Область техники
Настоящая заявка относится к соединению в качестве селективного ингибитора киназы PDGFR, а также к способу и применению для ингибирования активности киназы PDGFR и для лечения заболевания, ассоциированного с ингибированием активности киназы PDGFR с таким соединением..
Уровень техники
Тромбоцитарный фактор роста (ТФР, PDGF) представляет собой семейство эффективных митогенов почти для всех клеток, происходящих из мезенхимы. Существует четыре изоформы PDGF: А, В, С и D, которые образуют пять различных дисульфид-связанных димерных белков PDGF-AA, -ВВ, -АВ, -СС и- DD. Эти факторы роста оказывают свое клеточное действие через два структурно родственных рецептора рецептора а тирозинкиназы PDGF (PDGFRa) и рецептора PDGF р (PDGFRp) (Sandy, J. R. (1998) Br. J. Orthod. 25: 269-74; Betsholtz, C, et al., (2001) BioEssays 23: 494-507).
PDGFRα по своей структуре аналогичен PDGFRβ и может образовывать гетеродимеры и гомодимеры. PDGF-BB и PDGF-DD являются первичными активаторами гомодимеров ββ.PDGF-AA активирует только димеры рецептора αα, в то время как PDGF-AB, PDGF-BB и PDGF-CC активируют как димеры рецептора αα, так и димеры рецептора αβ. Молекула димерного лиганда связывается с двумя рецепторными белками одновременно и индуцирует димеризацию рецепторов, аутофосфорилирование конкретных остатков в цитоплазматическом домене рецептора и клеточную сигнализацию.
Патоморфологической основой хронической гипоксической легочной гипертензии является структурное ремоделирование легочных сосудов, проявляющееся в основном пролиферацией и миграцией гладкомышечных клеток средней оболочки. Пролиферация гладкомышечных клеток зависит от воздействия различных факторов роста, в частности тромбоцитарного фактора роста. Факторы роста регулируют пролиферацию клеток путем связывания с рецепторами фактора роста и активации тирозиновой протеинкиназы (ТПК) в рецепторах для фосфорилирования. Авторы Schermuly et al. сообщили в журнале JCI в 2005 г., что иматиниб в качестве ингибитора PDGFR может значительно улучшить симптомы легочной гипертензии (Schermuly, R.T., et al. 2005. Reversal of experimental pulmonary hypertension by PDGF inhibition. J. Clin. Invest. 115:2811-2821. doi:10.1172/JCI24838.). Авторы также исследовали легочную ткань пациентов с легочной гипертензией, проходящих трансплантацию легких, и наблюдали достоверно повышенный уровень экспрессии фактора роста тромбоцитов (PDGF) у пациентов с легочной гипертензией. Авторы считают, что ингибиторы PDGFR могут представлять собой новое средство клинической терапии легочной гипертензии.
Кроме того, хронический эозинофильный лейкоз (ХЭЛ) является типом гиперэозинофильного синдрома (ГЭС). Хронический эозинофильный лейкоз является редким и необъясненным заболеванием системы крови, характеризующимся постоянно повышенным уровнем эозинофильных гранулоцитов, осложненное полиорганным поражением. В 2001 году Schaller et al. впервые сообщили об иматиниба мезилате (торговое название: Gleevec, низкомолекулярный ингибитор тирозинкиназ ABL, KIT и PDGFR) в лечении 1 случая пациента с ГЭС со значительной эффективностью, и, таким образом, предположили, что ГЭС может быть присуща активация ABL, KIT, PDGFR или других неизвестных генов-мишеней (Schaller, J. L., & Burkland, G. A. (2001). Case report: rapid and complete control of idiopathic hypereosinophilia with imatinib mesylate. MedGenMed., 3(5), 9). В 2003 г. авторы Cools et al. обнаружили гибридный ген FIP1L1-PDGFRα у пациентов с ГЭС и в клетках EOL-1, культивируемых in vitro (линия клеток хронического эозинофильного лейкоза), что позволило не только идентифицировать молекулярную мишень лекарственного препарата Гливек для лечения ГЭС, чтобы обеспечить мощные молекулярные маркеры для диагностики и лечения ГЭС, но также выявить на молекулярном уровне, что ГЭС по существу является злокачественным клональным заболеванием системы кроветворения (Cools J., DeAngelo D. J., Gotlib J., A tyrosine kinase created by fusion of the PDGFRA and FIP1L1 genes as a therapeutic target of imatinib in idiopathic hypereosinophilic syndrome. N. Engl. J. Med. 2003, 348(13): 1201-14). Исследования авторов Cools et al. показали, что активатор транскрипции 5 (STAT5) является нижележащей мишенью эффекта слияния гена FIP1L1-PDGFRα, и активация STAT5 способствует пролиферации эозинофильных гранулоцитов.
Примеры известных в настоящее время селективных ингибиторов, как PDGFRα, так и PDGFRβ, включают: СР-673451 (регистрационный номер CAS: 343787-29-1; молекулярная масса: 417,5) и иматиниб (регистрационный номер CAS: 152459-95-5; молекулярная масса: 493,60), каждый из которых, однако, недостаточно хорош в плане селективности. В дополнение к ингибирующему воздействию на PDGFRα и β, данные селективные ингибиторы также ингибируют ингибирующий эффект в отношении cKIT, BCR-ABL и т.п.Следовательно, необходимо предоставить селективный ингибитор PDGFR, чтобы обеспечить исследовательскую основу для точной таргетной терапии.
Авторы настоящего изобретения посредством экспериментов обнаружили селективный ингибитор PDGFR, который может значительно ингибировать рост опухоли в модели опухоли на мышах с трансплантированными опухолевыми клетками EOL-1, а также может улучшить выживаемость крыс и облегчать состояния легочной гипертензии в крысиной модели легочной гипертензии.
Краткое описание изобретения
В настоящем изобретении предложен селективный ингибитор киназы PDGFR, содержащий соединение формулы (I) или его фармацевтически приемлемую соль, сольват, сложный эфир, кислоту, метаболит или пролекарство:
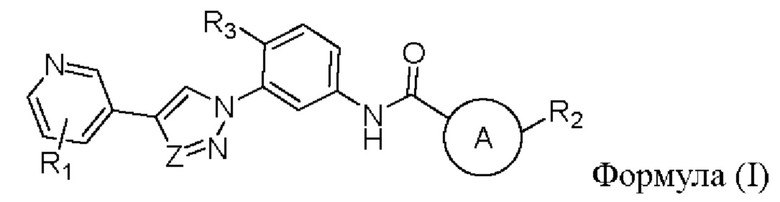
где,
кольцо А представляет собой пиридиновое кольцо;
Z выбран из группы, состоящей из N и СН;
R1 выбран из группы, состоящей из гетероциклоалкила, гетероциклоалкилокси, гетероциклоалкил C1-6 алкокси, гетер оциклоалкилами но, гетероспироциклоалкила, гетероспироциклоалкиламино, С3-6 циклоалкила, C1-6 алкокси, С3-6 циклоалкилокси, где гетероциклоалкил представляет собой 4-8-членный гетероциклоалкил, содержащий атом(ы) кислорода и/или азота, причем атом азота в гетероциклоалкиле необязательно замещен С1-6 алкилом;
R2 выбран из группы, состоящей из галогена и C1-6 галогеналкила;
R3 выбран из группы, состоящей из C1-6 алкила и галогена.
Предпочтительно, «гетероциклоалкил», описанный выше, представляет собой 4-6-членный гетероциклоалкил, содержащий атом(ы) кислорода и/или азота, такой как пирролидинил, морфолинил, пиперазинил, тетрагидропиранил, тетрагидрофуранил, оксетанил, азетидинил и т.п., и атом азота в этих гетероциклоалкильных группах необязательно замещен C1-6 алкилом. В другом аспекте «гетероспироциклоалкил», описанный выше, может быть выбран из 6-10-членных спироциклоалкильных групп, содержащих гетероатом(ы) кислорода и/или азота.
В предпочтительном варианте реализации кольцо А выбрано из группы, состоящей из 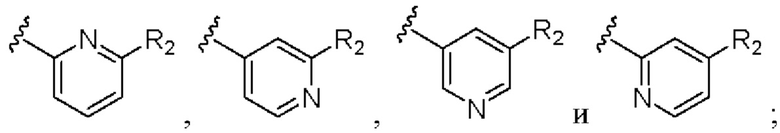 R2 выбран из группы, состоящей из фтора, хлора и трифторметила.
R2 выбран из группы, состоящей из фтора, хлора и трифторметила.
В другом предпочтительном варианте реализации R3 выбран из группы, состоящей из метила, фтора и хлора.
В одном аспекте в настоящем изобретении предложен селективный ингибитор киназы PDGFR, содержащий соединение формулы (Ia) или его фармацевтически приемлемую соль, сольват, сложный эфир, кислоту, метаболит или пролекарство:
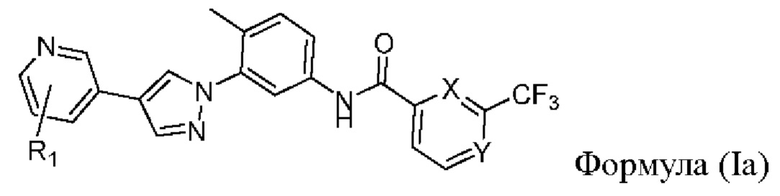
где
R1 выбран из группы, состоящей из гетероциклоалкила, гетероциклоалкилокси, гетероциклоалкил-C1-6-алкокси, гетероциклоалкиламино, гетероспироциклоалкила, гетероспироциклоалкиламино, С3-6-циклоалкил-С1-6-алкокси, С3-6-циклоалкилокси, где гетероциклоалкил представляет собой 4-6-членный гетероциклоалкил, содержащий атом(ы) кислорода и/или азота, причем атом азота в гетероциклоалкиле необязательно замещен C1-6-алкилом; и
один из X и Y представляет собой СН, а другой представляет собой N.
В этом варианте реализации изобретения «гетероциклоалкил» и «гетероспироциклоалкил» соответствуют приведенному выше описанию.
В предпочтительном варианте реализации настоящего изобретения R1 выбран из группы, состоящей из C1-6-алкилпиперазинила (такого как N-метилпиперазинил, например, 4-метилпиперазин-1-ил), морфолинила (такого как N-морфолинил), тетрагидропиранил-C1-6-алкокси (такого как тетрагидропиран-4-илметокси), оксетанилокси (такого как оксетан-3-илокси), морфолино-C1-6-алкокси (такого как 2-морфолиноэтокси), тетрагидрофуранил-С1-6-алкокси (такого как тетрагидрофуран-2-илметокси), С3-6-циклоалкил-С1-6-алкокси (такого как циклопентилметокси) и оксаазаспирогептил (такого как 2-окса-6-аза-спиро[3.3]гепт-6-ил).
Заместитель R1 предпочтительно замещен при атоме углерода в пара- или мета-положении относительно атома N в пиридиновом кольце, и более предпочтительно замещен при атоме углерода в мета-положении относительно атома N в пиридиновом кольце.
В другом аспекте настоящее изобретение также относится к фармацевтической композиции, содержащей соединение, описанное выше, или его фармацевтически приемлемую соль, сольват, сложный эфир, кислоту, метаболит или пролекарство, и фармацевтически приемлемый носитель или вспомогательное вещество и, необязательно, другое терапевтическое средство.
В еще одном аспекте настоящее изобретение также относится к способу или применению такого соединения или фармацевтической композиции для ингибирования активности тирозинкиназы (дикого типа или различных мутантов или их комбинации) и для лечения, предотвращения или уменьшения интенсивности заболевания, нарушения или состояния, которое модулируется активностью тирозинкиназы (дикого типа или различных мутантов или их комбинации), где тирозинкиназа может представлять собой PDGFR.
Настоящее изобретение также относится к ингибитору тирозинкиназы, который селективно оказывает более сильное ингибирующее действие на PDGFR по сравнению с одной или более мишенями cKIT, BCR-ABL, FLT3 и VEGFR2, а также к применению и ингибитора тирозинкиназы и способу с использованием ингибитора тирозинкиназы по настоящему изобретению для селективного ингибирования PDGFR.
Описание графических материалов
На фиг. 1а показано изменение средней массы тела мышей с течением времени в различных группах лечения с использованием соединения 1, иматиниба и носителя в мышиной модели опухоли с клетками хронического эозинофильного лейкоза человека EOL-1;
На фиг. 1b показано изменение среднего размера опухолей с течением времени в различных группах лечения с использованием соединения 1, иматиниба и носителя в мышиной модели опухоли с клетками хронического эозинофильного лейкоза человека EOL-1;
На фиг. 1с показана средняя масса опухолей и рассчитанная степень ингибирования опухоли у мышей на 14 сутки после введения в различных группах лечения с использованием соединения 1, иматиниба и носителя в мышиной модели опухоли с клетками хронического эозинофильного лейкоза человека EOL-1.
На фиг. 2а показано изменение выживаемости крыс с течением времени в различных группах лечения с использованием соединения 1, иматиниба, бозентана и носителя в крысиной модели легочной гипертензии;
На фиг. 2b показано систолическое артериальное давление правого желудочка у крыс в различных группах лечения с использованием соединения 1, иматиниба, бозентана и носителя в крысиной модели легочной гипертензии.
Подробное описание изобретения
Терминология
Если не указано иное, все технические и научные термины, применяемые в настоящем описании, имеют значение, обычно понимаемое специалистами в области, к которой относится заявленное изобретение.
Если не указано иное, в настоящем изобретении используются общепринятые методы масс-спектроскопии, ЯМР-спектроскопии, ВЭЖХ, химии белков, биохимии, технологий рекомбинантной ДНК и фармакологии, известные специалистам в данной области. Если конкретные определения не приведены, номенклатура, используемая в аналитической химии, синтетической органической химии и медицинской и фармацевтической химии, а также их лабораторные процедуры и методики, описанные в настоящем изобретении, представляют собой те, которые известны в данной области техники. В целом вышеупомянутые методики и процедуры могут быть выполнены традиционными способами, хорошо известными в данной области техники и описанными в различных общих и более конкретных источниках, которые цитируются и рассматриваются по тексту настоящего описания.
Термин «алкил» относится к алифатической углеводородной группе, которая может представлять собой алкильную группу с разветвленной или прямой цепью. В зависимости от структуры алкильная группа может представлять собой монорадикал или дирадикал (т.е., алкиленовую группу). В настоящем изобретении алкильная группа предпочтительно представляет собой алкил, содержащий от 1 до 8 атомов углерода, более предпочтительно «низший алкил», содержащий от 1 до 6 атомов углерода, и еще более предпочтительно алкил, содержащий от 1 до 4 атомов углерода. Типичные алкильные группы включают, но не ограничиваются ими, метил, этил, пропил, бутил, пентил, гексил и т.п. Следует понимать, что термин «алкил», указанный в настоящем тексте, включает все возможные конфигурации и конформации алкила, которые могут присутствовать. Например, термин «пропил», упоминаемый в настоящем изобретении, включает н-пропил и изо-пропил. Термин «бутил», упоминаемый в настоящем документе, включает н-бутил, изо-бутил и трет-бутил. Термин «пентил», упоминаемый в настоящем документе, включает н-пентил, изо-пентил, нео-пентил, трет-пентил, пент-3-ил и т.п.
Термин «алкокси» относится к -О-алкильной группе, где алкил соответствует определению в настоящем документе. Типичные алкоксигруппы включают, но не ограничиваются ими, метокси, этокси, пропокси, бутокси, пентилокси, гексилокси и т.п.
Термин «циклоалкил» относится к моноциклическому или полициклическому радикалу, который содержит только углерод и водород. Циклоалкильные группы включают группы, содержащие от 3 до 10 атомов в кольце. В зависимости от структуры циклоалкильная группа может представлять собой монорадикал или дирадикал (например, циклоалкиленовую группу). В настоящем изобретении циклоалкильная группа предпочтительно представляет собой циклоалкил, содержащий от 3 до 8 атомов углерода, и более предпочтительно «низший циклоалкил», содержащий от 3 до 6 атомов углерода. Примеры циклоалкилов включают, но не ограничиваются ими, циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклооктил, циклопентенил, циклогексенил, циклогептенил и диамантанил.
В данном документе термин «гетероциклоалкил» или «гетероциклил» относится к неароматическому кольцу, в котором один или более атомов, образующих кольцо, представляют собой гетероатом, выбранный из группы, состоящей из азота, кислорода и серы. Гетероциклоалкильное кольцо может представлять собой моноциклическое или полициклическое кольцо, образованное из трех, четырех, пяти, шести, семи, восьми, девяти или более чем девяти атомов. Гетероциклоалкильное кольцо может быть необязательно замещено. Примеры гетероциклоалкилов включают, но не ограничиваются ими, лактамы, лактоны, циклические имиды, циклические тиоимиды, циклические карбаматы, тетрагидротиопиран, 4Н-пиран, тетрагидропиран, пиперидин, оксетан, 1,3-диоксин, 1,3-диоксан, 1,4-диоксин, 1,4-диоксан, пиперазин, 1,3-оксатиан, 1,4-оксатиин, 1,4-оксатиан, тетрагидро-1,4-тиазин, 2Н-1,2-оксазин, малеимид, сукцинимид, барбитуровую кислоту, тиобарбитуровую кислоту, диоксопиперазин, гидантоин, дигидроурацил, морфолин, триоксан, гексагидро-1,3,5-триазин, тетрагидротиофен, тетрагидрофуран, пирролин, пирролидин, имидазолидин, пирролидон, пиразолин, пиразолидин, имидазолин, имидазолидин, 1,3-диоксол, 1,3-диоксолан, 1,3-дитиол, 1,3-дитиолан, изоксазолин, изоксазолидин, оксазолин, оксазолидин, оксазолидинон, тиазолин, тиазолидин и 1,3-оксатиолан. В зависимости от структуры гетероциклоалкильная группа может представлять собой монорадикал или дирадикал (т.е., гетероциклоалкиленовую группу).
