ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к новому типу соединения мочевины, способу его получения и содержащей его фармацевтической композиции, а также его применению в качестве ингибитора рецептора FMS-подобной тирозинкиназы третьего типа (FLT3), в частности в профилактике и(или) лечении злокачественного новообразования.
УРОВЕНЬ ТЕХНИКИ
Рак является одним из основных заболеваний, которые вызывают клиническую смерть у человека, особенно злокачественные опухоли, такие как рак легких, рак желудка, рак молочной железы, рак поджелудочной железы, рак печени, рак кишечника, рак пищевода и лейкемия, которые имеют чрезвычайно высокую смертность. Однако до настоящего времени не существует эффективных способов и лекарств для профилактики, лечения и искоренения рака. Таким образом, существует настоятельная потребность в высококачественных лекарствах и терапевтических способах против рака с хорошей специфичностью, высокой активностью, низкой токсичностью и отсутствием лекарственной устойчивости.
Лейкемия, также известная как рак крови, является клональной злокачественной болезнью гемопоэтических стволовых клеток. Поскольку лейкемические клетки теряют способность дифференцироваться в зрелые функциональные клетки крови и индуцируют злокачественную пролиферацию остановкой на разных этапах развития гемопоэтических клеток, они размножаются и накапливаются в костном мозге и других кроветворных тканях и проникают в другие органы и ткани, вызывая подавление нормального гематопоэза. Ее клинические проявления включают анемию, кровоизлияние, инфекцию и инфильтрацию различных органов и т.д. По частоте лейкемия занимает 6-е / 7-е место среди всех опухолей, особенно у детей и пожилых людей. Лейкемия представляет собой гетерогенную клеточную опухоль с множественными разновидностями, сложным патогенезом и различными клиническими особенностями. Некоторые лейкозы характеризуются быстрым началом, высокой смертностью, малым временем выживания, повышенной восприимчивостью к рецидиву, плохим прогнозом и являются трудноизлечимыми. Например, пятилетняя выживаемость для острого миелоидного лейкоза (АМЛ) у пациентов старше 60 лет составляет от 10 до 20%, а у пациентов в возрасте до 60 лет - от 40% до 50% (Dohner Н et al., Acute Myeloid Leukemia. N Engl J Med. 2015, 373 (12): 1136-52).
Несмотря на то, что рак крови можно лечить различными способами, такими как химиотерапия, лучевая терапия, иммунотерапия, таргетная терапия, индуцированная дифференцировочная терапия и трансплантация костного мозга/стволовых клеток, клиническая терапия для лечения АМЛ почти не изменилась за последние 40 лет, и в настоящее время ее стандартный способ лечения по-прежнему основан на индукции ремиссии, а именно «7+3» базовая терапия (даунорубицин 25-45 мг/м2, IV дни 1-3, цитарабин 100 мг/м2, IV дни 1-7, за исключением острого промиелоцитарного лейкоза APL). Хотя классические схемы химиотерапии могут эффективно вызывать ремиссию ОМЛ за короткий промежуток времени и подавлять или убивать раковые клетки, эти химиотерапевтические агенты имеют неблагоприятные побочные эффекты, слабую избирательность и легко вызывают рецидив и лекарственную устойчивость, в результате чего ОМЛ не может быть полностью излечим химиотерапией. Таргетная терапия и опухолевая иммунотерапия являются основным направлением развития для клинического лечения рака. Таргетная терапия обладает хорошей специфичностью, меньшим количеством побочных эффектов и очевидным лечебным эффектом. Многие целевые терапевтические препараты успешно применяются к различным видам рака, включая некоторые типы лейкемии, такие как Гливек для лечения CML. Однако до настоящего времени на мировой рынок не было введено никаких эффективных таргетных терапевтических препаратов, которые были одобрены для лечения ОМЛ. Поэтому существует настоятельная необходимость в большем количестве препаратов, лечащих ОМЛ в клинической практике.
В соответствии с анализом больших геномных данных по цитогенетике клинических образцов от пациентов с ОМЛ, частое появление мутаций генов является основной характеристикой ОМЛ. Например, частота генетических мутаций, относящихся к клеточным сигнальным путям, составляет около 50-60%, частота генетической аномалии, связанной с метилированием ДНК, составляет 44%, частота мутации генов хромосомной модификации составляет приблизительно 30%, частота аномалии гена миелоидного транскрипционного фактора составляют 20-25%, частота возникновения генов слияния транскрипционных факторов составляет приблизительно 18%, а частота мутации генов-супрессоров опухолей составляет 14%. У пациентов с ОМЛ наиболее распространенными генетическими аномалиями являются, например, FLT3-ITD (19%-28%), FLT3-TKD (5%-10%), NPM1 (частота мутации 27-35%), DNMTA (частота мутации 26%), NRAS (частота мутации 8%-9%)), ASXL1 (частота мутации 17%-19%), СЕВРА (частота мутации 4-6%), ТЕТ2 (частота мутации 8%-27%), WT1 (показатель аномалии гена 8%), IDH2 (точечная мутация 8%-9%), IDH1 (частота мутации 9%), KIT (частота мутации 2%-4%), RUNX1 (мутация частота 5-10%), MLL-PTD (5%), PHF6 (3%), KRAS (частота мутации 2-4%), ТР53 (частота мутации 2%-8%), EZH2 (частота мутации около 2%), JAK2 (частота мутации 1%-3%) (Coombs СС et al., Molecular therapy for acute myeloid leukaemia. Nat Rev Clin Oncol. 2016; 13, 305-318. Welch JS et al., The origin and evolution of mutations in acute myeloid leukemia. Cell. 2012; 150:264-278. Kandoth С et al., Mutational landscape and significance across 12 major cancer types. Nature 2013; 502(7471):333-339. Ding L et al., Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature. 2012; 481:506-510. Hanahan D et al., The hallmarks of cancer. Cell. 2000; 100: 57-70. The Cancer Genome Atlas Research Network Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013; 368:2059-2074).
Протеинкиназа представляет собой киназный фермент, который фосфорилирует белки и необходим для важных клеточных физиологических функций, таких как рост клеток, развитие, дифференцировка, метаболизм, старение и апоптоз. Существует два основных типа: трансмембранные протеинкиназы и цитозольные протеинкиназы. Аномалии протеинкиназ могут непосредственно приводить к клинически различным заболеваниям, таким как рак, воспаление, расстройствам иммунной системы и нервной системы, сердечно-сосудистым и цереброваскулярным заболеваниям. После десятилетий непрерывных усилий многие протеинкиназы, такие как EGFR, HER2 / 3/4, VEGFR, PDGFR, с-МЕТ, IGF-1R, FGFR, CSF-1R, TRK-рецепторы, эфриновые рецепторы, ТАМ-рецепторы, TIE-2, FLT-3, RET, ALK, BCR-ABL, JAKs, SRC, FAK, BTK, SYK, BLK, CDK, PI3K, MEK / RAS / RAF были идентифицированы как целевые белковые молекулы для различных заболеваний. Некоторые из этих ингибиторов протеинкиназ были успешно использованы в клинических применениях в качестве таргетных способов лечения и показали хорошие терапевтические эффекты, такие как BCR-ABL, EGFR / HER2, ALK, BTK, VEGFR, JAK и другие ингибиторы протеинкиназ. FMS-подобная тирозинкиназа 3 (FLT3), также известная как фетальная печеночная киназа-2 (FLK-2) или киназа-1 стволовых клеток человека (STK-1), относится к рецепторной тирозинкиназе типа III. Семейство киназ включает рецептор колониестимулирующего фактора 1 (CSF1R), тромбоцитарный рецептор фактора роста 1 (PDGFR α/β) и рецептор стволовых клеток TIP. При росте и развитии в нормальных физиологических условиях экспрессия гена FLT3 происходит главным образом при раннем развитии мозга, печени, плаценты, гонад и гемопоэтических клеток. Во время роста и развития миелоидных и лимфоидных стволовых клеток экспрессируется ген FLT3 и ген его лиганда FLT3-L. Когда FLT3-L связывается с FLT3, он индуцирует аутофосфорилирование белка FLT3 и активирует активность фермента FLT3 и его опосредованные нисходящие сигнальные пути, такие как PI3K, JAK / STAT и RAS, и участвует в биологических функциях, таких как рост клеток крови, развитие, пролиферация, и дифференцировка Drexler HG et al., FLT3: receptor and ligand. Growth Factors. 2004; 22(2)71-3. Review). Например, у мышей с дефицитом гена FLT3 число миелоидных и лимфоидных клеток-предшественников снижено. Однако, когда ген FLT3 аномально экспрессируется или мутирован, нормальные клетки крови становятся раковыми и развивается лейкемия. Например, около 30% пациентов с острой миелоидной лейкемией (АМЛ) проявляют мутации во внутреннем тандемном повторе (ITD, 19-28%) FLT3 и мутации доменов тирозинкиназы (TKD, 5%-10%). У пациентов с миелодиспластическим синдромом (МДС) с умеренными или серьезными рисками частота мутаций FLT3 составляет 2%; у пациентов с APL частота мутаций составляет менее 5%; заболеваемость для ALL составляет менее 1%, что в основном происходит в случаях с двойным фенотипом ALL.
Два вида мутаций FLT3 (FLT3-ITD и FLT3-TKD), включая двойную мутацию FLT3-ITD/FLT3-TKD, могут вызывать аутофосфорилирование белка FLT3, что приводит к независимой от FLT3 конститутивной активации лиганда и аномальной последующей трансдукции сигнала, тем самым способствуя злокачественной пролиферации лейкемических клеток и ингибированию нормального клеточного апоптоза. Конститутивная активная мутация FLT3-тирозинкиназы является одной из основных мутаций при ОМЛ и одной из основных причин ОМЛ. Поскольку клоны FLT3-ITD обладают селективными преимуществами роста, трудно вылечить этот тип лейкемии, используя только одни химиотерапевтические препараты. Кроме того, пациенты с этим типом лейкемии имеют более высокую толерантность к химиотерапевтическим препаратам и имеют плохой клинический прогноз. Пациенты склонны к развитию резистентности к химиотерапевтическим агентам и рецидиву, поэтому мутация активации тирозинкиназы FLT3 стала важной мишенью для целевой терапии ОМЛ (Gilliland DG, Griffin JD. The roles of FLT3 in hematopoiesis and leukemia. Blood. 2002; 100(5):1532-1542. Kiyoi H et al., Internal tandem duplication of the FLT3 gene is a novel modality of elongation mutation which causes constitutive activation of the product. Leukemia. 1998; 12(9): 1333-1337).
В области исследований ОМЛ в отношении разработки лекарств всегда больше всего интересовали ингибиторы FLT3-ITD и FLT3-TKD. На сегодняшний день доклинические исследования показали, что почти 100 различных типов низкомолекулярных соединений могут селективно/неселективно ингибировать или частично ингибировать активность протеинкиназы FLT3 и пролиферацию клеток in vitro, а также рост опухоли ксенотрансплантата in vivo с помощью экспресси мутанта FLT3 у положительных лейкозных клеток или пациентов с лейкемией. Некоторые из этих соединений участвуют в различных этапах клинических испытаний, такие как СЕР701, CHIR-258, PKC412, MLN-518, сунитиниб, АС220, XL-999, сорафениб, понатиниб, креноланиб, ASP 2215, AKN-028, TAK-659, Е6201, кабозантиниб, PLX 3387 и FLX 925 (Smith BD et al., Single-agent CEP-701, a novel FLT3 inhibitor, shows biologic and clinical activity in patients with relapsed or refractory acute myeloid leukemia. Blood. 2004; 103 (10):3669-76; Lopes de Menezes DE et al., CHIR-258: a potent inhibitor of FLT3 kinase in experimental tumor xenograft models of human acute myelogenous leukemia. Clin Cancer Res. 2005; 11(14):5281-91; Weisberg E et al., Inhibition of mutant FLT3 receptors in leukemia cells by the small molecule tyrosine kinase inhibitor PKC412. Cancer Cell. 2002; I (5):433-43; Zarrinkar PP et al., AC220 is a uniquely potent and selective inhibitor of FLT3 для лечения of acute myeloid leukemia (АМЛ). Blood. 2009; 114(14):2984-92; Kelly LM et al., CT53518, a novel selective FLT3 antagonist for the treatment of acute myelogenous leukemia (АМЛ). Cancer Cell. 2002; 1(5):421-32; Griswold IJ et al., Effects of MLN518, a dual FLT3 and KIT inhibitor, on normal and malignant hematopoiesis. Blood. 2004; 104(9):2912-8; Smith CC et al., Crenolanib is a selective type I pan-FLT3 inhibitor. Proc Natl Acad Sci USA. 2014; 111(14):5319-24; Zimmerman El et al., Crenolanib is active against models of drug-resistant FLT3-ITD-positive acute myeloid leukemia. Blood. 2013; 122 (22):3607-15; Safaian NN et al., Sorafenib (Nexavar) induces molecular remission and regression of extramedullary disease in a patient with FLT3-ITD+ acute myeloid leukemia. Leuk Res. 2009; 33 (2):348-50; Zhang W et al., Mutant FLT3: a direct target of sorafenib in acute myelogenous leukemia. J Natl Cancer Inst. 2008; 100(3): 184-98). Несмотря на то, что некоторые ингибиторы FLT3 демонстрируют обнадеживающие результаты на ранних стадиях клинических испытаний у пациентов с ОМЛ, демонстрирующих улучшение, большинство соединений не продемонстрировали ожидаемых клинических эффектов на более поздних стадиях клинических испытаний при использовании в качестве монотерапии или комбинированном лечение пациентов с ОМЛ, и в настоящее время ни один селективный ингибитор FLT3 не был одобрен для клинического лечения ОМЛ где-либо в мире.
Для характеристики болезни ОМЛ, обобщая и анализируя данные доклинических и клинических испытаний для существующих ингибиторов FLT3, мы обнаружили, что большинство ингибиторов FLT3 испытывают множество проблем, которые ограничивают их клинические эффекты. Эти проблемы включают: (1) серьезные побочные эффекты; (2) влияние на рост и развитие нормальных клеток крови и снижение иммунитета пациентов; (3) приобретение лекарственной устойчивости; (4) синдром лизиса опухоли; (5) низкая частота ответа пациента и (6) легко рецидивировать. Такие проблемы в основном связаны с селективностью, активностью, метаболизмом in vivo, токсичностью и эффективностью соединений. (Kadia ТМ et al., New Drugs in Acute Myeloid Leukemia (AML). Ann Oncol. 2016; 27 (5): 77-8. Stein EM et al., Emerging therapeutic drugs for AML. Blood. 2016; 127 (I):71-8. Stein EM, Molecularly targeted therapies for acute myeloid leukemia. Hematology Am Soc Hematol Educ Program. 2015; (I):579-83).
В настоящее время три линии клеток человеческого лейкоза MV4-11, MOLM-13 и MOLM-14 широко используются для исследований клеток, позитивных в отношении экспрессии FLT3-ITD в мире. Клетки MV4-11 содержат гомозиготную мутацию (+/+) FLT3 ITD и относятся к острому лимфобластному мононуклеарному лейкозу человека. MOLM-13 и MOLM-14 являются сестринскими клеточными линиями от одного и того же пациента и содержат гетерозиготную мутацию FLT3 ITD / WT (+/-), которая объясняется острой миелоидной лейкемией человека (Quentmeier Н et al., FLT3 mutations in acute myeloid leukemia cell lines. Leukemia. 2003; 17(1): 120-124). Многие исследования показали, что целевое ингибирование FLT3-ITD может эффективно ингибировать рост этих трех видов лейкемических клеток in vivo и in vitro. Эти клеточные линии, в частности MV4-11, стали распространенными клеточными моделями для скрининга и идентификации селективных ингибиторов FLT3-ITD.
В настоящем изобретении с использованием (1) клеточных линий MV4-11 и MOLM-13, позитивных в отношении экспрессии FLT3-ITD, (2) клеточной линии дикого типа RS4 11 с высокой экспрессией FLT3-гена, (3) других клинически распространенных онкоген- экспрессирующих клеточных линии лейкоза, а также различные типов клеточных линий солидных опухолей в качестве моделей клеток, автор(ы) разработали соединения нового типа с высокой активностью, высокой селективностью, хорошими фармакологическими эффектами и фармакокинетическими свойствами, а также слабыми побочными эффектами, которые могут быть использованы для лечения и/или профилактики рака, особенно лейкемии, в качестве эффективного избирательного ингибитора тирозинкиназы FLT3 (в частности, мутации активации FLT3).
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ
Настоящее изобретение относится к новому типу соединения мочевины, которое может эффективно ингибировать in vitro рост мутантных FLT3-ITD клеточных линий лейкемии MV4-11 и MOLM-13 и индуцировать клеточный апоптоз. GI50 находится в субнаномолярном диапазоне, без очевидного ингибирующего эффекта на рост линий раковых клеток с FLT3 дикого типа с высокой, нормальной или отсутствующей экспрессией. Кроме того, соединения настоящего изобретения, могут эффективно ингибировать рост опухолей у животных с ксенотрансплантатом MV4-11 клеток лейкемии FLT3-ITD эффективно, быстро и зависимым от концентрации образом. Дальнейшие фармакологические и фармакокинетические исследования показали, что соединения настоящего изобретения обладают хорошими фармацевтическими свойствами у крыс.
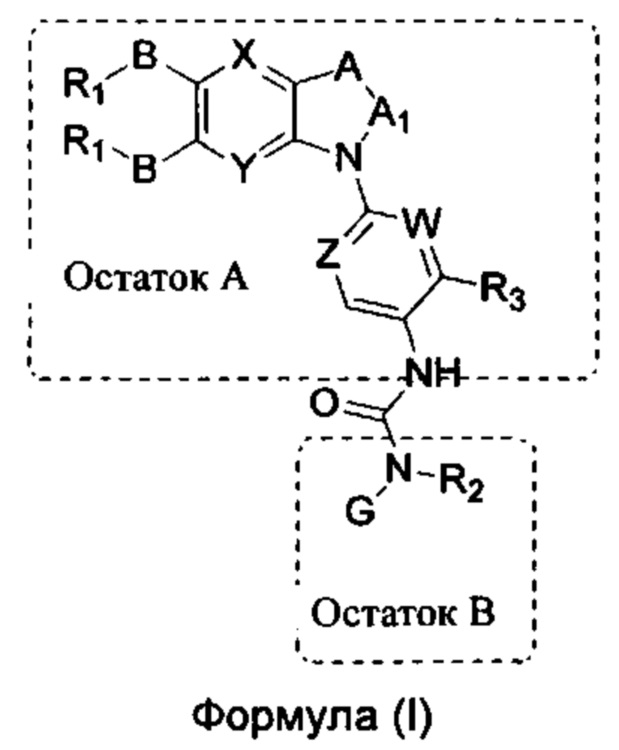
Таким образом, в настоящем изобретении предложено соединение общей формулы (I) или его фармацевтически приемлемые соли, сольваты или пролекарства,
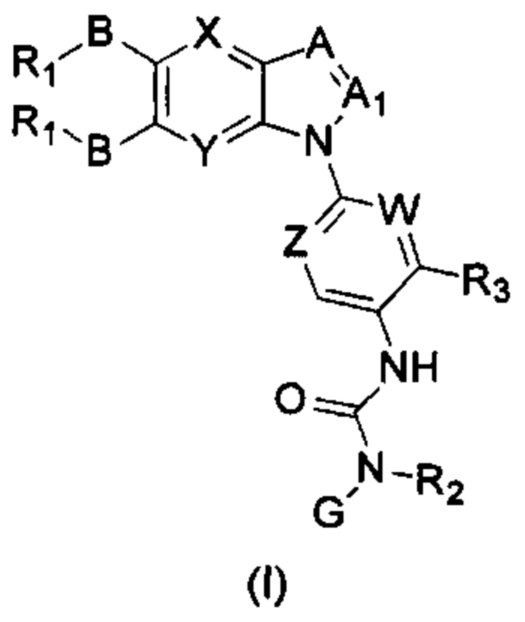
Где:
X и Y каждый независимо выбран из N и С-BR1;
A and A1 каждый независимо выбран из N и С-BR1;
W and Z каждый независимо выбран из N и С-BR1;
при отсутствии R1, В является идентичным или разным и каждый независимо выбран из группы, состоящей из водорода, галогена, алкила, алкенила, алкинила, циано, циклоалкила, гетероциклила, арила, или гетероарила, указанный алкил, алкенил, алкинил, циклоалкил, гетероциклил, арил, или гетероарил возможно дополнительно замещены одной или более групп Q;
в присутствии R1, В является идентичным или разным и каждый независимо выбран из группы, состоящей из -О- и -NR4-; и R1 является идентичным или разным и каждый независимо выбран из группы, состоящей из водорода, алкила, алкенила, алкинила, циклоалкила, гетероциклила, арила, гетероарила, -RuORx, -RuC(О)ORx, -RuN(Ry)(Rz), -C(O)N(Ry)(Rz), -RuS(O)nN(Ry)(Rz), -RuS(O)nRx; указанные группы алкил, алкенил, алкинил, циклоалкил, гетероциклил, арил, гетероарил возможно дополнительно замещены одной или более группами, выбранными из группы, состоящей из галогена, циано, гидрокси, алкила, алкокси, гидроксиалкила, гидроксиалкокси, амидо, циклоалкила, гетероциклила, арила, галоарила, гетероарила, циклоалкил-гетероарила; R4 выбран из группы, состоящей из водорода, алкила, алкенила, и алкинила, или R4 и R1 вместе с атомом азота, к которому они присоединены, образуют гетероциклильную или гетероарильную группу, и гетероциклильная или гетероарильная группа возможно дополнительно замещена одной или более группой, выбранной из группы, состоящей из галогена, алкила, галоалкила, алкокси, и галоалкокси;
где R2 представляет собой водород, G выбран из группы, состоящей из арила, гетероарила, и гетероциклила, и арил, гетероарил, или гетероциклил возможно дополнительно замещен одной или более группой, выбранной из группы, состоящей из галогена, алкила, алкенила, алкинила, алкокси, гидрокси, амино, ацила, циклоалкила, гетероциклила, арила, и гетероарила, где указанный алкил, алкенил, алкинил, алкокси, ацил, циклоалкил, гетероциклил, арил, или гетероарил возможно дополнительно замещен одной или более группой, выбранной из группы, состоящей из галогена, алкила, галоалкила, алкенила, алкинила, арила, гидрокси, алкокси, гало алкокси, циклоалкила, эфира и циано; или,
когда R2 не является водородом, G и R2 вместе с атомом азота, к которому они присоединены, образуют гетероциклильную или гетероарильную группу, гетероциклильная или гетероарильная группа возможно дополнительно замещена одной или более группой, выбранной из группы, состоящей из галогена, алкила, алкенила, алкинила, алкокси, гидрокси, амино, циклоалкила, гетероциклила, арила, и гетероарила, где указанный алкил, алкенил, алкинил, алкокси, циклоалкил, гетероциклил, арил, или гетероарил возможно дополнительно замещен одной или более группой, выбранной из группы, состоящей из галогена, алкила, галоалкила, алкенила, алкинила, арила, гидрокси, алкокси, галоалкокси, циклоалкила, эфира и циано;
R3 выбран из группы Q;
Ru выбран из связи, алкилена, алкенилена, или алкинилена;
Rx выбран из водорода, алкила, гидроксиалкила, галоалкила, алкенила, или алкинила; или
кислород в -RuORx- вместе с Ru и Rx, присоединенными к нему, образуют кислород-содержащее 3-7 членное гетероциклическое кольцо, которое возможно замещено одной или более группами Q;
Ry и Rz каждый независимо выбран из группы, состоящей из водорода, алкила, алкокси, алкенила, алкинила, циклоалкила, и галоалкила; или,
Ry и Rz вместе с атомом азота, к которому они присоединены, образуют гетероциклильную или гетероарильную группу, и гетероциклильная или гетероарильная группа возможно дополнительно замещена одной или более группой, выбранной из группы, состоящей из галогена, галоалкила, алкила, алкенила и алкинила;
Q выбран из группы, состоящей из водорода, галогена, гидрокси, амино, алкила, алкокси, циклоалкила, алкенила, алкинила, циано, арила, гетероциклила и гетероарила, и указанные амино, алкил, алкокси, циклоалкил, алкенил, алкинил, арил, гетероциклил, или гетероарил возможно дополнительно замещены одной или более группой, выбранной из группы, состоящей из гидрокси, галогена и алкила;
n представляет собой целое число от 0 до 2.
В предпочтительном воплощении настоящего изобретения, соединение общей формулы (I) или его фармацевтически приемлемая соль, сольват или пролекарство в соответствии с настоящим изобретением,
где:
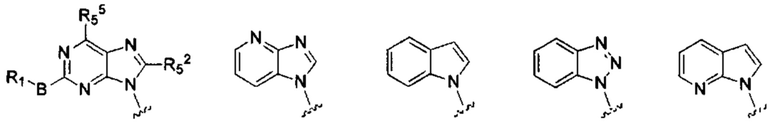
X, Y, А, и А1 выбраны из следующих структур:
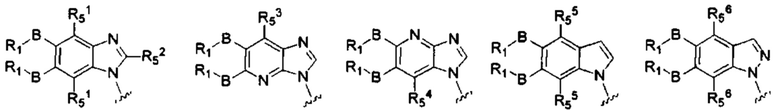
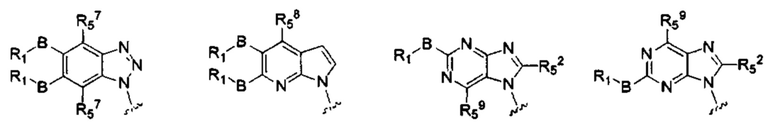
R51, R52, R53, R54, R55, R56, R57, R58 и R59 каждый независимо выбран из группы, состоящей из водорода, галогена, гидрокси, алкила, алкокси, алкенила, алкинила, -N(Ry)(Rz), циклоалкила, гетероциклила, арила, и гетероарила, где алкил, алкокси, алкенил, алкинил, -N(Ry)(Rz), циклоалкил, гетероциклил, арил или гетероарил возможно дополнительно замещены одной или более группами, выбранными из группы, состоящей из галогена, гидрокси, алкила, алкокси, галоалкила, галоалкокси, циклоалкила, эфирных групп;
RI, В, Ry, Rz являются такими, как определено в общей формуле (I).
В другом предпочтительном воплощении настоящего изобретения, соединение общей формулы (I) или его фармацевтически приемлемая соль, сольват или пролекарство в соответствии с настоящим изобретением,
где:
X, Y, А, и А1 выбраны из следующих структур:
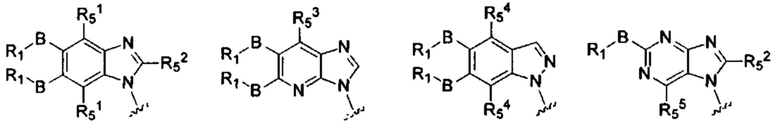
R51, R52, R53, R54, R55 каждый независимо выбран из группы, состоящей из водорода, галогена, алкила, алкокси, -N(Ry)(Rz), галоалкила и галоалкокси.
RI, В, Ry, Rz являются такими, как определено в общей формуле (I).
В другом предпочтительном воплощении настоящего изобретения, соединение общей формулы (I) или его фармацевтически приемлемая соль, сольват или пролекарство в соответствии с настоящим изобретением,
где:
X, Y, А, и А1 выбраны из следующих структур:
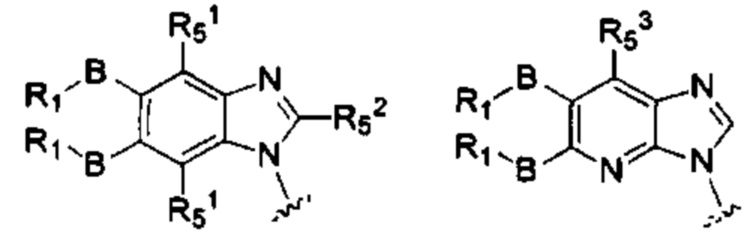
R51, R52, R53 каждый независимо выбран из группы, состоящей из водорода, галогена, алкила, алкокси, -N(Ry)(Rz), галоалкила, галоалкокси;
RI, В, Ry, Rz являются такими, как определено в общей формуле (I).
В другом предпочтительном воплощении настоящего изобретения, соединение общей формулы (I) или его фармацевтически приемлемая соль, сольват или пролекарство в соответствии с настоящим изобретением,
где,
R51, R52, R53 каждый независимо выбран из группы, состоящей из водорода, галогена, С1-С6 алкила, C1-C6 алкокси, -N(Ry)(Rz), С1-С6 галоалкила, С1-С6 галоалкокси, где Ry и Rz каждый независимо выбран из водорода и С1-С6 алкила.
В другом предпочтительном воплощении настоящего изобретения, соединение общей формулы (I) или его фармацевтически приемлемая соль, сольват или пролекарство в соответствии с настоящим изобретением, где W и Z выбраны из следующих четырех вариантов:
a) W и Z представляют собой CQ;
b) W и Z представляют собой N;
С) W представляет собой CQ и Z представляет собой N;
d) Z представляет собой CQ и W представляет собой N;
где, Q выбран из группы, состоящей из водорода, галогена, гидрокси, амино, циано, С1-С6 алкила, С1-С6 галоалкила, С1-С6 алкокси, С1-С6 галоалкокси, С3-С7 циклоалкила, С5-С7 арила, 5- до 7-членной гетероциклильной группы, и 5- до 7-членной гетероарильной группы.
В другом предпочтительном воплощении настоящего изобретения, соединение общей формулы (I) или его фармацевтически приемлемая соль, сольват или пролекарство в соответствии с настоящим изобретением где,
при отсутствии RI, В является идентичным или разным и каждый независимо выбран из группы, состоящей из водорода, галогена, С1-С6 алкила, С1-С6 алкокси, С1-С6 галоалкила и С1-С6 галоалкокси.
В другом предпочтительном воплощении настоящего изобретения, соединение общей формулы (I) или его фармацевтически приемлемая соль, сольват или пролекарство в соответствии с настоящим изобретением где,
при отсутствии RI, В выбран из группы, состоящей из водорода, галогена, С1-С6 алкила, С1-С6 галоалкила;
в присутствии RI, В является идентичным или разным и каждый независимо выбран из -О- и -NR4-, предпочтительно -О-; и RI является идентичным или разным и каждый независимо выбран из группы, состоящей из водорода, С1-С10 алкила, и указанный алкил возможно дополнительно замещен одной или более группой, выбранной из группы, состоящей из галогена, циано, гидрокси, С1-С6 алкокси, 4- до 6-членной гетероциклильной группы, С5-С7 арила, С5-С7 галоарила, 5- до 7-членной гетероарильной группы, С3-С6 циклоалкила, 5- или 7-членной гетероарильной группы, где 4- до 6-членная гетероциклильная группа предпочтительно выбрана из 4- до 6-членной гетероциклильной группы, содержащей кислород или азот; и указанные С5-С7 арил или С5-С7 галоарил предпочтительно представляют собой фенил или галофенил;
R4 является таким, как определено в общей формуле (I).
В другом предпочтительном воплощении настоящего изобретения, соединение общей формулы (I) или его фармацевтически приемлемая соль, сольват или пролекарство в соответствии с настоящим изобретением где,
при отсутствии RI, В выбран из группы, состоящей из водорода, галогена, С1-С6 алкила, C1-C6 галоалкила;
в присутствии RI, В является идентичным или разным и каждый независимо выбран из -О- и -NR4-, предпочтительно -О-; и RI является идентичным или разным и каждый независимо выбран из -RuORx-, где Ru выбран из C1-C6 алкилена, Rx выбран из водорода, C1-C6 алкила, C1-C6 гидроксиалкила, и C1-C6 галоалкила;
R4 является таким, как определено в общей формуле (I).
В другом предпочтительном воплощении настоящего изобретения, соединение общей формулы (I) или его фармацевтически приемлемая соль, сольват или пролекарство в соответствии с настоящим изобретением где,
при отсутствии RI, В выбран из группы, состоящей из водорода, галогена, C1-C6 алкила, С1-С6 галоалкила;
в присутствии RI, В является идентичным или разным и каждый независимо выбран из -О- и -NR4-, предпочтительно -О-; и RI является идентичным или разным и каждый независимо выбран из -C(O)N(Ry)(Rz), где Ry и Rz каждый независимо выбран из группы, состоящей из водорода, С1-С6 алкила, С1-С6 алкокси, C1-C6 галоалкила, С1-С6 галоалкокси, С3-С7 циклоалкила; или,
Ry и Rz вместе с атомом азота, к которому они присоединены, образуют 5- до 7-членную гетероциклильную группу или 5- до 7-членную гетероарильную группу, предпочтительно 6-членную гетероциклильную группу или 6-членную гетероарильную группу, более предпочтительно морфолинил, пиперидинил, пиперазинил, пиридинил, пиримидил, указанные 5- до 7-членная гетероциклильная группа или 5- до 7-членная гетероарильная группа возможно дополнительно замещена одной или более группой, выбранной из группы, состоящей из галогена, С1-С6 алкила, C1-C6 галоалкила;
R4 является таким, как определено в общей формуле (I).
В другом предпочтительном воплощении настоящего изобретения, соединение общей формулы (I) или его фармацевтически приемлемая соль, сольват или пролекарство в соответствии с настоящим изобретением, где,
при отсутствии RI, В выбран из группы, состоящей из водорода, галогена, C1-C6 алкила, С1-С6 галоалкила;
в присутствии RI, В является идентичным или разным и каждый независимо выбран из -О- или -NR4-, предпочтительно -О-; и RI является идентичным или разным и каждый независимо выбран из -RuN(Ry)(Rz), где Ru выбран из С1-С6 алкилена; Ry и Rz каждый независимо выбран из группы, состоящей из водорода, С1-С6 алкила, С1-С6 алкокси, С1-С6 галоалкила, С1-С6 галоалкокси, С3-С7 циклоалкила; или,
Ry и Rz вместе с атомом азота, к которому они присоединены, образуют 5- до 7-членную гетероциклильную группу или 5- до 7-членную гетероарильную группу, предпочтительно морфолинил, пиперидинил, пиперазинил, азепанил, пиридил, пиримидинил, и 5- до 7-членная гетероциклильная группа или 5- до 7-членная гетероарильная группа возможно дополнительно замещена одной или более группой, выбранной из группы, состоящей из галогена, С1-С6 алкила, и С1-С6 галоалкила;
R4 является таким, как определено в общей формуле (I).
В другом предпочтительном воплощении настоящего изобретения, соединение общей формулы (I) или его фармацевтически приемлемая соль, сольват или пролекарство в соответствии с настоящим изобретением где,
при отсутствии RI, В выбран из группы, состоящей из водорода, галогена, С1-С6 алкила, С1-С6 галоалкила;
в присутствии RI, В является идентичным или разным и каждый независимо выбран из -О- или -NR4-, предпочтительно -О-; и RI является идентичным или разным и каждый независимо выбран из -RuC(O)ORx, где, Ru выбран из C1-C6 алкилена; Rx выбран из группы, состоящей из водорода, C1-C6 алкила, C1-C6 гидроксиалкила, С1-С6 галоалкила;
R4 является таким, как определено в общей формуле (I).
В другом предпочтительном воплощении настоящего изобретения, соединение общей формулы (I) или его фармацевтически приемлемая соль, сольват или пролекарство в соответствии с настоящим изобретением где,
при отсутствии RI, В выбран из группы, состоящей из водорода, галогена, C1-C6 алкила, C1-C6 галоалкила;
в присутствии RI, В является идентичным или разным и каждый независимо выбран из -О- или -NR4-, предпочтительно -О-; и RI является идентичным или разным и каждый независимо выбран из 5- до 7-членного арила или 5- до 7-членной гетероарильной группы, предпочтительно тиадиазолила, пиразолила, оксазолила, оксадиазолила, имидазолила, триазолила, тиазолила, фурила, тиенила, пиридила, пирролила, N-алкилпирролила, пиримидила, пиразинила, имидазолила, тетразолила, фенила, пиридила, пиримидинила, и 5- до 7-членная арильная или 5-до 7-членная гетероарильная группа возможно дополнительно замещена одной или более группой, выбранной из группы, состоящей из С3-С6 циклоалкильных групп, 5-до 7-членных гетероциклильных групп, и амидогруппы;
R4 является таким, как определено в общей формуле (I).
В другом предпочтительном воплощении настоящего изобретения, соединение общей формулы (I) или его фармацевтически приемлемая соль, сольват или пролекарство в соответствии с настоящим изобретением где,
R4 выбран из водорода и С1-С6 алкила, или
R4 и R1 вместе с атомом азота, к которому они присоединены, образуют 5-до 7-членную гетероциклильную группу или 5- до 7-членную гетероарильную группу, предпочтительно пиперидинил, пиперазинил, морфолинил, пиридил, пиримидинил, и указанные 5- до 7-членная гетероциклильная группа или 5- до 7-членная гетероарильная группа возможно дополнительно замещена одной или более группой, выбранной из группы, состоящей из галогена, C1-C6 алкила, С1-С6 галоалкила, С1-С6 алкокси, C1-С6 галоалкокси.
В другом предпочтительном воплощении настоящего изобретения, соединение общей формулы (I) или его фармацевтически приемлемая соль, сольват или пролекарство в соответствии с настоящим изобретением где,
когда R2 представляет собой водород, G выбран из С5-С7 арила, 5- до 7-членной гетероарильной группы или 5- до 7-членной гетероциклильной группы, предпочтительно,
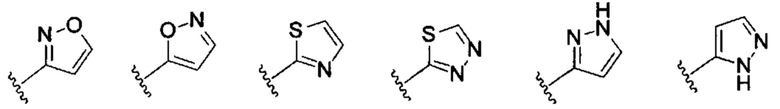
Укзанные С5-С7 арил, 5- до 7-членная гетероарильная группа или 5- до 7-членная гетероциклильная группа возможно дополнительно замещена одной или более группой, выбранной из группы, состоящей из галогена, C1-C6 алкила, C1-C6 галоалкила, C1-C6 алкокси, C1-C6 галоалкокси, гидрокси, амино, ацила, C3-C7 циклоалкила, 5- до 7-членной гетероциклильной группы, C5-C7 арила, 5- до 7-членной гетероарильной группы; C1-C6 алкил, C1-C6 алкоксил, C3-C7 циклоалкил, 5- до 7-членная гетероциклильная группа, C5-C7 арил, или 5- до 7-членная гетероарильная группа возможно дополнительно замещена одной или более группой, выбранной из группы, состоящей из галогена, гидрокси, C1-C6 алкила, C1-C6 алкокси, C1-C6 галоалкила, C1-C6 галоалкокси, эфира и циано; или
Когда R2 не является водородом, G и R2 вместе с атомом азота, к которому они присоединены, образуют 5- до 7-членную гетероциклильную группу или 5- до 7-членную гетероарильную группу, предпочтительно пирролил, пиразолил, имидазолил; указанные 5- до 7-членная гетероциклильная группа или 5- до 7-членная гетероарильная группа возможно дополнительно замещена одной или более группой, выбранной из группы, состоящей из галогена, C1-C6 алкила, C1-C6 галоалкила, C1-C6 алкокси, C1-C6 галоалкокси, гидрокси, амино, C3-C7 циклоалкила, 5- до 7-членной гетероциклильной группы, C5-C7 арильной группы и 5- до 7-членной гетероарильной группы.
В другом предпочтительном воплощении настоящего изобретения, соединение общей формулы (I) или его фармацевтически приемлемая соль, сольват или пролекарство в соответствии с настоящим изобретением где,
R3 выбран из группы, состоящей из водорода, галогена, гидрокси, амино, C1-C6 алкила, C1-C6 галоалкила, C1-C6 алкокси, C1-C6 галоалкокси, C3-C7 циклоалкила, циано, C5-C7 арила, 5- до 7-членной гетероциклильной группы или 5-до 7-членной гетероарильной группы.
Типичные соединения общей формулы (I) настоящего изобретения включают, без ограничения:
1-(4-бензимидазол-1-ил-фенил)3-изоксазол-3-ил-мочевина;
1-(4-бензимидазол-1-ил-фенил)-3-(5-метил-изоксазол-3-ил)-мочевина;
1-(4-бензимидазол-1-ил-фенил)-3-(5-трет-бутил-изоксазол-3-ил)-мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(5-гидрокси-бензимидазол-1-ил)-фенил]-мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(5-метоксил-бензимидазол-1-ил)-фенил]-мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(5-этоксил-бензимидазол-1-ил)-фенил]-мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(5-гексилоксил-бензимидазол-1-ил)-фенил]-мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(5-изопропокси-бензимидазол-1-ил)-фенил]-мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-{4-[5-(3-метил-оксетан-3-илметокси)-бензимидазол-1-ил]-фенил}-мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-{4-[5-(тетрагидрофуран-2-илметокси)-бензимидазол-1-ил]-фенил}-мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-{4-[5-(2-гидрокси-этоксил)-бензимидазол-1-ил]-фенил]мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-{4-[5-(2-метоксил-этоксил)-бензимидазол-1-ил]-фенил]мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-{4-[5-(2-этоксил-этоксил)-бензимидазол-1-ил]-фенил]мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-(4-{5-[2-(2-гидрокси-этоксил)-этоксил]-бензимидазол-1-ил}-фенил)-мочевина;
1-{4-[3-(5-трет-бутил-изоксазол-3-ил)-карбамидо]-фенил}-1Н-бензимидазол-5-илморфолин-4-карбоксиликат;
1-(5-трет-бутил-изоксазол-3-ил)-3-{4-[5-(2-морфолин-4-ил-этоксил)-бензимидазол-1-ил]-фенил}-мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-{4-[5-(2-пиперидин-1-ил-этоксил)-бензимидазол-1-ил]-фенил}-мочевина;
1-{4-[5-(2-азациклогептан-1-ил-этоксил)-бензимидазол-1-ил]-фенил}-3-(5-трет-бутил-изоксазол-3-ил)-мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-(4-{5-[3-(4-метил-пиперазин-1-ил)-пропоксил]-бензимидазол-1-ил}-фенил)-мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-{4-[5-(2-диметиламино-этоксил)-бензимидазол-1-ил]-фенил}-мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(5-трифторметокси-бензимидазол-1-ил)-фенил]-ure;
1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(5-фтор-бензимидазол-1-ил)-фенил]-мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(5-трифторметил-бензимидазол-1-ил)-фенил]-мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(5-метил-бензимидазол-1-ил)-фенил]-мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(6-метоксил-бензимидазол-1-ил)-фенил]-мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-{4-[6-(2-метоксил-этоксил)-бензимидазол-1-ил]-фенил]мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-{4-[6-(2-морфолин-4-ил-этоксил)-бензимидазол-1-ил]-фенил}-мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-(4-{6-[(2-диметиламино-этил)-метил-амино]-бензимидазол-1-ил}-фенил)-мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-{4-[6-(4-метил-пиперазин-1-ил)-бензимидазол-1-ил]-фенил}-мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-{4-[7-(2-морфолин-4-ил-этоксил)-бензимидазол-1-ил]-фенил}-мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(5,6-диметоксил-бензимидазол-1-ил)-фенил]-мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(5,6-диметил-бензимидазол-1-ил)-фенил]-мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(5-фтор-7-метил-бензимидазол-1-ил)-фенил]-мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(4-фтор-бензимидазол-1-ил)-фенил]-мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(2-метил-бензимидазол-1-ил)-фенил]-мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(2-хлоробензимидазол-1-ил)-фенил]-мочевина;
1-(4-бензимидазол-1-ил-3-метил-фенил)-3-(5-трет-бутил-изоксазол-3-ил)-мочевина;
1-(4-бензимидазол-1-ил-3-хлор-фенил)-3-(5-трет-бутил-изоксазол-3-ил)-мочевина;
1-(4-бензимидазол-1-ил-3-фтор-фенил)-3-(5-трет-бутил-изоксазол-3-ил)-мочевина;
1-(4-бензимидазол-1-ил-3,5-дифтор-фенил)-3-(5-трет-бутил-изоксазол-3-ил)-мочевина;
1-(4-бензимидазол-1-ил-2-хлор-фенил)-3-(5-трет-бутил-изоксазол-3-ил)-мочевина;
1-(6-бензимидазол-1-ил-пиридин-3-ил)-3-(5-трет-бутил-изоксазол-3-ил)-мочевина;
1-(2-бензимидазол-1-ил-пиримидин-5-ил)-3-(5-трет-бутил-изоксазол-3-ил)-мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-(4-индол-1-ил-фенил)-мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-(4-индазол-1-ил-фенил)-мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(6-фтор-индазол-1-ил)-фенил]-мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(7-фтор-индазол-1-ил)-фенил]-мочевина;
1-(4-бензотриазол-1-ил-фенил)-3-(5-трет-бутил-изоксазол-3-ил)-мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-(4-пирроло[2,3-b]пиридин-1-ил-фенил)-мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-(4-имидазо[4,5-b]пиридин-3-ил-фенил)-мочевина;
1-[4-(6-бромо-имидазо[4,5-b]пиридин-3-ил)-фенил]-3-(5-трет-бутил-изоксазол-3-ил)-мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(6-трифторметил-имидазо[4,5-b]пиридин-3-ил)-фенил]-мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(5-хлороимидазо[4,5-b]пиридин-3-ил)-фенил]-мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(5-метилимидазо[4,5-b]пиридин-3-ил)-фенил]-мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(7-метилимидазо[4,5-b]пиридин-3-ил)-фенил]-мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-(4-имидазо[4,5-b]пиридин-1-ил-фенил)-мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(6-метоксил-пурин-7-ил)-фенил]-мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(6-метоксил-пурин-9-ил)-фенил]-мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(6-диметиламино-пурин-7-ил)-фенил]-мочевина;
1-(4-бензимидазол-1-ил-фенил)-3-тиазол-2-ил-мочевина;
1-(4-бензимидазол-1-ил-фенил)-3-(4-метил-тиазол-2-ил)-мочевина;
1-(4-бензимидазол-1-ил-фенил)-3-[1,3,4]тиадиазол-2-ил-мочевина;
1-(4-бензимидазол-1-ил-фенил)-3-(5-трет-бутил-[1,3,4]тиадиазол-2-ил)-мочевина;
1-(4-бензимидазол-1-ил-фенил)-3-(5-метил-1Н-пиразол-3-ил)-мочевина;
1-(4-бензимидазол-1-ил-фенил)-3-(5-фенил-1Н-пиразол-3-ил)-мочевина;
1-(4-бензимидазол-1-ил-фенил)-3-(5-циклопропил-2Н-пиразол-3-ил)-мочевина;
1-(4-бензимидазол-1-ил-фенил)-3-(5-трифторметил-2Н-пиразол-3-ил)-мочевина;
1-(4-бензимидазол-1-ил-фенил)-3-(5-трет-бутил-2Н-пиразол-3-ил)-мочевина;
1-(5-трет-бутил-2Н-пиразол-3-ил)-3-[4-(5-фтор-бензимидазол-1-ил)-фенил]-мочевина;
1-(5-трет-бутил-2Н-пиразол-3-ил)-3-[4-(5-трифторметил-бензимидазол-1-ил)-фенил]-мочевина;
1-(5-трет-бутил-2Н-пиразол-3-ил)-3-[4-(5-метоксил-бензимидазол-1-ил)-фенил]-мочевина;
1-(5-трет-бутил-2Н-пиразол-3-ил)-3-[4-(5-этоксил-бензимидазол-1-ил)-фенил]-мочевина;
1-(5-трет-бутил-2Н-пиразол-3-ил)-3-[4-(5-гексилоксил-бензимидазол-1-ил)-фенил]-мочевина;
1-(5-трет-бутил-2Н-пиразол-3-ил)-3-[4-(5-изопропокси-бензимидазол-1-ил)-фенил]-мочевина;
1-[4-(5-втор-бутоксил-бензимидазол-1-ил)-фенил]-3-(5-трет-бутил-2Н-пиразол-3-ил)-мочевина;
1-(5-трет-бутил-2Н-пиразол-3-ил)-3-[4-(5-изобутокси-бензимидазол-1-ил)-фенил]-мочевина;
1-(5-трет-бутил-2Н-пиразол-3-ил)-3-{4-[5-(2-метоксил-этоксил)-бензимидазол-1-ил]-фенил}-мочевина;
1-(5-трет-бутил-2Н-пиразол-3-ил)-3-{4-[5-(2-этоксил-этоксил)-бензимидазол-1-ил]-фенил}-мочевина;
1-(5-трет-бутил-2Н-пиразол-3-ил)-3-{4-[5-(2-гидрокси-этоксил)-бензимидазол-1-ил]-фенил]-мочевина;
1-(5-трет-бутил-2Н-пиразол-3-ил)-3-{4-[5-(2-гидрокси-3-метоксил-пропоксил)-бензимидазол-1-ил]-фенил]мочевина;
1-(5-трет-бутил-2Н-пиразол-3-ил)-3-{4-[5-(3-диметиламино-пропоксил)-бензимидазол-1-ил]-фенил}-мочевина;
1-(5-трет-бутил-2Н-пиразол-3-ил)-3-{4-[5-(3-дибутиламинопропилоксил)-бензимидазол-1-ил]-фенил}-мочевина;
1-(5-трет-бутил-2Н-пиразол-3-ил)-3-[4-(5-цианометокси-бензимидазол-1-ил)-фенил]-мочевина;
1-(5-трет-бутил-2Н-пиразол-3-ил)-3-[4-(5-трифторметокси-бензимидазол-1-ил)-фенил]-мочевина;
1-(5-трет-бутил-2Н-пиразол-3-ил)-3-{4-[5-(3-метил-оксетан-3-илметокси)-бензимидазол-1-ил]-фенил]мочевина;
1-(5-трет-бутил-2Н-пиразол-3-ил)-3-{4-[5-(тетрагидрофуран-2-илметоксил)-бензимидазол-1-ил]-фенил}-мочевина;
1-(5-трет-бутил-2Н-пиразол-3-ил)-3-{4-[5-(тетрагидрофуран-2-илметоксил)-бензимидазол-1-ил]-фенил}-мочевина;
Этил
(1-{4-[3-(5-трет-бутил-2Н-пиразол-3-ил)-карбамидо]-фенил}-1Н-бензимидазол-5-илокси)-ацетат;
1-(5-трет-бутил-2Н-пиразол-3-ил)-3-{4-[5-(2-морфолин-4-ил-этоксил)-бензимидазол-1-ил]-фенил}-мочевина;
1-(5-трет-бутил-2Н-пиразол-3-ил)-3-{4-[5-(2-пиперидин-1-ил-этоксил)-бензимидазол-1-ил]-фенил}-мочевина;
1-{4-[5-(2-азациклогептан-1-ил-этоксил)-бензимидазол-1-ил]-фенил}-3-(5-трет-бутил-2Н-пиразол-3-ил)-мочевина;
1-(5-трет-бутил-2Н-пиразол-3-ил)-3-(4-{5-[3-(4-метил-пиперазин-1-ил)-пропоксил]-бензимидазол-1-ил}-фенил)-мочевина;
1-(5-трет-бутил-2Н-пиразол-3-ил)-3-{4-[5-(3-фтор-бензилокси)-бензимидазол-1-ил]-фенил]мочевина;
1-(5-трет-бутил-2Н-пиразол-3-ил)-3-(4-{5-[4-(1-циклогексил-1Н-тетразол-5-ил)-бутокси]-бензимидазол-1-ил}-фенил)-мочевина;
1-(5-трет-бутил-2Н-пиразол-3-ил)-3-{4-[5-(4-морфолин-4-ил-[1,2,5]тиадиазол-3-илокси)-бензимидазол-1-ил]-фенил]мочевина;
4-(1-{4-[3-(5-трет-бутил-2Н-пиразол-3-ил)-карбамидо]-фенил}-1Н-бензимидазол-5-илокси)-пиридин-2-карбоновой кислоты метиламин;
1-(5-трет-бутил-2Н-пиразол-3-ил)-3-[4-(5-фтор-7-метил-бензимидазол-1-ил)-фенил]-мочевина;
1-(5-трет-бутил-2Н-пиразол-3-ил)-3-{4-[6-(2-метоксил-этоксил)-бензимидазол-1-ил]-фенил}-мочевина;
1-(5-трет-бутил-2Н-пиразол-3-ил)-3-[4-(5,6-диметоксил-бензимидазол-1-ил)-фенил]-мочевина;
1-(4-бензимидазол-1-ил-фенил)-3-(5-трет-бутил-2-метил-2Н-пиразол-3-ил)-мочевина;
1-(5-трет-бутил-2-метил-2Н-пиразол-3-ил)-3-{4-[5-(2-метоксил-этоксил)-бензимидазол-1-ил]-фенил]мочевина;
1-(5-трет-бутил-2-метил-2Н-пиразол-3-ил)-3-{4-[5-(2-морфолин-4-ил-метоксил)-бензимидазол-1-ил]-фенил}-мочевина;
1-(4-бензимидазол-1-ил-фенил)-3-[5-трет-бутил-2-(2-гидрокси-этил)-2Н-пиразол-3-ил]-мочевина;
1-(4-бензимидазол-1-ил-фенил)-3-(5-трет-бутил-2-фенил-2Н-пиразол-3-ил)-мочевина;
1-(4-бензимидазол-1-ил-фенил)-3-(5-трет-бутил-2-р-толил-2Н-пиразол-3-ил)-мочевина;
1-(4-бензимидазол-1-ил-фенил)-3-[5-трет-бутил-2-(4-метоксил-фенил)-2Н-пиразол-3-ил]-мочевина;
1-(4-бензимидазол-1-ил-фенил)-3-[5-трет-бутил-2-(4-трифторметокси-фенил)-2Н-пиразол-3-ил]-мочевина;
1-(4-бензимидазол-1-ил-фенил)-3-[5-трет-бутил-2-(4-фтор-фенил)-2Н-пиразол-3-ил]-мочевина;
1-(4-бензимидазол-1-ил-фенил)-3-[5-трет-бутил-2-(2-фтор-фенил)-2Н-пиразол-3-ил]-мочевина;
1-(4-бензимидазол-1-ил-фенил)-3-(5-трет-бутил-2-пиридин-2-ил-2Н-пиразол-3-ил)-мочевина;
4-{5-[3-(4-бензимидазол-1-ил-фенил)-карбамидо]-3-трет-бутил-пиразол-1-ил}-бензоат;
1-(2-акрил-5-трет-бутил-2Н-пиразол-3-ил)-3-(4-имидазол-1-ил-фенил)-мочевина;
3-амино-5-метилпиразол-1-карбоновой кислоты (4-бензимидазол-1-ил-фенил)-амид;
5-амино-3-циклопропилпиразол-1-карбоновой кислоты (4-бензимидазол-1-ил-фенил)-амид;
5-амино-3-трифторметилпиразол-1-карбоновой кислоты (4-бензимидазол-1-ил-фенил)-амид;
5-амино-3-трет-бутил-пиразол-1-карбоновой кислоты (4-бензимидазол-1-ил-фенил)-амид;
5-амино-3-трет-бутил-пиразол-1-карбоновой кислоты [4-(5-гексилоксил-бензимидазол-1-ил)-фенил]-амид;
5-амино-3-трет-бутил-пиразол-1-карбоновой кислоты (4-{5-[3-(4-метил-пиперазин-1-ил)-пропоксил]-бензимидазол-1-ил}фенил)-амид;
5-амино-3-трет-бутил-пиразол-1-карбоновой кислоты {4-[5-(2-морфолин-4-ил-этоксил)-бензимидазол-1-ил]-фенил}-амид;
5-амино-3-трет-бутил-пиразол-1-карбоновой кислоты {4-[5-(2-метоксил-этоксил)-бензимидазол-1-ил]-фенил}-амид;
5-амино-3-трет-бутил-пиразол-1-карбоновой кислоты {4-[5-(2-гидрокси-этоксил)-бензимидазол-1-ил]-фенил}-амид;
5-амино-3-трет-бутил-пиразол-1-карбоновой кислоты {4-[5-(тетрагидрофуран-2-илметоксил)-бензимидазол-1-ил]-фенил}-амид;
5-амино-3-трет-бутил-пиразол-1-карбоновой кислоты {4-[5-(тетрагидрофуран-2-илметокси)-бензимидазол-1-ил]-фенил}-амид;
(1-{4-[(5-амино-3-трет-бутил-пиразол-1-карбонил)-амино]-фенил}-1Н-бензимидазол-5-илоксил)-уксусная кислота;
5-амино-3-трет-бутил-пиразол-1-карбоновой кислоты [4-(5-фтор-бензимидазол-1-ил)-фенил]-амид;
5-амино-3-трет-бутил-пиразол-1-карбоновой кислоты [4-(5-трифторметил-бензимидазол-1-ил)-фенил]-амид;
4-(1-{4-[(5-амино-3-трет-бутил-пиразол-1-карбонил)-амино]-фенил}-1Н-бензимидазол-5-илоксил)-пиридин-2-карбоновой кислоты метиламид
5-амино-3-трет-бутил-пиразол-1-карбоновой кислоты {4-[6-(2-метоксил-этоксил)-бензимидазол-1-ил]-фенил}-амид;
5-амино-3-трет-бутил-пиразол-1-карбоновой кислоты [4-(5,6-диметоксил-бензимидазол-1-ил)-фенил]-амид;
3-трет-бутил-пиразол-1-карбоновой кислоты (4-бензимидазол-1-ил-фенил)-амид;
1-(4-бензимидазол-1-ил-фенил)-3-(3,4-диметил-изоксазол-5-ил)-мочевина;
1-(4-бензимидазол-1-ил-фенил)-3-(3-изопропил-изоксазол-5-ил)-мочевина;
1-(4-бензимидазол-1-ил-фенил)-3-(3-трет-бутил-изоксазол-5-ил)-мочевина.
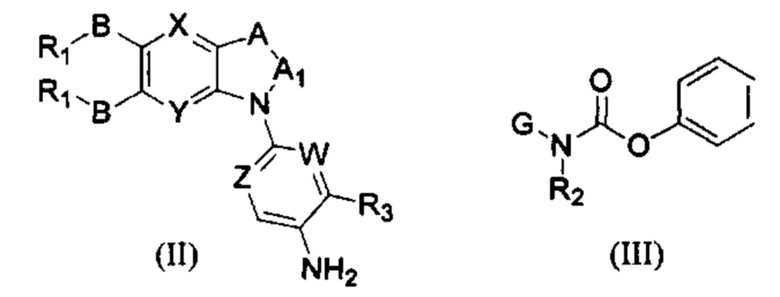
Другим аспектом настоящего изобретения является способ получения соединения общей формулы (I) или его фармацевтически приемлемой соли, сольвата или пролекарства, содержащий следующие стадии:
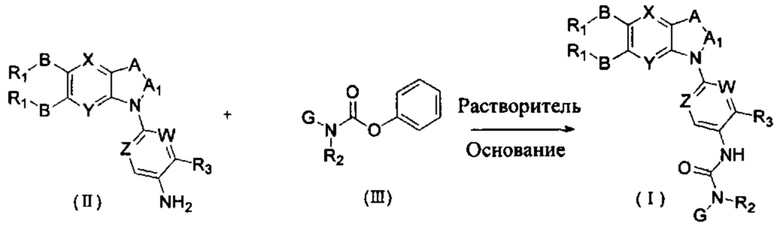
соединение формулы (II) взаимодействует с соединением формулы (III) в присутствии основания в подходящем растворителе при подходящей температуре и рН с получением соединения общей формулы (I);
растворитель предпочтительно представляет собой ТГФ, ацетонитрил, дихлорметан, или толуол, и основание предпочтительно представляет собой триэтиламин, N,N-диизопропилэтиламин, DMAP, или пиридин;
X, Y, A, Al, Z, W, RI R, В, R2, G, R3 являются такими, как определено в общей формуле (I).
В соответствии с другим аспектом настоящего изобретения предложен способ получения соединения общей формулы (I) или его фармацевтически приемлемой соли, сольваты или пролекарства, содержащий следующие стадии:
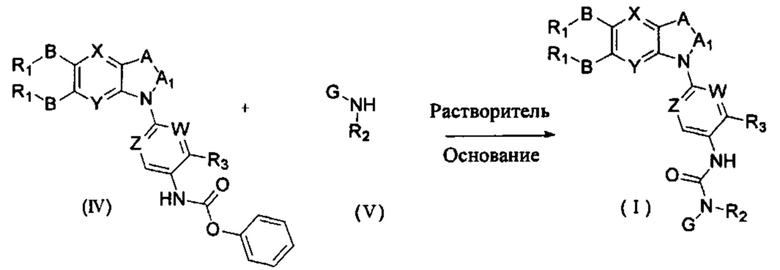
соединение формулы (IV) взаимодействует с соединением формулы (V) в присутствии основания в подходящем растворителе при подходящей температуре и рН с получением соединение общей формулы (I);
растворитель предпочтительно представляет собой ТГФ, ацетонитрил, дихлорметан, или толуол, и основание предпочтительно представляет собой триэтиламин, N,N-диизопропиламин, DMAP, или пиридин;
X, Y, A, Al, Z, W, RI, В, R2, G, R3 являются такими, как определено в общей формуле (I).
Настоящее изобретение также относится к фармацевтической композиции, содержащей терапевтически эффективное количество соединения общей формулы (I) или его фармацевтически приемлемую соль, сольват или пролекарство, а также один или более фармацевтически приемлемых носителей.
Настоящее изобретение также относится к применению соединения общей формулы (I) или его фармацевтически приемлемой соли, сольвата или пролекарства, или содержащей их фармацевтической композиции, для получения ингибиторов тирозиновой протеинкиназы FLT3.
Настоящее изобретение также относится к применению соединения общей формулы (I) или его фармацевтически приемлемой соли, сольвата или пролекарства, или фармацевтической композиции, их содержащей, для получения лекарственных средств для предотвращения и/или лечения рака у млекопитающих, включая человека. Упомянутый рак включает, без ограничения, несолидный опухоли, такие как лейкемия, солидные опухоли, например, рак кожи, меланому, рак легкого, желудка, груди, поджелудочной железы, печени и рак толстой кишки, и т.д.
Настоящее изобретение также относится к соединению формулы (I) или его фармацевтически приемлемой соли, сольвату или пролекарству, или фармацевтической композиции, их содержащей, для применения в качестве ингибиторов тирозиновой протеинкиназы FLT3.
Настоящее изобретение также относится к соединению общей формулы (I) или его фармацевтически приемлемой соли, сольвату или пролекарству, или фармацевтической композиции, их содержащей, для применения в качестве лекарственного средства для предотвращения и/или лечения рака у млекопитающих, включая человека. Такие опухоли включают, без ограничения, несолидный опухоли, такие как лейкемия, солидные опухоли, например, рак кожи, меланому, рак легкого, желудка, груди, поджелудочной железы, печени и рак толстой кишки.
Настоящее изобретение также относится к способу ингибирования тирозиновой протеинкиназы FLT3, содержащему введение нуждающемуся в этом пациента эффективную ингибирующую дозу соединения общей формулы (I) или его фармацевтически приемлемой соли, сольвата или пролекарства или фармацевтической композиции, их содержащей.
Настоящее изобретение также относится к способу предотвращения и/или лечения рака у млекопитающих, включая человека, который содержит введение нуждающемуся в этом пациента эффективной ингибирующей дозы соединения общей формулы (I) или его фармацевтически приемлемой соли, сольвата или пролекарства или фармацевтической композиции, их содержащей. Такие опухоли включают, без ограничения, несолидный опухоли, такие как лейкемия, солидные опухоли, например, рак кожи, меланому, рак легкого, желудка, груди, поджелудочной железы, печени и рак толстой кишки.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Если не указано иное, все технические и научные термины, используемые здесь, имеют то же значение, которое обычно понимается специалистом в данной области техники. Все патенты, заявки, опубликованные заявки и другие публикации включены сюда путем ссылки во всей их полноте. Если существует несколько определений для используемых здесь терминов, если не указано иное, главными являются понятия этого раздела. Если количество любых заданных замещенных групп не указано, может присутствовать одна или несколько замещенных групп. Например, «галоалкил» может содержать один или несколько одинаковых или разных галогенов. В приведенном здесь описании, если химическая структура не соответствует химическому названию, преобладает химическая структура. Как здесь используется, сокращения для любых защитных групп, аминокислот и других соединений указаны их общепринятыми сокращениями или указаны в соответствии с комиссией IUPAC-IUB по биохимической номенклатуре (см. Biochem., 1972, 77: 942-944), если указано иного.
Если не указано иное, следующие термины, используемые в описании и формуле изобретения, имеют следующие значения.
Термин «алкил» относится к насыщенной линейной или разветвленной алифатической углеводородной группе, содержащей 1-20 атомов углерода. Предпочтительно алкильная группа представляет собой алкил, имеющий от 1 до 18 атомов углерода, предпочтительно от 1 до 10 атомов углерода, еще более предпочтительно от 1 до 6 атомов углерода, наиболее предпочтительно от 1 до 4 атомов углерода. Типичные примеры включают, без ограничения, метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-пентил, изопентил, неопентил, гексил, изогексил, н-гептил, изогептил, н-октил, изооктил, н-децил, н-децил и тому подобное. В настоящем описании «алкил» дополнительно включает циклическую алкильную группу, содержащую от 3 до 10 атомов углерода, предпочтительно от 3 до 8 атомов углерода, предпочтительно от 4 до 6 атомов углерода, такую как циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклооктил, циклононил, циклодецил, декагидронафталинил, норборнан и адамантил. Алкил может быть замещенным или незамещенным. В случае замещения заместитель предпочтительно представляет собой одну или несколько групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклоалкокси, циклоалкилтио, гетероциклоалкилтио, оксо, амино, галогеналкила, гидроксиоксиалкила, карбокси или карбоксилата.
Термин «алкенил» относится к углеводородной группе с прямой или разветвленной цепью, состоящей из атомов углерода и водорода, содержащей по меньшей мере одну двойную связь, которая связана с остальной частью молекулы посредством одинарной связи или двойной связи. Он предпочтительно имеет от 2 до 10 атомов углерода, предпочтительно от 2 до 6 атомов углерода, еще более предпочтительно от 2 до 4 атомов углерода. Неограничивающие примеры включают винил, пропенил, бутенил, пентенил, пентадиенил, гексенил. Заместитель предпочтительно представляет собой одну или несколько групп, независимо выбранных из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, тиола, гидрокси, нитро, циано, циклоалкил, гетероциклоалкила, арила, гетероарила, циклоалкокси, гетероциклоалкокси, циклоалкилтио, гетероциклоалкилтио, оксо, амино, галогеналкила, гидроксиалкила, карбоксила или карбоксилаты.
Термин «алкинил» относится к углеводородной группе с прямой или разветвленной цепью, состоящей из атомов углерода и водорода, содержащих, по меньшей мере, одну тройную связь, которая соединена с остальной частью молекулы одинарной связью или тройной связью. Она предпочтительно имеет от 2 до 10 атомов углерода, предпочтительно от 2 до 6 атомов углерода, еще более предпочтительно от 2 до 4 атомов углерода. Неограничивающие примеры включают этинил, пропинил, бутинил, пентинил, гексинил. Алкинильная группа может быть замещенной или незамещенной, и, когда замещена, заместитель предпочтительно представляет собой одну или более групп, независимо выбранных из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклоалкила, арилгетероарила, циклоалкокси, гетероциклоалкокси, циклоалкилтио, гетероциклоалкилтио, оксо, амино, галоалкила, гидроксиалкила, карбоксила, или карбоксилатной группы.
Термин «циклоалкил» относится к насыщенной или частично ненасыщенной моноциклической или полициклической циклической углеводородной группе, содержащей от 3 до 20 атомов углерода, предпочтительно от 3 до 12 атомов углерода, более предпочтительно циклоалкильное кольцо, содержащее от 3 до 10 атомов углерода, наиболее предпочтительно циклоалкильное кольцо, содержащее от 3 до 7 атомов углерода, атомы. Неограничивающие примеры моноциклического циклоалкила включают циклопропил, циклобутил, циклопентил, циклопентенил, циклогексенил, циклогексил, циклогексадиенил, циклогептил, циклооктил, циклогептатриенил и т.д., предпочтительно циклопропил, циклогексенил. Полициклические циклоалкильные группы включают спиро, конденсированные и мостиковые циклоалкильные группы. Циклоалкил может быть необязательно замещенным или незамещенным. В случае замещения, заместитель предпочтительно представляет собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклоалкокси, циклоалкилтио, гетероциклоалкилтио, оксо, амино, галоалкила, гидроксиалкила, карбокси, или карбоксилатной групп.
Термин «гетероциклил» относится к насыщенной или частично ненасыщенной моноциклической или полициклической циклической углеводородной группе, содержащей от 3 до 20 кольцевых атомов, где один или несколько кольцевых атомов выбраны из азота, кислорода или S(O)m (m представляет собой целое число от 0 до 2), но не включает кольцевую часть -O-O-, -O-S- или -S-S-, а остальные атомы кольца - атомы углерода. Предпочтительно включают от 3 до 12 кольцевых атомов, из которых от 1 до 4 атомов являются гетероатомами, более предпочтительно гетероциклическое кольцо содержит от 3 до 10 кольцевых атомов, еще более предпочтительно гетероциклическое кольцо содержит от 5 до 7 кольцевых атомов. Неограничивающие примеры моноциклических гетероциклилов включают пирролидинил, пиперидинил, пиперазинил, морфолинил, тиоморфолинил, гомопиперазинил, пиранил, тетрагидрофуранил и тому подобное. Полициклические гетероциклические группы включают спиро, конденсированные и мостиковые гетероциклические группы. Гетероциклическая группа может быть возможно замещенной или незамещенной. При замещении заместитель предпочтительно представляет собой одну или несколько групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклоалкокси, циклоалкилтио, гетероциклоалкилтио, оксо, амино, галоалкила, гидроксиалкила, карбокси, или карбоксилатной групп.
Термин «арил» относится к полностью углеродной моноциклической или конденсированной полициклической (т.е. кольцам, которые имеют соседние пары атомов углерода) группам, имеющим сопряженную  -электронную систему, предпочтительно от 5 до 10 членов, более предпочтительно от 5 до 7 членов, даже более предпочтительно фенил и нафтил, наиболее предпочтительно фенил. Арильная группа может быть полностью ароматической, такой как фенил, нафтил, антрил или фенантрил. Арильная группа также может быть комбинацией ароматического кольца и неароматического кольца, например, индена, флуорена, аценафтенов. Арильное кольцо может быть слито в гетероарил, гетероциклил или циклоалкил, где кольцо, присоединенное к родительской структуре, является арильным кольцом. Неограничивающие примеры включают:
-электронную систему, предпочтительно от 5 до 10 членов, более предпочтительно от 5 до 7 членов, даже более предпочтительно фенил и нафтил, наиболее предпочтительно фенил. Арильная группа может быть полностью ароматической, такой как фенил, нафтил, антрил или фенантрил. Арильная группа также может быть комбинацией ароматического кольца и неароматического кольца, например, индена, флуорена, аценафтенов. Арильное кольцо может быть слито в гетероарил, гетероциклил или циклоалкил, где кольцо, присоединенное к родительской структуре, является арильным кольцом. Неограничивающие примеры включают:
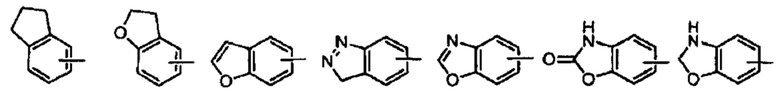
Арильная группа может быть замещенной или незамещенной. При замещении заместитель предпочтительно представляет собой одну или несколько групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклоалкокси, циклоалкилтио, гетероциклоалкилтио, оксо, амино, галоалкила, гидроксиалкила, карбокси, или карбоксилатной групп.
