Изобретение относится к некоторым новым производным аминопиразина или их фармацевтически приемлемым солям, которые обладают противораковой активностью и, соответственно, являются полезными в способах лечения человека или животного. Изобретение также относится к способам получения указанных производных аминопиразина, фармацевтических композиций, содержащих такие производные, и их применению в терапевтических способах, например, при изготовлении лекарственных средств для применения в предупреждении или лечении злокачественных новообразований у теплокровного животного, например, человека, включая применение в предупреждении или лечении рака.
Настоящее изобретение также относится к производным аминопиразина, которые являются селективными ингибиторами ферментов PI3-киназного семейства (альтернативно известного как фосфатидилинозитол-3-киназное семейство или PI3K-семейство), в частности, изоформ PI3K-α и PI3K-δ, и полезны, например, для противоопухолевой терапии.
В области онкологии в последние годы было обнаружено, что клетка может стать раковой в силу превращения части ее ДНК в онкоген, представляющий собой ген, который при активации приводит к образованию злокачественных опухолевых клеток (Bradshaw, Mutagenesis, 1986, 1, 91). Некоторые такие онкогены вызывают выработку пептидов, представляющих собой киназы, класс ферментов, которые способны фосфорилировать свои белковые или липидные субстраты. Существует несколько классов киназ.
Во-первых, тирозинкиназы, которые могут представлять собой рецепторные тирозинкиназы или нерецепторные тирозинкиназы. Известны различные классы рецепторных тирозинкиназ (Wilks, Advances in Cancer Research, 1993, 60, 43-73), соотнесенных с семействами факторов роста, которые могут связываться с внеклеточным доменом разных рецепторных тирозинкиназ; в качестве примера классификация включает рецепторные тирозинкиназы класса I, содержащего EGF(эпидермальный фактор роста)-семейство рецепторных тирозинкиназ. Нерецепторные тирозинкиназы располагаются внутри клетки; известны различные классы нерецепторных тирозинкиназ, включая Src-семейство киназ, таких как тирозинкиназы Src, Lyn, Fyn и Yes.
Во-вторых, некоторые киназы принадлежат к классу серин/треониновых киназ, которые также располагаются внутри клетки. Сигнальные пути с участием серин/треониновых киназ включают каскад Raf(киназа, индуцирующая быстро прогрессирующую фибросаркому)-MEK(киназа митоген-активируемой протеинкиназы)-ERK(внеклеточная сигнал-регулируемая киназа) и ферменты, расположенные в каскаде ниже PI3-киназы, такие как PDK-1 (PI3K-зависимая киназа-1), АКТ (протеинкиназа В) и mTOR (мишень рапамицина у млекопитающих) (Blume-Jensen and Hunter, Nature, 2001, 411, 355).
Также известно, что некоторые другие киназы принадлежат к классу липидных киназ, которые располагаются внутри клетки и, как и в случае вышеупомянутых киназ, вовлечены в передачу биохимических сигналов, таких как те, которые влияют на рост и инвазивность опухолевых клеток. Известны различные классы липидных киназ, включая вышеупомянутое семейство PI3-киназ.
В настоящее время хорошо известно, что дерегуляция онкогенов и генов-супрессоров опухолей способствует образованию злокачественных опухолей, например, путем усиления пролиферации клеток или повышения жизнеспособности клеток. Также в настоящее время известно, что сигнальные пути, опосредованные семейством PI3-киназ, играют центральную роль во многих клеточных процессах, включая пролиферацию и выживаемость, и дерегуляция этих путей является причинным фактором образования широкого спектра раковых и других заболеваний у человека (Katso et al., Annual Rev. Cell Dev. Biol., 2001, 17: 615-617 и Foster et al., J. Cell Science. 2003, 116: 3037-3040).
PI3-киназное семейство липидных киназ представляет собой группу ферментов, которые фосфорилируют 3-е положение кольца инозита фосфатидилинозита (PI). Известны три главные группы PI3-киназных ферментов, которые классифицируются в соответствии с их физиологической субстратной специфичностью (Vanhaesebroeck et al., Trends in Biol. Sci., 1997, 22, 267; Engleman et al., Nature Review Genetics. 2006, 7, 607). PI3-киназные ферменты класса III фосфорилируют исключительно PI. В отличие от них PI3-киназные ферменты класса II фосфорилируют и PI, и PI-4-фосфат (сокращенный ниже как PI(4)P). PI3-киназные ферменты класса I фосфорилируют PI, PI(4)P и PI-4,5-бифосфат (сокращенный ниже как PI(4,5)P2), несмотря на то, что только PI(4,5)P2, как полагают, является физиологическим клеточным субстратом. В результате фосфорилирования PI(4,5)P2 образуется липидный вторичный мессенджер PI-3,4,5-трифосфат (сокращенный ниже как PI(3,4,5)P3). Более отдаленно связанными членами этого суперсемейства являются киназы класса IV, такие как mTOR и ДНК-зависимая протеинкиназа, которые фосфорилируют остатки серина/треонина в белковых субстратах. Наиболее изученными и понятными из этих липидных киназ являются PI3-киназные ферменты класса I.
PI3-киназы класса I являются гетеродимерами, состоящими из каталитической субъединицы p110 и регуляторной субъединицы, и семейство далее разделено на ферменты класса Ia и класса Ib по признаку регуляторных партнеров и механизма регулирования (Engleman et al., Nature Review Genetics, 2006, 7, 607). Ферменты класса Ia содержат три разные каталитические субъединицы (p110α, p110β и p110δ; по номенклатуре изоформу PI3-киназы определяют как α, β или δ, соответственно), которые димеризуются с пятью разными регуляторными субъединицами (р85α, р55α, р50α, р85β и р55γ), причем все каталитические субъединицы способны вступать во взаимодействие со всеми регуляторными субъединицами с образованием разнообразных гетеродимеров. PI3-киназные ферменты класса Ia, как правило, активируются в ответ на стимуляцию рецепторных тирозинкиназ фактором роста, через взаимодействие SH2-доменов регуляторной субъединицы со специфическими фосфотирозиновыми остатками активированных рецепторных или адаптерных белов, таких как IRS-1. И p110α, и p110β широко экспрессируются во всех типах клеток и во всех тканях, тогда как экспрессия p110δ более ограничена популяциями лейкоцитов и некоторыми эпителиальными клетками. В противоположность этому, единственный фермент класса Ib состоит из каталитической субъединицы р110γ, которая взаимодействует с регуляторной субъединицей p101. Кроме того, этот фермент класса Ib активируется в ответ на системы G-белок-связанного рецептора (GPCR), так же как и механизмами, описанными выше.
В настоящее время имеется большое число данных, указывающих на то, что PI3-киназные ферменты класса Ia участвуют в онкогенезе широкого спектра раковых опухолей у человека, либо напрямую, либо опосредованно (Vivanco and Sawyers, Nature Reviews Cancer, 2002, 2, 489-501). В частности, ген PIK3CA, кодирующий каталитическую субъединицу p110α PI3-киназы, широко вовлечен в онкогенез. Активирующие точечные мутации, наиболее часто обнаруживаемые в спиральных или каталитических доменах p110α, повышают PI3-киназную активность холофермента и могут приводить к трансформации клеток. В частности, согласно опубликованным данным они представляют собой мутации в соматических клетках, возникающие с очень высокой частотой в широком диапазоне типов опухолей (Samuels et al., Science, 2004, 304, 554; Samuels et al., Cancer Cell, 2005, 7, 561; Engleman et al., Nature Review Genetics, 2006, 7, 607; Zhao L and Vogt PK, Oncogene, 2008, 27, 5486). Опухоль-ассоциированные мутации в p85α также были идентифицированы при таких видах рака, как рак яичника и толстой кишки (Philp et al., Cancer Research, 2001, 61, 7426-7429). Кроме того, субъединица p110α амплифицируется в некоторых опухолях, таких как опухоли яичника (Shayesteh et al., Nature Genetics, 1999, 21, 99-102) и шейки матки (Ma et al., Oncogene, 2000, 19, 2739-2744).
Полагают, что помимо прямого влияния активация PI3-киназы класса Ia вносит вклад в приводящие к образованию опухолей события, которые происходят выше в сигнальных путях, например, посредством лиганд-зависимой или лиганд-независимой активации рецепторных тирозинкиназ, GPCR систем или интегринов (Vara et al., Cancer Treatment Reviews, 2004, 30, 193-204). Примеры таких расположенных выше в сигнальных путях событий включают сверхэкспрессию рецепторной тирозинкиназы Erb2 в ряде опухолей, что приводит к активации PI3-киназа-опосредованных путей (Harari et al., Oncogene, 2000, 19, 6102-6114) и сверхэкспрессии онкогена Ras (Kauffmann-Zeh et al., Nature, 1997, 385, 544-548). Помимо этого, PI3-киназы класса Ia могут участвовать в онкогенезе, вызываемом различными событиями, расположенными ниже по пути передачи сигнала. Например, ослабление действия опухоль-супрессорной фосфатазы PTEN (гомолог фосфатазы и тензина с делецией по 10-й хромосоме), которая катализирует превращение PI(3,4,5)P3 обратно в PI(4,5)P2, связано с очень широким диапазоном опухолей через дерегуляцию PI3-киназа-опосредованного образования PI(3,4,5)P3 (Simpson and Parsons, Exp. Cell Res., 2001, 264, 29-41). Кроме того, считается, что усиление действия других PI3-киназа-опосредованных событий передачи сигнала, способствует возникновению многих раковых заболеваний, например, путем активации Akt (Nicholson and Anderson, CelluIar Signaling, 2002, 14, 381-395).
В связи с этим, общая дерегуляция PI3-киназы вместе с киназами, расположенными выше и ниже в сигнальных путях, в совокупности делают этот путь одним из наиболее часто разрегулированных путей при раковом заболевании у человека (Hennessey et al., Nature Reviews Drug Discovery, 2005, 4, 988).
Помимо участия в опосредовании передачи сигналов, касающихся пролиферации и жизнеспособности, в опухолевых клетках также имеются убедительные доказательства того, что PI3-киназные ферменты класса Ia также будут принимать участие в онкогенезе в результате их функционирования в опухоле-ассоциированных стромальных клетках. Например, известно, что ассоциированная с PI3-киназами передача сигнала играет важную роль в опосредовании ангиогенных событий в эндотелиальных клетках в ответ на проангиогенные факторы, такие как сосудистый эндотелиальный фактор роста (VEGF) (Abid et al., Arterioscler. Thromb. Vasc. Biol., 2004, 24, 294-300). Поскольку PI3-киназные ферменты класса I также вовлечены в подвижность и миграцию (Sawyer, Expert Opinion Investig, Drugs. 2004, 13, 1-19), ингибиторы PI3-киназ должны обеспечить терапевтическую пользу путем ингибирования инвазии опухолевых клеток и метастазирования.
Кроме того, PI3-киназные ферменты класса I играют важную роль в регуляции иммунных клеток с PI3-киназной активностью, способствуя проонкогенному действию воспалительных клеток (Coussens and Werb, Nature, 2002, 420, 860-867). В действительности, PI3-киназный фермент класса Ia, в частности, PI3-киназа 5, вовлечен в онкогенез при гематологических злокачественных новообразованиях, таких как хронический лимфолейкоз (CLL), острый лимфобластный лейкоз (ALL) и лимфома из клеток мантийной зоны (MCL). Сообщается об усилении передачи сигнала через PI3K (главным образом, p110δ) в целом ряде злокачественных лимфоидных клеток (Herman et al., Blood, 2010, 116, 2078; Ikeda et al., Blood, 2010, 116, 1460; Uddin et al., Blood, 2006, 108, 4178; Rudelius et al., Blood, 2006, 108, 1668; Garcia-Martinez., Br. J. Cancer, 2011, 104, 1116; Renne et al., Leukemia. 2007, 2, 780). Это стимулировало разработку агентов, направленно воздействующих на PI3-киназу δ, с многообещающими первоначальными клиническими результатами при гематологических злокачественных новообразованиях (Castillo et al., Expert Opinion on Investigational Drugs, 2012, 21, 15).
Эти данные наводят на мысль, что фармакологические ингибиторы PI3-киназных ферментов класса I должны обладать терапевтической ценностью для лечения различных форм заболевания раком, включающих солидные опухоли, такие как карциномы и саркомы, и лейкозы и лимфолейкозы.
В более ранних исследованиях, как доклинических, так и клинических, в которых изучали физиологическую и патологическую роли PI3-киназного фермента, были в основном использованы агенты с ограниченной селективностью в отношении ингибирования киназ, выбранных либо среди всех киназных семейств, либо среди PI3-киназного семейства, либо среди семейства PI3-киназ класса I. В связи с этим существует потребность в более селективных фармацевтических ингибиторах PI3-киназы класса I для получения полезных терапевтических агентов с возможностью достижения улучшенной широты терапевтического действия по сравнению с первоначальными агентами, которые уже введены в клиническую практику.
Как правило, соединения по настоящему изобретению обладают сильной ингибирующей активностью в отношении подгруппы PI3-киназных ферментов класса I, в частности, α- и δ-изоформ PI3-киназ класса Ia при относительном снижении в отношении γ- и, в особенности, β-изоформы. Данные соединения также селективны в отношении всего PI3-киназного семейства и кинома в целом. Такие соединения обладают достаточной эффективностью в отношении PI3-киназных ферментов класса I, так что их можно применять в количестве, достаточном для ингибирования подгруппы изоформ PI3-киназ класса I, в частности, для ингибирования α- и δ-изоформ PI3-киназных ферментов класса Ia, одновременно демонстрируя незначительную активность в отношении других киназ.
Понимание дерегуляции PI3-киназа-опосредованной передачи сигнала при раковых и других заболеваниях у человека открывает перспективу для направленного воздействия на подгруппу пациентов с наибольшей вероятностью получения пользы в результате лечения агентами, описанными в этой заявке, с использованием способа, известного как персонализированная медицинская помощь (РНС) или персонализированная медицина. Что касается этих агентов, то пациенты, заболевание у которых зависит от усиленной или иным образом измененной передачи сигнала посредством PI3K-α и/или передачи сигнала посредством PI3K-δ, могут, в частности, получить пользу в результате лечения. В данной области техники хорошо известно, что с целью получения показания для дающего предсказательный ответ биомаркера можно использовать диагностику. С помощью такой диагностики можно измерить одно или более показаний дерегуляции пути, таких как, но не ограничиваясь этим, мутация в генах PIK3CA, PTEN или р85 (PIK3R), амплификация или увеличение числа копий гена PIK3CA, сверхэкспрессия или повышение активности изоформы PI3K-α и/или -δ, либо использовать показание для фосфо-биомаркера данного пути, такого как фосфо-RTK (рецепторная тирозинкиназа) или фосфо-АКТ. Кроме того, определение мутационного статуса или активационного статуса дополнительных генов, таких как K-ras, потенциальный маркер устойчивости в опухолях с аберрантной или разрегулированной PIK3CA или PI3K-α (Engelman et al., Nature Medicine, 2008, 14, p. 1351-1355; Ihle et al., Cancer Research, 2009, 69, p. 143-160; Janku et al., MolecuIar Cancer Therapeutics, 2011, 10, p. 558-564), может помочь в повышении предсказательной способности персонализированного подхода в медицине. Альтернативно, в другом подходе с направленным действием, но с меньшей специфичностью, лечение может быть сфокусировано на разновидностях заболеваний, при которых, как известно, наиболее распространена дерегуляция релевантных изоформ PI3K.
Описанные соединения могут быть использованы для направленного воздействия на заболевание либо по отдельности, либо в комбинации с другим(ими) фармацевтическим(ими) агентом или агентами. Комбинируя ингибиторы PI3-киназ с другими терапиями, можно улучшить эффективность посредством преодоления механизмов устойчивости, или врожденной, или индуцированной в ответ на направленный на PI3-киназу агент. Имеются убедительные доклинические данные, подтверждающие такой подход (Courtney et al., J. Clin. Oncol., 2010, 28, 1075; Engleman et al., Nature Review Genetics, 2006, 7, 607). Один подход относится к комбинациям «внутри одного пути» ("intra-pathway") с агентами, модулирующими другие оси в PI3-киназа-опосредованных сигнальных путях (например, mTOR, АКТ, RTK, с другим направленным на PI3-киназу агентом). Второй подход относится к комбинациям с агентами «другого пути» ("inter-pathway"), при этом ингибирование более чем одного сигнального пути может быть полезно по сравнению с ингибированием одного пути (например, при комбинировании с ингибиторами MEK, ингибиторами Raf, модуляторами Bcl-семейства, ингибиторами RTK или модуляторами передачи сигнала при повреждении ДНК, такими как ингибиторы поли(АДФ-рибоза)-полимеразы (PARP). Другие подходы включают подходы, при которых ингибитор PI3-киназ объединяют с агентами или схемами лечения, уже принятыми в клинической практике, так называемые стандартные подходы к лечению (SoC), или включают комбинации с агентами, направленными на механизмы, не связанные с опухолевыми клетками, такими как стромальная клетка опухоли, или действующие посредством иммунной системы.
Помимо онкогенеза, имеются доказательства того, что PI3-киназные ферменты класса I играют роль в других заболеваниях (Wymann et al., Trends in Pharmacological Science, 2003, 24, 366-376). И PI3-киназные ферменты класса Ia, в частности, PI3K-δ, и единственный фермент класса Ib (PI3K-γ) играют важную роль в клетках иммунной системы (Koyasu, Nature Immunology, 2003, 4, 313-319), и таким образом они представляют собой терапевтические мишени в случае воспалительных и аллергических показаний. Ингибирование PI3-киназы, как описано ранее, также полезно для лечения сердечно-сосудистого заболевания благодаря противовоспалительному действию или непосредственному воздействию на кардиомиоциты (Prasad et al., Trends in CardiovascuIar Medicine, 2003, 13, 206-212). Таким образом, ингибиторы PI3-киназных ферментов класса I могут быть полезны в предупреждении и лечении целого ряда заболеваний, помимо рака.
Было установлено, что соединения по изобретению, т.е. производные аминопиразина, обладают сильной противоопухолевой активностью, являясь полезными для ингибирования неконтролируемой клеточной пролиферации, которая является результатом злокачественного заболевания. Не желая дать основания полагать, что соединения, описанные в настоящем изобретении, обладают фармакологической активностью только в силу воздействия на единственный биологический процесс, считают, что соединения обеспечивают противоопухолевое действие путем ингибирования PI3-киназных ферментов класса I, в частности путем ингибирования PI3-киназных ферментов класса Ia, более конкретно путем ингибирования α- и δ-изоформ PI3K.
Соединения по настоящему изобретению также могут быть полезны для ингибирования неконтролируемой клеточной пролиферации, которая является результатом различных незлокачественных заболеваний, таких как воспалительные заболевания (например, ревматоидный артрит и воспалительное заболевание кишечника), фибротические заболевания (например, цирроз печени и фиброз легких), гломерулонефрит, рассеянный склероз, псориаз, доброкачественная гипертрофия предстательной железы (BPH), реакции гиперчувствительности кожи, заболевания кровеносных сосудов (например, атеросклероз и рестеноз), аллергическая астма, инсулинозависимый диабет, диабетическая ретинопатия и диабетическая нефропатия.
Пролинамиды раскрыты в качестве селективных в отношении PI3K-α агентов в международных патентных заявках WO 2009/080705, WO 2010/029082 и WO 2011/000905 (Novartis). В WO 2011/143426 и WO 2010/071837 (Vertex) раскрыты аминопиразин-содержащие ингибиторы ATR(атаксия-телеангиэктазия+Rad3-родственной)-киназы.
Согласно одному из аспектов изобретения предложено соединение формулы (I)
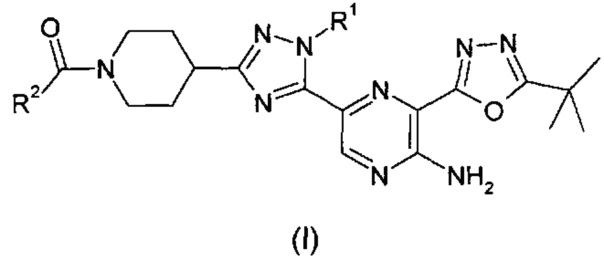 ,
,
где:
R1 представляет собой метил или этил; и
R2 представляет собой (C2-3)алкил, замещенный гидроксилом;
или его фармацевтически приемлемая соль.
Согласно другому аспекту изобретения предложено соединение формулы (I), которое определено выше.
Очевидно, что термин «(C2-3)алкил, замещенный группой гидрокси» включает в себя алкильные группы как с прямой цепью, так и разветвленной цепью, например такие, которые приведены ниже как группы от (1) до (11):
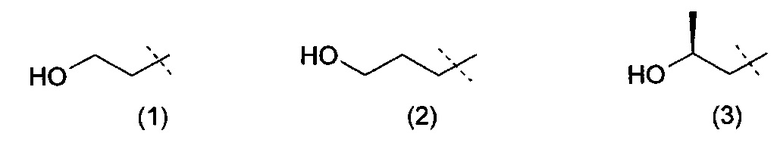
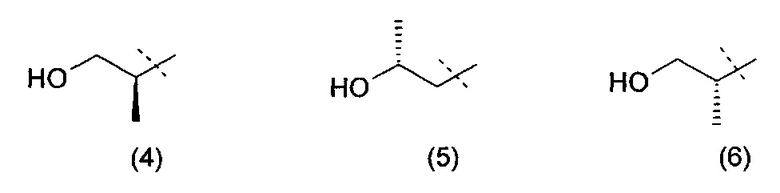

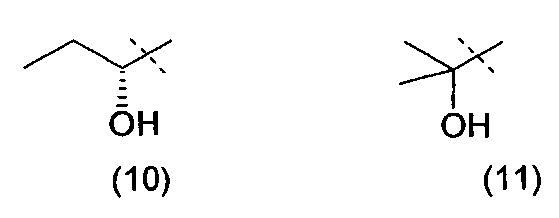
Следует понимать, что, поскольку некоторые из определенных выше соединений формулы (I) могут существовать в оптически активных или рацемических формах в силу наличия одного или более асимметрических атомов углерода, данное изобретение включает в своем определении любую такую оптически активную или рацемическую форму, которая обладает ингибирующей активностью в отношении PI3K-α и -δ. Синтез оптически активных форм может быть проведен с использованием стандартных методов органической химии, хорошо известных в данной области техники, например, путем синтеза из оптически активных исходных веществ или путем выделения из рацемической формы. Аналогично, вышеупомянутая активность может быть оценена с использованием стандартных лабораторных методов.
Конкретный энантиомер соединения, описанного в данной заявке, может быть более активным, чем другие энантиомеры того же соединения.
Согласно другому аспекту изобретения предложены соединение формулы (I) или его фармацевтически приемлемая соль, которое(ая) является отдельным энантиомером, находящимся в энантиомерном избытке (% ее), составляющим не менее 95, не менее 98% или не менее 99%. Подходящим образом отдельный энантиомер присутствует в энантиомерном избытке (% ее) не менее 99%.
Согласно другому аспекту изобретения предложена фармацевтическая композиция, содержащая соединение формулы (I), которое является отдельным энантиомером, находящимся в энантиомерном избытке (% ее) не менее 95, не менее 98% или не менее 99%, или его фармацевтически приемлемую соль вместе с фармацевтически приемлемым разбавителем или носителем. Подходящим образом отдельный энантиомер присутствует в энантиомерном избытке (% ее) не менее 99%.
Некоторые соединения формулы (I) могут быть кристаллическими и могут существовать более чем в одной кристаллической форме. Следует понимать, что настоящее изобретение охватывает любую кристаллическую либо аморфную форму или их смеси, при этом форма обладает свойствами, полезными в ингибировании активности PI3K-α и -δ, причем в данной области техники хорошо известно, как определить эффективность кристаллической или аморфной формы в отношении ингибирования активности PI3K-α и/или -δ, используя стандартные тесты, описанные далее.
Общеизвестно, что анализ кристаллических веществ можно провести, используя традиционные методы, такие как анализ с применением дифракции рентгеновских лучей на порошке (далее XRPD), дифференциальная сканирующая калориметрия (далее DSC), термогравиметрический анализ (далее TGA), инфракрасная спектроскопия диффузного отражения с преобразованием Фурье (DRIFT), спектроскопия в ближней инфракрасной области (NIR), спектроскопия ядерного магнитного резонанса в растворе и/или твердом состоянии. Содержание воды в таких кристаллических веществах можно определить посредством анализа по методу Карла Фишера.
В качестве примера, соединение из примера 1 демонстрирует кристалличность, и была идентифицирована одна его кристаллическая форма.
Соответственно, дополнительный аспект изобретения относится к Форме A 1-(4-(5-(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-метил-1H-1,2,4-триазол-3-ил)пиперидин-1-ил)-3-гидроксипропан-1-она.
Согласно дополнительному аспекту настоящего изобретения предложена кристаллическая форма, Форма А 1-(4-(5-(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-метил-1Н-1,2,4-триазол-3-ил)пиперидин-1-ил)-3-гидроксипропан-1-она, которая имеет картину дифракции рентгеновских лучей на порошке по меньшей мере с одним характерным пиком при 2-тета, приблизительно составляющем 5,1°.
Согласно дополнительному аспекту настоящего изобретения предложена кристаллическая форма, Форма А 1-(4-(5-(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-метил-1Н-1,2,4-триазол-3-ил)пиперидин-1-ил)-3-гидроксипропан-1-она, которая имеет картину дифракции рентгеновских лучей на порошке по меньшей мере с одним характерным пиком при 2-тета, приблизительно составляющем 18,0°.
Согласно дополнительному аспекту настоящего изобретения предложена кристаллическая форма, Форма А 1-(4-(5-(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-метил-1Н-1,2,4-триазол-3-ил)пиперидин-1-ил)-3-гидроксипропан-1-она, которая имеет картину дифракции рентгеновских лучей на порошке по меньшей мере с двумя характерными пиками при 2-тета, приблизительно составляющих 5,1 и 18,0°.
Согласно дополнительному аспекту настоящего изобретения предложена кристаллическая форма, Форма А 1-(4-(5-(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-метил-1H-1,2,4-триазол-3-ил)пиперидин-1-ил)-3-гидроксипропан-1-она, которая имеет картину дифракции рентгеновских лучей на порошке с характерными пиками при 2-тета, приблизительно составляющих 5,1; 18,0; 10,2; 11,7; 19,4; 18,5; 14,8; 26,7; 26,6; 17,8°.
Согласно настоящему изобретению предложена кристаллическая форма, Форма A, которая имеет картину дифракции рентгеновских лучей на порошке по существу аналогичную картине дифракции рентгеновских лучей на порошке, показанной на Фиг. 1.
Согласно дополнительному аспекту настоящего изобретения предложена кристаллическая форма, Форма А 1-(4-(5-(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-метил-1H-1,2,4-триазол-3-ил)пиперидин-1-ил)-3-гидроксипропан-1-она, которая имеет картину дифракции рентгеновских лучей на порошке по меньшей мере с одним характерным пиком при 2-тета, приблизительно составляющем 5,1°±0,2° 2-тета.
Согласно дополнительному аспекту настоящего изобретения предложена кристаллическая форма, Форма А 1-(4-(5-(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-метил-1H-1,2,4-триазол-3-ил)пиперидин-1-ил)-3-гидроксипропан-1-она, которая имеет картину дифракции рентгеновских лучей на порошке по меньшей мере с одним характерным пиком при 2-тета, приблизительно составляющем 18,0°±0,2° 2-тета.
Согласно дополнительному аспекту настоящего изобретения предложена кристаллическая форма, Форма A 1-(4-(5-(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-метил-1H-1,2,4-триазол-3-ил)пиперидин-1-ил)-3-гидроксипропан-1-она, которая имеет картину дифракции рентгеновских лучей на порошке по меньшей мере с двумя характерными пиками при 2-тета, приблизительно составляющих 5,1 и 18,0°±0,2° 2-тета.
Согласно дополнительному аспекту настоящего изобретения предложена кристаллическая форма, Форма А 1-(4-(5-(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-метил-1H-1,2,4-триазол-3-ил)пиперидин-1-ил)-3-гидроксипропан-1-она, которая имеет картину дифракции рентгеновских лучей на порошке с характерными пиками при 2-тета, приблизительно составляющих 5,1; 18,0; 10,2; 11,7; 19,4; 18,5; 14,8; 26,7; 26,6; 17,8°±0,2° 2-тета.
Соединение из примера 3 также является кристаллическим, и в данном изобретении описаны три формы (A, B и C).
Согласно настоящему изобретению предложена кристаллическая форма, Форма А 1-(4-(5-(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-этил-1H-1,2,4-триазол-3-ил)пиперидин-1-ил)-3-гидроксипропан-1-она, которая имеет картину дифракции рентгеновских лучей на порошке по меньшей мере с одним характерным пиком при 2-тета, приблизительно составляющем 4,8°.
Согласно настоящему изобретению предложена кристаллическая форма, Форма А 1-(4-(5-(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-этил-1Н-1,2,4-триазол-3-ил)пиперидин-1-ил)-3-гидроксипропан-1-она, которая имеет картину дифракции рентгеновских лучей на порошке по меньшей мере с одним характерным пиком при 2-тета, приблизительно составляющем 10,0°.
Согласно настоящему изобретению предложена кристаллическая форма, Форма А 1-(4-(5-(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-этил-1H-1,2,4-триазол-3-ил)пиперидин-1-ил)-3-гидроксипропан-1-она, которая имеет картину дифракции рентгеновских лучей на порошке по меньшей мере с двумя характерными пиками при 2-тета, приблизительно составляющих 4,8° и 10,0°.
Согласно настоящему изобретению предложена кристаллическая форма, Форма А 1-(4-(5-(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-этил-1H-1,2,4-триазол-3-ил)пиперидин-1-ил)-3-гидроксипропан-1-она, которая имеет картину дифракции рентгеновских лучей на порошке с характерными пиками при 2-тета, приблизительно составляющих 4,8; 10,0; 14,6; 5,2; 19,9; 10,4; 25,4; 23,6; 24,4; 16,2°.
Согласно настоящему изобретению предложена кристаллическая форма, Форма А 1-(4-(5-(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-этил-1H-1,2,4-триазол-3-ил)пиперидин-1-ил)-3-гидроксипропан-1-она, которая имеет картину дифракции рентгеновских лучей на порошке по существу аналогичную картине дифракции рентгеновских лучей на порошке, показанной на Фиг. 3.
Согласно настоящему изобретению предложена кристаллическая форма, Форма А 1-(4-(5-(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-этил-1H-1,2,4-триазол-3-ил)пиперидин-1-ил)-3-гидроксипропан-1-она, которая имеет картину дифракции рентгеновских лучей на порошке по меньшей мере с одним характерным пиком при 2-тета, составляющем 4,8° ± 0,2° 2-тета.
Согласно настоящему изобретению предложена кристаллическая форма, Форма А 1-(4-(5-(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-этил-1H-1,2,4-триазол-3-ил)пиперидин-1-ил)-3-гидроксипропан-1-она, которая имеет картину дифракции рентгеновских лучей на порошке по меньшей мере с одним характерным пиком при 2-тета, составляющем 10,0° ± 0,2° 2-тета.
Согласно настоящему изобретению предложена кристаллическая форма, Форма А 1-(4-(5-(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-этил-1H-1,2,4-триазол-3-ил)пиперидин-1-ил)-3-гидроксипропан-1-она, которая имеет картину дифракции рентгеновских лучей на порошке по меньшей мере с двумя характерными пиками при 2-тета, составляющих 4,8° и 10,0°, где указанные значения могут отклоняться на величину ± 0,2° 2-тета.
Согласно настоящему изобретению предложена кристаллическая форма, Форма А 1-(4-(5-(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-этил-1H-1,2,4-триазол-3-ил)пиперидин-1-ил)-3-гидроксипропан-1-она, которая имеет картину дифракции рентгеновских лучей на порошке с характерными пиками при 2-тета, составляющих 4,8; 10,0; 14,6; 5,2; 19,9; 10,4; 25,4; 23,6; 24,4; 16,2°, где указанные значения могут отклоняться на величину ± 0,2° 2-тета.
Согласно настоящему изобретению предложена кристаллическая форма, Форма В 1-(4-(5-(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-этил-1H-1,2,4-триазол-3-ил)пиперидин-1-ил)-3-гидроксипропан-1-она, которая имеет картину дифракции рентгеновских лучей на порошке по меньшей мере с одним характерным пиком при 2-тета, приблизительно составляющем 5,8°.
Согласно настоящему изобретению предложена кристаллическая форма, Форма В 1-(4-(5-(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-этил-1H-1,2,4-триазол-3-ил)пиперидин-1-ил)-3-гидроксипропан-1-она, которая имеет картину дифракции рентгеновских лучей на порошке по меньшей мере с одним характерным пиком при 2-тета, приблизительно составляющем 10,9°.
Согласно настоящему изобретению предложена кристаллическая форма, Форма В 1-(4-(5-(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-этил-1H-1,2,4-триазол-3-ил)пиперидин-1-ил)-3-гидроксипропан-1-она, которая имеет картину дифракции рентгеновских лучей на порошке по меньшей мере с двумя характерными пиками при 2-тета, приблизительно составляющих 5,8 и 10,9°.
Согласно настоящему изобретению предложена кристаллическая форма, Форма В 1-(4-(5-(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-этил-1H-1,2,4-триазол-3-ил)пиперидин-1-ил)-3-гидроксипропан-1-она, которая имеет картину дифракции рентгеновских лучей на порошке с характерными пиками при 2-тета, приблизительно составляющих 5,8; 10,9; 11,5; 25,9; 17,3; 24,0; 19,1; 12,9; 24,7; 27,2°.
Согласно настоящему изобретению предложена кристаллическая форма, Форма В 1-(4-(5-(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-этил-1H-1,2,4-триазол-3-ил)пиперидин-1-ил)-3-гидроксипропан-1-она, которая имеет картину дифракции рентгеновских лучей на порошке по существу аналогичную картине дифракции рентгеновских лучей на порошке, показанной на Фиг. 5.
Согласно настоящему изобретению предложена кристаллическая форма, Форма В 1-(4-(5-(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-этил-1H-1,2,4-триазол-3-ил)пиперидин-1-ил)-3-гидроксипропан-1-она, которая имеет картину дифракции рентгеновских лучей на порошке по меньшей мере с одним характерным пиком при 2-тета, составляющем 5,8° ± 0,2° 2-тета.
Согласно настоящему изобретению предложена кристаллическая форма, Форма В 1-(4-(5-(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-этил-1H-1,2,4-триазол-3-ил)пиперидин-1-ил)-3-гидроксипропан-1-она, которая имеет картину дифракции рентгеновских лучей на порошке по меньшей мере с одним характерным пиком при 2-тета, составляющем 10,9° ± 0,2° 2-тета.
Согласно настоящему изобретению предложена кристаллическая форма, Форма В 1-(4-(5-(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-этил-1H-1,2,4-триазол-3-ил)пиперидин-1-ил)-3-гидроксипропан-1-она, которая имеет картину дифракции рентгеновских лучей на порошке по меньшей мере с двумя характерными пиками при 2-тета, составляющих 5,8° и 10,9°, где указанные значения могут отклоняться на величину ± 0,2° 2-тета.
Согласно настоящему изобретению предложена кристаллическая форма, Форма В 1-(4-(5-(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-этил-1H-1,2,4-триазол-3-ил)пиперидин-1-ил)-3-гидроксипропан-1-она, которая имеет картину дифракции рентгеновских лучей на порошке с характерными пиками при 2-тета, составляющих 5,8; 10,9; 11,5; 25,9; 17,3; 24,0; 19,1; 12,9; 24,7; 27,2°, где указанные значения могут отклоняться на величину ± 0,2° 2-тета.
Согласно настоящему изобретению предложена кристаллическая форма, Форма С 1-(4-(5-(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-этил-1H-1,2,4-триазол-3-ил)пиперидин-1-ил)-3-гидроксипропан-1-она, которая имеет картину дифракции рентгеновских лучей на порошке по меньшей мере с одним характерным пиком при 2-тета, приблизительно составляющем 6,9°.
Согласно настоящему изобретению предложена кристаллическая форма, Форма С 1-(4-(5-(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-этил-1Н-1,2,4-триазол-3-ил)пиперидин-1-ил)-3-гидроксипропан-1-она, которая имеет картину дифракции рентгеновских лучей на порошке по меньшей мере с одним характерным пиком при 2-тета, приблизительно составляющем 12,3°.
Согласно настоящему изобретению предложена кристаллическая форма, Форма С 1-(4-(5-(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-этил-1H-1,2,4-триазол-3-ил)пиперидин-1-ил)-3-гидроксипропан-1-она, которая имеет картину дифракции рентгеновских лучей на порошке по меньшей мере с двумя характерными пиками при 2-тета, приблизительно составляющих 6,9 и 12,3°.
Согласно настоящему изобретению предложена кристаллическая форма, Форма С 1-(4-(5-(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-этил-1H-1,2,4-триазол-3-ил)пиперидин-1-ил)-3-гидроксипропан-1-она, которая имеет картину дифракции рентгеновских лучей на порошке по меньшей мере с характерными пиками при 2-тета, приблизительно составляющих 6,9; 12,3; 10,5; 21,0; 24,6; 13,6; 16,4; 19,6; 20,2; 22,5°.
Согласно настоящему изобретению предложена кристаллическая форма, Форма С 1-(4-(5-(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-этил-1Н-1,2,4-триазол-3-ил)пиперидин-1-ил)-3-гидроксипропан-1-она, которая имеет картину дифракции рентгеновских лучей на порошке по существу аналогичную картине дифракции рентгеновских лучей на порошке, показанной на Фиг. 7.
Согласно настоящему изобретению предложена кристаллическая форма, Форма С 1-(4-(5-(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-этил-1H-1,2,4-триазол-3-ил)пиперидин-1-ил)-3-гидроксипропан-1-она, которая имеет картину дифракции рентгеновских лучей на порошке по меньшей мере с одним характерным пиком при 2-тета, составляющем 6,9° ± 0,2° 2-тета.
Согласно настоящему изобретению предложена кристаллическая форма, Форма С 1-(4-(5-(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-этил-1H-1,2,4-триазол-3-ил)пиперидин-1-ил)-3-гидроксипропан-1-, которая имеет картину дифракции рентгеновских лучей на порошке по меньшей мере с одним характерным пиком при 2-тета, составляющем 12,3° ± 0,2° 2-тета.
Согласно настоящему изобретению предложена кристаллическая форма, Форма С 1-(4-(5-(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-этил-1H-1,2,4-триазол-3-ил)пиперидин-1-ил)-3-гидроксипропан-1-она, которая имеет картину дифракции рентгеновских лучей на порошке по меньшей мере с двумя характерными пиками при 2-тета, составляющих 6,9° и 12,3°, где указанные значения могут отклоняться на величину ± 0,2° 2-тета.
Согласно настоящему изобретению предложена кристаллическая форма, Форма С 1-(4-(5-(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-этил-1H-1,2,4-триазол-3-ил)пиперидин-1-ил)-3-гидроксипропан-1-она, которая имеет картину дифракции рентгеновских лучей на порошке по меньшей мере с характерными пиками при 2-тета, составляющих 6,9; 12,3; 10,5; 21,0; 24,6; 13,6; 16,4; 19,6; 20,2; 22,5°, где указанные значения могут отклоняться на величину ± 0,2° 2-тета.
Когда указано, что настоящее изобретение относится к кристаллической форме соединения по изобретению, такого как соединение из примера 1 или примера 3, степень кристалличности подходящим образом составляет более чем примерно 60%, еще более подходящим образом более чем примерно 80%, предпочтительно более чем примерно 90% и более предпочтительно более чем примерно 95%. Наиболее предпочтительно, если степень кристалличности составляет более чем примерно 98%.
Когда указано, что настоящее изобретение относится к кристаллической форме соединения по изобретению, такого как соединение из примера 1 или примера 3, то кристаллическая форма предпочтительно по существу не содержит других кристаллических форм или аморфной формы того же соединения. В этом контексте, термин «по существу не содержит» подходящим образом означает более чем примерно 60%, еще более подходящим образом более чем примерно 80%, предпочтительно более чем примерно 90%, более предпочтительно более чем примерно 95%, еще более предпочтительно более чем примерно 98% и наиболее предпочтительно более чем примерно 99% чистой отдельной кристаллической формы. Например, соединение из примера 3 может быть в виде Формы A и по существу не содержать Форм B и C; альтернативно, соединение из примера 3 может быть в виде Формы B и по существу не содержать Форм A и C; альтернативно, соединение из примера 3 может быть в виде Формы C и по существу не содержать Форм A и B. Аналогично, соединение из примера 3 может быть в виде Формы B и по существу не содержать альтернативных кристаллических или аморфных форм.
Будет понятно, что значения 2-тета на картинах дифракции рентгеновских лучей на порошке могут незначительно изменяться от одного устройства к другому или от одного образца к другому, и поэтому приведенные значения не следует истолковывать как абсолютные.
Известно, что можно получить картину дифракции рентгеновских лучей на порошке, которая имеет одну или более ошибок измерения в зависимости от условий измерения (таких как используемое оборудование или устройство). В частности, общеизвестно, что интенсивности на картине дифракции рентгеновских лучей на порошке могут колебаться в зависимости от условий измерения. Таким образом, следует понимать, что кристаллические формы по настоящему изобретению, описанные выше, если не указано иное, не ограничиваются кристаллами, которые обеспечивают получение картин дифракции рентгеновских лучей на порошке, идентичных картине дифракции рентгеновских лучей на порошке, показанной на Фиг. 1, 3, 5, и любые кристаллы, обеспечивающие получение картин дифракции рентгеновских лучей на порошке, по существу аналогичных показанным на этих Фигурах, попадают в объем настоящего изобретения. Специалист в области дифракции рентгеновских лучей на порошке в состоянии иметь суждение, насколько существенно идентичны картины дифракции рентгеновских лучей на порошке.
Специалистам в области дифракции рентгеновских лучей на порошке также понятно, что на относительную интенсивность пиков могут оказывать влияние, например, гранулы в размере свыше 30 микрон и с неунитарными соотношениями сторон, что может отразиться на анализе образцов.
Специалисту также понятно, что на положение отражений может влиять точность размещения по высоте, на которой образец находится в дифрактометре, и калибровка нуля дифрактометра. Плоскостность поверхности образца также может производить небольшой эффект. Следовательно, представленные данные дифракционных картин не должны приниматься за абсолютные значения (см. Jenkins, R. and Snyder, R.L. "Introduction to X-Ray Powder Diffractometry", John Wiley and Sons, 1996; Bunn, C.W. (1948), Chemical Crystallography, CIarendon Press, London; Klug, H.P. and Alexander, L.E. (1974), X-Ray Diffraction Procedures).
Обычно ошибка измерения угла дифракции на картине дифракции рентгеновских лучей на порошке составляет приблизительно±0,2° 2-тета, и такую величину ошибки измерения необходимо учитывать при рассмотрении данных дифракции рентгеновских лучей на порошке. Кроме того, следует понимать, что интенсивности могут колебаться в зависимости от условий эксперимента и подготовки образца (предпочтительной ориентации).
Конкретные соединения по изобретению представляют собой соединения из примеров, каждое из которых составляет дополнительный независимый аспект изобретения. Другими конкретными соединениями по изобретению являются фармацевтически приемлемая(ые) соль(и) тех соединений из примеров, каждое из которых обеспечивает дополнительный независимый аспект изобретения.
Согласно дополнительному аспекту изобретения, предложено соединение формулы (I), которое получают, следуя любому из примеров, описанных в данном изобретении.
Следующим признаком является любой из объемов изобретения, определенных в данном описании, при условии, что конкретные соединения из примеров, таких как пример 1, 3, 4 и т.д., каждый в отдельности, исключены из притязаний.
Специалистам в данной области техники будет очевидно, что определенные соединения формулы (I) содержат асимметрические замещенные атомы углерода и соответственно могут существовать и могут быть выделены в оптически активных и рацемических формах. Некоторые соединения формулы (I) могут проявлять полиморфизм. Следует понимать, что настоящее изобретение охватывает любую рацемическую, оптически активную, полиморфную или стереоизомерную форму или их смеси, при этом форма обладает свойствами, полезными в ингибировании активности PI3K-α и -δ, причем в данной области техники хорошо известно, как получить оптически активные формы (например, путем выделения из рацемической формы методами перекристаллизации, путем синтеза из оптически активных исходных веществ, посредством хирального синтеза, путем разделения с использованием ферментов, путем биотрансформации или путем хроматографического разделения с использованием хиральной неподвижной фазы) и как определить эффективность ингибирования активности PI3K-α и -δ, используя стандартные тесты, описанные далее
Следует понимать, что некоторые соединения формулы (I), определенные выше, могут демонстрировать явление таутомерии. Следует понимать, что настоящее изобретение включает в своем определении любую такую таутомерную форму или их смесь, которая обладает PI3K-ингибирующей активностью, и не должно быть ограничено только какой-либо одной таутомерной формой, использованной в изображениях формул или приведенной в названиях примеров. В общем случае, только одну из всех таких таутомерных форм приводят в названиях примеров, следующих далее, или представляют в любых релевантных изображениях формул, следующих далее.
Подразумевается, что настоящее изобретение включает все изотопы атомов, имеющихся в соединениях по настоящему изобретению. Понятно, что изотопы включают такие атомы, которые имеют один и тот же атомный номер, но разные массовые числа. Например, изотопы водорода включают тритий и дейтерий. Изотопы углерода включают 13C и 14C.
Подходящей фармацевтически приемлемой солью соединения формулы (I) является, например, полученная присоединением кислоты соль соединения формулы (I), например соль присоединения сильной неорганической или органической кислоты, такой как соляная, бромистоводородная, серная или трифторуксусная кислота. Другой подходящей фармацевтически приемлемой солью соединения формулы (I) является, например, соль, образуемая в организме человека или животного после введения соединения формулы (I).
Кроме этого следует понимать, что подходящий фармацевтически приемлемый сольват соединения формулы (I) также образует аспект настоящего изобретения. Подходящим фармацевтически приемлемым сольватом является, например, гидрат, как например полугидрат, моногидрат, дигидрат или тригидрат, или его альтернативное количество.
Кроме этого следует понимать, что подходящее фармацевтически приемлемое пролекарство соединения формулы (I) также образует аспект настоящего изобретения. Соответственно, соединения по изобретению можно вводить в форме пролекарства, представляющего собой соединение, которое распадается в организме человека или животного с высвобождением соединения по изобретению. Пролекарство можно применять для изменения физических свойств и/или фармакокинетических свойств соединения по изобретению. Пролекарство может образовываться, если соединение по изобретению содержит подходящую группу или подходящий заместитель, к которой (которому) может присоединяться группа, модифицирующая какое-либо свойство. Примеры пролекарств включают расщепляемые in vivo производные сложных эфиров, которые могут образовываться по гидроксильной группе в соединении формулы (I), и расщепляемые in vivo производные амидов, которые могут образовываться по аминогруппе в соединении формулы (I).
Соответственно, настоящее изобретение включает те соединения формулы (I), как определено выше, которые становятся доступными посредством органического синтеза и которые становятся доступными в организме человека или животного в результате расщепления их пролекарственной формы. Соответственно, настоящее изобретение включает те соединения формулы (I), которые получают методами органического синтеза, а также те соединения, которые образуются в организме человека или животного в ходе метаболизма соединения-предшественника, то есть соединение формулы (I) может представлять собой полученное синтетическим путем соединение или полученное метаболическим путем соединение.
Подходящим фармацевтически приемлемым пролекарством соединения формулы (I) является пролекарство, которое, согласно обоснованному медицинскому мнению считается подходящим для введения в организм человека или животного без нежелательных фармакологических активностей и без чрезмерной токсичности.
