Область техники, к которой относится настоящее изобретение
Настоящее изобретение относится к депо-препарату с замедленным высвобождением, включающему аналог GnRH в качестве фармацевтически активного вещества, а также к включающей его инъекционной композиции.
Предшествующий уровень техники настоящего изобретения
Препарат с замедленным высвобождением представляет собой препарат, который может непрерывно высвобождать фармакологически активное вещество при однократном введении, обеспечивая предотвращения побочных эффектов, которые могут возникнуть при повторных введениях, а также поддерживать эффективный диапазон концентраций фармакологически активного вещества в течение определенного времени или за определенный период или дольше.
Учитывая терапевтический механизм и физико-химические свойства фармакологически активного вещества, типичное фармакологически активное вещество, подходящее для разработки в качестве препарата с замедленным высвобождением, включает аналоги GnRH.
Гонадотропин-рилизинг-гормон (GnRH) или рилизинг-фактор лютеинизирующего гормона (LHRH) представляет собой нейроэндокринный пептид, синтезируемый в нервно-сосудистых окончаниях гипоталамуса. GnRH секретируется гипоталамусом, избирательно связывается со специфическим рецептором на мембране гонадотроф - клеток передней доли гипофиза, а затем стимулирует биосинтез и секрецию лютеинизирующего гормона (LH) и фолликулостимулирующего гормона (FSH). Продуцируемый фолликулостимулирующий гормон (FSH) и лютеинизирующий гормон (LH) играют определенную роль в регуляции продукции половых стероидов в мужских и женских половых железах. Благодаря биологическим функциям GnRH, аналоги GnRH могут применяться в лечении гормонозависимых заболеваний, таких как злокачественная опухоль предстательной железы, злокачественная опухоль молочной железы, злокачественная опухоль яичников, эндометриоз, миома матки, поликистоз яичников, гипертрихоз, преждевременное половое созревание, гонадотропная аденома гипофиза, синдром апноэ во сне, синдром раздраженного кишечника, предменструальный синдром, доброкачественная гиперплазия предстательной железы или бесплодие и т.д.
Среди широко представленных на рынке препаратов, препараты с замедленным высвобождением аналогов GnRH включают Lupron® Depot, содержащий в качестве фармакологически активного вещества лейпролида ацетат. Lupron® Depot широко применяется в качестве внутримышечного и подкожного инъекционного препарата с применением биоразлагаемых микросфер поли(молочно-гликолевой кислоты) (PLGA) в качестве субстрата с замедленным высвобождением. Обычно PLGA микросферы остаются в организме в течение определенного периода времени и распадаются на молочную кислоту и гликолевую кислоту с непрерывным высвобождением инкапсулированного фармакологически активного вещества, демонстрируя тем самым эффект замедленного высвобождения (патент США № 5480656). Однако способ получения PLGA микросфер имеет недостатки, заключающиеся в том, что он является сложным и трудоемким, причем эффективность инкапсулирования фармакологически активного вещества в значительной степени снижается. Кроме того, PLGA микросферы плохо поддаются фильтрации, а также расплавляются при температуре 40°С и выше, что делает невозможным применение метода, обычно применяемого для асептической обработки. Соответственно, существует проблема, заключающаяся в том, что способ необходимо осуществлять в высоко асептических условиях. Кроме того, необходимо дополнительно осуществить сложный процесс получения и смешивания двух или более различных микросфер для создания оптимальной схемы замедленного высвобождения (международная публикация № WO 2005/074896), и, соответственно, увеличиваются экономические затраты вследствие неэффективности способа. Кроме того, примеси уксусной кислоты и вещества кислотного распада PLGA микросфер вызывают воспалительную реакцию, снижение клеточной пролиферации и т.д. (K. Athanasiou, G. G. Niederauer, and C. M. Agrawal, Biomaterials, 17, 93 (1996)). Вследствие природы препарата, для которого требуется большой объем внутримышечного и подкожного введения путем суспендирования микросфер размером от 10 до 100 мкм в водном растворе, существует проблема, при которой возникает боль или повреждение тканей на участке инъекции. Инъекционный препарат с замедленным высвобождением аналога GnRH (лейпролида ацетата), введенный для устранения недостатка препарата на основе PLGA микросфер с замедленным высвобождением, включает ELIGARD Inj®. ELIGARD Inj широко представлен на рынке в качестве средства для подкожных инъекций, которое применяется путем растворения поли(DL-лактид-со-гликолида), содержащего защищенную концевую карбоксильную группу, и аналога GnRH (ацетат лейпролида) в N-метил-2-пирролидоне (NMP). В препарате ELIGARD Inj® устранены некоторые недостатки препарата на основе твердых PLGA микросфер путем растворения биоразлагаемого полимера в полярном апротонном растворе с получением текучей композиции и последующего подкожного ее введения (патент США № 6773714). Однако продукт не представляет собой полностью предварительно заполненное инъекционное устройство и, таким образом, имеет недостатки, заключающиеся в том, что он в значительной степени менее удобен и имеет низкую стабильность лекарственного средства в состоянии смешанного раствора. Набор, предоставляемый с продуктом, состоит из двух соединяемых шприцев, а также приспособлений для смешивания, приготовления и введения. В частности, приготовление конечного смешанного раствора представляет собой сложный процесс, состоящий примерно из 10 или более стадий, причем существуют затруднения, связанные с завершением приготовления и введением в течение 30 минут. Кроме того, для продукта требуется отдельное холодильное устройство, что обусловлено условиями хранения, и если конечный смешанный раствор не охлаждается, продукт нельзя применять в течение пяти суток или дольше. Кроме того, в препарате не смоли устранить проблему, связанную с высоким начальным высвобождением лекарственного средства, что является общим недостатком препаратов на основе PLGA, скорее напротив, препарат показал более высокую начальную концентрацию лекарственного средства, чем Lupron® Depot, препарат на основе микросфер PLGA (патент США № 6773714). Начальная концентрация лекарственного средства, значительно превышающая диапазон, в котором лекарственное средство может быть эффективным, нежелательна с функциональной или токсикологической точки зрения. В частности, с учетом того, что аналоги GnRH обладают механизмом, при котором секреция половых гормонов временно повышается в начале введения, а затем подавляется через определенный момент времени, избыточное начальное высвобождение лекарственного средства является фактором, которого следует избегать.
В качестве альтернативы для преодоления проблем, связанных с указанными препаратами на основе PLGA, в международной публикации № WO 2005/117830 описан исходный препарат, который включает, по меньшей мере, один нейтральный диациллипид и/или токоферол, по меньшей мере, один фосфолипид и, по меньшей мере, один органический растворитель, который биосовместим, содержит кислород и обладает низкой вязкостью. В международной публикации № WO 2006/075125 описан исходный препарат, который включает, по меньшей мере, один диацилглицерид, по меньшей мере, один фосфатидилхолин, по меньшей мере, один кислородсодержащий органический растворитель и, по меньшей мере, один аналог GnRH. Эти препараты не образуют продукты распада полимерной системы, молочную или гликолевую кислоты, и, таким образом, не вызывают боль или воспаление на участке инъекции, при этом непрерывно высвобождают фармакологически активные вещества in vivo в течение примерно четырех недель. Однако диациллипид, который является основным компонентом композиции препарата, представляет собой вещество, которое обычно не применяется в качестве наполнителя для фармацевтических препаратов вследствие недостаточной безопасности, и для которого требуется применение органического растворителя, вызывающего снижение активности некоторых фармакологически активных веществ (H. Ljusberg-Wahre, F. S. Nielse, 298, 328-332 (2005); H. Sah, Y. bahl, Journal of Controlled Release 106, 51-61(2005)).
Соответственно, авторы настоящего изобретения изобрели фармацевтическую композицию, которая включает а) сложный эфир сорбитана и ненасыщенной жирной кислоты; b) фосфолипид; c) отвердитель жидких кристаллов; и d) аналоги GnRH в качестве фармакологически активного вещества, представляет собой липидную жидкость с отсутствием жидкости на водной основе и образует жидкие кристаллы в жидкости на водной основе (публикация корейского патента № 10-2012-0157583). Было подтверждено, что исходный липидный препарат с замедленным высвобождением по настоящему изобретению обладает характеристиками безопасности и способности к биологическому разложению, которые аналогичны или превосходят указанные характеристики существующего исходного препарата, показанные в тестах на животных (in vivo), при этом фармацевтическая композиция, включающая фармакологически активное вещество, высвобождает лекарственное средство замедленным образом. Однако в исходном липидном препарате с замедленным высвобождением по настоящему изобретению высвобождение лекарственного средства прекращалось в течение двух недель вопреки ожиданиям в клинической практике (у человека). Это связано с тем, что исходный липидный препарат с замедленным высвобождением распространяется на подкожно-жировую клетчатку или ткани человеческого организма с низким содержанием влаги и подкожно-жировой клетчаткой высокой плотности, и, таким образом, жидкокристаллический гель распространяется или образуется в фрагментированной форме, что обеспечивает преждевременное прекращение замедленного высвобождения лекарственного средства.
Соответственно, авторы настоящего изобретения неоднократно проводили исследования путем предварительного добавления воды к исходному липидному препарату с замедленным высвобождением, содержащему аналоги GnRH и, таким образом, осуществили изобретение депо-препарата с замедленным высвобождением со значительно улучшенной первичной скоростью высвобождения и свойством замедленного высвобождения лекарственного средства по мере того, как жидкое инъекционное средство образует жидкокристаллический гель непосредственно после введения.
Далее рассмотрен предшествующий уровень техники, который может относиться к настоящему изобретению.
В патенте США № 7731947 описана композиция, в которой твердые частицы, состоящие из интерферона, сахарозы, метионина и цитратного буфера, диспергированы в органическом растворителе, таком как бензилбензоат и т.д., и поясняется, что аналоги GnRH могут применяться в качестве подходящего лекарственного средства. Кроме того, в некоторых примерах описано, что фосфатидилхолин можно растворять в органическом растворителе вместе с витамином Е (токоферолом) и применять в виде дисперсии твердых частиц. Однако композиция в соответствии с указанным выше патентом отличается от композиции по настоящему изобретению тем, что жидкие кристаллы не образуются, а композиция применяется для диспергирования твердых частиц.