В настоящем документе термин «спироциклоалкил» относится к 6 10-членной полициклической алифатической гидрокарбильной группе, в которой два отдельных кольца имеют общий атом углерода. Термин «гетероспироциклоалкил» относится к спироциклоалкилу, где один или более атомов, образующих кольцо, представляют собой гетероатом, выбранный из группы, состоящей из азота, кислорода и серы.
Термин «необязательный» означает, что одно или более описанных далее событий могут произойти (иметь место) или не произойти (не иметь места), и включает как происходящие события, так и события, которые не происходят. Термин «необязательно замещенный» или «замещенный» означает, что указанная группа может быть замещена одной или более дополнительной(ыми) группой(ами), каждая из которых независимо выбрана из группы, состоящей из алкила, циклоалкила, арила, гетероарила, гетероциклил а, гидрокси, алкокси, циано, галогена, амида, нитро, галогеналкила, амино, метилсульфонила, алкилкарбонила, алкоксикарбонила, гетероарилалкила, гетероциклоалкилалкилалкила, аминоацила, аминозащитной группы и т.п. Среди прочего, аминозащитная группа предпочтительно выбрана из группы, состоящей из пивалоила, трет-бутилоксикарбонила, бензилоксикарбонила, 9-флуоренилметоксикарбонила, бензила, п-метоксибензила, аллилоксикарбонила, трифторацетила и т.п.
В настоящем документе термин «тирозин-протеинкиназа» (ТПК) относится к классу киназ, которые катализируют перенос γ-фосфата из АТФ в тирозиновый остаток на белках и которые способны катализировать фосфорилирование тирозинового остатка различных белковых субстратов и, таким образом, оказывают важное влияние на рост, пролиферацию и дифференцировку клеток.
В настоящем документе термины «ингибировать», «ингибирующий» или «ингибитор», используемые в отношении киназы, относятся к ингибированию фосфотрансферазной активности.
«Метаболит» соединения, раскрытого в настоящем документе, представляет собой производное указанного соединения, которое образуется при метаболизме данного соединения. Термин «активный метаболит» относится к биологически активному производному соединения, которое образуется при метаболизме указанного соединения. В настоящем документе термин «метаболизм» относится к совокупности процессов (включая, но не ограничиваясь ими, реакции гидролиза и реакции, катализируемые ферментами, такие как реакции окисления), посредством которых организм изменяет конкретное вещество. Так, ферменты могут вызывать специфические структурные изменения, в результате чего образуется соединение. Например, цитохром Р450 катализирует различные окислительно-восстановительные реакции, а дифосфатглюкуронилтрансферазы катализируют перенос активированной молекулы глюкуроновой кислоты на ароматический спирт, алифатический спирт, карбоновую кислоту, амин и свободную меркаптогруппу. Дополнительную информацию о метаболизме можно почерпнуть в The Pharmacological Basis of Therapeutics, 9th Edition, McGraw-Hill (1996). Метаболиты соединения, раскрытого в настоящем документе, могут быть идентифицированы либо путем введения соединения хозяину и анализа образцов ткани хозяина, либо путем инкубации клеток печени с соединением in vitro и анализа полученного соединения. Оба способа хорошо известны в данной области техники. В некоторых вариантах реализации изобретения метаболиты соединения образуются в результате процессов окисления и соответствуют соответствующему гидроксисодержащему соединению. В некоторых вариантах реализации изобретения соединение метаболизируется до фармакологически активных метаболитов. В настоящем документе термин «модулировать» означает взаимодействовать с мишенью прямо или косвенно с изменением активности мишени, включая, только в качестве примера, усиление активности мишени, ингибирование (подавление) активности мишени, ограничение активности мишени или увеличение длительности активности мишени.
В настоящем документе термин «белок-мишень» относится к молекуле белка или части белка, которая может связываться селективно связывающим соединением. В некоторых вариантах реализации изобретения белок-мишень представляет собой тирозинкиназу PDGFR (включая ее дикий тип или различные мутанты или их комбинацию).
В настоящем документе GI50 относится к концентрации лекарственного средства, необходимой для 50% ингибирования роста клеток, т.е. к концентрации лекарственного средства, при которой рост 50% клеток (таких как раковые клетки) может ингибироваться или контролироваться лекарственным средством.
В настоящем документе IC50 относится к количеству, концентрации или дозе конкретного исследуемого соединения, при которой достигается 50% ингибирование максимального ответа в анализе, используемом для измерения такого ответа.
Новый ингибитор киназы согласно настоящему изобретению
В настоящем изобретении предложен селективный ингибитор киназы PDGFR, содержащий соединение формулы (I) или его фармацевтически приемлемую соль, сольват, сложный эфир, кислоту, метаболит или пролекарственый препарат:
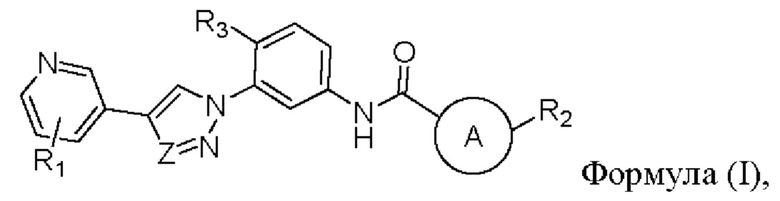
где
кольцо А представляет собой пиридиновое кольцо;
Z выбран из группы, состоящей из N и СН;
R1 выбран из группы, состоящей из гетероциклоалкила, гетероциклоалкилокси, гетероциклоалкил-С1-6-алкокси, гетероциклоалкиламино, гетероспироциклоалкила, гетероспироциклоалкиламино, С3-6-циклоалкил-С1-6-алкокси, С3-6-циклоалкилокси, где гетероциклоалкил представляет собой 4-8-членный гетероциклоалкил, содержащий атом(ы) кислорода и/ил и азота, и атом азота в гетероциклоалкиле необязательно замещен C1-6-алкилом;
R2 выбран из группы, состоящей из галогена и C1-6 галогеналкила;
R3 выбран из группы, состоящей из C1-6 алкила и галогена.
Предпочтительно кольцо А выбрано из группы, состоящей из 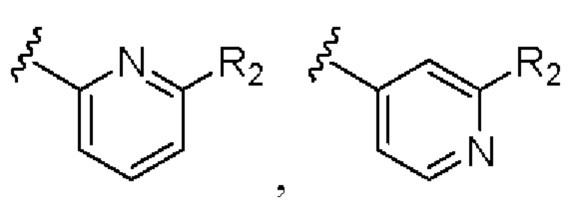
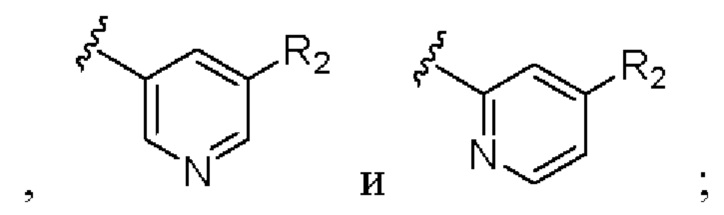 R2 выбран из группы, состоящей из фтора, хлора и трифторметила.
R2 выбран из группы, состоящей из фтора, хлора и трифторметила.
В другом случае R3 предпочтительно выбран из группы, состоящей из метила, фтора и хлора.
В одном варианте реализации настоящего изобретения предложен селективный ингибитор киназы PDGFR, содержащий соединение формулы (Ia) или его фармацевтически приемлемую соль, сольват, сложный эфир, кислоту, метаболит или пролекарство:
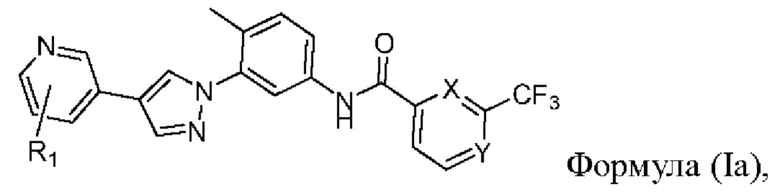
где
R1 выбран из группы, состоящей из гетероциклоалкила, гетероциклоалкилокси, гетероциклоалкил-С1-6-алкокси, гетероциклоалкиламино, гетероспироциклоалкила, гетероспироциклоалкиламино, С3-6-циклоалкил.С1-6-алкокси, С3-6-циклоалкилокси, где гетероциклоалкил необязательно замещен 1-6-алкилом;
один из X и Y представляет собой СН, а другой представляет собой N.
В предпочтительном варианте реализации «гетероциклоалкил», описанный выше, предпочтительно представляет собой 4-6-членный гетероциклоалкил, содержащий атом(ы) кислорода и/или азота, такой как пирролидинил, морфолинил, пиперазинил, тетрагидропиранил, тетрагидрофуранил, оксетанил, азетидинил и т.п., и атом азота в этих гетероциклоалкильных группах необязательно замещен C1-6 алкилом. Термин «гетероспироциклоалкил», описанный выше, предпочтительно представляет собой 6-10-членную спироциклоалкильную группу, содержащую гетероатом(а) кислорода и/или азота.
В предпочтительном варианте реализации R1 выбран из группы, состоящей из 1-6 алкилпиперазинила (такого как N-метилпиперазинил, например, 4-метилпиперазин-1-ил), морфолинила (такого как N-морфолинил), тетрагидропиранил-С1-6-алкокси (такого как тетрагидропиран-4-ил-метокси), оксетанилокси (такого как оксетан-3-илокси), морфолино-С1-6-алкокси (такого как 2-морфолиноэтокси), тетрагидрофуранил-С1-6-алкокси (такого как тетрагидрофуран-2-ил метокси), С3-6-циклоалкил-С1-6-алкокси (такого как циклопентилметокси) и оксаазаспирогептила (такого как, 2-окса-6-азаспиро[3.3]гепт-6-ил).
В другом предпочтительном варианте реализации заместитель R1 замещен при атоме углерода в пара- или положении относительно атома N в пиридиновом кольце, и более предпочтительно замещен при атоме углерода в мета-положении относительно атома N в пиридиновом кольце.
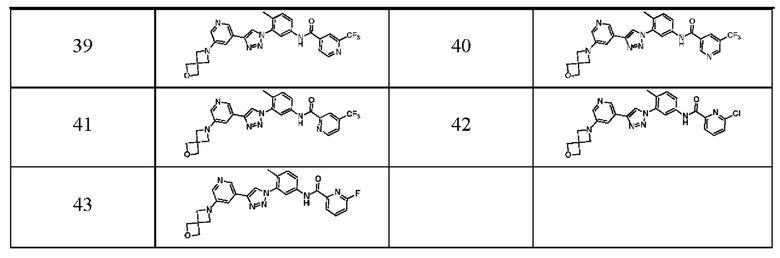
В предпочтительном варианте реализации ингибитор киназы PDGFR согласно настоящему изобретению выбран из группы, состоящей из следующих соединений или их фармацевтически приемлемых солей:
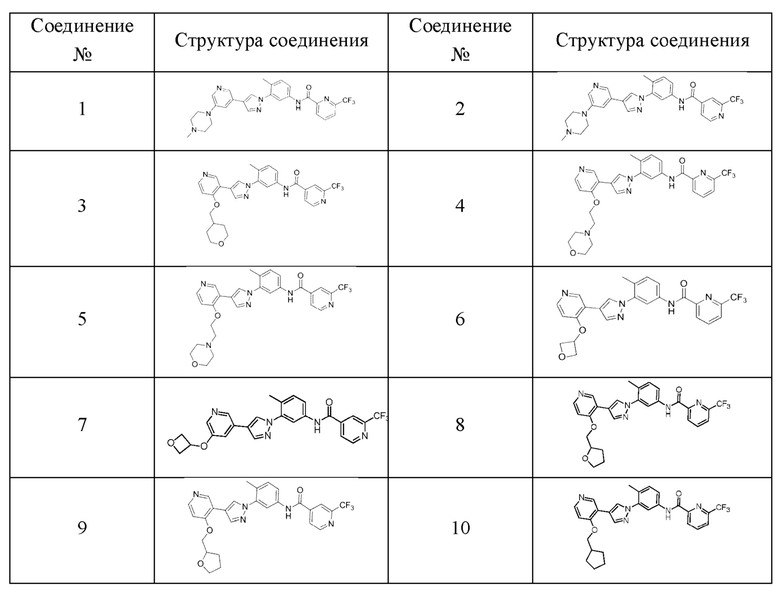

В настоящем документе рассматривается любая комбинация групп, описанных выше для различных переменных. Следует понимать, что заместители и схемы замещения в соединениях, представленных в настоящем документе, могут быть выбраны специалистом в данной области техники для получения химически стабильных соединений, которые могут быть синтезированы методами, известными в данной области, а также методами, указанными в настоящем документе
В настоящем документе описан новый ингибитор киназы. Фармацевтически приемлемые соли, сольваты, сложные эфиры, кислоты, фармацевтически активные метаболиты и пролекарства указанных соединений также описаны в настоящем документе.
В других или дополнительных вариантах реализации соединение, описанное в настоящем документе, метаболизируется при введении в организм, нуждающийся в этом, с образованием метаболита, который затем применяют для получения желаемого эффекта, включая желаемый терапевтический эффект.
Описанное в настоящем документе соединение может быть образовано и/или применено в виде фармацевтически приемлемой соли. Типы фармацевтически приемлемой соли включают, но не ограничиваются ими: (1) соли присоединения кислоты, образованные путем взаимодействия соединения в форме свободного основания с фармацевтически приемлемой неорганической кислотой, такой как хлористоводородная кислота, бромистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота, метафосфорная кислота или т.п.; или с органической кислотой, такой как уксусная кислота, пропионовая кислота, гексановая кислота, циклопентанпропионовая кислота, гликолевая кислота, пировиноградная кислота, молочная кислота, малоновая кислота, яблочная кислота, лимонная кислота, янтарная кислота, малеиновая кислота, винная кислота, фумаровая кислота, трифторуксусная кислота, бензойная кислота, 3-(4-гидроксибензоил)бензойная кислота, коричная кислота, миндальная кислота, метансульфоновая кислота, этансульфоновая кислота, 1,2-этандисульфоновая кислота, 2-гидроксиэтансульфоновая кислота, бензолсульфоновая кислота, толуолсульфоновая кислота, 4-метилбицикло-[2.2.2]окт-2-ен-1-карбоновая кислота, 2-нафталинсульфоновая кислота, трет -бутилуксусная кислота, глюкогептоновая кислота, 4,4'-метиленбис-(3-гидрокси-2-ен-1-карбоновая кислота), 3-фенилпропионовая кислота, триметилуксусная кислота, лаурилсерная кислота, глюконовая кислота, глутаминовая кислота, салициловая кислота, гидроксинафтойная кислота, стеариновая кислота, муконовая кислота или т.п.; (2) соли присоединения основания, образованные, когда кислотный протон, присутствующий в исходном соединении, либо замещен ионом металла, таким как ион щелочного металла (такой как, литий, натрий, калий), ионом щелочноземельного металла (таким как, магний или кальций), либо ионом алюминия; или координируется с органическим основанием или неорганическим основанием. Приемлемые органические основания включают этаноламин, диэтаноламин, триэтаноламин, триметиламин, N-метилглюкамин и тому подобное. Приемлемые неорганические основания включают алюминия гидроксид, кальция гидроксид, калия гидроксид, натрия карбонат, натрия гидроксид и т.п.
Соответствующие противоионы фармацевтически приемлемой соли могут быть исследованы и идентифицированы с применением различных методов, включая, но не ограничиваясь ими, ионообменную хроматографию, ионную хроматографию, капиллярный электрофорез, атомно-эмиссионную спектроскопию с индуктивно связанной плазмой, атомно-абсорбционную спектроскопию, масс-спектрометрию или любую их комбинацию.
Соли выделяют с использованием по меньшей мере одного из следующих методов: фильтрация, осаждение осадителем с последующей фильтрацией, выпаривание растворителя или, в случае водных растворов, лиофилизация.
Скрининг и определение характеристик фармацевтически приемлемых солей, полиморфных форм и/или сольватов могут быть осуществлены с применением различных методов, включая, но не ограничиваясь ими, термический анализ, рентгеновскую порошковую дифрактометрию, спектроскопию, микроскопию и элементный анализ. Различные применяемые спектроскопические методы включают, но не ограничиваются ими, рамановскую спектроскопию, ИК-спектрометрию с Фурье-преобразованием (FTIR), спектрофотометрию в УФ и видимой областях спектра и ЯМР-спектроскопию (в жидком и твердом состоянии). Различные методы микроскопии включают, но не ограничиваются ими, ИК-микроскопию и рамановскую микроскопию.
Фармацевтическая композиция согласно настоящему изобретению
В настоящем изобретении также предложена фармацевтическая композиция, содержащая по меньшей мере одно соединение формулы (I) или фармацевтически приемлемую соль, сольват, сложный эфир, кислоту, фармацевтически активный метаболит или пролекарство соединения и фармацевтически приемлемый носитель или вспомогательное вещество и необязательно другой терапевтический агент.
