Изобретение относится к медицинским токсикологическим исследованиям, в частности, к санитарной токсикологии и может быть использовано для количественного определения девяти N-нитрозоаминов: N-диметилнитрозоамин, N-метилэтилнитрозоамин, N-диэтилнитрозоамин, N-дибутилнитрозоамин, N-дипропилнитрозоамин, N-пиперидиннитрозоамин, N-пирролидиннитрозоамин, N-морфолиннитрозоамин, N-дифенилнитрозоамин, в продуктах питания, а именно, в копченых мясопродуктах, методом хромато-масс-спектрометрии.
Копченые мясные изделия, в отличие от не копченых мясопродуктов, имеют плотную эластичную консистенцию, острый солоноватый вкус и приятный аромат. Они обладают высокой питательностью, так как содержат много жира (25-60%), белковых веществ (21-22%). Но именно копченые изделия содержат повышенное количество канцерогенных нитрозоаминов широкого спектра.
Нитрозамины обладают широким спектром токсического действия на организм человека. В первую очередь, их токсичность направлена на печень и почки, в результате чего происходит нарушение их функции и некрозы. При хранении пищевых продуктов происходит существенное повышение концентрации некоторых аминов, что может служить признаком их порчи.
В связи с вышеизложенным, для решения проблемы канцерогенной безопасности пищи, необходимо усовершенствовать пищевые технологии, предупреждающие образование канцерогенных нитрозаминов в продуктах питания, а также обеспечить их надежный контроль. На этот контроль и направлен предлагаемый способ.
В настоящем описании использованы следующие сокращения для обозначения каждого из девяти определяемых предлагаемым способом N-нитрозоаминов:
N-диметилнитрозоамин - N-ДМНА
N-метилэтилнитрозоамин - N-МЭНА
N-диэтилнитрозоамин - N-ДЭНА
N-дибутилнитрозоамин - N-ДБНА
N-дипропилнитрозоамин - N-ДПНА
N-пиперидиннитрозоамин - N-ПИПНА
N-пирролидиннитрозоамин - N-ПИРНА
N-морфолиннитрозоамин - N-MOPHA
N-дифенилнитрозоамин - N-ДФНА.
Из уровня техники известен способ хроматографического определения летучих аминов и нитрозаминов в пищевых продуктах (Авт. свид-во СССР №1259187), включающий их экстракцию, отгонку растворителя, растворение остатка в соляной кислоте, деление на нескольких аликвотных частей, при этом обрабатывают их, кроме одной, щелочью до рН 8-14, упаривают досуха и растворяют с последующим использованием раствора необработанной части в качестве анализируемого, а растворов щелочных частей - в качестве стандартных после добавления к ним стандартных соединений.
Однако, при упаривании пробы возможны потери нитрозаминов, т.к. они являются высоколетучими соединениями, что существенно будет влиять на надежность и точность получаемого результата.
В статье (http://www.medved.kiev.ua/arh_nutr/art_2004/n04_2_l2.htm) «К вопросу об определении n-нитрозаминов в пищевых продуктах», авторы: О.С. Зульфигаров, В.В. Юрченко и др., Институт экогигиены и токсикологии им. Л.И. Медведя, Киев, Украина, также указано на то, что первым этапом определения нитрозоаминов является их выделение из анализируемых продуктов. Невысокие температуры кипения (меньше 200°С) и достаточная термическая стабильность нитрозоаминов обусловили возможность применения для их выделения отгонки с водяным паром. Простота в исполнении, универсальность по отношению к самым разным пищевым продуктам, в том числе, и мясным, полнота извлечения обусловили наиболее широкое применение этого подхода. Затем из водных отгонов нитрозамины выделяют методом экстракции, наилучшим экстрагентом для этого оказался хлористый метилен, который и находит наибольшее применение. Данная схема выделения эффективна как для жидких проб, так и для твердых. Далее рекомендуется проводить дериватизацию соответствующих вторичных аминов 4-нитрофенилдиазонием. Полученные при этом производные достаточно гидрофобны и легко могут быть сконцентрированы, а затем разделены методом обращено-фазной высокоэффективной жидкостной хроматографии. Сущность данного известного метода заключается в том, что неподвижная фаза - это неполярная (гидрофобные силикагели с привитыми группами С4, С8, С18 и др.) фаза; подвижная фаза - полярная (смеси воды и полярных растворителей: ацетонитрила, метанола, тетрагидрофурана и др.) фаза. Удерживание веществ растет с увеличением их гидрофобности (неполярности). Чем больше содержание
органического растворителя, тем выше элюирующая способность подвижной фазы.) Поскольку нитрофенилтриазены хорошо окрашены, чувствительность их детектирования на обычных, наиболее распространенных фотометрических детекторах, достаточно высока и составляет 2⋅10-7 моль/л.
Это позволяет определять нитрозамины в пищевых продуктах при их содержании на порядок ниже предельно-допустимой концентрации (ПДК).
Важным обстоятельством является возможность применения данной известной методики для определения соответствующих вторичных аминов. Однако и этот способ не лишен недостатков, т.к.
- много операций подготовки пробы к химическому анализу - перегонка с водяным паром, затем экстракция органическим растворителем и получение производных нитрозоаминов (дериватизация). Возможны большие потери целевых аналитов на указанных этапах пробоподготовки, если нитрозоамины в пробе содержатся в ультрамикроконцентрациях. Как результат, достаточно сложно получить достоверные данные по количественному содержанию нитрозоаминов в пробе.
- длительность проведения в серийных исследованиях, соэкстрагирование большого количества мешающих веществ, невысокая эффективность извлечения.
В настоящее время в РФ контроль содержания N-нитрозоаминов в продуктах питания осуществляется в соответствии с МУК 4.4.1.011-93 («Определение летучих N-нитрозаминов в продовольственном сырье и пищевых продуктах. Методические указания по методам контроля. - М.: Информационно-издательский центр Госкомсанэпиднадзора России, 1993. - 16 с.), в которых приведена методика, основанная на флуориметрическом и хемилюминисцентном определении летучих N-нитрозоаминов в продуктах питания.
Флуориметрический метод (полуколичественный) состоит в выделении летучих N-нитрозоаминов (НА) путем перегонки с паром или в вакууме; экстракции хлористым метиленом НА из водного дистиллята; концентрации экстракта; денитрозировании НА бромистым водородом в уксусной кислоте; алкилировании образовавшихся аминов 8-метокси-5-хинолинсульфонилазиридином (КАЗ), разделении и количественном определении образовавшихся флуоресцирующих 8-метокси-5-[N-(2-N-диэтиламино)] хинолинсульфонамидных производных (КАЭ-производные) в тонком слое силикагеля. Нижний предел определения летучих N-нитрозоаминов 1 мкг/кг продукта.
