.; Nat. Med. 2004, 10, 262-267.
22. Mortlock, A. A. et al.; J. Med. Chem. 2007, 50. 2213-2224.
23. Fancelli, D. et al.; J. Med. Chem. 2006, 49, 7247-7251.
24. Caprinelli. P. et al.; Mol. Cancer Ther. 2007, 6, 3158-3168.
25. Payton, M. et al.; Cancer Res 2010, 70, 9846-9854.
26. Bavetsias, V. et al.; J. Med Chem. 2010, 53, 5213-5228.
27. CLogP был вычислен с использованием ChemBioDraw Ultra 12 by CambridgeSoft (www.cambridgesoft.com).
28. Анализ киназ с использованием метода KINOMEScan™: www.kinomescan.com.
29. Mafredi, M. G. et al.; Proc. Natl. Acad. Sci. USA 2007, 104, 4106-4111.
30. Ikezoe, T. et al.; Mol Cancer Ther 2007, 6, 1851-1857.
31. Ochi. T. et al.; Blood 2009, 113, 66-74.
32. Huang, X.-F. et al.; Blood 2008, 111, 2854-2865.
33. Walsby, E. et al.; Haematologica 2008, 93, 662-669.
34. Meshinchi, S. et al.; Clin Cancer Res 2009, 15, 4263-4269.
35. Meshinchi, S. et al.; Blood 2006, 108, 3654-3661.
36. Chan, F. et al.; Mol Cancer Ther, 2007, 6, 3147-3157.
37. Stirewalt, D. L. et al.; Nat Rev Cancer 2003, 3, 650-665.
38. Kmdler, T. et al.; Blood 2010, 116, 5089-5102.
39. Levis, M. J.; Best Practice & Research Clinical Haematology 2010, 23, 489-494.
40. Bavetsias, V. et al., Bioorg. Med. Chem. Lett. 2007, 17, 6567-6571.
41. Ion Channel Cardiac Profiler; Millipore: Billerica, MA:
http://www.miHipore.com/life sciences/flx4/ld ion
42. hERG Safety Assay; Cyprotex pic, Cheshire, UK; www.cyprotex.com
43. Roden, D. M. N. Engl. J. Med. 2004, 350, 1013-1022.
| название | год | авторы | номер документа |
|---|---|---|---|
| КОМПОЗИЦИИ И СПОСОБЫ, КОТОРЫЕ МОЖНО ИСПОЛЬЗОВАТЬ ДЛЯ ЛЕЧЕНИЯ ПРОЛИФЕРАТИВНЫХ ЗАБОЛЕВАНИЙ | 2015 |
|
RU2711500C2 |