Во время лечения ее можно применять отдельно или в комбинации с одним или более другими терапевтическими агентами. Лекарственный препарат, содержащий соединение согласно настоящему изобретению, может быть введен пациенту посредством не менее одного способа, выбранного из инъекции, перорального введения, ингаляции, ректального и трансдермального введения. Другие терапевтические агенты могут быть выбраны из группы, состоящей из иммунодепрессантов (таких как такролимус, циклоспорин, рапамицин, метотрексат, циклофосфамид, азатиоприн, меркаптопурин, микофенолат или FTY720), глюкокортикоидов (таких как преднизон, кортизона ацетат, преднизолон, метилпреднизолон, дексаметазон, бетаметазон, триамцинолон, фторгидроксипреднизолон, беклометазон, фторгидрокортизона ацетат, дезоксикортикостерона ацетат, альдостерон), нестероидных противовоспалительных средств (таких как салицилаты, арилалкановые кислоты, 2-арилпропионовые кислоты, N-арилантранильные кислоты, оксикамы, коксибы или сульфонанилиды), аллерговакцины, антигистаминные средства, антилейкотриены, β-агонисты, теофиллин, антихолинергические средства или другие селективные ингибиторы киназ (таких как ингибиторы mTOR, ингибиторы с-Met) или агенты на основе her2-антител. Кроме того, указанные другие терапевтические агенты также могут представлять собой рапамицин, кризотиниб, тамоксифен, ралоксифен, анастрозол, эксеместан, летрозол, Герцептин™ (трастузумаб), Гливек™ (иматиниба мезилат), Таксол™ (паклитаксел), циклофосфамид, ловастатин, минозин, цитарабин, 5-фторурацил (5-ФУ), метотрексат, Таксотер™ (доцетаксел), Золадекс™ (гозерелин), винкристин, винбластин, нокодазол, тенипозид, этопозид, Гемзар™ (гемцитабин), эпотилон, навельбин, камптотецин, даунонибицин, дактиномицин, митоксантрон, амсакрин, доксорубицин (адриамицин), эпирубицин или идарубицин. В качестве альтернативы другие терапевтические агенты могут представлять собой, например, но не ограничиваясь ими, цитокины, такие как ГКСФ (гранулоцитарный колониестимулирующий фактор). В качестве альтернативы другими терапевтическими агентами могут быть, например, но не ограничиваясь ими, CMF (циклофосфамид, метотрексат и 5-фторурацил), CAF (циклофосфамид, адриамицин и 5-фторурацил), АС (адриамицин и циклофосфамид), FEC (5-фторурацил, эпирубицин и циклофосфамид), ACT или АТС (адриамицин, циклофосфамид и паклитаксел) или CMFP (циклофосфамид, метотрексат, 5-фторурацил и преднизолон).
В вариантах реализации настоящего изобретения при лечении пациента в соответствии с настоящим изобретением количество конкретного агента будет варьировать в зависимости от таких факторов, как конкретный режим дозирования, тип заболевания или состояния и его тяжесть, отличительные черты (например, масса тела) субъекта или хозяина, нуждающегося в лечении, но может быть легко определено способом, известным в данной области техники, в соответствии с конкретными обстоятельствами, включая, например, конкретный вводимый агент, способ введения, подвергаемое лечению состояние и подвергаемого лечению субъекта или хозяина. В целом дозы, используемые для лечения взрослых людей, обычно находятся в диапазоне 0,02-5000 мг в сутки, например около 1-1500 мг в сутки. Необходимая доза может быть удобным образом представлена в виде разовой дозы или отдельных доз, вводимых одновременно (или в течение короткого периода времени) или с соответствующими интервалами, например, в виде двух, трех, четырех или более субдоз в сутки. Специалистам в данной области техники будет понятно, что, хотя приведены вышеупомянутые диапазоны доз, конкретные эффективные количества могут быть соответствующим образом скорректированы в зависимости от состояния пациента и суждения практикующего врача.
Применение лекарственных препаратов согласно настоящему изобретению Соединение или его фармацевтически приемлемая соль, сольват, сложный эфир, кислота, метаболит или пролекарство или фармацевтическая композиция согласно настоящему изобретению способны селективно ингибировать активность тирозинкиназы PDGFR (дикого типа или различных мутантов или их комбинации), в частности, активность PDGFRα и PDGFRβ, и, более конкретно, активность PDGFRα. Соединение или его фармацевтически приемлемую соль, сольват, сложный эфир, кислоту, метаболит или пролекарство или фармацевтическую композицию согласно настоящему изобретению можно применять для лечения, предотвращения или облегчения одного или более заболеваний, нарушений или состояний, которые модулируются активностью PDGFR, или зависят от нее или участвуют в ней (в частности, PDGFRα и PDGFRβ), таких как заболевание, выбранное из группы, состоящей из легочной артериальной гипертензии, солидных опухолей (включая доброкачественные или злокачественные типы), саркомы, гастроинтестинальных стромальных опухолей, рака толстой кишки, острого миелобластного лейкоза (ОМЛ), хронического миелолейкоза (ХМЛ), неоплазии, карциномы щитовидной железы, системного мастоцитоза, синдрома эозинофилии, хронического эозинофильного лейкоза, фиброза, красной волчанки, болезни «трансплантат против хозяина», нейрофиброматозы, легочной гипертензии, болезни Альцгеймера, семиномы, дисгерминомы, опухоли тучных клеток, рака легкого, карциномы бронхов, внутриэпителиальной неоплазии яичка, меланомы, рака молочной железы, нейробластомы, папиллярной/фолликулярной карциномы щитовидной железы, злокачественной лимфомы, неходжкинской лимфомы, множественной эндокринной неоплазии 2 типа, феохромоцитомы, карциномы щитовидной железы, паращитовидной гиперплазии/аденомы, рака толстой кишки, колоректальной аденомы, рака яичников, рака предстательной железы, глиобластомы, опухоли головного мозга, злокачественной глиомы, рака поджелудочной железы, злокачественной мезотелиомы плевры, гемангиобластомы, гемангиомы, рака почки, рака печени, карциномы надпочечников, рака мочевого пузыря, рака желудка, рака прямой кишки, рака влагалища, рака шейки матки, рака эндометрия, мыножественной миеломы, опухоли шеи и головы, а также из других пролиферативных состояний или т.п., или их комбинации. Данное соединение особенно предпочтительно для лечения легочной артериальной гипертензии, хронического эозинофильного лейкоза или т.п.или их комбинации.
Соединение или его фармацевтически приемлемая соль, сольват, сложный эфир, кислота, метаболит или пролекарство или фармацевтическая композиция согласно настоящему изобретению пригодны для лечения, предотвращения или облегчения аутоиммунного заболевания, выбранного из группы, состоящей из артрита, ревматического артрита, волчанки, ревматоидного артрита, воспалительного заболевания кишечника, псориатического артрита, остеоартрита, болезни Стилла, ювенильного артрита, диабета, миастении гравис, тиреоидита Хашимото, тиреоидита Орда, болезни Грейвса, синдрома Шегрена, рассеянного склероза, синдрома Гийена-Барре, острого диссеминированного энцефаломиелита, болезни Аддисона, синдрома опсоклонус-миоклонус, анкилозирующего спондилита, синдрома антифосфолипидных антител, апластической анемии, аутоиммунного гепатита, целиакии, синдрома Гудпасчера, идиопатической тромбоцитопенической пурпури, неврита зрительного нерва, склеродермии, первичного билиарного цирроза, синдрома Рейтера, артериита Такаясу, височного артериита, аутоиммунной гемолитической анемии с тепловыми антителами, гранулематоза Вегенера, псориаза, алопеции универсальной, болезни Бехчета, хронической усталости, дизаутономии, эндометриоза, интерстициального цистита, нейромиотонии, склеродермы, вульводинии или их комбинации.
Получение соединения
Соединение по настоящему изобретению может быть синтезировано с применением стандартных способов синтеза, известных специалистам в данной области техники, или с применением способов, известных в данной области техники, в комбинации со способами, описанными в настоящем изобретении. Кроме того, растворители, температуры и другие условия реакций, представленные в настоящем изобретении, могут варьировать в зависимости от методик в данной области техники. В качестве дополнительного руководства также могут использоваться следующие способы синтеза.
Описанные реакции могут использоваться последовательно для получения соединений, описанных в настоящем изобретении, или они могут использоваться для синтеза строительных блоков, которые впоследствии соединяют способами, описанными в настоящем изобретении и/или известными в данной области техники.
В некоторых вариантах реализации в настоящем документе предложены способы получения и способы применения соединений, ингибиторов тирозинкиназы, описанных в настоящем изобретении. В определенных вариантах реализации соединения, описанные в настоящем документе, можно синтезировать с помощью следующих схем синтеза. Соединения могут быть синтезированы с применением методик, аналогичных описанным ниже, с применением соответствующего альтернативного сырья.
Сырье, используемое для синтеза соединений, описанных в настоящем изобретении, может быть синтезировано или получено на коммерческой основе. Соединения, описанные в настоящем изобретении, и другие родственные примеси, имеющие отличающиеся заместители, могут быть синтезированы с применением методов и материалов, известных специалистам в данной области техники. Общие способы получения соединений, описанных в настоящем изобретении, могут быть получены на основе реакций, известных в данной области, и реакции могут быть модифицированы с применением соответствующих реактивов и условий, как будет понятно специалисту в данной области техники, для введения различных фрагментов в молекулы, предложенные в настоящем изобретении.
Продукты реакции могут быть выделены и очищены при необходимости с применением традиционных методов, включая, но не ограничиваясь ими, фильтрацию, дистилляцию, кристаллизацию, хроматографию и т.п.Характеристики таких продуктов могут быть определены с применением традиционных средств, включая физические константы и спектральные данные.
Пример 1.
Синтез Н-(4-метил-3-(4-(5-(4-метилпиперазин-1-ил)пиридин-3-ил)-1Н-пиразол-1-ил)фенил)-6-(трифторметил)пиколинамида 1
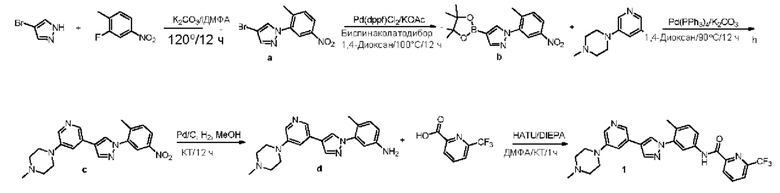
Этап 1. Синтез соединения 4-бром-1-(2-метил-5-нитрофенил)-1Н-пиразола а:
Соединения 4-бромпиразол (5 г, 1 экв.), 2-фтор-1-метил-4-нитробензол (5,5 г, 1,05 экв.) и калия карбоната (13,1, 3 экв.) смешивали в ДМФА (50 мл). Смесь перемешивали в течение ночи при температуре 120°С в атмосфере азота, далее охлаждали и концентрировали. В концентрат добавляли этилацетат (200 мл). Далее полученную смесь последовательно промывали водой и насыщенным солевым раствором, сушили над безводным натрия сульфатом и фильтровали. Фильтрат концентрировали, далее подвергали колоночной хроматографии с получением желтого продукта а (5,2 г).
Этап 2. Синтез соединения 1-(2-метил-5-нитрофенил)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)- 1Н-пиразола b:
Соединение а (5 г, 1 экв.), биспинаколатодибор (5,8 г, 1,3 экв.), калия ацетат (3,5 г, 2 экв.) и [1,1'-бис(дифенилфосфино)ферроцен]палладия дихлорид (0,72 г, 0,05 экв.) смешивали в 1,4-диоксане (50 мл). Смесь перемешивали в течение ночи при температуре 100°С в атмосфере азота и далее концентрировали. Концентрат подвергали колоночной хроматографии с получением желтого продукта b (4,0 г).
Этап 3. Синтез 1-метил-4-(5-(1-(2-метил-5-нитрофенил)-1Н-пиразол-4-ил)пиридин-3-ил)пиперазина с:
Соединение b (4,0 г, 1,1 экв.), 1-(5-бромпиридин-3-ил)-4-метилпиперазин (2,8 г, 1 экв.), калия карбонат (3,0 г, 2 экв.) итетракис(трифенилфосфин)палладий(0,6 г, 0,05 экв.) смешивали в 1,4-диоксане (40 мл) и воде (4 мл). Смесь перемешивали в течение ночи при температуре 90°С в атмосфере азота и далее концентрировали. Концентрат подвергали колоночной хроматографии с получением желтого продукта с (3,8 г).
Этап 4. Синтез 4-метил-3-(4-(5-(4-метилпиперазин-1-ил)пиридин-3-ил)-1Н-пиразол-1-ил)анилина d:
Соединение с (2,8 г, 1 экв.) и палладий на угле (0,5 г) смешивали в метаноле (30 мл). Смесь перемешивали в течение 2 ч при комнатной температуре в атмосфере водорода. Далее добавляли дихлорметан (100 мл) для разведения смеси. Полученную смесь фильтровали и концентрировали с получением бледно-зеленого продукта d (2,1 г).
Этап 5. Синтез соединения N-(4-метил-3-(4-(5-(4-метилпиперазин-1-ил)пиридин-3-ил)-1Н-пиразол-1-ил)фенил)-6-(трифторметил)пиколинамида 1:
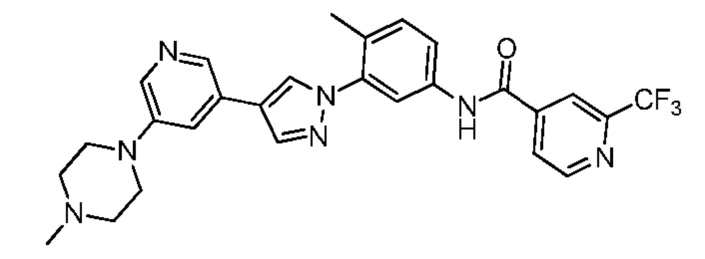
Соединение d (0,05 г, 1 экв.), 6-(трифторметил)пиридин-2-карбоновая кислота (0,27 г, 1 экв.), 2-(7-азабензотриазол)-N,N,N',N'-тетраметилурония гексафторфосфат HATU (0,072 г, 1,1 экв.) и диизопропилэтилендиамин (DIEPA) (0,22 г, 1 экв.) смешивали в N,N-диметилформамиде ДМФА (2 мл). Смесь перемешивали при комнатной температуре в течение 1 ч. Далее добавляли этилацетат (50 мл) для разведения смеси. Смесь последовательно промывали водой и насыщенным солевым раствором, сушили над безводным натрия сульфатом и фильтровали. Фильтрат концентрировали с получением продукта 1 (0,07 г). Точная масса (расчетная): 521,21; МС (ионизация электрораспылением) m/z (М+1)+: 522,21.
Пример 2.
Синтез N-(4-метил-3-(4-(5-(4-метилпиперазин-1-ил)пиридин-3-ил)-1Н-пиразол-1-ил)фенил)-2-(трифторметил)изоникотинамида 2
Соединение 2 синтезировали с применением этапов, аналогичных описанным в примере 1. Точная масса (расчетная): 521,21; МС (ионизация электрораспылением) m/z (М+1)+: 522,21.
Пример 3.
Синтез N-(4-метил-3-(4-(4-((тетрагидропиран-4-ил)метокси)пиридин-3-ил)-1Н-пиразол-1-ил)фенил)-2-(трифторметил)изоникотинамида 3
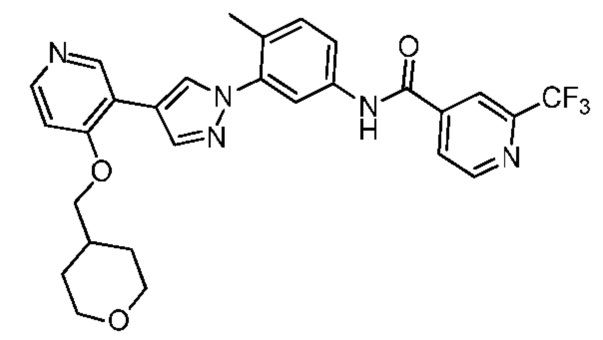
Соединение 3 синтезировали с применением этапов, аналогичных описанным в примере 1. Точная масса (расчетная): 537,19; МС (ионизация электрораспылением) m/z (М+1)+: 538,19.
Пример 4.
Синтез N-(4-метил-3-(4-(4-(2-морфолиноэтокси)пиридин-3-ил)-1Н-пиразол-1-ил)фенил)-6-(трифторметил)пиколинамида 4
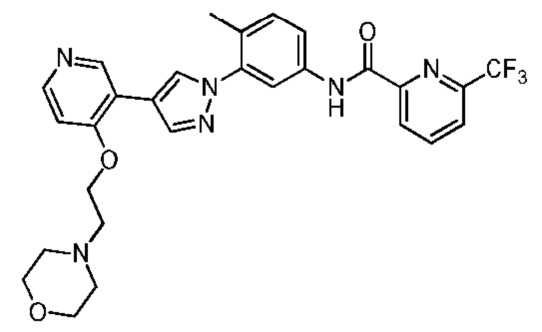
Соединение 4 синтезировали с применением этапов, аналогичных описанным в примере 1. Точная масса (расчетная): 552,20; МС (ионизация электрораспылением) m/z (М+1)+: 553,20.
Пример 5.
Синтез N-(4-метил-3-(4-(4-(2-морфолиноэтокси)пиридин-3-ил)-1Н-пиразол-1-ил)фенил)-2-(трифторметил)изоникотинамида 5
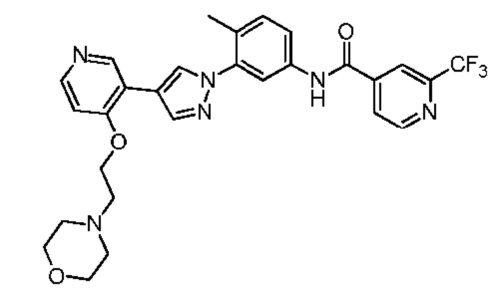
Соединение 5 синтезировали с применением этапов, аналогичных описанным в примере 1. Точная масса (расчетная): 552,20; МС (ионизация электрораспылением) m/z (М+1)+: 553,20.
Пример 6.
Синтез N-(4-метил-3-(4-(4-(оксетан-3-илокси)пиридин-3-ил)-Ш-пиразол-1-ил)фенил)-6-(трифторметил)пиколинамида 6
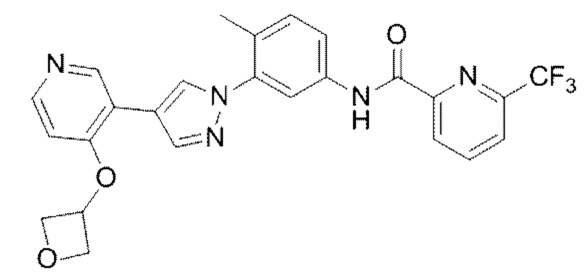
Соединение 6 синтезировали с применением этапов, аналогичных описанным в примере 1. Точная масса (расчетная): 495,15; МС (ионизация электрораспылением) m/z (М+1)+: 496,15.
Пример 7.