В патенте США № 7871642 описан способ получения дисперсии, в котором смесь фосфолипида, поверхностно-активного вещества, содержащего композиции полиоксиэтилена, триглицерида и этанола, диспергируется в воде, с целью доставки фармакологически активного вещества, включая гормональное средство, а также указано, что сложный эфир полиоксиэтиленсорбитана и жирной кислоты (полисорбат) и полиоксиэтиленовые производные витамина Е могут применяться в качестве одного из поверхностно-активных веществ, содержащих полиоксиэтилен. Однако сложный эфир полиоксиэтиленсорбитана и жирной кислоты и полиоксиэтиленовые производные витамина Е представляют собой вещества, в которых полиоксиэтилен, гидрофильный полимер, связан с эфиром сорбита и жирной кислоты, а также витамином Е соответственно, и являются веществами, имеющими структуру, полностью отличную от структуры исходного эфира сорбитана и жирной кислоты, а также витамина Е, и применяются в качестве гидрофильного поверхностно-активного вещества, использующего свойства полиоксиэтилена, и, таким образом, отличаются от составляющих компонентов по настоящему изобретению.
В патенте США № 5888533 описана композиция, которая обеспечивает свойство замедленного высвобождения фармакологически активных веществ, включая лейпролид, содержащая неполимерное водонерастворимое и биоразлагаемое вещество в качестве жидкой композиции для формирования имплантата, а также растворитель, который, по меньшей мере, частично растворяет вещество и смешивается или диспергируется водой или биологическими жидкостями, причем растворитель диффундирует и выделяется в биологическую жидкость при применении к организму человека, так что неполимерное водонерастворимое и биоразлагаемое вещество агрегирует или осаждается с образованием имплантата. В настоящем документе описано, что стерол, холестериловый эфир, жирная кислота, глицерид жирной кислоты, эфир сахарозы и жирной кислоты, эфир сорбитана и жирной кислоты, жирные спирты, продукты сложноэфирной связи жирного спирта и жирной кислоты, дегидратированный продукт жирной кислоты, фосфолипид, ланолин, ланолиновый спирт и т.д. могут применяться в качестве неполимерного, водонерастворимого и биоразлагаемого вещества, а этанол и т.д. описаны в качестве растворителя. Однако указанный выше патент отличается от настоящего изобретения не только тем, что он включает композицию для получения имплантата посредством простой агломерации или осаждения без образования жидких кристаллов, но и тем, что обязательно применяется большое количество органического растворителя.
В международной публикации № WO 2012/160212 описан исходный препарат, который включает 20-80%, по меньшей мере, одного диациллипида и/или токоферола, 20-80%, по меньшей мере, одного фосфолипида, 5-35%, по меньшей мере, одного биосовместимого спиртоорганического растворителя, 20% или меньше водного раствора, а также, по меньшей мере, одно лекарственное средство - двойной агонист амилинового рецептора/рецептора GLP-1, а в заявке США № 20070265190 описан исходный препарат, который включает, по меньшей мере, один диациллипид и/или токоферол, по меньшей мере, один фосфолипид, по меньшей мере, один кислородсодержащий органический растворитель с низкой вязкостью и, по меньшей мере, одно опиоидное лекарственное средство. Эти препараты не образуют продукты распада полимерной системы, молочную или гликолевую кислоты, и, таким образом, не вызывают какую-либо боль или воспаление на участке инъекции, при этом непрерывно высвобождают фармакологически активные вещества in vivo в виде лекарственного средства. Однако диациллипид, который является основным компонентом композиции препарата, представляет собой вещество, которое обычно не применяется в качестве вспомогательного вещества для фармацевтических препаратов вследствие отсутствия достаточной безопасности, и для него не удалось доказать характер высвобождения лекарственного средства в зависимости от содержания воды в те же условия, что и в организме человека с развитой подкожно-жировой клетчаткой. Кроме того, композиция отличается от композиции по настоящему изобретению тем, что она представляет собой композицию, обладающую свойством замедленного высвобождения лекарственного средства, полностью отличного от лекарственного средства по настоящему изобретению.
В международной публикации № WO 2005/074896 описана композиция, которая высвобождает агонист GnRH или его соли в течение длительного периода времени в виде PLGA микрокапсул, содержащих водный раствор. Кроме того, в патенте описано сохранение слабого побочного эффекта и свойство стабильного замедленного высвобождения для “сброса” лекарственного средства с высвобождением нулевого порядка без начального эффекта сброса лекарственного средства. Однако композиция, описанная в патенте, отличается от композиции по настоящему изобретению тем, что эта композиция представляет собой препарат с замедленным высвобождением, который полностью отличается от препарата по настоящему изобретению в виде препарата на основе PLGA микрочастиц, при этом водный канал внутри PLGA микрочастиц применяется для удержания водорастворимого лекарственного средства.
[Ссылка на предшествующий уровень техники]
[Патентные документы]
(Патентный документ 1) Международная публикация № WO 2005/074896
(Патентный документ 2) Международная публикация № WO 2005/117830
(Патентный документ 3) Международная публикация № WO 2006/075125
(Патентный документ 4) Международная публикация № WO 2012/160212
(Патентный документ 5) Патент США № 5480656
(Патентный документ 6) Патент США № 6773714
(Патентный документ 7) Патент США № 7731947
(Патентный документ 8) Патент США № 7871642
(Патентный документ 9) Патент США № 5888533
(Патентный документ 10) Заявка на патент США № 20070265190
Подробное раскрытие настоящего изобретения
Техническая задача
Целью настоящего изобретения является получение инъекционной композиции, которая обладает в значительной степени повышенной безопасностью вследствие образования в композиции сложного эфира сорбитана и ненасыщенной жирной кислоты, содержащего полярную головку с двумя или более -ОН (гидроксильными) группами, находится в жидкой фазе в фазе, где жидкость на водной основе отсутствует, чтобы ее можно было легко применять в отношении фармацевтических препаратов в лекарственных формах, и образует жидкие кристаллы в фазе жидкости на водной основе, благодаря чему свойство замедленного высвобождения аналогов GnRH, которые применяются в качестве фармацевтически активного вещества in vivo, может быть усилено.
Целью настоящего изобретения является получение инъекционной композиции, которая обладает в значительной степени повышенной безопасностью вследствие образования в композиции сложного эфира сорбитана и ненасыщенной жирной кислоты, содержащего полярную головку с двумя или более -ОН (гидроксильными) группами, находится в жидкой фазе в фазе, где жидкость на водной основе отсутствует, чтобы ее можно было легко применять в отношении фармацевтических препаратов в лекарственных формах, и образует жидкие кристаллы в фазе жидкости на водной основе, благодаря чему свойство замедленного высвобождения аналогов GnRH, применяемых в качестве фармацевтически активных веществ in vivo, может быть усилено.
Другой целью настоящего изобретения является получение инъекционной композиции, которую можно инъецировать в жидкой фазе и, таким образом, можно ослаблять боль при инъекциях, воспаление и понижать высокую начальную концентрацию высвобождения, что не может быть достигнуто посредством обычного препарата с замедленным высвобождением.
Решение технической задачи
Настоящее изобретение относится к инъекционной композиции, которая включает: а) сложный эфир сорбитана и ненасыщенной жирной кислоты, содержащий полярную головку с двумя или более -ОН (гидроксильными) группами; b) фосфолипид; c) отвердитель жидких кристаллов, который не содержит ионизированную группу, и гидрофобный фрагмент которого имеет триацильную группу, содержащую от 15 до 40 атомов углерода или структуру углеродного кольца; d) воду; и e) аналог гонадотропин-рилизинг-гормона (GnRH) в качестве фармацевтически активного вещества, причем композиция является жидкой перед инъекцией и образует жидкие кристаллы после инъекции.
Далее каждый компонент будет описан более подробно.
a) Сложный эфир сорбитана и ненасыщенной жирной кислоты
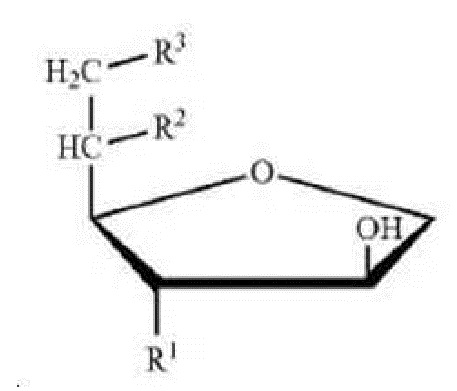
Сложный эфир сорбитана и ненасыщенной жирной кислоты, который представляет собой образующее жидкие кристаллы средство по настоящему изобретению, может относиться к соединению [формулы 1], содержащему полярную головку с двумя или более -ОН (гидроксильными) группами, в числе которых моноэфир сорбитана представляет собой R1=R2=OH, R3=R, и диэфир сорбитана представляет собой R1=OH, R2=R3=R или R3=OH, R1=R2=R. Здесь R относится к группе сложного алкилового эфира, содержащей от 4 до 30 атомов углерода и, по меньшей мере, одну двойную связь.
[Формула 1]
Это отличается от известного факта, что явление жидкой кристаллизации обычно вызывается олеилглицератом (OG), фитанилглицератом (PG), моноолеатом глицерина (GMO) и диолеатом глицерина (GDO), которые представлены [формулой 2], в предшествующем уровне техники. Другими словами, общеизвестные вещества для образования жидких кристаллов имеют общую структуру в том смысле, что все они имеют полярную “головку”, состоящую из глицерина или глицериновой кислоты, которая связана с “неполярным хвостом”, полученным из жирных спиртов или жирных кислот.
[Формула 2]
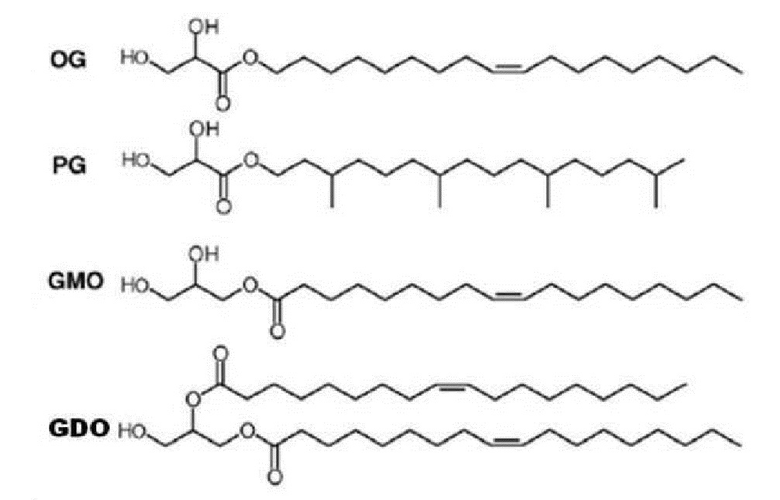
Однако обычные вещества для образования жидких кристаллов трудно применять для разработки лекарств вследствие каждого из следующих недостатков. Олейлглицерат (OG) и фитанилглицерат (PG) могут эффективно образовывать жидкие кристаллы, однако не применяются в качестве вспомогательных веществ для лекарственных средств вследствие относительно высокой токсичности. В то же время, моноолеат глицерина (GMO) можно применять в качестве фармацевтического вспомогательного вещества, однако его недостаток заключается в том, что он не может образовывать жидкие кристаллы с замедленным высвобождением, необходимые для фармацевтических препаратов, вследствие низкой способности образовывать жидкие кристаллы.