| СОЕДИНЕНИЯ ТИЕНОПИРИДИНОНА | 2019 |
|
RU2827549C2 |
| ХИМИЧЕСКИЕ СОЕДИНЕНИЯ - 759 | 2008 |
|
RU2481348C2 |
| ПРОИЗВОДНЫЕ БИЦИКЛИЧЕСКИХ АМИНОВ В КАЧЕСТВЕ ИНГИБИТОРОВ ТИРОЗИНКИНАЗЫ | 2007 |
|
RU2465275C2 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛА И ИХ ПРИМЕНЕНИЕ | 2020 |
|
RU2805207C1 |
| СОЕДИНЕНИЕ МОЧЕВИНЫ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО МЕДИЦИНСКОЕ ПРИМЕНЕНИЕ | 2017 |
|
RU2741596C2 |
| СОЕДИНЕНИЯ ПИРАЗОЛО[3,4-b]ПИРИДИНА В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗ ТАМ И МЕТ | 2019 |
|
RU2773611C1 |
| ИДЕНТИФИКАЦИЯ МУТАЦИИ LKB1 В КАЧЕСТВЕ ПРОГНОСТИЧЕСКОГО БИОМАРКЕРА ЧУВСТВИТЕЛЬНОСТИ К ИНГИБИТОРАМ TOR-КИНАЗЫ | 2011 |
|
RU2565034C2 |
| ПРОИЗВОДНЫЕ ИМИДАЗО [1,2-b]ПИРИДАЗИНА И ПИРАЗОЛО[1,5-a]ПИРИМИДИНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРА ПРОТЕИНКИНАЗ | 2007 |
|
RU2487875C2 |
| СОЕДИНЕНИЯ И КОМПОЗИЦИИ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕИНКИНАЗ | 2006 |
|
RU2387653C2 |
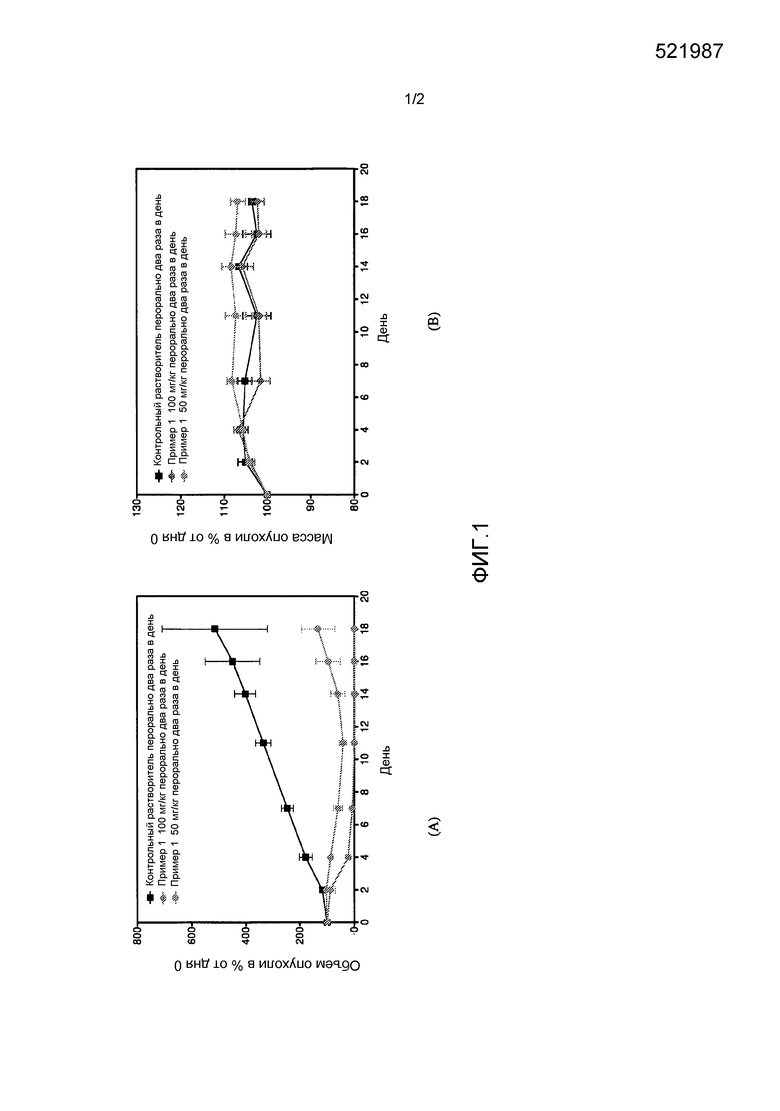
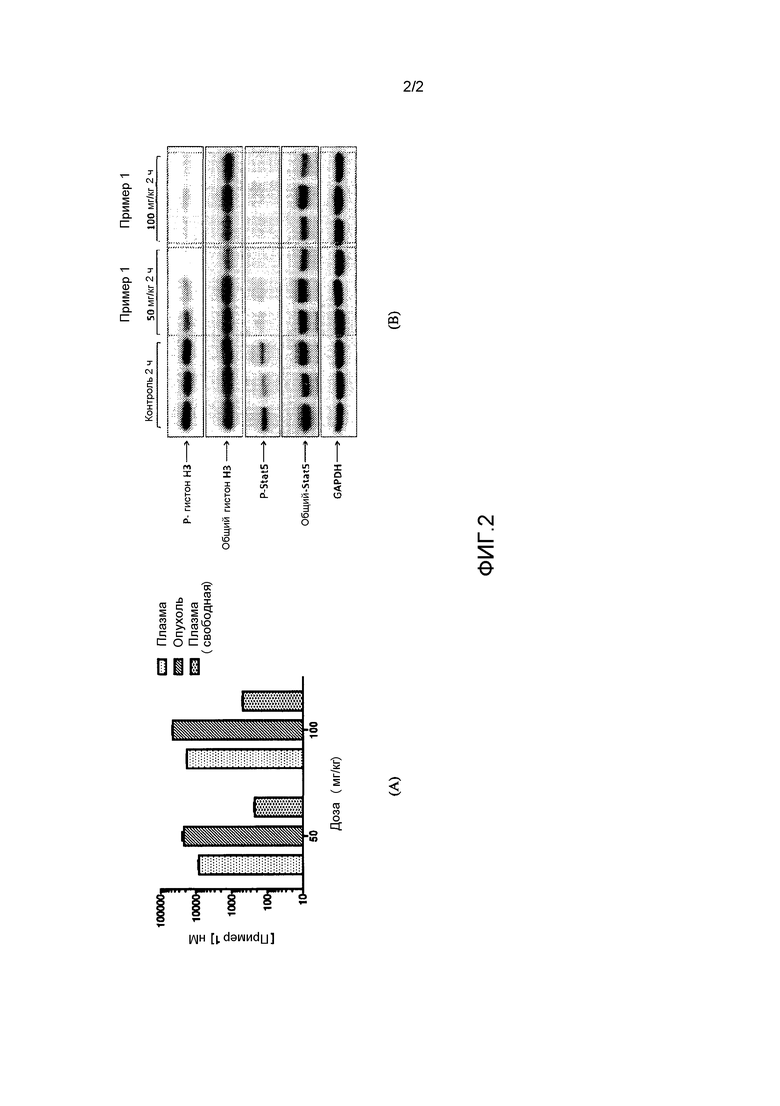
Изобретение относится к области органической химии, а именно к гетероциклическому соединению формулы I или его фармацевтически приемлемой соли, где R2 выбран из формулы II или формулы III, где Ra представляет собой водород или метил. Также изобретение относится к фармацевтической композиции на основе соединения формулы I и способу лечения пролиферативных расстройств и острого миелоидного лейкоза, основанного на использование соединения формулы I. Технический результат: получены новые производные имидазопиридина, обладающие полезными биологическими свойствами. 4 н. и 5 з.п. ф-лы, 2 ил., 5 табл., 3 пр.