Данная методика полуколичественного определения отличается продолжительностью синтеза производных N-нитрозоаминов и многостадийностью их хроматографического разделения, нижний предел определения составляет 1 мкг/кг продукта.
Хемилюминесцентный метод идентификации и количественного определения состоит в выделении летучих N-нитрозоаминов путем перегонки с паром или в вакууме, экстракции хлористым метиленом N-нитрозоаминов из водного дистиллята, концентрировании экстракта, разделении смеси методом газо-жидкостной хроматографии и количественном определении модифицированных летучих N-нитрозоаминов с помощью высокоселективного и высокочувствительного хемилюминисцентного (термоэнергетического) детектора ТЕА-502, производимого только компанией Agilent (USA).
Идентификацию N-нитрозоаминов осуществляют по временам удерживания в сравнении с параметрами удерживания стандартных N-нитрозоаминов. Количественное определение проводят методом абсолютной калибровки с систематическим контролем калибровочного коэффициента. Нижний предел определения - 0,1 мкг/кг.
Однако, указанная Методика МУК 4.4.1.011-93. позволяет выполнять либо полуколичественный анализ (флуориметрический метод), либо количественное определение модифицированных нитрозаминов с помощью хемилюминисцентного (термоэнергетического) детектора. При выполнении процедуры дериватизации возможны потери целевых аналитов, если матрица обладает сорбционными свойствами, что существенно будет влиять на надежность и точность определения.
Кроме того, эта известная Методика отличается многостадийностью их хроматографического разделения и при этом обеспечивается лишь полуколичественное определение нитрозаминов, нижний предел определения составляет 1 мкг/кг продукта.
Анализ зарубежной научной литературы по методикам определения N-нитрозоаминов в пищевых продуктах, в т.ч. копченых мясопродуктов, показал, что в настоящее время в химическом анализе зарубежных аналитических лабораторий широко используются хромато-масс-спектрометрические методы, которые являются наиболее эффективными в анализе сложных смесей органических соединений и позволяют все компоненты смеси не только разделить, но и подтвердить структуру определяемых компонентов методом идентификации с использованием данных библиотеки.
Вместе с тем, следует отметить, что в рассмотренных зарубежных высокочувствительных и селективных методиках хромато-масс-спектрометрии используется дорогостоящее приборное оснащение и длительная пробоподготовка, что требует использования в настоящих исследованиях дорогостоящего импортного оборудования и высокой квалификации специалистов, осуществляющих контроль качества пищевых продуктов.
Наиболее близким к предлагаемому способу является метод жидкостной хроматографии с использованием тандемной масс-спектрометрии с использованием химической ионизации при атмосферном давлении и электрораспылительной ионизации (Determination of Eight Volatile Nitrosamines in Meat Products by Ultrasonic Solvent Extraction and Gas Chromatography-Mass Spectrometry Method; Yuan Yuan, Wei Meng, Miao Yutian, Chen Fang, and Hu Xiaosong; International Journal of Food Properties, 18: 1181-1190, 2015). Это селективный метод для одновременного определения содержания (диапазона мкг/кг) как летучих, так и нелетучих N-нитрозоаминов в обработанных (копченых) мясных продуктах. Согласно этому методу определяют девять N-нитрозоаминов: N-ДМНА, N-МЭНА, N-ДЭНА, N-ДБНА, N-ДПНА, N-ПИПНА, N-ПИРНА, N-МОРНА, N-ДФНА. Маточные растворы в метаноле 1 мг/мл готовили для каждого из указанных N-нитрозоаминов. Смесь всех стандартов (стандартный раствор) затем была приготовлена путем объемного разбавления исходных растворов ацетонитрилом до 10 мкг/мл. Маточные растворы хранили при -60°С и стандартный раствор хранили при -18°С.
К 2,5 г гомогенизированного обработанного мясного продукта (Салями с жиром и без жира) добавляли N-нитрозодиметиламин-d6 (NDMA-d6) и N-нитрозопирролидин-d8 (NPYR-d 8) (оба образца 9 г/кг) и пипеколиновую кислоту (1 мг на г образца) в ацетонитриле. После дополнительного добавления 7,5 мл ацетонитрила с 1% раствором муравьиной кислотой в пробирки с образцом, последние энергично встряхивали в течение 10 мин с помощью мешалки. Далее центрифугировали в течение 10 мин при 4500 об/мин. 5 мл ацетонитрильной фазы переносили в стеклянную трубку Гамильтона и выпаривали в токе азота (при 30°С ± 0,5) до объема ~ 0,25 мл, объем доводили до 1,0 мл путем добавления воды Milli-Q. Аликвоту переносили в фильтровальные флаконы 0,45 мкм и разводили в соотношении 1:1 с Milli-Q. Образцы отфильтровывали и переносили в пробирки с автоматическим пробоотборником с дезактивированными стеклянными вставками для анализа LC-MS/MS (так сокращенно обозначается жидкостной хроматограф масс-спектрометр).
Разделение проводили с помощью LC на ВЭЖХ (высоко эффективный жидкостный хромато-масс-спектрометр) Agilent 1200 Series с колонкой Poroshell Phenyl-Hexyl 150×2,1 мм, колонкой 3 мкм (Agilent Technologies). Конечная подвижная фаза состояла из воды (элюент А) и метанола (элюент В) с 0,1% муравьиной кислотой. Градиентная программа для APCI была следующей: начальный процент В составлял 5%, увеличиваясь до 10% в течение 3,5 мин, затем до 20% в течение 2 мин, 80% в течение 4,5 мин и, наконец, до 90% в течение 1 мин и удерживался для 5 мин. Между каждым прогоном колонку уравновешивали в течение 6 мин 2% В.
Определение MS/MS проводилось на тройном квадруполе Agilent 6460 (Agilent Technologies), оборудованным либо APCI, либо источником потока Jet Stream. В качестве газа-реагента был использован азот. Валидация метода была выполнена в соответствии с ISO 5725-2, чтобы продемонстрировать специфичность, точность и нижний предел обнаружения (LOD). LOD рассчитывали как 3-кратное стандартное отклонение при самом низком принятом уровне сигнала. Точность метода определялась с использованием трех различных типов тестовых образцов для воспроизведения. Эта процедура выполняется на хромато-масс-спектрометре и для получения результатов используют градуировочную характеристику или график.
