Настоящее изобретение относится к стабилизированной самозатухающей полимерной композиции.
При получении полимерных композиций с огнестойкими свойствами, которые основаны на винилароматических полимерах, используются галогенированные органические присадки, галоген в которых представляет собой бром и/или хлор. Указанные присадки можно в расплавленном состоянии смешивать с полимерной массой, повышая ее огнестойкость.
Галогенированная органическая присадка представляет собой соединение, высвобождающее радикалы галогена при температуре воспламенения, в результате чего радикалы, образующиеся в ходе реакции с атмосферным кислородом, реагируют с указанными радикалами галогена, а не с винилароматическим полимерным соединением. Хорошая галогенированная огнестойкая присадка должна смешиваться с полимерной массой, должна быть стабильной при температуре смешивания (180°C - 230°C) и нестабильной при температуре воспламенения.
Галогенированные органические соединения, подходящие для использования в качестве огнестойких присадок, такие как гексабромциклододекан и бромированные стирол-бутадиеновые сополимеры, эффективно проходят стандартные тесты на огнестойкость и хорошо смешиваются с винилароматическим полимером; однако при температурах приготовления смесей с винилароматическими полимерами они могут высвобождать значительное количество галогенидов водорода.
Чтобы не допустить нестабильности галогенированных органических соединений, на стадии перемешивания с полимером можно добавить щелочные неорганические соединения, такие как оксиды или гидроксиды щелочных или щелочно-земельных металлов, а также содержащие эпоксиды органические вещества, которые перехватывают галогеноводород, образующийся при перешивании. Указанные соединения реагируют с образующимся галогеноводородом, формируя стабильные соли галогенов и галогидрины, которые, однако, уменьшают концентрацию галогена, доступную для реализации огнестойких свойств полимерной смеси.
Из уровня техники известно, что содержащие эпоксидные группы соединения можно использовать как дополнительные присадки, которые дополняют огнестойкие присадки, улучшая стабильность самозатухающих полимерных композиций. Известно также о возможности применения галогенированных замедляющих горение присадок, содержащих эпоксидные группы. Далее кратко описана некоторая патентная документация из уровня техники.
US 4,032,481 раскрывает процесс приготовления вспениваемого полистирола со сниженной горючестью. Процесс проводят в водной суспензии при температуре не выше 115°C, в присутствии галогенированных замедляющих горение агентов, эпоксидированного соевого масла, глицидилового эфира бисфенола А, бариевых солей производных фенола, кадмиевых солей органических кислот и фосфитов крезола.
EP 0066686 описывает полимерную композицию, содержащую вспениваемый полистирол и менее 0,1 масс.% эпоксидированных алифатических углеводородов, которые содержат от 6 до 18 атомов углерода, растворимых в винил-ароматическом мономере. Указанная композиция дает возможность увеличить минимальное время нахождения при температурах вспенивания, что позволяет охлаждать конечный продукт, не нарушая структуру ячеек, образующих формирующийся вспененный продукт. Смешиваемость эпоксидированных углеводородов в мономере не гарантирует его хорошей дисперсии в полимере, полученном путем полимеризации смеси мономеров.
DE 3402539 содержит описание полиолефинов, винилароматических полимеров и полиэфиров с повышенной стабильностью к деградации, содержащих замедлитель горения, такой как тетрабромбисфенола А бис(дибромпропиловый) эфир или гексабромциклододекан, а также эпоксидную смолу бисфенола А эпихлоргидрин.
EP 848727 относится к полимерным композициям, содержащим гексабромциклододекан и эпоксидную смолу в качестве температурного стабилизатора. Используемая эпоксидная смола содержит галоген, бром или хлор, а ее эпоксидный эквивалент находится в интервале от 150 до 800 г/экв. Конкретная описанная в EP 848727 композиция содержит полистирол или сополимеры стирола и не менее одной галогенированной эпоксидной смолы в количестве не менее 0,5 масс.%.
WO 2011/008417 (также US 2010331497) описывает бромированное и эпоксидированное органическое соединение, одновременно содержащее бром и не менее одной эпоксидной группы. Указанное соединение используется как замедляющий горение агент в составе полимеров и сополимеров стирола.
Это соединение получают, начиная от полимеров и сополимеров конъюгированного диенового мономера, предпочтительно бутадиена, таких как, например, стирол/бутадиеновых сополимеров и блок-сополимеров. Указанные полимеры и сополимеры в одну стадию бромируют подходящими агентами бромирования, а в еще одну стадию их эпоксидируют подходящим окислителем, порядок следования стадий произвольный.
Задача настоящего изобретения заключается в повышении термостабильности галогенированных замедляющих горение агентов в самозатухающих полимерных композициях, содержащих винилароматические полимеры, что обеспечивает эффективность замедляющих горение агентов в экономически выгодных концентрациях.
Чем более равномерно галогенированная присадка распространена в полимерной композиции, тем более эффективно она замедляет распространение пламени. Чем более равномерно в объеме полимерной композиции распределены эпоксидные группы, тем более эффективно они будут осуществлять стабилизацию замедляющих горение агентов в составе полимерной композиции.
Одним из способов гарантировать равномерное распределение эпоксидных групп в составе винилароматических полимеров может быть сополимеразиция винилароматических мономеров с одним или более виниловыми мономерами, содержащими эпоксидные группы, так что эпоксидные группы в образующихся полимерных цепях будут распределены статистически. Этот метод синтеза не является особенно гибким и требует модификации на этапе синтеза всей винилароматической полимерной композиции.
Обычно, можно считать, что смесь винилароматических сополимеров и винилароматических сополимеров, содержащих эпоксидные группы (получаемых путем статистической сополимеризации винилароматических мономеров и мономеров винила с эпоксидными группами или путем эпоксидирования реактивных винилароматических сополимеров), является достаточно однородной, если содержание эпоксидных групп винилароматического сополимера, выраженное через отношение массы оксиранового кислорода к общей массе содержащего его статистического сополимера, меньше 0,5%.
Во многих случаях, однако, проще и экономически более выгодно добавить к винилароматической композиции замедляющие горение присадки и содержащие эпоксидные группы стабилизаторы в концентрированной форме, это позволяет избежать необходимости модифицировать всю композицию винилароматических сополимеров в процессе синтеза.
Чтобы изделие могло эффективно пройти стандартные тесты на огнестойкость, замедляющие горение присадки и эпоксидные стабилизаторы должны быть равномерно распределены в винилароматической полимерной композиции, что позволяет избежать необходимости применять избыточные их количества и, кроме того, оказывает минимально возможное влияние на поведенческие свойства полимерной композиции.
