Область техники, к которой относится изобретение
Настоящее изобретение относится к липидному преконцентрату с замедленным высвобождением, содержащему аналог GnRH в качестве фармакологически активного вещества, и к содержащей его фармацевтической композиции.
Предшествующий уровень техники
Составы с замедленным высвобождением разработаны для высвобождения однократной дозы фармакологически активного вещества при заданной скорости для сохранения эффективной концентрации в кровотоке в течение определенного периода времени, со сведением к минимуму побочных эффектов, вызываемых многократными дозами.
При рассмотрении терапевтического механизма и их физических и химических свойств, производные гонадотропин-рилизинг гормона (GnRH) представляют собой фармакологически активное вещество, предназначенное для составов с замедленным высвобождением.
Гонадотропин-рилизинг гормон (GnRH) или рилизинг-гормон лютеинизирующего гормона (LHRH) представляет собой нейроэндокринный пептид, который синтезируется и высвобождается из нейронов в нейроваскулярном терминале гипоталамуса. Сразу после высвобождения из гипоталамуса GnRH селективно соединяется со специфическими рецепторами на мембране аденогипофизных гонадотропных клеток, чтобы вызвать биосинтез и высвобождение фолликулстимулирующего гормона (FSH) и лютеинизирующего гормона (LH). FSH и LH действуют, чтобы регулировать образование половых стероидов из половых желез у мужчин и женщин. Вследствие биологических функций GnRH, его аналоги могут применяться для лечения гормонозависимых заболеваний, таких как рак простаты, рак молочной железы, рак яичника, эндометриоз, фиброма матки, синдром поликистоза яичников, гипертрихоз, преждевременное половое созревание, гонадотропная аденома гипофиза, синдром апноэ во сне, синдром раздраженного кишечника, предменструальный синдром, доброкачественная гиперплазия предстательной железы и бесплодие.
Lupron® Depot представляет собой коммерчески доступный, инъекционный раствор для внутримышечного или подкожного введения с замедленным высвобождением леупролидацетатного аналога GnRH, с биоразлагаемыми PLGA-микрочастицами [поли(молочная-ко-гликолевая кислота)], поступающими в виде матрицы с замедленным высвобождением. Обычно, PLGA-микрочастицы разлагаются на молочную кислоту и гликолевую кислоту в течение определенного периода времени in vivo, высвобождая фармакологически активное вещество, обеспечивая возможность замедленного высвобождения (патент США 5480656). Однако процессы создания PLGA-микрочастиц являются не только сложными и трудными, но также фармакологически активные вещества отягощены в них в значительной степени слабой эффективностью. В дополнение, вследствие того что PLGA-микрочастицы трудны для фильтровальной стерилизации и плавятся при 40°C или выше, их стерилизация не может быть достигнута обычными способами, но требует более стерильных условий. Идеальный профиль для замедленного высвобождения получен при использовании двух или более разных типов PLGA-микрочастиц, которые, однако, дополнительно усложняют способы изготовления и перемешивания (WO 2005/074896), повышая стоимость производства. Кроме того, примеси уксусной кислоты и продукты кислотного разложения от PLGA-микрочастиц вызывают воспаление и снижение скорости роста клеток (K. Athanasiou, G.G. Niederauer and C.M. Agrawal, Biomaterials, 17, 93 (1996)). Для замедленного высвобождения, суспензию 10-100 мкм PLGA-микрочастиц в водном растворе инъектируют в значительном количестве, но это усиливает боль или поражение ткани в месте инъекции.
Eligard® был введен в качестве состава для инъекций с замедленным высвобождением для аналога GnRH (леупролидацетат), который компенсирует проблемы составов с замедленным высвобождением на основе PLGA-микрочастиц. Eligard® широко продается на рынке как препарат для подкожной инъекции, который получают растворением PLGA [поли(DL-лактид-ко-гликолид)], имеющим защищенную карбоксильную терминальную группу, и аналога GnRH (леупролидацетат) в N-метил-2-пирролидоне (NMP). Eligard® существует в виде текучей композиции, которую можно получить растворением способного к биоразложению полимера в полярном апротонном растворителе, и предназначен для подкожной инъекции с улучшением недостатков составов с твердыми PLGA-микрочастицами (патент США 6773714). Данный коммерческий продукт является неудачным в отношении способности к применению из-за отсутствия поставки полностью предварительно заполненного устройства со шприцами и проявляет низкую стабильность лекарственного средства в смесевом растворе. Устройство, обеспеченное для продукта, содержит два шприца, которые могут быть соединены друг с другом, и инструменты для смешивания, приготовления и инъекции. Конечный смесевой раствор не получают до тех пор, пока не выполнено более чем приблизительно 10 операций, и отводится только 30 мин для всего процесса от приготовления до инъекции. Кроме того, продукт должен храниться в холодильнике, и если не хранился в холодильнике, конечный смесевой раствор не может быть использован более 5 суток. Кроме того, в продукте не наблюдается никаких улучшений в отношении высокого начального всплеска, и это является недостатком, обычным для составов из PLGA-микрочастиц. Точнее, продукт проявляет более высокую концентрацию начального всплеска по сравнению с составом PLGA-микрочастиц Lupron® Depot (патент США 6773714). Концентрация начального всплеска, сильно превышающая аналогичную концентрацию, в которой лекарственное средство может функционировать, является нежелательной как в функциональном, так и в токсикологическом плане. Особенно при рассмотрении механизма аналога GnRH, в котором высвобождение полового гормона временно повышено на начальной стадии введения и затем менее регулируемо от определенной временной точки, следует избегать избыточной концентрации начального всплеска.
В качестве альтернативного решения проблем с составами PLGA-микрочастиц, международная патентная публикация WO 2005/117830 описывает пресостав, содержащий по меньшей мере один нейтральный диациллипид и/или по меньшей мере один токоферол, по меньшей мере один фосфолипид и по меньшей мере один биосовместимый, кислородсодержащий органический растворитель с низкой вязкостью. Другой вариант описан в международной патентной публикации WO 2006/075124, который относится к пресоставу, содержащему по меньшей мере один диацилглицерин, по меньшей мере один фосфатидилхолин, по меньшей мере один кислородсодержащий органический раствор и по меньшей мере один аналог GnRH. Данные пресоставы обеспечивают замедленное высвобождение фармакологически активного вещества in vivo в течение четырех недель и не образуют продукты разложения молочной кислоты или гликолевой кислоты из их полимерных систем, таким образом не вызывают боль или воспаление. Однако существует проблема с составами, в которых использование диациллипида, компонента, важного для пресоставов в качестве фармацевтического эксципиента, непригодно, и оно, как доказано, должно быть достаточно осторожным, и что их обязательный органический растворитель приводит к снижению активности некоторых фармакологически активных веществ (H. Ljuisberg-Wahre, F.S. Nielse, 298, 328-332 (2005); H. Sah, Y. Bahl, Journal of Controlled Release 106, 51-61 (2005)).
В завершении настоящего изобретения, авторы настоящего изобретения предложили липидный преконцентрат с замедленным высвобождением, содержащий: а) сложный сорбитановый эфир ненасыщенной жирной кислоты; b) фосфолипид; и с) отвердитель жидких кристаллов, и фармацевтическую композицию, содержащую преконцентрат (корейская патентная заявка 10-2012-0093677). Данный липидный преконцентрат с замедленным высвобождением проявляет in vivo безопасность и способность к биоразложению при тех же самых или более высоких уровнях по сравнению с обычными преконцентратами, и фармацевтическая композиция, как установлено, подходит для замедленного высвобождения фармакологически активного вещества, содержащегося в ней.
