ОБЛАСТЬ ТЕХНИКИ
[0001] Настоящее изобретение относится к покрытому металлическому листу для автомобиля, имеющему сопротивление скалыванию и обладающему превосходной стойкостью против ржавления в низкотемпературной среде эксплуатации.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
[0002] Далее будут описаны предпосылки создания настоящего изобретения.
[0003] Большинство элементов кузова автомобиля выполняют из металлических листов, таких как стальные листы, и производят с помощью [1] заготовочного процесса, в котором вырезается металлический лист заданного размера, [2] процесса масляной очистки, в котором металлический лист очищается с помощью масла, [3] процесса штамповки заготовки, [4] процесса соединения, в котором штампованный материал преобразуется в элемент с желаемой формой с помощью точечной сварки, адгезии и т.п., [5] процесса, в котором смазочное масло удаляется с поверхности элемента для его очистки, [6] процесса химической конверсионной обработки, и [7] процесса нанесения покрытия с помощью электролитического осаждения. Элемент кузова автомобиля, используемый в качестве внешнего листа, обычно дополнительно подвергается процессам нанесения покрытия, таким как [8] процесс нанесения промежуточного покрытия и [9] процесс нанесения поверхностного покрытия. Следовательно, в автомобильной промышленности высока потребность в снижении затрат за счет устранения или упрощения производственных процессов, в частности процесса химической конверсионной обработки и процесса нанесения покрытия.
[0004] В ответ на эти потребности были выполнены исследования использования покрытого металлического листа (листа металла с предварительным покрытием) для элементов кузова автомобиля с тем, чтобы исключить процесс химической конверсионной обработки, исключить или упростить процесс нанесения покрытия с помощью электролитического осаждения, а также исключить или уменьшить количество вспомогательных материалов во время производства автомобиля.
[0005] Одной из важных характеристик, требуемых от элементов кузова автомобиля, является сопротивление скалыванию. Скалывание относится к явлению, при котором камни и т.п., разлетающиеся во время движения автомобиля, сталкиваются с кузовом автомобиля, и в это время пленка покрытия и пленка металлизации разрушаются и отслаиваются, образуя сколы. Это явление представляет собой большую проблему в регионах с холодным климатом и называется явлением низкотемпературного скалывания. В регионах с холодным климатом пленка покрытия подвергается воздействию низких температур, что приводит к образованию внутреннего напряжения, работающего на сжатие. Когда летящий камень и т.п. ударяет по пленке покрытия, мало того, что повреждается пленка покрытия, но повреждается еще и нижележащая пленка металлизации, и, кроме того, могут образовываться трещины, доходящие до границы между пленкой металлизации и стальным листом. Считается, что это происходит вследствие того, что внутреннее напряжение в пленке покрытия воздействует на пленку металлизации. Отслоение части пленки металлизации немедленно приводит к снижению коррозионной стойкости и представляет собой серьезную проблему для системы покрытия кузова автомобиля.
[0006] Мера, которая была предпринята для того, чтобы купировать скалывание элементов кузова автомобиля, заключается во введении грунтовки против скалывания между пленкой электролитически осажденного покрытия и промежуточной пленкой покрытия. Задачей грунтовки против скалывания является смягчение воздействия на пленку покрытия во время столкновения с камнем за счет ее функционирования в качестве амортизирующего слоя. Следовательно, от грунтовки против скалывания требуются высокая эластичность пленки покрытия, большой коэффициент растяжения пленки покрытия и высокая прочность пленки покрытия.
[0007] В качестве грунтовки против скалывания с большим коэффициентом растяжения пленки покрытия в патентном документе 1 (JP 2003-251272A) описывается водная грунтовка против скалывания, у которой температура стеклования (Tс) регулируется в диапазоне от 0°C до -75°C.
[0008] С другой стороны, в автомобильной промышленности высока потребность в снижении затрат за счет исключения или упрощения производственных процессов, в частности процесса нанесения покрытия, как уже описано выше, и поэтому требуется такая система покрытия кузова автомобиля, с помощью которой можно было бы исключить дополнительный процесс, такой как нанесение грунтовки против скалывания.
[0009] Например, патентный документ 2 (JP 2003-245605A) и патентный документ 3 (JP 2005-15516A) описывают способ формирования пленки многослойного покрытия, в котором в промежуточную пленку покрытия помещают частицы резины, которые поглощают удар при скалывании, чтобы обеспечить сопротивление скалыванию, и таким образом нанесение грунтовки против скалывания может быть исключено.
[0010] Патентный документ 4 (JP 2003-253211A) раскрывает водную композицию промежуточного покрытия, которая состоит из пленкообразующей смолы, отвердителя, окрашивающего пигмента, талька и кремнийорганического аппрета, и обладает сопротивлением скалыванию.
[0011] Все патентные документы 2-4 нацелены на то, чтобы исключить применение грунтовки против скалывания с помощью способа, в котором после нанесения на автомобильный стальной лист грунтового материала, такого как электролитически осаждаемый материал покрытия, обеспечивается нанесение промежуточного слоя покрытия, обладающего сопротивлением скалыванию. В отличие от этого, пока еще не существует системы покрытия кузова автомобиля, в которой для элемента кузова автомобиля используется покрытый металлический лист, а сопротивлением скалыванию обладает сама пленка покрытия покрытого металлического листа, и таким образом исключается использование грунтовки против скалывания.
СПИСОК ЛИТЕРАТУРЫ
[0012] ПАТЕНТНАЯ ЛИТЕРАТУРА
Патентный документ 1: JP 2003-251272A
Патентный документ 2: JP 2003-245605A
Патентный документ 3: JP 2005-15516A
Патентный документ 4: JP 2003-253211A
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
ТЕХНИЧЕСКАЯ ПРОБЛЕМА
[0013] Настоящее изобретение было сделано с учетом упомянутой выше проблемы и относится к покрытому металлическому листу для автомобиля, имеющему сопротивление скалыванию и обладающему превосходной стойкостью против ржавления в низкотемпературной среде эксплуатации.
РЕШЕНИЕ ПРОБЛЕМЫ
[0014] Авторы настоящего изобретения нашли, что использование грунтовки против скалывания может быть исключено с помощью способа, в котором в качестве пленки покрытия покрытого металлического листа применяется используемая для обычных грунтовок против скалывания органическая смола, которая имеет высокий коэффициент растяжения и температуру стеклования Tс, равную 0°C или менее, и таким образом обеспечивается сопротивление скалыванию. Однако пленка покрытия, сформированная из органической смолы с температурой стеклования Tс 0°C или менее, обладает адгезионной способностью при обычной температуре, что создает проблему того, что когда покрытые металлические листы хранятся в сложенном в стопку состоянии, вышележащие и нижележащие покрытые металлические листы склеиваются. Авторы настоящего изобретения выполнили дополнительные исследования и решили эту проблему путем помещения имеющих конкретную твердость частиц в пленку покрытия и смогли получить покрытый металлический лист для автомобиля по настоящему изобретению, обладающий сопротивлением скалыванию.
[0015] Далее конкретно описывается настоящее изобретение.
[1] Покрытый металлический лист для автомобиля, содержащий:
металлический лист; и
пленку покрытия (α), присутствующую на по меньшей мере одной поверхности металлического листа,
причем пленка покрытия (α) содержит
органическую смолу (A),
электропроводящие пигменты (B) и
противокоррозионные пигменты (C), и
микротвердость по Мартенсу HM при -20°C поверхности пленки покрытия (α) составляет от 10 до 200 (мг/мм2) в 20 точках или более при измерении в 100 точках, а микротвердость по Мартенсу HM при 40°C поверхности пленки покрытия (α) составляет от 200 до 200000 (мг/мм2) в 5 точках или более при измерении в 100 точках.
[2] Покрытый металлический лист для автомобиля в соответствии с пунктом [1], в котором температура стеклования Tс органической смолы (A) составляет от -80°C до -20°C.
[3] Покрытый металлический лист для автомобиля в соответствии с пунктом [1], в котором органическая смола (A) выбрана из группы, состоящей из полиэфирной смолы, полиуретановой смолы, акриловой смолы и их модифицированного продукта.
[4] Покрытый металлический лист для автомобиля в соответствии с пунктом [1], в котором электропроводящие пигменты (B) представляют собой частицы неоксидной керамики с удельным электрическим сопротивлением при 25°C от 0,1×10-6 до 185×10-6 Ом⋅см, являющиеся по меньшей мере одним, выбираемым из борида, карбида, нитрида и силицида.
[5] Покрытый металлический лист для автомобиля в соответствии с пунктом [1], в котором пленка покрытия (α) содержит от 0,5 об.% до 65 об.% электропроводящих пигментов (B).
[6] Покрытый металлический лист для автомобиля в соответствии с пунктом [1], в котором противокоррозионные пигменты (C) содержат
одно или более соединений, выбираемых из соединений, способных выделять ион силиката, ион фосфата, ион ванадата, ион вольфрамата или ион молибдата,
одну или более частиц, содержащих элемент-металл, выбираемый из группы, состоящей из Si, Ti, Al и Zr, или
и то, и другое.
[7] Покрытый металлический лист для автомобиля в соответствии с пунктом [1], в котором пленка покрытия (α) содержит от 1 об.% до 40 об.% противокоррозионных пигментов (C).
[8] Покрытый металлический лист для автомобиля в соответствии с пунктом [1], содержащий в пленке покрытия зернистые частицы (D) с твердостью по Мартенсу при 40°C от 200 мг/мм2 до 200000 мг/мм2.
[9] Автомобильный компонент, выполненный путем обработки и формования покрытого металлического листа для автомобиля в соответствии с пунктом [1].
[10] Автомобильный компонент, выполненный путем дополнительного нанесения одного или более из слоя электролитически осаждаемого покрытия, слоя промежуточного покрытия и слоя поверхностного покрытия на автомобильный компонент в соответствии с пунктом [9].
ПОЛЕЗНЫЕ ЭФФЕКТЫ ИЗОБРЕТЕНИЯ
[0016] Поскольку в покрытом металлическом листе для автомобиля по настоящему изобретению сама пленка покрытия обладает сопротивлением скалыванию, процесс нанесения грунтовки против скалывания становится ненужным в процессе нанесения покрытия после того, как покрытый металлический лист обработан и сформирован в автомобильный компонент. Кроме того, сопротивление скалыванию пленки покрытия является эффективным особенно в низкотемпературной окружающей среде с температурой -15°C или менее, и поэтому может быть обеспечен покрытый металлический лист для автомобиля, обладающий превосходной коррозионной стойкостью.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
[0017] [Фиг. 1] Фиг. 1 показывает схематическое изображение в разрезе строения обычной пленки покрытия автомобиля, содержащей грунтовку против скалывания.
[Фиг. 2] Фиг. 2 показывает схематическое изображение разреза пленки покрытия в том случае, когда летящий объект сталкивается с элементом кузова автомобиля и поверхность металлического листа подвергается воздействию.
[Фиг. 3] Фиг. 3 показывает схематическое изображение разреза пленки покрытия в том случае, когда летящий объект сталкивается с элементом кузова автомобиля, который использует покрытый металлический лист для автомобиля по настоящему изобретению, и металлический лист подвергается воздействию, а затем антикоррозионный компонент, который растворяется из пленки покрытия (α) из-за смачивания водой, воздействует на открывшуюся поверхность металлического листа с тем, чтобы сформировать пленку защитного покрытия.
[Фиг. 4] Фиг. 4 показывает схематическое изображение разреза пленки покрытия в том случае, когда летящий объект сталкивается с элементом кузова автомобиля, который использует покрытый металлический лист для автомобиля, у которого свойства пленки покрытия (α) не соответствуют диапазону по настоящему изобретению, и поэтому вышележащая пленка покрытия, содержащая слой металлизации, в значительной степени отслаивается из-за большого внутреннего напряжения пленки покрытия (α), и поверхность металлического листа, даже при последующем смачивании водой, в недостаточной степени покрывается пленкой защитного покрытия, образующейся из антикоррозионного компонента, который получается из пленки покрытия (α), из-за большой открытой поверхности металлического листа.
[Фиг. 5] Фиг. 5 показывает схематическое изображение разреза покрытого металлического листа для автомобиля по настоящему изобретению в том случае, когда выполняется обработка грунтовкой.
[Фиг. 6] Фиг. 6 показывает схематическое изображение разреза покрытого металлического листа для автомобиля по настоящему изобретению в том случае, когда обработка грунтовкой не выполняется.
[Фиг. 7] Фиг. 7 показывает схематическое изображение, демонстрирующее состояния распределения частиц (P) в разрезе покрытого металлического листа для автомобиля по настоящему изобретению.
ОПИСАНИЕ ВАРИАНТОВ ОСУЩЕСТВЛЕНИЯ
[0018] Далее настоящее изобретение описывается более подробно.
<Металлический лист>
[0019] Покрытый металлический лист для автомобиля по настоящему изобретению представляет собой, например, снабженный пленкой металлического покрытия (пленкой металлизации) металлический лист, в котором по меньшей мере часть поверхности покрыта специальной пленкой электропроводящего покрытия. В этом металлическом листе, в зависимости от применения, пленкой электропроводящего покрытия могут быть покрыты обе поверхности металлического листа или только одна поверхность, а также может быть покрыта часть поверхности или вся поверхность. Часть металлического листа, покрытая пленкой электропроводящего покрытия, обладает превосходной свариваемостью и коррозионной стойкостью.
[0020] Примерами металла, составляющего снабженный пленкой металлизации металлический лист, который может использоваться для покрытого металлического листа по настоящему изобретению, являются алюминий, титан, цинк, медь, никель, сталь и т.п. Компоненты этих металлов конкретно не ограничены; например, в случае использования стали может использоваться обычная сталь или сталь, содержащая добавочный элемент, такой как хром. Однако, поскольку металлический лист по настоящему изобретению должен прессоваться, во всех случаях металлических листов предпочтительно подходящим образом управлять типом и количеством добавочных элементов и структурой металла так, чтобы при обработке обеспечивалась желаемая формуемость.
[0021] В том случае, когда в качестве металлического листа используется стальной лист, тип поверхностной пленки металлизации конкретно не ограничен. Примеры используемой пленки металлизации включают в себя металлическое покрытие, содержащее один из цинка, алюминия, кобальта, олова и никеля, металлическое покрытие из сплава, содержащего любой из этих элементов-металлов и другой элемент-металл и/или элемент-неметалл, и т.п. В частности, примеры пленки металлизации на основе цинка включают в себя покрытие из цинка, покрытие из сплава цинка и по меньшей мере одного из алюминия, кобальта, олова, никеля, железа, хрома, титана, магния и марганца, а также различные покрытия из сплава на основе цинка, дополнительно содержащие другой элемент-металл и/или элемент-неметалл (например, покрытие из четырехкомпонентного сплава цинка, алюминия, магния и кремния); причем компоненты сплава, отличающиеся от цинка, конкретно не ограничены. Кроме того, эти пленки металлизации могут содержать, в качестве другого элемента-металла или примеси в небольшом количестве, кобальт, молибден, вольфрам, никель, титан, хром, алюминий, марганец, железо, магний, свинец, висмут, сурьму, олово, медь, кадмий, мышьяк и т.п., а также могут содержать материал, в котором диспергировано неорганическое вещество, такое как кремнезем, глинозем или диоксид титана.
[0022] Примеры пленки металлизации на основе алюминия включают в себя металлическое покрытие из алюминия, металлическое покрытие из сплава алюминия и по меньшей мере одного из кремния, цинка и магния (например, покрытие из сплава алюминия и кремния, покрытие из сплава алюминия и цинка, а также покрытие из тройного сплава алюминия, кремния и магния), и т.п.
[0023] Кроме того, также может использоваться многослойная металлизация, в которой комбинируются упомянутое выше металлическое покрытие и другой тип металлического покрытия, такой как покрытие из железа, покрытие из сплава железа и фосфора, покрытие из никеля и покрытие из кобальта.
[0024] Способ формирования пленки металлического покрытия конкретно не ограничен. Примеры включают в себя электролитическое нанесение металлического покрытия, нанесение металлического покрытия методом химического восстановления, покрытие погружением в расплав, нанесение металлического покрытия осаждением из паровой фазы, дисперсионное нанесение металлического покрытия и т.п. Способ обработки металлического покрытия может представлять собой либо систему непрерывного действия, либо систему периодического действия. В случае использования стального листа обработка после металлизации может быть матовой обработкой, которая представляет собой обработку для достижения однородного внешнего вида после погружения в горячий расплав, отжиг, который представляет собой модификационную обработку пленки металлизации, дрессировку для корректировки состояния поверхности или качества материала, и т.д.; однако в настоящем изобретении эта обработка не ограничивается именно перечисленным и может использоваться любая подходящая обработка.
<Пленка покрытия (α)>
[0025] Пленка покрытия (α), которая покрывает металлический лист по настоящему изобретению, содержит органическую смолу (A), электропроводящие пигменты (B) и противокоррозионные пигменты (C), и микротвердость по Мартенсу HM при -20°C поверхности пленки покрытия (α) составляет от 10 до 200 (мг/мм2) в 20 точках или более из 100 измеряемых точек, а твердость по Мартенсу HM при 40°C поверхности пленки покрытия (α) составляет от 200 до 200000 (мг/мм2) в 5 точках или более из 100 измеряемых точек.
[0026] Микротвердость по Мартенсу HM обычно является показателем, указывающим твердость, и предписывает твердость поверхности пленки покрытия (α) в настоящем изобретении. Микротвердость по Мартенсу HM может быть измерена с использованием прибора Nanoindenter HM 500 производства компании Fischer Instruments K. K. при задании глубины углубления равной 5 мкм или менее в пленке покрытия с толщиной 10 мкм или более. В пленке покрытия с толщиной менее 10 мкм измерение может быть выполнено путем установки глубины углубления равной 1/5 толщины пленки покрытия; но поскольку в этом случае вариация при измерении является большой, число раз измерения увеличивается в соответствии с необходимостью, и среднее значение полученных измерений берется в качестве измеренного значения. В настоящем изобретении лист, который подпадает под оба из следующих случаев, берется в качестве покрытого металлического листа для автомобиля по настоящему изобретению: когда микротвердость по Мартенсу HM измеряется при -20°C в 100 случайных точках поверхности пленки покрытия (α) покрытого металлического листа, значение HM составляет от 10 до 200 (мг/мм2) в 20 точках или более из этих 100 точек; а когда микротвердость по Мартенсу HM измеряется при 40°C в 100 случайных точках, значение HM составляет от 200 до 200000 (мг/мм2) в 5 точках или более из этих 100 точек. Кроме того, предпочтителен случай, когда измерение микротвердости по Мартенсу HM при -20°C в 100 случайных точках дает HM от 10 до 200 (мг/мм2) в 40 точках или более из этих 100 точек, а измерение микротвердости по Мартенсу HM при 40°C в 100 случайных точках дает HM от 200 до 200000 (мг/мм2) в 10 точках или более из этих 100 точек, а более предпочтительным является случай, когда измерение микротвердости по Мартенсу HM при -20°C в 100 случайных точках дает HM от 10 до 200 (мг/мм2) в 60 точках или более из этих 100 точек, а измерение микротвердости по Мартенсу HM при 40°C в 100 случайных точках дает HM от 200 до 200000 (мг/мм2) в 20 точках или более из этих 100 точек.