Различные формы пролекарства описаны, например, в следующих документах:
а) Methods in Enzvmoloqy, Vol. 42, p. 309-396, под ред. К. Widder и др. (Academic Press, 1985);
б) Design of Pro-drugs, edited by H. Bundgaard, (Elsevier, 1985);
в) A Textbook of Drug Design и Development, под ред. Krogsgaard-Larsen and H. Bundgaard, Chapter 5 "Design and Application of Pro-drugs", by H. Bundgaard p. 113-191 (1991);
г) H. Bundgaard, Advanced Drug Delivery Reviews, 8, 1-38 (1992);
д) H. Bundgaard et al., Journal of Pharmaceutical Sciences, 77, 285 (1988);
е) N. Kakeya et al., Chem. Pharm. Bull., 32, 692 (1984);
ж) Т. Higuchi and V. StelIa, "Pro-drugs as Novel Delivery Systems", A.C.S. Symposium Series, Volume 14; и
з) E. Roche (редактор), "Bioreversible Carriers in Drug Design", Pergamon Press, 1987.
Подходящим фармацевтически приемлемым пролекарством соединения формулы (I), которое имеет гидроксильную группу, является, например, его сложный или простой эфир, расщепляемый in vivo. Расщепляемым in vivo сложным или простым эфиром соединения формулы (I), содержащего гидроксильную группу, является, например, фармацевтически приемлемый сложный или простой эфир, который расщепляется в организме человека или животного с получением исходного гидроксисоединения. Подходящие образующие фармацевтически приемлемый сложный эфир группы, в случае гидроксильной группы включают группы неорганических сложных эфиров, таких как сложные эфиры фосфорной кислоты (включая фосфорамидные циклические сложные эфиры). Другие подходящие образующие фармацевтически приемлемый сложный эфир группы, в случае гидроксильной группы включают (1-10C)алканоильные группы, такие как ацетильные, бензоильные, фенилацетильные и замещенные бензоильные и фенилацетильные группы, (1-10C)алкоксикарбонильные группы, такие как этоксикарбонильные, N,N-[ди-(1-4C)алкил]карбамоильные, 2-диалкиламиноацетильные и 2-карбоксиацетильные группы. Примеры заместителей в кольце фенилацетильной и бензоильной групп включают аминометил, N-алкиламинометил, N,N-диалкиламинометил, морфолинометил, пиперазин-1-илметил и 4-(1-4C)алкилпиперазин-1-илметил. Подходящие, образующие фармацевтически приемлемый простой эфир группы, в случае гидроксильной группы включают α-ацилоксиалкильные группы, такие как ацетоксиметильная и пивалоилоксиметильная группы.
Подходящим фармацевтически приемлемым пролекарством соединения формулы (I), которое имеет аминогруппу, является, например, его расщепляемое in vivo амидное производное. Подходящие фармацевтически приемлемые амиды, образующиеся из соединений с аминогруппой, включают, например, амид, образованный из соединений с (1-10C)алканоильными группами, такими как ацетильная, бензоильная, фенилацетильная и замещенные бензоильная и фенилацетильная группы. Примеры заместителей в кольце фенилацетильной и бензоильной групп включают аминометил, N-алкиламинометил, N,N-диалкиламинометил, морфолинометил, пиперазин-1-илметил и 4-(1-4C)алкилпиперазин-1-илметил.
Действия in vivo соединения формулы (I) могут проявляться частично посредством одного или более метаболитов, которые образуются в организме человека или животного после введения соединения формулы (I). Как указано ранее, действия in vivo соединения формулы (I) также могут проявляться в процессе метаболизма соединения-предшественника (пролекарства).
Соединения формулы (I) содержат субъединицу пиперидина, замещенную группой -C(O)R2, где R2 представляет собой (C2-3)алкил, замещенный гидроксилом. Один из возможных путей метаболизма этих соединений осуществляется посредством окисления гидроксильного заместителя на этой группе. У этих окисленных соединений обычно сохраняется некоторая ингибирующая PI3K-α и -δ активность.
Таким образом, согласно другому аспекту изобретения предложено соединение формулы (A):
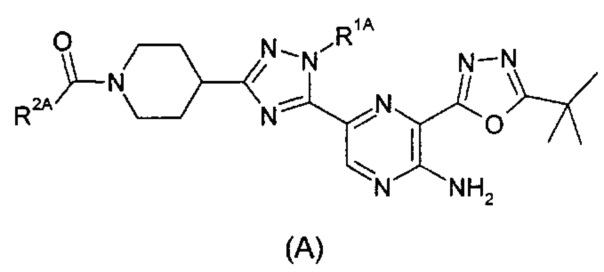 ,
,
где:
R1A представляет собой метил или этил; и
R2A представляет собой (С1-2)алкил, замещенный группой карбокси;
или его фармацевтически приемлемая соль.
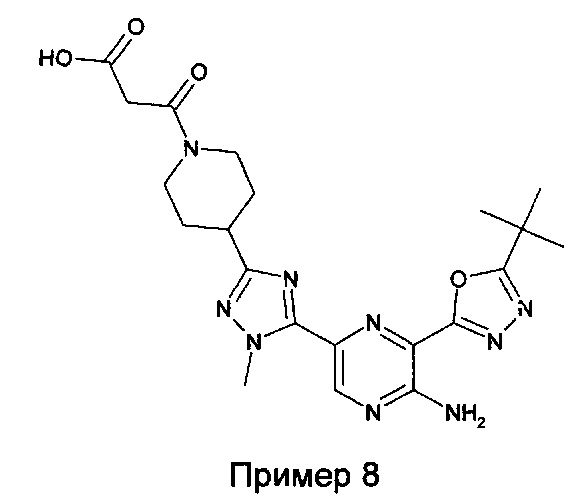
Примеры соединений формулы (А) включают соединение из примера 8, которое представляет собой идентифицированный метаболит соединения из примера 1,
и соединение из примера 9, которое представляет собой идентифицированный метаболит соединения из примера 3:
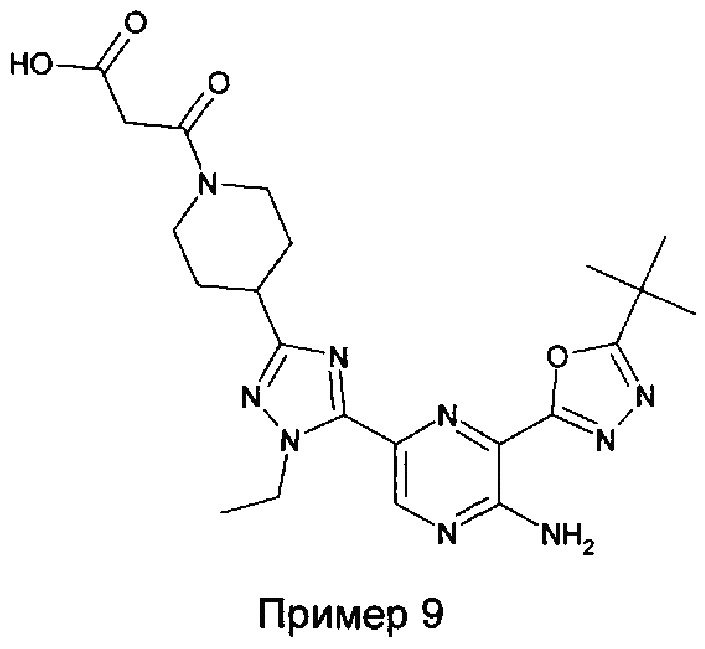 .
.
Другие возможные метаболиты соединения из примера 3 представляют собой два альтернативных продукта окисления, показанные ниже и описанные далее в примерах 10 и 11.
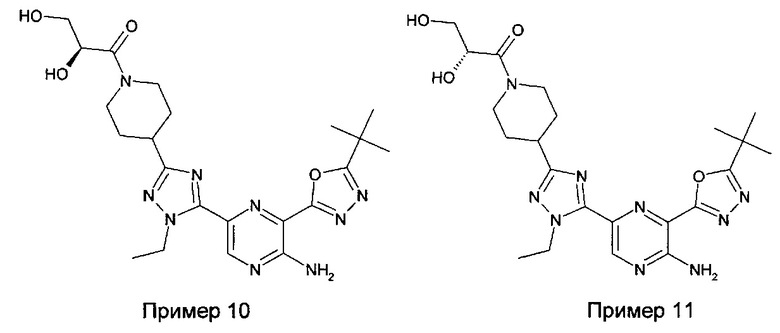
Подходящие фармацевтически приемлемые соли соединений формулы (А) включают, например соль щелочного или щелочноземельного металла, такую как соль кальция или магния, или соль аммония, или соль с органическим основанием, таким как метиламин, диметиламин, триметиламин, пиперидин, морфолин или трис-(2-гидроксиэтил)амин.
Во избежание неясности следует понимать, что там, где в данном описании группа квалифицируется как «ранее определенная» или «определенная ранее», указанная группа охватывает первое упоминаемое и самое широкое определение, а также каждое и все конкретные определения для этой группы.
Конкретные новые соединения по изобретению включают, например, соединения формулы (I) или их фармацевтически приемлемые соли, где, если не указано иное, каждый из R1 и R2 имеет любое из значений, определенных ранее или в приведенных ниже утверждениях:
R1 представляет собой метил;
R1 представляет собой этил;
R2 представляет собой любую из групп (1)-(11), которые определены ранее;
R2 представляет собой группы (1-(6), которые определены ранее;
R2 представляет собой группу (1).
Конкретную группу соединений по изобретению составляют соединения формулы (I), приведенной выше, где:
R1 представляет собой метил или этил,
R2 представляет собой группу (1):
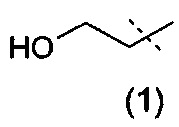 ,
,
или их фармацевтически приемлемая соль.
Конкретными соединениями по изобретению являются, например, соединения формулы (I), которые раскрыты в Примерах, которые приведены далее.
Например, конкретным соединением по изобретению является соединение формулы (I), выбранное из любого соединения, приведенного ниже:
1-[4-[5-[5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил]-1-метил-1,2,4-триазол-3-ил]-1-пиперидил]-3-гидрокси-пропан-1-она (пример 1 и 2);
1-[4-[5-[5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил]-1-этил-1,2,4-триазол-3-ил]-1-пиперидил]-3-гидрокси-пропан-1-она (пример 3);
(3R)-1-[4-[5-[5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил]-1-метил-1,2,4-триазол-3-ил]-1-пиперидил]-3-гидрокси-бутан-1-она (пример 4);
(3S)-1-[4-[5-[5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил]-1-метил-1,2,4-триазол-3-ил]-1-пиперидил]-3-гидрокси-бутан-1-она (пример 5);
(2R)-1-[4-[5-[5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил]-1-метил-1,2,4-триазол-3-ил]-1-пиперидил]-3-гидрокси-2-метил-пропан-1-она (пример 6);
1-[4-[5-[5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил]-1-метил-1,2,4-триазол-3-ил]-1-пиперидил]-2-гидрокси-2-метил-пропан-1-она (пример 7).
Согласно другому аспекту настоящего изобретения предложен способ получения соединения формулы (I) или его фармацевтически приемлемой соли. Подходящий способ проиллюстрирован следующими репрезентативными вариантами способа, в которых, если не указано иное, R1 и R2 принимают любое из значений, определенных ранее. Необходимые исходные вещества могут быть получены по стандартным методикам органической химии. Получение таких исходных веществ описано вместе со следующими далее репрезентативными вариантами способа и в сопроводительных примерах. Альтернативно, необходимые исходные вещества могут быть получены по методикам, аналогичным приведенным целью иллюстрации, которые известны специалисту в области органической химии.
Подходящие варианты способа включают, например, приведенные ниже.
(а) Взаимодействие, подходящим образом в присутствии подходящего активирующего реагента, соединения формулы II,
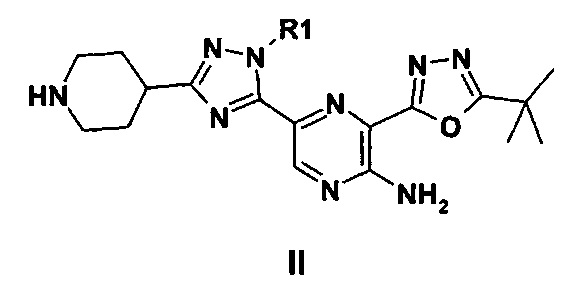
где R1 имеет любое из значений, определенных ранее, с карбоновой кислотой R2-COOH, за исключением того, что любая функциональная группа при необходимости защищена, в присутствии подходящего основания, после чего любую защитную группу, которая присутствует, удаляют.
Подходящие агенты сочетания для этой реакции включают, например гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония, TBTU (тетрафторборат 2-(1H-бензо[d][1,2,3]триазол-1-ил)-1,1,3,3-тетраметилизоурония) или гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида в присутствии N-оксида 2-гидрокси-пиридина.
Подходящим образом эту реакцию проводят в присутствии подходящего основания. Подходящим основанием является, например, основание органический амин, такое как, например, пиридин, 2,6-лутидин, коллидин, 4-диметиламинопиридин, триэтиламин, N-метилморфолин, диазабицикло[5.4.0]ундец-7-ен, диизопропилэтиламин, или, например, карбонат или гидроксид щелочного или щелочноземельного металла, например карбонат натрия, карбонат калия, карбонат кальция, гидроксид натрия или гидроксид калия; предпочтительно N-этил-N,N-диизопропиламин.
Подходящим образом эту реакцию проводят в присутствии подходящего инертного растворителя, такого как, например ацетонитрил, N,N-диметилформамид, N-метилпирролидон, тетрагидрофуран, 1,4-диоксан, 1,2-диметоксиэтан, бензол, толуол, ксилол, метанол, этанол, галогенированные растворители, такие как дихлорметан, хлороформ или четыреххлористый углерод, и при температуре в диапазоне, например, от -50°C до 100°C, предпочтительно в диапазоне от 0°C до 30°C.
Альтернативно, карбоновую кислоту R2-COOH можно перевести в активированную форму, которая затем может взаимодействовать с соединением формулы II в условиях, хорошо известных в данной области техники.
Подходящей защитной группой для гидроксильной группы является защитная группа тетрагидропирана, как описано в примере 2 и 3.
Подходящие условия для удаления этой группы включают слабокислотные условия в присутствии спирта, такого как метанол или этанол, в качестве растворителя при температуре от 20 до 70°C. Типичной используемой слабой кислотой является пиридин-п-толуолсульфонат.
Соединение формулы II может быть получено путем взаимодействия соединения формулы III:
 ,
,
где P представляет собой защитную группу, такую как трет-бутоксикарбонил,
с соединением формулы R1-L, где L представляет собой подходящую уходящую группу, такую как, например, галогенсодержащая группа, такая как бром, йод (подходящим образом йод), в присутствии подходящего основания, после чего любую защитную группу, которая присутствует, удаляют.
Подходящим основанием является, например, основание органический амин, такое как 1,8-диазабицикло[5.4.0]ундец-7-ен.
Подходящим образом эту реакцию проводят в присутствии подходящего инертного растворителя, такого как, например 2-метилтетрагидрофуран, тетрагидрофуран, 1,4-диоксан, 1,2-диметоксиэтан, бензол, толуол, ксилол, и при температуре в диапазоне, например, от -50°C до 60°C, предпочтительно в диапазоне от -10°C до 0°C.
Подходящие условия для удаления защитной трет-бутоксикарбонильной группы включают кислотные условия, такие как трифторуксусная кислота в инертном растворителе, таком как дихлорметан, при приблизительно комнатной температуре (20-25°С).
Соединение III может быть получено в результате реакции сочетания, в присутствии подходящего активирующего реагента, соединения формулы IV
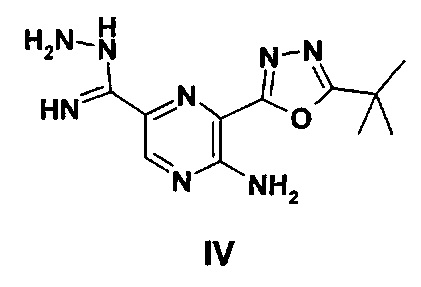
с соединением формулы V
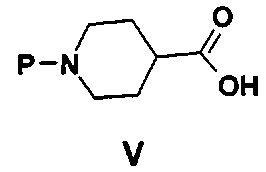 ,
,
предпочтительно в присутствии подходящего основания, с последующей реакцией циклизации в присутствии слабой кислоты.
Реакция сочетания может быть проведена в присутствии подходящего агента сочетания, такого как, например, гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония или TBTU (тетрафторборат 2-(1Н-бензо[d][1,2,3]триазол-1-ил)-1,1,3,3-тетраметилизоурония).
Подходящим образом реакцию сочетания проводят в присутствии подходящего основания. Подходящим основанием является, например, основание органический амин, такое как, например, пиридин, 2,6-лутидин, коллидин, 4-диметиламинопиридин, триэтиламин, N-метилморфолин, диазабицикло[5.4.0]ундец-7-ен, диизопропилэтиламин, или, например, карбонат или гидроксид щелочного или щелочноземельного металла, например карбонат натрия, карбонат калия, карбонат кальция, гидроксид натрия или гидроксид калия; предпочтительно N-этил-N,N-диизопропиламин.
Подходящим образом реакцию сочетания проводят в присутствии подходящего инертного растворителя, такого как, например N,N-диметилацетамид, N,N-диметилформамид, N-метилпирролидон, тетрагидрофуран, 1,4-диоксан, 1,2-диметоксиэтан, бензол, толуол, ксилол, метанол, этанол, галогенированные растворители, такие как дихлорметан, хлороформ или четыреххлористый углерод, и при температуре в диапазоне, например, от -50°C до 100°C, предпочтительно в диапазоне от 0°C до 30°C.
Циклизацию проводят в присутствии слабой кислоты, обычно уксусной кислоты. Подходящим образом эту реакцию проводят в присутствии подходящего инертного растворителя, такого как, например N,N-диметилацетамид, N,N-диметилформамид, N-метилпирролидон, тетрагидрофуран, 1,4-диоксан, 1,2-диметоксиэтан, бензол, толуол, ксилол, при температуре в диапазоне, например, от 50°C до 150°C, предпочтительно в диапазоне от 80°C до 100°C.
Соединение формулы IV может быть получено путем взаимодействия соединения формулы VI с гидразином.
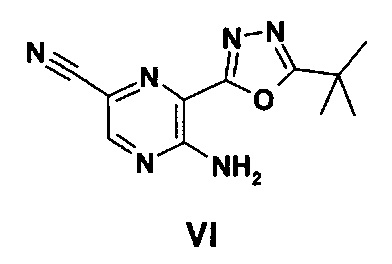
Подходящим образом эту реакцию проводят в присутствии подходящего инертного растворителя, такого как, например тетрагидрофуран, 1,4-диоксан, 1,2-диметоксиэтан, бензол, толуол, ксилол или спирт, такой как этанол или изопропанол, при температуре в диапазоне, например, от 20°C до 70°C, предпочтительно примерно 50°C.
Соединение формулы VI может быть получено в результате катализируемой металлами реакции, осуществляемой между соединением формулы VII и источником цианида, таким как дицианид цинка(II).
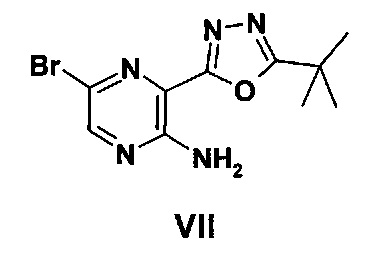
Подходящий для этой реакции катализатор включает, например, металлический катализатор, такой как палладий(0), например тетракис(трифенилфосфин)палладий(0); или катализатор, образованный in situ из соли палладия(II), например ацетата палладия(II), хлорида палладия(II), бромида палладия(II), хлорида бис(трифенилфосфин)палладия(II), [1,1'-бис(дифенилфосфино)ферроцен]-дихлорпалладия(II) или трис(дибензилиденацетон)дипалладия, и фосфинового лиганда, например, дициклогексил(2',4',6'-триизопропилбифенил-2-ил)фосфина. Подходящим образом эту реакцию проводят в подходящем растворителе, таком как N,N-диметилацетамид, N,N-диметилформамид, тетрагидрофуран, 1,4-диоксан, 1,2-диметоксиэтан, бензол, толуол или ксилол, и при температуре в диапазоне, например, от 20°C до 150°C, предпочтительно в диапазоне от 60°C до 120°C. Также подходящим образом реакцию проводят в присутствии дополнительного металла, такого как цинк.
Подходящие реакции этого типа описаны в "Metal-Catalyzed Cross-Coupling Reactions", второе издание, под ред. Armin Meijere, Francois Diederich, Wiley-VCH, 2004.
Синтез соединения VII описан в примерах 1 и 2.
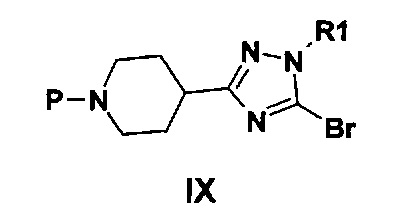
Альтернативно, соединение формулы II может быть получено в результате катализируемой металлами реакции, осуществляемой между соединением формулы VIII, где R представляет собой небольшую алкильную группу, и соединением IX, где P представляет собой защитную группу, такую как трет-бутокси карбонил.

Подходящий для этой реакции катализатор включает, например, металлический катализатор, такой как палладий(0), например тетракис(трифенилфосфин)палладий(0); или катализатор, образованный in situ из соли палладия(II), например ацетата палладия(II), хлорида палладия(II), бромида палладия(II), хлорида бис(трифенилфосфин)палладия(II), [1,1'-бис(дифенилфосфино)ферроцен]-дихлорпалладия(II) или трис(дибензилиденацетон)дипалладия, и фосфинового лиганда, например, дициклогексил(2',4',6'-триизопропилбифенил-2-ил)фосфина.
Подходящим образом эту реакцию проводят в подходящем растворителе, таком как N,N-диметилацетамид, N,N-диметилформамид, тетрагидрофуран, 1,4-диоксан, 1,2-диметоксиэтан, бензол, толуол, ксилол или спирт, такой как 4-метил-2-пентанол, и при температуре в диапазоне, например, от 50°C до 180°C, предпочтительно в диапазоне от 120°C до 150°C.
Также подходящим образом реакцию проводят в присутствии дополнительной соли, такой как хлорид лития.
Подходящие реакции этого типа описаны в "Metal-Catalyzed Cross-Coupling Reactions", второе издание, под ред. Armin Meijere, Francois Diederich, Wiley-VCH, 2004.
Соединение формулы VIII может быть получено в результате катализируемой металлами реакции, осуществляемой между соединением формулы VII и подходящим гексаалкил-дистаннаном. Подходящий для этой реакции катализатор включает, например, металлический катализатор, такой как палладий(0), например тетракис(трифенилфосфин)палладий(0); или катализатор, образованный in situ из соли палладия(II), например ацетата палладия(II), хлорида палладия(II), бромида палладия(II), хлорида бис(трифенилфосфин)палладия(II), [1,1'-бис(дифенилфосфино)ферроцен]-дихлорпалладия(II) или трис(дибензилиденацетон)дипалладия, и фосфинового лиганда, например, дициклогексил(2',4',6'-триизопропилбифенил-2-ил)фосфина.
Подходящим образом эту реакцию проводят в подходящем растворителе, таком как N,N-диметилацетамид, N,N-диметилформамид, тетрагидрофуран, 1,4-диоксан, 1,2-диметоксиэтан, бензол, толуол, ксилол или спирт, такой как 4-метил-2-пентанол, при температуре в диапазоне, например, от 50°C до 100°C, предпочтительно в диапазоне от 70°C до 80°C.
Соединение формулы IX может быть получено из имеющегося в продаже вещества за несколько стадий, как проиллюстрировано в примере 1 (при этом R1 представляет собой Me, а P представляет собой трет-бутоксикарбонил).
Следует понимать, что также возможны другие изменения стадий способа в вариантах способа, описанных выше.
Следует понимать, что любое соединение формулы (I), полученное посредством любого из способов, описанных ранее, при необходимости может быть превращено в другое соединение формулы (I).
Когда необходима фармацевтически приемлемая соль соединения формулы (I), например соль присоединения кислоты, она может быть получена путем, например, взаимодействия указанного соединения с подходящей кислотой.
Когда необходимо фармацевтически приемлемое пролекарство соединения формулы (I), оно может быть получено с использованием традиционной методики. Например, расщепляемый in vivo сложный эфир соединения формулы (I) может быть получен, например, путем взаимодействия соединения формулы (I), содержащего гидроксильную группу, с фармацевтически приемлемой карбоновой кислотой. Другая информация о пролекарствах приведена ранее.
Также будет очевидно, что в некоторых из реакций, упомянутых ранее, может оказаться необходимо или желательно защитить какие-либо чувствительные группы в соединениях. Случаи, где защита необходима или желательна, а также подходящие методы защиты известны специалистам в данной области техники. В соответствии с общепринятой практикой можно использовать традиционные защитные группы (для иллюстрации см. T.W. Green, Protective Groups in Organic Synthesis, John Wiley and Sons, 1991). Таким образом, если реагенты включают такие группы, как амино, карбокси или гидрокси, может оказаться необходимо или желательно защитить группу в некоторых из реакций, упомянутых в данном описании.
Подходящей защитной группой для амино- или алкиламиногруппы является, например, ацильная группа, например алканоильная группа, такая как ацетильная, алкоксикарбонильная группа, например метоксикарбонильная, этоксикарбонильная или трет-бутоксикарбонильная группа, арилметоксикарбонильная группа, например бензилоксикарбонильная, или ароильная группа, например бензоильная. Условия удаления защиты для вышеуказанных защитных групп несомненно меняются в зависимости от выбора защитной группы. Так, например, ацильная группа, такая как алканоильная или алкоксикарбонильная группа, или ароильная группа может быть удалена, например, путем гидролиза в присутствии подходящего основания, такого как гидроксид щелочного металла, например гидроксид лития или натрия. Альтернативно, ацильная группа, такая как трет-бутоксикарбонильная группа, может быть удалена, например, путем обработки подходящей кислотой, такой как соляная, серная или фосфорная кислота либо трифторуксусная кислота, а арилметоксикарбонильная группа, такая как бензилоксикарбонильная группа, может быть удалена, например, путем гидрирования над катализатором, таким как палладий-на-угле, или путем обработки кислотой Льюиса, например трис(трифторацетатом) бора. Подходящей альтернативной защитной группой для первичной аминогруппы является, например, фталоильная группа, которая может быть удалена путем обработки алкиламином, например диметиламинопропиламином, или гидразином.
Подходящей защитной группой для гидроксильной группы является, например, ацильная группа, например алканоильная группа, такая как ацетильная, ароильная группа, например бензоильная, или арилметильная группа, например бензильная. Условия удаления защиты для вышеупомянутых защитных групп несомненно будут меняться в зависимости от выбора защитной группы. Так, например, ацильная группа, такая как алканоильная или ароильная группа, может быть удалена, например, путем гидролиза в присутствии подходящего основания, такого как гидроксид щелочного металла, например гидроксид лития или натрия. Альтернативно, арилметильная группа, такая как бензильная группа, может быть удалена, например, путем гидрирования над катализатором, таким как палладий-на-угле.
Подходящей защитной группой для карбоксильной группы является, например, этерифицирующая группа, например метильная или этильная группа, которая может быть удалена, например, путем гидролиза в присутствии основания, такого как гидроксид натрия, или, например, трет-бутильная группа, которая может быть удалена, например, путем обработки кислотой, например органической кислотой, такой как трифторуксусная кислота, или, например, бензильная группа, которая может быть удалена, например, путем гидрирования над катализатором, таким как палладий-на-угле.
Защитные группы могут быть удалены на любой удобной стадии синтеза с использованием традиционных методов, хорошо известных в области химии.
Некоторые из промежуточных соединений (например, соединения формул II, III, IV, VI, VII, VIII), определенных в данном описании, являются новыми, и они предложены в качестве дополнительного признака данного изобретения.
Биологические анализы
Приведенные ниже анализы использовали для измерения эффектов соединений по настоящему изобретению: а) в качестве ингибиторов PI3-киназных ферментов в биохимических анализах, б) в качестве ингибиторов других киназ в биохимических анализах, в) в качестве ингибиторов in vitro фосфо-АКТ (Thr308) в клетках ВТ474, г) в качестве ингибиторов in vitro фосфо-АКТ (Ser473) в клетках MDA-MB-468, д) в качестве ингибиторов in vitro фосфо-АКТ (Ser473) в клетках JEKO, е) в качестве ингибиторов in vitro фосфо-Chk1 (Ser345) в клетках НТ29, ж) в качестве ингибиторов пролиферации клеток по всей панели линий опухолевых клеток, з и и) в качестве ингибиторов in vivo фосфо-АКТ (Ser473) или ингибиторов in vivo роста опухоли, соответственно, у мышей с тяжелым комбинированным иммунодефицитом (SCID), трансплантированных линией клеток аденокарциномы молочной железы человека, MCF7.
Сокращения, использованные в протоколах анализов:
(а) Анализ ингибирования ферментов in vitro
Ингибирование PI3K-β, PI3K-α, PI3K-γ и PI3K-δ оценивали на основе анализа ферментативной активности Kinase-Glo с использованием рекомбинантных ферментов человека. В основе анализа лежит опосредованное измерение истощения АТФ после инкубации с ферментом, субстратом PIP2, АТФ и соединением.
По завершении этой ферментативной реакции оставшийся АТФ использовали во вторичной ферментативной реакции, в которой люцифераза превращала люциферин жука в оксилюциферин с испусканием света. Между измеренной люминесценцией и АТФ, оставшимся в завершенной киназной реакции, существовала прямая зависимость. Таким образом, люминесценция обратно пропорциональна киназной активности. Обычно тестировали двенадцать разных концентраций соединения и строили графики зависимости начальных данных ингибирования PI3K-β, PI3K-α, PI3K-γ или PI3K-δ от концентрации ингибитора.
Подробное описание метода
Соединения в 100%-ном DMSO добавляли в используемые в анализе планшеты с помощью акустического диспенсера (acoustic dispensing). Фермент PI3K добавляли в трис-буфере (50 мМ трис, pH 7,4; 0,05% CHAPS, 2,1 мМ DTT и 10 мМ хлорид магния) и проводили преинкубацию с соединением в течение 20 минут, после чего добавляли раствор субстрата, содержащий PIP2 и АТФ. Ферментативную реакцию останавливали через 80 минут, добавляя раствор для обнаружения киназ Kinase-Glo, содержащий люциферин и люциферазу (из набора для анализа киназ люминесцентным методом Kinase-Glo® Plus (Promega, № V3772)). Планшеты оставляли на 30 минут при комнатной температуре, затем прочитывали на приборе PHERAstar со стандартным блоком люминесцентных светофильтров. Конечная концентрация DMSO, АТФ и PIP2 в данном анализе составляла 1%, 8 мкМ и 80 мкМ соответственно.
Анализ данных
Значения IC50 рассчитывали, используя log-аппроксимацию кривых методом нелинейной регрессии. Величина IC50 представляла собой концентрацию тестируемого соединения, которая ингибировала на 50% активность фермента.
(б) Оценка селективности в отношении киназ помимо PI3-киназных Ферментов класса I
Рядом коммерческих поставщиков, таких как Millipore, Invitrogen и ProQinase, предлагаются многочисленные панели киназных анализов. Такие панели дают возможность оценить общую селективность заданного соединения в отношении киназ. Точность методов/технологий будет варьировать в зависимости от поставщика.
Данные по селективности для некоторых соединений, описанных в данной заявке, получали с использованием ферментативных анализов, выполненных в Совете по медицинским исследованиям (MRC) - в Отделе терапии, направленной на пути передачи сигнала (Division of Signal Transduction Therapy, DSTT), отделении фосфорилирования белков MRC, г. Данди, Соединенное Королевство. Тестирование протеинкиназ осуществляли, используя формат радиохимического метода анализа. Анализы проводили в 384-луночных планшетах Multidrop при комнатной температуре в анализируемом объеме в общей сложности 25,5 мкл. Соединения предварительно инкубировали в присутствии фермента и пептидного/белкового субстрата в течение 5 минут, после чего реакцию инициировали добавлением 10 мкл раствора АТФ (конечная концентрация, выбранная для каждой киназы, составляла 5, 20 или 50 мкМ). Анализы выполняли при комнатной температуре, после чего реакцию останавливали, добавляя по 5 мкл раствора ортофосфорной кислоты. Содержимое используемого в анализе планшета затем собирали в фильтровальные планшеты Р81 Unifilter от Whatman с помощью харвестера от Packard (в качестве буфера для промывки использовали 50 мМ ортофосфорную кислоту) и сушили на воздухе. Сухие фильтровальные планшеты Unifilter затем герметично закрывали после добавления сцинтилляционной жидкости MicroScint O и считывание проводили на сцинтилляционных счетчиках Topcount NXT от Packard. Этот протокол охватывает обычный формат, подходящий для большинства киназ в данной панели, однако для незначительного количества киназ потребовались модификации к этим протоколам, которые известны специалистам в данной области техники.
Кроме того, примерно для 18 липидных киназ проводили анализы липидных киназ в DSTT. Все анализы липидных киназ проводили в 384-луночных планшетах при комнатной температуре в анализируемом объеме в общей сложности 40 мкл. Такой анализ выполняли в соответствии с протоколами, приведенными в анализе ADP-GLO (Promega, №V9101). Этот протокол охватывает обычный формат, подходящий для большинства киназ в данной панели, однако для незначительного количества киназ потребовались модификации к этим протоколам, которые известны специалистам в данной области техники.
Селективность в отношении киназ также оценивали с использованием платформы для скрининга KINOMEscan™, доступной от DiscoverX. В ней используется анализ активного сайт-специфического конкурентного связывания для количественного измерения взаимодействий между тестируемыми соединениями и более чем 450 киназами человека и мутантными вариантами, имеющими отношение к заболеваниям. Для анализов с применением KINOMEscan™ не требуется присутствия АТФ, и вследствие этого регистрируются истинные термодинамические аффинности взаимодействия, в отличие от величин IC50, которые могут зависеть от концентрации АТФ. Данная методология основывается на использовании соединений, которые связываются с активным центром киназ и непосредственно (стерически) или опосредованно (аллостерически) предотвращают связывание киназы с иммобилизованным лигандом, тем самым уменьшая количество киназы, захваченной на твердой подложке. В противоположность этому, тестируемые молекулы, которые не связываются с киназой, не оказывают никакого влияния на количество киназы, захваченной на твердой подложке. При скрининге выявляют соединения-«хиты», измеряя количество захваченной в тесте киназы против контрольных образцов, с использованием метода количественной полимеразной цепной реакции (колич. ПЦР), в котором осуществляют детектирование связанной с ДНК метки. Аналогичным образом рассчитывают константы диссоциации (Kd) для тестируемых взаимодействий соединение-киназа, измеряя количество киназы, захваченной на твердой подложке, как функцию от концентрации тестируемого соединения.
(в) Протокол анализа для измерения фосфорилированной АКТ (Tyr308) в клетках ВТ474
Этот анализ использовали для измерения ингибирования PI3K-α в клетках. Клетки ВТ474 (протоковая карцинома молочной железы человека, номер в Американской коллекции типовых культур (АТСС) НТВ-20) рассевали в черные 384-луночные планшеты (Costar, №3712) при плотности 5600 клеток/лунка в DMEM, содержащей 10% FBS и 1% глутамина, и оставляли прикрепляться в течение ночи.
На следующее утро в используемые в анализе планшеты добавляли соединения в 100%-ном DMSO с помощью ультразвукового дозирующего устройства. Через 2 часа инкубации при 37°C и 5% CO2 среду отсасывали и клетки подвергали лизису, используя буфер, содержащий 25 мМ трис, 3 мМ EDTA, 3 мМ EGTA, 50 мМ фторид натрия, 2 мМ ортованадат натрия, 0,27 М сахарозу, 10 мМ β-глицерофосфат, 5 мМ пирофосфат натрия, 0,5% тритона X-100 и таблетки полного коктейля ингибиторов протеаз (Roche, №04693116001, использовали 1 таблетку на 50 мл лизирующего буфера).
Через 20 минут клеточные лизаты переносили в планшеты для иммуноферментного твердофазного анализа (ELISA) (Greiner, №781077), которые предварительно были покрыты антителом к общей АКТ в PBS-буфере, и неспецифическое связывание блокировали 1%-ным BSA в PBS, содержащем 0,05% твина 20. Планшеты инкубировали в течение ночи при 4°C. На следующий день планшеты промывали буфером на основе PBS, содержащим 0,05% твина 20, и далее инкубировали с мышиным моноклональным антителом к фосфо-АКТ Т308 в течение 2 часов. Планшеты снова промывали, как указано выше, после чего добавляли вторичное лошадиное атимышиное антитело, конъюгированное с HRP. После 2 часов инкубации при комнатной температуре планшеты промывали и в каждую лунку добавляли рабочий раствор субстрата QuantaBlu (Thermo Scientific, №15169, приготовленный в соответствии с инструкцией поставщика). Образование флуоресцентного продукта останавливали через 60 минут, добавляя в лунки стоп-раствор. Планшеты прочитывали, используя планшетный ридер Safire от Tecan с применением возбуждения при длине волны 325 нм и испускания при длине волны 420 нм, соответственно. За исключением указанных отдельно случаев в этом ELISA-анализе использовали реагенты, содержащиеся в наборе для «сэндвич»-ELISA фосфо-АКТ (Thr308) Path Scan от Cell Signalling (№7144).
(г) Протокол для обнаружения фосфо-AKT (Ser473) в клетках MDA-MB-468 как показателя ингибирования PI3-киназы-бета
Этот анализ использовали для измерения ингибирования PI3K-β в клетках и вместе с приведенным выше анализом (в) использовали для определения селективности в отношении киназы-альфа в сравнении с киназой-бета в клетках. Клетки MDA-MB-468 (аденокарцинома молочной железы человека, № в АТСС НТВ 132) рассевали при плотности 1500 клеток/лунка в 40 мкл DMEM, содержащей 10% FBS и 1% глутамина, в 384-луночных черных плоскодонных планшетах Greiner. Планшеты с клетками инкубировали в течение 18 часов в инкубаторе при 37°C, после чего вводили соединения в 100%-ном DMSO, используя ультразвуковое дозирующее устройство.
Соединения вносили в диапазоне концентраций (12 точек) в планшет согласно рандомизированной схеме (randomised plate map). В контрольные лунки вносили либо 100%-ный DMSO (максимальный сигнал), либо соединение сравнения (ингибитор PI3K-β), которое полностью устраняло сигнал от pAKT (минимальный контрольный сигнал). Планшеты инкубировали при 37°C в течение 2 часов, затем клетки фиксировали, добавляя 10 мкл 3,7%-ного раствора формальдегида. Через 30 минут планшеты промывали PBS, используя устройство для отмывки планшетов PW384 от Tecan. Лунки блокировали и клетки пермеабилизировали, добавляя 40 мкл PBS, содержащего 0,5% твина 20 и 1% Marvel™ (порошковое сухое молоко), и инкубировали в течение 60 минут при комнатной температуре. Планшеты промывали PBS, содержащим 0,5% (об./об.) твина 20 и 20 мкл раствора антитела кролика к фосфо-АКТ Ser473 (Cell Signalling Technologies, №3787) в той же смеси PBS-твин с добавлением 1% Marvel™ и инкубировали в течение ночи при 4°С.
Планшеты промывали 3 раза смесью PBS+0,05% твина 20, используя PW384 от Tecan. В каждую лунку добавляли по 20 мкл раствора вторичного антикроличьего антитела, конъюгированного с Alexa Fluor 488 (MolecuIar Probes, №A11008), разбавленного в смеси PBS + 0,05% твина 20, содержащей 1% Marvel™, и инкубировали в течение 1 часа при комнатной температуре. Планшеты промывали три раза, как и ранее, затем в каждую лунку добавляли по 20 мкл PBS и планшеты герметично закрывали черной пленкой для заклеивания планшетов.
Планшеты прочитывали на планшетном ридере Acumen в возможно короткие сроки, измеряя зеленую флуоресценцию после возбуждения с использованием лазера на длине волны 488 нм. Применяя эту систему, получали значения IC50, и качество планшетов определяли по контрольным лункам. Для мониторинга выполнения анализа каждый раз использовали соединения сравнения.
(д) Протокол для обнаружения фосфо-АКТ (Ser473) в клетках JEKO
Этот анализ использовали для измерения ингибирования PI3K-δ в клетках. Соединения в 10х конечной концентрации в 10 мкл 1%-ного (об./об.) DMSO добавляли в лунки с V-образным дном 96-луночного планшета Greiner (Sigma, № М9686). Соединения вносили в диапазоне концентраций (10 точек), начиная с максимальной дозы, составляющей 1 мкМ или 10 мкМ; в один планшет вносили 8 соединений. В одном планшете было 8 контрольных лунок для максимального сигнала, в которые вносили антитело к иммуноглобулину М (IgM) (очищенный с использованием аффинной хроматографии (AffiniPure) F(ab')2-фрагмент антитела козы к IgM человека (Stratech, №109-006-129)) и разбавитель, и 8 контрольных лунок для минимального сигнала, в которые вносили антитело к IgM и используемый для сравнения ингибитор PI3K-δ. Конечная концентрация разбавителя DMSO составляла 0,1%. В каждое измерение включали селективное в отношение PI3K-δ соединение для получения полной кривой зависимости доза-ответ. В-клетки Jeko (лимфома из клеток мантийной зоны человека, № в АТСС CRL-3006) рассевали в лунки с V-образным дном 96-луночных планшетов Greiner, содержащие соединения. Клетки рассевали при плотности 100000 клеток/лунка в 70 мкл среды RPMI, содержащей 1% глутамина.
Планшеты с клетками инкубировали с соединением в течение 1 часа в инкубаторе при 37°С. По окончании этого времени преинкубации с соединением в планшеты добавляли вышеописанное антитело к IgM в 5х конечной концентрации в 20 мкл буфера для анализа (среда RPMI, содержащая 1% глутамина). Конечная концентрация антитела к IgM составляла 0,06 мкг/мл или дозу, эквивалентную EC90. Планшеты инкубировали при 37°С в течение 10 мин, затем планшеты сразу помещали на лед и центрифугировали при 12000 об./мин в течение 4 мин. На льду, осторожно удаляли супернатанты с помощью неавтоматической пипетки и добавляли 40 мкл лизирующего буфера. Планшеты инкубировали на льду в течение 5 мин и хранили при -80°C до проведения анализа с использованием набора для определения фосфо-(Ser473)/общей Akt в клеточном лизате в соответствии с инструкциями производителя (Mesoscale Diagnostics, № K11100D-3).
(е) Протокол для обнаружения фосфо-Chk1 (Ser 345) в клетках НТ29
ATR (атаксия-телеангиэктазия+Pad3-родственная киназа) представляет собой киназу, родственную PI3-киназе, которая фосфорилирует многочисленные субстраты по остаткам серина или треонина в ответ на повреждение ДНК или наличие блоков репликации. Chk1, протеинкиназа, расположенная ниже ATR в каскаде, играет ключевую роль в регулировании контрольных точек повреждения ДНК. Активация Chk1 вовлекает фосфорилирование Ser317 и Ser345 (последний остаток рассматривается как предпочтительная мишень для фосфорилирования/активации под действием ATR).
Далее представлен сам анализ, основанный на использовании клеток, для определения ингибирования киназы ATR посредством измерения уменьшения степени фосфорилирования Chk1 (Ser 345) в клетках НТ29 после обработки соединением и УФ-миметиком 4NQO (Sigma, №N8141). Клетки НТ29 (Европейская коллекция клеточных линий животных (ЕСАСС), №85061109) рассевали в 384-луночные используемые в анализе планшеты (Costar, №3712) при плотности 6000 клеток/лунка в 40 мкл среды ЕМЕМ, содержащей 1% L-глутамина и 10% FBS, и оставляли прикрепляться в течение ночи. На следующее утро соединения в 100%-ном DMSO добавляли в используемые в анализе планшеты с помощью ультразвукового дозирующего устройства. Через 1 час инкубации при 37°C и 5% CO2 во все лунки добавляли по 40 нл 3 мМ 4NQO в 100%-ном DMSO с помощью ультразвукового дозирующего устройства, за исключением контрольных лунок для минимального сигнала, которые оставляли без добавления 4NQO для получения нулевого ответа в качестве контроля. Планшеты возвращали в инкубатор еще на 1 час. Затем клетки фиксировали, добавляя 20 мкл 3,7%-ного формальдегида в растворе PBS и инкубируя в течение 20 мин при комнатной температуре. После этого добавляли 20 мкл 0,1%-ного тритона Х100 в PBS и инкубировали в течение 10 минут при комнатной температуре для пермеабилизации клеток. Затем планшеты промывали один раз PBS из расчета 50 мкл/лунка, используя устройство для отмывки планшетов EL405 от Biotek.
Антитело к фосфо-Chk1 Ser 345 (Cell Signalling Technology, №2348) разбавляли в 150 раз в PBS, содержащем 0,05% полисорбата/твина, в каждую лунку добавляли по 15 мкл и инкубировали в течение ночи при комнатной температуре.
На следующее утро планшеты промывали три раза PBS из расчета 50 мкл/лунка, используя устройство для отмывки планшетов EL405 от Biotek, и затем добавляли 20 мкл раствора вторичного Ab, содержащего разбавленное в 500 раз антитело козы к IgG кролика, конъюгированное с Alexa Fluor 488 (MolecuIar Probes, № A-11008), и краситель Хехст в концентрации 0,002 мг/мл (MolecuIar Probes, № Н-3570) в PBST. Через 2 часа инкубации при комнатной температуре планшеты промывали три раза PBS из расчета 50 мкл/лунка, используя устройство для отмывки планшетов EL405 от Biotek, и затем планшеты герметично закрывали черными пленками для заклеивания планшетов до прочтения. Планшеты прочитывали, используя устройство ArrayScan от VTI с применением фильтра XF53 с 10х объективом. Использовали две установки лазера для анализа окрашивания ядер красителем Хехст (405 нМ) и окрашивания pChk1 (фосфо-Chk1) вторичным антителом (488 нМ).
(ж) Анализы пролиферации клеток в линиях опухолевых клеток (использованные для демонстрации гипотезы персонализированной медицины)
Чувствительность панели линий раковых клеток человека к действию соединений определяли в стандартном анализе пролиферации. Клеточные линии получали через Банк клеток AstraZeneca. Большинство клеточных линий также доступны через депозитарии Банков клеток, известных специалистам, работающим в данной области техники, такие как, например АТСС, ЕСАСС, Немецкая коллекция микроорганизмов и клеточных культур (DMSZ; Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH), Институт физико-химических исследований (Rikagaku Kenkyusho; RIKEN), Корейский банк клеточных линий (KCLB; Korean Cell Line Bank), Японская коллекция биоресурсов для научных исследований (JCRB; Japanese Collection of Research Bioresources) (HSRRB; Health Science Research Resources Bank), Национальная лаборатория им. Лоуренса в Беркли (LBNL; Lawrence Berkeley National Laboratory), CLS (Cell line Service) и Межлабораторная коллекция клеточных линий (ICLC; Interlab Cell Line Collection).
Клетки высевали в 96-луночные планшеты при плотности 1000-6000 клеток на одну лунку в среде RPMI, содержащей 10% FBS. После инкубации при 37°C в течение 16 часов в используемые в анализе планшеты добавляли соединение в различных концентрациях. После инкубации в течение еще 72 ч жизнеспособные клетки определяли через 2 ч после добавления в каждую лунку реагента на основе MTS (Promega, №3582). MTS представляет собой соль тетразолия, которая биовосстанавливается метаболически активными клетками в присутствии реагента сочетания, осуществляющего перенос электронов, до формазана. Затем проводили количественное определение продукта формазана по поглощению при 490 нм, в качестве индикатора относительного количества живых клеток. Чтобы определить GI50 (концентрацию, при которой рост клеток ингибировался на 50%), определяли относительное количество клеток, присутствующих в случае добавления лекарственного средства, в сравнении с данными, полученными с использованием MTS перед добавлением лекарственного средства, и эту величину вычитали из величины, соответствующей 72 ч для необработанных клеток, в качестве показателя клеточного роста во время анализа.
