Область техники, к которой относится изобретение
Настоящее изобретение относится к способу получения 4-[5-(пиридин-4-ил)-1H-1,2,4-триазол-3-ил]пиридин-2-карбонитрила, являющегося полезным фармацевтическим средством, а также к новому промежуточному соединению, полезному в производстве указанного соединения.
Уровень техники
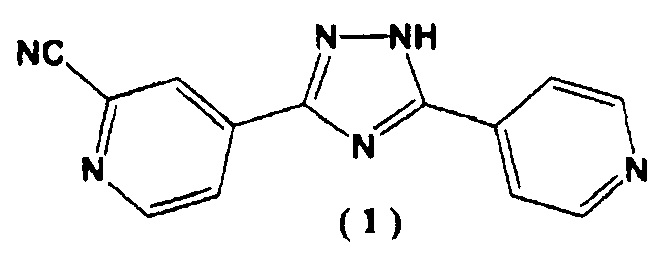
Известно, что соединение (1), 4-[5-(пиридин-4-ил)-1H-1,2,4-триазол-3-ил]пиридин-2-карбонитрил, может выступать в качестве лекарственного средства, которое обладает ксантиноксидазным ингибирующим действием и которое может снижать уровень мочевой кислоты в сыворотке крови (Патентный документ 1)
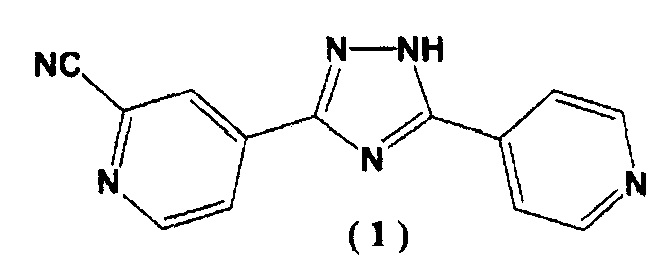 .
.
Известны несколько способов получения вышеуказанного соединения (1). В одном производственном способе метилизоникотинат N-оксид вводился в реакцию Рейссерта-Генце, приводя, тем самым, к образованию метил 2-цианоизоникотината, который превращался в гидразид, а гидразид конденсировался с 4-цианопиридином (Патентный документ 1, Пример 12). В другом производственном способе N-оксид изоникотиновой кислоты превращался в гидразид, в котором цианогруппа вводилась посредством реакции Рейссерта-Генце, и продукт конденсировался с 4-цианопиридином (Патентный документ 1, Пример 39). В альтернативном производственном способе исходный 4-цианопиридин-N-оксид конденсировался с гидразидом изоникотиновой кислоты, тем самым приводя к образованию триазольного кольца, которое затем защищалось (Патентный документ 2) или не защищалось (Патентный документ 3), а цианогруппа вводилась в продукт посредством реакции Рейссерта-Генце, тем самым приводя к образованию соединения (1).
Список цитирования
Патентный документ
Патентный документ 1: WO 2003/064410.
Патентный документ 2: WO 2005/009991.
Патентный документ 3: JP-A-2005-41802.
Сущность изобретения
Задачи, на решение которых направлено изобретение
Способ, раскрытый в Патентном документе 1, хотя и позволяет достигнуть удовлетворительного производственного результата лишь в малом масштабе, имеет проблемы. Например, производство замещенного или незамещенного гидразида 2-цианоизоникотиновой кислоты затруднительно и требует использования растворителя в реакции, подходящего к физическим свойствам соединений-продуктов на каждой стадии. К тому же, общий выход данного способа неудовлетворительный, что проявляет данный способ как непригодный в промышленном производстве. Способ, описанный в Патентном документе 2, включает в себя ряд стадий, связанных с защитой триазольного кольца, делая метод невыгодным в промышленном применении с точки зрения стоимости производства. Метод, раскрытый в Патентном документе 3, не подходит для промышленного применения, поскольку он требует множества стадий по очистке с целью обесцвечивания и удаления примесей.
Таким образом, объектом данного изобретения является промышленно полезный способ получения фармацевтически ценного 4-[5-(пиридин-4-ил)-1H-1,2,4-триазол-3-ил]пиридин-2-карбонитрила.