Недостатком указанного известного способа является большой разброс в количестве определяемых нитрозоаминов в параллельных пробах, т.е. способ характеризуется недостаточной воспроизводимостью, а значит - точностью. Нижний предел обнаружения составляет 1 мкг/кг продукта, в то время как по предлагаемому способу 0,2 мкг/кг.
Кроме того, в этом способе используется дорогостоящее приборное оснащение и длительная пробоподготовка.
Задача оценки селективного содержания N-нитрозоаминов в копченых пищевых мясопродуктах, остается весьма актуальной, поскольку до сих пор не предложено высокочувствительных и селективных методик определения большой группы этих токсичных соединений.
Технический результат, достигаемый предлагаемым способом заключается в обеспечении высокочувствительного и селективного хромато-масс-спектрометрического определения высокотоксичных и канцерогенных N-нитрозоаминов: N-ДМНА, N-МЭНА, N-ДЭНА, N-ДБНА, N-ДПНА, N-ПИПНА, N-ПИРНА, N-MOPHA, N-ДФНА, в пробах пищевой продукции - копченые мясные, мясо- и птице- продукты, при одновременном сокращении времени на пробоподготовку.
Поставленный технический результат достигается предлагаемым способом количественного определения N-нитрозоаминов: N-диметилнитрозоамин, N-метилэтилнитрозоамин, N-диэтилнитрозоамин, N-дибутилнитрозоамин, N-дипропилнитрозоамин, N-пиперидиннитрозоамин, N-пирролидиннитрозоамин, N-морфолиннитрозоамин, N-дифенилнитрозоамин, в пробах копченых мясопродуктов методом хромато-масс-спектрометрии, включающий пробоподготовку, проведение твердофазной экстракции (ТФЭ) и определение конкретных нитрозоаминов по градуировочному графику, при этом новым является то, что при пробоподготовке к 20 г пробы измельченных копченых мясопродуктов добавляют 200 мл предварительно нагретой до +55°С дистиллированной воды, производят настаивание в течение 30 минут и последующее фильтрование, к фильтрату добавляют 1,5 г калия гидрооксида, полученную смесь фильтрата и калия гидрооксида подвергают отгонке перегретым до tпарообразователя=100±5°С водяным паром с получением 70 см3 дистиллята, указанный дистиллят подвергают ТФЭ на приборе ТФЭ, содержащем угольный картридж, при которой вначале производят промывку картриджа прибора ТФЭ водой 2,5 мл, хлористым метиленом 2,5 мл, пропускают через него 2,5 мл этилацетата с его задержкой в течение 30 сек, далее производят промывку водой 4 мл; затем загружают ранее полученный дистиллят, производят сушку указанного картриджа в течение 20 минут, производят промывку картриджа водой объемом 2 мл, осуществляют элюирование водного слоя с картриджа хлористым метиленом объемом 3 мл, затем полученные элюаты анализируют методом хромато-масс-спектрометрии и с помощью градуировочного графика в режиме селективного ионного мониторинга определяют селективно в пробе копченых мясопродуктов количество N-диметилнитрозоамина, N-метилэтилнитрозоамина, N-диэтилнитрозоамина, N-дибутилнитрозоамина, N-дипропилнитрозоамина, N-пиперидиннитрозоамина, N-пирролидиннитрозоамина, N-морфолиннитрозоамина, N-дифенилнитрозоамина.
При хромато-масс-спектрометрии используют капиллярную колонку серии HP-FFAP 30 m⋅0,250 mm⋅0,250 μm длиной 30 метров, внутренним диаметром 0,25 мм и толщиной пленки неподвижной фазы 0,25 μm.
Указанный технический результат достигается за счет следующего.
Известно, что основным сырьем для изготовления колбасных изделий являются говядина, свинина, баранина, субпродукты, свиной шпик или грудинка, курдючное сало, мясо домашней птицы, кроликов. Вспомогательным сырьем являются яйца, молоко, сливочное масло, сыры, крахмал, мука пшеничная, крупа, колбасные оболочки. Для придания колбасам остроты, своеобразного вкуса и запаха в фарш добавляют соль, сахар, перец черный и душистый, мускатный орех, кардамон, гвоздику, фисташки, чеснок. В фарш некоторых колбас вносят вино (Мадеру или Кагор), коньяк. Для сохранения в изделиях розово-красной окраски, не разрушающейся при тепловой обработке, применяют нитрит натрия. В состав фарша колбас входят различные виды жиров. В основном это свиной жир межмышечный, шпик, жир-сырец говяжий и бараний, курдючный жир, внутренние жиры. Копченые продукты, в частности, колбасы, в отличие от вареных колбас, имеют плотную эластичную консистенцию, острый солоноватый вкус и приятный аромат. Они обладают высокой питательностью, так как содержат много жира (25-60%) и белковых веществ (21-22%).
Известные приемы и способы пробоподготовки такие как - экстракция пробы копченой мясной пищевой продукции органическим растворителем, дистилляция с перегретым водяным паром, статический и динамический анализ равновесной паровой фазы не позволяют достичь высокой полноты извлечения, а, следовательно, - необходимой точности определений для реальной оценки содержания девяти N-нитрозоаминов в пищевых продуктах на уровне гигиенического норматива, указанного в СанПиН 2.3.2. 560-96 на пищевые продукты питания.
Повышенное количество жирового компонента биологического происхождения мешает селективному извлечению N-нитрозоаминов из копченого пищевого продукта. Поэтому одним из этапов способа являлось удаление жира биологического происхождения из проб пищевой продукции (копченые мясные, мясо- и птице- продукты) на стадии пробоподготовки.
Для устранения влияния матричных эффектов на результаты хромато-масс-спектрометрического определения N-нитрозоаминов при их совместном присутствии: N-ДМНА, N-МЭНА, N-ДЭНА, N-ДБНА, N-ДПНА, N-ПИПНА, N-ПИРНА, N-MOPHA, N-ДФНА, в предлагаемом способе при пробоподготовке выполняют очистку образцов копченой мясной пищевой продукции от мешающих компонентов и жира путем выполнения нескольких операций: введения в измельченную пробу копченого продукта подогретой до +55°С воды из расчета 1 массовая доля пробы к 10 объемным пробам воды; выдержки, фильтрации и введения в фильтрат гидрооксида калия из расчета массового соотношения 13,3:1 соответственно, что обеспечивает увеличение полноты и селективности экстракционного извлечения аналита из образца.