Кроме того, диолеат глицерина (GDO), в представленной выше международной публикации № WO 2005/117830 описано применение глицерина в качестве полярной головки в форме глицерида, содержащего диацильную группу, однако проблема, заключается в том, что он обычно не применяется в качестве вспомогательное вещество для лекарственных средств вследствие недостаточной безопасности.
Авторы настоящего изобретения обнаружили, что, в отличие от обычных производных глицерина или глицериновой кислоты, жидкие кристаллы, образованные сложным эфиром сорбитана и ненасыщенной жирной кислоты, не только являются эффективными для замедленного высвобождения активного вещества, но также обладают более высокой безопасностью по сравнению с обычными веществами, образующими жидкие кристаллы, тем самым преодолевая недостатки традиционной технологии и делая ее доступной для разработки лекарственных средств. Для применения в качестве фармацевтической композиции должна быть обеспечена безопасность, при этом биоразлагаемость является важным элементом. Более того, в случае вещества, которое инъецируют in vivo и демонстрирует свойство замедленного высвобождения, биоразлагаемость является очень важным фактором. В случае PLGA, который применяется в качестве обычного инъекционного средства с замедленным высвобождением, если PLGA демонстрирует эффект замедленного высвобождения в течение одной недели, желательно, чтобы введенный PLGA разлагался in vivo и предпочтительно исчезал через одну неделю. Однако существует проблема, заключающаяся в том, что PLGA фактически сохраняется без разложения в течение периода от одного до нескольких месяцев даже после завершения функции замедленного высвобождения. Таким образом, становится ясно, что сложный эфир сорбитана и ненасыщенной жирной кислоты по настоящему изобретению обладает превосходными свойствами замедленного высвобождения, безопасностью и способностью к биологическому разложению и, таким образом, может представлять собой новое образующее жидкие кристаллы вещество, имеющее очень высокую промышленную ценность.
В частности, сложный эфир сорбитана и ненасыщенной жирной кислоты по настоящему изобретению может представлять собой, по меньшей мере, один эфир, выбранный из сложного моноэфира сорбитана, сесквиэфира сорбитана, диэфира сорбитана и их смесей, полученных из жирных кислот, которые могут быть получены из растительных масел (например, пальмового масла, касторового масла, оливкового масла, арахисового масла, рапсового масла, кукурузного масла, кунжутного масла, хлопкового масла, соевого масла, подсолнечного масла, сафлорового масла, льняного масла и т.д.), животных жиров и масел (например, молочный жир, свиной жир и говяжий жир), а также китового жира и рыбьего жира.
Сложный моноэфир сорбитана может представлять собой такое вещество, в котором одна группа жирной кислоты связана сложноэфирной связью с сорбитаном, и может представлять собой, по меньшей мере, одно вещество, выбранное из сорбитанмоноолеата, сорбитанмонолинолеата, сорбитанмонопальмитолеата, сорбитанмономиристолеата и их смесей.
Сорбитан сесквиэфир может представлять собой такое вещество, в котором в среднем 1,5 группы жирных кислот связаны сложноэфирной связью с сорбитаном, и может представлять собой, по меньшей мере, одно вещество, выбранное из сесквиолеата сорбитана, сесквилинолеата сорбитана, сесквипальмитолеата сорбитана, сесквимиристолата сорбитана и их смесей.
Сложный диэфир сорбитана может представлять собой такое вещество, в котором две группы жирной кислоты связаны сложноэфирной связью с сорбитаном, и может представлять собой вещество, выбранное из диолеата сорбитана, дилинолеата сорбитана, дипальмитолеата сорбитана, димиристолеата сорбитана и их смесей.
Сложный эфир сорбитана и ненасыщенной жирной кислоты по настоящему изобретению предпочтительно может быть выбран из моноолеата сорбитана, монолинолеата сорбитана, монопальмитолеата сорбитана, мономиристолата сорбитана, сесквиолеата сорбитана и их смесей.
b) Фосфолипид
Фосфолипид по настоящему изобретению представляет собой вещество, которое преимущественно применяется для формирования ламеллярной структуры, такой как липосома, в предшествующем уровне техники, но не может независимо образовывать жидкие кристаллы в виде неламеллярной фазовой структуры. Тем не менее, согласно настоящему изобретению фосфолипид играет определенную роль в стабилизации жидких кристаллов, принимая участие в формировании неламеллярной структуры, инициирумой сложным эфиром сорбитана и ненасыщенной жирной кислоты, который представляет собой образующее жидкие кристаллы средство по настоящему изобретению.
Фосфолипид по настоящему изобретению представляет собой форму, полученную из растений или животных, в частности, содержащую алкилэфирную группу, содержащую от 4 до 30 насыщенных или ненасыщенных атомов углерода, и которая представляет собой, по меньшей мере, одно соединение, выбранное из фосфатидилхолина, фосфатидилэтаноламина, фосфатидилсерина, фосфатидилглицерина, фосфатидилинозита и фосфатидной кислоты, сфингомиелина и их смесей в соответствии со структурой полярной головки.
Также фосфолипид представляет собой форму, полученную из растений или животных, таких как соевые бобы, яйца и т.п., причем алкилэфирная группа, связанная с фосфолипидом, может включать сложные эфиры насыщенных жирных кислот, такие как моно- и дипальмитоил, моно- и димиристоил, моно- и дилаурил, моно- и дистеарил и т.д., или сложные эфиры ненасыщенных жирных кислот, такие как моно- и дилинолеил, моно- и диолеил, моно- и дипальмитолеил, моно- и димиристоил и т.д., или могут включать форму в которой сложные эфиры насыщенных жирных кислот и сложные эфиры ненасыщенных жирных кислот присутствуют вместе.
c) Отвердитель жидких кристаллов
Отвердитель жидких кристаллов по настоящему изобретению не может независимо образовывать неламеллярную структуру, аналогичную образующему жидкие кристаллы средству, а также не может образовывать слоистую структуру, аналогичную липосомоподобному фосфолипиду. Однако отвердитель жидких кристаллов по настоящему изобретению может принимать участие в образовании непластинчатой структуры, инициируемом образующим жидкие кристаллы средством, чтобы увеличить искривление (деформацию) неламеллярной структуры, что приводит к дальнейшему увеличению степени регулярного перемешивания масла и воды. Для обладания таким действием, как отвердитель жидких кристаллов, предпочтительно, чтобы полярность в молекулярной структуре была в значительной степени ограничена, и в то же время неполярная область была большой.
Однако, в отличие от изложенного выше, в случае отвердителя жидких кристаллов по настоящему изобретению весьма необычно то, что вещество, которое вводится путем инъекции в организм человека и является биосовместимым, может быть выбрано только путем прямых и повторных экспериментов. В результате каждый из отвердителей жидких кристаллов, подходящих для композиции по настоящему изобретению, имеет отличную от других молекулярную структуру, и не может быть описан единой структурой. Однако при обнаружении отвердителя жидких кристаллов, подходящего для композиции по настоящему изобретению, и последующем наблюдении за его структурой можно было подтвердить, что отвердитель жидких кристаллов не содержит ионизированную группу, такую как карбоксильная группа или аминогруппа, а гидрофобный фрагмент содержит большую триацильную группу с общим числом атомов углерода от 15 до 40 или структуру углеродного кольца. Предпочтительно отвердитель жидких кристаллов не содержит ионизированную группу, такую как карбоксильная группа или аминогруппа, и содержит не более одной гидроксильной группы и структуру сложного эфира в качестве слабополярной части, а относительно гидрофобный фрагмент содержит объемную триацильную группу, имеющую суммарно от 20 до 40 атомов углерода или структуру углеродного кольца. Таким образом, в частности, отвердитель жидких кристаллов по настоящему изобретению может представлять собой, по меньшей мере, одно вещество, выбранное из, но ими не ограничиваясь, триглицерида, ретинилпальмитата, ацетата токоферола, холестерина, бензилбензоата, убихинона и их смесей. Предпочтительно отвердитель жидких кристаллов по настоящему изобретению может представлять собой ацетат токоферола, холестерин или их смеси.
d) Вода
Вода по настоящему изобретению применяется для быстрого образования жидкокристаллического геля непосредственно после введения жидкого инъекционного средства. Липидный исходный препарат, не содержащий воду, может впитывать влагу in vivo после инъекции и очень медленно образовывать жидкокристаллический гель. Поскольку исходный липидный препарат образует жидкокристаллический гель с низкой скоростью, исходный липидный препарат может широко распространяться на участке инъекции и образовывать в этом состоянии жидкокристаллический гель, тем самым образуя гель с большой площадью поверхности или фрагментированный гель. Жидкокристаллический гель, который имеет большую площадь поверхности или фрагментирован, может вызвать эффект “сброса” лекарственного средства на начальной стадии введения, что приводит к недостаточному высвобождению лекарственного средства на поздней стадии периода замедленного высвобождения, затрудняя таким образом поддержание свойства стабильного замедленного высвобождения в течение длительного периода времени. И наоборот, согласно настоящему изобретению исходный липидный препарат содержит воду и, таким образом, непосредственно после введения в ткани образует гель, что приводит к снижению начальной скорости высвобождения лекарственного средства, обеспечивая тем самым стабильное замедленное высвобождение лекарственного средства.
Согласно настоящему изобретению вода может добавляться в виде воды для инъекций, дистиллированной воды, буфера или смеси двух или более веществ, выбранных из указанных веществ.
e) Аналоги GnRH
Аналоги GnRH являются структурно сходными с GnRH, но по-разному действуют в организме. В общем случае GnRH выполняет биологическую функцию индукции продукции половых стероидов в организме посредством импульсной секреции, однако аналоги GnRH применяются для значительного ингибирования продукции половых стероидов в организме в течение определенного периода времени.