Чтобы повысить термостабильность галогенированных замедляющих горение агентов в полимерной композиции, заявитель обнаружил блок-сополимер, содержащий блоки сополимера, полученные из винилароматических мономеров, и блоки полимера, содержащие эпоксидные группы. Указанный блок-сополимер равномерно распределен в полимерной композиции и может стабилизировать галогениды водорода, выделяющиеся из галогенированной органической присадки, которую обычно используют в качестве замедляющего горение агента. В частности, почти каждая молекула галогенида водорода, образующаяся в самозатухающей полимерной композиции, реагирует с эпоксидной группой в отношении около 1:1, неожиданно не ограничивая эффективности действия галогенированной присадки при прохождении указанной композицией стандартных тестов на огнестойкость.
В целях настоящего изобретения блок-сополимер считается равномерно распределенным в полимерной композиции, если он измельчен настолько мелко, что образует однородную смесь, даже если эта смесь гетерогенная.
Важно поддерживать однородное распределение блок-сополимера в полимерной композиции, так как оно позволяет защитить всю композицию минимальным количеством эпоксидных групп.
Более того, галогенгидрины, образующиеся в ходе реакции эпоксидных групп с галогенидом водорода, также представляют собой замедляющие горение агенты.
Таким образом, объект настоящего изобретения относится к самозатухающей полимерной композиции, содержащей:
a. стабилизирующую полимерную композицию, содержащую:
i) от 80 масс.% до 99,5 масс.% одного или более совместимых друг с другом винилароматических полимеров; а также
ii) от 0,5 масс.% до 20 масс.% по меньшей мере одного блок-сополимера, содержащего:
1) по меньшей мере один блок винилароматического полимера, совместимый с (i) и в количестве не менее 15 масс.% по отношению к целому блок-сополимеру, а также
2) по меньшей мере один блок винилового сополимера, содержащий эпоксидные группы, так чтобы масса оксиранового кислорода составляла от 0,7 масс.% до 19 масс.%, по отношению к целому блок-сополимеру; а также
b) от 0,03 до 10 м.ч. относительно содержания компонента (а) по меньшей мере одного галогенированного замедляющего горение агента.
Присутствующие эпоксидные группы эффективно позволяют защитить всю самозатухающую полимерную композицию от деградации, индуцированной высвобождением галогенов, которые выделяются из галогенированных присадок при температурах, адаптированных для перемешивания этих присадок
Если содержание оксиранового кислорода в блок-сополимере составляет меньше 0,7 масс.%, использование последнего не является более выгодным, чем использование более простого статистического сополимера, так как в случае низкого содержания эпоксидных групп статистический сополимер равномерно распределяется в полимерной композиции винилароматических полимеров. Содержания оксиранового кислорода в блок-сополимере более 19 масс.% трудно добиться, и это экономически невыгодно.
Повышенная стабильность, которую удалось достичь одновременно с неожиданной эффективностью для обеспечения самозатухания, ведет к снижению требуемого количества галогенированных присадок и к меньшему образованию галогенидов водорода. Еще одно связанное с повышенной стабильностью преимущество заключается в возможности обрабатывать самозатухающие винилароматические полимерные композиции, характеризующиеся высокой вязкостью, при более высоких температурах, что позволяет получить конечные продукты с улучшенными эксплуатационными свойствами.
Подробное описание изобретения
В целях настоящего изобретения термин “содержит” или “включает” также включает случай “по существу состоит из” или “состоит только из”.
В целях настоящего изобретения, термин м.ч. (phr), определенный как “частей на 100 частей состава”, означает “частей на 100 частей компонента (а)”.
Настоящее изобретение относится к самозатухающей полимерной композиции, содержащей:
a) стабилизирующую полимерную композицию, содержащую:
i) от 80 масс.% до 99,5 масс% одного или более совместимых друг с другом винилароматических полимеров; а также
ii) от 0,5 масс.% до 20 масс.% по меньшей мере одного блок-сополимера, содержащего:
1) по меньшей мере один блок винилароматического полимера, совместимый с (i) и составляющий не менее 15 масс.% по отношению к целому блок-сополимеру, а также
2) по меньшей мере один блок винилового сополимера, содержащий эпоксидные группы, так чтобы масса оксиранового кислорода составляла от 0,7 масс.% до 19 масс.%, по отношению к целому блок-сополимеру; а также
b) от 0,03 до 10 м.ч. относительно компонента (а) по меньшей мере одного галогенированного замедляющего горение агента.
Винилароматические полимеры (i), используемые в композициях, являющихся объектом настоящего изобретения, могут представлять собой полимеры, получаемые путем гомо- или сополимеризации винилароматических мономеров общей формулы (I):

где R представляет собой водород или метильную группу, n означает ноль или целое число от 1 до 3, Y представляет собой галоген, такой как хлор или бром, или хлорметильную, алкильную группу или алкокси-группу, содержащую от 1 до 3 атомов углерода.
Предпочтительные винилароматические мономеры общей формулы (1) выбраны из стирола, α-метилстирола, изомеров винилтолуола, изомеров этилстирола, изомеров пропилстирола, изомеров хлорстирола, изомеров метилхлорстирола, изомеров метоксистирола, изомеров ацетоксистирола, гидроксистирола, изомеров метилгидроксистирола и их смесей. Указанные винилароматические мономеры могут быть, более предпочтительно, выбраны из стирола и α-метилстирола.
Винилароматические полимеры (i), используемые в составе композиций, которые являются объектом настоящего изобретения, могут представлять собой сополимеры, содержащие в качестве основных компонентов винилароматические мономеры формулы (1) и виниловые сомономеры, выбранные из диенов, нитрилов, производных C1-C8-алкильных эфиров акриловой и метакриловой кислоты, винилацетата и их смесей, более предпочтительно, из стирол-бутадиена, стирол-изопрена, гидрированного стирол-бутадиена, гидрированного стирол-изопрена, стирол-акрилонитрила, стирол-алкилакрилата, стирол-алкилметакрилата, стирол-бутадиен-алкилакрилата, стирол-бутадиен-алкилметакрилата, стирол-акрилонитрил-алкилакрилата, стирол-винилацетат.