Способы решения задач
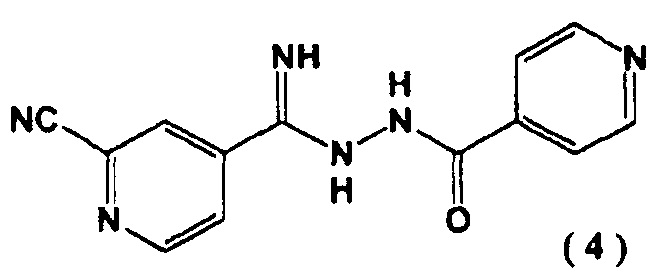
Авторы настоящего изобретения провели обширные исследования метода получения 4-[5-(пиридин-4-ил)-1H-1,2,4-триазол-3-ил]пиридин-2-карбонитрила и нашли, что целевое соединение может быть получено через новое промежуточное соединение, N’-(2-цианопиридин-4-карбонимидоил)гидразид 4-пиридинкарбоновой кислоты, тем самым предоставляя промышленно полезный производственный способ. Настоящее изобретение совершено на основе данного открытия.
Таким образом, данное изобретение предусматривает следующие аспекты [1]-[4]:
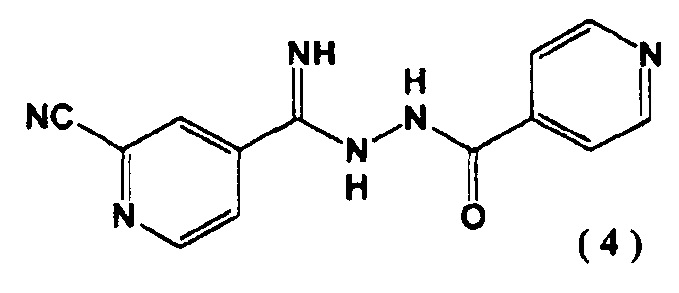
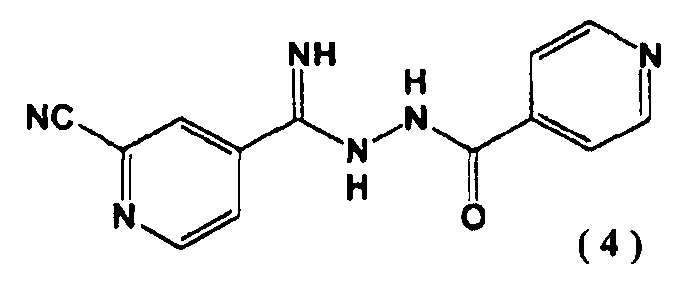
[1] N'-(2-цианопиридин-4-карбонимидоил)гидразид 4-пиридинкарбоновой кислоты, представленный следующей формулой (4):
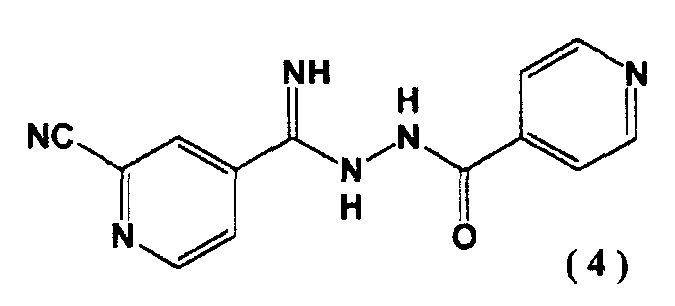 .
.