[0027] Здесь термин «случайный» относится к исключению при выборе 100 точек, которые являются точками измерения, такой произвольности, которая приводит к смещенному результату измерения. Например, могут быть заданы некоторые 2 точки, и 100 точек могут быть выбраны с равными или случайными интервалами между этими точками; а затем могут быть измерены микротвердость по Мартенсу HM при -20°C и микротвердость по Мартенсу HM при 40°C. В этом случае интервал между смежными точками измерения предпочтительно устанавливается так, чтобы точки измерения не находились под влиянием твердости друг друга. Хотя выше выбираются 100 точек, предполагается, что по мере того, как количество точек измерения увеличивается, измеренные значения усредняются все в большей степени, и точность измерения улучшается.
[0028] Авторы изобретения нашли, что, когда металлический лист для автомобиля, который содержит пленку покрытия (α) по настоящему изобретению и снабжен электролитически осаждаемым (гальваническим) покрытием, промежуточным покрытием и поверхностным покрытием, получает удар летящего камня в низкотемпературной окружающей среде, образование значительного дефекта вследствие воздействия камня, который может приводить к отслаиванию слоя металлизации, подавляется в том случае, когда пленка покрытия (α) является достаточно гибкой даже в низкотемпературной окружающей среде, по сравнению с другими случаями. Кроме того, авторы изобретения нашли, что образование значительного дефекта подавляется в том случае, когда микротвердость по Мартенсу пленки покрытия (α) в низкотемпературной окружающей среде является низкой, в диапазоне от 10 до 200 (мг/мм2).
[0029] В том случае, когда пленка покрытия (α) не является достаточно гибкой при низкой температуре, пленка поверхностного покрытия, промежуточная пленка покрытия и пленка электролитически осаждаемого покрытия разрушаются под воздействием удара камня, и в дополнение к этому разрушается сама пленка покрытия (α). Было найдено, что в этом случае усадочное напряжение этих пленок покрытия, высвобождаемое при разрушении, передается как напряжение, которое отслаивает слой металлизации, и, следовательно, слой металлизации сильно отслаивается. С другой стороны, было найдено, что в том случае, когда пленка покрытия (α) имеет достаточную гибкость при низкой температуре, даже когда пленка покрытия, находящаяся поверх пленки покрытия (α), разрушается под воздействием удара камня, усадочное напряжение поглощается за счет деформации пленки покрытия (α) и не передается к слою металлизации, и, следовательно, отслаивание слоя металлизации подавляется. Таким образом, было найдено, что в том случае, когда отслаивание слоя металлизации подавляется, даже когда вышележащая пленка покрытия портится, коррозия поверхностей слоя металлизации и нижележащего листа металла, открывшихся в разрушенной части, подавляется за счет действия противокоррозионных пигментов, содержащихся в пленке покрытия (α), и поэтому коррозионная стойкость при скалывании является высокой.
[0030] В соответствии с исследованием авторов изобретения было найдено, что пленка покрытия (α) была достаточно гибкой для того, чтобы в достаточной степени показать описанный выше эффект в том случае, когда микротвердость по Мартенсу HM, измеренная на поверхности пленки покрытия (α), составляла от 10 до 200 при -20°C в 20 точках или более из 100 случайных точек измерения. Было найдено, что если HM была больше, чем 200, пленка покрытия (α) не была гибкой, и эффект подавления переноса усадочного напряжения пленки покрытия к слою металлизации был недостаточным. Нижний предел HM при -20°C конкретно не установлен; но поскольку смола, которая придает пленке покрытия (α) микротвердость HM при -20°C менее 10, не может быть получена с разумными производственными затратами, это значение служит в качестве практического нижнего предела.
[0031] В случае пленки покрытия (α), которая является гибкой при низкой температуре до такой степени, чтобы иметь часть с HM при -20°C в 200 или менее, когда покрытые пленкой металлические листы находятся друг на друге в ситуациях хранения, транспортировки и т.д. при обычной температуре, т.е. приблизительно от -20°C до 40°C, есть вероятность того, что пленки покрытия (α) будут взаимно прилипать или слипаться, что будет мешать промышленному обращению с ними. В соответствии с исследованием авторов изобретения было найдено, что взаимная адгезия или слипание между вышеупомянутыми пленками покрытия (α) в достаточной степени подавлялась в том случае, когда микротвердость по Мартенсу HM, измеренная на поверхности пленки покрытия (α), составляла от 200 до 200000 при 40°C в 5 точках или более из 100 случайных точек измерения. Предполагается, что когда снабженные пленкой покрытия металлические листы удерживаются наложенные друг на друга в ситуации хранения, транспортировки и т.д. при обычной температуре, составляющей приблизительно от 20°C до 40°C, контакт частей с низкой HM при -20°C от 10 до 200, описанной выше, подавляется присутствием части с высокой HM при 40°C на поверхности пленки покрытия (α), и, следовательно, адгезия или слипание пленок покрытия (α) предотвращается. Описанный выше эффект уменьшается в том случае, когда количество точек, в которых микротвердость по Мартенсу HM при 40°C равна от 200 до 200000, составляет менее 5 из 100 случайных точек измерения.
[0032] Микротвердостью по Мартенсу HM при -20°C пленки покрытия (α) обычно можно управлять путем подходящего выбора органической смолы (A) и отвердителя композиции для пленки покрытия. Конкретные примеры этого способа включают в себя способ, в котором молекулярная структура смолы формируется так, чтобы она включала в себя легко деформируемую, гибкую структуру, в которой молекулярная масса части между точками сшивки является большой, способ, в котором тип и количество добавляемого отвердителя регулируются так, чтобы сохранить низкой плотность сшивающих связей между молекулярными цепями смолы, а также способ, в котором температура горячей сушки пленки покрытия уменьшается, или время горячей сушки сокращается, и тем самым реакция сшивки, производимая отвердителем, смягчается.
[0033] В дальнейшем в настоящем изобретении композиция покрытия для получения пленки покрытия (α) упоминается как композиция покрытия (β). Примеры композиции покрытия (β) включают композицию покрытия на водной основе и композицию покрытия на основе органического растворителя.
[0034] В настоящем изобретении «композиция покрытия на водной основе» относится к композиции, составленной с использованием «растворителя на основе воды», в котором вода составляет 50 мас.% или более всего растворителя. Кроме того, «композиция покрытия на основе органического растворителя» относится к композиции, составленной с использованием «растворителя на основе органического растворителя», в котором органический растворитель составляет 50 мас.% или более всего растворителя.
[0035] Примеры отличающегося от воды компонента упомянутого выше «растворителя на основе воды» включают минеральную кислоту, которая хорошо смешивается с водой, такую как серная кислота, азотная кислота, соляная кислота, фосфорная кислота, борная кислота и фтористоводородная кислота, растворимую в воде неорганическую соль, такую как растворимые в воде соли металла и соли аммония вышеупомянутых минеральных кислот, растворимое в воде неорганическое соединение, такое как силикаты, тиосульфаты и тиоцианаты (роданиды), а также органическое соединение, которое смешивается с водой. Кроме того, органический растворитель может быть добавлен к упомянутому выше «растворителю на основе воды» по мере необходимости. Однако в «композиции покрытия на водной основе» по настоящему изобретению с точки зрения трудовой гигиены предпочтительно, чтобы тип и количество добавляемого органического растворителя были отрегулированы так, чтобы получить композицию покрытия, которая не подпадает под органические растворители и т.д. (органические растворители класса 1, органические растворители класса 2, или органические растворители класса 3, или материалы, содержащие более 5 мас.% упомянутого выше органического растворителя), определяемые в Постановлении об осуществлении Закона о промышленной безопасности и здоровье (Постановление о предотвращении отравлений органическими растворителями, Глава 1, Раздел 1).
[0036] Предпочтительные примеры способа получения пленки на металлическом листе в случае композиции покрытия на водной основе или на основе органического растворителя включают способ, в котором композицию покрытия (β) наносят на металлический лист известным способом нанесения, таким как нанесение покрытия с помощью валика, нанесение покрытия с помощью валика с канавками, нанесение покрытия поливом, нанесение покрытия поливом с роликом, нанесение покрытия погружением или отжим воздушным шабером, а затем вода или растворитель удаляются из влажной пленки покрытия до сухости. Предпочтительные примеры способа отверждения этих высушенных пленок покрытия включают отверждение полимеризацией путем нагрева и термической обработки органической смолы в пленке покрытия; например, полимеризация или отверждение с помощью ультрафиолетового облучения могут использоваться в том случае, когда смола в пленке покрытия может полимеризоваться под воздействием ультрафиолетового излучения, и полимеризация или отверждение с помощью облучения электронным лучом могут использоваться в том случае, когда смола в пленке покрытия может полимеризоваться при воздействии электронным лучом.
[0037] Между пленкой покрытия (α) и поверхностью металлического листа может быть предусмотрена пленка грунтового покрытия для целей дополнительного улучшения адгезии к металлическому листу, коррозионной стойкости и т.д. пленки покрытия. В том случае, когда пленка грунтового покрытия предусмотрена, количество и состав этого слоя не ограничены; но для того, чтобы не ухудшить обрабатываемость и коррозионную стойкость пленки покрытия (α) во время формования металлического листа, необходимо, чтобы пленка грунтового покрытия обладала превосходной адгезией к нижележащему металлическому листу и к вышележащей пленке покрытия (α). С учетом совместимости с окружающей средой пленка грунтового покрытия предпочтительно имеет не содержащий хроматов состав. Кроме того, для того чтобы гарантировать достаточную электропроводность в направлении по толщине пленки покрытия, толщина пленки грунтового покрытия предпочтительно устанавливается равной 0,5 мкм или менее.
[0038] В том случае, когда предусматривается пленка грунтового покрытия, способ получения пленки грунтового покрытия не ограничивается при условии, что этот способ является промышленно применимым. Примеры способа получения пленки грунтового покрытия включают формирование пленки из композиции для грунтового покрытия путем ее нанесения, нанесения покрытия осаждением из паровой фазы, приклеивания пленки и т.д.; причем с точки зрения стоимости производства пленки (производительности), универсальности и т.д. предпочтителен способ, основанный на нанесении и сушке композиции для грунтового покрытия на водной основе или на основе растворителя. В случае использования композиции для грунтового покрытия на водной основе или на основе растворителя многослойная пленка покрытия может быть сформирована путем повторения нанесения и сушки каждого слоя от самого нижнего слоя до самого наружного слоя множества пленок покрытия, составляющих пленку грунтового покрытия (способ последовательного покрытия). Кроме того, в качестве простого и эффективного способа формирования пленки покрытия на поверхности металлического листа производство пленки может быть выполнено с помощью способа ламинирования, который содержит следующие процессы в указанном порядке: процесс, в котором множество пленок покрытия от самого нижнего слоя, находящегося в контакте с поверхностью металлического листа, до самого внешнего слоя наносят для последовательного или одновременного покрывания во влажном состоянии (процесс нанесения «влажный по влажному» или процесс одновременного многослойного нанесения композиции покрытия); процесс сушки, в котором вода или растворитель из находящихся во влажном состоянии пленок покрытия одновременно удаляются до сухости; и процесс получения пленки, в котором вышеупомянутая многослойная пленка покрытия отверждается. Здесь, способ нанесения «влажное по влажному» представляет собой способ, в котором покрывающую жидкость наносят на металлический лист, затем на эту покрывающую жидкость наносят другую покрывающую жидкость в содержащем растворитель состоянии, пока предыдущая покрывающая жидкость еще не высохла (во влажном состоянии), а затем растворители полученной многослойной покрывающей жидкости одновременно удаляют до сухости для отверждения, и таким образом получают пленку. Способ одновременного многослойного нанесения представляет собой способ, в котором с использованием машины для нанесения многослойных покрытий поливом скользящего типа, устройства для нанесения покрытия с помощью щелевой экструзионной головки и т.п. множество слоев покрывающих жидкостей одновременно наносят в состоянии «ламината» на металлический лист, затем растворители из покрывающих жидкостей этого ламината одновременно удаляют до сухости для отверждения, и таким образом получают пленка.
[0039] Средняя толщина пленки покрытия (α), которая покрывает металлический лист по настоящему изобретению, предпочтительно находится в диапазоне от 0,5 до 30 мкм, а более предпочтительно в диапазоне от 1 до 15 мкм. При толщинах менее чем 0,5 мкм пленка покрытия является слишком тонкой для того, чтобы удерживать достаточное количество противокоррозионных пигментов, и коррозионная стойкость не может быть получена. Если толщина пленки покрытия составляет более чем 30 мкм, используемое количество композиции покрытия (β) увеличивается, и увеличиваются производственные затраты, а кроме того, пленка покрытия может агрегироваться и ломаться или отслоиться во время прессования. В дополнение, из-за толстой пленки электрическая изоляция в направлении по толщине пленки увеличивается, и контактная электросварка становится затруднительной. Кроме того, в том случае, когда используется композиция покрытия на водной основе, весьма вероятно образование дефекта покрытия, такого как вспучивание, и становится довольно трудно стабильно получать внешний вид, необходимый для промышленного продукта.
[0040] Толщина пленки покрытия (α) может быть измерена с помощью наблюдения разреза пленки покрытия и т.п. Альтернативно, основываясь на том, что расчетная величина, полученная с помощью способа, в котором масса пленки покрытия, прикрепленной на единице площади металлического листа, делится на плотность пленки покрытия или плотность композиции покрытия (β) после сушки, ожидается близкой к значению измерения, полученному путем наблюдения разреза, возможен способ выполнения вычисления путем простого использования плотности. Способ определения массы пленки покрытия может быть подходящим образом выбран из существующих способов, таких как измерение разности масс между состояниями до и после нанесения покрытия, измерение разности масс между состояниями до и после отслаивания пленки покрытия после его нанесения или выполнение рентгеновского флуоресцентного анализа на пленке покрытия для того, чтобы измерить количество присутствующего элемента, содержание которого в пленке покрытия было найдено заранее. Способ определения плотности пленки покрытия или плотности композиции покрытия (β) после сушки может быть подходящим образом выбран из существующих способов, таких как измерение емкости и массы изолированной пленки покрытия, измерение объема и массы высушенной композиции покрытия (β), получаемой путем помещения подходящего ее количества в контейнер и выполнения сушки, или выполнение вычисления на основе количеств смешанных компонентов пленки покрытия и известной плотности каждого компонента.
<Органическая смола (A)>
[0041] Органическая смола (A) по настоящему изобретению является связующим компонентом пленки покрытия (α); путем ее подходящего выбора может быть получена микротвердость по Мартенсу HM при -20°C и температура стеклования Tс, необходимые пленке покрытия металлического листа для автомобиля по настоящему изобретению. Органическая смола (A) может представлять собой либо смолу на водной основе, либо смолу на основе органического растворителя, и в частности представляет собой смолу (A1), описываемую позже. Органическая смола (A) может дополнительно содержать реакционное производное (A2) смолы (A1).
[0042] Органическая смола (A) по настоящему изобретению предпочтительно имеет температуру стеклования Tс от -80°C до -20°C, как подробно описывается ниже.
[0043] Композиция покрытия (β), используемая для формирования пленки покрытия (α) в настоящем изобретении, может быть либо композицией на водной основе, либо композицией на основе органического растворителя и содержит от 50 до 100 мас.% описываемой позже смолы (A1) по ее нелетучему содержанию. Смола (A1) устойчиво присутствует в композиции покрытия (β). Когда такая композиция покрытия (β) наносится на металлический лист и выполняется нагревание, во многих случаях смола (A1) не реагирует, но высыхает в том состоянии, как она есть. В том случае, когда в композиции покрытия (β)содержится кремнийорганический аппрет, отвердитель, сшивающее средство и т.п., по меньшей мере часть смолы (A1) реагирует с ними с образованием производного (A2) смолы (A1). Таким образом, в этом случае материал, содержащий непрореагировавшую смолу (A1) и реакционное производное (A2) смолы (A1), служит в качестве органической смолы (A), которая является связующим компонентом пленки покрытия (α).
[0044] Тип смолы (A1) конкретно не ограничен, и она может быть, например, полиуретановой смолой, полиэфирной смолой, эпоксидной смолой, (мет)акриловой смолой или полиолефиновой смолой, их модифицированным продуктом и т.п., при условии, что она имеет необходимую микротвердость по Мартенсу HM и необходимую температуру стеклования Tс. Одно из этих веществ или смесь двух или более из них могут использоваться в качестве смолы (A1), или по меньшей мере одна органическая смола может быть модифицирована и одна или смесь двух или более из получаемых органических смол могут использоваться в качестве смолы (A1).
[0045] Предпочтительные примеры смолы (A1) включают полиуретановую смолу, модифицированный продукт полиуретановой смолы, композит полиуретановой смолы, их смесь с другой смолой и т.п. Уретановая группа (-NHCOO-) в полиуретановой смоле имеет более высокую энергию молекулярной агрегации (8,74 ккал/моль), чем многие другие органические группы; поэтому когда полиуретановая смола содержится в смоле (A1), адгезионная способность пленки покрытия увеличивается, отслаивание и задирание пленки покрытия происходят с меньшей вероятностью во время прессования, и, в дополнение к этому, проявляется эффект улучшения свойств блокирования коррозионных факторов (плотность пленки покрытия), улучшающий коррозионную стойкость за счет относительно высокой энергии агрегации. Энергии молекулярной агрегации органических групп, отличающихся от уретановой группы, например, группы метилена (-CH2-), простой эфирной группы (-O-), группы вторичного амина (иминогруппы, -NH-), сложноэфирной группы (-COO-) и бензольного кольца, составляют 0,68 ккал/моль, 1,00 ккал/моль, 1,50 ккал/моль, 2,90 ккал/моль и 3,90 ккал/моль соответственно; таким образом, энергия молекулярной агрегации уретановой группы (-NHCOO-) является намного более высокой. Следовательно, во многих случаях пленка покрытия, содержащая полиуретановую смолу, имеет более высокую адгезионную способность, чем пленка покрытия из многих других смол, таких как полиэфирная смола, (мет)акриловая смола и полиолефиновая смола, и имеет высокую коррозионную стойкость.
[0046] Как было описано выше, тип смолы (A1) конкретно не ограничен, при условии, что она имеет необходимую температуру стеклования Tс. Предпочтительно, чтобы смола (A1) представляла собой смолу, содержащую в своей структуре по меньшей мере одну функциональную группу, выбираемую из карбоксильной группы (-COOH), карбоксилатной группы (-COO-M+; где M+ представляет собой одновалентный катион), группы сульфокислоты (-SO3H), сульфонатной группы (-SO3-M+; где M+ представляет собой одновалентный катион), первичной аминогруппы (-NH2), вторичной аминогруппы (-NHR1; где R1 представляет собой углеводородную группу), третичной аминогруппы (-NR1R2; где R1 и R2 индивидуально представляют собой углеводородную группу), группу соли четвертичного аммония (-N+R1R2R3X-; где R1, R2 и R3 индивидуально представляют собой углеводородную группу, а X- представляет собой одновалентный анион), группу соли сульфония (-S+R1R2X-; где R1 и R2 индивидуально представляют собой углеводородную группу, а X- представляет собой одновалентный анион), и группу соли фосфония (-P+R1R2R3X-; где R1, R2 и R3 индивидуально представляют собой углеводородную группу, а X- представляет собой одновалентный анион). Подробности и конкретные примеры этого описываются позже.
[0047] Примеры смолы, используемой в композиции покрытия (β) для получения пленки покрытия (α) в настоящем изобретении, могут включать в себя водорастворимую смолу или растворимую в растворителе смолу, которая полностью растворяется в воде или органическом растворителе, а также смолу, которая равномерно и тонко диспергируется в воде или растворителе в виде эмульсии, суспензии или т.п. (диспергируемая в воде смола или диспергируемая в растворителе смола). Здесь термин «(мет)акриловая смола» относится к акриловой смоле и метакриловой смоле.