Синтез N-(4-метил-3-(4-(5-(оксетан-3-илокси)пиридин-3-ил)-1H-пиразол-1-ил)фенил)-2-(трифторметил)изоникотинамида 7
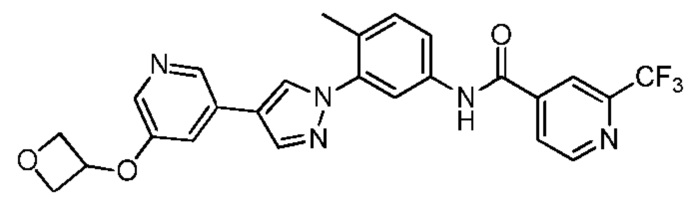
Соединение 7 синтезировали с применением этапов, аналогичных описанным в примере 1. Точная масса (расчетная): 495,15; МС (ионизация электрораспылением) m/z (М+1)+: 496,15.
Пример 8.
Синтез N-(4-метил-3-(4-(4-((тетрагидрофуран-2-ил)метокси)пиридин-3-ил)-1Н-пиразол-1-ил)фенил)-6-(трифторметил)пиколинамид а 8
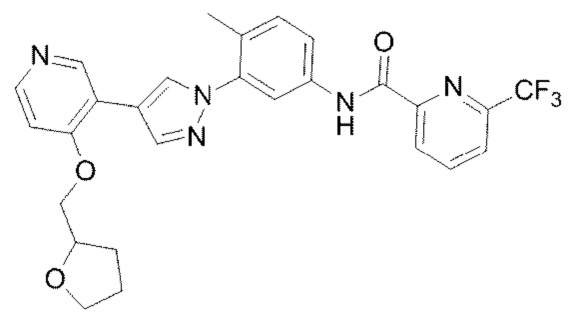
Соединение 8 синтезировали с применением этапов, аналогичных описанным в примере 1. Точная масса (расчетная): 523,18; МС (ионизация электрораспылением) m/z (М+1)+: 524,18.
Пример 9.
Синтез N-(4-метил-3-(4-(4-((тетрагидрофуран-2-ил)метокси)пиридин-3-ил)-1Н-пиразол-1-ил)фенил)-2-(трифторметил)изоникотинамида 9
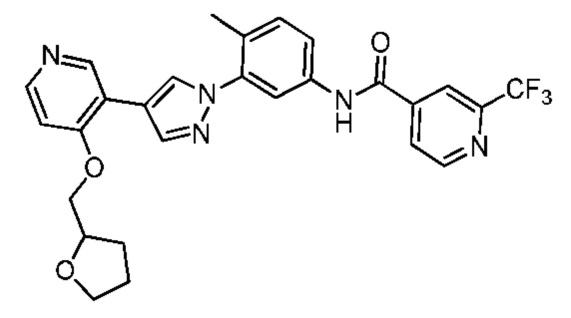
Соединение 9 синтезировали с применением этапов, аналогичных описанным в примере 1. Точная масса (расчетная): 523,18; МС (ионизация электрораспылением) m/z (M+l)+: 524,18.
Пример 10.
Синтез N-(3-(4-(4-(циклопентилметокси)пиридин-3-ил)-1H-пиразол-1-ил)-4-метилфенил)-6-(трифторметил)пиколинамида 10
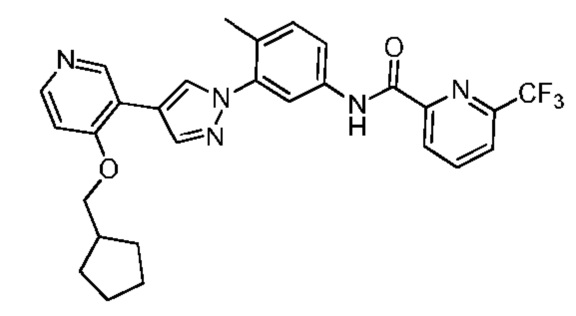
Соединение 10 синтезировали с применением этапов, аналогичных описанным в примере 1. Точная масса (расчетная): 521,20; МС (ионизация электрораспылением) m/z (М+1)+: 522,20.
Пример 11.
Синтез N-(3-(4-(4-(циклопентилметокси)пиридин-3-ил)-1H-пиразол-1-ил)-4-метилфенил)-2-(трифторметил)изоникотинамида 11
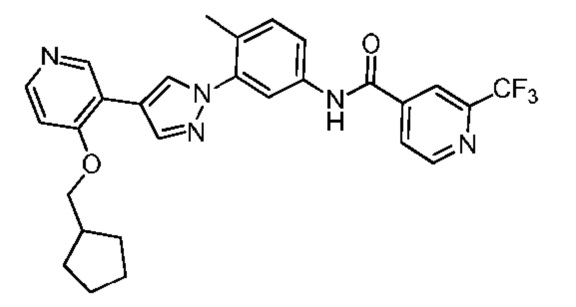
Соединение 11 синтезировали с применением этапов, аналогичных описанным в примере 1. Точная масса (расчетная): 521,20; МС (ионизация электрораспылением) m/z (М+1)+: 522,20.
Пример 12.
Синтез N-(4-метил-3-(4-(5-морфолинопиридин-3-ил)-1Н-пиразол-1-ил)фенил)-2(трифторметил)изоникотинамида 12
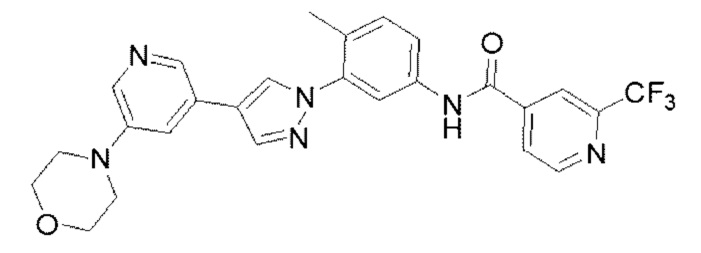
Соединение 12 синтезировали с применением этапов, аналогичных описанным в примере 1. Точная масса (расчетная): 508,18; МС (ионизация электрораспылением) m/z (М+1)+: 509,18.
Пример 13.
Синтез N-(4-метил-3-(4-(4-((тетрагидропиран-4-ил)метокси)пиридин-3-ил)-1Н-пиразол-1-ил)фенил)-6-(трифторметил)пиколинамида 13
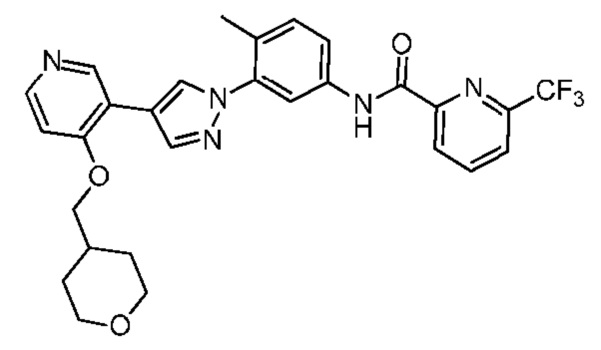
Соединение 13 синтезировали с применением этапов, аналогичных описанным в примере 1. Точная масса (расчетная): 537,19; МС (ионизация электрораспылением) m/z (М+1)+: 538,19.
Пример 14.
6-Фтор-N-(4-метил-3 -(4-(5-(4-метилпиперазин-1-ил)пиридин-3-ил)-1Н-пиразол-1-ил)фенил)пиколинамид 14
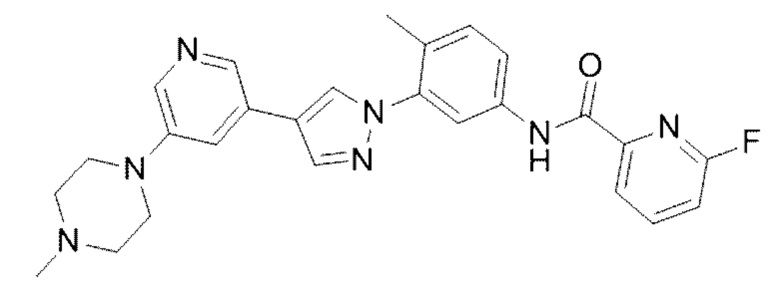
Соединение 14 синтезировали с применением этапов, аналогичных описанным в примере 1. Точная масса (расчетная): 471,21; МС (ионизация электрораспылением) m/z (М+1)+: 472,21.
Пример 15.
N-(4-Метил-3-(4-(5-(4-метилпиперазин-1-ил)пиридин-3-ил)-1Н-пиразол-1-ил)фенил)-4-(трифторметил)пиколинамид 15
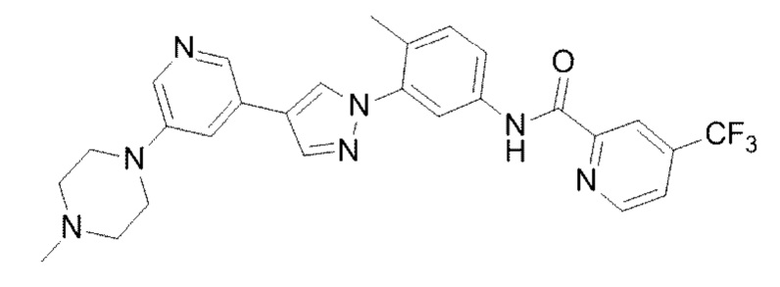
Соединение 15 синтезировали с применением этапов, аналогичных описанным в примере 1. Точная масса (расчетная): 521,21; МС (ионизация электрораспылением) m/z (М+1)+: 522,21.
Пример 16.
N-(4-Метил-3-(4-(5-(4-метилпиперазин-1-ил)пиридин-3-ил)-1Н-пиразол-1-ил)фенил)-5-(трифторметил)никотинамид 16
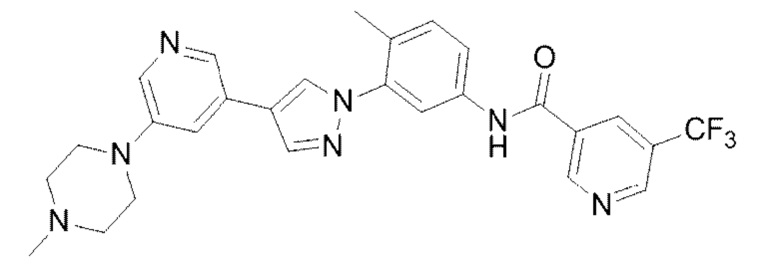
Соединение 16 синтезировали с применением этапов, аналогичных описанным в примере 1. Точная масса (расчетная): 521,21; МС (ионизация электрораспылением) m/z (М+1)+: 522,21.
Пример 17.
6-Хлор-N-(4-метил-3-(4-(5-(4-метилпиперазин-1-ил)пиридин-3-ил)-1H-пиразол-1-ил)фенил)пиколинамид 17
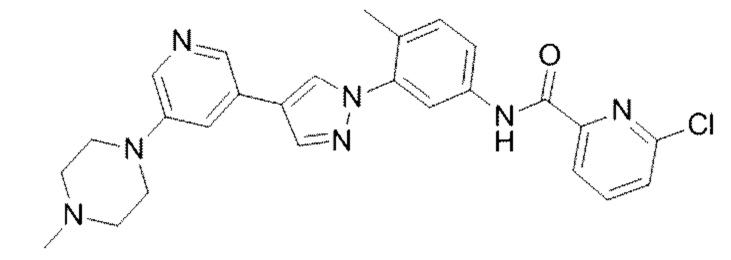
Соединение 17 синтезировали с применением этапов, аналогичных описанным в примере 1. Точная масса (расчетная): 487,18; МС (ионизация электрораспылением) m/z (М+1)+: 488,18.
Пример 18.
Синтез (4-метил-3-(4-(5-(4-метилпиперазин-1-ил)пиридин-3-ил)-1H-1,2,3-триазол-1-ил)фенил)-6-(трифторметил)пиколинамида 18
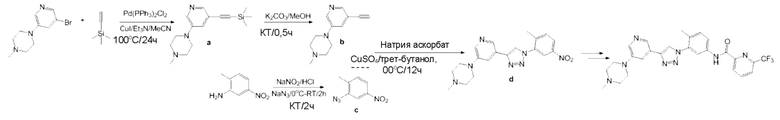
Этап 1. Синтез 1-метил-4-(5-(2-(триметилсилил)этинил)пиридин-3-ил)пиперазина а Смешивали 1-(5-бромпиридин-3-ил)-4-метилпиперазин (5 г, 1 экв), триметилсилилацетилен (5,7 г, 3 экв.), Pd(PPh3)2Cl2 (0,7 г, 0,05 экв.), Et3N (5,9 г, 3 экв.), CuI (0,18 г, 0,05 экв.) и ацетонитрил (50 мл). Смесь перемешивали в течение 24 часов при температуре 100°С при защите газообразным азотом, далее охлаждали. Твердое вещество фильтровывали. Фильтрат концентрировали и подвергали колоночной хроматографии с получением коричневого твердого вещества а (4,5 г).
Этап 2. Синтез 1-(5-этинилпиридин-3-ил)-4-метилпиперазина b
1-Метил-4-(5-(2-(триметилсилил)этинил)пиридин-3-ил)пиперазин а (4 г, 1 экв.), K2CO3 (4 г, 2 экв.) и метанол (20 мл) перемешивали в течение 0,5 ч при комнатной температуре. Далее добавляли этилацетат (20 мл) для разведения смеси. Полученную смесь последовательно промывали водой и насыщенным солевым раствором, сушили над безводным натрия сульфатом и фильтровали. Фильтрат концентрировали с получением черного масла b (2,4 г).
Этап 3. Синтез 2-азидо-1-метил-4-нитробензола с
2-Метил-5-нитро анилин (5 г, 1 экв.) растворяли в HCl (6,0 моль/л, 4,8 экв.). По каплям добавляли водный раствор NaNO2 (2,3 г, 1 экв.), далее водный раствор NaN3 (2,6 г, 1,2 экв.) при температуре 0°С. Смесь перемешивали в течение 2 ч при комнатной температуре. Далее добавляли воду (200 мл). Полученную смесь фильтровали. Осадок на фильтре промывали водой и сушили с получением желтого твердого вещества с (5,3 г).
Этап 4. Синтез 1-метил-4-(5-(1-(2-метил-5-нитрофенил)-1Н-1,2,3-триазол-4-ил)пиридин-3-ил)пиперазина d
1-(5-Этинилпиридин-3-ил)-4-метилпиперазин b (2 г, 1 экв.), 2-азидо-1-метил-4-нитробензол с (1,8 г, 1 экв.), натрия аскорбат (0,4 г, 0,2 экв.), CuSO4 (0,16 г, 0,1 экв.) и трет-бутанол/вода (1:1, 30 мл) перемешивали в течение ночи при температуре 90°С. Полученную смесь охлаждали и концентрировали. Концентрат подвергали колоночной хроматографии с получением желтого твердого вещества d (3,1 г).
Синтез соединения 18 завершили с применением этапов, аналогичных последним двум этапам, описанным в примере 1. Точная масса (расчетная): 522,21; МС (ионизация электрораспылением) m/z (М+1)+: 523,21.
Пример 19.
N-(4-Метил-3-(4-(5-(4-метилпиперазин-1-ил)пиридин-3-ил)-1Н-1,2,3-триазол-1-ил)фенил)-2-(трифторметил)изоникотинамид 19
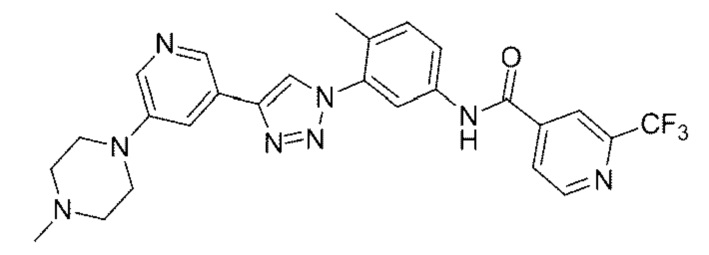
Соединение 19 синтезировали с применением этапов, аналогичных описанным в примере 18. Точная масса (расчетная): 522,21; МС (ионизация электрораспылением) m/z (М+1)+: 523,21.
Пример 20.
N-(4-Метил-3-(4-(5-(4-метилпиперазин-1-ил)пиридин-3-ил)-1Н-1,2,3-триазол-1-ил)фенил)-5-(трифторметил)никотинамид 20
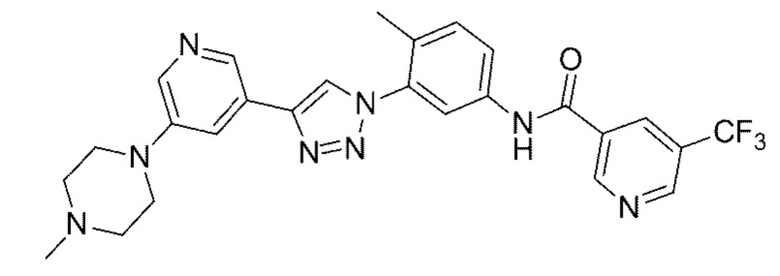
Соединение 20 синтезировали с применением этапов, аналогичных описанным в примере 18. Точная масса (расчетная): 522,21; МС (ионизация электрораспылением) m/z (М+1)+: 523,21.
Пример 21.
N-(4-Метил-3-(4-(5-(4-метилпиперазин-1-ил)пиридин-3-ил)-1Н-1,2,3-триазол-1-ил)фенил)-4-(трифторметил)пиколинамид 21
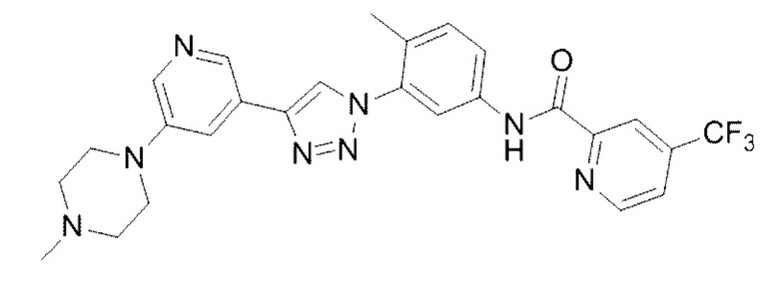
Соединение 21 синтезировали с применением этапов, аналогичных описанным в примере 18. Точная масса (расчетная): 522,21; МС (ионизация электрораспылением) m/z (М+1)+: 523,21.