[2] Способ получения соединения, указанного выше в пункте [1], включающий реакцию соединения, представленного следующей формулой (2):
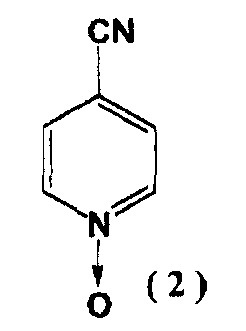
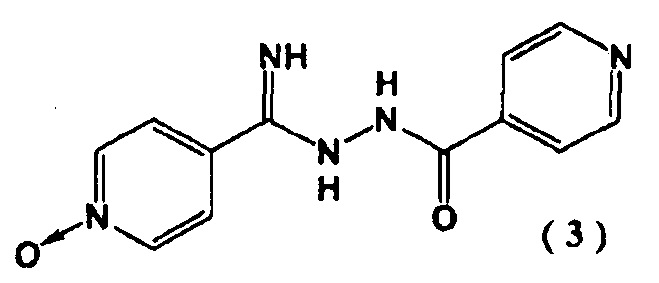
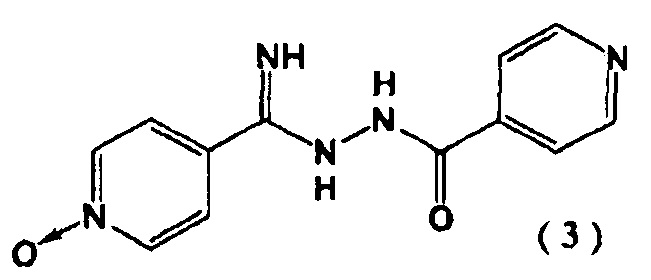
с гидразидом изоникотиновой кислоты в присутствии алкоксида щелочного металла с последующим образованием соединения, представленного следующей формулой (3):
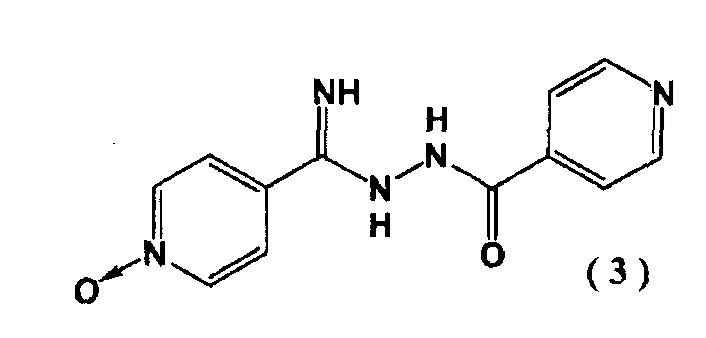
и введение цианогруппы в соединение (3) с помощью цианирующего агента.
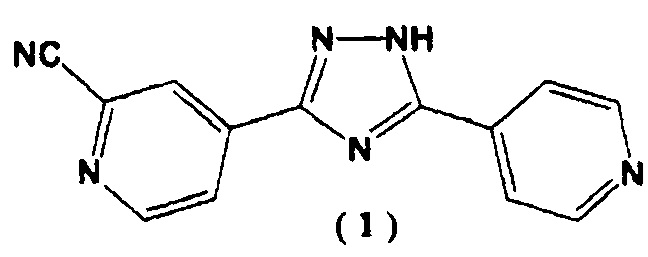
[3] Способ получения 4-[5-(пиридин-4-ил)-1H-1,2,4-триазол-3-ил]пиридин-2-карбонитрила, представленного следующей формулой (1):
,
включающий реакцию соединения, представленного следующей формулой (2):
с гидразидом изоникотиновой кислоты в присутствии алкоксида щелочного металла с последующим образованием соединения, представленного следующей формулой (3):
,
введение цианогруппы в соединение (3) с помощью цианирующего агента с последующим образованием соединения, представленного следующей формулой (4):
и введение соединения (4) в реакцию циклизации в присутствии кислотного катализатора.
[4] Способ получения 4-[5-(пиридин-4-ил)-1H-1,2,4-триазол-3-ил]пиридин-2-карбонитрила, представленного следующей формулой (1):
,
включающий введение соединения, представленного следующей формулой (4):
в реакцию циклизации в присутствии кислотного катализатора.
Результаты изобретения
В соответствии со способом получения в настоящем изобретении, в несколько простых стадий с высоким выходом и низким содержанием побочных продуктов может быть получен 4-[5-(пиридин-4-ил)-1H-1,2,4-триазол-3-ил]пиридин-2-карбонитрил, который может служить полезным лекарственным средством, обладая ксантиноксидазным ингибирующим действием.
Варианты осуществления изобретения
Далее настоящее изобретение будет описано более подробно.
Способ настоящего изобретения показан на следующей реакционной схеме.