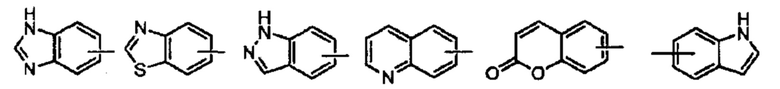
Термин «гетероарил» относится к гетероароматической системе, содержащей от 1 до 4 гетероатомов, от 5 до 14 кольцевых атомов, где гетероатом выбран из кислорода, серы и азота. Гетероарил предпочтительно включает от 5 до 10 членов, предпочтительно от 5 до 7 членов и еще более предпочтительно от 5 до 6 членов, например, тиадиазолил, пиразолил, оксазолил, оксадиазолил, имидазолил, триазолил, тиазолил, фурил, тиенил, пиридил, пирролил, пиримидил, пиразинил, имидазолил, тетразолил и т.п. Гетероарильное кольцо может быть слито с арильным, гетероциклильным или циклоалкильным кольцом, где кольцо, присоединенное к родительской структуре, представляет собой гетероарильное кольцо, а его неограниченные примеры включают:
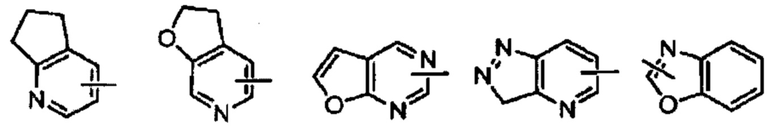
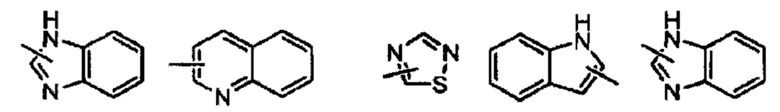
Гетероарил может быть возможно замещенным или незамещенным. При замещении заместитель предпочтительно представляет собой одну или несколько групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклоалкокси, циклоалкилтио, гетероциклоалкилтио, оксо, амино, галоалкила, гидроксиалкила, карбокси, или карбоксилатной групп.
Термин «алкокси» относится к -О-(алкил) и -O-(незамещенный циклоалкил), алкилу, циклоалкилу и т.п. Неограничивающие примеры включают метокси, этокси, пропокси, бутокси, циклопропилокси, циклобутилокси, циклопентилокси, циклогексилокси и тому подобное. Алкокси может быть возможно замещенным или незамещенным. При замещении заместитель предпочтительно представляет собой одну или несколько групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклоалкокси, циклоалкилтио, гетероциклоалкилтио, оксо, амино, галоалкила, гидроксиалкила, карбокси, или карбоксилатной групп.
Термин «галоалкил» относится к алкильной группе, в которой один или несколько атомов водорода замещены галогеном, где алкил имеет значения, определенные в выше. Неграничивающие примеры включают хлорметил, трифторметил, 1-хлор-2-фторэтил, 2,2-дифторэтил, 2-гидроксиэтил, 2-фторпроп-2-ил, 2,2,2-трифторэтил, 1,1-дифторэтил 1 3-дифтор-2-метилпропил, 2,2-дифторциклопропил, (трифторметил)циклопропил, 4,4-дифторциклогексил и 2, 2, 2-трифтор-1,1-диметил-этил.
Термин «галоген» включает фтор, хлор, бром и йод.
Термин «циано» относится к -CN.
Термин «гидрокси» относится к группе -ОН.
Термин «гидроксиалкил» относится к алкильной группе, замещенной гидроксилом, где алкил является таким, как определено выше.
Термин «гидроксиалкокси» относится к алкоксигруппе, замещенной гидроксигруппой, где алкоксигруппа определена выше.
Термин «ацил» относится к -С(О)R, где R обозначает алкил, циклоалкил, алкенил, алкинил, где алкил, циклоалкил, алкенил, алкинил определены выше. Неограничивающие примеры включают ацетил, пропионил, бутирил, пентаноил, гексаноил, винилацил и акрилоил.
Термин «амидо» относится к -NHC(О)OR, где R относится к алкил, алкенил, алкинил, алкил, алкенил и алкинил, таким, как определено выше. Неограничивающие примеры включают карбоксамидо, ацетамидо, пропионамидо, бутирил, пентамил, капроил, виниламидо и акриламидо.
Термин «эфирная группа» относится к -С(О)OR, где R относится к алкильной группе или циклоалкильной группе, где алкил и циклоалкил являются такими, как определено выше. Неограничивающие примеры включают этилэфирную группу, пропилэфирную группу, бутилэфирную группу, пентилэфирную группу, циклопропилэфирную группу, циклобутилэфирную группу, циклопентилэфирную группу, циклогексилэфирную группу.
«Возможно замещенный» в настоящем описании означает незамещенный или замещенный одним или несколькими (например, 2, 3, 4) заместителями. Где, заместитель выбран из группы, состоящей из галогена, алкила, алкенила, алкинила, галоалкила, алкокси, арила, галоарила, арилокси, арилалкила, аралкилокси, гетероциклоалкокси, галоарилалкилоксил, алкиламино, алкилацильной, циано и гетероциклила и т.д.. Эти заместители могут быть дополнительно замещены. Например, алкил в качестве заместителя также возможно замещен одной или несколькими группами, выбранными из группы, состоящей из галогена, гидрокси, алкокси, алкиламино, пирролидинила, фенила, пиридила или галогенфенила. Гетероциклическая группа в качестве заместителя также возможно замещена одной или несколькими группами, выбранными из галогена, алкила или алкоксигруппы.
Способ получения соединения общей формулы (I) настоящего изобретения.
Для достижения назначения соединений по настоящему изобретению в настоящем изобретении в основном применяют следующий путь синтеза и технические решения.
Первый способ синтеза настоящего соединения реализуется разделением структуры настоящего соединения общей формулы (I) на остаток А и остаток В, где остаток А представляет собой промежуточное соединение амина формулы (II), и остаток В представляет собой активный эфир промежуточного соединения формулы (III).
1. Способ синтеза промежуточного соединения амина формулы (II) является следующим:
Способ 1: Синтеза промежуточного соединения амина остатка А с помощью реакции замещения:
а) Синтез промежуточного соединения бензимидазола 1 или промежуточного соединения 2 или промежуточного соединения 3 показан на Схеме 1 ниже.
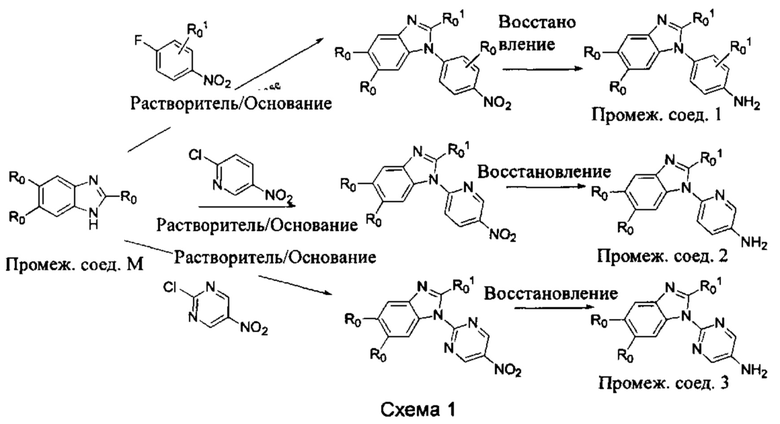
Сначала, промежуточное соединение получили реакцией замещения в присутствии щелочного катализатора в подходящем растворителе при подходящей температуре и рН с промежуточным бензимидазолом М в качестве исходного вещества; основанием может быть, например, карбонат калия, и карбонат цезия и т.д.; растворителем может быть, например, ДМФ или ацетонитрил и т.д. Затем, нитро группу промежуточного продукта восстановили до амино группы с получением промежуточных соединений 1, 2, или 3; восстановление нитро группы может быть достигнуто, например, в системе порошок железа - хлорид аммония или Н2/ палладий на угле.
b) Синтез других видов промежуточных соединений показан на Схеме 2 ниже.
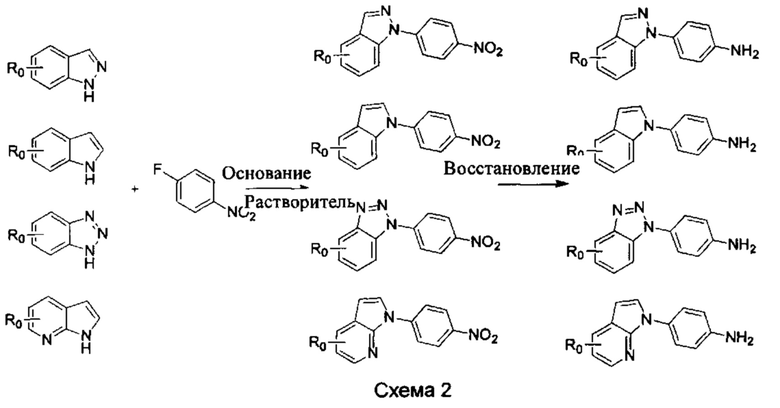
Способ синтеза является таким же, как для вышеуказанного промежуточного бензимидазола, за исключением того, что соответствующее исходное соединение использовали вместо промежуточного бензимидазола М с получением других промежуточных соединений соответствующего вида.
Способ 2: Синтез промежуточных соединений амина остатка А посредством реакции циклизации:
а) Синтез промежуточного бензимидазола 4, промежуточного соединения 5, промежуточного соединения 6, и промежуточного соединения 7 показан на Схеме 3 (способ 1) и Схеме 4 (способ 2) ниже.
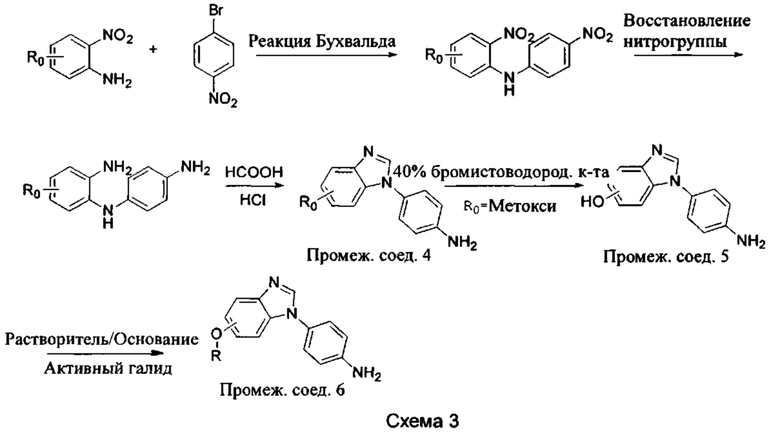
Стадия 1: Промежуточное соединение М1 и п-бромнитробензол подвергли реакции Бухвальда в присутствии основания, катализатора и лиганда в подходящем растворителе с получением промежуточного соединения М2; растворитель предпочтительно представляет собой диоксан, толуол; основание предпочтительно представляет собой трет-бутоксид натрия, трет-бутоксид калия, карбонат цезия; катализатор предпочтительно представляет собой (pd)2(dba)3, ацетат палладия, pd(dba)2; и лиганд предпочтительно представляет собой Xphos, BINAP;
Стадия 2: Две нитрогруппы Промежуточного соединения М2 восстановили с получением промежуточного соединения М3 в восстанавливающих условиях. Восстанавливающие условия могут представлять собой, например, систему порошка железа аммония или систему Н2/палладий на угле;
Стадия 3: Промежуточное соединение М3 и муравьиную кислоту подвергли реакции циклизации в кислой среде при высокой температуре с получением промежуточного соединения 4, кислая среда может представлять собой, например, соляную кислоту;
Стадия 4: Когда R0 выбран из метокси групп, промежуточное соединение 4 нагревают при кипении с обратным холодильником и в кислой среде с получением промежуточного соединения 5. Кислая среда может быть, например, в присутствии HBr;
Стадия 5: Промежуточное соединение 5 взаимодействует с соответствующим активным галидом в присутствии щелочного катализатора в подходящем растворителе при температуре нейтрализации, для получения промежуточного соединения 6. Основанием может быть, например, гидроксид натрия, карбонат калия, и т.д.. Растворитель может быть, например, ДМФ, и т.д..
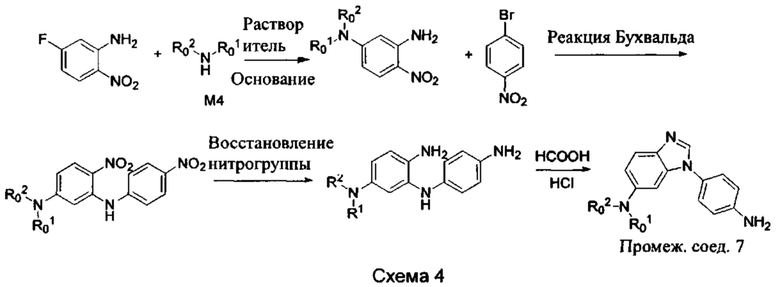
Стадия 1: 2-Нитро-5-фторанилин и промежуточное соединение М4 подвергли реакции замещения в присутствии основания в подходящем растворителе, с получением промежуточного соединения М5. Основанием может быть, например, карбонат калия, и т.д., и растворитель может быть, например, ДМФ, и т.д.;
Стадия 2: Промежуточное соединение М5 и п-бромнитробензол подвергли реакции Бухвальда в присутствии основания, катализатора и лиганда в подходящем растворителе, с получением промежуточного соединения М6; растворитель предпочтительно представляет собой диоксан или толуол; основание предпочтительно представляет собой натрия трет-бутоксид, калия трет-бутоксид, или карбонат цезия, и катализатор предпочтительно представляет собой (pd)2(dba)3, ацетат палладия, или pd(dba)2; лиганд предпочтительно представляет собой Xphos, или BINAP;
Стадия 3: Две нитрогруппы Промежуточного соединения М6 восстановили до аминогрупп в восстанавливающих условиях, с получением промежуточного соединения М7. Восстанавливающими условиями могут быть, например, система порошок железа хлорид аммония или система Н2/палладий на угле;
Стадия 4: Промежуточное соединение М7 и муравьиную кислоту подвергли реакции циклизации в кислой среде при высокой температуре с получением промежуточного соединения 7. Кислая среда может представлять собой, например, соляную кислоту.
b) Синтез промежуточных соединений других видов показан на Схеме 5 ниже.
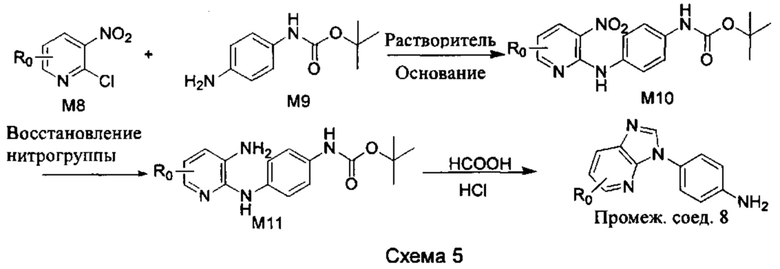
Стадия 1: Промежуточное соединение М8 и промежуточное соединение М9 подвергли реакции замещения в присутствии основания в подходящем растворителе с получением промежуточного соединения М10. Основанием может быть, например, триэтиламин, и растворитель может быть, например, ДМСО;
Стадия 2: Две нитрогруппы промежуточного соединения М10 восстановили в восстановительных условиях, с получением промежуточного соединения М11. Восстановительными условиями могут быть, например, система порошок железа хлорид аммония или Н2/палладий на угле;
Стадия 3: Промежуточное соединение М11 и муравьиную кислоту подвергли реакции циклизации в кислой среде при высокой температуре, с получением промежуточного соединения 8, т кислой средой может быть, например, в присутствии соляной кислоты.
2. Синтез остатка В активного сложного эфира промежуточного соединения В формулы (III)
1) Синтез пиразольного промежуточного соединения показан на Схеме 6 ниже.
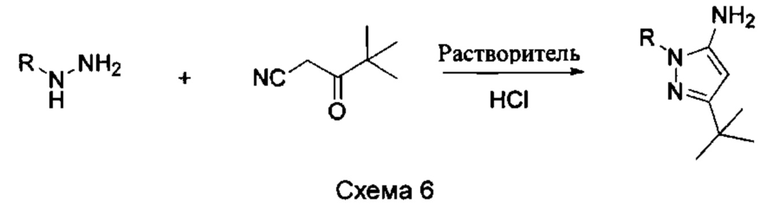
Пиразольное промежуточное соединение получили в присутствии кислотного катализа в подходящем растворителе при подходящей температуре и величине рН. Растворителем может быть, например, этанол, и кислотой может быть, например, соляная кислота.
2) Другие промежуточные соединения изоксазола, оксазола, тиазола, и тиадиазола являются коммерчески доступными.
3) Синтез активных сложных эфиров
Если остаток В промежуточного соединения обладает только одним участком образования мочевины, тогда он будет получен в активном сложном эфире, как показано на Схеме 7 ниже.
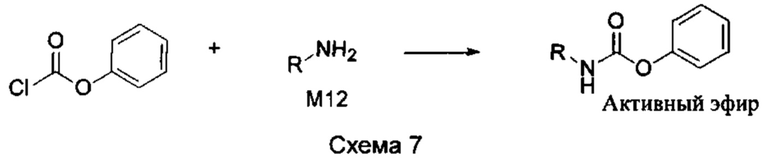
Фенилхлорформиат взаимодействует с соответствующим амином (промежуточное соединение М12) в присутствии щелочного катализа в подходящем растворителе при подходящей температуре и условиях рН, с получением соответствующего активного сложного эфира, где растворителем может быть, например, этилацетат, дихлорметан, тетрагидрофуран, ацетон, ацетонитрил, или вода, и т.д., и основанием может быть, например, пиридин, бикарбонат натрия, карбонат калия, триэтиламин, или гидроксид натрия и т.д.. Поскольку различия между группами, связанными с аминогруппами приводят к различной активности, выбираемые основания немного отличаются в процессе реакции. Квалифицированный специалист может провести рутинный выбор в соответствии с обычными техническими знаниями в уровне техники.
Если присутствует два участка, образующих мочевину (-NH- и -NH2-) в остатке В промежуточного соединения, например: 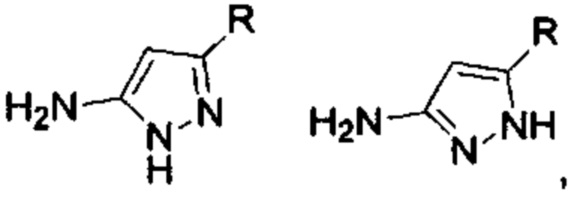 тогда промежуточное соединение амина остатка А получают в виде активных сложных эфиров. Существует второй способ синтеза соединений настоящего изобретения. Структура соединения общей формулы (I) разделяется на остатки А и остатки В, где остаток А представляет собой промежуточное соединение активного сложного эфира, представленное формулой (IV), и остаток В представляет собой промежуточное соединение амина, представленное формулой (V).
тогда промежуточное соединение амина остатка А получают в виде активных сложных эфиров. Существует второй способ синтеза соединений настоящего изобретения. Структура соединения общей формулы (I) разделяется на остатки А и остатки В, где остаток А представляет собой промежуточное соединение активного сложного эфира, представленное формулой (IV), и остаток В представляет собой промежуточное соединение амина, представленное формулой (V).
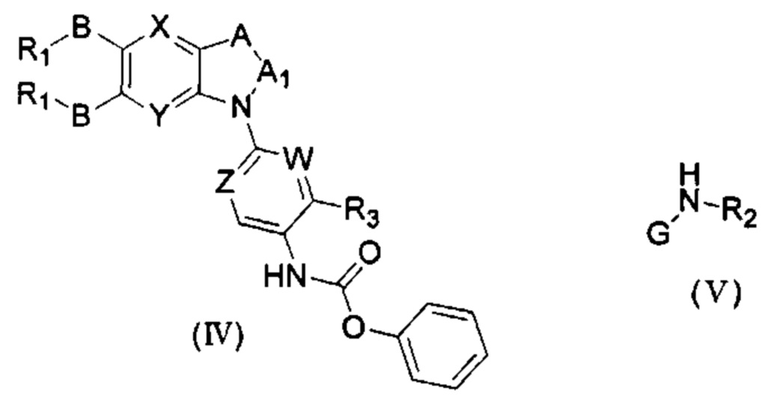
Для них способ получения промежуточного соединения активного сложного эфира, представленного формулой (IV) остатка А является аналогичным таковому для остатка В в качестве активного сложного эфира выше. То есть остаток А промежуточного соединения амина формулы (II) и фенилхлорформиат подвергабт реакции в присутствии щелочного катализа в подходящем растворителе при подходящей температуре и условиях рН, с получением промежуточного соединения активного эфира остатка А.
В заключение, как ранее описано, промежуточное соединение, представленное формулой (II) и промежуточное соединение, представленное формулой (III), или промежуточное соединение, представленное формулой (IV) и промежуточное соединение, представленное формулой (V), подвергают взаимодействию в присутствии основания в подходящем растворителе при подходящей температуре и условиях рН, таким образом активный сложный эфир элиминируется молекулой фенола с получением соответствующих изоцианатных промежуточных соединений, которые затем взаимодействуют с соответствующими аминами в присутствии основания с получением конечных соединений мочевины, т.е. соединения общей формулы (I). Растворитель предпочтительно представляет собой ТГФ, ацетонитрил, метиленхлорид, или толуол, и основание предпочтительно представляет собой триэтиламин, N,N-диизопропилэтиламин, DMAP, или пиридин.
Где, X, Y, A, A1, Z, W, R1, В, R2, G, R3 являются такими, как определено в общей формуле (I), и R0, R01, R02 определены в качестве -BR1 если не указано иного.
Для соединения общей формулы (I) в соответствии с настоящим изобретением его пролекарство должно следовать принципу пролекарственного дизайна и иметь возможность высвобождать исходное активное соединение формулы (I) путем ферментативного гидролиза, гидролиза, ацидолиза или метаболической деградации. Он включает, без ограничения, этерификацию гидроксильных групп (таких как образование фосфатных эфиров и карбонатных эфиров), защите аминогрупп и карбоксильных групп. Дизаин пролекарства относится к: (I) Karaman R, Prodrugs design based on inter- and intramolecular chemical processes. Chem Biol Drug Des. 82(6):643-158, 2013; (2) Rautio J et al., Prodrugs: design and clinical Applications. Nat Rev Drug Discov. 7(3): 255-70 2008; (3) Jampilek J, Prodrugs: pharmaceutical design and current perspectives. Curr Pharm Des. 17 (32): 3480-1, 2011; (4) Bundgaard H. Design of Progrugs Elservier, 1985.
Фармацевтически приемлемая соль соединения общей формулы (I) настоящего изобретения может представлять собой кислотно-аддитивную соль или основно аддитивную соль. Кислота может быть неорганической кислотой, включая, но не ограничиваясь, хлористоводородную кислоту, серную кислоту, фосфорную кислоту или бромистоводородную кислоту; или может быть органической кислотой, включая, но не ограничиваясь, лимонную кислоту, малеиновую кислоту, щавелевую кислоту, муравьиную кислоту, уксусную кислоту, пропионовую кислоту, валериановую кислоту, гликолевую кислоту, бензойную кислоту, фумаровую кислоту, трифторуксусную кислоту, янтарную кислоту, винную кислоту, молочную кислоту, глутаминовую кислоту, аспарагиновую кислоту, салициловую кислоту, пировиноградную кислоту, метансульфоновую кислоту, бензолсульфоновую кислоту, п-толуолсульфоновую кислоту и декстральную камфорсульфоновую кислоту и т.д.. Основанием может быть неорганическое основание, включая, но не ограничиваясь, гидроксид натрия, гидроксид калия, гидроксид магния или гидроксид кальция; или может быть органическим основанием, включая, но не ограничиваясь, гидроксид аммония, триэтиламин, N,N-дибензилэтилендиамин, хлоропрокаин, холин, аммиак, диэтаноламин и другие гидроксиалкиламины, этилендиамин, N-метилглюкозамин, прокаин, N-бензил фенилэтиламин, аргинин или лизин; или может быть солью щелочного металла, включая, но не ограничиваясь, соли лития, калия или натрия; или может быть солью щелочноземельного металла, включая, но не ограничиваясь, соли бария, кальция или магния; или соль переходного металла, включая, но не ограничиваясь ими, соль цинка; или других солей металлов, включая, но не ограничиваясь, фосфат натрия или динатрийгидрофосфат.
В другом месте настоящего изобретения соединение общей формулы (I) или его фармацевтически приемлемая соль или пролекарство готовят в клинически приемлемую фармацевтическую композицию. Согласно клиническим показаниям, пути и способу введения такие фармацевтические препараты включают в себя, без ограничения, пероральные препараты, такие как таблетки, гели, мягкие/твердые капсулы, эмульсии, диспергируемые порошки, гранулы и суспензионные эмульсии вода/масло; инъекции, в том числе внутривенные инъекции, внутримышечные инъекции, внутрибрюшинные инъекции, суппозитории ректального введения и внутричерепные инъекции, это могут быть водные растворы или масляные растворы; препараты для местного применения, включая кремы, мази, гели, водно-масляные растворы и препараты для включения; ингаляционные лекарственные формы, включая мелкие порошки, жидкие аэрозоли и различные лекарственные формы, подходящие для имплантации in vivo.
Фармацевтическая композиция может быть дополнена фармацевтически приемлемым носителем, разбавителем или эксципиентом по мере необходимости. Эти носители, разбавители или эксципиенты должны соответствовать правилам процесса фармацевтического приготовления и быть совместимыми с активными ингредиентами. Носители для твердых пероральных препаратов включают, без ограничения, маннит, лактозу, крахмал, стеарат магния, целлюлозу, глюкозу, сахарозу, циклодекстрин и витамин Е-ПЭГ 1000, молекулярный носитель для кишечной абсорбции. Пероральные препараты могут быть дополнены подходящими красителями, подсластителями, ароматизаторами и консервантами.
Соединение общей формулы (I) или его фармацевтически приемлемую соль или пролекарство вводят теплокровным животным в единичной дозе 0,01-100 мг/кг. Однако, как хорошо известно специалистам в данной области, доза вводимого лекарственного средства зависит от ряда факторов, включая, но не ограничиваясь, активность конкретного используемого соединения, возраст, вес, здоровье, поведение, и рацион пациента, время и способ введения, скорость экскреции, сочетание других лекарств и т.д. Таким образом, оптимальный режим лечения, такой как способ лечения, ежедневная доза введения соединения, представленного по общей формуле (I), или тип фармацевтически приемлемой соли может быть подтвержден в соответствии с обычным режимом лечения.
Соединение общей формулы (I) или его фармацевтически приемлемая соль или пролекарство могут быть использованы в качестве монотерапии или комбинированной терапии вместе с одним или несколькими из следующих средств: лучевая терапия, химиотерапия, иммунотерапия, терапия онколитическими вирусами, RNAi, адъювантная терапия рака, трансплантация костного мозга и трансплантация стволовых клеток, включая, но не ограничиваясь, следующие противоопухолевые препараты и способы лечения:
1) Алкилирующие агенты, такие как цисплатин, цисплатин, оксалиплатин, хлорамбуцил, циклофосфамид, азотная горчица, мелфалан, темозоломид, бусульфан и нитрозомочевины.
2) Противоопухолевые антибиотики, такие как доксорубицин, блеомицин, доксорубицин, даунорубицин, эпирубицин, идарубицин, митомицин С, актиномицин или митрамицин; антимитотические препараты, такие как винкристин, винбластин, виндезин, винорелбин, паклитаксел, таксотер и ингибиторы полокиназы.
3) Антиметаболиты и антифолаты, такие как фторпиримидин, метотрексат, цитарабин, азацитидин, децитабин, ральтитрекс, гидроксимочевина и ингибиторы мутантов IDH1 / IDH2.
4) Ингибиторы топоизомеразы, такие как эпиподофиллотоксин, камптотецин и иринотекан.
5) Ингибиторы клеточного роста, такие как антиэстрогенные / антиандрогенные лекарственные средства, такие как тамоксифен, фулвестрант, торемифен, ралоксифен, дролоксифен, идоксифен, бикалютамид, флутамид, нилутамид и ципротерон ацетат;
Антагонисты LHRH или агонисты LHRH, такие как гозерелин, лейпролид и бусерелин; прогестагены, такие как мегестролацетат;
Ингибиторы ароматазы, такие как анастрозол, летрозол, ворозол, экземестан и ингибиторы 5-редуктазы, такие как финастерид.
6) Антиинвазивные агенты, такие как ингибиторы семейства c-Src-киназ, ингибиторы металлопротеиназы, ингибиторы рецепторной функции активатора урокиназного плазминогена и гепариназные антитела.
7) Ингибиторы роста клеток, включая ингибиторы тирозинкиназы и ингибиторы сериновых / треонинкиназ, таких как ингибиторы сигнализации Ras / Raf, ингибиторы передачи клеток MEK и/или AKT киназы, ингибиторы c-Kit, ингибиторы c-Met, ингибиторы PDGFR, Ингибиторы киназы ABL, ингибиторы РI3-киназы, ингибиторы киназы CSF-1R, ингибиторы киназы семейства EGFR, ингибиторы киназы семейства FGFR, ингибиторы киназы рецептора IGF, ингибиторы синазомы ауры и ингибиторы циклинзависимой киназы, такие как ингибиторы CDK2 и/или CDK4, CDK6, ядерных ингибиторов CRM1 и ингибиторов Wnt / beta-catenin.
8) Ингибиторы антиапоптотических белков, таких как ингибиторы BCL2 (Venetoclax) и ингибиторы MCL1.
9) ингибиторы PARP, такие как Олапариб и Рукапариб.
10) Ингибиторы ангиогенеза, такие как ингибиторы VEGFR.
11) Эпигенетические ингибиторы, такие как ингибиторы гистондезацетилазы (HDAC) и ингибиторы ДНК-метилтрансферазы (DNMT).
12) Опухолевая иммунотерапия включает любые методы in vitro и in vivo для повышения иммуногенности пациента для опухолевых клеток. Например, трансфекции цитокинов IL-2, IL-4 или GM-CSF; способы снижения неэффективности Т-клеток, таких как mAb против PD-1 / PD-L; способы использования трансфицированных иммунных клеток, таких как дендритные клетки, трансфицированные цитокинами; способы использования линий опухолевых клеток, трансфицированных цитокинами; способы снижения функций иммуносупрессивных клеток, таких как регуляторные Т-клетки, миелоидные супрессорные клетки или дендритные клетки, экспрессирующие 2,3-дезоксигеназу индол амина; а также методы противораковых вакцин, состоящих из опухолеспецифических белков или пептидов.
13) Т-клеточная иммунотерапия химерного антигенного рецептора (CART).
14) Онкогеновая терапия, такая как CRISPR-Cas 9, RNAi и трансдукция генов.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Примеры
Настоящее изобретение будет подробно проиллюстрировано со ссылкой на следующие примеры. Однако следует понимать, что настоящее изобретение не ограничивается этими примерами.
Структура соединения определяется с помощью ядерного магнитного резонанса (ЯМР) или / и масс-спектрометрии (МС). ЯМР-сдвиг (δ) задается единицей 10-6 (ppm). ЯМР определяют с помощью спектрометра (Bruker AVANCE-400) ЯМР. Растворителями являются дейтерированный диметилсульфоксид (ДМСО-d6), дейтерированный хлороформ (CDC13), дейтерированный метанол (CD3OD), а внутренний стандарт - тетраметилсилан (ТМС).
МС определяют с использованием масс-спектрометра с жидкостной хроматографией (Thermo, Ultimate 3000 / MSQ).
ВЭЖХ проводят с помощью жидкостного хроматографа высокого давления (Agilent 1260 Infinity, Gemini С18 250×4,6 мм, колонка 5 мкм).
Пластина силикагеля HSGF245, используемая для тонкослойной хроматографии (ТСХ), имеет толщину от 0,15 до 0,2 мм. Характеристики для разделения и очистки продукта тонкослойной хроматографией составляют от 0,9 до 1,0 мм (Yantai Yellow Sea).
Колоночная хроматография обычно использует силикагель 200-300 меш в качестве носителя (силикагель Yantai Yellow Sea).
Известные исходные материалы настоящего изобретения, могут быть синтезированы с использованием или в соответствии со способами, известными в уровне техники или купленными в Shanghai Darui Fine Chemicals Co., Ltd., Shanghai Titan Technology Co., Ltd., Shanghai Runjie Chemical Reagent Co., Ltd., TCI или Aldrich
Chemical Company. Если экспериментальные условия не указаны в примерах, обычно принимаются стандартные условия или условия, рекомендованные производителями сырья или продукта. Реагенты, которые не указаны, являются обычными реагентами, купленными на рынке.
Если в примерах не указано иначе, все реакции могут проводиться в атмосфере аргона или азота. Аргонная или азотная атмосфера означает, что реакционная колба соединена с баллоном аргона или газообразного азота объемом приблизительно 1 л.
Если в примерах не указано иное, раствор относится к водному раствору.
Если в примерах не указано иное, температура реакции представляет собой комнатную температуру, приблизительно от 20 до 30°С.
Пример 1
Получение 1-(4-бензимидазол-1-ил-фенил)-3-изоксазол-3-ил-мочевины (Соединение 1)
Стадия 1: Получение 1-(4-нитро-фенил)-1Н-бензимидазола
При комнатной температуре, бензимидазол (23.6 г, 0.2 моль) и п-фторнитробензол (31.0 г, 0.22 моль) растворили в 300 мл ДМФ, и добавили безводный карбонат калия (69.0 г, 0.5 моль). Смесь нагрели до 90°С и дали прореагировать в течение 6 часов. Реакционный раствор охладили до комнатной температуры, медленно влили в воду, перемешивали при комнатной температуре в течение 0.5 часов, и затем отфильтровали. Получившийся осадок промыли водой и высушили под вакуумом в течение ночи. Получившийся осадок суспендировали с метил трет-бутиловым эфиром с получением 46 г 1-(4-нитро-фенил)-1Н-бензимидазола в виде желтого осадка.
Стадия 2: Получение 4-бензимидазол-1-ил-анилина
1-(4-нитро-фенил)-1Н-бензимидазол (46 г, 0.19 моль), полученный на Стадии 1, восстановленный порошок железа (53.9 г, 0.96 моль), хлорид аммония (81.3 г, 1.52 моль) добавили в этанол (500 мл)/вода (125 мл), и получившуюся смесь нагрели до 80°С и дали прореагировать в течение 4 часов. После охлаждения до комнатной температуры, реакционный раствор медленно влили в насыщенный водный раствор гидрокарбоната натрия (1 л), и экстрагировали этилацетатом (500 мл × 2). Органическую фазу промыли дважды с помощью насыщенного раствора NaCl, высушили над безводным сульфатом натрия, отфильтровали и фильтрат сконцентрировали при пониженном давлении с получением неочищенного продукта 37 г 4-бензимидазол-1-ил-анилина в виде желтого осадка. Указанный продукт использовали на следующей стадии без очистки.
Стадия 3: Получение фенил изоксазол-3-ил-карбамата (активный сложный эфир)
3-Аминоизоксазол (TCI) (840 мг, 10 ммоль) растворили в 20 мл ТГФ и добавили пиридин (2.37 г, 30 ммоль) при комнатной температуре. Смесь охладили в ванне со льдом до 0-5°С и фенилхлорформиат (2.34 г, 15 ммоль) медленно добавили по каплям. После добавления ледяную ванну убрали и температуру медленно повысили до комнатной температуры. После завершения реакции по данным ТСХ, реакционный раствор влили в воду и экстрагировали этилацетатом (100 мл × 2). Органическую фазу промыли с помощью насыщенного раствора NaCl (200 мл), высушили над безводным сульфатом натрия, отфильтровали, и фильтрат сконцентрировали при пониженном давлении. Остатки очистили с помощью колоночной хроматографии (элюент: петролейный эфир/этилацетат) с получением 1.5 г фенил изоксазол-3-ил-карбамата в виде белого осадка.
Стадия 4: Получение 1-(4-бензимидазол-1-ил-фенил)-3-изоксазол-3-ил-мочевины
4-бензимидазол-1-ил-анилин (100 мг, 0.478 ммоль), полученный на Стадии 2, фенил изоксазол-3-ил-карбамат (146.4 мг, 0.717 ммоль), полученный на Стадии 3, и триэтиламин (145 мг, 1.43 ммоль) растворили в 10 мл ТГФ и реакционную смесь кипятили с обратным холодильником в течение ночи. На следующий день реакционный раствор сконцентрировали при пониженном давлении, и остатки очистили с помощью колоночной хроматографии (элюент: дихлорметан/метанол) с получением 50 мг 1-(4-бензимидазол-1-ил-фенил)-3-изоксазол-3-ил-мочевины в виде белого осадка.
1HNMR (DMSO-d6, 400MHz) δ: 9.81 (s, IH), 9.36 (s, IH), 8.76-8.77 (d, IH), 8.52 (s, IH), 7.77-7.79 (m, IH), 7.70-7.72 (d, 2H), 7.60-7.63 (d, 2H), 7.57-7.60 (m, IH), 7.29- 7.36 (m, 2H), 6.88-6.89 (d, IH).
LC-MS (ESI): 320.2 (M+H)+.
Пример 2
Получение
1-(4-бензимидазол-1-ил-фенил)-3-(5-метил-изоксазол-3-ил)-мочевины (Соединение 2)
Способ получения был таким же, как в Примере 1, за исключением того, что 5-метил-3-аминоизоксазол (TCI) использовали вместо 3-аминоизоксазола на Стадии 3 с получением 1-(4-бензимидазол-1-ил-фенил)-3-(5-метил-изоксазол-3-ил)-мочевины.
1HNMR (DMSO-d6, 400MHz) δ: 9.55 (s, IH), 9.09 (s, IH), 8.51 (s, IH), 7.76-7.79 (m, IH), 7.68-7.71 (d, 2H), 7.60-7.62 (d, 2H), 7.57-7.60 (m, IH), 7.30-7.34 (m, 2H), 6.57 (d, IH), 2.38 (s, 3H).
LC-MS (ESI): 334.2 (M+H)+.
Пример 3
Получение 1-(4-бензимидазол-1-ил-фенил)-3-(5-трет-бутил-изоксазол-3-ил)-мочевины (Соединение 3)
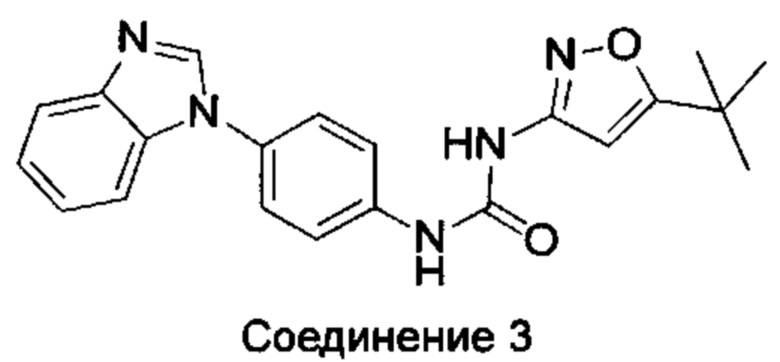
Способ получения был таким же, как в Примере 1, за исключением того, что 5-трет-бутил-3-аминоизоксазол (TCI) использовали вместо 3-аминоизоксазола на Стадии 3 с получением 1-(4-бензимидазол-1-ил-фенил)-3-(5-трет-бутил-изоксазол-3-ил)-мочевины в виде белого осадка.
1HNMR (DMSO-d6, 400MHz) δ: 9.73 (s, IH), 9.34 (s, IH), 8.51 (s, IH), 7.76-7.78 (m, IH), 7.69-7.71 (d, 2H), 7.57-7.62 (m, 3H), 7.30-7.35 (m, 2H), 6.54 (d, IH), 1.31 (s, 9H).
LC-MS (ESI): 376.1 (M+H)+.
Пример 4
Получение 1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(5-гидрокси-бензимидазол-1-ил)-фенил]-мочевины (Соединение 4)
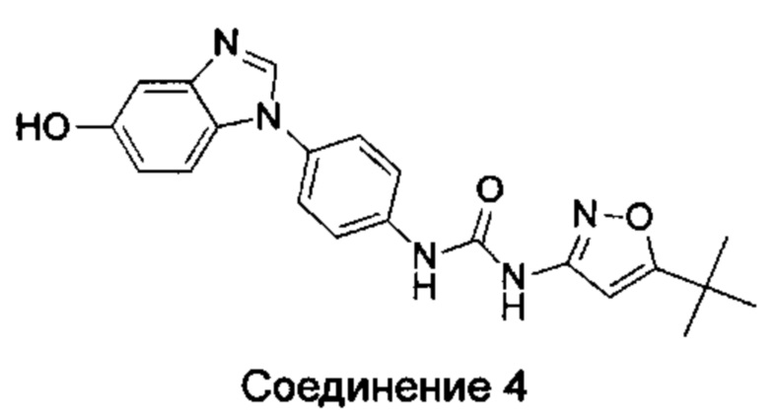
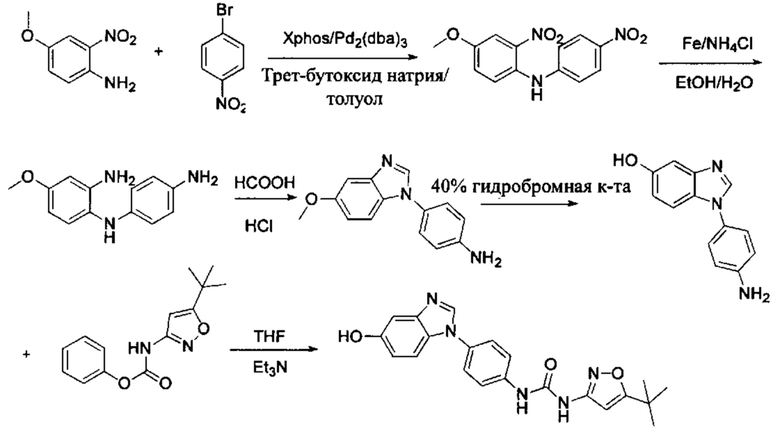
Стадия 1: Получение (4-метоксил-2-нитро-фенил)-(4-нитро-фенил)-амина
В атмосфере азота, 2-нитро-4-метоксиланилин (1.5 г, 8.92 ммоль, Darui), п-бромнитробензол (3.6 г, 17 ммоль), Xphos (425 мг), Pd2(dba)3 (408 мг) и трет-бутоксид натрия (1.71 г, 17.8 ммоль) растворили в 30 мл толуола и дали прореагировать при 90°С в течение 3 часов. Реакционный раствор охладили до комнатной температуры, и затем добавили 100 мл дихлорметана. Смесь перемешивали при комнатной температуре в течение 5 минут, и затем отфильтровали. Фильтрат сконцентрировали при пониженном давлении. Остатки очистили с помощью колоночной хроматографии (элюент: петролейный эфир/этилацетат) с получением 2.5 г (4-метокси-2-нитро-фенил)-(4-нитро-фенил)-амина в виде красно-черного осадка.
Стадия 2: Получение (4-метоксил-2-амино-фенил)-(4-амино-фенил)-амина
(4-метоксил-2-нитро-фенил)-(4-нитро-фенил)-амин (2.5 г, 8.65 ммоль), полученный на Стадии 1, восстановленный порошок железа (2.91 г, 52.0 ммоль) и хлорид аммония (4.62 г, 86.5 ммоль) добавили в этанол (50 мл)/вода (12.5 мл) и получившуюся смесь нагрели до 90°С и дали прореагировать в течение 1 часа. Затем реакционный раствор охладили до комнатной температуры, его медленно влили в насыщенный водный раствор бикарбоната натрия (150 мл), экстрагировали этилацетатом (100 мл × 2), и органическую фазу промыли дважды с помощью насыщенного раствора NaCl и высушили над безводным сульфатом натрия, отфильтровали, фильтрат сконцентрировали при пониженном давлении с получением 1.8 г неочищенного продукта (4-метоксил-2-амино-фенил)-(4-амино-фенил)-амина в виде желтого осадка. Продукт сразу использовали на следующей стадии без очистки.
Стадия 3: Получение 4-(5-метоксил-бензимидазол-1-ил)-анилина
(4-метоксил-2-амино-фенил)-(4-амино-фенил)-амин (1.8 г, 7.86 ммоль), полученный на Стадии 2 растворили в 60 мл соляной кислоты (4 моль/л) и 2 мл муравьиной кислоты добавили при комнатной температуре. Смесь нагрели до 120°С и дали прореагировать в течение 1 часа. Реакционный раствор охладили и довели до рН>9 с помощью водного аммония на ледяной бане и экстрагировали этилацетатом (80 мл × 2).Органическую фазу промыли с помощью насыщенного раствора NaCl, высушили над безводным сульфатом натрия, отфильтровали и фильтрат сконцентрировали при пониженном давлении. Остатки очистили с помощью колоночной хроматографии (элюент: дихлорметан/метанол) с получением 1.2 г 4-(5-метоксил-бензимидазол-1-ил)-анилина в виде желтого осадка.
Стадия 4: Получение 1-(4-амино-фенил)-1Н-бензимидазол-5-ола
4-(5-метоксил-бензимидазол-1-ил)-анилина (875 мг, 3.66 ммоль), полученный на Стадии 3 нагрели до 120°С в 15 мл 40% бромистоводородной кислоты и дали прореагировать в течение 6 часов. Реакционный раствор охладили до комнатной температуры и серо-белый осадок преципитировался. Осадок отфильтровали и затем растворили в 70 мл воды. Получившуюся смесь отфильтровали и фильтрат затем довели до приблизительно рН 7 с помощью водного аммония, и белый осадок преципитировался. Осадок отфильтровали и затем промыли водой и высушили под вакуумом с получением 600 мг 1-(4-амино-фенил)-1Н-бензимидазол-5-ола в виде серо-белого осадка.
1HNMR (DMSO-d6, 400MHz) δ: 9.13 (s, IH), 8.19 (s, IH), 7.23-7.25 (d, IH), 7.19-7.22 (d, 2H), 7.02 (d, IH), 6.76-6.79 (dd, IH), 6.70-6.73 (d, 2H), 5.41 (s, 2H).
LC-MS (ESI): 226.1 (M+H)+.
Стадия 5: Получение 1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(5-гидрокси-бензимидазол-1-ил)-фенил]-мочевины
1-(4-амино-фенил)-1Н-бензимидазол-5-ол (100 мг, 0.444 ммоль), полученный на Стадии 4, фенил(5-трет-бутил-изоксазол-3-ил)-карбамат (173.3 мг, 0.666 ммоль) и триэтиламин (137.5 мг, 1.332 ммоль) растворили в 10 мл ТГФ и реакционную смесь кипятили с обратным холодильником в течение ночи. На следующий день реакционный раствор сконцентрировали при пониженном давлении, и остатки очистили с помощью колоночной хроматографии (элюент: дихлорметан/метанол) с получением 80 мг 1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(5-гидрокси-бензимидазол-1-ил)-фенил]-мочевины в виде белого осадка.
1HNMR (DMSO-d6, 400MHz) δ: 9.60 (S, 1H), 9.27 (S, 1H), 9.11 (S, 1H), 8.38 (S, 1H), 7.66-7.68 (d, 2H), 7.56-7.58 (d, 2H), 7.37-7.39 (d, IH), 7.06-7.07 (d, IH), 6.81-6.84 (dd, IH), 6.52 (s, IH), 1.30 (s, 9H).
LC-MS (ESI): 392.1 (M+H)+.
Пример 5
Получение 1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(5-метоксил-бензимидазол-1-ил)-фенил]-мочевины (Соединение 5)
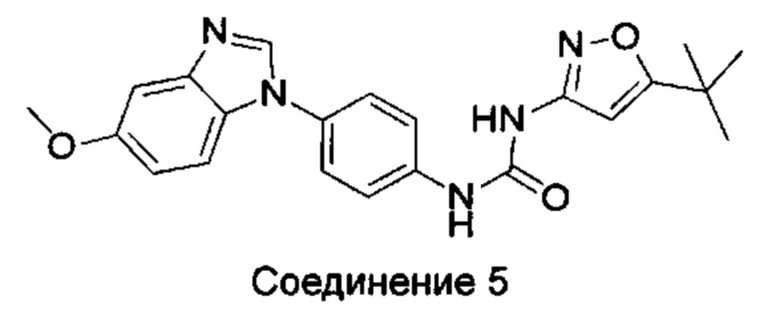
Способ получения был таким же, как в примере 4, за исключением того, что 4-(5-метоксил-бензимидазол-1-ил)-анилин использовали вместо 1-(4-амино-фенил)-1Н-бензимидазол-5-ола на Стадии 5, с получением 1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(5-метокси-бензимидазол-1-ил)-фенил]-мочевины.
1HNMR (DMSO-d6, 400MHz) δ: 9.59 (s, IH), 9.06 (s, IH), 8.44 (s, IH), 7.67-7.69 (d, 2H), 7.58-7.60 (d, 2H), 7.46-7.48 (d, IH), 7.29-7.30 (d, IH), 6.94-6.97 (dd, IH), 6.53 (s, IH), 3.82 (s, 3H), 1.30 (s, 9H).
LC-MS (ESI): 406.1 (M+H)+.
Пример 6
Получение 1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(5-этоксил-бензимидазол-1-ил)-фенил]-мочевины (Соединение 6)
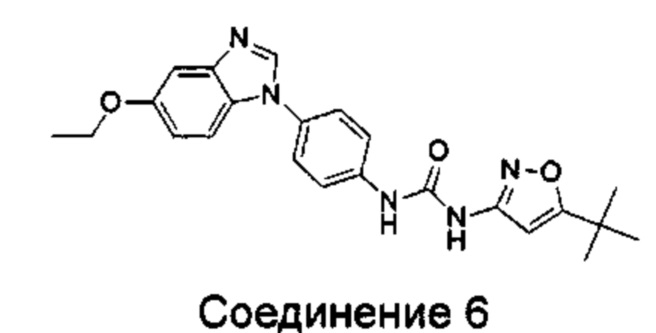
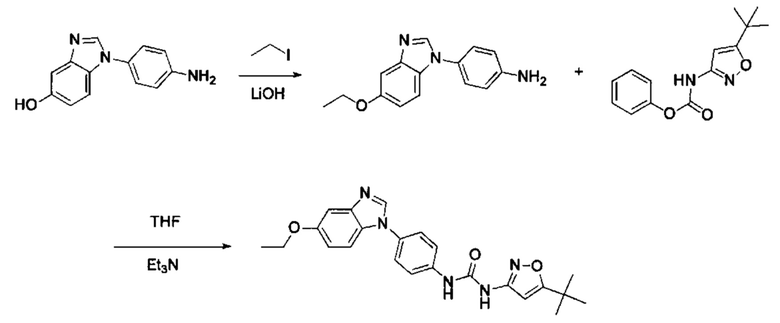
Стадия 1: Получение 4-(5-этоксил-бензимидазол-1-ил)-анилина
1-(4-амино-фенил)-1Н-бензимидазол-5-ол (синтезированный на Стадии 4 примера 4) (300 мг, 1.33 ммоль), йодоэтан (311 мг, 2.0 ммоль), и гидроксид лития (96 мг, 3.98 ммоль) добавили к 20 мл этанола и дали прореагировать при 60°С в течение ночи. На следующий день реакционный раствор охладили до комнатной температуры, и затем влили в воду (80 мл), и экстрагировали этилацетатом (50 мл × 2). Органическую фазу промыли дважды с помощью насыщенного раствора NaCl, высушили над безводным сульфатом натрия, отфильтровали, и фильтрат сконцентрировали при пониженном давлении. Остатки очистили с помощью колоночной хроматографии (элюент: дихлорметан/метанол) с получением 400 мг 4-(5-этоксил-бензимидазол-1-ил)-анилина в виде темно-желтого осадка.
Стадия 2: Методика была аналогична таковой, как на Стадии 5 в примере 4, за исключением того, что 4-(5-этоксил-бензимидазол-1-ил)-анилин использовали вместо 1-(4-амино-фенил)-1Н-бензимидазол-5-ола с получением 1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(5-этокси-бензимидазол-1-ил)-фенил]-мочевины.
1HNMR (DMSO-d6, 400MHz) δ: 9.60 (s, IH), 9.08 (s, IH), 8.44 (s, IH), 7.67-7.69 (d, 2H), 7.57-7.60 (d, 2H), 7.45-7.47 (d, IH), 7.27-7.28 (d, IH), 6.93-6.96 (dd, IH), 6.53 (s, IH), 4.05-4.1 I (q, 2H), 1.34-1.38 (t, 3H), 1.31 (s, 9H).
LC-MS (ESI): 420.2 (M+H)+.
Пример 7
Получение 1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(5-гексилоксил-бензимидазол-1-ил)-фенил]-мочевины (Соединение 7)
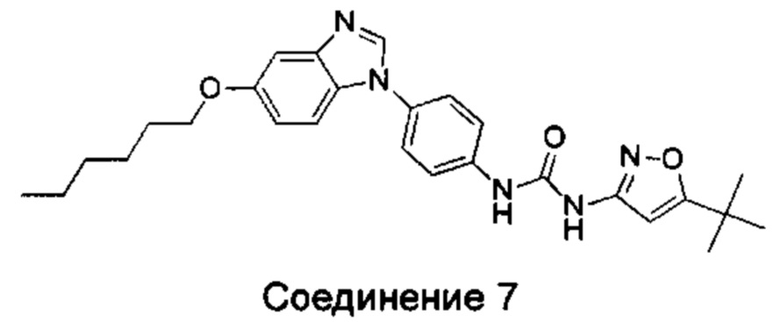
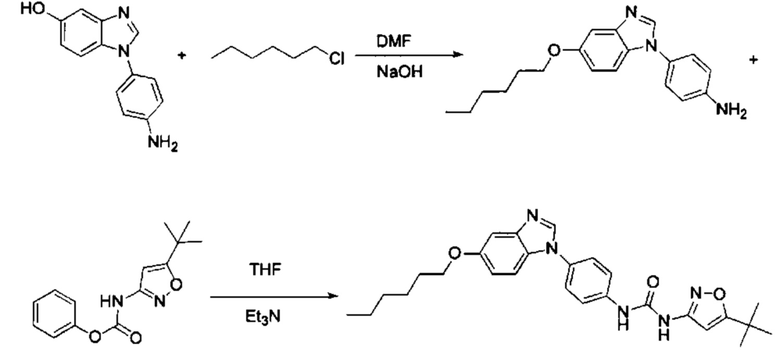
Стадия 1: Получение 4-(5-гексилоксил-бензимидазол-1-ил)-анилина
1-(4-амино-фенил)-1Н-бензимидазол-5-ол (синтезированный на Стадии 4 примера 4) (300 мг, 1.33 ммоль), 1-хлоргексан (240 мг, 2.0 ммоль, TCI) и гидроксид натрия добавили в 20 мл ДМФ, и смесь нагревали до 90°С в течение 2 часов. Реакционный раствор охладили до комнатной температуры, влили в воду (80 мл), и экстрагировали этилацетатом (50 мл × 2). Органическую фазу промыли дважды с помощью насыщенного раствора NaCl, высушили над безводным сульфатом натрия, и затем отфильтровали. Фильтрат сконцентрировали при пониженном давлении, с получением 400 мг неочищенного продукта 4-(5-гексилоксил-бензимидазол-1-ил)-анилина. Указанный продукт использовали сразу на следующей стадии без очистки.
Стадия 2: Методика была аналогична таковой, как на Стадии 5 в примере 4, за исключением того, что 4-(5-гексилоксил-бензимидазол-1-ил)-анилин использовали вместо 1-(4-амино-фенил)-1Н-бензимидазол-5-ола с получением 1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(5-гексилоксил-бензимидазол-1-ил)-фенил]-мочевины.
1HNMR (DMS0-d6, 400MHz) δ: 9.60 (s, IH), 9.07 (s, IH), 8.44 (s, IH), 7.67-7.69 (d, 2H), 7.58-7.60 (d, 2H), 7.45-7.47 (d, IH), 7.28-7.29 (d, IH), 6.94-6.96 (dd, IH), 6.53 (s, IH), 4.00-4.03 (t, 2H), 1.72-1.76 (m, 2H), 1.41-1.46 (m, 2H), 1.32-1.36 (m, 4H), 1.31 (s, 9H), 0.87-0.91 (t, 3H).
LC-MS (ESI): ESI 476.1 (M+H)+.
Пример 8
Получение 1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(5-изопропокси-бензимидазол-1-ил)-фенил]-мочевины (Соединение 8)
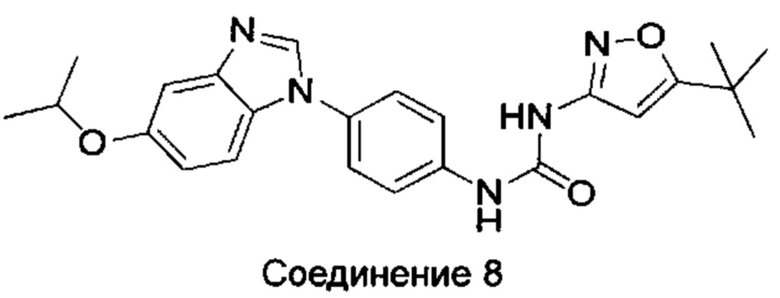
Способ получения был таким же, как в Примере 6, за исключением того, что йодизопропан (TCI) использовали вместо йодоэтана на Стадии 1, с получением 1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(5-изопропокси-бензимидазол-1-ил)-фенил]-мочевины.
1HNMR (DMSO-d6, 400MHz) δ: 9.60 (s, IH), 9.07 (s, IH), 8.46 (s, IH), 7.68-7.70 (d, 2H), 7.58-7.61 (d, 2H), 7.45-7.47 (d, IH), 7.31-7.32 (d, IH), 6.93-6.95 (dd, IH), 6.53 (s, IH), 4.61-4.67 (m, IH), 1.31 (s, 9H), 1.29-1.30 (d, 6H).
LC-MS (ESI): 434.1 (M+H)+
Пример 9
Получение 1-(5-трет-бутил-изоксазол-3-ил)-3-{4-[5-(3-метил-оксетан-3-илметокси)-бензимидазол-1-ил]-фенил]мочевины (Соединение 9)
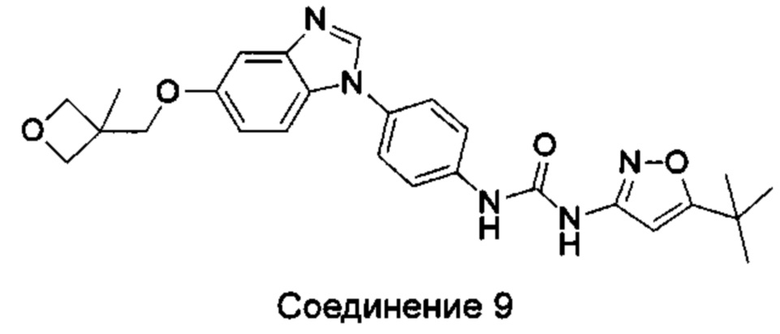
Способ получения был таким же, как в Примере 7, за исключением того, что 3-хлорметил-3-метилоксетан (Darui) использовали вместо 1-хлоргексана на Стадии 1, с получением 1-(5-трет-бутил-изоксазол-3-ил)-3-{4-[5-(3-метил-оксетан-3-илметоксил)-бензимидазол-1-ил]-фенил}мочевины.
1HNMR (DMSO-d6, 400MHz) δ: 9.59 (s, IH), 9.07 (s, IH), 8.45 (s, IH), 7.68-7.70 (d, 2H), 7.58-7.60 (d, 2H), 7.47-7.49 (d, IH), 7.35-7.36 (d, IH), 6.99-7.01 (dd, IH), 6.53 (s, IH), 4.52-4.54 (d, 2H), 4.32-4.34 (d, 2H), 4.11 (s, 2H), 1.40 (s, 3H), 1.31 (s, 9H).
LC-MS (ESI): ESI 476.1 (M+H)+.
Пример 10
Получение 1-(5-трет-бутил-изоксазол-3-ил)-3-{4-[5-(тетрагидрофуран-2-илметокси)-бензимидазол-1-ил]-фенил}-мочевины (Соединение 10)
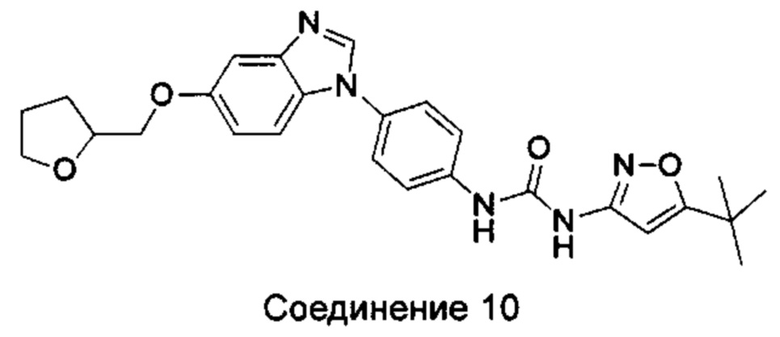
Способ получения был таким же, как в Примере 7, за исключением того, что 2-хлорметилтетрагидрофуран (Darui) использовали вместо 1-хлоргексана на Стадии 1, с получением 1-(5-трет-бутил-изоксазол-3-ил)-3-{4[5-(тетрагидрофуран-2-илметокси)-бензимидазол-1-ил]-фенил]мочевины.
1HNMR (DMSO-d6, 400MHz) δ: 9.59 (s, 1Н), 9.06 (s, 1Н), 8.44 (s, 1Н), 7.67-7.69 (d, 2Н), 7.58-7.60 (d, 2Н), 7.45-7.48 (d, 1H), 7.29-7.30 (d, 1H), 6.94-6.97 (dd, 1H), 6.53 (s, 1H), 4.16-4.22 (m, 1H), 3.95-4.04 (m, 2H), 3.78-3.83 (m, 1H), 3.67-3.72 (m, 1H), 1.98-2.05 (m, 1H), 1.81-1.93 (m, 2H), 1.68-1.73 (m, 1H), 1.31 (s, 9H).
LC-MS (ESI): ESI 476.1 (M+H)+.
Пример 11
Получение 1-(5-трет-бутил-изоксазол-3-ил)-3-{4-[5-(2-гидрокси-этоксил)-бензимидазол-1-ил]-фенил]-мочевины (Соединение 11)
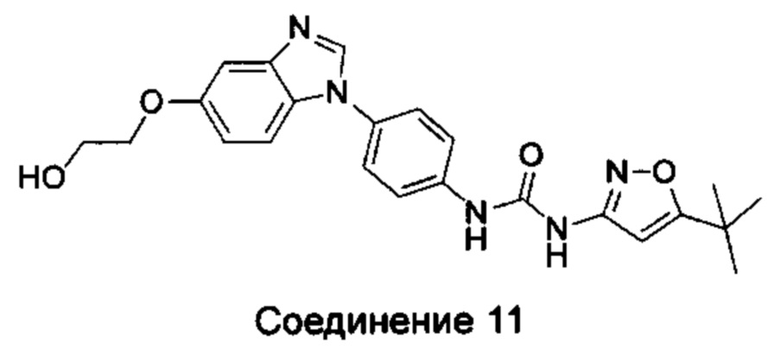
Способ получения был таким же, как в Примере 7, за исключением того, что бромэтанол (TCI) использовали вместо 1-хлоргексана на Стадии 1, с получением 1-(5-трет-бутил-изоксазол-3-ил)-3-{4-[5-(2-гидрокси-этоксил)-бензимидазол-1-ил]-фенил]-мочевины.
1HNMR (DMSO-d6, 400MHz) δ: 9.60 (s, 1Н), 9.07 (s, 1Н), 8.44 (s, 1Н), 7.67-7.69 20 (d, 2Н), 7.58-7.60 (d, 2Н), 7.46-7.48 (d, 1Н), 7.29-7.30 (d, 1Н), 6.95-6.98 (dd, 1Н), 6.53 (s, 1Н), 4.87-4.90 (t, 1Н), 4.02-4.06 (m, 2Н), 3.73-3.77 (m, 2H), 1.31 (s, 9H).
LC-MS (ESI): ESI 436.1 (M+H)+.
Пример 12
Получение 1-(5-трет-бутил-изоксазол-3-ил)-3-{4-[5-(2-метоксил-этоксил)-бензимидазол-1-ил]-фенил]-мочевины (Соединение 12)
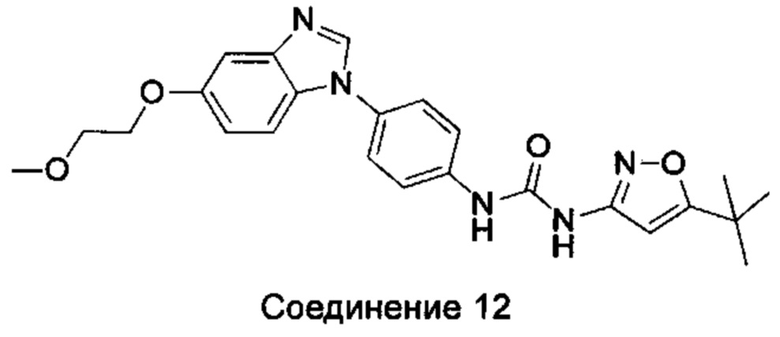
Стадия 1: Получение 4-[5-(2-метоксил-этоксил)-бензимидазол-1-ил]-анилина
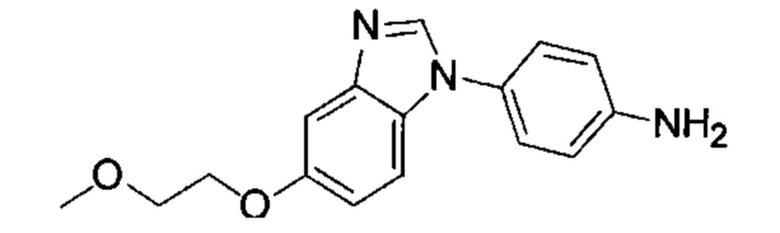
1-(4-амино-фенил)-1Н-бензимидазол-5-ол (полученный на Стадии 4 примера 4) (18.0 г, 0.08 моль) растворили в ДМФ (200 мл) при комнатной температуре, и добавили гидроксид натрия (9.6 г, 0.24 моль). Смесь перемешивали в течение 30 минут при комнатной температуре для преципитирования осадка. При комнатной температуре, добавили хлорэтилметиловый эфир (11.34 г, 0.12 моль, TCI) и смесь нагревали до 90°С в течение 1.5 часов. Реакционный раствор охладили до комнатной температуры, и медленно влили в 600 мл воды и осадок преципитировался. После перемешивания в течение 30 минут при комнатной температуре осадок отфильтровали и затем промыли водой и высушили под вакуумом с получением 20.5 г-[5-(2-метоксил-этоксил)-бензимидазол-1-ил]-анилина в виде розового осадка.
1HNMR (DMSO-d6, 400MHz) δ: 8.27 (s, 1H), 7.32-7.35 (d, 1H), 7.26 (d, 1H), 7.22-7.24 (d, 2H), 6.90-6.93 (dd, 1H), 6.72-6.74 (d, 2H), 5.41 (s, 2H), 4.12-4.15 (m, 10 2H), 3.67-3.70 (m, 2H), 3.33 (s, 3H).
LC-MS (ESI): 284.1 (M+H)+.
Стадия 2: Получение I-(5-трет-бутил-изоксазол-3-ил)-3-{4-[5-(2-метокси-этоксил)-бензимидазол-1-ил]-фенил}-мочевины
Методика была аналогична таковой, как на Стадии 5 в примере 4, за исключением того, что 4-[5-(2-метоксил-этоксил)-бензимидазол-1-ил]-анилин использовали вместо 1-(4-амино-фенил)-1Н-бензимидазол-5-ола с получением 1-(5-трет-бутил-изоксазол-3-ил)-3-{4-[5-(2-метокси-этоксил)-бензимидазол-1-ил]-фенил}-мочевины.
1HNMR (DMSO-d6, 400MHz) δ: 9.61 (s, 1H), 9.08 (s, 1H), 8.52 (s, 1H), 7.68-7.70 (d, 2H), 7.59-7.62 (d, 2H), 7.47-7.50 (d, 1H), 7.38 (d, 1H), 6.98-7.01 (dd, 1H), 6.53 (s, 1H), 4.13-4.15 (t, 2H), 3.68-3.71 (t, 2H), 3.33 (s, 3Н), 1.30 (s, 9H).
LC-MS (ESI): 450.1 (M+H)+.
Пример 13
Получение 1-(5-трет-бутил-изоксазол-3-ил)-3-{4-[5-(2-этоксил-этоксил)-бензимидазол-1-ил]-фенил]-мочевины (Соединение 13)
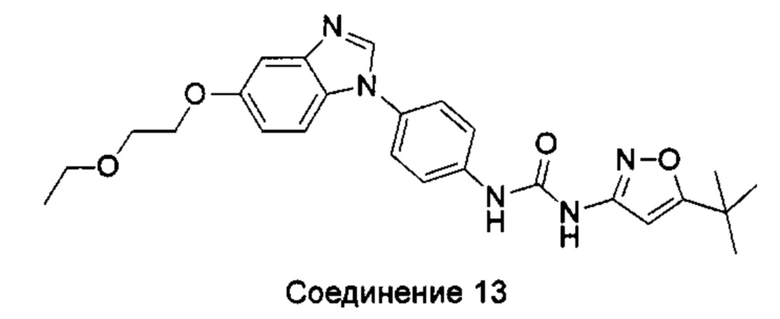
Способ получения был таким же, как в Примере 7, за исключением того, что 2-хлорэтилэтиловый эфир (Darui) использовали вместо 1-хлоргексана на Стадии 1 с получением 1-(5-трет-бутил-изоксазол-3-ил)-3-{4-[5-(2-этоксил-этоксил)-бензимидазол-1-ил]-фенил}-мочевины.
1HNMR (DMSO-d6, 400MHz) δ: 9.60 (s, 1H), 9.07 (s, 1H), 8.46 (s, 1H), 7.67-7.70 (d, 2H), 7.58-7.60 (d, 2H), 7.46-7.48 (d, 1H), 7.31 (d, 1H), 6.96-6.99 (dd, 1H), 6.53 (s, 1H), 4.13-4.16 (t, 2H), 3.72-3.74 (t, 2H), 3.50-3.55 (q, 2H), 1.31 (s, 9H), 1.13-1.17 (t, 3Н).
LC-MS (ESI): ESI 464.1 (M+H)+.