Кроме того, дополнительное исследование авторов настоящего изобретения привело к обнаружению того, что при применении липидного преконцентрата с замедленным высвобождением, аналог GnRH может высвобождаться замедленным образом в концентрации, достаточной для действия в виде фармакологически активного вещества in vivo, приводя к настоящему изобретению.
Ниже дано описание прототипов, релевантных настоящему изобретению.
Патент США 7731947 описывает композицию, содержащую: состав из частиц, включающий интерферон, сахарозу, метионин и цитратный буфер, и суспендирующий раствор, содержащий растворитель, такой как бензилбензоат, где состав из частиц диспергирован в суспендирующем растворе, и дополнительно разъяснение применения аналогов GnRH. В одном примере, описано, что фосфатидилхолин растворен вместе с витамином Е (токоферол) в органическом растворителе и использован для диспергирования в нем состава из частиц. Однако данная композиция отличается от настоящего изобретения тем, что композиция использована для диспергирования твердых частиц и не допускает возможности для формирования жидких кристаллов.
Патент США 7871642 описывает способ получения дисперсии для доставки фармакологически активного вещества, содержащего гормональный состав, диспергирование гомогенной смеси фосфолипида, полиоксиэтиленсодержащего соэмульгатора, трицлицерида и этанола в воде, где полиоксиэтиленсодержащее поверхностно-активное вещество выбрано из сложных полиоксиэтиленсорбитановых эфиров жирной кислоты (полисорбат) и полиэтоксилированных производных витамина Е. Сложные полиоксиэтиленсорбитановые эфиры жирной кислоты и полиэтоксилированные производные витамина Е, полученные присоединением гидрофильного полимерного полиоксиэтилена к сорбитановому сложному эфиру жирной кислоты и витамину Е, соответственно, полностью отличаются по структуре от сложного сорбитанового эфира жирной кислоты и витамина Е. Их обычно применяют в качестве гидрофильных поверхностно-активных веществ, используя свойства полиоксиэтилена, который отличается от компонента настоящего изобретения.
Патент США 5888533 описывает текучую композицию для формирования твердого биоразлагаемого имплантата in situ внутри организма, содержащую: неполимерный, водонерастворимый, биоразлагаемый материал; и биологически совместимый органический растворитель, который по меньшей мере частично делает растворимым неполимерный, водонерастворимый, биоразлагаемый материал и который способен к смешиванию или диспергированию в воде или в текучих средах организма и способен к диффундированию или вымыванию из композиции в текучую среду организма после размещения внутри организма, после чего неполимерный материал коагулирует или осаждается для формирования твердого имплантата. В данной композиции, стеролы, сложные холестериловые эфиры, жирные кислоты, глицериды жирных кислот, сложные эфиры сахарозы жирных кислот, сложные сорбитановые эфиры жирных кислот, жирные спирты, сложные эфиры жирных спиртов и жирных кислот, ангидриды жирных кислот, фосфолипиды, ланолин, ланолиновые спирты и их комбинации описаны в качестве неполимерного материала, и этанол использован как растворитель. Однако отличия от настоящего изобретения заключаются в том в том, что данная композиция не может формировать жидкие кристаллы и предназначена для образования твердых имплантатов посредством простой коагуляции или осаждения водонерастворимых материалов, и что необходимо использовать много органического растворителя.
РАСКРЫТИЕ ИЗОБРЕТЕНИЯ
ТЕХНИЧЕСКАЯ ПРОБЛЕМА
Следовательно, задача настоящего изобретения состоит в том, чтобы предоставить фармацевтическую композицию на основе ненасыщенного сложного сорбитанового эфира, имеющего полярную головку по меньшей мере с двумя ОН (гидроксильными) группами, которая имеет сравнительно высокую безопасность и биоразлагаемость и существует в жидком состоянии, предпочтительном для применений путем инъекций, с одновременным преобразованием в жидкий кристалл под воздействием водной текучей среды, таким образом повышается замедленное высвобождение аналога GnRH in vivo.
Другая задача настоящего изобретения состоит в том, чтобы предоставить фармацевтическую композицию, которая может быть введена путем инъекции не вызывая боли, воспалений, концентрата начального всплеска, проблем, о которых сообщается в отношении обычных составов.
РЕШЕНИЕ ПРОБЛЕМЫ
В соответствии с аспектом его осуществления, настоящее изобретение относится к фармацевтической композиции, содержащей: а) по меньшей мере один сложный сорбитановый эфир ненасыщенной жирной кислоты; b) по меньшей мере один фосфолипид; с) по меньшей мере один отвердитель жидких кристаллов; и d) по меньшей мере один аналог GnRH, в качестве фармакологически активного вещества, где композиция существует как жидкая фаза в отсутствие водной текучей среды и превращается в жидкий кристалл в присутствии водной текучей среды.
Ниже будет дано описание каждого компонента.
а) Сложный сорбитановый эфир ненасыщенной жирной кислоты
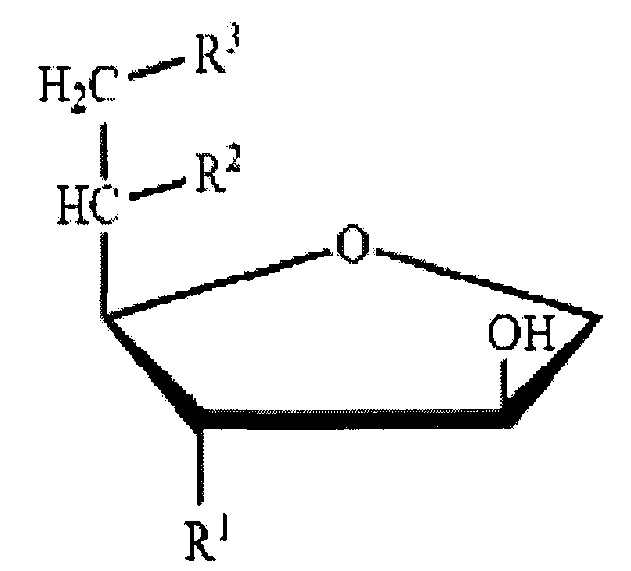
Для применения в качестве формирователя жидких кристаллов в настоящем изобретении, сложный сорбитановый эфир ненасыщенной жирной кислоты предпочтительно имеет две или более -ОН (гидроксильные) группы в полярной головке. Этот сложный сорбитановый эфир ненасыщенной жирной кислоты представлен следующей химической формулой 1. Соединение химической формулы 1 представляет собой сложный сорбитановый моноэфир, где R1=R2=OH, R3=R, R1=OH, R2=R3=R, R представляет собой алкиловый сложный эфир с 4-30 атомами углерода с по меньшей мере одной ненасыщенной связью.
[Химическая формула 1]
Сложный сорбитановый эфир жирной кислоты, который используется для формирования жидкого кристалла в настоящем изобретении, отличается от обычных противочастей, таких как олеилглицерат (OG), фитанилглицерат (PG) и глицеринмоноолеат (GMO), глицериндиолеат (GDO) следующей химической формулы 2. То есть, обычные молекулы, ответственные за жидкокристаллические фазы, участвуют в обычной структуре, состоящей из полярной головки, полученной из глицерина или глицериновой кислоты, и неполярного хвоста, полученного из липидного спирта или жирной кислоты.