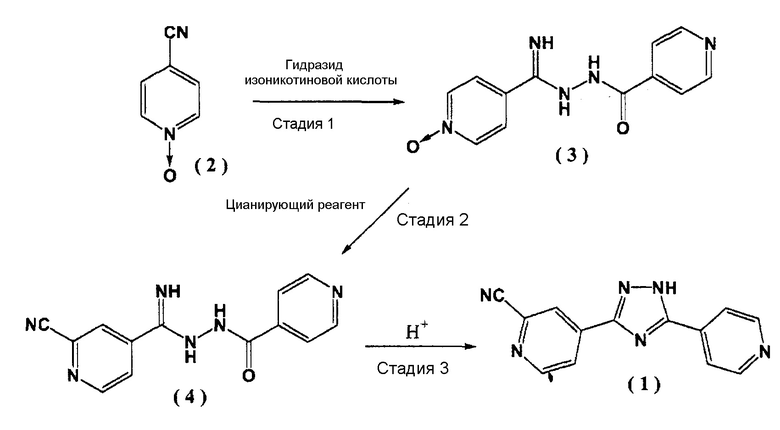 .
.
Первая стадия
На первой стадии, 4-цианопиридин-N-оксид (2) и гидразид изоникотиновой кислоты реагируют в присутствии алкоксида щелочного металла с образованием соединения (3).
Реагенты 4-циано-пиридин-N-оксид (2) и гидразид изоникотиновой кислоты являются известными соединениями и могут быть получены известными способами.
Алкоксидом щелочного металла, используемым в реакции, предпочтительно является C1-C6 алкоксид щелочного металла. Примерами таких алкоксидов являются метилат и этилат натрия. Данную реакцию предпочтительно проводят в растворителе, причем в качестве растворителя предпочтительно могут быть метанол или этанол, то есть спиртовые растворители.
В предпочтительном варианте вышеуказанной реакции, соединение (2) обрабатывают алкоксидом щелочного металла в растворителе, а затем гидразидом изоникотиновой кислоты. Реакцию между соединением (2) и алкоксидом щелочного металла проводят при охлаждении либо при нагревании, предпочтительно при температуре от 15 до 80°C. Время реакции обычно составляет от 30 минут до 12 часов, предпочтительно приблизительно от 1 до 4 часов. Последующую реакцию с гидразидом изоникотиновой кислоты проводят в аналогичных температурных условиях, с использованием эквивалентного, избыточного или недостаточного количества. Время реакции обычно составляет от приблизительно 30 минут до 12 часов, предпочтительно приблизительно от 1 до 5 часов.
Вторая стадия
На второй стадии в соединение (3) вводят цианогруппу посредством цианирующего реагента с последующим образованием соединения (4).
Примерами цианирующих агентов, используемых в реакции, являются такие цианиды щелочных металлов, как цианид натрия или калия; цианид цинка; триалкилцианиды, такие как триметилсилил цианид.
Предпочтительно, реакцию введения циано-группы проводят с помощью реакции Рейссерта-Генце (Heterocycles, Vol. 22, No. 5, 1994). В одном варианте проведения реакции введения цианогруппы, соединение (3) активируют алкилкарбамоилгалогенидом в органическом растворителе, а затем активированное соединение реагирует с цианирующим реагентом, образуя соединение (4). Алкилкарбамоилгалогенидом, который можно использовать при карбамоилировании на первой стадии в реакции Рейссерта-Генце, предпочтительно является ди(C1-C6-алкил)карбамоилгалогенид, например диметилкарбамоилхлорид. Из данного ряда диметилкарбамоилхлорид предпочтительнее. В реакции могут быть использованы, например, N,N-диметилформамид, N,N-диметилацетамид, N-метилпирролидон, тетрагидрофуран и ацетонитрил в качестве растворителей. N,N-диметилформамид предпочтительнее из вышеуказанного ряда. Температура реакции составляет предпочтительно от 15 до 60°C, более предпочтительно от 30 до 50°C. Время реакции составляет предпочтительно от 1 до 24 часов, более предпочтительно от 1 до 3 часов. В качестве цианирующих агентов можно использовать в последующей реакции введения цианогруппы те же вещества, которые приведены выше. К ним предпочтительно относятся цианид натрия, цианид калия, цианид цинка, триметилцианид, а более предпочтительным является цианид натрия для введения цианогруппы. Температура реакции составляет предпочтительно от -20 до 60°C, более предпочтительно от -10 до 40°C. Введение цианогруппы проводят при перемешивании от 1 до 4 часов.