[0048] Среди упомянутых выше примеров смолы (A1) примеры полиуретановой смолы включают материал, получаемый путем реакции соединения многоатомного спирта и соединения полиизоцианата, а затем выполнения удлинения цепи с использованием удлинителя цепи и т.п. Соединение многоатомного спирта конкретно не ограничено при условии, что оно представляет собой соединение, содержащее две или более гидроксильных группы на молекулу, и его примеры включают этиленгликоль, пропиленгликоль, диэтиленгликоль, 1,6-гександиол, неопентилгликоль, триэтиленгликоль, глицерин, триметилолэтан, триметилолпропан, поликарбонатный полиол, полиол сложного полиэфира, полиол простого полиэфира, такой как гидроксипропиловый эфир бисфенола, полиол полиамидоэфира, акриловый полиол, полиол полиуретана, а также смеси вышеперечисленного. Соединение полиизоцианата конкретно не ограничено при условии, что оно представляет собой соединение, содержащее две или более изоцианатных группы на молекулу, и его примеры включают алифатический изоцианат, такой как гексаметилендиизоцианат (HDI), алициклический диизоцианат, такой как изофорондиизоцианат (IPDI), ароматический диизоцианат, такой как толилендиизоцианат (TDI), ароматический алифатический диизоцианат, такой как дифенилметандиизоцианат (MDI), а также смеси вышеперечисленного. Удлинитель цепи конкретно не ограничен при условии, что он представляет собой соединение, содержащее один или более активных атомов водорода в молекуле, и в качестве удлинителя цепи могут использоваться вода или соединение амина. Примеры соединения амина включают в себя алифатический полиамин, такой как этилендиамин, пропилендиамин, гексаметилендиамин, диэтилентриамин, дипропилентриамин, триэтилентетрамин и тетраэтиленпентамин, ароматический полиамин, такой как толилендиамин, ксилилендиамин и диаминодифенилметан, алициклический полиамин, такой как диаминоциклогексилметан, пиперазин, 2,5-диметилпиперазин и изофорондиамин, соединение на основе гидразина, такое как гидразин, сукцинат дигидразида, адипат дигидразида и фталат дигидразида, алифатический аминоспирт, такой как гидроксиэтилдиэтилентриамин, 2-[(2-аминоэтил)амино]этанол и 3-аминопропандиол, и т.п.
[0049] В том случае, когда желательно получить полиуретановую смолу на водной основе, может использоваться, например, способ, в котором во время получения смолы по меньшей мере часть упомянутых выше соединений многоатомного спирта замещается содержащим карбоксильную группу соединением многоатомного спирта, содержащее карбоксильную группу соединение многоатомного спирта реагирует с соединением полиизоцианата, вводя карбоксильную группу в полимерную цепь, а затем карбоксильная группа нейтрализуется основанием с получением смолы на водной основе. Альтернативно, может использоваться способ, в котором во время получения смолы по меньшей мере часть упомянутых выше соединений многоатомного спирта заменяется соединением многоатомного спирта, имеющим вторичную аминогруппу или третичную аминогруппу в молекуле, соединение многоатомного спирта реагирует с соединением полиизоцианата, вводя вторичную аминогруппу или третичную аминогруппу в полимерную цепь, а затем выполняется нейтрализация кислотой, чтобы получить смолу на водной основе. В том случае, когда на полимерной цепи присутствует третичная аминогруппа, алкильная группа может быть введена в третичную аминогруппу для создания четвертичной аминогруппы, и тем самым может быть получена катионная смола на водной основе, имеющая группу соли четвертичного аммония. Эти соединения могут использоваться по отдельности или в виде смеси двух или более из них.
[0050] Как упомянуто выше, полиуретановая смола, которая может использоваться в качестве смолы (A1), предпочтительно является полиуретановой смолой, содержащей большое количество ароматических колец в своей молекулярной структуре. У такой полиуретановой смолы температура стеклования является более высокой, чем температура стеклования полиуретановой смолы, не имеющей ароматического кольца или имеющей ограниченное количество ароматических колец в своей молекулярной структуре, молекулярная цепь является жесткой, сопротивление деформации пленки покрытия – высоким, а коэффициент деформации растяжения пленки покрытия низок; следовательно, необходимая в настоящем изобретении твердость пленки покрытия (α) является более высокой, чем в полиуретановой смоле, не имеющей ароматического кольца или имеющей ограниченное количество ароматических колец. Таким образом, хотя нет никакого конкретного ограничения на соединение многоатомного спирта, соединение полиизоцианата и удлинитель цепи, используемые для получения смолы, предпочтительно использовать ароматическое алифатическое или ароматическое алициклическое соединение или т.п., содержащее большое количество ароматических колец.
[0051] Среди упомянутых выше примеров смолы (A1) полиэфирная смола конкретно не ограничена при условии, что она имеет необходимую HM и необходимую температуру стеклования Tс. Примеры включают материал, получаемый с помощью дегидратационной конденсационной полимеризации многоатомного спирта, такой как этиленгликоль, 1,3-пропандиол, 1,2-пропандиол, пропиленгликоль, диэтиленгликоль, 1,6-гександиол, неопентилгликоль, триэтиленгликоль, гидроксипропиловый эфир бисфенола, 2-метил-1,3-пропандиол, 2,2-диметил-1,3-пропандиол, 2-бутил-2-этил-1,3-пропандиол, 1,4-бутандиол, 2-метил-1,4-бутандиол, 2-метил-3-метил-1,4-бутандиол, 1,5-пентандиол, 3-метил-1,5-пентандиол, 1,4-циклогександиметанол, 1,3-циклогександиметанол, 1,2-циклогександиметанол, гидрированный бисфенол A, димерный диол, триметилолэтан, триметилолпропан, глицерин и пентаэритрит, а также многовалентная карбоновая кислота, такая как фталевая кислота, фталевый ангидрид, тетрагидрофталевая кислота, тетрагидрофталевый ангидрид, гексагидрофталевая кислота, гексагидрофталевый ангидрид, метилтетрафталевая кислота, метилтетрагидрофталевый ангидрид, изофталевая кислота, терефталевая кислота, янтарный ангидрид, адипиновая кислота, себациновая кислота, малеиновая кислота, малеиновый ангидрид, итаконовая кислота, фумаровая кислота, ангидрид Himic, тримеллитовая кислота, тримеллитовый ангидрид, пиромеллитовая кислота, пиромеллитовый ангидрид, азелаиновая кислота, янтарная кислота, янтарный ангидрид, молочная кислота, додеценилсукциновая кислота, додеценилсукциновый ангидрид, циклогексан-1,4-дикарбоновая кислота, а также ангидрид кислоты в эндо-форме. Также может использоваться смола на водной основе, получаемая путем их нейтрализации аммиаком, соединением амина или т.п.
[0052] Среди упомянутых выше примеров смолы (A1) эпоксидная смола конкретно не ограничена при условии, что она имеет необходимую HM и необходимую температуру стеклования Tс. Например, она может получаться путем реакции эпоксидной смолы, такой как эпоксидная смола типа бисфенола А, эпоксидная смола типа бисфенола F, эпоксидная смола типа резорцина, эпоксидная смола типа гидрированного бисфенола А, эпоксидная смола типа гидрированного бисфенола F, эпоксидная смола типа гидрированного резорцина и эпоксидная смола типа новолака, с соединением амина, таким как диэтаноламин и N-метилэтаноламин. Кроме того, могут использоваться смола на водной основе, получаемая путем нейтрализации вышеперечисленного органической кислотой или минеральной кислотой, и материал на водной основе, получаемый путем радикальной полимеризации акриловой смолы с высоким кислотным числом в присутствии вышеупомянутой эпоксидной смолы с последующей нейтрализацией аммиаком, соединением амина или т.п.
[0053] Среди упомянутых выше примеров смолы (A1) (мет)акриловая смола конкретно не ограничена при условии, что она имеет необходимую HM и необходимую температуру стеклования Tс. Примеры включают материал, получаемый путем радикальной полимеризации алкил(мет)акрилата, такого как этил(мет)акрилат, 2-этилгексил(мет)акрилат и н-бутил(мет)акрилат, гидроксиалкил(мет)акрилата, такого как 2-гидроксиэтил(мет)акрилат, или сложного эфира (мет)акриловой кислоты, такого как алкоксисилан(мет)акрилат, вместе с (мет)акриловой кислотой в воде с использованием инициатора полимеризации. Инициатор полимеризации конкретно не ограничен, и его примеры включают персульфат, такой как персульфат калия и персульфат аммония, азосоединение, такое как азобис(циановалериановая кислота) и азобисизобутиронитрил, и т.п. Здесь термин «(мет)акрилат» относится к акрилату и метакрилату, а термин «(мет)акриловая кислота» относится к акриловой кислоте и метакриловой кислоте.
[0054] Среди вышеупомянутых примеров смолы (A1) полиолефиновая смола конкретно не ограничена при условии, что она имеет необходимую температуру стеклования Tс. Примеры включают материал, получаемый путем радикальной полимеризации этилена и ненасыщенной карбоновой кислоты, такой как метакриловая кислота, акриловая кислота, малеиновая кислота, фумаровая кислота, итаконовая кислота или кротоновая кислота, при высокой температуре и высоком давлении. Кроме того, может использоваться смола на водной основе, получаемая путем нейтрализации аммиаком, соединением амина, основным металлическим соединением, таким как KOH, NaOH или LiOH, аммиаком, соединением амина или т.п., содержащим упомянутое выше соединение металла и т.п.
[0055] Вышеупомянутые примеры смолы (A1) могут использоваться по отдельности или в смесях двух или более из них. Кроме того, в качестве главного компонента композиции покрытия (β), один или более компонентов композитной смолы, которая получается путем модификации по меньшей мере части по меньшей мере одного из примеров смолы (A1) в присутствии той же самой смолы (A1), могут использоваться в качестве смолы (A1) в целом.
<Температура стеклования Tс органической смолы (A)>
[0056] Температура стеклования Tс органической смолы (A) предпочтительно составляет от -80°C до -20°C. Температура стеклования Tс может быть измерена с помощью способа, в котором органическая смола, которая образует пленку покрытия, отверждается путем нагревания при максимальной температуре нагрева 200°C, формируя пленку с толщиной 15 мкм, и максимальная температура нагрева дифференциального сканирующего калориметра (ДСК) или температура фазового перехода в устройстве измерения динамической вязкоупругости берется в качестве температуры фазового перехода Tс. Значение температуры стеклования Tс предпочтительно составляет не менее -80°C и не более -20°C. Смола с Tс выше чем -20°C имеет низкую гибкость и поэтому имеет ограниченную способность смягчать перенос к слою металлизации усадочного напряжения пленки покрытия, которое высвобождается при растрескивании пленки покрытия вследствие столкновения с летящим камнем. Нижний предел Tс конкретно не предписывается, но органическую смолу, имеющую Tс ниже чем -80°C, трудно получить в промышленном масштабе с низкими затратами. Более предпочтительно, Tс составляет не менее -60°C и не более -30°C.
<Электропроводящие пигменты (B)>
[0057] В качестве электропроводящих пигментов (B) предпочтительно используются один или более пигментов, выбираемых из металла, сплава, электропроводящего углерода, фосфида железа, карбида и полупроводникового оксида. Примеры включают металл, такой как цинк, никель, железо, алюминий, кобальт, марганец, медь, олово и хром, порошок их сплава, электропроводящий углерод, электропроводящий углеродный порошок, такой как порошок графита, порошок фосфида железа, порошок карбида, такого как карбид титана и карбид кремния, электропроводящий полупроводниковый порошок, керамические частицы и т.п. Среди них частицы неоксидной керамики являются особенно предпочтительными в покрытом металлическом листе по настоящему изобретению.
[0058] В том случае, когда используются частицы неоксидной керамики, даже когда композиция покрытия (β) для получения пленки покрытия (α) представляет собой композицию на водной основе, эти частицы неоксидной керамики не разлагаются в композиции и постоянно сохраняют высокую электропроводность. Следовательно, превосходная свариваемость контактной сваркой может поддерживаться в течение очень длительного промежутка времени по сравнению с электропроводящими частицами, которые разлагаются из-за воды, такими как частицы основного металла и частицы ферросилиция.
[0059] Неоксидная керамика, которая образует частицу неоксидной керамики, содержащуюся в пленке покрытия (α) по настоящему изобретению, представляет собой боридную керамику, карбидную керамику, нитридную керамику или силицидную керамику, удельное электрическое сопротивление которых (удельное объемное электрическое сопротивление, удельное сопротивление) при 25°C составляет в диапазоне от 0,1×10-6 до 185×10-6 Ом⋅см. Неоксидная керамика в настоящем документе представляет собой керамику, состоящую из элемента, отличающегося от кислорода, или из соединения, не содержащего кислород. Боридная керамика, карбидная керамика, нитридная керамика и силицидная керамика в настоящем документе представляют собой неоксидную керамику, содержащую соответственно бор B, углерод C, азот N и кремний Si в качестве главного составляющего элемента-неметалла. Среди них не нашлось ни одной, которая имела бы удельное электрическое сопротивление при 25°C менее чем 0,1×10-6 Ом⋅см. В том случае, когда удельное электрическое сопротивление (удельное объемное электрическое сопротивление, удельное сопротивление) при 25°C неоксидной керамики составляет больше, чем 185×10-6 Ом⋅см, ее необходимо добавлять к пленке покрытия в большом количестве для того, чтобы придать пленке полимерного покрытия достаточную электропроводность, и во время прессования покрытого металлического листа по настоящему изобретению происходят значительное отслаивание и задиры пленки покрытия, в результате чего коррозионная стойкость уменьшается; таким образом, это не является подходящим.
[0060] Поскольку частицы неоксидной керамики, содержащиеся в пленке покрытия (α) по настоящему изобретению, имеют высокую электропроводность, их количество, добавляемое для придания пленке полимерного покрытия достаточной электропроводности, может быть меньшим, а следовательно, плохое влияние на коррозионную стойкость и формуемость покрытого металлического листа уменьшается. Для сравнения, удельное электрическое сопротивление чистых металлов находится в диапазоне от 1,6×10-6 Ом⋅см (у простого вещества серебра, Ag) до 185×10-6 Ом⋅см (у простого вещества марганца, Mn), и можно заметить, что неоксидная керамика, используемая в качестве электропроводящих частиц в настоящем изобретении (удельное электрическое сопротивление: от 0,1×10-6 до 185×10-6 Ом⋅см), имеет превосходную электропроводность на уровне, по существу равном электропроводности чистых металлов.
[0061] Примеры неоксидной керамики, которая может использоваться в настоящем изобретении, включают в себя следующие. Так, примеры боридной керамики включают борид каждого переходного металла групп IV (Ti, Zr и Hf), V (V, Nb и Ta) и VI (Cr, Mo и W) Периодической таблицы, Mn, Fe, Co, Ni, редкоземельных элементов, а также щелочноземельных металлов (Ca, Sr и Ba), отличающихся от Be или Mg.
[0062] Некоторые бориды бериллия, имеющие удельное электрическое сопротивление при 25°C более 185×10-6 Ом⋅см (например, Be2B, BeB6 и т.д.) не являются подходящими для использования в настоящем изобретении, потому что их электропроводность является недостаточной. Кроме того, бориды Mg (Mg3B2, MgB2 и т.д.) не являются подходящими для использования в настоящем изобретении, потому что они неустойчивы в присутствии воды и кислоты.
[0063] Примеры карбидной керамики включают карбид каждого переходного металла групп IV, V и VI, Mn, Fe, Co и Ni. Карбиды редкоземельных элементов и щелочноземельных металлов (например, YC2, LaC2, CeC2, PrC2, Be2C, Mg2C3, SrC2 и т.д.), которые могут гидролизоваться во влажной атмосфере, не являются подходящими для использования в настоящем изобретении.
[0064] Примеры нитридной керамики включают в себя нитрид каждого переходного металла групп IV, V и VI, Mn, Fe, Co и Ni. Нитриды редкоземельных элементов и щелочноземельных металлов (например, LaN, Mg3N2, Ca3N2 и т.д.), которые могут гидролизоваться во влажной атмосфере, не являются подходящими для использования в настоящем изобретении. Примеры силицидной керамики включают в себя силицид каждого переходного металла групп IV, V и VI, Mn, Fe, Co и Ni. Силициды редкоземельных элементов и щелочноземельных металлов (например, LaSi, Mg2Si, SrSi2, BaSi2 и т.д.), которые могут реагировать с водой с образованием водорода во влажной атмосфере, не являются подходящими для использования в настоящем изобретении. Кроме того, примеры включают в себя смесь из двух или более веществ, выбираемых из этих боридов, карбидов, нитридов и силицидов, кермет, получаемый путем смешивания этих керамик с металлическим связующим материалом и выполнения спекания, и т.п.
[0065] В случае получения пленки покрытия (α) из композиции покрытия на водной основе стандартный электродный потенциал металла, составляющего часть кермета, предпочтительно равен -0,3 В или более для того, чтобы обеспечить устойчивость к разложению в воде. Причина этого заключается в том, что в том случае, когда стандартный электродный потенциал металла, составляющего часть кермета, имеет значение менее -0,3 В, когда частица кермета присутствует в композиции покрытия на водной основе в течение длительного периода времени, на поверхности этой частицы может образоваться слой ржавчины или толстый изолирующий слой оксида, и она может потерять электропроводность. Примеры устойчивых к разложению в воде частиц кермета включают WC-12Co, WC-12Ni, TiC-20TiN-15WC-10Mo2C-5Ni и т.п. Стандартные электродные потенциалы Co и Ni составляют -0,28 В и -0,25 В соответственно, оба из которых более благородны, чем -0,3 В, и поэтому оба металла являются устойчивыми к разложению в воде.
[0066] Среди упомянутых выше видов неоксидной керамики керамика на основе хрома (CrB, CrB2, Cr3C2, Cr2N, CrSi и т.д.) вызывает беспокойство по поводу вреда для экологии, а керамика на основе Hf (HfB2, HfC, HfN и т.д.) и большая часть керамики на основе редкоземельных элементов, которые являются более тяжелыми, чем Tb, является дорогой и коммерчески недоступна; следовательно, в настоящем изобретении предпочтительно использовать отличающуюся от них неоксидную керамику среди упомянутой выше группы или в смеси двух или более из вышеупомянутых керамик.