[Химическая формула 2]
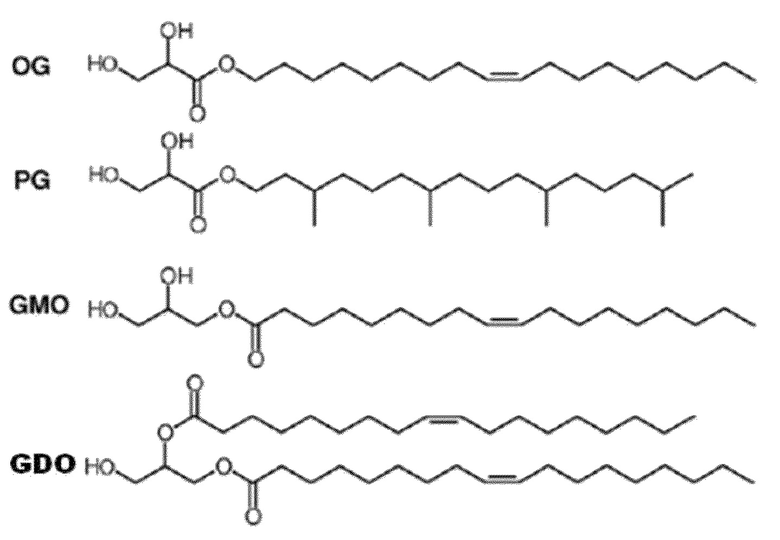
Однако, обычные молекулы, ответственные за жидкокристаллические фазы, до некоторой степени трудны для применения при разработке лекарственных средств из-за следующих недостатков. Олеилглицерат (OG) и фитанилглицерат (PG), хотя способны быстро образовывать жидкие кристаллы, редко используются как фармацевтически приемлемые эксципиенты для медицины человека из-за их относительно высокой токсичности. С другой стороны, глицеринмоноолеат (GMO) применим в качестве фармацевтически приемлемого эксципиента, но имеет слабую кристалличность для образования жидких кристаллов, необходимых для лекарственных средств с замедленным высвобождением.
Глицериндиолеат (GDO), который использован в международной патентной публикации WO 2005/117830, как описано выше, представляет собой диациллипид с глицерином, действующий в качестве полярной головки. Данную молекулу обычно не используют в качестве фармацевтического эксципиента, так как ее безопасность еще не была доказана.
После интенсивного и глубокого исследования авторы настоящего изобретения установили, что сложные сорбитановые эфиры ненасыщенных жирных кислот имеют преимущества над обычно применяемыми жидкокристаллическими молекулами, производными глицерина или глицериновой кислоты, в том, что они образуют жидкие кристаллы очень эффективно для замедленного высвобождения фармакологически активного вещества с превосходством в безопасности и в способности к биоразложению и применимы для разработки лекарственных средств, преодолевающих проблемы, возникающие на предшествующем уровне техники. Для применения в композициях лекарственных средств материалы должны быть гарантированы в отношении безопасности и биоразлагаемости. Кроме того, биоразлагаемость является очень важным фактором для материала, который отвечает за замедленное высвобождение в организме. Если инъекция с замедленным высвобождением с использованием PLGA предназначена для высвобождения фармакологически активного вещества в течение одной недели, идеально то, что PLGA подвергается разложению in vivo за одну неделю после инъекции. Фактически, однако, PLGA остается интактным в течение от одного до нескольких месяцев даже после того, как функция замедленного высвобождения закончена. Поэтому сложный сорбитановый эфир ненасыщенной жирной кислоты по настоящему изобретению, который имеет отличные характеристики в отношении замедленного высвобождения, безопасности и биоразлагаемости, применим для нового материала, индуцирующего жидкие кристаллы, имея большое значение в фармацевтической промышленности.
Более подробно, сложный сорбитановый эфир ненасыщенной жирной кислоты по настоящему изобретению может быть выбран из сложного сорбитанового моноэфира, сложного сорбитанового сесквиэфира, сорбитанового сложного диэфира и их комбинаций, которые могут быть получены из жирных кислот, получаемых из китовых жиров и рыбьих жиров, а также растительных масел (например, кокосового масла, касторового масла, оливкового масла, арахисового масла, рапсового масла, кукурузного масла, кунжутного масла, масла из семян хлопчатника, соевого масла, подсолнечного масла, сафлорового масла, льняного масла, и т.д.) и животных жиров и масел (например, молочного жира, свиного жира и говяжьего жира).
Сложный сорбитановый моноэфир представляет собой соединение, в котором одна группа жирной кислоты присоединена к сорбитану через сложноэфирную связь, и может быть выбран из сорбитанмоноолеата, сорбитанмонолинолеата, сорбитанмонопальмитолеата, сорбитанмономиристолеата, и их комбинации.
Сложный сорбитановый сесквиэфир представляет собой соединение, в котором 1,5 группы жирной кислоты, в среднем, присоединены к сорбитану через сложноэфирную связь, и может быть выбран из сорбитансесквиолеата, сорбитансесквилинолеата, сорбитансесквипальмитолеата, сорбитансесквимиристолеата, и их комбинации.
Сложный сорбитановый диэфир представляет собой соединение, в котором две группы жирной кислоты присоединены к сорбитану через сложноэфирную связь, и может быть выбран из сорбитандиолеата, сорбитандилинолеата, сорбитандипальмитолеата, сорбитандимиристолеата, и их комбинации.
Для применения в настоящем изобретении сложный сорбитановый эфир ненасыщенной жирной кислоты предпочтительно выбран из сорбитанмоноолеата, сорбитанмонолинолеата, сорбитанмонопальмитолеата, сорбитанмономиристолеата, сорбитансесквиолеата, и их комбинации.
b) Фосфолипид
Фосфолипиды неотъемлемы для конструкции слоистых структур, таких как липосомы, в обычных методиках, но не могут образовывать неслоистую фазовую структуру, такую как жидкий кристалл, самостоятельно. Однако, фосфолипиды могут участвовать в направленном образовании неслоистых фазовых структур с помощью формирователя жидких кристаллов, внося вклад в стабилизацию полученных жидких кристаллов.
Фосфолипид, используемый в настоящем изобретении, получают из растений или животных, и он содержит насыщенную или ненасыщенную алкильную сложноэфирную группу с 4-30 атомами углерода с полярной головкой. Фосфолипид может быть выбран из фосфатидилхолина, фосфатидилэтаноламина, фосфатидилсерина, фосфатидилглицерина, фосфатидилинозита, фосфатидной кислоты, сфингомиелина и их комбинации в соответствии со структурой полярной головки.
Фосфолипиды содержатся в растениях и животных, таких как соевые бобы и яйца. В фосфолипидах, длинные алкильные сложноэфирные группы, которые составляют гидрофобные хвосты, включают насыщенные жирнокислотные цепи, такие как моно- и дипальмитоил, моно- и димиристоил, моно- и дилаурил, и моно- и дистеарил, и ненасыщенные жирнокислотные цепи, такие как моно- и дилинолеил, моно- и диолеил, моно- и дипальмитолеил, и моно- и димиристолеил. Сложные эфиры насыщенных и ненасыщенных жирных кислот могут сосуществовать в фосфолипидах.