Анализ этих данных, описанный ниже под заголовком «Примеры персонализированной медицинской помощи/персонализированной медицины», иллюстрирует, как эти данные могут быть использованы для выявления того, что ингибиторы PI3Kα демонстрируют селективное ингибирование роста клеточных линий с мутацией в гене PIK3CA. Это иллюстрирует перспективы персонализированной медицинской помощи (РНС) или персонализированной медицины в случае, когда показание для дающего предсказательный ответ биомаркера могут быть использованы для идентификации пациентов с опухолями, содержащими мутацию в гене PIK3CA, и тех из них, которые с большей долей вероятности будут давать ответ на соединения, описанные в данной заявке.
Другие возможные маркеры ответа на соединения, описанные в данной заявке, включают, но не ограничиваются этим, увеличенное число копий, амплификацию или транслокацию гена PIK3CA и другие генетические, геномные или протеомные изменения, которые обеспечивают получение показаний, касающихся активации PI3-киназного пути или зависимости от PI3-киназного пути; например, но не ограничиваясь этим, таких показаний, как активация одной или более чем одной рецепторной тирозинкиназы либо мутация в генах или транслокация генов PIK3R, которые кодируют регуляторные субъединицы (р85) PI3-киназы, или фосфорилирование маркеров, расположенных ниже в пути передачи сигнала, таких как pAKT, pS6 или статус FOXO. Помимо этого, анализ других генов и/или их белковых продуктов, участвующих в передаче сигнала, например K-ras, может помочь в повышении предсказательной способности персонализированного подхода в медицине.
(з) Протокол для обнаружения фосфо-АКТ (Ser473), происходящей из опухолей MCF-7, развившихся у самцов SCID-мышей
Далее представлен фармакодинамический анализ для измерения ингибирования PI3K-α в животной модели. Самцам SCID-мышей (AZ, UK; также поставляемых Charles River, UK) трансплантировали подкожно (п.к.) линию клеток аденокарциномы молочной железы человека MCF7 (Имперский фонд исследования рака (ICRF), London; которую также можно получить в АТСС, № НТВ-22) для определения ингибирования фосфорилирования АКТ ингибиторами PI3-киназ. Мышам имплантировали гранулы эстрогена в дозе 0,5 мг (с высвобождением в течение 21 суток) (Innovative Research of America, № E121) за 24 часа до имплантации клеток. Животным в левый бок вводили п.к. 5x 106 клеток в 50%-ном матригеле (BD Bioscience). Животных рандомизировали в группы по 8 контрольных и 4 подвергаемых обработке животных, когда опухоли достигали объема 400 мм3, и введение доз начинали на следующий день. Опухоли извлекали в выбранные моменты времени и тогда также отбирали образцы крови для измерения фармакокинетических (ФК) параметров.
Вырезанные из мыши опухоли помещали в пробирку для FastPrep (в пробирки с заостренным дном емкостью 2 мл, содержащие лизирующий матрикс A, MP Biomedicals, №6910-500) и сразу же проводили быстрое замораживание. В каждую пробирку добавляли по 1 мл лизирующего буфера (25 мМ трис, 3 мМ EDTA, 3 мМ EGTA, 50 мМ фторид натрия, 2 мМ ортованадат, 0,27 М сахароза, 10 мМ бета-глицерофосфат, 5 мМ пирофосфат, 0,5% тритона x-100) плюс ингибиторы фосфатаз (№ по каталогу Sigma Р2850 и № по каталогу Sigma Р5726, разбавленные 1:100) и ингибиторы протеаз (№ по каталогу Sigma Р8340, разбавленные 1:200). Опухоли гомогенизировали в течение 1 минуты на устройстве FastPrep™ (MP Biomedicals, №116004500) и затем выдерживали на льду в течение 5 минут, далее проводили две дополнительные стадии гомогенизации, после каждой из которых следовала инкубация на льду в течение 5 минут. Образцы центрифугировали в течение 10 минут при 13000 об./мин в центрифуге с охлаждением. Осветленные лизаты затем помещали в свежие пробирки и использовали по 10 мкл в анализе для определения белка.
Обнаружение общей и фосфорилированной АКТ (Ser473) осуществляли, используя набор для многоточечного анализа от MSD (Meso Scale Discovery, №K1510OD-3). Каждая лунка планшета содержала 4 пятна; на два из них наносили мышиные моноклональные антитела, полученные вместе с набором; на одно наносили захватывающее антитело к общей АКТ, а на другое наносили антитело к фосфорилированной АКТ (Ser473). Содержимое планшетов блокировали в течение ночи в холодной комнате на шейкере, используя 150 мкл блокирующего раствора на одну лунку, который готовили, используя 20 мл 1х раствора для промывки плюс 600 мг блокатора A, поставляемого вместе с набором. Планшеты промывали три раза, используя по 0,3 мл раствора для промывки на одну лунку. Отбирали аликвоту лизата каждой опухоли и разбавляли лизирующим буфером до концентрации 2 мг/мл, затем в каждую лунку добавляли по 25 мкл разбавленного лизата, получая общее количество 50 мкг на одну лунку. Планшеты помещали на шейкер при комнатной температуре на один час, после чего планшеты промывали трижды. Раствор детектирующего антитела готовили, используя смесь блокирующего раствора и раствора для промывки плюс 50х раствор SULFO-TAG™ антитела к общей АКТ в разведении 1:50. Планшеты помещали на шейкер при комнатной температуре на один час, после чего планшеты промывали трижды. 150 мкл буфера для прочтения, поставляемого вместе с набором, разбавляли 1:4 деионизованной водой и добавляли в каждую лунку, затем планшет прочитывали на планшетном анализаторе от MSD. Буфер для прочтения создает корректное химическое окружение для электрохемилюминесценции, так что, когда в планшетном ридере подается напряжение на планшет, электроды, расположенные в основании планшета, вызывают эмиссию света меткой, связанной с детектирующим антителом. Интенсивность излученного света является количественной характеристикой присутствующей АКТ, или общей, или фосфорилированной. Для расчета отношения фосфорилированной АКТ к общей АКТ применяли расчет, предложенный Meso Scale: умноженный на 2х сигнал от фосфорилированной АКТ, деленный на сигнал от общей АКТ плюс фосфорилированной АКТ, далее умножали на 100, получая содержание фосфобелка в %. Эти значения преобразовывали к шкале log 10 и затем эти значения использовали для расчета среднего геометрического и стандартной ошибки для каждой группы. Затем применяли t-критерий Стьюдента, используя двустороннюю формулу и неодинаковую дисперсию для проверки значимости. Исследования показали, что контрольной группы из 8 животных вместе с подвергнутой обработке группой из 4 животных было достаточно для проведения исследования.
(и) Протокол для обнаружения ингибирования опухолевого роста в линии клеток аденокарциномы молочной железы человека MCF7, трансплантированных SCID-мышам
Этот метод предложен для оценки противоопухолевой эффективности ингибиторов PI3-киназ in vivo, в PI3K-α-зависимой модели. Как и в случае фармакодинамических (ФД) исследований, указанных выше, самцам SCID-мышей трансплантировали п.к. линию клеток аденокарциномы молочной железы человека, MCF7. Мышам имплантировали гранулы эстрогена в дозе 0,5 мг (с высвобождением в течение 21 суток) за 24 часа до имплантации клеток. Животным в левый бок вводили п.к. 5x 106 клеток в 50%-ном матригеле. Животных рандомизировали в группы по 10-15 штук, когда опухоли достигали объема примерно 200-300 мм3, и начинали обработку. Животным в течение 2-4 недель посредством перорального, внутривенного (в.в.) или внутрибрюшинного (в.б.) способов вводили соединение в разбавителе, подходящем для введения необходимым образом и отвечающем требованиям охраны здоровья (суспензию - для перорального введения в диапазоне pH 4-7, раствор - для в.б./в.в. введения в диапазоне pH 5,5-7,0), и в определенных дозах. Обычно опухоли измеряли два раза в неделю циркулем и объем опухолей рассчитывали, используя формулу для эллипсоида (π/6 × ширина × толщина × длина).
Хотя, как и ожидалось, фармакологические свойства соединений формулы (I) меняются в зависимости от структурных изменений, в большинстве случаев активность, которой обладают соединения формулы (I), может быть продемонстрирована для следующих концентраций или доз в одном или более чем одном из приведенных выше тестов (а) и (в):
тест (a): IC50 в отношении PI3K-α, для диапазона, например, 1 нМ-100 нМ;
тест (в): IC50 в отношении клеточной фосфо-АКТ (Tyr308) в клетках ВТ474, для диапазона, например, 10 нМ-1 мкМ.
Подходящим образом конкретные соединения по изобретению обладают активностью в следующих концентрациях или дозах в одном или более приведенных выше тестах (а) и (в):
тест (а): IC50 в отношении PI3K-α, для диапазона, например, 1 нМ-100 нМ;
тест (в): IC50 в отношении клеточной фосфо-АКТ (Tyr308) в клетках ВТ474, для диапазона, например, 10 нМ-1 мкМ.
Подходящим образом конкретные соединения по изобретению обладают активностью в следующих концентрациях или дозах в одном или более приведенных выше тестах (а), (в), (з) и (и):
тест (a): IC50 в отношении PI3K-α, для диапазона, например, 1 нМ-100 нМ;
тест (в): IC50 в отношении клеточной фосфо-АКТ (Tyr308) в клетках ВТ474, для диапазона, например, 10 нМ-1 мкМ.
тест (з): более 50% ингибирования фосфо-АКТ (Ser473) in vivo, для диапазона, например, 1-200 мг/кг/сутки;
тест (и): активность в модели ксенотрансплантатов, для диапазона, например, 1-200 мг/кг/сутки.
Для соединений из примеров получены следующие данные.
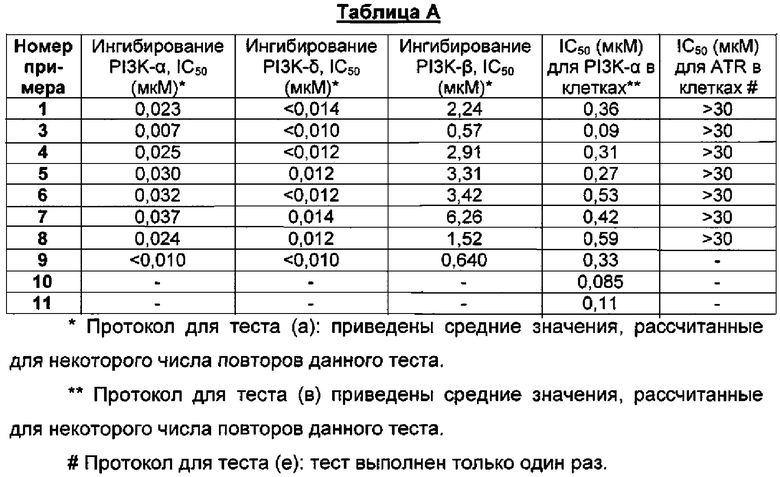
Исследование комбинаций
Материалы и методы
MCF7 представляет собой положительную по рецептору эстрогенов линию клеток опухоли молочной железы, несущую мутацию в гене PIK3CA (Е545К). Самцам SCID-мышей (AZ, UK) трансплантировали подкожно (п.к.) линию клеток аденокарциномы молочной железы человека MCF7 (ICRF, London) для определения противоопухолевой активности ингибиторов PI3-киназ. Мышам имплантировали гранулы эстрогена в дозе 0,5 мг с высвобождением в течение 21 суток (Innovative Research of America) за 24 часа до имплантации клеток. Животным в левый бок вводили п.к. 5x 106 клеток в 50%-ном матригеле (BD Bioscience).
ВТ474 представляет собой положительную по рецептору эстрогенов линию клеток опухоли молочной железы с усиленной экспрессией Her2 и несет мутацию в гене PIK3CA (K111N). Самкам бестимусных «голых» мышей линии Swiss (swiss nu/nu - AZ, UK) трансплантировали подкожно опухоли из линии эпителиальных клеток протоковой карциномы молочной железы человека ВТ474c (произведенной в AZ из ВТ474 - № в АТСС НТВ-20), привитые мышам. Мышам имплантировали гранулы эстрогена в дозе 0,36 мг с высвобождением в течение 60 суток (Innovative Research of America) за 24 часа до имплантации клеток. Животным в левый бок вводили п.к. 5x 106 клеток в 50%-ном матригеле (BD Bioscience).
НСС70 представляет собой линию опухолевых клеток молочной железы, дефицитную по экспрессии гена PTEN.
Самкам бестимусных «голых» мышей линии Swiss (swiss nu/nu - AZ, UK) трансплантировали подкожно клетки из линии опухолевых клеток эпителия протоков молочной железы НСС70 (№ в АТСС - CRL2315). Животным в левый бок вводили п.к. 1х 106 клеток в 50%-ном матригеле (BD Bioscience).
Животных рандомизировали в группы по 10-15 штук, когда опухоли достигали объема примерно 200-300 мм3, и начинали обработку. Животным в течение 3-4 недель пероральным путем вводили соединение в подходящем разбавителе в определенных дозах и по определенным схемам. Опухоли измеряли циркулем два-три раза в неделю и объем опухолей рассчитывали, используя формулу для эллипсоида (π/6 × ширина × толщина × длина).
При раздельном введении AZD5363 разводили в растворе, содержащем 10% DMSO, 25% клептозы. (Клептоза от Roquette-Pharma (зарегистрированный товарный знак) представляет собой гидроксипропилбетациклодекстрин, подходящий для применения in vivo и изготовления препаратов).
При совместном введении с соединением из примера 3 AZD5363 разводили в смеси НРМС/твин (0,5% метоцела (гидроксипропилметилцеллюлозы)/0,1% полисорбата 80). Суспензию измельчали в шаровой мельнице в течение ночи.
Соединение из примера 3 разводили в смеси НРМС/твин (0,5% метоцела (гидроксипропилметилцеллюлозы)/0,1% полисорбата 80).
AZD8186 разводили в смеси НРМС/твин (0,5% метоцела (гидроксипропилметилцеллюлозы)/0,1% полисорбата 80).
При совместном введении с соединением из примера 3 AZD8186 разводили в смеси НРМС/твин (0,5% метоцела (гидроксипропил-метилцеллюлозы)/0,1% полисорбата 80). Суспензию измельчали в шаровой мельнице в течение ночи.
Олапариб разводили в растворе, содержащем 10% DMSO/30% клептозы.
Ингибирование опухолевого роста под действием соединения из примера 3 в комбинации с ингибитором АКТ (AZD5363) последовательное введение
Исследования проводили на модели ксенотрансплантатов ВТ474. Соединение из примера 3 и AZD5363 вводили два раза в сутки (BID) с интервалом 6-8 часов согласно еженедельному циклу по схеме «2 суток вводят/5 суток не вводят», в такой последовательности: AZD5363 вводили на 1-е и 2-е сутки еженедельного цикла, а соединение из примера 3 вводили на 3-й и 4-е сутки еженедельного цикла. Соединение из примера 3 вводили BID в дозе 50 мг/кг, a AZD5363 вводили BID в дозе 170 мг/кг, в смесях НРМС/твин и DMSO/клептоза, соответственно.
Кривая роста опухолей (показанная на Фиг. 9) указывает на то, что попеременное введение или соединения из примера 3, или AZD5363 частично ингибировало рост опухолей по сравнению с введением в качестве контроля только разбавителя (НРМС/твин). Комбинация соединения из примера 3 плюс AZD5363 индуцировала регрессию опухоли.
Ингибирование опухолевого роста под действием соединения из примера 3 в комбинации с ингибитором АКТ (AZD5363) - совместное введение
Исследования проводили на модели ксенотрансплантатов ВТ474. Соединение из примера 3 и AZD5363 вводили два раза в сутки (BID) с интервалом 6-8 часов и одновременно согласно еженедельному циклу по схеме «2 суток вводят/5 суток не вводят». Соединение из примера 3 вводили BID в дозе 25 мг/кг, a AZD5363 вводили BID в дозе 100 мг/кг, оба в смеси НРМС/твин.
Кривая роста опухолей (показанная на Фиг. 10) указывает на то, что интермиттирующее введение соединения из примера 3 и AZD5363 частично ингибировало рост опухолей по сравнению с введением в качестве контроля только разбавителя (НРМС/твин). Комбинация соединения из примера 3 плюс AZD5363 индуцировала регрессию опухоли в течение всего периода введения, хотя затем наблюдали повторное увеличение опухоли в период времени, когда введение не осуществляли.
Ингибирование опухолевого роста под действием соединения из примера 3 в комбинации с ингибитором PARP (олапарибом)
Исследования проводили на модели ксенотрансплантатов ВТ474. Соединение из примера 3 и олапариб вводили каждый день в продолжение всего исследования, соединение из примера 3 два раза в сутки (BID) с интервалом 6-8 часов каждый раз в дозе 25 мг/кг, а олапариб один раз в сутки (QD) в дозе 100 мг/кг через 1 час после введения первой суточной дозы соединения из примера 3. Оба агента вводили в смеси НРМС/твин.
Кривая роста опухолей (Фиг. 11) указывает на то, что сам олапариб не оказывал никакого существенного эффекта на рост опухолей, само соединение из примера 3 ингибировало рост частично, зато комбинация соединения из примера 3 и олапариба индуцировала регрессию опухоли.
Ингибирование опухолевого роста под действием соединения из примера 3 в комбинации с ингибитором PARP (олапарибом)
Исследования проводили на модели ксенотрансплантатов MCF7. Соединение из примера 3 и олапариб вводили каждый день в продолжение всего исследования, соединение из примера 3 два раза в сутки с интервалом 6-8 часов каждый раз в дозе 25 мг/кг, а олапариб один раз в сутки (QD) в дозе 100 мг/кг через 1 час после введения первой суточной дозы соединения из примера 3. Оба агента водили в смеси НРМС/твин.
Кривая роста опухолей (Фиг. 12) указывает на то, что сам олапариб оказывал минимальный эффект на рост опухолей, само соединение из примера 3 вызывало некоторую регрессию опухоли, зато комбинация соединения из примера 3 и олапариба индуцировала более сильную регрессию опухоли.
Ингибирование опухолевого роста под действием соединения из примера 3 в комбинации с ингибитором PI3Kбета/дельта (AZD8186)
Исследования проводили на модели ксенотрансплантатов НСС70. В ходе всего исследования соединение из примера 3 и AZD8186 вводили каждые сутки, в режиме два раза в сутки (BID), соединение из примера 3 каждый раз в дозе 25 мг/кг, a AZD8186 каждый раз в дозе 50 мг/кг. Оба агента вводили в смеси НРМС/твин.
Кривая роста опухолей (Фиг. 13) указывает на то, что соединение AZD8186 ингибировало рост опухолей частично, само соединение из примера 3 ингибировало рост сильнее, а комбинация соединения из примера 3 плюс AZD8186 индуцировала регрессию опухоли.
Согласно другому аспекту изобретения предложена фармацевтическая композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль, которые определены ранее, вместе с фармацевтически приемлемым разбавителем или носителем.
Подходящие фармацевтически приемлемые эксципиенты для композиции в форме таблетки включают, например, инертные разбавители, гранулирующие и разрыхляющие агенты, связующие вещества, смазывающие вещества, консерванты и антиоксиданты. Композиции в форме таблетки могут быть без оболочки или покрыты оболочкой либо для модификации их распадаемости и последующего всасывания активного ингредиента в желудочно-кишечном тракте, либо для улучшения их стабильности и/или внешнего вида, в любом случае с использованием традиционных покрывающих агентов и методик, хорошо известных в данной области техники.
Композиции для перорального применения, альтернативно, могут быть представлены в форме твердых желатиновых капсул, в которых активный ингредиент смешан с инертным твердым разбавителем, или в форме мягких желатиновых капсул, в которых активный ингредиент смешан с водой или маслом.
Водные суспензии обычно содержат активный ингредиент в тонкоизмельченной форме вместе с одним или более чем одним суспендирующим агентом, диспергирующим или увлажняющим агентом. Водные суспензии также могут содержать один или более консервантов, антиоксидантов, красителей, корригентов и/или подсластителей.
Масляные суспензии могут быть изготовлены путем суспендирования активного ингредиента в растительном масле или минеральном масле. Кроме того, масляные суспензии также могут содержать загуститель. Для обеспечения приятного на вкус препарата для перорального приема можно добавить такие подсластители, как указанные выше, и корригенты. В эти композиции в качестве консерванта может быть добавлен антиоксидант.
Диспергируемые порошки и гранулы, подходящие для приготовления водной суспензии путем добавления воды, обычно содержат активный ингредиент вместе с диспергирующим или увлажняющим агентом, суспендирующим агентом и одним или более консервантами. Также могут присутствовать такие дополнительные эксципиенты, как подсластители, корригенты и красители.
Фармацевтические композиции по изобретению также могут быть представлены в форме эмульсий типа масло-в-воде. Масляной фазой может быть растительное масло или минеральное масло либо смесь любого из них. Эмульсии также могут содержать подсластители, корригенты и консерванты.
Сиропы и эликсиры могут быть изготовлены с использованием подсластителей и также могут содержать уменьшающее раздражение средство, консервант, корригент и/или краситель.
Фармацевтические композиции также могут быть представлены в форме стерильной водной или масляной суспензии для инъекций, которая может быть изготовлена согласно известным методикам с использованием одного или более чем одного соответствующего диспергирующего или увлажняющего агента и суспендирующего агента, которые упомянуты выше. Стерильный препарат для инъекций также может представлять собой стерильный раствор или стерильную суспензию для инъекций в нетоксичном приемлемом для парентерального введения разбавителе или растворителе.
Композиции для введения путем ингаляции могут быть представлены в форме традиционного находящегося под давлением аэрозоля, приспособленного для распределения активного ингредиента, либо в виде аэрозоля, содержащего тонкоизмельченное твердое вещество, либо в виде жидких капель. Можно использовать традиционные пропелленты для аэрозолей, как например, летучие фторированные углеводороды или углеводороды, и аэрозольное устройство удобным образом приспособлено для распределения отмеренного количества активного ингредиента.
Для дополнительной информации по технологии изготовления лекарственных средств читатель отсылается к главе 25.2 тома 5 "Комплексной медицинской химии" (Comprehensive Medicinal Chemistry; Corwin Hansch; Chairman of Editorial Board), Pergamon Press, 1990.
Количество активного ингредиента, комбинированного с одним или более чем одним эксципиентом для изготовления лекарственной формы для разового введения, обязательно будет меняться в зависимости от подвергаемого лечению пациента и конкретного пути введения. Например, для перорального введения людям обычно необходимо, например, от 1 мг до 2 г подлежащего введению активного агента (более приемлемо от 100 мг до 2 г, например, от 250 мг до 1,8 г, как например, от 500 мг до 1,8 г, в частности, от 500 мг до 1,5 г, подходящим образом от 500 мг до 1 г) в смеси с соответствующим и удобным количеством эксципиентов, которое может изменяться от примерно 3 до примерно 98 процентов от массы всей композиции. Будет очевидно, что в случае необходимости в большой дозировке может потребоваться много лекарственных форм, например две или более таблеток или капсул, содержащих активный ингредиент в дозе, удобно поделенной между ними. Подходящим образом твердая лекарственная форма для разового введения может содержать от 1 до 300 мг активного ингредиента.
Размер дозы соединения формулы (I) с точки зрения терапевтических или профилактических целей будет непременно меняться в зависимости от природы и тяжести болезненного состояния, возраста и пола животного или пациента и пути введения, согласуясь с хорошо известными принципами медицины.
В случае применения соединения формулы (I) для терапевтических или профилактических целей обычно его будут вводить так, чтобы получить суточную дозу в диапазоне, например, от 1 мг/кг до 100 мг/кг массы тела, введенную при необходимости разделенными дозами. В общем случае, при использовании парентерального пути будут введены более низкие дозы. Так, например, в случае внутривенного введения обычно будет применена доза в диапазоне, например, от 1 мг/кг до 25 мг/кг массы тела. Подобно этому, в случае введения посредством ингаляции будет применена доза в диапазоне, например, от 1 мг/кг до 25 мг/кг массы тела. Однако пероральное введение, в частности в форме таблетки, является предпочтительным. В типичном случае, стандартные лекарственные формы будут содержать примерно от 10 мг до 0,5 г соединения по данному изобретению.
Соединения по изобретению можно вводить один раз в сутки или более одного раз в сутки. Соединения по изобретению также можно вводить по соответствующей схеме дозирования, например, соединения по изобретению можно вводить один или несколько раз в сутки (например, один раз, два раза или три раза в сутки) в течение определенного числа суток, с последующим периодом времени, когда никакого введения не осуществляют. Такой цикл приема препарата (состоящий из дней введения препарата и дней, когда введения препарата нет) затем может быть повторен. Подходящим образом цикл приема препарата составляет период времени 5-14 суток, как например, 5, 7, 10 или 14 суток, более подходящим образом 7 суток. Согласно одному из аспектов, соединения формулы (I) вводят на протяжении 1 суток либо 2-х или 3-х следующих друг за другом суток, с последующими 3, 4, 5 или 6 сутками в цикле приема препарата, когда никакого введения не осуществляют.
Согласно одному из аспектов, соединение формулы (I) вводят в течение одних суток, после чего в течение 2, 3 или 4 суток никакого введения не осуществляют.
Согласно другому аспекту соединение формулы (I) вводят в течение 2-х суток, после чего в течение 4, 5 или 6 суток никакого введения не осуществляют.
Согласно другому аспекту соединение формулы (I) вводят в течение 3-х суток, после чего в течение 3, 4 или 5 суток никакого введения не осуществляют.
Согласно другому аспекту соединение формулы (I) вводят в течение 4-х суток, после чего в течение 2, 3 или 4 суток никакого введения не осуществляют.
Согласно другому аспекту соединение формулы (I) вводят в течение 5-ти суток, после чего в течение 1, 2 или 3 суток никакого введения не осуществляют.
Согласно другому аспекту соединение формулы (I) вводят через день.
Приведенные выше схемы дозирования удобно применять тогда, когда соединения по изобретению используют в качестве монотерапии. Дополнительные примеры возможных схем дозирования для введения соединений по изобретению в качестве комбинированной терапии, описаны далее.
Как указано выше, известно, что ферменты PI3K-α и -δ участвуют в онкогенезе путем оказания одного или более действий по опосредованию пролиферации раковых и других клеток, опосредованию ангиогенных событий и опосредованию подвижности, миграции и инвазивности раковых клеток. Авторы изобретения обнаружили, что соединения по настоящему изобретению обладают сильной противоопухолевой активностью, которая, как полагают, возникает в результате ингибирования ферментов PI3K-α и -δ, вовлеченных в стадии передачи сигнала, приводящие к пролиферации и выживаемости опухолевых клеток и инвазивной и миграционной способности метастазирующих опухолевых клеток.
Соответственно, соединения по настоящему изобретению полезны в качестве противоопухолевых агентов, в частности в качестве селективных ингибиторов пролиферации, выживаемости, подвижности, распространения и инвазивности раковых клеток млекопитающих, обуславливая ингибирование опухолевого роста и выживаемости и ингибирование роста метастатической опухоли. В частности, соединения по настоящему изобретению полезны в качестве антипролиферативных и антиинвазивных агентов для сдерживания распространения и/или лечения солидного опухолевого заболевания. В частности, ожидается, что соединения по настоящему изобретению будут полезны в предупреждении или лечении тех опухолей, которые чувствительны к ингибированию ферментов PI3K-α и/или -δ и которые вовлечены в стадии передачи сигнала, приводящие к пролиферации и выживаемости опухолевых клеток и миграционной способности и инвазивности метастазирующих опухолевых клеток. Кроме того, ожидается, что соединения по настоящему изобретению будут полезны для предупреждения или лечения тех опухолей, воздействие на которые опосредовано исключительно или частично ингибированием ферментов PI3K-α и/или -δ, т.е. соединения можно использовать для получения эффекта ингибирования фермента PI3K-α и/или -δ у теплокровного животного, нуждающегося в таком лечении.
Согласно другому аспекту изобретения предложены соединение формулы (I) или его фармацевтически приемлемая соль, которые определены ранее, для применения в качестве лекарственного средства у теплокровного животного, такого как человек.
Согласно другому аспекту изобретения, предложены соединение формулы (I) или его фармацевтически приемлемая соль, которые определены ранее, для применения в получении антипролиферативного эффекта у теплокровного животного, такого как человек.
Согласно другому признаку этого аспекта изобретения предложены соединение формулы (I) или его фармацевтически приемлемая соль, которые определены ранее, для применения у теплокровного животного, такого как человек, в качестве антиинвазивного агента для сдерживания распространения и/или лечения солидного опухолевого заболевания.
Согласно другому аспекту изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли, которые определены ранее, для получения антипролиферативного эффекта у теплокровного животного, такого как человек.
Согласно другому признаку этого аспекта изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли, которые определены ранее, в изготовлении лекарственного средства для применения в получении антипролиферативного эффекта у теплокровного животного, такого как человек.
Согласно другому признаку этого аспекта изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли, которые определены ранее, в изготовлении лекарственного средства для применения у теплокровного животного, такого как человек, в качестве антиинвазивного агента для сдерживания распространения и/или лечения солидного опухолевого заболевания.
Согласно другому признаку этого аспекта изобретения предложен способ получения антипролиферативного эффекта у теплокровного животного, такого как человек, нуждающегося в таком лечении, включающий введение указанному животному эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, как определено ранее.
Согласно другому признаку этого аспекта изобретения предложен способ получения антиинвазивного эффекта посредством сдерживания распространения и/или лечения солидного опухолевого заболевания у теплокровного животного, такого как человек, нуждающегося в таком лечении, включающий введение указанному животному эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, как определено ранее.
Согласно другому аспекту изобретения предложены соединение формулы (I) или его фармацевтически приемлемая соль, которые определены ранее, для применения в предупреждении или лечении рака у теплокровного животного, такого как человек.
Согласно другому аспекту изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли, которые определены ранее, в изготовлении лекарственного средства для применения в предупреждении или лечении рака у теплокровного животного, такого как человек.
Согласно другому признаку этого аспекта изобретения предложен способ предупреждения или лечения рака у теплокровного животного, такого как человек, нуждающегося в таком лечении, включающий введение указанному животному эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, как определено ранее.
Согласно другому аспекту изобретения предложены соединение формулы (I) или его фармацевтически приемлемая соль, которые определены ранее, для применения в предупреждении или лечении солидного опухолевого заболевания у теплокровного животного, такого как человек.
Согласно другому аспекту изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли, которые определены ранее, в изготовлении лекарственного средства для применения в предупреждении или лечении солидного опухолевого заболевания у теплокровного животного, такого как человек.
Согласно другому признаку этого аспекта изобретения предложен способ предупреждения или лечения солидного опухолевого заболевания у теплокровного животного, такого как человек, нуждающегося в таком лечении, включающий введение указанному животному эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, как определено ранее.
Согласно другому аспекту изобретения предложены соединение формулы (I) или его фармацевтически приемлемая соль, определенные ранее, для применения в предупреждении или лечении тех опухолей, которые чувствительны к ингибированию ферментов PI3K-α и/или -δ, вовлеченных в стадии передачи сигнала, приводящие к пролиферации, выживаемости, инвазивности и миграционной способности опухолевых клеток.
Согласно другому признаку этого аспекта изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли, определенных ранее, в изготовлении лекарственного средства для применения в предупреждении или лечении тех опухолей, которые чувствительны к ингибированию ферментов PI3K-α и/или -δ, вовлеченных в стадии передачи сигнала, приводящие к пролиферации, выживаемости, инвазивности и миграционной способности опухолевых клеток.
Согласно другому признаку этого аспекта изобретения предложен способ предупреждения или лечения тех опухолей, которые чувствительны к ингибированию ферментов PI3K-α и/или -δ, вовлеченных в стадии передачи сигнала, приводящие к пролиферации, выживаемости, инвазивности и миграционной способности опухолевых клеток, включающий введение указанному животному эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, как определено ранее.
Согласно другому аспекту изобретения предложены соединение формулы (I) или его фармацевтически приемлемая соль, которые определены ранее, для применения в получении эффекта ингибирования ферментов PI3K-α и -δ.
Согласно другому признаку этого аспекта изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли, которые определены ранее, в изготовлении лекарственного средства для применения в получении эффекта ингибирования ферментов PI3K-α и -δ.
Согласно другому аспекту изобретения также предложен способ получения эффекта ингибирования ферментов PI3K-α и -δ, включающий введение эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, как определено ранее.
Как указано ранее, некоторые соединения по настоящему изобретению обладают существенно лучшей эффективностью в отношении ферментов PI3K-α и -δ, чем в отношении других PI3-киназных ферментов или других киназ. Такие соединения обладают достаточной эффективностью в отношении ферментов PI3K-α и -δ, так что их можно применять в количестве, достаточном для ингибирования ферментов PI3K-α и -δ, одновременно демонстрируя незначительную активность в отношении фермента PI3K-β и в отношении большинства других киназных ферментов. Такие соединения вероятно будут использованы для селективного ингибирования ферментов PI3K-α и -δ и вероятно будут использованы для эффективного лечения опухолей, обуславливаемых, например ферментом PI3K-α и/или -δ.
Согласно этому аспекту изобретения предложены соединение формулы (I) или его фармацевтически приемлемая соль, которые определены ранее, для применения в получении эффекта, селективного в отношении ингибирования ферментов PI3K-α и -δ.
Согласно другому признаку этого аспекта изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли, которые определены ранее, в изготовлении лекарственного средства для применения в получении эффекта, селективного в отношении ингибирования ферментов PI3K-α и -δ.
Согласно другому аспекту изобретения также предложен способ получения эффекта, селективного в отношении ингибирования ферментов PI3K-α и -δ, включающий введение эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, как определено ранее.
Под «эффектом, селективным в отношении ингибирования ферментов PI3K-α и -δ» понимают, что соединения формулы (I) являются более сильнодействующими в отношении ферментов PI3K-α и -δ, чем в отношении других PI3-киназ класса I, и как правило демонстрируют хорошую селективность в отношении других членов всего PI3-киназного семейства и в отношении более широких классов киназных ферментов, содержащих тирозин- и ser/thr-киназы.
Согласно другому признаку данного изобретения предложены соединение формулы (I) или его фармацевтически приемлемая соль, которые определены ранее, для применения в лечении рака молочной железы, рака желудка и рака пищевода, немелкоклеточного рака легкого (NSCLC), включая плоскоклеточные карциномы (SCC) и аденокарциному, SCC головы и шеи (H&N), гинекологических раковых заболеваний (включая рак эндометрия, яичников и шейки матки), и гематологических раковых заболеваний, таких как множественная миелома, лимфомы и лейкозы (включая хронический лимфолейкоз (CLL), острый лимфобластный лейкоз (ALL) и лимфому из клеток мантийной зоны (MCL)).
Согласно другому признаку этого аспекта изобретения предложены соединение формулы (I) или его фармацевтически приемлемая соль, которые определены ранее, для применения в лечении рака мочевого пузыря, головного мозга/центральной нервной системы (ЦНС), ободочной и прямой кишки, легкого (всех возможных форм), желчного пузыря и желчных протоков и кожи.
Согласно другому признаку этого аспекта изобретения предложены соединение формулы (I) или его фармацевтически приемлемая соль, которые определены ранее, для применения в лечении рака предстательной железы, кости, почки, печени, меланомы, рака тканей желудочно-кишечного тракта, поджелудочной железы, яичек, щитовидной железы, пениса, вульвы и опухолей других типов с зависимостью от PI3-киназ, обусловленной мутацией, амплификацией или другими отклонениями.
Согласно другому признаку этого аспекта изобретения предложен способ лечения рака молочной железы, рака желудка и рака пищевода, NSCLC, включая SCC и аденокарциному, SCC головы и шеи, гинекологических раковых заболеваний (включая рак эндометрия, яичника и шейки матки) и гематологических раковых заболеваний, таких как множественная миелома, лимфомы и лейкозы (включая CLL, ALL и MCL), у теплокровного животного, такого как человек, нуждающегося в таком лечении, включающий введение эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, как определено ранее.
Согласно другому признаку этого аспекта изобретения предложен способ лечения рака мочевого пузыря, головного мозга/ЦНС, ободочной и прямой кишки, легкого (всех возможных форм), желчного пузыря и желчных протоков и кожи у теплокровного животного, например, человека, нуждающегося в таком лечении, включающий введение эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, как определено ранее.
Согласно другому признаку этого аспекта изобретения предложен способ лечения рака предстательной железы, кости, почки, печени, меланомы, рака тканей желудочно-кишечного тракта, поджелудочной железы, яичек, щитовидной железы, пениса, вульвы и опухолей других типов с зависимостью от PI3-киназ, обусловленной мутацией, амплификацией или другими отклонениями, у теплокровного животного, например, человека, нуждающегося в таком лечении, включающий введение эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, как определено ранее.
Согласно другому признаку данного изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли, которые определены ранее, в изготовлении лекарственного средства для применения в лечении рака молочной железы, рака желудка и рака пищевода, NSCLC, включая SCC и аденокарциному, SCC головы и шеи, гинекологических раковых заболеваний (включая рак эндометрия, яичника и шейки матки), и гематологических раковых заболеваний, таких как множественная миелома, лимфомы и лейкозы (включая CLL, ALL и MCL).
Согласно другому признаку этого аспекта изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли, которые определены ранее, в изготовлении лекарственного средства для применения в лечении рака мочевого пузыря, головного мозга/ЦНС, ободочной и прямой кишки, легкого (всех возможных форм), желчного пузыря и желчных протоков и кожи.
Согласно другому признаку этого аспекта изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли, которые определены ранее, в изготовлении лекарственного средства для применения в лечении рака предстательной железы, кости, почки, печени, меланомы, рака тканей желудочно-кишечного тракта, поджелудочной железы, яичек, щитовидной железы, пениса, вульвы и опухолей других типов с зависимостью от PI3-киназ, обусловленной мутацией, амплификацией или другими отклонениями.
Согласно одному признаку данного изобретения подлежащий лечению рак представляет собой рак молочной железы. Согласно другому аспекту этого признака рак молочной железы является раком, положительным (+ve) по рецептору эстрогенов. В одном из воплощений этого аспекта соединение формулы (I) или его фармацевтически приемлемую соль, вводят в комбинации с антигормональным агентом, как определено здесь. В другом воплощении этого аспекта соединение из примера 3 вводят в комбинации с антигормональным агентом, как определено здесь. В следующем воплощении этого аспекта соединение из примера 3 вводят в комбинации с олапарибом или его фармацевтически приемлемой солью и возможно дополнительно в комбинации с антигормональным агентом, как определено здесь. В следующем воплощении этого аспекта соединение из примера 3 вводят в комбинации с AZD5363 или его фармацевтически приемлемой солью и возможно дополнительно в комбинации с антигормональным агентом, как определено здесь.
Что касается одного из аспектов, где указывается лечение рака, то следует понимать, что оно может относиться к предотвращению появления метастазов и лечению метастазов, т.е. метастазирования. Поэтому соединения по настоящему изобретению могут быть использованы для лечения пациента, не имеющего метастазов, чтобы остановить их появление или чтобы удлинить период времени до их появления, и пациента, уже имеющего метастазы, для лечения самих метастазов. Кроме того, лечение рака может относиться к лечению установленной(ых) первичной(ых) опухоли или опухолей и развития первичной(ых) опухоли или опухолей. Таким образом, согласно одному из аспектов лечение рака относится к предотвращению появления метастазов. Согласно другому аспекту изобретения лечение рака относится к лечению метастазов. Согласно другому аспекту изобретения лечение рака относится к лечению установленной(ых) первичной(ых) опухоли или опухолей и развития первичной(ых) опухоли или опухолей.
Как указано ранее, эффекты, оказываемые соединением формулы (I) in vivo, частично могут быть обусловлены одним или более метаболитами (такими как соединения формулы A, определенные ранее), которые образуются в организме человека или животного после введения соединения формулы (I).
Конкретные соединения по изобретению обладают большей эффективностью в отношении ферментов PI3K-α и -δ, чем в отношении других изоформ PI3-киназ класса I, таких как -β и -γ. Согласно одному из аспектов, соединения по изобретению являются селективными в отношении PI3K-α и -δ в сравнении с PI3K-β или -γ.
Таким образом, согласно настоящему изобретению также предложен способ ингибирования PI3-киназы-α у пациента, включающий введение пациенту количества соединения формулы (I) или его фармацевтически приемлемой соли, эффективного для ингибирования фосфоинозитид-3-киназы-α у пациента.
Таким образом, согласно настоящему изобретению также предложен способ ингибирования PI3-киназы-α и -δ у пациента, включающий введение пациенту количество соединения формулы (I) или его фармацевтически приемлемой соли, эффективное для ингибирования PI3-киназы-α и -δ у пациента.
Соединение формулы (I) или его фармацевтически приемлемая соль, представляющие собой ингибитор PI3-киназы, также обладают потенциалом для применения в терапии ряда других болезненных состояний. Например, PI3-киназа играет важную роль в стимулировании пролиферации гладкомышечных клеток в сосудистой сети, т.е. гладкомышечных клеток сосудов (Thyberg, European Journal of Cell Biology, 1998, 76(1), 33-42), и в легких (гладкомышечных клеток дыхательных путей), Krymskaya, V.P., BioDrugs, 2007, 21(2), 85-95. Чрезмерная пролиферация гладкомышечных клеток сосудов играет важную роль в образовании атеросклеротических бляшек и в развитии гиперплазии неоинтимы после проведения инвазивных процедур на сосудах (Scwartz et al., Progress in CardiovascuIar Disease, 1984, 26, 355-372; Clowes et al., Laboratory Investigations, 1978, 39, 141-150). Кроме того, чрезмерная пролиферация гладкомышечных клеток дыхательных путей приводит к развитию хронической обструктивной болезни легких (COPD) на фоне астмы и хронического бронхита. Поэтому ингибиторы PI3-киназной активности можно использовать для предупреждения сосудистого рестеноза, атеросклероза и COPD.
PI3-киназа также играет важную роль в функционировании лейкоцитов (Fuller et al., The Journal of Immunology, 1999, 162(11), 6337-6340; Eder et al., The Journal of Biological Chemistry, 1998, 273(43), 28025-31) и функционировании лимфоцитов (Vicente-Manzanares et al., The Journal of Immunology, 1999, 163(7), 4001-4012). Например, адгезия лейкоцитов к эндотелию воспаленной ткани приводит к активации эндогенных лейкоцитарных интегринов посредством PI3-киназа-зависимого процесса передачи сигналов. Кроме того, окислительный взрыв (Nishioka et al., FEBS Letters, 1998, 441(1), 63-66 и Condliffe, A.M., et al., Blood. 2005, 106(4), 1432-40) и реорганизация цитоскелета (Kirsch et al., Proceedings National Academy of Sciences USA, 1999, 96(11), 6211-6216) в нейтрофилах, по-видимому, подразумевают PI3-киназные пути передачи сигналов. Миграция и направленное движение нейтрофилов также зависят от PI3-киназной активности (Camps, M., et al., Nat. Med., 2005, 11(9), 936-43 и Sadhu, С. et al., J. Immunol, 2003, 170(5), 2647-54). Так, ингибиторы PI3-киназы могут быть полезны для уменьшения адгезии и активации лейкоцитов в местах воспаления и поэтому могут быть применены для лечения острых и/или хронических воспалительных нарушений. PI3-киназа также играет важную роль в пролиферации и активации лимфоцитов, Fruman et al., Science, 1999, 283(5400), 393-397. В частности, PI3K-δ имеет принципиальное значение для развития и функционирования В-клеток, включая индуцированную IgM-специфическими антителами пролиферацию В-клеток (Okkenhaug K et al., Science. 2002, 297(5583), 1031-1034), индуцированные B-клеточным рецептором синтез ДНК и пролиферацию и индуцированную интерлейкином-4 (IL-4) выживаемость (BiIancio A et al., Blood, 2006, 107, 642-650). Результаты этих наблюдений указывают на то, что PI3K-δ играет весьма важную и незаменимую роль в функционировании В-клеток, которая не компенсируется другими PI3K класса I. Учитывая важную роль лимфоцитов в аутоиммунных заболеваниях, ингибитор PI3-киназной активности может быть применен для лечения таких расстройств (Rommel С., Camps М. and Ji Н., Nat. Rev. Immunol., 2007, 1038, 191-201).
В лечении рака, определенном ранее, можно применять монотерапию или можно, помимо введения соединения по изобретению, включать проведение стандартных хирургических операций, лучевую терапию или химиотерапию. Такая химиотерапия может включать введение одного или более противоопухолевых агентов следующих категорий:
(1) антипролиферативные/антинеопластические лекарственные средства и их комбинации, которые применяют в терапевтической онкологии, такие как алкилирующие агенты (например, цисплатин, оксалиплатин, карбоплатин, циклофосфамид, азотистый иприт, мелфалан, хлорамбуцил, бусульфан, темозоламид и нитрозомочевины); антиметаболиты (например, гемцитабин и антифолаты, такие как фторпиримидины типа 5-фторурацила и тегафура, ралтитрексед, метотрексат, цитозинарабинозид и гидроксимочевина); противоопухолевые антибиотики (например, антрациклины, такие как адриамицин, блеомицин, доксорубицин, дауномицин, эпирубицин, идарубицин, митомицин-C, дактиномицин и митрамицин); антимитотические агенты (например, алкалоиды барвинка, такие как винкристин, винбластин, виндезин и винорелбин, и таксоиды, такие как таксол и таксотер, и ингибиторы поло-киназы); и ингибиторы топоизомеразы (например, эпиподофиллотоксины, такие как этопозид и тенипозид, амсакрин, топотекан и камптотецин);
(2) антигормональные агенты, такие как антиэстрогены (например, тамоксифен, фулвестрант, торемифен, ралоксифен, дролоксифен и иодоксифен), антиандрогены (например, бикалутамид, флутамид, нилутамид и ципротерона ацетат), антагонисты рилизинг-фактора лютеинизирующего гормона (LHRH) или агонисты LHRH (например, гозерелин, лейпрорелин и бусерелин), прогестогены (например, мегестрола ацетат), ингибиторы ароматазы (например, такие как анастрозол, летрозол, воразол и эксеместан) и ингибиторы 5α-редуктазы, такие как финастерид;
(3) ингибиторы функции факторов роста и их расположенных ниже сигнальных путей: включены Ab модуляторы к любым целевым факторам роста или рецепторам факторов роста, обзор по которым представлен в Stern et al., Critical Reviews in Oncology/Haematology, 2005, 54, pp. 11-29; также включены низкомолекулярные ингибиторы таких мишеней, например, ингибиторы киназ; примеры включают антитело к erbB2 трастузумаб (Herceptin™), антитело к рецептору эпидермального фактора роста (EGFR) панитумумаб, антитело к EGFR цетуксимаб (эрбитукс, С225) и ингибиторы тирозинкиназ, в том числе ингибиторы erbB-семейства рецепторов, как например, ингибиторы тирозинкиназ семейства рецепторов эпидермального фактора роста (EGFR/erbB1), такие как гефитиниб или эрлотиниб, ингибиторы тирозинкиназы erbB2, такие как лапатиниб, и смешанные ингибиторы erb1/2, такие как афатиниб; подобные стратегии имеются для других классов факторов роста и их рецепторов, например, ингибиторов семейства факторов роста гепатоцитов или их рецепторов, включая c-met и ron; ингибиторы инсулина и семейства инсулиноподобных факторов роста или их рецепторов (IGFR (рецептор инсулиноподобного фактора роста), IR (инсулиновый рецептор)), ингибиторов семейства факторов роста тромбоцитов или их рецепторов (PDGFR (рецептор фактора роста тромбоцитов)) и ингибиторов передачи сигнала, опосредуемой другими рецепторными тирозинкиназами, такими как c-kit, киназа анапластической лимфомы (AnLK) и рецептор колониестимулирующего фактора-1 (CSF-1R);
также включены модуляторы, направленно воздействующие на белки передачи сигнала в более широком PI3-киназа-опосредованном сигнальном пути, например, ингибиторы других изоформ PI3-киназы, таких как PI3K-β, и ser/thr-киназ, таких как АКТ, mTOR, PDK, сыворотка- и глюкокортикоид-индуцибельная киназа (SGK), фосфатидилинозитол-4-киназа (PI4K) или фосфатидилинозитол-4-фосфат-5-киназа (PIP5K);
также включены ингибиторы серин/треониновых киназ, не приведенных выше, например, ингибиторы raf-киназ, такие как вемурафениб, ингибиторы МЕК, такие как селуметиниб (AZD6244), ингибиторы тирозинкиназы Абельсона (AbI), такие как иматиниб или нилотиниб, ингибиторы тирозинкиназы Бруттона (Btk), такие как ибрутиниб, ингибиторы тирозинкиназы из селезенки (Syk), такие как фостаматиниб, ингибиторы аврора-киназ (например, AZD1152), ингибиторы других ser/thr-киназ, таких как янус-киназа (JAK), передатчики сигнала и активаторы транскрипции (STAT) и киназа, ассоциированная с рецептором интерлейкина-4 (IRAK4), и ингибиторы циклин-зависимых киназ;
(4) модуляторы сигнальных путей повреждения ДНК, например ингибиторы PARP (например, олапариб), ингибиторы ATR или ингибиторы атаксия-телеангиэктазия-мутированной (ATM) киназы;
(5) модуляторы апоптотического и других путей гибели клетки, такие как модуляторы Bcl-семейства (например, АВТ-263/навитоклакс, АВТ-199);
(6) антиангиогенные агенты, например агенты, ингибирующие действие сосудистого эндотелиального фактора роста (например, антитело к фактору роста эндотелиальных клеток сосудов бевацизумаб (Avastin™) и, например, ингибитор рецепторной тирозинкиназы VEGF, такой как сорафениб, акситиниб, пазопаниб, сунитиниб и вандетаниб, и соединения, которые действуют по другим механизмам (например, линомид, ингибиторы функции интегрина avβ3 и ангиостатин));
(7) агенты, повреждающие сосуды, такие как комбретастатин A4;
(8) антиинвазивные агенты, например ингибиторы c-Src-киназного семейства, такие как дазатиниб (J. Med. Chem., 2004, 47, 6658-6661) и босутиниб (SKI-606), и ингибиторы металлопротеиназ, такие как маримастат, ингибиторы функции рецептора урокиназного активатора плазминогена или антитела к гепараназе;
(9) иммунотерапевтические подходы, включая, например, подходы для повышения иммуногенности опухолевых клеток пациента в условиях ex vivo и in vivo, такие как трансфекция цитокинами, такими как интерлейкин 2, интерлейкин 4 или гранулоцитарно-макрофагальный колониестимулирующий фактор, подходы для снижения T-клеточной анергии, подходы с использованием трансфицированных иммунных клеток, таких как трансфицированные цитокинами дендритные клетки, подходы с использованием трансфицированных цитокинами линий опухолевых клеток и подходы с использованием антиидиотипических антител. Конкретные примеры включают моноклональные антитела, направленно воздействующие на белок 1 программируемой (клеточной) смерти (PD-1) (например, BMS-936558) или цитотоксический Т-лимфоцитарный антиген 4 (CTLA4) (например, ипилимумаб и тремелимумаб);
(10) средства, используемые в антисмысловой терапии или терапии, основанной на РНК-интерференции (RNAi), например, такие, которые направлены на перечисленные выше мишени;
(11) генотерапевтические подходы, включая, например, подходы с заменой аберрантных генов, такие как подходы, касающиеся аберратного гена р53 или аберрантного гена 1 рака молочной железы (BRCA1) или BRCA2, с применением пролекарственной терапии ген-направленным ферментом (GDEPT), например, с использованием цитозиндезаминазы, тимидинкиназы или бактериального фермента нитроредуктазы, и подходы, применяемые для повышения толерантности пациента к химиотерапии или лучевой терапии, такие как терапия с использованием генов множественной лекарственной устойчивости.