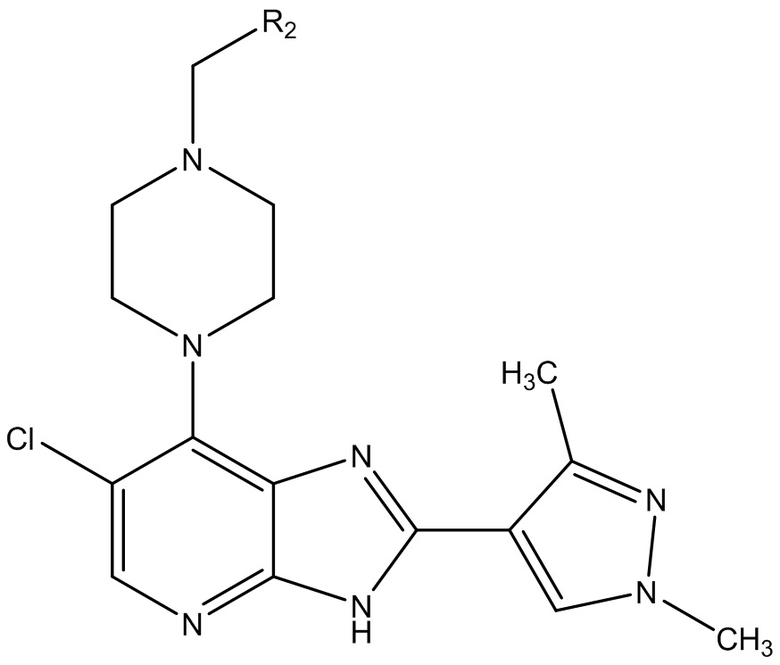 I ,
I ,
1. Соединение формулы I, представляющее собой:

I
где:
R2 выбран из формулы II или формулы III, показанных далее:

где Ra представляет собой водород или метил;
или его фармацевтически приемлемая соль.
2. Соединение по п.1, где R2 имеет формулу II.
3. Соединение по п.1, где R2 имеет формулу III.
4. Соединение по п.1, выбранное из следующих:
6-хлор-7-(4-(4-хлорбензил)пиперазин-1-ил)-2-(1,3-диметил-1H-пиразол-4-ил)-3H-имидазо[4,5-b]пиридин;
3-((4-(6-хлор-2-(1,3-диметил-1H-пиразол-4-ил)-3H-имидазо[4,5-b]пиридин-7-ил)пиперазин-1-ил)метил)-1,2,4-оксадиазол;
3-((4-(6-хлор-2-(1,3-диметил-1H-пиразол-4-ил)-3H-имидазо[4,5-b]пиридин-7-ил)пиперазин-1-ил)метил)-5-метил-1,2,4-оксадиазол;
или его фармацевтически приемлемая соль.
5. Фармацевтическая композиция для лечения пролиферативного расстройства, содержащая соединение по любому из пп.1-4 или его фармацевтически приемлемую соль и один или несколько фармацевтически приемлемых инертных наполнителей.
6. Соединение по п.1 или его фармацевтически приемлемая соль для применения при лечении пролиферативных расстройств, таких как злокачественное новообразование.
7. Соединение по п.6 или его фармацевтически приемлемая соль для применения при лечении острого миелоидного лейкоза.
8. Способ лечения пролиферативных расстройств, таких как злокачественное новообразование, где указанный способ включает введение субъекту при необходимости такого лечения терапевтически эффективного количества соединения по любому из пп.1-4 или его фармацевтически приемлемой соли.
9. Способ лечения острого миелоидного лейкоза, где указанный способ включает введение субъекту при необходимости такого лечения терапевтически эффективного количества соединения по любому из пп.1-4 или его фармацевтически приемлемой соли.
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| Колосоуборка | 1923 |
|
SU2009A1 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ ЯНУС-КИНАЗЫ 3 | 2006 |
|
RU2434013C2 |