Пример 14
Получение 1-(5-трет-бутил-изоксазол-3-ил)-3-(4-{5-[2-(2-гидрокси-этоксил)-этоксил]-бензимидазол-1-ил}-фенил)-мочевины (Соединение 14)
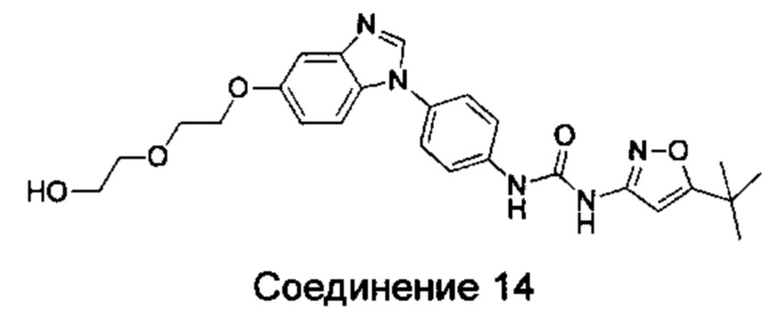
Способ получения был таким же, как в Примере 7, за исключением того, что 2-хлорэтоксиэтанол (Titan) использовали вместо 1-хлоргексана на Стадии 1, с получением 1-(5-трет-бутил-изоксазол-3-ил)-3-(4-{5-[2-(2-гидроксил-этоксил)-этоксил]-бензимидазол-1-ил}-фенил)-мочевины.
1HNMR (DMSO-d6, 400MHz) δ: 9.60 (s, 1Н), 9.07 (s, 1Н), 8.47 (s, 1Н), 7.68-7.70 (d, 2Н), 7.59-7.61 (d, 2Н), 7.47-7.49 (d, 1Н), 7.32-7.33 (d, 1Н), 6.96-6.99 (dd, 1Н), 6.53 (s, 1Н), 4.66 (br, 1H), 4.15-4.16 (t, 2H), 3.77-3.79 (t, 2H), 3.49-3.52 (m, 4H), 1.30 15 (s, 9H).
LC-MS (ESI): ESI 480.1 (M+H)+.
Пример 15
Получение 1-{4-[3-(5-трет-бутил-изоксазол-3-ил)-карбамидо]-фенил}-1Н-бензимидазол-5-ил морфолин-4-карбоксилата (Соединение 15)
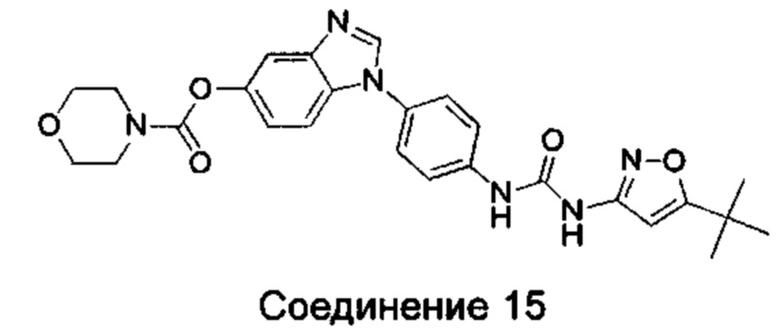
Соединение 7 (41 мг, 0.105 ммоль), триэтиламин (32 мг, 0.315 ммоль) и DMAP (3 мг) растворили в 10 мл ТГФ, и 4-морфолинкарбонилхлорид (19 мг, 0.126 ммоль, Darui) добавили при комнатной температуре. Смесь перемешивали при комнатной температуре в течение ночи. На следующий день реакционный раствор влили в воду (30 мл) и экстрагировали этилацетатом (30 мл × 2). Органическую фазу промыли дважды с помощью насыщенного раствора NaCl, высушили над безводным сульфатом натрия, отфильтровали и фильтрат сконцентрировали при пониженном давлении. Остатки очистили с помощью колоночной хроматографии (элюент: дихлорметан/метанол) с получением 20 мг белого осадка 1-{4-[3-(5-трет-бутил-изоксазол-3-ил)-карбамидо]-фенил}-1Н-бензимидазол-5-ил морфолин-4-карбоксилата.
1HNMR (DMSO-d6, 400MHz) δ: 9.61 (s, 1Н), 9.13 (s, 1Н), 8.55 (s, 1Н), 7.69-7.71 (d, 2Н), 7.60-7.63 (d, 2Н), 7.54-7.56 (d, 1Н), 7.52-7.53 (d, 1Н), 7.10-7.13 (dd, 1Н), 6.53 (s, 1Н), 3.58-3.62 (m, 8Н), 1.30 (s, 9Н).
LC-MS (ESI): 505.2 (М+Н)+.
Пример 16
Получение 1-(5-трет-бутил-изоксазол-3-ил)-3-{4-[5-(2-морфолин-4-ил-этоксил)-бензимидазол-1-ил]-фенил}-мочевины (Соединение 16)
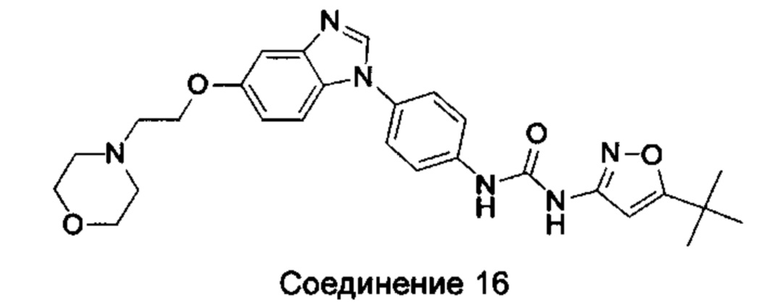
Стадия 1: Получение 4-[5-(2-морфолин-4-ил-этоксил)-бензимидазол-1-ил]-анилина
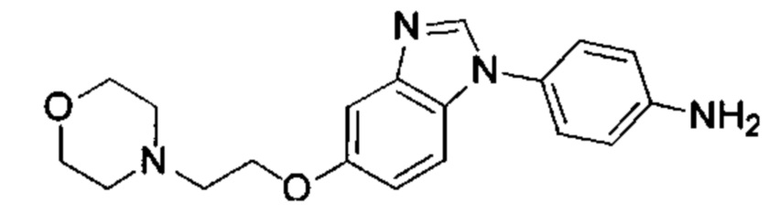
1-(4-аминофенил)-1Н-бензимидазол-5-ол (полученный на Стадии 4 примера 4) (3.0 г, 0.013 моль) растворили в ДМФ (200 мл), гидроксид натрия (1.60 г, 0.04 моль) и N-(2-хлорэтил) морфолина гидрохлорид (3.76 г, 0.020 моль, TCI) добавили при комнатной температуре и перемешивали в течение 30 минут. Смесь нагрели до 90°С и дали прореагировать в течение ночи. Реакционный раствор охладили до комнатной температуры и медленно влили в 100 мл воды для преципитирования осадка. После перемешивания при комнатной температуре в течение 30 минут осадок, полученный посредством фильтрации, промыли водой и высушили под вакуумом с получением 3.4 г 4-[5-(2-морфолин-4-ил-этоксил)-бензимидазол-1-ил]-анилина в виде желтого осадка.
1HNMR (DMSO-d6, 400MHz) δ: 8.27 (s, 1H), 7.32-7.34 (d, 1H), 7.27 (d, 1H), 20 7.22-7.24 (d, 2H), 6.89-6.92 (dd, 1H)), 6.72-6.74 (d, 2H), 5.41 (s, 2H), 4.11-4.14 (t, 2H), 3.58-3.60 (t, 4H), 2.70-2.73 (t, 2H), 2.48-2.50 (m, 4H).
LC-MS (ESI): 339.1 (M+H)+.
Стадия 2: Получение 1-(5-трет-бутил-изоксазол-3-ил)-3-{4-[5-(2-морфолин-4-ил-этоксил)-бензимидазол-1-ил]-фенил}-мочевины
Методика была аналогична таковой, как на Стадии 5 примера 4, за исключением того, что 4-[5-(2-морфолин-4-ил-этоксил)-бензимидазол-1-ил]-анилин использовали вместо 1-(4-амино-фенил)-1Н-бензимидазол-5-ола с получением 1-(5-трет-бутил-изоксазол-3-ил)-3-{4-[5-(2-морфолин-4-ил-этоксил)-бензимидазол-1-ил]-фенил}-мочевины.
1HNMR (DMSO-d6, 400MHz) δ: 9.61 (s, 1Н), 8.20 (s, 1Н), 8.06 (s, 1Н,), 7.75-7.77 (d, 2Н), 7.46-7.48 (d, 2Н), 7.38-7.41 (d, 1Н), 7.35-7.36 (d, 1Н), 6.99-7.02 (dd, 1Н), 30 5.94 (s, 1Н), 4.20-4.23 (t, 2Н), 3.76-3.79 (t, 4Н), 2.86-2.89 (t, 2Н), 2.62-2.64 (t, 4Н),
1.39 (s, 9Н).
LC-MS (ESI): 505.3 (M+H)+.
Пример 17
Получение 1-(5-трет-бутил-изоксазол-3-ил)-3-{4-[5-(2-пиперидин-1-ил-этоксил)-бензимидазол-1-ил]-фенил}-мочевины (Соединение 17)
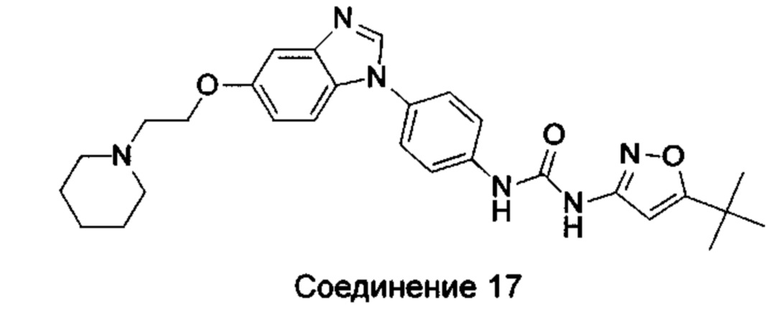
Способ получения был таким же, как в Примере 7, за исключением того, что 1-(2-хлор-этил)-пиперидина гидрохлорид (Runjie) использовали вместо 1-хлоргексана на Стадии 1 с получением 1-(5-трет-бутил-изоксазол-3-ил)-3-{4-[5-(2-пиперидин-1-ил-этоксил)-бензимидазол-1-ил]-фенил}-мочевины.
1HNMR (DMSO-d6, 400MHz) δ: 9.59 (s, 1H), 9.07 (s, 1H), 8.43 (s, 1H), 7.67-7.69 10 (d, 2H), 7.57-7.59 (d, 2H), 7.45-7.47 (d, 1H), 7.29-7.30 (d, 1H), 6.94-6.96 (dd, 1H), 6.52 (s, 1H), 4.11-4.14 (t, 2H), 2.67-2.70 (t, 2H), 2.45 (m, 4H), 1.48-1.54 (m, 4H), 1.38-1.41 (m, 2H), 1.30 (s, 9H).
LC-MS (ESI): 503.3 (M+H)+.
Пример 18
Получение 1-{4-[5-(2-азепан-1-ил-этоксил)-бензимидазол-1-ил]-фенил}3-(5-трет-бутил-изоксазол-3-ил)-мочевины (Соединение 18)
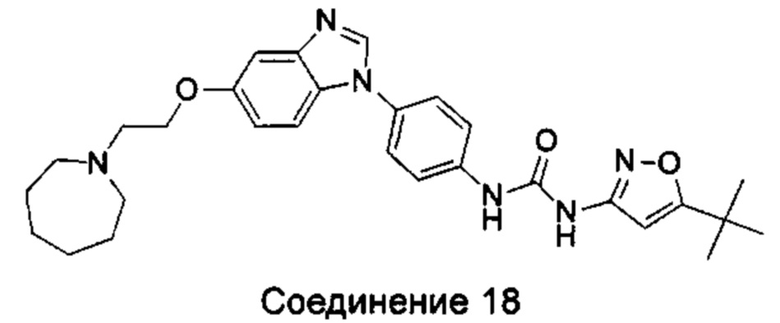
Способ получения был таким же, как в Примере 7, за исключением того, что 2-(азепанил)этилхлорида гидрохлорид (Runjie) использовали вместо 1-хлоргексана на Стадии 1 с получением 1-{4-[5-(2-азепан-1-ил-этоксил)-бензимидазол-1-ил]-фенил}3-(5-трет-бутил-изоксазол-3-ил)-мочевины.
1HNMR (DMSO-d6, 400MHz) δ: 9.62 (s, 1H), 9.13 (s, 1H), 8.43 (s, 1H), 7.67-7.69 (d, 2H), 7.57-7.59 (d, 2H), 7.45-7.48 (d, 1H), 7.30 (d, 1H), 6.94-6.97 (dd, 1H), 6.52 (s, 25 1H), 4.11-4.14 (t, 2H), 2.95 (m, 2H), 2.77 (m, 4H), 1.62 (m, 4H), 1.55 (m, 4H), 1.30 (s, 9H).
LC-MS (ESI): 517.3 (M+H)+.
Пример 19
Получение 1-(5-трет-бутил-изоксазол-3-ил)-3-(4-{5-[3-(4-метил-пиперазин-1-ил)-пропоксил]-бензимидазол-1-ил}-фенил)-мочевины (Соединение 19)
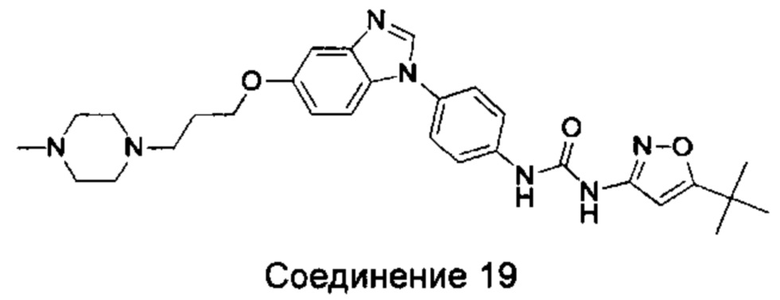
Способ получения был таким же, как в Примере 7, за исключением того, что 1-(3-хлорпропил)-4-метилпиперазина дигидрохлорид (TCI) использовали вместо 1-хлоргексана на Стадии 1 с получением 1-(5-трет-бутил-изоксазол-3-ил)-3-(4-{5-[3-(4-метил-пиперазин-1-ил)-пропоксил]-бензимидазол-1-ил}-фенил)-мочевины.
1HNMR (DMSO-d6, 400MHz) δ: 9.60 (s, 1H), 9.08 (s, 1H), 8.43 (s, 1H), 7.67-7.69 (d, 2H), 7.57-7.59 (d, 2H), 7.45-7.47 (d, 1H), 7.27-7.28 (d, 1H), 6.93-6.96 (dd, 1H), 6.53 (s, 1H), 4.04-4.07 (t, 2H), 2.43-2.46 (t, 2H), 2.35 (m, 8H), 2.16 (s, 3H), 1.87-1.90 (m, 2H), 1.30 (s, 9H).
LC-MS (ESI): 532.3 (M+H)+
Пример 20
Получение 1-(5-трет-бутил-изоксазол-3-ил)-3-{4-[5-(2-диметиламино-этоксил)-бензимидазол-1-ил]-фенил}-мочевины (Соединение 20)
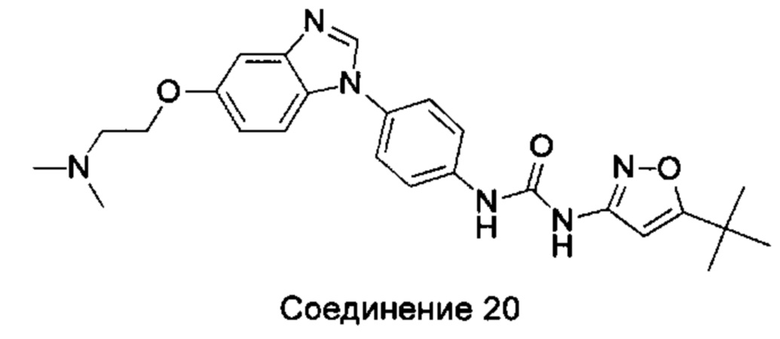
Способ получения был таким же, как в Примере 7, за исключением того, что диметиламинохлорэтана гидрохлорид (Runjie) использовали вместо 1-хлоргексана на Стадии 1 с получением 1-(5-трет-бутил-изоксазол-3-ил)-3-{4-[5-(2-диметиламино-этоксил)-бензимидазол-1-ил]-фенил}-мочевины.
1HNMR (DMSO-d6, 400MHz) δ: 9.56 (s, 1H), 8.89 (s, 1H), 8.04 (s, 1H), 7.71-7.73 (d, 2H), 7.41-7.43 (d, 2H), 7.34-7.36 (m, 2H), 6.99-7.02 (dd, 1H), 6.11 (s, 1H), 4.20-4.22 (t, 2H), 2.89-2.92 (t, 2H), 2.47 (s, 6H), 1.37 (s, 9H).
LC-MS (ESI): 463.2 (M+H)+.
Пример 21
Получение 1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(5-трифторметокси-бензимидазол-1-ил)-фенил]-мочевины (Соединение 21)
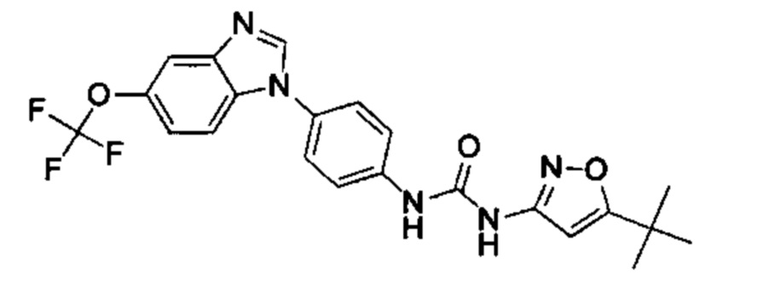
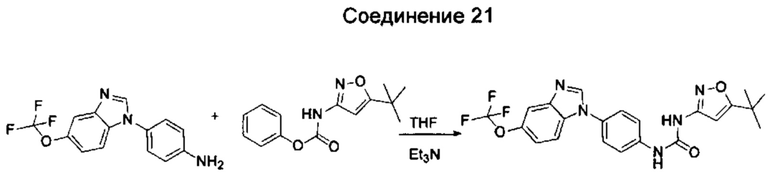
Стадия 1: Получение 4-(5-трифторметокси-бензимидазол-1-ил)-анилина
Методика была аналогична таковой, как на Стадиях 1-3 в примере 4, за исключением того, что 2-нитро-4-трифторметоксианилин использовали вместо 2-нитро-4-метоксиланилина на Стадии 1 с получением 4-(5-трифторметокси-бензимидазол-1-ил)-анилина.
Стадия 2: Получение 1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(5-трифторметокси-бензимидазол-1-ил)-фенил]-мочевины
Методика была аналогична таковой, как на Стадии 5 в примере 4, за исключением того, что 4-(5-трифторметокси-бензимидазол-1-ил)-анилин использовали вместо 1-(4-амино-фенил)-1Н-бензимидазол-5-ола с получением 1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(5-трифторметоксиI-бензимидазол-1-ил)-фенил]-мочевины.
1HNMR (DMSO-d6, 400MHz) δ: 9.62 (s, IH), 9.10 (s, IH), 8.66 (s, IH,), 7.79-7.80 (d, 1H), 7.70-7.72 (d, 2H), 7.61-7.64 (m, 3H), 7.32-7.35 (dd, 1H), 6.53 (s, 1H), 1.31 (s, 9H)
LC-MS (ESI): ESI 460.0 (M+H)+.
Пример 22
Получение 1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(5-фтор-бензимидазол-1-ил)-фенил]-мочевины (Соединение 22)
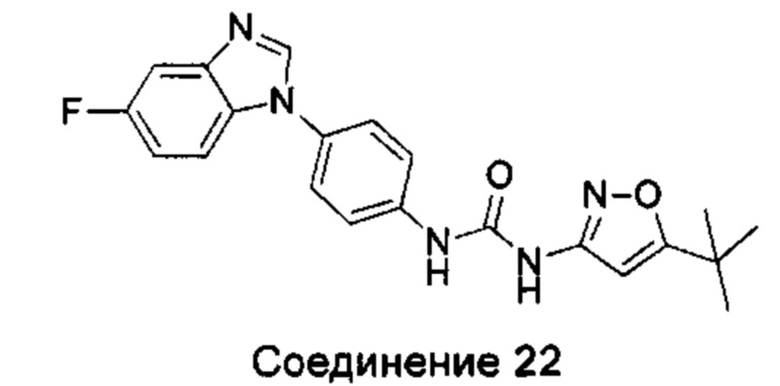
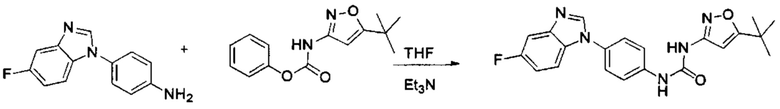
Стадия 1: Получение 4-(5-фтор-бензимидазол-1-ил)-анилина
Методика была аналогична таковой, как на Стадии 4 в примере 3, за исключением того, что 2-нитро-4-фторанилин использовали вместо 2-нитро-4-метоксиланилина на Стадии 1 с получением 4-(5-фторо-бензимидазол-1-ил)-анилина.
Стадия 2: Получение 1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(5-фтор-бензимидазол-1-ил)-фенил]-мочевины
Методика была аналогична таковой, как на Стадии 5 в примере 4, за исключением того, что 4-(5-фтор-бензимидазол-1-ил)-анилин использовали вместо 1-(4-амино-фенил)-1Н-бензимидазол-5-ола с получением 1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(5-фтор-бензимидазол-1-ил)-фенил]-мочевины.
1HNMR (DMSO-d6, 400MHz) δ: 9.61 (s, 1Н), 9.10 (s, 1Н), 8.57 (s, 1Н), 7.68-30 7.71 (d, 2Н), 7.56-7.62 (m, 4Н), 7.20-7.23 (m, 1Н), 6.52 (s, 1Н), 1.28 (s, 9Н).
LC-MS (ESI): 394.1 (M+H)+.
Пример 23
Получение 1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(5-трифторметил-бензимидазол-1-ил)-фенил]-мочевины (Соединение 23)
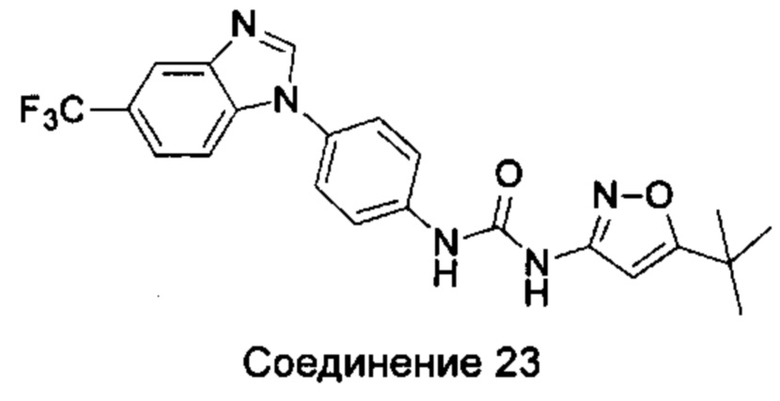
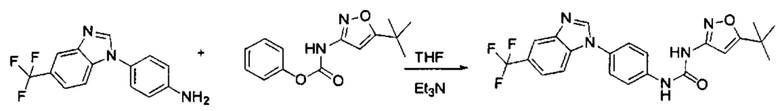
Стадия 1: Получение 4-(5-трифторметил-бензимидазол-1-ил)-анилина
Методика была аналогична таковой, как на Стадиях 1-3 примера 4, за исключением того, что 2-нитро-4-трифторметиланилин использовали вместо 2-нитро-4-метоксиланилина на Стадии 1 с получением 4-(5-трифторметил-бензимидазол-1-ил)-анилина.
Стадия 2: Получение 1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(5-трифторметил-бензимидазол-1-ил)-фенил]-мочевины
Методика была аналогична таковой, как на Стадии 5 в примере 4, за исключением того, что 4-(5-трифторметил-бензимидазол-1-ил)-анилин использовали вместо 1-(4-амино-фенил)-1Н-бензимидазол-5-ола с получением 1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(5-трифторметил-бензимидазол-1-ил)-фенил]-мочевины.
1HNMR (DMSO-d6, 400MHz) δ: 9.62 (s, 1Н), 9.12 (s, 1Н), 8.73 (s, 1Н), 8.16 (d, 1Н), 7.75-7.77 (d, 1H), 7.71-7.73 (d, 2H), 7.63-7.66 (m, 3Н), 6.53 (s, 1H), 1.30 (s, 9H).
LC-MS (ESI): 444.1 (M+H)+.
Пример 24
Получение 1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(5-метил-бензимидазол-1-ил)-фенил]-мочевины (Соединение 24)
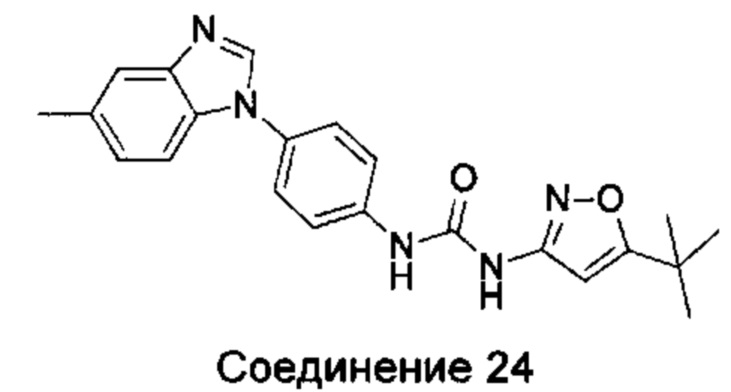
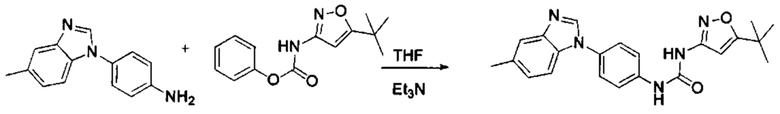
Стадия 1: Получение 4-(5-метил-бензимидазол-1-ил)-анилина
Методика была аналогична таковой, как на Стадиях 1-3 в примере 4, за исключением того, что 2-нитро-4-метиланилин использовали вместо 2-нитро-4-метоксиланилина на Стадии 1 с получением 4-(5-метил-бензимидазол-1-ил)-анилина.
Стадия 2: Получение 1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(5-метил-бензимидазол-1-ил)-фенил]-мочевины
Методика была аналогична таковой, как на Стадии 5 в примере 4, за исключением того, что 4-(5-метил-бензимидазол-1-ил)-анилин использовали вместо 1-(4-амино-фенил)-1Н-бензимидазол-5-ола с получением 1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(5-трифторметил-бензимидазол-1-ил)-фенил]-мочевины.
1HNMR (DMSO-d6, 400MHz) δ: 9.60 (s, 1H), 9.07 (s, 1H), 8.45 (s, 1H), 7.68-5 7.70 (d, 2H), 7.58-7.60 (d, 2H), 7.56 (m, 1H), 7.46-7.48 (d, 1H), 7.14-7.17 (dd, 1H), 6.53 (s, 1H), 2.45 (s, 3H), 1.31 (s, 9H).
LC-MS (ESI): 390.1 (M+H)+.
Пример 25
Получение 1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(6-метоксил-бензимидазол-1-ил)-фенил]-мочевины (Соединение 25)
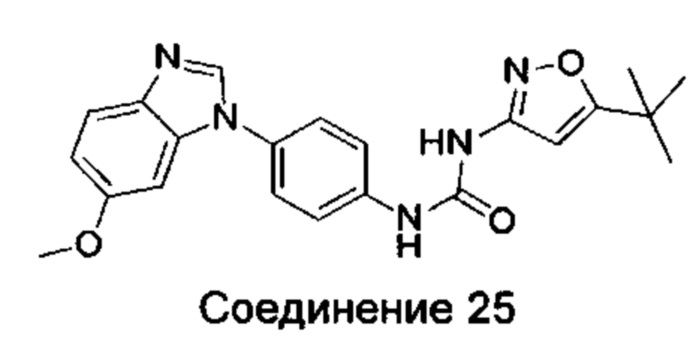
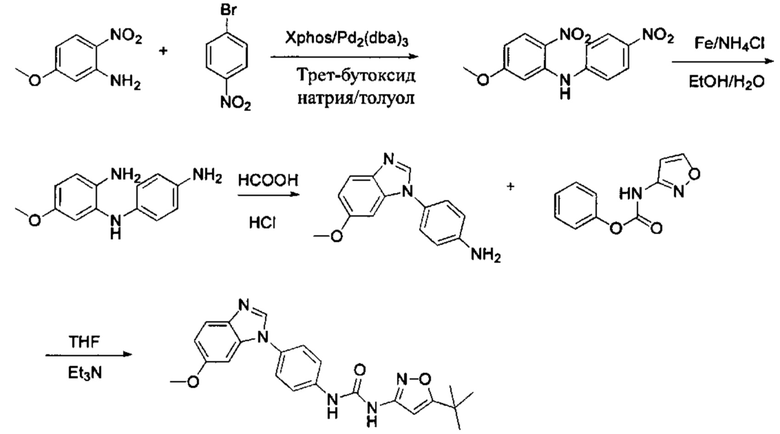
Стадия 1: Получение (5-метоксил-2-нитро-фенил)-(4-нитро-фенил)-амина
В атмосфере азота, 2-нитро-5-метоксиланилина (1.5 г, 8.92 ммоль), п-бромнитробензол (3.6 г, 17 ммоль), Xphos (425 мг), Pd2(dba)3 (408 мг) и трет-бутоксида натрия (1.71 г, 17.8 ммоль) растворили в 30 мл толуола, и смесь нагрели до 90°С и дали прореагировать в течение 3 часов. Затем реакционный раствор охладили до комнатной температуры, 100 мл дихлорметана добавили и смесь перемешивали при комнатной температуре в течение 5 минут, и затем отфильтровали и фильтрат сконцентрировали при пониженном давлении. Остатки очистили с помощью колоночной хроматографии (элюент: петролейный эфир/этилацетат) с получением 1.7 г (5-метоксил-2-нитро-фенил)-(4-нитро-фенил)-амина в виде красно-черного осадка.
Стадия 2: Получение (5-метоксил-2-амино-фенил)-(4-амино-фенил)-амина
(5-метоксил-2-нитро-фенил)-(4-нитро-фенил)-амин (1.7 г, 5.88 ммоль), полученный на Стадии 1, восстановленный порошок железа (2.63 г, 47.0 ммоль), хлорид аммония (3.15 г, 58.8 ммоль) нагревали до 90°С в этаноле (50 мл)/вода (12.5 мл) и дали прореагировать в течение 1 ч. Реакционный раствор охладили до комнатной температуры, и затем медленно влили в насыщенный водный раствор бикарбоната натрия (150 мл), экстрагировали этилацетатом (100 мл × 2), и органическую фазу промыли дважды с помощью насыщенного раствора NaCl, высушили над безводным сульфатом натрия, отфильтровали и фильтрат сконцентрировали при пониженном давлении с получением 1.3 неочищенного продукта, который сразу использовали на следующей стадии без очистки.
Стадия 3: Получение 4-(6-метоксил-бензимидазол-1-ил)-анилина
(5-метоксил-2-амино-фенил)-(4-амино-фенил)-амин, полученный на Стадии 2 (1.3 г, 5.67 ммоль) растворили в 50 мл соляной кислоты (4 моль/л), 1.5 мл муравьиной кислоты добавили при комнатной температуре, и смесь нагрели до 120°С и дали прореагировать в течение 1 часа. Реакционный раствор затем довели до рН>9 с помощью водного аммония в ванне со льдом, и экстрагировали этилацетатом (80 мл × 2).Органическую фазу промыли с помощью насыщенного раствора NaCl, высушили над безводным сульфатом натрия, отфильтровали, и фильтрат сконцентрировали при пониженном давлении. Остатки очистили с помощью колоночной хроматографии (элюент: дихлорметан/метанол) с получением 1.0 г 4-(6-метоксил-бензимидазол-1-ил)-анилина в виде темно-желтого осадка.
Стадия 4: Получение 1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(6-метоксил-бензимидазол-1-ил)-фенил]-мочевины
Методика была аналогична таковой, как на Стадии 5 в примере 4, за исключением того, что 4-(6-метоксил-бензимидазол-1-ил)-фениламин использовали вместо 1-(4-амино-фенил)-1Н-бензимидазол-5-ола с получением 1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(6-метоксил-бензимидазол-1-ил)-фенил]-мочевины.
1HNMR (DMSO-d6, 400MHz) δ: 9.61 (s, IH), 9.07 (s, IH), 8.35 (s, IH), 7.69-7.71 (d, 2H), 7.63-7.66 (d, 1H), 7.59-7.62 (d, 2H), 7.01-7.02 (d, 1H), 6.90-6.93 (dd, 1H), 6.53 (s, 1H), 3.79 (s, 3H), 1.31 (s, 9H).
LC-MS (ESI): 406.1 (M+H)+.
Пример 26
Получение I-(5-трет-бутил-изоксазол-3-ил)-3-{4-[6-(2-метоксил-этоксил)-бензимидазол-1-ил]-фенил]мочевины (Соединение 26)
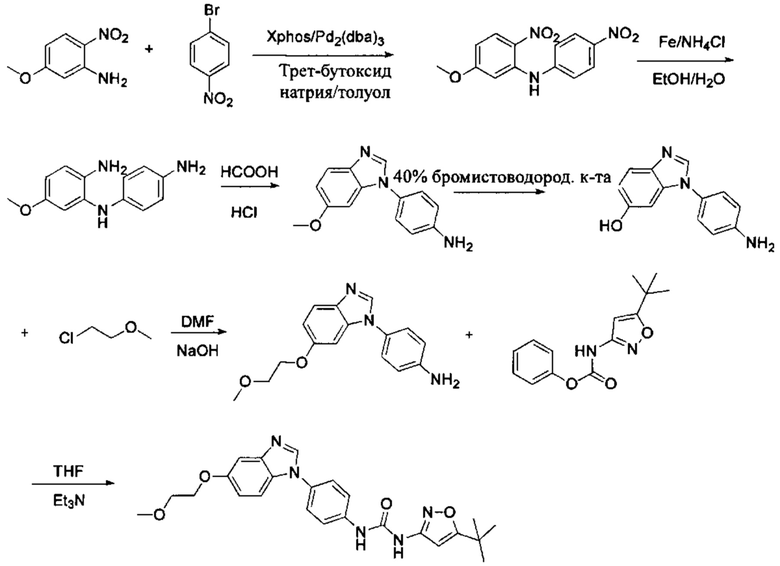
Стадия 1: Получение (5-метоксил-2-нитро-фенил)-(4-нитро-фенил)-амина
В атмосфере азота, 2-нитро-5-метоксиланилин (1.5 г, 8.92 ммоль), п-бромнитробензол (3.6 г, 17 ммоль), Xphos (425 мг), Pd2(dba)3 (408 мг), и трет-бутоксид натрия (1.71 г, 17.8 ммоль) растворили в 30 мл толуола, и смесь нагрели до 90°С и дали прореагировать в течение 3 часов. Затем реакционный раствор охладили до комнатной температуры, 100 мл дихлорметана добавили и перемешивали при комнатной температуре в течение 5 минут и затем отфильтровали. Фильтрат сконцентрировали при пониженном давлении. Остатки очистили с помощью колоночной хроматографии (элюент: петролейный эфир / этилацетат) с получением 1.7 г (5-метоксил-2-нитро-фенил)-(4-нитро-фенил)-амина в виде красно-черного осадка.
Стадия 2: Получение (5-метоксил-2-амино-фенил)-(4-амино-фенил)-амина
(5-метоксил-2-нитро-фенил)-(4-нитро-фенил)-амин (1.7 г, 5.88 ммоль), полученный на Стадии 1, восстановленный порошок железа (2.63 г, 47.0 ммоль), хлорид аммония (3.15 г, 58.8 ммоль) нагревали до 90°С в этаноле (50 мл)/ воде (12.5 мл) и дали прореагировать в течение 1 часа. Реакционный раствор охладили до комнатной температуры, медленно влили в насыщенный водный раствор бикарбоната натрия (150 мл), и экстрагировали этилацетатом (100 мл × 2). Органическую фазу промыли дважды с помощью насыщенного раствора NaCl, высушили над безводным сульфатом натрия, отфильтровали, и фильтрат сконцентрировали при пониженном давлении с получением 1.3 г неочищенного продукта, который сразу использовали на следующей стадии без очистки.
Стадия 3: Получение 4-(6-метоксил-бензимидазол-1-ил)анилина (1.3 г, 5.67 ммоль)
(5-метоксил-2-амино-фенил)-(4-амино-фенил)-амин (1.3 г, 5.67 ммоль), полученный на Стадии 2 растворили в 50 мл соляной кислоты (4 моль/л) и 1.5 мл муравьиной кислоты добавили при комнатной температуре. Смесь нагрели до 120°С и дали прореагировать в течение 1 часа, затем реакционный раствор довели до рН>9 с помощью водного аммония в ванне со льдом, экстрагировали этилацетатом (80 мл × 2), и органическую фазу промыли с помощью насыщенного раствора NaCl, высушили над безводным сульфатом натрия, и отфильтровали. Фильтрат сконцентрировали при пониженном давлении. Остатки очистили с помощью колоночной хроматографии (элюент: дихлорметан/метанол) с получением 1.0 г 4-(6-метоксил-бензимидазол-1-ил)-анилина в виде темно-желтого осадка.
Стадия 4: Получение 3-(4-амино-фенил)-3Н-бензимидазол-5-ола
4-(6-метоксил-бензимидазол-1-ил)-анилин (600 мг, 3.66 ммоль), полученный на Стадии 3 нагрели до 120°С в 15 мл 40% бромистоводородной кислоты и дали прореагировать в течение 6 часов. Затем реакционный раствор охладили до комнатной температуры, и преципитировался серо-белый осадок, и отфильтровали. Получившийся осадок растворили в 70 мл воды. Фильтрат довели до приблизительно рН=7 с помощью водного аммония, и белый осадок преципитировался. Осадок отфильтровали и промыли водой и высушили под вакуумом с получением 485 мг 3-(4-амино-фенил)-3Н-бензимидазол-5-ола в виде белого осадка.
Стадия 5: Получение 4-[6-(2-метоксил-этоксил)-бензимидазол-1-ил]-анилина
3-(4-амино-фенил)-1Н-бензимидазол-5-ол (300 мг, 1.33 ммоль), полученный на Стадии 4, 2-хлорэтилметиловый эфир (372.0 мг, 2.0 ммоль) и гидроксид натрия (160 мг, 4 ммоль) нагревали до 90°С в 20 мл ДМФ и дали прореагировать в течение 2 ч. Реакционный раствор охладили до комнатной температуры, влили в воду (80 мл) и экстрагировали этилацетатом (50 мл × 2).Органическую фазу промыли дважды с помощью насыщенного раствора NaCl, высушили над безводным сульфатом натрия, отфильтровали, и фильтрат сконцентрировали при пониженном давлении, с получением 360 мг неочищенного продукта, который сразу использовали в следующей реакции без очистки.
Стадия 6: Получение 1-(5-трет-бутил-изоксазол-3-ил)-3-{4-[6-(2-метоксил-этоксил)-бензимидазол-1-ил]-фенил}-мочевины
4-[6-(2-метоксил-этоксил)-бензимидазол-1-ил]-анилин (100 мг, 0.295 ммоль), полученный на Стадии 5, фенил (5-трет-бутил-изоксазол-3-ил)-карбамат (115.3 мг, 0.443 ммоль) и триэтиламин (90 мг, 0.888 ммоль) растворили в 10 мл ТГФ и реакционную смесь кипятили с обратным холодильником в течение ночи. На следующий день реакционный раствор сконцентрировали при пониженном давлении и остатки очистили с помощью колоночной хроматографии (элюент: дихлорметан/метанол) с получением 60 мг 1-(5-трет-бутил-изоксазол-3-ил)-3-{4-[6-(2-метоксил-этоксил)-бензимидазол-1-ил]-фенил}-мочевины в виде белого осадка.
1HNMR (DMSO-d6, 400MHz) δ: 9.60 (s, 1H), 9.07 (s, 1H), 8.36 (s, 1H), 7.68-7.70 25 (d, 2H,), 7.63-7.65 (d, 1H), 7.59-7.61 (d, 2H), 7.03-7.04 (d, 1H), 6.91-6.94 (dd, 1H), 6.53 (s, 1H), 4.11-4.13 (m, 2H), 3.65-3.68 (m, 2H), 3.31 (s, 3Н), 1.31 (s, 9H).
LC-MS (ESI): ESI 450.2 (M+H)+
Пример 27
Получение 1-(5-трет-бутил-изоксазол-3-ил)-3-{4-[6-(2-морфолин-4-ил-этоксил)-бензимидазол-1-ил]-фенил}-мочевины (Соединение 27)
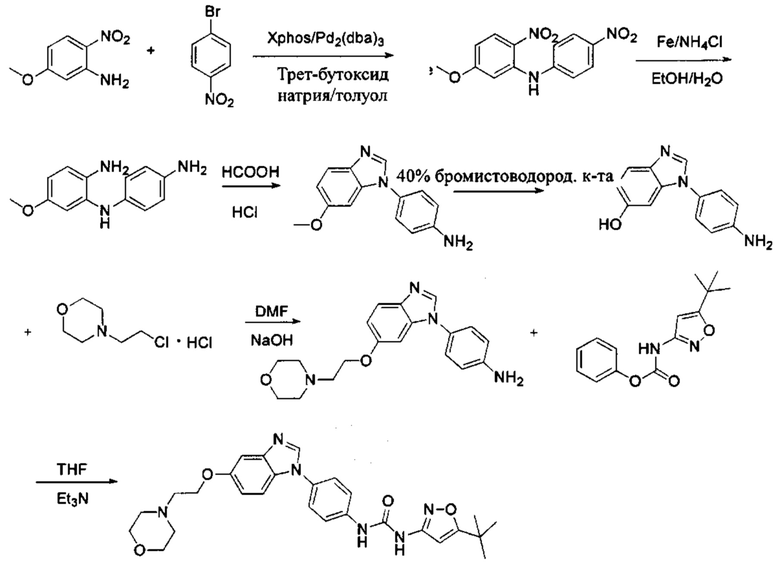
Стадия 1: Получение (5-метоксил-2-нитро-фенил)-(4-нитро-фенил)-амина
В атмосфере азота, 2-нитро-5-метоксиланилин (1.5 г, 8.92 моль, Darui), п-бромнитробензол (3.6 г, 17 ммоль), Xphos (425 мг), Pd2(dba)3 (408 мг), и трет-бутоксид натрия (1.71 г, 17.8 ммоль) растворили в 30 мл толуола. Смесь нагрели до 90°С и дали прореагировать в течение 3 часов. Затем реакционный раствор охладили до комнатной температуры, 100 мл дихлорметана добавили, и затем перемешивали при комнатной температуре в течение 5 минут. Смесь отфильтровали, и фильтрат сконцентрировали при пониженном давлении. Остатки очистили с помощью колоночной хроматографии (элюент: петролейный эфир/этилацетат) с получением 1.7 г (5-метоксил-2-нитро-фенил)-(4-нитро-фенил)-амина в виде красно-черного осадка.
Стадия 2: Получение (5-метоксил-2-амино-фенил)-(4-амино-фенил)-амина
(5-метоксил-2-нитро-фенил)-(4-нитро-фенил)-амин (1.7 г, 5.88 ммоль), полученный на Стадии 1, восстановленный порошок железа (2.63 г, 47.0 ммоль), и хлорид аммония (3.15 г, 58.8 ммоль) нагревали до 90°С в этаноле (50 мл)/вода (12.5 мл) и дали прореагировать в течение 1 часа. Реакционный раствор охладили до комнатной температуры и медленно влили в насыщенный водный раствор бикарбоната натрия (150 мл), экстрагировали этилацетатом (100 мл × 2), и затем органическую фазу промыли дважды с помощью насыщенного раствора NaCl, высушили над безводным сульфатом натрия, и отфильтровали. Фильтрат сконцентрировали при пониженном давлении, с получением 1.3 г неочищенного продукта, который сразу использовали на следующей стадии без очистки.
Стадия 3: Получение 4-(6-метоксил-бензимидазол-1-ил)-анилина
(5-метоксил-2-амино-фенил)-(4-амино-фенил)-амин (1.3 г, 5.67 ммоль), полученный на Стадии 2 растворили в 50 мл соляной кислоты (4 моль/л) и 1.5 мл муравьиной кислоты добавили при комнатной температуре. Смесь нагрели до 120°С и дали прореагировать в течение 1 часа. Затем реакционный раствор довели до рН>9 с помощью водного аммония в ванне со льдом, экстрагировали этилацетатом (80 мл × 2), и органическую фазу промыли с помощью насыщенного раствора NaCl, высушили над безводным сульфатом натрия, и отфильтровали. Фильтрат сконцентрировали при пониженном давлении. Остатки очистили с помощью колоночной хроматографии (элюент: дихлорметан/метанол) с получением 1.0 г 4-(6-метоксил-бензимидазол-1-ил)-анилина в виде темно-желтого осадка.
Стадия 4: Получение 3-(4-амино-фенил)-3Н-бензимидазол-5-ола
4-(6-метоксил-бензимидазол-1-ил)-анилин (600 мг, 3.66 ммоль), полученный на Стадии 3 нагрели до 120°С в 15 мл 40% бромистоводородной кислоты и дали прореагировать в течение 6 часов. Затем реакционный раствор охладили до комнатной температуры, и серо-белый осадок преципитировался. Осадок отфильтровали и затем растворили в 70 мл воды. Раствор отфильтровали и фильтрат довели до приблизительно рН 7 с помощью водного аммония, во время чего выпал белый осадок. Осадок затем отфильтровали, и промыли водой, высушили под вакуумом с получением 485 мг 3-(4-амино-фенил)-3Н-бензимидазол-5-ола в виде серо-белого осадка.
Стадия 5: Получение 4-[6-(2-морфолин-4-ил-этоксил)-бензимидазол-1-ил]-анилина
3-(4-амино-фенил)-1Н-бензимидазол-5-ол (300 мг, 1.33 ммоль), полученный на Стадии 4, 4-(2-хлор-этил)-морфолина гидрохлорид (372.0 мг, 2.0 ммоль), и гидроксид натрия (160 мг, 4 ммоль) нагревали до 90°С в 20 мл ДМФ и дали прореагировать в течение 2 часов. Реакционный раствор охладили до комнатной температуры, влили в воду (80 мл) и экстрагировали этилацетатом (50 мл × 2).Органическую фазу промыли дважды с помощью насыщенного раствора NaCl, высушили над безводным сульфатом натрия, и отфильтровали. Фильтрат сконцентрировали при пониженном давлении с получением 360 мг неочищенного продукта, который использовали сразу на следующей стадии без очистки.
Стадия 6: Получение 1-(5-трет-бутил-изоксазол-3-ил)-3-{4-[6-(2-морфолин-4-ил-этоксил)-бензимидазол-1-ил]-фенил}-мочевины
4-[6-(2-морфолин-4-ил-этоксил)-бензимидазол-1-ил]-анилин (100 мг, 0.295 ммоль), полученный на Стадии 5, фенил (5-трет-бутил-изоксазол-3-ил)-карбамат (115.3 мг, 0.443 ммоль) и триэтиламин (90 мг, 0.888 ммоль) растворили в 10 мл ТГФ и реакционную смесь кипятили с обратным холодильником в течение ночи. На следующий день реакционный раствор сконцентрировали при пониженном давлении, и остатки очистили с помощью колоночной хроматографии (элюент: дихлорметан/метанол) с получением 60 мг 1-(5-трет-бутил-изоксазол-3-ил)-3-{4-[6-(2-морфолин-4-ил-этоксил)-бензимидазол-1-ил]-фенил}-мочевины в виде белого осадка.
1HNMR (DMSO-d6, 400MHz) δ: 9.64 (s, 1Н), 8.04 (s, 1Н), 8.00 (s, 1Н,), 7.74-7.79 (m, 3Н), 7.45-7.49 (d, 2Н), 6.97-7.01 (m, 2Н), 5.93 (s, 1Н), 4.12-4.15 (t, 2Н), 3.74-3.77 (t, 4Н), 2.83-2.86 (t, 2Н), 2.60-2.62 (t, 4Н), 1.39 (s, 9Н).
LC-MS (ESI): 505.3 (M+H)+.
Пример 28 Получение
1-(5-трет-бутил-изоксазол-3-ил)-3-(4-{6-[(2-диметиламино-этил)-метил-амино]-бензимидазол-1-ил}-фенил)-мочевины (Соединение 28)
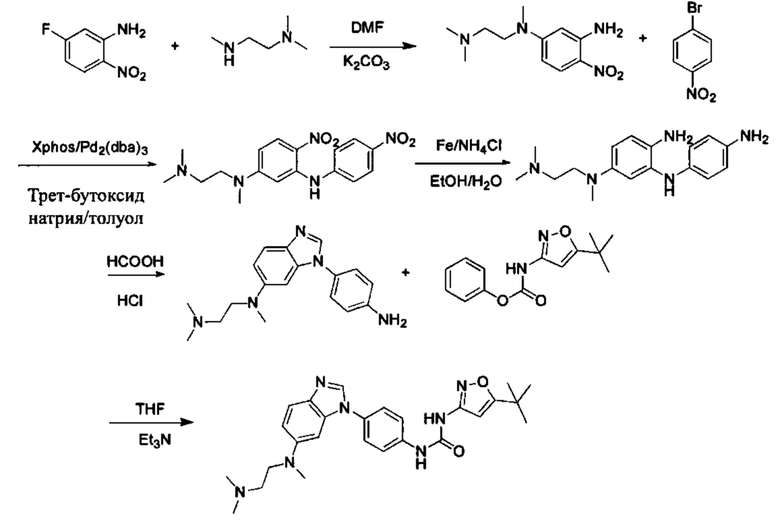
Стадия 1: Получение N1-(2-диметиламино-этил)-N1-метил-4-нитробензол-1,3-диамина
5-фтор-2-нитроанилин (2.5 г, 0.016 моль, TCI), N,N,N'-триметилэтилендиамин (2.45 г, 5 0.024 ммоль) и безводный карбонат калия (6.8 г, 0.048 моль) нагревали до 90°С в 50 мл ДМФ и дали прореагировать в течение 3 часов. Реакционный раствор охладили до комнатной температуры и влили в воду и экстрагировали этилацетатом (80 мл × 2). Органическую фазу промыли дважды с помощью насыщенного раствора NaCl, высушили над безводным сульфатом натрия, и отфильтровали. Фильтрат сконцентрировали при пониженном давлении с получением желтого осадка. Получившийся осадок суспендировали с метил трет-бутиловым эфиром с получением 2.66 г желтого осадка.
Стадия 2: Получение N1-(2-диметиламино-этил)-N1-метил-4-нитро-N3-(4-нитро-фенил)-бензол-1,3-диамина
В атмосфере азота продукт, полученный на Стадии 1 (2.66 г, 0.011 ммоль), п-бромнитробензол (4.70 г, 0.023 ммоль), Xphos (530 мг), Pd2(dba)3 (510 мг), и трет-бутоксид натрия (2.2 г, 0.023 ммоль) растворили в 80 мл толуола и перемешивали при 90°С в течение ночи. Реакционный раствор охладили до комнатной температуры, и 100 мл дихлорметана добавили, и перемешивали при комнатной температуре в течение 5 минут. Смесь отфильтровали, и фильтрат сконцентрировали при пониженном давлении. Остатки очистили с помощью колоночной хроматографии (элюент: дихлорметан/метанол) с получением 1.3 г красно-черного осадка.
Стадия 3: Получение N2-(4-амино-фенил)-N4-(2-диметиламино-этил)-N4-метил-фенил-1,2,4-триамина
Продукт (1.3 г, 3.62 ммоль), полученный на Стадии 2, восстановленный порошок железа (1.0 г, 18.1 ммоль), и хлорид аммония (1.55 г, 29.0 ммоль) нагревали до 90°С в этаноле (40 мл)/воде (10 мл) и дали прореагировать в течение 1 часа. Реакционный раствор охладили до комнатной температуры и медленно влили в насыщенный водный раствор бикарбоната натрия (150 мл) и экстрагировали этилацетатом (80 мл × 2). Органическую фазу промыли дважды с помощью насыщенного раствора NaCl, высушили над безводным сульфатом натрия, и отфильтровали. Фильтрат сконцентрировали при пониженном давлении, с получением 1.1 г неочищенного продукта, который сразу использовали на следующей стадии без очистки.
Стадия 4: Получение N-[3-(4-амино-фенил)-3Н-бензимидазол-5-ил]-N,N,N'-триметилэтан-1,2-диамина
Продукт (1.1 г, 4.37 ммоль), полученный на Стадии 3 растворили в 40 мл соляной кислоты (4 моль/л). 1.1 мл муравьиной кислоты добавили при комнатной температуре и смесь нагрели до 120°С и дали прореагировать в течение 1 часа. Реакционный раствор довели до рН>9 с помощью водного аммония в ванне со льдом и экстрагировали этилацетатом (60 мл × 2).Органическую фазу промыли с помощью насыщенного раствора NaCl, высушили над безводным сульфатом натрия, и отфильтровали. Фильтрат сконцентрировали при пониженном давлении. Остатки очистили с помощью колоночной хроматографии (элюент: дихлорметан/метанол) с получением 500 мг продукта.
Стадия 5: Получение 1-(5-трет-бутил-изоксазол-3-ил)-3-(4-{6-[(2-диметиламино-этил)-метил-амино]-бензимидазол-1-ил}-фенил)-мочевины.
N-[3-(4-амино-фенил)-3Н-бензимидазол-5-ил]N,N,N'-триметилэтан-1,2-диамин (100 мг, 0.323 ммоль), полученный на Стадии 4, фенил изоксазол-3-ил-карбамат (168 мг, 0.647 ммоль) и триэтиламин (100 мг, 0.99 ммоль) растворили в ТГФ и реакционную смесь кипятили с обратным холодильником в течение ночи. На следующий день реакционный раствор сконцентрировали при пониженном давлении, и остатки очистили с помощью колоночной хроматографии (элюент: дихлорметан/метанол) с получением 32 мг белого осадка.
1HNMR (DMSO-d6, 400MHz) δ: 9.60 (s, IH), 9.07 (s, IH), 8.18 (s, IH), 7.66-7.68 (d, 2H), 7.56-7.58 (d, 2H), 7.53-7.55 (d, 1H), 6.80-6.82 (dd, 1H), 6.67-6.68 (d, 1H), 6.52 (s, 1H), 3.42 (m, 2H), 2.90 (s, 3H), 2.37-2.40 (t, 2H), 2.16 (s, 6H), 1.30 (s, 9H).
LC-MS (ESI): 476.2 (M+H)+.
Пример 29
Получение 1-(5-трет-бутил-изоксазол-3-ил)-3-{4-[6-(4-метил-пиперазин-1-ил)-бензимидазол-1-ил]-фенил}-мочевины (Соединение 29)
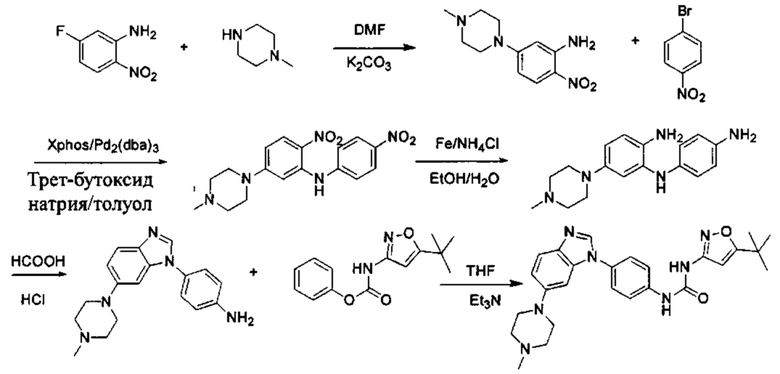
Стадия 1: Получение 5-(4-метил-пиперазин-1-ил)-2-нитро-анилина
5-фтор-2-нитроанилин (2.5 г, 0.016 моль), N-метилпиперазин (2.4 г, 0.024 ммоль, Darui) и безводный карбонат калия (6.8 г, 0.048 моль) нагревали до 90°С в 50 мл ДМФ и дали прореагировать в течение 3 часов. Реакционный раствор охладили до комнатной температуры и влили в воду и экстрагировали этилацетатом (80 мл × 2). Органическую фазу промыли дважды с помощью насыщенного раствора NaCl, высушили над безводным сульфатом натрия, и отфильтровали. Фильтрат сконцентрировали при пониженном давлении с получением желтого осадка. Получившийся осадок суспендировали с метил трет-бутиловым эфиром с получением 3 г 5-(4-метил-пиперазин-1-ил)-2-нитро-анилина в виде желтого осадка.
Стадия 2: Получение [5-(4-метил-пиперазин-1-ил)-2-нитро-фенил]-(4-нитро-фенил)-амина
В атмосфере азота 5-(4-метил-пиперазин-1-ил)-2-нитро-анилин (2.5 г, 0.011 ммоль), полученный на стадии 1, п-бромнитробензол (4.28 г, 0.021 ммоль), Xphos (480 мг), Pd2(dba)3 (460 мг) и трет-бутоксид натрия (2 г, 0.021 ммоль) растворили в 80 мл толуола, и перемешивали при 90°С в течение ночи. Реакционный раствор охладили до комнатной температуры, и 100 мл дихлорметана добавили. Смесь перемешивали при комнатной температуре в течение 5 минут, и отфильтровали. Фильтрат сконцентрировали при пониженном давлении. Остатки очистили с помощью колоночной хроматографии (элюент: дихлорметан/метанол) с получением 1.8 г [5-(4-метил-пиперазин-1-ил)-2-нитро-фенил]-(4-нитро-фенил)-амина в виде красно-черного осадка.
Стадия 3: Получение N2-(4-амино-фенил)-4-(4-метил-пиперазин-1-ил)-бензол-1,2- диамина
[5-(4-метил-пиперазин-1-ил)-2-нитро-фенил]-(4-нитро-фенил)-амин (1.8 г, 5.04 ммоль), полученный на Стадии 2, восстановленный порошок железа (2.26 г, 40.3 ммоль), и хлорид аммония (2.70 г, 50.4 ммоль) нагревали до 90°С в этаноле (40 мл)/воде (10 мл) и дали прореагировать в течение 1 часа. Реакционный раствор охладили до комнатной температуры и медленно влили в насыщенный водный раствор бикарбоната натрия (150 мл) и экстрагировали этилацетатом (80 мл × 2). Органическую фазу промыли дважды с помощью насыщенного раствора NaCl и высушили над безводным сульфатом натрия, и отфильтровали. Фильтрат сконцентрировали при пониженном давлении с получением 1.3 г неочищенного продукта, который сразу использовали на следующей стадии без очистки.
Стадия 4: Получение 4-[6-(4-метил-пиперазин-1-ил)-бензимидазол-1-ил]-анилина
N2-(4-амино-фенил)-4-(4-метил-пиперазин-1-ил)-бензол-1,2-диамин (1.3 г, 4.37 ммоль), полученный на Стадии 3 растворили в 50 мл соляной кислоты (4 моль/л), 1.5 мл муравьиной кислоты добавили при комнатной температуре, и смесь нагрели до 120°С и дали прореагировать в течение 1 часа. Реакционный раствор довели до рН>9 с помощью водного аммония в ванне со льдом, и экстрагировали этилацетатом (60 мл × 2). Органическую фазу промыли с помощью насыщенного раствора NaCl, высушили над безводным сульфатом натрия, и отфильтровали. Фильтрат сконцентрировали при пониженном давлении. Остатки очистили с помощью колоночной хроматографии (элюент: дихлорметан/метанол) с получением 600 мг 4-[6-(4-метил-пиперазин-1-ил)-бензимидазол-1-ил]-анилина.
Стадия 5: Получение 1-(5-трет-бутил-изоксазол-3-ил)-3-{4-[6-(4-метил-пиперазин-1-ил)-бензимидазол-1-ил]-фенил}-мочевины
4-[6-(4-метил-пиперазин-1-ил)-бензимидазол-1-ил]-анилин (100 мг, 0.326 ммоль) полученный на стадии 4, фенил изоксазол-3-ил-карбамат (127 мг, 0.490 ммоль) и триэтиламин (100 мг, 0.99 ммоль) растворили в ТГФ и реакционный растовр кипятили с обратным холодильником в течение ночи. На следующий день реакционный раствор сконцентрировали при пониженном давлении, и остатки очистили с помощью колоночной хроматографии (элюент: дихлорметан/метанол) с получением 1-(5-трет-бутил-изоксазол-3-ил)-3-{4-[6-(4-метил-пиперазин-1-ил)-бензимидазол-1-ил]-фенил}-мочевины в виде белого осадка.
1HNMR (DMSO-d6, 400MHz) δ: 9.61 (s, IH), 9.08 (s, IH), 8.27 (s, IH), 7.67-7.69 (d, 2H), 7.56-7.60 (m, 3H), 7.03-7.05 (dd, 1H), 6.91-6.92 (d, 1H), 6.52 (s, 1H), 3.13 (m, 4H), 2.51 (m, 4H), 2.25 (s, 3H), 1.30 (s, 9H).
LC-MS (ESI): 474.2 (M+H)+.
Пример 30
Получение 1-(5-трет-бутил-изоксазол-3-ил)-3-{4-[7-(2-морфолин-4-ил-этоксил)-бензимидазол-1-ил]-фенил}-мочевины (Соединение 30)
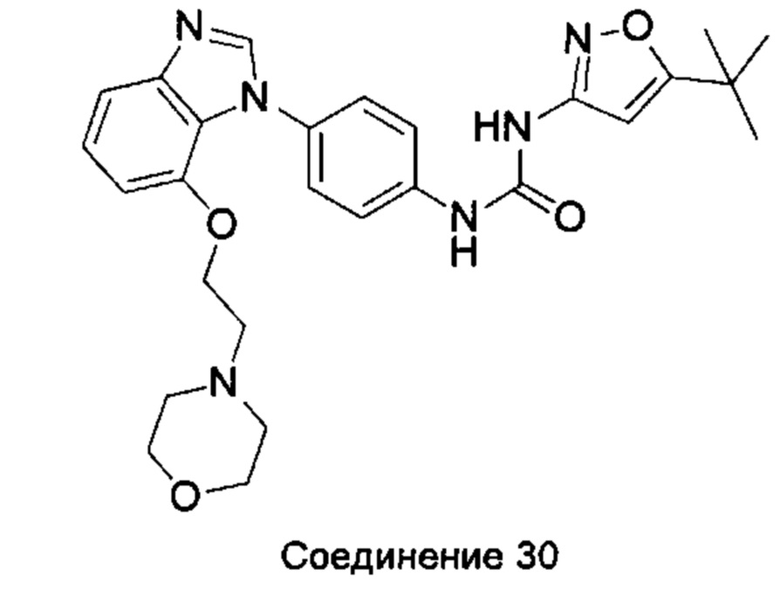
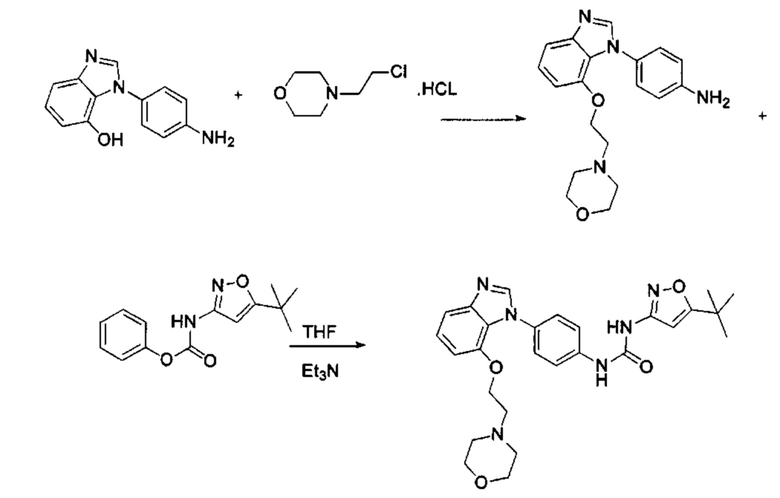
Стадия 1: Получение 3-(4-амино-фенил)-3Н-бензимидазол-4-ола
Методика была аналогична таковой, как на Стадиях 1-4 в примере 4, за исключением того, что 2-амино-3-нитрофенол использовали вместо 2-нитро-4-метоксиланилина на Стадии 1 с получением 3-(4-амино-фенил)-3Н-бензимидазол-4-ола.
Стадия 2: Получение 4-[7-(2-морфолин-4-ил-этоксил)-бензимидазол-1-ил]-анилина
Методика была аналогична таковой, как на Стадии 1 в примере 7, за исключением того, что 3-(4-амино-фенил)-3Н-бензимидазол-4-ол использовали вместо 1-(4-амино-фенил)-1Н-бензимидазол-5-ола, и N-(2-хлорэтил)морфолина гидрохлорид использовали вместо 1-хлоргексана, с получением 4-[7-(2-морфолин-4-ил-этоксил)-бензимидазол-1-ил]-анилина.
Стадия 3: Получение 1-(5-трет-бутил-изоксазол-3-ил)-3-{4-[7-(2-морфолин-4-ил-этоксил)-бензимидазол-1-ил]-фенил}-мочевины
Методика была аналогична таковой, как на Стадии 5 в примере 4, за исключением того, что 4-[7-(2-морфолин-4-ил-этоксил)-бензимидазол-1-ил]-анилин использовали вместо 1-(4-амино-фенил)-1Н-бензимидазол-5-ола, с получением 1-(5-трет-бутил-изоксазол-3-ил)-3-{4-[7-(2-морфолин-4-ил-этоксил)-бензимидазол-1-ил]-фенил}-мочевины.
1HNMR (DMSO-d6, 400MHz) δ: 9.57 (s, IH), 9.04 (s, IH), 8.22 (s, IH), 7.58-7.60 (d, 2H), 7.44-7.46 (d, 2H), 7.32-7.34 (d, 1H), 7.15-7.19 (t, 1H), 6.84-6.86 (d, 1H), 6.53 (s, 1H), 4.06-4.09 (t, 2H), 3.43-3.45 (t, 4H), 2.45-2.47 (t, 2H), 2.17 (m, 4H), 1.31 (s, 9H).
LC-MS (ESI): 505.2 (M+H)+.
Пример 31
Получение 1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(5,6-диметоксил-бензимидазол-1-ил)-фенил]-мочевины (Соединение 31)
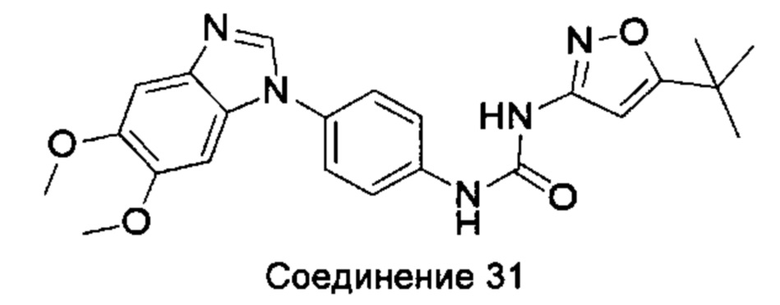
Стадия 1: Получение 4-(5,6-диметоксил-бензимидазол-1-ил)-анилина
Способ получения был такой же, как на Стадии 1-2 в примере 1, за исключением того, что 5,6-диметоксилбензимидазол (Darui) использовали вместо бензимидазола на Стадии 1 с получением 4-(5,6-диметоксил-бензимидазол-1-ил)-анилина.
Стадия 2: Получение 1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(5,6-диметоксил-бензимидазол-1-ил)-фенил]-мочевины
Методика была аналогична таковой, как на Стадии 5 в примере 4, за исключением того, что 4-(5,6-диметоксил-бензимидазол-1-ил)-анилин использовали вместо 1-(4-амино-фенил)-1Н-бензимидазол-5-ола с получением 1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(5,6-диметоксил-бензимидазол-1-ил)-фенил]-мочевины.
1HNMR (DMSO-d6, 400MHz) δ: 9.60 (s, IH), 9.06 (s, IH), 8.28 (s, IH,), 7.68-7.70 (d, 2H), 7.59-7.61 (d, 2H), 7.31 (s, 1H), 7.04 (s, 1H), 6.52 (s, 1H), 3.82 (s, 3H), 3.79 (s, 3H), 1.31 (s, 9H).
LC-MS (ESI): 436.1 (M+H)+.
Пример 32
Получение 1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(5,6-диметил-бензимидазол-1-ил)-фенил]-мочевины (Соединение 32)
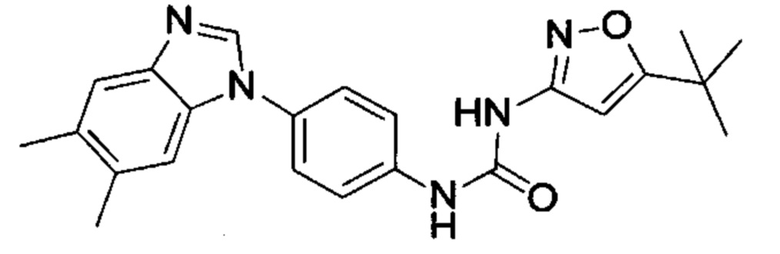
Соединение 32
Стадия 1: Получение 4-(5,6-диметил-бензимидазол-1-ил)-анилина
Способ получения был такой же, как на Стадии 1 to 2 в примере 1, за исключением того, что 5,6-dimethylбензимидазол использовали вместо бензимидазола на Стадии 1 с получением 4-(5,6-диметил-бензимидазол-1-ил)-анилина.
Стадия 2: Получение 1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(5,6-диметил-бензимидазол-1-ил)-фенил]-мочевины
Методика была аналогична таковой, как на Стадии 5 в примере 4, за исключением того, что 4-(5,6-диметил-бензимидазол-1-ил)-анилин использовали вместо 1-(4-амино-фенил)-1Н-бензимидазол-5-ола с получением 1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(5,6-диметил-бензимидазол-1-ил)-фенил]-мочевины.
1HNMR (DMSO-d6, 400MHz) δ: 9.60 (s, 1H), 9.08 (s, 1H), 8.36 (s, 1H), 7.67-7.70 (d, 2H), 7.57-7.59 (d, 2H), 753 (s, 1H), 7.37 (s, 1H), 6.53 (s, 1H), 2.34 (s, 6H), 1.31 (s, 9H)
LC-MS (ESI): 403.9 (M+H)+.