Согласно этому аспекту изобретения предложена комбинация, подходящая для применения в лечении рака, содержащая соединение формулы (I), определенное ранее, или его фармацевтически приемлемую соль и другой противоопухолевый агент, в частности любой из противоопухолевых агентов, перечисленных выше в пунктах (1)-(11). В частности, применение противоопухолевого агента, приведенного выше в пунктах (1)-(11), представляет собой стандартное лечение для конкретного подлежащего лечению рака; специалисту в данной области техники будет понятен смысл выражения «стандартное лечение».
Таким образом, согласно другому аспекту изобретения предложены соединение формулы (I) или его фармацевтически приемлемая соль в комбинации с другим противоопухолевым агентом, в частности противоопухолевым агентом, выбранным из агентов, перечисленных выше в данном описании в пунктах (1)-(11).
Согласно другому аспекту изобретения предложены соединение формулы (I) или его фармацевтически приемлемая соль в комбинации с другим противоопухолевым агентом, в частности противоопухолевым агентом, выбранным из агентов, перечисленных выше в пункте (1).
Согласно другому аспекту изобретения предложена комбинация, подходящая для применения в лечении рака, содержащая соединение формулы (I), определенное ранее, или его фармацевтически приемлемую соль и любой из противоопухолевых агентов, перечисленные выше в пункте (1).
Согласно другому аспекту изобретения предложена комбинация, подходящая для применения в лечении рака, содержащая соединение формулы (I), определенное ранее, или его фармацевтически приемлемую соль и таксоид, такой как, например таксол или таксотер, подходящим образом таксотер.
Согласно другому аспекту изобретения предложены соединение формулы (I) или его фармацевтически приемлемая соль в комбинации с другим противоопухолевым агентом, в частности противоопухолевым агентом, выбранным из агентов, перечисленных в данном описании выше в п. (2).
Согласно другому аспекту изобретения предложена комбинация, подходящая для применения в лечении рака, содержащая соединение формулы (I), определенное ранее, или его фармацевтически приемлемую соль и любой из антигормональных агентов, перечисленных выше в пункте (2), например, любой из антиэстрогенов, перечисленных выше в пункте (2).
Согласно другому аспекту изобретения предложены соединение формулы (I) или его фармацевтически приемлемая соль в комбинации с ингибитором mTOR, таким как ингибитор, раскрытый в WO 2008/023161, например,
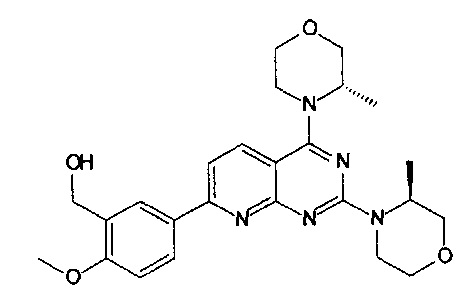 или
или 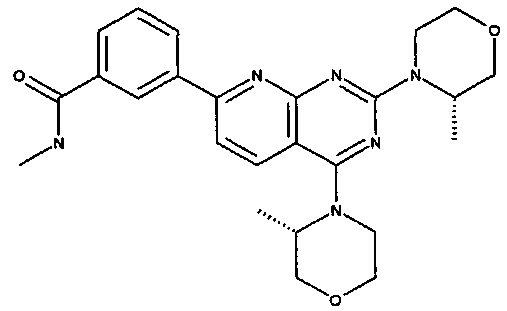 .
.
Согласно другому аспекту изобретения предложена комбинация, подходящая для применения в лечении рака, содержащая соединение формулы (I), определенное ранее, или его фармацевтически приемлемую соль и ингибитор mTOR, такой как ингибитор, раскрытый в WO 2008/023161, например,
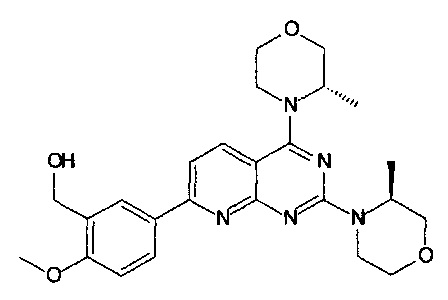 или
или 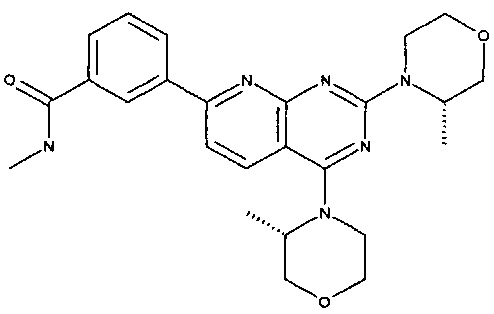 .
.
В частности, ингибитор mTOR представляет собой AZD2014, который имеет следующую структуру:
 .
.
Согласно одному из аспектов вышеупомянутая комбинация соединения формулы (I) и AZD2014 подходит для применения в лечении положительного по рецептору эстрогенов (ER+ve) рака молочной железы, возможно в комбинации со стандартным методом гормональной терапии.
Согласно другому аспекту изобретения предложены соединение формулы (I) или его фармацевтически приемлемая соль в комбинации с ингибитором PI3K-β.
Комбинация соединения формулы (I) с ингибитором PI3K-β может быть особенно полезна при лечении опухолей, например рака предстательной железы, молочной железы (например, трижды негативного рака молочной железы), плоскоклеточного NSCLC и рака почки, дефицитных по PTEN.
Согласно другому аспекту изобретения предложена комбинация, подходящая для применения в лечении рака, содержащая соединение формулы (I), определенное ранее, или его фармацевтически приемлемую соль и ингибитор PI3K-β.
Согласно одному из аспектов ингибиторы PI3K-β, описанные в данной заявке, также обладают некоторой PI3K-δ-ингибирующей активностью.
Согласно другому аспекту изобретения предложены соединение формулы (I) или его фармацевтически приемлемая соль в комбинации с ингибитором PI3K-β, таким как любой ингибитор из примеров, приведенных в международной патентной заявке WO 2011/051704.
Согласно другому аспекту изобретения предложена комбинация, подходящая для применения в лечении рака, содержащая соединение формулы (I), определенное ранее, или его фармацевтически приемлемую соль и ингибитор PI3K-β, такой как любой ингибитор из примеров, приведенных в международной патентной заявке WO 2011/051704.
Согласно другому аспекту изобретения предложены соединение формулы (I) или его фармацевтически приемлемая соль в комбинации с ингибитором PI3K-β и PI3K-δ, таким как 8-((1R)-1-(3,5-дифторфениламино)этил)-N,N-диметил-2-морфолино-4-оксо-4Н-хромен-6-карбоксамид (соединение из примера 3.06b в международной патентной заявке WO 2011/051704, также известное как AZD8186) или его фармацевтически приемлемая соль:
 .
.
Согласно другому аспекту изобретения предложена комбинация, подходящая для применения в лечении рака, содержащая соединение формулы (I), определенное ранее, или его фармацевтически приемлемую соль и ингибитор PI3K-β и PI3K-δ, такой как 8-((1R)-1-(3,5-дифторфениламино)этил)-N,N-диметил-2-морфолино-4-оксо-4Н-хромен-6-карбоксамид (соединение по примеру 3.06b из международной патентной заявки WO 2011/051704, также известное как AZD8186) или его фармацевтически приемлемая соль:
 .
.
Согласно другому аспекту изобретения предложены соединение формулы (I) или его фармацевтически приемлемая соль в комбинации с ингибитором киназы АКТ, таким как (S)-4-амино-N-(1-(4-хлорфенил)-3-гидроксипропил)-1-(7Н-пирроло[2,3-d]пиримидин-4-ил)пиперидин-4-карбоксамид (AZD5363) или его фармацевтически приемлемая соль (см., например WO 2009/047563).
Комбинация соединения формулы (I) и ингибитора АКТ может быть особенно полезна для лечения опухоли с более широкой распространенностью мутации в гене PIK3CA, такой как ER+ve-рак молочной железы, рак эндометрия, яичников, плоскоклеточный NSCLC, рак желудка, мочевого пузыря и желчных протоков.
Согласно другому аспекту изобретения предложена комбинация, подходящая для применения в лечении рака, содержащая соединение формулы (I), определенное ранее, или его фармацевтически приемлемую соль и ингибитор киназы АКТ, такой как (S)-4-амино-N-(1-(4-хлорфенил)-3-гидроксипропил)-1-(7Н-пирроло[2,3-d]пиримидин-4-ил)пиперидин-4-карбоксамид (AZD5363) или его фармацевтически приемлемая соль (см., например WO 2009/047563).
Согласно другому аспекту изобретения предложены соединение формулы (I) или его фармацевтически приемлемая соль в комбинации с олапарибом (4-[3-(4-циклопропанкарбонил-пиперазин-1-карбонил)-4-фтор-бензил]-2Н-фталазин-1-оном) или его фармацевтически приемлемой солью.
Комбинация соединения формулы (I) и олапариба может быть полезна, в частности, как в случае трижды негативного рака молочной железы, либо с геном BRCA дикого типа, либо дефицитного по нему, так и в случае положительного по рецептору эстрогенов (ER+ve) рака молочной железы, в частности, случаев рака с мутациями в гене PIK3CA.
Согласно другому аспекту изобретения предложена комбинация, подходящая для применения в лечении рака, содержащая соединение формулы (I), определенное ранее, или его фармацевтически приемлемую соль и олапариб (4-[3-(4-циклопропанкарбонил-пиперазин-1-карбонил)-4-фтор-бензил]-2Н-фталазин-1-он) или его фармацевтически приемлемую соль.
Конкретные комбинации по изобретению содержат любое из соединений из приведенных в данном описании примеров (или их фармацевтически приемлемую соль) и ингибитор mTOR, ингибитор PI3K-β, ингибитор киназы АКТ или олапариб, которые описаны выше. Другие конкретные комбинации по изобретению содержат соединение из примера 3 (или его фармацевтически приемлемую соль) и ингибитор mTOR, ингибитор PI3K-β, ингибитор киназы АКТ или олапариб, которые описаны выше. Другие конкретные комбинации по изобретению содержат соединение из примера 3 (или его фармацевтически приемлемую соль) и ингибитор PI3K-β, ингибитор киназы АКТ или олапариб (или фармацевтически приемлемую соль любого из этих трех), которые описаны выше. Следующие конкретные примеры комбинаций по изобретению содержат соединение из примера 3 (или его фармацевтически приемлемую соль) и любой из AZD8186, AZD5363 и олапариба (или фармацевтически приемлемой соли любого из этих трех). Другой пример комбинации по изобретению содержит соединение из примера 3 и AZD2014.
В случае всех вышеупомянутых комбинаций будет очевидно, что комбинация также может быть введена вместе со стандартным методом лечения, известным специалисту, таким как другие методы лечения из приведенных ранее в пунктах (1)-(11). Например, если предполагается применение любой из приведенных выше комбинаций для лечения ER+ve-рака молочной железы, то вместе с комбинацией по изобретению может быть применен стандартный метод гормональной терапии (например, агентами, перечисленными выше в пункте (2)). В других аспектах, соответственно, стандартное лечение может быть выбрано из приведенного выше пункта (1).
Таким образом, согласно дополнительному аспекту изобретения, предложена тройная комбинация, подходящая для применения в лечении рака, содержащая:
а) соединение формулы (I) (такое как соединение из примера 3) или его фармацевтически приемлемую соль;
б) ингибитор mTOR, ингибитор PI3K-β, ингибитор киназы АКТ или олапариб или их фармацевтически приемлемую соль; и
в) стандартное лечение для подлежащего лечению рака. Соответственно, стандартное лечение может осуществляться в дозах в соответствии с обычным режимом введения, который известен специалисту.
Согласно другому аспекту изобретения предложена фармацевтическая композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль в комбинации с противоопухолевым агентом, выбранным из агентов, перечисленных выше в данном описании в пунктах (1)-(11), вместе с фармацевтически приемлемым разбавителем или носителем.
Согласно другому аспекту изобретения предложена фармацевтическая композиция, содержащая соединение из примера 3 или его фармацевтически приемлемую соль в комбинации с противоопухолевым агентом, выбранным из агентов, перечисленных выше в данном описании в пунктах вместе с фармацевтически приемлемым разбавителем или носителем.
Согласно другому аспекту изобретения предложена фармацевтическая композиция, содержащая соединение из примера 3 или его фармацевтически приемлемую соль в комбинации с AZD5363, AZD8186 или олапарибом (или фармацевтически приемлемой солью любого из этих трех) вместе с фармацевтически приемлемым разбавителем или носителем.
Согласно другому аспекту изобретения предложена фармацевтическая композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль в комбинации с противоопухолевым агентом, выбранным из агентов, перечисленных в данном описании выше в пунктах (1)-(11), вместе с фармацевтически приемлемым разбавителем или носителем, для применения в лечении рака.
Согласно другому аспекту изобретения предложена фармацевтическая композиция, содержащая соединение из примера 3 или его фармацевтически приемлемую соль в комбинации с противоопухолевым агентом, выбранным из агентов, перечисленных в данном описании выше в пунктах (1)-(11), вместе с фармацевтически приемлемым разбавителем или носителем, для применения в лечении рака.
Согласно другому аспекту изобретения предложена фармацевтическая композиция, содержащая соединение из примера 3 или его фармацевтически приемлемую соль в комбинации с AZD5363, AZD8186 или олапарибом (или фармацевтически приемлемой солью любого из этих трех) вместе с фармацевтически приемлемым разбавителем или носителем, для применения в лечении рака.
Согласно другому признаку данного изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли в комбинации с противоопухолевым агентом, выбранным из агентов, перечисленных выше в данном описании в пунктах (1)-(11), в изготовлении лекарственного средства для применения при лечении рака у теплокровного животного, такого как человек.
Согласно другому признаку данного изобретения предложено применение соединения из примера 3 или его фармацевтически приемлемой соли в комбинации с противоопухолевым агентом, выбранным из агентов, перечисленных в данном описании выше в пунктах (1)-(11), в изготовлении лекарственного средства для применения при лечении рака у теплокровного животного, такого как человек.
Согласно другому признаку данного изобретения предложено применение соединения из примера 3 или его фармацевтически приемлемой соли в комбинации с AZD5363, AZD8186 или олапарибом (или фармацевтически приемлемой солью любого из этих трех) в изготовлении лекарственного средства для применения при лечении рака у теплокровного животного, такого как человек.
Таким образом, согласно дополнительному признаку данного изобретения предложен способ лечения рака у теплокровного животного, такого как человек, нуждающегося в таком лечении, включающий введение указанному животному эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли в комбинации с противоопухолевым агентом, выбранным из агентов, перечисленных в данном описании выше в пунктах (1)-(11).
Таким образом, согласно дополнительному признаку данного изобретения предложен способ лечения рака у теплокровного животного, такого как человек, нуждающегося в таком лечении, включающий введение указанному животному эффективного количества соединения из примера 3 или его фармацевтически приемлемой соли в комбинации с противоопухолевым агентом, выбранным из агентов, перечисленных в данном описании выше в пунктах (1)-(11).
Таким образом, согласно дополнительному признаку данного изобретения предложен способ лечения рака у теплокровного животного, такого как человек, нуждающегося в таком лечении, включающий введение указанному животному эффективного количества соединения из примера 3 или его фармацевтически приемлемой соли в комбинации с AZD5363, AZD8186 или олапарибом (или фармацевтически приемлемой солью любого из этих трех).
Согласно другому аспекту настоящего изобретения предложен набор, содержащий соединение формулы (I) или его фармацевтически приемлемую соль в комбинации с противоопухолевым агентом, выбранным из агентов, перечисленных выше в данном описании в пунктах (1)-(11).
Согласно другому аспекту настоящего изобретения предложен набор, содержащий:
а) соединение формулы (I) или его фармацевтически приемлемую соль в первой стандартной лекарственной форме;
б) противоопухолевый агент, выбранный из агентов, перечисленных выше в данном описании в пунктах (1)-(11), во второй стандартной лекарственной форме; и
в) контейнерные устройства для размещения в них указанных первой и второй лекарственных форм.
Согласно другому аспекту настоящего изобретения предложен набор, содержащий:
а) соединение формулы (I) или его фармацевтически приемлемую соль в первой стандартной лекарственной форме;
б) противоопухолевый агент, выбранный из агентов, перечисленных выше в данном описании в пунктах (1)-(11), во второй стандартной лекарственной форме;
в) контейнерные устройства для размещения в них указанных первой и второй лекарственных форм; и возможно
г) инструкции по применению.
Согласно другому аспекту настоящего изобретения предложен набор, содержащий:
а) соединение формулы (I) или его фармацевтически приемлемую соль в первой стандартной лекарственной форме;
б) ингибитор mTOR, такой как ингибитор, раскрытый в WO 2008/023161, например,
 или
или 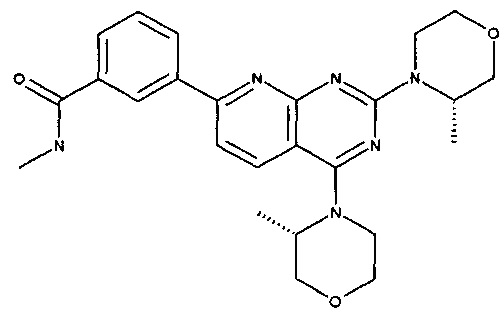 ,
,
во второй стандартной лекарственной форме; и
в) контейнерные устройства для размещения в них указанных первой и второй лекарственных форм.
Согласно другому аспекту настоящего изобретения предложен набор, содержащий:
а) соединение формулы (I) или его фармацевтически приемлемую соль в первой стандартной лекарственной форме;
б) ингибитор PI3K-β, например, любой ингибитор из примеров, приведенных в международной патентной заявке WO 2011/051704, или его фармацевтически приемлемую соль, во второй стандартной лекарственной форме; и
в) контейнерные устройства для размещения в них указанных первой и второй лекарственных форм.
Согласно другому аспекту настоящего изобретения предложен набор, содержащий:
а) соединение формулы (I) или его фармацевтически приемлемую соль в первой стандартной лекарственной форме;
б) ингибитор PI3K-β, например, любой ингибитор из примеров, приведенных в международной патентной заявке WO 2011/051704, или его фармацевтически приемлемую соль, во второй стандартной лекарственной форме;
в) контейнерные устройства для размещения в них указанных первой и второй лекарственных форм; и возможно
г) инструкции по применению.
Согласно другому аспекту настоящего изобретения предложен набор, содержащий:
а) соединение формулы (I) или его фармацевтически приемлемую соль в первой стандартной лекарственной форме;
б) ингибитор PI3K-β и PI3K-δ, представляющий собой 8-((1R)-1-(3,5-дифторфениламино)этил)-N,N-диметил-2-морфолино-4-оксо-4Н-хромен-6-карбоксамид (соединение из примера 3.06b, приведенного в международной патентной заявке WO 2011/051704, также известное как AZD8186), или его фармацевтически приемлемую соль, во второй стандартной лекарственной форме; и
в) контейнерные устройства для размещения в них указанных первой и второй лекарственных форм.
Согласно другому аспекту настоящего изобретения предложен набор, содержащий:
а) соединение формулы (I) или его фармацевтически приемлемую соль в первой стандартной лекарственной форме;
б) ингибитор PI3K-β и PI3K-δ, представляющий собой 8-((1R)-1-(3,5-дифторфениламино)этил)-N,N-диметил-2-морфолино-4-оксо-4Н-хромен-6-карбоксамид (соединение из примера 3.06b, приведенного в международной патентной заявке WO 2011/051704, также известное как AZD8186) или его фармацевтически приемлемую соль, во второй стандартной лекарственной форме;
в) контейнерные устройства для размещения в них указанных первой и второй лекарственных форм; и возможно
г) инструкции по применению.
Согласно другому аспекту настоящего изобретения предложен набор, содержащий:
а) соединение формулы (I) или его фармацевтически приемлемую соль в первой стандартной лекарственной форме;
б) ингибитор киназы АКТ, такой как (S)-4-амино-N-(1-(4-хлорфенил)-3-гидроксипропил)-1-(7Н-пирроло[2,3-d]пиримидин-4-ил)пиперидин-4-карбоксамид или его фармацевтически приемлемая соль (AZD5363, см., например WO 2009/047563), во второй стандартной лекарственной форме;
в) контейнерные устройства для размещения в них указанных первой и второй лекарственных форм; и возможно
г) инструкции по применению.
Согласно другому аспекту настоящего изобретения предложен набор, содержащий:
а) соединение формулы (I) или его фармацевтически приемлемую соль в первой стандартной лекарственной форме;
б) ингибитор киназы АКТ, такой как (S)-4-амино-N-(1-(4-хлорфенил)-3-гидроксипропил)-1-(7Н-пирроло[2,3-d]пиримидин-4-ил)пиперидин-4-карбоксамид или его фармацевтически приемлемая соль (AZD5363, см., например WO 2009/047563), во второй стандартной лекарственной форме; и
в) контейнерные устройства для размещения в них указанных первой и второй лекарственных форм.
Согласно другому аспекту настоящего изобретения предложен набор, содержащий:
а) соединение формулы (I) или его фармацевтически приемлемую соль в первой стандартной лекарственной форме;
б) ингибитор киназы АКТ, такой как (S)-4-амино-N-(1-(4-хлорфенил)-3-гидроксипропил)-1-(7Н-пирроло[2,3-d]пиримидин-4-ил)пиперидин-4-карбоксамид или его фармацевтически приемлемая соль (AZD5363, см., например WO 2009/047563), во второй стандартной лекарственной форме;
в) контейнерные устройства для размещения в них указанных первой и второй лекарственных форм; и возможно
г) инструкции по применению.
Согласно другому аспекту настоящего изобретения предложен набор, содержащий:
а) соединение формулы (I) или его фармацевтически приемлемую соль в первой стандартной лекарственной форме;
б) олапариб (4-[3-(4-циклопропанкарбонил-пиперазин-1-карбонил)-4-фтор-бензил]-2Н-фталазин-1-он) или его фармацевтически приемлемую соль, во второй стандартной лекарственной форме; и
в) контейнерные устройства для размещения в них указанных первой и второй лекарственных форм.
Согласно другому аспекту настоящего изобретения предложен набор, содержащий:
а) соединение формулы (I) или его фармацевтически приемлемую соль в первой стандартной лекарственной форме;
б) олапариб (4-[3-(4-циклопропанкарбонил-пиперазин-1-карбонил)-4-фтор-бензил]-2Н-фталазин-1-он) или его фармацевтически приемлемую соль, во второй стандартной лекарственной форме;
в) контейнерные устройства для размещения в них указанных первой и второй лекарственных форм; и возможно
г) инструкции по применению.
Во всех случаях комбинаций, применений, способов лечения и наборов, упомянутых выше, AZD5363, AZD8186 и олапариб могут быть в форме свободных оснований или в форме фармацевтически приемлемой соли. Таким образом, в одном из воплощений AZD5363 существует в форме свободного основания; в следующем воплощении AZD5363 существует в форме фармацевтически приемлемой соли. В другом воплощении AZD8186 существует в форме свободного основания; в следующем воплощении AZD8186 существует в форме фармацевтически приемлемой соли. В другом воплощении олапариб существует в форме свободного основания; в следующем воплощении олапариб существует в форме фармацевтически приемлемой соли.
Несмотря на то, что соединения формулы (I) прежде всего имеют значение как терапевтические агенты для применения в случае теплокровных животных (включая человека), они также являются полезными всякий раз, когда необходимо ингибировать действия PI3-киназы-α и -δ. Таким образом, они полезны в качестве фармакологических стандартов для применения в разработке новых биологических тестов и в поиске новых фармакологических агентов.
Там, где в данном описании используется термин «комбинация», следует понимать, что он относится к одновременному, раздельному или последовательному введению. Согласно одному из аспектов изобретения «комбинация» относится к одновременному введению. Согласно другому аспекту изобретения «комбинация» относится к раздельному введению. Согласно другому аспекту изобретения «комбинация» относится к последовательному введению. Если введение является последовательным или раздельным, то задержка во введении второго компонента не должна быть такой, чтобы привести к потере благоприятного эффекта.
В одном из воплощений последовательное лечение включает введение каждого компонента комбинации в пределах периода времени, составляющего 11 суток. В другом воплощении этот период составляет 10 суток. В другом воплощении этот период составляет 9 суток. В другом воплощении этот период составляет 8 суток. В другом воплощении этот период составляет 7 суток. В другом воплощении этот период составляет период в пределах 6 суток. В другом воплощении этот период составляет период в пределах 5 суток. В другом воплощении этот период составляет период в пределах 4 суток. В другом воплощении этот период составляет период в пределах 3 суток. В другом воплощении этот период составляет период в пределах 2 суток. В другом воплощении этот период составляет период в пределах 24 часов. В другом воплощении этот период составляет период в пределах 12 часов.
Последовательное и совместное введение в данном описании проиллюстрированы примерами в экспериментах с использованием комбинации соединения из примера 3 и AZD5363 в модели клеток ВТ474. Приведенное в этом примере последовательное введение иллюстрируется введением AZD5363 в течение 2-х суток, затем введением соединения из примера 3 в течение 2-х суток, затем в течение 3-х суток никакого введения ни того, ни другого агента не осуществляют, после чего схему повторяют («цикл приема препарата»). Совместное введение иллюстрируется режимом введения, когда в течение 2-х суток вводят как AZD5363, так и соединение из примера 3, затем в течение 5-ти суток никакого введения ни того, ни другого агента не осуществляют. Приведенное в этих двух примерах последовательное введение, по-видимому, является более эффективным для вызывания регрессии опухоли, иллюстрируя потенциальную важность оптимизации режима. Другие возможные режимы совместного введения включают:
1) цикл приема препарата, при котором как AZD5363, так и соединение из примера 3 вводят в течение 2-х суток, затем в течение 3-х суток никакого введения ни того, ни другого агента не осуществляют;
2) цикл приема препарата, при котором как AZD5363, так и соединение из примера 3 вводят в течение 3-х суток, затем в течение 4-х суток никакого введения ни того, ни другого агента не осуществляют;
3) цикл приема препарата, при котором как AZD5363, так и соединение из примера 3 вводят в течение 4-х суток, затем в течение 3-х суток никакого введения ни того, ни другого агента не осуществляют;
4) цикл приема препарата, при котором как AZD5363, так и соединение из примера 3 вводят в течение 5-ти суток, затем в течение 2-х суток никакого введения ни того, ни другого агента не осуществляют;
5) цикл приема препарата, при котором AZD5363 и соединение из примера 3 вводят через день;
6) цикл приема препарата, при котором AZD5363 и соединение из примера 3 вводят каждые трое суток;
7) цикл приема препарата, при котором AZD5363 и соединение из примера 3 вводят по еженедельной схеме с перерывами в 3-е и 4-е суток между введениями (например, по схеме понедельник/четверг);
8) цикл приема препарата, при котором AZD5363 и соединение из примера 3 вводят по еженедельной схеме с перерывами в 2-е и 3-е суток между введениями (например, по схеме понедельник/среда/пятница).
Комбинации соединений формулы (I), в частности, соединения из примера 3, с ингибитором mTOR, таким как AZD2014, или ингибитором PI3K-β (таким как ингибитор киназы-β/δ AZD8186), соответственно, могут быть введены в режиме, аналогичном описанным выше для комбинации соединения из примера 3 и AZD5363.
Комбинацию соединения формулы (I) и олапариба можно вводить согласно такому режиму, когда олапариб вводят один раз в сутки, а соединение формулы (I) вводят по схеме интермиттирующего приема препарата (как например, введение в течение 2-х суток, затем в течение 3-5 суток никакого введения не осуществляют).
Каждый из этих иллюстративных режимов введения составляет дополнительный аспект изобретения. Каждый из этих иллюстративных режимов введения также может быть применен к комбинациям с другими противоопухолевыми агентами, перечисленными выше в пунктах (1)-(11).
В рамках заданного цикла приема препарата может иметь преимущество введение одного конкретного компонента комбинации перед другим - т.е. последовательное введение.
Таким образом, в одном из воплощений последовательное введение включает последовательное введение соединения формулы (I) (в частности, соединения из примера 3) перед введением другого противоопухолевого агента из приведенного выше списка (1)-(11), в частности, противоопухолевого агента, выбранного из AZD5363, AZD8186 и олапариба, в рамках цикла приема препарата.
В другом воплощении последовательное введение включает последовательное введение противоопухолевого агента из приведенного выше списка (1)-(11), в частности, противоопухолевого агента, выбранного из AZD5363, AZD8186 и олапариба, перед введением соединения формулы (I) (в частности, соединения из примера 3) в рамках цикла приема препарата.
В одном из воплощений противоопухолевый агент из приведенного выше списка (1)-(11) и соединение формулы (I) вводят с интервалом до 2-х суток включительно. В другом воплощении противоопухолевый агент из приведенного выше списка (1)-(11) и соединение формулы (I) вводят с интервалом до 1-х суток включительно. В другом воплощении противоопухолевый агент, приведенный выше в пунктах (1)-(11), и соединение формулы (I) вводят с интервалом до 18-ти часов включительно. В другом воплощении противоопухолевый агент из приведенного выше списка (1)-(11) и соединение формулы (I) вводят с интервалом до 12-ти часов включительно. В другом воплощении противоопухолевый агент из приведенного выше списка (1)-(11) и соединение формулы (I) вводят с интервалом до 6-ти часов включительно. В другом воплощении противоопухолевый агент из приведенного выше списка (1)-(11) и соединение формулы (I) вводят с интервалом до 3-х часов включительно.
В других воплощениях продолжительность цикла введения может составлять от 5 до 10 суток.
В других воплощениях продолжительность цикла введения может составлять от 6 до 10 суток.
В других воплощениях продолжительность цикла введения может составлять от 7 до 9 суток.
В других воплощениях продолжительность цикла введения может составлять от 6 до 8 суток.
В других воплощениях продолжительность цикла введения может составлять 10 суток.
В других воплощениях продолжительность цикла введения может составлять 9 суток.
В других воплощениях продолжительность цикла введения может составлять 8 суток.
В других воплощениях продолжительность цикла введения может составлять 7 суток.
В других воплощениях продолжительность цикла введения может составлять 6 суток.
В других воплощениях продолжительность цикла введения может составлять 5 суток.
В других воплощениях цикл приема препарата может включать введение соединения формулы (I) (в частности, соединения из примера 3) в течение 2-4-х следующих друг за другом суток и отсутствие введения в остальные сутки в рамках цикла приема препарата продолжительностью от 6 до 9 суток.
В других воплощениях цикл приема препарата может включать введение соединения формулы (I) (в частности, соединения из примера 3) в течение 3-4-х следующих друг за другом суток и отсутствие введения в остальные сутки в рамках цикла приема препарата продолжительностью от 6 до 9 суток (например, продолжительностью 7 суток).
В других воплощениях цикл приема препарата может включать введение соединения формулы (I) (в частности, соединения из примера 3) в течение 3-5-ти следующих друг за другом суток и отсутствие введения в остальные сутки в рамках цикла приема препарата продолжительностью от 7 до 10 суток.
В других воплощениях цикл приема препарата может включать введение соединения формулы (I) (в частности, соединения из примера 3) в течение 5-ти следующих друг за другом суток и отсутствие введения в остальные сутки в рамках цикла приема препарата продолжительностью от 6 до 9 суток.
В других воплощениях цикл приема препарата может включать введение соединения формулы (I) (в частности, соединения из примера 3) в течение 4-х следующих друг за другом суток и отсутствие введения в остальные сутки в рамках цикла приема препарата продолжительностью от 6 до 9 суток (например, продолжительностью 7 суток).
В других воплощениях цикл приема препарата может включать введение соединения формулы (I) (в частности, соединения из примера 3) в течение 3-х следующих друг за другом суток и отсутствие введения в остальные сутки в рамках цикла приема препарата продолжительностью от 6 до 9 суток.
Циклы приема препаратов могут быть разделены некоторым количеством суток, когда ни один из активных компонентов комбинации не вводят.
Комбинированная терапия, описанная выше, может быть проведена в добавление к стандартному лечению, в типичном случае осуществляемому в соответствии с обычной предписанной схемой применения.
Персонализированная медицинская помощь
Другой аспект настоящего изобретения основан на идентификации связи между статусом гена, кодирующего полипептид - каталитическую субъединицу-альфа фосфоинозитид-3-киназы (PIK3CA), и чувствительностью к лечению соединением формулы (I). Таким образом, эта идентификация предоставляет возможности, способы и средства для отбора пациентов с целью лечения соединением формулы (I), в частности, раковых пациентов, и/или для избегания лечения пациентов, с меньшей вероятностью поддающихся данному лечению, что тем самым позволяет избежать нецелесообразного лечения и любых побочных эффектов, которые могут быть связаны с таким неэффективным лечением.
Настоящее изобретение относится к средствам и способам отбора пациентов (включая персонализированную медицину). Отбор основан на определении того, ген PIK3CA какого типа, дикого или мутантного, имеют подлежащие лечению опухолевые клетки. Таким образом, статус гена PIK3CA может быть использован в качестве биомаркера восприимчивости к лечению ингибитором PI3K-α и -δ.
Существует очевидная потребность в биомаркерах, с помощью которых будет расширен круг или будут отобраны пациенты, у которых опухоли будут поддаваться лечению ингибитором PI3K-α и -δ, таким как соединение формулы (I). Биомаркеры для отбора пациентов, с помощью которых идентифицируют пациентов, с наибольшей вероятностью поддающихся лечению агентом, являются идеальными биомаркерами для лечения рака, поскольку, применяя их, можно отказаться от нецелесообразного лечения пациентов, имеющих не поддающиеся лечению опухоли, и снизить возможные побочные эффекты таких агентов.
Биомаркер можно описать как «отличительный признак, который объективно определяют и оценивают в качестве индикатора нормальных биологических процессов, патогенных процессов или фармакологических ответов на терапевтическое вмешательство». Биомаркером является любой поддающийся определению и количественной оценке индикатор, ассоциированный с конкретным состоянием или заболеванием, где имеет место корреляция между наличием или уровнем этого биомаркера и некоторым аспектом данного состояния или заболевания (включая наличие, уровень или изменение уровня, тип, стадию состояния или заболевания, чувствительность к состоянию или заболеванию или восприимчивость к лекарственному средству, используемому для лечения данного состояния или заболевания). Корреляция может быть качественной, количественной или качественной и количественной. Обычно биомаркером является соединение, фрагмент соединения или группа соединений. Такими соединениями могут быть любые соединения, обнаруженные в организме или продуцируемые организмом, включая белки (и пептиды), нуклеиновые кислоты и другие соединения.
Биомаркеры могут обладать прогностической способностью, и в силу этого их можно использовать для предсказания или обнаружения присутствия, определения уровня, типа или стадии конкретных состояний или заболеваний (включая присутствие или уровень конкретных микроорганизмов или токсинов), предрасположенности (включая генетическую предрасположенность) к конкретным состояниям или заболеваниям или ответной реакции на конкретные виды лечения (включая лечения лекарственными средствами). Считается, что биомаркеры будут играть все более важную роль в будущем в открытии и разработке лекарственных средств, повышая эффективность программ научно-исследовательских работ и программ развития. Биомаркеры можно использовать в качестве диагностических агентов, средств мониторинга прогрессирования заболеваний, средств мониторинга лечения и средств прогноза клинического исхода. Например, различные научные проекты, касающиеся биомаркеров, представляют собой попытки идентификации маркеров конкретных видов рака и конкретных сердечно-сосудистых и иммунных заболеваний. Полагают, что разработка новых одобренных биомаркеров будет приводить к значительным сокращениям затрат в области здравоохранения и разработки лекарственных средств, а также к значительным улучшениям в лечении широкого спектра заболеваний и состояний.
Для оптимального планирования клинических испытаний и получения наиболее полной информации по результатам этих испытаний может потребоваться биомаркер. Маркер может поддаваться количественной оценке в суррогатных и опухолевых тканях. В идеале эти маркеры также будут коррелировать с эффективностью лечения и, таким образом, могут в конечном итоге быть использованы для отбора пациентов.
Таким образом, технической задачей, лежащей в основе этого аспекта настоящего изобретения, является идентификация средств стратификации пациентов с целью лечения соединением формулы (I). Данная техническая проблема решается путем применения воплощений, охарактеризованных в формуле изобретения и/или в описании данной заявки.
Как подробно описано в данной заявке в примерах, было обнаружено, что клетки, которые имеют мутацию в PIK3CA, как правило, более чувствительны к ингибированию роста соединением формулы (I).
Согласно изобретению предложен способ определения чувствительности клеток к соединению формулы (I). Способ включает определение статуса гена PIK3CA в указанных клетках. Клетки определяют как вероятно чувствительные к соединению формулы I, если эти клетки содержат подвергнутый мутации ген PIK3CA. Ввиду этого предполагается, что такие пациенты с подвергнутым мутации геном PIK3CA будут особенно чувствительны к лечению соединением формулы (I). Клетку определяют как чувствительную к соединению формулы (I), если оно ингибирует увеличение количества клеток в анализе клеточного роста (или посредством ингибирования клеточной пролиферации, и/или посредством усиления гибели клеток). Способы по изобретению полезны для предсказания того, какие клетки с большей долей вероятности реагируют ингибированием роста на соединение формулы (I).
Настоящее изобретение далее касается, в некоторой степени, способов, которые можно использовать для определения восприимчивости пациента к соединению формулы (I), включающих определение того, следует ли вводить соединение формулы (I). Конкретно, способы по настоящему изобретению включают определение статуса гена PIK3CA. Наличие подвергнутого мутации гена PIK3CA указывает на то, что опухолевые клетки с большей долей вероятности реагируют ингибированием своего роста при контакте с соединением формулы (I). Поэтому статус гена PIK3CA можно использовать для отбора пациентов с целью лечения соединением формулы (I).
Кроме того, описан способ in vitro идентификации пациента, который вероятно будет чувствительным к соединению формулы (I). Также описаны применения олиго- или полинуклеотидных праймеров или зондов, с помощью которых можно определить мутационный статус гена PIK3CA. Также описано применение наборов для обнаружения мутаций в PIK3CA, включая, но не ограничиваясь этим, наборы для обнаружения мутаций в PIK3CA, поставляемые на рынок диагностическими компаниями, включая Qiagen и Roche MolecuIar Systems. В другом воплощении изобретение относится к способу определения in vitro того, будет ли пациент, страдающий раковым заболеванием, с вероятностью отвечать на фармацевтическое лечение соединением формулы (I), где указанный способ включает стадии: (1) получения относящегося к опухоли образца, предварительно полученного от указанного пациента; и (2) определения того, содержат ли гены PIK3CA мутацию в указанном образце. Мутация в гене PIK3CA является указанием на повышение вероятности ответа на лечение соединением формулы (I). Как тест на биомаркер к единственному гену, идентификация опухолей, которые содержат мутацию в PIK3CA, будет повышать ответ на соединение формулы (I). Отдельные опухоли, которые содержат мутацию в PIK3CA, имеют наибольшую вероятность ответа на соединение формулы (I).
«Относящийся к опухоли» образец может представлять собой образец, непосредственно выделенный из опухоли, или может представлять собой образец, который был подвергнут дополнительной обработке, например, образец ПЦР-амллифицированной нуклеиновой кислоты из опухолевого образца.
Определения
В этом разделе под заголовком «Персонализированная медицинская помощь» использованы следующие термины.
«Аллель» относится к особой форме генетического локуса, отличающейся от других форм своей особой нуклеотидной или аминокислотной последовательностью.
«Реакции амплификации» представляют собой реакции с участием нуклеиновых кислот, которые приводят к специфическому многократному увеличению числа копий целевых нуклеиновых кислот по сравнению с нецелевыми нуклеиновыми кислотами. Полимеразная цепная реакция (ПЦР) представляет собой хорошо известную реакцию амплификации.
Термин «рак» используется в данном описании для обозначения неопластического роста, вызываемого трансформацией клеток с приобретением ими неопластического фенотипа. Такая трансформация клеток часто подразумевает генетическую мутацию.
«Ген» представляет собой сегмент ДНК, который содержит всю информацию относительно регулируемого биосинтеза продукта РНК, включая промотор, экзоны, интроны и другие элементы последовательности, которые могут быть локализованы между 5'- или 3'-фланкирующими участками (не между транскрибируемыми частями гена), которые регулируют экспрессию.
«Статус гена» относится к тому, является ли данный ген геном дикого типа или нет (т.е. мутантным геном).
«Метка» относится к композиции, способной давать детектируемый сигнал, указывающий на присутствие целевого полинуклеотида в анализируемом образце. Подходящие метки включают радиоактивные изотопы, нуклеотидные хромофоры, ферменты, субстраты, флуоресцентные молекулы, хемилюминесцентные группировки, магнитные частицы, биолюминесцентные группировки и тому подобное. Таким образом, метка представляет собой любую композицию, которую можно обнаружить спектроскопическим, фотохимическим, биохимическим, иммунохимическим, электрическим, оптическим или химическим способами.
«Несинонимичное изменение» относится к изменению (вариации) в кодирующей последовательности или к перекрыванию кодирующей последовательности гена, которое приводит к образованию отличающейся (измененной) полипептидной последовательности. Эти изменения могут влиять или могут не влиять на функционирование белка и включают миссенс-варианты (в результате замены одной аминокислоты на другую), нонсенс-варианты (в результате укорачивания полипептида ввиду образования преждевременного стоп-кодона) и варианты со вставкой/делецией.
«Синонимичное изменение» относится к изменению (вариации) в кодирующей последовательности гена, которое не влияет на последовательность кодируемого полипептида. Эти изменения могут влиять на функционирование белка опосредованно (например, путем изменения экспрессии гена), но, в отсутствие доказательств обратного, как правило, считаются безвредными.
Термин «нуклеиновая кислота» относится к одноцепочечным или двухцепочечным молекулам ДНК и РНК, в том числе обычным нуклеиновым кислотам, обнаруженным в природе, и/или модифицированным, искусственным нуклеиновым кислотам, имеющим модифицированные остовы или основания, которые известны в данной области техники.
«Праймер» относится к олигонуклеотидной последовательности одноцепочечной ДНК, способной действовать в качестве точки инициации синтеза продукта удлинения праймера, который комплементарен копируемой цепи нуклеиновой кислоты. Длина и последовательность праймера должны быть такими, чтобы способствовать праймированию синтеза продуктов удлинения. Типичный праймер содержит последовательности длиной по меньшей мере примерно 7 нуклеотидов, по существу комплементарные последовательности-мишени, однако предпочтительны несколько более длинные праймеры. Обычно праймеры содержат примерно 15-26 нуклеотидов, тем не менее также можно использовать более длинные или более короткие праймеры.
«Полиморфный сайт» представляет собой положение в пределах локуса, в котором в популяции обнаружены по меньшей мере две альтернативные последовательности.
«Полиморфизм» относится к изменению последовательности в полиморфном сайте, наблюдаемому у индивидуума. Полиморфизмы включают нуклеотидные замены, вставки, делеции и микросателлитные локусы и могут, но не обязательно, приводить к детектируемым различиям в экспрессии генов или функционировании белка. В отсутствие данных о влиянии на экспрессию или функционирование белка обычные полиморфизмы, включая несинонимичные варианты, обычно считаются включенными в определение последовательности гена дикого типа. Каталог полиморфизмов у человека и связанная с ним аннотация, включая валидацию, наблюдаемые частоты встречаемости и связь с заболеваниями, поддерживается Национальным центром биотехнологической информации (NCBI) (база данных по однонуклеотидным полиморфизмам (dbSNP): http://www.ncbi.nlm.nih.gov/proiects/SNP/). Необходимо отметить, что термин «полиморфизм» при его использовании в контексте последовательностей генов, не следует путать с термином «полиморфизм», когда его используют в контексте твердой формы соединения, то есть кристаллической или аморфной природы соединения. Специалисту будет понятно предполагаемое значение по его контексту.
«Зонд» относится к одноцепочечным олигонуклеотидам со специфической последовательностью, точно комплементарной последовательности-мишени аллеля, подлежащего обнаружению.
«Ответ» определяют по результатам измерений, проведенных в соответствии с «Критериями оценки ответа при солидных опухолях» (RECIST; Response evaluation criteria in solid tumours), включая разделение пациентов на две основные группы: на тех, которые демонстрируют частичный ответ или стабильное заболевание, и тех, которые демонстрируют признаки прогрессирующего заболевания.
Термин «строгие условия гибридизации» относится к инкубированию в течение ночи при 42°C в растворе, содержащем 50% формамида, 5х SSC (раствор хлорида и цитрата натрия) (750 мМ NaCl, 75 мМ трехзамещенный цитрат натрия), 50 мМ фосфат натрия (pH 7,6), 5x раствор Денхардта, 10% декстрансульфата и денатурированную, деградированную в результате гидродинамического сдвига ДНК из молок лососевых рыб в концентрации 20 пг/мл, с последующей промывкой фильтров в 0,1x SSC при примерно 65°C.