Жировой компонент биологического происхождения мешает селективному извлечению N-нитрозоаминов из копченого пищевого продукта. Для удаления из исследуемых образцов мясных пищевых продуктов (навеска 20 г) жиров биогенного происхождения и эффективного извлечения N-нитрозоаминов выполняли очистку образцов пищевой продукции от мешающих компонентов и жира методом дистилляции, что обеспечивало увеличение полноты и селективности экстракционного извлечения аналитов из образца. Для этого отрабатывали условия и параметры (природы и концентрации высаливателей, объем дистиллята) метода дистилляции N-нитрозоаминов.
В качестве высаливателей использовали, например, совместное введение сульфата натрия и хлорида натрия, а также гидрооксид калия.
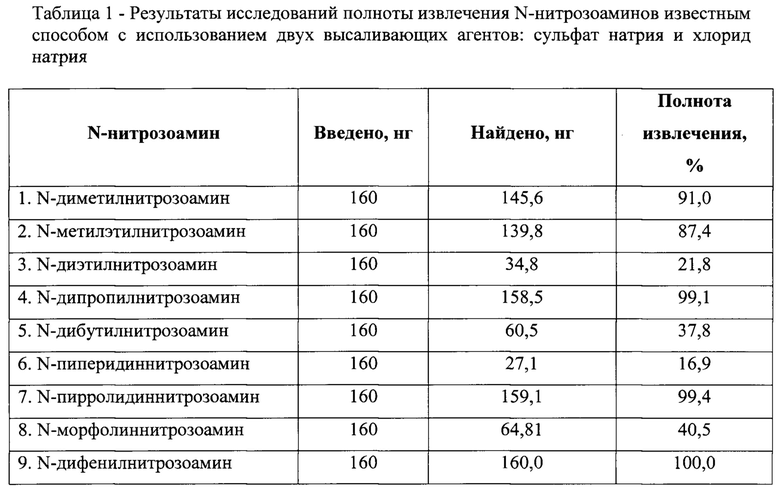
Алгоритм пробоподготовки по первому варианту высаливателей. В пробу пищевой продукции массой 20 г вводили высаливающие реагенты (в количестве 5 г сульфата натрия и 5 г хлорида натрия). С целью максимального извлечения N-нитрозоаминов и установления параметров экстракции стандартный образец N-нитрозоаминов подкисляли 2,5 см3 2%-ной сульфаминовой и 0,5 см3 60%-ным раствором серной кислоты до рН=3. С помощью перегретого водяного пара при tпарообразователя=100±5°С и tколбы со стандартным образцом=80±5°С отгоняли 70 см3 дистиллята и пропускали через угольный картридж Coconut 6 см3 системы твердофазной экстракции по оптимальной схеме элюирования (режим 1 таблица 4). Полученные элюаты были проанализированы хромато-масс-спектрометрическим методом. Результаты исследований полноты извлечения N-нитрозоаминов из пищевого продукта с применением стандартного образца методом дистилляции и ТФЭ на угольном картридже Coconut 6 см3 представлены в таблице 1.
Полнота извлечения летучих N-нитрозоаминов (таблица 1) из стандартного образца для N-диметилнитрозоамина 91,0%, N-метилэтилнитрозоамина 87,4%, N-диэтилнитрозоамина 21,8%, N-дипропилнитрозоамина 99,1%, N-дибутилнитрозоамина 37,8%, N-пиперидиннитрозоамина 16,9%, N-пирролидиннитрозоамина 99,4%, N-морфолиннитрозоамин 40,5% и N-дифенилнитрозоамин 100% установлена при использовании дистилляции с перегретым водяным паром (дистиллят объемом 70 см3) и элюировании летучей фракции отгона по оптимальной схеме элюирования методом твердофазной экстракции на картридже Coconut 6 см3.
Ввиду того, что полнота экстракции для 4-х нитрозоаминов при этих высаливателях не превышала 50%, в дальнейшем отрабатывали оптимальные параметры метода дистилляции N-нитрозоаминов с добавлением калия гидрооксида различной массы с последующей дистилляцией с перегретым водяным паром и концентрированием дистиллята на картриджах автоматической системы твердофазной экстракции (ТФЭ).
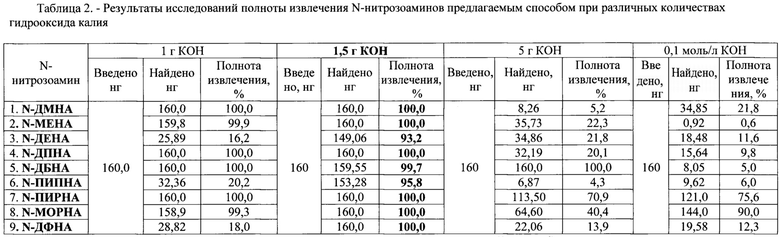
Готовили вытяжки из сырокопченых продуктов свинины, баранины, говядины и сырокопченых колбас. Для этого измельченную пробу пищевой продукции (колбаса салями) массой 20 г заливали 200 мл предварительно нагретой до +55°С дистиллированной водой, настаивали, периодически помешивая, в течение 30 мин. Затем вытяжку фильтровали через ватный фильтр, не перенося осадка на фильтр, и помещали в круглодонную колбу объемом 500 см3 для отгона. Добавляли гидроокись калия различной массы: 1 г, 1,5 г, 5 г и 0,1 моль/дм3. С помощью перегретого водяного пара при tпарообразователя=100±5°С и tколбы со стандартным образцом=80±5°С отгоняли 70 см3 дистиллята и пропускали через картридж Coconut 6 см3 системы твердофазной экстракции по оптимальной схеме элюирования.
Полученные элюаты были проанализированы хромато-масс-спектрометрическим методом. Результаты исследований полноты извлечения N-нитрозоаминов из пищевого продукта с применением стандартного образца методом дистилляции и ТФЭ на угольном картридже Coconut 6 см3 представлены в таблице 2.
Приготовление исходного раствора N-нитрозоаминов.
Исходный раствор N-нитрозоаминов готовят из mix ГСО С=2 мг/см3. В мерную колбу объемом 25 см3 дозатором добавляют метилового спирта в объеме 10 см3, вводят микрошприцем по 2 мм3 mix N-нитрозоаминов ГСО (С=2 мг/см3). Массовая концентрация N-нитрозоаминов в исходном растворе составляет 0,16 мкг/см3. Срок хранения раствора 5 часов.