Пример 22.
6-Хлор-N-(4-метил-3 -(4-(5-(4-метилпиперазин-1-ил)пиридин-3-ил)-1H-1,2,3-триазол-1-ил)фенил)пиколинамид 22
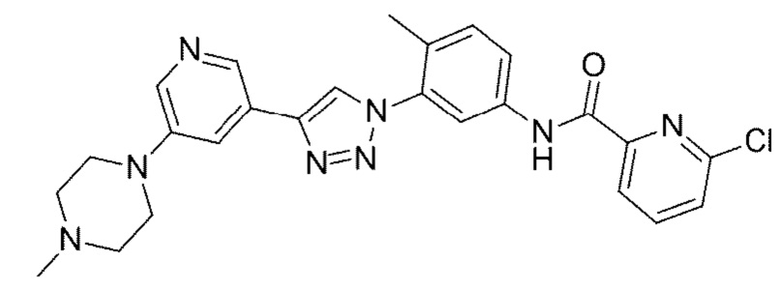
Соединение 22 синтезировали с применением этапов, аналогичных описанным в примере 18. Точная масса (расчетная): 488,18; МС (ионизация электрораспылением) m/z (М+1)+: 489,18.
Пример 23.
6-Фтор-N-(4-метил-3-(4-(5-(4-метилпиперазин-1-ил)пиридин-3-ил)-1H-1,2,3-триазол-1-ил)фенил)пиколинамид 23
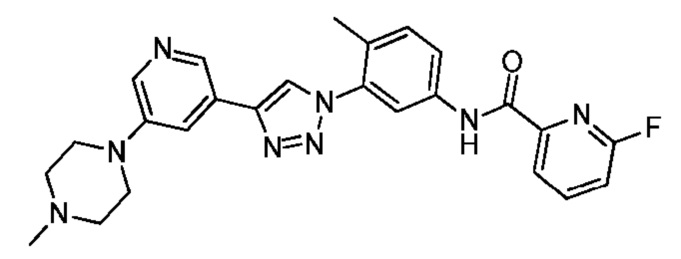
Соединение 23 синтезировали с применением этапов, аналогичных описанным в примере 18. Точная масса (расчетная): 472,21; МС (ионизация электрораспылением) m/z (М+1)+: 473,21.
Пример 24.
N-(4-Фтор-3-(4-(5-(4-метилпиперазин-1-ил)пиридин-3-ил)-1H-1,2,3-триазол-1-ил)фенил)-6-(трифторметил)пиколинамид 24
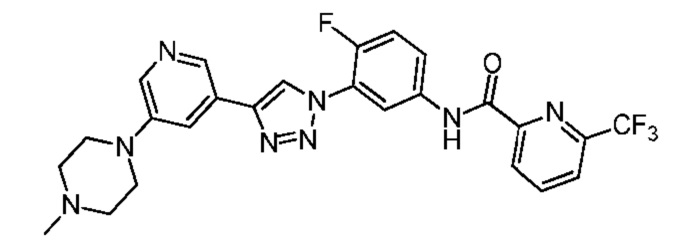
Соединение 24 синтезировали с применением этапов, аналогичных описанным в примере 18. Точная масса (расчетная): 526,18; МС (ионизация электрораспылением) m/z (М+1)+: 527,18.
Пример 25.
N-(4-Фтор-3-(4-(5-(4-метилпиперазин-1-ил)пиридин-3-ил)-1H-1,2,3-триазол-1-ил)фенил)-2-(трифторметил) изоникотинамид 25
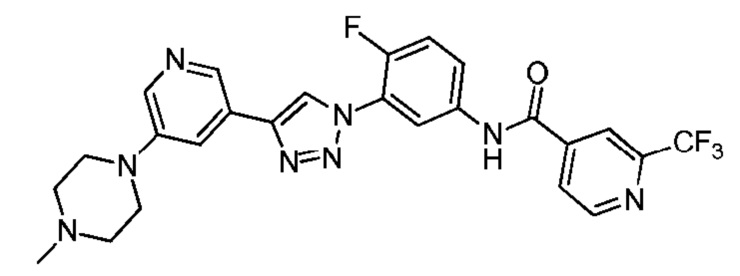
Соединение 25 синтезировали с применением этапов, аналогичных описанным в примере 18. Точная масса (расчетная): 526,18; МС (ионизация электрораспылением) m/z (М+1)+: 527,18.
Пример 26.
N-(4-Фтор-3-(4-(5-(4-метилпиперазин-1-ил)пиридин-3-ил)-1H-1,2,3-триазол-1-ил)фенил)-5-(трифторметил)никотинамид 26
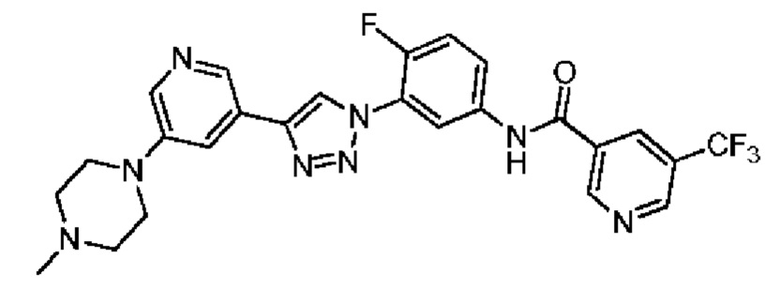
Соединение 26 синтезировали с применением этапов, аналогичных описанным в примере 18. Точная масса (расчетная): 526,18; МС (ионизация электрораспылением) m/z (М+1)+: 527,18.
Пример 27.
N-(4-Фтор-3-(4-(5-(4-метилпиперазин-1-ил)пиридин-3-ил)-1H-1,2,3-триазол-1-ил)фенил)-4-(трифторметил)пиколинамид 27
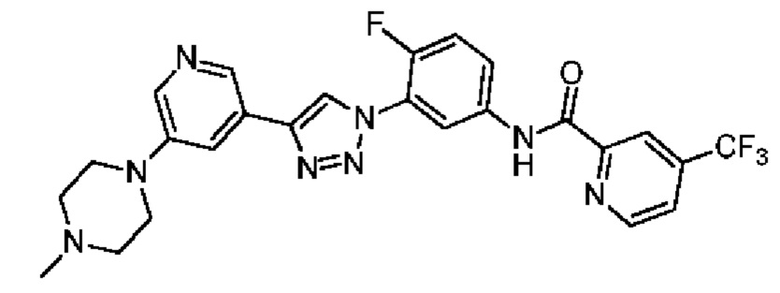
Соединение 27 синтезировали с применением этапов, аналогичных описанным в примере 18. Точная масса (расчетная): 526,18; МС (ионизация электрораспылением) m/z (М+1)+: 527,18.
Пример 28.
6-Хлор-N-(4-фтор-3-(4-(5-(4-метилпиперазин-1-ил)пиридин-3-ил)-1Н-1,2,3-триазол-1-ил)фенил)пиколинамид 28
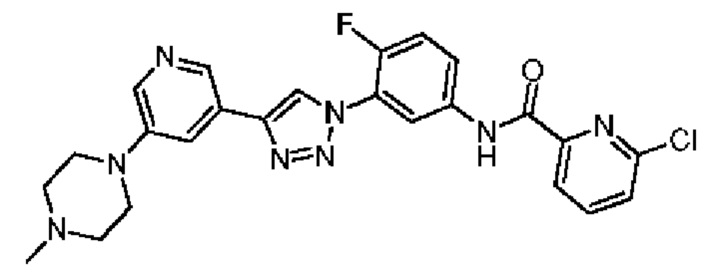
Соединение 28 синтезировали с применением этапов, аналогичных описанным в примере 18. Точная масса (расчетная): 492,15; МС (ионизация электрораспылением) m/z (М+1)+: 493,15.
Пример 29.
6-Фтор-N-(4-фтор-3-(4-(5-(4-метилпиперазин-1-ил)пиридин-3-ил)-1Н-1,2,3-триазол-1-ил)фенил)пиколинамид 29
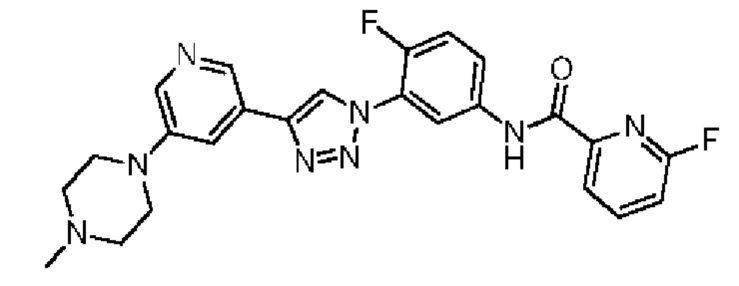
Соединение 29 синтезировали с применением этапов, аналогичных описанным в примере 18. Точная масса (расчетная): 476,18; МС (ионизация электрораспылением) m/z (М+1)+: 477,18.
Пример 30.
6-Хлор-N-(4-хлор-3-(4-(5-(4-метилпиперазин-1-ил)пиридин-3-ил)-1H-1,2,3-триазол-1-ил)фенил)пиколинамид 30
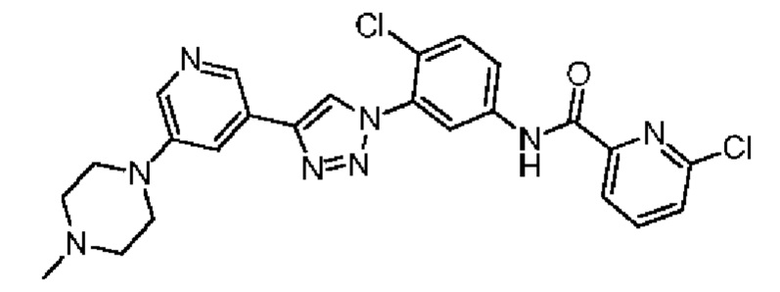
Соединение 30 синтезировали с применением этапов, аналогичных описанным в примере 18. Точная масса (расчетная): 508,12; МС (ионизация электрораспылением) m/z (М+1)+: 509,12.
Пример 31.
6-Фтор-N-(4-хлор-3-(4-(5-(4-метилпиперазин-1-ил)пиридин-3-ил)-1Н-1,2,3-триазол-1-ил)фенил)пиколинамид 31
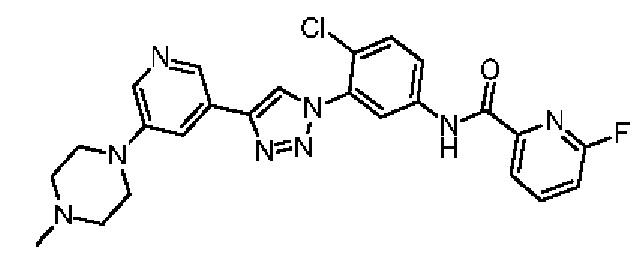
Соединение 31 синтезировали с применением этапов, аналогичных описанным в примере 18. Точная масса (расчетная): 492,15; МС (ионизация электрораспылением) m/z (М+1)+: 493,15.
Пример 32.
N-(3-(4-(5-(2-Окса-6-аза-спиро[3.3]гепт-6-ил]пиридин-3-ил)-1Н-пиразол-1-ил)-4-метилфенил)-6-(трифторметил)пиколинамид 32
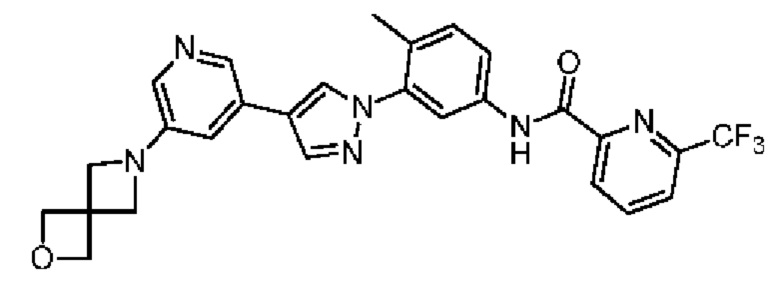
Соединение 32 синтезировали с применением этапов, аналогичных описанным в примере 1. Точная масса (расчетная): 520,18; МС (ионизация электрораспылением) m/z (М+1)+: 521,18.
Пример 33.
N-(3-(4-(5-(2-Окса-6-аза-спиро[3.3]гепт-6-ил]пиридин-3-ил)-1Н-пиразол-1-ил)-4-метилфенил)-2-(трифторметил)изоникотинамид 33
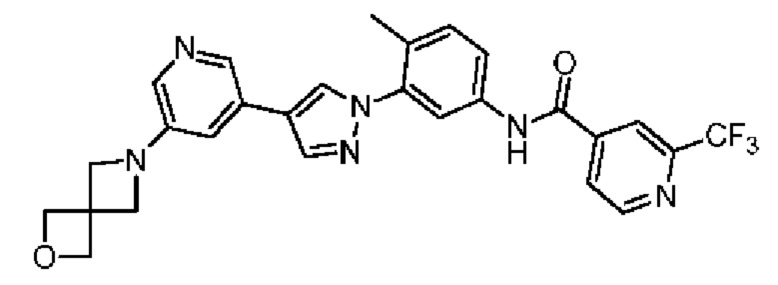
Соединение 33 синтезировали с применением этапов, аналогичных описанным в примере 1. Точная масса (расчетная): 520,18; МС (ионизация электрораспылением) m/z (М+1)+: 521,18.
Пример 34.
N-(3-(4-(5-(2-Окса-6-аза-спиро[3.3]гепт-6-ил]пиридин-3-ил)-1Н-пиразол-1-ил)-4-метилфенил)-5-(трифторметил)никотинамид 34
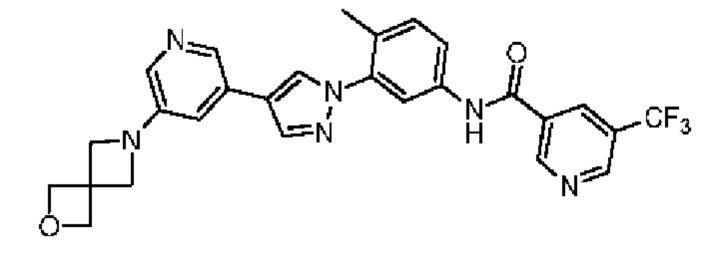
Соединение 34 синтезировали с применением этапов, аналогичных описанным в примере 1. Точная масса (расчетная): 520,18; МС (ионизация электрораспылением) m/z (М+1)+: 521,18.
Пример 35.
N-(3-(4-(5-(2-Окса-6-аза-спиро[3.3]гепт-6-ил]пиридин-3-ил)-1Н-пиразол-1-ил)-4-метилфенил)-4-(трифторметил)пиколинамид 35
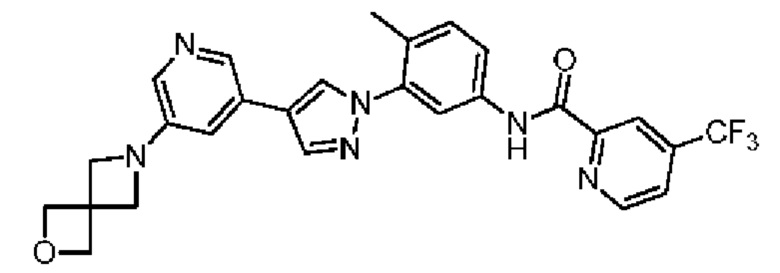
Соединение 35 синтезировали с применением этапов, аналогичных описанным в примере 1. Точная масса (расчетная): 520,18; МС (ионизация электрораспылением) m/z (М+1)+: 521,18.
Пример 36.
N-(3-(4-(5-(2-Окса-6-аза-спиро[3.3]гепт-6-ил]пиридин-3-ил)-1Н-пиразол-1-ил)-4-метилфенил)-6-хлорпиколинамид 36
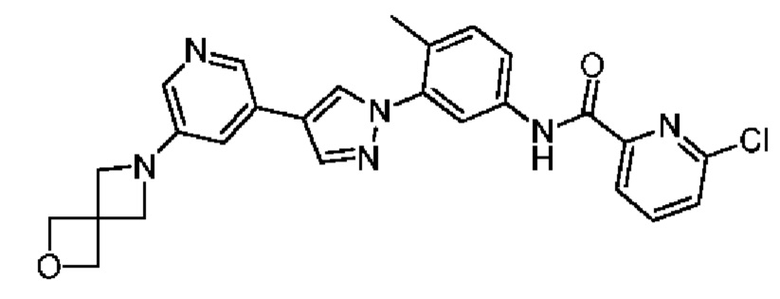
Соединение 36 синтезировали с применением этапов, аналогичных описанным в примере 1. Точная масса (расчетная): 486,15; МС (ионизация электрораспылением) m/z (М+1)+: 487,15.
Пример 37.
N-(3-(4-(5-(2-Окса-6-аза-спиро[3.3]гепт-6-ил]пиридин-3-ил)-1Н-пиразол-1-ил)-4-метилфенил)-6-фторпиколинамид 37
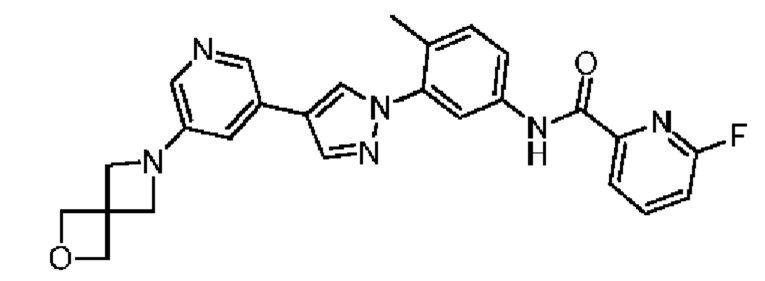
Соединение 37 синтезировали с применением этапов, аналогичных описанным в примере 1. Точная масса (расчетная): 470,18; МС (ионизация электрораспылением) m/z (М+1)+: 471,18.