Термин «выживаемость» охватывает общую выживаемость и выживаемость пациентов без прогрессирования заболевания.
«Общая выживаемость» (OS) определяется как промежуток времени от начала введения лекарственных средств до момента смерти по любой причине. «Выживаемость без прогрессирования заболевания» (PFS) определяется как промежуток времени от начала введения лекарственных средств до первого проявления прогрессирующего заболевания или смерти по любой причине.
Согласно одному аспекту изобретения предложен способ отбора пациента для лечения соединением формулы (I), включающий получение содержащего опухолевые клетки образца от пациента; определение того, является ли ген PIK3CA в содержащем опухолевые клетки образце от пациента геном дикого типа или мутантным геном; и на основании этого отбор пациента для лечения соединением формулы (I).
Способ может включать или не включать фактическую стадию выделения образца от пациента. Таким образом, согласно одному аспекту изобретения предложен способ отбора пациента для лечения соединением формулы (I), включающий определение того, является ли ген PIK3CA в содержащем опухолевые клетки образце, предварительно полученном от пациента, геном дикого типа или мутантным геном; и, исходя из этого, отбор пациентов для лечения соединением формулы (I).
В одном из воплощений пациентов отбирают для лечения соединением формулы (I), если ДНК из опухолевых клеток имеет мутантный ген PIK3CA. В других воплощениях, пациента, у которого ДНК из опухолевых клеток содержит ген PIK3CA дикого типа, не отбирают для лечения соединением формулы (I).
Согласно другому аспекту изобретения предложен способ прогнозирования восприимчивости пациента на соединение формулы (I), включающий определение того, является ли ген PIK3CA в опухолевых клетках пациента геном дикого типа или мутантным геном, и, на основании этого прогнозирование восприимчивости пациента на лечение соединением формулы (I).
Согласно другому аспекту изобретения предложен способ определения вероятности эффективности лечения соединением формулы I человека-пациента, пораженного раком, включающий: определение того являет(ют)ся ли ген(ы) PIK3CA в опухолевых клетках пациента геном(ами) дикого типа или мутантным(и) геном(ами), и, исходя из этого, прогнозирование восприимчивости пациента к лечению соединением формулы (I).
В контексте данного изобретения под статусом гена дикого типа подразумевается указание на обычную или соответствующую экспрессию гена и обычное функционирование кодируемого белка. И наоборот, под мутантным статусом подразумевается указание на аномальную или несоответствующую генную экспрессию или экспрессию белка с измененной функцией, соответствующую известной роли мутантной PIK3CA при раке (как описано в данной заявке). Любое число генетических или эпигенетических изменений, включая, но не ограничиваясь этим, мутацию, амплификацию, делецию, геномную перегруппировку или изменения в профиле метилирования, может приводить к формированию мутантного статуса. Однако, если такие изменения, тем не менее, приводят к соответствующей экспрессии нормального белка или функционально эквивалентного варианта, тогда статус данного гена считается статусом гена дикого типа. Примеры вариантов, которые, как правило, не приводят к формированию у гена функционального мутантного статуса, включают варианты, кодирующие синонимичные изменения, и общие полиморфизмы (синонимичные или несинонимичные). Как осуждается ниже, статус гена можно оценить в функциональном анализе, или он может быть выведен из природы детектируемых отклонений от ссылочной последовательности.
В некоторых воплощениях статус дикого типа или мутантный статус гена PIK3CA определяют по присутствию или отсутствию несинонимичных изменений нуклеиновой кислоты в генах. Наблюдаемые несинонимичные изменения, соответствующие известным общим полиморфизмам без каких-либо аннотированных функциональных эффектов, не придают гену статуса мутантного гена.
Другие изменения в гене PIK3CA, являющиеся признаком мутантного статуса, включают изменения в сайте сплайсинга, которые ухудшают распознавание мест соединения интрон/экзон во время процессинга пре-мРНК в мРНК. Это может приводить к пропуску/прерыванию экзона или включению в норме интронной последовательности в подвергнутую сплайсингу мРНК (удерживанию интрона или использованию скрытых границ сплайсинга). Это может, в свою очередь, приводить к получению аберрантного белка со вставками и/или делециями по сравнению с нормальным белком. Таким образом, в других воплощениях ген имеет мутантный статус, если имеется вариант, в котором изменена последовательность распознавания сайта сплайсинга в месте соединения интрон/экзон.
Кроме того, определение мутационного статуса или активационного статуса дополнительных генов, таких как K-ras, потенциальный маркер устойчивости в опухолях с аберрантной или разрегулированной PIK3CA или PI3K-α, может помочь в повышении предсказательной способности персонализированного подхода в медицине.
В ходе исследования, проведенного авторами изобретения в AstraZeneca на примере случаев рака молочной железы (на основе базы данных Каталога соматических мутаций при раковых заболеваниях (COSMIC; Catalog of Somatic Mutations in Cancer) (Благотворительный фонд института Сенждера (Welcome Trust Sanger Institute), сентябрь 2011 г.), было идентифицировано более 55 разных мутаций в гене PIK3CA в целом для набора данных, охватывающего более 5 тысяч опухолей человека. Большинство мутаций встречалось с частотой меньше 1%, 3 мутации встречались с частотой 1-3%, но 4 мутации составляли приблизительно 88% от общего числа мутаций PIK3CA. Это были миссенс-мутации киназного домена, расположенные в C-концевом домене киназы, H1047R (55%) и H1047L (5%), и в остатках спирального домена, Е545К (18%) и Е542К (11%). Более длинный список других превалирующих мутаций рака молочной железы, хотя подразумевается, что он не исчерпывается ими, охватывает R38H, R38C, R88Q, N345K, C420R, E453Q, P539R, Е542К, Е545К, Е545А, Q546K, Q546P, M1043I, M1043V, H1047R, H1047L, H1047Y. Следовательно, могут быть разработаны диагностические анализы, которые фокусируются на обнаружении наиболее часто встречающихся мутаций, тем самым позволяя идентифицировать большинство мутаций в PIK3CA. Например, тест для идентификации мутаций в PIK3CA Cobas (TM) от Roche MolecuIar Systems сконструирован для обнаружения 17 мутаций в экзонах 1, 4, 7, 9 и 20 гена PIK3CA (Е542К, Е545А, E545G, Е545К, E545D, Q546K, Q546R, Q546E, Q546L, N345K, C420R, R88Q, H1047L, H1047R, H1047Y, G1049R и M1043I) в ДНК, выделенной из фиксированных в формалине залитых парафином образцов опухолей. С помощью этого набора можно установить приблизительно до 95% мутаций при ER+ve-раке молочной железы. Распределение мутаций является неодинаковым среди других типов опухолей, и диагностическая стратегия может быть соответствующим образом адаптирована. Например, в случае рака эндометрия наблюдается более равномерное распределение мутаций по всей кодирующей ген PIK3CA последовательности и с большим количеством мутаций в N-концевом участке белка (согласно сообщению DougIas A., Levine, M.D. на 2-ом ежегодном симпозиуме TCGA, 28 ноября 2012 г.) по сравнению со случаями рака молочной железы.
В случае PIK3CA ссылочные последовательности для этого гена (номер доступа в Genbank: NG_012113), мРНК (номер доступа в Genbank: NM_006218) и белка (номер доступа в Genbank: NP_006209 или номер доступа в Swiss-Prot: Р42336) находятся в свободном доступе. Ссылочные последовательности гена (геномного участка) включают 5000 оснований расположенной в обратном направлении последовательности и 2000 оснований расположенной в прямом направлении последовательности. Мутации в PIK3CA хорошо известны (из базы данных COSMIC - Welcome Trust Sanger Institute), и специалист в данной области техники будет в состоянии определить статус гена PIK3CA, т.е. того, является ли конкретный ген PIK3CA геном дикого типа или мутантным геном, основываясь на сравнении последовательности ДНК или белка с последовательностями, отнесенными к дикому типу.
Очевидно, что каждая из последовательностей гена и мРНК, описанных для PIK3CA, и белковая последовательность каталитической субъединицы PI3-киназы-альфа, p110α, представляет собой репрезентативную последовательность. У здоровых индивидуумов имеется две копии каждого гена, материнская и отцовская копия, которые вероятно будут несколько отличаться по последовательности, кроме того, в пределах популяции будут существовать многочисленные аллельные варианты генной последовательности. Другие последовательности, отнесенные к дикому типу, включают последовательности, имеющие одно или более синонимичных изменений в нуклеиновокислотной последовательности (изменений, не влияющих на кодируемую белковую последовательность), несинонимичных общих полиморфизмов (например, полиморфизмов, относящихся к зародышевой линии), которые изменяют белковую последовательность, но не влияют на функцию белка, и изменений в интронах, не затрагивающих последовательность сайтов сплайсинга.
Согласно другому аспекту изобретения предложен способ определения вероятности эффективности лечения соединением формулы (I) человека-пациента, пораженного раком, включающий: обнаружение присутствия или отсутствия по меньшей мере одной несинонимичной вариации в нуклеиновой кислоте гена PIK3CA у указанного пациента по сравнению с геном дикого типа, причем присутствие по меньшей мере одной соматической несинонимичной вариации в нуклеиновой кислоте гена PIK3CA указывает на то, что лечение соединением формулы (I), по всей вероятности, будет эффективным.
Согласно другому аспекту изобретения предложен способ оценки чувствительности индивидуума к лечению соединением формулы (I), включающий:
(1) определение связанного с несинонимичным изменением мутационного статуса гена PIK3CA в ДНК из клеток опухоли индивидуума; и
(2) определение вероятной чувствительности индивидуума к лечению соединением формулы (I) с учетом связанного с несинонимичным изменением мутационного статуса гена PIK3CA в клетках опухоли.
Существуют многочисленные методы, доступные специалисту в данной области техники, для определения статуса гена PIK3CA. Статус гена может быть установлен посредством определения нуклеиновокислотной последовательности. Это можно осуществить путем прямого секвенирования полноразмерного гена или анализа специфических сайтов в данном гене, например, сайтов, чаще всего подвергающихся мутациям.
Альтернативное средство определения того, является ген PIK3CA геном дикого типа или мутантным геном или не является таковым, представляет собой оценку функции транскрибированного гена. При наличии в этом гене PIK3CA мутации, влияющей на функцию, образуется белок, обладающий повышенной активностью липидной киназы, что приводит к усиленной передаче сигнала ниже по пути в клетках, включая, но не ограничиваясь этим, активацию киназы Akt и S6. Такие анализы для оценки функционального статуса вариантов PIK3CA, в случае экспрессии в клетках, включают, но не ограничиваются этим:
(1) увеличенное образование продукта киназной активности гена PIK3CA, фосфатидилинозит-трифосфата (PI(3,4,5)P3);
(2) повышенные уровни фосфорилирования киназы АКТ или S6;
(3) усиленное образование фокусов и колоний клеток NIH-3T3, трансфицированных вариантами PIK3CA (Ikenoue Т. et al., Cancer Res., 2005, 65, 4562-4567).
Образцы
Образцом от пациента, предназначенным для тестирования на статус гена, может быть любая опухолевая ткань или образец, содержащий клетки опухоли, полученный или который может быть получен от индивидуума. Подходящим образом тестируемый образец представляет собой образец крови, мазка из ротовой полости, биоптата либо другой жидкости или ткани, полученный от индивидуума. Конкретные примеры включают: циркулирующие опухолевые клетки, циркулирующую в плазме или сыворотке крови ДНК, клетки, выделенные из асцитной жидкости пациентов с раком яичника, мокроту из легких от пациентов с опухолями в легких, тонкоигольный пунктат от пациента с раком молочной железы, мочу, периферическую кровь, клеточный соскоб, волосяной фолликул, взятый пункционной биопсией образец кожи или буккальный мазок.
Очевидно, что помимо этого тестируемый образец может содержать нуклеиновую кислоту с последовательностью, соответствующей последовательности в тестируемом образце, то есть весь участок или часть участка нуклеиновой кислоты в образце перед анализом можно сначала амплифицировать, используя любой удобный метод, например полимеразную цепную реакцию (ПЦР). Нуклеиновая кислота может представлять собой геномную ДНК либо фракционированную или суммарную клеточную РНК. В конкретных воплощениях РНК представляет собой суммарную клеточную РНК, и ее используют непосредственно в качестве матрицы для мечения первой цепи «ДНК с применением случайных праймеров или поли(A)-содержащих праймеров. Находящиеся в тестируемом образце нуклеиновую кислоту или белок можно экстрагировать из этого образца в соответствии со стандартными методологиями (см. Green & Sambrook, Eds., MolecuIar Cloning: A Laboratory Manual (2012, 4th edition, Vol. 1-3, ISBN 9781936113422), Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.).
Могут быть применены диагностические способы по изобретению с использованием образца, предварительно взятого у индивидуума или пациента. Такие образцы можно сохранять путем замораживания или можно фиксировать в формалине и заливать парафиновой или другими средами. Альтернативно, можно получить и использовать свежеприготовленный образец, содержащий опухолевые клетки.
Способы по изобретению могут быть применены для клеток из любой опухоли. Опухоли, подходящие для лечения соединением формулы (I), описаны ранее.
Мутации в PIK3CA обнаруживаются повсеместно в клинических опухолях, но распространенность мутаций в каждом гене значительно меняется в зависимости от типа опухолевой ткани. Например, мутации PIK3CA относительно часто встречаются в случае рака молочной железы, но относительно редко при опухолях в почках.
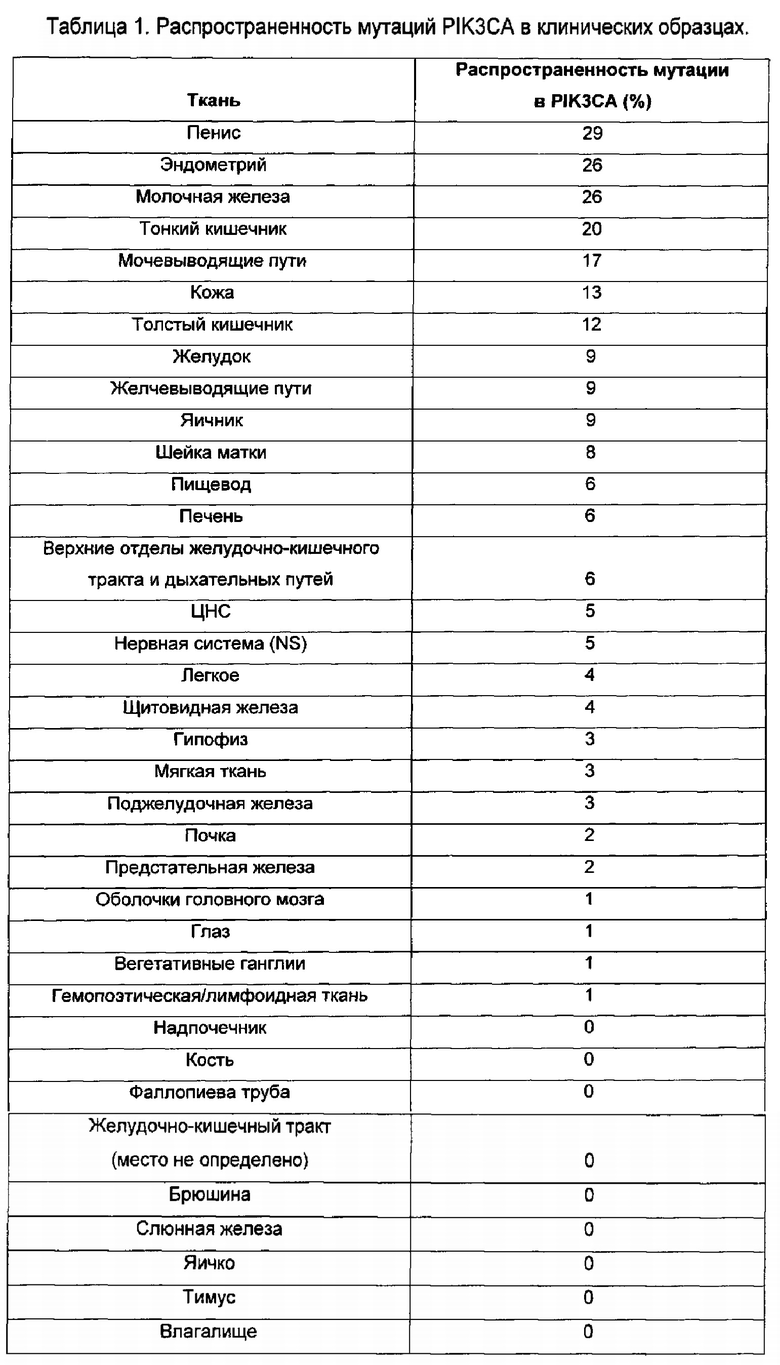
Источником информации относительно PIK3CA является база данных COSMIC (выпуск v.62). Применяемые для отбора пациентов способы по изобретению могут быть особенно полезны в отношении тех пораженных отделов (тканей), которые характеризуются высокой частотой встречаемости мутаций PIK3CA (например, молочной железы, мочевыводящих путей, эндометрия, толстого кишечника, шейки матки и т.д.).
Как будет ясно любому специалисту в данной области техники, эти данные по частоте встречаемости постоянно уточняются и обновляются по мере поступления новой и более полной информации от профилирующих консорциумов в области Генома рака человека (Human Cancer Genome), таких как TCGA (Атлас ракового генома) и Международный консорциум по изучению генома рака (ICGC; International Cancer Genome Consortium). Следовательно, могут быть идентифицированы дополнительные типы опухолей с PIK3CA-зависимостью и пригодные для лечения соединениями, описанными в данной заявке.
Способы обнаружения нуклеиновых кислот
Обнаружение мутантных по PIK3CA нуклеиновых кислот может быть применена, в контексте настоящего изобретения, для предсказания ответа на лечение лекарственными средствами. Поскольку мутации в этих генах присутствуют на уровне ДНК, способы по изобретению могут быть основаны на обнаружении мутаций или вариаций в геномной ДНК, а также в транскриптах и самих белках. Возможно будет необходимо подтвердить наличие мутаций в геномной ДНК путем анализа транскриптов и/или полипептидов, чтобы убедиться, что обнаруженная мутация на самом деле проявляется у субъекта.
Специалисту в данной области техники будет очевидно, что имеется большое число аналитических методик, которые можно использовать для обнаружения присутствия или отсутствия измененных нуклеотидов в одном или более положениях в гене. В общем случае, обнаружение аллельной вариации требует применения метода дискриминации мутаций, возможно реакции амплификации (например, основанной на полимеразной цепной реакции) и возможно системы генерирования сигнала. Существует большое число методов обнаружения мутаций, доступных в данной области техники, и они могут быть использованы в сочетании с системой генерирования сигнала, одной из многочисленных в данной области техники. Обзор многих методов обнаружения аллельной вариации приведен в NolIau et al., Clin. Chem., 1997, 43, 1114-1120; Anderson SM. Expert Rev. Mol. Diaqn., 2011, 11, 635-642; Meyerson M. et al., Nat. Rev. Genet., 2010, 11, 685-696 и в стандартных учебниках, например, "Laboratory Protocols for Mutation Detection" ("Лабораторные протоколы обнаружения мутаций"), под ред. U. Landegren, Oxford University Press, 1996 и "PCR", 2ое издание под ред. Newton & Graham, BIOS Scientific Publishers Limited, 1997.
Как отмечалось выше, определение присутствия или отсутствия конкретной вариации или множества вариаций в гене PIK3CA у пациента с раковым заболеванием может быть выполнено различными путями. Подобные тесты обычно проводят, используя ДНК или РНК, собранные из биологических образцов, например, биоптатов тканей, образцов мочи, кала, мокроты, крови, клеток, соскобов ткани, пунктатов молочной железы или другого клеточного материала, и они могут быть осуществлены различными способами, включая, но не ограничиваясь этим, ПЦР, гибридизацию с аллель-специфичными зондами, обнаружение мутаций с использованием ферментов, химическое расщепление ошибочных спариваний, масс-спектрометрию или секвенирование ДНК, в том числе минисеквенирование.
Подходящие методы обнаружения мутаций включают метод с использованием амплификационной системы рефракторных мутаций (ARMS™), амплификационной системы рефракторных мутаций с линейным расширением (ALEX™; amplification refractory mutation system linear extension), системы праймирования конкурентными олигонуклеотидами (COPS; competitive oligonucleotide priming system), Taqman, метод молекулярных маячков, метод оценки полиморфизма длин фрагментов рестрикции (RFLP; restriction fragment length polymorphism) и ПЦР с использованием сайтов рестрикции и метод резонансного переноса энергии флуоресценции (FRET).
В конкретных воплощениях метод, используемый для определения нуклеотида(ов) в гене-биомаркере, выбран из: аллель-специфической амплификации (аллель-специфической ПЦР), например, с использованием амплификационной системы для идентификации мутаций (ARMS), секвенирования, аллельного дискриминантного анализа, гибридизации, полиморфизма длины фрагментов рестрикции (RFLP) или лигирования олигонуклеотидных зондов (OLA).
В конкретных воплощениях гибридизация с аллель-специфическими зондами может быть проведена с применением: (1) аллель-специфических олигонуклеотидов, связанных с твердой фазой (например, стеклом, кремнием, нейлоновыми мембранами), с меченым образцом в растворе, например, как во многих системах использования ДНК-чипов; или (2) связанного образца (часто клонированной ДНК или ПЦР-амплифицированной ДНК) и меченых олигонуклеотидов в растворе (либо аллель-специфических, либо настолько коротких, что они позволяют осуществлять секвенирование посредством гибридизации). Диагностические тесты могут включать применение панели вариаций, часто на твердой подложке, что дает возможность для одновременного определения более чем одной вариации. Такие гибридизационные зонды хорошо известны в данной области техники (см. например, Green & Sambrook, Eds., MolecuIar Cloning: A Laboratory Manual (2012, 4th edition, Vol. 1-3, ISBN 9781936113422), Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.) и могут перекрывать два или более сайтов с вариациями.
Таким образом, в одном из воплощений обнаружение присутствия или отсутствия по меньшей мере одной мутации обеспечивается путем приведения в контакт нуклеиновой кислоты PIK3CA, содержащей сайт предполагаемой мутации, по меньшей мере с одним нуклеиновокислотным зондом. Зонд предпочтительно гибридизуется с нуклеиновокислотной последовательностью, содержащей сайт с вариацией и содержащей комплементарные нуклеотидные основания в сайте с вариацией, в выбранных условиях гибридизации. Обнаружение наличия гибридизации можно осуществить с помощью детектируемой метки, используя метки, известные специалисту в данной области техники. Такие метки включают, но не ограничиваются этим, радиоактивные, флуоресцентные метки, красители и ферментативные метки.
В другом воплощении обнаружение присутствия или отсутствия по меньшей мере одной мутации обеспечивается путем приведения в контакт нуклеиновой кислоты PIK3CA, содержащей сайт предполагаемой мутации, по меньшей мере с одним нуклеиновокислотным праймером. Праймер предпочтительно гибридизуется с нуклеиновокислотной последовательностью, содержащей сайт с вариацией и содержащей комплементарные нуклеотидные основания в сайте с вариацией, в выбранных условиях гибридизации.
Олигонуклеотиды, используемые в качестве праймеров для специфической амплификации, могут содержать нуклеотидное основание, комплементарное представляющей интерес мутации, в центре молекулы (так что амплификация зависит от дифференциальной гибридизации; см., например, Gibbs, et al., 1989, Nucl. Acids Res., 17, 2437-248) или на самом 3'-конце одного праймера, где в соответствующих условиях ошибочное спаривание может предотвращать или ослаблять удлинение под действием полимеразы (см., например, Prossner, 1993, Tibtech., 11, 238).
В еще одном воплощении обнаружение присутствия или отсутствия по меньшей мере одной мутации включает секвенирование по меньшей мере одной нуклеиновокислотной последовательности и сравнение полученной последовательности с известной нуклеиновокислотной последовательностью дикого типа.
Альтернативно, обнаружение присутствия или отсутствия по меньшей мере одной мутации включает масс-спектрометрическое определение по меньшей мере одной нуклеиновокислотной последовательности.
В одном из воплощений обнаружение присутствия или отсутствия по меньшей мере одной нуклеиновокислотной вариации включает проведение полимеразной цепной реакции (ПЦР). Нуклеиновокислотную последовательность-мишень, содержащую гипотетическую вариацию, амплифицируют и определяют нуклеотидную последовательность амплифицированной нуклеиновой кислоты. Определение нуклеотидной последовательности амплифицированной нуклеиновой кислоты включает секвенирование по меньшей мере одного сегмента нуклеиновой кислоты. Альтернативно, продукты амплификации можно анализировать, используя любой метод, с помощью которого можно провести разделение продуктов амплификации в соответствии с их размером, включая автоматизированный и ручной варианты гель-электрофореза и тому подобное.
Обнаружение мутаций в геномной нуклеиновой кислоте предпочтительно осуществляют методами, основанными на изменении подвижности амплифицированных фрагментов нуклеиновой кислоты. Например, в Chen et al., Anal. Biochem., 1996, 239, 61-9 описывается обнаружение точечных мутаций с использованием конкурентного анализа изменения подвижности (competitive mobility shift assay). Кроме того, коммерчески доступными являются анализы, основанные на методе Marcelino et al., BioTechniques, 1999, 26, 1134-1148.
В конкретным примере, для обнаружения присутствия мутаций можно использовать гетеродуплексный анализ с применением капиллярного электрофореза, основанного на изменении подвижности в капиллярных системах дуплексов нуклеиновых кислот, образованных в результате наличия ошибочных спариваний.
Получение нуклеиновых кислот из образцов для проведения анализа, как правило, требует амплификации нуклеиновой кислоты. Многие методы амплификации основаны на применении ферментативной цепной реакции (такой как полимеразная цепная реакция, лигазная цепная реакция или самоподдерживающаяся репликация последовательностей) или репликации всего или части вектора, использованного для клонирования. Предпочтительно, амплификация, соответствующая данному изобретению, представляет собой экспоненциальную амплификацию, как демонстрируется, например, в полимеразной цепной реакции.
Многие методы амплификации мишеней и усиления сигналов описаны в литературе, например, общие обзоры этих методов представлены в Landegren, U., et al., Science, 1988, 242, 229-237 и Lewis, R., Genetic Engineering News, 1990, 10, 54-55. Эти методы амплификации можно использовать в способах по настоящему изобретению, и они включают полимеразную цепную реакцию (ПЦР), ПЦР in situ, амплификацию в лигазной цепной реакции (LAR; ligase amplification reaction), лигазную гибридизацию, метод с применением репликазы бактериофага Qβ, систему транскрипционно-опосредованной амплификации (TAS; transcription-based amplification system), геномную амплификацию с секвенированием транскриптов (GAWTS; genomic amplification with transcript sequencing), амплификацию, основанную на последовательности нуклеиновых кислот (NASBA; nucleic acid sequence-based amplification) и гибридизацию in situ. Праймеры, подходящие для применения в различных методах амплификации, могут быть получены в соответствии со способами, известными в данной области техники.
Полимеразная цепная реакция (ПЦР) представляет собой метод амплификации нуклеиновых кислот, описанный среди прочего в патентах США №№4683195 и 4683202. ПЦР состоит из повторяющихся циклов генерируемых ДНК-полимеразой реакций удлинения праймеров. ДНК-мишень подвергают термоденатурации и осуществляют гибридизацию двух олигонуклеотидов, которые окружают последовательность-мишень на противоположных нитях предназначенной для амплификации ДНК. Эти олигонуклеотиды становятся праймерами для применения вместе с ДНК-полимеразой. ДНК копируют посредством удлинения праймеров, создавая вторую копию обеих нитей. В результате повторения цикла термоденатурации, гибридизации праймеров и удлинения ДНК-мишень может быть амплифицирована миллион или более раз в промежуток времени примерно от двух до четырех часов. ПЦР представляет собой инструмент молекулярной биологии, который должен быть использован совместно с методом обнаружения для определения результатов амплификации. Преимущество ПЦР заключается в том, что она повышает чувствительность посредством увеличения количества ДНК-мишени в число раз от 1 миллиона до 1 миллиарда приблизительно за 4 часа. ПЦР можно использовать в диагностике для амплификации любой известной нуклеиновой кислоты (Мок et al., Gynaecoloqic Oncology, 1994, 52: 247-252).
Метод аллель-специфической амплификации, такой как амплификационная система для идентификации мутаций (ARMS™) (Newton et al., Nucleic Acids Res., 1989, 17, 2503-2516), также может быть использован для обнаружения точечных мутаций. В соответствующих условиях ПЦР-амплификации ошибочного спаривания одного основания на 3'-конце праймера достаточно для предпочтительной амплификации полностью комплементарного аллеля (Newton и др., 1989, выше), что дает возможность для различения близкородственных видов. Основой амплификационной системы, использующей описанные выше праймеры, является то, что олигонуклеотиды с ошибочно спаренным 3'-остатком не будут действовать как праймеры в ПЦР в соответствующих условиях. Такая амплификационная система позволяет осуществлять генотипирование только путем исследования реакционных смесей после проведения электрофореза в агарозном геле.
Анализ продуктов амплификации может быть осуществлен с использованием любого метода, с помощью которого можно провести разделение продуктов амплификации в соответствии с их размером, включая автоматизированный и ручной варианты гель-электрофореза, масс-спектрометрию и тому подобное.
Методы выделения нуклеиновых кислот, амплификации и анализа являются рутинными для специалиста в данной области техники, и примеры протоколов можно найти, например, в Green & Sambrook, Eds., MolecuIar Cloning: A Laboratory Manual (2012, 4th edition, Vol. 1-3, ISBN 9781936113422), Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.) Особенно полезным источником протоколов для методов, используемых в ПЦР-амплификации, является PCR (Basics: From Background to Bench) под ред. M.J. McPherson, S.G. Mailer, R. Beynon, C. Howe, Springer VerIag, 1-е издание (15 октября 2000 г.), ISBN: 0387916008.
Согласно настоящему изобретению также предложены наборы для прогнозирования и диагностики, содержащие вырожденные праймеры для амплификации нуклеиновой кислоты-мишени в гене PIK3CA, и инструкции, содержащие протокол для амплификации и для анализа результатов. Набор также может альтернативно содержать буферы, ферменты и контейнеры для проведения амплификации и анализа продуктов амплификации. Данный набор также может являться компонентом набора для скрининга или диагностики, содержащего другие средства, такие как ДНК-микрочипы или другие подложки. Предпочтительно, в наборе также имеется одна или более чем одна контрольная матрица, например нуклеиновые кислоты, выделенные из нормального образца ткани и/или из серии образцов, представляющих разные вариации в генах сравнения (reference genes).
В одном из воплощений в наборе содержится две или более пар праймеров, каждая из которых позволяет амплифицировать свой участок в гене (PIK3CA) сравнения (каждый участок представляет собой сайт возможной вариации), тем самым предоставляется набор для анализа экспрессии нескольких генных вариаций в биологическом образце в одной реакции или нескольких параллельных реакциях.
Праймеры в наборах могут быть мечеными, например, меченными флуоресцентными метками, для облегчения обнаружения продуктов амплификации и проведения последующего анализа вариаций в нуклеиновой кислоте. С помощью этого набора также можно осуществлять обнаружение более чем одной вариации в одном анализе. Таким образом, комбинированный набор будет содержать праймеры, позволяющие осуществлять амплификацию разных сегментов гена сравнения. Праймеры могут быть помечены разными метками, например, с использованием разных флуоресцентных меток, чтобы различить такие вариации.
Также раскрыто применение наборов для обнаружения мутаций в PIK3CA, включая, но не ограничиваясь этим, наборы для обнаружения мутаций в PIK3CA, поставляемые на рынок диагностическими компаниями, включая Qiagen и Roche MolecuIar Systems.
Согласно другому аспекту изобретения предложен способ лечения пациента, страдающего раковым заболеванием, включающий: определение мутантного статуса гена PIK3CA или статуса дикого типа в опухолевых клетках пациента, и, если ген PIK3CA является мутантным, введение пациенту эффективного количества соединения формулы (I).
Использованные в данном описании термины «эффективный» и «эффективность» включают как фармакологическую эффективность, так и физиологическую безопасность. Фармакологическая эффективность относится к способности получения в результате такого лечения желаемого биологического эффекта у пациента. Физиологическая безопасность относится к уровню токсичности или другим неблагоприятным физиологическим эффектам на уровне клетки, органа и/или организма (часто называемым как побочные эффекты) в результате применения такого лечения. «Менее эффективный» означает, что в результате лечения наблюдается терапевтически значительно более низкий уровень фармакологической эффективности и/или терапевтически более высокий уровень неблагоприятных физиологических эффектов.
Согласно другому аспекту изобретения предложено применение соединения формулы (I) для лечения пациента с раковым заболеванием, у которого опухолевые клетки идентифицированы как содержащие мутантный ген PIK3CA.
Согласно другому аспекту изобретения предложено соединение формулы (I) для лечения случаев рака с опухолевыми клетками, идентифицированными как несущие мутантный ген PIK3CA.
В следующих дополнительных воплощениях изобретение относится к фармацевтической композиции, содержащей соединение формулы (I), для применения в предупреждении и лечении рака с опухолевыми клетками, идентифицированными как несущие мутантный ген PIK3CA.
Для всех изложенных выше аспектов
определенные/идентифицированные мутантные формы PIK3CA содержат мутации, расположенные в любых положениях по всему гену.
Для всех изложенных выше аспектов с использованием опухолей, таких как, в качестве примера, рак молочной железы, конкретные определенные/идентифицированные мутантные формы PIK3CA содержат мутации, расположенные в положениях R38, R88, N345, С420, Е453, Р539, Е542К, Е545К, Q546, М1043, М1043 и H1047R.
Для всех изложенных выше аспектов с использованием опухолей, таких как, в качестве примера, рак молочной железы, конкретные определенные/идентифицированные мутантные формы PIK3CA содержат мутации, расположенные в положениях Е542, Е545 и Н1047.
Примеры персонализированной медицинской помощи/персонализированной медицины
Анализ клеточной пролиферации в линиях опухолевых клеток
Чувствительность панели линий раковых клеток человека к эффектам соединений определяли в стандартном анализе пролиферации. Подробное описание протокола анализа приведено в разделе Биологические анализы (ж), приведенном выше.
Корреляционный анализ мутаций
Методы
Фармакологические данные, касающиеся измерения ингибирования роста клеток в ответ на обработку соединением из примера 3, получали для коллекции из 209 линий раковых клеток из различных тканей и большого числа источников. Каждую клеточную линию классифицировали как чувствительную (GI50 не больше 1,0 мкМ) или устойчивую (GI50 больше 1,0 мкМ).
Мутационный статус для генов в каждой клеточной линии определяли, обобщая результаты из внутренних (AstraZeneca) и открытых источников. Общедоступные данные включали данные по всем клеточным линиям из проекта «Геномика чувствительности к лекарственным средствам при раковых заболеваниях» (Genomics of Drug Sensitivity in Cancer Project), выпуск 3 (Garnett MJ, et al., Nature, 2012, Mar 483, 570-5), проекта «Энциклопедия линий раковых клеток» (Cancer Cell Line Encyclopedia) (Barretina J, et al., Nature 2012, 483, 603-7) и из базы данных Каталога соматических мутаций при раковых заболеваниях (COSMIC) (выпуск v. 61; http://www.sanger.ac.uk/genetics/CGP/cosmic/; Forbes SA, et al., Nucleic Acids Res., 2011, 39 (серия базы данных): D945-50; Forbes SA, et al., Curr. Protoc. Hum. Genet., 2008, Chapter 10: Unit 10.11.) и из отдельных журнальных статей. Молчащие мутации кодирующей области (синонимичные варианты) и несинонимичные полиморфизмы исключали, и для осуществления этого анализа игнорировали зиготность по мутациям. Для каждой комбинации клеточной линии и гена статус представляли как мутантный (MUT), дикого типа (WT) или противоречивый (INCON). В некоторых изначально противоречивых случаях (независимых наблюдений WT и MUT для одного и того же гена в одной и той же клеточной линии) решение принимали, исходя из собственных наблюдений и наблюдений, касающихся набора данных из проекта «Энциклопедия линий раковых клеток» (CCLP) в COSMIC, или путем выбора статуса после проверки вручную. В случаях, когда не удавалось принять решение на основании данных противоречивых наблюдений, сохраняли обозначение INCON, и статус генов при проведении анализа рассматривался как неизвестный.
Взаимосвязь между мутационным статусом и ответом идентифицировали, создавая таблицы сопряженности признаков для каждого гена и определяя соответствующие отношения шансов и p-значения двухстороннего точного критерия Фишера. Клеточные линии, классифицированные как неочевидные в отношении ответа, исключались из первоначальных анализов на предмет идентификации кандидатов-биомаркеров. При определении мутационного статуса подсчитывали данные для MUT или WT вариантов, и клеточные линии с генами, характеризующимися менее чем 4 WT или 4 MUT, также исключали. Результаты и обсуждение
Взаимосвязь между мутационным статусом и ответом идентифицировали, как описано в разделе «Методы». Ответ клеточных линий на соединение из примера 3 и соответствующий генетический статус для гена PIK3CA показаны в Таблице 3.
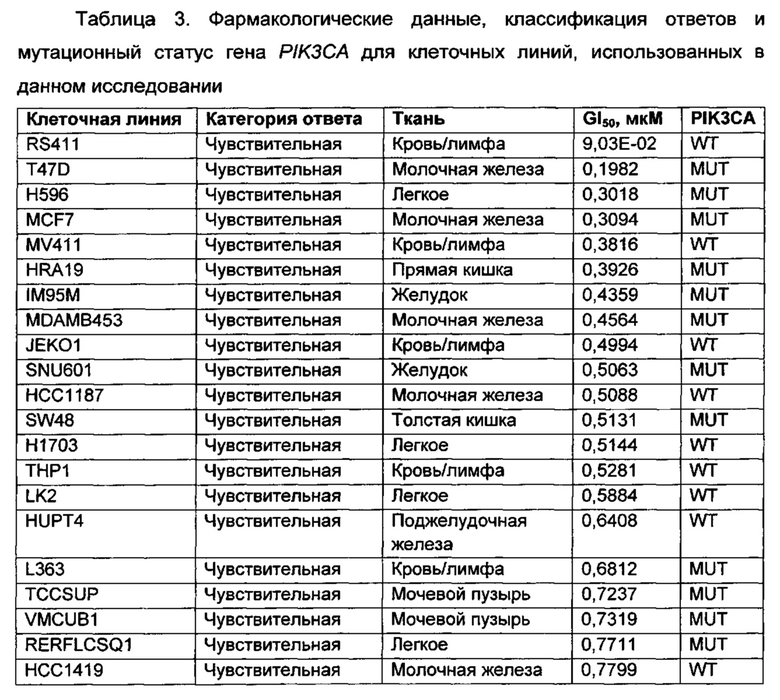
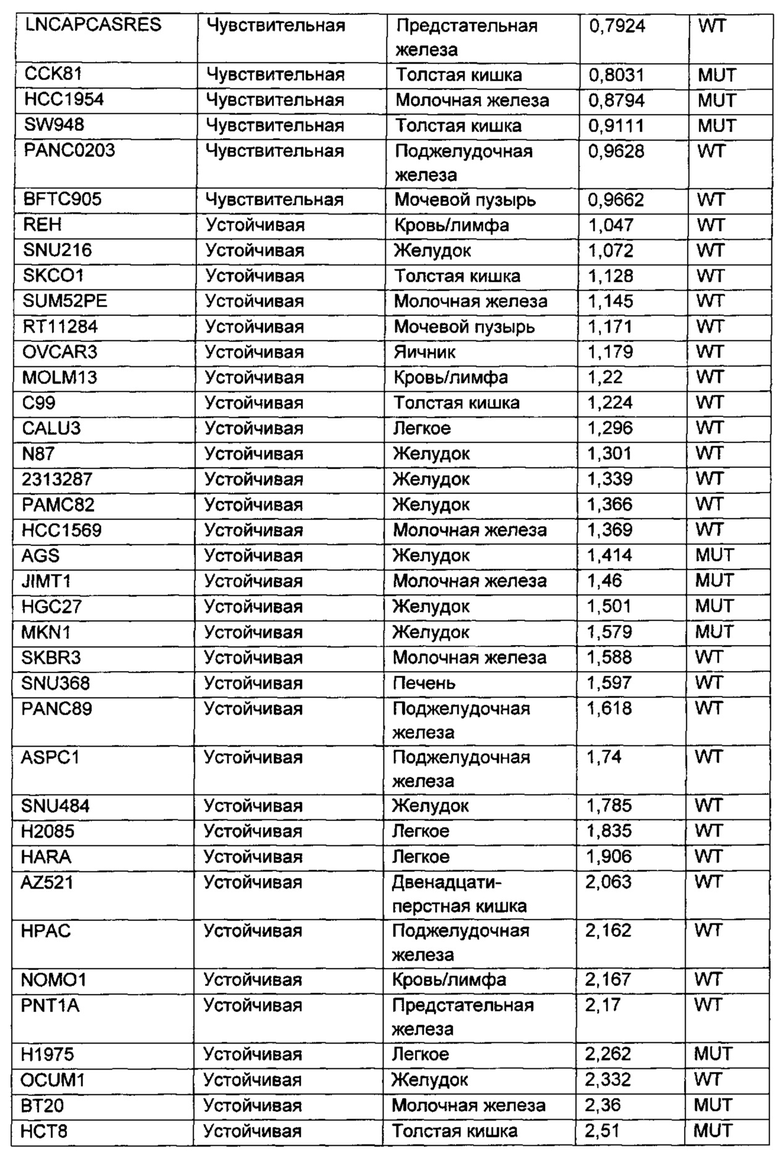
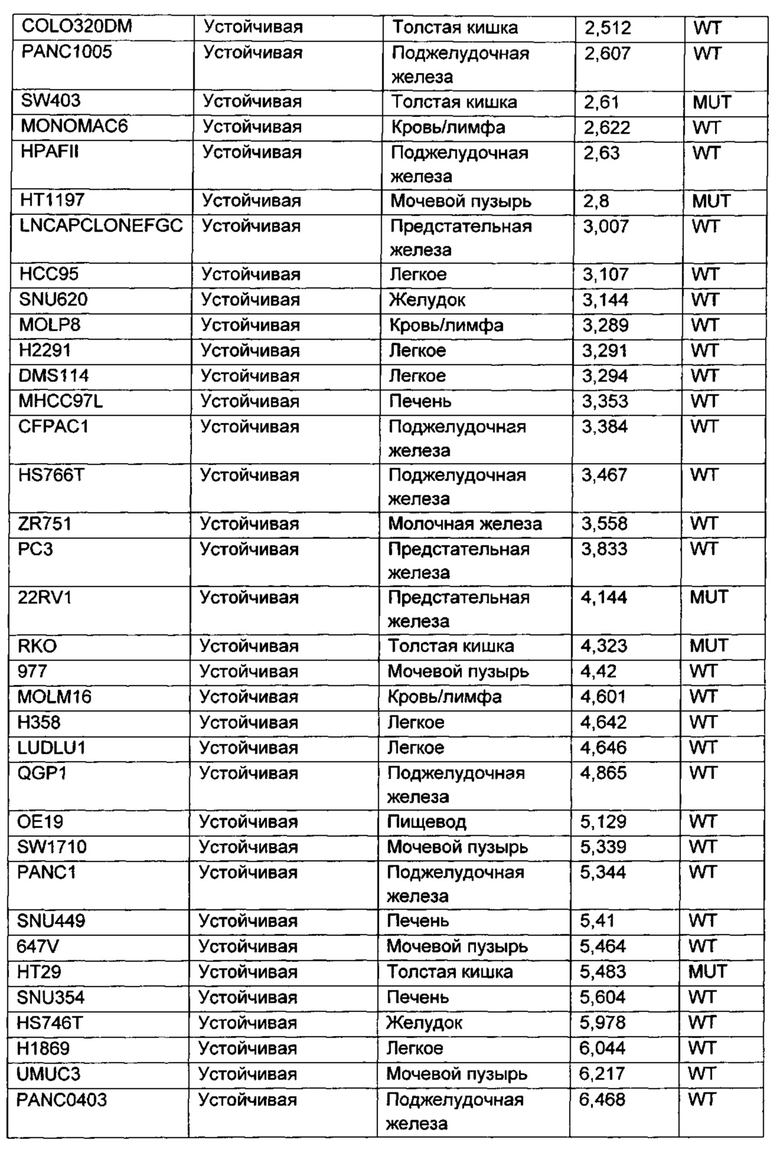
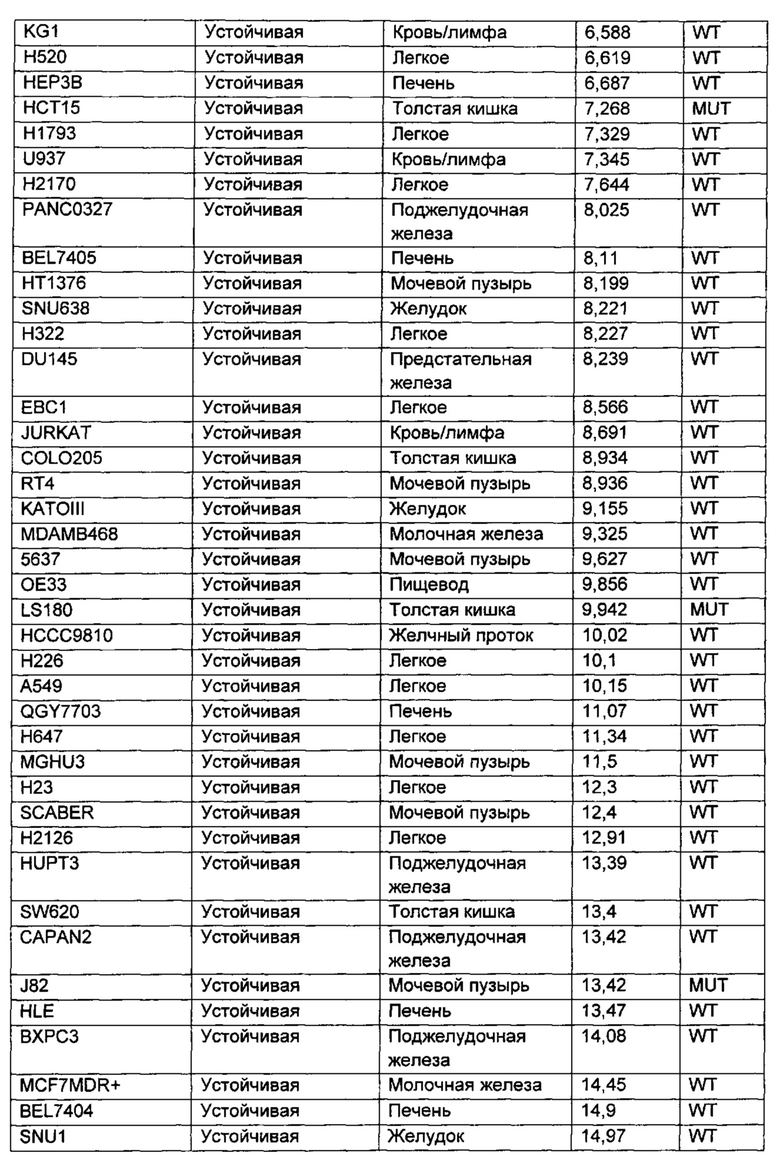
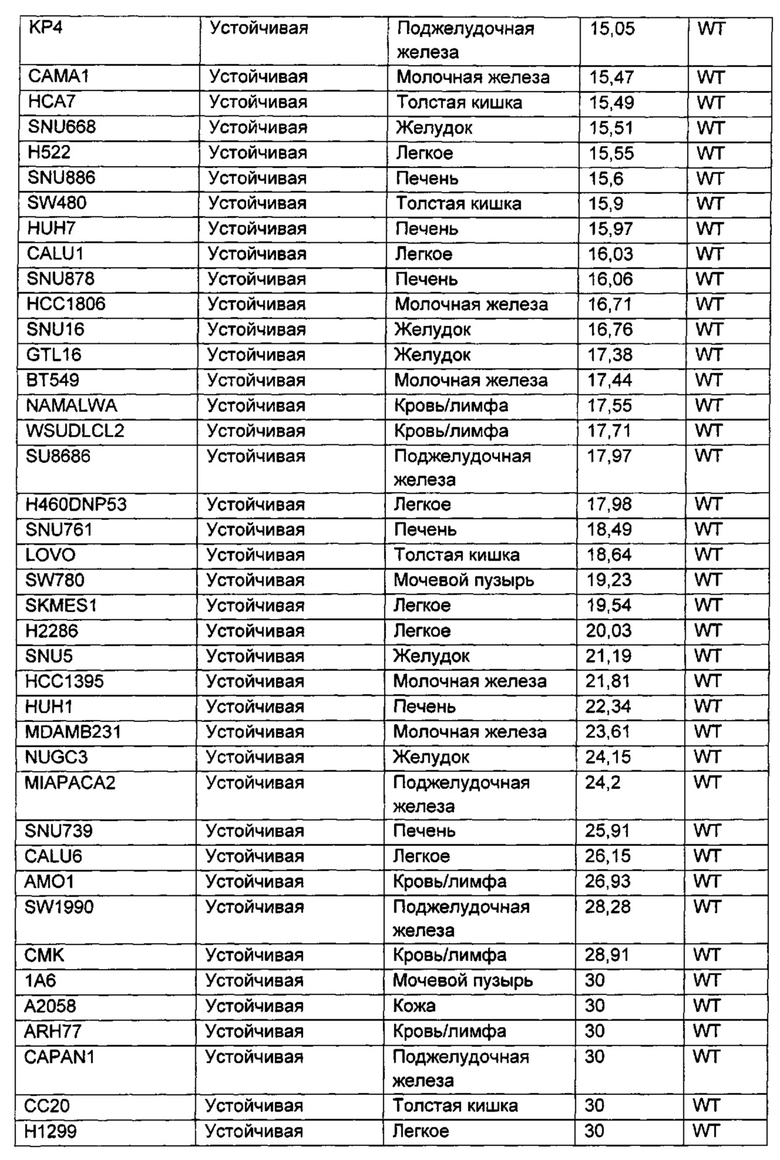
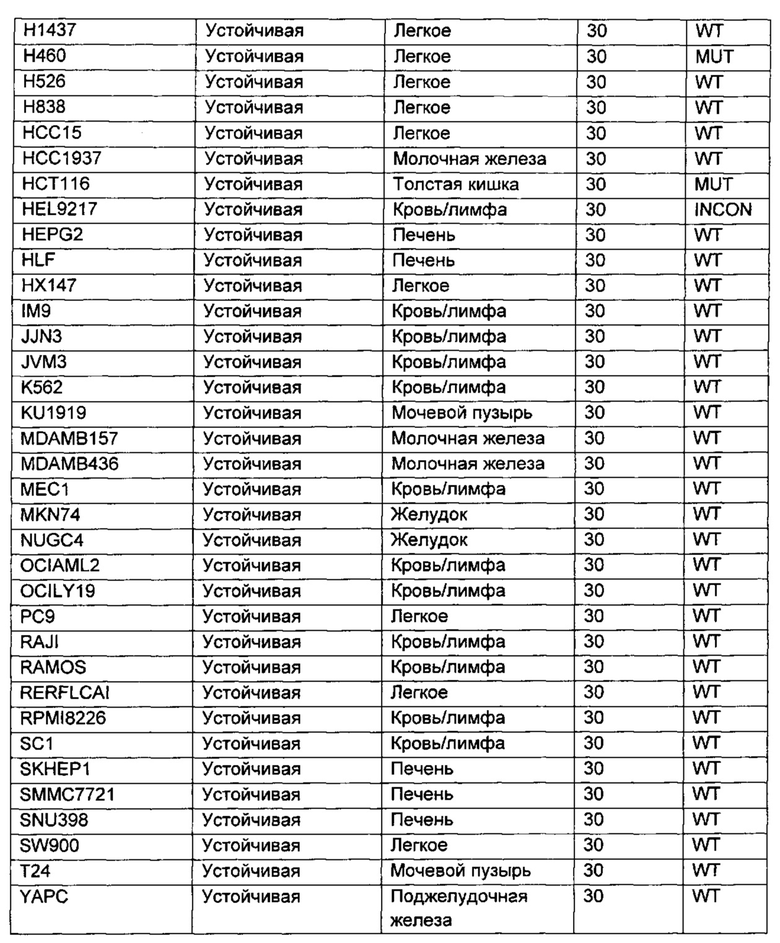
Геном, мутации в котором наиболее сильно коррелировали с чувствительностью к соединению из примера 3, был ген PIK3CA. Только 12 из 177 клеточных линий с геном PIK3CA WT (7,7%) были чувствительны к соединению из примера 3, в то время как из 32 клеточных линий, мутантных по PIK3CA, чувствительными были 15 линий (46,9%), что соответствует отношению шансов 12,1 и p-значению 1,2×10-7 (см. Таблицу 4).