Винилароматические полимеры (i), используемые в составе полимерной композиции, являющейся объектом настоящего изобретения, могут представлять собой смеси винилароматических сополимеров, прочность которых повышена другими сополимерами, предпочтительно, сополимерами полиэтиленов и этилен-винилацетата, диеновыми полимерами, терполимерами этилен-пропилен-диена, блок-полимерами, такими как стирол-бутадиен, стирол-изопрен, стирол-этилен-бутилен-стирол, гидрированный стирол-бутадиен, гидрированный стирол-изопрен.
Винилароматические полимеры (i), используемые в составе полимерной композиции, являющейся объектом настоящего изобретения, могут представлять собой винилароматические сополимеры, привитые на других полимерах, предпочтительно, получаемые из стирола, стирол-акрилонитрила, стирол-метилметакрилата, стирол-бутилакрилата, стирол-акрилонитрил-метил-метакрилата, стирол-акрилонитрил-бутилакрилат, стирол-акрилонитрил-малеимида, привитого на сополимерах полиэтиленов и этилен-винилацетата, диеновых полимерах, терполимерах этилен-пропилен-диена, блок-полимерах, таких как стирол-бутадиен, стирол-изопрен, стирол-этилен-бутилене-стирол, гидрированный стирол-бутадиен, гидрированный стирол-изопрен.
Блок-сополимеры (ii) можно получить путем контролируемой цепной радикальной сополимеризации, такой как полимеризация, опосредуемая нитрокси-группами (Nitroxy Mediated Polymerization (NMP)), радикальная полимеризация с переносом атома (Atom Transfer Radical Polymerization (ATRP)), обратимое присоединение с переносом фрагментов (Reversible Addition Fragmentation Transfer (RAFT)), как описано в книге Controlled and Living Polymerization Method and Materials (Материалы и методы контролируемой и живой полимеризации), под редакцией Axel H.E. Müller и Krzysztof Matyjaszewski, опубликованной Wiley-VCH Verlag GmbH & Co. Weinheim в 2009 году. В частности, блок-сополимеры винилароматических мономеров можно удобно синтезировать методом NMP, как описано в заявках EP 0960909 и WO 2004/005361.
Блок-сополимеры (ii) можно также получить, прививая содержащие эпоксиды сополимеры на сополимеры винилароматических мономеров и таких мономеров, как акриловая, метакриловая кислоты и малеиновый ангидрид, как описано в работе Xanthos M. и Dagli S.S. в журнале Polymer Engineering and Science 31(13), страницы 929-935 (1991).
Кроме того, блок-сополимеры (ii) можно получить, проводя эпоксидирование блок-сополимеров, содержащих ненасыщенные фрагменты, такие как, например, блок-сополимеры, стирол-бутадиена, стирол-бутадиен-стирола, которые можно эпоксидировать, как описано в US 4,051,199, US 4,131,725, US 6,576,692, US 6,903,164 и US 2010/0331497.
Кроме того, блок-сополимеры (ii) можно получить, проводя эпоксидирование с участием эпихлоргидрина винилароматических блок-сополимеров, содержащих заместители в бензольном цикле одного из блоков, такие как гидроксильные или гидроксиметильные группы, что описано в работе Ayres J.T. и Mann С.К. в Journal of Polymer Science, Часть B: Polymer Letters 3(6) страницы 505-508, (1965).
Для обеспечения однородного распределения (ii) в (i), блок-сополимеры (ii) должны содержать по меньшей мере один блок винилароматического сополимера, совместимого с (i).
Винилароматический блок полимер, совместимый с (i), должен составлять по меньшей мере 15 масс.% от блок-сополимера (ii), и его состав должен быть равным или, во всяком случае, совместимым с винилароматическими полимерами (i).
Мономеры винила, содержащие эпоксидные группы, которые используются в настоящем изобретении, можно выбрать из стиролов, замещенных по бензольному циклу, эпоксидированных производных гидроксистирола или гидроксиметилстирола, сложноэфирных производных акриловой и метакриловой кислоты и эпихлоргидрина или олигомеров бисфенола и эпихлоргидрина. Указанные мономеры винила, содержащие эпоксидные группы, могут быть более предпочтительно выбраны из глицидилметакрилата, глицидилоксистирола и 2,3-эпоксипропилвинилбензилового эфира, а также продуктов, образующихся в ходе реакции гидроксистирола, гидроксиметилстирола и метакриловой кислоты с эпоксидными смолами, состоящими из бисфенолов и эпихлоргидрина. Глицидилоксистирол можно получить, как описано в WO 2008/085513, а 2,3-эпоксипропилвинилбензиловый эфир - как описано в работе Tomoi М., Oda Н. и Kakiuchi в журнале Makromolekulare Chemie, Rapid Communications, том. 7, страницы 143-148 (1986). Содержащий эпоксидные группы наиболее распространенный и коммерчески доступный мономер винила представляет собой глицидилметакрилат.
Самозатухающие композиции, описанные в настоящем документе и формуле изобретения, могут включать в себя по меньшей мере один замедляющий горение агент, содержащий галогенированное соединение, основанное на алифатических углеводородах. Указанные замедляющие горение вещества выбраны из гексабромциклододекана, пентаброммонохлорциклогексана, пентабромфенилаллилового эфира, бромированного стирол-бутадиенового сополимера, тетрабромбисфенол А бис-дибромпропилового эфира. Галогенированные замедляющие горение вещества могут предпочтительно присутствовать количестве от 0,03 мас.ч. до 10 мас.ч. относительно компонента (а).
Самозатухающие композиции, являющиеся объектом настоящего изобретения, могут также включать синергетический агент в количестве в интервале от 0 мас.ч. до 4 мас.ч. относительно компонента (а), более предпочтительно, от 0,1 мас.ч. до 1 мас.ч. В частности, синергетический агент присутствует в самозатухающей полимерной композиции, являющейся объектом настоящего изобретения, которая также включает вспенивающий агент, из которого можно получать вспененные изделия и полимерные пены. Синергетический агент по определению является веществом, способным в точке воспламенения генерировать радикалы, которые индуцируют деградацию галогенированного замедляющего горения агента с образованием радикалов галогена.
Указанный синергетический агент может предпочтительно быть выбран из пероксида или нестабильного углеводорода. Синергетический агент, более предпочтительно, выбран из 2,3-диметил-2,3-дифенилбутана, 3,4-диметил-3,4-дифенилгексана, 3,6,9-триэтил-3,6,9-триметил-1,4,7-трипероксинонана.
Самозатухающие полимерные композиции, используемые при производстве вспененных изделий, то есть вспениваемые самозатухающие полимерные композиции, являющиеся объектом настоящего изобретения, могут также содержать вспенивающий агент в количестве от 0.2 мас.ч. до 10 мас.ч. относительно компонента (а), более предпочтительно, от 1 мас.ч. до 10 мас.ч.