с) Отвердитель жидких кристаллов
Отвердитель жидких кристаллов по настоящему изобретению сам по себе не может формировать ни неслоистую структуру, в отличие от формирователя жидких кристаллов, ни слоистую структуру, такую как липосома, в отличие от фосфолипидов. Однако отвердитель жидких кристаллов вносит свой вклад в направленное формирование неслоистых фазовых структур за счет увеличения кривизны неслоистых структур для увеличения упорядоченного сосуществования масла и воды. В интересах данной функции, отвердитель жидких кристаллов должен иметь очень ограниченный полярный фрагмент и большой неполярный фрагмент внутри своей молекулярной структуры.
На практике, однако, биосовместимые молекулы, которые инъецируют в организм, могут быть выбраны в качестве отвердителя жидких кристаллов по настоящему изобретению только посредством прямых и повторных экспериментов. В результате, отвердители жидких кристаллов, подходящие для композиции по настоящему изобретению, имеют молекулярные структуры, которые отличаются друга от друга и, таким образом, не могут быть истолкованы как имеющие только одну молекулярную структуру. Общий структурный признак, выведенный путем наблюдения всех отвердителей жидких кристаллов, состоял в том, что они свободны от ионизирующихся групп, таких как карбоксильные и аминогруппы, и имеют гидрофобные фрагменты, содержащие большую триацильную группу с 15-40 атомами углерода или углеродную циклическую структуру. Предпочтительные примеры отвердителя жидких кристаллов по настоящему изобретению могут быть свободны от ионизирующихся групп, таких как карбоксильные и аминогруппы, и имеют не более одного гидроксила и одной эфирной группы в качестве слабой полярной головки, с гидрофобными остатками, включающими большую триацильную группу с 20-40 атомами углерода или углеродную кольцевую структуру. Примеры отвердителя жидких кристаллов по настоящему изобретению могут включать, но не ограничиваясь ими, триглицерид, ретинилпальмитат, токоферолацетат, холестерин, бензилбензоат, убихинон и их комбинацию. Предпочтительно, отвердитель жидких кристаллов по настоящему изобретению может быть выбран из токоферолацетата, холестерина, и их комбинации.
d) Аналоги GnRH
Аналоги GnRH структурно похожи на GnRH, но действуют по-разному in vivo. В целом, после пульсирующей секреции GnRH выполняет биологическую функцию, состоящую в индуцировании выработки половых стероидов, тогда как аналоги GnRH используются, чтобы эффективно ингибировать выработку половых стероидов в течение определенного периода времени в организме.
В сооответствии с их действующими механизмами, аналоги GnRH могут быть классифицированы на агонисты и антагонисты. При введении терапевтической дозы в организм, агонист GnRH первоначально связывается с рецептором GnRH гипофиза, чтобы стимулировать биосинтез и секрецию фолликулостимулирующего гормона (FSH) и лютеинизирующего гормона (LH). Однако продолжение введения агониста GnRH приводит к истощению гонадотропина и ингибированию биосинтеза и секреции FSH и LH с одновременным снижением регулирования рецептора GnRH. На основании биологических функций ГнРг, аналоги GnRH могут быть применены для лечения половых гормонозависимых заболеваний, таких как рак простаты, рак молочной железы, рак яичника, эндометриоз, фиброма матки, синдром поликистоза яичников, гипертрихоз, преждевременное половое созревание, гонадотропная аденома гипофиза, синдром апноэ во сне, синдром раздраженного кишечника, предменструальный синдром, доброкачественная гиперплазия предстательной железы и бесплодие, и могут быть использованы в качестве противозачаточного средства.
Агонист GnRH в качестве фармакологически активного вещества по настоящему изобретению может быть выбран из леупролида, гозерелина, трипторелина, нафарелина, бусерелина, гистрелина, деслорелина, метерелина, гонадрелина и их фармацевтически приемлемой соли. Предпочтительно, фармацевтически активное вещество может быть выбрано из леупролида, гозерелина и его фармацевтически приемлемой соли.
С другой стороны, антагонист GnRH конкурирует с GnRH в отношении рецептора GnRH гипофиза, чтобы блокировать связывание GnRH с его рецептором, тем самым подавляя биосинтез и секрецию FSH и LH. Примеры антагониста GnRH в качестве фармакологически активного вещества по настоящему изобретению включают дегареликс, абареликс, ганиреликс, цетрореликс и его фармацевтически приемлемую соль. Предпочтительно, фармацевтически активное вещество может быть выбрано из леупролида, гозерелина и его фармацевтически приемлемой соли.
В фармацевтической композиции по настоящему изобретению массовое соотношение компонентов а) и b), подходящее для формирования жидких кристаллов, находится в интервале от 10:1 до 1:10, и предпочтительно в интервале от 5:1 до 1:5. Массовое соотношение а)+b) к с) находиться внутри интервала от 100:1 до 1:1 и предпочтительно внутри интервала от 50:1 до 2:1. С учетом данных интервалов для масс, компоненты эффективно гарантируют замедленное высвобождение жидких кристаллов, и характеристики замедленного высвобождения могут быть проконтролированы путем регулирования соотношения. Подходящее массовое соотношение а)+b+с) к d) для обеспечения замедленного высвобождения аналога GnRH находится в интервале от 10000:1 до 1:1 и предпочтительно от 1000:1 до 1:1.
Предпочтительно, фармацевтическая композиция по настоящему изобретению содержит а) в количестве 9-90% по массе; b) в количестве 9-90% по массе; с) в количестве 0,1-50% по массе; и d) в количестве 0,01-50% по массе.
В другом варианте осуществления, где фармакологически активное вещество представляет собой леупролид, фармацевтическая композиция содержит: а) в количестве 9-64% по массе; b) в количестве 18-76% по массе; с) в количестве 1-36% по массе; и d) леупролид или его фармацевтически приемлемую соль в количестве 0,1-50% по массе, но без ограничения только этим.
В другом варианте осуществления, где фармакологически активное вещество представляет собой гозерелин, фармацевтическая композиция содержит: а) в количестве 9-64% по массе; b) в количестве 18-76% по массе; с) в количестве 1-36% по массе; и d) гозерелин или его фармацевтически приемлемую соль в количестве 0,1-50% по массе, но не ограничиваясь эти этим.
В дополнительном варианте осуществления, где фармакологически активное вещество представляет собой дегареликс, фармацевтическая композиция содержит: а) в количестве 9-64% по массе; b) в количестве 18-76% по массе; с) в количестве 1-36% по массе; и d) дегареликс или его фармацевтически приемлемую соль в количестве 2-50% по массе, но не ограничиваясь этим.
С учетом интервалов по содержанию компонентов от а) до d), фармацевтическая композиция по настоящему изобретению показывает отличные характеристики в отношении замедленного высвобождения.
Как используется в настоящем описании, термин “водная текучая среда” предназначен для включения воды и текучей среды организма, такой как слизистый раствор, слезы, пот, слюна, желудочно-кишечная жидкость, внесосудистая жидкость, внеклеточная жидкость, интерстициальная жидкость и плазма. При внесении в поверхности организма, области или полости (например, внутрь организма), для которых внешняя среда создана с помощью водных текучих сред, фармацевтическая композиция по настоящему изобретению претерпевает переход из жидкой фазы в жидкокристаллическую фазу с полутвердым внешним видом. То есть, фармацевтическая композиция по настоящему изобретению существует в жидком состоянии перед применением в организме человека и переходит в жидкокристаллическую фазу с характеристиками замедленного высвобождения внутри организма.