Приготовление градуировочных растворов.
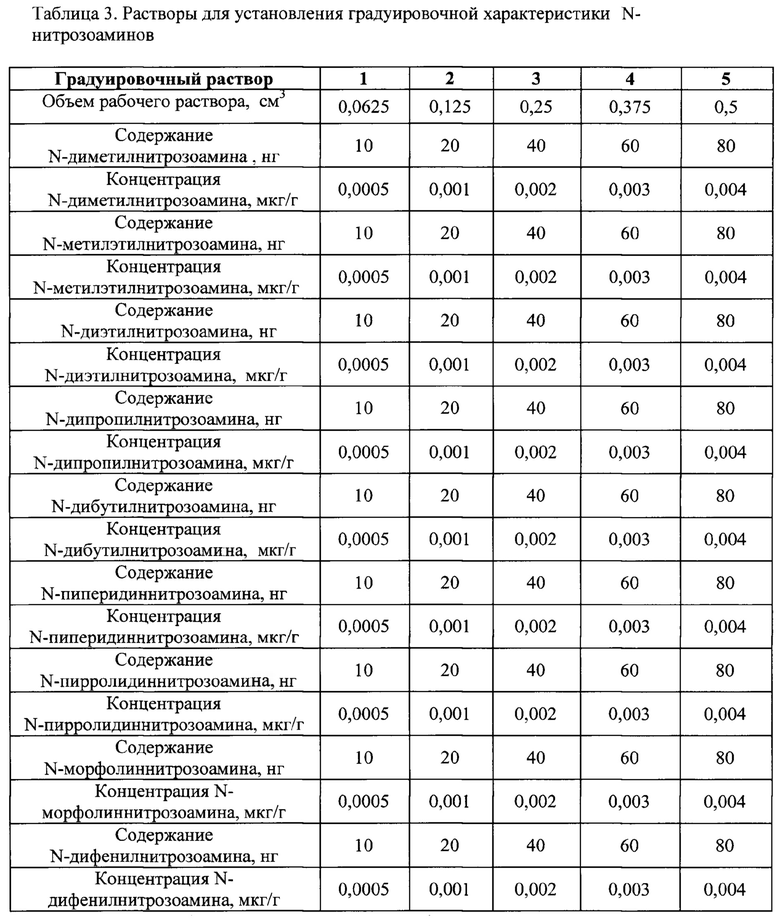
Градуировочные растворы N-нитрозоаминов готовят в мерных колбах объемом 100 см3. Для этого в каждую колбу вносят по 50 см3 бидистиллированной воды и добавляют исходный раствор для градуировки в соответствии с таблицей 3, содержимое колбы доводят до метки бидистиллированной водой и тщательно перемешивают (таблица 3).
В процессе выполненных экспериментальных исследований (таблица 2) установлено, что полнота экстракции N-нитрозоаминов из образцов пищевой продукции (копченые мясные, мясо- и птице- продукты) с добавлением калия гидрооксида массой 1,5 г и последующей дистилляцией с перегретым водяным паром, концентрированием дистиллята на картриджах Coconut 6 см3 и использованием оптимальной схемы элюирования автоматической системы твердофазной экстракции (ТФЭ) составила для N-диметилнитрозоамина, N-метилэтилнитрозоамина, N-дипропилнитрозоамина, N-пирролидиннитрозоамина, N-морфолиннитрозоамина и N-дифенилнитрозоамина 100,0%; для N-диэтилнитрозоамина 93,2%, N-дибутилнитрозоамина 99,7% и N-пиперидиннитрозоамина 95,8%. Это количество высаливателя гидрооксида калия, и было выбрано в предлагаемом способе.
Операция предлагаемого способа «полученную смесь фильтрата и калия гидрооксида подвергают отгонке перегретым до tпарообразователя=100±5°С водяным паром с получением 70 см3 дистиллята», необходима для обеспечения высокой полноты извлечения всех 9 нитрозоаминов из образцов пищевой продукции.
Благодаря использованию в предлагаемом способе операции твердофазной экстракции (далее - ТФЭ) при заявленных режимах и используемых реагентах также была обеспечена селективность и полнота извлечения всех девяти N-нирозоаминов из копченой мясной пробы.
С целью активации картриджа системы ТФЭ выполняется кондиционирование органическим растворителем - хлористым метиленом, затем осуществляется загрузка жидкого образца на угольный картридж с помощью азота. Во время этого, на сорбенте адсорбируются аналиты, а матрица идет в слив. На этой стадии сорбент удерживает целевые соединения до этапа элюирования. Мешающие компоненты пробы полностью удаляются на следующей стадии промывки картриджа органическим растворителем - хлористым метиленом.
Благодаря совокупности операций и их режимов обеспечивается, наряду с селективностью, и точность определения каждого из указанных N-нитрозоаминов.
Предлагаемый способ реализуется следующим образом по трем этапам:
- пробоподготовка,
- выполнение ТФЭ,
- анализ полученных элюатов хромато-масс-спектрометрическим методом по градуировочному графику в режиме селективного ионного мониторинга.
1. Пробоподготовка.
Готовили вытяжки из сырокопченых продуктов свинины, баранины, говядины и сырокопченых колбас. Для этого измельченную пробу пищевой продукции массой 20 г заливали 200 мл предварительно нагретой до +55,0°С дистиллированной водой, настаивали, периодически помешивая, в течение 30 мин. Затем получившуюся вытяжку фильтровали через ватный фильтр, не перенося осадка на фильтр, и помещали в круглодонную колбу объемом 500 см3 для отгона. Добавляли гидроокись калия в количестве 1,5 г. С помощью перегретого водяного пара при tпарообразователя=100±5°С и tколбы со стандартным образцом=80±5°С отгоняли 70 см3 дистиллята.
2. Выполнение ТФЭ.
Вначале производят стадию кондиционирования - активацию угольного картриджа Coconut 6 см3, для чего его промывают дистиллированной водой 2,5 мл и хлористым метиленом объемом 2,5 см3.
Затем через картридж пропускают растворитель - этилацетат, объемом 2,5 см3 с задержкой растворителя в течение 30 сек.
Для удаления остаточных количеств растворителей угольный картридж промывают водой объемом 4 мл.
Далее загружают ранее полученный дистиллят пробы - водный слой объемом 70 см3, и производят сушку картриджа в течение 20 минут.
Осуществляют элюирование дистиллята пробы с картриджа хлористым метиленом объемом 4 см3.