Указанные аналоги GnRH классифицируются на агонисты и антагонисты в зависимости от механизма действия. При введении в организм терапевтической дозы агониста GnRH сначала агонист GnRH связывается с рецептором GnRH гипофизарного гонадотропина и способствует биосинтезу и секреции фолликулостимулирующего гормона (FSH), а также лютеинизирующего гормона (LH). Однако постоянное введение агониста GnRH обедняет гонадотропин, при этом снижая экспрессию рецептора GnRH, тем самым ингибируя биосинтез и секрецию фолликулостимулирующего гормона (FSH) и лютеинизирующего гормона (LH). Благодаря биологическим функциям GnRH применяются аналоги GnRH для профилактики или лечения заболеваний, зависимых от половых гормонов, таких как злокачественная опухоль предстательной железы, злокачественная опухоль молочной железы, злокачественная опухоль яичников, эндометриоз, миома матки, поликистоз яичников, гипертрихоз, преждевременное половое созревание, гонадотропная аденома гипофиза, синдром апноэ во сне, синдром раздраженного кишечника, предменструальный синдром, доброкачественная гиперплазия предстательной железы или бесплодие или т.д., а также могут применяться в качестве контрацептивного средства.
Агонист GnRH, который можно применять в качестве фармакологически активного вещества по настоящему изобретению, может быть выбран из лейпролида, гозерелина, трипторелина, нафарелина, бусерелина, гистрелина, деслорелина, метерелина, гонадрелина или их фармакологически приемлемых солей. Предпочтительно агонистом GnRH может быть, по меньшей мере, одно фармакологически активное вещество, выбранное из лейпролида, гозерелина и их фармакологически приемлемых солей.
И наоборот, антагонист GnRH конкурентно реагирует на рецептор GnRH гипофизарного гонадотропина, блокируя связывание GnRH в организме, ингибируя тем самым биосинтез и секрецию фолликулостимулирующего гормона (FSH) и лютеинизирующего гормона (LH). Антагонист GnRH, который можно применять в качестве фармакологически активного вещества по настоящему изобретению, может быть выбран из дегареликса, абареликса, ганиреликса, цетрореликса и их фармакологически приемлемых солей. Предпочтительно антагонист GnRH может представлять собой, по меньшей мере, одно фармакологически активное вещество, выбранное из дегареликса и его фармакологически приемлемых солей.
В то же время, инъекционная композиция по настоящему изобретению может дополнительно включать растворитель в дополнение к пунктам а) - е). Примеры растворителя могут включать этанол, диметилсульфоксид (ДМСО), бензиловый спирт, бензилбензоат, метилпирролидон, пропиленгликоль и т.п., которые можно применять отдельно или в сочетании двух или более из указанных веществ.
Массовые соотношение компонента а) и компонента b), подходящее для желаемого жидкого кристалла инъекционной композиции по настоящему изобретению, может составлять от 10:1 до 1:10, и в частности от 5:1 до 1:5. Массовое соотношение компонентов а) + b) и компонента с) может составлять от 100:1 до 1:1, и может составлять, например, от 50:1 до 2:1. В пределах вышеуказанного диапазона эффект замедленного высвобождения желаемого жидкого кристалла по настоящему изобретению может быть удовлетворительным, при этом можно контролировать характер замедленного высвобождения путем регулирования указанного соотношения.
Кроме того, массовое соотношение компонентов а) + b) + c) и компонента d), подходящее для желаемого жидкого кристалла инъекционной композиции по настоящему изобретению, может составлять от 99:1 до 1:1 и может составлять, например, от 99:1 до 90:10 или от 75:25 до 62,5:37,5.
Кроме того, массовое соотношение компонентов а) + b) + c) + d) и компонента e), подходящее для желаемого жидкого кристалла инъекционной композиции по настоящему изобретению, может составлять от 10000:1 до 1:1 и может составлять, например, от 1000:1 до 1:1.
Согласно настоящему изобретению содержание одного компонента, выраженное в “% по массе”, может означать процент, на который приходится масса компонента, когда общая масса инъекционной композиции составляет 100%. Инъекционная композиция по настоящему изобретению может включать дополнительный растворитель вместе с компонентами а) - е).
Инъекционная композиция по настоящему изобретению может включать: от 9 до 90% по массе компонента а); 9-90% по массе компонента b); 0,1-50% по массе компонента с); 0,5-50% по массе компонента d); и 0,01-50% по массе компонента e).
Согласно одному примеру инъекционная композиция по настоящему изобретению может включать: 9-50% по массе компонента a); 18-60% по массе компонента b); 1-36% по массе компонента c); 0,5-50% по массе компонента d); и 0,1-45% по массе компонента e).
Согласно одному примеру инъекционная композиция по настоящему изобретению может включать: 9-50% по массе компонента a); 18-60% по массе компонента b); 1-36% по массе компонента c); 0,5-10,5% по массе компонента d); и 0,1-45% по массе компонента e).
Согласно одному примеру инъекционная композиция по настоящему изобретению может включать: 9-50% по массе компонента a); 18-60% по массе компонента b); 1-36% по массе компонента c); 2,5-10,5% по массе компонента d); и 0,1-45% по массе компонента e).
Согласно одному примеру инъекционная композиция по настоящему изобретению может включать: 9-50% по массе компонента a); 18-60% по массе компонента b); 1-36% по массе компонента c); 25-37,5% по массе компонента d); и 0,1-45% по массе компонента e).
Согласно одному варианту осуществления настоящего изобретения в случае, если фармакологически активным веществом является лейпролид, инъекционная композиция по настоящему изобретению может включать: 9-50% по массе компонента a); 18-60% по массе компонента b); 1-36% по массе компонента c); 0,5-10,5% по массе или 25-37,5% по массе компонента d); и 0,1-45% по массе лейпролида е) или его фармацевтически приемлемых солей.
Согласно одному варианту осуществления настоящего изобретения в случае, если фармакологически активным веществом является гозерелин, инъекционная композиция по настоящему изобретению может включать: 9-50% по массе компонента a); 18-60% по массе компонента b); 1-36% по массе компонента c); 0,5-10,5% по массе или 25-37,5% по массе компонента d); и 0,1-45% по массе гозерелина e) или его приемлемых солей для инъекций.
Согласно одному варианту осуществления настоящего изобретения в случае, если фармакологически активным веществом является дегареликс, инъекционная композиция по настоящему изобретению может включать: 9-50% по массе компонента a); 18-60% по массе компонента b); 1-36% по массе компонента c); 0,5-10,5% по массе или 25-37,5% по массе компонента d); и 0,1-45% по массе дегареликса e) или его фармацевтически приемлемых солей.
В случае, когда инъекционная композиция по настоящему изобретению содержит компоненты а) - е) в вышеуказанном составе, каждое из фармакологически активных веществ может обладать превосходным эффектом замедленного высвобождения.
В настоящем изобретении жидкий кристалл имеет неламеллярную структуру, в которой масло и вода упорядочно смешиваются и располагаются в очень ограниченных условиях, так что внутреннюю фазу и внешнюю фазу нельзя отличить друг от друга, и эта неламеллярная структура обладает свойствами мезофазы между жидкой фазой и твердой фазой благодаря специфическому упорядочному расположению масло-вода. Она имеет структуру, отличную от структуры, в которой мицеллы, эмульсии, микроэмульсии, липосомы, липидные бислои и т.д., которые широко применяются при разработке препаратов для инъекций, имеют общие свойства ламеллярной структуры, и эта ламеллярная структура образуется путем разделения внутренней фазы и внешней фазы в форме “масло в воде” (м/в) или “вода в масле” (в/м).
Согласно настоящему изобретению явление жидкой кристаллизации, представляющее “жидкий кристалл”, относится к явлению, при котором жидкий кристалл, структура неламеллярной фазы, образуется под воздействием избытка жидкости на водной основе из исходного препарата, как описано выше. “Избыток” жидкости на водной основе, в которой происходит явление кристаллизации жидкости, может означать превышение общей массы или объема инъекционной композиции по настоящему изобретению, по меньшей мере, в один или несколько раз.
Инъекционная композиция по настоящему изобретению может быть получена при комнатной температуре путем добавления а) по меньшей мере одного вещества, выбранного из сложного эфира сорбитана и ненасыщенной жирной кислоты, b) по меньшей мере одного соединения, выбранного из фосфолипидов, c) по меньшей мере одного соединения, выбранного из жидкокристаллических отвердителей, d) по меньшей мере одного вещества, выбранного из воды для инъекций, дистиллированной воды и буфера, и е) по крайней мере одного вещества, выбранного из аналогов GnRH, а также, при необходимости, может быть получена путем нагревания или с применением гомогенизатора. В данном случае применяемый в настоящем документе гомогенизатор может быть выбран из гомогенизатора высокого давления, ультразвукового гомогенизатора, измельчающего гомогенизатора и тому подобного.
Инъекционная композиция по настоящему изобретению может быть введена в любой форме подкожной инъекции или внутримышечной инъекции в качестве пути введения инъекционного средства, причем лекарственная форма может быть выбрана в зависимости от свойств каждого фармакологически активного вещества.
Инъекционная композиция по настоящему изобретению может быть получена путем добавления фармакологически активного вещества к исходному препарату по настоящему изобретению при комнатной температуре, а также, при необходимости, может быть получена путем нагревания или с применением гомогенизатора, однако настоящее изобретение этим не ограничивается.
Кроме того, настоящее изобретение может обеспечить способ профилактики или лечения заболеваний, зависимых от половых гормонов, или контрацепции, который включает: введение субъекту терапевтически эффективного количества инъекционной композиции по настоящему изобретению.
Согласно настоящему изобретению термин “субъект” может относиться к млекопитающим, включая человека, а термин “введение” может относиться к получению субъектом предопределенного вещества любым подходящим способом.
“Терапевтически эффективное количество” инъекционной композиции по настоящему изобретению может относиться к количеству активного вещества или фармацевтической композиции, которое вызывает у животных или человека биологические или терапевтические ответы, анализируемые исследователями, ветеринарами, врачами или клиницистами. Терапевтически эффективное количество может включать количество, вызывающее облегчение симптомов заболеваний или расстройств, подлежащих лечению, может быть аналогичным известной дозе фармакологически активного вещества, применяемого в настоящем документе, однако может изменяться в зависимости от типа заболевания пациента, степени его тяжести, симптома, возраста, пола и т.д., и может вводиться в любой форме подкожной инъекции или внутримышечной инъекции в зависимости от свойств фармакологически активного вещества и лекарственного средства. Настоящее изобретение может обеспечить применение инъекционной композиции по настоящему изобретению для профилактики или лечения заболеваний, зависимых от половых гормонов, или для контрацепции.