Жидкие кристаллы, образованные фармацевтической композицией по настоящему изобретению, имеют неслоистую фазовую структуру, в которой масло и вода находятся в упорядоченной смеси и расположении без разграничения между внутренними и внешними фазами. Упорядоченное расположение масла и воды делает неслоистую фазовую структуру мезофазной, что представляет собой промежуточное состояние вещества между жидким и твердым.
Фармацевтическая композиция по настоящему изобретению отличается от обычных композиций, которые являются слоистыми структурами, такими как мицеллы, эмульсии, микроэмульсии, липосомы и липидные бислои, которые широко используются в разработке фармацевтических композиций. Такие слоистые структуры представляют собой тип масло-в-воде (o/w) или вода-в-масле (w/o), в которых имеет место расположение внутренних и внешних фаз.
Термин “жидкая кристаллизация”, как используется в настоящем описании, относится к образованию жидких кристаллов, имеющих неслоистую фазовую структуру от преконцентрата под воздействием водной текучей среды.
Фармацевтическая композиция по настоящему изобретению может быть получена при комнатной температуре из: а) по меньшей мере одного формирователя жидких кристаллов, b) по меньшей мере одного фосфолипида; с) по меньшей мере одного отвердителя жидких кристаллов, d) по меньшей мере одного аналога GnRH, и, если необходимо, с помощью нагревания или использования гомогенизатора. Гомогенизатор может быть гомогенизатором высокого давления, ультразвуковым гомогенизатором, гомогенизатором с бисерной мельницей, и т.д.
Как описано выше, липидный преконцентрат с замедленным высвобождением по настоящему изобретению может представлять собой фармацевтическую композицию, которая существует в жидкой фазе в отсутствие водной текучей среды и преобразуется в жидкие кристаллы в присутствии водной текучей среды. Поскольку это относится к фармацевтической композиции, которая может применяться в организме при использовании способа, выбранного из инъекции, покрытия, капания, повязки, перорального введения и распыления, преконцентрат по настоящему изобретению может быть предпочтительно составлен в различные лекарственные формы, включающие инъекции, мази, гели, лосьоны, капсулы, таблетки, растворы, суспензии, спреи, средства для ингаляции, глазные капли, адгезивы, пластыри и чувствительные к давлению адгезивы.
В частности, когда выбирают инъекционный путь, фармацевтическая композиция по настоящему изобретению может быть введена путем подкожной или внутримышечной инъекции или другими способами впрыска в зависимости от свойств фармакологически активного вещества.
Фармацевтическая композиция по настоящему изобретению может быть предпочтительна в форме состава, выбранного из инъекций, мазей, гелей, лосьонов, капсул, таблеток, растворов, суспензий, спреев, средств для ингаляции, глазных капель, адгезивов, пластырей и чувствительных к давлению адгезивов, и более предпочтительно в в форме инъекций.
Фармацевтическая композиция по настоящему изобретению может быть получена путем добавления фармакологически активного вещества в преконцентрат по настоящему изобретению. При необходимости могут быть использованы нагревание или гомогенизатор при получении фармацевтической композиции по настоящему изобретению, но это не является ограничивающим фактором для настоящего изобретения.
Доза фармацевтической композиции по настоящему изобретению придерживается хорошо известной дозы используемого фармакологически активного вещества и может изменяться в зависимости от разных факторов, включающих состояние пациента, возраст и пол. Она может быть введена перорально или парентерально в зависимости от свойств фармакологически активного вещества.
В соответствии с еще одним аспектом, настоящее изобретение относится к способу сохранения фармацевтической эффективности посредством замедленного высвобождения фармакологически активного вещества путем введения фармацевтической композиции по настоящему изобретению млекопитающему, включая человека, и к применению фармацевтической композиции для замедленного высвобождения фармакологически активного вещества.
ПОЛЕЗНЫЕ ЭФФЕКТЫ ИЗОБРЕТЕНИЯ
Как описано прежде, фармацевтическая композиция по настоящему изобретению на основе сложного сорбитанового эфира ненасыщенной жирной кислоты в высшей степени безопасна и существует в жидкой фазе в отсутствие водной текучей среды, но быстро переходит в жидкие кристаллы под воздействием водной текучей среды в организме. Таким образом, фармацевтическая композиция может быть легко введена, проявляет отличное замедленное высвобождение аналога GnRH без побочных эффектов, таких как боль и воспаление, по сравнению с обычными составами с замедленным высвобождением в фазах твердых частиц.
Краткое описание чертежей
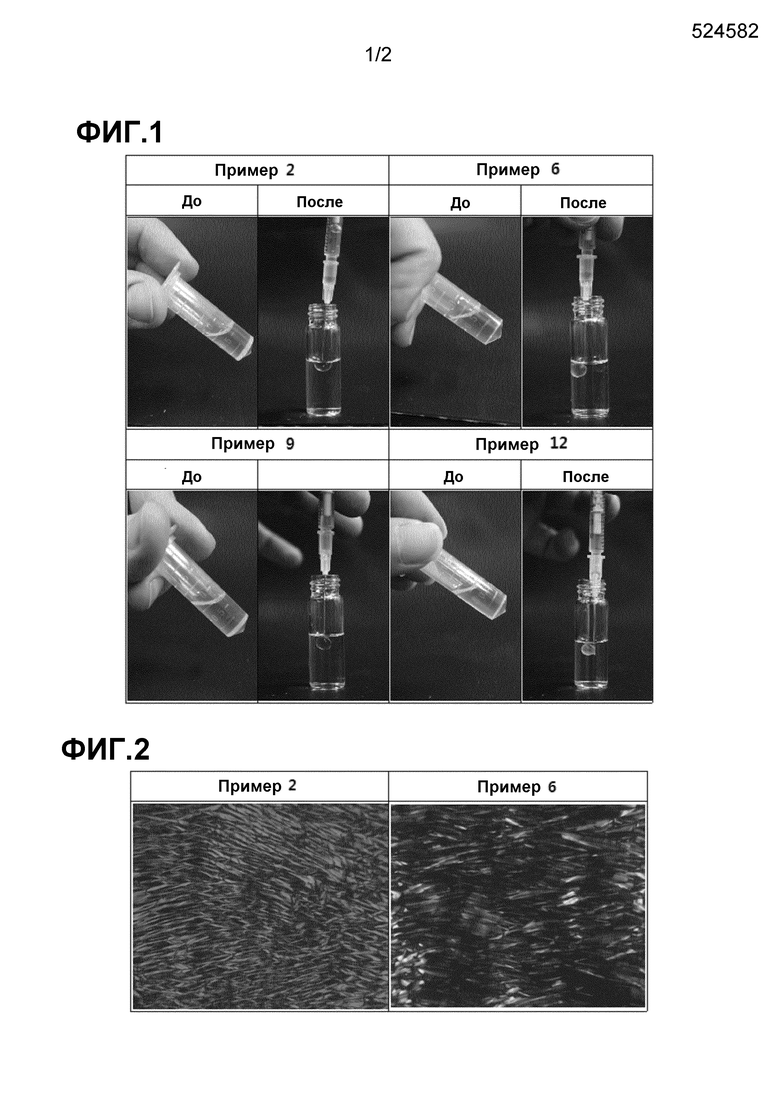
Фиг. 1 иллюстрирует фазы изменения характеристик для композиций примеров 2, 6, 9 и 12 под воздействием водной текучей среды.