Пример 33
Получение 1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(5-фтор-7-метил-бензимидазол-1-ил)-фенил]-мочевины (Соединение 33)
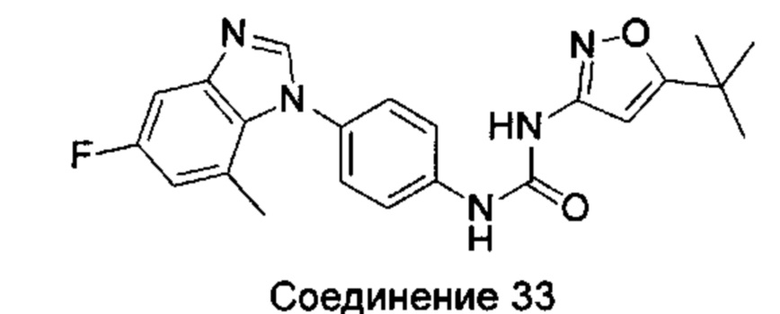
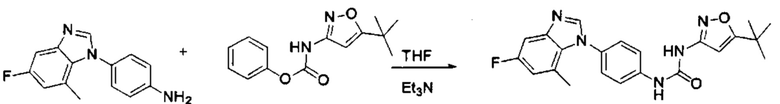
Стадия 1: Получение (5-фтор-7-метил-бензимидазол-1-ил)-анилина
Способ получения был такой же, как на Стадиях 1-3 в примере 4, за исключением того, что 2-нитро-4-фтор-6-метиланилин (TCI) использовали вместо 2-нитро-4-метоксиланилина на Стадии 1, с получением (5-фтор-7-метил-бензимидазол-1-ил)-анилина.
Стадия 2: Получение 1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(5-фтор-7-метил-бензимидазол-1-ил)-фенил]-мочевины
Методика была аналогична таковой, как на Стадии 5 в примере 4, за исключением того, что (5-фтор-7-метил-бензимидазол-1-ил)-анилин использовали вместо 1-(4-амино-фенил)-1Н-бензимидазол-5-ола с получением 1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(5-фтор-7-метил-бензимидазол-1-ил)-фенил]-мочевины.
1HNMR (DMSO-d6, 400MHz) δ: 9.64 (s, 1H), 9.12 (s, 1H), 8.30 (s, 1H), 7.62-7.65 (d, 2H), 7.49-7.51 (d, 2H), 7.37-7.40 (dd, 1H), 6.94-6.97 (dd, 1H), 6.53 (s, 1H), 2.03 (s, 3Н), 1.30 (s, 9H).
LC-MS (ESI): 408.1 (M+H)+.
Пример 34
Получение 1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(4-фтор-бензимидазол-1-ил)-фенил]-мочевины (Соединение 34)
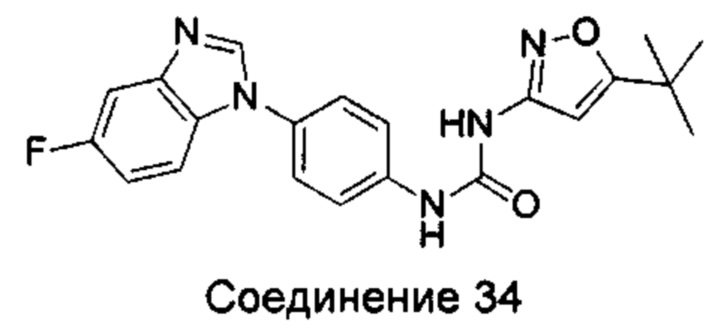
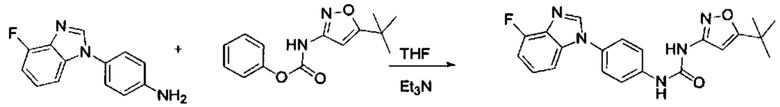
Стадия 1: Получение 4-(4-фтор-бензимидазол-1-ил)-анилина
Способ получения был такой же, как на Стадиях 1-3 в примере 4, за исключением того, что 2-нитро-3-фторанилин использовали вместо 2-нитро-4-метоксиланилина на Стадии 1, с получением 4-(4-фторо-бензимидазол-1-ил)-анилина.
Стадия 2: Получение 1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(4-фтор-бензимидазол-1-ил)-фенил]-мочевины
Методика была аналогична таковой, как на Стадии 5 в примере 4, за исключением того, что 4-(4-фтор-бензимидазол-1-ил)-анилин использовали вместо 1-(4-амино-фенил)1Н-бензимидазол-5-ола для получения 1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(4-фтор-бензимидазол-1-ил)-фенил]-мочевины.
1HNMR (DMSO-d6, 400MHz) δ: 9.62 (s, 1H), 9.08 (s, 1H), 8.46 (s, 1H), 7.55-10 7.65 (m, 5H), 7.25-7.30 (m, 1H), 7.12-7.17 (m, 1H), 6.53 (s, 1H), 1.30 (s, 9H).
LC-MS (ESI): 394.1 (M+H)+.
Пример 35
Получение 1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(2-метил-бензимидазол-1-ил)-фенил]-мочевины (Соединение 35)
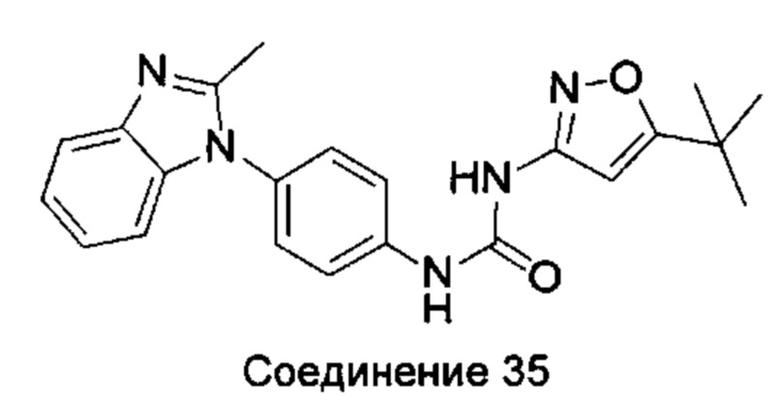
Стадия 1: Получение 4-(2-метилбензимидазол-1-ил)-анилина
Способ получения был такой же, как на Стадиях 1-2 в примере 1, за исключением того, что 2-метилбензимидазол использовали вместо бензимидазола на Стадии 1 с получением 4-(2-метилбензимидазол-1-ил)-анилина.
Стадия 2: Получение 1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(2-метил-бензимидазол-1-ил)-фенил]-мочевины
Методика была аналогична таковой, как на Стадии 5 в примере 4, за исключением того, что 4-(2-метилбензимидазол-1-ил)-анилин использовали вместо 1-(4-амино-фенил)-1Н-бензимидазол-5-ола с получением 1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(2-метил-бензимидазол-1-ил)-фенил]-мочевины.
1HNMR (DMSO-d6, 400MHz) δ: 9.63 (s, 1H), 9.12 (s, 1H), 7.69-7.71 (d, 2H), 7.61-7.63 (m, 1H), 7.46-7.48 (d, 2H), 7.16-7.23 (m, 2H), 7.10-7.12 (m, 1H), 6.53 (s, 1H), 2.43 (s, 3Н), 1.31 (s, 9H).
LC-MS (ESI): 389.9 (M+H)+.
Пример 36
Получение 1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(2-хлоробензимидазол-1-ил)-фенил]-мочевины (Соединение 36)
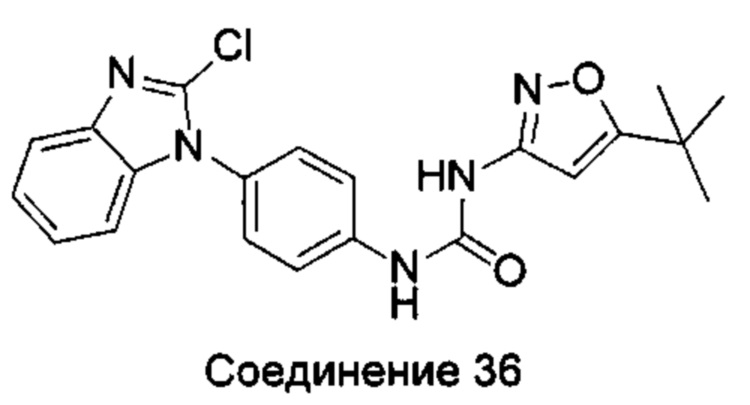
Стадия 1: Получение 4-(2-хлоробензимидазол-1-ил)-анилина
Способ получения был такой же, как на Стадиях 1-2 в примере 1, за исключением того, что 2-хлоробензимидазол использовали вместо бензимидазола на Стадии 1 с получением 4-(2-хлоробензимидазол-1-ил)-анилина.
Стадия 2: Получение 1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(2-хлоробензимидазол-1-ил)-фенил]-мочевины
Методика была аналогична таковой, как на Стадии 5 в примере 4, за исключением того, что 4-(2-хлоробензимидазол-1-ил)-анилин использовали вместо 1-(4-амино-фенил)-1Н-бензимидазол-5-ола, с получением 1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(2-хлоробензимидазол-1-ил)-фенил]-мочевины.
1HNMR (DMSO-d6, 400MHz) δ: 9.64 (s, 1H), 9.16 (s, 1H), 7.70-7.73 (m, 3Н), 7.50-7.53 (d, 2H), 7.29-7.34 (m, 2H), 7.17-7.19 (m, 1H), 6.54 (s, 1H), 1.31 (s, 9H).
LC-MS (ESI): 410.1 (M+H)+.
Пример 37
Получение 1-(4-бензимидазол-1-ил-3-метил-фенил)-3-(5-трет-бутил-изоксазол-3-ил)-мочевины (Соединение 37)
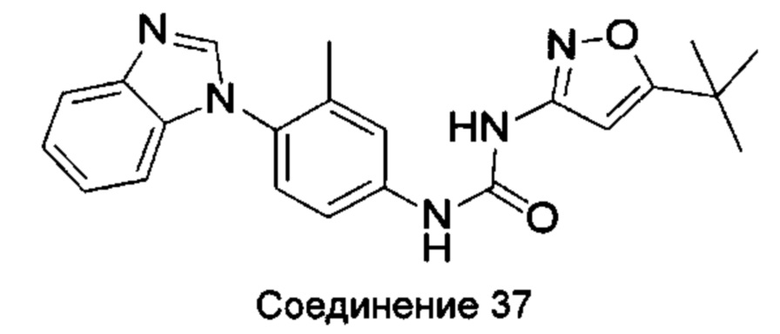
Стадия 1: Получение 4-бензимидазол-1-ил-3-метил-анилина.
Способ получения был такой же, как на Стадиях 1-2 в примере 1, за исключением того, что 3-метил-4-фторнитробензол (Runjie) использовали вместо п-фторнитробензола на Стадии 1, с получением 4-бензимидазол-1-ил-3-метил-анилина.
Стадия 2: Получение 1-(4-бензимидазол-1-ил-3-метил-фенил)-3-(5-трет-бутил-изоксазол-3-ил)-мочевины
Методика была аналогична таковой, как на Стадии 5 в примере 4, за исключением того, что 4-бензимидазол-1-ил-3-метил-анилин использовали вместо 1-(4-амино-фенил)-1Н-бензимидазол-5-ола, с получением 1-(4-бензимидазол-1-ил-3-метил-фенил)-3-(5-трет-бутил-изоксазол-3-ил)-мочевины.
1HNMR (DMSO-d6, 400MHz) δ: 9.63 (s, 1Н), 9.05 (s, 1Н), 8.41 (s, 1Н), 7.80-7.83 (m, 1Н), 7.61-7.62 (d, 1Н), 7.48-7.51 (dd, 1Н), 7.35-7.38 (d, 1Н), 7.29-7.31 (m, 2Н), 7.15-7.17 (m, 1Н), 6.54 (s, 1Н), 2.00 (s, 3Н), 1.31 (s, 9Н).
LC-MS (ESI): 390.2 (M+H)+.
Пример 38
Получение 1-(4-бензимидазол-1-ил-3-хлор-фенил)-3-(5-трет-бутил-изоксазол-3-ил)-мочевины (Соединение 38)
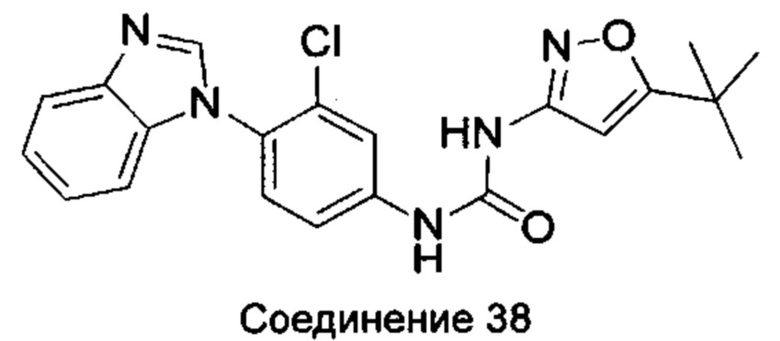
Стадия 1: Получение 4-бензимидазол-1-ил-3-хлор-анилина
Способ получения был такой же, как на Стадиях 1-2 в примере 1, за исключением того, что 3-хлор-4-фторнитробензол использовали вместо п-фторнитробензола на Стадии 1 с получением 4-бензимидазол-1-ил-3-хлор-анилина.
Стадия 2: Получение 1-(4-бензимидазол-1-ил-3-хлор-фенил)-3-(5-трет-бутил-изоксазол-3-ил)-мочевины
Методика была аналогична таковой, как на Стадии 5 в примере 4, за исключением того, что 4-бензимидазол-1-ил-3-хлор-анилин использовали вместо 1-(4-амино-фенил)-1Н-бензимидазол-5-ола с получением 1-(4-бензимидазол-1-ил-3-хлор-фенил)-3-(5-трет-бутил-изоксазол-3-ил)-мочевины.
1HNMR (DMSO-d6, 400MHz) δ: 9.77 (s, 1H), 9.29 (s, 1H), 8.40 (s, 1H), 8.03-8.04 (d, 1H), 7.76-7.79 (m, 1H), 7.60-7.63 (d, 1H), 7.51-7.54 (dd, 1H), 7.29-7.31 (m, 2H), 7.20-7.21 (m, 1H), 6.55 (s, 1H), 1.31 (s, 9H).
LC-MS (ESI): 410.1 (M+H)+.
Пример 39
Получение
1-(4-бензимидазол-1-ил-3-фтор-фенил)-3-(5-трет-бутил-изоксазол-3-ил)-мочевины (Соединение 39)
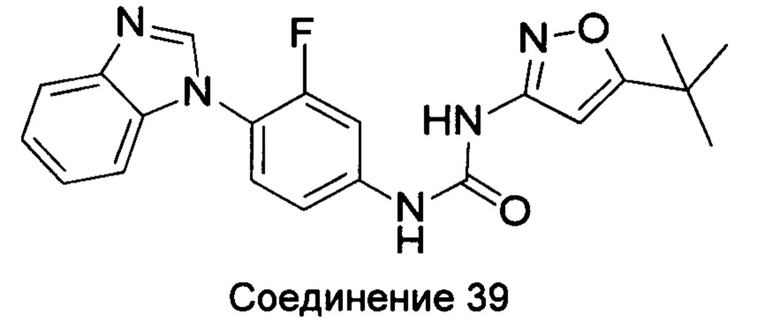
Стадия 1: Получение 4-бензимидазол-1-ил-3-фтор-анилина
Способ получения был такой же, как на Стадиях 1-2 в примере 1, за исключением того, что 3,4-дифторнитробензол использовали вместо п-фторнитробензола на Стадии 1 с получением 4-бензимидазол-1-ил-3-фтор-анилина.
Стадия 2: Получение 1-(4-бензимидазол-1-ил-3-фтор-фенил)-3-(5-трет-бутил-изоксазол-3-ил)-мочевины
Методика была аналогична таковой, как на Стадии 5 в примере 4, за исключением того, что 4-бензимидазол-1-ил-3-фтор-анилин использовали вместо 1-(4-амино-фенил)-1Н-бензимидазол-5-ола с получением 1-(4-бензимидазол-1-ил-3-фторо-фенил)-3-(5-трет-бутил-изоксазол-3-ил)-мочевины
1HNMR (DMSO-d6, 400MHz) δ: 9.74 (s, 1H), 9.30 (s, 1H), 8.45 (s, 1H), 7.80-7.84 (dd, 1H), 7.77-7.79 (m, 1H), 7.63-7.67 (t, 1H), 7.30-7.39 (m, 4H), 6.54 (s, 1H), 1.31 (s, 9H).
LC-MS (ESI): ESI 393.9 (M+H)+.
Пример 40
Получение
1-(4-бензимидазол-1-ил-3,5-дифтор-фенил)-3-(5-трет-бутил-изоксазол-3-ил)-мочевины (Соединение 40)
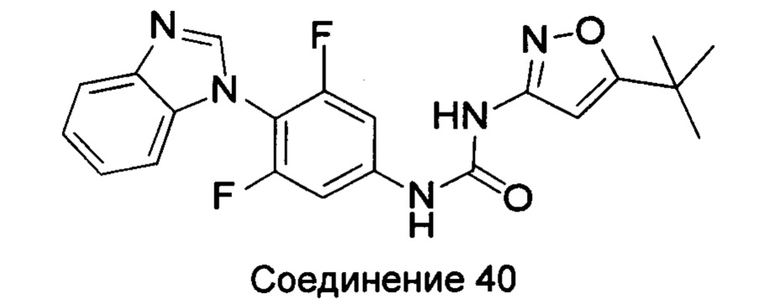
Стадия 1: Получение 4-бензимидазол-1-ил-3,5-дифтор-анилина
Способ получения был такой же, как на Стадиях 1-2 в примере 1, за исключением того, что 3,4,5-трифторнитробензол использовали вместо п-фторнитробензола на Стадии 1, с получением 4-бензимидазол-1-ил-3,5-дифторанилина.
Стадия 2: Получение 1-(4-бензимидазол-1-ил-3,5-дифтор-фенил)-3-(5-трет-бутил-изоксазол-3-ил)-мочевины
Методика была аналогична таковой, как на Стадии 5 в примере 4, за исключением того, что 4-бензимидазол-1-ил-3,5-дифтор-анилин использовали вместо 1-(4-амино-фенил)-1Н-бензимидазол-5-ола с получением 1-(4-бензимидазол-1-ил-3,5-дифтор-фенил)-3-(5-трет-бутил-изоксазол-3-ил)-мочевины.
1HNMR (DMSO-d6, 400MHz) δ: 9.91 (s, 1H), 9.46 (s, 1H), 8.48 (s, 1H), 7.78-7.81 (m, 1H), 7.55-7.58 (d, 2H), 7.31-7.35 (m, 3Н), 6.54 (s, 1H), 1.31 (s, 9H).
LC-MS (ESI): 412.1 (M+H)+.
Пример 41
Получение
1-(4-бензимидазол-1-ил-2-хлор-фенил)-3-(5-трет-бутил-изоксазол-3-ил)-мочевины (Соединение 41)
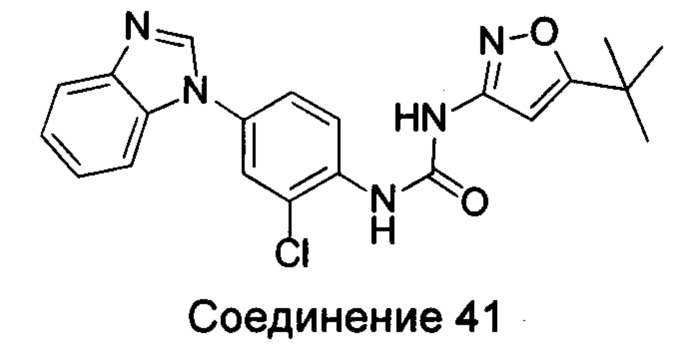
Стадия 1: Получение 4-бензимидазол-1-ил-2-хлор-анилина
Способ получения был такой же, как на Стадиях 1-2 примера 1, за исключением того, что 2-хлор-4-фторнитробензол использовали вместо п-фторнитробензола на Стадии 1 с получением 4-бензимидазол-1-ил-2-хлор-анилина.
Стадия 2: Получение 1-(4-бензимидазол-1-ил-2-хлор-фенил)-3-(5-трет-бутил-изоксазол-3-ил)-мочевины
Методика была аналогична таковой, как на Стадии 5 в примере 4, за исключением того, что 4-бензимидазол-1-ил-2-хлор-анилин использовали вместо 1-(4-амино-фенил)-1Н-бензимидазол-5-ола с получением 1-(4-бензимидазол-1-ил-2-хлор-фенил)-3-(5-трет-бутил-изоксазол-3-ил)-мочевины.
1HNMR (DMSO-d6, 400MHz) δ:10.32 (s, 1H), 8.89 (s, 1H), 8.56 (s, 1H), 8.39-8.41 (d, 1H), 7.90-7.91 (d, 1H), 7.77-7.79 (m, 1H), 7.67-7.69 (dd, 1H), 7.61-7.63 (m, 1H), 7.30-7.37 (m, 2H), 6.49 (s, 1H), 1.31 (s, 9H).
LC-MS (ESI): 410.1 (M+H)+.
Пример 42
Получение
1-(6-бензимидазол-1-ил-пиридин-3-ил)-3-(5-трет-бутил-изоксазол-3-ил)-мочевины (Соединение 42)
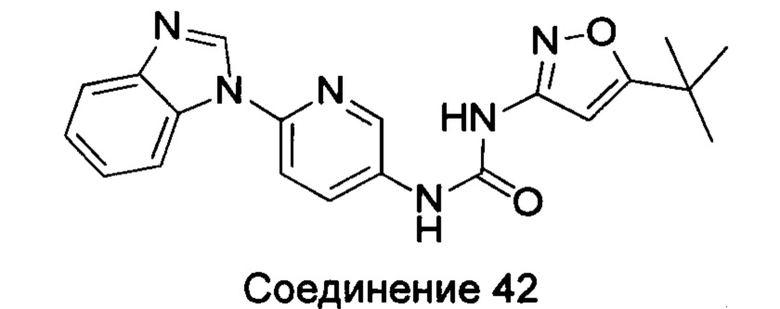
Стадия 1: Получение 6-бензимидазол-1-ил-пиридин-3-иламина
Способ получения был такой же, как на Стадиях 1-2 в примере 1, за исключением того, что 2-хлор-5-нитропиридин использовали вместо п-фторнитробензола на Стадии 1 с получением 6-бензимидазол-1-ил-пиридин-3-иламина.
Стадия 2: Получение 1-(6-бензимидазол-1-ил-пиридин-3-ил)-3-(5-трет-бутил-изоксазол-3-ил)-мочевины
Методика была аналогична таковой, как на Стадии 5 в примере 4, за исключением того, что 6-бензимидазол-1-ил-пиридин-3-иламин использовали вместо 1-(4-амино-фенил)-1Н-бензимидазол-5-ола с получением 1-(6-бензимидазол-1-ил-пиридин-3-ил)-3-(5-трет-бутил-изоксазол-3-ил)-мочевины.
1HNMR (DMSO-d6, 400MHz) δ: 9.80 (s, 1H), 9.20 (s, 1H), 8.88 (s, 1H), 8.68-8.69 (d, 1H), 8.19-8.22 (m, 2H), 7.89-7.91 (d, 1H), 7.76-7.78 (m, 1H), 7.33-7.38 (m, 2H), 6.53 (s, 1H), 1.31 (s, 9H).
LC-MS (ESI): 377.1 (M+H)+.
Пример 43
Получение 1-(2-бензимидазол-1-ил-пиримидин-5-ил)-3-(5-трет-бутил-изоксазол-3-ил)-мочевины (Соединение 43)
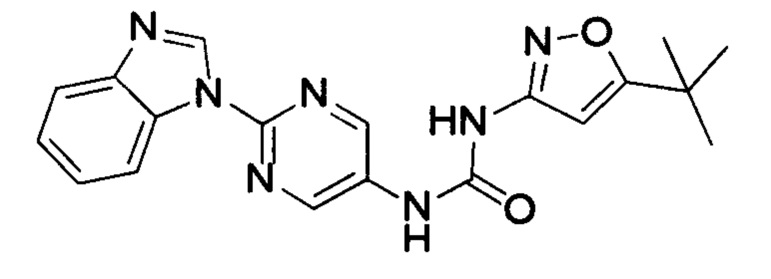

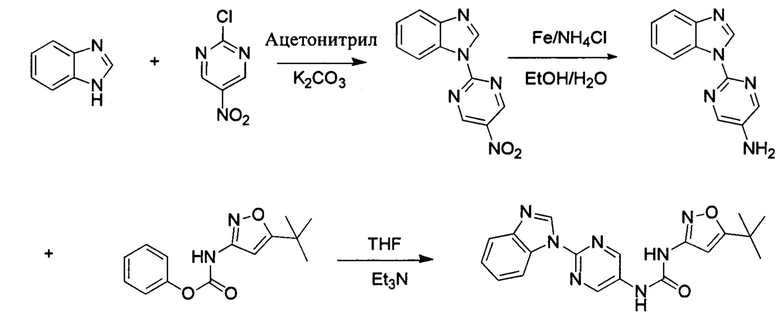
Стадия 1: Получение 1-(5-нитро-пиримидин-2-ил)-1Н-бензимидазола
При комнатной температуре, 2-хлор-6-нитропиримидин (118 мг, 1 ммоль) и бензимидазол (175.5 мг, 1.1 ммоль) растворили в 15 мл ацетонитрила и безводный калий (414 мг, 3 ммоль) добавили и дали прореагировать при комнатной температуре в течение ночи. На следующий день реакционный раствор влили в воду и экстрагировали этилацетатом (50×2). Органическую фазу промыли дважды с помощью насыщенного раствора NaCl, высушили над безводным сульфатом натрия и отфильтровали. Фильтрат сконцентрировали при пониженном давлении с получением желто-черного осадка, который использовали сразу на следующей стадии без очистки.
Стадия 2: Получение 2-бензимидазол-1-ил-пиримидин-5-иламина
Методика была аналогична таковой, как на Стадии 2 в примере 1, за исключением того, что 1-(5-нитро-пиримидин-2-ил)-1Н-бензимидазол использовали вместо 1-(4-нитро-фенил)-1Н-бензимидазола на Стадии 2 с получением 2-бензимидазол-1-ил-пиримидин-5-иламина.
Стадия 3: Получение 1-(2-бензимидазол-1-ил-пиримидин-5-ил)-3-(5-трет-бутил-изоксазол-3-ил)-мочевины
Методика была аналогична таковой, как на Стадии 5 в примере 4, за исключением того, что 2-бензимидазол-1-ил-пиримидин-5-иламин использовали вместо 1-(4-амино-фенил)-1Н-бензимидазол-5-ола с получением 1-(2-бензимидазол-1-ил-пиримидин-5-ил)-3-(5-трет-бутил-изоксазол-3-ил)-мочевины.
1HNMR (DMSO-d6, 400MHz) δ: 10.01 (s, 1H), 9.25 (s, 1H), 9.06 (s, 1H), 9.05 (s, 2H), 8.51-8.53 (d, 1H), 7.78-7.80 (d, 1H), 7.42-7.46 (m, 1H), 7.35-7.39 (m, 1H), 6.52 (s, 151H), 1.30 (s, 9H).
LC-MS (ESI): 378.2 (M+H)+.
Пример 44 Получение 1-(5-трет-бутил-изоксазол-3-ил)-3-(4-индол-1-ил-фенил)-мочевины (Соединение 44)
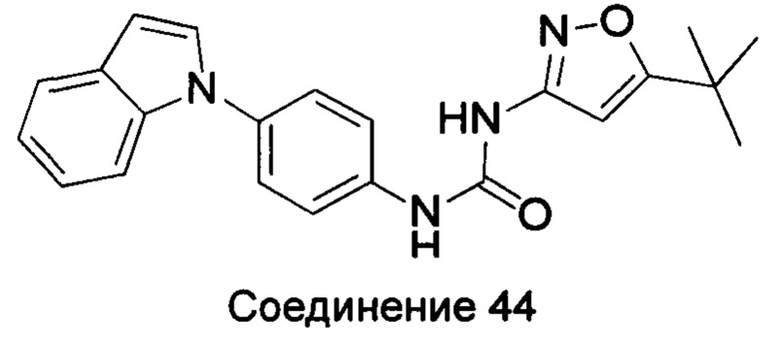
Стадия 1: Получение 4-индол-1-ил-анилина
Способ получения был такой же, как на Стадиях 1-2 в примере 1, за исключением того, что индол использовали вместо бензимидазола на Стадии 1 с получением 4-индол-1-ил-анилина.
Стадия 2: Получение 1-(5-трет-бутил-изоксазол-3-ил)-3-(4-индол-1-ил-фенил)-мочевины
Методика была аналогична таковой, как на Стадии 5 в примере 4, за исключением того, что 4-индол-1-ил-анилин использовали вместо 1-(4-амино-фенил)-1Н-бензимидазол-5-ола с получением 1-(5-трет-бутил-изоксазол-3-ил)-3-(4-индол-1-ил-фенил)-мочевины.
1HNMR (DMSO-d6, 400MHz) δ: 9.46 (s, 1H), 8.66 (s, 1H), 7.69-7.71 (m, 3H), 7.53-7.55 (d, 1H), 7.48-7.51 (d, 2H), 7.32-7.33 (d, 1H), 7.16-7.26 (m, 2H), 6.69-6.70 (d, 1H), 30 5.97(s, 1H), 1.39 (s, 9H).
LC-MS (ESI): 374.9 (M+H)+.
Пример 45 Получение 1-(5-трет-бутил-изоксазол-3-ил)-3-(4-индол-1-ил-фенил)-мочевины (Соединение 45)
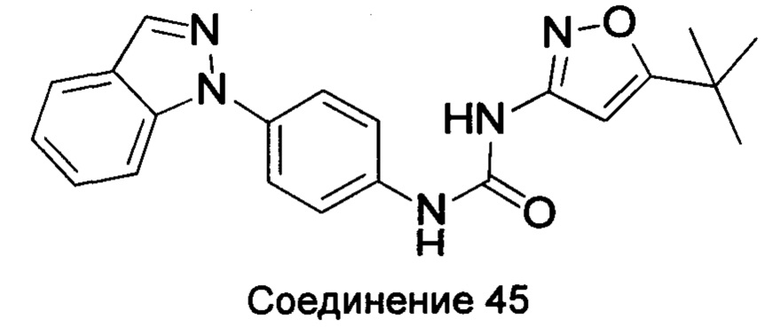
Стадия 1: Получение 4-индол-1-ил-анилина
Способ получения был такой же, как на Стадиях 1-2 в примере 1, за исключением того, что индазол использовали вместо бензимидазола на Стадии 1 с получением 4-индазол-1-ил-анилина.
Стадия 2: Получение 1-(5-трет-бутил-изоксазол-3-ил)-3-(4-индазол-1-ил-фенил)-мочевины
Методика была аналогична таковой, как на Стадии 5 примера 4 за исключением того, что 4-индазол-1-ил-анилин использовали вместо 1-(4-амино-фенил)-1Н-бензимидазол-5-ола с получением 1-(5-трет-бутил-изоксазол-3-ил)-3-(4-индазол-1-ил-фенил)-мочевины.
1HNMR (DMSO-d6, 400MHz) δ: 9.58 (s, 1H), 9.03 (s, 1H), 8.35 (d, 1H), 7.88-7.90 (d, 1H), 7.79-7.81 (d, 1H), 766-7.71 (m, 4H), 7.47-7.51 (m, 1H), 7.25-7.28 (m, 1H), 6.54 (s, 1H), 1.31 (s, 9H).
LC-MS (ESI): 376.0 (M+H)+.
Пример 46
Получение
1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(6-фтор-индазол-1-ил)-фенил]-мочевины (Соединение 46)
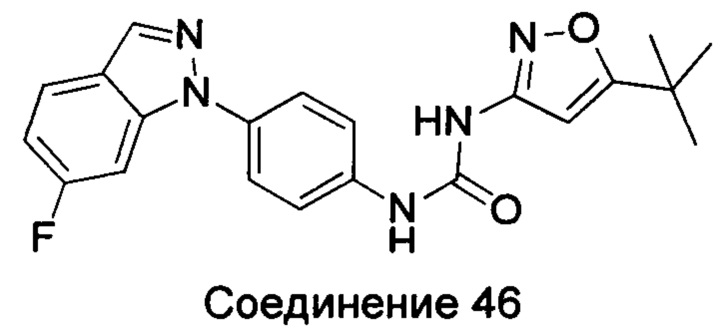
Стадия 1: Получение 4-(6-фтор-индазол-1-ил)-анилина
Способ получения был такой же, как на Стадиях 1-2 в примере 1, за исключением того, что 6-фториндазол использовали вместо бензимидазола на Стадии 1 для получения 4-(6-фтор-индазол-1-ил)-анилина.
Стадия 2: Получение 1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(6-фтор-индазол-1-ил)-фенил]-мочевины
Методика была аналогична таковой, как на Стадии 5 в примере 4, за исключением того, что 4-(6-фтор-индазол-1-ил)-анилин использовали вместо 1-(4-амино-фенил)-1Н-бензимидазол-5-ола с получением 1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(6-фтор-индазол-1-ил)-фенил]-мочевины.
1HNMR (DMSO-d6, 400MHz) δ: 9.58 (s, 1H), 9.04 (s, 1H), 8.37 (s, 1H), 7.92-7.95 (m, 1H), 7.65-7.70 (m, 4H), 7.57-7.59 (m, 1H), 7.13-7.18 (m, 1H), 6.53 (s, 1H), 1.30 (s, 9H).
LC-MS (ESI): 394.1 (M+H)+.
Пример 47
Получение
1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(7-фтор-индазол-1-ил)-фенил]-мочевины (Соединение 47)
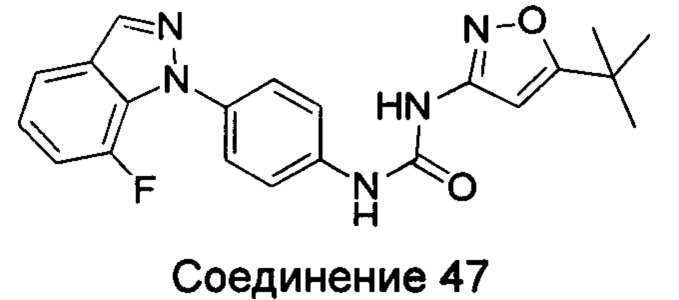
Стадия 1: Получение 4-(7-фтор-индазол-1-ил)-анилина
Способ получения был такой же, как на Стадиях 1-2 в примере 1, за исключением того, что 7-фториндазол использовали вместо бензимидазола на Стадии 1 для получения 4-(7-фтор-индазол-1-ил)-анилина.
Стадия 2: Получение 1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(7-фтор-индазол-1-ил)-фенил]-мочевины
Методика была аналогична таковой, как на Стадии 5 в примере 4, за исключением того, что 4-(7-фтор-индазол-1-ил)-анилин использовали вместо 1-(4-амино-фенил)-1Н-бензимидазол-5-ола для получения 1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(7-фтор-индазол-1-ил)-фенил]-мочевины.
1HNMR (DMSO-d6, 400MHz) δ: 9.63 (s, 1H), 9.16-9.17 (d, 1H), 9.08 (s, 1H), 8.03-8.05 (d, 2H), 7.67-7.69 (d, 2H), 7.59-7.61 (d, 1H), 7.05-7.13 (m, 2H), 6.54 (s, 1H), 1.31 (s, 9H)
LC-MS (ESI): 394.1 (M+H)+.
Пример 48
Получение 1-(4-бензотриазол-1-ил-фенил)-3-(5-трет-бутил-изоксазол-3-ил)-мочевины (Соединение 48)
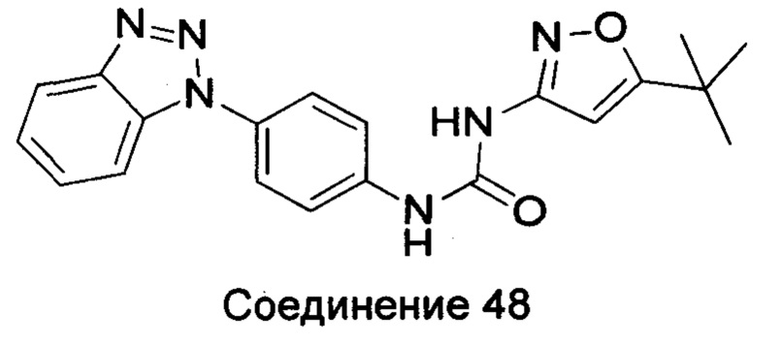
Стадия 1: Получение 4-бензотриазол-1-ил-анилина
Способ получения был такой же, как на Стадиях 1-2 в примере 1, за исключением того, что бензотриазол (TCl) использовали вместо бензимидазола на Стадии 1 для получения 4-бензотриазол-1-ил-анилина.
Стадия 2: Получение 1-(4-бензотриазол-1-ил-фенил)-3-(5-трет-бутил-изоксазол-3-ил)-мочевины
Методика была аналогична таковой, как на Стадии 5 в примере 4, за исключением того, что 4-бензотриазол-1-ил-анилин использовали вместо 1-(4-амино-фенил)-1Н-бензимидазол-5-ола для получения 1-(4-бензотриазол-1-ил-фенил)-3-(5-трет-бутил-изоксазол-3-ил)-мочевины.
1HNMR (DMSO-d6, 400MHz) δ: 9.65 (s, IH), 8.16-8.18 (d, IH), 8.12 (s, IH), 7.82-7.85 (d, 2H), 7.73-7.79 (m, 3Н), 7.55-7.59 (m, 1H), 7.44-7.48 (m, 1H), 5.94 (s, 1H), 1.40 (s, 9H).
LC-MS (ESI): 376.9 (M+H)+.
Пример 49
Получение
1-(5-трет-бутил-изоксазол-3-ил)-3-(4-пирроло[2,3-b]пиридин-1-ил-фенил)-мочевины (Соединение 49)
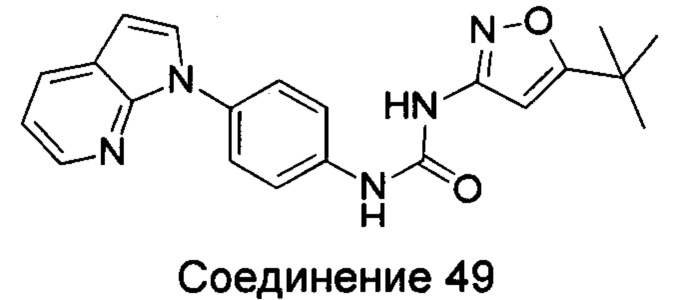
Стадия 1: Получение 4-пирроло[2,3-b]пиридин-1-ил-анилина
Способ получения был такой же, как на Стадиях 1-2 в примере 1, за исключением того, что 1Н-пирроло[2,3-b]пиридин (Runjie) использовали вместо бензимидазола на Стадии 1 с получением 4-пирроло[2,3-b]пиридин-1-ил-анилина.
Стадия 2: Получение 1-(5-трет-бутил-изоксазол-3-ил)-3-(4-пирроло[2,3-b]пиридин-1-ил-фенил)-мочевины
Методика была аналогична таковой, как на Стадии 5 в примере 4, за исключением того, что 4-пирроло[2,3-b]пиридин-1-ил-анилин использовали вместо 1-(4-амино-фенил)-1Н-бензимидазол-5-ола с получением 1-(5-трет-бутил-изоксазол-3-ил)-3-(4-пирроло[2,3-b]пиридин-1-ил-фенил)-мочевины.
1HNMR (DMSO-d6, 400MHz) δ: 9.55 (s, 1H), 8.98 (s, 1H), 8.30-8.32 (m, 1H), 8.07-8.09 (m, 1H), 7.90-7.91 (d, 1H), 7.80-7.82 (d, 2H), 7.61-7.63 (d, 2H), 7.18-7.22 (m, 1H), 6.70-6.71 (d, 1H), 6.53 (s, 1H), 1.30 (s, 9H).
LC-MS (ESI): 376.2 (M+H)+.
Пример 50
Получение
1-(5-трет-бутил-изоксазол-3-ил)-3-(4-имидазо[4,5-b]пиридин-3-ил-фенил)-мочевины (Соединение 50)
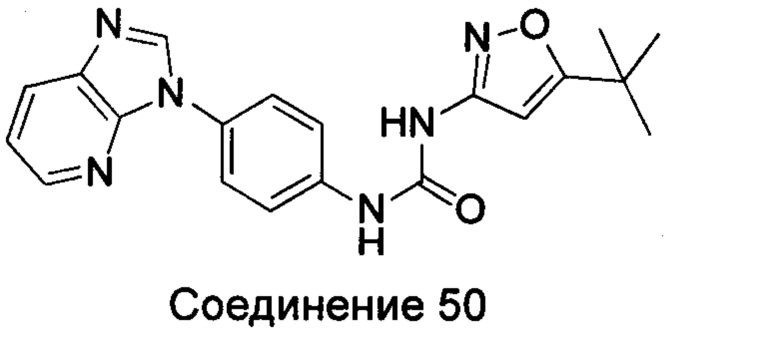
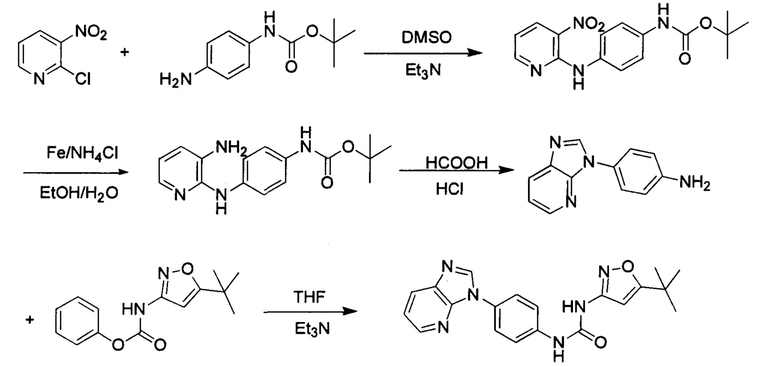
Стадия 1: Получение трет-бутил [4-(3-нитро-пиридин-2-иламино)-фенил]-карбамата 2-хлор-3-нитропиридин (1 г, 6.3 ммоль, TCl), трет-бутил(4-амино-фенил)-карбамат (Darui) (1.6 г, 7.56 ммоль), и триэтиламин (3.2 г, 31.5 ммоль) перемешивали в DMSO (30 мл) при 85°С в течение ночи. На следующий день реакционный раствор влили в воду (100 мг) и экстрагировали этилацетатом (60 мл×2). Органическую фазу промыли дважды с помощью насыщенного раствора NaCl, высушили над безводным сульфатом натрия, и отфильтровали. Фильтрат сконцентрировали при пониженном давлении. Остатки очистили с помощью колоночной хроматографии (элюент: петролейный эфир/этилацетат) с получением 1.25 г трет-бутил [4-(3-нитро-пиридин-2-иламино)-фенил]-карбамата.
Стадия 2: Получение трет-бутил [4-(3-амино-пиридин-2-иламино)-фенил]-карбамата
Трет-бутил [4-(3-нитро-пиридин-2-иламино)-фенил]-карбамат (1.25 г, 3.81 моль), восстановленный порошок железа (1.1 г, 19.05 ммоль), и хлорид аммония (1.63 г, 30.48 ммоль) перемешивали при кипении с обратным холодильником в этаноле (40 мл)/вода (10 мл) в течение 2 часов. Реакционный раствор охладили до комнатной температуры, влили в насыщенный водный раствор бикарбоната натрия, экстрагировали этилацетатом (50 мл×2), и органическую фазу промыли дважды с помощью насыщенного раствора NaCl, высушили над безводным сульфатом натрия, и отфильтровали. Фильтрат сконцентрировали при пониженном давлении с получением 1 г неочищенного продукта, который сразу использовали на следующей стадии без очистки.
Стадия 3: Получение 4-(имидазо[4,5-b]пиридин-3-ил)-анилина
При комнатной температуре, трет-бутил [4-(3-амино-пиридин-2-иламино)-фенил]-аминобензоат (1 г, 3.33 ммоль) растворили в 30 мл соляной кислоты (4 моль/л) и добавили 1 мл муравьиной кислоты. Смесь нагрели до 120°С в течение 1 часа. Реакционный раствор довели до рН>9 с помощью водного аммония в ванне со льдом, экстрагировали этилацетатом (50 мл×2),Органическую фазу промыли с помощью насыщенного раствора NaCl, высушили над безводным сульфатом натрия, и отфильтровали. Фильтрат сконцентрировали при пониженном давлении с получением 340 мг неочищенного продукта, который использовали сразу на следующей стадии без очистки.
Стадия 4: Получение 1-(5-трет-бутил-изоксазол-3-ил)-3-(4-имидазо[4,5-b]пиридин-3-ил-фенил)-мочевины
Методика была аналогична таковой, как на Стадии 5 в примере 4, за исключением того, что 4-(имидазо[4,5-b]пиридин-3-ил)-анилин использовали вместо 1-(4-амино-фенил)-1Н-бензимидазол-5-ола для получения 1-(5-трет-бутил-изоксазол-3-ил)-3-(4-имидазо[4,5-b]пиридин-3-ил-фенил)-мочевины.
1HNMR (DMSO-d6, 400MHz) δ:9.58 (s, IH), 9.05 (s, IH), 8.85 (s, IH), 8.43- 8.44 (dd, 1H), 8.20-8.22 (dd, 1H), 7.85-7.87 (d, 2H), 7.67-7.69 (d, 2H), 7.37-7.41 (q, 1H), 6.53 (s, 1H), 1.31 (s, 9H).
LC-MS (ESI): 377.2 (M+H)+.
Пример 51
Получение
1-[4-(6-бромо-имидазо[4,5-b]пиридин-3-ил)-фенил]-3-(5-трет-бутил-изоксазол-3-ил)-мочевины (Соединение 51)
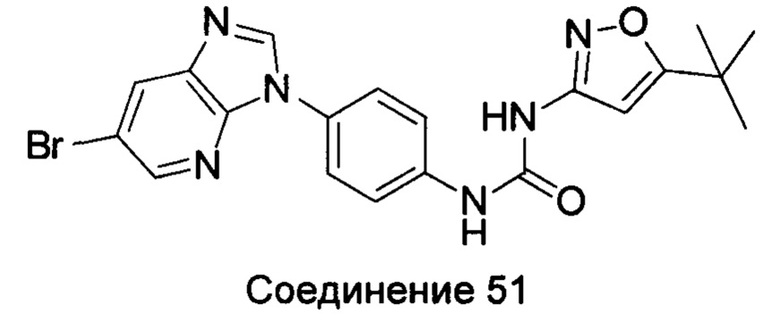
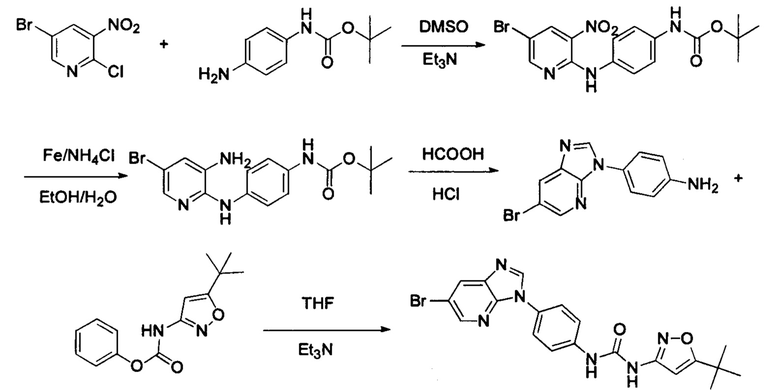
Стадия 1: Получение трет-бутил [4-(5-бромо-3-нитро-пиридин-2-иламино)-фенил]-карбамата
2-хлор-3-нитро-5-бромопиридин (1 г, 4.23 ммоль, TCl), трет-бутил (4-амино-фенил)-карбамат (1.06 г, 5.08 ммоль, Darui) и триэтиламин (2.14 г, 0.021 моль) перемешивали при 80°С в ДМСО (30 мл) в течение 4 часов. Реакционный раствор охладили до комнатной температуры, влили в воду (100 мг) и экстрагировали этилацетатом (60 мл×2). Органическую фазу промыли дважды с помощью насыщенного раствора NaCl, высушили над безводным сульфатом натрия, и отфильтровали. Фильтрат сконцентрировали при пониженном давлении. Остатки очистили с помощью колоночной хроматографии (элюент: петролейный эфир/этилацетат) с получением 700 мг трет-бутил [4-(5-бромо-3-нитро-пиридин-2-иламино)-фенил]-карбамата.
Стадия 2: Получение трет-бутил [4-(3-амино-5-бромо-пиридин-2-иламино)-фенил]-карбамата
Трет-бутил [4-(5-бромо-3-нитро-пиридин-2-иламино)-фенил]-карбамат (700 мг, 1.72 моль), полученный на Стадии 1, восстановленный порошок железа (480 мг, 8.58 ммоль), хлорид аммония (734 мг, 1.37 ммоль) кипятили с обратным холодильником в течение 1 часа в этаноле (30 мл)/вода (8 мл). Реакционный раствор охладили до комнатной температуры, влили в насыщенный водный раствор бикарбоната натрия, и экстрагировали этилацетатом (50 мл×2).Органическую фазу промыли дважды с помощью насыщенного раствора NaCl, высушили над безводным сульфатом натрия, и отфильтровали. Фильтрат сконцентрировали при пониженном давлении с получением 550 мг неочищенного продукта, который сразу использовали на следующей стадии без очистки.
Стадия 3: Получение 4-(6-бромо-имидазо[4,5-b]пиридин-3-ил)-анилина
Трет-бутил [4-(3-амино-5-бромо-пиридин-2-иламино)-фенил]-карбамат (550 мг, 1.45 ммоль), полученный на Стадии 2 растворили в 20 мл соляной кислоты (4 моль/л) при комнатной температуре, 0.5 мл муравьиной кислоты добавили, и смесь нагревали до 120°С в течение 1 часа. Реакционный раствор довели до рН>9 с помощью водного аммония в ванне со льдом, и экстрагировали этилацетатом (50 мл × 2).Органическую фазу промыли с помощью насыщенного раствора NaCl, высушили над безводным сульфатом натрия, и отфильтровали. Фильтрат сконцентрировали при пониженном давлении. Остатки очистили с помощью колоночной хроматографии (элюент: дихлорметан/метанол) с получением 230 мг 4-(6-бромо-имидазо[4,5-b]пиридин-3-ил)-анилина в виде желтого осадка.
Стадия 4: Получение 1-[4-(6-бромо-имидазо[4,5-b]пиридин-3-ил)-фенил]-3-(5-трет-бутил-изоксазол-3-ил)-мочевины
Методика была аналогична таковой, как на Стадии 5 в примере 4, за исключением того, что 4-(6-бромо-имидазо[4,5-b]пиридин-3-ил)-анилин использовали вместо 1-(4-амино-фенил)-1Н-бензимидазол-5-ола для получения 1-[4-(6-бромо-имидазо[4,5-b]пиридин-3-ил)-фенил]-3-(5-трет-бутил-изоксазол-3-ил)-мочевины.
1HNMR (DMSO-d6, 400MHz) δ: 9.59 (s, 1H), 9.06 (s, 1H), 8.90 (s, 1H), 8.51 (s, 1H), 8.52 (s, 1H), 7.80-7.82 (d, 2H), 7.67-7.69 (d, 2H), 6.54 (s, 1H), 1.30 (s, 9H).
LC-MS (ESI): 455.0\457.0 (M+H)+.
Пример 52
Получение
1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(6-трифторметил-имидазо[4,5-b]пиридин-3-ил)-фенил]-мочевины (Соединение 52)
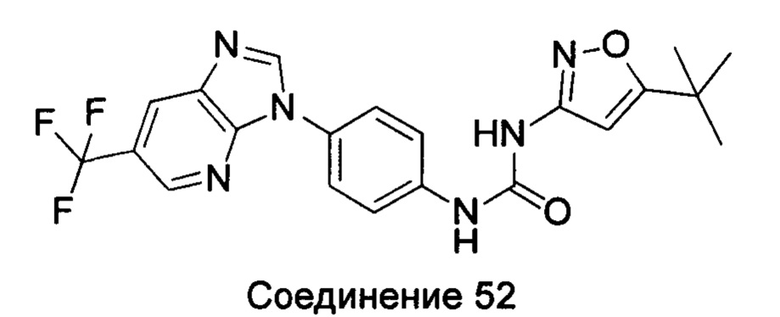
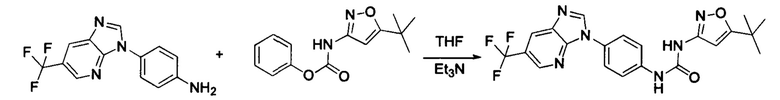
Стадия 1: Получение 4-(6-трифторметил-имидазо[4,5-b]пиридин-3-ил)-анилина
Способ получения был такой же, как на Стадиях 1-3 в примере 51, за исключением того, что 5-трифторметил-3-нитропиридин (Darui) использовали вместо 2-хлор-3-нитро-5-бромопиридина на Стадии 1 с получением 4-(б-трифторметил-имидазо[4,5-b]пиридин-3-ил)-анилина.
Стадия 2: Получение 1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(6-трифторметил-имидазо[4,5-b]пиридин-3-ил)-фенил]-мочевины
Методика была аналогична таковой, как на Стадии 5 в примере 4, за исключением того, что 4-(6-трифторметил-имидазо[4,5-b]пиридин-3-ил)-анилин использовали вместо 1-(4-амино)-фенил)-1Н-бензимидазол-5-ола с получением 1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(6-трифторметил-имидазо[4,5-b]пиридин-3-ил)-фенил]-мочевины.
1HNMR (DMSO-d6, 400MHz) δ: 9.61 (s, 1H), 9.09 (s, 1H), 9.05 (s, 1H), 8.82 (d, 20 1H), 8.67 (d, 1H), 7.82-7.84 (d, 2H), 7.69-7.71 (d, 2H), 6.53 (s, 1H), 1.30 (s, 9H).
LC-MS (ESI): 445.2 (M+H)+.
Пример 53
Получение
1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(5-хлороимидазо[4,5-b]пиридин-3-ил)-фенил]-мочевины (Соединение 53)
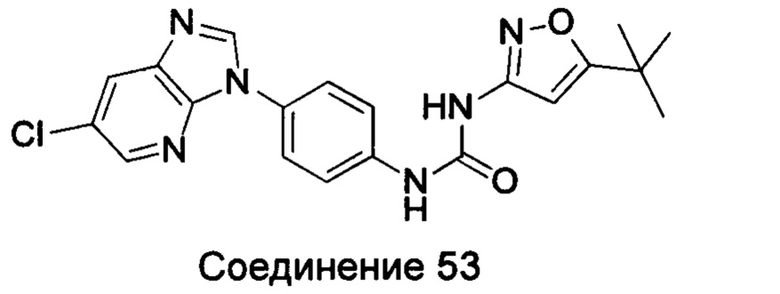
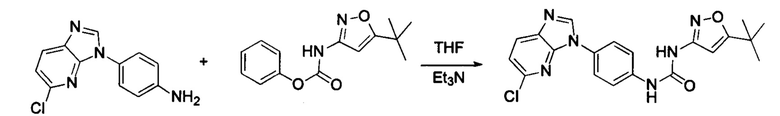
Стадия 1: Получение 4-(5-хлороимидазо[4,5-b]пиридин-3-ил)-анилина
Способ получения был такой же, как на Стадиях 1-3 в примере 51, за исключением того, что 2,6-дихлор-3-нитропиридин (TCl) использовали вместо 2-хлор-3-нитро-5-бромопиридина на Стадии 1 с получением 4-(5-хлороимидазо[4,5-b]пиридин-3-ил)-анилина.
Стадия 2: Получение 1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(5-хлороимидазо[4,5-b]пиридин-3-ил)-фенил]-мочевины
Методика была аналогична таковой, как на Стадии 5 в примере 4, за исключением того, что 4-(5-хлороимидазо[4,5-b]пиридин-3-ил)-анилин использовали вместо 1-(4-амино-фенил)-1Н-бензимидазол-5-ола для получения 1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(5-хлороимидазо[4,5-b]пиридин-3-ил)-фенил]-мочевины.
1HNMR (DMSO-d6, 400MHz) δ: 9.60 (s, 1H), 9.08 (s, 1H), 8.85 (s, 1H), 8.25-10 8.27 (d, 1H), 7.75-7.77 (d, 2H), 7.68-7.70 (d, 2H), 7.44-7.46 (d, 1H), 6.53 (s, 1H), 1.30 (s, 9H).
LC-MS(ESI):411.1 (M+H)+.
Пример 54
Получение
1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(5-метилимидазо[4,5-b]пиридин-3-ил)-фенил]-мочевины (Соединение 54)
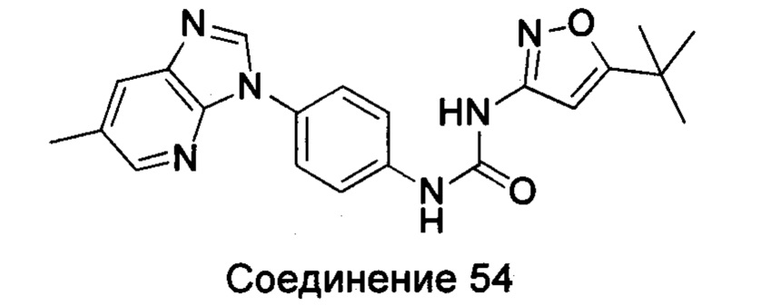
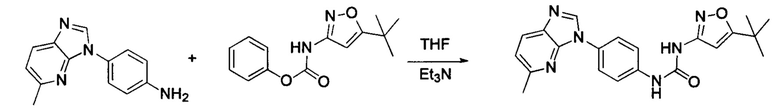
Стадия 1: Получение 4-(5-метилимидазо[4,5-b]пиридин-3-ил)-анилина
Способ получения был такой же, как на Стадиях 1-3 в примере 51, за исключением того, что 6-метил-3-нитропиридин (Darui) использовали вместо 2-хлор-3-нитро-5-бромопиридина на Стадии 1, с получением 4-(5-метилимидазо(4,5-b]пиридин-3-ил)-анилина.
Стадия 2: Получение 1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(5-метилимидазо [4,5-b] пиридин-3-ил)-фенил]-мочевины
Методика была аналогична таковой, как на Стадии 5 в примере 4, за исключением того, что 4-(5-метилимидазо[4,5-b]пиридин-3-ил)-анилин использовали вместо 1-(4-амино-фенил)-1Н-бензимидазол-5-ола для получения 1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(5-метилимидазо[4,5-b]пиридин-3-ил)-фенил]-мочевины.
1HNMR (DMSO-d6, 400MHz) δ: 9.59 (s, IH), 9.04 (s, IH), 8.71 (s, IH), 8.05-8.07 (d, 1H), 7.81-7.83 (d, 2H), 7.65-7.68 (d, 2H), 7.23-7.25 (d, 1H), 6.53 (s, 1H), 2.58 (s, 3H), 1.30(s, 9H).
LC-MS(ESI):391.2(M+H)+.
Пример 55
Получение
1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(7-метилимидазо[4,5-b]пиридин-3-ил)-фенил]-мочевины (Соединение 55)
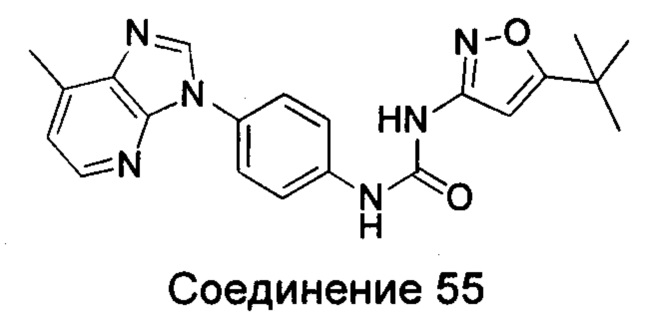
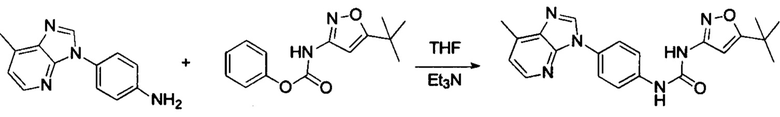
Стадия 1: Получение 4-(7-метилимидазо[4,5-b]пиридин-3-ил)-анилина
Способ получения был такой же, как на Стадиях 1-3 в примере 51, за исключением того, что 2-хлор-3-нитро-4-метилпиридин (Runjie) использовали вместо 2-хлор-3-нитро-5-бромопиридина на Стадии 1 с получением 4-(7-метилимидазо[4,5-b]пиридин-3-ил)-анилина.
Стадия 2: Получение 1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(7-метилимидазо[4,5-b]пиридин-3-ил)-фенил]-мочевины
Методика была аналогична таковой, как на Стадии 5 в примере 4, за исключением того, что 4-(7-метилимидазо[4,5-b]пиридин-3-ил)-анилин использовали вместо 1-(4-амино-фенил)-1Н-бензимидазол-5-ола для получения 1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(7-метилимидазо[4,5-b]пиридин-3-ил)-фенил]-мочевины.
1HNMR (DMSO-d6, 400MHz) δ: 9.58 (s, IH), 9.05 (s, IH), 8.78 (s, IH), 8.28-8.29 (d, 1H), 7.84-7.86 (d, 2H), 7.66-7.68 (d, 2H), 7.21-7.22 (m, 1H), 6.53 (s, 1H), 2.63 (s, 3H), 1.31 (s, 9H).
LC-MS(ESI): 391.1 (M+H)+.
Пример 56
Получение
1-(5-трет-бутил-изоксазол-3-ил)-3-(4-имидазо[4,5-b]пиридин-1-ил-фенил)-мочевины (Соединение 56)
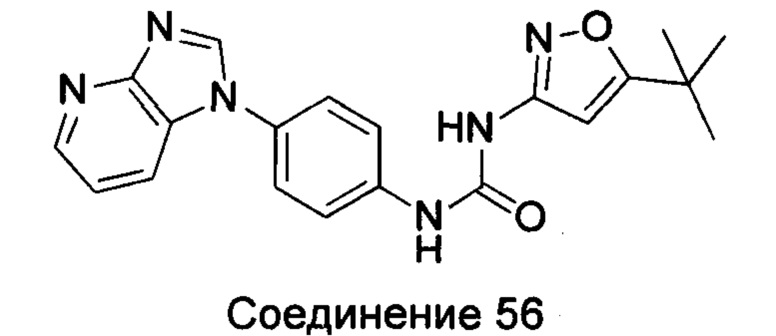
Стадия 1: Получение 4-имидазо[4,5-b]пиридин-1-ил-анилина
Способ получения был такой же, как на Стадиях 1-2 в примере 1, за исключением того, что 4-азабензимидазол (Darui) использовали вместо бензимидазола на Стадии 1 с получением 4-имидазо[4,5-b]пиридин-1-ил-анилина.
Стадия 2: Получение 1-(5-трет-бутил-изоксазол-3-ил)-3-(4-имидазо[4,5-b]пиридин-1-ил-фенил)-мочевины
Методика была аналогична таковой, как на Стадии 5 в примере 4, за исключением того, что 4-имидазо[4,5-b]пиридин-1-ил-анилин использовали вместо 1-(4-амино-фенил)-1Н-бензимидазол-5-ола для получения 1-(5-трет-бутил-изоксазол-3-ил)-3-(4-имидазо[4,5-b]пиридин-1-ил-фенил)-мочевины.
1HNMR (DMSO-d6, 400MHz) δ: 9.61 (s, IH), 9.09 (s, IH), 8.80 (s, IH), 8.51-8.52 (dd, 1H), 8.03-8.05 (dd, 1H), 7.70-7.72 (d, 2H), 7.63-7.66 (d, 2H), 7.35-7.38 (q, 1H), 6.53 (s, 1H), 1.31 (s, 9H).
LC-MS (ESI): 377.2 (M+H)+.
Пример 57
Получение
1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(6-метоксил-пурин-7-ил)-фенил]-мочевины (Соединение 57)
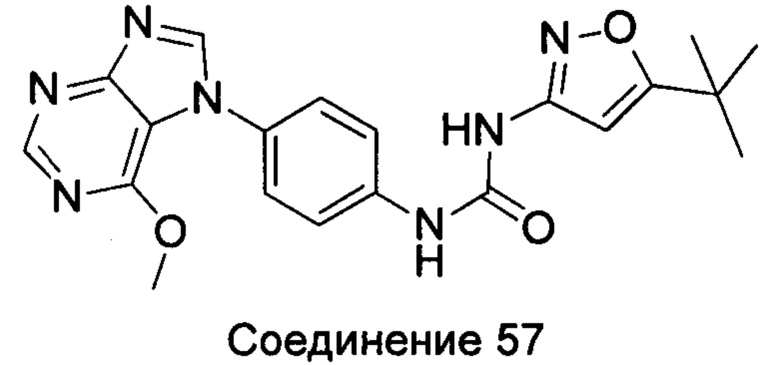
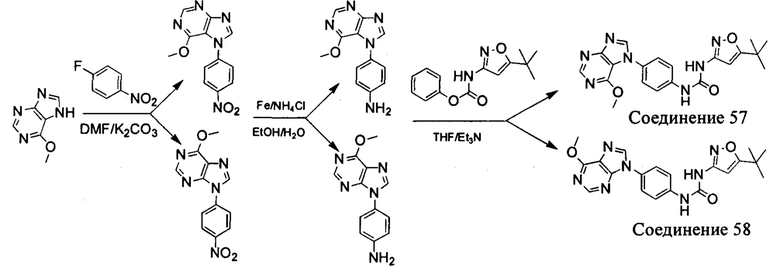
Стадия 1: Получение 4-(6-метоксил-пурин-7-ил)-анилина
Способ получения был такой же, как на Стадиях 1-2 в примере 1, за исключением того, что 6-метоксилпурин (Titan) использовали вместо бензимидазола на Стадии 1 с получением 4-(6-метоксил-пурин-7-ил)-анилина.
Стадия 2: Получение 1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(6-метоксил-пурин-7-ил)-фенил]-мочевины
Методика была аналогична таковой, как на Стадии 5 в примере 4, за исключением того, что 4-(6-метоксил-пурин-7-ил)-анилин использовали вместо 1-(4-амино-фенил)-1Н-бензимидазол-5-ола для получения 1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(6-метоксил-пурин-7-ил)-фенил]-мочевины.
1HNMR (DMSO-d6, 400MHz) δ: 9.60 (s, 1H), 9.08 (s, 1H), 8.76 (s, 1H), 8.60 (s, 1H), 7.79-7.81 (d, 2H), 7.67-7.69 (d, 2H), 6.53 (s, 1H), 4.14 (s, 3H), 1.31 (s, 9H).
LC-MS (ESI): 408.1 (M+H)+.
Пример 58
Получение
1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(6-метоксилпурин-9-ил)-фенил]-мочевины (Соединение 58)
Стадия 1: Способ получения был такой же, как на Стадиях 1-2 в примере 57, и получался другой продукт из такой же реакции, показанной на Стадии 1 - Стадии 2 примера 57.
1HNMR (DMSO-d6, 400MHz) δ: 9.63 (s, 1H), 9.07 (s, 1H), 8.70 (s, 1H), 8.63 (s, 10 1H), 7.62-7.64 (d, 2H), 7.56-7.58 (d, 2H), 6.53 (s, 1H), 3.98 (s, 3H), 1.31 (s, 9H).
LC-MS (ESI): 407.9 (M+H)+.
Пример 59
Получение
1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(6-диметиламино-пурин-7-ил)-фенил]-мочевины (Соединение 59)
Стадия 1: Получение [7-(4-амино-фенил)-7Н-пурин-6-ил]-диметил-амина
Способ получения был такой же, как на Стадиях 1-2 в примере 1, за исключением того, что 6-диметиламинопурин (Darui) использовали вместо бензимидазола на Стадии 1 с получением [7-(4-амино-фенил)-7Н-пурин-6-ил]-диметил-амина.
Стадия 2: Получение 1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(6-диметиламино-пурин-7-ил)-фенил]-мочевины
Методика была аналогична таковой, как на Стадии 5 в примере 4, за исключением того, что [7-(4-амино-фенил)-7Н-пурин-6-ил]-диметил-амин использовали вместо 1-(4-амино-фенил)-1Н-бензимидазол-5-ола для получения 1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(6-диметиламино-пурин-7-ил)-фенил]-мочевины.
1HNMR (DMSO-d6, 400MHz) δ: 9.57 (s, 1H), 9.05 (s, 1H), 8.52 (s, 1H), 8.27 (s, 1H), 7.76-7.78 (d, 2H), 7.64-7.66 (d, 2H), 6.53 (s, 1H), 3.49 (s, 6H), 1.30 (s, 9H).
LC-MS (ESI): 421.2 (M+H)+.
Пример 60
Получение 1-(4-бензимидазол-1-ил-фенил)-3-тиазол-2-ил-мочевины (Соединение 60)
Стадия 1: Получение фенил тиазол-2-ил-карбамата (активный сложный эфир)
Методика была аналогична таковой, как на Стадии 3 в примере 1, за исключением того, что 2-аминотиазол (TCl) использовали вместо 3-аминоизоксазола с получением фенил тиазол-2-ил-карбамата.
Стадия 2: Получение 1-(4-бензимидазол-1-ил-фенил)-3-тиазол-2-ил-мочевины
Методика была аналогична таковой, как на Стадии 4 в примере 1, за исключением того, что фенил тиазол-2-ил-карбамат (активный сложный эфир) использовали вместо фенил изоксазол-3-ил-карбамата (активный сложный эфир) с получением 1-(4-бензимидазол-1-ил-фенил)-3-тиазол-2-ил-мочевины.
1HNMR (DMSO-d6, 400MHz) δ: 10.65 (s, IH), 9.24 (s, IH), 8.52 (s, IH), 7.77-7.79 (m, 1H), 7.73-7.75 (d, 2H), 7.62-7.64 (d, 2H), 7.57-7.60 (m, 1H), 7.39-7.40 (d, 1H), 7.29-7.34 (m, 2H), 7.13-7.14 (d, 1H).
LC-MS (ESI): 336.1 (M+H)+.
Пример 61 Получение 1-(4-бензимидазол-1-ил-фенил)-3-(4-метил-тиазол-2-ил)-мочевины (Соединение 61)
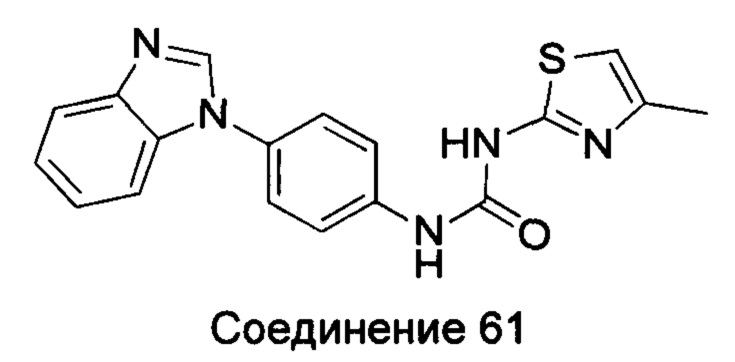
Стадия 1: Получение фенил (4-метил-тиазол-2-ил)-карбамата (активный сложный эфир)
Методика была аналогична таковой, как на Стадии 3 в примере 1, за исключением того, что 2-амино-4-метилтиазол (TCl) использовали вместо 3-аминоизоксазола с получением фенил (4-метил-тиазол-2-ил)-карбамата.