Настоящее изобретение может обеспечить применение инъекционной композиции по настоящему изобретению в изготовлении лекарственного средства для профилактики или лечения заболеваний, зависимых от половых гормонов, или для контрацепции.
Вещества, описанные в инъекционной композиции по настоящему изобретению, их применение и способ терапии, а также способ замедленного высвобождения фармакологически активного вещества применимы в равной степени, если не противоречат друг другу.
Положительные эффекты изобретения
Инъекционная композиция по настоящему изобретению может обладать значительно повышенной безопасностью вследствие включения сложного эфира сорбитана и ненасыщенной жирной кислоты, содержащего полярную головку с двумя или более -ОН (гидроксильными) группами, может находиться в жидкой фазе в фазе, где жидкость водной основе отсутствует, что позволяет легко применять ее в отношении фармацевтических препаратов в лекарственных формах, и образует жидкий кристалл в фазе жидкости на водной основе, что обеспечивает проявление эффектов замедленного высвобождения аналогов GnRH, которые применяются в качестве фармацевтически активного вещества in vivo. Кроме того, в инъекционной композиции по настоящему изобретению исходный липидный препарат с замедленным высвобождением, содержащий аналоги GnRH, предварительно включает воду, и, таким образом, жидкое инъекционное средство может образовывать жидкокристаллический гель непосредственно после введения, что может обеспечить проявление эффектов снижения начальной скорости высвобождения и значительного усиления свойств замедленного высвобождения лекарственного средства.
Краткое описание фигур
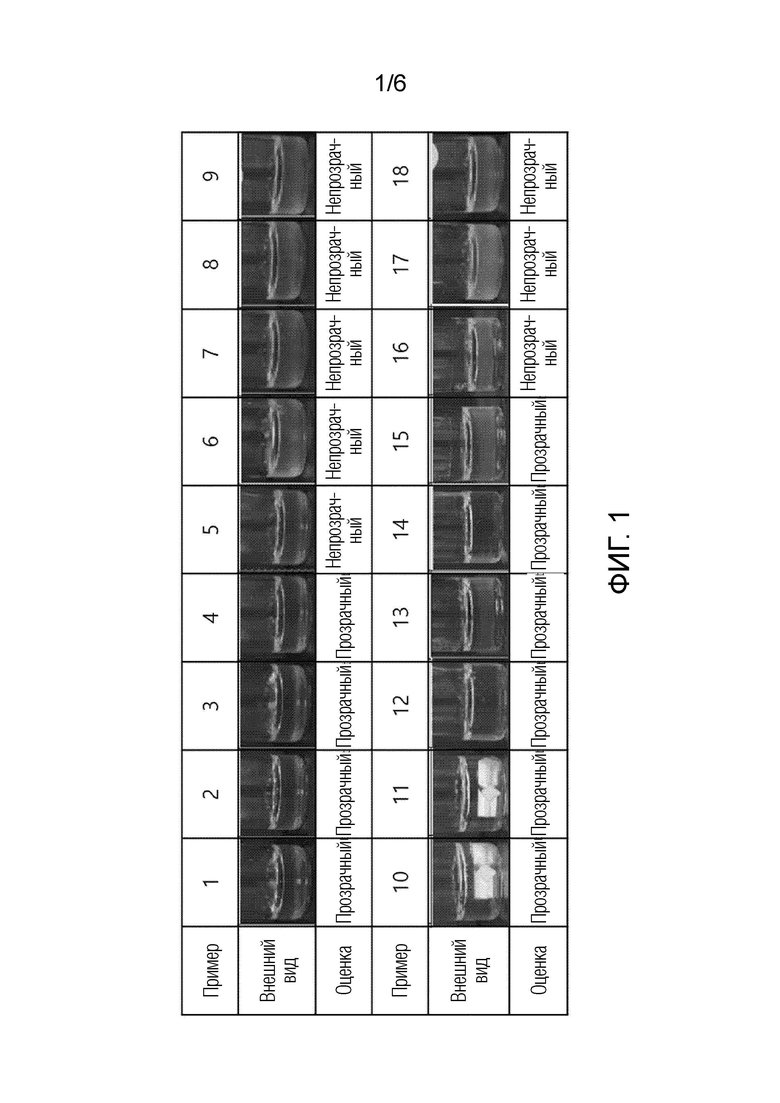
На фиг. 1 представлены изображения, демонстрирующие результаты исследований по определению внешнего вида композиций в соответствии с примерами 1-18.
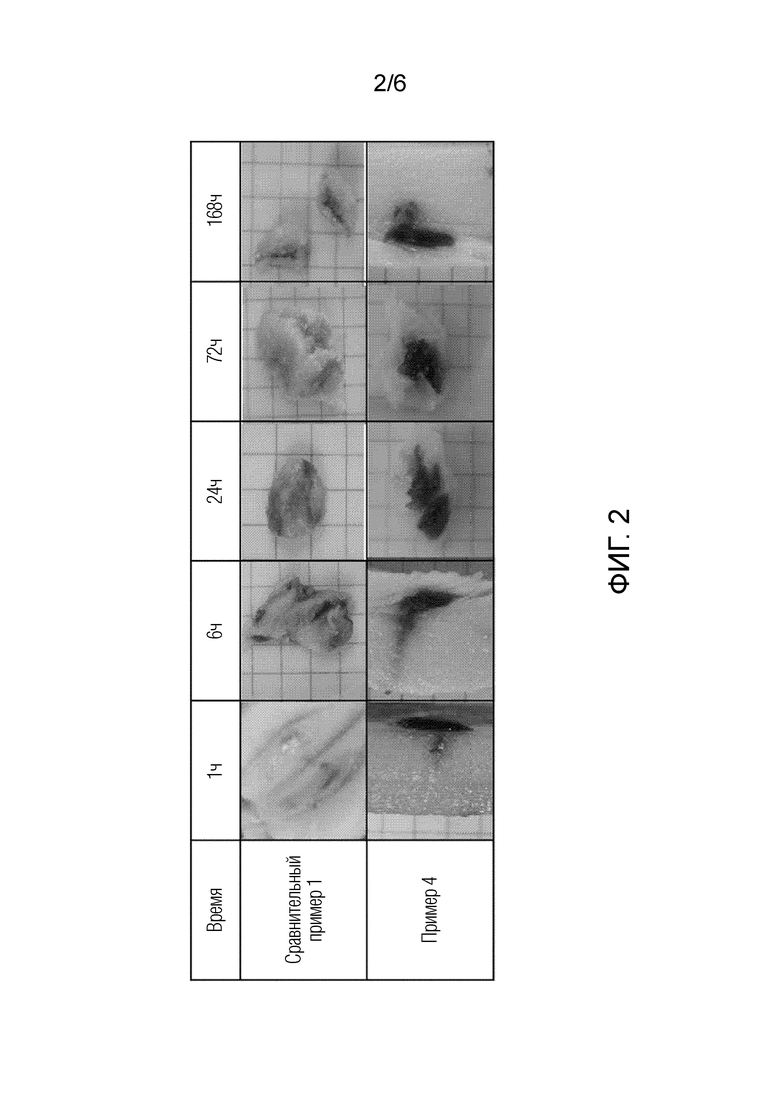
На фиг. 2 представлено изображение, подтверждающее золь-гелевое преобразование с течением времени в подкожном жире (ex-vivo) у свиней, которым вводили композиции сравнительных примеров 1 и примера 4 соответственно.
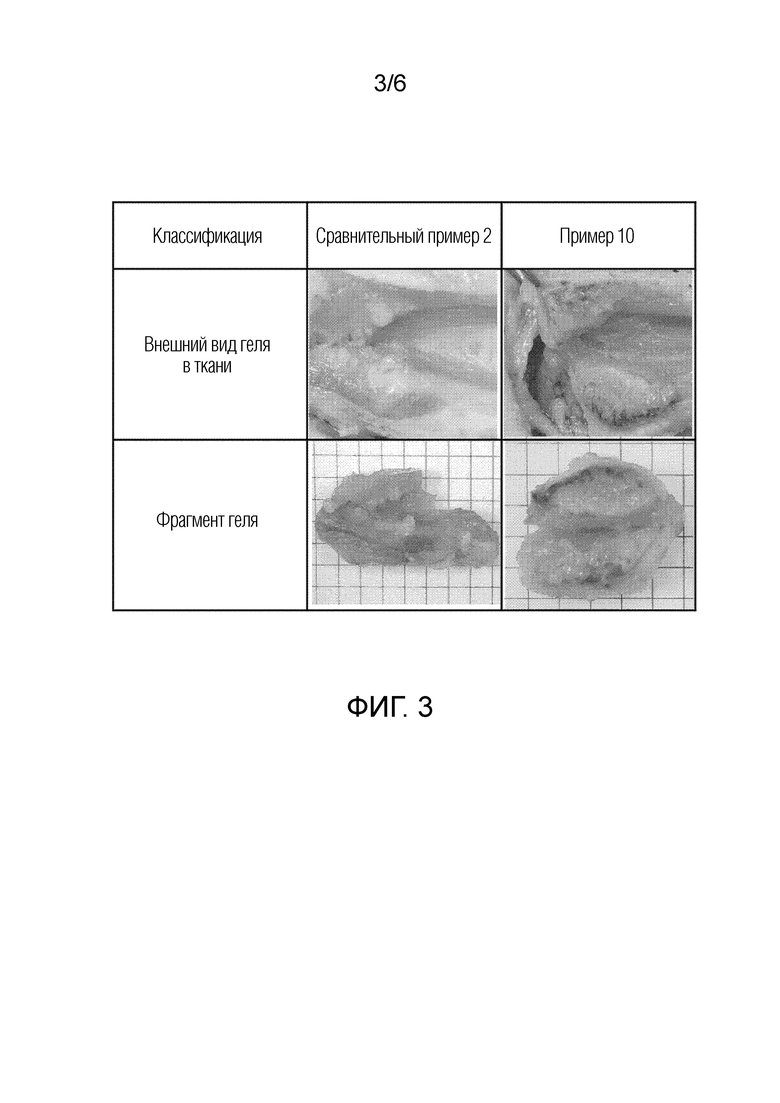
На фиг. 3 представлены изображения, показывающие внешний вид геля у мини-свиней, которым подкожно (in vivo) вводили композиции сравнительного примера 2 и примера 10 соответственно.