Фиг. 2 показывает жидкокристаллические структуры композиций примеров 2 и 6, образованных в водной текучей среде.
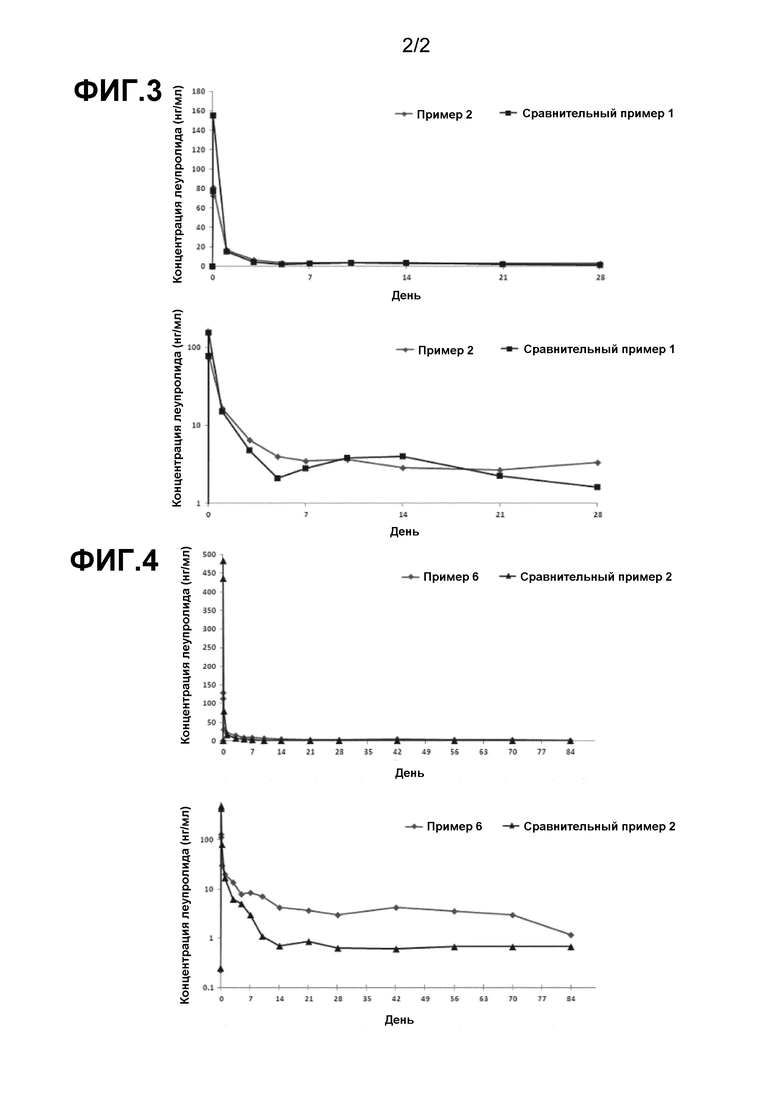
Фиг. 3 показывает характеристики высвобождения лекарственного средства in vivo для композиций примера 2 и сравнительного примера 1.
Фиг. 4 показывает характеристики высвобождения лекарственного средства in vivo для композиций примера 6 и сравнительного примера 2.
Способ изобретения
Следующие неограничивающие примеры служат для иллюстрации выбранных вариантов осуществления по настоящему изобретению. Следует иметь в виду, что изменения в пропорциях и альтернатив в элементах показанных компонентов будут очевидны специалистам в данной области техники в пределах объема вариантов осуществления по настоящему изобретению.
Добавки и эксципиенты, используемые в настоящем изобретении, удовлетворяли требованиям Фармакопеи и были приобретены у Aldrich, Lipoid, Croda и Seppic.
[Примеры 1-12] Получение фармацевтических композиций
Сложные сорбитановые эфиры ненасыщенных жирных кислот, фосфолипиды, отвердители жидких кристаллов и фармакологически активные вещества добавляли в массовых соотношениях, данных в таблице 1 ниже.
В примерах 1-12 вещества гомогенно смешивали на водяной бане, поддерживаемой при 20-75°C, с использованием гомогенизатора (PowerGen model 125, Fisher) в течение 0,5-3 ч при 1000-3000 об/мин. Полученные липидные растворы выдерживали при комнатной температуре, чтобы прийти к тепловому равновесию при 25°C, с последующим добавлением каждого из фармакологически активных веществ: леупролидацетата, гозерелинацетата и дегареликсацетата. Затем эти вещества гомогенизировали с помощью гомогенизатора в течение приблизительно 5-30 минут при 1000-3000 об/мин для получения фармацевтических композиций в жидкой фазе.
[Сравнительные примеры 1 и 2]
Для состава сравнительного примера 1, Leuplin DPS (CJ), содержащий леупролидацетат в виде фармакологически активного вещества, использовали в количестве 3,75 мг.
В качестве состава сравнительного примера 21, использовали 11,25 мг Leuplin DPS (CJ), содержащего фармакологически активное вещество, леупролидацетат.
[Экспериментальный пример 1]. Содержание фармакологически активных веществ в фармацевтических композициях
Чтобы определить, содержали ли фармацевтические композиции, полученные в примерах, фармакологически активные вещества в терапевтически эффективной концентрации, содержания леупролидацетата определяли с помощью ВЭЖХ, как следует.
Каждую из фармацевтических композиций растворяли в количестве, соответствующем 2,5 мг леупролидацетата в подвижной фазе (триэтиламиновый буфер:ацетонитрил:н-пропиловый спирт = 85:90:6), и центрифугировали в течение 10 мин при 1500 об/мин с последующей фильтрацией супернатанта тестируемого образца через фильтр 0,2 мкм. Для сравнения, стандартный образец с той же концентрацией, что в тестируемых образцах, получали из стандартного леупролидацетата. Стандартный образец и тестируемые образцы загружали в инъекционный объем 20 мкл при скорости потока из 1,0-1,5 мл/мин до 4,6×100 мм, 3 мкм заполнении колонки L1 или т.п., и количественно анализировали при 220 нм с использованием УФ-спектрометра. Средние содержания леупролидацетата в фармацевтической композиции получали из трех измерений (см. таблица 2).
Как можно видеть в таблице 2, все фармацевтические композиции, полученные в примерах 1-8, в идеале содержали леупролидацетат в количествах в пределах стандартного содержания (100%) ± 3%.
[Экспериментальный пример 2] Образование жидких кристаллов в водной текучей среде
Проводили исследование, чтобы подтвердить, образуют ли фармацевтические композиции, полученные в примерах, идеальные жидкие кристаллы в водной текучей среде. В связи с этим, композиции примеров 2, 6, 9 и 12, которые были в жидкой фазе, загружали в шприцы, а затем вводили в 2 г PBS (рН 7,4). Результаты показаны на Фиг. 1.
