ССЫЛКА НА АНАЛОГИ
[001] Прототипами настоящей заявки являются предварительная заявка США №61/880,974, поданная 22 сентября 2013, и предварительная заявка США №61/983,444, поданная 23 апреля 2014, обе упомянутые предварительные заявки полностью включены в настоящую заявку для справки.
ОБЛАСТЬ ИЗОБРЕТЕНИЯ
[002] Заявляются конкретные новые соединения, являющиеся ингибиторами киназы, также заявляются способы их изготовления, фармацевтические композиции, содержащие заявляемые соединения, и способы использования соединений или фармацевтических композиций для лечения различных расстройств. Более конкретно, заявляемые соединения являются ингибиторами активности или функциональности семейства фосфатидилинозитов 3-киназы (далее РI3-киназы), например PI3Kδ, PI3Kα, PI3Kβ и/или РI3Kγ.
[003] Поэтому заявляемые соединения потенциально эффективны для лечения широкого ряда нарушений, в частности нарушений, включая, но не ограничиваясь перечисленными далее, аутоиммунные нарушения, воспалительные заболевания, аллергические заболевания, заболевание или иммунопатологию инфекционного происхождения, заболевания дыхательных путей, такие как астма и COPD, (ХОБЛ), отторжение трансплантата, карциномы, например, гематопоэтического происхождения или солидные опухоли.
[004] Заявляемые соединения являются ингибиторами активности или функциональности РI3-киназ и могут использоваться в лечении общих воспалительных заболеваний, артрита, ревматизмов, остеоартрита, воспалительных болезней кишечника, воспалительных заболеваний глаз, воспалительных или лабильных заболеваний мочевого пузыря, псориаза, кожных заболеваний с воспалительными компонентами, хронических воспалительных состояний, включая, но не ограничиваясь аутоиммунными нарушениями, такими как системная красная волчанка (SLE), миастения (myasthenia gravis), ревматоидный артрит, острый диссеминированный энцефаломиелит, идиопатическая тромбоцитопеническая пурпура (ИТП, болезнь Верльгофа), рассеянный склероз, синдром Шегрена и аутоиммунная гемолитическая анемия, аллергические состояния, включая все формы гиперчувствительности; заболевания дыхательных путей, такие как астма, хроническая обструктивная болезнь легких (COPD, ХОБЛ) и идиопатический легочный фиброз (IPF); вирусных инфекций, включая вирусные инфекции дыхательных путей и вирусные обострения заболеваний дыхательных путей, такие как астма и COPD; инфекции дыхательных путей невирусного происхождения, включая аспергиллез и лейшманиоз, сердечно-сосудистые заболевания, включая тромбоз и атеросклероз; нейродегенеративные заболевания; панкреатит; синдром полиорганной недостаточности (СПОН, ПОН); нефропатия; седиментация тромбоцитов; подвижность сперматозоидов; отторжение трансплантата, отторжение ткани, поражение легких, и боль, вызываемые ревматоидным артритом, остеоартритом, боль в пояснице, боль, вызываемая общим воспалительным процессом, постгепатитная невралгия, диабетическая нейропатия, воспалительная невропатическая боль (травма), тригеминальная невралгия и центральная боль; гемобластозы, такие как острый миелоидный лейкоз (AML, ОМЛ), миело-дисплазированный синдром (MDS), миелопролиферативные заболевания (MPD), хронический миелоидный лейкоз (CML, ХМЛ), Т-клеточный острый лимфобластный лейкоз (T-ALL), В-клеточный острый лимфобластный лейкоз (B-ALL), Неходжкинские лимфомы (NHL, НХЛ), В-клеточная лимфома и солидные опухоли, такие как рак молочной железы.
ПРОТОТИПЫ
[005] Фосфоинозитиды 3-киназы (РI3-киназы или PI3Ks) являются семейством липидных киназ, которые, как обнаружилось, обладают главными регулирующими свойствами во многих процессах клеточного уровня, включая живучесть клетки, разрастание и дифференцировку. В качестве основных эффекторов, расположенных следом за клеточными рецепторами тирозинкиназ (RTK) и рецепторами, сопряженными с G-белком (GPCR), PI3K преобразуют сигналы от различных факторов роста и цитокинов во внутриклеточные сокращения посредством выработки фосфолипидов, которые активируют серин-треонин протеиновую киназу АКТ (известную также как протеиновая киназа В (РКВ)) и другие пути эффекторов, расположенных далее. Опухолевый супрессор или PTEN (гомолог фосфатазы и тензина) является наиболее важным отрицательным регулятором сигнального пути PI3K («Маломолекулярные ингибиторы цепи сигнальной сети PIK». Future Med Chem., 2011, 3, 5, 549-565).
[006] К настоящему времени установлено восемь PI3K млекопитающих, распределенных по трем основным классам (I, II и III), исходя из генной последовательности, структуры, молекул-адаптеров, проявления, режима активирования и предпочтительного субстрата. Из них I класс PI3K дополнительно подразделяется на класс IA и класс IB, исходя из сигнальных каналов и регуляторных белков. PI3K класса IA содержат три близкородственные киназы, PI3Kα, PI3Kβ и PI3Kδ, существующие как гетеродимеры, состоящие из каталитической подгруппы (р110α, р110β и p110δ соответственно) и р85 регулирующих адаптерных подгрупп (например, р85α, р85β, р55δ, р55α и р50α). Каталитическая подгруппа p110 использует АТР для фосфорилирования фосфатидилинозитола (РI, Ptdlns), PI4P и PI (4,5) P2. Эти ответы на сигналы обычно происходят посредством рецепторных тирозиновых киназ (RTK). Сигналы класса IB РI3Kγ проходят через рецепторы, сопряженные с G-белком (GPCR) и содержат каталитический домен р110γ, способный соединяться с регулирующими подгруппами, отличающимися от изоформ класса IA.
[007] В зависимости от функции и регулирования ферментов эффектора в фосфолипидных сигнальных путях, РI3-киназы класса I (например, PI3Kδ, PI3Kδ) создают вторичных посредников из мембранных фосфолипидных пулов. PDK класса I преобразуют мембранный фосфолипид PI(4,5) P2 в PI(3,4,5)Р3, который действует как вторичный посредник. PI и РI(4)Р являются также субстратами PI3K и могут быть фосфорилированы и преобразованы в PI3P и РI(3,4)Р2 соответственно. Кроме того, эти фосфоинозитиды могут быть преобразваны в другие фосфоинозитиды с помощью 5'-специфичных и 3'-специфичных фосфатаз. Таким образом, ферментная активность PI3K прямо или косвенно сказывается на формировании двух 3'-фосфоинозитидных субтипов, действующих как вторичные посредники в каналах внутриклеточной передачи сигналов (Nature Reviews Molecular Cell Biology, 2010, 11, 329).
[008] Формула изоформ PI3Kα и PI3Kβ широко распространена, тогда как модель формулы PI3Kδ и РI3Kγ кажется более ограниченной, причем обе изоформы первично обнаруживаются в лейкоцитах. Относительно ограниченная модель формулы PI3Kδ и РI3Kγ, в дополнение к данным, полученным из исследований на мышах, позволяет предположить, что эти две изоформы играют главную роль в адаптивных и врожденных иммунных системах (J. Med. Future Med Chem., 2012, 55, 20, 8559-8581).
[009] В В-клетках и Т-клетках РI3K-киназы играют важную роль за счет активации Tec-семейства протеинтирозинкиназ, в число которых входит тирозинкиназа Брутона (ВТК) в В-клетках и интерлейкин-2-индуцируемой Т-клеточной киназы (ITK) в Т-клетках. Когда PI3K активируется, ВТК или ITK перемещаются на плазменную мембрану, где они затем фосфорилирилируются Src киназами. Одной из главных целей активированной ITK является фосфолипаза С-гамма (PLCγ1), которая гидролизирует РI(4,5)Р2 в РI(3,4,5)Р3 и побуждает внутриклеточое увеличение в кальциевых уровнях и диаглицерол (DAG), который может активировать протеинкиназы C в активированных Т-клетках.
[010] PI3Kδ киназы мертвой мыши жизнеспособны, их фенотип ограничен нарушениями иммунной сигнализации (Okkenhaug и соавторы. Science, 2002, 297, р. 1031-4). Такие трансгенные мыши позволяют ознакомиться с функцией сигнализации PI3Kδ в В-клетке и Т-клетке. В частности, PI3Kδ требуется для формирования РI(3,4,5)Р3 следом за сигнализацией CD28 и/или рецептора Т-клетки (TCR). Главный эффект сигнализации PI3K следом за TCR заключается в активации Akt, который фосфорилирует анти-апоптичесие факторы, а также различные транскрипционные факторы для формирования цитокинов. Как следствие, Т-клетки с неактивной PI3Kδ имеют нарушения в пролиферации и секреции ТМ и Th2 цитокинов. Активирование Т-клеток посредством CD28 снижает порог активации TCR за счет антигена и увеличивает величину и продолжительность пролиферативного ответа. Эти эффекты опосредуются РI3Kδ-обусловленным увеличением в транскрибировании некоторого количества генов, включая including IL2, важного фактора роста Т-клетки.
[011] Поэтому ожидается, что PI3K ингибиторы обеспечат терапевтическое преимущество за счет их роли в опосредовании воспалительных реакций, опосредуемых Т-клетками, связанных с заболеваниями дыхательных путей, такими как астма, COPD и кистозный фиброз. Кроме того, имеется свидетельство, что направленная терапия Т-клетки может обеспечить экономию кортистероидов (Lancet, 1992, 339, р. 324-8), в предположении, что она может обеспечить практичное лечение автономно или в сочетании с ингаляцией или оральным приемом глюкокортикоидов при заболеваниях дыхательных путей. Ингибитор PI3K может также использоваться наряду с общепринятыми методами лечения, например, бета-антагонисты длительного действия (LABA)при астме.
[012] В системе кровообращения PI3Kδ проявляется посредством эндотелиальных клеток и участвует в нейтрофильной направленной миграции опосредованием проадгезивного состояния этих клеток в ответ на ТNFальфа (Blood, 2004, 103, 9, с.3448). Роль PI3Kδ в TNFальфа-индуцированной сигнализации эндотелиальных клеток демонстрируется фармакологическим подавлением фосфорилирования Akt и активности PDK1. Кроме того, PI3Kδ непосредственно связана с васкулярной проницаемостью и отеком тканей дыхательных путей через сигнальный путь VEGF (Allergy Clin. Immunol, 2006, 118, 2, с. 403). Эти наблюдения предполагают дополнительные преимущества PI3Kδ ингибирования в астме за счет комбинированного снижения диапедеза и васкулярной проницаемости, связанных с астмой. В дополнение к этому, активность PI3Kδ требуется для функционирования мастоцитов как in vitro, так in vivo (Nature, 2004, 431, p. 1007; J. Immunol, 2008, 180, 4, c. 2538), с дополнительным предположением, что PI3K ингибирование должно обладать терапевтическим преимуществом для аллергических проявлений, таких как астма, аллергические риниты и атопические дерматиты.
[013] Роль PI3Kδ в пролиферации В-клеток, секреции антител, антигена В-клетки и сигнализации IL-4 рецептора, функции представления антигена В-клетки также хорошо установлена (J. Immunol, 2007, 178, 4, р. 2328-35; Blood, 2006, 107, 2, с. 642-50) и обозначает роль в аутоиммунных заболеваниях, таких как ревматоидные артриты или системная красная волчанка. Поэтому PI3K ингибиторы могут также обладать преимуществами для этих показаний.
[014] Фармакологическое ингибирование PI3Kδ подавляет fMLP-зависимый нейтрофильный хемотаксис на покрытой ICAM агарозной матричной интегрин-зависимой смещенной системе (J. Immunol, 2003, 170, 5, с. 2647-54). Ингибирование PI3Kδ регулирует нейтрофильную активацию, адгезию и миграцию, на влияя на нейтрофильно опосредованный фагоцитоз и бактерициднцю активность золотистого стафилококка (Biochem. Biophys. Res. Commun, 2003, 308, 4, с. 764-9). В целом, данные позволяют предположить, что ингибирование PI3Kδ не должно полностью подавлять нейтрофильные функции, необходимые для защиты врожденного иммунитета. Роль PI3Kδ в нейтрофилах предоставляет дополнительную область для лечения воспалительных заболеваний, включая ремоделирование ткани, такое как COPD или ревматиоидные артриты.
[015] Установлено, что РI3Kγ представляет собой регулятор G бета-гамма-зависимого регулирования активности JNK, и G бета-гамма являются подгруппами гетеротриметрических G протеинов (J. Biol. Future Med Chem., 1998, 273, 5, 2505-8). Ранее упоминалось, что РI3Kγ передает воспалительные сигналы через различные G(i)-связанные рецепторы и является центральной по отношению к функционированию мастоцитов, возбудителем в контексте лейкоцитов, а также иммунологии, включая цитокины, хемокины, аденозины, антитела, интегрины, факторы седиментации, факторы роста, вирусы или гормоны, например (Immunity, 2002, 16, 3, p. 441-51; J. Cell Set, 2001, 114 (Pt 16), с. 2903-10 и Curr. Opinion Cell Biol, 2002, 14, 2, 203-13).
[016] В настоящее время хорошо известно, что дерегуляция онкогенов и опухолевых супрессорных генов способствует развитию злокачественных опухолей, например, за счет усиленного роста клеток и пролиферации или возросшего выживания клеток. Известно также, что сигнальные пути, опосредуемые семейством PI3K, играют основную роль в клеточных процессах, включая пролиферацию и выживание, и дерегуляция этих путей является этиологическим фактором широкого спектра злокачественных опухолей человека и других заболеваний (Annual Rev. Cell Dev. Biol, 2001, 17, с. 615-675 и J. Cell Science, 2003, 116, 15, c. 3037-3040).
[017] Имеется благоприятное проявление того, что ферменты PI3K I класса способствуют онкогенезу широкого спектра злокачественных опухолей человека, как прямо, так и косвенно (Nature Reviews Cancer, 2002, 2, 7, с. 489-501). Например, ингибирование PI3Kδ может играть терапевтическую роль в лечении гемобластозов, таких как острый миелоидный лейкоз (Oncogene, 2006, 25, 50, с. 6648-59). Более того, активирование внутренних мутаций p110α (ген PIK3CA) связывается с другими опухолями различной локализации, например в прямой кишке, грудной полости и легких (Science, 2004, 304, 5670, с. 554; Nature Reviews Cancer, 2009, 9, 551).
[018] Продемонстрировано также, что PI3K участвует в создании центральной сенсибилизации в болезненном воспалительном состоянии (J. of Neuroscience, 2008, 28, 16, с. 4261-4270).
[019] Широкий спектр ретровирусов и вирусов на основе ДНК активируют путь PI3K как путь предотвращения гибели клетки организма-носителя в процессе вирусной инфекции и полного использования механизма синтеза клетки организма-носителя для своей репликации (Virology, 2006, 344, 1, с. 131-8 и Nat. Rev. Microbiol, 2008, 6, 4, с. 265-75). Поэтому ингибиторы PI3K могут обладать противовирусными свойствами в добавление к более обоснованным онколитическим и противовоспалительным показаниям. Такие антивирусные эффекты увеличивают интересные перспективы в воспалительных обострениях, вызванных вирусами. Например, более чем 50% инфекционных респираторных заболеваний вызывается простудным риновирусом человека (HRV), однако осложнения этих инфекционных заболеваний могут быть значительными в определенных группах населения. Особенно это касается респираторных заболеваний, таких как астма или хроническая обструктивная болезнь легких (COPD, ХОБЛ). Риновирусное инфицирование эпителиальных клеток приводит к РI3K-зависимой секреции цитокинов или хемокинов (J. Biol. Chem., 2005, 280, 44, с. 36952). Эта воспалительная реакция коррелирует с ухудшением респираторных симптомов в ходе инфекционного заболевания. Поэтому ингибиторы PI3K способны ослабить преувеличенную иммунную реакцию на другой безвредный вирус. Большинство штаммов HRV инфицируют клетки бронхиального эпителия путем изначальной привязки к ICAM-1 рецептору. Затем связка HRV-ICAM-1 дополнительно интериоризируется посредством эндоцитоза, и, как уже показано, это явление требует активности PI3K (J. Immunol, 2008, 180, 2, с. 870-880). Поэтому ингибиторы PI3K в состоянии также блокировать вирусные инфекции путем ингибирования проникновения вируса в клетки организма-носителя.
[020] Ингибиторы PI3K могут оказаться полезными в сокращении респираторных инфекций других типов, включая грибковый инфекционный аспергиллез (Mucosal Immunol., 2010, 3, 2, с. 193-205). Кроме того, мыши с недостатком PI3Kδ более устойчивы к инфекциям, переносимым простейшим паразитом Leishmania major (J. Immunol, 2009, 183, 3, с. 1921-1933). С учетом воздействия на вирусные инфекции, эти свидетельства позволяют предположить, что ингибиторы PI3K могут быть полезными для лечения разнообразных вирусных инфекционных заболеваний.
[021] Ингибирование PI3K также продемонстрировано для стимулирования дифференцировки регуляторной Т-клетки (Proc. Natl Acad. Sci USA, 2008, 105, 22, с. 7797-7802) предполагая, что ингибиторы PI3K могут служить лечебным целям при аутоиммунных или аллергических показаниях посредством индуцирования иммунологической толерантности к собственному антигену или аллергену. В последнее время изоформу PI3Kδ также связывают с глюкокортикоидной невосприимчивостью, вызываемой табачным дымом (Am. J. Respir. Crit. Care Med., 2009, 179, 7, c. 542-548). Это наблюдение позволяет предположить, что пациенты COPD, которые в противном случае слабо реагируют на кортикостероиды, смогут воспользоваться преимуществом от сочетания ингибитора PI3K с кортикостероидом.
[022] PI3K также участвует в других респираторных состояниях, таких как идиопатический легочный фиброз (IPF). IPF является фиброзным заболеванием с прогрессивным угасанием функции легких и повышенной смертностью вследствие дыхательной недостаточности. При IPF циркулирующие фиброциты направляются в легкие через хемокинный рецептор CXCR4. PI3K требуется как для сигнализации, так и для проявления CXCR4 (Int. J. Biochem. and Cell Biol, 2009, 41, c. 1708-1718). Поэтому за счет снижения проявления CXCR4 и блокирования функции его эффектора, ингибитор PI3K должен подавлять восстанавливающее поступление фиброцитов в легкие и, как следствие, замедлять фибротический процесс, лежащий в основе идиопатического легочного фиброза (IPF), заболевания с высокой неудовлетворенной потребностью.
[023] РI3Kα и PI3Kβ играют важнейшую роль в поддержании гомеостаза и фармакологическом ингибировании этих молекулярных мишеней, связанных с лечением злокачественных опухолей (Maira и соавторы, Expert Opin. Ther. Targets, 2008, 12, 223).
[024] PI3Kα участвует в инсулиновой сигнализации и путях клеточного роста (Nature, 2006, 441, 366). Ожидается, что изоформ-селективное ингибирование PI3Kδ позволит избежать возможных побочных эффектов, таких как гипергликемия, и дисрегуляция метаболизма или роста.
[025] Селективные соединения для опосредования РВК[γ] вырабатываются несколькими группами как иммуносупрессоры аутоиммунных заболеваний (Nature Reviews, 2006, 5, 903-918). Известный AS 605240, селективный ингибитор РI3Кгамма, эффективно проявил себя на мышиной модели ревматоидного артрита (Nature Medicine, 2005, 11, 936-943) и при сдерживании начала заболевания на модели системной красной волчанки (Nature Medicine, 2005, 11, 933-935).
[026] Недавно появилось также описание селективных ингибиторов PI3Kδ. Самые селективные соединения включают в себя хинозолинон пуриновые ингибиторы (PIK39 и IC87114). IC87114 ингибирует PI3Kδ в верхнем наномолярном диапазоне (третий порядок) и более чем 100-кратно селективен против PI3Kδ, в 52 раз более селективен РI3Kβ, но не обладает селективностью против РI3Kγ (примерно 8-кратная селективность). Продемонстрирована активность против любой испытанной протеинкиназы (Cell, 2006, 125, 733-747). С помощью дельта-селективных соединений или генетически опосредуемых мышей (PI3KδD910A), было показано, что в дополнение к ключевой роли в активации В- и Т-клеток PI3Kδ также частично участвует в нейтрофильной миграции и готовит нейтрофильный респираторный всплеск, а также ведет к частичной блокировке антиген-IgE, управляемой дегрануляцией мастоцитов (Blood, 2005, 106, 1432-1440; Nature, 2002, 431, 1007-1011). Поскольку PI3Kδ проявляется как важный регулятор многих ключевых воспалительных реакций, известных участием в ложных воспалительных состояниях, включая, но не ограничиваясь аутоиммунными заболеваниями и аллергией. В поддержку этой идеи приводится увеличивающийся объем данных подтверждения наличия цели PI3Kδ, полученных в результате исследований с использованием как генетических инструментов, так и фармакологических агентов. Таким образом, используя дельта-селективное соединение IC87114 и мышь PI3KδD910A, автор Ali и его соавторы (Nature, 2002, 431, 1007-1011) продемонстрировал, что PI3Kδ играет критическую роль в мышиной модели аллергического заболевания. В отсутствие функциональной дельта, пассивная кожная анафилаксия (РСА) существенно уменьшается и может быть связана с уменьшением в аллерген-IgE индуцированной активации и дегрануляции мастоцитов. Кроме того, ингибирование дельта с помощью IС 87114 показало значительное улучшение воспаления и заболевания на мышиной модели астмы при помощи овальбумин-индуцированного воздушного раздражения (FASEB, 2006, 20: 455-465). Эти данные с использованием соединения были подтверждены на мутантной мыши PBK5D910A с применением той же модели аллергического воздушного раздражения другой группой (Ear. Immunol, 2007, 37, 416, с. - 424).
[027] Существует необходимость в новых ингибиторах PI3K, являющихся хорошим пропрепаратами. В частности, заявляемые соединения могут потенциально связываться с PI3K, выказывая невысокую аффинность к другим рецепторам и демонстрируя функциональную активность как ингибиторы. Они должны хорошо абсорбироваться из желудочно-кишечного тракта, быть метаболически стабильными и обладать благоприятными фармакокинетическими свойствами. При таргетировании против рецепторов в центральной нервной системе они должны свободно преодолевать гемоэнцефалический барьер, а при таргетировании селективно против рецепторов в периферийной нервной системе они не должны преодолевать гемоэнцефалический барьер. Они не должны быть токсичными и должны проявлять минимальные побочные эффекты. В дополнение к этому, идеальный пропрепарат будет существовать в стабильной, негигроскопичной и легко составляемой физической форме. Заявляемые соединения демонстрируют определенный уровень селективности против различных паралогов PI3K α, β, γ и δ. В частности, демонстрируется определенный уровень селективности против изоформы PI3Kδ.
[028] Поэтому заявляемые соединения потенциально эффективны для лечения широкого ряда нарушений, в частности, нарушений, включающих в себя, но не ограничивающихся перечисленным далее, аутоиммунные нарушения, воспалительные заболевания, аллергические заболевания, заболевание или иммунопатологию инфекционного происхождения, заболевания дыхательных путей, отторжение трансплантата, карциномы гематопоэтического происхождения или солидные опухоли.
[029] Заявляемое изобретение также связано с лечением, как автономным, так и в сочетании с одним или более фармакологически активным соединением, включает в себя способы лечения состояний, заболеваний или нарушений дыхательных путей, включая астму, хроническую обструктивную болезнь легких (COPD, ХОБЛ) и идиопатический легочный фиброз (IPF); вирусные инфекции, включая вирусные инфекции дыхательных путей и вирусные обострения заболеваний дыхательных путей, такие как астма и COPD; инфекции дыхательных путей невирусного происхождения, включая аспергиллез и лейшманиоз, аллергические риниты и атопические дерматиты, аутоиммунные заболевания, включая ревматоидные артриты и рассеянный склероз, воспалительные нарушения, включая воспалительную болезнь кишечника, сердечно-сосудистые заболевания, включая тромбоз и атеросклероз (Future Med. Chem., 2013, 5, 4, 479-492; Biochemical Society Transactions, 2004, 32, 378); гемобластозы; нейродегенеративные заболевания; панкреатит; синдром полиорганной недостаточности (СПОН, ПОН); нефропатия; седиментация тромбоцитов; подвижность сперматозоидов; отторжение трансплантата, отторжение ткани, поражение легких, и боль, вызываемые ревматоидным артритом, остеоартритом, боль в пояснице, боль, вызываемая общим воспалительным процессом, постгепатитная невралгия, диабетическая нейропатия, воспалительная невропатическая боль (травма), тригеминальная невралгия и центральная боль; гемобластозы, такие как острый миелоидный лейкоз (AML, ОМЛ), миело-дисплазированный синдром (MDS), миелопролиферативные заболевания (MPD), хронический миелоидный лейкоз (CML, ХМЛ), Т-клеточный острый лимфобластный лейкоз (T-ALL), В-клеточный острый лимфобластный лейкоз (B-ALL), Неходжкинские лимфомы (NHL, НХЛ), В-клеточная лимфома и солидные опухоли, такие как рак молочной железы.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
[030] Авторами настоящей заявки открыты новые соединения, являющиеся ингибиторами активности киназы, в частности, активности РI3-киназы. Соединения, являющиеся ингибиторами PI3-киназы, могут быть полезными в лечении нарушений, связанных с неадекватной активностью киназы, в частности, с неадекватной активностью РI3-киназы, например, в лечении и профилактике нарушений, опосредуемых механизмами РI3-киназы. К таким нарушениям относятся заболевания дыхательных путей, такие как астма, хроническая обструктивная болезнь легких (COPD, ХОБЛ) и идиопатический легочный фиброз (IPF); вирусные инфекции, включая вирусные инфекции дыхательных путей и вирусные обострения заболеваний дыхательных путей, такие как астма и COPD; инфекции дыхательных путей невирусного происхождения, включая аспергиллез и лейшманиоз, аллергические риниты и атопические дерматиты, аутоиммунные заболевания, включая ревматоидные артриты и рассеянный склероз, воспалительные нарушения, включая воспалительную болезнь кишечника, сердечно-сосудистые заболевания, включая тромбоз и атеросклероз; гемобластозы; нейродегенеративные заболевания; панкреатит; синдром полиорганной недостаточности (СПОН, ПОН); нефропатия; седиментация тромбоцитов; карцинома; подвижность сперматозоидов; отторжение трансплантата, отторжение ткани, поражение легких, и боль, вызываемые ревматоидным артритом, остеоартритом, боль в пояснице, боль, вызываемая общим воспалительным процессом, постгепатитная невралгия, диабетическая нейропатия, воспалительная невропатическая боль (травма), тригеминальная невралгия и центральная боль.
[031] В одной реализации заявляемые соединения могут выказывать селективность против РI3-киназ относительно других киназ.
[032] В другой реализации заявляемые соединения могут быть потенциальными ингибиторами PI3Kδ.
[033] В еще одной реализации заявляемые соединения могут выказывать селективность против PI3Kδ относительно других киназ.
[034] Заявляется соединение со скелетной формулой (I):
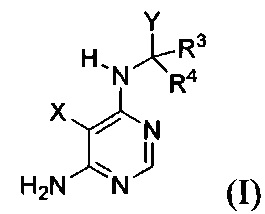 ,
,
или его стереоизомер, геометрический изомер, таутомер, N-оксид, гидрат, сольват, метаболит, фармацевтически приемлемая соль или их пропрепарат, где каждый из X, Y, R3 и R4 определен в настоящей заявке.
[035] В некоторых реализациях X представляет собой (C3-C7)гетероциклил, -(C1-C4)алкилен-(C3-C7)гетероциклил, (C6-C10)арил, -(C1-C4)алкилен-(C6-C10)арил, 5-10-членный гетероарил, или -(C1-C4)алкилен-(5-10-членный гетероарил), где X дополнительно замещен 1, 2, 3, 4 или 5 R1 группами;
Y представляет собой
 ,
,
где Y дополнительно замещен 1, 2, 3 или 4 R2 группами;
каждый R1 и R2 независимо представляет собой Н, F, Cl, Br, CN, NO2, оксо(=O), -C(=O)Ra, -C(=O)ORa, -C(=O)NRaRb, -OC(=O)NRaRb, -OC(=O)ORa, -N(Rc)C(=O)NRaRb, -N(Rc)C(=O)ORa, -N(Rc)C(=O)Ra, -S(=O)2NRaRb, -S(=O)2Ra, -N(Rc)S(=O)2Ra, -N(Rc)-(C1-C4)алкилен-S(=O)2Ra, -(C1-C4)алкилен-C(=O)NRaRb, -(C1-C4)алкилен-N(Rc)C(=O)NRaRb, -(C1-C4)алкилен-N(Rс)С(=O)ORа, -(C1-C4)алкилен-OС(=O)NRaRb, -(C1-C4)алкилен-S(=O)2NRaRb, -(C1-C4)алкилен-N(Rc)S(=O)2Ra, ORa, NRaRb, -(C1-C4)алкилен-ORa, -(C1-C4)алкилен-NRaRb, (С1-C6)алкил, (С2-C6)алкенил, (С2-C6)алкинил, (С3-C8)циклоалкил, -(C1-C4)алкилен-(С3-C8)циклоалкил, (С3-C7)гетероциклил, -(C1-C4)алкилен-(C3-C7)гетероциклил, (С6-C10)арил, -(C1-C4)алкилен-(C6-C10)арил, 5-10-членный гетероарил или -(C1-C4)алкилен-(5-10-членный гетероарил), где каждый из (С1-C6)алкил, (С2-C6)алкенил, (С2-C6)алкинил, (С3-C8)циклоалкил, -(C1-C4)алкилен-(С3-C8)циклоалкил, (С3-C7)гетероциклил, -(C1-C4)алкилен-(C3-C7)гетероциклил, (C6-C10)арил, -(C1-C4)алкилен-(C6-C10)арил, 5-10-членный гетероарил и -(C1-C4)алкилен-(5-10-членный гетероарил) дополнительно замещен 1, 2, 3 или 4 заместителями, независимо выбранными из F, Cl, Br, CN, ORa, NRaRb, (С1-C6)алкил, -(C1-C4)алкилен-ORа и -(C1-C4)алкилен-NRaRb;
каждый R3 и R4 независимо представляет собой Н, F, CN, -C(=O)Ra, -C(=O)ORa, -C(=O)NRaRb, -(C1-C4)aлкилeн-C(=O)NRaRb, -(C1-C4)алкилен-N(Rc)C(=O)NRaRb, -(C1-C4)алкилен-N(Rс)C(=O)ORа, -(C1-C4)алкилен-OC(=O)NRaRb, -(C1-C4)алкилен-S(=O)2NRaRb, -(C1-C4)алкилен-N(Rc)S(=O)2Rb, -(C1-C4)aлкилeн-ORa, -(C1-C4)aлкилeн-NRaRb, (С1-C6)алкил, (С2-C6)алкенил, (С2-C6)алкинил, (С3-C8)циклоалкил, -(С1-C4)алкилен-(С3-C8)циклоалкил, (С3-C7)гетероциклил, -(C1-C4)алкилен-(C3-C7)гетероциклил, (C6-C10)арил, -(C1-C4)алкилен-(C6-C10)арил, 5-10-членный гетероарил, или -(C1-C4)алкилен-(5-10-членный гетероарил), где каждый из: (С1-C6)алкил, (С2-C6)алкенил, (С2-C6)алкинил, (С3-C8)циклоалкил, -(C1-C4)алкилен-(С3-C8)циклоалкил, (С3-C7)гетероциклил, -(C1-C4)алкилен-(C3-C7)гетероциклил, (C6-C10)арил, -(C1-C4)алкилен-(C6-C10)арил, 5-10-членный гетероарил и -(C1-C4)алкилен-(5-10-членный гетероарил) дополнительно замещен 1 2, 3 или 4 заместителями, независимо выбранными из: F, Cl, Br, CN, ORa, NRaRb, (С1-C6)алкил, -(C1-C4)aлкилeн-ORa и -(C1-C4)алкилен-NRaRb; или R3 и R4, вместе с атомом углерода, к которому они присоединены, формируют дополнительно замещенный 3-8-членный карбоцикл или гетероцикл; и
каждый Ra, Rb и Rc независимо представляет собой Н, (С1-C6)алкил, (С2-C6)алкенил, (С2-C6)алкинил, (С3-C6)циклоалкил, -(C1-C4)алкилен-(С3-C6)циклоалкил, (С3-C6)гетероциклил, -(C1-C4)алкилен-(С3-C6)гетероциклил, (C6-C10)арил, -(C1-C4)алкилен-(C6-C10)арил, 5-10-членный гетероарил, или -(C1-C4)алкилен-(5-10-членный гетероарил), где каждый из (С1-C6)алкил, (С2-C6)алкенил, (С2-C6)алкинил, (С3-C6)циклоалкил, -(С1-C4)алкилен-(С3-C6)циклоалкил, (С3-C6)гетероциклил, -(C1-C4)алкилен-(С3-C6)гетероциклил, (C6-C10)арил, -(C1-C4)алкилен-(C6-C10)арил, 5-10-членный гетероарил и -(C1-C4)алкилен-(5-10-членный гетероарил) дополнительно замещен 1, 2, 3 или 4 заместителями, независимо выбранными из F, Cl, CN, N3, ОН, NH2, (С1-C6)алкил, (С1-C6)галоалкил, (С1-C6)алкокси и (С1-C6)алкиламино; или Ra и Rb, вместе с атомом азота, к которому они присоединены, формируют дополнительно замещенный 3-8-членный гетероцикл.
[036] В другой реализации X представляет собой (С3-C7)гетероциклил или 5-10-членный гетероарил, где X дополнительно замещен 1, 2, 3 или 4 R1 группами.
[037] В другой реализации каждый R1 и R2 независимо представляет собой H, F, Cl, CN, оксо (=O), -C(=O)ORa, -C(=O)NRaRb, -N(Rc)C(=O)NRaRb, -N(Rc)C(=O)ORa, -N(Rc)C(=O)Ra, -S(=O)2NRaRb, -N(Rc)S(=O)2Ra, -N(Rc)-(C1-C4)алкилен-S(=O)2Ra, -(C1-C4)алкилен-C(=O)NRaRb, -(C1-C4)алкилен-N(Rc)C(=O)NRaRb, -(C1-C4)алкилен-S(=O)2NRaRb, -(C1-C4)алкилен-N(Rc)S(=O)2Ra, ORa, NRaRb, -(C1-C4)алкилен-ORа, -(C1-C4)алкилен-NRaRb, (С1-C6)алкил, (С2-C6)алкенил, (С2-C6)алкинил, (С3-C8)циклоалкил, -(C1-C4)алкилен-(С3-C8)циклоалкил, (С3-C7)гетероциклил, -(C1-C4)алкилен-(C3-C7)гетероциклил, фенил, -(C1-C4)алкилен-фенил, или 5-6-членный гетероарил, где каждый из (С1-C6)алкил, (С2-C6)алкенил, (С2-C6)алкинил, (С3-C8)циклоалкил, -(С1-C4)алкилен-(С3-C8)циклоалкил, (С3-C7)гетероциклил, -(C1-C4)алкилен-(С3-C7)гетероциклил, фенил, -(C1-C4)алкилен-фенил и 5-6-членный гетероарил дополнительно замещен 1, 2, 3 или 4 заместителями, независимо выбранными из F, CN, ORa, NRaRb, (С1-C3)алкил, -(C1-C4)алкилен-ORа и -(C1-C4)алкилен-NRaRb.
[038] В другой реализации каждый R3 и R4 независимо представляет собой Н, F, CN, -C(=O)NRaRb, -(C1-C2)алкилен-C(=O)NRaRb, -(C1-C2)aлкилeн-N(Rc)C(=O)NRaRb, -(C1-C2)aлкилeн-N(Rc)C(=O)ORa, -(С1-C2)алкилен-OС(=O)NRaRb, -(C1-C2)алкилен-S(=O)2NRaRb, -(C1-C2)алкилен-N(Rc)S(=O)2Rb, -(C1-C2)aлкилeн-ORa, -(C1-C2)aлкилeн-NRaRb, (C1-C4)алкил, (С2-C4)алкенил, (С2-C4)алкинил, (С3-C6)циклоалкил, -(С1-C2)алкилен-(С3-C6)циклоалкил, (C3-C5)гетероциклил, -(С1-C2)алкилен-(C3-C5) гетероциклил, фенил, -(С1-C2)алкилен-фенил, 5-членный гетероарил, или -(С1-C2)алкилен-(5-членный гетероарил), где каждый из (C1-C4)алкил, (С2-C4)алкенил, (С2-C4)алкинил, (С3-C6)циклоалкил, -(С1-C2)алкилен-(С3-C6)циклоалкил, (С3-C6)гетероциклил, -(С1-C2)алкилен-(C3-C5)гетероциклил, фенил, -(С1-C2)алкилен-фенил, 5-членный гетероарил и -(С1-C2)алкилен-(5-членный гетероарил) дополнительно замещен 1, 2, 3 или 4 заместителями, независимо выбранными из F, Cl, Br, CN, ORa, NRaRb, (С1-C6)алкил, -(C1-C4)aлкилeн-ORa и -(C1-C4)алкилен-NRaRb; или R3 и R4, вместе с атомом углерода, к которому они присоединены, формируют дополнительно замещенный 3-8-членный карбоцикл или гетероцикл.
[039] В другой реализации каждый Ra, Rb и Rc независимо представляет собой Н, (C1-C6)алкил, (С2-C6)алкенил, (С2-C6)алкинил, (C3-C6)циклоалкил, -(C1-C4)алкилен-(C3-C6)циклоалкил, (C3-C6)гетероциклил, -(C1-C4)алкилен-(C3-C6)гетероциклил, или 5-10-членный гетероарил, где каждый из (C1-C6)алкил, (C2-C6)алкенил, (С2-C6)алкинил, (C3-C6)циклоалкил, -(С1-C4)алкилен-(C3-C6)циклоалкил, (C3-C6)гетероциклил, -(C1-C4)алкилен-(C3-C6)гетероциклил и 5-10-членный гетероарил дополнительно замещен 1, 2, 3 или 4 заместителями, независимо выбранными из F, CN, N3, ОН, NH2, (C1-C3)алкил, (C1-C3)галоалкил, (C1-C4)алкокси и (C1-C4)алкиламино.
[040] В другой реализации X представляет собой моновалентный гетероциклил или гетероарильную группу, полученную из одной из перечисленных ниже скелетных формул:
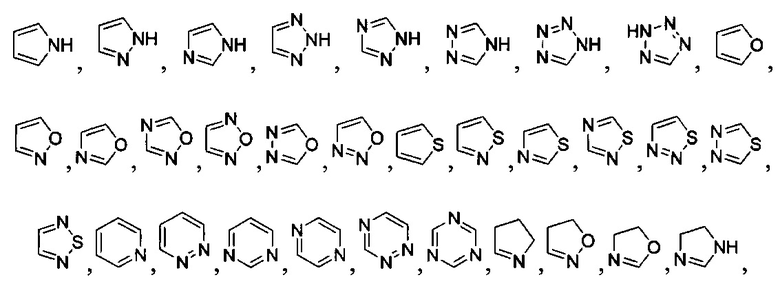
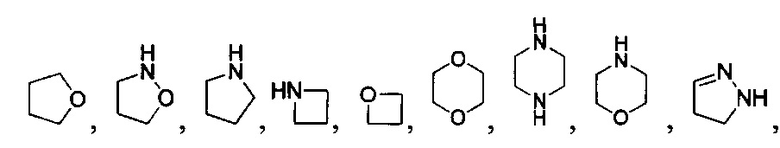 или
или  ;
;
и где Y дополнительно замещен 1, 2, 3 или 3 R1 группами;
[041] В другой реализации Y представляет собой
или  ;
;
и где Y дополнительно замещен 1 или 2 R2 группами;
[042] В другой реализации каждый R1 и R2 независимо представляет собой Н, F, Cl, CN, оксо(=O), -C(=O)ORa, -C(=O)NRaRb, -N(Rc)C(=O)NRaRb, -N(Rc)C(=O)ORa, -N(Rc)C(=O)Ra, -S(=O)2NRaRb, -N(Rc)S(=O)2Ra, ORa, NRaRb, (C1-C3)алкил, (С2-C4)алкенил, (С2-C4)алкинил, (C3-C6)циклоалкил, -(С1-C2)алкилен-(C3-C6)циклоалкил, (C3-C5)гетероциклил, -(С1-C2)алкилен-(C3-C5)гетероциклил, фенил, или -(C1-C2)алкилен-фенил, где каждый из (C1-C3)алкил, (С2-C4)алкенил, (С2-C4)алкинил, (С3-C6)циклоалкил, -(C1-C2)алкилен-(С3-C6)циклоалкил, (C3-C5)гетероциклил, -(С1-C2)алкилен-(C3-C5)гетероциклил, фенил и -(C1-C2)алкилен-фенил дополнительно замещен 1, 2, 3 или 4 заместителями, независимо выбранными из F, CN, ORa, NRaRb и (C1-C3)алкил.
[043] В другой реализации каждый R3 и R4 независимо представляет собой Н, F, CN, (C1-C3)алкил, (С3-C6)циклоалкил, (C3-C5)гетероциклил, или -(С1-C2)алкилен-(C3-C5)гетероциклил, где каждый из (C1-C3)алкил, (C3-C6)циклоалкил, (C3-C5)гетероциклил и -(C1-C2)алкилен-(C3-C5)гетероциклил дополнительно замещен 1, 2, 3 или 4 заместителями, независимо выбранными из: F, Cl, Br, CN, ORa, NRaRb, (С1-C6)алкил, -(C1-C4)алкилен-ORa и -(C1-C4)алкилен-NRaRb; или R3 и R4, вместе с атомом углерода, к которому они присоединены, формируют дополнительно замещенный 3-8-членный карбоцикл или гетероцикл.
[044] В другой реализации каждый Ra, Rb и Rc независимо представляет собой Н, (C1-C3)алкил, (С2-C4)алкенил, (С2-C4)алкинил, (C3-C6)циклоалкил, (C3-C5)гетероциклил, или 5-6-членный гетероарил, где каждый из (C1-C3)алкил, (С2-C4)алкенил, (С2-C4)алкинил, (С3-C6)циклоалкил, (C3-C5)гетероциклил и 5-6-членный гетероарил дополнительно замещен 1, 2, 3 или 4 заместителями, независимо выбранными из F, CN, ОН, NH2, (C1-C3)алкил, (C1-C3)галоалкил, (C1-C3)алкокси и (C1-C3)алкиламино.
[045] Заявляемое соединение или заявляемая фармацевтически приемлемая соль представлена для использования в качестве лекарственного средства.
[046] Заявляемая фармацевтическая композиция содержит фармацевтически приемлемый носитель, инертный наполнитель, разбавитель, активатор, основу или сочетание перечисленных компонентов, и соединение с формулой (I) или его фармацевтически приемлемую соль. В некоторых реализациях соединение содержит один или несколько лечебных агентов. В другой реализации соединение представлено в форме жидкости, твердого вещества, полумягкого вещества, геля или аэрозоля.
[047] Заявляется также способ опосредования активности ферментов PI3K, преимущественно PI3Kδ изоформы, в объекте лечения, заключающийся в приеме объектом лечения терапевтически эффективного количества заявляемого соединения, или его фармацевтически приемлемой соли.
[048] Заявляется также способ лечения нарушения, опосредованного неадекватной активностью РI3-киназы, заключающийся в приеме безопасного и эффективного количества заявляемого соединения, или его фармацевтически приемлемой соли пациентом, нуждающимся в лечении.
[049] Заявляется также способ лечения нарушения, опосредованного неадекватной активностью РI3-киназы, заключающийся в приеме безопасного и эффективного количества заявляемого соединения, или его фармацевтически приемлемой соли пациентом, нуждающимся в лечении.
[050] В некоторых реализациях нарушение, вызываемое неадекватной активностью PI3-киназы, представляет собой заболевание дыхательных путей, вирусную инфекцию, инфекцию дыхательных путей невирусного происхождения, аллергическое заболевание, аутоиммунное заболевание, воспалительное нарушение, сердечно-сосудистое заболевание, гемобластозы, нейродегенеративное заболевание, панкреатит, синдром полиорганной недостаточности (СПОН, ПОН), нефропатия, седиментация тромбоцитов, карцинома, подвижность сперматозоидов, отторжение трансплантата, отторжение ткани, поражение легких или боль.
[051] В других реализациях нарушением, вызванным неадекватной активностью РI3-киназы является астма, хроническая обструктивная болезнь легких (COPD, ХОБЛ), вирусные респираторные инфекции, вирусные обострения респираторных заболеваний, аспергиллез, лейшманиоз, аллергические риниты, атопические дерматиты, ревматоидные артриты, рассеянный склероз, воспалительная болезнь кишечника, тромбоз, атеросклероз, гемобластозы, нейродегенеративное заболевание, панкреатит, синдром полиорганной недостаточности (СПОН, ПОН), нефропатия, седиментация тромбоцитов, карцинома, подвижность сперматозоидов, отторжение трансплантата, отторжение ткани, поражение легких, боль, вызываемые ревматоидным артритом, остеоартритом, боль в пояснице, боль, вызываемая общим воспалительным процессом, постгепатитная невралгия, диабетическая нейропатия, воспалительная невропатическая боль (травма), тригеминальная невралгия или центральная боль.
[052] Заявляется также использование заявляемого соединения, или его фармацевтически приемлемой соли, или заявляемой фармацевтической композиции в производстве лекарственного средства для лечения нарушения или болезни, выбранного из следующего списка: астма, хроническая обструктивная болезнь легких (COPD, ХОБЛ), вирусные респираторные инфекции, вирусные обострения респираторных заболеваний, аспергиллез, лейшманиоз, аллергические риниты, атопические дерматиты, ревматоидные артриты, рассеянный склероз, воспалительная болезнь кишечника, тромбоз, атеросклероз, гемобластозы, нейродегенеративное заболевание, панкреатит, синдром полиорганной недостаточности (СПОН, ПОН), нефропатия, седиментация тромбоцитов, карцинома, подвижность сперматозоидов, отторжение трансплантата, отторжение ткани, поражение легких, боль, вызываемые ревматоидным артритом, остеоартритом, боль в пояснице, боль, вызываемая общим воспалительным процессом, постгепатитная невралгия, диабетическая нейропатия, воспалительная невропатическая боль (травма), тригеминальная невралгия или центральная боль.
[053] Заявляется способ ингибирования фосфатидилинозитол 3-киназы (PI3 киназа), заключающийся в следующем: контактирование с PI3 киназой эффективного количества заявляемого соединения. В некоторых реализациях этап контактирования включает в себя контактирование с клеткой, содержащей упомянутую PI3 киназу. В некоторых реализациях способа ингибирование происходит в организме субъекта лечения, страдающего от нарушения, связанного с нарушенным функционированием одного или более типов PI3 киназы. Некоторые показательные заболевания, включая нарушенное функционирование одного или более типов PI3 киназы, отобраны из группы, состоящей из аутоиммунных заболеваний, ревматоидного артрита, заболеваний дыхательных путей, аллергических реакций и карцином различного типа.
[054] В некоторых реализациях способ заключается в приеме второго лечебного агента объектом лечения.
[055] В определенных реализациях состояние или нарушение, опосредованное PI3K, отобрано из ревматоидного артрита, анкилозирующего спондилоартрита, остеоартроза, псориатического артрита, псориаза, воспалительных заболеваний и аутоиммунных заболеваний. В других реализациях состояние или нарушение, опосредованное PI3K, отобрано из следующего списка: сердечно-сосудистые заболевания, атеросклероз, гипертония, тромбоз глубоких вен, инсульт, инфаркт миокарда, нестабильная стенокардия, тромбоз, тромбоэмболия легочной артерии (ТЭЛА), тромболитические заболевания, острая артериальная ишемия, периферийные тромболитические закупоривания и коронарная недостаточность. В других реализациях состояние или нарушение, опосредованное PI3K, выбрано из следующих: карцинома, колоректальный рак, глиобластома, карцинома эндометрия, гепатоцеллюлярная карцинома, бронхогенная карцинома, меланома, почечно-клеточная карцинома, карцинома щитовидной железы, клеточная лимфома, лимфопролиферативные нарушения, мелкоклеточная карцинома легких, сквамозноклеточная карцинома легких, глиома, рак молочной железы, рак предстательной железы, рак яичников, рак шейки матки и лейкемия. В другой реализации состояние или нарушение, опосредованное PI3K, отобрано из диабета II типа. В других реализациях состояние или нарушение, опосредованное PI3K, выбрано из следующих: респираторные заболевания, бронхит, астма и хроническая обструктивная болезнь легких. В определенных реализациях объектом лечения является человек.
[056] Заявляется также способ лечения состояния или нарушения пациента, опосредованного PI3K, содержащий этап приема соединения по любой из приведенных выше реализаций.
[057] Заявляется также способ лечения ревматоидного артрита, анкилозирующего спондилоартрита, остеоартроза, псориатического артрита, псориаза, воспалительных заболеваний и аутоиммунных заболеваний, содержащий этап приема соединения пациентом по любой из приведенных выше реализаций.
[058] Заявляется также способ лечения заболеваний дыхательных путей, включая астму, хроническую обструктивную болезнь легких (COPD, ХОБЛ) и идиопатический легочный фиброз (IPF), содержащий этап приема соединения пациентом по любой из приведенных выше реализаций.
[059] Заявляется также способ лечения воспалительных болезней кишечника, воспалительных заболеваний глаз, воспалительных или лабильных заболеваний мочевого пузыря, псориаза, кожных заболеваний с воспалительными компонентами, хронических воспалительных состояний, системной красной волчанки (SLE), миастения (myestenia gravis), острого диссеминированного энцефаломиелита, идиопатической тромбоцитопенической пурпуры (ИТП, болезнь Верльгофа), рассеянного склероза, синдрома Шегрена и аутоиммунной гемолитической анемии, аллергических состояний и гиперчувствительности, содержащий этап приема пациентом соединения по любой из приведенных ниже реализаций.
[060] Заявляется также способ лечения злокачественных опухолей пациента, опосредованных, зависящих или связанных с активностью PI3K, в частности, с активностью РВKδ, содержащий этап приема пациентом соединения по любой из приведенных выше или ниже реализаций.
[061] Заявляется также способ лечения злокачественных опухолей, выбранных из следующих: острый миелоидный лейкоз, миело-дисплазированный синдром, миелопролиферативные заболевания, хронический миелоидный лейкоз, Т-клеточный острый лимфобластный лейкоз, В-клеточная острая лимфобластозная лейкемия, неходжкинская лимфома, В-клеточный острый лимфобластный лейкоз, солидные опухоли и рак молочной железы, содержащий этап приема пациентом соединения по любой из приведенных выше или ниже реализаций.
[062] Заявляется также использование соединения по любой из приведенных выше реализаций в качестве лекарственного средства.
[063] Заявляется также использование соединения по любой из приведенных выше реализаций в производстве лекарственного средства для лечения РI3K-опосредованного состояния или нарушения пациента.
[064] Заявляется также использование соединения по любой из приведенных выше реализаций в производстве лекарственного средства для лечения ревматоидного артрита, анкилозирующего спондилоартрита, остеоартроза, псориатического артрита, псориаза, воспалительных заболеваний, заболеваний дыхательных путей, включая астму, хроническую обструктивную болезнь легких (COPD, ХОБЛ) и идиопатический легочный фиброз (IPF), аутоиммунные заболевания и злокачественные опухоли.
[065] В отсутствие иных указаний, все стереоизомеры, геометрические изомеры, таутомеры, сольваты, гидраты, метаболиты, соли и фармацевтически приемлемые пропрепараты заявляемых соединений относятся к области заявляемого изобретения.
[066] В определенных реализациях соль является фармацевтически приемлемой солью. Фраза «фармацевтически приемлемая» означает, что вещество или соединение должно быть химически и/или токсикологически совместимо с другими ингредиентами препарата, и/или с млекопитающим, подвергающимся лечению этим препаратом.
[067] В состав заявляемых соединений должны также входить соли таких соединений, которые не обязательно являются фармацевтически приемлемыми солями и которые могу быть полезны как промежуточные продукты для приготовления и/или очистки соединений с формулой (I), и/или для отделения энантиомеров от соединений с формулой (I).
[068] Заявляемые соединения, включая их соли, могут быть также получены в форме их гидратов, или содержать другие растворители, используемые для их кристаллизации. Заявляемые соединения могут по своей природе или по расчету образовывать сольваты с фармацевтически приемлемыми растворителями (включая воду); поэтому заявляемое изобретение, по замыслу авторов, охватывает как сольватированные, так и не сольватированные формы.
[069] Заявляются также способы приготовления, способы сепарирования и способы очистки соединений с формулой (I). Заявляемые соединения могут, как правило, иметь несколько асимметричных центров, и обычно изображаются в форме рацемических смесей. Авторы заявляемого изобретения намереваются охватить рацемические смеси, частично рацемические смеси и отдельные энантиомеры и диастереомеры.
[070] Заявляемые соединения могут существовать в форме одного из возможных изомеров, ротамеров, атропоизомеров, таутомеров или их смесей. Авторы заявляемого изобретения намереваются охватить смеси изомеров, ротамеров, атропоизомеров, таутомеров, частично смешанных изомеров, ротамеров, атропоизомеров или таутомеров, и отдельные изомеры, ротамеры, атропоизомеры, таутомеры.
[071] Заявляемые соединения включают в себя изотопно меченые соединения, по определениям настоящей заявки, например, с наличием радиоактивных изотопов, таких как 3Н, 14С и 18F, или с наличием нерадиоактивных изотопов, таких как 2Н и 13С.
[072] Заявляются также способы приготовления, способы сепарирования и способы очистки соединений с формулой (I).
[073] Предшествующая часть объединяет некоторые аспекты заявляемого изобретения и не является ограничительной по своей сути. Эти и другие аспекты, а также реализации более подробно раскрыты ниже.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
ОПРЕДЕЛЕНИЯ И ОБЩАЯ ТЕРМИНОЛОГИЯ
[074] Ссылки будут приводиться на конкретные реализации заявляемого изобретения, примеры которых иллюстрированы прилагаемыми скелетными и аналитическими формулами. Предполагается, что заявляемое изобретение охватывает все альтернативы, модификации и эквиваленты, включенные в область заявляемого изобретения согласно пунктам патентной формулы. Специалисты ознакомятся со многими способами и материалами, сходными или эквивалентными заявляемым, что может быть использовано в реализации заявляемого изобретения. Заявляемое изобретение ни в коей мере не ограничивается описываемыми способами и материалами. Если один или более включенных в настоящую заявку источников, патентов или аналогичных материалов отличается или противоречит настоящей заявке, включая, но, не ограничиваясь оговоренными терминами, использованием терминов, описанными технологиями и тому подобными элементами, приоритет остается за настоящей заявкой.
[075] В отсутствие иных указаний все технические и научные термины настоящей заявки имеют то же самое значение, что и общепринятые у специалистов в данной области. Все упоминаемые в настоящей заявке патенты и публикации включены в качестве справочного материала.
[076] В отсутствие иных указаний в настоящей заявке следует пользоваться приведенными ниже определениями. В целях настоящей заявки на изобретение названия химических элементов приводятся в соответствии с периодической системой химических элементов, версия CAS, и «Справочником по химии и физике», 75 издание. 1994. Кроме того, основные принципы органической химии излагаются в монографии «Органическая химия», Томас Соррелл, University Science Books, Sausalito: 1999, и «Углубленный курс органической химии Марча», авторы Майкл Б. Смит и Джерри Марч, John Wiley & Sons, New York: 2007, ссылки приводятся на полные варианты перечисленных выше изданий.
[077] В количественных характеристиках и пунктах патентной формулы настоящей заявки единственное и множественное число тождественны, в отсутствие иных указаний или явного противоречия в тексте настоящей заявки.
[078] Термин «объект» в настоящей заявке относится к животному. Как правило, животным является млекопитающее. Также объектом (объектами) называются, например, приматы (например, человек, особи мужского или женского пола), коровы, овцы, козы, лошади, собаки, кошки, кролики, крысы, мыши, рыбы, птицы и тому подобные. В определенных реализациях объектом является примат. В других реализациях объектом является человек.
[079] В настоящей заявке термин «пациент» относится к человеку (взрослых и детей) или к другому животному. В одной реализации термин «пациент» относится к человеку.
[080] Заявляемое изобретение также включает в себя изотопно меченые соединения, идентичные перечисленным в настоящей заявке, но фактически с одним или более атомов, замещенным атомом с атомной массой или атомным номером, отличающимися от атомной массы или атомного номера, встречающимися в природе. К примерам изотопов, которые могут быть включены в состав заявляемых соединений, относятся изотопы водорода, углерода, азота, кислорода, фосфора, фтора и хлора, такие как 2Н, 3Н, 13С, 14С, 15N, 16О, 17О, 18О, 31Р, 32Р, 36S, 18F и 37Cl.
[081] Заявляемые соединения, содержащие упомянутые выше изотопы и/или другие изотопы других атомов, относятся к области заявляемого изобретения. Определенные изотопно меченые соединения настоящей заявки, например, с радиоактивными изотопами 3Н и 14С, полезны в испытаниях лекарственных средств и/или исследованиях распределения субстрата ткани. Изотопы, меченные тритием, например 3Н, и углерод-14, например 14С, особо предпочтительны ввиду простоты их приготовления. Кроме того, замещение более тяжелыми изотопами, такими как дейтерий, т.е. 2Н, может дать определенные терапевтические преимущества за счет повышенной метаболической стабильности, например, увеличенное in vivo время полураспада или сокращенные требования к дозировке, и поэтому такое замещение может оказаться более предпочтительным в определенных обстоятельствах.
[082] Используемые в настоящей заявке стереохимические определения и условные обозначения в основном заимствованы из С.П. Паркер, Издательство McGraw-Hill «Словарь химических терминов» (1984) McGraw-Hill Book Company, New York; и Элиэл Э. и Уилен С., «Стереохимия органических соединений», John Wiley & Sons, Inc., New York, 1994.
[083] Многие органические соединения существуют в оптически активных формах, т.е. обладают способностью вращать плоскость поляризации линейно поляризованного света. В описании оптически активных соединений используются префиксы D и L, или R и S для описания абсолютной конфигурации молекулы вокруг ее хирального центра (центров). Префиксы d и l или (+) и (-) используются для обозначения направления вращения плоскополяризованного света соединением, а именно (-) или (l) означает, что плоскость поляризации света поворачивается в соединении влево (левовращающее соединение). Соединение с префиксом (+) или d является правовращающим, плоскость поляризации света поворачивается в таком соединении вправо. С точки зрения конкретной химической структуры эти стереоизомеры идентичны, за исключением того, что представляют собой зеркальные отображения друг друга. Конкретный стереоизомер можно назвать энантиомером, а смесь таких изомеров часто называют энантиометрической смесью. Смесь энантиомеров в пропорции 50:50 называется рацемической смесью или рацематом, такая смесь может образоваться в отсутствие стереоселекции или стереоспецифичности в химической реакции или процессе.
[084] В зависимости от выбора исходных материалов и процедур соединения могут существовать в форме одного из возможных изомеров или их смесей, например, как чистые оптические изомеры, или как смеси изомеров, такие как рацематы или диастереоизомерные смеси в зависимости от количества асимметричных атомов углерода. Оптически активные (R)- и (S)- изомеры могут быть приготовлены с использованием хиральных синтонов или хиральных реагентов, или пептизированы при помощи общепринятых технологий. Если соединение содержит двойную связь, заместителем может быть Е или Z конфигурация. Если соединение содержит двузамещенный циклоалкил, циклоалкильный заместитель может иметь цис- или транс-конфигурацию.
[085] Заявляемые соединения могут содержать асимметричные или хиральные центры, и поэтому существуют в различных стереоизомерических формах. По замыслу авторов заявляемого изобретения, все стереоизотрические формы заявляемых соединений, включая, но не ограничиваясь диастереомерами, энантиомерами, атропоизомерами и геометрическими (или конформационными) изомерами, а также их смеси, такие как рацемические смеси, являются частью заявляемого изобретения.
[086] В отсутствие иных указаний, изображенные в настоящей заявке структуры также означают включение всех изомерных форм (т.е. энантиомерической, диастереомерической, атропоизомерической и геометрической (или конформационной) структуры; например, R и S конфигурации для каждого асимметричного центра, (Z) и (Е) изомеры с двойной связью, и (Z) и (Е) конформационные изомеры.
[087] Термин "таутомер" или "таутомерическая форма" относятся к структурным изомерам различной энергетики, взаимопревращаемым через низкоэнергетический барьер. Там, где возможна таутомеризация (например, в растворе), может быть достигнуто химическое равновесие таутомеров. Например, протонные таутомеры (известные также как протототропные таутомеры) включают взаимопревращение посредством миграции протона, такое как кето-энольная и имин-энаминная изомеризация. Валентные таутомеры включают взаимопревращение посредством реорганизации некоторых связующих электронов. Специфичным примером кето-энол таутомеризации является взаимопревращение пентан-2,4-дион и 4-гидроксипент-3-эн-2-один таутомеров. Другим примером таутомеризации является фенол-кето таутомеризация. Специфичным примером фенол-кето таутомеризации является взаимопревращение пиридин-4-ол и пиридин-4(1H)-один таутомеров. В отсутствие иных указаний все таутомерные формы заявляемых соединений относятся к области заявляемого изобретения.
[088] Любой асимметричный атом (например, углерод или подобного химического элемента) заявляемого соединения (соединений) может быть представлен в рацемической или энантиомерически обогащенной, например (R)-, (S)- или (R,S)-конфигурации. В некоторых реализациях каждый асимметричный атом имеет по меньшей мере 50% энантиомерного излишка, по меньшей мере 60% энантиомерного излишка, по меньшей мере 70% энантиомерного излишка, по меньшей мере 80% энантиомерного излишка, по меньшей мере 90% энантиомерного излишка, по меньшей мере 95% энантиомерного излишка, или по меньшей мере 99% энантиомерного излишка в (R)- или (S)-конфигурации. Заместители на атомах с ненасыщенными двойными связями могут быть представлены, по возможности, в цис- (Z)- или транс- (Е)-форме.
[089] Соответственно, любое заявляемое соединение может быть в форме одного или двух возможных изомеров, ротамеров, атропоизомеров, таутомеров или их смеси, например, в основном чистых (цис или транс) изомеров, диастереомеров, оптических изомеров (антиподов), рацематов или их смесей.
[090] Все готовые смеси изомеров можно разделить на основе физико-химических различий составляющих на чистые или в основном чистые геометрические или оптические изомеры, диастереомеры, рацематы, например, посредством хроматографии и/или фракционной кристаллизации.
[091] Любые готовые рацематы конечных или промежуточных продуктов можно пептизировать на оптические антиподы способами, известными специалистам в данной области, например, путем разделения их диастереомерических солей. Рацемические продукты также можно пептизировать с помощью хиральной хроматографии, например, путем высокопроизводительной жидкой хроматографии (HPLC) с использованием хирального абсорбента. Предпочтительные энантиомеры можно также приготовить посредством асимметричного синтеза. Можно обратиться, например, к изданию Ждакс и соавторы., «Энантиомеры, рацематы и критерии пептизации», Wiley Interscience, New York, 1981; Гаули и соавторы, «Принципы асимметричного синтеза», 2-е изд. Elsevier, Oxford, UK, 2012; Элиэл и соавторы, «Стереохимия углеродных соединений», McGraw-Hill, NY, 1962; Уилен и соавторы, «Таблицы агентов пептизации и оптическая пептизация» с.268 (EX. Eliel, изд. Univ. of Notre Dame Press, Notre Dame, IN, 1972) и Субраманиан и соавторы, «Технологии хирального разделения: практический поход» изд., Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2007.
[092] Заявляемые соединения могут быть дополнительно замещены одним или более заместителями, например приведенными ниже, или как проиллюстрировано конкретными классами, подклассами и реализациями изобретения. Фраза «дополнительно замещен» будет использоваться взаимозаменяемо с фразой «замещен или не замещен». В основном термин «замещен» относится к замене одного или более водородных радикалов в данной структуре радикалом заданного заместителя. Термин «дополнительный» или «дополнительно» означает, что описываемое в дальнейшем событие или обстоятельство может возникнуть, но может и не возникнуть, и что описание включает в себя факты, когда событие происходит, и факты, когда событие не происходит. В отсутствие иных указаний дополнительно замещенная группа может иметь заместитель на каждой замещаемой позиции группы. Когда боле чем одна позиция данной структуры может быть замещена более чем одним заместителем, выбранным из указанной группы, заместитель на каждой позиции может быть либо тем же самым, либо другим.
[093] Термин «алкил» или «алкильная группа» относится к насыщенному линейному или разветвленно-цепочечному моновалентному углеводородному радикалу, содержащему от 1 до 20 атомов углерода, где алкильный радикал может быть дополнительно замещен независимо одним или более приведенными ниже заместителями. В отсутствие иных указаний алкильная группа содержит 1-20 атомов углерода. В некоторых других реализациях алкильная группа содержит 1-12 атомов углерода. В других реализациях алкильная группа содержит 1-10 атомов углерода. В других реализациях алкильная группа содержит 1-8 атомов углерода. В других реализациях алкильная группа содержит 1-6 атомов углерода. В некоторых других реализациях алкильная группа содержит 1-4 атома углерода, а в других реализациях алкильная группа содержит 1-3 атома углерода.
[094] К некоторым неограничительным примерам алкильной группы относится метил (Me, -CH3), этил (Et, -CH2CH3), 1-пропил (n-Pr, n-пропил, -CH2CH2CH3), 2-пропил (i-Pr, i-пропил, -CН(CH3)2), 1-бутил (n-Bu, n-бутил, -CH2CH2CH2CH3), 2-метил-1-пропил (i-Bu, i-бутил, -CH2СН(CH3)2), 2-бутил (s-Bu, s-бутил, -CН(CH3)CH2CH3), 2-метил-2-пропил (t-Bu, t-бутил, -C(CH3)3), 1-пентил (n-пентил, -CH2CH2CH2CH2CH3), 2-пентил (-CН(CH3)CH2CH2CH3), 3-пентил (-CН(CH2CH3)2), 2-метил-2-бутил (-C(CH3)2CH2CH3), 3-метил-2-бутил (-CН(CH3)СН(CH3)2), 3-метил-1-бутил (-CH2CH2СН(CH3)2), 2-метил-1-бутил (-CH2СН(CH3)CH2CH3), 1-гексил (-CH2CH2CH2CH2CH2CH3), 2-гексил (-CН(CH3)CH2CH2CH2CH3), 3-гексил (-CН(CH2CH3)(CH2CH2CH3)), 2-метил-2-пентил (-C(CH3)2CH2CH2CH3), 3-метил-2-пентил (-CН(CH3)СН(CH3)CH2CH3), 4-метил-2-пентил (-CН(CH3)CH2СН(CH3)2), 3-метил-3-пентил (-C(CH3)(CH2CH3)2), 2-метил-3-пентил (-CН(CH2CH3)СН(CH3)2), 2,3-диметил-2-бутил (-C(CH3)2СН(CH3)2), 3,3-диметил-2-бутил (-CН(CH3)С(CH3)3, 1-гептил, 1-октил, и подобные.
[095] Префикс «алк-» относится как к прямой, так и к разветвленной насыщенной углеродной цепочке.
[096] Термин «алкилен» относится к насыщенной дивалентной углеводородной группе, полученной из прямой или разветвленной цепочки насыщенного углеводорода или разветвленной цепочки насыщенного углеводорода путем удаления двух атомов водорода. В отсутствие иных указаний алкиленовая группа содержит 1-6 атомов углерода. В некоторых реализациях алкиленовая группа содержит 1-4 атома углерода. В других реализациях алкиленовая группа содержит 1-2 атома углерода. К примерам алкиленовой группы относятся, не ограничиваясь перечисленным далее, метилен (-CH2-), этилиден (-CH2CH2-), изопропилиден (-CH(CH3)CH2-) и подобные.
[097] Термин «алкенил» относится к линейному или разветвленно-цепочечному моновалентному углеводородному радикалу, содержащему от 2 до 12 атомов углерода с по меньшей мере одним участком ненасыщенности, например, углерод-углерод, двойная связь sp2, где алкенильный радикал может быть дополнительно замещен независимо одним или более заявляемыми заместителями, и включать радикалы с ориентацией «цис» и «транс», или, альтернативно, с ориентациями «Е» и «Z». Алкенильная группа содержит предпочтительно от 2 до 8 атомов углерода, еще более предпочтительно от 2 до 6 атомов, и наиболее предпочтительно от 2 до 4 атомов углерода. К некоторым неограничительным примерам алкенильной группы относятся этиленил или винил (-CH=CH2), аллил (-CH2СН=CH2), и подобные.
[098] Термин «алкинил» относится к линейному или разветвленному моновалентному углеводородному радикалу, содержащему от 2 до 12 атомов углерода с по меньшей мере одним участком ненасыщенности, например, углерод-углерод, тройная связь sp, где алкинильный радикал может быть дополнительно замещен независимо одним или более заявляемыми заместителями. Алкинильная группа содержит предпочтительно от 2 до 8 атомов углерода, еще более предпочтительно от 2 до 6 атомов, и наиболее предпочтительно от 2 до 4 атомов углерода. К некоторым неограничительным примерам алкинильной группы относятся этинил (-C=СН), пропинил (пропаргил, -CH2С=СН), -C=С-CH3, и подобные.
[100] Термин «алкокси» относится к ранее определенной алкильной группе, присоединенной к главному атому углерода атомом кислорода. В отсутствие иных указаний алкоксильная группа содержит 1-20 атомов углерода. В некоторых реализациях алкоксильная группа содержит 1-10 атомов углерода. В других реализациях алкоксильная группа содержит 1-8 атомов углерода. В некоторых других реализациях алкоксильная группа содержит 1-6 атома углерода, а в других реализациях алкоксильная группа содержит 1-4 атома углерода. В других реализациях алкоксильная группа содержит 1-3 атома углерода.
[101] К некоторым неограничительным примерам алкокси группы относится метокси (МеО, -OCH3), этокси (EtO, -OCH2CH3), 1-пропокси (n-PrO, n-пропокси, -OCH2CH2CH3), 2-пропокси (i-PrO, i-пропокси, -OСН(CH3)2), 1-бутокси (n-BuO, n-бутокси, -OCH2CH2CH2CH3), 2-метил-1-пропокси (i-BuO, i-бутокси, -OCH2СН(CH3)2), 2-бутокси (s-BuO, s-бутокси, -OСН(CH3)CH2CH3), 2-метил-2-пропокси (t-BuO, t-бутокси, -OС(CH3)3), 1-пентокси (и-пентокси, -OCH2CH2CH2CH2CH3), 2-пентокси (-OСН(CH3)CH2CH2CH3), 3-пентокси (-OСН(CH2CH3)2), 2-метил-2-бутокси (-OС(CH3)2CH2CH3), 3-метил-2-бутокси (-OСН(CH3)СН(CH3)2), 3-метил-1-бутокси (-OCH2CH2СН(CH3)2), 2-метил-1-бутокси (-OCH2СН(CH3)CH2CH3), и подобные.
[102] Термин «галоалкил», «галоалкенил» или «галоалкокси» относится к алкилу, алкенилу или алкокси, в зависимости от ситуации, замещенному одним или более атомами водорода.
[103] Термин «карбоцикл», «карбоциклил» или «карбоциклическое кольцо» относится к моновалентному или мультивалентному неароматическому, насыщенному или частично ненасыщенному кольцу с 3-12 атомами углерода как моноцикличной, бицикличной или трицикличной системе. Карбоциклическая система включает в себя спиро карбоциклил или вплавленный карбоциклил. В некоторых реализациях карбоциклическая группа содержит от 3 до 8 атомов углерода. К некоторым неограничительным примерам карбоциклической группы относятся циклоалкил, циклоалкенил и циклоалкинил. Дополнительные неограничительные примеры карбоциклической группы включают в себя циклопропил, циклобутил, циклопентил, 1-циклопент-1-энил, 1-циклопент-2-энил, 1-циклопент-3-энил циклогексил, 1-циклогекс-1-энил, 1-циклогекс-2-энил, 1-циклогекс-3-энил, циклогексадиэнил, и подобные.
[104] Термин «циклоалкил» относится к моновалентному или мультивалентному насыщенному кольцу с 3-12 атомами углерода как моноцикличной, бицикличной или трицикличной системе. Бицикличная система включает в себя спиро бициклил или вплавленный бициклил. В некоторых реализациях циклоалкильная группа содержит от 3 до 10 атомов углерода. В других реализациях циклоалкильная группа содержит от 3 до 8 атомов углерода. В некоторых других реализациях циклоалкильная группа содержит от 3 до 6 атома углерода, а в других реализациях циклоалкильная группа содержит от 5 до 6 атома углерода. Циклоалкильная группа дополнительно замещена независимо одним или более приведенными в настоящей заявке заместителями.
[105] Термины «вплавленный бициклический», «вплавленный циклический», «вплавленный бициклил» и «вплавленный циклил», используемые как взаимозаменяемые, относятся к бициклической кольцевой системе, не являющейся ароматической. Такая система может содержать изолированные или сопряженные ненасыщенные, но не ароматические или гетероароматические кольца в корневой структуре (однако может вслед за тем иметь ароматическое замещение). Термины «спироциклил», «спироциклический», «спиробициклил» или «спиро бициклический», используемые как взаимозаменяемые, относятся к кольцу, возникшему из особого кольцевидного атома углерода другого кольца. Например, в изображенной ниже структуре а, насыщенная мостиковая кольцевая система (кольцо В и В') трактуется как «вплавленная бициклическая», тогда как кольцо А и кольцо В имеют один общий атом между двумя насыщенными кольцевыми системами, называемыми «спироциклил» или «спиробициклил». Каждое циклическое кольцо вплавленного бициклила или спиро бициклила может быть либо карбоциклилом, либо гетероциклилом.