Соединение (4), полученное на второй стадии, является новым соединением и может выступать в качестве промежуточного соединения при получении соединения (1). Соединение (4) может быть легко синтезировано с высоким выходом на второй стадии без проведения очистки. С помощью соединения (4) может быть синтезировано соединение (1) промышленным масштабом с высокой эффективностью.
Третья стадия
На третьей стадии, соединение (4) подвергают циклизации в присутствии кислотного катализатора, тем самым приводя к образованию соединения (1).
В реакции могут быть использованы как органические, так и неорганические кислоты, например фосфорная кислота, п-толуолсульфокислота, соляная кислота. Из этого ряда неорганические кислоты предпочтительнее, в частности фосфорная кислота. В качестве растворителя для реакции могут быть использованы вода, спирты, такие как 2-бутанол, 2-пропанол, этанол, или растворитель, полученный смешиванием воды и спирта. Из этого ряда предпочтительны смеси воды и 2-бутанола в соотношениях от 5:1 до 10:1. Температура реакции составляет от 60 до 100°C, предпочтительно от 70 до 90°C, а время реакции - от 2 до 12 часов, предпочтительно от 8 до 10 часов. Также предпочтительно проведение реакции при перемешивании.
Промежуточное соединение и соединение (1), полученные способом, входящим в настоящее изобретение, могут быть выделены из реакционной смеси и очищены посредством таких рутинных методов, как промывка, перекристаллизация или хроматографическими методами.
Примеры
Далее настоящее изобретение будет описано с помощью примеров, однако стоит понимать, что оно не ограничивается этими примерами.
В примерах используются следующие сокращения: 1 Н-ЯМР: спектр протонного ядерного магнитного резонанса, ДМСО-d6: дейтеродиметилсульфоксид, Гц: герц, J: константа спин-спинового взаимодействия, с: синглет, дд: дублет дублетов, д: дублет, уш: уширенный. Кроме того, «ЯМР» обозначает спектр ядерного магнитного резонанса при 270 МГц с использованием ТМС (тетраметилсилана) в качестве внутреннего стандартного вещества. «МС» обозначает масс-спектрометрию с ионизацией электрораспылением.
Пример 1. Синтез N''-(4-пиридинкарбонил)-4-пиридингидразидимид-1-оксида (3)
4-цианопиридин-N-оксида (2) (5,00 г) суспендировали в 40 мл метанола и добавили метилат натрия (22,4 мг). Перемешивали смесь в течение 2 часов при 40°C в атмосфере азота. Затем добавляли гидразид изоникотиновой кислоты (5,71 г) при 40°C и перемешивали полученную смесь 4 часа. Реакционную смесь охлаждали до комнатной температуры и выпавшие в осадок кристаллы выделяли фильтрованием. Кристаллы промывали метанолом (15 мл) и сушили при 80°C в течение 15 часов. Получали N"-(4-пиридинкарбонил)-4-пиридингидразидимид-1-оксид (3) с выходом 9,60 г.
1H-ЯМР (ДМСО-d6) δ (м.д.): 6,98 (ушир, 2H), 7,81 (д, 2H, J=5,77 Гц), 7,85 (д, 2H, J=7,09 Гц), 8,29 (д, 2H, J=7,09 Гц), 8,73 (д, 2H, J=5,77 Гц), 10,37 (ушир, 1H).
MS m/z: 256 [M-H]-.
Пример 2. Синтез N'-(2-цианопиридин-4-карбонимидоил)гидразида
4-пиридин карбоновой кислоты (4) N''-(4-пиридинкарбонил)-4-пиридингидразидимид-1-оксид (3) (10,0 г) суспендировали в N,N-диметилформамиде (48 мл) и добавляли к полученной суспензии в атмосфере азота диметилкарбамоил хлорид (9,20 г) при 40°C. Перемешивали реакционную смесь 1 час. Затем добавляли цианид натрия (2,48 г) при 40°C и перемешивали еще 1 час. Реакционную смесь охлаждали до ≤5°C и последовательно добавляли по каплям 5% водный раствор гидрокарбоната натрия (100 мл) и воду (100 мл). Выпавшие в осадок кристаллы выделяли фильтрацией, промывали водой (100 мл) и сушили при 80°C в течение 15 часов при пониженном давлении. N'-(2-цианопиридин-4-карбонимидоил)гидразид 4-пиридин карбоновой кислоты (4) был получен с выходом 9,28 г.