Фармацевтические композиции, полученные в примерах 1-12, существовали в жидкой фазе в отсутствие водной текучей среды. При введении в водную текучую среду (PSB), фармацевтические композиции в жидкой фазе преобразуются в сферические жидкие кристаллы, указывая на то, что аналог GnRH - фармакологически активное вещество не имеет никакого влияния на формирование фармацевтических композиций в жидкие кристаллы.
[Экспериментальный пример 3] Структурное определение жидких кристаллов в водной текучей среде
Жидкие кристаллы фармацевтических композиций примеров 2 и 6, образованные в водной текучей среде, наблюдали по структуре на поляризационном микроскопе (Motic, BA 300 Pol) (Фиг. 2).
Предметное стекло очень тонко покрывали каждой из фармацевтических композиций примеров 2 и 6 и оставляли на 4 ч в деионизированной воде в оболочке для образования жидких кристаллов. После накрытия покровным стеклом для предотвращения попадания воздуха, наблюдали тестовый образец на предметном стекле при увеличении в 200 раз, используя поляризационный микроскоп (Motic, BA 300 Pol). Как можно видеть на Фиг. 2, фармацевтические композиции примеров 2 и 6 были сформированы в жидкие кристаллы с типичными гексагональными кристаллическими структурами для превосходного замедленного высвобождения.
С учетом результатов экспериментальных примеров 1-3, фармацевтические композиции по настоящему изобретению могут образовывать физико-химически устойчивые, идеально жидкие кристаллы в присутствии водной текучей среды, даже если они содержат фармакологически активные вещества, которые имеют большие молекулярные массы и относительно высокую гидрофобность.
[Экспериментальный пример 4] In vivo PK профиль фармацевтических композиций
Поведения по высвобождению лекарственного средства из фармацевтических композиций по настоящему изобретению исследовали in vivo в следующем тесте.
Используя одноразовый шприц, каждую из фармацевтических композиций примеров 2 и 6 подкожно вводили при дозе леупролидацетата из 12,5 мг/кг (что соответствует 28-суточной дозе для людей) в спину 6 крысам SD (самцы) 9 недельного возраста со средней массой тела 300 г. Для сравнения с РК-профилями составов PLGA-микрочастиц, фармацевтические композиции сравнительных примеров 1 и 2 подкожно вводили в дозе леупролидацетата 12,5 мг/кг (что соответствует 28-суточной дозе для людей) в спину 6 крысам SD (самцы) 9 недельного возраста со средней массой тела 300 г.
Концентрации леупролидацетата в образцах плазмы, взятых у крыс SD, контролировали в течение 28 суток с помощью LC-MS/MS (жидкостная хроматография-масс-спектрометрия), чтобы вывести РК профили (фармакокинетические профили). Средние значения концентрации леупролидацетата, взятые у 6 крыс SD, приведены на графике каждой из Фиг. 3 и 4 и выражены в виде рассчитанных значений логарифма в нижнем графике каждой из Фиг. 3 и 4, чтобы исследовать различие в концентрации лекарственного средства в плазме крысы на поздней стадии.
РК-профили у крыс SD для фармацевтических композиций сравнительного примера 1 и примера 2 показаны на Фиг. 3. В качестве контроля (эталонное лекарственное средство) для примера 2, сравнительный пример содержал 3,75 мг Leuplin DPS(CJ), который широко используют в качестве 1-месячного состава леупролидацетата. По сравнению с контролем (эталонное лекарственное средство) из сравнительного примера 1, фармацевтическая композиция примера 2 проявляла идеальные рК-характеристики и превосходное замедленное высвобождение. Фармацевтическая композиция примера 2 имела концентрацию начального всплеска в 81 нг/мл, которая приблизительно наполовину снижена по сравнению с 155 нг/мл сравнительного примера 1, тем самым достигается исключительное улучшение в концентрации начального всплеска, типичной проблемы с композициями PLGA-микрочастиц. В отличие от композиции сравнительного примера 1, которая становилась нестабильной по РК-характеристикам от 5 суток после введения, фармацевтическая композиция примера 2 сохраняла очень стабильную эффективную концентрацию в плазме для леупролидацетата.
Фиг. 4 показывает РК-профили у крыс SD для фармацевтических композиций сравнительного примера 2 и примера 6. В качестве контроля (эталонное лекарственное средство) для примера 6, сравнительный пример 2 содержал 11,25 мг Leuplin DPS(CJ), который широко используют в качестве 3-месячного состава леупролидацетата. По сравнению с контролем (эталонное лекарственное средство) из сравнительного примера 2, фармацевтическая композиция примера 6 проявляла идеальные рК-характеристики, требуемые для композиций с замедленным высвобождением, и особенно с превосходным замедленным высвобождением в течение длительного периода. Композиция сравнительного примера 2 показала концентрацию начального всплеска, которая приблизительно в три раза выше аналогичной концентрации сравнительного примера 1, что, как полагают, обусловлено различием между ними по содержанию лекарственного средства. Хотя фармацевтическая композиция примера 6 имела в 3 раза большее содержание лекарственного средства, чем аналогичное содержание по примеру 2, никакие наблюдения не были сделаны по быстрому повышению концентрации начального всплеска, в отличие от композиции из сравнительного примера 2. Измеряли концентрацию начального всплеска, которая составляла 114 нг/мл, что приблизительно в 4 раза меньше, чем 484 нг/мл, которая была измерена в исходной фазе в композиции из сравнительного примера 2. Кроме того, композиция сравнительного примера 2 имела уровни леупролидацетата в крови от 10 суток после введения, которые были значительно ниже уровня по сравнению с композицией сравнительного примера 1 в средней до поздней фазы, с сохранением нестабильного РК-профиля. С другой стороны, фармацевтическая композиция примера 6, характеризующаяся кривой уровня леупролидацетата в крови, которая подобна кривой примера 2 в средней до поздней фазы, показывает, что фармацевтическая композиция по настоящему изобретению, даже при 3-кратном повышении содержания лекарственного средства, сохраняла превосходное долговременное замедленное высвобождение.
Изобретение относится к области фармацевтической промышленности, а именно к фармацевтической композиции, содержащей (a) по меньшей мере один сложный сорбитановый эфир ненасыщенной жирной кислоты, имеющий полярную головку по меньшей мере с двумя или более гидроксильными группами; (b) по меньшей мере один фосфолипид; (c) по меньшей мере один отвердитель жидких кристаллов, который свободен от способной к ионизации группы и имеет триацильную группу с 15-40 атомами углерода или углеродную кольцевую структуру в гидрофобной части; и (d) по меньшей мере один аналог GnRH в качестве фармакологически активного вещества, при этом указанная композиция существует в виде жидкой фазы в отсутствие водной текучей среды и преобразуется в жидкий кристалл в присутствии водной текучей среды. Изобретение обеспечивает высокую безопасность, способность к биодеградации и замедленное высвобождение аналога GnRH. 22 з.п. ф-лы, 4 ил., 2 табл., 18 пр.
1. Фармацевтическая композиция, содержащая:
a) по меньшей мере один сложный сорбитановый эфир ненасыщенной жирной кислоты, имеющий полярную головку по меньшей мере с двумя или более -ОН (гидроксильными) группами;
b) по меньшей мере один фосфолипид;
c) по меньшей мере один отвердитель жидких кристаллов, который свободен от способной к ионизации группы и имеет триацильную группу с 15-40 атомами углерода или углеродную кольцевую структуру в гидрофобной части: и
d) по меньшей мере один аналог GnRH (гонадотропин-рилизинг гормон) в качестве фармакологически активного вещества, где указанная композиция существует в виде жидкой фазы в отсутствие водной текучей среды и преобразуется в жидкий кристалл в присутствии водной текучей среды.