Любой распространенный вспенивающий агент, который можно включить в состав винилароматического полимера, можно использовать в композициях, являющихся объектом настоящего изобретения. Примерами используемых вспенивающих агентов являются алифатические углеводороды, фреон, диоксид углерода, спирты, такие как этиловый спирт, а также вода.
Для производства вспененных изделий или предназначенных для теплоизоляции полимерных пен, самозатухающие полимерные композиции, являющиеся объектом настоящего изобретения, могут также содержать непроницаемые для теплового излучения присадки в количестве от 0 мас.ч. до 25 мас.ч., более предпочтительно, от 0,2 мас.ч. до 25 мас.ч., еще более предпочтительно, от 1 мас.ч. до 20 мас.ч., относительно компонента (а). Непроницаемые для теплового излучения присадки, более предпочтительно, выбраны из сажи, кокса или графита, еще более предпочтительно, если это кокс.
Углеродный кокс (или просто кокс) доступен в виде мелко измельченного порошка с размером частиц (МТ50) в интервале от 0,5 до 100 мкм, предпочтительно, от 2 до 20 мкм. Размер частиц (МТ50 или d50) определяют лазерным гранулометром, он представляет собой диаметр, такой, что диаметр 50 масс.% частиц меньше этой величины, а диаметр других 50% частиц больше нее. Диаметр означает размер частиц, измеренный лазерным гранулометром, как описано выше.
Кокс образуется в результате пиролиза органического вещества и по крайней мере частично в процессе карбонизации переходит через жидкое или жидко-кристаллическое состояние. Исходное органическое вещество, предпочтительно, представляет собой нефть, уголь или лигнит.
Кокс, используемый при получении полимерных композиций в гранулированной форме, являющихся объектом настоящего изобретения, более предпочтительно, является продуктом карбонизации высококипящих фракций углеводородов, образующихся в ходе перегонки нефти, обычно эти фракции называют фракциями тяжелого остатка. В частности, кокс этого типа получают в ходе коксования фракции тяжелого остатка, это операция производится при высокой температуре, и при этом образуются дополнительные легкие фракции и твердый остаток (нефтяной кокс). Полученный таким способом нефтяной кокс кальцинируют при температуре от 1000 до 1600°C (обожженный кокс).
Если используется обогащенная ароматическими компонентами фракция тяжелого остатка, получаемый кокс имеет игловидную кристаллическую структуру (игольчатый кокс), он образуется при кальцинировании при температуре 1800-2200°C.
Дополнительная информация про кокс, способы его производства и характеристики коммерчески доступного кокса различного качества (зеленый кокс, получаемый из угля пековый (смолистый) кокс, отпущенный кокс, жидкий кокс, игольчатый кокс, кокс повышенного качества, прокаленный кокс, кокс с включениями, губчатый кокс и т.д.) содержится в интернете на сайте goldbook.iupac.org или в журнале Pure Appl. Chem., 1995, Том. 67, N. 3, страницы 473-506 “Recommended terminology for the description of carbon as a solid (Рекомендованная терминология для описания твердого углерода) (IUPAC Recommendations 1995)”.
Непроницаемые для теплового излучения присадки, в основном, используются в строительстве при изготовлении панелей, состоящих из винилароматических полимеров, вспененных под действием вспенивающих агентов, и, как правило состоят из углерода в форме кокса, природного или вспененного графита и сажи. Эти присадки снижают термостабильность галогенированных замедляющих горение агентов, что приводит к более низким температурам перемешивания винилароматических полимеров, а это затрудняет перемешивание в связи с большей вязкостью массы полимера. Другие неорганические присадки, поглощающие излучение в интервале от 100 до 20000 см-1 (по данным анализа в ближней и средней инфракрасной области), представляют собой титанаты, оксиды титана, оксиды кремния, оксиды и гидроксиды алюминия, сульфат бария, алюмосиликаты, силикаты кальция и магния, карбонаты кальция и магния, оксид кальция и цинка, а также бентонит.
Композиции, являющиеся объектом настоящего изобретения, особенно эффективны, если в качестве непроницаемой для теплового излучения присадки содержат кокс. Таким образом, указанные композиции особенно полезны при производстве вспененных изделий, применяемых для теплоизоляции. В таких случаях, фактически, наблюдается значительное снижение образования галогенидов водорода, которое индуцируется тепловой деградацией замедляющего горение вещества, что также проиллюстрировано примерами из настоящего документа.
Для получения компактных изделий самозатухающие композиции, описанные в настоящем документе и формуле изобретения, могут также содержать от 0 мас.ч. до 50 мас.ч., предпочтительно, от 0,01 мас.ч. до 10 мас.ч., относительно компонента (а), одной или более присадок, состоящих из лубрикантов, красителей, антистатических агентов, отслаивающих агентов, антиоксидантов.
Для компактных изделий, которые почти всегда обрабатывают при температуре выше 180°C, особого эффекта от введения замедляющих горение присадок не наблюдается. Преимущество сополимеров с эпоксидами заключается в возможности обойтись меньшим количеством замедляющих горение присадок и/или обойтись без использования матрицы низкой вязкости для обработки полимерных смесей при температурах ниже 200°C. Матрицы низкой вязкости характеризуются худшими механическими свойствами.
Указанное преимущество открывается в приводимых примерах УППС.
Самозатухающие полимерные композиции, описанные в настоящем документе и формуле изобретения, могут использоваться для производства компактных изделий, вспениваемых шариков, вспененных изделий и полимерных пен.
Композиции, являющиеся объектом настоящего изобретения, особенно хорошо подходят для применений, требующих в ходе своего получения или обработки по меньшей мере одного этапа, когда указанная огнестойкая композиция находится в статической или динамической мешалке при 180-230°C. Указанные композиции особенно удобны для получения содержащих вспенивающий агент шариков из винилароматического полимера посредством непрерывного процесса; для получения экструдированных пенопластов в присутствии вспенивающих агентов; для получения полимерных смесей, предназначенных для последующей трансформации путем горячего прессования (экструзии), термоотверждения или литья под давлением.
Несколько представленных далее иллюстративных примеров предназначены для лучшего понимания изобретения и области его применения, однако их не следует считать ограничивающими область настоящего изобретения.
Примеры
Примеры получения сополимеров винилароматических мономеров и мономеров винила со связанными эпоксидными группами.