[106] Термины «гетероцикл», «гетероциклил» или «гетероциклическое кольцо», используемые в настоящей заявке как взаимозаменяемые, относятся к моноциклической, бициклической или трициклической кольцевой системе, в которой один или более звеньев кольца независимо выбраны из гетероатомов и которая полностью насыщена, или содержит одну или более единицу ненасыщения, которая, однако, не является ароматической, при наличии одной или боле точек соединения с оставшейся частью молекулы. В состав бициклической кольцевой системы входит спиро бициклил или вплавленный бициклил, и одно из колец может быть либо монокарбоциклом, либо моногетероциклом. Один или более кольцевых атомов дополнительно замещен независимо одним или более приведенными в настоящей заявке заместителями. В некоторых реализациях «гетероцикличная», «гетероциклильная» или «гетероциклическая кольцевая» группа представляет собой моноцикл из 4-8 кольцевых звеньев (от 3 до 7 атомов углерода и от 1 до 3 гетероатомов, отобранных из N, О, P и S, где S или P дополнительно замещен одной или более оксо для получения группы S=O или SO2, PO или PO2). В других реализациях «гетероцикличная», «гетероциклильная» или «гетероциклическая кольцевая» группа представляет собой моноцикл из 4-7 кольцевых звеньев (от 3 до 6 атомов углерода и от 1 до 3 гетероатомов, отобранных из N, О, P и S, где S или P дополнительно замещен одной или более оксо для получения группы S=O или SO2, PO или PO2). В других реализациях «гетероцикличная», «гетероциклильная» или «гетероциклическая кольцевая» группа представляет собой моноцикл из 3-7 кольцевых звеньев (от 2 до 6 атомов углерода и от 1 до 3 гетероатомов, отобранных из N, О, P и S, где S или P дополнительно замещен одной или более оксо для получения группы S=O или SO2, PO или PO2). В других реализациях «гетероцикличная», «гетероциклильная» или «гетероциклическая кольцевая» группа представляет собой моноцикл из 4-6 кольцевых звеньев (от 3 до 5 атомов углерода и от 1 до 3 гетероатомов, отобранных из N, О, P и S, где S или P дополнительно замещен одной или более оксо для получения группы S=O или SO2, РО или PO2). В других реализациях «гетероцикличная», «гетероциклильная» или «гетероциклическая кольцевая» группа представляет собой моноцикл из 3-6 кольцевых звеньев (от 2 до 5 атомов углерода и от 1 до 3 гетероатомов, отобранных из N, O, P и S, где S или P дополнительно замещен одной или более оксо для получения группы S=O или SO2, РО или PO2), или бицикл, состоящий из 7-10 кольцевых звеньев (от 4 до 9 атомов углерода и от 1 до 3 гетероатомов, отобранных из N, O, P и S, где S или P дополнительно замещен одной или более оксо для получения группы S=O или SO2, PO или PO2).
[107] Гетероцикл может быть углеродным радикалом или гетероатомным радикалом. К некоторым неограничительным примерам гетероциклильной группы относятся пирролидинил, тетрагидрофуранил, дигидрофуранил, тетрагидротиенил, тетрагидропиранил, дигидропиранил, тетрагидротиопиранил, пиперидино, морфолино, тиоморфолино, тиоксанил, пиперазинил, гомо-пиперазинил, ацетидинил, оксетанил, тиетанил, гомопиперидинил, оксепанил, тиепанил, оксазепинил, диазепинил, тиазепинил, 4,5-дигидрооксазоли, 2-пирролинил, 3-пирролинил, индиолинил, 2H-пиранил, 4Н-пиранил, диоксанил, 1,3-диоксоланил, пиразолинил, дитианил, дитиоланил, дигидропиранил, дигидротиенил, дигидрофуранил, пиразолидинилимидазолинил, имидазолидинил, 1,2,3,4-тетрагидроизоквинолинил. Примерами гетероциклической группы, где 2 кольцевых атома углерода замещены оксо (=O) функциональными группами, являются пиримидиндионил и 1,1-диоксо-тиоморфолинил.
[108] Термин «гетероатом» относится к одному или более перечисленных далее химических элементов: кислород, сера, азот, фосфор или кремний, включая любую окисленную форму азота, серы или фосфора; кватернизованную форму любого азотистого основания; или замещаемый азот гетероциклического кольца, например, N (как в 3,4-дигидро-2H-пирролил), NH (как в пирролидинил) или NR (как в N-замещенном пирролидиниле).
[109] Термин «галоген» относится к фтору (F), хлору (Cl), брому (Br), или йоду (I).
[110] Термин «азидо» или «N3» относится к функциональной группе азотоводородной кислоты. Этот радикал может быть прикреплен, например, к метальной группе, создавая азидометан (метилазид, MeNs); или к фенильной группе, создавая фенилазид (PhN3).
[111] Термин «арил» используется автономно или как часть более крупной функциональной группы, как «арилалкил», «аралкокси» или «арилоксиалкил» и относится к моноциклическим, бициклическим и трициклическим карбоциклическим кольцевым системам с общим количеством звеньев цепи от 6 до 14, предпочтительно от 6 до 12 звеньев цепи, и наиболее предпочтительно от 6 до 10 звеньев цепи, где по меньшей мере одно кольцо системы является ароматическим, и где каждое кольцо системы содержит от 3 до 7 звеньев цепи и имеет по меньшей мере одну или больше точек крепления к остальной части молекулы. Термин «арил» допускается использовать взаимозаменяемо с термином «арильное кольцо» или «ароматический». К некоторым неограничительным примерам арильного кольца следует отнести фенил, нафтил и антраценил. Арильный радикал дополнительно замещен независимо одним или более приведенными в настоящей заявке заместителями.
[112] Термин «гетероарил» используется автономно или как часть более крупной функциональной группы, как «гетероарилалкил» или «гетероаралкокси» и относится к моноциклическим, бициклическим и трициклическим кольцевым системам с общим количеством звеньев цепи от 5 до 14, предпочтительно от 5 до 12 звеньев цепи, и наиболее предпочтительно от 5 до 10 звеньев цепи, где по меньшей мере одно кольцо системы содержит от 5 до 7 звеньев цепи и имеет по меньшей мере одну или больше точек крепления к остальной части молекулы. В некоторых реализациях гетероарил может быть 5-10-звенным гетероарил ом, содержащим 1, 2, 3 или 4 гетероатома, независимо отобранных из O, S и N. В других реализациях гетероарил может быть 5-6-звенным гетероарилом, содержащим 1, 2, 3 или 4 гетероатома, независимо отобранных из O, S и N. В других реализациях гетероарил может быть 5-звенным гетероарилом, содержащим 1, 2, 3 или 4 гетероатома, независимо отобранных из O, S и N. Термин «гетероарил» допускается использовать взаимозаменяемо с термином «гетероарильное кольцо» или с термином «гетероароматический». Гетероарильные радикалы дополнительно замещены независимо одним или более приведенными в настоящей заявке заместителями.
[113] К некоторым неограничительным примерам гетероарильного кольца относятся следующие моноциклы: 2-фуранил, 3-фуранил, N-имидазолил, 2-имидазолил, 4-имидазолил, 5-имидазолил, 3-изоксазолил, 4-изоксазолил, 5-изоксазолил, 2-оксазолил, 4-оксазолил, 5-оксазолил, N-пирролил, 2-пирролил, 3- пирролил, 2-пиридил, 3-пиридил, 4-пиридил, 2-пиримидинил, 4-пиримидинил, 5-пиримидинил, пиридазинил (например, 3-пиридазинил), 2-тиазолил, 4-тиазолил, 5-тиазолил, тетразолил (например, 5H-тетразолил и 2H-тетразолил), триазолил (например, 2-триазолил, 5-триазолил, 4H-1,2,4-триазолил, 1H-1,2,4-триазолил, и 1,2,3-триазолил), 2-тиенил, 3-тиенил, пиразолил (например, 2-пиразолил и 3-пиразолил), изотиазолил, 1,2,3-оксадиазолил, 1,2,5-оксадиазолил, 1,2,4-оксадиазолил, 1,3,4-оксадиазолил, 1,2,3-триазолил, 1,2,4-триазолил, 1,2,3-тиадиазолил, 1,3,4-тиадиазолил, 1,2,5-тиадиазолил, пиразинил, 1,3,5-триазинил и следующие бициклы: бензимидазолил, бензофурил, бензотиофенил, индолил (например, 2-индолил), пуринил, квинолинил (например, 2-квинолинил, 3-квинолинил, 4-квинолинил), и изоквинолинил (например, 1-изоквинолинил, 3-изоквинолинил или 4-изоквинолинил).
[114] Термины «карбокси» или «карбоксил», используемые как автономно, так и совместно с другими терминами, такими как «карбоксиалкил», относятся к -CO2H. Термин «карбонил», используемый как автономно, так и совместно с другими терминами, такими как «аминокарбонил», обозначается как -(C=O)-.
[115] Термин «алкиламино», охватывает «N-алкиламино» и «N,N-диалкиламино», где аминогруппы независимо замещены одним алкильным радикалом или, соответственно, двумя алкильными радикалами. Некоторыми неограничительными примерами алкиламино радикалов являются «нижние алкиламино» радикалы с одним или двумя алкил радикалами от одного до двух атомов углерода, прикрепленных к атому азота. Подходящими алкиамино радикалами могут быть моно или диалкиламино, такие как N-метиламино, N-этиламино, N,N-диметиламино, N,N-диэтиламино и тому подобные.
[116] Термин «ариламино» относится к аминогруппам, замещаемым одним или двумя арил радикалами, например, N-фениламино. Ариламино радикалы могут быть дополнительно замещены на ариловой части кольца радикала.
[117] Термин «аминоалкил» относится к линейным или разветвленным алкил радикалам, имеющим от одного до десяти атомов углерода, любой из которых может быть замещен одним или более амин радикалами. Наиболее предпочтительные аминоалкил радикалы являются «нижними аминоалкил» радикалами, имеющими от одного до шести атомов углерода и один или более амин радикалов. К примерам таких радикалов относятся аминометил, аминоэтил, аминопропил, аминобутил и аминогексил.
[118] Термин «n-звенный», где n является целым числом, обычно описывает количество кольцеобразующих атомов в функциональной группе, где количество кольцеобразующих атомов равно n. Скажем, пиперидинил является примером 6-звенного гетероциклоалкила и 1,2,3,4-тетрагидро нафталенил является примером 10-звенной карбоциклил группы.
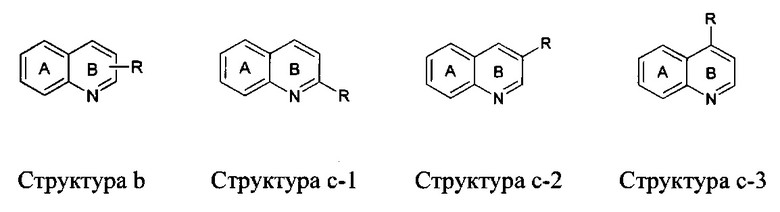
[119] В настоящей заявке связь, прочерченная от заместителя к центру одного кольца в пределах кольцевой системе (как показано ниже), представляет замещение заместителя на любой замещаемой позиции на кольце, к которому он прикреплен. Например, структура b представляет возможное замещение на любой из позиций кольца B, показанного на структуре с-1, с-2 и с-3.
[120] Термин «ненасыщенный» относится к функциональной группе с одной или более единицей ненасыщения.
[121] Термин «включающий в себя» означает открытость, то есть наличие указанного компонента, что не исключает присутствия других элементов.
[122] Термин «пропрепарат» в настоящей заявке используется для обозначения химического соединения, преобразуемого in vivo в соединение с формулой (I). Такое преобразование возможно, например, под действием гидролиза в крови или ферментного преобразования пропрепаратной формы в исходную форму, происходящего в крови или в ткани. Пропрепаратами заявляемых соединений могут быть, например, эфиры. Эфиры, которые допускается использовать в качестве пропрепаратов в заявляемом изобретении, представляют собой фенильные эфиры, алифатические (С1-C24) эфиры, ацилоксиметиловые эфиры, карбонаты, карбаматы и аминокислотные эфиры. Например, заявляемое соединение, содержащее группу ОН, может быть ацилировано на этой позиции в своей пропрепаратной форме. Другие пропрепаратные формы включают фосфаты, а именно, например фосфаты, возникающие из фосвфонации группы ОН на исходном соединении. Пропрепараты подробно рассмотрены в работах Хигучи и соавторов «Пропрепараты как новейшие системы доставки», том 14, A.C.S. Symposium Series; Роч и соавторы, «Биореверсивные носители в разработке лекарственных средств», American Pharmaceutical Association and Pergamon Press, 1987; Рауцио и соавторы, «Пропрепараты: Разработка и клинические прикладные задачи», Nat. Rev. Drug Discovery, 2008, 7, 255-270, и Хекер и соавторы, «Пропрепараты фосфатов и фосфонатов», J. Med. Chem., 2008, 51, 2328-2345, все упомянутые выше работы включены в настоящую заявку как справочный материал.
[123] «Метаболит» представляет собой продукт метаболизма заданного соединения или его соли в теле. Метаболит соединения может быть установлен с помощью традиционных известных способов, а его деятельность может быть определена посредством тестов, например, приведенных в настоящей заявке. Такие продукты могут быть, например, результатом окисления, редукции, гидролиза, амидирования, деамидирования, эстерификации, деэстерификации, ферментативного расщепления и тому подобных процессов принимаемого соединения. Соответственно, заявляемое изобретение включает в себя метаболиты заявляемых соединений, включая соединения, образованные процессом, включающим в себя контактирование заявляемого соединения с млекопитающим в течение времени, достаточного для образования метаболического продукта заявляемого соединения.
[124] Термин «фармацевтически приемлемая соль» относится к органическим или неорганическим солям заявляемого соединения. Фармацевтически приемлемые соли хорошо известны специалистам. Например, Берг и соавторы подробно описывают фармацевтически приемлемые соли в J. Pharm. Chem., 1977, 66, 1-19, эта работа включена в настоящую заявку как справочный материал. К некоторым неограничительным примерам фармацевтически приемлемой соли относятся соли аминогруппы, образованные неорганическими кислотами, такими как соляная кислота, бромистоводородная кислота, фосфорная кислота, серная кислота и перхлорная кислота, или органическим кислотами, такими как уксусная кислота, щавелевая кислота, малеиновая кислота, виннокаменная кислота, лимонная кислота, янтарная кислота или малоновая кислота, или другими известными способами, такими как ионный обмен.
[125] К другим примерам фармацевтически приемлемой соли относятся адипат, альгинат, аскорбат, аспартат, бензолсульфонат, бензоат, бисульфат, борат, бутират, камфарат, камфорсульфонат, цитрат, циклопентанпропионат, диглюконат, додецилсульфат, этансульфонат, формиат, фумарат, глюкогептонат, глицерофосфат, глюконат, гемисульфат, энантоат, гексаноат, йодгидрат, 2-гидрокси-этансульфонат, лакобионат, лактат, лаурат, лаурилсульфат, малат, малеат, малонат, метансульфонат, 2-нафталинсульфонат, никотинат, нитрат, олеат, оксалат, пальмитат, памоат, пектинат, персульфат, 3-фенилпропиноат, фосфат, пикрат, пивалат, пропионат, стеарат, сукцинат, сульфат, тартрат, тиоционат, p-толуолсульфонат, ундеканоат, соли валериановой кислоты и подобные. К солям, полученным из соответствующих оснований, относятся соли щелочных металлоа, щелочноземельных металлов, аммония и N+(C1-4alkyl)4. В заявляемом изобретении также предусматривается кватернизация любых щелочных азотсодержащих групп заявляемых соединений. Посредством такой кватернизации может быть получена вода или растворимые продукты. К показательным солям щелочей или щелочноземельных металлов относится натрий, литий, калий, кальций, марганец и подобные. К дополнительным примерам фармацевтически приемлемых солей относятся, по мере целесообразности, нетоксичный аммоний, кватернизованный аммоний и катионы амина, образованные с помощью противоионов, таких как галогенид, гидроксид, карбоксилат, сульфат, фосфат, нитрат, C1-8 сульфонат и арил сульфонат.
[126] Термин «сольват» относится к объединению или комплексу одной или более молекул растворителя и заявляемого соединения. К некоторым неограничительным примерам растворителей, образующих сольваты, относятся вода, изопропанол, этанол, метанол, DMSO, этил ацетат, уксусная кислота и этаноламин. Термин «гидрат» относится к комплексу, где молекулой растворителя является вода.
[127] В настоящей заявке термин «фармацевтически приемлемый носитель» охватывает любой и все вместе носители, диспергенты, покрытия, поверхностно-активные вещества (далее ПАВ), антиоксиданты, консерванты (например, антибактерицидные агенты, антигрибковые агенты), изотоничные агенты, агенты, задерживающие абсорбцию, соли, консерванты, стабилизаторы лекарственных средств, связующие вещества, инертные наполнители, агенты распада, лубриканты, подсластители, ароматизаторы, красители и тому подобные, а также их сочетания, известные специалистам (например, публикация Remington's Pharmaceutical Sciences, 18-е издание. Mack Printing Company, 1990, с. 1289-1329). За исключением явной несовместимости общераспространенного носителя с активным ингредиентом, предусмотрено применение такого носителя в лечебных или фармацевтических составах.
[128] Термин «терапевтически эффективное количество» заявляемого соединения относится к количеству заявляемого соединения, которое даст биологическую или медицинскую реакцию объекта, например, сокращение или ингибирование активности фермента или белка, или симптомы улучшения, облечение состояния, замедление или отсрочка прогрессирования заболевания, или профилактика заболевания, и так далее. В одной неограничительной реализации термин «терапевтически эффективное количество» относится к количеству заявляемого соединения, которое, будучи принятым объектом, оказывается эффективным для (1) по меньшей мере частичного облегчения, ингибирования, профилактики и/или улучшения состояния, или нарушения, или заболевания (i) опосредованного PI3K или (ii) связанного с активностью PI3K, или (iii) характеризуемого активностью (нормальной или аномальной) РВК, или (2) снижения или ингибирования активности PI3K или (3) снижения или ингибирования проявления РВК. В другой неограничительной реализации термин «терапевтически эффективное количество» относится к количеству заявляемого соединения, которое, при приеме в клетку, или в ткань, или в не-клеточный биологический материал, или в среду, оказывается эффективным для, по меньшей мере, частичного снижения или ингибирования активности РВК; или, по меньшей мере, частичного снижения или ингибирования проявления РВК. Значение термина «терапевтически эффективное количество», как показано в приведенной выше реализации для РВК, сохраняется и для любых других подходящих белков/пептидов/ферментов.
[129] В настоящей заявке термин «лечить», «лечебный процесс» или «лечение» любого заболевания или нарушения относится, в одной реализации к улучшению в ходе болезни или нарушения (например, замедление или прекращение, или сокращение развития заболевания или, по меньшей мере, одного из их клинических симптомов). В другой реализации термин «лечить», «лечебный процесс» или «лечение» относится к облегчению или к улучшению по меньшей мере одного физического параметра, включая не воспринимаемые пациентом. В другой реализации термин «лечебный процесс» или «лечение» относится к опосредованию течения заболевания или нарушения, либо физически (например, стабилизацией явного симптома), либо физиологически (например, стабилизацией физического параметра), или и тем, и другим. В другой реализации термин «лечебный процесс» или «лечение» относится к профилактике или отсрочке наступления, или развития, или прогрессирования заболевания или нарушения.
[130] Термин «защитная группа» или «PG» относится к заместителю, обычно используемому для блокирования или защиты конкретного функционального свойства в ходе реакции с другими функциональными группами соединения. Например, «амино-защитная группа» представляет собой заместитель, прикрепленный к аминогруппе и блокирующий или защищающий амин функциональность в соединении. К подходящим амино-защитным группам относится ацетил, трифторацетил, t-бутокси-карбонил (ВОС, Boc), бензилоксикарбонил (CBZ, CBz) и 9-фторэнилметилэнокси-карбонил (Fmoc). Аналогично, термин «гидрокси-защитная группа» относится к заместителю гидрокси группы, блокирующему или защищающему гидрокси функциональность. К подходящим защитным группам относится ацетил и силил. Аналогично, термин «карбокси-защитная группа» относится к заместителю карбокси группы, блокирующему или защищающему карбокси функциональность. К общераспространенным карбокси-защитным группам относится -CH2CH2SO2Ph, цианоэтил, 2-(триметилсилил)этил, 2-(триметилсилил)этокси-мети-1, 2-(р-толуолсульфонил) этил, 2-(р-нитрофенилсульфенил)-этил, 2-(дифенилфосфино)-этил, нитроэтил и подобные. Общее описание защитных групп и их применение приводится в издании Грин и соавторы. Защитные группы в органическом синтезе (Protective Groups in Organic Synthesis), John Wiley & Sons, New York, 1991 и Коцински и соавторы., Защитные группы (Protecting Groups), Thieme, Stuttgart, 2005.
ОПИСАНИЕ ЗАЯВЛЯЕМЫХ СОЕДИНЕНИЙ
[131] Авторами настоящей заявки открыты новые соединения, являющиеся ингибиторами активности киназы, в частности, активности РI3-киназы. Соединения, являющиеся ингибиторами PI3-киназы, могут быть полезными в лечении нарушений, связанных с неадекватной активностью киназы, в частности с неадекватной активностью РI3-киназы, например, в лечении и профилактике нарушений, опосредуемых механизмами РI3-киназы. К таким нарушениям относятся заболевания дыхательных путей, такие как астма, хроническая обструктивная болезнь легких (COPD, ХОБЛ) и идиопатический легочный фиброз (IPF); вирусные инфекции, включая вирусные инфекции дыхательных путей и вирусные обострения заболеваний дыхательных путей, такие как астма и COPD; инфекции дыхательных путей невирусного происхождения, включая аспергиллез и лейшманиоз, аллергические риниты и атопические дерматиты, аутоиммунные заболевания, включая ревматоидные артриты и рассеянный склероз, воспалительные нарушения, включая воспалительную болезнь кишечника, сердечно-сосудистые заболевания, включая тромбоз и атеросклероз; гемобластозы; нейродегенеративные заболевания; панкреатит; синдром полиорганной недостаточности (СПОН, ПОН); нефропатия; седиментация тромбоцитов; карцинома; подвижность сперматозоидов; отторжение трансплантата, отторжение ткани, поражение легких, и боль, вызываемые ревматоидным артритом, остеоартритом, боль в пояснице, боль, вызываемая общим воспалительным процессом, постгепатитная невралгия, диабетическая нейропатия, воспалительная невропатическая боль (травма), тригеминальная невралгия и центральная боль.
[132] В одной реализации заявляемые соединения могут выказывать селективность против РI3-киназ относительно других киназ.
[133] В другой реализации заявляемые соединения могут быть потенциальными ингибиторами PI3Kδ.
[134] В еще одной реализации заявляемые соединения могут выказывать селективность против PI3Kδ относительно других киназ.
[135] Заявляется соединение со скелетной формулой (I):
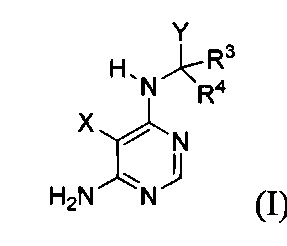 ,
,
или его стереоизомер, геометрический изомер, таутомер, N-оксид, гидрат, сольват, метаболит, фармацевтически приемлемая соль или их пропрепарат, где каждый из X, Y, R3 и R4 определяется в настоящей заявке.
[136] В некоторых реализациях X представляет собой (С3-C7)гетероциклил, -(С1-C4)алкилен-(C3-C7)гетероциклил, (C6-C10)арил, -(C1-C4)алкилен-(C6-C10)арил, 5-10-членный гетероарил, или -(C1-C4)алкилен-(5-10-членный гетероарил), где X дополнительно замещен 1, 2, 3, 4 или 5 R1 группами;
Y представляет собой
 ,
,
где Y дополнительно замещен 1, 2, 3 или 4 R2 группами;
каждый R1 и R2 независимо представляет собой Н, F, Cl, Br, CN, NO2, оксо(=O), -C(=O)Ra, -C(=O)ORa, -C(=O)NRaRb, -OC(=O)NRaRb, -OC(=O)ORa, -N(Rc)C(=O)NRaRb, -N(Rc)C(=O)ORa, -N(Rc)C(=O)Ra, -S(=O)2NRaRb, -S(=O)2Ra, -N(Rc)S(=O)2Ra, -N(Rc)-(C1-C4)алкилен-S(=O)2Ra, -(C1-C4)алкилен-C(=O)NRaRb, -(C1-C4)aлкилeн-N(Rc)C(=O)NRaRb, -(C1-C4)aлкилeн-N(Rc)C(=O)ORa, -(C1-C4)aлкилeн-OC(=O)NRaRb, -(C1-C4)алкилен-S(=O)2NRaRb, -(C1-C4)aлкилeн-N(Rc)S(=O)2Ra, ORa, NRaRb, -(C1-C4)aлкилeн-ORa, -(C1-C4)алкилен-NRaRb, (С1-C6)алкил, (С2-C6)алкенил, (С2-C6)алкинил, (С3-C8)циклоалкил, -(C1-C4)алкилен-(С3-C8)циклоалкил, (C3-C7)гетероциклил, -(C1-C4)алкилен-(C3-C7)гетероциклил, (C6-C10)арил, -(C1-C4)алкилен-(C6-C10)арил, 5-10-членный гетероарил или -(C1-C4)алкилен-(5-10-членный гетероарил), где каждый из (С1-C6)алкил, (С2-C6)алкенил, (С2-C6)алкинил, (С3-C8)циклоалкил, -(C1-C4)алкилен-(С3-C8)циклоалкил, (С3-C7)гетероциклил, -(C1-C4)алкилен-(С3-C7)гетероциклил, (C6-C10)арил, -(C1-C4)алкилен-(C6-C10)арил, 5-10-членный гетероарил и -(C1-C4)алкилен-(5-10-членный гетероарил) дополнительно замещен 1, 2, 3 или 4 заместителями, независимо выбранными из F, Cl, Br, CN, ORa, NRaRb, (С1-C6)алкил, -(C1-C4)aлкилeн-ORa и -(C1-C4)алкилен-NRaRb;
каждый R3 и R4 независимо представляет собой Н, F, CN, -C(=O)Rа, -C(=O)ORa, -C(=O)NRaRb, -(C1-C4)алкилн-C(=O)NRaRb, -(C1-C4)алкилен-N(Rc)C(=O)NRaRb, -(C1-C4)алкилен-N(Rc)С(=O)ORа, -(C1-C4)алкилен-OС(=O)NRaRb, -(C1-C4)алкилен-S(=O)2NRaRb, -(C1-С4)алкилен-N(Rc)S(=O)2Rb, -(C1-C4)aлкилeн-ORa, -(C1-C4)aлкилeн-NRaRb, (С1-C6)алкил, (С2-C6)алкенил, (С2-C6)алкинил, (С3-C8)циклоалкил, -(C1-C4)алкилен-(С3-C8)циклоалкил, (С3-C7)гетероциклил, -(C1-C4)алкилен-(С3-C7)гетероциклил, (C6-C10)арил, -(C1-C4)алкилен-(C6-C10)арил, 5-10-членный гетероарил, или -(C1-C4)алкилен-(5-10-членный гетероарил), где каждый из (С1-С6)алкил, (С2-C6)алкенил, (С2-C6)алкинил, (С3-C8)циклоалкил, -(C1-C4)алкилен-(С3-C8)циклоалкил, (С3-C7)гетероциклил, -(C1-C4)алкилен-(С3-C7)гетероциклил, (C6-C10)арил, -(C1-C4)алкилен-(C6-C10)арил, 5-10-членный гетероарил и -(C1-C4)алкилен-(5-10-членный гетероарил) дополнительно замещен 1, 2, 3 или 4 заместителями, независимо выбранными из F, Cl, Br, CN, ORa, NRaRb, (С1-C6)алкил, -(C1-C4)aлкилeн-ORa и -(C1-C4)aлкилeн-NRaRb; или R3 и R4, вместе с атомом углерода, к которому они присоединены, формируют дополнительно замещенный 3-8-членный карбоцикл или гетероцикл; и
каждый Ra, Rb и Rc независимо представляет собой Н, (С1-C6)алкил, (С2-C6)алкенил, (C2-C6)алкинил, (C3-C6)циклоалкил, -(С1-C4)алкилен-(C3-C6)циклоалкил, (C3-C6)гетероциклил, -(C1-C4)алкилен-(C3-C6)гетероциклил, (C6-C10)арил, -(C1-C4)алкилен-(C6-C10)арил, 5-10-членный гетероарил, или -(C1-C4)алкилен-(5-10-членный гетероарил), где каждый из (C1-C6)алкил, (С2-C6)алкенил, (С2-C6)алкинил, (С3-C6)циклоалкил, -(C1-C4)алкилен-(C3-C6)циклоалкил, (С3-C6)гетероциклил, -(C1-C4)алкилен-(C3-C6)гетероциклил, (C6-C10)арил, -(C1-C4)алкилен-(C6-C10)арил, 5-10-членный гетероарил и -(C1-C4)алкилен-(5-10-членный гетероарил) дополнительно замещен 1, 2, 3 или 4 заместителями, независимо выбранными из F, Cl, CN, N3, ОН, NH2, (C1-C6)алкил, (С1-C6)галоалкил, (С1-C6)алкокси и (С1-C6)алкиламино; или Ra и Rb, вместе с атомом азота, к которому они присоединены, формируют дополнительно замещенный 3-8-членный гетероцикл.
[137] В другой реализации X представляет собой (С3-C7)гетероциклил или 5-10-членный гетероарил, где X дополнительно замещен 1, 2, 3 или 4 R1 группами.
[138] В другой реализации каждый R1 и R2 независимо представляет собой Н, F, Cl, CN, оксо (=O), -C(=O)ORa, -C(=O)NRaRb, -N(Rc)C(=O)NRaRb, -N(Rc)C(=O)ORa, -N(Rc)C(=O)Ra, -S(=O)2NRaRb, -N(Rc)S(=O)2Ra, -N(Rc)-(C1-C4)алкилен-S(=O)2Ra, -(C1-C4)алкилен-C(=O)NRaRb, -(C1-C4)алкилен-N(Rc)C(=O)NRaRb, -(C1-C4)алкилен-S(=O)2NRaRb, -(C1-C4)aлкилeн-N(Rc)S(=O)2Ra, ORa, NRaRb, -(C1-C4)aлкилeн-ORa, -(C1-C4)aлкилeн-NRaRb, (С1-C6)алкил, (С2-C6)алкенил, (С2-C6)алкинил, (С3-C8)циклоалкил, -(С1-C4)алкилен-(С3-C8)циклоалкил, (С3-C7)гетероциклил, -(C1-C4)алкилен-(C3-C7)гетероциклил, фенил, -(C1-C4)алкилен-фенил, или 5-6-членный гетероарил, где каждый из (С1-C6)алкил, (С2-C6)алкенил, (С2-C6)алкинил, (С3-C8)циклоалкил, -(С1-C4)алкилен-(С3-C8)циклоалкил, (С3-C7)гетероциклил, -(C1-C4)алкилен-(С3-C7)гетероциклил, фенил, -(C1-C4)алкилен-фенил и 5-6-членный гетероарил дополнительно замещен 1, 2, 3 или 4 заместителями, независимо выбранными из F, CN, ORa, NRaRb, (C1-C3)алкил, -(C1-C4)алкилен-ORа и -(C1-C4)алкилен-NRaRb.
[139] В другой реализации каждый R3 и R4 независимо представляет собой Н, F, CN, -C(=O)NRaRb, -(С1-C2)алкилен-C(=O)NRaRb, -(C1-C2)алкилен-N(Rc)C(=O)NRaRb, -(C1-C2)алкилен-N(Rc)C(=O)ORa, -(C1-C2)алкилен-OС(=O)NRaRb, -(C1-C2)алкилен-S(=O)2NRaRb, -(C1-C2)алкилен-N(Rc)S(=O)2Rb, -(С1-C2)алкилен-ORа, -(C1-C2)aлкилeн-NRaRb, (C1-C4)алкил, (С2-C4)алкенил, (С2-C4)алкинил, (C3-C6)циклоалкил,-(С1-C2)алкилен-(C3-C6)циклоалкил, (C3-C5)гетероциклил, -(С1-C2)алкилен-(C3-C5) гетероциклил, фенил, -(C1-C2)алкилен-фенил, 5-членный гетероарил, или -(C1-C2)алкилен-(5-членный гетероарил), где каждый из: (C1-C4)алкил, (С2-C4)алкенил, (С2-C4)алкинил, (C3-C6)циклоалкил, -(С1-C2)алкилен-(C3-C6)циклоалкил, (С3-C6)гетероциклил, -(С1-C2)алкилен-(C3-C5)гетероциклил, фенил, -(С1-C2)алкилен-фенил, 5-членный гетероарил и -(C1-C2)алкилен-(5-членный гетероарил) дополнительно замещен 1, 2, 3 или 4 заместителями, независимо выбранными из F, Cl, Br, CN, ORa NRaRb, (С1-C6)алкил, -(C1-C4)алкилен-ORа и -(C1-C4)алкилен-NRaRb; или R3 и R4, вместе с атомом углерода, к которому они присоединены, формируют дополнительно замещенный 3-8-членный карбоцикл или гетероцикл.
[140] В другой реализации каждый Ra, Rb и Rc независимо представляет собой Н, (С1-С6)алкил. (C2-C6)алкенил, (С2-C6)алкинил, (С3-C6)циклоалкил, -(С1-C4)алкилен-(С3-C6)циклоалкил, (С3-C6)гетероциклил, -(C1-C4)алкилен-(C3-C6)гетероциклил, или 5-10-членный гетероарил, где каждый из (С1-C6)алкил, (С2-C6)алкенил, (С2-C6)алкинил, (C3-C6)циклоалкил, -(C1-C4)алкилен-(C3-C6)циклоалкил, (С3-C6)гетероциклил, -(C1-C4)алкилен-(C3-C6)гетероциклил и 5-10-членный гетероарил дополнительно замещенный 1, 2, 3 или 4 заместителями, независимо выбранными из F, CN, N3, ОН, NH2, (C1-C3)алкил, (C1-C3)галоалкил, (C1-C4)алкокси и (C1-C4)алкиламино.
[141] В другой реализации X представляет собой моновалентный гетероциклил или гетероарильную группу, полученную из одной из перечисленных ниже скелетных формул:
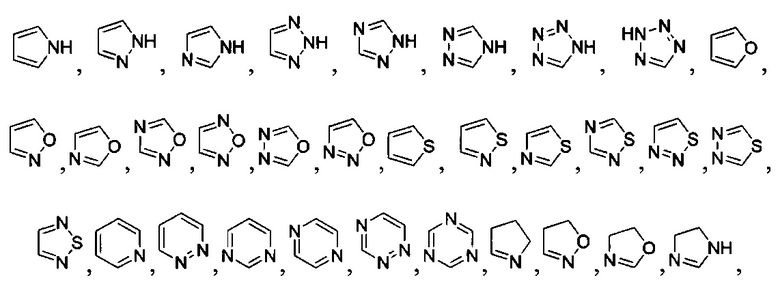
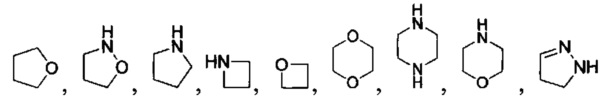 , или
, или  ;
;
и где Y дополнительно замещен 1, 2, 3 или 3 R1 группами;
[142] В другой реализации Y представляет собой
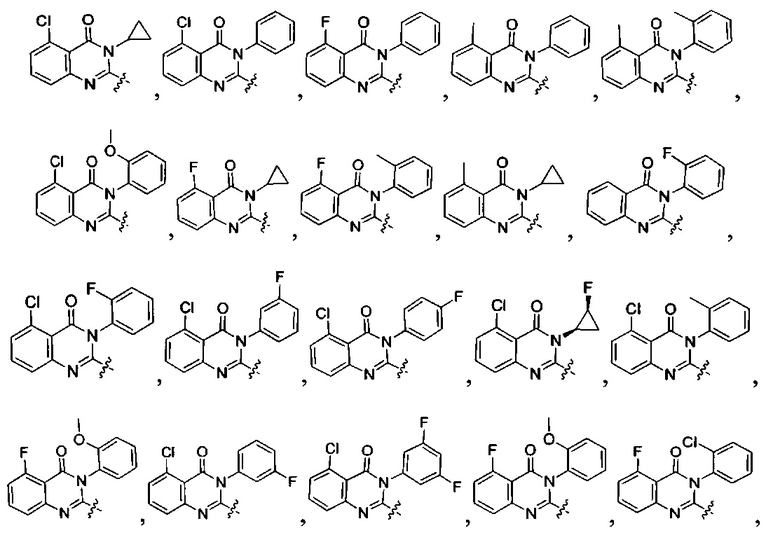
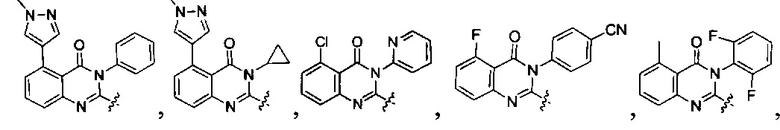 или
или
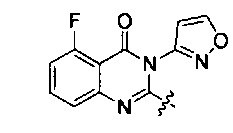 ;
;
и где Y дополнительно замещен 1 или 2 R2 группами;
[143] В другой реализации каждый R1 и R2 независимо представляет собой Н, F, Cl, CN, оксо (=O), -C(=O)ORa, -C(=O)NRaRb, -N(Rc)C(=O)NRaRb, -N(Rc)C(=O)ORa, -N(Rc)C(=O)Ra, -S(=O)2NRaRb, -N(Rc)S(=O)2Ra, ORa, NRaRb, (C1-C3)алкил, (С2-C4)алкенил, (С2-C4)алкинил, (С3-C6)циклоалкил, -(С1-C2)алкилен-(C3-C6)циклоалкил, (C3-C5)гетероциклил, -(C1-C2)алкилен-(C3-C5)гетероциклил, фенил, или -(C1-C2)алкилен-фенил, где каждый из (C1-C3)алкил, (С2-C4)алкенил, (С2-C4)алкинил, (С3-C6)циклоалкил, -(С1-C2)алкилен-(С3-C6)циклоалкил, (C3-C5)гетероциклил, -(C1-C2)алкилен-(C3-C5)гетероциклил, фенил и -(С1-C2)алкилен-фенил дополнительно замещен 1, 2, 3 или 4 заместителями, независимо выбранными из F, CN, 0Ra, NRaRb и (C1-C3)алкил.
[144] В другой реализации каждый R3 и R4 независимо представляет собой Н, F, CN, (C1-C3)алкил, (С3-C6)циклоалкил, (C3-C5)гетероциклил, или -(С1-C2)алкилен-(C3-C5)гетероциклил, где каждый из (С1-C3)алкил, (С3-C6)циклоалкил, (C3-C5)гетероциклил и -(С1-C2)алкилен-(C3-C5)гетероциклил дополнительно замещен 1, 2, 3 или 4 заместителями, независимо выбранными из: F, Cl, Br, CN, ORa, NRaRb, (С1-C6)алкил, -(C1-C4)aлкилeн-ORa и -(C1-C4)алкилен-NRaRb; или R3 и R4, вместе с атомом углерода, к которому они присоединены, формируют дополнительно замещенный 3-8-членный карбоцикл или гетероцикл.
[145] В другой реализации каждый Ra, Rb и Rс независимо представляет собой Н, (C1-C3)алкил, (С2-C4)алкенил, (С2-C4)алкинил, (С3-C6)циклоалкил, (C3-C5)гетероциклил, или 5-6-членный гетероарил, где каждый из (C1-C3)алкил, (С2-C4)алкенил, (С2-C4)алкинил, (C3-C6)циклоалкил, (C3-C5)гетероциклил и 5-6-членный гетероарил дополнительно замещен 1, 2, 3 или 4 заместителями, независимо выбранными из: F, CN, ОН, NH2, (C1-C3)алкил, (C1-C3)галоалкил, (С1-C3)алкокси и (C1-C3)алкиламино.
[146] Некоторые неограничительные примеры заявляемого соединения представлены ниже:
Таблица 1
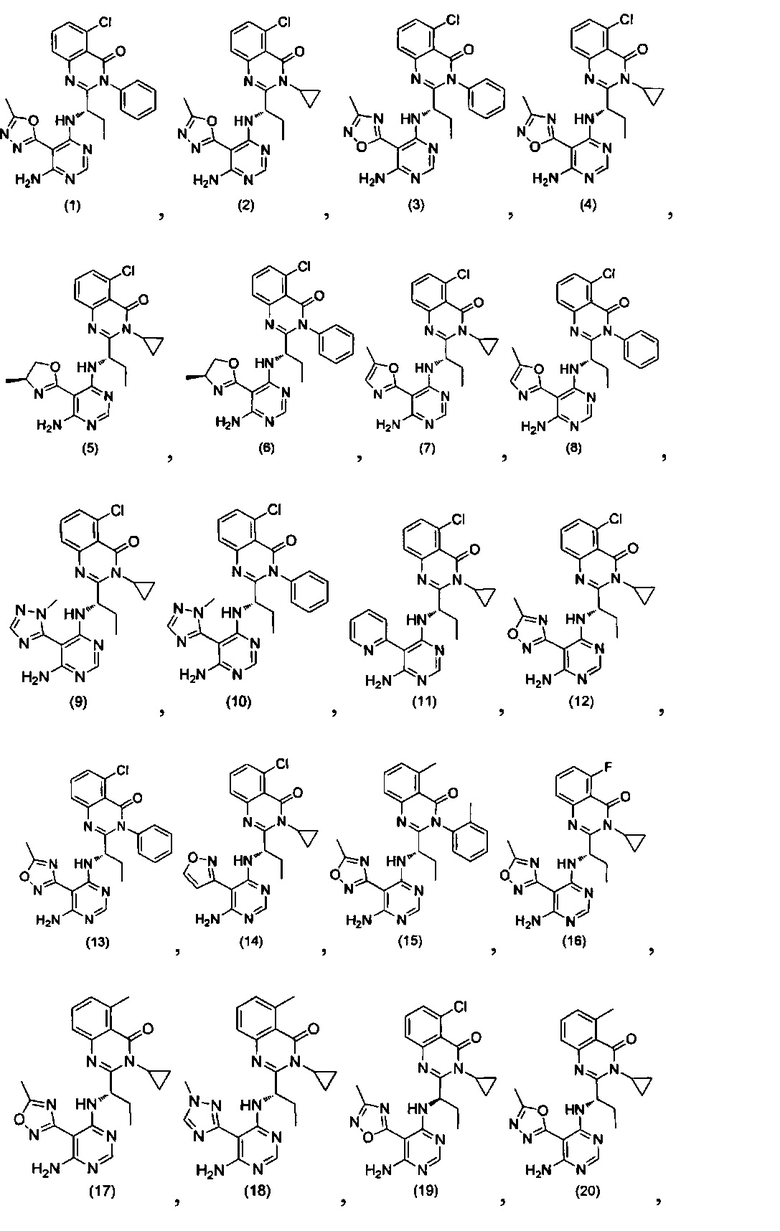
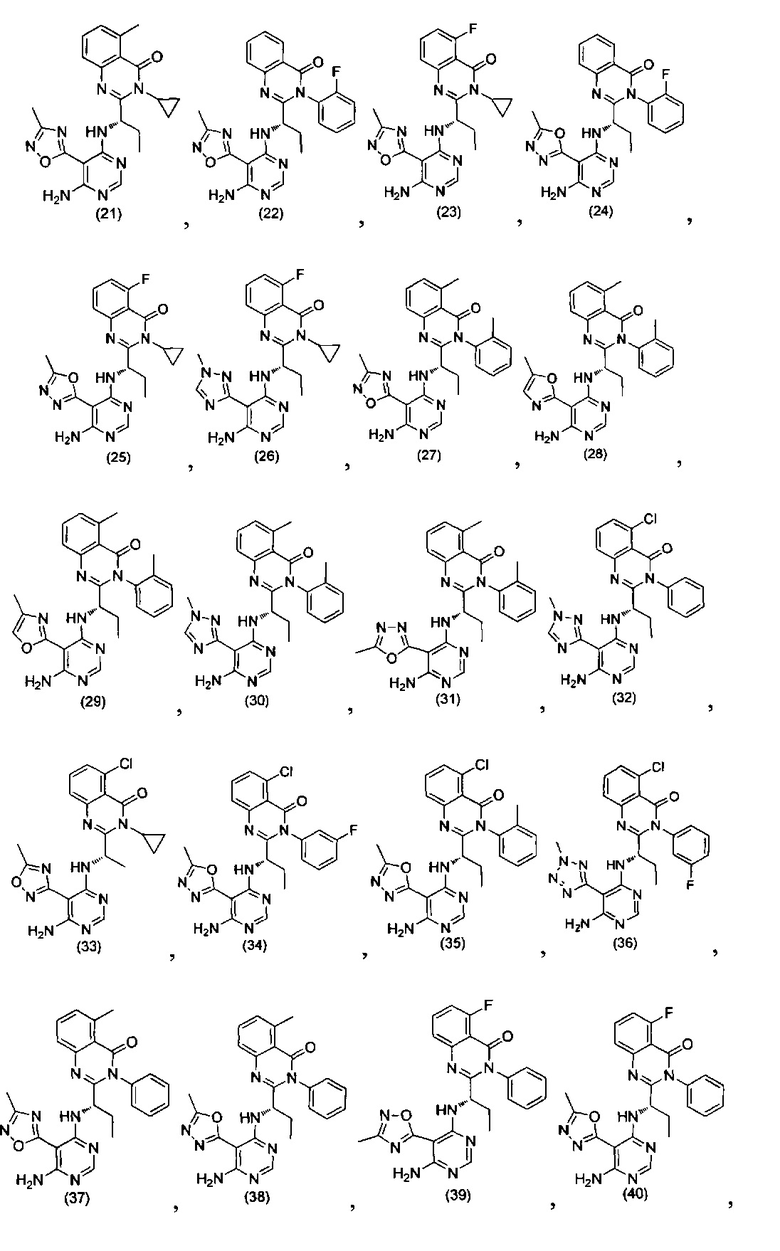
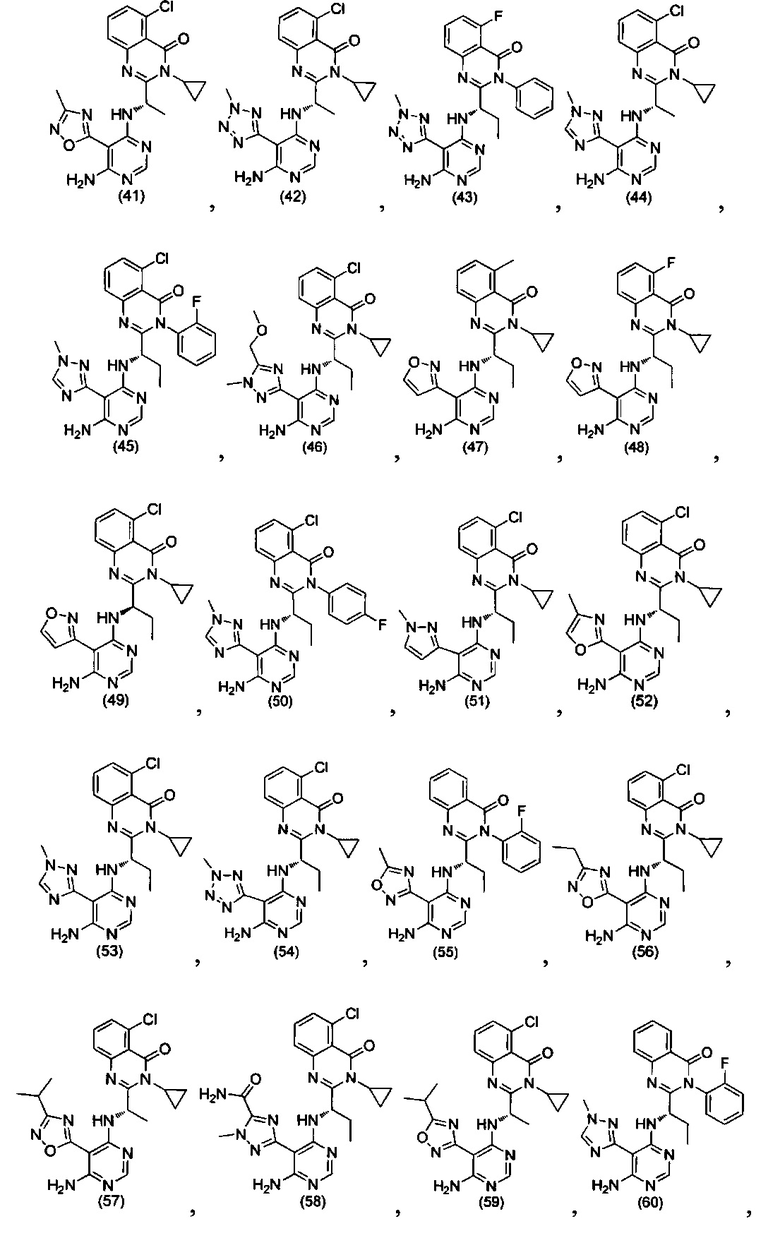
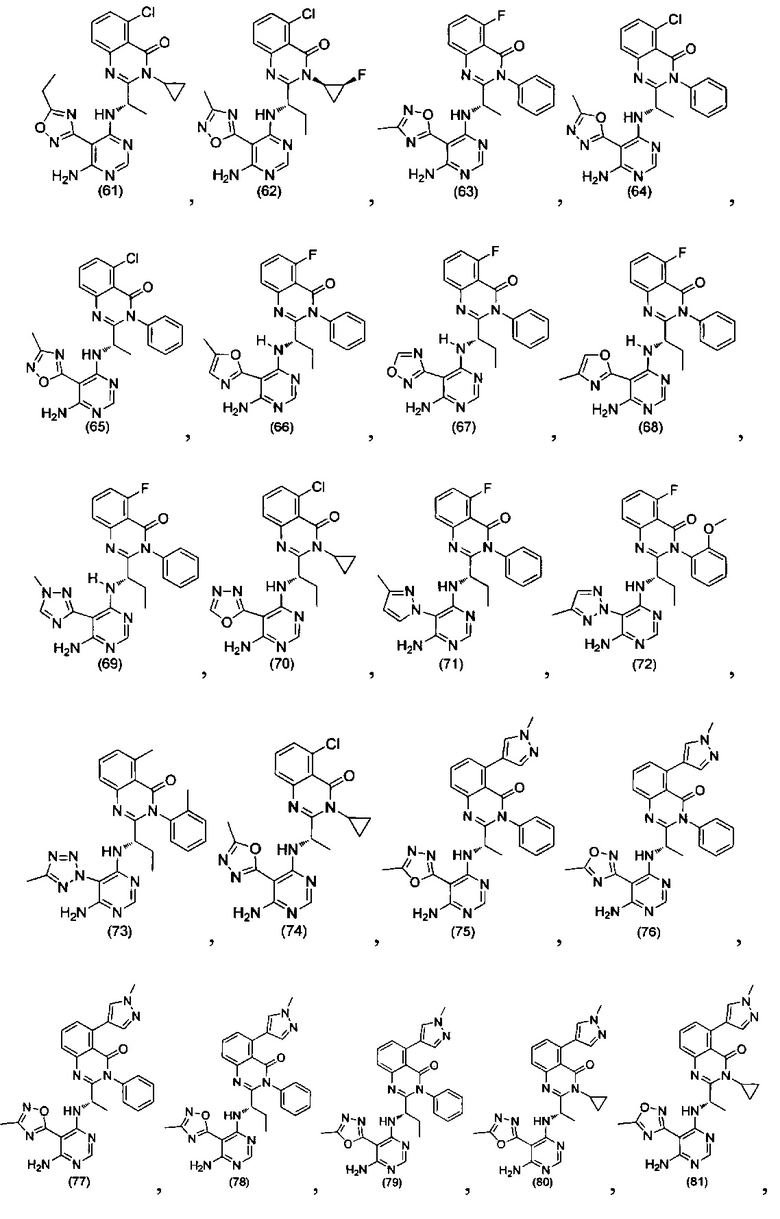