[0067] Кроме того, с точек зрения наличия или отсутствия промышленных продуктов, устойчивой дистрибуции на локальных и глобальных рынках, цен, удельного электрического сопротивления и т.д., более предпочтительна следующая неоксидная керамика. Так, предпочтительно использовать BaB6 (удельное электрическое сопротивление: 77×10-6 Ом⋅см), CeB6 (удельное электрическое сопротивление: 30×10-6 Ом⋅см), Co2B (удельное электрическое сопротивление: 33×10-6 Ом⋅см), CoB (удельное электрическое сопротивление: 76×10-6 Ом⋅см), FeB (удельное электрическое сопротивление: 80×10-6 Ом⋅см), GdB4 (удельное электрическое сопротивление: 31×10-6 Ом⋅см), GdB6 (удельное электрическое сопротивление: 45×10-6 Ом⋅см), LaB4 (удельное электрическое сопротивление: 12×10-6 Ом⋅см), LaB6 (удельное электрическое сопротивление: 15×10-6 Ом⋅см), Mo2B (удельное электрическое сопротивление: 40×10-6 Ом⋅см), MoB (удельное электрическое сопротивление: 35×10-6 Ом⋅см), MoB2 (удельное электрическое сопротивление: 45×10-6 Ом⋅см), Mo2B5 (удельное электрическое сопротивление: 26×10-6 Ом⋅см), Nb3B2 (удельное электрическое сопротивление: 45×10-6 Ом⋅см), NbB (удельное электрическое сопротивление: 6,5×10-6 Ом⋅см), Nb3B4 (удельное электрическое сопротивление: 34×10-6 Ом⋅см), NbB2 (удельное электрическое сопротивление: 10×10-6 Ом⋅см), NdB4 (удельное электрическое сопротивление: 39×10-6 Ом⋅см), NdB6 (удельное электрическое сопротивление: 20×10-6 Ом⋅см), PrB4 (удельное электрическое сопротивление: 40×10-6 Ом⋅см), PrB6 (удельное электрическое сопротивление: 20×10-6 Ом⋅см), SrB6 (удельное электрическое сопротивление: 77×10-6 Ом⋅см), TaB (удельное электрическое сопротивление: 100×10-6 Ом⋅см), TaB2 (удельное электрическое сопротивление: 100×10-6 Ом⋅см), TiB (удельное электрическое сопротивление: 40×10-6 Ом⋅см), TiB2 (удельное электрическое сопротивление: 28×10-6 Ом⋅см), VB (удельное электрическое сопротивление: 35×10-6 Ом⋅см), VB2 (удельное электрическое сопротивление: 150×10-6 Ом⋅см), W2B5 (удельное электрическое сопротивление: 80×10-6 Ом⋅см), YB4 (удельное электрическое сопротивление: 29×10-6 Ом⋅см), YB6 (удельное электрическое сопротивление: 40×10-6 Ом⋅см), YB12 (удельное электрическое сопротивление: 95×10-6 Ом⋅см), ZrB2 (удельное электрическое сопротивление: 60×10-6 Ом⋅см), MoC (удельное электрическое сопротивление: 97×10-6 Ом⋅см), Mo2C (удельное электрическое сопротивление: 100×10-6 Ом⋅см), Nb2C (удельное электрическое сопротивление: 144×10-6 Ом⋅см), NbC (удельное электрическое сопротивление: 74×10-6 Ом⋅см), Ta2C (удельное электрическое сопротивление: 49×10-6 Ом⋅см), TaC (удельное электрическое сопротивление: 30×10-6 Ом⋅см), TiC (удельное электрическое сопротивление: 180×10-6 Ом⋅см), V2C (удельное электрическое сопротивление: 140×10-6 Ом⋅см), VC (удельное электрическое сопротивление: 150×10-6 Ом⋅см), WC (удельное электрическое сопротивление: 80×10-6 Ом⋅см), W2C (удельное электрическое сопротивление: 80×10-6 Ом⋅см), ZrC (удельное электрическое сопротивление: 70×10-6 Ом⋅см), Mo2N (удельное электрическое сопротивление: 20×10-6 Ом⋅см), Nb2N (удельное электрическое сопротивление: 142×10-6 Ом⋅см), NbN (удельное электрическое сопротивление: 54×10-6 Ом⋅см), ScN (удельное электрическое сопротивление: 25×10-6 Ом⋅см), Ta2N (удельное электрическое сопротивление: 135×10-6 Ом⋅см), TiN (удельное электрическое сопротивление: 22×10-6 Ом⋅см), ZrN (удельное электрическое сопротивление: 14×10-6 Ом⋅см), CoSi2 (удельное электрическое сопротивление: 18×10-6 Ом⋅см), Mo3Si (удельное электрическое сопротивление: 22×10-6 Ом⋅см), Mo5Si3 (удельное электрическое сопротивление: 46×10-6 Ом⋅см), MoSi2 (удельное электрическое сопротивление: 22×10-6 Ом⋅см), NbSi2 (удельное электрическое сопротивление: 6,3×10-6 Ом⋅см), Ni2Si (удельное электрическое сопротивление: 20×10-6 Ом⋅см), Ta2Si (удельное электрическое сопротивление: 124×10-6 Ом⋅см), TaSi2 (удельное электрическое сопротивление: 8,5×10-6 Ом⋅см), TiSi (удельное электрическое сопротивление: 63×10-6 Ом⋅см), TiSi2 (удельное электрическое сопротивление: 123×10-6 Ом⋅см), V5Si3 (удельное электрическое сопротивление: 115×10-6 Ом⋅см), VSi2 (удельное электрическое сопротивление: 9,5×10-6 Ом⋅см), W3Si (удельное электрическое сопротивление: 93×10-6 Ом⋅см), WSi2 (удельное электрическое сопротивление: 33×10-6 Ом⋅см), ZrSi (удельное электрическое сопротивление: 49×10-6 Ом⋅см), ZrSi2 (удельное электрическое сопротивление: 76×10-6 Ом⋅см), или смесь двух или более из них.
[0068] Среди них особенно предпочтительна неоксидная керамика, удельное электрическое сопротивление которой при 25°C находится в диапазоне от 0,1×10-6 до 100×10-6 Ом⋅см. Причина этого заключается в том, что такая керамика имеет более высокую электропроводность, чем неоксидная керамика, имеющая удельное электрическое сопротивление при 25°C в диапазоне от более 100×10-6 Ом⋅см вплоть до 185×10-6 Ом⋅см; следовательно, количество частиц, добавляемых для придания пленке полимерного покрытия достаточной электропроводности, может быть меньшим, и поэтому формируется лишь ограниченное число путей проводимости тока коррозии, которые проникают через пленку покрытия, а следовательно, коррозионная стойкость почти не уменьшается. В дополнение к этому, причиной также является то, что вследствие ограниченного количества добавляемых частиц отслаивание и задиры пленки покрытия не образуются во время прессования, и формуемость практически не уменьшается.
[0069] Удельные электрические сопротивления упомянутой выше неоксидной керамики, дополнительно приведенные в круглых скобках, являются справочными значениями (литературными значениями) керамики, присутствующей на рынке и используемой в качестве промышленных материалов. Эти удельные электрические сопротивления увеличиваются или уменьшаются в зависимости от типа и количества загрязняющих элементов, которые входят в кристаллическую решетку неоксидной керамики; следовательно, в настоящем изобретении эти материалы могут использоваться только после проверки того, что их удельное электрическое сопротивление находится в диапазоне от 0,1×10-6 до 185×10-6 Ом⋅см, например, путем фактического измерения удельного электрического сопротивления при 25°C с использованием четырехполюсного четырехзондового метода и системы подачи тока постоянной величины в соответствии с японским промышленным стандартом JIS K7194, с использованием прибора для измерения удельного сопротивления Loresta EP (типа MCP-T360) и зондов ESP (диаметр части плоской головки: 2 мм) производства компании Mitsubishi Chemical Analytech Co., Ltd.
[0070] Форма частиц электропроводящих пигментов (B) предпочтительно является близкой к сфере формой, такой как сферическая частица или квазисферическая частица (например, эллипсоидальная форма, яйцевидная форма, форма, подобная мячу для игры в регби, и т.д.), а также многогранная частица (например, форма, подобная мячу для игры в футбол, форма, подобная игральной кости, формы огранки бриллианта и т.д.). Частицы длинной и тонкой формы (например, подобной прутку формы, игольчатой формы, волокнистой формы и т.д.) и плоской формы (например, подобной чешуйке формы, плоской листовой формы и т.д.) не являются подходящими для использования в настоящем изобретении, потому что в процессе нанесения покрытия они могут расположиться параллельно поверхности пленки покрытия или могут осаждаться около границы раздела между металлическим листом (или грунтовым слоем в том случае, когда выполняется обработка металлической поверхности грунтовкой), который является подложкой для покрытия, и самой пленкой покрытия, и это затрудняет формирование эффективного пути проникновения тока в направлении по толщине пленки покрытия.
[0071] Средний диаметр частиц электропроводящих пигментов (B) конкретно не ограничен; однако пигменты предпочтительно присутствуют в композиции покрытия (β) по настоящему изобретению в виде частиц со среднеобъемным диаметром от 0,2 до 20 мкм, более предпочтительно в виде частиц со среднеобъемным диаметром от 0,5 до 12 мкм, а особенно предпочтительно в виде частиц со среднеобъемным диаметром от 1 до 8 мкм. Диспергированная частица, имеющая среднеобъемный диаметр в вышеупомянутом диапазоне, может быть либо единой частицей, либо вторичной частицей, в которой множество единых частиц сильно агрегированы и устойчиво существуют в композиции покрытия (β) во время процесса получения, хранения и транспортировки композиции покрытия (β), в процессе нанесения на металлический лист, который является подложкой для покрытия (или на грунтовый слой в том случае, когда выполняется обработка металлической поверхности грунтовкой), или во время других подобных событий. В процессе нанесения композиции покрытия на подложку возможно агрегирование частиц (B) и увеличение их среднеобъемного диаметра в пленке покрытия во время сушки пленки покрытия и получения пленки.
[0072] Среднеобъемный диаметр в настоящем документе относится к среднему диаметру в расчете на объем, найденному из данных об распределении частиц по объему. Он может быть найден с использованием любого известного способа измерения распределения диаметра частиц, и предпочтительно использовать среднее значение распределения диаметра сфер эквивалентного объема, измеряемое с помощью способа Коултера (способ электрического сопротивления в отверстии). Причина этого заключается в том, что способ Коултера имеет небольшую разницу в измеряемом значении между различными изготовителями и типами измерительных приборов и позволяет делать высокоточные измерения по сравнению с другими способами измерения распределения диаметров частиц (например, (a) вычисление по объемному распределению, получаемому с помощью способа лазерного дифракционного рассеивания, (b) преобразование получаемого методом анализа изображений распределения диаметра окружностей с эквивалентной площадью в объемное распределение, (c) вычисление из распределения масс, получаемого способом осаждения центрифугированием, и т.д.). В способе Коултера испытываемые частицы суспендируют в водном растворе электролита, через отверстие стеклянной трубки пропускают фиксированный ток и устанавливают давление ниже атмосферного так, чтобы частицы проходили через отверстие. Когда частица проходит через отверстие, электрическое сопротивление отверстия увеличивается из-за объема водного раствора электролита, который вытесняется частицей (и который равен объему частицы). Когда прикладывается фиксированный ток, изменение сопротивления во время прохождения частицы отражается в импульсном изменении напряжения; таким образом, объем индивидуальной частицы может быть измерен напрямую путем измерения высоты импульса напряжения для каждой частицы. Поскольку частицы имеют во многих случаях неправильные формы, то они принимаются за сферическое тело с тем же самым объемом, что и частица, и размер частицы преобразуется в диаметр сферического тела с эквивалентным объемом. Такой способ измерения диаметра сферы с эквивалентным объемом с помощью способа Коултера известен; например, подробности описываются в литературе на официальном сайте компании Beckman Coulter, Inc., [http://www.beckmancoulter.co.jp/product/product03/Multisizer3.html (Multisizer 3, точный измерительный прибор для определения распределения частиц по размерам)].
[0073] Частицы неоксидной керамики со среднеобъемным диаметром менее 0,2 мкм обычно являются более дорогими, чем частицы неоксидной керамики со среднеобъемным диаметром более 0,2 мкм, и плохо представлены на рынках в качестве промышленных продуктов. Кроме того, поскольку площадь удельной поверхности является относительно большой, при приготовлении композиции покрытия на водной основе или на основе органического растворителя трудно диспергировать частицы так, чтобы смочить всю поверхность частицы, даже используя диспергатор влаги, и во многих случаях возникают нерастворенные или несмешанные комки, не совместимые с водой или органическими растворителями; следовательно, предпочтительно не использовать вышеупомянутые частицы в настоящем изобретении. Кроме того, частицы неоксидной керамики со среднеобъемным диаметром более 20 мкм осаждаются в композиции покрытия на водной основе или на основе органического растворителя быстрее, чем частицы неоксидной керамики со среднеобъемным диаметром менее 20 мкм (как это следует из уравнения Стокса). Следовательно, трудно гарантировать стабильность дисперсии, даже когда диспергирующий агент модифицирован, и могут возникать проблемы, такие как быстрое оседание частиц, их агрегирование и затвердевание и, следовательно, трудности с повторным диспергированием; а значит, предпочтительно не использовать вышеупомянутые частицы в настоящем изобретении.
[0074] Как правило, большинство доступных электропроводящих пигментов (B) готовятся с предписанным диаметром частиц путем распыления исходного материала и классификации получаемых частиц по мере необходимости, и поэтому имеют распределение диаметра частиц, в котором смешаны частицы с различными диаметрами. Следовательно, даже когда среднеобъемный диаметр находится в пределах вышеописанного диапазона диаметров частиц, свариваемость зависит от распределения диаметра частиц. Среди примеров электропроводящих пигментов (B) особенно хороший эффект свариваемости проявляет пигмент (B1), в котором среднеобъемный диаметр каждой частицы составляет от 0,25 до 24 мкм.
[0075] Количество электропроводящих пигментов (B), содержащихся в пленке покрытия (α) при 25°C, предпочтительно составляет от 0,5 об.% до 65 об.%, более предпочтительно от 1 об.% до 40 об.% с точки зрения электропроводности во время контактной сварки, обеспечения формуемости и увеличения стоимости из-за увеличения количества электропроводящих пигментов, а еще более предпочтительно составляет от 2 об.% до 20 об.%. Диапазон от 4 об.% до 20 об.% особенно предпочтителен с точки зрения обеспечения достаточной коррозионной стойкости и формуемости, а также обеспечения достаточной свариваемости контактной сваркой.
[0076] Причина, по которой пленка покрытия (α) показывает хорошую электропроводность в покрытом металлическом листе по настоящему изобретению, по-видимому заключается в том, что в пленке покрытия (α) электропроводящие пигменты (B), которые являются электропроводящими частицами, практически не агрегируются и достаточно равномерно диспергируются по всей поверхности пленки покрытия, и электропроводящие пути, ведущие к нижележащему металлическому листу, не существуют локально в пленке покрытия. Если электропроводящие частицы агрегировались в пленке покрытия, электропроводящие пути в состоянии равномерного рассеяния по всей поверхности пленки покрытия с меньшей вероятностью образуются в пленке покрытия, и вероятно образование в пленке покрытия области, которая не имеет никаких электропроводящих путей и препятствует контактной электросварке. В таком случае необходимо добавлять большее количество электропроводящего материала для того, чтобы гарантировать электропроводящие пути, и вероятность того, что хорошая коррозионная стойкость и формуемость не смогут быть обеспечены, увеличивается. В покрытом металлическом листе по настоящему изобретению вероятность возникновения такой проблемы является очень низкой.
[0077] Если количество электропроводящих пигментов (B), содержащихся в пленке покрытия (α), составляет более 65 об.%, может быть обеспечена достаточная электропроводность; но при этом вероятно образование отслаивания и задиров пленки покрытия во время прессования, и таким образом хорошая формуемость не может быть обеспечена, и коррозионная стойкость той части, где пленка покрытия отслоилась, может уменьшиться. Кроме того, если это количество составляет более 65 об.%, эффект улучшения свариваемости насыщается, а стоимость электропроводящих частиц при этом увеличивается.
[0078] Когда электропроводящие частицы добавляются в количестве не менее чем 0,5 об.% и менее чем 1 об.% пленки покрытия, способность проводить электричество во время контактной электросварки может оказаться недостаточной; а когда электропроводящие частицы добавляются в количестве не менее 40 об.% и не более 65 об.% пленки покрытия, формуемость и совместимость по стоимости могут оказаться недостаточными; таким образом, объемная доля добавляемых электропроводящих пигментов (B) более предпочтительно составляет не менее 1 об.% и менее 40 об.%. Кроме того, когда электропроводящие частицы добавляются в количестве не менее 1 об.% и менее 2 об.% пленки покрытия, способность проводить электричество во время контактной электросварки может быть несколько недостаточной; а когда электропроводящие частицы добавляются в количестве не менее 20 об.% и менее 40 об.% пленки покрытия, формуемость и совместимость стоимости могут оказаться несколько недостаточными; таким образом добавление электропроводящих частиц в количестве не менее 2 об.% и менее 20 об.% является еще более предпочтительным. Однако когда электропроводящие частицы добавляются в количестве не менее 2 об.% и менее 4 об.% пленки покрытия, может оказаться так, что высокоустойчивая свариваемость не сможет быть обеспечена при значительных изменениях условий контактной электросварки; и поэтому добавление в количестве не менее 4 об.% и менее 20 об.% является особенно предпочтительным.
[0079] В том случае, когда количество электропроводящих пигментов (B), содержащихся в пленке покрытия (α), составляет менее 0,5 об.%, хорошая электропроводность не может быть обеспечена, потому что количество диспергированных в пленке покрытия частиц неоксидной керамики мало, и есть опасность того, что пленке покрытия не может быть придана достаточная свариваемость контактной сваркой, в зависимости от толщины пленки покрытия (α).
<Противокоррозионные пигменты (C)>
[0080] Тип противокоррозионных пигментов (C), используемых в настоящем изобретении, конкретно не ограничен, но предпочтительно они представляют собой одно или более веществ, выбираемых из силикатного соединения, фосфатного соединения, ванадатного соединения и мелкодисперсных частиц оксида металла.
[0081] Силикатное соединение, фосфатное соединение и ванадатное соединение в композиции покрытия (β) или в пленке покрытия (α) могут высвобождать соответственно ион силиката, ион фосфата и ион ванадата, а также противокатионы этих анионов (например, ион щелочноземельного металла, ион Zn, ион Al и т.д.), в соответствии с изменениями состава воды в композиции или в пленке покрытия, контактом с другим веществом или поверхностью подложки, значением pH и т.д. Предполагается, что из этих ионов те ионы, которые уже были растворены в композиции покрытия (β), включаются в пленку покрытия (α) во время получения пленки; и в соответствии с увеличением или уменьшением количества воды в пленке покрытия, контактом с другим веществом или поверхностью подложки, изменением значения pH и т.д. эти ионы образуют покрывающую пленку из нерастворимой соли или оксида вместе с другим присутствующим атомом или атомной группой, и таким образом подавляют коррозию. Аналогичным образом предполагается, что в соответствии с изменением среды после формирования пленки покрытия, силикатное соединение, фосфатное соединение и ванадатное соединение, включенные в пленку покрытия (α), также постепенно высвобождают упомянутые выше анион и катион и образуют покрывающую пленку из нерастворимой соли или оксида, и таким образом подавляют коррозию. Кроме того, в том случае, когда пленка покрытия портится и пленка металлизации металлического листа или нижележащий металл становятся открытыми, вышеупомянутое действие производится ионами силиката, ионами фосфата и ионами ванадата, а также противокатионами этих высвобождаемых анионов, которые достигают открытой поверхности пленки металлизации или нижележащего металла. Это действие проявляется более эффективно в том случае, когда степень повреждения подавляется до низкого уровня, и открытая площадь пленки металлизации или нижележащего металла ограничивается небольшим участком.