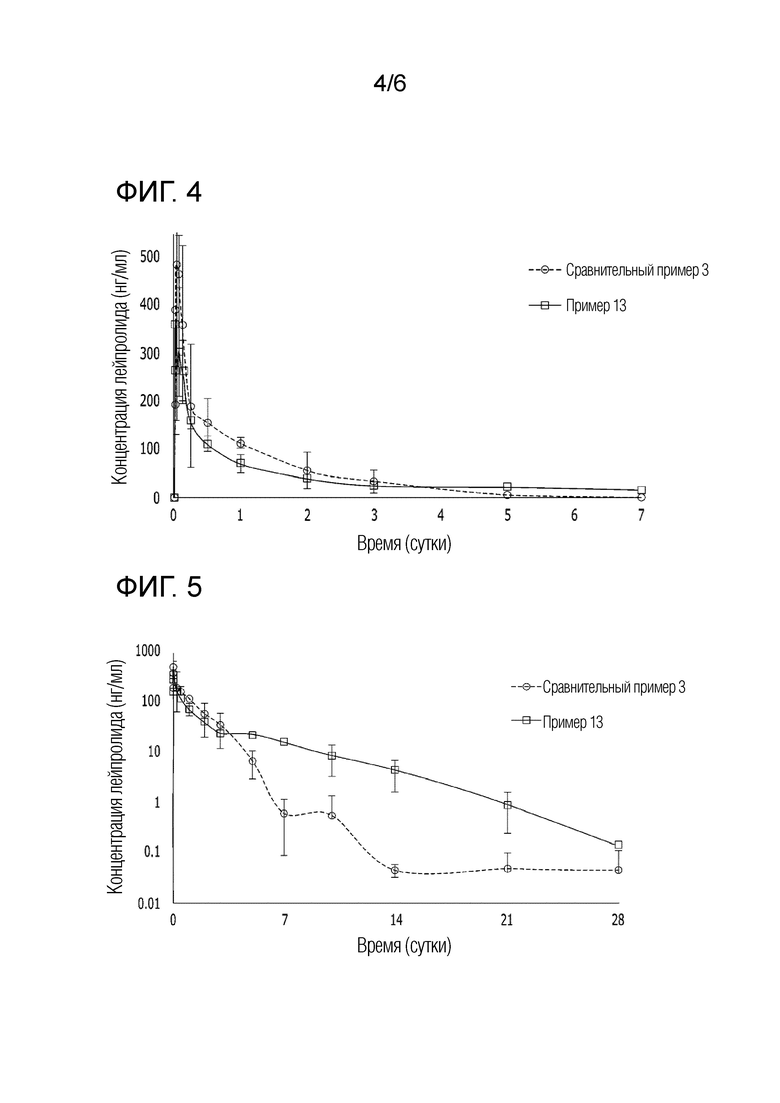
На фиг. 4 представлен график, показывающий изменение фармакокинетики в пределах до 7 суток у морских свинок, каждой из которых инъецировали композиции сравнительного примера 3 и примера 13.
На фиг. 5 представлен график, показывающий изменение фармакокинетики в пределах до 28 суток у морских свинок, каждой из которых инъецировали композиции сравнительного примера 3 и примера 13.
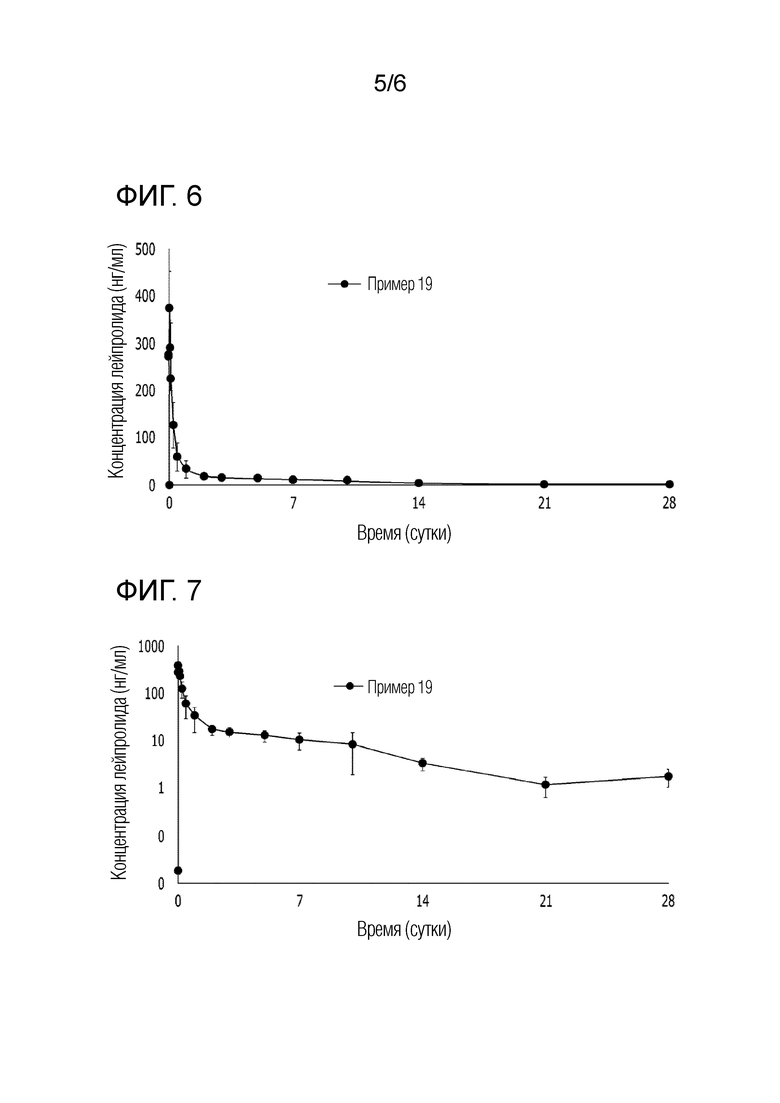
На фиг. 6 представляет собой график, показывающий изменение фармакокинетики начальной скорости высвобождения лекарственного средства у морских свинок, которым инъецировали композицию примера 19.
На фиг. 7 представляет собой график, показывающий изменение фармакокинетики в пределах до 28 суток у морских свинок, которым инъецировали композицию примера 19.
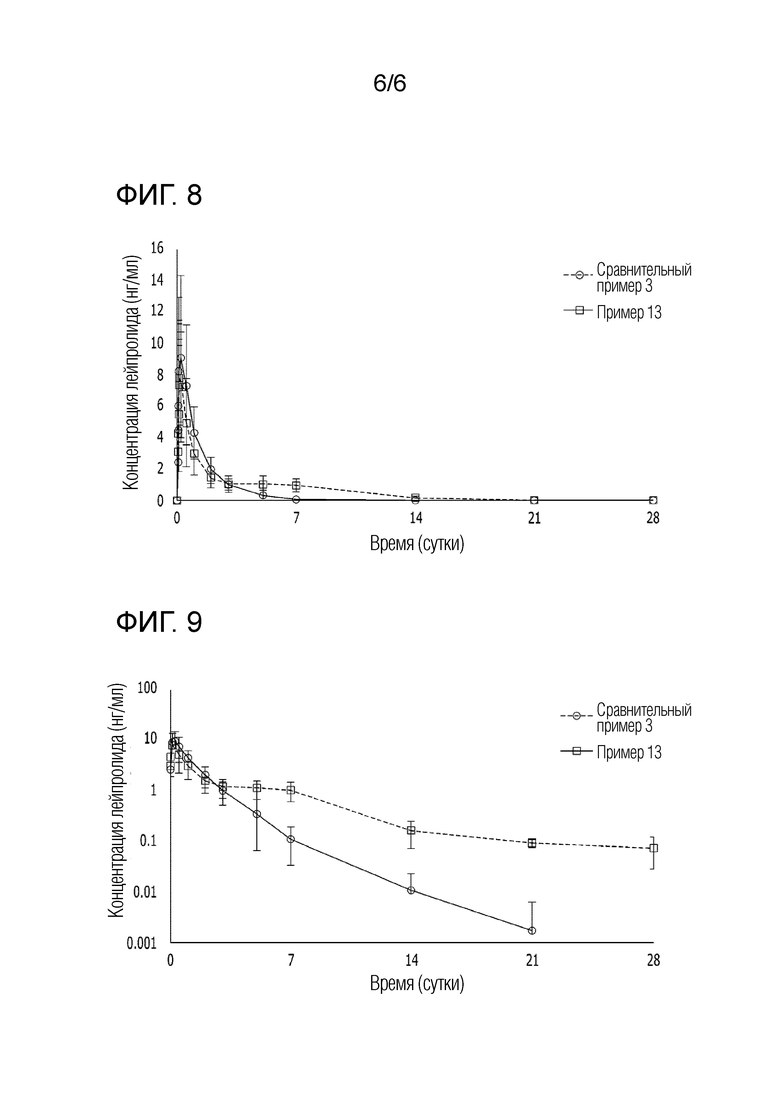
На фиг. 8 представляет собой график, показывающий изменение фармакокинетики в пределах до 7 суток у людей, каждому из которых инъецировали композиции сравнительного примера 3 и примера 13.
На фиг. 9 представляет собой график, показывающий изменение фармакокинетики в пределах до 28 суток у людей, каждому из которых инъецировали композиции сравнительного примера 3 и примера 13.
Принцип изобретения
Далее настоящее изобретение будет описано более подробно посредством следующих примеров и экспериментальных примеров. Однако следующие примеры и экспериментальные примеры представлены только с целью иллюстрации настоящего изобретения, и, соответственно, объем настоящего изобретения ими не ограничивается.
Добавки, применяемые в настоящем изобретении, представляли собой вспомогательные вещества в соответствии со стандартами фармакопеи, причем реагент приобретали у Aldrich, Lipoid, Croda и Seppic (названия компаний).
[Примеры 1-18] Получение инъекционной композиции по настоящему изобретению
Добавляли сложный эфир сорбитана и ненасыщенной жирной кислоты, фосфолипид, отвердитель жидких кристаллов, воду и фармакологически активные вещества массой, аналогичной представленной в таблице 1 ниже.
Соединения примеров 1-18 получали следующим образом. Сначала ацетат лейпролида, моноолеат сорбитана, фосфатидилхолин, ацетат токоферола, ДМСО и этанол смешивали с помощью гомогенизатора (PowerGenmodel125. Fisher) при комнатной температуре в течение 0,5 часов при скорости от 1000 до 3000 об/мин и гомогенизировали с получением липидного раствора. Затем к полученному липидному раствору добавляли соответствующее количество воды и гомогенизировали в течение примерно от 5 до 30 мин с помощью гомогенизатора при скорости примерно от 1000 до 3000 об/мин с получением инъекционной композиции в растворе.