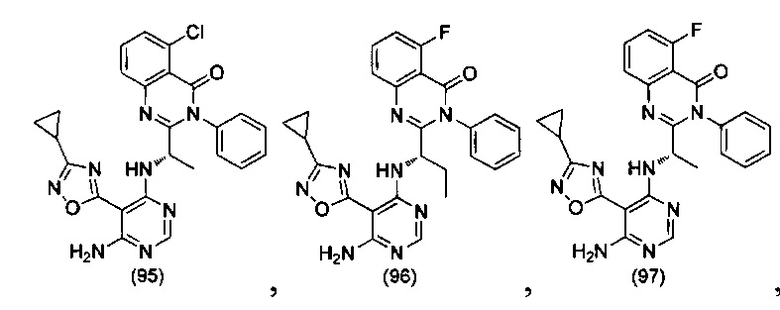 или
или 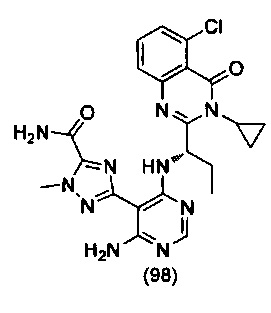 .
.
[147] Заявляемая фармацевтическая композиция содержит фармацевтически приемлемый носитель, инертный наполнитель, разбавитель, активатор, основу или сочетание перечисленных компонентов, и соединение с формулой (I) или его фармацевтически приемлемую соль. В некоторых реализациях соединение представлено в форме жидкости, твердого вещества, полумягкого вещества, геля или аэрозоля.
[148] Заявляется способ ингибирования фосфатидилинозитол 3-киназы (PI3 киназа), заключающийся в следующем: контактирование с PI3 киназой эффективного количества заявляемого соединения. В некоторых реализациях этап контактирования включает в себя контактирование с клеткой, содержащей упомянутую PI3 киназу. В некоторых реализациях способа ингибирование происходит в организме субъекта лечения, страдающего от нарушения, связанного с нарушенным функционированием одного или более типов PI3 киназы. Некоторые показательные заболевания, включая нарушенное функционирование одного или более типов PI3 киназы, отобраны из группы, состоящей из аутоиммунных заболеваний, ревматоидного артрита, заболеваний дыхательных путей, аллергических реакций и карцином различного типа.
[149] В некоторых реализациях способ заключается в приеме второго лечебного агента объектом лечения.
[150] В определенных реализациях состояние или нарушение, опосредованное PI3K, отобрано из ревматоидного артрита, анкилозирующего спондилоартрита, остеоартроза, псориатического артрита, псориаза, воспалительных заболеваний и аутоиммунных заболеваний. В других реализациях состояние или нарушение, опосредованное PI3K, отобрано из следующего списка: сердечно-сосудистые заболевания, атеросклероз, гипертония, тромбоз глубоких вен, инсульт, инфаркт миокарда, нестабильная стенокардия, тромбоз, тромбоэмболия легочной артерии (ТЭЛА), тромболитические заболевания, острая артериальная ишемия, периферийные тромболитические закупоривания и коронарная недостаточность. В других реализациях состояние или нарушение, опосредованное PI3K, отобрано из следующего списка: карцинома, колоректальный рак, глиобластома, карцинома эндометрия, гепатоцеллюлярная карцинома, бронхогенная карцинома, меланома, почечно-клеточная карцинома, карцинома щитовидной железы, клеточная лимфома, лимфопролиферативные нарушения, мелкоклеточная карцинома легких, сквамозноклеточная карцинома легких, глиома, рак молочной железы, рак предстательной железы, рак яичников, рак шейки матки и лейкемия. В другой реализации состояние или нарушение, опосредованное PI3K, отобрано из диабета II типа. В других реализациях состояние или нарушение, опосредованное PI3K, отобрано из респираторных заболеваний: бронхит, астма и хроническая обструктивная болезнь легких. В определенных реализациях объектом является человек.
[151] Заявляется также способ лечения состояния или нарушения пациента, опосредованного PI3K, содержащий этап приема соединения по любой из приведенных выше реализаций.
[152] Заявляется также способ лечения ревматоидного артрита, анкилозирующего спондилоартрита, остеоартроза, псориатического артрита, псориаза, воспалительных заболеваний и аутоиммунных заболеваний, содержащий этап приема соединения пациентом по любой из приведенных выше реализаций.
[153] Заявляется также способ лечения заболеваний дыхательных путей, включая астму, хроническую обструктивную болезнь легких (COPD, ХОБЛ) и идиопатический легочный фиброз (IPF), содержащий этап приема соединения пациентом по любой из приведенных выше реализаций.
[154] Заявляется также способ лечения воспалительных болезней кишечника, воспалительных заболеваний глаз, воспалительных или лабильных заболеваний мочевого пузыря, псориаза, кожных заболеваний с воспалительными компонентами, хронических воспалительных состояний, системной красной волчанки (SLE), миастения (myestenia gravis), острого диссеминированного энцефаломиелита, идиопатической тромбоцитопенической пурпуры (ИТП, болезнь Верльгофа), рассеянного склероза, синдрома Шегрена и аутоиммунной гемолитической анемии, аллергических состояний и гиперчувствительности, содержащий этап приема пациентом соединения по любой из приведенных ниже реализаций.
[155] Заявляется также способ лечения злокачественных опухолей пациента, опосредованных, зависящих или связанных с активностью PI3K, в частности, с активностью РВKδ, содержащий этап приема пациентом соединения по любой из приведенных выше или ниже реализаций.
[156] Заявляется также способ лечения злокачественных опухолей, выбранных из следующих: острый миелоидный лейкоз, миело-дисплазированный синдром, миелопролиферативные заболевания, хронический миелоидный лейкоз, Т-клеточный острый лимфобластный лейкоз, В-клеточная острая лимфобластозная лейкемия, неходжкинская лимфома, В-клеточный острый лимфобластный лейкоз, солидные опухоли и рак молочной железы, содержащий этап приема пациентом соединения по любой из приведенных выше или ниже реализаций.
[157] Заявляется также использование соединения по любой из приведенных выше реализаций в качестве лекарственного средства.
[158] Заявляется также использование соединения по любой из приведенных выше реализаций в производстве лекарственного средства для лечения РI3K-опосредованного состояния или нарушения пациента.
[159] Заявляется также использование соединения по любой из приведенных выше реализаций в производстве лекарственного средства для лечения ревматоидного артрита, анкилозирующего спондилоартрита, остеоартроза, псориатического артрита, псориаза, воспалительных заболеваний, заболеваний дыхательных путей, включая астму, хроническую обструктивную болезнь легких (COPD, ХОБЛ) и идиопатический легочный фиброз (IPF), аутоиммунные заболевания и злокачественные опухоли.
[160] В отсутствие иных указаний, все стереоизомеры, геометрические изомеры, таутомеры, сольваты, гидраты, метаболиты, соли и фармацевтически приемлемые пропрепараты заявляемых соединений относятся к области заявляемого изобретения.
[161] В определенных реализациях соль является фармацевтически приемлемой солью. Фраза «фармацевтически приемлемая» означает, что вещество или соединение должно быть химически и/или токсикологически совместимо с другими ингредиентами препарата, и/или с млекопитающим, подвергающимся лечению этим препаратом.
[162] В состав заявляемых соединений должны также входить соли таких соединений, которые не обязательно являются фармацевтически приемлемыми солями, и которые могу быть полезны как промежуточные продукты для приготовления и/или очистки соединений с формулой (I), и/или для отделения энантиомеров от соединений с формулой (I).
[163] Фармацевтически приемлемые кислотно-аддитивные соли могут образовываться с неорганическими и органическими кислотами, например, ацетат, аспарат, бензоат, безилат, бромид/гидробромид, бикарбонат/карбонат, бисульфат/сульфат, каморсульфанат, хлорид/гидрохлорид, хлортеофиллонат, цитрат, этандисульфонат, фумарат, глюцептат, глюконат, глюкуронат, гиппурат, гидройодид/йодид, изетионат, лактат, лактобионат, лаурилсульфат, малат, малеат, малонат, манделат, месилат, метилсульфат, нафтоат, напсилат, никотинат, нитрат, октадеканоат, олеат, оксалат, пальмитат, памоат, фосфат/водород фосфат/дигидроген фосфат, полигалатуронат, пропионат, стеарат, сукцинат, субсалицинате, тартрат, тосилат и трифторацетат соли.
[164] К неорганическим кислотам, из которых можно получить соли, относятся, например, соляная кислота, бромистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота, и подобные.
[165] К органическим кислотам, из которых можно получить соли, относятся, например, уксусная кислота, пропионовая кислота, гликолевая кислота, щавелевая кислота, малеиновая кислота, малоновая кислота, янтарная кислота, фумаровая кислота, виннокаменная кислота, лимонная кислота, бензойная кислота, миндальная кислота, метансульфоновая кислота, этансульфоновая кислота, толуолсульфоновая кислота, сульфосалициловая кислота, и подобные.
[166] Фармацевтически приемлемые щелочно-аддитивные соли могут быть образованы с неорганическими и органическими основаниями.
[167] К неорганическим основаниям, из которых такие соли могут образовываться, относятся, например, соли аммония и металлы из столбцов с I по XII периодической таблицы. В определенных реализациях соли образуются из натрия, калия, аммония, кальция, марганца, железа, серебра, цинка и меди; в частности, к подходящим солям относятся соли аммония, калия, натрия, кальция и марганца.
[168] К неорганическим основаниям, из которых такие соли могут образовываться, относятся, например, первичные, вторичные и третичные амины, замещенные амины, включая замещенные амины природного происхождения, циклические амины, щелочные ионообменные смолы и подобные. К определенным органическим аминам относятся изополиамин, бензатин, хоинат, диэтаноламин, диэтиламин, лизин, меглумин, пиперазин и трометамин.
[169] Заявляемые фармацевтически приемлемые соли могут быть синтезированы из щелочных или кислотных функциональных групп общепринятыми химическими способами. Как правило, такие сои могут быть приготовлены посредством реакции свободных кислотных форм этих химических соединений со стехиометрическим количеством подходящего основания (такого, как гидроксид, карбонат, бикарбонат Na, Са, Mg или K и подобные), посредством реакции свободных щелочных форм этих химических соединений со стехиометрическим количеством подходящей кислоты. Такие реакции обычно производят в воде или в органическом растворителе, или в их смеси. Как правило, желательно, по мере возможности, пользоваться безводной средой наподобие эфира, этилацетата, этанола, изопропанола или ацетонитрила. Перечни дополнительных подходящих солей приводятся, например, в публикации «Remington's Pharmaceutical Sciences», 20-е издание. Mack Publishing Company, Easton, PA, 1985; и в «Справочнике фармацевтических солей: свойства, выбор и применение», авторы Сталь и Вермут, Wiley-VCH, Weinheim, Germany, 2002.
[170] Кроме того, заявляемые соединения, включая их соли, могут быть также получены в форме их гидратов, или содержать другие растворители, используемые для их кристаллизации. Заявляемые соединения могут по своей природе или по расчету образовывать сольваты с фармацевтически приемлемыми растворителями (включая воду); поэтому заявляемое изобретение, по замыслу авторов, охватывает как сольватированные, так и не сольватированные формы.
[171] Заявляются также способы приготовления, способы сепарирования и способы очистки соединений с формулой (I). Заявляемые соединения могут, как правило, иметь несколько асимметричных центров, и обычно изображаются в форме рацемических смесей. Авторы заявляемого изобретения намереваются охватить рацемические смеси, частично рацемические смеси и отдельные энантиомеры и диастереомеры.
[172] Заявляемые соединения могут существовать в форме одного из возможных изомеров, ротамеров, атропоизомеров, таутомеров или их смесей. Авторы заявляемого изобретения намереваются охватить смеси изомеров, ротамеров, атропоизомеров, таутомеров, частично смешанных изомеров, ротамеров, атропоизомеров или таутомеров, и отдельные изомеры, ротамеры, атропоизомеры, таутомеры.
[173] Любая формула, приведенная в настоящей заявке, предназначена для представления как не меченых форм, так и изотопно меченых форм соединений. Структуры изотопно меченых химических соединений представлены формулами в настоящей заявке, за исключением того, что один или более атомов замещены атомом с выбранной атомной массой или массовым числом. К примерам изотопов, которые могут быть включены в состав заявляемых соединений, относятся изотопы водорода, углерода, азота, кислорода, фосфора, фтора и хлора, такие как 2Н, 3Н, 11C, 13C, 14С, 15N, 18F, 31Р, 32Р, 35S, 36Cl и 125I.
[174] Заявляемые соединения включают в себя изотопно меченые соединения, по определениям настоящей заявки, например, с наличием радиоактивных изотопов, таких как 3Н, 14С и 18F, или с наличием нерадиоактивных изотопов, таких как 2Н и 13С. Такие изотопно меченые соединения полезны в исследованиях метаболизма (с 14С), исследованиях химической кинетики (например, с 2Н или 3Н), в распознавании и обработке изображений, например, в позитронно-эмиссионной томографии (ПЭТ) или в однофотонной эмиссионной компьютерной томографии (ОФЭКТ), включая оценку тканевого распределения лекарственных препаратов и субстратов, или в радиотерапии пациентов. В частности, 18F или изотопно меченое соединение может оказаться особенно полезным для исследований ПЭТ или ОФЭКТ. Изотопно меченые соединения с формулой (I) можно, как правило, приготовить общепринятыми известными способами, или действуя по аналогии с прилагаемыми примерами и способами приготовления с применением изотопно-меченого реагента вместо немеченого реагента, использованного ранее.
[175] Кроме того, замещение более тяжелыми изотопами, такими как дейтерий (т.е. 2Н или D), может дать определенные терапевтические преимущества за счет повышенной метаболической стабильности, например, увеличенное in vivo время полураспада или сокращенные требования к дозировке, или улучшение в терапевтическом индексе.
Необходимо отметить, что дейтерий в этом контексте считается заместителем соединения с формулой (I). Концентрация такого более тяжелого изотопа, а именно дейтерия, может быть определена коэффициентом изотопного обогащения. Термин «коэффициент изотопного обогащения», используемый в настоящей заявке, означает отношение количества изотопа к природной распространенности конкретного изотопа. Если в качестве заместителя в заявляемом соединении указан дейтерий, коэффициент изотопного обогащения такого соединения для каждого обозначенного атома дейтерия составляет по меньшей мере 3500 (52,5% дейтерия, внедренного в каждый обозначенный атом дейтерия), по меньшей мере 4000 (60% внедренного дейтерия), по меньшей мере 4500 (67,5% внедренного дейтерия), по меньшей мере 5000 (75% внедренного дейтерия), по меньшей мере 5500 (82,5% внедренного дейтерия), по меньшей мере 6000 (90% внедренного дейтерия), по меньшей мере 6333,3 (95% внедренного дейтерия), по меньшей мере 6466,7 (97% внедренного дейтерия), по меньшей мере 6600 (99% внедренного дейтерия), или по меньшей мере 6633,3 (99,5% внедренного дейтерия). Фармацевтически приемлемые сольваты в соответствии с заявляемым изобретением включают такие, где растворитель кристаллизации может быть изотопно замещенным, например, D2O, ацетон-d6, или DMSO-d6.
СОСТАВ, ТЕХНОЛОГИИ ПРИГОТОВЛЕНИЯ И ПРИЕМ ЗАЯВЛЯЕМЫХ СОЕДИНЕНИЙ
[176] Заявляемые фармацевтические композиции включают в себя соединение с формулой (I), или соединение из перечисленных в таблице 1; и фармацевтически приемлемый носитель, активатор или основу. Количество соединения в заявляемой фармацевтической композиции обеспечивает ощутимое ингибирование протеинкиназы в биологическом образце или в организме пациента..
[177] Будет также учитываться, что определенное количество заявляемых соединений может существовать в свободной форме для лечения, или, где возможно, как их фармацевтически приемлемое производное. К некоторым неограничительным примерам фармацевтически приемлемых производных относятся фармацевтически приемлемые пропрепараты, соли, эфиры, соли таких эфиров, или другой аддукт или производное, которое, после приема нуждающимся пациентом, способно прямо или косвенно образовать соединение, иным способом описанное в настоящей заявке, или его метаболит, или его радикал.
[178] Согласно изложенному выше, заявляемые фармацевтические композиции или фармацевтически приемлемые композиции дополнительно включают в себя фармацевтически приемлемый носитель, активатор или основу, которые, по настоящей заявке, охватывает любой и все вместе растворители, разбавители или другую жидкую основу, диспергенты или средства создания суспензии, поверхностно-активные вещества, изотоничные агенты, загущающие или эмульгирующие вещества, консерванты, твердые связки, смазки и подобные, в зависимости от конкретной желаемой дозировочной формы.
В изданиях Remington: «Наука и практика фармакологии», 21-е издание, 2005, из-во D.B. Troy, Lippincott Williams & Wilkins, Philadelphia, и «Энциклопедия фармацевтической технологии», eds. J. Swarbrick and J.C. Boylan, 1988-1999, Marcel Dekker, New York, ссылка на которые содержится в настоящей заявке, приводятся различные носители, используемые в составлении фармацевтически приемлемых соединений, а также известные способы их приготовления. Исключая такой крайний случай, как несовместимость среды стандартного носителя с заявляемыми соединениями, например, возникновение нежелательного биологического эффекта или взаимодействие носителя с другим компонентом (компонентами), приводящее к ухудшению свойств фармацевтически приемлемого соединения, использование стандартного носителя предусмотрено областью заявляемого изобретения.
[179] Заявляемые фармацевтические композиции могут быть приготовлены и упакованы в нерасфасованной форме, причем безопасное и эффективное количество соединения с формулой (I) или его фармацевтически приемлемой соли может быть извлечено и затем выдано пациенту в виде порошков или сиропов. В качестве альтернативного варианта, заявляемые фармацевтические композиции могут быть приготовлены и упакованы расфасованными на дозы, причем отдельная физическая доза содержит соединение с формулой (I) или его фармацевтически приемлемую соль. Заявляемые лекарственные препараты, расфасованные на дозы, обычно могут содержать, например от 0,5 мг до 1 г, или от 1 мг до 700 мг, или от 5 мг до 100 мг соединения с формулой (I) или его фармацевтически приемлемой соли.
[180] Заявляемые фармацевтические композиции обычно содержат одно соединение с формулой (I) или его фармацевтически приемлемую соль.
[181] Термин «фармацевтически приемлемый инертный наполнитель» используемый в настоящей заявке, означает фармацевтически приемлемый материал, состав или основу, используемый для придания формы или консистенции фармацевтической композиции. Каждый инертный наполнитель должен быть совместим с другими ингредиентами фармацевтической композиции при смешивании, поскольку такие взаимодействия способны существенно снизить эффективность соединения с формулой (I) или его фармацевтически приемлемой соли в процессе приема пациентом, следует избегать взаимодействий, приводящих к образованию фармацевтически неприемлемых фармацевтических композиций. Кроме того, каждый инертны наполнитель должен быть, безусловно, фармацевтически приемлемым, то есть достаточно высокой степени химической чистоты. Соединение с формулой (I) или его фармацевтически приемлемая соль и фармацевтически приемлемый инертный наполнитель или наполнители будут, как правило, расфасовываться на дозы, рассчитанные на прием пациентом по назначенной схеме. Например, к расфасованным дозам относятся рассчитанные (1) на оральный прием, такие как таблетки, капсулы, таблетки-капсулы, пилюли, пастилки, порошки, сиропы, эликсиры, суспензии, растворы, эмульсии, саше, и крахмальные облатки; (2) на парэнтеральный прием, такие как стерильные растворы, суспензии и порошки для восстановления; (3) на трансдермальный прием, такие как трансдермальные терапевтические системы (далее ТТС); (4) на ректальный прием, такие как свечи; (5) на ингаляцию, такие как аэрозоли, растворы и сухие порошки; и (6) топический прием, такие как кремы, мази, лосьоны, растворы, пасты, спреи, пены и гели.
[182] Подходящие фармацевтически приемлемые инертные наполнители будут меняться в зависимости от конкретной расфасовки. Кроме того, подходящие фармацевтически приемлемые инертные наполнители можно выбирать, исходя из конкретной функции, предназначенной им в препарате. Например, определенные фармацевтически приемлемые инертные наполнители могут быть выбраны за их способность облегчения производства единообразной расфасовки. Определенные фармацевтически приемлемые инертные наполнители могут быть выбраны за их способность облегчения производства стабильной расфасовки. Определенные фармацевтически приемлемые инертные наполнители могут быть выбраны за их способность облегчения переноса или транспортирования соединения или соединений с формулой (I) или их фармацевтически приемлемых солей после приема их пациентом к органу, или к части тел, к другому органу, или к части тела. Определенные фармацевтически приемлемые инертные наполнители могут быть выбраны за их способность улучшать конформность пациента.
[183] К подходящим фармацевтически приемлемым инертным наполнителям относятся наполнители следующих типов: разбавители, наполнители, связки, дезинтегранты, смазки, глиданты (вещества, повышающие текучесть порошка), грануляционные вещества, покрывающие вещества, увлажняющие вещества, растворители, вспомогательные растворители, суспендирующие вещества, эмульгаторы, подсластители, ароматизаторы, дезодораторы, красители, ингибиторы комкования, гемектанты, хелатообразующие вещества, пластификаторы, вещества, увеличивающие вязкость, антиоксиданты, консерванты, стабилизаторы, ПАВ и буферные вещества. Специалисту будет очевидно, что определенные фармацевтически приемлемые инертные наполнители могут исполнять более чем одну функцию и могут обладать также альтернативными функциями в зависимости от количества данного наполнителя и присутствия других наполнителей в препарате.
[184] Знания и практический опыт позволяют специалистам выбрать соответствующее количество подходящих фармацевтически приемлемых инертных наполнителей для заявляемого изобретения. Помимо этого имеются источники, доступные специалистам, где приводятся описания фармацевтически приемлемых инертных наполнителей; эти источники могут оказаться полезными для выбора подходящих фармацевтически приемлемых инертных наполнителей. К примерам таких источников относятся: Remington's Pharmaceutical Sciences (Mack Publishing Company), «Справочник фармацевтических вспомогательных веществ» (Gower Publishing Limited), и «Справочник фармацевтических инертных наполнителей» (Американская фармацевтическая ассоциация и фармацевтическая пресса).
[185] Заявляемые фармацевтические композиции приготовляются способами, известными специалистам. Описания некоторых из этих общеизвестных способов приводятся в издании Remington's Pharmaceutical Sciences (Mack Publishing Company).
[186] Заявляется также способ приготовления фармацевтической композиции, содержащего соединение с формулой (I) или его фармацевтически приемлемую соль и один или более фармацевтически приемлемых инертных наполнителей, заключающийся в смешивании ингредиентов. Фармацевтическая композиции, содержащая соединение с формулой (I) или его фармацевтически приемлемую соль, может быть приготовлена, например, смешиванием при комнатной температуре и атмосферном давлении.
[187] В одной реализации соединения с формулой (I) или их фармацевтически приемлемые соли будут составляться для орального приема. В другой реализации соединения с формулой (I) или их фармацевтически приемлемые соли будут составляться для ингаляционного приема. В другой реализации соединения с формулой (I) или их фармацевтически приемлемые соли будут составляться для интраназального приема.
[188] В заявляемом изобретении предполагается твердая дозировочная форма для орального приема, такая как таблетка или капсула, содержащая безопасное и эффективное количество соединения с формулой (I) или его фармацевтически приемлемую соль, разбавитель или наполнитель. К подходящим разбавителям и наполнителям относится лактоза, сахароза, декстроза, маннитол, сорбит, крахмал (например, кукурузный крахмал, картофельный крахмал и предварительно желатинизированный крахмал), целлюлоза и ее производные (например, микрокристаллическая целлюлоза), сернокислый кальций и двухосновный фосфат кальция. Твердая дозировочная форма для орального приема может также содержать связку. К подходящим связкам относится крахмал (например, кукурузный крахмал, картофельный крахмал и предварительно желатинизированный крахмал), желатин, камедь, альгинат натрия, альгиновая кислота, трагакант, гуаровая камедь, повидон, а также целлюлоза и ее производные (например, микрокристаллическая целлюлоза). Твердая дозировочная форма для орального приема может дополнительно содержать дезинтегрант. К подходящим дезинтегрантам относятся кросповидон, глюконат натриевого крахмала, кроскармелоз, альгиновая кислота и карбоксиметилцеллюлоза натрия. Твердая дозировочная форма для орального приема может дополнительно содержать смазку. К подходящим смазкам относятся стеариновая кислота, стеарат магния, стеарат кальция и тальк.
[189] По возможности производится микрокапсулирование расфасованных доз для орального приема. Композиция может быть приготовлена с расчетом на прологированное или отложенное высвобождения, например, путем нанесения покрытия или внедрения метельчатого материала в полимеры, парафин и подобные элементы.
[190] Соединения с формулой (I) или их фармацевтически приемлемые соли можно также сочетать с растворимыми полимерами в качестве целевых носителей лекарстенных средств. В число таких полимеров может входить поливинилпирролидон, сополимер пирана, полигидроксипропилметакриламид-фенол, полигидроксиэтиласпартамидефенол, или поиэтиленоксид полилизин, замещенный осадками пальмитоила. Помимо этого, соединения с формулой (I) или их фармацевтически приемлемые соли можно также сочетать с полимерами биоразлагаемого класса, полезными для достижения регулируемого высвобождения лекарственного средства; к таким полимерам относятся, например, полимолочная кислота, поиэпсилон капролактон, полигидрокси масляная кислота, полиортоэфиры, полиацетали, полидигидропираны, полицианоакрилаты и перекрестно структурированные или амфипатические блочные сополимеры гидрогелей.
[191] Заявляется также жидкая дозировочная форма для орального приема. Жидкости для орального приема, такие как раствор, сиропы и эликсиры можно приготовить как однократными дозировочными формами так, чтобы конкретное количество такой формы содержало заданное количество соединения с формулой (I) или его фармацевтически приемлемой соли. Сиропы можно приготовить, диспергируя соединение с формулой (I) или его фармацевтически приемлемую соль в надлежащим образом ароматизированном водном растворе, тогда как эликсир приготовляется с использованием нетоксичной спиртовой основы. Суспензии можно составлять, диспергируя соединение с формулой (I) или его фармацевтически приемлемую соль в нетоксичной основе. Допускается также добавлять солюбилизаторы и эмульгаторы, такие как этоксилированные изостеариловые спирты и полиоксиэтиленсорбитоловые эфиры, консерванты, ароматизирующие добавки, такие как перечномятное масло или подсластители природного происхождения, или сахарин, или другие искусственные подсластители, и тому подобные.
[192] Заявляется также дозировочная форма, предназначенная для приема пациентом путем ингаляции, например, в виде сухого порошка, аэрозоля, суспензии или раствора. В одной реализации дозировочная форма, предназначенная для приема пациентом путем ингаляции, представляет собой сухой порошок. В другой реализации дозировочная форма, предназначенная для приема пациентом путем ингаляции, реализуется с помощью распылителя. Композиции в виде сухих порошков, предназначенные для доставки в легкие путем ингаляции, обычно содержат соединение с формулой (I) или его фармацевтически приемлемую соль в виде тонкодисперсного порошка вместе с одним или более нейтральным наполнителем в виде тонкодисперсных порошков. Фармацевтически приемлемые нейтральные наполнители, особо пригодные для использования в сухих порошках, известны специалистам и включают в себя лактозу, крахмал, маннитол и моно-, ди- и полисахариды. Тонкодисперсный порошок может быть приготовлен, например, посредством микронизации и помола. Как правило, измельченное (т.е. микронизированное) соединение характеризуется количественно параметром D50 величиной от 1 до примерно 10 микрон (например, по результатам измерения методом лазерной дифракции).
[193] Прием сухого порошка пациентом может осуществляться посредством ингалятора с резервуаром сухого порошка (RDPI), причем объем резервуара рассчитан на хранение нескольких доз (фасованных доз) лекарственного средства в форме сухого порошка. Аппараты RDPI обычно комплектуются средствами измерения каждой дозы лекарственного средства, подаваемого из резервуара. Например, средства измерения могут представлять собой мерную чашку, перемещаемую из первого положения туда, где происходит ее наполнение лекарственным средством из резервуара, и затем во второе положение, где отмеренная доза становится доступной для ингаляции пациентом.
[194] Как альтернативный вариант, для использования в многоразовом ингаляторе сухого порошка (MDPI) сухой порошок может быть упакован в капсулы (например, в желатиновые или пластиковые), картриджи или блистерные упаковки. Аппараты MDPI представляют собой ингаляторы, где лекарственное средство содержится в многоразовой упаковке (или подается иным способом), заряженной несколькими отмеренными дозами (или частичными дозами) лекарственного средства. Блистерная упаковка сухого порошка состоит из нескольких блистеров, содержащих лекарственное средство в виде сухого порошка. Блистеры обычно располагаются упорядочение, чтобы облегчить высвобождение лекарственного средства из них. Например, блистеры могут располагаться по кругу на блистерной упаковке в форме диска, или блистеры могут быть оформлены в виде полосы или ленты. В каждой капсуле, картридже или блистере может содержаться, например, от 20 мкг до 10 мг соединения с формулой (I) или его фармацевтически приемлемую соль.
[195] Аэрозоли могут быть приготовлены путем суспензии или диспергирования соединения с формулой (I) или его фармацевтически приемлемой соли в жидком пропелленте. В число подходящих пропеллентов входят галогенуглеводороды, углеводороды и другие сжиженные газы. К показательным пропеллентам относятся: трихлофторметан (пропеллент 11), дихлорфторметан (пропеллент 12), дихлортетрафторэтан (пропеллент 114), тетрафторэтан (HFA-134a), 1,1-дифторэтан (HFA-152a), дифторметан (HFA-32), пентафторэтан (HFA-12), гептафторпропан (HFA-227a), перфторпропан, перфторбутан, перфторпентан, бутан, изобутан и пентан. Прием пациентом аэрозолей, содержащих соединение с формулой (I) или его фармацевтически приемлемую соль, будет обычно осуществляться через дозирующий ингалятор (MDI). Такие устройства известны специалистам.
[196] В состав аэрозоля могут входить дополнительные фармацевтически приемлемые инертные наполнители, обычно применяемые совместно с MDI, такие как ПАВ, смазки, сорастворители и другие инертные наполнители, улучшающие физическую стабильность препарата, улучшающие производительность клапана, улучшающие растворимость или вкус.
[197] Исходя из сказанного выше, заявляется фармацевтический аэрозольный препарат, содержащий соединение с формулой (I) или его фармацевтически приемлемую соль и хлорфторуглерод, содержащий фторуглерод или водород, в качестве пропеллента, дополнительно в сочетании с ПАВ и/или сорастворителем.
[198] В соответствии с другим пунктом заявляемого изобретения, заявляется фармацевтический аэрозольный препарат, где пропеллент выбирается из 1,1,1,2-тетрафторэтана, 1,1,1,2,3,3,3-гептафтор-n-пропана и их смесей.
[200] Заявляемые препараты допускается забуферивать добавкой подходящих веществ буферизации.
[201] Капсулы и картриджи для ингалятора или инсуффлятора, например, желатиновые, могут содержать порошковую смесь для ингаляции, состоящую из соединения с формулой (I) или его фармацевтически приемлемой соли и подходящего порошкового основания, например, лактозы или крахмала. В каждой капсуле или картридже может содержаться, например, от 20 мкг до 10 мг соединения с формулой (I) или его фармацевтически приемлемую соль. Как альтернатива, соединение с формулой (I) или его фармацевтически приемлемая соль может быть представлено без инертных наполнителей, таких как лактоза.
[202] Пропорция активного соединения с формулой (I) или его фармацевтически приемлемой соли в конкретных заявляемых препаратах зависит от состава приготовляемого препарата, однако в основном будет находиться в диапазоне от 0,001 до 10% по весу. Как правило, для большинства препаратов используемая пропорция будет находиться в диапазоне от 0,005 до 1%, например, от 0,01 до 0,5%. Однако в порошках для ингаляции или инсуфляции используемая пропорция будет находиться в диапазоне от 0,1% до 5%.
[203] Аэрозольные препараты предпочтительно составляются так, чтобы в каждой отмеренной дозе, или «пшике» аэрозоля содержалось от 20 мкг до 10 мг, предпочтительно от 20 мкг до 2000 мкг, еще более предпочтительно от 20 мкг до 500 мкг соединения с формулой (I). Прием препарата может быть назначен один раз или несколько раз в день, например, 2, 3, 4 или 8 раз, например, по 1, 2 или 3 дозы за каждый прием. Общий объем дневной дозы аэрозоля будет находиться в диапазоне от 100 мкг до 10 мг, предпочтительно от 200 мкг до 2000 мкг. Общая дневная доза и отмеренная доза, доставляемая капсулами и картриджами ингалятора или инсуффлятора, будет обычно вдвое больше дозы, доставляемой посредством аэрозольных препаратов.
[204] Во взвешенных аэрозольных препаратах размер частицы метельчатого (т.е. измельченного) лекарственного средства должен обеспечивать ингаляцию практически всего лекарственного средства в легкие после приема аэрозольного препарата, поэтому размер частицы не будет превышать 100 микрон, предпочтительно частица должна быть меньше 20 микрон, в частности, размер частицы должен быть в диапазоне от 1 до 10 микрон, например, от 1 до 5 микрон, более предпочтительно от 2 до 3 микрон.
[205] Заявляемые препараты можно приготовить диспергированием или растворением лекарственного средства и соединения с формулой (I) или его фармацевтически приемлемой соли в избранном пропелленте в соответствующем контейнере, например, с помощью обработки ультразвуком или миксера с большими сдвиговыми усилиями. Процесс желательно производить в условиях регулируемой влажности.
[206] Химическую и физическую стабильность, а также фармацевтическую приемлемость аэрозольных препаратов по заявляемому изобретению можно определить известными способами. Так, например, химическую стабильность компонентов можно определить с помощью исследования HPLC, скажем, после длительного хранения продукта Параметры физической стабильности можно получить другими общепринятыми аналитическими способами, например, испытанием на утечку, исследованием производительности клапана (средняя масса выброса одного срабатывания), исследованием повторяемости дозы (количество активного ингредиента в оном срабатывании) и анализом распределения спрея.
[207] Стабильность взвешенных аэрозольных препаратов по заявляемому изобретению можно измерить известными способами, например, измерением распределения размера флоккуляции с помощью прибора отражения подсветки, или измерением распределения размера частицы последовательными импульсами, или аналитической процедурой «двойного импинжера». В настоящей заявке исследование с помощью «двойного импинжера» означает «Определение отложения выпущенной дозы в ингаляциях с избыточным давлением при помощи аппарата А», по определению Британской Фармакопеи 1988, страницы А204-207, Приложение XVII С. Такие способы позволяют вычислить «вдыхаемую фракцию» аэрозольных препаратов. Одним из способов вычисления «вдыхаемой фракции» является, согласно приведенным в настоящей заявке источникам, «фракция тонкой частицы», представляющая собой количество активного ингредиента, накопленного в нижней камере импинжера за каждое срабатывание, выраженного в процентах от общего количества активного ингредиента, выпущенного за одно срабатывание с помощью способа двойного импинжера, рассмотренного выше.
[208] Термин «дозирующий ингалятор» или MDI относится к устройству, состоящему из тары, зафиксированного колпачка, закрывающего тару, и дозирующего клапана препарата, расположенного на колпачке. В состав системы MDI входит подходящее направляющее устройство. Подходящее направляющее устройство содержит, например, привод клапана и цилиндрический или конический канал, через который лекарственное средство из заправленной тары через дозирующий клапан может быть подано в нос или в рот пациента, управляющего подачей лекарственного средства с помощью, например, мундштучного привода.
[209] Тара MDI обычно представляет собой контейнер, устойчивый к давлению паров пропеллента, такой как пластмассовая или стеклянная бутыль с пластиковым покрытием, или, предпочтительно, металлическая тара, изготовленная, например, из алюминия или алюминиевого сплава, поверхность которой может быть дополнительно анодирована, лакирована и/или покрыта пластиком (в качестве примера в настоящей заявке приводится ссылка на патентную публикацию WO 96/32099, где внутренние поверхности тары частично или полностью дополнительно покрыты одним или более фторуглеродными полимерами в сочетании с одним или более не-фторуглеродными полимерами), причем контейнер закрыт дозирующим клапаном. Колпачок может крепиться на таре с помощью ультразвуковой сварки, винтовым соединением или обжимным соединением. Тара MDI настоящей заявки может быть изготовлена известными способами (например, публикации Byron, выше и WO 96/32099). Предпочтительно состыковать тару с узлом колпачка, причем дозирующий клапан располагается в колпачке, а упомянутый колпачок зафиксирован по месту обжимом.
[210] В одной реализации заявляемого изобретения металлическая внутренняя поверхность тары покрыта фторполимером, более предпочтительно смешанным с не-фторполимером. В другой реализации заявляемого изобретения металлическая внутренняя поверхность тары покрыта полимерной смесью политетрафторэтилена (ПТФЭ) с полиэфирсульфоном (ПЭС). В дополнительной реализации заявляемого изобретения вся металлическая внутренняя поверхность тары покрыта полимерной смесью политетрафторэтилена (ПТФЭ) с полиэфирсульфоном (ПЭС). Дозирующие клапаны рассчитаны на подачу отмеренного количества препарата при каждом срабатывании и содержат прокладку для предотвращения утечки пропеллента из клапана. Прокладка может быть выполнена из любого подходящего эластомерного материала, например, из полиэтилена низкой плотности, хлорбутила, бромбутила, EPDM, черного и белого бутадиен-акрилнитрильного каучука, бутилового каучука и неопрена. Подходящие клапаны предлагаются на рынке изготовителями, хорошо известными в аэрозольной отрасли промышленности, например, Valois, Франция (скажем, DF10, DF30, DF60), Bespak. pic, Англия (например, ВK300, ВK357) и 3М-ТМ Neotechnic Ltd, Англия (например, Spraymiser).
[211] В различных реализациях MDI может также использоваться совместно с другими конструктивными элементами, например, не ограничиваясь перечисленным далее, оберточными упаковками для хранения и эксплуатации MDI, включая патенты США №№6,119,853; 6,179,118; 6,315,112; 6,352,152; 6,390,291; и 6,679,374, а также с блоками счетчика доз, например, не ограничиваясь перечисленным далее, как в патентах США №№6.360,739 и 6.431.168.
[212] Для приготовления с коммерческими целями крупных партий заправленной тары могут использоваться общепринятые способы производства нерасфасованной продукции и оборудование, хорошо известное специалистам аэрозольного производства. Так, например, по одному из способов производства нерасфасованной продукции для приготовления взвешенных аэрозольных препаратов дозирующий клапан крепится обжимом на алюминиевой таре, образуя пустую тару. Конкретное лекарственное средство вводится в заправочный резервуар, и сжиженный пропеллент вместе с дополнительными инертными наполнителями под давлением подается через заправочный резервуар в технологический резервуар Суспензия лекарственного средства перемешивается до рециркуляции к заправочной машине и затем вполне определенное количество суспензии лекарственного средства заправляется в тару через дозирующий клапан. В одном примере способов производства нерасфасованной продукции для приготовления взвешенных аэрозольных препаратов дозирующий клапан крепится обжимом на алюминиевой таре, образуя пустую тару. Сжиженный пропеллент вместе с дополнительными инертными наполнителями и растворенным лекарственным средством под давлением заправляется через заправочный резервуар в технологический резервуар.
[213] В альтернативном процессе определенное количество сжиженного препарата добавляется в открытую тару в окружающих условиях, достаточно холодных, чтобы исключить испарение препарата, после чего дозирующий клапан крепится обжимом на тару.
[214] Как правило, в партиях, изготовляемых для фармацевтического использования, производится контрольное взвешивание каждой заправленной тары, на таре проставляется номер партии и готовая продукция упаковывается на поддон для хранения перед испытанием выпуска. Прием пациентом суспензий и растворов, содержащих соединение с формулой (I) или его фармацевтически приемлемую соль, может также осуществляться с помощью распылителя. Раствор или агент суспензии, используемый для распыления, может быть любой фармацевтически приемлемой жидкостью, такой как вода, физиологический раствор, спирты или гликоли, например, этанол, изопропиловый спирт, пропиленгликоль и так далее, или их смеси. В физиологических растворах применяются соли, обнаруживающие малую физиологическую активность или вообще не обнаруживающие ее после приема. С этой целью допускается использовать как органические соли, например, соли щелочных металлов или аммоний галогенные соли, такие как хлористый натрий, хлористый калий или органические соли, такие как соли калия, натрия или аммония, так и органические кислоты, например, аскорбиновую кислоту, лимонную кислоту, уксусную кислоту, виннокаменную кислоту и так далее.
[215] К суспензии или раствору допускается добавлять другие фармацевтически приемлемые нейтральные наполнители. Стабилизация соединения с формулой (I) или его фармацевтически приемлемой соли может быть осуществлена добавкой неорганической кислоты, например соляной кислоты, азотной кислоты, серной кислоты и/или фосфорной кислоты; органической кислоты, например, аскорбиновой кислоты, лимонной кислоты, уксусной кислоты и виннокаменной кислоты, и так далее, комплексообразующего агента, такого как EDTA или лимонная кислота и ее соли; или антиоксиданта, такого как витамин Е или аскорбиновая кислота. Перечисленные выше вещества могут использоваться как по отдельности, так и вместе для стабилизации соединения с формулой (I) или его фармацевтически приемлемой соли. Допускается добавка консервантов, таких как бензалконий хлорид или бензойная кислота и их соли. Допускается добавка ПАВ, в частности, для улучшения физической стабильности суспензий. К числу ПАВ относится лецитин, динатриевый диоктилсульфосукцинат, олеиновая кислота и сорбитановые эфиры.
[216] Заявляется также дозировочная форма, предназначенная для интраназального приема пациентом.
[217] Препараты для приема в нос могут включать в себя аэрозольные препараты, распыляемые под давлением, и жидкие препараты, вводимые в нос насосом под давлением. Особый интерес представляют препараты без избыточного давления, пригодные для местного приема в носовую полость. Подходящие препараты содержат для этой цели воду в качестве растворителя или носителя. Жидкие препараты для приема в легкие или в нос могут обеспечиваться общепринятыми нейтральными наполнителями, такими как вещества буферизации, вещества изменения тонуса и тому подобными. Жидкие препараты можно также принимать в нос посредством распыления. Соединения с формулой (I) или их фармацевтически приемлемые соли могут быть составлены в виде жидкого препарата для доставки посредством распылителя жидкости, например, распылителя жидкости с соплом или отверстием, через которое отмеренная доза жидкого препарата под воздействие усилия пользователя подается в насосный механизм распылителя жидкости. Такие распылители жидкости обычно комплектуются резервуаром на несколько отмеренных доз жидкого препарата, дозы распыляются последовательными срабатываниями насоса. Конструкция распылительного сопла или отверстия может быть рассчитана на ввод в ноздри пользователя для распыления жидкого препарата в носовой полости. Распылитель жидкости упомянутого выше типа описан и проиллюстрирован в патентной публикации WO 05/044354, ссылка на полный текст которой имеется в настоящей заявке. Распределитель состоит из корпуса, в котором находится устройство выброса жидкости с насосом, смонтированном на таре с жидким препаратом. На корпусе имеется по меньшей мере один рычаг, рассчитанный на срабатывание от усилия пальца, перемещаемый внутрь относительно корпуса для переворота тары вверх внутри корпуса с тем, чтобы вызвать сжатие насоса и перекачивание отмеренной дозы препарата из штока насоса через носовое отверстие корпуса. В одной реализации распределитель жидкости относится к общераспространенному типу, иллюстрированному на фиг. 30-40 патентной публикации WO 05/044354.
[218] Фармацевтические композиции, рассчитанные на интраназальный прием, где носитель представляет собой твердое вещество, включая крупнодисперсный порошок с размером частиц, например, в диапазоне от 20 до 500 микрон, прием которых производится быстрой ингаляцией через носовой ход из тары с порошком, расположенной в непосредственной близости от носа. К подходящим препаратам, где носителем является жидкость, для приема в виде назального спрея или назальных капель, относятся водные или масляные растворы соединения с формулой (I) или его фармацевтически приемлемой соли.
[219] Фармацевтические композиции, рассчитанные на трансдермальный прием, могут быть представлены в виде отдельных ТТС, рассчитанных на непосредственный контакт с эпидермисом пациента в течение продолжительного времени. Например, активные ингредиенты могут доставляться из ТТС посредством ионтофореза, общее описание этого процесса приводится в публикации Pharmaceutical Research, 3(6), 318 (1986).
[220] Фармацевтические композиции, рассчитанные на топический прием, могут быть составлены в виде мазей, кремов, суспензий, лосьонов, порошков, растворов, паст, гелей, спреев, аэрозолей или масел. Мази, кремы и гели могут быть, например, составлены на жидкой или масляной основе с добавкой подходящего загустителя и/или гелеобразующего вещества и/или растворителей. В состав таких основ может входить, например, вода и/или масло, такое как жидкий парафин ил растительное масло, например, арахисовое масло или касторовое масло, или растворитель, такой как полиэтиленгликоль. Загустители и гелеобразующие вещества, которые можно использовать в зависимости от природы основы, включают в себя мягкий парафин, стеариновокислый алюминий, цетостеариловый спирт полиэтиленгликоль, ланолин, воск, карбоксиполиметилен и производные целлюлозы, и/или глицерил моностеарат, и/или неионогенные эмульгаторы.
[221] Лосьоны могут быть приготовлены на водной или масляной основе и, как правило, будут содержать один или более эмульгатор, стабилизатор, диспергент, суспендирующие вещества или загустители.
[222] Порошки для наружного применения могут быть изготовлены на любой подходящей порошковой основе, например, на тальке, лактозе или крахмале. Капли могут быть изготовлены на водной или безводной основе, содержащей также один или более диспергент, растворитель, суспендирующее вещество или консервант.
[223] Лекарственные препараты, рассчитанные на топический прием, допускается применять к пораженной зоне один или несколько раз в день; желательно пользоваться окклюзивной повязкой поверх участков кожи. Непрерывная или продолжительная доставка лекарственного препарата может быть достигнута за счет ТТС.
[224] Для лечения глаз или других наружных тканей, например, рта и кожи, препараты могут применяться как топическая мазь или крем. При составлении препарата в форме мази соединение с формулой (I) или его фармацевтически приемлемая соль может быть применена вместе с парафиновой или растворимой в воде основой мази. В качестве альтернативы, соединение с формулой (I) или его фармацевтически приемлемая соль может быть приготовлено в виде крема на масляно-водной основе или на водно-масляной основе.
[225] К фармацевтическим композициям, предназначенным для парэнтерального приема, относятся водные и безводные стерильные растворы для инъекций, которые могут содержать антиоксиданты, буферные вещества, бактериостаты и растворенные вещества, создающие изотоничность препарата с кровью целевого объекта; а также водные и безводные стерильные суспензии, которые могут содержать суспендирующие вещества и загустители. Препараты могут быть представлены в таре с однократной или многократной дозой, например, в герметичных ампулах, и могут храниться в охлажденно-осушенном состоянии (сублимированная форма), когда непосредственно перед применением требуется только добавить стерильный жидкий носитель, например, воду ль инъекций. Внеплановые растворы для инъекций и суспензии могут быть приготовлены из стерильных порошков, гранул или таблеток.
[226] Заявляемое соединение и лекарственные препараты могут использоваться в сочетании с одним или более другими лечебными агентами, например, выбранными из противовоспалительных агентов, антихолинергических агентов (в частности, рецепторный агонист М1/М2/М3), агонистов β2-андренорецептора, противоинфекционных агентов, таких как антибиотики и противовирусные препараты, или противогистаминные препараты. Таким образом, заявляется также сочетание соединения с формулой (I) или его фармацевтически приемлемой соли с одним или более других терапевтических агентов, например, выбранных из противовоспалительных агентов, таких как кортикостероид NSAID, антихолинергический агент, β2-андренорецептор агонист, противоинфекционный агент, такой как антибиотик и противовирусный препарат, или противогистаминный препарат. В одной реализации заявляемого изобретения реализуется сочетание, включающее в себя соединение с формулой (I) или его фармацевтически приемлемую соль и β2 андренорецептор агонист, и/или антихолинергический агент, и/или PDE-4 ингибитор, и/или противогистаминный препарат.
[227] В одной реализации заявляемого изобретения представлен способ лечения нарушения, опосредованного неадекватной активностью РI3-киназы, заключающийся в приеме безопасного и эффективного количества сочетания с формулой (I) или его фармацевтически приемлемой соли с одним или более терапевтически активных агентов.
[228] Определенные заявляемые соединения могут выказывать селективность против PI3Kδ относительно других РВ-киназ. Таким образом, заявляется также сочетание соединения с формулой (I) или его фармацевтически приемлемой соли, обладающего селективностью против PI3Kδ, с соединением или его фармацевтически приемлемой солью, обладающим селективность против другой РВ-киназы, например, РI3Kγ.
[229] В одной реализации заявляемого изобретения представлены сочетания из одного или двух других терапевтических агентов.
[230] Специалисту будет очевидно, что там, где это целесообразно, можно использовать другой терапевтический ингредиент (ингредиенты) в форме солей, например, солей щелочного металла или амина, или кислотно-аддитивных солей, или пропрепаратов, или эфиров, например, эфиров нижнего алкила, или сольватов, например, гидратов, для оптимизации активности и/или стабильности, и/или физических характеристик, таких как растворимость, терапевтического ингредиента. Будет очевидно также, что там, где это целесообразно, терапевтические ингредиенты могут использоваться в оптически чистой форме.
[231] В одной реализации заявляемого изобретения представлен продукт, содержащий соединение с формулой (I) и по меньше мере один терапевтический агент в виде комбинированного препарата для одновременного, раздельного или последовательного применения в терапии. В одной реализации терапия представляет собой лечение заболевания или состояния, опосредованного активностью PI3K ферментов. Продукты, представленные как комбинированный препарат, включают в себя препарат, содержащий соединение с формулой (I) и другой терапевтический агент (агенты) совместно в одной фармацевтической композиции, или соединение с формулой (I) другой терапевтический агент (агенты) по раздельности, то есть в виде набора.
[232] В одной реализации заявляемого изобретения представлена фармацевтическая композиция, содержащий соединение с формулой (I) и другой терапевтический агент (агенты). Фармацевтическая композиция может дополнительно содержать фармацевтически приемлемый носитель, описанный выше.
[233] В другой реализации заявляемого изобретения представлен набор, состоящий из двух или более фармацевтических композиций, по меньше мере один из которых содержит соединение с формулой (I). В одной реализации набор содержит средства раздельного хранения упомянутых выше препаратов, такие как контейнер, раздельная бутыль или раздельный фольговый пакет. Примером такого набора является блистерная упаковка, используемая, как правило, для упаковки таблеток, капсул и тому подобных форм.
[234] Набор заявляемого изобретения может использоваться для приема различных дозировочных форм, например, оральных или парэнтеральных, для приема отдельных препаратов с различным дозировочным интервалом, или для взаимного титрования отдельных препаратов. Для согласования набор заявляемого изобретения обычно комплектуется указаниями по применению.
[235] В комбинированной терапии заявляемого изобретения допускается использование заявляемого соединения и другого терапевтического агента, изготовленного и/или составленного разными изготовителями. Более того, заявляемое соединение и другой терапевтический агент допускается совмещать в комбинированной терапии: (i) до передачи комбинированного продукта врачам (т.е. в том случае, когда в наборе содержится заявляемое соединение и другой терапевтический агент); (ii) самими врачами (или под контролем врача) незадолго до приема; (iii) в организме самого пациента, например, в процессе последовательного приема заявляемого соединения и другого терапевтического агента.
[236] Соответственно, заявляется использование соединения с формулой (I) для лечения заболевания или состояния, опосредованного ферментами PI3K, где лекарственное средство приготовляется для приема с другим терапевтическим агентом. Заявляется также использование другого терапевтического агента для лечения заболевания или состояния, опосредованного ферментами PI3K, где лекарственное средство принимается с соединением с формулой (I).
[237] Заявляется использование соединение с формулой (I) для применения в способе лечения заболевания или состояния, опосредованного ферментами PI3K, где соединения с формулой (I) приготовляется для приема с другим терапевтическим агентом. Заявляется использование другого терапевтического агента для применения в способе лечения заболевания или состояния, опосредованного ферментами PI3K, где другой терапевтический агент приготовляется для приема с соединением с формулой (I). Заявляется использование соединения с формулой (I) для применения в способе лечения заболевания или состояния, опосредованного ферментами PI3K, где соединение с формулой (I) принимается с другим терапевтическим агентом. Заявляется также использование другого терапевтического агента для применения в способе лечения заболевания или состояния, опосредованного ферментами PI3K, где другой терапевтический агент принимается с соединением с формулой (I).
[238] Заявляется использование соединения с формулой (I) для лечения заболевания или состояния, опосредованного ферментами PI3K, где пациент предварительно (например, в пределах 24 часов) принимал лечение другим терапевтическим агентом Заявляется использование другого терапевтического агента для лечения заболевания или состояния, опосредованного ферментами PI3K, где пациент предварительно (например, в пределах 24 часов) принимал лечение соединением с формулой (I). Соединения с формулой (I) допускается принимать как самостоятельный активный ингредиент, или совместно, то есть как активатор, с другими лекарственными средствами, например, иммунодепрессивными или иммуномодулирующими агентами, или другими противовоспалительными агентами, например для лечения или профилактики острого или хронического отторжения алио- или ксенотрансплантата, или воспалительных или аутоиммунных нарушений, или химиотерапевтического агента, например, антипролиферантного агента гемобластозных клеток. Например, соединения с формулой (I) могут использоваться в сочетании с перечислеными далее веществами: ингибитор кальциневрина, например, циклоспорин А или FK 506; ингибитором mTOR, например, рапамицин, 40-O-(2-гидроксиэтил)рапамицин, CCI779, АВТ578, АР23573, TAFA-93, биолимус-7 или биолимус-9; аскомицин с иммунодепрессивными свойствами, например, АВТ-281, ASM981, и т.; кортикостероиды; циклофосфамид; азатиопрен; метотрексат; лефлимомид; мизорибин; микофенольная кислота или соль; микофенолат мофетил 15-деокисперквалин или иммунодепрессивный гомолог, аналог или его производная; ингибитор РКС, например, приведенный в WO 02/38561 или WO 03/82859, например, соединение примера 56 или 70; ингибитор JAK3 киназы, например, N-бензил-3,4-дигидрокси-бензилиден-цианоацетаамид-[α]-циано-(3,4-дигидрокси)-N-бензилциннамамид (Тирфостин AG 490), продигиозин 25-C (PNU156804), [4-(4'-гидроксифенил)амин-6,7-диметоксиквиназолин] (WHI-P131), [4-(3'-бром-4'-гидрокси[фенил)-амин-6,7-диметоксиквиназолин] (WHI-P154), [4-(3,5'-дибром-4'гидроксифенил)-амин-6,7-диметоксиквиназолин] WHI-P97, KRX-21 1,3-{(3R/4R)-4-метил-3-[метил-7H-пирроло[2,3-d]пиримин-4-ил)-амин]-пиперидин-1-ил}-3-оксо-пропионитрил, в свободной форме или в форме фармацевтически приемлемой соли, например, моно- цитрат (называемый также СР-690,550), или соединение, заявленное в WO 04/052359 или WO 05/066156; иммунодепрессивные моноклональные антитела, например, моноклональные антитела по отношению к лейкоцит рецепторам, например, МНС, CD2, CD3, CD4, CD7, CDS, CD25, CD28, CD40, CD45, CD52, CD58, CD80, CD86 или их лиганды; другие иммунодепрессивные соединения, например, рекомбинантная связующая молекула с по меньшей мере одной частью внеклеточного домена CTLA4 или ее мутант, например, по меньшей мере внеклеточная часть CTLA4 или ее мутант, соединенный с не-CTLA4 протеиновой последовательностью, например, CTLA41g (например, обозначенный как АТСС 68629) или его мутант, например, LEA29Y; ингибиторы адгезионной молекулы, например, LFA-1 антагонисты, ICAM-1 или -3 антагонисты, VCAM-4 антагонисты или YLA-4 антагонисты; или антигистамины; или противокашлевые агенты, или бронхолитический агент; или блокираторы ангиотензин рецептора; или противоинфекционный агент.
[239] Там, где соединения с формулой I принимаются вместе с другой иммунодепрессивной/иммуномодулирующей, противовоспалительной, химиотерапевтической или противоинфекционной терапией, дозы совместно принимаемого иммунодепрессивного, иммуномодулирующего, противовоспалительного, химиотерапевтического или противоинфекционного соединения будут, естественно, варьироваться в зависимости от принимаемого совместно лекарственного средства, например, является ли такое лекарственное средство стероидом или ингибитором кальциневрина, от конкретного используемого лекарственного средства, от состояния, подвергаемого лечению и так далее.
[240] В одной реализации заявляемого изобретения представлено сочетание соединения с формулой (I) или его фармацевтически приемлемой соли с агонистом β2-адренорецептора.
[241] К примерам агонистов β2-адренорецептора относится салметерол (который может быть рацематом или одиночным энантиомером, таким как R-энантиомер), сальбутамол (который может быть рацематом или одиночным энантиомером, таким как R-энантиомер), формотерол (который может быть рацематом или одиночным диастереомером, таким как R, R-диастереомер), салмефамол, фенотерол, кармотерол, этантерол, наминтерол, кленбутерол, пирбутерол, флербутерол, репротерол, бамбутерол, индакатерол, тербуталин и их соли, например, ксинафоат (1-гидрокси-2-нафталенкарбоксилат), соль салметерола, сульфатная соль или свободное основание сальбутамола или фумаровая соль форметерола. В одной реализации предпочтительны длительно действующие агонисты β2-адренорецептора, например, соединения, обеспечивающие эффективную бронходилатацию на протяжении 12 часов или дольше.
[242] Агонист β2-адренорецептора может быть представлен в форме соли, полученной с помощью фармацевтически приемлемой кислоты, выбранной из следующих кислот: серной, соляной, фумаровой, гидроксинафтойной (для примера 1- или 3-гидрокси-2-нафтойная), коричной, замещенной коричной, трифенилуксусной, сульфамидной, сульфаниловой, нафталакриловой, бензойной, 4-метоксибензойной, 2- или 4-гидроксибензойной, 4-хлорбензойной и 4-фенилбензойной.
[243] К подходящим антивоспалительным агентам относятся кортикостероиды. К подходящим кортикостероидам, которые можно применять в сочетании с соединениями с формулой (I) или их фармацевитчески приемлемыми солями, относятся кортикосероиды, рассчитанные на оральный или ингаляционный прием, а также их пропрепараты, обладающие антивоспалительным действием. Примеры: метил преднизолон, преднизолон, дексаметазон, флутиказон пропионат, 6α,9α-дифтор-11β-гидрокси-16α-метил-17α-(4-метил-1,3-тиазол-5-карбонил)окси-3-оксо-андроста-1,4-диен-17β-карботио кислоты S-фторметиловый эфир, 6α,9α-дифтор-17α-(2-фуранилкарбонил)окси-11β-гидрокси-16α-метил-3-оксо-андроста-1,4-диен-17β-карботио кислоты S-фторметиловый эфир (флутиказон фуркат), 6α,9α-дифтор-11β-гидрокси-16α-метил-3-оксо-17α-пропионилокси-андроста-1,4-диен-17β-карботио кислоты S-(2-оксо-тетраидро-фуран-3S-ил) эфир, 6α,9α-дифтор-11β-гидрокси-16α-метил-3-оксо-17α-(2,2,3,3 тетраметициклопропилкарбонил)окси-андроста-1,4-диен-. 17β-карботио кислоты S-цианметиловый эфир и 6α,9α-дифтор-11β-16α-метил-17α-(1-этициклопропилкарбонил)окси-3-оксо-андроста-1,4-диен-17β-карботио кислоты S-фторметиловый эфир, беклометазоновые эфиры (для примера 17-пропионат эфир или 17,21-дипропионат эфир), будезонид, флунизолид, мометазоновые эфиры для примера мометазон фуроат), триамцинолон ацетонид, рофлепонид, циклезонид (16α, 17-(R)-циклогексилметиленбис(окси)-11β,21-дигидрокси-прегна-1,4-диен-3,20-дион), бутиксокорт пропионат, RPR-106541, и ST-126. Предпочтительные корикостероиды: флутиказон пропионат, 6α,9α-дифтор-11β-гидрокси-16α-метил-17α-(4-метил-1,3-тиазол-5-карбонил)окси-3-оксо-андроста-1,4-диен-17β-карботио кислоты S-фторометиловый эфир, 6α,9α-дифтор-17α-(2-фупани 1 карбонил)окси-11β-гидрокси-16α-метил-3-оксо-андроста-1,4-диен-17β-карботио кислоты S-фторометиловый эфир, 6α,9α-дифтор-11β-гидрокси-16α-метил-3-оксо-17α-(2,2,3,3-тетраметициклопропилкарбонил)окси-андроста-1,4-диен-17β-карботио кислоты S-цианметиловый эфир и 6α,9α-11β-гидрокси-16α-метил-17α-(1-метициклопропилкарбонил)окси-3-оксо-андроста-1,4-диен-17β-карботио кислоты S-фторометиловый эфир. В одной реализации кортикостероидом является 6α,9α-дифтор-17α-(2-фуранилкарбонил)окси-11β-гидрокси-16α-метил-3-оксо-андроста-1,4-диен-17β-карботио кислоты S-фторометиловый эфир.
[244] Нестероидные соединения, обладающие глюкокортикоидным агонизмом, могут обладать избирательностью для трансрепресии над трансэтивацией, и могут оказаться полезными в комбинационной терапии, включая соединения, на которые распространяется действие следующих патентов: WO 03/082827, WO 98/54159, WO 04/005229, WO 04/009017, WQQ4/Q18429, WO 03/104195, WO 03/082787, WO 03/082280, WO 03/059899, WO 03/101932, WO 02/02565, WO 01/16128, WO 00/66590, WO 03/086294, WO 04/026248, WO 03/061651 и WO 03/08277. Дополнительные нестероидные соединения охватываются следующими патентами: WO 2006/000401, WO 2006/000398 и WO 2006/015870.
[245] К примерам противовоспалительных агентов относятся нестероидные и противовоспалительные лекарственные средства (NSAID).
[246] К примерам NSAID относятся натрий кромоглюцинат, недокромил нартрий, ингибиторы фосфодиэстеразы (PDE) (например, теофиллин, ингибиторы PDE4 или смешанные ингибиторы PDE3/PDE4, лейкотриен антагонисты, ингибиторы лейкотриен синтеза (например, монтелукаст), ингибиторы iNOS, ингибиторы триптазы и эластазы, антагонисты бета-2 интегрина и агонисты или антагонисты аденозин рецептора (к примеру, агонисты аденозин 2а), цитокин антагонисты (например, хемокин антагонисты, такие как антагонист CCR3) или ингибиторы цитокин синтеза, или ингибиторы 5-липоксигеназы. Для орального приема предпочтительным является iNOS (включая ингибитор синтазы окиси азота). Примеры ингибиторов iNOS приведены в патентных публикациях WO 93/13055, WO 98/30537, WO 02/50021, WO 95/34534 и WO 99/62875. Примеры ингибиторов CCR3 приведены в патентной публикации WO 02/26722.
[247] В одной реализации заявляемого изобретения представлено применение соединений с формулой (I) в сочетании с ингибитором фосфодиэстеразы 4 (PDE4), в частности, в случае препарата, специализированного для ингаляции. РDЕ4-специфичный ингибитор, полезный в этом аспекте заявляемого изобретения, может представлять собой соединение, известное ингибирующими свойствами по отношению к PDE4 ферменту, или обнаружившее свойства ингибитора PDE4, и соединения, являющиеся только ингибиторами PDE4, но не обладающие свойствами ингибитора по отношению к другим членам семейства PDE, таким как PDE3 и PDE5, а также PDE4. Соединения включают в себя цис-4-циано-4-(3-циклопентилокси-4-метоксифенил)циклогексан-1-карбоновая кислота, 2-карбоксиметокси-4-циано-4-(3-циклопропилметокси-4-дифторметоксифенил)циклогексан-1-один и цис-[4-циано-4-(3-циклопропилметокси-4-дифторметоксифенил)циклогексан-1-ол]. А также соединение цис циано-4-[3-(циклопентокси)-4-метоксифенил]циклогексан-1-карбоновая кислота (известное под названием киломиласт) и его соли, эфиры, пропрепараты или физические формы, приведенные в патенте США 5,552,438, выданном 03 сентября 1996; этот патент и заявленные в нем соединения полностью включены в список источников настоящей заявки.
[248] Примерами антихолинергических агентов, являются соединения, действующие как антагонисты на мускариновые рецепторы, в частности, соединения, являющиеся антагонистами Mi или Mb рецепторов, двойными антагонистами M1/M3 или М2/М3, рецепторов или пан-антагонистами M1/M2/M3 рецепторов. К показательным соединениям для приема путем ингаляции относятся ипратропий (например, в виде бромида, CAS 22254-24-6, имеющийся в продаже под названием Антровент), окситропий (например, в виде бромида CAS 30286-75-0) и тиотропий (например, в виде бромида, CAS 136310-93-5, имеющийся в продаже под названием Спирива).
Интерес представляет также реватропат (например, в виде гидробромида CAS 262586-79-8) и LAS-34273, заявляемый в патентной публикации WOO 1/04118. К показательным соединениям для орального приема относятся пирензепин (CAS 28797-61-7), дарифенацин (CAS 133099-04-4 или CAS 133099-07-7 для гидробромида, имеющийся в продаже под названием Энаблекс), оксибутинин (CAS 5633-20-5, имеющийся в продаже под названием Дитропан), теродилин (CAS 15793-40-5), толтеродин (CAS 124937-51-5, или CAS 124937-52-6 тартрата, имеющийся в продаже под названием Детрол), отилониум (например, в виде бромида, CAS 26095-59-0, имеющийся в продаже под названием Спасмомен), троспиум хлорид (CAS 10405-02-4) и солифенацин (CAS 242478-374, или CAS 242478-38-2 для сукцината, известный также как YM-905 и поступающий в торговые сети под названием Весикар).
[249] В одной реализации заявляемого изобретения представлено сочетание соединения с формулой (I) или его фармацевтически приемлемой соли и антагониста H1. К некоторым неограничительным примерам антагониста H1 относятся амелексанокс, астемизол, азатадин, азеластин, акривастин, бромфенирамин, цетиризин, левоцетиризин, эфлитиризин, хлорфенирарнин, клемастин, циклин, каребастин, ципрогептадин, карбиноксараин, дескарбоэтоксилоратадин, доксиламин, диметиден, эхастин, эпинастин, эфлетиризин, фексофенадин, гидроксизин, кетотифен, лоратадин, левокабастин, мизоластин, меквитазин, мианзерин, ноберастин, меклицин, норастемизол, олопатадин, пикумаст, пириламин, прометазин, терфенадин, трипеленнамин, темеластин, тримепразин и трипролидин, в частности, цетирин, левоцетиризин, эфлетиризин и фексофенадин. В дополнительной реализации заявляемого изобретения представлено сочетание соединения с формулой (I) или его фармацевтически приемлемой соли и антагониста Н3 (и/или инверсного агониста). К примерам антагонистов Н3 относятся, например, заявленные в патентных публикациях WO 2004/035556 и WO 2006/045416. К другим антагонистам гистаминного рецептора, которые могут быть использованы совместно с заявляемым соединением, относятся антагонисты (и/или инверсные агонисты) рецептора Н4, например, соединения, рассмотренные в статье Джабкмовски и соавторы, J. Med. Chem., 2003, 46, 3957-3960.
[250] Таким образом, заявляется сочетание соединения с формулой (I) или его фармацевтически приемлемой соли с ингибитором PDE4.
[251] Таким образом, заявляется сочетание соединения с формулой (I) или его фармацевтически приемлемой соли с агонистом β2 адренорецептора.
[252] Таким образом, заявляется сочетание соединения с формулой (I) или его фармацевтически приемлемой соли с кортикостероидом
[253] Таким образом, заявляется сочетание соединения с формулой (I) или его фармацевтически приемлемой соли с нестероидным агонистом GR.
[254] Таким образом, заявляется сочетание соединения с формулой (I) или его фармацевтически приемлемой соли с антихолинергиком.
[255] Таким образом, заявляется сочетание соединения с формулой (I) или его фармацевтически приемлемой соли с антигистамином.
[256] Таким образом, заявляется сочетание соединения с формулой (I) или его фармацевтически приемлемой соли с ингибитором PDE4 и агонистом β2 адренорецептора.
[257] Таким образом, заявляется сочетание соединения с формулой (I) или его фармацевтически приемлемой соли с антихолинергиком и ингибитором PDE4.
[258] Упомянутые выше сочетания могут быть удобно представлены для применения в качестве фармацевтической композиции и, таким образом, фармацевтических композиций, содержащих приведенное выше сочетание с фармацевтически приемлемым растворителем или носителем, что представляет собой дальнейшее расширение формулы заявляемого изобретения.
[259] Отдельные соединения таких сочетаний можно принимать либо последовательно, либо одновременно в самостоятельных или комбинированных фармацевтических композициях. В одной реализации отдельные соединения будут приниматься одновременно в составе одного лекарственного препарата. Соответствующие дозы известных терапевтических агентов будут заранее оценены специалистами.
[260] Таким образом, дополнительно заявляется фармацевтическая композиция, содержащая сочетание соединения с формулой (I) или его фармацевтически приемлемой соли с другим терапевтически активным агентом.
[261] Дополнительно заявляется фармацевтическая композиция, содержащая сочетание соединения с формулой (I) или его фармацевтически приемлемой соли с ингибитором PDE4.
[262] Дополнительно заявляется фармацевтическая композиция, содержащая сочетание соединения с формулой (I) или его фармацевтически приемлемой соли с агонистом β2 адренорецептора.
[263] Дополнительно заявляется фармацевтическая композиция, содержащая сочетание соединения с формулой (I) или его фармацевтически приемлемой соли с кортикостероидом.
[264] Дополнительно заявляется фармацевтическая композиция, содержащая сочетание соединения с формулой (I) или его фармацевтически приемлемой соли с нестероидным агонистом GR.
[265] Дополнительно заявляется фармацевтическая композиция, содержащая сочетание соединения с формулой (I) или его фармацевтически приемлемой соли с антихолинергиком.
[266] Дополнительно заявляется фармацевтическая композиция, содержащая сочетание соединения с формулой (I) или его фармацевтически приемлемой соли с антигистамином.
[267] Таким образом, фармацевтическая композиция, содержащая сочетание соединения с формулой (I) или его фармацевтически приемлемой соли с ингибитором PDE4 и агонистом β2 адренорецептора.
[268] Дополнительно заявляется фармацевтическая композиция, содержащая сочетание соединения с формулой (I) или его фармацевтически приемлемой соли с антихолинергиком и ингибитором PDE4.
[269] Соединение с формулой (I) может также использоваться с реализацией преимущества в сочетании с каждым по отдельности или со всеми вместе терапевтическими агентами, особенно с другими пролиферативными агентами. К таким пролиферативным агентам относятся, не ограничиваясь перечисленным далее списком, ингибиторы ароматазы; антиэстрогены; ингибиторы топоизомеразы; ингибиторы топоизомеразы II; микроканальные активные агенты; алкилирующие агенты; ингибиторы гистон дезацетилазы; соединения, индуцирующие процессы клеточной дифференцировки; ингибиторы циклооксигеназы; ингибиторы ММР; ингибиторы mTOR; антибластомные антиметаболиты; соединения платины; соединения, таргетирующие/снижающие активность белка или липидной киназы и дополнительные анти-ангиогенные соединения; соединения, таргетирующие, снижающие или ингибирующие активность белка или липидной фосфатазы, агонисты гонадорелина; анти-андрогены; ингибиторы метионин аминопептидазы; бифосфонаты; модификаторы биологического отклика; антипролиферативные антитела; ингибиторы гепараназы; ингибиторы Ras онкогенных изоформ; ингибиторы теломеразы; протеасомные ингибиторы; агенты, используемые в лечении гематологических злокачественных новообразований; оединения, таргетирующие, снижающие или ингибирующие активность Flt-3; Hsp90; темозолимид (TEMODAL®); и лейковорин.
[270] Термин «ингибитор ароматазы», используемый в настоящей заявке, относится к соединению, ингибирующему воспроизводство эстрогена, т.е. преобразование субстратов андростендиона и тестостерона в эстрон и эстрадиол, соответственно. Термин распространяется, не ограничиваясь перечисленным далее, на стероиды, в частности, атаместан, экземестан и форместан; и, в частности, нестероиды, особенно аминоглютетимид, роглетимид, пиридоглютемид, трилостан, тестолактон, кетоконазол, ворозол, фадрозол, анастрозол и летрозол. Экземестан можно принимать, например, в той форме, какая поступает в продажу, например, под товарным знаком AROMAS IN. Форместан можно принимать, например, в той форме, какая поступает в продажу, например, под товарным знаком LENTARON. Фадрозол можно принимать, например, в той форме, какая поступает в продажу, например, под товарным знаком AFEMA. Анастрозол можно принимать, например, в той форме, какая поступает в продажу, например, под товарным знаком ARIMIDEX. Летрозол можно принимать, например, в той форме, какая поступает в продажу, например, под товарным знаком FEMARA или FEMAR. Аминоглютетимид можно принимать, например, в той форме, какая поступает в продажу, например, под товарным знаком ORIMETEN. Сочетание заявляемого изобретения, содержащее хемотерапевтический агент, являющийся ингибитором ароматазы, особенно полезно для лечения гормональных рецепторных позитивных опухолей, например, опухолей молочной железы.
[271] Термин «анти-эстроген», используемый в настоящей заявке, относится к соединению, противодействующему влиянию эстрогенов на уровне эстрогенного рецептора. Термин распространяется, не ограничиваясь перечисленным далее, на тамоксифен, фкльвестрант, ралоксифен и гидрохлорид ралоксифена. Тамоксифен можно принимать, например, в той форме, какая поступает в продажу, например, под товарным знаком NOLVADEX, гидрохлорид ралоксифенаможно принимать, например, в той форме, какая поступает в продажу, например, под товарным знаком EVISTA. Фульвестрант может быть приготовлен по патенту США №4,659,516 например, в той форме, какая поступает в продажу, например, под товарным знаком FASLODEX. Сочетание заявляемого изобретения, содержащее хемотерапевтический агент, являющийся антиэстрогеном, особенно полезно для лечения эстрогенных рецепторных позитивных опухолей, например, опухолей молочной железы.
[272] Термин «анти-андроген», используемый в настоящей заявке, относится к любому веществу, способному ингибировать биологические эффекты андрогенных гормонов, к таким веществам относится, не ограничиваясь перечисленным далее, бикалутамид (CASODEX), который может быть приготовлен, например, по патенту США №4,636,505.
[273] Термин «агонист гонадорелина», используемый в настоящей заявке, включает в себя, не ограничиваясь перечисленным далее, абареликс, гозерелин и гозерелин ацетат. Гозерелин заявлен в патенте США №4,100,274, его можно принимать, например, в той форме, какая поступает в продажу, например, под товарным знаком ZOLADEX. Абареликс может быть приготовлен, например, по патенту США №5,843,901. Термин «ингибитор топоизомеразы 1», используемый в настоящей заявке, включает в себя, не ограничиваясь перечисленным далее, топотекан, гиматекан, иринотекан, камптотециан и его аналоги, 9-нитрокамптотецин и сложное соединение макромолекулярный камптотецин PNU-166148 (соединение А1 в WO 99/17804). Иринотекан можно принимать, например, в той форме, какая поступает в продажу, например, под товарным знаком CAMPTOSAR. Топотекан можно принимать, например, в той форме, какая поступает в продажу, например, под товарным знаком HYCAMTIN.
[274] Термин «ингибитор топоизомеразы II», используемый в настоящей заявке, включает в себя, не ограничиваясь перечисленным далее, антрациклины, такие как доксорубицин, включая липосомный препарат, например, CAELYX; даунорубицин; эпирубицин; идарубицин; неморубицин; антрахинонов митоксантрон и лозоксантрон; и подофилотоксинов этопозид и тенипозид. Этопозид можно принимать, например, в той форме, какая поступает в продажу, например, под товарным знаком ETOPOPBOS. Тенипозид можно принимать, например, в той форме, какая поступает в продажу, например, под товарным знаком VM 26-BRJ8TQL. Доксорубицин можно принимать, например, в той форме, какая поступает в продажу, например, под товарным знаком ADRIBLASTIN или ADRIAMYCIN.
[275] Эпирубицин можно принимать, например, в той форме, какая поступает в продажу, например, под товарным знаком FARMORUBICIN. Идарубицин можно принимать, например, в той форме, какая поступает в продажу, например, под товарным знаком ZAVEDOS. Митоксантрон можно принимать, например, в той форме, какая поступает в продажу, например, под товарным знаком NOVANTRON.
[276] Термин «микроканальный активный агент» относится к микроканальным стабилизирующим, микроканальным дестабилизирующим агентам и ингибиторам микроканальной полимеризаци, включая, но не ограничиваясь перечисленным далее, таксаны, например, паклитаксел и доцетаксел; vinca алкалоиды, например, винбластин, особенно винбластин сульфат; винкристин, особенно винкристин сульфат и винорелбин; дискодермолиды; кочицин; и эпотилоны и их производные, например, эпотилон В или D или их производные. Паклитаксе можно принимать, например, в той форме, какая поступает в продажу, например, под товарным знаком TAXOL. Доцетаксел можно принимать, например, в той форме, какая поступает в продажу, например, под товарным знаком TAXOTERE. Винбластин можно принимать, например, в той форме, какая поступает в продажу, например, под товарным знаком VINBLASTIN R.P. Винкристин можно принимать, например, в той форме, какая поступает в продажу, например, под товарным знаком FARMISTIN. Дискодермолид может быть приготовлен, например, по патенту США №5,010,099. Также включены производные эпотилона, заявленные в патентной публикации WO 98/10121, в патенте США №6,194,181, в патентных публикациях WO 98/25929, WO 98/08849, WO 99/43653, WO 98/22461 и WO 00/31247. Особое предпочтение отдается эпотилону А и/или В.
[277] Термин «алкилирующий агент», используемый в настоящей заявке, включает в себя, не ограничиваясь перечисленным далее, циклофосфамид, ифосфамид, мелфалан или нитрозомочевину (BCNU или Gliadel). Циклофосфамид можно принимать, например, в той форме, какая поступает в продажу, например, под товарным знаком CYCLOSTIN. Ифосфамид можно принимать, например, в той форме, какая поступает в продажу, например, под товарным знаком HOLOXAN.
[278] Термин «ингибиторы гистон дезацетилазы» или «HDAC ингибиторы» относится к соединениям, ингибирующим гистон дезацетилазу и проявляющим антипролиферативную активность. Сюда относятся соединения, заявленные в патентной публикации WO 02/22577, особенно, N-гидрокси-3-[4-[[(2-гидроксиэтил)[2-(1H-индол-3-ил)этил]-амин]метил]фенил]-2Е-2-пропенамид, N-гидрокси-3-[4-[[[2-(2-метил-1H-индол-3-ил)-этил]-амин]метил]фенил]-2Е-2-пропенамид и их фармацевтически приемлемые соли. Особо включена субериоланилид гидроксамовая кислота (SAHA).
[279] Термин «антибластомный антиметаболит» включает в себя, не ограничиваясь перечисленным далее, 5-фтороурацил или 5-FU; капецитабин; гемцитабин; DNA деметилирущие агенты, такие как 5-азацитидин и децитабин; меотрексат и эдатрексат; и антагонисты фолиевой кислоты, такие как пеметрексед. Капецитабин можно принимать, например, в той форме, какая поступает в продажу, например, под товарным знаком XELODA. Гемцитабин можно принимать, например, в той форме, какая поступает в продажу, например, под товарным знаком GEMZAR. Сюда также относится моноклональный антительный трастузумаб, который можно принимать, например, в той форме, какая поступает в продажу, например, под товарным знаком HERCEPTIN.
[280] Термин «соединения платины», используемый в настоящей заявке, включает в себя, не ограничиваясь перечисленным далее, карбоплатину, czs-платину, цисплатину и оксалиплатину. Карбоплатину можно принимать, например, в той форме, какая поступает в продажу, например, под товарным знаком CARBOPLAT. Оксалиплатину принимать, например, в той форме, какая поступает в продажу, например, под товарным знаком ELOXATIN. Выражение «соединения, таргетирующие/снижающие активность белка или липидной киназы, или активность белка или липидной фосфатазы, или дополнительные анти-ангиогенные соединения», используемое в настоящей заявке, охватывает, не ограничиваясь перечисленным далее, протеинтирозинкиназы и/или ингибиторы серин- и/или треонинкиназы, и/или ингибиторы липидной киназы, и т.д.
[281] а) соединения, таргетирующие, снижающие или ингибирующие активность роста вызванного тромбоцитами фактор-рецепторов (PDGFR), такие как соединения таргетирующие, снижающие или ингибирующие активность PDGFR, особенно соединения, ингибирующие рецептор PDGF, например, производное N-фенил-2-пиримидин-амин, скажем, иматиниб, SU101, SU6668 и GFB-111;
[282] b) соединения, таргетирующие, снижающие или ингибирующие активность фактор-рецепторов роста роста фибробласта (FGFR);
[283] с) соединения, таргетирующие, снижающие или ингибирующие активность инсулино-подобного фактор-рецептора роста I (IGF-IR), такие как соединения, таргетирующие, снижающие или ингибирующие активность IGF-IR, особенно соединения, ингибирующие рецептор IGF-IR, подобные соединениям из патентной публикации WO 02/092599;
[284] d) соединения, таргетирующие, снижающие или ингибирующие активность рецептора Trk семейства тирозинкиназы;
[285] е) соединения, таргетирующие, снижающие или ингибирующие активность рецептора Axl семейства тирозинкиназы;
[286] f) соединения, таргетирующие, снижающие или ингибирующие активность рецептора c-Met;
[287] g) соединения, таргетирующие, снижающие или ингибирующие активность рецептора Kit/SCFR семейства тирозинкиназы.
[288] h) соединения, таргетирующие, снижающие или ингибирующие активность C-kit рецептора тирозинкиназы - (часть семейства PDGFR), такие как соединения, таргетирующие, снижающие или ингибирующие активность c-Kit рецептора семейства тирозинкиназы, особенно соединения, ингибирующие c-Kit рецептор, например, иматиниб;
[289] i) соединения, таргетирующие, снижающие или ингибирующие активность членов семейства с-Abl и их генно-синтезированных продуктов, например, BCR-Abl киназы, например, соединения, таргетирующие снижение или ингибирующие активность членов семейства с-Abl и их генно-синтезированных продуктов, например, N-фенил-2-пиримидин-амин производной, такой как иматиниб, PD180970, AG957, NSC 680410 или PD173955 от ParkeDavis; j) соединения, таргетирующие, снижающие или ингибирующие активность членов протеинкиназы C (РКС) и семейства Raf серин/треонинкиназ, членов семейства МЕК, SRC, JAK, FAK, PDK и Ras/MAPK, или семейтсва Рl(3) киназы, или семейства, связанного с Рl(3)-киназой, и/или членов семейства циклин-зависимой киназы (CDK) и особенно производные стауроспорина из патента США №5,093,330, например, мидостаурин; примерами дополнительных соединений являются, например, UCN-01; сафингол; BAY 43-9006; Бриостатин 1; Перифозин; Илмофозин; RO 318220 и RO 320432; GO 6976; Исис 3521; LY333531/LY379196; изохинолиновы соединения, такие как в патентной публикации WO 00/09495; FTIs; PD184352; или QAN697 (ингибитор Р13К);
[290] k) соединения, таргетирующие, снижающие или ингибирующие активность ингибиторов протеин-тирозинкиназы, например, соединения таргетирующие, снижающие или ингибирующие активность ингибиторов протеин-тирозинкиназы, включая иматиниб, мезилат (GLEEVEC) или тирфостин. Тирпостин является предпочтительно соединением с малым молекулярным весом (Mr<1500) или его фармацевтически приемлемой солью, особенно соединение, выбранное из класса соединений бензилиденемалонитрила или класса S-арилбензенэмалонирила или бисубстрата квинолина, еще более конкретно любое соединение, выбранное из группы, состоящей из энантиомера Тирфостин A23/RG-50810, AG 99, Тирфостин AG 213, Тирфостин AG 1748, Тирфостин AG 490, Тирфостин В44, Тирфостин В44 (+), Тирфостин AG 555, AG 494, Тирфостин AG 556, AG957 и адафостин (4-([(2,5-дигидроксифенил)метил]амин}-бензойной кислоты адамантил эфир, NSC 680410, адапостин; и
[291] I) соединения, таргетирующие, снижающие или ингибирующие активность семейства фактора эпидермального роста рецептора тирозинкиназы (EGFR, ErbB2, ErbB3, ErbB4 как гомо- или гетеродимеры), например, соединения, таргетирующие, снижающие или ингибирующие активность семейства рецептора фактора эпидермального роста представляют собой особо соединения, белки или антитела, ингибирующие членов семейства рецептора тирозинкиназы EGF, например, EGF рецептор, ErbB2, ErbB3 и ErbB4, или связь с лигандами, относящимимся к EGF или EGF, в частности, соединения, белки или моноклональные антитела, в общем и в частности заявленные в патентной публикации WO 97/02266, например, соединения примера 39, или в патентных публикациях ЕР 0 564 409; WO 99/03854; ЕР 0520722; ЕР 0566226; ЕР 0787722; ЕР 0837063; в патенте США №5,747,498; в патентных публикациях WO 98/10767; WO 97/30034; WO 97/49688; WO 97/38983 и особенно в WO 96/30347, например, соединение, известное как CP 358774; WO 96/33980, например, соединение ZD 1839; и в патентной публикации WO 95/03283, например, соединение ZM105180, например, трастузуумаб (HERCEPTIN), сетуксимаб, Иресса, Таркева, OSI-774, ci-1033, ЕKВ-569, GW-2016, E1.1, Е2.4, Е2.5, Е6.2, Е6.4, Е2.1 1, Е6.3 или Е7.6.3; и 7Н-пиролло-[2,3-d]пиримидин производные, заявленные в патентной публикации WO 03/013541. К дополнительным ангиогенным соединениям относятся соединения с другим механизмом активности, например, не связанным с ингибированием протеина или киназы, например, талидомид (THALOMID) и TNP-470. Соединения, таргетирующие, снижающие или ингибирующие активность белка или липидной фосфатазы являются, например, иингибиторами фосфатазы 1, фосфатазы 2А, PTEN или CDC25, например, окадная кислота или их производные.
[292] Соединения, индуцирующие процессы клеточной дифференцировки, например, ретиноевая кислота, α- γ или δ- или α- γ или δ-токотриенол
[293] Термин «ингибитор циклооксигеназы», используемый в настоящей заявке, включает, не ограничиваясь перечисленным далее, например, Сох-2 ингибиторы, 5-алкил замещеннуую 2-ариламинофенилуксусная кислоту и производные, такие как целекоксиб (CELEBREX), рофекоксиб (VIOXX), эторикоксиб, валдекоксиб или 5-алкил-2-ариламинофенилуксусная кислоту, например, 5-метил-2-(2'-хлор-6'-фторанилино)фенильную кислоту или лумиракоксиб.
[294] Термин «бифосфонаты», используемый в настоящей заявке, включает, не ограничиваясь перечисленным далее, этридоновую, клодроновую, тилудоновую, памидроновую, алендроновую, ибандроновую, ризедроновую и золедроновую кислоту. «Этридоновую кислоту» можно принимать, например, в той форме, какая поступает в продажу, например, под товарным знаком DIDRONEL. «Клодроновую кислоту» можно принимать, например, в той форме, какая поступает в продажу, например, под товарным знаком BONEFOS. «Тилудоновую кислоту» можно принимать, например, в той форме, какая поступает в продажу, например, под товарным знаком SKELID. «Памидроновую кислоту» можно принимать, например, в той форме, какая поступает в продажу, например, под товарным знаком AREDIA™. «Алендроновую кислоту» можно принимать, например, в той форме, какая поступает в продажу, например, под товарным знаком FOSAMAX. «Ибандроновую кислоту» можно принимать, например, в той форме, какая поступает в продажу, например, под товарным знаком BONDRANAT. «Ризедроновую кислоту» можно принимать, например, в той форме, какая поступает в продажу, например, под товарным знаком ACTONEL. «Золедроновую кислоту» можно принимать, например, в той форме, какая поступает в продажу, например, под товарным знаком ZOMETA.
[295] Термин «ингибиторы mTOR inhibitors» относится к соединениям, ингибирующим млекопитающую цель рапамицина (mTOR) и демонстрирующим антипролиферативную активность, таким как сиролимус (RAPAMUNE®), эверолимус (CERTICAN™), CCI-779 и АВТ578.
[296] Термин «ингибитор гепараназы», используемый в настоящей заявке, относится к соединениям, таргетирующим, снижающим или ингибирующим гепаринсульфатную деградацию. Термин включает в себя, без ограничения перечисленным далее, PI-88.
[297] Термин «модификатор биологического отклика», используемый в настоящей заявке, относится к лимфокину или интерферонам, таким как интерферон [γ].
[298] Термин «ингибитор Ras онкогенных изоформ», например, H-Ras, K-Ras или N-Ras, используемый в настоящей заявке, относится к соединениям, таргетирующим, снижающим или ингибирующим онкогенную активность Ras, например, а «ингибитор фарнесилтрансферазы», например, L-744832, DK8G557 или R1 15777 (Зарнестра).
[299] Термин «ингибитор теломеразы», используемый в настоящей заявке, относится к соединениям, таргетирующим, снижающим или ингибирующим активность теломеразы. К соединениям, таргетирующим, снижающим или ингибирующим активность теломеразы относятся, в частности, соединения, ингибирующие рецептор теломеразы, например, теломестатин.
[300] Термин «ингибитор метионин аминопептидазы», используемый в настоящей заявке, относится к соединениям, таргетирующим, снижающим или ингибирующим активность метионин аминопептидазы. К соединениям, таргетирующим, снижающим или ингибирующим активность метионин аминопептидазы, относятся, в частности, бенгамид или его производная.
[301] Термин «протеасомный ингибитор», используемый в настоящей заявке, относится к соединениям, таргетирующим, снижающим или ингибирующим активность протеасомы. К соединениям, таргетирующим, снижающим или ингибирующим активность протеасомы, относятся, например, PS-341 и MLN 341.
[302] Термин «ингибитор матричной металлопротеиназы» или «ММР ингибитор», используемый в настоящей заявке, включает в себя, без ограничения перечисленным далее, коллаген пептидомиметичные и непептидомиметичны ингибиторы, производные тетрациклина, например, гидроксамат пептидомиметичный ингибитор батимастат и его биологически усваиваемый аналог маримастат (ВВ-2516), приномастат (AG3340), метастат (NSC 683551) BMS-279251, BAY 12-9566, TAA211, MMI270B или AAJ996.
[303] Термин «агенты, используемые в лечении гематологических злокачественных новообразований», используемый в настоящей заявке, включает в себя, без ограничения перечисленным далее, ингибиторы FMS-подобной тирозинкиназы, например, соединения, таргетирующие, снижающие или ингибирующие активность рецепторов FMS-подобной тирозинкиназы (Flt-3R); интерферон, 1-b-D-арабинофурансилцитозин ara-с) и бисульфан; а также ингибиторы ALK, например, соединения, таргетирующие, снижающие или ингибирующие киназу анапластической лимфомы.
[304] К соединениям, таргетирующим, снижающим или ингибирующим активность рецепторов FMS-подобной тирозинкиназы (Flt-3R) относятся, в частности, соединения, белки или антитела, ингибирующие членов семейства рецепторной киназы Flt-3R, например, РКС412, мидостаурин, производная стауорспорина, SU1 1248 и MLN518.
[305] Термин «Ингибиторы HSP90», используемый в настоящей заявке, включает в себя, без ограничения перечисленным далее, соединения, таргетирующие, снижающие или ингибирующие врожденную ATPase активность HSP90; разрушающие, таргетирующие, снижающие или ингибирующие клиентские протеины HSP90 убиквитиновым протеасомным путем. К соединениям, таргетирующим, снижающим или ингибирующим врожденную ATPase активность HSP90 относятся, в частности, соединения, белки или антитела, ингибирующие ATPase активность HSP90, например, 17-аллиламино, 17-деметоксигелданамицин (17AAG), производная гелданамицина, другие соединения, связанные с гелданамицином, радицикол и ингибиторы HDAC.
[306] Термин «антипролиферативные антитела», используемый в настоящей заявке, включает в себя, без ограничения перечисленным далее, трастузумаб (Herceptin™), Трастузумаб-DMl, эрлотиниб (Tarceva™), бевацизумаб (Avastin™), ритуксимаб (Rituxan®), PR064553 (анти-CD40) и антитело 2С4. Под антителами подразумеваются, например, непораженные моноклональные антитела, поликлональные антитела, полиспецифические антитела, сформированные как минимум из двух непораженных антител, и фрагменты антител, пока они еще сохраняют требуемую биологическую активность. Для лечения острого миелоидного лейкоза (ОМЛ) соединения с формулой (I) могут применяться в сочетании со стандартной терапией лейкемии, а именно, в сочетании с терапевтическими процедурами, используемыми для лечения ОМЛ. В частности, соединения с формулой (I) можно принимать в сочетании, например, с ингибиторами фарнесилтрансферазы и/или другими лекарственными средствами, полезными для лечения ОМЛ, такими как Даунорубицин, Адриамицин, Ara-C, VP-16, Тенипозид, Митоксатрон, Идарубицин, Карбоплатинум и РКС412.
[307] Соединение с формулой (I) может также использоваться с реализацией преимущества в сочетании с каждым по отдельности или со всеми вместе другими терапевтическими агентами, особенно с другими противомалярийными агентами. К таким противомалярийным агентам относятся, не ограничиваясь перечисленным далее, прогуанил, хлорпрогуанил, триметоприм, хлороквин, мелоквин, лумефантрин, атоваквон, пириметамин-сульфадоксин, пириметамин-дапсон, галофантрин, квинин, квинидин, амодиаквин, амопироквин, сульфонамиды, артемизинин, артефлен, артеметр, артесунат, примаквин, ингалируемая форма NO, L-аргинин, Дипропиленетри-амин NONOaт (донор NO), Розиглицон (агонист PPARy), активированный уголь, эритропоэтин, левамизол и пиронаридин.
[308] Соединение с формулой (I) может также использоваться с реализацией преимущества в сочетании с каждым по отдельности или со всеми вместе другими терапевтическими агентами, такими как применяемые для лечения лейшманиоза, трипаносомоза, токсоплазмоза и нейроцистицеркоза. К таким агентам относятся, не ограничиваясь перечисленным далее, хлороквин сульфат, атоваквон-прогуанил, артеметр-лумефантрин, квинин-сульфат, артесунат, квинин, доксициклин, клиндамицин, меглумин антимонат, натрия стибоглюконат, милтефозин, кетоконазол, пентамидин, амфотерицин В (AmB), липосомат-AmB, парамомицин, эфлорнитин, нифуртимокс, сурамин, меларсопрол, преднизолон, бензнидазол, сульфадиазин, пириметамин, клиндамицин, триметропим, сульфаметоксазол, азитромицин, атоваквон, дексаметазон, празиквантел, альбендазол, бета-лактамы, фторквинолоны, макролиды, аминоглюкозиды, сульфадиазин и пириметамин.
[309] Структура активных агентов определяется по кодовым номерам, оригинальным названиям или коммерческим обозначениям, с которыми можно ознакомиться в действующем издании стандартного однотомника «Мерк индекс» или в базах данных, например, международных патентов, например, IMS мировые публикации.
[310] Упомянутые выше соединения, которые можно использовать в сочетании с соединением с формулой (I), можно приготовлять и принимать известным предписанным образом, как, например, в процитированных выше документах.
[311] Соединение с формулой (I) может также использоваться с реализацией преимущества в сочетании с известными терапевтическими процедурами, такими как прием гормональных препаратов или, в частности, ионизирующее излучение.
[312] Соединение с формулой (I) может, в частности, использоваться как радиосенсибилизатор, особенно для лечения злокачественных опухолей, проявляющих слабую чувствительность к радиотерапии.
[313] Выражение «сочетание», используемое в настоящей заявке, означает либо фиксированное сочетание в однократной дозировочной форме, или набор частей для комбинированного приема, где соединение с формулой (I) и сочетающийся с ним элемент могут приниматься независимо в одно и то же время, или раздельно с разделением во времени, что особенно позволяет выявлять дополняющие свойства сочетающегося элемента, например, синергичность, эффект или любое сочетание перечисленных ранее свойств. Термины «соприем» или «совмещенный прием», или аналогичные, используемые в настоящей заявке, означают осуществление приема выбранного сочетающегося элемента одиночным нуждающимся объектом (например, пациентом); эти термины предназначены для охвата режимов лечения, когда агенты не обязательно принимаются одинаковым образом или одновременно. Термин «фармацевтическое сочетание», используемый в настоящей заявке, означает продукт, образующийся в результате смешивания или сочетания более одного активного ингредиента, и включает в себя как фиксированные, так и не фиксированные сочетания активных ингредиентов. Термин «фиксированное сочетание» означает, что активные ингредиенты, например, соединение с формулой I и сочетающийся элемент, принимаются пациентом одновременно единым целым или одной дозой. Термин «нефиксированное сочетание» означает, что активные ингредиенты, например, соединение с формулой (I) и сочетающийся элемент, принимаются пациентом как отдельные единицы, либо одновременно, параллельно или последовательно без ограничений по времени, причем такой прием обеспечивает терапевтически эффективные уровни двух соединений в теле пациента. Последнее замечание также относится к коктейльной терапии, то есть к приему трех или более активных ингредиентов.
ИСПОЛЬЗОВАНИЕ ЗАЯВЛЯЕМЫХ СОЕДИНЕНИЙ И ПРЕПАРАТОВ
[314] Заявляемые соединения являются ингибиторами активности киназы, в частности, активности РI3-киназы. Соединения, являющиеся ингибиторами РI3-киназы, могут оказаться полезными при лечении нарушений, где первопричина патологии (по меньшей мере частично) обусловлена неадекватной активностью PI3-киназы, примерами подобных нарушений могут служить астма и хроническая обструктивная болезнь легких (ХОБЛ). Фраза «Неадекватная активность РI3-киназы» относится к любой активности PI3-киназы, отклоняющейся от нормальной активности РI3-киназы, ожидаемой у конкретного пациента. Неадекватная активность PI3-киназы проявляться, например, в форме неадекватного возрастания активности, или в искажении синхронности или регулирования активности PI3-киназы. Подобная неадекватная активность может привести, например, за счет сверхсинтеза или мутации протеинкиназы к неадекватной или неуправляемой активации. Соответственно, заявляются также способы лечения подобных нарушений.
[315] К таким нарушениям относятся, не ограничиваясь перечисленным далее, заболевания дыхательных путей, такие как астма, хроническая обструктивная болезнь легких (COPD, ХОБЛ) и идиопатический легочный фиброз (IPF); вирусные инфекции, включая вирусные инфекции дыхательных путей и вирусные обострения заболеваний дыхательных путей, такие как астма и COPD; инфекции дыхательных путей невирусного происхождения, включая аспергиллез и лейшманиоз, аллергические риниты и атопические дерматиты, аутоиммунные заболевания, включая ревматоидные артриты и рассеянный склероз, воспалительные нарушения, включая воспалительную болезнь кишечника, сердечно-сосудистые заболевания, включая тромбоз и атеросклероз; гемобластозы; нейродегенеративные заболевания; панкреатит; синдром полиорганной недостаточности (СПОН, ПОН); нефропатия; седиментация тромбоцитов; карцинома; подвижность сперматозоидов; отторжение трансплантата, отторжение ткани, поражение легких, и боль, вызываемые ревматоидным артритом остеоартритом, боль в пояснице, боль, вызываемая общим воспалительным процессом, постгепатитная невралгия, диабетическая нейропатия, воспалительная невропатическая боль (травма), тригеминальная невралгия и центральная боль.
[316] В одной реализации к таким нарушениям относятся респираторные нарушения, в том числе астма, хроническая обструктивная болезнь легких (COPD, ХОБЛ), аллергические риниты и атопические дерматиты, аутоиммунные заболевания, включая ревматоидные артриты и рассеянный склероз, воспалительные нарушения, включая воспалительную болезнь кишечника, сердечно-сосудистые заболевания, включая тромбоз и атеросклероз; гемобластозы; нейродегенеративные заболевания; панкреатит; синдром полиорганной недостаточности (СПОН, ПОН); нефропатия; седиментация тромбоцитов; карцинома; подвижность сперматозоидов; отторжение трансплантата, отторжение ткани, поражение легких, и боль, вызываемые ревматоидным артритом остеоартритом, боль в пояснице, боль, вызываемая общим воспалительным процессом, постгепатитная невралгия, диабетическая нейропатия, воспалительная невропатическая боль (травма), тригеминальная невралгия и центральная боль.
[317] Заявляемые способы лечения заключаются в приеме безопасного и эффективного количества соединения с формулой (I) или его фармацевтически приемлемой соли пациентом, нуждающимся в таком лечении. Отдельные реализации заявляемого изобретения представляют способ лечения любого из упомянутых выше нарушений путем приема безопасного и эффективного количества соединения с формулой (I) или его фармацевтически приемлемой соли пациентом, нуждающимся в таком лечении.
[318] Прием соединений с формулой (I) или их фармацевтически приемлемых солей может осуществляться любым подходящим способом, включая как соматичесикй, так и топический прием. К соматическому способу приема от носится оральный, парэнтеральный, трансдермальный и ректальный. Парэнтеральный прием относится к другой категории приема, нежели энтеральный или трансдермальный, и обычно осуществляется инъекцией или инфузией. К парэнтеральному способу приема относится внутривенная, внтуримышечная и подкожная инъекция или инфузия. Топический способ приема заключается в нанесении на кожу, к нему относятся также внутриглазной, ушной, интравагинальный, ингаляционный и интраназальный прием. Ингаляция представляет собой прием препарата в легкие пациента либо ингаляцией ртом, либо ингаляцией через носовые ходы. В другой реализации соединения с формулой (I) или их фармацевтически приемлемые соли могут приниматься орально. В другой реализации соединения с формулой (I) или их фармацевтически приемлемые соли могут приниматься ингаляцией. В другой реализации соединения с формулой (I) или их фармацевтически приемлемые соли могут приниматься интраназально.
[319] Прием соединений с формулой (I) или их фармацевтически приемлемых солей может осуществляться однократно или в соответствии с дозировочным режимом, где несколько доз принимаются с различными интервалами в течение заданного отрезка времени. Например, дозы можно принимать один, два, три или четыре раза в день. В одной реализации прием производится один раз в день. В другой реализации прием производится дважды в день. Дозы могут приниматься до тех пор, пока не будет достигнут желаемый терапевтический эффект, или неограниченно для поддержания желаемого терапевтического эффекта. Подходящие режимы дозирования соединения с формулой (I) или его фармацевтически приемлемой соли зависят от фармакокинетических свойств этого соединения, таких как абсорбция, распределение и период полураспада, которые могут быть установлены специалистом. Помимо этого, подходящие режимы дозирования, включая продолжительность таких режимов, для соединения с формулой (I) или его фармацевтически приемлемой соли зависят от нарушения, которое подвергается лечению, от тяжести нарушения, которое подвергается лечению, от возраста и физического состояния пациента, от истории болезни пациента, от природы параллельной терапии, от желаемого терапевтического эффекта, и подобных факторов, находящихся в пределах знаний и опыта специалиста. Такому специалисту следует дополнительно иметь в виду, что подходящие режимы дозирования могут потребовать корректировки в зависимости от реакции конкретного пациента на режим дозировки, или с течением времени, по мере необходимости изменения пациента.
[320] Заявляемое соединение допускается принимать либо одновременно, либо перед, либо после одного или более другого терапевтического агента. Заявляемое соединение допускается принимать отдельно, тем же самым или другим способом, или совместно в одной фармацевтической композиции, как другие агенты.
[321] Заявляемая фармацевтическая композиция или сочетание может быть расфасовано на дозы порядка 1-1000 мг активного ингредиента (ингредиентов) для объекта весом порядка 50-70 кг, или порядка 1-500 мг, или порядка 1-250 мг, или порядка 1-150 мг, или порядка 0,5-100 мг, или порядка 1-50 мг активных ингредиентов. Терапевтически эффективная доза соединения, фармацевтической композиции или их сочетания зависит от биологического вида объекта, от массы тела, от возраста и индивидуального состояния, от нарушения или заболевания, или от их тяжести. Врач или ветеринар обычной квалификации может легко определить эффективное количество каждого из активных ингредиентов, необходимое для профилактики, лечения или ингибирования прогресса нарушения ли заболевания. Упомянутые выше свойства дозы наглядно демонстрируются тестами in vitro и in vivo на предпочтительных млекопитающих, например, мышах, крысах, собаках, обезьянах или на их отдельных органах, тканях и препаратах. Заявляемые соединения могут применяться in vitro в форме растворов, например, водных растворов, и in vivo либо энтерально, либо парэнтерально, предпочтительно внутривенно, например, в форме суспензии или водного раствора. Диапазон терапевтически эффективного количества для приема in vivo зависит от способа приема, между 0,01-500 мг/кг или между 1-100 мг/кг.
[322] Кроме того, соединения с формулой (I) могут приниматься как пропрепараты. В настоящей заявке термин «пропрепарат» соединения с формулой (I) означает функциональное производное препарата, который, будучи принятым пациентом, с течением времени высвобождает соединение с формулой (I) in vivo. Прием соединения с формулой (I) в виде пропрепарата может позволить опытному специалисту произвести одно или несколько из перечисленных ниже действий: (а) изменить наступление активности соединения in vivo; (b) изменить продолжительность действия соединения vivo; (с) изменить перенос или распределение соединения in vivo; (d) изменить растворимость соединения in vivo; и (е) преодолеть побочные эффекты или другие сложности, связанные с приемом соединения. Типовые функциональные производные, используемые для приготовления пропрепаратов, включают в себя модификации соединения, которые химически или ферментативно расщепляются in vivo. Такие модификации, к которым относятся приготовление фосфатов, амидов, эфиров, тиоэфиров, карбонатов и карбаматов, хорошо известны специалистам.
[323] Заявляется также способ лечения нарушения, опосредованного неадекватной активностью PI3-киназы, заключающийся в приеме безопасного и эффективного количества соединения с формулой (I) или его фармацевтически приемлемой соли пациентом, нуждающимся в лечении.
В одной реализации состояния, заболевания или нарушения, опосредованные неадекватной активностью PI3-киназы, выбраны из следующей группы: астма, хроническая обструктивная болезнь легких (COPD, ХОБЛ) и идиопатический легочный фиброз (IPF); вирусные инфекции, включая вирусные инфекции дыхательных путей и вирусные обострения заболеваний дыхательных путей, такие как астма и COPD; инфекции дыхательных путей невирусного происхождения, включая аспергиллез и лейшманиоз, аллергические риниты и атопические дерматиты, аутоиммунные заболевания, включая ревматоидные артриты и рассеянный склероз, воспалительные нарушения, включая воспалительную болезнь кишечника, сердечно-сосудистые заболевания, включая тромбоз и атеросклероз; гемобластозы; нейродегенеративные заболевания; панкреатит; синдром полиорганной недостаточности (СПОН, ПОН); нефропатия; седиментация тромбоцитов; карцинома; подвижность сперматозоидов; отторжение трансплантата, отторжение ткани, поражение легких, и боль, вызываемые ревматоидным артритом, остеоартритом, боль в пояснице, боль, вызываемая общим воспалительным процессом, постгепатитная невралгия, диабетическая нейропатия, воспалительная невропатическая боль (травма), тригеминальная невралгия и центральная боль.
[325] Заявляемые соединения могут быть полезны для лечения состояний, заболеваний или нарушений, включая заболевания или инфекционную иммунопатологию, в которых одна или более функций В-клеток, таких как выработка антитела, представление антигена, выработка цитокина или лимфоидный органогенез являются аномальными или нежелательными, включая ревматоидные артриты, обыкновенную пузырчатку и сопутствующие заболевания, идиопатическую тромбоцитопеническую пурпуру, системную красную волчанку, рассеянный склероз, миастения (myasthenia gravis), синдром Шегрена, аутоиммунную гемолитическую анемию, АНЦА (антинейтрофильные цитоплазматические антитела)-связанные васкулиты, криоглобулинемию, тромбоцитопеническую тромбогемолитическую пурпуру, хроническую аутоиммунную аллергическая сыпь, аллергию (атопические дерматиты, контактный дерматит, аллергический ринит), синдром Гудпасчера, AMR (отторжение трансплантата, опосредованное антителом), гиперострое, острое и хроническое отторжение трансплантата, опосредованное В-клетками и карциномы гематопоэтического происхождения, включая, без ограничения перечисленным далее, множественную миелому; острый миелоидный лейкоз (ОМЛ); хроническую миелоидную лейкемию; лимфолейкоз; миелоидную лейкемию; неходжкинскую лимфому; лимфому; истинную полицитемию; идиопатическую тромбоцитемию; миелофиброз с миелоидной метаплазией; и макроглобулинемию Вальденстрёма.
[326] Заявляются способы лечения состояний, заболеваний или нарушений, в которых одна или более функций нейтрофилов, таких как выпуск перекиси, стимулированный экзоцитоз или хемоаттракивная миграция являются аномальными или нежелательными, включая ревматоидный артрит, сепсис, пульмональные или респираторные заболевания, такие как астма, воспалительные дерматозы, такие как псориаз, а также в иммунопатологии, связанной с заболеванием или инфекцией, и в других.
[327] Заявляются способы лечения состояний, заболеваний или нарушений, в которых одна или более функций базофилов и мастоцитов, таких как хемоаттракивная миграция или аллерген-lgЕ-опосредованная дегрануляция являются аномальными или нежелательными, включая аллергические заболевания (атопические дерматиты, контактный дерматит, аллергический ринит), а также другие заболевания, такие как ХОБЛ, астма или эмфизема.
[328] Заявляются способы лечения состояний, заболеваний или нарушений, в которых одна или более функций Т-клеток, таких как выработка цитокинов или клеточно-обусловленная цитотоксичность, являются аномальными или нежелательными, включая ревматоидный артрит, рассеянный склероз, острое или хроническое отторжение клеточной ткани или трансплантата органа, или карциномы гематопоэтического происхождения, а иммунопатологию, связанную с заболеванием или инфекцией.
[329] Кроме того, заявляются способы лечения нейродегенеративных заболеваний, сердечно-сосудистых заболеваний и седиментации тромбоцитов.
[330] Помимо этого, заявляются способы лечения кожных заболеваний, таких как красная кожная порфира, полиморфное светлое высыпание, дерматомиозит, фотодерматоз, оральный красный плоский лишай, панникулит, склеродермия, крапивный васкулит.
[331] Кроме того, заявляются способы лечения хронических воспалительный заболеваний, таких как саркоидоз, анулярная гранулема.
[332] В других реализациях состояние или нарушение (например, РI3K-опосредованное) выбирается из следующей группы: истинная полицитемия, врожденная тромбоцитемия, миелофиброз с миелоидной метаплазией, астма, ХОБЛ, ОРДС (острый респираторный дистресс-синдром), синдром Лефлера, эозинофильная пневмония, паразитическая (в частности, многоклеточными организмами) инфестация (включая легочную субтропическую эозинофилию), бронхолегочный аспергиллез, узелковый периартериит, включая синдром Черджа-Строса, СЧС), эозинофильная гранулема, эозинофильно-связанные заболевания, сказывающиеся на дыхательных путях, обусловленные реакцией на лекарственные средства, псориаз, контактный дерматит, атопический дерматит, гнездная алопеция, полиморфная эритема, герпетиформный дерматит, склеродермия, витилиго, аллергические васкулиты, аллергическая сыпь, буллезный пемфигоид, красная волчанка, пемфигус, приобретенный буллезный эпидермолиз, аутоиммунные гематоидные нарушения (например, гемолитическая анемия, апластическая анемия, чистая эритроцитная анемия и идиопатическая тромбоцитопения), системная эритематозная волчанка, полихондрия, склеродермия, гранулематоз Вегенера, дерматомиозит, хронический активный гепатит, миастения (myasthenia gravis), синдром Стивенса-Джонсона, идиопатическая целиакия, аутоиммунная воспалительную болезнь кишечника (например, неспецифический язвенный колит и болезнь Крона), эндокринная офтальмопатия, болезнь Грейвса, саркоидоз, альвеолит, хронический аллергический пневмонит, рассеянный склероз, первичный билиарный цирроз печени, увеит (априорный и апостериорный), интерстициальный фиброз легких, псориатический артрит, гломерулонефрит, сердечно-сосудистые заболевания, атеросклероз, гипертония, тромбоз глубоких вен, инсульт, инфаркт миокарда, нестабильная стенокардия, тромбоэмболия, легочная эмболия, тромболитические заболевания, острая артериальная ишемия, периферийные тромболитические закупоривания и коронарная недостаточность, реперфузные повреждения, ретинопатия, такая как диабетическая ретинопатия или гипербарическая кислородно-индуцированная ретинопатия, а также условия, характеризуемые повышенным внутриглазным давлением или секрецией внутриглазной жидкости, например, глаукома.
[333] В другой реализации нарушением, опосредованным неадекватной активностью PI3-киназы, является боль.
[334] В другой реализации заявляемые соединения полезны в лечении состояний или нарушений, выбранных из следующей группы: первичная кожная В-клеточная лимфома, иммунобуллезное заболевание, обыкновенная пузырчатка, эксфолиативная пузырчатка, эндемичная форма бразильской пузырчатки (Fogo selvagem), паранеопластическая пузырчатка, буллезный пемфигоид, пемфигоид мембраны слизистой оболочки, приобретенный буллезный эпидермолиз, хроническое заболевание, вызванное трансплантом от генетически отличающейся особи, дерматомиозит, системная эритематозная волчанка, васкулит, малообъемный васкулит, кожный васкулит с синдромом гипокомплементемии, антинейтрофильный цитоплазматический антительный васкулит, криоглобулинемия, синдром Шнитцлера, макроглобулинемия Вальденстрема, ангионевротический отек, витилиго, системная эритематозная волчанка, идиопатическая тромбоцитопеническая пурпура, рассеянный склероз, синдром холодовой агглютинации, аутоиммунная гемолитическая анемия, антинейтрофильный цитоплазматический антительный васкулит, заболевание, вызванное трансплантом от генетически отличающейся особи, криоглобулинемия и тромбозный тромбоцитопенический.
[335] В другой реализации заявляемые соединения полезны в лечении, профилактике или улучшении аутоиммунного заболевания и воспалительных состояний, в частности, воспалительных состояний с этиологией, содержащей аутоиммунный компонент, таких как артрит (например, ревматоидный артрит, arthritis chronica progrediente и деформирующий артрит) и ревматических заболеваний, включая воспалительные состояния и ревматические заболевания, включающие остеопороз, воспалительную боль, спондилоартропатию, включая анкилозный спондилит, синдром Рейтера, реактивный артрит, псориатический артрит и энтерофатический артрит, гиперчувствительность (включая гиперчувствительность дыхательных путей и кожную гиперчувствительность), и аллергии. К специфическим аутоиммунным заболеваниям, для лечения которых могут быть использованы заявляемые антитела, относятся аутоиммунные гематоидные нарушения (например, гемолитическую анемию, апластическую анемию, чистую эритроцитную анемию и идиопатическую тромбоцитопению), приобретенная гемофилия A, cold agglutinin disease, синдром холодовой агглютинации, тромбоцитопеническая тромбогемолитическая пурпура, синдром Шегрена, системная эритематозная волчанка, воспалительные мышечные нарушения, полихондрия, склеродермия, антинейтрофильный цитоплазматический антительный васкулит, IgM-опосредованная невропатия, опсоклонический миоклонический синдром, гранулематоз Вегенера, дерматомиозит, хронический активный гепатит, миастения (myasthenia gravis), псориаз, синдром Стивенса-Джонсона, обыкновенная пузырчатка, эксфолиативная пузырчатка, идиопатическая целиакия, аутоиммунная воспалительную болезнь кишечника (например, неспецифический язвенный колит, болезнь Крона и синдром раздраженной толстой кишки), эндокринная офтальмопатия, болезнь Грейвса, саркоидоз, рассеянный склероз, оптикомиелит, первичный биллиарный цирроз, ювенильный диабет (сахарный диабет 1 типа), увеит (априорный, промежуточный и апостериорный, а также панувеит), сухой кератоконъюнктивит и весенний кератоконъюнктивит, интерстициальный фиброз легких, псориатический артрит и гломерулонефрит (вместе и без нефротического синдрома, например, включая идиопатический нефротический синдром или минимальное нефропатическое изменение), опухоли, воспалительное заболевание кожи и роговая оболочка глаза, миозит, ослабление костных имплантов, нарушения метаболизма, такие как атеросклероз, диабет и дислипидемия.
[336] В одной реализации заявляемого изобретения представлено использование соединения с формулой (I) в терапии. В другой реализации терапия выбирается из заболевания, которое может быть излечено ингибированием PI3K. В другой реализации заболевание выбирается из приведенного выше списка, подходит из аутоиммунных нарушений, воспалительных заболеваний, аллергических заболеваний, заболеваний дыхательных путей, таких как астма и ХОБЛ, отторжение трансплантата; выработка антитела, представление антигена, выработка цитокина или лимфоидный органогенез неадекватны или нежелательны, включая ревматоидный артрит, обыкновенную пузырчатку, идиопатическую тромбоцитопеническую пурпуру, системную красную волчанку, рассеянный склероз, синдром Шегрена, аутоиммунную гемолитическую анемию, АНЦА (антинейтрофильные цитоплазматические антитела)-связанные васкулиты, криоглобулинемия, тромбоцитопеническая тромбогемолитическая пурпура, хроническая аутоиммунная аллергическая сыпь, аллергия (атопические дерматиты, контактный дерматит, аллергический ринит), Синдром Гудпасчера, AMR (отторжение трансплантата, опосредованное антителом), гиперострое, острое и хроническое отторжение трансплантата, опосредованное В-клетками и карциномы гематопоэтического происхождения, включая, без ограничения перечисленным далее, множественную миелому; острый миелоидный лейкоз (ОМЛ); хроническую миелоидную лейкемию; лимфолейкоз; миелоидную лейкемию; неходжкинскую лимфому; лимфому; истинную полицитемию; идиопатическую тромбоцитемию; миелофиброз с миелоидной метаплазией; и макроглобулинемию Вальденстрема; больше подходит из: ревматоидного артрита (РА), обыкновенной пузырчатки (PV), идиопатической тромбоцитопенической пурпуры (ITP), тромбоцитопенической тромбогемолитической пурпуры (ТТР), аутоиммунной гемолитической анемии (AIHA), приобретенной гемофилии типа А (AHA), системной эритематозной волчанки (SLE), рассеянного склероза (MS), миастения (myasthenia gravis) (MG), синдрома Шегрена (SS), АНЦА (антинейтрофильные цитоплазматические антитела)-связанных васкулитов, криоглобулинемии, хронической аутоиммунной аллергической сыпи (CAU), аллергии (атопический дерматит, контактный дерматит, аллергический ринит), синдрома Гудпасчера, отторжения трансплантата и карцином гематопоэтического происхождения, а также в иммунопатологии, связанной с заболеванием или инфекцией, например, в тяжелой и церебральной малярии, трипаносомозе, лейшманиозе, токсоплазмозе и нейроцистицеркозе.
[337] Так, в еще одной реализации заявляемого изобретения представлено использование соединения с формулой (I) для производства лекарственного средства. В другой реализации лекарственное средство используется для лечения заболевания, которое может быть излечено ингибированием PI3K. В другой реализации заболевание выбирается из приведенного выше списка, подходит из аутоиммунных нарушений, воспалительных заболеваний, аллергических заболеваний, заболеваний дыхательных путей, таких как астма и ХОБЛ, отторжение трансплантата; выработка антитела, представление антигена, выработка цитокина или лимфоидный органогенез неадекватны или нежелательны, включая ревматоидный артрит, обыкновенную пузырчатку, идиопатическую тромбоцитопеническую пурпуру, системную красную волчанку, рассеянный склероз, синдром Шегрена, аутоиммунную гемолитическую анемию, АНЦА (антинейтрофильные цитоплазматические антитела)-связанные васкулиты, криоглобулинемия, тромбоцитопеническая тромбогемолитическая пурпура, хроническая аутоиммунная аллергическая сыпь, аллергия (атопические дерматиты, контактный дерматит, аллергический ринит), Синдром Гудпасчера, AMR (отторжение трансплантата, опосредованное антителом), гиперострое, острое и хроническое отторжение трансплантата, опосредованное В-клетками и карциномы гематопоэтического происхождения, включая, без ограничения перечисленным далее, множественную миелому; острый миелоидный лейкоз (ОМЛ); хроническую миелоидную лейкемию; лимфолейкоз; миелоидную лейкемию; неходжкинскую лимфому; лимфому; истинную полицитемию; идиопатическую тромбоцитемию; миелофиброз с миелоидной метаплазией; и макроглобулинемию Вальденстрема; больше подходит из: ревматоидного артрита (РА), обыкновенной пузырчатки (PV), идиопатической тромбоцитопенической пурпуры (ITP), тромбоцитопенической тромбогемолитической пурпуры (ТТР), аутоиммунной гемолитической анемии (AIHA), приобретенной гемофилии типа А (AHA), системной эритематозной волчанки (SLE), рассеянного склероза (MS), миастения (myasthenia gravis) (MG), синдрома Шегрена (SS), АНЦА (антинейтрофильные цитоплазматические антитела)-связанных васкулитов, криоглобулинемии, хронической аутоиммунной аллергической сыпи (CAU), аллергии (атопический дерматит, контактный дерматит, аллергический ринит), синдрома Гудпасчера, отторжения трансплантата и карцином гематопоэтического происхождения, а также в иммунопатологии, связанной с заболеванием или инфекцией, например, в тяжелой и церебральной малярии, трипаносомозе, лейшманиозе, токсоплазмозе и нейроцистицеркозе.
ОСНОВНЫЕ ПРОЦЕДУРЫ СИНТЕЗА
[338] Для иллюстрации заявляемого изобретения в заявку включены следующие примеры. Однако, следует иметь в виду, что эти примеры не ограничивают заявляемое изобретение, а означают только предлагаемый способ реализации заявляемого изобретения.
[339] Как правило, заявляемые соединения могут быть приготовлены заявляемыми способами, где заместители определены для приведенной выше формулы (I), за исключением дополнительных примечаний. Для дополнительного иллюстрирования заявляемого изобретения ниже приводятся схемы и примеры, не имеющие ограничительного характера. Специалистам будет очевидно, что приведенными химическими реакциями несложно воспользоваться для приготовления некоторого количества других заявляемых соединений, и альтернативные способы приготовления заявляемых соединений находятся в области заявляемого изобретения. Например, синтез соединений по заявляемому изобретению, не вошедших в приведенные примеры, может быть успешно осуществлен модификацией известных специалистам способов, скажем, соответствующей защитой реагирующих групп, использованием других подходящих реагентов, известных специалистам, но отличающихся от приведенных в настоящей заявке, и/или внесением общепринятых изменений в условия реакции. В качестве альтернативного варианта, как пригодные для приготовления других заявляемых соединений, будут установлены другие реакции, изложенные в настоящей заявке или известные специалистам.
[340] В отсутствие иных указаний все температуры приведенных ниже примеров указываются в градусах Цельсия. Реагенты были приобретены у коммерческих поставщиков, таких как Aldrich Chemical Company, Arco Chemical Company и Alfa Chemical Company, Shanghai Medpep. Co Ltd, Aladdin-Shanghai Jinchun Reagents, Ltd, и применялись без дополнительной очистки, если не указано иное. Общераспространенные растворители были приобретены у коммерческих поставщиков, таких как Shantou XiLong Chemical Factory, Guangdong Guanghua Reagent Chemical Factory Co. Ltd., Guangzhou Reagent Chemical Factory, Tainjin YuYu Fine Chemical Ltd., Qingdao Tenglong Reagent Chemical Ltd., и Qingdao Ocean Chemical Factory.
[341] Безводный THF, диоксан, толуол и эфир были получены возгонкой растворителя и натрия с обратным холодильником. Безводный CH2Cl2 и CHCl3 были получены возгонкой растворителя и СаН с обратным холодильником, EtOAc, РЕ, гексан, DMA и DMF были обработаны Na2SO4 перед применением.
[342] Изложенные ниже реакции производились, как правило, при избыточном давлении в атмосфере азота или аргона, или с помощью осушительной трубы (если не указано иное) в обезвоженных растворителях, и реакционные колбы обычно комплектовались каучуковой диафрагмой для ввода шприцом субстратов или реагентов. Стеклянная химическая посуда подвергалась сушке в печи и/или сушке нагреванием.
[343] Колонная хроматография производилась с помощью силикагельной колонны. Силикагель (300-400 меш) был приобретен у поставщика Qingdao Ocean Chemical Factory. 1H NMR спектры были записаны спектрометром Bruker 400 МГц или спектрометром Bruker 600 МГц при окружающей температуре. 1Н NMR спектры были получены как CDCl3, DMSO-d6, CD3OD или ацетон-d6 растворы (зафиксированы в ppm или мд, миллионные доли), с помощью TMS (0 ppm) или хлороформа (7,26 ppm) как эталонного стандарта. При указании кратности пиковых значений используются следующие аббревиатуры: s (однократная), d (двойная), t (тройная), m (многократная), br (расширенная), dd (удвоенная двойная), dt (удвоенная тройная). Константы взаимодействия (J), при наличии, измеряются в герцах (Гц).
[344] Масс-спектральные данные низкого разрешения (MS) обычно определялись прибором Agilent 6120 Quadrupole HPLC-MS (Zorbax SB-C18, 2,1×30 мм; 3,5 микрона, 6-минутный прогон; номинальный расход 0,6 мл/мин, 5%-95% (0,1% муравьиная кислота в CH3CN) в (0,1% муравьиная кислота в H2O)) с помощью УФ обнаружения на 210 нм/254 нм и в режиме электрораспылительной ионизации (ESI).
[345] Степень очистки компонентов анализировалась прибором Agilent 1260 Pre-HPLC или Calesep Pump 250 Pre-HPLC (колонна NOVASEP 50/80 мм DAC) с помощью УФ обнаружения на 210 нм/254 нм.
[346] В количественных характеристиках используются следующие аббревиатуры:
ATP аденозин трифосфат
АсОН, НАс, НО Ас, CH3СООН уксусная кислота
АсОК, СН3СООК уксуснокислый калий
AIBN азодиизобутиронитрил
BBr3 трехбромистый бор
BINAP 2,2'-бис(дифенилфосфино)-1,1'-бинафтил
Bu4NF тетрабутиламмоний фторид
Реактив Бёрджесса (карбоксисульфамоил)трииэтиламмоний гидрохлорид меотловый эфир внутренней соли
BSA альбумин бычьей сыворотки
ВОС, Boc бутилоксикарбонил
n-BuOH бутиловый спирт
n-BuLi n-бутиллитий
(n-Bu)3SnCl три-n-бутилтин хлорид
Ca(SO3CF3)2 кальций трифторметил сульфонат
CS2CO3 карбонат цезия
CCl4 тетрахлорид углерода
СН2Cl2, DCM метиленхлорид
CHCl3 хлороформ
CDCl3 дейтерированный хлороформ
CH3CN ацетонитрил
CH3CHCN пропионитрил
(CH3)2CHCN изобутиронитрил
CH3Cl метил хлорид
СН3I метил йoдид
C5F фторид цезия
CH3SO2Cl, MsCl метансульфонил хлорид
Cu медь
CuI йодид меди
DССN,N'-дициклогексилкарбодиимид
DBU 1,8-диазабицикло[5.4.0]undec-7-ene
D2 газообразный дейтерий
DIBAL гидрид диизобутилалюминия
DIAD диизопропил азодикарбоксилат
DIEA, DIPEA, iPR2Net N,N-диизопропилэтиламин
DEAD диметилазодикарбоксилат
DMF диметилформамид
DMAP 4-диметиламинопиридин
DMSO диметилсульфоксид
DMFDMA N,N-диметилформамид диметил ацетал
DPPA дифенилфосфорил азид
DTT DL-дитиотреитол
EDC, EDCI 1-(3-диметиламинопропил)-3-этилкарбодиимид гидрохлорид
EDTA этилендиаминетэтрауксусная кислота
Et3N, TEA триетиламин
EtOAc, ЕА, этилацетат
Et2O диэтиловый эфир
EtOH этанол
FBS эмбриональная бычья сыворотка
Fe железо
г грамм
h час (ч)
HATU 2-(7-аза-1H-бензотриазол-1-ил)-1,1,3,3-тетраметилурониум гексафторфосфат
HBr бромистоводородная кислота
HCl соляная кислота
HOAT 1-гидрокси-7-азабензотриазол
HOBt 1-гидроксибензотриазол гадрат
H2 водород
H2O вода
Н2O2 перекись водорода
H3PO4 ортофосфорная кислота
H2SO4 серная кислота
HNO3 азотная кислота
НСООК муравьинокислый калий
HCOONH4 муравьинокислый аммоний
HMDS гексаметилдисилазан
HPLC высокопроизводительная жидкостная хроматография жидкостная хроматография высокго давления
I2 йод
LiHMDS литий бис(триметилсилил)-амид
LDA диизопропиламид лития
МВР основной миелиновый белок
МСРВА мета-хлорпербензойная кислота
MeCN, CH3CN ацетонитрил
MgSO4 сульфат магния
MeOH, CH3OH метанол
MeI йодистый метил
MOPS 3-(N-морфолино)пропансульфоновая кислота
2-MeTHF 2-метил тетрагидрофуран
мл миллилитр
мин минута
N2 азот
NMP N-метилпирроидинон
NaHCO3 двууглекислый натрий
NaBH4 борогидрит натрия
NaBH3CN цианоборгидрид натрия
NaOtBu трет-бутоксид натрия
NaOMe, CH3ONa, NaOCH3 метоксид натрия
NaOH гидроокись натрия
NaClO2 хлористокислый натрий
NaClO гипохлорит натрия
NaCl хлористый натрий
NaH2PO4 мононатрийфосфат
NaH гидрид натрия
NaI йодистый натрий
Na2SO4 сульфат натрия
Na2O3 тиосульфат натрия
NBS N-бромсукцинимид
NIS N-йодсукцинимид
NCS N-хлорсукцинимид
NEt3 триэтиламин
NH3 аммиак
NH4Cl хлорид аммония
NH2OH-HCl гидроксиламина гидрохлорид
(NH4)2Ce(NO3)6 цериевый нитрат аммония
Pd/C палладий на углероде
Pd2(dba)3 бис(дибензилиденацетон) палладий
Pd(OAc)2 ацетат палладия
Pd(OH)2 гидроксид палладия
Pd(PPh3)4 тетракис трифенилфосфин палладия
Pd(PPh3)2Cl2 хлорид бис(трифенилфосфин)палладия(II)
Pd(dppf)Cl2 хлорид бис 1,1-бис(дифенилфосфин)ферроцен палладия
P(t-Bu)3 три(терт-бутил)фосфин
РЕ петролейный эфир (60-90°С)
PBS фосфат забуференный физиологаческий раствор
POCl3 хлорокись фосфора
PhI(ОАс)2 иодбензола диацетат
K2CO3 углекислый калий
КОН гидроокись калия
RT rt r.t. комнатная температура
Rt время выдержки
SOCl2 тионилхлорид
SO2Cl2 сульфурилхлорид
t-BuOK терт-бутанолат калия
TBTU О-бензотриазол-1-ил-N,N,N',N'-тетраметилурониум тетрафторборат
TBS трис забуференный физиологический раствор
THF тетрагидрофуран
TFA трифторуксусная кислота
ТЕАС бис(тетра-этиламмоний)карбонат
Трис тригидроксиметил аминометан
ТsСl4-толуол сульфонил хлорид
мкл микролитр
Zn цинк
[347] На следующих схемах представлены показательные процедуры синтеза для приготовления заявляемых соединений. В отсутствие иных указаний на каждый R1, R2, R3, R4 и X распространяются заданные выше определения в связи с формулой (I). «PG» является подходящей защитной алкиленовой группой.
Схема 1