В Примере 2 видно, что сополимер, содержащий 50% глицидилметакрилата, невозможно получить в виде термопластичного полимера (фактически, он нерастворим, и не поддается обработке). С другой стороны, Пример 3 показывает, что блок-сополимер, содержащий 50% полистирола и 50% стирол-глицидилметакрилата с 50% глицидил-метакрилата, поддается обработке и растворим. В Примере 1 описано получение статистического сополимера стирол-глицидилметакрилата и 14,8% глицидилметакрилата, не совместимого с полистиролом. Как показано в таблице 3, смесь ЭПС3, содержащая сополимер из Примера 1, не проходит теста на огнестойкость. Блок сополимер Примера 3, содержащий 25% глицидилметакрилата, по отношению к общей массе блок сополимера, совместим с полистиролом и в составе смеси ЭПС4 Таблицы 3 проходит тест на огнестойкость.
На основании Таблицы 3, можно видеть, что ЭПС1 проходит тест, но образует многочисленные бромиды, ЭПС2 снижает содержание бромидов, но не проходит теста, поскольку эпоксидированная присадка не распределена в общей композиции равномерно, ЭПС3 снижает содержание бромидов, но не проходит теста, поскольку сополимер Примера 1 не распределен в общей композиции равномерно, ЭПС4 - ЭПС8 снижают содержание бромидов и проходят тест на огнестойкость, показывая эффективность блок сополимеров Примеров 3-7, вероятно, из-за их равномерного распределения в полимерной композиции.
Пример 1: синтез статистического сополимера
1479,1 г стирола (Versalis S.p.A., Сан Донато Миланезе - Италия), 212,9 г глицидилметакрилата (Sigma-Aldrich, Милан - Италия), 108 г этилбензола (Versalis S.p.A., Сан Донато Миланезе - Италия) и 1,44 г трет-додецилмеркаптана (Sigma-Aldrich, Милан - Италия) в атмосфере азота загрузили при 20°C в стальной реактор вместимостью 2,4 л, снабженный лопастной мешалкой, погруженным в реакционную смесь температурным зондом и термостатичным кожухом, в котором циркулирует силиконовое масло, а температура последнего регулируется внешней термостатической баней. Нагревая поддерживающее температуру масло кожуха реактора, температуру реакционной смеси за 2 часа увеличили до 125°C и поддерживали ее при этой температуре в течение 5 часов. После этого смесь, содержащую около 50% полимера, выгрузили в стальные цилиндры по 120 г на цилиндр. Цилиндры поместили в печь с электроподогревом и 3 часа выдерживали при температуре 150°C. По завершении этого периода в печи с цилиндрами создали вакуум с остаточным давлением воздуха 20 мбар, затем приблизительно в течение часа температуру подняли до 220°C и поддерживали на этом уровне в течение 30 минут. После охлаждения печи полимер извлекли из цилиндров и измельчили на мельнице. Полученные гранулы полимера (1380 г) проанализировали и определили (путем анализа содержания эпоксида по методу ASTM D1652-04), что содержание глицидилметакрилата составляет 14,8% м/м, a MFR (melt flow rate, скорость потока расплата) (200°C/5 кг) соответствует 17,1 г/10 мин (метод ISO 1133).
Пример 2: синтез статистического сополимера
Была использована такая же процедура, что и в Примере 1, со следующим составом: 846 г стирола, 846 г глицидилметакрилата, 108 г этилбензола и 1,1 г ди-трет-додецилмеркаптана. Полученный полимер был нерастворим и не поддавался обработке.
Пример 3: синтез блок сополимера стирол-b-(стирол-глицидилметакрилата) посредством реакции контролируемой цепной полимеризации
1377 г стирола, 2,23 г ВРО (Luperox А75, ARKEMA), 1,55 г 4OH-ТЕМРО (Sigma-Aldrich) загрузили при 20°C в реактор Примеров 1-2. Нагревая поддерживающее температуру масло кожуха реактора, температуру реакционной смеси за 2 часа увеличили до 125°C, и поддерживали ее при этой температуре в течение 2 часов и 15 минут; после этого в реактор загрузили 423 г глицидилметакрилата и реакцию продолжали еще 2 часа. Через 2 часа после добавления глицидилметакрилата содержащую около 49% полимера реакционную смесь выгрузили в стальные цилиндры по 120 г на цилиндр. Цилиндры поместили в печь, создали вакуум с остаточным давлением 20 мбар, затем приблизительно в течение часа температуру подняли до 220°C и поддерживали на этом уровне в течение 30 минут. После охлаждения печи полимер извлекли из цилиндров и измельчили на мельнице. Полученные гранулы полимера (900 г) проанализировали и определили содержание глицидилметакрилата 25% м/м, a MFR (200°C/5 кг) соответствует 22 г/10 мин.
Пример 4: синтез привитого блок сополимера стирол-g-(стирол-глицидилметакрилата)
422,66 г стирола, 0,34 г малеинового ангидрида, 27 г этилбензола и 0,80 г ди-трет-додецилмеркаптана при 20°C загрузили в реактор Примеров 1-3. Нагревая поддерживающее температуру масло кожуха реактора, температуру реакционной смеси за 2 часа увеличили до 126°C и поддерживали ее при этой температуре в течение 6 часов. По достижении 126°C в реактор при помощи мембранного насоса ввели 450 г такой же композиции, что и первоначальная смесь, скорость подачи составляла 75 г/час. Через 6 часов, по окончании подачи раствора стирола, малеинового ангидрида, этилбензола и трет-додецилмеркаптана, в реактор загрузили смесь, содержащую 336 г стирола, 510 г глицидилметакрилат, 54 г этилбензола и 1,6 г трет-додецилмеркаптана, температуру реактора довели до 126°C и поддерживали на этом уровне 2 часа 40 минут. После этого смесь, содержащую около 50% полимера, выгрузили в стальные цилиндры по 120 г на цилиндр. Полученные гранулы полимера (920 г) проанализировали и определили, что содержание глицидилметакрилата составляет 24% м/м, a MFR (200°C/5 кг) соответствует 16,2 г/10 мин.