[0082] Примеры соединения силиката, которое может использоваться в настоящем изобретении, включают силикат щелочноземельного металла, такой как силикат магния и силикат кальция, силикат щелочного металла, такой как силикат лития, силикат натрия и силикат калия, силикат алюминия и т.п. Из них примеры силиката лития, силиката натрия и силиката калия включают силикат лития, в составе которого молярное отношение между оксидом кремния (SiO2) и оксидом лития (Li2O) составляет 0,5 ≤ (SiO2/Li2O) ≤ 8, силикат натрия, в составе которого молярное отношение между оксидом кремния (SiO2) и оксидом натрия (Na2O) составляет 0,5 ≤ (SiO2/Na2O) ≤ 4, и силикат калия, в составе которого молярное отношение между оксидом кремния (SiO2) и оксидом калия (K2O) составляет 0,5 ≤ (SiO2/K2O) ≤ 4, а также гидраты этих силикатов. Конкретные их примеры включают ортосиликат лития (Li4SiO4; 2Li2O⋅SiO2), диортосиликат гексалития (Li6Si2O7; 3Li2O⋅2SiO2), метасиликат лития (Li2SiO3; Li2O⋅SiO2), дисиликат лития (Li2Si2O5; Li2O⋅2SiO2), гептасиликат тетралития (2Li2O⋅7SiO2), тетрасиликат лития (Li2Si4O9; Li2O⋅4SiO2), нонасиликат тетралития (2Li2O⋅9SiO2), пентадекасиликат тетралития (2Li2O⋅15SiO2), ортосиликат натрия (Na4SiO4; 2Na2O⋅SiO2), метасиликат натрия (Na2SiO3; Na2O⋅SiO2), дисиликат натрия (Na2Si2O5; Na2O⋅2SiO2), тетрасиликат натрия (Na2Si4O9; Na2O⋅4SiO2), ортосиликат калия (K4SiO4; 2K2O⋅SiO2), метасиликат калия (K2SiO3; K2O⋅SiO2), дисиликат калия (K2Si2O5; K2O⋅2SiO2) и тетрасиликат калия (K2Si4O9; K2O⋅4SiO2), а также гидраты этих силикатов. Большинство гидратов этих силикатов легко превращается в гель в гидратированном состоянии из-за изменения pH среды, температуры и т.д., и часть из них может превращаться в макромолекулу с образованием полисиликата. Кроме того, такой полисиликат включается в силикатное соединение, которое может использоваться в настоящем изобретении.
[0083] Примеры фосфатного соединения, которое может использоваться в настоящем изобретении, включают соль металла и ортофосфорной кислоты, полифосфорной кислоты (простые линейные полимеры, в которых степень полимеризации ортофосфорной кислоты составляет вплоть до 6, или смесь двух или более из них), метафосфорной кислоты (простые циклические полимеры, в которых степень полимеризации ортофосфорной кислоты составляет от 3 до 6, или смесь двух или более из них), тетраметафосфорной кислоты, гексаметафосфорной кислоты и т.п., фосфатный минерал, такой как пентаоксид фосфора, монетит, трифилит, витлокит, ксенотим, стеркорит, струвит и вивианит, коммерчески доступные сложные фосфатные пигменты, такие как полифосфат кремнезема и триполифосфат, а также соль металла и фитиновой кислоты, фосфоновой кислоты (фосфористой кислоты), фосфиновой кислоты (фосфорноватой кислоты) и т.п., смесь двух или более из них и т.п. Ортофосфат в настоящем документе включает в себя однозамещенную кислую соль (HPO42-) и дигидросоль (H2PO4-) ортофосфорной кислоты. Кроме того, полифосфат включает в себя кислую соль. Тип катиона для образования фосфата конкретно не ограничен; примеры включают ионы Co, Cu, Fe, Mn, Nb, Ni, Sn, Ti, V, Y, Zr, Al, Ba, Ca, Mg, Sr, Zn и т.п., а также оксокатионы, такие как ванадил, титанил и цирконил; и предпочтительно используются Al, Ca, Mg, Mn и Ni. Упомянутые выше фосфатные соединения могут использоваться по отдельности или в сочетании двух или более из них.
[0084] Нежелательно использовать большое количество щелочного металла в качестве типа катиона для образования фосфата. В том случае, когда используется фосфат щелочного металла, продукт, получаемый путем обжига в процессе промышленного производства, имеет тенденцию к чрезмерному растворению в воде. Однако фосфат щелочного металла может использоваться в немного большем количестве, когда растворимостью в воде можно управлять во время получения противокоррозионных пигментов, получения композиции покрытия, получения пленки на металлическом листе, использования покрытого металлического листа и т.д. Примеры такого управления включают в себя способ, в котором противокоррозионные пигменты сосуществуют с другой добавкой, которая ограничивает растворимость в воде, или сосуществуют с макромолекулой на основе смолы или неорганики, которая имеет высокую степень сшивки, что позволяет управлять скоростью растворения в воде, и т.п.
[0085] Ванадатное соединение, которое может использоваться в настоящем изобретении, является соединением, в котором валентность ванадия равна 0, 2, 3, 4 или 5, или сложным соединением, имеющим две или более из этих валентностей, и примеры включают оксид, гидроксид, соль оксикислоты различных металлов, соединения ванадила, галогенид, сульфат, металлический порошок и т.д. ванадия, имеющего вышеперечисленные валентности. Эти соединения разлагаются во время нагревания или в присутствии воды и реагируют с присутствующим кислородом. Например, металлический порошок или двухвалентное соединение ванадия превращаются в соединение с валентностью 3, 4 или 5. Нуль-валентное соединение, например порошок металлического ванадия, может использоваться по упомянутой выше причине, но имеет такую проблему, как недостаточная реакция окисления, и поэтому не является предпочтительным с практической точки зрения. Соединение пятивалентного ванадия имеет ион ванадата и с большой вероятностью будет реагировать с ионом фосфата при нагревании, образуя гетерополимер, который способствует стойкости против ржавления; таким образом, содержание соединения пятивалентного ванадия в качестве компонента является предпочтительным. Конкретные примеры соединения ванадия включают соединение ванадия(II), такое как оксид ванадия(II) и гидроксид ванадия(II), соединение ванадия(III), такое как оксид ванадия(III), соединение ванадия(IV), такое как оксид ванадия(IV) и галогенид ванадила, и соединение ванадия(V), такое как оксид ванадия(V) и ванадат (ортованадат, метаванадат и пированадат различных металлов и т.д.), а также их смесь. Предпочтительный тип металла для образования ванадата является тем же самым, что и металлы, описанные для фосфата.
[0086] В том случае, когда используется ванадат щелочного металла, продукт, получаемый путем обжига в процессе промышленного производства, имеет тенденцию чрезмерно растворяться в воде; таким образом, как и в случае фосфата, нежелательно использовать большое количество ванадата щелочного металла. Однако он может использоваться, когда растворимостью в воде можно управлять, так же как и в том случае, когда используется фосфат щелочного металла. Это аналогичным образом применимо к случаям галогенида и сульфата ванадия.
[0087] В покрытом металлическом листе по настоящему изобретению общее количество упомянутых выше силикатного, фосфатного и ванадатного соединений составляет от 1 до 40 об.%, предпочтительно от 1 до 20 об.%, а более предпочтительно от 2 до 15 об.% пленки покрытия (α). Если это общее количество составляет менее 1 об.%, действие силикатного соединения, фосфатного соединения и ванадатного соединения становится недостаточным, и поэтому коррозионная стойкость может уменьшиться. Если это общее количество составляет более 20 об.%, пленка покрытия становится хрупкой; следовательно, за счет недостатка когезии пленки покрытия адгезионная способность и формуемость пленки покрытия во время прессования могут уменьшиться, и свариваемость может ухудшиться.
[0088] Противокоррозионные пигменты (C) предпочтительно содержат одно или более из силикатного соединения, фосфатного соединения и ванадатного соединения, и с точки зрения улучшения антикоррозионного эффекта более предпочтительно имеют состав, в котором сосуществуют фосфатное соединение (источник иона фосфата) и по меньшей мере одно из силикатного соединения (источника иона силиката) и ванадатного соединения (источника иона ванадата). Соотношение компонентов смеси между количеством источника иона фосфата и общим количеством источника иона силиката и источника иона ванадата [число молей P2O5]:[общее число молей SiO2 и V2O5] предпочтительно устанавливается равным от 25:75 до 99:1. Если молярная доля общего количества источника иона силиката и источника иона ванадата к общему количеству источника иона фосфата, источника иона силиката и источника иона ванадата составляет более 75%, антикоррозионный эффект за счет ионов фосфата может уменьшиться; а если молярная доля общего количества источника иона силиката и источника иона ванадата составляет менее 1%, эффект окисления и фиксации соседних химических компонентов ионами силиката (или ионами ванадата) может оказаться недостаточным.
[0089] Кроме того, в качестве используемых в настоящем изобретении противокоррозионных пигментов (C) могут применяться мелкодисперсные частицы оксида металла из одного или более элементов-металлов, выбираемых из группы, состоящей из Si, Ti, Al и Zr. Эти мелкодисперсные частицы оксида металла могут использоваться по отдельности, или могут быть смешаны вместе с силикатным соединением, фосфатным соединением и ванадатным соединением; тем самым коррозионная стойкость может быть дополнительно улучшена. Когда силикатное соединение, фосфатное соединение и ванадатное соединение, а также кремнезем сосуществуют, коррозионная стойкость улучшается еще больше; таким образом, это является предпочтительным. Примеры кремнезема включают пирогенный кремнезем, коллоидный кремнезем, агрегированный кремнезем и т.д. Также может использоваться карбонатный кремнезем.
[0090] Примеры вышеупомянутых мелкодисперсных частиц оксида металла, которые могут использоваться в настоящем изобретении, включают мелкодисперсные частицы кремнезема, мелкодисперсные частицы глинозема, мелкодисперсные частицы диоксида титана, мелкодисперсные частицы диоксида циркония и т.п., у которых среднеобъемный диаметр составляет приблизительно от 0,2 до 10 мкм; и мелкодисперсные наночастицы оксида металла, у которых среднеобъемный диаметр составляет приблизительно от 0,5 до 30 нм, являются более предпочтительными. Они могут использоваться по отдельности или в сочетании двух или более из них. Из них мелкодисперсные наночастицы кремнезема могут быть добавлены в том случае, когда необходимы одновременно улучшение коррозионной стойкости и повышение ударопрочности пленки покрытия.
[0091] В качестве оксида металла могут использоваться мелкодисперсные наночастицы с диаметром частицы не менее 0,5 нм и менее 30 нм, например, коллоидный кремнезем, коллоидный диоксид титана и коллоидный диоксид циркония. Они производятся различными способами из упомянутого выше оксида металла, который приготавливают в виде мелкодисперсных частиц распылением, и поэтому легко диспергируются в материале покрытия и в пленке покрытия, имея диаметр мелкодисперсных первичных частиц от 0,5 нм до 30 нм. Эти мелкодисперсные наночастицы оксида металла имеют более высокий антикоррозионный эффект, чем мелкодисперсные частицы оксида металла того же самого состава, имеющие больший диаметр частиц. Однако такие мелкодисперсные наночастицы оксида металла могут ухудшать свариваемость при контактной электросварке, при которой ток пропускается при приложении нагрузки электродами, и получаемое джоулево тепло используется для выполнения сварки, такой как точечная сварка.
[0092] Для количества мелкодисперсных наночастиц оксида металла предпочтительно, чтобы отношение суммарного объема мелкодисперсных наночастиц оксида металла в пленке покрытия к суммарному объему частиц неоксидной керамики (B) (мелкодисперсные наночастицы оксида металла/B) составляло 20 или менее. В том случае, когда важна свариваемость, это отношение более предпочтительно равно 10 или менее. Нижний предел отношения (мелкодисперсные наночастицы оксида металла/B) предпочтительно составляет 0,1 или более. Случай, когда значение отношения (мелкодисперсные наночастицы оксида металла/B) составляет менее 0,1, представляет собой состояние, в котором количество частиц неоксидной керамики (B) в пленке покрытия является слишком большим, или количество мелкодисперсных наночастиц оксида металла является слишком малым. В первом случае, поскольку количество частиц неоксидной керамики (B) в пленке покрытия является слишком большим, пленка покрытия охрупчивается, и может произойти растрескивание и отпадение пленки покрытия во время формования. Растрескивание и отпадение пленки покрытия приводит к снижению коррозионной стойкости, обеспечиваемой пленкой покрытия, а также к дефектному внешнему виду покрытого металлического листа. В последнем случае, поскольку количество мелкодисперсных наночастиц оксида металла в пленке покрытия является недостаточным, эффект улучшения коррозионной стойкости не может быть получен в достаточной степени. Стойкость против ржавления, которая уменьшается путем подавления количества мелкодисперсных наночастиц оксида металла для того, чтобы гарантировать свариваемость, может быть компенсирована за счет добавления противокоррозионных пигментов (C) с диаметром частиц 100 нм или более. Противокоррозионные пигменты (C) с диаметром частиц 100 нм или более с меньшей вероятностью попадают между электродом и (В), между (В) и (В) или между (В) и металлическим листом в таком состоянии, когда пленка покрытия наносится на металлический лист, или в таком состоянии, когда пленка покрытия деформируется под нагрузкой, прикладываемой сварочными электродами, и поэтому оказывает незначительное плохое влияние на контактную электросварку по сравнению с мелкодисперсными наночастицами оксида металла.
[0093] Предпочтительно, чтобы количество противокоррозионных пигментов (C) составляло от 1 до 40 об.% пленки покрытия (α) и чтобы общее количество противокоррозионных пигментов (C) и электропроводящих пигментов (B) не превышало 80 об.%. В том случае, когда важна коррозионная стойкость покрытого металлического листа, количество противокоррозионных пигментов (C) более предпочтительно составляет от 3 до 40 об.%, а еще более предпочтительно от 7,5 до 40 об.%. В том случае, когда важно достижение еще большей коррозионной стойкости покрытого металлического листа, количество противокоррозионных пигментов (C) более предпочтительно составляет от 13 до 40 об.%. Если это количество составляет менее 1 об.%, количество противокоррозионных пигментов (C) становится недостаточным, и поэтому эффект улучшения коррозионной стойкости не может быть получен в достаточной степени. Если это количество составляет более 40 об.%, пленка покрытия может делаться хрупкой, и адгезия пленки покрытия к металлическому листу может уменьшиться; следовательно, поверхность металлического листа может стать открытой из-за разрушения и отслаивания пленки покрытия во время формования, и внешний вид покрытого металлического листа может ухудшиться, и эффект улучшения коррозионной стойкости, обеспечиваемый пленкой покрытия, может уменьшиться.
[0094] Количество электропроводящих пигментов (B) и количество противокоррозионных пигментов (C) могут быть вычислены путем наблюдения разреза пленки покрытия под электронным микроскопом для того, чтобы идентифицировать каждую частицу, подсчета числа частиц в разрезе и преобразования полученного числа в число частиц в единице объема пленки покрытия. В этом случае каждая частица может быть идентифицирована с использованием EDX-спектрометра и т.п. по мере необходимости. Также возможно вычислить количество частиц в пленке покрытия исходя из количеств (B) и (C), содержащихся в материале покрытия до его нанесения, и количества пленки покрытия, нанесенной на металлический лист. Если найдены количества (B) и (C), включенных в материал покрытия, до нанесения, количество частиц в пленке покрытия может быть вычислено исходя из включенных количеств и количества материала покрытия, нанесенного на металлический лист. Когда включенные количества неизвестны, вычисление может быть сделано путем, например, идентификации и подсчета индивидуальных частиц в материале покрытия, который был разбавлен до подходящей концентрации, с использованием анализа изображения с помощью такого устройства, как Morphologi G3, анализатор изображений частиц производства компании Malvern Instruments Ltd. Этот способ может использоваться также в том случае, когда нанесенная на металлический лист пленка покрытия растворяется и подсчитывается количество частиц. Однако, основываясь на том факте, что расчетные значения количества электропроводящих пигментов (B) и количества противокоррозионных пигментов (C) в пленке покрытия (α), полученный с помощью вычислений, основанных на соотношении компонентов смеси между органической смолой (A), электропроводящими пигментами (B) и противокоррозионными пигментами (C) и их плотностях после сушки, как ожидается, будут близки к значениям измерений, полученным путем наблюдения разреза, возможен также способ выполнения вычислений просто на основе соотношения компонентов смеси.
[0095] Различные упомянутые выше противокоррозионные пигменты вводятся в органическую смолу (A) в пленке покрытия (α) предпочтительно путем заблаговременного растворения или диспергирования и стабилизации подходящего количества противокоррозионных пигментов в композиции покрытия (β).
<Частицы (D)>
[0096] В дополнение к электропроводящим пигментам (B) и противокоррозионным пигментам (C), в качестве частиц в пленке покрытия по настоящему изобретению могут содержаться частицы (D), такие как гранулированный воск или гранулированная смола, у которых твердость по Мартенсу при 40°C составляет от 200 мг/мм2 до 200000 мг/мм2. Гранулированный воск и гранулированная смола с твердостью по Мартенсу при 40°C от 200 мг/мм2 до 200000 мг/мм2 могут выбираться произвольно с учетом простоты добавления к материалу покрытия и т.д. Примеры таких частиц включают в себя полиолефиновый воск, полиэтиленовый воск, полипропиленовый воск, полибутиленовый воск, модифицированный полиолефиновый воск, частицы акриловой смолы, частицы силиконовой смолы, частицы фторкаучука, смолы полиакрилонитрила и т.п.
[0097] В том случае, когда твердость по Мартенсу при 40°C частиц (D) составляет менее 200 мг/мм2, когда поверхности покрытых металлических листов по настоящему изобретению входят в контакт друг с другом, или когда поверхность покрытого металлического листа входит в контакт с другим материалом, прибором или инструментом, эффект входящих в контакт частиц (D) является более предпочтительным, чем смолы (A), и тем самым предотвращение склеивания или слипания пленки покрытия (α) мало. В промышленном виде трудно найти частицы (D) с твердостью по Мартенсу при 40°C более чем 200000 мг/мм2, и это значение практически служит верхним пределом твердости HM. Диапазон значений твердости по Мартенсу более предпочтительно составляет не менее 300 мг/мм2 и не более 2000 мг/мм2.
[0098] Частицы, которые выбираются из упомянутых выше электропроводящих пигментов (B), противокоррозионных пигментов (C) и частиц (D) и имеют диаметр первичных частиц от 1 мкм до 10 мкм, определяются как частицы (P). Частицы (P) состоят из по меньшей мере одного из электропроводящих пигментов (B) и противокоррозионных пигментов (C) и при необходимости могут включать в себя частицы (D).
[0099] Состояние обнажения и диаметр частиц (P) могут быть найдены путем микроскопического наблюдения сверху покрытого металлического листа или микроскопического наблюдения разреза покрытого металлического листа. Также плотность обнаженных частиц (P) может быть найдена путем наблюдения сверху покрытого металлического листа. Альтернативно, когда диаметр и число частиц исходного материала частиц (P) известны, эта плотность может быть вычислена исходя из количества смешиваемого материала покрытия. Частицы (P), обнаженные на поверхности пленки покрытия (α), имеют микротвердость по Мартенсу HM при 40°C от 200 до 200000 (мг/мм2) и составляют по меньшей мере часть поверхности пленки покрытия (α). В соответствии с исследованиями, проведенными авторами изобретения, частицы (P), обнаженные на поверхности пленки покрытия (α), деформируются с меньшей вероятностью, чем смола (A), образующая пленку покрытия (α); следовательно, когда поверхности покрытых металлических листов по настоящему изобретению входят в контакт друг с другом, или когда поверхность покрытого металлического листа входит в контакт с другим материалом, прибором или инструментом, частицы (P) входят в контакт более предпочтительно, чем пленка покрытия (α), и могут тем самым предотвратить склеивание или слипание пленки покрытия (α).
[0100] Фиг. 7 показывает состояние, в котором частицы (P) обнажены, выступая из пленки покрытия (α), в виде схематического изображения. Для того, чтобы частицы (P) входили в контакт с другим материалом более предпочтительно, чем пленка покрытия (α), предпочтительно, чтобы соотношение между толщиной (T) пленки покрытия после сушки и диаметром (R) частиц частицы (P) удовлетворяло следующей формуле:
T/R = 0,6-2,5
(где R представляет собой среднеобъемный диаметр (мкм) частиц P).