[348] Промежуточное соединение (7) может быть приготовлено общепринятым способом, проиллюстрированным на схеме 1. Бензойная кислота (1) вначале реагирует с SOCl2 при температуре дистилляции в неполяризованном растворителе, таком как толуол, затем обрабатывается аминосоединением (2) для образования амида (3). Соединение (4) вначале реагирует с SOCl2 при температуре дистилляции в неполяризованном растворителе, таком как толуол, затем обрабатывается соединением (3) с образованием соединения (5). Восстановление и циклизация нитросоединения (5) в присутствии порошкообразного Zn и кислоты (такой как уксусная кислота) обеспечивает образование соединения (6). Снятие защиты аминогрупп соединения (6) при стандартных условиях, известное специалистам, например, без ограничения упомянутым далее, обработка кислотой для получения промежуточного соединения (7).
Схема 2
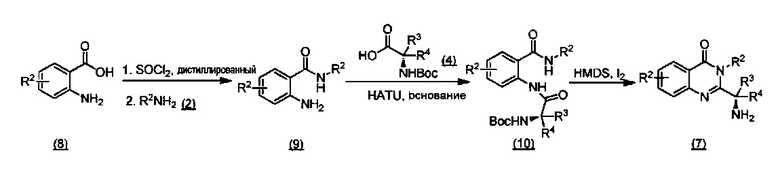
[349] Промежуточное соединение (7) может быть также приготовлено общепринятым способом, проиллюстрированным на схеме 2. Бензойная кислота (8) вначале реагирует с SOCl2 при температуре дистилляции в неполяризованном растворителе, таком как толуол, затем обрабатывается аминосоединением (2) для образования амида (9). Связывание соединения (9) с Вос-защищенной кислотой (4) в присутствии связующего реагента, такого как EDCI или HATU приводит к образованию соединения (10). Циклизация соединения (10) в присутствии катализатора 12 дает промежуточное соединение (7).
Схема 3
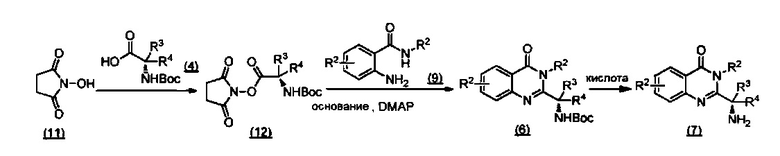
[350] На схеме 3 показан другой способ приготовления промежуточного соединения (7). Конденсация 1-гидроксипирролидина-2,5-диона (11) с Вос-защищенной кислотой (4) в присутствии основания, такого как DIPEA, приводит к образованию соединения (12), которое дополнительно циклируется с соединением (9), становясь бицикличным гетероароматическим (6). Снятие защиты аминогрупп соединения (6) при стандартных условиях, известное специалистам, такое, например, без ограничения упомянутым далее, как обработка кислотой для получения промежуточного соединения (7).
Схема 4
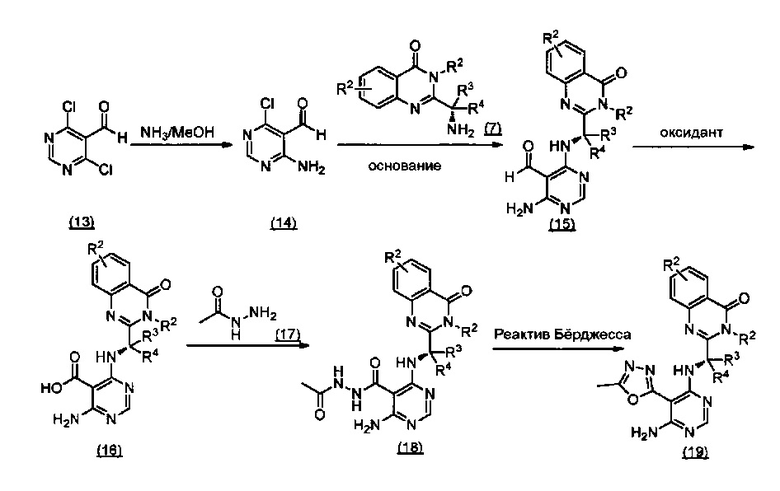
[351] Заявляемые соединения могут быть приготовлены общепринятыми способами синтеза, иллюстрированными на схеме 4 и подробно рассмотренными в примерах. Согласно схеме 4, вначале 4,6-дихлорпиримидин-5-карбальдегид (13) реагирует с раствором NH3 в МеОН для образования соединения (14). Конденсация соединения (14) с промежуточным соединением (7) в присутствии основания, такого как DIPEA, приводит к образованию соединения (15). Окисление соединения (15) в присутствии оксиданта, такого как NaClO2, приводит к образованию соединения (16). Затем соединение (16) реагирует с гидразидом уксусной кислоты (17) для образования соединения (18), которое может быть дополнительно преобразовано в соединение (9) с помощью реактива Бёрджесса в качестве желаемого ингибитора киназы.
Схема 5

[352] На схеме 5 показан другой способ приготовления желаемого ингибитора киназы. Йодирование (20) посредством N-йодсукцинимида при повышенной температуре приводит к образованию соединения (21). Соединение брома (22) реагирует с соединением (23) с помощью основания, такого как и-бутиллитий для образования соединения (24). Затем соединение (24) связывается с соединением (21) с помощью подходящего комплекса Pd в качестве катализатора, например, Pd(PPh3)2Cl2 для образования соединения (25). Наконец, соединение (25) реагирует с промежуточным соединением (7) в присутствии основания (такого как DIPEA) при температуре дистилляции для получения желаемого ингибитора киназы (26).
Схема 6
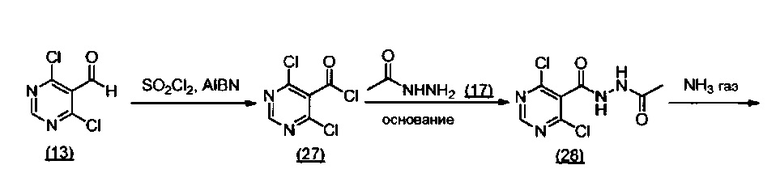
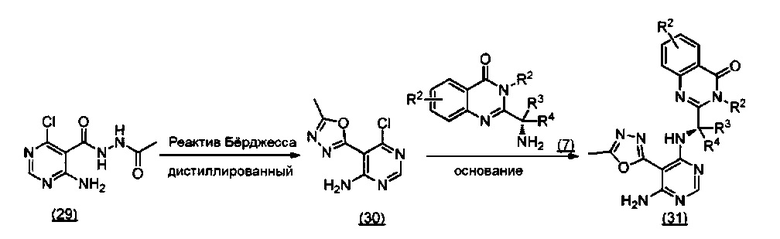
[353] Некоторые соединения структуры, определяемой формулой (I), можно также получить общепринятым способом, иллюстрированным на схеме 6. 4,6-дихлорпиримидин-5-карбальдегид (13) преобразуется в хлористый ацил (27) в присутствии SO2Cl2 и AIBN. Затем соединение (27) реагирует с гидразидом уксусной кислоты (17) для образования соединения (28). После чего соединение (28) обрабатывается газообразным NH3 для образования соединения (29), замещенного амином, затем следует реакция циклизации в условиях использования реактива Берджесса при температуре дистилляции для получения соединения (30). Соединение (30) реагирует с промежуточным соединением (7) в присутствии основания, такого как DIPEA, при температуре дистилляции для получения желаемого ингибитора киназы (31).
Схема 7
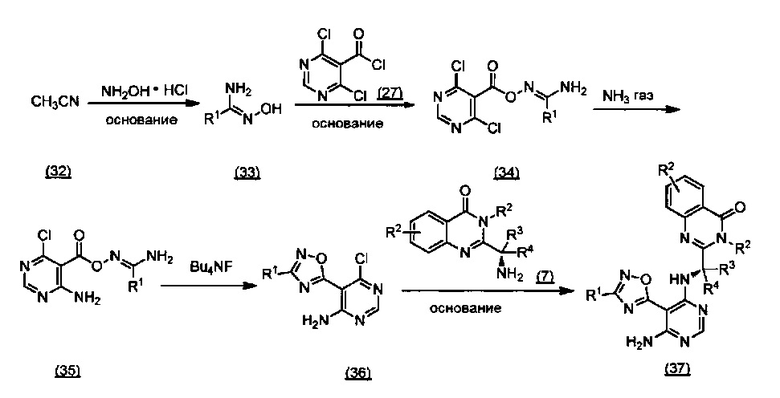
[354] На схеме 7 показан другой способ приготовления желаемого ингибитора киназы. Ацетонитрил (32) сначала реагирует с хлоргидратом гидроксиламина для получения (Z)-N'-гидроксиацетимид амида (33), который затем реагирует с хлористым ацилом (27) для получения соединения (34). Соединение (34) обрабатывается газообразным NH3 для образования соединения (35), затем следует реакция циклизации в условиях использования BU4NF при температуре дистилляции для получения соединения (36). Желаемый ингибитор киназы (37) получается в результате реакции соединения (36) с промежуточным соединением (7) в присутствии основания, такого как DIPEA.
Схема 8
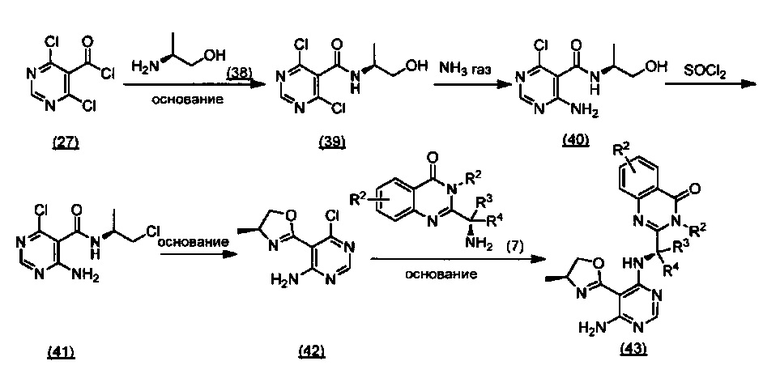
[355] На схеме 8 показан другой способ приготовления желаемого ингибитора киназы. Вначале хлористый ацил (27) обрабатывается соединением (38) для получения соединения (39), которое затем реагирует с газообразным NH3, образуя соединение (40). Гидроксигруппа соединения (40) преобразуется в Cl с помощью хлорирующего агента, такого как РОСl3 или SOCl2 при нагревании, затем следует реакция циклизации в присутствии основания, такого как NaH при комнатной температуре для получения соединения (42). Желаемый ингибитор киназы (43) получается в результате реакции соединения (42) с промежуточным соединением (7) в присутствии основания, такого как DIPEA.
Схема 9
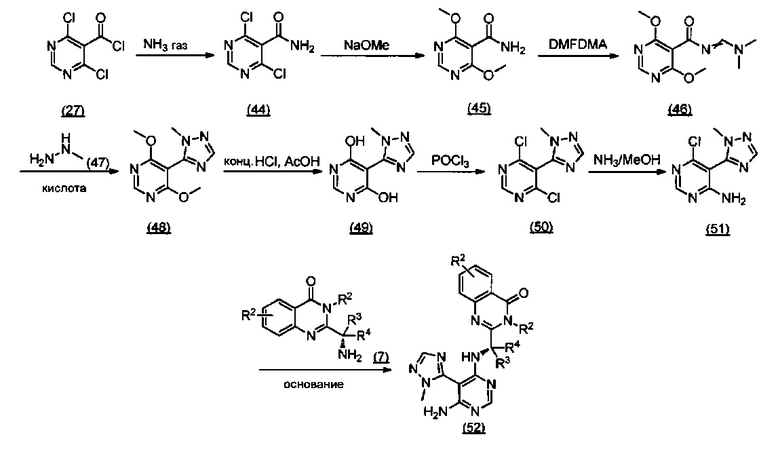
[356] В качестве альтернативного варианта, заявляемые соединения могут быть также приготовлены синтетическим способом, показанным на схеме 9. Вначале хлористый ацил (27) обрабатывается газообразным NH3 для получения амида (44). Затем амид (44) реагирует с метилатом натрия для получения соединения (45), которое дополнительно реагирует с DMFDMA для получения соединения (46). Циклизация соединения (46) с метилгидразином (47) при помощи кислоты (такой как уксусная кислота) приводит к образованию соединения (48). Далее соединение (48) реагирует с концентрированной соляной и уксусной кислотой для образования соединения (49), которое преобразуется в соединение хлора (50) с помощью хлорирующего агента, такого как РОСl3 или SOCl2 в условиях нагревания. После чего соединение (50) обрабатывается раствором NH3 в метаноле для образования соединения (51), замещенного амином. Соединение (51) реагирует с промежуточным соединением (7) в присутствии основания, такого как DIPEA, при температуре дистилляции для получения желаемого ингибитора киназы (52).
Схема 10
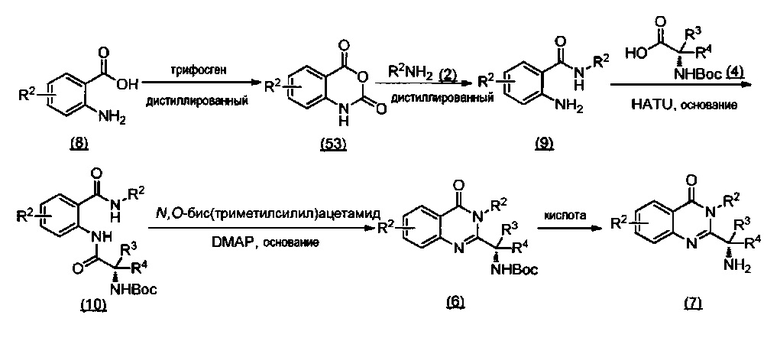
[357] На схеме 10 показан другой способ приготовления промежуточного соединения (7). Соединение (8) реагирует с трифосгеном при температуре дистилляции для получения соединения (53). Затем соединение (53) обрабатывается амином (2) при температуре дистилляции для получения соединения (9). Связывание соединения (9) с Boc-защищенной кислотой (4) в присутствии связующего реагента, такого как EDCI или HATU приводит к образованию соединения (10). Циклизация соединения (10) в присутствии N,O-бис(триметилсилил)ацетамида, DMAP и основания приводит к образованию соединения (6). Снятие защиты аминогрупп соединения (6) при стандартных условиях, известное специалистам, такое, например, без ограничения упомянутым далее, как обработка кислотой для получения промежуточного соединения (7).
Схема 11
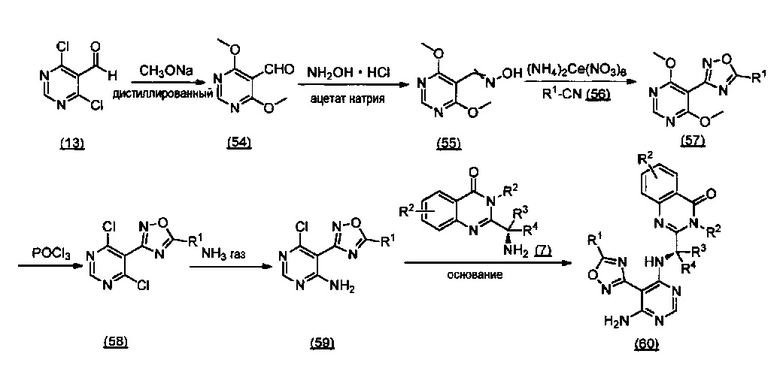
[358] Некоторые соединения структуры, определяемой формулой (I), можно также получить общепринятым способом, иллюстрированным на схеме 11. Соединение (13) обрабатывается CFbONa при температуре дистилляции для получения соединения (54). Затем соединение (54) реагирует с NH2OH-HCl для образования соединения (55). После чего производится циклизация соединения (55) с соединением (56)при условии использования (NH4)2Ce(NO3)6 для получения соединения (57). Вначале соединение (57) преобразуется в соединение хлора (58) с помощью хлорирующего агента, такого как РОСl3 или SOCl2 в условиях нагревания, затем соединение (58) стравливается через газообразный NH3 в течение суток для образования соединения (59), замещенного амином. Соединение (59) реагирует с промежуточным соединением (7) в присутствии основания, такого как DIPEA, при температуре дистилляции для получения желаемого ингибитора киназы (60).
Схема 12
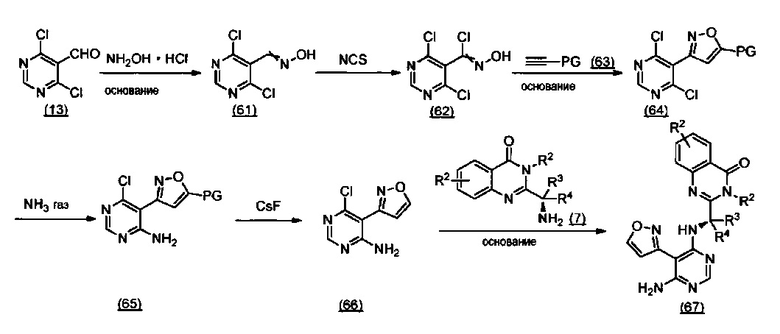
[359] Некоторые соединения структуры, определяемой формулой (I), можно также получить общепринятым способом, иллюстрированным на схеме 12. Соединение (13) реагирует с NFbOH-HCl в присутствии основания для получения формальдоксима (61). Затем соединение (61) обрабатывается NCS для получения соединения (62). В результате циклизации соединения (62) и соединения (63) в присутствии основания образуется соединение (64) с защитными алкин-группами PG. После чего соединение (64) стравливается через газообразный NH3 для образования соединения (65), замещенного амином. К подходящим защитным алкин-группам PG относятся, не ограничиваясь перечисленным далее, TMS, TES или TIPS. Защитные группы PG могут быть удалены при стандартных условиях, известных специалистам, таких, без ограничения перечисленным далее, как обработка гидратогенным основанием, TBAF или CsF для получения соединения (66). Соединение (66) реагирует с промежуточным соединением (7) в присутствии основания, такого как DIPEA, при температуре дистилляции для получения желаемого ингибитора киназы (67).
Схема 13
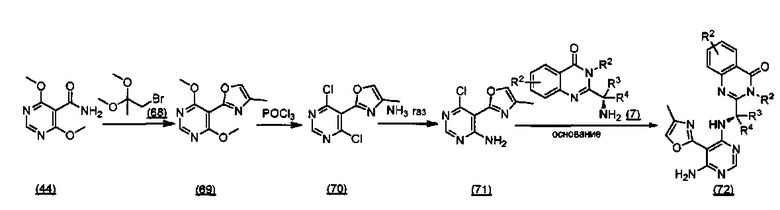
[360] Некоторые соединения структуры, определяемой формулой (I), можно также получить общепринятым способом, иллюстрированным на схеме 13. В результате циклизации соединения (44) и соединения (68) образуется соединение (69). Соединение (69) преобразуется в соединение хлора (70) с помощью хлорирующего агента, такого как РОСl3 или SOCl2 в условиях нагревания, затем соединение (70) стравливается через газообразный NH3 для образования соединения (71), замещенного амином. Наконец, соединение (71) реагирует с соединением (7) для получения желаемого ингибитора киназы (72).
Схема 14
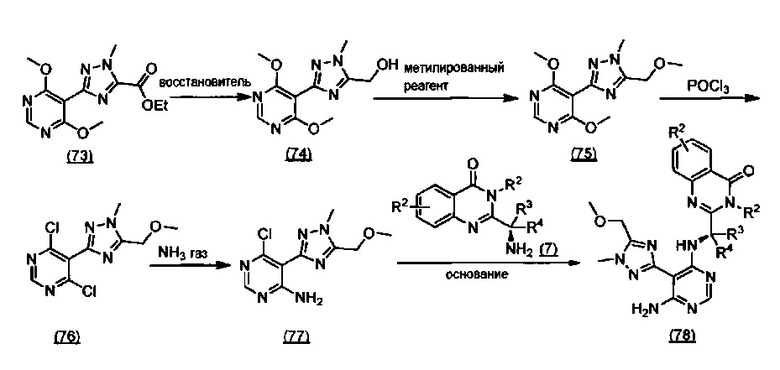
[361] Некоторые соединения структуры, определяемой формулой (I), можно также получить общепринятым способом, иллюстрированным на схеме 14. Восстановление соединения (73) с восстановителем, таким как LiAIH4, дает соединение (74). Соединение (74) обрабатывается метилированным реагентом, таким как СН3I для получения соединения (75). Соединение (75) преобразуется в соединение хлора (76) с помощью хлорирующего агента, такого как РОСl3 или SOCl2 в условиях нагревания, затем соединение (75) стравливается через газообразный NH3 для образования соединения (77), замещенного амином. Наконец, соединение (77) реагирует с соединением (7) для получения желаемого ингибитора киназы (78).
Схема 15

[362] Некоторые соединения структуры, определяемой формулой (I), можно также получить общепринятым способом, иллюстрированным на схеме 15. Соединение (79) обрабатывается раствором NH3 в метаноле для получения амида (80). Затем соединение (80) реагирует с соединением (7) для получения желаемого ингибитора киназы (81).
Схема 16

[363] Некоторые соединения структуры, определяемой формулой (I), можно также получить общепринятым способом, иллюстрированным на схеме 16. Реакция циклизации соединения (46) при условии использования хлоргидрата гидроксиламина и основания дает соединение (82). Вначале соединение (82) преобразуется в соединение хлора (83) с помощью хлорирующего агента, такого как РОСl3 или SOCl2 в условиях нагревания, затем соединение (83) стравливается через газообразный NH3 для образования соединения (84), замещенного амином. Соединение (84) реагирует с промежуточным соединением (7) в присутствии основания, такого как DIPEA, при температуре дистилляции для получения желаемого ингибитора киназы (85).
ПРИМЕРЫ
Пример 1
(S)-2-(1-((6-амин-5-(5-метил-1,3,4-оксадиазол-2-ил)пиримидин-4-ил)амин)-пропил)-5-хлор-3-фенилквиназолин-4(3H)-один
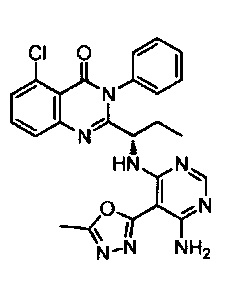
Шаг 1) 2-амин-6-хлор-N-фенилбензамид
[364] К раствору 2-амин-6-хлорбензойной кислоты (10,30 г; 60,0 моль) в толуоле (250 мл) добавлен по каплям SOCl2 (24 мл, 330,.4 ммоль) при rt. После добавления реагирующая смесь перемешивалась при 120°C в течение суток и была сконцентрирована in vacuo для получения коричневого масла, которое было непосредственно использовано в следующем шаге без дополнительной очистки.
К раствору хлорангидрида, приготовленному выше в СН3Cl (250 мл), был добавлен анилин (12 мл, 131,4 ммоль). После добавления реагирующая смесь перемешивалась при 80°C в течение 5 часов, затем была остужена до rt и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (PE/EtOAc (v/v)=10/1) для получения соединения, указанного в заголовке шага, в виде светло-желтого твердого вещества (10.66 г; 72.0%).
MS (ESI, pos, ion) m/z: 247,0 [M+H]+;
1H NMR (400 МГц; CDCl3) δ (ppm): 7,71 (br s; 1H); 7,65 (d; J=7,9 Гц; 2H); 7,41 (t; J=7,9 Гц; 2H); 7,21 (t; J=7,4 Гц; 1H); 7,13 (t; J=8,1 Гц; 1H); 6,80 (d; J=7,9 Гц; 1H); 6,66 (d; J=8,2 Гц; 1H).
Шаг 2) (S)-2,5-диоксопирролидин-1-ил 2-((терт-бутоксикарбонил) амин)бутанат
[365] К раствору 1-гидроксипирролидин-2,5-дион (5.80 г; 50,4 ммоль) и (S)-2-((терт-бутоксикарбонил)амин)бутановой кислоты (10.00 г; 49.2 ммоль) в THF (120 мл) был добавлен DCC (10.20 г; 49.4 ммоль) при 0°C. После добавления реагирующая смесь перемешивалась при 0°C в течение суток и была профильтрована. Осадок с фильтра был промыт с помощью EtOAc (50 мл×3) и фильтрат был сконцентрирован in vacuo. Осадок был растворен в EtOAc (500 мл), и полученная смесь была промыта насыщенным раствором NaHCO3 (100 мл) и насыщенным минеральным раствором (100 мл). Отсепарированная органическая фаза была высушена над обезвоженным Na2SO4, и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (РЕ/EtOAc (v/v)=3/1) для получения соединения, указанного в заголовке шага, в виде белого твердого вещества (10,66 г; 72,0%)
MS (ESI, pos. ion) m/z: 201.2 [M-Boc+H]+, 323.2 [M+Na]+;
1H NMR (400 МГц; CDCl3) δ (ppm): 5,03 (d; J=6,9 Гц; 1H); 4,66 (d; J=7,4 Гц; 1H); 2,86 (s; 4H); 2,02 (m; 1H); 1,90 (m; 1H); 1,48 (s; 9H); 1,09 (t; J=7,3 Гц; 3H).
Шаг 3) (S)-терт-бутил (1-(5-хлор-4-оксо-3-фенил-3,4-дигидроквиназолин-2-ил)пропил)-карбамат
[366] К раствору 2-амин-6-хло-N-фенилбензамида (4.58 г; 18.56 ммоль) и (5)-2,5-диоксопирролидин-1-ил 2-((терт-бутоксикарбонил)амин)бутаната (8.36 г; 27.85 ммоль) в толуоле (100 мл) были добавлены диметиламинопиридин (3,40 г; 27,85 ммоль) и диизопропилэтиламин (3,60 г; 27,85 ммоль). После добавления реагирующая смесь перемешивалась при 120°C в течение 24 часов, затем была остужена до rt и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (РЕ/EtOAc (v/v)=10/1) для получения соединения, указанного в заголовке шага, в виде желтого твердого вещества (1,90 г; 24,7%).
MS (ESI, pos, ion) m/z: 414,2 [M+H]+;
1H NMR (400 МГц; CDCl3) δ (ppm): 7,89 (br s; 1H); 7,67 (m; 2H); 7,57 (m; 3H); 7,47 (d; J=7,1 Гц; 1H); 7,30 (s; 1H); 5,86 (br s; 1H); 4,42 (m; 1H); 1,44 (s; 9H); 1,24 (q; J=8,0 Гц; 1H); 1,18 (q; J=8,0 Гц; 1H); 0,80 (t; J=7,4 Гц; 3H).
Шаг 4) (S)2-9-аминопропил)-5-хлор-3-фенилквиназолин-4(3H)-один
[367] К раствору (S)-терт-бутил (1-(5-хлор-4-оксо-3-фенил-3,4-дигидроквиназолин-2-ил)-пропил)карбамата (1.90 г; 4.6 ммоль) в DCM (50 мл) был медленно добавлен раствор HCl в EtOAc (0.5 М, 40 мл, 20 ммоль)при rt. После добавления реагирующая смесь перемешивалась при rt в течение суток и была сконцентрирована in vacuo. Осадок был растворен в воде (50 мл) и DCM (50 мл), затем смесь была оттитрована до рН=10 насыщенным гидратогенным раствором Na2CO3, после чего экстрактирована с помощью DCM (150 мл×3). Комбинированные органические фазы были промыты насыщенным минеральным раствором (100 мл×3), высушены над обезвоженным Na2SO4, и сконцентрированы in vacuo. Осадок был очищен хроматографией на силикагельной колонне (РЕ/EtOAc (v/v)=1/1) для получения соединения, указанного в заголовке шага, в виде желтого твердого вещества (550 мг; 38,2%).
MS (ESI, pos. ion) m/z: 314,2 [M+H]+;
1H NMR (400 МГц; CDCl3) δ (ppm): 7,60 (m; 5H); 7,49 (dt; J=7,5; 1,3 Гц; 1H); 7,34 (m; 1H); 7,30 (m; 1H); 3,55 (q; J=6,0 Гц; 1H); 1,82 (m; 1H); 1,56 (td; J=14,2; 7,1 Гц; 1H); 0,83 (t; J=7,4Hz; 3H).
Шаг 5) 4-амин-6-хлорпиримидин-5-карбальдегид
[368] К суспензии 4,6-дихлорпиримидин-5-карбальдегида (19,50 г; 110 ммоль) в толуоле (220 мл) был добавлен раствор NH3 в МеОН (7 М, 27 мл, 189 ммоль), и реагирующая смесь была нагрета до 60°C, затем дополнительно перемешивалась в течение 1 часа. После чего вновь был добавлен раствор NH3 в МеОН (7 М, 18 мл, 126 ммоль) и полученную смесь перемешивали при 60°C еще 3 часа. Смесь была остужена до rt и сконцентрирована in vacuo. Осадок был растворен с помощью EtOАс (50 мл). Полученную смесь перемешивали при rt течение 1 часа и профильтровали для получения соединения, указанного в заголовке шага, в виде желтого твердого вещества (20,50 г; 100%).
MS (ESI, pos. ion) m/z: 158.0 [M+H]+;
1H NMR (400 МГц; DMSO-d6) δ (ppm): 10,25 (s; 1H); 8,73 (br s; 1H); 8,57 (br s; 1H); 8,40 (s; 1H).
Шаг 6) (R)-4-амин-6-((1-(5-хлор-4-оксо-3-фенил-3,4-дигидроквиназолин-2-ил)пропил)амин)пиримидин-5-карбальдегид
[369] К раствору (S)-2-(1-аминопропил)-5-хлор-3-фенилквиназолин -4(3H)-один (530 мг; 1,69 ммоль) и 4-амин-6-хлорпиримидин-5-карбальдегид (320 мг; 2,03 ммоль) в n-BuOH (20 мл) был добавлен DIPEA (437 мг; 3,38 ммоль). После добавления реагирующая смесь перемешивалась при 120°C в течение 30 часов, затем была остужена до rt и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (РЕ/EtOAc (v/v)=1/1) для получения соединения, указанного в заголовке шага, в виде желтого твердого вещества (250 мг; 38,2%).
MS (ESI, pos. ion) m/z: 435,2 [M+H]+;
1HNMR (400 МГц; CDCl3) δ (ppm): 10,25 (s; 1H); 8,03 (s; 1H); 7,68 (s; 1H); 7,67 (d; J=4,0 Гц; 2H); 7,61 (m; 4H); 7,53 (dd; J=6,5; 2,5 Гц; 1H); 5,18 (td; J=1,1; 4,1 Гц; 1H); 1,78 (dd; J=14,6; 7,5 Гц; 2H); 0,90 (t; J=6,8 Гц; 3H).
Шаг 7) (R)-4-амин-6-((1-(5-хлор-4-оксо-3-фенил-3,4-дигидроквиназолин-2-ил)пропил)амин)пиримидин-5-карбоновая кислота
[370] К раствору (R)-4-амин-6-((1-(5-хлор-4-оксо-3-фенил-3,4-дигидроквиназолин-2-ил)пропил)амин) пиримидин-5-карбальдегида (240 мг; 0,55 ммоль) в DCM (8 мл) были добавлены DMSO (431 мг; 5,52 ммоль), Н3PO4 (0,75 М, 2 мл, 1,50 ммоль) и хлористокислый натрий (100 мг; 1,10 ммоль). Реагирующая смесь перемешивалась в течение 5 часов, затем были добавлены DMSO (500 мг; 6.40 ммоль), Н3РO4 (0.75 М, 6 мл, 4.50 ммоль) и хлористокислый натрий (200 мг; 2,20 ммоль). Реагирующая смесь перемешивалась при rt в течение 2 часов, была оттитрована до рН=5-6 насыщенным гидратогенным раствором NaHCO3 и экстрактирована с помощью СН2Cl2 (100 мл×3). Комбинированные органические фазы были промыты насыщенным минеральным раствором (100 мл×3), высушены над обезвоженным Na2SO4, и сконцентрированы in vacuo для получения соединения, указанного в заголовке шага, в виде желтого твердого вещества (300 мг; 100%), которое было непосредственно использовано в следующем шаге без дополнительной очистки.
MS (ESI, pos. ion) m/z: 451,2 [M+H]+.
Шаг 8) (S) N'-ацетил-4-амин-6-((1-(5-хлор-4-оксо-3-фенил-3,4-дигидроквиназолин-2-ил)пропил)амин)пиримидин-5-карбальдегид
[371] К раствору (R)-4-амин-6-((1-(5-хлор-4-оксо-3-фенил-3,4-дигидроквиназолин-2-ил)пропил)амин)пиримидин-5-карбоновой кислоты (373 мг; 0,828 ммоль) и гидразида уксусной кислоты (343 мг; 4,637 ммоль) в DCM (15 мл) были добавлены EDCI (317 мг; 1,656 ммоль) и НОAT (225 мг; 1,656 ммоль).
Реагирующая смесь перемешивалась в течение 24 часов при 45°C. Затем был добавлен раствор гидразида уксусной кислоты (343 мг; 4,637 ммоль) в DCM (15 мл), EDCI (317 мг; 1,656 ммоль) и НОАТ (225 мг; 1,656 ммоль). Реагирующая смесь перемешивалась при 45°C в течение суток, была резко охлаждена водой (20 мл), и получившаяся смесь была экстрактирована с помощью DCM (100 мл×3). Комбинированные органические фазы были промыты насыщенным минеральным раствором (100 мл×3), высушены над обезвоженным Na2SO4, и сконцентрированы in vacuo. Осадок был очищен подготовительным HPLC для получения соединения, указанного в заголовке шага, в виде желтого твердого вещества (130 мг; 31,0%).
MS (ESI, pos. ion) m/z: 507,0 [M+H]+.
Шаг 9) (S)-2-(1-((6-амино-5-(5-метил-1,3,4-оксадиазол-2-ил)пиримидин-4-ил)амино)пропил)-5-хлор-3-фенилквиназолин-4(3Н)-один
[372] К раствору (S)-N'-ацетил-4-амин-6-((1-(5-хлор-4-оксо-3-фенил-3,4-дигидроквиназолин-2-ил)пропил)амино)пиримидин-5-карбогидразида (40 мг; 0,08 ммоль) в THF (2 мл) был добавлен реактив Берджесса (40 мг; 0,16 ммоль). Реагирующая смесь была герметично закрыта в микроволновой печи и перемешивалась при 100°C в течение 1 часа, затем была сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (PE/EtOAc (v/v)=1/3) для получения соединения, указанного в заголовке шага, в виде бежевого твердого вещества (25 мг; 65,8%).
MS (ESI, pos. ion) m/z: 489,2 [M+H]+;
1H NMR (400 МГц; CDCl3) δ (ppm): 9,56 (s; 1H); 8,02 (s; 1H); 7,74 (m; 1H); 7,65 (d; J=4,5 Гц; 2H); 7,56 (m; 5H); 7,37 (m; 1H); 7,33 (m; 1H); 5,23 (td; J=7,7; 4,7 Гц; 1H); 2,77 (s; 3H); 1,85 (dd; J=14,6; 7,1 Гц; 2H); 1,01 (d; J=6,7 Гц; 3H).
Пример 2 (S)-2-(1-((6-амин-5-(5-метил-1,3,4-оксадиазол-2-ил)пиримидин-4-ил)амин)-пропил)-5-хлор-3-циклопропилквиназолин -4(3H)-один
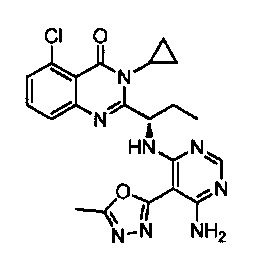
Шаг 1) 2-хлор-N-циклопропил-6-нитробензамид
[373] К суспензии 2-хлор-6-нитробензойной кислоты (10 г; 49,61 ммоль) в толуоле (50 мл) добавлен по каплям SOCl2 (5,28 мл, 74,41 ммоль) при комнатной температуре. После добавления реагирующая смесь перемешивалась при 110°C в течение суток и была сконцентрирована in vacuo. Осадок был растворен в 1,4-диоксане (30 мл), и по каплям была добавлена суспензия циклопропанамина (3,43 мл, 49,61 ммоль) и NaHCO3 (8.34 г; 99.22 ммоль) в 1,4-диоксане (30 мл) при 5°C. Затем полученная смесь перемешивалась при комнатной температуре в течение 24 часов, после чего была профильтрована. Фильтрат был сконцентрирован in vacuo для получения соединения, указанного в заголовке шага, в виде желтого порошка (11.66 г; 98%), который был непосредственно использован в следующем шаге без дополнительной очистки.
MS (ESI, pos. ion) m/z: 241,0 [M+H]+.
Шаг 2) (S)-терт-бутил(1-(2-хлор-N-циклопропил-6-нитробензоамидо)-1-оксобутан-2-ил)-карбамат
[374] К раствору 2-хлop-N-циклопропил-6-нитробензоамида (1.19 г; 4.9 ммоль) в толуоле (20 мл) добавлен по каплям SOCl2. (3,35 мл, 49,2 ммоль). После добавления реагирующая смесь перемешивалась при 120°C в течение суток, затем была сконцентрирована in vacuo для получения коричневого масла, которое было непосредственно использовано в следующем шаге без дополнительной очистки.
К раствору Boc-L-2-аминобутировой кислоты (1,50 г; 7,38 ммоль) и DIPEA (1,68 г; 12,98 ммоль) в дихлорметане (10 мл) при 0°C был добавлен раствор упомянутого выше коричневого масла в дихлорметане (30 мл). После добавления реагирующая смесь перемешивалась при комнатной температуре в течение 24 часов и была промыта 4% гидратогенной лимонной кислотой (100 мл), насыщенным гидратогенным раствором NaHCO3 (100 мл) и насыщенным минеральным раствором (30 мл). Отсепарированная органическая фаза была высушена над обезвоженным Na2SO4, и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (РЕ/EtOAc (v/v)=8/1) для получения соединения, указанного в заголовке шага, в виде желтого твердого вещества (1,41 г; 67,6%).
MS (ESI, pos. ion) m/z: 326,2 [M-Boc+H]+.
Шаг 3) (S)-терт-бутил(1-(5-хлор-3-циклопропил-4-оксо-3,4-дигидроквиназолин-2-ил)пропил)-карбамат
[375] К раствору (S)-терт-бутил (1-(2-хлор-N-циклопропил-6-нитробензоамидо)-1-оксобутан-2-ил)карбамат а (1,41 г; 3,31 ммоль) в уксусной кислоте (25 мл) был добавлен одной порцией порошкообразный цинк (1,13 г; 17,31 ммоль). Реагирующая смесь перемешивалась при rt в течение суток, была нейтрализована до рН=7-8 насыщенным гидратогенным раствором NaHCO3 и экстрактирована с помощью этилацетата (200 мл×3). Комбинированные органические фазы были высушены над обезвоженным Na2SO4, и сконцентрированы in vacuo. Осадок был очищен хроматографией на силикагельной колонне (РЕ/EtOAc (v/v)=50/6) для получения соединения, указанного в заголовке шага, в виде белого твердого вещества (863 мг; 69%).
MS (ESI, pos. ion) m/z: 378,1 [M+H]+.
Шаг 4) (S)-2-(1-аминопропил)-5-хлор-3-циклопропилквиназолин-4(3H)-один
[376] К раствору (S)-терт-бутил (1-(5-хлор-3-циклопропил-4-оксо-3,4-дигидроквиназолин-2-ил)пропил)карбамата (3,01 г; 7,97 ммоль) в этилацетате (11 мл) был одной порцией добавлен раствор PCl в EtOAc (3,5 М; 15 мл) при комнатной температуре. Смесь перемешивалась при комнатной температуре в течение 5,5 часов, затем была разведена водой (150 мл). Полученная смесь была экстрактирована этилацетатом (100 мл). Отсепарированная гидратогенная фаза была оттитрована до рН=6 порошком NaHCO3, и экстрактирована с помощью смеси EtOAc и МеОН (EtOAc/MeOH (v/v)=100/2, 200 мл×3). Комбинированные органические фазы были промыты насыщенным минеральным раствором (100 мл), высушены над обезвоженным Na2SO4, и сконцентрированы in vacuo для получения соединения, указанного в заголовке шага, в виде желтого твердого вещества (2,11 г; 96%).
MS (ESI, pos. ion) m/z: 278,2 [M+H]+;
1H NMR (400 МГц; DMSO-d6) δ (ppm): 7,67 (t; J=7,9 Гц; 1H); 7,52 (d; J=8,1 Гц; 1H); 7,47 (d; J=7,8 Гц; 1H); 4,43-4,30 (m; 1H); 3,06-2,95 (m; 1H); 1,88-1,74 (m; 1H); 1,62-1,49 (m; 1H); 1,27-1,15 (m; 2H); 1,00-0,90 (m; 4H); 0,73 (dd; J=8,5; 3,6 Гц; 1H).
Шаг 5) 4,6-дихлорпиримидин-5-карбонил хлорид
[377] Суспензия 4,6-дихлорпиримидин-5-карбальдегида (10 г; 56,5 ммоль), SO2Cl2 (11,44 г; 84,75 ммоль) и AIBN (0.464 г; 2.83 ммоль) в CCl4 (100 мл) перемешивалась при 80°C в течение 5 часов. Реагирующая смесь была охлаждена до rt, затем отфильтрована, и фильтрат был сконцентрирован in vacuo для получения соединения, указанного в заголовке шага, в виде желтого твердого вещества (11,9 г; 99,6%).
Шаг 6) N'-ацетил-4,6-дихлорпиримидин-5-карбогидразид
[378] К смеси гидразида уксусной кислоты (1,39 г; 18,74 ммоль) в CH2Cl2 (40 мл) был добавлен DIPEA (4,84 г; 37,48 ммоль) при 0°C, затем был добавлен раствор 4,6-дихлорпиримидин-5-карбонилхлорида (4 г; 18,74 ммоль) в CH2Cl2 (20 мл). Реагирующая смесь перемешивалась при 0°C в течение 30 минут, была разведена EtOAc (400 мл), и промыта насыщенным гидратогенным раствором NH4Cl (150 мл) и насыщенным минеральным раствором (100 мл). Отсепарированная органическая фаза была высушена над обезвоженным Na2SO4, и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (РЕ/EtOAc (v/v)=1/2) для получения соединения, указанного в заголовке шага, в виде белого твердого вещества (1,78 г; 38%).
MS (ESI, pos. ion) m/z: 246,9 [M-H]-;
1H NMR (600 МГц; DMSO-d6) δ (ppm): 10,92 (d; J=2,5 Гц; 1H); 10,48 (d; J=2,6 Гц; 1H); 9,03 (s; 1H); 1,93 (s; 3H).
Шаг 7) N-ацетил-4-амин-6-хлорпиримидин-5-карбогидразид
[379] Раствор N'-ацетил-4,6-дихлорпиримидин-5-карбогидразида (1,78 г; 7,20 ммоль) в THF (80 мл) был продут газообразным NH3. Реагирующая смесь перемешивалась при rt в течение 4 часов, затем была отфильтрована, и фильтрат сконцентрирован in vacuo. Осадок был очищен хроматографией на силикагельной колонне (РЕ/EtOAc (v/v)=1/2) для получения соединения, указанного в заголовке шага, в виде белого твердого вещества (1,06 г; 64,2%).
MS (ESI, pos. ion): 230,0 [M+H]+.
Шаг 8) 6-хлор-5-(5-метил-1,3,4-оксадиазол-2-ил)пиримидин-4-амин
[380] К раствору N'-ацетил-4-амин-6-дихлорпиримидин-5-карбогидразида (1,08 г; 4,7 ммоль) в толуоле (50 мл) был добавлен реактив Берджесса (2,41 г; 10,11 ммоль). Реагирующая смесь была нагрета для дистилляции и дополнительно перемешивалась в течение 1 часа, затем была остужена до rt и поделена на две части: жидкий супернатант и коричневый сироп. Отсепарированный жидкий супернатант был сконцентрирован in vacuo. Осадок был разведен EtOAc (50 мл), промыт водой (20 мл) и насыщенным минеральным раствором (20 мл). Органическая фаза была высушена над обезвоженным Na2SO4, и сконцентрирована in vacuo. Сироп был очищен хроматографией на силикагельной колонне (PE/EtOAc (v/v)=2/1). Осадок жидкого супернатанта и очищенный сироп были объединены и вновь очищены хроматографией на силикагельной колонне (PE/EtOAc (v/v)=2/1) для получения соединения, указанного в заголовке шага, в виде белого твердого вещества (242 мг; 24,3%).
MS (ESI, pos. ion): 211,9 [M+H]+;
1HNMR (600 МГц; DMSO-d6) δ (ppm): 8,44 (s; 1H); 8,30 (s; 1H); 7,85-7,71 (m; 1H); 2,65 (s; 3H).
Шаг 9) (S)-2-(1-((6-амин-5-(5-метил-1,3,4-оксадиазол-2-ил)пиримидин-4-ил)амин)пропил)-5-хлор-3-циклопропилквиназолин-4(3H)-один
[381] К суспензии 6-хлор-5-(5-метил-1,3,4-оксадиазол-2-ил)пиримидин-4-амин (50 мг; 0,24 ммоль) и (S)-2-(1-аминопропил)-5-хлор-3-циклопропилквиназолин-4(3H)-один (68,9 мг; 0,25 ммоль) в n-BuOH (4 мл) был добавлен DIPEA (61 мг; 0,.470 ммоль).
Реагирующая смесь была нагрета для дистилляции и перемешивалась в течение 6 часов. Реагирующая смесь остужена до rt и сконцентрирована in vacuo. Осадок был разведен EtOAc (20 мл), промыт насыщенным гидратогенным раствором NH4Cl (5 мл) и насыщенным минеральным раствором (5 мл). Органическая фаза была высушена над обезвоженным Na2SO4, и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (PE/EtOAc (v/v)=1/2) для получения соединения, указанного в заголовке шага, в виде белого твердого вещества (48 мг; 44,9%).
MS (ESI, pos. ion): 453,1 [M+H]+;
1H NMR (400 МГц; DMSO-d6) δ (ppm): 8,80 (d; J=7,6 Гц; 1H); 8,04 (s; 1H); 7,68 (t; J=8,0 Гц; 1H); 7,54-7,43 (m; 2H); 7,27 (s; 2H); 6,09 (td; J=7,8; 4,8 Гц; 1H); 3,21-3,09 (m; 1H); 2,61 (s; 3H); 2,17-2,03 (m; 1H); 1,96-1,81 (m; 1H); 1,32-1,26 (m; 2H); 1,16-1,07 (m; 1H); 0,98 (t; J=7,4 Гц; 3H); 0,89-0,81 (m; 1H).
Пример 3 (S)-2-(1-((6-амин-5-(3-метил-1,2,4-оксадиазол-5-ил)пиримидин-4-ил)амин)-пропил)5-хлор-3-фенилквиназолин-4(3H)-один
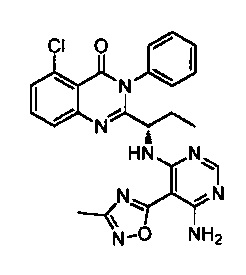
Шаг 1) (Z)-N'-гидроксиацетимидамид
[382] Суспензия гидроксиламин гидрохлорида (10,16 г; 146,16 ммоль) и гидратогенного углекислого калия (20,20 г; 146,16 ммоль) в EtOH (40 мл) перемешивалась при комнатной температуре в течение 1 часа. Затем был добавлен ацетонитрил (2,00 г; 48,72 ммоль), и реагирующая смесь была нагрета для дистилляции и дополнительно перемешивалась в течение 17 часов. Реагирующая смесь отфильтрована, и фильтрат был сконцентрирован in vacuo для получения соединения, указанного в заголовке шага, в виде бледно-желтого твердого вещества (2,72 г; 25%).
MS (ESI, pos. ion): 75,2 [M+H]+.
Шаг 2) (E)-N'-(4,6-дихлорпиримидин-5-карбонил)окси)ацетимидамид
[383] К суспензии 4,6-дихлорпиримидин-5-карбонилхлорида (3,62 г; 17,4 ммоль) в CH2Cl2 (20 мл) была добавлена смесь (Z)-N'-гидроксиацетимидамида (1,27 г; 17,14 ммоль) и DIPEA (4.43 г; 34. 28 ммоль) в CH2Cl2 (20 мл) при 0°C. Реагирующая смесь перемешивалась при 0°C в течение 1 часа и была разведена водой (40 мл). Отсепарированные органические фазы были промыты насыщенным гидратогенным раствором NaHCO3 (40 мл) и насыщенным минеральным раствором (40 мл), высушены над обезвоженным Na2SO4, и сконцентрированы in vacuo. Осадок был очищен хроматографией на силикагельной колонне (CH2Cl2/МеОН (v/v)=250/1) для получения соединения, указанного в заголовке шага, в виде светло-желтого твердого вещества (2,45 г; 57,5%).
MS (ESI, pos. ion): 249,8 [M+H]+;
1H NMR (600 МГц; CDCl3) δ (ppm): 8,87 (s; 1H); 4,95 (s; 2H); 2,05 (s; 3H).
Шаг 3) (E)-N'-(4-амин-6-хлорпиримидин-5-карбонил)окси)ацетимидамид
[384] Раствор (E)-N'-((4,6-дихлорпиримидина-5-карбонил)окси)ацетимидамида (3,1 г; 12,45 ммоль) в THF (50 мл) был продут газообразным NH3. Реагирующую смесь перемешивали в течение суток при комнатной температуре. Смесь была профильтрована, и фильтрат перемешивался со смешанным раствором EtOH/H2O (1/5 (v/v), 12 мл) в течение 6 часов. Фильтрация была повторена для получения соединения, указанного в заголовке шага, в виде серо-белого твердого вещества (1,9 г; 66,6%).
MS (ESI, pos. ion): 230,2 [M+H]+;
1H NMR (600 МГц; DMSO-d6) δ (ppm): 9,07 (s; 1H); 8,27 (s; 1H); 7,55 (s; 2H); 6,44 (s; 2H); 1,79 (s; 3H).
Шаг 4) 6-хлор-5-(3-метил-1,2,4-оксадиазол-5-ил)пиримидин-4-амин
[385] К суспензии (E)-N'-((4-амин-6-хлорпиримидин-5-карбонил)окси)ацетамидамида(200 мг; 0,87 ммоль) in DMSO (4 мл) был добавлен BU4NF (1 M в THF, 2,61 мл, 2,61 ммоль), и смесь перемешивалась при rt в течение суток. Осадок был разведен EtOAc (30 мл), промыт водой (15 мл×2) и насыщенным минеральным раствором (15 мл). Отсепарированная органическая фаза была высушена над обезвоженным Na2SO4, и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (РЕ/EtOAc (v/v)=4/1) для получения соединения, указанного в заголовке шага, в виде белого твердого вещества (21 мг; 11,4%).
MS (ESI, pos. ion): 212,0 [M+H]+;
1HNMR (600 МГц; DMSO-d6) δ (ppm): 8,41 (s; 1H); 8,03 (s; 1H); 2,47 (s; 3H).
Шаг 5) 2-амин-6-хлор-N-фенилбензамид
[386] К суспензии 2-амин-6-хлорбензойной кислоты (10 г; 58,28 ммоль) в толуоле (60 мл) был добавлен SOCl2 (17 мл, 233,1 ммоль) при rt. Реагирующая смесь была нагрета для дистилляции и перемешивалась в течение 4 часов, затем была остужена до rt и сконцентрирована in vacuo. Осадок был растворен в DCM (100 мл), затем был добавлен раствор анилина (4,8 мл, 52,45 ммоль) и Et3N (15,5 мл, 116,56 ммоль) в DCM (50 мл) при 0°C. Полученная смесь перемешивалась при rt в течение суток, затем была промыта минеральным раствором (100 мл) и насыщенным гидратогенным раствором NaHCO3 (100 мл). Отсепарированная органическая фаза была сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (PE/EtOAc (v/v)=1/1) для получения соединения, указанного в заголовке шага, в виде желтоватого твердого вещества (2,6 г; 18%).
MS (ESI, pos. ion) m/z: 247,0 [M+H]+;
1H NMR (600 МГц; CDCl3) δ (ppm): 7,71 (br s; 1H); 7,63 (d; J=8,0 Гц; 2H); 7,38 (t; J=7,8 Гц; 2H); 7,18 (t; J=1А Гц; 1H); 7,10 (t; J=8,1 Гц; 1H); 6,77 (d; J=7,9 Гц; 1H); 6,63 (d; J=8,2 Гц; 1H); 4,68 (s; 2H).
Шаг 6) (S)-терт-бутил(1-((3-хлор-2-(фенилкарбамоил)фенил)амин)-1-оксобутан-2-ил)-карбамат
[387] К раствору (S)-2-((терт-бутоксикарбонил)амин)бутановой кислоты (2,3 г; 11,19 ммоль), 2-амин-6-хлор-N-фенилбензамида (2.6 г; 10.66 ммоль) и DIPEA (5,5 мл, 31,62 ммоль) в DCM (40 мл) был добавлен HATU (4,81 г; 12,65 ммоль) при -10°C. Смесь перемешивалась при -10°C в течение 1 часа, затем была нагрета до rt, нагрета для дистилляции и дополнительно перемешивалась в течение 24 часов. Затем реагирующая смесь была остужена до rt, промыта H2O (200 мл×2) и насыщенным гидратогенным раствором NaHCO3 (200 мл). Отсепарированная органическая фаза была очищена хроматографией на силикагельной колонне (РЕ/EtOAc (v/v)=4/1) для получения соединения, указанного в заголовке шага, в виде желтоватого твердого вещества (3,3 г; 72%).
MS (ESI, neg. ion) m/z: 430,0 [M-H]-;
1H NMR (600 МГц; CDCl3) δ (ppm): 9,43 (br s; 1H); 8,13-8,11 (m; 1H); 7,95 (m; 1H); 7,64-7,62 (d; J=7,8 Гц; 2H); 7,39-7,37 (t; J=7,8 Гц; 2H); 7,35-7,32 (t; J=7,8 Гц; 1H); 7,21-7,18 (m; 2H); 4,99 (br s; 1H); 4,15 (br s; 1H); 1,91-1,90 (m; 1H); 1,67-1,63 (m; 1H); 1,39 (s; 9H); 0,94-0,91 (t; , 7=7,2 Гц; 3H).
Шаг 7) (S)2-(1-аминопропил)-5-хлор-3-фенилквиназолин-4(3H)-один
[388] К раствору (S)-терт-бутил(1-((3-хлор-2-(фенилкарбамоил)фенил)амин)-1-оксобутан-2-ил)карбамата (2,0 г; 4,63 ммоль) в DCM (50 мл) был добавлен шприцом йод (823 мг; 3,24 ммоль) и HMDS (2,9 мл, 13,89 ммоль) в атмосфере N2. Полученная смесь перемешивалась в течение суток при rt, затем была резко охлаждена насыщенным гидратогенным раствором тиосульфата натрия (200 мл). Отсепарированная органическая фаза была сконцентрирована in vacuo и очищена хроматографией на силикагельной колонне (DCM/MeOH (v/v) для получения соединения, указанного в заголовке шага, в виде желтоватого твердого вещества (500 мг; 34%).
MS (ESI, pos. ion.) m/z: 314,0 [M+H]+;
1H NMR (400 МГц; CDCl3) δ (ppm): 10,41 (br s; 1H); 9,11 (br s; 1H); 8,37-8,34 (d; J=8,4 Гц; 1H); 8,27-8,25 (d; J=8,0 Гц; 1H); 7,62-7,35 (m; 4H); 7,28-7,26 (d; J=8,4 Гц; 1H); 7,20-7,18 (d; J=8,0Hz; 11H); 3,44-3,41 (m; 1H); 1,96-1,90 (m; 1H); 1,66-1,60 (m; 1H); 0,99-0,96 (t; J=6,8 Гц; 3H).
Шаг 8) (S)-2-(1-((6-амин-5-(3-метил-1,2,4-оксадиазол-5-ил)пиримидин-4-ил)амин)пропил)-5-хлор-3-фенилквиназолин-4(3H)-один
[389] К суспензии 6-хлор-5-(3-метил-1,2,4-оксадиазол-5-ил)пиримидин-4-амин (30 мг; 0,142 ммоль) и (S)-2-(1-аминопропил)-5-хлор-3-фенилквиназолин-4(3H)-один (47 мг; 0,149 ммоль) в n-BuOH (2 мл) был добавлен DIPEA (37 мг; 0,284 ммоль). Реагирующая смесь была нагрета для дистилляции и перемешивалась в течение 12 часов, затем была остужена до rt и сконцентрирована in vacuo. Осадок был растворен в EtOAc (15 мл), и полученная смесь была промыта насыщенным гидратогенным раствором NH4Cl (10 мл) и насыщенным минеральным раствором (10 мл). Отсепарированная органическая фаза была высушена над обезвоженным Na2SO4, и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (РЕ/EtOAc (v/v)=2/1) для получения соединения, указанного в заголовке шага, в виде светло-желтого твердого вещества (47 мг; 67,8%).
MS (ESI, pos. ion) m/z: 489,1 [M+H]+;
1H NMR (600 МГц; DMSO-d6) δ (ppm): 9,04 (d; J=7,0 Гц; 1H); 7,99 (s; 1H); 7,80 (t; J=8,0 Гц; 1H); 7,71-7,43 (m; 7H); 4,96-4,87 (m; 1H); 2,50 (s; 3H); 1,96-1,86 (m; 1H); 1,70-1,59 (m; 1H); 0,76 (t; J=7,4 Гц; 3H).
Пример 4 (S)-2-(1-((6-амин-5-(3-метил-1,2,4-оксадиазол-5-ил)пиримидин -4-ил)амин)-пропил)-5-хлор-3-циклопропилквиназолин-4(3H)-один
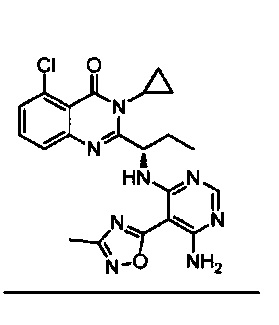
[390] К суспензии (S)-2-(1-аминопропил)-5-хлор-3-циклопропилквиназолин-4(3H)-один (110 мг; 0.396 ммоль) и 6-хлор-5-(3-метил-1,2,4-оксадиазол-5-ил)пиримидин-4-амин (92 мг; 0,436 ммоль) в n-BuOH (5 мл) был добавлен DIPEA (102 мг; 0,792 ммоль). Полученная смесь была подвергнута дистилляции, затем остужена до rt и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (PE/EtOAc (v/v)=2/1) для получения соединения, указанного в заголовке шага, в виде белого твердого вещества (168 мг; 93,7%).
MS (ESI, pos. ion) m/z: 453,1 [M+H]+;
1H NMR (600 МГц; DMSO-d6) δ (ppm): 9,17 (d; J=7,5 Гц; 1H); 8,10 (s; 1H); 7,72 (t; J=8,0 Гц; 1H); 7,59-7,47 (m; 2H); 6,16 (td; J=7,2; 5,3 Гц; 1H); 3,14 (ddd; J=11,2; 7,2; 4,2 Гц; 1H); 2,50 (s; 3H); 2,12 (ddt; J=14,7; 12,4; 7,4 Гц; 1H); 1,91 (tt; J=14,5; 7,3 Гц; 1H); 1,31-1,25 (m; 2H); 0,94 (t; J=7,4 Гц; 3H); 0,88-0,76 (m; 2H).
Пример 5 2-((S)-1-((6-амин-5-((S)-4-метил-4,5-дигидрооксазол-2-ил)пиримидин-4-ил)амин)пропил)-5-хлор-3-циклопропилквиназолин-4(3H)-один
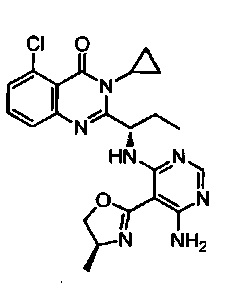
Шаг 1) (S)-4,6-дихлор-N-(1-гидроксипропан-2-ил)пиримидин-5-карбоксамид
[391] К суспензии 4,6-дихлорпиримидин-5-карбонилхлорида (1,63 г; 7,71 ммоль) в CH2Cl2 (4 мл) был добавлен раствор (5)-2-аминопропан-1-ол (0,579 г; 7,71 ммоль) и DIPEA (1,50 г; 11,57 ммоль) в CH2Cl2 (6 мл) при 0°C.Реагирующая смесь перемешивалась при 0°C в течение 1 часа, затем была сконцентрирована in vacuo и осадок был очищен хроматографией на силикагельной колонне (CH2Cl2/МеОН (v/v)=50/1) для получения сиропа. Сироп был разведен EtOAc (15 мл) и промыт насыщенным гидратогенным раствором HCl (1 М, 15 мл). Водосодержащий слой был экстрактирован посредством EtOAc (10 мл×4). Комбинированные органические фазы были высушены над обезвоженным Na2SO4, и сконцентрированы in vacuo для получения соединения, указанного в заголовке шага, в виде желтого твердого вещества (1,28 г; 66,5%).
MS (ESI, pos. ion) m/z: 249.9 [M+H]+;
lH NMR (600 МГц; CDCl3) δ (ppm): 8,81 (s; 1H); 6,56 (d; J=6,6 Гц; 1H); 4,38-4,22 (m; 1H); 3,82 (dd; J=11,1; 3,7 Гц; 1H); 3,69 (dd; J=11,1; 4,7 Гц; 1H); 2,37 (s; 1H); 1,33 (d; J=6,8 Гц; 3H).
Шаг 2) (S)-4-амин-6-хлор-N-(1-гидроксипропан-2-ил)пиримидин-5-карбоксамид
[392] Раствор (S)-4,6-дихлор-N-(1-гидроксипропан-2-ил)пиримидин-5-карбоксамид (1,3 г; 5,20 ммоль) в THF (20 мл) был продут газообразным NH3. Реагирующая смесь перемешивалась при rt в течение 6 часов, была профильтрована, и фильтрат промыт EtOAc (10 мл). Фильтрат был сконцентрирован in vacuo для получения соединения, указанного в заголовке шага, в виде желтого твердого вещества (1,125 г, 93.8%).
MS (ESI, pos. ion) m/z: 231,1 [M+H]+;
1H NMR (600 МГц; DMSO-d6) δ (ppm): 8,40 (d;7=8,4 Гц; 1H); 8,21 (s; 1H); 7,15 (s; 2H);4,92 (t; J=5,5 Гц; 1H); 3,98 (dt; J=14,7; 6,6 Гц; 1H); 3,38 (t; J=5,9 Гц; 2H); 1,08 (d; J=6,8 Гц; 3H).
Шаг 3) (S)-4-амин-6-хлор-N-(1-хлорпропан-2-ил)пиримидин-5-карбоксамид
[393] К суспензии (S)-4-амин-6-хлор-.N-(1-гидроксипропан-2-ил)пиримидин-5-карбоксамида (500 мг; 2,17 ммоль) в CHCl3 (10 мл) был добавлен SOCl2 (0,78 мл, 10,84 ммоль). Реагирующая смесь была нагрета для дистилляции и перемешивалась в течение 2 часов, затем была остужена до комнатной температуры и сконцентрирована in vacuo. Осадок был растворен в смеси CH2Cl2 и МеОН (CH2Cl2/МеOН (v/v)=25/1, 15 мл), профильтован через силикагельную прокладку, и фильтрат был промыт смесью CH2Cl2 и МеОН (CH2Cl2/МеОН (v/v)=20/1, 200 мл). Фильтрат был сконцентрирован in vacuo для получения соединения, указанного в заголовке шага, в виде желтого твердого вещества (0,54 г; 100%).
MS (ESI, pos. ion) m/z: 249,0 [M+H]+;
1H NMR (600 МГц; DMSO-d6) δ (ppm): 8,76 (d; J=7,7 Гц; 1H); 8,22 (s; 1H); 7,15 (s; 2H); 4,20 (ddd; J=19,5; 13,1; 6,7 Гц; 1H); 3,73 (dd; J=10,7; 4,9 Гц; 1H), 3,67 (dd; J=10,7; 6,1 Гц; 1H); 1,21 (d; J=6,7 Гц; 3Н).
Шаг 4) (S)-6-хлор-5-(4-метил-4,5-дигидрооксазол-2-ил)пиримидин-4-амин
[394] К суспензии (S)-4-амин-6-хлор-N-(1-хлорпропан-2-ил)пиримидин-5-карбаксамида (690 мг; 2.77 ммоль) в осушенном THF (20 мл) была добавлена смесь NaH (222 мг; 5,54 ммоль, 60% диспергировано в минеральном масле) в THF (5 мл) при 0°C. Реагирующая смесь была нагрета до rt и дополнительно перемешивалась в течение 30 минут, разведена с помощью EtOAc (50 мл), и промыта водой (20 мл) и насыщенным минеральным раствором (20 мл). Органическая фаза была высушена над обезвоженным Na2SO4, и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (CH2Cl2/МеОН (v/v)=100/1) для получения соединения, указанного в заголовке шага, в виде желтого твердого вещества (438 мг; 74,4%).
MS (ESI, pos. ion) m/z: 213.0 [M+H]+;
1H NMR (600 МГц; CDCl3) δ (ppm): 9,15 (br s; 1H); 8,30 (s; 1H); 6,03 (br s; 1H); 4,55 (dd; J=9,4; 8,3 Гц; 1H); 4,46-4,36 (m; 1H); 4,00 (t; J=8,1 Гц; 1H); 1,38 (d; J=6,6 Гц; 3H).
Пример 5
2-((S)-1-((6-амин-5-((S)-4-метил-4,5-дигидрооксазол-2-ил)пиримидин-4-ил)амин)пропил)-5-хлор-3-циклопропилквиназолин-4(3H)-один
[395] К суспензии (S)-6-хлор-5-(4-метил-4,5-дигидроксадиазол-2-ил)пиримидин-4-амина (50 мг; 0.235 ммоль) и (S)-2-(1-аминопропил)-5-хлор-3-циклопропилквиназолин-4(3H)-один (72 мг; 0,259 ммоль) в n-BuOH (2 мл) был добавлен DIPEA (61 мг; 0,470 ммоль).
Реагирующая смесь была нагрета для дистилляции и перемешивалась в течение 8 часов, затем была остужена до комнатной температуры и сконцентрирована in vacuo. Осадок был растворен в EtOAc (15 мл), и полученная смесь была промыта водой (10 мл) и насыщенным минеральным раствором (10 мл). Отсепарированная органическая фаза была высушена над обезвоженным Na2SO4, и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (РЕ/EtOAc (v/v)=1/1) для получения соединения, указанного в заголовке шага, в виде грязно-белого твердого вещества (85 мг; 79,7%).
MS (ESI, pos. ion) m/z: 454,2 [M+H]+;
1H NMR (600 МГц; CDCl3) δ (ppm): 9,37 (d; J=7,4 Гц; 1H); 8,02 (s; 1H); 7,57-7,49 (m; 2H); 7,42 (d; J=7,4 Гц; 1H); 6,16 (dd; J=13,0; 7,3 Гц; 1H); 4,58-4,50 (m; 1H); 4,47-4,37 (m; 1H); 3,96 (t; J=7,8 Гц; 1H); 3,14-3,05 (m; 1H); 2,06-2,03 (m; 1H); 1,98-1,91 (m; 1H); 1,44-1,40 (m; 2H); 1,39 (d; J=6,6 Гц; 3H); 1,03 (t; J=7,4 Гц; 3H); 0,93-0,85 (m; 2H).
Пример 6
2-((S)-1-((6-амин-5-((S)-4-метил-4,5-дигидрооксазол-2-ил)пиримидин-4-ил)амин)пропил)-5-хлор-3-фенилквиназолин-4(3H)-один
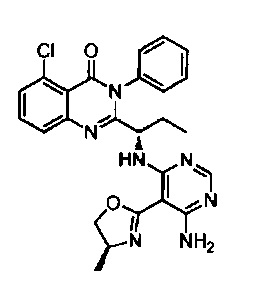
[396] К суспензии (S)-6-хлор-5-(4-метил-4,5-дигидроксадиазол-2-ил)пиримидин-4-амина (30 мг; 0,141 ммоль) и (S)-2-(1-аминопропил)-5-хлор -3-фенилквиназолин-4(3H) -один (47 мг; 0,148 ммоль) в n-BuOH (2 мл) был добавлен DIPEA (36 мг; 0,282 ммоль). Реагирующая смесь была нагрета для дистилляции и перемешивалась в течение 8 часов, затем была остужена до комнатной температуры, разведена в EtOAc (15 мл) и промыта водой (10 мл) и концентрированным минеральным раствором (10 мл). Отсепарированная органическая фаза была высушена над обезвоженным Na2SO4, и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (РЕ/EtOAc (v/v)=3/2) для получения соединения, указанного в заголовке шага, в виде грязно-белого твердого вещества (54 мг; 78,1%).
MS (ESI, pos. ion) m/z: 490,1 [M+H]+;
1H NMR (600 МГц; DMSO-d6) δ (ppm): 9,41 (d; J=6,8 Гц; 1H); 7,86 (s; 1H); 7,76 (t; J=8,0 Гц; 1H); 7,62-7,49 (m; 7H); 7,45 (s; 1H); 7,17 (s; 1H); 4,66 (td; J=7,8; 4,2 Гц; 1H); 4,57-4,52 (m; 1H); 4,38-4,29 (m; 1H); 3,94 (t; J=8,0 Гц; 1H); 1,90-1,82 (m; 1H); 1,64-1,57 (m; 1H); 1,27 (d; J=6,6 Гц; 3H); 0,74 (t, J=7,4 Гц; 3H).
Пример 7 (S)-2-(1-((6-амин-5-((5-метилоксазол-2-ил)пиримидин-4-ил)амин)пропил)-5-хлор-3-цилкопропилквиназолин-4(3H)-один
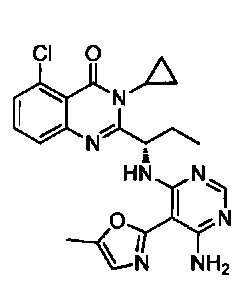
Шаг 1) 4,6-дихлор-N-(2-оксопропил)пиримидин-5-карбоксамид
[397] К раствору 4,6-дихлорпиримидин-5-карбонилхлорида (4.0 г; 18,92 ммоль) в CH2Cl2 (30 мл) был по каплям добавлен DIPEA (7.34 г; 56,76 ммоль) при 0°C, затем была добавлена суспензия 1-аминопропан-2-один водородхлорида (2.28 г; 20.81 ммоль) в CH2Cl2 (20 мл). Реагирующая смесь перемешивалась при 0°C в течение 1 часа, была промыта насыщенным гидратогенным раствором NH4Cl (40 мл) и насыщенным минеральным раствором (40 мл). Отсепарированная органическая фаза была высушена над обезвоженным Na2SO4, и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (РЕ/EtOAc (v/v)=5/1) для получения соединения, указанного в заголовке шага, в виде желтого твердого вещества (2.53 г; 53,9%).
MS (ESI, pos. ion) m/z: 247,9 [M+H]+;
1H NMR (600 МГц; CDCl3) δ (ppm): 8,85 (s; 1H); 6,76 (s; 1H); 4,43 (d; J=4,4 Гц; 2H); 2,33 (s; 2H).
Шаг 2) 4-амин-6-хлор-N-(2-оксопропил)пиримидин-5-карбоксамид
[398] Раствор 4,6-дихлор-N-(2-оксопропил)пиримидин-5-карбоксамида (2,57 г; 10,36 ммоль) в THF (50 мл) был продут газообразным NH3. Смесь перемешивалась при rt в течение 4 часов, затем была сконцентрирована in vacuo и осадок был очищен хроматографией на силикагельной колонне (CH2Cl2/МеОН (v/v)=50/1) для получения соединения, указанного в заголовке шага, в виде желтого твердого вещества (1,71 г; 72%).
MS (ESI, pos. ion) m/z: 229,1 [M+H]+.
Шаг 3) 6-хлор-5-(5-метилоксазол-2-ил)пиримидин-4-амин
[399] К раствору 4-амин-6-хлор-N-(2-оксопропил)пиримидин-5-карбоксамида (1,44 г; 6,30 ммоль) в толуоле (40 мл) был добавлен реактив Берджесса (3.23 г; 13.55 ммоль) при rt. Реагирующая смесь была нагрета до 120°C и перемешивалась в течение 2,5 часов, затем была остужена до комнатной температуры и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (CH2Cl2/МеОН (v/v)=250/1) для получения соединения, указанного в заголовке шага, в виде светло-желтого твердого вещества (450 мг; 34,3%).
MS (ESI, pos. ion) m/z: 211,0 [M+H]+;
1HNMR (600 МГц; CDCl3) δ (ppm): 9,11 (s; 1H); 8,32 (s; 1H); 6,95 (d; J=0,7 Гц; 1H); 5,96 (s; 1H); 2,47 0,7 Гц; 3H).
Шаг 4) (S)-2-(1-((6-амин-5-(5-метилоксазол-2-ил)пиримидин-4-ил)амин)пропил)-5-хлор-3-циклопропилквиназолин-4(3H)-один
[400] К суспензии 6-хлор-5-(5-метилоксазол-2-ил)пиримидин-4-амина (50 мг; 0,237 ммоль) и (S)-2-(1-аминопропил)-5-хлор-3-циклопропилквиназолин-4(3H)-один (69 мг; 0,249 ммоль) в n-BuOH (5 мл) был добавлен DIPEA (61 мг; 0.475 ммоль). Полученная смесь была перемешивалась в течение 18 часов, затем была остужена до комнатной температуры и сконцентрирована in vacuo. Осадок был растворен в EtOAc (15 мл), и полученная смесь была промыта насыщенным гидратогенным раствором NH4Cl (15 мл) и насыщенным минеральным раствором (15 мл). Отсепарированная органическая фаза была высушена над обезвоженным Na2SO4, и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (РЕ/EtOАс (v/v)=1/1) для получения соединения, указанного в заголовке шага, в виде светло-желтого твердого вещества (66 мг; 61,5%).
MS (ESI, pos. ion) m/z: 452,1 [M+H]+;
1Н NMR (400 МГц; CDCl3) δ (ppm): 8,97 (d; J=7,9 Гц; 1H); 8,10 (s; 1H); 7,62-7,37 (m; 3H); 6,92 (s; 1H); 6,31 (dd; J=13,2; 7,5 Гц; 1H); 3,19-3,05 (m; 1H); 2,50 (s; 3H); 2,10-1,98 (m; 2H); 1,48-1,40 (m; 2H); 1,06 (t; J=7,4 Гц; 3H); 0,95-0,84 (m; 2H).
Пример 8
(S)-2-(1-((6-амин-5-((5-метилоксазол-2-ил)пиримидин-4-ил)амин)пропил)-5-хлор-3-фенилквиназолин-4(3H)-один
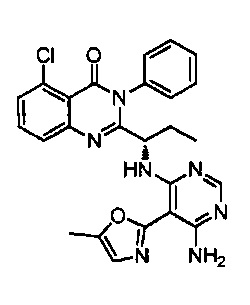
[401] К суспензии 6-хлор-5-(5-метилоксазол-2-ил)пиримидин-4-амина (40 мг; 0,19 ммоль) и (S)-2-(1-аминопропил)-5-хлор-3-фенилквиназолин-4(3H)-один (62 мг; 0,20 ммоль) в n-BuOH (5 мл) был добавлен DIPEA (49 мг; 0,38 ммоль). Реагирующая смесь была нагрета до 120°C и перемешивалась дополнительно в течение 9 часов. Реагирующая смесь была остужена до rt, сконцентрирована in vacuo, и осадок был разведен в EtOAc (20 мл). Полученная смесь была промыта насыщенным гидратогенным раствором NH4Cl (5 мл) и насыщенным минеральным раствором (5 мл). Отсепарированная органическая фаза была высушена над обезвоженным Na2SO4, и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (РЕ/EtOAc (v/v)=1/1) для получения соединения, указанного в заголовке шага, в виде светло-желтого твердого вещества (17 мг; 18,5%).
MS (ESI, pos. ion) m/z: 488,11 [M+H]+;
1H NMR (600 МГц; DMSO-d6) δ (ppm): 9,17 (d; J=7,5 Гц; 1H); 8,10 (s; 1H); 7,72 (t; J=8,0 Гц; 1H); 7,59-7,47 (m; 2H); 6,16 (td; J=7,2; 5,3 Гц; 1H); 3,14 (ddd; J=11,2; 7,2; 4,2 Гц; 1H); 2,50 (s; 3H); 2,12 (ddt; J=14,7; 12,4; 7,4 Гц; 1H); 1,91 (tt; J=14,5; 7,3 Гц; 1H); 1,31-1,25 (m; 2H); 0,94 (t; J=7,4 Гц; 3H); 0,88-0,76 (m; 2H).
Пример 9 2-((1S)-1-((6-амин-5-(1-метил-1H,2,4-триазол-5-ил)пиримидин-4-ил)амин)-пропил)-5-хлор-3-циклопропилквиназолин-4(3H)-один
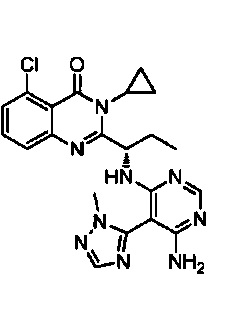
Шаг 1) 4,6-дихлорпиримидин-5-карбоксамид
[402] Раствор 4,6-дихлорпиримидин-5-карбонилхлорида (2,11 г; 10,0 ммоль) в THF (20 мл) был продут газообразным NH3. Реагирующая смесь перемешивалась при rt в течение 5 минут, затем была отфильтрована, и фильтрат сконцентрирован in vacuo. Осадок был очищен хроматографией на силикагельной колонне (чистый EtOAc) для получения соединения, указанного в заголовке шага, в виде белого твердого вещества (1,43 г; 74%).
MS (ESI, pos. ion) m/z: 192,0 [M+H]+;
1H NMR (600 МГц; CDCl3) δ (ppm): 8,85 (s; 1H); 6,23 (br s; 1H); 5,96 (br s; 1H).
Шаг 2) 4,6-диметоксипиримидин-5-карбоксамид
[403] К раствору 4,6-дихлорпиримидин-5-карбоксамида (1,65 г; 8,.6 ммоль) в метаноле (30 мл) был добавлен метилат натрия (1,15 г; 21,3 ммоль), затем смесь была нагрета до 40°C и дополнительно перемешивалась в течение 12 часов. Смесь была отфильтрована и фильтрат сконцентрирован in vacuo. Осадок был очищен хроматографией на силикагельной колонне (чистый EtOAc) для получения соединения, указанного в заголовке шага, в виде белого твердого вещества (1,30 г; 83%).
MS (ESI, pos. ion) m/z: 184,1 [M+H]+;
1H NMR (600 МГц; CDCl3) δ (ppm): 8,47 (s; 1H); 6,30 (br s; 1H); 5,98 (br s; 1H); 4,07 (s; 6H).
Шаг 3) N-((диметиламино)метилен)-4,6-диметоксипиримидин-5-карбоксамид
[404] Смесь 4,6-диметоксипиримидин-5-карбоксамида (732,6 мг; 4,0 ммоль) и N,N-диметилформамид диметил ацетата (10 мл) перемешивалась при комнатной температуре в течение 1 часа. Реагирующая смесь была сконцентрирована in vacuo для получения соединения, указанного в заголовке шага, в виде белого твердого вещества (952,0 мг; 100%), которое было непосредственно использовано в следующем шаге без дополнительной очистки.
MS (ESI, pos. ion) m/z: 239,0 [M+H]+.
Шаг 4) 4,6-диметокси-5-(1-метил-1H-2,4-триазол-5-ил)пиримидин
[405] К раствору N-((диметиламино)метилен)-4,6-диметоксипиримидин-5-карбоксамида (952 мг; 4,0 ммоль) в уксусной кислоте (15 мл) был добавлен метилгидразин (1,84 г; 40 ммоль). Смесь перемешивалась при комнатной температуре более 2 часов, затем была сконцентрирована in vacuo, и осадок был растворен в этилацетате (100 мл). Полученная смесь была промыта насыщенным гидратогенным раствором NH3Cl (20 мл) и насыщенным минеральным раствором (30 мл). Отсепарированная органическая фаза была сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (чистый EtOAc) для получения соединения, указанного в заголовке шага, в виде светло-желтого твердого вещества (0,65 г; 74%).
MS (ESI, pos. ion) m/z: 222,0 [M+H]+;
1HNMR (600 МГц; CDCl3) δ (ppm): 8,57 (s; 1H); 8,04 (s; 1H); 4,00 (s; 6H); 3,75 (s; 3H);
13C NMR (150 МГц; CDCl3) δ (ppm): 168,6; 158,8; 151,3; 146,6; 93,3; 54,9; 35,9;
2D-NMR (HMBC): (8,57; 168,6); (8,04; 146,6); (4,00; 168,6); (3,75; 146,6).
Шаг 5) 5-(1-метил-1H-1,2,4-триазол-5-ил)пиримидин-4,6-диол
[406] Смесь 4,6-диметокси-5-(1-метил-1Н-1,2,4-триазол-5-ил)иримидин (650 мг; 2,94 ммоль), концентрированной соляной кислоты (6 мл) и уксусной кислоты (6 мл) была нагрета 100°C и перемешивалась в течение последующих 3 часов. Реагирующая смесь остужена до комнатной температуры и сконцентрирована in vacuo для получения соединения, указанного в заголовке шага, в виде белого твердого вещества (568 мг; 100%), которое было использовано в следующем шаге без дополнительной очистки.
MS (ESI, pos. ion) m/z: 194,0 [M+H]+.
Шаг 6) 4,6-дихлор-5-(1-метил-1H-1,2,4-триазол-5-ил)пиримидин
[407] К суспензии 5-(1-метил-1H-1,2,4-триазол-5-ил)пиримидин-4,6-диола (568 мг; 2,94 ммоль) и DMF (1.0 мл) в толуоле (15 мл) был добавлен РОСl3 (901,6 мг; 5,88 ммоль). Реагирующая смесь была нагрета до 100°C и перемешивалась в течение последующих 3,5 часов, затем была остужена до комнатной температуры, сконцентрирована in vacuo и растворена в этилацетате (100 мл). Полученная смесь была промыта насыщенным гидратогенным раствором NH3Cl (20 мл) и насыщенным минеральным раствором (30 мл). Отсепарированный органический слой был высушен над обезвоженным Na2SO4, и сконцентрирован in vacuo. Осадок был очищен хроматографией на силикагельной колонне (чистый EtOAc) для получения соединения, указанного в заголовке шага, в виде светло-желтого масла (650 мг; 96%).
MS (ESI, pos. ion) m/z: 229,9 [M+H]+.
Шаг 7) 6-хлор-5-(1-метил-1H-1,2,4-триазол-5-ил)пиримидин-4-амин
[408] Смесь 4,6-дихлор-5-(1-метил-1H-1,2,4-триазол-5-ил)пиримидина (650 мг; 2,8 ммоль) и раствора NH3 в метаноле (7 М, 20 мл) перемешивалась при комнатной температуре в течение 24 часов. Реагирующая смесь была отфильтрована и фильтрат сконцентрирован in vacuo. Осадок был очищен хроматографией на силикагельной колонне (DCM/MeOH (v/v)=20/1) для получения соединения, указанного в заголовке шага, в виде белого твердого вещества (180 мг; 31%).
MS (ESI, pos. ion) m/z: 211,.0 [M+H]+.
Пример 8
2-((1S)-1-((6-амин-5-(1-метил-1H-1,2,4-триазол-5-ил)пиримидин-4-ил)амин)пропил-5-хлор-3-циклопропилквиназолин-4-(3Н)-один
[409] Смесь 6-хлор-5-(1-метил-1Н-1,2,4-триазол-5-ил)пиримидин-4-амина (80 мг; 0,38 ммоль), (S)-2-(1-аминопропил)-5-хлор-3-циклопроилквиназолин-4(3H)-один (116.1 мг; 0.42 ммоль) и N,N-диизопропилэтиламина (147.3 мг; 1.14 ммоль) в n-бутаноле (3 мл) была нагрета до 120°C и подвергалась дистилляции в течение 24 часов. Реагирующая смесь была сконцентрирована in vacuo и осадок был растворен в этилацетате (100 мл). Полученная смесь была промыта насыщенным гидратогенным раствором NH4Cl (20 мл) и насыщенным минеральным раствором (20 мл). Отсепарированная органическая фаза была высушена над обезвоженным Na2SO4, и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (EtOAc) для получения соединения, указанного в заголовке шага, в виде белого твердого вещества (30 мг; 17,5%).
MS (ESI, pos. ion) m/z: 452,1 [M+H]+;
1H NMR (600 МГц; CDCl3) δ (ppm): 8,24 (s; 1H); 8,18 (s; 1H); 7,54 (t; J=7,8 Гц; 1H); 7,43 (d; J=7,8 Гц; 1H); 7,36 (d; J=7,8 Гц; 1H); 6,13 (s; 2H); 5,98 (s; 1H); 3,89 (s; 3H); 3,10-2,99 (m; 1H); 2,05-1,97 (m; 1H); 1,84-1,75 (m;2H); 1,23-1,15 (m; 2H); 0,98 (t; J=12 Гц; 3Н); 0,95-0,90 (m; 2H).
Пример 10 2-((1S)-1-((6-амин-5-(1-метил-1H,4-триазол-5-ил)пиримидин-4-ил)амин)-пропил)-5-хлор-3-фенилквиназолин-4(3H)-один
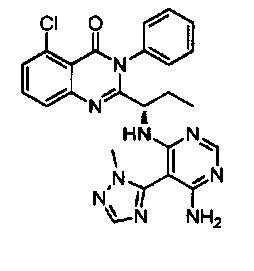
[410] Смесь 6-хлор-5-(1-метил-1Н-1,2,4-триазол-5-ил)пиримидин-4-амина (40 мг; 0,19 ммоль), (S)-2-(1-аминопропил)-5-хлор-3-фенилквиназолин-4(3H)-один (59.6 мг; 0.19 ммоль) и N,N-диизопропилэтиламина (73.6 мг; 0,57 ммоль) в n-бутаноле (2 мл) была нагрета до 120°C и подвергалась дистилляции в течение 40 часов. Реагирующая смесь была профильтрована, и фильтрат промыт метанолом (10 мл) и этилацетатом (10 мл). Фильтрат был сконцентрирована in vacuo и осадок был растворен в этилацетате (20 мл). Полученная смесь была промыта насыщенным гидратогенным раствором NH4Cl (10 мл) и насыщенным минеральным раствором (10 мл). Отсепарированная органическая фаза была высушена над обезвоженным Na2SO4, и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (EtOAc) для получения соединения, указанного в заголовке шага, в виде белого твердого вещества (20 мг; 22%).
MS (ESI, pos. ion) m/z: 488,2 [M+H]+;
1HNMR (600 МГц; CDCl3) δ (ppm): 8,16 (s; 1H); 8,13 (s; 1H); 7,67-7,54 (m; 5H); 7,54-7,42 (m; 5H); 3,90 (s; 3H); 1,89-1,77 (m; 1H); 1,64-1,51 (m; 2H); 0,77 (t; J=7,2 Гц; 3H).
Пример 11 (S)-2-(1-((6-амин-5-(пиридин-2-ил)пиримидин-4-ил)амин)пропил)-5-хлор-3-циклопропилквиназолин-4(3H)-один
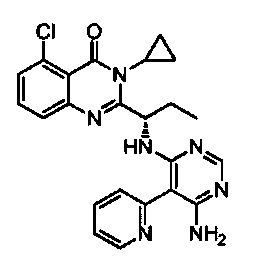
Шаг 1) 6-хлор-5-(5-йодпиримидин-4-амин
[411] К раствору 6-хлорпиримидин-4-амина (1,29 г; 10 ммоль) в DMF (8 мл) был добавлен одной порцией N-йодсукцинимид (2.25 г; 10 ммоль). Реагирующая смесь перемешивалась при 100°C в течение 8 часов, затем была остужена до комнатной температуры и сконцентрирована in vacuo. Осадок был растворен в EtOAc (300 мл), и полученная смесь была промыта смесью насыщенного гидратогенного раствора Na2S2O3 и насыщенного гидратогенного раствора NaHCO3 (1/2 (v/v), 100 мл×3), водой (150 мл×3) и насыщенным минеральным раствором (100 мл).
Отсепарированная органическая фаза была высушена над обезвоженным Na2SO4, и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (PE/EtOAc (v/v)=5/1) для получения соединения, указанного в заголовке шага, в виде белого твердого вещества (1,20 г; 46,97%).
MS (ESI, pos. ion.) m/z: 255,8 [M+H]+.
Шаг 2) 2-(трибутилстаннил)пиридин
[412] 2-бромпиридин (2.09 г; 13.33 ммоль) был растворен в обезвоженном THF (10 мл) для получения бесцветного раствора. Раствор был дегазирован и трижды прокачан N2, перемешан при -78°C в течение 30 минут, затем был медленно добавлен n-бутиллитий (2.4 М, 6 мл, 14.4 ммоль) в течение 15 минут. Реагирующая смесь была нагрета до комнатной температуры и перемешивалась в течение последующих 30 минут, затем была вновь охлаждена до -78°C. К реагирующей смеси был медленно добавлен три-и-бутилин хлорид (3,67 г; 13,33 ммоль). Реагирующая смесь перемешивалась при -78°C в течение 1 часа, затем перемешивалась при комнатной температуре в течение суток. Реагирующая смесь была разведена водой (200 мл) и экстрактирована EtOAc (100 мл×3). Комбинированные органические фазы были промыты водой (100 мл×2) и насыщенным минеральным раствором (100 мл), высушены над обезвоженным Na2SO4, и сконцентрированы in vacuo. Осадок был очищен хроматографией на силикагельной колонне (РЕ/EtOAc (v/v)=10/1) для получения соединения, указанного в заголовке шага, в виде светло-желтого твердого вещества (2,92 г; 59,53%).
MS (ESI, pos. ion.) m/z: 370,1 [M+H]+.
Шаг 3) 6-хлор-5-(пиридин-2-ил)пиримидин-4-амин
[413] К суспензии 6-хлор-5-йодпиримидин-4-амина (511 мг; 2,0 ммоль), Pd(PPh3)2Cl2 (140 мг; 0,2 ммоль) и Cul (38 мг; 0,2 ммоль) в DMF (10 мл) был добавлен триэтиламин (405 мг; 4,0 ммоль), затем был добавлен раствор 2-(трибутилстаннил)пиридина (1.47 г; 4.0 ммоль) в DMF (5 мл). Реагирующая смесь перемешивалась при 120°C в течение 2,5 часов, затем остужена до комнатной температуры, разведена с EtOAc (300 мл) и промыта водой (200 мл×3). Отсепарированная органическая фаза была сконцентрирована in vacuo, и осадок был очищен хроматографией на силикагельной колонне (PE/EtOAc (v/v)=10/1) для получения соединения, указанного в заголовке шага, в виде желтоватого твердого вещества (58,9 мг; 14,25%).
MS (ESI, pos. ion.) m/z: 207,0 [M+H]+;
1H NMR (400 МГц; CDCl3) δ (ppm): 8,72 (dd; J=4,9; 0,8 Гц; 1H); 8,36 (s; 1H); 7,87 (td; J=7,8; 1,8 Гц; 1H); 7,79-7,74 (m; 1H); 7,36 (ddd; J=7,5; 4,9; 1,1 Гц; 1H); 6,12 (s; 2H).
Пример 4
(S)-2-(1-((6-амин-5-(пиридин-2-ил)пиримидин-4-ил)амин)пропил)-5-хлор-3-циклопропилквиназолин-4(3H)-один
[414] К смеси (S)-2-(1-аминопропил)-5-хлор-3-циклопропилквиназолин-4(3H)-один (93,56 мг; 0,34 ммоль) и 6-хлор-5-(пиридин-2-ил)пиримидин-4-амин (34,8 мг; 0,17 ммоль) в n-BuOH (2 мл) был добавлен диизопропилэтиламин (148 мг; 1.14 ммоль). Смесь была подвергнута дистилляции при 130°C в течение суток, затем остужена до комнатной температуры и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (PE/EtOAc (v/v)=2/3) для получения соединения, указанного в заголовке шага, в виде желтого твердого вещества (21 мг; 27,.6%).
MS (ESI, pos. ion.) m/z: 448,1 [M+H]+;
1H NMR (600 МГц; CDCl3) δ (ppm): 8,81 (d; J=4,8 Гц; 1H); 8,14 (s; 1H); 7,95-7,84 (m; 1H); 7,55-7,51 (m; 1H); 7,42 (d; J=7,8 Гц; 1H); 7,37 (d; J=8,1 Гц; 1H); 7,34-7,29 (m; 1H); 6,99 (d; J=8,2 Гц; 1H); 5,64 (s; 2H); 4,18-4,06 (m; 1H); 3,13-2,99 (m; 1H); 2,05-1,94 (m; 1H); 1,85-1,74 (m; 1H); 1,50-1,40 (m; 2H); 1,05-0,99 (m; 1H); 1,00 (t; J=6,8 Гц; 3H); 0,94-0,90 (m; 1H).
Пример 12
(S)-2-(1-((6-амин-5-(5-метил-1,2,4-оксадиазол-3-ил)пиримидин-4-ил)амин)-пропил)-5-хлор-3-циклопропилквиназолин-4(3H)-один
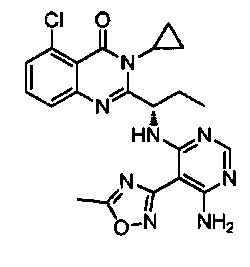
Шаг 1) 4,6-диметоксипиримидин-5-карбальдегид
[415] К суспензии 4,6-дихлорпиримидин-5-карбаальдегида (20 г; 113,0 ммоль) в осушенном МеОН (100 мл) был медленно добавлен раствор метанолата натрия (27,47 г; 508,5 ммоль) в осушенном МеОН (100 мл) при 0°C. Реагирующая смесь была нагрета до 70°C и перемешивалась на протяжении последующих 2 часов, затем вновь была охлаждена до 0°C, и был добавлен гидратогенный раствор HCl (1 М, 300 мл) для резкого охлаждения реагирующей смеси, после чего полученная смесь была нейтрализована насыщенным гидратогенным раствором NaHCO3 до pH=7. Смесь была экстрактирована посредством EtOAc (500 мл×3). Комбинированные органические фазы были высушены над обезвоженным Na2SO4 и сконцентрированы in vacuo для получения осадка, который был очищен хроматографией на силикагельной колонне (эфир/EtOAc (v/v)=4/1) для получения указанного в заголовке соединения в белого твердого вещества (8,37 г; 44%).
MS (ESI, pos. ion) m/z: 169,1 [M+H]+.
Шаг 2) 4,6-диметоксипиримидин-5-карбальдегид оксим
[416] К раствору 4,6-диметоксипиримидин-5-карбальдегида (3,0 г; 17,8 ммоль) в этилацетате (50 мл) был добавлен раствор NH2OH-HCl (1,24 г; 17,8 ммоль) в воде (30 мл), затем был добавлен уксуснокислый натрий (1,46 г; 17,8 ммоль) при комнатной температуре. После перемешивания в течение 2 часов при rt, реагирующая смесь была промыта водой (100 мл×2). Отсепарированная органическая фаза была высушена над обезвоженным Na2SO4, и сконцентрирована in vacuo для получения соединения, указанного в заголовке шага, в виде белого твердого вещества (3.2 г; 97%).
MS (ESI, pos. ion) m/z: 184,1 [M+H]+;
1HNMR (600 МГц; DMSO-d6) δ (ppm): 11,47 (s; 1H); 8,47 (s; 1H); 8,08 (s; 1H); 3,96 (s; 6H).
Шаг 3) 3-(4,6-диметоксипиримидин-5-ил)-5-метил-1,2,4-оксадиазол
[417] В трехгорлую химическую колбу, заправленную 4,6-диметоксипиримидин-5-карбальдегид оксимом (3,0 г; 16,4 ммоль) и (NH4)2Ce(NO3)6 (18,0 г; 32,8 ммоль) был добавлен CH3CN (100 мл) при rt в защитной атмосфере N2. Реагирующая смесь была нагрета до 70°C и перемешивалась в течение последующих 4 часов, затем была профильтрована, и фильтрат был сконцентрирован при пониженном давлении для получения желтого осадка, который был очищен хроматографией на силикагельной колонне (PE/EtOAc (v/v)=4/1) для получения соединения, указанного в заголовке шага, в виде белого твердого вещества (0,58 г; 16%).
MS (ESI, pos. ion) m/z: 223,2 [M+H]+;
1H NMR (400 МГц; DMSO-d6) δ (ppm): 8,68 (s; 1H); 3,93 (s; 6H); 2,67 (s; 3H).
Шаг 4) 3-(4,6-дихлорпиримидин-5-ил)-5-метил-1,2,4-оксадиазол
[418] К суспензии 3-(4,6-диметоксипиримидин-5-ил)-5-метил-1,2,4-оксадиазола (391 мг; 1,76 ммоль) и DMF (3.0 мл) в толуоле (20 мл) был добавлен РОСl3 (2.04 г; 13.3 ммоль), затем реагирующая смесь была нагрета для дистилляции, мониторинг осуществлялся посредством TLC (PE/EtOAc, v/v, 4/1). После завершения процесса смесь была сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (РЕ/EtOAc (v/v)=8/1) для получения соединения, указанного в заголовке шага, в виде белого твердого вещества (0,36 мг; 89%).
MS (ESI, pos. ion) m/z: 231,1 [M+H]+;
1H NMR (600 МГц; DMSO-d6) δ (ppm): 9,16 (s; 1H); 2,78 (s; 3H).
Шаг 5) 6-хлор-5-(5-метил-1,2,4-оксадиазол-3-ил)пиримидин-4-амин
[419] Раствор 3-(4,6-дихлорпиримидин-5-ил)-5-метил-1,2,4-оксадиазола (355 мг; 1,54 ммоль) в THF (25 мл) был продут газообразным NH3. Реагирующая смесь перемешивалась при rt, мониторинг осуществлялся посредством TLC (РЕ/EtOAc, v/v, 4/1)). После завершения смесь была профильтрована, и фильтрат был сконцентрирован in vacuo для получения соединения, указанного в заголовке шага, в виде белого твердого вещества (325 мг; 100%).
MS (ESI, pos. ion) m/z: 212,05 [M+H]+;
1H NMR (600 МГц; DMSO-d6) δ (ppm): 8,35 (s; 1H); 7,88 (br s; 1H); 7,06 (br s; 1H); 2,67 (s; 3H).
Шаг 6) (S)-2-(1-((6-амин-5-(5-метил-1,2,4-оксадиазол-3-ил)пиримидин-4-ил)амин)пропил)-5-хлор-3-циклопропилквиназолин-4(3H)-один
[420] Смесь 6-хлор-5-(5-метил-1,2,4-оксадиазол-3-ил)пиримидин-4-амина (42,3 мг; 0,2 ммоль), (S)-2-(1-аминопропил)-5-хлор-3-циклопропилквиназолин-4(3H)-один (55.5 мг; 0.2 ммоль) и N,N-диизопропилэтиламина (77,5 мг; 0,.6 ммоль) в n-бутаноле (2,5 мл) была нагрета до 125°C и подвергалась дистилляции в течение 12 часов. Смесь была сконцентрирована in vacuo для получения осадка, который был очищен хроматографией на силикагельной колонне (РЕ/EtOAc (v/v)=1/1) для получения соединения, указанного в заголовке шага, в виде белого твердого вещества (78 мг; 86%).
MS (ESI, pos. ion) m/z: 453,2 [M+H]+; HPLC: 98%;
1H NMR (400 МГц; CDCl3) δ (ppm): 8,89 (d; J=7,7 Гц; 1H); 8,13 (s; 1H); 7,61-7,48 (m; 2H); 7,46-7,38 (m; 1H); 6,30 (td; J=7,7; 5,3 Гц; 1H); 3,20-3,04 (m; 1H); 2,74 (s; 3H); 2,18-2,05 (m; 1H); 2,04-1,95 (m; 1H); 1,49-1,39 (m; 2H); 1,25-1,19 (m; 1H); 1,06 (t; J=7,4 Гц; 3H); 0,96-0,91 (m; 1H);
13C NMR (100 МГц; CDCl3) δ (ppm): 174,2; 166,0; 161,6; 161,5; 160,3; 159,7; 149,3; 133,8; 133,2; 129,2; 126,4; 118,3; 82,7; 52,7; 28,1; 27,0; 12,4; 10,6; 10,3; 10,2.
Пример 13. (S)-2-(1-((6-амин-5-(5-метил-1,2,4-оксадиазол-3-ил)пиримидин-4-ил)амин)-пропил)-5-хлор-3-фенилквиназолин-4(3H)-один
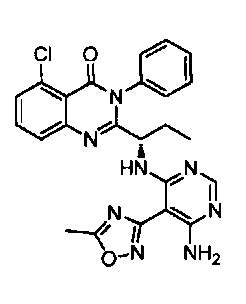
[421] Смесь 6-хлор-5-(5-метил-1,2,4-оксадиазол-3-ил)пиримидин-4-амина (35,0 мг; 0,165 ммоль), (S)-2-(1-аминопропил)-5-хлор-3-фенилквиназолин-4(3H)-один (52.0 мг; 0.165 ммоль) и N,N-диизопропилэтиламина (60,6 мг; 0,47 ммоль) в п-бутаноле (2,5 мл) была нагрета до 125°C и подвергалась дистилляции в течение 20 часов. В результате был получен белый осадок. Реагирующая смесь была профильтрована. Фильтрат был промыт n-бутанолом (4 мл) и этанолом (4 мл) для получения соединения, указанного в заголовке шага, в виде белого твердого вещества (50 мг; 62%).
MS (ESI, pos. ion) m/z: 489,1 [M+H]+; HPLC: 99,7%;
1H NMR (400 МГц; DMSO-d6) δ (ppm): 8,73 (d; J=7,1 Гц; 1H); 7,96 (s; 1H); 7,76 (t; J=8,0 Гц; 1H); 7,64-7,54 (m; 7H); 7,51 (s; 2H); 4,85 (td; J=7,5; 4,3 Гц; 1H); 2,76 (s; 3H); 2,04-1,82 (m; 1H); 1,72-1,59 (m; 1H); 0,77 (t; J=7,4 Гц; 3H);
13C NMR (100 МГц; DMSO-d6) δ (ppm): 175,8; 165,8; 161,7; 159,8; 159,4; 158,7; 158,6; 149,8; 136,8; 135,0; 133,3; 130,0; 129,8; 129,71; 129,67; 129,5; 127,1; 118,2; 81,6; 53,8; 10,3.
Пример 14 (S)-2-(1-((6-амин-5-(изоксазол-3-ил)пиримидин-4-ил)амин)пропил)-5-хлор-3-циклопропилквиназолин-4(3H)-один
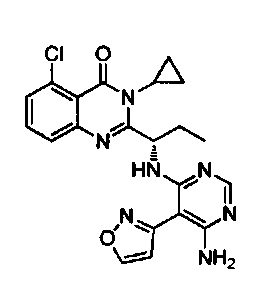
Шаг 1) 4,6-дихлорпиримидин-5-карбальдегид оксим
[422] К раствору 4,6-дихлорпиримидин-5-карбальдегида (7,20 г; 40,6 ммоль) в этилацетате (100 мл) был добавлен раствор NH2OH•HCl (2.82 г; 40.6 ммоль) в воде (30 мл), затем был добавлен раствор уксуснокислого натрия (3,34 г; 40,6 ммоль) в воде (30 мл) при комнатной температуре. Смесь перемешивалась при комнатной температуре в течение 4 часов, затем была промыта водой (100 мл×2). Отсепарированная органическая фаза была высушена над обезвоженным Na2SO4, и сконцентрирована in vacuo для получения соединения, указанного в заголовке шага, в виде светло-желтого твердого вещества (6,0 г; 77%).
MS (ESI, pos. ion) m/z: 192,1 [M+H]+.
Шаг 2) 4,6-дихлор-N-гидроксипиримидин-5-карбимидоил хлорид
[423] К раствору 4,6-дихлорпиримидин-5-карбальдегид оксима (4,0 г; 20,8 ммоль) в DMF (18 мл) был медленно добавлен N-хлорсукцинимид (3.05 г; 22.9 ммоль) при 0°C, затем смесь была перемещена в условия комнатной температуры и перемешивалась в течение 1 часа, после чего была разведена этилацетатом (100 мл). Смесь была промыта насыщенным минеральным раствором (50 мл×4). Отсепарированная органическая фаза была высушена над обезвоженным Na2SO4, и сконцентрирована in vacuo для получения соединения, указанного в заголовке шага, в виде светло-желтого твердого вещества (4,7 г; 100%).
MS (ESI, pos. ion) m/z: 225.9 [M+H]+;
1HNMR (400 МГц; DMSO-d6) δ (ppm): 13,14 (s; 1H); 9,08 (s; 1H).
Шаг 3) 3-(4,6-дихлорпиримидин-5-ил)-5-(триметилсилил)изоксазол
[424] К смеси 4,6-дихлор-N-гидроксипиримидин-5-карбимидоил хлорида (4,71 г; 20,8 ммоль) в THF (60 мл) был добавлен этинилтриметилсилан (6,13 г; 62,4 ммоль), затем был медленно добавлен раствор триэтиламина (4,2 г; 41,6 ммоль) в THF (5 мл) при rt. Реагирующая смесь перемешивалась в течение суток при rt, затем была профильтрована. Фильтрат был сконцентрирован in vacuo, и осадок был очищен хроматографией на силикагельной колонне (РЕ/EtOAc (v/v)=4/1) для получения соединения, указанного в заголовке шага, в виде светло-желтого твердого вещества (4,30 г; 72%).
MS (ESI, pos. ion) m/z: 288,1 [M+H]+;
1HNMR (600 МГц; CDCl3) δ (ppm): 8,86 (s; 1H); 6,60 (s; 1H); 0,43 (s; 9H).
Шаг 4) 6-хлор-5-(5-(триметилсилил)изоксазол-3-ил)пиримидин-4-амин
[425] Раствор 3-(4,6-дихлорпиримидин-5-ил)-5-(триметилсилил)изоксазола (2.0 г; 6.94 ммоль) в THF (50 мл) был продут газообразным NH3 при rt. Реагирующая смесь перемешивалась в течение 5 часов при rt, затем была профильтрована. Фильтрат был сконцентрирован in vacuo для получения соединения, указанного в заголовке шага, в виде белого твердого вещества (1,88 г; 100%).
MS (ESI, pos. ion) m/z: 269, [M+H]+;
1H NMR (600 МГц; DMSO-d6) δ (ppm): 8,29 (s; 1H); 7,78 (br s; 1H); 7,04 (s; 1H); 6,88 (br s; 1H); 0,37 (s; 9H).
Шаг 5) 6-хлор-5-(изоксазол-3-ил)пиримидин-4-амин
[426] Смесь 6-хлор-5-(5-(триметилсилил)изоксазол-3-ил)пиримидин-4-амина (1,0 г; 3,72 ммоль) и CsF (1,13 г; 7,44 ммоль) в DMF/H2O (5 мл/1 mL) перемешивалась при rt в течение 30 минут, затем смесь была разведена этилацетатом (100 мл) и промыта насыщенным минеральным раствором (50 мл×4). Органический слой был высушен над обезвоженным Na2SO4, и сконцентрирован in vacuo. Осадок был очищен хроматографией на силикагельной колонне (РЕ/EtOAc (v/v)=2/1) для получения соединения, указанного в заголовке шага, в виде белого твердого вещества (210 мг; 29%).
MS (ESI, pos. ion) m/z: 197,0 [M+H]+;
1H NMR (600 МГц; DMSO-d6) δ (ppm): 9,14 (d; J=1,6 Гц; 1H); 8,31 (s; 1H); 7,75 (br s; 1H); 6,94 (br s; 1H); 6,89 (d; J=1,6 Гц; 1H).
Пример 6 (S)-2-(1-((6-амин-5-(изоксазол-3-ил)пиримидин-4-ил)амин)пропил)-5-хлор-3-циклопропилквиназолин-4(3Н)-один
[427] Смесь 6-хлор-5-(1-метил-3Н-1,2,4-триазол-5-ил)пиримидин-4-амина (35,0 мг; 0,178 ммоль), (S)-2-(1-аминопропил)-5-хлор-3-циклопроилквиназолин-4(3H)-один (59,3 мг; 0,214 ммоль) и N,N-диизопропилэтиламина (69,0 мг; 0,534 ммоль) в n-бутаноле (2,0 мл) была нагрета до 125°C и подвергалась дистилляции в течение 45 часов, затем была сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (РЕ/EtOAc (v/v)=1/1) для получения соединения, указанного в заголовке шага, в виде белого твердого вещества (28 мг; 36%).
MS (ESI, pos. ion) m/z: 438,1 [M+H]+;
1H NMR (600 МГц; CDCl3) δ (ppm): 8,65 (d; J=1,5 Гц; 1H); 8,17 (s; 1H); 7,55 (t; J=8,0 Гц; 1H); 7,44 (d; J=7,7 Гц; 1H); 7,43 (d; J=1,1 Гц; 1H); 7,04 (d; J=1,5 Гц; 1H); 6,73 (d; J=8,3 Гц; 1H); 6,23 (td; J=8,0; 4,7 Гц; 1H); 5,66 (br s; 2H); 3,11-3,05 (m; 1H); 2,11-2,01 (m; 1H); 1,91-1,80 (m; 1H); 1,50-1,41 (m; 2H); 1,02 (t; J=7,4 Гц; 3H); 0,96-0,87 (m; 2H). 13C NMR (150 МГц; CDCl3): δ (ppm): 161,5; 160,4; 160,3; 159,4; 158,8; 158,1; 156,6; 148,8; 133,9; 133,4; 129,4; 126,0; 118,2; 104,2; 86,1; 52,2; 27,9; 26,8; 10,7; 10,2; 10,1.
Пример 15 (S)-2-(1-((6-амин-5-(5-метил-1,2,4-оксадиазол-3-ил)пиримидин -4-ил)амин)-пропил)-5-метил-3-(о-толил)квиназолин-4(3H)-один
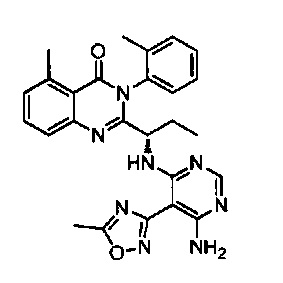
Шаг 1) 2-метил-6-нитро-N-(о-толил)бензамид
[428] К суспензии 2-метил-6-нитробензойной кислоты (5,43 г; 30 ммоль) в толуоле (45 мл) был добавлен одной порцией SOCl2 (4.5 мл, 60 ммоль) при комнатной температуре. После добавления реагирующая смесь перемешивалась при дистилляции в течение суток и была сконцентрирована in vacuo. Осадок был растворен в 1,4-диоксане (30 мл), и к полученному раствору была добавлена по каплям суспензия о-толуидина (3,22 г; 30 ммоль) и NaHCO3 (6,34 г; 75 ммоль) в 1,4-диоксане(30 мл) при 0°C. Полученная смесь была разведена EtOAc (400 мл) и водой (200 мл). Отсепарированная органическая фаза была промыта водой (200 мл) и насыщенным минеральным раствором (300 мл), высушена над обезвоженным Na2SO4, и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (PE/EtOAc (v/v)=3/2) для получения соединения, указанного в заголовке шага, в виде розового порошка (7,44 г; 92%).
MS (ESI, pos. ion) m/z: 271,1 [M+H]+;
1H NMR (600 МГц; CDCl3) δ (ppm): 8,08 (d; J=8,2 Гц; 1H); 7,94 (d; J=8,0 Гц; 1H); 7,62 (d; J=7,6Hz; 1H); 7,52 (t;, 7=7,9 Гц; 1H); 7,33 (t;, 7=7,6 Гц; 1H); 7,27 (d; J=7,5 Гц; 1H); 7,19 (dd; J=14,1; 6,6 Гц; 1H); 7,15 (s; 1H); 2,61 (s; 3H); 2,31 (s); 3H).
Шаг 2) (S)-терт-бутил(1-(2-метил-6-нитро-N-(о-толил) бензоамидо)-1-оксобутан-2-ил)-карбамат
[429] К суспензии 2-метил-6-нитро-N-(о-толил)бензамида (7,44 г; 27,5 ммоль) в толуоле (150 мл) добавлен по каплям DMF (0.5 мл) и SOCl2 (15,84 мл; 220 ммоль). После добавления реагирующая смесь перемешивалась при дистилляции в течение суток и была сконцентрирована in vacuo. Осадок был растворен в DCM (100 мл) для получения бледно-желтого раствора А.
К раствору (S)-2-((терт-бутоксикарбонил)амин)бутановой кислоты (5,6 г; 27,5 ммоль) и DIPEA (10 мл, 60.5 ммоль) в безводном DCM (100 мл) был медленно добавлен раствор A при 0°C. Полученная смесь перемешивалась при комнатной температуре в течение 24 часов, затем была промыта СН3СООН/Н2O (1/100 (v/v), 100 мл×3), насыщенным гидратогенным раствором NaHCO3 (100 мл) и насыщенным минеральным раствором (100 мл). Отсепарированная органическая фаза была высушена над обезвоженным Na2SO4, и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (РЕ/EtOAc (v/v)=10/1) для получения соединения, указанного в заголовке шага, в виде светло-желтого твердого вещества (11,35 г; 91%).
MS (ESI, pos. ion) m/z: 478,1 [M+Na]+.
Шаг 3) терт-бутил(1-(5-метил-4-оксо-3-(о-толил)-3,4-дигидроквиназолин-2-ил)пропил)-карбамат
[430] К раствору (S)-терт-бутил(1-(2-метил-6-нитро-N-(о-толил)бензоамидо)-1-оксобутан-2-ил)карбамата (11,2 г; 24,6 ммоль) в уксусной кислоте (60 мл) был добавлен одной порцией порошкообразный цинк (6,43 г; 98,4 ммоль). Реагирующая смесь перемешивалась в течение 24 часов при 35°C, затем была профильтрована. Фильтрат был сконцентрирован in vacuo, осадок был растворен в EtOAc (300 мл), и полученная смесь была промыта насыщенным гидратогенным раствором NaHCO3 (00 мл×2) и насыщенным минеральным раствором (100 мл). Органический слой был высушен над обезвоженным Na2SO4, и сконцентрирован in vacuo. Осадок был очищен хроматографией на силикагельной колонне (PE/EtOAc (v/v)=50/3) для получения соединения, указанного в заголовке шага, в виде белого твердого вещества (5,4 г; 54%).
MS (ESI, pos. ion) m/z: 408,2 [M+H]+.
Шаг 4) 2-(1-аминопропил)-5-метил-3-(о-толил)квиназолин-4(3H)-один
[431] К раствору терт-бутил(1-(5-метил-4-оксо-3-(о-толил)-3,4-дигидроквиназолин-2-ил)пропил)карбамата (5,4 г; 13,3 ммоль) в EtOAc (20 мл) был добавлен одной порцией раствор HCl в EtOAc (3,5 М, 70 мл) при комнатной температуре. Смесь перемешивалась при комнатной температуре на протяжении 3,5 часов. Полученная суспензия была растворена в воде (400 мл). Отсепарированная гидратогенная фаза была экстрактирована посредством EtOAc (100 мл×2), затем нейтрализована до pH=7 с помощью порошка NaHCO3 и вновь экстрактирована с помощью EtOAc/MeOH (6/1 (v/v), 100 мл×3). Комбинированные органические фазы были промыты насыщенным минеральным раствором (100 мл), высушены над обезвоженным Na2SO4, и сконцентрированы in vacuo для получения соединения, указанного в заголовке шага, в виде коричневого масла (4,09 г; 100%), состоящего из двух изомеров в пропорции 4/7 (изомер А/изомер В).
MS (ESI, pos. ion) m/z: 308,2 [M+H]+;
Изомер А: 1Н NMR (400 МГц; DMSO-d6): δ (ppm): 7,72-7,66 (m; 1Н); 7,57-7,52 (m; 1H); 7,48-7,43 (m; 2H); 7,43-7,36 (m; 1H); 7,36-7,26 (m; 2H); 3,13-3,05 (m; 1H); 2,74 (s; 3H); 2,02 (s; 1H); 1,79-1,63 (m; 1H); 1,50-1,27 (m; 1H); 0,75-0,67 (m; 3H).
Изомер В: 1H NMR (400 МГц; DMSO-d6) δ (ppm): 7,72-7,66 (m; 1H); 7,57-7,52 (m; 1H); 7,48-7,43 (m; 2H); 7,43-7,36 (m; 1H); 7,36-7,26 (m; 2H); 2,99-2,92 (m; 1H); 2,74 (s; 3H); 2,08 (s; 2H); 1,79-1,63 (m; 1H); 1,50-1,27 (m; 1H); 0,75-0,67 (m; 3H).
Пример 5 (S)-2-(1-((6-амин-5-(5-метил-1,2,4-оксадиазол-3-ил)пиримидин-4-ил)амин)пропил)-5-метил-3-(о-толил)квиназолин-4(3H)-один
[432] Смесь 6-хлор-5-(5-метил-1,2,4-оксадиазол-3-ил)пиримидин-4-амина (31,0 мг; 0,15 ммоль), 2-(1-аминопропил)-5-метил-3-(о-толил)квиназолин-4(3H)-один (50.0 мг; 0.16 ммоль) и N,N-диизопропилэтиламина (57,4 мг; 0,44 ммоль) в n-бутаноле (2,0 мл) была нагрета до 125°C и подвергалась дистилляции в течение 12 часов. После завершения процесса реагирующая смесь была сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (PE/EtOAc (v/v)=1/1) для получения соединения, указанного в заголовке шага, в виде светло-желтого твердого вещества (50,0 мг; 70%), состоящего из двух изомеров в пропорции 10/9 (изомер А/изомер В).
MS (ESI, pos. ion) m/z: 483,2 [M+H]+; HPLC: 92% (общая степень чистоты изомера А и В);
Изомер А: 1Н NMR (400 МГц; DMSO-d6): δ (ppm): 8,93 (d; J=8,3 Гц; 1H); 7,92 (s; 1H); 7,66-7,58 (m; 2H); 7,48-7,35 (m; 3H); 7,28-7,21 (m; 2H); 5,26-5,14 (m; 1H); 2,98 (s; 3H); 2,85 (s; 3H); 2,21 (s; 3H); 1,83-1,67 (m; 4H); 0,91-0,83 (m; 3Н);
Изомер В: 1H NMR (400 МГц; DMSO-d6): δ (ppm): 8,76 (d; J=7,5 Гц; 1H); 7,95 (s; 1H); 7,66-7,58 (m; 2H); 7,48-7,35 (m; 3H); 7,28-7,21 (m; 2H); 5,26-5,14 (m; 1H); 2,91 (s; 3H); 2,85 (s; 3H); 2,07 (s; 3H); 1,83-1,67 (m; 4H); 0,91-0,83 (m; 3H).
Пример 16
(S)-2-(1-((6-амин-5-(5-метил-1,2,4-оксадиазол-3-ил)пиримидин-4-ил)амин)-пропил)-3-циклопропил-5-фторквиназолин-4(3H)-один
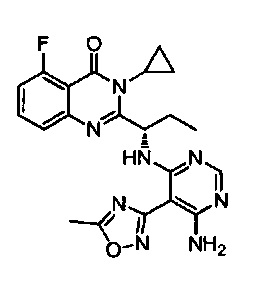
Шаг 1) 2-фтор-N-циклопропил-6-нитробензамид
[433] К желтой суспензии 2-фтор-6-нитробензойной кислоты (2,0 г; 10,8 ммоль) в толуоле (11 мл) добавлен по каплям SOCl2 (3,0 мл, 32,4 ммоль) при комнатной температуре. После добавления реагирующая смесь перемешивалась при 110°C в течение суток и была сконцентрирована in vacuo для получения коричневого масла для использования в следующем шаге без дополнительной очистки.
К раствору циклопропанамина (1,2 мл, 16,2 ммоль) и NaHCO3 (1,8 г; 21,6 ммоль) в 1,4-диоксане (7 мл) был добавлен раствор упомянутого выше коричневого масла в 1,4-диоксане (7 мл) при 5°C, затем полученная смесь перемешивалась в течение суток при комнатной температуре, после чего была профильтрована. Фильтрат был сконцентрирован in vacuo. Осадок был очищен хроматографией на силикагельной колонне (РЕ/EtOAc (v/v)=1/1) для получения соединения, указанного в заголовке шага, в виде светло-коричневого твердого вещества (2,44 г; 100%).
MS (ESI, pos. ion) m/z: 225,1 [M+H]+;
1H NMR (400 МГц; DMSO-d6) δ (ppm): 8,78 (s; 1H); 8,05-7,98 (m; 1H); 7,81-7,68 (m; 2H); 2,79 (m; 1H); 0,76-0,69 (m; 2H); 0,53-0,48 (m; 2H).
Шаг 2) (S)-терт-бутил(1-(N-циклопропил-2-фтор-6-нитробензоамидо)-1-оксобутан-2-ил)карбамат
[434] К раствору 2-фтор-N-циклопропил-6-нитробензоамида (1,0 г; 4.46 ммоль) в толуоле (15 мл) добавлен по каплям SOCl2 (3,0 мл, 40,14 ммоль) и DMF (0,5 мл). После добавления реагирующая смесь перемешивалась при 120°C в течение суток и была сконцентрирована in vacuo для получения коричневого масла для использования в следующем шаге без дополнительной очистки.
К раствору Boc-L-2-аминобутировой кислоты (1,20 г; 5,58 ммоль) и DIPEA (1.73 г; 13.38 ммоль) в дихлорметане (8 мл) при 0°C был добавлен раствор упомянутого выше коричневого масла в дихлорметане (23 мл). После добавления реагирующая смесь перемешивалась при комнатной температуре в течение суток и была промыта 1% гидратогенной уксусной кислотой (100 мл), насыщенным гидратогенным раствором NaHCO3 (100 мл) и насыщенным минеральным раствором (100 мл). Отсепарированный органический слой был высушен над обезвоженным Na2SO4, и сконцентрирован in vacuo. Осадок был очищен хроматографией на силикагельной колонне (PE/EtOAc (v/v)=5/1) для получения соединения, указанного в заголовке шага, в виде желтого масла (1,72 г; 94%).
MS (ESI, pos. ion) m/z: 310,1 [M-Boc+H]+.
Шаг 3)
(S)-терт-бутил(1-(5-фтор-3-циклопропил-4-оксо-3,4-дигидроквиназолин-2-ил)пропил)-карбамат
[435] К раствору ((S)-терт-бутил(1-(N-циклопропил-2-фтор-6-нитробензоамидо)-1-оксобутан-2-ил)карбамата (3,88 г; 9,48 ммоль) в уксусной кислоте (52 мл) был добавлен одной порцией порошкообразный цинк (2,50 г; 38,23 ммоль). Реагирующая смесь перемешивалась при комнатной температуре на протяжении 11 часов. Реагирующая смесь была отфильтрована и фильтрат сконцентрирован in vacuo. Осадок был растворен в EtOAc (150 мл) и нейтрализован до pH=7-8 насыщенным гидратогенным раствором NaHCO3. Органическая фаза была промыта насыщенным минеральным раствором (150 мл×2), высушена над обезвоженным Na2SO4, и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (РЕ/EtOAc (v/v)=5/1) для получения соединения, указанного в заголовке шага, в виде коричневого масла (2,87 г; 83%).
MS (ESI, pos. ion) m/z: 362,2 [M+H]+.
Шаг 4)
(S)-2-(1-аминопропил)-3-циклопропил-5-фторквиназолин-4(3H)-один
[436] К раствору (S)-терт-бутил(1-(5-фтор-3-циклопропил-4-оксо-3,4-дигидроквиназолин-2-ил)пропил)кар бамата (2,87 г; 7,94 ммоль) в EtOAc (12 мл) был добавлен одной порцией раствор HCl в EtOAc (3.5 М, 20 мл) при комнатной температуре. Смесь перемешивалась при комнатной температуре в течение 2 часов, затем была разведена водой (200 мл). Смесь была нейтрализована до pH=7-8 насыщенным гидратогенным раствором NaHCO3, и отсепарированная гидратогенная фаза была экстрактирована смесью EtOAc/МеОН (50/1 (v/v), 100 мл×3). Комбинированные органические фазы были промыты насыщенным минеральным раствором (150 мл), высушены над обезвоженным Na2SO4, и сконцентрированы in vacuo для получения соединения, указанного в заголовке шага, в виде светло-коричневого масла (1,79 г; 86%).
MS (ESI, pos. ion) m/z: 262,2 [M+H]+;
1H NMR (600 МГц; CDCl3) δ (ppm): 7,62 (td; J=8,1; 5,5 Гц; 1H); 7,41 (d; J=8,2 Гц; 1H); 7,11-7,03 (m; 1H); 4,62-4,55 (m; 1H); 2,95-2,88 (m; 1H); 1,95-1,84 (m; 1H); 1,73-1,61 (m; 1H); 1,44-1,36 (m; 1H); 1,36-1,29 (m; 1H); 1,03 (t; J=7,4 Гц; 3H); 0,97-0,89 (m; 2H).
Пример 5
(S)-2-(1-((6-амин-5-(5-метил-1,2,4-оксадиазол-3-ил)пиримидин-4-ил)амин)пропил)-3-циклопропил-5-фторквиназолин-4(3Н)-один
[437] Смесь 6-хлор-5-(5-метил-1,2,4-оксадиазол-3-ил)пиримидин-4-амина (25,0 мг; 0,118 ммоль), (S)-2-(1-аминопропил)-3-циклопропил-5-фторквиназолин-4(3H)-один (33.9 мг; 0.130 ммоль) и N,N-диизопропилэтиламина (45.8 мг; 0.354 ммоль) в n-бутаноле (2,0 мл) была нагрета до 125°C и подвергнута возгоне с обратным холодильником в течение 22 часов, после чего реагирующая смесь была сконцентрирована in vacuo, и осадок был очищен подготовительным TLC (DCM/MeOH (v/v)=50/1) для получения соединения, указанного в заголовке шага, в виде светло-желтого твердого вещества (32,0 мг; 62%).
MS (ESI, pos. ion) m/z: 437,2 [M+H]+; HPLC: 98,7%;
1H NMR (400 МГц; CDCl3) δ (ppm): 8,92 (d; J=7,7 Гц; 1H); 8,13 (s; 1H); 7,61 (td; J=8,2; 5,4 Гц; 1H); 7,42 (d; J=8,2 Гц; 1H); 7,28 (s; 1H); 7,07 (dd; J=10,1; 8,6 Гц; 1H); 6,32 (td; J=7,7; 5,4 Гц; 1H); 3,15-3,07 (m; 1H); 2,74 (s; 3H); 2,16-2,06 (m; 1H); 2,05-1,96 (m; 1H); 1,26-1,19 (m; 2H); 1,06 (t; J=7,4 Гц; 3H); 0,97-0,86 (m; 2H). 13C NMR (151 МГц; CDCl3) δ (ppm): 174,2; 165,9; 161,9; 161,4; 160,6; 160,54; 160,52; 160,1; 159,7; 158,4; 148,9; 134,22; 134,15; 123,0; 122,9; 113,1; 113,0; 110,9; 110,8; 82,6; 52,7; 28,2; 26,7; 12,4; 10,6; 10,3; 10,2.
Пример 17
(S)-2-(1-((6-амин-5-(5-метил-1,2,4-оксадиазол-3-ил)пиримидин-4-ил)амин)пропил)-3-циклопропил-5-фторквиназолин-4(3H)-один
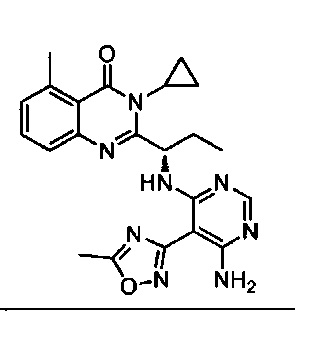
Шаг 1) 2-метил-N-циклопропил-6-нитробензамид
[438] К перемешиваемой желтой суспензии 2-метил-6-нитробензойной кислоты (3,0 г; 16,6 ммоль) в толуоле (17 мл) добавлен по каплям SOCl2 (3,7 мл, 49,7 ммоль) при комнатной температуре. После добавления реагирующая смесь перемешивалась при 110°C в течение суток и была сконцентрирована in vacuo для получения коричневого масла для использования в следующем шаге без дополнительной очистки.
К суспензии цилопропиламина (1,8 мл, 24,8 ммоль) и NaHCO3 (2,.8 г; 33,1 ммоль) в 1,4-диоксане (10 мл) при 5°C был добавлен раствор упомянутого выше коричневого масла в 1,4-диоксане (10 мл). Полученная смесь перемешивалась при комнатной температуре в течение 10 часов, затем была профильтрована, и фильтрат сконцентрирован in vacuo. Осадок был очищен хроматографией на силикагельной колонне (РЕ/EtOAc (v/v)=2/1) для получения соединения, указанного в заголовке шага, в виде светло-коричневого твердого вещества (3,2 г; 88%).
MS (ESI, pos. ion) m/z: 221,2 [M+H]+;
1H NMR (400 МГц; DMSO-d6) δ (ppm): 8,55 (s; 1H); 7,95 (d; J=8,1 Гц; 1H); 7,67 (d; J=7,5 Гц; 1H); 7,54 (t; J=7,9 Гц; 1H); 2,87-2,64 (m; 1H); 2,32 (s; 3H); 0,75-0,66 (m; 2H); 0,52-0,45 (m; 2H).
Шаг 2) (S)-терт-бутил(1-(2-метил-N-циклопропил-6-нитробензоамидо)-1-оксобутан-2-ил)-карбамат
[439] К раствору 2-метил-N-циклопропил-6-нитробензоамида (3,2 г, 14,5 ммоль) в толуоле (48 мл) добавлен по каплям SOCl2 (10 мл, 130,8 ммоль) и DMF (0.5 мл). После добавления полученная смесь перемешивалась при 120°C в течение суток и была сконцентрирована in vacuo для получения коричневого масла для использования в следующем шаге без дополнительной очистки.
К раствору Boc-L-2-аминобутировой кислоты (3,.8 г; 18,8 ммоль) и DIPEA (8 мл, 43,6 ммоль) в дихлорметане (26 мл) при 0°C был добавлен раствор упомянутого выше коричневого масла в дихлорметане (73 мл). После добавления реагирующая смесь перемешивалась при комнатной температуре в течение 5 часов и была промыта 1% гидратогенным раствором уксусной кислоты (150 мл×2), насыщенным гидратогенным раствором NaHCO3 (150 мл×2) и насыщенным минеральным раствором (150 мл×2). Отсепарированная органическая фаза была высушена над обезвоженным Na2SO4, и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (РЕ/ЕtOAc (v/v)=5/1) для получения соединения, указанного в заголовке шага, в виде коричневого масла (5,9 г; 99%).
MS (ESI, pos. ion) m/z: 306,1 [M-Boc+H]+.
Шаг 3) (S)-терт-бутил(1-(5-метил-3-циклопропил-4-оксо-3,4-дигидроквиназолин-2-ил)пропил)-карбамат
[440] К раствору (S)-терт-бутил(1-(2-метил-N-циклопропил-6-нитробензоамидо)-1-оксобутан-2-ил)карбама та (5,9 г; 14,5 ммоль) в уксусной кислоте (72 мл) был добавлен одной порцией порошкообразный цинк (3.8 г; 58.0 ммоль). Реагирующая смесь перемешивалась при комнатной температуре в течение суток, затем была профильтрована, и фильтрат сконцентрирован in vacuo. Осадок был растворен в EtOAc (150 мл) и полученная смесь нейтрализована до pH=7-8 насыщенным гидратогенным раствором NaHCO3. Отсепарированная органическая фаза была промыта насыщенным минеральным раствором (150 мл×2), высушена над обезвоженным Na2SO4, и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (PE/EtOAc (v/v)=5/1) для получения соединения, указанного в заголовке шага, в виде коричневого масла (3,9 г; 75,8%).
MS (ESI, pos. ion) m/z: 358,2 [M+H]+.
Шаг 4) (S)-2-(1-аминопропил)-3-циклопропил-5-метилквиназолин-4(3Н)-один
[441] К перемешиваемому раствору (S)-терт-бутил(1-(5-метил-3-циклопропил-4-оксо-3,4-дигидроквиназолин-2-ил)пропил)карбамата (3,9 г; 11,0 ммоль) в этилацетате (15 мл) был одной порцией добавлен раствор HCl в EtOAc (3,5 М;, 50 мл) при комнатной температуре. Смесь перемешивалась при комнатной температуре на протяжении 2 часов, затем была разведена водой (100 мл) и EtOAc (200 мл), и полученная смесь была нейтрализована до рН=7-8 насыщенным гидратогенным раствором NaHCO3. Водосодержащая фаза был экстрактирована посредством смеси EtOAc/MeOH (50/1 (v/v), 100 мл×3) Комбинированные органические фазы были промыты насыщенным минеральным раствором (150 мл×2), высушены над обезвоженным Na2SO4, и сконцентрированы in vacuo для получения соединения, указанного в заголовке шага, в виде светло-коричневого масла (2,83 г; 100.0%).
MS (ESI, pos. ion) m/z: 258,2 [M+H]+;
1HNMR(600 МГц; CDCl3) δ (ppm): 7,54-7,49 (m; 1H); 7,46 (d; J=8,0 Гц; 1H); 7,19 (d; J=7,3 Гц; 1H); 4,80 (t; J=6,0 Гц; 1H); 2,95-2,87 (m; 1H); 2,84 (s; 3H); 2,00-1,94 (m; 1H); 1,86-1,77 (m; 1H); 1,44-1,36 (m; 1H); 1,33-1,27 (m; 1H); 1,09-1,04 (m; 1H); 1,03 (t; J=7,4 Гц; 3H); 0,94-0,87 (m; 1H).
Шаг 5 (S)-2-(1-((6-амин-5-(5-метил-1,2,4-оксадиазол-3-ил)пиримидин-4-ил)амин)-пропил)-3-циклопропил-5-фторквиназолин-4(3H)-один
[442] Смесь 6-хлор-5-(5-метил-1,2,4-оксадиазол-3-ил)пиримидин-4-амина (27,7 мг; 0,130 ммоль), (S)-2-(1-аминопропил)-3-циклопропил-5-метилквиназолин-4(3H)-один (37,0 мг; 0,144 ммоль) и N,N-диизопропилэтиламина (50.0 мг; 0.390 ммоль) в n-бутаноле (2,0 мл) была нагрета до 125°C и подвергалась дистилляции в течение 20 часов. После завершения реагирующая смесь была сконцентрирована in vacuo, и осадок был очищен подготовительным TLC (дихлорметан/метанол (v/v)=50/1) для получения соединения, указанного в заголовке шага, в виде светло-желтого твердого вещества (36,0 мг; 64%).
MS (ESI, pos. ion) m/z: 433,3 [M+H]+; HPLC: 96%;
1H NMR (600 МГц; CDCl3) δ (ppm): 9,03 (d; J=7,8 Гц; 1H); 8,13 (d; J=1A Гц; 1H); 7,54 (t; J=7,7 Гц; 1H); 7,48 (d; J=8,0 Гц; 1H); 7,19 (d; J=7,2 Гц; 1H); 6,35-6,30 (m; 1H); 3,09 (ddd; J=11,1; 7,1; 4,2 Гц; 1H); 2,86 (s; 3H); 2,74 (s; 3H); 2,15-2,07 (m; 1H); 2,05-1,98 (m; 1H); 1,19-1,11 (m; 2H); 1,04 (t; 7=7,4 Гц; 3H); 0,94-0,83 (m; 2H);
13C NMR (151 МГц; CDCl3) δ (ppm): 174,1; 165,9; 164,1; 161,4; 159,6; 158,9; 158,3; 148,3; 140,8; 133,0; 129,2; 125,2; 119,6; 82,5; 52,6; 28,1; 26,6; 23,0; 12,3; 10,6; 10,2; 10,0.
Пример 18 (S)-2-(1-((6-амин-5-(1-метил-1H-1,2,4-триазол-3-ил)пиримидин-4-ил)амин)пропил)-3-циклопропил-5-метилквиназолин-4(3H)-один