Стадия 2: Получение 1-(4-бензимидазол-1-ил-фенил)-3-(4-метил-тиазол-2-ил)-мочевины
Методика была аналогична таковой, как на Стадии 4 в примере 1, за исключением того, что фенил (4-метил-тиазол-2-ил)-карбамат (активный сложный эфир) использовали вместо фенил изоксазол-3-ил-карбамата (активный сложный эфир) с получением 1-(4-бензимидазол-1-ил-фенил)-3-(4-метил-тиазол-2-ил)-мочевины.
1HNMR (DMSO-d6, 400MHz) δ:10.50 (s, IH), 9.27 (s, IH), 8.51 (s, IH), 7.76-7.79 (m, 1H), 7.73-7.75 (d, 2H), 7.61-7.63 (d, 2H), 7.57-7.59 (m, 1H), 7.29-7.35 (m, 2H), 6.66 (s, 1H), 2.24 (s, 3H).
LC-MS (ESI): 350.1 (M+H)+.
Пример 62
Получение 1-(4-бензимидазол-1-ил-фенил)-3-[1,3,4]тиадиазол-2-ил-мочевины (Соединение 62)
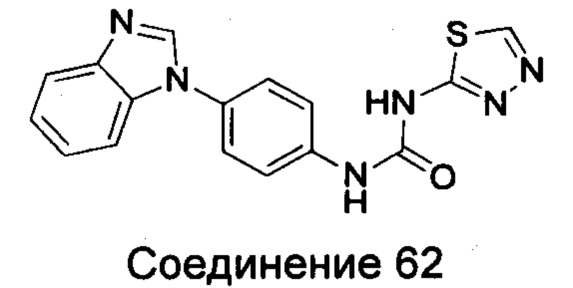
Стадия 1: Получение фенил[1,3,4] тиадиазол-2-ил-карбамата (активный сложный эфир)
Методика была аналогична таковой, как на Стадии 3 в примере 1, за исключением того, что 2-аминотиадиазол (Titan) использовали вместо 3-аминоизоксазола с получением фенил [1,3,4] тиадиазол-2-ил-карбамата.
Стадия 2: Получение 1-(4-бензимидазол-1-ил-фенил)-3-[1,3,4] тиадиазол-2-ил-мочевины
Методика была аналогична таковой, как на Стадии 4 в примере 1, за исключением того, что фенил [1,3,4]тиадиазол-2-ил-карбамат (активный сложный эфир) использовали вместо фенил изоксазол-3-ил-карбамата (активный сложный эфир) с получением 1-(4-бензимидазол-1-ил-фенил)-3-[1,3,4] тиадиазол-2-ил-мочевины.
1HNMR (DMSO-d6, 400MHz) δ: 11.10 (s, 1H), 9.36 (s, 1H), 9.08 (s, 1H), 8.53 (s, 1H), 7.73-7.79 (m, 3H), 7.58-7.65 (m, 3H), 7.29-7.36 (m, 2H).
LC-MS (ESI): 337.1 (M+H)+.
Пример 63
Получение
1-(4-бензимидазол-1-ил-фенил)-3-(5-трет-бутил-[1,3,4]тиадиазол-2-ил)-мочевины (Соединение 63)
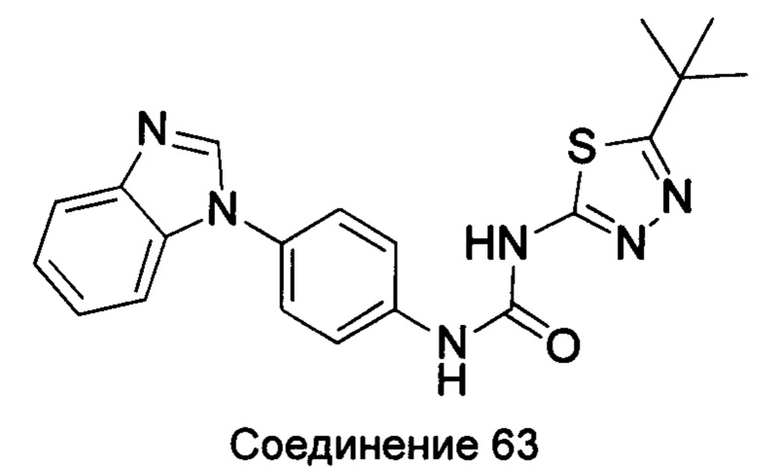
Стадия 1: Получение (5-трет-бутил-[1,3,4] тиадиазол-2-ил)-фенил карбамата (активный сложный эфир)
Методика была аналогична таковой, как на Стадии 3 в примере 1, за исключением того, что 2-амино-5-трет-бутил тиадиазол (Darui) использовали вместо 3-аминоизоксазола с получением 1-(4-бензимидазол-1-ил-фенил)-3-(5-трет-бутил-[1,3,4] тиадиазол-2-ил)-мочевины.
Стадия 2: Получение 1-(4-бензимидазол-1-ил-фенил)-3-(5-трет-бутил-[1,3,4]тиадиазол-2-ил)-мочевины
Методика была аналогична таковой, как на Стадии 4 в примере 1, за исключением того, что фенил (5-трет-бутил-[1,3,4]тиадиазол-2-ил)-карбамат (активный сложный эфир) использовали вместо фенил изоксазол-3-ил-карбамата (активный сложный эфир) с получением 1-(4-бензимидазол-1-ил-фенил)-3-(5-трет-бутил-[1,3,4]тиадиазол-2-ил)-мочевины.
1HNMR (DMSO-d6, 400MHz) δ: 11.09 (s, IH), 9.35 (s, IH), 8.56 (s, IH), 7.80-7.82 (m, 1H), 7.75-7.77 (d, 2H), 7.61-7.64 (d, 2H), 7.57-7.60 (m, 1H), 7.30-7.36 (m, 2H), 1.37 (s, 9H).
LC-MS (ESI): 393.1 (M+H)+.
Пример 64
Получение
1-(4-бензимидазол-1-ил-фенил)-3-(5-метил-1Н-пиразол-3-ил)-мочевины (Соединение 64)
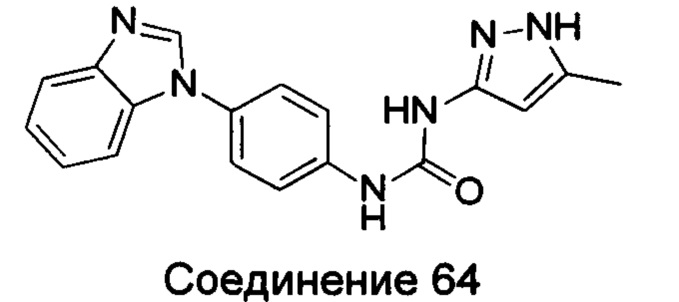
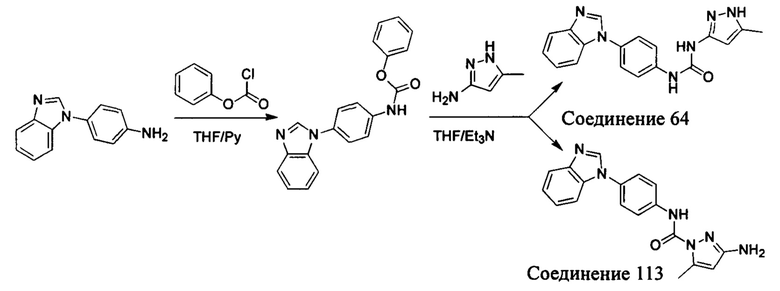
Стадия 1: Получение фенил (4-бензимидазол-1-ил-фенил)-карбамата
4-Бензимидазол-1-ил-анилин (синтезированный на Стадии 2 примера 1) (10.0 г, 0.048 моль) и пиридин (11.33 г, 0.143 моль) растворили в 100 мл ТГФ. Фенилхлорформиат (11.23 г, 0.072 моль) медленно добавили по каплям в ванне со льдом. После добавления, ледяную ванну убрали и реакционную смесь медленно нагрели до комнатной температуры и перемешивали в течение 3 часов. Реакционный раствор влили в воду, экстрагировали этилацетатом (100 мл×2), и органическую фазу промыли дважды с помощью насыщенного раствора NaCl, высушили над безводным сульфатом натрия, и отфильтровали. Фильтрат сконцентрировали при пониженном давлении с получением коричневого осадка. Получившийся осадок затем суспендировали с метил трет-бутиловым эфиром/петролейный эфир (1:1) с получением 14 г фенил (4-бензимидазол-1-ил-фенил)-карбамата в виде желтого осадка.
Стадия 2: Получение 1-(4-бензимидазол-1-ил-фенил)-3-(5-метил-1Н-пиразол-3-ил)-мочевины
Фенил (4-бензимидазол-1-ил-фенил)-карбамат (100 мг, 0.304 ммоль), полученный на Стадии 1, триэтиламин (92 мг, 0.912 ммоль), и 3-амино-5-метилпиразол (50.7 мг, 0.365 ммоль, TCl) растворили в 10 мл ТГФ. Реакционный раствор кипятили с обратным холодильником в течение ночи. На следующий день реакционный раствор сконцентрировали при пониженном давлении, и остатки очистили с помощью колоночной хроматографии (элюент: дихлорметан/метанол) с получением 40 мг 1-(4-бензимидазол-1-ил-фенил)-3-(5-метил-1Н-пиразол-3-ил)-мочевины в виде белого осадка и 10 мг Соединения 113 (см. Пример 113).
1HNMR (DMSO-d6, 400MHz) δ: 11.97 (s, 1H), 9.42 (s, 1H), 8.96 (s, 1H), 8.49 (s, 20 1H), 7.76-7.78 (m, 1H), 7.67-7.70 (d, 2H), 7.56-7.59 (m, 3H), 7.28-7.35 (m, 2H), 6.02 (s, 1H), 2.20 (s, 3H).
LC-MS (ESI): 333.1 (M+H)+.
Пример 65
Получение
1-(4-бензимидазол-1-ил-фенил)-3-(5-фенил-1Н-пиразол-3-ил)-мочевины (Соединение 65)
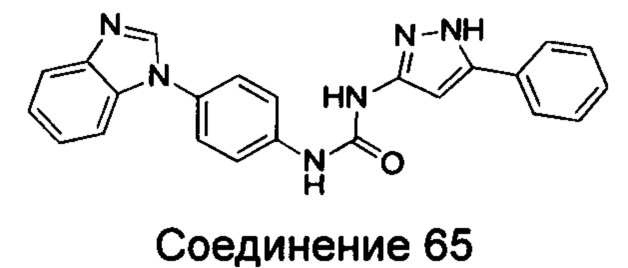
Способ получения был таким же, как в Примере 64, за исключением того, что 3-амино-5-фенил пиразол (Darui) использовали вместо 3-амино-5-метилпиразола на Стадии 2 с получением 1-(4-бензимидазол-1-ил-фенил)-3-(5-фенил-1Н-пиразол-3-ил)-мочевины.
1HNMR (DMSO-d6, 400MHz) δ: 12.80 (s, 1H), 9.27 (s, 1H), 9.13 (s, 1H), 8.51 (s, 5 1H), 7.71-7.79 (m, 5H), 7.58-7.61 (m, 3H), 7.45-7.48 (m, 2H), 7.29-7.38 (m, 3H), 6.72 (s, IH).
LC-MS (ESI): 395.1 (M+H)+.
Пример 66
Получение
1-(4-бензимидазол-1-ил-фенил)-3-(5-циклопропил-2Н-пиразол-3-ил)-мочевины (Соединение 66)
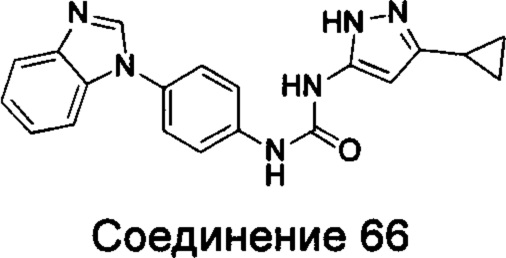
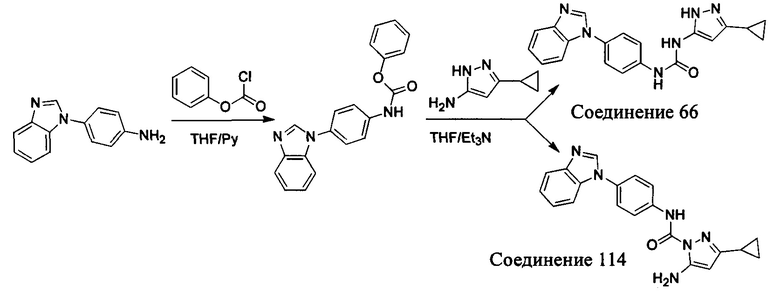
Способ получения был таким же, как в Примере 64, за исключением того, что 3-циклопропил-1Н-пиразол-5-амино (Darui) использовали вместо 3-амино-5-метилпиразола на Стадии 2 с получением 1-(4-бензимидазол-1-ил-фенил)-3-(5-циклопропил-2Н-пиразол-3-ил)-мочевины и Соединения 114 (см. Пример 114).
1HNMR (DMSO-d6, 400MHz) δ: 12.03 (s, 1H), 9.37 (s, 1H), 8.96 (s, 1H), 8.50 (s, 1H), 7.76-7.79 (m, 1H), 7.67-7.70 (d, 2H), 7.56-7.59 (m, 3H), 7.30-7.34 (m, 2H), 5.92 20 (s, 1H), 1.85-1.89 (m, 1H), 0.91-0.95 (m, 2H), 0.66-0.70 (m, 2H).
LC-MS (ESI): 359.2 (M+Н)+.
Пример 67
Получение
1-(4-бензимидазол-1-ил-фенил)-3-(5-трифторметил-2Н-пиразол-3-ил)-мочевины (Соединение 67)
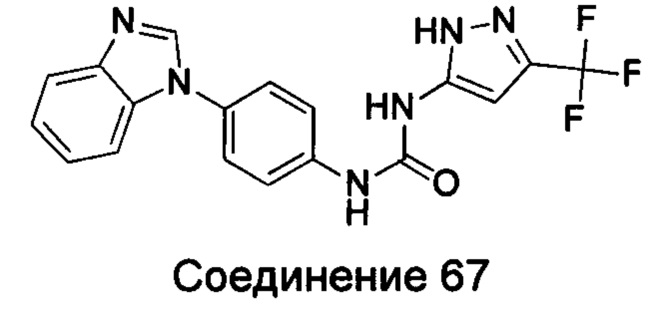
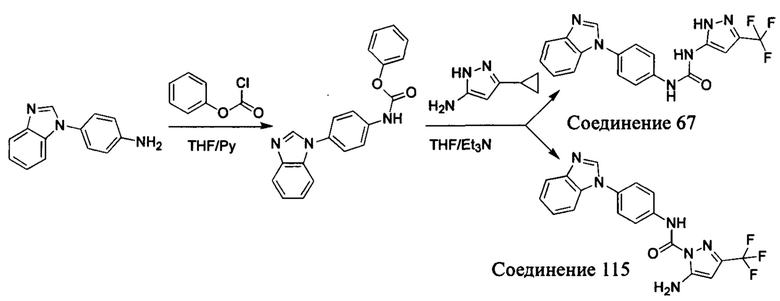
Способ получения был таким же, как в Примере 64, за исключением того, что 5-амино-3-трифторметилпиразол (Darui) использовали вместо 3-амино-5-метилпиразола на Стадии 2 с получением 1-(4-бензимидазол-1-ил-фенил)-3-(5-трифторметил-2Н-пиразол-3-ил)-мочевины и Соединения 115 (см. Пример 115).
1HNMR (DMSO-d6, 400MHz) δ: 13.30 (s, 1H), 9.33 (s, 1H), 9.33 (s, 1H), 8.50 (s, 1H), 7.76-7.78 (m, 1H), 7.70-7.72 (d, 2H), 7.57-7.62 (m, 3H), 7.28-7.36 (m, 2H), 6.42 (s, IH).
LC-MS (ESI): 387.1 (M+H)+.
Пример 68
Получение
1-(4-бензимидазол-1-ил-фенил)-3-(5-трет-бутил-2Н-пиразол-3-ил)-мочевины (Соединение 68)
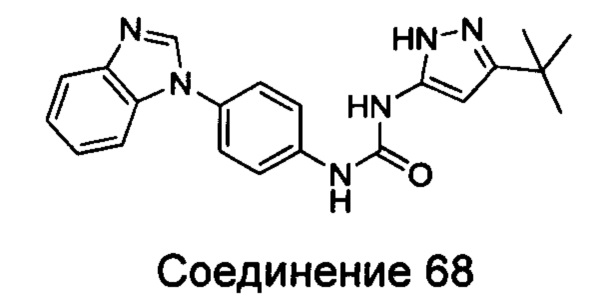
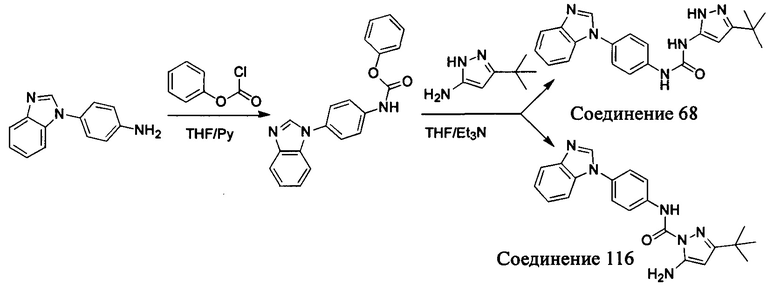
Стадия 1: Получение фенил (4-бензимидазол-1-ил-фенил)-карбамата
4-Бензимидазол-1-ил-анилин (синтезированный на Стадии 2 примера 1) (10.0 г, 0.048 моль), и пиридин (11.33 г, 0.143 моль) растворили в 100 мл ТГФ. Фенилхлорформиат (11.23 г, 0.072 моль) медленно добавили по каплям в ванне со льдом. После добавления, ледяную ванну убрали и раствор медленно нагрели до комнатной температуры и дали прореагировать в течение 3 часов. Реакционный раствор влили в воду и экстрагировали этилацетатом (100 мл×2). Органическую фазу промыли дважды с помощью насыщенного раствора NaCl, высушили над безводным сульфатом натрия, и отфильтровали. Фильтрат сконцентрировали при пониженном давлении с получением серого осадка. Получившийся осадок суспендировали с метил трет-бутиловым эфиром/петролейный эфир (1:1) с получением 14 г фенил (4-бензимидазол-1-ил-фенил)-карбамата в виде желтого осадка.
Стадия 2: Получение 1-(4-бензимидазол-1-ил-фенил)-3-(5-трет-бутил-2Н-пиразол-3-ил)-мочевины
Фенил (4-бензимидазол-1-ил-фенил)-карбамат (100 мг, 0.304 ммоль), полученный на Стадии 1, триэтиламин (92 мг, 0.912 ммоль), и 3-трет-бутил-пиразол-5-амин (50.7 мг, 0.365 ммоль) растворили в 10 мл ТГФ и реакционный раствор кипятили с обратным холодильником в течение ночи. На следующий день реакционный раствор сконцентрировали при пониженном давлении, и остатки очистили с помощью колоночной хроматографии (элюент: дихлорметан/метанол) с получением 40 мг 1-(4-бензимидазол-1-ил-фенил)-3-(5-трет-бутил-2Н-пиразол-3-ил)-мочевины в виде белого осадка и 12 мг Соединения 116 (см. Пример 116).
1HNMR (DMSO-d6, 400MHz) δ: 12.03 (s, 1H), 9.41 (s, 1H), 9.00 (s, 1H), 8.50 (s, 1H), 7.76-7.78 (m, 1H), 7.67-7.70 (d, 2H), 7.57-7.59 (m, 3H), 7.28-7.35 (m, 2H), 6.03 10 (s, 1H), 1.27 (s, 9H).
LC-MS (ESI): 375.2 (M+H)+.
Пример 69
Получение
1-(5-трет-бутил-2Н-пиразол-3-ил)-3-[4-(5-фтор-бензимидазол-1-ил)-фенил]-мочевины (Соединение 69)
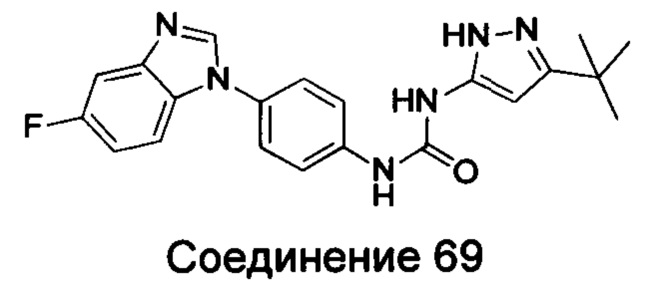
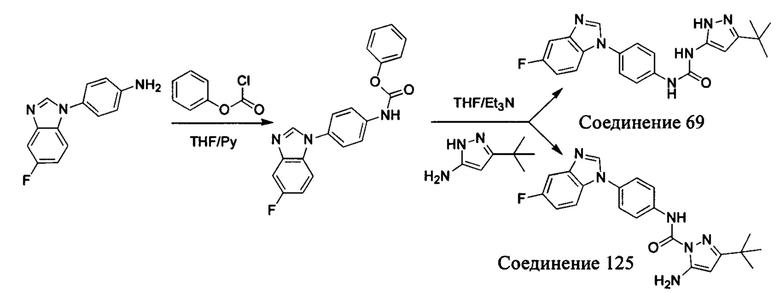
Способ получения был таким же, как в Примере 68, за исключением того, что 4-(5-фтор-бензимидазол-1-ил)-анилин (синтезированный на Стадии 1 примера 22) использовали вместо 4-бензимидазол-1-ил-анилина на Стадии 1 с получением 1-(5-трет-бутил-2Н-пиразол-3-ил)-3-[4-(5-фтор-бензимидазол-1-ил)-фенил]-мочевины и Соединения 125 (см. Пример 125).
1HNMR (DMSO-d6, 400MHz) δ: 12.03 (s, 1H), 9.43 (s, 1H), 8.99 (s, 1H), 8.56 (s, 1H), 7.67-7.70 (d, 2H), 7.55-7.60 (m, 4H), 7.17-7.22 (m, 1H), 6.03 (s, 1H), 1.27 (s, 9H).
LC-MS (ESI): 393.1 (M+H)+.
Пример 70
Получение
1-(5-трет-бутил-2Н-пиразол-3-ил)-3-[4-(5-трифторметил-бензимидазол-1-ил)-фенил]-мочевины (Соединение 70)
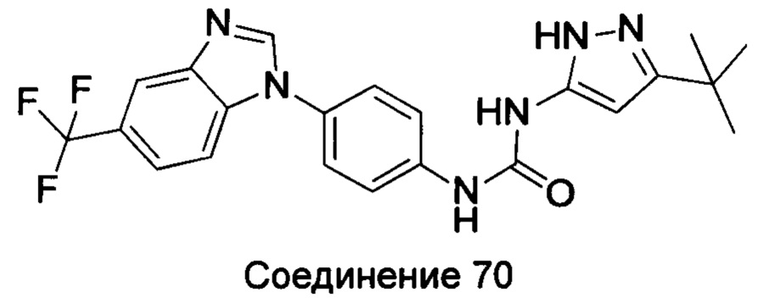
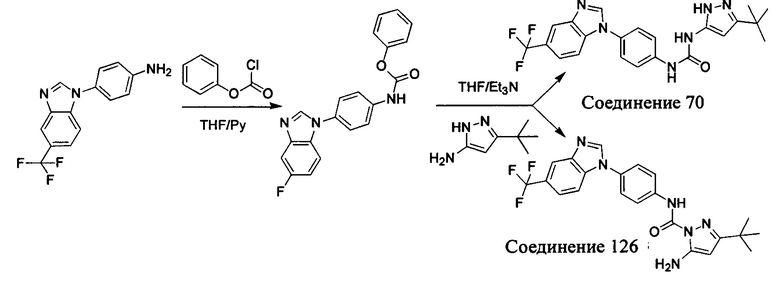
Способ получения был таким же, как в Примере 68, за исключением того, что 4-(5-трифторметил- бензимидазол-1-ил)-анилин (синтезированный на Стадии 1 примера 23) использовали вместо 4-бензимидазол-1-ил-анилина на Стадии 1 с получением
1-(5-трет-бутил-2Н-пиразол-3-ил)-3-[4-(5-трифторметил-бензимидазол-1-ил)-фенил]-мочевины и Соединения 126 (см. Пример 126).
1HNMR (DMSO-d6, 400MHz) δ: 12.03 (s, 1H), 9.44 (s, 1H), 9.00 (s, 1H), 8.72 (s, 1H), 8.15 (d, 1H), 7.75-7.77 (d, 1H), 7.70-7.72 (d, 2H), 7.64-7.67 (dd, 1H), 7.60-7.63 (d, 2H), 6.03(s, 1H), 1.27 (s, 9H).
LC-MS (ESI): 443.1 (М+Н)+.
Пример 71
Получение
1-(5-трет-бутил-2Н-пиразол-3-ил)-3-[4-(5-метоксил-бензимидазол-1-ил)-фенил]-мочевины (Соединение 71)
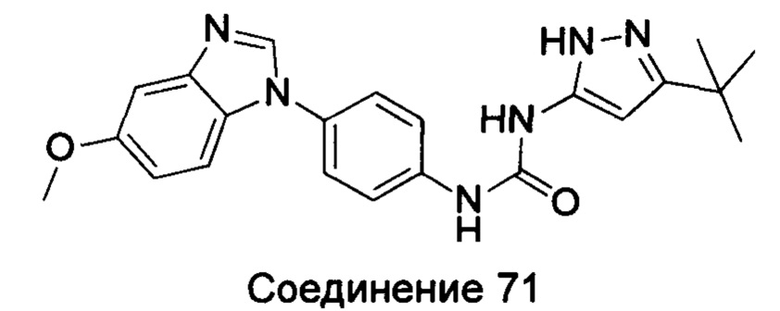
Способ получения был таким же, как в Примере 68, за исключением того, что 4-(5-метоксил-бензимидазол-1-ил)-анилин (синтезированный на Стадии 3 примера 4) использовали вместо 4-бензимидазол-1-ил-анилина на Стадии 1 с получением 1-(5-трет-бутил-2Н-пиразол-3-ил)-3-[4-(5-метоксил-бензимидазол-1-ил)-фенил]-мочевины.
1HNMR (DMSO-d6, 400MHz) δ: 12.02 (8, 1H), 9.41 (8, 1H), 8.99 (s, 1H), 8.43 (8, 25 1H), 7.66-7.68 (d, 2H), 7.55-7.57 (d, 2H), 7.46-7.48 (d, 1H), 7.29-7.30 (d, 1H), 6.94-6.96 (dd, 1H), 6.02 (8, 1H), 3.82 (s, 3H), 1.27 (8, 9H).
LC-MS (ESI): 405.1 (М+Н)+.
Пример 72
Получение
1-(5-трет-бутил-2Н-пиразол-3-ил)-3-[4-(5-этоксил-бензимидазол-1-ил)-фенил]-мочевины (Соединение 72)
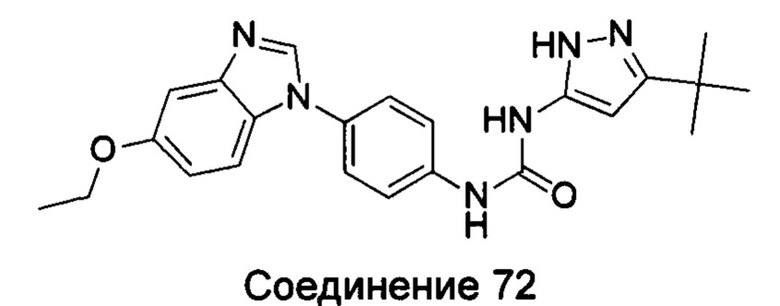
Способ получения был таким же, как в Примере 68, за исключением того, что 4-(5-этоксил-бензимидазол-1-ил)-анилин (синтезированный на Стадии 1 примера 6) использовали вместо 4-бензимидазол-1-ил-анилина на Стадии 1 с получением 1-(5-трет-бутил-2Н-пиразол-3-ил)-3-[4-(5-этоксил-бензимидазол-1-ил)-фенил]-мочевины.
1HNMR (DMSO-d6, 400MHz) δ: 12.02 (s, 1H), 9.40 (s, 1H), 8.98 (s, 1H), 8.42 (s, 10 1H), 7.66-7.68 (d, 2H), 7.55-7.57 (d, 2H), 7.44-7.47 (d, 1H). 7.27 (d, 1H), 6.92-6.95 (dd, 1H), 6.02 (s, 1H), 4.06-4.11 (q, 2H), 1.34-1.38 (t, 3H), 1.27 (s, 9H).
LC-MS (ESI): 419.2 (M+Н)+.
Пример 73
Получение 1-(5-трет-бутил-2Н-пиразол-3-ил)-3-[4-(5-гексилоксил-бензимидазол-1-ил)-фенил]-мочевины (Соединение 73)
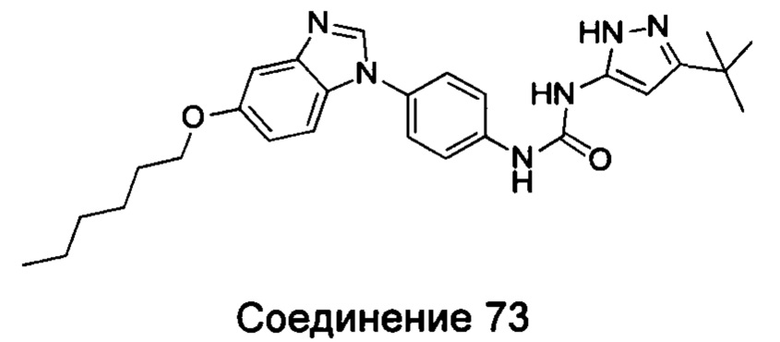
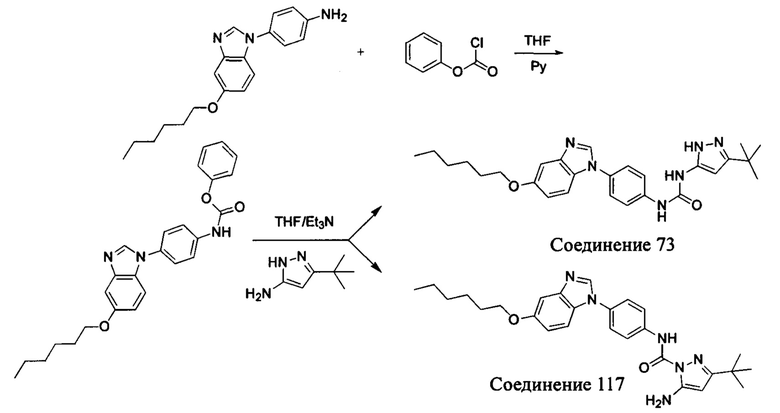
Способ получения был таким же, как в Примере 68, за исключением того, что 4-(5-гексилоксил-бензимидазол-1-ил)-анилин (синтезированный на Стадии 1 примера 7) использовали вместо 4-бензимидазол-1-ил-анилина на Стадии 1 с получением
1-(5-трет-бутил-2Н-пиразол-3-ил)-3-[4-(5-гексилоксил-бензимидазол-1-ил)-фенил]-мочевины и Соединения 117 (см. Пример 117).
1HNMR (DMSO-d6, 400MHz) δ:12.02 (s, 1H), 9.41 (s, 1H), 8.99 (s, 1H), 8.42 (s, 1H), 7.66-7.68 (d, 2H), 7.55-7.57 (d, 2H), 7.44-7.46 (d, 1H), 7.27-7.28 (d, 1H), 6.92-5 6.95 (dd, 1H), 6.02 (s, 1H), 4.00-4.03 (t, 2H), 1.70-1.76 (m, 2H), 1.41-1.47 (m, 2H), 1.30-1.35 (m, 4H), 1.27 (s, 9H), 0.87-0.91 (t, 3H).
LC-MS (ESI): 475.2 (M+H)+.
Пример 74
Получение
1-(5-трет-бутил-2Н-пиразол-3-ил)-3-[4-(5-изопропокси-бензимидазол-1-ил)-фенил]-мочевины (Соединение 74)
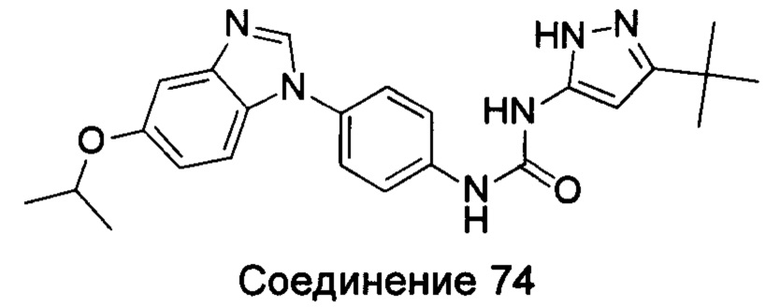
Способ получения был таким же, как в Примере 68, за исключением того, что 4-(5-изопропокси-бензимидазол-1-ил)-анилин (синтезированный в примере 8) использовали вместо 4-бензимидазол-1-ил-анилин на Стадии 1 с получением 1-(5-трет-бутил-2Н-пиразол-3-ил)-3-[4-(5-изопропокси-бензимидазол-1-ил)-фенил]-мочевины.
1HNMR (DMSO-d6, 400MHz) δ:12.03 (s, 1H), 9.41 (s, 1H), 8.99 (s, 1H), 8.42 (s, 1H), 7.66-7.68 (d, 2H), 7.55-7.57 (d, 2H), 7.44-7.46 (d, 1H), 7.27-7.28 (d, 1H), 6.91-6.94 (dd, 1H), 6.02 (s, 1H), 4.61-4.67 (m, 1H), 1.28-1.30 (d, 6H), 1.27 (s, 9H).
LC-MS (ESI): 433.1 (M+H)+.
Пример 75
Получение
1-[4-(5-втор-бутокси-бензимидазол-1-ил)-фенил]-3-(5-трет-бутил-2Н-пиразол-3-ил)-мочевины (Соединение 75)
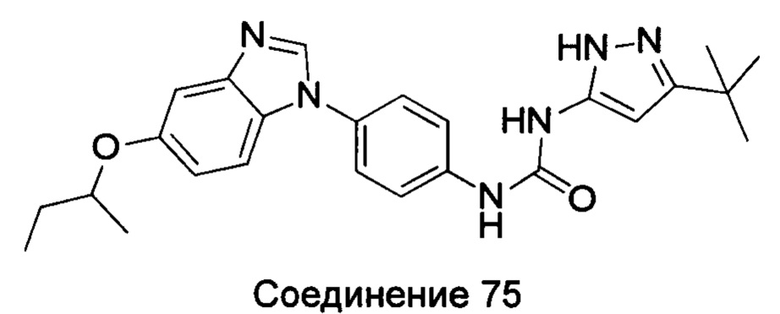
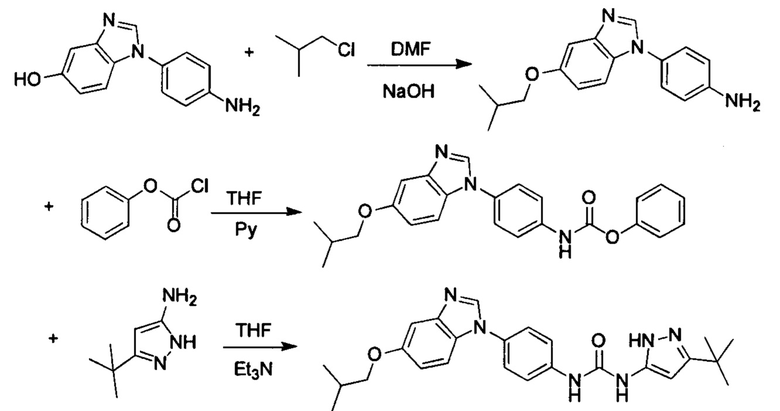
Стадия 1: Получение 4-(5-втор-бутокси-бензимидазол-1-ил)-анилина
1-(4-амино-фенил)-1Н-бензимидазол-5-ол (полученный на Стадии 4 примера 4) (300 мг, 1.22 моль) растворили в ДМФ (20 мл) при комнатной температуре и гидроксид натрия (98 мг, 2.44 ммоль) добавили и перемешивали при комнатной температуре в течение 30 минут во время чего преципитировался осадок. При комнатной температуре, хлорбутан (TCl) (170 г, 1.83 ммоль) добавили и смесь нагрели до 90°С и дали прореагировать в течение 1.5 часов. Реакционный раствор охладили до комнатной температуры, медленно влили в 100 мл воды и экстрагировали этилацетатом (60 мл×2). Органическую фазу промыли с помощью насыщенного раствора NaCl дважды, высушили над безводным сульфатом натрия, и отфильтровали. Фильтрат сконцентрировали при пониженном давлении, с получением 350 мг желто-черного маслянистого вещества, которое сразу использовали на следующей стадии без очистки.
Стадия 2: Получение фенил [4-(5-втор-бутокси-бензимидазол-1-ил)-фенил]-карбамата
4-(5-втор-бутокси-бензимидазол-1-ил)-анилин (350 мг, 1.24 ммоль), полученный на Стадии 1 и пиридин (294 мг, 3.72 ммоль) растворили в ТГФ (100 мл), и фенилхлорформиат (291 мг, 1.86 ммоль) добавили по каплям в ванне со льдом, и осадок преципитировался во время процесса. После добавления, реакционную смесь нагревали до комнатной температуры, дали прореагировать в течение 1.5 часов и затем погасили 100 мл воды, экстрагировали этилацетатом (60 мл×2), и органическую фазу промыли с помощью насыщенного раствора NaCl, высушили над безводным сульфатом натрия, и отфильтровали. Фильтрат сконцентрировали при пониженном давлении. Остатки очистили с помощью колоночной хроматографии (элюент: дихлорметан/метанол) с получением 350 мг фенил [4-(5-втор-бутокси-бензимидазол-1-ил)-фенил]-карбамата.
Стадия 3: Получение 1-[4-(5-втор-бутокси-бензимидазол-1-ил)-фенил]-3-(5-трет-бутил-2Н-пиразол-3-ил)-мочевины
Методика была аналогична таковой, как на Стадии 2 в примере 68, за исключением того, что фенил [4-(5-втор-бутокси-бензимидазол-1-ил)-фенил]-карбамат использовали вместо фенил (4-бензимидазол-1-ил-фенил)-карбамата с получением 1-[4-(5-втор-бутокси-бензимидазол-1-ил)-фенил]-3-(5-трет-бутил-2Н-пиразол-3-ил)-мочевины.
1HNMR (DMSO-d6, 400MHz) δ: 12.02 (s, 1H), 9.41 (s, 1H), 8.99 (s, 1H), 8.42 (s, 1H), 7.66-7.68 (d, 2H), 7.55-7.57 (d, 2H), 7.44-7.46 (d, 1H), 7.27-7.28 (d, 1H), 6.92-6.95 (dd, 1H), 6.02 (s, 1H), 4.38-4.45 (m, 1H), 1.54-1.74 (m, 2H), 1.27 (s, 9H), 1.25-25 1.26 (d, 3H), 0.94-0.97 (t, 3H).
LC-MS (ESI): 447.2 (M+H)+.
Пример 76
Получение
1-(5-трет-бутил-2Н-пиразол-3-ил)-3-[4-(5-изобутокси-бензимидазол-1-ил)-фенил]-мочевины (Соединение 76)
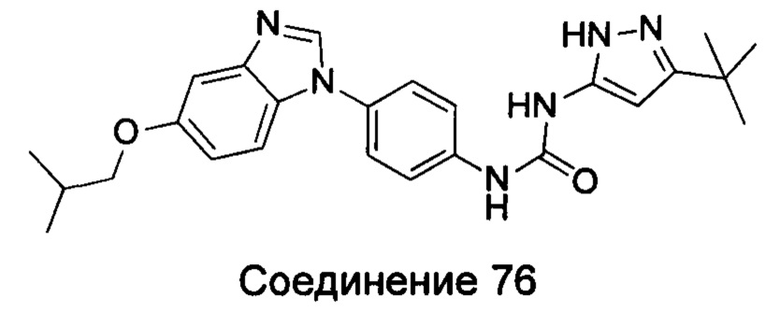
Способ получения был таким же, как в Примере 75, за исключением того, что 1-хлор-2-метилпропан (TCl) использовали вместо хлорбутана на Стадии 1 с получением
1-(5-трет-бутил-2Н-пиразол-3-ил)-3-[4-(5-изобутокси-бензимидазол-1-ил)-фенил]-мочевины.
1HNMR (DMSO-d6, 400MHz) δ: 12.03 (s, 1H), 9.41 (s, 1H), 8.99 (s, 1H), 8.42 (s, 1H), 7.66-7.68 (d, 2H), 7.55-7.57 (d, 2H), 7.45-7.47 (d, 1H), 7.26-7.27 (d, 1H), 6.94-6.97 (dd, 1H), 6.02 (s, 1H), 3.80-3.81 (d, 2H), 2.02-2.08 (m, 1H), 1.27 (s, 9H), 1.00-1.02 (d, 6H).
LC-MS (ESI): 447.3 (M+H)+.
Пример 77
Получение
1-(5-трет-бутил-2Н-пиразол-3-ил)-3-{4-[5-(2-метоксил-этоксил)-бензимидазол-1-ил]-фенил}-мочевины (Соединение 77)
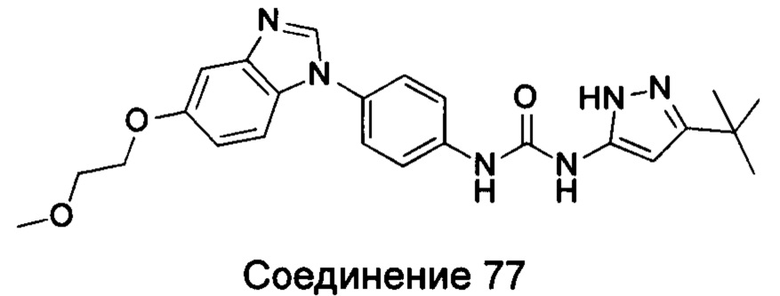
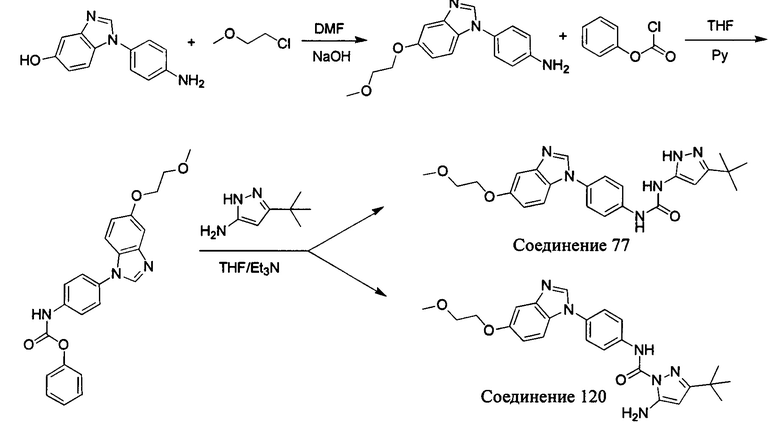
Стадия 1: Получение 4-[5-(2-метоксил-этоксил)-бензимидазол-1-ил]-анилина При комнатной температуре, 1-(4-аминофенил)-1Н-бензимидазол-5-ол (18.0 г, 0.08 моль) (полученный на Стадии 4 примера 4) растворили в ДМФ (200 мл), гидроксид натрия (9.6 г, 0.24 моль) добавили, и смесь перемешивали при комнатной температуре в течение 30 минут, во время чего преципитировался осадок. При комнатной температуре, хлорэтилметиловый эфир (11.34 г, 0.12 моль) добавили и смесь нагрели до 90°С в течение 1.5 часов. Реакционный раствор охладили до комнатной температуры, медленно влили в 600 мл воды, и осадок преципитировался. Смесь перемешивали при комнатной температуре в течение 30 минут. После фильтрации, получившийся осадок промыли водой и высушили под вакуумом с получением 20.5 г 4-[5-(2-метоксил-этоксил)-бензимидазол-1-ил]-анилина в виде розового осадка.
Стадия 2: Получение фенил {4-[5-(2-метоксил-этоксил)-бензимидазол-1-ил]-фенил}-карбамата
4-[5-(2-метоксил-этоксил)-бензимидазол-1-ил]-анилин (10.0 г, 0.035 моль), полученный на Стадии 1 и пиридин (8.37 г, 0.106 моль) растворили в ТГФ (100 мл). Фенилхлорформиат (6.63 г, 0.042 моль) добавилипо каплям в ванне со льдом и осадок преципитировался во время процесса. После добавления, реакционную смесь нагревали до комнатной температуры в течение 1.5 часов, затем погасили 200 мл воды, экстрагировали этилацетатом (100 мл×2), и органическую фазу промыли с помощью насыщенного раствора NaCl, высушили над безводным сульфатом натрия, и отфильтровали. Фильтрат сконцентрировали при пониженном давлении. Остатки очистили с помощью колоночной хроматографии (элюент: дихлорметан/метанол) с получением 15 г фенил {4-[5-(2-метоксил-этоксил)-бензимидазол-1-ил]-фенил}-карбамата. LC-MS (ESI): 404.1 (М+Н)+.
Стадия 3: 1-(5-трет-бутил-2Н-пиразол-3-ил)-3-{4-[5-(2-метоксил-этоксил)-бензимидазол-1-ил]-фенил}-мочевины
Методика была аналогична таковой, как на Стадии 2 в примере 68, за исключением того, что фенил {4-[5-(2-метоксил-этоксил)-бензимидазол-1-ил]-фенил}-карбамат использовали вместо фенил (4-бензимидазол-1-ил-фенил)-карбамата на Стадии 2 с получением 1-(5-трет-бутил-2Н-пиразол-3-ил)-3-{4-[5-(2-метоксил-этоксил)-бензимидазол-1-ил]-фенил}-мочевины и Соединения 120 (см. Пример 120).
1HNMR (DMSO-d6, 400MHz) δ: 12.03 (s, 1H), 9.41 (s, 1H), 8.99 (s, 1H), 8.43 (s, 20 1H), 7.65-7.68 (d, 2H), 7.55-7.57 (d, 2H), 7.45-7.48 (d, 1H), 7.30-7.31 (d, 1H), 6.95-6.98 (dd, 1H), 6.02 (s, 1H), 4.14-4.16 (m, 2H), 3.68-3.71 (m, 2H), 3.33 (s, 3Н), 1.27 (s, 9H).
LC-MS (ESI): 449.2 (M+H)+.
Пример 78
Получение
1-(5-трет-бутил-2Н-пиразол-3-ил)-3-{4-[5-(2-этоксил-этоксил)-бензимидазол-1-ил]-фенил}-мочевины (Соединение 78)
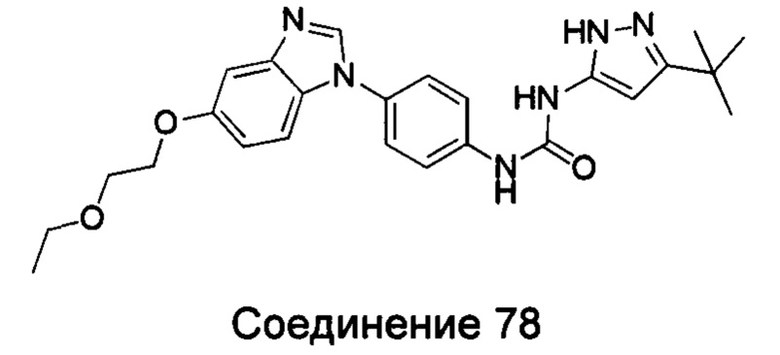
Методика была аналогична таковой, как на Стадиях 1-3 в примере 77, за исключением того, что 2-этоксил-хлорэтан использовали вместо 2-хлорэтилметилового эфира на Стадии 1 с получением 1-(5-трет-бутил-2Н-пиразол-3-ил)-3-{4-[5-(2-этоксил-этоксил)-бензимидазол-1-ил]-ф енил}-мочевины.
1HNMR (DMSO-d6, 400MHz) δ: 12.02 (s, 1H), 9.40 (s, 1H), 8.98 (s, 1H), 8.43 (s, 1H), 7.66-7.68 (d, 2H), 7.55-7.57 (d, 2H), 7.45-7.47 (d, 1H), 7.30 (d, 1H), 6.95-6.98 (dd, 1H), 6.02 (s, 1H), 4.13-4.16 (m, 2H), 3.72-3.74 (m, 2H), 3.50-3.55 (q, 2H), 1.27 (s, 9H), 1.13-1.17 (t, 3H).
LC-MS (ESI): 463.2 (M+H)+.
Пример 79
Получение 1-(5-трет-бутил-2Н-пиразол-3-ил)-3-{4-[5-(2-гидрокси-этоксил)-бензимидазол-1-ил]-фенил] мочевины (Соединение 79)
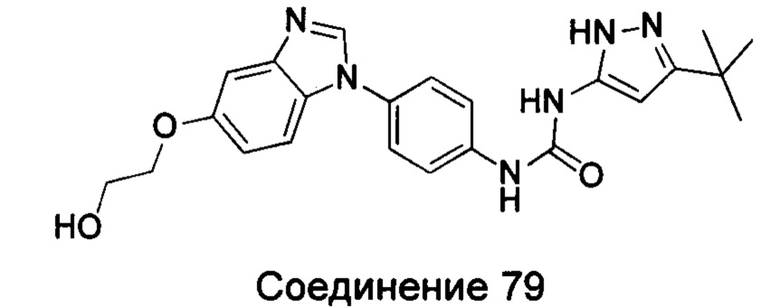
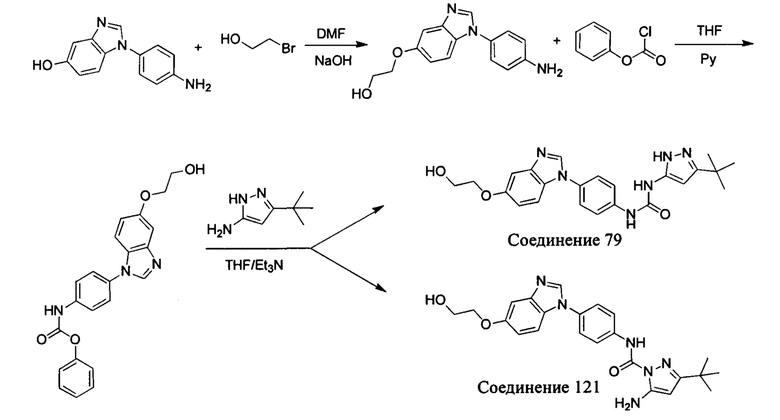
Методика была аналогична таковой, как на Стадиях 1-3 в примере 77, за исключением того, что бромэтанол использовали вместо 2-хлорэтилметилового эфира на Стадии 1 с получением 1-(5-трет-бутил-2Н-пиразол-3-ил)-3-{4-[5-(2-гидрокси-этоксил)-бензимидазол-1-ил]-фенил]-мочевины и Соединения 121 (см. Пример 121).
1HNMR (DMSO-d6, 400MHz) δ: 12.03 (s, 1H), 9.42 (s, 1H), 8.99 (s, 1H), 8.43 (s, 1H), 7.66-7.68 (d, 2H), 7.55-7.58 (d, 2H), 7.46-7.48 (d, 1H), 7.28-7.29 (d, 1H), 6.95-6.98 (dd, 1H), 6.02 (s, 1H), 4.90 (m, 1H), 4.03-4.06 (t, 2H), 3.75-3.76 (m, 2H), 1.27 (s, 9H).
LC-MS (ESI): 435.2 (M+H)+.
Пример 80
Получение
1-(5-трет-бутил-2Н-пиразол-3-ил)-3-{4-[5-(2-гидрокси-3-метоксил-пропоксил)-бензим идазол-1-ил]-фенил}-мочевины (Соединение 80)
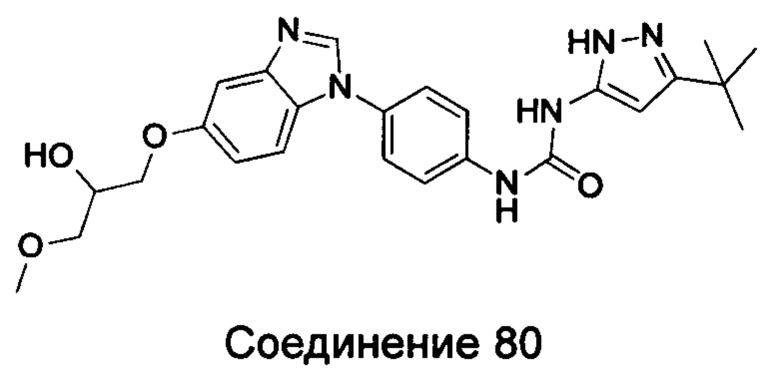
Способ получения был такой же, как на Стадиях 1-3 в примере 77, за исключением того, что 1-хлор-3-метоксил-2-пропанол (Darui) использовали вместо 2-хлорэтилметилового эфира на Стадии 1 с получением 1-(5-трет-бутил-2Н-пиразол-3-ил)-3-{4-[5-(2-гидрокси-3-метоксил-пропоксил)-бензимидазол-1-ил]-фенил}-мочевины.
1HNMR (DMSO-d6, 400MHz) δ: 12.03 (s, 1H), 9.44 (s, 1H), 9.00 (s, 1H), 8.46 (s, 1H), 7.67-7.69 (d, 2H), 7.55-7.58 (d, 2H), 7.46-7.48 (d, 1H), 7.28 (d, 1H), 6.96-6.99 (dd, 1H), 6.03 (s, 1H), 5.14 (s, 1H), 3.93-4.02 (m, 3H), 3.42-3.48 (m, 2H), 3.30 (s, 3Н), 1.27 (s, 9H).
LC-MS (ESI): 479.1 (M+H)+.
Пример 81
Получение
1-(5-трет-бутил-2Н-пиразол-3-ил)-3-{4-[5-(3-диметиламино-пропоксил)-бензоимидазол-1-ил]-фенил}-мочевины (Соединение 81)
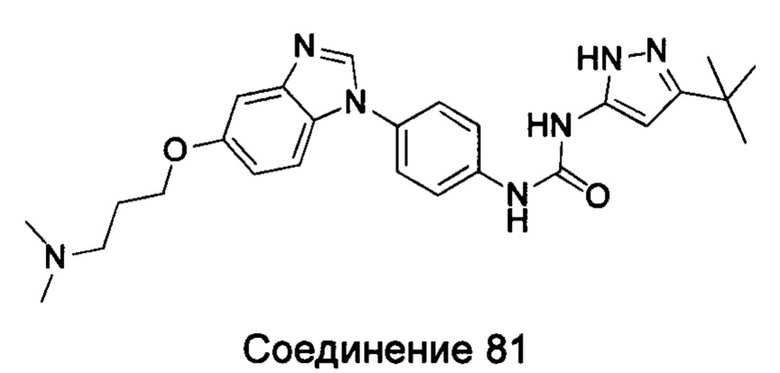
Способ получения был такой же, как на Стадиях 1-3 в примере 77, за исключением того, что N,N-диметиламинохлорпропана гидрохлорид (Titan) использовали вместо 2-хлорэтилметилового эфира на Стадии 1 с получением 1-(5-трет-бутил-2Н-пиразол-3-ил)-3-{4-[5-(3-диметиламино-пропоксил)-бензоимидазол-1-ил]-фенил}-мочевины.
1HNMR (DMSO-d6, 400MHz) δ: 12.04 (s, 1H), 9.59 (s, 1H), 9.09 (s, 1H), 8.42 (s, 1H), 7.66-7.69 (d, 2H), 7.54-7.57 (d, 2H), 7.45-7.47 (d, 1H), 7.27 (d, 1H), 6.94-6.96 (dd, 1H), 6.04 (s, 1H), 4.04-4.07 (t, 2H), 2.41-2.45 (t, 2H), 2.19 (s, 6H), 1.87-1.90 (m, 2H), 1.27 (s, 9H).
LC-MS (ESI): 476.2 (M+H)+.
Пример 82
Получение
1-(5-трет-бутил-2Н-пиразол-3-ил)-3-{4-[5-(3-дибутиламинопропокси)-бензимидазол-1-ил]-фенил}-мочевины (Соединение 82)
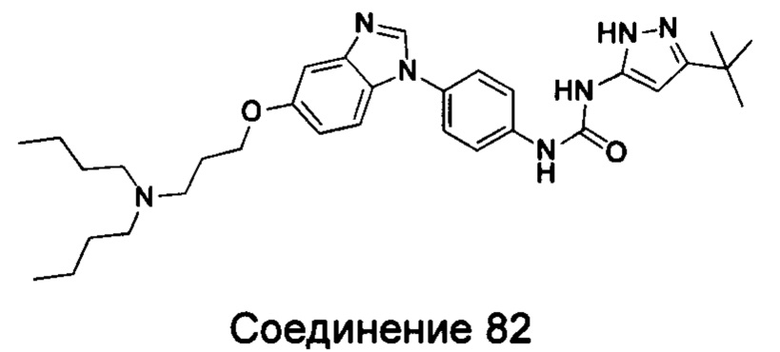
Способ получения был такой же, как на Стадиях 1-3 в примере 77, за исключением того, что N-(3-хлорпропил)-дибутиламин (Titan) использовали вместо 2-хлорэтилметилового эфира на Стадии 1 с получением 1-(5-трет-бутил-2Н-пиразол-3-ил)-3-{4-[5-(3-дибутиламинопропокси)-бензимидазол-1-ил]-фенил}-мочевины.
1HNMR (DMSO-d6, 400MHz) δ: 12.02 (s, 1H), 9.52 (s, 1H), 9.02 (s, 1H), 8.44 (s, 1H), 7.67-7.69 (d, 2H), 7.54-7.56 (d, 2H), 7.47-7.50 (d, 1H), 7.32 (d, 1H), 6.95-6.97 (dd, 1H), 6.03 (s, 1H), 4.11-4.14 (t, 2H), 3.12 (m, 2H), 2.95 (m, 4H), 2.10 (m, 2H), 1.58 (m, 4H), 1.29-1.35 (m, 4H), 1.27 (s, 9H), 0.88-1.92 (t, 6H).
LC-MS (ESI): 560.2 (M+H)+.
Пример 83
Получение
1-(5-трет-бутил-2Н-пиразол-3-ил)-3-[4-(5-цианометокси-бензимидазол-1-ил)-фенил]-мочевины (Соединение 83)
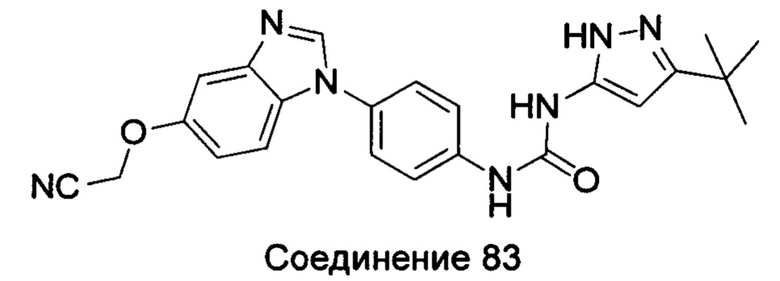
Способ получения был такой же, как на Стадиях 1-3 в примере 77, за исключением того, что хлорацетонитрил использовали вместо 2-хлорэтил метилового эфира на Стадии 1 с получением 1-(5-трет-бутил-2Н-пиразол-3-ил)-3-[4-(5-цианометокси-бензимидазол-1-ил)-фенил]-мочевины.
1HNMR (DMSO-d6, 400MHz) δ: 12.02 (s, 1H), 9.41 (s, 1H), 8.99 (s, 1H), 8.51 (s, 1H), 7.67-7.69 (d, 2H), 7.57-7.59 (d, 2H), 7.53-7.55 (d, 1H), 7.51-7.52 (d, 1H), 7.06-7.08 (dd, 1H), 6.02 (s, 1H), 5.24 (s, 2H), 1.27 (s, 9H).
LC-MS (ESI): 430.2 (M+H)+.
Пример 84 Получение 1-(5-трет-бутил-2Н-пиразол-3-ил)-3-[4-(5-трифторметокси-бензимидазол-1-ил)-фенил]-мочевины (Соединение 84)
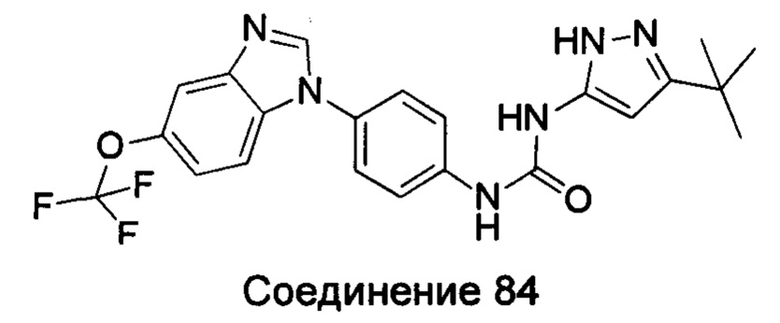
Способ получения был таким же, как в Примере 68, за исключением того, что 4-(5-трифторметокси-бензимидазол-1-ил)-анилин (синтезированный в примере 21) использовали вместо 4-бензимидазол-1-ил-анилина с получением 1-(5-трет-бутил-2Н-пиразол-3-ил)-3-[4-(5-трифторметокси-бензимидазол-1-ил)-фенил]-мочевины.
1HNMR (DMSO-d6, 400MHz) δ: 12.03 (s, 1H), 9.42 (s, 1H), 9.01 (s, 1H), 8.65 (s, 1H), 7.79 (d, 1H), 7.69-7.71 (d, 2H), 7.64-7.67 (d, 1H), 7.59-7.61 (d, 2H), 7.32-7.35 (dd, 30 1H), 6.03 (s, 1H), 1.27 (s, 9H).
LC-MS (ESI): 459.1 (M+H)+.
Пример 85
Получение
1-(5-трет-бутил-2Н-пиразол-3-ил)-3-{4-[5-(3-метил-оксетан-3-илметокси)-бензимидазол-1-ил]-фенил] мочевины (Соединение 85)
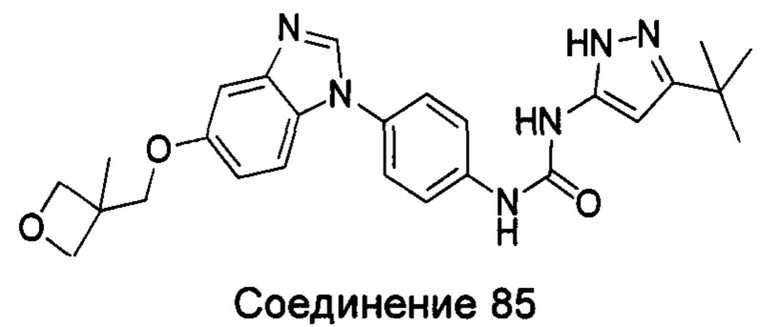
Способ получения был такой же, как на Стадиях 1-3 в примере 77, за исключением того, что 3-хлорметил-3-метилоксетан (Darui) использовали вместо 2-хлорэтилметилового эфира с получением 1-(5-трет-бутил-2Н-пиразол-3-ил)-3-{4-[5-(3-метил-оксетан-3-илметокси)-бензимида зол-1-ил]-фенил]-мочевины.
1HNMR (DMSO-d6, 400MHz) δ:12.02 (s, 1H), 9.41 (s, 1H), 8.99 (s, 1H), 8.44 (s, 1H), 7.67-7.69 (d, 2H), 7.55-7.58 (d, 2H), 7.47-7.49 (d, 1H), 7.35-7.36 (d, 1H), 6.98-10 7.01 (dd, 1H), 6.02 (s, 1H), 4.52-4.54 (d, 2H), 4.32-4.34 (d, 2H), 4.11 (s, 2H), 1.40 (s, 3H), 1.27 (s, 9H).
LC-MS (ESI): 475.2 (M+H)+.
Пример 86
Получение
1-(5-трет-бутил-2Н-пиразол-3-ил)-3-{4-[5-(тетрагидрофуран-2-илметокси)-бензимидазол-1-ил1-фенил}-мочевины (Соединение 86)
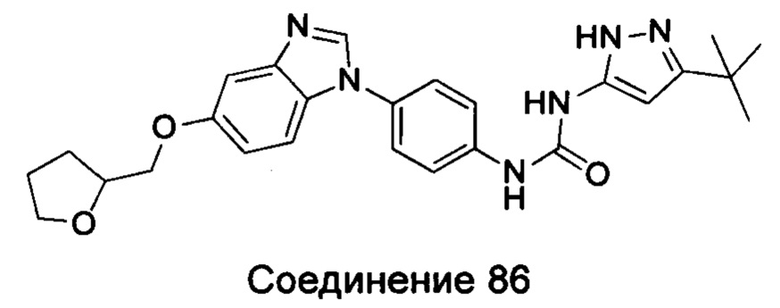
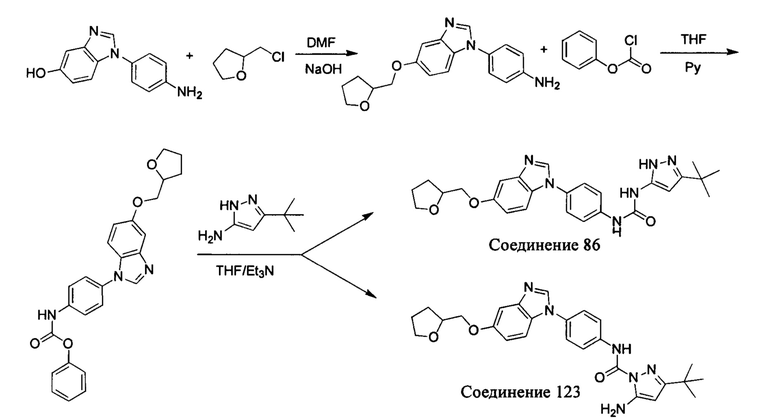
Способ получения был такой же, как на Стадиях 1-3 в примере 77, за исключением того, что 2-хлорметилтетрагидрофуран (Darui) использовали вместо 2-хлорэтилметилового эфира на Стадии 1 с получением 1-(5-трет-бутил-2Н-пиразол-3-ил)-3-{4-[5-(тетрагидрофуран-2-илметокси)-бензимидазол-1-ил]-фенил}-мочевины, и Соединения 123 (см. Пример 123).
1HNMR (DMSO-d6, 400MHz) δ: 12.02 (s, 1H), 9.41 (s, 1H), 8.99 (s, 1H), 8.43 (s, 1H), 7.66-7.69 (d, 2H), 7.55-7.57 (d, 2H), 7.45-7.47 (d, 1H), 7.29-7.30 (d. 1H), 6.94-6.97 (dd, 1H), 6.02 (s, 1H), 4.16-4.22 (m, 1H), 3.95-4.04 (m, 2H), 3.78-3.84 (m, 1H), 3.67-3.72 (m, 1H), 1.98-2.07 (m, 1H), 1.81-1.95 (m, 2H), 1.66-1.75 (m, 1H), 1.27 (s, 5 9H)
LC-MS (ESI): 475.2 (M+H)+.
Пример 87
Получение
1-(5-трет-бутил-2Н-пиразол-3-ил)-3-{4-[5-(тетрагидро-пиран-2-илметокси)-бензимидазол-1-ил]-фенил}-мочевины (Соединение 87)
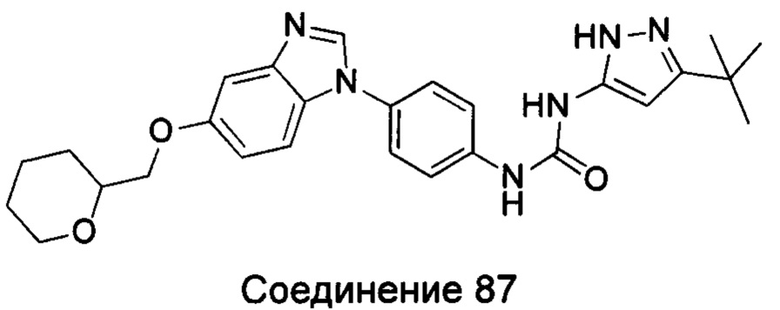
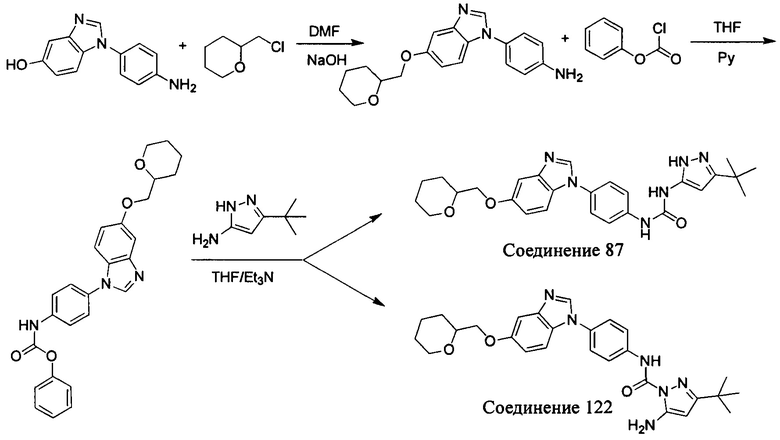
Способ получения был такой же, как на Стадиях 1-3 в примере 77, за исключением того, что 2-(хлорметил)тетрагидропиран (Darui) использовали вместо 2-хлорэтилметилового эфира на Стадии 1 с получением 1-(5-трет-бутил-2Н-пиразол-3-ил)-3-{4-[5-(тетрагидропиран-2-илметокси)-бензимидазол-1-ил]-фенил}-мочевины и Соединения 122 (см. Пример 122).
1HNMR (DMSO-d6, 400MHz) δ: 12.03 (s, 1Н), 9.42 (s, 1Н), 8.99 (s, 1Н), 8.42 (s, 20 1Н), 7.66-7.68 (d, 2Н), 7.55-7.57 (d, 2Н), 7.45-7.47 (d, 1Н), 7.27-7.28 (d, 1Н), 6.94-6.97 (dd, 1Н), 6.02 (s, 1Н), 3.89-3.97 (m, 3Н), 3.63-3.69 (m, 1Н), 3.47-3.41 (m, 1H), 1.82-1.84 (m, 1H), 1.67-1.70 (m, 1H), 1.46-1.54 (m, 4H), 1.27 (s, 9H).
LC-MS (ESI): 489.2 (M+H)+.