[Пример 19] Получение инъекционной композиции по настоящему изобретению
В примере 19 в стеклянный сосуд добавляли 292 мг ацетата лейпролида, 430 мг ДМСО, 1034 мг сорбитанмоноолеата, 2195 мг фосфатидилхолина, 697 мг ацетата токоферола и 452 мг безводного спирта и перемешивали при комнатной температуре с получением прозрачного липидного раствора. Полученный липидный раствор фильтровали через капсульный фильтр из ПВДФ с диаметром ячейки 0,2 мкм, а затем измеряли массу. В оцениваемый липидный раствор вводили воду с получением прозрачного раствора, в котором содержание воды может составлять 10,5% (масса/масса) в расчете на 100% по массе прозрачного раствора, полученного путем введения воды, потом дополнительно перемешивали при комнатной температуре для подтверждения прозрачности раствора, и затем, наконец, фильтровали через капсульный фильтр из ПВДФ с диаметром ячейки 0,2 мкм с получением инъекционного жидкого препарата.
[Сравнительные примеры 1-3]
Добавляли сложный эфир сорбитана и ненасыщенной жирной кислоты, фосфолипид, отвердитель жидких кристаллов и фармакологически активное вещество массой, аналогичной представленной в таблице 2 ниже.
В сравнительных примерах 1-3 смешивали ацетат лейпролида, моноолеат сорбитана, фосфатидилхолин, ацетат токоферола, ДМСО и этанол с помощью гомогенизатора (PowerGenmodel125. Fisher) при комнатной температуре в течение 0,5 часов при скорости от 1000 до 3000 об/мин и гомогенизировали.
[Экспериментальный пример 1] Тест на определение внешнего вида
Определяли внешний вид препаратов, полученных в соответствии с примерами 1-18 настоящего изобретения. Можно подтвердить, что образцы примеров 1-4 имеют прозрачный внешний вид, когда содержание воды находится в диапазоне от 0,5 до 10% по массе в расчете на 100% по массе всей композиции. Кроме того, можно подтвердить, что образцы примеров 10-15 с содержанием воды от 25 до 37,5% по массе являются прозрачными. Эта часть, в которой вода, обладает физическими свойствами, отличными от липидов, стабильно растворяется без отделения и суспендирования в липидном растворе.
[Экспериментальный пример 2] Тест на золь-гелевое преобразование в подкожной жировой ткани свиньи (ex-vivo)
Тест на золь-гелевое преобразование проводили в подкожной жировой ткани свиньи с применением композиций в соответствии со сравнительным примером 1 и примером 4 по настоящему изобретению следующим образом.
После медленного инъецирования 100 мкл композиций в соответствии со сравнительным примером 1 и примером 4 в подкожный жир свиньи рассекали участок инъекции через 1, 6, 24, 72 и 168 часов после инъекции для наблюдения его поперечного сечения. Результаты наблюдения представлены на [фиг. 2].
Со ссылкой на фиг. 2, в случае сравнительного примера 1, в котором вода не добавлялась в подкожные жировые ткани свиньи, можно подтвердить, что жидкий препарат вытягивает влагу из тканей и очень медленно превращается в гель. В случае примера 4, содержащего воду, можно подтвердить, что жидкий препарат уже содержит воду и, таким образом, быстро превращается в гель непосредственно после введения.
Аналогично сравнительному примеру 1, лекарственное средство быстро высвобождается в организме в виде золя. В примерах по настоящему изобретению лекарственное средство проходит через многочисленные липидные решетчатые структуры и, таким образом, очень медленно высвобождается после превращения в гель, имеющий жидкокристаллическую структуру.
[Экспериментальный пример 3] Тест на определение внешнего вида геля под кожей у минисвиней (in vivo)
Для сравнительного примера 2 и примера 10 настоящего изобретения определяли внешний вид геля в подкожных тканях минисвиньи. Эксперимент осуществляли путем введения 0,3 мл каждой из композиций сравнительного примера 2 и примера 10 с помощью одноразового шприца под кожу спинки минисвиньи (самца) массой около 15 кг. Через семь суток после введения участок введения иссекали, и результаты сравнительного наблюдения внешнего вида геля представлены на фиг. 3.
Со ссылкой на фиг. 3, композиция в соответствии со сравнительным примером 2 представляет собой липидный жидкий препарат без добавления воды, и при превращении из золя в гель композиция превращается в гель с очень низкой скоростью при медленном втягивании жидкости организма в ткань. Поскольку композиция превращается в гель очень медленно, золь, не превратившийся в гель, проникает между подкожными жирами и образует гель в фрагментированном состоянии. Можно подтвердить, что площадь поверхности геля увеличивается, когда гель образуется не в результате агломерации, а образуется в фрагментированном состоянии. Это может быть причиной высокой начальной скорости высвобождения лекарственного средства.
С другой стороны, поскольку композиция примера 10 по настоящему изобретению представляет собой форму, уже содержащую воду, можно подтвердить, что композиция быстро превращается в гель в ткани и агломерируется, в результате чего гель не образуется. Таким образом композиция примера 10 по настоящему изобретению может обеспечить низкую начальную скорость высвобождения лекарственного средства.
[Экспериментальный пример 4] Подтверждение фармакокинетики инъекционной композиции у морской свинки
[Экспериментальный пример 4-1] Подтверждение фармакокинетики инъекционной композиции примера 13 и сравнительного примера 3 у морской свинки
Фармакокинетику композиций по настоящему изобретению подтверждали на морских свинках в следующем эксперименте. С помощью одноразового шприца вводили подкожно композиции сравнительных примеров 3 и примера 13 в спинку шестинедельным самцам морских свинок (n=4), которые имели в среднем массу 500 г, так что масса вводимого лейпролида могла достигать 3,75 мг/животное (эквивалентно месячной дозе для человека). Аналогично человеку, морская свинка является животным с развитым подкожной жировой клетчаткой, и была предпринята попытка подтвердить ФК профиль (фармакокинетический профиль) в жировых тканях.
Концентрацию лейпролида в образце плазмы морских свинок анализировали в отношении фармакокинетического профиля в течение 28 суток посредством жидкостной хроматографии в сочетании с масс-спектрометрией (ЖХ-МС). Результаты, представленные на [фиг. 4] и [фиг. 5], показывают среднее значение для четырех морских свинок, использованных в эксперименте. На [фиг. 4] представлено подтверждение разницы в скорости начального высвобождения лекарственного средства, при этом на [фиг. 5] показан результат логарифмического преобразования для подтверждения разницы в концентрации лекарственного средства в крови морских свинок во второй половине.
Со ссылкой на фиг. 4, в случае композиции примера 13, содержащей воду, было подтверждено, что жидкий препарат быстро превращается в гель in vivo, тем самым значительно снижая начальную скорость высвобождения. Для композиции сравнительного примера 3 может быть подтверждено, что площадь под кривой (AUC) до трех суток составляет 7864 мкг*ч/мл. Однако для композиции примера 13 также может быть подтверждено, что AUC в пределах до трех суток составляет 5528 мкг*ч/мл, что примерно на 30% меньше по сравнению со сравнительным примером 3.
Со ссылкой на фиг. 5, может быть подтверждено, что композиция примера 13 демонстрирует уменьшение эффекта начального сброса лекарственного средства и поддерживает концентрацию лекарственного средства в крови выше, чем композиция сравнительного примера 3, в течение одного месяца, при этом лекарственное средство остается в гелеобразном препарате.
[Экспериментальный пример 4-2] Подтверждение фармакокинетики инъекционной композиции примера 19 у морской свинки
При применении жидкого инъекционного препарата, полученного в примере 19 по настоящему изобретению, была подтверждена фармакокинетика примера 19 на морских свинках. С помощью одноразового шприца композицию примера 19 вводили подкожно в спинку шестинедельным самцам морских свинок (n=4), которые в среднем имели массу 500 г, так что масса вводимого лейпролида могла достигать 3,75 мг/животное (эквивалентно месячной дозе для человека). В данном случае морская свинка является животным с развитой подкожной жировой клетчаткой, аналогичной человеку, и была предпринята попытка подтвердить ФК профиль (фармакокинетический профиль) в жировых тканях.
Концентрацию лейпролида в образце плазмы морских свинок анализировали в отношении фармакокинетического профиля в течение 28 суток посредством жидкостной хроматографии в сочетании с масс-спектрометрией (ЖХ-МС). Результаты, представленные на [фиг. 6] и [фиг. 7], показывают среднее значение для четырех морских свинок, использованных в эксперименте. На [фиг. 6] представлено подтверждение разницы в скорости начального высвобождения лекарственного средства, при этом на [фиг. 7] показан результат логарифмического преобразования для подтверждения разницы в концентрации лекарственного средства в крови морских свинок во второй половине.
Результаты, представленные на [фиг. 6] и [фиг. 7], показывают результаты исследования фармакокинетики морской свинки примера 19 для жидкого препарата, содержащего 10,5% воды, и было подтверждено, что начальный выброс лекарственного средства является низким по сравнению со значением результата фармакокинетики сравнительного примера 3 экспериментального примера 4-1, причем было подтверждено, что концентрация лекарственного средства сохраняется более высокой, чем в сравнительном примере 3, даже через 28 суток.
В вышеприведенных экспериментальных примерах 4-1 и 4-2, в случае жидких инъекционных препаратов, содержащих воду, из примеров 19 и 13, полагают, что начальная концентрация лекарственного средства ниже, чем у препарата сравнительного примера 3, который не содержит воды, и поэтому безопасность является более высокой. Также было подтверждено, что свойство замедленного высвобождения у лекарственного средства улучшается, поскольку концентрация лекарственного средства сохраняется выше, чем у композиции сравнительного примера 3, даже через 28 суток.
[Экспериментальный пример 5] Подтверждение фармакокинетики инъекционной композиции у человека
Для каждой из композиций сравнительного примера 3 и примера 13 была подтверждена фармакокинетика примера 13 у человека. Здоровым женщинам в постменопаузе, шести пациентам на каждую испытуемую группу, вводили подкожно в брюшную полость 3,75 мг сравнительного соединение примера 3 и примера 13 в виде лейпролида. Концентрацию лейпролида в образце плазмы человека анализировали в отношении фармакокинетического профиля в течение 28 суток посредством жидкостной хроматографии в сочетании с масс-спектрометрией (ЖХ-МС). ФК профили сравнительного примера 3 и примера 13 у человека были такими, как показано на [фиг. 8] и [фиг. 9]. На [фиг. 8] представлено подтверждение разницы в скорости начального высвобождения лекарственного средства, а на [фиг. 9] показано логарифмическое преобразование для подтверждения разницы в концентрации лейпролида в крови во второй половине.