Как указано в данном описании, сообщалось, что определение мутационного статуса или активационного статуса дополнительных генов, таких как K-RAS, потенциальный маркер устойчивости в опухолях с аберрантной или разрегулированной PIK3CA или PI3K-α, может помочь в повышении предсказательной способности персонализированного подхода в медицине.
Авторы изобретения продемонстрировали на примере это положение для приведенного выше набора данных путем сравнения обогащения мутациями К-RAS мутантных по PIK3CA клеток с ответом клеточной линии на ингибирование. Анализ ограничивали клеточными линиями, содержащими мутации в «горячих точках» этих двух генов (в кодонах Е542, Е545 и Н1047 для PIK3CA и в кодонах К12, 13 и Q61 для K-RAS). Анализ продемонстрировал, что в мутантных по PIK3CA клеточных линиях, мутации в гене K-RAS придавали устойчивость к ингибированию соединением из примера 3.
- Двадцать восемь клеточных линий содержали активирующие мутации в PIK3CA.
- 6 из 19 клеточных линий (31,6%), содержащих активирующую мутацию в PIK3CA и ген K-RAS дикого типа, были устойчивы к соединению из примера 3.
- 7 из 9 мутантных по PIK3CA клеточных линий (77,8%), одновременно содержащих мутации K-RAS, были устойчивы к соединению из примера 3.
Из этих данных получается отношение шансов 7,5 и p-значение 0,042 (см. Таблицу 5).
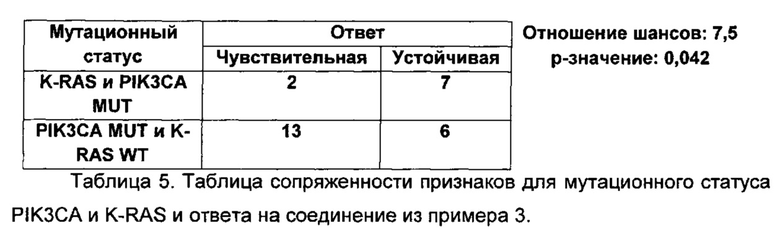
Примеры
Теперь изобретение будет проиллюстрировано следующими далее примерами, в которых, как правило:
(1) операции осуществляли при температуре окружающей среды, т.е. в диапазоне 17-25°C, и в атмосфере инертного газа, такого как азот, если не указано иное;
(2) упаривание осуществляли на роторном испарителе или с использованием оборудования Genevac в вакууме, и процедуры обработки выполняли после удаления фильтрованием оставшихся твердых веществ;
(3) флэш-хроматографические очистки осуществляли на автоматизированной системе Spot II Ultimate с программным обеспечением Armen Glider FIash (Armen Instrument, Saint-Ave, France), используя предварительно упакованные картриджи с нормальной фазой из диоксида кремния Si60 от Merck (гранулометрия: 15-40 или 40-63 мкм), полученные от Merck, Darmstad, Germany;
(4) препаративную хроматографию осуществляли на приборе от Waters (600/2700 или 2525), оснащенном масс-спектрометрами ZMD или ZQ ESCi и колонкой для обращенно-фазовой хроматографии Waters X-Тегга или Waters X-Bridge или Waters SunFire (диоксид кремния C-18, 5 микрон; диаметр 19 мм, длина 100 мм, скорость потока 40 мл/минута) с использованием в качестве элюента смесей воды (содержащей 0,2% карбоната аммония) и ацетонитрила с уменьшающейся полярностью;
(5) выходы, где они приведены, необязательно являются максимально достижимыми;
(6) в общем случае структуры конечных продуктов формулы I подтверждали с применением спектроскопии ядерного магнитного резонанса (ЯМР); величины химического сдвига при проведении ЯМР измеряли по шкале дельта (спектры протонного магнитного резонанса определяли, используя прибор Avance 500 (500 МГц) от Bruker); измерения проводили при температуре окружающей среды, если не указано иное; использовали следующие сокращения: s, синглет; d, дублет; t, триплет; q, квартет; m, мультиплет; dd, дублет дублетов; ddd, дублет дублетов дублетов; dt, дублет триплетов; bs, уширенный сигнал;
(7) в общем случае конечные продукты формулы I также охарактеризовывали посредством жидкостной хроматографии с последующей масс-спектроскопией (LCMS); LCMS осуществляли, используя прибор Alliance НТ (2790 и 2795) от Waters, оснащенный масс-спектрометром ZQ ESCi или ZMD ESCi от Waters и колонкой С-18 XBridge, 5 мкм (2,1×50 мм), при скорости потока 2,4 мл/мин, применяя систему растворителей от 95% A + 5% C до 95% B + 5% C в течение 4 минут, где A представляет собой воду, B представляет собой метанол, С представляет собой смесь 1:1 метанол:вода (содержащая 0,2% карбоната аммония);
(8) промежуточные соединения, как правило, не охарактеризовывали полностью, и чистоту оценивали посредством анализа с применением тонкослойной хроматографии, масс-спектрального анализа, высокоэффективной жидкостной хроматографии (HPLC) и/или ЯМР-анализа;
(9) спектры дифракции рентгеновских лучей на порошке определяли (используя аналитический прибор Bruker D4) путем помещения образца кристаллического вещества на держатель пластины из монокристалла кремния (SSC) от Bruker, и распределения образца в виде тонкого слоя с помощью предметного стекла микроскопа. Образец вращали при 30 оборотах в минуту (для улучшения статистики счета) и облучали рентгеновскими лучами, генерируемыми длиннофокусной трубкой с медным анодом, работающей при 40 кВ и 40 мА, с длиной волны 1,5418 ангстрем. Коллимированный пучок рентгеновских лучей пропускали через автоматически изменяющуюся щель расходимости, установленную на V20, а отраженное излучение направляли через антирассеивающую щель 5,89 мм и приемную щель 9,55 мм. Образец подвергали воздействию в течение 0,03 секунды с шагом 0,00570° 2-тета (непрерывный режим сканирования) в диапазоне от 2 градусов до 40 градусов 2-тета в режиме тета-тета. Продолжительность испытания составляла 3 минуты и 36 секунд. Прибор был оснащен позиционно-чувствительным детектором (Lynxeye). Получение контрольных и экспериментальных данных осуществляли с помощью дисплейного терминала (Workstation) Dell Optiplex 686 NT 4.0, работающего с программным обеспечением Diffrac+. Специалисты в области дифракции рентгеновских лучей на порошке также осведомлены, что на относительную интенсивность пиков могут оказывать влияние, например, гранулы в размере свыше 30 микрон и с неунитарными соотношениями сторон, что может отразиться на анализе образцов. Специалисту также понятно, что на положение отражений может влиять точность размещения по высоте, на которой образец находится в дифрактометре, и калибровка нуля дифрактометра. Плоскостность поверхности образца также может производить небольшой эффект. Следовательно, представленные данные дифракционных картин не должны приниматься за абсолютные значения;
(10) дифференциальную сканирующую калориметрию проводили, используя прибор для DSC Q1000 от TA Instruments. Обычно менее 5 мг вещества, содержащегося в стандартном алюминиевом тигле, снабженном крышкой, нагревали в температурном диапазоне от 25°C до 300°C с постоянной скоростью нагревания 10°С в минуту. В качестве продувочного газа использовали азот при скорости потока 50 мл в минуту; и
(11) использованы следующие сокращения:
Пример 1
1-(4-(5-(5-Амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-метил-1H-1,2,4-триазол-3-ил)пиперидин-1-ил)-3-гидроксипропан-1-он
3-Гидроксипропановую кислоту (30%-ный об./об. раствор в воде) (200 мкл; 47,0 мг; 0,52 ммоль) упаривали досуха, затем азеотропно отгоняли с толуолом. Кислоту растворяли в NMP (1 мл) и добавляли молекулярные сита (100 мг; 0,26 ммоль), N-этил-N-изопропилпропан-2-амин (0,136 мл; 0,78 ммоль), затем добавляли тетрафторборат 2-(1H-бензо[d][1,2,3]-триазол-1-ил)-1,1,3,3-тетраметилизоурония (209 мг; 0,65 ммоль). После перемешивания в течение 30 минут добавляли 3-(5-трет-бутил-1,3,4-оксадиазол-2-ил)-5-(1-метил-3-(пиперидин-4-ил)-1Н-1,2,4-триазол-5-ил)пиразин-2-амин (100 мг; 0,26 ммоль) и смесь перемешивали в течение 2 часов. Реакционную смесь очищали препаративной HPLC, используя колонку для обращенно-фазовой хроматографии X-Bridge от Waters (диоксид кремния C-18, 5 микрон; диаметр 19 мм, длина 100 мм, скорость потока 40 мл/минута) и в качестве элюента смеси воды (содержащей 0,2% карбоната аммония) и ацетонитрила с уменьшающейся полярностью.
Фракции, содержащие желаемое соединение, упаривали досуха, получая 1-(4-(5-(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-метил-1Н-1,2,4-триазол-3-ил)пиперидин-1-ил)-3-гидроксипропан-1-он (45,0 мг; 37,9%) в виде ярко-желтого твердого вещества: 1Н ЯМР-спектр (CDCl3) 1.52 (9Н, s), 1.79-1.94 (2Н, m), 2.07-2.15 (2Н, m), 2.58 (2Н, t), 2.84-2.94 (1Н, m), 3.00-3.10 (1Н, m), 3.17-3.26 (1Н, m), 3.53 (1Н, t), 3.86-3.94 (3H, m), 4.30 (3H, s), 4.56-4.62 (1Н, m), 9.02 (1Н, s); масс-спектр: [М+Н]+ = 456.
3-(5-трет-Бутил-1,3,4-оксадиазол-2-ил)-5-(1-метил-3-(пиперидин-4-ил)-1Н-1,2,4-триазол-5-ил)пиразин-2-амин (пример 1.1) получали так, как указано ниже.
При 20°C трет-бутил-4-карбамоилпиперидин-1-карбоксилат (47 г; 205,88 ммоль) в дихлорметане (500 мл) по каплям добавляли к перемешиваемому раствору гексафторфосфата(V) триэтилоксония (56,2 г; 226,47 ммоль) в дихлорметане (500 мл) в течение 45 минут в атмосфере азота. Полученный раствор перемешивали при 20°C в течение ночи.
Затем добавляли насыщенный водный раствор Na2CO3 до получения pH 8. Фазы декантировали и водную фазу экстрагировали еще раз, используя 200 мл CH2Cl2, затем органические фазы сушили над MgSO4, фильтровали и концентрировали, получая трет-бутил-4-(этокси(имино)метил)пиперидин-1-карбоксилат (51,0 г; 97%) в виде бесцветной жидкости: 1Н ЯМР-спектр (CDCl3) 1.28 (3H, t), 1.46 (9Н, s), 1.47 (2Н, m), 1.79-1.93 (2Н, m), 2.28 (1Н, m), 2.73 (2Н, m), 4.10 (2Н, q), 4.13-4.18 (2Н, m); масс-спектр: [М+Н]+ = молекулярный ион - нет данных (no mass ion).
К перемешиваемому раствору трет-бутил-4-(этокси(имино)метил)пиперидин-1-карбоксилата (51 г; 198,95 ммоль) в диоксане (500 мл) добавляли гидразид муравьиной кислоты (17,92 г; 298,43 ммоль).
Этот раствор оставляли перемешиваться при 40°C в течение ночи в атмосфере N2, что приводило к выпадению в осадок белого твердого вещества (промежуточного гидразида). Затем реакционную смесь нагревали до 80°C в течение 6 ч, охлаждали до комнатной температуры и концентрировали. Остаток растворяли в 500 мл CH2Cl2 и добавляли 300 мл воды. Фазы декантировали и органическую фазу промывали рассолом, сушили над MgSO4, фильтровали и концентрировали, получая трет-бутил-4-(1H-1,2,4-триазол-3-ил)пиперидин-1-карбоксилат (46,0 г; 92%) в виде белого твердого вещества: 1Н ЯМР-спектр (CDCl3) 1.47 (9Н, s), 1.76 (2Н, m), 1.98-2.11 (2Н, m), 2.91 (2Н, s), 2.97-3.08 (m, 1Н), 4.06-4.23 (2Н, m), 8.05 (1Н, s); масс-спектр: [М+Н]+ = молекулярный ион - нет данных.
К перемешиваемому раствору трет-бутил-4-(1H-1,2,4-триазол-3-ил)пиперидин-1-карбоксилата (22 г; 87,19 ммоль) в дихлорметане (250 мл) добавляли 2 н. раствор гидроксида натрия (131 мл; 261,58 ммоль). Реакционную смесь энергично перемешивали с использованием механической мешалки и затем по каплям добавляли раствор трибромида бензилтриметиламмония (37,4 г; 95,91 ммоль) в дихлорметане (250 мл), поддерживая температуру около 15°C.
Реакционную смесь оставляли перемешиваться при комнатной температуре в течение 1 ч и добавляли 2 н. HCl до получения pH 5 (поддерживая температуру около 15°C). Фазы декантировали, и органическую фазу промывали H2O (2×1 л), сушили над MgSO4, фильтровали и концентрировали, получая трет-бутил-4-(5-бром-1H-1,2,4-триазол-3-ил)пиперидин-1-карбоксилат (25,00 г; 87%) в виде беловатого твердого вещества: 1Н ЯМР-спектр (CDCl3) 1.46 (9Н, s), 1.67-1.84 (2Н, m), 1.90-2.13 (2Н, m), 2.77-2.96 (2Н, m), 2.98-3.10 (1Н, m), 3.94-4.35 (2Н, m); масс-спектр: [М+Н]+ = молекулярный ион - нет данных.
К перемешиваемой суспензии трет-бутил-4-(5-бром-1H-1,2,4-триазол-3-ил)пиперидин-1-карбоксилата (26 г; 78,50 ммоль) в толуоле (200 мл) и метаноле (50 мл) по каплям добавляли 2 М раствор (диазометил)триметилсилана в гексане (43,2 мл; 86,35 ммоль) в атмосфере N2, поддерживая температуру около 20°C: наблюдали выделение газа и небольшой экзотермический эффект. Полученный желтый раствор перемешивали при комнатной температуре в течение 1 ч. Растворитель выпаривали и полученное масло очищали на диоксиде кремния с элюированием 40%-ным EtOAc в петролейном эфире, получая трет-бутил-4-(5-бром-1-метил-1H-1,2,4-триазол-3-ил)пиперидин-1-карбоксилат (15,00 г; 55,3%) в виде масла: 1Н ЯМР-спектр (CDCl3) 1.46 (9Н, s), 1.65-1.78 (2Н, m), 1.90-2.01 (2Н, m), 2.68-3.02 (3H, m), 3.83 (3H, s), 3.94-4.31 (2H, m); масс-спектр: [M+H]+ = молекулярный ион - нет данных.
Моногидрат гидразина (34 мл; 1094,95 ммоль) порциями добавляли к перемешиваемой суспензии метил-3-аминопиразин-2-карбоксилата (21,3 г; 139,09 ммоль) в этаноле (65 мл) при кт. Полученную суспензию перемешивали при 60°C в течение 2 часов, охлаждали до комнатной температуры и фильтровали. Твердое вещество промывали холодным этанолом (2×25 мл) и сушили до постоянной массы, получая 3-аминопиразин-2-карбогидразид (20,75 г; 97%) в виде бежевого твердого вещества: 1Н ЯМР-спектр (DMSO-d6) 4.49 (2Н, d), 7.46 (2Н, br s), 7.78 (1Н, d), 8.17 (1Н, d), 9.79 (1Н, t); масс-спектр: [М+Н]+ = 154.
Тетрафторборат 2-(1Н-бензо[d][1,2,3]триазол-1-ил)-1,1,3,3-тетраметилизоурония (47,7 г; 148,69 ммоль) порциями добавляли в течение 15 минут к перемешиваемой суспензии N-этил-N-изопропилпропан-2-амина (70,6 мл; 405,51 ммоль), пивалевой кислоты (17,08 мл; 148,69 ммоль) и 3-аминопиразин-2-карбогидразида (20,7 г; 135,17 ммоль) в ацетонитриле (350 мл) и реакционную смесь перемешивали при 80°C в течение 20 минут (получали раствор). Реакционную смесь охлаждали до 0°C и добавляли N-этил-N-изопропилпропан-2-амин (70,6 мл; 405,51 ммоль), затем 4-метилбензол-1-сульфонилхлорид (77 г; 405,51 ммоль) в течение 15 минут. Реакционную смесь (желтую суспензию) доводили до кипения с использованием обратного холодильника (для солюбилизации) и затем оставляли перемешиваться при комнатной температуре в течение 14 часов, получая темно-оранжевый раствор. Раствор концентрировали. Остаток разбавляли дихлорметаном, промывали водой, рассолом, сушили над сульфатом магния и концентрировали. Неочищенный продукт очищали флэш-хроматографией на силикагеле, элюируя 0-40%-ным этилацетатом в дихлорметане. Растворитель выпаривали досуха. Полученную смесь растирали с эфиром (100 мл), фильтровали, промывали минимальным количеством эфира и сушили, получая 3-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-амин (20,8 г; 70,2%) в виде бледно-желтого твердого вещества: 1Н ЯМР-спектр (CDCl3) 1.53 (9Н, s), 1.58-1.68 (2Н, m), 6.67 (2Н, s), 8.13 (2Н, dt); масс-спектр: [М+Н]+ = 220.
1-Бромпирролидин-2,5-дион (18,57 г; 104,36 ммоль) порциями добавляли к раствору 3-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-амина (20,8 г; 94,87 ммоль) в THF (320 мл) и раствор перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь концентрировали, и остаток растворяли в дихлорметане (300 мл), промывали водой (2×150 мл), рассолом, сушили над сульфатом магния и концентрировали. Растворитель выпаривали, и неочищенный продукт очищали флэш-хроматографией на силикагеле, элюируя 0-10%-ным этилацетатом в дихлорметане. Растворитель выпаривали досуха, получая 5-бром-3-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-амин (25,5 г; 90%) в виде бежевого твердого вещества: 1Н ЯМР-спектр (CDCl3) 1.52 (9Н, s), 8.23 (1Н, s); масс-спектр: [М+Н]+ = 300.
К суспензии 5-бром-3-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-амина (45 г; 150,94 ммоль) в толуоле (450 мл) добавляли 1,1,1,2,2,2-гексаметилдистаннан (37,6 мл; 181,12 ммоль) и хлорид бис(трифенилфосфин)палладия(II) (5,30 г; 7,55 ммоль). Реакционную смесь продували аргоном и нагревали при 80°C в течение 2 часов (при нагревании солюбилизация до оранжевого раствора, затем повторное выпадение в осадок и приобретение черной окраски, указывающей на завершение реакции). Реакционную смесь охлаждали, концентрировали, остаток растворяли в CH2Cl2 и фильтровали на декалите для удаления нерастворенных примесей. Фильтрат концентрировали и очищали на диоксиде кремния, элюируя 0-10%-ным EtOAc в CH2Cl2. Растворитель концентрировали, получая 3-(5-трет-бутил-1,3,4-оксадиазол-2-ил)-5-(триметилстаннил)пиразин-2-амин (22,63 г; 39,2%) в виде оранжевого твердого вещества: 1Н ЯМР-спектр (CDCl3) 0.38 (9Н, s), 1.53 (9Н, s), 6.49 (2Н, br s), 8.13 (1Н, s); масс-спектр: [М+Н]+ = 384.
К перемешиваемой суспензии трет-бутил-4-(5-бром-1-метил-1Н-1,2,4-триазол-3-ил)пиперидин-1-карбоксилата (2700 мг; 7,82 ммоль) и 3-(5-трет-бутил-1,3,4-оксадиазол-2-ил)-5-(триметилстаннил)пиразин-2-амина (2988 мг; 7,82 ммоль) в 4-метил-2-пентаноле (28 мл) добавляли хлорид лития (995 мг; 23,46 ммоль) и хлорид бис(трифенилфосфин)палладия(II) (220 мг; 0,31 ммоль). Смесь продували аргоном и нагревали при 140°C в течение 2 ч. Реакционную смесь охлаждали, полученный осадок собирали фильтрованием, промывали изопропанолом (25 мл), водой (25 мл) и сушили отсасыванием. Содержащую изопропанол органическую фракцию концентрировали, сформировавшийся осадок собирали и объединяли с основным осадком, получая трет-бутил-4-(5-(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-метил-1Н-1,2,4-триазол-3-ил)пиперидин-1-карбоксилат (3,0 г; 79%): 1Н ЯМР-спектр (DMSO-d6) 1.41 (9Н, s), 1.45 (9Н, s), 1.50-1.68 (2Н, m), 1.95 (3H, dd), 2.78-3.05 (1Н, m), 3.96 (3H, d), 4.21 (3H, s), 8.86 (1Н, s); масс-спектр: [М+Н]+ = 484.
Раствор трет-бутил-4-(5-(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-метил-1H-1,2,4-триазол-3-ил)пиперидин-1-карбоксилата (3 г; 6,20 ммоль) в TFA (15 мл) и CH2Cl2 (15 мл) перемешивали при 25°C в течение 1 часа. Смесь азеотропно отгоняли с толуолом, добавляли 7 н. раствор аммиака в метаноле и дихлорметане и смесь адсорбировали на силикагеле. Неочищенный продукт очищали флэш-хроматографией на силикагеле, элюируя 0-8%-ным метанолом в дихлорметане, затем 10%-ным метанольным аммиаком в дихлорметане. Растворитель выпаривали досуха, получая 3-(5-трет-бутил-1,3,4-оксадиазол-2-ил)-5-(1-метил-3-(пиперидин-4-ил)-1H-1,2,4-триазол-5-ил)пиразин-2-амин (2,040 г; 86%) в виде желтого кристаллического вещества: 1Н ЯМР-спектр (DMSO-d6) 1.45 (9Н, s), 1.55-1.66 (2Н, m), 1.86 (2Н, dd), 2.52-2.61 (2Н, m), 2.69-2.78 (1Н, m), 2.95-3.02 (2Н, m), 4.20 (3H, s), 8.86 (1Н, s); масс-спектр: [М+Н]+ = 384.
Выделение монокристаллической формы соединения из примера 1
Спектры дифракции рентгеновских лучей на порошке для выделенного выше вещества показали, что данное вещество является кристаллическим, но представляет собой смесь полиморфных форм. Это вещество имело точку плавления 226,4°C (начало).
Форму A соединения получали путем суспендирования исходного вещества в ацетонитриле при 25°C. Приблизительно 20 мг исходного вещества помещали во флакон со стержнем для перемешивания на магнитной мешалке и добавляли приблизительно 2 мл ацетонитрила, затем флакон плотно закрывали крышкой и оставляли перемешиваться на столе магнитной мешалки. Приблизительно через 5 суток образец убирали со стола, крышку открывали и суспензию оставляли сушиться в окружающих условиях, после чего ее анализировали посредством XRPD и DSC. С использованием XRPD определяли, что эта форма (Форма A) является кристаллической. Это вещество имело точку плавления 227,2°C (начало).
Такую же кристаллическую форму можно получить в результате перемешивания неочищенного вещества в ацетонитриле в течение ночи при комнатной температуре, затем фильтрования полученного твердого вещества, промывки холодным ацетонитрилом и сушки.
Согласно одному из аспектов изобретения предложен способ получения кристаллической формы соединения из примера 1 (Формы А) путем суспендирования образца соединения в ацетонитриле.
Десять пиков дифракции рентгеновских лучей на порошке показаны в приведенной ниже таблице.
Десять пиков дифракции рентгеновских лучей на порошке для Формы A соединения из примера 1
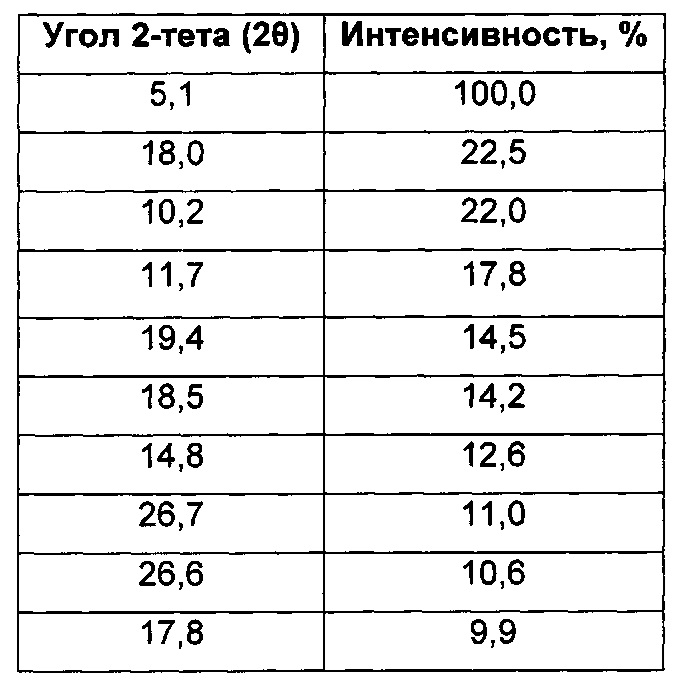
Спектр дифракции рентгеновских лучей на порошке для Формы A соединения из примера 1 показан на Фиг. 1.
DSC-анализ Формы A соединения из примера 1 показывает эндотерму плавления с началом при 227,2°C и пиком при 228,6°С (Фиг. 2).
Таким образом, DSC-анализ показывает, что Форма A соединения из примера 1 представляет собой высокоплавкое твердое вещество с началом плавления при примерно 227,2°С и пиком при 228,6°С.
Данные DSC для Формы A соединения из примера 1 показаны на Фиг. 2.
Дифракция рентгеновских лучей на порошке
Аналитический прибор: Bruker D4.
Картину дифракции рентгеновских лучей на порошке определяли путем помещения образца кристаллического вещества на держатель пластины из монокристалла кремния (SSC) от Bruker, и распределения образца в виде тонкого слоя с помощью предметного стекла микроскопа. Образец вращали при 30 оборотах в минуту (для улучшения статистики счета) и облучали рентгеновскими лучами, генерируемыми длиннофокусной трубкой с медным анодом, работающей при 40 кВ и 40 мА, с длиной волны 1,5418 ангстрем. Коллимированный пучок рентгеновских лучей пропускали через автоматически изменяющуюся щель расходимости, установленную на V20, а отраженное излучение направляли через антирассеивающую щель 5,89 мм и приемную щель 9,55 мм. Образец подвергали воздействию в течение 0,03 секунды с шагом 0,00570° 2-тета (непрерывный режим сканирования) в диапазоне от 2 градусов до 40 градусов 2-тета в режиме тета-тета. Продолжительность испытания составляла 3 минуты и 36 секунд. Прибор был оснащен позиционно-чувствительным детектором (Lynxeye). Получение контрольных и экспериментальных данных осуществляли с помощью дисплейного терминала Dell Optiplex 686 NT 4.0, работающего с программным обеспечением Diffrac+. Специалисты в области дифракции рентгеновских лучей на порошке осведомлены, что на относительную интенсивность пиков могут оказывать влияние, например, гранулы в размере свыше 30 микрон и с неунитарными соотношениями сторон, что может отразиться на анализе образцов. Специалисту также понятно, что на положение отражений может влиять точность размещения по высоте, на которой образец находится в дифрактометре, и калибровка нуля дифрактометра. Плоскостность поверхности образца также может производить небольшой эффект. Следовательно, представленные данные дифракционных картин не должны приниматься за абсолютные значения.
Дифференциальная сканирующая калориметрия
Аналитический прибор для DSC: Q1000 от ТА Instruments.
Обычно менее 5 мг вещества, содержащегося в стандартном алюминиевом тигле, снабженном крышкой, нагревали в температурном диапазоне от 25°C до 300°C с постоянной скоростью нагревания 10°C в минуту. В качестве продувочного газа использовали азот при скорости потока 50 мл в минуту.
Альтернативный синтез соединения из примера 1 приведен ниже в Примера 2.
Пример 2
1-(4-(5-(5-Амино-6-(5-трего-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-метил-1H-1,2,4-триазол-3-ил)пиперидин-1-ил)-3-гидроксипропан-1-он
4-Метилбензолсульфонат пиридина (3,58 г; 14,25 ммоль) добавляли к суспензии 1-(4-(5-(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-метил-1H-1,2,4-триазол-3-ил)пиперидин-1-ил)-3-(тетрагидро-2Н-пиран-2-илокси)пропан-1-она (37 г; 68,57 ммоль) в метаноле (275 мл) в атмосфере азота. Смесь перемешивали при 60°C в течение 1,5 часа. Через 5 минут смесь превращалась в раствор. Смесь выдерживали при 50°C в течение ночи, за это время образовывался осадок. Реакционную смесь растворяли в дихлорметане (400 мл), промывали водой (300 мл) и рассолом (100 мл). Водные экстракты снова промывали DCM (100 мл) и объединенные органические слои сушили над MgSO4 и концентрировали. Неочищенный продукт очищали флэш-хроматографией на силикагеле с элюированием от 100%-ного этилацетата до смеси 10:50:40 метанол/этилацетат/DCM. Фракции, содержащие продукт, упаривали досуха, получая бежевое твердое вещество (24,5 г). Это твердое вещество суспендировали в течение ночи в ацетонитриле (500 мл), фильтровали и сушили под высоким вакуумом, получая 1-(4-(5-(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-метил-1H-1,2,4-триазол-3-ил)пиперидин-1-ил)-3-гидроксипропан-1-он (пример 2) (24 г; 78%) в виде кремового твердого вещества: 1Н ЯМР-спектр (DMSO-d6) 1.51 (9Н, s), 1.55-1.68 (1Н, m), 1.68-1.84 (1Н, m), 1.96-2.13 (2Н, m), 2.78-2.93 (1Н, m), 2.98-3.1 (1Н, m), 3.19-3.3 (1Н, m), 3.71 (2Н, q), 3.93-4.04 (1Н, m), 4.27 (3H, s), 4.35-4.48 (1Н, m), 4.54 (1Н, t), 7.96 (2Н, s), 8.92 (1Н, s); масс-спектр: [М+Н]+ = 456.
(4-(5-(5-Амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-метил-1Н-1,2,4-триазол-3-ил)пиперидин-1-ил)-3-(тетрагидро-2Н-пиран-2-илокси)пропан-1-он (пример 2.1) получали так, как указано ниже.
Гидрат гидразина (23,59 мл; 480,75 ммоль) по каплям добавляли к перемешиваемой смеси метил-3-амино-6-бромпиразин-2-карбоксилата (100 г; 418,04 ммоль) в ЕtOН (2 л). Смесь нагревали при 50°C в атмосфере азота. Полученную густую суспензию перемешивали при 50°C в течение 16 часов. Добавляли дополнительно гидразин (2,5 мл) в виде одной порции, и суспензию перемешивали при 50°C в течение еще 24 часов. В густую реакционную смесь загружали этанол (500 мл), и смесь оставляли охлаждаться до комнатной температуры. Полученную суспензию фильтровали, твердое вещество промывали этанолом (1 л) и сушили в вакууме, получая 3-амино-6-бромпиразин-2-карбогидразид (98 г; количественный выход) в виде кремового твердого вещества: 1Н ЯМР-спектр (DMSO-d6) 4.52 (2Н, s), 7.59 (2Н, s), 8.30 (1Н, s), 9.74 (1Н, s); масс-спектр: [М+Н]+ = 232.
Ангидрид пивалевой кислоты (165 мл; 815,38 ммоль) добавляли к перемешиваемой смеси 3-амино-6-бромпиразин-2-карбогидразида (172 г; 741,26 ммоль) в ацетонитриле (1,8 л) и смесь нагревали при 80°С в течение 1 часа. Реакционную смесь оставляли перемешиваться в течение 16 часов. Необходимое желтое твердое вещество выделяли фильтрованием. Фильтрат распределяли между EtOAc (2 л) и водным раствором бикарбоната натрия (2 л). Органический слой промывали насыщенным рассолом и сушили над MgSO4.
Раствор фильтровали и концентрировали, получая оранжевое вязкое твердое вещество, которое растирали с МТВЕ (250 мл). Нерастворенное желтое твердое вещество выделяли фильтрованием, и было показано, что это вещество идентично первому твердому веществу. Объединенные твердые вещества сушили в вакуумном сушильном шкафу при 50°C в течение 3 суток, получая 3-амино-6-бром-N'-пивалоилпиразин-2-карбогидразид (224 г; 96%) в виде желтого твердого вещества: 1Н ЯМР-спектр (DMSO-d6) 1.17 (9Н, s), 7.62 (2Н, s), 8.37 (1Н, s), 9.42-9.56 (1Н, m), 10.09-10.23 (1Н, m); масс-спектр: [М+Н]+ = 318.
п-Толуолсульфонилхлорид (164 г; 861,60 ммоль) порциями добавляли к суспензии 3-амино-6-бром-N'-пивалоилпиразин-2-карбогидразида (227 г; 718,00 ммоль) и N,N-диизопропилэтиламина (297 мл; 1795,01 ммоль) в ацетонитриле (2200 мл). Смесь перемешивали в течение 2 часов при 70°С. Реакционную смесь оставляли охлаждаться до комнатной температуры в течение ночи. Реакционную смесь распределяли между этилацетатом (2 л) и раствором бикарбоната натрия (2 л). Органический слой промывали насыщенным рассолом, сушили с использованием сульфата магния, фильтровали и концентрировали при пониженном давлении. Полученное бежево-коричневое твердое вещество растирали с горячим МТВЕ (1000 мл), выделяли фильтрованием и сушили, получая 5-бром-3-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-амин в виде желтого твердого вещества (187 г; 87%). Маточные жидкости упаривали досуха. Неочищенное твердое вещество растирали с МТВЕ (500 мл), фильтровали и промывали 100 мл МТВЕ. Полученное твердое вещество сушили на воздухе в течение ночи, получая вторую порцию 5-бром-3-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-амина (36 г; 17%): 1Н ЯМР-спектр (DMSO-d6) 1.43 (9Н, s), 7.70 (2Н, s), 8.39 (1Н, s); масс-спектр: [М+Н]+ = 298.
При альтернативном получении к 3-амино-6-бром-N'-пивалоилпиразин-2-карбогидразиду (2301 г; 7,28 моль) в MeCN (10,8 л) добавляли DIPEA (3,044 л; 17,48 моль) и п-толуолсульфонилхлорид (1665 г; 8,73 моль) порциями (приблизительно по 280 г × 6) при 50°C в течение 30 мин. Температуру реакционной смеси поддерживали в диапазоне 65-70°C, контролируя скорость добавления. По завершении добавления реакционную смесь перемешивали при 70°C в течение 1 ч. Смесь охлаждали до комнатной температуры и гасили 5%-ным NaHCO3 (водным; 24,2 л). Полученную суспензию перемешивали в течение 30 мин, затем фильтровали. Продукт промывали водой (14,8 л), подсушивали и сушили при 50°C в течение 16 ч. Продукт растворяли в DCM (12 л) и проводили разделение фаз. Органическую фазу загружали на набивку диоксида кремния (6 кг) и продукт элюировали 20% EtOAc/DCM (8×10 л). После концентрирования фракций, содержащих продукт, получали 1987 г (выход 92%) с чистотой 99,8% по данным HPLC.
Через раствор 5-бром-3-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-амина (89,35 г; 239,75 ммоль) в DMA (1,2 л) в течение 20 минут пропускали струю газообразного азота. К перемешиваемой смеси последовательно добавляли дициклогексил(2',4',6'-триизопропилбифенил-2-ил)фосфин (11,43 г; 23,98 ммоль), трис(дибензилиденацетон)дипалладий(0) (5,49 г; 5,99 ммоль), цинк (1,568 г; 23,98 ммоль) и дицианоцинк (16,89 г; 143,85 ммоль). Смесь нагревали до 100°C и перемешивали в течение 1 часа. Смесь охлаждали и распределяли между DCM (3 л) и водой (1 л). Черную смесь фильтровали через целит, и органический слой отделяли. Раствор промывали водой, затем рассолом. Раствор сушили, используя сульфат магния, и концентрировали при пониженном давлении. Остаток растирали с МТВЕ и выделяли фильтрованием, промывая МТВЕ. Осадок на фильтре сушили в вакууме, получая 5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-карбонитрил (55,7 г; 95%) в виде бледно-оранжевого твердого вещества: 1Н ЯМР-спектр (DMSO-d6) 1.46 (9Н, s), 6.02 (1Н, s), 8.38 (2Н, s); масс-спектр: [M-H]-=242.
Продукт можно суспендировать в гептанах, затем фильтровать и сушить, в качестве альтернативы растиранию с МТВЕ.
Гидрат гидразина (82 мл; 1,69 моль) добавляли к 5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-карбонитрилу (55 г; 225,18 ммоль) в IPA (200 мл) и смесь нагревали при 50°C в атмосфере азота в течение 16 часов. Смесь охлаждали в ледяной бане. Полученный осадок собирали фильтрованием, промывали IPA и диэтиловым эфиром и сушили до постоянной массы, получая (Z)-5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-карбогидразонамид (49,2 г; 79%) в виде желтого твердого вещества: 1Н ЯМР-спектр (DMSO-d6) 1.45 (9Н, s), 5.26 (2Н, s), 5.58 (2Н, s), 7.56 (2Н, s), 8.75 (1Н, s); масс-спектр: [М+Н]+ = 277.
Гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (74,3 г; 195,44 ммоль) добавляли к раствору N-Boc-изонипекотиновой кислоты (41,1 г; 179,15 ммоль) и 4-метилморфолина (35,9 мл; 325,74 ммоль) в DMA (800 мл). Смесь перемешивали в течение 10 минут, затем к раствору добавляли (Z)-5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-карбогидразонамид (45 г; 162,87 ммоль) в виде одной порции (наблюдали экзотермический эффект в диапазоне от 22°C до 27°C). Через несколько минут продукт кристаллизовали из реакционной смеси. Реакционную смесь извлекали из сосуда и фильтровали через фильтр из пористого стекла. Чтобы смыть продукт со стенок сосуда, добавляли дополнительное количество DMA (150 мл) и все это выливали на осадок на фильтре. В сосуд добавляли изопропанол (600 мл), и оставшуюся в сосуде часть продукта суспендировали в этом растворителе, используя энергичное перемешивание. После удаления DMA отсасыванием эту суспензию в изопропаноле использовали для промывки осадка на фильтре. Осадок на фильтре сушили отсасыванием, затем промывали МТВЕ и еще раз подсушивали отсасыванием. Осадок на фильтре сушили в вакууме, получая (Z)-трет-бутил 4-(2-(амино(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)метилен)гидразинкарбонил)пиперидин-1-карбоксилат (76 г; 95%) в виде желтого твердого вещества: 1Н ЯМР-спектр (DMSO-d6) 1.40 (9Н, s), 1.46 (9Н, s), 1.63-1.9 (2Н, m), 2.33-2.6 (2Н, m), скрыт сигналом от DMSO), 2.63-3.03 (2Н, m), 3.18-3.48 (4Н, m, скрыт сигналом от воды), 3.88-4.11 (2Н, m), 6.43 (2Н, s), 7.76 (2Н, br), 8.84 (0,5Н, s), 8.87 (0,5Н, s), 9.85 (1Н, s): масс-спектр: [М+Н]+ = 488.
При альтернативном получении N-Boc-изонипекотиновая кислота может быть получена in situ так, как указано ниже. Изонипекотиновую кислоту (858 г; 3,74 моль) растворяли в DMA (25,3 л) и добавляли 4-метилморфолин (393 мл; 3,74 моль). Перемешивали в течение 5 мин и добавляли изобутилхлорформиат (489 мл; 3,74 моль). Реакционную смесь перемешивали при 25°С в течение 2 ч и охлаждали до 15°С, после чего порциями в течение 10 мин добавляли (Z)-5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-карбогидразонамид (940 г; 3,4 моль). Реакционную смесь перемешивали в течение 1-2 ч при 15°С. Добавляли порциями в течение 1 ч воду (20,5 л) и перемешивали в течение еще 1 ч, после чего фильтровали. Затем осадок на фильтре промывали водой (4×4 л) и подсушивали на фильтре, после чего сушили досуха в вакуумном сушильном шкафу при 50°C, получая желаемый продукт.
В закрепленный сосуд с двойной рубашкой емкостью 3 л, содержащий диоксан (500 мл), добавляли уксусную кислоту (200 мл) и раствор нагревали до 70°C в атмосфере азота. К этой нагретой смеси порциями добавляли (Z)-трет-бутил-4-(2-(амино(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)метилен)-гидразинкарбонил)-пиперидин-1-карбоксилат (74,5 г; 152,80 ммоль). Через 10 минут температуру повышали до 100°C (слабое кипение с обратным холодильником). Реакционную смесь перемешивали при 100°C в течение 1,5 часа (суспензию), затем выдерживали при 80°C в течение ночи (после выдерживания в течение ночи образовывался раствор). Полученный раствор концентрировали при пониженном давлении, затем разбавляли толуолом, упаривали досуха, переносили в толуол и концентрировали еще раз. Оставшееся масло смешивали с некоторым количеством этилацетата и концентрировали досуха. Из раствора кристаллизовалось твердое вещество, которое растирали с МТВЕ (200 мл) и выделяли фильтрованием. Осадок на фильтре промывали водой и МТВЕ, получая трет-бутил-4-(5-(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1H-1,2,4-триазол-3-ил)пиперидин-1-карбоксилат (50 г; 70%) в виде серого твердого вещества.
Фильтрат концентрировали при пониженном давлении, получая желтое твердое вещество. Это вещество растирали с МТВЕ и фильтровали. Осадок на фильтре промывали этилацетатом и затем МТВЕ, получая вторую порцию в виде бледно-желтого твердого вещества (4,93 г; 7%). Это вещество было идентично веществу из первой порции: 1Н ЯМР-спектр (DMSO-d6) 117 (9Н, s), 1.22 (9Н, s), 1.29-1.47 (2Н, m), 1.67-1.78 (2Н, m), 2.57-2.87 (3H, m), 3.57-3.92 (2Н, m), 7.56 (2Н, br), 8.56 (1Н, s), 13.47 (2Н, br s); масс-спектр: [M+H]+ = 470.
1,8-Диазабицикло[5.4.0]ундец-7-ен (19,87 мл; 132,90 ммоль) добавляли к суспензии трет-бутил-4-(5-(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1Н-1,2,4-триазол-3-ил)пиперидин-1-карбоксилата (48 г; 102,23 ммоль) в 2-метил-ТНР (300 мл). Через 5 минут получали темный раствор, который обрабатывали активированным углем и фильтровали через набивку целита, промывая активированный уголь дополнительным количеством 2 метил-THF (100 мл). Фильтрат перемешивали, используя воздушную мешалку, при -5°С в закрепленном сосуде с рубашкой емкостью 3 л в атмосфере азота. 2-Метил-THF (100 мл) добавляли, чтобы облегчить перемешивание желтой суспензии. По каплям в течение 15 минут добавляли подметан (7,96 мл; 127,78 ммоль).