[443] К суспензии 6-хлор-5-(1-метил-1H-1,2,4-триазол-3-ил)пиримидин-4-амина (35 мг; 0,165 ммоль) и (S)-2-(1-аминопропил)-3-циклопропил-5-метилквиназолин-4(3H)-один (55 мг; 0,214 ммоль) в n-BuOH (2 мл) был добавлен DIPEA (43 мг; 0,331 ммоль). Реагирующая смесь была нагрета для дистилляции и перемешивалась в течение последующих 36 часов. Реакция контролировалась посредством TLC (PE/EtOAc, v/v, 1/3). После завершения реагирующая смесь была остужена до rt и сконцентрирована in vacuo для удаления растворителя. Осадок был растворен в EtOAc (15 мл), и полученная смесь была промыта водой (15 мл) и насыщенным минеральным раствором (10 мл). Отсепарированная органическая фаза была высушена над обезвоженным Na2SO4, и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (РЕ/EtOAc (v/v)=20/1), затем подготовительным TLC (DCM/MeOH (v/v)=20/1) для получения соединения, указанного в заголовке шага, в виде светло-желтого твердого вещества (36 мг; 50,2%).
MS (ESI, pos. ion): 432,3 [M+H]+;
1H NMR (600 МГц; CDCl3) δ (ppm): 9,67 (s; 1H); 8,16 (s; 1H); 8,13 (s; 1H); 7,61-7,39 (m; 2H); 7,18 (d; J=6,3 Гц; 1H); 6,40-6,25 (m; 1H); 4,06 (s; 3H); 3,09 (s; 1H); 2,86 (s; 3H); 2,13-2,00 (m; 2H); 1,46-1,38 (m; 2H); 1,05 (t; J=6,3 Гц; 3H); 0,91-0,83 (m; 2H).
Пример 19 (S)-2-(1-((6-амин-5-(3-метил-1,2,4-оксадиазол-5-ил)пиримидин-4-ил)амин)-пропил)-5-хлор-3-циклопропилквиназолин-4(3H)-один
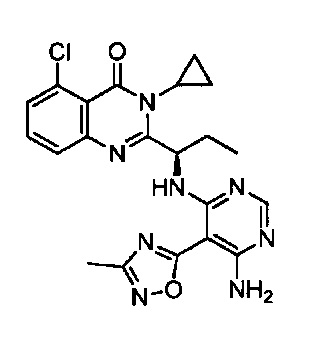
Шаг 1) 2-хлор-N-циклопропил-6-нитробензамид
[444] К желтой суспензии 2-хлор-6-нитробензойной кислоты (5,0 г; 24,8 ммоль) в толуоле (25 мл) добавлен по каплям SOCl2 (5,5 мл, 74,4 ммоль), затем DMF (1 мл) при комнатной температуре. После добавления реагирующая смесь перемешивалась при 110°C в течение суток и была сконцентрирована in vacuo для получения коричневого масла для использования в следующем шаге без дополнительной очистки.
К суспензии циклопропанамина (2,6 мл, 37,2 ммоль) и NaHCO4 (4.2 г; 49.6 ммоль) в 1,4-диоксане (15 мл) был добавлен раствор упомянутого выше коричневого масла в 1,4-диоксане (15 мл) при 5°C, затем полученная смесь перемешивалась в течение суток при комнатной температуре, после чего была профильтрована. Фильтрат был сконцентрирован in vacuo для получения соединения, указанного в заголовке шага, в виде коричневого твердого вещества (5,2 г; 87,0%).
MS (ESI, pos. ion) m/z: 241,1 [M+H]+;
1H NMR (400 МГц; DMSO-d6) δ (ppm): 8,78 (s; 1H); 8,05-7,98 (m; 1H); 7,81-7,68 (m; 2H); 2,79 (m; 1H); 0,76-0,69 (m; 2H); 0,53-0,48 (m; 2H).
Шаг 2) (S)-терт-бутил(1-(2-хлор-N-циклопропил-6-нитробензоамидо)-1-оксобутан-2-ил)-карбамат
[445] К раствору 2-хлор-N-циклопропил-6-нитробензоамида (5,2 г; 21,6 ммоль) в толуоле (72 мл) добавлен по каплям SOCl2 (14,5 мл, 194,5 ммоль). После добавления реагирующая смесь была нагрета до 120°C и перемешивалась при этой температуре в течение суток, затем была сконцентрирована in vacuo для получения коричневого масла для использования в следующем шаге без дополнительной очистки.
К перемешиваемому раствору Boc-D-2-аминобутировой кислоты (5,7 г; 28,1 ммоль) и DIPEA (8.4 г; 64.8 ммоль) в дихлорметане (36 мл) был медленно добавлен раствор упомянутого выше коричневого масла в дихлорметане (110 мл) при 0°C. Полученная смесь перемешивалась при комнатной температуре в течение суток, затем была промыта 1% гидратогенным раствором уксусной кислоты (150 мл), насыщенным гидратогенным раствором NaHCO3 (100 m×2) и насыщенным минеральным раствором (150 мл). Отсепарированный органический слой был высушен над обезвоженным Na2SO4, и сконцентрирован in vacuo для получения соединения, указанного в заголовке шага, в виде желтого масла (7,7 г; 83,4%).
MS (ESI, pos. ion) m/z: 326,1 [M-Boc+H]+.
Шаг 3) (R)-терт-бутил(1-(5-хлор-3-циклопропил-4-оксо-3,4-дигидроквиназолин-2-ил)пропил)карбамат
[446] К раствору (R)-терт-бутил(1-(2-хлор-N-циклопропил-6-нитробензоамидо)-1-оксобутан-2-ил)карбамата (7,6 г; 18,0 ммоль) в уксусной кислоте (72 мл) был добавлен одной порцией порошкообразный цинк (4,7 г; 72,1 ммоль). Реагирующая смесь перемешивалась в течение суток при комнатной температуре, затем была профильтрована. Фильтрат был сконцентрирован in vacuo для получения соединения, указанного в заголовке шага, в виде бледно-коричневого твердого вещества. Осадок был растворен в этилацетате (200 мл) и полученная смесь нейтрализована до pH=7-8 насыщенным гидратогенным раствором NaHCO3. Отсепарированная органическая фаза была промыта насыщенным минеральным раствором (150 мл×2), высушена над обезвоженным Na2SO4, и сконцентрирована in vacuo для получения соединения, указанного в заголовке шага, в виде бледно-коричневого твердого вещества (5,3 г; 78,0%).
MS (ESI, pos. ion) m/z: 378,2 [M+H]+.
Шаг 4) (R)-2-(1-аминопропил)-5хлор-3-циклопропилквиназолин-4(3H)-один
[447] К раствору (R)-терт-бутил(1-(5-хлор-3-циклопропил-4-оксо-3,4-дигидроквиназолин-2-ил)пропил)карбамата (5,2 г; 13,7 ммоль) в этилацетате (30 мл) был добавлен одной порцией раствор HCl в этилацетате (3.5 М, 35 мл). Полученная смесь перемешивалась при комнатной температуре на протяжении 3 часов, затем была разведена эфиром уксусной кислоты (100 мл) и водой (80 мл), и полученная смесь была нейтрализована до pH=7-8 насыщенным гидратогенным раствором NaHCO3. Отсепарированная водосодержащая фаза был экстрактирована смесью ацетат/метанол (50/1 (v/v), 100 мл×2). Комбинированные органические фазы были промыты насыщенным минеральным раствором (200 мл×2), высушены над обезвоженным Na2SO4, и сконцентрированы in vacuo для получения соединения, указанного в заголовке шага, в виде коричневого масла (3,2 г; 83,7%).
MS (ESI, pos. ion) m/z: 278,2 [M+H]+;
1H NMR (600 МГц; CDCl3) δ (ppm): 7,58-7,50 (m; 2H); 7,42 (dd; J=12; 1,5 Гц; 1H); 4,56 (dd; J=7,3; 5,4 Гц; 1H); 3,00-2,88 (m; 1H); 1,95-1,83 (m; 1H); 1,73-1,61 (m; 1H); 1,44-1,37 (m; 1H); 1,37-1,30 (m; 1H); 1,03 (t; J=7,4 Гц; 3H); 0,98-0,87 (m; 2H).
Шаг 5) (R)-2-(1-((6-амин-5-(3-метил-1,2,4-оксадиазол-5-ил)пиримидин-4-ил)амин) пропил)-5-хлор-3-пиклопропилквиназолин-4(3Н)-один
[448] К суспензии 6-хлор-5-(3-метил-1,2,4-оксадиазол-5-ил)пиримидин-4-амина (32 мг; 0,151 ммоль) и (R)-2-(1-аминопропил)-5-хлор-3-циклопропилквиназолин-4(3H)-один (47 мг; 0,169 ммоль) в n-BuOH (3 мл) был добавлен DIPEA (39 мг; 0,302 ммоль). Реагирующая смесь была нагрета для дистилляции и перемешивалась в течение последующих 24 часов. Реакция контролировалась посредством TLC (PE/EtOAc, v/v, 1/3). Смесь была остужена до комнатной температуры и сконцентрирована in vacuo. Осадок был растворен в EtOAc (15 мл), и полученная смесь была промыта водой (15 мл) и насыщенным минеральным раствором (10 мл). Отсепарированная органическая фаза была высушена над обезвоженным Na2SO4, и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (РЕ/EtOAc (v/v)=3/2) для получения соединения, указанного в заголовке шага, в виде бледно-желтого твердого вещества (66 мг; 96,4%).
MS (ESI, pos. ion) m/z: 453,1 [M+H]+;
1H NMR (600 МГц; CDCl3) δ (ppm): 8,93 (d; J=6,2 Гц; 1H); 8,16 (s; 1H); 7,69 (br s; 1H); 7,65-7,51 (m; 2H); 7,44 (s; 1H); 6,40-6,24 (m; 1H); 5,92 (s; 1H); 3,10 (s; 1H); 2,60 (d; J=63,7 Гц; 3H); 2,20-2,06 (m; 1H); 2,06-1,93 (m; 1H); 1,45 (s; 2H); 1,05 (t; J=6,7 Гц; 3H); 0,96-0,81 (m; 2H).
Пример 20 (S)-2-(1-((6-амин-5-(5-метил-1,3,4-оксадиазол-2-ил)пиримидин-4-ил)амин)пропил)-3-циклопропил-5-метилквиназолин-4(3H)-один
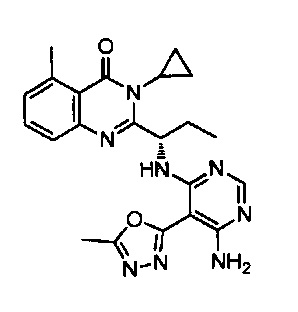
[449] К суспензии 6-хлор-5-(5-метил-1,3,4-оксадиазол-2-ил)пиримидин-4-амина (32 мг; 0,151 ммоль), (S)-2-(1-аминопропил)-3-циклопропил-5-метилквиназолин-4(3H)-один (42 мг; 0,163 ммоль) в n-BuOH (2 мл) был добавлен DIPEA (39 мг; 0,302 ммоль). Реагирующая смесь была нагрета для дистилляции и перемешивалась в течение последующих 24 часов. Реакция контролировалась посредством TLC (PE/EtOAc, v/v, 1/3). Реагирующая смесь была остужена до rt и сконцентрирована in vacuo. Осадок был растворен в EtOAc (15 мл), и полученная смесь была промыта водой (15 мл) и насыщенным минеральным раствором (10 мл). Отсепарированная органическая фаза была высушена над обезвоженным Na2SO4, и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (РЕ/EtOAc (v/v)=1/1) для получения соединения, указанного в заголовке шага, в виде бледно-желтого твердого вещества (44 мг; 67,3%).
MS (ESI, pos. ion) m/z: 433,2 [M+H]+; HPLC: 96%;
1H NMR (400 МГц; CDCl3) δ (ppm): 8,89 (d; J=7,6 Гц; 1H); 8,19 (s; 1H); 7,56 (t; J=7,7 Гц; 1H); 7,46 (d; J=8,0 Гц; 1H); 7,22 (d; J=7,3 Гц; 1H); 6,39-6,30 (m; 1H); 3,10-3,01 (m; 1H); 2,87 (s; 3H); 2,77 (s; 3H); 2,18-2,08 (m; 1H); 2,05-1,95 (m; 1H); 1,49-1,41 (m; 2H); 1,11-1,04 (m; 1H); 1,02 (t; 7=7,4 Гц; 3H); 0,93-0,89 (m; 1H).
Пример 21 (S)-2-(1-((6-амин-5-(3-метил-1,2,4-оксадиазол-5-ил)пиримидин-4-ил)амин)пропил)-3-циклопропил-5-метилквиназолин-4(3H)-один
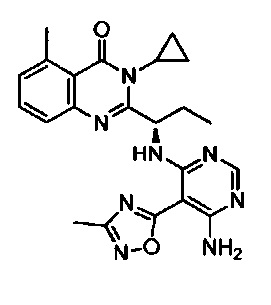
[450] К суспензии 6-хлор-5-(3-метил-1,2,4-оксадиазол-5-ил)пиримидин-4-амина (32 мг; 0,151 ммоль) и (S)-2-(1-аминопропил)-3-циклопропил-5-метилквиназолин-4(3H)-один (42 мг; 0,163 ммоль) в n-BuOH (2 мл) был добавлен DIPEA (39 мг; 0,302 ммоль). Реагирующая смесь была нагрета для дистилляции и перемешивалась в течение последующих 22 часов. Реакция контролировалась посредством TLC (PE/EtOAc, v/v, 1/3). Реагирующая смесь была остужена до rt и сконцентрирована in vacuo. Осадок был растворен в EtOAc (15 мл), и полученная смесь была промыта водой (15 мл) и насыщенным минеральным раствором (10 мл). Отсепарированная органическая фаза была высушена над обезвоженным Na2SO4, и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (РЕ/EtOAc (v/v)=2/1) для получения соединения, указанного в заголовке шага, в виде бледно-желтого твердого вещества (56 мг; 85,6%).
MS (ESI, pos. ion) m/z: 433,3 [M+H]+; HPLC: 98%;
1H NMR (400 МГц; CDCl3) δ (ppm): 9,05 (d; J=7,6 Гц; 1H); 8,17 (s; 1H); 7,63-7,45 (m; 2H); 7,20 (d; J=7,0 Гц; 1H); 6,33 (td; J=7,3; 5,5 Гц; 1H); 3,13-3,01 (m; 1H); 2,87 (s; 3H); 2,55 (s; 3H); 2,19-2,10 (m; 1H); 2,05-1,95 (m; 1H); 1,45-1,40 (m; 2H); 1,15-1,07 (m; 1H); 1,02 (t; J=7,4 Гц; 3H); 0,93-0,89 (m; 1H).
Пример 22 (S)-2-(1-((6-амин-5-(3-метил-1,2,4-оксадиазол-5-ил)пиримидин-4-ил)амин)пропил)-3-(2-фторфенил)квиназолин-4(3H)-один
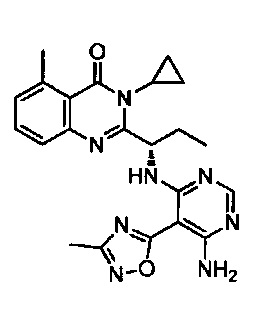
[451] К суспензии 6-хлор-5-(3-метил-1,2,4-оксадиазол-5-ил)пиримидин-4-амина (31 мг; 0,147 ммоль), (S)-2-(1-аминопропил)-3-(2-фторфенил)квиназолин-4(3H)-один (48 мг; 0,161 ммоль) в n-BuOH (2 мл) был добавлен DIPEA (38 мг; 0,293 ммоль). Реагирующая смесь была нагрета для дистилляции и перемешивалась в течение последующих 13 часов. Реакция контролировалась посредством TLC (РЕ/EtOAc, v/v, 2/3). Реагирующая смесь остужена до rt и сконцентрирована in vacuo. Осадок был растворен в EtOAc (15 мл), и полученная смесь была промыта водой (15 мл) и насыщенным минеральным раствором (10 мл). Отсепарированная органическая фаза была высушена над обезвоженным Na2SO4, и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (РЕ/EtOAc (v/v)=3/1) для получения соединения, указанного в заголовке шага, в виде грязно-белого твердого вещества (65 мг; 93,9%), состоящего из двух изомеров (изомер А и изомер В) в пропорции 5/4 (А/В).
MS (ESI, pos. ion) m/z: 473,3 [M+H]+; HPLC: 98% (общая степень чистоты изомера А и В);
Изомер A: 1H NMR (400 МГц; CDCl3) δ (ppm): 8,79 (d; J=7,5 Гц; 1H); 8,31 (d; J=7,8 Гц; 1H); 8,04 (s; 1H); 7,90-7,69 (m; 2H); 7,66-7,46 (m; 3H); 7,45-7,30 (m; 2Н); 5,36-5,16 (m; 1Н); 2,55 (s; 3H); 2,00-1,78 (m; 2H); 0,93-0,86 (m; 3H);
Изомер В: 1H NMR (400 МГц; CDCl3) δ (ppm): 8,79 (d; J=7,5 Гц; 1H); 8,31 7,8 Гц; 1H); 7,94 (s; 1H); 7,90-7,69 (m; 2H); 7,66-7,46 (m; 3H); 7,45-7,30 (m; 2H); 5,36-5,16 (m; 1H); 2,53 (s; 3H); 2,00-1,78 (m; 2H); 0,93-0,87 (m; 3H).
Пример 23 (S)-2-(1-((6-амин-5-(3-метил-1,2,4-оксадиазол-5-ил)пиримидин-4-ил)амин)-пропил)-3-циклопропил-5-фторквиназолин-4(3H)-один
[452] К суспензии 6-хлор-5-(3-метил-1,2,4-оксадиазол-5-ил)пиримидин-4-амина (31 мг; 0,147 ммоль), (S)-2-(1-аминопропил)-5-фтор-3-циклопропилквиназолин-4(3H)-один (40 мг; 0,154 ммоль) в n-BuOH (2 мл) был добавлен DIPEA (38 мг; 0,294 ммоль).
Реагирующая смесь была нагрета для дистилляции и перемешивалась в течение последующих 13 часов. Реакция контролировалась посредством TLC (PE/EtOAc, v/v, 2/3). Реагирующая смесь была остужена до rt и сконцентрирована in vacuo. Осадок был растворен в EtOAc (15 мл), и полученная смесь была промыта водой (15 мл) и насыщенным минеральным раствором (10 мл). Отсепарированная органическая фаза была высушена над обезвоженным Na2SO4, и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (РЕ/EtOAc (v/v)=2/1) для получения соединения, указанного в заголовке шага, в виде бледно-желтого твердого вещества (49 мг; 76,6%).
MS (ESI, pos. ion) m/z: 437,2 [M+H]+; HPLC: 99%;
1HNMR (600 МГц; CDCl3) δ (ppm): 9,50 (d; J=6,1 Гц; 1H); 8,21 (s; 1H); 7,67 (dd; J=13,1; 7,8 Гц; 1H); 7,47 (d; J=8,1 Гц; 1H); 7,18-7,04 (m; 1H); 6,37 (dd; J=11,9; 6,6 Гц; 1H); 3,11-3,00 (m; 1H); 2,58 (s; 3H); 2,21-2,12 (m; 1H); 2,11-1,96 (m; 1H); 1,53-1,40 (m; 2H); 1,14-1,06 (m; 1H); 1,03 (t; ,7=7,3 Гц; 3H); 1,01-0,95 (m; 1H).
Пример 24
(S)-2-(1-((6-амин-5-(5-метил-1,3,4-оксадиазол-2-ил)пиримидин-4-ил)амин)пропил)-3-(2-фторфенил)квиназолин-4(3H)-один