2. Фармацевтическая композиция по п. 1, где сложный сорбитановый эфир ненасыщенной жирной кислоты выбран из группы, состоящей из сорбитанмоноолеата, сорбитанмонолинолеата, сорбитанмонопальмитолеата, сорбитанмономиристолеата, сорбитансесквиолеата, сорбитансесквилинолеата, сорбитансесквипальмитолеата, сорбитансесквимиристолеата, сорбитандиолеата, сорбитандилинолеата, сорбитандипальмитолеата, сорбитандимиристолеата и их комбинации.
3. Фармацевтическая композиция по п. 1, где сложный сорбитановый эфир ненасыщенной жирной кислоты выбран из группы, состоящей из сорбитанмоноолеата, сорбитанмонолинолеата, сорбитанмонопальмитолеата, сорбитанмономиристолеата, сорбитансесквиолеата и их комбинации.
4. Фармацевтическая композиция по п. 1, где фосфолипид содержит насыщенную или ненасыщенную алкильную сложноэфирную группу из 4-30 атомов углерода и выбран из группы, состоящей из фосфатидилхолина, фосфатидилэтаноламина, фосфатидилсерина, фосфатидилглицерина, фосфатидилинозита, фосфатидной кислоты, сфингомиелина и их комбинации.
5. Фармацевтическая композиция по п. 4, где фосфолипид представляет собой фосфатидилхолин.
6. Фармацевтическая композиция по п. 1, где отвердитель жидких кристаллов выбран из группы, состоящей из триглицерида, ретинилпальмитата, токоферолацетата, холестерина, бензилбензоата, убихинона и их комбинации.
7. Фармацевтическая композиция по п. 1, где отвердитель жидких кристаллов представляет собой токоферолацетат, холестерин и их комбинацию.
8. Фармацевтическая композиция по п. 1, где аналог GnRH представляет собой агонист GnRH или антагонист GnRH.
9. Фармацевтическая композиция по п. 8, где агонист GnRH выбран из группы, состоящей из леупролида, гозерелина, трипторелина, нафарелина, бусерелина, гистрелина, деслорелина, метерелина, гонадрелина, их фармацевтически приемлемой соли и их комбинации.
10. Фармацевтическая композиция по п. 8, где антагонист GnRH выбран из группы, состоящей из дегареликса, абареликса, ганиреликса, цетрореликса, их фармацевтически приемлемой соли и их комбинации.
11. Фармацевтическая композиция по п. 1, где аналог GnRH выбран из группы, состоящей из леупролида, гозерелина, трипторелина, дегареликса, абареликса, их фармацевтически приемлемой соли и их комбинации.
12. Фармацевтическая композиция по п. 1, где аналог GnRH представляет собой леупролид или его фармацевтически приемлемую соль.
13. Фармацевтическая композиция по п. 1, предназначенная для профилактики или лечения полового гормонозависимого заболевания или в качестве контрацептива.
14. Фармацевтическая композиция по п. 13, где половое гормонозависимое заболевание выбрано из группы, состоящей из рака простаты, рака молочной железы, рака яичника, эндометриоза, фибромы матки, синдрома поликистоза яичников, преждевременного полового созревания, гипертрихоза, гонадотропной аденомы гипофиза, синдрома апноэ во сне, синдрома раздраженного кишечника, предменструального синдрома, доброкачественной гиперплазии предстательной железы и бесплодия.
15. Фармацевтическая композиция по п. 1, где массовое отношение а) к b) находится в интервале от 10:1 до 1:10.
16. Фармацевтическая композиция по п. 1, где массовое отношение а) + b) к с) находится в интервале от 1000:1 до 1:1.
17. Фармацевтическая композиция по п. 1, где массовое отношение а) + b) + с) к d) находится в интервале от 10000:1 до 1:1.
18. Фармацевтическая композиция по п. 1, содержащая:
a) по меньшей мере один сложный сорбитановый эфир ненасыщенной жирной кислоты, имеющий полярную головку по меньшей мере с двумя или более -ОН (гидроксильными) группами в количестве 9-90% по массе;
b) по меньшей мере один фосфолипид в количестве 9-90% по массе;
c) по меньшей мере один отвердитель жидких кристаллов, который свободен от способной к ионизации группы и имеет триацильную группу с 15-40 атомами углерода или углеродную кольцевую структуру в гидрофобной части в количестве 0,1-50% по массе; и
d) по меньшей мере один аналог GnRH (гонадотропин-рилизинг гормон) в количестве 0,01-50% по массе.
19. Фармацевтическая композиция по п. 1, содержащая:
a) по меньшей мере один сложный сорбитановый эфир ненасыщенной жирной кислоты, имеющий полярную головку по меньшей мере с двумя или более -ОН (гидроксильными) группами в количестве 9-64% по массе;
b) по меньшей мере один фосфолипид в количестве 18-7 6% по массе;
c) по меньшей мере один отвердитель жидких кристаллов, который свободен от способной к ионизации группы и имеет триацильную группу с 15-40 атомами углерода или углеродную кольцевую структуру в гидрофобной части в количестве 1-36% по массе; и
d) леупролид или его фармацевтически приемлемую соль в количестве 0,1-50% по массе.
20. Фармацевтическая композиция по п. 1, содержащая:
а) по меньшей мере один сложный сорбитановый эфир ненасыщенной жирной кислоты, имеющий полярную головку по меньшей мере с двумя или более -ОН (гидроксильными) группами в количестве 9-64% по массе;
b) по меньшей мере один фосфолипид в количестве 18-76% по массе;
c) по меньшей мере один отвердитель жидких кристаллов, который свободен от способной к ионизации группы и имеет триацильную группу с 15-40 атомами углерода или углеродную кольцевую структуру в гидрофобной части в количестве 1-36% по массе; и
d) гозерелин или его фармацевтически приемлемую соль в количестве 0,1-50% по массе.
21. Фармацевтическая композиция по п. 1, содержащая:
a) по меньшей мере один сложный сорбитановый эфир ненасыщенной жирной кислоты, имеющий полярную головку по меньшей мере с двумя или более -ОН (гидроксильными) группами в количестве 9-64% по массе;
b) по меньшей мере один фосфолипид в количестве 18-76% по массе;
c) по меньшей мере один отвердитель жидких кристаллов, который свободен от способной к ионизации группы и имеет триацильную группу с 15-40 атомами углерода или углеродную кольцевую структуру в гидрофобной части в количестве 1-36% по массе; и
d) дегареликс или его фармацевтически приемлемую соль в количестве 2-50% по массе.
22. Фармацевтическая композиция по п. 1, находящаяся в составе, причем указанный состав выбран из группы, состоящей из инъекции, мази, геля, лосьона, капсулы, таблетки, раствора, суспензии, спрея, средства для ингаляции, глазных капель, адгезива, пластыря и адгезива, чувствительного к давлению.
23. Фармацевтическая композиция по п. 1, где состав представляет собой инъекцию.
| WO 9847487 A1, 29.10.1998 | |||
| US 20070080323 A1, 12.04.2007 | |||
| US 20120269772 A1, 25.10.2012. |