Если значение T/R составляет менее 0,6, большая часть отдельной частицы (P) выступает из пленки покрытия (α); следовательно частицы (P) с большой вероятностью будут выпадать, и частицы (P) не будут показывать эффект в достаточной степени, либо частицы (P), которые выпали, с большой вероятностью будут смешиваться в процессе и вызывать проблемы с качеством; следовательно, это не является предпочтительным. Если значение T/R составляет более 2,5, выступание частиц (P) из пленки покрытия (α) является недостаточным, и эффект предотвращения склеивания или слипания становится незначительным; следовательно, это не является предпочтительным.
[0101] Было найдено, что этот эффект становится значительным в том случае, когда диаметр первичных частиц (P) составляет от 1 мкм до 10 мкм, а плотность обнаженных частиц составляет от 100 до 2,0×106/мм2. Случай, в котором плотность обнаженных частиц составляет от 1,0×103 до 2,0×105/мм2, является предпочтительным, а более предпочтителен случай, в котором плотность обнаженных частиц составляет от 5,0×103 до 2,0×104/мм2. В том случае, когда диаметр первичных частиц составляет менее 1 мкм, частицы (P) находятся полностью внутри пленки покрытия (α), и поэтому трудно обеспечить действие частиц (P), предпочтительным образом входящих в контакт. В том случае, когда диаметр первичных частиц составляет более 10 мкм, возникает проблема устойчивого существования частиц (P) в материале покрытия для образования пленки покрытия (α), и экономическая эффективность хранения материала покрытия и самого покрытия становится низкой. В том случае, когда плотность обнаженных частиц (P) составляет менее 100/мм2, эта плотность слишком низка, и поэтому трудно обеспечить действие частиц (P), предпочтительным образом входящих в контакт. В том случае, когда плотность обнаженных частиц (P) является достаточно большой и составляет более 2,0×106/мм2, количество частиц (P) в пленке покрытия (α) слишком велико, и поэтому могут возникать проблемы отслаивания пленки покрытия, проблемы с нанесением покрытия и т.д.
<Приготовление композиции покрытия (β)>
[0102] Способ получения композиция покрытия (β), используемой для формирования пленки покрытия (α) по настоящему изобретению, конкретно не ограничен. Примеры включают способ, в котором компоненты для формирования пленки покрытия (α) добавляют в воду или органический растворитель и выполняют перемешивание с помощью диспергирующей машины, такой как диспергатор, чтобы выполнить растворение, диспергирование или распыление. В случае композиции покрытия на водной основе при необходимости может быть добавлен известный гидрофильный растворитель и т.п. для того, чтобы улучшить растворимость или диспергируемость компонентов с тем, чтобы сформировать пленку покрытия (α).
[0103] В частности, в случае композиции покрытия (β) на водной основе, в дополнение к частицам (D), к смоле (A1), электропроводящим пигментам (B) и противокоррозионным пигментам (C) могут быть при необходимости добавлены различные водорастворимые или диспергируемые в воде добавки в такой степени, чтобы водная природа и укрывистость материала покрытия не ухудшались. Например, могут быть добавлены различные водорастворимые или диспергируемые в воде антикоррозионные средства, не имеющие формы пигментов, поверхностно-активное вещество, такое как пеногаситель, средство против оседания, выравниватель, а также диспергатор влаги, загуститель, модификатор вязкости и т.д. Кроме того, для целей стабилизации составляющих компонентов композиции покрытия (β), таких как смола или другое органическое соединение и т.д., может быть добавлено небольшое количество органического растворителя в такой степени, чтобы получаемая композиция не подпадала под ограничения на органические растворители и т.д. (органические растворители класса 1, органические растворители класса 2 или органические растворители класса 3, или материалы, содержащие более 5 мас.% упомянутого выше органического растворителя), определяемые в Постановлении об осуществлении Закона о промышленной безопасности и здоровье (Постановление о предотвращении отравлений органическими растворителями, Глава 1, Раздел 1).
[0104] В том случае, когда пленка покрытия (α) по настоящему изобретению формируется из композиции покрытия (β) на водной основе, вследствие свойств водной основы поверхностное натяжение является высоким по сравнению с композициями покрытия на основе органического растворителя; таким образом смачиваемость металлического листа, который является подложкой (или грунтового слоя в том случае, когда выполняется обработка грунтовкой), электропроводящих пигментов (B), противокоррозионных пигментов (C), частиц (D) и т.д. является недостаточной, и однородное покрытие и диспергируемость частиц не могут быть получены, когда предписанное количество покрытия наносится на подложку. В таком случае могут быть добавлены диспергатор влаги и упомянутый выше загуститель. В качестве диспергатора влаги может использоваться поверхностно-активное вещество, которое уменьшает поверхностное натяжение, а предпочтительно используется макромолекулярное поверхностно-активное вещество с молекулярной массой 2000 или более (макромолекулярный диспергатор). Низкомолекулярное поверхностно-активное вещество может относительно легко перемещаться через пленку полимерного покрытия, содержащую влагу, и поэтому может переносить к металлической поверхности воду, адсорбированную на полярной группе поверхностно-активного вещества, и коррозионные факторы, такие как находящиеся в воде растворенный кислород и растворенные соли, и, кроме того, сами по себе могут выпотевать и растворяться, что может зачастую приводить к ухудшению стойкости против ржавления пленки покрытия. В отличие от этого, макромолекулярное поверхностно-активное вещество может адсорбироваться на поверхности металла, керамической частицы и пигмента за счет многоточечной адсорбции, и поэтому практически не отделяется, когда оно адсорбировано; таким образом, оно является эффективным для улучшения смачиваемости даже в низкой концентрации. В дополнение к этому, его молекулы являются объемными, и поэтому будут с меньшей вероятностью перемещаться через пленку полимерного покрытия и с меньшей вероятностью будут переносить факторы коррозии к поверхности металла. Некоторые из акриловых смол, добавление которых к органической смоле (A) рекомендуется выше в разделе <Органическая смола (A)>, выполняют функцию вышеупомянутого макромолекулярного поверхностно-активного вещества, а также имеют эффекты ограничения седиментации электропроводящих пигментов (B), противокоррозионных пигментов (C), частиц (D) и т.д., и однородного их диспергирования в композиции покрытия на водной основе.
[0105] Загуститель может добавляться в качестве меры в том случае, когда диспергатор влаги сам по себе не может обеспечить отталкивающую часть поверхности подложки с достаточной площадью покрытия поверхности, или в том случае, когда вязкость композиции покрытия на водной основе является слишком низкой для того, чтобы гарантировать необходимую толщину пленки покрытия. Многие загустители имеют молекулярную массу от нескольких тысяч до нескольких десятков тысяч; и молекулы загустителя адсорбируются на поверхности пигмента и т.д. за счет многоточечной адсорбции и связываются друг с другом, образуя слабую сетевую структуру, и могут таким образом улучшать вязкость композиции покрытия.
[0106] В том случае, когда композиция покрытия (β) на водной основе содержит электропроводящие пигменты (B), противокоррозионные пигменты (C) и частицы (D) с высокой плотностью, к материалу покрытия при необходимости может быть добавлен модификатор вязкости, который может обеспечивать тиксотропные свойства (тиксотропию). Как и в случае вышеупомянутого загустителя, молекулы модификатора вязкости адсорбируются на поверхности пигмента и т.д. за счет многоточечной адсорбции в композиции покрытия на водной основе и образуют сетевую структуру. Молекулярная масса такого модификатора вязкости составляет от нескольких сотен тысяч до нескольких миллионов, то есть является очень высокой, и поэтому модификатор вязкости образует в композиции покрытия (β) на водной основе прочную сетевую структуру, имеющую большой предел текучести; таким образом композиция покрытия (β) с меньшей вероятностью деформируется при низкой скорости сдвига и имеет высокую вязкость. Когда к композиции покрытия (β) прикладывается большое напряжение сдвига, большее, чем предел текучести, сетевая структура разрушается и вязкость быстро уменьшается. Таким образом, когда добавляется модификатор вязкости, проявляются следующие эффекты: во время хранения и транспортировки, при которых композиция покрытия (β) на водной основе обычно сохраняет стационарное состояние, вязкость композиции покрытия (β) улучшается, и седиментация тяжелых пигментов ограничивается; а когда прикладывается высокое напряжение сдвига (высокая скорость сдвига), например, когда композиция течет через трубы на установке для нанесения покрытия, и когда композиция наносится на подложку, вязкость композиции покрытия (β) уменьшается, и ее течение становится более легким.
[0107] В случае композиции покрытия (β) на основе органического растворителя, композиция покрытия, в которой смола растворена в органическом растворителе, имеет относительно высокую вязкость, и эта вязкость легко регулируется. Следовательно, вязкость композиции покрытия может легко и устойчиво поддерживаться на уровне 100 мПа⋅с или более, что является выгодным для подавления седиментации пигментов. Кроме того, неоксидная керамика, используемая в качестве электропроводящего материала, является веществом, имеющим также гидрофобную часть на своей поверхности, и поэтому в большинстве случаев легко диспергируется в композиции покрытия (β) на основе органического растворителя; следовательно, покрытие может быть выполнено без седиментации электропроводящих пигментов (B) в композиции покрытия (β), а значит, это является предпочтительным.
[0108] Когда композиция покрытия, в которой композиция покрытия (β) на основе органического растворителя, образующая пленку покрытия, имеет вязкость от 100 до 2000 мПа⋅с, наносится на металлический лист с помощью устройства для нанесения покрытия валиком или устройства для нанесения покрытия поливом, а затем выполняются сушка и спекание, вероятность седиментации электропроводящих пигментов (B) уменьшается, а значит, это является более предпочтительным. Если вязкость композиции покрытия (β) составляет менее 100 мПа⋅с, вероятность седиментации электропроводящих пигментов (B) увеличивается, а если эта вязкость составляет более 2000 мПа⋅с, она является слишком высокой, что может вызывать дефектный внешний вид покрытия, обычно называемый волнистостью или полосатостью. Эта вязкость более предпочтительно составляет от 250 до 1000 мПа⋅с. Вязкость композиции покрытия (β) на основе органического растворителя может быть измерена с использованием вискозиметра Брукфилда при той же самой температуре, что и температура композиции покрытия во время нанесения покрытия с помощью устройства для нанесения покрытия валиком или устройства для нанесения покрытия поливом.
[0109] Эта вязкость может регулироваться за счет типа используемого органического растворителя и его количества. В качестве органического растворителя обычно может использоваться любой известный растворитель, но предпочтителен органический растворитель с высокой температурой кипения. Поскольку время горячей сушки в линии по производству металлического листа по настоящему изобретению является коротким, использование растворителя с низкой температурой кипения может вызвать образование дефекта покрытия, обычно называемого пузырем. Предпочтительно используется растворитель с температурой кипения 120°C или более. В качестве органических растворителей с высокой температурой кипения может использоваться известный растворитель, такой как циклогексан или Solvesso (название продукта компании Exxon Mobile Yugen Kaisha), который является органическим растворителем на основе ароматического углеводорода.
<Формирование пленки покрытия (α)>
[0110] Как описано в разделе <Пленка покрытия (α)>, в том случае, когда композиция покрытия (β) является композицией на водной основе или на основе органического растворителя, пленку покрытия (α) по настоящему изобретению предпочтительно получают таким способом получения пленки, в котором композиция покрытия (β) наносится на металлический лист с использованием известного способа нанесения покрытия, такого как нанесение покрытия с помощью валика, нанесение покрытия с помощью валика с канавками, нанесение покрытия поливом, нанесение покрытия поливом с роликом, нанесение покрытия погружением или отжим воздушным шабером, а затем вода или растворитель удаляются из влажной пленки покрытия до сухости. В случае отверждаемой ультрафиолетовым излучением или отверждаемой электронным лучом композиции на водной основе или на основе органического растворителя предпочтительно наносить композицию на металлический лист с помощью вышеупомянутого способа нанесения покрытия, затем удалять воду или растворитель до сухости и применять ультрафиолетовое излучение или облучение электронным лучом для того, чтобы выполнить полимеризацию.
[0111] Далее будет конкретно описан способ горячей сушки в том случае, когда композиция покрытия (β) является термоотверждаемой композицией на водной основе или на основе органического растворителя. В том случае, когда композиция покрытия (β) является термоотверждаемой композицией на водной основе или на основе органического растворителя, способ горячей сушки конкретно не ограничен; металлический лист может быть нагрет заранее, или металлический лист может быть нагрет после нанесения, или эти два способа могут быть скомбинированы для выполнения сушки. Способ нагревания конкретно не ограничен; нагревание горячим воздухом, индукционный нагрев, нагрев ближним инфракрасным светом, нагрев открытым огнем и т.д. могут использоваться по отдельности или в сочетании.
[0112] В том случае, когда композиция покрытия (β) является термоотверждаемой композицией на водной основе, температура горячей сушки предпочтительно составляет от 120°C до 250°C в качестве максимальной температуры нагрева поверхности металлического листа. Если максимальная температура нагрева составляет менее 120°C, отверждение пленки покрытия может быть недостаточным, и коррозионная стойкость может уменьшиться; а если максимальная температура нагрева составляет более 250°C, термоотверждение может быть чрезмерным, и коррозионная стойкость и формуемость могут уменьшиться. Время горячей сушки предпочтительно составляет от 1 до 60 секунд, и более предпочтительно от 3 до 20 секунд. Если это время составляет менее 1 секунды, термоотверждение может оказаться недостаточным, и коррозионная стойкость может уменьшиться, а если это время составляет более 60 секунд, может уменьшиться производительность.
[0113] В том случае, когда композиция покрытия (β) является термоотверждаемой композицией на основе органического растворителя, максимальная температура нагрева поверхности металлического листа предпочтительно составляет от 180°C до 260°C. Если максимальная температура нагрева составляет менее 180°C, отверждение пленки покрытия может быть недостаточным, и коррозионная стойкость может уменьшиться, а если максимальная температура нагрева составляет более 260°C, термоотверждение может оказаться чрезмерным, и коррозионная стойкость и формуемость могут уменьшиться. Время горячей сушки предпочтительно составляет от 10 до 80 секунд, а более предпочтительно от 40 до 60 секунд. Если это время составляет менее 10 секунд, термоотверждение может оказаться недостаточным и коррозионная стойкость может уменьшиться, а если это время составляет более 80 секунд, может уменьшиться производительность.
[0114] Далее будет конкретно описан способ получения пленки в том случае, когда композиция покрытия (β) является отверждаемой ультрафиолетовым излучением или отверждаемой электронным лучом композицией на водной основе или на основе органического растворителя. Любую из этих композиций наносят способом, аналогичным упомянутому выше случаю композиции на водной основе или на основе органического растворителя, затем воду или растворитель удаляют из влажной пленки покрытия до сухости, и после этого применяют ультрафиолетовое излучение или электронный луч. Пленку покрытия получают путем инициирования отверждения главным образом за счет радикалов, образующихся при облучении ультрафиолетовым излучением или электронным лучом; следовательно температура сушки может быть более низкой, чем в случае термоотверждаемой композиции. Облучение ультрафиолетовым излучением или электронным лучом предпочтительно выполняют после того, как большая часть воды или растворителя улетучится в процессе сушки, при относительно низкой максимальной температуре нагрева металлической поверхности, составляющей приблизительно от 80°C до 120°C.
[0115] Облучение ультрафиолетовым излучением для радикальной полимеризации и отверждения отверждаемой ультрафиолетом смолы в пленке покрытия обычно выполняется в воздушной атмосфере, в атмосфере инертного газа, в смешанной атмосфере из воздуха и инертного газа, и т.п. При ультрафиолетовом отверждении по настоящему изобретению облучение ультрафиолетовым излучением выполняется предпочтительно в смешанной атмосфере из воздуха и инертного газа, в которой концентрация кислорода поддерживается равной 10 об.% или менее, или в атмосфере инертного газа. Поскольку кислород действует как ингибитор радикальной полимеризации, в том случае, когда концентрация атмосферного кислорода во время облучения ультрафиолетовым излучением низка, дезактивация и ингибирование реакции сшивки из-за присоединения кислорода к образующимся радикалам подавляются, и отверждаемая ультрафиолетом композиция, используемая в настоящем изобретении, претерпевает радикальную полимеризацию и сшивку и в достаточной степени превращается в макромолекулы. Следовательно, адгезия электропроводящих пигментов (B) и поверхности металлического листа улучшается, и в результате улучшается коррозионная стойкость пленки покрытия по сравнению со случаем отверждения ультрафиолетовым излучением в воздушной атмосфере. Примеры используемого при этом инертного газа включают газообразный азот, газообразный диоксид углерода и газообразный аргон, смеси этих газов и т.п.
[0116] Ультрафиолетовое излучение может быть применено путем использовании источника ультрафиолетового излучения, такого как ртутная лампа высокого давления системы с разрядом в парах металла, металло-галогенная лампа и т.п., ксеноновая лампа и подобные ей системы с разрядом в инертном газе, или безэлектродная лампа, использующая микроволновое излучение. При покрытом металлическом листе по настоящему изобретению может использоваться любая лампа при условии, что отверждаемая ультрафиолетом пленка покрытия может быть в достаточной степени отверждена и могут быть получены желаемые свариваемость при контактной сварке, коррозионная стойкость и формуемость. В большинстве случаев пиковая освещенность и интегральное количество ультрафиолетового излучения, получаемого пленкой покрытия, влияют на отверждаемость пленки покрытия, но условия облучения ультрафиолетовым излучением конкретно не ограничены, если только отверждаемая ультрафиолетом пленка покрытия может быть в достаточной степени отверждена и могут быть получены желаемые коррозионная стойкость и формуемость.
[0117] В том случае, когда композиция покрытия (β) является отверждаемой электронным лучом композицией, обычное устройство для облучения электронным лучом, используемое в областях печати, покрытия, пленочного покрытия, упаковки, стерилизации и т.д., может использоваться для отверждения электронным лучом. Такое устройство представляет собой аппарат, который прикладывает высокое напряжение к генерируемым из горячего волокна в глубоком вакууме термоэлектронам для их ускорения, извлекает получаемый электронный ток в атмосферу инертного газа и подает этот ток к полимеризующемуся веществу. При покрытом металлическом листе по настоящему изобретению может использоваться любое такое устройство при условии, что отверждаемая электронным лучом пленка покрытия может быть в достаточной степени отверждена и могут быть получены желаемые свариваемость при контактной сварке, коррозионная стойкость и формуемость. В общем, ускоряющее напряжение электронного луча, поглощаемого пленкой покрытия, влияет на глубину, на которую электронный луч проникает в пленку покрытия, а поглощенная доза влияет на скорость полимеризации (отверждаемость пленки покрытия); но условия облучения электронным лучом конкретно не ограничены при условии, что отверждаемая электронным лучом пленка покрытия может быть в достаточной степени отверждена и могут быть получены желаемые коррозионная стойкость и формуемость. Однако в случае радикальной полимеризации с помощью электронного луча присутствие даже небольшого количества кислорода вызывает ингибирование реакции дезактивации и сшивки из-за присоединения кислорода к образующимся радикалам, что делает отверждение недостаточным; следовательно, облучение электронным лучом предпочтительно выполняют в атмосфере инертного газа, в которой концентрация кислорода составляет 500 миллионных долей (млн-1) или менее. Примеры используемого при этом инертного газа включают газообразный азот, газообразный диоксид углерода и газообразный аргон, смесь этих газов и т.п.
[ПРИМЕРЫ]
Пример I
[0118] Настоящее изобретение будет теперь описано конкретнее с помощью Примера I, использующего композицию покрытия на водной основе.