Смесь перемешивали в течение 2 часов и реакционную смесь нагревали до комнатной температуры. Через 16 часов добавляли дополнительное количество подметана (6 мл) и DBU (20 мл), и перемешивание продолжали в течение 16 часов. Смесь выливали в воду и перемешивали в течение 5 минут. Нерастворенное вещество выделяли в виде бежевого твердого вещества и сушили в вакууме, получая трет-бутил-4-(5-(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-метил-1Н-1,2,4-триазол-3-ил)пиперидин-1-карбоксилат (24,77 г; 50,1%). Маточные жидкости концентрировали при пониженном давлении, и остаток очищали колоночной флэш-хроматографией на диоксиде кремния, используя МТВЕ в качестве элюента. Таким образом получали вторую порцию желаемого продукта (13,04 г; 26%) в виде желтого твердого вещества: 1Н ЯМР-спектр (DMSO-d6) 1.47 (9Н, s), 1.51 (9Н, s), 1.57-1.76 (2Н, m), 1.94-2.1 (2Н, m), 2.87-3.09 (3H, m), 3.9-4.08 (2Н, m), 4.26 (3H, s), 7.97 (2Н, br, s), 8.92 (1Н, s); масс-спектр: [М+Н]+ = 484.
трет-Бутил-4-(5-(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-метил-1H-1,2,4-триазол-3-ил)пиперидин-1-карбоксилат (36,81 г; 76,12 ммоль) добавляли к раствору 2,2,2-трифторуксусной кислоты (100 мл; 1305,87 ммоль) в DCM (100 мл). Смесь перемешивали в течение 3 часов при комнатной температуре. Смесь концентрировали при пониженном давлении. Остаток растворяли в DCM (1,5 л) и добавляли к энергично перемешиваемому концентрированному раствору аммиака (150 мл) в воде (400 мл). Водную фазу промывали DCM (400 мл) и объединенные органические растворы сушили с использованием сульфата магния, фильтровали и концентрировали досуха, получая 3-(5-трет-бутил-1,3,4-оксадиазол-2-ил)-5-(1-метил-3-(пиперидин-4-ил)-1Н-1,2,4-триазол-5-ил)пиразин-2-амин (30,0 г; 103%) в виде желтого твердого вещества: 1Н ЯМР-спектр (DMSO-d6) 1.44 (9Н, s), 1.54-1.69 (2Н, m), 1.8-1.92 (2Н, m), 2.53-2.63 (2Н, m), 2.68-2.83 (1Н, m), 2.93-3.05 (2Н, m), 4.19 (3H, s), 7.89 (2Н, br), 8.85 (1Н, s); масс-спектр: [М+Н]+ = 384.
Гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (30,4 г; 80,04 ммоль) порциями добавляли при 25°С к перемешиваемому раствору 3-(тетрагидро-2Н-пиран-2-илокси)пропановой кислоты (12,67 г; 72,76 ммоль) и N-этил-N-изопропилпропан-2-амина (25,3 мл; 145,52 ммоль) в ацетонитриле (200 мл). Полученный раствор перемешивали при 25°С в течение 20 минут, затем порциями добавляли 3-(5-трет-бутил-1,3,4-оксадиазол-2-ил)-5-(1-метил-3-(пиперидин-4-ил)-1H-1,2,4-триазол-5-ил)пиразин-2-амин (30 г; 72,76 ммоль), вливая последнюю порцию в эту смесь в виде суспензии в ацетонитриле (100 мл). После перемешивания в течение 1 часа осадок собирали фильтрованием, промывали ацетонитрилом и сушили в вакууме, получая 1-(4-(5-(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-метил-1H-1,2,4-триазол-3-ил)пиперидин-1-ил)-3-(тетрагидро-2Н-пиран-2-илокси)пропан-1-он (35,0 г; 89%) в виде бежевого твердого вещества. Фильтрат разбавляли DCM (600 мл), промывали водой, сушили над сульфатом магния и концентрировали. Остаток очищали флэш-хроматографией на силикагеле, элюируя градиентом 2-2,5% (7 н. аммиака в метаноле) в дихлорметане. Вторую порцию продукта (3,31 г; 6,13 ммоль; 8,43%) также получали в виде кремового твердого вещества. Оба образца объединяли, получая бежевое твердое вещество: 1Н ЯМР-спектр (DMSO-d6) 1.44 (9Н, s), 1.52-1.79 (4Н, m), 1.88-2.04 (2Н, m), 2.53-2.73 (2Н, m), 2.73-2.87 (1Н, m), 2.91-3.05 (1Н, m), 3.13-3.24 (1Н, m), 3.37-3.47 (1Н, m), 3.53-3.65 (1Н, m), 3.7-3.8 (1Н, m), 3.81-3.89 (1Н, m), 3.89-3.99 (1Н, m), 4.20 (3H, s), 4.29-4.4 (1Н, m), 4.54-4.61 (1Н, m), 7.60-8.20 (2Н, br), 8.85 (1Н, s); масс-спектр: [М+Н]+ = 540.
Пример 3
1-(4-(5-(5-Амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-этил-1H-1,2,4-триазол-3-ил)пиперидин-1-ил)-3-гидроксипропан-1-он
4-Метилбензолсульфонат пиридина (11,62 г; 46,24 ммоль) добавляли к суспензии 1-(4-(5-(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-этил-1Н-1,2,4-триазол-3-ил)пиперидин-1-ил)-3-(тетрагидро-2Н-пиран-2-илокси)пропан-1-она (128 г; 231,19 ммоль) в метаноле (1 л) в атмосфере азота. Смесь перемешивали при 60°C в течение 1,5 часа. Через 5 минут смесь превращалась в раствор. Смесь выдерживали при 50°C в течение ночи, за это время образовывался осадок. Твердое вещество выделяли фильтрованием и промывали водой и ацетонитрилом. Это вещество все еще содержало небольшое количество примесей с предыдущей стадии и требовало дополнительной очистки. Вещество растворяли в дихлорметане и очищали флэш-хроматографией на силикагеле (от 0% метанол/DCM до 10% метанол/DCM). Таким образом выделяли желаемый продукт, 1-(4-(5-(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-этил-1H-1,2,4-триазол-3-ил)пиперидин-1-ил)-3-гидроксипропан-1-он (пример 3) (92 г; 85%), в виде кремового твердого вещества (Форма А): 1Н ЯМР-спектр (DMSO-d6) 1.4-1.51 (12Н, m), 1.51-1.78 (2Н, m), 1.89-2.05 (2Н, m), 2.72-2.86 (1Н, m), 2.91-3.05 (1Н, m), 3.12-3.24 (1Н, m), 3.64 (2Н, q), 3.83-4.01 (1Н, m), 4.29-4.41 (1Н, m), 4.47 (1Н, t), 4.58 (2Н, q), 8.26 (2Н, s), 8.85 (1Н, s); масс-спектр: [М+Н]+ = 470.
1-(4-(5-(5-Амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-этил-1Н-1,2,4-триазол-3-ил)пиперидин-1-ил)-3-(тетрагидро-2Н-пиран-2-илокси)пропан-1-он (пример 3.1) получали так, как указано ниже.
1,8-Диазабицикло[5.4.0]ундец-7-ен (76 мл; 511,14 ммоль) добавляли к суспензии трет-бутил-4-(5-(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1H-1,2,4-триазол-3-ил)пиперидин-1-карбоксилата (150 г; 319,46 ммоль) в 2-Memn-THF (1,2 л). Добавляли иодэтан (46 мл; 575,03 ммоль) и смесь перемешивали в течение 16 часов при 35°С. Еще раз добавляли иодэтан (46 мл; 575,03 ммоль) и 1,8-диазабицикло[5.4.0]ундец-7-ен (76 мл; 511,14 ммоль) и перемешивание продолжали в течение 24 часов при 35°С.Смесь выливали в воду, и нерастворенное вещество выделяли фильтрованием, промывали водой и МТВЕ и сушили в вакууме, получая трет-бутил-4-(5-(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-этил-1Н-1,2,4-триазол-3-ил)пиперидин-1-карбоксилат (116 г; 73,0%) в виде желтого твердого вещества. Фильтрат экстрагировали DCM и органический раствор сушили, используя сульфат магния, фильтровали и концентрировали при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на диоксиде кремния, используя градиентное элюирование (от 30% МТВЕ/гептан до 100% МТВЕ). Таким образом выделяли вторую порцию желаемого продукта (12 г; 24,12 ммоль; 7,55%) в виде желтого твердого вещества, которое позже объединяли с первой порцией: 1Н ЯМР-спектр (DMSO-d6) 1.41 (9Н, s), 1.44 (9Н, s), 1.48 (3H, t), 1.52-1.69 (2Н, m), 1.87-2.04 (2Н, m), 2.79-3.03 (3H, m), 3.86-4.03 (2Н, m), 4.59 (2Н, q), 7.89 (2Н, s), 8.85 (1Н, s); масс-спектр: [М+Н]+ = 498.
Подходящим растворителем для приведенной выше реакции также может быть THF.
TFA (400 мл) порциями добавляли к раствору трет-бутил-4-(5-(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-этил-1H-1,2,4-триазол-3-ил)пиперидин-1-карбоксилата (126 г, 253,22 ммоль) в DCM (400 мл). Смесь перемешивали в течение 16 часов при комнатной температуре. Смесь концентрировали при пониженном давлении, растворяли в DCM (1 л) и медленно добавляли к энергично перемешиваемому раствору концентрированного водного раствора аммиака (500 мл) в воде при 0°С. Органический раствор отделяли от водного и концентрировали при пониженном давлении*, получая 3-(5-трет-бутил-1,3,4-оксадиазол-2-ил)-5-(1-этил-3-(пиперидин-4-ил)-1Н-1,2,4-триазол-5-ил)пиразин-2-амин (101 г; 100%) в виде желтого твердого вещества: 1Н ЯМР-спектр (DMSO-d6) 1.4-1.52 (12Н, m), 1.57-1.73 (2Н, m), 1.83-1.93 (2Н, m), 2.57-2.7 (2Н, m), 2.71-2.84 (1Н, m), 2.96-3.09 (2Н, m), 4.58 (2Н, q), 8.06 (2Н, s), 8.84 (1Н, s); масс-спектр: [М+Н]+ = 398.
*В другом эксперименте подобного масштаба (приблизительно 170 г исходного вещества) использовали следующую методику выделения: слои разделяли и верхний слой (эмульсию с твердым веществом) фильтровали. Твердое вещество промывали DCM (0,5 л) и фильтрат переносили в делительную воронку. Слои разделяли, и водный слой экстрагировали DCM (0,5 л). Органические слои сушили над MgSO4, фильтровали и концентрировали. Продукт сушили при 50°C в течение ночи (81,75 г). Твердое вещество, полученное в результате экстракции, суспендировали в воде (200 мл) в течение 30 мин при комнатной температуре и отфильтровывали. Продукт сушили при 50°C в вакууме (61,8 г).
Ниже приведен другой вариант.
Суспензию трет-бутил-4-(5-(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-этил-1Н-1,2,4-триазол-3-ил)пиперидин-1-карбоксилата (3009,5 г; 6,05 моль) в DCM (9 л) охлаждали до 5-10°C в атмосфере N2. К суспензии порциями добавляли TFA (9 л), поддерживая температуру меньше 30°С. Реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Смесь концентрировали, полученный остаток растворяли в воде (30 л) и медленно добавляли к 35%-ному водному раствору аммиака (12 л) при 0-5°С. Суспензию перемешивали в течение 30 мин, затем продукт отфильтровывали и промывали водой (2×6 л). Продукт сушили при 50°C в вакууме в течение 2 суток (2496 г).
Гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N,N'-тетраметилурония (HATU; 106 г; 279,51 ммоль) порциями добавляли при 25°C к перемешиваемому раствору 3-(тетрагидро-2Н-пиран-2-илокси)пропановой кислоты (44,3 г; 254,10 ммоль) и N-этил-N-изопропилпропан-2-амина (89 мл; 508,21 ммоль) в ацетонитриле (600 мл). Полученный раствор перемешивали при 25°С в течение 20 мин, затем порциями добавляли 3-(5-трет-бутил-1,3,4-оксадиазол-2-ил)-5-(1-этил-3-(пиперидин-4-ил)-1Н-1,2,4-триазол-5-ил)пиразин-2-амин (101 г; 254,10 ммоль), промывая последнюю порцию в эту смесь в виде суспензии в ацетонитриле (300 мл). После перемешивания в течение 1 часа осадок собирали фильтрованием, промывая ацетонитрилом, и сушили в вакууме, получая 1-(4-(5-(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-этил-1Н-1,2,4-триазол-3-ил)пиперидин-1-ил)-3-(тетрагидро-2Н-пиран-2-илокси)пропан-1-он (128 г; 91%) в виде бежевого твердого вещества. Фильтрат разбавляли DCM (600 мл), промывали водой, сушили над сульфатом магния и концентрировали. Остаток очищали флэш-хроматографией на силикагеле, элюируя градиентом 2-2,5% (7 н. аммиака в метаноле) в дихлорметане. Вторую порцию желаемого продукта (40 г; 72,2 ммоль; 28,4%) получали в виде кремового твердого вещества, которое объединяли с первой порцией: 1Н ЯМР-спектр (DMSO-d6) 1.29-1.48 (16Н, m), 1.48-1.75 (4Н, m), 1.83-1.99 (2Н, m), 2.48-2.68 (2Н, m), 2.68-2.79 (1Н, m), 2.87-2.99 (1Н, m), 3.07-3.19 (1Н, m), 3.32-3.42 (1Н, m), 3.47-3.6 (1Н, m), 3.64-3.75 (1Н, m), 3.75-3.84 (1Н, m), 3.84-3.95 (1Н, m), 4.24-4.39 (1Н, m), 4.47-4.6 (3H, m), 7.84 (2Н, s), 8.79 (1Н, s); масс-спектр: [M+Na]+ = 577.
Альтернативное получение
К раствору 3-(тетрагидро-2Н-пиран-2-илокси)пропановой кислоты (48,80 г; 0,2774 моль) и N-этил-N-изопропилпропан-2-амина (86,96 мл; 0,4993 моль) в THF (552 мл) порциями при кт в атмосфере азота добавляли гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (115,73 г; 0,3051 моль). Полученную смесь перемешивали в течение 20 мин, затем порциями в течение 1 ч добавляли 3-(5-трет-бутил-1,3,4-оксадиазол-2-ил)-5-(1-этил-3-(пиперидин-4-ил)-1H-1,2,4-триазол-5-ил)пиразин-2-амин (122,5 г (110,25 г активного вещества); 0,2774 моль). Через 3,5 ч смесь концентрировали и остаток суспендировали в MeCN (275 мл) в течение 15 мин при комнатной температуре. Продукт отфильтровывали, промывали MeCN (3×110 мл) и сушили в течение ночи при 50°C в вакууме. В результате получали 1-(4-(5-(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-этил-1Н-1,2,4-триазол-3-ил)пиперидин-1-ил)-3-(тетрагидро-2Н-пиран-2-илокси)пропан-1-он (131,9 г; 96%).
В другом альтернативном способе получения в качестве агента сочетания вместо HATU можно использовать HBTU (гексафторфосфат O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония) в THF.
Альтернативное получение соединения из примера 3
К суспензии 1-(4-(5-(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-этил-1H-1,2,4-триазол-3-ил)пиперидин-1-ил)-3-(тетрагидро-2Н-пиран-2-илокси)пропан-1-она (131,9 г; 0,2382 моль) в метаноле (1045 мл) добавляли п-толуолсульфонат пиридиния (11,97 г; 47,7 ммоль) в атмосфере N2. Реакционную смесь перемешивали при 60°C в течение 5,5 ч, затем при 50°C в течение ночи. Реакционную смесь охлаждали до 0°C и твердое вещество отфильтровывали. Продукт суспендировали в воде (250 мл) в течение 20 мин при комнатной температуре, отфильтровывали, промывали водой (3×40 мл) и сушили при 50°C в вакууме. В результате получали 1-(4-(5-(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-этил-1H-1,2,4-триазол-3-ил)пиперидин-1-ил)-3-гидроксипропан-1-он (21,4 г) в виде Формы A (см. ниже).
Концентрировали метанольные растворы и полученное твердое вещество суспендировали в воде (0,6 л) в течение 20 мин при комнатной температуре. Твердое вещество выделяли фильтрованием и промывали водой (3×100 мл). Осадок на фильтре суспендировали второй раз в воде (0,5 л) в течение еще 20 минут. Продукт выделяли фильтрованием, промывали водой (100 мл) и сушили при 50°С в вакууме. В результате получали 81,9 г 1-(4-(5-(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-этил-1H-1,2,4-триазол-3-ил)пиперидин-1-ил)-3-гидроксипропан-1-она (81,9 г) в виде Формы А.
Обе порции объединяли (103,3 г), вносили затравку Формы B (16,68 г) и суспендировали в MeCN (826 мл) при комнатной температуре в течение ночи. В результате получали 117,4 г 1-(4-(5-(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-этил-1H-1,2,4-триазол-3-ил)пиперидин-1-ил)-3-гидроксипропан-1-она в виде бледно-желтого твердого вещества (117,4 г), Форму B (см. ниже). Это вещество далее очищали, суспендируя в гептане (7,5 относительных объемов) в течение 1 часа. Смесь фильтровали, подсушивали на фильтре и сушили при 50°С в вакуумном сушильном шкафу в течение ночи, получая 1-(4-(5-(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-этил-1H-1,2,4-триазол-3-ил)пиперидин-1-ил)-3-гидроксипропан-1-он (102,5 г) в виде Формы В.
Форма B также может быть получена путем суспендирования Формы A в MeCN без внесения затравки.
Форма A или B также может быть превращена в Форму C, как указано ниже.
Суспензию 1-(4-(5-(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-этил-1H-1,2,4-триазол-3-ил)пиперидин-1-ил)-3-гидроксипропан-1-она (например, Формы B, полученной способами, приведенными выше) в IPA (12 об.) нагревали при температуре дефлегмации до растворения твердого вещества. Раствор фильтровали в теплом состоянии, затем охлаждали до комнатной температуры. В результате получали 1-(4-(5-(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-этил-1Н-1,2,4-триазол-3-ил)пиперидин-1-ил)-3-гидроксипропан-1-он в виде бледно-желтого твердого вещества (99,3 г; 97%) в виде Формы С.
Форма C также может быть превращена в Форму B, как указано ниже.
В колбе с патрубком емкостью 10 л нагревали до 110-115°C Форму С (1 порцию 377,8 г) в MIBK (7900 мл), получая раствор. Раствор оставляли охлаждаться до 97-103°C и незамедлительно выполняли осветляющую фильтрацию в сосуд емкостью 50 л, содержащий затравку Формы B (0,8 г) в ацетонитриле (8220 мл), с перемешиванием при -15°C. Во время добавления температуру в сосуде емкостью 50 л поддерживали в диапазоне от -15 до 25°C, используя для охлаждения рубашку. Три следующих порции соединения, растворенного в MIBK, добавляли тем же самым способом. К полученной суспензии добавляли затравку Формы B (0,8 г) и затем смесь перемешивали при 10-20°C в течение ночи. Анализом в ходе процесса подтверждали наличие желаемой формы (Формы В) без заметных количеств Формы C или аморфной формы. Смесь фильтровали и промывали ацетонитрилом (3340 мл). Твердое вещество сушили в сушильном шкафу в течение 2 суток (твердое вещество измельчали во время сушки до порошка и смеси небольших комков размеров приблизительно от 1 мм до 3-4 мм) до получения постоянной массы. Выход 1532,8 г (93,5%).
3-(Тетрагидро-2Н-пиран-2-илокси)пропановую кислоту получали так, как указано ниже.
К перемешиваемому раствору метанола (2,4 л) и концентрированной серной кислоты (44,4 мл; 832,61 ммоль) при 0°C в атмосфере азота по каплям добавляли бета-пропиолактон (175 мл; 2,78 моль). Этот раствор оставляли перемешиваться при комнатной температуре в течение 2 суток. Реакционную смесь охлаждали до 10°C, затем порциями добавляли бикарбонат натрия (145 г; 1,72 моль), полученную суспензию оставляли перемешиваться при комнатной температуре в течение 75 минут. Этот раствор фильтровали, осадок на фильтре промывали метанолом (800 мл). Фильтрат упаривали до состояния масла, которое перерастворяли в дихлорметане (1,2 л) и перемешивали в течение 60 минут, после чего повторно фильтровали. Этот раствор фильтровали, после чего упаривали, получая метил-3-гидроксипропаноат (219 г; 76%) в виде масла: 1Н ЯМР-спектр (CDCl3) 2.50 (2Н, t), 3.63 (3H, s), 3.78 (2Н, t).
п-Толуолсульфонат пиридиния (7,65 г; 30,45 ммоль) добавляли к прозрачному раствору метил-3-гидроксипропаноата (63,4 г; 609,00 ммоль) и 3,4-дигидро-2Н-пирана (78 мл; 852,60 ммоль) в дихлорметане (650 мл) при комнатной температуре в атмосфере азота, получая мутный раствор. Его оставляли перемешиваться при комнатной температуре в течение ночи. Реакционную смесь промывали водой (250 мл) и рассолом (250 мл), после чего сушили (MgSO4) и упаривали до состояния масла. Этот неочищенный продукт очищали флэш-хроматографией на диоксиде кремния, элюируя градиентом от 15 до 30% EtOAc в гептане. Фракции чистого продукта упаривали досуха, получая метил-3-(тетрагидро-2Н-пиран-2-илокси)пропаноат (67,7 г; 59,0%) в виде бесцветного масла: 1Н ЯМР-спектр (CDCl3) 1.47 (4Н, dddd), 1.55-1.84 (2Н, m), 2.55 (2Н, t), 3.33-3.53 (1Н, m), 3.53-3.7 (4Н, m), 3.78 (1Н, ddd), 3.93 (1Н, dt), 4.42-4.72 (1Н, m); масс-спектр: [МН]+ = 189.
Гидроксид натрия (2 М; 349 мл; 697,58 ммоль) добавляли к раствору метил-3-(тетрагидро-2Н-пиран-2-илокси)пропаноата (67,68 г; 359,58 ммоль) в THF (680 мл) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 3 часов. THF удаляли в вакууме, затем водный слой промывали этилацетатом (260 мл), после чего охлаждали до 0°C и осторожно подкисляли до pH 5, добавляя соляную кислоту (2 М). Продукт экстрагировали этилацетатом (3×250 мл), после чего сушили (MgSO4) и упаривали, получая 3-(тетрагидро-2Н-пиран-2-илокси)пропановую кислоту (57,0 г; 91%) в виде прозрачного масла. Это вещество растворяли в этилацетате (750 мл), затем промывали водой (3×250 мл) и рассолом (250 мл) для удаления оставшейся уксусной кислоты. Органический раствор сушили (MgSO4) и упаривали, получая 3-(тетрагидро-2Н-пиран-2-илокси)пропановую кислоту (45,67 г; 72,9%) в виде бесцветного масла: 1Н ЯМР-спектр (CDCl3) 1.43-1.67 (4Н, m), 1.65-1.95 (2H, m), 2.68 (2H, t), 3.48-3.58 (1H, m), 3.73 (1H, dt), 3.88 (1H, ddd), 4.02 (1H, dt), 4.59-4.7 (1H, m); масс-спектр: [M-H]- = 173.
Соединение из примера 3, выделенное так, как описано выше, представляло собой кристаллическое вещество в трех разных кристаллических формах, описанных в данной заявке как Формы A, B и C.
Кристаллическую структуру Формы A соединения из примера 3 можно охарактеризовать с использованием XRPD и DSC.
Способы осуществления этих методов являются такими, как описано для соединения из примера 1.
Десять пиков дифракции рентгеновских лучей на порошке для Формы А соединения из примера 3
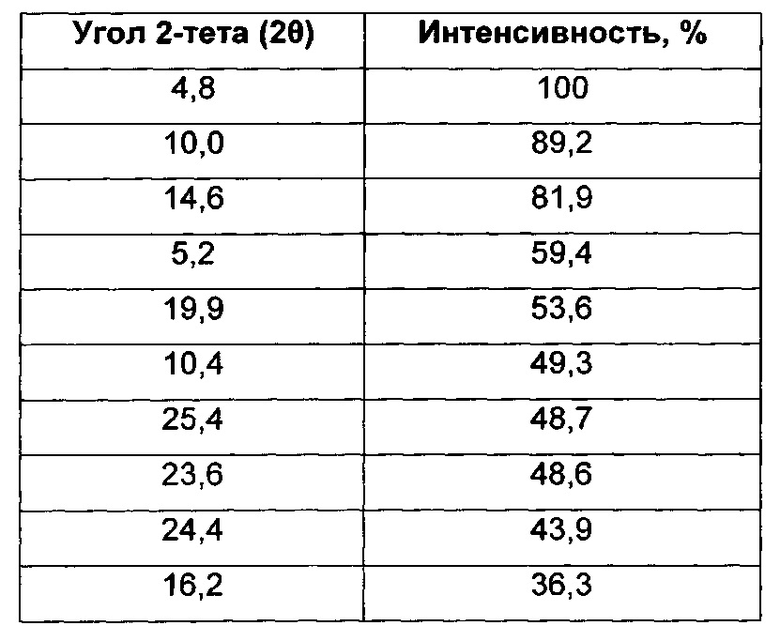
Спектр XRPD для Формы A соединения из примера 3 показан на Фиг. 3.
DSC-анализ Формы A соединения из примера 3 показывает первоначальную эндотерму с началом при 27,0°C и пиком при 63,0°C, далее видны эндотермические сдвиги с началами и пиками при следующих температурах: 166,5°C и 168,7°C, 172,2°C и 173,2°C, и окончательное плавление при 174,8°C и с пиком при 175,7°C (Фиг. 4).
Таким образом, DSC-анализ показывает, что Форма A соединения из примера 3 представляет собой сольватированное вещество с началом десольватации при примерно 27,0°C и пиком при примерно 63,0°C.
Спектры дифракции рентгеновских лучей на порошке для соединения из примера 3 (Формы А) показали, что данное вещество является кристаллическим. Это вещество имело точку десольватации при 28,0°C (начало).
Соединение из примера 3 также может существовать в альтернативной полиморфной форме, обозначенной в данном описании как Форма B. Получение Формы B описано выше.
Это вещество имело точку плавления 172,5°C (начало).
Согласно следующему аспекту изобретения предложен способ получения Формы B соединения из примера 3 путем суспендирования образца соединения из примера 3 в ацетонитриле. Согласно другому аспекту изобретения предложен способ получения Формы B соединения из примера 3 из раствора Формы C соединения из примера 3 в MIBK.
Десять пиков дифракции рентгеновских лучей на порошке для Формы B соединения из примера 3
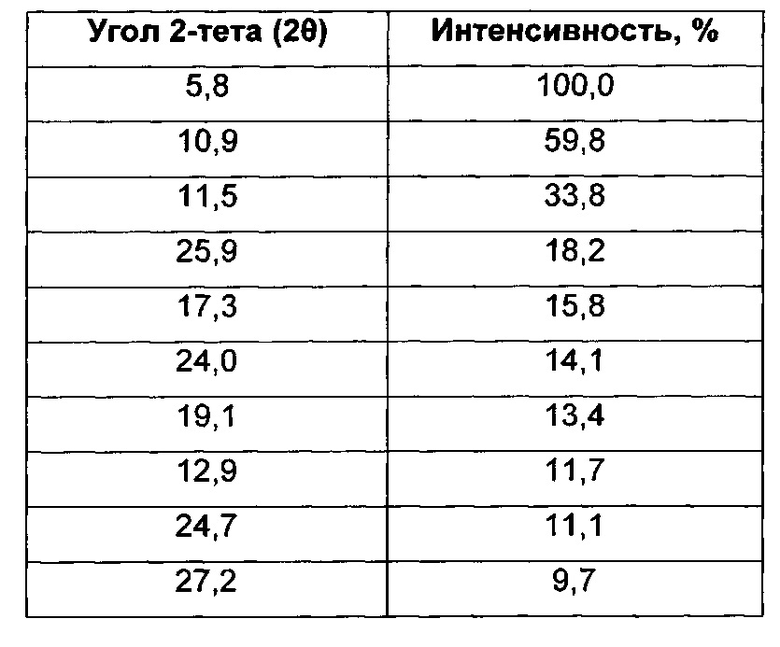
Спектр XRPD для Формы B соединения из примера 3 показан на Фиг. 5.
DSC-анализ Формы B соединения из примера 3 показывает эндотерму плавления с началом при 172,5°C и пиком при 174,2°C (Фиг. 6).
Таким образом, DSC-анализ показывает, что Форма B соединения из примера 3 представляет собой высокоплавкое твердое вещество с началом плавления при примерно 172,5°C и пиком при примерно 174,2°С.
Соединение из примера 3 также может существовать в третьей кристаллической форме, обозначенной в данном описании как Форма С.Способ получения соединения в виде Формы C, например, из соединения в виде Формы B посредством кристаллизации из изопропилового спирта (IPA), описан выше.
Таким образом, согласно другому аспекту изобретения предложен способ получения Формы C соединения из примера 3 посредством кристаллизации соединения примера 3 из IPA.
Форма C соединения из примера 3 характеризуется наличием по меньшей мере одного из следующих значений 20, измеренных с использованием CuKa-излучения: 6,9 и 12,3. Форма C соединения из примера 3 характеризуется наличием картины дифракции рентгеновских лучей на порошке, по существу такой, как показано на Фиг. 7. Десять пиков дифракции рентгеновских лучей на порошке показаны ниже.
Десять пиков дифракции рентгеновских лучей на порошке для Формы C соединения из примера 3
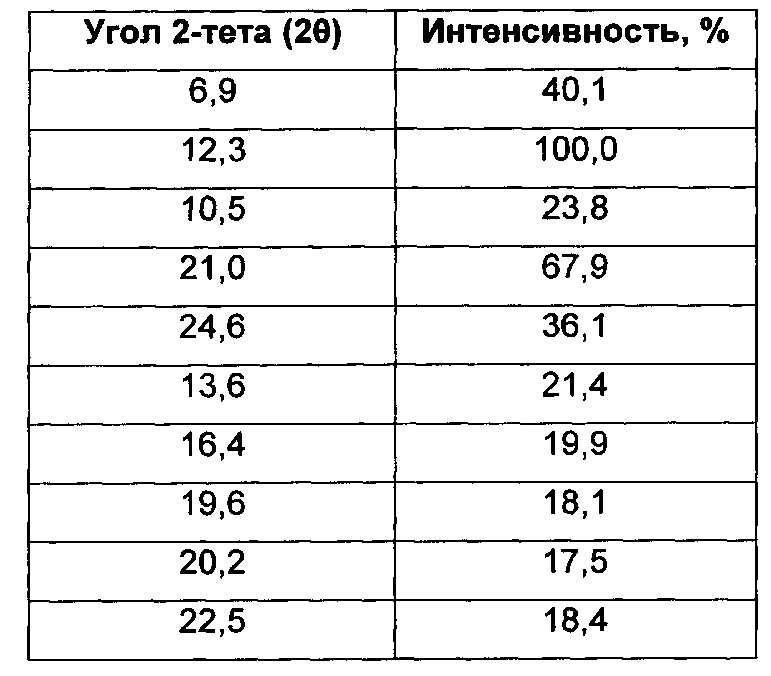
DSC-анализ Формы C соединения из примера 3 показывает эндотерму плавления с началом при 183,0°C и пиком при 185,6°C (Фиг. 8).
Таким образом, DSC-анализ показывает, что Форма C соединения из примера 3 представляет собой высокоплавкое твердое вещество с началом плавления при примерно 183,0°C и пиком при примерно 185,6°C.
Подробное описание методов, использованных для анализа Формы C
Дифракция рентгеновских лучей на порошке
Аналитический прибор: Panalytical Cubix.
Картину дифракции рентгеновских лучей на порошке определяли путем помещения образца кристаллического вещества на держатель пластины из монокристалла кремния (SSC) от Panalytical, и распределения образца в виде тонкого слоя с помощью предметного стекла микроскопа. Образец вращали при 30 оборотах в минуту (для улучшения статистики счета) и облучали рентгеновскими лучами, генерируемыми длиннофокусной трубкой с медным анодом, работающей при 45 кВ и 40 мА, с длиной волны 1,5418 ангстрем. Пучок рентгеновских лучей пропускали через щель Соллера 0,04 радиана, затем автоматически изменяющуюся щель расходимости, установленную на 20 мм, и окончательно через отверстие 20 мм для диафрагмирования пучка (beam mask). Отраженное излучение направляли через антирассеивающую щель 20 мм и щель Соллера 0,04 радиана. Образец подвергали воздействию в течение 1,905 секунды с шагом 0,0025067° 2-тета (непрерывный режим сканирования) в диапазоне от 2 градусов до 40 градусов 2-тета в режиме тета-тета. Прибор был оснащен детектором X-Celerator. Получение контрольных и экспериментальных данных осуществляли с помощью дисплейного терминала) Dell Pentium 4НТ, работающего с программным обеспечением X'Pert Industry. Специалисты в области дифракции рентгеновских лучей на порошке осведомлены, что на относительную интенсивность пиков могут оказывать влияние, например, гранулы в размере свыше 30 микрон и с неунитарными соотношениями сторон, что может отразиться на анализе образцов. Специалисту также понятно, что на положение отражений может влиять точность размещения по высоте, на которой образец находится в дифрактометре, и калибровка нуля дифрактометра. Плоскостность поверхности образца также может производить небольшой эффект. Следовательно, представленные данные дифракционных картин не должны приниматься за абсолютные значения.
Дифференциальная сканирующая калориметрия
Аналитический прибор для DSC: Q1000 от ТА Instruments.
Обычно менее 5 мг вещества, содержащегося в стандартном алюминиевом тигле, снабженном крышкой, нагревали в температурном диапазоне от 25°C до 300°C с постоянной скоростью нагревания 10°C в минуту. В качестве продувочного газа использовали азот при скорости потока 50 мл в минуту.
Пример 4
(3R)-1-[4-[5-[5-Амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил]-1-метил-1,2,4-триазол-3-ил1-1-пиперидил1-3-гидрокси-бутан-1-он
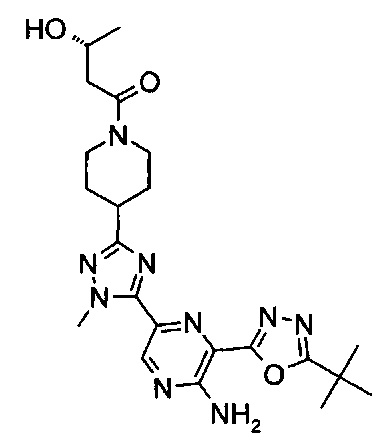
Тетрафторборат 2-(1H-бензо[d][1,2,3]триазол-1-ил)-1,1,3,3-тетраметилизоурония (201 мг; 0,63 ммоль) добавляли к перемешиваемой суспензии 3-(5-трет-бутил-1,3,4-оксадиазол-2-ил)-5-(1-метил-3-(пиперидин-4-ил)-1Н-1,2,4-триазол-5-ил)пиразин-2-амина (200 мг; 0,52 ммоль; описанного в примере 1), N-этил-N-изопропилпропан-2-амина (0,273 мл; 1,56 ммоль) и (R)-3-гидроксибутановой кислоты (65,2 мг; 0,63 ммоль) в N,N-диметилформамиде (3 мл). Полученную суспензию перемешивали при комнатной температуре в течение 2 часов. Полученную смесь очищали препаративной HPLC, используя колонку для обращенно-фазовой хроматографии X-Bridge от Waters (С-18, диоксид кремния, 5 микрон, диаметр 30 мм, длина 150 мм, скорость потока 60 мл/минута) с применением изократической смеси 31% ацетонитрила в воде (содержащей карбонат аммония (2 г/л)). Фракции, содержащие желаемое соединение, упаривали досуха, получая бледно-желтое твердое вещество. Это твердое вещество перемешивали в ацетонитриле (3 мл) при комнатной температуре. Полученное твердое вещество отфильтровывали, промывали холодным ацетонитрилом и сушили, получая указанное в заголовке соединение (125 мг; 51,0%) в виде бледно-желтого твердого вещества.
1Н ЯМР-спектр: (CDCl3 1.24 (3H, d), 1.52 (9Н, s), 1.85 (2Н, m), 2.10 (2Н, m), 2.35 (1Н, dd), 2.55 (1Н, d), 2.90 (1Н, m), 3.05 (1Н, m), 3.20 (1Н, m), 3.90 (1Н, m), 4.25 (1Н, m), 4.31 (3H, s), 4.6 (1Н, m), 9.03 (1Н, s); масс-спектр: [М+Н]+ = 470.
Пример 5
(3S)-1-[4-[5-[5-Амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил]-1-метил-1,2,4-триазол-3-ил]-1-пиперидил]-3-гидрокси-бутан-1-он
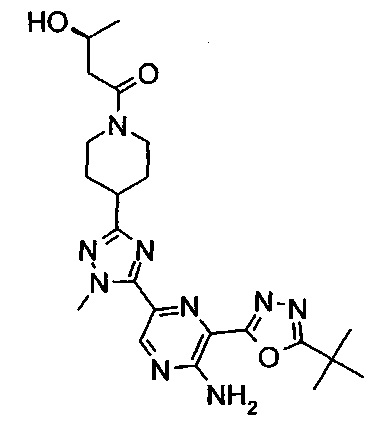
Используя методику, аналогичную методике из примера 4, приводили во взаимодействие 3-(5-трет-бутил-1,3,4-оксадиазол-2-ил)-5-(1-метил-3-(пиперидин-4-ил)-1H-1,2,4-триазол-5-ил)пиразин-2-амин с (S)-3-гидрокси-бутановой кислотой, получая указанное в заголовке соединение (167 мг; 68,2%) в виде бледно-желтого твердого вещества.
1Н ЯМР-спектр: (CDCl3) 1.24 (3H, d), 1.52 (9Н, s), 1.85 (2Н, m), 2.10 (2Н, m), 2.35 (1Н, dd), 2.55 (1Н, d), 2.90 (1Н, m), 3.05 (1Н, m), 3.20 (1Н, m), 3.90 (1Н, m), 4.25 (1Н, m), 4.31 (3H, s), 4.6 (1Н, m), 9.03 (1Н, s); масс-спектр: [М+Н]+ = 470.
Пример 6
(2R)-1-[4-[5-[5-Амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил]-1-метил-1,2,4-триазол-3-ил]-1-пиперидил]-3-гидрокси-2-метил-пропан-1-он
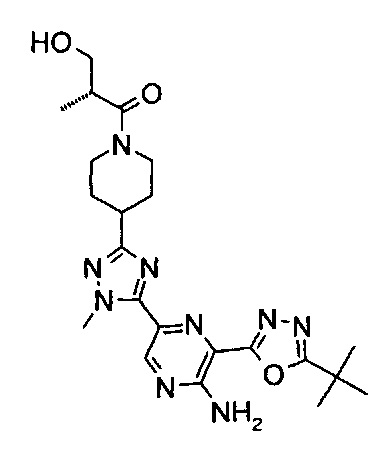
Используя методику, аналогичную методике из примера 4, приводили во взаимодействие 3-(5-трет-бутил-1,3,4-оксадиазол-2-ил)-5-(1-метил-3-(пиперидин-4-ил)-1H-1,2,4-триазол-5-ил)пиразин-2-амин с (R)-3-гидрокси-2-метилпропановой кислотой, получая указанное в заголовке соединение (87 мг; 47,4%) в виде бледно-желтого твердого вещества.
1Н ЯМР-спектр: (CDCl3) 1.55 (9Н, s), 1.61 (3H, s br), 1.8-2.0 (2Н, m), 2.10-2.25 (2Н, m), 2.90 (2Н, m), 3.10 (1Н, m), 3.3 (2Н, m), 3.77 (2H, m), 4.33 (3H, s), 4.6 (1H, m), 9.05 (1H, s); масс-спектр: [M+H]+ = 470.
Пример 7
1-[4-[5-[5-Амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил]-1-метил-1,2,4-триазол-3-ил]-1-пиперидил]-2-гидрокси-2-метил-пропан-1-он
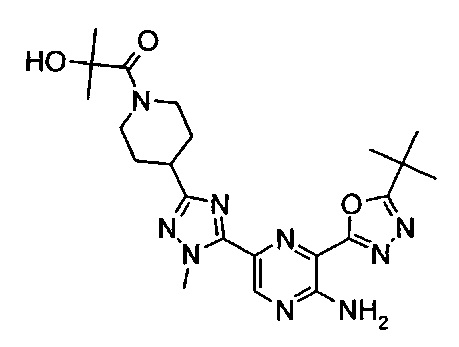
Гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (100 мг; 0,52 ммоль) добавляли в виде одной порции в атмосфере аргона к 2-гидрокси-2-метилпропановой кислоте (38,0 мг; 0,37 ммоль), 3-(5-трет-бутил-1,3,4-оксадиазол-2-ил)-5-(1-метил-3-(пиперидин-4-ил)-1Н-1,2,4-триазол-5-ил)пиразин-2-амину (100 мг; 0,26 ммоль) и N-оксиду 2-гидроксипиридина (57,9 мг; 0,52 ммоль), растворенным в NMP (1,2 мл). Полученный раствор перемешивали при 25°C в течение 3 часов. Добавляли пиридин (100 мкл; 1,24 ммоль) и смесь перемешивали в течение 18 часов. Добавляли дополнительное количество 1-оксида 2-гидроксипиридина (57,9 мг; 0,52 ммоль) и гидрохлорида 1-(3-диметиламинопропил)-3-этилкарбодиимида (100 мг; 0,52 ммоль). Затем смесь нагревали до 70°C в течение 48 часов, добавляли еще раз 2-гидрокси-2-метилпропановую кислоту (15 мг; 0,14 ммоль), гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (50,0 мг; 0,26 ммоль) и 1-оксид 2-гидроксипиридина (25,0 мг; 0,23 ммоль) и далее смесь выдерживали при 70°C в течение 8 часов. Раствор очищали препаративной HPLC с использованием колонки для обращенно-фазовой хроматографии X-Bridge от Waters (диоксид кремния С-18, 5 микрон; диаметр 19 мм, длина 100 мм, скорость потока 40 мл/минута) и смесей воды (содержащей 0,2% карбоната аммония) и ацетонитрила с уменьшающейся полярностью в качестве элюента, получая указанное в заголовке соединение (71 мг; 58%) в виде бледно-желтого твердого вещества.
1Н ЯМР-спектр: (CDCl3) 1.55 (15Н, s br), 1.90 (2Н, m), 2.15 (2Н, m), 3.05-3.3 (4Н, m), 4.32 (3H, s), 4.6 (1Н, m), 9.03 (1H, s); масс-спектр: [M+H]+ = 470.
Пример 8
3-[4-[5-[5-Амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил]-1-метил-1,2,4-триазол-3-ил]-1-пиперидил]-3-оксо-пропановая кислота
Этил-3-хлор-3-оксопропаноат (0,037 мл; 0,29 ммоль) по каплям добавляли к перемешиваемому раствору 3-(5-трет-бутил-1,3,4-оксадиазол-2-ил)-5-(1-метил-3-(пиперидин-4-ил)-1Н-1,2,4-триазол-5-ил)пиразин-2-амина (100 мг; 0,26 ммоль) и триэтиламина (0,047 мл; 0,34 ммоль) в CH2Cl2 (1,5 мл), в течение 2 минут при 0°C в атмосфере азота. Смесь перемешивали при 0°C в течение 10 минут, затем оставляли нагреваться до комнатной температуры и перемешивали в течение 1 часа. Смесь упаривали, растворяли в DMF; белое твердое вещество отфильтровывали и фильтрат очищали препаративной HPLC, используя колонку для обращенно-фазовой хроматографии X-Terra от Waters, с элюированием смесью воды (содержащей 0,2% карбоната аммония) и ацетонитрила, получая этил-3-(4-(5-(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-метил-1Н-1,2,4-триазол-3-ил)пиперидин-1-ил)-3-оксопропаноат (80 мг; 61,7%) в виде желтого твердого вещества. Это вещество суспендировали в THF (2 мл). Добавляли 2 н. гидроксид натрия (0,235 мл; 0,47 ммоль) и воду (0,5 мл). Смесь перемешивали при комнатной температуре в течение ночи. К смеси добавляли 2 н. соляную кислоту (230 мкл). Растворители выпаривали. Остаток разбавляли CH2Cl2 (30 мл) и водой (5 мл). Органическую фазу промывали рассолом и сушили над MgSO4. Растворители выпаривали. Полученную пену растирали в эфире. Полученное желтое твердое вещество отфильтровывали, сушили, растирали в ацетонитриле (3 мл). Желтое твердое вещество собирали фильтрованием, сушили при 40°C, получая указанное в заголовке соединение (50 мг; 68%) в виде желтого твердого вещества.
1Н ЯМР-спектр: (DMSO-d6) 1.46 (9Н, s), 1.58 (1Н, m), 1.74 (1Н, m), 1.98 (2Н, m), 2.84 (1Н, m), 3.0 (1Н, m), 3.21 (1Н, m), 3.46 (2Н, m), 3.83 (1Н, m), 4.22 (3H, s), 4.34 (1Н, m), 7.8-8.2 (1Н, m), 8.87 (1Н, s); масс-спектр: [М+Н]+ = 470.
Пример 9
3-[4-[5-[5-Амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил]-1-этил-1,2,4-триазол-3-ил]-1-пиперидил]-3-оксо-пропановая кислота
Гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (474 мг; 1,25 ммоль) порциями в течение 30 секунд добавляли к перемешиваемому раствору 3-этокси-3-оксопропановой кислоты (150 мг; 1,13 ммоль), N-этил-N-изопропилпропан-2-амина (0,394 мл; 2,26 ммоль) и 3-(5-(трет-бутил)-1,3,4-оксадиазол-2-ил)-5-(1-этил-3-(пиперидин-4-ил)-1H-1,2,4-триазол-5-ил)пиразин-2-амина (450 мг; 1,13 ммоль) в DMF (20 мл), при 50°C. Через 1 мин из полученного раствора отбирали образец (реакция завершилась) и незамедлительно оставляли охлаждаться до температуры окружающей среды. Реакционную смесь концентрировали, разбавляли EtOAc (100 мл) и промывали последовательно водой (20 мл) и насыщенным рассолом (20 мл). Органический слой сушили над MgSO4, фильтровали и упаривали, получая неочищенный этил-3-(4-(5-(5-амино-6-(5-(трет-бутил)-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-этил-1H-1,2,4-триазол-3-ил)пиперидин-1-ил)-3-оксопропаноат (850 мг).
Часть этого вещества (780 мг) растворяли в THF (20 мл). К этому раствору добавляли 2 н. водный раствор гидроксида натрия (2,3 мл; 4,57 ммоль) и воду (5 мл), затем метанол (5 мл), получая прозрачный раствор. Смесь перемешивали при комнатной температуре в течение 3 часов. THF выпаривали. Водный слой подкисляли до pH 3, используя 2 н. водную соляную кислоту (2,5 мл). Добавляли дихлорметан (50 мл) и органическую фазу экстрагировали. Органическую фазу промывали рассолом (10 мл) и сушили над MgSO4. Растворители выпаривали. Полученное смолообразное вещество очищали препаративной HPLC (препаративная колонка С18 OBD X-Bridge от Waters, диоксид кремния, 5 мкм; диаметр 50 мм, длина 100 мм), используя смеси воды (содержащей 1% аммиака) и ацетонитрила с уменьшающейся полярностью в качестве элюентов. Фракции, содержащие желаемое соединение, упаривали досуха, получая чистую соль аммония. Ее растворяли в воде и подкисляли до pH 3, используя 2 н. соляную кислоту (приблизительно 0,3 мл). Добавляли дихлорметан (50 мл) и органическую фазу отделяли, промывали рассолом (5 мл) и сушили над MgSO4. Полученный после фильтрования раствор упаривали досуха, остаток растирали с диэтиловым эфиром (5 мл) и проводили фильтрование, получая 3-(4-(5-(5-амино-6-(5-(трет-бутил)-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-этил-1H-1,2,4-триазол-3-ил)пиперидин-1-ил)-3-оксопропановую кислоту (195 мг; 26,5%) в виде кремового твердого вещества.