Производят промывку картриджа водой объемом 2 мл и осуществляют элюирование водного слоя с картриджа хлористым метиленом объемом 3 мл.
3. Анализ полученных элюатов хромато-масс-спектрометрическим методом.
Полученные элюаты анализируют известным хромато-масс-спектрометрическим методом, устанавливая по хроматограммам количественное содержание каждого из указанных нитрозоаминов.
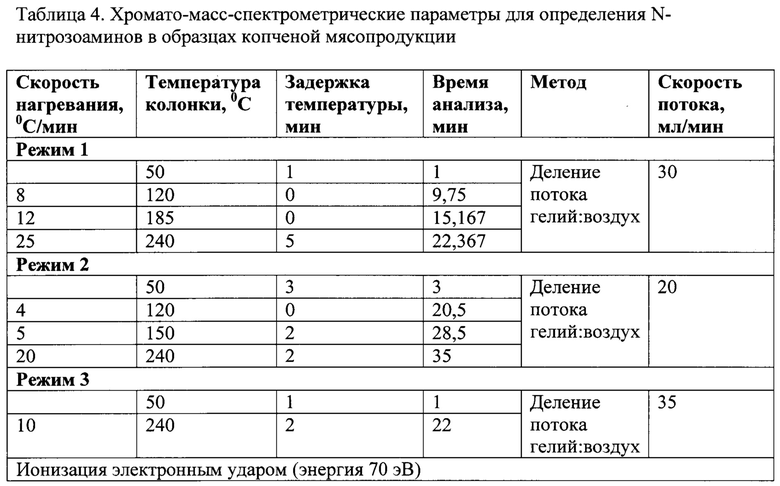
Отработка оптимальных хромато-масс-спектрометрических параметров определения N-нитрозоаминов в копченых пищевых образцах осуществлялась с использованием газовой хроматографии и масс-спектрометрии (далее - ГХ/МС) на газовом хроматографе Agilent 7890А (производство США) с квадрупольным масс-спектрометрическим детектором (MCD) 5975С. Режим ионизации электронным ударом при 70 эВ.
Для количественного селективного определения содержания девяти указанных нитрозоаминов выполняли хромато-масс-спектрометрический анализ стандартных растворов и на основе результатов измерений строили градуировочную зависимость в режиме селективного ионного детектирования (SIM) по характеристичным ионам соединения в диапазоне концентраций 0,0002-0,0016 мкг/г (0,2-1,6 мкг/кг). Для этого в хромато-масс-спектрометрическом анализе в режиме селективного ионного мониторинга строится градуировочный график по выбранным ионам вещества и выполняется количественный анализ в режиме регистрации индивидуальных ионов (Agilent GC/MSD ChemStation - Agilent Technologies: H4043A, том 2, Учебное пособие).
В процессе экспериментальных исследований были изучены следующие капиллярные колонки различной длины и толщины пленки неподвижной фазы: DB-624, HP-FFAP, HP-Plot/U, HP-VOC.
Качественное разделение девяти N-нитрозоаминов: N-дифенилнитрозоамина, N-диметилнитрозамина, N-метилэтилнитрозоамина, N-диэтилнитрозоамина, N-пирролидиннитрозоамина, N-морфолиннитрозоамина, N-дибутилнитрозоамина, N-дипропилнитрозоамина, N-пиперидиннитрозоамина, с близкими физико-химическими свойствами было достигнуто на капиллярной колонке серии HP- FFAP 30 m⋅0,250 mm⋅0,250 μm длиной 30 метров, внутренним диаметром 0,250 мм и толщиной пленки неподвижной фазы 0,250 μm.
Использовали режим программирования колонки: начальная температура 50°С, повышение температуры до 120°С со скоростью 8°С/мин; от 120°С до 185°С со скоростью 12°С/мин и от 185°С до 240°С со скоростью 25°С/мин. с выдержкой при конечной температуре 5 мин. В качестве газа-носителя применяли гелий; скорость газа-носителя 1,0 мл/мин в режиме постоянного потока. Температура аналитического интерфейса 220°С. Ввод пробы осуществляли с помощью автосамплера Agilent ALS (Agilent Technologies, США) в режиме pulsed/splitless; объем пробы 2 мкл.
Режимы хромато-масс-спектрометрических параметров представлены в таблице 4.
Данные, приведенные в таблице 4, показывают, что в режимах 2 и 3 не наблюдалось достаточно эффективного разделения летучих N-нитрозоаминов (такие результаты показала хроматограмма). Качественное разделение было достигнуто в режиме 1, который и был выбран для дальнейшей работы.
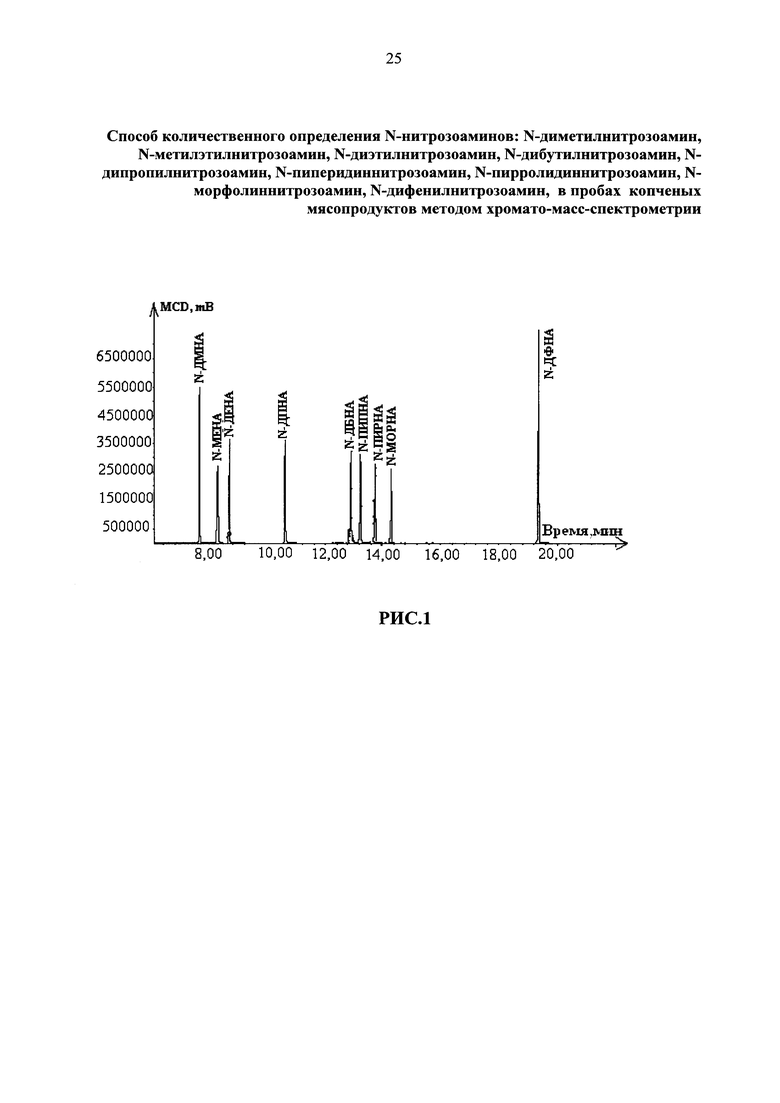
В качестве доказательства оптимальности этого режима, на рис. 1 приведена хроматограмма стандартного раствора разделения N-нитрозоаминов при оптимально подобранных условиях хромато-масс-спектрометрического анализа (режим 1 таблица 4), зарегистрированная по полному ионному току (TIC).