Пример 38.
N-(3-(4-(5-(2-Окса-6-аза-спиро[3.3]гепт-6-ил]пиридин-3-ил)-1Н-1,2,3-триазол-1-ил)-4-метилфенил)-6-(трифторметил)пиколинамид 38
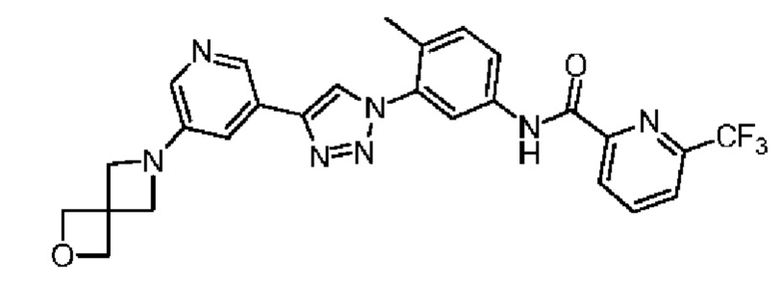
Соединение 38 синтезировали с применением этапов, аналогичных описанным в примере 18. Точная масса (расчетная): 521,17; МС (ионизация электрораспылением) m/z (М+1)+: 522,17.
Пример 39.
N-(3-(4-(5-(2-Окса-6-аза-спиро[3.3]гепт-6-ил]пиридин-3-ил)-1Н-1,2,3-триазол-1-ил)-4-метилфенил)-2-(трифторметил)изоникотинамид 39
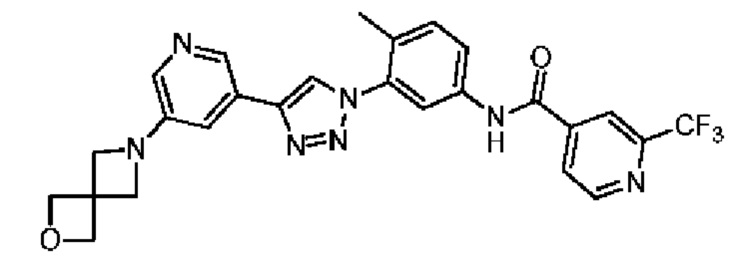
Соединение 39 синтезировали с применением этапов, аналогичных описанным в примере 18. Точная масса (расчетная): 521,17; МС (ионизация электрораспылением) m/z (М+1)+: 522,17.
Пример 40.
N-(3-(4-(5-(2-Окса-6-аза-спиро[3.3]гепт-6-ил]пиридин-3-ил)-1Н-1,2,3-триазол-1-ил)-4-метилфенил)-5-(трифторметил)никотинамид 40
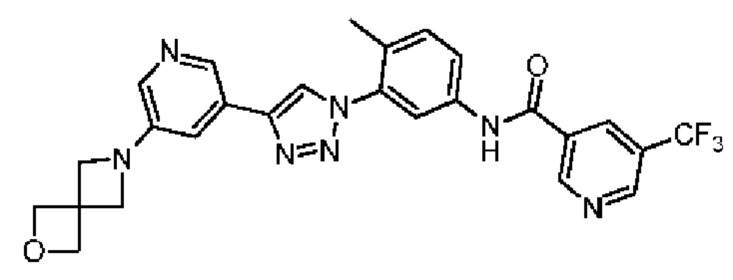
Соединение 40 синтезировали с применением этапов, аналогичных описанным в примере 18. Точная масса (расчетная): 521,17; МС (ионизация электрораспылением) m/z (М+1)+: 522,17.
Пример 41.
N-(3-(4-(5-(2-Окса-6-аза-спиро[3.3]гепт-6-ил]пиридин-3-ил)-1Н-1,2,3-триазол-1-ил)-4-метилфенил)-4-(трифторметил)пиколинамид 41
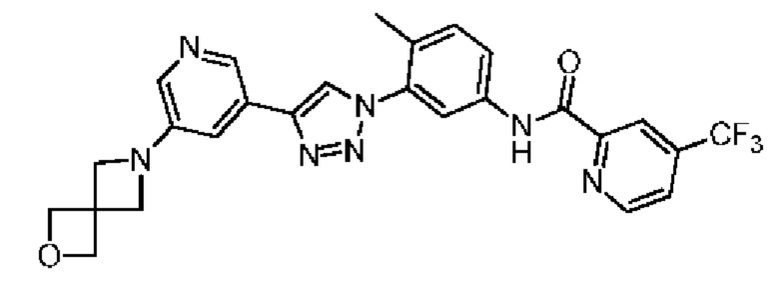
Соединение 41 синтезировали с применением этапов, аналогичных описанным в примере 18. Точная масса (расчетная): 521,17; МС (ионизация электрораспылением) m/z (М+1)+: 522,17.
Пример 42.
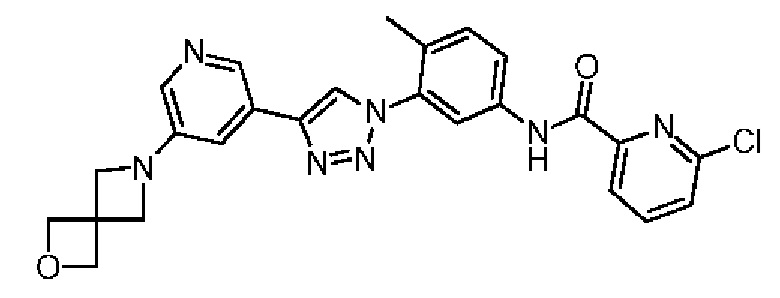
N-(3-(4-(5-(2-Окса-6-аза-спиро[3.3]гепт-6-ил]пиридин-3-ил)-1Н-1,2,3-триазол-1-ил)-4-метилфенил)-4-хлорпиколинамид 42
Соединение 42 синтезировали с применением этапов, аналогичных описанным в примере 18. Точная масса (расчетная): 487,15; МС (ионизация электрораспылением) m/z (М+1)+: 488,15.
Пример 43.
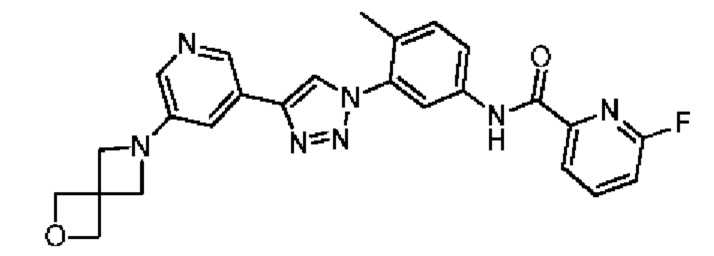
N-(3-(4-(5-(2-Окса-6-аза-спиро[3.3]гепт-6-ил]пиридин-3-ил)-1Н-1,2,3-триазол-1-ил)-4-метилфенил)-4-фторпиколинамид 43
Соединение 43 синтезировали с применением этапов, аналогичных описанным в примере 18. Точная масса (расчетная): 471,18; МС (ионизация электрораспылением) m/z (М+1)+: 472,18.
Пример 44. Влияние нового ингибитора киназы на рост раковых клеток В этом примере использовали первичную В-клетку мыши BaF3 (приобретенную у компании АТСС, США). Кроме того, в этом примере также использовали мышиную BaF3-tel-PDGFRα (стабильно экспрессирующую киназу PDGFRα), мышиную BaF3-tel-PDGFRβ (стабильно экспрессирующую киназу PDGFRβ), BaF3-P210 (стабильно экспрессирующую киназу ABL), BaF3-P210-T315I (стабильно экспрессирующую киназу ABL-T315I), BaF3-FL-BRAF-V600E (стабильно экспрессирующую киназу BRAF-V600E), BaF3-TEL-cKIT (стабильно экспрессирующую киназу cKIT), BaF3-TEL-VEGFR2 (стабильно экспрессирующую киназу VEGFR2), BaF3-TEL-FGFR2 (стабильно экспрессирующую киназу FGFR2). Все вышеуказанные линии клеток были сконструированы в нашей лаборатории следующим способом. Последовательности BCR-ABL человека (Р210 или P210/T315I с мутацией), полноразмерного фрагмента BRAF-V600E, cKIT, VEGFR2, FGFR2, PDGFRα, области PDGFRβ киназы амплифицировали, соответственно, методом ПЦР, и включали, соответственно, в MSCV-Puro вектор (приобретенный у компании Clontech), содержащий N-концевой фрагмент TEL и/или NPM фрагмент, и/или TPR фрагмент, и стабильно переносили в клетки BaF3 мыши методом с использованием ретровируса, а фактор роста ИЛ-3 удаляли. В итоге были получены линии клеток, которые переносятся в белки в зависимости от PDGFRα, PDGFRβ.
В этом примере к указанным клеткам добавляли растворы исследуемого соединения при разных концентрациях (0,000508 мкМ, 0,00152 мкМ, 0,00457 мкМ, 0,0137 мкМ, 0,0411 мкМ, 0,123 мкМ, 0,370 мкМ, 1,11 мкМ, 3,33 мкМ, 10 мкМ). Клетки инкубировали в течение 72 ч. Инкубированные клетки детектировали с помощью набора для определения жизнеспособности клеток Cell Titer-Glo (приобретенного в компании Promega, США) (при использовании набора Cell Tier-Glo жизнеспособность клеток рассчитывают путем измерения значения люминесценции, которое пропорционально количеству АТФ, которое положительно связано с количеством клеток во флаконе, следовательно, жизнеспособность клеток может быть получена путем определения количества АТФ), для количественного определения жизнеспособных клеток с помощью микропланшетного анализатора. Рассчитывали медианную ингибирующую концентрацию GI50 соответствующих соединений и контрольных соединений по сравнению с пролиферацией соответствующих линий клеток (с результатами, показанными в таблицах 1 и 2). Результаты показывают, что исследованные соединения обладают очень сильным ингибирующим действием в отношении каждого из PDGFRα и PDGFRβ, соединение 1 не обладает ингибирующим действием или относительно слабым ингибирующим действием в отношении других киназных мишеней, таких как BRAF-V600E, ABL, ABL-T315I, cKIT, VEGFR2, FGFR2.
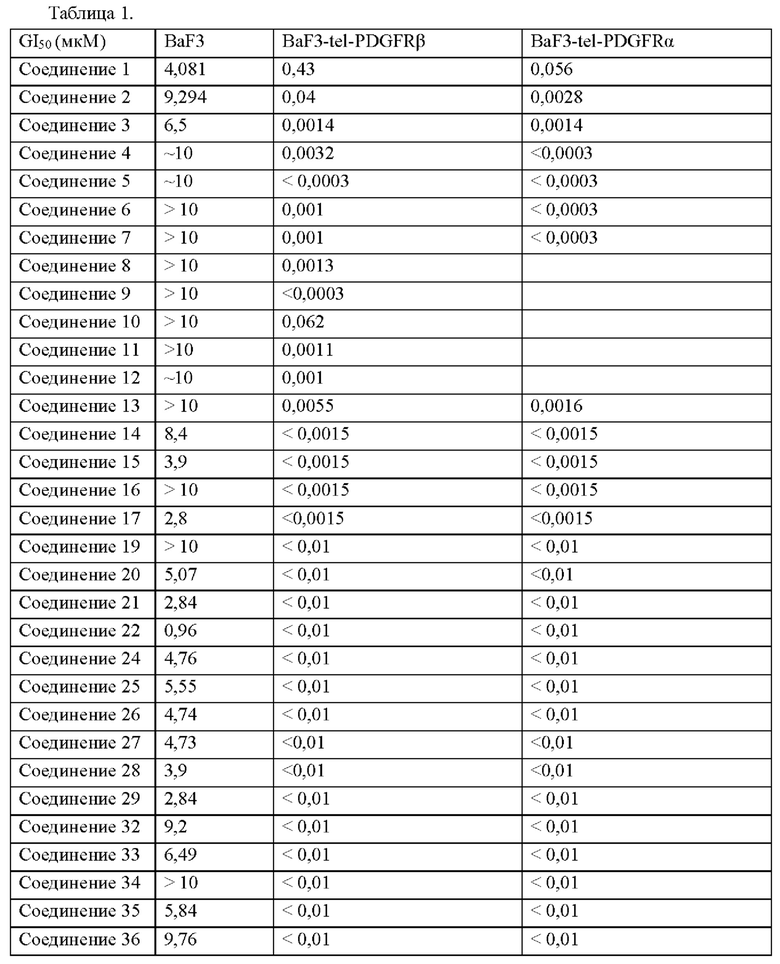

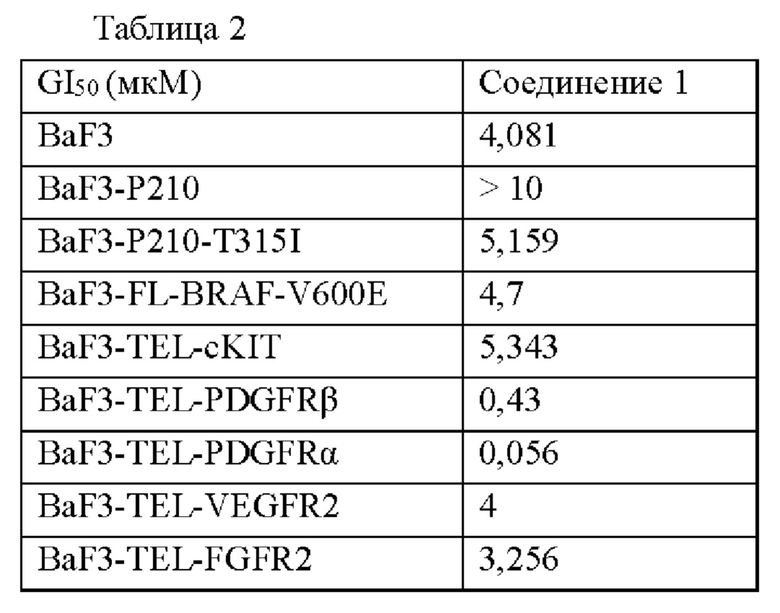
Пример 45. Экспериментальные результаты для соединения 1 на мышиных моделях с клетками хронического эозинофильного лейкоза человека EOL-1 (экспрессирующими PDGFRα)
1) Самок мышей линии Bal b/с, 46 недель, приобретали в компании Shanghai SLAC Laboratory Animal Co., Ltd. и растили в лаборатории SPF; питьевую воду и подстилку стерилизовали автоклавированием; и все операции с участием мышей выполняли в асептических условиях;
2) В нулевой день вводили 1 × 107 клеток хронического эозинофильного лейкоза человека EOL-1 (экспрессирующих PDGFRα) (приобретенных в АТСС (Американской коллекции типовых клеточных культур)) подкожно в левый бок каждой из мышей;
3) На 15 день мышей случайным образом разделили на четыре группы по пять мышей на группу и вводили соответственно в течение 14 дней. Мышам в группе 1 внутрибрюшинно вводили носитель на основе метилцеллюлозы (приобретенный у компании Sangon); мышам в группах 2 и 3 вводили соединение 1 в дозе 1 мг/кг массы тела мыши и 5 мг/кг массы тела мыши, соответственно; мышам в группе 4 вводили иматиниб в дозе 25 мг/кг (приобретенный в компании МСЕ, Шанхай);
4) С 15-го дня ежедневно измеряли длину/ширину подкожных опухолей с помощью штангенциркуля, и ежедневно регистрировали массу тела мышей, чтобы определить влияние соединения 1 на массу тела мышей;
5) На 29-й день мышей умерщвляли углекислым газом, а подкожные опухоли извлекали и взвешивали для сравнения;
6) Тенденцию роста подкожной опухоли в течение 15-29 дней анализировали статистически. Объем опухоли рассчитывали как длина × ширина × ширина/2 мм3.
Результаты показаны на фигурах 1а-1с. На фиг. 1а показано изменение средней массы тела мышей с течением времени в различных группах лечения (показано на фигуре в виде относительной массы тела: процент, рассчитанный на основе массы тела мыши в начале введения) в мышиной модели опухоли из клеток хронического эозинофильного лейкоза человека EOL-1; на фиг. 1b показано изменение среднего размера опухолей с течением времени в различных группах лечения (показано на фиг. в виде относительного размера опухоли: процент, рассчитанный на основе размера опухоли, имеющейся у мыши в начале введения) в мышиной модели опухоли клетки хронического эозинофильного лейкоза человека EOL-1; на фиг. 1с показано среднее значение массы опухоли и рассчитанная скорость ингибирования роста опухоли у мышей в различных группах лечения через 14 дней после введения в мышиной модели с клетками хронического эозинофильного лейкоза человека EOL-1.
Экспериментальные результаты на фиг. 1b продемонстрировали, что группа, которой вводили соединение 1 в дозе 5 мг/кг, показала превосходный эффект ингибирования опухоли у мышей в мышиной модели опухоли из клеток хронического эозинофильного лейкоза человека EOL-1 (экспрессирующей PDGFRα). Экспериментальные результаты на фиг. 1с показали, что скорость ингибирования роста опухоли достигала 96% через 14 дней после введения в мышиной модели с клетками хронического эозинофильного лейкоза человека EOL-1 для группы, которой вводили соединение 1 в дозе 5 мг/кг (см. рисунок 1с), где степень ингибирования роста опухоли (TGI) = (масса опухоли в контрольной группе - масса опухоли в испытуемой группе)/масса опухоли в контрольной группе. Это указывает на то, что соединение 1 согласно настоящему изобретению может значительно ингибировать рост опухоли в животной модели с клетками хронического эозинофильного лейкоза человека EOL-1 (экспрессирующей PDGFRα). Кроме того, результаты на фиг. 1а также продемонстрировали, что соединение 1 не только эффективно ингибировало рост опухоли у мыши, но и мало влияло на массу тела мыши, что позволяет предположить, что соединение 1 пригодно для введения животному.