Пример 5: синтез блок сополимера стирол-b-(стирол-глицидилоксистирола) посредством реакции контролируемой радикальной цепной полимеризации
352 г п-гидроксикоричной кислоты (Sigma-Aldrich, Милан - Италия) и 1112 г N,N-диметилацетамида (Sigma-Aldrich, Милан - Италия) в атмосфере азота при перемешивании и при 20°C загрузили в стальной реактор вместимостью 6 л с выгрузкой со дна, оборудованный магнитной системой перемешивания и лопастной мешалкой, обратным холодильником, погруженным в реакционную смесь температурным зондом и термостатичным кожухом, в котором циркулирует силиконовое масло, а температура последнего регулируется внешней термостатической баней. Через 10 минут в атмосфере азота добавили еще 2,94 г ацетата калия (Sigma-Aldrich, Милан - Италия), после чего, нагревая поддерживающее температуру масло кожуха реактора, температуру реакционной смеси за 2 часа увеличили до 150°C и поддерживали ее при этой температуре в течение 3 часов 30 минут, за это время п-гидроксикоричная кислота превратилась в п-гидроксистирол. Охладив реакционную смесь до 20°C, обратный холодильник заменили насадкой для перегонки, которая через холодильник соединяется с колбой для сбора конденсата и вакуумным насосом. Реакционную смесь сконцентрировали при 40°C и остаточном давлении 33 Па, удалив 556 г N,N-диметилацетамида, и выгрузили в бак в атмосфере азота. 132 г гидрата калия (Sigma-Aldrich, Милан - Италия) мелко измельчили в сухой коробке, смесь п-гидроксистирола и N,N-диметилацетамида вынули из реактора, после чего 2927 г эпихлоргидрина (Sigma-Aldrich, Милан - Италия) загрузили в реактор с холодильником, промыли N,N-диметилацетамидом в атмосфере азота. Полученную таким способом реакционную смесь нагрели до 90°C, нагревая поддерживающее температуру масло кожуха реактора, поддерживали при этой температуре один час, после чего охладили до 25°C. Полученную реакционную смесь выгрузили из нижней части реактора в стеклянные колбы. В те же колбы поместили промывочную жидкость, полученную путем опрыскивания стенок реактора и вкладышей приблизительно 500 мл N,N-диметилацетамида. Содержимое колбы охладили до 4°C, профильтровали через пористую прокладку со средней пористостью и затем на фильтре из политетрафторэтилена со средним диаметром пор 0,45 микрон, после чего промыли 100 мл N,N-диметилацетамида. Профильтрованную жидкость сконцентрировали в том же стальном реакторе вместимостью 6 л, который использовали в предыдущих реакциях. Заменив холодильник наконечником для конденсации и применив вакуум, удалили избыток эпихлоргидрина и часть N,N-диметилацетамида. В конце получили раствор, содержащий около 351 г п-глицидилоксистирола и приблизительно 355 г N,N-диметилацетамида.
249 г стирола, 2,23 г ВРО, 1,55 г 4OH-ТЕМРО в атмосфере азота добавили в тот же реактор вместимостью 6 литров, содержащий п-глицидилоксистирол и N,N-диметилацетамид. Нагревая поддерживающее температуру масло кожуха реактора, температуру реакционной смеси за 2 часа увеличили до 125°C и поддерживали ее при этой температуре в течение 6 часов 30 минут; в реактор загрузили 4350 г стирола; температуру реакции снова подняли до 125°C и продолжали реакцию один час. Смесь затем охладили до 95°C, после чего, применив вакуум, отогнали 3050 г стирола. По завершении отгонки реакционную смесь, содержащую около 50% полимера, выгрузили в стальные цилиндры по 120 г на цилиндр. Цилиндры поместили в печь, применили вакуум с остаточным давлением воздуха 20 мбар, затем приблизительно в течение часа температуру подняли до 220°C и 30 минут поддерживали на этом уровне. После охлаждения печи полимер извлекли из цилиндров и измельчили на мельнице. Полученные гранулы полимера (910 г) проанализировали и определили, что содержание п-глицидилоксистирола составляет 32,5% м/м, a MFR (200°C/5 кг) соответствует 20 г/10 мин.
Пример 6: синтез блок сополимера стирол-b-(стирол-2,3-эпоксипропилвинилбензилового эфира) посредством реакции контролируемой радикальной цепной полимеризации
В сухой коробке мелко измельчили 133 г гидрата калия, затем 338 г гидроксиметилстирола (Sigma-Aldrich, Милан - Италия) и 2940 г эпихлоргидрина загрузили в атмосфере азота в реактор Примера 5. Полученную таким способом реакционную смесь довели до 90°C, нагревая поддерживающее температуру масло кожуха реактора, поддерживали при этой температуре один час, после чего охладили до 25°C. Полученную реакционную смесь выгрузили из нижней части реактора в стеклянные колбы. В те же колбы поместили промывочную жидкость, полученную путем опрыскивания стенок реактора и вкладышей приблизительно 500 мл N,N-диметилацетамида. Содержимое колбы охладили до 4°C, профильтровали через пористую прокладку со средней пористостью и затем на фильтре из политетрафторэтилена со средним диаметром пор 0,45 микрон, после чего промыли 50 мл N,N-диметилацетамида. Профильтрованную жидкость сконцентрировали в том же стальном реакторе вместимостью 6 л, который использовали в предыдущей реакции. Заменив холодильник наконечником для конденсации и применив вакуум, удалили избыток эпихлоргидрина и часть N,N-диметилацетамида. В конце получили раствор, содержащий около 384 г 2,3-эпоксипропилвинилбензилового эфира и приблизительно 390 г N,N-диметилацетамида.
216 г стирола, 2,23 г ВРО, 1,55 г 4OH-ТЕМРО в атмосфере азота добавили в тот же реактор вместимостью 6 литров, содержащий 2,3-эпоксипропилвинилбензиловой эфир и N,N-диметилацетамид. Нагревая поддерживающее температуру масло кожуха реактора, температуру реакционной смеси за 2 часа увеличили до 125°C, и поддерживали ее при этой температуре в течение 7 часов; в реактор загрузили 4350 г стирола; температуру реакции снова подняли до 125°C и продолжали реакцию 1 час 15 минут. Смесь затем охладили до 95°C, после чего, применив вакуум, отогнали 3050 г стирола. По завершении отгонки реакционную смесь, содержащую около 50% полимера, выгрузили в стальные цилиндры по 120 г на цилиндр. Цилиндры поместили в печь, применили вакуум с остаточным давлением воздуха 20 мбар, затем приблизительно в течение часа температуру подняли до 220°C и 30 минут поддерживали на этом уровне. После охлаждения печи полимер извлекли из цилиндров и измельчили на мельнице. Полученные гранулы полимера (905 г) проанализировали и определили, что содержание 2,3-эпоксипропилвинилбензилового эфира составляет 35,2% м/м, а MFR (200°C/5 кг) соответствует 24 г/10 мин.