1. Подготовка металлического листа
[0119] Следующие пять типов оцинкованных стальных листов подготовили и погружали в водный раствор при 40°C, содержащий 2,5 мас.% щелочного обезжиривающего средства на водной основе (FC-301 производства компании Nihon Parkerizing Co., Ltd.) на 2 минуты, чтобы обезжирить поверхность, а затем выполняли промывку водой и сушку; таким образом сформировали металлические листы для покрытия.
[0120] EG: покрытый гальванически цинком стальной лист (толщина листа: 0,8 мм; плотность покрытия: 30 г/м2)
ZL: покрытый гальванически сплавом Zn-10% Ni стальной лист (толщина листа: 0,8 мм; плотность покрытия: 30 г/м2)
GI: покрытый цинком методом погружения в расплав стальной лист (толщина листа: 0,8 мм; плотность покрытия: 40 г/м2)
SD: покрытый сплавом Zn-11% Al-3% Mg-0,2% Si методом погружения в расплав стальной лист (толщина листа: 0,8 мм; плотность покрытия: 40 г/м2)
GA: покрытый цинковым сплавом методом погружения в расплав стальной лист (толщина листа: 0,8 мм; 10 мас.% Fe; плотность покрытия: 45 г/м2)
2. Получение пленки грунтового покрытия
[0121] Как описано в разделе <Пленка покрытия (α)>, в настоящем изобретении пленка грунтового покрытия не обязательно должна предусматриваться между пленкой покрытия (α) и поверхностью металлического листа, но может использоваться для того, чтобы дополнительно улучшить адгезию к металлическому листу, коррозионную стойкость и т.д. пленки покрытия (α). Здесь часть металлических листов для покрытия снабдили пленкой грунтового покрытия и выполнили их оценку.
[0122] Композицию покрытия на водной основе, состоящую из полиэфирной смолы, мелкодисперсных частиц кремнезема и кремнийорганического аппрета, приготовили в качестве композиции покрытия для получения пленки грунтового покрытия.
[0123] Упомянутую выше композицию нанесли на упомянутый выше металлический лист для покрытия путем нанесения покрытия с помощью стержневого устройства так, чтобы толщина покрывающей пленки составляла 0,08 мкм, и деталь высушили в сушильном шкафу при максимальной температуре нагрева металлической поверхности 70°C, т.е. высушили на воздухе.
3. Приготовление композиции покрытия на водной основе и получение пленки
[0124] Для того, чтобы приготовить композицию покрытия на водной основе, сначала приготовили смолу (A), электропроводящие пигменты (B) и противокоррозионные пигменты (C).
(1) Смола (A1)
[0125] Приготовили коммерчески доступные смолы A1-A8, показанные в Таблице 1, в качестве смол для Примеров (Примеров по изобретению) и Сравнительных примеров.
[0126] [Таблица 1]
(2) Электропроводящие пигменты (B)
[0127] Использовали коммерчески доступные мелкодисперсные частицы (реагенты), показанные в Таблице 2 (Примеры). Среднеобъемный диаметр частиц измеряли с использованием прибора Multisizer 3 производства компании Beckman Coulter, Inc. (прибор для точного измерения распределения размеров частиц в соответствии с принципом Коултера). Удельное электрическое сопротивление измеряли путем превращения каждого типа мелкодисперсных частиц в спеченный лист с длиной 80 мм, шириной 50 мм и толщиной 2-4 мм и использования четырехполюсного четырехзондового способа и системы подачи тока постоянной величины при 25°C в соответствии с JIS K7194, с использованием прибора для измерения удельного сопротивления Loresta EP (типа MCP-T360) и зондов ESP (диаметр части плоской головки: 2 мм) производства компании Mitsubishi Chemical Analytech Co., Ltd.
[0128] [Таблица 2]
[0129] TiB: мелкодисперсные частицы TiB2 (TII11PB производства компании Kojundo Chemical Lab. Co., Ltd.; среднеобъемный диаметр: 2,9 мкм; удельное электрическое сопротивление: 30×10-6 Ом⋅см)
ZrB: мелкодисперсные частицы ZrB2 (производства компании Wako Pure Chemical Industries, Ltd.; среднеобъемный диаметр: 2,2 мкм; удельное электрическое сопротивление: 70×10-6 Ом⋅см)
MoB: мелкодисперсные частицы Mo2B (борид димолибдена производства компании Mitsuwa Chemicals Co., Ltd.; среднеобъемный диаметр: 5,2 мкм; удельное электрическое сопротивление: 30×10-6 Ом⋅см)
LaB: мелкодисперсные частицы LaB6 (гексаборид лантана производства компании Soekawa Rikagaku K. K.; среднеобъемный диаметр: 2,8 мкм; удельное электрическое сопротивление: 20×10-6 Ом⋅см)
VC: мелкодисперсные частицы VC (производства компании Wako Pure Chemical Industries, Ltd.; среднеобъемный диаметр: 2,3 мкм; удельное электрическое сопротивление: 140×10-6 Ом⋅см)
TiC: мелкодисперсные частицы TiC (производства компании Wako Pure Chemical Industries, Ltd.; среднеобъемный диаметр: 3,2 мкм; удельное электрическое сопротивление: 180×10-6 Ом⋅см)
TiN: мелкодисперсные частицы TiN (производства компании Wako Pure Chemical Industries, Ltd.; среднеобъемный диаметр: 1,6 мкм; удельное электрическое сопротивление: 20×10-6 Ом⋅см)
NiSi: мелкодисперсные частицы Ni2Si (NII11PB производства компании Kojundo Chemical Lab. Co., Ltd. добавили в воду и суспендировали путем перемешивания, тонкодисперсные частицы, все еще всплывающие после истечения 5 минут, отфильтровали и использовали получившееся вещество; среднеобъемный диаметр: 4,8 мкм; удельное электрическое сопротивление: 40×10-6 Ом⋅см)
SUS: частицы SUS304 (среднеобъемный диаметр: 3,3 мкм; удельное электрическое сопротивление: 70×10-6 Ом⋅см)
ZrB+VC: смесь вышеупомянутого ZrB и вышеупомянутого VC (объемное отношение: 1:1)
ZrB+TiC: смесь вышеупомянутого ZrB и вышеупомянутого TiC (объемное отношение: 1:1)
VC+TiN: смесь вышеупомянутого VC и вышеупомянутого TiN (объемное отношение: 1:1)
(3) Противокоррозионные пигменты (C)
[0130] Использовали коммерчески доступные реагенты или промышленные продукты, или их смеси, как показано в Таблице 3 и Таблице 4 (Примеры).
[0131] [Таблица 3]
[0132] [Таблица 4]
(4) Частицы (D)
[0133] Использовали коммерчески доступные промышленные продукты, показанные в Таблице 5 (Примеры).
[0134] [Таблица 5]
[0135] Затем использовали вышеупомянутые смолу (A), электропроводящие пигменты (B), противокоррозионные пигменты (C) и частицы (D), а также дистиллированную воду для того, чтобы приготовить композиции покрытия на водной основе с различными соотношениями компонентов смеси.
[0136] Электропроводящие пигменты (B), противокоррозионные пигменты (C) и частицы (D) смешивали в желаемом объемном отношении к общему количеству смолы (A), электропроводящих пигментов (B), противокоррозионных пигментов (C) и частиц (D), содержащихся в сухом остатке композиции покрытия на водной основе. Концентрацию сухого остатка композиции покрытия на водной основе регулировали при изменении количества добавляемой воды по мере необходимости для того, чтобы получить целевое количество нанесенной пленки покрытия и хорошую укрывистость. Здесь «сухой остаток» относится к компонентам, которые остаются после испарения воды или органического растворителя из материала или композиции покрытия.
[0137] Таблица 6 показывает состав и количества (в объемной доле, %) компонентов, содержащихся в пленке покрытия (α), полученной с использованием вышеупомянутых составляющих компонентов пленки покрытия.
[0138] [Таблица 6-1]
[Таблица 6-2]
[0139] Приготовили упомянутую выше композицию покрытия на водной основе, каждый компонент равномерно диспергировали, затем полученное вещество нанесли с использованием устройства для нанесения покрытия роликом на металлический лист для покрытия или на металлический лист, снабженный вышеупомянутой пленкой грунтового покрытия, тестовый образец высушили в сушильном шкафу при максимальной температуре нагрева металлической поверхности 200°C, после чего выполняли охлаждение водой и сушку на воздухе. Таблица 7 показывает толщину пленки покрытия (единица: мкм) после получения пленки. Толщину пленки покрытия вычисляли путем деления разности масс между состояниями до и после отслаивания пленки покрытия на плотность пленки покрытия. Плотность пленки покрытия вычисляли из количеств составляющих компонентов пленки покрытия и известных плотностей каждого компонента.
[0140] Таблица 6 (Таблица 6-1 и Таблица 6-2) показывает распределение микротвердости HM при -20°C и распределение микротвердости HM при 40°C поверхности пленки покрытия (α) на поверхности покрытого металлического листа. Каждое из состояний распределения описывается ниже.
(1) Распределение HM при -20°C
Состояние 1: имеется от 0 до 19 точек измерения, в которых микротвердость по Мартенсу HM при -20°C составляет от 10 до 200 мг/мм2, из 100 случайных точек измерения
Состояние 2: имеется от 20 до 39 точек измерения, в которых HM составляет от 10 до 200 мг/мм2
Состояние 3: имеется от 40 до 59 точек измерения, в которых HM составляет от 10 до 200 мг/мм2
Состояние 4: имеется 60 или более точек измерения, в которых HM составляет от 10 до 200 мг/мм2
(2) Распределение HM при 40°C
Состояние 1: имеется от 0 до 4 точек измерения, в которых микротвердость по Мартенсу HM при 40°C составляет от 200 до 200000 мг/мм2, из 100 случайных точек измерения
Состояние 2: имеется от 5 до 9 точек измерения, в которых HM составляет от 200 до 200000 мг/мм2
Состояние 3: имеется от 10 до 19 точек измерения, в которых HM составляет от 200 до 200000 мг/мм2
Состояние 4: имеется 20 или более точек измерения, в которых HM составляет от 200 до 200000 мг/мм2
4. Оценка характеристик
[0141] Используя покрытые металлические листы, полученные вышеупомянутым способом 3, была выполнена оценка свариваемости, коррозионной стойкости, стойкости к скалыванию, коррозионной стойкости после скалывания и устойчивости к слипанию. Каждое испытание и способ оценки описаны ниже.
(1) Свариваемость
[0142] Используя электроды Cr-Cu типа CF, имеющие наконечник с диаметром 5 мм и радиусом кривизны 40 мм, выполняли испытание на последовательную точечную сварку при прикладываемом давлении 1,96 кН, токе сварки 8 кA и времени подачи питания 12 циклов/50 Гц, и нашли число сварных точек непосредственно перед тем, как диаметр ядра сварной точки станет меньше чем 3√t (где t – толщина листа). Характеристику свариваемости при точечной сварке оценивали с использованием следующих оценочных баллов.
[0143] 4: число сварных точек равно 1000 или более
3: не менее 200 и менее 1000
2: менее 200
1: ядро сварной точки не образовалось и не удалось сварить ни одну точку
(2) Коррозионная стойкость
[0144] Вырезали прямоугольный тестовый образец с размером 150×70 мм из покрытого металлического листа, полученного вышеупомянутым способом 3, и его концевую часть запечатали смолой; таким образом сформировали тестовый образец для испытания на коррозионную стойкость участка плоской поверхности.
[0145] Эти тестовые образцы подвергли циклическому коррозионному испытанию, в котором каждый цикл длился в общей сложности 8 часов и состоял из 2 часов распыления соленой воды, 4 часов сушки и 2 часов увлажнения. Условия распыления соленой воды соответствовали JIS-Z2371. Условия сушки были установлены такими, что температура составляла 60°C, а относительная влажность (RH) составляла 30% или менее, и условия увлажнения были установлены такими, что температура составляла 50°C, а относительная влажность (RH) составляла 95% или более. Исследовали условия образования красной ржавчины и оценивали совершенство или несовершенство коррозионной стойкости обработанного участка с использованием следующих оценочных баллов.
[0146] 4: красная ржавчина не образовывалась за 450 циклов
3: красная ржавчина не образовывалась за 300 циклов
2: красная ржавчина не образовывалась за 150 циклов
1: красная ржавчина образовывалась за 150 циклов
(3) Сопротивление скалыванию
[0147] Вырезали прямоугольный тестовый образец с размером 150×70 мм из покрытого металлического листа, полученного вышеупомянутым способом 3, и использовали его в качестве образца для испытания на сопротивление скалыванию. Этот тестовый образец подвергали электролитическому осаждению покрытия (толщина пленки: 15 мкм), промежуточному покрытию (толщина пленки: 30 мкм) и поверхностному покрытию (толщина пленки: 30 мкм). 100 кусочков щебня (базальт; диаметр частиц: 5-7,5 мм), ускоренные давлением воздуха до скорости от 30 до 60 км/ч в помещении с температурой -15°C, выбрасывались и сталкивались с вышеупомянутым покрытым стальным листом, с плоской поверхностью тестового образца, наклоненной под углом 15 градусов к направлению прилета щебня. Наблюдали область размером 20×20 мм вокруг центра столкновения щебня с образцом и оценивали совершенство или несовершенство коррозионной стойкости участка скола с использованием следующих оценочных баллов.
[0148] 4: нижележащее железо покрытого стального листа не обнажено
3: обнажено одно место на нижележащем железе покрытого стального листа с размером 100 мкм или более, определяемым как среднее из длинного диаметра и короткого диаметра
2: обнажено от двух до четырех мест на нижележащем железе покрытого стального листа с размером 100 мкм или более, определяемым как среднее из длинного диаметра и короткого диаметра
1: обнажено пять мест или более на нижележащем железе покрытого стального листа с размером 100 мкм или более, определяемым как среднее из длинного диаметра и короткого диаметра
(4) Коррозионная стойкость после скалывания
[0149] Покрытый металлический лист, испорченный упомянутым выше способом (4), использовали в качестве образца для испытания на коррозионную стойкость после скалывания.
[0150] Эти тестовые образцы подвергали циклическому коррозионному испытанию, в котором каждый цикл длился в общей сложности 8 часов и состоял из 2 часов распыления соленой воды, 4 часов сушки и 2 часов увлажнения. Условия распыления соленой воды соответствовали JIS-Z2371. Условия сушки были установлены такими, что температура составляла 60°C, а относительная влажность (RH) составляла 30% или менее, а условия увлажнения были установлены такими, что температура составляла 50°C, а относительная влажность (RH) составляла 95% или более. Исследовали условия образования красной ржавчины и оценивали совершенство или несовершенство коррозионной стойкости участка скола с использованием следующих оценочных баллов.
[0151] 4: красная ржавчина, в которой визуально идентифицируется ржавая жидкость, не образовывалась за 60 циклов
3: красная ржавчина, в которой визуально идентифицируется ржавая жидкость, образовывалась за 30-59 циклов
2: красная ржавчина, в которой визуально идентифицируется ржавая жидкость, образовывалась за 13-29 циклов
1: красная ржавчина, в которой визуально идентифицируется ржавая жидкость, образовывалась за 12 циклов
(5) Устойчивость к слипанию
[0152] Переднюю поверхность и противоположную, заднюю поверхность полученного покрытого металлического листа приводили в поверхностный контакт друг с другом под давлением и оценивали устойчивость к слипанию.
[0153] В качестве способа испытаний вырезанные тестовые образцы размером 50 мм × 50 мм укладывали друг на друга таким образом, чтобы передняя поверхность и задняя поверхность находились в поверхностном контакте друг с другом, затем выполняли горячее прессование при 40°C и давлении 100 кг/см2 в течение 24 часов, после чего оценивали степень слипания (адгезии) пленок покрытия с помощью следующих критериев.
[0154] 3: имелось незначительное слипание пленок покрытия
2: пленки покрытия были немного склеены, но достаточно легко разделялись вручную
1: пленки покрытия были склеены слишком прочно для того, чтобы разделить их вручную
[0155] Таблица 7 (Таблица 7-1 и Таблица 7-2) показывает результаты оценки.