Пример 46. Экспериментальные результаты для соединения 1 в крысиной модели легочной артериальной гипертензии (ЛАГ)
120 самцов крыс линии SD массой 180±20 г было предоставлено Центром животноводства Цинлуншань с лицензией №SCXK(SU)2017-0001. Этих крыс кормили обычными гранулами (Jiangsu Xietong Bio. Co., Ltd.), и выращивали в чистом помещении для животных с циклом света/темноты 12 часов/12 часов. Крысы получали пищу и питьевую воду без ограничений. Температуру поддерживали на уровне 20-26°С, а относительная влажность составляла 40-70%.
120 крыс линии SD распределили в 24 клетки по 5 крыс на группу. После адаптивного роста в течение 7 дней без каких-либо аномальных условий 110 крыс использовали для создания модели легочной артериальной гипертензии, а остальных 10 крыс использовали для нормального контроля. С животными обращались в строгом соответствии с правилами этики в отношении животных на протяжении всего эксперимента.
Согласно способу, описанному в «Фармацевтических экспериментальных моделях животных: Изготовление и применение» ("Pharmaceutical experimental animal models: Fabrication and application") и стандартной операционной процедуре для построения модели ЛАГ в модельном центре для животных крысам внутрибрюшинно вводили раствор 1% монокроталина (МСТ, приобретенный у компании Sigma, США) один раз в дозе 35 мг/кг. На 7-й день после первой инъекции МСТ снова вводили МСТ в дозе 20 мг/кг. Крысам в нормальной контрольной группе внутрибрюшинно вводили эквивалентное количество воды в качестве холостого растворителя. Конкретные этапы представлены ниже.
После того, как крысы в каждой из клеток голодали в течение 8 ч, каждую из крыс взвешивали и регистрировали исходную массу тела после голодания; исходя из измеренной иходной массы тела каждой из крыс, количество МСТ, необходимое для инъекции каждой из крыс, рассчитывали в соответствии с модельной дозой 35 мг/кг; исходя из количества МСТ, необходимого для введения каждой крысе, рассчитывали дозу для инъекции 1% раствора МСТ; крыс фиксировали в держателе и внутрибрюшинно вводили 1% раствор МСТ в расчетной дозе; после инъекции крыс возвращали в клетки для обычного кормления.
Кровь из хвостовой артерии брали для анализа газа крови на 3-й и 4-й неделях, соответственно, после инъекции МСТ. 0,5 мл крови из хвостовой артерии отбирали медленно, переносили в пробирку с антикоагулянтом и загружали в анализатор газов крови для определения индексов парциального давления кислорода (рО2), парциального давления углекислого газа (pCO2) и насыщения крови кислородом (SaO2) в крови. Анализатор газов крови работал в соответствии со стандартной операционной процедурой. На основании измеренных результатов крысы с легочной гипертензией были случайным образом разделены на следующие группы (10 крыс на группу): группа отрицательного контроля (то есть, группа носителя), группа 50 мг/кг бозентана, лекарственный препарат для клинического лечения легочной гипертензии (приобретенный у компании МСЕ, Шанхай), группа 50 мг/кг иматиниба, группа 45 мг/кг соединения 1, группа 30 мг/кг соединения 1 и группа 15 мг/кг соединения 1. Каждой из крыс вводили вещества через желудочный зонд один раз в день, начиная со дня перегруппировки на неделе 4. Крысам в группе отрицательного контроля ежедневно вводили через желудочный зонд с равным объемом метилцеллюлозы в качестве носителя. Крысам в каждой из групп вводили через желудочный зонд в течение 4 последовательных недель (т.е. 28 дней). Для каждой из крыс в соответствующих группах состояние, появление симптомов одышки, снижение активности, ускорение сердцебиения и т.п.наблюдали одновременно с ежедневным введением через желудочный зонд. Крыс взвешивали после голодания в течение ночи два раза в неделю. Дозировку введения рассчитывали на основании результатов взвешивания.
Определение артериального давления в легких и систолического давления в правом желудочке крыс: в конце эксперимента (через 28 дней после введения через зонд) крыс взвешивали и анестезировали путем внутрибрюшинной инъекции 10% хлоралгидрата (приобретенного у компании Sangon) (0,3 мл/100 г). После того, как крысы находились под наркозом, измеряли легочное артериальное давление и систолическое давление правого желудочка крыс. Метод измерения можно найти в стандартных операционных процедурах функциональной экспериментальной системы. Следующие этапы представлены ниже.
Катетер №3.5 для пупочной вены подключали к датчику давления системы. Готовый раствор гепарина натрия (приобретенный у компании Sangon) заливали в датчик и катетер, и выпускали пузырьки. Анестезированную крысу помещали на хирургическую анатомическую пластину, регулируемую по температуре. Температуру пластины регулировали таким образом, чтобы поддерживать ее на уровне около 37°С. Крысу фиксировали в положении лежа на спине. Ножницами разрезали кожу шеи до края ключицы с последующим тупым рассечением подкожных тканей и мышц, обнажая правую наружную яремную вену. Жировую ткань на поверхности удаляли офтальмологическими хирургическими ножницами. Наружную яремную вену лигировали на телецентрическом конце хирургической нитью и делали свободный узел на проксимальном конце для резервирования. Наружную яремную вену осторожно приподнимали офтальмологическим пинцетом и разрезали офтальмологическими ножницами, чтобы сделать V-образное отверстие. Катетер быстро вставляли, и ослабленный узел на проксимальном конце слегка затягивали, чтобы предотвратить кровотечение. Изгиб катетера в переднем сегменте поддерживали в направлении влево, и на расстоянии около 1-1,5 см катетер далее вводился в положение 2 см, при этом подмышечную вену крысы отодвигали, чтобы приблизиться к правой ушной раковине. В это время катетер осторожно вращали по часовой стрелке на 100-180°, удерживая правую ушную раковину на расстоянии. На расстоянии около 3 см конец катетера вошел в правое предсердие и его ввели дальше, чтобы достичь атриовентрикулярного отверстия на расстоянии около 4-4,5 см. В это время катетер осторожно вращали против часовой стрелки на 90-180°, чтобы зацепить атриовентрикулярное отверстие и войти в правый желудочек, при этом наблюдалась волна правого желудочка с относительно большой амплитудой. Катетер далее вводили медленно вперед и вводили в легочную артерию на расстоянии около 5 см.
Ключевые точки измерения: катетер вводили на 1-2 см, чтобы достичь верхней полой вены, на 2-3 см, чтобы достичь правого предсердия, на примерно 4 см, чтобы войти в правый желудочек, и на около 5 см, чтобы войти в легочную артерию. Давление правого предсердия было близко к нулю, а давление легочной артерии было самым высоким.
После измерения легочной артерии брюшную полость крыс вскрывали, и брюшную аорту осторожно отделяли. Медленно отбирали 3 мл крови из аорты путем введения иглы, направленной к проксимальному концу брюшной аорты, используя шприц объемом 5 мл, инфильтрированный раствором натрия гепарина. Кровь переносили в пробирку с антикоагулянтом и загружали в анализатор газов крови для определения индексов парциального давления кислорода (pO2), парциального давления углекислого газа (рСО2) и насыщения крови кислородом (SaO2) в крови.
В конце эксперимента крыс забивали и извлекали их сердца. Правый желудочек (RV) и левый желудочек и перегородку (LV+S) разделяли, соответственно, промывали физиологическим раствором, и влагу абсорбировали фильтровальной бумагой. Взвешивали RV и LV+S, соответственно. В качестве оценочного показателя гипертрофии правого сердца использовали правый желудочковый индекс (RVI), полученный по следующей формуле: RVI=RV/(LV+S).
Результаты показаны на фиг. 2а-2b. На фиг. 2а показано изменение выживаемости крыс с течением времени в различных группах лечения (показано на фигуре как относительная выживаемость: процент, рассчитанный на основе количества крыс в начале эксперимента) в модели легочной гипертензии крыс; и на фиг. 2b показано систолическое давление в правом желудочке в различных группах лечения в модели легочной гипертензии крыс.
Как можно видеть из анализа значимых различий среднего давления в легочной артерии (mPAP) в соответствующих группах по сравнению с нормальной группой, группа носителя крайне значимо отличалась (р<0,001); группа иматиниба (n=10, 27,27+2,02) с самым низким mPAP примерно в 1,5 раза превосходила нормальную группу (n=10, 18,33+0,23); по сравнению с группой носителя каждая из двух групп лекарственного средства положительного контроля, группа 45 мг/кг соединения 1, группа 30 мг/кг соединения 1 и группа 15 мг/кг соединения 1 крайне значимо отличались (р<0,001). Группа высокой дозы 45 мг/кг соединения 1 и группа средней дозы 30 мг/кг соединения 1 не показали значимых отличий по сравнению с каждой из группы бозентана и группы иматиниба, и продемонстрировали крайне значимое отличие от каждой из других групп (р<0,001).
Как видно из анализа значимых различий в систолическом давлении в правом желудочке (RVSP) в соответствующих группах по сравнению с нормальной группой, группа носителя крайне значимо отличалась (р<0,001); группа иматиниба с самой низкой RVSP (n=10, 40,84+1,49) примерно в 1,8 раза превосходила нормальную группу (n=10, 22,44+1,09); каждая из двух групп средства положительного контроля, группа высокой дозы 45 мг/кг соединения 1 и группа средней дозы 30 мг/кг соединения 1 была чрезвычайно значительно различна по сравнению с группой носителя (Р<0,001). Группа высокой дозы 45 мг/кг соединения 1 и группа средней дозы 30 мг/кг соединения 1 не показали значимых отличий по сравнению с каждой из группы иматиниба и группы бозентана, и продемонстрировали крайне значимое отличие от каждой из других групп (р<0,001).
Парциальное давление кислорода (рО2) в артерии, которое отражает поглощение кислорода легочными капиллярами, является показателем, отражающим состояние дыхания, и является наиболее чувствительным показателем гипоксии организма. РО2 в нормальных условиях составляет около 80-110 мм рт. ст. РО2 ниже 80 мм рт. ст. свидетельствует о гипоксии организма. Парциальное давление углекислого газа в артериальной крови является важным показателем, отражающим состояние респираторного кислотно-щелочного баланса, и составляет около 35-45 мм рт. ст. в нормальных условиях. В случае нарушения функции легких и недостаточной вентиляции парциальное давление СО2 увеличивается по таким причинам, как чрезмерно низкое выделение СО2, которое представляет собой респираторный ацидоз. Насыщение крови кислородом SaO2, представляющее собой показатель, отражающий процентную долю емкости оксигемоглобина (HbO2) к общей емкости гемоглобина (Hb), доступной для связывания кислорода, является важным физиологическим параметром дыхательного кровообращения. Если наблюдается патологическое изменение функции легких, то возникает гипоксия, приводящая к снижению насыщения крови кислородом. В нормальных условиях SaO2≥90%.
После вмешательства путем введения парциальное давление кислорода, парциальное давление углекислого газа и насыщение крови кислородом изменялись в различной степени в каждой из групп. Анализ данных парциального давления кислорода показал, что по сравнению с группой носителя, группы средства положительного контроля и группа высокой дозы соединения 1, крайне значимо отличались (Р<0,001), и группа средней дозы соединения 1 также продемонстрировала крайне значимое отличие (Р<0,01). Сравнение крыс в каждой из групп показало, что парциальное давление кислорода у крыс в части групп находилось в диапазоне группы нормального контроля, что указывает на то, что лечение лекарственным средством играет определенную роль в поддержании и восстановлении парциального давления кислорода.
Анализ данных по парциальному давлению диоксида углерода для соответствующих групп показал, что по сравнению с группой носителя группы средства положительного контроля и группа высокой дозы соединения 1 крайне значимо отличались (Р<0,001), и группа средней дозы соединения 1 крайне значимо отличалась (Р<0,01). Сравнение крыс в каждой из групп показало, что парциальное давление углекислого газа в части групп находилось в диапазоне группы нормального контроля, что указывает на то, что лечение лекарственным средством играет определенную роль в восстановлении вентиляции легких у крыс с легочной гипертензией.
Анализ данных насыщения крови кислородом для соответствующих групп показал, что по сравнению с группой носителя группы средства положительного контроля и группа высокой дозы соединения 1 значительно отличались (р<0,05). Сравнение крыс в каждой из групп показало, что насыщение крови кислородом части групп находилось в пределах диапазона группы нормального контроля.
RVI относится к измерению индекса гипертрофии правого желудочка у крыс. Измеренные результаты показали, что после вмешательства путем введения индекс гипертрофии правого желудочка в каждой из групп изменялся в различной степени, при этом RVI группы бозентана был снижен на 15,7% по сравнению с группой отрицательного контроля, a RVI группы иматиниба был снижен на 17,8% по сравнению с группой отрицательного контроля, RVI группы высокой дозы 45 мг/кг соединения 1 был снижен на 29,6% по сравнению с группой отрицательного контроля, RV1 группы средней дозы 30 мг/кг соединения 1 был снижен на 9,4% по сравнению с группой отрицательного контроля, a RVI группы низкой дозы 15 мг/кг соединения 1 был снижен на 5,5% по сравнению с группой отрицательного контроля.
Анализ значимых различий RVI для соответствующих групп показал, что по сравнению с нормальной группой группа носителя была крайне значимо отлична (р<0,001); группа иматиниба с наименьшим RVI (n=10, 0,403+0,016) примерно в 1,4 раза превосходила нормальную группу (n=10, 0,279+0,16); каждая из двух групп средства положительного контроля и группа высокой дозы 45 мг/кг соединения 1 была крайне значимо отличалась от группы носителя (Р<0,001). Группа высоких доз 45 мг/кг соединения 1 не показала значимого отличия по сравнению с каждой из групп иматиниба и группы бозентана и продемонстрировала крайне значимое отличие от с каждой из других групп (р<0,001).
Промышленная применимость
В настоящем изобретении предложен селективный ингибитор киназы PDGFR, который можно применять для ингибирования активности киназы PDGFR и для лечения заболевания, расстройства или состояния, связанного с ингибированием активности киназы PDGFR. Таким образом, он может быть изготовлен в виде соответствующего лекарственного средства и имеет промышленную применимость.