Шаг 1) N-(2-фторфенил)-2-нитробензамид
[453] К желтой суспензии 2-нитробензойной кислоты (5,01 г; 30 ммоль) в толуоле (60 мл) был добавлен одной порцией SOCl2 (4,9 мл, 67,5 ммоль) при комнатной температуре. Реагирующая смесь перемешивалась в течение 10 часов при 110°C, затем была сконцентрирована in vacuo. Осадок был растворен в 1,4-диоксане (30 мл), и по каплям была добавлена суспензия 2-фторанилина (3,36 г; 30 ммоль) и NaHCO3 (6,34 г; 75,5 ммоль) в 1,4-диоксане (30 мл) при 5°C. Полученная смесь перемешивалась при комнатной температуре в течение 24 часов. Затем было добавлено 200 мл воды для резкого охлаждения реакции, и полученная смесь была профильтрована. Фильтрат был промыт водой (100 мл×2) и высушен in vacuo при 50°C для получения соединения, указанного в заголовке шага, в виде бледно-желтого твердого вещества (7,55 г; 97%).
MS (ESI, pos. ion.) m/z: 261,1 [M+H]+;
1HNMR (400 МГц; DMSO-d6) δ (ppm): 10,52 (s; 1H); 8,22-8,10 (m; 1H); 7,94-7,83 (m; 2H); 7,83-7,69 (m; 2H); 7,37-7,17 (m; 3H).
Шаг 2) (S)-tert-butyl(1-(N-2-фторфенил)-2-нитробензоамидо)-1-оксобутан-2-ил)карбамат
[454] К суспензии N-(2-фторфенил)-2-нитробензоамида (5,20 г; 20 ммоль) в толуоле (60 мл) были добавлены одной порцией DMF (146 мг) и SOCl2 (18,88 г; 160 ммоль). После добавления реагирующая смесь перемешивалась при дистилляции в течение суток и была сконцентрирована in vacuo для получения бледно-коричневого масла для использования в следующем шаге без дополнительной очистки.
К раствору (S)-2-((терт-бутоксикарбонил)амин)бутановой кислоты (4,08 г; 20 ммоль) и DIPEA (7 мл, 42 ммоль) в безводном DCM (100 мл) была медленно добавлена суспензия упомянутого выше бледно-коричневого масла в DCM (100 мл) при 0°C. Полученная смесь перемешивалась при комнатной температуре в течение 24 часов, затем была промыта СН3СООН/Н2O (1/100 (v/v), 100 мл×3), насыщенным гидратогенным раствором NaHCO3 (100 мл) и насыщенным минеральным раствором (100 мл). Органическая фаза была высушена над обезвоженным Na2SO4, и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (PE/EtOAc (v/v)=5/1) для получения соединения, указанного в заголовке шага, в виде бледно-желтого твердого вещества (5,1 г; 57%).
MS (ESI, pos. ion.) m/z: 468,1 [M+Na]+.
Шаг 3) (S)-терт-бутил(1-(3-(2-фторфенил)-4-оксо-3,4-дигидроквиназолин-2-ил)пропил)-карбамат
[455] К раствору ((S)-терт-бутил (1-(N-(2-фторфенил)-2-нитробензоамидо)-1-оксобутан-2-ил)карбамата (5,1 г; 11,4 ммоль) в уксусной кислоте (58 мл) был добавлен одной порцией порошкообразный цинк (3,0 г; 45,8 ммоль). Реагирующая смесь перемешивалась в течение 24 часов при комнатной температуре, затем была профильтрована. Фильтрат был сконцентрирован in vacuo. Осадок был растворен в EtOAc (100 мл), и полученная смесь была промыта насыщенным гидратогенным раствором NaHCO3 (100 мл×2) и насыщенным минеральным раствором (100 мл×2). Органический слой был высушен над обезвоженным Na2SO4, и сконцентрирован in vacuo. Осадок был очищен хроматографией на силикагельной колонне (РЕ/EtOAc (v/v)=10/1) для получения соединения, указанного в заголовке шага, в виде желтого твердого вещества (2,7 г; 59%).
MS (ESI, pos. ion.) m/z: 398,2 [M+H]+.
Шаг 4) (S)-2-(1-аминопропил)-3-(2-фторфенил)квиназолин-4(3H)-один
[456] К раствору (S)-терт-бутил (1-(3-(2-фторфенил)-4-оксо-3,4-дигидроквиназолин-2-ил)пропил)карбамата (2,87 г; 7,94 ммоль) в EtOAc (10 мл) был добавлен одной порцией раствор HCl в EtOAc (3,5 М; 50 мл) при комнатной температуре.
Смесь перемешивалась при комнатной температуре на протяжении 2 часов. Полученная суспензия была растворена в воде (200 мл). Отсепарированная гидратогенная фаза была экстрактирована посредством EtOAc (100 мл×2), затем нейтрализована до рН=7 с помощью порошка NaHCO3 и вновь экстрактирована с помощью EtOAc/метанол (6/1 (v/v), 100 мл×3). Комбинированные органические фазы были промыты насыщенным минеральным раствором (100 мл), высушены над обезвоженным Na2SO4, и сконцентрированы in vacuo. Осадок был очищен хроматографией на силикагельной колонне (PE/EtOAc (v/v)=1/20) для получения соединения, указанного в заголовке шага, в виде желтого масла (2,02 г; 100%).
MS (ESI, pos. ion.) m/z: 298,2 [M+H]+;
1H NMR (400 МГц; DMSO-d6) δ (ppm): 8,14 (dd; J=7,9; 1,1 Гц; 1H); 7,90 (ddd; J=8,6; 7,3; 1,6 Гц; 1H); 7,75 (d; J=7,7 Гц; 1H); 7,67-7,60 (m; 2H); 7,60-7,48 (m; 2H); 7,46-7,40 (m; 1H); 3,16 (dd; J=7,5; 4,8 Гц; 1H); 1,85-1,73 (m; 1H); 1,47-1,33 (m; 1H); 0,75 (t; J=7,4 Гц; 3H).
Пример 5 (S)-2-(1-((6-амин-5-(5-метил-1,3,4-оксадиазол-2-ил)пиримидин-4-ил)амин)пропил)-3-(2-фторфенил)квиназолин-4(3H)-один
[457] К суспензии 6-хлор-5-(5-метил-1,3,4-оксадиазол-2-ил)пиримидин-4-амина (30 мг; 0,142 ммоль) и (S)-2-(1-аминопропил)-3-(2-фторфенил)квиназолин-4(3H)-один (44 мг; 0,149 ммоль) в n-BuOH (2 мл) был добавлен DIPEA (37 мг; 0,284 ммоль). Реагирующая смесь была нагрета для дистилляции и перемешивалась в течение последующих 19 часов. Реакция контролировалась посредством TLC (PE/EtOAc, v/v, 1/3). Реагирующая смесь остужена до it и сконцентрирована in vacuo. Осадок был растворен в EtOAc (15 мл), и полученная смесь была промыта водой (15 мл) и насыщенным минеральным раствором (10 мл). Органическая фаза была высушена над обезвоженным Na2SO4, и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (РЕ/EtOAc (v/v)=1/1) для получения соединения, указанного в заголовке шага, в виде бледно-желтого твердого вещества (42 мг; 62,7%), состоящего из двух изомеров (А и В) в пропорции 7/5 (изомер А/изомер В).
MS (ESI, pos. ion) m/z: 473,2 [M+H]+; HPLC: 97,1% (общая степень чистоты изомера А и В);
Изомер А: 1H NMR (600 МГц; DMSO-d6) δ (ppm): 8,74 (d; J=6,8 Гц; 1H); 8,15 (d; J=7,6 Гц; 1H); 7,96 (s; 1H); 7,91 (s; 1H); 7,75-7,68 (m; 2H); 7,68-7,50 (m; 3H); 7,49-7,35 (m; 1H); 7,21 (br s; 2H); 5,08-4,97 (m; 1H); 2,62 (s; 3H); 1,88-1,78 (m; 1H); 1,72-1,59 (m; 1H); 0,78 (t; J=6,9 Гц; 3H);
Изомер В: 1HNMR (600 МГц; DMSO-d6) δ (ppm): 8,63 d; J=7,8 Гц; 1H); 8,15 (d; J=7,6 Гц; 1H); 7,91 (s; 1H); 7,77 (s; 1H); 7,75-7,68 (m; 2H); 7,68-7,50 (m; 3H); 7,49-7,35 (m; 1H); 7,21 (br s; 2H); 5,08-4,97 (m; 1H); 2,59 (s; 3H); 1,88-1,78 (m; 1H); 1,72-1,59 (m; 1H); 0,84 (t; J=6,9 Гц; 3H).
Пример 25
(S)-2-(1-((6-амин-5-(5-метил-1,3,4-оксадиазол-2-ил)пиримидин-4-ил)амин)-пропил)-3-циклопропил-5-фторквиназолин-4(3H)-один
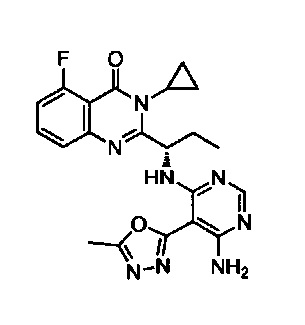
[458] К суспензии 6-хлор-5-(5-метил-1,3,4-оксадиазол-2-ил)пиримидин-4-амина (30 мг; 0,142 ммоль) и (S)-2-(1-аминопропил)-5-фтор-3-циклопропилквиназолин-4(3H)-один (40 мг; 0,153 ммоль) в n-BuOH (2 мл) был добавлен DIPEA (37 мг; 0,284 ммоль). Реагирующая смесь была нагрета для дистилляции и перемешивалась в течение последующих 23 часов. Реакция контролировалась посредством TLC (РЕ/EtOAc, v/v, 1/3). Реагирующая смесь была остужена до rt и сконцентрирована in vacuo. Осадок был растворен в EtOAc (15 мл), и полученная смесь была промыта водой (15 мл) и насыщенным минеральным раствором (10 мл). Органическая фаза была высушена над обезвоженным Na2SO4, и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (PE/EtOAc (v/v)=3/5) для получения соединения, указанного в заголовке шага, в виде грязно-белого твердого вещества (45 мг; 72,7%).
MS (ESI, pos. ion) m/z: 437,1 [M+H]+; HPLC: 98%;
1H NMR (400 МГц; CDCl3) δ (ppm): 8,54 (d; J=8,0 Гц; 1H); 8,16 (s; 1H); 7,63 (td; J=8,2; 5,4 Гц; 1H); 7,41 (d; J=8,2 Гц; 1H); 7,08 (dd; J=10,1; 8,6 Гц; 1H); 6,35 (td; J=7,6; 5,3 Гц; 1H); 3,16-3,04 (m; 1H); 2,74 (s; 3H); 2,18-2,06 (m; 1H); 2,05-1,94 (m; 1H); 1,49-1,39 (m; 2H); 1,22-1,13 (m; 1H); 1,05 (t; J=7,4 Гц; 3H); 0,94-0,81 (m; 1H).
Пример 26
(S)-2-(1-((6-амин-5-(1-метил-1H-1,2,4-триазол-3-ил)пиримидин-4-ил)амин)пропил)-3-циклопропил-5-фторквиназолин-4(3H)-один
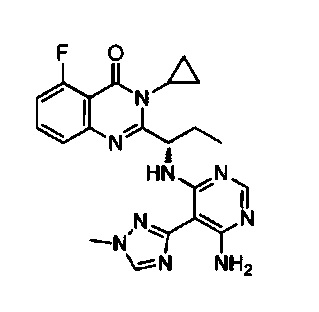
[459] К суспензии 6-хлор-5-(1-метил-1H-1,2,4-триазол-3-ил)пиримидин-4-амина (31 мг; 0,147 ммоль) и (S)-2-(1-аминопропил)-5-фтор-3-циклопропилквиназолин-4(3H)-один (41 мг; 0,157 ммоль) в n-BuOH (2 мл) был добавлен DIPEA (38 мг; 0,294 ммоль). Реагирующая смесь была нагрета для дистилляции и перемешивалась в течение последующих 29 часов. Реакция контролировалась посредством TLC (DCM/MeOH, v/v, 25/1). Реагирующая смесь была остужена до rt и сконцентрирована in vacuo. Осадок был растворен в EtOAc (15 мл), и полученная смесь была промыта водой (15 мл) и насыщенным минеральным раствором (10 мл). Органическая фаза была высушена над обезвоженным Na2SO4, и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (РЕ/EtOAc (v/v)=1/3) для получения соединения, указанного в заголовке шага, в виде бледно-желтого твердого вещества (49 мг; 76,5%).
MS (ESI, pos. ion) m/z: 436,2 [M+H]+; HPLC: 98,5%;
1H NMR (400 МГц; CDCl3) δ (ppm): 9,50 (d; J=7,5 Гц; 1H); 8,15 (s; 1H); 8,09 (s; 1H); 7,59 (dd; J=13,2; 7,7 Гц; 1H); 7,40 (d; J=8,1 Гц; 1H); 7,10-7,01 (m; 1H); 6,36-6,25 (m; 1H); 4,05 (s; 3H); 3,14 (s; 1H); 2,13-2,01 (m; 2H); 1,47-1,41 (m; 2H); 1,08 (t; J=7,2 Гц; 3H); 0,96-0,89 (m; 2H).
Пример 27
(S)-2-(1-((6-амин-5-(3-метил-1,2,4-оксадиазол-5-ил)пиримидин-4-ил)амин)-пропил)-5-метил-3-(о-толил)квиназолин-4(3H)-один
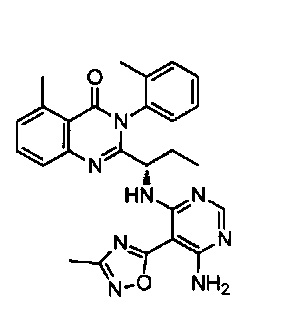
[460] К суспензии 6-хлор-5-(3-метил-1,2,4-оксадиазол-5-ил)пиримидин-4-амина (30 мг; 0,142 ммоль) и 2-(1-аминопропил)-5-метил-3-(о-толил)квиназолин-4(3H)-один (44 мг; 0,142 ммоль) в n-BuOH (2 мл) был добавлен DIPEA (37 мг; 0,284 ммоль). Реагирующая смесь была нагрета для дистилляции и перемешивалась в течение 22 часов, затем была остужена до rt и сконцентрирована in vacuo. Осадок был растворен в EtOAc (15 мл), и полученная смесь была промыта водой (15 мл) и насыщенным минеральным раствором (10 мл). Отсепарированная органическая фаза была высушена над обезвоженным Na2SO4, и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (РЕ/EtOАс (v/v)=5/1) для получения соединения, указанного в заголовке шага, в виде грязно-белого твердого вещества (56 мг; 81,9%), состоящего из двух изомеров (А и В) в пропорции 3/2 (изомер А/изомер В).
MS (ESI, pos. ion) m/z: 483,3 [M+H]+; HPLC: 97,0% (общая степень чистоты изомера А и В);
Изомер А: 1Н NMR (400 МГц; DMSO-d6) δ (ppm): 9,13 (d; J=7,9 Гц; 1H); 7,89 (s; 1H); 7,78-7,70 (m; 1H); 7,60-7,54 (m; 1H); 7,50-7,42 (m; 3H); 7,42-7,27 (m; 2H); 4,94 (td; J=7,5; 4,7 Гц; 1H); 2,73 (s; 3H); 2,49 (s; 3H); 1,96 (s; 3H); 1,89-1,43 (m; 2H); 0,74 (t; J=7,4 Гц; 3H).
Изомер В: 1H NMR (400 МГц; DMSO-d6) δ (ppm): 9,07 (d; 7=7,3 Гц; 1H); 7,95 (s; 1H); 7,78-7,70 (m; 1H); 7,60-7,54 (m; 1H); 7,50-7,42 (m; 3H); 7,42-7,27 (m; 2H); 5,17-5,07 (m; 1H); 2,73 (s; 3H); 2,48 (s; 3H); 2,10 (s; 3H); 1,89-1,43 (m; 2H); 0,74 (t;J=7,4 Гц; 3H).
Пример 28
(S)-2-(1-((6-амин-5-(5-метил-2,4-оксадиазол-3-ил)пиримидин-4-ил)амин)-пропил)-5-метил-3-(о-толил)квиназолин-4(3H)-один
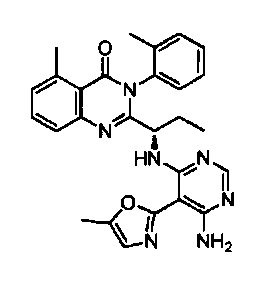
[461] К суспензии 6-хлор-5-(5-метилоксазол-2-ил)пиримидин-4-амина (30 мг; 0,142 ммоль) и 2-(1-аминопропил)-5-метил-3-(о-толил)квиназолин-4(3H)-один (44 мг; 0,142 ммоль) в n-BuOH (2 мл) был добавлен DIPEA (37 мг; 0.284 ммоль). Реагирующая смесь была нагрета для дистилляции в течение 21 часа и контролировалась посредством TLC (PE/EtOAc, v/v, 1/1). Реагирующая смесь была остужена до rt и сконцентрирована in vacuo. Осадок был растворен в EtOAc (15 мл), и полученная смесь была промыта водой (15 мл) и насыщенным минеральным раствором (10 мл). Отсепарированная органическая фаза была высушена над обезвоженным Na2SO4, и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (РЕ/EtOAc (v/v)=2/1) для получения соединения, указанного в заголовке шага, в виде грязно-белого твердого вещества (48 мг; 70,0%), состоящего из двух изомеров (А и В) в пропорции 4/5 (изомер А/изомер В).
MS (ESI, pos. ion) m/z: 482.2 [M+H]+; HPLC: 94,3% (общая степень чистоты изомера А и В);
Изомер А: 1Н NMR (400 МГц; DMSO-d6) δ (ppm): 9,19 (d; J=8,2 Гц; 1H); 7,84 (s; 1H); 7,75-7,67 (m; 1H); 7,59-7,49 (m; 1H); 7,48-7,38 (m; 3H); 7,38-7,27 (m; 2H); 7,25 (br s; 2H); 7,10 (s; 1H); 5,07-4,98 (m; 1H); 2,73 (s; 3H); 2,44 (s; 3H); 2,09 (s; 3H); 1,81-1,47 (m; 2H); 0,80-0,70 (m; 3H).
Изомер В: 1H NMR (400 МГц; DMSO-d5) δ (ppm): 9,19 (d; J=8,2 Гц; 1H); 7,77 (s; 1H);
7,75-7,67 (m; 1H); 7,59-7,49 (m; 1H); 7,48-7,38 (m; 3H); 7,38-7,27 (m; 2H); 7,25 (br s; 2H); 7,10 (s; 1H); 4,96-4,87 (m; 1H); 2,73 (s; 3H); 2,44 (s; 3H); 1,97 (s; 3H); 1,81-1,47 (m; 2H); 0,80-0,70 (m; 3H).
Пример 29
(S)-2-(1-((6-амин-5-(4-метил-2,4-оксадиазол-3-ил)пиримидин-4-ил)амин)-пропил)-5-метил-3-(о-толил)квиназолин-4(3H)-один
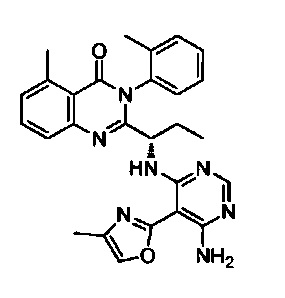
Шаг 1) 4,6-дихлорпиримидин-5-карбоксамид
[462] Раствор 4,6-дихлорпиримидин-5-карбонилхлорида (9,9 г; 46,82 ммоль) в THF (100 мл) был продут газообразным NH3. Реагирующая смесь перемешивалась при комнатной температуре на протяжении 20 минут. Реакция контролировалась посредством TLC (PE/EtOAc, v/v, 1/1). После завершения смесь была профильтрована и промыта THF (20 мл). Фильтрат был сконцентрирован in vacuo. Осадок был очищен хроматографией на силикагельной колонне (PE/EtOAc (v/v)=1/1) для получения соединения, указанного в заголовке шага, в виде бледно-желтого твердого вещества (6,2 г; 69,0%).
MS (ESI, pos. ion) m/z: 192,1 [M+H]+.
Шаг 2) 4,6-диметоксипиримидин-5-карбоксамид
[463] К раствору 4,6-дихлорпиримидин-5-карбоксамида (5,5 г; 28,65 ммоль) в безводном CH3OH (65 мл) был добавлен раствор метоксида натрия в CH3OH (30%, 12,9 г; 71,61 ммоль). Реагирующая смесь была нагрета для дистилляции и перемешивалась в течение 5 часов, затем была сконцентрирована in vacuo. Осадок был переведен в водную суспензию (50 мл) и полученная смесь нейтрализована до рН=6-7 гидратогенным раствором 4 М НСl. Реагирующая смесь была профильтрована. Фильтрат был промыт EtOAc (10 мл) и EtOH (5 мл) и высушен in vacuo для получения соединения, указанного в заголовке шага, в виде желтого твердого вещества (3,9 г; 74,3%).
MS (ESI, pos. ion) m/z: 184,1 [M+H]+.
Шаг 3) 2-(4,6-диметоксипиримидин-5-ил)-5-метил-4-метилоксазол
[464] Суспензия 4,6-диметоксипиримидин-5-карбоксамида (500 мг; 2,73 ммоль) в 1-бром-2,2-диметоксипропане (5 мл) была нагрета до 130°C и перемешивалась в течение последующих 1,5 часов. Реакция контролировалась посредством TLC (РЕ/EtOAc, v/v, 2/1). После завершения смесь был разведена в EtOAc (30 мл), и полученная смесь была промыта водой (30 мл) и насыщенным минеральным раствором (30 мл). Органическая фаза была высушена над обезвоженным Na2SO4, и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (РЕ/EtOAc (v/v)=8/1) для получения соединения, указанного в заголовке шага, в виде белого твердого вещества (229 мг; 37,9%).
MS (ESI, pos. ion): 222,2 [M+H]+;
1HNMR (600 МГц; CDCl3) δ (ppm): 8,51 (s; 1H); 7,53 (d; J=1,0 Гц; 1H); 7,28 (s; 1H); 4,04 (s; 6H); 2,29 (s; 3H);
13C NMR (151 МГц; CDCl3) δ (ppm): 168,74 (s); 157,80 (s); 154,00 (s); 137,37 (s); 135,04 (s); 94,71 (s); 54,97 (s); 11,72 (s).
Шаг 4) 2-(4,6-дихлорпиримидин-5-ил)-4-метоксазол
[465] К суспензии 2-(4,6-диметоксипиримидин-5-ил)-4-метилоксазол (549 мг; 2,48 ммоль) в безводном толуоле (20 мл) были добавлены РОСl3 (2.3 мл, 24.8 ммоль) и DMF (0,5 мл, 6,46 ммоль). Реагирующая смесь была нагрета для дистилляции и перемешивалась в течение последующих 18 часов, затем была сконцентрирована in vacuo. Осадок был растворен в EtOAc (100 мл), и полученная смесь была промыта водой (100 мл). Отсепарированная водосодержащая фаза была экстрактирована посредством EtOAc (100 мл×2). Комбинированные органические фазы были промыты насыщенным минеральным раствором (100 мл), высушены над обезвоженным Na2SO4, и сконцентрированы in vacuo. Осадок был очищен хроматографией на силикагельной колонне (PE/EtOAc (v/v)=1/4) для получения соединения, указанного в заголовке шага, в виде грязно-белого твердого вещества (257 мг; 45,1%).
MS (ESI, pos. ion) m/z: 230,1 [M+H]+;
1H NMR (400 МГц; CDCl3) δ (ppm): 8,89 (s; 1H); 7,64 (d; J=1,2 Гц; 1H); 2,34 (d; J=1,2 Гц; 3H).
Шаг 5) 6-хлор-5-(4-метилоксазол-2-ил)пиримидин-4-амин
[466] Раствор 2-(4,6-дихлорпиримидин-5-ил)-4-метилоксазола (550 мг; 2,39 ммоль) в безводном THF (15 мл) был продут газообразным NH3. Реагирующая смесь перемешивалась при комнатной температуре на протяжении 1 часа. Реакция контролировалась посредством TLC (PE/EtOAc, v/v, 2/1). После завершения смесь была профильтрована, фильтрат промыт THF (20 мл). Фильтрат был сконцентрирован in vacuo. Осадок был очищен хроматографией на силикагельной колонне (РЕ/EtOAc (v/v)=5/1) для получения соединения, указанного в заголовке шага, в виде грязно-белого твердого вещества (331 мг;65,7%).
MS (ESI, pos. ion) m/z: 211,1 [M+H]+;
1HNMR (400 МГц; CDCl3) δ (ppm): 9,16 (s; 1H); 8,33 (s; 1H); 7,56 (d; J=1,2 Гц; 1H); 5,93 (s; 1H); 2,29 (d;, 7=1,2 Гц; 3H).
Пример 6
(S)-2-(1-((6-амин-5-(4-метилоксазол-2-ил)пиримидин-4-ил)амин)пропил)-5-метил-3-(о-толил)квиназолин-4(3H)-один
[467] К суспензии 6-хлор-5-(4-метилоксазол-2-ил)пиримидин-4-амина (31 мг; 0,147 ммоль) и 2-(1-аминопропил)-5-метил-3-(о-толил)квиназолин-4(3H)-один (48 мг; 0,155 ммоль) в n-BuOH (3 мл) был добавлен DIPEA (38 мг; 0,294 ммоль). Реагирующая смесь была нагрета для дистилляции и перемешивалась в течение последующих 22 часов. Реакция контролировалась посредством TLC (PE/EtOAc, v/v, 1/1). После завершения реагирующая смесь была остужена до rt и сконцентрирована in vacuo. Осадок был растворен в EtOAc (15 мл), и полученная смесь была промыта водой (15 мл) и насыщенным минеральным раствором (10 мл). Отсепарированная органическая фаза была высушена над обезвоженным Na2SO4, и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (РЕ/EtOAc (v/v)=2/1) для получения соединения, указанного в заголовке шага, в виде бледно-желтого твердого вещества (55 мг; 77,6%), состоящего из двух изомеров (А и В) в пропорции 3/2 (изомер А/изомер В).
MS (ESI, pos. ion) m/z: 482,3 [M+H]+; HPLC: 93,1% (общая степень чистоты изомера А и В);
Изомер А: 1Н NMR (400 МГц; DMSO-d6) δ (ppm): 9,32 (d; J=8,1 Гц; 1H); 7,95 (d;, 7=1,3 Гц; 1Н); 7,80 (s; 1H); 7,76-7,67 (m; 1H); 7,58 (t; J=7,8 Гц; 1H); 7,50-7,38 (m; 3H); 7,37-7,27 (m; 2H); 4,96-4,87 (m; 1H); 2,74 (s; 3H); 2,25 (d; J=1,1 Гц; 3H); 1,97 (s; 3H); 1,83-1,48 (m; 2H); 0,76 (t; J=7,3 Гц; 3H).
Изомер В: 1H NMR (400 МГц; DMSO-d6) δ (ppm): 9,23 (d; J=7,2 Гц; 1H); 7,97 (d;,7=1,3 Гц; 1H); 7,87 (s; 1H); 7,76-7,67 (m; 1H); 7,58 (t; J=7,8 Гц; 1H); 7,50-7,38 (m; 3H); 7,37-7,27 (m; 2H); 5,13-5,02 (m; 1H); 2,74 (s; 3H); 2,24 (d; J=1,1 Гц; 3H); 2,10 (s; 3H); 1,83-1,48 (m; 2H); 0,76 (t; J=7,3 Гц; 3H).
Пример 30 (S)-2-(1-((6-амин-5-(1-метил-H-1,2,4-триазол-3-ил)пиримидин-4-ил)амин)-пропил)-5-метил-3-(о-толил)квиназолин-4(3H)-один
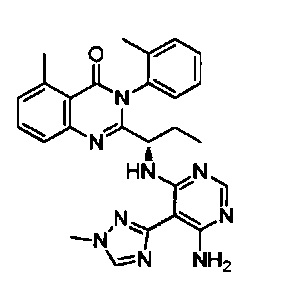
[468] К суспензии 6-хлор-5-(1-метил-1H-1,2,4-триазол-3-ил)пиримидин-4-амина (30 мг; 0,142 ммоль) и (S)-2-(1-аминопропил)-5-метил-3-(о-толил)квиназолин-4(3H)-один (46 мг; 0,150 ммоль) в n-BuOH (2 мл) был добавлен DIPEA (37 мг; 0,284 ммоль). Реагирующая смесь была нагрета для дистилляции и перемешивалась в течение последующих 22 часов. Реакция контролировалась посредством TLC (DCM/MeOH, v/v, 100/3). После завершения реагирующая смесь была остужена до rt и сконцентрирована in vacuo. Осадок был растворен в EtOAc (15 мл), и полученная смесь была промыта водой (15 мл) и насыщенным минеральным раствором (10 мл). Органическая фаза была высушена над обезвоженным Na2SO4, и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (РЕ/EtOAc (v/v)=1/1) для получения соединения, указанного в заголовке шага, в виде белого твердого вещества (53 мг; 77%), состоящего из двух изомеров (А и В) в пропорции 3/2 (изомер А/изомер В).
MS (ESI, pos. ion) m/z: 482.2 [M+H]+; HPLC: 95,4% (общая степень чистоты изомера А и В);
Изомер А: 1Н NMR (400 МГц; DMSO-d6) δ (ppm): 9,53 (d; J=8,2 Гц; 1H); 8,74 (s; 1Н); 8,13 (s; 1H); 7,74 (s; 1H); 7,72-7,65 (m; 1H); 7,55 (s; 1H); 7,49-7,37 (m; 3H); 7,38-7,24 (m; 2H); 7,06 (s; 1H); 4,96-4,88 (m; 1H); 4,01 (s; 3H); 2,73 (s; 3H); 1,98 (s; 3H); 1,86-1,51 (m; 2H); 0,81-0,73 (m; 3H).
Изомер В: 1H NMR (400 МГц; DMSO-d6) δ (ppm): 9,49 (d; J=7,5 Гц; 1H); 8,74 (s; 1H); 8,13 (s; 1H); 7,82 (s; 1H); 7,72-7,65 (m; 1H); 7,53 (s; 1H); 7,49-7,37 (m; 3H); 7,38-7,24 (m; 2H); 7,06 (s; 1H); 5,06-4,98 (m; 1H); 4,00 (s; 3H); 2,73 (s; 3H); 2,09 (s; 3H); 1,86-1,51 (m; 2H); 0,81-0,73 (m; 3H).
Пример 31
(S)-2-(1-((6-амин-5-(5-метил-1,3,4-оксадиазол-2-ил)пиримидин-4-ил)амин)пропил)-5-метил-3-(о-толил)квиназолин-4(3H)-один
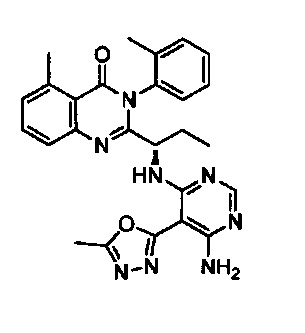
[469] К суспензии 6-хлор-5-(5-метил-1,3,4-оксодиазол-2-ил)пиримидин-4-амина (30 мг; 0,142 ммоль) и (S)-2-(1-аминопропил)-5-метил-3-(о-толил)квиназолин-4(3H)-один (46 мг; 0,149 ммоль) в n-BuOH (2 мл) был добавлен DIPEA (37 мг; 0.284 ммоль). Реагирующая смесь была нагрета для дистилляции и перемешивалась в течение последующих 15 часов. Реакция контролировалась посредством TLC (PE/EtOAc, v/v, 1/3). После завершения реагирующая смесь была остужена до rt и сконцентрирована in vacuo.
Осадок был растворен в EtOAc (15 мл), и полученная смесь была промыта водой (15 мл) и насыщенным минеральным раствором (10 мл). Отсепарированная органическая фаза была высушена над обезвоженным Na2SO4, и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (РЕ/EtOAc (v/v)=2/1) для получения соединения, указанного в заголовке шага, в виде белого твердого вещества (53 мг; 77,5%), состоящего из двух изомеров (А и В) в пропорции 4/5 (изомер А/изомер В).
MS (ESI, pos. ion) m/z: 483,2 [M+H]+; HPLC: 97,3% (общая степень чистоты изомера А и В);
Изомер A: 1H NMR (400 МГц; DMSO-d6) δ (ppm): 8,75 (d; J=8,3 Гц; 1H); 7,89 (s; 1H); 7,76-7,67 (m; 1H); 7,58-7,49 (m; 1H); 7,49-7,39 (m; 3H); 7,37-7,31 (m; 1H); 7,30-7,21 (m; 1H); 5,05-4,98 (m; 1H); 2,73 (s; 3H); 2,61 (s; 3H); 2,10 (s; 3H); 1,90-1,48 (m; 2H); 0,79-0,70 (m; 3H).
Изомер В: 1H NMR (400 МГц; DMSO-d6) δ (ppm): 8,73 (d; J=7,6 Гц; 1H); 7,83 (s; 1H); 7,76-7,67 (m; 1H); 7,58-7,49 (m; 1H); 7,49-7,39 (m; 3H); 7,37-7,31 (m; 1H); 7,30-7,21 (m; 1H); 4,94-4,87 (m; 1H); 2,73 (s; 3H); 2,61 (s; 3H); 1,98 (s; 3H); 1,90-1,48 (m; 2H); 0,79-0,70 (m; 3H).
Пример 32
(S)-2-(1-((6-амин-5-(1-метил-1H-1,2,4-триазол-3-ил)пиримидин-4-ил)амин)-пропил)-5-хлор-3-фенилквиназолин-4(3H)-один
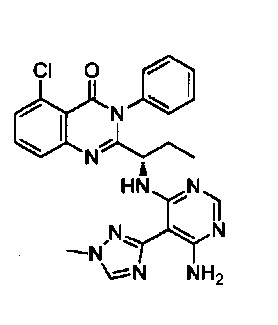
[470] К суспензии 6-хлор-5-(1-метил-1H-1,2,4-триазол-3-ил)пиримидин-4-амина (30 мг; 0,142 ммоль) и (5)-2-(1-аминопропил)-5-хлор-3-фенилквиназолин-4(3H)-один (47 мг; 0,150 ммоль) в n-BuOH (1,5 мл) был добавлен DIPEA (37 мг; 0.285 ммоль). Полученная смесь нагревалась при дистилляции в течение 24 часов. Реакция контролировалась посредством TLC (CH2Cl2/МеОН, v/v, 100/3). После завершения смесь была остужена до комнатной температуры и сконцентрирована in vacuo. Осадок был растворен в EtOAc (20 мл), и полученная смесь была промыта водой (10 мл) и насыщенным минеральным раствором (10 мл). Органическая фаза была высушена над обезвоженным Na2SO4, и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (PE/EtOAc (v/v)=1/4) для получения соединения, указанного в заголовке шага, в виде белого твердого вещества (41 мг; 59,0%).
MS (ESI, pos. ion): 488,1 [M+H]+; HPLC: 97,8%;
1H NMR (600 МГц; CDCl3) δ (ppm): 9,35 (s; 1H); 8,14 (s; 1H); 7,96 (s; 1H); 7,63-7,48 (m; 6H); 7,48-7,31 (m; 2H); 5,02 (s; 1H); 4,09 (d; J=59,2 Гц; 3H); 2,01-1,92 (m; 2H); 0,92 (t; J=7,1 Гц; 3H).
Пример 33
(S)-2-(1-((6-амин-5-(5-метил-1,2,4-оксадиазол-3-ил)пиримидин-4-ил)амин)-этил)-5-хлор-3-циклопропилквиназолин-4(3H)-один
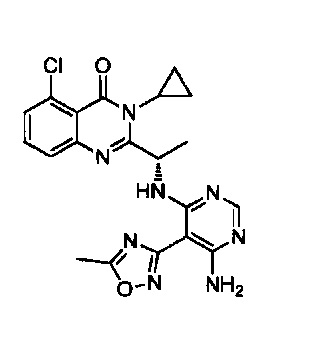
Шаг 1) 2-хлор-N-циклопропил-6-нитробензамид
[471] К суспензии 2-метил-6-нитробензойной кислоты (5,0 г; 24.8 ммоль) в толуоле (50 мл) был добавлен одной порцией SOCl2 (5,85 г; 49,6 ммоль) при комнатной температуре. После добавления реагирующая смесь перемешивалась при 110°C в течение 6 часов и была сконцентрирована in vacuo для получения желтого масла, которое было растворено в 1,4-диоксане (30 мл) для получения бледно-желтой суспензии. Полученная бледно-желтая суспензия была по каплям добавлена к суспензии циклопропанамина (1,42 г; 24.8 ммоль) и NaHCO3 (4,17 г; 49,6 ммоль) в 1,4-диоксане (30 мл) при 0°C. Полученная смесь перемешивалась при комнатной температуре в течение суток, затем медленно была добавлена вода (350 мл) для образования суспензии. Осадок был собран фильтрацией, промыт водой (100 мл) и высушен in vacuo при 50°C для получения соединения, указанного в заголовке шага, в виде бледно-желтого твердого вещества (3,9 г; 65.36%).
MS (ESI, pos. ion.) m/z: 241,0 [M+H]+;
1H NMR (600 МГц; CDCl3) δ (ppm): 8,09 (d; J=8,2 Гц; 1H); 7,73 (d; J=7,8 Гц; 1H); 7,52 (t; J=8,2 Гц; 1H); 5,95 (s; 1H); 2,95 (m; 1H); 0,92 (m; 2H); 0,76 (m; 2H).
Шаг 2) (S)-терт-бутил(1-(2-хлор-N-циклопропил-6-нитробензоамидо)-1-оксопропан-2-ил)-карбамат
[472] К суспензии 2-хлор-N-циклопропил-6-нитробензойной кислоты (5,31 г; 22,1 ммоль) в толуоле (50 мл) был добавлен одной порцией SOCl2 (6.4 мл, 88.4 ммоль) при комнатной температуре. Реагирующая смесь перемешивалась при 120°C в течение суток и была сконцентрирована in vacuo для получения бледно-коричневого масла, которое было растворено в безводном DCM (50 мл) для получения бледно-желтого раствора.
К раствору (S)-2-((терт-бутоксикарбонил)амин)пропановой кислоты (4,18 г; 22,1 ммоль) и DIPEA (5,71 г; 44,2 ммоль) в 100 мл безводного DCM был медленно добавлен упомянутый выше бледно-желтый раствор при 0°C. Полученный раствор перемешивался при комнатной температуре в течение 24 часов, затем был промыт водой (100 mL) и насыщенным минеральным раствором (100 мл). Органическая фаза была высушена над обезвоженным Na2SO4, и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (РЕ/EtOAc (v/v)=7/1) для получения соединения, указанного в заголовке шага, в виде желтого твердого вещества (6,58 г; 72,4%).
MS (ESI, neg. ion.) m/z: 410,0 [M-H]-.
Шаг 3) (S)-терт-бутил(1-(5-хлор-3-циклопропил-4-оксо-3,4-дигидроквиназолин-2-ил)пропил)-карбамат
[473] К раствору (S)-терт-бутил (1-(2-хлор-N-циклопропил-6-нитробензоамидо)-1-оксопропан-2-ил)карбамата (6,58 г; 16,0 ммоль) в уксусной кислоте (32 мл) был добавлен одной порцией порошкообразный цинк (4,16 г; 63,6 ммоль). Реагирующая смесь перемешивалась в течение 24 часов при 35°C, затем была профильтрована. Фильтрат был сконцентрирован in vacuo. Осадок был растворен в EtOAc (300 мл), и полученная смесь была промыта насыщенным гидратогенным раствором NaHCO3 (100 мл×2) и насыщенным минеральным раствором (200 мл). Отсепарированный органический слой был высушен над обезвоженным Na2SO4, и сконцентрирован in vacuo. Осадок был очищен хроматографией на силикагельной колонне (PE/EtOAc (v/v)=8/1) для получения соединения, указанного в заголовке шага, в виде бледно-желтого твердого вещества (4,74 г; 81,25%).
MS (ESI, pos. ion.) m/z: 364,2 [M+H]+.
Шаг 4) (S)-2-(1-аминоэтил)-5-хлор-3-циклопропилквиназолин-4(3H)-один
[474] К раствору (S)-терт-бутил (1-(5-хлор-3-циклопропил-4-оксо-3,4-дигидроквиназолин-2-ил)этил)карбамата (4,7 г; 12,9 ммоль) в EtOAc (10 мл) был добавлен одной порцией раствор HCl в EtOAc (10 мл; 3,5 M) при комнатной температуре. Реагирующую смесь перемешивали в течение суток при комнатной температуре. Полученная суспензия была разведена в воде (150 мл). Отсепарированная гидратогенная фаза была экстрактирована посредством EtOAc (30 мл×3), затем нейтрализована до pH=8 с помощью порошка NaHCO2 и вновь экстрактирована с помощью EtOAc (100 мл×4). Комбинированные органические фазы были промыты насыщенным минеральным раствором (200 мл), высушены над обезвоженным Na2SO4, и сконцентрированы in vacuo для получения соединения, указанного в заголовке шага, в виде бледно-желтого твердого вещества (2,57 г; 75,6%).
MS (ESI, pos. ion.) m/z: 264,2 [M+H]+;
1H NMR (400 МГц; CDCl3) δ (ppm): 7,54 (m; 2H); 7,43 (m; 1H); 4,87 (dd; J=13,0; 6,4 Гц; 1H); 2,94 (m; 1H); 1,52 (d, J=6,6 Гц, 3H), 1,38 (m, 2H), 0,95 (m, 2H).
Шаг 5 (S)-2-(1-((6-амин-5-(5-метил-1,2,4-оксадиазол-3-ил)пиримидин-4-ил)амин)этил)-5-хлор-3-циклопропилквиназолин-4(3H)-один
[475] К суспензии (S)-2-(1-аминоэтил)-5-хлор-3-циклопропилквиназолин -4(3H)-один (80 мг; 0.30 ммоль) и 6-хлор-5-(5-метил-1,2,4-оксадиазол-3-ил)пиримидин-4-амина (60 мг; 0,28 ммоль) в n-BuOH (5 мл) был добавлен DIPEA (90 мг; 0,70 ммоль). Смесь нагревалась при дистилляции в течение 24 часов. Реакция контролировалась посредством TLC (PE/EtOAc, v/v, 1/2). Смесь была остужена до комнатной температуры и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (PE/EtOAc (v/v)=1/2) для получения соединения, указанного в заголовке шага, в виде белого твердого вещества (67 мг; 51%).
MS (ESI, pos. ion): 439,2 [M+H]+; HPLC: 98,1%;
1H NMR (400 МГц; CDCl3) δ (ppm): 9,01 (d; J=6,7 Гц; 1H); 8,15 (s; 1H); 7,55 (d; J=3,9 Гц; 2H); 7,43 (d; J=3,9 Гц; 1H); 6,39-6,22 (m; 1H); 3,11 (s; 1H); 2,74 (s; 3H); 1,78 (s; 2H); 1,67 (d; J=6,4 Гц; 3H); 1,44 (m; 2H); 0,96 (m; 2H).
Пример 34 (S)-2-(1-((6-амин-5-(5-метил-1,3,4-оксадиазол-2-ил)пиримидин-4-ил)амин)пропил)-5-хлор-3-(3-фторфенил)квиназолин-4(3H)-один
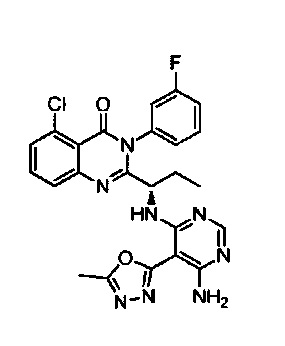
Шаг 1) 2-хлор-N-(3-фторфенил)-6-нитробензамид
[476] К суспензии 2-хлор-6-нитробензойной кислоты (5,0 г; 24,8 ммоль) в толуоле (25 мл) был добавлен SOCl2 (5,5 мл, 74,4 ммоль), и смесь перемешивалась при 110°C в течение суток, затем была сконцентрирована in vacuo для получения коричневого масла. К суспензии 3-фторанилина (4,1 г; 37,2 ммоль) и NaHCO3 (4.2 г; 49.6 ммоль) в 1,4-диоксане (15 мл) при 5°C был медленно добавлен раствор упомянутого выше коричневого масла в 1,4-диоксане (15 мл). Полученная смесь перемешивалась в течение суток при комнатной температуре и была сконцентрирована in vacuo. Осадок был разведен этилацетатом (200 мл) и водой (80 мл). Отсепарированная органическая фаза была промыта насыщенным минеральным раствором (150 мл×2), высушена над обезвоженным Na2SO4, и сконцентрирована in vacuo для получения соединения, указанного в заголовке шага, в виде бежевого твердого вещества (7,3 г; 100%).
MS (ESI, pos. ion) m/z: 295,1 [M+H]+;
1H NMR (600 MГц; CDCl3) δ (ppm): 8,18 (d; J=18,6 Гц; 1H); 7,86-7,73 (d; 1H); 7,70-7,46 (m; 3H); 7,36 (dd; J=14,6; 8,0 Гц; 1H); 6,92 (dd; J=25,7; 16,2; 1H); 3,70 (s; 1H).
Шаг 2) 2-амин-6-хлор-N-фторфенил)бензамид
[477] К суспензии 2-хлор-N-(3-фторфенил)-6-нитробензамида (4,0 г; 13,6 ммоль) в безводном этаноле (70 мл) были добавлены порошок Fe (3.8 г; 67.8 ммоль) и раствор HCOONH4 (8,23 г; 130,57 ммоль) в воде (14 мл). Полученная смесь перемешивалась в течение суток при 95°C, затем была профильтрована. Фильтрат был сконцентрирован in vacuo, и осадок был очищен хроматографией на силикагельной колонне (РЕ/EtOAc (v/v)=5/1) для получения соединения, указанного в заголовке шага, в виде бежевого твердого вещества (1,5 г; 42%).
MS (ESI, pos. ion) m/z: 265,1 [M+H]+;
1H NMR (600 МГц; DMSO-d6) δ (ppm): 10,65 (s; 1H); 7,74 (d; J=11,7 Гц; 1H); 7,45 (d; J=8,2 Гц; 1H); 7,37 (dd; J=15,1; 8,0 Гц; 1H); 7,10 (t; J=8,1 Гц; 1H); 6,93 (dd; J=11,9;4,9 Гц; 1H); 6,67 (dd; J=30,2; 7,9 Гц; 1H); 5,36 (s; 2H).
Шаг 3) (S)-терт-бутил-1-((3-хлор-2-((3-фторфенил)карбамоил)фенил)амин)-1-оксобутан-2-ил)-карбамат
[478] К раствору 2-амин-6-хлор-N-(3-фторфенил)бензамида (1,5 г; 5,67 ммоль) и Boc-L-2-аминобутировой кислоты (1.21 г; 5.95 ммоль) в DCM (20 мл) при -10°C были добавлены DIPEA (2,20 г; 17,01 ммоль) и HATU (2,59 г; 6,80 ммоль). Спустя 1 час смесь была нагрета для дистилляции и перемешивалась в течение суток, затем была промыта водой (150 мл×2) и насыщенным гидратогенным раствором NaHCO3 (150 мл×2). Отсепарированная органическая фаза была сконцентрирована in vacuo, и осадок был очищен хроматографией на силикагельной колонне (РЕ/EtOAc (v/v)=5/1) для получения соединения, указанного в заголовке шага, в виде бледно-желтого твердого вещества (1,7 г; 66,7%).
MS (ESI, pos. ion) m/z: 350,2 [M-Boc+H]+.
Шаг 4) (S)-терт-бутил(1-(5-хлор-3-(3-фторфенил)-4-оксо-3,4-дигидроквиназолин-2-ил)пропил)карбамат
[479] К раствору (S)-терт-бутил(1-((3-хлор-2-((3-фторфенил)карбамоил)фенил)амин)-1-оксобутан-2-ил)карбамата (1,6 г; 3,60 ммоль) и триэтиламина (15 мл, 108 ммоль) в CH3CN (100 мл) был добавлен шприцем N,O-бис(триметилсилил)ацетамид (13 мл, 36 ммоль) в атмосфере азота. Полученная смесь была нагрета до 85°C и перемешивалась в течение 33 часов, затем была остужена до комнатной температуры и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (РЕ/EtOAc (v/v)=8/1) для получения соединения, указанного в заголовке шага, в виде бледно-желтого твердого вещества (1,28 г; 82,3%).
MS (ESI, pos. ion) m/z: 432.1 [M+H]+.
Шаг 5) (S)-2-(1-аминопропил)-5-хлор-3-(3-фторфенил)квиназолин-4(3H)-один
[480] К суспензии (S)-терт-бутил(1-(5-хлор-(3-фторфенил)-4-оксо-3,4-дигидроквиназолин-2-ил)пропил)карб амата (1,28 г; 2,96 ммоль) в EtOAc (10 мл) был добавлен раствор HCl в EtOAc (3.0 М; 8 мл; 24,00 ммоль), смесь перемешивалась при комнатной температуре в течение 4 часов и была сконцентрирована in vacuo. Осадок был растворен в EtOAc (50 мл) и полученная смесь нейтрализована до рН=7-8 насыщенным гидратогенным раствором NaHCO3. Отсепарированная органическая фаза была промыта насыщенным минеральным раствором (100 мл×2), высушена над обезвоженным Na2SO4, и сконцентрирована in vacuo для получения соединения, указанного в заголовке шага, в виде грязно-белого твердого вещества (982 мг; 100%).
MS (ESI, pos. ion) m/z: 332,1 [M+H]+;
1H NMR (400 МГц; DUSO-d6) δ (ppm): 7,79 (t; J=8,0 Гц; 1H); 7,67-7,61 (m; 2H); 7,57 (d; J=1,1 Гц; 1H); 7,50 (d; J=9,6 Гц; 1H); 7,46-7,36 (m; 2H); 7,33 (d; J=1,9 Гц; 1H); 3,17-3,15 (s; 1H); 1,40 (m; 2H); 0,72 (t; J=7,3 Гц; 3H).
Пример 6 (S)-2-(1-((6-амин-5-(5-метил-1,3,4-оксадиазол-2-ил)пиримидин-4-ил)амин)пропил)-5-хлор-3-(3-фторфенил)квиназолин-4(3H)-один
[481] К суспензии (S)-2-(1-аминопропил)-5-хлор-3-(3-фторфенил)квиназолин-4(3H)-один (98.9 мг; 0.298 ммоль) и 6-хлор-5-(5-метил-1,3,4-оксадиазол-2-ил)пиримидин-4-амина (60 мг; 0,284 ммоль) в n-BuOH (5 мл) был добавлен DIPEA (0.15 мл, 0.568 ммоль). Смесь перемешивалась при 120°C в течение суток, затем была профильтрована, и фильтрат промыт 1 мл этанола получения указанного в заголовке соединения в виде белого твердого вещества (40 мг; 27,7%).
MS (ESI, pos. ion) m/z: 507,1 [M+H]+; HPLC: 99,8%;
1H NMR (600 МГц; DMSO-d6) δ (ppm): 8,64 (d; J=7,1 Гц; 1H); 7,95 (d; J=15,8 Гц; 1Н); 7,82-7,73 (m; 1H); 7,72-7,54 (m; 3H); 7,44 (m; 2H); 7,21 (s; 2H); 4,87-4,76 (m; 1H); 2,62 (s; 3H); 2,03-1,92 (m; 2H); 0,87-0,85 (t; J=12 Гц; 3H).
Пример 35 (S)-2-(1-((6-амин-5-(5-метил-1,3,4-оксадиазол-2-ил)пиримидин-4-ил)амин)пропил)-5-хлор-3-(о-толил)квиназолин-4(3H)-один
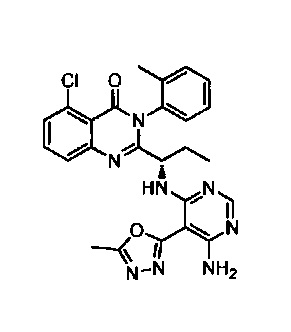
Шаг 1) 2-хлор-6-нитро-N-(о-толил)бензамид
[482] К суспензии 2-хлор-6-нитробензойной кислоты (5,0 г; 24,8 ммоль) в толуоле (25 мл) добавлен по каплям SOCl2 (5,5 мл, 74,4 ммоль) и DMF (3 мл) при комнатной температуре. После добавления реагирующая смесь перемешивалась при 110°C в течение суток и была сконцентрирована in vacuo для получения светло-коричневого масла. К суспензии о-толуидина (4.0 г; 37.2 ммоль) и NaHCO3 (4,2 г; 49,6 ммоль) в 1,4-диоксане (15 мл) при 5°C был добавлен раствор упомянутого выше светло-коричневого масла в 1,4-диоксане (15 мл). Смесь перемешивалась при комнатной температуре в течение 7 часов, затем была разведена этилацетатом (300 мл) и водой (300 мл). Отсепарированные органические фазы были промыты насыщенным минеральным раствором (200 мл), высушены над обезвоженным Na2SO4 и сконцентрированы in vacuo для получения бежевого твердого вещества, которое было очищено хроматографией на силикагельной колонне (DCM) для получения соединения, указанного в заголовке шага, в виде бледно-белого твердого вещества (6,3 г; 87,4%).
MS (ESI, pos. ion) m/z: 291,0 [M+H]+.
Шаг 2) 2-амин-6-хлор-N-(о-толил)бензамид
[483] К суспензии 2-хлор-6-нитро-N-(о-толил)бензамид (4,0 г; 13,7 ммоль)) в безводном этаноле (70 мл) были добавлены порошок Fe (3.8 г; 67.9 ммоль) и раствор HCOONH4 (8,7 г; 137,6 ммоль) в воде (14 мл). Полученная смесь была нагрета до 95°C и перемешивалась в течение суток, затем была разведена этилацетатом (400 мл) и водой (20 мл), и гидратогенная фаза была экстрактирована этилацетатом (100 мл×2). Комбинированные органические фазы были промыты насыщенным минеральным раствором (150 мл×3), высушены над обезвоженным Na2SO4, и сконцентрированы in vacuo. Осадок был очищен хроматографией на силикагельной колонне (РЕ/EtOAc (v/v)=5/1) для получения соединения, указанного в заголовке шага, в виде белого твердого вещества (2,7 г; 75,8%).
MS (ESI, pos. ion) m/z: 261,1 [M+H]+;
1H NMR (600 МГц; DMSO-d6) δ (ppm): 9,89 (s; 1H); 7,51 (d; J=7,8 Гц; 1H); 7,36-7,17 (m; 2H); 7,14 (t; J=7,4 Гц; 1H); 7,09 (t; J=8,0 Гц; 1H); 6,71 (d; J=8,2 Гц; 1H); 6,64 (d; J=18,0 Гц; 1H); 5,33 (s; 2H); 2,30 (s; 3H).
Шаг 3) (S)-терт-бутил(1-((3-хлор-2-(о-толилкарбамоил))фенил)амин)-1-оксобутан-2-ил)-карбамат
[484] К суспензии 2-амин-6-хлор-N-(о-толил)бензамида (2,7 г; 10,4 ммоль). Вос-L-2-аминобутировой кислоты (2,2 г; 10,8 ммоль) и DIPEA (5,4 мл, 31,1 ммоль) в DCM (40 мл) был добавлен HATU (4,7 г; 12,4 ммоль) при -10°C. Смесь перемешивалась при -10°C в течение 1 часа, была нагрета для дистилляции и перемешивалась в течение суток. Смесь была промыта водой (200 мл×2) и насыщенным гидратогенным раствором NaHCO3 (200 мл×2). Отсепарированная органическая фаза была высушена над обезвоженным Na2SO4, и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (EtOAc) для получения соединения, указанного в заголовке шага, в виде светло-желтого твердого вещества (3,25 г; 70,3%).
MS (ESI, pos. ion) m/z: 346,2 [M-Boc+H]+.
Шаг 4) (S)-терт-бутил(1-(5-хлор-4-оксо-3-(о-толил)-3,4-дигидроквиназолин-2-ил)пропил)-карбамат
[485] К раствору (S)-терт-бутил(1-(3-хлор-2-(о-толилкарбамоил)фенил)амин)-1-оксобутан-2-ил)карбамата (1,7 г; 3,80 ммоль) в CH3CN (120 мл) был добавлен триэтиламин (27 мл, 190 ммоль). Смесь была продута азотом, затем шприцем был добавлен N,O-бис(триметилсилил)ацетамид (19 rnL, 76.25 ммоль). После добавления смесь была нагрета до 90°C и перемешивалась в герметичной трубке в атмосфере азота в течение 3 дней, затем была сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (РЕ/EtOAc (v/v)=10/1) для получения соединения, указанного в заголовке шага, в виде желтого твердого вещества (1,26 г; 77%).
MS (ESI, pos. ion) m/z: 428,1 [M+H]+;
1H NMR (600 МГц; DMSO-d6) δ (ppm): 7,85-7,75 (m; 1H); 7,66 (dd; J=12,9; 8,2 Гц; 1H); 7,59 (dd; J=11,0; 8,0 Гц; 1H); 7,53-7,29 (m; 4H); 7,19 (dd; J=40,9; 7,9 Гц; 1H); 3,91-3,74 (m; 1H); 2,09(s; 3H); 1,56-1,46 (m; 2H); 1,35 (s; 9H); 0,66 (m; 3H).
Шаг 5) -2-(1-аминопропил)-5-хлор-3-(о-толил)квиназолин-4(3H)-один
[486] К суспензии (S)-терт-бутил(1-(5-хлор-4-оксо-3-(о-толил)-3,4-дигидроквиназолин-2-ил)-пропил)карбамата (1,24 г; 2,90 ммоль) в EtOAc (10 мл) был добавлен раствор HCl в EtOAc (3 М; 8 мл; 24,00 ммоль). Полученная смесь перемешивалась в течение суток при комнатной температуре и была сконцентрирована in vacuo. Осадок был растворен в EtOAc (100 мл) и полученная смесь нейтрализована до pH=7-8 насыщенным гидратогенным раствором NaHCO3. Отсепарированная гидратогенная фаза была экстрактирована с помощью EtOAc (100 мл×2), и комбинированные органические фазы были промыты насыщенным минеральным раствором (150 мл×2), затем сконцентрированы in vacuo для удаления примерно 90% растворителя с тем, чтобы получить концентрированный раствор, после чего в раствор по каплям был добавлен РЕ (50 мл). После добавки смесь перемешивалась при комнатной температуре в течение 30 минут, была профильтрована, и фильтрат был сконцентрирован in vacuo для получения соединения, указанного в заголовке шага, в виде бледно-коричневого твердого вещества (850 мг; 89%).
MS (ESI, pos. ion) m/z: 328,1 [M+H]+;
1H NMR (400 МГц; DMSO-d6) δ (ppm): 7,81-7,77 (t; J=8,0 Гц; 1H); 7,68-7,66 (m; 1H); 7,57-7,55 (m; 1H); 7,50-7,34 (m; 4H); 3,10-3,06 (t; J=6,6 Гц; 1H); 2,09 (s; 3H); 1,49-1,27 (m; 2H); 0,72 (m; 3H).
Пример 6 (S)-2-(1-((6-амин-5-(5-метил-1,3,4-оксадиазол-2-ил)пиримидин-4-ил)амин)пропил)-5-хлор-3-(о-толил)квиназолин-4(3H)-один
[487] К суспензии (S)-2-(1-аминопропил)-5-хлор-3-(3-(о-толил))квиназолин-4(3H)-один (98,9 мг; 0,298 ммоль) и 6-хлор-5-(5-метил-1,3,4-оксадиазол-2-ил)пиримидин-4-амина (60 мг; 0,284 ммоль) в n-BuOH (5 мл) был добавлен DIPEA (0.15 мл, 0.568 ммоль). Смесь была нагрета до 120°C и перемешивалась в течение суток, затем была профильтрована, и фильтрат промыт 1 мл этанола для получения соединения, указанного в заголовке шага, в виде белого твердого вещества (38,8 мг; 27,2%).
MS (ESI, pos. ion) m/z: 503,1 [M+H]+; HPLC: 99,0%;
1H NMR (400 МГц; DMSO-d6) δ (ppm): 8,93 (d; J=8,0 Гц; 1H); 7,92 (s; 1H); 7,84-7,78 (m; 1H); 7,69-7,57 (m; 2H); 7,54-7,38 (m; 5H); 4,93-4,88 (m; 1H); 2,61 (s; 3H); 2,01 (s; 3H); 1,87-1,54 (m; 2H); 0,75 (t; J=7,4 Гц; 3H).
Пример 36 (S)-2-(1-((6-амин-5-(2-метил-2H-тетразол-5-ил)пиримидин-4-ил)амин)пропил)-5-хлор-3-(3-фторфенил)квиназолин-4(3H)-один
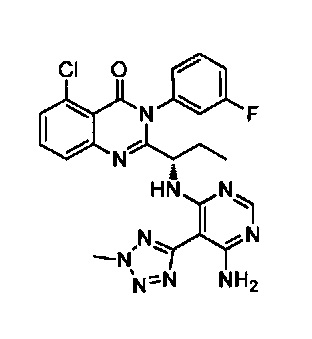
[488] Смесь (S)-2-(1-аминопропил)-5-хлор-3-(3-фторфенил)квиназолин-4(3H)-один (30 мг; 0,09 ммоль), 6-хлор-5-(2-метил-2H-тетразол-5-ил)пиримидин-4-амин (19 мг; 0,09 ммоль) и DIPEA (41 мг; 0.31 ммоль) в n-BuOH (1 мл) была нагрета до 130°C и перемешивалась в течение последующих 24 часов, затем была остужена до rt и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (DCM/MeOH (v/v)=100/1) для получения полуфабриката, затем был дополнительно очищен подготовительным TLC (DCM/MeOH, v/v, 25/1) для получения соединения, указанного в заголовке шага, в виде грязно-белого твердого вещества (27 мг; 59%).
MS (ESI, pos. ion) m/z: 503,1 [M+H]+; HPLC: 90%;
1H NMR (400 МГц; DMSO-d6) δ (ppm): 8,79-8,77 (d; J=6,8 Гц; 1H); 7,97-7,95 (d; J=8,8 Гц; 1H); 7,78-7,74 (dd; J=8,0; 7,8 Гц; 1H); 7,627,43 (m; 6H); 4,88-4,85 (m; 1H); 4,52 (s; 2H); 4,43 (s; 1H); 2,02-1,99 (m; 1H); 1,75-1,66 (m; 1H); 0,82-0,79 (dd; J=6,8; 6,0 Гц; 3H).
Пример 37 (S)-2-(1-((6-амин-5-(3-метил-1,2,4-оксадиазол-5-ил)пиримидин-4-ил)амин)-пропил)-5-метил-3-фенилквиназолин-4(3H)-один
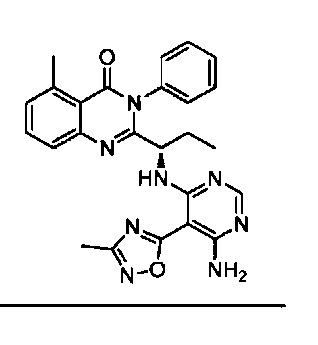
Шаг 1) 2-амин-6-нитро-N-фенилбензамид
[489] К желтой суспензии 2-метил-6-нитробензойной кислоты (5 г; 27,6 ммоль) в толуоле (50 мл) был добавлен одной порцией SOCl2 (6.51 г; 54.7 ммоль) при комнатной температуре. После добавления реагирующая смесь перемешивалась при 110°C в течение суток и была сконцентрирована in vacuo для получения желтого масла, которое было растворено в 1,4-диоксане (30 мл) для получения раствора. Раствор был по каплям добавлен к суспензии анилина (2,51 г; 27,6 ммоль) и NaHCO3 (5,85 г; 69,6 ммоль) в 1,4-диоксане (30 мл) при 5°C. Затем полученная смесь перемешивалась при комнатной температуре в течение 24 часов, после чего к смеси была добавлена вода (200 мл) для резкого охлаждения реакции. Смесь была экстрактирована посредством EtOAc (200 мл×3). Комбинированные органические фазы были промыты насыщенным минеральным раствором (200 мл) и сконцентрированы in vacuo. Осадок был очищен хроматографией на силикагельной колонне (PE/EtOAc (v/v)=8/3) для получения соединения, указанного в заголовке шага, в виде бледно-желтого твердого вещества (6,5 г; 92%).
MS (ESI, pos. ion) m/z: 257,1 [M+H]+;
1H NMR (600 МГц; CDCl3) δ (ppm): 7,98 (d; J=8,2 Гц; 1H); 7,75 (s; 1H); 7,56 (d; J=19,6 Гц; 3H); 7,45 (t; J=7,9 Гц; 1H); 7,37 (t; J=7,7 Гц; 2H); 7,20 (t; J=7,4 Гц; 1H);2,49(s; 3H).
Шаг 2) (S)-терт-бутил(1-(2-метил-6-нитро-N-фенилбензамидо)-1-окосбутан-2-ил)карбамат
[490] К суспензии 2-метил-6-нитро-N-фенилбензамида (6,5 г; 25,4 ммоль) в толуоле (100 мл) был добавлен по каплям SOCl2 (7,3 мл, 101,6 ммоль). После добавления реагирующая смесь перемешивалась при 120°C в течение 12 часов, затем была сконцентрирована in vacuo для получения коричневого масла, которое было непосредственно использовано в следующем шаге без дополнительной очистки.
К раствору Boc-L-2-аминобутировой кислоты (5,17 г; 25,4 ммоль) и DIPEA (9,85 г; 76,2 ммоль) в DCM (50 мл) был добавлен раствор упомянутого выше коричневого масла в DCM (50 мл) при 0°C. После добавления реагирующая смесь перемешивалась при rt в течение 24 часов и была промыта 4% гидратогенным раствором лимонной кислоты (100 мл×3), насыщенным гидратогенным раствором NaHCO3 (100 мл×2) и насыщенным минеральным раствором (100 мл). Отсепарированный органический слой был высушен над обезвоженным сульфатом натрия и сконцентрирован in vacuo. Осадок был очищен хроматографией на силикагельной колонне (РЕ/EtOAc (v/v)=9/1) для получения соединения, указанного в заголовке шага, в виде желтого масла (7,47 г; 66,54%).
MS (ESI, pos. ion) m/z: 464.,52 [M+Na]+.
Шаг 3) (S)-терт-бутил(1-(5-метил-4-оксо-3-фенил-3,4-дигидроквиназолин -2-ил)пропил)-карбамат
[491] К раствору (S)-терт-бутил (1-(2-метил-6-нитро-N-фенилбензоамидо)-1-оксобутан-2-ил)карбамата (7,37 г; 16,7 ммоль) в уксусной кислоте (30 мл) был добавлен одной порцией порошкообразный цинк (4,37 г; 66,8 ммоль). После добавления реагирующая смесь перемешивалась при 35°C в течение суток. Реагирующая смесь была отфильтрована и фильтрат сконцентрирован in vacuo. Осадок был растворен в EtOAc (200 мл), и полученная смесь была промыта насыщенным гидратогенным раствором NaHCO3 (200 мл×2), водой (200 мл) и насыщенным минеральным раствором (100 мл). Отсепарированный органический слой был высушен над обезвоженным сульфатом натрия и сконцентрирован in vacuo. Осадок был очищен хроматографией на силикагельной колонне (РЕ/EtOAc (v/v)=25/2) для получения соединения, указанного в заголовке шага, в виде бледно-желтого твердого вещества (2,82 г; 43%).
MS (ESI, pos. ion) m/z: 394.,2 [M+H]+;
1H NMR (600 МГц; DMSO-d6) δ (ppm): 7,68 (t; J=1,1 Гц; 1H); 7,63-7,55 (m; 2H); 7,52 (dd; J=17,9; 7,8 Гц; 2H); 7,46 (d; J=1,1 Гц; 1H); 7,38 (d; J=7,6 Гц; 1H); 7,29 (d; J=7,3 Гц; 1H); 7,20 (d; J=7,6 Гц; 1H); 3,95 (td; J=9,1; 3,8 Гц; 1H); 2,72 (s; 3H); 1,71 (m; 1H); 1,54 (m; 1H); 1,34 (s; 9H); 0,63 (t; 3H).
Шаг 4) (S)-2-(1-аминопропил)-5-метил-3-фенилквиназолин-4(3H)-один
[492] К раствору (S)-терт-бутил (1-(5-метил-4-оксо -3-фенил-3,4-дигидроквиназолин-2-ил)пропил)карбамата (2,82 г; 7,2 ммоль) в EtOAc (80 мл) был добавлен одной порцией раствор НС1 в EtOAc (3,5 М; 21 мл) при комнатной температуре. Реагирующую смесь перемешивали в течение суток при комнатной температуре.
Полученная суспензия была растворена в воде (300 мл). Отсепарированная гидратогенная фаза была экстрактирована посредством EtOAc (100 мл), затем нейтрализована до рН=8 с помощью порошка NaHCO3 и вновь экстрактирована с помощью EtOAc (150 мл×3). Комбинированные органические фазы были промыты насыщенным минеральным раствором (100 мл), высушены над обезвоженным Na2SO4, и сконцентрированы in vacuo для получения соединения, указанного в заголовке шага, в виде желтого порошка (2,05 г; 97,2%).
MS (ESI, pos. ion) m/z: 294,2 [M+H]+;
1H NMR (600 МГц; CDCl3) δ (ppm): 7,62 (dd; J=13,5; 6,0 Гц; 1H); 7,61-7,56 (m; 3H); 7,53 (t; J=7,4 Гц; 1H); 7,33-7,27 (m; 2H); 7,25 (d; J=7,2 Гц; 1H); 3,41 (dt; J=63,0; 31,5 Гц; 1H); 2,84 (s; 3H); 1,93-1,81 (m; 1H); 1,61-1,44 (m; 1H); 0,82 (t; J=7,4 Гц; 3H).
Пример 5 (S)-2-(1-((6-амин-5-(3-метил-1,2,4-оксадиазол-5-ил)пиримидин-4-ил)амин)-пропил)-5-метил-3-фенилквиназолин-4(3H)-один
[493] К суспензии (S)-2-(1-аминопропил)-5-метил-3-фенилквиназолин-4(3H)-один (59 мг; 0,2 ммоль) и 6-хлор-5-(3-метил-1,2,4-оксадиазол-5-ил)пиримидин-4-амин (42 мг; 0,2 ммоль) в n-BuOH (2 мл) был добавлен DIPEA (52 мг; 0,4 ммоль). Реагирующая смесь была нагрета для дистилляции и перемешивалась в течение последующих суток. Реакция контролировалась посредством TLC (PE/EtOAc, v/v, 1/3). Реагирующая смесь была остужена до rt и сконцентрирована in vacuo. Осадок был разведен EtOAc (10 мл), промыт водой (10 мл×2) и насыщенным минеральным раствором (10 мл). Отсепарированная органическая фаза была высушена над обезвоженным Na2SO4, и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (РЕ/EtOAc (v/v)=2/1) для получения соединения, указанного в заголовке шага, в виде грязно-белого твердого вещества (79 мг; 84,3%).
MS (ESI, pos. ion): 469,2 [M+H]+;
1H NMR (400 МГц; CDCl3) δ (ppm): 9,37 (d; J=7,3 Гц; 1H); 8,06 (s; 1H); 7,69-7,53 (m; 5H); 7,35 (dd; J=17,7; 9,4 Гц; 2H); 7,28 (m; 1H); 5,23 (m; 1H); 2,85 (s; 3H); 2,58 (s; 3H); 2,23 (m; 6,4 Гц; 1H); 1,65 (m; 1H); 0,86 (t; J=7,4 Гц; 3H).
Пример 38 (S)-2-(1-((6-амин-5-(5-метил-1,3,4-оксадиазол-2-ил)пиримидин-4-ил)амин)-пропил)-5-метил-3-фенилквиназолин-4(3H)-один
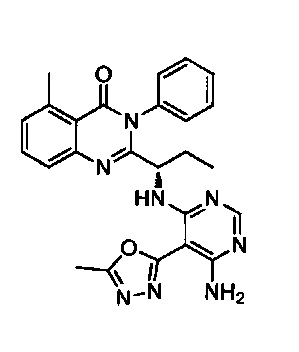
[494] К суспензии (S)-2-(1-аминопропил)-5-метил-3-фенилквиназолин-4(3H)-один (40 мг; 0.2 ммоль) и 6-хлор-5-(5-метил-1,3,4-оксадиазол-2-ил)пиримидин-4-амин (28,9 мг, 0,2 ммоль) в n-BuOH (2 мл) был добавлен DIPEA (36,2 мг; 0.4 ммоль). Реагирующая смесь была нагрета для дистилляции и перемешивалась в течение последующих суток. Реакция контролировалась посредством TLC (PE/EtOAc, v/v, 1/4). Реагирующая смесь была остужена до rt и сконцентрирована in vacuo. Осадок был растворен в EtOAc (10 мл), и полученная смесь была промыта водой (10 мл×2) и насыщенным минеральным раствором (10 мл). Органическая фаза была высушена над обезвоженным Na2SO4, и сконцентрирована in vacuo. Осадок был очищен подготовительным TLC (РЕ/EtOAc (v/v)=2/1) для получения соединения, указанного в заголовке шага, в виде грязно-белого твердого вещества (45 мг; 68,8%).
MS (ESI, pos. ion): 469,2 [M+H]+;
1H NMR (400 МГц; DMSO-d6) δ (ppm): 8,69 (d; J=7,3 Гц; 1H); 7,95 (s; 1H); 7,67 (t; J=7,8 Гц; 1H); 7,63-7,43 (m; 6H); 7,29 (d; J=7,3 Гц; 1H); 4,89-4,76 (m; 1H); 2,72 (s; 3H); 2,61 (s; 3H); 1,97-1,84 (m; 1H); 1,74-1,60 (m; 1H); 0,76 (t; J=7,3 Гц; 3H).
Пример 39 (S)-2-(1-((6-амин-5-(3-метил-1,2,4-оксадиазол-5-ил)пиримидин-4-ил)амин)-пропил)-5-фтор-3-фенилквиназолин-4(3H)-один
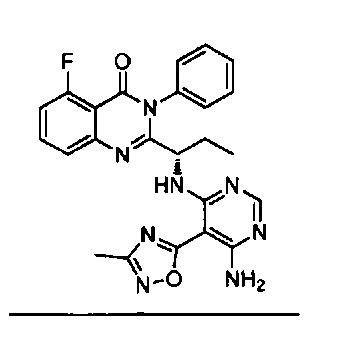
Шаг 1) 2-амин-6-нитро-N-фенилбензамид
[495] К желтой суспензии 2-фтор-6-нитробензойной кислоты (5,55 г; 30 ммоль) в толуоле (50 мл) был добавлен одной порцией SOCl2 (7,08 г; 60 ммоль) при комнатной температуре. После добавления реагирующая смесь перемешивалась при 110°C в течение 8 часов и была сконцентрирована in vacuo для получения желтого масла, которое было растворено в 1,4-диоксане (40 мл), к полученному раствору была добавлена по каплям суспензия анилина (2.79 г; 30 ммоль) и NaHCO3 (5.04 г; 60 ммоль) в 1,4-диоксане (40 мл) при 5°C. Затем полученная смесь перемешивалась при комнатной температуре в течение суток, затем была добавлена вода (350 мл) для резкого охлаждения реакции. Смесь была профильтрована, фильтрат был промыт водой (100 мл×2) и высушен in vacuo при 50°C для получения соединения, указанного в заголовке шага, в виде бледно-коричневого твердого вещества (5,6 г; 71,6%).
MS (ESI, pos. ion) m/z: 261,1 [M+H]+;
1HNMR (600 МГц; CDCl3) δ (ppm): 8,02 (d; J=8,2 Гц; 1H); 7,63 (dd; J=13,8; 8,2 Гц; 4H); 7,52 (dd; J=111; 9,0 Гц; 1H); 7,42 (t; J=7,8 Гц; 2H); 7,23 (t; J=7,4 Гц; 1H).
Шаг 2) 2-амин-6-хлор-N-фенилбензамид
[496] К раствору 2-фтор-6-нитро-N-фенилбензамида (5,6 г; 21,5 ммоль) в этиловом спирте (150 мл) был добавлен порошок Fe (6,08 г; 107,5 ммоль), затем был добавлен одной порцией раствор HCOONH4 (13,55 г; 215 ммоль) в воде (30 мл). Реагирующая смесь перемешивалась в течение 7 часов при 80°C, затем была профильтрована горячей. Фильтрат был сконцентрирована in vacuo и осадок был растворен в EtOAc (300 мл). Полученная смесь была промыта водой (200 мл×2) и насыщенным минеральным раствором (200 мл). Отсепарированный органический слой был высушен над обезвоженным сульфатом натрия и сконцентрирован in vacuo. Осадок был очищен хроматографией на силикагельной колонне (PE/EtOAc (v/v)=40/1) для получения соединения, указанного в заголовке шага, в виде белого твердого вещества (2,7 г; 54,4%).
Шаг 3) (S)-терт-бутил(1-((3-фтор-2-(фенилкарбамоил)фенил)амин)-1-оксобутан-2-ил)-карбамат
[497] К суспензии 2-амин-6-фтор-N-фенилбензамида (700 мг; 3,0 ммоль) и (5)-2-((терт-бутоксикарбонил)амин)бутановой кислоты (S)0 мг; 3.0 ммоль) в DCM (11 мл) был добавлен HATU (1,37 г; 3,6 ммоль) и DIPEA (1.16 г; 9.0 ммоль) при -10°C. Полученная смесь перемешивалась при -10°C в течение 1 часа, затем была нагрета для дистилляции и перемешивалась в течение суток. Смесь была охлаждена до комнатной температуры, промыта водой (20 мл×2) и насыщенным гидратогенным раствором NaHCO3 (20 мл×2). Отсепарированная органическая фаза была высушена над обезвоженным сульфатом натрия и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (PE/EtOAc (v/v)=10/1) для получения соединения, указанного в заголовке шага, в виде желтого твердого вещества (1,14 г; 91,46%).
MS (ESI, neg. ion) m/z: 414,2 [M-H]+;
1HNMR(400 МГц; CDCl3) δ (ppm): 11,73 (s; 1H); 8,55 (d; J=8,5 Гц; 1H); 8,40 (d; J=14,4 Гц; 1H); 7,64 (d; J=8,2 Гц; 1H); 7,47 (dd; J=16,8; 9,4Hz; 1H); 7,42 (t; J=7,7 Гц; 2H); 7,27(m; 1H); 7,23 (t; J=1A Гц; 1H); 6,93 (dd; J=12,3; 8,4 Гц; 1H); 5,13 (s; 1H); 4,27 (s; 1H); 1,77 (m; 1H); 1,47 (m; 1H); 1,44 (s; 9H); 1,02 (t; J=7,4 Гц; 3H).
Шаг 3) (S)-терт-бутил(1-(5-фтор-4-оксо-3-фенил-3,4-дигидроквиназолин-2-ил)пропил)карбамат
[498] Раствор (S)-терт-бутил(1-((3-фтор-2-(фенилкарбамоил)фенил)амин)-1-оксобутан-2-ил)карбамата (4,0 г; 9,63 ммоль) в ацетонитриле (250 мл) и триэтиламине (48,72 г; 481,5 ммоль) был продут азотом. К раствору при комнатной температуре был добавлен N,O-бис(триметилсилил)ацетамид (29,38 г; 144,.45 ммоль). Реагирующая смесь была подвергнута дистилляции при 90°C в течение суток, и реакция контролировалась посредством LC-MS. После завершения реагирующая смесь была сконцентрирована in vacuo. Осадок был растворен в EtOAc (100 мл), и полученная смесь была промыта водой (100 мл×2) и насыщенным минеральным раствором (100 мл). Органическая фаза была высушена над обезвоженным Na2SO4, и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (РЕ/EtOAc (v/v)=20/1) для получения соединения, указанного в заголовке шага, в виде белого твердого вещества (2,57 г; 67%).
MS (ESI, pos. ion) m/z: 398,2 [M+H]+;
1H NMR (600 МГц; CDCl3) δ (ppm): 7,72 (m; 1H); 7,62 (t; J=7,2 Гц; 1H); 7,55 (m; 3H); 7,39 (d; J=7,1 Гц; 1H); 7,29 (m; 1H); 7,14 (m; 1H); 5,47 (d; J=8,3 Гц; 1H); 4,43 (m; 1H); 1,75 (m; 1H); 1,53 (m; 1H); 1,45 (s; 9H); 0,78 (t; J=7,4 Гц; 3H).
Шаг 4) (S)-2-(1-аминопропил)-5-фтор-3-фенилквиназолин-43H)-один
[499] К раствору (S)-терт-бутил(1-(5-фтор-4-оксо-3-фенил-3,4-дигидроквиназолин-2-ил)пропил)карбамата (220 мг; 0,55 ммоль) в EtOAc (2 мл) был добавлен одной порцией раствор HCl в EtOAc (2,5 мл; 3,5 М) при комнатной температуре. Реагирующая смесь перемешивалась в течение суток при комнатной температуре. Полученная суспензия была растворена в воде (20 мл). Отсепарированная гидратогенная фаза была экстрактирована посредством EtOAc (20 мл), затем нейтрализована до pH=8 с помощью порошка NaHCO2 и вновь экстрактирована с помощью EtOAc (20 мл×3). Комбинированные органические фазы были промыты насыщенным минеральным раствором (20 мл), высушены над обезвоженным Na2SO4, и сконцентрированы in vacuo для получения соединения, указанного в заголовке шага, в виде белого порошка (163 мг; 100%).
MS (ESI, pos. ion) m/z: 298,1 [M+H]+;
1H NMR (400 МГц; DMSO-d6) δ (ppm): 7,83 (td; J=8,2; 5,6 Гц; 1H); 7,56 (m; 5H); 7,43 (m; 1H); 7,28 (dd; J=11,0; 8,2 Гц; 1H); 3,14 (dd; J=7,5; 5,4 Гц; 1H); 1,72 (m; 1H); 1,36 (m; 1H); 0,69(t; J=7,4 Гц; 3H).
Пример 5
(S)-2-(1-((6-амин-5-(3-метил-1,2,4-оксадиазол-5-ил)пиримидин-4-ил)амин)-пропил)-5-фтор-3-фенилквиназолин-4(3H)-один
[500] К суспензии (S)-2-(1-аминопропил)-5-фтор-3-фенилквиназолин-4(3H)-один (42 мг; 0,14 ммоль) и 6-хлор-5-(3-метил-1,2,4-оксадиазол-5-ил)пиримидин-4-амин (30 мг; 0,14 ммоль) в n-BuOH (2 мл) был добавлен DIPEA (36 мг; 0,28 ммоль). Реагирующая смесь была нагрета для дистилляции и перемешивалась в течение последующих суток. Реакция контролировалась посредством TLC (РЕ/EtOAc, v/v, 1/4). Затем реагирующая смесь была охлаждена до комнатной температуры и профильтрована. Фильтрат был промыт EtOH (10 мл×2) для получения соединения, указанного в заголовке шага, в виде белого твердого вещества (45,3 мг; 68,5%).
MS (ESI, pos. ion): 473,1 [M+H]+;
1H NMR (600 МГц; DMSO-d6) δ (ppm): 9,04 (d; J=6,9 Гц; 1H); 7,99 (s; 1H); 7,86 (dd; J=13,5;8,1 Гц; 1H); 7,58 (m; 5H); 7,52 (d; J=8,2 Гц; 1H); 7,31 (dd; J=10,3; 8,6 Гц; 1H); 4,93 (dd; J=11,4; 6,9 Гц; 1H); 2,50 (s; 3H); 1,92 (m; 1H); 1,66 (m; 1H); 0,76 (t; J=7,4 Гц; 3H).
Пример 40
(S)-2-(1-((6-амин-5-(5-метил-1,3,4-оксадиазол-2-ил)пиримидин-4-ил)амин)-пропил)-5-фтор-3-фенилквиназолин-4(3H)-один
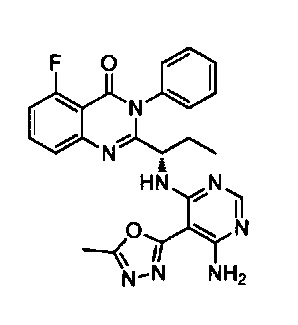
[501] К суспензии (S)-2-(1-аминопропил)-5-фтор-3-фенилквиназолин-4(3H)-один (50,5 мг; 0.17 ммоль) и 6-хлор-5-(5-метил-1,3,4-оксадиазол-2-ил)пиримидин-4-амин (35,6 мг; 0,17 ммоль) в n-BuOH (2 мл) был добавлен DIPEA (44 мг; 0,34 ммоль). Реагирующая смесь была нагрета для дистилляции и перемешивалась в течение последующих суток. Реакция контролировалась посредством TLC (РЕ/EtOAc, v/v, 1/4). Реагирующая смесь была охлаждена до комнатной температуры для образования белой суспензии. Фильтрат был собран и промыт EtOH (10 мл×2) для получения соединения, указанного в заголовке шага, в виде белого твердого вещества (49 мг; 61%).
MS (ESI, pos. ion): 473,2 [M+H]+;
1H NMR (400 МГц; DMSO-d6) δ (ppm): 8,66 (d; J=7,2 Гц; 1H); 7,96 (s; 1H); 7,82 (td; J=8,2; 5,6 Гц; 1H); 7,58 (m; 5H); 7,46 (d; 7=8,1 Гц; 1H); 7,29 (dd; J=10,7; 8,3 Гц; 1Н); 7,19 (s; 2H); 4,81 (td; J=7,8; 4,2 Гц; 1H); 2,61 (s; 3H); 1,94 (m; 1H); 1,69 (m; 1H); 0,76 (t; J=7,3 Гц; 1H).
Пример 41
(S)-2-(1-((6-амин-5-(3-метил-1,2,4-оксадиазол-5-ил)пиримидин-4-ил)амин)-этил)-5-хлор-3-циклопропилквиназолин-4(3H)-один
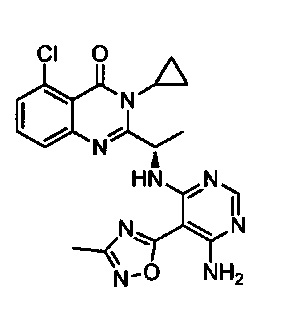
[502] К суспензии (S)-2-(1-аминоэтил)-5-хлор-3-циклопропилквиназолин-4(3H)-один (37 мг; 0,14 ммоль) и 6-хлор-5-(3-метил-1,2,4-оксадиазол-5-ил)пиримидин-4-амина (30 мг; 0,14 ммоль) в n-BuOH (2 мл) был добавлен DIPEA (36,2 мг; 0,28 ммоль). Реагирующая смесь была нагрета для дистилляции и перемешивалась в течение последующих суток. Реакция контролировалась посредством TLC (РЕ/EtOAc, v/v, 1/4). Затем реагирующая смесь была остужена до комнатной температуры и сконцентрирована in vacuo. Осадок был растворен в EtOAc (20 мл), и полученная смесь была промыта водой (10 мл×2) и насыщенным минеральным раствором (10 мл). Отсепарированная органическая фаза была высушена над обезвоженным Na2SO4, и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (РЕ/EtOAc (v/v)=1/1) для получения соединения, указанного в заголовке шага, в виде бледного твердого вещества (45,3 мг; 68,5%).
MS (ESI, pos. ion): 439,1 [M+H]+;
1H NMR (400 МГц; DMSO-d6) δ (ppm): 9,32 (d; J=6,8 Гц; 1H); 8,12 (s; 1H); 7,74 (t; J=8,0 Гц; 1H); 7,54 (dd; J=9,5; 8,1 Гц; 1H); 6,11 (p; J=6,5 Гц; 1H); 3,14 (m; 1H); 2,51 (s; 3H); 1,59 (d; J=6,6 Гц; 3H); 1,26 (d; J=6,8 Гц; 2H); 0,85 (d; J=6,9 Гц; 2H).
Пример 42
(S)-2-(1-((6-амин-5-(2-метил-2H-тетразол-5-ил)пиримидин-4-ил)амин)-этил)-5-хлор-3-циклопропилквиназолин-4(3H)-один
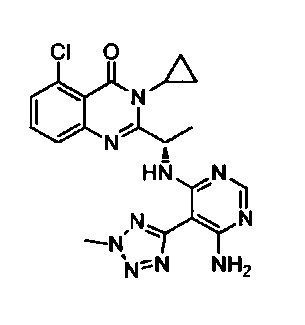
[503] К суспензии (S)-2-(1-аминоэтил)-5-хлор-3-циклопропилквиназолин-4(3H)-один (52,7 мг; 0,2 ммоль) и 6-хлор-5-(2-метил-2Н-тетразол-5-ил)пиримидин-4-амина (42,3 мг; 0,2 ммоль) в n-BuOH (2 мл) был добавлен DIPEA (51.7 мг; 0.4 ммоль). Реагирующая смесь была нагрета для дистилляции и перемешивалась в течение суток. Реакция контролировалась посредством TLC (PE/EtOAc, v/v, 1/4). Затем реагирующая смесь была остужена до комнатной температуры и сконцентрирована in vacuo. Осадок был растворен в EtOAc (15 мл), и полученная смесь была промыта водой (10 мл×2) и насыщенным минеральным раствором (10 мл). Отсепарированная органическая фаза была высушена над обезвоженным Na2SO4, и сконцентрирована in vacuo. Осадок был очищен подготовительным TLC (DCM) для получения соединения, указанного в заголовке шага, в виде бледно-желтого твердого вещества (23 мг; 26,2%).
MS (ESI, pos. ion): 439,2 [M+H]+;
1H NMR (400 МГц; CDCl3) δ (ppm): 9,78 (d; J=7,0 Гц; 1H); 8,22 (s; 1H); 7,58 (m; 2H); 7,46 (m; 1H); 6,36 (p; J=6,7 Гц; 1H); 4,56 (s; 3H); 3,08 (m; 1H); 1,71 (d; J=6,6 Гц; 3H); 1,10 (m; 2H); 0,99 (m; 2H).
Пример 43
(S)-2-(1-((6-амин-5-(2-метил-2H-тетразол-5-ил)пиримидин-4-ил)амин)-пропил)-5-фтор-3-фенилквиназолин-4(3H)-один
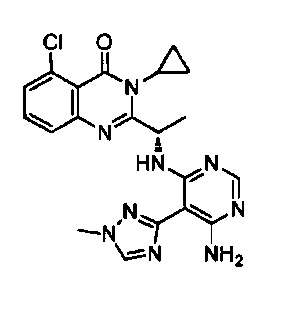
[504] К суспензии 6-хлор-5-(2-метил-2H-тетразол-5-ил)пиримидин-4-амина (30 мг; 0,142 ммоль) и (5)-2-(1-аминопропил)-5-хлор-3-фенилквиназолин-4(3H)-один (44 мг; 0,148 ммоль) в n-BuOH (3 мл) был добавлен DIPEA (37 мг; 0,284 ммоль). Полученная смесь нагревалась при дистилляции в течение 25 часов. Реакция контролировалась посредством TLC (PE/EtOAc, v/v, 1/4). Смесь была остужена до комнатной температуры и сконцентрирована in vacuo. Осадок был переведен в суспензию в EtOH (1.5 мл) и профильтрован. Фильтрат был промыт EtOAc (10 мл) и EtOH (1 мл) и высушен in vacuo для получения соединения, указанного в заголовке шага, в виде желтоватого твердого вещества (38 мг; 56,7%).
MS (ESI, pos. ion): 473,2 [M+H]+; HPLC: 93,6%;
1H NMR(400 МГц; DMSO-d6) δ (ppm): 8,83 (d; J=7,2 Гц; 1H); 7,98 (s; 1H); 7,86-7,78 (m; 1H); 7,65-7,52 (m; 5H); 7,48 (d; J=7,9 Гц; 1H); 7,29 (dd; J=10,7; 8,4 Гц; 1H); 4,89 (td; J=7,6; 4,3 Гц; 1H); 4,54 (s; 3H); 2,04-1,89 (m; 1H); 1,75-1,61 (m; 1H); 0,78 (t; J=7,4 Гц; 3H).
Пример 44
(S)-2-(1-((6-амин-5-(1-метил-1H-триазол-3-ил)пиримидин-4-ил)амин)-этил)-5-хлор-3-циклопропилквиназолин-4(3H)-один
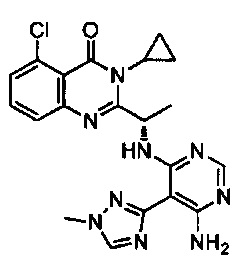
[505] К суспензии 6-хлор-5-(1-метил-1H-1,2,4-триазол-3-ил)пиримидин-4-амина (30 мг; 0,142 ммоль) и (S)-2-(1-аминоэтил)-5-хлор-3-циклопропилквиназолин-4(3H)-один (40 мг; 0,150 ммоль) в n-BuOH (3 мл) был добавлен DIPEA (37 мг; 0,285 ммоль). Полученная смесь нагревалась при дистилляции в течение 21 часа. Реакция контролировалась посредством TLC (CH2Cl2/МеОН, v/v, 25/1). Смесь была остужена до комнатной температуры и сконцентрирована in vacuo. Осадок был растворен в EtOAc (15 мл), и полученная смесь была промыта водой (15 мл) и насыщенным минеральным раствором (10 мл). Отсепарированная органическая фаза была высушена над обезвоженным Na2SO4, и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (DCM/MeOH (v/v)=200/3) для получения соединения, указанного в заголовке шага, в виде бледно-желтого твердого вещества (33 мг; 52,9%).
MS (ESI, pos. ion): 438,2 [M+H]+; HPLC: 97,4%;
1H NMR (400 МГц; CDCl3) δ (ppm): 9,76 (d; J=7,3 Гц; 1H); 8,18 (s; 1H); 8,12 (s; 1H); 7,61-7,49 (m; 2H); 7,43 (dd; J=6,1; 2,8 Гц; 1H); 6,43-6,24 (m; 1H); 4,07 (s; 3H); 3,18-3,04 (m; 1H); 1,68 (d; J=6,6 Гц; 3H); 1,21-1,11 (m; 1H); 1,02-0,80 (m; 3H).
Пример 45 (S)-2-(1-((6-амин-5-(1-метил-1H-1,2,4-триазол-3-ил)пиримидин-4-ил)амин)пропил)-5-хлор-3-(2-фторфенил)квиназолин-4(3H)-один
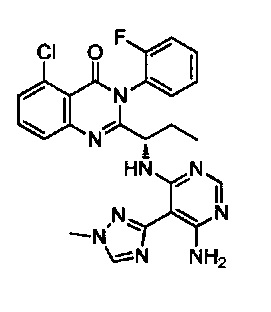
Шаг 1) 2-хлор-N-(2-фторфенил)-6-нитробензамид
[506] К суспензии 2-хлор-6-нитробензойной кислоты (5,0 г; 24,8 ммоль) в толуоле (25 мл) был добавлен SOCl2 (5,5 мл, 74,4 ммоль), и смесь перемешивалась при 110°C в течение суток, затем была сконцентрирована in vacuo для получения коричневого масла. К суспензии 2-фторанилина (4,1 г; 37,2 ммоль) и NaHCO3 (4,2 г; 49,6 ммоль) в 1,4-диоксане (15 мл) при 5°C был медленно добавлен раствор упомянутого выше коричневого масла в 1,4-диоксане (15 мл). Смесь перемешивалась при комнатной температуре в течение суток, затем была разведена EtOAc (200 мл) и водой (80 мл). Отсепарированная органическая фаза была промыта насыщенным минеральным раствором (150 мл×2), высушена над обезвоженным Na2SO4, и сконцентрирована in vacuo для получения соединения, указанного в заголовке шага, в виде бежевого твердого вещества (8,0 г; 100%).
MS (ESI, pos. ion) m/z: 295,1 [M+H]+;
1H NMR (600 МГц; DMSO-d6) δ (ppm): 10,65 (s; 1H); 8,26 (dd; J=8,3; 0,7 Гц; 1H); 8,10-7,97 (m; 2H); 7,77 (t; J=8,2 Гц; 1H); 7,40-7,16 (m; 3H).
Шаг 2) 2-амин-6-хлор-N-(2-фторфенил)бензамид
[507] К суспензии 2-хлор-N-(2-фторфенил)-6-нитробензамида (4,0 г; 13,6 ммоль) в безводном этаноле (70 мл) были добавлены порошок Fe (3,8 г; 67,8 ммоль) и раствор HCOONH4 (8,23 г; 130,57 ммоль) в воде (14 мл). Полученная смесь перемешивалась в течение суток при 95°C, затем была профильтрована. Фильтрат был сконцентрирован in vacuo, и осадок был очищен хроматографией на силикагельной колонне (РЕ/EtOAc (v/v)=5/1) для получения соединения, указанного в заголовке шага, в виде бежевого твердого вещества (1,51 г; 42%).
MS (ESI, pos. ion) m/z: 265,1 [M+H]+;
1H NMR (600 МГц; DMSO-d6) δ (ppm): 10,29 (s; 1H); 7,83 (td; J=7,8; 1,7 Гц; 1H); 7,33-7,15 (m; 3H); 7,10 (t; J=8,0 Гц; 1H); 6,68 (dd; J=30,4; 8,0 Гц; 2H); 5,34 (s; 2H).
Шаг 3) (S)терт-бутил-((3-хлор-2-((2-фторфенил)карбамоил)фенил)амин)-1-оксобутан-2-ил)-карбамат
[508] К раствору 2-амин-6-хлор-N-(2-фторфенил)бензамида (1,5 г; 5,67 ммоль) и 2-((терт-бутоксикарбонил)амин)бутановой кислоты (1,21 г; 5,95 ммоль) в DMF (20 мл) были добавлены DIPEA (2,20 г; 17,01 ммоль) и HATU (2.59 г; 6.80 ммоль) при комнатной температуре. Смесь перемешивалась в течение суток при rt, затем была промыта водой (150 мл×2) и насыщенным гидратогенным раствором NaHCO3 (150 мл×2). Отсепарированная органическая фаза была сконцентрирована in vacuo, и осадок был очищен хроматографией на силикагельной колонне (PE/EtOAc (v/v)=5/1) для получения соединения, указанного в заголовке шага, в виде бледно-желтого твердого вещества (0,72 г; 28%).
MS (ESI, pos. ion) m/z: 350,2 [M-Boc+H]+.
Шаг 4) (S)-терт-бутил(1-(5-хлор-3-(2-фторфенил)-4-оксо-3,4-дигидроквиназолин-2-ил)пропил)карбамат
[509] К раствору (S)-терт-бутил(-1-((3-хлор-2-((2-фторфенил)карбамоил)фенил)амин)-1-оксобутан-2-ил)карбамата (1,6 г; 3,60 ммоль) и триэтиламина (15 мл, 108 ммоль) в CH3CN (100 мл) был добавлен шприцем N,0-бис(триметилсилил)ацетамид (13 мл, 36 ммоль) в атмосфере азота. Полученная смесь была нагрета до 85°C и перемешивалась в течение последующих 33 часов, затем была остужена до комнатной температуры и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (PE/EtOAc (v/v)=8/1) для получения соединения, указанного в заголовке шага, в виде бледно-желтого твердого вещества (0,63 г; 41%).
MS (ESI, pos. ion) m/z: 432,1 [M+H]+.
Шаг 5) (S)-2-(1-аминопропил)-5-хлор-3-(2-фторфенил)квиназолин--4(3H)-один
[510] К суспензии (S)-терт-бутил (1-(5-хлор-3-(2-фторфенил)-4-оксо-3,4-дигидроквиназолин-2-ил)-пропил)карбамата (1,24 г; 2,90 ммоль) в EtOAc (10 мл) был добавлен раствор HCl в EtOAc (3,0 М; 8 мл; 23,68 ммоль). Полученная смесь перемешивалась в течение 4 часов при комнатной температуре и была сконцентрирована in vacuo. Осадок был растворен в EtOAc (50 мл) и полученная смесь нейтрализована до pH=7-8 насыщенным гидратогенным раствором NaHCO3. Комбинированная органическая фаза была промыта насыщенным минеральным раствором (100 мл), высушена над обезвоженным Na2SO4, и сконцентрирована in vacuo для получения соединения, указанного в заголовке шага, в виде грязно-белого твердого вещества (0,81 г; 81%), состоящего из двух изомеров (А и И) в пропорции 2/1 (изомер А/изомер В).
MS (ESI, pos. ion) m/z: 332,1 [M+H]+;
Изомер A: 1H NMR (400 МГц; DMSO-d6) δ (ppm): 7,85-7,77 (m; 1H); 7,73-7,56 (m; 4H); 7,56-7,48 (m; 1H); 7,48-7,40 (m; 1H); 3,22 (t; J=6,4 Гц; 1H); 1,90 (s; 2H); 1,82-1,57 (m; 1H); 1,48-1,29 (m; 1H); 0,69 (t;J=7,3 Гц; 3H).
Изомер В: 1H NMR (400 МГц; DMSO-d6) δ (ppm): 7,85-7,77 (m; 1H); 7,73-7,56 (m; 4H); 7,56-7,48 (m; 1H); 7,47-7,39 (m; 1H); 3,11 (dd; J=7,5; 4,8 Гц; 1H); 1,90 (s; 2H); 1,82-1,57 (m; 1H); 1,48-1,29 (m; 1H); 0,75 (t; J=7,4 Гц; 3H).
Шаг 6 (S)-2-(1-((6-амин-5-(1-метил-1H-1,2,4-триазол-3-ил)пиримидин-4-ил)амин)пропил)-5-хлор-3-(2-фторфенил)квиназолин-4(3H)-один
[511] К суспензии 6-хлор-5-(1-метил-1H-1,2,4-триазол-3-ил)пиримидин-4-амина (30 мг; 0,142 ммоль) и (S)-2-(1-аминопропил)-5-хлор-3-(2-фторфенил)квиназолин-4(3H)-один (50 мг; 0,150 ммоль) в n-BuOH (3 мл) был добавлен DIPEA (37 мг; 0,285 ммоль).
Полученная смесь нагревалась при дистилляции в течение 29 часов. Реакция контролировалась посредством TLC (DCM/MeOH, v/v, 25/1). Реагирующая смесь была остужена до it и сконцентрирована in vacuo. Осадок был растворен в EtOAc (15 мл), и полученная смесь была промыта водой (15 мл) и насыщенным минеральным раствором (10 мл). Органическая фаза была высушена над обезвоженным Na2SO4, и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (DCM/MeOH (v/v)=200/3) для получения соединения, указанного в заголовке шага, в виде белого твердого вещества (35 мг; 48,6%), состоящего из двух изомеров (А и В) в пропорции 3/2 (изомер А/изомер В).
MS (ESI, pos. ion): 506,1 [M+H]+; HPLC: 93,6% (общая степень чистоты изомера А и В);
Изомер A: 1H NMR (400 МГц; CDCl3) δ (ppm): 9,35 (s; 1Н); 8,14 (s; 1H); 7,95 (s; 1H); 7,70-7,43 (m; 5H); 7,35-7,29 (m; 2H); 5,09 (td; J=7,9; 5,2 Гц; 1H); 4,05 (s; 3H); 2,11-1,94 (m; 2H); 0,99-0,89 (m; 5H).
Изомер В: 1H NMR (400 МГц; CDCl3) δ (ppm): 9,35 (s; 1H); 8,10 (s; 1H); 7,83 (s; 1H); 7,70-7,43 (m; 5H); 7,35-7,29 (m; 2H); 5,28 (td; J=7,5; 6,6 Гц; 1H); 4,02 (s; 3H); 2,11-1,94 (m; 2H); 0,99-0,89 (m; 5H).
Пример 46 (S)-2-(1-((6-амин-5-(метоксиметил)-1-метил-1H-1,2,4-триазол-3-ил)пиримидин-4-ил)амин)-пропил)-5-хлор-3-циклопропилквиназолин-4(3H)-один
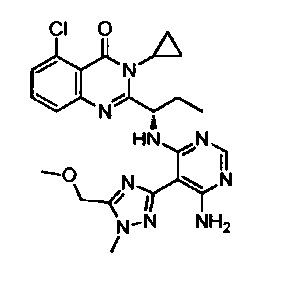
Шаг 1) (3-(4,6-диметоксипиримидин-5-ил)-1-метил-1Н-1,2,4-триазол-5-ил)метанол
[512] К суспензии этил 3-(4,6-диметоксипиримидин-5-ил)-1-метил-1Н-1,2,4-триазол-5-карбоксилата (380 мг; 1,30 ммоль) в THF (8 мл) при 0°C был частями добавлен LiAIH4 (49 мг; 1,30 ммоль). Реагирующая смесь перемешивалась при 0°C в течение 1 часа, затем была медленно добавлена Н2O (50 мг). Смесь перемешивалась при 0°C в течение 15 минут, затем бело добавлено немного Na2SO4. Смесь перемешивалась в течение последующих 15 минут, после чего было добавлено 5 М гидратогенного раствора NaOH (0,04 мл). Смесь перемешивалась в течение последующих 30 минут, затем была профильтрована. Фильтрат был сконцентрирован in vacuo, и осадок был очищен хроматографией на силикагельной колонне (CH2Cl2/МеОН (v/v)=50/1) для получения части указанного в заголовке соединения в виде белого твердого вещества (79 мг). Фильтрат был переведен в суспензию в EtOAc (20 мл) и перемешивался при rt в течение 30 минут, полученная смесь была профильтрована, и фильтрат был сконцентрирован in vacuo для получения другой части указанного в заголовке соединения в виде белого твердого вещества (153 мг; общий выход: 71,3%).
MS (ESI, pos. ion) m/z: 252,1 [M+H]+;
1HNMR (400 МГц; CDCl3) δ (ppm): 8,51 (s; 1H); 4,85 (s; 2H); 4,02 (s; 3H); 3,99 (s; 6H).
Шаг 2) 4,6-диметокси-5-(5-(метоксиметил)-1-метил-1Н-1,2,4-триазол-3-ил)пиримидин
[513] К суспензии (3-(4,6-диметоксипиримидин-5-ил)-1-метил-1Н-1,2,4-триазол-5-ил)метанола (232 мг; 0,92 ммоль в THF (15 мл) при 0°C был добавлен NaH (60% раствор в минеральном масле, 55 мг; 1,39 ммоль). Полученная смесь перемешивалась при 0°C в течение 40 минут, затем был добавлен раствор СН3I (157 мг; 1,11 ммоль) в THF (1 мл). Полученная смесь была остужена до rt и перемешивалась в течение последующих 4 засов, затем была резко охлаждена насыщенным гидратогенным раствором NH4Cl (15 мл) и экстрактирована посредством EtOAc (10 мл×3). Водосодержащая фаза была окислена до pH=2-3 гидратогенным раствором 4 М HCl и экстрактирована посредством EtOAc (10 мл×3). Комбинированные органические фазы были очищены хроматографией на силикагельной колонне (PE/EtOAc (v/v)=1/10) для получения соединения, указанного в заголовке шага, в виде белого твердого вещества (49 мг; 19,9%).
MS (ESI, pos. ion) m/z: 266,2 [M+H]+;
1H NMR (400 МГц; CDCl3) δ (ppm): 8,51 (s; 1H); 4,70 (s; 2H); 4,02 (s; 3H); 3,98 (s; 6H); 3,46 (s; 3H).
Шаг 3) 4,6-дихлор-5-(5-(метоксиметил)-1-метил-1H-1,2,4-триазол-3-ил)пиримидин
[514] К суспензии 4,6-диметокси-5-(5-(метоксиметил)-1-метил-1Н-1,2,4-триазол-3-ил)пирими дина (56 мг; 0,21 ммоль) в безводном толуоле (5 мл) были добавлены РОСl3 (0,2 мл, 2,11 ммоль) и DMF (0,2 мл). Реагирующая смесь была нагрета для дистилляции и перемешивалась в течение последующих 22 часов. Супернатант был отсепарирован и сконцентрирован in vacuo для получения желтого осадка. Темно-коричневый осадок был разведен H2O (10 мл) и экстрактирован посредством EtOAc (10 мл×3). Комбинированная органическая фаза была промыта насыщенным минеральным раствором (10 мл) и сконцентрированы in vacuo для получения другого желтого осадка. Оба желтых осадка были использованы на следующем шаге без дополнительной очистки.
MS (ESI, pos. ion) m/z: 274,0 [M+H]+.
Шаг 4) 6-хлор-5-(5-(метоксиметил)-1-метил-1H-1,2,4-триазол-3-ил)пиримидин-4-амин
[515] Раствор желтого осадка, полученного на предыдущем шаге в THF (5 мл) перемешивался при 30°C в атмосфере газообразного NH3 на протяжении 18 часов. Реакция контролировалась посредством TLC (РЕ/EtOAc, v/v, 1/4). Смесь была профильтрована, фильтрат промыт EtOAc (15 мл). Фильтрат был сконцентрирован in vacuo для получения соединения, указанного в заголовке шага, в виде желтого твердого вещества (52 мг; выход 95,6% за два шага).
MS (ESI, pos. ion) m/z: 255,1 [M+H]+;
1HNMR (400 МГц; CDCl3) δ (ppm): 8,33 (s; 1H); 4,71 (s; 2H); 4,05 (s; 3H); 3,47 (s; 3H).
Шаг 5 (S)-2-(1-((6-амин-5-(метоксиметил)-1-метил-1H-1,2,4-триазол-3-ил)пиримидин-4-ил)амин)-пропил)-5-хлор-3-циклопропилквиназолин-4(3H)-один
[516] К суспензии 6-хлор-5-(5-(метоксиметил)-1-метил-1Н-1,2,4-триазол-3-ил)пиримидин-4-амин (26 мг; 0,102 ммоль) в n-BuOH (2 мл) был добавлен DIPEA (26 мг; 0,204 ммоль). Полученная смесь нагревалась при дистилляции в течение 22 часов. Реакция контролировалась посредством TLC (CH2Cl2/МеОН, v/v, 25/1). Смесь была остужена до комнатной температуры и сконцентрирована in vacuo. Осадок был растворен в EtOAc (15 мл), и полученная смесь была промыта водой (15 мл) и насыщенным минеральным раствором (10 мл). Отсепарированная органическая фаза была высушена над обезвоженным Na2SO4, и сконцентрирована in vacuo. Осадок был очищен подготовительным TLC (CH2Cl2/МеОН (v/v)=25/1) для получения соединения, указанного в заголовке шага, в виде бледно-желтого твердого вещества (31 мг; 61,2%).
MS (ESI, pos. ion) m/z: 496,2 [M+H]+; HPLC: 96,1%;
1H NMR (400 МГц; CDCl3) δ (ppm): 9,50 (d; J=7,6 Гц; 1H); 8,10 (s; 1H); 7,58-7,46 (m; 2H); 7,42 (dd; J=6,9; 2,0 Гц; 1H); 6,37-6,24 (m; 1H); 4,71 (s; 2H); 4,04 (s; 3H); 3,45 (s; 3H); 3,18-3,05 (m; 1H); 2,12-1,99 (m; 2H); 1,45-1,41 (m; 2H); 1,07 (t; J=7,4 Гц; 3H); 0,90-0,86 (m; 2H).
Пример 47
(S)-2-(1-((6-амин-5-(изоксазол-3-ил)пиримидин-4-ил)амин)пропил)-3-хлорпропил-5-метилквиназолин-4(3H)-один
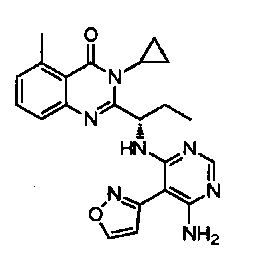
[517] Смесь 6-хлор-5-(изоксазол-3-ил)пиримидин-4-амина (50 мг; 0,25 ммоль), (S)-2-(1-аминопропил)-3-циклопропил-5-метилквиназолин-4(3H)-один (78,5 мг; 0,31 ммоль) и N,N-диизопропилэтиламин (98,5 мг; 0,76 ммоль) в и-бутаноле (3,.0 мл) нагревалась до 150°C в герметичной колбе течение 36 часов. После завершения реагирующая смесь была сконцентрирована in vacuo, и осадок был очищен хроматографией на силикагельной колонне (PE/EtOAc (v/v)=1/2) для получения соединения, указанного в заголовке шага, в виде светло-желтого твердого вещества (45 мг; 42%).
MS (ESI, pos. ion) m/z: 418,2 [M+H]+; HPLC: 99,5%;
1H NMR (600 МГц; CDCl3) δ (ppm): 8,66 (d; J=1,5 Гц; 1H); 8,17 (s; 1H); 7,54 (t; J=7,7 Гц; 1H); 7,38 (d; J=8,0 Гц; 1H); 7,20 (d; J=7,3 Гц; 1H); 7,10 (d; J=1,5 Гц; 1H); 6,75 (d; J=8,2 Гц; 1H); 6,24 (td; J=7,8; 4,8 Гц; 1H); 5,71 (br s; 2H); 3,05 (ddd; J=11,1; 7,2; 4,2 Гц; 1H); 2,86 (s; 3H); 2,10-2,01 (m; 1H); 1,90-1,80 (m; 1H); 1,50-1,39 (m; 2H); 1,00 (t; J=7,4 Гц; 3H); 0,94-0,85 (m; 2H);
13C NMR (151 МГц; CDCl3) δ (ppm): 164,0; 160,2; 159,4; 159,0; 158,7; 158,1; 156,6; 147,7; 140,8; 133,2; 129,3; 124,9; 119,4; 104,2; 86,1; 51,9; 27,9; 26,4; 23,0; 10,6; 10,1; 9,9.
Пример 48
(S)-2-(1-((6-амин-5-(изоксазол-3-ил)пиримидин-4-ил)амин)пропил)-3-циклопропил-5-фторквиназолин-4(3H)-один
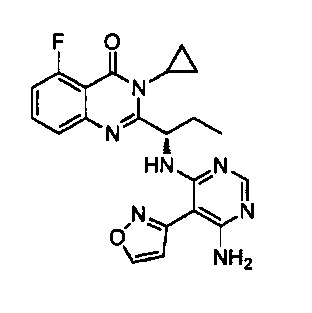
[518] Смесь 6-хлор-5-(изоксазол-3-ил)пиримидин-4-амина (39,3 мг; 0,20 ммоль),
(S)-2-(1-аминопропил)-5-фтор-3-циклопропилквиназолин-4(3H)-один (62,7 мг; 0,24 ммоль) и N,N-диизопропилэтиламин (77,5 мг; 0,6 ммоль) в n-бутаноле (2,5 мл) нагревалась до 150°C в герметичной колбе течение 36 часов. После завершения реагирующая смесь была сконцентрирована in vacuo, и осадок был очищен хроматографией на силикагельной колонне (РЕ/EtOAc (v/v)=1/2) для получения соединения, указанного в заголовке шага, в виде белого твердого вещества (30 мг; 36%).
MS (ESI, pos. ion) m/z: 422,2 [M+H]+; HPLC: 94,1%;
1H NMR (600 МГц; CDCl3) δ (ppm): 8,65 1,2 Гц; 1H); 8,16 (s; 1H); 7,62 (td; J=8,1; 5,4 Гц; 1H); 7,34 (d; J=8,2 Гц; 1H); 7,08 (dd; J=10,0; 8,5 Гц; 1H); 7,04 (d; J=1,2 Гц; 1H); 6,72 (d; J=8,3 Гц; 1H); 6,24 (td; J=8,1; 4,7 Гц; 1H); 5,67 (br s; 2H); 3,07 (ddd; J=11,1; 7,2; 4,2 Гц; 1H); 2,09-2,00 (m; 1H); 1,90-1,80 (m; 1H); 1,51-1,40 (m; 2H); 1,03 (t; J=1A Гц; 3H); 0,98 -0,85 (m; 2H);
13C NMR (151 МГц; CDCl3) δ (ppm): 161,9; 160,7; 160,41; 160,36; 160,3; 160,1; 159,4; 158,8; 158,0; 156,6; 148,5; 134,4; 134,3; 122,7; 122,6; 113,3; 113,2; 110,8; 110,7; 104,1; 86,4; 52,2; 28,0; 26,5; 10,6; 10,2; 10,1.
Пример 49
(S)-2-(1-((6-амин-5-(изоксазол-3-ил)пиримидин-4-ил)амин)пропил)-5-хлор-3-циклопропилквиназолин-4(3H)-один
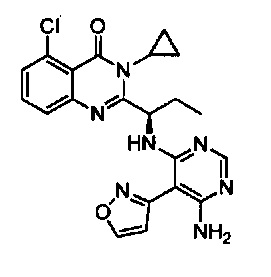
[519] Смесь 6-хлор-5-(изоксазол-3-ил)пиримидин-4-амина (39,3 мг; 0,2 ммоль), (S)-2-(1-аминопропил)-5-хлор-3-циклопропилквиназолин-4(3H)-один (66,7 мг; 0,24 ммоль) и N,N-диизопропилэтиламин (77,5 мг; 0,60 ммоль) в n-бутаноле (2,5 мл) нагревалась до 150°C в герметичной колбе течение 30 часов. После завершения реагирующая смесь была сконцентрирована in vacuo, и осадок был очищен хроматографией на силикагельной колонне (PE/EtOAc (v/v)=1/2) для получения соединения, указанного в заголовке шага, в виде белого твердого вещества (47 мг; 54%).
MS (ESI, pos. ion) m/z: 438,2 [M+H]+; HPLC: 99,51%;
1H NMR (600 МГц; CDCl3) δ (ppm): 8,65 (d; J=1,6 Гц; 1H); 8,16 (s; 1H); 7,54 (t; J=8,0 Гц; 1H); 7,46-7,40 (m; 2H); 7,04 (d; J=1,6 Гц; 1H); 6,70 (d; J=8,4 Гц; 1H); 6,23 (td; J=8,1; 4,7 Гц; 1H); 5,63 (br s; 2H); 3,08 (ddd; J=11,1; 7,1; 4,2 Гц; 1H); 2,11-2,00 (m; 1H); 1,89-1,80 (m; 1H); 1,50-1,41 (m; 2H); 1,03 (t; J=7,4 Гц; 3H); 0,95-0,88 (m; 2H);
13C NMR (151 МГц; CDCl3) δ (ppm): 161,5; 160,38; 160,35; 159,4; 158,8; 158,1; 156,6; 148,8; 133,9; 133,4; 129,3; 125,9; 118,1; 104,1; 86,3; 52,1; 27,8; 26,8; 10,7; 10,2; 10,1.
Пример 50 (S)-2-(1-((6-амин-5-(1-метил-1Н-1,2,4-триазол-3-пиримидин-4-ил)амин)пропил)-5-хлор-3-(4-фторфенил)квиназолин-4(3H)-один
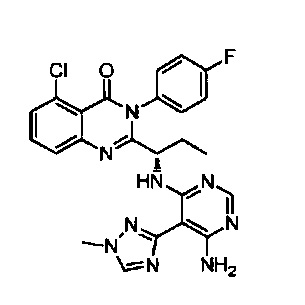
Шаг 1) 2-хлор-N-(4-фторфенил)-6-нитробензамид
[520] К желтой суспензии 2-хлор-6-нитробензойной кислоты (5,0 г; 24,8 ммоль) в толуоле (50 мл) был добавлен одной порцией SOCl2 (5,85 г; 49,6 ммоль) при комнатной температуре. После добавления реагирующая смесь перемешивалась при 120°C в течение суток и была сконцентрирована in vacuo. Осадок был растворен в 1,4-диоксане (30 мл), и к полученному раствору была добавлена по каплям суспензия 4-фторанилина (2,76 г; 24,8 ммоль) и NaHCO3 (6,34 г; 49,6 ммоль) в 1,4-диоксане (30 мл) при 5°C. Затем полученная смесь перемешивалась при комнатной температуре в течение 7 и была резко охлаждена водой (250 мл) для получения желтой суспензии. Осадок был собран фильтрацией, промыт водой (100 мл ×) и высушен in vacuo при 50°C для получения соединения, указанного в заголовке шага, в виде бледно-коричневого твердого вещества (6.37 г; 87,1%).
MS (ESI, pos. ion) m/z: 295,0 [M+H]+.
Шаг 2) 2-амин-6-хлор-N-(4-фторфенил)бензамид
[521] К раствору 2-хлор-N-(4-фторфенил)-6-нитробензоамида (5,78 г; 19,6 ммоль) этиловом спирте (145 мл) был добавлен при перемешивании порошок Fe (5,47 г; 98 ммоль). К упомянутой выше суспензии был добавлен одной порцией раствор HCOONH4 (12,3 г; 196 ммоль) в 30 мл воды. Полученная смесь перемешивалась в течение 7 часов при 80°C, затем была профильтрована горячей. Фильтрат был сконцентрирован in vacuo. Осадок был растворен в EtOAc (300 мл), и полученная смесь была промыта водой (200 мл×2) и насыщенным минеральным раствором (200 мл). Отсепарированная органическая фаза была высушена над обезвоженным сульфатом натрия и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (РЕ/EtOAc (v/v)=10/1) для получения соединения, указанного в заголовке шага, в виде белого твердого вещества (2,85 г; 55,1%).
MS (ESI, pos. ion) m/z: 265,1 [M+H]+;
1H NMR (600 МГц; DMSO-d6) δ (ppm): 10,49 (s; 1H); 7,75 (m; 2H); 7,18 (t; J=8,9 Гц; 2H); 7,09 (t; J=8,1 Гц; 1H); 6,70 (d; J=8,2 Гц; 1H); 6,64 (m; 1H); 5,36 (s; 2H).
Шаг 3) (S)-терт-бутил-((3-хлор-2-((4-фторфенил)карбамоил)фенил)амин)-1-оксобутан-2-ил)-карбамат
[522] К суспензии 2-амин-6-хлор-N-(4-фторфенил)бензамида (3,57 г; 13,5 ммоль) и (S)-2-((терт-бутоксикарбонил)амин)бутановой кислоты (3,03 г; 3,.0 ммоль) в DCM (50 мл) был добавлен HATU (5,67 г; 14,9 ммоль) и DIPEA (5,23 г; 40,5 ммоль) при -10°C. Полученная смесь перемешивалась при -10°C в течение 1 часа, затем дистиллировалась при 45°C в течение суток. Реагирующая смесь была охлаждена до комнатной температуры и промыта насыщенным гидратогенным раствором NaHCO3 (200 мл×2), водой (150 мл) и насыщенным минеральным раствором (200 мл). Отсепарированная органическая фаза была высушена над обезвоженным сульфатом натрия и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (PE/EtOAc (v/v)=4/1) для получения соединения, указанного в заголовке шага, в виде белого твердого вещества (2,78 г; 45,9%).
MS (ESI, neg. ion) m/z: 448,1 [M-H]+;
1H NMR (600 МГц; DUSO-d6) δ (ppm): 10,65 (s; 1H); 9,44 (s; 1H); 7,84 (d; J=8,1Hz; 1H); 7,74 (m; 2H); 7,47 (t; J=8,1 Гц; 1H); 7,36 (&J=8,0 Гц; 1H); 7,19 (t;J=8,8 Гц; 2H); 7,11 (d; J=7,2 Гц; 1H); 4,00 (dd; J=12,8; 7,5 Гц; 1H); 1,68 (m; 1H); 1,51 (m; 1H); 1,33 (s; 9H); 0,81 (t;, 7=7,4 Гц; 3H).
Шаг 4) (S)-терт-бутил(1-(5-хлор-3-(4-фторфенил)-4-оксо-3,4-дигидроквиназолин-2-ил)пропил)карбамат
[523] Раствор (S)-терт-бутил(1-((3-хлор-2-((4-фторфенил)карбамоил)фенил)амин)-1-оксобутан-2-ил)карбамата (2,779 г; 6,2 ммоль) в смеси безводного ацетонитрила (250 мл) и триэтиламина (18,92 г; 186 ммоль) был продут газообразным N2. К раствору при комнатной температуре был добавлен N,O-бис(триметилсилил)ацетамид (18,92 г; 93 ммоль). Реагирующую смесь дистиллировали в течение суток при температуре 90°C. Реакция контролировалась TLC вплоть до завершения. Полученная смесь была сконцентрирована in vacuo. Осадок был растворен в EtOAc (300 мл), и полученная смесь была промыта водой (100 мл×2) и насыщенным минеральным раствором (100 мл). Отсепарированная органическая фаза была высушена над обезвоженным Na2SO4, и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (РЕ/EtOAc (v/v)=10/1) для получения соединения, указанного в заголовке шага, в виде бледно-желтого твердого вещества (2.51 г; 93,5%).
MS (ESI, pos. ion) m/z: 432,2 [M+H]+.
Шаг 5) (S)-2-(1-аминопропил)-5-хлор-3-(4-фторфенил)квиназолин--4(3H)-один
[524] К раствору (S)-терт-бутил (1-(5-хлор-3-(4-фторфенил)-4-оксо-3,4-дигидроквиназолин-2-ил)пропил)карбамат (2,51 г; 5,8 ммоль) в EtOAc (10 мл) был добавлен одной порцией раствор HCl в EtOAc (3,88 М; 8,3 мл) при комнатной температуре. Реагирующая смесь перемешивалась в течение суток при комнатной температуре. Полученная суспензия была разведена в воде (200 мл). Отсепарированная гидратогенная фаза была экстрактирована посредством EtOAc (100 мл), затем нейтрализована до pH=8 с помощью порошка Na2СО3 и вновь экстрактирована с помощью EtOAc (100 мл). Комбинированные органические фазы были промыты насыщенным минеральным раствором (50 мл), высушены над обезвоженным Na2SO4, и сконцентрированы in vacuo для получения соединения, указанного в заголовке шага, в виде белого порошка (1,7 г; 87,9%).
MS (ESI, pos. ion) m/z: 332,0 [M+H]+;
1H NMR (400 МГц; CDCl3) δ (ppm): 7,65 (m; 2H); 7,50 (m; 1H); 7,28 (m; 4H); 3,42 (m; 1H); 1,81 (m; 1H); 1,54 (tt; J=14,7; 7,4 Гц; 1H); 0,84 (t; J=7,4 Гц; 3H).
Шаг 6 (S)-2-(1-((6-амин-5-(1-метил-1H-1,2,4-триазол-3-ил)пиримидин-4-ил)амин)пропил)-5-хлор-3-(4-фторфенил)квиназолин-4(3H)-один
[525] К суспензии (S)-2-(1-аминопропил)-5-хлор-3-(4-фторфенил)квиназолин-4(3H)-один (66 мг; 0.2 ммоль) и 6-хлор-5-(1-метил-1H-1,2,4-триазол-3-ил)пиримидин-4-амин (42 мг; 0,2 ммоль) в n-BuOH (2 мл) был добавлен DIPEA (51.7 мг; 0.4 ммоль). Реагирующая смесь была нагрета для дистилляции и перемешивалась в течение суток. Реакция контролировалась посредством TLC (DCM/MeOH, v/v, 10/1). После завершения смесь была остужена до комнатной температуры и сконцентрирована in vacuo. Осадок был растворен в EtOAc (20 мл), и полученная смесь была промыта водой (20 мл×2) и насыщенным минеральным раствором (10 мл). Отсепарированная органическая фаза была высушена над обезвоженным Na2SO4, и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (CH2Cl2/МеОН (v/v)=100/1) для получения соединения, указанного в заголовке шага, в виде белого твердого вещества (35 мг; 34,6%).
MS (ESI, pos. ion) m/z: 506,1 [M+H]+;
1H NMR (400 МГц; DMSO-d6) δ (ppm): 9,25 (d; J=7,1 Гц; 1H); 8,24 (s; 1H); 7,83 (s; 1H); 7,50 (dd; J=11,2; 7,2 Гц; 3H); 7,36 (d; J=6,8 Гц; 1H); 7,24 (s; 1H); 7,17 (dd; J=15,0; 7,5 Гц; 2H); 4,84 (d; J=5,0 Гц; 1H); 3,96 (s; 3H); 1,84 (m; 1H); 1,77 (m; 1H); 0,85 (t; J=7,3 Гц; 3H).
Пример 51
(S)-2-(1-((6-амин-5-(1-метил-1H-пиразол-3-ил)пиримидин-4-ил)амин)пропил)-5-хлор-3-циклопропилквиназолин-4(3H)-один
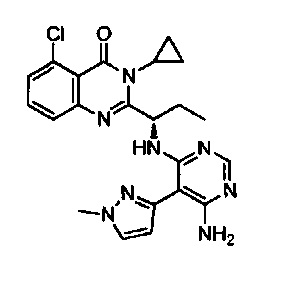
[526] К суспензии 6-хлор-5-(1-метил-1H-пиразол-3-ил)пиримидин-4-амин (30 мг; 0,143 ммоль) и (S)-2-(1-аминопропил)-5-хлор-3-циклопропилквиназолин-4(3H)-один (42 мг; 0.150 ммоль) в n-BuOH (1,5 мл) был добавлен DIPEA (74 мг; 0,572 ммоль). Полученная смесь нагревалась при дистилляции в течение 48 часов. Реакция контролировалась посредством TLC (CH2Cl2/МеОН, v/v, 125/3). После завершения смесь была остужена до комнатной температуры и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (РЕ/EtOAc (v/v)=1/3) для получения соединения, указанного в заголовке шага, в виде бледно-желтого твердого вещества (24 мг; 37,2%).
MS (ESI, pos. ion) m/z: 451,1 [M+H]+; HPLC: 99,5%;
1H NMR (600 МГц; DMSO-d6) δ (ppm): 7,92 (d; J=6,8 Гц; 2H); 7,76-7,62 (m; 1H); 7,64-7,38 (m; 3H); 6,67 (s; 1H); 6,35 (s; 2H); 6,00 (s; 1H); 3,97 (s; 3H); 3,12 (s; 1H); 2,05-1,96 (m; 1H); 1,80 (d; J=6,2 Гц; 1H); 1,35-1,27 (m; 2H); 1,09 (s; 1H); 0,93 (s; 3H); 0,87-0,81 (m; 1H).
Пример 52
(S)-2-(1-((6-амин-5-((4-метилоксазол-2-ил)пиримидин-4-ил)амин)пропил)-5-хлор-3-цилкопропилквиназолин-4(3H)-один
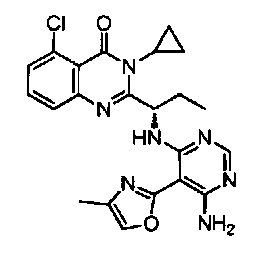
[527] К суспензии 6-хлор-5-(4-метилоксазол-2-ил)пиримидин-4-амина (31 мг; 0,147 ммоль) и (S)-2-(1-аминопропил)-5-хлор-3-циклопропилквиназолин-4(3H)-один (50 мг; 0,180 ммоль) в n-BuOH (3 мл) был добавлен DIPEA (38 мг; 0.294 mmoll). Полученная смесь нагревалась при дистилляции в течение 16 часов. Реакция контролировалась посредством TLC (PE/EtOAc, v/v, 1/1). После завершения смесь была остужена до комнатной температуры и сконцентрирована in vacuo. Осадок был растворен в EtOAc (15 мл), и полученная смесь была промыта водой (15 мл) и насыщенным минеральным раствором (10 мл). Отсепарированная органическая фаза была высушена над обезвоженным Na2SO4, и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (РЕ/EtOAc (v/v)=2/1) для получения соединения, указанного в заголовке шага, в виде бледно-желтого твердого вещества (64 мг; 96,2%).
MS (ESI, pos. ion) m/z: 452,1 [M+H]+; HPLC: 96,2%;
1H NMR (400 МГц; DMSO-d6) δ (ppm): 9,32 (d; J=1,5 Гц; 1H); 8,00 (s; 1H); 7,97 (d; J=1,3 Гц; 1H); 7,73-7,64 (m; 1H); 7,51 (td; J=8,4; 1,0 Гц; 2H); 7,37 (s; 2H); 6,12 (td; J=7,2; 5,0 Гц; 1H); 3,18-3,08 (m; 1H); 2,25 (d; J=1,1 Гц; 3H); 2,16-2,02 (m; 1H); 1,93-1,83 (m; 1H); 1,31-1,26 (m; 2H); 1,11-1,03 (m; 1H);0,96 (t; J=7,4 Гц; 3H); 0,85-0,82 (m; 1H).
Пример 53 (S)-2-(1-((6-амин-5-(1-метил-1H-1,2,4-триазол-3-ил1)пиримидин-4-ил)амин)-пропил)-5-хлор-3-циклопропилквиназолин-4(3H)-один
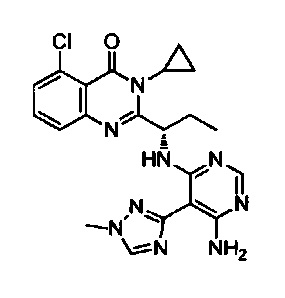
[528] К суспензии 6-хлор-5-(1-метил-1H-1,2,4-триазол-3-ил)пиримидин-4-амина (30 мг; 0,142 ммоль) и (S)-2-(1-аминопропил)-5-хлор-3-циклопропилквиназолин -4(3H)-один (42 мг; 0,150 ммоль) в n-BuOH (1,5 мл) был добавлен DIPEA (37 мг; 0.285 ммоль). Полученная смесь нагревалась при дистилляции в течение 24 часов. Реакция контролировалась посредством TLC (CH1Cl2/MeOH, v/v, 125/3). После завершения смесь была остужена до комнатной температуры и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (РЕ/EtOAc (v/v)=1/3) для получения соединения, указанного в заголовке шага, в виде бледно-желтого твердого вещества (35 мг; 54,4%).
MS (ESI, pos. ion) m/z: 452,1 [M+H]+; HPLC: 93,4%;
1H NMR (600 МГц; DMSO-d6) δ (ppm): 9,56 (d; J=7,5 Гц; 1H); 8,76 (s; 1H); 8,16 (s; 1H); 7,94 (s; 1H); 7,65 (t; J=8,0 Гц; 1H); 7,48 (dd; J=8,3; 1,7 Гц; 2H); 7,10 (s; 1H); 6,13-5,96 (m; 1H); 4,02 (s; 3H); 3,21-3,11 (m; 1H); 2,15-2,02 (m; 1H); 1,96-1,82 (m; 1H); 1,32-1,24 (m; 2H); 1,17-1,07 (m; 1H); 1,01 (t; J=7,3 Гц; 3H); 0,88-0,81 (m; 1H).
Пример 54 (S)-2-(1-((6-амин-5-(2-метил-2H-тетразол-5-ил)пиримидин-4-ил)амин)пропил)-5-хлор-3-циклопропилквиназолин-4(3H)-один
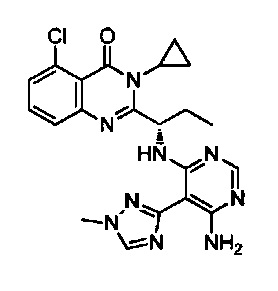
[529] К суспензии 6-хлор-5-(2-метил-2H-тетразол-5-ил)пиримидин-4-амина (31 мг; 0,147 ммоль) и (S)-2-(1-аминопропил)-5-хлор-3-циклопропилквиназолин-4(3H)-один (43 мг; 0,154 ммоль) в n-BuOH (3 мл) был добавлен DIPEA (38 мг; 0.293 ммоль). Полученная смесь нагревалась при дистилляции в течение 29 часов. Реакция контролировалась посредством TLC (PE/EtOAc, v/v, 1/4). После завершения смесь была остужена до комнатной температуры и сконцентрирована in vacuo. Осадок был растворен в EtOAc (15 мл), и полученная смесь была промыта водой (15 мл) и насыщенным минеральным раствором (10 мл). Органическая фаза была высушена над обезвоженным Na2SO4, и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (РЕ/EtOAc (v/v)=2/1) для получения соединения, указанного в заголовке шага, в виде грязно-белого твердого вещества (39 мг; 58,8%).
MS (ESI, pos. ion) m/z: 453,2 [M+H]+; HPLC: 98,3%;
1H NMR (400 МГц; CDCl3) δ (ppm): 9,02 (d; J=7,6 Гц; 1H); 8,17 (s; 1H); 7,57-7,48 (m; 2H); 7,42 (dd; J=6,4; 2,6 Гц; 1H); 6,34 (td; J=7,7; 5,3 Гц; 1H); 4,52 (s; 3H); 3,17-3,05 (m; 1H); 2,20-2,08 (m; 1H); 2,06-1,97 (m; 1H); 1,48-1,40 (m; 2H); 1,27-1,21 (m; 1H); 1,08 (t; J=7,4 Гц; 3H); 0,91-0,86 (m; 1H).
Пример 55 (S)-2-(1-((6-амин-5-(5-метил-1,2,4-оксадиазол-3-ил)пиримидин-4-ил)амин)-пропил)-3-циклопропил-5-фторквиназолин-4(3H)-один
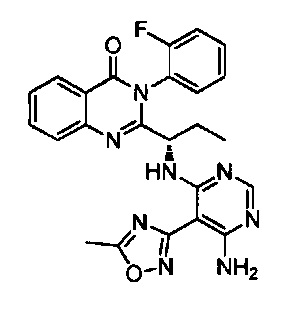
[530] Смесь 6-хлор-5-(5-метил-1,2,4-оксадиазол-3-ил)пиримидин-4-амина (23,0 мг; 0,11 ммоль), (S)-2-(1-аминопропил)-3-(2-фторфенил)квиназолин-4(3H)-один (38,8 мг; 0,13 ммоль) и N,N-циизопропилэтиламина (42,3 мг; 0,33 ммоль) в n-бутаноле (2,0 мл) была нагрета до 125°C и подвергалась дистилляции в течение последующих 24 часов. После завершения реагирующая смесь была сконцентрирована in vacuo, и осадок очищен подготовительным TLC (DCM/MeOH, v/v, 50/1) для получения соединения, указанного в заголовке шага, в виде светло-желтого твердого вещества (20 мг; 39%), состоящего из двух изомеров (А и В) в пропорции 3/4 (изомер А/изомер В).
MS (ESI, pos. ion) m/z: 473,2 [M+H]+; HPLC: 99.1% (общая степень чистоты изомера А и В);
Изомер A: 1H NMR (600 МГц; CDCl3) δ (ppm): 8,76 (d; J=7,3 Гц; 1H); 7,88 (s; 1H); 7,84-7,69 (m; 2H); 7,60-7,44 (m; 3H); 7,41-7,30 (m; 2H); 7,20 (t; J=8,8 Гц; 1H); 5,80 (br s; 1H); 5,32-5,26 (m; 1H); 2,71 (s; 3H); 2,16-2,08 (m; 1H); 2,02-1,88 (m; 1H); 0,95 (t; J=7,4 Гц; 3H).
Изомер В: 1H NMR (600 МГц; CDCl3) δ (ppm): 8,31 (d; J=7,8 Гц; 1H); 7,99 (s; 1H); 7,84-7,69 (m; 3H); 7,60-7,44 (m; 3H); 7,41-7,30 (m; 2H); 5,18 (td; J=7,7; 5,0 Гц; 1H); 2,74 (s; 3H); 2,45 (brs; 1H); 2,02-1,88 (m; 1H); 1,81 (tt; J=14,9; 7,4 Гц; 1H); 0,91 (t; J=7,3 Гц; 3H).
Пример 56 (S)-2-(1-((6-амин-5-(3-этил-1,2,4-оксадиазол-5-ил)пиримидин-4-ил)амин)-пропил)-5-хлор-3-циклопропилквиназолин-4(3H)-один
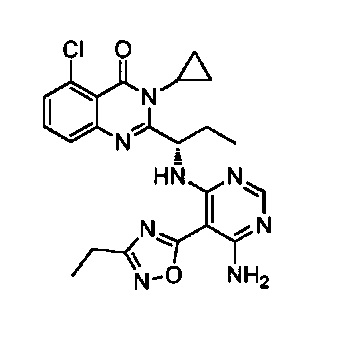
Шаг 1) N'-гидроксипропионимидамид
[531] Гидроксиламин гидрохлорид (18,9 г; 272,7 ммоль) и безводный карбонат калия (37,6 г; 272,7 ммоль) были приготовлены в виде суспензии в EtOH/H2O (200 мл/50 мл), и смесь перемешивалась при комнатной температуре в течение 1 часа, затем был добавлен пропиононитрил (10,0 г; 181,8 ммоль), и полученная смесь была нагрета для дистилляции, и перемешивалась в течение последующих 20 часов. Реакция контролировалась посредством TLC (CH2Cl2/МеОН, v/v, 50/1). После завершения реакции неорганическая соль была отфильтрована, и фильтрат был сконцентрирован in vacuo для получения соединения, указанного в заголовке шага, в виде бледно-желтого твердого вещества (13,5 г, 81%).
Шаг 2) N'-(4,6-дихлорпиримидин-5-карбонил)окси)пропионимидамид
[532] К суспензии 4,6-дихлорпиримидин-5-карбонил хлорида (20,0 г; 94,8 ммоль) в CH2Cl2 (100 мл) при 0°C была добавлена смесь N-гидроксипропионимидамида (8,3 г; 94,8 ммоль) и DIPEA (25,1 г; 190,6 ммоль) в CH2Cl2 (100 мл). Реагирующая смесь перемешивалась в течение 2 часов при 0°C. Реакция контролировалась посредством TLC (CH2Cl2/МеОН, v/v, 100/1). После завершения реакции реагирующая смесь была разведена водой (100 мл). Отсепарированная органическая фаза была промыта насыщенным гидратогенным раствором NaHCO3 (100 мл) и насыщенным минеральным раствором (100 мл), высушена над обезвоженным Na2SO4, и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (DCM/MeOH (v/v)=500/1) для получения соединения, указанного в заголовке шага, в виде желтого твердого вещества (13,7 г; 55%).
MS (ESI, pos. ion) m/z: 263,1 [M+H]+.
Шаг 3) N'-(4-амин-6-хлорпиримидин-5-карбонил)окси)пропионимидамид
[533] Раствор N'-((4,6-дихлорпиримидин-5-карбонил)окси)пропионимидамида (3,1 г; 11,8 ммоль) в THF (30 мл) был продут газообразным NH3. Реагирующую смесь перемешивали в течение суток при комнатной температуре. Реакция контролировалась посредством TLC (CH2Cl2/МеОН, v/v, 100/1). После завершения реакции смесь была сконцентрирована in vacuo. Осадок был разведен смесью EtOH (2 мл) и воды (10 мл). Полученную смесь перемешивали в течение 1 часа и профильтровали для получения соединения, указанного в заголовке шага, в виде белого твердого вещества (2,5 г; 88%).
MS (ESI, pos. ion) m/z: 244,1 [M+H]+.
Шаг 4) 6-хлор-5-(3-метил-1,2,4-оксадиазол-5-ил)пиримидин-4-амин
[534] К суспензии N-((4-амин-6-хлорпиримидин-5-карбонил)окси)пропионимидамида (7,5 г; 30,8 ммоль) в DMSO (50 мл) был добавлен BU4NF (1 M в THF, 60 мл; 60,0 ммоль), и смесь перемешивалась при rt в течение суток. Реакция контролировалась посредством TLC (PE/EtOAc, v/v, 1/1). После завершения реакции смесь был разведена в EtOAc (100 мл), и полученная смесь была промыта водой (50 мл×2) и насыщенным минеральным раствором (100 мл). Отсепарированная органическая фаза была высушена над обезвоженным Na2SO4, и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (РЕ/EtOAc (v/v)=6/1) для получения соединения, указанного в заголовке шага, в виде белого твердого вещества (1,1 г; 16%).
MS (ESI, pos. ion) m/z: 226,1 [M+H]+;
1H NMR (400 МГц; DMSO-d6) δ (ppm): 8,41 (s; 1H); 2,84 (q; J=7,5 Гц; 2H); 1,30 (t; J=7,5 Гц; 3H).
Шаг 5) (S)-2-(1-((6-амин-5-(3-этил-1,2,4-оксадиазол-5-ил)пиримидин-4-ил)амин)пропил)-5-хлор-3-циклопропилквиназолин-4(3H)-один
[535] К суспензии (S)-2-(1-аминопропил)-5-хлор-3-циклопропилквиназолин-4(3H)-один (70 мг; 0,252 ммоль) и 6-хлор-5-(3-этил-1,2,4-оксадиазол-5-ил)пиримидин-4-амина (55 мг; 0,243 ммоль) в n-BuOH (4 мл) был добавлен DIPEA (75 мг; 0.580 ммоль). Полученная смесь нагревалась при дистилляции в течение 12 часов. Реакция контролировалась посредством TLC (РЕ/EtOAc, v/v, 1/1). После завершения смесь была остужена до комнатной температуры и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (РЕ/EtOAc (v/v)=2/1) для получения соединения, указанного в заголовке шага, в виде белого твердого вещества (74 мг; 65%).
MS (ESI, pos. ion) m/z: 467,1 [M+H]+; HPLC: 98,9%;
1H NMR (600 МГц; CDCl3) δ (ppm) 8,96 (d; J=7,3 Гц; 1H); 8,15 (s; 1H); 7,54 (d; J=4,2 Гц; 2H); 7,47-7,38 (m; 1H); 6,33 (m 1H); 3,10 (m; 1H); 2,90 (q; J=15,0; 7,5 Гц; 2H); 2,16 (m; 1H); 1,99 (m 1H); 1,44 (t; J=7,5 Гц; 3H); 1,27 (m; 2H); 1,05 (t; J=7,3 Гц; 3H); 0,96-0,86 (m; 2H)
Пример 57
(S)-2-(1-((6-амин-5-(3-изопропил-1,2,4-оксадиазол-5-ил)пиримидин-4-ил)амин)-этил)-5-хлор-3-циклопропилквиназолин-4(3H)-один
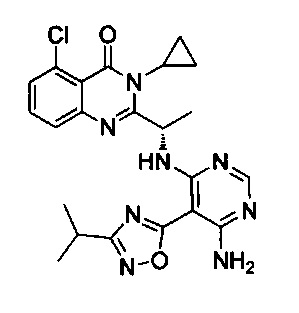
Шаг 1) N'-гидроксиизобутиримидамид
[536] Суспензия гидроксиламин гидрохлорида (11,1 г; 159,7 ммоль) и безводного карбоната калия (23,9 г; 173,7 ммоль) в EtOH/H2O (150 мл/50 мл) перемешивалась при комнатной температуре в течение 1 часа, затем был добавлен изобутиронитрил (10,0 г; 144,7 ммоль)), и полученная смесь была нагрета для дистилляции, и перемешивалась в течение последующих 20 часов. Реакция контролировалась посредством TLC (CH2Cl2/МеОН, v/v, 50/1). После завершения неорганическая соль была отфильтрована. Фильтрат был сконцентрирован in vacuo для получения соединения, указанного в заголовке шага, в виде бледно-желтого твердого вещества (11,8 г, 80%).
MS (ESI, pos. ion) m/z: 103,2 [M+H]+;
Шаг 2) N'-(4,6-дихлорпиримидин-5-карбонил)изобутиримидамид
[537] К суспензии 4,6-дихлорпиримидин-5-карбонилхлорида (21,1 г; 100,1 ммоль) в CH2Cl2 (100 мл) при 0°C была добавлена смесь N-гидроксиизобутиримидамида (10,3 г; 100,1 ммоль) и DIPEA (25,8 г; 200,2 ммоль) в CH2Cl2 (100 мл). Реагирующая смесь перемешивалась в течение 2 часов при 0°C.Реакция контролировалась посредством TLC (CH2Cl2/МеОН, v/v, 100/1). После завершения реакции реагирующая смесь была разведена водой (100 мл). Отсепарированная органическая фаза была промыта насыщенным гидратогенным раствором NaHCO3 (100 мл) и насыщенным минеральным раствором (100 мл), высушена над обезвоженным Na2SO4, и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (PE/EtOAc (v/v)=500/1) для получения соединения, указанного в заголовке шага, в виде желтого масла (12,4 г; 45%).
MS (ESI, pos. ion) m/z: 277,1 [M+H]+.
Шаг 3) N'-(4-амин-6-хлорпиримидин-5-карбонил)окси)изобутиримидамид
[538] Раствор N'-((4,6-дихлорпиримидин-5-карбонил)окси)изобутиримидамида (12,4 г; 44,7 ммоль) в THF (100 мл) был продут газообразным NH3. Реагирующую смесь перемешивали в течение суток при комнатной температуре. Реакция контролировалась посредством TLC (DCM). После завершения реагирующая смесь была сконцентрирована in vacuo. Осадок был разведен смесью EtOH (2 мл) и воды (50 мл). Полученную смесь перемешивали в течение 1 часа и профильтровали для получения соединения, указанного в заголовке шага, в виде белого твердого вещества (9,8 г; 85%).
MS (ESI, pos. ion) m/z: 258,1 [M+H]+;
1H NMR (400 МГц; DMSO-d6) δ (ppm): 8,27 (s; 1H); 7,55 (s; 2H); 6,32 (s; 2H); 2,40 (m; 1H); 1,13 (d; J=7,0Hz; 6H).
Шаг 4) 6-хлор-5-(3-метил-1,2,4-оксадиазол-5-ил)пиримидин-4-амин
[539] К суспензии N'-((4-амин-6-хлорпиримидин-5-карбонил)окси)изобутиримид амида (7.5 г; 30.8 ммоль) в DMSO (20 мл) был добавлен BU4NF (1 M в THF, 60 мл; 60,0 ммоль), и полученная смесь перемешивалась при rt в течение суток. Реакция контролировалась посредством TLC (РЕ/EtOAc, v/v, 1/1). После завершения реагирующая смесь была разведена EtOAc (100 мл), промыта водой (100 мл) и насыщенным минеральным раствором (100 мл). Отсепарированная органическая фаза была высушена над обезвоженным Na2SO4, и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (РЕ/EtOAc (v/v)=7/1) для получения соединения, указанного в заголовке шага, в виде белого твердого вещества (0,9 г; 14%).
MS (ESI, pos. ion) m/z: 240,1 [M+H]+;
1HNMR (400 МГц; DMSO-d6) δ (ppm): 8,47 (s; 1H); 8,43 (s; 1H); 8,10 (s; 1H); 3,18 (m; 1H); 1,34 (d; J=6,9 Гц; 6H).
Шаг 5) (S)-2-(1-((6-амин-5-(3-изопропил-1,2,4-оксадиазол-5-ил)пиримидин-4-ил)амин)этил)-5-хлор-3-циклопропилквиназолин-4(3H)-один
[540] К суспензии (S)-2-(1-аминоэтил)-5-хлор-3-циклопропилквиназолин -4(3H)-один (70 мг; 0,27 ммоль) и 6-хлор-5-(3-изопропил-1,2,4-оксадиазол-5-ил)пиримидин-4-амина (65 мг; 0,28 ммоль) в n-BuOH (4 мл) был добавлен DIPEA (75 мг; 0,58 ммоль). Полученная смесь нагревалась при дистилляции в течение 20 часов. Реакция контролировалась посредством TLC (РЕ/EtOAc, v/v, 1/1). После завершения смесь была остужена до комнатной температуры и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (DCM/MeOH (v/v)=100/1) для получения соединения, указанного в заголовке шага, в виде белого твердого вещества (82 мг; 65%).
MS (ESI, pos. ion) m/v: 467,1 [M+H]+; HPLC: 99,2%;
1H NMR (400 МГц; DMSO-d6) δ (ppm): 9,24 (d; J=7,1 Гц; 1H); 8,12 (s; 1H); 7,72 (t; J=8,0 Гц; 1H); 7,52 (m; 4H); 6,19 (m; 1H); 3,20 (m; 1H); 3,13 (m; 1H); 1,59 (d; J=6,6 Гц; 3H); 1,36 (d; J=6,9 Гц; 6H); 1,26 (m; 1H); 1,03 (m; 1H); 0,91-0,78 (m; 2H).
Пример 58 (S)-3-(4-амин-6-((1-(5-хлор-3-циклопропил-4-оксо-3,4-дигидроквиназолин-2-ил)пропил)амин)пиримидин-5-ил)-1-метил-1H-1,2,4-триазол-5-карбоксамид
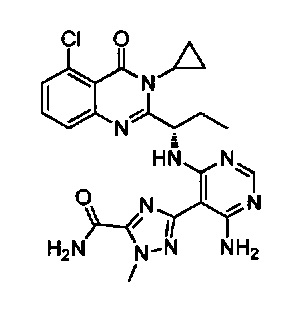
Шаг 1) 3-(4-амин-6-хлорпиримидин-5-ил)-1-метил-1Н-1,2,4-триазол-5-карбоксамид
[541] Смесь этил 3-(4-амин-6-хлорпиримидин-5-ил)-1-метил-1Н-1,2,4-триазол-5-карбоксилата (100 мг; 0,354 ммоль) и раствор NH3 в метаноле (10 мл) были герметизированы в колбе и перемешивались при 65°C в течение 5 часов. Затем смесь была профильтрована, и фильтрат был сконцентрирован in vacuo для получения соединения, указанного в заголовке шага, в виде белого твердого вещества (85 мг; 94%).
MS (ESI, pos. ion) m/z: 254,1 [M+H]+.
Пример 2 (S)-3-(4-амин-6-((1-(5-хлор-3-циклопропил-4-оксо-3,4-дигидроквиназолин-2-ил)пропил)амин)пиримидин-5-ил)-1-метил-1H-1,2,4-триазол-5-карбоксамид
[542] К смеси (S)-2-(1-аминопропил)-5-хлор-3-циклопропилквиназолин-4(3H)-один (97,7 мг; 0,352 ммоль) и 3-(4-амин-6-хлорпиримидин-5-ил)-1-метил-1Н-1,2,4-триазол-5-карбоксамида (85 мг; 0,335 ммоль) в 5 мл n-BuOH был добавлен DIPEA (86,6 мг; 0,670 ммоль), и смесь была нагрета до 125°C и перемешивалась в течение суток. Затем смесь была остужена до комнатной температуры, профильтрована, и фильтрат промыт 2 мл этанола для получения соединения, указанного в заголовке шага, в виде белого твердого вещества (30 мг; 18%).
MS (ESI, pos. ion) m/z: 495,2 [M+H]+; HPLC: 95%;
1H NMR (400 МГц; DMSO-d6) δ (ppm): 9,17 (d; J=7,5 Гц; 1H); 8,63 (s; 1H); 8,32 (s; 1H); 8,08 (s; 1H); 7,96 (s; 1H); 7,67 (t; J=8,0 Гц; 1H); 7,49 (m; 2H); 7,07 (s; 1H); 6,15 (m; 1H); 4,29 (s; 3H); 3,21-3,13 (m; 1H); 2,06 (m; 1H); 1,94 (m; 1H); 1,28 (m; 2H); 1,07 (m; 1H); 0,99 (t; J=7,3 Гц; 3H); 0,90-0,78 (m; 1H).
Пример 59
(S)-2-(1-((6-амин-5-(5-изопропил-1,2,4-оксадиазол-3-ил)пиримидин-4-ил)амин)этил)-5-хлор-3-циклопропилквиназолин-4(3H)-один
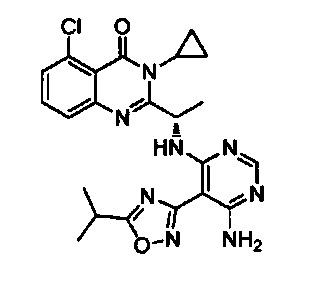
Шаг 1) 4,6-диметоксипиримидин-5-карбальдегид
[543] К суспензии 4,6-дихлорпиримидин-5-карбальдегида (5 г; 28,25 ммоль) в осушенном МеОН (100 мл) был добавлен K2CO3 (7,8 г; 56,5 ммоль). Реагирующая смесь была нагрета до 90°C и перемешивалась дополнительно в течение 1 часа. Затем смесь была сконцентрирована in vacuo, и осадок был растворен в смешанном растворе воды (100 мл) и DCM (100 мл). Отсепарированная гидратогенная фаза была экстрактирована посредством DCM (50 мл×3), и комбинированные органические фазы были сконцентрированы in vacuo. Осадок был разведен смешанным растворителем из РЕ (20 мл) и DCM (1 мл), полученная смесь перемешивалась при rt в течение 3 часов, затем была профильтрована для получения желтого твердого вещества, которое было очищено хроматографией на силикагельной колонне ((РЕ/EtOAc (v/v)=5/1) для получения соединения, указанного в заголовке шага, в виде белого твердого вещества (2,2 г; 46,3%).
MS (ESI, pos. ion) m/z: 169,1 [M+H]+.
Шаг 2) 4,6-диметоксипиримидин-5-карбальдегид оксим
[544] К раствору 4,6-диметоксипиримидин-5-карбальдегида (2,2 г; 13,08 ммоль) в этилацетате (45 мл) был добавлен раствор NH2OH-HCl (910 г; 13,08 ммоль) в воде (18 мл), затем был добавлен уксуснокислый натрий (1,07 г; 13,08 ммоль) при комнатной температуре. Смесь перемешивалась при 28°C в течение 3 часов, затем была профильтрована, и фильтрат промыт 30 мл воды, высушен при 60°C в вакуумной сушилке для получения соединения, указанного в заголовке шага, в виде белого твердого вещества (2,2 г; 91,8%).
MS (ESI, pos. ion) m/z: 184,1 [M+H]+;
1HNMR (600 МГц; DMSO-d6): δ 11,47 (s; 1H); 8,47 (s; 1H); 8,08 (s; 1H); 3,96 (s; 6H).
Шаг 3) 3-(4,6-диметоксипиримидин-5-ил)-5-изопропил-1,2,4-оксадиазол
[545] В трехгорлую химическую колбу, заправленную 4,6-диметоксипиримидин-5-карбальдегид оксимом (50 мг; 0,273 ммоль) и (NH4)2Ce(NO3)6 (300 мг; 0,546 ммоль) был добавлен CH3CN (3 мл) при rt в защитной атмосфере N2. Реагирующая смесь была нагрета до 70°C и перемешивалась в течение 4 часов, затем была профильтрована и сконцентрирована in vacuo. Осадок был растворен в EtOAc (10 мл), и полученная смесь была промыта насыщенным гидратогенным раствором NH2Cl (10 мл) и насыщенным минеральным раствором (10 мл).
Отсепарированная органическая фаза была сконцентрирована in vacuo, и осадок был очищен хроматографией на силикагельной колонне (РЕ/EtOAc (v/v)=3/1) для получения соединения, указанного в заголовке шага, в виде желтого твердого вещества (24 мг; 35,1%).
MS (ESI, pos. ion) m/z: 251,1 [M+H]+;
1H NMR (400 МГц; CDCl3) δ (ppm): 8,54 (s; 1H); 4,01 (s; 6H); 3,33 (m; 1H); 1,50 (d; J=7,0 Гц; 6H).
Шаг 4) 3-(4,6-диметоксипиримидин-5-ил)-5-изопропил-1,2,4-оксадиазол
[546] К суспензии 3-(4,6-диметоксипиримидин-5-ил)-5-изопропил-1,2,4-оксадиазола (573 мг; 2,29 ммоль) и DMF (6.0 мл) в толуоле (50 мл) был добавлен РОСl3 (6 мл, 62,64 ммоль). Реагирующая смесь была нагрета для дистилляции и перемешивалась в течение последующих 24 часов. Реагирующая смесь была сконцентрирована in vacuo, и осадок был очищен хроматографией на силикагельной колонне (РЕ/EtOAc (v/v)=10/1) для получения соединения, указанного в заголовке шага, в виде желтого масла (350 мг; 58,9%).
Шаг 5) 6-хлор-5-(5-метил-1,2,4-оксадиазол-3-ил)пиримидин-4-амин
[547] Раствор 3-(4,6-дихлорпиримидин-5-ил)-5-изопропил-1,2,4-оксадиазола (350 мг; 1,35 ммоль) в THF (10 мл) был продут газообразным NH3. Реакция контролировалась посредством TLC (РЕ/EtOAc, v/v, 3/1). После завершения реагирующая смесь была профильтрована, и фильтрат был сконцентрирован in vacuo для получения соединения, указанного в заголовке шага, в виде желтого твердого вещества (325 мг; 100%).
MS (ESI, pos. ion) m/z: 240,1 [M+H]+;
1H NMR (400 МГц; DMSO-d6) δ (ppm): 8.35 (s; 1H); 7.11 (s; 2H); 3.37 (m; 1H); 1.38 (d;J=7.0 Гц; 6H).
Шаг 6) (S)-2-(1-((6-амин-5-(5-изопропил-1,2,4-оксадиазол-3-ил)пиримидин-4-ил)амин)этил)-5-хлор-3-циклопропилквиназолин-4(3H)-один
[548] К суспензии (S)-2-(1-аминоэтил)-5-хлор-3-циклопропилквиназолин-4(3H)-один (50 мг; 0,19 ммоль) и 6-хлор-5-(5-изопропил-1,2,4-оксадиазол-3-ил)пиримидин-4-амина (45,5 мг; 0,19 ммоль) в n-BuOH (4 мл) был добавлен DIPEA (49.2 мг; 0.38 ммоль). Смесь была нагрета до 120°C и перемешивалась в течение суток, затем была сконцентрирована in vacuo. Осадок был растворен в EtOAc (10 мл), и полученная смесь была промыта водой (10 мл). Отсепарированная органическая фаза была сконцентрирована in vacuo, и осадок был очищен хроматографией на силикагельной колонне (PE/EtOAc (v/v)=1/3) для получения соединения, указанного в заголовке шага, в виде грязно-белого твердого вещества (35 мг; 39,5%).
MS (ESI, pos. ion) m/z: 467,2 [M+H]+;
1H NMR (400 МГц; DMSO-d6) δ (ppm): 8,95-8,93 (d; J=7,3 Гц; 1H); 8,06 (s; 1H); 7,72-7,68 (t; J=8,0 Гц; 1H); 7,54 (s; 2H); 7,50 (m; 2H); 6,21-6,15 (m; 1H); 3,43-3,40 (m; 1H); 3,16-3,13 (m; 1H); 2,03-1,97 (m; 1H); 1,59-1,57 (d; J=6,6 Гц; 3H); 1,43-1,42 (d; J=7,0 Гц; 6H); 1,06-1,04 (m; 1H); 0,88-0,84 (m; 2H).
Пример 60
(S)-2-(1-((6-амин-5-(1-метил-1Н-1,2,4-триазол-3-ил)римидин-4-ил)амин)пропил)-3-(2-фторфенил)квиназолин-4(3H)-один
[549] К суспензии 6-хлор-5-(1-метил-1H-1,2,4-триазол-3-ил)пиримидин-4-амина (30 мг; 0,142 ммоль) и (S)-2-(1-аминопропил)-3-(2-фторфенил)квиназолин-4(3H)-один (44 мг; 0,150 ммоль) в n-BuOH (1 мл) был добавлен DIPEA (37 мг; 0.284 ммоль). Реагирующая смесь была нагрета для дистилляции и перемешивалась в течение 22 часов, контролировалась посредством TLC (DCM/MeOH, v/v, 100/3), затем была остужена до rt и сконцентрирована in vacuo. Осадок был растворен в EtOAc (15 мл), и полученная смесь была промыта водой (15 мл) и насыщенным минеральным раствором (10 мл). Отсепарированная органическая фаза была высушена над обезвоженным Na2SO4, и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (РЕ/EtOAc (v/v)=1/1) для получения соединения, указанного в заголовке шага, в виде белого твердого вещества (15 мг; выход 22,3%), состоящего из двух изомеров (А и В) в пропорции 3/2 (изомер А/изомер В).
MS (ESI, pos. ion): 472,2 [M+H]+; HPLC: 95,4% (общая степень чистоты изомера А и В);
Изомер A: 1HNMR (400 МГц; CDCl3) δ (ppm): 8,31 (d; J=7,7 Гц; 1H); 8,15 (s; 1H); 7,95 (s; 1H); 7,82-7,68 (m; 2H); 7,58-7,44 (m; 3H); 7,39-7,30 (m; 2H); 5,21-5,12 (m; 1H); 4,05 (s; 3H); 3,68 (t; J=2,5 Гц; 2H); 0,99-0,93 (m; 3H).
Изомер В: 1H NMR (400 МГц; CDCl3) δ (ppm): 8,31 (d; J=7,7 Гц; 1H); 8,10 (s; 1H); 7,84 (s; 1H); 7,82-7,68 (m; 2H); 7,58-7,44 (m; 3H); 7,39-7,30 (m; 2H); 5,35-5,26 (m; 1H); 4,02 (s; 1H); 3,68 (t; J=2,5 Гц; 2H); 0,99-0,93 (m; 3H).
Пример 61
(S)-2-(1-((6-амин-5-(5-этил-1,2,4-оксадиазол-3-ил)пиримидин-4-ил)амин)-этил)-5-хлор-3-циклопропилквиназолин-4(3H)-один