Со ссылкой на фиг. 8, в фармакокинетическом профиле у человека, аналогичном профилю морской свинки, было подтверждено, что эффект сброса лекарственного средства непосредственно после введения заметно снижается в композиции примера 13 по сравнению с композицией сравнительного примера 3.
В отношении композиции сравнительного примера 3 было обнаружено, что площадь под кривой (AUC) в пределах до трех суток после введения составляет 276 мкг*ч/мл. Однако для композиции примера 13 было обнаружено, что AUC в пределах до трех суток составляет 209 мкг*ч/мл, что примерно на 25% ниже, чем у композиции сравнительного примера 3.
Со ссылкой на фиг. 9, было обнаружено, что композиция примера 13 демонстрирует снижение эффекта начального сброса лекарственного средства и поддерживает концентрацию лекарственного средства в крови выше, чем у композиции сравнительного примера 3 в течение одного месяца, при этом лекарственное средство остается в гелеобразном препарате.
Группа изобретений относится к композиции для профилактики или лечения заболеваний, зависимых от половых гормонов, или для контрацепции, содержащей: а) сложный эфир сорбитана и ненасыщенной жирной кислоты, содержащий две или более -ОН (гидроксильных) групп в группе полярной головки; b) фосфолипид; c) отвердитель жидких кристаллов, который не содержит ионизированную группу, и гидрофобный фрагмент которого имеет триацильную группу, содержащую от 15 до 40 атомов углерода или структуру углеродного кольца; d) воду; и e) аналог гонадотропин-рилизинг-гормона (GnRH) в качестве фармацевтически активного вещества, также относится к способу профилактики или лечения заболеваний, зависимых от половых гормонов, или для контрацепции, включающему введение терапевтически эффективного количества композиции субъекту, и также относится к применению композиции для профилактики или лечения заболеваний, зависимых от половых гормонов, или для контрацепции и к применению композиции в производстве лекарственного средства для профилактики или лечения заболеваний, зависимых от половых гормонов, или для контрацепции. Группа изобретений обеспечивает значительное снижение начальной скорости высвобождения и улучшение профиля замедленного высвобождения лекарственного средства за счет предварительного добавления воды к исходному липидному препарату с замедленным высвобождением (композиции по настоящему изобретению), содержащему аналоги GnRH, позволяющие жидкому инъекционному составу образовывать жидкокристаллический гель сразу после введения, и также обеспечивает значительную безопасность. 4 н. и 16 з.п. ф-лы, 9 ил., 2 табл., 28 пр.
1. Композиция для профилактики или лечения заболеваний, зависимых от половых гормонов, или для контрацепции, содержащая:
а) сложный эфир сорбитана и ненасыщенной жирной кислоты, содержащий две или более -ОН (гидроксильных) групп в группе полярной головки;
b) фосфолипид;
c) отвердитель жидких кристаллов, который не содержит ионизированную группу, и гидрофобный фрагмент которого имеет триацильную группу, содержащую от 15 до 40 атомов углерода или структуру углеродного кольца;
d) воду; и
e) аналог гонадотропин-рилизинг-гормона (GnRH) в качестве фармацевтически активного вещества.
2. Композиция по п. 1, причем сложный эфир сорбитана и ненасыщенной жирной кислоты выбран из группы, состоящей из моноолеата сорбитана, монолинолеата сорбитана, монопальмитолеата сорбитана, мономиристолеата сорбитана, сесквиолеата сорбитана, сесквилинолеата сорбитана, сесквипальмитолеата сорбитана, сесквимиристолеата сорбитана, диолеата сорбитана, дилинолеата сорбитана, дипальмитолеата сорбитана, димиристолеата сорбитана и их смесей.
3. Композиция по п. 1, причем сложный эфир сорбитана и ненасыщенной жирной кислоты выбран из группы, состоящей из сорбитана моноолеата, сесквиолеата сорбитана, монолинолеата сорбитана, монопальмитолеата сорбитана, мономиристолеата сорбитана, сесквиолеата сорбитана и их смесей.
4. Композиция по п. 1, причем фосфолипид выбран из группы, состоящей из фосфатидилхолина, фосфатидилэтаноламина, фосфатидилсерина, фосфатидилглицерина, фосфатидилинозита, фосфатидной кислоты, сфингомиелина и их смесей, которые содержат от 4 до 30 насыщенных или ненасыщенных атомов углерода.
5. Композиция по п. 1, причем отвердитель жидких кристаллов выбран из группы, состоящей из триглицерида, ретинилпальмитата, ацетата токоферола, холестерина, бензилбензоата, убихинона и их смесей.
6. Композиция по п. 1, причем вода добавляется в виде, по меньшей мере, одного вещества, выбранного из группы, состоящей из воды для инъекций, дистиллированной воды и буфера.
7. Композиция по п. 1, причем аналогом GnRH является агонист GnRH или антагонист GnRH, где агонист GnRH выбран из группы, состоящей из лейпролида, гозерелина, трипторелина, нафарелина, бусерелина, гистрелина, деслорелина, метрелина, гонадрелина, их фармацевтически приемлемых солей и их смесей, и где антагонист GnRH выбран из группы, состоящей из дегареликса, абареликса, ганиреликса, цетрореликса, их фармацевтически приемлемые соли и их смесей.
8. Композиция по п. 1, причем массовое соотношение компонента а) и компонента b) составляет от 10:1 до 1:10.
9. Композиция по п. 1, причем массовое соотношение компонентов а) + b) и компонента c) составляет от 1000:1 до 1:1.
10. Композиция по п. 1, причем массовое соотношение компонентов а) + b) + c) и компонента d) составляет от 99:1 до 1:1.
11. Композиция по п. 1, причем массовое соотношение компонентов а) + b) + c) + d) и компонента e) составляет от 10000:1 до 1:1.
12. Композиция по п. 1, содержащая:
а) от 9 до 90% по массе сложного эфира сорбитана и ненасыщенной жирной кислоты, содержащего две или более -ОН (гидроксильных) групп в группе полярной головки;
b) от 9 до 90% по массе фосфолипида;
c) от 0,1 до 50% по массе отвердителя жидких кристаллов, который не содержит ионизированную группу, и гидрофобный фрагмент которого имеет триацильную группу, содержащую от 15 до 40 атомов углерода или структуру углеродного кольца;
d) от 0,5 до 50% по массе воды; и
e) от 0,01 до 50% по массе аналога гонадотропин-рилизинг-гормона (GnRH).
13. Композиция по п. 1, содержащая:
а) от 9 до 50% по массе сложного эфира сорбитана и ненасыщенной жирной кислоты, содержащего две или более -ОН (гидроксильные) группы в группе полярной головки;
b) от 18 до 60% по массе фосфолипида;
c) от 1 до 36% по массе отвердителя жидких кристаллов, который не имеет ионизированную группу, и гидрофобный фрагмент которого имеет триацильную группу, содержащую от 15 до 40 атомов углерода или структуру углеродного кольца;
d) от 0,5 до 10,5% по массе или от 25 до 37,5% по массе воды; и
e) от 0,1 до 45% по массе лейпролида или его фармацевтически приемлемых солей.
14. Композиция по п. 1, содержащая:
а) от 9 до 50% по массе сложного эфира сорбитана и ненасыщенной жирной кислоты, содержащего две или более -ОН (гидроксильные) группы в группе полярной головки;
b) от 18 до 60% по массе фосфолипида;
c) от 1 до 36% по массе отвердителя жидких кристаллов, который не имеет ионизированную группу, и гидрофобный фрагмент которого имеет триацильную группу, содержащую от 15 до 40 атомов углерода или структуру углеродного кольца;
d) от 0,5 до 10,5% по массе или от 25 до 37,5% по массе воды; и
e) от 0,1 до 45% по массе гозерелина или его фармацевтически приемлемых солей.
15. Композиция по п. 1, содержащая:
а) от 9 до 50% по массе сложного эфира сорбитана и ненасыщенной жирной кислоты, содержащего две или более -ОН (гидроксильные) группы в группе полярной головки;
b) от 18 до 60% по массе фосфолипида;
c) от 1 до 36% по массе отвердителя жидких кристаллов, который не имеет ионизированную группу, и гидрофобный фрагмент которого имеет триацильную группу, содержащую от 15 до 40 атомов углерода или структуру углеродного кольца;
d) от 0,5 до 10,5% по массе или от 25 до 37,5% по массе воды; и
e) от 0,1 до 45% по массе дегареликса или его фармацевтически приемлемых солей.
16. Способ профилактики или лечения заболеваний, зависимых от половых гормонов, или для контрацепции, включающий введение терапевтически эффективного количества композиции по п. 1 субъекту.
17. Способ по п. 16, причем заболеванием, зависимым от половых гормонов, является злокачественная опухоль предстательной железы, злокачественная опухоль молочной железы, злокачественная опухоль яичников, эндометриоз, миома матки, поликистоз яичников, преждевременное половое созревание, гирсутизм, гонадотропная аденома гипофиза, синдром апноэ во сне, синдром раздраженного кишечника, предменструальный синдром, доброкачественная гиперплазия предстательной железы или бесплодие.
18. Применение композиции по п. 1 для профилактики или лечения заболеваний, зависимых от половых гормонов, или для контрацепции.
19. Применение композиции по п. 1 в производстве лекарственного средства для профилактики или лечения заболеваний, зависимых от половых гормонов, или для контрацепции.
20. Применение композиции по п. 18 или 19, причем заболеванием, зависимым от половых гормонов, является злокачественная опухоль предстательной железы, злокачественная опухоль молочной железы, злокачественная опухоль яичников, эндометриоз, миома матки, поликистоз яичников, преждевременное половое созревание, гирсутизм, гонадотропная аденома гипофиза, синдром апноэ во сне, синдром раздраженного кишечника, предменструальный синдром, доброкачественная гиперплазия предстательной железы или бесплодие.
| ЛИПИДНЫЙ ПРЕКОНЦЕНТРАТ АНАЛОГОВ GnRH С ЗАМЕДЛЕННЫМ ВЫСВОБОЖДЕНИЕМ И СОДЕРЖАЩАЯ ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2013 |
|
RU2646487C2 |
| Установка для формования изделий из бетонных смесей | 1986 |
|
SU1404357A1 |
| WO 9847487 A1, 29.10.1998 | |||
| US 2012269772 A1, 25.10.2012 | |||
| US 2014348903 A1, 27.11.2014. | |||