1H-ЯМР (ДМСО-d6) δ (м.д.): 7,15 (ушир, 2H), 7,82 (д, 2H, J=5,61 Гц), 8,14 (д, 1H, J=5,11 Гц), 8,37 (с, 1H), 8,75 (д, 2H, J=5,61 Гц), 8,86 (д, 1H, J=5,11 Гц), 10,47 (ушир, 1H).
MS m/z: 265 [M-H]-.
Пример 3. Синтез 4-[5-(пиридин-4-ил)-1Н-1,2,4-триазол-3-ил]пиридин-2-карбонитрила (1)
К N'-(2-цианопиридин-4-карбонимидоил)гидразиду 4-пиридин карбоновой кислоты (4) (9,25 г) добавляли воду (82 мл), 2-бутанол (8,2 мл) и фосфорную кислоту (4,00 г), затем перемешивали полученную смесь при 80°C 8 часов. Реакционную смесь охлаждали до комнатной температуры, а выпавшие в осадок кристаллы отфильтровывали и промывали смесью вода:2-бутанол (10:1) (92,5 мл). Полученные кристаллы сушили при пониженном давлении в течение 13 часов при 80°C. 4-[5-(пиридин-4-ил)-1Н-1,2,4-триазол-3-ил]пиридин-2-карбонитрил (1) был получен с выходом 7,89 г.
1H-ЯМР (ДМСО-d6) δ (м.д.): 8,02 (дд, 2H, J=4,59, 1,62 Гц), 8,32 (дд, 1H, J=5,13, 1,62 Гц), 8,55 (дд, 1H, J=1,62, 1,08 Гц), 8,80 (дд, 2H, J=4,59, 1,62 Гц), 8,93 (дд, 1H, 5,13, 1,08 Гц).
MS m/z: 247 [M-H]-.
| название | год | авторы | номер документа |
|---|---|---|---|
| КРИСТАЛЛИЧЕСКИЙ ПОЛИМОРФ 4-[5-(ПИРИДИН-4-ИЛ)-1Н-1,2,4-ТРИАЗОЛ-3-ИЛ]-ПИРИДИН-2-КАРБОНИТРИЛА И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2013 |
|
RU2639149C2 |
| ПРОИЗВОДНЫЕ 1,2,4-ТРИАЗОЛА ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ | 1992 |
|
RU2095358C1 |
| 4-(ИМИДАЗО[1,2-а]ПИРИДИН-3-ИЛ)-ПИРИМИДИНОВЫЕ ПРОИЗВОДНЫЕ | 2020 |
|
RU2822388C2 |
| НОВЫЕ 1,2,4-ТРИАЗОЛЬНЫЕ СОЕДИНЕНИЯ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И СОДЕРЖАЩИЕ ИХ ЛЕКАРСТВЕННЫЕ СРЕДСТВА | 2002 |
|
RU2293733C2 |
| ПРОИЗВОДНЫЕ ПИРИДИН-4-ИЛА | 2010 |
|
RU2547098C2 |
| ТРИЦИКЛИЧЕСКИЕ 5,6-ДИГИДРО-9H-ПИРАЗОЛО[3,4-С]-1,2,4-ТРИАЗОЛО [4,3-А] ПИРИДИНЫ, СПОСОБ ИНГИБИРОВАНИЯ ФОСФОДИЭСТЕРАЗЫ ТИПА IУ И СПОСОБ ИНГИБИРОВАНИЯ ПРОДУЦИРОВАНИЯ ФАКТОРА ОПУХОЛЕСПЕЦИФИЧЕСКОГО НЕКРОЗА | 1996 |
|
RU2161158C2 |
| АЗОТСОДЕРЖАЩИЕ КОНДЕНСИРОВАННЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОДУКЦИИ БЕТА-АМИЛОИДА | 2010 |
|
RU2515976C2 |
| ЗАМЕЩЕННЫЕ ПИРИДИНОВЫЕ И ПИРАЗИНОВЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ PDE4 | 2014 |
|
RU2802185C2 |
| ПИРИДИН-2-ИЛЬНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИММУНОМОДУЛИРУЮЩИХ АГЕНТОВ | 2009 |
|
RU2494099C2 |
| ГЕТЕРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ | 2019 |
|
RU2808435C2 |