Шаг 1) 4,6-диметоксипиримидин-5-карбальдегид
[550] К суспензии 4,6-дихлорпиримидин-5-карбальдегида (5 г; 28,25 ммоль) в осушенном МеОН (100 мл) был добавлен K2CO3 (7,8 г; 56,5 ммоль). Реагирующая смесь была нагрета до 90°C и перемешивалась в течение последующего 1 часа, затем была сконцентрирована in vacuo. Осадок был растворен в смешанном растворе воды (100 мл) и DCM (100 мл), смесь была экстрактирована посредством DCM (50 мл×3). Отсепарированные органические фазы были сконцентрирована in vacuo. Осадок перемешивался с DCM/PE (10 мл, v/v, 1/20) при rt в течение 3 часов, и был профильтрован с получением полуфабриката соединения в виде желтого твердого вещества. Полуфабрикат был очищен хроматографией на силикагельной колонне (PE/EtOAc (v/v)=5/1) для получения соединения, указанного в заголовке шага, в виде белого твердого вещества (2,2 г; 46,3%).
MS (ESI, pos. ion) m/z: 169,1 [M+H]+.
Шаг 2) 4,6-диметоксипиримидин-5-карбальдегид оксим
[551] К раствору 4,6-диметоксипиримидин-5-карбальдегида (2,2 г; 13,08 ммоль) в этилацетате (45 мл) был добавлен раствор NH2OH-HCl (910 г; 13,08 ммоль) в воде (18 мл), затем был добавлен уксуснокислый натрий (1.07 г; 13.08 ммоль) при комнатной температуре. Смесь перемешивалась при 28°C в течение 3 часов, затем была профильтрована, и фильтрат промыт 30 мл воды, высушен при 60°C в вакуумной сушилке для получения соединения, указанного в заголовке шага, в виде белого твердого вещества (2,2 г; 91,8%).
MS (ESI, pos. ion) m/z: 184,1 [M+H]+;
1HNMR (600 МГц; DMSO-d6): δ (ppm): 11.47 (s; 1H); 8.47 (s; 1H); 8.08 (s; 1H); 3.96 (s; 6H).
Шаг 3) 3-(4,6-диметоксипиримидин-5-ил)-5-метил-1,2,4-оксадиазол
[552] В трехгорлую химическую колбу, заправленную 4,6-диметоксипиримидин-5-карбальдегид оксимом (2,0 г; 10,93 ммоль) и (NH4)2Ce(NO3)6 (12,0 г; 21,86 ммоль) был добавлен CH3CHCN (37 мл) при rt в защитной атмосфере N2. Реагирующая смесь была нагрета до 70°C и перемешивалась в течение 6 часов, затем была профильтрована и сконцентрирована in vacuo. Осадок был растворен в EtOAc (50 мл), и полученная смесь была промыта насыщенным гидратогенным раствором Na2CO3 (50 мл×2) и насыщенным минеральным раствором (50 мл×2). Отсепарированная органическая фаза была сконцентрирована in vacuo, и осадок был очищен хроматографией на силикагельной колонне (РЕ/EtOAc (v/v)=10/1) для получения соединения, указанного в заголовке шага, в виде желтого твердого вещества (805 мг; 31,0%).
MS (ESI, pos. ion) m/z: 237,2 [M+H]+.
Шаг 4) 3-(4,6-дихлорпиримидин-5-ил)-5-этил-1,2,4-оксадиазол
[553] К суспензии 3-(4,6-диметоксипиримидин-5-ил)-5-этил-1,2,4-оксадиазола (650 мг; 2,75 ммоль) и DMF (6,0 мл) в толуоле (14 мл) был добавлен POCl3 (6 мл, 62,75 ммоль). Реагирующая смесь была нагрета до 120°C и перемешивалась в течение последующих 10 часов, затем была сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (РЕ/EtOAc (v/v)=10/1) для получения соединения, указанного в заголовке шага, в виде желтого сиропа (320 мг; выход 51,4%).
MS (ESI, pos. ion) m/z: 245,1 [M+H]+.
Шаг 5) 6-хлор-5-(5-этил-1,2,4-оксадиазол-3-ил)пиримидин-4-амин
[554] Раствор 3-(4,6-дихлорпиримидин-5-ил)-5-этил-1,2,4-оксадиазола (320 мг; 1,31 ммоль) в THF (10 мл) был продут NH3 (газ) при rt в течение 1 часа, реагирующая смесь была профильтрована, и фильтрат был сконцентрирован in vacuo для получения соединения, указанного в заголовке шага, в виде бледно-желтого твердого вещества (330 мг; выход 100%).
MS (ESI, pos. ion) m/z: 226.1 [M+H]+;
1H NMR (400 МГц; DMSO-d6) δ (ppm): 8,35 (s; 1H); 7,85 (s; 1H); 7,07 (s; 1H); 3,05-2,99 (q; J=7,6 Гц; 2H); 1,36-1,32 (t; J=7,6 Гц; 3H).
Шаг 6)
(S)-2-(1-((6-амин-5-(5-этил-1,2,4-оксадиазол-3-ил)пиримидин-4-ил)амин)пропил)-5-хлор-3-циклопропилквиназолин-4(3H)-один
[555] К суспензии (S)-2-(1-аминоэтил)-5-хлор-3-циклопропилквиназолин-4(3H)-один (50 мг; 0.19 ммоль) и 6-хлор-5-(5-этил-1,2,4-оксадиазол-3-ил)пиримидин-4-амина (45,5 мг; 0,19 ммоль) в n-BuOH (4 мл) был добавлен DIPEA (49,2 мг; 0,38 ммоль). Смесь была нагрета до 120°C и перемешивалась в течение суток, затем была сконцентрирована in vacuo. Осадок был растворен в EtOAc (15 мл), и полученная смесь была промыта водой (10 мл×2). Отсепарированная органическая фаза была высушена над обезвоженным Na2SO4, и сконцентрирована in vacuo. Осадок был очищен моментальной хроматографией на силикагельной колонне (PE/EtOAc (v/v)=1/3) для получения соединения, указанного в заголовке шага, в виде желтого твердого вещества (31 мг; выход 36%).
MS (ESI, pos. ion) m/z: 453,3 [M+H]+;
1HNMR (400 МГц, CDCl3) δ (ppm): 9,01 (d, J=7,4 Гц, 1H), 8,14 (s, 1H), 7,53 (m, 2H), 7,46-7,39 (m, 1H), 6,33 (m, 1H), 6,01-5,39 (s, 2H), 3,12 (m,lH), 3,04 (q, J=7,6 Гц, 2H), 1,65 (d, J=9,6 Гц, 3H), 1,50 (t, J=7,6 Гц, 3H), 1,46-1,39 (m, 2H), 0,90 (m, 2H).
Пример 62
(S)-1-(1-((6-амин-5-(3-метил-1,2,4-оксадиазол-5-ил)пиримидин-4-ил)амин)пропил)-5-хлор-3-((1R,2S)-2-фторциклопропил)квиназолин-4(3H)-один
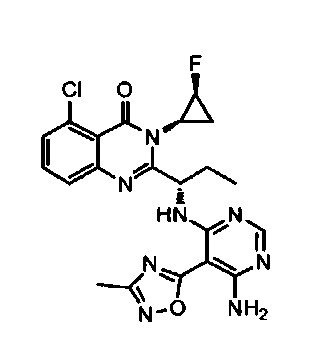
Шаг 1) 2-хлор-N-((1R,2S)-2-фторциклопропил)-6-нитробензамид
[556] К суспензии 2-метил-6-нитробензойной кислоты (806 мг; 4,0 ммоль) в толуоле (10 мл) был добавлен одной порцией SOCl2 (952 мг; 8,0 ммоль) при комнатной температуре. После добавления реагирующая смесь перемешивалась при 110°C в течение 9 часов и была сконцентрирована in vacuo для получения желтого масла, которое было растворено в 1,4-диоксане (10 мл) для получения бледно-желтой суспензии. Полученная бледно-желтая суспензия была по каплям добавлена к суспензии (1R,2S)-2-фторциклопропиламин тосилата (1,0 г; 4,0 ммоль) и NaHCO4 (1.34 г; 16.0 ммоль) в 1,4-диоксане (10 мл) при 5°C. Реагирующая смесь перемешивалась при комнатной температуре в течение суток, затем разведена водой (100 мл). Полученная смесь была экстрактирована посредством EtOAc (50 мл×3). Комбинированные органические фазы были промыты насыщенным минеральным раствором (100 мл), высушены над обезвоженным Na2SO4, и сконцентрированы in vacuo для получения соединения, указанного в заголовке шага, в виде бледно-желтого твердого вещества (940 мг; выход 90,86%).
MS (ESI, pos. ion) m/z: 259,0 [M+H]+;
1H NMR (600 МГц; CDCl3) δ (ppm): 8,12 (d; J=8,3 Гц; 1H); 7,75 (d; J=8,0 Гц; 1H); 7,55 (t; J=8,2 Гц; 1H); 6,08 (s; 1H); 4,80 (ddd; J=63,6; 8,8; 5,7 Гц; 1H); 3,08 (td; J=9,4; 5,3 Гц; 1H); 1,32 (dt; J=14,9; 8,3 Гц; 1H); 1,24 (m; 1H).
Шаг 2) терт-бутил ((S)-1-(2-хлор-N-((1R,2S)-2-фторциклопропил)-6-нитробензоамидо)-1-оксобутан-2-ил)карбамат
[557] К суспензии 2-хлор-N-((1R,2S)-2-фторциклопропил)-6-нитробензоамида (940 мг; 3,63 ммоль) в толуоле (20 мл) был добавлен одной порцией SOCl2 (2 мл, 27,57 ммоль) при комнатной температуре. Реагирующая смесь перемешивалась при 120°C в течение 10 часов и была сконцентрирована in vacuo для получения бледно-коричневого масла, которое было растворено в безводном DCM (10 мл) для получения бледно-желтого раствора.
К раствору (S)-2-((терт-бутоксикарбонил)амин)бутановой кислоты (734 мг; 3,63 ммоль) и DIPEA (1,41 г; 10,89 ммоль) в 10 мл безводного DCM был добавлен упомянутый выше бледно-желтый раствор при 0°C. Полученная реагирующая смесь перемешивалась при комнатной температуре в течение 24 часов, затем была промыт водой (30 мл×3) и насыщенным минеральным раствором (10 мл). Отсепарированная органическая фаза была высушена над обезвоженным Na2SO4, и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (PE/EtOAc (v/v)=10/1) для получения соединения, указанного в заголовке шага, в виде желтого масла (0,83 г; выход 52,34%).
MS (ESI, pos. ion) m/z: 466,1 [M+Na]+.
Шаг 3) терт-бутил((S)-1-(5-хлор-3-((1R,2S)-2-фторциклопропил)-4-оксо-3,4-дигидроквиназолин-2-ил)пропил)карбамат
[558] К раствору (терт-бутил((S)-1-(2-хлор-N-((1R,2S)-2-фторциклопропил)-6-нитробензоамидо)-1-оксобутан-2-ил)карбамата (730 мг; 1,64 ммоль) в уксусной кислоте (5 мл) была одной порцией добавлена цинковая пыль (420 мг; 6,58 ммоль). Реагирующая смесь перемешивалась при 35°C в течение 24 часов, затем была отфильтрована, и фильтрат сконцентрирован in vacuo. Осадок был растворен в EtOAc (30 мл), и полученная смесь была промыта насыщенным раствором NaHCO3 (10 мл) и насыщенным минеральным раствором (10 мл). Отсепарированная органическая фаза была высушена над обезвоженным сульфатом натрия и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (РЕ/EtOAc (v/v)=10/1) для получения соединения, указанного в заголовке шага, в виде бледно-желтого твердого вещества (226 мг; выход 34,81%).
MS (ESI, pos. ion) m/z: 396,2 [M+H]+.
Шаг 4) -1-(1-аминопропил)-5-хлор-3-((1R,2S)-2-фторциклопропил)квиназолин-4(3H)-один
[559] К раствору терт-бутил-((S)-1-(5-хлор-3-((1R,2S)-2-фторциклопропил)-4-оксо-3,4-дигидроквиназолин-2-ил)пропил)карбамата (233 мг; 0,59 ммоль) в EtOAc (6 мл) был добавлен одной порцией раствор HCl в EtOAc (3.5 М, 6 мл) при комнатной температуре. Смесь перемешивалась при комнатной температуре на протяжении 6 часов. Полученная суспензия была разведена в воде (50 мл). Отсепарированная гидратогенная фаза была экстрактирована посредством EtOAc (20 мл×3), затем нейтрализована до рН=8 с помощью порошка Na2CO3 и вновь экстрактирована с помощью EtOAc (30 мл×4). Комбинированные органические фазы были промыты насыщенным минеральным раствором (50 мл), высушены над обезвоженным Na2SO4, и сконцентрированы in vacuo для получения соединения, указанного в заголовке шага, в виде бледно-желтого твердого вещества (151 мг; 86,54%).
MS (ESI, pos. ion) m/z: 296,1 [M+H]+;
1H NMR (400 МГц; CDCl3) δ (ppm): 7,61-7,53 (m; 2H); 7,45 (dd; J=6,7; 2,3 Гц; 1H); 4,97 (ddd; J=62,8; 8,7; 5,4 Гц; 1H); 4,28 (t; J=6,2 Гц; 1H); 3,04 (dd; J=13,9; 5,4 Гц; 1H); 2,04-1,94 (m; 1H); 1,75 (dt;J=18,0; 5,4 Гц; 2H); 1,55-1,42 (m; 1H); 1,02 (t; J=7,4T4; 3H).
Пример 5
(S)-1-(1-((6-амин-5-(3-метил-1,2,4-оксадиазол-5-ил)пиримидин-4-ил)амин)пропил)-5-хлор-3-((1R,2S)-2-фторциклопропил)квиназолин-4(3H)-один
[560] К суспензии 2-((S)-1-аминопропил)-5-хлор-3-((1R,2S)-2-фторциклопропил)квиназолин-4(3H)-один (59 мг; 0,2 ммоль) и 6-хлор-5-(3-метил-1,2,4-оксодиазол-5-ил)пиримидин-4-амина (42 мг; 0,2 ммоль) в n-BuOH (10 мл) был добавлен DIPEA (52 мг; 0,4 ммоль). Реагирующая смесь была нагрета для дистилляции и перемешивалась в течение последующих 6 часов. Реакция контролировалась посредством TLC (РЕ/EtOAc, v/v, 2/1). Затем реагирующая смесь была остужена до комнатной температуры и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (РЕ/EtOAc (v/v)=1/4) для получения соединения, указанного в заголовке шага, в виде белого порошка (58 мг; выход 61,58%).
MS (ESI, pos. ion): 471,2 [M+H]+;
1HNMR (400 МГц; CDCl3) δ (ppm): 8,70 (s; 1H); 8,15 (s; 1H); 7,58 (d; J=4,3 Гц; 2H); 7,45 (m; 1H); 6,10 (d;, 7=5,1 Гц; 1H); 5,07 (ddd; J=63,2; 8,7; 5,5 Гц; 1H); 3,17 (dd; J=13,4; 6,3 Гц; 1H); 2,52 (s; 3H); 2,16 (m; 1H); 2,06 (m; 1H); 1,84 (m; 1H); 1,69 (m; 1H); 1,07 (t; J=7,4 Гц; 3H).
Пример 63 (S)-2-(1-((6-амин-5-(3-метил-1,2,4-оксадиазол-5-ил)пиримидин-4-ил)амин)этил)-5-фтор-3-фенилквиназолин-4(3H)-один
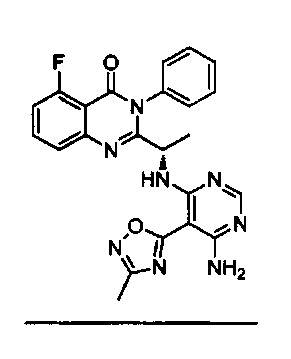
Шаг 1) (S)-терт-бутил(1-((3-фтор-2-(фенилкарбамоил)фенил)амин)-1-оксопропан-2-ил)-карбамат
[561] К суспензии 2-амин-6-хлор-N-фенилбензамида (2,31 г; 10 ммоль). и (5)-2-((терт-бутоксикарбонил)амин)пропановой кислоты (2,08 г; 11 ммоль) в DCM (40 мл) был добавлен HATU (4,56 г; 12 ммоль) и DIPEA (3,88 г; 30 ммоль) при -10°C.Полученная реагирующая смесь перемешивалась при -10°C в течение 1 часа, затем дистилировалась в течение суток. Смесь была охлаждена до комнатной температуры, промыта водой (200 мл×2) и насыщенным гидратогенным раствором NaHCO3 (200 мл×2). Отсепарированная органическая фаза была высушена над обезвоженным сульфатом натрия и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (РЕ/EtOAc (v/v)=8/1) для получения соединения, указанного в заголовке шага, в виде белого твердого вещества (1,14 г; выход 67,8%).
MS (ESI, neg. ion) m/z: 400,1 [M-H]-.
Шаг 2) (S)-терт-бутил(1-(5-фтор-4-оксо-3-фенил-3,4-дигидроквиназолин-2-ил)пропил)карбамат
[562] Раствор (S)-терт-бутил(1-((3-фтор-2-(фенилкарбамоил)фенил)амин)-1-оксопропан-2-ил)карбамата (2,85 г; 7,1 ммоль) в меси ацетонитрила (180 мл) и триэтиламина (21,55 г; 481,5 ммоль) был дегазирован и накачан N2, затем к раствору при комнатной температуре был добавлен N,O-бис(триметилсилил)ацетамид (29,38 г; 213 ммоль). Реагирующая смесь была подвергнута дистилляции при 90°C в течение суток, мониторинг производился посредством LC-MS вплоть до завершения реакции. После завершения реакции смесь была сконцентрирована in vacuo. Осадок был растворен в EtOAc (70 мл), и полученная смесь была промыта водой (70 мл×2) и насыщенным минеральным раствором (50 мл). Отсепарированная органическая фаза была высушена над обезвоженным Na2SO4, и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (РЕ/EtOAc (v/v)=10/1) для получения соединения, указанного в заголовке шага, в виде белого твердого вещества (1,0 г; выход 36,6%).
MS (ESI, pos. ion) m/z: 384,2 [M+H]+.
Шаг 3) (S)-2-(1-аминоэтил)-5-фтор-3-фенилквиназолин-4(3H)-один
[563] К раствору (S)-терт-бутил(1-(5-фтор-4-оксо-3-фенил-3,4-дигидроквиназолин-2-ил)пропил)карбамата (1,0 г; 2,61 ммоль) в EtOAc (8 мл) был добавлен одной порцией раствор HCl в EtOAc (11 мл, 3,88 М) при комнатной температуре. Смесь перемешивалась в течение суток при комнатной температуре. Полученная суспензия была разведена в воде (20 мл). Отсепарированная гидратогенная фаза была экстрактирована посредством EtOAc (20 мл), затем нейтрализована до pH=8 с помощью порошка NaHCO2 и вновь экстрактирована с помощью EtOAc (20 мл×3). Комбинированные органические фазы были промыты насыщенным минеральным раствором (20 мл), высушены над обезвоженным Na2SO4, и сконцентрированы in vacuo для получения соединения, указанного в заголовке шага, в виде белого порошка (440 мг; 59,4%).
MS (ESI, pos. ion) m/z: 284,2 [M+H]+;
1H NMR (400 МГц; DMSO-d6) δ (ppm): 7,89 (td; J=8,2; 5,6 Гц; 1H); 7,66-7,46 (m; 6H); 7,34 (dd; J=10,8; 8,3 Гц; 1H); 5,92 (s; 2H); 3,63 (q; J=6,6 Гц; 1H); 1,23 (d; 7=6,7 Гц; 3H).
Шаг 4
(S)-2-(1-((6-амин-5-(3-метил-1,2,4-оксадиазол-5-ил)пиримидин-4-ил)амин)этил)-5-фтор-3-фенилквиназолин-4(3H)-один
[564] Смесь (S)-2-(1-аминоэтил)-5-фтор-3-(3-фенилквиназолин-4(3H)-один (30 мг; 0,105 ммоль), 6-хлор-5-(3-метил-1,2,4-оксадиазол-5-ил)пиримидин-4-амин (22 мг; 0,105 ммоль) и DIPEA (27 мг; 0,211 ммоль) в n-BuOH (1 мл) была нагрета до 125°C и перемешивалась в течение последующего 1 часа, затем была остужена до rt и профильтрована. Фильтрат был промыт EtOH (2 мл), высушена в вакууме для получения соединения, указанного в заголовке шага, в виде белого твердого вещества (32,7 мг; 67%).
MS (ESI, pos. ion) m/z: 459,0 [M+H]+; HPLC: 99%;
1H NMR (400 МГц; DMSO-d6) δ (ppm): 9,23-9,21 (d; J=6,5 Гц; 1H); 7,99 (s; 1H); 7,92-7,86 (ddd; J=8,4; 8,4; 6,4 Гц; 1H); 7,60-7,54 (m; 8H); 7,35-7,30 (dd; J=10,4; 8,4 Гц; 1H); 4,93 (m; 1H); 2,50 (s; 3H); 1,36 (d; J=6,6 Гц; 3H).
Пример 64 (S)-2-(1-((6-амин-5-(5-метил-1,3,4-оксадиазол-2-ил)пиримидин-4-ил)амин)этил)-5-хлор-3-фенилквиназолин-4(3H)-один
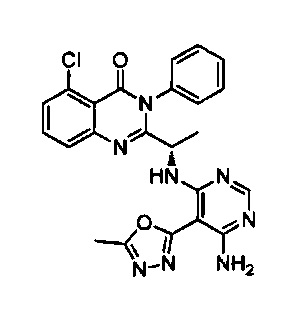
Шаг 1) 5-хлор-1H-бензо[d][1,3]оксазин-2,4-дион
[565] К суспензии 2-амин-6-хлорбензойной кислоты (1,00 г; 5,83 ммоль) в 1,4-диоксане (20 мл) был добавлен трифосген (605 мг; 2,04 ммоль). Реагирующая смесь дистиллировалась в течение 3 часов, затем была остужена до rt и профильтрована. Фильтрат был промыт 50 мл РЕ, высушен в вакууме для получения соединения, указанного в заголовке шага, в виде светло-коричневого твердого вещества (954 мг; 83%).
MS (ESI, pos. ion) m/z: 198,0 [M+H]+;
1H NMR (600 МГц; DMSO-d6) δ (ppm): 11,85 (s; 1H); 7,67-7,64 (dd; J=7,8; 8,4 Гц; 1H); 7,31-7,30 (d; J=7,8 Гц; 1H); 7,11-7,10 (d; J=8,4 Гц; 1H).
Шаг 2) 2-амин-6-хлор-N-фенилбензамид
[566] К суспензии 5-хлор-1H-бензо[d][1,3]оксазин-2,4-диона (1,00 г; 5,06 ммоль) в 1,4-диоксане (20 мл) был добавлен анилин (471 мг; 5,06 ммоль). Полученная смесь дистиллировалась в течение 12 часов, затем была остужена до rt и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (РЕ/EtOAc (v/v)=1/2) для получения соединения, указанного в заголовке шага, в виде светло-желтоватого твердого вещества (1,12 г; 90%).
MS (ESI, pos. ion) m/z: 247,0 [M+H]+;
1H NMR (600 МГц; CDCl3) δ (ppm): 7,71 (br s; 1H); 7,63 (d; J=8,0 Гц; 2H); 7,38 (t; J=7,8 Гц; 2H); 7,18 (t; J=7,4 Гц; 1H); 7,10 (t; J=8,1 Гц; 1H); 6,77 7,9 Гц; 1H); 6,63 (d; J=8,2 Гц; 1H); 4,68 (s; 2H).
Шаг 3) (S)-терт-бутил (1-((3-фтор-2-(фенилкарбамоил)фенил)амин)-1-оксопропан-2-ил)-карбамат
[567] К смеси 2-амин-6-хлор-N-фенилбензамида (600 мг; 2,43 ммоль) и (S)-2-((терт-бутоксикарбонил)амин)пропановой кислоты (552 мг; 2,92 ммоль) в DCM (10 мл) был добавлен DIPEA (1,27 мл, 7,30 ммоль) и HATU (1,11 г; 2,92 ммоль) при 0°C. Реагирующая смесь перемешивалась при 0°C в течение 1 часа, затем была нагрета для дистилляции и перемешивалась в течение суток, после чего была охлаждена до rt и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (РЕ/EtOAc (v/v)=40/7) для получения соединения, указанного в заголовке шага, в виде белого твердого вещества (934 мг; 92%).
MS (ESI, neg. ion) m/z: 416,0 [M-H]-;
1H NMR (400 МГц; CDCl3) δ (ppm): 9,49 (s; 1H); 8,15 (br s; 1H); 8,13-8,11 (d; J=8,4 Гц;1Н); 7,68-7,66 (d; J=7,6 Гц; 2H); 7,41-7,37 (t; J=7,6 Гц; 2H); 7,37-7,33 (t; J=8,4 Гц; 1H); 7,23-7,19 (m; 2H); 5,03-5,02 (d; J=4,8 Гц; 1H); 4,29 (m; 1H); 1,40 (s; 9H); 1,40-1,39 (d; J=5,6 Гц; 3H).
Шаг 4) (S)-терт-бутил(1-(5-фтор-4-оксо-3-фенил-3,4-дигидроквиназолин-2-ил)пропил)карбамат
[568] К раствору (S)-терт-бутил(1-((3-хлор-2-((2-фенилкарбамоил)фенил)амин)-1-оксопропан-2-ил)карбамата (400 мг; 0,96 ммоль), DMAP (117 мг; 0,96 ммоль) и DIPEA (0,33 мл, 1,91 ммоль) в CH3CN (3 мл) был добавлен N,O-бис(триметилсилил)ацетамид (2,34 мл, 9,6 ммоль) при rt. Реагирующая смесь была нагрета для дистилляции и перемешивалась в течение 4 часов, затем была остужена до rt и резко охлаждена насыщенным гидратогенным раствором NaHCO4. Полученная смесь была сконцентрирована in vacuo, и осадок был очищен хроматографией на силикагельной колонне (PE/EtOAc (v/v)=10/1) для получения соединения, указанного в заголовке шага, в виде желтоватого твердого вещества (253 мг; 66%).
MS (ESI, pos. ion) m/z: 400,0 [M+H]+;
1H NMR (400 МГц; CDCl3) δ (ppm): 7,63-7,61 (m; 2H); 7,59-7,57 (m; 1H); 7,55-7,51 (m; 2H); 7,48-7,46 (dd; J=6,4; 3,6 Гц; 1H); 7,39-7,37 (d; J=7,2 Гц; 1H); 7,29-7,27 (m; 1H); 5,59-5,57 (d; J=7,2 Гц; 1H); 4,52-4,49 (m; 1H); 1,42 (s; 9H); 1,26-1,26 (d; J=6,8 Гц; 3H).
Шаг 5) (S)-2-(1-аминоэтил)-5-фтор-3-фенилквиназолин-4(3H)-один
[569] К раствору (S)-терт-бутил(1-(5-хлор-4-оксо-3-фенил-3,4-дигидроквиназолин-2-ил)этил)карбамата (367 мг; 0,92 ммоль) в EtOAc (10 мл) был добавлен раствор HCl в EtOAc (8 мл, 24 ммоль, 3 М) при комнатной температуре. Реагирующая смесь перемешивалась в течение 40 часов, затем была сконцентрирована in vacuo. Осадок был растворен в Н2O (26 мл), и полученная смесь была экстрактирована EtOAc/РЕ (10 мл/5 мл). Основность водосодержащей фазы была доведена до pH=8.5 порошком NaHCO3, затем была произведена экстракция посредством DCM (75 мл×2). Комбинированная органическая фаза была промыта насыщенным минеральным раствором (50 мл), высушена над обезвоженным Na2SO4, и сконцентрирована in vacuo для получения соединения, указанного в заголовке шага, в виде желтоватого твердого вещества (253 мг; 92%).
MS (ESI, pos. ion) m/z: 300,0 [M+H]+;
1H NMR (400 МГц; CDCl3) δ (ppm): 7,63-7,61 (m; 2H); 7,59-7,51 (m; 3H); 7,47-7,45 (dd; J=6,6; 2,5 Гц; 1H); 7,28-7,26 (m; 2H); 3,68 (q; J=6,6 Гц; 1H); 1,28-1,26 (d; J=6,6 Гц; 3H).
Шаг 6)
(S)-2-(1-((6-амин-5-(5-метил-1,3,4-оксадиазол-2-ил)пиримидин-4-ил)амин)этил)-5-хлор-3-фенилквиназолин-4(3H)-один
[570] Смесь (S)-2-(1-аминоэтил)-5-хлор-3-(3-фенилквиназолин-4(3H)-один (35 мг; 0,116 ммоль), 6-хлор-5-(5-метил-1,3,4-оксадиазол-2-ил)пиримидин-4-амин (24,7 мг; 0,116 ммоль) и DIPEA (30 мг; 0,233 ммоль) в n-BuOH (1 мл) была нагрета до 125°C и перемешивалась в течение последующих 4 часов, затем была остужена до rt и профильтрована. Осадок был переведен в суспензию с EtOAc (2 мл) и Н2O (2 мл), и соединение, указанное в заголовке шага, было собрано фильтрацией в виде белого твердого вещества (42 мг; 76%).
MS (ESI, pos. ion) m/z: 475,0 [M+H]+; HPLC: 99%;
1H NMR (400 МГц; CDCl3) δ (ppm): 8,54 (m; 1H); 8,01 (s; 1H); 7,59-7,45 (m; 6H); 7,33-7,26 (m; 1H); 6,40 (br s; 2H); 5,13 (m; 1H); 2,71 (s; 3H); 1,46 (m; 3H).
Пример 65 (S)-2-(1-((6-амин-5-(3-метил-1,2,4-оксадиазол-5-ил)пиримидин-4-ил)амин)этил)-5-хлор-3-фенилквиназолин-4(3H)-один
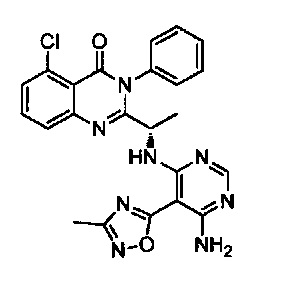
[571] Смесь (S)-2-(1-аминоэтил)-5-хлор-3-фенилквиназолин-4(3H)-один (35 мг; 0,116 ммоль), 6-хлор-5-(3-метил-1,2,4-оксадиазол-5-ил)пиримидин-4-амин (24,7 мг; 0,116 ммоль) и DIPEA (30 мг; 0.233 ммоль) в n-BuOH (1 мл) была нагрета 125°C и перемешивалась в течение последующих 4 часов, затем была остужена до rt и сконцентрирована in vacuo. Осадок был переведен в суспензию с EtOAc (2 мл) и Н2O (2 мл), и соединение, указанное в заголовке шага, было собрано фильтрацией в виде белого твердого вещества (25 мг; 45%).
MS (ESI, pos. ion) m/z: 475,0 [M+H]+; HPLC: 98%;
1H NMR (400 МГц; CDCl3) δ (ppm): 8,80-8,78 (d; J=6,9 Гц; 1H); 8,03 (s; 1H); 7,66-7,51 (m; 5H); 7,48-7,43 (m; 2H); 7,35-7,33 (m; 1H); 5,14 (dddd; J=6,4; 6,4; 6,4; 6,4 Гц; 1H); 2,52 (s; 3H); l,46 (d; J=6,7Hz; 3H).
Пример 66 (S)-2-(1-((6-амин-5-(3-метил-1,2,4-оксадиазол-5-ил)пиримидин-4-ил)амин)этил)-3-циклопропил-5-(1-метил-1H-пиразол-4-ил)квиназолин-4(3H)-один
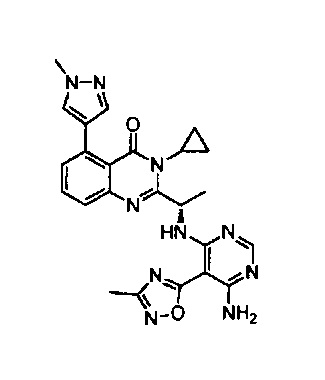
Шаг 1) (S)-терт-бутил(3-циклопропил-5-(1-метил-1H-пиразол-4-ил)-4-оксо-3,4-дигидроквиназол ин-2-ил)этил)карбамат
[572] К смеси (S)-терт-бутил(1-(5-хлор-3-циклопропил-4-оксо-3,4-дигидроквиназолин-2-ил)этил)скарбамата (500 мг; 1,37 ммоль) и 1-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразола (429 мг; 2,06 ммоль) в DMAC (10 мл) был добавлен Pd(dppf)Cl2•CH2Cl2 (113 мг; 0,14 ммоль) и раствор Na2CO3 (437 мг; 4,12 ммоль) в воде (4.0 мл), затем смесь была продута азотом в течение 2 минут и перемешивалась при 120°C в течение 4 часов. Смесь была остужена до rt и резко охлаждена 10 мл воды, затем профильтрована через прокладку CELITE®, и фильтрат был экстрактирован EtOAc (20 мл×3). Комбинированные органические фазы были высушены над обезвоженным Na2SO4, и сконцентрированы in vacuo. Осадок был очищен хроматографией на силикагельной колонне (DCM/MeOH (v/v)=50/1) для получения соединения, указанного в заголовке шага, в виде бледно-желтого масла (507 мг; 90,0%).
MS (ESI, pos. ion) m/z: 410,3 [M+H]+;
1H NMR (400 МГц; DMSO-d6) δ (ppm): 7,85 (s; 1 H); 7,69-7,65 (t; J=7,8 Гц; 1 H); 7,54 (s; 1 H); 7,46-7,44 (dd; J=8,0; 0,9 Гц; 1 H); 7,34-7,29 (t; J=8,8 Гц; 2 Н); 3,88 (s; 3 Н); 2,98-2,91 (m; 1 Н); 1,99-1,96 (m; 1 Н); 1,49-1,30 (m; 12 Н); 0,99-0,91 (m; 2Н); 0,86-0,84 (m; 2 Н).
Шаг 2) (S)-2-(1-аминоэтил)-3-циклопропил-5-(1-метил-1H-пиразол-4-ил)квиназолин-4(3H)-один
[573] К раствору (S)-терт-бутил(1-(5-хлор-3-циклопропил-4-оксо-3,4-дигидроквиназолин-2-ил)этил)карбамата (507 мг; 1,24 ммоль) в EtOAc (6 мл) был добавлен раствор HCl в EtOAc (3,0 М; 4,0 мл), смесь перемешивалась при rt в течение 5 часов, затем была сконцентрирована in vacuo. Осадок был переведен суспензию в DCM (20 мл) и полученная суспензия нейтрализована насыщенным гидратогенным раствором NaHCO3. Отсепарированная водосодержащая фаза была экстрактирована посредством DCM (10 мл×2). Комбинированная органическая фаза была высушена над обезвоженным Na2SO4, и сконцентрирована in vacuo для получения соединения, указанного в заголовке шага, в виде бледно-коричневого масла (322 мг; 84,0%).
MS (ESI, pos. ion) m/z: 310,2 [M+H]+.
Шаг 3)
(S)-2-(1-((6-амин-5-(3-метил-1,2,4-оксадиазол-5-ил)пиримидин-4-ил)амин)этил)-3-циклопропил-5-(1-метил-1H-пиразол-4-ил)квиназолин-4(3H)-один
[574] К смеси (S)-2-(1-аминоэтил)-3-циклопропил-5-(1-метил-1H-пиразол-4-ил)квиназолин-4(3H)-один (113 мг; 0,36 ммоль) и 6-хлор-5-(3-метил-1,2,4-оксадиазол-5-ил)пиримидин-4-амина (81,1 мг; 0,38 ммоль) в n-BuOH (1.5 мл) был добавлен N,N-диизопропилэтиламин (0,15 мл; 0,85 ммоль), температура смеси поддерживалась равной 120°C в течение 3 часов, затем смесь была охлаждена до rt и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (DCM/MeOH (v/v)=50/1) для получения соединения, указанного в заголовке шага, в виде бледно-желтого твердого вещества (150 мг; 84,7%).
MS (ESI, pos. ion) m/z: 485,8 [M+H]+; HPLC: 98,2%;
1H NMR (400 МГц; DMSO-d6) δ (ppm): 9,37-9,36 (d; J=6,8 Гц; 1 H); 8,13 (s; 1 H); 7,87 (s; 1 H); 7,75-7,72 (t; J=7,8 Гц; 1 H); 7,63 (s; 2H); 7,55 (s; 1 H); 7,53-7,51 (m; 1 H) 7,36-7,31 (m; 1 H); 6,15-6,08 (m; 1 H); 3,88 (s; 3 H); 3,11-3,06 (m; 1 H); 1,61-1,59 (d; J=6,5 Гц; 3 H); 1,04-1,01 (m; 2H); 0,87-0,84 (m; 2H).
1H NMR (400 МГц; CDCl3) δ (ppm): 9,21 (d; J=7,1 Гц; 1 H); 8,18 (s; 1 H); 7,71-7,60 (m; 4 H); 7,34 (dd; J=6,7; 1,9 Гц; 1 H); 6,30 (m; 1 H); 3,99 (s; 3 H); 3,05 (m; 1H);2,56 (s;3H); 1,68 (d; J=6,6 Гц; 3 H); 1,09-1,01 (m; 2 H); 0,88 (m; 2H).
Пример 67 (S)-2-(1-((6-амин-5-(5-метил-1,3,4-оксадиазол-2-ил)пиримидин-4-ил)амин)этил)-3-циклопропил-5-(1-метил-1H-пиразол-4-ил)квиназолин-4(3H)-один