Для выбора оптимальных режимов и используемых реагентов предлагаемого способа были проведены лабораторные исследования по установлению необходимых марок картриджей в приборе ТФЭ, отработаны условия пробоподготовки и схемы элюирования ТФЭ и дистилляции на стандартных образцах N-нитрозоаминов, полученных путем общеизвестного приема построения градуировочной зависимости методом абсолютной градуировки. Для этого приготовленные стандартные растворы хроматографируют на капиллярной колонке. На полученной хроматограмме определяют площади пиков молекулярных ионов компонентов и по средним результатам измерений по 5 концентрациям для градуировки строят градуировочную характеристику. Она выражает зависимость площади пика конкретного молекулярного иона на хроматограмме (мВ - при автоматическом обсчете с использованием программно-аппаратного комплекса) от содержания (нг).
Для получения экстрактов необходимой чистоты для хромато-масс-спектрометрического анализа и установления достаточной степени концентрирования N-нитрозоаминов, были апробированы сменные картриджи прибора ТФЭ, заполненные различными сорбентами, органические растворители для элюирования N-дифенилнитрозоамина с картриджей и изучены условия оптимальной схемы элюирования. Селективность проведения процесса ТФЭ достигнута подбором картриджа с соответствующей фазой.
Для этого апробированы следующие типы картриджей: угольный Coconut 6 мл; заполненные октадецилом Chromabond С18 на 100 и 500 мг; картриджы на полимерной основе Strata на 200 мг. Анализ полученных результатов показал, что на угольном картридже Coconut 6 см3 при использовании экспериментальной схемы элюирования (режим 1 таблица 4) наблюдалась наибольшая полнота извлечения N-нитрозоаминов.
На следующем этапе были проведены исследования по отработке оптимальных условий процесса элюирования всех девяти N-нитрозоаминов с угольного картриджа Coconut 6 мл автоматической системы твердофазной экстракции с применением различных растворителей. Для этого варьировали параметрами: объемами растворителей на стадии кондиционирования, объемами образца на стадии загрузки, временем сушки картриджа, объемом слива.
Отработанная оптимальная схема селективного элюирования включала несколько стадий и характеризуется следующими параметрами:
1. Стадия кондиционирования - активация картриджа водой 2,5 мл, затем хлористым метиленом объемом 2,5 мл, затем этилацетатом объемом 2,5 мл с задержкой растворителя в течение 30 сек. Для удаления остаточных количеств растворителей картридж промывали водой объемом 4 мл;
2. Стадия адсорбции целевых компонентов на картридже при загрузке дистиллята пробы объемом 70 см3;
3. Сушка картриджа в течение 20 минут для удаления остаточных количеств образца;
4. Заключительная стадия - элюирование целевых аналитов с картриджа хлористым метиленом объемом 3 мл и продувка автоматической системы азотом в течение 2 минут.
Стандартный образец (объемом 70 см3) пропускали через картридж Coconut 6 см3 системы твердофазной экстракции по оптимальной схеме элюирования (режим 1 таблица 4).
Полученные элюаты были проанализированы хромато-масс-спектрометрическим методом. Результаты исследований полноты извлечения N-нитрозоаминов из пищевого продукта с применением стандартного образца методом дистилляции и ТФЭ на угольном картридже Coconut 6 см3 представлены в таблице 2.
Данные, приведенные в таблицах 1 и 2, показывают, что предлагаемый способ по сравнению с известным, обеспечивает практически 100%-ную полноту извлечения каждого конкретного N-нитрозоамина: N-ДМНА, N-МЭНА, N-ДЭНА, N-ДБНА, N-ДПНА, N-ПИПНА, N-ПИРНА, N-MOPHA, N-ДФНА, из пробы копченого пищевого продукта, в то время как в известном способе (с двумя высаливателями: сульфата натрия и хлорид натрия) наблюдается нестабильность в полноте извлечения, которая составляет от 16,9 до 100% нитрозоамина.
Таким образом, предлагаемый способ имеет следующие преимущества:
- обеспечивает практически полное извлечение каждого конкретного N-нитрозоамина: N-ДМНА, N-МЭНА, N-ДЭНА, N-ДБНА, N-ДПНА, N-ПИПНА, N-ПИРНА, N-MOPHA, N-ДФНА, из исследуемой пробы (93,2-100%), что свидетельствует о точности и достоверности предлагаемого способа;
- позволяет значительно снизить нижний предел определения N-нитрозоаминов (до 0,0002 мг/кг продукта), в то время как в известных способах этот показатель вообще не определялся или был значительно больше;
- предел обнаружения нитрозоаминов предлагаемым способом составляет 0,0002-0,0016 мкг/г или мг/кг, что соответствует 0,2-1,6 мкг/кг.
Примечание: При опытах было введено 1 см3 стандартного раствора нитрозоаминов (160 нг). Стандартные растворы нитрозоаминов представляли собой смесь указанных N-нитрозоаминов.
Концентрация (мкг/г) рассчитана на навеску колбасного продукта 20 г.