Пример 7: синтез блок сополимера полистирол-b-Pbu, эпоксидирование которого осуществляли путем эпоксидирования блок сополимера PS-b-PBu
3000 г циклогексана (Versalis S.p.A., San Donato Milanese - Италия), 2000 г этилацетата (Sigma-Aldrich, Milan - Италия) и 1000 г EUROPRENE SOL В 183 (блок сополимер стирол - бутадиена с 11% м/м бутадиена, Versalis SpA, San Donato Milanese - Италия) в атмосфере азота при перемешивании и при 20°C загрузили в стальной реактор вместимостью 15 л с выгрузкой со дна, оборудованный магнитной системой перемешивания и лопастной мешалкой, насадкой для перегонки, подключенной холодильник к баку для сбора, который, в свою очередь, соединен с вакуумным насосом, погруженным в реакционную смесь температурным зондом и термостатичным кожухом, в котором циркулирует силиконовое масло, а температура последнего регулируется внешней термостатической баней. Нагревая поддерживающее температуру масло кожуха реактора, температуру реакционной смеси увеличили до 40°C и два часа поддерживали ее при этой температуре до полного растворения эластомера. По окончании растворения в течение 2 часов при помощи дозировочного насоса загрузили 540 г смеси этилацетата и 30% перуксусной кислоты (Sigma-Aldrich, Милан - Италия) и температуру еще час поддерживали на уровне 40°C. По завершении эпоксидирования смесь охладили до 4°C и в реактор при перемешивании загрузили 5 литров холодной деминерализованной воды. Через 15 минут перемешивание прервали и из нижней части реактора выгрузили около 5 литров водной фазы. Промывание 5 литрами воды повторяли еще два раза (всего три промывания). Смесь из реактора затем нагрели до 75°C, применили вакуум для отгонки растворителей и такого концентрированния раствора, чтобы получить смесь из 40 частей эластомера и 60 частей растворителя. Раствор 40% стирол-бутадиенового сополимера, который эпоксидировали в остаточном растворителе реакции, выгрузили в стальные цилиндры по 120 г на цилиндр. Цилиндры поместили в печь, применили вакуум с остаточным давлением воздуха 20 мбар, затем приблизительно в течение часа температуру подняли до 180°C и 15 минут поддерживали на этом уровне. После охлаждения печи полимер извлекли из цилиндров и измельчили на мельнице. Полученные гранулы полимера (990 г) проанализировали и определили, что содержание оксиранового кислорода составляет 2,8% м/м.
Примеры, подтверждающие улучшенную термостабильность ГБЦД (примеры для ЭПС и УППС
Указанные в Таблице 1 смеси (числа означают количество частей каждого компонента в смеси) были приготовлены в безводных условиях во вращающемся цилиндре (миксере), пропущены через двухшнековый экструдер Вернера при температуре 180°C - 220°C и времени нахождения там 1-2 минуты и гранулированы:

EDISTIR N2982 (Versalis S.p.A., Сан Донато Миланезе - Италия) представляет собой гомополимер полистирола общего применения (ГППС) с MFR (200°C/5 кг) 25 г/10 мин.
DGEBPA (Sigma-Aldrich, Милан - Италия) представляет собой диглицидиловый эфир бисфенола А.
Смеси MIX СВ1 - СВ8 используют для приготовления других смесей, указанных в таблице 2, которые получают во вращающемся цилиндре (миксере), пропускают через экструдер Брабендера при температуре 190°C и времени нахождения там 3 минуты и гранулируют.

EDISTIR N1782 (Versalis S.p.A., Сан Донато Миланезе - Италия) представляет собой гомополимер полистирола общего применения (ГППС) с MFR (200°C/5 кг) 8 г/10 мин.
PK30 (Akzo-Nobel) представляет собой 2,3-диметил-2,3-дифенилбутан, Перкадокс 30.
ГБЦД (Albemarle) представляет собой гексабромциклододекан, НР900.
Получение ЭПС в непрерывном режиме
77 частей полистирола EDISTIR N1782 в виде гранул и 23 части гранул, состоящих из МВ1 - МВ8, в безводных условиях смешивают во вращающемся цилиндре (ЭПС1 - ЭПС8 в Таблице 3) и передают в одношнековый экструдер со временем нахождения 7 минут при давлении 260 бар и температуре около 190°C; из него смесь поступает в статический миксер, на входе которого добавляют 5 частей смеси н-пентана (75%) и изопентана (25%). Полученную таким способом смесь распределяют через отверстия диаметром 0,5 мм, немедленно охлаждают потоком воды и нарезают последовательностью вращающихся ножей, как описано в патенте US 7,320,585. Давление в камере для гранулирования составляет 5 бар, а сдвиговое напряжение подбирают так, чтобы получить гранулы среднего диаметра 1,2 мм. Для охлаждения жидкость опрыскивают водой, а в качестве носителя гранул используют азот, который перемещает гранулы в центрифужную сушилку, где их сушат, затем в непрерывном шнековом миксере к смеси добавляют 3 части глицерина моностеарата, одну часть стеарата цинка и 0,2 части глицерина на 1000 частей гранул. Полученные таким способом гранулы предварительно вспенивают паром при 100°C, оставляют на один день, после чего изготавливают из них цилиндры диаметром 260 мм и высотой 40 мм, плотность которых находится в интервале от 14 до 16 г/дм3. Цилиндры на два дня оставляют в термостатически регулируемой камере, температуру в которой поддерживают равной 70°C. Из таких цилиндров с регулируемой температурой готовят затем испытуемые образцы размером 90×190×20 мм, которые подвергают тесту на огнестойкость в соответствии со стандартом DIN 4102 и определяют содержание бромидов. Результаты исследований представлены в Таблице 3.

В соответствии со стандартом DIN 4102, высота пламени не должна превышать 15 см.
Метод определения бромидов
Взвешивают 0,1-1,5 г образца (основываясь на предполагаемом содержании бромидов), полимерная композиция которого содержит бромированное соединение, помещают его в предназначенную для испытаний пробирку вместимостью 50 мл и растворяют в 15 мл хлороформа. По завершении растворения добавляют 25 мл элюента для ионной хроматографии (водный раствор 0,0020 M NaHCO3 и 0,0013 M Na2CO3), пробирку закрывают и перемешивают смесь 20 минут. По завершении перемешивания водную и органическую фазу оставляют, чтобы они разделились, и анализируют водную фазу методом ионной хроматографии Metrohm Mod. Compact 761 IC на колонке Metrohm Dual2 (код 6.1006.100) с суппрессором проводимости, а также с пре-колонкой Metrosep A Supp 4/5 (код 6.1006.500). На основании хроматограммы рассчитывают концентрацию раствора, выраженную в м.д. м/м.