Пример 88
Получение этил (1-{4-[3-(5-трет-бутил-2Н-пиразол-3-ил)-карбамидо]-фенил}-1Н-бензимидазол-5-илокси)-ацетата (Соединение 88)
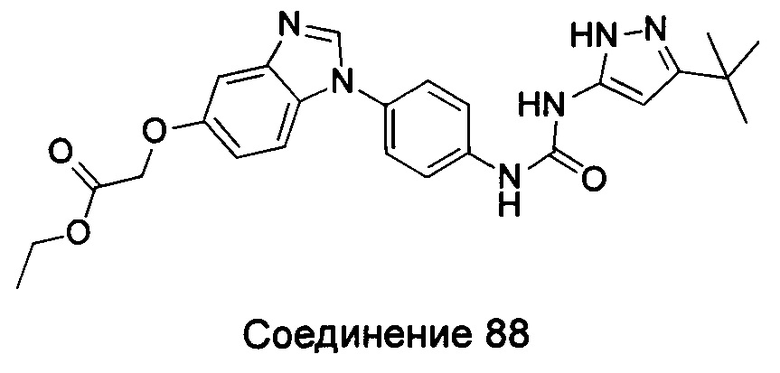
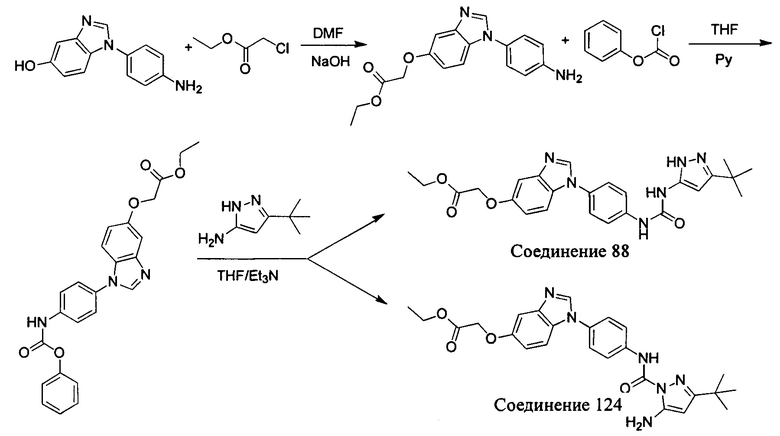
Способ получения был такой же, как на Стадиях 1 - 3 в примере 77, за исключением того, что этилхлорацетат (Darui) использовали вместо 2-хлорэтилметилового эфира на Стадии 1 с получением (1-{4-[3-(5-трет-бутил-2Н-пиразол-3-ил)-карбамидо]-фенил}-1Н-бензимидазол-5-илокси)-этилацетата и Соединения 124 (см. Пример 124).
1HNMR (DMSO-d6, 400MHz) δ:12.02 (s, 1H), 9.41 (s, 1H), 8.99 (s, 1H), 8.44 (s, 1H), 7.66-7.68 (d, 2H), 7.55-7.57 (d, 2H), 7.47-7.49 (d, 1H), 7.26-7.27 (d, 1H), 6.98-10 7.01 (dd, 1H), 6.02 (s, 1H), 4.84 (s, 2H), 4.16-4.21 (q, 2H), 1.27 (s, 9H), 1.21-1.25 (t, 3Н).
LC-MS (ESI): 477.2 (M+H)+.
Пример 89
Получение 1-(5-трет-бутил-2Н-пиразол-3-ил)-3-{4-[5-(2-морфолин-4-ил-этоксил)-бензимидазол-1-ил]-фенил}-мочевины (Соединение 89)
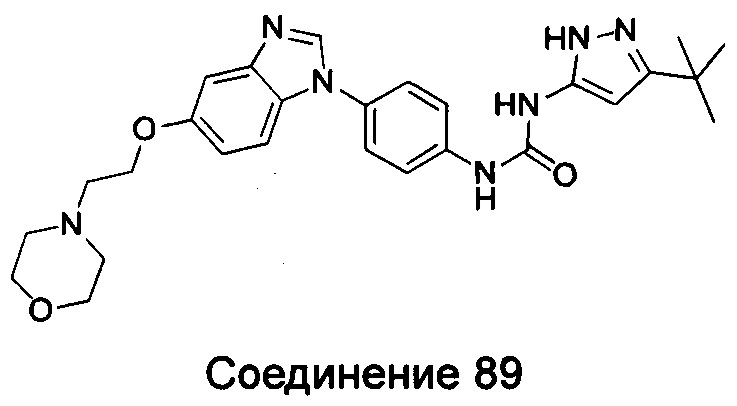
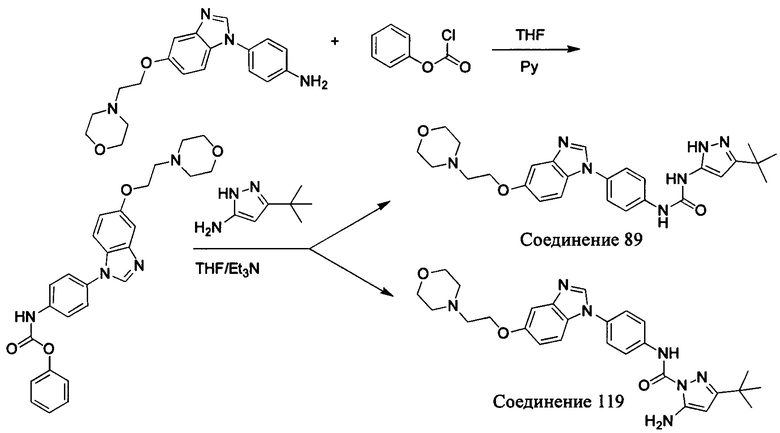
Стадия 1: Получение фенил {4-[5-(3-морфолин-4-ил-этоксил)-бензимидазол-1-ил]-фенил}-карбамата
4-[5-(3-морфолин-4-ил-этоксил)-бензимидазол-1-ил]-анилин (синтезированный на Стадии 1 примера 16) (300 мг, 0.887 ммоль) и пиридин (210 мг, 2.66 ммоль) растворили в ТГФ (20 мл), и фенилкарбамат (277.6 мг, 1.74 ммоль) добавили по каплям в ванне со льдом, во время чего преципитировался осадок. После добавления, реакционную смесь нагревали до комнатной температуры и затем перемешивали в течение 2 часов. Реакционную смесь погасили 50 мл воды, и экстрагировали этил ацетатом (30 мл × 2). Органическую фазу промыли с помощью насыщенного раствора NaCl, высушили над безводным сульфатом натрия, отфильтровали и фильтрат сконцентрировали при пониженном давлении. Остатки очистили с помощью колоночной хроматографии (элюент: дихлорметан/метанол) с получением 280 мг фенил {4-[5-(3-морфолин-4-ил-этоксил)-бензимидазол-1-ил]-фенил}-карбамата. L-MS (ESI): 459.2 (М+Н)+.
Стадия 2: Получение 1-(5-трет-бутил-2Н-пиразол-3-ил)-3-{4-[5-(2-морфолин-4-ил-этоксил)-бензимидазол-1-ил]-фенил}-мочевины
Способ получения был такой же, как на Стадии 2 в примере 68 за исключением того, что фенил 4-[5-(3-морфолин-4-ил-этоксил)-бензимидазол-1-ил]-фенил}-карбамат использовали вместо фенил (4-бензимидазол-1-ил-фенил)-карбамата с получением 1-(5-трет-бутил-2Н-пиразол-3-ил)-3-{4-[5-(2-морфолин-4-ил-этоксил)-бензимидазол-1-ил]-фенил}-мочевины и Соединения 119 (см. Пример 119).
1HNMR (DMSO-d6, 400MHz) δ: 12.03 (s, 1Н), 9.41 (s, 1Н), 8.99 (s, 1Н), 8.43 (s, 1Н), 7.66-7.68 (d, 2H), 7.55-7.57 (d, 2H), 7.45-7.47 (d, 1H), 7.31-7.32 (d, 1H), 6.94-6.97 (dd, 1H), 6.02 (s, 1H), 4.14-4.17 (m, 2H), 3.60 (m, 4H), 2.73 (m, 2H), 2.51 (m, 4H), 20 1.27 (s, 9H).
LC-MS (ESI): 504.1 (M+H)+.
Пример 90 Получение
1-(5-трет-бутил-2Н-пиразол-3-ил)-3-{4-[5-(2-пиперидин-1-ил-этоксил)-бензимидазол-1-ил]-фенил}-мочевины (Соединение 90)
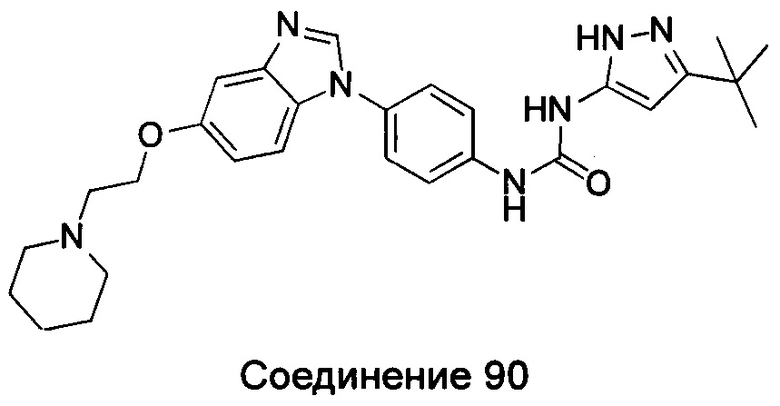
Способ получения был такой же, как на Стадиях 1-3 в примере 77, за исключением того, что 1-(2-хлорэтил)пиперидина гидрохлорид использовали вместо 2-хлорэтилметилового эфира на Стадии 1 с получением 1-(5-трет-бутил-2Н-пиразол-3-ил)-3-{4-[5-(2-пиперидин-1-ил-этоксил)-бензимидазол-1-ил]-фенил}-мочевины.
1HNMR (DMSO-d6, 400MHz) δ: 12.02 (s, 1Н), 9.41 (s, 1Н), 8.99 (s, 1Н), 8.42 (s, 1Н), 7.66-7.68 (d, 2Н), 7.53-7.57 (d, 2Н), 7.45-7.47 (d, 1Н), 7.30 (d, 1H), 6.94-6.96 (dd, 1H), 6.02 (s, 1H), 4.12-4.14 (t, 2H), 2.70 (m, 2H), 2.47 (m, 4H), 1.49-1.54 (m, 4H), 1.39-1.42 (m, 2H), 1.27 (s, 9H).
LC-MS (ESI): 502.2 (M+H)+.
Пример 91
Получение 1-{4-[5-(2-азепан-1-ил-этокси)-бензимидазол-1-ил]-фенил}-3-(5-трет-бутил-2Н-пиразол-3-ил)-мочевины(Соединение 91)
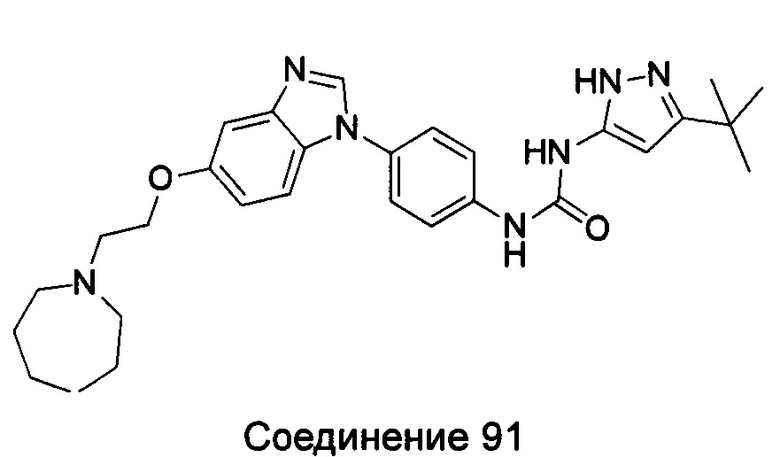
Способ получения был такой же, как на Стадиях 1 - 3 в примере 77, за исключением того, что 2-(азепанил)этилхлорид (Runjie) использовали вместо 2-хлорэтилметилового эфира на Стадии 1 с получением 1-{4-[5-(2-азепан-1-ил-этокси)-бензимидазол-1-ил]-фенил}3-(5-трет-бутил-2Н-пиразол-3-ил)-мочевины.
1HNMR (DMSO-d6, 400MHz) δ: 12.03 (s, 1Н), 9.44 (s, 1Н), 9.00 (s, 1Н), 8.43 (s, 20 1Н), 7.66-7.68 (d, 2Н), 7.55-7.57 (d, 2H), 7.45-7.47 (d, 1H), 7.30-7.31 (d, 1H), 6.94-6.97 (dd, 1H), 6.02 (s, 1H), 4.12-4.15 (t, 2H), 2.97 (m, 2H), 2.79 (m, 4H), 1.63 (m, 4H), 1.55 (m, 4H), 1.27 (s, 9H).
LC-MS (ESI): 516.3 (M+H)+.
Пример 92
Получение 1-(5-трет-бутил-2Н-пиразол-3-ил)-3-(4-{5-[3-(4-метил-пиперазин-1-ил)-пропоксил]-бензимидазол-1-ил}-фенил)-мочевины (Соединение 92)
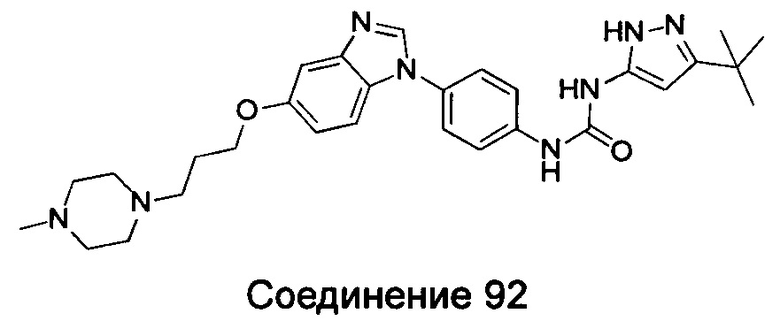
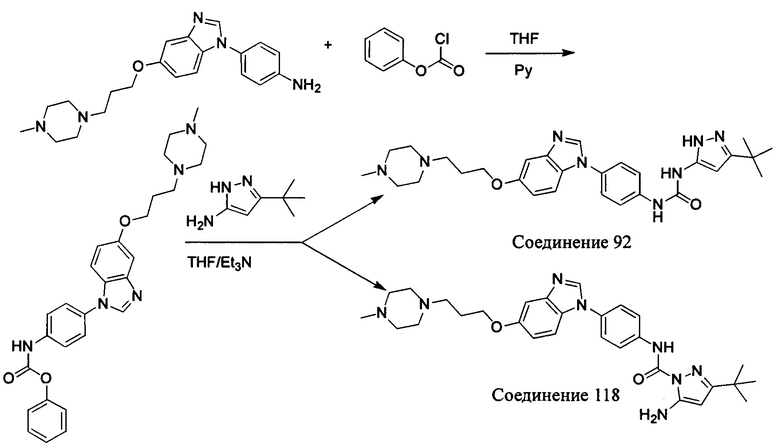
Способ получения был такой же, как на Стадиях 1-3 в примере 77, за исключением того, что 1-(3-хлорпропил)-4-метилпиперазин (Runjie) использовали вместо 2-хлорэтилметилового эфира на Стадии 1 с получением 1-(5-трет-бутил-2Н-пиразол-3-ил)-3-(4-{5-[3-(4-метил-пиперазин-1-ил)-пропоксил]-бензимидазол-1-ил}-фенил)-мочевины и Соединения 118 (см. Пример 118).
1HNMR (DMSO-d6, 400MHz) δ: 12.03 (s, 1Н), 9.50 (s, 1Н), 9.03 (s, 1Н), 8.42 (s, 1Н), 7.67-7.69 (d, 2Н), 7.54-7.56 (d, 2Н), 7.44-7.46 (d, 1Н), 7.27 (d, 1Н), 6.92-6.95 (dd, 1Н), 6.03 (s, 1Н), 4.03-4.06 (t, 2Н), 2.25-2.46 (m, 10Н), 2.17 (s, 3Н), 1.85-1.92 (m, 2Н), 1.27 (s, 9Н).
LC-MS (ESI): 531.3 (M+H)+.
Пример 93
Получение 1-(5-трет-бутил-2Н-пиразол-3-ил)-3-{4-[5-(3-фтор-бензилокси)-бензимидазол-1-ил]-фенил]-мочевины (Соединение 93)
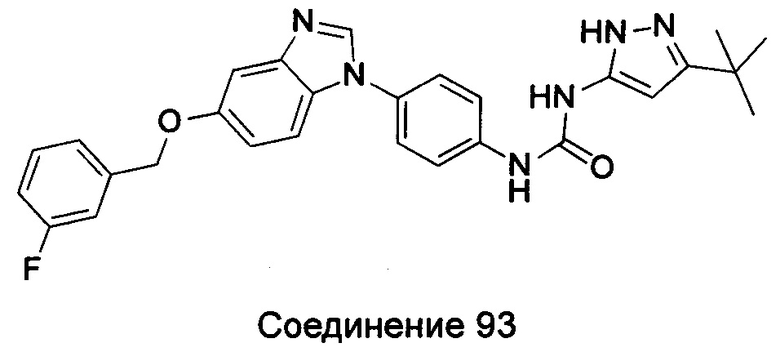
Способ получения был такой же, как на Стадиях 1-3 в примере 77, за исключением того, что 3-фторобензилбромид (Darui) использовали вместо 2-фторэтилметилового эфира на Стадии 1 с получением 1-(5-трет-бутил-2Н-пиразол-3-ил)-3-{4-[5-(3-фтор-бензилокси)-бензимидазол-1-ил]-фенил]-мочевины.
1HNMR (DMSO-d6, 400MHz) δ: 12.02 (s, 1H), 9.50 (s, 1H), 9.02 (s, 1H), 8.47 (s, 1H), 7.67-7.69 (d, 2H), 7.55-7.57 (d, 2H), 7.42-7.50 (m, 2H), 7.31-7.38 (m, 3Н), 7.14-7.18 (m, 1H), 7.05-7.07 (m, 1H), 6.04 (s, 1H), 5.22 (s, 2H), 1.27 (s, 9H).
LC-MS (ESI): 499.2 (M+H)+.
Пример 94
Получение 1-(5-трет-бутил-2Н-пиразол-3-ил)-3-(4-{5-[4-(1-циклогексил-1Н-тетразол-5-ил)-бутокси]-бензимидазол-1-ил}-фенил)-мочевины (Соединение 94)
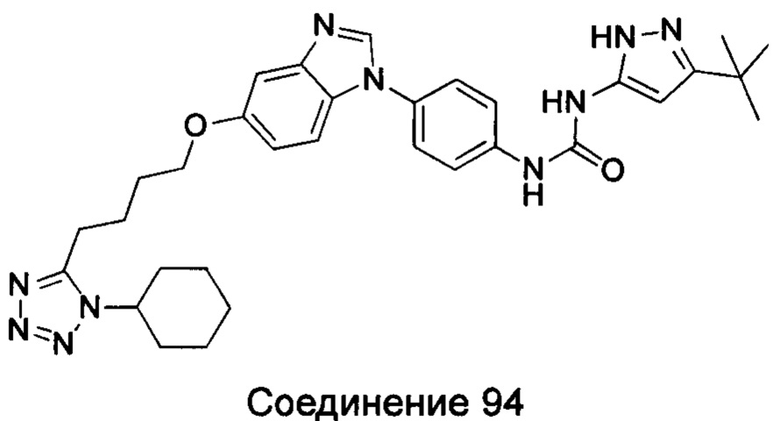
Способ получения был такой же, как на Стадиях 1-3 в примере 77, за исключением того, что 1-циклогексил-5-(4-хлорбутил)-тетразол (Runjie) использовали вместо 2-хлорэтилметилового эфира с получением 1-(5-трет-бутил-2Н-пиразол-3-ил)-3-(4-{5-[4-(1-циклогексил-1Н-тетразол-5-ил)-бутокси]-бензимидазол-1-ил}-фенил)-мочевины.
1HNMR (DMSO-d6, 400MHz) δ:12.03 (s, 1H), 9.41 (s, 1H), 8.99 (s, 1H), 8.42 (s, 10 1H), 7.66-7.68 (d, 2H), 7.55-7.57 (d, 2H), 7.45-7.47 (d, 1H), 7.30-7.31 (d, 1H), 6.93-6.96 (dd, 1H), 6.02 (s, 1H), 4.38-4.45 (m, 1H), 4.08-4.11 (t, 2H), 2.99-3.02 (t, 2H), 1.66-1.98 (m, 12H), 1.39-1.48 (m, 2H), 1.27 (s, 9H).
LC-MS (ESI): 597.3 (M+H)+.
Пример 95
Получение 1-(5-трет-бутил-2Н-пиразол-3-ил)-3-{4-[5-(4-морфолин-4-ил-[1,2,5]тиадиазол-3-илокси)-бензимидазол-1-ил]-фенил}-мочевины (Соединение 95)
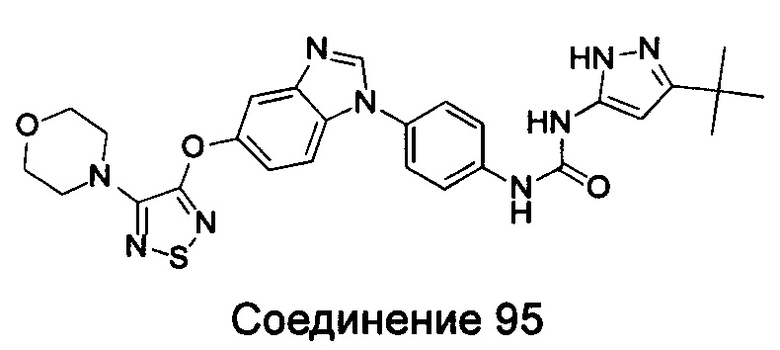
Способ получения был такой же, как на Стадиях 1-3 в примере 77, за исключением того, что 3-хлор-4-морфолинил-1,2,5-тиадиазол (Runjie) использовали вместо 2-хлорэтилметилового эфира на Стадии 1 с получением 1-(5-трет-бутил-2Н-пиразол-3-ил)-3-{4-[5-(4-морфолин-4-ил-[1,2,5]тиадиазол-3-илокси)-бензимидазол-1-ил]-фенил}-мочевины.
1HNMR (DMSO-d6, 400MHz) δ:12.03 (s, 1H), 9.43 (s, 1H), 9.00 (s, 1H), 8.58 (s, 1H), 7.75-7.76 (d, 1H), 7.69-7.71 (d, 2H), 7.60-7.63 (m, 3Н), 7.29-7.32 (dd, 1H), 6.03 25 (s, 1H), 3.76-3.78 (t, 4H), 3.55-3.57 (t, 4H), 1.27 (s, 9H).
LC-MS (ESI): 560.1 (M+H)+.
Пример 96
Получение
4-(l-{4-[3-(5-трет-бутил-2Н-пиразол-3-ил)-карбамидо]-фенил}-1Н-бензимидазол-5-илокси)-пиридин-2-карбоксиметиламин (Соединение 96)
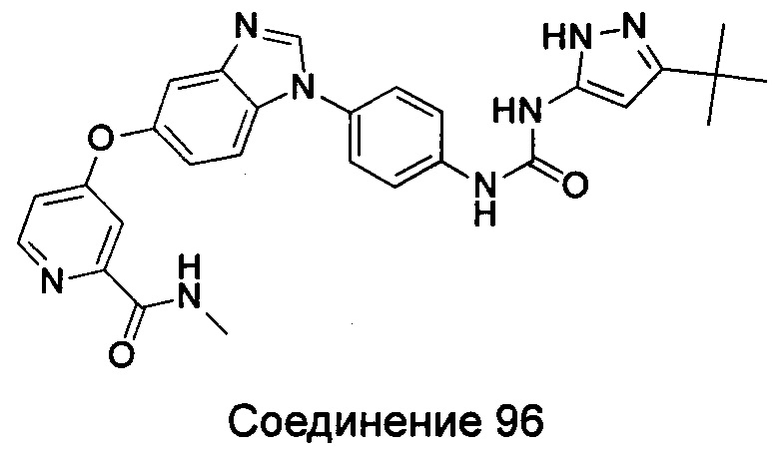
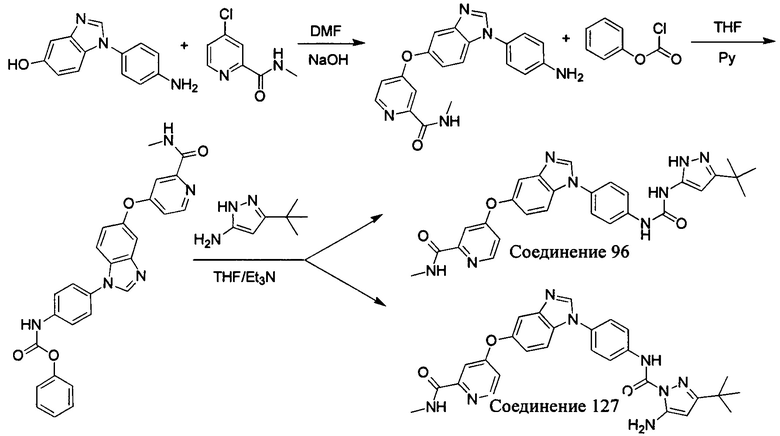
Способ получения был такой же, как на Стадиях 1 - 3 в примере 77, за исключением того, что N-метил-4-хлор-2-пиридинкарбоксамид (Titan) использовали вместо 2-хлорэтилметилового эфира на Стадии 1 с получением 4-(l-{4-[3-(5-трет-бутил-2Н-пиразол-3-ил)-карбамидо]-фенил}-1Н-бензимидазол-5-илокси)-пиридин-2-карбоксиметиламина и Соединения 127 (см. Пример 127).
1HNMR (DMSO-d6, 400MHz) δ: 12.03 (s, 1Н), 9.44 (s, 1Н), 9.01 (s, 1Н), 8.77-10 8.80 (m, 1Н), 8.62 (s, 1Н), 8.51-8.52 (d, 1Н), 763-7.72 (m, 6Н), 7.39-7.40 (d, 1Н), 7.18-7.21 (d, 2Н), 6.03 (s, 1Н), 2.77-2.79 (d, 3Н), 1.27 (s, 9Н).
LC-MS (ESI): 525.2 (M+H)+.
Пример 97
Получение 1-(5-трет-бутил-2Н-пиразол-3-ил)-3-[4-(5-фтор-7-метил-бензимидазол-1-ил)-фенил]-мочевины (Соединение 97)
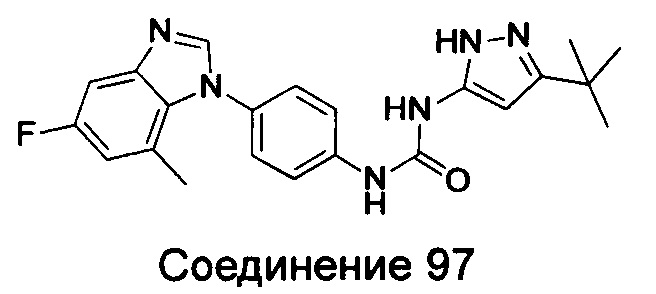
Способ получения был таким же, как в Примере 68, за исключением того, что 4-(5-фтор-7-метил-бензимидазол-1-ил)-анилина (синтезированный на Стадии 1 примера 33) использовали вместо 4-бензимидазол-1-ил-анилина на Стадии 1 с получением 1-(5-трет-бутил-2Н-пиразол-3-ил)-3-[4-(5-фтор-7-метил-бензимидазол-1-ил)-фенил]-мочевины.
1HNMR (DMSO-d6, 400MHz) δ: 12.03 (s, 1H), 9.42 (s, 1H), 9.02 (s, 1H), 8.27 (s, 1H), 7.61-7.64 (d, 2H), 7.45-7.48 (d, 2H), 7.36-7.39 (dd, 1H), 6.93-6.96 (dd, 1H), 6.03 (s, 1H), 2.04 (s, 3H), 1.27 (s, 9H).
LC-MS (ESI): 407.1 (M+H)+.
Пример 98
Получение 1-(5-трет-бутил-2Н-пиразол-3-ил)-3-{4-[6-(2-метоксил-этоксил)-бензимидазол-1-ил]-фенил}-мочевины (Соединение 98)
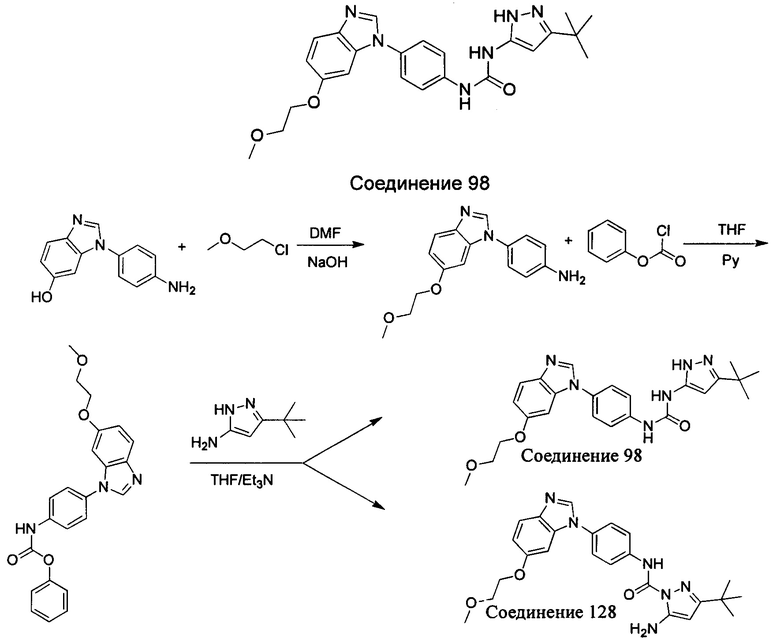
Стадия 1: Получение 4-[6-(2-метоксил-этоксил)-бензимидазол-1-ил]-анилина
Способ получения был такой же, как на Стадиях 1-5 в примере 27, за исключением того, что 2-хлорэтилметиловый эфир использовали вместо 4-(2-хлор-этил)-морфолина гидрохлорида на Стадии 5 с получением 4-[6-(2-метоксил-этоксил)-бензимидазол-1-ил]-анилина.
Стадия 2: Получение 1-(5-трет-бутил-2Н-пиразол-3-ил)-3-{4-[6-(2-метоксил-этоксил)-бензимидазол-1-ил]-фенил}-мочевины
Способ получения был таким же, как в Примере 68, за исключением того, что 4-[6-(2-метоксил-этоксил)-бензимидазол-1-ил]-анилин использовали вместо 4-бензимидазол-1-ил-анилина на Стадии 1, с получением 1-(5-трет-бутил-2Н-пиразол-3-ил)-3-{4-[6-(2-метоксил-этоксил)-бензимидазол-1-ил]-фенил}-мочевины и Соединения 128 (см. Пример 128).
1HNMR (DMSO-d6, 400MHz) δ: 12.03 (s, 1Н), 9.40 (s, 1H), 9.00 (s, 1H), 8.35 (s, 25 1H), 7.67-7.69 (d, 2H,), 7.63-7.65 (d, 1H), 7.56-7.59 (d, 2H), 7.03-7.04 (d, 1H), 6.91-6.94 (dd, 1H), 6.02 (s, 1H), 4.11-4.13 (m, 2H), 3.65-3.68 (m, 2H), 3.31 (s, 3Н), 1.27 (s, 9H).
LC-MS (ESI): ESI 449.1 (M+H)+.
Пример 99
Получение 1-(5-трет-бутил-2Н-пиразол-3-ил)-3-[4-(5,6-диметоксил-бензимидазол-1-ил)-фенил]-мочевины (Соединение 99)
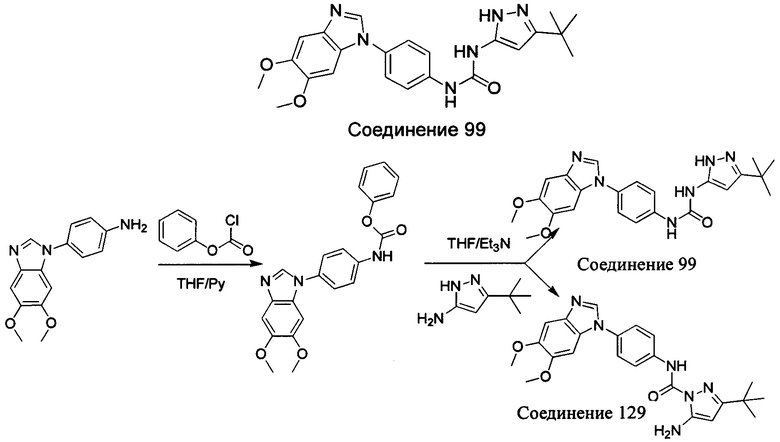
Способ получения был таким же, как в Примере 68, за исключением того, что 4-(5,6-диметоксил-бензимидазол-1-ил)-анилин (синтезированный на Стадии 1 примера 31) использовали вместо 4-бензимидазол-1-ил-анилина на Стадии 1 с получением 1-(5-трет-бутил-2Н-пиразол-3-ил)-3-[4-(5,6-диметоксил-бензимидазол-1-ил)-фенил]-мочевины и Соединения 129 (см. Пример 129).
1HNMR (DMSO-d6, 400MHz) δ: 12.02 (s, 1Н), 9.40 (s, 1Н), 9.00 (s, 1Н,), 8.27 (s, 1Н), 7.67-7.69 (d, 2Н), 7.56-7.58 (d, 2Н), 7.31 (s, 1Н), 7.04 (s, 1Н), 6.02 (s, 1Н), 3.82 (s, 3Н), 3.79 (s, 3Н), 1.27 (s, 9Н).
LC-MS (ESI): ESI 435.1 (M+H)+
Пример 100
Получение 1-(4-бензимидазол-1-ил-фенил)-3-(5-трет-бутил-2-метил-2Н-пиразол-3-ил)-мочевины (Соединение 100)
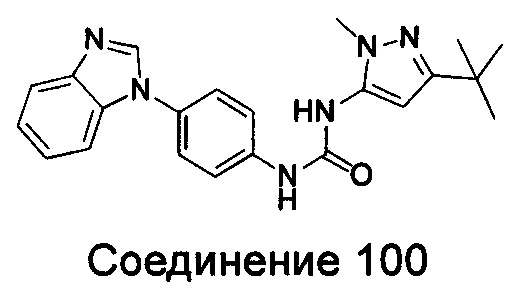
Стадия 1: Получение фенил (5-трет-бутил-2-метил-2Н-пиразол-3-ил)-карбамата (активный сложный эфир)
Методика была аналогична таковой, как на Стадии 3 в примере 1, за исключением того, что 5-амино-3-трет-бутил-1-метилпиразол (Darui) использовали вместо 3-аминоизотиазола на Стадии 3 с получением фенил (5-трет-бутил-2-метил-2Н-пиразол-3-ил)-карбамата.
Стадия 2: Получение 1-(4-бензимидазол-1-ил-фенил)-3-(5-трет-бутил-2-метил-2Н-пиразол-3-ил)-мочевины
Методика была аналогична таковой, как на Стадии 4 в примере 1, за исключением того, что фенил (5-трет-бутил-2-метил-2Н-пиразол-3-ил)-карбамат (активный сложный эфир) использовали вместо фенил изоксазол-3-ил-карбамата (активный сложный эфир) с получением 1-(4-бензимидазол-1-ил-фенил)-3-(5-трет-бутил-2-метил-2Н-пиразол-3-ил)-мочевин ы.
1HNMR (DMSO-d6, 400MHz) δ: 9.17 (s, IH), 8.58 (s, IH), 8.51 (s, IH), 7.77-7.79 (m, 1H), 7.69-7.72 (d, 2H), 7.57-7.60 (m, 3H), 7.29-7.36 (m, 2H), 6.08 (s, 1H), 10 3.63 (s, 3H), 1.23 (s, 9H).
LC-MS (ESI): 389.2 (M+H)+.
Пример 101
Получение 1-(5-трет-бутил-2-метил-2Н-пиразол-3-ил)-3-{4-[5-(2-метоксил-этоксил)-бензимидазол-1-ил]-фенил}мочевины (Соединение 101)
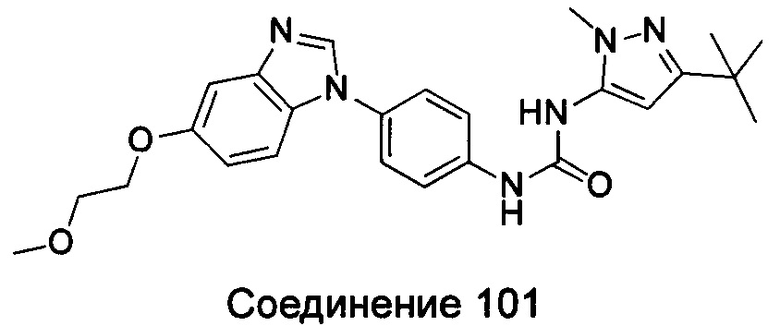
Стадия 1: 1-(5-трет-бутил-2-метил-2Н-пиразол-3-ил)-3-{4-[5-(2-метоксил-этоксил)-бензимидазол-1-ил]-фенил]мочевины
4-[5-(2-метоксил-этоксил)-бензимидазол-1-ил]-анилин (как получено на Стадии 1 примера 77) (107 мг, 0.378 ммоль), фенил (5-трет-бутил-2-метил-2Н-пиразол-3-ил)-карбамат (как получено на Стадии 1 примера 100) (156.0 мг, 0.571 ммоль) и триэтиламин (115 мг, 1.14 ммоль) растворили d 10 мл ТГФ и реакционный раствор кипятили с обратным холодильником в течение ночи. На следующий день реакционный раствор сконцентрировали при пониженном давлении, и остатки очистили с помощью колоночной хроматографии (элюент: дихлорметан/метанол) с получением 56 мг 1-(5-трет-бутил-2-метил-2Н-пиразол-3-ил)-3-{4-[5-(2-метоксил-этоксил)-бензимидазол-1-ил]-фенил]-мочевины в виде белого осадка.
1HNMR (DMSO-d6, 400MHz) δ: 9.16 (s, IH), 8.57 (s, IH), 8.43 (s, IH), 7.67-7.69 (d, 2H,), 7.56-7.58 (d, 2H), 7.45-7.47 (d, 1H), 7.29-7.30 (d, 1H), 6.94-6.97 (dd, 1H), 6.08 (s, 1H), 4.14-4.16 (t, 2H), 3.68-3.70 (t, 2H), 3.62 (s, 3H), 3.33 (s, 3H), 1.22 (s, 9H).
LC-MS: ESI 463.3 (M+H)+.
Пример 102
Получение 1-(5-трет-бутил-2-метил-2Н-пиразол-3-ил)-3-{4-[5-(2-морфолин-4-ил-метоксил)-бензимидазол-1-ил]фенил}-мочевины (Соединение 102)
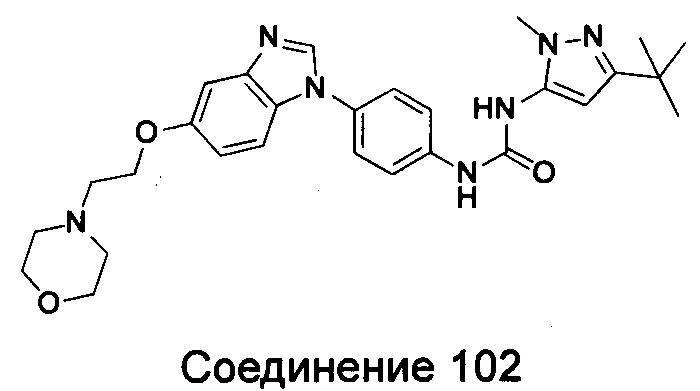
Стадия 1: Получение 1-(5-трет-бутил-2-метил-2Н-пиразол-3-ил)-3-{4-[5-(2-морфолин-4-ил-метоксил)-бензимидазол-1-ил]фенил}-мочевины
4-[5-(2-морфолин-4-ил-метоксил)-бензимидазол-1-ил]-анилин (полученный на Стадии 1 примера 16) (125 мг, 0.368 ммоль), фенил (5-трет-бутил-2-метил-2Н-пиразол-3-ил)-карбамат (полученный на Стадии 1 примера 100) (152.0 мг, 0.555 ммоль) и триэтиламин (112 мг, 1.11 ммоль) растворили в 10 мл ТГФ и кипятили с обратным холодильником в течение ночи. На следующий день реакционный раствор сконцентрировали при пониженном давлении, и остатки очистили с помощью колоночной хроматографии (элюент: дихлорметан/метанол) с получением 60 мг 1-(5-трет-бутил-2-метил-2Н-пиразол-3-ил)-3-{4-[5-(2-морфолин-4-ил-метоксил)-бензимидазол-1-ил]-фенил}-мочевины в виде белого осадка.
1HNMR (DMSO-d6, 400MHz) δ: 9.17 (s, 1Н), 8.57 (s, 1Н), 8.43 (s, 1Н), 7.67-7.70 (d, 2Н,), 7.55-7.57 (d, 2Н), 7.45-7.47 (d, 1Н), 7.30-7.31 (d, 1Н), 6.94-6.96 (dd, 1Н), 6.08 (s, 1Н), 4.13-4.16 (t, 2Н), 3.62 (s, 3Н), 3.58-3.60 (t, 4Н), 2.71-2.74 (t, 2H), 2.49-2.51 (m, 4H), 1.22 (s, 9H).
LC-MS: ESI 518.2 (M+H)+.
Пример 103
Получение 1-(4-бензимидазол-1-ил-фенил)-3-[5-трет-бутил-2-(2-гидрокси-этил)-2Н-пиразол-3-ил]-мочевины (Соединение 103)
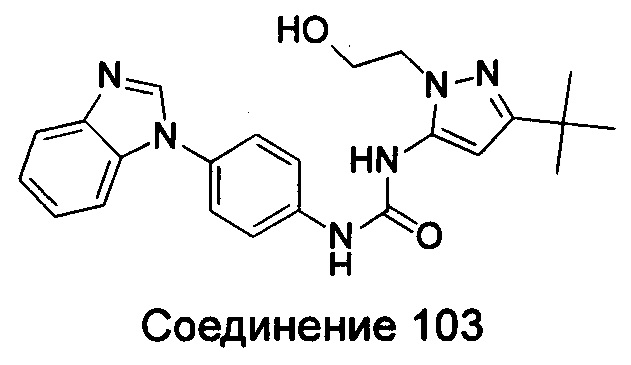
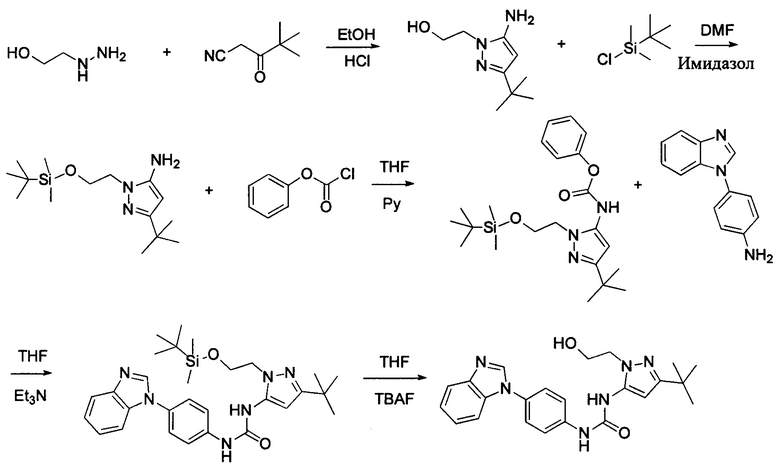
Стадия 1: Получение 2-(5-амино-3-трет-бутил-пиразол-1-ил)-этанола
2-Гидразинэтанол (6.69 г, 0.088 моль, Titan), пивалоилацетонитрил (10 г, 0.08 моль, Darui), и концентрированную соляную кислоту (0.2 мл) кипятили с обратным холодильником в 80 мл этанола в течение 5 часов. Реакционный раствор сконцентрировали при пониженном давлении, и получившееся масло оставили отстаиваться в течение ночи. На следующий день преципитировался осадок. Получившийся осадок суспендировали с метил трет-бутиловым эфиром с получением 12 г 2-(5-амино-3-трет-бутил-пиразол-1-ил)-этанола в виде желто-белого осадка.
Стадия 2: Получение 5-трет-бутил-2-[2-(трет-бутил-диметил-силокси)-этил]-2Н-пиразол-3-иламина
2-(5-амино-3-трет-бутил-пиразол-1-ил)-этанол (6.6 г, 0.0361 моль), полученный на Стадии 1, TBDMSCI (6.49 г, 0.0433 ммоль), и имидазол (6.13 г, 0.09 моль) растворили в 100 мл ДМФ и дали прореагировать в течение ночи при комнатной температуре в атмосфере азота. На следующий день реакционный раствор влили в воду и экстрагировали этилацетатом (80 мл × 2). Органическую фазу промыли дважды с помощью насыщенного раствора NaCl, высушили над безводным сульфатом натрия, отфильтровали, и фильтрат сконцентрировали при пониженном давлении с получением 6 г неочищенного продукта, который сразу использовали на следующей стадии без очистки.
Стадия 3: Получение фенил {5-трет-бутил-2-[2-(трет-бутил-диметил-силокси)-этил]-2Н-пиразол-3-ил}-карбамата
Способ получения был такой же, как на Стадии 3 в примере 1, за исключением того, что 5-трет-бутил-2-[2-(трет-бутил-диметил-силокси)-этил]-2Н-пиразол-3-иламин использовали вместо 3-аминоизоксазола на Стадии 3 с получением фенил {5-трет-бутил-2-[2-(трет-бутил-диметил-силокси)-этил]-2Н-пиразол-3-ил}-карбамата.
Стадия 4: Получение 1-(4-бензимидазол-1-ил-фенил)-3-(5-трет-бутил-диметил-силокси)-этил]-2Н-пиразол-3-ил}-мочевины
Способ получения был такой же, как на Стадии 4 в примере 1, за исключением того, что фенил {5-трет-бутил-2-[2-(трет-бутил-диметил-силокси)-этил]-2Н-пиразол-3-ил}-карбамат использовали вместо фенил изоксазол-3-ил-карбамата на Стадии 4 с получением 1-(4-бензимидазол-1-ил-фенил)-3-{5-трет-бутил-2-[2-(трет-бутил-диметил-силокси)-л]-мочевины
1-(4-Бензимидазол-1-ил-фенил)-3-{5-трет-бутил-2-[2-(трет-бутил-диметил-силокси)-этил]-2Н-пиразол-3-ил}-мочевину (1.05 г, 1.97 ммоль), полученную на Стадии 4 растворили в 30 мл ТГФ, 3 мл TBAF (1М раствор в ТГФ) добавили по каплям, и смесь реагировала при комнатной температуре в течение 15 минут. Реакционный раствор влили в воду и экстрагировали этилацетатом (80 мл × 2).Органическую фазу промыли дважды с помощью насыщенного раствора NaCl, высушили над безводным сульфатом натрия, отфильтровали, и фильтрат сконцентрировали при пониженном давлении. Остатки очистили с помощью колоночной хроматографии (элюент: дихлорметан/метанол) с получением 930 мг 1-(4-бензимидазол-1-ил-фенил)-3-[5-трет-бутил-2-(2-гидрокси-этил)-2Н-пиразол-3-ил]-мочевины.
1HNMR (DMSO-d6, 400MHz) δ: 9.38 (s, IH), 8.51 (s, IH), 8.50 (s, IH), 7.76-7.78 (m, 1H), 7.69-7.71 (d, 2H), 7.57-7.60 (m, 3H), 7.31-7.33 (m, 2H), 6.12 (s, 1H), 5.11-5.13 (t, 1H), 4.00-4.02 (m, 2H), 3.68-3.73 (m, 2H), 1.23 (s, 9H).
LC-MS (ESI): 418.9 (M+H)+.
Пример 104
Получение 1-(4-бензимидазол-1-ил-фенил)-3-(5-трет-бутил-2-фенил-2Н-пиразол-3-ил)-мочевины (Соединение 104)
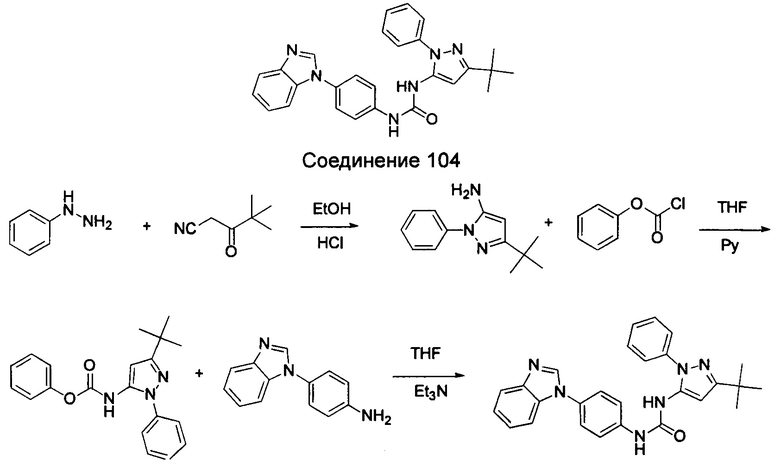
Стадия 1: Получение 5-трет-бутил-2-фенил-2Н-пиразол-3-иламина
Методика была аналогична таковой, как на Стадии 1 в примере 103, за исключением того, что фенилгидразин использовали вместо 2-гидразиноэтанола с получением 5-трет-бутил-2-фенил-2Н-пиразол-3-иламина.
Стадия 2: Получение фенил (5-трет-бутил-2-фенил-2Н-пиразол-3-ил)-карбамата
Методика была аналогична таковой, как на Стадии 3 в примере 1, за исключением того, что 5-трет-бутил-2-фенил-2-Н-пиразол-3-иламин использовали вместо 3-аминоизоксазола с получением фенил (5-трет-бутил-2-фенил-2Н-пиразол-3-ил)-карбамата.
Стадия 3: Получение 1-(4-бензимидазол-1-ил-фенил)-3-(5-трет-бутил-2-фенил-2Н-пиразол-3-ил)-мочевины
Методика была аналогична таковой, как на Стадии 4 в примере 1, за исключением того, что фенил (5-трет-бутил-2-фенил-2Н-пиразол-3-ил)-карбамат использовали вместо фенил изоксазол-3-ил-карбамата с получением 1-(4-бензимидазол-1-ил-фенил)-3-(5-трет-бутил-2-фенил-2Н-пиразол-3-ил)-мочевины.
1HNMR (DMSO-d6, 400MHz) δ: 9.30 (s, IH), 8.50 (s, IH), 8.50 (s, IH), 7.76-7.78 (m, 1H), 7.64-7.66 (d, 2H), 7.54-7.58 (m, 7H), 7.41-7.44 (m, 1H), 7.30-7.35 (m, 2H), 6.41 (s, 1H), 1.29 (s, 9H).
LC-MS (ESI): 451.2 (M+H)+.
Пример 105
Получение 1-(4-бензимидазол-1-ил-фенил)-3-(5-трет-бутил-2-р-толил-2Н-пиразол-3-ил)-мочевины (Соединение 105)
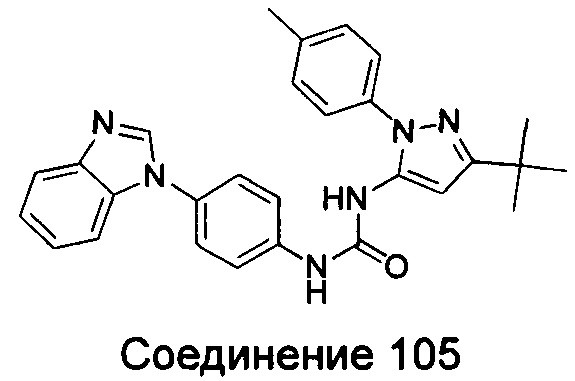
Способ получения был такой же, как на Стадиях 1-3 в примере 104, за исключением того, что 4-метилфенилгидразин использовали вместо фенилгидразина на Стадии 1, с получением 1-(4-бензимидазол-1-ил-фенил)-3-(5-трет-бутил-2-р-толил-2Н-пиразол-3-ил)-мочевины.
1HNMR (DMSO-d6, 400MHz) δ: 9.29 (s, IH), 8.49 (s, IH), 8.43 (s, IH), 7.76-7.78 (m, 1H), 7.63-7.66 (d, 2H), 7.55-7.59 (m, 3H), 7.41-7.43 (d, 2H), 7.29-7.36 (m, 4H), 6.39 (s, 1H), 2.39 (s, 3H), 1.29 (s, 9H).
LC-MS (ESI): 465.2 (M+H)+.
Пример 106
Получение 1-(4-бензимидазол-1-ил-фенил)-3-[5-трет-бутил-2-(4-метоксил-фенил)-2Н-пиразол-3-ил]-мочевины (Соединение 106)
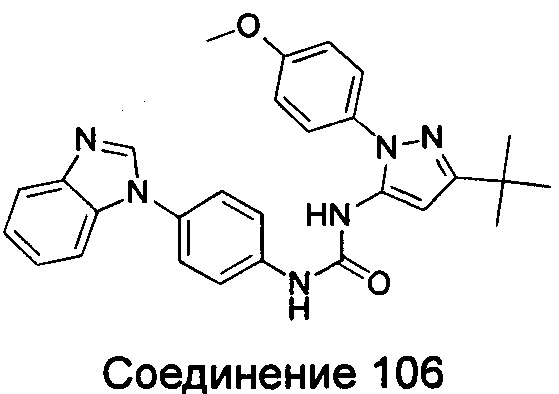
Способ получения был такой же, как на Стадиях 1-3 в примере 104, за исключением того, что 4-метоксил фенилгидразин использовали вместо фенилгидразина на Стадии 1 с получением 1-(4-бензимидазол-1-ил-фенил)-3-[5-трет-бутил-2-(4-метоксил-фенил)-2Н-пиразол-3-ил]-мочевины.
1HNMR (DMSO-d6, 400MHz) δ: 9.28 (s, 1Н), 8.50 (s, 1Н), 8.39 (s, 1Н), 7.76-20 7.78 (m, 1Н), 7.63-7.66 (d, 2Н), 7.55-7.59 (m, 3Н), 7.42-7.46 (d, 2Н), 7.28-7.34 (m, 1Н), 7.08-7.12 (d, 2Н), 6.38 (s, 1Н), 3.83 (s, 3Н), 1.29 (s, 9Н).
LC-MS (ESI): 481.2 (M+H)+.
Пример 107
Получение 1-(4-бензимидазол-1-ил-фенил)-3-[5-трет-бутил-2-(4-трифторметокси-фенил)-2Н-пиразол-3-ил]-мочевины (Соединение 107)
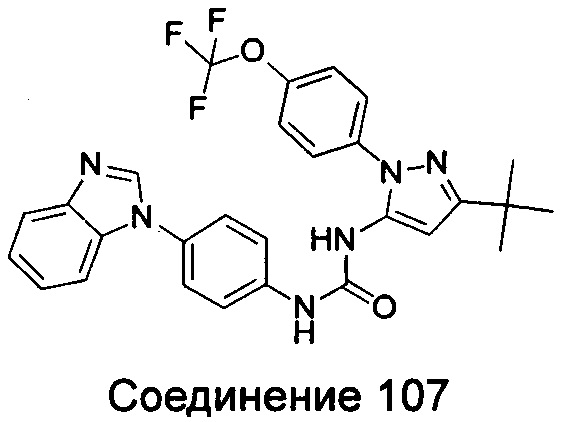
Способ получения был такой же, как на Стадиях 1-3 примера 104, за исключением того, что 4-трифторметокси фенилгидразин использовали вместо фенилгидразина на Стадии 1 с получением 1-(4-бензимидазол-1-ил-фенил)-3-[5-трет-бутил-2-(4-трифторметокси-фенил)-2Н-пиразол-3-ил]-мочевины.
1HNMR (DMSO-d6, 400MHz) δ: 9.29 (s, 1H), 8.58 (s, 1H), 8.51 (s, 1H), 7.78- 5 7.80 (m, 1H), 7.65-7.72 (m, 4H), 7.54-7.59 (m, 5H), 7.29-7.36 (m, 2H), 6.43 (s, 1H), 1.30 (s, 9H).
LC-MS (ESI): 535.2 (M+H)+.
Пример 108
Получение 1-(4-бензимидазол-1-ил-фенил)-3-[5-трет-бутил-2-(4-фтор-фенил)-2Н-пиразол-3-ил]-мочевины (Соединение 108)
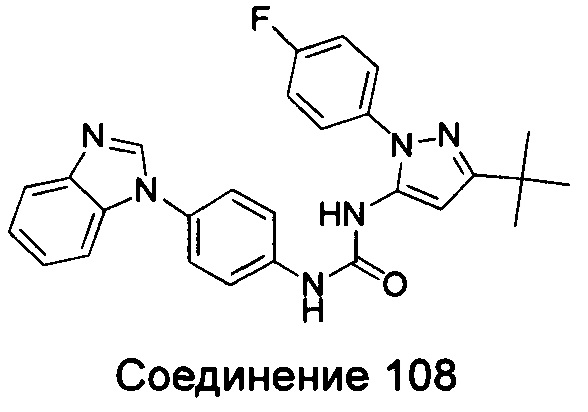
Способ получения был такой же, как на Стадиях 1-3 в примере 104, за исключением того, что 4-фторофенилгидразин использовали вместо фенилгидразина на Стадии 1 с получением 1-(4-бензимидазол-1-ил-фенил)-3-[5-трет-бутил-2-(4-фтор-фенил)-2Н-пиразол-3-ил]-мочевины.
1HNMR (DMSO-d6, 400MHz) δ: 9.27 (s, IH), 8.49 (s, IH), 8.47 (s, IH), 7.76-7.78 (m, 1H), 7.64-7.66 (d, 2H), 7.55-7.61 (m, 5H), 7.37-7.41 (m, 2H), 7.28-7.34 (m, 2H), 6.40 (s, 1H), 1.29 (s, 9H).
LC-MS (ESI): 469.2 (M+H)+.
Пример 109
Получение 1-(4-бензимидазол-1-ил-фенил)-3-[5-трет-бутил-2-(2-фтор-фенил)-2Н-пиразол-3-ил]-мочевины (Соединение 109)
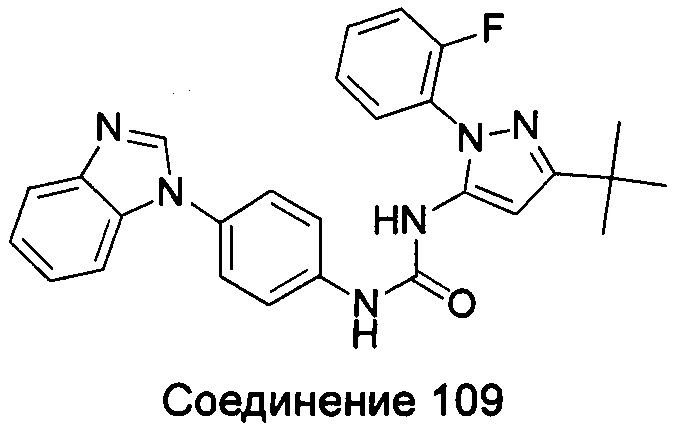
Способ получения был такой же, как на Стадиях 1-3 в примере 104, за исключением того, что 2-фторфенилгидразин использовали вместо фенилгидразина на Стадии 1 с получением 1-(4-бензимидазол-1-ил-фенил)-3-[5-трет-бутил-2-(2-фтор-фенил)-2Н-пиразол-3-ил]-мочевины.
1HNMR (DMSO-d6, 400MHz) δ: 9.17 (s, IH), 8.49 (s, IH), 8.47 (s, IH), 7.76- 7.78 (m, 1H), 7.63-7.65 (d, 2H), 7.55-7.60 (m, 5H), 7.53-7.48 (m, 1H), 7.39-7.43 (m, 1H), 7.28-7.35 (m, 2H), 6.42 (s, 1H), 1.28 (s, 9H).
LC-MS (ESI): 469.2 (M+H)+.
Пример 110
Получение 1-(4-бензимидазол-1-ил-фенил)-3-(5-трет-бутил-2-пиридин-2-ил-2Н-пиразол-3-ил)-мочевины (Соединение 110)
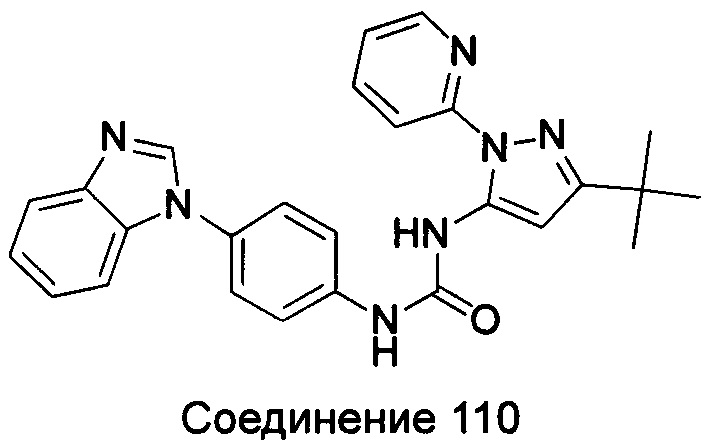
Способ получения был такой же, как на Стадиях 1-3 в примере 104, за исключением того, что 2-гидразинопиридин (Darui) использовали вместо фенилгидразина на Стадии 1 с получением 1-(4-бензимидазол-1-ил-фенил)-3-(5-трет-бутил-2-пиридин-2-ил-2Н-пиразол-3-ил)-мочевины.
1HNMR (DMSO-d6, 400MHz) δ: 11.32 (s, 1H), 10.22 (s, 1H), 8.53 (s, 1H), 8.50-8.52 (m, 1H), 8.01-8.05 (m, 1H), 7.92-7.94 (d, 1H), 7.77-7.79 (m, 3Н), 7.59-7.64 (m, 3Н), 7.29-7.37 (m, 3Н), 6.65 (s, 1H), 1.31 (s, 9H).
LC-MS (ESI): 452.2 (M+H)+.
Пример 111
Получение этил 4-{5-[3-(4-бензимидазол-1-ил-фенил)-карбамидо]-3-трет-бутил-пиразол-1-ил}-бензоата (Соединение 111)
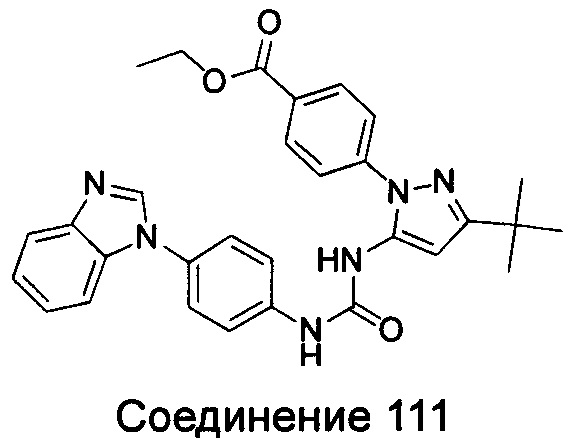
Способ получения был такой же, как на Стадиях 1-3 в примере 104, за исключением того, что этил 4-гидразино-бензоат (Darui) использовали вместо фенилгидразина с получением этил 4-{5-[3-(4-бензимидазол-1-ил-фенил)-карбамидо]-3-трет-бутил-пиразол-1-ил}-бензоата.
1HNMR (DMSO-d6, 400MHz) δ:9.32 (s, IH), 8.64 (s, IH), 8.49 (s, IH), 8.09-8.11 (d, 2H), 7.75-7.78 (m, 3H), 7.64-7.67 (d, 2H), 7.55-7.58 (m, 3H), 7.28-7.34 (m, 2H), 6.45 (s, 1H), 4.32-4.37 (q, 2H), 1.32-1.36 (t, 3H), 1.31 (s, 9H).
LC-MS (ESI): 523.2 (M+H)+.
Пример 112
Получение 1-(2-акрил-5-трет-бутил-2Н-пиразол-3-ил)-3-(4-имидазол-1-ил-фенил)-мочевины (Соединение 112)
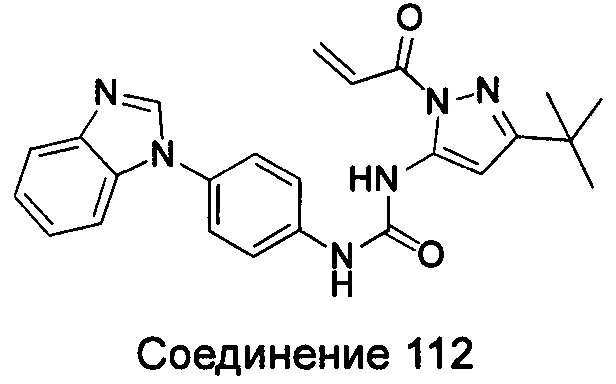
Стадия 1: Получение 1-(2-акрил-5-трет-бутил-2Н-пиразол-3-ил)-3-(4-имидазол-1-ил-фенил)-мочевины
1-(4-Бензимидазол-1-ил-фенил)-3-(5-трет-бутил-2Н-пиразол-3-ил)-мочевину (Соединение 68) (150 мг, 0.40 ммоль), и N,N-диизопропилэтиламин (77.4 мг, 0.60 ммоль) растворили в 10 мл ТГФ, и акрилоилхлорид (43.4 мг, 0.48 ммоль) добавили по каплям в ванне со льдом (0-5°С). После добавления реакционную смесь перемешивали в течение 30 минут на ледяной бане, и реакционную смесь погасили водой (30 мл), экстрагировали этилацетатом (50×2), и органическую фазу промыли дважды с помощью насыщенного раствора NaCl, высушили над безводным сульфатом натрия и сконцентрировали при пониженном давлении. Остатки очистили с помощью колоночной хроматографии (элюент: дихлорметан/метанол) с получением 80 мг 1-(2-акрил-5-трет-бутил-2Н-пиразол-3-ил)-3-(4-имидазол-1-ил-фенил)-мочевины в виде белого осадка.
1HNMR (DMSO-d6, 400MHz) δ: 10.36 (s, 1H), 10.13 (s, 1H), 8.52 (s, 1H), 7.77-15 7.79 (m, 1H), 7.72-7.75 (d, 2H), 7.61-7.63 (d, 2H), 7.58-7.60 (m, 1H), 7.48-7.55 (q, 1H), 7.31-7.34 (m, 2H), 6.72 (s, 1H), 6.61-6.66 (m, 1H), 6.20-6.23 (m, 1H), 1.29 (s, 9H).
LC-MS (ESI): 429.1 (M+H)+.
Пример 113
Получение 3-амино-5-метилпиразол-1-карбоновой кислоты (4-бензимидазол-1-ил-фенил)-амида (Соединение 113)
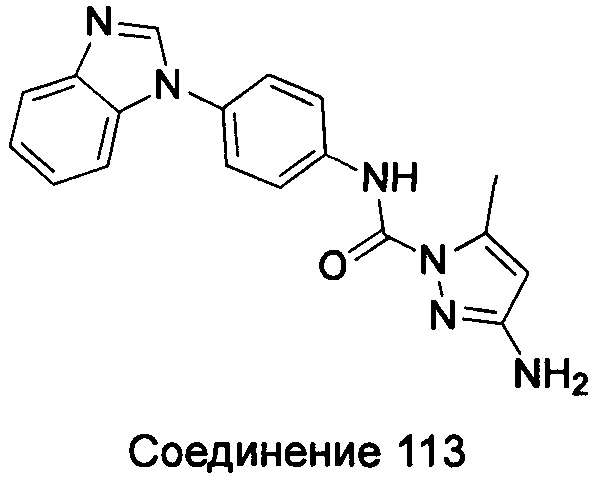
Это соединение синтезировали одновременно в примере 64.
1HNMR (DMSO-d6, 400MHz) δ: 9.91 (s, 1H), 8.53 (s, 1H), 7.92-7.94 (d, 2H), 25 7.77-7.79 (m, 1H), 7.62-7.65 (d, 2H), 7.59-7.61 (m, 1H), 7.31-7.34 (m, 2H), 5.71 (d, 1H), 5.34 (s, 2H), 2.48 (d, 3Н).
LC-MS (ESI): 333.1 (M+H)+
Пример 114
Получение 5-амино-3-циклопропилпиразол-1-муравьиной кислоты (4-бензимидазол-1-ил-фенил)-амида (Соединение 114)
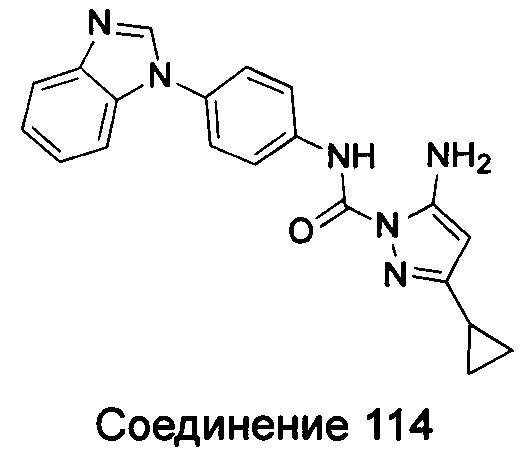
Это соединение синтезировали одновременно в примере 66.
1HNMR (DMSO-d6, 400MHz) δ: 10.07 (s, 1Н), 8.54 (s, 1Н), 7.94-7.96 (d, 2H), 7.77-7.79 (m, 1H), 7.65-7.67 (d, 2H), 7.61-7.63 (m, 1H), 7.31-7.34 (m, 2H), 6.46 (s, 2H), 5.06 (s, 1H), 1.83-1.88 (m, 1H), 0.88-0.93 (m, 2H), 0.70-0.74 (m, 2H).
LC-MS (ESI): 359.1 (M+H)+.
Пример 115
Получение 5-амино-3-трифторметилпиразол-1-карбоновой кислоты (4-бензимидазол-1-ил-фенил)-амида (Соединение 115)
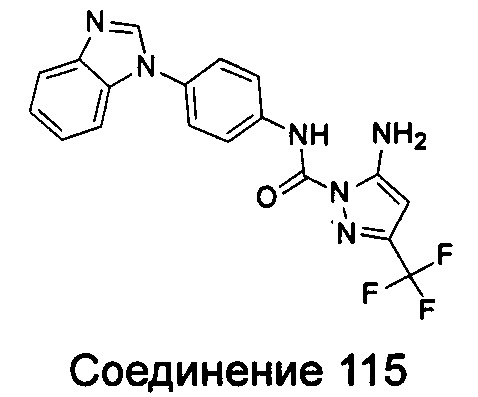
Это соединение синтезировали одновременно в примере 67.
1HNMR (DMSO-d6, 400MHz) δ: 10.40 (s, 1H), 8.55 (s, 1H), 7.92-7.95 (d, 2H), 7.77-7.79 (m, 1H), 7.68-7.71 (d, 2H), 7.62-7.64 (m, 1H), 7.30-7.37 (m, 2H), 6.90 (s, 2H), 5.76 (s, IH).
LC-MS (ESI): 387.0 (M+H)+.
Пример 116
Получение 5-амино-3-трет-бутил-пиразол-1-карбоновой кислоты (4-бензимидазол-1-ил-фенил)-амида (Соединение 116)
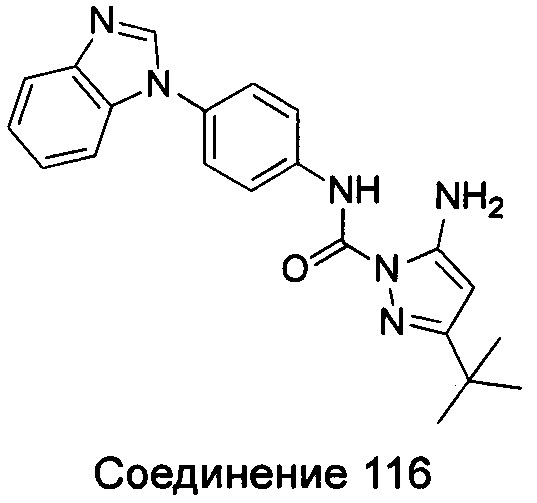
Это соединение синтезировали одновременно в примере 68.