Несмотря на то, что изобретение было подробно описано в настоящем документе, изобретение не ограничивается описанием, и специалисты в данной области техники могут вносить модификации на основе принципов изобретения, и, таким образом, следует понимать, что все модификации в соответствии с принципами изобретения находятся в рамках объема охраны изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЙ ИНГИБИТОР КИНАЗЫ PAN-RAF И ЕГО ПРИМЕНЕНИЕ | 2019 |
|
RU2792626C1 |
| ИНГИБИТОР КИНАЗЫ PAN-KIT, ИМЕЮЩИЙ СТРУКТУРУ ХИНОЛИНА, И ЕГО ПРИМЕНЕНИЕ | 2018 |
|
RU2789405C2 |
| ПРОИЗВОДНЫЕ ДИГИДРОИЗОХИНОЛИНОНА И ИЗОИНДОЛИНОНА И ИХ ПРИМЕНЕНИЕ | 2021 |
|
RU2833422C1 |
| Соединения пиразола, их фармацевтические композиции и их применение | 2019 |
|
RU2770835C1 |
| ГЕТЕРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ, ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2019 |
|
RU2800153C2 |
| ПРОИЗВОДНЫЕ ПИРРОЛИДИНА В КАЧЕСТВЕ АГОНИСТОВ PPAR | 2017 |
|
RU2711991C1 |
| ПИКОЛИНАМИДНЫЕ И ПИРИМИДИН-4-КАРБОКСАМИДНЫЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ВКЛЮЧАЮЩАЯ ИХ | 2011 |
|
RU2566827C2 |
| ФЕНИЛ [A]ИНДОЛ [2,3-G] ХИНОЛИЗИНЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ, СОДЕРЖАЩИЕ ИХ КОМПОЗИЦИИ И ВАРИАНТЫ ПРИМЕНЕНИЯ | 2017 |
|
RU2756197C2 |
| СОЕДИНЕНИЯ И КОМПОЗИЦИИ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕИНКИНАЗ | 2006 |
|
RU2368602C2 |
| АМИДНЫЕ ПРОИЗВОДНЫЕ И ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, СПОСОБ ИХ ПОЛУЧЕНИЯ И МЕДИЦИНСКОЕ ПРИМЕНЕНИЕ | 2015 |
|
RU2681537C2 |
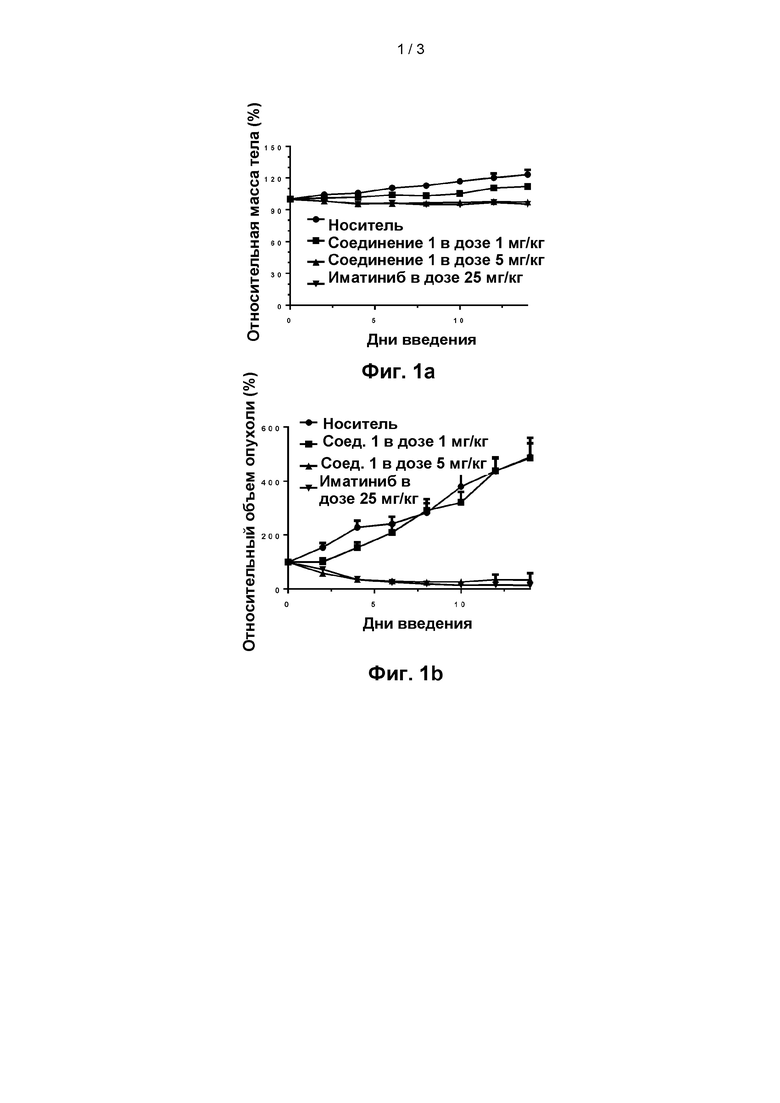
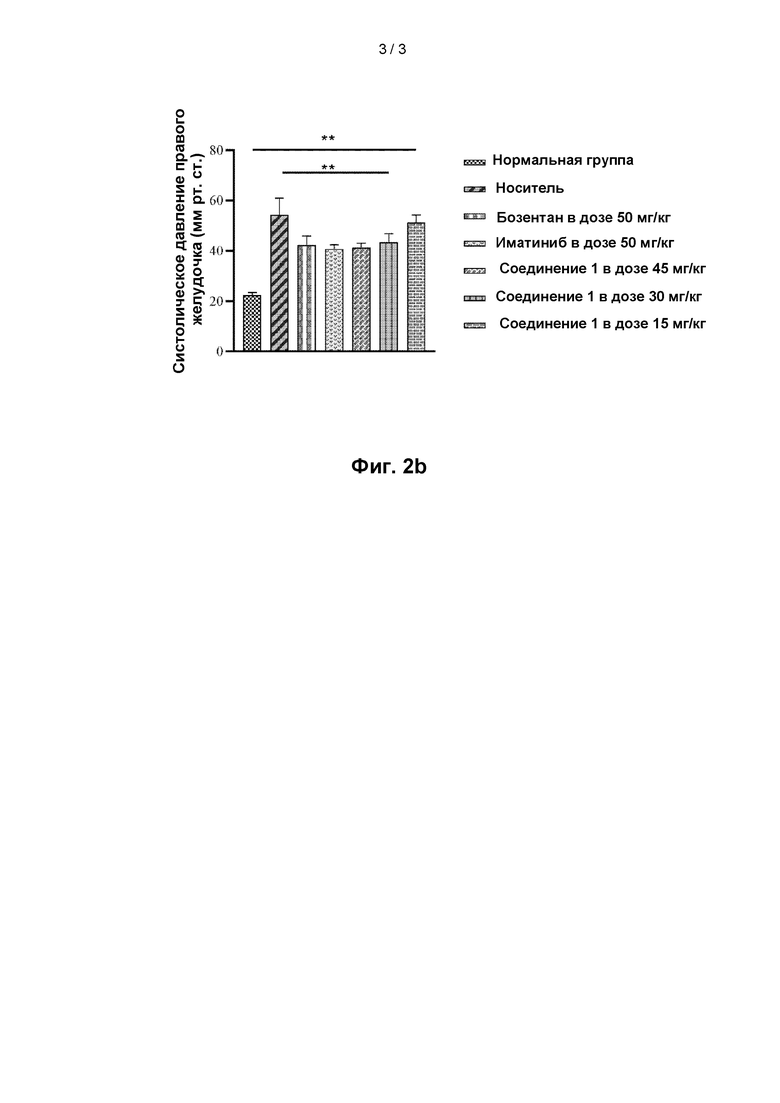
Группа изобретений относится к фармацевтической химии и включает соединение формулы (I) или его фармацевтически приемлемую соль, а также фармацевтическую композицию на его основе. В формуле (I) A представляет собой пиридиновое кольцо; Z представляет собой CH; R1 выбран из группы, состоящей из гетероциклоалкила, гетероциклоалкилокси, гетероциклоалкил-C1-6-алкокси, 6-10-членного гетероспироциклоалкила, содержащего 1-2 гетероатома, выбранных из кислорода и азота, C3-6-циклоалкил-C1-6-алкокси, где гетероциклоалкил представляет собой 4-6-членный гетероциклоалкил, содержащий 1-2 гетероатома, выбранных из кислорода и азота, и атом азота в гетероциклоалкиле необязательно замещен C1-6-алкилом; R2 выбран из группы, состоящей из галогена и C1-6-галогеналкила; R3 представляет собой C1-6-алкил. Технический результат - соединение формулы (I) в качестве ингибитора активности киназы PDGFR, в частности киназ PDGFRα и/или PDGFRβ. 2 н. и 10 з.п. ф-лы, 5 ил., 2 табл., 45 пр.
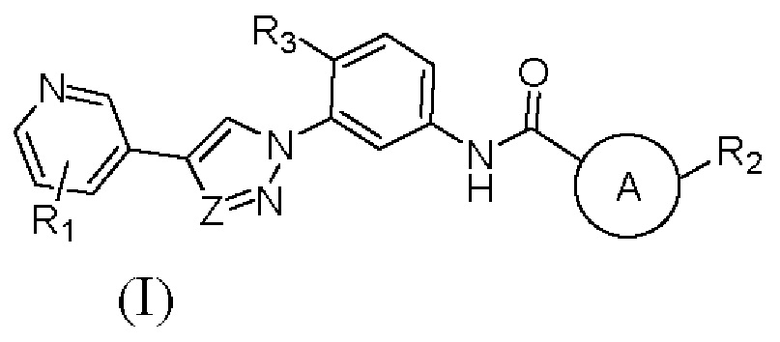
1. Ингибитор киназы PDGFR, представляющий собой соединение формулы (I) или его фармацевтически приемлемую соль
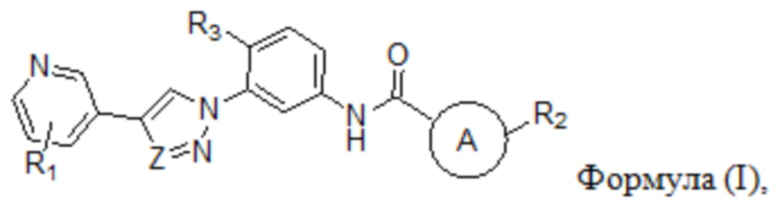
где кольцо A представляет собой пиридиновое кольцо;
Z представляет собой CH;
R1 выбран из группы, состоящей из гетероциклоалкила, гетероциклоалкилокси, гетероциклоалкил-C1-6-алкокси, 6-10-членного гетероспироциклоалкила, содержащего 1-2 гетероатома, выбранных из кислорода и азота, C3-6-циклоалкил-C1-6-алкокси, где гетероциклоалкил представляет собой 4-6-членный гетероциклоалкил, содержащий 1-2 гетероатома, выбранных из кислорода и азота, и атом азота в гетероциклоалкиле необязательно замещен C1-6-алкилом;
R2 выбран из группы, состоящей из галогена и C1-6-галогеналкила;
R3 представляет собой C1-6-алкил.
2. Ингибитор киназы PDGFR по п. 1, характеризующийся тем, что
кольцо A выбрано из группы, состоящей из
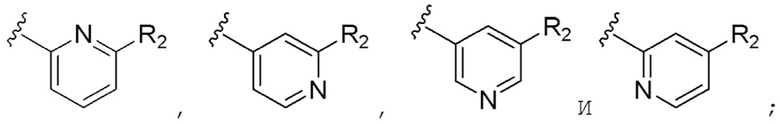
R2 выбран из группы, состоящей из фтора, хлора и трифторметила.
3. Ингибитор киназы PDGFR по п. 1, характеризующийся тем, что R3 представляет собой метил.
4. Ингибитор киназы PDGFR по п. 1, представляющий собой соединение формулы (Ia) или его фармацевтически приемлемую соль
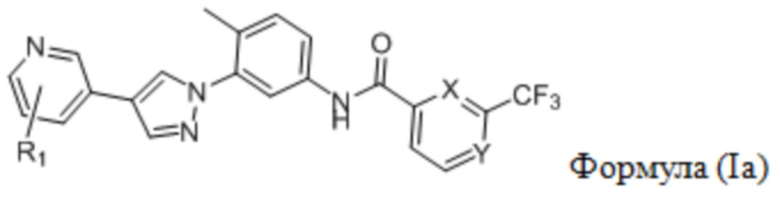
где R1 выбран из группы, состоящей из гетероциклоалкила, гетероциклоалкилокси, гетероциклоалкил-C1-6-алкокси, 6-10-членного гетероспироциклоалкила, содержащего 1-2 гетероатома, выбранных из кислорода и азота, C3-6-циклоалкил-C1-6-алкокси, где гетероциклоалкил представляет собой 4-6-членный гетероциклоалкил, содержащий 1-2 гетероатома, выбранных из кислорода и азота, и атом азота в гетероциклоалкиле необязательно замещен C1-6-алкилом; и
один из X и Y представляет собой CH, а другой представляет собой N.
5. Ингибитор киназы PDGFR по любому из пп. 1-4, характеризующийся тем, что заместитель R1 замещен при углероде в пара- или мета-положении относительно атома N в пиридиновом кольце.
6. Ингибитор киназы PDGFR по любому из пп. 1-4, характеризующийся тем, что гетероциклоалкил выбран из группы, состоящей из пирролидинила, морфолинила, пиперазинила, тетрагидропиранила, тетрагидрофуранила, оксетанила и азетидинила.
7. Ингибитор киназы PDGFR по любому из пп. 1-4, характеризующийся тем, что R1 выбран из группы, состоящей из C1-6-алкилпиперазинила, морфолинила, тетрагидропиранила C1-6-алкокси, оксетанилокси, морфолино C1-6-алкокси, тетрагидрофуранила C1-6-алкокси, C3-6-циклоалкил-C1-6-алкокси и окса-аза-спирогептила.
8. Ингибитор киназы PDGFR по любому из пп. 1-4, где R1 выбран из группы, состоящей из N-метилпиперазин-1-ила, N-морфолинила, тетрагидропиран-4-илметокси, оксетан-3-илокси, 2-морфолиноэтокси, тетрагидрофуран-2-ил-метокси, циклопентилметокси и 2-окса-6-аза-спиро[3.3]гепт-6-ила.
9. Ингибитор киназы PDGFR по любому из пп. 1-4, который представляет собой соединение, выбранное из группы, состоящей из:
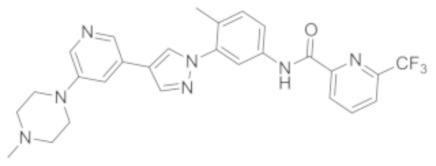
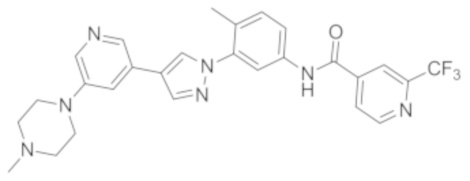
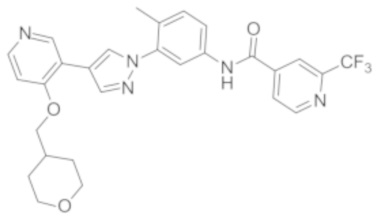
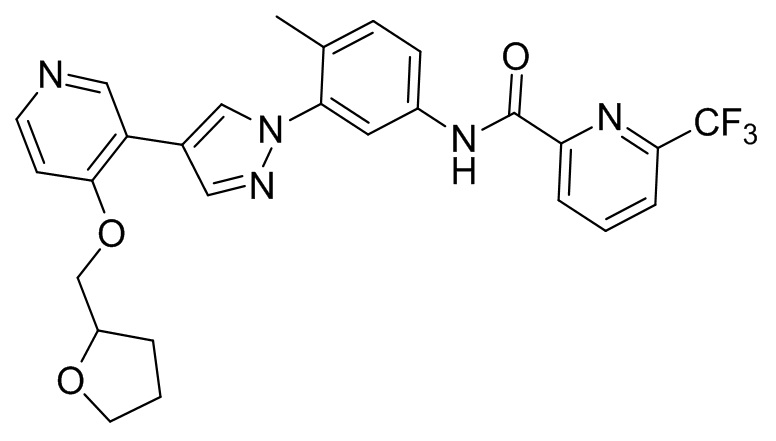
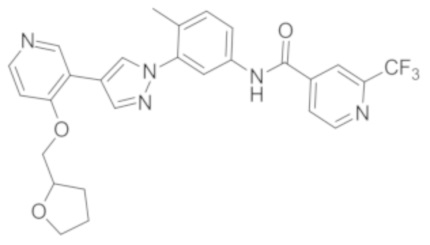
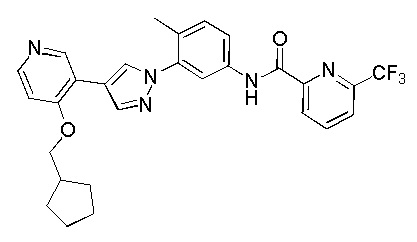
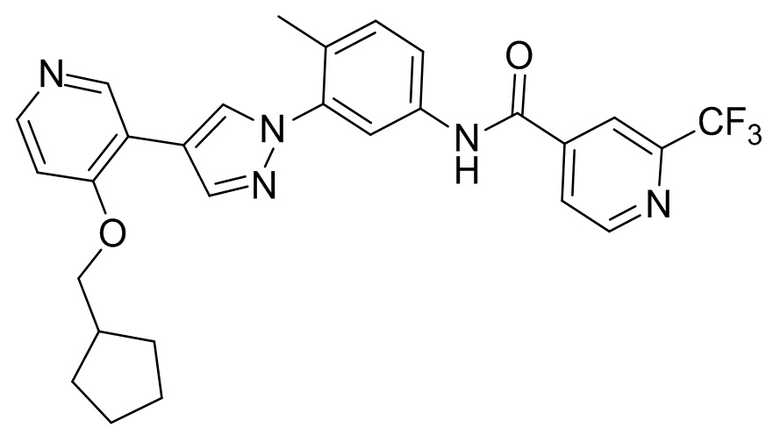
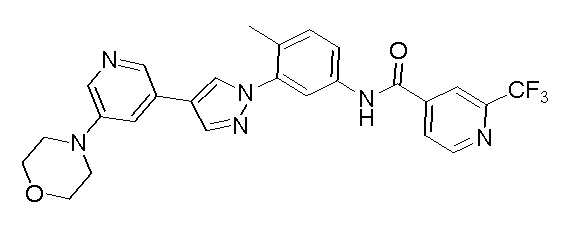
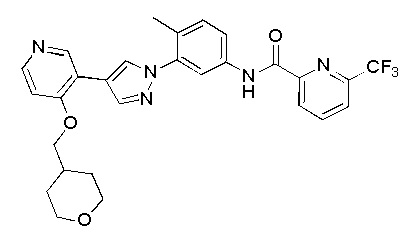
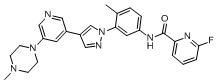
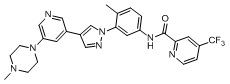
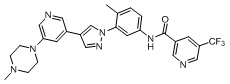
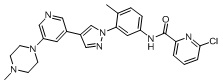
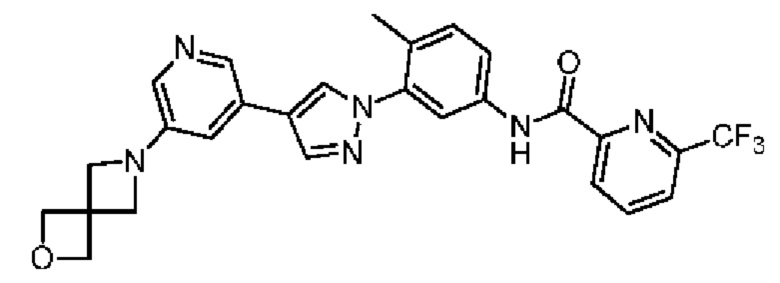
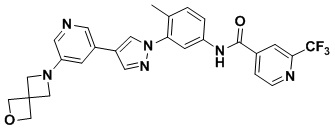
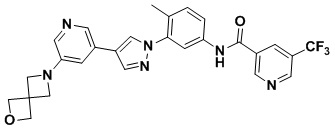
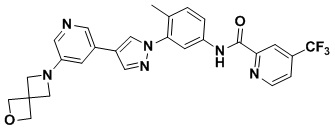
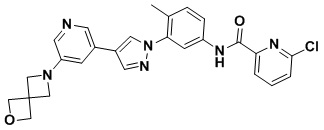
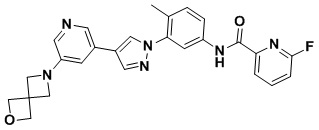
10. Фармацевтическая композиция для ингибирования киназы PDGFR, содержащая эффективное количество ингибитора киназы PDGFR по любому из пп. 1-9 и фармацевтически приемлемый носитель или вспомогательное вещество.
11. Ингибитор киназы PDGFR по любому из пп. 1-9 для применения для ингибирования активности PDGFRα и/или PDGFRβ.
12. Ингибитор киназы PDGFR по любому из пп. 1-9 для применения в лечении или облегчении заболевания, нарушения или состояния, ассоциированного с ингибированием PDGFRα и/или PDGFRβ, где указанное заболевание, нарушение или состояние представляет собой легочную артериальную гипертензию, хронический эозинофильный лейкоз или их комбинацию.
| ЕА 200900072 А1, 30.06.2009 | |||
| WO 2011117381 A1, 29.09.2011 | |||
| US 7531560 B2, 12.05.2009 | |||
| Двигатель внутреннего горения | 1928 |
|
SU26152A1 |