Примеры приготовления УППС
Описанные в таблице 4 смеси готовят в безводной среде во вращающихся цилиндрах.
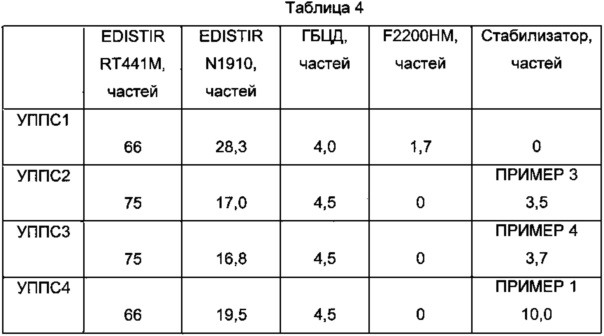
EDISTIR RT441M (Versalis S.p.A., Сан Донато Миланезе - Италия) представляет собой ударопрочный полистирол (УППС) с MFR (200°C/5 кг) 8 г/10 мин.
EDISTIR N1910 (Versalis S.p.A., Сан Донато Миланезе - Италия) представляет собой гомополимер полистирола общего применения (ГППС), смазанный парафиновым маслом, с MFR (200°C/5 кг) 20 г/10 мин.
F2200HM представляет собой диглицидиловый эфир тетрабромбисфенола А производства ICL-Industrial Products.
Полученные таким способом смеси выдавливают в одношнековый экструдер AMUT при 190°C с временем нахождения 1-2 минуты и гранулируют. Гранулы плавят на прессе Battenfeld 350 при 190°C, получая тестовые образцы размером 127×12,7×1,59 мм, предназначенные для испытаний на огнестойкость в соответствии с методом UL-94 V, описанным в работе “Standard For Tests for Flammability of Plastic Materials For Parts in Devices and Appliances” (Стандарты испытаний на воспламенимость пластиковых материалов, предназначенных для деталей устройств и приспособлений), 3 Издание, 28 января 1980 года. Смеси УППС1-3 принадлежат классу V2 и, в особенности, смеси УППС2 и УППС3, характеризуются более низким общим содержанием брома и более высоким содержанием ударопрочного винилароматического полимера, по сравнению с УППС1. Смесь УППС4, с такой же долей ГБЦД, даже если ее содержание оксиранового кислорода приблизительно в два раза превышает смесь УППС2, не проходит тест на огнестойкость.
Изобретение относится к самозатухающей полимерной композиции. Описана самозатухающая полимерная композиция, содержащая: a) стабилизирующую полимерную композицию, содержащую: i) от 80 до 99,5 мас.% одного или более совместимых друг с другом винилароматических полимеров; и ii) от 0,5 до 20 мас.% по меньшей мере блок-сополимера, содержащего: 1) по меньшей мере блок винилароматического полимера, совместимый с (i), в количестве не менее 15 мас.%, по отношению к целому блок-сополимеру, а также 2) по меньшей мере блок винилового сополимера, содержащий эпоксидные группы, так чтобы масса оксиранового кислорода составляла от 0,7 до 19 мас.% по отношению к целому блок-сополимеру; и b) от 0,03 до 10 мас.ч. относительно компонента (а), по меньшей мере галогенированного замедляющего горение агента. Также описаны вспениваемые шарики, прессованные изделия и полимерные пены. Технический результат: получена самозатухающая полимерная композиция, содержащая галогенированные замедляющие агенты горения с повышенной термостабильностью, а также винилароматические полимеры, что обеспечивает эффективность замедляющих горение агентов в экономически выгодных концентрациях. 4 н. и 8 з.п. ф-лы, 4 табл.
1. Самозатухающая полимерная композиция, содержащая:
a) стабилизирующую полимерную композицию, содержащую:
i) от 80 до 99,5 мас.% одного или более совместимых друг с другом винилароматических полимеров; и
ii) от 0,5 до 20 мас.% по меньшей мере блок-сополимера, содержащего:
1) по меньшей мере блок винилароматического полимера, совместимый с (i), в количестве не менее 15 мас.% по отношению к целому блок-сополимеру, а также
2) по меньшей мере блок винилового сополимера, содержащий эпоксидные группы, так чтобы масса оксиранового кислорода составляла от 0,7 до 19 мас.% по отношению к целому блок-сополимеру; и
b) от 0,03 до 10 мас.ч. относительно компонента (а) по меньшей мере галогенированного замедляющего горение агента.
2. Самозатухающая полимерная композиция по п. 1, дополнительно содержащая от 0 до 4 мас.ч. относительно компонента (а) синергетического агента.
3. Самозатухающая полимерная композиция по п. 2, дополнительно содержащая от 0,1 до 1 мас.ч. относительно компонента (а) синергетического агента.
4. Самозатухающая полимерная композиция по любому из пп. 1-3, дополнительно содержащая от 0,2 до 10 мас.ч. относительно компонента (а) вспенивающего агента.
5. Самозатухающая полимерная композиция по п. 4, дополнительно содержащая от 1 до 10 мас.ч. относительно компонента (а) вспенивающего агента.
6. Самозатухающая полимерная композиция по п. 1, дополнительно содержащая от 0 до 25 мас.ч. относительно компонента (а) не проницаемой для теплового излучения присадки.
7. Самозатухающая полимерная композиция по п. 6, дополнительно содержащая от 0,2 до 25 мас.ч. относительно компонента (а) не проницаемой для теплового излучения присадки.
8. Самозатухающая полимерная композиция по п. 7, дополнительно содержащая от 1 до 20 мас.ч. относительно компонента (а) не проницаемой для теплового излучения присадки.
9. Самозатухающая полимерная композиция по любому из пп. 6-8, где не проницаемая для теплового излучения присадка представляет собой кокс.
10. Вспениваемые шарики, содержащие самозатухающую полимерную композицию по любому из пп. 4-9.
11. Прессованные изделия, содержащие самозатухающую полимерную композицию по любому из пп. 1-3.
12. Полимерные пены, содержащие самозатухающую полимерную композицию по любому из пп. 2-9.
| RU 2010139633 A 20.06.2012 | |||
| US 20100041800 A1 18.02.2010 | |||
| JP 2000103952 A 11.04.2000. |