Изобретение относится к соединению формулы (4) и к промышленно полезному способу получения фармацевтически ценного 4-[5-(пиридин-4-ил)-1H-1,2,4-триазол-3-ил]пиридин-2-карбонитрила (1) и промежуточного соединения (4) для получения этого соединения. Способ получения соединения представлен следующей схемой реакции:
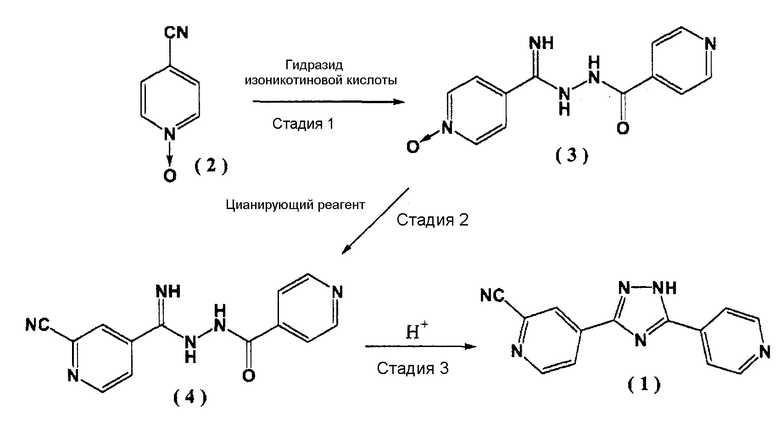 .
.
4 н.п. ф-лы, 3 пр.
1. N'-(2-цианопиридин-4-карбонимидоил)гидразид 4-пиридин карбоновой кислоты, представленный следующей формулой (4):
2. Способ получения соединения, представленного в п. 1, включающий реакцию соединения, представленного формулой (2)

с гидразидом изоникотиновой кислоты в присутствии алкоксида щелочного металла, выбранного из метилата натрия и этилата натрия, с последующим образованием соединения, представленного следующей формулой (3):
и введение цианогруппы в соединение (3) посредством цианирующего реагента, выбранного из цианида щелочного металла, цианида цинка и триалкилцианида.
3. Способ получения 4-[5-(пиридин-4-ил)-1Н-1,2,4-триазол-3-ил]пиридин-2-карбонитрила, представленного следующей формулой (1):
включающий реакцию соединения, представленного следующей формулой (2):
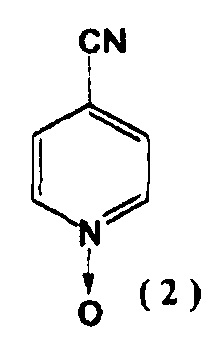
с гидразидом изоникотиновой кислоты в присутствии алкоксида щелочного металла, выбранного из метилата натрия и этилата натрия, с последующим образованием соединения, представленного следующей формулой (3):
введение цианогруппы в соединение (3) посредством цианирующего реагента, выбранного из цианида щелочного металла, цианида цинка и триалкилцианида, с образованием соединения представленного следующей формулой (4):
и введение соединения (4) в реакцию циклизации в присутствии кислотного катализатора, выбранного из органического кислотного катализатора и неорганического кислотного катализатора.
4. Способ получения 4-[5-(пиридин-4-ил)-1Н-1,2,4-триазол-3-ил]пиридин-2-карбонитрила, представленного следующей формулой (1):
включающий введение соединения, представленного следующей формулой (4):
в реакцию циклизации в присутствии кислотного катализатора, выбранного из органического кислотного катализатора и неорганического кислотного катализатора.
| НОВЫЕ 1,2,4-ТРИАЗОЛЬНЫЕ СОЕДИНЕНИЯ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И СОДЕРЖАЩИЕ ИХ ЛЕКАРСТВЕННЫЕ СРЕДСТВА | 2002 |
|
RU2293733C2 |
| WO 2000024735 A1, 04.05.2000 | |||
| US 4097599 A1, 27.06.1978 | |||
| ПРОИЗВОДНОЕ ТРИАЗОЛА, СПОСОБЫ ЕГО ПОЛУЧЕНИЯ И АФИЦИДНОЕ СРЕДСТВО НА ЕГО ОСНОВЕ | 1993 |
|
RU2101282C1 |