[575] К смеси (S)-2-(1-аминоэтил)-3-циклопропил-5-(1-метил-1H-пиразол-4-ил)квиназолин-4(3H)-один (113 мг; 0,36 ммоль) и 6-хлор-5-(5-метил-1,3,4-оксадиазол-2-ил)пиримидин-4-амина (81,1 мг; 0,38 ммоль) в n-BuOH (1,5 мл) был добавлен N,N-диизопропилэтиламин (0.15 мл, 0.85 ммоль), температура смеси поддерживалась равной 120°C в течение 4 часов, затем смесь была охлаждена до rt и сконцентрирована in vacuo. Осадок был очищен хроматографией на силикагельной колонне (DCM/MeOH (v/v)=50/1) для получения соединения, указанного в заголовке шага, в виде бледно-желтого твердого вещества (154 мг; 87,0%).
MS (ESI, pos. ion) m/z: 485,3 [M+H]+; HPLC: 98,0%;
1H NMR (400 МГц; DMSO-d6) δ (ppm): 8,97 (d; J=7,1 Гц; 1 H); 8,08 (s; 1 H); 7,86 (s; 1 H); 7,70 (t; J=7,8 Гц; 1 H); 7,55 (s; 1H); 7,46 (d; J=7,5 Гц; 1 H); 7,32 (d; J=7,4 Гц; 2H); 6,12 (m; 1 H); 3,88 (s; 3 H); 3,64-3,57 (m; 1 H); 2,63 (s; 3 H); 1,60 (d; J=6,6 Гц; 3 H); 1,10-1,00 (m; 2 H); 0,92-0,80 (m; 2 H).
Пример 68 (S)-2-(1-((6-амин-5-(3-метил-1,2,4-оксадиазол-5-ил)пиримидин-4-ил)амин)этил)-5-(1-метил-1H-пиразол-4-ил)-3-фенилквиназолин-4(3H)-один

Шаг 1) (S)-терт-бутил(1-(5-(1-метил-1H-пиразол-4-ил)-4-оксо-3-фенил-3,4-дигидроквиназолин-2-ил)этил)карбамат
[576] К смеси (S)-терт-бутил(1-(5-хлор-4-оксо-3-фенил-3,4-дигидроквиназолин-2-ил)этил)карбамата (100 мг; 0,25 ммоль) в DMAC (1,2 мл) и воды (0,7 мл) был добавлен 1-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол (80 мг; 0,375 ммоль), Na2CO3 (81 мг; 0,755 ммоль) и Pd(dppf)Cl2•CH2Cl2 (23 мг; 0,025 ммоль), реагирующая смесь перемешивалась при 120°C в течение 3 часов, затем была остужена до rt и был добавлен DCM (20 мл). Полученная смесь была профильтрована через прокладку CELITE®, фильтрат был сконцентрирован in vacuo для получения полуфабриката. Полуфабрикат был очищен хроматографией на силикагельной колонне (CH2Cl2/МеОН (v/v)=30/1) для получения соединения, указанного в заголовке шага, в виде белого твердого вещества (97,8 мг; 87%).
MS (ESI, pos. ion) m/z: 446,1 [M+H]+.
Шаг 2) (S)-(1-аминоэтил)-5-(1-метил-1H-пиразол-4-ил)-3-фенилквиназолин-4(3H)-один
[577] К раствору HCl в EtOAc (3 М, 2 мл) был добавлен (S)-терт-бутил(1-(5-(1-метил-1H-пиразол-4-ил)-4-оксо-3-фенил-3,4-дигидроквиназолин-2-ил)этил)карбамат (100 мг; 0,225 ммоль), реагирующая смесь перемешивалась при rt в течение 12 часов, затем была резко охлаждена добавлением насыщенного гидратогенного раствора NaHCO3 (15 мл), и полученная смесь была экстрактирована CH2Cl2 (20 мл×3). Комбинированные органические слои были высушены над обезвоженным Na2SO4 и сконцентрированы in vacuo для получения полуфабриката, который был очищен хроматографией на силикагельной колонне (CH2Cl2/МеОН (v/v)=20/1) для получения указанного в заголовке соединения в белого твердого вещества (71 мг; 91%).
MS (ESI, pos. ion) m/z: 346,2 [M+H]+;
1R NMR (400 МГц; DMSO-d6) δ (ppm): 7,84-7,72 (m; 2H); 7,62 (d; J=7,3 Гц; 1H); 7,60-7,46 (m; 4H); 7,44 (t; J=8,0 Гц; 2H); 7,36 (d; J=7,4 Гц; 1H); 3,82 (s; 3H); 3,42 (dd; J=13,2; 6,6 Гц; 1H); 1,16 (d; J=6,6 Гц; 3H).
Шаг 3 (S)-2-(1-((6-амин-5-(3-метил-1,2,4-оксадиазол-5-ил)пиримидин-4-ил)амин)этил)-5-(1-метил-1H-пиразол-4-ил)-3-фенилквиназолин-4(3H)-один
[578] К раствору (5)-2-(1-аминоэтил)-5-(1-метил-1H-пиразол-4-ил)-3-фенилквиназолин-4(3H)-один (50 мг; 0,145 ммоль) в бутан-1-ол (0,5 мл) был добавлен N-этил-N-изопропил-пропан-2-амин (51 мкл, 0,29 ммоль) и 6-хлор-5-(3-метил-1,2,4-окадиазол-5-ил)пиримидин-4-амин (38 мг; 0,180 ммоль), реагирующая смесь перемешивалась при 100°C в течение 5 часов, затем была резко охлаждена добавлением насыщенного гидратогенного раствора NaHCO3 (15 мл), и полученная смесь была экстрактирована посредством DCM (20 мл×3). Комбинированные органические слои были промыты насыщенным минеральным раствором, высушены над обезвоженным Na2SO4, и сконцентрированы in vacuo для получения полуфабриката. Полуфабрикат был очищен хроматографией на силикагельной колонне (CH2Cl2/МеОН (v/v)=30/1) для получения соединения, указанного в заголовке шага, в виде белого твердого вещества (67,8 мг; 90%).
MS (ESI, pos. ion) m/z: 521,80 [M+H]+;
1H NMR (400 МГц; DMSO-d6) δ (ppm): 9,28 (d; J=6,5 Гц; 1H); 9,28 (d; J=6,5 Гц; 1H); 7,99 (s; 1H); 7,99 (s; 1H); 7,87-7,77 (m; 2H); 7,94-7,62 (m; 5H); 7,78-7,43 (m; 9H); 7,59-7,30 (m; 7H); 7,40 (d; J=7,3 Гц; 1H); 5,33-4,67 (m; 3H); 4,98-4,89 (m; 1H); 3,82 (s; 3H); 2,52 (s; 18H); 2,52 (s;3H); 1,37 (d; J=6,5 Гц; 3H); 1,37 (d; J=6,5 Гц; 10Н).
Пример 69 (S)-2-(1-((6-амин-5-(5-метил-1,3,4-оксадиазол-2-ил)пиримидин-4-ил)амин)этил)-5-(1-метил-1H-пиразол-4-ил)-3-фенилквиназолин-4(3H)-один
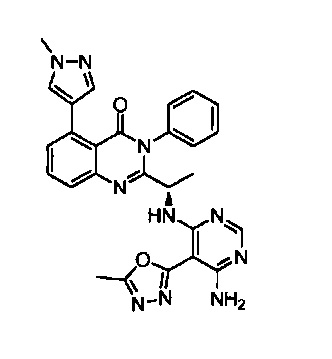
[579] К раствору (S)-2-(1-((6-амин-5-(5-метил-1,3,4-оксадиазол-2-ил)пиримидин-4-ил)амин)этил)-5-хлор-3-фенилквиназолин-4(3H)-один (50 мг; 0,105 ммоль) в DMAC (0.5 мл) и воды (0,3 мл) были добавлены 1-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол (33 мг; 0,157 ммоль), Na2CO3 (34 мг; 0,306 ммоль) и Pd(dppf)Cl2-CH2Cl2 (9 мг; 0.011 ммоль), реагирующая смесь перемешивалась при 120°C в течение 3 часов, затем была остужена до rt, и к смеси был добавлен DCM (15 мл). Смесь была профильтрована через прокладку CELITE®, фильтрат был сконцентрирован in vacuo для получения полуфабриката. Полуфабрикат был очищен хроматографией на силикагельной колонне (CH2Cl2/МеОН (v/v)=30/1) для получения соединения, указанного в заголовке шага, в виде белого твердого вещества (47 мг; 85%).
MS (ESI, pos. ion) m/z: 521,8 [M+H]+;
1H NMR (400 МГц; DMSO-d6) δ (ppm): 8,81 (d; J=6,8 Гц; 1H); 7,95 (s; 1H); 7,79 (dd; J=10,2; 5,3 Гц; 2H); 7,62-7,46 (m; 7H); 7,37 (d; J=7,5 Гц; 1H); 7,25 (s; 2H); 4,92-4,87 (m; 1H); 3,81 (s; 3H); 2,62 (s; 3H); 1,37 (d; J=6,6 Гц; 3H).
Пример 70
(S)-2-(1-((6-амин-5-(5-метил-1,3,4-оксадиазол-2-ил)пиримидин-4-ил)амин)этил)-5-хлор-3-циклопропилквиназолин-4(3H)-один
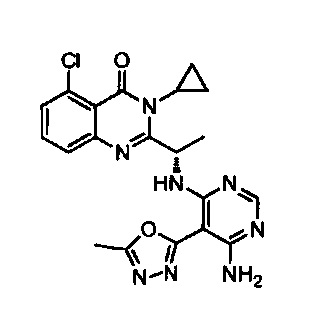
[580] Смесь (5)-2-(1-аминоэтил)-5-хлор-3-циклопропилквиназолин-4(3H)-один (100,3 мг; 0,379 ммоль), 6-хлор-5-(5-метил-1,2,4-оксадиазол-2-ил)пиримидин-4-амина (89,6 мг; 0.417 ммоль) и DIPEA (99,8 мг; 0,379 ммоль) в n-BuOH (4 мл) была нагрета до 125°C и перемешивалась в течение последующих 6 часов. Смесь была остужена до rt, сконцентрирована in vacuo, и осадок был очищен хроматографией на силикагельной колонне (EtOAc/PE (v/v)=1/10) для получения соединения, указанного в заголовке шага, в виде белого твердого вещества (160 мг; 95%).
MS (ESI, Pos. ion.), m/z: 439,2 [M+H]+;
1H NMR (600 МГц; DMSO-d6) δ (ppm): 8,88 (d; J=7,0 Гц; 1H); 8,06 (s; 1H); 7,70 (t; J=8,0 Гц; 1H); 7,50 (d; J=8,0 Гц; 2H); 7,29 (s; 2H); 6,13-6,05 (m; 1H); 3,16 (m; 1H); 2,62 (s; 3H); 1,59 (d; J=6,6Hz; 3H); 1,26 (m; J=5,6 Гц; 2H); 1,07 (m; J=6,8 Гц; 1H); 0,87 (m; J=10,1 Гц; 1H).
БИОЛОГИЧЕСКОЕ ТЕСТИРОВАНИЕ
[581] Система LC/MS/MS, применяемая для анализа, состоит из вакуумного дегазатора серии Agilent 1200, бинарного насоса, автоматического устройства отбора проб луночно-пластинчатого типа, термостатированного отсека колонны, трехполюсного квадрупольного масс-спектрометра Agilent G6430 с источником электроионизационного распыления (ESI). Количественный анализ выполнялся в режиме MRM. Параметры переходов MRM приведены в таблице А.

[582] Для анализа использовалась колонна Agilent XDB-C18, 2,1×30 мм, 3.5 мкМ.
Было впрыснуто 5 мкл проб. Условия анализа: Мобильная фаза представляла собой 0,1% раствор муравьиной кислоты в воде (А) и 0.1% раствор муравьиной кислоты в метаноле (В). Расход равнялся 0,4 мл/мин. Значения градиента мобильной фазы приведены в таблице В.

[583] В качестве альтернативного оборудования для анализа использовался спектрометр LC/MS/MS серии Agilent 6330, оснащенный бинарными насосами G1312A, автоматическим устройством отбора проб G1367A и УФ детектором G1314C. На спектрометре LC/MS/MS использовался источник ESI. Анализ проводился в режиме позитивных ионов, как подходящем, и переход MRM для каждого аналита был оптимизирован с помощью стандартного раствора. В процессе анализа использовалась 5 мкМ колонна Capcell МР-С18 100×4,6 мм (внутренний диаметр), 5 mkM (поставщик Phenomenex, город Torrance, Калифорния, США). Мобильная фаза представляла собой 5 мМ нашатырный спирт; 0,1% раствор МеОН в воде (А): 5 мМ нашатырный спирт, 0Б1% раствор МеОН в ацетонитриле (В) (70:30, v/v). Расход равнялся 0,6 мл/мин. Температура колонны поддерживалась равной окружающей температуре. Было впрыснуто 20 мкл проб.
Пример A: Стабильность соединения в микросомах печени человека и крысы
[584] Инкубирование микросом печени человека и крысы было произведено с дублированием в полипропиленовых пробирках. Типовые инкубационные смеси содержали микросомы печени человека и крысы (0,5 мг белка/мл), исследуемые соединения (5 (мкМ) и NADPH (1,0 мМ) в общем объеме 200 мкл буфера фосфорнокислого калия (PBS, 100 мМ, рН 7,4). Соединения были растворены в DMSO и разведены посредством PBS так, что окончательная концентрация DMSO была равна 0,05%. Ферментативные реакции производились с добавкой белка после 3-минутной предварительной инкубации, инкубирование производилось в открытой водяной бане при 37°C. Реакции прекращались на разных отсчетах времени (0, 5, 10, 15, 30, 60 мин) посредством добавки равного объема ледяного ацетонитрила. Пробы хранились при -80°C вплоть до исследования LC/MS/MS.
[585] Концентрации соединений в инкубационных смесях микросом печени человека или крысы определялись способом LC/MS/MS. Границы линейности диапазона концентрации определялись для каждого тестируемого соединения.
[586] Производилась также параллельная инкубация с использованием денатурированных микросом в качестве подтверждающего контроля, и реакции прекращались на разных отсчетах времени (0, 15, 60 мин) после инкубирования при 37°C.
[587] В качестве подтверждающего регулятора был выбран декстрометорфан (70 (мкМ), и реакции прекращались на разных отсчетах времени (0, 5, 10, 15, 30, 60 мин) после инкубирования при 37°C. Как основные, так и подтверждающие пробы были включены в каждое исследование для обеспечения единства микросомной инкубационной системы.
Анализ данных
[588] Концентрации соединений в инкубированных микросомах печени человека или крысы были представлены в виде процента соответствующего нулевого контрольного отсчета времени каждой реакции. Значения in vivo CLint были экстраполированы (литература: Наритоми, Я.; Терашита, С.; Кимура, С.; Сузуки, А.; Кагаяма, А.; и Сугияма, Я.; Прогноз печеночного клиренса человека по результатам экспериментов in vivo на животных и по метаболическим исследованиям in vitro микросом печени животных и людей. DrugMetab. Dispos., 2001, 29: 1316-1324).
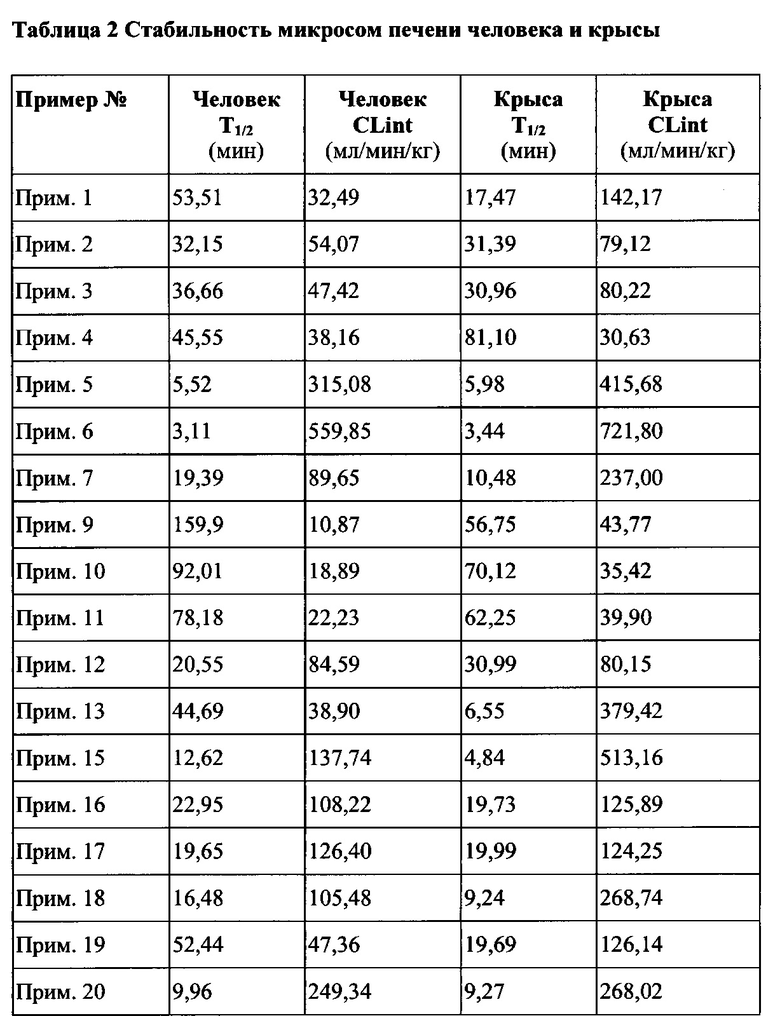
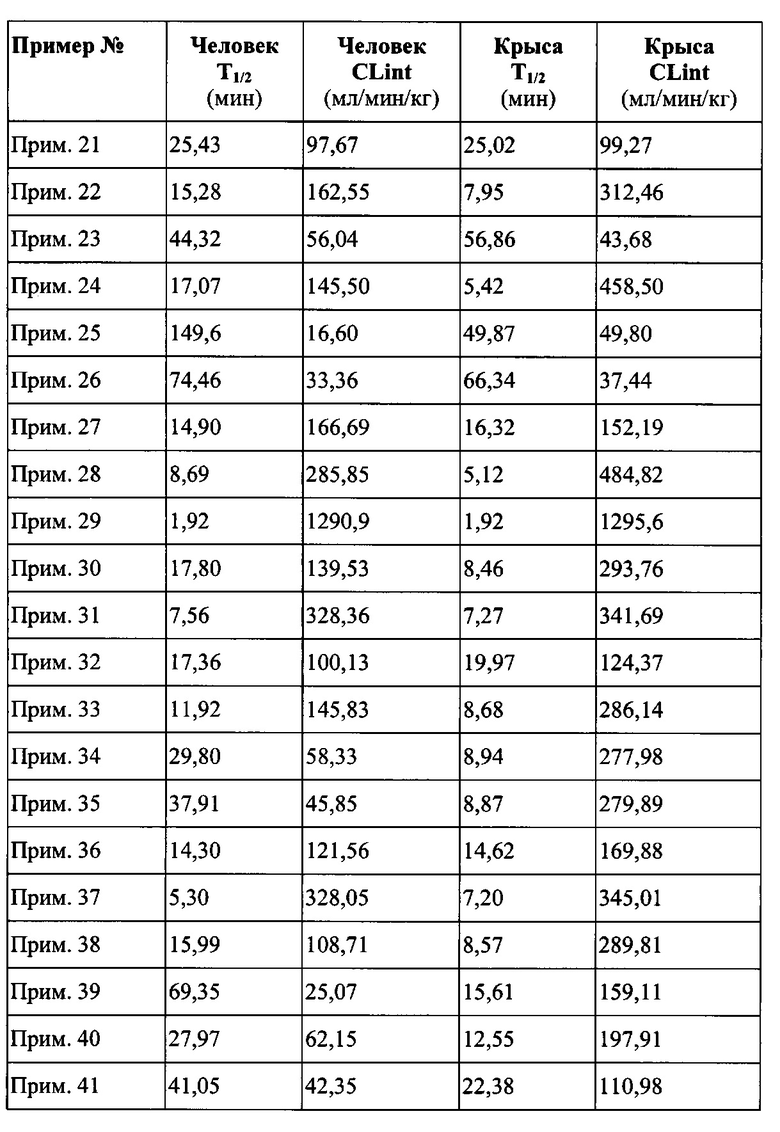

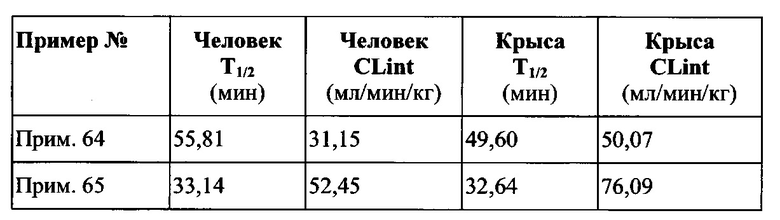
Пример В: Оценка фармакокинетики в мышах, крысах, собаках и обезьянах после внутривенного или орального приема заявляемых соединений
[589] Фармакокинетические исследования заявляемых соединений производились на мышах, крысах, собаках и обезьянах. Соединения принимались в виде водного раствора, 2% НРМС + 1% TWEEN®80 в водном растворе, 5% DMSO + 5% концентрация в физиологическом растворе, 4% МС суспензия или капсула. Доза внутривенной инъекции животным производилась обычно из расчета 1 или 2 мг/килограмм веса. Доза орального приема (р.о.) для мышей и крыс составляла обычно 5 или 10 мг/килограмм веса, доза для собак и обезьян составляла, как правило, 10 мг/килограмм веса. Пробы крови (0,3 мл) отбирались с интервалом 0.25, 0.5, 1.0, 2.0, 3.0,4.0, 6.0, 8.0, 12 и 24 часа или 0.083, 0.25, 0.5, 1.0, 2.0, 4.0, 6.0, 8.0 и 24 часа и обрабатывались на центрифуге при 3000 или 4000 об/мин от 2 до 10 мин. Растворы плазмы собирались и хранились при -20°C или -70°C вплоть до вышеупомянутого анализа LC/MS/MS.

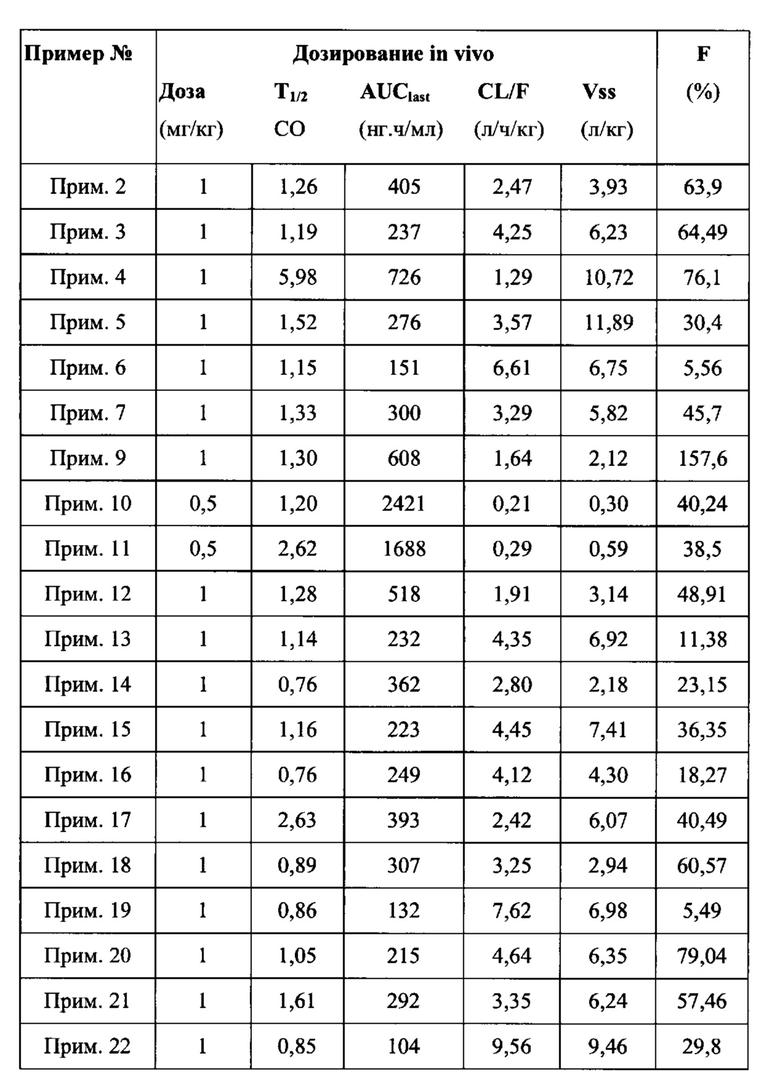

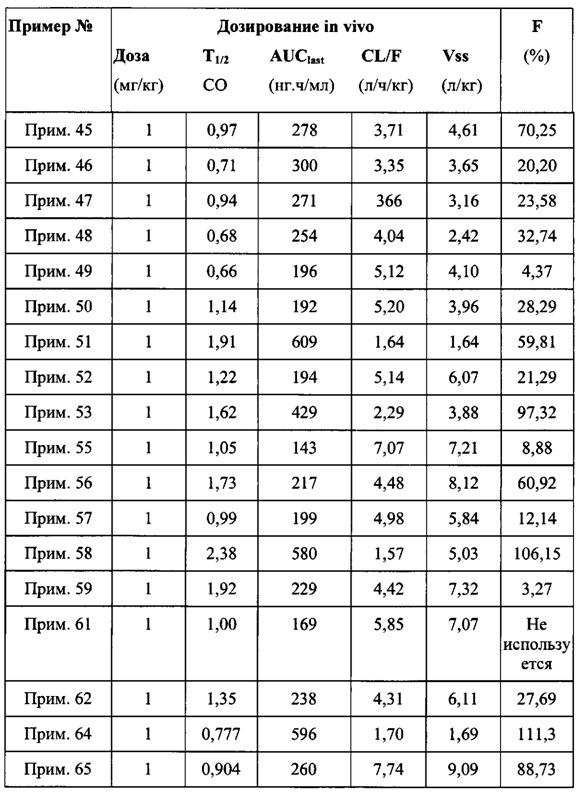
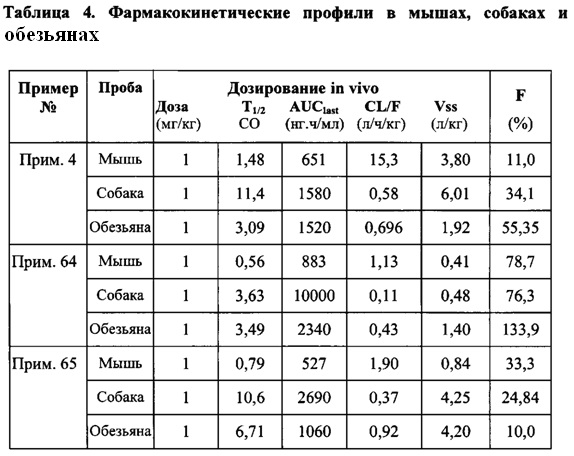
Пример C: Исследование активности киназы
[590] Эффективность заявляемых соединений в качестве ингибиторов РI3-киназ и mTOR-киназ может быть оценена следующим образом.
Общее описание анализа киназы
[591] Анализ киназы может быть выполнен измерением встраивания γ-33Р АТР в иммобилизированный основной белок миелина (МБР). Высоковяжущие белые 384 пластины с лунками (Greiner) покрыты МВР (Sigma #М-1891) инкубацией 60 мкл/лунка 20 мкг/мл МВР в физрастворе тройной буферизации (TBS; 50 мМ Tris рН 8.0, 138 мМ NaCl; 2,7 мМ KCl) на 24 ч при 4°C. Пластины промыты 3 раза 100 мкл TBS. Реакции киназы осуществлялись в общем объеме 34 мкл в буфере киназы (5 мМ Hepes pH 7,6; 15 мМ NaCl; 0,01% бычий гамма-глобулин (Sigma #1-5506), 10 мМ MgCl2, 1 мМ DTT; 0,02% TritonX-100). Растворы соединений приготовлялись в DMSO и были добавлены в аналитические лунки в окончательной концентрации DMSO 1%. Данные в каждой исследуемой точке измерялись дважды, и по меньшей мере два дублирующих исследования производились для определения каждого соединения. Окончательная концентрация добавляемых ферментов равнялась, например, 10 нМ или 20 нМ. Смесь не-меченого АТР и [γ]-33Р АТР была добавлена, чтобы начать реакцию (2×106 см/мин γ-33Р АТР на лунку (3000 Ci/mmole) и 10 мкМ не-меченого, как правило, АТР. Реакции производились в течение 1 часа при rt со встряхиванием. Пластины были промыты 7 раз с помощью TBS, затем была добавлена сцинтилляционная жидкость из расчета 50 мкл/лунка (Wallac). Пластины были считаны счетчиком Wallac Trilux. Это единственно возможный вариант такого исследования; возможны другие варианты, известные специалистам.
[592] Рассмотренная выше процедура исследования может быть использована для определения IC50 для ингибирования и/или константы ингибирования К. Параметр IC50 представляет собой концентрацию соединения, требуемую для снижения на 50% активности фермента в условиях исследования. Значение IC50 определяется построением 10-точечной кривой с помощью серий разведения в 1/2-логарифмическом масштабе (например, типовая кривая может быть построена с использованием следующих концентраций соединения: 10 мкМ, 3 мкМ, 1 мк; 0,3 мкМ; 0,1 мкМ; 0,03 мкМ; 0,01 мкМ; 0,003 мкМ; 0,001 мкМ и 0 мкМ).
ОБЩИЙ ПРОТОКОЛ ИССЛЕДОВАНИЯ PI3 КИНАЗЫ
PI3K (p110α/р85α) (h) [не-радиологическое исследование]
[593] PI3K (р110α/р85α) (ч) инкубирована в исследовательском буфере, содержащем 10 мкМ фосфатидилинозитол 4,5-бифосфата и MgATP (концентрация по необходимости). Реакция начинается добавлением раствора АТР. После 30-минутной инкубации при комнатной температуре реакция прекращается добавлением стоп-раствора, содержащего EDTA биотинилированный фосфатидилинозитол-3,4,5-трифосфат.
Под конец добавляется детектирующий буфер, содержащий меченое европием анти-GST моноклональное антитело, GST-меченый GRP1 РН домен и стрептавидин аллофикоцианин. Затем пластина считывается во флюоресцентном режиме с разрешением по времени и гомогенный флюоресцентный сигнал с разрешением по времени (HTRF) определяется по формуле HTRF = 10000 × (Em665 нм / Em620 нм).
PI3K (p110β/р85α) (ч) [не-радиологическое исследование]
[594] PI3K (р110β/р85α) (ч) инкубирована в исследовательском буфере, содержащем 10 мкМ фосфатидилинозитол-4,5-бифосфата и MgATP (концентрация по необходимости). Реакция начинается добавлением смеси MgATP. После 30-минутной инкубации при комнатной температуре реакция прекращается добавлением стоп-раствора, содержащего EDTA биотинилированный фосфатидилинозитол-3,4,5-трифосфат. Под конец добавляется детектирующий буфер, содержащий меченое европием анти-GST моноклональное антитело, GST-меченый GRP1 РН домен и стрептавидин аллофикоцианин. Затем пластина считывается во флюоресцентном режиме с разрешением по времени и гомогенный флюоресцентный сигнал с разрешением по времени (HTRF) определяется по формуле HTRF = 10000 × (Em665 нм/Em620 нм).
PI3K (pi 108/р85а) (ч) [не-радиологическое исследование]
[595] PI3K (p110δ/p85α) (h) инкубирована в исследовательском буфере, содержащем 10 мкМ фосфатидилинозитол-4,5-бифосфата и MgATP (концентрация по необходимости). Реакция начинается добавлением смеси MgATP. После 30-минутной инкубации при комнатной температуре реакция прекращается добавлением стоп-раствора, содержащего EDTA биотинилированный фосфатидилинозитол-3,4,5-трифосфат. Под конец добавляется детектирующий буфер, содержащий меченое европием анти-GST моноклональное антитело, GST-меченый GRP1 РН домен и стрептавидин аллофикоцианин. Затем пластина считывается во флюоресцентном режиме с разрешением по времени и гомогенный флюоресцентный сигнал с разрешением по времени (HTRF) определяется по формуле HTRF = 10000 × (Em665 нм/Em620 нм).
PI3K (р120γ) (ч) [не-радиологическое исследование]
[596] PI3K (р120γ) (h) инкубирована в исследовательском буфере, содержащем 10 мкМ фосфатидилинозитол-4,5-бифосфата и MgATP (концентрация по необходимости). Реакция начинается добавлением смеси MgATP. После 30-минутной инкубации при комнатной температуре реакция прекращается добавлением стоп-раствора, содержащего EDTA биотинилированный фосфатидилинозитол-3,4,5-трифосфат. Под конец добавляется детектирующий буфер, содержащий меченое европием анти-GST моноклональное антитело, GST-меченый GRP1 РН домен и стрептавидин аллофикоцианин. Затем пластина считывается во флюоресцентном режиме с разрешением по времени и гомогенный флюоресцентный сигнал с разрешением по времени (HTRF) определяется по формуле HTRF = 10000 × (Em665 нм/Em620 нм).
mTOR (ч)
[597] mTOR (ч) инкубируется с 50 мМ HEPES pH 7,5; 1 мМ EDTA; 0,01% TWEEN®20, 2 мг/мл субстратом, 3 мМ хлористым марганцем и γ-33Р-АТР (специфическая активность примерно 500 см в мин/pmol, концентрация по необходимости). Реакция начинается добавлением смеси MgATP. После 40-минутной инкубации при комнатной температуре реакция прекращается добавлением 3% раствора фосфорной кислоты. 10 мкл реагирующей смеси наносится затем на Р30 фильтровальный материал и промывается три раза по 5 минут в 75 мМ фосфорной кислоты и один раз в метаноле до сушки и подсчета сцинтилляций.
[598] Изложенное исследование киназы было выполнено на базе Millipore UK Ltd, Dundee Technology Park, Dundee DD2 1SW, UK.
[599] Заявляемые соединения продемонстрировали убедительную активность в исследованиях РI3Kα (ч) и mTOR (ч).
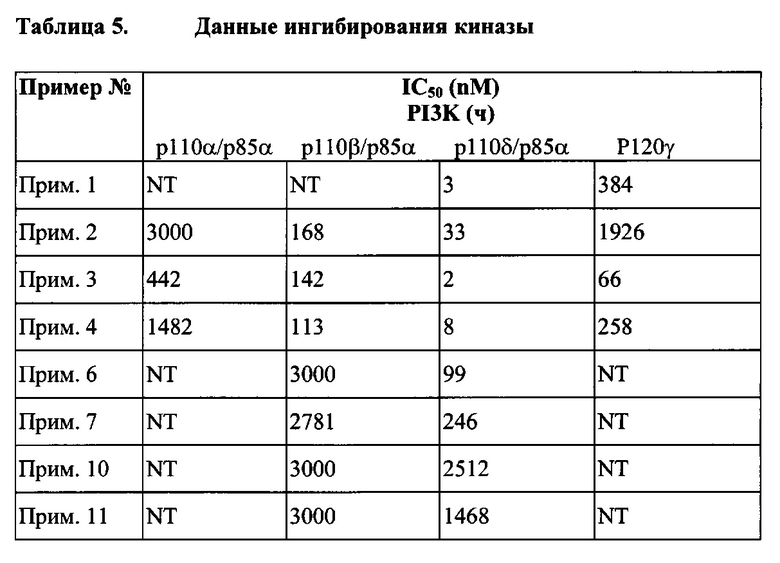
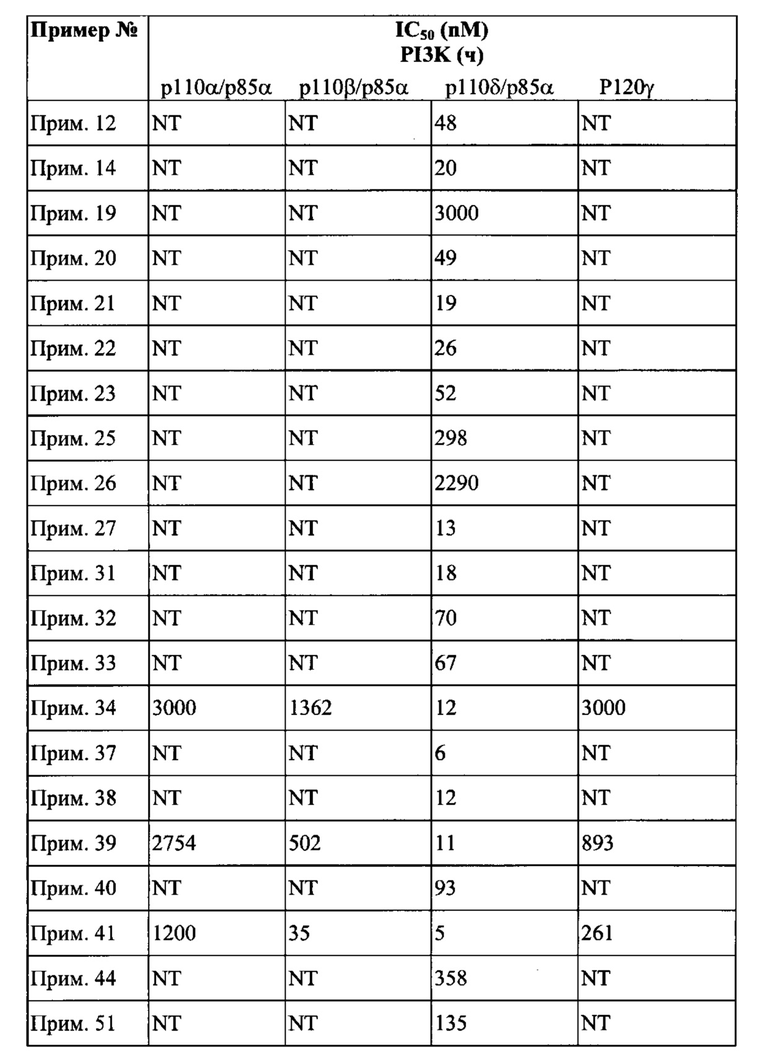
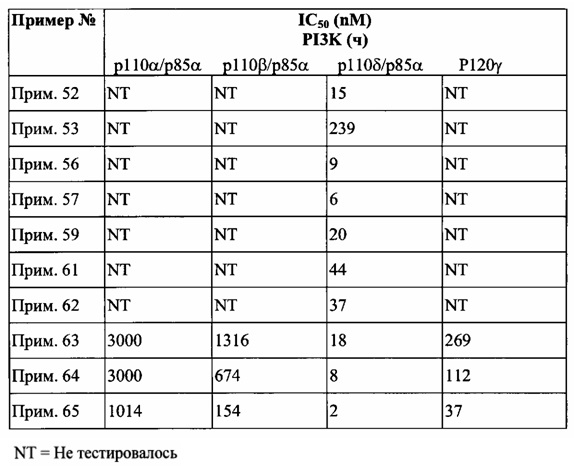
[600] В качестве альтернативного варианта воздействие соединений на активность киназы может быть измерена с помощью KJNOMEscan™, основанном на исследовании конкурирующего связывания, в котором количественно измеряется способность соединения конкурировать с иммобилизованным, направленным на активный участок лигандом. Исследование выполняется сочетанием трех компонентов: DNA-меченая киназа; иммобилизованный лиганд; и тестируемое соединение. Способность тестируемого соединения конкурировать с иммобилизованным лигандом измеряется количественным PCR метки DNA.
[601] Для большинства исследований, меченые киназой штаммы бактериофагов Т7 были приготовлены в Е. coli host, выведенном их штамма BL21. E.coli выращивались до логарифмической фазы и были инфицированы фагом Т7, и инкубированы со встряхиванием при 32°C вплоть до лизиса.
Лизаты были обработаны на центрифуге и профильтрованы для удаления продуктов разрушения клеток. Оставшиеся киназы были воспроизведены в НЕК-293 клетках, а затем помечены DNA для qPCR обнаружения. Магнитные бусины, покрытые стрептавидином, были обработаны биотинилированными маломолекулярными лигандами в течение 30 минут при комнатной температуре для формирования аффинных смол для исследования киназы. Лигандиованные бусины были блокированы избыточным биотином и промыты блокирующим буфером (SEABLOCK™ (Pierce), 1% BSA; 0,05% TWEEN®20, 1 мМ DTT) для удаления несвязанного лиганда и для уменьшения неспецифического связывания. Реакции связывания были собраны комбинированием киназ, лигандированных аффинных бусин и тестируемыми соединениями в 1х связывающем буфере (20% SEABLOCK™; 0,17 × PBS; 0,05% TWEEN®20; 6 мМ DTT). Все реакции производились в полистиреновых 96-луночных пластинах в окончательном объеме 0,135 мл. Исследовательские пластины были инкубированы при комнатной температуре со встряхиванием в течение 1 часа и аффинные бусины были промыты промывочным буфером (1 х PBS; 0,05% TWEEN®20). Затем бусины были повторно помещены в суспензию буфера элюцирования (1 × PBS; 0,05% TWEEN®20; 0,5 мкМ не-биотинилированный аффинный лиганд) и инкубированы при комнатной температуре со встряхиванием в течение 30 минут. Концентрация киназы в элюатах измерялась посредством qPCR.
[602] Изложенные исследования киназы производились с использованием KINOMEscan™ Profiling Service компании DiscoveRx Corporation, 42501 Albrae St. Fremont, CA 94538, USA.
[603] В завершение следует отметить наличие альтернативных способов внедрения заявляемого изобретения. Соответственно, представленные реализации следует считать иллюстративными и не ограничивающими область заявляемого изобретения изложенными деталями, однако эти детали могут быть модифицированы без нарушения области заявляемого изобретения и эквивалентов пунктов патентной формулы. Все упоминаемые в настоящей заявке патенты и публикации включены в качестве справочного материала.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЗАМЕЩЕННЫЕ АМИНОПИРИМИДИНОВЫЕ СОЕДИНЕНИЯ И СПОСОБЫ ИХ ИСПОЛЬЗОВАНИЯ | 2014 |
|
RU2683793C2 |
| АЗОТСОДЕРЖАЩИЕ ГЕТЕРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ В ЛЕКАРСТВЕННЫХ ПРЕПАРАТАХ | 2014 |
|
RU2694254C1 |
| АЗОТСОДЕРЖАЩИЕ ГЕТЕРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ В ФАРМАЦЕВТИЧЕСКИХ ПРЕПАРАТАХ | 2013 |
|
RU2701156C2 |
| БИЦИКЛИЧЕСКИЕ НИТРОИМИДАЗОЛЫ, КОВАЛЕНТНО СОЕДИНЕННЫЕ С ЗАМЕЩЕННЫМИ ФЕНИЛОКСАЗОЛИДИНОНАМИ | 2009 |
|
RU2504547C2 |
| Новые соединения пиридопиримидинона для модулирования каталитической активности гистонлизиндеметилаз (KDMS) | 2015 |
|
RU2684396C2 |
| ПРОТИВОВИРУСНЫЕ СОЕДИНЕНИЯ | 2006 |
|
RU2441010C2 |
| ПРОИЗВОДНЫЕ [1,8]НАФТИРИДИНА, ПОЛЕЗНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ РЕПЛИКАЦИИ ВИРУСА HCV | 2006 |
|
RU2467007C2 |
| ИНГИБИТОРНЫЕ СОЕДИНЕНИЯ | 2013 |
|
RU2673079C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ МИНДАЛЬНОЙ КИСЛОТЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ТРОМБИНА | 2001 |
|
RU2300521C2 |
| АЗАБИЦИКЛИЧЕСКИЕ И ДИАЗЕПИНОВЫЕ ПРОИЗВОДНЫЕ ДЛЯ ЛЕЧЕНИЯ ОФТАЛЬМОЛОГИЧЕСКИХ НАРУШЕНИЙ | 2018 |
|
RU2795521C2 |
Изобретение относится к новым аминопиримидиновым соединениям формулы (I), обладающим свойствами ингибитора PI3-киназы, в частности PI3Kδ. Соединения предназначены для регулирования, лечения или облегчения тяжести заболевания, вызванного неадекватной активностью PI3-киназы, путем введения терапевтически эффективного количества соединения. Соединения могут быть использованы для лечения таких заболеваний, как астма, хроническая обструктивная болезнь легких (COPD, ХОБЛ), вирусные респираторные инфекции, вирусные обострения респираторных заболеваний, аспергиллез, лейшманиоз, аллергический ринит, атопический дерматит, ревматоидный артрит, рассеянный склероз, воспалительная болезнь кишечника, гемобластозы, панкреатит, нефропатия, карцинома, отторжение трансплантата, отторжение ткани, поражение легких, боли, вызываемые ревматоидным артритом, остеоартритом, боль в пояснице, боль, вызываемая общим воспалительным процессом, постгепатитная невралгия, диабетическая нейропатия, воспалительная нейропатическая боль (травма), тригеминальная невралгия или центральная боль. В соединении формулы (I)
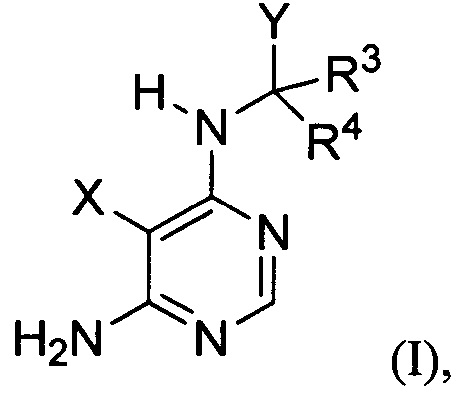
X представляет собой моновалентную гетероарильную группу, выбранную из структур:

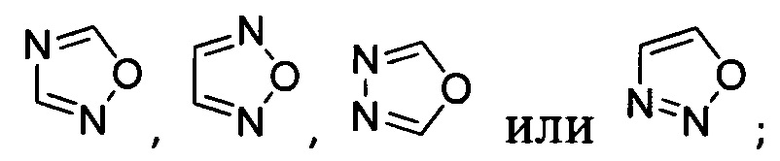
и где X замещен 1 или 2 R1 группами; Y представляет собой
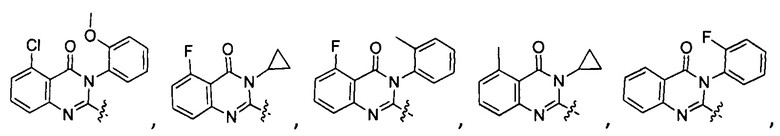
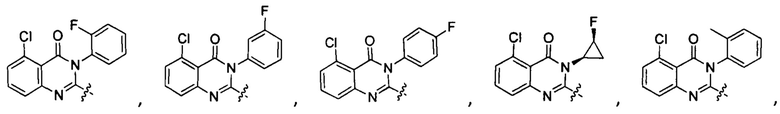
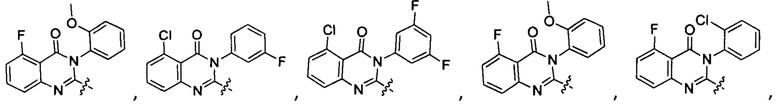
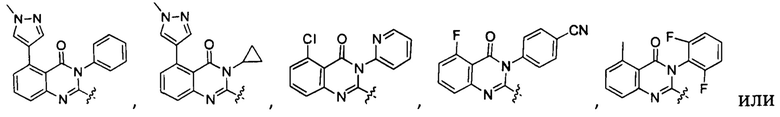
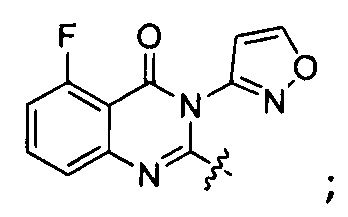 каждый R1 независимо является Н, -C(=O)NRaRb, (С1-С6)алкилом или (С3-С8) циклоалкилом; R3 представляет собой Н; R4 представляет собой (С1-С6)алкил; и Ra и Rb независимо являются Н, (С1-С3)алкилом, (С2-С4)алкенилом, (С2-С4)алкинилом, (С3-С6)циклоалкилом, (С3-С5)гетероциклилом или 5-6-членным гетероарилом, где каждый из (С1-С3)алкила, (С2-С4)алкенила, (С2-С4)алкинила, (С3-С6)циклоалкила, (С3-С5)гетероциклила и 5-6-членного гетероарила замещен 1, 2, 3 или 4 заместителями, независимо выбранными из F, CN, ОН, NH2, (С1-С3)алкила, (С1-С3) галоалкила, (С1-С3)алкокси и (С1-С3)алкиламино. 3 н. и 9 з.п. ф-лы, 5 табл., 70 пр.
каждый R1 независимо является Н, -C(=O)NRaRb, (С1-С6)алкилом или (С3-С8) циклоалкилом; R3 представляет собой Н; R4 представляет собой (С1-С6)алкил; и Ra и Rb независимо являются Н, (С1-С3)алкилом, (С2-С4)алкенилом, (С2-С4)алкинилом, (С3-С6)циклоалкилом, (С3-С5)гетероциклилом или 5-6-членным гетероарилом, где каждый из (С1-С3)алкила, (С2-С4)алкенила, (С2-С4)алкинила, (С3-С6)циклоалкила, (С3-С5)гетероциклила и 5-6-членного гетероарила замещен 1, 2, 3 или 4 заместителями, независимо выбранными из F, CN, ОН, NH2, (С1-С3)алкила, (С1-С3) галоалкила, (С1-С3)алкокси и (С1-С3)алкиламино. 3 н. и 9 з.п. ф-лы, 5 табл., 70 пр.
1. Соединение по формуле (I):
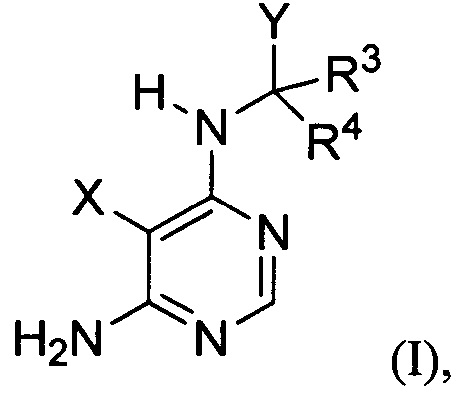
где
X представляет собой моновалентную гетероарильную группу, выбранную из структур

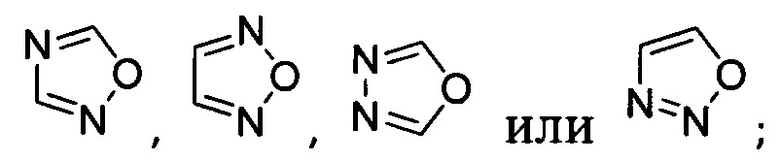
и где X замещен 1 или 2 R1 группами;
Y представляет собой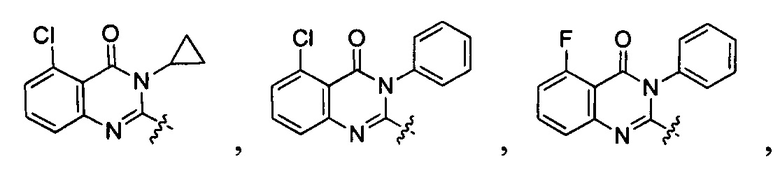
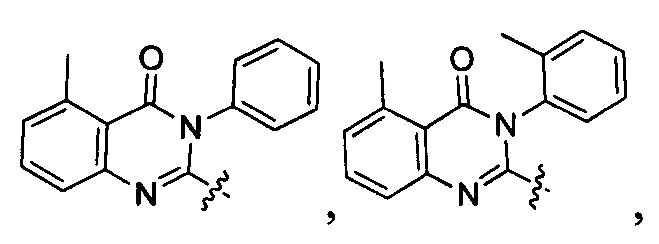
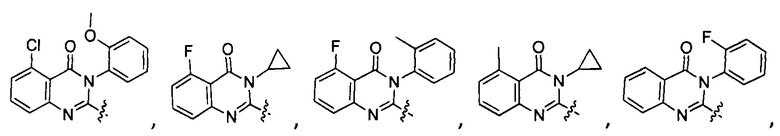
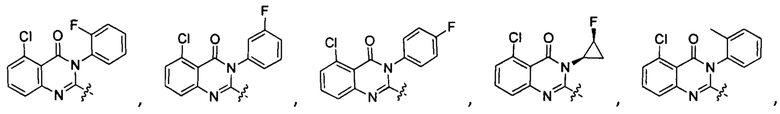
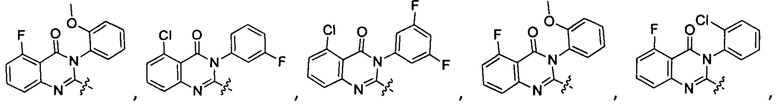
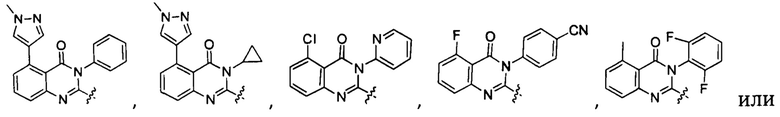
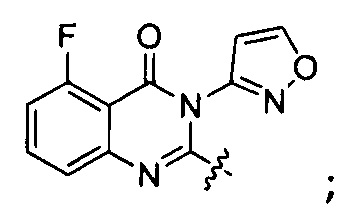
каждый R1 независимо является Н, -C(=O)NRaRb, (С1-С6)алкилом или (С3-С8) циклоалкилом;
R3 представляет собой Н;
R4 представляет собой (С1-С6)алкил; и
Ra и Rb независимо являются Н, (С1-С3)алкилом, (С2-С4)алкенилом, (С2-С4)алкинилом, (С3-С6)циклоалкилом, (С3-С5)гетероциклилом, или 5-6-членным гетероарилом, где каждый из (С1-С3)алкила, (С2-С4)алкенила, (С2-С4)алкинила, (С3-С6)циклоалкила, (С3-С5)гетероциклила и 5-6-членного гетероарила замещен 1, 2, 3 или 4 заместителями, независимо выбранными из F, CN, ОН, NH2, (С1-С3)алкила, (С1-С3)галоалкила, (С1-С3)алкокси и (С1-С3)алкиламино.
2. Соединение по п. 1, где R4 является (С1-С4)алкилом.
3. Соединение по п. 1, где каждый R1 независимо представляет собой Н, -C(=O)NRaRb, (С1-С3)алкил или (С3-С6)циклоалкил.
4. Соединение по п. 1, где R4 представляет собой (С1-С3)алкил.
5. Соединение по п. 1 с одной из приведенных ниже структурных формул
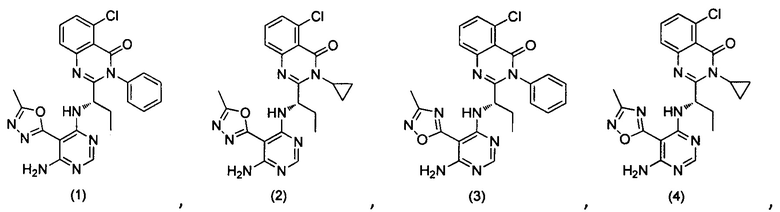

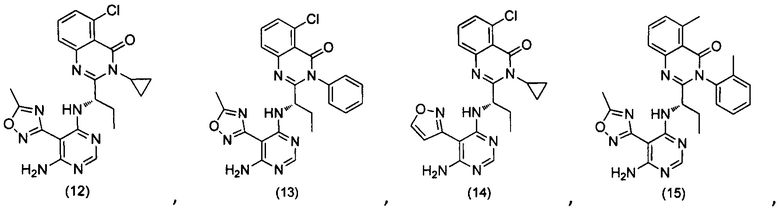
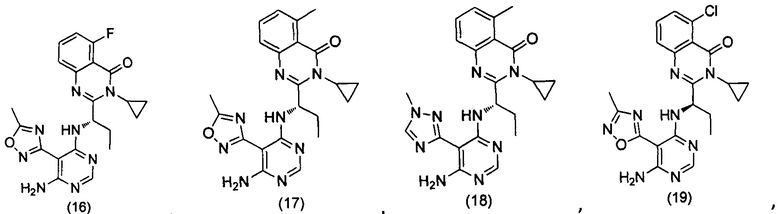
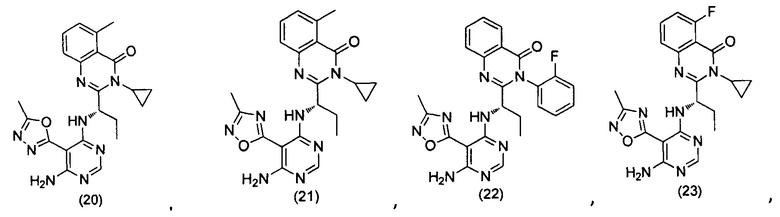
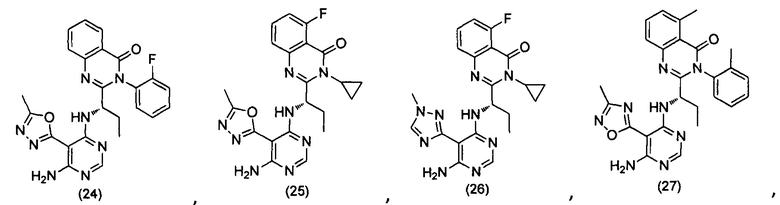
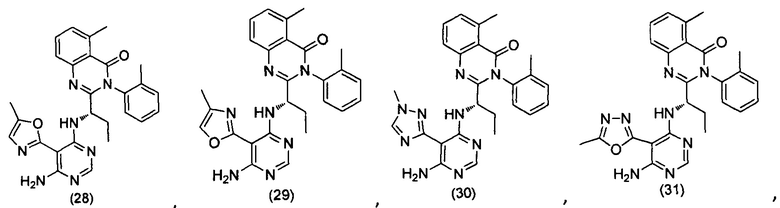
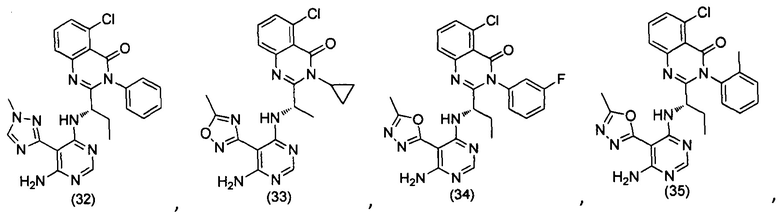


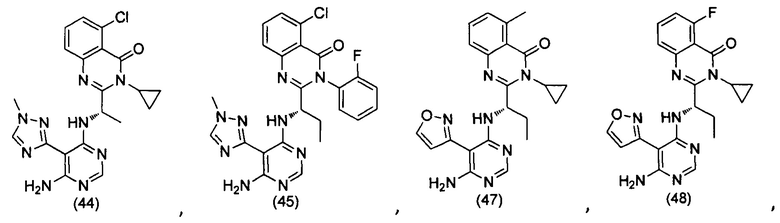
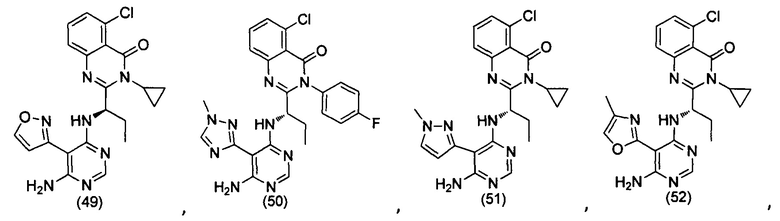
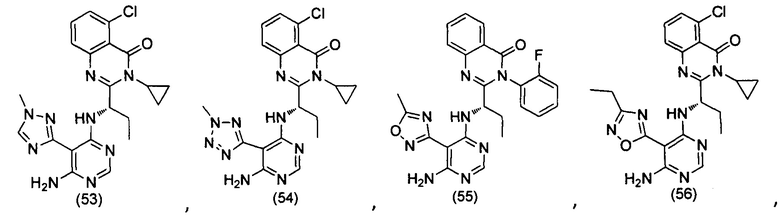
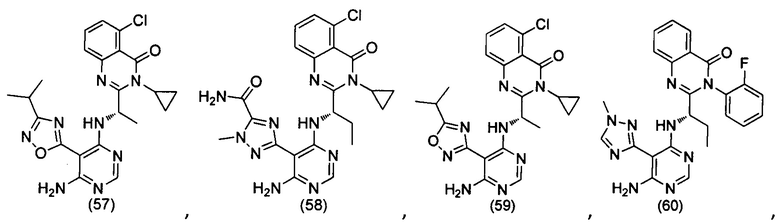
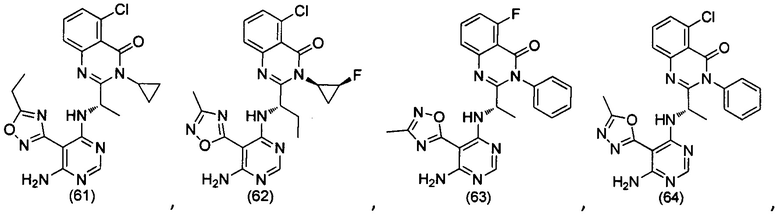
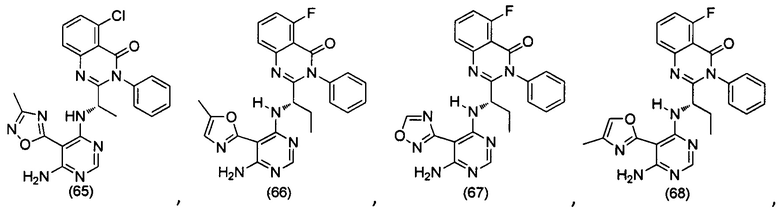
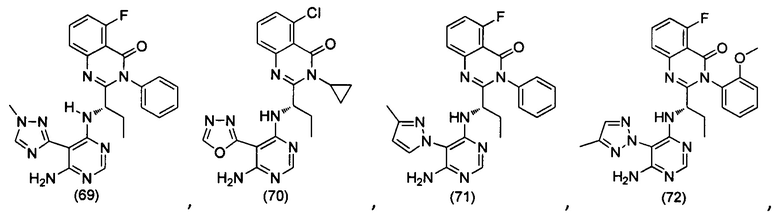
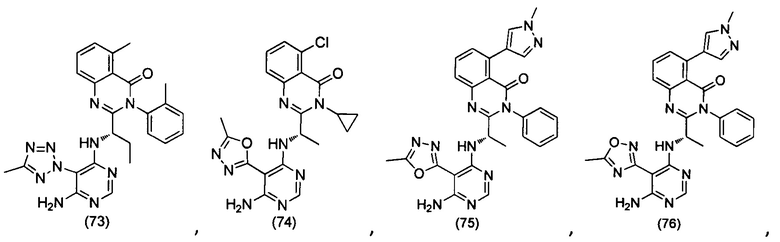
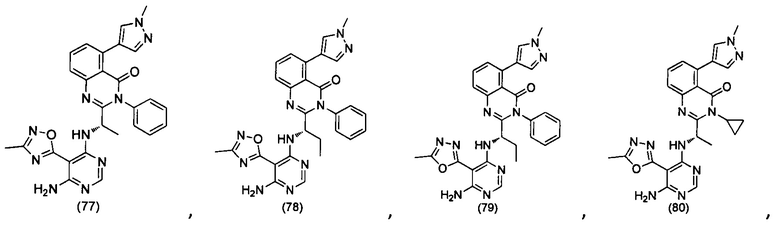
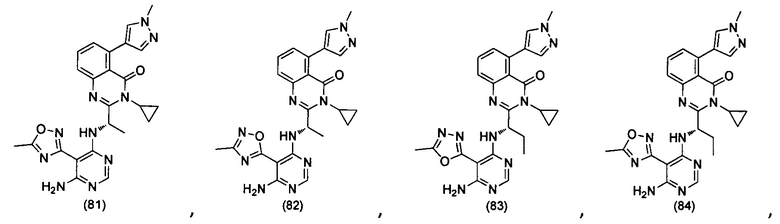

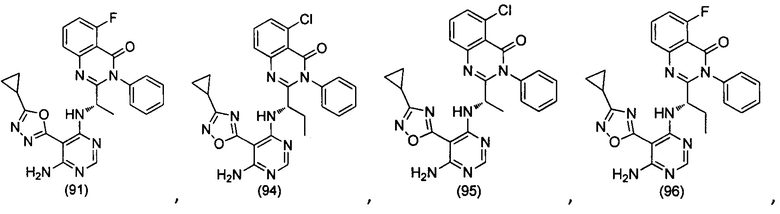
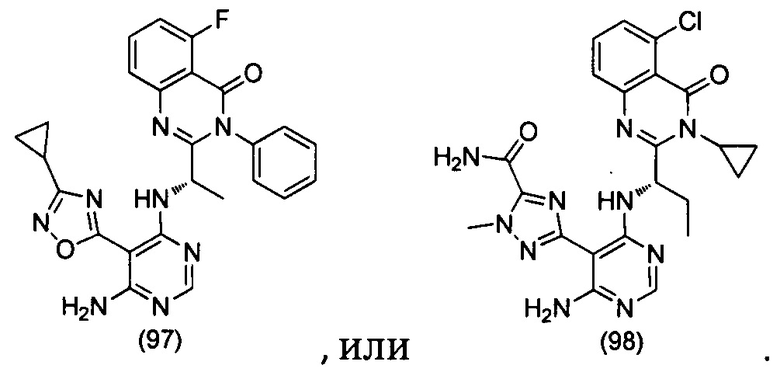
6. Фармацевтическая композиция, имеющая свойства ингибитора PI3-киназы, содержащая терапевтически эффективное количество соединения по любому из пп. 1-5 и один или более фармацевтических приемлемых носителей, инертных наполнителей, разбавителей, активаторов, основ или их сочетание.
7. Соединение по любому из пп. 1-5, имеющее свойства ингибитора PI3-киназы, используемое для профилактики, регулирования, лечения или облегчения тяжести заболевания, вызванного неадекватной активностью PI3-киназы в организме пациента, где заболеванием является астма, хроническая обструктивная болезнь легких (COPD, ХОБЛ), вирусные респираторные инфекции, вирусные обострения респираторных заболеваний, аспергиллез, лейшманиоз, аллергический ринит, атопический дерматит, ревматоидный артрит, рассеянный склероз, воспалительная болезнь кишечника, гемобластозы, панкреатит, нефропатия, карцинома, отторжение трансплантата, отторжение ткани, поражение легких, боли, вызываемые ревматоидным артритом или остеоартритом, боль в пояснице, боль, вызываемая общим воспалительным процессом, постгепатитная невралгия, диабетическая нейропатия, воспалительная нейропатическая боль (травма), тригеминальная невралгия или центральная боль.
8. Фармацевтическая композиция по п. 6, используемая для профилактики, регулирования, лечения или облегчения тяжести заболевания, вызванного неадекватной активностью PI3-киназы, где заболеванием является астма, хроническая обструктивная болезнь легких (COPD, ХОБЛ), вирусные респираторные инфекции, вирусные обострения респираторных заболеваний, аспергиллез, лейшманиоз, аллергические риниты, атопические дерматиты, ревматоидные артриты, рассеянный склероз, воспалительная болезнь кишечника, гемобластоз, панкреатит, нефропатия, карцинома, отторжение трансплантата, отторжение ткани, поражение легких, боли, вызываемые ревматоидным артритом или остеоартритом, боль в пояснице, боль, вызываемая общим воспалительным процессом, постгепатитная невралгия, диабетическая нейропатия, воспалительная нейропатическая боль (травма), тригеминальная невралгия или центральная боль.
9. Соединение по любому из пп. 1-5, имеющее свойства ингибитора PI3-киназы, предназначенное для регулирования активности PI3-киназы в объекте путем введения терапевтически эффективного количества соединения.
10. Соединение по п. 9, где PI3-киназа представляет собой PI3Kδ.
11. Способ ингибирования РI3-киназы, состоящий во введении объекту терапевтически эффективного количества композиции по п. 6.
12. Способ по п. 11, где РI3-киназа представляет собой PI3Kδ.
| WO 2013116562 A1, 08.08.2013 | |||
| US 2009012060 A1, 08.01.2009 | |||
| US 2010249155 A1, 30.09.2010 & EA 019499 B1, 30.04.2014 | |||
| RU 2016115082 A, приоритет 22.09.2013, опубл | |||
| Видоизменение пишущей машины для тюркско-арабского шрифта | 1923 |
|
SU25A1 |
| Приспособление к чесальным маши нам для останова при обрыве или утонении вы пускаемой ленты | 1929 |
|
SU19499A1 |