1HNMR (DMSO-d6, 400MHz) δ: 9.80 (s, 1H), 8.56 (s, 1H), 7.91-7.94 (d, 2H), 7.77-7.79 (m, 1H), 7.68-7.70 (d, 2H), 7.62-7.64 (m, 1H), 7.31-7.35 (m, 2H), 6.42 (s, 2H), 5.32 (s, 1H), 1.27 (s, 9H).
LC-MS (ESI): 375.0 (M+H)+.
Пример 117
Получение 5-амино-3-трет-бутил-пиразол-1-карбоновой кислоты [4-(5-гексилоксил-бензимидазол-1-ил)-фенил]-амида (Соединение 117)
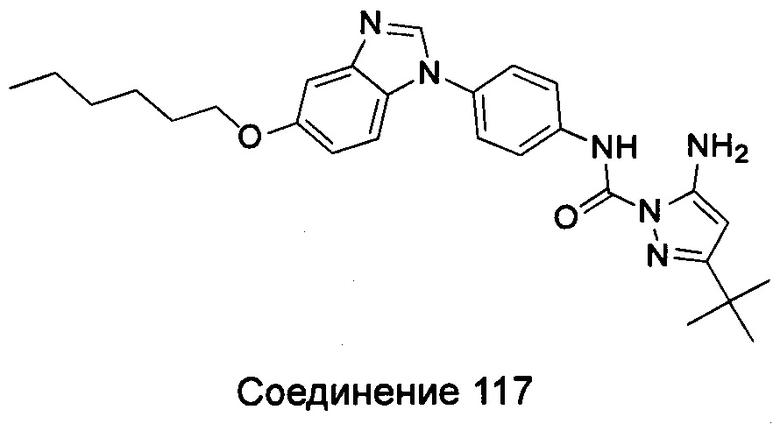
Это соединение синтезировали одновременно в примере 73.
1HNMR (DMSO-d6, 400MHz) δ: 9.79 (s, 1H), 8.48 (s, 1H), 7.90-7.92 (d, 2H), 7.65-7.67 (d, 2H), 7.49-7.51 (d, 1H), 7.28-7.29 (d, 1H), 6.94-6.96 (dd, 1H), 6.41 (s, 2H), 5.32 (s, 1H), 4.01-4.04 (t, 2H), 1.71-1.76 (m, 2H), 1.43-1.47 (m, 2H), 1.31-1.35 (m, 4H), 1.27 (s, 9H), 0.87-0.91 (t, 3Н).
LC-MS (ESI): 475.2 (M+H)+.
Пример 118
Получение 5-амино-3-трет-бутил-пиразол-1-карбоновой кислоты (4-{5-[3-(4-метил-пиперазин-1-ил)-пропоксил]-бензимидазол-1-ил}-фенил)-амида (Соединение 118)
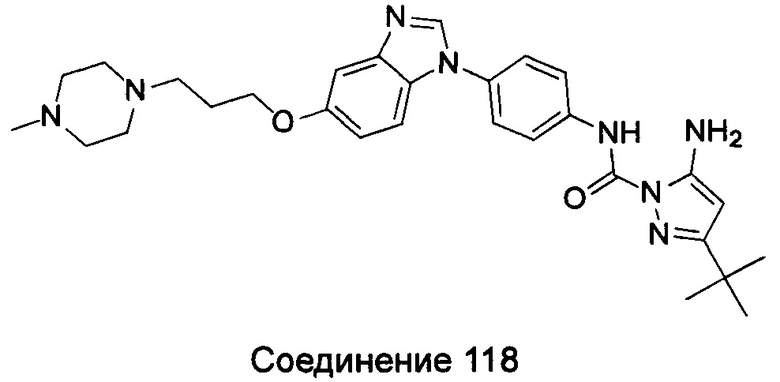
Это соединение синтезировали одновременно в примере 92.
1HNMR (DMSO-d6, 400MHz) δ: 9.28 (s, IH), 8.06 (s, IH), 7.78-7.80 (d, 2H), 7.49-7.52 (d, 2H), 7.39-7.41 (d, 1H), 7.34-7.35 (d, 1H), 6.97-7.00 (dd, 1H), 5.44 (s, 2H), 4.10-4.13 (t, 2H), 2.50-2.70 (m, 10H), 2.37 (s, 3H), 2.01-2.08 (m, 2H), 1.31 (s, 9H).
LC-MS (ESI): 531.2 (M+H)+.
Пример 119
Получение 5-амино-3-трет-бутил-пиразол-1-карбоновой кислоты {4-[5-(2-морфолин-4-ил-этоксил)-бензимидазол-1-ил]-фенил}-амида (Соединение 119)
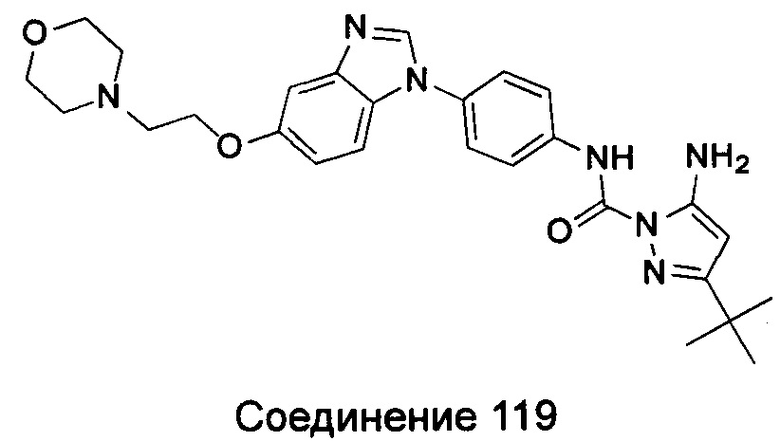
Это соединение синтезировали одновременно в примере 89.
1HNMR (DMSO-d6, 400MHz) δ: 9.79 (s, 1H), 8.49 (s, 1H), 7.90-7.92 (d, 2H), 5 7.65-7.67 (d, 2H), 7.50-7.52 (d, 1H), 7.32-7.33 (d, 1H), 6.96-6.98 (dd, 1H), 6.42 (s, 2H), 5.32 (s, 1H), 4.15-4.17 (t, 2H), 3.59-3.61 (t, 4H), 2.73-2.76 (t, 2H), 2.49-2.51 (m, 4H), 1.27 (s, 9H).
LC-MS (ESI): 504.2 (M+H)+.
Пример 120
Получение 5-амино-3-трет-бутил-пиразол-1-карбоновой кислоты {4-[5-(2-метоксил-этоксил)-бензимидазол-1-ил]-фенил}-амида (Соединение 120)
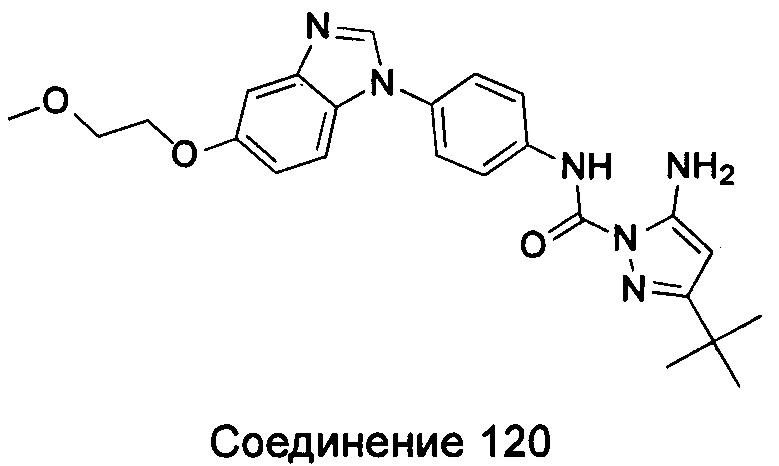
Это соединение синтезировали одновременно в примере 77.
1HNMR (DMSO-d6, 400MHz) δ: 9.79 (s, 1Н), 8.49 (s, 1Н), 7.90-7.92 (d, 2Н), 7.65-7.68 (d, 2Н), 7.51-7.53 (d, 1Н), 7.31-7.32 (d, 1Н), 6.96-6.99 (dd, 1Н), 6.42 (s, 2Н), 5.32 (s, 1Н), 4.15-4.17 (m, 2Н), 3.69-3.71 (m, 2Н), 3.33 (s, 3Н), 1.27 (s, 9Н).
LC-MS (ESI): 449.2 (M+H)+.
Пример 121
Получение 5-амино-3-трет-бутил-пиразол-1-карбоновой кислоты {4-[5-(2-гидрокси-этоксил)-бензимидазол-1-ил]-фенил}-амида (Соединение 121)
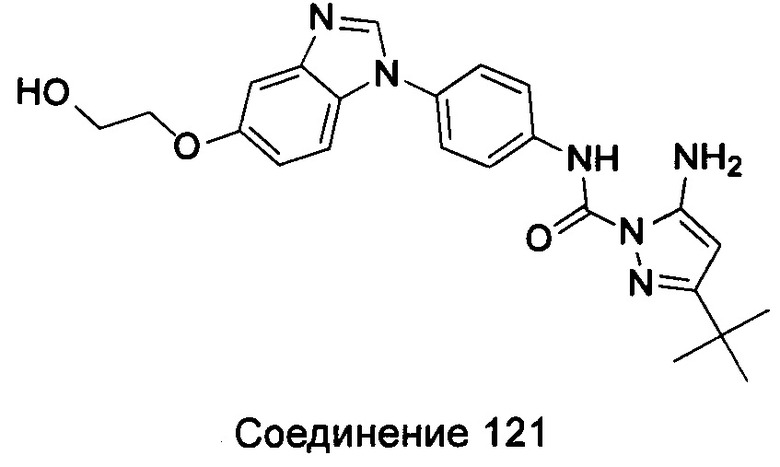
Это соединение синтезировали одновременно в примере 79.
1HNMR (DMSO-d6, 400MHz) δ: 9.79 (s, 1Н), 8.49 (s, 1Н), 7.90-7.92 (d, 2Н), 7.65-7.68 (d, 2Н), 7.51-7.53 (d, 1H), 7.30 (d, 1H), 6.97-6.99 (dd, 1H), 6.41 (s, 2H), 5.32 (s, 1H), 4.89-4.92 (t, 1H), 4.04-4.06 (t, 2H), 3.74-3.78 (m, 2H), 1.27 (s, 9H).
LC-MS (ESI): 435.2 (M+H)+.
Пример 122
Получение 5-амино-3-трет-бутил-пиразол-1-карбоновой кислоты {4-[5-(тетрагидро-пиран-2-илметокси)-бензимидазол-1-ил]-фенил}-амида (Соединение 122)
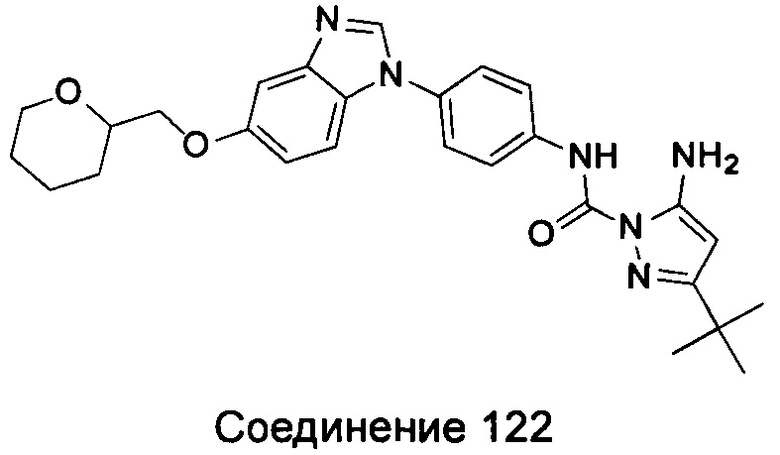
Это соединение синтезировали одновременно в примере 87.
1HNMR (DMSO-d6, 400MHz) δ: 9.79 (s, 1H), 8.48 (s, 1H), 7.89-7.91 (d, 2H), 10 7.65-7.67 (d, 2H), 7.50-7.52 (d, 1H), 7.28-7.29 (d, 1H), 6.96-6.98 (dd, 1H), 6.41 (s, 2H), 5.32 (s, 1H), 3.89-3.98 (m, 3Н), 3.63-3.69 (m, 1H)), 3.34-3.42 (m, 1H), 1.82-1.84 (m, 1H), 1.67-1.70 (m, 1H), 1.46-1.54 (m, 4H), 1.26 (s, 9H).
LC-MS (ESI): 489.2 (M+H)+.
Пример 123
Получение 5-амино-3-трет-бутил-пиразол-1-карбоновой кислоты {4-[5-(тетрагидро-фуран-2-илметокси)-бензимидазол-1-ил]-фенил}-амида (Соединение 123)
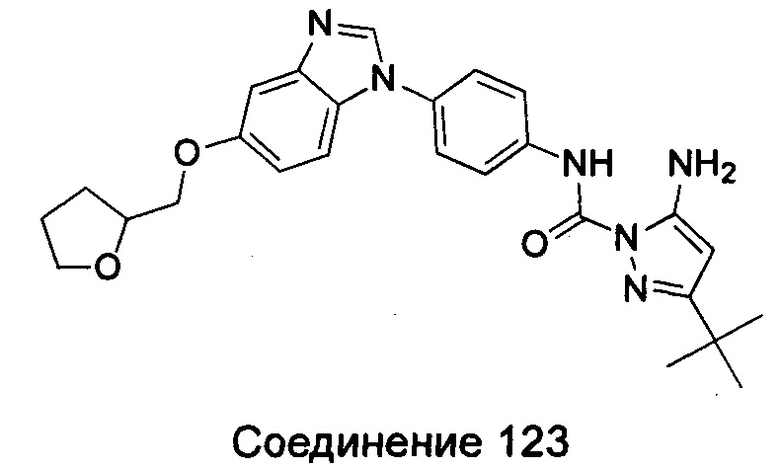
Это соединение синтезировали одновременно в примере 86.
1HNMR (DMSO-d6, 400MHz) δ: 9.79 (s, 1Н), 8.49 (s, 1Н), 7.90-7.92 (d, 2Н), 7.65-7.68 (d, 2Н), 7.50-7.53 (d, 1Н), 7.31-7.32 (d, 1Н), 6.96-6.99 (dd, 1Н), 6.41 (s, 2Н), 5.32 (s, 1Н), 4.18-4.20 (m, 1Н), 3.98-4.02 (m, 2Н), 3.78-3.82 (m, 1Н), 3.69-3.71 (m, 1Н), 1.67-2.07 (m, 4Н), 1.27 (s, 9Н).
LC-MS (ESI): 475.1 (M+H)+.
Пример 124
Получение этил (1-{4-[(5-амино-3-трет-бутил-пиразол-1-карбонил)-амино]-фенил}-1Н-бензимидазол-5-илокси)-ацетата (Соединение 124)
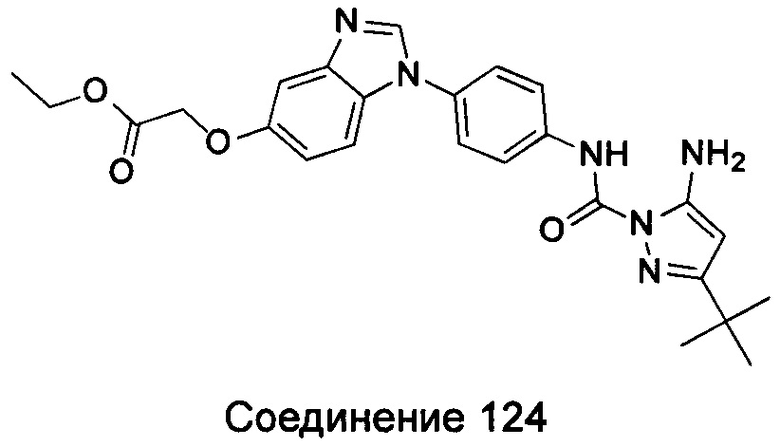
Это соединение синтезировали одновременно в примере 88.
1HNMR (DMSO-d6, 400MHz) δ: 9.79 (s, 1H), 8.50 (s, 1H), 7.90-7.92 (d, 2H), 7.65-7.67 (d, 2H), 7.52-7.54 (d, 1H), 7.28 (d, 1H), 6.99-7.02 (dd, 1H), 6.41 (s, 2H), 5.32 (s, 1H), 4.84 (s, 2H), 4.16-4.21 (q, 2H), 1.27 (s, 9H), 1.21-1.25 (t, 3Н)
LC-MS (ESI): 477.1 (M+H)+.
Пример 125
Получение 5-амино-3-трет-бутил-пиразол-1-карбоновой кислоты [4-(5-фтор-бензимидазол-1-ил)-фенил]-амида (Соединение 125)
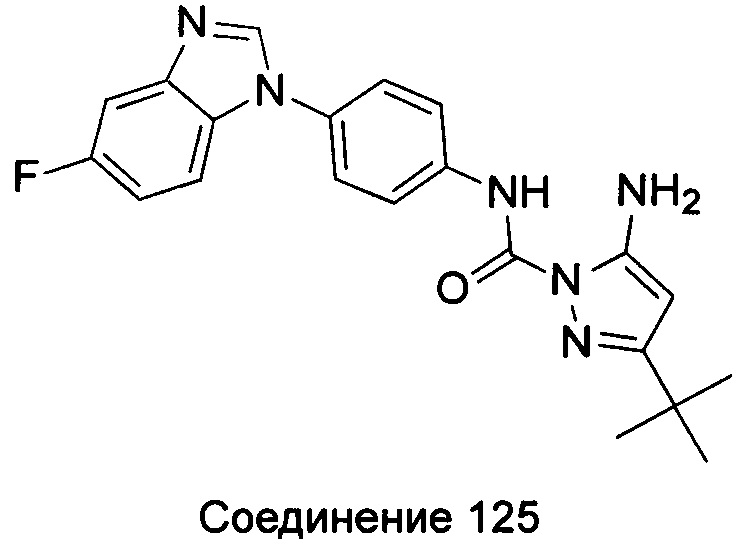
Это соединение синтезировали одновременно в примере 69.
1HNMR (DMSO-d6, 400MHz) δ: 9.81 (s, 1Н), 8.61 (s, 1Н), 7.91-7.93 (d, 2Н), 7.67-7.69 (d, 2Н), 7.58-7.64 (m, 2Н), 7.19-7.25 (m, 1Н), 6.41 (s, 2Н), 5.33 (s, 1Н), 1.26 (s, 9Н).
LC-MS (ESI): 393.2 (M+H)+.
Пример 126
Получение 5-амино-3-трет-бутил-пиразол-1-карбоновой кислоты [4-(5-трифторметил-бензимидазол-1-ил)-фенил]-амида (Соединение 126)
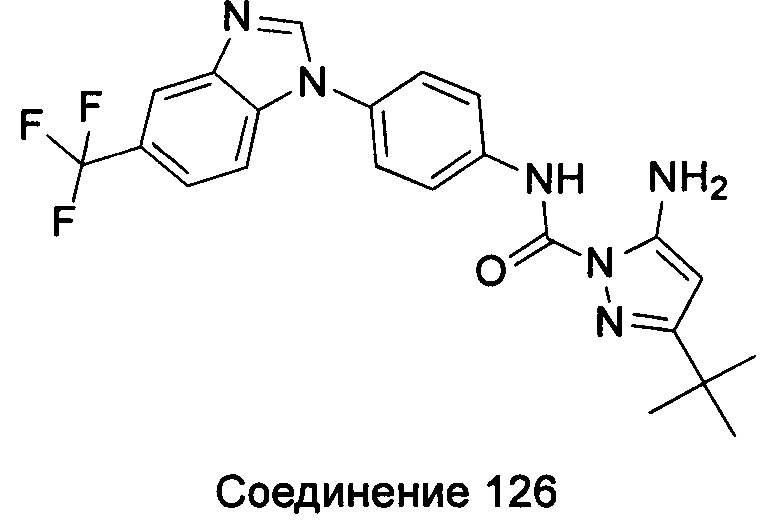
Это соединение синтезировали одновременно в примере 70.
1HNMR (DMSO-d6, 400MHz) δ: 9.83 (s, 1Н), 8.77 (s, 1Н), 8.17 (d, 1Н), 7.94-7.96 (d, 2Н), 7.80-7.82 (d, 1Н), 7.71-7.73 (d, 2Н), 7.66-7.68 (dd, 1Н), 6.41 (s, 2Н), 5.33 (s, 1Н), 1.27 (s, 9Н).
LC-MS (ESI): 443.1 (M+H)+.
Пример 127
Получение 4-(1-{4-[(5-амино-3-трет-бутил-пиразол-1-карбонил)-амино]-фенил}-1Н-бензимидазол-5-илокси)-пиридин-2-карбоновой кислоты метиламида (Соединение 127)
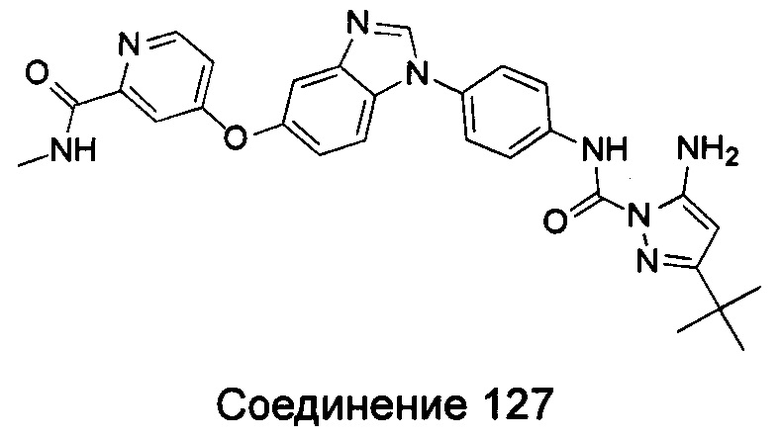
Это соединение синтезировали одновременно в примере 96.
1HNMR (DMSO-d6, 400MHz) δ: 9.82 (s, 1Н), 8.78-8.79 (m, 1Н), 8.68 (s, 1Н), 8.50-8.52 (d, 1Н), 7.93-7.95 (d, 2Н), 7.72-7.75 (m, 3Н), 7.67 (d, 1Н), 7.39-7.40 (d, 1Н), 7.18-7.23 (m, 2Н), 6.42 (s, 2Н), 5.33 (s, 1Н), 2.77-2.79 (d, 3Н), 1.27 (s, 9Н).
LC-MS (ESI): 525.1 (M+H)+.
Пример 128
Получение 5-амино-3-трет-бутил-пиразол-1-карбоновой кислоты {4-[6-(2-метоксил-этоксил)-бензимидазол-1-ил]-фенил}-амида (Соединение 128)
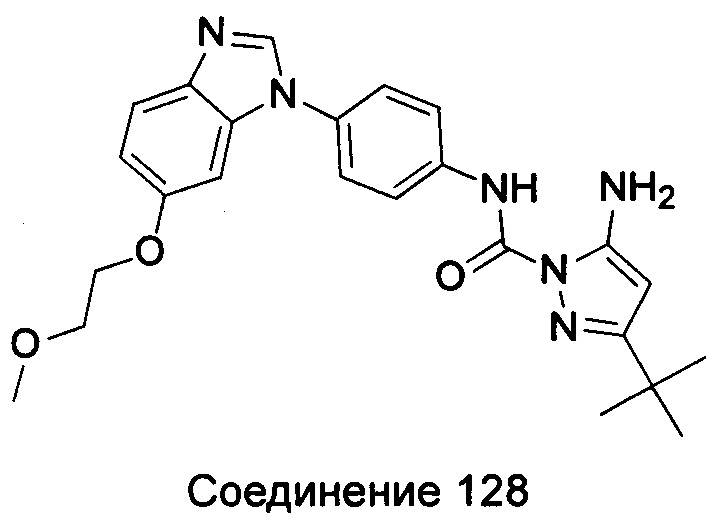
Это соединение синтезировали одновременно в примере 98.
1HNMR (DMSO-d6, 400MHz) δ: 9.80 (s, 1Н), 8.45 (s, 1Н), 7.91-7.93 (d, 2Н,), 7.68-7.70 (m, 3Н), 7.08 (d, 1Н), 6.95-6.98 (dd, 1Н), 6.42 (s, 2Н), 5.33 (s, 1Н), 4.13-20 4.15 (m, 2Н), 3.66-3.68 (m, 2Н), 3.31 (s, 3Н), 1.27 (s, 9Н).
LC-MS (ESI): ESI 449.1 (M+H)+.
Пример 129
Получение 5-амино-3-трет-бутил-пиразол-1-карбоновой кислоты [4-(5,6-диметоксил-бензимидазол-1-ил)-фенил]-амида (Соединение 129)
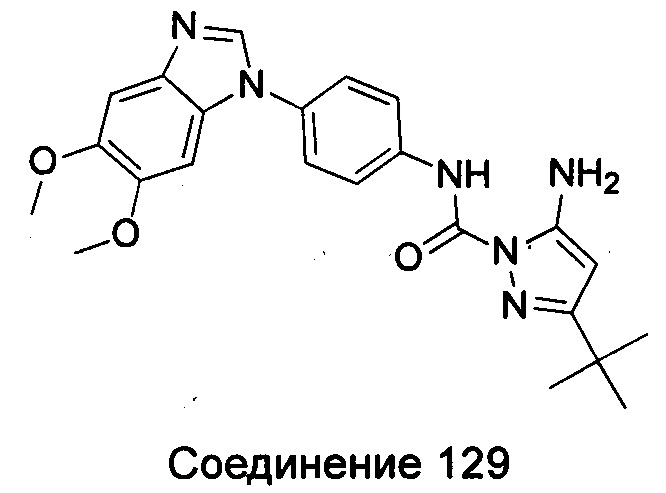
Соединение синтезировали одновременно в Примере 98.
1HNMR (DMSO-d6, 400MHz) δ: 9.78 (s, 1H), 8.33 (s, 1H), 7.90-7.92 (d, 2H,), 7.67-7.69 (d, 2H), 7.32 (s, 1H), 7.08 (s, 1H), 6.41 (s, 2H), 5.32 (s, 1H), 3.83 (s, 3Н), 3.81 (s, 3Н), 1.27 (s, 9H).
LC-MS (ESI): ESI 435.1 (M+H)+.
Пример 130
Получение 3-трет-бутил-пиразол-1-карбоновой кислоты (4-бензимидазол-1-ил-фенил)-амида (Соединение 130)
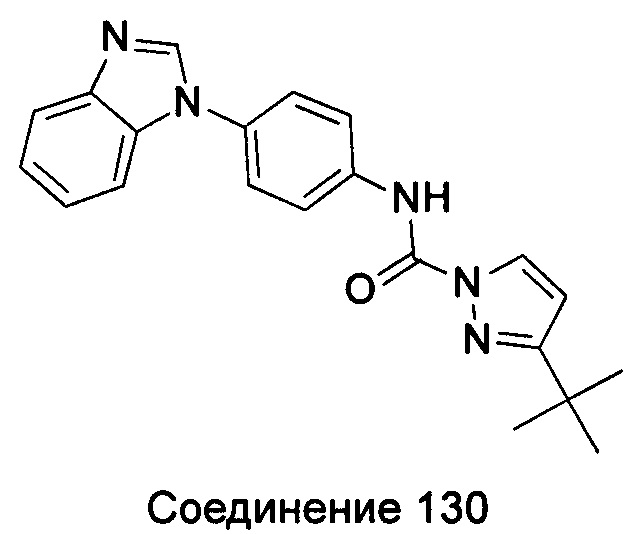
Способ получения был аналогичным таковому в примере 68, за исключением того, что 3-(трет-бутил)-1Н-пиразол использовали вместо 3-трет-бутил-пиразол-5-амина на Стадии 2 с получением 3-трет-бутил-пиразол-1-карбоновой кислоты (4-бензимидазол-1-ил-фенил)-амида.
1HNMR (DMSO-d6, 400MHz) δ: 9.26 (s, IH), 8.23-8.24 (d, IH), 8.14 (s, IH), 7.89-7.92 (m, 1H), 7.86-7.88 (d, 2H), 7.54-7.57 (m, 3H), 7.36-7.38 (m, 2H), 6.40-6.41 (d, 1H), 1.39 (s, 9H).
LC-MS (ESI): 360.1 (M+H)+.
Пример 131
Получение 1-(4-бензимидазол-1-ил-фенил)-3-(3,4-диметил-изоксазол-5-ил)-мочевины (Соединение 131)
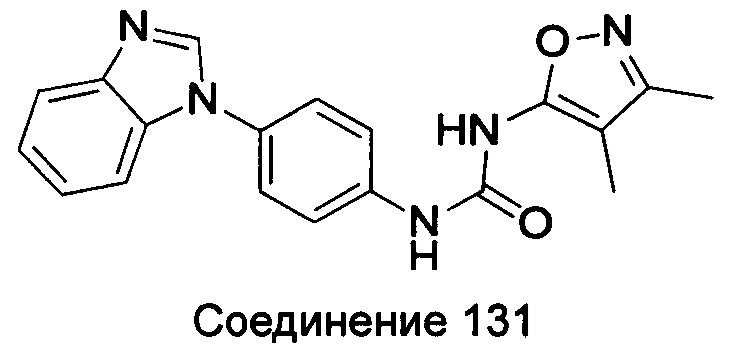
Стадия 1: Получение фенил (3,4-диметил-изоксазол-5-ил)-карбамата (активный сложный эфир)
Способ получения был такой же, как на Стадии 3 в примере 1, за исключением того, что 3,4-диметил-5-аминоизоксазол использовали вместо 3-аминоизоксазола с получением фенил (3,4-диметил-изоксазол-5-ил)-карбамата.
Стадия 2: 1-(4-бензимидазол-1-ил-фенил)-3-(3,4-диметил-изоксазол-5-ил)-мочевины
Способ получения был такой же, как на Стадии 4 в примере 1, за исключением того, что фенил (3,4-диметил-изоксазол-5-ил)-карбамат использовали вместо изоксазол-3-ил-фенилкарбамата с получением 1-(4-бензимидазол-1-ил-фенил)-3-(3,4-диметил-изоксазол-5-ил)-мочевины.
1HNMR (DMSO-d6, 400MHz) δ: 9.22 (s, 1H), 9.18 (s, 1H), 8.50 (s, 1H), 7.76-7.78 (m, 1H), 7.69-7.71 (d, 2H), 7.57-7.61 (m, 3H), 7.30-7.33 (m, 2H), 2.16 (s, 3H), 1.85 (s, 3H).
LC-MS (ESI): 348.1 (M+H)+.
Пример 132
Получение 1-(4-бензимидазол-1-ил-фенил)-3-(3-изопропил-изоксазол-5-ил)-мочевины (Соединение 132)
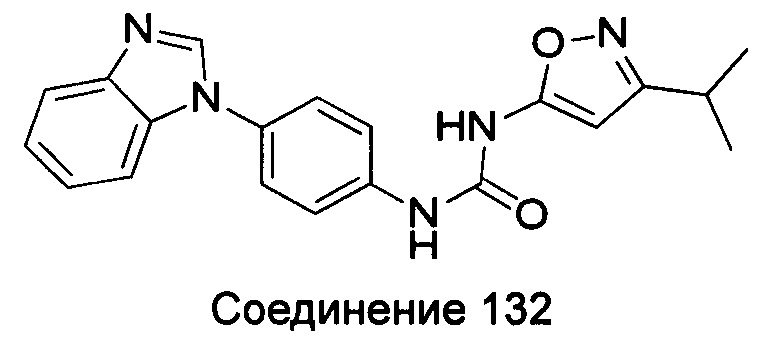
Стадия 1: Получение фенил (3-изопропил-изоксазол-5-ил)-карбамата (активный сложный эфир)
Способ получения был такой же, как на Стадии 3 в примере 1, за исключением того, что 3-изопропил-5-аминоизоксазол использовали вместо 3-аминоизоксазола с получением фенил (3-изопропил-изоксазол-5-ил)-карбамата.
Стадия 2: 1-(4-бензимидазол-1-ил-фенил)-3-(3-изопропил-изоксазол-5-ил)-мочевина
Способ получения был такой же, как на Стадии 4 в примере 1, за исключением того, что фенил (3-изопропил-изоксазол-6-ил)-карбамат использовали вместо фенил изоксазол-3-ил-карбамата с получением 1-(4-бензимидазол-1-ил-фенил)-3-(3-изопропил-изоксазол-5-ил)-мочевины.
1HNMR (DMSO-d6, 400MHz) δ: 10.21 (s, IH), 9.13 (s, IH), 8.51 (s, IH), 7.76- 7.78 (m, 1H), 7.70-7.72 (d, 2H), 7.61-7.63 (d, 2H), 7.57-7.59 (m, 1H), 7.28-7.36 (m, 2H), 6.05 (s, 1H), 2.90-2.96 (m, 1H), 1.21-1.23 (d, 6H).
LC-MS (ESI): 362.1 (M+H)+.
Пример 133
Получение 1-(4-бензимидазол-1-ил-фенил)-3-(3-трет-бутил-изоксазол-5-ил)-мочевины (Соединение 133)
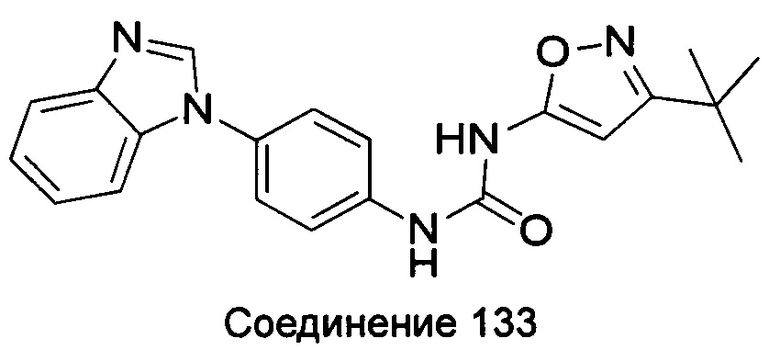
Стадия 1: Получение фенил (3-трет-бутил-изоксазол-5-ил)-карбамата (активный сложный эфир)
Способ получения был такой же, как на Стадии 3 в примере 1, за исключением того, что 3-трет-бутил-5-аминоизоксазол использовали вместо 3-аминоизоксазола с получением фенил (3-трет-бутил- изоксазол-5-ил)-карбамата.
Стадия 2: Получение 1-(4-бензимидазол-1-ил-фенил)-3-(3-трет-бутил-изоксазол-5-ил)-мочевины
Способ получения был такой же, как на Стадии 4 в примере 1, за исключением того, что фенил (3-трет-бутил-изоксазол-5-ил)-карбамат использовали вместо phenyl изоксазол-3-ил-карбамата с получением 1-(4-бензимидазол-1-ил-фенил)-3-(3-tеrt-бутил-изоксазол-5-ил)-мочевины.
1HNMR (DMSO-d6, 400MHz) δ: 10.21 (s, IH), 9.12 (s, IH), 8.51 (s, IH), 7.76-7.79 (m, 1H), 7.70-7.73 (d, 2H), 7.61-7.63 (d, 2H), 7.57-7.60 (m, 1H), 7.29-7.35 (m, 2H), 6.10 (s, 1H), 1.27 (s, 9H).
LC-MS (ESI): 376.1 (M+H)+.
Пример 134 Тест на образование соли
(1) от 100 мг до 1000 мг соединения настоящего изобретения взвесили в 50 мл одногорлую бутылку;
(2) 2-12 мл метанола добавили и перемешивали при комнатной температуре с образованием мутной дисперсионной системы;
(3) 3 эквивалента соответствующей кислоты растворили в небольшом количестве метанола и затем добавили по каплям в указанную систему, во время которой система стала прозрачной;
(4) Перемешивание продолжили при комнатной температуре в течение 2 часов;
(5) растворение:
1) Если осадок выпадал, этилацетат добавляли в 10-кратном объеме метанола, и перемешивали в течение 30 мин, осадок отфильтровали и промыли этилацетатом, затем высушили для получения продукта.
2) Если осадок не выпадал, реакционную систему сконцентрировали для получения неочищенного продукта, неочищенный продукт растворили в небольшом количестве метанола (полностью растворенного), затем этилацетат или метил трет-бутиловый эфир в 10-кратном объеме метанола добавили по каплям для осаждения твердых веществ, и затем перемешивали в течение 1-2 часов, после чего осадок отфильтровали и промыли этилацетатом и высушили для получения продукта.
3) Если осадок не выпадал, реакционную систему сконцентрировали для получения неочищенного продукта, неочищенный продукт растворили в небольшом количестве метанола (полностью растворенного), затем этилацетат или метил трет-бутиловый эфир в 10-кратном объеме метанола добавили по каплям; когда осадок не осаждался или твердые свойства были плохими (вязкими), тогда его концентрировали и затем закачивали в течение 30 мин до 1 часа масляным насосом и полученный сырой продукт медленно измельчали в гранулы этилацетатом или метил трет-бутиловым эфиром, и затем суспендировали в однородную дисперсионную систему; затем добавили метанол в объеме 1/20 этил ацетата, и перемешивали в течение 30 минут; осадок отфильтровали и затем промывали этилацетатом и высушили для получения продукта.
Пример 135 Испытание на растворимость и стабильность
(1) Тест на растворимость
Мелкий порошок испытуемого образца взвешивали в соответствующее количество воды при 25°С±2°С и встряхивали энергично в течение 30 секунд каждые 5 минут, чтобы он полностью растворился в течение 30 минут. Если не было обнаружено видимых частиц растворенного вещества, это считалось полностью растворенным. «Легко растворимый» означает, что 1 г испытуемого образца можно растворить в 1-10 мл воды; и «растворимый» означает, что 1 г испытуемого образца можно растворить в 10-30 мл воды; «Слегка растворимый» означает, что 1 г испытуемого образца можно растворить в 30-100 мл воды; и «умеренно растворимый» означает, что 1 г испытуемого образца можно растворить в 100-1000 мл воды; «Почти нерастворимый или нерастворимый» означает, что 1 г испытуемого образца не может быть полностью растворен в 10000 мл воды.
(2) Тест на стабильность
1) Высокотемпературный тест
Испытуемые образцы помещали в интегрированную испытательную камеру стабильности лекарственного средства (YSEI) при открытом действии при 60°С в течение 10 дней. Образцы были взяты на 5-й и 10-й дни и протестированы в соответствии с показателями теста на стабильность лекарств.
2)Тест высокой влажности
Испытуемые образцы помещали в интегрированную испытательную камеру стабильности лекарственного средства (YSEI) при открытом действии при температуре 25°С и относительной влажности 90±5% в течение 10 дней. Образцы были взяты на 5-й и 10-й дни и протестированы в соответствии с показателями теста на стабильность лекарств.
3) Интенсивный световой тест Испытуемые образцы помещали в интегрированную испытательную камеру стабильности лекарственного средства (YSEI) под воздействием света под 4500LX±500LX в течение 10 дней. Образцы были взяты на 5-й и 10-й дни и протестированы в соответствии с показателями теста на стабильность лекарств.
(3) Обнаружение ВЭЖХ
Соответствующее количество пробных образцов брали и растворяли в ацетонитриле-воде (1 : 1) и количественно разбавляли раствором примерно 0,3 мг/мл. 20 мкл раствора вводили в жидкостный хроматограф (Agilent 1260 Infinity), затем хроматограмму регистрировали и вычисляли в соответствии с методом процентной площади. Подвижная фаза представляет собой фосфатный буфер и водный раствор ацетонитрила, и затем проводят градиентное элюирование.
В таблице 1 приведены результаты испытаний на растворимость и стабильность солей настоящих Соединений.
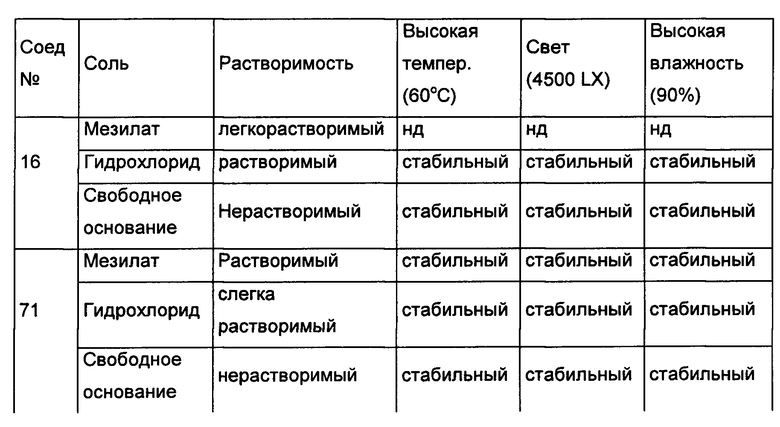
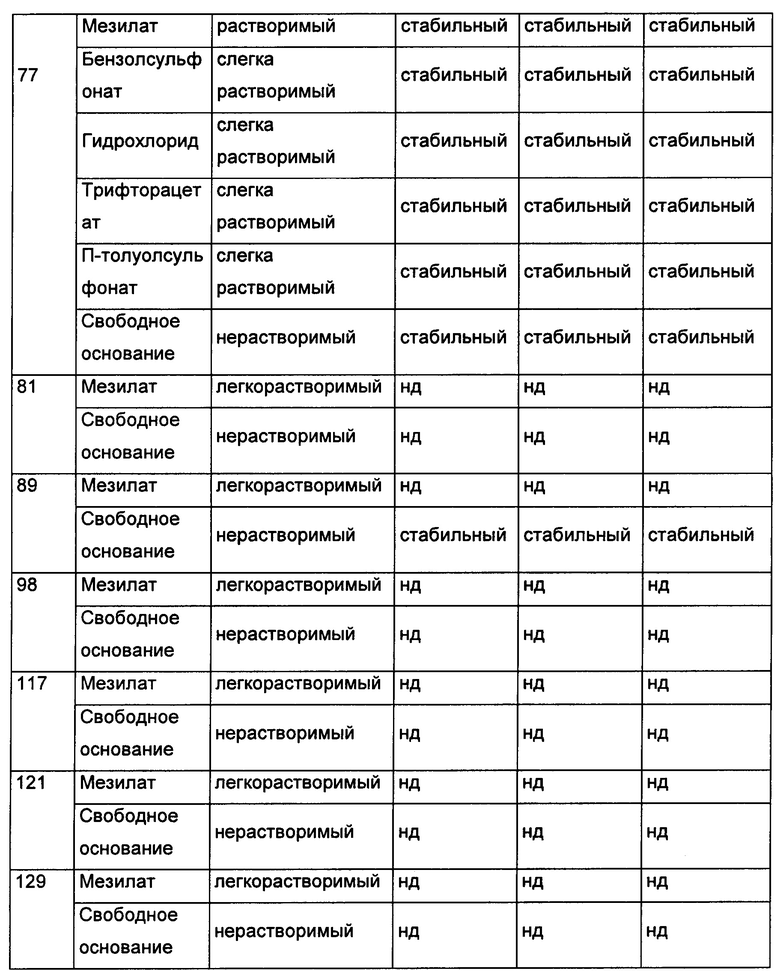
*нд: нет данных.
Тестовый Пример 1
Определение концентрации ингибирования in vitro 50% (GI 50) настоящих соединений в клеточных линиях, экспрессирующих дикий тип FLT3 и внутренний тандемный повтор (ITD)
Экспериментальные материалы и методы 1.
Линии клеток и клеточные культуры
Линия опухолевых клеток является эффективной клеточной моделью для изучения ингибирования роста опухолевых клеток или пролиферации in vitro. В настоящем изобретении была выбрана репрезентативная линия клеток опухоли для определения активности ингибирования роста клеток настоящими соединениями. Все используемые клеточные линии были получены от АТСС, DSMZ и клеточного банка Китайской академии наук. Условия и способы культивирования клеток соответствовали требованиям для каждой клеточной линии. Клетки субкультивировали in vitro не более чем на 3 пассажа каждый раз, и, при необходимости, можно было проводить однократную очистку и идентификацию клеточных линий.
В качестве среды для культивирования клеток использовали RPMI1640 (Gibco), MEM (Gibco),  (Gibco) и IMDM (Gibco). Полную среду получали с добавлением 5-20% фетальной телячьей сыворотки (Gibco), 1% двойных антибиотиков (10000 единиц / мл пенициллина и 10000 единиц / мл), 2 мМ глутамина или 1 мМ пирувата натрия, соответственно.
(Gibco) и IMDM (Gibco). Полную среду получали с добавлением 5-20% фетальной телячьей сыворотки (Gibco), 1% двойных антибиотиков (10000 единиц / мл пенициллина и 10000 единиц / мл), 2 мМ глутамина или 1 мМ пирувата натрия, соответственно.
(1) Линии клеток, экспрессирующие дикий тип FLT3 (WT) и внутренний тандемный повтор (ITD).
Линию клеток клеточной острой лимфобластной лейкемии млекопитающих MV4-11 (FLT3 ITD +/+ мутантный тип, АТСС) культивировали в полной среде, содержащей I × IMDM и 10% FBS. Линия клеток острой миелоидной лейкемии человека MOLM-13 (FLT3 ITD +/- мутантный тип, DSMZ), клеточная линия острой лимфобластной лейкемии человека RS4; 11 (FLT3 дикого типа, АТСС), клеточная линия хронической гранулоцитарной лейкемии человека K562 (отрицательная экспрессия FLT3, положительная экспрессия слитого белка BCR-ABL, АТСС), клеточная линия промиелоцитарной лейкемии человека HL-60 (FLT3 дикого типа, АТСС), человека В лимфобластическая лейкозная линия RAMOS (положительная экспрессия белковой тирозинкиназы Брутона, АТСС), клеточная линия острой миелоидной лейкемии человека Kasumi-1 (мутантный тип c-Kit N822K, АТСС), клеточная линия острой моноцитарной лейкемии человека U937 (дикого типа FLT3, АТСС), клеточная линия острой миелоидной лейкемии человека OCI-ОМЛ3 (NPMc + мутантный тип, АТСС), линия клеточной линии миелоидного лейкоза человека KG-1 (экспрессия слитого белка FGFR1OP2-FGFR1, АТСС) культивировали в полной среде, содержащей 1 × RPMI1640 и 10% FBS, соответственно.
(2) EGFR дикого типа (WT) или мутантные линии опухолевых клеток Линия клеток эпидермального рака человека А431 (EGFR WT, амплификация и высокая экспрессия, из Шанхайского клеточного банка Китайской академии наук и АТСС), клеточная линия рака легких человека NCI-H292 (EGFR WT, приобретенная у Шанхайского клеточного банка Китайской академии наук), клеточные линии NSCLC РС-9 и НСС827 (удаление EGFR экзона 19 Е746-А750, две клеточные линии чувствительны к первому поколению ингибиторов EGFR РТК), клеточная линия NSCLC NCI-H1975 (EGFR L858R / Т790М и устойчивость к ингибиторам EGFR первого поколения, АТСС) культивировали в полной среде 1 × RPMI1640 (добавка с 10% FBS) соответственно.
(3) Линии опухолевых клеток с амплификацией/сильной экспрессией или мутацией гена HER2 / ErbB2/
Линия клеток рака желудка человека NCI-N87 (HER2 / ErbB2 amp, АТСС), клеточная линия рака молочной железы человека НСС1954 (HER2 / ErbB2 amp, АТСС) и ZR-75-30 (HER2 / ErbB2 amp, АТСС), клеточная линия аденокарциномы человека AU565 (HER2 / ErbB2 amp, АТСС), клеточная линия карциномы легких легких человека NCI-H2170 (HER2 / ErbB2 amp, АТСС), линия клеток бронхоальвеолярной аденокарциномы человека NCI-H1781 (HER2 / ErbB2 Ins G776V, С-мутантный тип, АТСС) культивировали с полной средой 1 × RPMI1640 (10% FBS), соответственно. Линию клеток аденокарциномы легкого человека Calu-3 (HER2 / ErbB2 amp, АТСС) культивировали с 1 × MEM, 10% FBS, 1% двойным антителом, 1% NEAA (Gibco), 2 мМ глутамина и 1 мМ пируватом натрия. Линию клеток рака молочной железы человека SK-BR-3 (HER2 / ErbB2 amp, АТСС) культивировали с полной средой (10% FBS).
(4) Линии опухолевой клетки с амплификацией/ сверхэкспрессией гена тирозин-белкин-киназы с-МЕТ Линию клеток рака желудка человека MKN-45 (с-МЕТ amp, АТСС) и линию клеток NCI-H1993 немелкоклеточного рака легких (с-МЕТ amp, АТСС) культивировали в полной среде, содержащей 1 × RPMI1640 и 10% FBS, соответственно.
(5) Линии опухолевой клетки, экспрессирующие слитый белок ALK и матунтные по гену ALK
Клеточная линия немелкоклеточного рака легкого NCI-H2228 человека (экспрессирующая геном слияния EML4-ALK, АТСС) и клеточная линия клеточной лимфатической анапластической клетки человека Karpas-299 (экспрессирующая ген слияния NPM-ALK, АТСС) культивировали в полной среде, содержащей 1 × RPMI1640 (10% FBS), соответственно. Клеточная линия нейробластомы SH-SY5Y (экспрессирующий мутантный белок ALK F1174L, клеточный банк Шанхая, Китайская академия наук) культивировали в полной среде MEM (I × MEM, 10% FBS, 1% NEAA, 1 мМ пирувата натрия).
6) Линии опухолевых клеток с амплификацией гена FGFR1 / сильной экспрессией
Клеточная линия немелкоклеточного рака легкого человека NCI-H1581 (FGFR1 amp, АТСС) представляет собой линию опухолевых клеток с амплифицированным/сверхэкспрессированным FGFR1. Ее культивировали в полной среде, содержащей 1 × RPMI1640 с 10% FBS.
(7) Линии опухолевых клеток, экспрессирующих мутации RAS и RAF Клеточная линия крупноклеточного рака легких человека NCI-H460 (мутантный тип KRAS G61H, АТСС), клеточная линия немелкоклеточного рака легкого человека Н1299 (тип мутантного рака NRAS Q61K, АТСС), клеточная линия меланомы человека А375 (тип мутантов BRAF V600E, Шанхайский клеточный банк Китайской академии наук), клеточная линия рака толстой кишки НСТ116 (тип мутантов KRAS G13D, Шанхайский клеточный банк, Китайская академия наук) культивировали в полной среде 1 × RPMI1640 (10% FBS) соответственно. Клеточная линия немелкоклеточного рака легких человека А549 (мутантный тип KRAS G12S, Шанхайский клеточный банк, Китайская академия наук) культивировали в полной среде I × Ham с добавлением 10% FBS, 1% двойного антитела и 2 мМ глутамина.
2. Обработка лекарственным средством
Адгезионные клетки отщепляли 0,25% панкреатин-ЭДТА (Gibco). Суспензию клеток собрали центрифугированием. Супернатант отбрасывали и осадок клеток ресуспендировали и подсчитывали. Различные концентрации клеток (5 ~ 10 × 104 клеток / мл) получали в соответствии с циклом роста каждой клеточной линии и затем высевали на 96-луночный планшет (Corning), 100 мкл / лунку. Клетки инкубировали в течение ночи при 37°С, 5% CO2. Соединения добавили к клеткам на второй день с двумя лунками параллельно. Конечная концентрация органического растворителя не должна превышать 1%о. Клетки непрерывно инкубировали в течение от 3 до 5 дней и затем подвергали анализу МТТ.
Соединение настоящего изобретения и контрольное соединение растворили в ДМСО (Sigma) соответственно, с чистотой более 98%. Соединение хранили в концентрации 10 мМ при -20оС и разбавляли до 2- или 10-кратной серии перед использованием.
В настоящем изобретении АС220, селективный ингибитор FLT3, был выбран в качестве контрольного соединения и синтезирован нашей компанией в соответствии с методом подготовки оригинальной компании (Chao Q et al., Identification of N-(5-tert-butyl-isoxazol-3-yl)-N'-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,I-b][I,3]benzothia zol-2-yl]phenyl}urea dihydrochloride (AC220), a uniquely potent, selective, and efficacious FMS-like tyrosine 5 kinase-3 (FLT3) inhibitor. J Med Chem. 2009; 52 (23): 7808-16).
3. МТТ-детектирование и расчет GI50
Набор реагентов Dojindo ССК8 использовался в качестве теста МТТ. В качестве ридера микропланшетов использовался измеритель THERMO MULTISKAN FC.
Для прикрепленных клеток культуральную среду удалили и немедленно заменили свежей подготовленной средой, которая содержит 10% реагента CCK8 (100 мкл/лунку). Для суспензионных клеток CCK8 был непосредственно добавлен до 10% от конечной концентрации. Клетки непрерывно инкубировали в течение 1-4 ч. Когда темно-желтый цвет можно было наблюдать в лунках для контроля растворителей, измеряли значение оптической плотности при OD450 нм и скорость роста клеток рассчитывали по следующей формуле:
Скорость роста клеток (%)=100*(Т-Т0)/(С-Т0)
Т - оптическая плотность лунки с клетками, обработанная лекарственным средством; Т0 = оптическая плотность лунки с клетками до лечения лекарственным средством - оптическая плотность пустой контрольной лунки; С = оптическая плотность контрольной лунки группы растворителя - оптическая плотность пустой контрольной лунки. Значение концентрации 50% ингибирования роста клеток (GI50) рассчитывали в соответствии с концентрацией лекарственного средства и кривой роста клеток. Эксперимент был независимо повторен 1~3 раза и подвергался биологическому статистическому анализу.
4. Экспериментальные результаты
В таблице 2 суммированы результаты диапазонов концентрации GI50 для ингибирующей рост активности настоящих соединений на клеточных линиях, экспрессирующих мутантный тип FLT3-ITD и дикого тип лейкемии. Чем меньше значение GI50, тем сильнее активность ингибирования роста клеток. Соединение, обладающее сильной ингибирующей активностью роста (т.е. низким GI50) на FLTS-ITD-экспрессирующих клетках (MV4-11 и MOLM-13), но слабой или относительно неэффективной активностью ингибирования роста клеток на клетках с высокой экспрессией дикого типа FLT3 (например, RS4; 11) или клеток с низкой экспрессией или отсутствием экспрессии (например, K562) (т.е. высокая концентрация GI50), указывает, что этот тип соединения обладает высокой селективностью в отношении мутаций активации FLT3-ITD и имеет потенциальную ценность для разработки лечения связанных с FLT3-ITD заболеваний.
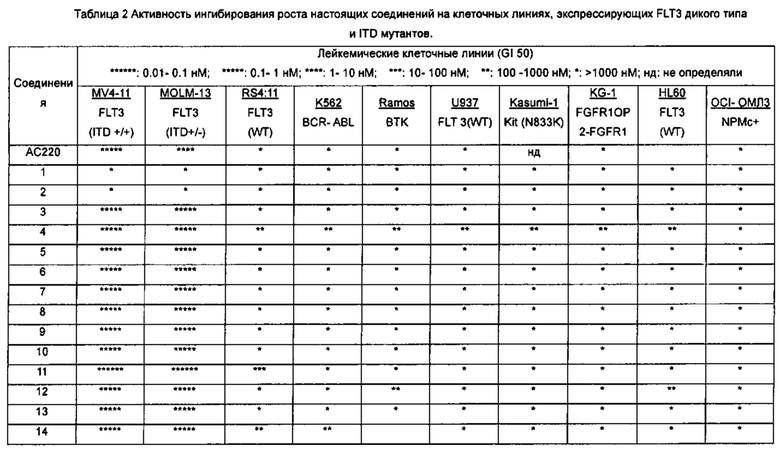
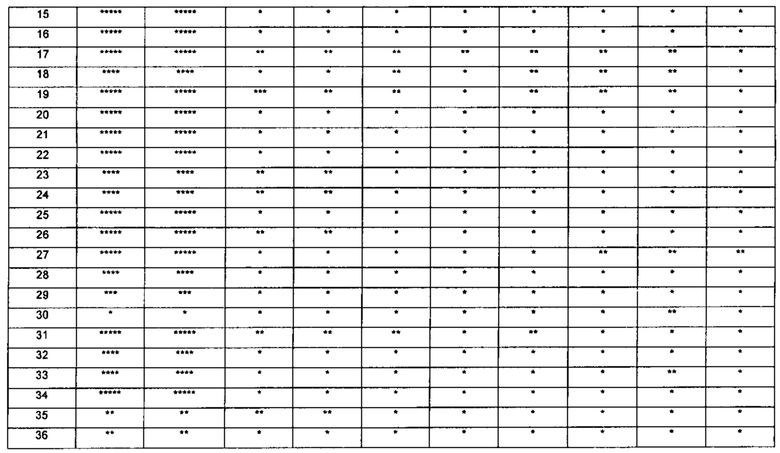
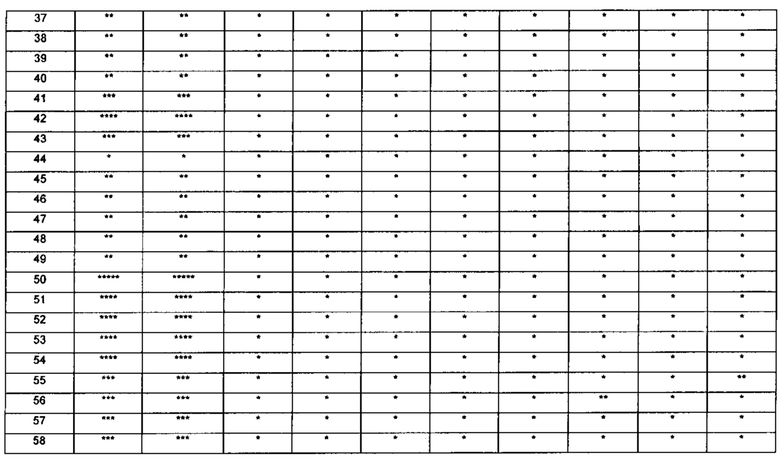
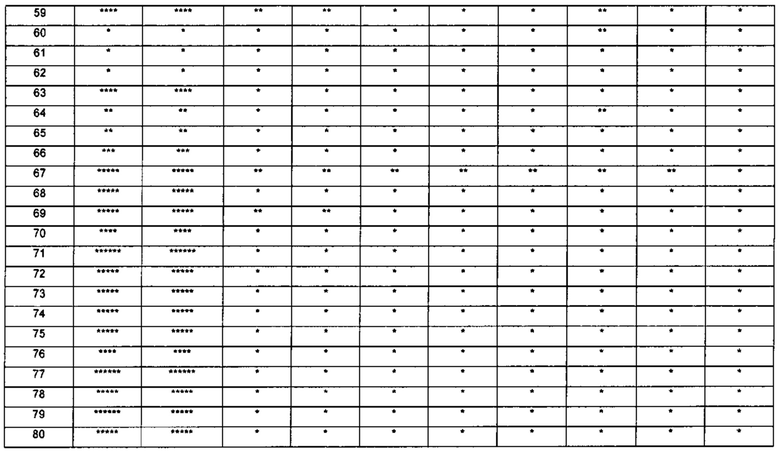
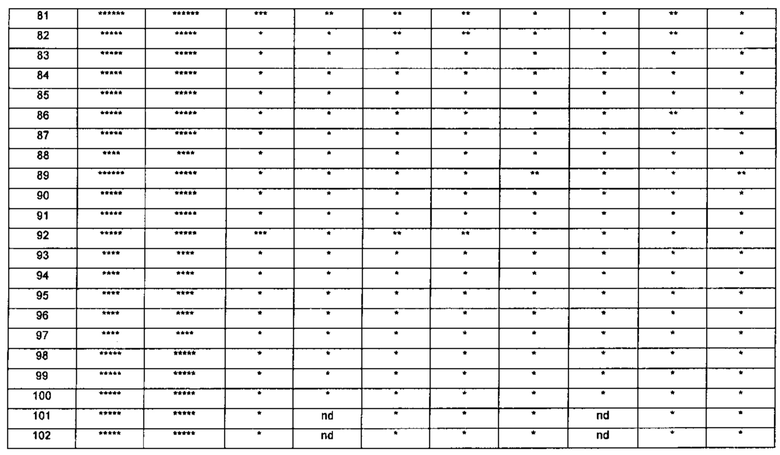
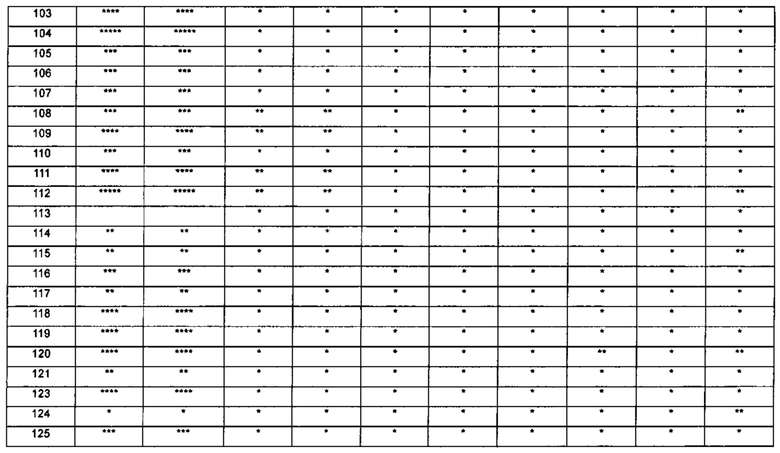
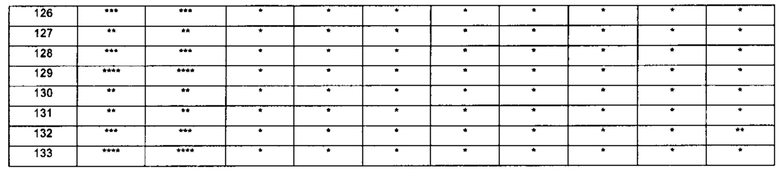
Из приведенной выше таблицы 2 настоящие соединения могут эффективно ингибировать рост (или индукцию апоптоза) FLT3-ITD-положительных экспрессирующих лейкемических клеток, MV4-11 и MOLM-13, а их значение GI50 может находиться на субнаномолярном уровне. По сравнению с контрольным соединением АС220 настоящие соединения (например, соединения 11, 71, 77, 79, 81 и 89) обладают более сильной ингибирующей активностью против роста клеток MV4-11 и MOLM-13. Кроме того, настоящие соединения обладают слабым или отсутствующим ингибирующим действием на рост клеток экспрессирующих FLT3 дикого типа (RS4; 11) или нормально экспрессирующих клеток (HL-60, Ramos, U937, Kasumi-1, KG-1 и OCI-ОМЛЗ) или не экспрессирующих клеток (K562), что указывает на то, что настоящие соединения обладают высокой селективностью и высокой активностью в отношении лейкемических клеток, экспрессирующих мутацию FLT3-ITD, и они являются своего рода новыми потенциальными селективными ингибиторами FLT3-ITD с высокой активностью.
Тестовый Пример 2
Определение 50% концентрации ингибирования роста in vitro (GI50) настоящих соединений в разных опухолевых клетках
Тест на ингибирование роста опухолевых клеток проводили путем выбора репрезентативных соединений (соединения 4, 11, 12, 16, 27, 68, 71, 77, 79, 89 и 116 настоящего изобретения). Начальная концентрация соединения составляла 5000 нМ, затем его серийно разбавляли в 2 раза или 10 раз до 0,01 нМ. Значение GI50 каждого испытуемого соединения рассчитывали в соответствии с методом анализа и расчета GI50. Эксперименты повторялись от 1 до 3 раз, и данные подвергались биологическому статистическому анализу. Результаты показаны в таблице 3.
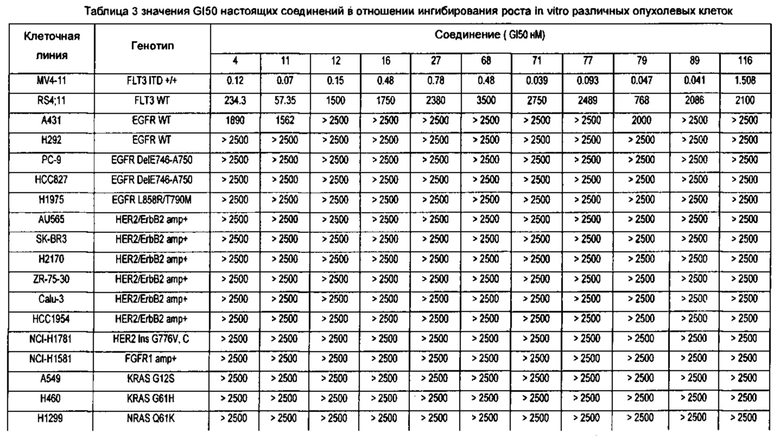
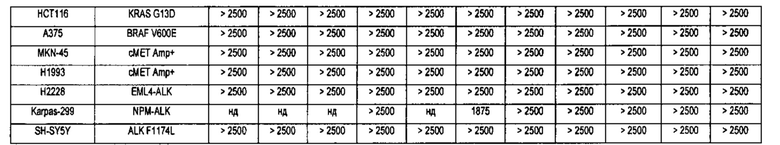
*нд: не определяли.
Результаты, приведенные в таблице 3, показывают, что соединения 12, 16, 27, 68, 71, 77, 89 и 116 настоящего изобретения обладают высокой ингибирующей активностью роста в отношении FLV3-ITD-положительно экспрессирующей клеточной линии MV4-11, Значение GI50 в субнаномолярном диапазоне (0,039-0,8 нМ), но они не проявляют явного ингибирующего эффекта на клетки, высоко экспрессирующие FLT3 дикого типа RS4; 11 в высоких концентрациях с величиной GI50 в микромолярном диапазоне. Испытуемые соединения 4, 11 и 79 оказывают сильное ингибирующее действие на рост FLV3-ITD-положительной клеточной линии MV4-11, также обладают определенной ингибирующей активностью роста на линию клеток, высоко экспрессирующей FLT3 дикого типа RS4; 11 с более высокой ингибирующей концентрацией и значение GI50 в диапазоне 50~800 нМ. Результаты в таблице 3 также показывают, что настоящие соединения (соединения 12, 16, 27, 68, 71, 77, 89 и 116) не оказывают значительного ингибирующего действия на рост опухолевых клеток с аномальными экспрессиями EGFR, HER2/ERBB2, FGFR1, RAS, BRAF, сМЕТ и ALK, соответственно. Значение GI50 находится в микромолярном диапазоне, который более чем в 1000 раз превышает ингибирующую рост концентрацию GI50 для клеток MV4-11, высокоэкспрессирующих ген FLT3-ITD.
Тестовый Пример 3
Эксперимент по ингибированию роста опухоли in vivo
Ксенотрансплатация у иммунодефицитных мышей является эффективной моделью для тестирования противоопухолевой активности in vivo соединений у животных. Bab/c иммунодефицитные мыши являются одним из наиболее часто используемых ксенотрансплантатов опухолевых клеток. Чтобы проверить, может ли данное соединение эффективно ингибировать рост лейкозных клеток in vivo с положительной экспрессией FLT3-ITD, для теста использовалась модель опухоли мышиной мыши Bab4C MV4-11. Клетки MV4-11 в лог-фазе собрали в 50 мл центрифужные пробирки (Corning), центрифугировали при 1700 об/мин в течение 3 минут и супернатант отбрасывали. Клетки суспендировали в 50 мл 1 × RPMI 1640 бессывороточной среды и центрифугировали, надосадочную жидкость отбрасывали и проводили ресуспензию. Клетки подсчитывали (Countess® Automated Cell Counting Apparatus, Invitrogen), готовили в 10×107 клеток/мл, помещали на лед и инокулировали подкожно 10×106 (0,2 мл) в правый бок 6-8 недельных самок иммунодефицитных мышей Bab/c (вес около 20 г) (приобретается у Shanghai Sippr-BK Experimental Animal Co., Ltd., Центр животных Шанхайского университета традиционной китайской медицины, утвержденный Этическим комитетом Шанхайского университета традиционной китайской медицины). Когда опухоль вырастала до 100-200 мм3, животные были случайным образом сгруппированы и отобраны и взвешены. Группа лечения состояла из 3-6 голых мышей в каждой группе. Препараты получали в разной концентрации молочной суспензии с раствором растворителя РТР (30% полиэтиленгликоль 400, 0,5% Tween-80, 2,5% пропиленгликоля). Животные получали перорально через желудочный зонд непрерывно при концентрации 0,1 мл/10 г массы тела с диапазоном доз 0,1-200 мг/кг (чистота испытуемого соединения была равна 99% или более, а доля одной примеси не более 0,1%). В контрольной группе от 3 до 6 голых мышей в каждой группе давали тот же объем (0,1 мл/10 г массы тела) раствора РТР растворителя, а опухоли измеряли от 3 до 4 раз в неделю. Когда средний объем опухоли в контрольной группе достиг 1500 мм3 или на 28-й день введения, эксперимент закончился, затем опухоль взвешивалась и подвергалась анализу молекулярной патологии.
Скорость ингибирования опухоли = (1 - относительный объем опухоли группы лечения TRTV / относительный объем опухоли контрольной группы CRTV) × 100%. Объем опухоли рассчитывали как V=I/2 a×b2, где а - длина, a b - ширина. Относительный объем опухоли (RTV) рассчитывали на основе измеренных результатов, а формула была RTV=Vt/V0, где V0 - измеренный объем опухоли при введении в разделенных клетках (т.е. d0), a Vt - объем опухоли при каждом измерении. Конкретные результаты показаны в таблице 4.
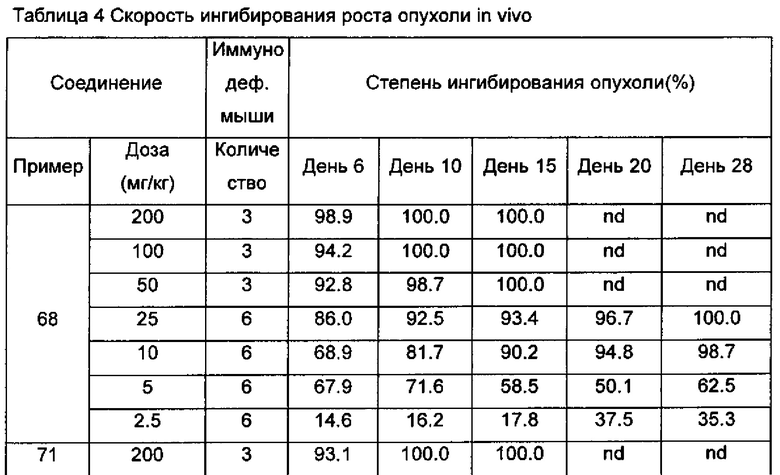
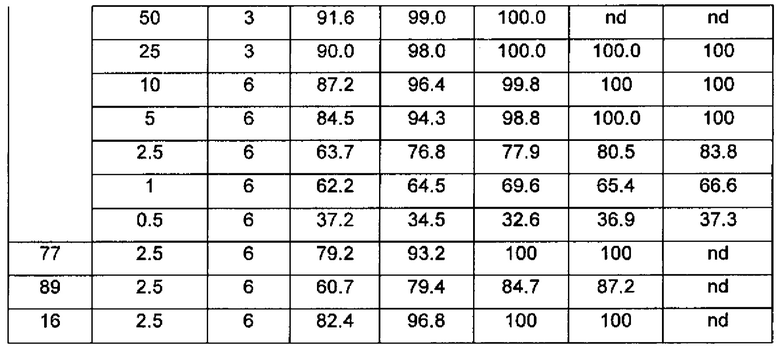
*nd: не определяли.