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ количественного определения N-нитрозаминов в детских кашах | 2015 |
|
RU2613303C1 |
| Способ количественного определения N-дифенилнитрозамина в мясных пробах пищевой продукции методом хромато-масс-спектрометрии | 2017 |
|
RU2626601C1 |
| Способ количественного определения фурана и метилфурана в детских кашах | 2022 |
|
RU2782424C1 |
| Способ подготовки пробы мочи для определения монометилфталата, моноэтилфталата, монобутилфталата, монобензилфталата, моноэтилгексилфталата методом высокоэффективной жидкостной хроматографии/масс-спектрометрии | 2019 |
|
RU2687738C1 |
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ N-НИТРОЗОДИМЕТИЛАМИНА И N-НИТРОЗОДИЭТИЛАМИНА В КРОВИ МЕТОДОМ КАПИЛЛЯРНОЙ ГАЗОВОЙ ХРОМАТОГРАФИИ | 2015 |
|
RU2578026C1 |
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ БЕНЗ(А)ПИРЕНА В КРОВИ МЕТОДОМ ЖИДКОСТНОЙ ХРОМАТОГРАФИИ | 2014 |
|
RU2546530C1 |
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ N-НИТРОЗОДИМЕТИЛАМИНА И N-НИТРОЗОДИЭТИЛАМИНА В МОЧЕ МЕТОДОМ ГАЗОХРОМАТОГРАФИЧЕСКОГО АНАЛИЗА | 2013 |
|
RU2521711C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ СОДЕРЖАНИЯ ФУМАРОВОЙ И МАЛЕИНОВОЙ КИСЛОТ В ПЛАЗМЕ КРОВИ МЕТОДОМ ВЫСОКОЭФФЕКТИВНОЙ ЖИДКОСТНОЙ ХРОМАТОГРАФИИ | 2018 |
|
RU2677341C1 |
| Способ количественного определения гексахлорбензола в крови методом газохроматографического анализа | 2016 |
|
RU2613306C1 |
| Способ количественного определения фурана и метилфурана в детских кашах на основе зерна газохроматографическим методом с использованием парофазного анализа | 2022 |
|
RU2798667C1 |
Изобретение относится к медицинским токсикологическим исследованиям. Способ количественного определения N-нитрозоаминов включает проведение пробоподготовки, твердофазную экстракцию (ТФЭ) и выполнение определения конкретных нитрозоаминов по градуировочному графику, при пробоподготовке к 20 г пробы измельченных копченых мясопродуктов добавляют 200 мл предварительно нагретой до +55°С дистиллированной воды, производят настаивание в течение 30 минут и последующее фильтрование, к фильтрату добавляют 1,5 г калия гидрооксида, полученную смесь фильтрата и калия гидрооксида подвергают отгонке перегретым до tпарообразователя=100±5°С водяным паром с получением 70 см3 дистиллята, указанный дистиллят подвергают ТФЭ, полученные элюаты анализируют методом хромато-масс-спектрометрии с селективным выделением девяти N-нитрозоаминов. 1 з.п. ф-лы, 4 табл., 1 ил.
1. Способ количественного определения N-нитрозоаминов: N-диметилнитрозоамин, N-метилэтилнитрозоамин, N-диэтилнитрозоамин, N-дибутилнитрозоамин, N-дипропилнитрозоамин, N-пиперидиннитрозоамин, N-пирролидиннитрозоамин, N-морфолиннитрозоамин, N-дифенилнитрозоамин, в пробах копченых мясопродуктов методом хромато-масс-спектрометрии, включающий пробоподготовку, проведение твердофазной экстракции (ТФЭ) и определение конкретных нитрозоаминов по градуировочному графику, отличающийся тем, что при пробоподготовке к 20 г пробы измельченных копченых мясопродуктов добавляют 200 мл предварительно нагретой до +55°C дистиллированной воды, производят настаивание в течение 30 минут и последующее фильтрование, к фильтрату добавляют 1,5 г калия гидрооксида, полученную смесь фильтрата и калия гидрооксида подвергают отгонке перегретым до tпарообразователя=100±5°C водяным паром с получением 70 см3 дистиллята, указанный дистиллят подвергают ТФЭ на приборе ТФЭ, содержащем угольный картридж, при которой вначале производят промывку картриджа прибора ТФЭ водой 2,5 мл, хлористым метиленом 2,5 мл, пропускают через него 2,5 мл этилацетата с его задержкой в течение 30 сек, далее производят промывку водой 4 мл; затем загружают ранее полученный дистиллят, производят сушку указанного картриджа в течение 20 минут, производят промывку картриджа водой объемом 2 мл, осуществляют элюирование водного слоя с картриджа хлористым метиленом объемом 3 мл, затем полученные элюаты анализируют методом хромато-масс-спектрометрии и с помощью градуировочного графика в режиме селективного ионного мониторинга определяют селективно в пробе копченых мясопродуктов количество N-диметилнитрозоамина, N-метилэтилнитрозоамина, N-диэтилнитрозоамина, N-дибутилнитрозоамина, N-дипропилнитрозоамина, N-пиперидиннитрозоамина, N-пирролидиннитрозоамина, N-морфолиннитрозоамина, N-дифенилнитрозоамина.
2. Способ по п. 1, отличающийся тем, что при хромато-масс-спектрометрии используют капиллярную колонку серии HP-FFAP 30 м⋅0,250 мм⋅0,250 мкм длиной 30 метров, внутренним диаметром 0,25 мм и толщиной пленки неподвижной фазы 0,25 мкм.
| Mohammad Al-Kaseem et al | |||
| Rapid and Simple Extraction Method for Volatile N-Nitrosamines in Meat Products / Pharmacology & Pharmacy, 2013, 4, pages 611-618 | |||
| Marina Bergoli Scheerena et al | |||
| Determination of N-nitrosamines in processed meats by liquid extraction combined with gas chromatography-methanol chemical ionisation/mass spectrometry / Food Additives & Contaminants: Part A, 2015 [Найдено в Интернете он-лайн 19.04.2018 https://www.researchgate.net/profile/Joseph_Arul3/publication/280581839_Determination_of_N_-nitrosamines_in_processed_meats_by_liquid_extraction_combined_with_gas_chromatography-methanol_chemical_ionisationmass_spectrometry/links/55ce965408aee19936fc5cdc/Determination-of-N-nitrosamines-in-processed-meats-by-liquid-extraction-combined-with-gas-chromatography-methanol-chemical-ionisation-mass-spectrometry.pdf?origin=publication_detail] | |||
| И.Н.Ким и др | |||
| Исследование содержания канцерогенных соединений в копченой рыбе промышленной выработки / Гигиена и санитария, 2012, 2, стр | |||
| Механический грохот | 1922 |
|
SU41A1 |