1Н ЯМР-спектр: (DMSO-d6) 1.45 (9Н, s), 1.48 (3H, m), 1.55-1.62 (1Н, m), 1.70-1.80 (1Н, m), 1.95-2.05 (2Н, m), 2.80-2.90 (1Н, m), 2.95-3.05 (1Н, m), 3.15-3.25 (1Н, m), 3.45 (2Н, s), 3.78-3.85 (1Н, m), 4.30-4.40 (1Н, m), 4.55-4.65 (2Н, m), 7.80-8.00 (2Н, brs), 8.88 (1Н, s), 12.60 (1Н, s); масс-спектр: [М+Н]+ = 484.
Пример 10
(2S)-1-[4-[5-[5-Амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил]-1-этил-1,2,4-триазол-3-ил]-1-пиперидил]-2,3-дигидрокси-пропан-1-он
К смеси 3-(5-(трет-бутил)-1,3,4-оксадиазол-2-ил)-5-(1-этил-3-(пиперидин-4-ил)-1Н-1,2,4-триазол-5-ил)пиразин-2-амина (257 мг; 0,50 ммоль; соль TFA), (S)-2,2-диметил-1,3-диоксолан-4-карбоксилата калия (101 мг; 0,55 ммоль) и EDCI (105 мг; 0,55 ммоль) в DCM (5 мл) добавляли гидрат 1-гидрокси-1H-бензотриазола (85 мг; 0,56 ммоль) и DIPEA (194 мг; 1,50 ммоль). Смесь перемешивали в течение 16 часов при комнатной температуре. К смеси добавляли воду и смесь экстрагировали, используя DCM. Органические слои промывали рассолом и сушили над Na2SO4, фильтровали и концентрировали, получая (S)-(4-(5-(5-амино-6-(5-(трет-бутил)-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-этил-1H-1,2,4-триазол-3-ил)пиперидин-1-ил)(2,2-диметил-1,3-диоксолан-4-ил)метанон (320 мг). Масс-спектр: [М+Н]+ = 526. К смеси (S)-(4-(5-(5-амино-6-(5-(трет-бутил)-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-этил-1H-1,2,4-триазол-3-ил)пиперидин-1-ил)(2,2-диметил-1,3-диоксолан-4-ил)метанона (320 мг) в DCM (10 мл) при кт по каплям добавляли TFA (1,6 мл; 20,77 ммоль). Смесь перемешивали в течение 16 ч при кт, концентрировали и очищали препаративной HPLC (препаративная колонка С18 OBD X-Bridge от Waters, диоксид кремния, 5 мкм; диаметр 19 мм, длина 100 мм), используя смеси воды (содержащей 0,1% NH3) и MeCN с уменьшающейся полярностью в качестве элюента. Фракции, содержащие желаемое соединение, упаривали досуха, получая указанное в заголовке соединение (142 мг; 48%) в виде белого твердого вещества: 1Н ЯМР-спектр (400 Гц, DMSO-d6, 30°C) 1.45 (12Н, m), 1.56 (1Н, m), 1.70 (1Н, m), 1.98 (2Н, m), 2.85 (1Н, m), 3.00 (1Н, m), 3.20 (1Н, m), 3.45 (1Н, s), 3.55 (1Н, s), 4.05 (1Н, m), 4.35 (2Н, m), 4.60 (2Н, m), 4.70 (1Н, m), 4.85 (1Н, m), 7.90 (2Н, m), 8.85 (1Н, s); масс-спектр: [М+Н]+ = 486.
Пример 11
(2R)-1-[4-[5-[5-Амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил]-1-этил-1,2,4-триазол-3-ил]-1-пиперидил]-2,3-дигидрокси-пропан-1-он
3-(5-(трет-Бутил)-1,3,4-оксадиазол-2-ил)-5-(1-этил-3-(пиперидин-4-ил)-1Н-1,2,4-триазол-5-ил)пиразин-2-амин приводили во взаимодействие с (R)-2,2-диметил-1,3-диоксолан-4-карбоксилатом калия, используя методику, аналогичную описанной в примере 10, с получением указанного в заголовке соединения (0,145 г; 40%) в виде твердого вещества. 1Н ЯМР-спектр (400 Гц, DMSO-d6, 30°C) 1.45 (12Н, m), 1.60 (2Н, m), 1.98 (2Н, m), 2.85 (1Н, m), 3.00 (1Н, m), 3.17 (1Н, m), 3.45 (1Н, s), 3.55 (1Н, s), 4.05 (1Н, m), 4.35 (2Н, m), 4.60 (2Н, m), 4.70 (1Н, m), 4.85 (1Н, m), 7.90 (2Н, m), 8.85 (1Н, s); масс-спектр: [М+Н]+ = 486.
Краткое описание Фигур
На Фиг. 1 показана картина дифракции рентгеновских лучей на порошке для Формы A соединения из примера 1.
На Фиг. 2 показана термограмма DSC для Формы A соединения из примера 1.
На Фиг. 3 показана картина дифракции рентгеновских лучей на порошке для Формы A соединения из примера 3.
На Фиг. 4 показана термограмма DSC для Формы A соединения из примера 3.
На Фиг. 5 показана картина дифракции рентгеновских лучей на порошке для Формы B соединения из примера 3.
На Фиг. 6 показана термограмма DSC для Формы B соединения из примера 3.
На Фиг. 7 показана картина дифракции рентгеновских лучей на порошке для Формы C соединения из примера 3.
На Фиг. 8 показана термограмма DSC для Формы C соединения из примера 3.
На Фиг. 9 показано ингибирование опухолевого роста под действием соединения из примера 3 в комбинации с ингибитором АКТ (AZD5363) - последовательное введение.
На Фиг. 10 показано ингибирование опухолевого роста под действием соединения из примера 3 в комбинации с ингибитором AKT (AZD5363), совместное введение.
На Фиг. 11 показано ингибирование опухолевого роста под действием соединения из примера 3 в комбинации с ингибитором PARP (олапарибом) в модели ксенотрансплантатов ВТ474.
На Фиг. 12 показано ингибирование опухолевого роста под действием соединения из примера 3 в комбинации с ингибитором PARP (олапарибом) в модели ксенотрансплантатов MCF7.
На Фиг. 13 показано ингибирование опухолевого роста под действием соединения из примера 3 в комбинации с AZD8186.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЗАМЕЩЕННЫЕ АМИНОПИРИМИДИНОВЫЕ СОЕДИНЕНИЯ И СПОСОБЫ ИХ ИСПОЛЬЗОВАНИЯ | 2014 |
|
RU2683793C2 |
| ПРОИЗВОДНЫЕ ТЕТРАГИДРОПИРИДО[3,4-b]ИНДОЛА, ПОЛЕЗНЫЕ ПРИ ЛЕЧЕНИИ РАКА | 2014 |
|
RU2664109C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ МОДУЛЯТОРЫ АКТИВНОСТИ HIF ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ | 2013 |
|
RU2698197C2 |
| ЗАМЕЩЕННЫЕ АМИНОПИРИМИДИНОВЫЕ СОЕДИНЕНИЯ И СПОСОБЫ ИХ ИСПОЛЬЗОВАНИЯ | 2014 |
|
RU2675105C9 |
| НОВОЕ ПРОИЗВОДНОЕ ПИПЕРИДИНА И СОДЕРЖАЩАЯ ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ИНГИБИРОВАНИЯ АУТОТАКСИНА | 2022 |
|
RU2834850C2 |
| КОНФОРМАЦИОННО ОГРАНИЧЕННЫЕ ИНГИБИТОРЫ PI3K И mTOR | 2014 |
|
RU2669696C2 |
| ИНГИБИТОРЫ АКТИВНОСТИ АКТ | 2006 |
|
RU2421454C2 |
| СПИРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ | 2016 |
|
RU2745069C2 |
| ИНГИБИТОРЫ ДЕМЕТИЛАЗЫ LSD1 НА ОСНОВЕ АРИЛЦИКЛОПРОПИЛАМИНА И ИХ ПРИМЕНЕНИЕ В МЕДИЦИНЕ | 2011 |
|
RU2611437C2 |
| ПИРАЗОЛО[3,4-b]ПИРИДИНОВЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ФОСФОДИЭСТЕРАЗ | 2003 |
|
RU2348633C2 |
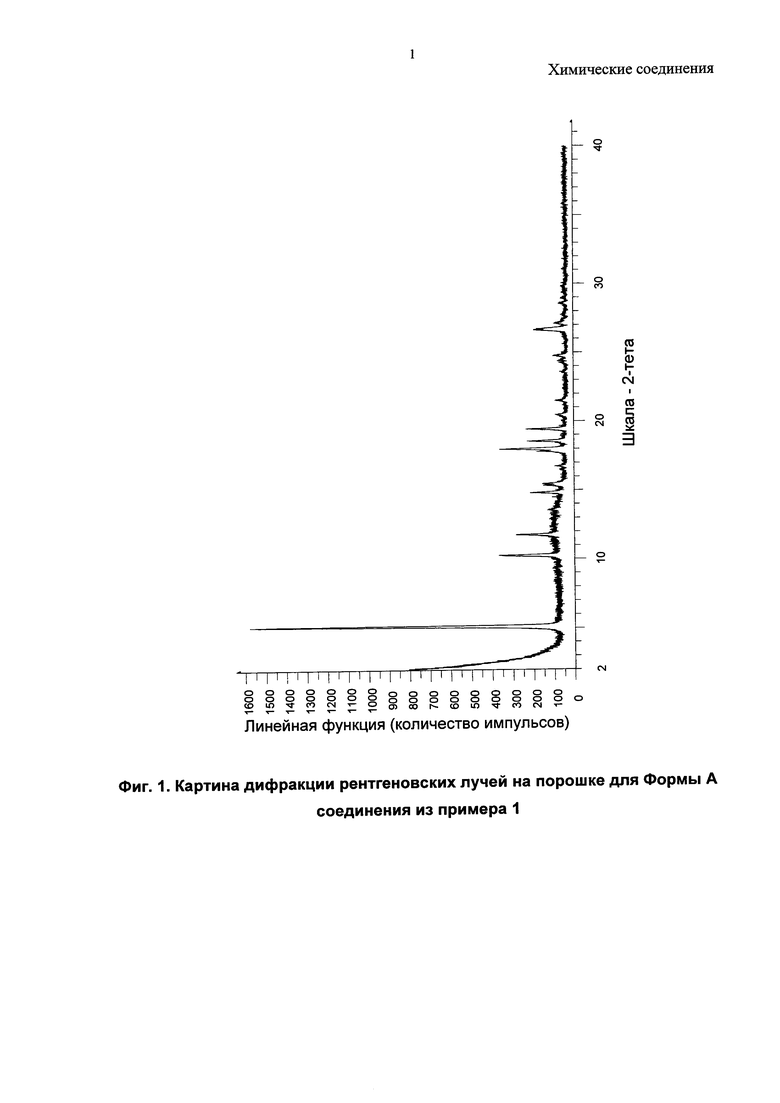
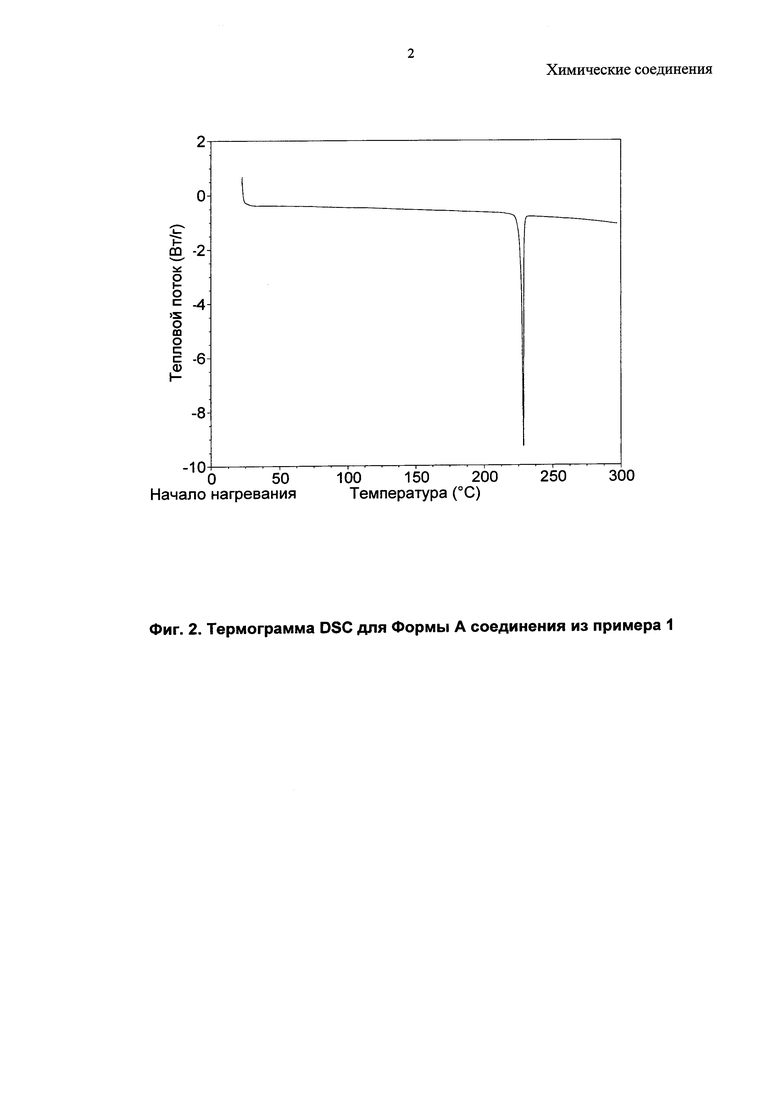
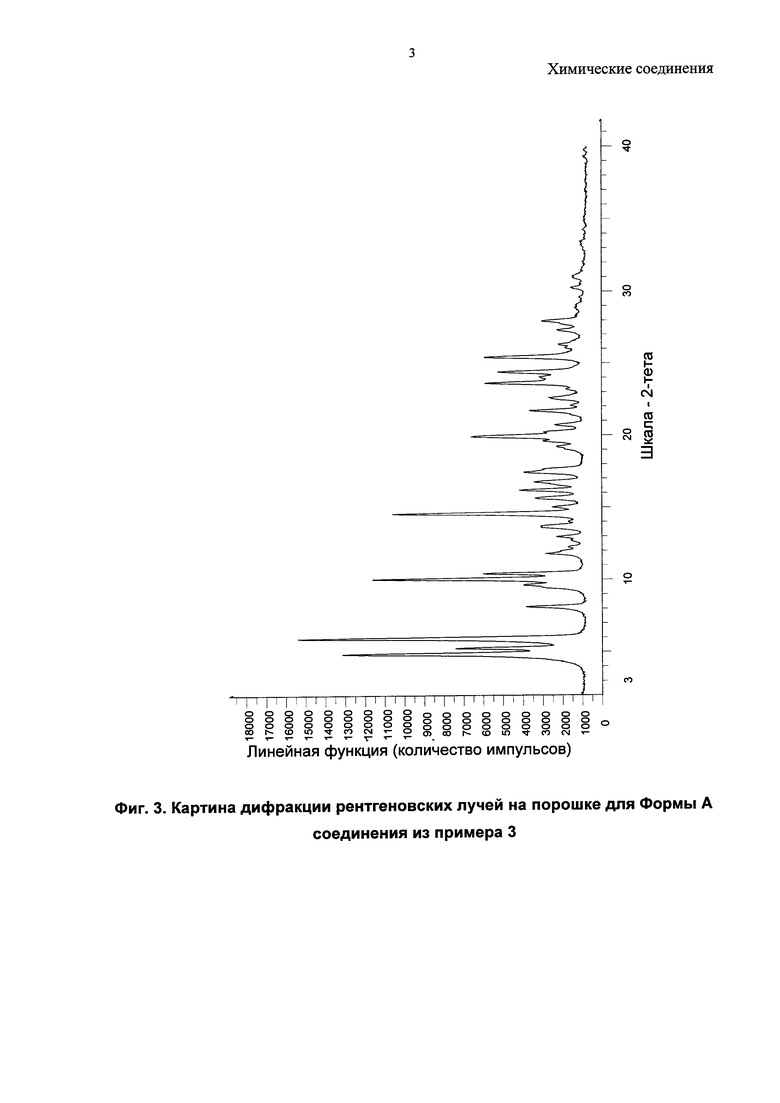
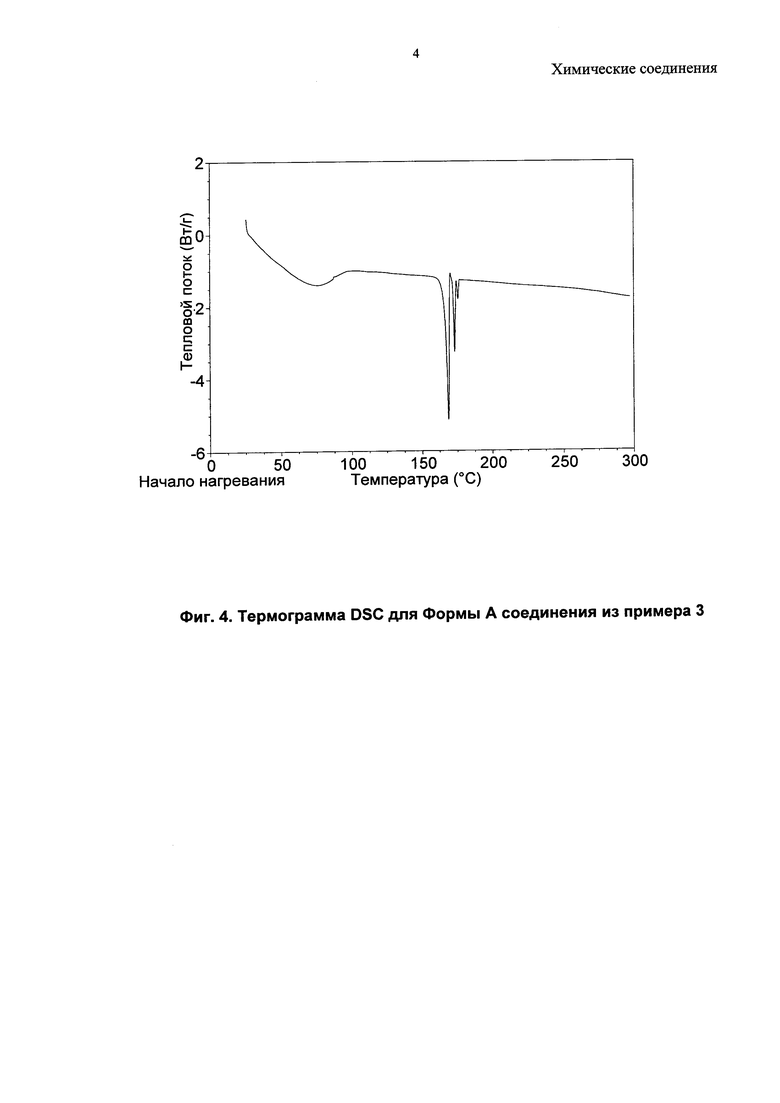
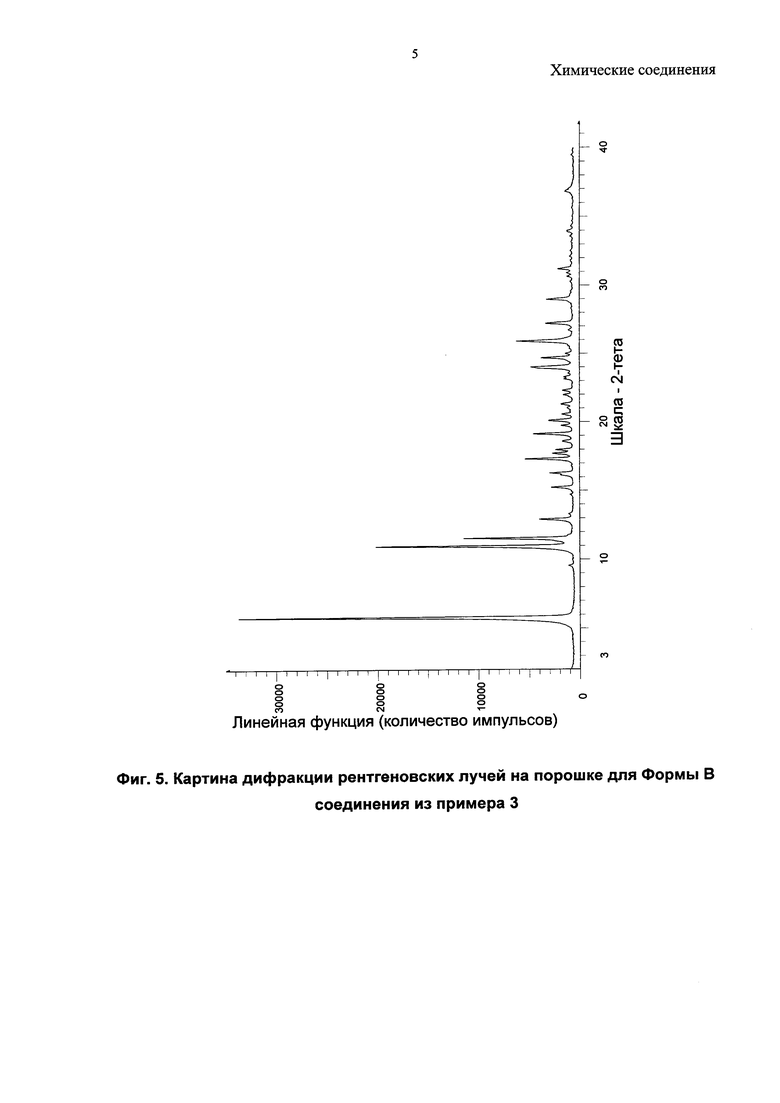
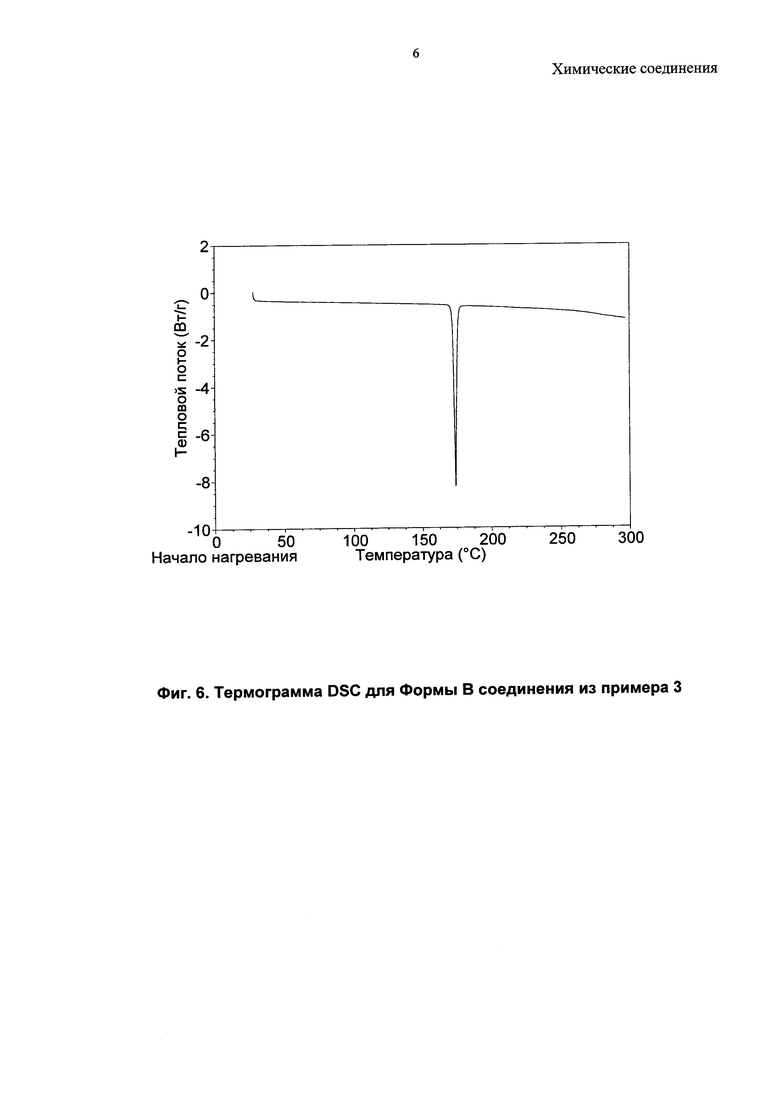
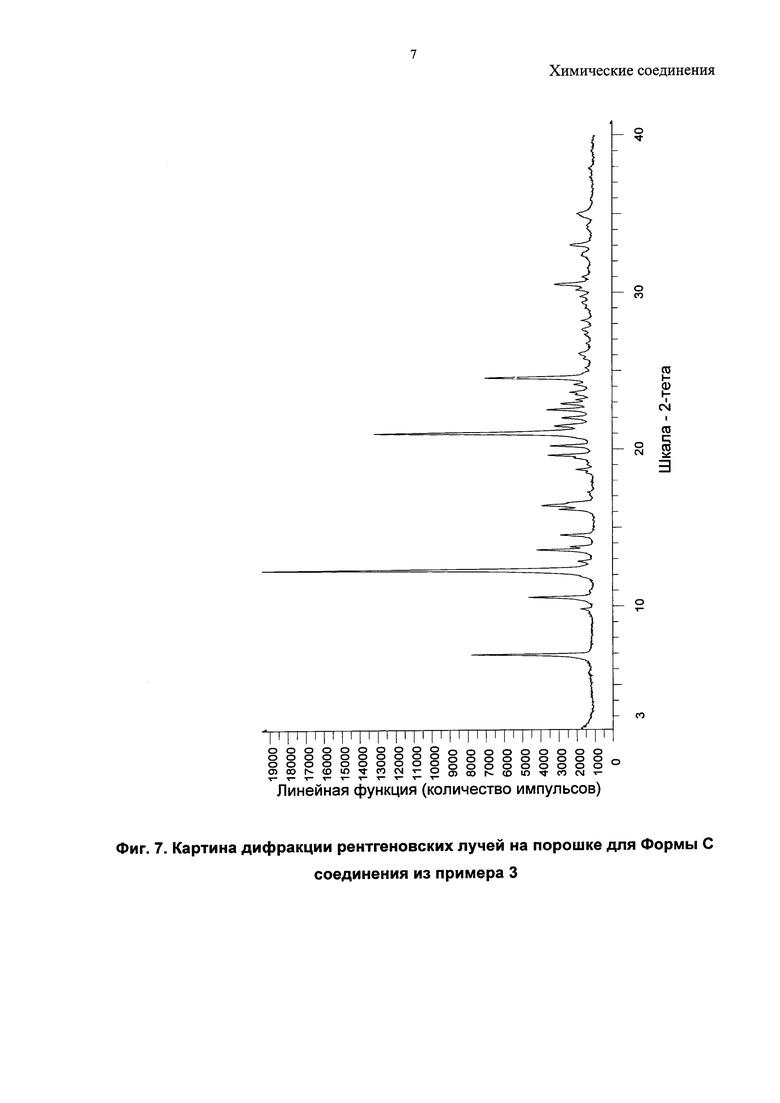
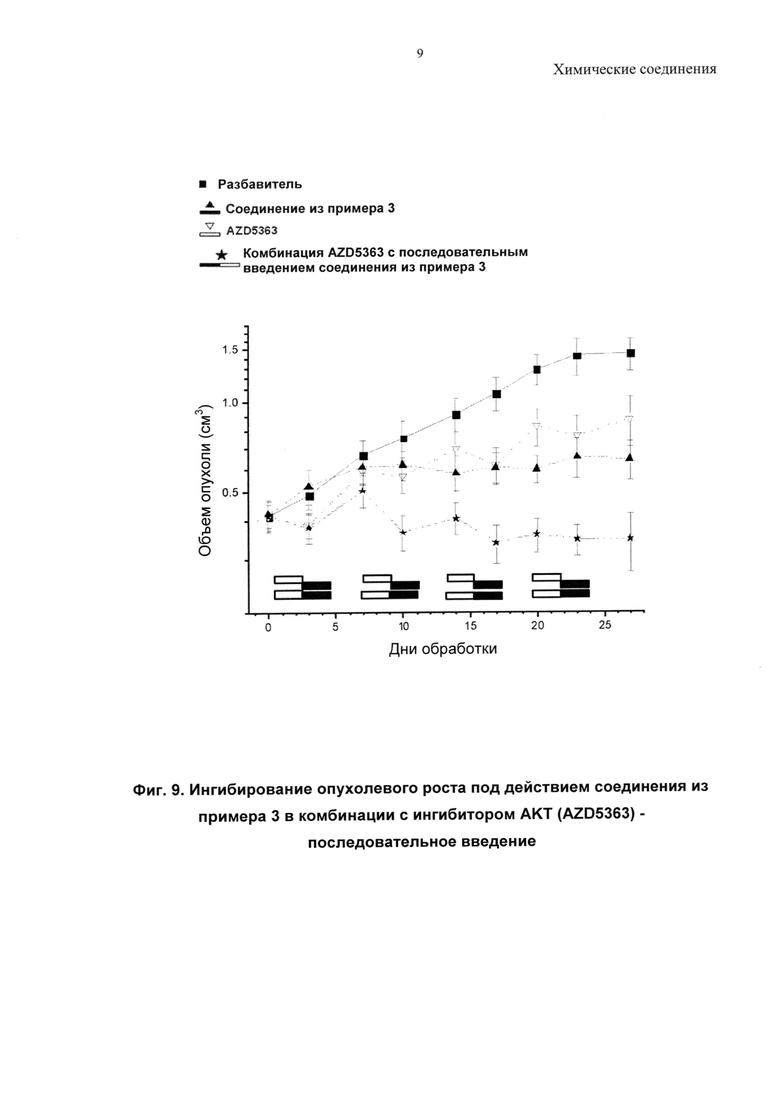
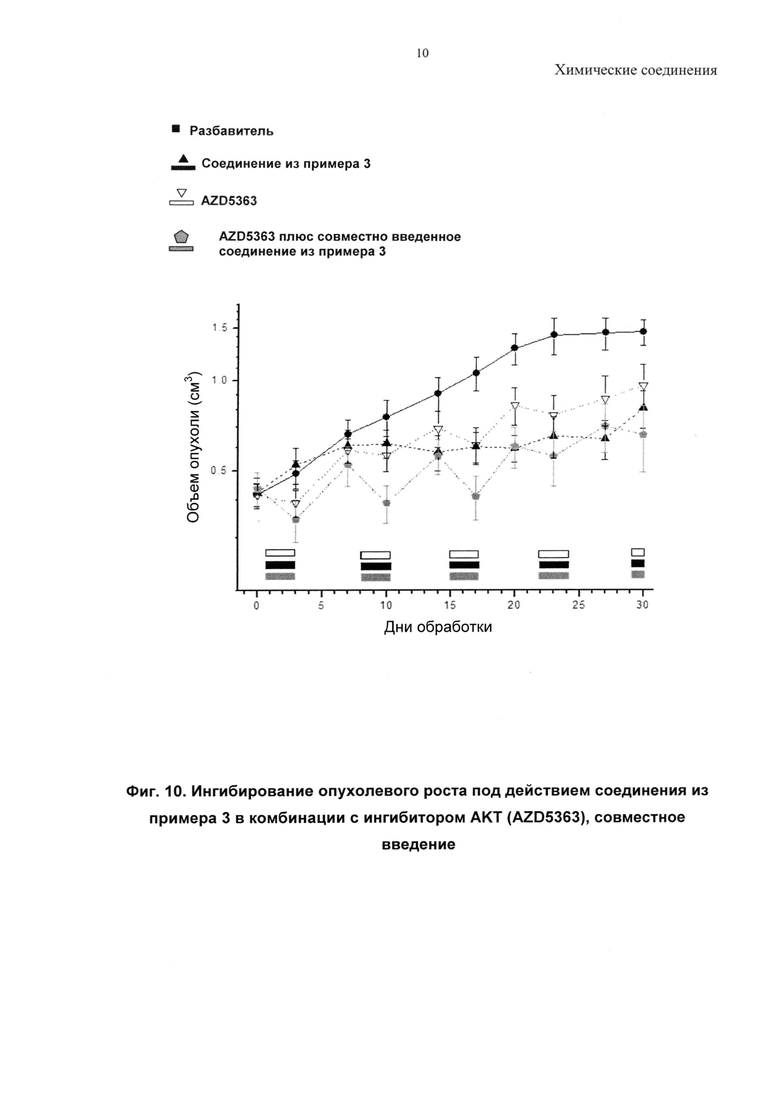
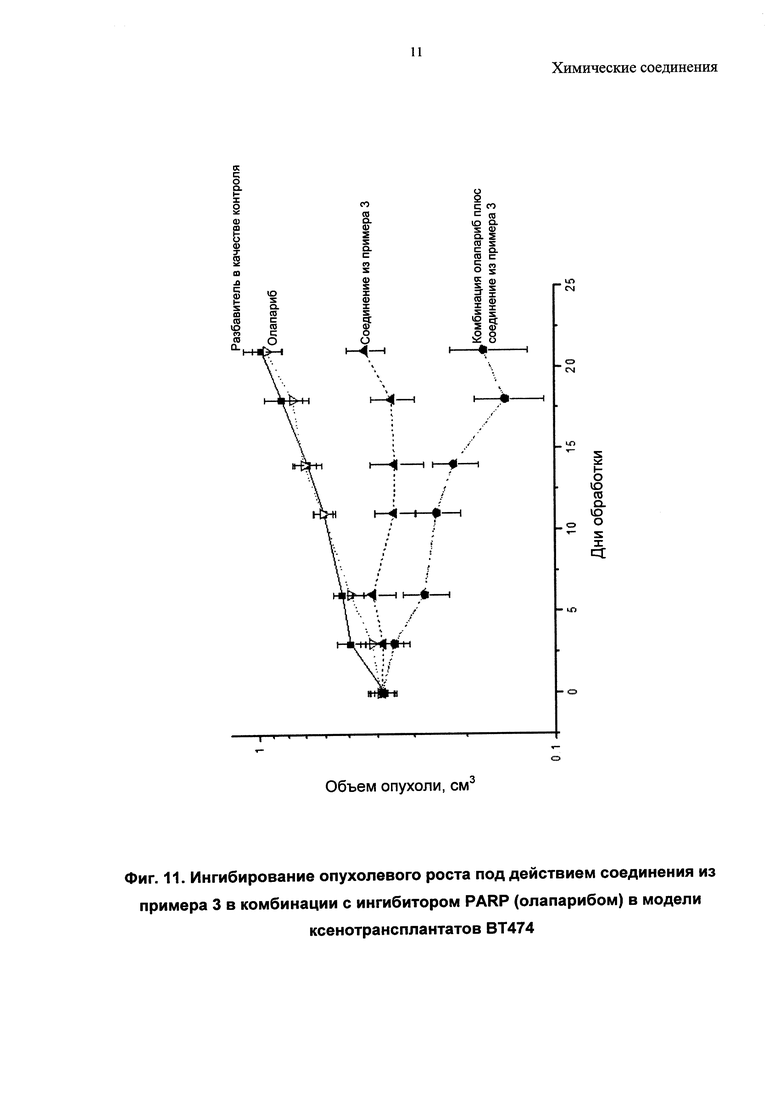
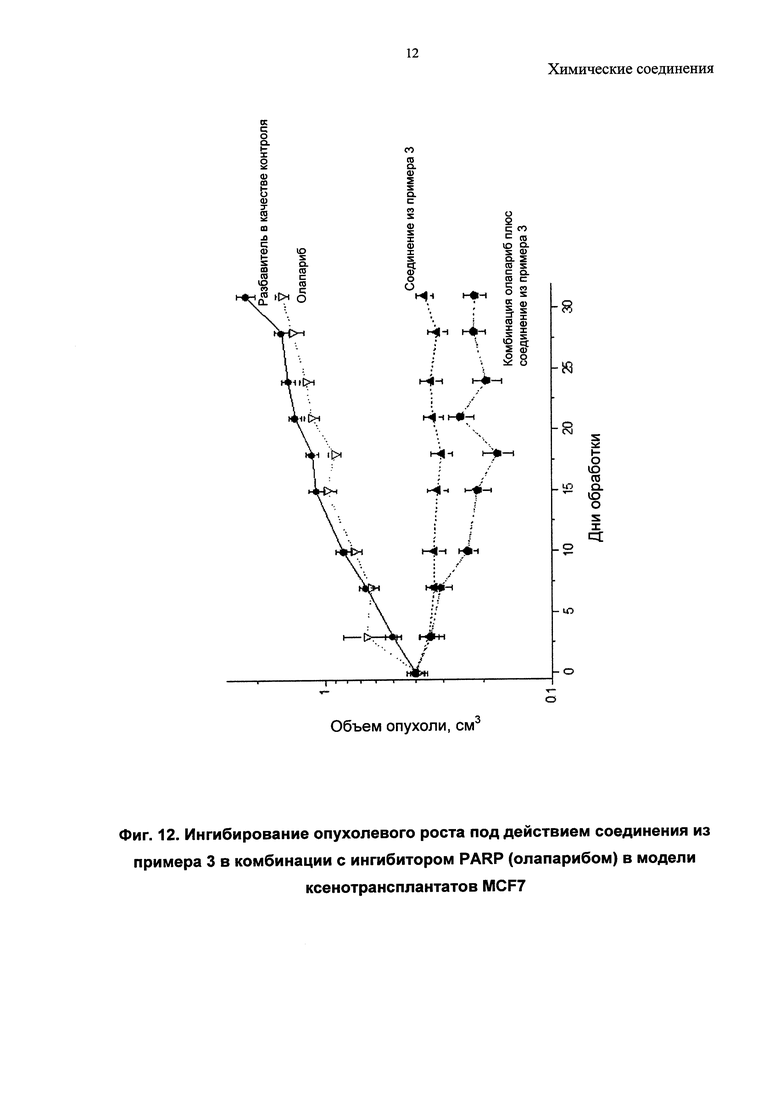
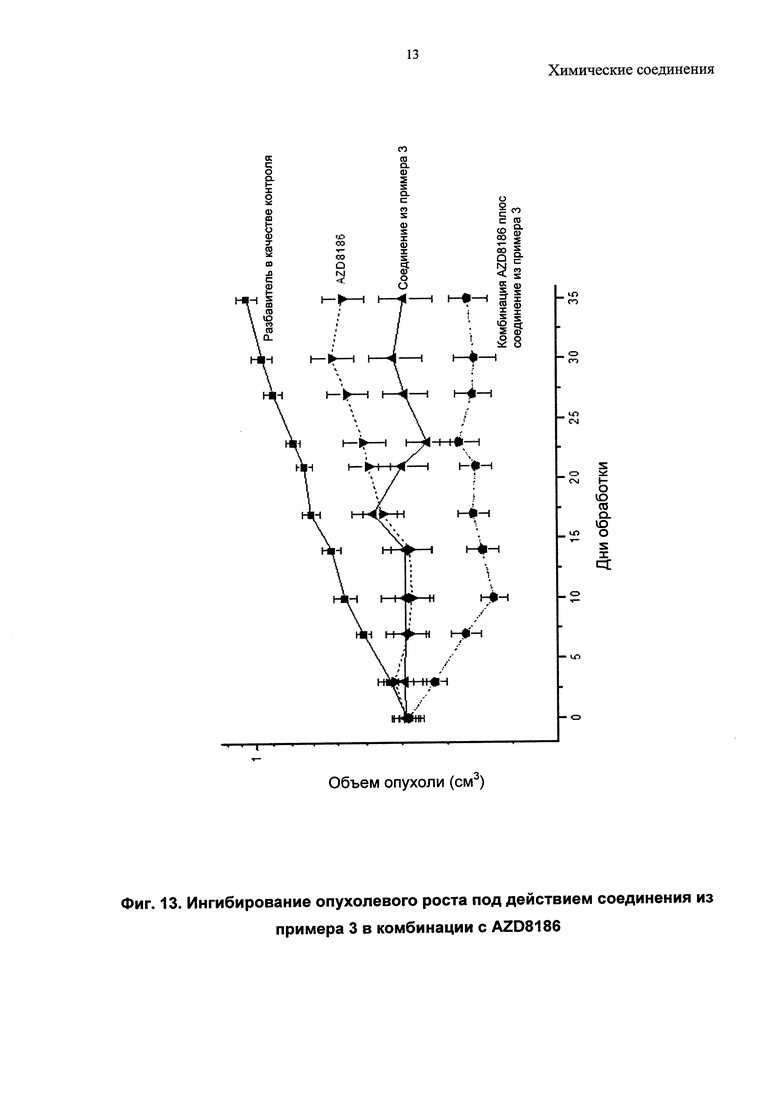
Изобретение относится к соединению формулы (I)
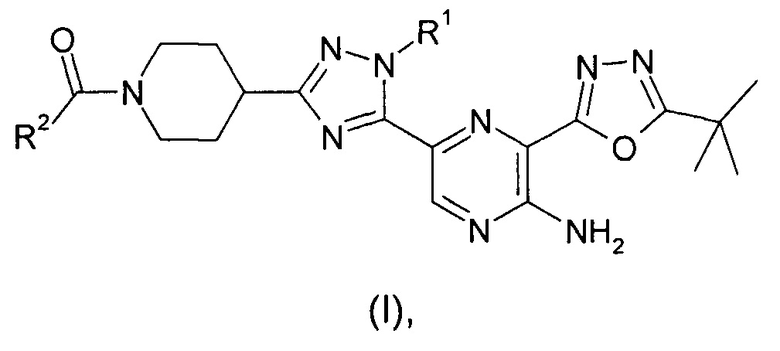
в которой R1 представляет собой метил или этил и R2 представляет собой (С2-3)алкил, замещенный группой гидрокси; или его фармацевтически приемлемой соли. Также изобретение относится к соединению, представляющему собой 1-[4-[5-[5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил]-1-этил-1,2,4-триазол-3-ил]-1-пиперидил]-3-гидроксипропан-1-он, к комбинации, подходящей для применения в лечении рака, опосредованного PI3K-α или PI3K-δ, к фармацевтической композиции, к способу отбора пациента для лечения рака и к способу лечения пациента, страдающего раком. Технический результат: получены новые соединения, обладающие свойствами ингибитора киназы PI3K-α или PI3K-δ, которые могут быть полезны при лечении рака. 6 н. и 11 з.п. ф-лы, 13 ил., 9 табл., 11 пр.
1. Соединение формулы (I)

где
R1 представляет собой метил или этил; и
R2 представляет собой (С2-3)алкил, замещенный группой гидрокси;
или его фармацевтически приемлемая соль.
2. Соединение по п. 1 или его фармацевтически приемлемая соль, где R2 выбран из групп (1)-(11):
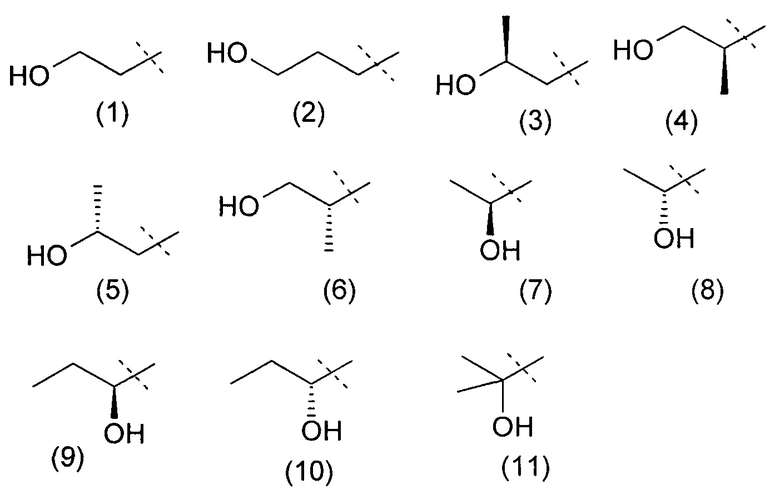
3. Соединение по п. 2 или его фармацевтически приемлемая соль, где R2 выбран из групп (1)-(6), как они определены в п. 2.
4. Соединение по п. 3 или его фармацевтически приемлемая соль, где R2 представляет собой группу (1), как она определена в п. 2.
5. Соединение по п. 1 или его фармацевтически приемлемая соль, где R1 представляет собой метил.
6. Соединение по п. 1 или его фармацевтически приемлемая соль, где R1 представляет собой этил.
7. Соединение по п. 1 или его фармацевтически приемлемая соль, где соединение выбрано из:
1-[4-[5-[5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил]-1-метил-1,2,4-триазол-3-ил]-1-пиперидил]-3-гидроксипропан-1-она;
1-[4-[5-[5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил]-1-этил-1,2,4-триазол-3-ил]-1-пиперидил]-3-гидроксипропан-1-она;
(3R)-1-[4-[5-[5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил]-1-метил-1,2,4-триазол-3-ил]-1-пиперидил]-3-гидроксибутан-1-она;
(3S)-1-[4-[5-[5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил]-1-метил-1,2,4-триазол-3-ил]-1-пиперидил]-3-гидроксибутан-1-она;
(2R)-1-[4-[5-[5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил]-1-метил-1,2,4-триазол-3-ил]-1-пиперидил]-3-гидрокси-2-метил-пропан-1-она;
1-[4-[5-[5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил]-1-метил-1,2,4-триазол-3-ил]-1-пиперидил]-2-гидрокси-2-метил-пропан-1-она.
8. Соединение, представляющее собой 1-[4-[5-[5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил]-1-этил-1,2,4-триазол-3-ил]-1-пиперидил]-3-гидроксипропан-1-он, или его фармацевтически приемлемая соль.
9. Соединение по п. 8, представляющее собой 1-[4-[5-[5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил]-1-этил-1,2,4-триазол-3-ил]-1-пиперидил]-3-гидроксипропан-1-он.
10. Соединение по п. 1 или его фармацевтически приемлемая соль в кристаллической форме.
11. Соединение по п. 1 или его фармацевтически приемлемая соль для применения в качестве лекарственного средства для лечения заболеваний, опосредованных фосфатидилинозитол-3-киназой альфа (PI3K-α) или фосфатидилинозитол-3-киназой дельта (PI3K-δ).
12. Соединение по п. 1 или его фармацевтически приемлемая соль для применения в предупреждении или лечении рака, опосредованного PI3K-α или PI3K-δ, у теплокровного животного, такого как человек.
13. Комбинация, подходящая для применения в лечении рака, опосредованного PI3K-α или PI3K-δ, содержащая соединение формулы (I) по п. 1 или его фармацевтически приемлемую соль и другой противоопухолевый агент, выбранный из ингибитора АКТ-протеинкиназы AZD5363, ингибитора поли(АДФ-рибоза)полимеразы (PARP) олапариба и ингибитора РI3Kбета/дельта AZD8186.
14. Фармацевтическая композиция для ингибирования киназы PI3K-α или PI3K-δ, содержащая эффективное количество соединения формулы (I) или его фармацевтически приемлемой соли по п. 1 и фармацевтически приемлемый разбавитель или носитель.
15. Способ отбора пациента для лечения рака, опосредованного PI3K-α или PI3K-δ, соединением формулы (I), включающий получение от пациента образца, содержащего опухолевую ДНК или опухолевые клетки; определение того, является ли ген PIK3CA в опухолевых клетках пациента или в опухолевой ДНК пациента геном дикого типа или мутантным геном; и на основании этого отбор пациента для лечения соединением формулы (I).
16. Способ лечения пациента, страдающего раком, опосредованным PI3K-α или PI3K-δ, включающий: получение содержащего опухолевые клетки образца от пациента; определение того, является ли ген PIK3CA в опухолевых клетках пациента геном дикого типа или мутантным геном; и введение пациенту эффективного количества соединения формулы (I) по п. 1, если опухолевые клетки содержат мутантный ген PIK3CA.
17. Соединение формулы (I) по п. 1 для лечения раковых заболеваний, опосредованных PI3K-α или PI3K-δ, с опухолевыми клетками, идентифицированными как несущие мутантный ген PIK3CA.
| EA 201100447 A1, 31.10.2011 | |||
| EA 201100778 A1, 30.01.2012 | |||
| WO 2010071837 A1, 24.06.2010 | |||
| WO 2010091824 A1, 19.08.2010 | |||
| WO 2012037226 A1, 22.03.2012. |