Результаты, приведенные в таблице 4 выше, показывают, что настоящие соединения проявляют концентрационную и зависящую от времени активность ингибирования роста против опухолей in vivo в клетках MV4-11. Когда доза соединения 68 составляет ≥25 мг/кг, скорость ингибирования роста опухоли составляет более 80% на 6-й день после введения, а на 28-й день опухоль полностью исчезает. Соединение 71 обладает более сильной ингибирующей активностью роста опухоли, чем активность соединения 68, и проявляет явную активность ингибирования роста опухоли в дозе от 1 до 2,5 мг/кг. Кроме того, по сравнению с контрольной группой с растворителем соединения 68 и 71 не оказывают существенного влияния на массу тела иммунодефицитных мышей при дозе ≤25 мг/кг в группе лечения; и когда доза ≥50 мг/кг, масса тела иммунодефицитных мышей уменьшается на 10-20%. Тестируемые соединения 77 и 16 демонстрируют более сильную ингибирующую активность роста опухоли in vivo в клетках MV4-11. Когда доза препарата составляет 2,5 мг/кг, опухоль полностью исчезает на 15-й день введения, и они не оказывают существенного влияния на массу тела голых мышей по сравнению с таковой в контрольной группе с растворителем.
Тестовый Пример 4
Тест hERG
Тест цельноклеточного hERG проводили с использованием соединения 77 в качестве типичного примера, в качестве положительного контроля использовали 30 мкМ хинидина (Sigma).
Методика: HERG-калий-ионный канал HEK-293 (Krius Biology) был сверхэкспрессирован и культивирован при 37°C, 5%-ном CO2-инкубаторе. Культуральной средой был DMEM, дополненный 15% эмбриональной бычьей сывороткой и 1% пенициллин-стрептомицином. Стабильно трансфицированные клетки инокулировали на слайды с плотностью клеток менее 50% и культивировали в течение ночи. Экспериментальные клетки переносили в ванну с емкостью приблизительно 1 мл, встроенную в платформу с инвертированным микроскопом (Diaphot, Nikon), а внеклеточную жидкость перфузировали с перфузией 2,7 мл/мин. Эксперимент может быть начат после стабилизации в течение 5 минут и осаждения клеток. Мембранный ток регистрировался патч-кламп усилителем HEKAEPC-10 и системой сбора PATCHMASTER (HEKA Instruments Inc., D-67466 Lambrecht / Pfalz Germany). Все эксперименты были выполнены при комнатной температуре (22-24°C). Во время эксперимента использовалось устройство для управления микроэлектродом Р-97 (Sutter Instrument Company, One Digital Drive, Novato, CA 94949), предназначенное для выпрямления электродов (BF150-110-10). Внутренний диаметр электрода составлял 1-1,5 мм, а входное сопротивление после заполнения жидкостью составляло 2-4 MQ. Электрофизиологическая стимуляция калийного канала hERG: во-первых, напряжение мембраны было установлено при -80 мВ, и клеткам давали непрерывную стимуляцию напряжения 2 с +20 мВ для активации канала ионов калия HERG и реполяризации до -50 мВ в течение 5 с, для генерации внешнего хвостового тока. Частота стимуляции составляла каждые 15 секунд. Текущее значение было пиковым значением следового тока. Измеряя максимальное значение тока группы лечения и контрольной группы, было рассчитано отношение максимального значения тока группы лечения к максимальному значению тока контрольной группы (среднее ± SE) для оценки влияния теста соединений на канал ионов калия HERG при контрольной концентрации. Статистика и анализ экспериментальных данных были выполнены на Origin 8.5 (OriginLab Corporation, Northampton, MA). Результаты соотношений соединения 77, блокирующего канал ионов калия HERG в различных концентрациях, показаны в таблице 5.
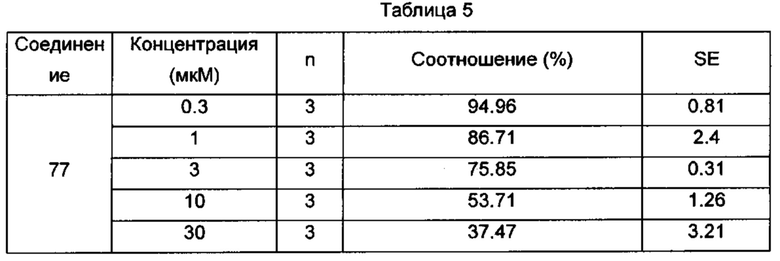
Результаты показывают, что соединение 77 настоящего изобретения оказывает определенное блокирующее действие на канал ионов калия HERG в диапазоне тестовых концентраций.
Тестовый Пример 5
Стабильность настоящих соединений в гепатоцитах крыс
Эксперимент по стабильности гепатоцитов крыс проводили с использованием соединений примера 77 в качестве типичного примера.
Методика: Смешанный реакционный раствор William's Medium Е гепатоцитов крысы (2*106 клеток/мл, 25 BD Gentest) помещали в инкубатор с диоксидом углерода и определенный объем испытуемого вещества или эталонного вещества добавляли в реакционную систему для инициирования реакции. В конечной реакционной системе концентрации тестируемого вещества или тестостерона с положительным контролем были равны 1 мкМ. Образцы брали через 0, 5, 15, 30, 45, 60 и 120 минут после реакции в новую центрифужную пробирку (по 3 порции параллельно в каждый момент времени) и 3-кратный объем ледяного раствора метанола, содержащего внутренний стандарт добавили для немедленного прекращения реакции, затем после центрифугирования в течение 5 мин при 15000 об/мин для осаждения белка 100 мкл надосадочной жидкости выливали в пробирку для автосамплера для анализа LC-MS/MS (Waters Corp. ACQUITY UPLC, USA Applied Biosystems Inc., масс-спектрометр API 400). Было рассчитано отношение средней площади пика соединения 77 к средней площади пика внутреннего стандарта в каждый момент времени и рассчитано процентное отношение этого отношения к таковому в момент времени 0, и затем изменение в этом проценте использовали для обозначения метаболической стабильности соединения 77. Система обработки данных представляла собой программу Analyst 1.5 от Applied Biosystems Inc., США.
Результаты показывают, что соединение 77 имеет период полураспада более 120 мин в гепатоцитах крыс.
Тестовый Пример 6
Исследование метаболической стабильности соединений настоящего изобретения в микросомах печени крыс
Эксперимент по стабильности соединения в микросомах печени крысы проводили с использованием соединений примера 77 в качестве типичного примера.
Методика. Смешанный реакционный раствор, содержащий восстановительный кофермент II (1 мг/мл, Roche 10621706001), микросомы печени крысы (0,5 мг/мл, BD Gentest), фосфатный буфер (0,1 М) и ионизированную воду, получали в соответствии с определенным соотношением, и затем реакционный раствор помещали в водяную баню с температурой 37°C и предварительно нагревали в течение 2 минут. Некоторое количество соединения 77 или эталонного вещества добавляли к приготовленному смешанному реакционному раствору для начала реакции. В конечной реакционной системе концентрация соединения 77 или положительного контрольного мидазолама (Национальные институты по контролю за продуктами и лекарствами) составляла 2 мкМ, а воду использовали вместо восстановленного коэнзима II как отрицательный контроль. Образцы (50 мкл) отбирали из реакционного раствора через 0, 10, 15, 30, 45 и 60 мин после реакции в новую центрифужную пробирку, две порции получали параллельно в каждый момент времени, затем сразу же добавляли в 3-кратный объем ледяного раствора метанола, содержащего внутренний стандарт для прекращения реакции, затем после центрифугирования в течение 5 мин при 15000 об/мин для осаждения белка, 100 мкл надосадочной жидкости отбирали в автосамплеровую пробирку для анализа LC-MS/MS (Waters Corp. ACQUITY UPLC, USA Applied Biosystems Inc., масс-спектрометр API 400). Было рассчитано отношение средней площади пика соединения 77 к средней площади пика внутреннего стандарта в каждый момент времени и рассчитано процентное отношение этого отношения к таковому в момент времени 0, и затем изменение в этом проценте использовали для обозначения метаболической стабильности соединения 77. Система обработки данных была программой Analyst 1.5 от Applied Biosystems Inc., США.
Результаты показывают, что подходящая кривая устойчивости соединения 77 в микросоме печени крыс составляла y=-0,0031x-2,3407 (R2=0,9454), константа скорости элиминации составляла 0,0031, а период полувыведения составлял 224 мин.
Тестовый пример 7
Испытание скорости связывания данного соединения с белками плазмы крыс
Скорость связывания соединения 77 в соответствии с изобретением с белками плазмы при концентрации в плазме крысы 2 мкМ оценивали методом диализа.
Методика: исходный раствор ДМСО соединения 77 (10 мМ) или положительный контроль пропранолола (USP) добавили к 500 мкл пустой плазмы. Конечная концентрация тестируемого вещества в плазме составляла 2 мкМ, а конечная концентрация положительного контроля составляла 1 мкМ в трех повторениях. 300 мкл приготовленных образцов плазмы, добавленных в плазменную камеру, и 500 мкл буфера для диализа, добавленного в буферную камеру (фосфатный буферный раствор PBS содержал 100 мМ фосфата натрия и 150 мМ хлорида натрия, РН 7.2), герметизировали и инкубировали в течение 4 ч при встряхивании при 100 об/мин, 37°С. После инкубации из плазменной камеры и буферной камеры брали 50 мкл образца и 50 мкл PBS добавляли к образцам плазменной камеры и 50 мкл пустой плазмы добавляли к образцам буферной камеры. Сразу же 3-кратный раствор метанола, содержащий внутренний стандарт, добавили, чтобы осадить белки плазмы. После центрифугирования супернатант брали для анализа LC/MS/MS. Скорость связывания белков плазмы рассчитывали по следующей формуле: % свободного состояния = (отношение площади пика в буферной камере / отношение площади пика в плазменной камере) × %, % связанного состояния = 100% - % свободного состояния. Скорости связывания белков плазмы соединения 77 и пропранолола у крыс показаны в таблице 6.
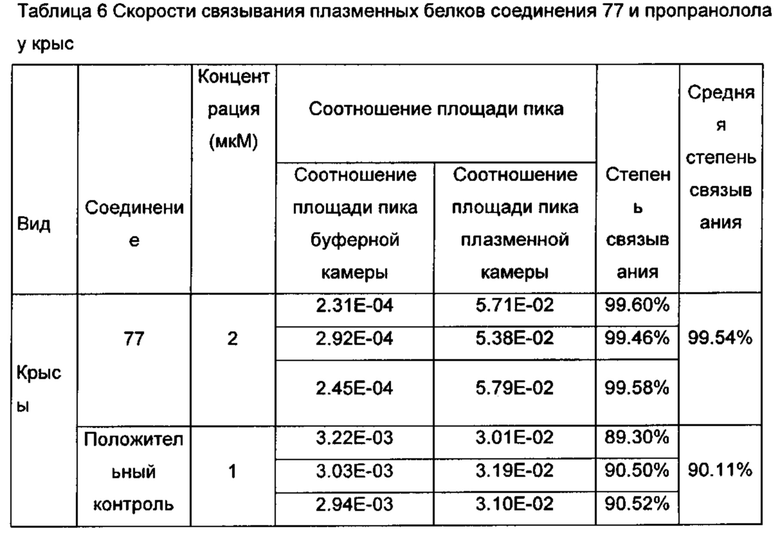
Результаты показывают, что скорость связывания соединения 77 с белком плазмы крысы составляет 99,54%, а скорость связывания положительного контрольного пропранолола с белком плазмы крысы составляет 90,11%.
Тестовый Пример 8
Определение ингибирующей активности Фермента Р450 соединениями изобретения
Восемь реакций ингибирования фермента цитохрома р450 на микросомах печени человека проводили с использованием соединений примера 77 в качестве типичного примера.
Основные реакционные системы и процессы: в водяной бане 37°С, тестируемые соединения (7 концентраций, 0, 0,25, 0,5, 5, 10, 25 и 50 мкМ, конечная концентрация метанола составляла 1% в системе), микросомы печени человека (BD Gentest) (0,5 мг/мл) и восстановленный коэнзим II (Roche 10621706001) (1 мг/мл) и соответствующий субстрат ферментативного зонда цитохрома р450 были совместно инкубированы в течение 5-20 минут. Среди них: (1) зондовый субстрат для CYP1A2 был фенацетином (20 пМ) (Sigma), а селективный ингибитор альфа-нафтофлавона (2 мкМ) (Sigma) был для параллельного обнаружения в качестве положительного контроля, и они инкубировались в течение 20 минут, (2) Зондовой субстрат для CYP2b6 был Бупропион (40 мкМ) (Sigma), а селективный ингибитор тиклопидин (10 мкМ) (Sigma) был для параллельного обнаружения в качестве положительного контроля, и они инкубировались в течение 20 минут. (3) Зондовой субстрат CYP2C8 был амодиаквин (1 мкМ) (Sigma), а селективный ингибитор монтелукаст (10 мкМ) (Sigma) был для параллельного обнаружения в качестве положительного контроля, и они инкубировались в течение 20 минут. (4) Зондовой субстрат для CYP2C9 был диклофенак (2,5 мкМ) (Sigma), а селективный ингибитор сульфафеназол (10 мкМ) (Sigma) был для параллельного обнаружения в качестве положительного контроля, и они инкубировались в течение 20 минут. (5) Зондовой субстрат для CYP2C19 был мефенитоин (40 мкМ) (Sigma), а селективный ингибитор тиклопидин (10 мкМ) (Sigma) был для параллельного обнаружения в качестве положительного контроля, и они инкубировались в течение 20 минут. (6) Зондовой субстрат для CYP2D6 был Декстрометорфан (5 мкМ) (Sigma), а селективный ингибитор Хинидин (10 мкМ) (Sigma) был для параллельного обнаружения в качестве положительного контроля, и они инкубировались в течение 20 минут. (7) Зондовые субстраты для CYP3A4-T и CYP3A4-M были тестостероном (5 мкМ) (сигма) и мидазоламом (5 мкМ) (Sigma) соответственно, а селективным ингибитором был кетоконазол (1 мкМ) (Sigma), они были обнаружены параллельно в качестве положительного контроля и инкубированы в течение 5 минут. Реакционную систему гасили добавлением метанола, содержащего соответствующий внутренний стандарт. Внутренним стандартным соединением был толбутамид (Sigma). Концентрацию соответствующих метаболитов определяли с помощью ЖХ/МС/МС. Для анализа данных уменьшение площади пика метаболита образца после добавления соединения 77 относительно площади пика метаболита контрольного образца растворителя использовалось для расчета IC50 (концентрация испытуемого вещества при 50% ингибировании), Проценты пиковых областей метаболитов зондов CYP1A2, CYP2B6, CYP2C8, CYP2C9, 15CYP2C19, CYP2D6, CYP3A4-T и CYP3A4-M после добавления различных концентраций соединения 77 и селективных ингибиторов по сравнению с областями пика контроля растворителя показаны в Таблице 7.
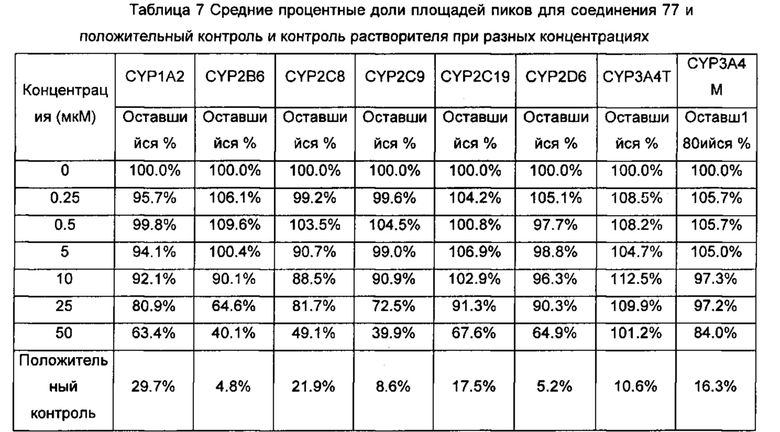
Как показано в таблице 7, пиковые области метаболитов в положительном контроле восьми ферментов значительно ниже, чем в контроле растворителя, что указывает на ингибирование активности фермента. Когда концентрация соединения 77 составляет 50 мкМ, значительного ингибирующего эффекта не наблюдается на CYP3A4-T и CYP3A4-M; полуингибирующие концентрации CYP1A2, CYP2C19 и CYP2D6 (IC50) составляют >50 мкМ, a IC50 CYP2C8 больше или близко к 50 мкМ. Полуингибирующая концентрация (IC50) CYP2B6 составляет 38,98 мкМ, тогда как половина ингибирующей концентрации (IC50) CYP2C9 составляет 41,56 мкМ.
Тестовый Пример 9
Исследование двунаправленного трансмембранного транспорта и свойств тока P-gp соединений в моделях монослоев Сасо-2
Эксперимент переноса клеток Сасо-2 проводили с использованием соединений примера 77 в качестве типичного примера.
Основная методика: клетки Сасо-2 (АТСС, НТВ-37) культивировали в культуральной среде с высоким содержанием глюкозы (Hyclone), содержащей 10% сыворотки плода теленка (GIBCO) и пенициллина-стрептомицина (по 100 единиц/мл каждый) (Sigma). Клетки культивировали при 37°С в инкубаторе, содержащем 5% CO2. В эксперименте переноса клетки Сасо-2 инокулировали на фильтр 12-луночного планшета Transwell с плотностью 2×105 клеток/лунку (Coming Costar, каталог №3401, 1,12 см2, 0,4 мкм размер пор). После инокуляции клетки Сасо-2 формируют интактные клеточные монослои через 21 день, включая экспрессию транспортера тока Р-gp и формирование резистентности трансепителиальных клеток (TEER). В течение периода культуральная среда заменялась один раз в день. Объем культуральной среды на трансвельберной мембране Apical Side (А) и Basolateral Side (В) составлял 0,5 мл и 1,5 мл, соответственно. Перед экспериментом по транспортировке (день 21) предварительно нагретый буфер HBSS (137 мМ NaCl, 4,17 мМ NaHCO3, 0,34 мМ Na2HPO4, 5,37 мМ KCl, 0,44 мМ KH2PO4, 1,26 мМ CaCl2, 0,49 мМ MgCl2, 0,41 мМ MgSO4, 5,55 Мм D-Глюкоза, 10 мМ HEPES, pH 7,4) использовали сначала для промывки монослоев Сасо-2 клеток три раза и помещали их при 37°С для инкубации в течение 30 минут, затем значения TEER определяли с использованием клеточного резистометра (Millicell-ERS2) для подтверждают целостности и компактности монослоев клеток. Когда буферный раствор HBSS на трансмембранной стороне А или стороне В был заменен испытуемым соединением, начался транспортный эксперимент. Наконец, после инкубации при 37°С в течение 2 ч, эксперимент переноса прекращали и 50 мкл раствора образца брали с обеих сторон мембраны фильтра Transwell отдельно и добавляли к 100 мкл ацетонитрила, содержащего внутреннее стандартное соединение, и хорошо перемешивали, затем центрифугировали при 13000 об/мин, 4°С в течение 10 мин и, наконец, 10 мкл (или удельный объем) супернатанта для анализа ЖКТ-МС/МС. Для каждого соединения эксперименты по транспортировке в направлениях А-В и В-А имели три повтора, то есть n=3.
Анализ образцов LC-MS/MS: высокоэффективной жидкостная хроматография (ВЭЖХ), используемая в этом исследовании, состояла из двух насосов (Shimadzu LC-20AD) и автосамплера (Shimadzu SIL-20AC). Система ВЭЖХ была соединена последовательно с тройным квадрупольным масс-спектрометром API 4000 (Applied Biosystems). Тройной квадрупольный масс-спектрометр API 4000 был оснащен источником ионизации электрораспылением (ESI). Ультраулучший азот использовался в качестве занавеса газа, газа GAS1, GAS2 и столкновений, при скорости потока 20 л/ч, 55 л/ч, 65 л/ч и 6 л/час, соответственно. Температура распыления источника ионов была установлена равной 500°С. Данные собрали с использованием программного обеспечения Analyst 1.5.
Анализ данных: Коэффициент кажущейся проницаемости (Рарр) может быть рассчитан по скорости, с которой соединение проникает в монослой клеток Сасо-2.
Его значение связано с поглощением соединения in vivo. После того, как концентрации со стороны А и В определяли с помощью ЖХ-МС/МС, значение Рарр в направлении А-В или В-А рассчитывали по следующей формуле:
Рарр (А→В) или Рарр (В→А)=(dQ/dt)/(А*С0)=(C2h*V)/(t*πr2*С0)
В приведенной выше формуле dQ/dt представляет собой скорость проницаемости, то есть количество соединения, которое проникает в течение dt времени, С0 представляет собой начальную концентрацию лекарственного средства на стороне введения, а A - площадь поверхности монослоя клетки, т.е. площадь мембраны.
После получения значений Рарр (А→В) и Рарр (В→А) соединения отношение тока (ER) соединения может быть рассчитано по следующей формуле: Соотношение тока (ER)=Рарр (В-А)/Рарр (А-В).
Когда отношение тока было >2, соединение можно рассматривать как субстрат транспортера оттока.
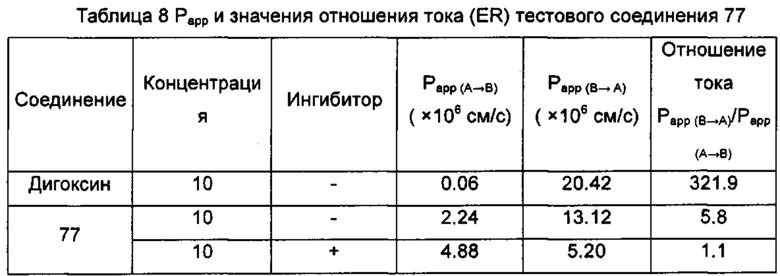
Выводы: (1) Контрольное соединение Дигоксин демонстрирует более низкую трансмембранную проницаемость в модели монослоя Сасо-2 (значение Рарр (А→В) составляет менее 1⋅10-6 см/с) в качестве субстрата тока Р-gp, его значение ER равно 321,9, что подтверждает точность настоящего исследования. (2) Соединение 77 показывает умеренную трансмембранную проницаемость в модели монослоя Сасо-2 (значение Рарр (А→В) находится в диапазоне 1-10×10-6 см/с), а субстрат для транспортера тока Р-gp (значение ER составляет 5.8 без добавления ингибитора Р-gp GF120918, тогда как оно равно 1,1 после добавления ингибитора Р-gp GF120918).
Тестовый Пример 10
Изучение Фармакокинетики in vivo настоящих соединений у крыс
Фармакокинетические эксперименты на крысах проводили с использованием соединений примера 77 в качестве типичного примера.
Методика: 6 крыс самцов SD (~200 г, приобретенных у Shanghai Sippr-BK Experimental Animal Co., Ltd) были отобраны и разделены на две группы в соответствии с экспериментальной таблицей дизайна исследования (таблица 9). Животным вводили внутривенно и перорально. Дозирование проводили один раз в день лечения, а дозы составляли 5 мг/кг и 10 мг/кг соответственно.
Способ приготовления лекарственного средства: необходимое количество испытуемого образца добавили к определенному объему 40% (HP-β-CD) водного раствора, и затем перемешивали при нагревании (температура 25-45°С) нерастворимого растворения, затем тот же объем деионизированной воды добавили и довели до pH 5 с помощью NaOH, для получения 20% чистого водного раствора НР-β-CD.
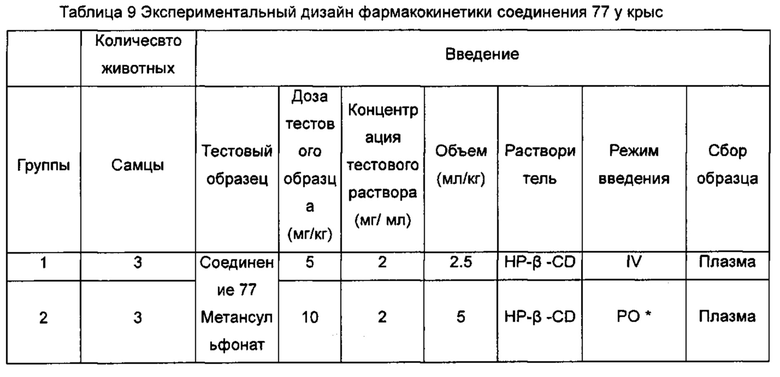
* До перорального приема все животные голодали в течение ночи (10-14 часов) и кормили 4 часа после дозирования.
Для группы внутривенного введения образцы крови брали из яремных вен перед введением и через 2 мин, 5 мин, 15 мин, 30 мин, 1 ч, 2 ч, 4 ч, 6 ч, 8 ч и 24 ч после введения; для группы перорального введения образцы крови брали из яремных вен перед введением и через 5 мин, 30 мин, 1 ч, 2 ч, 4 ч, 6 ч, 8 ч, 10 ч и 24 ч. Собранные образцы крови центрифугировали для отделения плазмы в течение 30 мин (условия центрифугирования: 8000 об/мин, 6 минут, 2-8°С). Образцы плазмы хранили при -80°С в морозильной камере перед анализом. Образцы плазмы использовались для разработки метода LC-MS/MS и тестирования образцов.
Фармакокинетические параметры (AUC (0-t), AUC (0-∞), Т1/2, MRT (0-∞), Cmax, Tmax и F) рассчитывались с использованием WinNonlin Professional v5.2 (Фарсигл США). Кроме того, биодоступность (F) рассчитывалась по следующей формуле.
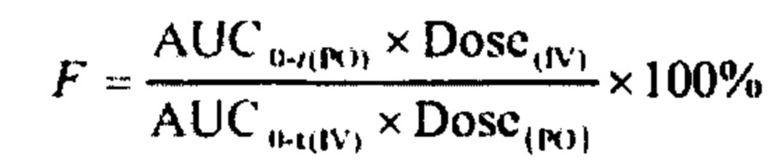
Фармакокинетические параметры после введения крыс с помощью метансульфоната соединения 77 показаны в таблице 10 и таблице 11.
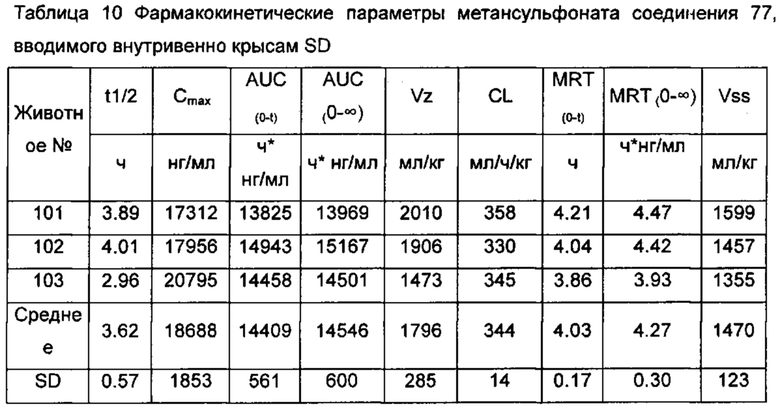
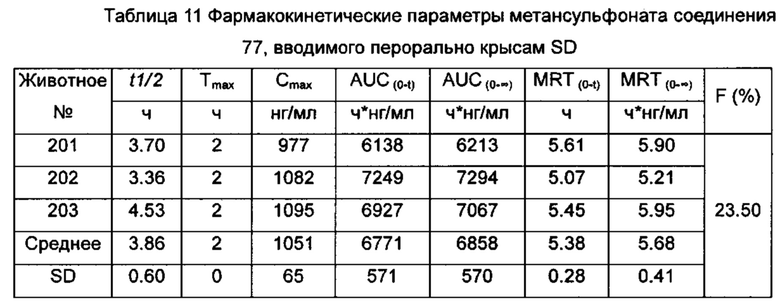
Результаты показывают, что после того, как крысам SD вводят внутривенно метансульфонатом соединения 77 при 5 мг/кг, Cmax составляет 18688 нг/мл, а AUC (0-t) составляет 14409 ч*нг/мл; после того, как крысы SD вводят перорально метансульфонат соединения 77 при 10 мг/кг, Cmax составляет 1051 нг/мл, AUC (0-t) составляет 6771 ч*нг/мл, а биодоступность составляет 23,50%.
Тестовый Пример 11
Влияние настоящих соединений на клетки крови
Исследование долгосрочной оральной токсичности у мышей проводили с примерами изобретения 77 в качестве типичного примера.
Методика: 6-недельные мыши ICR, приобретенные у Shanghai Sippr-BK Experimental Animal Co., Ltd., были отобраны половина самцов и половина самок, и животных случайным образом разделяли на 6 мышей каждой группы. Испытуемые и контрольные группы получали перорально один раз в день в течение 40 последовательных дней, соответственно. Вес тела и физиологические и патологические особенности мышей наблюдались и регистрировались каждый день. Дозы препарата испытуемой группы составляли 500 мг/кг, 350 мг/кг и 250 мг/кг метансульфоната соединения 77 в 20%-ном прозрачном водном растворе HP-β-CD соответственно (метод приготовления был одинаковым как тест примера 10), а контрольная группа представляет собой 20%-ный водный раствор НР-β-CD.
Выводы: после того, как метансульфонат соединения 77 непрерывно вводится на 40-й день, смертность для трех доз препарата при 500 мг/кг, 350 мг/кг и 250 мг/кг составляет 66,7%, 16,7% и 0%, соответственно. По сравнению с контрольной группой их вес восстановлен на 34,7%, 23,1% и 8,2% соответственно. Анализ параметров клеток крови показывает, что метансульфонат соединения 77 оказывает влияние на ретикулоциты (уменьшены на 10-30%), но не оказывает существенного влияния на количество других клеток крови.
| название | год | авторы | номер документа |
|---|---|---|---|
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2015 |
|
RU2667498C2 |
| ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛА, СОДЕРЖАЩИЕ ИХ КОМПОЗИЦИИ, ИХ ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ | 2004 |
|
RU2346938C2 |
| ПИРАЗОЛО[3,4-С]ПИРИДИНЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2012 |
|
RU2638116C2 |
| ГИДРОКСИПУРИНОВЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2016 |
|
RU2685417C1 |
| СПОСОБ ПОДАВЛЕНИЯ РОСТА ОПУХОЛЕВЫХ КЛЕТОК, ОПОСРЕДОВАННОГО КИНАЗОЙ RAF, ГЕТЕРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ МОЧЕВИНЫ (ВАРИАНТЫ), ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ (ВАРИАНТЫ) | 1998 |
|
RU2232015C2 |
| Соединения 1-циано-пирролидинов в качестве ингибиторов USP30 | 2016 |
|
RU2717238C2 |
| ПОЛИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И СПОСОБЫ ЦЕЛЕНАПРАВЛЕННОЙ ДЕГРАДАЦИИ ПОЛИПЕПТИДОВ БЫСТРО УСКОРЕННОЙ ФИБРОСАРКОМЫ | 2019 |
|
RU2830173C2 |
| ЗАМЕЩЕННЫЙ 2-АМИНОПИРИДИН В КАЧЕСТВЕ ИНГИБИТОРА ПРОТЕИНКИНАЗЫ | 2014 |
|
RU2671212C2 |
| ПРОИЗВОДНОЕ АМИНОПИРАЗОЛА | 2010 |
|
RU2580543C9 |
| НОВЫЕ ПИРРОЛЬНЫЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2014 |
|
RU2607788C2 |
Изобретение относится к соединению общей формулы (I) или его фармацевтически приемлемым солям, где группа
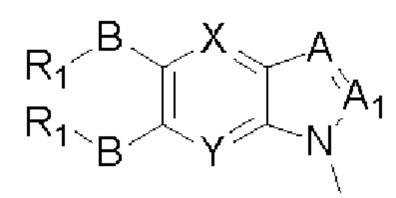 выбрана из следующей структуры:
выбрана из следующей структуры: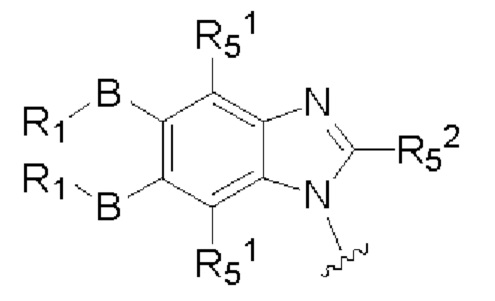 , каждый из R51 и R52 независимо выбран из группы, состоящей из водорода; W и Z - каждый независимо выбран из N и C-BR1; при отсутствии R1, В являются одинаковыми или разными и каждый независимо выбран из группы, состоящей из водорода, галогена, С1-С6 алкила, причем указанный С1-С6 алкил возможно дополнительно замещен одной или более группами Q; в присутствии R1, В являются одинаковыми или разными и каждый независимо выбран из группы, состоящей из -О- и -NR4-; и R1 являются одинаковыми или разными и каждый независимо выбран из группы, состоящей из водорода, С1-С6 алкила, гетероарила, который представляет собой гетероароматическую систему, содержащую от 1 до 2 гетероатомов, от 5 до 6 кольцевых атомов, где гетероатом выбран из серы и азота, -RuORx, -RuC(О)ORx, -RuN(Ry)(Rz), -C(O)N(Ry)(Rz); указанные алкил и гетероарил возможно дополнительно замещены одной, двумя или тремя группами, выбранными из группы, состоящей из галогена, циано, гидрокси, С1-С6 алкила, С1-С6 алкокси, С1-С6 гидроксиалкокси, -C(O)NHCH3, гетероциклила, который представляет собой насыщенную моноциклическую углеводородную группу, содержащую от 4 до 6 кольцевых атомов, 1 или 2 из которых могут быть заменены кислородом и/или азотом, галофенила, С3-С6 циклоалкил-тетразолила; R4 выбран из группы, состоящей из С1-С6 алкила, или R4 и R1 вместе с атомом азота, к которому они присоединены, образуют пиперазинил, который дополнительно замещен одной группой, выбранной из группы, состоящей из С1-С6 алкила; R2 представляет собой водород, G выбран из группы:
, каждый из R51 и R52 независимо выбран из группы, состоящей из водорода; W и Z - каждый независимо выбран из N и C-BR1; при отсутствии R1, В являются одинаковыми или разными и каждый независимо выбран из группы, состоящей из водорода, галогена, С1-С6 алкила, причем указанный С1-С6 алкил возможно дополнительно замещен одной или более группами Q; в присутствии R1, В являются одинаковыми или разными и каждый независимо выбран из группы, состоящей из -О- и -NR4-; и R1 являются одинаковыми или разными и каждый независимо выбран из группы, состоящей из водорода, С1-С6 алкила, гетероарила, который представляет собой гетероароматическую систему, содержащую от 1 до 2 гетероатомов, от 5 до 6 кольцевых атомов, где гетероатом выбран из серы и азота, -RuORx, -RuC(О)ORx, -RuN(Ry)(Rz), -C(O)N(Ry)(Rz); указанные алкил и гетероарил возможно дополнительно замещены одной, двумя или тремя группами, выбранными из группы, состоящей из галогена, циано, гидрокси, С1-С6 алкила, С1-С6 алкокси, С1-С6 гидроксиалкокси, -C(O)NHCH3, гетероциклила, который представляет собой насыщенную моноциклическую углеводородную группу, содержащую от 4 до 6 кольцевых атомов, 1 или 2 из которых могут быть заменены кислородом и/или азотом, галофенила, С3-С6 циклоалкил-тетразолила; R4 выбран из группы, состоящей из С1-С6 алкила, или R4 и R1 вместе с атомом азота, к которому они присоединены, образуют пиперазинил, который дополнительно замещен одной группой, выбранной из группы, состоящей из С1-С6 алкила; R2 представляет собой водород, G выбран из группы:
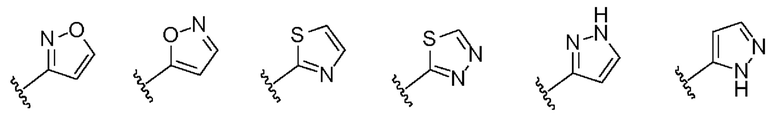 , которая возможно дополнительно замещена одной или двумя группами, выбранными из группы, состоящей из С1-С6 алкила, амино, -С(O)-СН=СН2, С3-С6 циклоалкила, фенила и пиридинила; причем указанные С1-С6 алкил или фенил могут быть дополнительно замещены одной или тремя группами, выбранными из группы, состоящей из галогена, гидрокси, С1-С6 алкила, С1-С6 алкокси, С1-С6 галоалкила, -С(О)ОС2Н5; R3 выбран из группы Q; Ru выбран из связи или С1-С6 алкилена; Rx выбран из С1-С6 алкила; Ry и Rz каждый независимо выбран из группы, состоящей из С1-С6 алкила; или Ry и Rz вместе с атомом азота, к которому они присоединены, образуют гетероциклильную группу, содержащую от 6 до 7 кольцевых атомов, которая дополнительно содержит 1 гетероатом, выбранный из азота или кислорода, и гетероциклильная группа возможно дополнительно замещена одной группой, выбранной из группы, состоящей из С1-С6 алкила; Q выбран из группы, состоящей из водорода и галогена. Изобретение также относится к способам получения указанных соединений формулы (I). Технический результат – получены новые соединения, которые могут найти применение в медицине в качестве ингибитора тирозиновой протеинкиназы FLT3 для предотвращения и/или лечения злокачественного новообразования. 6 н. и 6 з.п. ф-лы, 11 табл., 134 пр.
, которая возможно дополнительно замещена одной или двумя группами, выбранными из группы, состоящей из С1-С6 алкила, амино, -С(O)-СН=СН2, С3-С6 циклоалкила, фенила и пиридинила; причем указанные С1-С6 алкил или фенил могут быть дополнительно замещены одной или тремя группами, выбранными из группы, состоящей из галогена, гидрокси, С1-С6 алкила, С1-С6 алкокси, С1-С6 галоалкила, -С(О)ОС2Н5; R3 выбран из группы Q; Ru выбран из связи или С1-С6 алкилена; Rx выбран из С1-С6 алкила; Ry и Rz каждый независимо выбран из группы, состоящей из С1-С6 алкила; или Ry и Rz вместе с атомом азота, к которому они присоединены, образуют гетероциклильную группу, содержащую от 6 до 7 кольцевых атомов, которая дополнительно содержит 1 гетероатом, выбранный из азота или кислорода, и гетероциклильная группа возможно дополнительно замещена одной группой, выбранной из группы, состоящей из С1-С6 алкила; Q выбран из группы, состоящей из водорода и галогена. Изобретение также относится к способам получения указанных соединений формулы (I). Технический результат – получены новые соединения, которые могут найти применение в медицине в качестве ингибитора тирозиновой протеинкиназы FLT3 для предотвращения и/или лечения злокачественного новообразования. 6 н. и 6 з.п. ф-лы, 11 табл., 134 пр.
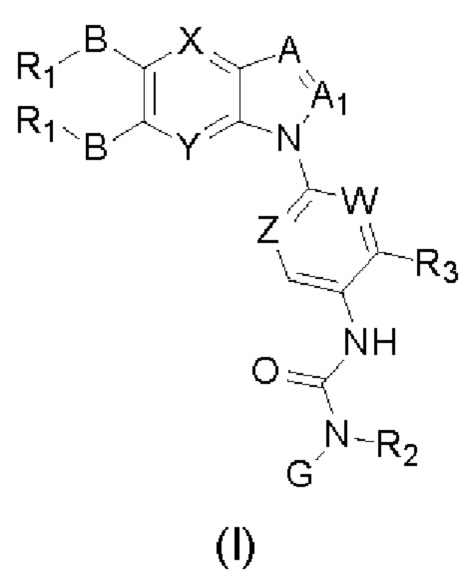
1. Соединение общей формулы (I) или его фармацевтически приемлемые соли,
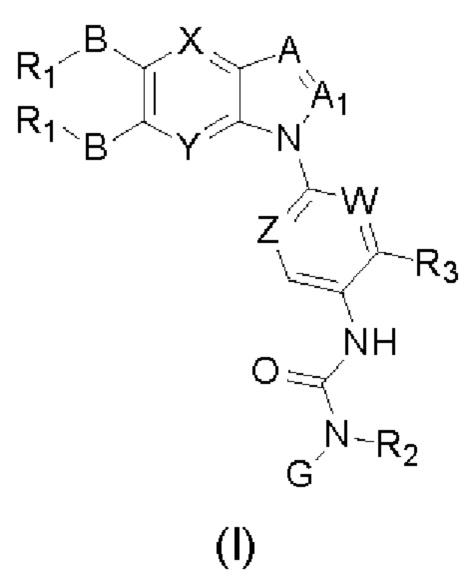
где:
группа 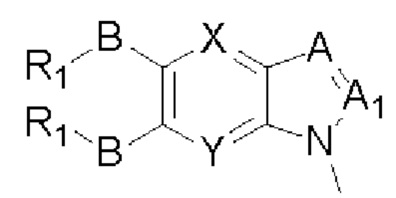 выбрана из следующей структуры:
выбрана из следующей структуры:
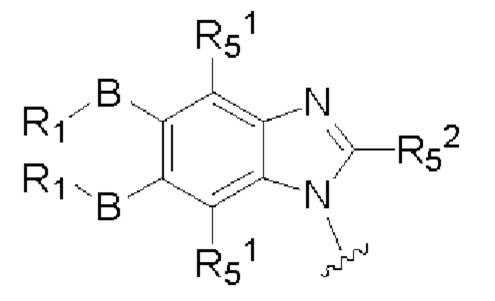
каждый из R51 и R52 независимо выбран из группы, состоящей из водорода;
W и Z - каждый независимо выбран из N и C-BR1;
при отсутствии R1, В являются одинаковыми или разными и каждый независимо выбран из группы, состоящей из водорода, галогена, С1-С6 алкила, причем указанный С1-С6 алкил возможно дополнительно замещен одной или более группами Q;
в присутствии R1, В являются одинаковыми или разными и каждый независимо выбран из группы, состоящей из -О- и -NR4-; и R1 являются одинаковыми или разными и каждый независимо выбран из группы, состоящей из водорода, С1-С6 алкила, гетероарила, который представляет собой гетероароматическую систему, содержащую от 1 до 2 гетероатомов, от 5 до 6 кольцевых атомов, где гетероатом выбран из серы и азота, -RuORx, -RuC(О)ORx, -RuN(Ry)(Rz), -C(O)N(Ry)(Rz); указанные алкил и гетероарил возможно дополнительно замещены одной, двумя или тремя группами, выбранными из группы, состоящей из галогена, циано, гидрокси, С1-С6 алкила, С1-С6 алкокси, С1-С6 гидроксиалкокси, -C(O)NHCH3, гетероциклила, который представляет собой насыщенную моноциклическую углеводородную группу, содержащую от 4 до 6 кольцевых атомов, 1 или 2 из которых могут быть заменены кислородом и/или азотом, галофенила, С3-С6 циклоалкил-тетразолила;
R4 выбран из группы, состоящей из С1-С6 алкила,
или R4 и R1 вместе с атомом азота, к которому они присоединены, образуют пиперазинил, который дополнительно замещен одной группой, выбранной из группы, состоящей из С1-С6 алкила;
R2 представляет собой водород, G выбран из группы:
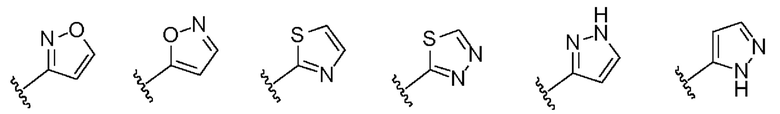
которая возможно дополнительно замещена одной или двумя группами, выбранными из группы, состоящей из С1-С6 алкила, амино, -С(O)-СН=СН2, С3-С6 циклоалкила, фенила и пиридинила; причем указанные С1-С6 алкил или фенил могут быть дополнительно замещены одной или тремя группами, выбранными из группы, состоящей из галогена, гидрокси, С1-С6 алкила, С1-С6 алкокси, С1-С6 галоалкила, -С(О)ОС2Н5;
R3 выбран из группы Q;
Ru выбран из связи или С1-С6 алкилена;
Rx выбран из С1-С6 алкила;
Ry и Rz каждый независимо выбран из группы, состоящей из С1-С6 алкила;
или
Ry и Rz вместе с атомом азота, к которому они присоединены, образуют гетероциклильную группу, содержащую от 6 до 7 кольцевых атомов, которая дополнительно содержит 1 гетероатом, выбранный из азота или кислорода, и гетероциклильная группа возможно дополнительно замещена одной группой, выбранной из группы, состоящей из С1-С6 алкила;
Q выбран из группы, состоящей из водорода и галогена.
2. Соединение общей формулы (I) или его фармацевтически приемлемая соль по п. 1, где W, Z выбраны из следующих четырех вариантов:
a) W и Z представляют собой CQ;
b) W и Z представляют собой N;
с) W представляет собой CQ и Z представляет собой N;
d) Z представляет собой CQ и W представляет собой N;
где Q выбран из группы, состоящей из водорода и галогена.
3. Соединение общей формулы (I) или его фармацевтически приемлемая соль по п. 1 или 2, где
при отсутствии R1, В выбран из группы, состоящей из водорода, галогена, С1-С6 алкила и С1-С6 галоалкила;
в присутствии R1, В является идентичным или разным и каждый независимо выбран из -О- и -NR4-, предпочтительно -О-; и R1 является идентичным или разным и каждый независимо выбран из -RuORx-, где Ru выбран из С1-С6 алкилена, Rx выбран из С1-С6 алкила;
R4 является таким, как определено в п. 1.
4. Соединение общей формулы (I) или его фармацевтически приемлемая соль по п. 1 или 2, где
при отсутствии R1, В выбран из группы, состоящей из водорода, галогена, С1-С6 алкила, С1-С6 галоалкила;
в присутствии R1, В является идентичным или разным и каждый независимо выбран из -О- и -NR4-, предпочтительно -О-; и R1 является идентичным или разным и каждый независимо выбран из -C(O)N(Ry)(Rz), где Ry и Rz каждый независимо выбран из группы, состоящей из С1-С6 алкила; или
Ry и Rz вместе с атомом азота, к которому они присоединены, образуют морфолинил, пиперидинил, пиперазинил, пиридинил или пиримидил, который возможно дополнительно замещен одной группой, выбранной из группы, состоящей из С1-С6 алкила;
R4 является таким, как определено в п. 1.
5. Соединение общей формулы (I) или его фармацевтически приемлемая соль по п. 1 или 2, где
при отсутствии R1, В выбран из группы, состоящей из водорода, галогена, С1-С6 алкила, С1-С6 галоалкила;
в присутствии R1, В является идентичным или разным и каждый независимо выбран из -О- или -NR4-, предпочтительно -О-; и R1 является идентичным или разным и каждый независимо выбран из -RuN(Ry)(Rz), где Ru выбран из С1-С6 алкилена; Ry и Rz каждый независимо выбран из группы, состоящей из С1-С6 алкила; или
Ry и Rz вместе с атомом азота, к которому они присоединены, образуют морфолинил, пиперидинил, пиперазинил, азепанил, пиридил или пиримидинил, который возможно дополнительно замещен одной группой, выбранной из группы, состоящей из С1-С6 алкила;
R4 является таким, как определено в п. 1.
6. Соединение общей формулы (I) или его фармацевтически приемлемая соль по п. 1 или 2, где
при отсутствии R1, В выбран из группы, состоящей из водорода, галогена, С1-С6 алкила, С1-С6 галоалкила;
в присутствии R1, В является идентичным или разным и каждый независимо выбран из -О- или -NR4-, предпочтительно -О-; и R1 является идентичным или разным и каждый независимо выбран из -RuC(O)ORx, где, Ru выбран из С1-С6 алкилена; Rx выбран из группы, состоящей из С1-С6 алкила;
R4 является таким, как определено в п. 1.
7. Соединение общей формулы (I) или его фармацевтически приемлемая соль по п. 1 или 2, где
при отсутствии R1, В выбран из группы, состоящей из водорода, галогена, С1-С6 алкила, С1-С6 галоалкила;
в присутствии R1, В является идентичным или разным и каждый независимо выбран из -О- или -NR4-, предпочтительно -О-; и R1 является идентичным или разным и каждый независимо выбран из пиразолила, имидазолила, тиазолила, фурила, тиенила, пиридила, пирролила, N-алкилпирролила, пиримидила, пиразинила, имидазолила, пиридила и пиримидинила, который возможно дополнительно замещен одной, двумя или тремя группами, выбранными из группы, состоящей из гетероциклила, который представляет собой насыщенную моноциклическую углеводородную группу, содержащую от 4 до 6 кольцевых атомов, 1 или 2 из которых могут быть заменены кислородом и/или азотом, и группы -C(O)NHCH3;
R4 является таким, как определено в п. 1.
8. Соединение или его фармацевтически приемлемая соль, где соединение выбрано из группы, состоящей из:
1-(4-бензимидазол-1-ил-фенил)3-изоксазол-3-ил-мочевина;
1-(4-бензимидазол-1-ил-фенил)-3-(5-метил-изоксазол-3-ил)-мочевина;
1-(4-бензимидазол-1-ил-фенил)-3-(5-трет-бутил-изоксазол-3-ил)-мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(5-гидрокси-бензимидазол-1-ил)-фенил]-мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(5-метоксил-бензимидазол-1-ил)-фенил]-мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(5-этоксил-бензимидазол-1-ил)-фенил]-мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(5-гексилоксил-бензимидазол-1-ил)-фенил]-мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(5-изопропокси-бензимидазол-1-ил)-фенил]-мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-{4-[5-(3-метил-оксетан-3-илметокси)-бензимидазол-1-ил]-фенил}-мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-{4-[5-(тетрагидрофуран-2-илметокси)-бензимидазол-1-ил]-фенил}-мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-{4-[5-(2-гидрокси-этоксил)-бензимидазол-1-ил]-фенил]мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-{4-[5-(2-метоксил-этоксил)-бензимидазол-1-ил]-фенил]мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-{4-[5-(2-этоксил-этоксил)-бензимидазол-1-ил]-фенил]мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-(4-{5-[2-(2-гидрокси-этоксил)-этоксил]-бензимидазол-1-ил}-фенил)-мочевина;
1-{4-[3-(5-трет-бутил-изоксазол-3-ил)-карбамидо]-фенил}-1Н-бензимидазол-5-ил морфолин-4-карбоксиликат;
1-(5-трет-бутил-изоксазол-3-ил)-3-{4-[5-(2-морфолин-4-ил-этоксил)-бензимидазол-1-ил]-фенил}-мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-{4-[5-(2-пиперидин-1-ил-этоксил)-бензимидазол-1-ил]-фенил}-мочевина;
1-{4-[5-(2-азациклогептан-1-ил-этоксил)-бензимидазол-1-ил]-фенил}-3-(5-трет-бутил-изоксазол-3-ил)-мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-(4-{5-[3-(4-метил-пиперазин-1-ил)-пропоксил]-бензимидазол-1-ил}-фенил)-мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-{4-[5-(2-диметиламино-этоксил)-бензимидазол-1-ил]-фенил}-мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(5-трифторметокси-бензимидазол-1-ил)-фенил]-мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(5-фтор-бензимидазол-1-ил)-фенил]-мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(5-трифторметил-бензимидазол-1-ил)-фенил]-мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(5-метил-бензимидазол-1-ил)-фенил]-мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(6-метоксил-бензимидазол-1-ил)-фенил]-мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-{4-[6-(2-метоксил-этоксил)-бензимидазол-1-ил]-фенил]мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-{4-[6-(2-морфолин-4-ил-этоксил)-бензимидазол-1-ил]-фенил}-мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-(4-{6-[(2-диметиламино-этил)-метил-амино]-бензимидазол-1-ил}-фенил)-мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-{4-[6-(4-метил-пиперазин-1-ил)-бензимидазол-1-ил]-фенил}-мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-{4-[7-(2-морфолин-4-ил-этоксил)-бензимидазол-1-ил]-фенил}-мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(5,6-диметоксил-бензимидазол-1-ил)-фенил]-мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(5,6-диметил-бензимидазол-1-ил)-фенил]-мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(5-фтор-7-метил-бензимидазол-1-ил)-фенил]-мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(4-фтор-бензимидазол-1-ил)-фенил]-мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(2-метил-бензимидазол-1-ил)-фенил]-мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(2-хлоробензимидазол-1-ил)-фенил]-мочевина;
1-(4-бензимидазол-1-ил-3-метил-фенил)-3-(5-трет-бутил-изоксазол-3-ил)-мочевина;
1-(4-бензимидазол-1-ил-3-хлор-фенил)-3-(5-трет-бутил-изоксазол-3-ил)-мочевина;
1-(4-бензимидазол-1-ил-3-фтор-фенил)-3-(5-трет-бутил-изоксазол-3-ил)-мочевина;
1-(4-бензимидазол-1-ил-3,5-дифтор-фенил)-3-(5-трет-бутил-изоксазол-3-ил)-мочевина;
1-(4-бензимидазол-1-ил-2-хлор-фенил)-3-(5-трет-бутил-изоксазол-3-ил)-мочевина;
1-(6-бензимидазол-1-ил-пиридин-3-ил)-3-(5-трет-бутил-изоксазол-3-ил)-мочевина;
1-(2-бензимидазол-1-ил-пиримидин-5-ил)-3-(5-трет-бутил-изоксазол-3-ил)-мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-(4-индазол-1-ил-фенил)-мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(6-фтор-индазол-1-ил)-фенил]-мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(7-фтор-индазол-1-ил)-фенил]-мочевина;
1-(4-бензотриазол-1-ил-фенил)-3-(5-трет-бутил-изоксазол-3-ил)-мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-(4-пирроло[2,3-b]пиридин-1-ил-фенил)-мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-(4-имидазо[4,5-b]пиридин-3-ил-фенил)-мочевина;
1-[4-(6-бромо-имидазо[4,5-b]пиридин-3-ил)-фенил]-3-(5-трет-бутил-изоксазол-3-ил)-мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(6-трифторметил-имидазо[4,5-b]пиридин-3-ил)-фенил]-мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(5-хлороимидазо[4,5-b]пиридин-3-ил)-фенил]-мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(5-метилимидазо[4,5-b]пиридин-3-ил)-фенил]-мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(7-метилимидазо[4,5-b]пиридин-3-ил)-фенил]-мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-(4-имидазо[4,5-b]пиридин-1-ил-фенил)-мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(6-метоксил-пурин-7-ил)-фенил]-мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(6-метоксил-пурин-9-ил)-фенил]-мочевина;
1-(5-трет-бутил-изоксазол-3-ил)-3-[4-(6-диметиламино-пурин-7-ил)-фенил]-мочевина;
1-(4-бензимидазол-1-ил-фенил)-3-тиазол-2-ил-мочевина;
1-(4-бензимидазол-1-ил-фенил)-3-(4-метил-тиазол-2-ил)-мочевина;
1-(4-бензимидазол-1-ил-фенил)-3-[1,3,4]тиадиазол-2-ил-мочевина;
1-(4-бензимидазол-1-ил-фенил)-3-(5-трет-бутил-[1,3,4]тиадиазол-2-ил)-мочевина;
1-(4-бензимидазол-1-ил-фенил)-3-(5-метил-1Н-пиразол-3-ил)-мочевина;
1-(4-бензимидазол-1-ил-фенил)-3-(5-фенил-1Н-пиразол-3-ил)-мочевина;
1-(4-бензимидазол-1-ил-фенил)-3-(5-циклопропил-2Н-пиразол-3-ил)-мочевина;
1-(4-бензимидазол-1-ил-фенил)-3-(5-трифторметил-2Н-пиразол-3-ил)-мочевина;
1-(4-бензимидазол-1-ил-фенил)-3-(5-трет-бутил-2Н-пиразол-3-ил)-мочевина;
1-(5-трет-бутил-2Н-пиразол-3-ил)-3-[4-(5-фтор-бензимидазол-1-ил)-фенил]-мочевина;
1-(5-трет-бутил-2Н-пиразол-3-ил)-3-[4-(5-трифторметил-бензимидазол-1-ил)-фенил]-мочевина;
1-(5-трет-бутил-2Н-пиразол-3-ил)-3-[4-(5-метоксил-бензимидазол-1-ил)-фенил]-мочевина;
1-(5-трет-бутил-2Н-пиразол-3-ил)-3-[4-(5-этоксил-бензимидазол-1-ил)-фенил]-мочевина;
1-(5-трет-бутил-2Н-пиразол-3-ил)-3-[4-(5-гексилоксил-бензимидазол-1-ил)-фенил]-мочевина;
1-(5-трет-бутил-2Н-пиразол-3-ил)-3-[4-(5-изопропокси-бензимидазол-1-ил)-фенил]-мочевина;
1-[4-(5-втор-бутоксил-бензимидазол-1-ил)-фенил]-3-(5-трет-бутил-2Н-пиразол-3-ил)-мочевина;
1-(5-трет-бутил-2Н-пиразол-3-ил)-3-[4-(5-изобутокси-бензимидазол-1-ил)-фенил]-мочевина;
1-(5-трет-бутил-2Н-пиразол-3-ил)-3-{4-[5-(2-метоксил-этоксил)-бензимидазол-1-ил]-фенил}-мочевина;
1-(5-трет-бутил-2Н-пиразол-3-ил)-3-{4-[5-(2-этоксил-этоксил)-бензимидазол-1-ил]-фенил}-мочевина;
1-(5-трет-бутил-2Н-пиразол-3-ил)-3-{4-[5-(2-гидрокси-этоксил)-бензимидазол-1-ил]-фенил]мочевина;
1-(5-трет-бутил-2Н-пиразол-3-ил)-3-{4-[5-(2-гидрокси-3-метоксил-пропоксил)-бензимидазол-1-ил]-фенил]мочевина;
1-(5-трет-бутил-2Н-пиразол-3-ил)-3-{4-[5-(3-диметиламино-пропоксил)-бензимидазол-1-ил]-фенил}-мочевина;
1-(5-трет-бутил-2Н-пиразол-3-ил)-3-{4-[5-(3-дибутиламинопропилоксил)-бензимидазол-1-ил]-фенил}-мочевина;
1-(5-трет-бутил-2Н-пиразол-3-ил)-3-[4-(5-цианометокси-бензимидазол-1-ил)-фенил]-мочевина;
1-(5-трет-бутил-2Н-пиразол-3-ил)-3-[4-(5-трифторметокси-бензимидазол-1-ил)-фенил]-мочевина;
1-(5-трет-бутил-2Н-пиразол-3-ил)-3-{4-[5-(3-метил-оксетан-3-илметокси)-бензимидазол-1-ил]-фенил]мочевина;
1-(5-трет-бутил-2Н-пиразол-3-ил)-3-{4-[5-(тетрагидрофуран-2-илметоксил)-бензимидазол-1-ил]-фенил}-мочевина;
1-(5-трет-бутил-2Н-пиразол-3-ил)-3-{4-[5-(тетрагидрофуран-2-илметоксил)-бензимидазол-1-ил]-фенил}-мочевина;
этил (1-{4-[3-(5-трет-бутил-2Н-пиразол-3-ил)-карбамидо]-фенил}-1Н-бензимидазол-5-илокси)-ацетат;
1-(5-трет-бутил-2Н-пиразол-3-ил)-3-{4-[5-(2-морфолин-4-ил-этоксил)-бензимидазол-1-ил]-фенил}-мочевина;
1-(5-трет-бутил-2Н-пиразол-3-ил)-3-{4-[5-(2-пиперидин-1-ил-этоксил)-бензимидазол-1-ил]-фенил}-мочевина;
1-{4-[5-(2-азациклогептан-1-ил-этоксил)-бензимидазол-1-ил]-фенил}-3-(5-трет-бутил-2Н-пиразол-3-ил)-мочевина;
1-(5-трет-бутил-2Н-пиразол-3-ил)-3-(4-{5-[3-(4-метил-пиперазин-1-ил)-пропоксил]-бензимидазол-1-ил}-фенил)-мочевина;
1-(5-трет-бутил-2Н-пиразол-3-ил)-3-{4-[5-(3-фтор-бензилокси)-бензимидазол-1-ил]-фенил]мочевина;
1-(5-трет-бутил-2Н-пиразол-3-ил)-3-(4-{5-[4-(1-циклогексил-1Н-тетразол-5-ил)-бутокси]-бензимидазол-1-ил}-фенил)-мочевина;
1-(5-трет-бутил-2Н-пиразол-3-ил)-3-{4-[5-(4-морфолин-4-ил-[1,2,5]тиадиазол-3-илокси)-бензимидазол-1-ил]-фенил]мочевина;
4-(1-{4-[3-(5-трет-бутил-2Н-пиразол-3-ил)-карбамидо]-фенил}-1Н-бензимидазол-5-илокси)-пиридин-2-карбоновой кислоты метиламин;
1-(5-трет-бутил-2Н-пиразол-3-ил)-3-[4-(5-фтор-7-метил-бензимидазол-1-ил)-фенил]-мочевина;
1-(5-трет-бутил-2Н-пиразол-3-ил)-3-{4-[6-(2-метоксил-этоксил)-бензимидазол-1-ил]-фенил}-мочевина;
1-(5-трет-бутил-2Н-пиразол-3-ил)-3-[4-(5,6-диметоксил-бензимидазол-1-ил)-фенил]-мочевина;
1-(4-бензимидазол-1-ил-фенил)-3-(5-трет-бутил-2-метил-2Н-пиразол-3-ил)-мочевина;
1-(5-трет-бутил-2-метил-2Н-пиразол-3-ил)-3-{4-[5-(2-метоксил-этоксил)-бензимидазол-1-ил]-фенил]мочевина;
1-(5-трет-бутил-2-метил-2Н-пиразол-3-ил)-3-{4-[5-(2-морфолин-4-ил-метоксил)-бензимидазол-1-ил]-фенил}-мочевина;
1-(4-бензимидазол-1-ил-фенил)-3-[5-трет-бутил-2-(2-гидрокси-этил)-2Н-пиразол-3-ил]-мочевина;
1-(4-бензимидазол-1-ил-фенил)-3-(5-трет-бутил-2-фенил-2Н-пиразол-3-ил)-мочевина;
1-(4-бензимидазол-1-ил-фенил)-3-(5-трет-бутил-2-р-толил-2Н-пиразол-3-ил)-мочевина;
1-(4-бензимидазол-1-ил-фенил)-3-[5-трет-бутил-2-(4-метоксил-фенил)-2Н-пиразол-3-ил]-мочевина;
1-(4-бензимидазол-1-ил-фенил)-3-[5-трет-бутил-2-(4-трифторметокси-фенил)-2Н-пиразол-3-ил]-мочевина;
1-(4-бензимидазол-1-ил-фенил)-3-[5-трет-бутил-2-(4-фтор-фенил)-2Н-пиразол-3-ил]-мочевина;
1-(4-бензимидазол-1-ил-фенил)-3-[5-трет-бутил-2-(2-фтор-фенил)-2Н-пиразол-3-ил]-мочевина;
1-(4-бензимидазол-1-ил-фенил)-3-(5-трет-бутил-2-пиридин-2-ил-2Н-пиразол-3-ил)-мочевина;
4-{5-[3-(4-бензимидазол-1-ил-фенил)-карбамидо]-3-трет-бутил-пиразол-1-ил}-бензоат;
1-(2-акрил-5-трет-бутил-2Н-пиразол-3-ил)-3-(4-имидазол-1-ил-фенил)-мочевина;
3-амино-5-метилпиразол-1-карбоновой кислоты (4-бензимидазол-1-ил-фенил)-амид;
5-амино-3-циклопропилпиразол-1-карбоновой кислоты (4-бензимидазол-1-ил-фенил)-амид;
5-амино-3-трифторметилпиразол-1-карбоновой кислоты (4-бензимидазол-1-ил-фенил)-амид;
5-амино-3-трет-бутил-пиразол-1-карбоновой кислоты (4-бензимидазол-1-ил-фенил)-амид;
5-амино-3-трет-бутил-пиразол-1-карбоновой кислоты [4-(5-гексилоксил-бензимидазол-1-ил)-фенил]-амид;
5-амино-3-трет-бутил-пиразол-1-карбоновой кислоты (4-{5-[3-(4-метил-пиперазин-1-ил)-пропоксил]-бензимидазол-1-ил}фенил)-амид;
5-амино-3-трет-бутил-пиразол-1-карбоновой кислоты {4-[5-(2-морфолин-4-ил-этоксил)-бензимидазол-1-ил]-фенил}-амид;
5-амино-3-трет-бутил-пиразол-1 -карбоновой кислоты {4-[5-(2-метоксил-этоксил)-бензимидазол-1-ил]-фенил}-амид;
5-амино-3-трет-бутил-пиразол-1-карбоновой кислоты {4-[5-(2-гидрокси-этоксил)-бензимидазол-1-ил]-фенил}-амид;
5-амино-3-трет-бутил-пиразол-1-карбоновой кислоты {4-[5-(тетрагидрофуран-2-илметоксил)-бензимидазол-1-ил]-фенил}-амид;
5-амино-3-трет-бутил-пиразол-1 -карбоновой кислоты {4-[5-(тетрагидрофуран-2-илметокси)-бензимидазол-1-ил]-фенил}-амид;
(1-{4-[(5-амино-3-трет-бутил-пиразол-1-карбонил)-амино]-фенил}-1Н-бензимидазол-5-илоксил)-уксусная кислота;
5-амино-3-трет-бутил-пиразол-1-карбоновой кислоты [4-(5-фтор-бензимидазол-1-ил)-фенил]-амид;
5-амино-3-трет-бутил-пиразол-1-карбоновой кислоты [4-(5-трифторметил-бензимидазол-1-ил)-фенил]-амид;
4-(1-{4-[(5-амино-3-трет-бутил-пиразол-1-карбонил)-амино]-фенил}-1Н-бензимидазол-5-илоксил)-пиридин-2-карбоновой кислоты метиламид;
5-амино-3-трет-бутил-пиразол-1-карбоновой кислоты {4-[6-(2-метоксил-этоксил)-бензимидазол-1-ил]-фенил}-амид;
5-амино-3-трет-бутил-пиразол-1-карбоновой кислоты [4-(5,6-диметоксил-бензимидазол-1-ил)-фенил]-амид;
3-трет-бутил-пиразол-1-карбоновой кислоты (4-бензимидазол-1-ил-фенил)-амид;
1-(4-бензимидазол-1-ил-фенил)-3-(3,4-диметил-изоксазол-5-ил)-мочевина;
1-(4-бензимидазол-1-ил-фенил)-3-(3-изопропил-изоксазол-5-ил)-мочевина;
1-(4-бензимидазол-1-ил-фенил)-3-(3-трет-бутил-изоксазол-5-ил)-мочевина.
9. Способ получения соединения общей формулы (I) или его фармацевтически приемлемой соли по любому из пп. 1-7, содержащий следующие стадии:
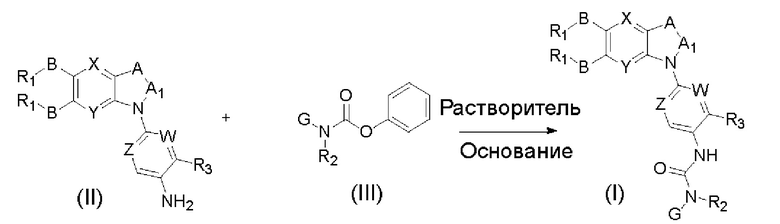
соединение формулы (II) взаимодействует с соединением формулы (III) в присутствии основания в подходящем растворителе с получением соединения общей формулы (I);
растворитель предпочтительно представляет собой ТГФ (тетрагидрофуран), ацетонитрил, дихлорметан или толуол, и основание предпочтительно представляет собой триэтиламин, N,N-диизопропилэтиламин, DMAP (4-диметиламинопиридин) или пиридин;
X, Y, A, Al, Z, W, R1 В, R2, G, R3 являются такими, как определено в п. 1.
10. Способ получения соединения общей формулы (I) или его фармацевтически приемлемой соли по любому из пп. 1-7, содержащий следующие стадии:
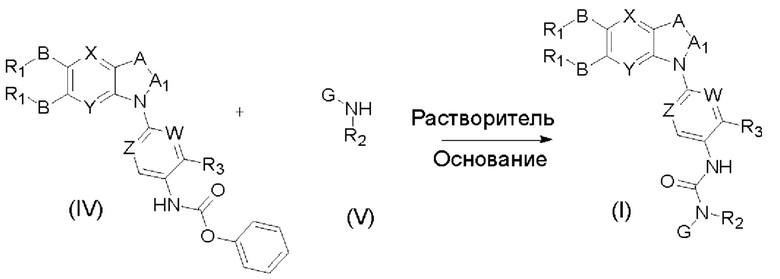
соединение формулы (IV) взаимодействует с соединением формулы (V) в присутствии основания в подходящем растворителе с получением соединения общей формулы (I);
растворитель предпочтительно представляет собой ТГФ (тетрагидрофуран), ацетонитрил, дихлорметан или толуол, и основание предпочтительно представляет собой триэтиламин, N,N-диизопропиламин, DMAP (4-диметиламинопиридин) или пиридин;
X, Y, A, Al, Z, W, R1 В, R2, G, R3 являются такими, как определено в п. 1.
11. Применение соединения общей формулы (I) или его фармацевтически приемлемой соли по любому из пп. 1-8 при получении ингибиторов тирозиновой протеинкиназы FLT3.
12. Применение соединения общей формулы (I) или его фармацевтически приемлемой соли по любому из пп. 1-8 для получения лекарственного средства для лечения злокачественного новообразования у млекопитающих, включая человека, где злокачественное новообразование включает лейкемию, рак кожи, меланому, рак легкого, рак желудка, рак груди, рак поджелудочной железы, рак печени или рак толстой кишки.
| Способ лечения больных отечным экзофтальмом и псевдотумором орбиты | 1989 |
|
SU1724258A1 |
| ZHAO CUI-RONG ET AL, Bioorganic & Medicinal Chemistry Letters, vol | |||
| Прибор для равномерного смешения зерна и одновременного отбирания нескольких одинаковых по объему проб | 1921 |
|
SU23A1 |
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| WO 2007112093 A2, 04.10.2007 | |||
| WO 2005042495 A1, 12.05.2005 | |||
| CN 103450152 B, 18.11.2015 | |||
| CN 102319242 A, 18.01.2012 | |||
| CN 102153551 B, 25.04.2012 | |||
| WO 2015043492 A1, 02.04.2015 | |||
| CN 103864792 B, 18.01.2017 | |||
| Способ получения производных фенилмочевины | 1987 |
|
SU1491333A3 |