[0156] [Таблица 7-1]
[Таблица 7-2]
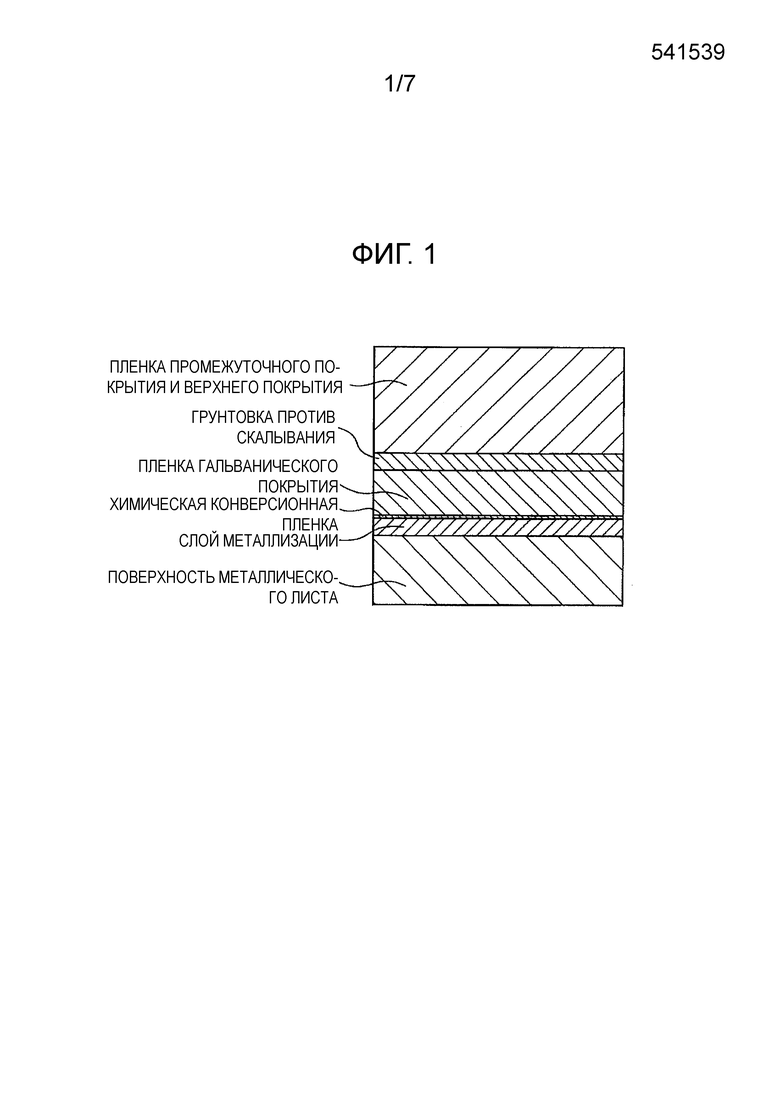
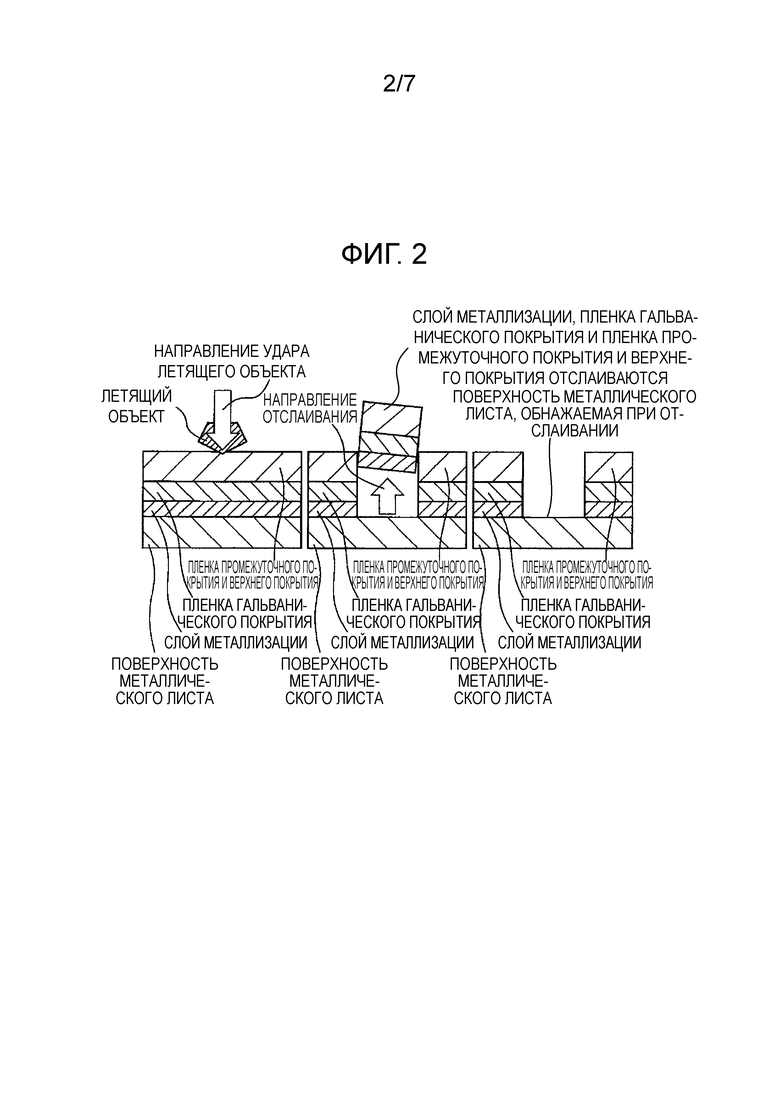
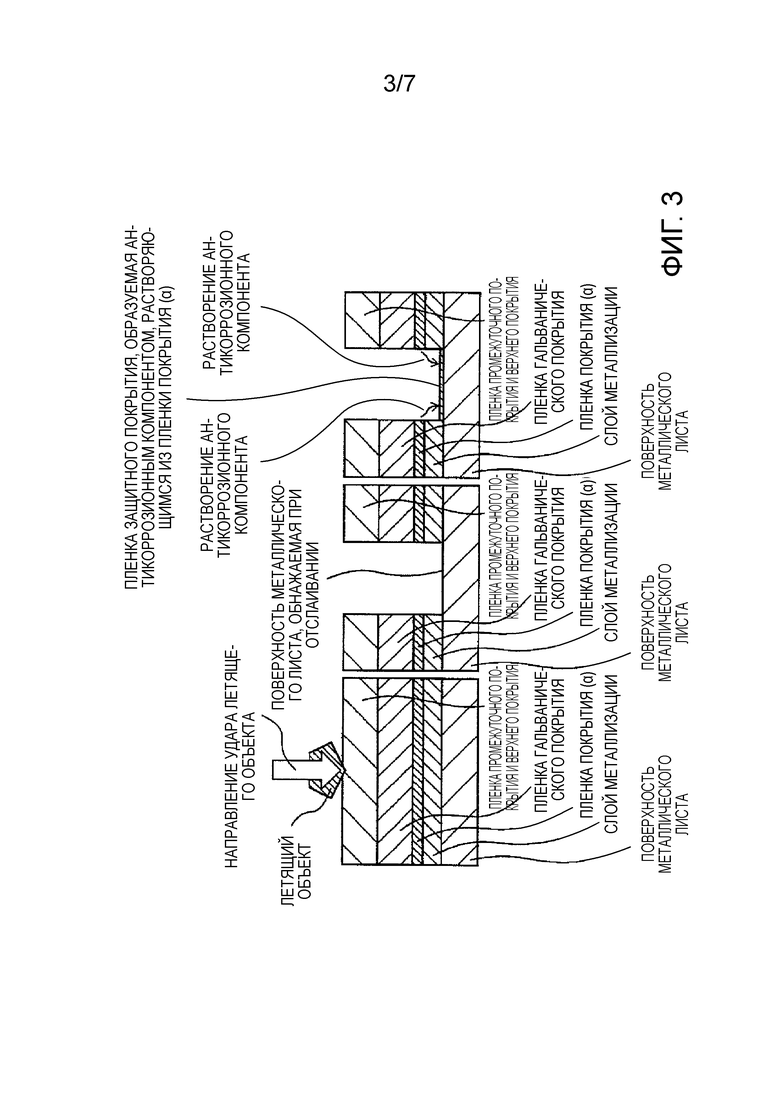
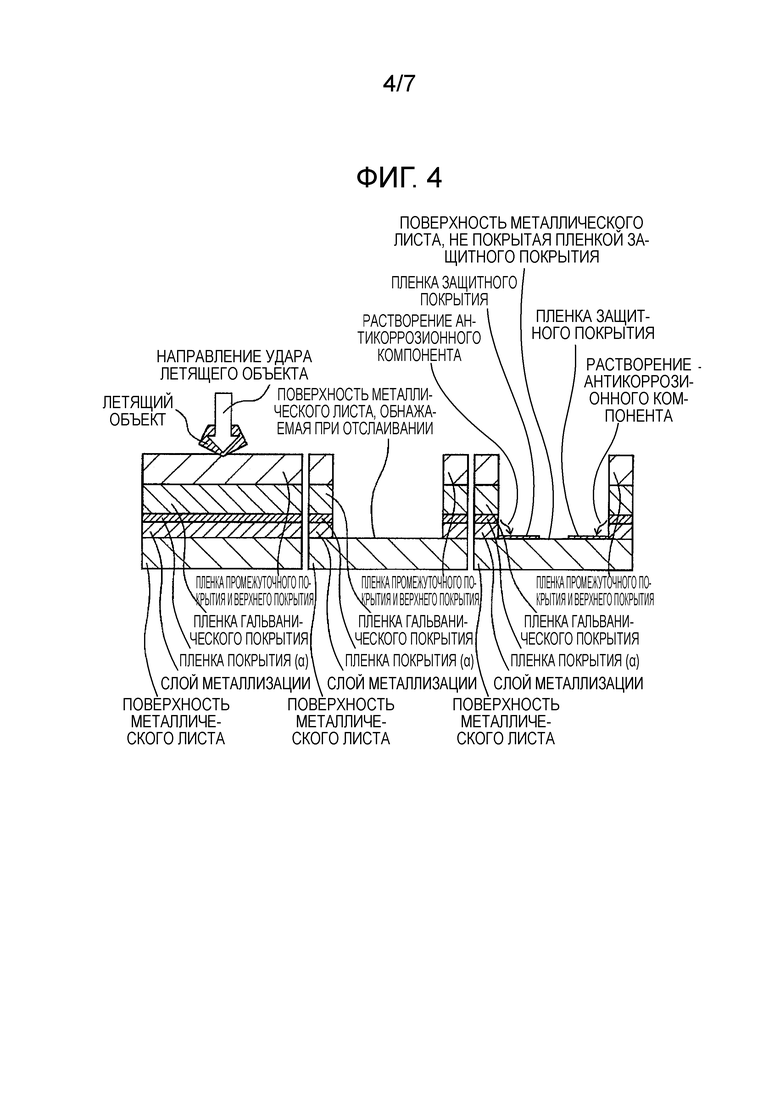
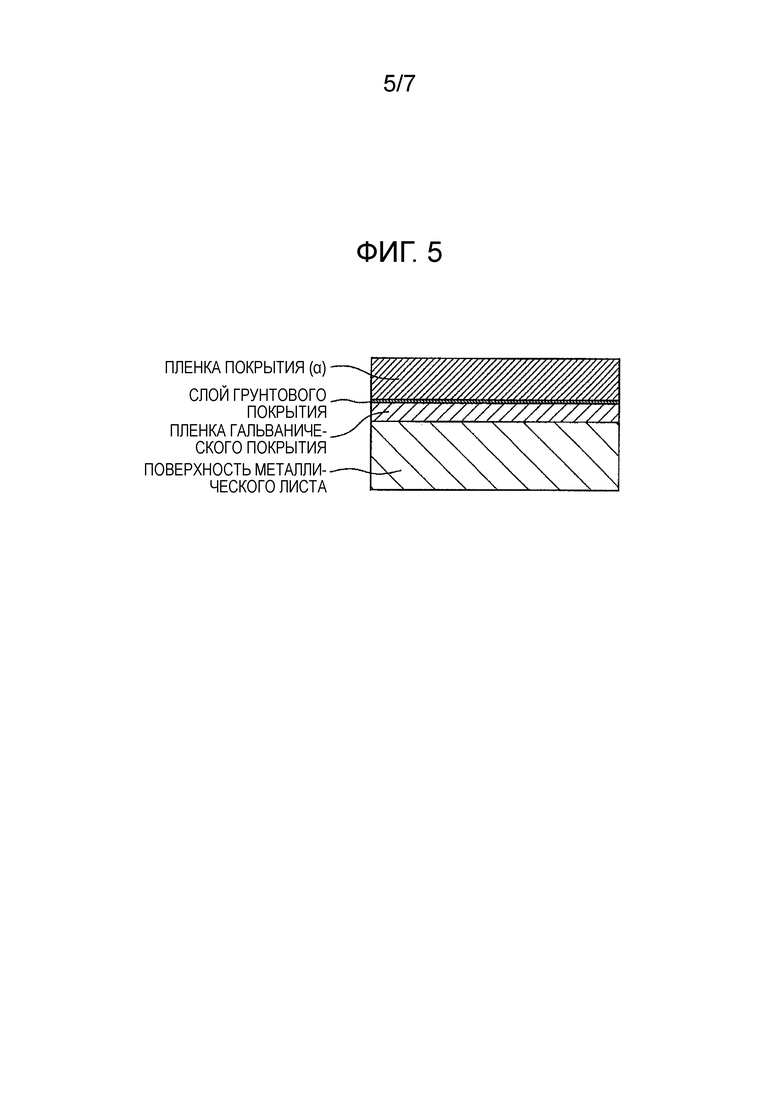
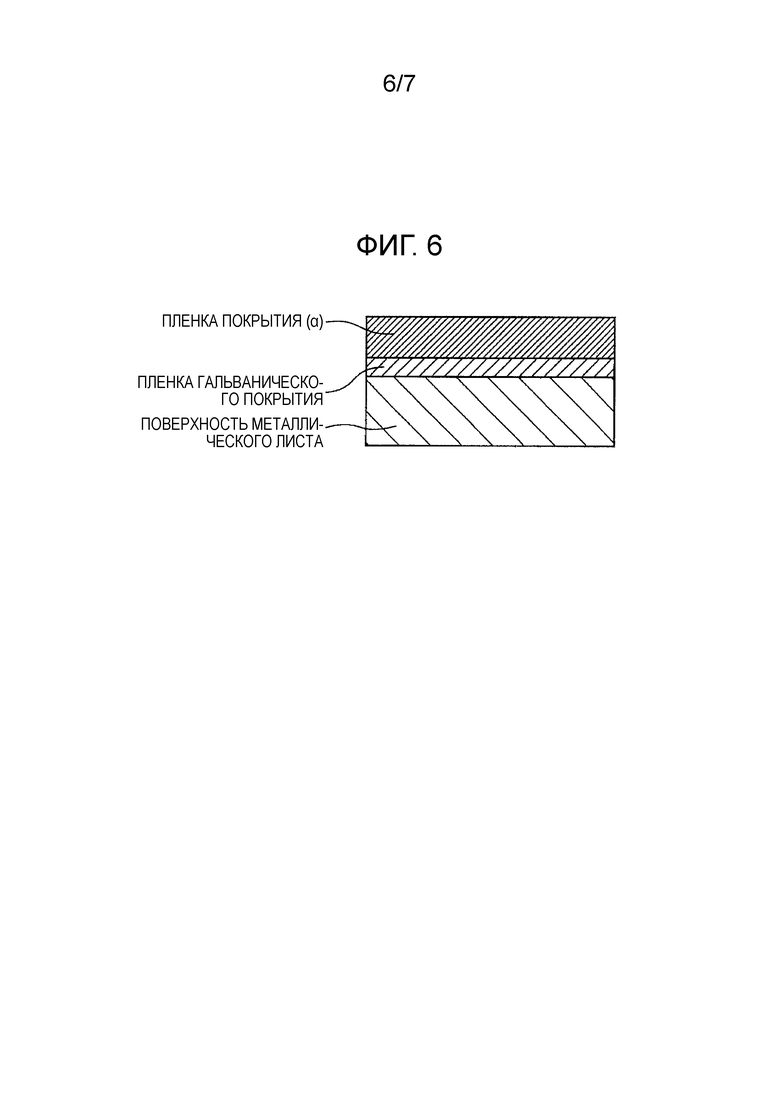
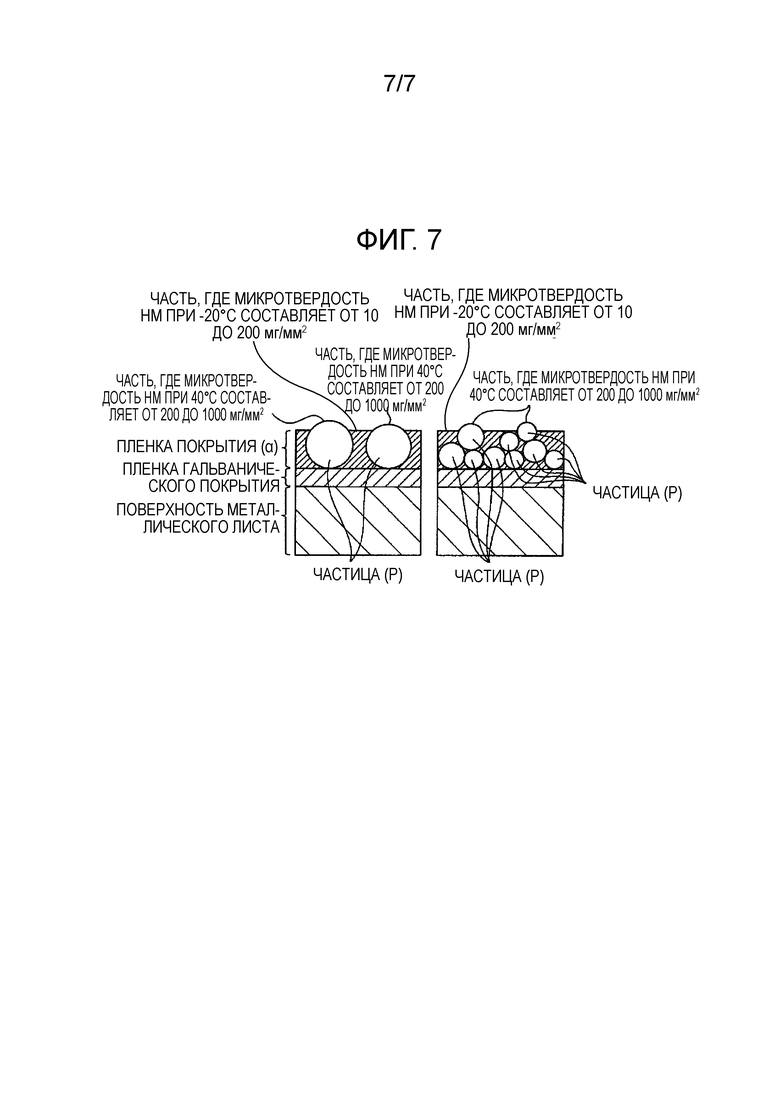
Изобретение относится к области химии для использования в автомобилестроении и касается покрытого металлического листа для автомобиля. Лист содержит: металлический лист и пленку покрытия , присутствующую на по меньшей мере одной поверхности этого металлического листа. Пленка покрытия содержит органическую смолу, электропроводящие пигменты и противокоррозионные пигменты, и микротвердость по Мартенсу HM при -20°C поверхности пленки покрытия составляет от 10 до 200 (мг/мм2) в 20 точках или более при измерении в 100 точках, а микротвердость по Мартенсу HM при 40°C поверхности пленки покрытия составляет от 200 до 200000 (мг/мм2) в 5 точках или более при измерении в 100 точках. Изобретение обеспечивает создание системы покрытия, в которой для элемента кузова автомобиля используется покрытый металлический лист, а сопротивлением скалыванию обладает сама пленка покрытия, что исключает использование грунтовки против скалывания. 3 н. и 7 з.п. ф-лы, 9 табл., 7 ил.
1. Покрытый металлический лист для автомобиля, содержащий:
металлический лист; и
пленку покрытия (α), присутствующую на по меньшей мере одной поверхности металлического листа,
причем пленка покрытия (α) содержит
органическую смолу (A),
электропроводящие пигменты (B) и
противокоррозионные пигменты (C), и
микротвердость по Мартенсу HM при -20°C поверхности пленки покрытия (α) составляет от 10 до 200 (мг/мм2) в 20 точках или более при измерении в 100 точках, а микротвердость по Мартенсу HM при 40°C поверхности пленки покрытия (α) составляет от 200 до 200000 (мг/мм2) в 5 точках или более при измерении в 100 точках.
2. Покрытый металлический лист для автомобиля по п. 1, в котором температура стеклования Tс органической смолы (A) составляет от -80°C до -20°C.
3. Покрытый металлический лист для автомобиля по п. 1, в котором органическая смола (A) выбрана из группы, состоящей из полиэфирной смолы, полиуретановой смолы, акриловой смолы и их модифицированного продукта.
4. Покрытый металлический лист для автомобиля по п. 1, в котором электропроводящие пигменты (B) представляют собой частицы неоксидной керамики с удельным электрическим сопротивлением при 25°C от 0,1×10-6 до 185×10-6 Ом⋅см, являющиеся по меньшей мере одним, выбираемым из борида, карбида, нитрида и силицида.
5. Покрытый металлический лист для автомобиля по п. 1, в котором пленка покрытия (α) содержит от 0,5 об.% до 65 об.% электропроводящих пигментов (B).
6. Покрытый металлический лист для автомобиля по п. 1, в котором противокоррозионные пигменты (C) содержат
одно или более соединений, выбираемых из соединений, способных выделять ион силиката, ион фосфата, ион ванадата, ион вольфрамата или ион молибдата,
частицы, содержащие элемент-металл, выбираемый из группы, состоящей из Si, Ti, Al и Zr, или
и то, и другое.
7. Покрытый металлический лист для автомобиля по п. 1, в котором пленка покрытия (α) содержит от 1 об.% до 40 об.% противокоррозионных пигментов (C).
8. Покрытый металлический лист для автомобиля по п. 1, содержащий в пленке покрытия зернистые частицы (D) с твердостью по Мартенсу при 40°C от 200 мг/мм2 до 200000 мг/мм2.
9. Автомобильный компонент, выполненный путем обработки и формования покрытого металлического листа для автомобиля по п. 1.
10. Автомобильный компонент, выполненный путем дополнительного нанесения одного или более из слоя электролитически осаждаемого покрытия, слоя промежуточного покрытия и слоя поверхностного покрытия на автомобильный компонент по п. 9.
| JP 2010065254 A, 25.03.2010 | |||
| JP 2003245605 A, 02.09.2003 | |||
| JP 2964943 B2, 18.10.1999 | |||
| WO 2007072830 A1, 28.06.2007. |

